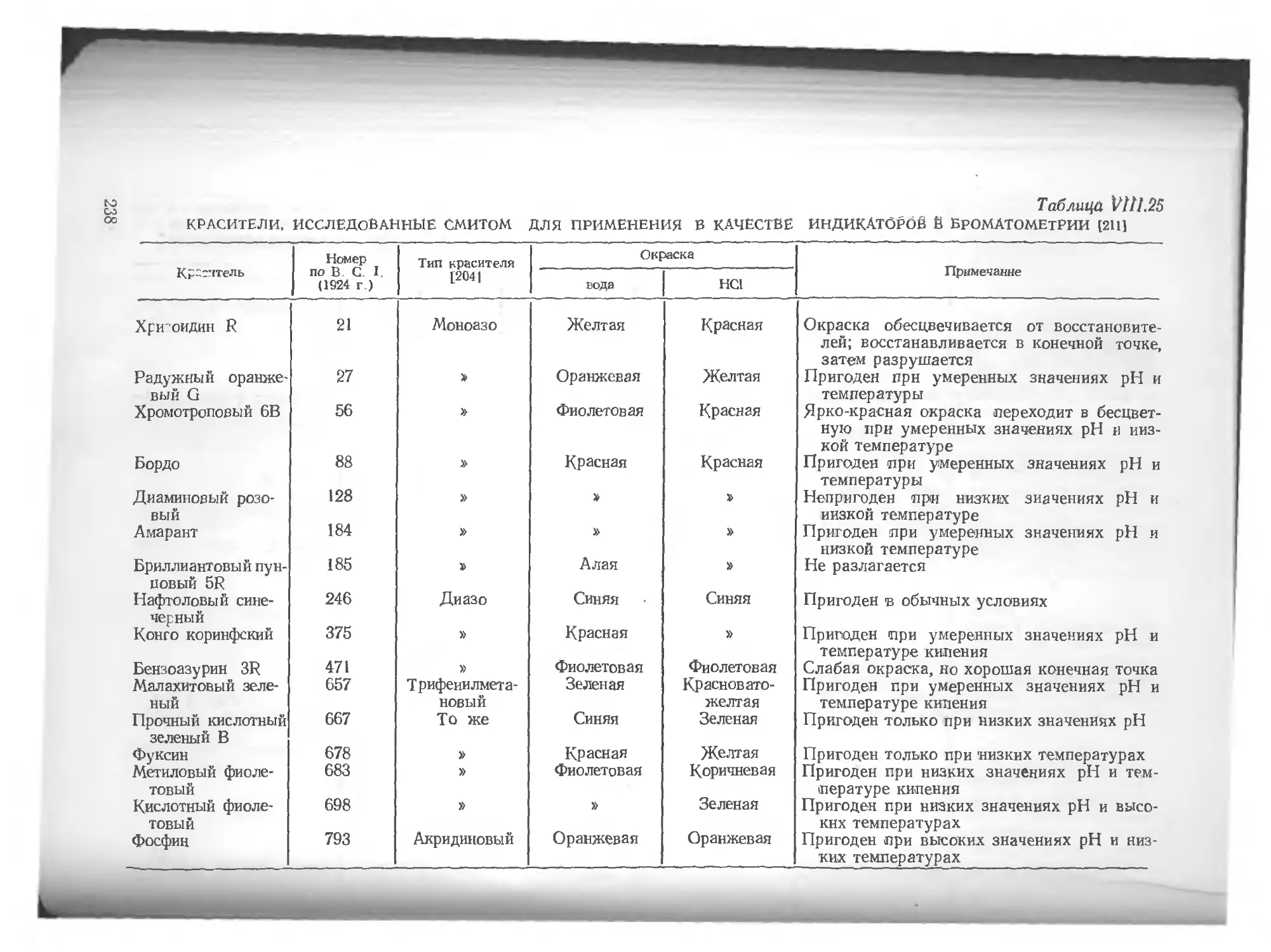
/





























































































































































































































































































































































































































































Text
INDICATORS
Edited by
EDMUND BISHOP
Department of Chemistry, University of Exeter
PERGAMON PRESS
Oxford-New York-Toronto Sydney-Braunschweig 1972
ИНДИКАТОРЫ
ТОМ 2
Под редакцией Э. Бишопа
Перевод с английского
И. В. Матвеевой
Под редакцией
доктора хим. наук И. Н. Марова
Издательство «Мир» Москва 1976
УДК 543-4
Редакция литературы по химии
20506-109
И 041{01)-76
109-76 © Перевод на русский язык, «Мир», 1976
ГЛАВА VII
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
Е. Пунгор, Е. Шулек
ИСТОРИЧЕСКИЙ ОБЗОР
Адсорбционные индикаторы были предложены Фаянсом ['25, 26] для определения конечной точки при титровании по методу осаждения. Фаянс и Хассел [26] отметили, что электронная оболочка красителя, адсорбированного на поверхности осадка, может легко искажаться. Поэтому, подобрав подходящий индикатор, можно определять конечную точку титрования. Новый принцип установления конечной точки вскоре утвердился в аналитической химии благодаря работам Фаянса и Хассела [26], Фаянса и Вольфа ['27], а также главным образом в результате работ Кольтгофа с сотр. [37—44]. За последние годы исследовались возможности использования ряда красителей для определения конечной точки при титровании по методу осаждения ['1—4, 7-—9, 11—14, 22—24, 28—36, 45, 50, 62—67, 69, 72—78, 101—104, 107—118].
Механизм установления конечной точки титрования с помощью п-этоксихризоидина ['79] не согласовывался с механизмом действия адсорбционных индикаторов, предложенным Фаянсом, а затем подробно изученным Кольтгофом. Поэтому вместо качественного метода описания, введенного Фаянсом, Шулек и Пунгор [73] предложили количественный метод описания теории адсорбционных индикаторов. При помощи этого метода механизм действия адсорбционных индикаторов объясняется одновременно происходящими изменениями физико-химических свойств адсорбционных красителей вследствие адсорбции. Такой подход открывает новые пути исследования в области адсорбционных индикаторов [59—61]. Применяя этот метод, Богнар с сотр. [12—21] получил целую серию новых индикаторов. В то же самое время удалось расширить основные группы уже известных адсорбционных индикаторов. Применяя к индикаторам терминологию приведенной ниже классификации, надо сказать, что исследования в области поверхностных кислотно-основных и поверхностных окислительно-восстановительных индикаторов в значительной мере продвинулись наряду с исследованиями поверхностных адсорбционных индикаторов. В этой
6
ГЛАВА VII
области больших успехов достигли Асенси Мора [5], Сьерра Хименес [80—92] и Барриель-Марти [86], а затем Сьерра и Санчес-Педрено [84, 85, 87—89], а также Богнар с сотр. ['17, 18].
Следуя по пути, намеченному Богнаром [16], Пунгор и Холлос [53, 54] разработали далее теорию адсорбционных индикаторов. Эти авторы установили, что при варьировании условий один и тот же индикатор может действовать вследствие адсорбции по нескольким механизмам при установлении конечной точки. Механизм действия метанилового желтого представляет собой типичный пример полифункциональных адсорбционных индикаторов.
Взамен адсорбционной теории Шулека и Пунгора Сьерра и Санчес-Педрено [87] разработали новую теорию, в основе которой лежит теория Льюиса о кислотах и основаниях. Новая теория предлагает единое решение всех проблем в области адсорбционных индикаторов.
Однако формальное обобщенное рассмотрение поведения индикаторов с сильно различающимися механизмами скорее мешает развитию в этой области, как правильно заметили Пунгор и Кон-коль Тэге [58] в случае поверхностных адсорбционных индикаторов. Поэтому чисто формальное обобщение интерпретации механизма действия адсорбционных индикаторов, предложенное Сьер-рой и Санчес-Педрено, сопряжено с трудностями.
Меротра и Тандон [48] попытались разработать далее теорию адсорбционных индикаторов, опираясь на результаты опытов, ранее проведенных Мерогрой [47]. Эти авторы заметили, что некоторые адсорбционные индикаторы, не принадлежащие к группе производных флуоресцеина, тоже образуют соединения с ионами серебра [97, 98]. Одни соединения серебра оказались устойчивыми в водных средах, а другие неустойчивыми. При растворении в воде неустойчивые соединения гидролизуются, и их можно хранить только в неводных средах. Тандон и Меротра получили соединения серебра с конго красным, n-этоксихризоидином и некоторыми другими индикаторами, соединения которых с серебром не были известны.
Наблюдения указанных авторов, по-видимому, подтверждаются отчасти тем фактом, что изменение окраски кислотно-основных адсорбционных индикаторов не соответствует в точности изменениям, которые они проявили бы в гомогенном растворе как простые кислотно-основные индикаторы. Кроме того, в теории Шулека и Пунгора трудно понять (по словам Тандона и Меротры), каким образом катионы красителя могут притягиваться положительно заряженным осадком галогенида серебра. Однако объяснение Тандона и Меротры не согласуется с результатами, которые получили Пунгор и Холлос-Рокосиньи [52], так как соединения серебра с адсорбционными индикаторами совсем не образуются в водных средах, а если и образуются, то лишь в малом количестве. Что каса
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
7
ется этого факта, то не приводится никаких оснований, почему для объяснения поведения адсорбционных индикаторов надо учитывать образование соединений с серебром. По теории Шулека и Пунгора не требуется, чтобы происходила особого вида адсорбция анионов или катионов красителя на поверхности положительно заряженного осадка, должны адсорбироваться только нейтральные молекулы красителя. При адсорбции ионов красителя нарушается равновесие красителя в растворе, и поэтому на положительно заряженной поверхности осадка адсорбируются нейтральные молекулы красителя. Этот вопрос изложен в работе Пунгора и Холлоса-Роко-синьи [52].
КЛАССИФИКАЦИЯ И КРАТКАЯ ХАРАКТЕРИСТИКА АДСОРБЦИОННЫХ ИНДИКАТОРОВ
Приводимая здесь классификация адсорбционных индикаторов основывается на теории Шулека и Пунгора. Различные индикаторы можно классифицировать по механизму их действия в следую-щ е группы:
1) поверхностные индикаторы для титрования по методу осаждения («адсорбционные индикаторы», введенные в практику Фаянсом, принадлежат главным образом к этой группе);
2) поверхностные кислотно-основные индикаторы;
3) поверхностные окислительно-восстановительные индикаторы, называемые также окислительно-восстановительными адсорбционными индикаторами;
4) поверхностные флуоресцентные индикаторы;
5) поверхностные комплексометрические индикаторы.
В табл. VII. 1 приведены соединения, часто применяемые в качестве адсорбционных индикаторов, а также даны их структурные формулы и механизм действия.
Поверхностные индикаторы для титрования по методу осаждения характеризуются образованием слаборастворимого осадка, который состоит из красителя, применяемого в качестве индикатора, и одного из ионов, участвующих в титровании. В оригинальных и остроумных исследованиях Фаянса [25] показано в случае эозина, что’ образовавшийся эозинат серебра заметно хуже растворяется в присутствии галогенида серебра, чем в его отсутствие. Для этой группы индикаторов можно определить так называемое кажущееся произведение растворимости, которое меньше, а в ряде случаев гораздо меньше, чем в отсутствие посторонних поверхностноактивных веществ.
Исследования Пунгора и Конколя Тэге [58] показали, однако, что для этой группы индикаторов предположение о том, что растворимость индикатора уменьшается из-за адсорбции, неправомерно. В случае эозината серебра эти исследования показали, что
оо
Таблица VI 1.1
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ, КЛАССИФИЦИРОВАННЫЕ ПО МЕХАНИЗМУ ИХ ДЕЙСТВИЯ
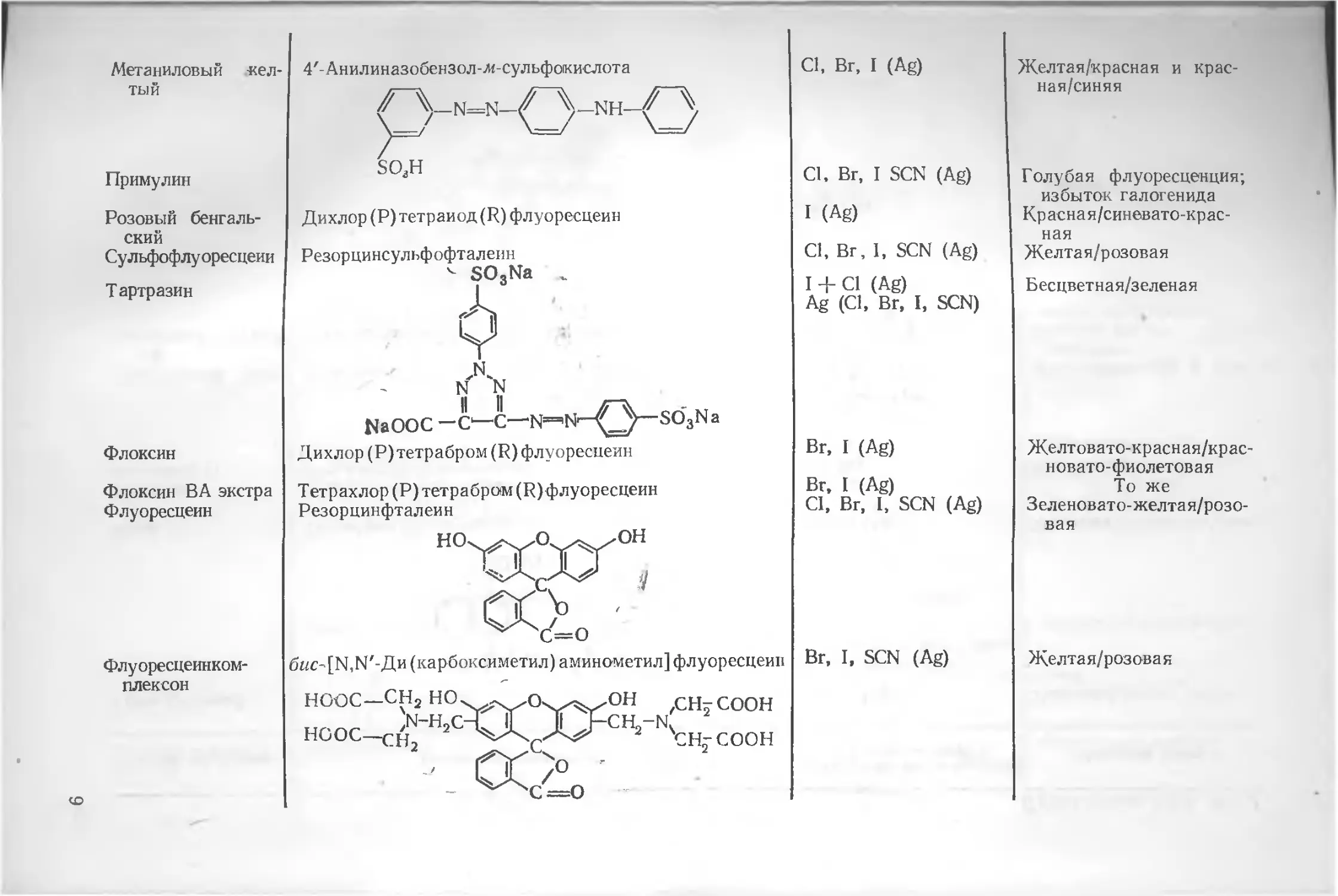
Тривиальное название Химическое название или формула Титруемый ион (в скобках указан титрант) Изменение окраски
Ализариновый красный Бриллиантовый орсейль С Бромфе./оловый синий 1. Индикаторы для титрования по мета Ализаринсул_1фокислота О ОН цпгт О , NH N"~N II °->n <_/ NaOaS^^^^SOgNa Тетрабромфснолсульфофталеин Дибром (Р)сульфофталеин Цибром (R) флуоресцеин Динод (R) сульфофталеин Диметил (Р)дииод(Р) флуоресцеин Диметил (R) флуоресцеин Дихлор (Р) флуоресцеин Дихлор (R) сульфофталеин Дпхлор (R) флуоресцеин ду осаждения SCN (Ag) Cl, Br, I (Ag), Ag (Br) Cl, I (Ag) Cl, Br, I, SCN (Ag) Br, I (Ag) Cl, Br, I, SCN (Ag) I (Ag) Cl, Br, I (Ag) Cl, Br, I (Ag) Cl, Br, I SCN (Ag) Cl, Br, I (Ag) Желтая/красная Красная/синевато-зеленая Желтовато-зеленая/зеле-ная и желточато-зеле-ная/синяя Желтая/густорозовая Желтовато-красная/крас-новато-фиолетовая Желтая/кр асновато-фиолетовая Желтовато-красная/ красновато-фиолетовая Зеленовато-желтая/розо-вая То же Желтая/розовая Зеленовато-желтая/ /красная
Метаниловый желтый 4'-Анилиназобензол-л«-сульфокислота /~~y_N=N—NH—
Примулин so3h
Розовый бенгальский Сульфофлуоресцеии Т артразин Дихлор (Р) тетраиод (R) флуоресцеин Резорцинсульфофталеин < SO,Na „ ф н N N ЦаООС — С1—С—N=^N'—SO3Na
Флоксин Д ихлор (Р)тетрабром (R) флуоресцеин
Флоксин ВА экстра Флуоресцеин Тетрахлор (Р)тетрабром(Р) флуоресцеин Резорцинфталеин НО^л^О^^/ОН XjQu J
Флуоресцеинком-плексон СО бис- [N.N'-Ди (карбоксиметил) аминометил] флуоресцеин НООС— СН2 НО. Os^^OH .СН-СООН НГПС 'N~H=C -V X JLJ~CH2-NS НООС—сНз 'сн-соон СК > '
ci, Br, I (Ag) Желтая/красная и крас-ная/синяя
Cl. Br, I SCN (Ag) Голубая флуоресценция; избыток галогенида
I (Ag) Красная/синевато-крас-ная
Cl, Br, I, SCN (Ag) I + Cl (Ag) Ag (Cl, Br, I, SCN) Желтая/розовая Бесцветная/зеленая
Br, I (Ag) Желтовато-красная/крас-новато-фиолетовая
Br, I (Ag) CI, Br, I, SCN (Ag) То же Зеленовато-желтая/розо-вая
Br, I, SCN (Ag) Желтая/розовая
о
Продолжение табл. VII. 1
Тривиальное название Химическое название или формула Титруемый ион (в скобках указан титрант) Изменение окраски
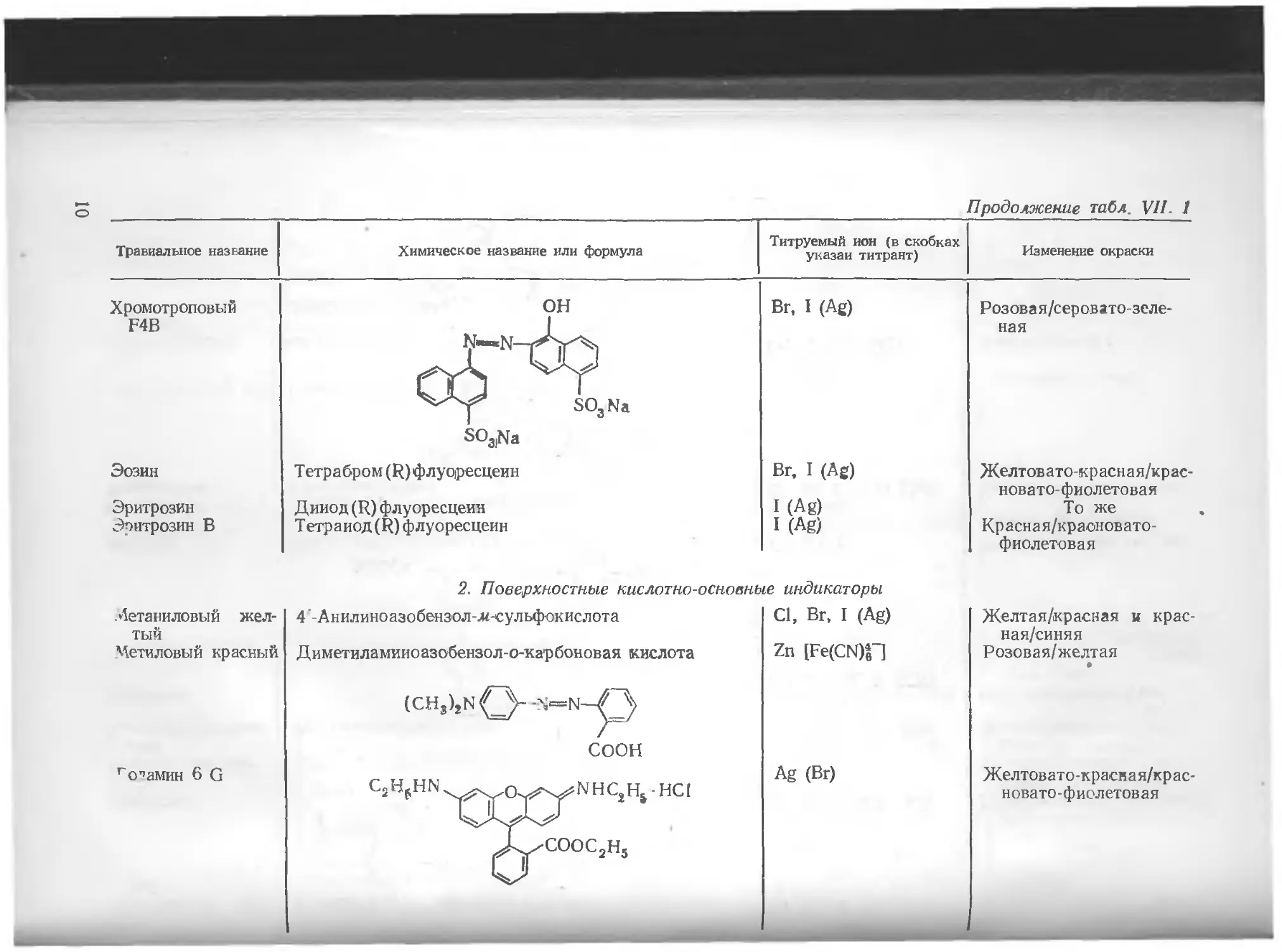
Хромотроповый F4B он SO3Na SO3|Na Br, I (Ag) Розовая/серовато зеленая
Эозин T етрабром (R) флуо|ресцеин Br. I (Ag) Желтовато-красная/крас-новато-фиолетовая
Эритрозин Динод (R) флуоресцеин I (Ag) То же
Эритрозин В Тетраиод(Р) флуоресцеин I (Ag) Красная/краоновато-фиолетовая
2. Поверхностные кислотно-основные индикаторы
Детаниловый желтый
Цетиловый красный
’'самин 6 G
4’-Анилиноазобензол-л<-сульфокислота
Диметиламииоазобензол-о-карбоновая кислота
(chs)2nQ
Cl, Br, I (Ag)
Zn [Fe(CN)n
Ag (Br)
Желтая/красная и крас-ная/синяя
Розовая/желтая
Желтовато-красная/крас-новато-фиелетовая
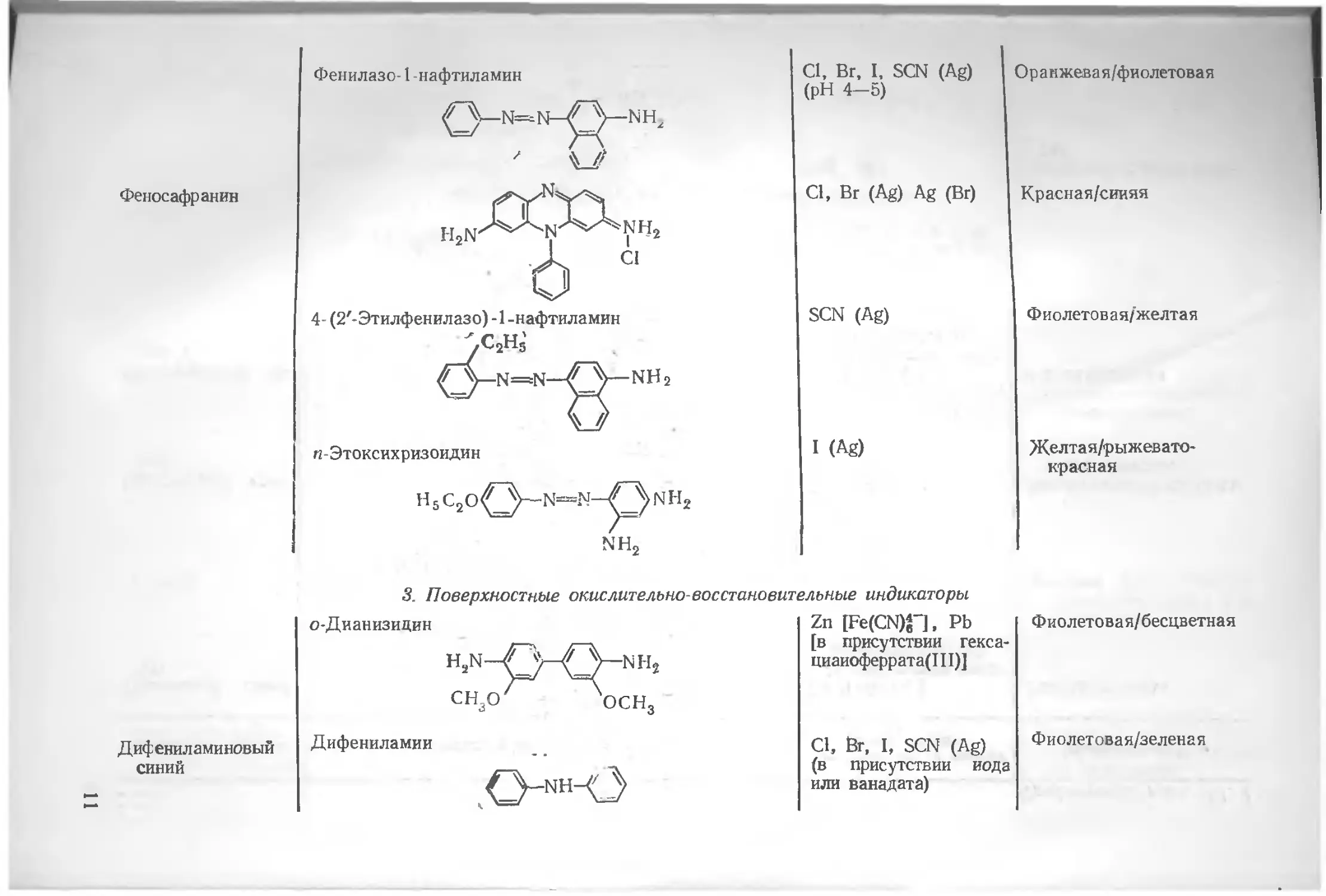
Феносафранин
Фенилазо-1 -нафтиламин
н-Этоксихризоидин
Cl, Br, I, SCN (Ag) (pH 4—5)
Орамжевая/фиолетовая
Cl, Br (Ag) Ag (Br) Красная/сиияя
SCN (Ag)
I (Ag)
Фиолетовая/’желтая
Желтая/рыжевато-красная
3. Поверхностные окислительно-восстановительные индикаторы
Дифениламиновый синий
Zn [Fe(CN)Jl. Pb
[в присутствии гекса-циаиоферрата(111)]
Фиолетовая/бесцветная
Cl, Br, I, SCN (Ag) (в присутствии иода или ванадата)
Фиолетовая/зеленая
Тривиальное название
Химическое название или формула
Титруемый иои (в скобках указан титрант)
Продолжение табл, VII. 1
Изменение окраски
Ксиленовый синий
VB
Нейтральный красный
Патентованный синий V
Zn lFe(CN)J"J
[в присутствии гекса-цианоферрата(Ш)]
Br, I (Ag)
Zn [Fe(CN)n
[в присутствии гекса-цианоферрата(1 II)]
4. Поверхностные комплексометрические индикаторы
п-Диметиламинобензилиденроданин
Н—N—С=О
SC^UcH-Q- N(CH3)2
Розовая/зеленая
Фиолетаво-красная/жел-товато-красная
Розовая/зеленая
Hgt (Br)
Фиолетово красная/голу-бая
PAN
Ауразин
Индигозоль
IBC
синий
Дифенплкарбазид
Дифенилкарбазои
Hgl+(Cl + Br),Cl(Hg2)
Синевато-фиолетовая/ /бесцветная
ОС. -х
'NH-N1I
/N=N
0С\ z-x
'nh-nh< \
1(1 -Пиридилазо) -2-на<Ьтол
N=N
Cl, Br, I, SCN (Ag) (в присутствии HgCl2)
1 (Ag) или Си (ЭДТА) (в присутствии Cu2+)
5. Поверхностные флуоресцентные индикаторы
Диаминодиметилаклидинформиат
Фиолетовая
Красновато-фиолетовая/ /бледно-зеленая
Ag (SCN)
Br, I, SCN (Ag)
Зеленая флуоресценция/ /оранжево-красиая флу оресценция
Зеленовато-желтая флуоресценция при избытке галогенида
Продолжение Кбл.УП. 1
Тривиальное название Химическое название или формула Титруемый ион (в скобках указан титрант) Изменение окраски
Лактофлавин Магдала красный сносной -снон-сноч-сн2он CH3y^>NYNC==O С Нз^^-N^C' N Н II о (XnUO ТТ Т I 00 Ag (SCN) (Ag) Зеленовато-желтая флуоресценция при избытке SCN Соломенно-желтая флуо-ресценция/Лиолетовая
Приму ЛИН zTiPT4) 1 Н3С q г>я2 Cl, Br, I, SCN (Ag) Голубая флуоресценция при избытке галогенида
Тиофлавин S —N —N x^/NH2 Г я_Г Г“ и Г JT I-I3C-^^s'C"^^S'C-so, N а CI, Br, I, SCN (Ag) То же
Трипановый красный «л'-Диаминодифенил-ти-сульфокислота-диазо-бис-2-нафтиламин-3,6-дисульфокислота Cl Br, I, SCN (Ag) » »
Трипаф лавин
Хинин
Этоксидиаминоакридинлактат
ОС2Н5
НОиССНОН-СНд
SCN (Ag) Зеленая флуоресценция
Cl, Br, 1, SCN (Ag) Желтовато-зеленая флуоресценция
Cl, Br, I, SCN (Ag) Зеленоватая флуоресценция при избытке серебра
16
ГЛАВА VII
при избытке серебра концентрация этого металла на поверхности твердой фазы галогенида серебра возрастает вследствие адсорбции настолько, что, хотя в гомогенной фазе не достигается предел растворимости эозината серебра, на поверхности гетерогенной фазы концентрация серебра легко превышает этот предел. Поэтому, зная скорость адсорбции ионов, входящих в состав осадка, можно рассчитать, когда проявится изменение индикатора при данной концентрации красителя. Таким образом, действие поверхностных адсорбционных индикаторов однозначно определяется адсорбцией ионов, которые образуют осадок с индикатором.
Группа поверхностных кислотно-основных индикаторов наиболее типична для адсорбционных индикаторов [78]. Она характеризуется тем, что, хотя адсорбция красителя не изменяется быстро в конечной точке титрования, значение рК красителя существенно изменяется в зависимости от природы и количества избытка собственных ионов [59, 73]. В благоприятном случае индикатор действует также в нейтральной среде. Связывание или отщепление протона красителем можно установить с помощью рН-метра [73]. Однако если среда сильнокислая, то таким способом определять изменение концентрации протонов, вызванное красителем, нельзя [52]. Недавно были сделаны попытки объяснить поведение поверхностных кислотно-основных индикаторов исходя из образования соответствующих соединений серебра [48]. Эти объяснения еще не подтвердились, и следует продолжить исследования в этой области. Однако некоторые поверхностные кислотно-основные индикаторы ведут себя аномально при титровании галогенидов. Например, 4-(2-этилфенилазо)-1-нафтиламин пригоден для определения хлорид-ионов, но не годится для титрования бромид- или иодид-ионов [46].
Поверхностные окислительно-восстановительные индикаторы [5, 15, 17, 18, 80—82, 86] представляют собой недавно разработанную группу адсорбционных индикаторов. Механизм их действия основывается в принципе на том, что окислительно-восстановительный потенциал индикатора, адсорбированного на поверхности осадка, изменяется под влиянием избытка собственных ионов. За последнее время проведено значительное количество исследований в области разработки поверхностных окислительно-восстановительных индикаторов. В поисках новых индикаторов этого типа тщательно исследовалось их поведение, в результате чего оказалось, что индикаторы можно с успехом применять в аналитической химии [94].
Для определения конечной точки титрования на основе адсорбции можно, помимо выше рассмотренных случаев, использовать и другие явления, например флуоресценцию [12, 14] и смещение равновесия при комплексообразовании [83, 84, 90]. В общем следует сказать, что, за исключением поверхностных адсорбционных
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
17
индикаторов, установление конечной точки титрования при помощи адсорбции можно объяснить изменениями физико-химических свойств индикатора-носителя на поверхности различных осадков. В соответствии с этим существенно изменяются константы диссоциации, окислительно-восстановительные потенциалы и другие показатели, причем эти изменения зависят от ирироды и количества собственных ионов, адсорбированных на поверхности осадка.
МЕТОДЫ ИССЛЕДОВАНИЯ АДСОРБЦИОННЫХ ИНДИКАТОРОВ
ИНДИКАТОРЫ, ОБРАЗУЮЩИЕ ОСАДКИ
Исследование ранее рассмотренных различных типов адсорбционных индикаторов представляет собой трудную задачу. Для этого нужно проводить многосторонние исследования, с тем чтобы
Концентрация ионоб, 10 холь • л
Рис. VII 1. Адсорбция эозина на бромиде серебра в присутствии собственных ионов.
(По данным Фаянса [24].)
установить механизм действия индикатора. В этом разделе мы предполагаем рассмотреть главные принципы, которых надо придерживаться в исследованиях адсорбционных индикаторов.
В первую очередь нужно установить, как индикатор-краситель адсорбируется при избытке обоих видов собственных ионов осадка [58]. Если адсорбция при избытке собственных ионов происходит так, как показано на рис. VII. 1, рассматриваемый индикатор относится, вообще говоря, к индикаторам типа Фаянса, т. е. к поверхностным адсорбционным (осаждаемым) индикаторам.
В этом случае кажущееся произведение растворимости индика-Ра определяют следующим образом. Готовят водный раствор ИН-2-913
18
ГЛАВА VII
дикатора-красителя соответствующей концентрации (10~2—10“5М). Затем получают осадок в присутствии известного избытка собственных ионов. (Обычно надо брать в избытке тот ион, для которого предварительный опыт показал, что краситель хорошо адсорбируется.) Затем титруют раствор, содержащий осадок, раствором красителя в качестве титранта до изменения окраски на поверхности осадка. Операцию повторяют с различными количествами избытка собственного иона. Зная, какое соединение образует краситель с собственным ионом, находящимся в избытке, можно рассчи
Рис. VII.2. Кондуктометрическое титрование б мл 0,5 М нитрата серебра эозинатом натрия.
(По данным Пуигора и Конколя Тэге [53].)
нии следует выбирать такую
тать кажущееся произведение растворимости на основе избытка собственного иона и количества красителя, добавленного при титровании.
Вообще говоря, поверхностные флуоресцентные индикаторы, а отчасти также поверхностные индикаторы для комплексометрии обладают свойствами индикаторов Фаянса.
Степень адсорбции красителя можно определить фотометрически, для чего отфильтровывают раствор от осадка и берут для измерения аликвотную часть фильтрата. Так как частицы осажденного галогенида рассеивают свет, то измеренное поглощение в отсутствие красителя надо вычесть из значения поглощения в присутствии красителя. (Это составляет, например, 0,08 в случае метанилового желтого.)
При фотометрическом определе-длину волны, которая соответствует
максимуму светопоглощения красителя, находящегося в растворе: краситель, адсорбированный осадком, отличается по интенсивности
от красителя в растворе.
Если краситель дает осадок с одним из компонентов реакции титрования по методу осаждения, например с ионами серебра в случае аргентометрии, рекомендуется определять произведение растворимости кондуктометрически. На рис. VII.2 изображена кривая кондуктометрического титрования эозината серебра. Реальная кривая титрования наряду с кривыми, представляющими идеальную реакцию осаждения и полное отсутствие реакции осаждения, служат основанием для кондуктометрического определения произведения растворимости. Полученные этим путем значения произведения растворимости заметно больше значений кажущегося про
адсорбционные индикаторы
19
изведения растворимости, которое было определено в присутствии постороннего поверхностноактивного вещества с находящейся на нем в виде осадка слаборастворимой соли.
Для определения произведения растворимости соли красителя, образовавшейся на постороннем поверхностноактивном веществе, надо измерить адсорбцию собственных ионов, участвующих в образовании соли. Так, например, Пунгор и Конколь Тэге показали, что растворимость эозината серебра, находящегося на поверхности осадка хлорида серебра, такая же, как растворимость эозината серебра, осажденного из гомогенного раствора. Кажущееся произведение растворимости имеет заметно меньшее значение, чем измеренное в гомогенном растворе, потому что произведение растворимости соли красителя достигается быстрее, чем в отсутствие посторонних поверхностноактивных веществ; причина этого заключается в высокой концентрации собственного иона, адсорбированного на поверхности осадка.
ПОВЕРХНОСТНЫЕ КИСЛОТНО-ОСНОВНЫЕ ИНДИКАТОРЫ
В случае поверхностных кислотно-основных индикаторов при условии, что конечная точка может находиться в нейтральной, слабокислой или слабощелочной среде [60, 61], с помощью рН-метра можно определить, связываются или отщепляются протоны красителем, если концентрация красителя находится в соответственно подобранных пределах [74]. На рис. VII.3 и VII.4 представлены
Кривая титрования иодида Л М нитратом серебра по индикатору я-этоксихризоидину при помощи стеклянного электрс да.
° Данным Шулека и Пунгора [67].)
Рис. VI 1.4. Кривая титрования серебра 0,1 М иодидом калия по индикатору п этоксихризоидину при помощи стеклянного электрода.
(По данным Шулека и Пунгора [67].)
20
ГЛАВА VII
такие кривые для п-этоксихризоидина при титровании иодида ионами серебра и соответственно серебра иодид-ионами. Однако имеются случаи, когда при кислотно-основном определении по изменениям на поверхности осадка заранее известно, что раствор имеет кислую реакцию. Так как в этом случае связывание или отщепление протонов адсорбционных индикаторов невозможно определить pH-метрически, используют фотометрический метод [52]. Для этого получают кривые поглощения света красителем в кислом и щелочном растворах, а затем строят кривые поглощения красителя в присутствии коллоидного осадка в зависимости от избыточных количеств обоих участвующих ионов.
Как уже говорилось, для поверхностных кислотно-основных индикаторов огромное значение имеет величина р/С Ее определяют следующим образом [75]. Путем взбалтывания или добавления хлопьеобразующего реагента (например, нитрата калия) получают хлопьевидный адсорбат красителя. Жидкость отфильтровывают и тщательно промывают осадок дистиллированной водой. Затем, добавляя кислоту и щелочь, устанавливают с помощью рН-метра нижний и верхний пределы значений pH, в которых изменяется окраска. Среднее из значений pH, установленных таким способом, принимается за реальное значение р/С Обычно ширина интервала перехода окраски зависит от числа протонов, участвующих в установлении кислотно-основных свойств молекулы. Таким образом можно определить, например, для n-этоксихризоидина, что интервал перехода окраски равен приблизительно одной рВ единице. Зная число протонов, участвующих в реакции присоединения или в реакции отщепления, а также количество адсорбированного красителя, можно установить, что n-этоксихризоидин присоединяет два протона при адсорбции его осажденным иодидом серебра. В отсутствие осадка получается большая разница в значениях pH при присоединении этих двух протонов. В то время как один из протонов захватывается в отсутствие осадка при pH 4,5, связывание второго протона происходило только лишь в среде концентрированной серной кислоты. В случае адсорбированного красителя не наблюдается заметной разницы между значениями pH при связывании обоих протонов.
В табл. VII.2 приведено несколько значений рК для п-этокси-хризоидина. По этим результатам видно, что изменения значений р/С адсорбированного индикатора зависят от степени деформации галогенид-иона в галогениде серебра. Аналогично в случае метани-лового желтого два протона связываются при избытке галогенид-ионов. В этих условиях, неблагоприятных для определения pH, механизм действия индикатора устанавливают следующим способом. Определяют спектр поглощения метанилового желтого, находящегося в виде адсорбата. Затем строят кривую светопоглоще-ния красителя в гомогенной системе при pH 1,1. Если переход
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
21
Таблица VI 1.2
ЗНАЧЕНИЯ рК л-ЭТОКСИХРИЗОИДИНА, АДСОРБИРОВАННОГО НА ПОВЕРХНОСТИ ГАЛОГЕНИДОВ СЕРЕБРА (ПО ДАННЫМ ШУЛЕКА И ПУНГОРА [67])
Адсорбент Избыток собственного иона, % (по экв.) Интервал перехода, pH pK
Agl 50% Ag 3,30—4,58 3,90
10% Ag 3,90—5,20 4,68
Точка эквивалентности 5,50—8,50 7,00
10% I 7,40—8,70 8,15
50% I 7,70—8,90 8,29
Agl 50% Ag 3,30—4,50 3,90
Ag I 50% I 7,70—8,90 8,29
AgBr 50% Ag 4,20—5,30 4,75
AgBr 50% Br 5,20—6,30 5,75
Agd 50% Ag 4,50—5,50 5,00
AgCl 50% Cl 4,60—5,90 5,65
окраски метанилового желтого происходит при pH 1,2, то краситель в растворе с pH 1,1 содержит протоны. Протоны присоединены к сульфогруппе. Это доказывается существованием почти полного сходства окраски протонированного красителя с окраской его соли с серебром. В данном случае переход окраски в растворе концентрированной серной кислоты занимает промежуточное место между двумя вышеуказанными кривыми. После определения с по
мощью этих кривых соотношения основных окрасок в растворе концентрированной серной кислоты при данной длине волны (в нашем исследовании синего соединения было 86%, а красного — 14%) можно рассчитать точки для кривой серной кислоты, исходя из найденного процентного соотношения. Полученные результаты представлены на рис. VII.5; они хорошо совпадают с предположе-
ниями, высказанными выше.
В области конечной точки титрования не происходит быстрого изменения адсорбции поверхностных кислотно-основных индикаторов [76]. Кривые адсорбции n-этоксихризоидина и метанилового желтого показаны на рис. VII.6 и VII.7 соответственно. В последнем случае среда имеет кислую реакцию. Интересно отметить, что в нейтральной среде тот же метаниловый желтый [52] дает кривую адсорбции, совершенно отличную от кривой в кислой среде, кривая в нейтральной среде однозначно характеристична для по-
22
ГЛАВА VII
верхностных осажденных индикаторов. Так, исходя из кривой адсорбции, можно легко обнаружить изменение механизма действия индикатора. В связи с этим очень важно отметить, что в случае
Рис VII.5. Кривые поглощения света метаниловым желтым в различных условиях:
W—♦ при pH 1,1 в гомогенной среде, □—П—*□ при pH 1,1 в присутствии иодида серебра в избытке иодида; X—X—X в растворе концентрированной серной кислоты; О—0—0 вычисленное поглощение красителя в концентрированной серной кислоте. Концентрация метани-лового желтого 10-5 моль-л-1. (По данным Пунгора и Холлоса Рокоснньи [47] )
метанилового желтого при обоих механизмах действия индикатора почти нет разницы между окраской протонированного красителя и окраской его соли с серебром. Ион серебра вследствие его не-
Рис. VII.6. Адсорбция п-этоксихризоиди-на на иодиде серебра в присутствии собственных ионов.
(По данным Пунгора и Шулека [55].)
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
23
большого ионного объема оказывает на молекулу красителя почти такое же влияние, как и протон. Этот факт служит доводом против выводов Сьерры и Санчес-Педрено.
Рис. VII.7. Адсорбция метанилового желтого на иодиде серебра в присутствии собственных ионов при pH 1,1.
Концентрация метанилового желтого 10-s моль-л—1. (По данным
Пунгора и Шулека [55].>
ПОВЕРХНОСТНЫЕ ИНДИКАТОРЫ ДРУГИХ ТИПОВ
В области исследований поверхностных окислительно-восстановительных и просто окислительно-восстановительных индикаторов [59] значительные результаты получили Сьерра и Барриель-Мар-ти [86], Асенси Мора [5] и Богнар [15]. На основании их работ можно считать, что окисление с помощью НЮ происходит в случае адсорбированного красителя главным образом при потенциале заметно более отрицательном, чем потенциал в гомогенном растворе, что и показано на рис. VII.8 [74]. Ясно видно, что потенциал кривой титрования в присутствии одной капли 1%-ного раствора иода в этиловом спирте заметно возрастает по сравнению с потенциалом того же раствора, но содержащего также п-этоксихри-зоидин.
Для определения изменения окислительно-восстановительного потенциала осажденного индикатора, связанного с поверхностью, практически удобно использовать зависимость этого изменения от природы и количества собственных ионов; для этого поступают так же, как при определении кислотно-основных свойств, которое было рассмотрено выше [59]. После получения адсорбата красителя маточник сливают, осадок взбалтывают с дистиллированной водой, снова сливают и только тогда используют как индикатор окислительно-восстановительного титрования. Во время титрования замеряют потенциал. Таким способом при цсриметрическом титро-
г MB \
700
600
500
400
jo—о—° 2 ✓
300
Рис. VII.8. Кривая потенциометрического титрования иодида нитратом серебра при добавлении 1 капли 1 %-кого иода
Кривая 1 — титрование в отсутствие красителя; кривая 2— титрование при добавлении 8 капель 0,2%-ного л-этоксихризондииа. (По данным Сьерры и Асенси [74].)
-100 -
-200 -
-3001 I I I I I I I I I | I | I |
о ^2 4 6 8 10 12 14
0,1 М AgNO3, мл
Рис VII.9. Кривые титрования бромида нитратом серебра.
Кривая 1 — титрование в отсутствие красителя; кривая 2— титрование в темноте при добавлении 2 капель 1 %-кого дифенилкарбазида; кривая 3 — титрование на свету при добавлении 2 капель 1%-иого дифенилкарбазида; кривая 4 — титрование на свету при добавлении 2 капель 1%-иого дифенилкарбазида и 2 капель 0.005 М хлорида ртути (II). (По данным Сьерры и Санчес Педрено [831.)
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
25
вании ионов Fe(II) установили, что потенциал адсорбата красителя, состоящего из n-этоксихризоидина и иодида серебра, полученного при избытке серебра, составляет 0,85 ±0,02 В, тогда как адсорбат красителя, приготовленный в избытке иодида, имеет потенциал 0,98+0,02.
В области исследований поверхностных флуоресцентных индикаторов Богнар [12—14] занимает ведущее место. В результате его исследований было установлено, что в случае поверхностных флуоресцентных индикаторов, чувствительных к кислотам и основаниям, протон, который может быть связан с красителем, не играет никакой роли. В настоящее время не имеется данных о корреляции между структурой и флуоресцентными свойствами красителей, применяемых в качестве индикаторов. Поэтому для группы поверхностных флуоресцентных индикаторов приходится удовлетвориться определением вида собственного иона, при избытке которого адсорбция индикатора становится значительной. На основе этих результатов можно сделать вывод о комплексе, образовавшемся на поверхности с участием данного иона.
В области поверхностных комплексометрических индикаторов проведены обширные исследования [82, 84, 90]. Отмечается [89], что в случае комплексов, образованных на поверхности осадков, исследование изменений pH раствора можно использовать для изучения реакции образования поверхностных комплексов. Результаты одного из исследований такого рода показаны на рис. VII.9 для случая образования комплексов дифенилкарбазида и дифенил-карбазона со ртутью(II) на поверхности осадка галогенида серебра. Эти исследования были проведены с осадком бромида серебра.
ТЕОРИЯ АДСОРБЦИОННЫХ ИНДИКАТОРОВ
ПОВЕРХНОСТНЫЕ ИНДИКАТОРЫ ДЛЯ ТИТРОВАНИЯ ПО МЕТОДУ ОСАЖДЕНИЯ
По способу действия адсорбционные индикаторы, приведенные в табл. VII. 1, можно разделить на две группы. К первой группе относятся так называемые индикаторы Фаянса, которые характеризуются образованием соли индикатора на поверхности исследуемого осадка. Механизм действия этих индикаторов исключительно прост. Согласно законам коллоидной химии, собственные ионы, находящиеся в избытке, в значительной степени адсорбируются осадком, образующимся в процессе титрования. Концентрация собственных ионов на поверхности (внутри слоя раствора, Динамически связанного с осадком) повышается вследствие ад сорбции по обе стороны от точки эквивалентности, превышая кон
26
ГЛАВА VII
центрации в растворе. Исследования Пунгора и Конколя Тэге [58] доказали, что в случае хлорида серебра увеличение концентрации очень значительно.
Из различных компонентов, находящихся в растворе, наиболее прочно адсорбируются собственные ионы, за ними следуют близкие им ионы, а затем ионы наибольшего размера. Поэтому в принципе надо учитывать адсорбцию всех ионов индикатора на поверхности осадка [76]. Однако наши исследования показали, что в пределах ошибки опыта непосредственная адсорбция красителя на поверхности частиц осадка меньше по сравнению со значительной адсорбцией собственных ионов и поэтому не имеет решающего значения для образования окрашенного осадка. Механизм остается одним и тем же как в случае сильнодиспергированного осадка (подобного галогениду серебра), так и в случае более компактного осадка (подобного сульфату бария). Адсорбция собственных ионов ппеобладает на поверхности частиц, что приводит к тому, что локальное увеличение их концентрации превышает произведение растворимости соли, образованной собственными ионами и ионами индикатора. Следовательно, например, соль иона серебра и аниона красителя осаждается на поверхности частиц и задерживается благодаря продолжающейся адсорбции собственных ионов.
К индикаторам Фаянса относится та группа индикаторов, для которой адсорбция собственных ионов является решающим фактором. Для этих индикаторов ионы образца наравне с ионами индикатора образуют осадок с ионами титранта, поэтому отклонение конечной точки от точки эквивалентности можно определить путем варьирования относительных концентраций реагирующих частиц. Если индикатор и образец образуют с титрантом осадки, растворимости которых сопоставимы, то осадок может окраситься еще до того, как будет достигнута точка эквивалентности.
Используя поверхностные индикаторы для титрования по методу осаждения, можно расширить возможности определения конечной точки путем применения различных растворителей. Растворимость красителей, которые образуют осадки, можно изменять, используя неводные растворители, в первую очередь растворители с низкой диэлектрической постоянной. Богнар и Шароши [19] показали в обширных исследованиях, что так действует диоксан на производные флуоресцеина. В табл. VII.3 приведены данные о пригодности двенадцати производных флуоресцеина для использования их в качестве индикаторов при аргентометрическом титровании галогенидов в водной среде. Исключением является последний пример, где показано, что слаборастворимая серебряная соль эритрозина В не годится для использования в качестве адсорбционного индикатора в водных растворах для титриметриче-ского определения ни одного из трех галогенид-ионов. Однако установлено, что в кислой среде, содержащей избыток диоксана,
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
27
Таблица VI 1.3
ПРИМЕНЕНИЕ ФЛУОРЕСЦЕИНА И ЕГО ПРОИЗВОДНЫХ В КАЧЕСТВЕ ИНДИКАТОРОВ в АРГЕНТОМЕТРИЧЕСКОМ ТИТРОВАНИИ ГАЛОГЕНИДОВ (ПО ДАННЫМ ФАЯНСА [24»
Название3
Аргентометрическое титрование иоиов^
ci- | Вг— | I-
Флморесцеин
Диметил (R) флуоресцеин
Дихлор (Р) флуоресцеин
Дихлор (R) флуоресцеин
Тетрахлор (Р)тетрабром (R) флуоресцеин (флоксин)
Дибром (R) флуоресцеин
Дихлор (Р) тет рс бром (R) флуоресцеин
Тетрабром (R) флуоресцеин (эозин)
Дииоц (R) флуоресцеин
Диметил (R) дииод (R) флуоресцеин
Дихлор (Р) тетраиод (R) флуоресцеин (беггальский оозовый)
Тетраиод(Р)флусресцеин (эритрозин В)
а К означает, что заместитель (CI, Вг, I) находится у кольца резорцина, Р означает, что заместитель (Cl, Br, I) находится у кольца бензола.
® Знак «+» означает, что индикатор пригоден для аргентометрического титрования; знак «—» означает, что индикатор не пригоден для аргентометрического титрования.
можно применять эритрозин В для титрования даже хлорид-ионов и получать требуемую точность. Подробности, касающиеся применения эритрозина В в титровании галогенидов, приведены в табл. VII.4 [19].
В настоящее время известно, что механизм действия адсорбционного индикатора отличается от механизма поверхностных индикаторов для титрования по методу осаждения и заключается в изменении физических констант индикатора, когда он адсорбируется на поверхности.
ПОВЕРХНОСТНЫЕ КИСЛОТНО-ОСНОВНЫЕ ИНДИКАТОРЫ
Действие поверхностных кислотно-основных индикаторов зависит от изменения константы кислотно-основной диссоциации красителя после его адсорбции. Адсорбция происходит как до наступления точки эквивалентности, так и после нее, а изменение константы диссоциации зависит от природы адсорбированных собственных ионов. На адсорбцию красителя, которая обусловливается его свойствами, природа собственных ионов оказывает совсем незначительное влияние [52—54, 59, 74]. Следовательно, ион титранта ведет себя по отношению к одному и тому же красителю в од-
28
ГЛАВА VII
Таблица VII. 4
ПРИМЕНЕНИЕ ЭРИТРОЗИНА ПРИ ТИТРОВАНИИ ГАЛОГЕНИДОВ РАСТВОРОМ НИТРАТА СЕРЕБРА (ПО ДАННЫМ БОГНАРА И ШАРОШИ [19])
Галогеннд-нон Состав растворителя Ошибка, %
Вода, % Кислота Органический растворитель
I- 37 13% СН8СООН 50% диоксана 0,0
36 14% СН3СООН 50% диоксана +0,4
30 20% СН3СООН 50% ацетона —0,1
48 2% H2SOa 50% диоксана +0,1
48 2% H2SOa 50% ацетона +0,1
48 2% HNO3 50% ацетона 0,0
30 7% НСЮа 63% ацетона —0,1
44 6% НС1Оа 50% этанола +0,1
Вг- 25 25% СН3СООН 50% диоксана 0,0
49 1% H2SOa 50% ацетона +0,2
48 2% H2SOa 50% ацетона +0,3
ci- 11 22% CH3COOH 67% диоксана +0,2
ном случае так, как если бы это был кислый раствор, а в другом случае так, как если бы это был щелочной, в зависимости от природы различных собственных ионов. Вообще говоря, изменения значений р/( индикатора всегда связаны с присоединением или отщеплением протонов, как вторичным эффектом заряда на поверхности Однако этот эффект можно заметить только в том случае, если титрование проводится в почти нейтральном растворе, не содержащем буферирующих веществ.
Применение неводных растворителей по способу, описанному для титрования индикаторов по методу осаждения, по-видимому, не является перспективным для поверхностных кислотно-основных индикаторов, если учесть их механизм действия. Известно, что при понижении диэлектрических постоянных константы ионного равновесия уменьшаются пропорционально кубическому корню от значения диэлектрической постоянной. Так, Богнар и Шароши [19] показали, что для n-этоксихризоидина точность определения конечной точки не улучшается при использовании органических растворителей, а, наоборот, конечные точки становятся нечеткими. Это явление, однако, кроме влияния диэлектрической постоянной,
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
29
обусловливается еще тем, что адсорбция красителя уменьшается в присутствии органических растворителей, легко адсорбирующихся на поверхности осадка.
Краситель n-этоксихризоидин пригоден главным образом для установления конечной точки при аргентометрическом титровании иодида и, наоборот, при титровании ионов серебра раствором иодида [72, 78]. В результате исследований Пунгора [51], а также Пунгора и Конколя Тэге [56, 57] удалось установить, что точка эквивалентности при титровании иодидов смещается (в смысле количества израсходованного титранта), а степень смещения зависит от вида и концентрации ионов в растворе. Если присутствует только один вид посторонних ионов (любые анионы, содержащиеся в исследуемом растворе наряду с иодидом, считаются посторонними ионами), то количество 'перерасходованного титранта увеличивается с ростом концентрации постороннего иона до некоторого предельного значения. В табл. VII.5 приведены данные, показывающие, как изменяется количество перерасходованного титранта в зависимости от концентрации хлорида. Кривая потенциометрического титрования изображена на рис. VII. 10. На основе полученных результатов Пунгор и Конколь Тэге сделали вывод, что изменение окраски n-этоксихризоидина фактически происходит тогда,
Таблица VI 1.5
ПЕРЕРАСХОД НИТРАТА СЕРЕБРА ПРИ ТИТРОВАНИИ ИОДИДА В ПРИСУТСТВИИ ХЛОРИДА (ПО ДАННЫМ ПУНГОРА) [46])
К шцентрация С1“, моль - л—1 Перера сход 0 1 М AgNO3 мл Концентрация CI , моль л—1 Перерасход 0,1 М AgNO3, мл
1(Г* 0,18 0,90
5 • 10-« 0,48 3 10-3 1,02
10-» 0,81 0,97
0,79 5 • IO-» 1,06
0,74 1,04
0,76 1,08
0,75 1,03
0,94 1,06
2 • Ю-з 0,88 io-2 1,08
0,92 1,08
0,97
Адсорбируется 10—* моля Agl
30
ГЛАВА VII
когда закончится образование серебряной соли родственного иона (в данном случае хлорида), адсорбированного на поверхности иодида серебра, а вообще иона с отрицательным зарядом, адсор-
рн
Рис. VII.10. Кривые потенциометрического титрования иода и смешанного раствора иодида с хлоридом.
Кривая I — раствор иодида; кривая 2 — раствор иодида и хлорида (10: I); кривая 3 — раствор иодида и хлорида (2 1); кривая 4 — смесь растворов иодида и хлорида (1 : 1). Концентра ция n-этоксихризоидина 3-10-5 моль-л-1; стеклянный рабочий электрод. (По данным Пуи-гора [46].)
5,5 _L_ I । | | | । | । | , |
° 2 4 6 8 10 12
О 01 М AgNOs, мл
бированного на поверхности. Рассчитанные на основе перерасхода титранта значения ионных радиусов даны в табл. VII.6. Результаты проведенных опытов показывают, что в случае галогенидов адсорбируются несольватированные ионы, а в случае сульфатов и фосфатов — гидратированные ионы.
Таблица VI 1.6
ИОННЫЕ РАДИУСЫ, ВЫЧИСЛЕННЫЕ ПО ПЕРЕРАСХОДУ НИТРАТА СЕРЕБРА ПРИ ТИТРОВАНИИ ИОДИДА
В ПРИСУТСТВИИ ИССЛЕДУЕМЫХ ИОНОВ (ПО ДАННЫМ ПУНГОРА И КОНКОЛЯ ТЭГЕ [51])
Исследуемые ноны Перерасход 0,1 М AgNOg, мл Ионный радиус, вычисленный на основе ионного радиуса С1—, равного о о 1.81 1 (А)
С1- 1,06 —
SCN- 0,46 2,75
so|- 0,12 5,4
рог 0,11 5,5
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
31
ПОВЕРХНОСТНЫЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
Механизм действия поверхностных окислительно-восстановительных индикаторов следует объяснить изменениями окислительно-восстановительного потенциала индикатора, обусловленными избытком собственных ионов на поверхности частиц [59]. Типичными примерами являются ксиленовый синий VS и патентованный синий V, тщательно исследованные Богнаром и Надем [18]. Эти авторы использовали оба индикатора при титровании цинка по методу осаждения раствором гексацианоферрата(II) калия. Ясно, что титрант должен содержать некоторое количество гексацианоферрата (III) калия.
Группа окислительно-восстановительных индикаторов, изменяющихся под влиянием положительно заряженного иода, действует по этому механизму, осложненному дополнительной реакцией. Индикаторы этого типа предложены Богнаром и Елинеком [15], а также Сьеррой и Асенси [82]. Механизм действия таких индикаторов Сьерра и Асенси объяснили на примере нейтрального красного следующим образом:
+ I- + H+
i
Согласно этой схеме, протон кислотного красителя замещается ионом иода, имеющим положительный заряд. Для этого необходимо, чтобы иодид связывался в растворе путем превращения его в соль серебра. Следовательно, конечная точка титрования может проявиться только лишь в присутствии избытка серебра, который способствует диспропорционированию иода.
ПОВЕРХНОСТНЫЕ КОМПЛЕКСОМЕТРИЧЕСКИЕ ИНДИКАТОРЫ
В случае поверхностных комплекссметрических индикаторов изменяется константа устойчивости комплекса металла, адсорбированного на поверхности осадка [83, 85, 90, 91]. Сьерра и Асенси отмечали, что в некоторых случаях механизм действия индикатора очень сложен. Типичным примером применения индикаторов этой группы является определение меди(II) в присутствии серебра (83]. Сначала серебро оттитровывают раствором иодида, а затем
32
ГЛАВА VII
титруют этилендиаминтетрауксусной кислотой медь в присутствии комплекса медь — PAN [1-(2-пиридилазо)-2-нафтол] в качестве индикатора. Переход окраски этого индикатора на поверхности осажденного иодида серебра распознается очень легко.
ПОВЕРХНОСТНЫЕ ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
В настоящее время еще нельзя классифицировать в определенные группы поверхностные флуоресцентные индикаторы, которые изучал Богнар [12—14]. Однако исследования адсорбции, проведенные Богнаром, по-видимому, показывают, что эти индикаторы относятся к последней группе адсорбционных индикаторов.
ВЛИЯНИЕ АДСОРБЦИИ КРАСИТЕЛЯ НА ОСАДОК
В этом разделе говорится об индикаторах, которые не осаждаются на поверхности. Такие индикаторы характеризуются значительной адсорбцией красителя по обе стороны от точки эквива
Рис VII.11 Золь иодида серебра, скоагулированный нитратом аммония при избытке ионов серебра (увеличение в 12 000 раз)
(По данным Шулека, Пунгора и Tj6a [71].)
Рис VII 12 Золь иодида серебра, скоагулированный нитратом аммония при избытке ионов серебра. Осадок получен в присутствии п этоксихризоидина (увеличение в 14 000 раз).
(По да(.’!им Ш, пека. П>н:ора и Губа |Н] )
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
33
лентности. Рассмотрим 1несколько случаев влияния адсорбированного красителя
В первую очередь рассмотрим вопрос о повышении фоточувствительности как таковой [73]. Это явление, изученное детально в технической фотографии, наблюдается при адсорбции индикаторов и задерживает, а в некоторых случаях препятствует установлению конечной точки. В ходе наших исследований была изучена адсорбция ряда производных хризоидина. Оказалось, что п-этоксихризо-идин адсорбируется этоксигруппой. Известно, что при наличии неблокированного фенольного гидроксила галогенид серебра вступает в фотохимическую реакцию, которая идет с очень большой скоростью на рассеянном солнечном свету. Светочувствительностью обладает и n-этоксихризоидин, хотя и в небольшой степени. Ее можно уменьшить, применяя нитрат калия в качестве коагулирующего агента [84].
Адсорбированный краситель оказывает большое влияние на образование осадка [55, 76, 77]. Так, адсорбция красителя на образовавшемся зародыше кристалла мешает его росту (рис. VII.11 и VII. 12). При коагуляции нитратом аммония получаются различные по структуре осадки в зависимости от присутствия или отсутствия п-этоксихризоидина. Снимки, сделанные с помощью электронного микроскопа, показали, что в присутствии п-этоксихризо-идина осадок имеет очень тонкую структуру, а в отсутствие красителя структура осадка более грубая. Адсорбция красителя значительна (см. результаты, приведенные в табл. VII.7). Кроме того, адсорбция красителя не зависит от pH, а зависит только от природы присутствующих собственных ионов.
Таблица VII.7 АДСОРБЦИЯ п-ЭТОКСИХРИЗОИДИНА (ММОЛЬ НА 1 МОЛЬ Agl) НА ОСАЖДЕННОМ И0ДИДЕ СЕРЕБРА В МОМЕНТ ОБРАЗОВАНИЯ В ПРИСУТСТВИИ СОБСТВЕННЫХ ИОНОВ И ПРИ РАЗЛИЧНЫХ ЗНАЧЕНИЯХ pH (ПО ДАННЫМ ШУЛЕКА И ПУНГОРА [70])
pH Концентрация собственного иона при перетитровании, % Концентрация красителя, мкмоль • л—1
16,67 33,51 83,52 166.7
1 50% Ag 21,0 36,8 60,8 66,1
2 50% Ag 22,0 37,3 58,0 68,5
4,8 50% Ag 22,7 37,2
1,3 10% Ag 22,9 38,7 65,3 86,7
1.3 10% I 24,5 48,3 82,3 100,0
1,3 50% I 24,4 48,6 85,1 122,5
1,3 100% I 25,3 48,6 86,5 126,5
3-S18
34
ГЛАВА VII
Интересно заметить, что после того, как закончится осаждение, добавленный к раствору индикатор адсорбируется в очень незначительной степени. Это видно по результатам (см. табл. VIII.8), полученным при изучении адсорбции красителя предварительно осажденным иодидом серебра. По-видимому, это явление находится в согласии с наблюдением, сделанным в ходе электронномикроскопических исследований; краситель оказывает защитное действие на коагуляцию осадка.
Таблица VII.8
АДСОРБЦИЯ п-ЭТОКСИХРИЗОИДИНА (ММОЛЬ НА 1 МОЛЬ Agl) НА ПРЕДВАРИТЕЛЬНО ОСАЖДЕННОМ ИОДИДЕ СЕРЕБРА В ЗАВИСИМОСТИ ОТ ИЗБЫТКА СОБСТВЕННЫХ ИОНОВ ПРИ pH 1,3 (ПО ДАННЫМ ШУЛЕКА И ПУНГОРА (10])
Концентрация собственного иона при перетитровании, % Концентрация красителя, мкмоль - л—1
3,35 8,35 16,67 33,51
10% Ag 3,6 6,1
10% I 1,о 2,1 5,5
50% I 2,9 5,0 8,2
100% I 2,7 5,0 7,4 Н,4
Интересные исследования по люминесценции цианиновых красителей провели Базиньски и др. [6]. Они нашли, что 1,1-диметил-бензоксацианиниодид и 1,1-диметилбензотиацианиниодид адсорбируются на осадке иодида серебра и при УФ облучении сильно флуоресцируют. Затем свечение тушится иодидом серебра. Флуоресценция адсорбированных красителей очень избирательна, потому что осаждение бромида серебра в присутствии этих индикаторов приводит к флуоресценции различного цвета, а осадок хлорида серебра флуоресцирует совсем слабо.
ПРИМЕНЕНИЕ АДСОРБЦИОННЫХ ИНДИКАТОРОВ ОПРЕДЕЛЕНИЕ ХЛОРИДА
Из числа поверхностных осажденных индикаторов флуоресцеин [27, 41] можно применять при аргентометрическом определении хлорида в нейтральной водной среде. Обычно считают, что этот индикатор наиболее типичен для титрования хлоридов. Одно из производных флуоресцеина — дихлор (R) флуоресцеин — было предложено Кольтгофом [44] с указанием, что его надо применять при рН<4. Согласно данным Фаянса, хлориды можно также титровать в присутствии диметил (R) флуоресцеина и дихлор (Р)флуорес-
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
35
ценна [25], тогда как в присутствии других производных флуоресцеина нельзя определять хлориды в нейтральной водной среде.
Сьерра и Санчес-Педрено [88] показали в своих опытах, что определение хлоридов в присутствии производных флуоресцеина возможно, если раствор хлорида подкислить азотной кислотой до „pj J_2. Так как эти индикаторы являются слабыми кислотами,
то подкисление благоприятно отражается на образовании соли серебра на границе гетерогенной фазы; в результате повышения концентрации кислоты и последующего удаления анионов красителя образование серебряной соли происходит при более высокой концентрации серебра. Таким образом, можно гарантировать, что при аргентометрическом титровании хлорида не идет одновременно другая реакция, т. е. при незначительном избытке хлорида соль эозина с серебром не осаждается одновременно с хлоридом. Подобные свойства наблюдали Тандон и Меротра [99] у сульфофлуоресцеина и его галогенпроизводных при использовании их в титровании. Эти авторы показали, что если сила индикатора-кислоты повышается, то в случае титрования одного и того же галогенида значение pH титруемою раствора следует понизить.
Явление, обнаруженное испанскими исследователями при повышении концентрации кислоты, наблюдал также Богнар с сотр. [19—21] при понижении диэлектрической постоянной среды. В обоих случаях это влияние зависит от степени подавления диссоциации индикатора. Изучая многие производные флуоресцеина, Богнар с сотр. нашел, что эозин можно использовать как индикатор при определении хлоридов в этаноле или ацетоне при условии, что раствор подкислен уксусной кислотой. Кроме того, они показали, что хлорид можно определять, используя в качестве индикатора дииодфлуоресцеин в среде, содержащей ацетальдегид или диоксан. В водном растворе, содержащем 50% ацетона и 15% ледяной уксусной кислоты, хлорид легко оттитровывается в присутствии индикатора дихлор (Р)тетраиод (К) флуоресцеина. Наконец, Богнар показал, что хлорид можно определять даже в присутствии эритрозина, если проводить титрование в растворе диоксана, содержащем ледяную уксусную кислоту.
Возможность столь широкого применения производных флуоресцеина позволила испанским авторам [87] распространить теорию адсорбционных индикаторов, предложенную авторами настоящей главы, по аналогии с теорией Льюиса. Однако это не способствовало улучшению теории и не принесло облегчения в предвидение и интерпретацию рассмотренных выше явлений.
Сьерра с сотр. [84] предложил использовать в качестве индикатора при титровании хлоридов п-этоксихризоидин. В простой водной среде не удается установить конечную точку титрования, но если скоагулировать осадок нитратом калия, то наблюдается переход окраски. Это можно объяснить увеличением адсорбции 3*
36
ГЛАВА VII
ионов калия или нитрат-ионов, а также заменой одного иона на другой вследствие изменения знака заряда осадка при переходе через точку эквивалентности.
В области поверхностных окислительно-восстановительных индикаторов [80] Асенси [5], а также Сьерра и Асенси [81] предложили для определения хлоридов систему дифениламин+ванадат. Тогда аргентометрическое титрование хлорида проводится в сернокислой среде (примерно 0,5 М раствор) в присутствии нескольких капель 2%-ных растворов дифениламина и ванадата калия.
Из поверхностных флуоресцентных индикаторов некоторые были предложены Богнаром [12—14] для определения хлоридов, как, например, магдала красный, или лактат этоксидиаминоакридина (в присутствии декстрина); можно применять также трипановый красный в растворах, pH которых больше 1.
Из поверхностных комплексометрических индикаторов Сьерра Хименес и Санчес-Педрено [89] предложили для титрования хлоридов комплексы дифенилкарбазона или дифенилкарбазида со ртутью(И). К раствору, содержащему хлорид, добавляют немного дифенилкарбазона или дифенилкарбазида и несколько капель 0,005 М нитрата ртути (II). В конечной точке титрования комплекс хлорида с ртутью(II) разлагается от избытка нитрата серебра, и на поверхности осадка образуется новый окрашенный комплекс ртути [90].
Хлориды можно также определять меркурометрически [68, 105, 106]. Конечная точка легко наблюдается в кислой среде в присутствии индикатора эритрозина В [91]. Хлорид можно титровать в азотнокислой среде раствором нитрата ртути (I), используя в качестве индикатора дифенилкарбазид [89].
При аргентометрическом определении хлорид-ионов в качестве индикаторов пригодны также бриллиантовый орсейль С [7], феносафранин [10], дифениламиновый синий [45] и бромфеноловый синий [33]. Для определения хлорид-ионов в нейтральной среде были предложены [71, 49] в качестве адсорбционных индикаторов триазиновые соединения, ацетилтетрахлорбензойная кислота, производные резорцина и орсина. Для предотвращения коагуляции рекомендовалось добавление декстрина. При определении хлорида в области значений pH 2,8—3,3 предлагалось использовать [46] 4-(2-этилфенилазо)-1-нафтиламин солянокислый в качестве адсорбционного индикатора.
ОПРЕДЕЛЕНИЕ БРОМИДОВ
Те же индикаторы, которые рекомендуются для титрования хлоридов, пригодны для определения бромидов. В случае поверхностных индикаторов для титрования по методу осаждения не требуется изменять условия определения. Поскольку бромид се-
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
37
ебца растворяется труднее хлорида серебра, нет необходимости Увеличивать растворимость серебряной соли индикатора путем ^одкисления или уменьшения диэлектрической постоянной среды. Так по данным Фаянса [25], можно использовать флуоресцеин и его производные, имеющие четыре атома брома в кольце резорцина (эозин), для определения бромидов в слегка подкисленной водопроводной воде. К подходящим для этой цели производным флуоресцеина относятся диметил (R)дихлор (Р)-, дихлор (R)-, тетрахлор (Р)тетрабром(Е)-, дибром (R)- и дихлор (Р)тетрабром (R) флуоресцеины, а также сам флуоресцеин и тетрабром (R) флуоресцеин (эозин). Полигалогенпроизводные и другие производные флуоресцеина тоже можно применять как индикаторы, если понизить диэлектрическую постоянную среды [19] или понизить pH раствора [88].
Поверхностные окислительно-восстановительные, поверхностные флуоресцентные и поверхностные комплексометрические индикаторы, которые применяются в титровании хлоридов, в равной мере пригодны для определения бромидов. Метаниловый желтый [16, 52] используют в условиях, характерных для определения хлоридов: его поверхностные кислотно-основные индикаторные свойства проявляются при значениях pH <2.
В меркурометрическом определении бромидов можно использовать те же индикаторы, что и в случае титрования хлоридов [89, 91].
Индикатор и-диметиламинобензилиденроданин [92] используется при титровании бромидов нитратом ртути (I) >в среде примерно 0,1 М азотной кислоты. Так как при этом образуется комплекс ртути, чувствительный к свету, титрование надо проводить на рассеянном свету.
В последнее время для определения бромид-ионов были предложены в качестве адсорбционных индикаторов [71, 49] производные триазина, ацетилтетрахлорбензойной кислоты, резорцина и орсина в нейтральной среде, а 4-(2-этилфенилазо)-1-нафтиламингидро-хлорид в кислой среде.
ОПРЕДЕЛЕНИЕ ИОДИДОВ
Иодид дает с нитратом серебра в водной среде осадок, наименее растворимый по сравнению с другими галогенидами. Соли производных флуоресцеина с серебром обладают большей раствори-ью, чем иодид серебра. Поэтому в принципе все производные Флуоресцеина, перечисленные Фаянсом [25], пригодны для приме-2?Н45 70]В качестве индикаторов при определении иодида [7—9, поп^3 ПОвеРХНОСТНЬ1Х кислотно-основных индикаторов наиболее ллодящнм
для работы в нейтральной среде оказывается п-эток-
38
ГЛАВА VII
сихризоидин [72, 78]. Метаниловый желтый ['16, 50], действующий как поверхностный кислотно-основной индикатор, тоже можно использовать при титровании иодида в условиях рН<3,5; в нейтральной среде этот индикатор действует как поверхностный индикатор для титрования по методу осаждения.
Малые количества иодида легко определяются с индикатором феносафранином в присутствии нитрата калия [84] в качестве коагулирующего реагента.
В области поверхностных окислительно-восстановительных индикаторов заслуживает внимания определение иодида по методу Богнара и Елинека [15], а затем доработанному Сьеррой и Асен-си [81].
Богнар использовал в качестве индикаторов ксиленовый синий VS и патентованный синий V. Для выделения свободного иода к раствору иодида добавляются следовые количества йодата; если раствор достаточно кислый, чтобы могла пройти эта реакция, pH не имеет решающего значения. Краситель адсорбируется на осажденном иодиде серебра, а при прохождении точки эквивалентности прочно адсорбируются также ионы серебра. На поверхности частиц ионы серебра способствуют диспропорционированию иода путем отрыва иодид-ионов, так что образуются ионы 1+ или гидроокись иода и окисляется адсорбированный краситель. Сьерра и Асенси [80] использовали подобным образом п-этоксихризоидин в 0,05 М серной кислоте и нашли, что этот метод пригоден для определения иодида в присутствии хлорида.
Если в исследуемом растворе иодида содержался бромид, эти авторы работали со смесью ванадата и n-этоксихризоидина; при этом они рекомендовали, чтобы содержание хлорида или бромида в растворе было бы того же порядка, что и определяемого иодида. В присутствии следов иода индикаторами для определения иодида могут быть нейтральный красный [82] в 0,5 М кислоте или дифениламин [81] в 0,5 М серной кислоте. Если присутствуют также хлорид или бромид, рекомендуется применять ванадат в сильнокислой среде и дифениламин [81]. Также применяют лей-косоединение малахитового зеленого вместе с иодом или ванадатом, причем иодид можно определять в азотнокислом растворе.
Для определения иодида в качестве поверхностных флуоресцентных индикаторов находят применение магдала красный [12] и лактат этоксидиаминоакридина. По существу, все перечисленные Богнаром поверхностные флуоресцентные индикаторы позволяют определить конечную точку при аргентометрическом титровании роданида или любого галогенида [13].
Сьерра Хименес и Санчес-Педрено [89] приводят пример установления конечной точки при титровании иодида с помощью поверхностного комплексометрического индикатора. Предложенный ими метод подобен методу определения хлорида, а именно титру
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
39
ют иодид ИЛИ смесь галогенидов в присутствии хлорида ртути(II) дифенилкарбазона или дифенилкарбазида. Под влиянием избытка серебра в конечной точке титрования происходит превращение комплекса галогенида со ртутью(II) в окрашенный комплекс красителя.
ОПРЕДЕЛЕНИЕ РОДАНИДА
Для определения роданидов можно использовать производные Флуоресцеина в качестве поверхностных индикаторов для титрования по методу осаждения [27, 41] в соответствии с принципами, описанными в случае определения галогенидов. Установлено, что в списке индикаторов, составленном Фаянсом [25], все индикаторы, перечисленные до эозина, включая сам эозин, пригодны для установления конечной точки титрования.
Предложенные Богнаром поверхностные флуоресцентные индикаторы [12—14] с успехом могут быть использованы и в этом случае.
ОПРЕДЕЛЕНИЕ СЕРЕБРА
Аргентометрические методы титрования, рассмотренные выше, могут быть применимы для определения ионов серебра, если титровать раствором, содержащим роданид- или галогенид-ионы. По методу Берри и Дюрранта [10] можно с достаточной точностью определять серебро даже в присутствии свинца и кадмия, если проводить титрование в среде 0,5 М азотной кислоты, используя тартразин в качестве индикатора.
Надежным индикатором в титровании серебра является п-эток-сихризоидин [16]. В светло-розовом растворе, не изменяющем свою окраску до самой конечной точки, поверхность осадка имеет желтый оттенок. При избытке иодида индикатор быстро превращается в фиолетово-красный. Такого типа установление конечной точки с успехом можно использовать при проведении обратного титрования в нейтральной среде.
ОПРЕДЕЛЕНИЕ ЦИНКА [18, 96]
Богнар [18] титровал цинк раствором гексацианоферрата(II) калия в присутствии небольшого количества гексацианоферра-та(Ш), используя в качестве индикатора ксиленовый синий VS ИЛИ патентованный синий V. Резкое изменение окраски в конечной точке титрования цинка обусловливается наличием избытка гек-сацианоферрата(П) калия. Однако вблизи конечной точки рекомендуется вести титрование очень медленно, так как слишком Острое титрование дает ошибочные результаты. Указанные инди
40
ГЛАВА VII
каторы действуют как поверхностные окислительно-восстановительные индикаторы.
В области адсорбционных индикаторов сделано значительное улучшение тем, что разработан новый метод определения цинка [93, 94] Сьеррой Хименес и Санчесом-Педрено, которые назвали этот метод «гексацианоферратометрией». Эти авторы использовали кислотно-основные индикаторы, такие, как, например, конго красный и n-этоксихризоидин, и поверхностные окислительно-восстановительные индикаторы, такие, как о-фенетидин. Опыты, проведенные с этими индикаторами, подтвердили ожидаемые результаты. Дальнейшие исследования позволили распространить этот метод на определение свинца.
ОПРЕДЕЛЕНИЕ СУЛЬФАТА
Для титрования сульфат-ионов раствором соли бария Цомбори 117] предложил использовать в качестве поверхностного индикатора для титрования по методу осаждения ализаринсульфонат натрия. Однако этот метод сопряжен с трудностями из-за быстрого разложения раствора индикатора.
ОПРЕДЕЛЕНИЕ РТУТИ
Для определения ртути (I) раствором хлорида или бромида Цомбори [‘117] и Кольтгоф [43] предложили использовать в качестве индикатора бромфеноловый синий, а Сьерра и Монтанер [96] разработали метод титрования ртути бромидом в присутствии и-диметиламинобензилиденроданина. Те же авторы предложили [91] применять эритрозин В в качестве поверхностного индикатора для титрования по методу осаждения при определении ртути(I) раствором хлорида или бромида. Для этой же цели подходит ди-фенилкарбазид.
ПРИМЕНЕНИЕ АДСОРБЦИОННЫХ ИНДИКАТОРОВ В ДРУГИХ ОБЛАСТЯХ АНАЛИТИЧЕСКОЙ ХИМИИ
Исследования Пунгора и Шулека [75] показали, что краситель п-этоксихризоидин, адсорбированный на осадке иодида серебра, может быть использован как кислотно-основной индикатор. Интервал перехода окраски этого индикатора близок к интервалу перехода метилового красного, если на поверхности осадка имеется избыток ионов серебра; при избытке иодид-ионов интервал перехода приближается к интервалу перехода фенолфталеина. Применяя соответствующим образом приготовленные адсорбаты индикаторов, можно оттитровывать кислоты различной силы.
АДСОРБЦИОННЫЕ ИНДИКАТОРЫ
41
Сьерра и Асенси ['83] при комплексометрическом определении меди использовали в качестве индикатора комплекс меди(II) с 1-(1-пиридилазо) -2-нафтолом (CuPAN), адсорбированный на поверхности осажденного иодида серебра. По этому методу можно определять серебро и медь при их одновременном присутствии. В кислой среде раствором иодида оттитровывают сначала ионы серебра, затем продолжают титрование комплексоном для определения содержания меди.
СПИСОК ЛИТЕРАТУРЫ
I. Akiyama Т., J. Pharm. Soc. Japan, 55, 224 (1935).
2. Akiyama T., Mine У., J. Pharm. Soc. Japan, 55, 226 (1935).
3. Akiyama T., Yabe S., J. Pharm. Soc. Japan, 55, 22 (1935).
4. Akiyama T., Yabe S., J. Pharm. Soc. Japan, 55, 23 (1935).
5 Asensi Mora G., Dissertation, Publicaciones de la Universidad de Murcia 1957.
6. Basinski A., Kiciak K., Taianta, 11, 1459 (1964).
7. Belladen L., Piazza G., Ann. chim. appl., 22, 631 (1932).
8. Berry A. J., Analyst, 57, 511 (1932).
9. Berry A. J., Analyst, 61, 315 (1936).
10. Berry A. J., Durrant P. J., Analyst, 55, 613 (1930).
11. Bobranski B., Z. anal. Chem., 99, 108 (1934).
12. Bognar J., Acta Chim. Hung., 19, 433 (1959).
13. Bognar J., Acta Chim. Hung., 20, 103 (1959).
14. Bognar J., Acta Chim. Hung., 20, 193 (1959).
15. Bognar J., Jellinek O., Acta Chim. Hung., 10, 125 (1956).
16. Bognar J., Murguly K., Magy. Kern. Folyoirat, 60, 45 (1954).
17. Bognar J., Nagy L., Acta Chim. Hung., 10, 259 (1956).
18. Bognar J., Nagy L., Acta Chim. Hung., 16, J (1958).
19. Bognar J., Sarosi Sz., Acta Chim. Hung., 7, 361 (1955).
20. Bognar J., Vereskoi J., Acta Chim. Hung., 5, 91 (1954).
21. Bognar J., Vereskoi J., Acta Chim. Hung., 5, 105 (1954).
22. Burstein R., Z. anorg. allg. Chem.. 164, 219 (1927).
23. Candea C., Murgulescu I. G., Ann. chim. anal. chim. appl., 3, 33 (1936).
24. Chirnuaga E., Z. anal. Chem., 101, 31 (1935).
25. Fajans K., «Adsorptionsindikatoren fur Fallungstitrationen», Die chemische Analyse, Vol. XXXIII, F. Enke, Stuttgart, 1937.
26. Fajans K, Hassel O., Z. Elektrochem. 29, 495 (1923).
27. Fajans K., Wolff»H., Z. anorg. allg. Chem., 137, 221 (1923).
28. Fearon W. R., Gillespie W. A., Biochem. J., 28, 1629 (1934).
29. Fleck H. R„ Holnes R. F. G., Ward A. M„ Analyst, 60, 32 (1935).
30. Hassel O., Koll. Z„ 34, 304 (1924).
31. Henkel H., Z. anal. Chem., 119, 326 (1940).
32. Hodakow J., Z. Phys. Chem., 127, 43 (1927).
33. Hok W., Svensk Farmaceutisk Tidskrift, 34, 121 (1930).
34. Hok W., Svensk Farmaceutisk Tidskrift, 34, 141 (1930).
35. Holscher F., Z. anal. Chem., 96, 308 (1934).
36. Kocsis E. A., Z. anorg. allg. Chem., 221, 318 (1935).
37- Kolthoff I. M., Z. anal. Chem., 70, 95 (1927).
3». Kolthoff I. M., Z. anal. Chem., 71, 235 (1927).
39- Kolthoff I. M., Koll. Z., 68, 190 (1934).
Kolthoff I. M„ Chem. Rev., 16, 87 (1935).
42
ГЛАВА VII
41. Kolthoff I. M., Von Berk L. M„ Z. anal. Chem., 70, 369 (1927).
42. Kolthoff I. M., Von Fischer W., Rosenblum Ch., J. Am. Chem. Soc., 56, 832 (1934).
43. Kolthoff I. M., Larson W. D., J. Am. Chem. Soc., 56, 1881 (1934).
44. Kolthoff I. M., Lauer W. M., Sunde C. J., J. Am. Chem. Soc., 51, 3273 (1929).
45. Lang R., Messinger J., Ber., 63, 1429 (1930).
46. Legradi L., Acta Chim. Hung., 42, 107 (1964).
47. Mehrotra R. C., D. Phil. Thesis, Univ, of Allahabad, 1948.
48. Mehrotra R. C., Tandon K- N., Taianta, 11, 1093 (1964).
49. Mushran S. P., Sanyal P., Chim. anal., 48, 160 (1966).
50. Pieters FL A. L, Chem. Weekbl., 26, 6 (1929).
51. Pungor E., Acta Chim. Hung., 12, 265 (1957).
52. Pungor E., Hollds-Rokosinyi E., Z. anal. Chem., 156, 161 (1957).
53. Pungor E., Hollds-Rokosinyi E., Acta Chim. Hung., 22, 69 (1960).
54. Pungor E., Hollds-Rokosinyi E., Annales Univ. Eotvos Budapest, Sect. Chim., 2, 17 (1960).
55. Pungor E., Hollds-Rokosinyi E., Acta Chim. Hung., 27, 63 (1961).
56. Pungor E., Konkoly Thege /„Acta Chim. Hung., 17, 113 (1958).
57. Pungor E., Konkoly Thege L, Annales Univ. Eotvos Budapest, Sect. Chim., 2, 31 (1960).
58. Pungor E„ Konkoly Thege I., Taianta, 10, 1211 (1963).
59. Pungor E., Schulek E., Z. anal. Chem., 150, 166 (1956).
60. Pungor E., Schulek E., Annales Univ. Eotvos Budapest, Sect. Chim., 1, 109 (1959).
61. Pungor E., Schulek E., Annales Univ. Eotvos Budapest, Sect. Chim., 2, 13 (1960).
62. Райхинштейн P., Коробов H., Ж0Х, 2, 661 (1932).
63. Райхинштейн P., Коробов M., Ж- прикл. хим., 8, 154 (1935).
64. Ricci J. E., Ind. Eng. Chem. Anal. Ed., 8, 130 (1936).
65. Ripan R., Z. anal. Chem., 94, 335 (1933).
66. Ripan-Tilici R., Z. anal. Chem., 102, 32 (1935).
67. Ripan-Tilici R., Z. anal. Chem., 104, 16 (1936).
68. Roberts L., Ind. Eng. Chem. Anal. Ed., 8, 365 (1937).
69. Roy S. N., J. Indian Chem. Soc., 12, 584 (1931).
70. Руденко H. П., Ж- физ. хим., 62, 505 (1930).
71. Sanyal P., Mushran S. P., Chim. Anal., 46, 391 (1964).
72. Schulek E., Pungor E., Anal. Chim. Acta, 4, 109 (1950).
73. Schulek E„ Pungor E., Anal. Chim. Acta, 4, 213 (1950).
74. Schulek E., Pungor E., Anal. Chim. Acta, 5, 422 (1951).
75. Schulek E., Pungor E., Anal. Chim. Acta, 7, 446 (1952).
76. Schulek E., Pungor E., Anal. Chim. Acta, 7, 243 (1952).
77. Schulek E., Pungor E., Guba F., Anal. Chim. Acta, 8, 261 (1953).
78. Schulek E., Rozsa P., Z. anal. Chem., 115, 195 (1938). t
79. Schulek E., Somogyi Z., Z. anal. Chem., 115, 185 (1938).
80. Sierra F., Asensi G., Anales real soc. esp. fis. у quim., B, 53, 625 (1957).
81. Sierra F., Asensi G., Anales real soc. esp. fis. у quim., B, 54, 13 (1958).
82. Sierra F., Asensi G., Anales real soc. esp. fis. у quim., B, 55, 377 (1959).
83. Sierra F„ Asensi G„ Anales real soc. esp. fis. у quim., B, 55, 797 (1959).
84. Sierra F., Asensi G., Sanchez-Pedreno C., Anales real. soc. esp. fis. у quim., B, 53, 607 (1957).
85. Sierra F., Asensi G., Sanchez-Pedreno C., Anales real soc. esp. fis. у quim., B, 55, 365 (1959).
86. Sierra Jimenez F., Burriel-Marti F., Anales real. soc. esp. fis. у quim., B, 52, 459 (1956).
87. Sierra F., Sanchez-Pedreno C., Informacion de quimica analitica, 91.
88. Sierra F., Sanchez-Pedreno C., Anales real. soc. esp. fis. у quim., B, 53, 429 (1957).
адсорбционные индикаторы
43
Ъ'егга Jimenez F., Sanchez-Pedreno С., Anales real. soc. fis. у quim., В, 89 54 541 (1958).
Sierra F., Montaner L., Anales real soc. esp. fis. у quim., B, 54, 745 (1958).
Qi Sierra F.’, Montaner L., Anales real soc. esp. fis. у quim., B, 55, 321 (1959).
Q9 Sierra F.’, Montaner L., Anales real soc. esp. fis. у quim., B, 55, 571 (1959).
03 Sierra f', Sanchez-Pedreno C., Inform, quim. anal., 18, 129, 194 (1964).
or Sierra F., Sanchez-Pedreno C., Inform, quim. anal., 19, 1 (1965).
05 Strebinger R-, Zombory L., Z. anal. Chem., 79, 1 (1929); 105, 346 (1936).
Q6 Tananaiev E, Georgobiani M. J., Z. anal. Chem., 107, 92 (1937).
07 Tandon K. N., Mehrotra R. C Anal. Chim. Acta, 27, 15 (1962).
08 Tandon K. N-> Mehrotra R. C., Anal Chim. Acta, 27, 97 (1962).
09 Tandon K- N-, Mehrotra R. C., Anal. Chim. Acta, 27, 198 (1962).
100 Tandon K. N., Mehrotra R. C., Anal. Chim. Acta, 30, 407 (1964).
[01 Tomicek O., Chemical Indicators, Butterworths, London, 1951.
[02 Tomicek O., Casopis Ceskoslov, Lekarnictva, 5, 1 (1926).
103 Tomicek O., Casopis Ceskoslov. Lekarnictva, 5, 15 (1926).
104. Tomicek O., Coll. Czech. Chem. Comms., 3, 116 (1931).
105. Trtilek J-, Chem. Obzor., 8, 3 (1933).
106. Trtilek J., Z. anal. Chem., Ill, 10 (1937).
107. Uzumasa Y., Miyake Y., J. Chem. Soc. Japan, 53, 904 (1932).
108. Uzumasa R., Miyake Y., J. Chem. Soc. Japan, 54, 624 (1933).
199 Uzumasa Y., Miyake Y., J. Chem. Soc. Japan, 54, 1043 (1933).
110 Uzumasa Y., Miyake Y., J. Chem. Soc. Japan, 55, 627 (1934).
111. Verwey E. J. W., Koll. Z„ 72, 187 (1935).
112. Wellings A. W., Trans. Faraday Soc., 28, 561 (1932).
113. Wellings A. W., Trans. Faraday Soc., 28, 565 (1932).
114. Wellings A. W., Trans. Faraday Soc., 28, 665 (1932).
115. Wellings A. W., Analyst, 58, 331 (1933).
116. Wellings A. W„ Analyst, 60, 316 (1935).
117. Zombory L., Z. anorg. allg. Chem., 184, 237 (1929).
118. Zombory L., Pollak L., Z. anorg. allg. Chem., 215, 255 (1933).
ГЛАВА VIII
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
А. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
ИНДИКАТОРЫ, ИМЕЮЩИЕ ПОТЕНЦИАЛ Е'о <0,76 В
Дж. Оттоуэй
1. ВВЕДЕНИЕ
Многие индикаторы этого класса хорошо известны и довольно подробно изучены. Однако применение их в титриметрии ограниченно по сравнению с другими используемыми индикаторами. Область рассматриваемых формальных потенциалов такова, что эти индикаторы применяются только с сильными восстановителями, такими, как хром(II), ванадий (II), титан (III), олово (II) и аскорбиновая кислота, растворы которых неустойчивы в присутствии кислорода. Титрование этими восстановителями надо проводить в отсутствие кислорода, что сопряжено со многими трудностями. По этой причине такие методы стараются по возможности не использовать. Например, хотя железо(III) можно непосредственно титровать ванадием (II), хромом (II) или титаном (Ш) , чаще восстанавливают его до железа (II) и затем оттитровывают сильным окислителем, например церием (IV) или марганцем (VII). Были предложены многие методы, в которых используются индикаторы, имеющие формальный потенциал ниже 0,76 В; некоторые из них будут описаны. Эти индикаторы находят применение в биологических исследованиях для измерения окислительно-восстановительных потенциалов в конкретных системах. Кларк с сотр. [1—19] предпринял первое систематическое исследование в этой области, получив большое число окислительно-восстановительных индикаторов, потенциалы которых находились в области потенциалов биологических систем. В настоящее время имеются индикаторы, почти полностью принадлежащие этой области потенциалов.
Обзоры по этому вопросу сделаны Кольтгофом и Стенгером [20], Томичеком [21], Певцовым [22—24] и Баньяи [25]. Наиболее значительные работы, опубликованные после этих обзоров, касаются открытия новых соединений, пригодных для использования их в качестве индикаторов (например, производные вариами-нового синего), а также расширения области применения новых и старых индикаторов. В результате этого более широко стали использоваться сильные восстановители, такие, как ванадий (II),
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
45
ом(П) и аскорбиновая кислота. Однако внимание, по-видимо-XPv было направлено на индикаторы более доступные, чем те, ко-ооые обладают лучшими свойствами в отношении растворимости и устойчивости. Например, нейтральный красный, феносафранин, метиленовый голубой и 2,6-дихлориндофенол широко применяются
в качестве индикаторов, несмотря на некоторые их ограничения.
Кольтгоф и Стенгер [20] предложили сделать систематический обзор по индикаторам в этой области. До сих пор такая работа не была осуществлена. В прекрасной книге Кларка «Окислительно-восстановительные потенциалы органических систем» [26] содержится масса полезных сведений в виде таблиц. Эта монография очень ценна для исследователя, работающего в данной области, но она не предназначена быть источником информации для химика-аналитика. Эта глава предполагает связать проблемы, описанные в книге Кларка [26], с задачами аналитика. Однако предло
жение Кольтгофа и Стенгера остается в силе и в настоящее время,
поскольку существует недостаток в информации, необходимой химику-аналитику. В качестве примера ограниченности представляемых данных можно привести работы, в которых дано большое число индикаторов, применяемых в кислых растворах при рН~0. Однако для этой области pH дается очень немного потенциометрических данных; большинство их относится к нейтральной области значений pH. В некоторых случаях это объясняется неустойчивостью соединений в кислом растворе, но чаще причиной является недостаток экспериментальных исследований. Основная часть приведенных формальных потенциалов для более высоких значений pH получена путем экстраполяции данных. Такое положение со-
здалось потому, что толчком для исследования окислительно-восстановительных потенциалов послужило применение их в биологических системах, где область нейтральных значений pH имеет большое значение. Поэтому необходимо располагать потенциометрическими сведениями о сильнокислых растворах многих веществ, особенно таких, которые наиболее часто используются как индикаторы.
В этой главе индикаторы классифицированы по их химической природе и каждый класс рассматривается отдельно. Первый раздел «Вопросы теории» не охватывает полностью теорию окислительно-восстановительных индикаторов, которая рассматривается в другом месте (см. гл. II и VIIIB). И все же этот раздел необходим для введения специальной терминологии, используемой в последующих разделах этой главы. Кроме того, нужно разобраться в вопросе, который имеет огромное и особое значение для рассматриваемых индикаторов, касающемся взаимосвязи формального потенциала индикатора и pH раствора.
В дальнейшем рассмотрении отдельных индикаторов (в зависимости от их принадлежности к различным классам химических
46
ГЛАВА VIII
соединений) имеется необычный класс, о включении которого надо, может быть, дать дополнительное объяснение. Вариаминовый синий и его производные фактически являются производными дифениламина. Почти все дифениламины имеют формальный потенциал выше 0,76 В и рассматриваются в гл. VIПБ, тогда как формальные потенциалы наиболее важных производных вариаминового синего ниже 0,76 В. Поэтому решено включить производные вариаминового синего в виде отдельного раздела в эту главу, несмотря на некоторую некорректность такого подхода.
2. ВОПРОСЫ ТЕОРИИ
Прежде чем приступить к рассмотрению отдельных классов индикаторов, следует вкратце объяснить смысл применяемых терминов и дать основы теории для вывода уравнений, характеризующих индикатор; эти уравнения будут использованы и в последующих разделах.
Рассматриваемые классы индикаторов почти все являются органическими красителями, участвующими в обратимых окислительно-восстановительных реакциях. Если представить половину реакции окисления-восстановления в виде
Ох-\-пе < > Red,
где Ох — окисленные частицы, Red — восстановленные частицы, а п — число перенесенных электронов е, то электродный потенциал окислительно-восстановительной пары дается уравнением Нернста
EhF=E°+^-ln-^, (1)
где tzox и tired — активности окисленной и восстановленной форм соответственно, Т — абсолютная температура, R и F имеют обычное значение, Ей — потенциал, отнесенный к нормальному водородному электроду, а Е° — константа, характерная для пары окислитель— восстановитель. Е° соответствует значению Еъ при активностях окисленной и восстановленной форм, равных единице, и называется стандартным потенциалом пары окислитель — восстановитель.
ФОРМАЛЬНЫЕ ПОТЕНЦИАЛЫ
Если химик-аналитик располагает довольно ограниченными сведениями о коэффициентах активности растворов, с которыми он имеет дело, то приведенное выше уравнение почти неприменимо. Однако активность каждого иона можно заменить произведением
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 47
оэффициента активности и концентрации. Например, активность окисленных частиц можно представить как
fox [Ох] ,
тогда уравнение (I) можно переписать в следующем виде:
Е—Е° I RT In ?ох 4- RT In (2}
t nF In fred + nF m [Red] (Z)
Если объединить первые два члена в правой части уравнения, то можно написать
Eh=E 4 In (3)
Ch nF 1П [Red] ’ W
где g0—значение потенциала при молярных концентрациях окисленной и восстановленной частиц, равных единице, и называется нормальным потенциалом пары окислитель — восстановитель. Если [Ox]=[Red] = l, уравнение (3) становится
Еъ=Е0
Таким образом получаем приведенное выше определение Ео. Однако нормальный потенциал Ео может изменяться в зависимости от коэффициентов активности и, значит, от ионной силы раствора, а следовательно, от природы и концентрации других электролитов, присутствующих в растворе.
Следует отметить, что даже если
[Ох] = [Red] ф 1, то Eh = Ео,
но это не дает то же самое значение Ео, как в том случае, когда [Ох] = [Red] = 1, так как в уравнение для Ео входят коэффициенты активности:
Е.=£" + -^ m-fe- (4>
Здесь коэффициенты активности будут, несомненно, изменяться при варьировании концентраций. Однако в этом случае любое изменение Ео будет небольшим просто потому, что мы использовали отношение коэффициентов активности, а изменения, вызванные присутствием различных электролитов и изменением их концентрации, могут быть значительны, 1как это наблюдается для иона водорода (см. ниже).
Для учета всех изменений используют величину Ео, называемую формальным потенциалом. Это потенциал раствора, содержащего в Равных концентрациях (не обязательно молярных) окислитель и восстановитель, при точно определенных условиях в отношении Других электролитов. Значит, для любой окислительно-восстановительной системы индикатора надо знать формальный потенциал
48
ГЛАВА VIII
при различных условиях эксперимента. Для многих неорганических окислительно-восстановительных систем такие данные имеются [27], но для органических систем, рассматриваемых в этой главе, таких сведений, к сожалению, нет и единственным источником служит работа Кларка [26]. В тех случаях, когда в последующих разделах этой главы даны значения формальных потенциалов, необходимо будет указать на условия, при которых проводились измерения, а также привести данные о концентрации использованного индикатора. Все эти сведения будут по возможности приводиться в соответствующих местах.
ЗАВИСИМОСТЬ £о ОТ ЗНАЧЕНИЯ pH
Наиболее важным случаем применения формальных потенциалов, который будет рассмотрен здесь, является изменение Е'о в зависимости от значения pH раствора. Оба потенциала Ео и Ео относятся к рН=0, т. е. к активности иона водорода, равной 1. Для значений Ео при других значениях pH вводится обозначение Ет, впервые предложенное Михаэлисом; чтобы показать, при каком значении pH проводились измерения, в верхней части буквы Е пишут соответствующее число. Например, Ет соответствует значению формального потенциала при pH 7. Обозначение Е'о всегда относится к рН = 0.
Очень важно помнить о различии между Е’о и Ет при рассмотрении окислительно-восстановительных индикаторов, так как их формальные потенциалы зависят от pH раствора. Соотношение между Ет и pH многих индикаторов исследовал Кларк с сотр. [1—19, 26]. Для наглядности приведем один пример из их работы.
СИСТЕМА 2,6-ДИХЛОРИНДОФЕНОЛА
Реакцию восстановления 2,6-дихлориндофенола можно представить следующим образом:
С1
ОН2Н++ 2е:
окисленные частицы
С1
он—
Н
(5)
С1 восстановленные частицы
>-ОК
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
4S,
Ох + 2Н+ + 2е <—> H2Red (6)
При диссоциации всех фенольных групп и отщеплении ионов водорода имеем следующую реакцию:
Ох- + 2е + Н+ > Red2- (7)
Потенциал этой системы, согласно уравнению Нернста,
Еъ=Е'о
RT . [Ox ] [Н+] 2F [Red2-]
(8)
Это уравнение будет справедливо до тех пор, пока единственными частицами в условиях эксперимента будут Ох- и Red2-. Пренебрегая влиянием основности атома азота (справедливость этого станет очевидной позже), все же нужно учитывать диссоциацию обеих гидроксильных групп восстановленных частиц и гидроксильной группы окисленных частиц, которые следует принимать во внимание. Они определяются уравнениями
H2Red
HRed- + Н+,
HRed" Red2--f-H+,
HOx <—» Ox- + H<-
Константы диссоциации этих реакций будут к _[HRed"] [Н+] ri [H2Red] ’
к _ [Red2-] [Н+] гг [HRed-] ’
К __ [Ох~][Н+] [НОх]
(9)
(Ю)
(И)
где Г] соответствует первой диссоциации восстановителя и т. д. Следовательно, получаются три восстановленные частицы, и если Сг — общая концентрация всех таких частиц, то
Cr=[H2Red] + [HRed-] + [Red2-]
Подставляя вместо [HaRed] и [HRed-] выражения из уравнений (9) и (10), получаем
[Red2-]= [H+]2 + KrJH+]+КГ1КГ2 (12)
получаются также две окисленные частицы, и, если Со —общая концентрация окисленных частиц, тогда
Со=[НОх] + [Ох-]
4- 918
50
ГЛАВА VIII
И
[Ox^=wfer (13)
Подставляя вместо [Red2-] и [Ох-] выражения из уравнения (8), получаем
Г? , RT 1 С° _j 1 *o^H+P + KrJH^+tfriKr2[H+]) Eh— Ео+ 2F In Cr + 2F In ([Н+] + КО1)КГ1КГ2
и поэтому
P P' , RT 1 C° -£h=£0 + -2y- ln-^— +
, RT [H+P + K [Н+Р + КГ1КГ2[Н+]
+ T^ta---------— <14)
где
RT Ko,
E;==Eo + —ln-^- (15)
Если поддерживать постоянной [H+], то последний член в уравнении (14) тоже постоянная величина при постоянной температуре. При комбинации с Ео получается
Eh=Em+-g^ln-£-
Кроме того, если С0/Сг = 1, то Еъ=Ет. Тогда уравнение (14) становится
m—2f m [Н+] + КО1
В этом уравнении показана взаимосвязь Ет и pH, так как все другие члены его — постоянные величины. Оно справедливо для 2,6-дихлориндофенола, а также для любого другого индикатора реакции следующего типа:
Ox- -f- Н+ + 2е < » Red2-
Сюда относятся многие индофенолы, используемые в качестве индикаторов.
В случае 2,6-дихлориндофенола постоянными величинами в уравнениях (14) и (16) являются Еб=0,668 В, КО1=2-10-6, КГ1 = = 10—7 и Кг2=7,4- 10-11. Подставляя эти величины в уравнение (16), можно построить кривую, описывающую зависимость Ет от pH (это сплошная линия на рис. VIII.1). Там же представлен ряд значений Ет, определенных Гиббсом, Коеном и Каннаном [7]; эти значения хорошо совпадают со значениями, вычисленными по уравнению (16), подтверждая тем самым справедливость исход
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
51
ных предположений об ионизации окисленных и восстановленных чястиц*
Из рис. VIII. 1 видна также строгая зависимость значений £т типичной индикаторной системы от значения pH. Очевидна важность этого при выборе индикатора для титрования, не идущего
Рис. VIII.1. Зависимость Ет от ipH для 2,6-дихлориндофенола: кривая, построенная иа основе уравнения (16) с использованием постоянных величин (см. текст); 0—0 кривая, построенная по данным работы [7].
при pH 0, которому соответствует Е'о. Почти все индикаторы, которые будут рассмотрены, проявляют подобную строгую зависимость Ет от значения pH.
Однако надо рассмотреть и другие факторы. Например, крайние участки сплошной линии на рис. VIII. 1 построены в результате теоретических расчетов и не получили экспериментального подтверждения, так как 2,6-дихлориндофенол нерастворим в кислом растворе и неустойчив в сильнощелочном растворе.
На кривой Ет — pH (рис. VIII. 1) имеются два линейных участ-Ка при рН<5 и рН>11,5. В первом случае при рН<5 [Н+]з>кГ1[Н^»ЛГ1ЛГ2[Н+] и [Н+]> £01,
4*
52
ГЛАВА VIII
и поэтому уравнение (16) упрощается до
Дп=Д; + -^-1п[Н+] (17)
При постоянных величинах и 7=298 К уравнение (17) становится
Дт =0,668—0,06рН
Тогда тангенс угла наклона линейной части при pH<5 будет
-fe-= -0,06 (18)
арН v 7
Если рН> 11,5,
[Н+] > КТ1 [Н+р» [Н+]« и кО1 > [Н+]
и уравнение (16) упрощается до
Ьт=Ьо + 2F 1П---------- (19)
Откуда
—0,03 (20)
dpH ’ v ’
Для значений pH 8—9 тоже имеется короткий линейный участок. Тогда ДГ[Н+]2 является самым большим членом в числителе второго члена уравнения (16), которое поэтому упрощается до
, ВТ , Л'г, [Н+]2
Дт=^ + -2Г In-^2- (21)
и
-^- =—0,06 (22)
арН ' '
Очевидно, что если кривая Дт— pH проходит через pH, равное рДГ2 (или рДГ1), тангенс угла наклона, определяемый —dEmfdpH, уменьшится на 30 мВ (при 30 °C) в зависимости от увеличения pH. Подобным образом при прохождении через pH, равное рД0, тангенс угла наклона увеличивается на 30 мВ.
В точке пересечения прямых линий, описанных уравнениями (19) и (21), Дт и pH имеют одинаковые значения. Сочетание уравнений (19) и (21) для точки пересечения приводит к равенству рН=рДГ2
Следовательно, измерением значений Дт в широкой области значений pH можно довольно просто определить значения рД многих обратимых окислительно-восстановительных систем; этот метод использовал Кларк с сотр. [1—17]. В приведенном примере изменения тангенса угла наклона, обусловленные рДо и рДГ1, близки
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
53
их трудно объяснить, исходя из кривой. В этом случае можно Иппеделить константы диссоциации либо спектрофотометрически, либо если в области значений pH 5—8 получено достаточно много точек на кривой Ет—pH, можно проводить вычисления с некоторыми предполагаемыми значениями р/С0 и р/СГ1 до тех пор, пока расчетная кривая не совпадет с экспериментальной кривой. В любом случае нужно знать величину р/С, чтобы правильно применять 2 6-дихлориндофенол или любую другую подобную окислительно-восстановительную систему в качестве индикатора. Например, если известны значения Ео, p/G,, p/G2. рЛо, то можно вычислить Ет для любого значения pH, при котором придется проводить титрование.
СИСТЕМА ВАРИАМИНОВОГО СИНЕГО
Второй пример взаимосвязи Ет — pH приводит Эрдей с сотр. [28], который изучал индикатор вариаминовый синий и его производные. Окислительно-восстановительную реакцию этих соединений можно проиллюстрировать на реакции самого вариаминового синего
HN=<^ \=N—^~^-ОСН3 + 2Н+ + 2е окисленные частицы
(23)
Оба вида частиц подвергаются двухступенчатой диссоциации
RedHi+ =«=> RedH++H+, [RedH+] [Н+] [RedHf+] ’
RedH+ =«=> Red + Н+, [Red] [Н+] [RedH+]
ОхН1+ ОхН+4-Н+, [ОхН+] [Н+] [Ох^+] ’
ОхН+ Ох + Н+, [Ox] [Н+] [ОхН+]
Если принять во внимание только полностью ионизованные формы, то реакция имеет следующий вид:
ОхН|+ + 2Н+ + 2е <—» RedHfb, (24)
Для которой справедливо уравнение Нернста
£ £' л- In [Н+]2[ОхН|+] ,25х
Eh Ео+ 2F Ш [RedH|+] ( О)
54
ГЛАВА VIII
Подставляя вместо [ОхН1+] и [RedHa+] соответствующие им выражения (как в предыдущем примере), получаем уравнение для окислительного потенциала
(26)
Р _ р, , РТ , Со RT [Н+]4 + Кгг[Н+]3 + /<Г1Кг.,[Н+]2 h 2F cr + 2Р ln [Н+]2 + Ко+ КО1ХОа
Рис. VIII.2. Зависимость Ет от pH.
Кривая А — для 2-метилварнаминового синего; кривая Б — для 2-метоксивариаминового синего [28].
Зависимость £т от pH выражается следующим образом: р - Р' щ [Н+]* + [Н+Р + tH+P
£»+ 2F ln [Н+]2 + Д01 [Н+] + КО1Ко2 ( '
На рис. VIII.2 построена кривая зависимости Дщ для 2-метил- и 2-метоксивариаминового синего от pH
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
55
2-метилвариаминовый синий
2-метоксивариаминовый синий
На основе этих кривых были получены все четыре значения p/Cs для этих индикаторов. Замечено, что при диссоциации восстановленной формы уменьшается тангенс угла наклона кривой £т — pH, а при диссоциации окисленной формы имеется тенденция к его увеличению.
Этих двух примеров вполне достаточно, чтобы показать общий метод выявления зависимости £т— pH для большинства веществ, рассматриваемых в этой главе. Со многими из этих соединений работали Кларк и Коен [2, 26], для других даны ссылки на литературу при рассмотрении отдельных соединений. В дальнейшем изложении уравнения для конкретных индикаторов даются одновременно со значениями тех констант, которые были определены. За интересующими подробностями рекомендуется обращаться к соответствующим литературным источникам.
3. ИНДИГОСУЛЬФОКИСЛОТЫ
Индиго почти совсем не растворяется в водных средах, а растворимость солей его сульфопроизводных с калием повышается с увеличением степени сульфирования, так что моно-, ди-, три- и тетрасульфонаты растворяются достаточно хорошо, чтобы их можно было использовать в качестве индикаторов. Если их восстановить до бесцветной лейкокислоты, они ведут себя как полностью обратимые окислительно-восстановительные системы. Их реакцию на примере индигодисульфокислоты, известной под названием индигокармин, можно представить следующим образом:
восстановленная лейкоформа
(28)
56
ГЛАВА VIII
В водном растворе окисленная индигомоносульфокислота имеет темно-синюю окраску, а по мере увеличения степени сульфирования окраска соединений становится более красновато-синей. Спектры поглощения водных растворов, снятые Холмсом [29], показали смещение максимума поглощения с 608 нм для моносульфоната до 590 нм для тетрасульфоната. Восстановленные формы бесцветны или окрашены в слабый бледно-желтый цвет.
Причину интенсивной окраски индигокрасителей объяснили Клезингер и Лютке [30]. Для окисленной формы возможна цис— транс-изомерия по центральной С = С связи. У транс-формы наблюдается мезомерия по водородной связи между атомом водорода, связанного с азотом, и противоположным атомом кислорода:
При восстановлении исчезает двойная связь между центральными атомами углерода, а с этим и возможность образования мезомер-ной транс-формы. Рентгеноструктурным анализом доказано, что индиго существует в транс-форме.
Первые исследования формальных потенциалов этих систем были проведены в работе [4]. Анализируя уравнение 28, можно, по-видимому, ожидать, что сульфогруппы полностью ионизированы в обеих формах, однако обе гидроксильные группы восстановленных частиц следует принимать во внимание при любой формулировке зависимости Ет — pH. Поэтому индикаторы относятся к такому типу, для которого в силе реакция
Ох + 2е < Red2-,
(29)
а это приводит [2] к теоретической зависимости, описываемой уравнением
Ет=Е'о + In ([НЧ2 + КГ1 [НЧ + КГ1ХГ2),
(30)
где КТ1 и Аг2 —константы диссоциации гидроксильных групп восстановленной формы.
Экспериментальные исследования не представили доказательства существования второй ступени диссоциации, и, не считая некоторых отклонений при высоких значениях pH, полученные значения Em удовлетворяют уравнению
Ет=Е'в + In ([НЧ2 + КТ1 [НЧ)
(31)
Таблица VIII. 1
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ рК ПРИ 30 'С ДЛЯ ИНДИГО- И ТИОИНДИГОСУЛЬФОНАТОВ
Название £о Е7 m РКгх gpKr2 ркО1 ₽КО2 PKS Область PH Литература
Индиго-5-моносульфокислота 0,262 —0,157 7,8[4] 7,5[32] 12,4 — — — <9 4, 32
Индиго-5,5'-дисульф оки глота (Индигокармин, C.I. 73015 [80]) 0,291 —0,125 7,3 7,5 12,1 12,3 12,8 13,9 <9 4, 31, 32
И,щигс-5,5',7-трисульфокислота 0,332 —0,081 7,1 7,3 12,1 11,7 12,8 13,3 <9 4, 32
Индиго-5,5', 7,7'-тетрасульфокислота (С I. 73020 [80]) 0,365 —0,046 6,95 7,2 11.8 11,9 12,9 13,6 <9 4, 32
Тиоиндиго-5,5'-дисульфокислота 0,347 0,014 4,27 — — — — 0,5—10,5 31
Тиоиндиго-5,5', 7,7'-тетрасульфокислота 0,409 0,063 4,5 — — — — 0—11,5 50
58
ГЛАВА VIII
Значения Eh даются уравнением
In + 1и ([И+р + ЛГ1 [Н+])
(32)
Значения £т и р/Сп приведены в табл. VIII. 1 [4]. Для растворов, имеющих рН<8, уравнение (31) еще более упрощается:
£т=^+^1п[Н+] (33)
Так что значение £т можно очень легко получить. Это исследование повторили Мейер и Тридвел [51] для индигокармина, а Прис-лер с сотр. [32] для всех четырех индикаторов. Было установлено, что в щелочных растворах при pH 10,5—12,5 образуется семихинон. Реакция характеризуется появлением промежуточной красной окраски при переходе от синей окисленной формы к желтой восстановленной. Описано также использование спектрофотометрического и потенциометрического методов для оценки констант диссоциации восстановленных и окисленных частиц и семихинона, образующихся при этом синтезе. Значения рК, приведенные в табл. VIII. 1, объясняют, почему уравнение (31) непригодно для рН> 11.
ОБЛАСТИ ПРИМЕНЕНИЯ
ИндигосульФокислоты являются исключительно хорошими индикаторами; особенно часто их используют в кислых растворах. При pH >9 их надо применять с предосторожностью из-за того, что образуется семихинон и усложняются выражения для констант диссоциации; в таких случаях следует обращаться к работе Прислера с сотр. [32]. В пределах pH от 10,5 до 12,5 окраска переходит от синей к красной и затем к желтой. При более высоких значениях pH семихинон не образуется, а только окраска изменяется от желтой до бледно-желтой.
Нормальные потенциалы этих систем низки (см. табл. VIII. 1) и составляют 4-0,262 В для моносульфоната. По мере повышения степени сульфирования они возрастают и достигают 4“0,365 В для тетрасульфоната. Поэтому их используют в качестве индикаторов только при работе с такими сильными восстановителями, как олово (II), титан (III) и хром (II). Раньше [33, 34] индигосульфо-кислоты применяли при титровании сурьмы(III) броматом калия [34]. В работе [35] использован индигокармин при определении солей олова (II) хлоридом железа (III). Титрование проводилось в среде, содержащей не менее 50% соляной кислоты, при комнатной температуре и в атмосфере двуокиси углерода. Однако в другой работе [36] недавно установлено, что для полного протекания реакции необходима высокая температура и что индигокармин не
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
59
совсем пригоден для определения олова(II), поскольку последнее не восстанавливает его с достаточной скоростью. Для этого титрования предпочтительнее применять какотелин (см. разд. 10 настоящей главы). Индигокармин использовали также в качестве индикатора при титровании рения (VII) оловом (II) ['37]. В 4—12 М соляной кислоте рений(VII) восстанавливается оловом(II) до рения (V), а в 10—12 М соляной кислоте восстановление продолжается до рения(IV). Обе реакции дают четкое изменение потенциала Этот метод предложен [37] для определения рения в виде перрената, но восстановление идет только до рения (V). Титрование проводили в 5 М соляной кислоте при комнатной температуре в атмосфере двуокиси углерода и при добавлении трех капель 0,2%-ного водного раствора индигокармина в качестве индикатора.
Холнес и Корниш [38] использовали индигокармин при титровании сурьмы(V) титаном(III). Сначала сурьму окисляли до пятивалентного состояния бромом, избыток которого удаляли кипячением растзора в течение 10 мин, а затем титровали сурьму (V) сульфатом титана(III) в атмосфере двуокиси углевода при 60°C в присутствии 5 капель 0,1%-ного водного раствора индигокармина в качестве индикатора.
Индигокармин использовали также при титровании железа (III) хромом (II) ['39, 40]. Эту реакцию следует проводить в атмосфере двуокиси углерода; в конечной точке окраска меняется из синей на зеленую, что обусловливается присутствием хрома (III). Другим (несколько неожиданным, если обратить внимание на формальные потенциалы) является использование индикатора при титровании железа(II) хлорамином-Т [41]. Титрование идет в среде 2—3 М соляной кислоты при 40 °C и в присутствии 0,1 М фторида натрия. Фторид образует комплексы преимущественно с железом (III) и, по видимому, понижает потенциал реакции железо (III)/железо (II) ниже потенциала индикатора. Афанасьев также сообщает [42] об использовании индигокармина при титровании многих органических соединений хлорамином-Т.
Индигокармин или индигодисульфонат является единственным «з индигосульфокислот, который широко был использован в качестве индикатора; это не удивительно, так как только индигокармин имеется в продаже и его легко приобрести. Однако по-видимому, высокий окислительно-восстановительный потенциал тетрасульфоната благоприятно влиял бы на некоторые случаи титрования, например на титрование олова (II) железом (III).
Индигокармин использовали также как восстановитель, причем сам же он служил и индикатором ['например, титрование сульфата Церия (IV) [43], перманганата [44], гексацианоферрата (III) [44] и железа (III) [’44] ]. Реакция с церием(IV) [43] нестехиометрична, поскольку индикатор лишь частично окисляется до изатина, особенно если индиго добавляется к раствору церата; но в растворе
€0
ГЛАВА VIII
2,5 М серной кислоты реакция вполне надежна. Медленную реакцию индигокармина с йодатом калия [45] можно ускорить добавлением хлорид-ионов. Прямое титрование индигокармина йодатом возможно в 6—8 М соляной кислоте, причем окраска в конечной точке изменяется от синей до желтой. Реакция идет по уравнению (34), а не по уравнению (28), как это показано в работе ['45].
изатин
N-иодизатин
(34)
При более низких концентрациях соляной кислоты реакция слишком медленна для прямого титрования, поэтому индигокармин определяют следующим методом [46]. Добавляют иодат до исчезновения синей окраски индигокармина, а затем определяют избыток йодата путем добавки иодида и титрования выделившегося иода тиосульфатом в присутствии крахмала.
Другой интересный случай применения индигокармина касается определения микроколичеств кислорода в воде ['47—49]. Восстановленный индигокармин быстро реагирует с кислородом при pH 11. Лумис [47, 48] использовал эту реакцию для определения количества растворенного кислорода посредством измерения концентрации образовавшегося окисленного индигокармина спектрофотометрическим методом при длине волны 586 нм. (Это не означает, что индигокармин железа нельзя определить титриметриче-ски с помощью олова (II), титана (III) и т. д.) Джон с сотр. [49] показал, что образование семихинона мешает проведению анализа по методу Лумиса при pH 11, и предложил определять кислород по уменьшению поглощения (при 410 нм), вызванному восстановленными частицами. При этой длине волны ни окисленные частицы, ни семихинон не поглощают света.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
61
ТИОИНДИГОСУЛЬФОКИСЛОТЫ
Прислер и Хемпельман [50] исследовали родственное индигокармину соединение — тиоиндиготетрасульфокислоту [50].
О
SO3H О SO3H тиоиндиготетрасульфокислота
Они измеряли значения Еш в области pH 0—11,5; в табл. VIII.I даны значения Ео, Еш и рКг,. Красный семихинон образуется при pH 8—11,5. Авторы установили, что тиоиндиготетрасульфокислота пригодна в качестве окислительно-восстановительного индикатора только при pH<2,5, где окраска изменяется от оранжевокрасной (окисленная форма) до желтой. Был исследован также тиоиндигодисульфонат [31, 51], и в результате оказалось [51], что его нельзя применять как индикатор из-за аномального концентрационного эффекта. Однако Мейер и Тридвел [31] не наблюдали такого отклонения и отрицают образование семихинона в пределах pH от 0,5 до 10,5. Их данные, приведенные в табл. VIII.1, соответствуют значениям других индигосульфонатов, и они ничего не сообщают о степени пригодности тиоиндигодисульфоната в качестве индикатора, хотя из последней работы этих же авторов [52] видно, что окраска этого соединения намного интенсивнее индигокармина.
4. ИНДОФЕНОЛЫ
Многие производные индофенола (или фенолиндофенола) были исследованы Кларком с сотр. [3, 5—7, 13, 14] в качестве возможных индикаторных систем. Наиболее широко используемые соединения приведены в табл. VIII.2. Нумерация положения заместителей предложена Кларком [26] и приводится ниже для простых индофенолов
индофенол
62
ГЛАВА VIII
Таблица VIII.2
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ рК ПРИ 30 "С НЕКОТОРЫХ ИНДОФЕНОЛОВ
Название Ео Е1 m РКО1 рКГ1 РКГ 2 Область pH Литература
Индофенол 0,649 0,228 8,1 9,4 10,6 6,3—12,3 5, 13
2-Хлориндофенол 0,663 0,233 7,0 8,4 10,3 4—12,3 5, >3
2- Броминдофенол 0,659 0,231 7,1 8,5 10,24 5,8—11,7 5, 13
2.6-Дихлориндофенол 0,668 0,217 5,7 7,0 Ю,1 6,3—11,4 7, 13
2,6-Дибро.миндофенол 0,668 0,216 5,7 7,0 10,05 7,0-12,3 7, 13
2,6-Дихлор-3'-метилиндофенол 0,639 0,181 5,5 7,1 10,4 5,7-11,4 7, 13
2.6,3'-Трихлориндофенол 0,668 0,219 5,8 7,1 8,75 5,7—11,4 7, 13
2,6 Диброминдофенол-З'-суль-фонат иатрия 0,683 0,242 6,1 7,0 10,2 1,1—11,5 13, и
2,6-Диброминдофенол-2'-суль-фонат натрия 0,691 0,273 7,4 7,1 8,76 3,8—11,5 13, 14
Тимолиндофенол 0,592 0,171 8,8 9,85 10,8 5,7—12,3 5, 13
1 -Нафтанон-2->сульфонатиндо-фенол 0,544 0,123 8,7 9,1 10,7 3,9—12,6 3, Ц
1 -Нафтанон-г-сульфонат-З'Д'-дихлориндофенол 0,563 0,119 6,1 7,45 9,3 5-11,4 7, 14
и для нафтанониндофенолов
1 - нафтанониндофенол
Для индофенолов общей реакцией является реакция (5). Для! нафтанониндофенолов, например, для 1-нафтанон-2-сульфонат-3',5/-дихлориндофенола это следующая реакция:
0=
HSO3
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
63
(35)
Кларк и ДР- [3, 5—7, 13, 14] показали, что окислительно-восстановительные потенциалы этих систем определяются уравнением (14) а зависимость Ет— pH — уравнением (16), где KOl—константа диссоциации фенольной группы окисленных частиц, — константа диссоциации восстановленных частиц, а Кг2— константа писсоциации фенольной группы, образовавшейся в результате восстановления.
Большое число индофенолов было получено и исследовано Кларком с сотр. [3, 5—7, 13, 14]. Однако многие из этих соединений оказались неподходящими для использования их в качестве индикаторов из-за их плохой растворимости или неустойчивости. Поэтому Гиббс, Холл и Кларк [13] отобрали наиболее пригодные из них. Таким индофенолам соответствует область потенциалов в единицах Em от +0,119 до +0,273 В. Эти соединения приведены в табл. VIII.2; их можно применять только в нейтральной области значений pH. В кислых растворах при pH<4—5 индофенолы име ют тенденцию выпадать в осадок, а в щелочных растворах при рН^Н происходит разложение, в результате чего изменяются их потенциалы. Область значений pH, в которой индофенолы пригодны для использования их в качестве индикаторов, различна для отдельных индофенолов. Наибольшую область pH от 1,1 до 11,5 приписывают 2,6-диброминдофенол-3/-сульфонату натрия:
Следует заметить, что значения Е6, приведенные в табл. VIIL2, получены путем экстраполяции кривой Ет — pH или путем вычисления только но уравнению (16), а поэтому не имеют практическс г« значения.
Восстановленные формы всех этих индикаторов бесцветны, а окисленные формы действуют как кислотно-основные индикаторы и окрашены в красный цвет в кислом растворе (или при рН<рК01) и в синий в щелочном растворе. Синяя щелочная окраска гораздо ивтенсивнее красной кислой. Кларк с сотр. ['5, 6] нашел, что при ВеДении атомов галогена в молекулу индофенола понижается
64
ГЛАВА VIII
значение pKoi, так что эти производные сохраняют синюю окраску в довольно сильнокислых растворах.
Возможно, по этой причине чаще всего применяли в качестве индикатора 2,6-дихлориндофенол. Примером неустойчивости этих соединений может служить тот факт, что 2,6-дихлориндофенол медленно разлагается даже в нейтральном водном растворе. В работе [53] изучалось влияние света и температуры на устойчивость растворов 2,6-дихлориндофенола и было найдено, что они разлагаются как в темноте, так и на свету, а особенно быстро при повышенных температурах, даже при 30 °C. В другой работе [54] сообщается, что профильтрованный 0,1%-ный водный раствор устойчив только в течение 3 недель, поэтому желательно работать с твердыми смесями индикатора с чистым хлоридом натрия (1 :500 вес. ч.). Эти смеси устойчивы; для титрования достаточно брать 0,5 г. Стоун [55] тоже сообщил, что растворы 2,6-дихлорин-дофенола устойчивы, если их готовить в свежеперегнанном диоксане, содержащем 1 мл ледяной уксусной кислоты в 100 мл. Тил-манс с сотр. [56, 57] нашел, что устойчивы также 0,29 %-ные растворы в фосфатном буфере с pH 7, и использовал 2,6-дихлориндофенол в качестве индикатора-восстановителя при обнаружении восстанавливающихся веществ в пищевых продуктах, например в лимонном соке.
ОБЛАСТИ ПРИМЕНЕНИЯ
2,6-Дихлориндофенол чаще всего использовали в определениях аскорбиновой кислоты, применяя его либо в качестве титранта, когда он является одновременно и индикатором, либо просто в качестве индикатора. Например, Берч с сотр. ['58] использовал окисленный 2,6-дихлориндофенол для титрования им аскорбиновой кислоты в трихлоруксусной кислоте до исчезновения красной окраски. Титрование проводилось за 2—3 мин, так как краситель легко разрушается. Эммери и др. [59, 60] предложили модифицировать этот метод, сущность которого состоит в удалении мешающих примесей путем осаждения ацетатом ртути(II). Титр растворов 2,6-дихлориндофенола для титрования аскорбиновой кислоты можно установить по реакции с иодидом в растворе разбавленной серной кислоты [61]. Иодид количественно окисляется 2,6-дихлории-дофенолюм до иода, который затем оттитровывают тиосульфатом натрия в присутствии крахмала.
Использование 2,6-дихлориндофенола как индикатора при титровании гексацианоферрата(III) калия аскорбиновой кислотой привлекло внимание многих исследователей ['54, 62—67]. Эту реакцию можно представить уравнением
2[Fe(CN)6]3" + CeHgOe -> 2[Fe(CN)e]4- + 2Н+ + С6Н6О6 (36)
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
65
т пование проводилось при pH 4,7, так как при более низких зна-ениях pH формальный потенциал индикатора выше, чем у системы гексацианоферрат(III)—гексацианоферрат(II). Для титрования примерно 200 мл достаточно взять 1 мл 0,1%-ного водного раствора индикатора. Окраска раствора изменяется от синей до бес-цве™ ~ и др [54, 63—67] использовали эту реакцию для проведения ряда других определений. Количество гексацианоферрата (II) можно установить [63] путем окисления его нитритом до гексацианоферрата (III). Избыток нитрита разлагают сульфаминовой кислотой и титруют гексацианоферрат(III) аскорбиновой кислотой в присутствии индикатора 2,6-дихлориндофенола. Бихромат и перманганат можно определить [63] при помощи гексацианоферрата (II) с последующим титрованием образовавшегося гексацианоферрата (III) аскорбиновой кислотой по индикатору 2,6-дихлорин-'I ^фенолу. Например, для перманганата
МпСД + 5[Fe(CN)6]4- + 8Н+ -> Мп2+ + 5[Fe(CN)6]3- + 4Н2О
Подобный метод [64] был применен для определения хлора, брома, иода, гипохлорита, гипобромита, хлорита, бромата, йодата, хлорамина Т и персульфата [54]. Для этого вводили галогены в реакцию с гексацианоферратом (II) калия в растворе бикарбоната, и в той же среде оттитровывали гексацианоферрат(Ш). Другие окислители реагировали с гексацианоферратом(II) в солянокислом раствоое; после окончания реакции кислоту нейтрализовали бикарбонатом прежде, чем проводить титрование.
Восстановители, такие, как перекись водорода [54], гидроксиламин [65] и гидразин [66], можно также определять, используя реакцию между гексацианоферратом(III) и аскорбиновой кислотой. В каждом случае восстановитель взаимодействует с избытком Щелочного гексацианоферрата(III) калия, избыток которого оттит-ровывается аскорбиновой кислотой, используя в качестве индикатора 2,6-дихлориндофенол. Интересны следующие окислительно-восстановительные реакции: •
2[Fe(CN)6]3-+ Н2О2 --> 2[Fe(CN)6p-+ 2Н++ О2,
pH 8—10
2NH2OH + 2[Fe(CN)6]3- + 2ОН----> N2 + 2[Fe(CN)e]4- + 4H2O.
pH 8—10
N2H4 + 4[Fe(CN)f]3- + 2OH~ ---> N2 + 4[Fe(CN)6]4- + 4H2O
Реакция с гидроксиламином стехиометрична только при постоянном избытке гексацианоферрата(III). Поэтому надо приливать Раствор гидроксиламина к раствору, содержащему избыток гекса-Цйаноферрата(Ш), а не наоборот. Реакция заканчивается через 6 мин, если проводить ее в буферном растворе из борной кислоты бУры. В этих же условиях реакция с гидразином тоже протекает
66
ГЛАВА VIII
медленно (30 мин). В качестве примера приведем методику опре-деления перекиси водорода, принятую Эрдеем с сотр. [54].
Смешивают 25 мл 0,1 М. гексацианоферрата (III) калия с 20 мд 5 М гидроокиси натрия и вносят в смесь примерно 10—20 мг перекиси водорода. Перемешивают, оставляют в покое на 4—5 мин и затем добавляют индикатор (1 мл 0,1%-ного водного раствора 2,6-дихлориндофенола). Осторожно нейтрализуют разбавленной соляной кислотой (соотношение 1:1) до перехода окраски от зеленой до красно-бурой. Тогда раствор имеет слабокислую реакцию; его нейтрализуют, внося 1 г твердого бикарбоната калия и оттптровывают избыток гексацианоферрата(III) 0,05 М аскорбиновой кислотой. В ходе титрования окраска постепенно изменяется на синюю, но в конечной точке становится бесцветной от одной капли титранта.
Те же авторы разработали метод определения серебра [67]. I В этом случае серебро осаждается из нейтрального или азотнокислого раствора в виде гексацианоферрата(III) серебра. На осадок действуют аммиачным раствором гексацианоферрата (II) калия, и происходит обменная реакция с осадком:
Ag3[Fe(CN)6] + KJFe(CN)6] -> KAg3[Fe(CN)J + K3[Fe(CN)6]
В результате этой реакции образуется гексацианоферрат(II) серебра в виде осадка, а гексацианоферрат(Ш) переходит снова в раствор. Раствор отфильтровывают и титруют аскорбиновой кислотой гексацианоферрат(III), количество которого прямо пропорционально количеству серебра в исходном образце. Точно такие же методы разработаны [68] для определения висмута, цинка и марганца.
2,6-Дихлориндофенол находит применение как индикатор и в других методах титрования аскорбиновой кислотой [69, 70]. Эрдей и др. [69] использовали его при титровании меди (II) аскорбиновой кислотой. Титрование проводится при высокой концентрации галогенид-ионов, хлорида или бромида для большей устойчивости образующейся меди(1) и повышения формального потенциала системы медь(1)—медь (II) до такой степени, чтобы аскорбиновая кислота могла восстанавливать медь (II). При этом pH среды должно быть 5—6, и, так как нужно работать при высокой концентрации индикатора, необходимо провести холостой опыт в отсуг ствие меди. 2,6-Дихлориндофенол применяют также при йодометрическом титровании аскорбиновой кислотой [70]. Многие окисли тели, например бромат, иодат, перманганат, бихромат, гипохлорй1 и др., можно определять путем добавки иодида и титрования вь1’ деленного иода в среде бикарбоната аскорбиновой кислотой в прИ’ сутствии индикатора 2,6-дихлориндофенола. Ряд восстановителе! также определялся путем добавления избытка иода и титровали* избытка по тому же методу.
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
67
В одной из работ [71] описано применение других производных фенола, например 2,6-диброминдофенола, 2 броминдофенола ИНД°мо индофенола, для титриметрического определения хлорида И ™ (I) сульфатом железа (III). Для титрования обычно берут п?мл 0,2%-ного спиртового раствора индикатора. Для первых °’ vx индикаторов окраска переходит от светло-розовой к светло-А еной а для тимолиндофенола от светло-оранжевой к свегло-зеленой’ Для растворов не указывается значение pH, но они долж-11Ы быть, по-видимому, довольно кислыми
5. ИНДОАНАЛИНЫ И ИНДАМИНЫ
Ряд соединений, названных индоаналинами или аминоиндофенолами и имеющих общую формулу
r2N=/ \=N-f Л_он
исследовался [15] в качестве индикаторов. Применимость этих соединений ограничена по той же причине, что и индофенолов, т. е. из-за неустойчивости. Окисленные частицы становятся неустойчивыми при крайних значениях pH, но могут найти некоторое применение в нейтральной среде.
Феноловый синий, у которого в общей формуле R = CH3, неустойчив как в кислой, так и в щелочной среде. При низких значениях pH он быстро разлагается с образованием бензохинона, (который удалось выделить и идентифицировать [15]) и диметил-n-фенилендиамина. В щелочном растворе полярная диметиламино-группа легко замещается кислородом с образованием индофенола, причем этот процесс идет все быстрее по мере повышения щелочности раствора. Обе эти реакции вызывают резкое изменение потенциалов за пределами pH 2,5—9,7. В области этих значений изменение значительно медленнее.
Исходя из реакции
(CH8)2N=
—ОН + н+ + 2е
(37)
которая относится к типу реакций [2]
Ох+ + 2е + Н+ HRed,
5*
68
ГЛАВА VIII
Коен и Филлипс [15] вывели следующее уравнение
£h=^' + <ln-^- +
. 1п [Н+]3 + Kri [Н+]2 + ^гЛгг [Н+] 2F 1п [Н+1 + К0]
где /<01 и Кп — константы диссоциации фенольного иона водорода окислителя и восстановителя соответственно, а Кг2 — константа диссоциации иона водорода аминогруппы восстановителя. Значения рК даны в табл. VIII.3. Эту работу повторили Шварценбах и Михаэлис [72], которые, кроме того, доказали, что образуется семихинон.
Таблица VII 1.3
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ рК ПРИ 30 °C ДЛЯ НЕКОТОРЫХ ИНДОАНАЛИНОВ И ИНДАМИНОВ
Название Е'о Е7 ГЛ Р^о, РКо2 рКГ1 РКГ 2 PS Область PH Литература
Феноловый синий 0,677 0,224 4,86 — 5,96 9,89 — 1,1—12,6 15, 72
л-Т олуилендиами-_нивдофенол 0,567 0,125 8,07 11,42 2,72 4,96 10,32 1,1—12,6 15
Зеленый Биндшед-, лера (С. I. 49405 f [80]) 0,680 0,224 3,27 — 5,1 6,46 *— 2—9,5 11, 72
Толуиленовый синий (С. 1.49410 [80]) 0,601 0,115 3,80 10,48 2,14 4,40 6,57 1—12 11
Коен и Филлипс [15] изучили также два других родственных соединения — о-толуидиновый синий и .м-толуилендиамининдофе-нол:
СН3
о-толуидиновый синий
т-толуилендиамининдофенол
о-Толуидиновый синий оказался исключительно неустойчивым, особенно в избытке кислоты или щелочи и не мог быть индикатором л-Толуилендиамининдофенол гораздо более устойчив, даже устой-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
69
фенолового синего, и его можно применять как индикатор. ^Целительный потенциал этой системы подчиняется уравнению
£=£' + RT In —С° 4
£ 2F Ст 4
RT , [Н+Г + [Н+р^1 + [Н+рКГ1^2 + [Н+]КГ1КГ2КГз + 2F 1П [Н+Р-н£01[Н+] + К0^02
(39)
где Кп, Кг2 и Koi означают то же, что и в уравнении для фено-ловог» :инего, а КТа и Ко2 — константы диссоциации ионов водорода край116® амино! руппы восстановителя и окислителя соответственно. Значения р£ приведены в табл. VIII.3. Во всех трех случаях восстановленные формы гораздо устойчивее окисленных форм, они были использованы Коеном и Филлипсом [15] в их исследованиях.
Филлипс, Кларк и Коен [11] изучили также два других родственных соединения, известных как индамины; в них вторая фенольная группа в индофеноле замещена диметиламиногруппой. В одном или двух случаях их применили в качестве индикаторов. Эти соединения— зеленый Биндшедлера и толуиленовый синий:
зеленый Биндшедлера
Зеленый Биндшедлера дает устойчивый потенциал в пределах pH 2—9,5. При более высоких значениях pH окислительные частицы разлагаются с образованием сначала фенолового синего, а затем индофенола. Свободное основание очень плохо растворяется, и его приходилось применять в разбавленных растворах, чтобы избежать выпадения осадка. Тем не менее Филлипс, Кларк и Коен [И] нашли, что для зеленого Биндшедлера в пределах pH 2—9,5 справедливо следующее уравнение:
£h=£« + <-ln-f^ +
RT [Н+Р + КГ1[Н+Р + £Г1£Г [Н+]
~---In----------1---------Ь—£-----
2F [Н+] + Ко
(40)
Константы диссоциации КО1, КГ1 и Кг2 соответствуют диссоциации °На водорода диметиламиногруппы в окисленных частицах и двух
70
ГЛАВА VIII
диметиламино^рупп в восстановленных частицах. Реакцию можно написать в следующем виде:
(CH3)2N=
(CHs)2N-
Значения р/С, определенные Филлипсом и др. [11], приведены в табл. VIII.3 наряду со значением и значением Ео, полученным путем экстраполяции. Эту работу перепроверили Шварценбах и Михаэлис [72], которые и в этом случае нашли доказательство в пользу образования семихинона.
Шайн с сотр. [73] исследовал свойства зеленого Биндшедлера. По методу Виланда [74] получается зеленое твердое вещество, устойчивое в течение долгого времени. Из водных растворов зеленого твердого вещества при добавлении концентрированного раствора хлорида цинка осаждается красное вещество. Элементным анализом установлены следующие формулы для зеленого и красного соединений:
красное твердое соединение
[ZnCij;
2
(CH3)2N-'^_.n= н
[ZnCIJ
зеленое твердое соединение
Четкую конечную точку можно получить только в том случае, если зеленое твердое соединение в ходе его получения тщательно про мыть кислым бихроматом, а затем водой (или этанолом) и ди этиловым эфиром. Если же промывать только водой, то конечная точка титрования не ясная из-за коричневого оттенка. Торговые препараты зеленого Биндшедлера всегда имеют коричневый этте-нок. Эти препараты [73] дали удовлетворительные конечные точки I при титровании титана(III) бихроматом или в тех случаях, когда 1 само вещество титровали титаном(III).
Зеленый Биндшедлера можно применять в более широкой области значений pH, чем большинство индофенолов, но и этот индикатор неустойчив при крайних значениях pH. Он плохо растворяется,., а интенсивность его окраски слаба. Однако его использовали [75— 78] в качестве окислительно-восстановительного индикатора при комплексометрическом титровании железа (III) раствором ЭДТА-Титрование происходит при pH 2,5—3,5, а окраска изменяется от
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
71
ой до оранжевой. Индикатор готовили в виде твердой смеси 3еЛеНосстановленного зеленого Биндшедлера [78] с 0,1 г сафрани-1 г ® ЮО г хлорида натрия. Для титрования достаточно брать Нл г смеси. Хром (III) можно определять посредством реакции
^бытком ЭДТА при pH 3, избыток ЭДТА оттитровывают железом (Ш) в присутствии зеленого Биндшедлера в качестве инди-КЭТНекоторый интерес вызывает также толуиленовый синий. Установлено, что для него справедливо следующее уравнение:
£h=£;+^-ln-g- +
RT , [H+F + ^JH+p + XrjXrJH+F + ^^KrJH+l2
+ -2?-1п-------гн+Р-^гн+1д-ТГк,-----------• (42)
[н+р + кО1 [н+] + ЯоЛ>2
в котором константы диссоциации имеют такие же значения, что и для зеленого Биндшедлера, а дополнительные константы диссоциации для окислителя и восстановителя относятся к дополнительной аминогруппе молекулы. Толуиленовый синий отличается более интенсивной окраской по сравнению с зеленым Биндшедлера, но неустойчив при нагревании, превращаясь при этом в толуиленовый красный, поэтому его можно использовать только при комнатной температуре. По-видимому, он разлагается очень медленно даже и в этих условиях. Несмотря на это, толуиленовый синий применяли [79] как индикатор при титровании железа (II) церием (IV) в растворе разбавленной серной кислоты.
6. АЗИНЫ
Окислительные свойства ряда производных феназина тщательно изучены. Эти соединения нашли широкое применение как окислительно-восстановительные индикаторы. Их потенциалы Е'о обычно низкие и находятся в интервале 0,05—0,30 В (значительно ниже, чем у соответствующих оксазинов и тиазинов). Основная структура восстановленного феназина и нумерация положений в его молекуле следующая:
Н
Н феназин
72
ГЛАВА VIII
Первое подробное потенциометрическое исследование соедине ния этой группы было проведено для пиоцианина Фридгеймом и Михаэлисом [81]. Структуру и реакцию восстановления пиоциани-на можно описать уравнением
Н
пиоцианин бесцветное соединение
Окраска пиоцианина синяя в щелочном растворе и красная в кис-лом (pKOl=4,85) из-за основных свойств одного из атомов азота. При восстановлении обе формы образуют бесцветное лейкосоединен ие, которое легко окисляется на воздухе. Фридгейм и Михаэлис [81] нашли, что при восстановлении красной кислой формы появляется промежуточное зеленое соединение. Это было первое сообщение об образовании семихинона. Авторы считали, что в кислой среде при рН<6 восстановление происходит в две стадии
Ind -|- Н+ + в --> IndH (зеленый семихинон)
IndH -f- Н+ + е -> IndH2 (бесцветное соединение)
Изучение пиоцианина проводилось в пределах значений pH 1—12. При рН>12 окисляемая форма медленно и необратимо окисляется, а в области pH 1—12 потенциалы подчиняются уравнению
Р IH+F + ^rJH+I
Ет—Ео+ 2F 1п [н+] + лО1 •
где КГ1—константа диссоциации фенольной группы, образовавшейся при восстановлении, а Лох—константа диссоциации гидроксиль ной группы окисленных частиц. Семихиноидная или свободноради кальная природа зеленого интермедиата подтверждена исследовв пием методом ЯМР спектроскопии [82], формула семихинона им! ет следующий вид:
Михаэлис [83] изучал также окислительно-восстановительны' свойства другого азинового соединения, розиндулина 2G. Этот ив
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
73
,ятоп является натриевой солью моносульфированного красите-ДИКпозиндона. Хотя положение сульфогруппы неизвестно, в общем виде краситель имеет следующую формулу
розиндулин 2G
Окисленная форма ярко-красного цвета; при восстановлении, которое вызывает точно такое же изменение как и в случае пио-цианина, появляется желтая или бесцветная лейкоформа. Этот индикатор устойчив в обоих состояниях даже в сильнощелочном растворе. Восстановленная форма очень чувствительна к кислороду, что следовало ожидать от системы со столь низким нормальным потенциалом. Все опыты поэтому проводились в атмосфере азота. Михаэлис [83] изучал потенциалы в области pH 4—12 и не нашел доказательства тому, что окисленная форма ионизируется. Потенциал для этого индикатора определяется следующим простым уравнением:
Ет=Е'0 + In ([Н+]2 + ДГ] [Н+]) (45)
Потенциалы розницу лина оказались очень низкими, и поэтому его лучше использовать в качестве сильного восстановителя [81], а не индикатора.
Два вещества, нейтральный красный и простой нейтральный красный (или толуиленрот), были предметом исследований Кларка и Перкинса [17]:
N
N
СИ-
СН3
простой нейтральный красный нейтральный красный
Центральный красный ведет себя отчасти как обратимая система, ° в некоторых условиях подвергается второму изменению, кото-. е Уже необратимо. Бесцветная восстановленная форма легко овь окисляется на воздухе; если же ее хранить без доступа воз-
Цн aJIPK PH 5,3, появляется быстро желто-зеленая флуоресцен-та реакция зависит от pH, протекает в слабой степени при и почти совсем не идет при pH 8,2. Образование вещества
74
ГЛАВА VIII
с зеленой флуоресценцией, которое Кларк и Перкинс выделили но не идентифицировали, приводит к ошибке в определении потен’ циалов вследствие их быстрого изменения, так что нейтральный красный фактически непригоден для использования его в качестве индикатора в области pH, в которой он получается. Любопытно отметить, что само флуоресцирующее вещество не окисляется легко на воздухе. Потенциалы в щелочном растворе очень устойчивы. Простой нейтральный красный тоже флуоресцирует в восстановленных растворах, его потенциалы гораздо устойчивее, чем у нейтрального красного. Для обеих систем справедливо уравнение
Ет~ Ее+ 2р 1п [Н+] + КО1
где Ап и Кг.2 —константы, которые относятся к присоединению ионов водорода к двум крайним аминогруппам, а К01— подобная диссоциация окисленных форм. Мейер и др. [31] исследовали соединение, называемое нейтральным красным, которое, по-видимому, отличалось от указанного выше. Метильная группа находилась! орто-положении к диметиламиногруппе, а не к аминогруппе. Их константы диссоциации тоже сильно отличаются от соответствующих констант, установленных Кларком и Перкинсом, в частнофЛ Р Ко ,=8,00.
Серия соединений, содержащих фенильную группу у мостикового атома азота, исследовалась в работе [18]. Это сафранины и феносафранины:
сафранин Т феносафранин
Сафранин Т (или сафранин О) является, по-видимому, смесью из двух красителей, из которых у одного имеется, а у другого отсутствует метильная группа; в приведенной формуле эта группа заключена в скобки. К феносафранинам же относятся сам феносафранин, диметилфеносафранин (с одной диметиламиногруппой), тетраметилфеносафранин (две диметиламиногруппы) и тетраэтил-, аминосафранин (две диэтил аминогруппы), который известен поД названием аметистовый фиолетовый. Сафранины, подобно нейтральному красному, сначала образуют обратимую систему, которая затем при некотором значении pH претерпевает второй необратимый процесс, приводящий к смещению потенциалов и необрЗ' тимости системы. Если сафранины восстановить и затем хранить
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
75
ме или в инертной среде при pH 3—7,5, то наблюдается в вакуУ^ изменение окраски и появление флуоресценции, как у За"16пального красного. Флуоресцирующее вещество не удалось НеИелить. Потенциалы очень неустойчивы в этой области, но при ®Ь1Д нИзких или более высоких значениях pH они значительно С° ойчивее. Во всех случаях зависимость Еш — pH описывается уравнением
Ет=Е'0+-^~ In([Н+]3 + КГ1 [Н+]2 + /<Г1КГ2[Н+])
(47)
Некоторые затруднения встретились при оценке КГ1 и КГ2 на основе пот< тонометрических данных вследствие близости их значений, так что не было заметного промежуточного наклона кривой, равного 0 06 В/pH, и потому обе константы диссоциации проявляются в области смещенных и неустойчивых потенциалов. Поэтому значения рЛг1 и рМ2 в табл. VIII.4 хотя и находятся в правой области, до некоторой степени гипотетичны [18]. Кроме того, отмечалось [18] небольшое концентрационное влияние даже в кислых растворах. Иными словами, потенциалы смещаются максимально на 6 мВ при 10-кратном изменении концентрации красителя; в габл. VIII.4 значения потенциалов даны для 10~4 М. растворов. В случае тетраэтилфеносафранина свободное основание восстановителя выделяется из нейтрального или щелочного раствора при концентрациях ниже 10-5 моль-л-1. Стайлер с сотр. [18] утверждает, что значения рК в этом случае даны с очень грубым приближением. Мейер и Тридвел [31] тоже изучали сафранин Т и нашли, что он представляет собой смесь двух неидентифицированных соединений. Эти авторы определили индивидуальные константы для каждого вещества, но ничего не сообщили об их химической формуле.
Стайлер и Кларк [19] исследовали также серию родственных соединений, названнных апосафранинами. Нейтральный синий и изорозиндулины имеют в общем виде формулу с заместителями:
Название R R' R" Ео |
Нейтральный синий СН3 н Н 0,170
Изорозиндулин №1 CSH5 н н 0,199
Изорозиндулин №2 С2Н5 н сн3 0,195
Изорозиндулин №3 с2н5 сн3 н 0,202
Эт
и соединения совсем нерастворимы в растворах, pH которых ачительно выше 4,5—5,0, и поэтому авторы не смогли определить
I
76 ГЛАВА VIII
Таблица VHl.q |
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ г НЕКОТОРЫХ АЗИНОВ К ПРИ 31) °<j ||
Название Е’о В7 рк01 ркг 1 Область р % pH С.1 180]
Индулиновый ярко-красный 0,047 —0,299 — 4,5 — 3,0—8,6 50 080 19
Розиндулин 2G 0,139 —0,281 — 9,5 — 4,8—11,4 — 83
Нейтральный синий 0,170 — — 5,0 — 1—4,2 50 150 19 ।
Лиссамнновый синий BF (25 °C) 0,196 —0,253 3,5 5,0 7,3 1—11 50 230 89
М-Метил-2-оксифеназин 0,197 — 0,167 3,0 1,0 10,1 0,1—11,7 — 84
Изорозиндулины 0,200 — — — — 3,5 — 19
Сафранин Т 0,235 —0,289 — 4,7 5,75 1—12 50 240 18
Пиоцианин 0,235 —0,034 4,85 9,5 — 1—12 — 81
Простой нейтральный красный 0,237 —0,323 6,32 4,95 5,96 2,1—11 — 17
Нейтральный красный 0,240 —0,325 6,80 5,30 6,16 2,1 — 11 50 040 17 1
Сульфированный розин-дон 0,247 —0,385 — 7,5 9,5 6,1-10,7 — 19 1
Хлорорафин 0,247 —0,115 — — — 0—10,4 — 86
Феносафрании 0,280 —0,252 — 4,96 5,78 1,1-11 50 200 18
Диметилфеносафранин 0,286 —0,260 — 4,89 6,33 1,0-12 50 205 18
Гетраметилфеносафранин 0,288 —0,273 — 5,32 6,45 3,0—8,2 — 18
Тетраэтилфеносафранин 0,355 —0,254 — 6,4 7,7 2,2—9,8 50 225 18
Kn> Кг2 или Em этих красителей во всех случаях, кроме КТ1 для нейтрального синего. Предполагая, что в исследуемой области pH Д-
все окислители имеют сильную полярную группу=NR2 и что нужно рассматривать только диссоциацию восстановленной формы, Стай-лер и Кларк получили следующую формулу для потенциалов этих красителей:
Ет=Е'в + ()Н+)2 + КГ1 [Н+] + КгЛ? <48)
Для нейтрального синего величину КГ2 не удается определить ® доступной для измерений области pH, и поэтому уравнение (48) упрощается до уравнения (45). Для изорозиндулинов не удается определить и величину /СГ1. Поэтому уравнение (48) еще более упрощается;
£m=£' + -^Lln[H+] Н
1
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
77
Значения Е'о в табл. VIII.4 экстраполированы с помощью этих УР&Стайлер и Кларк [19] исследовали два других апосафрани-на__индулиновый ярко-красный и сульфированный розиндон:
и сульфированный розиндон:
индулиновый ярко-красный
Индулиновый ярко-красный отличается исключительно низким нормальным потенциалом, самым низким из азинов. При pH 1—5 формальный потенциал его немного выше потенциала водородного электрода в атмосфере водорода. В изученной области значений pH его потенциалы выражаются уравнением (45). Уравнение для формального потенциала сульфированного розиндоча исключительно сложно, так как этот краситель, по-видимому, обратимо ассоциирует в водных растворах, но все же формальный потенциал удовлетворяет зависимости
Е -Е' I RT In 2К 4-
4- ([Н+]3 4- КГ1 [Н+]2 4- КГ1Кг2 [ НЧ) (50)
где К—константа диссоциации для равновесия между моно- и бимолекулярными частицами
Ох2 < > 2Ох
Установлено, что К. имеет значение 5-10-5 в фосфатных буферах и 1 • 10~4 5 в боратных буферах. Потенциалы были определены только в пределах pH 6—10,7 и экстраполировались до pH 0. Для Различных буферных систем получались различные значения Ео. *ак, в фосфатном буфере £6=0,243 В, а в боратном 0,251 В. В табл. VIII.4 приведены средние значения потенциалов.
Простые оксифеназины, как, например, 1-оксифеназин, 2-окси-феназин и хлорорафин, хотя и очень интересны (они способны образовывать семихиноны [29]), однако непригодны для использования их в качестве индикаторов из-за их плохой растворимости и очень близкой окраски обеих форм [84—86].
78
ГЛАВА VIII
Например, хлорорафин, который может при определенных условиях быть индикатором, меняет окраску в пределах pH 4—10 от оранжево-желтой до желтой [86]. Ниже pH 4 образуется зеленый промежуточный семихинон. Прислер и Хемпелман [84], однако, сообщили, что в области pH 5,5—12 М-метил-2-оксифеназин дает более четкое изменение окраски: от красной до бесцветной или светло-желтой.
N
N
Ан3
N-метил-2-оксифеназин
Окислительно-восстановительные потенциалы этого соединения изучены в пределах pH от 0 до 12,6 и описываются уравнением
F P'<RT Щ [Н+]3 + [W + [Н-ь]
Е™= Ео + In---------[H+I4-K”--------' >
где Koi и Ктг — константы диссоциации от катиона к свободному основанию, а КГ2—константа диссоциации от свободного основания к аниону. Синтезирован ряд других феназинов [87, 88], которые использовались как переносчики электрона в биологических системах [87], но не имеется данных об их потенциометрических свойствах и не рассмотрена возможность их применения в качестве индикаторов.
Интересное исследование было проведено [89] с другим азином, называемым лиссаминовый синий BF. Это соединение имеет сложное строение. Его окисленной форме соответствует следующая формула:
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
79
v эти авторы представили результаты в несколько ином виде, ^°Табл VIII-4 приведены формальные потенциалы относительно В пмал'ьного водородного электрода, вычисленные по уравнению и значения рК. Окисленный лиссаминовый синий BF имеет инюю окраску в растворах при pH>5, но при pH <5 становится Соасным, а при pH 1 — фиолетовым. Восстановленная форма блед-о-желтая. Потенциалы, измеренные в пределах pH 1—11, усюй-чивы и легко получаются. Не наблюдалось смещения потенциалов, о котором сообщали Кларк и др. [17, 18] для нейтрального красного и более простых сафранинов. В этом отношении лиссаминовый синий BF должен был бы обладать значительным преимуществом по сравнению с обычно используемыми индикаторами нейтральным красным, сафранином Т и феносафранином. Из данных табл. VIII.4 видно, что его потенциалы находятся приблизительно в той же области потенциалов.
ОБЛАСТИ ПРИМЕНЕНИЯ
Вообще говоря, потенциалы азинов, используемых в качестве индикаторных систем, исключительно низки, так что их можно применять только при работе с сильными восстановителями, такими, как титан (HI), хром (II) и ванадий (II). Восстановленные формы легко окисляются атмосферным кислородом, поэтому титрование нужно проводить в инертной атмосфере. Почти во всех случаях применения, которые будут описаны ниже, индикаторы применялись в сильнокислых растворах, т. е. за пределами области, в которой проводились теоретические исследования.
Из соединений, приведенных в табл. VIII.4. только нейтральный красный, сафранин Т и феносафранин широко применялись как индикаторы. Все три были использованы в методе титрования титаном(III). Монье [90] титровал медь(П) раствором титана(Ш) в сернокислом или солянокислом растворе в присутствии сафранина Т, а Холнес и Корниш [38] сообщают об исследовании всех индикаторов для титрования сурьмы(V) титаном(III), которое они проводили в 4—8 М соляной кислоте при 60 °C и в инертной атмосфере, 1 мл индикатора добавляли непосредственно перед конечной точкой. Окраска в этих условиях изменялась от синей до бесцветной (при использовании нейтрального красного), от фиоле-81 юй до бесцветной (в присутствии сафранина Т) и от фуксиновокрасной до бесцветной (при добавлении феносафранина). Опре-Д| ние конечной точки ib присутствии феносафранина не так четко. как с остальными двумя. Мурти и Рао [91] титровали ти-тан(1Ц) метаванадатом натрия, используя все три красителя в качестве индикаторов. Они сообщают, что восстановленные частицы 1^° окисляются следовыми количествами метаванадата, но в М соляной кислоте окисленные частицы реагируют более медлен
80
ГЛАВА VIII
но с титаном (III). Однако последнюю реакцию катализирует щавелевая кислота, а все три вещества являются хорошими индикаторами для реакции
2Ti(III) + V(V) > 2Ti(IV) + V(III) (52)
Окрашенные частички появляются сразу при избытке ванадата. Обычно используют свежеприготовленные 0,1%-ные водные растворы каждого из индикаторов; 0,1 мл раствора достаточен для 50 мл титруемого раствора. Рао Н. и Рао Д. [92] тоже титровали железо(III) титаном(III). Все три индикатора показывали четкую конечную точку, но необратимую в 1 М соляной кислоте. И в этом случае необходимо присутствие щавелевой кислоты, чтобы катализировать индикаторную реакцию.
Нейтральный красный и феносафранин были использованы при титровании хромом(II). Так, феносафранин применялся [93, 94] для установления конца реакции восстановления железа (III) [93] или урана (VI) хромом (II) в 1,5 М серной кислоте. Для количественного определения любого из этих ионов добавляли в избытке хлорид хрома (II), тогда в присутствии феносафранина раствор из розового становился светло-зеленым, как только заканчивалась реакция восстановления до урана (IV) или железа (II). Избыток хрома (II) окисляют, пропуская через раствор кислород до появления розовой окраски окисленного феносафранина. Затем железо(II) титровалось непосредственно бихроматом, в случае урана (IV) сначала проводят реакцию его с избытком железа (III), после чего образовавшееся железо (II) оттитровывают бихроматом. Тандон и Меротра [39, 40, 95, 96] провели ряд определений раствором хрома (II) в присутствии этих индикаторов. Железо(III) определяли ['39, 40] в инертной атмосфере в 1,5 М серной кислоте с использованием нейтрального красного или феносафранина. Окраска изменялась от розовато-фиолетовой до зеленой (нейтральный красный) и от фиолетовой до зеленой (феносафранин); зеленая окраска в конечной точке обусловливается присутствием хрома(III). Этот метод применялся для определения персульфата и перекиси водорода [95]. Железо (II) вводили в реакцию с персульфатом или перекисью водорода в атмосфере двуокиси углерода и образовавшееся железо (П1) оттитровывали сульфатом хрома (II) в присутствии нейтрального красного в качестве индикатора. Олово(II) также определяли путем реакции с избытком железа (III) в ~1 М соляной кислоте; избыток железа (III) титровали хромом(II), используя один из тех же индикаторов. Тандон и Меротра [96] проводили прямое титрование уранилацетата и установили, что нейтральный красный и феносафранин слишком быстро обесцвечивались и поэтому не так хороши как метиловый красный или n-этоксихризоидин. Описано [39, 40] также определение меди(II) методом титрования хромом(II) в присутствии нейтрального крас-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 81
го или феносафранина. Титрование проводится в большом избытке хлорид-иона (например, в 3 М соляной кислоте) и идет по ре-
[CuClJ2- + Сг2+ -> [СиС13]2- + С1- + Сг3+
Кроме этого, был выполнен ряд определений ванадия (II) с использованием нейтрального красного или сафранина Т в качестве индикатора. Например, определялся [97, 98] ванадий(II) путем титрования его перйодатом калия [97] или броматом калия [98]. Реакция ванадия (II) с перйодатом калия в 0,5—2 М серной кислоте проходит через две стадии:
8V2+ + IO4 + 8Н+ > 8V3+ +!“ + 4Н2О, (53)
8V3+ + 107 + 8Н+ > 8V4+ +I- + 4Н2О (54)
Для установления конечной точки на первой стадии можно использовать сафранин Т или нейтральный красный. Для титрования ванадия (П) в 0,5 М. серной кислоте раствором бромата калия [98] можно также применять сафранин Т, а при титровании раствором йодата калия пригодны и сафранин Т, и нейтральный красный. Ванадий (II) можно титровать, кроме того, хлоратом калия в присутствии следов смеси иод+иодид в качестве катализатора и в среде 0,5 М серной кислоты. Полнота окисления ванадия (II) в ванадий (III) устанавливается по нейтральному красному.
Проведено также титрование ванадия (II) железом (III) с использованием в качестве индикатора феносафранина [99], сафранина Т [100] или нейтрального красного [100, 101]. Потенциометрическое титрование [100] в 0,5 М серной кислоте идет через четко разграниченные две стадии
V2+ 4- Fe3+-> V3+ 4- Fe2+, (55)
V3+4-Fes+ --> V4+4-Fe2+ (56)
Несмотря на различие в шкалах потенциалов, ясно, что нейтральный красный и сафранин Т показывают конечную точку реакции (55), а роданид аммония — окончание реакции (56). Нейтральный красный и феносафранин использовали также [101] для установления конечной точки как реакции (55), так и реакции окисления ванадия(II) до ванадия (III) раствором меди (II). Нитраты тоже определяли [99] посредством реакции с избытком ванадия (II), который титровали затем железом (HI), используя феносафранин в качестве индикатора.
7. ОКСАЗИНЫ
Были исследованы окислительно-восстановительные свойства Ряда оксазинов, и в нескольких случаях оксазины нашли примене-ие в качестве индикаторов. Формула в общем виде для фенокса-6-918
82
ГЛАВА VIII
зина следующая:
фенокср&ин
В табл. VIII.5 приведены окислительно-восстановительные потенциалы и значения р/С ряда оксазинов. Нормальные потенциалы их значительно выше, чем у азинов, но ниже, чем у многих других систем, например у индофенолов.
Предметом тщательного потенциометрического исследования, проведенного Михаэлисом и др. [102], были галлоцианин и гал-лофенин
галлофенин
Реакцию восстановления галлоцианина можно представить в сле
дующем виде:
соон
| соон
N I
(57)
Галлоцианин—амфотерное соединение, поэтому легко растворяется в щелочных и сильнокислых растворах, но умеренно растворяется в области pH 3,5—5,5. Таким образом, он не очень хорошо растворим в воде и удовлетворительно растворим в буферных растворах, pH которых выходит за пределы 4—5. При рН<4 и pH>8 окраска растворов красно-фиолетовая, а при pH от 5,5 до 8 синяя. Красная форма при рН<4 является ионом с положительным за-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
83
Таблица VIII.5
ЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ рК ПРИ 30 °C
ОКИСЛИ 1Ь НЕКОТОРЫХ ОКСАЗИНОВ
Название Ео m ркО1 РКГ Г2 Область pH С. I. [80] Литература
Бриллиантовый крезиловый синий 0,583 0,047 10,74 4,60 6,30 0-11 51 010 16, 31
Метилкапри синий 0,477 —0,061 — 4,85 6,10 1,0—12,3 51 000 16
Этилкапри синий 0,540 —0,072 — 6,70 7,14 1,0—10,0 —• 16
Нильский синий 0,406 —0,119 9,70 3,92 6,90 1,4—12,3 51 180 16
Галлоцианин (25 °C) — 0,021 10,1 9,1 — 5,5—10 51 030 102
Галлофеиин (25 °C) — -0,142 — — — 4,5—9 51 125 102
G-Кислота фенонафтоксазина (25 °C) 0,491 0,115 — 5,73 — 1—7 51 1759 104
R-Кислота фенонафтоксазина (25 °C) 0,471 0,105 — 5,34 — 0,5—7,5 51 175а 104
Прюн 0,528 0,058 3.6 Р£о,= =8,05 5,35 7,44 0—10 51 040 103
Резоруфин 0,380 —0,051 6,93 9,26 10,00 0,2—12,0 — 105
аПод номером C.I. 51175 даны производные сульфокислот.
рядом, образовавшимся при диссоциации диметиламиногруппы, .а синяя форма соответствует аниону, который получается при диссоциации карбоксильной группы. Красная форма при рН>8 представляет собой двухвалентный анион, образовавшийся в результате дополнительной диссоциации гидроксильной группы. Галло-Цианин, как и галлофенин, представляет собой полностью обратимую систему; значения потенциалов галлоцианина близки к потенциалам метиленового голубого, о котором пойдет речь в следующем разделе и который обладает рядом недостатков, мешающих его использованию. К сожалению, Михаэлис и др. [102] проводили исследования в пределах pH 5,5—10 и не представили данных об £0, поэтому нельзя получить уравнение зависимости потенциала от pH для галлоцианина. Однако кривая Ет—pH имеет тангенс угла наклона —0,059 В/pH в пределах pH от 5,5 до 9.
1 аллофенин растворяется при любом значении pH, и обычно Растворы его окрашены в синий цвет. Потенциалы его иногда бо-е отрицательны, чем у галлоцианина, и подобны потенциалам дигомоносульфоната. Исследования галлофенина проводились в ласти pH 4,5—9, тангенс угла наклона кривой Ет—pH состав-6*
84
ГЛАВА VIII
ляет —0,059 В/pH для pH 6—9. При рН<6 тангенс угла наклона увеличивается. Галлоцианин и галлофенин, по-видимому, можно применять в качестве индикаторов, но были бы очень ценны сведения об их поведении в области кислых значений pH.
Коен и Прислер [16] исследовали свойства четырех оксазиновых красителей: нильского синего, бриллиантового крезилового синего, метил- и этилкапри синего.
нильский синий
бриллиантовый крезиловый синий
метилкапри синий этилкапри синий
Данные, полученные для нильского синего и бриллиантового крезилового синего, удовлетворяют уравнению
F - F' д_ In mW + W*!8 ,
Ео+ 2F П [Н+] + К01 ^581
Данные для обоих капри синих удовлетворяют уравнению
Ет=Е' + In ([Н+]Э + [Н+]2 + ЕГ1ЛГ2 [ Н+]) (59)
где Koi— константа диссоциации полярной аминогруппы окисленных частиц (но ее не определяли для обоих капри синих), а /СГ1 и /Сг2 — константы диссоциации обеих аминогрупп восстановленных частиц. Считается, что один атом водорода в молекуле восстановителя прочно связан с мостиковым атомом азота. Мейер и Тредвелл ['31] также получили результаты для крезилового синего, в основном совпадающие с результатом Коена и Прислера для восстановителя. Они даже обнаружили, что окислитель диссоциирует при очень низких значениях pH и р/С=0,62, что соответствует потенциалу Е6~О,55 В, т. е. эта величина меньше той, которую получили Коен и Прислер (табл. VIII.5). Мейер и Тредвелл установили также образование семихинона, но не сообщают об этом никаких подробностей.
Нильский синий умеренно растворяется в воде с образованием коллоидных растворов, Коен и Прислер ['16] обнаружили заметный солевой и концентрационный эффекты электродного потенциала. Только при концентрации меньше 10-7 моль-л-1 нильский синий ведет себя подобно истинному раствору. Три других красителя
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
85
меют небольшой концентрационный эффект. Нильский синий в кислом и нейтральном растворах (при рН<8,4) окрашен в синий цвет а в щелочном — в красный. Продукт его восстановления бесцветный при pH 1—8,4 и красноватый при более высоких значениях pH. Оба капри синих и крезиловый синий используются в качестве индикаторов [16], так как в пределах pH 1—10 растворы их окрашены в синий цвет, а восстановленные формы бесцветны.
В качестве окислительно-восстановительного индикатора предложен краситель прюн, который является метиловым эфиром гал-лоцианина ['103]:
(CH3)2N-
(6О>
Прюн разлагается до диметиланилина при pH>10. Его электродные потенциалы были измерены в пределах pH 0—10 [103]. Они удовлетворяют уравнению
[Н+Г + КГ1[Н+]з + кГ1КГ2[Н+р
Ео + 2F In [Н+]2 + К01 [Н+] + КО1Ло2 ’
где КО1 и Кп — константы щелочной диссоциации диметиламиногруппы окислителя и восстановителя соответственно, а Кг2 и /<О2 константы диссоциации фенольной группы. Константа диссоциации Кг3 второй фенольной группы не фигурирует в изучаемой области pH. Прюн имеет интенсивную фуксиново-красную окраску в кислом растворе, глубокую синюю при pH 4—8 и пурпурную при рН>8. Этот краситель легко растворяется в воде, причем синие частицы постепенно образуют агрегаты, а пурпурные неустойчивы. Восстановленная форма имеет желтый цвет в кислом и нейтральном растворах; в кислом растворе она окисляется на воздухе медленнее, чем в нейтральном или щелочном. Концентрационный эффект этого красителя сравним с аналогичным эффектом нильского синего. Данные табл. VIII.5 для этого красителя получены при работе с 1,5 • 10-4 М. растворами. Мелвил и Ричардсон [103] с успехом использовали для определения аскорбиновой кислоты прютт Как титрант и как индикатор, а при титровании аскорбиновой кислоты гексацианоферратом(III) прюн служил только индикатором.
86
ГЛАВА VIII
Два кислотных оксазина были предложены в качестве индикаторов [104]—G-кислота фенонафтоксазина и R-кислота фено-.нафтоксазина:
G-кислота фенонафтоксазина
R-кислота фенонафтоксазина
Оба красителя легко растворяются; изучение их проводилось в кислых растворах. Реакции окисления и восстановления их идут с большой скоростью и обратимо. В изученной области pH была обнаружена одноступенчатая ионизация восстановителя. Зависимость Ет—pH в каждом отдельном случае представляется уравнением
Ет=Е'0 + -^- ln([H+]2 + ^ [Н+]), (62)
где Лп — константа щелочной диссоциации диметиламиногруппы. Установлено образование семихинона, которое достигает максимума при pH 6—7 для R-кислоты фенонафтоксазина и при pH 4 для G-кислоты фенонафтоксазина. Обе окисленные формы дают красно-фиолетовые водные растворы, которые становятся синими в сильной серной кислоте (>3,5 моль-л-1)- Восстановленные формы желтоватые, а промежуточный семихинон зеленовато-желтый. Метод синтеза этих красителей приведен в работе [Ю4].
Окислительно-восстановительные свойства резоруфина описаны в работе [105]. Этот краситель является продуктом восстановления резазурина. Реакция восстановления резазурина в резоруфин необратима, а реакция дальнейшего восстановления резоруфина обратима. Эти реакции можно представить следующим образом:
дигидрорезоруфин
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
87
Обратимая система соответствует реакции Ох + 2е < > Red2-,
а электродное уравнение для потенциала, удовлетворяющее этой „еакции и экспериментальным результатам, полученным при концентрации красителя 1,5-10~5 моль-л-1 и в отсутствие кислорода, имеет следующий вид:
р г- RT in + +
Е»+ 2F 111 [H+J + X01 ’
где К01, Кп и ЛГ2—константы фенольной диссоциации. Резазурин имеет синюю окраску, резоруфин — розовую, а продукт восстановления резоруфина — бесцветную. При восстановлении резоруфина титаном (III) или окислении дигидрорезоруфина бихроматом в сильнокислой среде (рН<3) образуется сине-зеленое промежуточное соединение. Резоруфин и резазурин наряду с другими оксазинами используются как индикаторы в броматометрии [106]. К другим оксазинам относятся феноксазоны: 2-амино-З-феноксазон, 7-амино-З-феноксазон, 7-амино-1-метил-3-феноксазон и галлоцианин. Многие методы титрования были выполнены в присутствии этих соединений в качестве индикаторов; сюда относится определение раствором бромата калия соединений мышьяка(III), сурьмы (III), гидразина, аскорбиновой кислоты, титана(III), олова(II) и железа (II). Под действием бромата калия солянокислый раствор этих красителей становится вместо красного или оранжевого желтым или желто-зеленым, по-видимому, вследствие бромирования. Под действием сильных восстановителей окраска изменяется при титровании от бесцветной до желтой. Косвенное определение таких окислителей, как перекись водорода, персульфат калия и бромат калия, проводили путем добавления избытка мышьяка (III) и конечного титрования броматом калия.
В качестве индикаторов предложен также незамещенный феноксазон [Ю7]:
Н аЖ—оэХ о о
феноксазон
О нем почти не имеется потенциометрических данных, но установлено, что нормальный потенциал равен 0,35 В. Окисленная форма Красная, а восстановленная синяя. Феноксазон применяли для титрования олова (II) железом (Ш) в растворе соляной кислоты и получили точные результаты.
«8
ГЛАВА VIII
Пять других оксазинов исследовались [108] в узкой области значений pH от 3 до 8. Имеются только данные о значениях Е1*™.
прочный синий В для хлопка +0,08; С. I. 51190
новый метиленовый голубой NGG £’1= +0,025, С. I. 51195
мускарин DH
Е^ = +0,05; С. I. 51205
Изучались также окислительно-восстановительные свойства ряда .других оксазинов; отмечено значительное образование семихинона {109].
Оксазины нашли сравнительно малое применение как индикаторы в титриметрии. Это кажется неожиданным, если учесть преимущества, например, галлоцианина перед метиленовым голубым. Помимо сказанного выше, бриллиантовый крезиловый синий использовался при титровании сурьмы (V) раствором титана (III) [38], а галлоцианин применялся в ряде определений с участием .ванадия(П) ['101, НО, 111]. Галлоцианин и метиленовый голубой указывают на окончание реакции (64) при титровании ванадия (II)
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 8fi>
и железа(Ш):
V2+ + 2Fe3+ -> V4+ + 2Fe2+ (64>
Как и следовало ожидать, эта реакция проходит через промежуточную стадию образования ванадия (III); последний окисляется в. ванадий (IV) медленно. Для облегчения процесса титрования необходимо нагревание (80°С) [101] или добавка меди(П) [НО] в качестве катализатора. Восстановители, например олово(II), и окислители, например хлорат, персульфат и перекись водорода, можно определить [111] при помощи реакции с избытком железа (Ш) (Для олова) или избытком ванадия(II) (для окислителей)г а затем избыток оттитровать по реакции (64) в присутствии галлоцианина или метиленового голубого в качестве индикатора.
8. ТИАЗИНЫ
Эти соединения сходны с азинами и оксазинами с той лишь разницей, что в их молекуле мостиковый азот или кислород замещен на серу. Формула фенотиазина в общем виде
И
фенотиази*
Производные фенотиазина, используемые как индикаторы, имеют заместители, в основном в положениях 3 и 7, а в некоторых случаях и в других местах. Нормальные потенциалы составляют примерно 0,5 В по отношению к нормальному водородному электроду, и, следовательно, они значительно выше потенциалов азинов и ин-дигосульфонатов, но ниже потенциалов индофенолов, т. е. занимают промежуточное положение.
Первыми тиазинами, подвергнутыми детальному потенциометрическому изучению, были метиленовый голубой и тионин (известный также под названием фиолетовый Лаута) [8]. Окисленные формы имеют вид
метиленовый голубой
тионин (фиолетовый Лаута)
so
ГЛАВА VIII
Реакция восстановления метиленового голубого:
(65)
При определении окислительно восстановительных потенциалов метиленового голубого встретились трудности [8]. Метиленовый голубой плохо поддавался очистке, при сушке выше ПО °C разлагался. Анализ тщательно перекристаллизованных образцов показал в них высокое содержание хлора и серы. Предполагалось неполное метилирование и в связи с этим наличие электроактивных примесей. Для устранения этих помех приходилось [8] доставать ряд торговых препаратов и сопоставлять все результаты, полученные для каждого из образцов. С тиазинами возникает другая трудность, которая состоит в том, что они как основания стремятся образовать нерастворимые соли с некоторыми из кислотных окислителей или восстановителей, используемых в потенциометрических исследованиях; например, сюда относятся гексацианоферрат (III) и индигокармин. Поэтому, чтобы получить значения Е'о, авторы вынуждены были использовать метод смесей, а не титри-метрические методы, хотя восстановитель можно определять тит-риметрически с помощью бензохинона. Следующая трудность состоит в том, что восстановленные частицы (метиленовый белый) оказались чувствительными к свету; растворы этих частиц, оставленные на свету в отсутствие кислорода, быстро приобретают синий цвет метиленового голубого. Так как метиленовый белый растворяется максимально до концентрации 5-10-4 моль-л-1 в кислых растворах и до 2-10-5 моль-л-1 в щелочных растворах, то все потенциометрические измерения проводились в очень разбавленных растворах. Это, конечно, усиливает эффекты, обусловленные адсорбцией частиц на поверхности стекла; для метиленового голубого это хорошо известный эффект [8]. Кларк с сотр. [8] нашел также, что смеси метиленового голубого и метиленового белого дают потенциалы, величина которых варьирует с изменением концентрации красителя. Эта зависимость гораздо больше той, которая была обнаружена для сафранинов. При 10-кратном изменении концентрации изменение потенциала равно 10—12 мВ. Зависимость
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
91
т адсорбции в той работе не рассматривалась. Трудности, воз-°икшие при первоначальном изучении метиленового голубого, описаны подробно, и эти сведения могут оказаться ценными при попытке использовать метиленовый голубой в качестве индикатора (в прошлом этот краситель широко применялся для этой цели). Главным преимуществом метиленового голубого является его интенсивная окраска (намного более яркая, чем, например, окраска индигосульфонатов), и поэтому его можно использовать в разбавленных растворах. Кроме того, он находится в обычной области потенциалов, где выбор небогат.
Полученные результаты [8], несмотря на указанные трудности, описываются уравнением для области pH от 1 до 13
„ , RT 1 [Н+Г + КГ1 [Н+р + КТ1КГ, [№]2
Ет=Ев + -^^п -
(66)
[Н+] + Л,
где Koi—константа диссоциации иона водорода полярной аминогруппы в окислителе. Константа диссоциации второй аминогруппы пренебрежимо мала. У восстановителя атом водорода фиксируется мостиковым атомом азота, поэтому Кг, и Кг2— константы щелочной диссоциации иона водорода каждой аминогруппы. Уравнение (66) относится к тионину. Для метиленового голубого К01 тоже очень небольшая и не поддается определению, поэтому уравнение (66) упрощается до уравнения
Ет= Е'о + Ш ([Н+р + КГ1 [Н+]2 + КГ1КГ2 [Н+]) (67)
Это уравнение удовлетворяет данным, согласно которым при низких значениях pH (<5) тангенс угла наклона составляет —0,09 В/pH, а при высоких pH (>6) составляет —0,03 В/рН. Тем не менее оно не согласуется с уравнением, предложенным Кларком [26], в которое, как полагают, вкралась опечатка. Другие авторы [31, 112] измерили формальные потенциалы и константы диссоциации метиленового голубого [31, 112] и тионина [31] и получили результаты, по существу совпадающие с результатами Кларка с сотр. [8]. Михаэлис и др. [113] попытались идентифицировать семихинон в тиазиновых системах. Результаты показали, что семихинон может существовать в равновесии с красителем и его лейкоформой, и в лучшем случае концентрация семихинона составляет несколько процентов от общей концентрации красителя в области pH значений, соответствующих обычным буферным растворам; в случае тионина присутствие желтого семихинона было обнаружено в концентрированных растворах серной кислоты (5— 13 моль-л-1). Для метиленового голубого присутствие семихинона Установлено только при концентрациях серной кислоты выше ' моль-л-1, а поэтому практически при использовании метиле
92
ГЛАВА VIII
нового голубого в качестве индикатора образованием семихинона можно пренебречь.
Краситель бриллиантовый ализариновый синий изучали Михаэлис с сотр. [102]. Исследования проводились при pH от 4,5 до 11 л не касались области низких значений pH, где можно было бы получить значения Ео и рЕг(. Этот краситель является сульфопроизводным гиазина и имеет формулу
бриллиантовый ализариновый синий
Растворимость его невысока, но достаточна, чтобы можно было использовать его как индикатор. Раствор в чистой воде дает осадок, который отфильтровывают и получают устойчивый синий раствор. Теоретически этот краситель должен удовлетворять уравнению (67), но так как неизвестны значения рКГ1 и Ео, то это бесполезно. В области pH 6—-11 тангенс угла наклона кривой равен —0,06 В/pH, за исключением небольшого участка, соответствующего pH—8, где проявляются рЕо, и рМ2. Типичные значения Ет. будут Ет=—0,040 В, Ет=—0,173 В и Ет=—0,337 В.
Описано [114] приготовление тионола из фенотиазина и замерены его формальные потенциалы при pH от 1,42 до 9,90 и температуре 21 °C. Окислительно-восстановительная реакция тионола:
ТИОНОЛ
В другой работе [115] излагается подробное потенциометрическое изучение этого красителя, которым установлено, что Ет =+0,160 В, и показано, что тангенс угла наклона кривой Ет—pH изменяется при pH от 2 до 5, но объяснения этому не дается. Граник и Михаэлис [116] провели более тщательное потенциометрическое исследование этой системы при значении Ет — +0,037 и не обнаружили каких-либо особенностей при низких значениях pH. Резуль-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
93
Таблица VIII.6
ОКИСЛИТЕЛЬНО
ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ рК НЕКОТОРЫХ ТИАЗИНОВ
Название £о m Температура, °C ркО1 VKr 1 Область PH С. I. |ьо| Литература
Тионин Толуидиновый синий 0,563 0,064 30 11,0 4,38 5,30 1—13 52 000 8, 31
0,534 0,034 20 11,04 4,81 5,41 1—13 52 040 112, 117
Тиазиновый синий 0.537 0,027 25 —• PKr.+PKr,= = 10,30 0,5-12 52 025 31
Метиленовый голубой 0,532 0,011 30 — 4,52 5,85 1—13 52 015 8, 31, 112
Новый метиленовый голубой 0,46 —0,021 25 —’ 3,54 4,82 0,5-12 52 030 31
Ализариновый бриллиантовый синий — —0,173 25 8,2 <6 7,8 4,5—11 52 055 102
Тиокол 0,53 0,037 21 5,8 9,2 10,5 1—13 — 114—116
тэты этих авторов находятся в согласии с уравнением (69), а значения Ео, рК01, рДц и рКГ2 приведены в табл. VIII.6.
2Р 1П [Н+]4-/С01
Здесь КТ1, Ег2 и /Сох—константы диссоциации фенольных групп. Их определяли в области pH от 1,46 до 12,67 и обнаружили, что при рЙ<2,3 образуется значительное количество семихинона, т. е. при гораздо меньшей концентрации кислоты, чем в случае тиони-на или метиленового голубого. Определения проводились в кислом растворе, содержащем 20% этанола, так как краситель плохо растворяется в кислой среде. Расхождения результатов в двух работах-— [115] и [116]—можно, вероятно, объяснить различной степенью чистоты препарата. В работе ['116] описан способ получения чистого тионола.
Мейер и Тредвелл [31] проводили потенциометрическое исследование двух других тиазинов — тиазинового синего и нового метиленового голубого:
N
N(C2H6)
тиазиновый синий
6Н новый метиленовый голубой
2
94
ГЛАВА VIII
Ни у одного из этих красителей не обнаружено образования семихинона в пределах pH от 0,5 до 12. Окислительно-восстановительные потенциалы обоих красителей соответствуют уравнению (67). Однако в случае тиазинового синего трудно раздельно определить /Сп и КГ2 и для этого красителя приводится среднее значение рК: PS+P^=5J51 _ I
которое не подходит для уравнения (67); но если p/Cr2=pKr1 =5,15, то тогда уравнение (67) годится для всех значений pH, за исключением области pH от 4 до 6.
Никольский и Пальчевский [112, 117] сообщают результаты изучения толуидинового синего:
толуидиновый синий
Формальные потенциалы этой системы определялись в пределах от 1 до 13 и хорошо согласуются с уравнением (66). Полученные результаты приведены в табл. VIII.6.
ОБЛАСТИ ПРИМЕНЕНИЯ
Несмотря на трудности использования метиленового голубого в качестве индикатора, его широко применяли в различных методах титрования. Другие тиазины тоже иногда применяются. Как можно было ожидать, в основном с ними работали при титровании сильными восстановителями. Метиленовый голубой использовали при определении железа (III) и хрома(VI) титаном (III); титрование проводилось в солянокислой среде и в отсутствие воздуха [90]. В другой работе [118] метиленовый голубой использовался для установления конечной точки титрования титана (III) перманганатом. Можно определять титан и железо последовательно в их смеси путем титрования перманганатом. Оба металла восстанавливают восстановителем Джонса, а затем титруют при СО °C перманганатом, причем первую конечную точку, соответствующую титану, устанавливают при помощи метиленового голубого, а вторую, соответствующую железу, с помощью трис- (о-фенантролин) железа (II). Подобный метод был еще раньше предложен Смитом и Куртцем [119], которые использовали в качестве титранта раствор церия (IV).
Титан в присутствии ниобия можно определить, титруя титан (III) раствором железа (III) [120]. Титан восстанавливают цинковой амальгамой в 25 %-ной винной кислоте; в этих условиях ниобий не восстанавливается. Поэтому титан можно определить
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
95
становлением его до титана (III) и титрованием последнего ВОСооидом железа(III) в присутствии метиленового голубого.
ХЛ Ружичка и Адамек [121] использовали тионол, тионолин и онин в качестве индикаторов при титровании раствором титана (Ш) Формула тионолина, о котором до сих пор не упоминалось, следующая:
тионолин
В концентрированном растворе серной кислоты образуется семихинон тионолина [109], однако тщательное потенциометрическое исследование при низких концентрациях кислоты не проводилось. В работе [122] сообщаются некоторые потенциометрические результаты: Ео для тионолина равно 0,570 В, а Ет =+0,126 В; оба значения получены при 40 °C. Тионолин плохо растворяется (за исключением сильной сернокислой среды); концентрация насыщенного водного раствора менее 10-4 моль-л-1, поэтому при измерениях использовался 96%-ный этанольный раствор [122]. Тионолин применяли [122] в качестве титранта для хрома (II), олова (II) и титана (III) (причем он восстанавливался до лейкотионолина), а также для определения хлора и брома (причем продуктом реакции был не лейкотионолин, а тетрагалогенотионолин, например тетрабромтионолин):
Н I N
лейкотионолин
Вг Вг
тетрабромтионолин
В работе [121] использовался тионолин, а также тионол и тионин, Для титрования бихромата, железа (III), ванадата, хлорида, золота (III), платины (IV), гексацианоферрата (III) и осмиевой кислоты раствором титана (III). Титрование проводилось в среде 4 М соляной кислоты при 60—70 °C в инертной атмосфере. Единственным ограничением было то, что тионолин непригоден для титрования хлорида золота(Ш), а тионол — для титрования платины(1У) или осмиевой кислоты.
Для определения окислителей титаном (III) был предложен метод [123], по которому метиленовый голубой реагирует как интермедиат. Раствор метиленового голубого непосредственно восстанавливают до метиленового белого титаном (П1) в растворе со
96
ГЛАВА VIII
ляной или серной кислоты. Затем вводят окислитель [хром (VI) или ванадий (V)] и образовавшийся вновь метиленовый голубой оттитровывают титаном (III). Подобный метод был предложен с использованием хрома (II) [124], и по нему определяли хром(VI), персульфат, перйодат, железо (III), перекись натрия и перекись водорода. Необходимо работать в инертной атмосфере. Тандон ц Меротра [’39] тоже показали, что можно прямо титровать железо (III) хромом (II) в присутствии индикатора метиленового голубого. Затем Миттел, Тандон и Меротра [101] нашли, что в качестве восстановителя можно использовать ванадий (II). Тогда при помощи метиленового голубого устанавливается конечная точка реакции окисления ванадия (II) до ванадия (IV), а вторая стадия титрования от -J-3B до +4В соответствует медленной реакции при комнатной температуре; для ускорения реакции нужно повысить температуру до 80°С [101] или проводить титрование в присутствии катализатора сульфата меди(II) [НО].
Метиленовый голубой был использован в качестве индикатора при титровании железа (III) тиосульфатом натрия ['125] или хлоридом олова (II) [125], а также как титрант для определения олова ['126]. Из многочисленных случаев применения приведем только несколько. Определение молибдена (V) с помощью раствора железа (Ш) выполнялось в присутствии метиленового голубого или тионина [127]. Так как вблизи конечной точки идет медленная реакция метиленового голубого с молибденом (V), конечную точку устанавливают преждевременно. Реакцию проводят в инертной атмосфере при 90 °C, в среде 0,5 М соляной кислоты и в присутствии лимонной или винной кислоты.
Железо (II) можно применять как сильный восстановитель при высокой концентрации- фосфорной кислоты, например для титрования урана (VI) ['128], молибдена (VI) [129] и ванадия (IV) [130] в фосфорной кислоте, концентрация которой больше 10 моль-л-1, используя метиленовый голубой или тионин в качестве индикатора. Необходимо работать в инертной атмосфере. Метиленовый голубой использовали также при титровании ванадия (IV) гексацианоферратом (III) калия в среде 2,5—4 М гидроокиси калия ['131].
В качестве индикатора предложен [132, 133] хлорпромазингид-рохлорид. Его нормальный потенциал (0,70 В) значительно выше потенциала других тиазинов, и случаи его применения совершенно другие. Формула его следующая:
(CH2)3N(CH3)2 -
хлорпромазингидрохлорид
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
9/
и окислении хлорпромазингидрохлорида образуется ярко-крас-I “ вободный радикал или семихинон, который устойчив в кис-ны,и де Реакция окисления обратима, а дальнейшее необрати-Л0И окисление красного свободного радикала сильными окисли-М°еями приводит к бесцветному сульфоксиду. Эти реакции можно представить в следующем виде:
(СН^СНзЬ (СН^СН^а
ОН хлорпромазин- свободный радикал
гидрохлорид красного цвета
использовали как индикатор при тит-
Хлорпромазингидрохлорид
ровании железа (II), аскорбиновой кислоты, гидрохинона и мышьяка (III) раствором церия (IV) ,[132]. Первые три вещества титруют в емеси серной и фосфорной кислот в присутствии небольшого количества метиленового голубого. Окраска изменяется от бледно-голубой до фиолетовой. Разбавленные растворы церия (IV) (0,001—0,005 моль-л-1) дают очень незначительную индикаторную ошибку. Можно проводить определение и без добавления метиленового голубого, но тогда конечная точка (менее четкая. Мышь-як(Ш) реагирует очень медленно с церием (IV), поэтому для его определения поступают следующим образом. В раствор мышьяка (Ш)
вносят иодид и в избытке приливают раствор церия(IV). Спустя 30 мин добавляют в избытке железо (II) и оттитровывают его раствором церия (IV), как описано выше. Вместо церия (IV) ельзя использовать бихромат, поскольку получаются ошибочные Результаты для железа и аскорбиновой кислоты. При работе с растворами церия (IV), концентрация которых больше 0,01 моль-л-’, ельзя^применять индикатор, так как в этих условиях красный сво-°дный радикал хлорпромазина разлагается.
7—Э18
98
ГЛАВА VIП
Хлорпромазингидрохлорид использовали также при титровании ванадатов [133]. В этом случае нормальный потенциал был 0,736 В в 1 М серной кислоте при 28 °C. Для предотвращения разложения применялся 0,1%-ный раствор индикатора с добавлением 1% лимонной кислоты [133]. При хранении в темной склянке такой раствор устойчив в течение долгого времени. Титрование железа (II) и молибдена (V) проводилось в смеси серной и фосфорной кислот. Окраска изменялась от синевато-зеленой до фиолетовой, что обусловливалось присутствием ванадия (IV). Окраска в конечной точке устойчива в течение 15 мин. Для титрования достаточно 1 мл раствора индикатора, что дает поправку на индикаторную ошибку в количестве 0,04 мл 0,01 М ванадата.
Метиленовый голубой был также предложен в качестве индикатора в иодометрии [134]. Иод обесцвечивает синюю окраску метиленового голубого вследствие образования гидроиодида тетра-иодметиленового синего. Восстановители, такие, как олово (II), сульфит, тиосульфат и мышьяк(III), разрушают синюю окраску метиленового голубого, и поэтому его можно использовать вместо крахмала в йодометрических методах. Имеются данные о том, что работать с метиленовым голубым так же легко, как с крахмалом, а в разбавленных растворах изменение окраски еще более четкое. Метиленовый голубой можно также использовать для определения растворенного кислорода [135]. Для этого 5-10-5 М метиленовый белый вводят в реакцию с кислородом, а затем титруют тиосульфатом образовавшийся метиленовый голубой.
В качестве метода анализа можно использовать каталитические реакции, в которых участвует метиленовый голубой. Файгль и Уэст [136] разработали метод определения селена, который катализирует реакцию восстановления метиленового голубого сульфидами, а Богнар ['137] определял молибден по его каталитическому действию на реакцию метиленового голубого с гидразином.
9. ПРОИЗВОДНЫЕ ВАРИАМИНОВОГО СИНЕГО
В новую группу окислительно-восстановительных индикаторов, введенных в практику за последние годы, входят производные ва-риаминового синего, или 4-амино-4,-метоксидифениламина. Структурная формула в общем виде:
1
Это наиболее важный и широко используемый индикатор группы. Его производные [28, 138, 189], обладающие индикаторными свойствами, имеют различные заместители в положении 2. Эти соединения являются производными дифениламина (см. гл. VIIIB), но
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
99
Таблица VIII7
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ И ЗНАЧЕНИЯ РК ПРИ 30 °C
0К НЕКОТОРЫХ ПРОИЗВОДНЫХ ВАРИАМИНОВОГО СИНЕГО
Вариаминовый синий, или 4-амино-4'-метоксидифениламин
Название £о Е7 m рдО1 РКо2 ркГ1 ркг 2 Область PH Литература
о Аминов ар и аминовый синий 0,532 0,172 — 11,5 5,0 — 2—14 28
2-Метоксивариаминовый синий 0,636 0,267 7,5 10,6 5,2 9,6 0—14 28
2-Метилвариаминовый синий 0,686 0,297 7,0 10,5 5,7 9,4 0—14 28
Вариаминовый синий (C.L 37255 [80]) 0,712 0,310 6,6 — 5,9 — 1,5—6,3 142
Вариаминовый синий анилид 2-карбоновой кислоты 0,754 —— ~5,5 — ~4,7 — 2-5 28
Вариаминовый синий 2-сульфокислота 0,776 — 4,1 — — •— 1-6 28
Вариаминовый синий анизидид 2-сульфокислоты 0,780 0,335 5,0 —- 5,9 •— 1—7 28
Вариаминовый синий метиловый эфир 2-суль-фокислоты 0,786 — 5,1 — — — 3—5 28
Вариаминовый синий аиилид 2-сульфокисло-ты 0,788 0,338 4,7 — 5,7 — 1—7 28
их формальные потенциалы (0,532—0,788 В) лежат в более низкой области, чем потенциалы дифениламинов (см. табл. VIII.7). Имеется много предложений об использовании этих соединений в качестве индикаторов в окислительно-восстановительных, комплексе метрических и аргентометрических методах титрования, а также в качестве реагентов в спектрофотометрии. Эрдей [140] написал обзор всех случаев их применения, опубликованных до 1958 г.
Эрдей и Бодор [141] впервые в 1953 г. предложили использовать вариаминовый синий как окислительно-восстановительный индикатор в растворах с pH 1,5—6,3. Эти авторы сообщают, что вариаминовый синий восстанавливается и окисляется обратимо при Резком изменении окраски. В кислой среде он восстанавливается 7*
100
ГЛАВА VIII
с образованием бесцветного катиона, который можно окислить, например, бромом или хлором с отрывом двух электронов и превращением в иминохинон фиолетово-красного цвета. Эту реакцию можно представить в следующем виде [142]:
H2N=/ \=N—/~А-ОСН3 + 2Н+ + 2е
(70)
Иминохинон реагирует обратимо с другой восстановленной молекулой и образует ярко-синий хингидрон по реакции
R + Q D,
где К— восстановленная молекула, Q — иминохинон, a D — синий хингидрон. Последний частично диссоциирует, и получается синефиолетовый свободный радикал семихинона S,
D 2S,
который идентифицировали методом ЭПР спектроскопии [143] при титровании вариаминового синего. Концентрация свободного радикала достигает максимума в момент титрования, соответствующий присоединению одного электрона, и спадает до нуля в стадии окисления двумя электронами. Установлено, что образование семихинона идет энергично в четыреххлористом углероде или бензоле и достигает максимума, когда диссоциировано 25% хингидрона. Свободному радикалу соответствует следующая формула:
Его образование, т. е. диссоциация хингидрона, значительно меньше в водных растворах и достигает максимума 0,66% в области pH от 2 до 3.
При значениях рН<1,5 хингидрон разлагается на аммиак и соединение, которое не обладает свойствами индикатора, по реакции [142]:
h2^=Q=n_0-och3 + Н2О >
--> О=/ S=N— осн3 + NH3
При рН>6,5 иминсхинон тоже диссоциирует с образованием бесцветного продукта, и поэтому из-за этих двух реакций разложения можно применять вариаминовый синий только в ограниченной об-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
101
(71)
пН Ряд производных вариаминового синего оказался устой-лаСТИ рв 'значительно более широкой области pH, и их следует чИрппочесть самому вариаминовому синему.
П Бее .соединения -в табл. VII1.7 подвергаются обратимым двух-ктоонным окислительно-восстановительным реакциям по меха-эле р соответствующему реакциям вариаминового синего {28, 138, ^391 В реакции 2-метил- и 2-|Метоксипроизводных вариаминового щего установлено образование промежуточного семихинона [28].
Подобно другим группам индикаторов, значения потенциала Ео производных вариаминового синего сильно зависят от pH среды. Диссоциацию обоих атомов азота в окисленных и восстановленных частицах следует рассматривать в строгой зависимости от взаимосвязи Ещ — pH, данной ранее уравнением (27), и этому уравнению удовлетворяют результаты, полученные для 2-метокси- и 2-метил-вариаминового синего (табл. VIII.7). Из других соединений этой группы, включая сам вариаминовый синий, константы диссоциации окисленной и восстановленной форм были определены только для 4-аминопроизводного ['138, 142, 144, 145], а уравнение (27) упрощается до следу ющего вида:
RT [Н+р + КГ1[Н+]2
m ^0 I 2F [Н+Ц-К01
где Koi и КГ1 имеют значения, которые можно определить из табл. VIII.7.
Вариаминовый синий является легкодоступным торговым препаратом; методы получения его производных описаны подробно [138]. Обычно вариаминовый синий используют в виде гидрохлорида. Работают с 1%-ными водными растворами, которые устойчивы только в течение недели, тогда как твердые смеси с хлоридом натрия или безводным сульфатом натрия (соотношение 1 : 100) устойчивы, особенно при хранении в темных склянках. Твердый гидрохлорид чувствителен к действию света, поэтому препараты, имеющие первоначальный белый цвет, вскоре синеют. Растворы также становятся при хранении синими, а через несколько дней в них выпадает осадок. Поэтому торговый препарат гидрохлорида вариаминового синего В тоже синий вследствие образования про-эти™ окисления за время хранения. Описаны методы очистки
х ^веществ [140]. Один из них приводится ниже.
Доба СТвоРЯЮт Ю г вариаминового синего в 200 мл горячей воды. чивак)ЛЯ|К? 0’4 Г дитионита натрия. Полученный раствор обесцве-НИ[1 3 г активированного угля и фильтруют в горячем состоя-50 .. j Оесйветному или светло-желтому фильтрату добавляют фи.1ЬтпОНась1щенного раствора хлорида натрия. Охлаждают, от-мываг— ?1Вают выпавшие белые игольчатые кристаллы, их про-г' I" ' хп°ЛОДНОЙ ВОДой- сушат в вакууме при комнатной темпера-Ранят в склянке из темного стекла.
102
ГЛАВА VIII
Метод получения сульфата вариаминового синего предложен g работе [146]. Это соединение несколько более устойчиво; его мои! но применять в тех случаях, когда нежелательно присутстви хлоридов. Твердые смеси с безводным сульфатом натрия (соотн! шение 1:400) устойчивы в течение неограниченного времени прц хранении в склянках из темного стекла. Для титрования достаток, но брать 200 мг такой смеси. Для работы использовался та кд 0,2%-ный водный раствор или 0,4%-ный в 10 %-ной уксусной кис лоте [146], но эти растворы вскоре становятся синими и vepej 4—5 дней дают осадок.
Окисленные формы вариаминового синего и большая часть его производных имеют синюю окраску только в кислых раствооах При pH 5—7 они желтеют и поэтому неудобны для использованц I в качестве индикаторов. Изменение окраски соответствует измен нию формы индикатора вследствие диссоциации при рД'.( (табл. VIII.7) [142]. Исключение составляет 2-аминовариамиж вый синий, который остается красным вплоть до pH 12 [28], а также ранее описанный 4-амино-2-окси-4'-метоксидифениламин илв 2-оксивариаминовый синий [147—150]; это соединение остает фиолетовым до pH 12 [148] и почти устойчиво в области pi от 2 до 12. Следовательно, его можно использовать как окислительно-восстановительный индикатор как в кислых, так и в щело1. ных средах, например при титровании аскорбиновой кислоты ра твором гексацианоферрата (III) при pH 6—8. Формальный поте циал Ет = 0,565В [147] и на 35 мВ ниже, чем для самого вар аминового синего. При окислении 2-оксивариаминового синего возможно получение двух продуктов окисления, которые могут имег о- или n-хиноидную структуру:
‘ОН
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
103
лено, что именно n-хиноидная структура действительно Устинов СИне-фиолетовую окраску окисленной формы ['149]. п*еСПеспользовании окислителя умеренной силы реакция обратима, ПРИ Икак сильные окислители, как, например, бром, приводят к необратимым изменениям окраски.
ОБЛАСТИ ПРИМЕНЕНИЯ
Несмотря на то что вариаминовый синий устойчив и пригоден как индикатор в несколько ограниченной области значений pH, установлено, что его можно применять в самых разнообразных вариантах. Наряду с обычными методами окислительно-восстановительного титрования его используют также в ряде случаев титрования по методу осаждения и комплексометрического титрования, при которых для установления конечной точки используют способность самого малого избытка одного из реагентов окислять или восстанавливать вариаминовый синий. Производные вариаминового синего, за исключением 2-оксипроизводного, применяются редко, хотя, учитывая ограничения в используемой области pH и небольшие сдвиги Ео, можно было бы предполагать, что эти производные вполне заменят вариаминовый синий.
Окисленная форма вариаминового синего разлагается в растворах, pH которых ниже 1,5, тогда как в этих условиях восстановленная форма устойчива. Следовательно, такие восстановители, как железо(II), можно титровать в концентрированных растворах соляной кислоты сильным окислителем, как например, церием (IV) и бихроматом, в присутствии вариаминового синего [140—141]. Образующееся железо(III) следует связывать в комплекс с фторидом или пирофосфатом, а синяя окраска, появляющаяся в конечной точке, обычно очень неустойчива. Железо(II) в среде пирофосфата можно титровать раствором ванадия(V) [151], причем вариаминовый синий добавляют незадолго до наступления конечной точки. Однако, несомненно имеются многие гораздо более подходящие индикаторы для этих случаев титрования, а вариамино-выи синий находит более значительное применение в растворах, которые имеют более высокие pH и которые титруют такими титрантами, как аскорбиновая кислота, нитрат серебра, иод, нитраты Ртути(Ц) и ртути(I).
Вариаминовый синий используют в качестве индикатора при оп-П?*Г?ении железа(Ш) [144], ванадия(У) [152—153], серебра(1)
Ь Ртути (II) [155], церия (IV) [156] и иода [157], титруя их КоРбиновой кислотой. Так как сильные окислители разлагают добИКатоР’ то ПРИ титровании аскорбиновой кислотой индикатор g авляют в систему незадолго до наступления конечной точки. ШаТИх же случаях рекомендуется [140] к концу титрования повыть температуру до 50—60°C. Прямое титрование ванадия^)
104
ГЛАВА VIII
[152] может не давать точных результатов, а размер ошибки зависит от скорости процесса титрования. При медленном титровании продукт окисления аскорбиновой кислоты — дегидроаскорбиновая кислота вступает в реакцию с непрореагировавшим ванадием (V). Для определения ванадия (V) был предложен вариант косвенного метода [153], по которому сначала восстанавливают ванадий (V) до ванадия(IV) железом(II), а затем оттитровывают образованию-еся железо (III) аскорбиновой кислотой в присутствии вариаминового синего до бледно-голубой окраски, характерной для ванадия (IV).
Иод [157] окисляет вариаминовый синий при рН>1,5, поэтому в присутствии этого индикатора можно титровать иод аскорбиновой кислотой. Многие окислители, которые переводят иодид в иод, определяются титрованием иода аскорбиновой кислотой [157]. Определение иода раствором тиосульфата можно проводить [140], используя в качестве индикатора вариаминовый синий. При тит-| ровании аскорбиновой кислотой или тиосульфатом этот индикатор добавляют, как и крахмал, незадолго до наступления конечной точки.
Вариаминовый синий использовали также в качестве индикатора при прямом титровании таллия(III) аскорбиновой кислотой [158, 159]. Таллий(I) окисляют избытком брома в растворе муравьиной кислоты, а после удаления избытка брома титруют таллий (III) аскорбиновой кислотой в присутствии ацетата вариаминового синего как индикатора [158].
ТР+ + С6Н8Ов -> Т1+ + С6Н6О6 + 2Н+ (72)
Эту же реакцию использовали также для косвенного определения гипохлоритов и гипобромитов [159]. Таллий (I) добавляют к гипо-галогениту и окисляют до таллия (III) согласно реакции
СЮ- + Т1+ + 2Н+--> Т13+ + С1- + Н2О (73)
Образовавшийся таллий(III) титруют затем аскорбиновой кислотой по вариаминовому синему. Методика определения представлена ниже.
К раствору 25—250 мг гипохлорита добавляют избыток сульфа,-та таллия (I). Перемешивают и приливают 15 мл 2 М соляной кислоты. Снова тщательно перемешивают и добавляют 2 М гидрч окись натрия до начала образования осадка, затем 10 мл буфер* ной смеси ацетат+уксусная кислота (pH 4,1) и 0,2 г индикаторной смеси, состоящей из вариаминового синего и хлорида натрия (1 :400), после чего титруют 0,05 М аскорбиновой кислотой Д° обесцвечивания. Для определения гипобромита поступают так же, только добавляют немного больше соляной кислоты для полного восстановления гипобромита до брома.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
105
п шенение вариаминового синего в аскорбинометрических ме-- возможно лишь в кислых растворах, но для ряда определе-тоД^ щелочных растворах использован 2-оксивариаминовый си-НИ" Например, гексацианоферрат (III) .количественно восстанавли-НИется до гексацианоферрата (II) (уравнение 36) аскорбиновой кис-ВЯ ой (1481 и можно использовать 2-оксивариаминовый синий для ^становления конечной точки в бикарбонатной среде. Методика состоит в следующем [148]. К нейтральному раствору 100—500 мг гексацианоферрата (III) добавляют 1 г бикарбоната калия и в 80— 100 мл конечного объема вносят 0,3—0,5 г твердой индикаторной смеси из 2-оксивариаминового синего и хлорида натрия (1 :500); титруют стандартной 0,05 М аскорбиновой кислотой. Раствор, вначале окрашенный в красно-коричневый цвет, становится синим, затем при приближении к конечной точке сине-фиолетовым, а в самой конечной точке обесцвечивается, так как индикатор восстанавливается избытком аскорбиновой кислоты.
Гексацианоферрат (II) можно тоже определять по этому методу, предварительно окислив его нитритом до гексацианоферра-та(Ш) [148]. Такие окислители, как иод, бром, иодат, бромат и бихромат, можно определять [149], введя их во взаимодействие с гексацианоферратом (II) и титруя образовавшийся гексацианоферрат (III) аскорбиновой кислотой в присутствии 2-оксивариаминового синего. Семикарбазид окисляют до азота гексацианоферратом (III). Избыток последнего оттитровывают аскорбиновой кислотой по 2-оксивариаминовому синему, как описано выше. Для полного окисления семикарбазида гексацианоферратом(III) в растворе бикарбоната требуется около 25—30 мин.
В нейтральных или слабокислых растворах серебро(I) окисляет вариаминовый синий в синюю окисленную форму; эта реакция была использована для установления конечной точки в ряде случаев титрования раствором серебра (I). Например, определяли аскорбиновую кислоту [154] и железо(П) [161] при 60°C в ацетатном буфере, причем при титровании железа (II) добавляли в раствор фторид. Вариаминовый синий применяли также в качестве индикатора при аргентометрическом определении хлорида, бромида, иодида, роданида и цианида [162]. С помощью вариаминового синего обнаруживается малейший избыток нитрата серебра в конечной точке. При определении хлорида оптимальное pH flRoi Для ДРУГИХ анионов 4,6. Можно также определять серебро I 62] посредством титрования иодидом лри pH 4,6—6. Для определения калия и аммония предложены методы аргентометрическо-в титрования [162—164] с применением вариаминового синего Мо^Честве индикатора. Для этого тетрафенилборат калия или ам-пп0Ия осаждают в уксуснокислой среде, осадок отфильтровывают, затр1ЫВают и ’Растворяют в смеси ацетона и уксусной кислоты, а м титруют тетрафенилборат раствором нитрата серебра по ва-
106
ГЛАВА VIII
риаминовому синему:
[В(С6Н5) J- + Ag+ -> Ag[B(C6H5)J
Нитраты можно определять [164] в кислой или щелочной среде после предварительного восстановления до аммиака с последую щим титрованием, как описано выше.
Вариаминовый синий нашел применение как индикатор в ряде методов титрования нитратом ртути(II) [155, 165, 166] и нитратом ртути(I) [167]. Аскорбинометрическое определение [155] ртути(II) в виде нитрата можно осуществить при pH 2—3 в азотнокислом растворе и при температуре 60°C с использованием вариаминового синего в качестве индикатора. Роданид [165] и цианид [166] оп ределяли титрованием нитратом ртути (II) в ацетатном буфере (pH 4—6). Вариаминовый синий добавляют незадолго до наступ, ления конечной точки, на которую указывает появление фиолето] вой окисленной формы индикатора. При наличии следов нитрата ртути(I) нужно работать при повышенной температуре (60°С), В растворах нитрата аммония нитрат ртути (I) тоже окисляет вариаминовый синий в фиолетовую окисленную форму; эту реакции использовали в разработке методов определения хлорида, бромида и самой ртути (I) [167]. Хлорид и бромид остаются в растворе нитрата аммония и медленно оттитровываются нитратом ртути (I) при энергичном перемешивании до первой неисчезающей фиолетовой окраски индикатора. Для определения ртути (I) добавляю! избыток хлорида, затем этот избыток определяют, титруя нитрат ртути (I), как описано выше.
Вариаминовый синий применяли не только в аргентометрических методах, но также в титровании по методу осаждения свинца и цинка раствором гексацианоферрата (II) калия [168]. Здесь снова проявляется важное значение окислительно-восстановительны! свойств индикатора, так как даже незначительный избыток гексацианоферрата (II) калия восстанавливает его, и в конечной точке раствор из фиолетового становится бесцветным. Для проведения этих титрований необходимо повышение температуры раствора до 60 °C и наличие в нем следов гексацианоферрата (III) калия. Определение свинца по этому методу привело к разработке косвенного определения анионов, как, например, хромата [169, 170], сульфата [169—171] и фосфата [169, 172]. В каждом случае к раствору аниона добавляют избыток нитрата свинца(II), осадок отфильтровывают, промывают и т. д. и титруют в фильтрате непрореагировавший свинец(II) раствором гексацианоферрата (II) калия по ва-риаминовому синему. При определении фосфата были получены точные результаты и при титровании без отделения осадка.
Применение окислительно-восстановительных индикаторов в мФ тодах комплексометрического титрования стало общепризнанным [173, 174], и вариаминовый синий используют при титровании
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 107
(III) [175—178] и меди(П) [179] раствором ЭДТА. Вариами-ле3а'й синий находит также широкое применение как реагент в Н°ектрофотометрии для определения окислителей [140], а также пида цианида и роданида [180]. Максимум поглощения окис-Й иного вариаминового синего находится при 570 нм. Определение ^лида, цианида и роданида основывается на том, что в присут-И° ии этих ионов медь (II) окисляет вариаминовый синий, а степень окисления прямо пропорциональна концентрации
присутству-
ющих ионов.
Вариаминовый синии применяется также в качестве индикатора в каталитических реакциях, используемых в анализе [181—183]. Например, молибден катализирует реакцию перекиси водорода с иодидом, при которой выделяется иод:
Н2О2 + 2Н+ + 21- --> 2Н2О + 12
К исследуемому раствору добавляют точно определенное количество аскорбиновой кислоты, которая вначале связывает образующийся иод. Когда израсходуется аскорбиновая кислота, окисляется индикатор вариаминовый синий и появляется сине-фиолетовая окраска. Момент, в который проявляется индикатор, зависит от нескольких факторов, в том числе от концентрации молибдена; этот факт использовали для определения молибдена в концентрации 0,1—1,0 мкг-мл-1.
10. КАКОТЕЛИН И БРУЦИН
Оба эти соединения, нашедшие широкое применение в качестве индикаторов, являются алкалоидами чилибухи. Фактически какотелин представляет собой производное бруцина, и обычно его получают действием концентрированной азотной кислоты на бруцин по реакции
(74)
обпап СОТР’ [1^4] повторили эту реакцию и обнаружили, что при ной сГКе ^Рудина 3,5 М азотной кислотой, нагревании реакцион-нием п6СИ На водян°й бане в течение 2 ч с последующим охлажде-котелин^тГ В течение 8 4 получается чистый (99—99,5%-ный) ка-• в продаже имеются препараты какотелина и бруцина.
108
ГЛАВА VIII
Какотелин
Какотелин известен давно как реагент для определения оло ва(П) [185—189]. Критическое изучение [186, 187] этой реакции показало, что ярко-фиолетовая окраска, образующаяся при реакции, обусловливается продуктом восстановления какотелина. Таким образом, эту цветную реакцию дают также другие восстановители, такие, как сульфаты, сероводород, титан(Ш), уран(1Ц)> родий(III) и железо(П) в присутствии фосфорной кислоты или фторида, а также медь(1) и т. д. Восстановление какотелина в фиолетовое соединение — обратимый процесс, но избыток сильного восстановителя, такого, как олово в соляной кислоте, восстанавливает какотелин до бесцветного соединения, и эта реакция уже необратима. Изменения, которые происходят в молекуле какотелина при этих реакциях, рассматривались в работах [190, 191, 192]. Эти изменения можно представить себе так:
Известно, что интенсивно окрашенным соединением является нитрогидрохинон [189], что было доказано химически и с помощью УФ-спектроскопии [193]. Интенсивная окраска приписывается [191] таутомерной форме,
которая, естественно, не может образоваться при необратимом восстановлении нитрогруппы.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
109
Применение какотелина в качестве реагента или индикатора зависит от обратимой реакции какотелин^дигидрокакотелин. Фиолетовая окраска дигидрокакотелина разрушается под действием очень сильных восстановителей. Сильные окислители стремятся разрушить хиноидное кольцо [190, 192], а это значит, что какотелин нельзя применять для титрования сильных окислителей таким jo'с^ановителем, как, например, хлорид олова (II). В подобных случаях надо титровать восстановитель окислителем.
К сожалению, проводилось очень мало исследований [184, 194] для определения нормального 'потенциала этого индикатора. Имеющиеся данные, которые относятся к очень узкой области pH, плохо согласуются. Некоторые из них приведены в табл. VIII.8 [184].
Таблица VIII.8
ФОРМАЛЬНЫЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ КАКОТЕЛИНА В РАСТВОРЕ СОЛЯНОЙ КИСЛОТЫ (КОНЦЕНТРАЦИИ КАКОТЕЛИНА И ДИГИДРОКАКОТЕЛИНА 1,225-10-’ моль-л-1; (=28 °C)
Концентрация соляной кислоты 0,499 0,996 1,990 2,990 5,000
^т» В 0,463 0,482 0,495 0,513 0,534
Области применения какотелина
Какотелин рекомендуется в качестве реагента для определения не только олова(II), но также железа (II) [195, 196] и селена [136]. Железо (II) обнаруживают в присутствии комплексообразующих реагентов, которые предпочтительно образуют комплексы с железом (III) и поэтому понижают формальный потенциал системы Fe(II)—Fe(III). Обнаружение селена основывается на свойстве свободного селена катализировать восстановление какотелина сульфидами щелочных металлов. Для определения молибдена [137] предложен каталитический метод с участием какотелина. В основе его лежит каталитическое действие молибдена на реакцию между какотелином и гидразином при 100 °C.
Первое сообщение о применении какотелина в качестве индикатора сделал, по-видимому, Боргман в 1894 г. [197]: он титровал олово(II) спиртовым раствором иода. С тех пор предложено много методов применения какотелина при титровании олова (II). Для титрования железа(III) предлагался в качестве титранта хлорид олова(II) в 2 М соляной кислоте [198]. Какотелин дает четкую фиолетовую окраску в конечной точке (ошибка <0,25%), но титрование надо проводить при кипении раствора, поскольку при комнатной температуре реакция железа (III) с оловом (II) идет
110
ГЛАВА VIII
медленно и не полностью. Обычный метод определения железа (Ш) или общего содержания железа был усовершенствован [199, 200] тем, что железо (III) восстанавливалось до железа (II) избытком хлорида олова (II) с последующей обработкой хлоридом ртути (II) и титрованием железа (II) бихроматом. Отмечалось, что избыток олова (II) можно оттитровать непосредственно бихроматом до конечной точки по какотелину, когда фиолетовая окраска переходит в желтую или зеленовато-желтую. Затем можно титровать железо (II), как обычно, бихроматом в присутствии индикатора дифениламиносульфокислоты. В этом случае титрование тоже надо проводить при температуре 70—100 °C и добавить фторид аммония, вероятно для снижения потенциала системы Fe(II)—Fe(III) во второй части титрования. Хьюм и Кольтгоф [36] получили хорошие результаты по этому методу, используя церий (IV) в качестве окислителя вместо бихромата. Они отмечают, что высокая температура необходима для титрования до первой конечной точки, так как только тогда можно получить по отдельности потенциалы олова (II) и железа (II). При комнатной температуре выписывается плавная линия с одной волной. Описание этого метода приводится ниже.
Образец железа растворяют, разбавляют до 20—25 мл и добавляют 5 мл концентрированной соляной кислоты. Нагревают смесь почти до кипения, приливают по каплям 0,5 М хлорид олова (II) до перехода желтой окраски в бледно-зеленую и тогда добавляют еще 1—2 капли раствора. В горячем состоянии титруют избыток олова (II) 0,02 М раствором церия (IV) по какотелинх (3—5 капель 1 %-него раствора) до перехода от фиолетовой к желтой окраске. Приливают 200 мл воды, 6 мл 9 М серной кислоты и титруют железо(П) 0,1 М раствором церия(IV), используя трис-(о-фенантролин)железо(П) в качестве индикатора.
Есть сообщение [201] об использовании какотелина при определении с помощью хлорида олова (II) железа(III), бихромата, церия (IV), ванадата и гексацианоферрата (III) и об использовании какотелина в смеси с дифениламином для последовательного титрования железа(III) и бихромата, а также железа(III) и ванадата. В работе [202] говорится о применении какотелина для титрования церия (IV) раствором олова (II) и наоборот; ошибка при этом обычно меньше 0,5%, но при 10-кратном избытке свинца (II) она может быть больше 1%. С участием какотелина в качестве индикатора проводилось прямое титрование олова (II) гексацианоферратом (III) калия при pH 11—12,5 в присутствии пирофосфата Натрия [203].
Какотелин использовали как индикатор также при титровании титана(III) церием (IV) [204] и для титрования до первой конечной точки смесей титайа(Ш) и железа (II) тем же титрантом [205], а фенилантраниловую кислоту применили для установления
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
111
торой конечной точки. Сырокомский и Силаева [204] установили что титрование титана(III) можно проводить в присутствии -лорода, если добавить уксусную кислоту или сульфат аммония к кислому раствору, для того чтобы связать в комплекс титан (ПО-
Наиболее важным применением какотелина являются, несомненно, методы титрования с участием олова (II) и титана(III), однако имеются сообщения об одном или двух методах еще. Например какотелин использовали как окислительно-восстановительный индикатор в комплексонометрическом титровании железа (III) раствором ЭДТА [206]. В присутствии ЭДТА формальный потенциал системы железо (II)—железо (III) существенно понижается вслед ствие того, что железо (III) образует более прочный комплекс с ЭДТА, чем железо (II), и при pH 4—5 потенциал равен 0,12 В ['207]. Следовательно, в этих условиях железо (II) более сильный восстановитель и восстанавливает какотелин. Титрование железа (III) раствором ЭДТА при pH 4—5 проводится в присутствии небольших количеств железа (II) и какотелина в атмосфере двуокиси углерода. В конечной точке индикатор восстанавливается комплексом железа (II) с ЭДТА, который дает розовую окраску.
Какотелин использовали также [208] для титрования гексацианоферрата (II) калия раствором церия (IV). Сьерра и др. [209—211] применяли какотелин при титровании тиосульфатом натрия. Железо (III) ['209] титровали в солянокислом растворе в присутствии сульфата меди(II) в качестве катализатора. Раствор кипятили для удаления кислорода и поддерживали кипячение во время титрования. Медь определялась по методу, описанному ниже [210]
Растворяют образец в серной кислоте, подщелачивают аммиаком до pH 5—6, затем добавляют 1 мл 10 М серной кислоты и добавляют необходимое количество смеси роданида со щавелевой кислотой, чтобы избежать образования осадка. Добавляют сульфат цинка (катализатор) и 2 мл насыщенного раствора какотелина. Кипятят и титруют тиосульфатом до перехода изумрудно-зеленой окраски в фиолетовую.
Медь и железо можно определять в смесях [211] путем титрования последней по методу, описанному для меди, а железо находят, осадив его в виде гидроокиси железа (III), которую отфильтровывают в горячем состоянии, и затем титруя железо по методу, Указанному выше [209].
Бруцин
бпрБРУцин тоже использовали в качестве индикатора. Окисление РУЦина в мягких условиях (5 М азотная кислота) при 0—5 °C
112
ГЛАВА VIII
[191, 212] приводит к образованию бруцихинона
Бруцихинон — ярко-красное соединение; при сильном окислении хиноидное кольцо разрывается и окраска исчезает. При восстановлении бруцихинона сернистой кислотой получается бесцветный гидрохинон ['213]
гидрохинон х (бесцветный)
В этом случае окрашенной является окисленная форма индикатора, что обусловливается, по-видимому, хиноидной структурой кольца, причем отсутствие нитрогруппы в восстановленной форме не приводит к образованию таутомера, как это имеет место у вое становленного какотелина.
Данных о формальном потенциале бруцина не имеется, но его потенциал должен быть гораздо выше потенциала какотелина, так как бруцин впервые применили в титровании железа (II) би-хроматом [214—216].
Окраска переходит от бесцветной к красной; используют 1%-ный раствор бруцина в концентрированной [214] или 1,5 М серной кислоте [215]. Довольно большие количества (10—20 капель) этого раствора не приводят к индикаторной ошибке. Алексеева и Андроникова [216] утверждают, что при титровании в присутствии соляной кислоты предпочтительнее работать с бруцином, чем < дифениламином.
О некоторых интересных наблюдениях относительно бруцина сообщили Рао и Шастри [217]. Скорость окисления бруцина бихроматом калия небольшая в 1 М серной кислоте, но возрастает, если реакцию проводить в присутствии железа (II). Поэтому смесь бруцина с бихроматом не окрашивается в красный цвет, но эта окраска сразу же появляется в присутствии следов железа (II). Соли хрома (III) замедляют реакцию, а соли железа (III) ускоря-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
113
ее Авторы [217] утверждают, что при титровании бихроматом ^присутствии железа (II) концентрация серной кислоты должна быть 1 5__2 моль-л-1, так как при более низких концентрациях
реакция с индикатором медленная. Сообщается также [217] о прямом титровании урана (IV) в холодной 2 М серной кислоте бихроматом с применением 1 мл 0,1% бруцина и о подобном титровании гидрохинона в 2,5 М серной кислоте. Бруцин был индикатором при титровании олова(II) бихроматом [218].
Рао и Шастри изучали также возможность применения бруцина в периметрии [218, 219] для титрования железа(II) [218], гексацианоферрата (II) [219], молибдена (V) [219] и гидрохинона [219]. Оказывается, что 0,1%-ный раствор бруцина в 2—3 М уксусной кислоте более устойчив, чем в 1,5 М серной кислоте, которую обычно применяли, и что такой раствор устойчив в течение 9 месяцев. Реакция с молибденом (V) вблизи конечной точки замедляется, но добавка фосфорной кислоты (10 мл концентрированной фосфорной кислоты на 100 мл анализируемого раствора) способствует ее ускорению. Мышьяк(III) и уран(IV) нельзя определять по этому методу, так как они слишком медленно взаимодействуют с окисленной формой индикатора, поэтому конечная точка устанавливается раньше времени.
Серьезным недостатком бруцина является неустойчивость его красной окраски. При титровании церием (IV) в серной кислоте [218] окраска в конечной точке устойчива только в течение минуты. В 1—2 М соляной кислоте ['218] окраска исчезает примерно через 20 с, а при более высоких концентрациях совсем нельзя установить конечную точку. По-видимому, в присутствии избытка окислителя это объясняется разрушением окрашенных частиц на меньшие бесцветные фрагменты.
11. ВИОЛОГЕНЫ
Раствор 4,4'-дипиридила (или у,у'-дипиридила) в разбавленной уксусной кислоте при восстановлении хлоридом хрома (III) приобретает темно-фиолетовую окраску. Образующееся фиолетовое соединение очень устойчиво, но легко окисляется до дипиридила, например кислородом воздуха. Эту реакцию и подобную реакцию ряда М,М'-замещенных дипиридилов изучали Михаэлис и Аилл [220—221] с целью возможного использования их в качестве индикаторной системы. Замещенные соединения оказались олее устойчивыми при различных значениях pH, чем исходное соединение; указанные авторы титровали диметилпроизводные в ределах pH от 10 до 13. Кривые титрования подтвердили, что ч Реносится один электрон, а значение Ет не зависит от pH в изу-пОдМом нрсДбле значений pH. Михаэлис и Хилл ['220—222] пред-0>кили, что фиолетовый продукт является семихиноном или Ь- SIU
114
ГЛАВА VIII
свободным радикалом, а реакция при этом следующая:
R—=«=* R—/N—R семихинои
Если проводить восстановление сильным восстановителем, например порошком цинка, то реакция проходит через «фиолетовую стадию» ко второй бесцветной стадии, связанной, по-видимому, с образованием хинона. Вторая стадия восстановления необратима.
Эти выводы позже были подтверждены [223], так как возрос интерес к применению дипиридилов в качестве селективных средств для уничтожения водорослей. Бун [223] установил, что только те соединения, которые не имеют заместителя у атомов углерода, соседних к межкольцевой связи, можно восстановить до бесцветного свободного радикала, т. е. только у таких соединений, в которых оба кольца могут иметь копланарную конфигурацию, получены свободные радикалы. Как раз эти соединения обладают гербицидной активностью. Свободный радикал, по-видимому, стабилизируется вследствие мезомерии из-за полной делокализации неспаренного электрона, а копланарная конфигурация необходима для образования нескольких мезомерных гибридов.
Михаэлис и Хилл [220—221] обнаружили, что потенциалы таких систем не зависят от pH, как показала реакция (75), и поэтому окислительно восстановительный потенциал этих систем дается простым уравнением Нернста:
Еь=Е0 + 4-1пЛ-§7- (76)
практически же потенциалы гораздо ниже; некоторые из них, определенные Михаэлисом и Хиллом [220—221], приведены в табл. VIII.9. В нейтральном растворе потенциалы очень близки к потенциалу водородного электрода при том же значении pH, и при более низких значениях pH нельзя получить полные кривые
Таблица VJII.9
ЗНАЧЕНИЯ Ет ПРИ 30 °C ДЛЯ ЗАМЕЩЕННЫХ ВИОЛОГЕНОВ
Заместитель PH
Ы.К'-Диметил —0,446 8,4-13
М.Ы'-Диэтил —0,449 И
Ы.М'-Дибетаин —0,444 11
N.N'-Дибензил —0,359 8
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
115
пования, так как нормальный потенциал будет ниже потенциа-™ водородного электрода. Однако в щелочном растворе достига-ла устойчивости .свободного радикала, титровали бисульфитом ЛдТрИя и получали очень четкие кривые титрования. Наблюдалось Неболыпое смещение потенциала даже в сильнощелочных раствс-пах (смещение в кислом растворе гораздо больше). Михаэлис и Хилл^ объясняли это либо присутствием следов кислорода в очищенном азоте, используемом для удаления кислорода из раствора, либо неустойчивостью самого свободного радикала. Вследствие такого смещения точность определения потенциалов, приведенных в табл. VIII.9, составляет примерно ±0,002 В.
В отличие от большинства окислительно-восстановительных индикаторов у виологенов окрашена восстановленная форма. Окраска замещенных виологенов синяя (максимум поглощения М,Г4'-ди-Метил-4,4'-дипиридила находится при длине волны 602 нм); они исключительно чувствительны к кислороду. О применении этих соединений в качестве индикатора нет ни одного сообщения. Они могут быть пригодны, в частности, для определения хрома (II), так как почти не существует обратимых индикаторных систем, с которыми можно было бы паботать в этой области потенциалов. Методы их получения из 4,4'-дипиридила дали Михаэлис и Хилл [221]. Бун [223], кроме упомянутых выше, исследовал ряд других производных и определил их формальные потенциалы, но ничего не говорит о возможности использования их в качестве индикаторов.
12. РАЗНЫЕ ИНДИКАТОРЫ
К этому разделу относится ряд индикаторов, которые не принадлежат ни к одной из перечисленных групп. Ни для одного из них не имеется данных о точных потенциометрических измерениях или же об области их применения.
биг-(Диметилглиоксимат)железо(11]
Этот комплекс железа (II) ведет себя в слабощелочном растворе как обратимый окислительно-восстановительный индикатор [224— 449]. Реакция аналогична реакции комплекса железа (II) с о-фе-нантролином:
/ СН3—C=NOH\
Fe2+| I I
\CH3—C=NO /2
/ CH3—C=NOH\ 1+
Fe3+ [ 4-e
\CH3— C=NO /,
KPa „ка изменяется от красной, которая соответствует восстанов-«н°й Форме, до бесцветной, но это относится только к нейтраль-или слабощелочным растворам, т. е. для pH 6—10. В кислых
8»
116
ГЛАВА VIII
растворах окраска исчезает, а в сильнощелочных разрушается само соединение. Этот комплекс при pH 9,4 имеет потенциал Ет= =0,155 В ['225] и используется в качестве индикатора для титрования железа (II) [224], бисульфита {226, 229] и сульфида {227] раствором гексацианоферрата (III) в щелочной среде.
Fe2++(Fe(CN)6l3- --> Fe3+ + [Fe(CN)6]4-
S2O1~ 4- 2[Fe(CN)6]3- + 4OH- -> 2SO3- + 2[Fe(CN)6]4~ + 2H2O
S2-4-2[Fe(CN)6]3- -> S| + 2[Fe(CN)6]4-
Определение бисульфита проводилось путем титрования гекса-цианоферратом(П) в присутствии буфера из аммиака и хлорида аммония (pH 9,4) в атмосфере аргона [229]. По сравнению с иодо-метрическим методом ошибка составляла меньше 1%.
Меконовая кислота
Кольтгоф [230] предложил использовать комплекс железа (III) с меконовой кислотой в качестве индикатора для титрования железа (III) раствором титана (III):
н /СООН
/С=С\
О=С' V)
\с=с( он Хсоон
меконовая кислота
В кислом растворе красновато-бурый меконат железа (III) образуется при добавлении меконовой кислоты к раствору железа(III). При титровании этого раствора хлоридом титана(III) происходит изменение окраски через оранжево-коричневую до желтовато-коричневой, которая в конечной точке становится бледно-желтой. Конечную точку нельзя четко определить, поэтому сопоставляют окраску анализируемого раствора с раствором сравнения. Меконо-вую кислоту используют также как индикатор при титровании железа (III) тиосульфатом в присутствии меди как катализатора [231]. Однако этот метод неудобен из-за медленного протекания реакции, особенно вблизи конечной точки.
Хиноны
Потенциометрические данные, полученные для ряда бензохинонов, нафтохинонов и антрахинонов, представляют значительный интерес для теоретической органической химии. Все данные собрал Кларк ([26] стр. 362 и сл.), однако очень немногие из этих соеди
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 117
нений использовались или рекомендуются как окислительно-восстановительные индикаторы.
В качестве окислительно-восстановительного индикатора для щелочного раствора можно применять фтиокол или 2-метил-З-окси-1 4-нафтохинон [232]. Реакция обратима и идет с переносом двух электронов:
В растворах с рН>6 фтиокол растворяется, давая темно-красную окраску, а в более кислых растворах малорастворим, окрашивая раствор в бледно-желтый цвет. Поэтому он непригоден как индикатор для .работы с кислыми растворами. Изменение формального потенциала от pH было тщательно изучено [232]. Эта зависимость подчиняется уравнению
„ , RT [Н+Р+КГ1[Н+Р + ^Г1КГг[Н+]
Ет=Е0 + In-------------;11+] + /<о -1- -, (77)
когда .Ео=0,299 В, р/Со = 5,08, р/СГ1 = 8,90, р/СГ2 =11,46. Ко — константа диссоциации гидроксильной группы окислителя, а £Г1 и Кг, — константы диссоциации гидроксильных групп восстановителя. Диссоциация третьего гидроксила в молекуле восстановителя несущественна в изучаемом пределе значений pH (1,1—12,6). В одной из работ [233] описано применение ряда аминоантрахинонов в качестве окислительно-восстановительных индикаторов при титровании аскорбиновой кислоты, феназона и мышьяка (III) броматом калия, а также при титровании железа (II) бихроматом. Нормальный потенциал этих индикаторов равен 0,70 В; индикаторы обратимы. Предполагается следующий механизм реакции индикатора:
В табл. VII 1.10 приведены данные о производных этих индикаторов. Все индикаторы очень четко изменяют свою окраску и дают удовлетворительные результаты при броматометрическом титровании аскорбиновой кислоты и феназона. Однако для определения этим методом мышьяка (III) подходят только первые два антрахинона, а Для титрования железа (II) бихроматом пригодны все, за исключением целлитонового прочного синего FFB и целлитонового
118
ГЛАВА VIII
Таблица VIII.10
ИЗМЕНЕНИЯ ОКРАСКИ НЕКОТОРЫХ ЗАМЕЩЕННЫХ АНТРАХИНОНОВ, ПРИГОДНЫХ В КАЧЕСТВЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ИНДИКАТОРОВ
Название Ri R2 Rs Изменение окраски
окисление восстановление
Целлитоновый синий FR прочный Н Н —^~^>-ОСН3 Синяя Красная
Целлитоновый синий FFR прочный СН3 Н — СН2СН2ОН » »
Целлитоновый синий FFB прочный Н — conh2 сн3 » »
Целлитоновый синий FFG прочный Н - conh2 — » »
Целлитоновый фиолетовый В прочный Н Н н » »
Целлитоновый тем но-зеленый прочный в Не указаны » »
прочного фиолетового. В последнем случае титрования играет значительную роль присутствие фосфорной кислоты. При избытке окислителя все красители разлагаются, и поэтому их нельзя применять в тех случаях, когда титрование проводится восстановителем.
Кремниймолибденовая кислота
Ряд авторов [234, 235] предложил использовать кремниймолибденовую кислоту как индикатор для определения первой конечной точки при титровании смеси олова(II) и железа(II) раствором бихромата [234] или церия (IV) [235]. Исследователи усовершенствовали обычный метод титрования хлорида олова (II) раствором хлорида ртути(II) для определения железа (III) подобно методу с применением какотелина. Хлорид олова (II) восстанавливает кремниймолибденовую кислоту до молибденовой сини. При добавлении избытка олова(II) для восстановления железа (III) в железо (И) и в присутствии индикатора синяя окраска исчезает. Затем раствор титруют бихроматом или церием (IV) до возобновления синей окраски, что указывает на окончание титрования избытка олова (II). Тогда можно уже титровать железо (II), используя в качестве индикатора N-фенилантраниловую кислоту (при титровании бихроматом) или же грцс-(о-фенантролин)железо(П) [при титровании раствором церия (IV)].
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
119
Дифенил карбазид
Чувствительным реагентом для обнаружения хромата является ^„.дифенилкарбаэид (C6H5NHNH)2CO [236]. Позднее его рекомендовали в качестве индикатора при титровании железа (II) бихроматом [237—241]. Определению не мешают ни олово (II), ни ртуть (II)- Дифенилкарбазид используют [238] как индикатор при титровании железа (II) бихроматом, предварительно восстановив железо(Ш) Д° железа (II) обычным способом с помощью хлорида олова(П) и хлорида ртути(II). В конечной точке окраска раствора становится фиолетовой, а затем зеленой, т. е. приобретает цвет хрома(III). Титрование до получения зеленой окраски создает большую индикаторную ошибку вследствие расхода бихромата на реакцию с индикатором. Брандт [238] предположил, что индикаторную ошибку можно определить, добавив еще небольшое количество индикатора к раствору, титрование которого закончено, и продолжить титрование. (Проведение холостого опыта не приводило к точным результатам.) Он использовал растворы дифенилкарбазида в уксусной кислоте, но они, к сожалению, устойчивы не более недели. Большая индикаторная ошибка была подтверждена другими авторами [240, 241], которые предложили ряд методов для точного определения этой ошибки.
Комплекс ванадия(Н) с 1,10-фенантролином
В качестве индикатора для титрования железа(III) хромом(II) был предложен [242] комплекс ванадия с 1,10-фенантролином; точный состав комплекса неизвестен. Его получают, смешивая ва-надилхлорид (IV) с 1,10-фенантролином в растворе гидроокиси натрия. Титрование проводили в 1 М серной кислоте, добавив 3 капли раствора индикатора на 50 мл титруемого раствора. Кислород удаляли, продувая азотом. Окраска индикатора изменяется от бесцветной до синей (восстановленная форма); изменение происходит приблизительно при потенциале +0,35 В относительно нормального водородного электрода. Получаются удовлетворительные результаты, но в сообщении не приводятся подробности об используемом индикаторе.
СПИСОК ЛИТЕРАТУРЫ
1. Clark W. М., Public Health Reports. (U. S.), 38, 443 (1923).
2. Clark W. M„ Cohen B., Public Health Reports (U.S.), 38, 666 (1923).
3. Clark W. M., Cohen B., Public Health Report (U. S.), 38, 933 (1923).
4. Sullivan M. X., Cohen B, Clark W. M„ Public Health Reports (U.S.), 38, 1669 (1923)
5. Cohen B, Gibbs H. D„ Clark W. M„ Public Health Reports (U.S.), 39, 381 (1924).
120
ГЛАВА VIII
6. Cohen В., Gibbs Н. D., Clark W. M., Public Health Reports (U.S.), 39, 804 (1924).
7. Gibbs H. D., Cohen B., Cannan R. K., Public Health Reports (U.S.), 40, 649 (1925).
8. Clark W. M., Cohen B., Gibbs H. D., Public Health Reports (U.S.), 40, 1131 (1925).
9. Clark U7. M., Cohen B., Gibbs H. D., Public Health Reports (U.S.), Suppl № 54 (1926).
10. Cannan R. K-, Cohen B., Clark W. M.. Public Health Reports (U.S.), Suppl № 55 (1926).
11. Phillips'M., Clark U7. M., Cohen B., Public Health Reports (U S), Suppl. № 61 (1927).
12. Clark W. M., Cohen B., Sullivan M. X., Public Health Reports (U.S.), Suppl. № 66 (1927).
13. Gibbs H. D., Hall W. L., Clark W. M., Public Health Reports (U.S.), Suppl. № 69 (1928).
14. Hall W. L., Preisler P. W., Cohen B., Public Health Reports (U.S.), Suppl. № 71 (1928).
15. Cohen B„ Phillips M., Public Health Reports (U.S.), Suppl. № 74 (1929).
16. Cohen B., Preisler P. W., Public Health Reports (U.S.), Suppl. № 92 (1931).
17. Clark W. M., Perkins M. E., J. Am. Chem. Soc., 54, 1228 (1932).
18. Stiehler R. D., Chen T.-T., Clark W. M„ J. Am. Chem. Soc., 55, 891 (1933).
19. Steihler R. D„ Clark W. M„ J. Am. Chem. Soc., 55, 4097 (1933).
20. Кольтгоф И. M., Стенгер В. А., Объемный анализ, т. 1, Госхимиздат, М.-Л.. 1950. ~
21. Tomicek О., Chemical Indicators, Butterworths, London, 1951.
22. Певцов Г. А., Труды ин-та чистых хим. реагентов, 16, 125 (1939).
23. Певцов Г. А., Труды комиссии по анал хим., 1, 53 (1947).
24. Певцов Г. А., Труды Всесоюзн. науч.-исслед. ин-та хнм., 22, 95 (1958).
25. Banyai E., Kemiai indikatorok, Miiszaki Konyvkiado, Budapest, 1961.
26. Clark W. M., Oxidation-Reduction Potentials of Organic Systems, Williams & Wilkins, Baltimore, 1960.
27. Chariot G., Selected Constants: Oxidation-Reduction Potentials, Pergamon Press, New York, 1958.
28. Erdey L„ Banyai E., Gere E. B., Talanta, 3, 54 (1959).
29. Holmes W. C., J. Am. Chem. Soc., 46, 208 (1924).
30. Hiesinger M., Luettke W., Tetrahedron, 19, Suppl. 2, 315 (1963).
31. Meyer H. W., Treadwell W. D., Helv, Chim. Acta, 35, 1444 (1952).
32. Preisler P. W., Hill E. S., Loeffel R. G., Shaffer P. A., J. Am. Chem. Soc., 81, 1991 (1959).
33. Rielbasinski V., Z. Farben. u. Textilchem., 2, 114 (1903).
34. Nissenson H., Siedler P., Chem. Ztg., 27, 749 (1903).
35. Schluttig W„ Z. anal. Chem., 70, 55 (1927).
36. Hune D. N., Kolthoff I. M., Anal. Chim. Acta, 16, 415 (1957).
37. Henze G., Geyer R., Z. anal. Chem., 200, 434 (1964).
38. Holness H., Cornish G., Analyst, 67, 221 (1942).
39. Tandon J. P., Mehrotra R. C., Z. anal. Chem., 158, 20 (1957).
40. Tandon J. P., Mehrotra R. C., Z. anal. Chem., 187, 410 (1962).
41. Афанасьев Б. H., Уральская А. В., Завод, лаб., 15, 407 (1949).
42. Афанасьев Б. Н., Завод, лаб., 15, 1271 (1949).
43. Vanossi R., Ferramola R„ Ann. chim. anal, appl., 15, 529 (1934).
44. Korenman I. M., Mikrochemie, 18, 31 (1935).
45. Rao G. G„ Rao N. V., Taianta, 8, 539 (1961).
46. Rao G. G., Rao N. V., Acta Chim. Acad. Sci. Hung., 26, 489 (1961).
47. Loomis W. F., Anal. Chem., 26, 402 (1954).
48. Loomis W. F., Anal. Chem., 28, 1347 (1956).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
121
49 St John Р. A., Winefor drier J. D., Silver W. S., Anal. Chim. Acta, 30, 49 />964).
rn Preisler P. W., Hampelmann L. H., J. Am. Chem. Soc., 58, 2305 (1936).
51 Remick A. E., J. Am. Chem. Soc., 58, 733 (1936).
52 Metier H. W., Treadwell W. D„ Helv. Chim. Acta, 35, 1460 (1952).
53 Lojander W., Suomen Kemistilehti, 14A, 26 (1941).
54 Erdey L., Svehla G., Koltai L., Anal. Chim. Acta, 27, 164 (1962).
55 Stone I., Ind. Eng. Chem. Anal. Ed., 12, 415 (1940).
56 Tillmans J., Hirsh P., Reinshagen E., Z. Untersuch. Lebensm., 56, 272 (1928).
57 Tillmans J., Z. Untersuch. Lebensm., 60, 34 (1930).
58' Birch T„ Harris L., Ray S., Biochem. J., 27, 590 (1933).
59 Emmerie A., Van Eekelen M., Biochem. J., 28, 1153 (1934).
60^ Emmerie A., Van Eekelen M., Biochem. J., 30, 25 (1936).
6L Menacher M. H„ Guerrant N. B., Ind. Eng. Chem., Anal. Ed.. 10. 25 (1938).
62* Erdey L., Svehla G., Z. anal. Chem., 150, 407 (1956).
63. Erdey L., Svehla G., Z. anal. Chem., 163, 6 (1958).
64^ Erdey L., Svehla G., Z. anal. Chem., 167, 164 (1959).
65^ Erdey L., Koltai L., Svehla G„ Anal. Chim. Acta, 27, 363 (1962).
66. Erdey L., Svehla G., Koltai L., Anal. Chim. Acta, 28, 398 (1963).
67. Erdey L., Koltai L., Svehla G., Anal. Chim. Acta, 27, 498 (1962).
68. Erdey L., Khalifa H., Svehla G., Anal. Chim. Acta, 32, 88 (1965).
69. Erdey L„ Siposs G., Z. anal. Chim., 157, 166 (1957).
70. Svehla G., Koltai L., Erdey L., Anal. Chim. Acta, 29, 442 (1963).
71. Misra G. J., Tandon J. P., Z. anal. Chem., 214, 94 (1965).
72. Schwarzenbach G., Michaelis L., J. Am. Chem. Soc., 60, 1667 (1938).
73. Shine H. J., Snell R. L., Trisler J. G., Anal. Chem., 30, 383 (1958).
74. Wieland H., Ber., 48, 1078 (1915).
75. Wehber P., Z. anal. Chem., 149, 161 (1956).
76. Wehber P., Z. anal. Chem., 149, 241 (1956).
77. Wehber P., Z. anal. Chem., 150, 186 (1956).
78. Wehber P., Z. anal. Chem., 151, 276 (1956).
79. Villegas de Bollini L. S., Anales farm, bioquim. (Buenos Aires), 18, 115 (1947).
80. Society of Dyers and Colourists, British Colour Index, Rowe F. M. (ed.), 2nd ed., Bradford, 1956. Suppl. 1963.
81. Friedheim E., Michaelis L„ J. Biol. Chem., 91, 355 (1931).
82. Dufraisse C., Etienne A., Toromanoff E., Fellion Y., Compt. rend., 242, 854 (1956).
83. Michaelis L., J. Biol. Chem., 91, 369 (1931).
84. Preisler P. W., Hempelmann L. H., J. Am. Chem. Soc., 59, 141 (1937).
85. Elema B„ Rec. Trav. Chim., 52, 569 (1933).
86. Michaelis L„ J. Biol. Chem., 92, 211 (1931).
87. Dickens F., McIlwain H., Biochem. J., 32, 1615 (1938).
88. Albert A., Duewell H., J.S.C.I., 66, 11 (1947).
89. Horrobin S., Dennison S. R., Holmes W. S., Pritchard J., Thacker R., Trans. Far. Soc., 60, 803 (1954).
90. Monnier A., Ann. chim. anal., 21, 109 (1916).
91. Murthy В. V. S. R., Rao G. G., Taianta, 8, 438 (1961).
92. Rao N. V. K., Rao G. G„ Anal. Chim. Acta, 29, 82 (1963).
93. Cooke W. D., Hazel F., McNabb W. M„ Anal. Chem., 21, 643 (1949).
94. Cooke W. D„ Hazel F., McNabb W. M„ Anal. Chim. Acta, 3, 656 (1949).
95. Tandon J. P„ Mehrotra R. C., Z. anal. Chem., 159, 422 (1957).
E P-, Mehrotra R. C., Z. anal. Chem., 164, 314 (1958).
97. Mazor L., Erdey L., Acta Chim. Acad. Sci. Hung., 2, 331 (1952).
on L’ Mazor L> Acta Chim. Acad. Sci. Hung., 3, 469 (1953).
inn C- M- V°Eel A. I-, Analyst, 81, 693 (1956).
n. ,,°trka M" SaSner Z., Chem. Premysl., 9, 526 (1959).
iui Mittal R. K., Tandon J. P.. Mehrotra R. C.. Z. anal. Chem., 189. 330 (1962).
122
ГЛАВА VIII
102. Michaelis L., Eagle H., J. Biol. Chem., 87, 713 (1930).
103. Melville J., Richardson G. M., Biochem. J., 28, 1565 (1934).
104. Eggers H., Dieckmann H., Biochem. Z., 310, 233 (1942).
105. Twigg R. S., Nature, 155, 401 (1945).
106. Ruzicka E., Coll. Czech. Chem. Comms., 29, 2244 (1964).
107. Musha S., Kitagawa T., J. Chem. Soc. Japan, Pure Chem. Sect., 76, 1289 (1955).
108. Letort M., Compt. rend., 194, 711 (1932)..
J09 Granick S., Michaelis L., Schubert M. P., J. Am. Chem. Soc., 62, 1802 (1940).
110. Chawla K. L., Tandon J. P., Taianta, 12, 665 (1965).
111. Mittal R. K-, Tandon J. P., Mehrotra R. C., Z. anal. Chem., 189, 406 (1962).
112. Никольский Б. П., Пальчевский В. В., Изв. АН СССР, отд. хим. наук, 547 (1957).
113. Michaelis L., Schubert М. Р., Granick S., J. Am. Chem. Soc., 62, 204 (1940).
114. De Eds F., Eddy C. W., J. Am. Chem. Soc., 60, 1446 (1938).
115. De Eds F., Eddy C. W., J. Am. Chem. Soc., 60, 2079 (1938).
116. Granick S., Michaelis L., J. Am. Chem. See., 69, 2983 (1947).
117. Никольский Б. П., Пальчевский В. В., Ж- физ. хим., 32, 1280 (1958).
118. Shippy В. A., Anal. Chem., 21, 698 (1949).
119. Smith G. F., Kurtz L. T., Ind. Eng. Chem., Anal. Ed., 14, 854 (1942).
120. Tomicek O., Spumy K, Jerman L., Holecek V., Coll. Czech. Chem. Comms., 18, 757 (1953).
121. Ruzicka E., Adamek J., Z., anal. Chem., 195, 411 (1963).
122. Ruzicka E., Kotoucek M., Z. anal. Chem., 183, 351 (1961).
123. Atack F. W., Analyst, 38, 99 (1913).
124. Tandon J. P., Mehrotra R. C., Z. anal. Chem., 162, 31 (1958).
125. Jellinek K., Winogradoff L„ Z. anorg. Chem., 129, 15 (1923).
126. Leutwein F„ 1. anal. Chem., 120, 233 (1940).
127 Sagi S. R., Rao G. G., Acta. Chim. Acad. Sci. Hung., 38, 89 (1963).
128. Rao G. G., Sagi S. R., Taianta, 9, 715 (1962).
129. Rao G. G., Sagi S. R„ Taianta, 10, 169 (1963).
130. Rao G. G., Dikshitulu L. S. A., Taianta, 10, 295 (1963).
131. Basinska H., Wisniewski W., Chem. Anal. (Warsaw), 10, 1007 (1965).
132. Кит-Tatt L., Tong H. K, Anal. Chim. Acta, 26, 583 (1962).
133 Gowda H. S., Shakunthala R., Taianta, 13, 1375 (1966).
134. Gautier J. A., Ann. pharm. Franc., 6, 171 (1948)
135. Marsh G., J. Biol. Chem., 95, 25 (1932).
136. Feigl F„ West P. W., Anal. Chem., 19, 351 (1947).
137. Bognar J., Magy. Kcm. Folyoirat., 69, 320 (1963).
138. Erdey L., Banyai Ё., Zalay E., Tesy M., Acta Chim. Acad. Sci. Hung., 15, 65 (1958).
139. Erdey L., Zalay E., Bodor E., Acta Chim. Acad. Sci. Hung., 12, 251 (1957).
140. Erdey L., Chemist-Analyst, 48, 106 (1959).
141. Erdey L., Bodor E., Z. anal. Chem., 137, 410 (1953).
142. Ba.nyai Ё., Erdey L., Szabadvary F„ Acta Chim. Acad. Sci. Hung., 20, 307 (1959).
143. Erdey L., Pink R. C., Taianta, 9, 329 (1962).
144. Banyai Ё., Zuman P., Coll. Czech. Chem., Comm., 24, 522 (1959).
145. Erdey L., B&nyai Ё., Koloriszt. Ertesito., 6, 343 (1964).
146. Erdey L., Gere E. B., Banyai E., Taianta, 3, 209 (1959).
147. Erdey L., Kasa J., Klatsmanyi I., Meisel T., Acta Chim. Acad. Sci. Hung., 26,
53 (1961).
148. Erdey L., Kasa J., Meisel T., Taianta, 10, 471 (1963).
149. Erdey L., Kasa I., Taianta, 10, 1273 (1963).
150. Erdey L., Kasa I., Acta Chim. Acad. Sci. Hung., 41, 59 (1964).
151. Erdey L., Vigh K, Bodor E., Acta Chim. Acad. Sci. Hung., 7, 293 (1955).
152. Erdey L., Bodor E., Buzas /., Acta Chim. Acad. Sci. Hung., 7, 277 (1955).
153. Erdey L., Buzas /., Bodor E., Acta Chim. Acad. Sci. Hung., 7, 287 (1955).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
123
Erdeu L., Buzas I., Acta Chim. Acad. Sci. Hung., 4, 195 (1954).
; r-' Erdeu L., Buzas I., Acta Chim. Acad. Sci. Hung., 8, 263 (1955).
146 Lach Z., Pacovsky J., Svach M., Z. anal. Chem., 154, 185 (1957).
147 Erdeu L., Bodor E., Papay M., Acta Chim. Acad. Sci. Hung., 5, 235 (1955).
148* Erdeu L Vigh K., Buzas L, Acta Chim. Acad. Sci. Hung., 26, 93 (1961).
49 Erdeu L., Vigh Taianta, 10, 439 (1963).
160 Kasa I-, Erdey L., Acta Chim. Acad. Sci. Hung., 39, 21 (1963).
61 Erdey L., Vigh K., Z. anal. Chem., 157, 184 (1957).
162’ Erdey L„ Buzas I., Vigh K., Taianta, 1, 377 (1958).
16з’ Erdey L., Vigh K... Polos L., Taianta, 3, 1 (1959).
164^ Erdey L„ Polos L., Gregorowicz Z., Taianta, 3, 6 (1959).
165. Gregorowicz Z., Buhl F., Piwowarska B., Z. anal. Chem., 188, 2 (1962).
166^ Gregorowicz Z., Buhl F., Z. anal. Chem., 173, 115 (1960).
167. Gregorowicz Z., Stoch J., Z. anal. Chem., 173, 383 (1960).
168* Erdey L., Polos L., Z. anal. Chem., 153, 411 (1956).
169. Gregorowicz Z., Anal. Chim. Acta, 23, 299 (1960).
170. Gregorowicz Z., Stoch J., Chem. Anal. (Warsaw), 6, 457 (1961).
171. Gregorowicz Z., Buhl F., Z. anal. Chem., 177, 91 (1960).
172. Gregorowicz Z., Mazo ns ka D., Prajsnar D., Anal. Chim. Acta, 24, 546 (1961).
173 Schwarzenbach G., Complexometric Titrations, 2nd ed., Methuen, London, 1957, p. 47.
174. West T. S., Sykes A. S., Analytical Applications of Diamino-ethane-tetra-acetic Acid, 2nd ed., 1960. British Drug Houses Ltd.
175. Wehber P., Mikrochim. Acta, 1955, 812.
176. Erdey L., Rady G., Z. anal. Chem., 149, 250 (1956).
177. "Fischer W., Z. anal. Chem., 215, 251 (1965).
178. Wehber P„ Z. anal. Chem., 158, 321 (1957).
179. Wehber P., Z. anal. Chem., 149, 244 (1956).
180. Gregorowicz Z., Buhl F., Z. anal. Chem., 187, 1 (1962).
181. Erdey L., Svehla G., Acta Chim. Acad. Sci. Hung., 26, 77 (1961).
182. Svehla G., Erdey L., Mikrochem. J., 7, 206 (1963).
183. Svehla G., Erdey L., Mikrochem. J., 7, 221 (1963).
184. Rao G. G., Krishnamurty N., Rao V. N., Taianta, 12, 243 (1965).
185. Gutzeit G„ Helv. Chim. Acta, 12, 720 (1929).
186. Newell I. L., Ficklen J. B., Maxfield L. S., Ind. Eng. Chem., Anal. Ed., 7, 26 (1935).
187. Feigl F., Spot Tests, Vol. 1. Elsevier Pub. Co., Amsterdam, 1954, p. 104
188. Lang R., Z. anal. Chem., 128, 167 (1948).
189. Rosenthaler L., Mikrochim. Acta, 3, 190 (1938).
190. Leuchs H., Kahrn FL, Ber., 55, 724 (1922) .
191. The Alkaloids, Manske R. H. F., Holmes H. L. (eds.), vol. 1. Acad. Press Inc., N. Y., 1950.
192. The Alkaloids, Manske R. H. F. (ed.), vol. 8, Acad. Press Inc., N. Y„ 1965.
193. Gallo N„ Ann. chim., 50, 1693 (1960).
194. Szarvas P„ Lantos J., Taianta, 10, 477 (1963).
195. Rao G. G., Rao V. N., Somidevamma G., Lalita K-, Anal. Chim. Acta, 12, 271 (1955).
196. Rao G. G., Rao V. N., Somidevamma G., Lalita K., Taianta, 1, 169 (1958).
197. Borgman P. J. S., Ned. Tijdschr. Pharm., 8, 140 (1896).
198. Кухмент M. Л., Генгринович А. И., Завод, лаб., 11, 268 (1945).
199. Wehber P., Angew. Chem., 66, 271 (1954).
200. Yoshimura T., J. Chem. Soc. Japan, Pure Chem. Sect., 74, 116 (1953).
on ’ Szarvas P-> Lantos J., Magy. Kem. Folyoirat, 65, 145 (1959).
"2. Wawrzyczek W., Wishiewska K., Z. anal. Chem., 207, 182 (1965).
опи Japan Analyst, 10, 19 (1961).
on!' СыР0К°мский В. С., Силаева Е. В., Завод, лаб., 15, 1015 (1949).
205. Веселого Л. И., Ж. анал. хим., 12, 381 (1957).
124
ГЛАВА VIII
206. Rao G. G., Rao V. N., Somidevamma G., Z. anal. Chem., 152, 346 (1956).
207. Belcher R., Gibbons D., West T. S., Anal. Chim. Acta, 12, 107 (1955).
208. Yoshimura T., J. Chem. Soc. Japan, Pure Chem. Sect., 74, 407 (1953,
209. Sierra F., Monllor E., Anal. fis. quim., 46B, 319 (1950).
210. Sierra F., Monllor E., Anal. fis. quim., 46B, 505 (1950).
211. Sierra F., Monllor E., Anal. fis. quim., 47 B, 239 (1951).
212. Leuchs H., Seeger H., Jaegers K., Ber., 71, 2023 (1938).
213. Leuchs H., Fricker K., Ber., 55, 1244 (1922).
214. Miyagi S., J. Soc. Chem. Ind. Japan, 36, 146 (1933).
215. N arayanamurthi D. S., Seshadri T. R., Proc. Ind. Acad. Sci., ЗА, 38 (1936).
216. Алексеева E. А., Андроникова H. H., Ж. приклад, хим., 11, 1024 (1938).
217. Rao G. G., Sastri T. P., Z. anal. Chem., 172, 28 (1960).
218. Rao G. G., Sastri T. P., Taianta, 1, 213 (1958).
219. Sastri T. P., Rao G. G., Z. anal. Chem., 169, 422 (1959)
220. Michaelis L., Hill E. S , J. Am. Chem. Soc., 55, 1481 (1933).
221. Michaelis L., Hill E. S., J. Gen. Physiol., 16, 859 (1933).
222. Michaelis L., Chem. Rev., 16, 243 (1935).
223. Boon W. R., Chem. and Ind., 1965, 782.
224. Komarek K, Coll. Czech. Chem. Comms., 9, 247 (1937).
225. Chariot G., Bull. soc. chim. France, Mem.. 6, 970 (1939).
226. Chariot G., Bull. soc. chim. France, Mem., 6, 977 (1939).
227. Chariot G., Bull. soc. chim. France, Mem., 6, 1447 (1939).
228. Chariot G., Bull. soc. chim. France, Mem., 7, 144 (1940).
229. Wawrzyczek W., Rychcik W., Z. anal Chem., 202, 21 (1964).
230. Kolthoff I. M., Rec. Trav. Chim., 43. 816 (1924).
231. Kolthoff I. M„ Tomicek O., Pharm. Weekblad, 61, 1205 (1924).
232. Ball E. G„ J. Biol. Chem., 106, 515 (1934).
233. Krauz I., Endroi-Havas A., Proc. S.A. C. Conference Nottingham, 1965, 568.
234. Titus A. C„ Sill C. W., Ind. Eng. Chem., Anal. Ed., 13, 416 (1941).
235. Titus A. C., Sill C. W., Ind. Eng. Chem., Anal. Ed., 14. 121 (1942).
236. Cazeneuve P., Compt. rend., 131, 346 (1900).
237. Brandt L., Z. anal. Chem., 45, 95 (1906).
238. Brandt L., Z. anal. Chem., 53, 1 (1914).
239. Brandt L., Z. anal. Chem., 53, 729 (1914).
240. Barneby O. L., Wilson S. R., J. Am. Chem. Soc., 35, 156 (1913).
241. Crossley H. E., Analyst, 61, 164 (1936).
242. Schaffer W. P.. Anal. Chem., 35, 1746 (1963).
Б. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ С ВЫСОКИМ ФОРМАЛЬНЫМ ПОТЕНЦИАЛОМ
Е. Бишоп
ВВЕДЕНИЕ
формальные потенциалы большинства неорганических титрантов-окислителей, за исключением иода и меди (II), превышают О 76 В. Таков формальный потенциал гидратированной системы железо(III)—железо (II), а поэтому более сильными окислителями являются такие, которые окисляют количественно железо (II), иногда с помощью таких лигандов, как фосфаты, фториды и хлориды, до более низкого условного потенциала системы железо (HI) —железо(II). К области низких потенциалов, представляющей интерес для аналитической химии, рассмотренной Оттоуэйем в гл. VIIIA, относятся более сильные восстановители, которые количественно окисляются железом(III) в подходящих условиях. Такое подразделение по потенциалам имеет огромное практическое значение, однако оно не совсем точное, и многие реагенты и индикаторы относятся к обеим группам. Так, например, весьма произвольно относят вариаминовый синий и его производные к группе соединений с низким потенциалом [1], а дифениламин, дифенил-бензидин и их производные к группе соединений с высоким потенциалом. Многие из индикаторов с высоким потенциалом часто используются при титровании сильными восстановителями. В части Б этой главы будут также приведены случаи применения индикаторов с низким потенциалом при титровании сильными окислителями. К группе индикаторов, обладающих высоким потенциалом, относятся индикаторы, формальные потенциалы которых находятся в области от 0,76 В (например, для дифениламина) до 1,3 В [например, для иона трис- (5-нитро-о-фенантролин) железа (II)]; такие индикаторы используются с окислителями, начиная от ванадата (V)
кончая перхлоратом церия (IV). В эту группу входят также мно-1а пСОединения> К0Т°Рые обладают индикаторными свойствами из-друго спос°бности изменять окраску или претерпевать какое-либо •овнь ”ИЗМеНение’ но К0Т0Рые не могут иметь формальный или ус-I _ 1 ПОтенциал, так как при реакции происходит их разложение всем необратимое изменение структуры. Сюда же относятся
126
ГЛАВА VIII
и другие соединения, например крахмал, розанилин и некоторые хемилюминесцентные индикаторы, которые являются чувствительными специфическими реагентами по отношению к частицам окислителя.
История истинных внутренних индикаторов, имеющих высокий формальный потенциал, сравнительно коротка. Раньше для установления конечной точки титрования использовали окраску реагента (например, перманганата), внешние индикаторы [например гексацианоферрат(III) для железа (II)] или потенциометрические методы. Брандт [2] был первым, применившим в 1906 г. внутренний цветной индикатор. Это был дифенилкарбазид при титровании железа (II) хромом (VI). Действие дифенилкарбазида в значительной мере зависит от условий титрования; обратимость его довольно плохая, и, хотя в течение следующих 30 лет [3—10] он продолжал применяться, его нельзя считать хорошим индикатором для окислительно-восстановительного титрования. Среди других ранних исследований можно указать на работы Кильбазинского по индиго-сульфокислоте [11], Монье по индулину, сафранину и метиленовому голубому [12], Руссо [13, 14] и Синат [15, 16] по метиленовому голубому. Все эти индикаторы правильнее отнести к группе соединений с низким потенциалом, и они рассмотрены в гл. VIIIA. Изящные классические исследования Кларка с сотр. [1] были начаты в этот же период и внесли большой вклад в понимание лей ствия индикаторов. В 1924 г. Кноп [17] описал первый случай успешного применения внутреннего цветного индикатора дифениламина. Индикаторные свойства этого соединения еще раньше исследовали Керманн с сотр. [18—20], Маркейроль и Мюраур [21], а также Виланд [22]. Вскоре было предпринято исследование других подобных соединений, и в течение следующих десяти лет к ним прибавились две большие группы индикаторов, которые нашли широкое применение: трифенилметановые красители и комплексы о-фенантролина. С тех пор были введены в практику другие важные индикаторы или группы индикаторов, но то, что было проделано за первые десять лет, легло в основу метода, применяемого и в настоящее время.
Начальный период развития индикаторов прекрасно описан Бренке [23], а в 1941 г. — Уайтхидом и Уилсом [24], которые провели также синтез многих индикаторов. В монографии Кольт-J гофа и Стенгера [25] рассмотрены индикаторы с низким и высоким потенциалом, а в последнем томе этой монографии, авторами которого являются Кольтгоф и Белчер [26] и который был опубликован 15 годами позже, даны подробные указания по ПРЙ' менению большей части известных в настоящее время индикатО| ров. В монографию Томичека [27] включены окислительно-восстановительные индикаторы, описанные в литературе по 1950 г. В РЯ1 де других общих изданий [28, 29] имеются отделы по теории
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
127
пименению окислительно-восстановительных индикаторов, а в бо-ее поздней монографии [30] рассматриваются индикаторы, дей-ЛтВУющие как реагенты, в менее распространенных окислительно-восстановительных процессах.
Окислительно-восстановительные индикаторы сами по себе являются окислительно-восстановительными системами, и, хотя в -ледующем разделе рассматриваются окислительно-восстановительные реакции вообще, все это целиком относится и к реакциям индикаторов. При рассмотрении процессов окислительно-восстановительного титрования внимание сконцентрировано на факторах, от которых зависит выбор индикатора и создание условий для хорошего проявления действия индикатора. В следующем разделе дается общее представление об индикаторах, а в последующих разделах индикаторы рассматриваются по отдельности.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ
Следует заметить, что окислительно-восстановительные реакции часто более сложны, менее понятны и не так гладко протекают, как большинство реакций простого взаимодействия. Механизм реакций может быть сложным, может отклоняться от прямого пути и включать много стадий, а кроме того, эти реакции часто бывают очень чувствительны к экспериментальным условиям. Только не давно серьезные попытки уяснить себе эти процессы привели к существенным результатам. Монография Тернея [31] не критична, многие вопросы изложены слишком упрощенно в ней, но все же она дает некоторое представление о состоянии этой проблемы. Рех-ниц дал содержательные, но слишком краткие комментарии в своей недавней монографии [32] по кинетике в аналитической химии и сам провел некоторые очень полезные исследования. Очень своевременна книга Пардю с сотр. [33] по катализу реакции церия(1У) с мышьяком (III) при помощи соединений осмия. Это богатая, но еще сравнительно мало изученная область, в которой надо еще много поработать. Исследования механизма окислительно-восстановительных реакций обобщил Левич [34]; многие из этих исследований относятся к окислительно-восстановительному титрованию [35].
За исключением некоторых процессов обмена лигандами, встречающихся в комплексометрии, самыми медленными из титримет-Рических реакций являются окислительно-восстановительные, по-тому часто надо учитывать их кинетику. Медленное протекание Реакций может привести к нежелательным явлениям; к ним отно-щиТСЯ РеакЦии, в которых могут участвовать индикаторы. Боль-ДостСТВ° окислителей, рассматриваемых в этом разделе, обладает Во*ТО™° высоким формальным потенциалом, чтобы окислить
У и другие растворители. Действительно, такие окислители, как
128
ГЛАВА VIII
серебро(II), перхлорат церия (IV), феррат(У1) и др., именно так реагируют. Другие же реагенты, как, например, бихромат, сульфат церия(IV), бромат и т. д., устойчивы в 1водном растворе в течение долгого времени, и их растворы даже можно кипятить без заметного их разложения. Это объясняется просто тем, что скорость реакции реагента с растворителем исключительно низкая: тем не менее эту реакцию можно катализировать, что приведет к росту ошибки. И наоборот, многие окислители с переносом нескольких электронов не действуют как обычные окислители до тех пор, пока не разложатся при таких жестких условиях, как нагревание в сильнокислой среде. Бромат [36] и хлорамин Т [37] служат примером окислителей, которые не действуют в кислых растворах, не способных восстанавливать, как, например, серная кислота. Эти окислители специфически взаимодействуют с галогенидами, так же как перманганат реагирует с марганцем (II), а бихромат с ионом хрома (III) с образованием частиц интермедиатов, которые являются сильными окислителями в титровании. В этом случае большое значение имеет условный потенциал интермедиатов. Хотя в таблицах приводятся значения стандартных потенциалов для пер бромат—бромид и перманганат—марганец(П), они не имеют практического применения. Любая попытка измерить эти потенциалы приведет к установлению потенциала интермедиата, если условия допускают протекание реакции. В условиях, не ведущих к образованию интермедиата, значения потенциалов будут колебаться из-за малой величины переменного тока у электрода.
Здесь нецелесообразно останавливаться подробнее на этом вопросе, но необходимо, чтобы химик знал об его существовании и мог бы понять его смысл. Это означает, что нужно критически подходить к каждому конкретному случаю; общие рассуждения относительно окислительно-восстановительной реакции дают не больше информации, чем простой случай идеальной стехиометрии, и значения стандартных электродных потенциалов в таблицах мо гут почти не иметь смысла в экспериментальных условиях. Чем больше аналитик знает о действительном механизме реакции (а количество литературы по этому вопросу быстро растет), тем лучше он сможет контролировать, регулировать и интерпретировать полу ченные данные. Поэтому, не вникая в тонкости отдельных реакций, выражают равновесное состояние на любой стадии реакции, используя обычное теоретическое описание с применением понятия электродных потенциалов и уравнения Нернста [38]. Это уравне ние можно вывести и использовать с одинаковым успехом на ос новании как термодинамики [39], так и кинетики, исходя из тео рии абсолютной скорости реакции [40]. Термодинамический метод очень хорошо описывает истинное равновесное состояние и совсем не подходит для неравновесных состояний [41], а кинетический метод имеет более общее применение. Кинетический метод пока
окислительно-восстановительные индикаторы
129
ывает, что уравнение Нернста относится к гетерогенному процессу обмена электронами, происходящему на границе раздела плотного слоя раствора и металлического электрода, и только недавно было отмечено [42], что число электронов п в уравнении Нернста
относится к электронам, которые участвуют в стадии, определяющей скорость гетерогенного электродного процесса. Это число электронов не обязательно совпадает с числом электронов, которые переносятся в гомогенной реакции титрования. Кроме того, константа скорости гетерогенного процесса на электродах может
отличаться на несколько порядков от константы скорости гомогенного процесса в растворе: реакция мышьяк(У) — мышьяк(Ш) идет на электроде исключительно медленно, однако она легко используется в прямой и обратной титриметрических реакциях [26]. Если гетерогенная [43] и гомогенная [32] реакции идут с большой скоростью, т. е. если переменный ток электродного процесса большой, то реакции в этом случае можно описать уравнением Нернста и результаты будут близки к истинным. Если нет необходимости перечислять все отдельные реакции, тогда надо использовать уравнение Нернста.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ
(2)
Рассмотрим простую реакцию первого порядка
Ох + пе ч Red (1)
Потенциал £eq равновесной смеси окисленной Ох и восстановленной Red форм дается без вывода формулы уравнением (2):
Е —Е~ \ Р' Г In °ох
£eq-£ + пр 1П ,
где/? газовая постоянная (8,3143 Дж-град-1-моль-1); —абсолютная температура; F — число Фарадея (9,64872-104 А-с-экв-1), аох и nre(j — активности обеих частиц. Если flOx=£red= 1,0 моль-л' 3, тогда Eeq=E°, где £°— стандартный потенциал системы. Исходя из теории абсолютной скорости реакции очевидно, что стандартный потенциал зависит от соотношения констант скорости прямой и обратной реакций (1) при электродном потенциале, равном нулю в шкале водородного электрода,
г, RT
Е~
,,-ln-^-, лД kb
ответе И констан™ скорости прямой и обратной реакций со-твенно. А если исходить из термодинамики, стандартный по-
9~»18
(3)
130
ГЛАВА VIII
тенциал зависит от изменений свободной энергии образования активированного состояния в обоих направлениях:
AG* - AG*
Е =——
(4)
Тогда п связано со стадией, определяющей скорость реакции. Определение активностей частиц не является простым делом, особенно в среде с высокой ионной силой, в которой обычно работают при окислительно-восстановительном титровании, и поэтому вводятся концентрации частиц. Если [ох и [red— коэффициенты активности частиц, концентрации которых [Ох] и [Red] выражены в моль-л"1 (в дальнейшем обозначаются М), тогда
аох=/ох[Ох] и nred=/red[Red] (5)
Подстановка в уравнение (2) дает
Р р— I In f°x [Ох! /д\
E^=E (6)
или
р __р- I /ох , ВТ i„ [Ох]
E*t~E + nF In + nF In [Redj (7)
Путем объединения первых двух членов в правой части уравнения и обозначения их получаем
£ea==£'o + ^^ln’^df. (8|
где Ео — так называемый «нормальный потенциал» или концентрационный потенциал, который при предположении, что Ох и Red — нейтральные молекулы или простые сольватированные ионы, можно определять в любом растворе при условии, что [Ох] = [Red] = = 1,0 М. Теперь это уже условная константа, потому что fox и [red в значительной степени зависят от условий, а Ох и Red будут находиться в состоянии различной степени окисления, проявляя различную зависимость коэффициентов активности от ионной силы. Поэтому необходимо оговорить, что концентрации Ох и Red равны 1,0 М, а не просто одинаковые.
Маловероятно, чтобы Ох и Red были простыми сольватированными ионами; почти наверняка другие лиганды входят в их состав, и, следовательно, их концентрация изменяется вследствие комплексообразования. Например, предположим, что присутствующий в растворе лиганд L образует комплексы OxLa и RedL, константы образования которых и Тогда
[°х]=гЙ- и ^--ПЕГ (9)
Ра 1LJ Pi
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 131
Подставив эти значения в уравнение (8), получаем
Е —Е + - R—~ In-IOxL2^ Ш ПО)
req—-Co-t- пр р2 [Ц2 [RedL]
Это положение можно объяснить различными способами, а среди лучших надо признать объяснение, базирующееся на введенных рингбомом условных константах (ср. гл. VIA и [44]). Таким образом, [Ох] можно рассматривать как сумму концентраций всех частиц, в состав которой входит [Ох] (аналогично для ['Red]), а дополнительный член, введенный в уравнение, можно объяснить комплексообразованием наряду с членом для концентрации лиганда. Точное выражение первого члена несущественно, предположим, что это Aox^red, но знак второго члена зависит от реакций комплексообразования, так что уравнение (10) становится
£,eq==£0 + AOx^red ± In [LP + -§1 In -g- (11)
При определенной концентрации лиганда можно объединить первые три члена в виде Ед, который является формальным или условным потенциалом:
+ (12)
Это выражение означает, что «константа» Ед фактически зависит от экспериментальных условий, но тем не менее ее наиболее часто используют аналитики. Считается, что перхлорат-анион обладает минимальной способностью образовывать комплексы, так что измерения в среде перхлората обычно больше всего приближаются к условиям простого сольватированного иона в уравнении (8). Даже простая замена среды на сульфатную или хлоридную может сильно повлиять на кажущийся потенциал системы. Например, для системы церий(1У) — церий(Ш) Ео, в 1,0 М хлорной кислоте равную 1,70 В, он смещается до 1,44 В в 1,0 М серной кислоте и до 1,28 В в 1,0 М соляной кислоте.
Во многих реакциях типа (1) участвуют ионы водорода (или ионьт гидроксила) и их потенциалы в значительной мере зависят °Т Р рСРеды- По всей вероятности, это входит в условный потенциал Е' так же, как L в уравнение (11), где х обозначает число ионов водорода, участвующих в реакции при условии, что реакция ксиТ ВЫС0К0Й концентрации ионов водорода (или ионов гидро-ла) или в буферной среде, в которой значение pH постоянно, мож’1ОИЗВеСТНО для данного значения pH. Члены в [L] и [Н+] нить о П11ЯМ0 включить в уравнение для потенциала, чтобы пом-влияниаИХ; °НИ дают представление о степени их влияния, но это редко бывает непосредственным.
9*
132
ГЛАВА VIH
Выражения типа уравнения Нернста указывают на два момента. Во-первых, потенциал зависит от фактора, который по своей природе является константой равновесия и характеризует данную систему (1); это либо стандартный потенциал Е~, представляющий собой истинную термодинамическую константу при выбранной температуре, либо условный или формальный потенциал Ею дополнительно характеризующий данные экспериментальные условия, которые должны всегда быть тщательно оговорены, если надо использовать значения Е’о. Можно, конечно, вывести значение Е’о из Е~, если располагать достаточно точной информацией о системе и о коэффициентах активности, константах образования и т. д. Такой исчерпывающей информацией пока еще располагают редко, а вычисления очень большие и сложные. Когда собрана необходимая информация, легко провести расчеты на компьютере с помощью программ, подобных программе Халтафала [45, 46]. Во-вторых, потенциал зависит от соотношения активностей или концентраций окислителя и восстановителя.
УСТАНОВЛЕНИЕ РАВНОВЕСИЯ
Уравнения (2) или (12) описывают равновесное состояние реакций типа (1). Для таких реакций концентрационный член выражается в виде отношения и не зависит от абсолютной концентрации [47]. Поскольку рассматривается равновесие, то для данного соотношения величин ['Ох] и [Red] не имеет значения, будут ли абсолютные концентрации порядка 0,1 М или 10-12 М; потенциал будет одинаков во всех случаях. Однако скорость установления равновесия после некоторого его нарушения, например от добавки инкремента титранта, пропорциональна абсолютной концентрации или зависит от нее. Так, хотя рассчитан потенциал системы в равновесии, кривая титрования, например, для окисления железа (II) раствором церия(IV) одинакова при концентрациях реагента 1,0 М и 10-6 М, абсолютная скорость реакции более разбавленного раствора будет в 1012 меньше скорости при концентрации 1,0 М. Далее, если время релаксации в концентрированном растворе составляет 1 мкс, т. е. реакция идет с очень большой скоростью, в растворе с низкой концентрацией требуется срок 10 дней и поэтому титрование при низких концентрациях не имеет практического смысла. Хотя эти примеры достаточно наглядны, следует подчеркнуть, что, несмотря на то что реакция термодинамически удовлетворительна для использования ее, равновесие может устанавливаться так медленно, что она теряет практическое значение. Даже при разумных концентрациях реагентов реакция может иметь медленную стадию, которую приходится ускорить, чтобы можно было применять реакцию на практике. Например, при окислении броматом таких частиц, как мышьяк(Ш), сурьма(Ш) или гидразин,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
133
бщая реакция является реакцией нулевого порядка в отношении °осстановителя, первого порядка по бромату или галогениду (бромиду или хлориду) и второго порядка по иону водорода. В отсутствие бромида или хлорида реакция совсем не идет, как не идет и при низкой концентрации кислоты [36]. Стадией, определяющей скорость реакции, является стадия образования активного окислителя (брома или монохлорида брома), которая зависит от реакции четвертого порядка, происходящей между парами ионов Н+ВгО7 и Н Вг \ Полное уравнение для скорости имеет следующий вид:
- d =k* IВг°П lBr~l lH+i2 (13)
Выше говорилось о переменном токе гетерогенного электродного процесса, используемого для измерения Е при равновесии. И в этом случае, не вдаваясь в подробности вывода уравнения ['48], для реакции
Ox -f- пе ч — > Red, (14)
где ic — катодный ток (ток восстановления), ia — анодный ток (ток окисления), уравнение переменного тока i0, протекающего в системе в состоянии равновесия, т. е. когда ic—ia и Е=Ее(1, имеет следующий вид:
i0=nFAk [Ox 1( ’-“I [Red]a (15)
В этом выражении k — кажущаяся или общая константа скорости переноса заряда, а а—коэффициент переноса заряда, являющийся частью приложенного потенциала, который обеспечивает протекание прямой (катодной) реакции. Чем больше значение io, тем быс-
трее наступает равновесное состояние на электроде после его нарушения. Независимо от значения a io имеет максимальную величину, когда [Ох] = [Red], т. е. когда Ее<1~Е0, и возрастает с увеличением концентрации. Включение площади электрода А (в см2) в выражение показывает, что это реакция гетерогенного типа, т. е. подчеркивается преимущество работы с не слишком большими электродами. Если общая концентрация Ох и Red больше 10~7 моль-л-1 в k раз, то реакция идет достаточно быстро, для того чтобы можно было с уверенностью использовать понятие электродного потенциала для объяснения гомогенного равновесно-° состояния в растворе.
РЕАКЦИИ ТИТРОВАНИЯ И РАВНОВЕСИЯ
гУт^1°М0ГеННаЯ Реакйия (О или гетерогенная реакция (14) не мо-злект)0ТеКаТЬ сами по себе. Для прямой реакции необходим донор рона, а для обратной реакции — акцептор электрона. Для
134
ГЛАВА VIII
гетерогенной реакции (14) это может быть электрический ток, протекающий через электрод (на этом основывается метод кулонометрического титрования ['49]). Для гомогенных процессов это может быть система, подобная реакции (1), но с достаточно более низким Ео в случае прямой реакции или достаточно более высоким Е'о для обратной реакции. Обозначим индексом 1 систему окислителя, а индексом 2 систему восстановителя в титриметрической реакции
aOXj -f-ще <- _> cRedx; dOx2-f-n2e <—> fcRed2
Система 1 соответствует реакции в прямом направлении за счет электронов, производимых системой 2, соответствующей реакции в обратном направлении
аОхг + гце < cRedj
bRed2 ч —*- n2e-(-dOx2 (16)
ап^Отц + fen1Red2 ч—CTi2Redj -f- dn1Ox2
Уравнение (16) представляет общую стехиометрию реакции титрования. Смесь любого числа таких систем, включая индикаторы, будет взаимодействовать путем обмена электронами так, что концентрации всех присутствующих в системе частиц изменяются до установления равновесия, а потенциал становится одним и тем же для всех индивидуальных систем. Если частицы реакционноспособны, то константу равновесия реакции типа (16) можно вычислить методом, изложенным в гл. II. Величина К' по-прежнему представляет собой условную константу равновесия:
[Red1Jcfl2 [Ох/"1 [Ox/"2 [Red]2ni
=ехр
' n1n2(Ef>1— E'Ov )F
. RT
(17)
На основании этого выражения и соотношений потенциалов для отдельных реакций
nrF [Red/ 03 n2F [Red/
(18)
можно математически ['47, 50, 51] рассчитать любые данные о процессе титрования, например получить кривую титрования, Q-функ-ции, точность, надежность (отклонение конечной точки от точки эквивалентности), количественную оценку протекания реакции, потенциалы точки эквивалентности и конечной точки и т. д. Реакции титрования, подобные реакции (16), подразделяют на три группы на основании их стехиометрических коэффициентов, сложности или простоты математического расчета [47, 52].
1) Реакции с неодинаковыми коэффицентами, когда а#=с и (или) b=£d. В этом случае необходимо использовать точные выражения (17) и (18). Потенциал, в который входит потенциал точки
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
135
эквивалентности, зависит от абсолютных концентраций различных частиц ['47, 50].
2) Реакции с одинаковыми коэффициентами, когда а=с и h=d обычно все равны единице, но П1=#П2- В этом случае необхо-имо только, чтобы соотношение ['Ox]/[Red] было известно и чтобы система не зависела от абсолютных концентраций, поскольку печь идет о состоянии равновесия. Точные решения для количественной оценки протекания реакции и для потенциала точки эквивалентности можно получить в простом виде [47]. Кривые титрования имеюч асимметричную форму.
3) Реакции с симметричными коэффициентами, когда а=с, а П1=П2- Система также ие зависит от абсолютной концентрации, и можно написать простые выражения для критериев. Кривые титрования в этом случае симметричны.
При использовании реакции титрования для определения одной из участвующих частиц и применении визуального индикатора для установления конечной точки необходимо знать о реакции следующее: а) потенциал точки эквивалентности, для того чтобы выбрать индикатор с нужным E'oini, и б) значение Q (см. гл. II). Эти данные нужны, чтобы узнать, достаточно ли ясно изменение окраски индикатора и можно ли получить требуемую точность определения. Конечно, затем надо провести опыт, чтобы убедиться, не влияют ли кинетические факторы, не учитываемые в расчетах равновесий.
ПОТЕНЦИАЛ ТОЧКИ ЭКВИВАЛЕНТНОСТИ
Потенциал точки эквивалентности для реакций с симметричными или одинаковыми коэффициентами не зависит от абсолютного потенциала и дается следующими уравнениями (без вывода уравнений [47, 53]):
и1=п2=п, £cq.pt= '£о1^~£°а (19)
^eq.pt— „1 + па
Для реакций с неодинаковыми коэффициентами потенциал точки ю вивалентности зависит от абсолютных концентраций реагиру-метоХ веществ> Для точного расчета этого потенциала требуются котопЫ С пРименением чисел. Рассмотрим случай титрования, в станет система окислителя 1 является титрантом, а система вос-ет£Я 0«Теля — титруемым веществом. Предположим, что титру-а титраЪеМгЛй МЛ вос'с'ганови'геля Red2, молярность которого Cd, Р нт Oxi имеет концентрацию тогда для того, чтобы до
136
ГЛАВА VHI
стигнуть точки эквивалентности, требуется израсходовать объем Vt мл окислителя Oxi:
у/___
l~ bn^Ct
(21)
В этой точке концентрация окислителя Ох2 выражается уравнением
0X2 ~ +
(22)
а количество восстановителя Red2, остающегося в растворе в этой точке из-за того, что реакция не протекает количественно (законченность реакции [47, 50] определяется концентрацией 6red2), выражается уравнением
(23)
где К дано уравнением (17). Хорошее приближение для 6red2 можно получить, пренебрегая членом d-6red2/b по отношению к Сох,, а если реакция позволяет использовать визуальные индикаторы, такое упрощение определенно допустимо. Это дает возможность написать точное решение, которое, несмотря на его сложный вид, легко решить без помощи компьютера, так как a, b, с, d, щ и п2 целые числа:
; —f-L[/_LV ( с \с( ъ \а-с1П21 red2_) J 0X2 J
(24)
Тогда потенциал точки эквивалентности получается путем подстановки в уравнение (18):
_ RT fCox2- (d/b) 6red2]d
^eq.pt —С02+ &b
ured2
(25)
ЗНАЧЕНИЕ ВЕЛИЧИНЫ Q ТИТРОВАНИЯ
Функцию Q титрования обычно рассматривают, исходя из потенциала окислительно-восстановительных реакций. Если через Р обозначить требуемую точность определения в процентах, то это означает, что конечную точку титрования следует определять в пределах инкремента титранта, выраженного Ди мл при объеме Vt мл в точке эквивалентности,
р_ЮОДч
~~vT
(26)
137
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ ------'
Гпрповательно, если титрование 25 мл (У<) надо осуществить с точностью до 0,1% (т- е- ±0,05%), то Ди=0,025 мл. Необходимо паспределить этот инкремент поровну по обе стороны это положение, между прочим, дает максимальное изменение потенциала при добавке инкремента До мл в ходе титрования. Тогда Q будет изменением получающего потенциала:
Р=£(при 24,9875 мл)—£ (при 25,0125 мл)
Это дает отрицательное значение Q для титранта-окислителя и положительное значение Q для титранта-восстановителя. Если нужна только абсолютная величина, то берут модуль, и тогда общее выражение будет
Q=|£(npn V=Vt—0,5Ди)—£(при V—Е/ + 0,5Дц)| (27
Если для установления конечной точки нужно работать с индикатором, Q должно быть существенно большим, особенно в случае окислительно-восстановительных реакций, которые имеют тенденцию протекать кинетически медленно. Однако нет необходимости в точном расчете [54] значения Q. Если потребуется вычисление, то или титрование невозможно, или нужную точность слишком трудно получить. Следовательно, можно использовать наиболее упрощенное выражение для Q, особенно потому, что оно дает более низкое значение, чем точные выражения. Тем не менее этого вполне достаточно для данного случая. Когда ответ меньше 0,12/nindK тогда титрование вряд ли будет успешным, даже если E6i nd точно равно £eq.pt- Поэтому разумнее принять, что реакция заканчивается в любой точке, т. е. величина потенциала в точке эквивалентности, выражающая степень законченности реакции [50], равна нулю. В общем случае титранта-окислителя и при использовании обозначений, принятых для уравнений (21) и (22), можно вычислить кажущиеся концентрации реагирующих веществ на основе значений Cd, Ct и стехиометрии реакции (16) после добавки выбранного объема титранта при предположении, что реакция прошла полностью. Эти значения можно подставить в уравнение (28), которое представляет собой развернутое уравнение 1 /) для самого общего случая, и получить простые точные выра-ения для Q, исходя из изменения потенциала; эти выражения для вполне пригодны для использования их с индикаторами:
9=Д£=£' КГ ]п [Охх(при yz + 0,5Au)]Q _
01 °2 niF [Redj (при yz + O,5A0]c
_ RT In [0*2 (при —0,5Au)]d дач nzF [Red2 (при Vt — 0,5Av)]6
138
ГЛАВА VIII
Переведя для удобства натуральные логарифмы в десятичные и используя для упрощения стехиометрическое соотношение (29)
(29)
получают уравнение (28) в другом виде для реакций с неодинаковыми коэффициентами:
л___р' р' । 2,303/??’ ( а ] ______Cj\v_______
Ч—£01 £оя-г р | П1 2(yz + rd + 0,5Av) +
с 1_ а(У, + 1^ + О,5Д0 b . _____________ЬгцС^у________
•" гц о Cthv "г „2 ® %ап2 (И, + Va — О,5Д0 ’
d 1 в anslVt + Vd— 0,5Др) |
+ пя ё dritCtlVt — 0,5 Дк) J
Величину Q легко вычислить для специального случая, и уравнение (30) можно упростить при некоторых условиях, показанных ниже, для выяснения влияния отдельных параметров.
В случае одинаковых коэффициентов, когда a=b=c=d~\, уравнение (30) упрощается и принимает следующий вид:
г» г?» о' । 2,303т??1 ( 1 1 „ Дс 1 Дс ) zq|,
E«i~F ('nTlg'2l7’+_«rlg 2(FZ —О.5Д0 /
Последнее выражение не зависит от концентраций реагентов, за исключением случаев, когда они определяют значение Vt.
В случае симметричных коэффициентов, когда ni = n2, выражение упрощается еще больше:
л__r-z j-iz . 2,303/??’ 1 Дс2
'£<>1 ^<>2 -1 (Гд 2УД^ — 0,5Дг) (3 )
Если значение Q становится меньше 0,12//iindK тогда реакция в значительной степени остается незаконченной и надо использовать более точные уравнения с привлечением значений незаконченности реакции 6red2 до точки эквивалентности и fioxj после точки эквивалентности [47,50].
ВЫБОР УСЛОВИЙ ТИТРОВАНИЯ
Необходимо всегда обращать внимание не только на состояние равновесия, но и на другие факторы. Однако, если скорость реакции удовлетворительна, то очевидно, что, чем больше значение Qz тем лучше идет процесс титрования и тем шире выбор подходящего индикатора. При работе с обратимыми индикаторами реакция должна достигать равновесия в течение нескольких секунд, а в случае необратимых индикаторов требуется намного более быстрое достижение равновесия, чтобы избежать преждевременного установления конечных точек. Для всех типов реакций титрования
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
139
Функция Q прямо пропорциональна разности между формальными потенциалами окислителя и восстановителя, а поэтому приходится подбирать такие реагенты, чтобы разница между потенциалами была возможно больше. Эти формальные потенциалы являются условными константами, и поэтому их можно в большей или меньшей степени регулировать, изменяя pH и концентрацию лиганда. Идеальным примером был бы окислитель, присоединяющий ион водорода, в комбинации с восстановителем, который отдавал бы ион водорода при своем окислении. При высокой концентрации кислоты получается большая разность между формальными потенциалами. Такой же эффект можно получить при работе с лигандом, который образует комплекс преимущественно с окисленной формой восстановителя и восстановленной формой окислителя.
Вообще, Q растет с увеличением числа участвующих электронов, а также возрастает логарифмически с увеличением Av и уменьшением объема Vt в точке эквивалентности. Иными словами, Q пропорционально значению lgiAo/V«, поэтому при менее строгих требованиях к точности метода определения труднее добиться лучшего титрования и лучшей точности, чем требуется в данных условиях. Если требуемая точность составляет 1 % или выше, а значение Q больше 120 мВ, то можно упростить приближенные уравнения (30—32), чтобы яснее выявить влияние параметров в них, пренебрегая для этого значением Av по отношению к значениям Vt или Vt+Va- Это оправдано только в том случае, если соблюдены оба условия, а именно Av<g.Vt и Q>120 мВ. Упрощение и преобразование уравнений приводит к следующим результатам.
Реакции с неодинаковыми коэффициентами. Если разделить члены уравнения на целые коэффициенты и обозначить результат через константу, то
2,зозет awntMd-bm^,bb/n^ п[ь-<ы к нстанта— р 1g ^а/пмь/ъП Л<Иъ
Получаем
Q=С01—константа -Т г.зоз/гт
+ р
[(a-c/ni) + (b-d/n2)} Ap[W«l)+(W] |
1 + J ' ’
единственный тип реакции, который явно зависит от концент-млс™ Реагентов (Cd вводится косвенно вследствие взаимозависи-чаети величин Cd, Vd, Ct и У<). Точная зависимость в данном слу-°ьУСловливается значением показателя степени концентрационного члена.
Если -а с &—d
ni + п2 >0, то Q увеличивается с повышением кон-Ц нтРации; если эта сумма равна 0, то Q не зависит от концентра
140
ГЛАВА VIII
ции, а если сумма меньше нуля, Q растет с уменьшением концентрации. Последний, довольно странный вывод был подтвержден теоретически и экспериментально на примере галогенов в качестве окислителей. Этот тип реакции представляет собой наиболее общий случай, хотя снижение скорости реакции накладывает ограничения на ее практическое применение. Зависимость Q от объемов растворов является тоже функцией показательной степени. Но если предположить, что значения Vt и Vj одного порядка, тогда зависимость приближается к выражению
/ а Ь X , 1
\пГ+~пГ)
Этим подтверждается, что Q улучшается с уменьшением объемов.
Реакции с одинаковыми коэффициентами. Реакция в этом случае не зависит от концентрации, a Q зависит только от точности и значений п\, п2 и формальных потенциалов:
р' । ni + n2 2,303/??’ . Ач
(34)
Если значения п\ и п2 больше единицы, создается более выгодное положение.
Реакции с симметричными коэффициентами. В этом случае выражение для Q похоже на уравнение (34), за исключением того, что Q возрастает линейно с увеличением п:
q-£;-^+A-.
2,303т??1 F
1g
Ди 2V7
(35)
Для увеличения Q с ростом п в уравнениях (34) и (35) необходимо, чтобы &v<giVt и последний член был бы отрицательным. Оба эти уравнения ясно показывают величину возрастания До и уменьшения Vt-
Выводы. Для данного случая титрования, чтобы получить четкую конечную точку и наиболее высокое значение Q в сочетании с требуемой точностью, надо подобрать наиболее подходящие условия для объемов и концентраций реагентов и величину инкремента титранта, добавляемого при приближении к конечной точке. Для этой цели инкремент Ди должен быть большим, объем в точке эквивалентности малым, а концентрации реагентов, кроме случая, указанного для неодинаковых коэффициентов, должны быть высокими для обеспечения быстрого протекания реакции. Реагенты следует подбирать так, чтобы значение п и разность между фор* мальными потенциалами были большими.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ индикаторы
141
ВЫБОР СРЕДЫ
Уже говорилось о влиянии pH и концентрации лиганда на ус-овные потенциалы. Для того чтобы медленные реакции смогли найти применение, необходимо увеличить разность потенциалов Е' __£q2 и эффективную скорость реакции, используя pH, концентрации Вигандов, а также другие внешние условия. Все эти факторы следует учитывать при выборе индикатора и рассматривать их влияние на сам индикатор, если таковое имеется. Индикаторы почти всегда бывают чувствительны к pH среды и иногда могут образовывать осадки или комплексы с отдельными частицами, участвующими в реакции титрования. Применение некоторых фак торов среды будет кратко рассмотрено при обсуждении этих вопросов.
Концентрация иона водорода
Ионы водорода могут оказывать влияние тремя путями. Во-первых, они могут образовать комплексы или протонированные соединения с одной или несколькими реагирующими частицами либо частицами окружающей среды, а часто и с индикатором. Во-вто рых, ионы водорода могут принимать существенное участие как в реакциях окисления или восстановления, так и в реакции с индикатором. В ходе реакции эти ионы могут либо присоединяться, либо отщепляться. В-третьих, они могут участвовать или катализировать стадию, определяющую скорость реакции образования активных частиц одного из реагентов. Нередко ионы водорода участвуют в реакции по всем этим трем путям, и тогда зависимость системы от их концентрации может усложниться. Обычно концентрацию ионов водорода поддерживают постоянной во время титрования, используя для этого сильнокислые, сильнощелочные или буферные растворы, так что во время титрования pH не изменяется, хотя оно является определяющим параметром, выступающим иногда в качестве постоянной величины в уравнениях равновесия или, когда рассматривается третий путь влияния ионов водорода, в Уравнениях скорости. Если точно известен характер участия иона водорода, то можно его ввести в виде отдельного члена или нескольких членов в выражения для потенциала и Q-функции, а затем использовать при количественном выборе оптимальных усло-и титрования. Иными словами, концентрацию ионов водорода СПрЖно включать в выражение для условного потенциала Ео при 28-^4ofbHbIX услсвиях- Можно привести множество примеров [26, ного ° Б П°ЛЬЗУ РегУлиР°вания реакций путем выбора определен-в нейЗНаЧеНИ? PH- Мышьяк(Ш) количественно окисляется иодом навл ТРаЛЬН0Й сРеде> ТОГДЗ как мышьяк(У) количественно восста-
ивается иодом в сильнокислой среде; изменение pH на 6 еди
142
ГЛАВА VIII
ниц вызывает количественные изменения в направлении этой реакции. В броматометрии активный окислитель — бром или монохлорид брома — реагирует независимо от pH при условии, что значение pH не настолько высоко, чтобы вызвать гидролиз; однако реакция образования активного окислителя зависит от квадрата концентрации ионов водорода и становится слишком медленной, если эту концентрацию уменьшить ниже некоторого не слишком высокого значения, зависящего от природы и концентрации присутствующего галогенид-иона.
Реакции комплексообразования и концентрация комплексообразующих лигандов
Если лиганд образует более прочный комплекс с окислителем, чем с восстановителем, отношение концентраций свободного окислителя и восстановителя [Ox]/[Red] уменьшается тем больше, чем больше концентрация лиганда. В результате внесения лиганда в раствор понизится потенциал смеси. Выше приводился пример влияния неорганических ионных лигандов на систему церий (IV) — церий(Ш). В качестве таких лигандов для системы железо(Ш)— железо (II) используют фосфаты, фториды и этилендиаминтетраацетат, хотя ион водорода конкурирует с железом(III) в отношении к лиганду, и влияние в данном случае уменьшается, если повышается концентрация ионов водорода. Обычно и окислитель, и восстановитель образуют комплексы с лигандом, а так как окислитель находится в более высокой степени окисления и, значит, имеет больше вакансий на орбиталях, он гораздо чаще образует устойчивые комплексы. В приведенном примере интересно влияние ЭДТА: потенциал может уменьшиться до такой степени, что раствор становится неустойчивым к ионам водорода, которые восстанавливаются до газообразного водорода. Почти не имеется случаев противоположного влияния, когда восстановитель образует более устойчивый комплекс: в частности, таким примером является о-фенантролин в качестве лиганда, который образует ион 7/шс-(о-фенантролин) железо (II), очень ценный как индикатор. Однако, если реакции с лигандом полностью поняты, их влияние можно отразить в виде специального члена или нескольких членов в уравнениях либо их можно ввести в уравнение условного потенциала Е'о для специфических постоянных условий.
Реакции осаждения
На соотношение [Ox]/[Red] можно повлиять путем добавления к раствору иона или лиганда, образующего малорастворимые соединения с частицами Ох и Red. Потенциал сместится в сторону более растворимых частиц. Примером может служить система гек-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
143
пианоферрат(Ш)—гексацианоферрат (II); ионы таких металлов, как цинк, кадмий и др., образуют гексацианоферраты (II), которые К паздо труднее растворяются, чем соответствующие гексациано-Аеораты(Ш). Хотя стандартный потенциал этой системы существенно ниже, чем у системы иод — иодид, она может окислить иодид, если добавить ионы цинка для осаждения образующегося гексацианоферрата (II). Противоположное действие облегчает окисление таллия(I) в нейтральной или щелочной среде, потому что образующийся таллий(III) сразу же гидролизуется и осаждается в виде нерастворимого осадка. Зная произведение растворимости и концентрации, можно ввести в уравнения новые члены или отразить влияние в условной константе.
Интермедиаты реакции, катализаторы и кинетически затрудненные реакции
Многие реакции с кажущимся высоким Q или протекают слишком медленно, чтобы можно было титровать прямо без преждевременного определения конечной точки, или кинетически затруднены и вообще не идут с видимой скоростью. Такие реакции часто можно сделать применимыми, используя методы обратного титрования либо добавив интермедиат реакции или носитель электронов. С точки зрения индикаторов, если бы это было осуществимо, то означало бы изменение природы реакции титрования в зависимости от следующей новой серии параметров, feq.pt и Q, на основании которых был бы произведен выбор индикатора.
Если реакция просто слишком медленна, ее можно ускорить, добавив избыток титранта А и увеличив время, чтобы реакция могла закончиться. Затем избыток титранта определяют одним из Двух методов: 1) титруют обратно вторым титрантом Б или 2) добавляют избыток реагента В (или Б), который быстро взаимодействует с избытком А, а оставшийся избыток В оттитровывают затем титрантом А. Потом титрованием того же количества В титрантом А находят поправку на холостой опыт, вычитают из общего израсходованного количества титранта А и получают титр образца. По методу 1 титруют А раствором Б, а по методу 2 титруют В дикаВО^ОМ Для КОТОРОГО наД° знать Eeq.pt и Q для выбора ин-
Если окислитель с условным потенциалом E'oL не в состоянии кислить восстановитель с условным потенциалом Е'о2, то можно ставить идти реакцию окисления путем внесения интермедиата ин7ЛОВНЬШ потенЦиал°м Еоз при условии, что E’ot >Е'о3>Е'о, и что н еРмедиат быстро окисляет восстановитель и быстро восста-рий(1\пСТ окислитель ПРИ соответствующем Q. Например, це-иесмо Не БЗаимо Действует в заметной степени с мышьяком (III), три на большую разницу между Е^ и Е'о2. Однако в подхо
144
ГЛАВА VIII
дящих условиях монохлорид иода легко и количественно окисляет мышьяк(Ш), при этом сам восстанавливается до иода. При этих же условиях церий(IV) легко окисляет иод до монохлорида иода. Следовательно, добавка монохлорида иода создает условия, при которых можно титровать мышьяк(Ш) раствором церия(IV). Для выбора индикатора имеет значение реакция между иодом и церием (IV). Некоторые элементы, которые способны ib разной степени окисляться, быстро переходя из одного состояния в другое путем различных быстрых реакций, могут служить носителями электронов, Их обычно добавляют в небольших, каталитически действующих количествах, сравнимых с количеством используемого индикатора. При этом их следует добавлять в том состоянии окисления, в котором они будут находиться в конце титрования. Это d-элементы, из которых чаще всего используется осмий. Обычно осмий (VIII) используют в реакции церия(IV) с мышьяком(III) и предпочитают этот способ методу с применением монохлорида иода. Механизм этой реакции недавно изучен [33], и индикатор выбирают с учетом реакции церий(IV)—осмий(VII).
Если условия влияют на реакцию титрования и на реакцию индикатора (или только на последнюю), в любом случае следует обращать внимание на правильный выбор индикатора. Это проще всего сделать с помощью условных констант, если их можно определить.
ИНДУЦИРОВАННЫЕ РЕАКЦИИ
Возможно, что рассмотрение этого вопроса более уместно в титриметрическом анализе, однако это очень важный вопрос, до сих пор недостаточно освещенный [53], а индикаторы подвергаются индуцированным реакциям [55] и иногда зависят от индуцирования ([26], в гл. III, т. 1). Эти реакции известны на протяжении ста лет [56] и изучались в некоторой степени [57, 58] на рубеже двух столетий. Более тщательное исследование провел Скрэ-бел, который опубликовал в 1908 г. монографию на эту тему. В книге Кольтгофа ([25], в гл. III, т. 1) лишь дан краткий обзор, так что очень необходимо было бы иметь современную монографию по этому вопросу.
Аналитик должен ожидать возникновения индуцированных реакций при окислительно-восстановительном титровании, а также подобных явлений среди индикаторов. Сравнительно недавно почти не обращалось внимания на индукцию [60—63], когда Кшаньи с сотр. [65—71] и другие авторы [36, 55, 72—74] провели количественные исследования. Бишоп [53, 72] классифицировал индукционные процессы и рассмотрел некоторые новые и необычные примеры [72].
Не вдаваясь в подробности, надо сказать, что индуцированные
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 14S
акции бывают двух типов. К первому типу относятся такие реакции при которых окислитель А взаимодействует с восстановите-аем g’ но ни А, ни Б не вступают по отдельности в реакцию с В, а окисление Б посредством А в присутствии В индуцирует реакцию В с А (окисление В) или с Б (восстановление В). Ко второму типу относятся такие реакции, при которых окисление Б веществом' А индуцирует совсем обособленную реакцию В с Г, которая нормально не идет. Такие реакции обычно происходят в тех случаях, когда реагент А является одним из реагентов, подобных перманганату, бихромату, бромату, хлорамину-Т и т. д., которые реагируют с образованием промежуточных частиц, являющихся сильным окислителем, и эта реакция является медленной стадией и-ли индукционным периодом этого процесса. Любое вещество, которое замедляет образование реакционноспособных промежуточных частиц, приводит к возникновению неожиданных индуцированных реакций. Этот эффект можно существенно уменьшить или устранить путем подбора подходящих условий, при которых стадия, определяющая скорость реакции, пойдет с большой скоростью.
Перманганат вызывает появление многих таких реакций. Например, реакция перманганата А с оксалатом Б индуцирует образование перекиси водорода путем окисления воды В растворенным кислородом Г. Перманганат участвует также и в процессе первого типа: окисление железа(II) Б перманганатом А индуцирует окисление хлорида В до хлора. В частности, общим и трудным примером является окисление восстановителей (Б), таких, как иодид, мышьяк(Ш), сурьма(Ш), олово(П), железо(П), сульфит и др., такими окислителями А, как бромат, бихромат, перманганат и др., которые индуцируют окисление Б растворенным молекулярным кислородом В. Обычно медленно протекающая реакция вещества Б с кислородом значительно ускоряется, если замедлить стадию, определяющую скорость реакции с окислителем А. Примером служит титрование бихроматом железа (II) в присутствии сахаров или многоатомных спиртов с использованием дифениламиновых индикаторов [35, 75]. Окисление железа (II) бихроматом индуцирует окисление сахара с высоким результатом, а окисление сахара индуцирует окисление индикатора с последующей деструкцией последнего. По этой причине изменение окраски наблюдается очень корот кое время. F
ОБЩИЕ ПРИНЦИПЫ ДЕЙСТВИЯ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ИНДИКАТОРОВ
окисли Т°’ ЧТ° было сказано в предыдущем разделе относительно нимо кТельно-восстановительных реакций, в равной мере приме-реакциям окислительно-восстановительных индикаторов, в
10- i1R
146
ГЛАВА VIII
частности потому, что они подобны окислительно-восстановительным системам. Индикаторные системы претерпевают реакцию
Iadox -f- Indred, (36)
и часто на стадии их действия как индикаторов они вступают во взаимодействие с ионом водорода:
Indox + ninde + хН+ Indred, (37)
где х может иметь положительный или отрицательный знак. Кроме того, lndox (или Indred) может образоваться на обратимой или необратимой стадии, в которой может участвовать также и ион водорода. Это может не влиять на поведение веществ как индикаторов, но может расходовать титрант.
Возможно, что реакция индикатора в большей степени, чем реакции, рассмотренные в предыдущем разделе, чувствительны к условиям, к кинетическим факторам, к побочным и индуцированным реакциям, обмену лигандами, промежуточным стадиям и разложению, особенно в избытке окислителя. Вследствие всего этого выбор подходящего индикатора менее прост, чем для реакций простого взаимодействия ионов. Более того, область применения и количество окислительно-восстановительных индикаторов гораздо ограниченнее, чем, например, для кислотно-основных индикаторов. Наиболее удобными для практических целей в окислительно-восстановительном титровании являются индикаторные системы, имеющие высокие значения Q, так как в этом случае уменьшаются указанные трудности; больше сложностей создается кинетическими факторами.
КОНСТАНТЫ ИНДИКАТОРА
Здесь можно использовать обычные рассуждения, исходя из электродного потенциала, при тех же оговорках о различии между гетерогенным электродным процессом и гомогенной реакцией раствора. Потенциал, возникший в результате реакции (36), можно выразить несколькими способами, рассмотренными выше:
£ind =£."nd + In (38)
«1ПСВ Ure^ind
где Ем — стандартный электродный потенциал, a tzOxind и Credjnd"~ активности обеих форм индикатора в логарифмическом члене.
Из-за отсутствия точных сведений об активностях окисленной и восстановленной форм индикатора можно вместо активностей подставить концентрации
Р . ,==Е । In IIndoxl (39)
£md + Пыр ш [Indred] ,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЬПЕ ИНДИКАТОРЫ
147
где Ео а__нормальный потенциал для единицы концентрации обеих
форм индикатора.
Обычно нельзя получить растворы, в которых активность или онцентрация обеих форм индикатора была бы равна единице, а также не реально использовать такие низкие концентрации индикатора где эти условия были бы реализованы. Поэтому на практике почти не применяют стандартный и нормальный потенциалы, даже если они известны. Гораздо больше подходит условный или формальный потенциал, который можно использовать в экспериментальных условиях титрования, где концентрации индикатора такого порядка, который применяется на практике. За исключением реакций с образованием семихинона, реакции индикатора относятся к классу реакций с одинаковыми стехиометрическими коэффициентами и поэтому не зависят от концентрации, так что для данного отношения [Indox]/[Indred] потенциал не зависит от абсолютной концентрации. Образование семихинона при стехиометрическом соотношении 2: 1 будет зависеть от концентрации, но такие реакции представляют собой нежелательные побочные реакции, которые можно подавить путем создания подходящих условий. Поэтому условный потенциал можно отнести к низким концентрациям индикатора без каких-либо осложнений:
р ____р' I RT [Indox] /доч
Eojnd+„indF ’ <40'
где £oind—условный потенциал для эквимолярных концентраций обеих форм индикатора в определенных условиях титрования; а Е<>та> кроме того, является обычно используемой формой константы для окислительно-восстановительных индикаторов.
Обычно ион водорода участвует в реакции, подобной реакции (37), и, хотя его влияние можно ввести в условную константу индикатора, часто бывает полезно включить его в уравнение потенциала индикатора
RT
In [Н+] + -5Z- In t[TIn,d°-x-j- (41)
£ind-fo,nd4 -p
BFC иый потенциал £ojnd определяется тогда для концентрации сво^ В0ДоР°Аа’ равной единице, и многие индикаторы показывают HeofiCTBa’ близкие свойствам, описанным уравнением (41). Однако траци°ДИМ° помнить ° других факторах и сознавать, что концен-Бишо Кислоты не единственный фактор, влияющий на потенциал. пРосаП[б43]Л исчеРПЬ1БаюЩУю математическую обработку этого во-
Ю*
148
ГЛАВА VIII
ИНТЕРВАЛ ПЕРЕХОДА
(ИНТЕРВАЛ ИЗМЕНЕНИЯ ОКРАСКИ)
Область потенциалов, в которой индикатор полностью изменяет окраску одной формы на окраску другой формы, субъективна и условна и зависит, между прочим, от длины волны (максимума поглощения (Хтах) и коэффициента молярной экстинкции окисленной и восстановленной форм.
Двухцветные индикаторы
Если и окисленная, и восстановленная формы индикатора визуально окрашены, а коэффициенты молярной экстинкции не слишком отличаются, тогда считают, что одна окраска становится неразличимой в присутствии 10-кратной концентрации другой. Из уравнения (40) видно, что индикатор ^находится полностью в восстановленной форме при потенциале
£ind=£,oind + n.ndZ? In Iq- (42)
и полностью в окисленной форме при потенциале
£ind— £oind+ П:пйр In 1 (43)
Значит, индикатор полностью изменяет окраску в области потенциала
г г?' . ЯГ , ,2,3RT
Ejnd;=Eo j i----рг In 10 = Eg. , i-рг (44)
lnd nnd — nindF uind — niniF v ’
При 30 °C одноэлектронный индикатор изменяет полностью свою окраску в пределах 120 мВ, двухэлектронный индикатор — при 60 мВ и т. д. Если коэффициенты молярной экстинкции обеих форм не одинаковы, интервал изменения окраски не распределится равномерно около значения Eoind , а сместится в сторону меньшего значения коэффициента молярной экстинкции. Следовательно, если 6red>e0I, интервал перехода в большей степени сместится в сторону высокого потенциала Eoxind; кроме того, если разница большая, окраска восстановленной формы в этом примере будет продолжать усиливаться после полного исчезновения окисленной формы. Примером является система трис- (о-фенантролин)железо (II)—трис-(°' фенантролин)железо(III), где ered= 1,11 • Ю4 при 510 нм, тогда как Еох=6-102 при 590 нм, так что, хотя формальный потенциал в 1 серной кислоте равен 1,06 В, интервал перехода окраски находится в пределах от 1,08 до 1,20 В. Это приводит к изменению значения формального потенциала индикатора [76], которому раньше Дава' лось значение 1,14 В. Несоответствие в коэффициентах молярн°
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
149
стинкции можно учесть путем введения еще одного члена в уравнение (44), тогда интервал перехода становится
г-. ry I 2,3/^Т1 , Bred 2,3RT /лкл
Е-,пй—Eoind + nindF 1g — _ -^р (45)
Это уравнение предполагает, что субъективная чувствительность глаза оператора одинакова для обеих окрасок и поэтому дает только приблизительную оценку. Когда коэффициент молярной экстинкции одной формы в 50 раз больше, чем другой (или даже в 20 раз больше, если работать с низкой концентрацией индикатора), тогда индикатор считается одноцветной системой, а не двухцветной.
Одноцветные индикаторы
Если одна из форм индикатора бесцветная, т. е. не поглощает свет в видимой области спектра, или коэффициент молярной экстинкции ее значительно ниже, чем у другой формы, тогда константой индикатора является условный потенциал, при котором окраска индикатора образовалась наполовину. Поглощение раствора в этой точке составит половину от максимума поглощения. Чувствительность глаза будет зависеть от оттенка (Хтах) и интенсивности, которая обусловливается коэффициентом молярной экстинкции и концентрацией окрашенной формы. Поэтому не так легко определить интервал перехода, начиная от первой едва заметной окраски до полного образования окраски, потому что этот интервал будет изменяться в зависимости от %шах, е и концентрации. Однако опыт автора и литературные данные свидетельствуют о том, что интервал перехода этих индикаторов приблизительно подобен интервалу перехода двухцветных индикаторов. Так, если окисленная форма окрашена, а восстановленная форма бесцветна, окраска, по-видимому, достигнет максимума, когда
Р ______р' । RT [Indpx] р/
ind toind + n,ndF 1П [Indre<3] h°ind
RT
In 10,
а индикатор бесцветен, если с с , RT 1 1
£>nd—Еоы+ „.ndF ln 10
Таким образом, интервал перехода при обычных концентрациях Дикатора приближается к условию
р с' _^2,3RT ..„
= (4б)
Ес пц
пот,„?КРашеннь1е частицы соответствуют восстановленной форме, - ется тот же интервал перехода [64].
150
ГЛАВА VIH
Хотя интервалы перехода, выведенные выше, дают довольно точные показания, действительный интервал перехода для данного индикатора носит субъективный характер и различен для каждого аналитика, а для одного и того же исследователя меняется от цвета к цвету. Обычно рабочие условия дают достаточно высокое значение Q, чтобы обеспечить полное изменение окраски для выбранного индикатора при добавлении последнего инкремента титранта, а небольшие отклонения в интервале перехода не имеют большого зачения. Одноцветные индикаторы подробно рассмотрены Бишопом в его книге [64].
НЕОБРАТИМЫЕ ИНДИКАТОРЫ И ИНДИКАТОРЫ ОСОБОГО ДЕЙСТВИЯ
Некоторые вещества изменяют свою окраску необратимо (обычно при окислении) или же разрушаются с образованием бесцветных продуктов. Такие вещества применяются в ограниченной степени в качестве индикаторов из-за их очень быстрого изменения окраски или отсутствия обратимости; однахо о них нельзя строго сказать, что они не обладают константой индикатора или интервалом перехода и что добавка избытка исходного титруемого восстановителя не приводит к обратимости измененной окраски. Подобными процессами является окисление гидразина до азота или сульфита в сульфат. И гидразин, и сульфит не окисляются до тех пор, пока потенциал раствора не достигнет определенного значения, величина которого зависит от условий; однако, если после окисления понизить потенциал даже гораздо ниже этого значения, нельзя восстановить первоначальную окраску. Тем не менее этот критический потенциал существует, и, хотя он не очень воспроизводим, может служить вместо константы индикатора. Это не обычный окислительно-восстановительный потенциал в принятом смысле. Скорее это потенциал, при котором освобождается энергия активации данной реакции, и реакция, начавшись, идет до конца. Если энергия активации высока, реакция может идти за счет окисленной формы титруемого вещества (Ох2) и потенциал раствора понизится, как только появится вещество Red2. Кроме того, титрант (Oxi) может попросту расходоваться на окисление индикатора, и потенциал раствора будет поддерживаться постоянным до тех пор, пока не окислится весь индикатор. Хотя интервал перехода можно вывести, исходя из объема титранта, интервал перехода, рассчитанный по данным для потенциала, не имеет смысла. Скорость реакции индикатора или продолжительность индукционного периода зависит от потенциала раствора.
Другим типом индикатора, который может быть обратимым или необратимым, является не окислительно-восстановительная система, а чувствительный, специфический реагент для одного вида
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
151
тип участвующих в титровании; такими частицами обычно бывает ион титранта или вещества Oxi из реакции (16), а может 6iTb какой-нибудь интермедиат. Индикаторы этого типа зависят потенциала (имеется в виду, что частицы, присутствующие в пастворе и реагирующие с индикаторами, зависят от потенциала). Примером обратимой системы такого рода является обнаружение свободного иода с помощью крахмала и других подобных реагентов Появление синей окраски зависит не от потенциала раствора, а от присутствия иода в достаточной концентрации, чтобы образовалась заметная окраска с крахмалом. Это зависит от концентрации иода [12] (или [1з]) и не зависит от концентрации иодида, тогда как потенциал зависит от соотношения [I2]/[I-] (правильнее сказать, от соотношения [П]/[1-]3). В этом примере «константа индикатора» зависит от концентрации иода S (в моль-л-1), при которой появляется заметная синяя окраска; здесь концентрация должна быть порядка 10-5—10-6 моль-л-1, а потенциал системы иод—иодид при соответствующих условиях будет
£°ind ~ Е'о12/1- + ~2F~ [I-]3
Если индикатор используется для установления конечной точки окисления иода до 1+ (например, в виде IC1), тогда «константа индикатора» будет
<48>
Интервал перехода не имеет существенного значения в данной ситуации. Примером необратимой системы этого типа может быть обнаружение свободного брома при бромировании парарозанилина, розанилина и родственных им соединений, механизм действия индикатора будет рассмотрен ниже. Бронированный индикатор не может дебромироваться путем понижения потенциала раствора, но образование пурпурной окраски зависит от потенциала; потенциал должен быть довольно высоким, чтобы образовалось достаточное Для обнаружения количество свободного брома. Если S моль-л-1— чувствительность реагента на бром, «константа индикатора» бу-
^°ind = £°вг2/вг- + 2F [Вг—]3 (4^
В данном случае S имеет значение порядка 10-6—10-7 моль-л-1.
ОПРЕДЕЛЕНИЕ КОНСТАНТЫ ИНДИКАТОРА
х^омоА ПОМО1ЦИ некоторых индикаторов, особенно такого типа, как тодов п дииминожелезо(П), можно с помощью подходящих ме-епосредственно определить E'oinil; однако такие индикаторы
152
ГЛАВА VIII
скорее исключение, а не правило. Константу индикатора для индикаторов реакций простого взаимодействия ионов можно определить сравнительно легко, но окислительно-восстановительные индикаторы в гораздо меньшей степени пригодны для прямых методов и в значительной мере зависят от условий. Помимо их зависимости от ионов водорода и обмена лигандов, которую можно рассматривать по крайней мере полуколичественно, окислительно-восстановительные индикаторы выдвигают ряд важных проблем касающихся скоростей реакции, плохой растворимости, неустойчивости к свету, кислотам, щелочам, избытку окислителя или восстановителя, побочных реакций, таких, как образование семихинона и автоокисление, и индуцированных реакций. Некоторые индикаторы не взаимодействуют с такими окислителями, как бихромат или даже церий(IV), хотя восстановитель, такой, как железо (II), одновременно окисляется и вызывает появление индуцированной реакции. Некоторые индикаторы, хотя для них не удается определить константу индикатора, довольно хорошо проявляют себя при титровании, и поэтому их применение оправдано. Имеются и такие индикаторы, которые при различных методах титрования показывают различные потенциалы в точке перехода '[77]. Константу индикатора надо определять с помощью возможно большего числа различных методов, с тем чтобы обнаружить любую особенность и сравнить поведение индикатора в различных случаях титрования, а также чтобы исследовать влияние концентрации кислоты и других факторов на поведение индикатора. Все методы, которые кратко будут описаны в дальнейшем изложении, имеют недостатки и ограничения в их применении; часть из них указывается. Однако эти методы обладают также рядом достоинств. В некоторых случаях полезно провести обычное хроматографическое или спектрофотометрическое исследование индикатора.
ПРЕДВАРИТЕЛЬНОЕ ИССЛЕДОВАНИЕ
Хроматография. Методами колоночной, бумажной, тонкослойной, а также ионообменной хроматографии можно исследовать индикатор в его исходном состоянии, полностью восстановленном и полностью окисленном. Это позволяет установить, является ли индикатор индивидуальным соединением или смесью из нескольких соединений. Если это смесь, что часто случается, или краситель, или соединение со многими заместителями, тогда его можно элюировать или разделить компоненты смеси и установить, какой из них обладает нужными индикаторными свойствами. Хроматографируя окисленную и восстановленную формы, можно обнаружить присутствие побочных продуктов или продуктов распада.
Спектрофотометрия. По спектрам исходной, восстановленной и окисленной форм можно получить данные о длине волны макси
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
153
мума поглощения Х1Пах и о химической идентичности, а также ко-(ЬсЬициент молярной экстинкции, в случае если индикатор накопится в чистом состоянии и можно приготовить его раствор из-вестной концентрации. За неимением данных о коэффициентах молярной экстинкции по спектрам раствора окисленной и восстановленной форм одинаковой, но неизвестной концентрации получают соотношение ered/eOx- По изменению поглощения во времени можно определить устойчивость обеих форм индикатора. Спектры, снятые при различных концентрациях иона водорода и лиганда, дают сведения о смещении максимумов вследствие протонирования и обмена лиганда. Этим методом можно обнаружить образование семихинона и другие побочные реакции.
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД
Чтобы можно было применять этот метод, индикатор должен быть устойчивым при высоких и низких потенциалах; необходимо, чтобы eox И еге<1 не очень сильно различались, иначе точность будет низкой; различие между еох и ered .должно быть не более 5:1, предпочтительнее не более 2:1.
Подготовка опыта. Готовят разбавленный раствор индикатора, лучше в среде, в которой будет проводиться титрование (например, в 1,0 М серной кислоте). Переносят равные аликвотные части по 5—10 мл в две мерные колбы на 100 мл и доливают 50 мл среды, в которой проводится титрование. Одну порцию титруют 0,01 М восстановителем [например, оловом(II) или железом(II)] до постоянной окраски и добавляют 1,0 мл в избытке. Вторую порцию титруют таким же образом 0,01 М окислителем [например, церием (IV)]. Доливают обе колбы до 100 мл раствором среды для титрования. Снимают спектры относительно раствора среды. Если максимальное поглощение более сильно поглощающего раствора обозначить А, тогда или готовят новый раствор индикатора в концентрации, составляющей 1/Л от предыдущей, или умножают величину аликвотных частей на 1/А. Аликвотные части должны быть достаточно большими, чтобы их точно распределить. Раствор среды, в которой проводится титрование, восстановитель, окислитель и их продукты не должны поглощать свет, в противном случае на-Д° д?ове?ти холостые опыты как можно более точно.
Методика определения. Принцип ее состоит в использовании рех растворов и измерений при двух длинах волн; но лучше брать или три смешанных раствора Мд, Мг и т. д. и снимать спектры г™-ТРех Различных длинах волн в любой стороне от изобестиче-скои точки.
Указа)ИГ°ТаВЛИВаЮТ РаствоР индикатора, концентрация которого нения Э Быше’ в сРеде Для титрования. В случае чистого соеди-готовят раствор количественно, чтобы можно было опреде
154
ГЛАВА VIII
лить коэффициент молярной экстинкции. Отбирают три равные аликвотные части в мерные колбы на 100 мл, добавляют 60 мл среды для титрования. Затем раствор О титруют окислителем и добавляют 1 мл избытка. Раствор R титруют восстановителем и тоже добавляют 1 мл избытка. К раствору 14 добавляют несколько капель окислителя или восстановителя, чтобы получить промежуточную или смешанную окраску. Все три колбы доливают до метки средой для титрования. Измеряют потенциал Е раствора М, используя для этого чистый активный платиновый электрод и каломельный электрод сравнения. Приготавливают соответственно растворы для холостых опытов и снимают спектры на одном и том
Рис. VIII.3. Спектры окисленной (О) и восстановленной (R) форм индикатора и раствора (М) со смешанной окраской.
же графике. На рис. VIII.3 показаны спектры для частного случая, когда восстановленная форма индикатора поглощает сильнее при большей длине волны. Тщательно проверяют, является ли место пересечения I настоящей изобестической точкой или это треугольник. Если же получится треугольник, значит, концентрация индикатора в трех растворах неодинакова или же одна из форм индикатора неустойчива. В последнем случае при повторном снятии спектра поглощение должно уменьшаться. Предположим, что все идет нормально, тогда при длине волны Zo будет находиться максимум поглощения окисленной формы, а при Zr—максимум поглощения восстановленной формы. Если раствор индикатора приготовлен количественно, тогда при Zo
еох=-4г~ л-моль^-см-1, (50)
ох Ьс ’
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
155
Ь___толщина кюветы с образцом, см; с — концентрация раство-
ГДе индикатор а, моль-л-1. Значение ered вычисляют аналогично при Ра прИ длинах волн, меньших длины волны изобестической точки, например при Яо,
(51)
тогда как при более длинных волнах, например при XR, р___________________р' । 2,3RT . Лм Ao /соч
Эти уравнения не зависят от абсолютной концентрации, а поэтому их можно использовать и не зная концентрации. Если возможно, лучше снимать спектры на двухлучевом самопишущем спектрофотометре, чтобы получить спектр, подобный показанному на рис. VIII. 1. На таком приборе получаются результаты достаточной точности. Более высокой точности можно добиться путем измерения трех растворов при избранных длинах волн на ручном спектрофотометре. Если неизвестно значение Пш<ь решают уравнения (51) и (52) при п=1, 2 и т. д., пока не получится близкое значение для £oind, а затем по нему находят значение пща-
ПОТЕНЦИОМЕТРИЧЕСКИЙ МЕТОД
В принципе все, что надо знать, — это потенциал эквимолярной смеси окисленной и восстановленной форм индикатора в среде для титрования. Это возможно только в том случае, если обе формы индикатора устойчивы и находятся в чистом состоянии. Тогда останется измерить скорость процесса обмена электрона в гомогенном растворе [32, 78, 79] и силу тока i0, при котором происходит обмен (или кинетические параметры электрода Айа для гетерогенного электродного процесса [48]). Потенциометрические методы можно разделить на четыре группы: 1) потенциометрические измерения индикатора; 2) визуальный колориметрический метод с использованием серии растворов с известными потенциалами, так же как индикаторы на pH, оцениваются с помощью серии буферных растворов; 3) потенциометрическое титрование окислительно-восстановительных систем, содержащих индикатор, и 4) определение потенциала известной точки эквивалентности по изменению окраски при титровании.
1] Потенциометрические измерения индикатора
Фо ^TOT метод применяют только в случае устойчивости обеих Ф рм индикатора, а результаты сопоставляют с результатами, поученными по методу 2 или 3.
156
ГЛАВА VIII
а) Если обе формы индикатора имеются в чистом состоянии а плотность тока обмена больше 10-2 с, где с — численное значение концентрации индикатора, тогда готовят 10~3 М. растворы каждой формы в среде для титрования, смешивают равные объемы и измеряют потенциал на активном платиновом электроде. Потенциал должен достигнуть равновесия за секунды; его обозначают Eci,nd. Готовят смесь из 1 объема раствора окисленной формы индикатора IndOx и 10 объемов раствора восстановленной формы Indred и повторяют измерение. Потенциал будет Eoind —0,06/ntad в. Смесь из 10 объемов Indox и 1 объема Indred даст потенциал Eoind +0,06/nind В. Ни один из растворов не должен изменять свою окраску при стоянии. Для проверки электродной реакции погружают в эквимолярную смесь два платиновых электрода и пропускают ток 0,1 мА-см~2. Потенциал любого электрода ори энергичном перемешивании раствора должен отклоняться от Eollld не более чем на 10 мВ. В таком случае считают, что реакция идет с большой скоростью. Если отклонение больше 0,06 В для обоих электродов, то при катодном потенциале £Cat вольт и анодном Еап вольт можно приблизительно определить кинетические параметры электродов (в пределах одного порядка величины для k), используя следующие приблизительные соотношения [80], которые основываются на значении £/бзс=1'0~5 в перемешиваемом растворе и значении предельного тока i— 1 мА-см-2,
а ; (53)
£an £cat
1ел^КПм-(£;*07-£°м) -6, (54)
lg k « " nd--£an) — 6 (55)
Если заметны отклонения, вызываемые проходящим током, надо исследовать скорость обмена электронов в гомогенной среде [32, 78, 79], так как в этом случае действие индикатора должно быть медленным.
б) Если располагают только одной формой индикатора в чистом состоянии, а плотность тока обмена больше, чем в случае а, тогда готовят 2-10~3 М. раствор чистой формы в среде для титрования. Выбирают такой восстановитель или окислитель, который реагирует количественно и быстро с индикатором, и готовят стандартный раствор, концентрация которого эквивалентна 5-кратной концентрации индикатора. К трем порциям раствора индикатора по 100 мл добавляют соответственно 1,82, 10 и 18,18 мл реагента или тайие его количества, чтобы соотношение [IndOx]/['Indred] было 1 : 10; 1 : 1 и 10: 1. Измеряют потенциалы на активном платино-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
157
ом электроде. Эти потенциалы быстро уравновешиваются и соответствуют £6nd —0,06/nmd, Eoind и Eoind +0,06/п1па; потенциалы и окраски должны быть устойчивыми. Погружают второй электрод в раствор состава 1:1, пропускают ток 0,2 мА-см~2 и замеряют потенциал анода (при работе с восстановителем) или катода (при работе с окислителем). Согласно случаю а смещение от E6ind не должно превышать 10 мВ для быстрой индикаторной реакции. Добавленный реагент мешает определению кинетических параметров электрода простым способом, описанным в а.
в) Если чистота индикатора сомнительна, но он не содержит активных примесей, или в случае, чем-то иным отличающемся от случая б, готовят от 2-10-3 М до 10~2 14 раствор в среде для титрования, а затем титруют аликвотную часть в 100 мл потенциометрически раствором подходящего окислителя или восстановителя, концентрация которого равна 10-кратной эквивалентной концентрации раствора индикатора. По окраске определяют интервал перехода. Равновесие потенциала и изменения окраски должно устанавливаться быстро, за исключением области вблизи точки эквивалентности. После добавления избытка реагента необходимо проверить устойчивость потенциала и окраски. Потенциал в точке полутитрования дает Eoind , по тангенсу угла наклона находят величину Щпа, но значение EOind следует проверить путем вычисления, используя величины «ы и уравнение Нернста для нескольких точек на кривой.
2] Растворы, поддерживающие постоянным значение потенциала
Обычные колориметрические методы для исследования кислотно-основных индикаторов охватывают определение ряда растворов с постоянным pH или буферных растворов, которые относятся к интервалу перехода, равному 0,2 pH единицы; для этого к растворам добавляют одинаковые количества индикатора и наблюдают окраску (см. гл. III, т. 1). Подобным образом можно оценить окислительно-восстановительные индикаторы, сопоставляя серию буферных в отношении потенциала растворов или растворов с постоянным значением измеряемого потенциала [81]. Кислотно-основные реакции относятся к наиболее быстрым, взаимодействия между кислотно-основными индикаторами и средой очень редки, а буферные по pH растворы бесцветны. Не так обстоит дело с кислительно-восстановительными индикаторами. Изменение окра-и индикатора может происходить медленно. Обычно индикатор щ аим°Действует с различными соединениями в растворе. Боль-длНСТво окислительно-восстановительных систем, используемых приготовления растворов с постоянным потенциалом, окраше-
158
ГЛАВА VIII
Таблица VI 11.11
ОБЪЕМ ОКИСЛИТЕЛЯ, КОТОРЫЙ НАДО РАЗБАВИТЬ ДО 100 мл ЭКВИМОЛЯРНЫМ РАСТВОРОМ ВОССТАНОВИТЕЛЯ, ЧТОБЫ ПОЛУЧИТЬ РЯД РАСТВОРОВ С ПОТЕНЦИАЛАМИ, РАЗЛИЧАЮЩИМИСЯ НА 10 мВ ДЛЯ ОДНОЭЛЕКТРОННОИ РЕАКЦИИ
Объем раствора окислителя, мл Е — мВ Объем раствора окислителя, мл Е — Eq, мВ
9,1 —60 59,4 10
12,7 —50 68,2 20
17,7 —40 76,0 30
24,0 —30 82,3 40
31,8 —20 87,3 50
40,6 —10 90,9 60
50,0 0
но в состоянии одной или обеих форм. Хотя в разбавленных растворах окраски очень слабо сравнимы с окраской индикаторов, все же использование разбавленных растворов приводит к медленным реакциям. Тем не менее метод используется или сам по себе, или в подтверждение результатов, полученных другими методами.
Должна быть выбрана окислительно-восстановительная система типа
Ох + пе < -> Red
со значением Е'о, близким к E'oin6, и приготовлены эквимолярные растворы окислителя и восстановителя в среде для титрования; концентрации этих растворов должны быть максимально возможными, не вызывая при этом изменения окраски; однако если концентрация этих растворов не менее чем в десять раз больше концентрации индикатора, то окраска изменяется. Раствор индикатора готовят в той же среде для титрования и такой концентрации, чтобы при десятикратном разбавлении его получалось четкое изменение окраски: Затем готовят серию смесей растворов окислителя и восстановителя с различными потенциалами в пределах определенного интервала потенциалов. В табл. VIII.11 показано, какие объемы окислителя надо разбавить до 100 мл восстановителем, чтобы получить серию растворов, отличающихся друг от друга на 10 мВ при п=1. Маловероятно, чтобы одна окислительно-восстановительная система могла охватить интервал перехода одноэлектронного индикатора; в идеальном случае для этого нужна серия систем, различающихся значением ^oind в 120/п мВ, например 0,'» 0,82; 0,94; 1,06; 1,18 и 1,30 В. В редких случаях, когда на индика-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
159
не оказывают влияния ион водорода и лиганд, можно исполь-Т пять эти системы, чтобы сместить потенциал Е'о окислительно-вос-3?ановительной системы. Когда готова подходящая серия раство-с0,в проверяют значения их потенциалов, добавляют 5 мл раствора Индикатора к 50 мл каждого раствора серии и наблюдают окра-Затем интерполируют интервал перехода. При одинаковых ко-щиентах молярной экстинкции обеих форм индикатора (что маловероятно) значение Eoind будет средним; в противном случае в это значение надо внести поправку
___р | 0 >06 1 п еох
°ind lnd"H nind 6 ered
(56)
Очевидно, этот метод имеет ряд недостатков и связан с трудностями. Смит и Баник [81] использовали влияние иона водорода и сульфата в качестве лиганда на простую окислительно-восстановительную систему, с тем чтобы легче получить ряд потенциалов от 0,9 до 1,23 В при сохранении максимального постоянства потенциалов. Такая система состояла из ванадия (V)—ванадия (IV) в растворе серной кислоты, концентрация которой варьировала от 0,5 до 6,0 моль-л-1. Концентрация растворов ванадия (V) и ванадия (IV) была 0,0014 моль-л-1, а изменение потенциала составляло около 47 мВ на единицу изменения молярности серной кислоты, так что фактически потенциал зависит от восьмой степени концентрации серной кислоты в противоположность формальной зависимости от квадрата концентрации иона водорода. Хотя эти авторы утверждают, что существует линейная связь между потенциалом и концентрацией серной кислоты, кривая результатов показывает четыре волны, соответствующие большим изменениям реагирующих частиц. Это не важно при условии, что потенциалы измеряются экспериментально. Эти измерения имеют воспроизводимость ±3 мВ; равновесное значение потенциала достигается очень медленно. Это не удивительно, если учесть сложность реакций такой системы с ионом водорода {82, 83] и сульфат-ионом [83] и ее различную кинетику на электродах [83, 84]. В 2М серной кислоте, например, константа скорости гетерогенной реакции на платиновых электродах находится в пределах от 2-Ю-8 до 4-10-8 л-см-2-с-1, а коэффициент переноса равен 0,22 [83, 84]. Смит и Баник с ус-ехом применяли этот метод для комплексов ионов металлов с иро-зводными о-фенантролина [81], однако надо учитывать влияние топ^ы™ И меня1°Щихся концентраций серной кислоты на индика-да^ Необходимо оценивать такие факты, как участие иона водоро-с цВ Реакции индикатора [уравнение (37)], или конкуренцию его °ном металла по отношению к лиганду, осаждение слаборас-римых сульфатов индикатора или даже его сульфирование, а
160
ГЛАВА VIII
также ионную силу раствора. Этот метод может дать интервал перехода индикатора и значение п1па, а для определения Eo.rd над0 знать соотношение ered/eOx.
3) Потенциометрическое титрование растворов в присутствии индикаторов
Возможно, что наиболее практичным, если не наиболее точным методом является потенциометрическое титрование восстановителя раствором окислителя в обычных экспериментальных условиях-предложено использовать при этом индикатор, вводя известное количество его в титруемый раствор [85]. Если количество индикатора достаточно для того, чтобы израсходовать на себя определенный объем титранта, то на кривой титрования появится волна, по которой можно определить E6ind. Сопоставление окраски с потенциалом позволит определять интервал перехода. Титрование раствора с низким значением Q 0,1 М растворами одноэлектронных реагентов будет способствовать повышению постоянства потенциалов, но может внести ошибку при определении волны. Использование же при титровании более разбавленных растворов с увеличенными Q улучшает определения волны индикатора [84]. Этого же можно достигнуть, если при переходе области точки эквивалентности заменить титрант на раствор, разбавленный в 10 раз [77]. Для проведения титрования в пределах 5% от точки эквивалентности индикатор должен быть довольно устойчивым.
При начальном исследовании выбирают по возможности простую окислительно-восстановительную систему, одноэлектронную и симметричную, у которой электродные процессы идут с большой скоростью; примером может служить титрование железа (II) церием (IV) в солянокислой, сернокислой или азотнокислой среде; Eeq.pt этих сред увеличивается в том же порядке, и выбор может быть любым. Готовят 0,02 М или 0,01 14 стандартные растворы и титруют потенциометрически 25 мл аликвоты восстановителя в подходящей среде раствором окислителя. Повторяют титрование после добавки к титруемому раствору индикатора в количестве, превышающем обычное в 5—10 раз; это количество должно быть таким, чтобы на него расходовалось 0,2—0,5 мл титранта; как только индикатор начнет изменять свою окраску, надо отмечать окраску после каждого добавления инкремента титранта. На кривой титрования получается перегиб или волна, где идет титрование индикатора, и средняя точка дает значение Eoind - Наблюдение за окраской позволяет определить интервал перехода, который не располагается симметрично вокруг E6ind, если коэффициенты молярных экстинкций обеих форм индикатора разные; на основе этих ДаН' ных и тангенса угла наклона перегиба индикатора получают зна-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
161
е /Zjnd- Кроме того, сопоставление с кривой титрования в от-чеТСтвиёП индикатора дает значение поправки на индикатор. По-„дующие титрования в условиях различных концентраций иона %дорода и лиганда определяют влияние этих факторов, а исследо-вание с применением различных окислителей и восстановителей показывает, для каких реакций титрования и в каких условиях титрования пригоден данный индикатор.
Если индикатор не устойчив к избытку окислителя, данным методом можно получить некоторые полезные сведения; для этого либо проводят быстрое титрование (если потенциалы после точки эквивалентности не находятся в равновесии), либо добавляют немного индикатора и определяют потенциал, при котором происходит изменение окраски. В последнем случае не получают очень надежных значений, так как в области точки эквивалентности плохо поддерживается постоянным потенциал раствора.
Обратимость изменения окраски и воспроизводимость значения Е'отй следует проверить путем повторной добавки небольшого избытка восстановителя и повторного перехода через область точки эквивалентности. Иногда после первого процесса в обратном направлении наблюдается заметное изменение £oind.
4) Оценка индикаторов на основе их применения
Иногда индикатор проявляет себя в процессе титрования устойчивым к избытку реагента и обратимым после нескольких переходов через точку эквивалентности, хотя при исследовании по одному из описанных методов он не показал удовлетворительных свойств. Этому может быть много причин, как, например, крайне низкий ток обмена, взаимодействие со средой, экранирующая адсорбция на электроде, каталитические или индуцированные реакции и т. д. В'се же индикатор хорошо проявляет себя в титровании, и его рекомендуют вполне оправданно. Можно определить его условный потенциал на основе одного из ряда различных титрований. Например, при титровании железа(II) ванадатом или бихроматом окраска может не изменяться, а изменится при титровании сульфатом иерия(IV), при условии что £6ind находится между 1,0—1,1 В. Параллельно проводимое титрование потенциометрическим методом или с применением визуального индикатора служит для определе-чения°Е'аВКИ На индикатоР и для установления более точного зна-£'°ind •
ОШИБКИ ТИТРОВАНИЯ И ПОПРАВКИ НА ИНДИКАТОР сост°оЩИбКа титР°вания при использовании визуальных индикаторов конец11 *ИЗ ТРеХ частея: О химическая ошибка из-за несовпадения нои точки с точкой эквивалентности; 2) визуальная ошибка 11-918
162
ГЛАВА VIII
из-за неспособности глаза точно различать окраски; 3) индикатор ная ошибка вследствие расхода титранта на индикатор.
Первая ошибка может стать незначительно малой, если правильно выбрать индикатор и условия титрования, о чем речь пой дет ниже. Визуальная ошибка небольшая, порядка 5—10 мВ. Индикаторная ошибка зависит от количества индикатора, которое дает различимое изменение окраски, иногда, возможно, из-за присутствия окрашенных частиц, таких, как ион хрома (III), комплексы хлорферрата(Ш), гексацианоферрата (III) и т. д. В связи с этим коэффициент молярной экстинкции становится важным фактором, а длины волн максимумов поглощения Ятят индикатора и других окрашенных частиц должны быть различными. Чем больше коэффициент молярной экстинкции е, тем меньше концентрация индикатора, при которой получается видимая окраска, а значит, и меньше расход титранта на индикатор. Если е превышает К)4, одна капля 0,005 М раствора индикатора окрасит 50--100 мл титруемого раствора, а расход составит 0,0025 мл 0,1 М титранта. Тогда индикаторной ошибкой можно пренебречь. Однако при работе с более разбавленным титрантом или индикаторами с низким коэффициентом молярной экстинкции (либо с менее различимой окраской) нужно брать индикатор в большем количестве и расход титранта будет больше. Визуальное наблюдение за изменение’ I окраски субъективно, поэтому индикаторную ошибку надо опреде-
лять экспериментально; лучше всего использовать потенциометрическое титрование по методу (3) для определения Eoind, который был описан выше; прямое титрование аликвоты индикатора редко дает удовлетворительный результат.
Принято концентрацию раствора индикаторов выражать в % массы/объем, но если вещество содержит примеси или представляет собой смесь, как в случае многих торговых препаратов красителей, то обычный способ выражения для чистых веществ здесь неприменим. Для таких растворов надо определять молярность, и она носит значительно более информативный характер. Часто считают разбавленным 1%-ный раствор, но для вещества с молекулярным весом 200 раствор, концентрация которого 0,05 моль-л'1, вовсе не будет разбавленным.
Химическая ошибка
Потенциал в точке эквивалентности Eeq.pt представляет собой потенциал, при котором добавлено стехиометрически точное коли* чество титранта. Потенциал конечной точки является потенциалом индикатора Еща, при котором происходит избранное аналитиком изменение индикатора. Если разность между потенциалами Eeq.pf" —Eind превышает Q/2, химическая ошибка появляется вследствие того, что значения этих потенциалов не совпадают. Если титров3'
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
163
вещества Red2 раствором Oxi имеет одинаковое Q, то ошибку НИе ем легко рассчитать. Потенциал в точке эквивалентности выделяют, как было показано раньше; тогда, если Eind<Eeq. pt, расчет производится для системы восстановителя
Е а—Е’ I- 0,06 1ц
«2
(57)
На основании этого уравнения приблизительно находят ошибку, выражая ее в процентах:
100[Red2] „ , (
[<ЭхйГ ~ I00c/?«lg[
^2 (Ей* ^ind)
(58)
В этом случае конечная точка определяется преждевременно и ошибка имеет отрицательный знак. Если Etad имеет более положительное значение, чем Eeq.pt, то расчет производится для системы
окислителя
Ejnd—
0.06 ; [OxJ «1 ‘HRedJ
(59)
Ошибка в процентах приблизительно равна
100 [OxJ [Redj]
= lOOanti lg
П1 (£jnd Eq.)
' 0fi6
(60)
Если ошибка больше 1%, то может быть нужна более точная оценка ошибки. В этом случае надо работать с другим, более подходящим индикатором.
Точку Е1п(1 можно так выбрать, чтобы она соответствовала первому заметному изменению окраски при титровании в избранном направлении либо соответствовала E^nd (когда половина индикатора претерпела изменение или же индикатор изменился полностью). Последняя точка достаточно хорошо определяется, если известен Ди (Дп/Vt — требуемая точность определения) и до конечной точки остается добавить инкремент титранта величиной Ди (в мл). Интервал перехода индикатора дает значение потенциалов, соответствующих первому заметному изменению окраски и ее полному изменению. Когда известно только EJjind, то для оценки интервала перехода с достаточной точностью можно использовать сравнение (45).
Рассмотрим случай титрования железа(II) в 1,0 М серной кис-вОте раствором церия(1У). Здесь Е^ =1,44В, Eq2 = 0,695B, следо-0 01^°’ £e<TPt= 1»06 В, a Q для титрования 25 мл с точностью до ц’иа Мл .составляет 0,319 В. Любой индикатор с условным потен-нило°М ’06 В Даст правильный результат, например N-фенилантра-но няЗЯ кислота или сульфат трис- (1,10-фенантролин)железа (II), помним, что Ered/Eox=20 для последнего индикатора поэтому И*
164
ГЛАВА VIII
лучше брать его 5-метилпроизводное (£6ind =0,98В). Любой индикатор с условным потенциалом в пределах 0,93—1,16 В полностью изменяется от добавки одного инкремента титранта.
В случае 4,7-диметоксиферроина, формальный потенциал которого 0,82 В, подстановка этого значения в уравнение (58) даст ошибку — 0,8%, тогда как в случае 5-нитроферроина (формальный потенциал 1,25 В) расчет по уравнению (60) дает ошибку +0,08%. Ошибка обоих индикаторов находится вне допустимого предела точности при титровании (0,04%), но все же лучшим индикатором оказывается 5-нитроферроин. Так обстоит дело с учетом формальных потенциалов, но если принимать во внимание соотношение Ered/вох, то ошибки становятся —0,04 и +1,26%, так что в действительности лучшим индикатором оказывается 4,7-диметоксиферроин.
ВЫБОР ПОДХОДЯЩЕГО ИНДИКАТОРА
Главными факторами при выборе подходящего индикатора для данного титрования являются: а) условный потенциал индикатора £oind ; б) коэффициенты молярной экстинкции обеих форм индикатора Еох и Егеа; в) цветовые оттенки обеих форм, определенные дифференциально посредством Яшах; г) потенциал в точке эквивалентности Eeq.pt и значение Q титрования. Другие общие необходимые характеристики будут рассмотрены в следующем разделе.
На первый взгляд может показаться, что индикатор надо так выбирать, чтобы Eojnd =Eeq.pt, но это правильно только в том случае, когда fiox=Ered и Q>0,12//2ind В. При правильном выборе индикатора учитываются формальный потенциал, коэффициент молярной экстинкции и Q-функция титрования, а также потенциал в точке эквивалентности в условиях процесса титрования
р,, . 2,3/?Г , ered р I Q 2 ART \
=£eq.pt±(-2— (61)
или, считая 2,3 RT/F=0,06,
£' =£ J______°’06 lg । fQ _ 0,06 (62)
Ojnd eq.pt + nJnd g gred nind }
Так подобранный индикатор изменит полностью свою окраску пр’1 добавлении одного инкремента Av мл, а конечная точка совпадет с точкой эквивалентности. Если ошибка индикатора больше отношения Av/4 мл или Q<0,12/Пта В, то окраска индикатора изменится лишь частично. В первом случае (когда ошибка болыпе Av/4 мл) титрование до первого заметного изменения окраски д301 правильный результат, а в случае О<0,12/щпаВ требуемую точность нужно либо уменьшить, чтобы Q стало равно 0,12//ш+’|
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
165
бо выбрать другие реагенты для той же цели, либо вести титро-Л ние до стандартной установленной окраски, либо заменить ме-ва на метод спектрофотометрического или потенциометрического Т°Трования. Высокое значение «ща, по-видимому, способствует более четкому изменению окраски в зависимости от области потенциалов.
В области, определенной по описанному методу уравнением /62) может оказаться несколько индикаторов; предпочтение надо отдать тем, у которых выше значения еох и Bred, так чтобы можно было работать с низкими концентрациями индикатора и получать меньшую индикаторную ошибку. Выбор оттенка индикатора зависит от оператора и от окраски титруемого раствора. Дополнительные цвета дают резкий нейтральный оттенок в момент перехода окраски, положение которого зависит от соотношения коэффициентов молярной экстинкции. Для бесцветных сред предпочтительнее работать с одноцветными индикаторами. При использовании окислительно-восстановительных индикаторов выбор оттенка очень ограничен: индикаторы группы дифениламина большей частью из бесцветных становятся синими или красными, комплексы дииминожелеза (II) меняют окраску от оранжево-красной до бледно-голубой, а трифенилметановые красители чаще всего от желто-зеленой до оранжево-красной. Оказывается, что очень многие люди страдают в некоторой степени цветовой слепотой, часто не различают красного и зеленого цветов, и многие из них обнаруживают этот недостаток только к концу жизни. Этот недостаток можно установить с помощью нескольких простых опытов с индикаторами. Таким людям легче работать с одноцветными индикаторами или меняющими желтую окраску на пурпурную. Другим фактором, о котором нужно помнить при выборе оттенка, является различие в спектральном составе света используемого источника. Следует избегать прямого солнечного света. Кроме того, дневной свет при ясном или облачном небе, искусственный свет от лампы с вольфрамовой нитью и различные виды флуоресцентного освещения различаются по спектральному составу, из-за чего бывает трудно применять некоторые индикаторы при данных условиях освещения.
ДРУГИЕ КРИТЕРИИ ИНДИКАТОРА
Помимо главных требований, касающихся точности определения^ с помощью визуальных индикаторов, существует ряд других Сч°иИСТВ’ К0Т0Рые имеют большое значение для оценки индикатора. н Тается> что многие из них присущи индикаторам для титрова-ппа С ПРОСТЫМ взаимодействием ионов, но их искали, часто пона-ствяН^’ У окислительно-восстановительных индикаторов. Эти свой-можно обобщить следующим образом:
166
ГЛАВА X III
1) индикатор должен легко и дешево получаться в чистом состоянии; •
2) индикатор должен хорошо растворяться в воде, разбавленных кислотах и других обычных средах для титрования;
3) раствор индикатора должен быть устойчив к свету, воздуху и экстрагируемым из стекла веществам, а также не должен разлагаться при длительном хранении;
4) реакция индикатора должна быть быстрой и полностью обратимой, на нее не должны влиять побочные и индуцированные реакции;
5) окисленная форма индикатора должна быть устойчивой к избытку окислителя, так чтобы титрование в любом направлении шло без распада или деструкции индикатора;
6) индикатор не должен взаимодействовать с частицами, встречающимися при проведении титрования, и образовывать с ними соединения или комплексы, которые помешали бы его действию [например, индикатор не должен подвергаться влиянию хлорида ртути (II)];
7) индикатор должен быть устойчивым в присутствии высококонцентрированных сильных кислот и при температуре вплоть до точки кипения.
К сожалению, в настоящее время ни один из индикаторов еще не отвечает большей части этих требований, но то, что индикаторы в некотором смысле несовершенны, не мешает их успешному применению на практике. Следует помнить об ограничениях и давать соответствующие указания о применении индикаторов.
КЛАССИФИКАЦИЯ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ИНДИКАТОРОВ
Для удобства индикаторы подразделяются на классы по сходству их состава и по механизму реакции. Распределяют их в порядке увеличения формального потенциала и тогда применяют другую подходящую классификацию:
1. Серия дифениламина—дифенилбензидина.
2. Трифенилметановые и другие красители.
3. Хелатные комплексы дииминожелеза (II).
4. Индикаторы особого действия.
5. Необратимые индикаторы, подвергающиеся деструкции.
6. Различные соединения.
7. Смешанные индикаторы.
Каждый из этих классов будет подробно рассмотрен.
Наиболее важным из этих классов и ближе всех приближающимся к идеальным свойствам является класс 3 (см., например, трпс-(1,10-фенантролин)железо(П). По этому классу имеется значительное количество фундаментальной информации, и поэтому он
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИН ДИКАТОРЫ
167
будет рассмотрен особенно тщательно. Следующим по важности, по-видимому, может быть класс 1, для которого дано много при-
->в и который исторически является самым старым. Основные вопросы, касающиеся этого класса, еще не разрешены, и поэтому следует наметить пути дальнейших исследований, несмотря на огромные экспериментальные трудности. Класс 2 по количеству самый большой, хотя многие индикаторы не используются. Кроме того, попытки объяснить их действие неудовлетворительны. Остальные классы малы по численности, но к ним относятся некоторые индикаторы, имеющие огромное значение.
1. БЕНЗИДИН, ЕГО ПРЕДШЕСТВЕННИКИ И ГОМОЛОГИ И ИХ ПРОИЗВОДНЫЕ
Хотя сам бензидин не представляет собой подходящий окислительно-восстановительный индикатор, некоторые N-, 3,3'-замещен-ные бензидины, нафтидин и З.З'-замещенные нафтидины, а также различные производные этих соединений четко изменяют окраску в области потенциалов 0,7—1,1 В по н. в. э. Вариаминовый синий и его производные тоже близки по структуре и механизму действия к этому классу индикаторов, но они рассмотрены Оттоуэем в гл. VI ПА.
Для удобства соединения этого класса подразделены на сле
дующие подклассы:
А. Дифениламин и его производные, окисляющиеся, по-видимому, до соответствующих дифенилбензидинов, обладающих индикаторными свойствами.
Б. Дифенилбензидин и его производные.
В. N-Алифатические соединения бензидина, аналогичные группе Б, как, например, К,М'-диметил- и К.ЬУ-тетраметилбензидины.
Г. З,3'-Дизамещенные бензидины, например диметил- или диметоксисоединения.
Д. Нафтидины и их 3,3'-дизамещенные производные.
Многие, если не все, из этих соединений проявляют некоторые общие свойства в реакциях:
а) бесцветная восстановленная форма индикатора окисляется в глубоко-окрашенную форму; подклассы А, Б и Д дают синий или фиолетовый продукт окисления, подкласс В — желтый, а подкласс желтый или красный; коэффициент молярной экстинкции находится в пределах 104—6-Ю4 л-моль-1-см-1;
„ ' часто наблюдается образование интермедиата между вос-
в?ВбеНН°Й И окисленной формами;
чивы б0льшинст„в° окисленных частиц или сами по себе неустой-шаютс!ЛИ неУСТ0ЙЧИВЫ к избытку окислителя и необратимо разрулены* Я С образованием нерастворимых продуктов, желтых или зе-Для подклассов А и Б и коричневых для подкласса Г.
168
ГЛАВА VIII
Эти свойства не обсуждались и требуют объяснения. Классической теорией является теория Кольтгофа и Сарвера [86]. которой руководствовались в течение 40 лет лишь с некоторыми оговорками. Она считалась применимой ко всем индикаторам этого класса. По этой теории соединения подкласса А сначала окисляются до соответствующих соединений подкласса Б, как это показано на исходном соединении дифениламине (ДФА):
Н
;ифениламин
-----------> необратимый
+ 2Н+ + 2г
дифегилбечзидин
(63)
Окисление дифениламина до дифенилбензидина (ДФБ) происходит необратимо, и на него расходуется окислитель. Затем дифенилбен-зидин, или какой-либо другой N-замещенный бензидин, или бензидин с заместителем в ядре окисляется обратимо с образованием продукта с глубокой окраской, который называют «голохиноидным» дихинондиимином. В случае Н,ЬУ-дифенилпроизводного бензидина образовавшийся продукт обычно известен как дифенилбензидиновый фиолетовый (ДФБФ). Зеленый интермедиат, который часто образуется при низких концентрациях кислоты, называют «семихиноном» или «мерихиноидом» [87—90] и считают, что это комплекс дифенилбензидина с продуктом его окисления молекулярного состава 1:1. Дальнейшее окисление этого интермедиата может привести к образованию «голохиноида»:
Н Н
Зеленый «семихинон» или «мерихиноид» (ДФБ-ДФБ1)
дифенилбензидиновый фиолетовый (ДФБФ)
«голохиноид»
Наконец, окрашенный продукт окисления может необратимо разрУ' шаться при дальнейшем окислении или путем другой реакции с об-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
169
азованием желтого или зеленого нерастворимого вещества неизвестного состава. Суммарно реакция выглядит так:
__2НХ—~ 7
2ДФА —------> ДФБ < * ДФБФ ----->- продукт дестр^ кции
ДФБ-ДФБФ
Эта теория, несомненно, ближе к истине, чем, например, теория Мэйдел^нга, Рейса и Гера [91], которые считали, что окрашенное соединение является комплексом Вернера и состоит из амина и металла в 1высшей стадии окисления, хотя это как раз скрывает объяснение эффекта присутствия хлорида ртути(II) или эффекта Вейтца и Швехтена [92, 93], которые утверждали, что образуется комплекс с азотом в качестве центрального атома. Однако эта теория не лишена недостатков. В первую очередь можно было бы ожидать, что «голохиноидный» дихинондиимин ДФБФ будет желтым, а коэффициент молярной экстинкции его около 103. Сиджвик [94] сообщает, что свободное основание и протонированный катион слабо окрашены. Фактически же продукт окисления получается синий или пурпурный, а коэффициент молярной экстинкции порядка 5-Ю4.
Несомненно, что экспериментальное исследование этих индикаторов исключительно трудно. Плохая растворимость, медленные реакции, плохая обратимость, неустойчивость продуктов окисления, очень низкий ток обмена, побочные и индуцированные реакции, необходимость в некоторых случаях в индукторах и ряд дру-
гих вопросов затрудняют получение однозначных результатов и четкую интерпретацию. Об этом свидетельствует имеющаяся литература, изобилующая описанием применения индикаторов и отсутствием основных объяснений: конечно, многие авторы в первых своих работах обещали дать формальные потенциалы и объяснить механизм действия индикаторов, но затем признавали себя не способными преодолеть эти трудности. В этом отношении заслуживают похвалы исследования, проведенные школой Белчера над рядом нафтидинов.
Физер [95] показал, что диамины ряда бензидина не окисляются прямо до дихинондииминов, а Бренке [23] сомневался в правильности того, что образуется промежуточный «семихинон». Первая часть теории Кольтгофа, касающаяся окисления дифениламина в дифенилбензидин, по-видимому, правильна, потому что, если дифениламин окислить в синее соединение, а затем восстановить цинковой пылью, получается дифенилбензидин [94], а не дифениламин. В остальном, по-видимому, разумнее обратиться к работе
Урстера [95] конца 1870 г., который получил соединения HN=/ 5=^(СН3)2Вг- ,
170
ГЛАВА VIII
известные под названием солей Вурстера и которые теперь считаются ион-радикалами. Хаусер [96] подтвердил радикальную природу солей Вурстера посредством измерений магнитной восприимчивости. Цвет этих соединений можно объяснить на основе теории резонанса или существования ряда мезомерных форм соединения, а в тех случаях, когда можно подразумевать существованье триплетного состояния, можно использовать для той же цели теорию резонанса между мезомерными формами иона. В современной интерпретации окраску приписывают значительной делокализации л-электронов в кольцевых системах. Свойства четырех подклассов индикаторов (от Б до Д) рассматриваются именно с этой точки зрения. Будут даны некоторые объяснения о превращении соединений подкласса А в производные дифенилбензидина.
А. ОКИСЛЕНИЕ ДИФЕНИЛАМИНА И ЕГО ПРОИЗВОДНЫХ
Окисление фенил- и алкилфениламинов и полиаминов, а также аминофенолов является очень сложным процессом из-за неустойчивости и высокой реакционной способности продуктов, в результате чего образуются свободные радикалы или полимерные вещества [97].
Окисление простейшего первичного фениламина—анилина [98, 100] окислителями, которые фактически отнимают водород (а это характерно для большинства окислителей), приводит к образованию радикалов CeH5NH« и C6H5N:, которые среди других продуктов образуют N-фенилхинондиимин. Это соединение полимеризуется по кольцу с образованием азофенина, нигранилинов и полифеназинового красителя анилинового черного. Присоединение п-аминогруппы в пара-положении к ароматическому кольцу N-фенилхинондиимин а дает индамин.
Большинство работ по реакциям окисления вторичных и третичных аминов проведено чисто качественно, и в них рассматривают главные реакции, не обращая внимания на побочные реакции. Некоторые теоретические объяснения, конечно, приводятся, однако не разрешают противоречий в экспериментальных наблюдениях. По-видимому, третичные фениламипы окисляются сначала до ка-тион-радикалов, тогда как первичной стадией окисления вторичных фениламинов является образование свободных радикалов, которые могут затем превращаться в бензидин или гидразин. Последний может перейти в бензидин или в свободные радикалы. Если соединение содержит азот, непосредственно соединенный с ароматической системой, радикал или ион-радикал будет мезомерным соединением, которое сильно поглощает в видимой области спектра и поэтому имеет окраску. Окраски, получающиеся при окислении вторичных и третичных фениламинов или от фенилгидразинов (или
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
171
фенилтетразанов), очень похожи, хотя не указываются их количественные спектральные характеристики. Поэтому имеет смысл вкратце рассмотреть существующее положение:
а) Дифениламин (CeH5)2NH окисляется в нейтральной среде до тетрафенилгидразина, который дает пурпурную окраску с концентрированными кислотами, но неспособен к бензидиновой перегруппировке. Восстановление пурпурного вещества приводит к образованию тетрафенилгидразина, который при последующем восстановлении дает исходное вторичное основание.
б) При окислении (CeH5)2NH в кислой среде образуется пурпурная окраска, а при последующем восстановлении получается ди-фенилбензидин.
в) При окислении третичных оснований (CeH5)2NCH3 и (CeHshN полх чается пурпурная окраска, но гидразины не образуются. При восстановлении окрашенного вещества получаются соответственно П.Ы'-диметил-П.Ы'-дифенилбензидин и N,N,N',N'-TeT-рафенилбензидин.
г) Окисление ди-и-толиламина (n-CH3C6H4)2NH может дать гидразин, который не подвергается перегруппировке в щи'-бензи-дин вследствие блокировки метиловыми группами. При окислении в кислой среде получается желтая окраска, а не пурпурная, однако в концентрированной серной кислоте тетрафенилгидразин дает пурпурную окраску.
д) Третичное основание три-и-толиламин (и-СН3СбН4)3П не может дать ни гидразин, ни бензидин, но при окислении в кислой среде получается обычная пурпурная окраска; после восстановления опять возвращается в исходное третичное основание.
Следовательно, здесь имеют значение свойства фенилгидрази-нов и бензидиновая перегруппировка.
Дифенилгидразины
Простейшим из дифенилгидразинов является гидразобензол C6H5NHNHC6H5, который очень легко окисляется до азобензола [105—107]. Механизм этого процесса [106] состоит, вероятно, из отщепления двух протонов с образованием реакционноспособного Дианиона, который очень легко отдает электроны окислителю. Без добавки окислителя гидразобензол диспропорционирует с образованием азобензола и анилина в результате мономолекулярной реакции [108], что предполагает на первой стадии диссоциацию сво-одних радикалов; однако это предположение не подтверждается >а людениями Виланда [109] и, по-видимому, истинный механизм ю Кции заключается в переносе водорода [110]. Так, например, теп диФенилгидРазинь1 не образуют свободных радикалов, а пре-рпевают бензидиновую перегруппировку.
172
ГЛАВА VIII
Бензидиновая перегруппировка
Это явление хорошо известно, но еще не вполне понятно. Перегруппировка простейшего дифенилгидразина— гидразобензола дает преимущественно щи'-диаминодифенил или бензидин, по которому названы перегруппировка и класс соединения. и.п'-Диаминоди-фенил не является единственно возможным продуктом реакции— образуются также о,о'- и о,п'-диаминодифенилы вместе с двумя се-мидинами:
Хотя из гидр азобензо та образуется меньше всего о,о'-диаминоди-фенила [Ш], последний является главным продуктом окисления Й,М'-динафтилгидразина; это стоит запомнить. Количества продуктов зависят от условий эксперимента и от природы и положения заместителей в бензольных кольцах [П2]. Поэтому выход бензидина не получается количественным, в нем содержатся примеси ряда других соединений с близкими свойствами. В случае когда и,и'-диаминодифенил является главным продуктом, заместители в дифениламине или гидразине в пара-положении относительно атома азота могут блокировать последующее превращение (как в случае алкильной группы) или привести к образованию о-семидина как главного продукта (в случае заместителей СН3О и NH2) или же заместители могут отделяться (например, С1, СООН, SO3H и ОСОСНз).
Диссоциацией гидразина на свободные радикалы можно пренебречь как предварительной стадией реакции, потому что тетра-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
173
(Ьенилгидразин, который образует свободные радикалы, не подвергается дальнейшему превращению [ИЗ]. Кроме того, опыты с 2-метилпроизводными, меченными 14С [114], показали, что во время реакции не происходит распада молекул. Внутримолекулярная перегруппировка все еще является темой для обсуждения [115, 116], но, видимо, она охватывает «складывание» молекулы, образование новой связи между атомами углерода в орто- или пара-тю-ложении к атомам азота, разрыв связи N—N и отщепление ионов водорода [97]. Реакция катализируется кислотой, она идет по первому порядку по гидразобензолу и по второму порядку по иону водорода для образования самого бензидина, но кинетический порядок различен:
Поэтому образование бензидина и его производных по такому пути ограничивается гидразинами, которые не дают свободных радикалов при разрыве связи N—N. Сюда относятся гидразобензол и другие дифенилгидразины, за исключением таких тетрафенилгид-разинов, которые образуются из дифениламина.
Моноалкил-М,Ь1'-дифенилгидразины подвергаются бензидиновой перегруппировке [117], так же как некоторые М.М'-диалкил-М.ЬГ-> дифенилгидразины, например Ы.ЬГ-диметил-Г^Ы'-дифенилгидразин, из которого образуется N.N'-диметилбензидин [118].
ь
Трифенилгидразины
Эти соединения не являются сильными восстановителями; их можно окислить до тетразанов и гексафенилтетразанов (С6Н5) 2NNC6H5NC6H6N (С6Н5) 2; они диссоциируют с образованием темно-синих, зеленых или фиолетовых гидразил-радикалов (C6H5)2N—NC6H5 [119].
Тетрафенилгидразины
Тетрафенилгидразин легко получается в реакции дифенилами-на натрия с иодом [120] или при окислении дифениламина в нейральном растворе [113]. На тетрафенилгидразин и подобные ему
174
ГЛАВА VIII
соединения не действуют щелочи, но они могут легко восстанавливаться до исходного вторичного амина и образовывать два вида радикалов. Эти гидразины диссоциируют в растворе и дают радикалы состава (C6H6)2N« [121—122] аналогично диссоциации гекса-фенилэтана, который дает трифенилметильные радикалы (СеН5)3С>. Диссоциация может пройти в значительной степени, и растворы приобретают глубокую окраску [123]. Химические свойства этих веществ соответствуют свойствам радикала, а не недиссоциирован-ного гидразина. Тетрабензилгидразин не образует радикалов, потому что азэт связан с бензольным кольцом не прямо, а посредством метиленовой группы. Необходима непосредственная связь СеН5—N, и радикалы образуются вследствие того, что делокализация неспаренного электрона стабилизирует продукт, это происходит только в том случае, когда радикал имеет по меньшей мере две ароматические группы. Влияние заместителей можно коррелировать посредством их ориентации относительно бензольного кольца в реакциях замещения [124]. Растворы свободного радикала дифениламина неустойчивы из-за реакционной способности радикала, который в отсутствие других, способных к взаимодействию с ним веществ реагирует со вторым свободным радикалом с образованием феназина и дифениламина.
Второй тип радикала ответствен за пурпурную окраску, которая получается при действии концентрированной серной кислоты (или эфирного раствора хлористого водорода) на тетрафенилгид-разин, который реагирует с кислотой по аналогии с благородным металлом, таким, как медь. Однако, хотя медь образует катионы при обработке кислотой только в присутствии окислителя, тетра-фенилгидразин может действовать как автоокислитель, восстанавливаясь в ходе процесса до вторичного амина. Ион-радикал имеет формулу [(CeH5)2N—1\т(СбН5)2]+; он образуется вследствие отщепления электрона [125], а не протонирования. Следует отметить связь его с гидразил-радикалом (C6H5)2N—NC6H5, который является, как показано выше, продуктом диссоциации тетразана.
Вернемся к дифениламину. Окисление его в нейтральных средах приводит к образованию радикала (CeH5)2N«, который имеет глубокую окраску [123] и образует тетрафенилгидразин. Последний не подвергается бензидиновой перегруппировке [ИЗ], а диссоциирует до того же радикала (C^HshN-, который диспропорцио-нирует в растворе до дифениламина и феназина. Феназины тоже индикаторы [1], и при окислении сами дают красную или синюю окраску. Наконец, тетрафенилгидразин в концентрированной серной кислоте легко окисляется до катион-радикала
[(CeH5)2N—N(CeH5)2] + пурпурного цвета. На первый взгляд здесь имеется достаточно соединений с пурпурной окраской и побочных
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
175
продуктов, которые бы объяснили все индикаторные свойства дифениламина, если его применять в нейтральной или щелочной среде Однако имеются некоторые неясности: во-первых, пурпурная окраска, получаемая при применении дифениламина в качестве индикатора, появляется в разбавленной (около 1—2 моль-л-1) кислой среде, а не в нейтральном растворе или концентрированной серной кислоте; во-вторых, при восстановлении этого пурпурного вещества образуется М,ЬГ-дифенилбензидин. Тому, что любая из пурпурных частиц, о которых говорилось выше, даст при восстановлении различные соединения (кроме тетрафенилгидразина или дифениламина), не имеется доказательств. При восстановлении радикал (CeHshN* может участвовать в какой-то реакции, до сих пор еще неясной. Поэтому образование бензидинов в кислых средах происходит, по-видимому, по какому-то другому механизму, а не в результате внутримолекулярной перегруппировки арилгид-разинов; пурпурная окраска индикатора обусловливается соединением, которое является производным бензидина [101—104]. Прежде чем рассматривать этот вопрос, надо напомнить еще об одном классе фениламинов.
Третичные фениламины
При окислении третичных фениламинов, имеющих в составе кольца по крайней мере два атома азота, связанных непосредст-
венно друг с другом, также получается пурпурная окраска и, по-видимому, окрашенные вещества образуются по двум путям. Если имеется хотя бы один углерод в пара-положении без заместителя, свойства такого соединения кажутся подобными свойствам дифениламина, т. е. окрашенное вещество является производным бензидина. Восстановление пурпурного продукта окисления тетрафенил-амина (C6H5)3N, например, дает М.М.Й'.М'-тетрафенилбензидин.
Однако Виланд [103—104] нашел, что такая же синяя или пурпурная окраска появляется при окислении три-п-толиламина в растворе разбавленной кислоты. Здесь группы метила в пара-положении мешают образованию п,п'-бензидина, и при восстановлении окрашенного вещества получается исходный трифениламин, а не бензидин. Вейц и Швехтен [92] показали, что в этом случае ок-
раска обусловливается катион-радикалом (С6Н5)зП+, который образуется путем отделения одного электрона от третичного основания (C6H5)3N: при его окислении. Эту частицу они назвали ионом аминия, но это не четвертичный ион; координационное число азота 3, а не 4, и поэтому этот ион образует соли, например перхлорат
и трибромид (Вг3). Катион-радикал (C6H5)3Nt изоэлектронен с нейтральным радикалом трифенилметила (С6Н5)зС*.
176
ГЛАВА VIII
ОКИСЛЕНИЕ ВТОРИЧНЫХ ФЕНИЛАМИНОВ В КИСЛЫХ СРЕДАХ
За исключением редко применяемых третичных фениламинов, все соединения, используемые в качестве индикаторов, являются вторичными фениламинами, у которых в пара-положении по отношению к атому азота не имеется заместителя или же находится группа, которую можно легко заменить, например сульфогруппа. Здесь уместно обобщить данные по первой стадии окисления вторичных фениламинов, соответствующего общему уравнению (63). Керманн [101, 102] и Виланд [103, 104] показали, что воздействию подвергается водород в пара-положении, а не в иминогруппе, тем не менее прямое окисление его кажется маловероятным. По-видимому, нельзя считать, что радикал дифенилимина (возможно, протонированный) является первым продуктом окисления. В кислом растворе тетрафенилгидразин не образуется, так что радикалы не комбинируют посредством атомов азота, возможно из-за протонирования; но эти радикалы представляют собой мезо-мерные соединения, и неспаренный электрон может появиться у атомов углерода в орто- или пара-положении по отношению к азоту, Если отсутствует заместитель, на который можно было бы воздействовать, вторичные фениламины являются очень слабыми основаниями, но тем не менее, вероятно, протонируются в обычных 1—2 М растворах кислот. Сначала образуется радикал
который может иметь четыре мезомерные формы, согласно теории (если же кольца различаются между собой каким-нибудь образом, то число мезомеров возрастает до семи),
Затем, по-видимому, из двух радикалов с неспаренным электроном у углерода в пара-положении образуется дифенилбензидин:
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
177
Учитывая различные продукты бензидиновой перегруппировки, показанные уравнением (65), и считая, что ни одному мезомеру в уравнении (67) не дается предпочтение в его образовании, можно сказать, что уравнение (68) представляет собой одну из пяти возможностей образования различных продуктов, а количество их [они те же, что и в уравнении (65)] будет зависеть от эксперимен тальных условий, природы и положения заместителя в ароматических кольцах, так же как при бензидиновой перегруппировке. Однако при помощи этого механизма снимаются ограничения внутримолекулярной перегруппировки и, если окисляется смесь из двух дифениламинов, может образоваться больше двух продуктов. Далее, если в одном дифениламине оба кольца различаются, можно ожидать, что получатся три основных продукта.
Установлено, что дифенилбензидин является продуктом восстановления пурпурного соединения цинковой пылью после разбавления кислого раствора и, несомненно, является главным продуктом окисления дифениламина; о проведении исследований в отношении образования других продуктов сведения не имеются. Весьма вероятно, что образуются различные количества некоторых других соединений, обладающих довольно близкими свойствами. Такое положение может привести к некоторой путанице в определении индикаторных свойств дифениламина, хотя не обязательно считать, что индикатор непригоден для применения.
Б. ОКИСЛЕНИЕ ДИФЕНИЛБЕНЗИДИНА И ЕГО ПРОИЗВОДНЫХ
Окисление дифенилбензидина и его производных, как таковых или потученных путем окисления соответствующих дифениламинов протекает, по всей вероятности, по уравнению (64). Виланд и другие авторы показали, однако, что соединение, названное дифенилбензидиновым фиолетовым, и его соль
HBC6NH=^^=^2y=-NHC6Hd
окрашены совсем слабо. Они также показали, что соль дихинондиимина является продуктом дальнейшего окисления окрашенного вещества, и утверждают, что синее соединение образуется на промежуточной стадии при переходе от бензоидного дифенилбензидина к хиноидному дихинондиимину вследствие отрыва одного электрона от дифенилбензидина. По их мнению, окрашенное вещество подобно неустойчивым красителям, которые получаются при окислении кислых растворов n-фенилендиамина, т. е. подобны так называемым солям Вурстера [95]. Из-за плохой растворимости различных частиц, побочных реакций и неустойчивости продуктов окисления исключительно трудно получить однозначные данные о •2—'48
178
ГЛАВА VIII
числе электронов, участвующих в реакциях индикатора. Нельзя быть уверенным, что один и тот же механизм применим ко всем производным соединений этого ряда. Однако данные показывают, что скорее идет одноэлектронная реакция, а не двухэлектронная, как утверждает Кольтгоф [86].
Степень протонирования восстановленных и окисленных частиц не легко определить, но при предположении полного протонирования можно представить одноэлектронное окисление в следующем виде:
(69)
Бикатион-монорадикал ДФБФ является мезомерным соединением. Если одно из колец имеет заместители, так что атомы азота будут отличаться один от другого, тогда получатся две мезомерные формы, различающиеся азотом с семью электронами. Волновая функция мезомерного соединения ДФБФ будет суммой волновых функций отдельных мезомеров пропорционально их вероятностям, т. е. обратно пропорционально энергии отдельных форм:
ф=пфя + йфь+сфсН------|-пф„ (70
Не вдаваясь в математические расчеты, надо сказать, что волновые функции мезомеров, у которых неспаренный электрон находится в орто- или пара-положении к атомам азота, составляют существенную часть общей волновой функции. Доля волновых функций других возможных мезомеров настолько мала, что ими можно пренебречь. Кроме мезомеров, показанных уравнением (69), можно» добавить еще следующие:
2
4
6
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
179
Таким образом, симметричный незамещенный дифенилбензидин имеет семь мезомеров. Это число удваивается при несимметричном замещении у атомов азота, а кроме того, увеличивается еще, если заместитель находится в одном из орто-положений (в положении 1 или 3) или в положении 5.
Хотя нельзя количественно и точно предсказать максимум поглощения Zmax и коэффициент молярной ЭКСТИНКЦИИ Е, исходя из структуры соединения, тем не менее можно с определенной уверенностью сказать, что, согласно теории, значение е возрастает с увеличением числа мезомеров. В то же самое время повысится устойчивость мезомерного соединения. Замещение водорода в иминогруппе или при активированных атомах углерода в мезомерном радикале, т. е. в положениях, отмеченных знаком «х», на сопряженные группы, такие, как фенильные, нафтильные, нитро, карбокси, нитрозил, азо, азокси и т. д., может повысить значение коэффициента молярной экстинкции продукта, и этим улучшатся индикаторные свойства.
Заместители, конечно, влияют на формальный окислительный потенциал индикатора; группы, подобные алкильной, понижают, а такие группы, как нитро или карбоксильная, повышают потенциал.
ИНТЕРМЕДИАТЫ И ПРОДУКТЫ ДЕСТРУКЦИИ
Бикатион-монорадикал является, по-видимому, наиболее вероятным продуктом обратимого изменения окраски индикатора на синюю или фиолетовую, причем точная окраска зависит от концентрации кислоты. Это соединение по современной терминологии называется семихиноном, а по старой мерихиноидом, но оно не может быть интермедиатом из схемы уравнения (64). Образование труднорастворимого, по-видимому, необратимого продукта деструкции тоже требует объяснения. На основании реакционной способности свободных радикалов можно ожидать, что произойдет диспропорционирование:
(71)
с образованием дпхинондиимина и дифенилбензидина, которые, по-
12*
180
ГЛАВА VIII
видимому, имеют слабую тенденцию к симметричному диспропорционированию. Виланд показал, что дальнейшее окисление пурпурного соединения дает «соль» дихинондиимина. Это легко представить в виде реакции
Н+ + е
(72)
Виланд сообщает, что протонированная форма и свободное основание окрашены слабо. Вероятно, это справедливо для свободного основания, но протонированная форма может обладать мезомерией, заряды могут перейти на кольца и, следовательно, в растворе она будет довольно сильно окрашена. При обоих процессах окисления по уравнениям (69) и (72) образуется ион водорода, а формальная зависимость потенциала равна —60 pH мВ. Надо отметить, что избыток окислителя разрушает индикатор гораздо быстрее, если повышается pH среды [23]; образование желтого продукта деструкции следует приписать свободному основанию, а красного продукта— протонированной форме дихинондиимина. Неустойчивость окисленной формы индикатора в отсутствие избытка окислителя объясняется, «по-видимому, реакцией диспропорционирования по уравнению (71). Тогда интермедиат, который, как считали раньше, является семихиноном, должен появляться, вероятнее всего, при низких концентрациях иона водорода, например в ацетатной среде, и его можно приписать дихинондиимину, а зеленый цвет может быть результатом смешения синего монорадикала и желтого непротонированного дихинондиимина.
Наиболее простым и кратким объяснением поведения таких индикаторов является отнесение синей или пурпурной окраски одноэлектронному окислению дифенилбензидина до монорадикала, а образование интермедиата и продуктов деструкции — диспропорционированию или дальнейшему окислению монорадикала до дихи-нондйимина. Существуют еще и другие возможности. Если индикатор используют в форме диариламина, а не в 'бензидиновой форме, интермедиатом будет радикал (CeHajsN», особенно при высоких значениях pH. Реакционная способность монорадикала увеличивает возможность протекания реакций конденсации, которые могут быть довольно сложными. Отделение интермедиата в виде зеленого осадка заставляет предположить, что образовался полимерный хингидрон посредством водородных связей. Водородные связи не могут образоваться при полностью протонированных частицах, а зеленый осадок образуется при низкой концентрации кислоты, когда преобладает свободное основание или монопротонированные часги-пы. По-видимому, возможно образование полимера в твердом со-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
18Г-
стоянии, как, например,
из бензидина и дихинондиимина. Допустимо частичное протонирование, вследствие чего может измениться окраска. Полимер не может существовать в растворе.
ДВУХЭЛЕКТРОННОЕ ОКИСЛЕНИЕ ИНДИКАТОРА
Если индикаторная реакция является действительно двухэлектронным окислением, как в уравнении (64), то для получения интенсивной окраски окисленная форма должна быть мезомерным соединением. Совсем не просто представить себе удовлетворительную формулу такого продукта, который может окисляться дальше с образованием продуктов деструкции. Двухэлектронное окисление свободного основания или протонированной формы дифенилбензидина может дать или бикатион-бирадикал, что сомнительно, или протонированный дихинондиимин, который, по утверждению Виланда, был им выделен и оказался слабо окрашенным. Ни один из этих продуктов не дает при депротонировании свободное основание дихинондиимина, показанное в уравнении (64). Сказанное-можно изобразить схематически:
свободное основание
R R
протонированная форма
свободное основание
182
ГЛАВА VIII
Бикатион-бирадикал сильно отличается от монорадикала, образование которого предполагается при одноэлектронном окислении, потому что бирадикал имеет четное число электронов, и если их спины противоположны, то делокализация спинов сразу же приводит к образованию дихинондиимина, не являющегося свободным радикалом. Для того чтобы мог существовать радикал, неспаренные электроны у атомов азота должны иметь параллельные спины, а это приводит к их возбужденному состоянию, которое, вероятно, имеет короткое время релаксации. Даже если допускается существование триплетного состояния для увеличения времени релаксации, довольно трудно согласиться с тем, что окраска может быть устойчива в течение многих часов или даже дней. Однако если предположить, что бирадикал существует, то любой электрон или «дырка» (положительный заряд), локализованный на азоте [см. уравнение(76)], могли бы делокализоваться. Изобразив структурные формулы специфических мезомеров, относящихся к классическим мезомерным соединениям, и не указывая отдельных мезомеров, можно принять, что электроны или положительный заряд локализованы в местах, указанных точками или знаком + по скелету кольцевых систем, причем азот, находящийся справа, считается инициатором.
Соответствующая схема справедлива для левого атома азота в качестве инициатора, кроме того, возможны еще 22 формы, если электрон или положительный заряд будет мигрировать одновременно от обоих атомов азота. Естественно, было бы неправильно представить наглядно такие отвлеченные структуры или считать, что они находятся в равновесии друг с другом, но их отдельные волновые функции могли бы участвовать в общей волновой функции мезомерного бирадикала, и чем ближе друг к другу находятся заряды (положительный и отрицательный), тем меньше будет их у частие. Очень большое число мезомерных форм предполагает очень высокий коэффициент молярной экстинкции в видимой области спектра Даже если не проводить количественного квантовомеханического анализа системы, вероятность такой схемы, по-видимому, мала.
Кроме того, хотя и кажется, что свободное основание дихиноп-диимина имеет слабую окраску вследствие его хромофорно-хромогенного сочетания, как сообщил Виланд, протонированная форма должна быть совершенно другой. Несмотря на то что она совсем
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
183
отличается от бирадикала, который имеет только семь электронок на каждом атоме азота (тогда как атом азота дихинондиимина имеет восемь электронов), а положительный заряд возникает вследствие протонирования (т. е. вследствие координации протона с изолированной парой электронов с противоположными спинами, находящихся у атомов азота), тем не менее, по-видимому, такое соединение может быть мезомерным из-за делокализации положительного заряда [126]. Это соединение может также иметь много мезомеров. Интересно, в противоположность утверждению Виланда о том, что свободное основание и протонированные частицы слабо окрашены, привести сообщение Керманна, который наблюдал си-нию окраску монобисульфата дихинондиимина [101].
Ни одна из этих гипотез не является удачной или удовлетворительной, и стадию дальнейшего окисления с разложением продукта с трудом можно представить иначе, чем отрыв электрона от атома азота дихинондиимина в состоянии свободного основания. При этом должен образоваться катион-радикал. Легко представить себе, что он обладает высокой реакционной способностью и действует на дихинондиимин с образованием крупных, слаборастворимых молекул.
Уже говорилось, что трудно получить однозначные данные о числе электронов, участвующих в реакции. Это объясняется и плохой растворимостью индикаторов, и неустойчивостью окисленных форм, и низким током обмена на электродах. Если восстановленная форма слабо растворяется, тогда концентрация ее во время титрования будет поддерживаться постоянной, а изменяется только концентрация окисленной формы. Взаимозависимость
р__₽' I ° >06 щ 10х!
п е [Red]
показывает, что при окислении должно происходить уменьшение-изменения потенциала и поэтому наклон ошибочно покажет более высокое значение п, чем оно в действительности. То же самое касается случая, если окисленная форма слабо растворяется и выпадает в осадок во время титрования или же разрушается (а разрушение ее может регенерировать восстановленную форму). Кроме того, изменение потенциала уменьшается и получается ошибочновысокое значение п. Если обе формы слаборастворимы или восстановленная форма плохо растворяется, а окисленная разрушается, то получается еще более сильный эффект. Формальный потенциал тоже устанавливается с небольшой ошибкой. В этом случае равновесие получается независимо от времени, которое требуется, чтобы достигнуть этого равновесия. Многие из индикаторных реакций идут медленно, а ток обмена электродных процессов низкий, что еще более понижает скорость установления равновесия. Поэтому возможно, что развитие окраски можно ускорить в ши-
^84
ГЛАВА VIII
.рокой области потенциалов вследствие кинетических факторов и, следовательно, могут появиться ошибочно большие значения тангенса угла наклона и неправильные низкие значения п. Плохая растворимость считается [128] причиной широких интервалов перехода. Даже предполагалось [132], что индикатор с более низким потенциалом, чем требуется, может дать удовлетворительные результаты при титровании, если растворимость его будет достаточно низкой Если имеется индикатор высокой степени чистоты и при титровании стандартным окислителем он не разлагается, тогда можно определить число п по количеству израсходованного окислителя определенной концентрации на потенциометрическое титрование известного количества индикаторов. Это редко бывает возможно и даже тогда не всегда бывает однозначно.
Известны только три случая, в которых точно доказано, что окисление идет с переносом двух электронов. Это окисление 3,3'-диметоксибензидина [127], 3,3'-диметилнафтидина [в смеси уксусной и4М серной кислот (соотношение 1:1)] и нафтидин-3,3'-ди-сульфокислоты [‘128]. Следует отметить, что все эти соединения не имеют заместителей у атомов азота [’R = H в формуле (73)]. Тут возможны два механизма процесса окисления. Во-первых, об-.щий механизм процесса тот же, что и для одноэлектронного окисления, за малым исключением в случае бензидина или нафтидина, но не в случае производных дифенилбензидина; во-вторых, все случаи окисления идут с переносом двух электронов, как ранее считал Кольтгоф. Экспериментальные трудности не позволяют получить четкий ответ, и поэтому следует рассмотреть оба механизма.
В пользу теории окисления с переносом двух электронов свидетельствуют следующие факты: во-первых, большее число мезомеров обусловливает высокий коэффициент молярной экстинкции и, во-вторых, окисление с переносом одного электрона, в результате которого образуется монорадикал, можно использовать для объяснения промежуточной стадии окисления. Поэтому более вероятной схемой является
С6Н5 CgHj,
н N—NH
I продукт деструкции
v полимера
интермедиат —
с6н NH
(74)
Только с помощью количественного квантовомеханического исследования можно выяснить реальность этого предположения.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 18&
ИНДИКАТОРЫ И ИХ ПРИМЕНЕНИЕ
В связи с тем, что существует начальная стадия окисления дифениламина и его производных до дифенилбензидина, последний-фактически является индикатором. Однако самому дифенилбензи-дину и его производным уделялось мало внимания, а большинстве работ было посвящено предшественникам, т. е. дифениламину и его производным. Интересно заметить, что 4,4,-дизамещенные ди-фениламины, у которых заместители не отщепляются, не могут образовать дифенилбензидины. К таким заместителям относятся амино- и метоксигруппы, при участии которых главным продуктом окисления является семидин. Однако 4,4,-диаминофениламин окисляется в темно-синее соединение, а формальный потенциал в 50%-ном ацетоне составляет 0,75В [129]. Вариаминовый синий представляет собой 4-амино-4,-метоксидифениламин; механизм era окисления рассмотрен в части А этой главы. Следует также заметить, что промежуточное производное дифенилбензидина никогда-не было выделено при изучении индикаторов, хотя имеются исследования, в которых использовали некоторые предполагаемые интермедиаты в качестве исходных реагентов [130], а Виланд выделил дифенилбензидин из фиолетового продукта окисления путем восстановления его цинковой пылью после разбавления. Трудности, связанные с определением формальных потенциалов, интервалов перехода и спектральных характеристик, привели к некоторой путанице и к очевидным противоречиям в литературе. Белчер рассмотрел создавшееся положение и способствовал его разрешению [131]. Не всегда бывает ясно, идет ли речь о стандартных пли формальных потенциалах, а иногда не приводятся условия их определения. Редко исследовалось влияние концентрации ионов водорода, а там, где это делалось, снова получалась путаница в результатах. Такие трудности, по-видимому, привели к тому, что стали сообщать значение потенциала, при котором наблюдается первое заметное изменение окраски, и иногда такой потенциал называли формальным потенциалом; поскольку наблюдение субъективно, неизбежны некоторые различия в полученных значениях. По этой причине, возможно, появилась мысль об определении «чувствительности» индикатора в отношении специфического реагента вместо определения формального потенциала. Однако это тоже субъективное определение, и, чтобы его можно было применять, нужно полностью уточнить условия. Это, конечно, должны ыть условия, подходящие для изучения индикаторов в качестве чувствительных колориметрических реагентов на следовые количества окислителей [130, 132], а в отсутствие более прямых сведений эти условия можно использовать в титриметрии. За исключением некоторых случаев упоминания о замедленном действии или о слабой реакции индикатора, почти не имеется сведений, и чието случайно [130] проведено количественное исследование ки-
Таблица VIII. 12
ДИФЕНИЛБЕНЗИДИН, ДИФЕНИЛАМИН И ИХ ПРОИЗВОДНЫЕ3
Соединение Потенциал® относительно нвэ, В \пах* нм [130] Еох, МОЛЬ—1 л - см—1 [130]
\| N'-Дифснилбснзидии N-Замещенные ^trails 0,76 [86] 0,76-0-94 [133] 560 [134] 5,0 • 104
М,М'-Диметил-М,М'- дифенил бензидин — 525—532 4,5 • 104
Замещенные в кольце
Ы,М'-Дифенилбензидиндекасульфо- 0,80 [135] — —
кислота
Ы,М'-Ди 12-карбоксифснил) бензидин — 556-562 4,27 • 104
N.N'-Д и- (2-карбоксифенил) -3,3'-ди — 564—574 5,2 • 104
метилбензидин 6,36 - 104
N.N'-Ди- (2-карбоксифенил) -3,3'-ди- — 618—630
метоксибензидин
Дифениламин 0,76 [86] 0,76—0,86 [133] 565 [134] 4,5 • 104 [134]
Монозамещенные в кольце
2-Карбоксидифениламин (N-фенил-антраниловая кислота) [135] [131] 0,94—1,00 [137] — —
2-Метилдифениламин (о-толилфе- —— — —
ни ла мин) [137]
З-Метилдифениламин (ж-толилфенил- — —— —
амин) i[138]
4-Метилдифенила мин (п-толилфенил-амип) [138] 4-Нитродифениламин Е'о 1,03 [141] —
4-Аминодифениламин Е- 0,76 [129] — —
4- (Ацетиламино) дифениламин Е'о 0,70 [140] — —
Дифениламин-4-сульфокислота Д' 0,85 [142] 0,83—0,91 [133,144] 593 [143] —
Изомер дифениламиносульфокислоты —• — —
[137] Двузамещенные в одном кольце
2,4-Диаминодифениламин Е'о 0,7 [140] — —
З-Метокси-4-аминодифениламин Др 0,64 [129] — —
Двузамещенные в разных кольцах
2-Метилдифенчламин-4-сульфокисло- — — —
та (о-толилфениламиномоиосульфо-нат)
2-Аминодифениламин-4-сульфокис- — — —
лота
2,2'-Дикарбоксидифениламин е'о 0,88 [130] 1,26 [145] 608—612 2,31 • 104
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
187
Продолжение табл. VIII. 12
Соединение Потенциал^ относительно нвэ, В \пах» нм [130] Fox, МОЛЬ-1 . л см—1 [130]
2 З'-Дикарбоксидифениламин £0 1.12 [145] 575-582 1,04 104
2,4'-Дикарбоксидифен иламип — 565—572 1,8 • 104
2-Карбокси-2'-метилдифениламин (N-о-толилантраниловая кислота) Е'о 0,92 [130] Е'о 1,01 [137] 0,81—1,01 [137] 554—560 2,86 • 104
2-Карбокси-3'-метилдифеииламин (ЬГ-лг-толилантраниловая кислота) Е'о 0,85 [137] — —
2-Карбокси-4'-метилдифеииламин (N-ai толилаитраниловая кислота) Ео 0,85 [137] — —
2-Карбокси-2'-метоксидифениламин Е'о 1,00 [130] 622—624 3,13 10*
2-Карбокси-2'-нитродифениламин Е'о 0,90 [130] 592—598 1,85,- 104
2-Карбоксидифениламин-4-сульфо-кислота — 560—570 1,6 • 1()4
4,4'-Диаминодифениламин 4-Амино-4'-метоксидифениламин (вариаминовый синий, см. часть А этой главы) Тризамещенные в кольце (2-замеш.енный вариаминовый синий, см. часть А) Е- 0,75 [129]
2,4,4'-Тр иаминодифениламин [140] .— — —
2,4-Диа мино-4'- метокси дифенила мин [140] (2-аыиновариам.иновый синий, см. часть А) — — —‘
Гриамино- и тетрааминодифенилами-ны [1403 N-Замещенные
№Метилдифениламин-4-сульфокис-лота Е' 0,86 [130] Е' 0,80 [146) 520—525 511 [134] 2,64 104 4,4 104 [134J
а п
б ь ^квадратных скобках дана литература
условный потенциал; Е- — стандартный потенциал; без обозначения — природа енциала неизвестна; интервал значений (напр., 0,76—0,94) — это нижний и верхний потенциалы перехода (интервал перехода).
нетики реакции. Основные сведения об этих индикаторах очень отрывочны, и их не легко собрать.
Соединения, которые были исследованы как индикаторы, обобщены в табл. VIII.12; там же приведены их известные физические свойства. Надо заметить, что условия определения потенциалов не оговорены; причина этому — неуверенность в точном смысле ука
188
ГЛАВА VIII
занного потенциала. Часто считают более низкий потенциал перехода формальным потенциалом и не делают разницы между «окислительно-восстановительным потенциалом», потенциалом перехода, потенциалом первого заметного изменения окраски (более низкий потенциал перехода), формальным потенциалом и стандартным потенциалом; особенно это касается более ранних работ. Более поздние работы, и в особенности те, в которых приводятся интервалы перехода, более надежны [128, 129, 131—133, 144]. Если не указан потенциал, то можно считать, что индикатор был испытан в титровании обычно железа (II) бихроматом, но потенциалы не были определены.
Из перечисленных индикаторов только четыре нашли широкое применение. Это дифениламин, дифенилбензидин, дифениламин-сульфокислота (и ее соли) и 2-карбоксидифениламин (т. е. N-фе-ннлантраниловая кислота). Незамещенные соединения представляют собой исключительно слабые основания и очень плохо растворяются. Для дифенилбензидика Кь=2-10-14, а ‘растворимость в воде при 25—50 °C составляет 0,06 мг-л-1 (5-1О-7 моль-л-1); эта растворимость в пять раз больше, чем в 2 М соляной кислоте, и в 2—3 раза больше, чем в 1 М. серной кислоте. Для дифениламина Ль=7,6 • 10-14, а растворимость в воде [147] при 15 °C составляет 0,0316 г-л-1 (т. е. 2-10-4 моль-л-1) Плохая растворимость является причиной многих экспериментальных трудностей при исследовании и использовании этих индикаторов. Обычно исходные растворы индикатора готовят в концентрированной серной кислоте, которая не должна содержать соединений азота. Принято готовить 1%-ный или 0,1%-ный растворы, либо 1%-ный раствор в концентрированной серной кислоте, который разбавляют ледяной уксусной кислотой в соотношении 1 :9. Две капли 0,1 % -ного дифенил-бензидина, добавленные к 100 мл титруемого раствора, дают перенасыщенный раствор индикатора (10-кратный избыток сверх значения, соответствующего насыщению). Но фактически сразу же после добавления раствора индикатора образуются хлопья осадка еще до смешивания с титруемым раствором и этот осадок, в частности для случая с дифенилбензидином, коагулирует и больше не растворяется. Окраска такого разбавленного раствора индикатора кинетически медленно изменяется, несмотря иа высокий коэффициент молярной экстинкции продукта окисления. В результате этого Кольтгоф попытался увеличить растворимость индикатора путем введения сульфогруппы в дифениламин [142], а Сарвер [135] синтезировал декасульфопроизводное дифенилбензидина. Это соединение в дальнейшем не было исследовано, а дифенил-амин-4-сульфокислота и ее бариевая и натриевая соли, которые легче выделить, стали наиболее популярными из этих индикаторов. Дифениламин-4-сульфокислота и ее соли растворяются в воде, и, таким образом, трудности при работе с ними преодолены. Пред
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
189
полагается, что механизм окисления такой же, как и для незамещенного соединения; введение такой сильнополярной группы должно оказать значительное влияние на плотность распределения электронов в молекуле, и, действительно, из-за этого формальный потенциал и интервал перехода более положительны примерно на 100 мВ. На окисление дифениламина в дифенилбензидин должен расходоваться окислитель, и это приводит к индикаторной ошибке, которую можно уменьшить, если вместо дифениламина брать в качестве индикатора дифенилбензидин. Несмотря на ранее существовавшие сомнения ['23], теперь установлено, что величины поправок для ряда дифенилбензидин—дифениламин—дифениламиносульфокислота относятся как 1:2:3. Причина того, что поправка для сульфокислоты больше, еще совсем не ясна, но вполне возможно, что она вызвана эффектом растворимости. Однако выгода от применения дифенилбензидина вместо дифениламина ограниченна, потому что в гомогенном растворе из дифениламина образуется дифенилбензидин в виде коллоидной дисперсии, которая гораздо быстрее взаимодействует с окислителем, тогда как коагулированный осадок, полученный при непосредственном использовании дифенилбензидина, с трудом растворяется и медленно вступает в реакции. Теперь уже известно [134], что дифениламин р его производные окисляются непосредственно в синее соединение, минуя промежуточную стадию образования производного дифенилбензидина (см. ниже).
Этот подкласс индикаторов можно использовать и используется при работе с такими сильными окислителями, как, например, церий (IV) или перманганат. Применение этих индикаторов при восстановительном титровании ограничивается только такими восстановителями, формальный потенциал которых ниже 0,72 В (за исключением использования нитропроизводных и карбоновых кислот индикаторов этого подкласса). Чаще всего эти индикаторы применяются при титровании бихроматом и другими менее сильными окислителями, как, например, ванадий (V), а в отдельных случаях титрования по методу осаждения — гексацианоферрат(Ш). В качестве индикаторов при титровании более сильными окислителями лучше подходят комплексы металлов с лигандами а,</-ди-иминогруппы. Их можно использовать вместо производных бензидина даже при титровании бихроматом, правильно подобрав концентрацию кислоты или выбрав подходящий заместитель (например, 5,6-диметил-1,10-фенантролин). Тем не менее благодаря небольшому расходу и очень четкому изменению окраски производные бензидина продолжают вызывать к себе интерес, особенно при титровании бихроматом. Последний, несмотря на ряд недостатков, удобен тем, что бихромат калия легко получить очень чистым и из него можно непосредственно готовить растворы для определения.
190
ГЛАВА VIII
Бихромат как окислитель отличается особой кинетикой и ме-ханпзмО1М реакции и вообще не может прямо окислять индикаторы этого подкласса, если в растворе отсутствует железо(III). Иногда такое явление называют индукционным эффектом, и особенно ярко оно выражено у метилпроизводнь^х дифениламина. Более сильные окислители или окислители с высокой скоростью обмена электронами окисляют эти индикаторы непосредственно. Другой особенностью бихромата является то, что при титровании бихромата раствором железа(II) изменение потенциала (и значение Q) (от 1,22 В до 0,85 В) больше, например, чем при титровании железа (II) бихроматом (от 0,85 до 1,0 В) в тех же условиях [148]. Поэтому такой метод титрования ставит свои собственные проблемы, одной из которых является наличие зеленой окраски от ионов хрома (III). Потенциал системы дифениламин—дифенилбензидин, определенный любым методом, низок, и можно было бы ожидать, что индикатор изменит окраску значительно раньше, чем окислится железо (II). Поэтому с самого начала добавляют фосфорную кислоту, чтобы связывать в комплекс железо (III) и этим понизить потенциалы системы железо (III)—железо (II). Расчет констант комплексообразования может выявить неосновательность такого подхода; Стокдаль [133] утверждает, что выгода от этого только кажущаяся. Из своей практики Бишоп заметил, что, чем больше промежуток времени между добавлением инкрементов бихромата по мере приближения к конечной точке, тем раньше наступает конечная точка, так что это, видимо, просто вопрос кинетики. Как утверждает 'Стокдаль, при титровании с нормальной скоростью конечная точка почти не отличается от точки эквивалентности.
Вольфрамат и еще один или два аниона мешают титрованию, так как осаждают даже более труднорастворимые соли индикатора. Вследствие этого свойства индикаторы подкласса бензидина широко применяются в качестве органических осадителей. Присутствие меди(II) не сказывается на индикаторе. Но мешающее действие ее проявляется при окислении железа(II) атмосферным кислородом: прямая реакция идет довольно медленно, и ею можно пренебречь, но медь (II) частично восстанавливается железом (II) до меди(1), которая очень энергично реагирует с кислородом с образованием меди (II), являющейся, следовательно, катализатором. Удаление кислорода предупреждает появление ошибки, обусловленной присутствием меди. «Эффект хлорида ртути(II)», как его часто называют, усугубляет медленные реакции многих из индикаторов этого подкласса. При определении железа(III) часто используют метод, который состоит в восстановлении железа (III) хлоридом олова (II) в соляной кислоте при повышенной температуре, охлаждении раствора, содержащего небольшой избыток олова (II), уничтожении избытка олова (II) при помощи хлорида ртути (II) [при этом образуется осадок хлорида ртути(I)] и, наконец.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
191
титровании железа(II) бихроматом. Этот метод требует умения и критического подхода, но при известной опытности дает очень хорошие результаты. Однако в присутствии ртути (II) незамещенные индикаторы и многие из замещенных, в частности метилпроиз-водные, действуют крайне медленно и окраска получается нечеткой из-за образования продуктов разложения. Причина этого заключается в сродстве ртути к азотсодержащим лигандам и в связывании индикатора в виде комплекса
HgCl2 + Ind HglndCH + С1-, который очень медленно освобождает индикатор для окисления титрантом. Отрицательного действия хлорида ртути (II) можно избежать, если использовать какотелин в качестве индикатора для титрования в первую очередь избытка олова (II) (происходит изменение фиолетовой окраски на бесцветную), а затем, как обычно, титровать железо(II).
Многих трудностей, встречающихся при использовании незамещенных индикаторов (как, например, плохая растворимость, медленная индикаторная реакция, осаждение вольфраматов и в некоторой степени влияние полярного заместителя на плотность электронов атома азота, эффект хлорида ртути(II)), можно избежать, если работать с сульфокислотами индикаторов. Появляющиеся при этом небольшие недостатки (иногда более высокая поправка на индикатор и повышение неустойчивости окисленной формы, особенно в среде высокой концентрации кислоты) компенсируются тем, что у сульфокислот растворимость лучше, реакция идет быстрее и потенциал окисления выше. Соли бария и натрия легко выделить; они растворяются в воде, поэтому растворы индикатора можно готовить на воде. В табл. VIII. 13 приведены потенциалы перехода (определяемые в данном случае при первом появлении окраски) по данным Белчера [144] и Стефена [149].
Таблица VIII. 13
ПОТЕНЦИАЛЫ ПЕРЕХОДА (ПЕРВОГО ПОЯВЛЕНИЯ ОКРАСКИ) ДИФЕНИЛАМИН-4-СУЛЬФОКИСЛОТЫ В РАСТВОРЕ СЕРНОЙ КИСЛОТЫ
[H2so4], моль л—1 £trans 11441 £trans 1,491 [H2so4], моль • л—1 brans'1441 р cfrans [149]
0,1 0,853 0,81 2,0 0,800 0,80
0,5 0,850 0,82 4,0 0,770 0,75
1,0 0,830 0,83 8,0 0,710
Обычно с индикаторами работают в 1—2 М соляной или серной кислоте. Достаточно взять 1—2 капли 0,1%-ного раствора ин
192
ГЛАВА VIII
дикатора на 100 мл титруемого раствора, чтобы в конечной точке получилось резкое изменение окраски. Из-за окраски раствора ионами хрома (III) трудно заметить первое изменение окраски индикатора на искусственном свету и приходится брать большее количество индикатора. Поправку на индикатор следует определять в условиях проведения эксперимента; на каждый миллилитр 1%-ного раствора индикатора поправка, выраженная в объеме 1/60 М бихромата, составляет 0,25 мл для дифенилбензидина, а 0,5 мл для дифениламина и дифениламиносульфоната бария, но эти числа только приблизительны. Титрование по мере приближения к конечной точке должно идти достаточно медленно, особенно в случае незамещенных индикаторов; рекомендуется выдерживать интервал в 1 мин между последними двумя-тремя добавками инкремента титранта. Однако при обратном титровании бихромата железом (II) задержка в добавке титранта приведет к деструкции индикатора, поэтому лучше вносить индикатор не в начале титрования, а при приближении к конечной точке; само же титрование надо вести с разумной скоростью, особенно если использовать сульфокислоты. Белчер [150] провел тщательное исследование реакции титрования системы бихромат—железо(II).
Уже говорилось об индукционном эффекте: сам бихромат не окисляет дифениламин или дифенилбензидин, поэтому нельзя получить величину поправки на индикатор путем прямого титрования холостого раствора. Для образования синей окраски необходимо присутствие железа. Уже упоминалось о том, что железо (III) нужно для окисления дифениламина в дифенилбензидин, однако тот же эффект наблюдался при работе с самим дифенилбензидином. Белчер [26] утверждает, что окисление индикатора обычно идет медленно, но его индуцирует реакция между бихроматом и железом (II). Для этого не требуется специально наличия железа(III) или железа (II), а достаточно присутствие восстановителя, у которого скорость обмена электрона высока и который при реакции с бихроматом обеспечивает энергией активации стадию, определяющую скорость реакции. Такие индикаторы подвергаются и другим индукционным реакциям [151]. В присутствии многоатомных спиртов и сахаров окисление индикаторов (включая N-фенилантра-ниловую кислоту) и их необратимая деструкция настолько ускоряются, что конечная точка смещается и определяется с запозданием. Если вместо бихромата работать с ванадием (V) в тех же условиях, смещений не наблюдается. Следовательно, недостатки связаны с механизмом реакции бихромата, а не с индикаторами, хотя их подверженность деструкции усугубляет протекание процесса.
Единственным производным дифениламина, широко применяемым в практике, является 2-карбоксидифениламин, называемый еще N-фенилантраниловой кислотой [136]. Долго считалось, что
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
193
ее потенциал равен 1,08 В, но, как установил Белчер [131], это противоречит поведению индикатора при титрованиях системы бихромат—железо (II) и потенциалам перехода [137]. Формальный потенциал системы т’рцс-(1,10-фенантролин)железо(11)—трис--(1,10-фенантролин)железо(Ш) тоже равен 1,08В, но этот индикатор нельзя использовать для титрования бихроматом в 1 М серной кислоте. Это не является противоречием, потому что в таком случае интенсивно окрашена восстановленная форма индикатора и ее потенциал перехода выше формального потенциала более чем на 60 мВ, тогда как в случае N-фенилантраниловой кислоты окрашена окисленная форма и первое заметное изменение окраски на-бподается при потенциале по крайней мере на 60 мВ ниже формального потенциала. При коэффициенте молярной экстинкции около 2-104 л-моль-1 см-1 первое заметное изменение окраски будет в области обычного титрования 1/60 М раствором бихромата. Тем не менее Белчер показал, что истинный потенциал перехода (определяемый в момент первого появления окраски) в среде 0,5 М серной кислоты равен 0,89 В, в 1,0 М — 0,87 В и в 2,0 М кислоте 0,88 В [131]. Предполагаемым бесцветным интермедиатом окисления этого индикатора является М,М'-ди-(2-карбоксифенил)-бензидин. Фрумина с сотр. [130] исследовала этот продукт в качестве реагента для колориметрического определения ванадия (V) (табл. VIII.12). В таблице не приведены потенциалы, но, вероятно, они такие же, как и для N-фепилантраниловой кислоты. Высокая концентрация кислоты в титруемой среде способствует, по-видимому, образованию карбоновых кислот ['130, 145], стабилизирует окисленную форму; так же [130] действует и на присутствующую восстановленную форму. Имеются сообщения, что N-фенилантра-ниловая кислота быстро дает устойчивую окраску в серной кислоте при концентрации ее больше 6 моль-л-1; при более низких концентрациях окраска появляется медленно, а при более высоких снижается ее интенсивность. Белчер [131] рекомендует при титровании железа (II) раствором церия (IV) работать с 0,5—2,0 М серной кислотой, но в случае титрования бихроматом не советует использовать N-фенилантраниловую кислоту как индикатор, хотя результаты получаются довольно точными; Белчер считает, что в последнем случае необходимо добавлять фосфорную кислоту при титровании в среде 2—4 М серной кислоты. Обычно используемый раствор индикатора готовят путем нейтрализации свободной кислоты эквивалентным количеством карбоната натрия и разбавлением водой до получения 0,1%-ного раствора [136]. Такой раствор очень неустойчив, так как N-фенилантраниловая кислота является гормоном для роста растений и в ней быстро размножаются плесени и водоросли [152]. Хотя свойства индикатора ухудшаются не сразу, все же более устойчивый и активный раствор получают, растворив 0,1 г индикатора в 5 мл 0,1 М гидроокиси натрия и раз
13—518
194
ГЛАВА VIII
бавив до 100 мл. Поправка на индикатор при работе с 1/60 М бихроматом или другим равноценным окислителем незначительна [26]. N-Фенилантраниловая кислота—хороший индикатор для периметрии, хотя из-за неустойчивости его окисленной формы в серной кислоте, концентрация которой ниже 6 моль-л-1, предпочтительнее применять трис-(1,10-фенантролин) железо (II).
Главной областью применения этих индикаторов является титрование бихроматом и ванадатом. Это все хорошо описано в учебниках и периодической научной литературе [23, 25—30, 131—133, 150]. Их часто употребляют для определения некоторых двухвалентных металлов, например цинка, кадмия и свинца, осаждая их гексацианоферратом (II) в виде осадка состава K2M,3I[Fe(CN)6]2. Гексацианоферраты (III) этих металлов гораздо лучше растворяются, и поэтому добавка небольшого количества гексацианоферрата (Ш) в титруемую среду создает окислительно-восстановительную систему, которую можно использовать для установления момента полного осаждения. Если раствор иона металла использовать в качестве титранта, то устранение гексацианоферрата (II) из титруемого раствора вызывает резкое повышение потенциала и окисление индикатора, тогда как обратное титрование приводит к падению потенциала и восстановлению индикатора. При этом титровании можно использовать дифениламин-дифенилбензидино-вые индикаторы, но они хуже 3,3'-ди метоксибензидин а (или о-ди-анизидина) [85], который в свою очередь хуже некоторых производных нафтидина [153] (о них речь будет ниже). Появление хелатообразующих титрантов, например ЭДТА, уменьшило интерес к этим методам: их избирательность в отношении металлов, образующих гексацианоферраты(II), нерастворяющиеся в сильнокислых средах, поддерживала некоторое время интерес к ним, пока избирательно маскирующие процессы не развились до такой степени, что методы с образованием хелатов стали даже более чувствительными [44].
Остальные индикаторы, перечисленные в табл. VII 1.12, не подвергались тщательному изучению. Некоторые из них были испытаны в ходе общего исследования индикаторов [130, 140], а не в процессе титрования [133, 150]. Другие были применены только в одном или нескольких случаях. Использование индикатора в окисленной форме, как в случае сульфированного дифенилбензидина [135] и как рекомендуется для дифениламин-4-сульфокислоты [154], вряд ли выгодно из-за неустойчивости окисленной формы.
Единственным производным дифенилбензидина, используемым в качестве индикатора, является его сульфокислота. Сарвер и Фишер [135] синтезировали одно соединение, анализ которого показал, что оно содержит 10 сульфогрупп. После сульфирования изменение окраски происходит при несколько более высоком потенциале. При частичном окислении получается зеленая окраска, а
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
195
при полном — фиолетовая, которая постепенно исчезает. Окисление бромом приводит к появлению быстро исчезающей синей окраски. Белчер [150] упоминает о дисульфонате, но, по-видимому, подразумевается интермедиат, получающийся из дифениламиномоносульфоната. Остальные соединения, перечисленные в табл. VIII. 12 (за исключением сведений о том, что они, по-видимому, образуются в виде интермедиатов из соответствующих производных дифениламина), не были исследованы как индикаторы. Их использовали в 9—12 М серной кислоте в качестве реагентов для колориметрического определения ванадия (V) ['130]. Имеются некоторые указания, что карбоксильные группы находятся на крайних фенильных кольцах [137], но, если, например, N.N'-ди- (2-карбокси-фенил)-3,3/-диметилбензидин [т. е. 2,2,-дикарбокситолидин или точнее И,М/-ди-(2-карбоксифенил)-4,4'-диамино-3,3'-диметил-1,1'-дифе-нил] получается при окислении о-толилантраниловой кислоты (2-карбокси-2'-метилдифениламина), тогда возможно образование пятнадцати продуктов; три из них получаются, если окисление произойдет только по углероду в /шра-положении.
Довольно трудно с уверенностью установить влияние заместителей в дифениламине, потому что сведения об этом очень редки и условия, в которых проводились эти исследования, весьма различны. Можно сказать, что такие заместители, как карбокси-, нитро-, метокси- и сульфогруппы, повышают окислительные потенциалы; наибольшее влияние оказывают заместители в положении 2, а наименьшее — в положении 4; алкильная и аминогруппа понижают окислительный потенциал. Алкилирование по азоту, видимо, немного снижает значение окислительного потенциала. Влияние заместителей на максимум поглощения можно увидеть из данных табл. VIII. 12, но эти сведения недостаточны, чтобы можно было сделать определенные выводы.
Монометилпроизводные дифениламина (т. е. толилфениламины) исследовал Еспер [138, 139], который нашел, что они обладают большим индукционным эффектом при титровании бихроматом и сильным эффектом хлорида ртути(II). При титровании бихроматом раствора железа(II) они действуют как обратимые индикаторы; получающаяся синяя окисленная форма устойчива в течение 10— 15 мин, что позволяет проводить прямое титрование. Хотя монометилпроизводные дифениламина предложены в качестве индикаторов в перманганатометрии, они не были испытаны в этом методе, а исследованы только при титровании железа(II) бихроматом.
4-Нитродифениламин имеет слишком высокий потенциал, чтобы его можно было использовать в титровании бихроматом [140], и легко разрушается при глубоком окислении. При титровании церием (IV) он необратимо окисляется, но результаты получаются удовлетворительные. 4-Нитродифениламин не имеет преимуществ перед трцс-(1,Ю-фенантролин)железом(П) в этом методе титро-13*
196
ГЛАВА VIII
вания. Полинитропроизводные дифениламина непригодны в качестве индикатора, так как их формальные потенциалы слишком высоки.
4-Аминодифениламин окисляется с образованием красно-фиолетовой окраски [140], но легко разрушается от избытка окислител? и не имеет преимуществ перед незамещенным дифениламином.
4-(Ацегиламино)дифениламин окисляется до зеленовато-синего продукта; более устойчив к деструкции в избытке окислителя, чем 4-аминодифепиламин [140]; потенциал, по-видимому, несколько
ниже потенциала 4-аминодифенила; не считается хорошим индикатором [140].
2,4-Диаминодифениламин довольно хорошо растворяется в воде и разбавленных кислотах, как в окисленной, так и в восстановленной формах [140, 141], и окисляется до красного соединения. Последнее устойчивее дифенилбензидинового фиолетового и труднее разрушается избытком окислителя. Сообщается, что потенциал 2,4-диаминодифениламина в 1 М серной кислоте при появлении первой заметной окраски равен 0,7 В; это слишком низкий потенциал для титрования железа (II) даже в присутствии фосфорной кислоты; этот индикатор можно было бы применять в титровании более сильных восстановителей.
З-Метокси-4-аминодифениламин [129] при окислении дает синюю окраску, но его стандартный потенциал, равный 0,64 В, слишком низок для титрования железа(II).
2-Метилдифениламин-4'-сульфокислота. Исследуя изомерные толилфениламины, Коен и Еспер [138] изучали также сульфокислоты и пришли к заключению, что только 2-метил-изомер (т. е. о-толилфениламин) обладает свойствами индикатора. Сульфокислота окисляется с образованием красной окраски; на нее не оказывает влияния серная кислота; но в присутствии соляной кислоты в концентрации выше 0,1 М определение конечных точек и изменения окраски дают ошибку.
2-Аминодифениламин-4'-сульфокислота, по данным Вилларда и Манало [155], изучавших производные дифениламина, является лучшим индикатором в титровании гипобромитом. При этом титровании индикатор разрушается, но определение конечной точки четкое, а поправка на индикатор небольшая. Дифениламин-4-суль-фокислота окисляется гипобромитом обратимо, но изменение окраски менее четкое, а поправка на индикатор сравнительно большая.
Дикарбоновые кислоты дифениламина. Кирсанов и Черкасов [145] исследовали индикаторные свойства 2,2'-, 2,3'- и 2,4'-дикар-боновых кислот и нашли, что окраска четко изменяется с зеленой на синюю или фиолетовую в среде 7,5—10 М серной кислоты. В растворах соляной кислоты окраска не изменяется. Продукт окисления 2,2'-дикарбоновой кислоты очень устойчив, 2,4'-дикарбо-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
197
новой кислоты менее устойчив, а 2,3'-изомер неустойчив. Окислительный потенциал равен 1,26 В для 2,2'-изомера и 1,12 В для 2,3'-изомера. Относительно 2,4'-изомера не имеется данных, но можно предпсложить, что его потенциал будет самым низким из трех. Поправка на индикатор очень большая. Фрумина с сотр. [130] исследовала эти соединения в качестве реагентов для колориметрического определения ванадия(V) в 12 М серной кислоте и в противоположность более ранней работе сообщает, что в 9 М серной кислоте потенциал 2,2'-дикарбоновой кислоты равен 0,88 В.
N-Толилантраниловые кислоты. Ледерер и Уорд [137] исследовали три изомерных толилпроизводных N-фенилантраниловой кислоты. Из них о-изомер (2-карбокси-2-метилдифениламин), по-видимому, наиболее эффективный, дает при окислении довольно устойчивую пурпурную окраску. Окислительный потенциал по первым сообщениям был равен 1,01 В, но, видимо, это слишком высокое значение; изменение окраски происходит в пределах 0,81— 1,01В [132]. Белчер с сотр. [131] повторно определял потенциал перехода (т. е. первого появления розовой окраски) и нашел, что его значение немного понижается с повышением концентрации серной кислоты; так, в 0,5 М серной кислоте потенциал равен 0,89 В, в 1,0 М — 0,88 В, а в 2,0 М — 0,87 В. Фрумина с сотр. [130] нашла, что в 0,5 М серной кислоте формальный потенциал составляет 0,92 В. 2-Ка1рбокси-3'-метилдифениламин (т. е. N-лг-толилан-траниловая кислота), потенциал которого 0,85 В, дает очень неустойчивый продукт окисления [137], а 2-карбокси-4'-метилдифе-ниламин (т. е. N-n-толилантраниловая кислота) не дает окрашенного продукта при окислении.
2-Карбокси-2'-метоксидифениламин не был испытан в качестве индикатора, но его использовали как реагент для колориметрического определения ванадия (V). Формальный потенциал его 1,00 Б в 0,5 М серной кислоте [130], а максимум поглощения окисленной формы находится при значительно большей длине волны, чем у большинства производных дифениламина.
2-Карбокси-2'-нитродифениламин должен был бы иметь очень высокий потенциал, но, по данным Фруминой и др. [130], он равен 0,90 В в 9М серной кислоте. В отношении индикаторных свойств исследования не проводились.
2-Дарбоксидифениламин-4'-сульфокислота является сульфокислотой N-фенилантраниловой кислоты; ее использовали как реагент Для колориметрического определения ванадия (V) [130]; сведений об окислительном потенциале и индикаторных свойствах не имеется.
4,4'-Диаминодифениламин окисляется в темно-синий продукт; в водно-ацетоновой смеси (1:1) стандартный потенциал равен 0,75 В [129]. Это соединение интересно в том отношении, что содержит неотделимые блокирующие группы у обоих углеродов в
198
ГЛАВА VIII
пара-положении, поэтому его нельзя окислить до производного дифенилбензидина. Возможно при этом следующее: а) окисление идет по одному из направлений реакции (68), например с образованием о,о'-изомера; б) окраска обусловливается образованием радикала дифенилимина как в реакции (66); в) механизм окисления такой же, как для вариаминового синего (т. е. 4-амино-4'-ме-токсидифениламина), рассмотренного в главе VIIIA. Последнее, видимо, более вероятно.
Полизамещенные производные дифениламина. Многие производные вариаминового синего с заместителем в положении 2 уже были рассмотрены [1]. Гаммет, Вальден и Эдмондс [140] изучили ряд полиаминосоединений, полученных путем восстановления соответствующих нитросоединений, в том числе 2,4,4'-триаминодифе-ниламин и различные три- и тетр а а минопроизводные, и установили, что ни одно из этих соединений не обладает свойствами окислительно-восстановительных индикаторов и не имеет никаких преимуществ. Наряду с этими соединениями эти авторы изучали 2,4-диамино-4'-метоксидифениламин, который является 2-аминова-риаминовым синим и нашли, что он не имеет преимуществ по сравнению с дифениламином.
^-Замещенные дифениламины. Кноп [146] исследовал N-метил-дифениламин-4-сульфокислоту и нашел, что продукт ее окисления устойчивее аналогичного продукта дифениламин-4-сульфокислоты, хотя потенциал немного ниже. Фрумина с сотр. [130] сообщает о формальном потенциале 0,86 В в 0,5 М серной кислоте. Хотя этот индикатор мало применяется, он, видимо, так же хорош, как незамещенная сульфокислота, и, возможно, имеет преимущества, заслуживающие тщательного изучения.
В. N-АЛИФАТИЧЕСКИЕ БЕНЗИДИНЫ
Замена фенильной группы на алкильную у атомов азота в бензидине ограничивает эффект делокализации в кольцевой системе бензидина, а также уменьшает коэффициент молярной экстинкции и сдвигает максимум поглощения в сторону более коротких волн в видимой области спектра. Окисленным формам N-алкилбензиди-нов приписывается желтый или интенсивный желтый цвет, хотя имеются 'некоторые указания на то, что на промежуточной стадии окисления появляется розовая, малоинтенсивная окраска. Более высокая степень алкилирования, видимо, улучшает устойчивость окисленной формы при избытке окислителя, но в то же самое время замедляет реакцию окисления и обратимость изменения окраски. О механизме реакции соединений, имеющих лишь заместители у атома азота, ничего не известно, но окраска и устойчивость тет-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
199
раалкилпроизводных могут дать некоторую информацию. Возможный механизм окисления свободного основания выглядит так:
Восстановленная форма может быть моно- и дипротонированной, интермедиат — монопротонированным, а протонированию дихинондиимина мешают алкильные группы. По сравнению со схемой (74) для дифенилбензидина атомы азота здесь полностью замещены, и не имеется больше электронов, на которые мог бы воздействовать окислитель. Это, по-видимому, находится в согласии с наблюдаемой устойчивостью окисленной формы N-тетраалкилбензидинов. Исследованные соединения приведены в табл. VIII.14. Алкил- и фенилбензидины получаются, вероятно, из N-алкилдифениламинов; о метилпроизводных упоминалось в предыдущем разделе.
Таблица VIII.14
N-АЛКИЛБЕНЗИДИНЫ
Соединение Потенциал перехода F R rans’ D Максимум поглощения \nax- ™ Литература
N-Метил бензидин N-Этилбензидин N.N'-Диметил бензидин НЫ'-Диэтилбензидин N М.Ы'.И'-Тетраметилбензидин 0,86 (1 М H2SO4) 470 156 156 156 156 156-158
|'7,1'1,1']',Ь1,-Тетраэтил бензидин 1 '^'-Тетраметилбензидин-З-суль- <Ьок.иелота ^М,№,№4етраметил<Ц£'-диметил-оензидин 0,90 (1 М НСЮ^ 0,88 (IM H2SO4) 0,91 (M HC1OJ Е- 0,948 485 156, 157 157 159
200
ГЛАВА VIII
Белчер с сотр. [156] исследовал моно- и симметричные ди-N алкилпроизводные и сообщил о высокой чувствительности их к окислителям и повышенной устойчивости к избытку окислителя Большое внимание было уделено тетра-Ы-алкилированным соеди нениям. Адамс с сотр. [157] исследовал тетраметил- и тетраэтил соединения, а также 3-сульфокислоту тетраметилпроизводного. Он определил потенциалы в момент первого появления желтой окра ски (см. табл. VIII. 14), но заметил также, что примерно за 0,5 мл дэ наступления конечной точки появляется слабая розовая окраска которая служит указанием на приближение конечной точки. По явление желтой окраски происходило, однако, медленно и приво дило к перетитрованию. Тетраэтилпроизводные настолько медленно изменяли окраску, что Адамс с сотр. пришел к выводу о бесполез-ности этого соединения как индикатора. Обратное изменение окра ски происходило тоже медленно, но в случае N,N,N',N'-тетраметил-бензидин-3-сульфокислсты наблюдалось быстрое изменение окраски, а также очень высокий потенциал в момент перехода окраски. Индикаторная ошибка очень незначительна (составляет около> 0,5 мл IO-3 М раствора церия(IV) на 2 капли 0,2%-ного индикатора в 1 М соляной кислоте). Так как окраска окисленной формы этих индикаторов интенсивно-желтая, Адамс отметил, что их нельзя использовать в титровании гексацианоферрата (II), а также солянокислых растворов железа (II). Кульберг и Борзова, используя это соединение в качестве реагента для колориметрического определения хлора, сообщили [158], что максимум поглощения Атят тетраметилпроизводного находится при длине волны 470 нм. Белчер с сотр. [156] подтвердил исследования Адамса, но считает, что-медленное изменение окраски настолько увеличивает степень пере-титрсвания, что этот индикатор нельзя рекомендовать для титрования церий (IV)—железо(II). Однако устойчивость окрашенного продукта окисления заставила их испытать этот индикатор в реакциях титрования церий (IV)—оксалат, но снова медленное изменение окраски оказалось слишком большим недостатком.
М,М,№,М'-Тетраметилбензидин-3-сульфокислота является производным с заместителем в кольце; как и ожидалось, она имеет высокий потенциал. Иорданов и Даев [159] провели исследование аналога толидина И.М.М'.М'-тетраметил-З.З'-здиметилбензидина и сопоставили его с незамещенным толидином (3,3'-диметилбензиди-ном) в качестве реагента для колориметрического определения следовых количеств окислителей. Эти авторы установили, что в реакции окисления обоих соединений участвуют два электрона и два иона водорода, а окисленная и восстановленная формы несут на себе два положительных заряда. К сожалению, в их сообщении отсутствуют существенные данные, так что нельзя оценить справедливость их утверждений. Из приведенных результатов ясно, что в реакции участвует не более 20% от двух ионов водорода, а число
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
201
электронов очень близко к 1,5. Однако они нашли, что наличие четырех метильных групп у атомов азота повышает потенциал индикатора до 140 мВ, а экстраполяция их результатов позволяет определить потенциал индикатора из выражения:
£=l,023 + ^§^lg
[Indox] [Indred]
2,ЗЯТ тт
-^Рн
хотя тангенсы углов наклона существенно отличаются от теоретических значений и не указан состав среды, за исключением того, что это буферный раствор. И окисленная, и восстановленная формы реагента проходят через анионообменную колонку без каких-либо изменений, но прочно удерживаются на катионообменной колонке; это указывает на то, что обе фэрмы должны иметь положительный заряд. Иорданов и Даев определили величину заряда, измерив изменение pH раствора при прохождении через катионообменную колонку, и пришли к выводу, что обе формы несут двойной положительный заряд. Реакцию окисления они описали так:
оранжевая
Значит, восстановленная форма протонируется даже при значениях рН>4. Эти авторы не объясняют свое представление об окисленной форме, но, как показано выше, оба атома азота имеют восемь электронов и все они участвуют в образовании связей. Положительный заряд этой структуры не может образоваться вследствие протонирования или какого-либо другого способа, за исключением разрыва связи. Поэтому надо принять, что это формальное представление бикатион-бирадикала, в котором два электрона с параллельными спинами (или две положительные «дырки») делокализованы по кольцевой системе. Если атомы азота полностью алкилированы, окрашенный продукт окисления долгое время устойчив, так что если бы он был бикатион-бирадикалом, то должен был бы быть исключительно устойчивым и образовываться также в случае других производных бензидина. Те же авторы определили коэффициент молярной экстинкции окисленных форм в зависимости от окислителя; он может быть функцией окислительно-восста
202
ГЛАВА VIII
новительного потенциала окислителя и числа электронов, участвующих в реакции, но приводимые зна“ения е не обоснованы. Значения коэффициентов молярной экстинкции приведены в табл. VIII.15. Опыты, по-видимому, проводились в разбавленной
Таблица VIII. 15
ЗНАЧЕНИЯ КОЭФФИЦИЕНТА МОЛЯРНОЙ ЭКСТИНКЦИИ ОКИСЛЕННЫХ ФОРМ М,М,Ь1',Ь1'-ТЕТРАМЕТИЛ-З.З'-ДИМЕТИЛБЕНЗИДИНА В ЗАВИСИМОСТИ ОТ ОКИСЛИТЕЛЯ
Окислитель В, Л • МОЛЬ—1 • СМ-1 Окислитель В, Л - МОЛЬ—1 • СМ—1
Аи3+ 5- 104 Сг2О?- 4,5 • 103
Се4+ 1,75- 104 С1О3 102
МпОГ 1,75 • 104 ВЮ7 3,3 • ю4
vo; 8 • 103 ю; 4 • 103
серной кислоте, а время проявления окраски в значительной степени зависит от свойств и концентрации окислителя, от pH и температуры и колеблется от 30 до 600 с.
Несмотря на то что индикаторы этого подкласса устойчивы к избытку окислителя, медленное изменение окраски не позволяет использовать их в титриметрии.
Г. БЕНЗИДИНЫ С ЗАМЕСТИТЕЛЯМИ В КОЛЬЦЕ
Исходное соединение бензидин, или 4,4'-диаминодифенил, Кларк [160] включил в ряд исследуемых им диаминосоединений. В области значений pH от 0 до 3 окислительный потенциал бензидина близок к выражению
Е=0,9214- 0,03 Ig-JJ-S!---0,06рН
llncire(jj
Однако поведение бензидина [25] настолько усложняется побочными реакциями, что нельзя оценить его пригодность в качестве индикатора. Введение заместителей в орто-положение к аминогруппе создает некоторую выгоду, несмотря на большую неустойчивость. Время от времени появлялся интерес к 3-моно- и З.З'-ди-замещенным бензидинам. Систематическое изучение замещенных бензидинов провел Белчер с сотр. [156, 161—167]; Рис и Стефен исследовали алкоксипроизводные [149], а Лиль [132] написал обзор по этому вопросу. Большинство определений потенциалов индикаторов было проведено при добавлении значительного коли
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
203
чества индикатора к раствору железа(II), титровании почти до точки изменения потенциала 0,1 М церием (IV) или 0,01667 М бихроматом, дотитровании до конечной точки титрантом, разбавленным в 10 или в 100 раз; за ходом титрования следили потенциометрически, а за изменением окраски индикатора наблюдали визуально. Метод был предложен Кнопом [168] и разработан далее Адамсом с сотр. [157]. Используя два различных окислителя при определении потенциала индикатора, Белчер наблюдал значительную разницу в значениях (более 10 мВ). Соединения, которые были исследованы в отношении индикаторных свойств, приведены в табл. VIII. 16. Потенциалы были установлены при работе с 1,0 М серной кислотой; подробные исследования влияния концентрации кислоты будут даны ниже.
Таблица VIII.16
ИНДИКАТОРНЫЕ СВОЙСТВА БЕНЗИДИНОВ С ЗАМЕСТИТЕЛЯМИ В КОЛЬЦЕ (В КИСЛОЙ СРЕДЕ)
Соединение Потенциал Е, В Окраска окислений формы3 Литература
Бензидин 0,921 В нейтральной среде си- 160
няя
В кислой среде (426) 134
З-Метилбензидин — Желтая 162-165
З-Метокси бензидин 0,82 Красная (435—440) 149
З-Этоксибензидин 0,80 Красная (435—440) 149
З-Хлорбензпдин —• Желтая 167
3 Бромбензидип 0,90 » 167
3,3'-ДиметилбеИзидин 0,873 » 160
0,883 (435) 134, 159
(437) 169
З,3'-Диэтил бензидин •—- Желтая 162—165
З,3'-Диметоксибензидин (см. £о=0,81 Красная (450) 149
табл. VIII.22)
3,3'-Диэтокси бензидин 0,79 Красная (455) 149
З,3'-Дифенилбензидин •— Фиолетово-синяя 170
3,3-Диамино бензидин — Желто-оранжевая (470; 171
3,3'-Дихлорбензидин З.З'-Дибромбензидин е 3,31-103)
0,936 Желто- оранжева я Оранжево-желтая 167 167
2,7-Диаминофлуорен — Желтая 162, 165
К]-12
\=/ \==/
\cZ Н2-
а р
скобках указана длина волны максимума поглощения
204
ГЛАВА VIII
Следует заметить, что все перечисленные в таблице производные имеют заместители в положении 3, т. е. в орто-положении относительно атома азота, за исключением диаминофлуорена, в котором кольца блокированы мостиковой группой. Замещение в положении 2, т. е. в орто-положении к дифенильной связи, приводит к образованию соединений с высокими окислительными потенциалами, окисляющихся с трудом даже в нейтральных средах [162, 172], вероятно из-за того, что заместители в положении 2 мешают окислению вследствие копланарности бензоидного кольца. Эгертсон и Вейс [173] изучали окисление производных бензидина 4-аминодифениламина и n-фенилендиамина при pH 9,4 в водно-ацетоновой среде (1 :1), определяя полярографически потенциалы полуволн. Для окисления брали перекись водорода или гемин — перекись водорода и установили, что нарастание образующейся окраски обратно пропорционально величине потенциала полуволны индикатора, что совершенно ясно. Эти авторы нашли, что заместители повышают потенциал полуволны, особенно если заместитель подвергается воздействию электронов. Однако анализ табл. VIII.16 показывает, что большинство заместителей в бензидине понижает потенциал; только дибромпроизводное имеет более высокий потенциал, чем бензидин.
Совсем не так просто определить механизм окисления. Раньше Белчер [162], так же как и Шленк [174], предполагал, что синий продукт окисления бензидина является молекулярным комплексом мерихиноида и основания (в соотношении 1:1), а впоследствии [144], ссылаясь на Физера [95], стал считать, что диамины ояда бензидинов не подвергаются прямому окислению до дихинондииминов. Затем [167] он сообщил, что бромпроизводные непригодны в титровании оксалата раствсром церия(IV) из-за окисления, которое идет вне стадии образования мерихиноида. Это подтверждает предположение об одноэлектронном окислении до окрашенной формы, и особенно в нейтральной или слабокислой среде. Однако в обширных исследованиях нафтидинов [128, 144] Белчер склоняется к мнению, что существование широкого интервала перехода (120—180 мВ) обусловливается плохой растворимостью индикатора, а повторное обратное изменение окраски сужает интервал перехода [144]; в то же время повышение растворимости индикатора путем сульфирования или замены растворителя приводит к интервалу перехода, соответствующему двухэлектронному окислению. Очень трудно точно установить интервал перехода или число электронов, участвующих в реакции окисления индикатора при методе титрования железа (II) бихроматом или церием (IV) из-за низких переменных токов на электродах в критической области. Прямое титрование раствора индикатора стандартным раствором окислителя связано с еще большими трудностями: даже если индикатор легко растворим, разрушение продукта окисления является огра
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
205
ничивающим фактором. Иорданов и Даев [159] смогли использовать этот метод для полностью N-алкилированного производного, потому что оно оказывается единственным устойчивым соединением. Из этой группы индикаторов наибольшее внимание привлекает 3,3'-диметоксибензидин, или о-дианизидин. Рассмотрение предыдущих работ [85, 127] показало, что окисление в кислых средах идет, несомненно, с участием двух электронов и нет доказательств образования продукта одноэлектронного окисления, значит, оба электрона отрываются одновременно. Кроваво-красный продукт окисления неустойчив, окраска при стоянии постепенно исчезает независимо от наличия избытка окислителя. Имеются некоторые указания, что разложение идет автокаталитически. Хотя у индикатора, перекристаллизованного из воды, не содержащей кислорода, почти не изменяется точка плавления, продукт окисления такого индикатора получается немного бледнее и более устойчивым к дальнейшему окислению. Красная окраска исчезает через 15 ч, и если количество индикатора достаточно, из раствора выпадает шоколадно-коричневый осадок. Бишоп также наблюдал, что исходная восстановительная форма индикатора снова образуется в процессе этого разрушения: после исчезновения красной окраски полностью окисленного индикатора добавка новой порции окислителя приводит к образованию гой же кроваво-красной окраски. По-видимому, в этом процессе снова образуется около одной трети исходной восстановленной формы индикатора. На основании поведения индикатора в ацетатном буферном растворе, химических свойств коричневого осадка и некоторых предыдущих наблюдений по окислению дианизидина, главным образом в неводных средах [175], был предположен механизм реакции разложения. В основном это реакция диспропорционирования, и в принципе она возможна. Однако при детальном рассмотрении первой стадии, в которой подразумевается присутствие неокислен-ного индикатора, реакция диспропорционирования может не идти. Теперь это кажется невероятным, хотя в принципе диспропорционирование остается возможным. Ни го одному процессу регенерации исходного продукта не имелось столько сообщений; по-видимо-му, это явление не ограничивается только о-дианизидином.
При изучении зависимости потенциалов от концентрации ионов водорода [127] было найдено, что формальный потенциал в среде серной кислоты с данными значениями pH имеет следующие значения при 18 °C:
pH 0 0,3 0,74 0,81 1,39
В 0,849 0,829 0,797 0,795 0,755
2Л6
СЛАБА VIII
При предположении, что в окислении участвует только один протон, зависимость потенциала от значения pH изображается прямой линией с тангенсом угла наклона 68 мВ/pH. Рис и Стефен [149] дали потенциалы перехода (первое появление окраски) ряда алкоксипроизводных бензидина в растворах серной кислоты различной концентрации (табл. VIII.17).
Таблица VIII.17 ПОТЕНЦИАЛЫ ПЕРЕХОДА Е ( (В ВОЛЬТАХ) АЛКОКСИБЕНЗИДИНОВ ПРИ 18—20 °C [1491
Соединение [H2SO4]. МОЛЬ Л—1
0.1 0,5 1,0 2,0 4,0
Титрование железа(П) бихроматом
З-Метокси бензидин 0,79 0,83 0,82 0,83 0,84
3-Этоксибензидин 0,78 0,82 0,80 0,82 0,83
3,3'-Диметоксибензидин 0,75 0,79 0,78 0,80 0,81
3,3' Диэтоксибензидин 0,76 0,79 0,79 0,81 0,81
Титрование железа(П) церием (IV)
З-Метоксибензидин — 0,81 0,80 0,83 0,85
3- Этокси бенз идин — 0,80 0,80 0,81 0,85
З.З'-Диметоксибензидин — 0,77 0,76 0,79 0,82
3,3'-Диэтоксибензидин — 0,78 0,75 0,80 0,83
В табл. VIII.18 приведены значения потенциалов перехода для бромпроизводных, полученные Белчером с сотр. [167].
Очень мало индикаторов этой группы нашло широкое применение. 3,3'-Диметилбензидин, или о-толидин, долгое время использовался для колориметрического определения хлора в воде, и интерес к нему продолжается; несмотря на известную канцероген-
Таблица VIII. 18
ПОТЕНЦИАЛЫ ПЕРЕХОДА EtraIK (в ВОЛЬТАХ) БРОМВЕНЗИДИНОВ ПРИ 18 °C [167]
Соединение [H2SO4I, моль л—1
0,5 1.0 2.0 4.0 8.0
З-Бромбензидин 0,892 0,900 0,912 0,920 —
3,3' Дибромбензидин 0,920 0,936 0,954 0,960 0,965
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
207
ность, даже был получен патент на применение его в виде таблеток [176], однако он не был одобрен как индикатор. Белчер с сотр. [165] нашел_ что 2,7-диаминофлуорен лучше о-дианизидина для титрования золота (III) гидрохиноном, но он отдавал предпочтение 3-метил- и 3,3'-диэтилбензидинам. Титрование проводилось в среде соляной кислоты в присутствии KHF2, причем изменение окраски индикатора от желтой или зеленой до бледно-фиолетовой восстановленной формы происходило быстрее при низких концентрациях соляной кислоты, а очень быстро в 0,025 М соляной кислоте. Белчер с сотр. [167] сообщил, что 3-хлор-, 3-бром-, 3,3'-ди-хлор- и 3,3'-дибромпроизводные ведут себя обратимо при титровании железа(II) бихроматом и церием (IV), окисленная форма индикатора получается ярко-желтая или оранжево-желтая, но только бромпроизводные дают четкую конечную точку, а производные хлора действуют слишком медленно. Окраска иона гексахлорфер-рата(Ш) мешает наблюдать конечную точку в среде концентрированной соляной кислоты. Добавка фосфорной кислоты излишняя, результаты получаются с точностью ±0,02%.
З.З'-Диметоксипроизводное (о-дианизидин) легко доступен и применяется чаще других соединений этой группы. Сначала его использовали в титровании железа (II) бихроматом по методу Уикса [177], а затем в определении золота (III) с помощью гидрохинона по методам Поларда [178] и Джемисона и Уотсона [179]. Бишоп [85, 127] тщательно исследовал его применение при титровании цинка гексацианоферратом (II), для тех же целей этот индикатор рекомендован также Фростом [180]; со временем этот индикатор был заменен на 3,3'-диметилнафтидин (см. ниже). Бишоп исследовал также возможность применения о-дианизидина в методах титрования железа (II) бихроматом или церием (IV) и гидрохинона раствором церия (IV). Четкие конечные точки и хорошие результаты были получены при титровании церием (IV), а с бихроматом конечные точки были хуже. При титровании иодида раствором серебра в присутствии следов иода с успехом применялся о-дианизидин [127]. Рис и Стефен [149] исследовали все моно- и дизамещенные метокси- и этоксибензидины, и ни один из них не оказался вполне удовлетворительным при титровании железа (II) бихроматом; поэтому эти авторы предпочли монозамещен-ные производные для работы в среде высоких концентраций серной кислоты; монозамещенные производные оказались лучше, чем дифенил амин-4-сульфокислота в 4 М серной кислоте. При титровании раствором церия (IV) получались гораздо более четкие конечные точки, особенно в случае З^'-диэтоксибензидина. Этот индикатор, по сообщению тех же авторов, дает самую четкую конечную точку в системе титрования цинк—гексацианоферрат(II), а для системы кадмий—гексацианоферрат(II) не пригодно ни одно из алкоксипроизводных, так как они не дают четкую конечную точку.
208
ГЛАВА VIII
Д. НАФТИДИН и ЕГО ПРОИЗВОДНЫЕ
Нафталиновый аналог ‘бензидина 4,4'-диамино 1,1'-динафтил Ьпервые был исследован Еспером [139], который рекомендовал использовать его при титровании железа (II) бихроматом, так как красный продукт окисления резко отличался от зеленой окраски солей хрома(III) и давал более различимую конечную точку. Этот индикатор обратимый, пригодный для титрования в любом направлении, и на него не оказывает влияние присутствие хлорида ртути (II), хотя красная окраска исчезает менее чем за две минуты. Другие случаи применения не исследовались. Однако использованные случаи синтеза [181], представляющие собой усовершенствование старых методов, сомнительны; по ним получали большие количества 2,2'чдиамино-1,1'-динафтил а, из которого путем отщепления аммиака образовывался динафтокарбазол. Окраска окисленной формы позволяет предположить, что копланарность циклической системы невозможна.
На эту систему не обращалось внимания до тех пор, пока Белчер не предпринял тщательного исследования нафтидина [164] и его производных {128, 144, 153, 182—184], из которых лучшим индикатором для титоования гексацианоферратом(II) по методу осаждения оказался 3,3'-диметилнафтцдин. Однако Белчер пришел к выводу, что в других методах титрования, например при титровании раствором бихромата или церия (IV), индикаторы ряда нафтидина не имеют существенного преимущества перед дифенил-амин-4-сульфокислотой, за исключением, может ‘быть, сульфопроизводных нафтидинов при высоких концентрациях кислоты в титровании раствором ванадия (V) [128]. В обширной статье Белчер с сотр. [185] сделал обзор методов синтеза нафтидина и его 3,3'-дизамещенных производных и рассмотрел механизм образования этих соединений из 1-нафтиламинов. Не имея возможности сделать окончательный вывод, он склонен считать, что димеризация идет через образование мезомерной формы свободных радикалов с неспаренным электроном, локализованным у атома углерода в пара-положении по отношению к аминогруппе, аналогично способу, показанному в реакции (68). Поэтому синтезы не однозначны, но конечными продуктами, по-видимому, являются 3,3'-ди-замещенные 4,4'-Д'иамино-1,1'-динафтилы. Хотя возможность участия изомерных соединений в реакциях и возможность индуцирования неустойчивости маловероятны, их нельзя полностью исключить.
Белчер [144] нашел, что окисленные формы этих индикаторов слишком неустойчивы, чтобы можно было определить формальные потенциалы прямым титрованием индикатора стандартным раствором окислителя. Однако 3,3'-диметилпроизводное и дисульфо-нат можно титровать до окисления на 95% и 75—80% соответст
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 209
венно прежде чем наступит глубокое разрушение индикатора Путем прямого титрования раствором церия (IV) при 18 °C удалось получить формальные потенциалы. которые приведены в табл. VIII. 19. Белчер определял также потенциалы первого по-
Таблица VIII.19
ФОРМАЛЬНЫЕ ПОТЕНЦИАЛЫ ИНДИКАТОРА, ПОЛУЧЕННЫЕ ПРЯМЫМ ТИТРОВАНИЕМ ЦЕРИЕМЦУ) ПРИ 18 °C [144]
Соединение pH
0 0.28 0,50 0,79 1,0
3,3'-Диметилнафтидин 0,776 0,755 0,726 0,714 0,711
3,3/-ДиметилиаФтиДинДисУльФ°’ кислота 0,838 0,796 0,802 0,806 0,784
явления окраски по методу Адамса [157] и нашел, что при титровании железа (II) бихроматом они на 40—60 мВ ниже. Белчер пришел к заключению, что сульфирование повышает формальный потенциал и потенциал перехода, тогда как метилирование понижает их значения; на основании этого он предположил, что нафти-динмоносульфокислота должна быть лучшим индикатором в условиях высоких концентраций серной кислоты, чем дифениламин-4-сульфокислота, хотя окраска нафтидинмоносульфокислоты изменяется медленнее, а окисленная форма ее быстро разрушается.
В работе [128] Белчер с сотр. повторил некоторые определения потенциалов перехода, а также изучил значительное число других производных, о получении которых говорилось выше [185]. Для окисленных форм соединений, имеющих общую структурную формулу
характерны следующие окраски:
R Окраска R Ок раска
Н СН3, С2Н5, я-С3Н7, СРп'СзН” S°3H Бордово- красная Пурпурно-красная Зеленовато-синяя с6нБ no2 nh2 Фиолетовая Реакция не идет Переходящая зеленая, коричневый осадок
14—818
210
ГЛАВА VIII
Из этих соединений 3,3'-динитропроизводное не окислялось даже церием (IV) в хлорной кислоте, а 3,3'-диаминопроизводное слишком неустойчиво к окислению, поэтому в дальнейшем эти два соединения не исследовались. 3,3'-Дисульфокислота оказалась достаточно устойчивой при проведении исследований лишь в серной кислоте, концентрация которой превышает 2 моль-л-1, в которой она не диссоциирует. Потенциал перехода в момент первого появления окраски и при полном проявлении окраски (по Стокдалю [133]) были определены при помощи метода титрования железа (II) бихроматом и церием (IV). Часто получалось значительное
Таблица VIII.20
ПОТЕНЦИАЛЫ ПЕРЕХОДА Efr.,Ils (В ВОЛЬТАХ ОТНОСИТЕЛЬНО Н. В. Э.) НАФТИДИНА И ЕГО ПРОИЗВОДНЫХ ПРИ 20 "С
Соединение* [H2SO4], моль л—1
0,1 0.5 1.0 2,0 4,0 8.0
первая окраска полная окраска ДЕ, В
Нафтидин (а) 0,80 0,81 0,79 0,83 0,04 0,78 0,76 0,74**
(б) 0,79 0,79 0,76 0,74
Нафтидинмоносульфокио лота** (а) 0,84 0,84 0,84 0,82 0,81 0,79
З.З'-Диметилнафтндин (а) 0,69 0,71 0,71 0,85 0,14 0,71 0,70 0,70**
(б) 0,70 0,70 0,70 0,67
З.З'-Димстилиафтидинди-сульфокислота** (а) 0,82 0,81 0,80 0,79 0,77 0,75
З.З'-Диэтилнафтидин (а) 0,70 0,72 0,72 0,90 0,18 0,72 0,72
(б) 0,72 0,71 0,72 0,69
3,3'-Ди н пропилиафтидин (а) 0,70 0,72 0,72 0,90 0,18 0,72 0,72
(б) 0,72 0,71 0,72 0,69
3,3' Диизопропилнафтидин (а) 0,73 0,75 0,75 0,91 0,16 0,75 0,74
(б) 0,74 0,74 0,74 0,72
3,3'-Дифенилнафтидин (а) 0,75 0,76 0,76 0,94 0,18 0,76 0,76
(б) 0,76 0,76 0,75 0,73
Нафтидин-3,3'-дисульфо- (а) — — 0,83 0,91 0,08 0,83 0,83
кислота (б) 0,86 0,86 0,85 0,85
З.З'-Диметсчсннафтидин (а) —
(б) 0,74 0,74 0,88 0,14 0,73 0,70
* (а) — Титрование железа(П) раствором бихромата; (б) — титрование железа(П) раствором церия (IV)
** При 18 °C.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
211
расхождение результатов определения двумя титрантами. При титровании бихроматом проявляется в различной степени индукционный эффект, который совсем отсутствует при титровании церием (IV), так же как в случае применения производных дифениламина. В табл. VIII.20 приведены полученные результаты наряду с более ранними результатами для других производных [144]. Интервал перехода (ДЕ для 1 М серной кислоты) от первого появления окраски до ее полного развития варьирует в пределах от 40 до 180 мВ. При быстром повторном обратном изменении окраски интервал перехода уменьшается на несколько милливольт [144], а Белчер предполагает [128], что большие значения интервалов перехода определяются растворимостью индикатора, и приводит в качестве доказательства то, что более растворимой нафтидинди-сульфокислоте соответствует больший интервал перехода. Прямое титрование 3,3'-диметилнафтидина в среде, в которой он хорошо растворяется (смесь из 4 М серной кислоты и ледяной уксусной кислоты в соотношении 1:1), соответствует двухэлектронному окислению, а интервал перехода равен 50 мВ [128]. Таким образом, Белчер считает, что в окислении этого подкласса индикаторов участвуют два электрона. Автор не касается механизма окисления, но предполагает, что окраска появляется вследствие делокализации, и ссылается на пространственные факторы. На основании результатов нельзя сделать вывод о том, что в реакции окисления участвуют ионы водорода, но, по-видимому, за исключением сульфокислот, по меньшей мере один ион водорода участвует в реакции.
Белчер с сотр. провел также спектрофотометрическое исследование некоторых нафтидиновых индикаторов, но при использовании прибора с ручным управлением разложение индикатора приводит к серьезной ошибке. Окисление проводилось гипохлоритом, который добавлялся в сернокислый раствор индикатора. Установлено, что спектры не зависят от природы окислителя, а следовательно, и от значения коэффициента молярной экстинкции; повышение концентрации серной кислоты тоже не влияет на коэффициент молярной экстинкции, но смещает максимум поглощения в сторону более длинных волн; особенно заметно это смещение окраски при визуальном наблюдении. Сам нафтидин дает окраску, которая за 10 мин полностью исчезает, а окраска 3,3'-ди-метоксинафтидина — за 20 мин. 3,3'-Диметоксинафтидин обладает чувствительностью к свету, длина волны .которого находится в области максимума поглощения, поэтому трудно определить его максимум поглощения, а коэффициент молярной экстинкции — просто невозможно. При концентрации серной кислоты выше 4 моль-л-1 окраска нафтидиндисульфокислоты вдвое неустойчивее окрасок Диалкил- и дифенилнафтидина; при более низких концентрациях серной кислоты окраска нафтидинсульфокислоты тоже очень не-Г4*
212
ГЛАВА VIII
устойчива. При длине волны максимума поглощения исходные значения оптической плотности растворов диалкил- и дифенилпро-изводных, равные соответственно 0,2 и 0,8, понижаются на 0,025— 0,045 за 5—6 ч. В табл. VIII.21 приведены некоторые коэффициенты молярной экстинкции, вычисленные при предположении, что молярная масса соединений не изменяется при окислении.
Таблица VИ 1.21
СПЕКТРАЛЬНЫЕ ХАРАКТЕРИСТИКИ ОКИСЛЕННЫХ ФОРМ НЕКОТОРЫХ ПРОИЗВОДНЫХ НАФТИДИНА [128]
Соединение е, Л-МОЛЬ—1 см—1
1 М H2SO4 [ 4 М H2SO4
Нафтидин 530 535 2,9-104
Метилнафтидин 550 560 3,0.104
Этилнафтидин 550 560 3,0-Ю4
н-Пропилнафтидин 550 560 3,1-Ю4
Изопролилнафтидин 550 560 2,6.10s
Метоксинафтидин 615 — —
Фени^нафтидин 570 575 2.1.104
Нафтидинсульфокислота — 560 —
Приведенные значения ниже значений, найденных для коэффициентов молярной экстинкции производных бензидина [134], что свидетельствует о меньшей делокализации электрона, чем ожидалось. Низкий коэффициент молярной экстинкции окисленного ди-фенилнафтидина находится в согласии с экспериментально установленной чувствительностью в отношении окислителей [128], так что, как ни странно, делокализация снова ограничена. Эти наблюдения позволяют предположить, что пространственные факторы препятствуют копланарности кольцевой системы. Стерическое влияние приводится также [128] для объяснения большей устойчивости 3,3'-дизамещенных производных по сравнению с устойчивостью самого нафтидина: по-видимому, заместители экранируют аминогруппы, которые чувствительны к окислению.
Результаты исследования индикаторов ряда нафтидина, очевидно, находятся в согласии с механизмом окисления, по которому два электрона участвуют в одной стадии для образования окрашенного продукта, который, возможно, является бикатион-биради-калом. Точно еще не выяснена роль иона водорода. Отсутствие данных об интермедиате и продуктах разложения приводит к тому,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
213
что рассмотрение реакции разложения имеет чисто теоретический1 характер.
Белчер [164] применил сначала нафтидин при титровании цинка гексацианоферратом (II) и нашел, что этот индикатор лучше 3,3'-диметоксибензидина (о-дианизидина), а затем испробовал в этом же методе титрования 3,3'-диметилнафтидин [153]. Он сопоставил различные индикаторы в титровании кадмия, кальция и индия гексацианоферратом (II) [182] и установил, что 3,3'-ди-метилнафтидин лучше остальных индикаторов; за ним следуют нафтидин, о-дианизидин и дифенилбензидин. Наконец, он изучил нафтидинмоносульфонат и 3,3'-диметилнафтидиндисульфонат [183]’ в методах титрования цинка и кадмия. В то же самое время Белчер применил все указанные выше индикаторы в других методах титрования и нашел, что конечные точки при титровании железа (II) бихроматом получаются менее четкими, чем с дифениламином, не так хороши, как в случае дифениламина-сульфокислоты при титровании железа (II) ванадием (V), и что эти индикаторы почти непригодны для титрования церием (IV). Диметилнафтидиц и диметилнафтидиндисульфокислота были использованы для титрования малых количеств галлия гексацианоферратом(II) [184].
Нафтидин-3,3'-дисульфокислота совсем не ведет себя как индикатор при титровании гексацианоферратом (II), а в случае ди-фенилчафтидина изменение окраски идет медленно и нечетко [128]. Лучшим индикатором является 3,3'-диметилнафтидин, особенно при титровании по методу осаждения раствором гексацианоферрата (II).
ЗАКЛЮЧЕНИЕ
В первом варианте рукописи в этом месте было дано обобщение свойств и механизмов реакций производных бензидина. Из-за недостатка однозначных данных все, касающееся механизма, носило теоретииеский характер, но вывод был сделан в пользу двухэлектронного процесса окисления, несмотря на большую привлекательность теории одноэлектронного процесса, который был описан в общих чертах в начале раздела. Однако после этого П| шилась возможность провести фундаментальное исследование [134], в котором был уточнен вопрос о числе электронов, участвующих в стадии окисления индикатора, и выяснена дальнейшая стадия окисления или деструкции. Эти сведения поступили слишком поздно, чтобы можно было переделать написанное; поэтому вместо обычного заключения предлагается следующее изложение.
В то время, когда проводилось большинстве цитированных ра-бот (например, [149] >и [128]), скоростные двухлучевые спектрографы с автоматической записью еще не были широко распространены, а построение спектра от руки для определения максимума
214
ГЛАВА VIII
поглощения и коэффициента молярной экстинкции было сопряжено с большими трудностями. Не были известны также спектрометры электронного парамагнитного резонанса (ЭПР). Вариаминовый синий и его производные составляют одну из недавно разработанных групп индикаторов, и этому способствовали исследования методом ЭПР, проведенные Эрдеем с сотр., как сообщил Оттоуэй в первой части этой главы. Исследование проводилось с очищенными и индивидуальными соединениями типа
и с аналогом динафтила при помощи скоростной сканирующей спектрофотометрии, определения поглощения окисленной формы во времени при длине волны максимума поглощения и методом электронного парамагнитного резонанса.
Число электронов, участвующих в реакции индикатора, установили с помощью серии растворов, содержащих равные количества индикатора с добавкой церия (IV), количество которого варьировало от 0 до 4 эквивалентов. Спектры этих растворов снимали сразу же после смешивания, а затем через различные промежутки времени, чтобы обнаружить изменение спектров во времени. В качестве среды использовали серную кислоту в различных концентрациях. Установлено, что при увеличении количества окислителя от нуля до двух эквивалентов интенсивность полосы поглощения индикатора в ультрафиолетовой области понижается до нуля, а в видимой области одновременно возрастает от нуля до максимума; полоса поглощения растворенного церия (III) в ультрафиолетовой области тоже возрастает до максимума. Изменение в поглощении находится в точно линейной зависимости от количества добавленного окислителя. При добавлении более двух эквивалентов окислителя поглощение индикатора остается постоянным, а появляется полоса поглощения, обусловленная растворенным церием (IV), которая постепенно увеличивается. Во всех случаях максимум поглощения окисленной формы индикатора достигается при двух эквивалентах окислителя с ошибкой ±1%. Ни в одном случае не наблюдалось на спектрах появления интермедиата или продукта одноэлектронного окисления индикатора. Не доказано также образование продукта дальнейшего окисления в присутствии избытка окислителя. Неизбежен вывод о том, что окисление в среде серной ки'слоты представляет собой одностадийный двухэлектронный процесс. Результаты спектрального исследования обобщены в табл. VIII.22.
Полосы поглощения восстановленной формы в УФ-области и окисленной формы в видимой области близки по форме. Когда
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
215
Таблица VI 11.22
СПЕКТРАЛЬНЫЕ ХАРАКТЕРИСТИКИ НЕКОТОРЫХ ПРОИЗВОДНЫХ БЕНЗИДИНА И БЛИЗКИХ ЕМУ СОЕДИНЕНИИ В 2 М СЕРНОЙ КИСЛОТЕ
Соединение Indred Indox Смещение спектра, ккал моль-1 "ф ю X О ю
\пах» нм е, л моль—1-СМ—1 \пах> нм Е, Л МОЛЬ—1- СМ— 1
Дифенил 248 1,2-Ю4
Бензидин 2,03-104 426 6,95-104 50,8 3,3
3,3' Диметилбензидина 250 1.8-104 435 6,5-Ю4 48,6 3,6
248 1,8-104 438 49
3,3'-Диметоксибензидин 252 1,28-Ю4 454 3,1-104 49 2,4
287 8,1.10s 510 2,3-104 41,5 2,8
М.М'-Дифевилбензидин 2536 3.12.1046 560 5,0-Ю4 61,9 1,6
Дифениламин 220° Разрушается 565 4,5-104 — —
N-Фенилантраниловая 256° 524 3,0-104 — —
кислота
N-Метилдифениламин- 320г 2,4-Ю4 511 4,4-Ю4 62 1,7
4-сульфокислота 294° 1,4-IO4®
Нафтидин 282 400 7,0-10» 80
294 1,1-104д 527 1.8-104 43 1,6
3,3'-Диметилнафтидип Сложный спектр 543 3,5-104 — —
а В 10-* М серной кислоте.
б В концентрированной серной кислоте.
®В виде фениламина.
ГВ бензидиновой форме после восстановления Ind ох цинковой пылью.
д УФ-спектр с двумя максимумами, е при 294 нм.
заместители X=Y=H, то в большинстве случаев при окислении наблюдается батохромный сдвиг около 17 000 см-1 и увеличение коэффициента молярной экстинкции примерно в три раза. Для N-фе-нилшроизводных батохромный сдвиг возрастает до 21 500 см-1, а коэффициент молярной экстинкции уменьшается вдвое. Это свидетельствует о том, что при окислении структура молекулы не претерпевает существенного изменения, но .средняя энергия возбужденного состояния уменьшается или, что более вероятно, увеличивается средняя энергия основного состояния на 50 ккалХ Хмоль-1 в случае X=Y=H и на 62 ккал-моль-1 для N-фенилпро-извсдных. Эти данные подтверждают представление о том, что окисленной формой является бикатион-бирадикал в триплетном состоянии. Все индикаторы проявляют батохромный сдвиг от повышения концентрации серной кислоты без изменения коэффициента молярной экстинкции, что уже показал Белчер для соединений ряда нафтидина [128].
Повторное снятие спектров (через различные промежутки времени) растворов, содержащих только два эквивалента окислителя,
-'216
ГЛАВА VIII
показало, что поглощение окисленной формы понижается и вновь появляется спектр восстановленной формы, хотя не всегда его можно отделить от спектра церия (IV). Имеется некоторое указание на существование индукционного периода и совсем точное доказательство того, что разрушение ускоряется во времени, т. е. происходит автокаталитически, и гораздо быстрее в растворе, на который воздействовали одним эквивалентом окислителя, чем в полностью окисленном растворе. Во всех случаях раствор в конце концов становится фактически бесцветным, а при использовании дифеничбензидина и N-фенилантраниловой кислоты образуется зеленый осадок. Если на такой разложившийся раствор снова подействовать окислителем, то сразу же появляется спектр, идентичный по виду исходному с1вежеокисленн'ому раствору, но поглощение будет меньше; это доказывает, что вновь образовалась исходная восстановленная форма индикатора.
Повторное снятие спектров растворов, обработанных более чем двумя эквивалентами окислителя, приводит к подобным результатам, нс разложение окисленной формы идет медленнее из-за того, что происходит окисление вновь образующейся восстановленной формы. Однако поглощение церия(IV) уменьшается сильнее, чем можно было бы объяснить повторным окислением; следовательно, окислитель расходуется на другую реакцию. Более того, если к растворам, обработанным избытком окислителя и доведенным до полного разрушения видимой окраски, добавить еще окислитель, то сразу же появляется окраска индикатора; это означает, что восстановленная форма образуется вновь даже при наличии избытка окислителя. Это явление наблюдается до тех пор, пока количество добавляемого окислителя не достигнет двадцати двух эквивалентов. Обычно при втором проведении окисления индикатора окислитель расходуется быстрее, чем при первом, а по израсходованному количеству видно, что должен был произойти разрыв кольцевой системы индикатора.
Все индикаторы, следовательно, неустойчивы по своей природе, т. е. разлагаются в отсутствие избытка окислителя, а также и при его избытке. Если характерное для окисленного индикатора разложение представить в виде
mbidox----*- D + nbidred,
тде D — главный продукт разложения, то избыток окислителя расходуется на три реакции:
а) повторное окисление вновь образовавшейся восстановленной формы (Indred) до окисленной формы (Indox);
б) прямое окисление Indox до D;
в) воздействие на D с образованием продуктов расщепления.
Следовательно, нельзя определить стехиометрию непосредственного разложения от добавки известного избытка окислителя,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
217
соответствующего различным соотношениям (например, четыре эквивалента окислителя при т=2 и п=1), так чтобы после разложения в растворе не было бы вновь образовавшегося Indred; в этом случае преобладает расход окислителя по реакции (в). Указания относительно стехиометрии можно получить, если разложить полностью раствор индикатора путем обработки двумя эквивалентами окислителя, разделить этот раствор на аликвотные части, добавить к ним ряд инкрементов окислителя, долить до определенного объема и измерить поглощение при длине волны максимума. Кривая поглощения, построенная относительно количества окислителя, выраженного в эквивалентах, достигает максимума, когда вся вновь образовавшаяся восстановленная форма индикатора Indred будет окислена и останется постоянной. Хотя окисление Indred до Indox идет быстро, присутствие в растворе продукта разложения D вносит некоторую ненадежность, которая обусловливается также реакцией (.в) и, возможно, реакцией (б). В целом, однако, отдается предпочтение соотношению 2:1 (т=2, п=1), нс совсем не исключается другое соотношение, как, например, 5:2 или 3: 1. Использование более мягкого окислителя, чем церий(1У) например ванадия (V), может уменьшить или элиминировать протекание реакции (в) и этим упростить положение.
Скорость разложения Indox изучали по изменению максимума поглощения во времени; результаты представлены в табл. VIII.23.
В отсутствие восстановленной формы Indred или избытка окислителя скорость разложения зависит от природы заместителей X, Y, Z и от концентрации серной кислоты. Во всех случаях скорость разложения понижается с увеличением концентрации серной кислоты. Для дифенилбензидина, например, эта зависимость следую щая:
[H2SO1], % масса/объем нм Период полураспада, мин
18 560 188
22 565 200
36 570 445
63 585 1300
Понижение скорости разложения можно объяснить или уменьшением концентрации воды, или увеличением степени протонирования частиц индикатора с повышением концентрации серной кислоты. В ряде работ, рассмотренных в предыдущих разделах предполагалось, что повышение концентрации серной кислоты оказывает стабилизирующее действие.
218
ГЛАВА VIII
Таблица VIИ.23
ПЕРИОД ПОЛУРАСПАДА Indox В ОТСУТСТВИЕ ИЗБЫТКА ОКИСЛИТЕЛЯ
И ПРИ НАЧАЛЬНОМ ОТСУТСТВИИ Indred В 2 М СЕРНОЙ КИСЛОТЕ ПРИ 26 "С И НОВОЕ ОБРАЗОВАНИЕ Indred ПОСЛЕ ПОЛНОГО ЕГО РАЗЛОЖЕНИЯ
Соединение Период полураспада, мин Вновь образовавшийся Indred. %
Бензидин 500 80
3,3'-Диметилбензидин 200а 264 46
3,3'-Диметоксибензидин 190 60
N,N'-Дифенилбензидин 188 46, 50, 51
Дифениламин 96
N-Фенилантраниловая кислота (начальное разложение до зеленой окраски) 6—10 100
М-Метилдифениламин-4-сульфокислота 1706 55°
Нафтидин 23 40
З.З'-Диметилнафтидин 80 35, 40
а В 10-4 М серной кислоте.
® Третичный амин сначала окисляют полностью, сразу Же восстанавливают цинковой пылью и тогда считают, что это М.Ы'-диметнл-М.Ы'-днфенилбензндннсульфокислота.
Примечание: Период полураспада бензнднна и нафтнднна намного короче прн дневном освещении (50 и 2,5 мин соответственно).
Присутствие Indred ускоряет разрушение окраски, так что частично окисленный индикатор разлагается быстрее, чем полностью окисленный, и наиболее четко это выражено, когда окислена половина всего количества индикатора. Этот факт можно объяснить влиянием образующегося молекулярного комплекса Indox-Indred, семихинона. Кроме того, когда окислена половина индикатора, интенсивность спектра ЭПР медленно возрастает до максимума, g-фактор этого спектра равен почти точно 2, что указывает на наличие свободного радикала с одним неспаренным электроном; затем интенсивность спектра ЭПР уменьшается приблизительно с такой же скоростью, как и скорость уменьшения светопоглощения. Эти выводы, однако, следует рассматривать с большой осторожностью. Существует также некоторое доказательство в пользу иного механизма окисления в средах, содержащих уксусную кислоту, который, возможно, включает стадию одноэлектронного окисления: это толуе служит указанием на специфичность механизма в уксусной кислоте; однако, прежде чем сделать какие-либо оп-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
219
ределепные выводы, надо провести более тщательные исследования.
Скорость разложения Indox, а значит, устойчивость окисленной формы зависят от природы заместителей X, Y и Z. Сульфирование приводит к заметному снижению устойчивости, как было отмечено в предыдущих разделах. Вообще говоря производные динафтила (или нафтидина) менее устойчивы, чем производные дифенила (бензидина), и, чем больше у них заместителей, тем они устойчивее. Устойчивость повышается в порядке X=Y=Z=H<X=Y= = Н<Х = Н<полностью замещенные.
Разложение не соответствует точно кинетическому порядку реакции, так что стехиометрия реакции разложения не поддается кинетическим измерениям. Разложение 'ближе всего к реакции первого порядка, но эта реакция автокаталитична; один из продуктов реакции, видимо, действует как катализатор. Предположение, что автокатализатором является Indre(j, подкрепляется тем, что скорость реакции разложения заметно возрастает при добавке Indred к полностью окисленному раствору; однако это определение не однозначно. Реакцию разложения можно описать следующим уравнением:
d [Indox] [][nd^ + [продукт] _|_ k2X
В случае дифенилбензидина в 2 М серной кислоте k\ = 0,0023 мин-1, а й2 = 0,0082 мин-1. Это не может соответствовать приемлемой стехиометрии для реакции разложения, и поэтому возможно, что в разложение входит мономолекулярная стадия, определяющая скорость реакции, или же эта стадия предшествует реакции разложения. По-видимому, можно предположить, что этой стадией является релаксация триплетного бикатион-бирадикала в дихинондиимин
и что дихинондиимин участвует в процессе разложения. Спектры Дают слабое и сомнительное доказательство образования дихинон-Диимина; разложенному раствору соответствует очень широкая полоса поглощения в видимой области, совсем немного поднимающаяся над нулевой линией (как при образовании очень малого количества диспергированного осадка). При концентрациях, применяемых в спектрофотометрии (обычно около 10-5 М, за исключением труднооастворимых соединений, подобных дифенилбензиди-ну и диметилнафтидину и странному продукту из N-фенилантра-
220
ГЛАВА VIII
ниловой кислоты), не получаются видимые осадки, они образуются в более концентрированных растворах.
Сигнал ЭПР полуокисленного индикатора появляется медленно и затем исчезает по мере разложения Indox, как уже было отмечено. Этот сигнал имеет вид необычно широкой полосы, вероятно, из-за влияния растворителя. Если количество окислителя, добавленного к образцу, увеличивается до двух эквивалентов, этот сигнал исчезает и вместо него появляется слабый сложный спектр, который можно приписать, но не с полной уверенностью, бирадикалу с двумя отдельными электронами с параллельными спинами. Этот сигнал изменяется, но не пропадает при исчезновении окраски, и это до некоторой степени доказывает, что продукт разложения является свободным радикалом. Большая часть этой работы была проведена с очищенным дифенилбензидином в среде концентрированной серной кислоты.
Исследование в основном касалось бензидина и его производных, но, так как исследователи не располагали К.И.И'.М'-тетраза-мещенным соединением, опыты проводились с его предшественником N-замещенным дифениламином, поскольку ожидалось, что из него может образоваться производное бензидина при добавлении в раствор первых двух эквивалентов окислителя (на два моля третичного амина). Это оказалось не так. При окислении серии растворов N-метилдифениламин-1-сульфокислоты посредством увеличения инкрементов церия (IV) непрерывно образовывалась конечная окраска без какого-либо намека на промежуточное образование производного бензидина. Поглощение N-метилдифениламина в УФ-области уменьшалось, а поглощение Indox увеличивалось линейно с количеством добавленного окислителя, достигая соответственно нуля и постоянного максимума после добавки двух эквивалентов окислителя на один моль третичного амина. Спектры имели характерную изобестическую форму. И в этом случае окисление является одностадийной двухэлектронной реакцией (эквивалентной четырем электронам на две молекулы при предположении, что бензидин образуется в качестве интермедиата), и не имеется никакого доказательства существования промежуточной стадии. Будет показано, что при восстановлении окрашенного соединения образуется N-тетразамещенный бензидин, а не третичный амин; однако сильный окислитель прямо окисляет третичный амин до бикатион-бирадикала.
Точно такие же явления наблюдались в случае дифениламина и N-фенилантраниловой кислоты; оба эти соединения окислялись непосредственно до бирадикала без промежуточного образования производного бензидина. Были испробованы более слабые окислители с тем чтобы убедиться, не ограничивается ли такое прямое окисление окислителями вроде церия (IV) или марганца(VII). В присутствии железа (III) окраска не наблюдалась, а ванадий(V)
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
221
окислял N-метилдифениламин в одну двухэлектронную стадию до окрашенной окисленной формы Indox. Поэтому следует сделать вывод, что это — общее явление для производных дифениламина (и третичного амина). Следует еще раз подчеркнуть, что окисление симметричных ди- и трифениламинов может дать пять возможных продуктов: о,о'-, о,п'- и щп'-бензидины и два семидина, а если фе-нлламин несимметричен (например, дифениламин-2-карбоновая кислота, N-фенилантраниловая кислота), то заместители могут появиться у обоих конечных фенилов, по одному на конечном фениле и на кольце бензидина или оба на кольце бензидина, давая в общем пятнадцать возможных продуктов, хотя в любом случае один из них может преобладать. Энергичное восстановление цинковой пылью окрашенного lndox сразу же после его образования приводит к образованию продукта, отличного от предшествующего фениламина: в УФ-области наблюдается батохромный сдвиг спектра. Однако этот продукт окисляется путем одноэлектронной реакции до исходного фениламина или путем двухэлектронной реакции до предполагаемого бензидина, и при этом получается такой же спектр, как для Indox. Восстановление Indox, полученного любым способом (или из фениламина, или из бензидина), приводит к образованию бензидина. Этот факт объясняет, почему, против ожидания, поправки на индикатор, например на дифенилбензидин, не так малы, как на дифениламин. Уже подчеркивалось выше, что отсутсгвуют сведения о выделении промежуточного бензидина, за исключением случая восстановления конечного продукта окисления и то только для дифенилбензидина.
Стоит рассмотреть странное поведение N-фенилантраниловой кислоты. Она непосредственно окисляется двумя эквивалентами окислителя в синевато-красную форму Indox, но сразу же начинает разлагаться с образованием зеленого продукта, который в конце концов выпадает в осадок. На спектре наблюдается увеличение полосы при длине волны 436 нм при одновременном уменьшенш-i полосы Indox при 520 нм; это изменение идет быстро, период полураспада Indox составляет 30 мин. Спектры дают довольно хорошую изобестическую точку, которая смазывается вследствие полного разложения зеленых и синевато-красных частиц; этот процесс идет значительно медленнее. Следует отметить, что N-фенилантраниловая кислота содержит группы
I I I, II
—N—С— С—С— О-, I I
которые могут образовывать шестичленные устойчивые циклы хелатов с ионами металлов.
222
ГЛАВА VIII
Основную реакцию окисления, проводимую в сернокислой среде, можно представить «следующим образом:
где Indox представляет собой бикатион-бирадикал в триплетном состоянии. В случае незамещенных бензидинов (X=Y=Z = H) коэффициент молярной экстинкции для Indox имеет более высокое значение (табл. VIII.22); заместители в положении Z вызывают слабый батохромный сдвиг и небольшое снижение е; если только заместитель Z не содержит кислорода. Если же Z — алкокси- или карбоксигруппа, тогда батохромный сдвиг ясно выражен, а спектры алкоксипроизводных в виде форм Indred и Indox расщепляются на два пика. Заместители в положении Z, по наблюдению Белчера, экранируют чувствительную к окислению аминогруппу и этим значительно повышают устойчивость. Заместители в положении X и/или У еще больше увеличивают устойчивость. Продукт окисления окрашен в желтый или желто-оранжевый цвет; если же Z не содержит кислорода, он становится красным, или, если имеется ароматический заместитель с сопряженными связями, окраска становится фиолетовой или синей. Такое же смещение происходит в ультрафиолетовой области спектра восстановленной формы.
Реакция разложения регенерирует Indred и образует слабоокра-шенный труднорастворимый продукт разложения. Регенерация Indred является обратной реакцией окислению, если пренебречь участием иона водорода:
Indox4-2e Indred
Электроны, необходимые для этой реакции, должны откуда-то отрываться, и, так как Indox может расходовать па себя окислитель, он, очевидно, и является источником электронов:
xlndox---> j/D + ze,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
223
где D главный продукт деструкции, а при окислении его самого избытком окислителя образуются бесцветные продукты распада. Следовательно, самопроизвольное разложение является диспропорционированием, несколько примеров которого были приведены ранее:
plndox---> qD + rlndred
Поскольку значение р минимально равно 2 для реакции диспропорционирования, а разложение не идет по второму порядку, мо-номолекулярная стадия, определяющая скорость реакции, должна предшествовать диспропорционированию. В связи с этим уже говорилось о релаксации триплетного состояния, и, возможно, этот процесс будет ускорен путем обмена энергией с Indred.'
дихинондиимин (ДХДИ)
Окончательную реакцию можно тогда представить в виде
alndox + адхДИ --> D + clndred,
где соотношение коэффициентов должно быть (а+Ь)/с. Соотношения 2:1, 5:2, 3:1 и 4:1 приведут к регенерации соответственно 50, 40, 33 и 25% Indred- Основной продукт разложения D не был выделен и исследован, но, даже если бы это удалось сделать, трудно представить себе, что D не претерпевает дальнейших изменений в ходе исследования. Поэтому любое предположение о природе D носит, безусловно, теоретический характер, но, так как существует некоторая связь между природой заместителей X, Y, Z и устойчивостью окрашенного Indred, может быть стоит пересмотреть имеющиеся возможности.
Заместители в положении Z, по-видимому, экранируют аминогруппу до некоторой степени, но при попытке сформулировать процесс разложения надо учитывать заместители X и Y. Стоит также напомнить, что скорости разложения уменьшаются с повышением концентрации серной кислоты, так что протонирование может иметь известное влияние. Более того, сам дифенил не окисляется, а распадается деструктивно, так что реакционными центрами этих индикаторов являются атомы азота, что не могло бы иметь места при делокализации.
Если заместители в положении X и У не являются атомами водорода, то продукт окисления в виде бикатион-бирадикала или в виде дихинондиимина не имеет локализованных пар, доступных протонированию, и нельзя сформулировать дальнейшее окисление
224
ГЛАВА VIII
любой формы без разрыва связей или отрыва одного из заместителей X или Y:
Адамс с сотр. [157], Белчер с сотр. [156] и Иорданов с сотр. [159] придерживаются единого мнения об устойчивости окисленных тетраалкилбензидинов, но устойчивость не определена количественно, так что подразумевается некоторая неопределенная устойчивость. Кноп [146] сообщил, что продукт окисления N-метилди-фениламин-4-сульфойислоты более устойчив, чем продукт окисления неметил-ированной сульфокислоты дифениламина. Если продукт этого индикатора имеет заместители Х = СН3, Y=C6H4-p-SO3H и Z = H, тогда его устойчивость сопоставима с устойчивостью о-ди-анизидина (X=Y=H, Z = OCH3) и при его разложении регенерирует восстановленная форма. Это, по-видимому, обусловливается либо влиянием сульфогруппы, которая понижает устойчивость, либо каким-то другим влиянием, например отрывом метильной группы (окраска показывает, что ароматическая группа в положении Y сохраняется). Тогда легко понять, почему N-алкилированные производные устойчивы и дальнейшее окисление их невозможно. Трудно найти приемлемое объяснение разложению N-алкилфенил-производного.
Если в положениях X или Y имеется водород, тогда бикатионбирадикал и дихинондиимин могут протонироваться и дать электроны, способные окисляться, особенно дихинондиимин. Частицы Indred, Indox И дихинондиимин могут существовать в виде свободных оснований или могут быть моно- или дипротонированы:
дипротонированный бикатион-бирадикал протонированный
Ind red или протонированный дихинондиимин
Indox
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
225
Если на основании регенерации Indre(i реакция диспропорционирования является 2: 1 диспропорционированием, то стадией, определяющей скорость этой реакции, может быть стадия образования основания дихинондиимина, а диспропорционирование можно выразить следующим уравнением:
Indox + дхди-------->
где первичный продукт разложения D является бикатион-биради-калом дихинондиимина. Эта частица, имеющая такое невероятное строение, должна быть очень реакционноспособной и будет полимеризоваться путем воздействия на кольца, но маловероятно, чтобы образовывались линейные полимеры, если учесть отталкивание зарядов. Немного легче представить себе диспропорционирование 3: 1, но оно противоречит регенерации Indred, идущей на 50%. Это может произойти посредством отрыва единственного электрона от дихинондиимина с образованием монорадикала, который димеризуется, и получается группа
II II YN—NY,
но место нахождения заряда остается спорным. Однако 2 Indox могут сочетаться с образованием гидразина, но он будет тетраза-мещенным и не сможет окислиться, если не отделится заместитель Y.
Если в положениях X и Y находится водород, то для диспропорционирования в соотношении 2: 1 снова требуется образование бикатион-бирадикала дихинондиимина при Y, замещенном на Н, а это открывает ряд других возможностей. Диспропорционирование в соотношении 3: 1 допускает образование гидразина, в данном случае окисляющегося до азосоединения, которое может изомеризоваться в димер дихинондиимина, или окисляющегося до азоксисоединения. Образование линейного тримерного бис-гидразина может дать соотношение 5:2 и на 40% регенерировать Indred- Полимеризация может идти также по типу феназина.
'Вряд ли нужно перечислять все возможности, так как вопрос Должен рассматриваться только теоретически до тех пор, пока не будут проведены дальнейшие исследования продукта D. Тем не менее следует отметить, что устойчивость окисленной формы повышается, если атомы водорода в положениях Z, Y и X замещаются последовательно; кроме того, устойчивость повышается с увеличением концентрации серной кислоты и регенерация Indred достигает 50%. Все это указывает на то, что окисление Indox до продукта D является двухэлектронным пооцессом.
15~ 918
226
ГЛАВА VIII
2. ТРИФЕНИЛМЕТАНОВЫЕ И ДРУГИЕ КРАСИТЕЛИ
Многие из применяемых кислотно-основных, металлохромных и окислительно-восстановительных индикаторов можно найти среди торговых препаратов красителей различного типа. Однако эти вещества производятся из-за их красящих свойств, а не для применения в качестве химических реагентов. Следовательно, они бывают очень редко чистыми соединениями точно известного состава. Часто они представляют собой либо смеси изомерных соединений (или различных соединений), либо соли. Кроме того, они могут содержать неорганические вещества или органические добавки. Аналитику, использующему их в качестве реагентов в колориметрии, хорошо известно, что они могут в значительной мере отличаться составом от партии к партии. Несмотря на это, можно эмпирически применять индикаторы и получать удовлетворительные результаты, но по указанным выше причинам трудно или даже невозможно научно исследовать их свойства. Что касается более фундаментальных вопросов, то в современных руководствах по органической химии умалчивается важная тема красителей. В книгах тридцатых годов [94] проводилось обсуждение на основе новых в то время концепций резонанса, но известно, что эта теория и теория валентных связей являются в лучшем случае лишь некоторым приближением. Рилей [186] описал трифенилметановые и ксантеновые красители и слегка затронул основные вопросы, но нужно изучить более глубоко литературу, чтобы получить данные об окраске и строении красителей, а многое из того что имеется в литературе, не касается самих красителей. Уотсон, который разработал [187] хиноидную теорию Армстронга [188], вкратце изложил предыдущие теории цвета вплоть до 1918 г. [189]. Прейфер [190] в 1910 г. установил, что главным хромофором является трехвалентный углерод; исследование этого вопроса продолжил Дилтей [191]. Уизингер [192] отнес ауксохромы к катионным хромогенам, а антиауксохромы — к анионным хромогенам. Интересно отметить, что по мнению Штиглица [193], краситель является промежуточным окисленным состоянием, восстановление разрушает окраску, мягкое окисление снова восстанавливает окраску, а жесткое окисление часто разрушает его снова. Эту активность Штиглиц приписывает активности ауксохромных и хромофорных групп в молекуле. Шако [194] подчеркивает важность интенсивности и длины волны поглощения в развитии классической теории поглощения света. Бари [195] первый описал старые представления с позиции квантово-механического резонанса; Склар [196] показал важность возбужденных состояний. Эти исследования продолжил Ферстер [197]. Полинг [198] описал квантово-механический метод 'исследования цвета красителей в 1939 г., и тогда же Льюис и Кальвин [199] дали впервые окончательную квазиклас-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
227
сическую трактовку, на основе которой развиваются современные теории. Обзор работ по этим вопросам вплоть до 1947 г. написал Маколл [200], а Хамер [201] сделал в 1950 г. обзор по цианиновым красителям. Работа Маррела [202] основывается на теории молекулярных орбиталей. Больше не имеется литературы по красителям, которую можно было бы рекомендовать. Ни в одной из более поздних монографий по этим вопросам [203] не упоминается об окраске и ее происхождении. Обычно пользуются снабженной примечаниями классификацией красителей, так чтобы их можно было идентифицировать и сопоставлять с другими соединениями, имеющими подобное строение. Обычно для этой цели используют «Британский указатель красителей» (В. С. I.) [204—206]. Однако при исследовании современных торговых препаратов красителей часто оказывается, что среди перечисленных красителей, представляющих собой одно определенное соединение (кроме соединений с неизвестной структурой и смесей) имеются различные образцы иногда даже одного и того же производителя, которые имеют совершенно различные свойства и окраски. Как покажет краткое знакомство с такими указателями, название и марка качества красителя не дают сведений о его строении, но такие указатели о строении красителей необходимы. Беглый просмотр описанных методов приготовления показывает, что немногие из них обеспечивают получение чистого продукта, и, хотя приводится единая структура, продукты могут оказаться смесями. Если данный торговый образец красителя при испытаниях покажет, что обладает свойствами индикатора, нельзя с уверенностью принять, что все красители, перечисленные в указателе под одним и тем же номером, имеют те же свойства; также нельзя утверждать, что другие продукты под тем же номером не обладают свойствами индикатора, если у одного из них таких свойств не оказалось. В этом можно убедиться при просмотре прилагаемого списка красителей. Следовательно, нельзя давать только номер или обозначение соединения в указателе: надо указать также фактическое название, фабричную марку и производителя, а в некоторых случаях желательно даже номер партии.
Учитывая сказанное, было бы нецелесообразно заниматься подробным обсуждением механизма реакции индикаторов. По окислительному механизму индикаторов с высоким потенциалом имеется очень мало работ. Многие красители принадлежат к классам соединений, рассмотренных Оттоуэем в части А этой главы, и поэтому нет необходимости рассматривать их снова. К ним относятся производные индиго, азины, оксазины, тиазины и другие соединения. Многие индикаторы, хотя и не включены в классификацию индикаторов, являются производными замещенных бензидинов или дифениламинов, а механизм их реакций может частично или полностью зависеть от поведения этих частей в красителе, возможно., 15*
228
ГЛАВА VIII
в частности, в азокрасителях, в которых азогруппы экранированы от окислительного воздействия. То, что азосоединения можно окислить с уничтожением окраски, как, например, случай с метиловым оранжевым, понимается как окислительное воздействие на азогруппу с образованием сначала азоксигруппы, а затем нитрозопроизводных фрагментов молекулы вследствие разложения. Если X, Y и Z — ароматические части, тогда для диазосоединений возможна последовательность
О t X—N=N—Y—N=N—z ----> X—N=N—Y—N=N—Z ---->
О О о
t t t
---> X—N=N— Y— N=N—Z -> X-NO+ ONY—N=N—Z >
---> X—NO + ON—Y—NO 4- Z—NO
Это необратимый процесс; имеется несколько из применяемых индикаторов, которые ведут .себя подобным образом. Однако некоторые красители, принадлежащие к азосоединениям, оказываются обратимыми. Это можно иногда объяснить присутствием других групп, как, например, части дифениламина в мстаниловом желтом:
Однако соединение, которое считается только диазокрасителем и не имеет других функциональных групп,
обладает полной обратимостью в одних случаях, необратимостью в других, а в третьих совсем не реагирует. Реакции простого взаимодействия ионов как раз проявляют высокую специфичность, так что, если исходить из чистых веществ, краситель может содержать 90% или более продукта и 10% изомера, в котором присоединение происходит в орто- вместо пара-положения к аминогруппе. Если принять, что данные указателя правильны, тогда или сведения, представляемые производителем, должны быть ошибочны, или некоторые образцы содержали много примесей различного характера, как, например, трифенилметановый краситель. Такие свойства
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
229
наблюдались у ряда красителей: диаминогеновый синий NA (С) изменяет окраску от бихромата, церия (IV) и брома; диаминогеновый темно-синий (С) обратимо изменяет окраску ют церия (IV), необратимо от брома, не окисляется бихроматом; диаминогеновый синий ВВ (С) изменяет окраску от бихромата, необратимо изменяет окраску от церия (IV), разлагается бромом; диазоиндиго синий 2RL (By) плохо изменяет окраску от бихромата, церия (IV) и брома; диазоиндиго синий BR (By) совсем не окисляется; хлоразол диазо-синий 2RS (ICI) обратимо изменяет окраску от перманганата и брома, слабо изменяется от церия (IV), не окисляется бихроматом.
Трудно также представить себе, как антрахиноны могут проявлять обратимые или необратимые индикаторные свойства сами по себе если они не были сначала восстановлены: возможно, что из-за заместителей система хинон—анион-радикал—гидрохинон относится к области потенциалов, в которых идет титрование железа (II) церием (IV). Некоторые из реакционноспособных антрахиноновых красителей содержат вторичные фениламиногруппы, фе-нилантрахинониламины, которые могут вступать в реакции как дифениламин. Здесь опять же нельзя исключить наличие активных примесей.
Важнейшей группой красителей, обладающих свойствами индикаторов с высоким потенциалом, является группа производных ди-и трифенилметанов. Часто эти исходные соединения бесцветны, но при окислении образуют дифенилметил-, дифенилметилен- или три-фенилметил-радикалы, которые являются цветными красителями. Некоторые из этих соединений изменяют окраску при дальнейшем окислении, а некоторые также изменяются в зависимости от значения pH. Кноп [207—210] был первым, кто использовал индикаторные свойства этих красителей, но он не рассматривал механизм окисления, а только предположил на основании подобия окраски некоторых три- и дифенилметилпроизводных, что трифенилметил-соединения при окислении разрушаются с отрывом одного кольца и образованием соответствующего дифенилметиленового соединения. Смит [2П] исследовал некоторые из них в качестве индикаторов в броматометрии наряду с другими красителями, неустойчивыми к действию хлора. Другие исследователи тоже проявили известный интерес [23—30, 212—217] а Адамс с сотр. [157] начал исследование альфазурина G [С. 1.712 (NAC)], набросав схему изучения механизма, которое он не закончил. Брэзайер и Стефен [218] провели систематическое исследование и попытались увязать свои наблюдения со структурой индикаторов, но не касались механизма окисления, отметив, что данные об этом процессе неодинаковы. Не давая никаких оценок этой мастерски выполненной работе, особенно учитывая предыдущие замечания о чистоте и идентичности рассматриваемых веществ, целесообразно пред-
230
ГЛАВА VIII
ставить образование красителя в следующем виде: (С6Н6)3СН (С6Н6)3С + Н+ + е
лейкосоединение краситель
Краситель (С6Н5)зС по своей природе является радикалом, а ароматические группы включают по меньшей мере один, а обычно не менее двух таких радикалов, которые имеют аминогруппу или гидроксил в na^a-положении. Брэзайер и Стефен [218] отметили, что соединения с гидроксильными группами в таком положении не являются хорошими индикаторами, что подтверждается автором этой главы; поэтому внимание будет обращено на красители с двумя n-аминогруппами. Радикал красителя можно представить в виде двух предельных форм: радикала трехзалентного углерода, который не несет заряда, но имеет неспаренный электрон, и его ме-зомерной формы, в которой электрон отрывается от атома азота, чтобы насытить формальную валентность, оставляя при этом азот с неспаренным электроном:
Фактически, конечно, это модельные формы валентных связей, а электроны делокализованы по молекулярным орбиталям. Если, однако, один из атомов азота протонировать, делокализация радикала будет ограничена, и можно ожидать, что появится гипсохром-ный сдвиг, который обычно наблюдается в красителях, изменяющих свою окраску при подкислении раствора. Поскольку центральный атом углерода окислен полностью, более глубокое окисление должно задеть атомы азота. В результате реакции одноэлектронного окисления получится бикатион-бирадикал:
«основание»
протонированная форма
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
231
Можно ожидать, что увеличится степень делокализации и появится батохромный сдвиг, который часто наблюдается. В некоторых красителях одна или две фенильные группы бывают замещены на дифениламиногруппу, которая, как в случае азокрасителей, участвует, по-видимому, в механизме реакции окисления; установлено, что такие красители пригодны для иодатометрических методов титрования [218]; мы даже нашли, что спирторастворимый синий 2В (BDC) (С. 1.689), который содержит две такие группы, совсем не дает индикаторных реакций. Брэзайер и Стефен [218] высказали мнение, что соединения с двумя аминогруппами в пара-положении и простые заместители в кольцах, по всей вероятности, обладают приемлемыми индикаторными свойствами; этот вывод требует подтверждения, однако соединение с пара-аминогруппами на всех трех кольцах обычно понижает устойчивость к окислению; этому имеются некоторые доказательства. Но простейшее из таких соединений, у которого аминогруппы совсем не защищены, три-(п-аминофеиил) метил (парарозанилин) почти не реагирует или совсем не реагирует с сильными окислителями. Следовательно, довольно трудно объяснить наблюдения при проведении опытов, не пытаясь представить механизм реакции.
ХАРАКТЕРИСТИКА КРАСИТЕЛЕЙ В КАЧЕСТВЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ИНДИКАТОРОВ
Сначала красители, и в частности класс трифенилметанов (ТФМ) как индикаторов, использовались для титрования церием (IV) и для получения четкой конечной точки в перманганато-метрии; однако ни один из них, по наблюдению Бишопа, не был пригоден в методе титрования бихроматом. После применения трис-(1,1С фенантролин)железа(II) для периметрии дешевые красители вызывали постоянный интерес. Возможность применять их при тигровании церием (IV) в 'среде хлорной кислоты стимулировала дальнейшие исследования [217]. Однако, помимо описаний применения, было проведено только четыре основных исследования красителей в качестве индикаторов. В хронологическом порядке это работы Кнопа, Смита, неопубликованная работа Бишопа, а также работы Белчера и Стефена. Первые три из них удобнее привести в виде таблиц и комментариев, тогда как последняя, опубликованная позднее предыдущих работ и выполненная на более высоком научном уровне, будет рассмотрена более подробно. Хотя многие красители реагируют обратимо с окислителями, их окисленная форма либо редко остается постоянной значительное время, либо неустойчива к избытку окислителя. Поэтому определение формальных потенциалов и спектрохимических характеристик связано с трудностями. Точное определение формального потенциала посредством тщательного (а потому медленного) прямого
ТРИФЕНИЛМЕТАНОВЫЕ КРАСИТЕЛИ, ИССЛЕДОВАННЫЕ КНОПОМ [209]
Таблица VIII.24
Тривиальные названия3 Номер по' В. С. I. (1924 г.) £trans, В Окраска^ Примечание
нейтральная среда кислая среда окисленная форма
Бриллиантовый темносиний (J) Темно-синий (J) (В) Акроноловый бриллиантовый синий (BDC) 664 1.01 Синяя (614) Зелено-желтая [слабая полоса в фиолетовой области] Желто-красная (490) [несимметричная полоса в фиолетовой области] Хорошая обратимость. Четкое изменение окраски, устойчивое
Ксиленовый синий AS (S) Азуровый синий Z (К) 673 1,02 Синяя (638) То же Розовая (517) Хорошая обратимость. Очень четкое изменение окраски, быстрое возвращение к исходной; титровать до первого изменения окраски
Ксиленовый синий VS (S) Эриоглауцнн супра (G) Азуровый синий VX (К) 672 1,09 Синяя (639) Желтая [слабая полоса в фиолетовой области] Розовая (509) Хорошая обратимость. Очень четкое изменение окраски, устойчивое
Ксиленовый цианол FF (S) Цианол FF, АВ, GG, С, V VN, BN, ВВ, BSB,H(C) Циашхл экстра FF (StD) Кислотный синий 6G (С) 715 0,99 Синяя (615) То же Розовая (496) [465] Хорошая обратимость. Очень четкое изменение окраски, устойчивое
Патентованный синий А (М) Альфазурин A (NAC) Патентованный синий А (А) (К) Бриллиантовый кислотный синий А (By) 714 0,99 Синяя (637) Зеленовато-желтая [слабая несимметричная полоса] Розовая (515) Хорошая обратимость. Четкое изменение окраски, быстрое возвращение к исходной; титрование до первого изменения окраски
Прочный кислотный синий A (GrE)
Дисульфииовый синий А (BDC)
Карминово-синий А (М) Китоновый синий N (J) Нептуновый синий В (В) Тетрацианол А (С) Ксиленовый синий A (S)
Сетоглауцин О (G) Сетоглауцин (G) (StD) Новый прочный зеленый
ЗВ (J)
Родулиновый синий 6G (By)
Сетопалин конц. (G)
Сетоцианин супра (G)
Сетоцианин О (G)
Цианин В (М)
Цианоловый прочный зе-
658
713
1,00 Синяя [631]
1,06 Синяя [635]
0,99 Синяя [614]
1,01 Синяя (632)
1,03 Сине-зеленая (642)
Желтая [слабая по- Желто-красная лоса в фиолето- (499) вой области]
Очень хорошая обратимость. Четкое изменение окраски
Желтая Оранжевая (509) [485] Хорошая обратимость. Четкое изменение окраски, устойчивое
Зеленовато-желтая [614] [слабая полоса в фиолетовой области] Желто-красная (490) Хорошая обратимость. Четкое изменение окраски, устойчивое
Желтая [слабая несимметричная полоса в фиолетовой области] Оранжевая (505) Очень хорошая обратимость. Четкое изменение окраски, устойчивое
Желтая [слабая полоса в фиолетовой области] Розовая (506) [477] Очень хорошая обратимость. Четкое изменение окраски, устойчивое
ьэ
Тривиальные названия3 Номер по В. С. I. (1924 г.) Е trans. В
нейтральная среда
Эриоглауцин A (G) Эриоглауцин G, RB, В, JB, V, Р, супра X, коиц. (G) Альфазурин FG (NAG) Азуровый синий AEG (К) Нептуновый зеленый BR (В) Патентованный кармнно-во синий АЕ, АЕ экстра (М) Кислотный синий ЕС (L) Кислотный бриллиантовый синий EG (By) Тетрацианол 1570J (С) (М) 671 0,99 Синяя (631)
Эриозеленый В (G) Эриозеленый экстра (G) Основной прочный зеленый V (К) Этонический прочный зеленый 3G (JC) Китоновый прочный зеле ный V (М) Ксиленовый прочный зе леный В (S) 735 1,0 Синевато-зеленая (644)
Продолжение табл. VIII. 24
Окраска® Примечание
кислая среда окисленная форма
Зеленая (6Q1) [ела- Синевато-красная Очень хорошая обрати
бая полоса в сине-фиолетовой области] (518) [631] мость. Четкое изменение окраски, довольно устойчивое
Желтая (несимметричная полоса в сине-фиолетовой области) Оранжевая (506) Очень хорошая обратимость. На изменение окраски в конечной точке требуется не- сколько секунд; затем окраска устойчива
Трифенилметановые красители, имеющие меньшее применение
Бирюзовый синий ВВ 661 (By)
Виктория синий В высококонцентрированный (В)
729
Синяя (627)
Зеленовато-желтая
Красно-желтая (503)
Четкое изменение окраски, очень хорошая обратимость, но окраска малоустойчива, в конечной точке быстро возвращается к исходной. Хороший индикатор
Виктория синий 4R высококонцентрированный (В)
690
Зеленый для шерсти S (G)
737
Синяя (619) [568] Зеленая Коричнево-красная (неустойчивая полоса в зеленой области) Четкое изменение окраски, но неустойчивое, переходит в серо-зеленую. Во время титрования подвергается сильному воздействию. Почти не применяется
— Фиолетово-синяя (593) [539] Сине-зеленая (630) Красноватая, потом коричнево-желтая (неустойчивая полоса в зеленой области) Хорошее н обратимое изменение окраски, но при титровании подвергается сильному воздействию. Почти не применяется
— Зеленовато-синяя (638) Желтоватая Красно-желтая (502) Хорошая обратимость. Четкое изменение окраски, неустойчивое. Довольно хороший индикатор
ьэ оо о Тривиальные названия3 Номер по В. С. I. (1924 г.) £trans> В
нейтральная среда
Кислотный зеленый GV (М) 666 — Сине-зеленая (620)
Кислотный фиолетовый 5В (G) 698 — Фиолетовая (592) (541]
Кислотный фиолетовый 6BN (В) 717 — Сине-фиолетов ая (619) [532]
Кислотный фиолетовый 7В (В) 702 — Фиолетово-синяя (605) [551]
Метиловый фиолетовый В чистый (G) 680 — Фиолетовая (587) [535]
Продолжение табл. VIII.24
Окраска^ Примечание
кислая среда окисленная форма
Слабая зеленовато-желтая Оранжево-желтая (491) Четкое изменение окраски, неустойчивое. Сильно окисляется во время титрования. Довольно хороший индикатор
Снне-зеленая (633) Оранжево-желтая (482) Четкое изменение окраски; после окисления обратимость теряется
Голубая Синевато-красная Быстро окисляется при титровании после четкого изменения окраски. Слабая обратимость
Сине-зеленая (642) Фиолетово-красная (неустойчивая полоса в зеленой области) Сильно окисляется во время титрования. Окраска быстро становится серой. Обратимость слабая. Почти не применяется
Зеленовато-желтая Оранжево-желтая Четкое изменение окраски. Очень сильно окисляется во время титрования. Окраска в конечной точке быстро исчезает и необратима
Прочный зеленый экстра (By)
691
Прочный кислотный синий В (By)
733
Эриовнридин В супра (G)
667
— Синяя (622) Желто-зеленая Красновато-желтая Четкое изменение окраски, но окраска быстро возвращается к исходной. Постепенно окисляется во время титрования
— Синяя (617) Голубая Сине-красная (очень быстро исчезающая полоса в зеленой области) Сильно окисляется во 'время титрования. Если вносить в раствор перед самой точкой эквивалентности, дает резкое изменение окраски
— Зелено-синяя (637) Травянисто-зеленая (638) Коричнево-красная (нечеткие полосы в зеленой и синефиолетовой областях) Хорошая устойчивость во время титрования. Четкое изменение окраски, медленное возвращение к исходной. Довольно
хороший индикатор
Эриоцианин А супра (G) 699
— Синяя (614) Т равянисто-зеленая Коричнево-красная
(636) (5Ю)
Хорошая устойчивость во время титрования. Четкое изменение окраски, неустойчивое. Довольно хороший индикатор
а Сокращенные названия производителей приведены согласно приложению к табл. VIII.26. ьэ ® В круглых скобках даны значения длин волн максимумов поглощения интенсивных по-лос, а в квадратных скобках — более слабых полос.
м Таблица VI11.25
КРАСИТЕЛИ, ИССЛЕДОВАННЫЕ СМИТОМ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНДИКАТОРОВ Ё БРОМАТОМЕтРИИ [211]
Кргсятель Номер по В. С. I. (1924 г ) Тип красителя [2041 Окраска Примечание
вода НС1
Хри-оидин R 21 Моноазо Желтая Красная Окраска обесцвечивается от восстановителей; восстанавливается в конечной точке, затем разрушается
Радужный оранжевый G 27 Оранжевая Желтая Пригоден прн умеренных значениях pH и температуры
Хромотроповый 6В 56 » Фиолетовая Красная Ярко-красная окраска переходит в бесцветную при умеренных значениях pH и низкой температуре
Бордо 88 » Красная Красная Пригоден при умеренных значениях pH и температуры
Диаминовый розовый 128 » Непригоден при низких значениях pH и низкой температуре
Амарант 184 » » » Пригоден при умеренных значениях pH и низкой температуре
Бриллиантовый пун- 185 Алая Не разлагается
новый 5R
Нафтоловый сине- 246 Диазо Синяя Синяя Пригоден в обычных условиях
черный Красная Пригоден при умеренных значениях pH и температуре кипения
Конго коринфский 375 » »
Бензоазурин 3R 471 » Фиолетовая Фиолетовая Слабая окраска, но хорошая конечная точка
Малахитовый зеленый 657 Трифеиилмета-новый Зеленая Красновато- желтая Пригоден при умеренных значениях pH и температуре кипения
Прочный КИСЛОТНЫЙ 667 То же Синяя Зеленая Пригоден только при низких значениях pH
зеленый В
Фуксин 678 » Красная Желтая Пригоден только при низких температурах
Метиловый фиолетовый 683 » Фиолетовая Коричневая Пригоден при низких значениях pH и температуре кипения
Кислотный фиолетовый 698 » » Зеленая Пригоден при низких значениях pH и высоких температурах
Фосфин 793 Акридиновый Оранжевая Оранжевая Пригоден при высоких значениях pH и низких температурах
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
239
титрования потенциометрическим методом невозможно, поэтому прибегали к методу Адамса и Хамакера, рассмотренному в разделе о производных бензидина. Однако даже этот метод имеет свои трудности, так как Белчер с сотр. показал, что сообщенные Кно-пом значения потенциалов имеют значительную ошибку. Индикаторные ошибки при прямом титровании красителя очень малы; их можно определить путем сопоставления с результатами потенциометрического титрования в точно определенных экспериментальных условиях.
Кноп [207—210] первый провел систематическое исследование индикаторных свойств красителей и на основании предварительных данных отобрал двадцать семь трифенилметановых соединений для более тщательного изучения [209]. Из них он выделил в качестве лучших первые двенадцать, которые обобщены в табл. VIII.24. Опыты проводились титрованием 0,01667 М раствора железа(II) перманганатом в 0,65 М серной кислоте. В таблице первое название красителя соответствует тому индикатору, с которым проводили опыты; другие названия, по-видимому, заимствованы в качестве синонимов из книги Шульца «Таблицы красителей», изданной в 1918 г.
Надо отметить, что Кноп использовал очень сильный окислитель, поэтому экстраполяция на другие образцы кроме фактически испытанных, сомнительна, а спектральные данные и значения формальных потенциалов следует считать приблизительными. Совсем недавно использование формальных потенциалов вызвало недоверие, хотя эти величины широко применялись в течение многих лет; в этом направлении работали Белчер и Стефен, о чем будет сказано ниже.
Следующее систематическое изучение провел Смит [211] в поисках внутренних индикаторов для броматометрии [219—222]. Так как при прямом титровании мышьяка (III) или сурьмы(III) в соляной кислоте броматом гелиантин разлагался, Смит искал краситель, менее устойчивый к хлорированию. Установлено, что красители с номером 5 по B.C.I. [204] легко разрушаются, и предпочтительнее работать с красителями, имеющими красную и желтую окраску. Подробные данные об этих красителях приведены в табл. VIII.25.
Из этих красителей Смит и Блис [211] отдавали предпочтение бриллиантовому пунцовому 5R (185), нафтоловому сине-черному (246) и фуксину (678), но использовали фуксин, хризоидин R(21), бордо (88) и нафтоловый сине-черный при титровании основным роматом ртути(II) [219]. Смит и Мей [220] предпочитали растить с красителями 88, 185 и 246 при титровании мышьяка(III) и сурьмы(III) броматом, причем указывали на необратимость индикаторов и советовали вносить больше индикатора незадолго До установления конечной точки. Смит и Уилкокс [221] исполь
240
ГЛАВА VIII
зовали амарант, бриллиантовый пунцовый и нафтоловый сине-черный при титровании йодатом по методу Эндрьюса и утвержда--ли, что ни иод, ни монохлорид иода не действуют на индикатор, который разр} шается от избытка йодата в следовых количествах. В частности, амарант и бордо, а в некоторой степени бриллиантовый пунцовый и нафтоловый сине-черный, стали считать (наряду с гметиловым красным) индикаторами, которые разлагаются при использовании их в броматометрии; позже в их число вошел п-эток-сихризоидин [223], а фуксином пренебрегали до тех пор, пока в 1944 г. Бишоп не ввел в практику розанилин. Все эти индикаторы, за исключением n-этоксихризоидина, необратимы. Юзл [224] и Шулек [225] нашли что а-нафтофлавон, который не является красителем, представляет собой обратимый индикатор для броматометрии; это соединение образует коричневатый осадок со свободным бромом, похожий на систему крахмал—иод. Белчер предложил [226] применять краситель хинолиновый желтый (С. I. 801) в качестве обратимого индикатора на бром, но, хотя он и обратим, окраска получается очень бледная и изменяется два раза, а поправка на индикатор очень высокая. Тем не менее его применяют как индикатор для предварительного и приблизительного определения конечной точки перед точным титрованием в присутствии розанилина. Можно сделать так, чтобы розанилин стал обратимым индикатором [227, 228], но тогда конечная точка не такая четкая и поправка на индикатор больше, чем в случае необратимого индикатора.
Шулек и Шомодьи [229] синтезировали и испытали ряд азосоединений, подобных n-этоксихризоидину, и установили некоторую взаимосвязь между структурой и индикаторными свойствами; однако, хотя некоторые из соединений обладали такими свойствами, ни одно из них не оказалось лучше п-этоксихризоидина.
В 1944 г. мы получили доступ в большую библиотеку красителей, которая впоследствии была уничтожена. Нам удалось изучить многие сотни соединений [230]. Хотя мы по возможности не занимались теми красителями, которые были уже испытаны раньше другими исследователями, многие красители были неизбежно вновь открыты под различными именами. При дальнейшей проверке в «Указателе красителей» [204] было обнаружено, что часто несколько исследованных различных образцов, иногда до двадцати, являются одним и тем же соединением. Иногда некоторые из этих образцов давали те же или почти те же реакции, но в большинстве случаев различия были значительны: пример этому уже был описан. Готовили примерно 0,1%-ные растворы красителей и наблюдали изменение окраски при подкислении соляной и серной кислотами. Затем проводили грубое титрование в присутствии 0,5 мл раствора красителя (см. рубрику в табл. VIII.27). Исследовали также прямое воздействие различных окислителей на краситель
ОКИСЛ ИТЕЛЬНО-ВОССТАНОВ ИТЕЛЬНЫЕ ИНДИКАТОРЫ
241
в кислой среде и в нескольких случаях наблюдали индукционный эффект при титровании бихроматом. Лучшие индикаторы были отобраны для дальнейшего исследования их поведения в условиях тщательного и точного титрования; при этом в качестве контроля использовали потенциометрию. Чтобы исследование было полным, постарались получить точные значения формальных потенциалов, но, как и ожидалось, их получить не удалось. Такие работы не были опубликованы, в надежде что позже методы будут усовершенствованы для проведения таких измерений и тогда можно будет дать полную оценку красителям. До сих пор эта надежда не осуществилась. Исследованные красители обобщены в два списка: в первый (табл. VIII.26) входят красители, не проявляющие окислительно-восстановительных индикаторных свойств, во втором (табл. VIII.27) содержатся красители изменяющие свою окраску при добавке одного или более окислителей. Вместо описания окрасок в таблице дано субъективное суждение о качестве изменения окраски с указанием устойчивости и обратимости индикатора; об устойчивости индикатора судят по изменению степени окрашивания или по обесцвечиванию. Однако в случае, например, хинолинового желтого [226] вывод о разрушении индикатора по обесцвечиванию ошибочен. Между реакциями с перманганатом и церием (IV) наблюдалось странное несоответствие: очень многие соединения не вступают в реакцию с перманганатом даже в присутствии железа(III) или железа(II), а реагируют с церием(IV).
Все эти исследования проводились с тем, чтобы найти во-первых, индикатор для метода титрования бихроматом и во-вторых, для метода титрования броматом, в то время как несколько индикаторов Кнопа были пригодны для титрования церием (IV), хотя ни один из них не был особенно примечательным. Из данных табл. VIII.27 видно, что многие красители при титровании железа (II) бихроматом в большинстве случаев изменяют свою окраску плохо; можно считать, что это изменение не так хорошо выражено как окажем у эриозеленого при титровании церием (IV). Необратимые или обратимые в слабой степени индикаторы не представляют интереса для титрования бихроматом. Из оставшихся были выбраны шесть лучших индикаторов и испробованы в нескольких титрованиях бихроматом, включая сюда определение железа (Ш) после обработки оловом (II) и хлоридом ртути(II). Из этих индикаторов пять показали преимущества в титровании при большом разбавлении (0,001667 М. бихромат), но не в такой степени, как в случае производных бензидина. Такими индикаторами оказались нафтоловый зеленый В9211К (BDC) (определенный П0 „С I- П°Д номером 5, что сомнительно) дураноловый бриллиантовый синий В 300 порошок (ICI), дураноловый бриллиантовый фиолетовый В 300 порошок (ICI), дураноловый бриллиантовый сини СВ 300 порошок (ICI) (ни один из них не указан в В. С. I. за
16—818
Таблица VII 1.26
КРАСИТЕЛИ, ОБЛАДАЮЩИЕ СЛАБЫМИ ИЛИ ВООБЩЕ НЕ ОБЛАДАЮЩИЕ СВОЙСТВАМИ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ИНДИКАТОРОВ ПРИ ВЫСОКИХ ПОТЕНЦИАЛАХ
(Красители, отмеченные звездочкой, изменяют окраску как кислотно-основные индикаторы. В скобках сокращенно указаны фнрмы-производнтели, список которых дан в приложении к таблице)
Краситель Номер по В. С. I. (1924 г.)
* Азидиновый бордо (CJ) 376
* Азидиновый желтый СР (CJ) 365
Азидиновый желтый 5G (CJ) 813
Азидиновый зеленый 2В (CJ) 593
Азидиновый зеленый 2G (CJ) 594
Азидиновый коричневый М (СJ) 420
Азидиновый оранжевый G (СJ) 478
Азидиновый прочный желтый G (CJ) 620
Азидиновый синий BA (CJ) 502
Азидиновый синий ВХ (CJ) 472
Азидииовый черный BHN (СJ) 401
Азиновый синий паста (WDC) 860
Азо-желтый RMC (МС) 145
Азо-оранжевый NA (MLB) 69
Азо4>убиновый(Ьеу), (ААР), (CD), (SCI), ,(FB), (GrE); 179
A i(C); экстра (NAC); G экстра конц. R (NCW); S (Sch); SI кони (StD)
Азодиаминовый желтый R (MLy) 812
Азокислотный рубиновый (CJ), i(WDC); R (K) 179
Азокислотный фуксин (RF) 179
Азоминовый прочный желтый D, 2F (JBS) 814
Азофлавин (MC); RS (В); 2R (WDC); 3R конц. (tM) 145
* Акридиновый оранжевый RS (ICI) 788
Акроноловый желтый Т (BDC) 815
Акроиоловый желтый TCS (ICI) 815
Ализадиновый коричневый М паста (BDC) 101
Ализантреновый оливковый G (ВАС) 1167
Ализантреновый фиолетовый RR экстра (ВАС) 1104
Ализарин-дельфииол SEN (BDC) 1053
Ализарин-рубинол 3G (By) 1091
* Ализарин-сафирол SE (By) 1053
Ализариновый желтый L (BDC) Азо-протравной
Ализариновый коричневый В паста (ССС); G паста (ААР) 101
Ализариновый красный IP порошок (ICI)
Ализариновый оранжевый (MDW); А паста, АО порошок, G, 1033
GG, N паста (CN); NL паста (MLB); Р, R, RD, RP паста (NAC); RR (DH); SW порошок, W паста (В): W паста (Bv)
Ализариновый светло-синий SE (S) 1053
242
Продолжение табл. VIII.26
Краситель Номер по В. С. I. (1924 г.)
Ализароловый коричневый RB (NAC) Алый DH для валки, порошок (DH) Амаииловый зеленый G (ААР) Аманиловый зеленый LT экстра (ААР) Амаииловый коричневый М (CD) Аманиловый кубовый оливковый (ААР) Амиииловый прочный желтый А (ААР) Аманиловый прочный оранжевый 3G (ААР) Амаииловый рубиновый R (ААР) Аманиловый черный ВН (ААР) Аманиловый черный R экстра (ААР) Амидиновый зеленый В супра (JC) Амидиновый коричневый М (JC) Амидиновый оранжевый GG (JC) Амидиновый прочный желтый 4G (JC) Амидиновый прочный оранжевый i(JC) Амидиновый черный ВН (JC) Амидиновый черный RB коиц. (JC) Анилиновый синий 2В (МС) Анилиновый синий (НМ); В, ЗВ, RN (tM); спирторастворимый Анилиновый розовый Антра красный В (By) Аитрациаииновый коричневый 2RL (By) Аурамин (различные сорта и марки) Ауреолин (Н), (DH) Аурофеиии О (MLB) Аурофосфин G (А) Афгаиовый желтый (Н) Ацетиндулин R паста; растворимый (С) Ацетиновый синий R (В) Бенгальский синий R (Gy) Беизаминовый коричневый М (WDC) Беизаминовый синий ВХ (WDC) Бензиловый фиолетовый 5BN (SCI) Бензо бордо 6В (By) 101 443 594 593 420 1167 620 621 376 401 582 593 420 478 620 621 401 582 861 689 841 1207 655 812 365 подобен № 789 620 860 860 909 402 472 698 Азо-прямой
16*
243
Продолжение табл. VIII.26
Краситель Номер по В С. I (1924 г.)
Бензо зеленый С (By) Бензо коричневый МС (By) * Бензо прочный алый 4ВА, 4BS (By) Бензо прочный алый 8ВА, 5BS, 7BS, 8BS, 4FB, GS (By) * Бензо прочный гелиотроп BL, 2RL (By) * Бензо прочный красный L (By) Бензо прочный оранжевый S (By) Бензо прочный фиолетовый R (By) Бензо пурпуриновый 10В (различные марки) Бензо родулиновый красный В (By) Бензо синий ВХ (LDC) Бензо синий RW (By) Бензоиновый темно-зеленый (ВК) Бисмарк коричневый (А) Бисмарк коричневый (различные сорта и марки) Брентаминовый прочный алый 2GS основание (ICI) Брентаминовый прочный бордо GP паста (ICI) Брентаминовый прочный красный TR (ICI) Брентаминовый прочный фиолетовый В основание (ICI) Брентол AS (ICI) Брентол AT (ICI) Брентол СТ (ICI) Брентол FR (ICI) Бриллиант-индулин (К) Бриллиант-кармоизин О (MLB) Бриллиант-кроцеин ЗВА (By) Бриллиант сафранин BR и др.; G |(А) Бриллиант фосфин G (А) * Бриллиантовый акридиновый оранжевый А порошок (DH) Бриллиантовый бензо прочный фиолетовый BL, 2RL (By) Бриллиантовый дельфиновый синий В, ЗС (S) Бриллиантовый диазолевый оранжевый NJ (CN) * Бриллиантовый дианиловый синий R (MLB) Бриллиантовый зеленый крист. (By) Бриллиантовый индиго BASF/4B (В) 593 420 327 326 319 Азо-прямой 326 Азо-прямой 495 Азо-прямой 472 512 594 331 332 861 179 Азо-прямой 841 789 788 319 878 478 319 662 1184
244
Продолжение табл. VIII.26
Краситель Номер по В С. I. (1924 г )
.—— —
Бриллиантовый малиновый (САС) 179
Бриллиантовый масляно-черный основной (LBH) 864
* Бриллиантовый прочный синий В, 2G, 5G (By) 319
Бриллиантовый прямой оранжевый J (MLy) 478
* Бриллиантовый синий для хлопка R (К) 319
Бриллиантовый синий В экстра для шерсти (By) 733
* Бриллиантовый синий FFR экстра для шерсти (By) —
Броминдиго (MDW); FB паста (By) 1184
Буффало кардинал 7В (Sch) 495
Буффало коричневый 53 (Sch) 332
Вакансеиновый синий (Н) 135
Везувин (В) 331
Везувин NR коиц. (CN); В, BLP, ВРХ (В); R (By) 332-
Везувин ООО экстра (В) —
Гелиндоновый фиолетовый J2R (MLB) 1104
Гелио красный RL (Gy) 69
Гелио прочный красный RL (LDC), (Maj); RL порошок и 69
паста (By)
Гельвеция коричневый J (SCI) 331
Гельвеция коричневый R (SCi) 332
Генциаиовый голубой 6В (А) 689
Гидрантреновый оливковый R порошок (LBH) 1167
Глубокий хромовый коричневый М для хлопка (StCI) 420
Графитоловый ирочный красный GAERR (GrE) 69
Дельфин синий (ААР); В (By) 878
Диазиновый желтый R (BEL) 620
Диазиновый зеленый В (BEL) 593
Диазиновый коричневый М (BEL) 420'
Диазиновый синий BR (К) 135
Диазиновый черный (К) 134
Диазииовый черный Н экстра (NAC), (Sch) 401
Диазо бриллиантовый алый S4B (By) Азо-прямой
Диазо геранин В экстра (By) Азо-прямоь
Диазо желтый R (By) —
Диазо индиго синий BR (By) 316
Диазо коричневый 6G (By) Азо-прямой.
245
Продолжение табл. VIII.26
Краситель Номер по В. С. I (J924 г.)
Диазо оливковый G (By) Диазо рубиновый В (By) Диазо прочный алый BL (By) Диазо прочный бордо BL (By) Диазо прочный желтый 2G (By) Диазо прочный красный 5BL (By) Диазо прочный красный 7BL (By) Диазо прочный черный MG (By) Диазо прочный фиолетовый BL (By) Диазо прочный фиолетовый 3RL (By) Диазо светло-желтый 2G (By) Диазо синий 3R (А), (By) Диазо черный В экстра, R экстра (StCl); BHL (JWL) Диазолевый зеленый NB (CN) Диазолевый зеленый NJ (CN) Диазолевый коричневый NM (CN) Диазолевый оранжевый N2R (CN) Диазолевый прочный желтый NA (CN) Диазолевый прочный желтый NB (CN) * Диазолевый пурпурин N10B (CN) * Диазолевый светло-фиолетовый NB (CN) Диазолевый синий МЗХ (CN) Диазолевый синий NRW (CN) Диазолевый черный NBH (MLy), (В) * Диамииовый желтый СР (MLy), (С) Диаминовый зеленый В (LDC), (MLy), (С); BN (StD) Диаминовый зеленый G (LDC), (MLy), (С) Диаминовый коричневый М (MLy), (С) * Диаминовый красный 10В (С) Диамииовый красный D (С) Диаминовый иитразоловый коричневый Т (С) Диаминовый оранжевый (С) * Диаминовый прочный бриллиантовый синий R (С) Диамииовый прочный желтый A, AR (MLy), (С) Диаминовый прочный желтый В, С, FF (С) Диаминовый розовый (С) 595 Аэо-прямой Азо-прямой 654 Азо-прямой Азо-прямой Азо-прямой Азо-прямой Азо-прямой 656 512 401 593 594 420 621 620 814 495 319 472 512 401 365 593 594 420 495 Азо-прямой Подобен 621 319 620 814 128
246
Продолжение табл. VIII.26
Краситель Номер по В. С. К (1924 г.)
— Диамииовый розовый В экстра, BD, BG, GD, GGN, FFB (С) Диаминовый синий ВХ (MLy), (С) Диаминовый синий RW (С) Диаминовый черный ВН (MLy), (С) Дианиловый зеленый В (MLB) Дианиловый зеленый С, BBN (MLB) Дианиловый красный 10В (MLB) Дианиловый коричневый МН (MLB) Дианиловый оранжевый N (MLB) Дианиловый розовый BD (MLB) Дианиловый синий G (MLB) Дианиловый синий HG (MLB) Дианиловый черный ES (MLB) Дианиловый чисто-желтый HS (MLB) Дианоловый зеленый В (Lev) Дианоловый зеленый G (Lev) Дианоловый черный ВН (LBH) Диантреновый синий 2В (SCI) Дислерсоловый желтый 3G 150 порошок (ICI) Диоперсоловый прочный оранжевый G 150 порошок (ICI) Диуриндоновый алый R (BDC), (Lev) Дифениловый зеленый (Gy) Дифениловый зеленый 3GC (Gy) Дифениловый коричневый BVV (Gy) Дифенилхлориновый желтый FF (Gy) Дифенилхризоин 3G (Gy) Дуразоловый прочный фиолетовый 2RS (ICI) Дураноловый бриллиантовый желтый 6G 300 тонкий порошок (ICI) Дураноловый коричневый BR тонкий порошок (ICI) Дураноловый красный 2В 300 тонкий порошок (ICI) Дураноловый оранжевый G 300 тонкий порошок ДУРантРено₽ый бриллиантовый фиолетовый R порошок (BDC) Дурантреновый оливковый GL порошок (BDC) Дуриидоновый коричневый G 400 порошок (ICI) Дуриндоновый красный В 400 порошок (ICI) 128 472 512 401 593 594 495 420 478 128 508 472 401 816 593 594 401 1184 1225 593 594 420 814 365 1135 1167 1207
247
Продолжение табл. Vlll.26
Краситель Номер по В С I (1924 г.)
Дуриндоиовый красный ЗВ 400 порошок (ICI) Дуриндоновый оранжевый R 400 порошок (ICI) Дуриндоиовый синий 4ВС 250 порошок (ICI) Жасмин (Gy); SF конц. (Gy) Желтый PR (By) Желтый для хлопка (Н) Желтый для хлопка S (Вт) * Желтый для хлопка CH (SCI) Желтый окисный L 117 (ICI) Жирорастворимый желтый A (SCI) Жирорастворимый индулин R (SCI) * Жирорастворимый нигрозин В (SCI) Жирофле (DH) Замбези черный (А) Замбези черный ВН (А) Замбези черный BR (А) Золотисто-коричневый (А) Иммедчальный бордо G (С) Иммедиальный каштановый В (С) Иидаитреновый бриллиантовый фиолетовый RK порошок, RK паста (By) Иидаитреновый бриллиантовый фиолетовый RR порошок, RR паста (В) Инлантренсзый красно-фиолетовый RH порошок (MLB) Индиановый желтый R (ACC), (Н), (LBH), (Mo), (By), (С); NR (CN); 15 (LJ) Индигеи D, F (By) Индиго КВ (К); MLB/4B (MLB) Индиго LL порошок (BDC) Индиго LL порошок (ICI) Индиго N в зернах 60% (ICI) Индиновый синий 2RD (BDC); 2RS (ICI) Индогеиии (NF) Индоин порошок В, 2В, R, 2R (Gy) Индоиновый синий (LBH) Индоиновый синий R, ВВ, BR порошок и паста (В ) Индоловый синий В, R (A); F, L, 4В (L) 1212 1184 145 812 813 620 365 655 860 864 852 514 331 1012 1012 1135 1104 1212 145 860 1184 1177 1177 909 135 135 135 135 135
248
Продолжение табл. VIII.26
Краситель Номер по В. С. Е (1924 г )
Индоловый синий R (А) 135
Иидоновый синий 2В, 2R (By) 135
Индофениновый экстра (By) 860
Индофеновый синий (MLB) 135
Индулин 5В (BDC) 861
Индулии основание 2В (различные марки) 860
Индулин (САС), (ICA); ЗВ (СНС), (А), (By); В, R (By), 961
(К); 5В крист. (LBH); NBS (CN); ВВ (МС); BS (StCl); 6В (By); W (WDC)
Индулин (CD), (S) 860
Индулин 2BS (ICI) 861
Индулии для печати В, L (MLB) 860
Индулии спирторасгворимый (различные марки) 860
Интенсивный синий (By) 733
Каледоновый бриллиантовый пурпурный RR (SDC) 1104
Каледоновый бриллиантовый фиолетовый R 300 тонкий по- 1135
рошок (ICI)
Каледоновый зеленый XN 300 тонкий порошок <(1С1) 1101
Каледоновый золотисто-оранжевый 3G 300 тонкий порошок —
(ICI)
Каледоновый синий RC 300 тонкий порошок (ICI) 1114
Канареечно-желтый (GrE) 655
Карбидный черный ER (SCI) 58^
Карболановый желтый 3GS (ICI) —
Кардинал ЗВ (BDC) 179
Кармоизин (СНС), (ЛВС), (JWL), (LDC), (W), (WSS), (S); 179
A ^C^^<SCC)> (GCC). (By); LAS (JCO); NS (CN);
Кармоизин WS (BDC) 179
Кармотин (CAC) 812
Кислотный азо-рубиновый (StCl) 179
Кислотный антраценовый красный G (A), (By) 443
* Кислотный желтый (DH), (SCI), .(LP); С (MLy); NY (ССС) 10
Кислотный красный AR (АЛ) 179
Клайтон желтый (САС); G (САС) 813
Колумбия желтый (А) 814
Колумбия зеленый G (А) 594
Колумбия коричневый М (А) 420
24D
Продолжение табл. VIII.26
Краситель Номер по в. с I (1924 г.)
Колумбия черный F экстра (А) * Конго рубиновый (А) * Конго рубиновый A (S); В (К) * Конго рубиновый (BDC) Коричневый AT (Gy); С, N (StD); R (Н) Коричневый BGN (Gy) Коричневый Y (Н) Коричневый для кожи А (С) Крампсол желтый (Lev) Крамлсол прочный синий (Lev) Красный для лака С (ААР) * Красный для хлопка 10В (SCI), (StD) Крезиловый коричневый экстра (StCl); С (StD) Кромеко коричневый В паста (JC) Ксиленовый желтый 3G (S) Ксиленовый светло-желтый 2G (САС), (S); 3GS, R (S) Ксиленовый хромии 5G (S) Кубовый фиолетовый BR (GrE) Кумасси алый 5BS (ICI) Кумасси коричневый GS (ICI) Кумасси фиолетовый 2RS (ICI) Куркумин экстра (А), (ВК) Лак красный С (CD); NC (CN); С паста (SAPC), (MLB) Лак красный R (САС) Лак красный R порошок и паста (SAPC) Лиссаминовый прочный желтый 2GS (ICI) Литоль красный (LDC); R (ААР), (В); NR (CN) Литоль прочный алый NRN (CN); R, RPN (В) Мадрасский синий Р (StD) Малиновый (FA) Манчестерский коричневый (CV), (Lev), (RD), (W) Манчестерский коричневый ЕЕ (Lev), (С); PS (С) Марс красный G (В) Мелантерин ВН (САС), (SCI) Мельдола синий М (StD) Мельдолин синий (BSS) 582 376 1376 376 332 420 331 331 197 878 165 495 332 101 639 639 639 1135 443 698 145 165 69 189 69 135 179 331 332 179 401 909 909
250
Продолжение табл. VII1.26
Краситель Номер по В. С. I. (1924 г.)
— Метаминовый синий В, G (L) 910
Метаминовый синий М (L) 909
Метахромовый коричневый В (АСС); В, BR (Вг); В (САС); 101
L (JWL)
Метахромовый фиолетовый В (А); В паста (Вт), (А) —
Метахромовый фиолетовый 2R (А) —
* Метиленовый голубой ВХ (В) 922
Метиленовый желтый Н (MLB) 815
Метиленовый зеленый В (By) 924
Метиленовый зеленый (различные сорта и марки) 924
Микадо оранжевый различный (А) (By) 621
Мимоза (РСС), (Gy); Z (Gy) 813
Моиастралевый прочный зеленый GS порошок (ICI) —
Моиастралевый прочный синий BS порошок (ICI) —
Моиастралевый прочный синий GS порошок (ICI) —
Монозоль карминовый LS (ICI) —
Монозоль оранжевый OS (ICI) —
Монолитный бордо BLS порошок (ICI) —
Монолитный красный CNS порошок (ICI) —
Монолитный красный R (BDC) 189
Монолитный красный 4RS порошок (ICI) —
Монолитный прочный алый RNS порошок (ICI) Подобен 69
Монолитный прочный желтый GNS порошок (ICI) —
Монолитный прочный желтый 10 GS порошок (ICI) —
Монолитный прочный зеленый BNS порошок (ICI) —
Монолитный прочный красный 2G NNS порошок (ICI) —
Монолитный прочный красный 2RS порошок (ICI) —
Монолитный прочный синий BS порошок (ICI) —
Монолитный рубиновый GS порошок (ICI) .—
Монохромовый коричневый Н паста (LBH) 101
Накарат (StD) 179
Нафталиновый оранжевый GS (ICI) —
Нафтаминовый бриллиантовый синий G (К) 508
Нафтаминовый желтый G (К) 620
Нафтаминовый желтый N (К) 814
Нафтаминовый зеленый AN (К) 594
251
Продолжение табл. VHL.2б
Краситель Номер по В. С I (1924 г.)
Нафтаминовый зеленый В (К) 593
Нафтаминовый оранжевый 2R (К) 621
Нафтаминовый оранжевый TG (К) 478
* Нафтаминовый прочный алый Е4В (К) 327
Нафтаминовый прочный черный KS (К) —
Нафтаминовый прямой черный RWK экстра (К) 582
Нафтаминовый синий ВХ (К), (OeV) 472
* Нафтаминовый темно-зеленый В (К) —
Нафтаминовый черный СЕ (К) 401
Нафтамнновый чисто-желтый G (К) 813
Нафтиламиновый синий R крист. (By) 909
Нафтиндон ВВ, ВВ паста; BR, Т (MLy), (С) 135
* Нафтоловый желтый (различные марки) 10
* Нафтоловый желтый FY (BDC) 10
* Нафтоловый желтый S (различные марки); FYAS (Lev), 10
NS экстра (CN); S экстра (StCl); OS (StD); S экстра
конц. (tM)
Нафтоловый синий В (R) 910
Нафтоловый синий 3R, R (С) 909
Нерастворимый глубокий синий 860
Нерастворимый коричневый DLG (StCl) 331
Ниагара синий BR (NAC); ВХ (Sch) 472
Ниагара синий RW (NAC) 512
* Нигоозин SS (BDC) 864
Нигрозин G крист. (BDC) 865
Новый бриллиантовый желтый L для хлопка конц. (РСС) 813
Новый желтый (tM) 145
Новый желтый LV (S) 620
* Новый желтый для хлопка (WDC) 365
Новый метиленовый голубой F (By) —
Новый прочный синий для хлопка RS (SCI) 909
Новый синий (С) 1290
Новый синий В (С) 910
Новый синий R (САС), (By) 909
Новый фосфин G (С) 71
Оксаминовый зеленый В (В) 593
252
Продолжение табл. VHI.26
Краситель Номер по В. С. I, (1924 г.)
Оксаминовый зеленый G (В) Оксаминовый коричневый R (В) Оксаминовый черный BHN (В) Оксидиазолевый черный NER (С) Оксидиаминовый желтый TZ (С) Оксидиаминовый оранжевый G (MLy), (С) Оксиднаминовый синий R (С) Оксидиаминовый черный (С) Оксидиаминовый черный JB, JW (С) Оксиднаминовый черный JEI (С) Оксидианиловый желтый G, О (MLB) Оксифенин (различные сорта и марки) Оксифениновый золотой (САС) Опаловый голубой (С) * Оранжевый для кожи A (StD) Орион зеленый В (JBS) Орион зеленый G, GG (JBS) Орион синий ВХ (JBS) Орион черный RW экстра (JBS) Основной глубокий черный конц. i(WDC) Основной желтый RN (WDC) Основной зеленый (WDC) Основной зеленый D (WDC) Основной коричневый BR, BXN (DuP) Основной коричневый G, GX (DuP) Основной оранжевый GT (WDC) Основной прочный синий В (Ш) * Основной пурпурово-красный 10В (WDC) * Основной рубиновый (WDC) Основной сине-черный (WDC) Основной синий N2R (CN) Основной синий для кожи N2B (CN) Пакко зеленый В (РАС) Пакко прямой коричневый М (РАС) Пара алый 6В экстра (By) Пара коричневый G (MLB) 594 420 401 582 813 478 512 582 814 814 814 689 788 593 594 472 582 582 620 593 594 332 331 478 135 495 376 401 909 910 593 420 331
253
Продолжение табл. VI1I.26
Краситель j Номер по в. с. I (1924 г.)
Паральдегид RW экстра конц. (LBH) 582
Парамиловый черный ВН (LBH) 401
Параминовый желтый G, R (LBH) 820
Параминовый зеленый G (LBH) 594
Параминовый прочный коричневый М (LBH) 420
Параминовый прочный оранжевый GD (LBH) 621
Параниловый желтый G (А) —
Параниловый зеленый В (А) —
Парафениновый желтый G (LBH) 314
Патентованный диапиловый черный EBV экстра конц 582
(MLB)
Пигмент прочный алый 3L экстра (JWL) 69
Пигмент прочный красный HL (MLB) 69
* Пираминовый желтый G, GX (В) 365
Пираминовый оранжевый 2G (В) 478
Плуто-коричневый R (By) —
Плуте оранжевый G (By) 478
Полихромии (Gy) 812
Понсслевый фиолетовый RR паста; RRD паста (DuP) 1104
Понтаминовый диазо черный ВН конц. BHSW конц. (DuP) 401
* Понтаминовый желтый CH (DuP) 365
Понтаминовый желтый SX (DuP) 620
Понтаминовый зеленый ВХ (DuP) 593
Понтаминовый зеленый GX (DuP) 594
Понтаминовый коричневый R (DuP) 420
Понтаминовый прочный алый 4ВА, 4BS, 8BS (DuP) 326
Понтаминовый синий RW (DuP) 512
Понтаминовый черный RX (DuP) 582
Понтахромовый коричневый MW (DuP) 101
Понтациловый рубиновый G (DuP) 179
Примулин (различные сорта и марки) 812
Примулиновый желтый (By) 812
Протравной зеленый 2G конц. (BDC) Сульфид.
Прочный бременский синий G супра (ICI) —
Прочный желтый G супра порошок (ICI) —
Прочный желто-зеленый G супра порошок HCI) —
254
Продолжение табл. VIII.26
Краситель Номер по В. С. I. (1924 г.)
* Прочный зеленый крист. 0 (С) Прочный зеленый G супра порошок (ICI) Прочный кислотный синий В (By) Прочный красный G (GDC), (В), (ВК) Прочный красный 6В супра порошок (ICI) Прочный лаковый красный R паста (StD) Прочный нейтральный синий крист. (А) Прочный прямой черный ВН (ICA) * Прочный розовый В супра порошок (ICI) Прочный светло-желтый 2G (ААР) Прочный синевато-серый конц. (Gy) Прочный синий (С) Прочный синий В (Gy) Прочный синий В (tM) Прочный синий В, R (А) Прочный синий В (Gy), (А), (В); R (А), (В); 3R (MLy); ЗВ, 6В (А) Прочный синий для хлопка В (GrE) Прочный синий для хлопка В (MLB) Прочный синий для хлопка В (S), (А) Прочный синий 3R для хлопка (В), (By) Прочный синий R, RR для хлопка (MLB) Прочный синий R для хлопка крист. (А) Прочный синий основной (GrE) Прочный синий RRB спирторастворимый (Gy) Прочный синий В супра порошок (ICI) Прочный темно-синий R (MLB) Прочный фиолетовый R супра порошок (ICI) Прочный флотский серый J, JB (SCI) Прочный флотский синий G (GrE) Прочный флотский синий R (СгЕ); ММ (К) Прямой бриллиантовый желтый С (ССС) Прямой бриллиантовый розовый В экстра (РСС) Прямой бриллиантовый синий G (NCW) Прямой бриллиантовый флавин S (РСС) ямой бриллиантовый флавин Т (РСС) Прямой Гарнета (ССС) 733 179 189 909 401 639 861 860 861 135 860 861 135 910 910 909 909 909 860 860 861 861 910 909 365 128 508 816 815 376
255
Продолжение табл. VII 1.26
Краситель Номер по В. С I (1924 г.) '
Прямой глубокий черный Е экстра (By) 582
Прямой глубокий черный RW (МС); RW экстра (By) 582
Прямой диазочерный ВН (StD) 401
* Прямой желтый CH (Lev); С (S); CRG (L) 365
Прямой желтый F, G, J, R, Т (различные марки) 620
Прямой желтый R ,(StCl) 814
Прямой зеленый В (многие); BL (JWL); FE (S); BN (StD) 593
Прямой зеленый G (AJ), (SCC), (S), (ICA), (JDC); BG .(JCO) (StCl) 594
* Прямой золотисто-желтый (LJ) 365
Прямой коричневый (AJ); М (САС), (SCI), (LJ), (StD); MB (StCl); 3RB (Sell) 420
* Прямой красный 10В (ССС), (StD) 495
* Прямой малиновый В (Sch) 376
Прямой оранжевый G (ICA) 620
Прямой оранжевый 2R, 2RG (NCW) 621
Прямой оранжевый Y (Sch); G (S) 478
Прямой прочный алый BE '(САС); 4BS (StD); SE (САС), (SCI) 326
Прямой прочный желтый (различные марки) 814
Прямой прочный желтый (различные марки) 620
Прямой прочный коричневый М (ICA); MB (NCW) 420
Прямой прочный оранжевый RL, 2RL (JWL); G (ССС) 621
Прямой прочный оранжевый SE (САС), (SCI) 326
Прямой прочный синий RW (NCW) 512
Прямой прочный черный ВН (РСС) 401
Прямой розовый В (SCI) 130
Прямой синий RW (SCI) 512
Прямой синий ВХ i(AAP), (NCW), (SCI); ВХХ (StD) 472
Прямой темно-коричневый М (L) 420
Прямой хризофенин (StD) 365
Прямой черный BD (StD); ВН (AJ), (JDC); НВ (L) 401
Прямой черный EL (JLW); RE, RXX i(NCC); 2R (StD); RW (SCC), (NCW) 582
Пурпурин Дюпон 10B (DuP) 495
Растворимый индулин ЗВ (WSS) 861
Реноламиновый черный ВН, BHN (tM) 401
256
Продолжение табл. УШ.26
Краситель Номер по В. С. I. (1924 г.)
Реноловый желтый R (tM) Реноловый зеленый В экстра (tM) Реноловый зеленый G экстра (tM) Реноловый коричневый МВ конц. (tM) Реноловый оранжевый G (tM) * Реноловый рубиновый экстра (tM) Реноловый светло синий (tM) Реноловый черный R экстра (tM) Родулиновый желтый 24496 (By) Родулиновый желтый S (By) Родулиновый желтый Т (By) * Родулиновый красный G (By) * Родулиновый оранжевый N, NO (By) Родулиновый синий R (By) * Родулиновый фиолетовый (By) Ротор бриллиантовый красный R (ICI) Ротор желтый (ICI) Ротор зеленый Y (ICI) Ротор красный S (ICI) Ротор синий В (ICI) * Рубиновый для хлопка (В) Рубиновый красный A (MLy) Рубиновый мелкокристаллический (А) Салициновый сине-«ерный АЕ (К) Сафранин (различные марки) * Сафранин FF экстра (By) Сафранин S № 150 (С) * Сафранин Т конц. (BDC) Сафранин Т экстра (В) Сафранин TS (ICI) Сафраниновый синий R (StD) Световой красный A (SAPC) Световой синий (StD), (tM) Светопрочный красный для лаков С-Рнистый желтый S (FB), (К) Сигнальный красный (САС) 620 593 594 420 478 376 518 582 816 815 844 788 844 376 179 677 841 841 811 841 841 841 135 69 689 69 10 189
P-S18 257
Продолжение табл. VIII.26
Краситель Номер по В. Q. 1 (1924 г.)
Синий СВ водорастворимый (DH) Синий для печатания (MLy), (А) Синий для хлопка В (В) Синий для хлопка NVB, VB (L) Синий для хлопка R (В); RR (В), (By) Синий II спирторастворимый (MI R) Синий N экстра для шерсти (By) Синяя паста (NF) Ситара прочный красный RL (tiM) Слолин RS (WSS) Солаичиновый красный 8PL (А) Солаце~овый прочный алый BS (ICI) Солацетовый прочный желтый GS (ICI) Солацетовый прочный кпасный 3RS (ICI) Солацетовый прочный малиновый BS (ICI) Солацетовый прочный фиолетовый 4RS (ICI) Соледоновый бриллиантовый пурпуровый 2R250 порошок (ICI) * Соледоновый желтый GS паста (ICI) 861 860 910 861 909 689 733 135 69 861 878 Производное от 1104 Производное от 1104
Соледоновый оранжевый R (ICI) * Соледоновый синий 2RCS (ICI) Солохромовый азуриновый BS (ICI) Солохромовый желтый 2GS (ICI) Солохромовый желтый Y (BDC) Солохромовый коричневый MGS (ICI) Солохромовый оранжевый GPS (ICI) Солохромфлавин RS (ICI) Соль коричневая G (MLB) Соль темно-коричневая G (MLB) Сольвей синий SE (SDC) Спирторастворимый индулин (различные марки) * Спирторастворимый нигрозин (BDC), (JBS); С, G, LM (BDC) Спирторастворимый нигрозин R (NAC) Спирторастворимый розанилиновый синий (StD) Спирторастворимый синий В (С) 197 101 Азо протравной 331 331 1053 860 864 860 689 689
258
Продолжение табл. VIH.26
—
Краситель Номер по В С. I. (1924 г )
Спирторастворимый синий 2В (BDC) 689
Спирторастворимый синий СВ >(DH) 860
* Спирторастворимый синий Купьера (МС) 864
Спирторастворимый синий (различные марки В и R) 689
* Спирторастворимый черный Р (BDC) 864
Стильбеновый желтый 3 G (В) 621
Стильбеновый желтый экстра (StCl) 620
Стильбеновый оранжевый 4R (САС) 621
* Султан 10В (Н) 495
* Султан желтый G (Н) 365
Сульфин A, N (By) 812
Суперхромовый желтый RN (NAC) 197
* Равнинный гелиотроп (С) 852
Таннинный оранжевый R (С) 72
Таннинный оранжевый R порошок (С) 72
Таннофлавин Т (S) 815
Тиоиндиго алый R (К) 1225
Тиоиндиго алый R паста (К) 1225
Тиоиндиго красный В (К) 1207
Тиоиндиго красный ЗВ (К) 1212
Тиоиндигофлавин Т (С) 815
Тиоиндигофлавиь ТСА (С) —
Тионоловый бриллиантовый бордо ZR (LBH) 1012
Тиоголовый желтый GR (BDC) —
Тионоловый желтый R (BDC) —
Тионоловый коринфский RBX (BDC) —
Тионоловый коричневый О (BDC) —
Тионоловый коричневый GM (BDC) —
Тионоловый небесно-голубой PRX (BDC) —
Тионоловый черный RS (ICI) —
Тиофлавин S (С) 816
Тиофлавин Т (С), (К) 815
Тиофосфин J (LP) 814
Тиохромоген (WDC) 812
Титановый желтый G (BDC) 813
Толуидиновый красный (ААР) 69
17*
259
Продолжение табл. VII1.2fi
Краситель Номер по В С I (1924 г.)
Толуиленовый коричневый М (By) Толуиленовый оранжевый G (A), (By), (S) Толуиленовый прочный оранжевый LX (By) * Триазоловый желтый G (GrE) Триазоловый зеленый 2G (GrE) Триазоловый прочный желтый G, 2G, GN (GrE) Триазоловый темно-синий ВН (GrE) Трисульфоновый коричневый В (S) Тсуйя индиго (MDW) Турмерин (BSS) Ультрачерный ВН экстра конц. (NCW), RH (ССС) Фенилиновый синий (ВК) * Фениновый желтый (StD) Флотский серый BE экстра (StD) Хлоразолевый бордо 6BS (ICI) Хлоразолевый бриллиантовый желтый 3GS (ICI) Хлоразолевый голубой 2R (BDC) Хлсразолевый голубой RW (BDC) Хлоразолевый желтый СХ (BDC); GS (ICI) Хлоразолевый зеленый BN (BDC) Хлоразолевый зеленый BNS (ICI) Хлоразолевый зеленый GS (ICI) Хлоразолевый коричневый LF (BDC); LFS (ICI) Хлоразолевый коричневый MS (ICI) Хлоразолевый оранжево-коричневый XI50 (ICI) * Хлоразолевый прочный алый 4BS (BDC) Хлоразолевый прочный алый 4BS (ICI) * Хлоразолевый прочный гелио BKS (ICI) Хлоразолевый прочный желтый В (BDC) Хлоразолевый пробный желтый BS (ICI) Хлоразолевый прочный оранжевый AG (BDC) Хлоразолевый прочный оранжевый AG125 (ICI) Хлоразолевый прочный оранжевый D (BDC) Хлоразолевый прочный оранжевый RS (ICI) Хлоразолевый прочный синий FFKS (ICI) Хлоразолевый розовый BS (ICI) 478 620 365 594 814 401 561 813 813 401 909 365 861 472 512 620 593 593 594 561 420 Азо прямой 327 327 319 814 814 Азо-прямой Азо-прямой 621 326
260
Продолжение табл. VIII.26
Краситель Номер по В. С I. (1924 г.)
Хлоразолевый тускло-коричневый RH (BDC) Хлоразолевый черный BHS (ICI) Хлоразолевый черный DV (BDC) Хлоразолевый черный LF (BDC) Хлораминовый желтый (By) Хлораминовый желтый (различные марки G и F) * Хлораминовый желтый W экстра (By) Хлораминовый оранжевый G (By) Хлораминовый прочный алый 2В, 4BS (S) Хлораминовый синий ВХ, BXR (S) Хлора «новый черный ВН, BHR (S) Хлорантиновый оранжевый JR (SCI) Хлорофенин Y (САС) Хлорофосфин V (САС) Хокусетцу зеленый G (MDW) * Хризобарин G экстра конц. (tM) Хризоидин А (В) Хризоидин А крист., СЕЕ крист.. G, 2G экстра конц., 2G экстра крист., GN. GS, J, J экстра, JE. JE70, JEE, N, R. RE, Y, Y конц., Y экстра, Y порошок, YRP (различные Азокраситель 401 Азокраситель 582 814 814 365 621 326 472 401 621 814 814 594 365 220 20
марки) * Хризоидин YRP (BDC) * Хризофенин G (By) (различные сорта и марки) Хромин GS (MLB) Хромовый коричневый (ССС) Хромовый синий GD (NAC) Хромовый цианин (DH) Централиновый зеленый (CD) Централиновый коричневый В (CD) Централиновый оранжевый G (CD) Централиновый черный ВН (CD) Циба розовый В (SCI) Циба синий 2В (САС). (SCI); 2BD (SCI) Цибаноновый фиолетовый R (SCI) * Цитромин A (L) Цитромин (САС). (S); ООО (SCI); R конц. (BDC) Черный для хлопка RW экстра (В) 20 365 816 101 878 812 594 561 478 401 1207 1184 1104 10 145 582
2(1
Продолжение табл. VIII.26
Краситель Номер по в. с. I. (1924 г.)
Чикаго синий RW (А) Чистый светло-зеленый брауншвейгский, порошок (ICI) Чистый синий без бронзового оттенка, 16266 порошок (ТС1) Чистый ярко-красный 725 SW (ICI) Эболи новый синий 2В (L) Эксельсиор коричневый (WDC) Энбико прямой зеленый В (NBC) Энкисей коричневый (MDW) * Эозин YS (ICI) Эри желтый F, 2RF (NAC) * Эри желтый Y l(NAC) Эри зеленый МТ (NAC) * Эри кардинал 7В (NAC) Эри конго 4В (NAC) Эри оранжевый Y (NAC) Эри прочный алый 7А, 4ВА, 8ВА (NAC) Эри прочный желтый WB (NAC) Эри прочный коричневый 3RB (NAC) Эри прочный оранжевый CG (NAC) Эри прямой зеленый ВТ, МТ (Sch) Эри прямой черный RX (Sch) Эри розовый 2В (NAC) Эри черный RX (NAC) Эрика BN (А); В экстра, BN (Lev),.(RF), (A), (L); В (S); Z (А) * Этиловый зеленый крист. (А) Эухризин 2G, R, RR '(В) Эухризин GGD (В) * Эухризин 3R (В) Юнион синий Н (S) Янусовый синий G, R (MLB) Янусовый темно-синий В, R (MLB) Янусовый черный (ML3) Яркий желтый (Gy); 3GC, G, BG (Gy) и т. д. Яркий желтый (MLB) Яркий желтый 2RS (S) 512 472 331 593 331 768 620 365 593 495 370 478 326 813 420 621 594 582 128 582 130 662 797 797 788 135 135 135 134 620 814 621
262
(Приложение к табл. VII 1.26)
(А) /ААР) (АСС) (AJj (В) (ВАС) (BDC) (BEL) (ВК) (Вг) (BSS) (By) (С) (САС) (ССС) (СНС) (CJ) (CN)
(CV) (DH) (DPC) (DuP) (FA) (FB) (GCC) (GDC) (GrE) (Gy)
(H) (HM) (HP) (ICA) (ICI) (JBC) (JC) (JCO)
ФИРМЫ — ПРОИЗВОДИТЕЛИ красителей
A-G fur Anilin-Fabrikation, Berlin.
American Aniline Prods. Inc.
Alliance Colour and Chemical Co. Ltd., Manchester.
Ajax Aniline Dye Manufacturing Co. Ltd., London
Badische Anilin-u.-Soda-Fabrik, Ludwigshafen
British Alizarine Co. Ltd., Manchester
British Dyestuffs Corporation, Huddersfield
J. C. Bottomley and Emerson Ltd., Huddersfield
Leipziger Anilinfabrik Beyer und Kugel G.m.b.H., Fiirstenberg/Oder
Brotherton and Co. Ltd., Birkenhead
Brooke, Simpson and Spiller Ltd., London
Farbenfabriken vorm. Fried. Bayer, Leverkusen
Leopold Cassella & Co., Frankfurt/Main
Clayton Aniline Co. Ltd., Manchester
Calco Chemical Co. Bound Brook, N. J.
Cutler Hill Colours and Chemicals Ltd., Manchester
Carl Jager G.m.b.H. Anilinfarbenfabrik, Diisseldorf
Compagnie National de Matieres Colorantes et de Produits Chimiques, Paris.
Colne Vale Chemical Co., Huddersfield.
Durand, Huguenin and Co., Basel
Dye Products and Chemical Co. Inc., Newark, N. J.
Du Pont de Nemours and Co., Delaware
Farbwerk Amersfoort, Amersfoort, Holland.
Fabrique Beige de Couleurs d’Aniline Paul Entrop., Haren, Belgium
Grasselli Chemical Co., Albany, N. Y.
Gray’s Dyes and Colours Ltd., Grays, Essex.
Chemische Fabrik Griesheim-Elektron, Frankfurt/Main
J. R. Geigy S. A., Basel.
Read, Holliday and Sons Ltd., Huddersfield
Heller and Merz Co., Newark, N. J.
Hickson and Partners Ltd., Castleford.
Societa Italiana di Colori artificiali, Milan
Imperial Chemical Industries Ltd., Dyestuffs Division.
J. B. and W. R. Sharp Ltd., Edenfield.
J. Campbell and Co., New York.
J- C. Oxley’s Dyes and Chemicals Ltd., Dewsbury.
263
(JDC) Japan Dyestuff Manufacturing Co., Osaka.
(JWL) J. W. Leitch and Co. Ltd., Huddersfield.
(K) Kalle und Co. A-G., Biebrich/Rhein.
(L) Farbwerk Miihlheim vorm. A. Leonhardt and Co., Miihlheim.
(LBH) L. B. Holliday and Co. Ltd., Huddersfield.
(LDC) London Dye Manufacturing Co., London.
(Lev) Levinstein Ltd., Manchester.
(LJ) Laroche and Juillard, Lyon.
(LP) Lucien Picard et Cie, Lyon.
(Maj) Major and Co., Hull.
(MC) Mabboux and Cammell, Lyon-Vaise.
(MDW) Mitsui and Co., Omuta, Japan.
(MLB) Farbwerke vorm. Meister, Lucius u. Bruning, Hochst/Main.
(MLy) Manufacture Lyonnaise de Matieres Colorantes S. A., Lyon.
(Mo) Societe Chimique des Usines du Rb6ne.
(NAC) National Aniline and Chemical Co. Inc., N. Y.
(NBC) North British Chemical Co., Manchester.
(NCC) Noil Chemical and Colour Works Inc., N. Y.
(NCW) Newport Chemical Works Inc., Passaic, N. J.
(NF) Niederlandische Farben- u. Chemikalien-Fabrik, Delft.
(NI) Chemikalienwerk Griesheim G.m.b.H.
(OeV) Oesterreichischer Verein fur Chemische u. Metallurgische Produktion, Hruschau.
(РАС) Palatine Aniline and Chemical Corp., Poughkeepsie N. Y.
(PCC) Peerless Colour Co. Inc., Bound Brook, N. J.
(R) Chemische Fabriken, Worms
(RD) Roberts, Dale and Co., Manchester.
(RF) J. Ruch et Fils, Pantin, Seine.
(S) Sandoz Chemical Works, Basel.
(SAPC) Societe Alsacienne de Produits Chimiques, Mulhouse.
(SCC) Southdown Chemical Co., Birkenhead.
(Sch) Schoeikopf, Hartford and Hanna Co., Buffalo, N. Y.
(SCI) Society of Chemical Industry, Basel.
(StCl) Compagnie Fran^aise de Pioduits Chimiques et Matieres.
Colorantes de StCloud du Rhone.
(StD) S. A des Matieres Colorantes et Produits Chimiques de St. Denis.
(T) Tarrasa Chemical Co., Tarrasa, Spain.
(tM) Chemische Fabtiken vorm. Weiler-ter-Meer, Uerdingen/Rhein.
(TMC) Tower Manufacturing Co. Inc., Brooklyn, N. Y.
(VSt) Victor Steiner, Vernon (Eure).
(W) Williams Bros, and Co., Hounslow.
(WDC) Wolfing, Dahl and Co. A.-G., Barmen.
(WSS) W. S. Simplon and Co., London.
(Z) Zinsser and Co. Inc.. Hastings-on-Hudson, N. Y.
264
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
265
1924 г.) и ализаринцианин 2R600 порошок (ICI) (определенный по Б.С. 1. под номером 1050, что сомнительно).
Возможно, что последнее соединение — бирюзово-синий G (BDC) (В.С.1. 661) —является тем же соединением, что и бирюзово-синий ВВ(Ву), мо бирюзово-синий G(By) (B.C.I. 661) и .некоторые другие марки тех же производителей не реагировали с бихроматом, а с броматом вступали в слабую реакцию (см. табл. VIII.27). Использованные образцы представляли смесь красителя с хлоридом цинка, но это не влияло на индикаторные свойства красителя. Изменение окраски происходило от померанцевозеленого до огненно-красного; в присутствии хлорида ртути(II) окраска немного тускнеет и быстро исчезает в присутствии фосфорной кислоты, добавка которой излишня. Индикаторная ошибка составляет 0,001 мл 0,01667 М раствора бихромата. Изменение окраски и обратная реакция происходят очень быстро, поэтому для титрования бихроматом можно спокойно рекомендовать бирюзово-синий G (BDC). Попытки точно определить формальный потенциал не удались из-за деструктивного окисления индикатора с последующим образованием зеленого осадка; однако методом Адамса и Хамакера было установлено, что интервал перехода находится в области от 0,930 до 1,06 В.
Поскольку в методах титрования раствором церия (IV) уже применяют в качестве индикаторов ряд красителей, не было смысла рассматривать все соединения, которые изменяют свою окраску при взаимодействии с этим реагентом. Чтобы составить шкалу субъективной оценки в табл. Vlll.27, индикатор Кнопа эриозеленый был принят за плохой, но обратимый индикатор. Многие соединения были затем идентифицированы по указателю для красителей и оказались формально идентичны красителям, которые уже были исследованы Клопом или Смитом. Однако между различными образцами, по-видимому одного и того же соединения, часто наблюдалось большое расхождение и нередки были небольшие или существенные отклонения от наблюдений Кнопа. Несколько из наиболее обещающих соединений были испытаны в жестких условиях титрования церием (IV) для определения конечной точки потенциометрическим методом и трис- (1,10-фенантролин)железом (II) для сравнения. В итоге такого тщательного исследования было исключено значительное число индикаторов, так как они оказались не лучше эриозеленого Кнопа (или медленно обратимыми, или давали индикаторную ошибку более 0,1%, или, наконец, окисленная форма была неустойчивой). Остальные красители описаны подробнее в табл. VIII.28. Большая часть их опять является трифенилметано-выми соединениями, которые, как уже было отмечено, дают синюю или пурпурную окраску в нейтральных растворах, при подкислении проявляют гипсохромный сдвиг в сторону зеленой или желтой окраски, а при окислении — батохромный сдвиг к оранжевой или
Таблица VIII.27
ТЕХНИЧЕСКИЕ КРАСИТЕЛИ, ПРОЯВЛЯЮЩИЕ ОПРЕДЕЛЕННЫЕ ОКИСЛИТЕЛЬНЫЕ ИНДИКАТОРНЫЕ СВОЙСТВА ПРИ ВЫСОКИХ ПОТЕНЦИАЛАХ
Принятые сокращения
В. С. I. — номер по Британскому указателю красителей pH — изменение окраски от кислотности среды Мп7 — титрование железа (II) перманганатом в среде 1 М серной кислоты Се4 —титрование железа (II) раствором церия (IV) в среде 1 М серной кислоты
Сгв —титрование железа (II) бихроматом <в среде 2 М серной кислоты
Вга —титрование мышьяка (III) броматом в среде 2 М HCl/0,05 М КВг П - плохое изменение цвета или интенсивности окраски X — хорошее изменение окраски
О — обратимое (4—6-разовое) изменение окраски
Н — необратимое изменение окраски или плохая обратимость
Р --индикатор разрушается
— означает отсутствие изменения окраски
В графе B.C.I: «•—» означает отсутствие данных; Б — указан я указателе, но не имеет номера После названия красителя в скобках приведены сокращенные обозначения фирм-производителей (см приложение к табл. VIII.26)
Название (фирма) Тип красителя Номер по В. С. I. (1924 г ) PH МП? Се4 Сг« Вга
Агалма черный 10В, 12В (В) Диазо 246 П п
Азидиновый голубой (CJ) Триазо 520 НП — П
Азидиновый голубой FF (CJ) Диазо 518 —. — Р П
Азидиновый синий 2В (CJ) » 406 ПР Р ПР
Азидиновый фиолетовый DV (CJ) » 394 п — НП — р
Азо-малиновый S (By) Азо 54 —. —- ПР
Азо-синий для шерсти (С) » 53 X по —~ —.
Азо-темно-синий S (S) Диазо 246 п — п
Азо-фиолетовый 4BS (StD) Азо 53 — ПР ПР —. —
Азо-фиолетовый для шерсти 4В (NAC); 2R (С) » 53 п п НП — .—
Азогеранин 2CG (ICI) » 31 — — ПР — —
Азогренадин S (By)
Азокислотный красный В (MLB); L (S) Азокислотный фиолетовый AL (By)
Азокислотный фиолетовый A2R, А2, В R экстра, 4R (By)
экстра,
Азомин прочный розовый В (JBS) Азонафтареновыи красный G (SCC) Азонафтоловый красный 6В (StD) Азонафтоловый красный J (StD) Азородин 2В (S); 6В (S)
Азородин 2G (S)
Азофлоксин GA (GCC); 2G (By) Азофуксин В (By)
Азофуксип ЗВ (LBH) Азофуксин &В (GCC)
Азофуксин G (By)
Азоцианин GR (К)
ко
о
Азоцианин 5R (К) Азоэозин SL экстра Азурин (JDC) Азуровый синий V (К)
Акроноловый бриллиантовый синий (BDC) Акроноловый бриллиантовый синий AS (ICI) Ализарин геранол (By)
Ализарин геранол В (By)
Ализарин дельфинол В, BDN (BDC)
Ализарин сафирол В (By)
Ализарин сафирол В (By)
Ализарин сафирол В порошок (By)
Ализарин уранол 2В (By)
Ализарин ураиол ВВ, R (By)
Ализарин цианин 2R600 порошок (ICI)
Ализариновый бриллиантовый зеленый EF (LBH)
Ализариновый бриллиантовый синий В (LBH)
Ализариновый голубой В (By)
Ализариновый голубой 3R (By)
Ализариновый коричневый R (Т)
Ализариновый красный AS порошок (ICI)
»
Диазо
»
Азо
»
»
»
»
»
»
»
»
»
Диазо
»
Диазо ТФМ
»
»
Антрахиноновый
»
»
»
»
»
»
»
»
Антрахиноновый
»
Азо
54 ПР
54 — — — ПР
255 — — — п
255 — — — п
225 —— — ПР
31 — — ПР —.
57 — .— р — ПР
31 .— — ПР .—
57 — .— р — ПР
31 — — ПР —
31 — — ПР —
66 — нп .—- п
54 — .— — ПР
57 — — р —. ПР
153 — — .— р
288 — нп — ПР
289 п п п — ПР
— — — — ХР
502 — — — ПР
712 X 0 хо п п
664 п хо хо — —
664 п хо хо —
1092 •— нп п ПР
1092 — ПР п ПР
1054 — хо п п
1054 — п п п
1054 — хо п п
1054 — п п п
1058 — п — ПР
1058 — п — ПР
1050 — О о
1078 — п п п
1054 — — хо п п
1088 п HP п ПР
1088 п HP п ПР
98 п — по —
— п — on — —
ОО
Название (фирма) Тип красителя
Ализариновый рубиновый G, 3G, 5G, R (By) Ализариновый рубиновый R (Зу) Ализариновый рубиновый R (Ву) Ализариновый сапфирово синий (NAC) Ализариновый сапфирово-синий В (SCI) Ализариновый светло синий В (S) Ализариновый сине-черный A (MLA); BN (ССС)
Ализариновый синий RBN (ССС) Ализариновый синий SAP (GCC) Ализариновый фиолетовый (Т) Ализариновый фиолетовый ZBS (Z) Ализариновый флотский синий Z2R экстра (Z) Ализариновый цианиновый зеленый Е (Ву) Ализариновый цианиновый зеленый различных сортов (Ву)
Ализариновый циановый зеленый (Ву) Ализариновый чистый синий В (By); 2R (Ву) Ализуроловый рубиновый (В4С) Ализуроловый сапфировый В (ВАС) Ализуроловый цианиновый зеленый Е, G экстра, К (ВАС)
Алмазный зеленый R ВХ (В)
Алмазный зеленый В, 3G, SS (Ву) Алмазный маджента (В)
Алмазный сине-черный ЕВ (By); RB (ССС)
Алмазный фуксин (В), (By), (С); различные сорта, крупные крист. (Ву)
Алмазный черный F (CD), (В), (By), (L) Алый для бумаги (JBS), (MLB) Алый для хлопка (В); ЗВ конц. (К) Альфазурин A (NAC) Альфазурин G (NAC) Альфаноловый синий 5RN (С)
Антрахиноновый »
»
» »
» Азо
Оксазиновый Антрахиноновый Азо Ксантеновый Оксазиновый Антрахиноновый »
»
»
» »
»
ТФМ Диазо ТФМ Азо ТФМ
Диазо
»
»
ТФМ
» Диазо
Продолжение табл. VIII.27
Номер по В С. I (1924 г.) PH Мп? Cel С г6 ВГ2
1091 HP
1091 — П п
1091 — HP — —
1054 —. — О — —
1054 .—. — О — п
1054 — хо п п
202 п п п — —
883 п р о —.
1054 — —. хо п п
169 X р р •— —
781 п р ПР — ПР
883 п р о —
1078 —. — — п
1078 — — — п
1078 — п п п
1088 п HP п ПР
1091 — п 11
1054 — —. о — —
1078 — п п п
657 п о —
302 —. — пн — —
677 п — — — X
202 — ПР ПР — —
677 п п — р
299 -— п п —.
252 — пн пн
252 — — пн пн —
714 п п хо — ПР
712 X хо п п
289 — МП — ПР
Аманиловый бриллиантовый синий 6В (ААР) Аманиловый голубой (ААР) Аманиловый зеленый 2G (ЛАР) » Триазо »
Аманиловый прочный оранжевый R (ААР) Диазо
Аманиловый синий 2В (ААР) »
Аманиловый фиолетовый NC конц. (ААР) »
Аманиловый черный G экстра (ААР) Триазо
Амацидный оранжевый Y (ААР) Азо
Амацидный прочный красный А конц. (ЛАР) »
Амацидный сине-черный G экстра (АЛР) Диазо
Амацидный флоксин (ААР) Азо
Амацидный фуксиновый 10В (ААР) »
Амидиновый синий 2В (JC) Диазо
Амидиновый темно-зеленый N (JC) Триазо
Амидиновый фиолетовый В (JC) Диазо
Амидиновый черный NH конц. (JC) Триазо
Амидо черный 10ВО (MLB) Диазо
Амидонафтоловый NJ (CN), красный G (AJ), Азо
(JBS), (CD), (MLB)
Амидонафтоловый красный 2В (MLB); 6В »
(AJ), (JBS), (CD), (MLB); N6B (CN)
Амидона фтюловый синий R (StD) »
Аминовый черный 10В (А) Диазо
Анилиновый коричневый (С) ТФМ
Анилиновый серый В (А) Азиновый
Антразурен В (К) Антрахиноновый
Антраценовый кислотный черный DSF (MLy), Диазо
(С); DSN (С)
Антраценовый сине черный (MLy), BE (С) Азо
Антраценовый хромовый фиолетовый В (С) »
Антрацианин 3GL (By) Антрахиноновый
Антрацианин RL (By) Смесь
Антрацианиновый серый GL (By) —
Атлас оранжевый (BSS) Азо
Аутохромовый красный G (К) »
Ацеко оранжевый II (JC) Моно
Ацеко прочный красный A (JC) Азо
Ацеко прочный красный Е (JC) »
Ацеко прочный синий 2R (JC) »
518 Н - ПР
520 —— НП — I Р
583 Il — РП ——
415 X — н П 1 —
406 — ПР ПР ПР
394 п — HP —. р
581 — — р — —.
151 — — ПР - --
176 — п НРХ п —
246 — — п — П
31 — .— ПР — —
57 — — р — РП
406 — ПР р ПР
583 п — ПР —
387 — — п — -
581 — -— р — —
246 — — п .— —
31 — — ПР — —
57 — — р — ПР
208 п п по — по
246 — — п .— п
677 и — — О
865 — НП —
1054 — п п п
299 — п п — —
202 п п п
169 X р р —.
1053 — — — ПР
Б — п —
— — п —.
151 — — ПР .—
216 — ПР ПР .— ПР
151 — — р —
176 — п НРХ п
182 -— ПР —. Р
208 п п по — ПО
bO
о Название (фирма) Тип красителя
Ацеко цианин 3R экстра, 5R экстра (JC) Диазо
Ацеко черный 10В (JC) »
Ацетиловый розовый 2BL, 6BL (LBH) Азо
Ацетиловый розовый 2GL (LBH) »
Ацетиловый красный G (В); J (VSt) »
Ацетофениленовый красный R (StD) »
Ацидоловый синий G (tM) ТФМ
Ацидоловый хромовый черный FF (tM) Диазо
Ацидоловый черный 6G экстра конц. (tM)
Бенгальский зеленый ОО (GrE) ТФМ
Бенгальский синий (К) Азиновый
Бенгальский синий G (GrE) Диазо
Бензаминовый голубой (WDC) Триазо
Бензаминовый голубой FF (WDC) Диазо
Бензаминовый синий 2В (WDC) »
Бензиловый зеленый В (SCI) ТФМ
Бензиловый фиолетовый различных сортов и »
марок
Бензо голубой (LDC) (By) Триазо
Бензо оранжевый R (JBC), (LDC), (S), (А), Диазо
(Ву)
Бензо прочный красный 8BL (Ву) »
Бензо синий 2В (Ву) »
Бензо фиолетовый R (А), (Ву) »
Бензо черно-синий G >(Ву) Триазо
Бензоазурин G (Ву) Диазо
Бензоазурин G различных марок »
Бензоиловый зеленый различных сортов и ма- ТФМ
рок
Бензоиновый голубой (ВК) Триазо
Бензоиновый прочный красный АЕ (ВК) Азо
Бензоиновый синий GN, 2GN, 5GN (ВК) Диазо
Бензопурпурин 4В '(Ву) »
Беизопурпурин 10В (Ву) »
Продолжение табл. VIII.27
Номер по В С. I. (1924 г.) pH Мп? Се* Сгв ВГ2
289 П п п ПР
246 — — п п
57 — — р ПР
31 — — ПР
31 — — ПР — —
54 —— — — ПР
712 X О хо п п
299 —- п п — —
246 — — п — п
657 п о —.
865 — — нп
502 —- — — ПР
520 — нп р
518 — — р п
406 — ПР р ПР
667 п по хо — ПР
683 п — по по —
520 —— нп р
415 X — пн п ПР
278 — ПР ПР ПР
406 —. ПР р ПР
388 — п XP п
578 — нп п п
502 — — — ПР
502 — — — ПР
657 X хо — —
520 нп р
225 —. НРП — .
502 — — — ПР
448 X — ПР
495 X — ПР
Бензопурпурин 4BS (ICI) Бетанафтоловый оранжевый Бибрихский кислотный красный В (К) Бирюзовый голубой G (Ву) Бирюзовый голубой G (BDC) Бирюзовый голубой G, В, ВВ, GC, CL экстра (Ву)
Биттер Олмонд масляно-зеленый (tM) Блеклый алый (Lev)
Бриллиант кроцеин различных сортов и марок Бриллиантовый ланафуксин 2G (С) Бриллиант сафранин G (А)
Бриллиант фосфин 'G, 3G, 5G (САС), (SCI) Бриллиантовый ализариновый зеленый (WDC) Бриллиантовый ализариновый синий (GCC) Бриллиантовый ализариновый синий R450
(ICI)
Бриллиантовый бензо синий иВ (Ву)
Бриллиантовый зеленый Y кристаллический (BDC)
Бриллиантовый зеленый YS кристаллический (BDC)
Бриллиантовый зеленый различных сортов и марок
Бриллиантовый кислотный зеленый 6В (Ву) Бриллиантовый кислотный • карминовый 6В (GrE)
Бриллиантовый кислотный карминовый 2G ( Е Бриллиантовый кислотный розаминовый G, 2G (JCO)
Бриллиантовый кислотный синий А (Ву) Бриллиантовый кислотный синий V (Ву) Бриллиантовый ледяной голубой (SCI) Бриллиантовый прочный желтый С (SCI) Бриллиантовый розовый В (L) Бриллиантовый розовый 5G (L) Бриллиантовый синий 6В (JDC) Бриллиантовый фиолетовый 6В, 8В (SCI) Бриллиантовый хромо® яй синий Р (S)
» Азо Азо ТФМ
»
»
»
Диазо
»
Азо Азиновый Акридиновый ТФМ Тиазиновый
»
Диазо ТФМ
»
»
» Азо
»
»
ТФМ
»
» Хинолиновый Ксантеновый
»
Диа о ТФМ Оксазиновый
448 151
54 661 661 661
657 252 252
31 841 789 667 931 931
518 662
662
661
667
57
31
31
714 712
664 800 749 752 518
683 883
X ПР
— — ПР 1 — —
— — ПР
п — —. п
п хо хо по
п — п
п О
— — пн пн
— — пн пн .
— —- ПР
— — п — —
— — — — п
п п хо —
— п — п
— — п — п
— — н р
X п п —
X по по — —
п О хо — .—
п п хо
— — р — РП
— ПР
— — р — РП
п хо ПР
X хо п П
п хо хо —,
——• — р — П
по р
пн — п
— — н — р
п по по
п р О
ЬФ к>
Название (фирма)
Тип красителя
Бриллиантовый черный для шляр В (By) Буффало хромовый черный F, 2BR, 2BN (NAC) Буффало черный MBR (NAC); NB (Sch) Виктория желтый О двойной конц (MLB) Виктория зеленый (DuP), (В), (tM) Виктория зеленый I (В) Виктория прочный фиолетовый 2R (By) Виктория синий (FB) Виктория синий BS (BDC) Виктория синий В (Н), (ССС), (S), (SCI), (StD), (А), (В), (By), (MLB), (tM); В конц., ВХ (DuP); NB (CN) Виктория BS спирторастворимый (SCI) Виктория фиолетовый 4BS (САС), (S), (А), (By), (MLB); N4BS (CN); S4B (GrE); L (SCI), 5J (By); RL, 8BS (MLB) Вири дан FF (MLB) Витолин желтый (tM) Вишневый (DH), (SCI), (StD), (A), (NI), (tM); DN, DII, DIV (B); laN (С); L (K); G (MLB), (tM); R (MLB) Вишневый В, BB, YS (HP) Вишневый 79456 (BDC) Водорастворимый синий различных сортов и марок Галлеин (Bv); паста (В); MS раств. порошок (DH), W порошок (В), (By) Галлеин L порошок (BDC) Галлоцианин (ААР), (GCC), (S), (В), (By), (С); W (DuP) Галлоцианин BD порошок (BDC) Гамбии (А), (К) Гамбин Y (Н). (JBS) G зкстра паста (LBH) Гелиотроп ‘2В (Lev), (A), (By), (L) Диазо » Азо ТФМ » Азо ТФМ » » » Азо Нитрозо Акридиновый ТФМ » » » Ксантеновый » Оксазиновый » Нитрозо » Диазо
Продолжение табл. V1II.27
Номер по В. С. I. (1924 г.) pH Мп? Cel Сг« ВГ<?
— НП П
299 — п п — —
246 — — п — п
138 X О О
657 п О — —
657 X хо —. —
53 — ПР ПР —
729 п хо хо — —
729 п хо хо —. —
729 п хо хо —
729 п хо хо -
53 X п н — —
2 - - РП
793 —. .— - п
677 п — — — X
677 п X
677 п — — п
707 — п — п
781 п р ПР ПР
781 п р п ПР
883 п р О —' —
883 п р О _—
2 — — — ПР
2 — — ПР
386 — 1 •— ПР
£ Гелиотроп BB (By)
" Гераниум GN (С)
Германский оранжевый (JDC) Гернзеиский синий О (MLB) Глубокий черный экстра копц. (JDC) Далия В (WDC)
Дельтапурпурин 5В (А), (Ву), (К) Диадемовый хромовый сине-черный Р6В (LBH) Диадемовый хромовый черный Г новый (LBH) Диазаминовый синий BR (S), NA (CN) Диазаниловый синий BB (MLB)
Диазинавый прямой черный G конц. (BEL) Диазиновый темно-зеленый G (BEL) Диазо индиго синий В (Ву)
Диазо индиго синий BR экстра 2RL (Ву) Диазо ичдиго синий 4GL экстра (Ву) Диазо прочный зеленый BL (Ву) Диазо прочный зеленый GF (Ву) Диазо синии 3R (Ву) Диазолевый пурпурин N4B (CN) Диазолевый синий N2B (CN); 2В (YSt) Диазолевый фиолетовый NN (CN) Диазолевый чистый синий N (CN) Диазолевый чистый синий NFF (CN) Диаминеральный синий В (С) Диаминовый гелиотроп (С) Диаминовый голубой A (MLy), i(C) Диаминовый голубой FF (MLy), (С) Диаминовый красный В (RF), (А) Диаминовый красный 4В i(C) Диаминовый новый синий (С)
Диаминовый синий BB, 2В (LDC), (CD), (MLy), (С)
Диаминовый фиолетовый N (JC), (MLy), (С) Диаминоген В (С)
Диаминогеновый голубой N (С) Диаминогеновый синий BB (С); NB (С) ю Диаминогеновый синий NA (С) w Диаминогеновый темно-синий (С)
»
ТФМ Азо ТФМ Триазо ТФМ Диазо Азо Диазо » »
Триазо »
Диазо
t
Азо прямой Триазо
»
Азо примой Диазо
»
» Триазо Диазо
»
»
Триазо Диазо
» »
Диазо
»
»
»
386 677 П П —
151 — — ПР
707 — П
581 — — p
680 П 0 П
451 — p ox
202 П П П
299 — П П
316 — П П
316 — П 0 П
581 —~ — p
583 П — РП
316 — П П
316 — on П
Б —• on
532 — ox on
532 — HX П
Б —- —" НП
448 X
406 — ПР p
394 П — HP
520 — НП
518 — p
316 — 0 П
406 — __
520 — НП
518 — — p
451 — P ox
448 X
— — НП
406 — ПР p —
394 П — HP
—
— П .—,
316 — no
316 — П П
316 — 0 П
gx ]C |Cft I ICC | laCCCC 1ЁЁалСлЁаЁЙсСС ftCCCCft
Название (фирма) Тип красителя
Диаминонитразол G (С) Диаминонитразоловый зеленый ВВ GF (С) Дианилазурин G (MLB) Дианиловый голубой PH (MLB) Дианиловый красный 4В (MLB) Дианиловый красный R (MLB) Дианиловый синий H2G (MLB) Дианиловый синий H6G (MLB) Дианиловый фиолетовый Н (MLB) Дианоловый бриллиантовый синий 6В (Lev) Дианоловый фиолетовый N (Lev)
Дисперсоловый диазо черный В150 порошок (ICI)
Дисперсоловый прочный красный R150 порошок (ICI)
Дисперсоловый прочный малиновый В150 порошок (ICI)
Дисульфиновый зеленый В (BDC) Дисульфиновый зеленый BS (ICI) Дисульфиновый синий A (BDC) Дисульфиновый синий V (BDC) Дисульфиновый синий VS (ICI) Дифениловый бриллиантовый синий FF (Gy) Дифениловый красный 4В (Gy) Дифениловый фиолетовый BV (Gy) Дифеновый синий В (А) Дифеновый синий R i(A) Дифеновый синий основной (А) Доминго фиолетовый A (L)
Дуразоловый кислотный синий В (BDC) Дуразоловый кислотный синий В (Lev) Дураноловый бриллиантовый синий В300 порошок (ICI)
иазо
»
»
»
»
»
» Триазо Диазо
»
»
ТФМ
»
»
»
»
Диазо
»
Диазо
Азиновый
»
»
Азо
Антрахиноновы й
»
20—1
Продолжение табл. VIII.27
Номер no B. C. I. (1924 r.) pH Mn? Cel Сг6
418 X П НИ
418 X П п
502 —. —
518 — — H .—
448 X
370 X — ПР _
406 —- ПР p —.
520 —. I in
394 П — HP —.
518 — — H
394 П — HP —
— X П ox —
— — П П —
— П ПР —
667 П
667 П no xo
714 П 0 0
712 X ox п
712 X о о
518 — — H
448 X П нп
394 П — HP
851 — П
851 П
951 — П
53 X no
1054 —- — о
1054 — П п
X ПР ПР ОР
CCgcg Iga&CaC I g & | |C |сиСилла I |EC
Вг2
Дураноловый бриллиантовый синий СВ300 тонкий порошок (ICI)
Дураноловый бриллиантовый фиолетовый В300 тонкий порошок (ICI)
Дураноловый фиолетовый 2R300 тонкий порошок (1CI)
Дуроловый черный В, 2В (NAC) Железисто-зеленый для печати (Gy) Желтый JJ (StD)
Желтый М (StD)
Желтый для кожи различных сортов и марок Жирорастворимый красный A (SCI) Жирорастворимый синий В (SCI) Замбези черный V ,(А) Зеленые кристаллы (Н) Зеленый PL (Вт), (В) Зеленый для печати BW (L) Зернистый синий BN (WDC)
Золотисто-оранжевый (ВК), (By), (tM), (WDC) Изумрудный зеленый крист. (В), (By) Индиго дисульфонат Cert (NAC)
I [ндиго экстра 20 (StCl)
Индию экстракт L паста (BDC); порошок (JCO)
Индигокармин (САС), (SCI); X (BDC); D паста (В)
Индигокармин XS (JCI)
Индиготин (NAC); la (ТМС)
Индоловый синий R (А)
Индохромин RR, Т i(S)
Индулин А крист. (BDC)
Камелия (А)
Камелия В (А)
Карбидный черный Е (САС), (SCI) Карболановый зеленый GS (ICI) Карболановый синий BS (ICI) Карболановый фиолетовый 2RS (ICI) to Кардинал (A); G, R i(MLB) сл Кардинал красный (SCI)
Диазо Нитрозо Акридиновый Азо Акридиновый Ксантеновый ТФМ
ТФМ Нитрозо ТФМ
» Азо ТФМ Индигоидный
»
»
»
Индигоидный Азо Тиазиновый Азиновый ТФМ
»
Триазо
ТФМ »
П 0 0 1 о
— П on p on
— П П ПН no
307 П П П
2 — 1 —
793 — —
138 X 0 0 .—
793 — — — —
749 — no
729 П ox ox —
— — П ——
657 X — ox —
5 — 0 0 0
667 П 0 —
729 П xo xo —
151 .—- — ПР
662 X П П
1180 — ПР ПР ПР
1180 -— ПР ПР ПР
1180 — ПР ПР ПР
1180 — ПР ПР ПР
— ПР ПР ПР
1180 —• ПР ПР ПР
135 — .— — П
931 П no no no
861 -— — П
677 П — —
677 П — —
581 — — p —.
— — p p no
— no no no
— —— П
677 П — П
677 П — — —
р
ПР
g cgc|СьI I I IЁI I 1ЁЁЁ Ё ЁЁI I |CICXX
ьо
О')
Название (фирма)
Тип красителя
Кардинал красный J (ВВС)
Карминовый синий A (MLB)
Карбиновый синий V, VX (MLB)
Кар-моизнн (CD)
Кармоизип WS (ICI)
Картамин В (tM)
Каштановый (С); R (К)
Кислотный ализариновый красный В (AALB)
Кислотный ализариновый сине-черный A (MLB)
Кислотный желтый R (ССС)
Кислотный зеленый О (ССС)
Кислотный карминово-синий A (StD) Кислотный карминово-синий V (StD) Кислотный карминовый (Вт) Кислотный кармоизин В (ВК)
Кислотный оранжевый A (Gy); G (BDC); Y (ССС), II'(AJ)
Кислотный сине-черный (AJ), (Br), (JDC); 6В (JCO); экстра конц. (NCW)
Кислотный синий G >(tM)
Кислотный синий 5RS (SAPC)
Кислотный фиолетовый (FB), (JDC); 4В экстра (В); 5В (By); 5ВК (К); 4BS (L), (WDC)
Кислотный фиолетовый 4BS (NCW), (DH)
Кислотный фиолетовый IV (Н); 6В (Gy);
4BNS (S)
Кислотный фиолетовый для печати К (АСС)
Кислотный фиолетовый 4В экстра (Ву)
Кислотный хромовый красный NB (CN)
Кислотный черный В (LDC); 10В, 10ВХ (NCC);
НА (САС), (SCI)
Кислотный черный для печати В (Gy) Китайский желтый (С)
Китайский зеленый (Ву)
Азо ТФМ
» Диазо Азо Ксантеновый ТФМ Азо
»
» Нитрозо ТФМ
» Азо
»
»
Диазо
ТФМ Диазо ТФМ
Азо ТФМ
Азо ТФМ Азо Диазо
» Хинолиновый ТФМ
Продолжение табл. VJII.27
Номер по В. С. I. (1924 г.) pH Мп? Се* Сг6 В?2
176 п НРХ п —
714 п ХО — ПР
712 X ХО п п
252 — —— пн пн —
179 — -— Р — Р
749 — по — Р
677 п — — — X
216 —- ПР ПР -—. ПР
202 п по п — —
138 X О О — —
5 — О о О —
714 п хо — ПР
712 X хо п п
31 — — ПР —
182 — ПР — Р
151 — — ПР — —
246 — — п — п
712 X хо — ПР
289 —. нп — ПР
698 — п — п
53 — ПР ПР — —
698 X — п — п
169 X р Р — —'
698 .—. п — п
216 ПР ПР —- ПР
246 — — п — п
307 п п п — п
801 —• — Р — п
657 п о — —*
Китайский зеленый крист. (Ву)
Китайский синий различных сортов и марок Китоновый красный 6В (САС), (SCI) Китоновый красный G (САС), (SCI) Кетоновый красный 6 (САС), (SCI) Китоновый прочный зеленый V (SCI) Китоновый прочный синий V (SCI) Китоновый синий N (SCI) Кокцеин ЗВ, 3BN (StD) Колумбия зеленый В (А) Колумбия фиолетовый R (Л) Колумбия черный EAW экстра (А) Конго (А) Конго голубой (Lev), (А)
Конго красный (многие); R (Н), (CNW); R экстра (LBH); SD (SCC); W (BDC); WS (ICI)
Конго синий 2ВХ (Lev), (А) Конго экстра (S) Коричневый для кожи Коричневый AL, OF (В) Космический красный экстра (В) Красный I (MLy) Красный 4В для хлопка (В), (К) Красный В для хлопка (К); 4В (GrE); С (SCI), (StD); конц. (tM)
Красный для бумаги PSN (SCI) Красный D для хлопка Красный для шерсти SB, SG (GrE) Крезоловый сине-черный (GrE) Кроцеин ЗВ (StD)
Кроцеиновый алый ЗВ (BDC); MOO (CD), (NAC)
Кроцеиновый алый 3BS (ICI) Ксантин (SCI), (StD), (GrE) Ксиленовый прочный зеленый В (S) Ксиленовый синий A (S) Ксиленовый синий V (S) ю Ксиленовый фиолетовый RL (S) Кумасси зеленый TS (ICI)
ТФМ
» Азо
»
» ТФМ
»
» Диазо Триазо Диазо Триазо Диазо Триазо Диазо
» » Акридиновый
»
Диазо Азо Диазо »
»
» Азо Диазо
»
»
» Акридиновый ТФЛ1
»
» Азо Диазо
657 n 1 1 о ‘
707 П ' —.
57 — — p —
31 ** — ПР —•
54 — — —
735 X no no —
712 X — xo П
714 П — xo П
252 — .— ПН ПН
593 П — ПР
394 П — HP —
581 — — P —.
370 X — ПР .—
520 — ПН —
370 X — HP —
406 — ПР p ,
370 X —- ПР —,
793 — —<• — —
793 — — —- —
370 X — HP —
176 — П HPX П
448 X —
370 X — ПР —
252 — ПН ПН
451 — p ox
54 — — —
246 — — П —.
252 — ПН ПН
252 — — ПН ПН
252 — ПН ПН
793 — — —
735 X no no —
714 П xo —
712 X xo П
53 X no НП .—.
302 — — пн —
I labial I I д?* I I I I a a I 1'п1|'о||аа|91?д1
ьо
00
Название (фирма) Тип красителя
Кумасси кислотный синий RL (BDC) Кумасси прочный черный В (BDC) Кумасси прочный черный В150 (ICI) Кумасси сине-черный (BDC); G, NN, X (Lev) Кумасси фиолетовый AV (BDC) Кумасси фиолетовый R (BDC) Кумасси фиолетовый RS (ICI) Кумасси флотский синий GNX (BDC) Кумасси флотский синий 2RNX (BDC) Кумасси черный для шерсти ESN (BDC) Купраминовый бриллиантовый синий RB (К) Лак алый ЗВ (BDC) Ланафуксин 2BS (С) Ланафуксин 6В, SB, SG (MLy), (С) Ланафуксин NSJ (CN) Ледяной синий (SCI), (В) Лионский синий 0 (MLB) Лиссаминовый зеленый VS (ICI) Лиссаминовый красный 6В (BDC) Лиссаминовый синий В (BDC) Лиссаминовый фиолетовый AVS (ICI) Лиссаминовый фиолетовый RR (BDC) Маджента PS (ICI) Маджента алый G (В) Маджента наилучший (tM) Маджента порошок А (В) Малахитовый зеленый крист (А) Малахитовый зеленый различных сортов и марок Малхаус зеленый (SAPC) Мандариновый G экстра (А), (ВК), (By) Маяковый хромовый красный В (JCO) Маяковый хромцианин R (JCO) Медно-красный N (MLB) Азо Диазо » » Азо ТФМ » Диазо » » Азо » » » » ТФМ » » Азо » » » ТФМ ТФМ ТФМ » Нитрозо Азо » »
Продолжение табл. VIII27
Номер no В C. I. (1924 r.) pH Mn? Cel Cr6
208 П П no
307 П П n —
307 П П П П
246 — П —
53 X PO — —
698 X —- 0 —
698 П no no —
289 П П П —
289 П П p —.
Б П — П —
—- • — П —
216 — IIP ПР —
57 — — p —
54 — — —
54 — ——- —_
664 П xo xo —
707 — П —
735 X no no —
57 .— — p —
Б X — n —
53 П П НП —
53 — ПР ПР —
667 П — — —
— X — НП —
677 П —
— П П —
657 П no —
657 П 0 —
2 _
151 — — ПР —
216 — ПР ПР .—-
202 П no П —
169 X p p —
gc |С I Ig^ccIccccIе lgC I icxxx|| g lg I |
ВГ2
to to
Метаниловый желтый Y (BDC)
Метаниловый желтый многих сортов и марок 'Иетафениленовый синий R (С) Метахромовый сине-черный (Вг) Метиленовый голубой 2В (^)
Метиленовый голубой 2В, ВВ и др. различные сорта и марки
Метиленовый голубой 2В порошок экстра (А) Метиловый синий сверхтонкий (Gy) Метиловый фиолетовый В (В) Метиловый фиолетовый IB (Ву)
Метиловый фиолетовый 2В (В)
Метиловый фиолетовый В, 2В, ЗВ, 4В и др различные марки
Метиловый фиолетовый 2BS (ICI)
Метиловый фиолетовый 5В, 6В, 7В 10В различных марок
Метиловый фиолетовый 6В (Ву)
Монозоль прочный синий 2GS (ICI)
Монохромовый фиолетовый В порошок (LBH) Мышиный серый (А)
Нафтазиновый сине-черный 6В (BEL)
Нафталиновый зеленый V 1SCI), (MLB); NV (CN)
Нафталиновый кислотный зеленый J (StD)
Нафталиновый черный 12В (BDC)
Нафтаминовый голубой I (OeV)
Нафтаминочый прочный черный SCE (К)
Нафтаминовый прямой черный ERK экстра (К)
Нафтаминовый синий 7В (К)
Нафтаминовый синий 12В (К)
Нафтаминочый синий 2ВХ (К), (OeV)
Нафтаминовый синий S3B (К)
Нафтаминовый темно-зеленый В (К)
Нафтаминовый темно-зеленый 2G (К)
Нафтаминовый фиолетовый N (К); R (OeV)
Нафтаминовый черный BE 1К)
Нафтиламиновый черный 10В (GCC), (Ву) Нафтин S (StD)
»
» Азиновый Азо Тиазиновый
»
ТФМ
»
»
»
»
»
»
»
Азо Азиновый Диазо ТФМ
» Диазо Триазо
Триазо
» Диазо
»
Диазо
Диазо Нитрозо
138 X 0 О
138 X о о —— ——
851 .—- п —- р
202 п по п
922 — п
922 — п — п
922 — п п
707 — п — п
680 п по 1 — п
680 п о п п
680 п по по п
680 п п — п
680 п 0 о о нп
683 п оп оп —
683 п оп оп
— п п по .
169 X р р
865 —- нп
246 — — п п
735 X по по —
735 X по по
246 — — п — п
520 — нп — р
— п нп — р
581 — —- р
520 — нп — р
518 -— —— н п
406 — ПР р — ПР
— — по нп —
— — нп -— РП
-—• -— нп —_
394 п — HP р
• — р
246 — —. п — п
2 — — — — ПР
Назьание (фирма) Тип красителя
Нафтионовый красный A (StD) Нафтогеновый голубой 4В (А) Нафтогеновый индиго синий В (А) Нафтогеновый индиго синий R (А) Нафтогеновый синий 2В, 2R (А) Нафтогеновый синий 6R (А) Нафтоловый зеленый 500 (DPC) Нафтоловый зеленый В, В порошок, В паста (многие) Нафтоловый зеленый В9211К (BDC) Нафтоловый зеленый BNS (ICI) Нафтоловый зеленый (JBS), (StD), (tM) Нафтоловый зеленый Y (BEL) Нафтоловый красный A (StD) Нафтоловый сине-черный (ACC), (JWL), (NBC), (CD), (С); S конц. (JBS); 10В (LBH), 10BG, 10BDB (SCC); В конц. (Gy); S (С), В (L) Нейтральный синий R (SCI) Неохромовый сине-черный 6В, G (SCC) Неохромовый черный Т (SCC), NT (CN) Нептуновый зеленый SG (В) Нептуновый синий В (В) Нептуновый синий ВХ, BGX (В) Ниагара голубой (NAC) Ниагара голубой 6В (NAC) Ниагара синий 2В (NAC), (Sch) Ниагара синий 4В (Sch) Ниагара синий G конц. (NAC) Нигрозин различных сортов и марок Нигрозин SC (BDC) Нигрозин WH (В) Нильский синий А (В), (Ву) Нильский синий А готовый (В) Азо Диазо Нитрозо » » » » Азо Диазо Азо » » ТФМ » Триазо Диазо » Триазо Диазо Азиновый » » Оксазиновый »
Продолжение табл. VIII.27
Номер по В С I (1924 г.) PH Мп? Cel с.6 ВГ£
182 — ПР Р
— — О п п
-— — — —’ п
—- — —— п
316 — п п п
—— — — — п
5 — о о о —
5 — О о о —
5 —. п по по ПР
5 — п по по ПР
5 -— О о о —
2 — — ПР
182 — ПР — р
246 — — П — П
208 п п по по
202 -— ПР ПР -—. —
203 п по по — п
667 п — ПР
714 п хо — ПР
712 X хо п п
520 — нп — р
518 — —. н - — п
406 >—. ПР р — ПР
520 — нп .—- р
502 — —- — ПР
865 -—- нп — —
865 — п п — —
865 — нп — —
913 п оп нп РП
913 п оп нп РП
/
to
00
Нигрозин NX (DH) Нитрозо р-нафтол (SCC) Нитрозопрочный зеленый L (JWL) Новый виктория зеленый экстра, различные марки (В)
Новый прочный кислотный зеленый (JC) Ночной зеленый A (tM)
Оксаминовый голубой 5В (В) Оксаминовый голубой 6В (В) Оксидиазоловый синий NJ (CN) Оксидиазоловый черный NJE, NJEE (CN) Оксидиаминовый синий G (MLy), (С) Оксидиаминовый черный JE (С) Оксихромовый черный F (GrE) Омегахромовый цианин R (S) Омегахромовый черный F (S) Омегахромовый черный S, Т (S) Опаловый голубой, синеватый (MLB) Оранжевый MN0 (САС), (SCI); MW (SCI) Оранжевый А для шерсти коиц. (NAC) Оранжевый 2 многих сортов и марок Орион голубой (JBS) Орион синий A (JBS) Орион синий 2В конц. (JBS) Орион темно-зеленый ЕТ (JBS) Орион черный EW экстра (JBS) Ортовишчевый В (А) Основной азурин G (WDC) Основной прочный зеленый V (К) Основной пурпурово-красный НВ (WDC) Основной глубокий черный J конц. 540 (WDC) Осфалиновый оранжевый BRN (OeV) Пакко прямой черный Е экстра (РАС) Палатиновый хромовый коричневый 2G (В) Палатиновый хромовый красный G (В) Палатиновый хромовый черный 6В (YDC), (В) Палахромовый < иний В порошок (РАС) Параминовый голубой 6В (DuP) Параминовый оранжевый R (LBH)
Нитрозо » »
ТФМ
Нитрозо ТФМ Триазо Диазо
» Триазо Диазо Триазо Диазо Азо Диазо Азо ТФМ Азо
»
»
Триазо Диазо
»
Три азо
» Азо Диазо ТФМ Диазо Триазо Диазо Триазо Азо
»
»
»
Диазо »
2 — — .—
2 •—- — —
2 —— -—• —
657 П — no —
5 .— 0 0 0
667 П —
520 — НП —
518 -— .— p 1 -—
502 .—- .—
581 — p —- —
502 — —
581 — .— p —
299 — n П .—
202 П ПР ПР —
299 .— n П —
203 П no no .—
707 ——. П —- —
138 X 0 0 —
151 -—. —> ПР —
151 — — ПР -—
520 — НП —.
502 — — —.
406 .— ПР p —
583 П .—. РП -—
581 1— —> p
169 X p p —
502 —. — —
735 X no no —
448 X -—. ПР —
581 —. — p —
415 X — H П
581 -—. -—. p .—-
98 П -— no —.
216 — ПР ПР .—
202 П no П —
202 П П П —
518 —. .— H —
415 X — H П
ПР
1^1 I I I I I ? I I I I I I I ДД I I I I
Название (фирма) Тип красителя
Параминовый синий 2В, 2В новый (LBH) Параминовый прочный фиолетовый N (LBH) Параминовый фиолетовый В (LBH) Параминовый черный В экстра, BR экстра, Е экстра (LBH) Параниловый зеленый G (А) Параниловый черный Т (А) Парижский фиолетовый (StD) Парижский фиолетовый 6В, 7В (R) Паровой зеленый G (В) Патентованный дианиловый черный ЕВ конц. (MLB) Патентованный зеленый AGL (MLB) Патентованный синий А (А), (К), (MLB) Патентованный синий V '(А) Пигмент алый ЗВ [(разные марки) Плутоформовый черный 3GL (Ву) Плутоформовый черный L, 3GL (Ву) Понсо ВО экстра (А) Понтаминовый голубой 5ВХ (DuP) Понтаминовый голубой 6В (DuP) Понтаминовый оранжевый R (DuP) Понтаминовый синий АХ (DuP) Понтаминовый синий BBF (DuP) Понтаминовый фиолетовый N (DuP) Понтаминовый черный ЕХ, ЁХХ, ЕХ конц. (DuP) Понтациловый карминовый 6В (DuP) Понтациловый карминовый 2G (DuP) Понтациловый сине-черный SX '(DuP) Понтациловый сульфановый синий 5R конц. (DuP) Понтациловый фиолетовый С4В (DuP) Примулин AS (ICI) Диазо » » Триазо ТФМ » Нитрозо Триазо ТФМ » » Азо Триазо » Диазо Триазо Диазо » » » » Триазо Азо » Диазо » ТФМ Тиазоловый
Продолжение табл. VIII.27
Номер по В. С. I. (1924 г.) pH Мп? Cel Сг6 Bl'2
406 _ ПР Р ПР
394 п — HP — р
388 —- п РХ —.
581 — — р — —
— — нп — РП
— — нп п РП
680 п О п п
683 п оп оп —
2 — —> . ПР
581 — — р — —
667 п ПР
714 п хо —. ПР
712 X хо п П
216 —. ПР ПР ПР
545 — п п р
545 — п п Р
252 —• — ПН пн .—.
520 — нп р
518 —. — н п
415 X — н п
502 — .—. .— ПР
406 —. ПР р ПР
394 п —» HP - р
581 — — р — —
57 — — р РП
31 — — ПР — .
246 — — п п
289 п п ПР — ПР
698 п — п
812 — — п —
Примулин экстра (BDC)
Прочный зеленый JJO (SCI)
Прочный зеленый SD (StD)
Прочный зеленый для печати (BDC), (Ву), (К)
Прочный зеленый С для печати конц. (К) Прочный зеленый для шерсти BW (К) Прочный зеленый различных сортов и марок Прочный кислотный зеленый В (NAC) Прочный кислотный красный А (ССС) Прочный кислотный синий A (GrE) Прочный кислотный синий RH (BDC) Прочный кислотный синий X (GrE)
Прочный красный различных сортов и марок Прочный красный различных сортов и марок Прочный красный Е, EAS, EL различных марок Прочный красный экстра (А)
Прочный малиновый 6BL (NAC)
Прочный малиновый GR (NAC)
Прочный паровой зеленый S порошок (SAPC)
Прочный протравной черный В, Т (MLB) Прочный прямой фиолетовый N '(ICA) Прочный сине-черный О (MLB) Прочный синий SLV (StD)
Прочный синий для одежды В, BR, G, GTB (SCI)
Прочный синий для одежды R (САС), (SCI) Прочный синий для шерсти R (NAC); NR (CN) Прочный фиолетовый (DH)
Прочный фуксин G i(Sch)
Прочный хромовый синий G (BDC)
Прочный хромовый синий R (BDC)
Прочный цианоновый для шерсти 3R (NAC) Прочный черный для одежды В i(CAC), (SCI) Прочный черный для шерсти N2B (CN) Прямой алый 6ВХ (NCW)
Прямой бензоазурин J (StD)
Прямой бриллиантовый фиолетовый R конц.
(NCW)
Прямой голубой (StD) 6В конц., FF (NCW)
ТМФ
»
Нитрозо
»
ТФМ
Азо Т<ТМ Азо ТФМ Азо » » » » »
Нитрозо Диазо Азо Азиновый ТФМ Диазо
Азо Оксазиновый Азо
»
»
Диазо
»
»
Азо
Диазо
»
»
812 662 667 X П — оп \ 111
2 — — —
2 — —- —
667 п по О —
657 п по —
667 п —-
176 — П • НРХ п
714 п хо —
208 п п по .—
712 X хо п
176 — п НРХ
182 — ПР .—.
182 .— ПР —
182 —• ПР —.
57 — — р ——
31 .— — ПР —
2 — —' —
299 — п п —
394 п — HP —
865 —— нп —
712 X хо п
289 п п ПР ПР
288 —> нп
208 п п по -—
883 п р о —
153 —. -— —
208 п — ПР —
202 — ПР ПР —
289 .— нп
307 п п п —.
307 п п п —
225 — — ПР _
502 — — —
387 — j — п —
518 — — р —
5 1^1дд^|^д1д^ ^дIДII?IД??^1
Название (фирма) Тип красителя
Прямой голубой различных марок Триазо
Прямой глубокий черный EW (МС); EW экст- »
pa (SCC), i(By)
Прямой конго красный (StD) Диазо
Прямой красный Y (ССС); С (S), (StD), (VSt), »
(FA), (F|B)
Прямой красный 4В различных марок »
Прямой оранжевый R (JDC) »
Прямой прочный синий (SCI) Триазо
Прямой розовый 2В (NCN) Азо
Прямой сине-черный В (By) Триазо
Прямой синий 65 (StD); RBA (L) »
Прямой синий 2В (многие), 2BL (JWL) 2ВХ Диазо
(NCC); BR (КО"Ц. (NCW)
Прямой синий G экстра (Sch); В (StD) »
Прямой синий экстра синеватый (LJ) Триазо
Прямой темно-зеленый (AJ) »
Прямой фиолетовый (StCl); J (StD); С (VSt) Диазо
Прямой фиолетовый К (AJ); J\l (J\ICW), (SCI), »
VMC); R (Sch)
Прямой фиолетовый R (ССС) »
Прямой черный Е (NCC); Е экстра (StCl); ЕЕ Триазо
экстра конц. (NCW); экстра конц. ( СС), GX iNCC); MS (AJ); RL (JWL); 2V (StD)
Прямой чистый синий различных марок »
Прямой яркий прочный синий конц. (ССС) Диазо
Пурпурин 4В (DuP) »
Пурпуровый 5В для хлопка (В) »
Растворимый желтый OL i(StCl) Азо
Растворимый нигрозин (WSS) Азиновый
Растворимый синий ЗМ, 2R (BDC) ТФМ
Растворимый синий для хлопка 4В (StD); »
BJOO (GrE)
Продолжение таьл. V11I.27
Номер по В. С. I. (1924 г.) pH Мп7 Cel Сгв Вг2
520 ПН Р
581 — — Р — —
370 X — ПР — —
370 X — ПР — —
448 X ПР — ПР
415 X п н —. ПР
520 — —— НП — р
225 — — ПР — —
575 — НП п п
520 .— НП р
406 — ПР р — ПР
502 .— ПР
520 —. НП — р
583 п — ПР —► —.
394 п —. Н •— р
394 п — р — р
387 —. — п — —
581 — — р —. —
520 ПН — р
502 — —. —- ПР
448 X — •— ПР
451 — р ох ХР
138 X О О — —
865 — НП — —
707 -— п — п
707 п п
Растворимый синий для шелка ЗВ, 6В, R (StD) Растворимый синий G и R различных марок Реноловый голубой (tM) Реноловый синий В (tM)
Реноловый черный G, G экстра (tM)
Родамин В (В)
Родамин BS (ICI)
Родамин 6G (В)
Родамин 6G (Ву)
Родамин различных сортов и марок
Родамин 6G различных марок
Родил 4G (BDC)
Родулин гелиотроп (Ву)
Родулин розовый SG (Ву)
Розазеин В (MLB)
Розазеин 6G '(MLB)
Розалин, основание (HP)
Розанол 10В (К)
Розеин (FB)
Розеин кристаллы, порошок различных марок
Розеин плитки (BSS)
Розофенин 10В (САС)
Розофениновый розовый (САС)
Рокалин многих сортов и марок
Ротор бриллиантовый синий 2В (ICI)
Рубиновый (FB), (А)
Рубиновый N (В)
Рубиновый 1а мелкие кристаллы (tM)
Рубиновый мелкие кристаллы (А)
Русский красный (А)
Салициновый сине-черный В (К)
Салициновый сине-черный В (К)
Салициновый черный D (К), (OeV)
Салициновый черный U, UL (К)
Сансей красный A (MDW)
Сансей оранжевый II (MDW)
Сансей сине-черный (MDW)
to Сафранилин (Ву)
сп Сафранолин 5G (L)
ТФМ
»
Триазо Диазо Триазо Ксантеновый
»
»
»
»
»
»
Ксантеновый
ТФМ
Азо
ТФМ
»
»
Азо
»
»
ТФМ
ТФМ
»
»
Азо
»
Диазо
Азо
»
»
Диазо Ксантеновый
707 707 — П П 1 —
520 —. НП
502 — — -—
581 p —
749 —. no —
749 .—- no no —
752 —. ПН —
752 — — —
749 — no —
752 — — —
752 —• nil —
— — —— П —
—. •— ox —
749 no —
752 — nn —
677 П — —
225 — РП —
677 П —. — —
677 П -— — —
677 П — —
255 — РП —
225 — НП —.
176 — П HPX П
—- П «' * П —•
677 П —~- — —
— X —. —— ——
677 П n —.
677 П П —.
677 П - —
202 П П П —
202 — ПР ПР —.
299 — П П —.
202 П о П —
176 — П HPX П
151 —• ——“~ ПР —
246 ‘ ~ П —
749 —- no —
752 — П —
СС(Х^|Л|С^аСи|ллиХ|СХХ| I [ I СХХХХ I I I I [ |СЛС
Название (фирма) Тип красителя
Световой синий 550 .(SCI) Световой синий сверхтонкий водорастворимый (MLB) Серебристо-серый N (А), (С) Серый В, ВВ, R (SCI) Синечерный NB (К); Noe (OeV) Сине-черный N для шерсти (CN) Синий МТ1 (DH) ' Синий С4В, 3BS (StD) Синий для одежды R (StD) Синий 1 для хлопка (Ву) Синий N для хлопка (SCI) Синий для хлопка различных сортов и марок Синий для шелка (MLB); BTSL (GrE) Синий для шерсти ЗЕ (BDC) Синий Купьера ВВ (МС) Синий 5R экстра для печати (А) Соланиновый синий В (А) Соланиновый синий BF '(А) Солацетовый синий 4BS (ICI) Солацетовый прочный рубиновый 3BS (ICI) Соледон блекло-зеленый (ICI) Соледон желтый G (ICI) Соледон розовый FF (ICI) Соледон синий 4BCS порошок (ICI) Солохромовый коричневый RHS (ICI) Солохромовый красный В (BDC) Солохромовый красный ERS (ICI) Солохромовый темно-синий BS (ICI) Солохромовый фиолетовый RS (ICI) Солохромовый черный F (BDC); FS (ICI) Солохромовый черный Т (BDC) Солохромовый черный WDFA (ICI) Триазо 1ФМ Азиновый » Диазо » Тиазиновый ТФМ Диазо ТФМ Диазо ТФМ » Азо Азиновый Диазо Азо » » Азо Диазо Азо »
Продолжение табл. VIII.27
Номер по В. С. I. (1924 г.) рн Мп7 Се! Сг6 Вг2
520 . нп Р
707 — п —• п
865 — нп —
865 — нп —. —
246 — — п — п
246 — п —— п
922 — п — п
707 — п —• п
289 п п ПР —- ПР
707 п — п
502 — — — ПР
707 — п — п
707 — п —- п
Б — п по —. п
865 — — нп — —
289 п п ПР —. ПР
— — но п —.
— — по — —
— — р р р —
— п — р — ПР
— — п пн п —.
— п — п — —
— — п —— п
——— — нх нх нх нх
98 п — по — —
216 — ПР ПР — ПР
216 — ПР ПР — ПР
— п п п —. п
169 X р р — —
299 — п п — .—.
203 п по по — п
203 п по по —~ п
Сольвей зеленый Е, EF, GM (SDC) Сольвей зеленый GS (ICI) Сольвей рубиновый S (ICI) Солывей синий Ь (SDC) Сольвей синий BS >(1С1) Сорбиновый красный (В) Спокойный красный (АСС) Стальной серый (А) Стеллахромовый сине-черный R (JBS) Стеллахромовый черный Т (JBS) Султан 4В (Н) Сульфамин (WDC) Сульфаниновый синий D (WDC) Сульфониновый синий 5R экстра (S) Сульфониновый черный В (S) Сульфоновый кислотный синий R (NBC), (GCC), (S), (By); RB (S)
Сульфоновый флотский синий 5RX (NBC) Сульфонцианин G, GR экстра (By) Сульфонцианин 5R (GCC); 3R, 5R экстра
(В), (By)
Сульф шцианиновый черный BA (GCC); В. 2В (By)
Сульфоциан G (CD)
Сунхроминовый зеленый (JDC) Сунхроминовый черный D (JDC) Суперхромовый красный В (NAC) Суперхромовый синий В экстра (NAC) Суперхромовый фиолетовый В (NAC) Таннинный розовый С (BDC) Тетрацианол A (MLy), (С) Тетрацианол V (С) Тиазиновый красный R (В)
Толиловый синий (CD); GR экстра (MLB) Толиловый синий SR (MLB) Толиловый синий 5R экстра (MLB) Толиловый черный В, ВВ (MLB) Триазоловый синий 4В (G.E) Триазоловый синий 2ВХ (GrE) Трианизолин (Мо)
Антр ахиноновы й
Антрахиноновы й »
Азо
» Азиновый Азо
»
Диазо Нитрозо Диазо
»
»
Азо
Диазо
» »
»
»
»
» Азо
»
»
ТФМ
» Азо Диазо Азо Диазо
»
Триазо Диазо
^Ксантеновый
1078 — п п
— — по по по
— — пн пн —-
1054 —— хо п
1054 — — о
54 — — •—-
31 — — ПР —
865 — НП .—
202 — ПР ПР
203 п по по
448 X —
2 —. .
288 — НП
289 -—- — НП —
307 п п п п
208 п п по
289 п п
288 — НП
289 п п ПР —
307 п п П —
288 п п
302 - пн
299 —. п п
216 . ПР ПР
202 п ПР п
169 X р р —
— — —. по
714 п — хо .
712 X хо п
225 НРП
288 — НП .—
208 п п по —
289 п п п
307 п п п -—
520 — НП -
406 — ПР р
752 — пн —
| д? I I I ? I I д л ?gl g I I II дДЭ 1
Название (фирма) Тип красителя
Трисульфоновый фиолетовый (LDC); В (JBS), (S) Трисульфоновый фиолетовый N (S) Тропеолин G (С) Фенохромовый синий 2R (К) Филадельфия желтый G, 2G (А) Фиолетовый ЗВ (CD) Фиолетовый 5В, 6В, 7В (tM) Фиолетовый 4BN для шерсти (NAC) Флотский синий V (MLB) Флотский синий NJ экстра для шерсти (CN) Формиловый фиолетовый NS4B (CN) S4B, 5BN (С) Фосфин 3R (А) Фосфин различных сортов и марок Фуксин RS (ССС) Фуксин различных сортов и марок Хинальдиновый желтый (SCI) Хинолиновый желтый AS (ICI) Хлоразолевый голубой FF (BDC) Хлоразолевый голубой FFS (ICI) Хлоразолевый диазосиний 2RS (ICI) Хлоразолевый коричневый М (BDC) Хлоразолевый оранжевый RN (BDC) Хлоразолевый оранжевый RNS (ICI) Хлоразолевый прочный красный К (BDC) Хлоразолевый прочный красный KS (ICI) Хлоразолевый прочный черный BGS (ICI) Хлоразолевый розовый Y (BDC) Хлоразолевый розовый YS (ICI) Хлоразолевый синий BS (ICI) Хлоразолевый темно-зеленый BN (BDC) Хлоразолевый фиолетовый N (BDC) Диазо » Азо Акридиновый ТФМ » » » Диазо ТФМ Акридиновый » ТФМ » Хинолиновый » Диазо » » » » » » » Азо прямой Диазо » » Триазо Диазо
Номер по В. С. I. (1924 г.) PH Мп7 Се*
387 — — П
394 п — нп
138 X О о
— п нх
793 —
680 п о
683 п оп
698 — п
707 — п
289 п п HP
698 — п
793 —.
793 —.
677 п — —
677 п —
801 р
801 — — р
518 — — н
518 — —— р
316 —. по п
420 X — н
415 X .— н
415 п нп нп
278 — IIP ПР
278 — ПР ПР
Б — п п
225 X н
225 п нп нп
406 — ПР р
583 п — ПР
394 п •— HP
П ОП
Продолжение табл. VIII.27
IjCCJCCgC
"Тх_ф—R(BDC) 2 Хлоразолевый фиолетовый RS (ICI) “ Хлоразолевый фиолетовый WBX (BDC) Хлоразолевый черный ВН (BDC) Хлоразолевый черный Е200 (ICI) Хлораминовый голубой A (S) Хлораминовый голубой FF (S) Хлораминовый синий 2В, 2ВЕ (S) Хлораминовый черный ЕС экстра, Ех экстра (S) Хокусетсу красный (MDW) Хокусетсу красный 4В (WDC) Хокусетсу оранжевый R (MDW) Хокусетсу синий 2В (MDW) Хокусетсу черный W конц., экстра (MDW) Хризоидин экстра (А) Хромовый глубокий черный (IM) Хромовый коричневый R (К); TV (StD) Хромовый прочный черный В (Sch); FN экстра (ААР); FRW (SCI); FW (САС) Хромовый прочный черный CAT (САС) Хромовый прочный черный PV, PW (SCI) Хромовый прочный фиолетовый В (Br), (SCI) Хромовый сине-черный BV конц. (NCW); NR (CN); R (АСС) Хромовый синий GCB (NAC) Хромовый синий N (StD) Хромовый фиолетовый (StD) Хромовый черный '(CD); MCA (МС); WC (ААР) Хромовый черный AN (StD); GT (ААР); Т (АСС) Хромовый черный F (StD); J (Н) Хромовый черный для кожи (StCl) Хромцианин прочный В (САС), (SCI) Централиновый оранжевый R (CD) Централиновый синий 2В (CD) Централиновый фиолетовый N (CD) Церазин (CJ) Цианиновый флотский синий GP (LBH) Диазо » » » Триазо » Диазо » Триазо Диазо » v> » Триазо Диазо Азо Диазо Азо » » Оксазиновый Азо » » » Диазо Триазо Азо Диазо » Азо Диазо
388 п РХ п
388 — пн ПР
387 — —. п
401 —• —• р , п
581 —- — р ——
520 .— нп Р
518 —• — р - ПР
406 —• ПР р ПР
581 — р
*
370 X — ПР
448 X ПР
415 X —— н п
406 —- ПР р ПР
581 —• — р
— - -
—iк х
299 — п п
98 299 п п по п — —
203 п по по п
202 169 п X п р п р —
202 — ПР ПР — —
883 п р п
202 — ПР ПР
169 X Р Р
202 п п п — —
203 п по по — п
299 — п п
581 • р
*
202 п ПР ПР
415 X НП нп ПР
406 — ПР р ПР
394 п HP р
176 —• П ПРО п
288 — нп ПР
СО . -- о
Название (фирма) Тип красителя
Цианиновый флотский синий 2RN, 2RNX (LBH) Черный CBRS (StD) Черный КВ для джута (А) Черный для кашемира ЗВХ (By)
Черный для полушерстяных тканей (By) Черный Е экстра для хлипка (В)
Черный 10В экстра для шерсти (StCl); 6ВА (NI); 6G экстра (tM)
Чикаго синий 6В (А) Чистый желтый (МС) Чистый растворимый синий (BDC), (MLy), (FB)
Чистый синий DSG (BDC) Швейцарский синии (Н) Экстра лондонский синий (B3S) Эльзасский зеленый J (SAPC) Эмпайер кислотный черный (РАС) Эмпайер прочный красный (РАС) Энбико прямой черный Е экстра ^NBC) Энбико хромовый черный F (NBC) Эра хромовый темно-синий В (BDC) Эра хромовый темпо-синий R (ICI) Эра хромовый черный J конц. (BDC) Эра черный F (Lev) Эри зеленый WT (NAC) Эри красный 4В (NAC) Эри прямой зеленый ЕТ (Sch)
Диазо Азиновый Смесь Смесь азо Триазо
»
Диазо
» Акридиновый ТФМ
» Тиазиновый ТФМ Нитрозо Диазо
»
Триазо Диазо Азо
» Диазо
» Триазо Диазо Триазо
Продолжение табл. VIII.27
Номер по В. С. I. (1924 г.) PH Мп7 Се4 Сгв Вг2
289 П п ПР ПР
865 — -— ПН —. .—
Б п -— — — п
Б —-. —— НП —— п
581 - -— р
581 ~~ — р — ——
246 — — п — п
518 — —. р ПР
793 ~~ —. — п
707 — п — п
707 — п п
922 ~~ — — п
707 — п —. п
2 — — — - ПР
246 —• — п — п
176 — п НРХ п
581 — —. р
299 — п п —
202 — п п —. —
202 п по п —.
299 — и п — -
299 — п п .
583 п —. РП —
448 X —. — ПР
583 п РП
Эри прямой черный GX (Teh) Эри фиолетовый BW (NAC) Эри фиолетовый 3R (NAC) Эри черный GXOO (NAC) Эриовиридин В (Gy) Эриозеленый В (Gy) Эриорубин G (Gy) Эриофиолетовый В, RL (Gy) Эриофлоксин 2G, 2GI i(Gy) Эриохромовый красный PEI (Gy) Эриохромовый сине-черный R (Gy) Эриохромовый черный Т (Gy) Этиленовый синии (GrE) Этиловый зеленый (А)
Этиловый кислотный фиолетовый S4B (В)
Этиловый пурпуровый (В)
Этиловый пурпуровый 6В (В)
Этиловый фиолетовый (В)
Этоновый прочный зеленый 3G (JC)
Этоновый прочный карминовый 5В (JC) Этоновый прочный красный G (JC) Японский голубой (JDC) Японский оранжевый R (JDC) Японский темно-синий (JDC)
Триазо Диазо
» Триазо ТФМ ТФМ Азо Азо
»
»
»
»
Тиазиновый ТФМ Азо
ТФМ
»
»
» Азо
» Триазо Диазо
»
581 1 р 1
387 -— — п -
394 п — ПР п
581 —. р —
667 п по хо ПР
735 X по по —
54 -—- — — ПР
53 — по ПР .
31 — — ПР
216 "—- ПР ПР .— ПР
202 п п II
202 -— ПР ПР
922 ——— П
662 X п —
53 X по —
682 X по п .
682 X по п —
682 X по п
735 X по по
57 — — р — РП
31 —1 - —— ПР
520 нп р
415 X нп н п ПР
406 ПР р ПР
Таблица VII1.28
СВОЙСТВА НЕКОТОРЫХ КРАСИТЕЛЕЙ, ИСПОЛЬЗУЕМЫХ ПРИ ТИТРОВАНИИ ЦЕРИЕМ (IV) В КАЧЕСТВЕ ИНДИКАТОРА
(Среда: 2 М серная кислота, 0,5 мл 0,1%-ного красителя на 100 мл титруемого раствора. Ошибки относятся к потенциометрическому методу титрования [230])
Тривиальное название, номер по B.C.I. [204] и тип соединения Окраска Ошибка, % Свойства индикатора
вода кислота
Акроноловый бриллиантовый синий (BDC) 664; ТФМ Синяя Зеленов ато-же л-тая 0 От желтой до ярко-желтой. Четкая конечная точка
Акроноловый бриллиантовый синий AS (ICI) 664(?); ТФМ(?) Ярко-синяя То же 0 +0,2 От желтой до оранжевой и далее до ярко-оранжевой. Фосфорная кислота не оказывает влияния
Ализариновый красный AS, порошок (ICI); тип неизвестен Красная Лимонно-желтая 0 От желтой до бесцветной. Хороший 1 ндикатор, но окраска слабая. Непригоден для [FeCle]3- или [Fe(CN)e]3-
Ализарин сафирол В (By) 1054. Антрахиноновый (ср. с сольвей синим) Синяя Синяя 0 Постепенно приобретает пурпурную и далее розовую окраску, в большом количестве индикатор разрушается до сине-пурпурного соединения. В конечной точке становится бледно-серым. При обратном титровании красно-пурпурная окраска переходит в конечной точке в кремовую. При повторении обратимых процессов окраска бледнеет
Ализарин цианин 2R600 порошок (IC1) 1050(?). .Антрахиноновый (?) Пурпурная Оранжево-розовая + 1,0 От коричневато-розовой до кремовато-розовой. При обратном процессе розовая окраска переходит в кремовую. Разрушаетси при повторных обратных процессах. Похож на 1054. Плохой индикатор
Бирюзовый голубой В (BDC) 66\; ТФМ Синяя Лиственно-зеленая 0 От лиственно-зеленой до оранжевой и далее до ярко-красной. Прекрасный индикатор
Виктория зеленый 1 (В) 657; ТФМ Ярко-синяя Бледно-желтая
Виктория синий BS (ICI) 729 (?); ТФМ (?) Ярко-синяя Тусклая оливко во-зеленая
Диазо прочный зеленый BL (Ву) 532; триазо Синяя Синяя
Дисульфиновый зеленый BS (ICI) 667 (?); ТФМ(?) » Зеленая
Дисульфиновый синий A (BDC) 714; ТфМ » Лиственно-зеленая
Дисульфиновый синий VS (ICI) 712 (?); ТФМ (?) Желтая
Дуразоловый кислотный синий В (BDC) 1054 (?). Антрахиноновый (?) (ср. с сольвей синим) » Синяя
Дураноловый бриллиантовый синий, В300 порошок (ICI), тип неизвестен Ярко-синяя Розовая
Дураноловый бриллиантовый синий CD 300 порошок (ICI), тип неизвестен То же »
Дураноловый бриллиантовый ьэ фиолетовый В 300 порошок S (ICI), тип неизвестен Синяя Розовая
О От желтой до ярко-красной. Очень медленно обесцвечивается. Превосходный индикатор, особенно для •[Fe(CN)6]4-
-f-0,05 От ярко-зеленой до розовой, устойчивой в течение 1 мин, потом становится от 0,01 мл окислителя серой и далее розовой в течение 2 мин; от следующего 0,01 мл—оранжевая, устойчивая. Превосходная конечная точка
0
—0,04 0
—0 8
От индиго синей до серовато-красной. Медленная обратимость
От лиственно-зеленой до оранжевой и далее до ярко-оранжевой. Медленно возвращается к исходной окраске. Небольшая капля титранта уничтожает ярко-оранжевую окраску
От зеленовато-желтой до оранжевой. В присутствии фосфорной кислоты преждевременная конечная точка
—0,02 От яркой лимонно-желтой до ярко-оранжевой
—0,02
Синяя переходит в красную вблизи конечной точки, затем снова становится синей. Эта последовательность повторяется бесконечно
—0,1 0
—0,1 о
о
От розовой до голубовато-розовой и далее до синей. В фосфорной кислоте от розовой до синей; четкая конечная точка
В присутствии и в отсутствие фосфорной кислоты. Окраски хуже, чем у марки В
Очень похож на дураноловый бриллиантовый синий В, но не дает ошибки; фосфорная кислота улучшает четкость конечной точки
Продолжение табл. VIII.28
Тривиальное название, номер по В.С.1- [204] и тип соединения Окраска Ошибка, % Свойства индикатора
вода | кислота
Карболановый фиолетовый 2RS (ICI), тип неизвестен Китайский зеленый (Ву) 657; ТФМ Пурпурная Пурпурная +0,1 Синяя перед самой конечной точкой, затем бесцветная См виктория зеленый
Кумасси фиолетовый R (BDC) 698; ТФМ » Зеленая —0,02 От желтой до оранжевой; конечная точка несколько преждевременная
Кумасси фиолетовый RS (ICI) 698 (?): ТФМ (?) Сине-фиолетовая Изумрудная +0,1 От желтой до оранжевой Постепенно обесцвечивается
Лиссаминовый зеленый VS (ICI)) 735 (?); ТФМ (?) Малахитовый зеленый, кристаллический (А) 657; ТФМ Сине-зеленая Желтая 0 От желтой до оранжевой. Похож на эриозеленый См. виктория зеленый
Метениловый желтый Y (BDC) 138; моноазо Оранжево-желтая Алая -1-0,1 Становится пурпурной. Пурпурная резко переходит в серую. Обратимость немного замедленная
Родамин BS (ICI) 749 (?). Ксантеновый (?) Розовая флуоресценция Розовая —0,04 0 От ярко-оранжевой до чисто-оранжевой и далее до розовой. Странное, но четкое изменение окраски
Сольвей синий BS (ICI) 1054 (?). Антрахиноновый (ср. ализарин сафирол и дуразо-ловый кислотный синий) Синяя Синяя —0,02 От королевской сини до пурпурной и далее красной вблизи конечной точки, затем до синей. При обратном процессе красная. Последовательность повторяется
Фиолетовый Н для печати .(BDC) ТФМ Фиолетовая Ярко-зеленая 0 +0,1 От зеленовато-желтой до оранжевой и далее до ярко-оранжевой. Не очень устойчив к избытку окислителя
Китайский зеленый (Ву) 657 (ср. с виктория зеленым 1 (В) 657; ТФМ) Сине-пурпурная Яблочно-зеленая 0 От желтой до ярко-оранжевой, далее разрушается, приобретая оранжевую окраску Лучше малахитового зеленого, но хуже виктория зеленого
Малахитовый зеленый кристаллический (А) 657 (ср с виктория зеленым 1 (В) 657; ТФМ) Синяя Зеленовато-желтая 0 От зелено-оранжевой до малиновой, далее разрушается, приобретая оранжевую окраску. Хуже китайского зеленого и виктория зеленого .
окислительно-восстановительные индикаторы
295
красной окраске (см. также табл. VIII.24). Метаниловый желтый "зляется простым производным дифениламина и действует как дифениламин, а не как азосоединение. Соединения антрахинона (при предположении, что идентификация правильна) включены по причине их любопытного поведения. Ализаринцианин не применяется в качестве индикатора, но три соединения, идентифицированные по В. С. I. под номером 1054, определенно хорошие индикаторы. Два соединения имеющие, согласно «Указателю красителей» [204], следующие формулы
ОН О ОН NH2 О ОН
1050 1054
очевидно, можно окислить с образованием ряда анион-радикалов,
хинонов и хинониминов. Высокая устойчивость соединения № 1054, возможно, обусловливается отсутствием гидроксильной группы в орто-положении и наличием анионной сульфогруппы. По-видимому, окисление идет в две стадии. Первая стадия заканчивается совсем близко от конечной точки и приводит к гипсохромному сдвигу; возможно, что это стадия одноэлектронного окисления до анион-радикала. Вторая стадия соответствует конечной точке и приводит к батохромному сдвигу; исходная окраска разрушается, вероятно, вследствие дальнейшего одноэлектронного окисления с образованием хинона. Наблюдается расхождение в реакциях соединений, которым приписывается одинаковая структура по «Указателю красителей» (ср. также табл. VIIL24). Кноп часто пишет, что индикатор вновь приобретает свою первоначальную окраску (табл. VIII.24); то же самое не раз отмечал и Бишоп. Теперь это можно объяснить диспропорционированием окисленной формы индикатора с образованием продукта разложения и исходной восстановленной Фермы индикатора. Ясно, что механизмы реакций трифенилметановых красителей и производные бензидина имеют много общего.
Бишоп установил, что трис- (1,10-фенантролин)железо (II) не является хорошим индикатором в титровании гексацианоферра-та(П) раствором церия(IV), особенно при высоких концентрациях кислоты, и приписывает это обмену лигандов между цианидом и фенантролином. В опытах, результаты которых приведены в табл. VIII.28, очень хорош виктория зеленый 1(B), и поэтому его тщательно проверяли в этом титровании. В среде 0,25—7 М серкой кислоты виктория зеленый дает четкие конечные точки, а индикаторную ошибку не удается обнаружить (меньше 0,01 мл на
296
ГЛАВА VIII
25 мл титруемого раствора). Любопытно, что виктория зеленый 1(B) должен быть гораздо лучше китайского зеленого (Ву) и намного лучше, чем малахитовый зеленый кристаллический (А), если все три, как предполагается, являются одним и тем же соединением. Однако именно виктория зеленый 1(B) рекомендуется для указанных выше методов титрования.
По другим данным табл. VIIL28 видно, что трифенилметилнро-изводные ведут себя в отношении перманганата как и в реакции с церием (IV), но надо отметить, что имеются соединения, которые реагируют только с одним окислителем [обычно с церием (IV)], а с другими не вступают в реакцию. Среди соединений, которые окисляются броматом, одно оказалось особенно устойчивым. Это розанилин, который будет рассмотрен в следующем разделе, где говорится об индикаторах, участвующих в специфических реакциях.
Эриоглауцин А и эриозеленый В исследовались в процессе титрования железа(II) церием (IV) [213]. Себеледь с сотр. [231] нашел, что трифенилметановый краситель руброфен не обладает быстрой обратимостью и довольно легко разлагается при титровании броматом. Кноп [232] применял этот индикатор в микрометодах титрования железа (II) перманганатом и установил, что их можно проводить в среде соляной кислоты, если предварительно ввести в титруемый раствор достаточное количество марганца (II). Рекомендации к некоторым красителям, подобные рекомендациям Смита и Блиса [219], сделал Сотджиа-Ровели [233], но, по-видимому, это не дает никаких преимуществ. Белчер и Кларк [234] нашли, что п-этоксихризоидин, предложенный Шулеком для титрования броматом, оказался ценным обратимым индикатором в титровании йодатом по методу Эндрьюса. Белчер [235] предложил тартразин в качестве обратимого индикатора в методах титрования гипогалогенитами; необходимо, чтобы титруемый раствор содержал бромид. Он также исследовал несколько других пиразолоновых красителей, но оказалось, что они хуже тартразина. В этой же работе Белчер показал, что хинолиновый желтый, который вполне пригоден для методов титрования гипохлоритом в кислой среде и в присутствии бромида, становится необратимым в среде бикарбоната, не содержащей бромида, и разрушается гипохлоритом в противоположность утверждениям Сина [236] поддерживаемым Янгом и Дас Гуптой [237]. Необратимо разрушающиеся красители, которые применяются в качестве индикаторов, будут рассмотрены в следующем разделе.
Адамс и Хамакер [157] провели систематическое исследование влияния природы и концентрации кислоты в среде титрования на потенциал перехода трифенилметанового красителя альфазурина G (NAC) (В. С. I. 712); потенциал перехода, по-видимому, является потенциалом первого четко различимого изменения окраски.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
297
Возвращение в исходное состояние и повторное проведение реакции не влияет на потенциал перехода, но изменение окраски получается более четкое, чем было до этого. По методу Циммермана—Рейнхардта были получены явно заниженные потенциалы перехода по сравнению с методом прямого титрования железа(II) церием (IV), а осадок хлорида ртути (II) не мешал изменению окраски. Результаты этих исследований приведены в табл. VIII.29. При титровании 0,1 М растворами получается незначительная индикаторная ошибка.
Таблица VIII.29
ПОТЕНЦИАЛЫ ПЕРЕХОДА АЛЬФАЗУРИНА G (NAC) [157]
Концентрация кислоты, ^trans’ В
прямое титрование по методу Циммермана— Рейнхардта
МОЛЬ-Л—1
1,0 (H2SO4) 1,03 0,92
2,0 (H2so4) 1,02 0,93
4,0 (H2SO4) 1,02 0,93
1,0 (НС1) 1,02 0,96
2,0 (HC1) 1,02 0,95
4,0 (HC1) 1,01 0,93
1,0 (HC1O4) 1,07 1,02
2,0 (HC1O4) 1,06 1,02
4,0 (HC1O4) 1,07 1,03
6,0 (HC1O4) 1,06 1,03
Титрование гексацианоферрата(11)
1,0 (H2SO4) 0,99
1,0 (НС1) 0,98
Больше всего сведений об индикаторах содержится в работе Белчера и Стефена. Брэзайер и Стефен [218] провели систематическое исследование трифенилметановых красителей и некоторых фталоцианинов. Белчер, Брэзайер и Стефен [238] внесли поправки в значения потенциалов перехода и интервалов перехода для некоторых индикаторов, которые были даны Кнолом [209]. Белчер с сотр. [239] исследовал сульфофталеины в качестве окислительно-восстановительных индикаторов для титрования гипохлоритом. Б работах [218, 238] использована нумерация индикаторов по второму изданию «Указателя красителей» (1956 г.) [205]. Поскольку мы не имели возможности использовать это издание, ну-
Таблица VIII30
ТРИФЕНИЛМЕТАНОВЫЕ КРАСИТЕЛИ ИМЕЮЩИЕ НЕУДОВЛЕТВОРИТЕЛЬНЫЕ ИНДИКАТОРНЫЕ СВОЙСТВА [218]
(Звездочкой отмечены индикаторы, которые не выдержали более строгих испытаний)
В.С I. (1956 г,)
Торговое название® номер [205] название
* Малахитовый зеленый (Chem) 42000 Основной зеленый 4
* Бриллиантовый зеленый (Chem) 42040 Основной зеленый 1
* Гвинейский прочный зеленый G (FDN) 42055 Кислотный зеленый 7
* Кислотный зеленый A (FDN) 42095 Кислотный зеленый 5
Бриллиантовый индийский синий 5G (FH) 42120 Кислотный синий 103
* Асилановый прочный зеленый 10G (Ву) 42170 Кислотный зеленый 22
Регина пурпуровый (WSS) 42515 —
Рубиновый HF для кожи конц. (FH) 42520 Основной фиолетовый 2
* Асилановый фиолетовый 4BL (Ву) 42580 Кислотный фиолетовый 21
* Метиловый зеленый (Chem) 42585 Основной синий 20
* Этиловый фиолетовый (Chem) 42600 Основной фиолетовый 4
* Бриллиантовый синий для печати FF (YDC) 42645 Кислотный фиолетовый 15
Бриллиантовый кислотный синий G (YDC) 42655 Кислотный зеленый 90
* Бензиловый цианин 6В (САС) 42660 Кислотный синий 83
Супраноловый цианин 7BF (САС) 42675 Кислотный синий 100
Фуксин кислотный (Chem); кислотный маджента (CV) 42685 Кислотный фиолетовый 19
Бриллиантовый чистый синий 8G (FDN) 42700 Прямой синий 41
* Основной фиолетовый 3ROO (В) 42710 Кислотный фиолетовый 38
* Асилановый бриллиантовый синий R (Ву) 42730 Кислотный синий 24
Асилановый бриллиантовый синий FFB (Ву) 42740 Кислотный синий 109
Основной синий (WSS) 42750 Кислотный синий НО
Растворимый синий (Chem); растворимый синий ЗВ (CV) 42755 Кислотный синий 22
Опаловый голубой SS (WSS) 42760 Растворимый синий 23
Основной синий 6В (Chem) 42765 Кислотный синий 119
Растворимый метиловый синий (WSS) 42770 Кислотный синий 48
Спирторастворимый синий (Chem) (CV) 42775 Растворимый синий 3
Темно синий (Chem) 42780 Кислотный синий 93
* Монокромсвый бриллиантовый фиолетовый 5В (Ву) 43550 Протравной фиолетовый И
298
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
299
Продолжение табл VIII.30
Торговое название2 В C.I. (1956 г.)
номер [205] название
Бриллиантовый монохромовый фиолето- 43565 Протравной фиолетовый
вый 2В (LBH) 1
Нафтохромовый фиолетовый 2В (САС) 43570 Протравной фиолетовый 28
Эриохромовый цианин R (Chem) 43820 Протравной синий 3
Хромазурол S (Chem) 43825 Протравной синий 29
Дурохромазурол В (YDC) 43830 Протравной синий 1
Алмазный синий FBC (Ву) 43855 Протравной синий 47
Омегахромовый фиолетовый R (S) 43865 Протравной синий 16
Бензиловый фиолетовый 5BN (САС) 44055 Кислотный синий 24
Базолановый синий R (В) 44060 Кислотный синий 88
Базолановый синий FG (В) 44075 Кислотный синий 86
Темно синий (Chem) 44085 Основной синий 15
Базолановый синий G (В) 44095 Кислотный синий 97
Прочный синий для шерсти FBL (FH) 44510 Кислотный синий 123
Нафтохромазурин В (САС) 44535 Протравной синий 28
а (Chem) = Laboratory Chemical suppliers:
(FDN) = Franken-Donders’ n. v. Tilburg, Holland: (FH) = Hoechst-Cassella Dyestiffs Ltd., Leeds.
Об остальных сокращениях см. приложение к табл. VIII. 26.
мерация красителей приведена нами по первому изданию 1924 г. [204]. В работе [238] приведены поправки, которые возникли из-за несоответствия между поведением некоторых из этих индикаторов при титровании церием (IV) в среде хлорной кислоты (наблюдавшемся Д. Рао и В. Рао [217]) и значениями потенциалов перехода (приведенными Кнопом). Это входит в предварительные сообщения с комментариями и составляет часть обширного исследования Брэзайера и Стефена [218].
Большинство исследованных красителей первый раз изменяет свою окраску при подкислении, а затем еще раз от избытка пе-Pnn(IV); у многих окраска изменяется плохо или же изменение окраски устойчиво только в течение нескольких секунд. Ряд красителей оказался непригодным из-за их плохой обратимости, медленного изменения окраски и нечеткой конечной точки, а поэтому Дальше не исследовался. В табл. VIII.30 эти красители помечены звездочкой.
Таблица VII 1.31
ИНТЕРВАЛ ПЕРЕХОДА ОБРАТИМЫХ ТРИФЕНИЛМЕТАНОВЫХ ИНДИКАТОРОВ В 1.0 М СЕРНОЙ КИСЛОТЕ И В 1,0 М ХЛОРНОЙ КИСЛОТЕ [218]
Торговое название В. С. I. (1956 г.) Интервал переходя, В
номер [205] название серная кислота хлорная кислота
Асилановый прочный зеленый BBF (By) 42050 Кислотный зеленый 8 0,945—1,005 1,145—1,185
Патентованный синий V (FDN) [Альфазурин G, B.C.I. (1924 г.) 712] 42051 Кислотный синий 3 1,105—1,145 1,120-1,170
Эриоглауцин (Chem) [эриоглауцин A (Gy) B.C.I. (1924 г.) 671] 42090 Кислотный синий 9 1,095—1,155 1,120-1,190
Ксиленовый пианол FF (Chem); [ксиленовый ци-анол FF (S) B.C.I. (1924 г.) 715] 42135 Кислотный синий 147 1,085—1,145 1,125—1,195
Лиссаминовый фиолетовый 10В (ICI); [ксиленовый прочный синий 10В (S)] 42571 Кислотный синий 13 1,090—1,150 1,075—1,140
Виктория чистый голубой ВО (FDN) 42595 Основной синий 7 1,065—1,110 1,110-1,180
Астрацианин В (FH) 42705 Основной синий 18 1,075—1,115 1,090—1,130
Ксиленовый бриллиантовый синий FBR экстра (S) 42735 Кислотный синий 104 1,125—1,165 1,145—1,165
Эриозеленый (Chem) [эриозеленый В, B.C.I. (1924 г.) 735] 44025 Кислотный зеленый 16 1,115—1,155 1,135—1,195
Виктория лак синий R (ICI); [виктория голубой R (LBH)] 44040 Основной синий 11 1,040—1,085 1,060—1,125
Сетзпалин (Chem) (вероятно, сетопалин конц. (Gy) не указан) — Основной синий 23 1,105—1,150 1,120—1,180
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
301
Таким образом, число хороших индикаторов уменьшилось до одиннадцати, пять из которых были, по-видимому, исследованы Кнопом или Адамсом и Хамакером. Попытки определить их формальные потенциалы оказались безуспешными. Неудачны были и попытки применить метод Смита и Баника [81] (по этому методу работают с растворами ванадия (V) и ванадия (IV) в серной кислоте при постоянном потенциале), так как этому мешала окраска реагента и индикаторов, а также высокие потенциалы индикатора. Поэтому потенциалы перехода и интервалы перехода определяли по видоизмененному методу титрования Адамса и Хамакера. Результаты приведены в табл. VIII.31. Видно, что они значительно выше ранее приводимых значений, причем в среде хлорной кислоты они существенно выше, чем в серной кислоте. По утверждениям авторов, область потенциалов одинакова независимо от направления, в котором проводилось титрование, за исключением красителя С. I. 42595; избыток в одну каплю 10-3 М раствора церия (IV) не вызывает существенного разложения индикатора в течение 10 мин.
Изменение окраски наблюдалось также в среде азотной кислоты, но оно было в значительной степени смещено от точки эквивалентности, так что не рекомендуется применять эти индикаторы при работе в среде азотной кислоты.
При титровании 0,01 М реагентами достаточно брать 0,5 мл 0,1%-ного раствора индикатора на 50—100 мл титруемого раствора. В присутствии индикатора виктория чисто-синего ВО (FDN) 42595 использовали 0,15 мл индикатора. Индикатор надо добавлять в раствор незадолго до установления конечной точки, особенно при обратном титровании церия (IV) железом (II). При условии, что избыток окислителя не превышает 0,1 мл 0,01 М раствора, обратимость очень хорошая, воспроизводимость результатов находится в пределах 0,02 мл для 25 мл титрования, а индикаторные ошибки (по данным потенциометрического титрования) не превышают 0,15 мл 0,01 М раствора церия (IV).
При использовании 0,01 М титрантов окраска индикатора изменяется медленно, а при 0,1 М — быстро и с малой индикаторной ошибкой. Однако авторы утверждают без каких-либо объяснений, что получались ошибки индикаторов вплоть до 1% по сравнению с потенциометрическим методом, и считают, что все индикаторы хуже индикаторов группы трнс-(1,10-фенантролин)железа(Н). Все они разрушаются избытком окислителя, и ни один из новых индикаторов не показал преимуществ перед индикаторами Кнопа. При титровании бихроматом ни один из индикаторов не дает удовлетворительных результатов даже в условиях высоких концентраций серной кислоты.
Поскольку ничего или почти ничего не известно о механизме окисления этих соединений, Брэзайер и Стефен попытались собрать
302
ГЛАВА VIII
и объяснить экспериментальные наблюдения и пришли к следую-щим выводам:
а) Два ароматических кольца должны иметь аминогруппы в пара-положении; предпочтительнее, чтобы они имели алкильные или бензильные заместители; третье кольцо не должно иметь в пара-положении ни аминогруппы, ни гидроксила. Лучшие индикаторы имеют такое строение.
б) Наличие простых заместителей в кольце, как, например, метила или галогена, должно, по всей вероятности, привести к приемлемым свойствам индикатора.
в) Если соединение имеет во всех трех кольцах незамещенные или замещенные аминогруппы в пара-положении, то оно менее устойчиво к деструктивному окислению. Исключение составляют три-(п-аминофенил)'метилпарарозанилин и его .м-метилпроизводное розанилин, которое не изменяет своей окраски от действия окислителя.
г) Если вместо аминогрупп в пара-положении находятся гидроксильные группы, то получается краситель три-(n-оксифенил) метил, который непригоден в качестве индикатора.
Те же авторы исследовали также все трифенилметановые красители в методах титрования галогенсодержащими окислителями, включая гипохлорит, хлорамин-Т, бромат, иодат, бром и иод, и нашли, что некоторые из этих красителей четко, хотя и необратимо, изменяют свою окраску. Об этом пойдет речь в соответствующем разделе.
Таблица VII 1.32
ФТАЛОЦИАНИНОВЫЕ КРАСИТЕЛИ, ИССЛЕДОВАННЫЕ В КАЧЕСТВЕ ИНДИКАТОРОВ [218]
В. С. I. (1956 г.)
Торговое название номер [205] название
Индантреновый бриллиантовый синим 4G (Ву) Монастралевый прочный синий В (ICI) Дуразоловый синий 8G (ICB Синий для бумаги 10GS (ICI) Альциановый синий 8GX (ICI) Монастралевый прочный синий RFS (ICI) Монастралевый прочный зеленый GNS (ICI) Цапоновый прочный синий HFL (В); метазоловый прочный синий 2G (ICI) 74140 74160 74180 74200 74240 74250 74260 74350 Кубовый синий 29 Пигментный синий 15 Прямой синий 86 Прямой синий 87 Играиновый синий 1 Пигментный синий 15 Пигментный зеленый 7 Растворимый синий 25
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
303
Рао обнаружил, что медная [240—242] и никелевая [243] соли фталоцианинсульфокислоты могут быть использованы в качестве индикаторов в цериметрии. Исходя из этого, Брэзайер и Стефен [218] исследовали ряд других фталоцианиновых красителей, которые перечислены в табл. VIII.32. В большинстве случаев оказалось, что обратимость этих индикаторов плохая и они хуже трифениловых красителей. Единственным хорошим индикатором является фталоцианинсульфонат кобальта, имеющий номер 74140. Области потенциалов перехода составляют 0,935—0,990 В в 1,0 М серной кислоте (предполагается, что этот индикатор можно использовать при титровании бихроматом) и 0,985—1,035 В в 1,0 М хлорной кислоте. В серной кислоте изменение окраски происходит от бирюзово-синего цвета до пурпурного, но вскоре пурпурная окраска обесцвечивается вследствие деструктивного окисления. В хлорной кислоте бирюзово-синий цвет медленно переходит в сине-розовый и быстро обесцвечивается.
ВЫВОДЫ
Низкая стоимость и довольно хорошее изменение окраски позволяет использовать некоторые красители, в частности трифенилметановые соединения, в качестве индикаторов в методах титрования церием(IV); это особенно относится к виктория зеленому I для титрования гексацианоферрата (II) в тех случаях, когда мешает присутствие высших хелатных комплексов, о которых речь пойдет в следующем разделе. Установлено, что только один индикатор— бирюзово-синий G — имеет преимущество перед производными бензидина в титровании бихроматом. Вопрос о механизме окисления индикаторов не ясен, и, по-видимому, это положение сохранится до тех пор, пока не будут располагать чистыми индивидуальными соединениями с установленной структурой. Между производными бензидина, содержащими простые единицы дифениламина или бензидина, и трифенилметановыми соединениями существует близкая аналогия. Неустойчивость к избытку окислителя и малочисленность соединений с очень четким изменением окраски составляют главные недостатки обратимых индикаторов.
3. ДИИМИНОЖЕЛЕЗО(П) И БЛИЗКИЕ ЕМУ ХЕЛАТНЫЕ КОМПЛЕКСЫ
ВВЕДЕНИЕ
О свойствах дииминожелеза (II) и близких ему хелатных комплексов известно больше, чем о какой-либо другой группе индикаторов, несмотря на то что и там есть области, сведения о которых отсутствуют или неполные. Литература по этим соединениям ис
304
ГЛАВА VIII
ключительно обширна, но более подробное рассмотрение ее выходит за рамки данной монографии; поэтому она будет приведена только в общих чертах. Такое изобилие литературы является следствием огромного интереса к этим соединениям в спектрофотометрии, в процессах экстракции растворителями, в реакциях маскирования и в других областях аналитической химии, теоретической неорганической и физической химии, не говоря уже о применении их в качестве окислительно-восстановительных индикаторов. Конечно, столь широкий интерес способствовал пониманию их поведения. Появление краткой, но прекрасной монографии Шильта [244] позволяет существенно сократить наш очерк; мы отсылаем читателя к этой книге, где он сможет получить больше подробностей и сведений.
Общей для всех соединений этого рода является а,а'-диимино-группа
I I
—N=C—C=N—
которая обычно, хотя иногда и в измененном виде, составляет часть сопряженной ароматической системы; эта группа действует как бидендатный лиганд, образуя устойчивые пятичленные хелатные циклы с ионами металлов. В целом класс этих соединений представлен обычным и широко применяемым 1,10-фенантролином и его гексакоординационным трис-хелатом с железом (II), который имеет оранжево-красный цвет и окисляется в бледно-голубой комплекс железа (III).
Скорость обмена электроном между ионами комплексов железа (II) и железа (III) очень велика (константа скорости больше 105 л-моль-1-с-1 при 25 °C [245]). Она намного больше скорости обмена между акво-ионами (4,0 л • моль-1 • с-1 при 25 °C [246]) или между ионами аквожелеза (II) и аквомонооксожелеза(Ш) (3,2-103 л-моль-1-с-1 при 25°С [246]). Скорость диссоциации комплекса трис-(1,10-фенантролин)железа(II) очень мала (1СНс-1 при 25°C [247]), как и скорость обмена железом(II) между ионом аквокомплекса и ионов хелатного комплекса, что было доказано методом изотопной метки. Константы скорости реакции окисления хелата железа (II) раствором церия(IV) и реакции восстановления хелата железа (III) раствором железа (II) превышают 104 л-моль-1-с-1 при 25 °C [248, 249]. Следовательно, в механизм
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
305
окисления не входят ни предварительная стадия диссоциации, ни прямая координация окислителя с железом(II) в комплексе. Структуры обоих комплексных ионов очень близки, поэтому не требуется большой перегруппировки, помимо небольшого смещения плотности л-электронов, которому соответствует очень короткое время релаксации. Механизм обмена электронами почти наверняка сводится к прямому тоннельному механизму продвижения электрона по ароматическим лигандам.
Окраски обоих комплексных ионов очень различаются по интенсивности. Окраска оранжево-красного комплекса железа (II) очень интенсивна (Z.max=510 нм, е=1,1Ы04 л-моль-1-см-1) [250], тогда как бледно-голубая окраска комплекса железа (III) слаба (Ятах= =590 нм, е=6-103 л-моль-1-см-1) [251]. Оба комплекса находятся в низкоспиновом магнитном состоянии. Большее поглощение иона трис-(1,10-фенантролин)железа(II) обусловливается, вероятно, переносом заряда от металла к лиганду. Поглощение иона комплекса железа (III), возможно, является результатом d—d'-перехода или переноса заряда от лиганда к металлу либо, что менее вероятно, результатом Мл*-перехода.
1,10-Фенантролин, 2,2'-дипиридил и другие лиганды, содержащие а,а'-дииминогруппу, образуют простые или смешанные комплексы со многими металлами [244], но интерес, обусловленный окислительно-восстановительными индикаторными свойствами, сосредоточен на триаде железо—рутений—осмий и на ближайших к ним металлах, у которых интенсивности окраски и формальные потенциалы наиболее благоприятны. Последующее изложение касается веществ, используемых или предложенных в качестве индикаторов. Однако не следует забывать, что многие другие ионы тоже образуют комплексы и могут участвовать в конкурирующих реакциях с металлом индикатора.
ИСТОРИЧЕСКИЙ ОБЗОР
В 1889 г. Гердисен [252] синтезировал 2-метил-1,10-фенантро-лин, который не дает устойчивого комплекса с железом (II) и поэтому теряет свое значение в качестве хромогенной группы. Блау наблюдал интенсивную красную окраску комплекса 2,2'-дипириди-ла и применял его [253, 254], но только в 1898 году он опубликовал замечательное полное исследование 1,10-фенантролина и его комплексов [255]. Он показал сходство между 2,2'-дипиридилом и 1,10-фенантролином и то, что другие дипиридилы, исключая 2,2'-Дипиридил, не вступают в реакцию с солями железа (II). Блау синтезировал и охарактеризовал комплексы железа (II) и железа (III) с 2,2 -дипиридилом и 1,10-фенантролином и показал обратимость окислительно-восстановительных реакций. Он получил также комплексы с двухвалентными металлами — никелем, медью, кобальтом, 20- -918
306
ГЛАВА VIII
цинком и кадмием — и с трехвалентным хромом и показал, что это — комплексы Вернера; однако их октаэдрическая конфигурация не была доказана до тех пор, пока в 1912 г. Вернер не разделил оптические изомеры комплекса 2,2/-дипиридила с железом (II).
Очень странно, что прошло более 40 лет, прежде чем работа Блау нашла применение в аналитической химии; фактически такое положение существовало до 1928 года, когда были получены комплексы серебра [256, 257], а при изучении реакции железа (II) с перекисью водорода был использован 2,2'-дипиридил [258]. Впервые этот реагент применили в количественной колориметрии для определения железа в пиве [259]; это случилось как раз после проявления большого интереса к индикаторным свойствам химических соединений. Поиски новых индикаторов начались в 1931 г. после работы Вальдена, Гаммета и Чепмена [260] по применению 1,10-фенантролинжелеза (II) в качестве индикатора в периметрии. Это было очень своевременно, так как Атанасиу [261], Фурман [262] и Виллард [263] начали систематическое изучение церия (IV) в качестве титранта, что и обеспечило широкое распространение этого реагента в титриметрии. Вальден с сотр. [264] продолжил их работу и предложил 5-нитропроизводное 1,10-фенантролина [265], которое имеет гораздо более высокий формальный потенциал; предполагалось, что введение заместителей настолько изменит формальный потенциал, что можно будет применять индикатор в условиях особых требований. Обширные систематические исследования проводили Смит (аналитические работы) и Кэйс (синтез соединений); в результате этих исследований появились индикаторы с формальными потенциалами в пределах 0,84—1,33 В, а также несколько важных колориметрических и маскирующих реагентов. Печальным фактом является то, что из-за изобилия индикаторов производители (за исключением собственной компании Смита) производят только комплексы 1,10-фенантролина и иногда 2,2'-дипи-ридила и не используют никакие другие в аналитической химии.
ЛИГАНДЫ, СОДЕРЖАЩИЕ ад/-ДИ ИМИ НО ГРУППУ
Исходные лиганды 1,10-фенантролин и 2,2'-дипиридил уже широко исследованы, получены многие их производные (более 150 производных 1,10-фенантролина) и проведены исследования по выяснению влияния заместителей. Тридентатный лиганд 2,2',2"-терпи-ридил, который образует бис-хелатные комплексы, имел такие же свойства, как и 2,27-дипиридил, и тоже привлекал внимание исследователей [266]. Включение одного дополнительного углерода между атомами азота
ОКИСЛИТЕЛЬНО ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
307
с образованием шестичленного хелатного цикла, как, например, 2,2'-дихинолилметана [267], приводит к менее устойчивым комплексам, тогда как а-имино-а'-аминогруппа
I I
—N=C—С—NH, I
дает хелаты, которые больше напоминают комплексы типа этилендиамина, чем типа фенантролина; в качестве примера были исследованы 8-аминохинолин [268] и 2-пиколиламин [269]. Менее устойчивые и более слабые по окраске комплексы получаются в тех случаях, когда а,а'-дииминогруппа не является частью ароматической системы, как, например, глиоксаль-бнс-метилимин [270], 2,3'-дипиперидеин-1 [270] или диацетилгидразон [271].
Очень заманчиво было бы связать степень делокализации электронов в кольцевых системах, в которые входят иминогруппы, с устойчивостью и коэффициентом молярной экстинкции получающегося комплекса, но для этого не имеется еще исчерпывающих количественных данных. Если одна иминогруппа ароматическая, например 2-пиридил, а другая алифатическая, как в 2 пиридиналь-метилимине [271], такой комплекс устойчивее комплекса с полностью алифатическим глиоксаль-бнс-метилимином [270], но гораздо менее устойчив, чем совсем ароматический 2,2'-дипиридил. В свою очередь дипиридильные комплексы, в которых ароматические кольца отделены друг от друга связью, менее устойчивы, чем комплексы 1,10-фенантролина, у которого ароматические кольца слиты [272]. Кроме того, доказано, что пятичленные гетероциклические кольца, как, например, тиазол и имидазол, в которых делокализация в большей степени ограничена, образуют менее устойчивые комплексы, чем система из двух ароматических колец, подобных 2,2/-дипиридилу, но более устойчивые, чем система из ароматического кольца и алифатического заместителя типа 2-пириди-нальметилимина. Примерами могут быть 2-(2-пиридил)-имидазолин и 2-(2-пиридил) бензимидазол [273], 2-(2-пиридил)-тиазол [274, 275] и 4-(2-пиридил) тиазол [275]. Структура этих соединений показана на рис. VIII.4; нумерация положений заместителей соответствует в них нумерации в фенантролиновых и дипиридиловых кольцах.
Несмотря на то что наиболее устойчивые комплексы должны были бы получаться с полностью ароматическими высоко делокализованными соединениями, заместители, которые могут увеличивать степень делокализации, не обязательно повышают устойчивость комплекса или интенсивность их поглощения. Верно, что 4,7-дифенил- 1,10-фенантролин образует с железом (II) комплекс, У которого коэффициент молярной экстинкции в два раза больше по сравнению с незамещенным фенантролином и константы комп-20*
308
ГЛАВА VIII
1,10-фенантролин
Н\ /н
Н3С—W 'vQ—СН3
глиоксаль-бис-метилимин
Н3Ск ,СН3
H2N—W 4SI—NH3
2,2'-дипиридил
2-пиридинальметилимин
2,2'-би-(3,4,5,6-тетрагидропиридин)
диацетилгидразон
Н
2-(2-пиридил) бензимидазол
2-(2-пиридил) имидазолин
2-(2-пиридил) тиазол
тиазол
Рис. VIII. 4. а, а'-Дииминовые лиганды.
лексообразования находятся примерно в том же соотношении. Однако Ирвинг [276, 277] показал, что заместители, которые уменьшают копланарность колец и этим ограничивают делокализацию (как в случае 1,1'-диизохинолина— нафталинового аналога дипиридила [277]), образуют комплексы со значительно меньшим коэффициентом молярной экстинкции, чем у незамещенного дипиридила. Примерами такого npoi транственного эффекта являются 3-фенил- [274] и 3,3'-диметил-1,2,2'-дипиридилы [278]. При образовании комплексов возникают еще другие стерические эффекты в тех случаях, когда все заместители пространственно препятствуют расположению двух или более лигандов вокруг иона металла. Это происходит с заместителями, даже с метильными группами, в положениях 6 и 6' у 2,2'-дипиридила [279] и в положениях 2 и 9 у 1.10-фенантролина [280]; в каждом случае диметилпроизводные
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
3091
не образуют трис-хелатных комплексов с железом(II), несмотря на то что могут образовать тетраэдрические бис-хелатные комплексы с медью(1). Подобным образом стерический эффект препятствует образованию поликомплексов [281], хотя делокализация в 2,2'-Дихинолине очень, сильна. На рис. VIII.5 показаны структуры упомянутых соединений.
Н3С СН3
6,6'-диметил-
2,2'-дипиридил
3,3'-диметил-
-2,2'-дипиридил
1,1 '-дни зохинолин
2,2' -дихинолин
3-фенил- 2,2'-дипиридил копланарные кольца
2,9-диметил-1, 10-фенантролин затрудненное полихелатирование
Рис. VIII. 5. Стерические эффекты.
Прежде чем закончить рассмотрение систем, содержащих а,а'-дииминогруппу, стоит остановиться на немногих лигандах, которые еще не были исследованы в отношении индикаторных свойств. Комплекс бис- (2,2',2"-терпиридил)железо (II) имеет коэффициент молярной экстинкции 1,25-104 л-моль^-см-1. При фени-лировании каждого из трех пиридиновых колец в положении 4 получается лиганд [282, 283], бис-хелатный комплекс которого с железом (II) имеет самый высокий коэффициент молярной экстинкции (3,02-104 л• моль^1 • см-1 при 583 нм), отмеченную до сих пор Для этого класса соединений. Хотя в этом комплексе не имеется слитых колец, делокализация по всем шести кольцам очень высокая. Структурным элементом является группа
II II I
—N=C—C=N—С—C=N—
310
ГЛАВА VIII
и ее можно обнаружить в соединениях, кроме терпиридила. Например, 2,6-пиридиндиамидоксим дает бис-хелатный комплекс железа (II), коэффициент молярной экстинкции которого 1,68-104 л-моль-1-см-1 при 523 нм [284].
4,4',4"-трифенил-2,2',2"-терпиридил 2,6-пиридиндиамидоксим
Триазины, в частности симм.- 1,3,5-триазин и его производные, еще более интересны. В молекуле трис- (2'-11Иридил)-1,3,5-триазина имеются три отдельные а,а'-дииминогруппы, расположенные симметрично (структура I). Пиридильные группы, конечно, могут свободно вращаться и ориентироваться таким образом, чтобы атомы азота с отрицательным зарядом были по возможности предельно отдалены. Тем не менее этот лиганд образует бис-хелат с железом (II) [285—287], коэффициент молярной экстинкции которого в водном растворе 2,26-104 л-моль_1-см-1 при 593 нм, так что предпочтение надо отдать структуре II.
/и/шс-(2'-пиридил)-1,3,5-триазин
Следовательно, это соединения одного типа с терпиридилом; фе-нилирование в трех положениях 4, 4' и 4" приводит к повышению коэффициента молярной экстинкции до 2,52-104 л-моль-1-см-1 при 505 нм. Однако до сих пор не используют потенциал а,а'-диимино-группы в соединении II. Если соединение I или II способно образовать трис-хелатный комплекс типа дипиридила, можно предвидеть, что образуется полимер. Производные 1,2,4-триазина образуют трис-хелаты, как и фенил-2-пиридилкетоксим. Последний можно экстрагировать; коэффициент молярной экстинкции его изменяется
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
311
в зависимости от pH. Однако после экстракции амиловым спиртом изменение не наблюдается (е=1,56-104 л-моль-1-см-1 при 550 нм) [288]. 3-(2-Пиридил) -5,6-дифенил-1,2,4-триазин дает трис-хелат с железом (II); коэффициент молярной экстинкции комплекса 2,4-104 л-моль-1-см-1 при 555 нм, а при введении фенила в положение 4 пиридинового кольца возрастает до 2,87-104 л-моль-1-см-1 [289]
фенил-2-пиридилкетоксим
3-(2- пиридил)-5,6-дифенил-
-1,2,4-триазин
В молекулах бис-3,3'- (5,б-дичиетил-1,2,4-триазина) плоскости колец стремятся стать под прямым углом друг к другу. Эти лиганды образуют трис-хелаты с железом (II), коэффициент молярной экстинкции которых 1,5-104 л-моль-1-см-1 при 493 нм; можно предположить, что в соединении III имеются пространственные препятствия образованию хелата, так что соединение IV в этом отношении лучше:
III
IV
бис-3,3'-(5,6-диметил-1,2,4-триазин)
До сих пор не обращалось внимания на исследования различных типов лигандов в качестве индикаторов, хотя их изучали очень тщательно в других отношениях [290—296]. В дальнейшем мы будем рассматривать незамещенные и замещенные соединения МО-фенантролина, 2,2'-дипиридила и 2,2'2"-терпиридила, используя сокращенные обозначения phen, bipy и terpy соответственно.
ОБРАЗОВАНИЕ КОМПЛЕКСОВ
Здесь будет приведено значительное количество данных в подтверждение-трехстадийного механизма образования комплекса между бидентатным лигандом L—L и сольватированным ионом металла М(Н2О)в (обозначения зарядов упускаются); в этом ме-
312
ГЛАВА VHI
жанизме отрыв первой молекулы воды является стадией, определяющей скорость реакции:
(H2O)SM(H2O) + (L-L) (H2O)SM(H2O) (L-L)
(H2O)„M(H2O)(L-L) =₽=* (HaO)6M(L-L) + Н2О
,L
(H2O)6M(L-L) (Н2О)4д/ | + H2O
Холиер и др. [297, 298] нашли, что константа скорости второго порядка для образования комплексов состава 1 : 1 уменьшается в следующем ряду:
Hg2+ > Ag+ > Cd2+ ~ Cu2+ > Zn2+ > Mn2+~ Fe2+ > Co2+ > Ni2+
Образование соли в случае двух указанных выше оксимов [284, 288] и образование смешанных комплексов, в которых некоторые лиганды являются анионами (например, цианид), вызовут изменения заряда, но большинство лигандов являются нейтральными молекулами, и если не обращать внимания на заряды, можно составить следующие уравнения равновесий, где (L—L—L) представляет тридентатный лиганд:
Мл+ +L—L ч=* M(L— L)"+,
к [M(L—ЬГ+]
1 [Mn+][L—L] ’
M(L—L)n+ +L— L M(L—L)2 + ,
M(L-L)”+ + L—L (L—L)"+,
[M(L—L)2+]
[M(L—L)"+][L—L] ’
[M(L-L)g+]
[M(L—L)2+][L—L] ’
₽2=«=
[M(L-L)g+]
[M"+][L—L]2’
[M(L-L)g+V L]3
Mn+ +L-L—L M(L—L-L)n+,
„ __ [M(L—L—L)n+]
1 [Mn+][L—L—L] :
+L-L-L М<Ь-Ь_ЦГ,
₽2=KA=
[M(L—L—L)2+] [Mn+][L—L—L]2
Понятно, что эта схема применима для обычного координационного числа 6 и что в координационных местах, не занятых хелатны-
Таблица VIII.33
КОНСТАНТЫ СКОРОСТИ ПРЯМОЙ И ОБРАТНОЙ РЕАКЦИЙ ОБРАЗОВАНИЯ ХЕЛАТНЫХ КОМПЛЕКСОВ
Лиганд и ион Температура, CC ie*f Среда Литература
1,10-Фенантролин
Мп2* 11 5,1 1,5 297
Fe phen2+ 25 17,3 —4,1 299
100,7 —0,1 2 M H2SO4 300
100,7 —3,5 83% H2SO4 300
100,7 -5,8 98% H2SO4 300
Со2* 25 5,5 —1,8 297, 301
Ni2* 25 3,5 —5,0 297, 302
N phen2* 25 —4,75—5,11 303
24,5 —5,05 3 M HC1 304
24,5 —5,15 5 M HC1 304
34,5 —4,60 5 M HC1 304
Cu2* 25 >7,0 —1,4 297
Cu phen2+ 25 1,8 297
Cu phenf* 25 >2,3 297
Zn2* 25 6,3 0,6 297
Cd2* 25 7,0 1,6 297
Ag* 25 >6,5 >2,3 297
Ag phen*j 25 2,5 297
Дипиридил
Fe2* 25 5,2 297
Fe bipy2* 25 5,1 298
Fe bipy|* 25 —1,9 305-
100,7 —3,7 87% H2SO4 300
Co2+ 25 4,8 297
Ni2* 25 4,8 297
Ni bipy|+ 25 3,3 —2,5 297
10,5 —2,8 1 M HC1 304-
10,5 —2,7 2 M HC1 304.
13 —2,4 5 M HC1 304
Cu2* 25 >7,0 —0,7 297
Zn2* 25 6,0 1,2 297
Hg2+ 25 1,4 297
Терпиридил
Mn2+ 25 5,0 0,6 298
Fe terpy2* 5 7,0 298
Co2+ 25 4,4 —4,0 298
Co terpy2+ 5 6,7 —3,2 298
Ni2+ 25 3,1 —7,6 298
313
314
ГЛАВА VIII
Продолжение табл. VIII. S3
Лиганд и ион Температу pa, °C 1g >gfeb Среда Литература
Ni terpy2+ 25 5,3 —5,8 298
Cu2+ 5 7,3 298
Zn2+ 25 6,1 0,1 298
Cd2+ 25 6,5 1,4 298
ми лигандами, находятся молекулы растворителя. Размерность констант равновесия составляет л-моль-1 для К\, К2 и К3, а л2-молы-2 для Р2 и л3-моль-3 для р3. Константа равновесия — это отношение констант скоростей прямой и обратной реакций
ki
Ион Ц- лиганд < *~- [ион (лиганд)],
*Ь
где kj—-константа скорости прямой реакции второго порядка, выраженная в л-моль-1-с-1, kb—константа скорости обратной реакции первого порядка, выраженная в с-1. Константы скорости, определенные Холиером и др., наряду с другими данными обобщены в табл. VIII.33; они даны в десятичных логарифмах. Константы устойчивости комплекса сами по себе не показывают, насколько легко лиганды обмениваются с другими лигандами. Гораздо больше информации дают константы скорости прямой и обратной реакций наряду со знанием фактического механизма рассматриваемой реакции обмена лигандами. Анализ значений kb в табл. VIII.33 особенно интересен. Известно, что комплексы фенантролина и дипиридила с двухвалентными марганцем, медью, цинком, кадмием и ртутью и одновалентными медью и серебром неустойчивы, тогда как с двухвалентными железом, никелем, рутением, палладием, осмием и платиной, трехвалентными хромом и кобальтом и четырехвалентным ванадием устойчивы.
Имеется значительное количество данных об основных свойствах лигандов и комплексов, но только в редких случаях известны все свойства какого-нибудь из комплексов. Часто исследовалось только одно из свойств, например коэффициент молярной экстинкции или окислительно-восстановительный потенциал. В обширных таблицах, хотя и удобных для справки, содержится больше пустых мест, чем приводится значений. Поэтому физико-химические свойства даны в отдельных таблицах; сопоставление их даст те значения, которые еще не были известны во время их составления. Константы комплексообразования интересующих нас комплексов металлов приведены в табл. VIII.34, где указаны источники и методы
Таблица V1U.34
ЛОГАРИФМЫ КОНСТАНТ КОМПЛЕКСООБРАЗОВАНИЯ ПРИ 25 °C И ИОННОИ СИЛЕ ц=0,1 М (если иет других указаний)
Металл Метод 1g К1 1g Яг 1g Кз 1g ₽з Примечание Литература
1,10-Фенантролин
Mg2+ Потенциометрический 1,2 306
Са2+ » 0,7 306
v«+ — 5,47 4,22 р = 0,082 307
Мп2+ Экстракционный 4,10 3,10 3,20 10,40 308
Потенциометрический 3,88 3,16 3,07 10,11 309
» 4,13 3,48 2,7 10,3 20 °C 306
Fe2* Спектрофотометрический 5,85 21,3 299
Потенциометрический 21,3 20 °C 306
Fes+ » 14,1 299
Со2+ Экстракционный 6,96 6,73 6,08 19,77 310
Потенциометрический 7,25 6,70 5,95 19,90 20 °C 306
Ni2+ Экстракционный 8,0 8,0 7,9 23,9 308
8,8 8,3 7,7 24,8 20 °C 306
Cu2+ Метод Бьеррума3 6,3 6,15 5,5 17,95 р = 0,4 311
» » 9,15 6,65 5,25 21,05 312
Экстракционный 8,82 6,57 5,02 20,41 308
Потенциометрический 9,25 6,75 5,35 21,35 20 °C ЗОБ
Zn2+ Экстракционный 6,43 5,69 5 17 313
Метод Бьеррумаа 6,36 5,64 5,2 17,0 312
Экстракционный 6,30 5,65 5,10 17,05 308
Потенциометрический 6,83 5,22 4,87 16,92 309
» 6,55 5,80 5,20 17,55 306
Ag+ » lgfe=13.4 этанол 314
» 5,02 7,05 309
Cd2+ Экстракционный 5,17 4,83 4,26 14,26 308
Потенциометрический 5,78 5,04 4,10 14,92 20 °C 306
» 5,93 4,59 3,78 14,3 309
Hg2+ » 1g Р2 = =19,65 3,7 23,35 20 °C 306
T1+ » 1g ₽2 = = 4 315
T]s+ » 11,57 6,73 (i= 1,0 315
Pb2+ » 4,65 20 °C 306
2-Метил-1,10-фенантролин
Mn2+ Экстракционный 3,0 2,5 2,40 7,9 316
Fe2+ » 4,2 3,6 з,о 10,8 316
Co2+ » 5,1 4,9 3,9 13,8 316
№2+ » 5,95 6,85 4,9 16,7 316
Cu2+ » 7,4 6,45 316
Zn2+ » 4,96 4,40 3,9 12,7 316
Cd2+ 5,15 4,50 3,65 13,3 316
315
П родолжение табл. VIII.34
Металл Метод 1g 1g Kz 1g Кз 1g ₽з Примечание Литера тура
5-Метил-1,1О-фенантролин
Мп2+ Экстракционный 4,28 3,33 3,60 11,21 310
Fe2+ 6,46 7 8,40 21,94 310
Со2+ 7,14 6,86 6,60 20,60 310
Ni2+ 8,30 8,65 7,73 24,68 310
Cu2+ 8,55 6,47 5,10 20,12 310
Zn2+ 6,62 5,96 5,67 18,25 310
Cd2+ 6,13 4,90 5,00 16,03 310
S-Фенил-1,10-фенантролин
•pe2+ | Спектрофотометрический | 21,1 317
5-Нитро-1,10-фенантролин
Fe2+ Спектрофотометрический 17,8 317
Со2+ Экстракционный 6,25 5,41 4,63 16,3 312
Ni2+ » 7,0 6,4 7,0 20,4 312
/п2+ Полярографический 5,6 312
5-Хлор-1,10-фенантролин
рег+ Полярографический 19,7 317
Zn2+ - 5,85 312
2,9-Д иметил-1,10-фенантролин
Мп2+ Экстракционный «4 316
ре2+ » «4 316
Со2+ 4,2 2,8 316
Ni2+ » 5,0 3,5 316
Cu2+ » 5,2 5,8 316
Zn2+ 4,1 3,6 316
Cd2+ 4,1 3,3 3 10,4 316
4,7-Диметил-1,10-фенантролин
ре2* Экстр акционный 5,60 23,05 318
Со2+ 8,08 8,00 8,43 24,51 318
Ni2+ 8,44 8,20 8,40 25,04 318
Cu2+ 8,08 8,00 8,43 24,51 318
Zn2+ ъ 6,90 6,18 6,04 19,12 318
4,7-Дифенил-1,10-фенантролин
fe2+ | Спектрофотометрический 1 | 1 | 21,8 | 18 °C | 319
316
Продолжение табл. VIII.34
Металл Метод 1g Ki 1g Кг lg K3 lg ₽3 Примечание Литература
5 6-Диметил-1,10-фенантролин
Fe2+ Экстракционный 6,37 23,00 318
Со2+ 7,47 8,00 8,14 23,62 318
Ni2+ » 8,25 8,30 8,21 24,76 318
Cu2+ » 7,47 8,00 8,14 23,62 318
Zn2+ 6,87 6,02 5,71 18,60 318
2,2'-Дипиридил
v»+ Потенциометрический 4,91 4,67 3,85 13,43 320
Сг’+ » 4 6,4 3,5 14 320
Мп2+ Метод Бьеррума® 4,06 3,78 3,63 11,47 p = 1,0 321
Потенциометрический 2,72 1,99 322
Экстракционный 2,62 2,00 1,1 5,6 308
Fe2+ » 4,20 3,70 9,55 17,45 308
Потенциометрический 17,45 20 °C 306
Fes+ » 12,0 323
Со2+ Экстракционный 5,65 5,60 4,80 16,05 308
Потенциометрический 5,73 15,18 322
6,06 5,36 4,60 16,02 20 °C 306
Ni2+ Метод Бьеррума® 6,80 6,46 5,20 18,46 p = 1,0 321
Экстракционный 7,07 6,86 6,20 20,13 309
Потенциометрический 7,17 6,88 6,53 20,54 306
Cu+ Полярографический lg₽2 = = 14,20 324
Потенциометрический lg₽2 = = 12,95 20 °C 306
Cu2+ Метод Бьеррума® 6,94 5,74 p = 1,0 321
Потенциометрический 8,47 5,58 3,51 17,56 325
Экстракционный 8,15 5,50 3,30 16,95 309
Потенциометрический 8,0 5,60 3,48 17,0 20 °C 306
Zn2+ Метод Бьеррума® 5,39 4,74 4,01 14,14 10 °C 326
» » 5,20 4,50 3,85 13,35 20 °C 326
5,16 4,46 3,74 13,36 326
» 5,4 4,4 3,5 13,3 328
4,89 4,58 4,27 13,74 p = 1,0 321
4,86 4,24 3,56 12,66 40 °C 326
Экстракционный 5,04 4,35 3,57 12,96 309
Потенциометрический 5,30 4,53 3,80 13,63 20 °C 306
Ag+ Ъ lg₽2 = 8,89 Этанол 314
Aga+ lg₽2 = 6,8 327
Cd2+ Метод Бьеррума® 4,5 3,5 2,5 10,5 328
Экстракционный 4,12 3,50 2,60 10,24 309
Потенциометрический 4,25 3,60 2,70 10,55 20 °C 306
Hg*+ 9,64 7,10 2,8 19,54 20 °C 306
11+ » lg₽2 = = 3 p = 1,0 315
T13+ 9,40 6,70 p = 1,0 315
Pb2+ » 2,9 20 °C 306
317
318
ГЛАВА VIII
Продолжение табл. VIII.S4
Металл Метод 1g К1 1g Кг 1g Кз 1g fe Примечание Литература
4,4' -Диметил-2,2' -дипиридил
Zn2+ Метод Бьеррума8 6,0 5,0 4,0 15,0 329
Cd2+ » » 4,9 3,8 3,0 Н,7 329
2,2',2''-Т ерпиридил
Fe2+ Спектрофотометрический 1g ₽г = 18 330
» Ig₽2 = = 20,1 20,4 331
Со2+ 8,8 9,9 298
Ni2+ 10,7 11,1 298
а Автор допускает неточность там, где в столбце «Метод» пишет «метод Бьеррума» Как из-вестно, экспериментального метода Бьеррума не существует. Очевидно, имеется в виду измерение pH со стеклянным электродом и последующий расчет констант с использованием функции Бьеррума. — Прим. ред.
определения, а длины волн максимумов поглощения в видимой области спектра и коэффициенты молярной экстинкции полностью координированных комплексов незамещенных лигандов даны в табл. VIII.35. Спектральные свойства комплексов железа (II) и меди(1) с некоторыми замещенными лигандами обобщены в табл. VIII.36. Формальные потенциалы комплексов меди(1) находятся в области низких потенциалов (около 0,06—0,6 В), но из-за плохой растворимости и способности легко разрушаться в кислых средах они непригодны для использования в качестве индикаторов; они включены в табл. VIII.36 из-за стерических факторов и поскольку представляют интерес для спектрофотометрии, хотя там не приводятся производные дихинолина [244].
Формальные потенциалы окисления, кинетика реакций обмена электронов и окисления, хотя и относятся к свойствам комплексов, будут рассмотрены не здесь, а отдельно в одном из следующих разделов.
Таблица VIII.35
Спектральные характеристики в видимой области полностью КООРДИНИРОВАННЫХ КОМПЛЕКСОВ МЕТАЛЛОВ С НЕЗАМЕЩЕННЫМИ ЛИГАНДАМИ
Металл 1,10-Фенатролин 2,2'-Дипиридил 2,2' ,2"-ТерпиридиЛ
Xmax- ™ E, л моль—1 CM—1 Литерату pa ’"max' ™ 8, Л МОЛЬ—1-CM—1 Литература ’"max- ™ E, л моль—1 см—1 Литература
V2+ 645 8-10s 332
Fe2+ 510 1,11-104 333 522 8,65-10s 334 552 1,15-Ю4 334
Fe3+ 590 600 335 610 330 244
Со2+ 4256 100 244 4356 90 244 505 1,36-Ю3 336
Ni2+ 519 11,9 337 521 11,6 337
Cu+a 435 7-10® 338 455 4,5-103 338 430 3,25-Ю3 336
Cu2+ 700 60 311
Ru2+ 448 1,85-104 339 455 1,46-Ю4 340
Ru3+ 418 8,1-103 340
Ag2+ 454 l,6-103 341
Os2+ 477 1.37-104 342
а После экстракции З-метилбутанолом-1.
& Плечо на полосе поглощения в УФ-области, вытянутое в видимую область.
Со
О
Таблица VII 1.3Ь
СПЕКТРАЛЬНЫЕ ХАРАКТЕРИСТИКИ В ВИДИМОЙ ОБЛАСТИ КОМПЛЕКСОВ МЕДИ(1) И ЖЕЛЕЗА(П) С ЗАМЕЩЕННЫМИ ЛИГАНДАМИ
Железо(П) Медь(1)
Лигавд ^шах» им е, Л МОЛЬ—1-см—1 \пах» нм 8, л-моль—1 см—1
1,10-Фенантролин
-З-Сульфокислота 517 1,084-Ю4
5-Фенил- 522 1,27-10* 440 8,6-103
5-Амино- 524 1,21-10*
5-Нитро- 510 1,15-10* 441 7,9-103
5-Фтор- 509 1,2-10* 439 6,87-103
5-Бром- 515 1,25-10* 442 7,7-10s
-5-Суль фо кислота 512 1,224-10*
2,9-Диметил- — — 455 7.95-103
2,9-Дифенил- — — 441 3,62-10s
3,8-Диметил- 496 1,15-10* 431 7,99-103
3,8-Дифенил- 525 7,02-103 441 6,78-103
4,7-Диметил- 512 1,4-10* 438 8.98-103
4,7-Дифенил- 533 2,24-10* 457 1,21-10*
4,7-Диметокси- 500 1,13-10*
4,7-Дифенокси- 500 1,47-10* 425 8-103
4,7-Диокси- 520 1,48-10*
5,6-Диметил- 520 1,26-10* 445 8,22-103
5,6-Диметокси- 518 1,2-10* 441 7,5-Ю3
2,9-Диметил-4,7-дифенил- — — 479 1,42-10*
[Fe phen2(CN)2] 597
[Fe phenfCNJJ2- 462
2,2'-Дипиридил:
4,4'-Диметил- 528 9,3-103
4,4'-Дифеиил- 552 2,11-10* 463 9,6-Ю3
4,4'-Дикарбокси- 540 1,48-10*
4,4'-Диамиио- 569 1,36-10*
4,4'-Дихлор- 532 8.3-103
2,2',2"-Терпиридил:
4-Фенил- 569 2,2-10*
4,4"-Диметил-4'-фенил- 575 2,63-10*
320
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
321
Продолжение табл. VIII.36
Железо (II) Медь (I)
Лиганд ^гпзх» нм е, Л МОЛЬ—1 СМ—1 \пах» нм е, Л - МОЛЬ—1- СМ—1
4,4" Диэтил-4,-фенил- 570 2,71-10»
4,4',4"-Трифенил- 583 3,02-10»
Т риазины
2,4,6-Три- (2'-пиридил) -1,3,5-триазин 594 2,26-10»
2,4,6-Т ри- (4'-фенилпиридил) -1,3,5-триазин 605 2,52-10»
бис-3,3'- (5,6- Диметил-1,2,4-триазин) 493 1,50-10» 444 9,7-103
3- (2-Пиридил) -5,6-дифенил- 1,2,4-триазин 555 2,4-10» 478 4,7-103
3- (4-Фенил-2-пиридил) -5,6-дифенил-1,2,4-триазин 561 2,87-10» 480 7,9-Ю3
Оксимы
Фенил-2 пиридилкетоксим 550 1,56-10»
2 6 Пиридиндиамидоксим 523 1,68-10»
ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ
Обычными опытами доказано влияние заместителей на растворимость, хотя, по-видимому, не проводилось количественное исследование этого влияния. Алкил- и арилгруппы в качестве заместителей понижают растворимость лиганда и комплекса в водной среде, но повышают растворимость их в смешанных с водой растворителях, и поэтому их можно экстрагировать такими смесями. Полярные заместители, такие, как карбокси-, амино-, нитро- или оксигруппы, оказывают обратное влияние. Сульфирование является испытанным средством для повышения растворимости органических молекул и с успехом применяется для устранения этого недостатка У некоторых индикаторов. Комплексы т/?ыс-(1,10-фенантролин)же-леза(П) образуют умеренно растворимые перхлораты, которые осаждаются из раствора хлорной кислоты, и поэтому в такой среде трудно проводить титрование. Блаир и Дил [343, 344] применили сульфопроизводные этих индикаторов и провели титрование в среде хлорной кислоты без мешающего осаждения.
Поскольку число производных лигандов и основные количественные сведения о них и об их комплексах постоянно росли, то
21—918
322
ГЛАВА VIII
вполне логично было попытаться сперва собрать данные, а затем объяснить взаимосвязи между природой и положением заместите ля, с одной стороны, и свойствами лигандов и их комплексов -с другой. Ивенс [345], по видимому, был первым, кто попытался связать окислительно-восстановительный потенциал комплексов 1,10-фенантролинжелеза(П) с кислотностью лиганда; затем после довала работа [346], в которой исследовали комплексы руте ния(II); Смит [347] распространил это представление на комплек сы железа(II), а также исследовал смещение Хтах вследствие ме тилирования, считая, что это влияние аддитивное; позже он подтвердил существование такой же взаимосвязи для комплексов меди(1) [348]. Брандт и Галлстром [317] связывали константы комплексообразования со степенью кислотности лиганда и другими свойствами, в частности со скоростью реакции. Таким образом
кислотные свойства лиганда удобно использовать для сравнения и корреляции с главным свойством, т. е. распределением электро нов в молекуле лиганда. Непосредственно или благодаря связи с кислотностью можно объяснить влияние заместителей на такие свойства, как кислотность или основность лиганда, кинетику комплексообразования и константы комплексообразования, константы скорости реакции обмена электронов и окислительно-вбсстанови-тельной реакции, формальные и стандартные окислительно восстановительные потенциалы комплексов металлов различных степеней окисления и спектральные характеристики комплексов. Уже наблюдались корреляции, подобные указанным, и были попытки объяснить их, но все это еще находится в начальной стадии. Достаточно информации имеется о целенаправленном синтезе лигандов, обладающих специфическими свойствами [349, 350]. Для объяснения влияния заместителей [302, 352, 353] было использовано уравнение Гаммета [351].
Для расчета относительных плотностей л-электронов в незамещенной молекуле фенантролина были использованы теория возмущения первого порядка и теория молекулярных орбиталей; результаты были выражены величиной зарядов [354]:
0,015—.
q.720—/ \
УЧ
0,138 N
' _ Л 377
Распределение электронов симметрично, поскольку оба атома азота и положения 2 и 9, 3 и 8, 4 и 7, 5 и 6 эквивалентны. Корреляция плотностей электронов с поведением в реакциях замещения в близких структурах позволила предсказать, что положения 2 и 4 (а также 9 и 7) в 1,10-фенантролине должны быть нуклеофильны, а положения 5 и 6 электрофильны. Влияние метилирования
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
323
на основность получающихся лигандов [329, 352] находится в тесной связи с плотностью электронов, показанной выше. Метилирование в положении 2 или 9 приводит к наибольшему увеличению основности, в положении 4 или 7 к очень малому влиянию и совсем незначительному в других положениях. Данные по влиянию заместителей обобщены в табл. VIII.37, где основность выражена, как общепринято, константой диссоциации в виде десятичного показателя.
Анализ приведенных в таблице значений позволяет обнаружить некоторые факторы, влияющие на плотность электронов вокруг атомов азота. Например, метильная группа производит индукционный, но не таутомерный эффект, а при полиметилировании повышается основность [352]. Фенилирование в положении 2 или 4 повышает вследствие таутомерного эффекта плотность электпонов у атомов азота, но электрофильный эффект более значителен в положениях 3 и 5, поэтому основность уменьшается. Сильный индукционный, но небольшой таутомерный эффект при введении галогенов выражается в предельном уменьшении основности атомов азота. Были проведены некоторые исследования влияния заместителей на образование и свойства комплексов и получены многообещающие результаты.
В отношении образования комплексов можно указать на работу Уилкенса с сотр. [302] по диссоциации комплексов никеля (II) с замещенными фенантролинами и дипиридилами. Реакции диссоциации (kb в табл. VIII.33) способствует наличие заместителей, уменьшающих плотность электронов у атомов азота. В согласии с этим kb значительно снижается в случае 4,7-диметил-1,10-фенан-тролина и 4,4'-диметил-2,2'-|дипиридила по сравнению со значениями kb для незамещенных лигандов. Те же авторы нашли, что существует линейная зависимость между kb и рАа Для комплексов никеля(II) с фенантролином, имеющим заместитель в положении 5; они обнаружили также пространственный и электронный эффекты в возросшей скорости диссоциации комплексов с заместителем в положении 2: в случае метила скорость диссоциации возрастает в 50 раз, а в случае хлора — в 10С0 раз. Исследование обмена лиганда на растворитель проводилось позже [353], но прежде было изучено влияние заместителя на растворитель при кислотном [360] и щелочном [361] расщеплениях комплексов трис-(1,10-фенантролин)железа (II). Были объяснены зависимости между кислотной константой лиганда и устойчивостью комплексов железа (II) для фенантролинов с заместителем в положении 5 без учета стерических факторов; для них зависимости lgP3=2,5 рКа+ +9,15 [317] и lg/G = 0,596 рАа+2,93 [312] дают хорошее совпадение.
21*
Таблица VI11.37
КОНСТАНТЫ КИСЛОТНОЙ ДИССОЦИАЦИИ 1,10-ФЕНАНТРОЛИНА, 2,2'-ДИПИРИДИЛА И ИХ ПРОИЗВОДНЫХ ПРИ 25 "С
(ЕСЛИ НЕ ИМЕЕТСЯ ДРУГИХ УКАЗАНИЙ)
Соединение Метод3 JX, моль л—I Р*а Литература
1,10-Фенантролин т 0 4,77 299
к 0,001 4,96 299
с 0,2 4,92 (20 °C) 355
pH 0,002 4,96 317
т 0,002 4,86 352
т 0,1 4,92 328
т 0 4,857 356
т 0,1 4,95 (20 °C) 306
с 0,1 4,93 318
2-Метил- т 0,1 5,45 316
с 0,1 5,35 316
З-Метил- pH 0,002 5,00 352
З-Этил- pH 0,002 4,98 352
З-Фенил- pH 0,002 4,82 352
З-Хлор- pH 0,002 3,99 352
4-Этил- pH 0,002 5,44 352
4-Пропил- pH 0,002 5,45 352
4-Фенил- pH 0,002 4,90 352
4-Бром- pH 0,002 4,03 352
5-Метил- pH 0,002 5,23 317
т 0,1 5,26 329
с 0,1 5,28 Ло
5-Фенил- pH 0,002 4,80 317
pH 0,002 4,72 352
5-Нитро- pH 0,1 3,57 317
с 0 4,18 312
с 0,002 3,23 357
5-Хлор- pH 0,002 4,26 317
с 0,1 3,33 312
5-Бром- с 0,1 4,20 312
5-Сульфо- с 0,1 5,60 353
324
Продолжение табл. VIII.37
Соединение ’ Метод8 JLI, МОЛЬ Л—1 РКа Литература
2,9-Диметил- т о,1 6,15 329
pH 0,002 6,17 352
т 0,1 5,85 316
с 0,1 5,79 318
3,8-Диметил- pH 0,002 5,23 352
3,8-Дибром- pH 0,002 3,90 352
4,7-Диметил- т 0,1 5,94 329
с 0,1 5,95 318
4,7-Диэтил- pH 0,002 5,60 352
4,7-Дифенил- pH 0,002 4,84 352
с 10% С2Н6ОН 4,80 (18 °C) 319
4,7-Диметокси- pH 0,002 6,45 352
4,7-Дифеноксн- pH 0,002 5,34 352
4,7-Дихлор- pH 0,002 3,03 352
5,6-Диметил- с 0,1 5,60 318
с 0,1 5,6 353
5,6-Диметокси- т 0,1 4,42 352
Циклогексен-5,6 pH 0,002 5,30 352
5,6-Дихлор- pH 0,002 3,47 352
2,4-Диметил- pH 0,002 5,96 352
Циклопентен-3,4 pH 0,002 5,78 352
Циклогексен-3,4 pH 0,002 5,66 352
3,7-Диметил- pH 0,002 5,57 352
4,6-Диметил- pH 0,002 5,71 352
4,6-Дифенил- pH 0,002 4,69 352
З,4,6-Тр1пметил- pH 0,002 5,93 352
3,4,7-Триметил- pH 0,002 5,99 352
3,5,6-Трпметил- pH 0,002 5,34 352
3,4,7-Триметил pH 0,002 5,90 352
3,5,8-Триметил- pH 0,002 5,27 352
2.4,7,9-Т етра мети л- pH 0,002 6,50 352
3,4,6,7-Тетраметил- pH 0,002 6,45 352
3.4,6,8-Тетраметил- pH 0,002 6,07 352
3,4,7,8-Т етр а метил- pH 0,002 6,31 352
3,5,6,8-Тетраметил- pH 0,002 5,54 352
325
326
ГЛАВА VIII
Продолжение табл VIII.37
Соединение Метода JLL, МОЛЬ Л—1 Р«а Литература
Дициклогеисен-3,4,7,8 pH 0,002 6,23 352
2,2' Дьпиридил т 0 4,34 358
т 0,025 4,33 323
с 0,2 4,44 (20 °C) 355
т 0,005 4,33 359
т 0,1 4,44 328
т 0,1 4,49 (20 °C) 306
4,4'-Диметил- т 0,1 5,32 329
а Т — потенциометрическое титрование, стеклянный электрод; pH — измерение pH в вод-но-диоксановых растворах, экстраполяция до нулевой концентрации диоксана; С — спектрофотометрическое определение типа изобестической точки; К — измерение электропроводности.
До сих пор нельзя вычислить интенсивности светопоглощения, исходя из структуры молекулы, и, хотя метилирование увеличивает в некоторой степени коэффициент молярной экстинкции, по данным табл. VIII.36 для комплексов железа(II) и меди(1) видно, что между е и распределением плотности электронов нельзя вывести простую зависимость. Однако Смит нашел, что в случае комплексов фенантролина с медью(I) [348] и железом(II) [347] под влиянием метилирования происходит смещение максимума поглощения; то же относится и к формальным потенциалам [347]:
Положение метила нм max Д£ , мВ [комплексы Fe(II)/Fe( III)]
комплекс с Cu(I) комплекс с Fe( 11)
2,9 + 10 — —
3,8 —2 —8 —30
4,7 + 1 + 1 —но
5,6 +5 +5 —40
Доказано, что в отсутствие пространственных затруднений существует линейная зависимость между формальными потенциалами комплексов, состоящих из замещенных фенантролинов с желе
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
327
зом(П)/железом(Ш) [317, 345, 347] и рутением (Н)/рутением(Ш) [346], и значением рДа лиганда. Влияние заместителей на кинетику окислительно-восстановительных реакций будет рассмотрено в следующем разделе.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ СВОЙСТВА
КИНЕТИКА РЕАКЦИИ
Следует различать два вида реакций и констант скорости реакций. Во-первых, это реакции обмена электроном, и для них константа скорости относится к скорости обмена электронами между двумя частицами с общим центральным атомом, например Fe phen|+ и Fe phen ®+, и, во-вторых, перенос электрона или, вернее, окислительно-восстановительные реакции, для которых константа скорости является по меньшей мере константой второго порядка и относится к скорости переноса электрона от одной частицы к другой, иной частице [например, при окислении Fe рЬепз+ церием (IV) ].
Реакция обжена электронами
Главным фактором окислительно-восстановительных процессов является скорость обмена электроном между двумя состояниями окисления данной частицы [78, 79], которая может существенно зависеть от условий и особенно от лигандов, присоединенных к центральному атому. Хорошо известно положительное влияние сульфат-иона на титрование железа (II) церием(IV) и на половину из числа участвующих в нем реакций. Для подробного исследования этих вопросов теперь имеются подходящие методы [32] и стоит привести один из примеров [362]. С помощью изотопной метки была исследована система Fe(II)—Fe(III) в серной кислоте при условии, что координационные места, оставшиеся свободными, заполнены молекулами растворителя; наиболее вероятными этапами процесса обмена являются следующие:
ко
Fe2+-p*Fe3+ --> Fe3+ V *Fe2+,
kt Fe2+4-*FeOH2+ ----> FeOH2++ *Fe2+,
^2
FeHSOj + *FeSOj --► FeSOj + *FeHSOt,
. ks
Fe2+ + *FeSO4 ->- FeSOj + *Fe2+,
kt
Fe2+-|-*Fc(SO4)7 ->- Fe(SO4)7 + *Fe2+,
*5
FeSO„ -f- *FeOH2+ -> FcOH2+ -f- *FeSO4
328
ГЛАВА VIH
Из всех констант скорости реакции k0 и kz очень малы, и эти реакции в незначительной степени участвуют в общем процессе, который почти полностью основывается на последних трех реакциях; константы скорости этих реакций при ионной силе раствора 0,25 моль-л-1 и температуре 25 °C представляют &3 = = 692 л-моль-1-с-1. &4=1,94-104 л-моль-1-с-1 и k5=
= 1,3-106 л-моль"1-с-1. Интересно сравнить для системы Fe(II) — Fe(III) некоторые из комплексных индикаторов, имеющих анионные комплексы с полностью сольватированной системой. Эти значения приведены в табл. VIII.38.
Таблица VI 11.38
СОПОСТАВЛЕНИЕ СКОРОСТЕЙ ОБМЕНА ЭЛЕКТРОНАМИ ДЛЯ СИСТЕМЫ ЖЕЛЕЗО(П) — ЖЕЛЕЗО(Ш) В ВОДНЫХ РАСТВОРАХ ПРИ 25 °C.
НЕУКАЗАННЫЕ КООРДИНАЦИОННЫЕ МЕСТА ЗАНЯТЫ МОЛЕКУЛАМИ
РАСТВОРИТЕЛЯ
Вещества Константа скорости k, л моль—I CM—1 Литература
Fe(CN)e-/Fe phenf >10® 363
FeSO4/FeOH2+ 1,3-10е 362
Fe phen|+/Fe phenjp >103 364
Fe2+/Fe tripy|+ 8,5-Ю4 365
Fe2+/Fe phen|+ 3,7-Ю4 366
Fe2+/Fe dipy|+ 2,7-104 365
Fe^/FefSO^ 1,94-104 362
Fe^/FelQO^ 4250 367
Fe'^/FelQO^ 3300 (20 °C) 368
Fe2+/FeOH2+ 3200 369
Fe(CN)r/Fe(CN)g- 740 370
Fe2+/Fe(HPO4)+ 520 (0°C) 368
Fe2+/FeSO4+ 692 362
295 371
Fe2+/FeCl2+ 65 369
Fe2+/FeC12 38 369
Fe2+/Fe3+ 4 369
Fe2+/Fe ЭДТА- 4-JO"4 372
Форд-Смит и Сатин [365] исследовали скорости восстановления хелатов железа (III) с помощью железа (II) в хлорной и серной кислотах и нашли, что константы скорости в сульфатной среде примерно в 8 раз больше, чем в среде перхлората. Они также
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
329
установили, что существует линейная зависимость между константами скорости для комплексов фенантролина с заместителем в положении 5 и значениями рКа лиганда и что свободная энергия активации для всех реакций находится в линейной зависимости от стандартной свободной энергии каждой реакции. Эти результаты позволили внести поправку в формальный потенциал 4,7-ди-фенилпроизводного. Некоторые из результатов этих авторов приведены в табл. VIII.39.
Таблица VII 1.39
КОНСТАНТЫ СКОРОСТИ ВОССТАНОВЛЕНИЯ ХЕЛАТОВ ЖЕЛЕЗА(Ш) ЖЕЛЕЗОМ(П) В СРЕДЕ ХЛОРНОЙ И СЕРНОЙ КИСЛОТ ПРИ 25 °C [365?а
Лиганд Константа скорости fe, л-моль—1-с—1 ftH2S04/?,HClO4
0,5 М НСЮ4 0,5 М H2SO4
1,10-Фенантролин 3.7-104 3-105 8,1
5-Метил- 2-Ю4 1,5-Ю5 7,5
5-Нитро- 1,1.10» — —
5-Хлор- 2,1.10» 1,5-Ю6 7,1
5-Феиил- — 3,2-Ю5 —
5,6-Диметил- 7,8-103 6,9-Ю4 8,8
4,7-Ди фенил- — 3,3-Ю4 —
3,4,7,8-Т етраметил- — 1,9-10» —
2,2'-Дипиридил 2,7-10* 2,2-Ю5 8,2
4,4х-Диметил- 6-Ю2 5,9-103 9,8
2,2',2"-Терпир идил 8,5.10* 7,4-Ю5 8,7
а Ирвии сообщает [373], что реакция Fe phen3+/Fe bipy|+ идет с большой скоростью (fe > 105 л МОЛЬ—1-с—1).
Реакция окисления-восстановления
Скорость, с которой происходит окисление индикатора, зависит по крайней мере от трех факторов: от скорости обмена электронами индикаторной системы, от скорости обмена электронами системы окислителя и от разности формальных потенциалов ДЕо обеих систем. Если какая-нибудь система проходит через образование активного интермедиата (как, например, бромат дает бром или монохлорид брома), то могут влиять и другие факторы. Ирвин [374], например, предложил такую зависимость:
lg k =константа + х&Е'о,
Таблица VI11.40
КОНСТАНТЫ СКОРОСТИ ОКИСЛЕНИЯ КОМПЛЕКСОВ ИНДИКАТОРОВ ПРИ 25 °C
Металл Лигадд Окислитель Среда k, л моль—1 - C—1 Литература
Fe2+ Растворитель Ces+ 0,5 M H2SO4 1,3-10® 375
Ru bipy|+ 0,001 M HNO3 >106 373
CN- Ce4+ 0,5 M H2SO4 1,9-10® 376-
Ru bipy?+ 0,001 M HNO3 >10® 373
1,Ю-Фенантролин Ce4+ 0,5 M H2SO4 1,42-10® 376
Mn3+ a 72 377
Co3* 3 M HC1O4 1,4-lC4 376
ЦЗ+ 0,001 M HNO3 7-10-3 373
5-Метил- s2o|- 6 0,3 374
Ce4+ 0,5 M H2SO4 2,2-10® 375
Mn3+ a 1,4-lC2 377
Co3+ 3 M HC1O4 1.5-104 376
S2O2- 6 0,11 374
5-Фенпл- Ce4+ 0,5 M H2SO4 1,2-10® 375
Mn3+ a 68 377
5-Нитро- Ce4+ 0,5 M H2SO4 3,9-103 375
Co3* 3 M HC1O4 1,49-Ю3 376
5-Хлор- Ce4+ 0,5 M H2SO4 2,5-104 375
Co3+ 3 M HC1O4 5.02-103 376
4,7-Диметил- Mn3+ a 1,4-103 377
5,6-Диметил- Ce4+ 0,5 M H2SO4 4,3-10® 375
Mn3+ a 3,9-10a 377
3,4,7,8-Тетрамстил- Ce4+ 0,5 M H2SO4 1.6-106 375
Mn3+ a 2,3-103 377
3,5,6,8-Тетраметил- Mns+ a 4.3-102 377
phen2(CN)2 Ce4+ 0,5 M H2SO4 7,11-10® 376
phen (CN)|- Ce4+ 0,5 M H2SO4 8,88-10® 376
2,2'-Дипиридил Ce4+ 0,5 M H2SO4 1,96-10® 376
s2o2- 6 0,55 374
Ru bipy> 0,001 M HNO3 * >10® 373
Ru2+ Ce4+ 1,0 M HC1O4 >10® 373
Ce4+ 1,0 M H2SO4 4,10s 373
s2o2- 6 9-10-3 374
Os2+ S2O2- 6 49 374
Fe2+ 4,4'-Диметил- S2O2- 6 6,68 374
bipy2 (CN)2 Ce4+ 0,5 M H2SO4 8,4-10® 376
bipy (CN)2' Ce4+ 0,5 M H2SO4 1.25-107 376
а Среда пирофосфата при рН=1,0 и ц=0,5 моль-л-1.
6 Ионная сила 0,3—1,0 моль-л-’; экстраполирована до нуля для указанных значений k; в среде NaClOj.
330
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
331
и нашел, что значение константы составляет —6,4, х=7,0 при 25 °C в случае окисления раствором пероксидисульфата ряда комплексов индикаторов с двухвалентными железом, рутением и осмием. Было проведено ‘множество прямых и косвенных (посредством соотношения Маркуса) исследований по окислению индикаторов различными окислителями; сюда относятся, в частности, работы Сатина, Ирвина и Уола с сотр. Некоторые результаты обобщены в табл. VIII.40.
СВОЙСТВА ИНДИКАТОРОВ
Остается рассмотреть главное свойство индикаторов — их окислительно-восстановительные потенциалы. В большинстве случаев индикаторы применяются в кислой среде, иногда в почти концентрированных растворах, как, например, в 10 М серной кислоте, а поэтому гораздо интереснее иметь дело с формальными потенциалами, чем со стандартными потенциалами. При работе с формальными потенциалами необходимо подробно описывать используемую среду, но из-за этого таблицы становятся громоздкими. За исключением нейтральных соединений рутения (II) и осмия (II) с дициано-бнс-дипиридилом, формальные потенциалы значительно ниже стандартных потенциалов и уменьшаются с ростом ионной силы и концентрации иона водорода. Обычно чаще всего работают в среде серной кислоты, и ,в ней определяют формальный потенциал. В табл. VIII.41 приведены значения формальных потенциалов для трис-хелатных систем с незамещенными лигандами и металлами: железом, рутением и осмием в двух- и трехвалентном состоянии.
Был проведен ряд исследований, касающихся комплексов замещенных фенантролинов, но совсем или почти не обращалось внимания на влияние заместителей у лиганда. Во многих работах приводятся данные определения или повторной проверки значения формального потенциала одного какого-нибудь соединения в данных условиях; однако приводимые значения редко отличаются существенно от значений, полученных при более тщательных и систематических исследованиях. Большинство опытов проводили в серной кислоте; результаты суммированы в табл. VIII.42.
ПРИМЕНЕНИЕ
Приведенные в таблицах индикаторы содержат только один тип лиганда; о смешанных комплексах смотри ниже. Эти индикаторы находятся в очень необычном положении: теоретические вопросы, хотя и далеко не полные, опережают их применение. Из многочисленных указанных индикаторов только небольшая часть используется в титриметрическом анализе, большее применение
Таблица VIII.4t
ФОРМАЛЬНЫЕ ПОТЕНЦИАЛЫ НЕЗАМЕЩЕННЫХ ТР.ИС-ХЕЛАТНЫХ СИСТЕМ В СЕРНОЙ КИСЛОТЕ ПРИ 25 °C
[H2SO4], моль л—1 Формальные потенциалы Е$, В
1,10-Фенантролнн 2,2'-Дипирндил 2,2' ,2"-Терпиридил
• }Келезо(11)1железо(Ш )
0 1,120 [378] 1,141 [379] 1,096 [378]
0,005 1,112 [378]
0,01 1,084 [378] 1,069 [380]
0,05 1,102 [378] 1,10 [382]
0,10 1,076 [381]
0,11 1,062 [380]
0,20 1,060 [381]
0,25 1,086 [378]
0,50 1,054 [381]
1,00 1,06 [383] 1,026 [380] 0,927 [381]
1,06 [384] 1,057 [378] 1,06 [382] 1,023 [385]
2,0 1,028 [378] 1,03 [382] 1,00 [380]
2,5 1,015 [378]
з.о 0,996 [378] 1,00 [382]
3,5 0,977 [378]
4,0 0,96 [382] 0,95 [380]
6,0 0,89 [382] 0,88 [380]
8,0 0,76 [382]
10,0 0,80 [380]
Рутений(Н)1рутений(Ш) в серной кислоте
0,01 1,274 [380]
0,05 0,11 1,273 [380] 1,262 [380] 1,281 [387]
1,0 1,236 [380] 1,202 [387]
332
Продолжение табл. VIII.41
[H2SO4], моль-л—1 Формальные потенциалы Eq, В
1,10-Фенантролин 2,2'-Дипиридил 2,2' ,2"-Терпиридил
2,0 1,20 [380] 1,175 [387]
2,5 1,22а [386]
3,55 1,205а [386]
4,0 1,15 [380]
5,0 1,16а [386]
6,0 1,15а [386] 1,09 [380]
6,75 1,105а [386]
10,0 0,88 [380]
12,0 0,76 [380]
Рутений(П)1рутений(Ш) в азотной кислоте
0,002 1,316 [381] 1,3036 [381]
0,1 1,306 [381] 1,2886 [381]
о.з 1,296 [381] 1,2796 [381]
0,5 1,286 [381] 1,2706 [381]
1,0 1,26б [381] 1,2576 [381]
1,29а [386]
2,0 1,246 [381] 1,2406 [381]
1,26а [386]
3,0 1,226 [381] 1,2226 [381]
5,0 1,196 [381]
1,22а [386]
Осмий(П)1осмий(Ш) в серной кислоте
0 0,877 [388] 0,877 [388] 0,885 [389] 0,9866 [389]
0,01 0,858 [380]
0,1 0,859 [388] 0,855 [388] 0,951 [387]
0,11 0,841 [380]
0,2 0,941 [387]
0,5 0,822 [388] 0,819 [388] 0,925 [387]
1,0 0,802 [388] 0,907 [387]
0,803 [380]
1,5 0,775 [388]
2,0 0,777 [380] 0,884 [387]
2,5 0,727 [388]
4,0 0,723 [380]
6,0 0,63 [380]
10,0 0,44 ,380]
12,0 0,37 [380]
а При J5 °C.
6 При О °C.
333
Таблица VIII.42
ФОРМАЛЬНЫЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ ТТИС-ХЕЛАТПЫХ КОМПЛЕКСОВ ЖЕЛЕЗО(П)/ЖЕЛЕЗО(1П) С ЗАМЕЩЕННЫМИ 1,10 ФЕНАНТРОЛИНАМИ В СЕРНОЙ КИСЛОТЕ ПРИ 25 °C
Заместитли [H2so4], моль л -1 Е , В 0 Литература Заместители [H2SO4J, моль л -1 е' , в 0 Литература
З-Метил- 0,1 1,07 382 4,7-Дифенилдисульфо- 1,0 1,09 343
1,0 1,03 382 кислота в 1 М НС1О4 1,01 343
З-Этил- 2,1 1,00 81 4,7-Диметокси- 0,1 0,85 81
3-Фенил- 2,4 1,02 81 4,7-Дифенокси- 2,9 1,04 81
3 Хлор- 4,4 1,11 81 5,6-Диметил- 0,1 1,00 382
ЗСульфокислота 1,0 1,23 344 0,5 0,97 382
в 1 М НС1О4 1,21 344 5,6-Диэтил- 1,6 0,98 81
4-Метил- 1,0 1,02 384 5,6-Диметокси- 2,4 1,02 81
2,0 1,00 384 Циклогексен 5,6- 1,4 0,96 81
4,0 0,93 384 5,6-Дихлор- 4,4 1,Н 81
4-Этил- 1,4 0,96 81 5 Нитро-6-метил- 1,0 1,23 384
4-Пропил 1,4 0,96 81 3,4-Диметил- 0,1 0,97 382
4-Фенил- 1,9 0,99 81 1,0 0,93 382
4-Бром- 2,9 1,04 81 Циклопентен-3,4- 1,4 0,96 81
5-Нитро- 0,5 1,26 384 Циклогексен-3,4- 0,9 0,94 81
1,0 1,25 384 Циклооктен-3,4- 1,0 0,95 81
2,0 1,22 384 3,7-Диметил- 1,4 0,96 81
4,0 1,12 384 4,5-Диметил- 0,1 0,95 382
5-Фтор- 3,2 1,05 81 4,6-Диметил- 0,1 0,95 382
5-Хлор- 1,0 1,П 384 4,6-Дифенил- 4,5 1,12 81
2,0 1,10 3,4,6-Триметил- 0,1 0,92 382
4,0 1,04 3,4,7-Триметил- 0,1 0,88 382
5-Бром- 0,5 1,13 384 3,5,6-Триметил- 1,0 0,95 81
5-Сульфокислота 1,0 1,20 344 3,5,7-Триметил- 0,5 0,93 382
в 1 М НС1О4 1,16 344 0,1 0,89 382
3,8-Диметил- 0,1 1,03 382 3,5,8-Триметил- 0,1 0,99 382
3,8-Дибром- 5,4 1,21 81 3,4,6,7-Тетраметил- 0,1 0,84 382
4,7-Диметил- 0,1 0,88 382 3,4,6,8-Тетраметил- 0,1 0,89 382
0,5 0,87 382 3,4,7,8-Тетраметил- 0,1 0,85 382
4,7-Диэтил- 0,9 0,94 81 Дициклогексен-3,4,7,8- 0,3 0,87 81
4,7-Дпфепил- 4,6 1,13 81
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
335
они находят в спектрофотометрии. Правда, многие из индикаторов в недостаточной степени отличаются от исходного комплекса трис-(1,10-федантролин)железа(П) по формальному потенциалу или коэффициенту молярной экстинкции, чтобы привлекать к себе особый интерес. При выборе индикатора для данного метода титрования следует помнить, что во всех случаях точка перехода индикатора находится при более высоком значении потенциала, чем указано в таблицах для формального потенциала, потому что Bred гораздо выше, чем eox-
Из комплексов с незамещенными лигандами чаще всего применяют [244, 390] исходную систему (Fe phen3)2+/(Fe phen3)3+ [260], особенно при титровании церием (IV). Эта система пригодна также при титровании перманганатом и другими сильными окислителями [30], но из-за более низких стандартных потенциалов бихромата и ванадата надо работать в среде очень высоких концентраций кислоты, например в 6—10 М серной кислоте, чтобы поднять их потенциал и понизить формальный потенциал индикатора; тогда получатся хорошие конечные точки. Однако при такой высокой концентрации кислоты индикатор проявляет некоторую неустойчивость, особенно в окисленной форме. Поскольку комплексы железа инертны, тенденция к обмену железа на другой металл в исследуемом растворе не выражена сильно, кроме как в экстремальных условиях, однако может произойти обмен лигандами, и этого надо остерегаться. Цианид обменивается с фенантролином очень быстро при высоких значениях pH, что служит основанием для хорошо известного метода определения следов цианида [244]; у получающегося комплекса формальный потенциал ниже. Было обнаружено, что индикатор ведет себя неудовлетворительно при титровании гексацианоферрата (II) церием (IV) в средах различных кислот, и это может быть результатом обмена лигандами. Однако в данном случае простой комплекс фенантролина является одним из лучших и наиболее удобных из имеющихся индикаторов.
Дипиридильный комплекс железа (II) ведет себя подобно комплексу фенантролинжелеза (II) и имеет несколько более низкий формальный потенциал. Его легче синтезировать, и он дешевле, но гораздо менее устойчив в растворах сильных кислот. Это приводит к ошибочным значениям формального потенциала и поправки, объяснение которым дал Белчер [385]. Вальден, Гаммет и Чепмен [264] сообщили, что комплекс дипиридила менее устойчив, чем комплекс фенантролина, а Каджл и Смит [391] не советовали оставлять его продолжительное время в контакте с избытком окислителя. Комплекс дипиридила использовали при титровании раствором церия(IV) железа (II) [264, 391, 392] и гидрохинона [393]; в последнем случае необходимо определить поправку на индикатор. Однако считают [381], что, несмотря на четкое изменение окраски от фиолетовой на зеленую при окислении, комплекс 2,2',2"-
336
ГЛАВА VIII
терпиридила имеет серьезные недостатки из-за неустойчивости окисленной формы. Ни один из лигандов не имеет существенных преимуществ перед фенантролином.
Внимание исследователей привлекали также замещенные фенантролины. Уже говорилось о линейной зависимости между р/Са лиганда и формальным потенциалом индикатора, имеющим заместителей в положении 5 [317]. Некоторые авторы [394] подчеркивали также влияние пространственных факторов, мезомерны.х свойств групп-заместителей и таутомерии замещенной молекулы. Однако наибольшее влияние на окислительно-восстановительные свойства индикатора оказывает плотность электронов у атомов азота; в случае нуклеофильных заместителей значение формального потенциала повышается, а при электрофильных понижается. Метилирование производит аддитивное влияние и, следовательно, является подходящим средством для доведения индикаторов до требуемого значения формального потенциала.
Первым производным фенантролина, привлекшим внимание исследователей, было 5-нитропроизводное [265]; Смит с сотр. [395] показал, что благодаря высокому потенциалу перехода это соединение пригодно в качестве индикатора в методах титрования церием (IV) в среде хлорной или азотной кислоты. Высокий формальный потенциал церия (IV) в хлорной кислоте обеспечивает стехиометрическое окисление ряда органических соединений [396] и оксалатов [397-—399]. При использовании катализатора можно также титровать ртуть(I) [400]. Следовательно, 5-нитропроизводное занимает определенное место в методах титрования церием (IV) в хлорной кислоте. Используют также 5-сульфонат и 3-сульфонат при титровании в среде, содержащей перхлорат, с которым большинство других индикаторов образует труднорастворимые соли [343, 344]. Подобными преимуществами обладает дисульфонат 4,7-дифенилпроизводного [343].
Как видно из таблиц, метилпроизводные были тщательно исследованы, но их почти не применяют в титриметрии. Смит и Брандт [401] первыми внедрили применение 5,6-диметилпроизвод-ного в титровании железа (II) в среде 1—2 М соляной или серной кислоты раствором бихромата, для которого потенциал перехода комплекса незамещенного фенантролина слишком высок. В сильно разбавленной кислоте требуется еще более низкий формальный потенциал индикатора, и Смит [402] предложил использовать 4,7-ди-метилпроизводное для 0,5 М. кислоты, а 3,4,7,8-тетраметилпроиз-водное для 0,1 М кислоты.
Описаны также комплексы со многими другими металлами [244], а из комплексов с одним лигандом, обладающих потенциальными индикаторными свойствами, наиболее замечательны, возможно, комплексы с рутением, которые флуоресцируют. Формаль
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
373
ные потенциалы комплексов рутения и осмия с незамещенными лигандами включены в табл. VI 11.41. Ниже приводятся сведения о комплексах этих и некоторых других металлов.
Комплексы рутения
При окислении этих комплексов окраска .резко и четко изменяется от оранжево-красной до бледно-зеленой. Значения формальных потенциалов высокие, но уменьшаются с повышением концентрации кислоты. Комплексы почти не применяются в титриметрии, так как потенциал системы грнс-(5-нитро-1,10-фенантролин)желе-зо(П)—грис-(5-нитро-1,10-фенантролин)железо(Ш) находится в той же области потенциалов, а изменение окраски этой системы более четкое. Дуиер [381] исследовал влияние заместителей в комплексах рутения 1,10-фенантролина и 2,2'-дипиридила. Влияние метилирования на комплексы рутения подобно влиянию на комплексы железа. Система трис- (5-бром-1,10-фенантролин)руте-ний(П)—т/шс-(5-бром-1,10-фенантролин)рутений(Ш) имеет самый высокий формальный потенциал: 1,41 В в 0,002 М азотной кислоте и 1,36 в 0,1 М азотной кислоте. Стейгман с сотр. [403] использовал комплексы дипиридила при титровании оксалата в 2 М хлорной кислоте раствооом церия (IV). Однако интереснее всего то, что эти комплексы флуоресцируют. Были исследованы [404] флуоресцентные индикаторные свойства нескольких комплексов дипиридила и обнаружено, что комплексы незамещенного 2,2'-ди-пиридила наиболее подходят для титрования церием (IV) в среде хлорной кислоты, а комплекс 4,4'-диметил-2,2'-дипиридила с рутением— лучший индикатор для титрования перманганатом или церием (IV) в среде серной кислоты. Тушение оранжево-красной флуоресценции при окислении идет быстро и обратимо. Максимум поглощения находится при длине волны 450—465 нм, а максимум флуоресценции в области 575—590 нм.
Комплексы осмия
При окислении комплексов осмия с 1,10-фенантролином или 2,2'-дипиридилом окраска изменяется от зеленой до бледно-красной; потенциалы этих индикаторов находятся в удобной для работы области. Дуиер с сотр. [387] сообщает, что комплекс (2,2',2"-терпиридил) осмия (III) неустойчив, и считает его не подходящим Для применения в качестве индикатора. Дуиер и Гибсон [405] с успехом использовали комплекс осмия с фенантролином при титровании железа (II) бихроиатом в 0,5 М серной кислоте в присутствии фосфорной кислоты, а комплекс с дипиридилом считали неподходящим (объяснения не давались). Однако комплекс с ди-пирилом оказался хорошим индикатором для титрования желе-
22—918
Таблица VI11.43
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ
КОМПЛЕКСОВ МЕДИ(1)/МЕДИ(П) В ВОДНЫХ И ВОДНОДИОКСАНОВЫХ РАСТВОРАХ (СООТНОШЕНИЕ 1:1) [408]
Лиганд Е. В
1,10-Фенантролин в водном растворе
Незамещенный 0,174
2-Метил- 0,337
2-Хлор- 0,400
2,9-Диметил- 0,594
1,10-Фенантролин в водно-диоксановом растворе
Незамещенный 0,296
5-Метил- 0,299
5-Фенил- 0,291
5-Нитро- 0,379
5-Амино- 0,248
5-Хлор- 0,321
4,7-Диметил- 0,220
4,7-Дифенил- 0,222
4,7-Дихлор- 0,387
5,6-Диметил- 0,250
3,5,6.8-Тетраметил- 0,268
2,2'-Дипиридил в водном растворе
Незамещенный 0,120
4,4' -Диметил- 0,091
4,4'-Дикар бокси- 0,150
3,3'-Дикарбокси- 0,213
2,2'-Дипиридил в водно-диоксановом растворе
Незамещенный 0,251
4,4'-Диметил- 0,204
4,4'-Диэтил- 0,215
4,4'-Дифенил- 0,269
4,4'-Ди (фенилэтил) - 0,243
4,4'-Дикарбоксилат 0,234
Этиловый эфир 4,4'-дикарбоновой кислоты 0,357
5,5'-Диметил- 0,251
5,5'-Дикарбоксилат 0,234
Этиловый эфир 5,5'-дикарбоновой кислоты 0,380
6,6'-Диметил- 0,750
5,5'-Диэтил-4,4'-диметил- 0,210
Для 2,2',2"-терпиридилпроизводных Е = —
—0,80 В
338
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
339
за (II), гидрохинона и гексацианоферрата(II) раствором церия(IV) в среде серной кислоты [380]. Формальные потенциалы комплексов осмия (см. табл. VIII.41) показывают, что имеет смысл продолжить их исследования.
Комплексы других металлов
Коэффициент молярной экстинкции комплекса ванадия (II) с 1,10-фенантролином составляет 8-103 л • моль-1 • см-1 при 645 нм. Формальный потенциал, по-видимому, равен 0,25 В в разбавленной соляной кислоте. Этот комплекс использовался при титровании железа(III) раствором хрома(II) [332]. Гаммет с сотр. [265] исследовал комплекс хрома (II) с 1,10-фенантролином и нашел, что формальный потенциал его подобен потенциалу сольватированной пары железо(Н)—железо(Ш), но изменение окраски нечеткое. Слабое окрашивание раствора характерно также для комплексов кобальта; потенциал этих комплексов с фенантролином равен 0,37 В [406]. Комплексы меди окрашены интенсивно, но труднорастворимы в водной среде и неустойчивы в условиях сильных кислот или щелочей. Формальные потенциалы их низки. Однако возможно, что они найдут применение в смешанных или неводных средах. Значения стандартных потенциалов для комплексов медь (I)/медь(II) в водных средах следующие [407]:
Лиганд Стандартный потенциал при
10 °C 20 °C 30 °C
2-Хлор-1,10-фенантролин 0,4668 0,4668 0,4666
2,9-Диметил-1,10-фенантролин 0,6233 0,6240 0,6248
Джеймс и Уильямс [408] изучали влияние заместителей в комплексах на формальные потенциалы в водных средах и водно-диоксановых растворах (соотношение 1:1) (табл. VIII.43).
СМЕШАННЫЕ КОМПЛЕКСЫ
До сих пор рассматривались комплексы, в которых металл был координирован лигандом одного вида. Если комплекс содержит лиганды более одного вида, перестановок становится огромное шсло. Достаточно привести два примера: в первом незамещенный бидентатный лиганд замещается цианид-ионами, а во втором чередуется множество лигандов, являющихся производными пиридина.
29*
240
ГЛАВА VIII
Смешанные цианидные комплексы
Замещение бидентатных лигандов парами цианид-ионов снижает формальные потенциалы получающихся индикаторов, но меняет при обратной реакции зависимость потенциала от значения pH. Формальные потенциалы этих индикаторов увеличиваются с повышением концентрации кислоты и ионной силы в отличие от потенциалов комплексов с одним видом лиганда. Было проведено исследование [380, 409] поведения нескольких дициановых комплексов в серной кислоте; результаты обобщены в табл. VIII.44.
Таблица VIII.44
ФОРМАЛЬНЫЕ ПОТЕНЦИАЛЫ ДИЦИАНКОМПЛЕКСОВ В СЕРНОЙ КИСЛОТЕ
ПРИ 25 °C [380, 409]
[H2SO4], моль л—1 £o,TF
[Fe phen2(CN)2] [Fe bipy2(CN)2] [Ru bipy2(CN)2l 1 j [Os bipy2(CN)2]
0,01 0,781 1,12 0,778
0,05 0,777 1,11 0,783
0,11 0,776 1,10 0,791
0,50 0,776 1,12 0,804
1,0 0,810
2,0 0,806 0,786 1,13 0,836
4,0 0,818 0,787 1,16 0,89
6,0 0,852 0,820 1,22 0,95
10,0 0,925 0,901 1,26 1,01
12,0 0,990 0,979 1,30 1,02
Дицианкомплексы с рутением отличаются плохой обратимостью. Комплексы с другими металлами—хорошие индикаторы; их окраска при окислении изменяется от оранжево-красной до бледно-фиолетовой. Тетрацианкомплексы имеют, однако, более низкие формальные потенциалы. Ирвин с сотр. [379] сообщает, что стандартный потенциал системы тетрацианмонодипиридилжелезо (II) —тет-рацианмонодипиридилжелезо(Ш) равен 0,541В при экстраполяции ионной силы до нуля, а тангенс угла наклона прямой Дебая—Хюккеля составляет -J-0,085 и находится в хорошем соответствии с теорией. Он нашел, что в среде 0,197 М соляной кислоты формальный потенциал равен 0,563 В. Шилт [410] исследовал систему тетрацианмонофенантролинжелезо (И) — тетрацианмонофе-нантролинжелезо(Ш) (Лтах=462 нм, е = 4,45-103 л-моль-1-см-1 для комплекса железа (II) и 7тах=500 нм, е=350 л • моль-1 • см-1
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
34 Р
при pH 7 для железа (III), как рассчитано по данным Шилта). Формальный потенциал увеличивается с повышением концентрации кислоты; полученные Шилтом значения приведены в табл. VIII.45.
Таблица VII1.45
ФОРМАЛЬНЫЕ ПОТЕНЦИАЛЫ СИСТЕМЫ ТЕТРАЦИАНМОНОФЕНАТРОЛИН ЖЕЛЕЗО(П)/ТЕТРАЦИАНМОНОФЕНАНТРОЛИНЖЕЛЕЗО(Ш) В 1,0 М ХЛОРИДЕ
КАЛИЯ ПРИ 25 °C
pH 0,5 0,7 0,9 1,2 1,6 1,9 2,2 3,0—12,0
Е'в, В 0,67 0,66 0,65 0,64 0,63 0,62 0,62 0,61
Комплекс дициан-бпс-(фенантролин) железо (II) применяли в некоторых методах титрования [409, 411]; из них наиболее примечательно титрование при проведении реакций диазотитрования и определения азидов в 6 М соляной кислоте посредством раствора нитрита натрия.
Смешанные хелатные комплексы
Букингем с сотр. [389] изучил комплексы осмия со смешанными лигандами путем измерения потенциалов растворов, приготовленных из навесок данных комплексов в обоих состояниях окисления, и расчета стандартных потенциалов, экстраполированных, до нулевой ионной силы. Были проведены термодинамические и квантовомеханические расчеты, а также анализ электронных спектров в рамках теории поля лигандов для объяснения наблюдаемых эффектов. Ряд этих объяснений был лишь предварительным, но некоторые результаты стоит отметить как указание на природу и степень наблюдаемых эффектов (см. табл. VIII.46). Уменьшение заряда комплекса, происходящее при замещении нейтральной молекулы пиридина на галогенид-ион, обычно приводит к тому, что новый комплекс легче окисляется. Увеличение степени сопряженности повышает способность образовывать л-связи между металлом и лигандом и таким образом стабилизирует комплекс осмия (III) при одновременном увеличении стандартного потенциала. Небольшое влияние природы галогенид-иона указывает, что потенциал системы зависит от остающихся ароматических лигандов. Замещение пиридина на различные его производные приводит к нелинейному изменению потенциала в зависимости от основности замещенного пиридина, но согласуется с эффектом сверхсопряжения.
Таблица VIII.46
ВЛИЯНИЕ ПРИРОДЫ ЛИГАНДА НА СТАНДАРТНЫЕ ПОТЕНЦИАЛЫ КОМПЛЕКСОВ ОСМИЯ(П)/ОСМИЯ(И1) [389]
Комплекс осмия(П) Ео ДЕ0
Влияние общего заряда комплекса
Терпиридил — дипиридил — пиридин 0,8713 —0,3085
Терпиридил — дипиридил — хлор 0,5628
(Дипиридил) 2— (пиридин) 2 0,8345 —0,3509
(Дипиридил) 2 — пиридин — хлор 0,4836
Дипиридил— (пиридин) 4 0,8052 —0,3797
Дипиридил — (пиридин) 3 — хлор 0,4255
(Дипиридил) 3 0,8847 —0,7317
(Дипиридил) 2 — пентандион-2,4 0,1530
Влияние сопряжения
(Дипиридил) з 0,8847 —0,0502
(Дипиридил) 2— (пиридин) 2 0,8345 —0,0293
Дипиридил— (пиридин) 4 0,8052
(Терпиридил)2 0,9866 —0,1153
Терпиридил — дипиридил — пиридин 0,8713 —0,0714
Терпиридил— (пиридин) з 0,7999
Терпиридил — дипиридил — хлор 0,5628 —0,0792
(Дипиридил) 2 — пиридин — хлор 0,4836 —0,0581
Дипиридил— (пиридин) 3 — хлор 0,4255
Влияние природы галогенид-иона
Терпиридил — дипиридил — иод 0,5666 +0,0009 —0,0047
Терпиридил — дипиридил — бром 0,5675
Терпиридил — дипиридил — хлор 0,5628
(Дипиридил) 2 — пиридин — иод 0,4888 —0,0017
(Дипиридил) 2 — пиридин — бром 0,4871 —0,0035
(Дипиридил) 2 — пиридин — хлор 0,4836
Дипиридил — (пиридин) 3 — иод 0,4508 —0,0071
Дипиридил— (пиридин) 3 бром 0,4437 —0,0182
Дипиридил— (пиридин) 3 — хлор 0,4255
Влияние заместителей в одном из лигандов
Терпиридил — дипиридил — пиридин 0,8713
Терпиридил — дипиридил — 3-метплпиридин 0,8672
Терпиридил — дипиридил — 4-изопропилпиридин 0,8609
Терпиридил — дипиридил —4-этилпиридин 0,8587
Терпиридил — дипиридил - -4-метилпиридин 0,8506
342
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
343.
ДРУГИЕ СЛУЧАИ ПРИМЕНЕНИЯ КОМПЛЕКСОВ В КАЧЕСТВЕ ИНДИКАТОРОВ
Интересно отметить, что применение рассматриваемых комплексов в качестве индикаторов не ограничивается окислительно-восстановительным титрованием. В частности, комплексы дициан-бис- (1,10-фенантролин)железо (II) и дициан-бпс- (2,2'-дипиридил)-железо(П) обладают хорошими кислотно-основными индикаторными свойствами, особенно в неводных и полуводных средах; комплексы рутения (II) образуют труднорастворимые трииодиды, так что по этой именно причине происходит резкое тушение флуоресценции в методах титрования иодом, а не вследствие окисления индикатора. Некоторые двухкомнонентные и трехкомпонентные комплексы имеют значение в аргентометрии и в качестве металло-хромных индикаторов. По всем этим вопросам был написан обзор [244] с большим знанием дела, так что нет необходимости рассматривать их здесь.
4. ИНДИКАТОРЫ СПЕЦИФИЧЕСКОГО ДЕЙСТВИЯ
Бывает, что один или два реагента дают характерную реакцию с данным веществом, но чаще встречаются реагенты, которые обладают высокой избирательностью и для которых можно разработать такие условия, чтобы реакции с ними стали специфическими. Если реакция вызывает, например, четкое визуальное изменение окраски, если реакция очень чувствительна и если вещество, взаимодействующее с реагентом, является титрантом или титруемым соединением, то такой реагент может быть индикатором, особенно1 в тех случаях, когда реакция обратима. Из окислительно-восстановительных индикаторов используют два таких реагента: один — обратимый дисперсный коллоид, образующий адсорбаты с иодом, и второй — необратимый розанилин, у которого гипсохромный сдвиг от протонирования становится обратимым в результате бромирования.
ИНДИКАТОРЫ НА ИОД
Естественный цвет иона 1з можно обнаружить визуально в обычных условиях титрования. Например, в 100 мл титруемого раствора, находящегося в конической колбе емкостью 250 мл, можно установить присутствие иода вплоть до концентрации 2,5-10-5 М. Это соответствует примерно одной капле 0,05 М раствора иода в 100 мл воды [412]. Экстрагирование иода в небольшом объеме органического растворителя, хлороформа или четыреххлористого углерода, в которых иод, так же как в парах, имеет пурпурную окраску, приводит к увеличению чувствительности вследствие механического повышения концентрации: 5 мл раство
344
ГЛАВА VIII
рителя, добавленные к 100 мл титруемого раствора, дают повышение чувствительности в 20 раз, а 1 мл растворителя — в 100 раз. Чувствительность конечной точки в экстракте немного улучшается .вследствие более четкой различимости окраски, но может и понизиться при неполной экстракции, так что увеличение чувствительности надо оценивать практически. Установление конечной точки в экстракте обычно применяется при титровании йодатом по методу Эндрьюса [23, 26]. В противоположность этому коллоидные адсорбирующие индикаторы, такие, как крахмал, повышают чувствительность определения конечной точки не более чем в 2—3 раза, но они удобны для работы, так как гораздо легче различается изменение их окраски. Установление конечной точки в экстракте вошло в практику до применения крахмала [413]. Однако оно имеет тот недостаток, что титрование надо проводить в колбе с притертой пробкой, содержимое колбы надо энергично встряхивать после добавки каждой порции титранта для установления равновесия экстракции или реэкстракции. Следовательно, это медленный процесс.
Крахмал издавна применяют в иодометрии, и ему посвящено много работ. Вначале по механизму, предложенному Милиусом [114 115], предполагалось образование соединения определенного -состава, но в более поздних работах эта мысль не нашла подтверждения; появление синей окраски объясняется теперь адсорбцией молекул иода или, что более вероятно, трииодид-ионов коллоидными макромолекулами крахмала [416—419]. Тон окраски зависит от свойств крахмала: чем больше длина основной цепи, тем больше сродство к трииодиду, а разветвленные цепи отличаются меньшим сродством. Таким образом, неразветвленные фракции амилозы дают привычную чистую синюю окраску [420], а так как картофельный крахмал содержит много амилозы (20%), его издавна используют для приготовления индикаторных растворов. Фракции разветвленного амилопектина дают пурпурно-красную окраску, а эритроамилоза и эритрюгранулоза — красную. Сильно разветвленный гликоген дает красновато-коричневую окраску, а очень разнообразные остатки декстрина из гликогена окрашиваются в желтокоричневый цвет, почти не отличающийся по чувствительности от окраски свободного трииодид-иона [421]. Следовательно, лучшим крахмальным индикатором является амилоза; ее выделение описал Шох [422], а Лаиберт рекомендовал ее применение [423].
Чистый молекулярный иод не дает окраски с крахмалом. Присутствующий иодид в концентрации, немного большей концентрации иода, сразу же дает синюю окраску; чувствительность составляет 2-10_5 М. Чувствительность несколько возрастает с увеличением концентрации иодида и, кроме того, увеличивается в присутствии сильной кислоты (соляной или серной), достигая 5-10-6 М при рН = 0. При высоких температурах чувствительность заметно
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
34L
понижается, а такие органические вещества, как спирты, тоже понижают ее; в 50 %-ном этаноле синяя окраска совсем не образуется. В присутствии спиртов желтая окраска триодид-иона является более чувствительной индикацией, чем окраска с крахмалом. Обычно распределение крахмальных зерен в растворе довольно быстро ухудшается, поэтому за последнее столетие было предложено множество рецептов для приготовления более устойчивых растворов. Некоторые из них приводятся в современных руководствах [26]. Мы предпочитаем готовить свежий раствор каждый день, добавляя 100 мл кипящей воды к смеси 1 г торгового препарата «растворимого крахмала» с 1—2 мл холодней воды. Цул-ковски [424] впервые получил «растворимый крахмал» в 1874 г.; процесс состоит, 'в сущности, в обработке рисовою или картофельного крахмала горячей разбавленной щелочью, подкислении и осаждении спиртом. Платнер [425], по-видимому, вторично обнаружил, что такой способ обработки приводит к хорошим результатам, и утверждал, что раствор устойчив в течение года. Было предложено [424] производное крахмала, а именно гликолят крахмала натрия, однако чувствительность его меньше, чем крахмала [427]. Другие коллоиды тоже дают синие окраски с трииодид-ионом. Баргер, например, сообщил об окраске с диспергированными производными ксантона, флавона, тиофлавона и а-пирона и с неорганическими золями, состоящими из основных ацетатов лантанидов, а также с кумарином и ацетилкумарином [428—430]; однако окраска образуется только в том случае, когда вещества находятся в коллоидном состоянии. Хан с сотр. [431] наблюдал подобное явление с а-нафтофлавоном, а Рейт [432] провел систематическое исследование его индикаторных свойств, которые проявлялись опять-таки только в коллоидном состоянии. а-Нафтофла-вон оказался несколько более чувствительным индикатором по сравнению с крахмалом, причем чувствительность его в противоположность крахмалу увеличивалась при незначительном повышении температуры. а-Еафтолфлавон ведет себя подобным образом как обратимый индикатор на бром, окраска тускло-коричневая [224, 225]. Коллоидные поливиниловые спирты тоже окрашиваются иодом [433]. Миллер с сотр. [434] показал, что полностью деацетилированный полимер дает синюю окраску; при содержании 10 мол. % ацетата окраска становится красной, а максимум чувствительности получается при 20 мол. % остаточных ацетатных групп. При сильном разбавлении окраска становится коричневато-желтой, а чувствительность в присутствии иодида в 10 раз больше, чем с крахмалом.
В иодометрии использовались и другие окислительно-восстановительные индикаторы, имеющие низкие потенциалы; например, раньше находил применение метиленовый голубой [12—16], а в последние годы вариаминовый синий [1, 435].
346
ГЛАВА VIII
ИНДИКАТОРЫ НА БРОМ
Смит первым использовал парарозанилин в качестве индикатора в броматометрии [211], но не указал, для какой цели это было сделано. Он ошибочно принял реакцию за деструктивное окисление; пурпурная окраска небольшого количества бромсодержащего соединения, образующегося непосредственно перед точкой эквивалентности, была дополнительным цветом ik желтой окраске индикатора, и в итоге раствор становится бесцветным. Истинный механизм действия этого индикатора состоит в бромировании ортоположений к аминогруппам, что вызывает батохромный сдвиг, сильно нарушающий существующее положение перед протонированием аминогрупп, которое приводит к гипсохромному сдвигу окраски свободного основания. Бромирование необратимо, но идет, к большому удобству, медленно, а окраска изменяется от бледной лимоннс-желтой до глубокой чисто-пурпурной. Последняя окраска устойчива в течение длительного времени, но если перейти конечную точку, то бромирование, видимо, продолжается и окраска изменяется на красную. Красное вещество окисляется затем в оранжевое [227], и, хотя эта окраска обратима и очень четкая, одновременно происходят деструкция и осаждение, поэтому такое определение конечной точки нежелательно [228]: получаются завышенные результаты титрования. Такое же красное или оранжевое вещество получается в том случае, если индикатор добавлять слишком рано во время титрования, потому что в месте приливания титранта (бромата) в титруемый раствор образуется местный избыток брома, который действует на индикатор. Если индикатор добавлять позже, то этого удается избежать и появление глубокой красно-пурпурной окраски служит указанием конечной точки; из-за преждевременной деструкции появляется небольшая поправка на индикатор. Обычно удобно представлять парарозанилин и розанилин следующими формулами:
но, конечно, структуры могут быть делокализованными или мезо-мерными. В водных растворах эти индикаторы имеют фуксиновокрасный цвет. При подкислении аминогруппы протопируются, так что число мезомеров ограничивается и увеличивается средняя энергия переноса электрона, в результате чего происходит гипсо-хромный сдвиг к желтой области. Бромирование в орточположении
окислительно-восстановительные индикаторы
347
по отношению к протонированной аминогруппе уменьшает основность аминогруппы вследствие индукционного эффекта и превращает —NHs в —NH2, возвращая, таким образом, молекулу в прежнее .мезомерное состояние и уменьшая энергию переноса электрона до ее прежнего уровня. Поэтому батохромный сдвиг доводит окраску до ее исходного оттенка, и, кроме того, наблюдается слабый дополнительный сдвиг из-за присутствия брома. Следовательно, индикатор-основание совеем слабо изменяет окраску при бромировании. Помимо этого, существует низший предел концентрации кислоты, ниже которой изменение окраски недостаточно четкое; на практике это соответствует pH 2. Чем кислее раствор, тем желтее становится индикатор и тем более четко изменение окраски. Но существует также верхний предел концентрации кислоты, когда уменьшение основности аминогрупп при бромировании в орто-положении преодолевается увеличением концентрации протонов, а изменение окраски становится слабее при концентрации ионов водорода выше 2,5 моль-л-1. Изменение окраски при 3,0 М концентрации кислоты становится слишком слабым, чтобы его можно было использовать [74, 436]. Бишоп [230] исследовал многие модификации фуксина (С. I. 678) и розанилина (С. I. 677) и пришел к вызоду, что, хотя свободный незамещенный фуксин дает наиболее интенсивные окраски, он очень быстро бромируется, и поэтому нужно быть очень внимательным, чтобы не определить конечную точку преждевременно. Монометилпроизводное розанилин дает довольно хорошую окраску, но наличие одной метильной группы в активном положении в кольце замедляет в достаточной степени бромирование, что позволяет избежать преждевременного определения конечной точки. После добавления конечного инкремента титранта, составляющего часть капли, окраска достигает максимальной интенсивности за каких-нибудь 10—20 с. Более того, реакция эта специфична: хлор и бром не оказывают влияния. Замещение водородов аминогрупп дает продукт, который сохраняет реакционную способность розанилина, хотя окраска его совершенно иная. Метилирование до пентаметилпроизводного [метиловый фиолетовый 2В (С. I. 680)], этилирование до гексаэтилпроиз-водного [этиловый фиолетовый (С. I. 682)] или введение алкилфенильных групп, как, например, бензильная [метиловый фиолетовый 6В (C.I. 683)], снижают реакционную способность и ухудшают изменение окраски. Сульфирование дает продукт такой же реакционной способности, что и у исходного вещества, но он не имеет никаких преимуществ Замещение одной из аминогрупп путем диазотирования или сочетания, например для получения нафтолов, снижает реакционную способность.
Поскольку бромат был все же до некоторой степени непонятным реагентом, Бишоп [230] критически рассмотрел большое число определений, в которых использовался в качестве индикатора
348
ГЛАВА VIII
розанилин. Мышьяк(Ш), сурьму (III) и олово(II) можно титровать непосредственно, хотя обратное титрование с двойным избытком лучше и легче в случае олова (II). Наиболее подходящей средой оказалась среда 2 М по соляной кислоте и 0,02—0,1 М по бромиду, хотя бромидом можно пренебречь, работая с соляной кислотой. В последней среде сурьма давала отрицательную индукционную ошибку Позже были подробно исследованы реакции с мышьяком, сурьмой и гидразином [36, 73, 74]. Гидразин гоже давал отрицательную индукционную ошибку при прямом титровании, но при обратном титровании с двойным избытком получались превосходные результаты. По этому методу к образцу добавляют избыток стандартного раствора бромата (бромид необходим при работе в среде серной кислоты или для процесса бромирования), выжидают нужное для протекания реакции время, затем добавляют аликвотное количество мышьяка (III) или сурьмы(III) в избытке по отношению к количеству, которое должно прореагировать со свободным бромом, выделяемым в титруемом растворе, и, наконец, титруют избыток мышьяка(III) или сурьмы(III) оставшимся в бюретке раствором бромата по розанилину в качестве индикатора. Холостое титрование такой же аликвотной части мышьяка (III) или сурьмы (III) после вычета из общего количества израсходованного бромата дает титр образца по бромату: можно использовать раствор как мышьяка(III), так и сурьмы(III). Этот метод был применен с успехом для определения гексацианоферрата (II), олова (II), гидразина, гидроксиламина и семикарбазида. Ароматические окси- и аминосоединения определяли бромированием по этому способу, устанавливая в то же время критические интервалы бромирования. Таким путем были определены фенол, анилин, о-толуидин, о-кре-зол, лг-толуидин, n-крезол, п-юлуидин, фенилгидразин, 8-оксихи-нолин (и цинк, никель, магний и алюминий после осаждения их комплексов), .м-нитроанилин, п-нитроанилин, n-нитрофенол, аце-тил-п-фенетидин, ацетилсалициловая кислота, салициловая кислота, n-хлоранилин, 2,4-динитрофенол, о-хлорфенол, 2-хлор-4-нитро-анилин, сульфаниловая кислота, n-оксибензонитрил, п-оксибензаль-дегид, З-бром-4-оксибензальдегид, о-метоксифенол и п-метоксифе-нол. Антраниловая кислота не дает удовлетворительного результата, для фенилгидразина надо контролировать условия определения, а муравьиная кислота и формальдегид не реагируют при этом. Во многих случаях титруемый раствор был сильно окрашен, и в нем находился осадок во взвешенном состоянии, тоже окрашенный, однако определение конечной точки было четким.
На холостой опыт с индикатором расходуется менее 0,1 мл 6-10-3 М бромата. Так как индикатор необратим, его следует вносить в титруемый раствор по возможности ближе к конечной точке, предпочтительно в пределах одной капли 0,01667 М титранта бромата Для этого, если приходится титровать неизвестный раствор,
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
349
надо провести ориентировочное грубое титрование. В случае бесцветных растворов это можно сделать в присутствии хинолинового желтого [226] и из результата вычесть 0,1 мл. Конечно, ориентировочное титрование можно провести и с розанилином. Титровать -ледует в колбе со стеклянной притертой пробкой. Бромат приливают, вращая колбу, до тех пор, пока бром в месте его образования не начнет рассеиваться более медленно; тогда добавляют 2 капли 0,1 %-него розанилина в 1 М соляной кислоте и продолжают внимательно титровать при энергичном перемешивании. Если индикатор становится красным или оранжевым, надо добавить еще и еще, если это необходимо. Вблизи конечной точки после каждой добавки инкремента титранта колбу закрывают пробкой и энергично встряхивают, затем оставляют в покое на 10—15 с перед добавлением следующего инкремента. В конечной точке окраска углубляется и значительно темнеет в течение 10 с. Для точного титрования 2 капли того же раствора индикатора следует добавить, не доходя на 0,1 мл до ожидаемой конечной точки, и закончить титрование, приливая титрант не каплями, а частями капли, закрывать колбу пробкой и энергично встряхивать содержимое после каждой добавки инкремента, которые надо производить через интервалы в 10—20 с.
При любом другом окислительном титровании, где бром присутствует или образуется в конечной точке из бромида, можно с успехом использовать в качестве индикатора розанилин: минимальная индикаторная ошибка, четкое и интенсивное изменение окраски заставляют предпочесть его даже обратимым индикаторам. Так, Бишоп с сотр. [437] использовал его при точном установлении титра хлорамина-Т путем титрования мышьяка (III). Те же авторы критически сопоставили розанилин с четырьмя лучшими необратимыми и пятью обратимыми индикаторами в этом методе титрования [438], используя в качестве метода сравнения потенциометрическое титрование [37]. При титровании сурьмы (III) хлорами-ном-Т в соответствующих условиях [439] розанилин не дает такую хорошую конечную точку, как при титровании броматом; это несоответствие существует также при титровании гидразина [440]. Наблюдаемые недостатки зависят от реакции с хлорамином-Т, а не от индикатора, который дает очень хорошие результаты при использовании метода обратного титрования с двойным избытком, заканчивающегося титрованием мышьяка (III) хлорамином-Т в присутствии бромида [441 j. Сопоставление с другими индикаторами будет проведено в следующем разделе.
Реакции дисперсных коллоидных индикаторов на иод были изучены также и в применении к брому [442]. Из этих индикаторов только а-нафтофлавон дал положительные результаты. Впервые его предложили Шулек [225] и Юзел [224]. Этот индикатор обладает обратимостью, в бесцветной среде изменение окраски происхо
350
ГЛАВА VIII
дит от белой опалесценции до желтой или коричневато-желтой опалесценции, четко различимой. Применение его в средах, содержащих бромид, при титровании хлорамином-Т изучал Бишоп с сотр. [437—442]; об этом будет сказано в следующем разделе, где он будет сопоставлен с другими индикаторами. Хан [443] использовал бромирование флуоресцеина, чтобы получить эозин для определения конечной точки в титровании броматом; этот автор утверждает, что медленное бромирование помогает избежать трудностей, возникающих вблизи конечной точки из-за необратимости индикатора; однако бромирование не так медленно, чтобы определение конечной точки было легким или четким.
5. НЕОБРАТИМЫЕ И РАЗРУШАЮЩИЕСЯ ИНДИКАТОРЫ
Возможно, самым ранним окислительно-восстановительным индикатором был индиго; Гей-Люссак нашел, что он разрушался от действия гипохлоритов, и использовал его в титровании мышьяка (III) гипохлоритом [444], самом раннем из всех титриметриче-ских методов. Гексацианоферрат(Ш), впервые использованный в качестве внешнего индикатора Пенни [445] при титровании железа (II) бихроматом, является, конечно, необратимым индикатором. Через сто лет его снова исследовал Стокдаль [133], который нашел, что чувствительность его по отношению к железу(II) составляет 1 часть на тысячу, причем он не окисляется в обычных условиях титрования. Фордос с сотр. [446] впервые использовал гипохлорит в качестве титранта и обесцвечивание индиго для установления конечной точки в титровании тиосульфата и политионатов; он также предсказывал возможность применения гипохлорита для других определений. При титровании мышьяка (III) гипобромитом также использовали индиго в качестве индикатора. К концу XIX в. был предложен [447] метиловый оранжевый, как деструктивный индикатор в титровании мышьяка (III) и сурьмы(III) броматом. Использовались также индигокармин и метиловый красный. Кроме того, в связи с применением гипохлорита натрия как титранта в слабокислых средах исследовались и сопоставлялись [448] индиго, метиловый оранжевый, метиловый красный и анилингидрсхторид в качестве необратимых индикаторов, а также использовалась окраска свободного галогена, образующегося в конечной точке в случае присутствия бромида или иодида. Был сделан вывод: лучшими индикаторами являются иодид калия и крахмал [449].
К тому времени появились обратимые окислительно-восстановительные индикаторы. Несомненно, что обратимые индикаторы следует предпочитать необратимым, даже жертвуя в некоторой степени яркостью изменения окраски. Однако интерес к необратимым и деструктивным индикаторам продолжает сохраняться по
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
351
двум причинам. Во-первых, изменение их окраски часто бывает резким и, во-вторых, имеющиеся обратимые индикаторы для работы с галогенными окислителями (галогены, гипогалогениты, галогенаты, хлорамин-Т, N-бромсуксинимид и пр.) часто бывают неудовлетворительны; изменение их окраски слабое или плохое, дают высокую и колеблющуюся индикаторную ошибку или же бывают недостаточно устойчивы. Для титрования указанными реагентами все еще недостает хороших обратимых индикаторов. Несмотря на то что дифениламин-4-сульфокислота обратима при титровании гипохлоритом, Вилард и Манало [450] считают ее хуже 4'-амино-дифениламин-4-сульфокислоты, которая необратимо разрушается. К таким индикаторам сохраняется интерес в методах титрования церием(1У): Вилард и Янг [451] использовали деструкцию 'метиленового голубого для определения конечной точки в титровании оксалата в присутствии монохлорида иода. Ратсбург [452] тоже использовал метиленовый голубой наряду с метиловым красным, метиловым оранжевым и конго красным в титровании сурьмы(III) раствором церия(IV), тогда как Фурман с сотр. [213] работал с метиловым красным при титровании гидрохинона и железа (II) раствором церия (IV). Метиленовый голубой можно использовать несколькими способами; в начале главы говорилось о давнем его применении в иодометрии; восстановленный до лейкосоединения, он служит в качестве индикатора с низким потенциалом; в виде трифенилметанового красителя — это обратимый индикатор, хотя и низкого качества, и его использовали в таком состоянии, например, для определения влаги по методу Карла Фишера [453]; его можно также деструктивно окислять [451, 452] броматом при плохом изменении окраски [230]. Метиленовый голубой использовали в качестве титранта [454, 455], а нафтиловый красный (бензоазо-а-нафтиламин) использовали для титрования избытка гипобромита после проведения с его помощью окисления [456].
Смит [2И] провел первое систематическое исследование некоторых красителей, обесцвечивающихся под действием хлора, которые могли бы служить индикатор ими, подходящими для бромато-метрии; этот вопрос рассмотрен подробно в разд. 2 о красителях. На основании работы Смита и ряда последующих работ [219—222] предложено использовать амарант (В. С. I. 184 [204]), бриллиантовый пунцовый 5R (185) и бордо (88), окраска которых изменяется от красной до бесцветной, а также нафтоловый сине-черный (246) с изменением окраски от синей до бесцветной. Все они являются азосоедикениями. Ошибочно считалось, что фуксин (678) разрушается, однако оранжево-желтая окраска связана фактически не с разрушением [230], а с изменением окраски при образовании пурпурного бромпроизводного, имеющего дополнительный цвет. Все эти индикаторы пригодны для применения в броматометрии, а амарант, бриллиантовый пунцовый 5R и нафтоловый сине-чер
352
ГЛАВА VIII
ный были затем предложены для иодатометрии [221]; остальные исследованные соединения [2П] оказались неподходящими.
Среди красителей, исследованных Бишопом [230] и приведенных в табл. VIII.27, имеется много таких, которые можно использовать в качестве индикаторов в броматометрии. Большинство их необратимо или разрушается. К сожалению, проведенное исследование не охватило методы титрования йодатом и .гипохлоритом, но вряд ли можно получить какие-либо дополнительные сведения. Кроме того, по данным таблицы .видно, что бордо является плохим и необратимым, а п-этоксихризоидин— плохим индикатором, поскольку его обратимость не использовалась из-за ее ограниченности. У хинолинового желтого не было замечено обратимости, но позже Белчер показал [235], что лучшими обратимыми индикаторами для .броматометрии являются а-нафтофлавон, п этоксихризоидин и хинолиновый желтый. Из-за выдающихся свойств розанилина, который является специфическим и чувствительным реагентом на свободный бром, Бишоп не исследовал подробно другие подходящие для броматометрии индикаторы, помещенные в табл. VIII.27: очень немногие из них были лучше .индикаторов Смита, а розанилин мы рассматривали в предыдущем разделе.
Кольтгоф и Стенгер [457] исследовали обычные окислительно-восстановительные-индикаторы в титриметрии мышьяка (III) в присутствии бромида раствором гипохлорита кальция и нашли, что они непригодны для этой цели; эти же авторы испробовали ряд красителей и пришли к выводу, что лучшим из них оказался бордо (В. С. I. 88 [204]). Для метода титрования олова (II) броматом был рекомендован родамин В [458] или бензопурпурин В и 4В [459]. Сотджиа-Ровели [233] исследовал несколько красителей, в том числе некоторые из изученных Смитом [211], но ни один не оказался лучше метилового оранжевого. Шулек и Шомодьи [229] синтезировали ряд аналогов п-этоксихризоидина, но не нашли, чтобы какой-нибудь из них имел преимущество перед исходным соединением. Как уже было отмечено, Себелед с сотр. [231] исследовал трифенилметановый краситель руброфен, но его обратимость оказалась плохой. Среди разрушающихся красителей, предложенных для титрования гипохлоритом или гипобромитом, находятся крезиловый фиолетовый [460], бразилин и сантолин [461], кошениль [462] и амарант [235]. Флуоресцеин Хана [443], конечно, тоже необратимый индикатор на бром; Бишоп не дает ему высокой оценки [230].
Белчер считает амарант (В. С. I. 184 [204]) лучшим из разрушающихся индикаторов в методах титрования гипогалогенитами [235], а тартразин (В.С. I. 640) лучшим обратимым индикатором, но отмечает, что важно, чтобы в титруемом растворе находился бромид, иначе индикатор разрушается необратимо. Были использованы и другие пиразолоновые красители; только желтые соедп-
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
353
нения обладали обратимостью, но ни очно из них не было лучше тартразина. Такое же изменение окраски от желтого до бесцветного показывает хинолиновый желтый [236]. Белчер предложил добавлять в титруемый раствор бромид и показал, что индикатор необратимо разрушается в бикарбонатных средах, если в них не содержится бромид [463]
В дополнение к индикаторам, предложенным Смитом и Уилкоксом для титрования йодатом [221], Белчер и Кларк нашли, что для этой цели пригоден п-этоксихризоидин, обладающий обратимостью, но заметили, что многие торговые препараты содержат примеси и, несмотря на то, что можно получить четкую конечную точку, изменение окраски может быть необратимым. Они также сообщают о различных значениях поправки на холостой опыт и о том, что обратимую реакцию нельзя проводить более двух раз из-за деструкции. Брэзайер и Стефен [218] провели систематическое исследование большого числа трифенилметановых красителей в титрованиях йодатом по методу Эндрьюса и установили, что два из них дают превосходное, но необратимое изменение окраски при титровании иодида, гидроксиламина, мы1шьяка(Ш), сурымы(Ш) и олова(II). Это ретина пурпуровый (WSS) (В. С. I. 42515 i[205]), изменяющийся от красновато-коричневого до зеленого и далее до пурпурного, и темно-синий (Chem) (B.C.I. 44085 [205]), изменяющийся от желтого до оранжевого и далее до зеленого; лучшим является регина пурпуровый. Брэзайер и Стефен предположили, что замещение фенильной группы в красителе на замещенный дифениламин, присоединяющийся в лшра-положении, сможет придать красителю индикаторные свойства в методах титрования по Эндрьюсу. Эти же два индикатора показали приемлемые, но необратимые изменения окраски от действия бромата, однако не имели преимуществ по сравнению с розанилином, который, как нашли эти же авторы, оказался лучшим индикатором при титровании ги похлоритом в среде, содержащей бромид. Рубиновый HF конц. для кожи (HF) (В. С. I. 42520 [205]), который изменяет окраску от малиновой до пурпурной в пределах значений pH 2—3, тоже считался заслуживающим рекомендаций. Брэзайер и Стефен, кроме того, исследовали трифенилмета новые красители в методах тит-ования иода тиосульфатом [218] и нашли несколько таких, которые изменяли окраску. Виктория лак синий R (ICI) (В.С. I. 44040 205]), меняющий желто-зеленую окраску на синюю, получил вы сокую оценку; темно-синий (Chem) (В. С. I. 44085 [205]), переходящий из желто-зеленого в синий, и метиловый зеленый (Chem) (B.C.I. 42585 [205]) с изменением окраски из бирюзовой на голубую— все эти индикаторы, так же как и крахмал, одинаково чувствительны к иоду. В спиртовых растворах или при высоких концентрациях солей действие крахмала несовершенно [464], а малахитовый зеленый (В. С. I. 42000 [205]) дает в этих же условиях
23—918
354
ГЛАВА VIII
хорошую конечную точку. Брэзайер и Стефен нашли, что показание трех красителей улучшается ib 50%-ном спирте, и считают, что оно лучше, чем в случае малахитового зеленого. В 0,5 М серной кислоте наилучшее изменение окраски дает этиловый фиолетовый (Chem) (B.C.I. 42600 [205]) от сине-зеленой до желто-зеленой, а также'виктория лаковый синий R (ICI) (В.С. I. 44040 [205]) от оливково-желтой до бирюзовой. Об обратимости ничего не сказано.
В работах по использованию метода титрования хлорамином Т Бишоп с сотр. критически оценил обратимые и необратимые индикаторы. Он установил [438], что прямое титрование растворов индикатора в различных средах дает такие значения индикаторной ошибки, которые соответствуют ошибке при сопоставлении потенциометрического и визуального методов титрования, но посчитал, что эти значения имеют только лишь качественный смысл. В практике индукция имеет тенденцию уменьшать эту ошибку, и, если главная реакция титрования идет медленно, происходит заметное ухудшение окраски индикатора до наступления конечной точки, а легче разрушающиеся индикаторы дают преждевременную козеч-ную точку. Указанные авторы сообщают, что для каждого особого определения и экспериментальных условий следовало бы отдельно определять индикаторную ошибку и сравнивать с потенциометрическим титрованием, проведенным в таких условиях, которые, как известно, дают точные результаты. Хотя это замечание учитывается в данном обсуждении, в табл. VIII.47 приводятся ошибки пяти обратимых и пяти необратимых индикаторов в методах титрования мышьяка (III) ib различных средах раствором 0,05 М хлорамина Т; для обратимых индикаторов приводятся вторые конечные точки, получаемые после добавления еще одной аликвоты мышьяка (III). Следует помнить об особенностях титранта: например, в отсутствие бромида главная реакция становится слишком медленной и ее нельзя использовать в соляной кислоте слабее 0,5 моль-л-1, так что большую положительную ошибку в этой среде нельзя отнести за счет индикатора.
В соляной кислоте, не содержащей бромиды, нельзя использовать реагенты, специфические для свободного брома, а хинолиновый желтый быстро разрушается. Тартразин становится необратимым после первого изменения окраски, а и-этоксихризоидин не дает второй конечной точки. о-Дианизидин обладает хорошей обратимостью и дает прекрасную вторую конечную точку при более высоких концентрациях соляной кислоты, поэтому его можно рекомендовать для применения в этих средах. Метиловый оранжевый, метиловый красный и тартразин необратимы и дают значительные индикаторные ошибки, но в 1,0 М и 2,0 М соляной кислоте ошибка незначительна в случае бордо, амаранта и п-этоксихризоидина, а также обратимого о-дианизидина.
о? ' Таблица VIII.47 ИНДИКАТОРНЫЕ ОШИБКИ, ПОЛУЧЕННЫЕ ПРИ ТИТРОВАНИИ МЫШЬЯКАЦП) в различных средах и отнесенные К 100 МЛ ТИТРУЕМОГО РАСТВОРА В ТОЧКЕ ЭКВИВАЛЕНТНОСТИ ПРИ ТИТРОВАНИИ ХЛОРАМИНОМ-Т, РАСХОД КОТОРОГО
СОСТАВЛЯЕТ 25 МЛ ПРИ КОНЦЕНТРАЦИИ 0.5 МОЛЬ-Л-,а
Индикатор Ошибка, мл титранта
Концентрация НС1 Концентрация НС1 (плюс 0,1 М КВг) 1,0 М H2SO4 + 0.1 М КВг Ацетатный буфер (pH 4,1) + 0,1 М КВг
0,5 моль-л—1 1,0 моль л—1 2,0 моль л—1 0,5 моль-л—1 1,0 МОЛЬ-л-1 2,0 моль л—1
Розанилин — —- — 0,00 0,00 0,00 0,00 —
Метиловый оранжевый +0,06 +0,02 +0,02 -[-0.02 +0,04 +0,06 +0,04 +0,06
Метиловый красный +0,05 +0,02 +0,02 4 0,02 +0,04 +0,06 +0,05 +0,04
Бордо +0,03 0,00 0,00 +0,02 +0,02 +0,04 0,00 +0,06
Амарант +0,03 0,00 0,00 0,00 0,00 +0,02 0,00 +0,04
п-Этоксихризоидин +0,05 0,00 0,00 +0,05 +0,03 +0,02 +0,01 —
(2-я конечная точка) — 0,00 0,00 +0,02 +0,01 0,00 +0,02 —
Хинолиновый желтый — — — +0,06 +0,04 +0,06 +0,03 +0,06
(2-я конечная точка) — — — +0,02 +0,01 — +0,06 +0,05
о-Дианизидин +0,04 0,00 0,00 +0,02 +0,03 +0,03 0,00 —
(2-я конечная точка) — 0,00 0,00 0,00 +0,03 +0,02 — —
Тартразин +0,06 +0,02 +0,02 — — — — +0,06
(2-я конечная точка) — — — — — — т- +0,05
а Нафтофлавон — — — 0,00 0,00 +0,01 0,00 —
(2-я конечная точка) — —• — —0,01 —0,01 —0,01 +0,03 —
а Две капли 0,1%-ного водного раствора индикатора, кроме а-нафтофлавона (1 мл которого берут в виде 0,01%-ного этанольного раствора).
со сл сл дианизндина (который берут в виде 0.2%-него раствора в 2 М уксусной кислоте) и хинолинового желтого (который берут в виде водного раствора) 0,5%-ного
356
ГЛАВА VIII
В соляной кислоте, содержащей бромид, тартразин не окисляется и не определяет конечную точку. Реагенты, специфические для свободного брома, дают превосходные результаты, свободные от ошибки; а-нафтофлавон — обратимый индикатор, но его можно использовать только в бесцветных средах, розанилин дает прекрасное изменение окраски. Другие индикаторы в присутствии бромида показывают растущую положительную ошибку, которая становится больше с повышением концентрации кислоты; исключение составляет только и-этоксихризоидин. Кроме розанилина и а-нафто-флавона, прекрасные и точные конечные точки получаются с амарантом и бордо. Несмотря на то что п-этоксихризоидин, хинолиновый желтый (в 1,0 М соляной кислоте, так как в 2,0 М кислоте он теряет обратимость) и о-дианизидин дают большую индикаторную ошибку и изменение окраски плохое (особенно у хинолинового желтого), эти индикаторы имеют преимущество перед остальными благодаря своей обратимости.
В среде серной (или какой-либо другой) кислоты, не содержащей галогениды, хлорамин Т не является практически удобным окислителем, но в присутствии бромида и при концентрациях серной кислоты 0,25—2,0 моль-л-1 розанилин показывает четкие безошибочные конечные точки (оптимальная концентрация серной кислоты 1,0—1,5 моль-л-1). Бордо и амарант тоже дают прекрасные конечные точки, свободные от ошибок. Метиловый оранжевый и метиловый красный, хотя и отличаются четкой конечной точкой, показывают высокую положительную ошибку. Из двух обратимых индикаторов — хинолинового желтого и и-этоксихризоидина— лучшие результаты получаются с первым из них; о-дианизидин и а-нафтофлавон не проявляют хорошей обратимости.
При pH 4 только в присутствии бромида можно проводить титрование с аналитической целью [37]; в этой среде розанилин, и-этоксихризоидин, о-дианизидин и а-нафтофлавон изменяют окраску задолго до точки эквивалентности. Метиловый оранжевый, метиловый красный, бордо и амарант постепенно блекнут, а поэтому определение конечной точки нечеткое и запаздывающее Хинолиновый желтый и тартразин дают обратимую, но запаздывающую конечную точку.
Исследования Бишопа с corp. [37, 437—'441, 465, 466] показали, что нельзя переносить результаты определения мышьяка (III), как показано выше, на определение сурьмы(III), или результаты реакций с броматом на реакцию с хлорамином-Т в среде бромида, или результаты, полученные с гипохлоритом на реакцию с хлорамином Т, и, наконец, результаты реакций с йодатом на хлорамин Т в среде, содержащей монохлорид иода. Титрование сурьмы [439] дает точные результаты в 1—3 М соляной кислоте по амаранту с добавкой винной кислоты до 6% (вес/об); большее количество вносит положительную ошибку в определение. Амарант и бордо осо
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
357
бенно хороши как индикаторы ib 2 М соляной кислоте, и результаты получаются без ошибки, тогда как тартразин, и-этоксихризо-идин и метиловый оранжевый, несмотря на свою пригодность, дают высокую положительную ошибку. Хинолиновый желтый, метиловый красный и о-дианизидин преждевременно изменяют свою окраску; тартразин и и-этоксихризоидин необратимы. Добавка бромида в раствор соляной кислоты не дает никаких преимуществ: розанилин можно использовать, и хотя ошибка меньше 0,1%, изме-ение окраски получается нечетким. Серная кислота, в присутствии или в отсутствие добавленного галогенида, совсем непригодна в качестве среды для проведения титрования. Лучшим методом титриметрического определения сурьмы хлорамином Т является буферный раствор с pH 6,5—7,5, содержащий 5-10-4—5-10~3 М иодида и около 1 г винной кислоты; в качестве индикатора используют крахмал. Гидразин нельзя точно определить ни при каких условиях и ни в какой среде, если титровать хлорамином Т [440, 466], несмотря на то что ошибки получаются меньше 0,4% в 10-4 — 2,0 М соляной кислоте, содержащей бромид, с амарантом в качестве индикатора, а в 2,0—4,0 М соляной кислоте с розанилином, в 0,025 М иодиде при оН 7,0 + 0,1 ошибка определения по крахмалу составляет 0,2%. Определение таллия (I) и роданида [441] одинаково неудовлетворительно, за исключением результатов, полученных по методу Эндрьюса [466]. С помощью этого метода, используя монохлорид иода в качестве интермедиата реакции, можно точно определять мышьяк (III), роданид и таллий (I) по четыреххлористому углероду в качестве индикатора, титруя хлорамином Т. Гидразин дает по этому методу ошибку до 0,2%, а в случае присутствия винной кислоты нельзя использовать четыреххлористый углерод в качестве индикатора; винную кислоту приходится добавлять, чтобы устранить мешающее действие сурьмы при использовании этого индикатора [466]. Сурьму определяют по амаранту, который дает в этом случае прекрасные результаты. В других методах определения ни амарант, ни н-этоксихризоидин не имеют преимущества перед ’.етыреххлорлстым углеродом.
Исследования Белчера производных сульфофталеина [239], используемых в качестве индикаторов в методах титрования гипохлоритом, охватывают несколько соединений, которые разрушаются при окислении. Сюда относятся феноловый красный, хлорфеноловый красный, бромфеноловый красный, тетрабромфеноловый синий, катехиновый фиолетовый, крезоловый пурпуровый, бромкрезоловый зеленый, пирогаллоловый красный и бромпирогаллоловый красный; некоторые из них совсем слабо изменяют свою окраску. Эти соединения были отвергнуты как индикаторы в пользу бромтимолового синего и бромкрезолового пурпурового, которые обладают обратимостью; они рассматриваются подробнее в следующем разделе.
358
ГЛАВА VIII
6. РАЗЛИЧНЫЕ СОЕДИНЕНИЯ
Имеется несколько небольших групп соединений в .несколько отдельных соединений, которые можно использовать или которые уже используются как окислительно-чосста1новителыные индикаторы, но их нельзя включать ни в одну из рассмотренных групп индикаторов. Многие соединения были исследованы и отклонены по разным причинам как неудовлетворительные, кроме большого числа красителей и производных бензидина, перечисленных ранее. Страка и Еспер [139], например, исследовали и нашли непригодными еще больше соединений, чем указано в разделе о производных бензидина. Сюда надо отнести ^Д'ДД'-тетрабром-, 2,4-дихлор-, 2,2',4,4'-тетрахлор-, гД'ДД'Дб'-гексахлор, 2-нитро-, 2,4-динитро-, 2,4-динитро-4/-амино-, 2,4-динитро-4'-метокси-, 2,4,6-тринитро- и 2,4,6,3/-тетранитропроизводные дифениламина, которые или не окислялись или образовывали неокрашенные продукты окисления. Эти авторы отклонили также метил- и этилдифениламинсульфонат и М.М'-диметил-М.М'-дифенилбензидин, как. образующие несколько различных окрасок или мало изменяющих свою окраску, и высказались против применения о-тслилфениламина и фенилгидразонафталина. Те же авторы [139] установили, что хромотроповая кислота, 1-нафтиламин, р,р'-динафтиламин, At-толил-1-нафтиламин, о-, м- и п-толил-2-нафтиламины, диаминодифенетол, о- и At-толидины и тетрабром-А1-толидин дают бесцветные продукты окисления.
Ароматические полиамины, как видно на примере п-фенилен-диамина и его N-алкил- и N-фенилпроизводных, несомненно, дают окрашенные продукты окисления; эти реакции сложны и, вероятно, слишком упрощенно рассматривались в более ранних работах. Вильштетер с сотр. [467—469] изучал окисление и-фенилендиамина и упоминал о продуктах окисления гидрохинона в виде мерихино-идных и голохиноидных аналогов. Кларк, Коен и Гиббс [470] показали сложность реа кций в проводимых ими потенциометрических и спектрофотометрических исследованиях, а Михаэлис [471] доказал образование семихинонов или свободных радикалов N.N'-Ди-метил- и М,М,Ь1',М'-тетраметил-и-фенилендиамины окисляются соответственно в красный Вурстера и синий Вурстера [472], которые можно окислить дальше. Были измерены потенциалы этих соединений [473], но сами соединения оказались непригодными для использования их в качестве индикаторов. Михаэлис и Хилл [473] исследовали Ы.Ы'-дифенил-п-фенилендиамин [474] и нашли, что окисление его идет в две стадии в присутствии уксусной кислоты при формальных потенциалах на каждой стадии 0,592 и 0,795 В соответственно.
В качестве индикаторов были предложены и другие органические системы. Для титрования железа(II) бихроматом [475] был рекомендован бруцин [475] или пирогаллол [476]. Томичек [477]
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
359
использовал хинализарин для окислительного титрования гидрохинона в среде ледяной уксусной кислоты раствором ацетата свинца (IV); механизм реакции не обсуждался. Себелед с сотр. [479] предложил апоморфин для титрования сурьмы(III) броматом; свободный бром вызывает появление розовой окраски, индикатор обратимый. Однако Белчер [228] считает, что изменение окраски слабое и индикатор слишком легко окисляется, чтобы его можно было использовать.
Порфирексид и порфириндин
Системы порфирексид — порфирексчн и порфириндин — лейко-порфириндин могут представлять интерес как индикаторы, но до сих пор еще они не были исследованы в этом отношении. Формальные потенциалы их высокие и не зависят от значения pH. Кун с сотр. [478] замерил эти потенциалы и предложил следующий механизм реакций окисления этих соединений:
“а /ОН Н3С—С—N
'\=NH
HN=C—N
порфирексин
Indre(i красный
СНз /О
Н3С—С—N
'4C=NH + Н+ + е HN=C—bK
\н
порфирексид
Indox бесцветный
СНз /ОН НО/ СИ» Н3С—С—N N—С—СН3
ХчС=Н—N=C//
лейкопорфириндин Indred синий
сн3 0 О/ СН3
Н3С— C-N N—С—СН3
'\=N— N=C// + 2Н+ + 2e
HN=C-N// C=NH
\н н/
порфириндин
Indox бесцветный
С60
ГЛАВА VIII
Для системы порфирексид—порфирексин формальный потенциал равен 1,34 В при pH 0 и 0,97 В при pH 7, а для порфириндин—лей-копорфириндин формальный потенциал составляет 1,2 В при pH 0 и 0,81 В при pH 7.
Неорганические системы
Себелед с сотр. первым обнаружил существование обратимых индикаторов для броматометрии и на их основе разработал группу неорганических индикаторов [479—485]. Первым был селен [479]; селенит, внесенный в кислый раствор 1мышьяка(Ш), восстанавливается до красного коллоидного селена, который исчезает при окислении броматом; индикаторной ошибки не получается. Аналогично [480, 481] осмиевая кислота восстанавливается до темно-синего коллоидного осмия раствором мышьяка (III) в среде серной кислоты, содержащей бромид, и снова окисляется в конечной точке броматом. Система бесцветного дибромаурата(1) и желтого тетра-бромаурата(Ш) обратима [482, 483], и ее можно использовать в титровании мышьяка(III) раствором церия(1У) в отсутствие катализатора или же раствором бромата. Гидразин восстанавливает молибденфосфорную кислоту до 'молибденовой сини, которая снова окисляется броматом. Предполагается, что подобным образом можно определять и другие восстановители, которые взаимодействуют с молибденфосфорной кислотой.
Сульфофталеины
С'ерана [486] заметил, что бромтимоловый синий является обратимым индикатором в методах титрования гипохлоритом. При окислении этот индикатор изменяется от темно-синего до зеленовато-желтого, и это можно наблюдать визуально при титровании иодида [487], которое до сих пор должны были проводить потенциометрически. Такие наблюдения [488] заставили Белчера с сотр. [239] исследовать другие сульфофталеины в отношении индикаторных свойств. Большинство исследованных соединений подвергалось деструкции или давало совсем слабое изменение окраски, о них шла речь в разд. 5 этой главы. Однако было установлено, что, помимо бромтимолового синего Сераны, бромкрезоловый пурпуровый, крезоловый красный, ксиленоловый оранжевый, тимоловый синий и метилтимоловый синий обладают ожидаемыми индикаторными свойствами. Из этих соединений тимоловый синий и метилтимоловый синий изменяют свою окраску на более темную синюю перед самой конечной точкой и после обратной реакции дают более глубокую синюю окраску; крезоловый красный и ксиленоловый оранжевый становятся пурпурными непосредственно перед конечной точкой и после обратной реакции снова дают такую же пурпурную ок
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
361
раску. Установлено, что необходим избыток бромида, чтобы индикатор не разрушился необратимо. По-видимому, происходит бромирование, и на основании спектрофотометрических данных можно сказать, что тимоловый синий и метилтимоловый синий превращаются в бромтимоловый синий, а крезоловый красный и ксиленоловый оранжевый — в бромкрезоловый пурпуровый. Белчер с сотр. считает [239], что почти нет смысла применять эти соединения, разве только бромтимоловый синий и бромкрезоловый пурпуровый, причем второй иногда оказывается лучшим индикатором по сравнению с первым.
Все эти индикаторы проверялись при работе с другими титрантами. В случае бромата оказались подходящими только тимоловый синий, метилтимоловый синий и катехиновый фиолетовый; изменение окраски происходило от красной до желтой. Лишь метилтимо-повый синий был с самого начала обратимым; в случае других требовалось нагревание. Конечные точки были удовлетворительно четкими только в том случае, когда в титруемом растворе не было вначале брома; тогда катехиновый фиолетовый становится необратимым. Растворы метилтимолового синего следует выдержать некоторое время, пока первоначальная оранжевая окраска не станет красной. Эти же три индикатора оказались единственными подходящими для титрования раствором хлорамина-Т. В отсутствие бромида только метилтимоловый синий обратим, а >в присутствии бромида катехиновый фиолетовый обратим при нагревании; тимоловый синий необратим во всех условиях. Для титрования йодатом калия подходящими индикаторами являются только ме-тилтимоловый синий и тимоловый синий. Тимоловый синий необратим, а метилтимоловый синий может быть обратимым только один раз.
Флуоресцентные индикаторы
Бишоп [230], исследуя красители, обнаружил несколько флуоресцентных окислительно-восстановительных индикаторов и провел критическое изучение лучших из них. Однако он не опубликовал результаты из-за того, что во время войны появились японские работы [489, 490]. Во многих случаях флуоресценция затухает при подкислении, так что эти соединения нельзя применять в кислых средах. Родамин 6G (Ву) очень слабо изменяет окраску, тогда как родамин 6G (А) дает хорошее изменение, оба они необратимы в методе титрования железа (II) церием(IV), а при титровании бихроматом или броматом нельзя установить конечную точку с помощью родамина. Однако родамин В (А), несмотря на то, что при подкислении флуоресценция спабее, дает неплохую и обратимую конечную точку при титровании железа (II) церием (IV); совсем непригоден при титровании бихроматом или броматом. Лучшим инди
362
ГЛАВА VIII
катором считается {230] родулиновый розовый 5G (By), который дает хорошую и обратимую конечную точку в методах титрования церием (IV), индикаторная ошибка при этом небольшая. Этот индикатор можно использовать при обычном дневном сзете, но флуоресценция значительно усиливается при УФ-облучении, в темной комнате. Если титровать бихроматом, флуоресценция постепенно ослабевает и не получается четкой конечной точки. Такие индикаторы имеют преимущество в окрашенных растворах.
Хемилюминесцентные индикаторы
Впервые Кенни и Курц [491] предложили использовать сило-ксен в качестве хемилюминесцентного индикатора при методах окислительного титрования. Хемилюминесцентные индикаторы рассматриваются в специальной главе.
7. СМЕШАННЫЕ ИНДИКАТОРЫ
Смешанные индикаторы в обычном смысле слова почти не применяются в окислительно-восстановительном титровании. В титровании олова (II) раствором ванадия (V) была предложена [492] смесь метиленового голубого и 3,3'-дифенилбензидина. В конечной точке окраска изменялась дважды от темно-синей через бесцветную до фиолетово-синей. Обычно для успешного титрования сильного или слабого восстановителя можно использовать индикатор с низким потенциалом и бесцветной окисленной формой в смеси с индикатором, имеющим высокий потенциал и бесцветную восста новлеяную форму. Например, после восстановления железа (III) избытком олова(II) сначала можно оттитровать олово(II) в присутствии индикатора какотелина (изменяет фиолетовую окраску на бесцветную), а затем титруют железо(II) по подходящему индикатору с высоким потенциалом из серии бензидиновых индикаторов; разница между обеими конечными точками даст объем титранта, эквивалентный количеству железа (II). В качестве окислителей здесь подходят растворы церия(IV) и бихпомата.
СПИСОК ЛИТЕРАТУРЫ
1. Оттоуэй Дж. М., Эта книга, гл. VIIIA.
2. Brandt L., Z. anal. Chem., 45, 95 (1906).
3. Barnebey О. L., Wilson S. R., J. Am. Chem. Soc., 35, 156 (1913).
Brandt L., Z. anal. Chem., 53, 1 (1914).
5. Brandt L., Z. anal. Chem., 53, 729 (1914).
6. Etheridge A. T., Analyst, 48, 588 (1923).
7. Koch B., Chem. Ztg., 49, 479 (1925).
8. Brandt L., Stahl u. Eisen, 46, 976 (1926).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
363
9 Thompson Р. F., Alabaster Е. С., Proc. Soc. Chem. Ind. Victoria, 33, 810 ’ (1933).
10. Crossley H. E., Analyst, 61, 164 (1936).
11. Kielbasinski V, Z. Farben u. Textilchem., 2, 114 (1903).
12. Monneir A., Ann. chim. anal, applic., 21, 109 (1916).
13. Russo C., Gazz. chim. itaL, 44, 1, (1914).
14 Russo C., Sensi G., Gazz. chim. Ital., 44. 9 (1914).
15’ Sinatt F. S., Analyst, 35, 309 (1910).
16. Sinatt F. S., Analyst, 37, 252 (1912).
17. Knop J., J. Am. Chem. Soc., 46, 263 (1924).
18. Kehrmann F., Mieciwicz, Ber., 45, 2641 (1912).
19. Kehrmann F., Mieciwicz, Helv. Chim. Acta, 4, 949 (1921).
20. Kehrmann F., Roy G., Ber., 55, 156 (1922).
21. Marqueyrol M., Muraour H., Bull. soc. chim., France, 15, 186 (1914).
22. Wieland H., Ber., 46, 3296 (1913).
23. Brennecke E., in «Neuer Methoden der MaBanalyse», ch. 6, Bottger W. (ed.), Enke, Stuttgart, 1938.
24. Whitehead T. H., Wills С. C., Chem. Rev., 26, 69 (1941).
25. Кольтгоф И. M., Стенгер В. А., Объемный анализ, т. 1. М.-Л., Госхимиздат, 1950.
26. Кольтгоф И. М., Белчер Р., Стенгер В. А., Матсуяма Дж., Объемный анализ, т. 3, М., Госхимиздат, 1961.
27. Tomicek О., Chemical Indicators, Butterworths, London, 1951.
28. Wilson C. L., Wilson D. W. (eds-), Comprehensive Analytical Chemistry, Vol. Ib, Elsevier, Amsterdam, 1960.
29. Kolthoff I. M., Elving P. J., Treatise on Analytical Chemistry.
30. Berka A., Vulterin J., Zyka J., Newer Redox Titrants, Pergamon. London, 1965.
31. Turney T. A., Oxidation Mechanisms, Butterworths, London, 1965.
32. Mark H. B., Rechnitz G. A., Kinetics in Analytical Chemistry, Interscience, New York, 1968, chapter 8.
33. Habig R. L., Pardue H. L., Worthington J. B., Anal. Chem., 39, 600 (1967).
34. Levich V. G., Advanced in Electrochemical Engineering, Delahay P. (ed.), Vol. 4, Interscience, New York, 1966, p. 249.
35. Rosseinsky D. R., Nicol M. J., Trans. Farad. Soc., 61, 2718, (1965).
36. Ottaway J. M., Bishop E., Anal. Chim. Acta, 39, 153 (1965).
37. Bishop E., Jennings V. J., Taianta, 8, 22 (1961).
38. Nernst W., Z. physik. Chem., 4, 129 (1889).
39. Rosotti H. S., Chemical Applications of Potentiometry, Van Nostrand, London, 1969.
40. Glasstone S., Laidler K. J-, Eyting H., «The Theory of Rate Processes», McGraw-Hill, New York, 1941, ch. X.
41. Van Rysselberg P., Some Aspects of the Termodynamic Structure of Electrochemistry, in «Modern Aspects of Electrochemistry», Bockris J.O’M. (ed.), Vol. 4, Butterworths, London, 1966.
42. Hitchcock P. H., Dissertation, University of Exeter, 1969.
43. Tanaka N., Tamamushi R., Electrochim. Acta, 1964, 9, 963.
44. Ringbom A., Complexation in Analytical Chemistry, Interscience, New York, 1963.
45. Ingri N., KakoXowicz W., Sillen L. G., Warnqvist B., Taianta, 14, 1261 (1967)
46. Dyrssen D., Jagner D, Wengelin F., Computer Calculation of Ionic Equilibria and Titration Procedures, Almqvist & Wiksell, Stockholm, Wiley, London, 1969.
47. Bishop E., Anal. Chim. Acta, 27, 253 (1962).
48. Делахей П., Двойной слой и кинетика электродных процессов, М., «Мир»,
364
ГЛАВА VIII
49. Bishop Е., Coulometric Analysis, in «Comprehensive Analytical Chemistry Wilson C. L., Wilson D. W. (eds.), Vol. I ID, Elsevier, Amsterdam, 1960.
50. Bishop E., Anal. Chim. Acta, 26, 397 (1962)
51. Bishop E., Algol programme COMBOXTIT, English Electric 4-50 programme EXCHO511A, University of Exeter.
52. Goldman J. A., J. Electroanal. Chem., 11, 416 (1966).
53. Bishop E., Theory and Principles of Titrimetric Analysis in «Comprehensive Analytical Chemistry», Wilson C. L., Wilson D. W. (ed.), Vol. Ib. Elsevier, Amsterdam, 1960.
54. Bishop E., in «Proceedings of the SAC Conference», Shallis P. W. (ed.), Nottingham 1965, Heffer, Cambridge, p. 291.
55. Bishop E., Crawford A. B., Analyst, 75, 273 (1950).
56. Kessler F„ Pogg, Ann., 195, 218 (1863).
57. Engler C., Ber., 33, 1097 (1900).
58. Luther R., Schilov N., Z. physik. Chem., 46, 777 (1903).
59. Skrabal A., Die induzierten Reaktionen, Enke, Stuttgart, 1908.
60. Oberhauser F., Hensinger W., Ber., 61, 521 (1928).
61. Oberhauser F., Schornmuller J., Ann., 470, 111 (1929).
62. Lang R., Z. anal. Chem., 97, 395 (1934).
63. Lang R., Fauda E., Z. anal. Chem., 108, 258 (1937).
64. Bishop E., Analyst, 86, 537 (1971).
65. Csanyi L. J., Solomosi F., Acta Chim. Hung., 4, 445 (1954).
66. Csanyi L. J., Solomosi F., Acta Chim. Hung., 13, 19 (1957).
67. Csanyi L. J., Solomosi F., Acta Chim. Hung., 13, 257 (1958).
68. Csanyi L. J., Szabo M., Taianta, 1, 359 (1958).
69. Csanyi L. J., Disc. Faraday Soc., 29, 146 (1960).
70. Csanyi L. J., Kaszai S„ Molnar J., in press.
71. Csanyi L. J., Acta Chim. Hung., 26, 29 (1961).
72. Bishop E., Acta Chim. Hung., 35, 273 (1963).
73. Bishop E., Ottaway J. M., Short G. D., Anal. Chim. Acta, 27, 529 (1962).
74. Ottaway J. M., Bishop E., in «Proceedings of the SAC Conference», Shallis P. W. (ed.), Nottingham 1965, Heffer, Cambridge, p. 316.
75. Rao G. G., Ramanjaneyulu J. V. S., Current Sci. (India), 18, 72 (1949).
76. Walden G. H., Hammett L. P„ Chapman R. P., J. Am. Chem. Soc., 53, 3908 (1931).
77. Rees D. L, Stephen W. L, Taianta, 2, 361 (1959).
78. Marcus R. A., Ann. Rev. Phys. Chem., 15, 155 (1964).
79. Marcus R. A., J. Phys. Chem., 67, 853 (1963).
80. Bishop E., Observations on the «Pattern» Region in Voltammetry, University of Exeter, 1968.
81. Smith G. F„ Banick W. M., Taianta, 2, 348 (1959).
82. Pope M. T., Dale B. W„ Quarter. Revs., 22, 527 (1968).
83. Hitchcock P. H., Ph. D. Thesis, University of Exeter, 1968.
84. Hitchcock P. H., Bishop E., Paper presented at the 2nd SAC Conference, Nottingham, 1968.
85. Bishop E., Dissertation, University of Glasgow, 1942.
86. Kolthoff I. M., Sarver L. A., J. Am. Chem. Soc., 52, 4179 (1930).
87. Michaelis L., Naturwiss., 19, 461 (1931).
88. Michaelis L., Oxydation-Reduktion Potentiate, Springer, Berlin, 1933.
89. Pfeiffer P., Organische Molekfllverbindungen, Stuttgart, 1927.
90. Weitz E., Z. Elektrochem., 34, 538 (1928).
91. Madelung W., Reiss E„ Herr E., Ann., 454, 7 (1927).
92. Weitz E., Schwechten H. W„ Ber., 59, 2307 (1926).
93. Weitz E„ Schwechten H. W., Ber., 60, 545 (1927).
94. Taylor T. W. J., Baker W., «Sidgwick’s Organic Chemistry of Nitrogen», 2nd ed , Oxford University Press, 1936.
95. Wurster C. et al., Ber., 12, 1803, 1807, 2071 (1879); 19, 3195, 3217 (1883).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
365
96. Hausser К. Н., Naturwiss., 43, 15 (1956).
97. Millar I. Т., Springall Н. D., «The Organic Chemistry of Nitrogen by N. V. Sidgwick», 3rd ed., Oxford University Press, 1966.
98. Goldschmidt S., Ber., 53, 28 (1920).
99. Goldschmidt S„ Wurzschmitt B., Ber., 55, 3216 (1922).
100. Goldschmidt S Wurzschmitt B., Ber., 55, 3216 (1922).
101. Kehrmann F., Micewicz, Ber., 45, 2641 (1912).
102. Kehrmann F., Roy G., Ber., 55, 156 (1922).
103. Wieland H„ Ber., 46, 3296 (1913).
104. Wieland H„ Ber., 52, 886 (1919).
105. Walton J. H., Filson G. W., J. Am. Chem. Soc., 54, 3228 (1932).
106. Blackadder D. A., Hinshelwood C., J. Chem. Soc., 1957, 2898.
107. Blackadder D. A., Hinshelwood C., J. Chem. Soc., 1957, 2904.
108. Curme G. O., J. Am. Chem. Soc., 35, 1143 (1913).
109. Wieland H„ Ber., 48, 1098 (1915).
110. Holt P. F., Hughes B. P„ J. Chem. Soc., 1953, 1666.
111. Vecera M. et al., Chem. Listy, 51, 911, 1690 (1957).
112. Jacobsen P. et al., Ann., 428, 76 (1922).
113. Wieland H., Gambarjan S„ Ber., 39, 1499 (1906).
114. Smith D. H., Schwartz J. R., Wheland G. W., J. Am. Chem. Soc., 74, 2282 (1952).
115. Ingold С. K. et al., J. Chem. Soc., 1962, 2386.
116. Ingold С. K. et al., J. Chem. Soc., 1964, 2864.
117. Clemo G. R., Lee T. B., J. Chem. Soc., 1954, 2417.
118. Wittig G., loos W., Rathfelder P., Ann., 610, 180 (1957).
119. Goldschmidt S. et al., Ber., 53, 44 (1920); 55, 616, 628 (1922); Ann., 437, 194 (1924); 473, 137 (1929).
120. Chattaway F. D., Ingle H., J. Chem. Soc., 67, 1090 (1895).
121. Wieland H„ Ann., 381, 212 (1911).
122. Wieland H., Lecher H., Ann., 392, 156 (1912).
123. Wieland H„ Ber., 48, 1091 (1915).
124. Ingold C. K, Trans. Faraday Soc., 30, 55 (1934).
125. Weitz E.. Schwechten H. W., Ber., 60, 1203 (1927).
126. Schofield K., personal communication.
127. Crawford A. B., Bishop E., J. Roy. Tech. Coll., 5, 52 (1950).
128. Belcher R., Lyle S. J., Stephen W. L, J. Chem. Soc., 1958, 4454.
129. Oldfield L. F., Bockris J. O’M., J. Phys. Colloid Chem., 55, 1255 (1951).
130. Frumina N. S., Mustafin I. S„ Nikurashina M. L., Vechera M. K., Taianta, 16,
138 (1969).
131. Belcher R., Rees D. L, Stephen W. L, Chimie analytique, 10, 397 (1959).
132. Lyle S. J., Taianta, 2, 293 (1959).
133. Stockdale D., Analyst, 75, 150 (1950).
134. Bishop E., Hartshorn L„ Analyst, 96, 26 (1971).
135. Sarver L. A., von Fischer W., Ind. Eng. Chem. Anal. Ed., 7, 271 (1935).
136. Сырокомский В. С., Степин В. В., Завод, лаб., 5, 144 (1936).
137. Lederer М„ Ward F. L., Anal. Chim. Acta, 6, 1 (1952).
138. Cohen S„ Oesper R. E., Ind. Eng. Chem. Anal. Ed., 8, 364 (1936).
139. Straka L. E., Oesper R. E., Ind. Eng. Chem. Anal. Ed., 6, 465 (1934).
140. Hammett L. P., Walden G. H., Edmonds S. M., J. Am. Chem. Soc., 56, 1092 (1934).
141. Walden G. H., Edmonds S. M„ Chem. Rev., 16, 81 (1935).
142. Sarver L. A., Kolthoff I. M., J. Am. Chem. Soc., 53, 2902 (1931).
143. Bovee H. H., Robinson R. J., Anal. Chem , 33, 1115 (1961).
144. Belcher R., Nutten A. J., Stephen W. L, J. Chem. Soc., 1952, 3857.
145. Kirsanov A. V., Tscherkassow V. P., Bull. soc. chim., France 3, 2037 (1936).
146. Knop J., Kubelkova-Knopova 0., Z anal. Chem., 122, 183 (1941).
147. Thiele J., Z. Elektrochem., 35, 274 (1929).
366
ГЛАВА VIII
148. Smith G. F„ Brandt I. W., Anal. Chem., 21, 948 (1949).
149. Hees D. I., Stephen W. I., Lalanta, 2, 361 (1959).
150. Belcher R., Chakrabarti C. L., Stephen W. I., Analyst, 94, 20 (1969).
151. Bishop E., Crawford A. B., Analyst, 75, 273 (1950).
152. Bishop E., Crawford A. B., Analyst, 74, 365 (1949).
153. Belcher R., NuttenA. J., Stephen IF. I., J. Chem. Soc., 1951, 1520.
154. Willard H. H., Young P., Ind. Eng. Chem. Anal. Ed., 5, 154 (1933).
155. Willard H. H., Manalo G. D., Ind. Eng. Chem. Anal. Ed., 19, 167 (1947).
156. Belcher R., Nutten A. J., Stephen W. I., J. Chem. Soc., 1954, 2543.
157. Adams R. N., Hammaker E. M., Anal. Chem., 23, 744 (1951).
158. Кульберг JI. M., Борзова Л. Д., Завод, лаб., 21, 920 (1955).
159. Jordanov N., Daiev Ch., Taianta, 10, 163 (1963).
160. Clark W. M., Cohen B., Gibbs H. D., U.S. Pub. Health Repts., Supplement No. 54, 1926
161. Belcher R., Nutten A. J., J. Chem. Soc., 1951, 544.
162. Belcher R., Nutten A. J. Chem. Soc., 1951, 546.
163. Belcher R., Nutten A. J, J. Chem. Soc., 1951, 547.
164. Belcher R., Nutten A. J., J. Chem. Soc., 1951, 548.
165. Belcher R., Nutten A. J., J. Chem. Soc., 1951, 550.
166. Belcher R., Nutten A. J., J. Chem. Soc., 1951, 1516.
167. Belcher R., Nutten A. J., Stephen W. I., J. Chem. Soc., 1958, 2336.
168. Knop J., Z. anal. Chem., 85, 253 (1931).
169. Clabaugh W. S., J. Res. N.B.S., 36, 119 (1946).
170. Wawrzyczek W., Wisniewska K„ Chemia Analit, 10, 1287 (1965).
171. Cheng K. L., Taianta, 8, 658 (1961).
172. Bilbo A. J., Wyman G. M., J. Am. Chem. Soc., 75, 5312 (1953).
173. Eggertson F. T.. Weiss F. T., Anal. Chem., 28, 1008 (1956).
174. Schlenk A., Ann., 363, 313 (1908).
175. Moir J., S. African J. Sci., 1913, 163.
176. Leitch J. L., пат. США 2436814, 1948.
177. Weeks M. E„ Ind. Eng. Chem. Anal. Ed., 4, 127 (1932).
178. Pollard W. B., Analyst, 62, 597 (1937).
179. Jamieson A. R., Watson R. S., Analyst, 63, 702 (1938).
180. Frost H. F., Analyst, 68, 51 (1943).
181. Cohen S., Oesper R. A., Ind. Eng. Chem. Anal. Ed., 8, 306 (1936).
182. Belcher R., Nutten A. J., Stephen W. I., J. Chem. Soc., 1951, 3444.
183. Belcher R., Nutten A. J., Stephen W. I., J. Chem. Soc., 1952, 1269.
184. Belcher R., Nutten A. J., Stephen W. I., J. Chem. Soc., 1952, 2438.
185. Belcher R., Lyle S. J., Stephen W. I., J. Chem. Soc., 1958, 3243.
186. Riley A., «Triarylmethane and Xanthen Dyes», in «Thorpes Dictionary of Applied Chemistry», 4th ed., Vol. XI, 1954, p. 689.
187. Watson E. R., J. Chem. Soc., 105, 759 (1914); 107, 1567 (1915).
188. Armstrong H. E., Ber., 9, 522 (1876).
189. Watson E. R., «Colour in Relation to Chemical Constitution», Longmans Green, London, 1918.
190 Pfeiffer P., Ann . 376, 292 (1910).
191. Dilthey W., Ber., 53, 261 (1920).
192. Wizinger R. K., Organische Farbstoffe, Dummler, Berlin, 1933.
193. Stieglitz J., Proc. Nat. Acad. Sci., 9, 303 (1923).
194. Chaho N. Q„ J. Chem. Phys., 2, 644 (1934).
195. Bury C. R., J. Am. Chem. Soc., 57, 2115 (1935).
196. Sklar A. L., J. Chem. Phys., 5, 669 (1937).
197. Forster T., Z physik. Chem., B41, 287 (1938).
198. Pauling L., Proc. Nat. Acad. Sci., 25, 577 (1939).
’99. Lewis G. N., Calvin M., Chem. Rev., 25, 273 (1939).
200. Maccoll A., Quarter. Rev. Chfcm. Soc., 1, 16 '’'947).
201. Hamer F. M., Quarter. Rev. Chem. Soc., 4, 327 (1950).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
367
202. Murrell J. N. «The Theory of the Electronic Spectra of Organic Molecules», Methuen, London, 1963.
203. Bradley W., «Recent Progress in the Chemistry of Dyes and Pigments, RIC Lectures, monographs and Reports», 1958, No. 5.
204. Society of Dyers and Colorists, British Colour Index, 1st ed. 1924; Supplement, 1928.
205. Society of Dyers and Colorists, British Colour Index, 2nd ed., 1956.
206. Society of Dyers and Colorists, British Colour Index, 3rd ed., scheduled for publication 1972.
207. Knop J., Z. anal. Chem., 77, 111 (1929).
208. Knop J., Kubelkova O., Z. anal. Chem., 77, 125 (1929).
209. Knop J., Z. anal. Chem., 85, 253 (1931).
210. Knop J., Kubelkova O., Z. anal. Chem., 85, 401 (1931)
211. Smith G. F., Bliss H. H., J. Am. Chem. Soc., 53, 2091 (1931).
212. Whitmoyer R. B., Ind. Eng. Chem. Anal. Ed., 6, 268 (1934).
213. Furman N. H., Wallace J. H., J. Am. Chem Soc., 52, 2347 (1930).
214. Yoe J. H., Boyd Q. R., Ind. Eng. Chem. Anal. Ed., 3, 492 (1931).
215. De Beer E. J., Hjort A. M., Ind. Eng. Chem. Anal. Ed., 7, 120 (1935).
216. Tomlinson H. M., Aepli О. T., Ebert H. M., Anal. Chem., 23, 286 (1951).
217 Rao G. G., Rao V. P., Z. anal. Chem., 168, 172 (1959).
218. Brazier J. N., Stephen W. I., Anal. Chim. Acta, 33, 625 (1965).
219. Smith G. F., Bliss H. FL, J. Am. Chem. Soc., 53, 4291 (1931).
220. Smith G. F., May R. L., Ind. Eng. Chem. Anal. Ed., 13, 460 (1941).
221. Smith G. F., Willcox C. S., Ind. Eng. Chem. Anal. Ed., 14, 49 (1942).
222. Smith G. F., May R. L„ J. Am. Ceram. Soc., 22, 31 (1939).
223. Schulek E., Rozsa P., Z. anal. Chem., 115, 185 (1939).
224. Uzel R., Coll. Czech. Chem. Comms., 7, 380 (1935).
225. Schulek E., Z. anal. Chem., 102, 111 (1935).
226. Belcher R., Anal. Chim. Acta, 5, 30 (1951).
227. Райхинштейн Ц. Г., Кочерыгина T. В., Ж анал. хим., 2, 173 (1947).
228. Belcher R., Anal. Chim. Acta, 3, 578 (1949).
229. Schulek E., Somogyi Z., Z. anal. Chem., 128, 398 (1948).
230. Bishop E., unpublished work, 1944, 1946.
231. Szebelledy L„ Madis W., Z. anal. Chem., 114, 197 (1938/9).
232. Knop J., Z. Anal. Chem., 100, 161 (1935).
233. Sotgia-Rovelli T., Boll. chim. farm., 74, 265 (1935).
234. Belcher R., Clark S. J., Anal. Chim. Acta, 4, 580 (1950).
235. Belcher R., Anal. Chim. Acta, 4, 468 (1950).
236. Sinn V., Chim. Anal., 29, 58 (1947).
237. Young J. H., Das Gupta R. N., Analyst, 74, 367 (1949).
238. Belcher R., Brazier J. N., Stephen W. I., Taianta, 12, 963 (1965).
239. Belcher R„ El-Khiami I., Stephen W. I., Taianta, 12, 775 (1965).
240. Rao G. G., Sastri T. P., Z. anal. Chem., 160, 109 (1958).
241. Rao G. G„ Sastri T. P., Z. anal. Chem., 163, 1, 263, 26. (1958).
242. Rao G. G., Sastri T. P., Z. anal. Chem., 167, 1 f 1959).
243. Rao G. G., Rao N. V., Z. anal. Chem., 190, 213 (1962).
244. Schilt A. A., «Analytical Applications of 1,10-Phenanthroline and Related Compounds», Pergamon, Oxford, 1969.
245. Eichler E„ Wall A. C., J. Am. Chem. Soc., 80, 4145 (1958).
246. Silverman J., Dodson R. W., J. Phys. Chem., 56, 846 (1952).
247. Lee T. S., Kolthoff I. M., Leussing D. L., J. Am. Chem. Soc., 70, 3596 (1948).
248. Campion R. J , Purdie N., Sutin N., Inorg. Chem., 3 1091 (1964).
249. Sutin N„, Gordon В. M., J. Am. Chem. Soc., 83, 70 (1961).
250. Fortune W. B., Mellon M. G., Ind. Eng. Chem. Anal. Ed., 10 эС (1938).
251. Harvey A. E., Manning D. L., J. Am. Chem. Soc., 74, 4744 (1952).
252. Gerdeissen, Ber., 22, 245 (1889).
253 Blau F„ Ber , 21, 1077 (1888)
368
ГЛАВА VIII
254. Blau F., Monatsh., 10, 375 (1889).
255. Blau F„ Monatsh., 19, 647 (1898).
256. Hieber W., Muhlbauer F., Ber., 61, 2149 (1928).
257. Morgan G. T„ Burstall F. H., J. Chem. Soc., 1930, 2594.
258. Manchot W., Lehmann G., Ann., 460, 191 (1928).
259. Bode B„ Wochschr. Brau., 50, 321 (1933).
260. Walden G. FL, Hammett L. P., Chapman R. P., J. Am. Chem. Soc., 53, 3908 (1931).
261. Atanasiu J. A., Bull. Soc. Romane, 30, 73 (1927); Atanasiu J. A., Stefanescu V., Ber., 61, 1343 (1928).
262. Furman N. H., J. Am. Chem. Soc., 50, 755 (1928); Furman N. H., Evans О. M., ibid., 51, 1129 (1929); Furman N. H.. Wallace J. H„ ibid., 52, 1443 (1930).
263. Willard H. H., Young P., J. Am Chem. Soc., 50, 1322, 1334, 1368, 1372, 1379 1928); 51, 139, 149 (1929).
264. Walden G. H., Hammett L. P., Chapman R. P., J. Am. Chem. Soc., 55, 2649 (1933).
265. Hammett L. P.. Walden G, H., Edmonds S. M., J. Am. Chem. Soc., 56, 1099 (1934).
266. Moss M. L., Mellon M. G., Ind. Eng. Chem. Anal. Ed., 15, 74 (1943).
267. Schiebe G., Friedrich H. J., Z. Elektrochem., 64, 720 (1960).
268. Fanning J. C., Taylor L. T„ J. Inorg. Nucl. Chem., 27, 2217 (1965).
269. Sutton G. J., Australian J. Chem., 14, 550 (1961).
270. Krumholz P., J. Am. Chem. Soc., 75, 2163 (1953).
271. Busch D. H., Bailor J. C., J. Am. Chem. Soc., 78, 1137 (1956).
272. Anderegg G., Helv. Chim. Acta, 46, 2813 (1963).
273. Walter J. L., Freiser H., Anal. Chem., 26, 217 (1954).
274. Knott R., Breckenridge J., Can. J. Chem., 35, 512 (1954).
275. Kahmann K., Sigel H., Erlenmeyer H., Helv. Chim. Acta, 48, 295 (1965).
276. Irving H. M. N. H., Cabell M. J., Mellor D. H., J. Chem. Soc., 1953, 3417.
277. Irving H. M. N. H., Hampton A., J. Chem. Soc., 1955, 430.
278. Cagle F. W., Smith G. F., J. Am. Chem. Soc., 69, 1860 (1947).
279. Burstall F. H., J. Chem. Soc., 1938, 1664.
280. McCurdy W. H., Smith G. F., Anal. Chem., 24, 371 (1952).
281. Hoste J., Anal. Chim. Acta, 4, 23, 1950.
282. Case F. H., Kasper T., J. Am. Chem. Soc., 78, 5824 (1956).
283. Schilt A. A., Smith G. F., Anal. Chim. Acta, 15, 567 (1956).
28ч. Wehking M. W., Pflaum R. T., Tucker E. S., Anal. Chem., 38, 1950 (1966).
285. Case F. H., Koft E., J. Am. Chem Soc., 81, 905 (1959).
286. Collins P., Diehl H., Smith G. F.. Anal. Chem., 31, 1862 (1959).
287. Buchanan E. B., Crichton D., Bacon J. R., Taianta, 13, 903 (1966).
288. Trusell F., Diehl H., Anal. Chem., 31, 1978 (1959).
289. Schilt A. A., Hoyle W. C., Anal. Chem., 39, 114 (1967).
290. Schilt A. A., Kluge K. R„ Taianta, 15, 475 (1968).
291. Schilt A. A., Hoyle I/. C., Taianta, 15, 582 (1968).
292. Schilt A. A., Kluge K. R., Taianta, 15, 1055 (1968).
293. Schilt A. A., Taylor P. J., Taianta, 16, 448 (1969).
294. Schilt A. A., Dunbar W. E., Taianta. 16. 519 (1969).
2'?". Stephen W. I, Tala.ita, 16, 939 (1969).
296. Pemberton J. R., Diehl H., Taianta, 16, 542 (1969).
297. Holyer R. H., Hubbard C. D., Kettle S. F. A., Wilkens R. G., Inorg Chem., 4,
929 (1965).
298. Holyer R. H., Hubbard C. D., Kettle S. F. A., Wilkens R. G., Inorg. Chem, 5 622 (1966).
299. Lee T. S., Kolthoff I. M., Leussing D L., J. Am. Chem. Soc., 79, 2348 (1948).
300. Richards A. F., Ridd J. H., Tobe M. L., Chem Ind., 1963, 1276.
301. Ellis P.. Wilkens R. G„ J. Chem. Soc., 1959, 299.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
369
302. Ellis Р., Hogg R., Wilkens R. G., J. Chem. Soc., 1959, 3308.
303. Wilkens R. G., Williams M. J. G., J. Chem. Soc., 1957, 4515.
304. Basolo F„ Hayes J. C„ Neumann H. M„ J. Am. Chem. Soc., 76, 3807 (1954).
305. Baxendale J. H., George P., Trans. Faraday Soc., 46, 736 (1950).
306. Anderegg G., Helv. Chim. Acta, 46, 2397 (1963).
307. Trujillo R., Brito F., Anales real soc. espan. fis. у quim. (Madrid), 53B, 249 (1957).
308. Irving H. M. N. H., Mellor D. H., J. Chem. Soc., 1962, 5222.
309. Dale J. M., Banks С. V., Inorg. Chem., 2, 591 (1963).
310. McBryde W. A. E., Brisbin D. A., Irving H. M. N. H., J. Chem. Soc., 1962, 5245.
311. Pflaum R. T., Brandt W. W., J. Am. Chem. Soc., 76, 6215 (1954).
312. Banks С. V., Bystroff R. I., J. Am. Chem. Soc., 81, 6153 (1959).
313. Kolthoff I. M., Leussing D. L., Lee T. S., J. Am. Chem. Soc., 73, 390 (1951).
314. Peard W. J., Pflaum R. T., J. Am. Chem. Soc., 80, 1593 (1958).
315. Кульба Ф. Я., Макашев Ю. А., Миронов В. Е., Ж. неорг. хим., 6, 630 (1961).
316. Irving Н. М. N. Н., Mellor D. Н., J. Chem. Soc., 1962, 5237.
317. Brandt W. W., Gullstr от D. K-, J. Am. Chem. Soc., 74, 3532 (1952).
318. Brisbin D. A., McBryde W. A. E., Can. J. Chem., 41, 1135 (1965).
319. Nakashima F., Sakai K-, Bunseki Kagaku, 10, 94 (1961).
320. Crabtree J. M., Marsh D. W., Tomkinson J. C., Williams R. J. P., Ferne-lius W. C., Proc. Chem. Soc., 1961, 336.
321. Atkinson G., Bauman J. E„ Inorg. Chem., 1, 900 (1962).
322. Cabani S., Landucci M., Ann. Chim., 52, 1030 (1962).
323. Baxendale J. H., George P., Trans. Farad. Soc., 46, 55 (1950).
324. Onstott E. I., Laitinen H. A., J. Am. Chem. Soc., 72, 4724 (1950).
325. Cabani S., Moretti G., Scrocco E., J. Chem. Soc., 1962, 88.
326. Cabani S., Landucci M., J. Chem. Soc., 1962, 278.
327. Scrocco E., Salvetti 0., Boll. Sci. Fac. Chim. Ind. Bologna, 12, 98 (1954).
328. Yamasaki K-, Yasuda M„ J. Am. Chem. Soc., 78, 1324 (1956).
329. Yasuda M, Sone K., Yamasaki K., J. Phys. Chem., 60, 1667 (1956).
330. Brandt W. W., Wright J. P., J. Am. Chem. Soc., 76, 3082 (1954).
331. Martin R. B„ Liss felt J. A., J. Am. Chem. Soc., 78, 938 (1956).
332. Schaeffer W. P., Anal. Chem., 35, 1746 (1963).
333. Fortune W. B., Mellon M. G., Ind. Eng. Chem. Anal. Ed., 10, 60 (1938).
334. Moss M. L., Mellon M. G., Ind. Eng. Chem. Anal. Ed., 14, 862 (1942).
335. Harvey A. E., Manning D. L., J. Am. Chem. Soc., 74, 4744 (1952).
336. Moss M. L., Mellon M. G., Ind. Eng. Chem. Anal. Ed., 15, 74 (1943).
337. 'Jorgensen С. K-, Acta Chem. Scand., 9, 1362 (1955).
338. Pflaum R. T., Brandt W. W., J. Am. Chem. Soc., 77, 2019 (1955).
339. Banks С. V., O'Laughlin J. W„ Anal. Chem., 29, 1412 (1957).
340. Miller R. R„ Brandt W. W., Puke Sr. M., J. Am. Chem. Soc., 77, 3178 (1955>
341. Banjerjee R. S., Basu S., J. Inorg. Nucl. Chem., 27, 2217 (1965).
342. Burstall F. H., Dwyer F. P., Gyarfas E. C., J. Chem. Soc., 1950, 953.
343. Blair D., Diehl H„ Taianta, 7, 163 (1961).
344. Blair D., Diehl H., Anal. Chem., 33, 867 (1961).
345. Ewens R. V. G„ Nature, 155, 398 (19451
346. Dwyer F. P., J. Proc. Roy. Soc. N. S. Wales, 83, 134 (1949).
347. Brandt W. W., Smith G. F., Anal. Chem., 21, 1313 (1949).
348. McCurdy W. H., Smith G. F., Analyst, 77, 846 (1952).
349. Brandt W. W., Dwyer F. P„ Gyarfas E. C., Chem. Rev., 54, 959 (1954).
350. Smith G. F„ Anal. Chem., 26, 1534 (1954).
361. Jaffe H. H., Chem. Rev., 53, 191 (1953).
352. Schilt A. A., Smith G. F„ J. Phys. Chem., 60, 1546 (1956).
S?3. Steinhaus R. K-, Margerum D. W., J. Am. Chem. Soc., 88, 441 (1966).
354. Longuet-Higgins H. C„ Coulson C. A., J. Chem. Soc.. 1954, 1534.
370
ГЛАВА VIII
355. Krumholz Р., J. Am. Chem. Soc., 73, 3487 (1951).
356. Nasanen R., Uusitalo E., Suomen Kemistilehti, 29B, 11 (1956).
357. Lahiri S. C., Aditya S., J. Indian Chem. Soc., 41, 469 (1964).
358. Krumholz P„ J. Am. Chem. Soc., 71, 3654 (1949).
359. Harkins T. R., Freiser H„ J. Am. Chem. Soc., 77, 1374 (1955).
360. Burgess J., Prince R. H., J. Chem. Soc., 1963, 5752.
361. Burgess J., Prince R. H., J. Chem. Soc., 1965, 4697.
362. Reynolds W. L., Fukushima S., Inorg. Chem., 2, 176 (1963).
363. Gordon В. M., Williams L. L., Sutin N., J. Am. Chem. Soc., 83, 2064 (1961).
364. Eichler E„ Wahl A. C., J. Am. Chem. Soc., 80, 4145 (1958).
365. Ford-Smith M. H., Sutin N., J. Am. Chem. Soc., 83, 1830 (1961).
366. Sutin N., Gordon В. M., J. Am. Chem. Soc., 83, 70 (1961).
367. Horne R. A., J. Phys. Chem., 64, 1512 (1960).
368. Sheppard J. C., Brown L C., J. Phys. Chem., 67, 1025 (1963).
369. Silverman J., Dodson R. W., J. Phys. Chem., 56, 846 (1952).
370. Wahl A. C., Deck C. F., J. Am. Chem. Soc., 76, 4054 (1954).
371. Bachmann K, Lieser К. H., Z. physik. Chem., 36, 236 (1963).
372. Reynolds W. L., Lin N., Mickus J., J. Am. Chem. Soc., 83, 1078, (1961).
373. George P., Irvine D. H., J. Chem. Soc., 1954, 587.
374. Irvine D. H., J. Chem. Soc., 1959, 2977.
375. Dulz G., Sutin N., Inorg Chem., 2, 917 (1963).
376. Campion R. J., Purdie N., Sutin N., Inorg. Chem., 3, 1091 (1964).
377. Diebier H., Sutin N., J. Phys. Chem., 68, 174 (1964).
378. Dwyer F. P., McKenzie H. A., J. Proc. Roy. Soc. N. S. Wales, 81, 93 (1947).
379. George P., Hanania G. I. H., It vine D. FL, J. Chem. Soc., 1959, 2548.
380. Schilt A. A., Anal. Chem., 35, 1599 (1963).
381. Dwyer F. P., J. Proc. Roy. Soc. N. S. Wales, 83, 134 (1949).
382. Brandt W. W„ Smith G. F., Anal. Chem., 21, 1313 (1949).
383. Hume D. N., Kolthoff I. M., J. Am. Chem. Soc., 65, 1895 (1943).
384. Smith G. F., Richter F. P., Ind. Eng. Chem. Anal. Ed., 16, 580 (1944).
385. Belcher R., Brazier J. N., Stephen W. L, Taianta, 12, 778 (1965).
386. Dwyer F. P„ Humpoletz J. E., Nyholm R. S., J. Proc. Roy. Soc. N. S. Wales, 80, 212 (1947).
387. Dwyer F. P., Gyarfas E. C., J. Am. Chem. Soc., 76, 6320 (1954).
388. Dwyer F. P„ Gibson N. A., Gyarfas E. C., J. Proc. Roy. Soc. N. S. Wales, 84, 80 (1950).
389. Buckingham D. A., Dwyer F. P., Sargeson A. M., Inorg. Chem., 5, 1243 (1966).
390. Smith G. F., Cerate Oxidimetry, 2nd ed., G. Frederick Smith Chemical Co., Columbus, Ohio, 1964.
391. Cagle F. W., Smith G. F., Anal. Chem., 19, 384 (1947).
392. van Nieuwenburg C. J., Blumendal H. B., Mikrochemie, 18, 39 (1935).
393. Sakai T., Nippon Kagaku Zasshi, 83, 1078 (1962).
394. Hawkins C. J., Duewell H., Pickering W. F., Anal. Chim. Acta, 25, 257 (1961).
395. Smith G. F., Getz C. A., Ind. Eng. Chem. Ana1. Ed., 10, 304 (1938).
396. Smith G. F., Duke F. R., Ind. Eng. Chem. Anal. Ed., 15, 120 (1943).
397. Smith G. F., Fritz J. S., Anal. Chem., 20, 874 (1948).
398. Salomon K-, Gabrio B. W., Smith G. F., Archiv. Biochem., 11, 433 (1946).
399. Rao К. B., Z. Anal. Chem., 184, 171 (1961).
400. Guilbault G. G., McCurdy W. H., Anal. Chim. Acta, 24, 214 (1961).
401. Smith G. F., Brandt W. W, Anal. Chem., 21, 948 (1949).
402. Smith G. F., Anal. Chem., 23, 925 (1951).
403. Steigman J., Birnbaum N., Edmonds S. M., Ind. Eng. Chem. Anal. Ed., 14, 30 (1942).
404. Kratochvil B., Zatko D. A., Anal. Chem., 36, 52' (1964).
405. Dwyer F. P., Gibson N. A., J. Proc. Roy. Soc. N.S. Wales, 84, 83 (1950).
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ
371
406. Paglia Е., Sironi С., Gazz. Chim. Ital., 87, 1125 (1957).
407- Hawkins C. J., Perrin D. D., J. Chem. Soc., 1963, 2996.
408. James B. R., Williams R. J. P., J. Chem. Soc., 1961, 2007.
409. Schilt A. A., Anal. Chim. Acta, 26, 134 (1962).
410. Schilt A. A., Cresswell A. M., Taianta, 13, 911 (1966).
411. Schilt A. A., Sutherland J. W., Anal. Chem., 36, 1805 (1964).
412. Anhorn V. J., Hunt H., Ind. Eng. Chem. Anal. Ed., 9, 591 (1937).
413. Rabourdin, Compt. rend., 31, 784 (1850).
414. Mylius F., Ber., 20, 688 (1887).
415. Mylius F., Bei 28, 385 (1896).
416. Freudenberg IL, Boppel H., Ber., 71B, 2505 (1938).
417. Rundle R. E., Baldwin R. R., J. Am. Chem. Soc., 65, 554 (1943).
418. Stein R. S., Rundle R. E., J. Chem. Phys., 16, 195 (1948).
419. Gilbert G. A., Marriott J. V. R., Trans. Faraday Soc., 44, 84 (1948).
420. Patterson A. M., Chem. Eng. News, 30, 3024 (1952); J. Polymer Sci., 8, 269 (1952).
421. Meyer К. H., «Advances in Colloid Science», Kraemer O. (ed.), Vol. I. Interscience, New York, 1942, p. 179.
422. Schoch T. J., J. Am. Chem. Soc., 64, 2957 (1942).
423. Lambert J. L., Anal. Chem., 25, 984 (1953).
424. Zulkowsky K-, Ber., 13, 1395 (1874).
425. Plainer W. S., Ind. Eng. Chem. Anal. Ed., 16, 369 (1944).
426. Peat S., Bourne E. J., Thrower R. D., Nature, 159, 810 (1947).
427. Knowles G., Lowden G. F., Analyst, 78, 159 (1953).
428. Barger G., Field E., J. Chem. Soc., 101, 1394 (1912).
429. Barger G., Starling W., J. Chem. Soc., 107, 411 (1915).
430. Barger G., Eaton F. J., J. Chem. Soc., 1924, 2407.
431. Hahn M., Schutz F., Pavlide F., Z. Hyg. Infektionskrankh., 108, 439 (1928).
432. Reith J. F„ Pharm. Weekblad., 65, 1097 (1929).
433. Staudinger H., Frey K., Starck W., Ber., 60, 1782 (1927).
434. Miller S. A., Bracken A., J. Chem. Soc., 1951, 1933.
435. Erdey L., Bodor E., Papay Al., Acta Chim. Acad. Sci. Hung., 5, 235 (1955).
436. Ottaway J. M., Ph. D. Thesis, University of Exeter, 1965.
437. Bishop E., Jennings V. J., Taianta, 1, 197 (1958).
438. Bishop E., Jennings V. J., Taianta, 8, 34 (1961).
439. Bishop E., Jennings V. J., Taianta, 9, 593 (1962).
440. Bishop E., Jennings V. J., Taianta, 9, 603 (1962).
441. Bishop E., Jennings V. J., Taianta, 9, 679 (1962).
442. Jennings V. J., Ph. D. Thesis, University of Exeter, 1956.
443. Hahn F. L., Ind. Eng. Chem., Anal. Ed., 14, 571 (1942).
444. Gay-Lussac J. L., Ann. chim. et phys., 60, 225 (1835).
445. Penny F., Brit. Assoc. Adv. Sci., Report, 18 [2], 58 (1850).
446. Quinquaud E., Z. anal. Chem., 21, 605 (1882).
447. Gyory S., Z. anal. Chem., 32, 415 (1893).
448. Jellinek K., Krestejf W., Z. anal. Chem., 137, 333 (1924).
449. Jellinek K., Kiihn W., Z. anal. Chem., 138, 99 (1924).
450. Willard H. H., Manalo G. D., Ind. Eng. Chem. Anal. Ed., 19, 167 (1947).
451. Willard H H„ Young P„ J. Am. Chem. Soc., 50, 1322 (1928).
452. Rathsburg H., Ber., 61, 1663 (1928).
453. Fischer E., Angew. Chem., 64, 592 (1952).
454. Knecht E., Hibbert E„ Ber., 36, 1549 (1903).
455. Knecht E., Hibbert E., «New Reduction Methods in Volumetric Analysis», 2nd ed., Longmans Green, London, 1925.
456. Teorell T., Biochem. Z., 248, 246 (1932).
457. Kolthoff I. M., Stenger V. A., Ind. Eng. Chem. Anal. Ed., 7, 79 (1935).
458. Goto H„ Sci. Repts. Tohoku Imp. Univ. Ser. I, 29, 446 (1940).
459. Райхинштейн Ц. Г., Ж. приклад, химии, 8, 1470 (1935).
372
ГЛАВА VIII
460. Шейнцис О. Г., Завод, лаб., 12, 930 (1946).
461. Bitskei J., Petrich К-, Magyar К cm. Lapja, 2, 230 (1947).
462. Bitskei J., Forhencz M., Magyar К cm. Lapja, 2, 117 (1947).
463. Belcher R., Anal. Chim. Acta, 5, 27 (1951).
464. Meditsch J. O., Anal. Chim. Acta, 31, 286 (1964).
465. Bishop E., Jennings V. J., Taianta, 8, 697 (1961).
466. Bishop E., Jennings V. J., Taianta, 9, 581 (1962).
467. Willst otter R., Piccard J., Ber., 41, 1485 (1908).
468. Wills tatter R., Mayer R., Ber., 37, 1495 (1904).
469. Willstatter R., Pfannenstiehl A., Ber., 38, 4605 (1905).
470. Clark W. M., Cohen B., Gibbs H. D., Pub Health Repts. U.S.A., Supplement 54, 1926.
471. Michaelis L., J. Am. Chem. Soc., 53, 2953 (1931).
472. Wurster E., Sendtner R., Ber., 12, 1803 (1879).
473. Michaelis L., Hill E. S., J. Am. Chem. Soc., 55, 1481 (1933).
474. von Bandrowski E., Monatsh., 8, 475 (1887).
475. Miyagi S., J. Soc. Chem. Ind. Japan, 36 Supp., 146 (1933).
476. Dutta A. B., Current Sci. (India), 24, 97 (1955).
477. Tomtcek O., Valcha J., Coll. Czech. Chem. Comms., 16/17, 113 (1951/2); Chem. Listy, 44, 283 (1950).
478. Kuhn R., Franke W., Ber., 68, 1528 (1935).
479. Szebelledy L., Schick K, Ber. ungar. pharm. Ges., 9, 449 (1933); Z. anal. Chem , 97, 186 (1934).
480. Szebelledy L., Mikrochim. Acta, 1, 226 (1937).
481. Szebelledy L., Mikrochim. Acta, 2, 56 (1938).
482. Szebelledy L., Madis W., Mikrochim. Acta, 3, 1 (1938).
483. Szebelledy L., Madis W., Ber. ungar/chem. Ges., 13, 81 (1937).
484. Szebelledy L., Sik K., Z. anal. Chem., 108, 81 (1937).
485. Szebelledy L., Tannay K-. Pharm. Zentralh., 79, 441, 383 (1938).
486. Cerana L. A., Rev. Fac. Ing. Quim. Univ. Nac. Lit. Santa Fe, Argentina, 29, 105 (1960).
487. Cerana L. A., Primer Congreso Interamericancf de Ing. Quim., Puerto Rico, 1961.
488. Belcher R., Wilson C. L., «New Methods of Analytical Chemistry», Chapman & Hall, London, 1964, p. 36.
489. Goto Ji., Kakita Y., J. Chem. Soc. Japan, 63, 120 (1942).
490. Goto H., Kakita Y., J. Chem. Soc. Japan, 64, 515 (1943).
491. Kenny F., Kurtz R. B., Anal. Chem., 22, 693 (1950).
492. Wawrzyczek W., Wisniewska K., Chemie Analit., 10, 1287 (1965)
ГЛАВА IX
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
Г. Ф. Киркбрайт
ВВЕДЕНИЕ
Когда молекула при поглощении энергии переходит из основного состояния в возбужденное, она может терять избыточную колебательную энергию в результате столкновений с другими молекулами; при этом наблюдается эмиссия энергии, если молекула возвращается из возбужденного состояния к колебательно-возбужденным уровням основного состояния. Процесс испускания энергии света, который сопровождает прямой переход возбужденных частиц из возбужденного состояния к основному за очень короткое время (порядка 10~8 сек), называется «флуоресценцией». Частота колебаний испускаемого излучения ниже частоты поглощенного излучения.
Возбуждающее и испускаемое излучения могут находиться в ультрафиолетовой области спектра, и поэтому они не видимы глазом. Подобное явление характерно для паров бензола 'и других простых органических молекул. Многие органические вещества разлагаются так быстро после поглощения возбуждающего излучения, что степень флуоресценции совсем незначительна. В чистом жидком состоянии или в растворе простые неорганические и органические вещества редко флуоресцируют, вероятно, потому, что в них происходит множество дезактивирующих столкновений, в результате которых энергия почти полностью рассеивается в виде тепловой энергии. Боуэн [1], а также Паркер и Рис [2] изучали явление флуоресценции и различные причины тушения флуоресценции. Многие комплексные органические соединения, в которых перенос л-электрона при переходе в более высокое энергетическое состояние не оказывает большого влияния на структуру и устойчивость молекулы, а приводит к образованию относительно устойчивых возбужденных частиц, флуоресцируют в растворе в видимой области спектра. Чтобы молекула флуоресцировала, ей необходимо иметь определенную электронную конфигурацию, а для этого она должна находиться в точно определенном электрическом состоянии (быть знионом, катионом или нейтральной молекулой). Имеются соеди
374
ГЛАВА IX
нения, представляющие интерес для химика-аналитика вследствие того, что на их флуоресценцию сильно влияют химические элементы и изменения в физической среде, которые вызывают изменения в интенсивности, положении или ширине полос флуоресцентного излучения.
Во многих веществах, флуоресцирующих при облучении УФ-светом, изменяется окраска раствора либо происходит усиление или тушение флуоресценции, в зависимости от природы и концентрации посторонних ионов, содержащихся в растворе. Вещества, флуоресцентные свойства которых в растворе зависят от концентрации ионов водорода, окислительного потенциала или концентрации иона металла, можно использовать ,в качестве «флуоресцентных индикаторов» в методах титрования, при которых в точке эквивалентности происходит четкое изменение одного из этих свойств. Очень немногие флуоресцентные индикаторы дают изменение в точке эквивалентности, если титрование проводится при дневном или искусственном свете, в то время как при УФ-эблуче-нии наблюдаются резкие изменения флуоресценции. Так как многие индикаторы флуоресцируют довольно интенсивно, не всегда надо работать в полной темноте; достаточно бывает вести титрование в полузатемненной комнате или боксе.
До недавних пор применение флуоресцентных индикаторов ограничивалось установлением конечной точки в ацидиметрии и алкалиметрии, и даже еще в настоящее время только немногие из них рекомендованы для применения в окислительно-восстановительной титриметрии. Область применения флуоресцентных индикаторов значительно расширилась в результате поисков более совершенных индикаторов для установления конечной точки в комп-лексометрическом титровании. За последнее время предложено много новых соединений в качестве флуоресцентных индикаторов для комплексометрического титрования некоторых ионов металлов.
Источником УФ-света может быть ртутная, дуговая или электрическая лампа из подходящего стекла, фильтрующего нежелательные лучи. Подходящие фильтры должны пропускать пучок света с длиной волны от 3000 А до 4000 А. Чаще всего работают со ртутной лампой, которая имеет фильтр. Примером простой аппаратуры для флуоресцентного фильтрования [3] может послужить схема, показанная на рис. IX. 1. Для точных измерений интенсивности флуоресценции при длине волны максимума излучения используют флуориметр. Для титримстрического анализа, который будет описан ниже, обычно бывает достаточно прямого визуального определения цвета или интенсивности флуоресценции.
Методика титрования очень проста. Бюретка и другая стеклянная посуда, в которой проводится титрование, должны быть из нефлуоресцирующего стекла. Титрование проводят в конической колбе из тонкого стекла. В этих условиях нет необходимости исполь
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
375
зовать кварцевое стекло. Колбу для титрования устанавливают непосредственно на пути УФ-лучей, комнату затемняют. Титруют как обычно. Нижний конец бюретки смазывают вазелином (который сильно флуоресцирует) для того, чтобы следить за вытеканием титранта из бюретки. Деления на бюретке тоже делают видимыми в темноте, смазав их вазелином, а чтобы освещать мениск раствора, помещают на поверхность жидкости в бюретке несколько крупинок твердого флуоресцирующего вещества. Грант [4] описал поплавковое устройство для освещения мениска и делений бюретки. В полузатемненной комнате предпочтительно использовать дополнительную лампу с вольфрамовой нитью, чтобы производить отсчет по бюретке.
Рис. IX. 1. Аппарат для титрования с применением флуоресцентных индикаторов.
Ультрафиолетовый свет опасен для зрения, поэтому надо защищать глаза от прямого излучения.
ФЛУОРЕСЦЕНТНЫЕ КИСЛОТНО-ОСНОВНЫЕ ИНДИКАТОРЫ
Флуоресцентные индикаторы, чувствительные к концентрации ионов водорода, можно использовать для определения pH растворов, если зависимость интенсивности или цвета флуоресценции индикатора от pH раствора определить флуориметрически в буферном растворе. Затем сопоставляют интенсивность и цвет флуоресценции раствора, pH которого неизвестно, с данными стандартного раствора. Коломбье [5] исследовал, как зависит от pH цвет флуоресценции растворов эозина, эритрозина, флуоресцеина, акридина, умбеллиферона и р-нафтола, и пришел к выводу, что ни одно из этих 'соединений не пригодно для определения величины pH. Однако Линзер [6], исследуя эскулин (pH 1,5—2,0 и pH 5,5—6,5), флуоресцеин (pH 3,4—4,1) и умбеллиферон (pH 6,5—8,0), нашел, что г помощью этих соединений можно определять pH растворов с
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ ДЛЯ АЦИДИМЕТРИИ И АЛКАЛИМЕТРИИ
co о
Таблица IX.1
Номер Название Формула Изменение окраски Область pH Раствор индикатора Литература
1 2 3 4 Бензофлавии 3,6-Диоксифталимид Эозин (тетрабромфлуо-ресцеин) 4-Этокеиакридон (TJ H2N ОН сУ°>н у\ох он вг Вг О<уЛ^°ч}Ду'ОН вгААсЛаВг „А^сооч чХ 0 офр и ос2н5 Желтая/зеленая Синяя/зеленая Зеленая/желто-зеленая Не флуоресцирует/зеле-ная Зелеиая/сиияя 0,3-1,7 0—2,4 6,0—8,0 0-3,0 1,2—3,2 1 %-ный в этаноле То же 1 %-ный водный раствор соли натрия 1 %-ный в этаноле 12 12, 13 7, 14, 15 12
5 3,6-Тетраметилдиами-ноксантон О II Iff нт
L. И JI (Ch3 )2 (сн3)2
6 Эскулин НО^^г^О^О । т т
7 Антраниловая кислота ^xNH2
^^соон
8 З-Амино-1-нафтойная кислота соон
см X Z 5
9 1 -Нафтиламино-6-суль-фамид nh2
H2NO28
10 1 -Нафтиламино-7-суль-фамид N’Hj
H2NO1Sx%^v-4.1
MJ
оэ
» »
1,2—3,4 То же
12
Голубая/ярко-синяя
1,5—2,0
Не флуоресцирует/голу-бая
Голубая/темпо-снняя
Темно-синяя/не флуоресцирует
Не флуоресцирует/зеле ная
Зелена я/синяя
Синяя/не флуоресцирует
Не флуоресцирует/зеле-ная
Зеленая/не флуоресциру ет
1,5-3,0 4,5—6,0 12,5—14 1,5—3,0 4,0—6,0 11,6—13,0 0,1 %-ный в 50%-ном этаноле 0,1 %-ный раствор сульфата в 50%-ном этаноле 8, 16 8, 16
1,9—3,9 0,05%-ный в 16, 17
90%-ном эта-
9,6—13,0 ноле
Не флуоресцирует/зеле- 1,9—3,9 То же 16, 17
ная
Зеленая/не флуоресциру 9,6—13,0
ет
w oo
Название
Формула
2-Нафтиламино-6-суль-фамид
2-Нафтиламино-8-суль-фамид
1-Нафтиламино-5-суль-фамид
14 1-Нафтойная кислота
15 Салициловая кислота
Продолжение табл. 1Х.1
Изменение окраски Область pH Раствор индикатора Литература
Не флуор есцнрует/темно- синяя 1,9—3,9 0,05%-ный в 90 %-ном эта- 16, 17
Темно-синяя/не флуорес цирует 9,6—13,0 ноле 16, 17
Не флуоресцирует/голу-бая Голубая/не флуоресцирует 1,9—3,9 9,6—13,9 То же 16, 17
Не флуоресцирует/жел-то-оранжевая Желто-оранжевая/не флуоресцирует 2,0—4,0 9,5—13,0 » 16, 17
Не флуоресцирует/синяя 2,5—3,5 Раствор соли натрия в дистиллированной воде 7
Не флуоресцирует/тгмно-синяя 2,5—4,0 0,5%-ный водный раствор соли натрия 14
16
Флоксин ВА экстра (тетр ахлортетрабром-флуоресцеин)
Вг Вт
Эритрозин В (тетраиод-флуоресцеин)
18 2-Нафтиламин
19 Магдала красный
и-Аминофенилбензосуль-фамид
h2n^O-s°2nh2
Не флуоресцирует/темно-синяя 2,5—4,0 0,1 %-ный в этаноле 14, 18
Не флуоресцирует/свет-ло-зеленая 2,5—4,0 0,2 %-ный раствор соли натрия в дистиллированной воде 7, 14, 19
Не флуоресцирует/фио-летовая 2,8—4,4 0,5%-ный в этаноле
Не флуор есцирует/пур пурная 3,0-4,0
12
20
Не флуоресцирует/голу- 3,0—4,0 0,05%-ный в 16, 17
бая 90%-ном эта-
ноле
§
Название
21
2-Окси-З-нафтойная кислота
22
Хромотроповая кислота
23
1-Нафтионовая кислота
24
1-Нафтиламин
х
25
5-Аминосалициловая кислота
Продолжение табл. IX.1
Изменение окраски Область pH Раствор инднка тора Литература
Г олубая/зеленая 3,0—6,8 0,1%-ный раствор соли натрия в дистиллированной воде 21
Не флуоресцирует/голу-бая 3,1—4,4 5%-ный водный 22
Не флуоресцирует/синяя Синяя/желто-зеленая 3,0—4 10—12 Раствор соли натрия в воде 15, 23
Не флуоресцирует/синяя 3,4—4,8 0,5%-ный в этаноле 12
Не флуоресцирует/свет-ло-зеленая 3,1—4,4 Свежий 0,2%-ный в этаноле 24
26
Хинин
27 о-Метоксибензальдегид
28 о-Фенилендиамин
29 я-Фенилендиамин
30 Морин (2/14,,315,7-пента оксифлавон)
Синяя/светло-фиолетовая
Светло-фиолетовая/не флуоресцирует
3,0—5,0 I
9,5—10,0
0,1 %-ный в эта-17, 15, иоле I 21,
23, 25
Не флуоресцирует/зеле ная 3,1—4,4 0,2 %-ный водный 19
Зеленая/не флуоресцирует 3,1—4,4 0,2 %-ный в 70%-ном этаноле 19
Не флуоресцирует/оран жево-желтая 3,1—4,4 То же 19
Не флуоресцирует/зеле ная
Зелеиая/желто-зелеиая
3.1—4,4
8- 9,8
0,2%-ный в дистиллированной воде
27
ф s о К Название Формула
31 Тиофлавин S С. 1.а 49010. Прямой желтый 7. Метилированный и сульфированный примуЛИН
32 Флуоресцеин Г II 1 1
33 Эозин+ксиленоловый ци-анол FF
34 Дихлорфлуоресцеин НО 1 X XX civXXc=o I J
35 Р-Метилэскулетин ЦДосн H
36 Хининовая кислота соон ОНзСЧ^уХ
Продолжение табл. IX.1
Изменение окраски Область PH Раствор индикатора Литература
Темно-синяя/голубая 3,1—4,4 0,2%-ный в дистиллированной воде 28
Розово-зеленая/зеленая 4,0— 4,5/6,0 1 %-ный раствор соли натрия в дистиллированной воде 7, 14, 15, 18
Не флуоресцирует/зеле ная 4,0—5,0 1 %-ный в этаноле и н-бутано-ле 29
Сине-зеленая/зеленая 4,0—6,6 I %-ный в этаноле 14, 15, 18
Не флуоресцирует/синяя 4,0—6,2 То же 12, 13
Синяя/светло-зеленая 9,0—10,0
Желтая/синяя 4,0—5,0 Насыщенный водный 30
’ 37 ft-Нафтохинолин
38 Резоруфин (7-оксифеноксазон)
39 Акридин
40 3,6-Диоксиксантон
41 5,7-Диокси-4-метилкума-рин
42 co ОС co Динитрил-3,6-диокси-фталевой кислоты
Синяя/не флуоресцирует 4,4—6,3 0,05%-ный в 50%-ном эта- ноле 16
Желтая/оранжевая 4,4—6,4 Совсем слабо флуоресцирует 12
Зеленая/фиолетовая 5,2—6,6 0,1 %-ный в этаноле 7, 15, 26
Не флуоресцирует/сине фиолетовая
5,4—7,6
1 %-ный в этаноле
12
Голубая/темно-синяя 5,5—5,8
Синяя/зеленая
5,8—8,2
1 %-ный в этаноле
12
ОЭ СО
1 Номер 1 Название Формула
43 1,4-Диоксибензодисульфокислота он Х/З°зн г у HO3S'Xy он
44 Люминол <TV_ >=< ® / \ К5 г> о ? ? Z—Z ж а
45 2-Нафтол-6-сульфокис-лота (кислота Шеффе- хЛ^х/ОЕ Г у Т
ра)
46 Хинолин OQ
47 1 -Нафтол-5-сульфокис- он
лота (кислота Клеве) Од SO3H
48 Умбеллиферон НОх^х о 0 1 L JL У
Продолжение табл. IX.1
Изменение окраски Область PH Раствор индикатора Литература
11е флуоресцирует/голу-бая 6—7 0,1 %-ный раствор соли калия в дистиллированной воде 32
Не флуоресцирует/синяя 6—7 См, гл. X о хемилюминесцентных индикаторах 33
Не флуоресцирует/синяя 5/7—8/9 Водный раствор соли натрия 7, 15
Синяя/не флуоресцирует 6,2—7,2 Насыщенный водный 52
Не флуоресцирует/зеле ная 6,5-7,5 Водный раствор соли натрия 15
Не флуоресцирует/синяя 6,5—8,0 —ш 5, 7, 15, 19, 26, 34, 35
25—418
49 8 Оксихинолинат магния
50 Орсинаурин
51
52
53
Диазо бриллиантовый желтый
Кумариновая кислота
Р-Метилумбеллиферон
Гармин
Не флуоресцирует/жел-тая о,5- -7,5 0,1 %-ный раствор комплекса в 0,01 М НС1
Не флуоресцирует/зеле-иая 6,5-8,0 0,03%-ный в дистиллированной воде
36
30
Не флуоресцирует/синяя 6,5-7,5 15
Не флуоресцирует/зеле-ная 7,2—9,0 1 %-ный в этаноле 12
Не флуоресцирует/синяя >7,0 0,3%-ный в 50 % -ном этаноле 13, 19 35, 37
Синяя/желтая
7,2-8,9
38
Номер
Название
Формула
2-Нафтол-6,8-дисульфо-кислога (G-кислота)
56 Салицилальдегидсеми-
карбазон
1 -Нафтол-2-сульфокис-
лота
Н~N-NH-CONH?
Салицилальдегидаце-тилгидразон
Салицилальдегидтиосе-микарбазои
C№=N-NH-COCH3
ОН
Продолжение табл. IXЛ
Изменение окраски Область pH Раствор индика тора Литература
Синяя/голубая 7,5—9,1 Водный раствор соли натрия 15
Желта я/синяя 7,6—8,0 0,1 %-ный спиртовой 50, 54
Темно-синяя/голубая 8,0—9,0 Водный раствор соли натрия 7, 23
Не флуоресцирует/зеле но-синяя 8,3 0,1 %-ный в этаноле 54
Не флуоресцирует/сине зеленая 8,4 То же 54
Темно-синяя/голубая 8,2 Водный раствор 23
соли натрия
Не флуоресцирует/желто- 8,2—10,3 То же 15
зеленая
Не флуоресцирует/синяя 8,5—9,5 0,1%-ный в этаноле 7, 39
Не флуоресцирует/желто зеленая 8,4—10,4 1 %-ный в этаноле 12, 15
Не флуоресцирует/жел-тая 8,6— 10.С 40
Темно синяя/голубая 9,0—9,5 Водный раствор соли натрия 15, 41
Зеленая/не флуоресцирует 9-11 1 %-ный в этаноле 12
CO
Номер Название Формула
67 68 о-Оксифенил бензотиазол о-Оксифенил бензоксазол R Q Т = Т в а а°
69 о-Оксифенилбензими-дазол чч» он
70 Кумарин о , II л? о
71 6,7 -Диметоксиизохинолин-1-карбоновая кислота сН,о.Г1А CH3O'x^xyN соон
Продолжение табл. IX 1
Изменение окраски Область 3 pH Раствор индикатора Литература
Не флуоресцирует/сине-зеленая 9,3 0,1 %-ный в этаноле 54
Не флуоресцирует/сине-фиолетовая 9,3 То же 54
Не флуоресцирует/сине-фиолетовая 9,9 » 54
Не флуоресцирует/свет-ло-зеленая 9,5—10,5 7, 14
Желтая/синяя 9,5—11,0 0,1 %-ный в глицерине, разбавленном » равным объемом этанола и 9 объемами дистиллированной воды 30
Темно-синяя/бело-синяя
Фиолетовая/желто-зеле-ная
Желтая/белая
9,5—13,0
10—12
>12,5
0,05%-ный
в 90%-ном спирте
16, 17
Водный раствор соли натрия
15
23
Синяя/фиолетовая 12—13 1%-ный водный 1 раствор соли натрия 23
Синяя/желто-розовая 12—14 1%-ный водный раствор соли натрия 15
390
ГЛАВА IX
точностью около 0,05 pH единиц при концентрации раствора индикатора 10-5%. Так как большинство флуоресцентных индикаторов проявляет изменения во флуоресценции в относительно широкой области значений pH, то визуальное наблюдение довольно трудно. В настоящее время имеются флуориметры, которые позволяют определять цвет и интенсивность флуоресценции в растворах по спектрам флуоресценции. Гото [7] исследовал, как изменяется в зависимости от pH флуоресценция следующих индикаторов: салициловой кислоты, эритрозина, умбеллиферона, флоксина, акридина, Р-нафтола, р-нафтол-6-сульфокислоты, хинина, кумарина и 1-наф-тойной кислоты.
Томичек [8] и Деман [9] написали обзоры по флуоресцентным индикаторам и их применению. В табл IX. 1 приведено 76 флуоресцентных индикаторов, .которые рекомендованы для применения в ацидиметрии и алкалиметрии; для каждого из них дана формула, область pH и цветная контрастность изменения флуоресценции. Все индикаторы расположены в порядке возрастания значений pH, при которых происходит изменение флуоресценции. Если флуоресценция какого-нибудь соединения изменяется несколько раз, положение такого соединения в таблице определяется значением pH, при котором происходит 1-е изменение. Небольшое расхождение в данных для некоторых индикаторов объясняется различиями в методике получения этих соединений и их степенью чистоты.
Индикаторы, изменяющие флуоресценцию в пределах pH от 3 до 10, можно использовать для титрования при соответствующих концентрациях сильных кислот сильными основаниями и наоборот. Для титрования слабых оснований сильными кислотами наиболее подходят индикаторы, флуоресценция которых изменяется при низких значениях pH, например р-нафтиламин (pH 2,8—4,4), бензофлавин (pH 0,3—1,7), 4-этоксьакридон (pH 1,2—3,2) или 3,6 тет-раметилдиаминоксантон (pH 1,2—3,4). Такие индикаторы, как 1-нафтионовая кислота (2-е изменение при pH 10—12), 2-нафтионовая кислота (pH 12—13), кумарин (pH 9,5—10,5), морин (2-е изменение при pH 8—9,8) и хинин (2-е изменение при pH 9,5—10,0), которые изменяют флуоресценцию в области высоких значений pH, следовало бы использовать для титрования слабых кислот сильными основаниями. Хинин, который дважды изменяет флуоресценцию (при pH 3,0—5,0 и 9,5—10,0), использовали при титровании первых двух ступеней дисссциации фосфорной кислоты; в особенности он пригоден для работы с мутными растворами.
Себелед и Шик [10] исследовали ряд хорошо известных обычных кислотно-основных индикаторов в качестве флуоресцентных индикаторов. Они установили, что феноловый красный, тимоловый синий и ализариновый красный изменяют флуоресценцию приблизительно в той же области значений pH, в которой изменяется и их
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
391
окраска, а поэтому их можно использовать как флуоресцентные индикаторы для титрования сильно окрашенных растворов.
Некоторые из флуоресцентных индикаторов, приведенных в табл. IX.1 (соединения 9—13, 20), можно применять вместо обычных индикаторов в реакциях нейтрализации, проводимых в неводной среде [11]. Нафтиламиносульфамиды пригодны в качестве индикаторов для титрования хлорной кислотой в среде безводной уксусной кислоты.
Как уже говорилось, флуоресцентные кислотно-основные индикаторы с успехом можно использовать в титровании сильно окрашенных или мутных растворов. Для работы в этих условиях с обычными индикаторами приходится значительно разбавлять раствор, тогда как применение подходящих флуоресцентных индикаторов позволяет в большинстве случаев легко титровать эти жидкости при УФ-облучении. Сама титруемая жидкость не должна сильно флуоресцировать. В присутствии флуоресцентных индикаторов с успехом можно титровать вина [42—45], пиво [14], синтетические смолы [46], эфирные масла [47], воски [48], поверхностноактивные вещества [49], растительные экстракты и экстракты почв. Индикатор берется в очень малой концентрации, и это является преимуществом большинства титрований. Кроме того, в отличие от многих индикаторов в титриметрии флуоресцентные индикаторы действуют косвенно и не участвуют в самой реакции.
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ В ТИТРОВАНИИ ПО МЕТОДУ ОСАЖДЕНИЯ
Некоторые из флуоресцентных кислотно-основных индикаторов, приведенных в табл. IX. 1 (№ 3, 16, 17, 26, 31, 32, 34, 47, 52, 56), и такие флуоресцирующие красители, как примулин, трипафлавин и родамин 6G, широко используются в качестве адсорбционных индикаторов при титровании по методу осаждения. Эти индикаторы прменялись в аргентометрическом титровании галогенидов [7, 28, 35, 36]; они пригодны также (за исключением трипафлавина и родамина 6G) для титрования роданида раствором серебра. Умбелли-ферон и p-метилумбеллиферон нашли применение при титровании серебра бихроматом и свинца хроматом калия, гексацианоферратом (II) калия, метаванадатом аммония, фосфатом натрия и оксалатом натрия. Умбеллиферон пригоден также для титрования ортовольфраматов [53] нитратом свинца и ионов ртути(I) гидроокисью натрия или роданидом калия. Хинин можно использовать при титровании гексацианоферрата (II) нитратом свинца [53] и лонов ртути(I) хроматом калия или гексацианоферратом(II) калия. Конечную точку титрования устанавливают по появлению или исчезновению флуоресценции, а в некоторых случаях происходит резкое изменение интенсивности флуоресценции.
392
ГЛАВА IX
По поводу более детального описания областей применения большинства этих индикаторов в качестве адсорбционных индикаторов рекомендуем обратиться к главе, в которой рассматриваются адсорбционные индикаторы.
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ ДЛЯ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯ
Хотя флуоресцентные индикаторы широко использовались в кислотно-основном и комплексометрическом титровании, а также в титровании по методу осаждения, только немногие из них предложены для методов окислительного и восстановительного титрования.
В 1938 г. Гото использовал неочищенную смесь хризанилина и его гомологов в титровании перманганатом [55], но при этом он не выделял флуоресцирующие частицы. Позднее он предложил родамин В и флуоресцеин для оксидиметрического титрования , 56]. При добавлении свободного иода к кислому раствору родамина В-исчезает оранжевонкрасная флуоресценция, а при удалении иода появляется снова. Значит, родамин Е можно использовать в качестве индикатора при йодометрическом титровании олова(II), а также при титровании олова (II) броматом в условиях, аналогичных титрованию в присутствии метилового оранжевого. При добавлении к оттитрованному раствору хлорида олова(II) флуоресценция вновь не появляется. Этот индикатор можно также использовать при титровании мышьяка(III) броматом или перманганатом в присутствии катализатора четырехокиси осмия. При титровании броматом можно вместо родамина В работать с флуоресцеином. Гото [57] предложил применять в качестве флуоресцентных индикаторов риванол, гармин и фосфин. Фосфин можно использовать при титровании железа (II) перманганатом в фосфорнокислой среде, а также для титрования перекиси водорода в 0,1—0,15 М серной кислоте раствором перманганата. Риванол, гармин и фосфин пригодны при титровании мышьяка(III) и олова(II) хлорамином и для йодометрического титрования тиосульфата и арсенита.
Недавно было описано [58] применение некоторых 3,6-диокси-фталевых кислот в качестве обратимых флуоресцентных индикаторов для окислительно-восстановительных методов титрования. Вос становительные потенциалы этих соединений находятся в области 0,85—0,95 В относительно нормального водородного электроде Следовательно, эти индикаторы надо применять в той области потенциалов, где дифениламиносульфокислота действует в бесцветных растворах, но потенциал перехода этих индикаторов недостаточно высок для использования их в реакциях с такими сильными окислителями, как сульфат или перхлорат церия (IV).
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
393
Рао с сотр. предложил флуоресцентный индикатор родамин 6G для титрования урана (IV) раствором железа (III) [59] и ванадия (IV) раствором сульфата церия (IV) [60]. При титровании урана первый избыток железа (III) тушит флуоресценцию индикатора, а при титровании ванадия индикатор окисляется до формы, которая не флуоресцирует. Значит, родамин 6G окисляется в две стадии, из которых первая обратима, а вторая необратима. Рао и др. [61] предложили родамин 6G для титрования молибдена^) сульфатом ферроаммония в 2,5—3 М соляной кислоте при 100 °C. Присутствие небольших количеств ванадия (IV) не мешает определению.
Обратимыми флуоресцентными индикаторами для окислительно-восстановительных методов титрования оказались комплексы рутения с 2,2'-дипиридилом, 1,10-фзнантролином и некоторыми их метилпроизводными [62]. Комплексы рутения (II) флуоресцируют оранжево-красным цветом, а комплексы рутения (III) не флуоресцируют. Комплекс рутения (II) с 2,2'-дипиридилом лучше всего подходит для титрования перхлоратом церия (IV), а комплекс рутения (II) с 4,4/-диметил-2,2'-дипиридилом для титрования сульфатом церия (IV) и перманганатом. Конечная точка устанавливается по исчезновению оранжево-красной флуоресценции. Индикатор действует быстро, обладает хооошей чувствительностью и обратимостью; его применяют в микротитровании мышьяка(III) раствором сульфата церия (IV). Комплексы рутения с метилзамещенными фенантролинами и а,а'-дипиридилом также используются [91] в качестве индикаторов при титровании раствором иода. От добавления иода к раствору комплекса рутения выпадает в осадок три-иодид комплекса и тушится оранжево-красная флуоресценция. Комплекс рутения с 4,4'-диметилпиридилом применяется в качестве индикатора для титрования тиосульфата или арсенита раствором иода, а также и для обратного титрования.
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ ДЛЯ КОМПЛЕКСОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Органические соединения, образующие комплексы с катионами в растворе с последующим тушением или появлением флуоресценции, были использованы как индикаторы в комплексометрическом титровании. Они действуют подобно металлохромным индикаторам, т. е. в конечной точке титрования комплексообразующим реагентом происходит изменение флуоресценции, вызываемое разрушением комплекса металла с индикатором. В случае обратного титрования изменение ф чуоресценции в конечной точке (появление или исчезновение) обусловливается образованием комплекса металла с индикатором. ИЮПАК принял термин «металлофлуорес-
о
Таблица IX.2
Название
1 Морин
(2',4',3,5,7-пентаокси-флавон)
2 Салицилальде-
1гидацетил-гидразон
3 Салицилиден-о-аминофе-нол
З-Окси-2-наф-тойная кислота
5 8-Оксихинолин
МЕТАЛЛОФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
Формула
CH=N-NH— COCH
Титруемые ионы3 Область рн Изменение флуоресценции Раствор индикатора Литература
Ga8+ In3+ 3,8 5,0 Зеленая/не флуоресцирует Зеленая/не флуоресцирует 0,5%-ный в метаноле или 0,2% в смеси этанол 4~ +вода (1:1) 65
Zn2+ 4,5 (ацетат) Синяя/не флуоресцирует 0,1%-ный в этаноле 67
Al3+, титрование раствором ЭДТА или F“ 4-5 (ацетат) Желто-зеле-ная/не флуоресцирует Свежий 0,05%-ный в этаноле 68
А13+ 3,0 Синяя/зеленая 0,1%-ный водный раствор соли натрия 72
Ga3+ 2,5—3,5 (фталат) Желто-зеле-ная/не флуоресцирует 0,05 %-ный в 0,5%-ной уксусной кислоте 69
6 8-Оксихинолин-5-сульфокис-лота SO,H ФЬ он
7 Кальцеин Н О х^ххОх^-х,/ о н ноос—сн2 Wxcj^A сн-соон n-ch2 Ic'o h2c-n НООС—СН2 сн—соон
8
Кальцеиновый синий
НООС—СН2 СНз
1 ill
НООС—СН2 он
Zn2+ Zn2+, Cd2+, Mg2+ + ЦДТА 10 7 To же ъ 0,2 %-ный 1 водный ' 70 92
Cu2+ Mn2+ 10 (NH40H— NH4C1) 8—10 (пиридин— ацетат или NH40H) Не флуоресци-рует/желто-зеленая То же 74—80
Ca2+ Cr3+, Co2+, обратное титрование +Cu2+ >12 (KOH) 10 (NH4OH— NH4C1) Желто-зеле-ная/не флуоресцирует То же 1%-ная дисперсия в порошке КС!
Cu2+ Ca2+, Ba2+, Sr2+, Cr3+, Ni2+, Co2+, Fe3+, Ti4+ Al3+, Mn2+, Zn2+, Hg2+, обратное титрование + Cu2+ 4,8 (ацетат— CH3COOH) >12 (KOH) 4,8 (ацетат— СН3СООН) Не флуоресцирует/синяя Синяя/не флуоресцирует То же 0,1 %-ный водный 81, 82
Название
Формула
Ксантонком-плексан
СН2—СООН
Метилкальце-ин
Метилкальцеи-новый синий
12
о-Дианизидин-N,N,N',N'-тетрауксус-ная кислота
НООС—СН2 СН3—СООН
НООС- СН °СНз нзсо СН2—СООН
Продолжение табл. IX.2
Титруемые ионыа Область pH Изменение флуоресценции Раствор индикатора Литература
Г Свойства подобны c войствам кальцеи нового синего 81
Мп2* Си2+, ОЭТА Си2+, ЦДТА 9,5 5 5-5,5 Не флуоресци-рует/желто-зеленая То же » 0,1 %-ный водный 83, 84
Мп2+ Си2+, ОЭТА Си2+, ЦДТА 9,5 5 5—5,5 Не флуоресци-рует/синяя То же » То же 83, 84
Си2+ 4-10 Не флуоресци-рует/синяя 1 %-ная дисперсия со- 85, 86
Hg2+ 4 То же ли натрия в порошке
Bi8+, Cd2+, Се8+, Fe2+, Fe8+, 1п8+, La8+, Ni2+, Pb2+, Th4+, Tl8+, Sn2+, Zr4+, обратное титрование |- Cu2+ 6 Синяя/не флуоресцирует KNO3
13 о-Дифенети-ДИН-N,N,N',N'-тетрауксус-ная кислота ноос—сн N— НООС—СН; 2 ос2н5 сн,—соон Q-N Н5С2°СН2—соон Cu2+ Hg2+ 4—10 4 He флуоресци-рует/сиияя То же То же 85, 86
14 З.З'-Диокси-бензидин-N,N,N'N'-тетрауксус-ная кислота ноос—сн 1 N— 1 ноос—сн 2 он 2 СН,—СООН Q-i НО сн— СООН Си2+ РЬ2+ Cd2+, Со2+, Мп2+, Zn2+, в смеси с родамином 6-8 6 6 » Розовая/синяя 88
15 3,3'-Дикарбо-ксибензидин-N,N,N',N'-тетрауксус-ная кислота ноос—н2с N-1 ноос- н2с 6 \ —V V" соон сн,—соон \=/ । ноос сн2 соон Ca2+ 10—13 Не флуоресци-рует/зеленая 85, 86
16 4,4'-Диамино-стильбен-N,N,N',N'-тетрауксус-ная кислота ноос—сн2 ноос—сн2 ^-СНг СН2— COOI -ch-q^A Chy—COOi Cu2+ Bi3+, Co21-, Fe2+, Ni2+, Pb2+, In3+, V4+, обратное титрова-ние+Си2+ 6 6 Не флуоресци-рует/синяя Синяя/не флуоресцирует 0,1%-ная дисперсия соли натрия в порошке KNO3 87
17 со со 4,4'-Диаминостильбен-2,2'-дисульфо-N.N.N'.N'-тетрауксус-ная кислота ноос— сн2 и 1 ноос— сн2 Vch= so3h сн,—соон 1 =СН-^_У“ N SO3H СН2—соон Си2+ Bi3+, Со2+, Fe2+, Ni2+, Pb2+, In3+, 1 V4+, обратное титрование Cu2+ 6 6 Не флуоресци-рует/синяя Синяя/не флуоресцирует То же 87
Если не имеется других указаний, титрование проводится раствором натриевой соли этилендиаминотетрауксусиой кислоты (ЫаЭДТА). ОЭТА — оксиэтилэтнлентриуксусная кислота; ЦДТА — трснс-циклогексан-1,2-диаминотетрауксусная кислота.
398
ГЛАВА IX
центные индикаторы», предложенный Кёрблем [63] для такого рода соединений.
Как и в случае флуоресцентных кислотно-основных индикаторов, для титрования мутных или окрашенных растворов предпочтительнее работать не с обычными, а с металлофлуоресцентными индикаторами. Их можно с успехом использовать, например, при определении некоторых ионов металлов (кобальта, меди), которые образуют интенсивно окрашенные комплексы с ЭДТА и другими комплексообразователями. Если используется обычный металло-хромный индикатор, то очень трудно определить конечную точку комплексометрического титрования ионов указанных металлов раствором ЭДТА, так как приходится работать с сильно разбавленными растворами.
В табл. IX.2 перечислены органические комплексообразующие реагенты, рекомендуемые в качестве металлофлуоресцентных индикаторов. Там же приведены условия процесса титрования, в которых их следует применять. Большинство индикаторов используется лишь при комплексометрическом титровании раствором ЭДТА, хотя некоторые соединения применяются и в случае других титрантов, нагример фторида.
Патровски [65, 66] впервые применил металлофлуоресцентные индикаторы в комплексометрии; он титровал галлий при pH 3,8 по тушению зеленой флуоресценции комплекса галлия с морином избытком ЭДТА. Индикатор был использован также при титровании индия раствором ЭДТА при pH 5.
Хольцбехер [67] применил ацетилгидразон салицилальдегида в комплексометрическом титровании пинка раствором ЭДТА в ацетате. Конечная точка устанавливалась по тушению синей флуоресценции комплекса цинка с ацетилгидразоном салицилальдегида. Мешающие ионы ртути и меди можно маскировать добавкой тиосульфата, a Al, Fe(III), Ti, Zr, La, Be, Ce(III) и Th — добавкой фторида. Хольцбехер [68] предложил также салицилиден-о-амино-фенол в качестве металлсфлуоресцентного индикатора в титровании алюминия раствором ЭДТА или фторида; в присутствии двух предложенных им индикаторов определялись одновременно цинк и алюминий в сплавах.
8-Оксихинолин был применен как металлофлуоресцентный индикатор для титрования галлия раствором ЭДТА при pH 2,5—3,5 в среде фталатного буфера [69]. Конечную точку определяли по исчезновению желто-зеленой флуоресценции ксмплекса галлия с 8-оксихинолином. Бадринас [70] использовал тушение желто-зеленой флуоресценции комплекса цинка с 8-оксихинолин-5-сульфокис-лотой при титровании пинка раствором ЭДТА. Бишоп [92] тоже использовал этот индикатор при титровании 1О-3 М растворов магния, кадмия и цинка 1,2-диаминоциклогексан-М,М,М',М'-тетраук-сусной кислотой (ДЦИТА) при pH 7. Этот же автср отмечает, что
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
399
комплексы кадмия и цинка с 8-оксихинолин-5-сульфокислотой можно использовать в качестве флуоресцентных кислотно-основных индикаторов в титровании 0,01 М гидроокиси аммония соляной кислотой. Бадринас с сотр. [71] предложил 7-(2-окси-4-сульфонаф-тилазо)-8-окскх'Инолин и его аналоги как металлофлуоресцентные индикаторы для определения ряда ионов металлов.
Черкесов [72] сообщил, что алюминий дает селективную и чувствительную реакцию с З-окси-2-нафтойной кислотой при pH 3, сопровождающуюся флуоресценцией. Свободная кислота флуоресцирует зеленым цветом; а комплекс ее с алюминием — синим. Этот реагент был использован [73] для ступенчатого титрования железа (III) и алюминия раствором ЭДТА. Сначала при pH 2 и комнатной температуре титруют железо(III). В конечной точке синяя окраска раствора переходит в желтую вследствие разложения синего комплекса индикатора с железом(III). Затем при pH 3 и температуре 50 °C алюминий титруют раствором ЭДТА при УФ-облучении до перехода синей флуоресценции в зеленую.
При обработке флуоресцеина формальдегидом и иминодиуксус-ной кислотой образуется смесь, называемая кальцеином [74], которая и используется в качестве металлохромного индикатора для титрования кальция; об изменении флуоресценции при образовании комплекса ничего не известно. Более чистый препарат кальцеина, переименованный в флуорексон [75], действует в конечной точке как флуоресцентный индикатор, а не как визуальный. При высоких значениях pH (>12) индикатор не флуоресцирует, а образует флуоресцирующие комплексы с кальцием, барием, стронцием и магнием. Сам индикатор может флуоресцировать, если его раствор довести до сильнощелочнсй реакции при титровании щелочноземельных металлов. Гидроокись калия вызывает совсем слабую флуоресценцию, поэтому ее можно использовать вместо гидроокиси натрия для подщелачивания раствора [76]. При рН<10 свободный индикатор флуоресцирует желто-зеленым светом; многие ионы металлов тушат эту флуоресценцию [77]. Магний можно титровать при pH 8 в пиридине с добавкой ацетатного буфера или при pH 10 в аммиачном буфере; медь оттитровывают подобным образом при pH 10. Уилкинс [78] применял кальцеин к титрованию сильно окрашенных растворов и определял хром (III), медь(II) и кобальт обратным титрованием избытка ЭДТА стандартным раствором меди. Природу двух, по-видимому, различных типов комплексов, образованных кальцеином, обсуждали Уилкинс [78], а также Кёрбль и Свобода [80]. Уилкинс считает, что тушение флуоресценции кальцеина ионами меди(II) указывает на образование «нормального комплекса», в котором кислород фенольной группы, ближайшей к дикарбоксиметиламинометильной группе, координирует с ионом металла. Предложено флуоресцирующий комплекс кальция, образующийся при высоком значении pH, называть «обратимым
•400
ГЛАВА IX
комплексом индикатора». Считается, что в этом комплексе кислород фенольной группы не присоединен к иону металла. Кёрбль и Свобода [80] предложили другое объяснение образования двух типов хелата. Они предполагают, что тушение флуоресценции при низких pH является результатом образования комплексов металлов, в которых катион непрочно связан ионной связью с фенольным кислородом. При высоком значении pH они объясняют образование флуоресцирующих комплексов с Щелочноземельными металлами возникновением ковалентной связи между катионом и фенольным атомом кислорода.
Эггерс [81] получил умбеллиферонкомплексан конденсацией 4-метилумбеллиферона с формальдегидом и иминодиуксусной кислотой. Уилкинс [82] тоже получил это соединение и назвал его кальцеиновым синим, так как он дает ярко-синюю флуоресценцию при pH ниже 12, а при более высоких значениях pH не флуоресцирует. Флуоресценцию тушит медь(II) (при pH 4—10), а также кобальт, магний и никель (в щелочном растворе). В присутствии кальцеинового синего при избытке ЭДТА и обратном титровании этого избытка раствором меди можно с успехом определять никель, кобальт, медь, железо(III), титан (в присутствии перекиси водорода), алюминий, магний, цинк и ртуть. Уилкинс [82] описал ступенчатое титрование никеля и хрома. Хром реагирует очень медленно с ЭДТА при pH 4,8 и комнатной температуре, а никель в тех же условиях реагирует практически мгновенно. Поэтому добавляют избыток ЭДТА и быстро определяют содержание никеля в растворе методом обратного титрования; снова добавляют избыток ЭДТА, кипятят раствор для полного взаимодействия с хромом и определяют хром обратным титрованием непрореагировавшего ЭДТА. При высоком pH (>12) сам индикатор не флуоресцирует, а образует флуоресцирующие «обратимые комплексы индикатора» с щелочноземельными металлами. Кальций, барий и стронций можно прямо титровать при рН>12; конечную точку устанавливают по тушению флуоресценции комплексов индикатора с щелочноземельными металлами. В качестве .металлофлуоресцентного индикатора кальцеиновый синий эффективнее кальцеина, потому что максимум его флуоресценции (при 445 нм) получается вследствие возбуждения светом с длиной волны 370 нм. Величина 370 нм ближе к максимуму излучения источников УФ-света со ртутными лампами (366 нм), чем длина полны возбуждения кальцеина.
Уилкинс [83] описал применение двух новых индикаторов: ме-тилкальцеина и метилкальцеинового синего в методе хелометриче-ского определения алюминия, никеля и марганца без их предварительного разделения. Эти индикаторы являются продуктами конденсации флуоресцеина и 4-метилумбеллиферона с формальдегидом и N-метилглицином. В сообщении не имеется сведений о ме
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
401
таллофлуоресцентных свойствах этих индикаторов. Их использовали Для установления конечной точки при титровании оксиэтил-этилентриуксусной кислоты раствором меди при pH 5 и при титровании магния раствором ЭДТА в условиях pH 9,5. Замещение одной из карбоксильных групп в кальцеине или кальцеиновом синем на метильную группу, которая не образует хелаты, понижает константу устойчивости комплексов индикатора с медью; вследствие этого предпочтительнее применять кальцеин и кальцеиновый синий при титровании комплексанами, которые образуют более слабые хелаты с металлами, чем ЭДТА. В работе [84] предложен улучшенный вариант метода селективного определения титана в присутствии ниобия и тантала, состоящий в обратном титровании избытка транс-циклогексанНД-диамино-М.М.Й'.М'-тетрауксусной кислоты раствором меди при участии перекиси водорода с использованием метилкальцеина или метилкальцеинового синего в качестве металлофлуоресцентных индикаторов [84].
Эггерс [81] синтезировал ксантонкомплексан конденсацией 3,6-диоксиксантона, Формальдегида и иминодиуксусной кислоты Ксантонкомплексан обладает свойствами металлофлуоресцентных индикаторов подобно кальцеиновому синему; синюю флуоресценцию при pH 9—9,5 в боратном буфере тушат медь(II), никель, кобальт, железо(II), магний, ртуть(II) и висмут. При рН~ 13 в 0,5 М КОН не флуоресцирующий индикатор образует флуоресцирующие комплексы с щелочноземельными металлами и слабо флуоресцирующие комплексы с цинком, кадмием и свинцом.
Белчер и др. [85, 86] предложили о-дианизидин-М,М,М',М'-тет-рауксусную кислоту в качестве флуоресцентного индикатора для титрования меди (II) при pH 4—10 и ртути(II) при pH 4—6 раствором ЭДТА. Катионы этих металлов тушат голубую флуоресценцию индикатора, которая снова появляется в точке эквивалентности при титровании раствором ЭДТА. В случае определения меди (II) получается очень четкая конечная точка, а для ртути(II) она несколько хуже. Титрованию мешают Bi3+, Cd2+, Се2+, Са2+, Fe2+, Fe3+, Hg2+,'ln3+, La3+, Ni2+, Pb2+, Th4+, Tl3+, Sn2+ и Zr4 так как на них расходуется титрант. Однако эти ионы не оказывают влияния на четкость определения конечной точки; их можно определить, добавив в избытке ЭДТА в раствор и титруя избыток стандартным раствором меди(II). Эти же исследователи синтезировали также о-дифенетидин-М.Н.Н'.Н'-тетрауксусную кислоту, которая проявляет свойства металлофлуоресцентного индикатора подобно о-дианизидинпроизводному; новое соединение рекомендовали в качестве флуоресцентного индикатора для титрования меди (II) и ртути(II) раствором ЭДТА. В тех же сообщениях [85, 86] авторы описывают синтез 3,3'-дикарбоксибензидин-М,М,М',М'-тетрауксусной кислоты и ее 'использование при титровании кальция. В щелочном Растворе (pH 10—13) флуоресценция этого индикатора тушится
26—918
402
ГЛАВА IX
кальцием, а в точке эквивалентности при титровании раствором ЭДТА она снова появляется.
Киркбрайт и др. [87] получили 4,4/-диаминостильбен-М,М,М',М/-тетрауксусную кислоту и ее 2,2'-дисульфопроизводное. Интенсивная синяя флуоресценция этих соединений тушится медью(II) при титровании раствором ЭДТА в условиях pH 6. Обратным титрованием избытка ЭДТА стандартным раствором меди(II) определяли Bi3+, Со3-1-, Fe2+, Ni2+, Pb2+, 1п3+ и V4+. В водном растворе эти индикаторы флуоресцируют гораздо интенсивнее, чем 3,3'-дизамещен-ные бензидины, по-видимому, вследствие большей сопряженности молекулы стильбена. Высокая чувствительность рассматриваемых индикаторов и возможность применения их в низких концентрациях позволяют работать с очень разбавленными титруемыми растворами. Даже в случае 0,001 М растворов меди(II) и ЭДТА точность результатов совсзм незначительно отличается от точности при работе с 0,01 М растворами, если учитывать поправку на холостой опыт в присутствии индикатора, тогда как при использовании визуального индикатора бывает исключительно трудно определить с любой степенью точности положение конечнсй точки.
ЗД'-Диокси'бензидин-Г^М.ЬГ.М'-тетрауксусную кислоту [88] можно использовать как индикатор при прямом титровании меди (II) раствором ЭДТА в области pH 4—10 и свинца при pH 6. Наиболее четкая конечная точка определения меди(II) получается при pH 6—8. Кадмий, кобальт(П), марганец(П) и цинк можно также титровать непосредственно при pH 6, хотя конечные точки менее четки из-за остаточной флуоресценции раствора перед наступлением конечной точки. Добавкой нескольких капель 0,01 %-кого раствора родамина В можно экранировать остаточную флуоресценцию. Тогда конечная точка распознается по изменению цвети флуоресценции раствора от розового до синего.
Киркбрайт и др. [90] исследовали реакции ионов металлов с флуоресцирующими производными М,М,М',М'-тетрауксусной кислоты, а именно с пятью 4,4'-дизамещенными 2,2'-диаминодифенилами. Эти соединения 'взаимодействуют с ионами металлов с образованием устойчивых нефлуоресцирующих хелатов состава 1:1. Была исследована возможность их использования в качестве металлофлуоресцентных индикаторов для титрования растворами ЭДТА и транс-циклогексан- 1,2-дигмино-К,М,М/,М'-тетрауксус11ОЙ кислоты ШДТА) и для определения никеля с помощью флуориметра. Высокая устойчивость образованных хелатов металлов позволяет использовать некоторые из этих соединений в качестве автоиндика-торных титрантов, хотя при этом получаются нечеткие конечные точки и неизбежно происходит перетитрование.
Установлено, что в комплексометрическом титровании у многих из исследованных соединений, предложенных в качестве металлофлуоресцентных индикаторов, наблюдается остаточная флуоресцен
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
403
ция перед наступлением конечной точки или после нее. Это ухудшает определение конечной точки Остаточная флуоресценция может обусловливаться присутствием неактивных флуоресцирующих примесей в индикаторе, которые могли появиться как загрязнения в процессе синтеза индикатора или образоваться при разложении индикатора во время его хранения. Слабая флуоресценция комплексов кальцеина с ионами щелочных металлов при высоких значениях pH также может быть причиной остаточной флуоресценции. Киркбрайт [89] предложил остаточную флуоресценцию устранять при помощи нейтрального (не образующего комплексы) флуоресцирующего красителя, флуоресценция которого отличается от остаточной флуоресценции. Так, при титровании кальция раствором ЭДТА в присутствии кальцеина при pH 12 или выше в конечной точке наблюдается желто-зеленая флуоресценция. Добавка нескольких капель 0,01%-ного водного раствора акридина в начале титрования обеспечивает четкое изменение окраски в конечной точке, а флуоресценция раствора изменяется от светлой желто-зеленой до ясно-синей. Аналогично, если использовать кальцеиновый синий в том же процессе титрования, остаточную синюю флуоресценцию можно эффективно экранировать при помощи родамина В. Конечная точка отмечается четким изменением флуоресценции раствора от светло-синей до оранжево-розовой. Как говорилось выше, процесс экранирования можно также использовать для расширения области применения 3,3'-диоксибензидин-М,М,М',М'-тетрауксусной кислоты в качестве металлофлуоресцентного индикатора.
СПИСОК ЛИТЕРАТУРЫ
1. Bowen Е. J., Quart. Revs., 1, 1 (1947).
2. Parker С. A., Rees Г. Т„ Analyst, 85, 587 (1960); 87, 83 (1962).
3. Belcher R., Nutten A. J., Quantitative Inorganic Analysis, 2nd ed., Vol. IID, Butterworths Scientific Publ., London, 1975, p. 174.
4. Grant J., J. Sci. Inst., 9, 359 (1932).
5. Colombier L„ Ann. Falsif., 24, 89 (1931).
6. Linser H., Biochem. Z., 244, 157 (1934).
7. Goto H., Sci. Rep. Tohoku Imp. Univ. Ser. I, 28, 458, 465, 513 (1940); 29, 1 (1940).
8. Tomicek O., Chemical Indicators, Butterworths Scientific Publ., London, p. 203.
9. de Ment J., J. Chem. Ed., 30, 145 (1953).
10. Szebelledy L., Sik K., Magyar Gyogy. Tarsasag Ert., 14, 383, 387, 391, 394, 397, 401, 405, 409, 413, 418 (1938).
11. Tomicek O., Coll. Czech. Chem. Comms., 13, 116 (1948).
12. Jensen R. A., Z. anal. Chem., 94, 177 (1933).
13. Jensen J\. A., Z. anal. Chem., 117, 50 (1939).
14. Nasini A. G., de Cori P., Atti III Congr. Naz. Chim. pure ed appl., Firence a Toscana, 1929, 698; 1930, 668.
15. Deribere M., Ann. chim. anal., 18, 37, 120, 173, 289 (1936); 19, 262, 290 (1937); Ind. Chim., 24, 163 (1937); Z. anal. Chem., 116, 341 (1939).
16. Suk V., Thesis, Charles Univ., Prague, Czechoslovakia, 1950.
26*
404
ГЛАВА IX
17. Tomicek О., Col. Czech Chem. Comms., 13, 116 (1948).
18. Bravo A. G„ Ind. Chimica, 4, 8 (1929).
19. Rocsis E. A., Pettko E., Z. anal. Chem., 124, 45 (1942).
20. Salm E., Z. phys. Chem., 67, 471 (1901).
21. Marinelli G., Ann. chim. appl., 31, 415 (1941); C. A., 37, 5331 (1943).
22. I\ocsis E. A., Nagy Z. Sv., Z. anal. Chem., 108, 317 (1937).
23. Eisenbrand J., Pharm. Z., 75, 1033 (1930).
24. I\ocsis E. A., Biro K., Z. anal. Chem., 122, 94 (1942).
25. Mellet A., Bischoff M. A., C.r. acad. sei., Paris, 182, 1616 (1926).
26. Volmar M. У., Widder E., Bull. soc. chim. France, 43, 813 (1928); 45, 130, 696 (1929).
27. Nocsis E. A., Zador G., Z. anal. Chem., 42, 124 (1942).
28. Kocsis E. A., Zador G.. Z. anal. Chem., 124, 274 (1942).
29. Marshall T. M. B., J. Soc. Dyers and Col., 52, 299 (1936).
30. Szebelledy L, Jonas J., Z. anal. Chem., 113, 266, 326, 422 (1938).
31. Podall H., Anal. Chem., 24, 424 (1952).
32. Kostir J. V., Nature, London, 157, 586 (1946).
33. Briner E., Perrottet E., Helv. Chim. Acta, 23, 1253 (1940).
34. Robl R., Ber., 59, 1725 (1926).
35. Kocsis E. A., Zador G., Kallos J. F., Z. anal. Chem., 126, 138, 177 (1943).
36. Fleck H. R., Holness R. F. G., Ward A. M., Analyst, 60, 32 (1935).
37. Billow C., Dieck W., Z. anal. Chem., 75, 81 (1928).
38. Ismailov N. A., Schraiber M. S., Farmaz. Farmakol., 4, 8 (1938).
39. Eisenbrand J., Z. phys. Chem., A., 144, 441 (1929).
40. Clark A. B„ Lubbs H. A., J. Bacteriol., 2, 1, 109, 171 (1917).
41. Deska L. J., Sherill R. E., Harrison L. M., J. Am. Chem. Soc., 48, 1493 (1926).
42. Volmar Y., Clavera J. M., J. Pharm. Chim., 13, 561 (1931).
43. Volmar Y., Clavera J. M., Anales Fisica у Quimica, 29, 247 (1931); Brit. C. A., B., 693 (1931).
44. Gallart J. M., Anales Fisica у Quimica, 29, 490 (1931); Brit. С. A., B, 1117 (1931).
45. Clavera J. M., Anales fisica у quimica, 29, 484 (1931).
46. Borgialli A., Bull. uff. r. staz. sperim. ind. pelli mat concianti, 13, 245 (1935).
47. Maloman S., Chem. Ztg., 57, 755 (1933).
48 Hessler W., Marsen H., Fette, Seifen, Anstrichmittel, 62, 574 (1960); 63, 924 (1961); C.A., 55, 3094a (1961).
49. Dolezil M., Bulander J., Chem. Listy, 51, 255 (1957); C. A., 51, 6425g (1957).
50. Божеволнов E. А., Серебрякова Г. В., Труды Всесоюзн. научн.-исслед. ин-та химии реактивов, № 23, 116 (1959).
51. Michalski Е., Turowska М., Chem. Anal. (Warsaw), 3, 599 (1958); С. А., 54, 3047i (1960).
52. Sljivic S., Baric I., Nikolic K., Z. anal. Chem., 145, 16 (1955).
53. Del Campo A., Sierra F., Ann. soc. espan, fisica quim., 33, 364 (1935); Z. anat Chem., 104, 279 (1937).
54. Holzbecher Z., Chem. Listy, 52, 425 (1958); Coll Czech. Chem. Comms., 24, 1451 (1959).
55. Goto H„ J. Chem. Soc. Japan, 59, 1357 (1938); C. A., 33, 2062 (1939).
56. Goto H„ Sci. Reps. Tohoku. Univ., (1), 29, 204, 446 (1940).
57. Goto H., J. Chem. Soc. Japan, 63, 470 (1942).
58. Geyer R., Steinmetzer H., Angew. Chem., 72, 634 (1960).
59. Sagi S., Rao G. G., Taianta, 5, 154 (1960).
60. Dikshitulu L. S. A., Rao G. G„ Taianta, 9, 289 (1962).
61. Rao G. G., Sagi S., Z. anal. Chem., 188, 164 (1962).
62. Kratochvil B., Zatko D. A., Anal. Chem., 36, 527 (1964).
63. Forb! J., Svoboda V., Taianta, 3, 370 (1960
64 Wilkins D. H., Taianta, 4. 80 (I960).
ФЛУОРЕСЦЕНТНЫЕ ИНДИКАТОРЫ
405.
65. Patrovsky V., Chem. Listy, 47, 676, 1338 (1953).
66. Dolezal J., Patrovsky V., Sulcek Z., Svasta J., Chem. Listy, 49, 1517 (1955)" Coll. Czech. Chem. Comms., 21, 979 (1956).
67. Holzbecher Z., Chem. Listy, 52, 430 (1958).
68. Holzbecher Z., Chem. Listy, 52, 1822 (1958); Coll. Czech. Chem. Comms., 24, 1457 (1959).
69. Crawley R. H. A., Anal. Chim. Acta, 19, 540 (1958).
70. Badrinas A., Publ. Inst. Biol. Appl. Barcelona, 28, 75 (1958); C. A., 53, 18754г (1959).
71. Martinez F. D., Badrinas A., Prieto Bouza A., Inform, quim. anal., 14, 151 (1960).
72. Черкесов А. И., ДАН СССР, 118, 309 (1958).
73. Kristiansen H., Anal. Chim. Acta, 25, 513 (1961).
74. Diehl H., Ellingboe J. L., Anal. Chem., 28, 882 (1956).
75. Kbrbl J., Vydra F., Chem. Listy, 51, 1457, (1957); Coll. Czech. Chem. Comms.,.
23, 622 (1958).
76. Korbl J., Vydra F., P^ibil R., Taianta, 1, 281 (1958).
77. Vydra F., Pribil R., Korbl J., Col. Czech. Chem. Comms., 24, 2623 (1958).
78. Wilkins D. H., Anal. Chim. Acta, 20, 323 (1959).
79. Wilkins D. H., Tai anta, 2, 277 (1959).
80. Korbl J., Svoboda V., Taianta, 3, 370 (1960).
81. Eggers J. H., Taianta, 4, 38 (1960).
82. Wilkins D. H.. Taianta, 4, 182 (1960).
83. Wilkins D. H., Anal. Chim. Acta, 23, 309 (1960).
84. Lassner E., Scharf R., Chemist-Analyst, 51, 49 (1962).
85. Belcher R., Rees D. I., Stephen IT. I., Taianta, 4, 78 (1960).
86. Rees D. I., Stephen W I., J. Chem. Soc., 1961, 5101.
87. Kirkbright G. F., Rees D. I., Stephen W. I., Anal. Chim. Acta, 27, 558 (1962)..
88. Kirkbirght G. F., Stephen W. I., Anal. Chim. Acta, 28, 327 (1963).
89. Kirkbright G. F, Stephen W. I., Anal. Chim. Acta, 27, 294 (1962).
90. Kirkbright G. F., Stephen U7 I., Anal. Chim. Acta, 32, 544 (1965).
91. Kratochvil B., White M. C., Anal. Chim. Acta, 31, 528 (1964).
92. Bishop J. A., Anal. Chim. Acta, 29, 178 (1963).
ГЛАВА X
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
Л. Эрдеи
ВВЕДЕНИЕ
Излучение света, сопровождающее химические реакции, называется хемилюминесценцией. В этих реакциях часть освобождающейся энергии выделяется из системы в виде протонов, хотя в то же самое время температура системы существенно отличается от соответствующей температуры светового излучения. Поэтому хемилюминесценция— это «холодный свет», т. е. она вызывается нетепловой релаксацией возбуждения. С точки зрения химических индикаторов наиболее важными хемилюминесцентными процессами являются процессы, происходящие в водных растворах. К хемилюминесцентным системам относятся сложные молекулы, содержащие один электрон на молекулярной орбитали, который способен легко возбуждаться. В таких молекулах либо самопроизвольно, либо под влиянием сильных парамагнитных ион-радикалов (например, О- или О2), либо под влиянием растворенного кислорода одна из пар электронов, образующих двойную связь, возбуждается и переходит из синглетного в возбужденное триплетное состояние Это значит, что пара электронов, имеющих первоначально анти-параллельный спин, переходит в триплетное состояние, которому соответствует параллельный спин. В таком возбужденном триплетном состоянии электрон гораздо устойчивее, чем в возбужденном синглетном состоянии, потому что по принципу Паули вероятность возвращения электрона из триплетного состояния в синглетное очень мала. Устойчивость возбужденной молекулы еще более возрастает вследствие того, что она образует трансаннулярные перекиси с газообразным кислородом, перекисями или свободными радикалами кислорода, присутствующими в системе, и при этом освобождается очень немного энергии. Затем образовавшиеся перекиси, расходуя энергию активации при столкновениях молекул, разрушаются, а возбужденный электрон в триплетном состоянии возвращается в невозбужденное состояние, что сопровождается испусканием хемилюминесцентного холодного света. В зависимости ют экспериментальных условий, таких, как присутствие катализате-
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
407
ра или веществ, блокирующих свободные радикалы, возможен перенос электрона без светового излучения.
Химические процессы, приводящие к испусканию энергии хемилюминесценции, обычно являются окислительно-восстановительными реакциями, а энергия реакции окисления переводит молекулу в состояние возбуждения. Такой молекулой может быть соединение» не участвующее в реакции, или периодически регенерируемое вещество, или же продукт окисления, который обладает свойством испускать свет. Свечение, т. е. хемилюминесценция, происходит в присутствии данного окислителя, если при изменении pH начинается реакция. В таком случае эту реакцию можно использовать в качестве кислотно-основного индикатора. Однако часто хемилюминесцентная реакция начинается только тогда, когда при данном значении pH стандартный раствор окислителя добавлен в тако л количестве, что окислительно-восстановительный потенциал раствора возрос до нужного значения. Отсюда следует, что хемилюминесцентную реакцию 'можно использовать также в качестве окислительно-восстановительного индикатора. Если окислительно-восстановительную систему использовать в качестве титранта, который образует осадок с веществом, участвующим в титровании, то появление и исчезновение хемилюминесценции можно использовать для установления конечной точки, т. е. можно применять хемилюминесцентные вещества в качестве индикаторов при титровании по> методу осаждения. Аналогично если происходят реакции комплексообразования, то хемилюминесцентные вещества могут служить индикаторами в комплексометрическом титровании. Хемилюминесцентные реакции идут до тех пор, пока не израсходуется индикатор или окислитель. Следовательно, процессы с участием индикатора часто бывают необратимыми.
Из механизма хемилюминесцентных реакций следует, что они очень чувствительны к присутствию посторонних веществ, которые действуют как катализаторы или ингибиторы. Эти вещества чаете могут вызывать очень сильное свечение, поэтому могут быть полезны, но во многих случаях они тушат хемилюминесценцию. Чувствительность хемилюминесцентных систем к катализаторам чаете используют для целей каталитического анализа [1—6].
Хемилюминесцентные индикаторы, используемые в объемном анализе, имеют преимущества перед флуоресцентными индикаторами, так как для их применения нет необходимости в источниках УФ-света или какой-либо другой специальной аппаратуре. Конечная точка титрования, устанавливаемая по появлению или прекращению свечения, легко наблюдается в темной комнате без помощи специального прибора, если глаз привык к темноте. Глаз должен привыкнуть к темноте в течение нескольких минут, особенно если-наблюдатель входит из ярко освещенной комнаты в темную, пото
408
ГЛАВА X
му что глаз начинает различать фиолетовый цвет только через некоторое более длительное время.
Титрование в темноте, а также точное определение конечной точки из-за неизвестной скорости добавления титранта составляют известные трудности. Чтобы можно было легко добавлять титрант
Рис. Х.1. Бюретка с краном, снабженным ограничителем.
по каплям, рекомендуется работать с краном, снабженным специальным ограничителем (рис. Х.1). При положении А крана раствор
вытекает струйкой, а при положении Б медленно вытекает по кап-
Рис. Х.2. Титриметр с темной камерой.
лям. Поэтому в начале титрования кран устанавливают в положение А, а вблизи конечной точки — в положение Б. Ограничитель вырезают из 2-миллиметровой алюминиевой полоски и укрепляют двумя болтами на кране бюретки. Рекомендуется сделать под металлом тонкую резиновую прокладку.
Титрование следует проводить в темной комнате. Вместо этого можно работать с темной камерой [7, 8], удобная форма которой показана на рис. Х.2. Бокс
можно сделать из картона,
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
409
а через его светонепроницаемую скользящую дверцу вносят внутрь колбу с титруемым раствором. В колбу помещают небольшой кусочек железа, запаянный в стеклянную трубку, которым перемешивают раствор с помощью магнитной мешалки, находящейся под боксом. Нижняя часть бюретки, входящая в бокс, должна быть зачернена. Точное определение конечной точки требует известного опыта.
ФОТОМЕТРИЧЕСКОЕ И АВТОМАТИЧЕСКОЕ ТИТРОВАНИЕ
Наблюдение световых эффектов, происходящих в конечной точ-
ке при хемилюминесцентном титровании, будет более эффективным,
если вместо визуального способа использовать фотовольтовый при-
бор [7], т. е. комбинированный прибор фотоумножителя с фотометром. В таком случае устраняется субъективная ошибка, а кроме того, появляется возможность работать с меньшими количествами индикаторов, экономя довольно дорогостоящее хемилюминесцентное вещество.
Применение хемилюминесцентных индикаторов позволило сконструировать автоматические титриметры, в которых нет необходимости погружать в исследуемый раствор индикаторные электроды или зонды для
220 У
Рис. Х.З. Блок-схема устройства для титро-
вания.
измерения электропроводности Можно создать безэлек-
тродный автоматический ап-
парат для титрования. Если хемилюминесцентное свечение, появляющееся в конечной точке титрования, регистрировать при помо-
щи фотоэлемента или фотоумножителя, чувствительного в данной области спектра, ток на выходе можно усилить и сработает система реле, контролирующая цепь высокого напряжения. Электромагнит в этой цепи открывает или перекрывает кран бюретки. Блок-схема такого устройства [45] показана на рис. Х.З. Этот аппарат пригоден для титрования в присутствии люцигенина. Для синевато-зеленой хемилюминесценции люцигенина наиболее чувствителен фото-
элемент из сурьмы — окиси цезия, поскольку кривая чувствительности такого элемента имеет максимум при 420 нм, что близко
410
ГЛАВА X
к длине волны максимума интенсивности синего хемилюминесцентного свечения люцигенина. Кислоту наливают в колбу 5 и титруют основанием, которое находится в бюретке 1. Под колбой помещается сурьмяный фотоэлемент 6, фототок которого трансформируется в переменный ток при помощи мультивибратора; этот ток можно усилить в усилителе 7, а затем выпрямить. Усиленный выходной ток заставляет действовать реле 4, которое включает сильный ток через спираль 3, чтобы закрыть магнитный кран 2 бюретки. Если нет хемилюминесцентного свечения, кран 2 открыт и стандартный раствор течет в колбу до тех пор, пока в конечной точке не установится стойкая хемилюминесценция, после чего магнитный кран бюретки перекроется. Чтобы аппарат работал, естественно, нужна минимальная интенсивность хемилюминесценции, которая соответствует небольшой 'индикаторной ошибке. Чувствительность аппарата можно регулировать потенциометром, и с его помощью можно наладить аппарат на такую малую интенсивность света, чтобы он давал сигнал, надежно отличающийся от шумовых помех. Индикаторная ошибка такого аппарата соответствует +0,09 мл 0,1 н. стандартного раствора; ее следует вычитать из конечного значения. При использовании люцигенина рекомендуется добавлять к титруемому раствору этанол в качестве катализатора, потому что он смещает максимум интенсивности испускаемого света в сторону синей области, в которой чувствительность слоя сурьмы в фотоэлементе значительно выше. Аппарат для автоматического титрования можно снабдить прибором для прямого отсчета показаний, самописцем или прибором для дистанционного наблюдения.
ПРИМЕНЕНИЕ ХЕМИЛЮМИНЕСЦЕНТНЫХ ИНДИКАТОРОВ В КИСЛОТНО-ОСНОВНОМ ТИТРОВАНИИ
В качестве хемилюминесцентных кислотно-основных индикаторов применяются те системы, которые хемилюминесцируют только при данном значении pH, а ниже этого значения свечение прекращается. Для этих целей наиболее подходящими оказываются лю-цигенин, люминол и лофин [9], окисление которых при данных значениях pH вызывает явление хемилюминесценции. При окислении этих восстановителей высвобожденная энергия будет возбуждать молекулу или один из продуктов реакции. При стабилизации эта энергия частично испускается в виде хемилюминесцентного излучения. Свечение продолжается, естественно, до тех пор, пока не израсходуется весь индикатор или окислитель. Поэтому индикатор обратим только в очень ограниченной степени. Хемилюминесцентные реакции очень чувствительны к каталическому действию ионов тяжелых металлов Во многих случаях они катализируют одну ста
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
411
дию хемилюминесцентной реакции, вызывающей очень сильное свечение, тогда как в других случаях они разрушают промежуточные переходные перекиси и таким образом тушат люминесценцию.
ЛЮЦИГЕНИН (ДИМ :ТИЛДИАКРИДИЛДИНИТРАТ) В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО КИСЛОТНО-ОСНОВНОГО ИНДИКАТОРА
Хемилюминесценция люцигенина была тщательно изучена. Ее можно объяснить следующим образом [10]: карбинольное основа-
метилакридо»
ние I [уравнение (1)] образует с азотной кислотой растворимую в воде соль. Водный раствор люцигенина проявляет интенсивную зеленую флуоресценцию в УФ-свете [11, 12]; его можно хранить в течение многих дней. В щелочном растворе с добавкой перекиси водорода люцигенин испускает зеленоватый хемилюминесцентный свет в продолжении двух часов. Это явление можно повторить, если прилить к раствору еще немного перекиси водорода. Другие окислители не вызывают хемилюминесценции*. В зависимости от кон
* При автоокислении некоторых восстановителей (аскорбиновая кислота, сернистая кислота, дитионит натрия) тоже образуется перекись водород?, и ее можно обнаружить в щелочных растворах с помощью люцигенина [13, 14]. В сильно-Щелочных растворах (pH > 12) люцигенин сам окисляется кислородом воздуха с образованием перекиси водорода, следовательно, такие растворы имеют очень слабую хемилюминесценцию даже без добавки перекиси водорода.
"412
ГЛАВА X
центрации и свойств присутствующих в системе катализаторов можно наблюдать хемилюминесценцию при pH 8,6—9,4. При рН>12 она становится особенно интенсивной. Усиление хемилюминесценции с ростом pH связано с повышением скорости диссоциации перекиси водорода:
Н2О2 =?==>: Н+ + ООН- (КР~10-1г)
Интенсивность хемилюминесценции пропорциональна концентрации пергидроксильных ионов*. При высоких концентрациях лю-хигенина, а также в присутствии катализаторов (этанол, четырех-•окись осмия) первоначальное желтовато-зеленое свечение становится синим. Исследование спектра излучаемого света [11, 12, 16— 19] показало, что спектр желтовато-зеленого света идентичен спектру излучаемой флуоресценции люцигенина, а спектр синей люминесценции подобен спектру метилакридона, который является продуктом окисления люцигенина. Поэтому можно предположить, что эти два вида излучаемого света испускаются двумя типами молекул в результате возбуждения энергией, выделяемой при окислительно-восстановительной реакции. Согласно уравнению (1), реакция начинается с карбинольного основания I, которое при взаимодействии с пергидроксильными ионами образует перекись люци-тенина II. Соединение II выделяет кислород в ходе медленной реакции и превращается в возбужденную молекулу III, в которой оба неспаренных электрона находятся в триплетном состоянии. Затем эти электроны переходят в синглетное состояние и испускается желтовато-зеленое флуоресцирующее излучение. Образовавшийся диакриден окисляется затем пергидроксильными ионами до окиси люцигенина, которая присоединяет воду и переходит в исходное карбонильное основание I. Серия реакций начинается снова, и никаких новых изменений не происходит, кроме того, что разлагаются два пергидроксильных иона, а высвобождаемая при этом энергия выделяется частично в виде хемилюминесцентного излучения. Следовательно, люцигенин действует как катализатор реакции разложения перекиси и одновременно испускает свет. Свечение продолжается долго, а количество света зависит практически от количества присутствующей перекиси водорода. Это обстоятельство позволяет обратимо использовать люцигенин в качестве индикатора. При высоких концентрациях люцигенина и пергидроксильных ионов, а также в присутствии катализаторов (спирт, четырехокись осмия) происходит необратимая реакция. В этих условиях возрастает возможность столкновений молекул перекиси люцигенина II.
* Это явление привело к правильному объяснению саморазложения перекиси водорода « люцигенина [15]. Саморазложение перекиси водорода можно объяснить реакцией Н2О2+ООН_=Н2О+О2+ОН_. Поэтому максимальное саморазложение перекиси водорода происходит при pH 12. Реакция разложения проходит через стадию образования комплекса Н2О2-ООН_.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
413
При этом атом кислорода переходит от одной молекулы к другой. В результате образуются бирадикал III и молекула с избыточным кислородом, которая разлагается на трансаннулярную перекись V и метилакридон VII. Перекись V превращается в возбужденный бирадикал VI при отщеплении кислорода и, испуская синее хемилюминесцентное излучение, характерное для метилакридона, переходит в конце концов полностью в метилакридон VII. Последний является нехемилюминесцирующим неактивным конечным продуктом. Побочная реакция вызывает в присутствии катализаторов смещение спектра хемилюминесценции в сторону синей области, а при наличии соответствующего количества перекиси водорода весь люцигенин превращается в метилакридон. В этих реакциях не участвуют свободные радикалы, образующиеся из перекиси водорода, а поэтому все катализаторы, способствующие образованию радикалов (подобно промежуточным комплексам тяжелых металлов), катализируют не хемилюминесценцию люцигенина, а только разложение перекиси. Недавние исследования реакции разложения люцигенина [47, 48] и некоторых его производных [49] доказали правильность этого механизма.
ЛЮЦИГЕНИН В КАЧЕСТВЕ ОБРАТИМОГО КИСЛОТНО-ОСНОВНОГО ХЕМИЛЮМИНЕСЦЕНТНОГО ИНДИКАТОРА [20]
Люцигенин отличается следующими преимуществами перед другими хемилюминесцентными кислотно-основными индикаторами: 1) его можно применять для прямого титрования, так как процесс с индикатором обратим; 2) с ним можно работать в отсутствие катализатора; 3) индикаторная смесь не имеет буферного действия; 4} на одно титрование достаточно очень небольшого количества люцигенина (0,5—5 мг). Переход индикатора наблюдается при значении pH 9,5, а в растворах, содержащих 5% этанола, это значение снижается до 8,5. (Следовательно, люцигенин можно использовать во всех тех случаях, для которых подходит фенолфталеин.) Он чувствителен к двуокиси углерода, поэтому необходимо работать со стандартными растворами щелочей, не содержащими карбонатов. В нагретых растворах интенсивность хемилюминесценции гораздо сильнее. Интенсивность свечения можно усилить, повысив концентрацию перекиси водорода. Разумеется, что кислоту, добавляемую к перекиси водорода для его стабилизации, необходимо предварительно нейтрализовать.
Приготовление растворов
Люцигенин. Приготавливают 0,5%-ный водный раствор. На одно титрование берут 0,5—1,0 мл раствора. Если к титруемому образцу добавить 5--10 мл этанола, то достаточно 3—10 капель индикатора.
414
ГЛАВА X
Перекись водорода. Приготавливают 3%-ный раствор путем разбавления продажной 30%-ной перекиси водорода в соотношении 1 :9. Содержание свободной кислоты в растворе определяют на большем образце и учитывают в виде поправки при титровании. На одно титрование надо брать 5 мл 3%-ной перекиси водорода. Содержание свободной кислоты в этом количестве перекиси дает ошибку не более 0,03 мл нормального раствора титранта.
Методика определения
К исследуемому раствору сильной или слабой кислоты либо сильного основания добавляют 0,5—1,0 мл 0,5%-ного индикатора люцигенина и 5 мл раствора перекиси водорода, затем титруют его в темноте из бюретки с ограничителем на кране. Во время титрования кислот наблюдается появление коротких вспышек света, которые все медленнее исчезают по мере приближения к конечной точке. В самой конечной точке весь раствор светится равномерно. В ходе титрования оснований светится исходный раствор, его свечение постепенно пропадает, а в конечной точке, даже если добавить часть капли, свечение исчезает. В приблизительно 1 н. щелочах люцигенин испускает свет еще до прибавления перекиси водорода. В таких случаях обязательно надо титровать быстро, потому что диакриден [соединение III в уравнении (1)] выпадает из раствора в виде желтого осадка. Однако это не сказывается на точности результатов.
Для экономного расходования достаточно добавлять только 3— 10 капель индикатора, но в таком случае к титруемому раствору надо прилить 5—10 мл этанола.
Индикатор можно с успехом использовать для титрования 1,0— 0,01 н. щелочей, насыщенного раствора гидроокиси кальция, соляной, серной, азотной, лимонной и винной кислот, борной кислоты (в виде комплекса с инвертным сахаром), уксусной и щавелевой кислот, а также для определения других слабых кислот, даже если они содержат так много красителя (крезолового красного, нафтолового зеленого, метилово"о фиолетового, флуоресцеина и т. д.), что совершенно невозможно определять их визуально. Индикаторная ошибка никогда не превышает ошибки соответствующего титрования в присутствии фенолфталеина. При титровании аскорбиновой кислоты нет необходимости добавлять перекись водорода, потому что вследствие автоокисления в растворе последняя всегда об' разуется в количестве, достаточном для того, чтобы произошло хемилюминесцентное излучение.
Хорошие результаты получаются при определении кислотности молока. Люцигенин в этом случае гораздо чувствительнее обычных индикатосов-красителей. Он пригоден также для других анализов: для определения содержания кислот в патоке, красном вине, фрук
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
415
товых соках, горчице; для определения ацетилсалициловой и салициловой кислот в спиртовых растворах; для титрования с замещением фосфатов и карбонатов. Если надо определить общую кислотность растворов окиси ванадия(V), то перекись водорода следует добавлять в избытке сверх количества, которое расходуется на образование пероксованадиевой кислоты, для того чтобы наблюдалась хемилюминесценция. В растворах солей тяжелых металлов, таких, как сульфаты меди(II), никеля(II), железа (П1) или кобальта (II), нельзя определить содержание кислот, потому что в присутствии этих катионов перекись водорода разлагается еще до наступления конечной точки титрования.
Определение кислотного числа жиров и масел
Хемилюминесцентные индикаторы особенно подходят для определения содержания кислот в темно-окрашенных жирах и маслах. Однако их нельзя титровать в совершенно безводных растворителях, так как вода нужна для регенерации люцигенина. Поэтому для титрования следует боать такие растворители, в которых в достаточной степени растворяются жиры, масла и вода.
Методика определения. Растворяют точно отвешенный образец жира или масла в 30 мл амилового спирта, добавляют 1—2 капли 30%-ной перекиси водорода и 1 каплю 0,5%-ного индикатора люцигенина и титруют раствор 0,5 М. раствором гидроокиси калия в этаноле. Индикаторная ошибка этого метода титрования ниже, чем получаемая при титровании в присутствии индикатора щелочного синего 6 В.
При анализе минеральных масел на одно титрование надо брать 20 г. Образец растворяют в нейтрализованной смеси 30 мл бензола и 20 мл этанола. Затем добавляют 0,2 мл 30 %-ной перекиси водорода и 1 мл насыщенного на холоду раствора люцигенина в этаноле и титруют в темноте 0,1 М спиртовым раствором гидроокиси калия. В некоторых случаях предпочитают работать со смесью метанол — бензол (в объемном отношении 1:2) [49].
В отработанных минеральных маслах, содержащих примеси металлов, перекись водорода разлагается быстро, поэтому в этих случаях титрование имеет ряд трудностей.
Некоторые аналоги люцигенина, такие, как динитраты N,NZ-бпс-(о-хлорфенил)- или Ы,Ы'-бпс-(л1-хлорфенил)диакридила, позволяют определять конечную точку с большей точностью [50]. Другие производные люцигенина (Ы.Ы'-дипропил-, М,Ы'-дитолил-, N,NZ-Дифенил- и N.N'-диаллилдиакридин) пригодны в качестве индикаторов для определения содержания кислот в пиве [51, 52], а также алифатических спиртов [53] в неводных растворителях.
416
ГЛАВА X
ЛЮМИНОЛ [ГИДРАЗИД 3-АМИНОФТАЛЕВОЙ КИСЛОТЫ) В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО КИСЛОТНО-ОСНОВНОГО ИНДИКАТОРА
Люминол можно применять в присутствии перекиси водорода или катализаторов [гемина, гексацианоферрата(Ш), флуоресцеина] в качестве необратимого кислотно-основного индикатора [21— 23]. В сильнощелочных растворах (рН>8) можно использовать люминол также как окислительно-восстановительный индикатор даже в отсутствие катализаторов. Люминол VIII [уравнение (2)] флуоресцирует синим светом в щелочных растворах. При добавке сильного окислителя происходит быстрая реакция, сопровождаемая
N—н +о N-Н
о
VIII
ЛЮМИНОЛ гидразид 3-аминофтал ев ой кислоты
XI
анион амииофталевой кислоты
синей хемилюминесценцией. В результате реакции сначала получается красный хинон IX, который затем взаимодействует со свободными радикалами О- или Оз, образующимися из окислителя. На основании этого сначала получается трансаннулярная перекись, подобная соединению V [уравнение (1)], которая затем превращается в неактивный продукт (анион амииофталевой кислоты XI) [9, 10, 24—26]. Дальнейшие исследования доказали этот механизм реакции [54] и показали, что гидроксильные ионы играют важную роль в хемилюминесцентной реакции [55]. Люминол расходует эквивалентное количество окислителя, и поэтому его можно титровать, например, раствором гипобромита натрия. С гипохлоритами реакция идет медленнее, поэтому разбавленный раствор гипохлорита натрия дает более продолжительную, но менее интенсивную хемилюминесценцию.
Приготовление индикаторного 0,01%-ного раствора люминола. Вносят 0,1 г люминола в 500 мл воды и подщелачивают 5 мл 1,0 М гидроокиси натрия. Когда все растворится, доливают до 1 л водой. При работе с 0,1 М стандартными растворами достаточно брать на одно титрование 3 мл раствора индикатора.
ТИТРОВАНИЕ СЛАБЫХ КИСЛОТ В ПРИСУТСТВИИ ЛЮМИНОЛА
В присутствии перекиси водорода в щелочных растворах (pH 8—8,5) люминол (гидразид амииофталевой кислоты) медленно окисляется с излучением синего хемилюминесцентного света.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
417
Реакция настолько медленна, что надо применять катализатор [гемин, гемоглобин, комплексные соединения железа(III)]. Во время реакции люминол окисляется и расходуется, поэтому конечную точку можно определить по моменту, когда впервые весь раствор будет излучать свет. После этого свечение медленно исчезает и при добавлении следующей капли щелочи не наблюдается больше хемилюминесценции. Из-за того, что индикатор расходуется, обнаружение конечной точки необратимо, а потому нельзя прямо титровать щелочь в присутствии этого индикатора, так как он разлагается еще до наступления конечной точки. Следовательно, индикатор можно применять в прямом титровании сильных и слабых кислот. Интервал перехода находится в пределах значений pH 8,0—8,5. Индикатор чувствителен к двуокиси углерода. Рабочий раствор индикатора содержит 10 мг люминола в 1 л; его можно хранить в течение нескольких лет, и при этом с ним не происходит никаких изменений.
Методика определения
К Ю мл титруемого кислого раствора добавляют 3 капли 0,1 %-ной перекиси водорода, 4 капли 5%-ного раствора гексацианоферрата (III) калия и 6 капель индикаторного раствора (10 мг люминола в 1 л) и затем осторожно титруют в темноте 0,1 М гидроокисью натрия или калия. Когда капли стандартного раствора достигают смеси, замечается слабое излучение, которое исчезает все медленнее по мере приближения к конечной точке. После добавления последней капли происходит свечение всего раствора, которое продолжается около 0,5 мин. После записи показаний бюретки к раствору добавляют еще одну каплю. Если конечная точка установлена правильно, то свечение больше не должно наблюдаться.
Щелочи можно определять путем добавления к образцу известного избыточного количества сильной кислоты и последующим обратным титрованием по методике, описанной выше. Прямое определение щелочей тоже возможно, если люминол раствсрить в стандартном растворе кислоты и титровать им после добавки к образцу перекиси водорода и гексацианоферрата (III) калия.
ЛОФИН (2,4,5-ТРИФЕНИЛИМИДАЗОЛ) В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО КИСЛОТНО-ОСНОВНОГО ИНДИКАТОРА [27]
В присутствии перекиси водорода и подходящего катализатора лофин излучает в щелочном растворе желтовато-белый хемилюминесцентный свет. Механизм хемилюминесцентной реакции подобен
27—918
418
ГЛАВА X
механизму реакции с люминолом [уравнение (3)].
При окислении лофина I сначала получается свободный радикал II, который становится устойчивым, перейдя в перекись III. Если перекись III нагреть, она расщепляется с образованием возбужденных мономерных свободных радикалов, которые отрывают протон, димеризуются и снова возвращаются в исходное состояние. По сравнению с другими хемилюминесцентными индикаторами [28, 29] лофин получается очень легко. Синтезирован ряд производных лофина, использовавшихся в качестве хемилюминесцентных индикаторов [56]. Лофин нерастворим в воде, но легко растворяется в спирте, поэтому к титруемым растворам надо добавлять 30— 40% этанола. В присутствии катализатора гексацианоферрата(III) калия излучение света происходит при pH 8,9—9,4. Так, индикатор можно использовать во всех тех случаях, где используются фенолфталеины и люцигенины. Индикатор чувствителен к двуокиси углерода. Так как реакция с лофином идет с большой скоростью, индикатор непригоден для прямого титрования щелочей. В 1 М растворах титрование можно проводить на холоду, но при работе с 0,1 М растворами реакционную смесь надо нагревать до 60 °C.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
419
Методика определения
К 5—50 мл раствора сильной или слабой кислоты добавляют 30 мл этанола, 5 мл 3%-ной перекиси водорода и 3 мл 5 %-кого раствора гексацианоферрата(III) калия. Если определяемая кислота разбавлена (около 0,1 моль-л-1), раствор надо нагреть до 60°C. Затем добавляют 1 мл 1%-ного ацетонового или 0,45%-ного спиртового раствора лофина и титруют в темноте стандартной 0,1 М гидроокисью натрия, не содержащей карбонатов, до тех пор пока не появится люминесценция всего раствора, сохраняющаяся в течение нескольких минут. Более концентрированные кислоты (около 1 моль-с-1) следует титровать на холоду 1 М. гидроокисью натрия. Иногда в титруемом растворе появляется белый осадок.
Среднее из двенадцати параллельных определений [27] 20,00 мл 1,0 М соляной кислоты совпадает с истинным значением в пределах ±0,05%) при стандартном отклонении ±0,021 мл. При титровании 0,1 М соляной кислоты отклонение от истинного значения составляет ±0,25%, а стандартное отклонение равно ±0,057 мл. Подобную точность можно получить, работая с 1,0 н. растворами, при титровании серной, азотной, уксусной, лимонной, щавелевой, винной и фосфорной (как двухосновной) кислот, а также комплекса борной кислоты с инвертированным сахаром.
ХЕМИЛЮМИНЕСЦЕНТНЫЙ СМЕШАННЫЙ ИНДИКАТОР [30]
Если нагревать флуоресцеин с перекисью водорода в сильнощелочной среде, то наблюдается слабое хемилюминесцентное свечение, подобное по спектральному составу флуоресцентному излучению. Если в раствор добавить немного люминола, интенсивность зеленого хемилюминесцентного свечения сильно возрастает, т. е. люминол катализирует хемилюминесценцию флуоресцеина. Люминол LU, в сущности, сенсибилизирует хемилюминесцентную реакцию флуоресцеина FLU тем, что способствует образованию промежуточной трансаннулярной перекиси. На первой стадии люминол образует перекись люминола LUO2 согласно уравнению (4а). Затем перекись люминола LUO2 дает с флуоресцеином FLU переходный комплекс LUO2-FLU„ который распадается на люминол LU и перекись флуоресцеина FLUO2 по уравнению (46).
LU +[H2O2O0Hj—* LUO2+ ОН- +Н2О------4а
I
FLUO2+LU—[LUO2FLU] — FLU _________________4 б
Oa + FLU* —*- hv (зеленый)—* FLU _______4e
(4)
27*
420
ГЛАВА X
При нагревании перекись флуоресцеина разлагается с выделением кислорода. Одновременно образуется возбужденная молекула флуоресцеина FLU*, которая возвращается в основное состояние после излучения зеленого хемилюминесцентного свечения [уравнение (4е)]. Из этого механизма следует, что можно ожидать максимальную интенсивность свечения в том случае, если люминол и флуоресцеин содержатся в растворе в эквимолярных количествах н если pH ~12; при этом значении pH концентрация комплекса перекиси водорода с пергидроксильным ионом Н2О2-ООН_ имеет максимум*.
Смесь индикаторов, состоящая из флуоресцеина и люминола, в процессе титрования излучает при pH ~8,9 хемилюминесцентное свечение, если в растворе содержится перекись водорода. В присутствии больших количеств катализаторов, т. е. в присутствии спиртов и ионов тяжелых металлов, хемилюминесцентное свечение становится синим, а это показывает, что в таком случае флуоресцеин не участвует в реакции и хемилюминесценцию вызывает только люминол.
Приготовление хемилюминесцентного смешанного индикатора флуоресцеина с люминолом
Растворяют 0,15 г флуоресцеина и 0,1 г люминола в 10 мл 0,1 М. гидроокиси натрия и разбавляют водой до 100 мл. Для того чтобы этот раствор проявлял индикаторные свойства, надо в титруемый раствор добавить 6%-ную перекись водорода в объеме, равном объему смешанного индикатора. Однако, так как кислотность препаратов перекиси водорода обычно высокая, рекомендуется определять отдельно поправку на индикатор или индикаторную ошибку. Для этого смешивают 5 мл индикатора с 5 мл 6%-ной перекиси водорода, разбавляют 20 мл горячей воды и титруют 0,01 М раствором гидроокиси натрия в темноте. Данные этого титрования надо выразить в количестве реагента, используемого для титрования исследуемого образца. Индикаторная ошибка редко достигает 0,4 мл 0,01 М. раствора гидроокиси натрия, и поэтому в большинстве случаев ею можно пренебречь.
Кислотно-основное титрование с применением хемилюминесцентного смешанного индикатора флуоресцеина с люминолом
К 20 мл раствора сильной кислоты или сильного основания добавляют 1 мл 6%-ной перекиси водорода и 1 мл смешанного индикатора, нагревают раствор до 60 °C и титруют в темноте стан-
ем. сноску на стр. 412.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
421
дартным раствором кислоты или щелочи (0,1—0,001 М.) до исчезновения или появления хемилюминесценции соответственно.
Из значения pH, соответствующего переходу индикатора (pH 8,9), следует, что индикатор чувствителен к кислотам и поэтому его можно применять для установления конечной точки титрования сильных и слабых кислот и сильных оснований, не содержащих карбонаты. Так как индикатор чувствителен к двуокиси углерода, то даже в случае определения сильных кислот надо проводить титрование щелочами, не содержащими карбонатов. При титровании сильных кислот и оснований можно пренебречь индикаторной ошибкой даже в 0,01 М. растворах. Хорошие результаты при титровании уксусной, щавелевой и винной кислот получаются, если титровать 5 мл образца при концентрации 0,1 моль-л-1. В опытах с лимонной кислотой определение конечной точки ненадежное, чувствительность хемилюминесцентной реакции понижается, и поэтому получается значительная ошибка, достигающая 6%. При 60 °C индикатор действует обратимо до тех пор, пока перекись водорода не распадется полностью.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ В ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОМ ТИТРОВАНИИ
Хемилюминесцентные реакции, о которых говорилось до сих пор, происходят в щелочных растворах; поэтому, если их применить в окислительно-восстановительных реакциях, то можно рассматривать только те из них, которые идут в щелочных растворах. Действительно, как будет показано ниже, люминол и люцигенин пригодны для определения конечной точки ряда окислительно-восстановительных титрований в щелочных растворах. Если титровать сильный восстановитель окислителем в присутствии этих индикаторов, окислительно-восстановительный потенциал раствора в конечной точке неожиданно смещается в сторону более положительных значений и происходит хемилюминесцентная реакция. При участии люцигенина хемилюминесценция наблюдается только тогда, когда после достижения эквивалентной точки в растворе содержится перекись водорода. Отчасти это достигается путем титрования стандартным раствором перекиси водорода или путем использования таких соединений, которые образуют перекись водорода вследствие автоокисления, например разбавленный водный раствор гидразина. В кислых растворах с рН<3,5 можно использовать в качестве окислительно-восстановительного индикатора силоксен. В среде сильного окислителя, потенциал которой выше 1,17 В, нерастворимый в воде силоксен хемилюминесцирует ярко-красным светом. Благодаря кислой среде и подходящему потенциалу в точке перехода можно проводить многие окислительно-восстановительные титрования.
422
ГЛАВА X
СИЛОКСЕН В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ИНДИКАТОРА
Впервые силоксен был синтезирован Велером [31], а название ему дал Кени [32]. Это вещество можно получить из силицида кальция, который разлагают спиртом и соляной кислотой. Каутски [33] установил общую формулу продукта, полученного на холоду и в темноте (51бН60з)т, и структуру
Силиконовые кольца содержат по шесть атомов Si и соединяются друг с другом посредством кислородных мостиков. К каждому атому Si подсоединен один атом водорода. Если силоксен получен в горячем растворе на дневном свету и с помощью концентрированной соляной кислоты, то, по данным Кени, часть атомов водорода в кольцах замещается на атомы хлора или гидроксильные группы, а в составе некоторых колец между атомами кремния имеются атомы кислорода. Индикатор силоксен излучает красное свечение в кислой среде, содержащей сильный окислитель, и одновременно поглощает кислород.
ОКСИДИМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ЖЕЛЕЗА(П] ПО МЕТОДУ КЕНИ И КУРЦА [32]
Если подкислить серной или азотной кислотой раствор сульфата или хлорида железа (II) и добавить к нему 0,1 г силоксена, его можно титровать стандартным раствором сульфата церия (IV). На рис. Х.4 показано изменение окислительно-восстановительного по-
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
423
тенциала при титровании вблизи конечной точки; там же приведено значение потенциала, которое можно замерить в присутствии больших количеств солей кобальта (II). В последнем случае рас-
твор сильно окрашен из-за содержания кобальта, и поэтому конечную точку нельзя определять посредством визуального индикатора. Конечные точки, получаемые в присутствии или в отсутствие кобальта, почти одинаковы и соответствуют потенциалу перехода 1,17 В при pH 0,23. Отклонение от стехиометрической конечной точки вызывает практически незначительную индикаторную ошибку (0,08 мл).
Титрование можно проводить также в присутствии ионов хрома (III) в концентрации 0,1 моль-л-1; индикаторная ошибка тогда тоже незначительна (0,13— 0,07%). На окисление сило-ксена всегда расходуется известное количество титранта. На 100 мг силоксена идет обычно 0,04 мл 0,02 М.
Объем раствора церия(!У)-, хл
Рис. Х.4. Кривая титрования Fe(II) раствором сульфата церия (IV).
стандартного раствора перманганата калия или 0,1 М. раствора сульфата церия (IV). В присутствии иодид-ионов индикаторная ошибка возрастает до 0,7 мл. Силоксен как индикатор можно использовать только для сильнокислых растворов (рН<3,5).
ДРУГИЕ ОКСИДИМЕТРИЧЕСКИЕ МЕТОДЫ ТИТРОВАНИЯ В ПРИСУТСТВИИ ХЕМИЛЮМИНЕСЦЕНТНОГО ИНДИКАТОРА СИЛОКСЕНА
Силоксен можно применять для перманганатометрического определения [34] железа(II), титана(III), олова(II), молибдена (III), мышьяка (III), иодид- и оксалат-ионов, а также перекиси водорода. Цериметрическим методом с использованием силоксена были проведены определения [35] железа (II), мышьяка (III), оксалат- и иодид-ионов, а также перекиси водорода и гидрохинона.
424
ГЛАВА X
Гидрохинон и ионы железа (II) можно титровать и бихроматсм калия [36]. В сильных солянокислых растворах можно титровать ионы таллия(I) бихроматом калия в присутствии силоксена [37].
Определение иодид-ионов
Среди всех методов перманганатометрическое и периметрическое определения иодида следует рассмотреть отдельно, потому что эти реакции до сих пор нельзя было использовать просто из-за того, что выделяющийся при окислении иод мешает визуальному определению конечной точки. Однако хемилюминесценция си-локсена хорошо заметна в коричневом растворе даже в случае выпадения иода в осадок.
Методика определения
Раствор, содержащий 50—500 мг иодид-ионов, подкисляют 15 мл 1,0 М. раствора серной кислоты, разбавляют до 100 мл, добавляют 60 мг силоксена и титруют в темноте стандартным 0,02 М раствором перманганата калия или 0,1 М. сульфата церия (IV). Конечную точку устанавливают по появлению устойчивой красной хемилюминесценции во всем объеме раствора. Из израсходованного объема стандартного раствора следует вычесть 0,04 мл, что соответствует индикаторной ошибке.
По этому методу получают действительные значения с точностью ±0,06%. Иодид-ионы в растворе иодида калия и иода можно определять прямым титрованием. Иод и иодат определяют после предварительного восстановления металлическим цинком. Этот метод пригоден также для косвенного определения ионов серебра. Если к раствору ионов серебра добавить известное избыточное количество иодида калия, то избыток иодид-ионов можно оттитровать в растворе, содержащем осадок. Ионы иодида можно определять избирательно в присутствии других ионов, которые не образуют осадка с серебром. Первую аликвотную часть раствора следует титровать без серебра, а вторую — после добавления избытка нитрата серебра. Разность между обоими результатами соответствует содержанию иодида в растворе.
ЛЮЦИГЕНИН В КАЧЕСТВЕ ОБРАТИМОГО ХЕМИЛЮМИНЕСЦЕНТНОГО ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ИНДИКАТОРА
Если в щелочном растворе люцигенина содержится вещество, которое реагирует с перекисью водорода, то хемилюминесценция наблюдается только в том случае, если перекись водорода нахо
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
425
дится в избытке. Поэтому люцигенин можно использовать в качестве окислительно-восстановительного индикатора для титрования' стандартным раствором перекиси водорода [38]. Установление конечной точки полностью обратимо, а точность титрования почти такая же, как при классических методах титрования.
Приготовление стандартного 0,05 М раствора перекиси водорода. Для приготовления раствора надо брать воду, перегнанную в стеклянном аппарате, которая содержит 10 мл 0,5 М. серной кислоты в 1 л. Титр такого раствора перекиси понижается не более чем на 0,1% в день, но его можно использовать даже через 6 месяцев. Удобнее всего определять титр раствора по мышьяковистой кислоте и вполне достаточно делать это один раз в день.
Подготовка раствора перед титрованием. Титруемый раствор следует разбавить до 100 мл и добавить такое количество 1,0 М. гидроокиси натрия, чтобы содержание свободной щелочи было эквивалентно 5—10 мл 1,0 М. гидроокиси натрия. Раствор не должен содержать большее количество щелочи, потому что в сильнощелочных растворах люцигенин хемилюминесцирует даже в отсутствие перекиси водорода. Если неизвестно содержание кислоты или щелочи в растворе, то его сначала нейтрализуют по фенолфталеину и затем добавляют 5—10 мл 1,0 М. гидроокиси натрия.
Методика определения
К раствору гексацианоферрата (III) калия, мышьяковистой кислоты или гипогалогенита щелочного металла, соответствующему 5—50 мл 0,05 М перекиси водорода, добавляют 5—10 мл 1,0 М. гидроокиси натрия и 1 мл 0,5%-ного люцигенина; разбавляют раствор до 100 мл. Гипобромиты и гипоиодиты щелочных металлов можно определять на холоду, а растворы гексацианоферрата (III) и арсенита щелочных металлов надо титровать при 80 °C вплоть до появления постоянного свечения. Гипохлориты щелочных и щелочноземельных металлов следует титровать на холоду до установления стойкой хемилюминесценции. Затем раствор нагревают до 80 °C, причем свечение прекращается, и титрование продолжают до тех пор, пока вновь не появится постоянное свечение.
Если к щелочному раствору гипохлорита добавить 2—3 г бромида калия, через несколько минут можно начинать титрование на холоду. Гипоиодиты щелочных металлов окисляются очень быстро в ходе титрования йодата, поэтому результат соответствует только одному моментальному значению. Содержание свободного галогена в хлорной и бромной воде можно точно так же определять по этому методу. Точность этого метода можно оценить на основе следующих данных. Если израсходовано 20 мл стандартного раствора, то ошибка и стандартные отклонения, вычисленные на основе десяти параллельных 'измерений, следующие:
426
ГЛАВА X
Титруемое соединение Ошибка, % Стандартное отклонение, мл
K3[Fe(CN)6] —о,1 ±0,014
+0,05 ±0,008
NaOCl —0,04 ±0,002
NaOBr —0,04 ±0,002
Определение ионов хролш(Ш). Так как люцигенин чувствителен только к перекиси водорода и не хемилюминесцирует с другими окислителями, гипогалогениты щелочных металлов можно титрова гь даже в присутствии хроматов. Это явление можно использовать для определения ионов хрома(III). Гипобромиты щелочных металлов окисляют ионы хрома (III) до хромат-ионов в течение 5—10 мин, а затем избыток гипобромита оттитровывают 0,05 М перекисью водорода. При определении 10—100 мг хрома (III) результаты приближаются к действительным значениям в пределах от —0,08 до + 0,7%.
Гипогалогениты можно титровать также стандартным раствором гидразинсульфата в щелочной среде, используя люцигенин в качестве индикатора. Проявление индикатора облегчается еще тем, что щелочные растворы гидразинсульфата окисляются атмосферным кислородом, который восстанавливается до перекиси водорода. Перекись водорода, т. е. избыток гидразина, обнаруживается по хемилюминесценции люцигенина [39]. Метод дает точные результаты для гипохлоритов при ошибке +0,15% и стандартном отклонении ±0,05 мл. Результаты определения гипобро-митов иногда менее точны.
ЛЮМИНОЛ В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ИНДИКАТОРА
Люминол можно использовать в качестве окислительно-восстановительного индикатора чаще, чем люцигенин, потому что он не специфичен только в отношении перекиси водорода; все остальные окислители вызывают его хемилюминесценцию, если pH раствора выше 8 (т. е. раствор в достаточной степени щелочен). Дальнейшие исследования показали, что явление хемилюминесценции связано с промежуточным образованием свободных радикалов кисло рода (О-, Ог), как это видно из механизма реакции [см. уравнение (2) данной главы]. Люминол в отличие от люцигенина расходует на себя окислитель, поэтому в ходе титрования появляется более или менее точно определяемая индикаторная ошибка. Так,
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
427
например, люминол можно титровать етехиометрически раствором гипобромита натрия, тогда как реакция с гипохлоритом идет медленно. Поэтому гипохлориты дают с люминолом устойчивую хемилюминесценцию.
Приготовление 0,01 %-кого индикаторного раствора люминола
Вносят 0,1 г люминола в 500 мл воды и подщелачивают 5 мл 1,0 М. гидроокиси натрия. После растворения разбавляют раствор водой до 1 л. Если работают с 0,1 н. стандартным раствором, достаточно брать 3 мл этого 0,01%-ного раствора индикатора. Индикаторная ошибка составляет 0,07 мл. При использовании 0,001 н. стандартного раствора получается большое разбавление и реакция с индикатором так медленна, что даже одна капля титранта вызывает появление устойчивой хемилюминесценции; вследствие этого не создается индикаторной ошибки и не требуется вносить поправку в результаты.
ТИТРОВАНИЕ СТАНДАРТНЫМ РАСТВОРОМ ГИГОБРОМИТА НАТРИЯ [40]
Стандартным раствором гипобромита натрия в присутствии люминола как индикатора можно титровать ионы мышьяка(III), сурьмы (III), роданида, цианида, тиосульфата, сульфита и сульфида. Большим преимуществом является низкий эквивалентный вес анионов, в состав которых входит сера, так как гипобромит окисляет серу во всех случаях до 6-валентного электроположительного состояния, а это означает, что сульфиды переносят в реакции 8 электронов.
Аммиак и соли аммония мешают титрованию гипогалогенитами, потому что на них расходуется стандартный раствор.
Приготовление стандартного 0,05 М раствора гипобромита натрия. При встряхивании насыщают бромом 500 мл воды, перегнанной в стеклянном аппарате (т. е. не содержащей следов тяжелых металлов). Бромную воду сливают от нерастворившегося брома и смешивают с 500 мл 1,0 М. гидроокиси натрия, приготовленной на такого же качества воде. Титр устанавливают по мышьяковистой кислоте по методике, описанной ниже. Если раствор слишком концентрированный, его следует разбавить 0,5 М гидроокисью натрия. При хранении в темном прохладном месте раствор окисляется совсем медленно. Титр его уменьшается за 3 месяца на 17,7%. Старые растворы, содержащие много бромата, непригодны для определения сульфидов и сульфитов.
Стандартный, не содержащий бромата, 0,05 М. раствор гипобромита можно приготовить также, исходя из гораздо более устойчивого раствора гипохлорита кальция или щелочных металлов, ко
428
ГЛАВА X
торый берут в эквивалентном количестве и добавляют к нему 10 г бромида калия на 1 л.
Методика определения
К почти нейтральному раствору определяемого вещества добавляют от 5 до 20 мл 1,0 М. гидроокиси натрия и 3,0 мл люминола как индикатора, разбавляют водой до 100 мл и титруют в темноте стандартным 0,05 М раствором гипобромита натрия, используя для этой цели бюретку с ограничителем на кране. В месте приливания стандартного раствора появляются небольшие вспышки света, а по мере приближения к конечной точке люминесценция становится все более устойчивой. Конечную точку устанавливают по появлению постоянной люминесценции всего раствора в течение нескольких секунд. Считается что конечная точка определена точно, если следующая капля стандартного раствора не вызывает усиления хемилюминесценции.
При титровании солей сурьмы (III) к раствору добавляют 1 г сегнетовой соли (тартрат .калия-натрия), чтобы помешать образованию осадка. Растворы сульфидов и тиосульфатов перед титрованием следует довести до щелочной реакции, добавив 5 мл 1,0 М. гидроокиси натрия. Титрование сульфидов и сульфитов может быть точным только при работе с 0,05 М гипобромитом натрия, не содержащим бромата. Однако гораздо удобнее добавлять исследуемые образцы к известному избытку гипобромита натрия, затем восстановить его избытком мышьяковистой кислоты и титровать эту кислоту обратно раствором гипобромита натрия по данной методике.
По этому методу получаются точные результаты даже при работе с 0,0005 М. концентрациями, и поэтому его можно использовать в микроанализе. Ниже приводятся средняя точность и среднее стандартное отклонение из двадцати параллельных определений каждого образца при расходе 20 мл стандартного 0,05 М. раствора:
Титруемое соединение Относительная точность, % Стандартное отклонение, мл
As2O3 —0,05 ±0,04
Sb2O3 +0,05 ±0,01
KCN +0,15 ±0,018
KSCN +0,75 ±0,015
N22S2O3 +0,05 ±0,023
Na2SO3 +0,2 ±0,020
Na2S +0,1 ±0,026
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
429
Хотя индикаторный процесс необратим, титрование можно проводить точно, потому что конечная точка получается четко.
ТИТРОВАНИЕ РАСТВОРОМ ГИПОХЛОРИТА НАТРИЯ ИЛИ ХЛОРНОЙ ИЗВЕСТИ [41]
Гипохлориты взаимодействуют с индивидуальными восстановителями обычно медленнее, чем гипобромиты, поэтому их нельзя использовать в методах прямого титрования. Однако большим преимуществом является то, что гипохлориты медленно реагируют с самим люминолом, а значит, появление постоянной хемилюминесценции дает конечную точку без индикаторной ошибки. Медленная реакция с индикатором позволяет проводить обратное титрование.
Аммиак и соли аммония мешают титрованию. Этот метод в первую очередь пригоден для титрования ионов мышьяка (III), а вследствие обратимости определения конечной точки получается такая же точность, как и при других методах определения мышьяка.
Приготовление устойчивого стандартного 0,05 М раствора гипохлорита натрия
При охлаждении насыщают воду хлором (воду надо предварительно перегнать в стеклянном аппарате, для того чтобы она не содержала примеси тяжелых металлов), затем приливают 5,0 М гидроокиси натрия до полного исчезновения желтого цвета хлора. Добавляют еще этого же раствора гидроокиси натрия до концентрации 0,5 моль-л-1. Для стабилизации приливают к раствору 20 мл 1,0 М хлорида магния на 1 л и сливают раствор от осевше го осадка через стеклянный пористый фильтр. С гидроокисью магния выпадают в осадок следы ионов тяжелых металлов, которые ускоряют разложение гипохлорита. Затем растьор разбавляют 0,5 М. гидроокисью натрия, чтобы довести концентрацию гипохлорита до 0,05 моль-л-1.
Методика определения
К почти нейтральному раствору 20—200 мг As2O3 добавляют 10—30 мл 1,0 М. гидроокиси натрия, 10 мл 0,01%-ного люминола и доливают водой до 100 мл; нагревают до 80 °C и титруют в темноте стандартным 0,05 М. раствором гипохлорита натрия до тех пор, пока от добавки одной капли раствора не появится хемилюминесценция, которая продержится по крайней мере в течение 2 мин.
430
ГЛАВА X
Метод отличается высокой точностью; среднее значение шести параллельных определений, на каждое из которых расходуется 20 мл стандартного раствора, отклоняется от действительного значения в пределах ±0,05% при стандартном отклонении ±0,012 мл. Метод дает точные результаты также и при работе с концентрациями 0,005 моль-л-1.
ТИТРОВАНИЕ ГИПОГАЛОГЗДИТОВ 0,05 М СТАНДАРТНЫМ РАСТВОРОМ АРСЕНИТА НАТРИЯ [42]
Так как люминол при добавлении к щелочному раствору гипохлорита окисляется в самом начале титрования и может израсходоваться еще до конца титрования, то предлагается вносить немного индикатора в стандартный раствор и им титровать.
Приготовление 0,05 М стандартного раствора арсенита натрия, содержащего люминол. Навеску 4,9455 г х. ч. трехокиси мышьяка сушат при 130 °C и растворяют при нагревании в растворе 5 г гидроокиси натрия. После частичного разбавления вносят в этот раствор 0,01 г люминола и доливают водой до 1 л.
Методика определения
К исследуемому раствору гипохлорита или гипобромита добавляют 2—10 мл 1,0 М гидроокиси натрия, разбавляют водой примерно до 100 мл и титруют в темноте 0,05 М раствором арсенита, содержащим люминол, пока от добавки одной капли не прекратятся вспышки свечения.
В случае гипохлоритов можно добавить к исследуемому раствору 2 мл 0,01%-ного люминола, чтобы еще с самого начала получилось четкое свечение. Точность результатов колеблется в пределах ±0,1% при титровании 20—50 мл 0,05 М гипохлорита и 5—50 мл 0,05 М гипобромита; стандартное отклонение ±0,04 мл.
Определению не мешают даже большие количества хлоратов, броматов и хлоритов. Для хлорной воды, бромной воды и выбеливающих растворов гипохлорита этот метод дает лучшие результаты, чем йодометрический. Присутствие железа (III) и хлорит-ио-нов не мешает определению.
КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
С ИСПОЛЬЗОВАНИЕМ ХЕМИЛЮМИНЕСЦЕНТНЫХ ИНДИКАТОРОВ
Хемилюминесцентные индикаторы можно с успехом применять для комплексометрического определения ионов меди (II) или для обратного процесса титрования этилендиаминтетраацетата раствором ионов меди (П) [10]. Можно также проводить косвенное определение других ионов металлов посредством использования основ
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
431
ной реакции. Сущность установления конечной точки с помощью хемилюминесценции состоит в следующем. Известно, что двунатриевая соль этилендиаминтетрауксусной кислоты стабилизирует раствор перекиси водорода тем, что образует устойчивые комплексы с ионами тяжелых металлов, которые катализируют распад перекиси водорода так же, как и ионы меди (II), являющиеся особенно эффективным катализатором. В присутствии свободных, не связанных .в комплекс, ионов меди (II) перекись водорода быстро разлагается с образованием промежуточных свободных радикалов кислорода (О~, Ог). В присутствии ЭДТА каталитическое действие ионов меди (II) прекращается, так как комплекс не обладает каталитическими свойствами. Известно также, что люминол не излучает света в щелочных растворах перекиси водорода до тех пор, пока имеются ионы тяжелых металлов, как, например, ионы меди (II), которые вызывают^ образование свободных радикалов кислорода в растворе (О-, О2). Опыты показали, что от введения в раствор ЭДТА в количестве, достаточном для связывания ионов меди (II) в комплекс, прекращается хемилюминесценция люминола, вызываемая ионами меди (II) и перекисью водорода. В противоположность люминолу люцигенин светится в щелочном растворе перекиси водорода, содержащем комплекс меди(II) с ЭДТА, потому, что в этом случае нужны не свободные радикалы кислорода, а только молекулы Н2О2. Однако, если к щелочному раствору перекиси водорода, содержащему люцигенин, добавить ионы меди (II), произойдет быстрый распад перекиси водорода и исчезнет хемилюминесценция. Поэтому когда титруют аммиачный раствор меди (II) в присутствии люминола стандартным раствором ЭДТА, в котором имеется 0,1% перекиси водорода, раствор люминесцп-рует, хотя ионы меди (II) находятся в избытке. В точке эквивалентности свечение прекращается. Если же это титрование проводить в присутствии люцигенина, то до наступления точки эквивалентности наблюдаются лишь короткие вспышки, а после точки эквивалентности свечение становится постоянным, так как комплекс меди (II) с ЭДТА не разлагает перекись водорода а, значит, может идти хемилюминесцентная реакция. Несколько лет тому назад был опубликован обзор по применению флуоресцентных и хемилюминесцентных индикаторов [57].
Приготовление стандартного 0,02 М раствора ЭДТА. Растворяют в воде 3,8 г кристаллической двунатриевой соли этилендиаминтетрауксусной кислоты, имеющей 2 молекулы кристаллизационной воды, и доливают до 0,5 л. Титр устанавливают по стандартному раствору сульфата магния. Из этого раствора готовят 0,01 М раствор ЭДТА путем разбавления.
Приготовление 0,01 М раствора ЭДТА, содержащего перекись водорода. Для титрования ионов меди (II) готовят 0,02 М раствор ЭДТА, к которому добавляют 2 мл 30%-ной перекиси водорода, а
432
ГЛАВА X
затем разбавляют до 0,01 М концентрации. Титр раствора понижается на 0,5% за одну неделю.
Приготовление 0,01 М раствора сульфата меди(П). Растворяют 2,497 г кристаллического сульфата меди (II) в воде и доливают до 1 л. Титр раствора устанавливают по 0,01 М стандартному раствору ЭДТА, содержащему перекись водорода.
Приготовление индикаторов. 1) 0,5%-ный люцигенин: растворяют 1,0 г люцигенина в 200 мл воды при нагревании; 2) 0,01 %-ный люминол: растворяют 0,1 г люминола в 5 мл 1,0 М гидроокиси натрия и доливают водой до 1 л.
Комплексометрическое определение ионов меди(11)
К нейтральному или нейтрализованному раствору ионов меди (II) добавляют 20 мл 4 М гидроокиси аммония, 1 мл 0,5%-ного люцигенина (или 3 мл 0,01%-ного люминола), нагревают до 90°C и титруют в темноте 0,01 М раствором ЭДТА, содержащим перекись водорода. В присутствии люминола конечную точку определяют по прекращению хемилюминесценции, а при работе с люци-генином — по ее появлению.
Точность метода так высока, что индикаторная ошибка в случае люцигенина меньше ±0,14%, а для люминола меньше ±0,16%). Кроме того, люминол дает индикаторную ошибку ±0,06 мл. Преимущество метода в том, что можно определять медь(II) даже в присутствии ионов серебра. Определению мешают сильные окислители и комплексообразователи. Метод можно применять также для анализа микроколичеств меди (6—600 мкг).
Определение раствора ЭДТА
Метод, описанный выше, можно использовать также в обратном варианте. К 0,01 М раствору ЭДТА добавляют 20 мл 4 М гидроокиси аммония, 1 мл 3%>-ной перекиси водорода и 3 мл индикаторного раствора люминола (или 1 мл люцигенина). Нагревают до 90°C и титруют раствором сульфата меди (II) в темноте. Конечную точку устанавливают в случае люцигенина по исчезновению свечения, а в случае люминола — по появлению зеленого свечения. Этот метод можно использовать и для определения титра раствора ЭДТА. При работе с люцигенином стандартное отклонение составляет ±0,026 мл (±0,13%), а результат отличается от действительного значения на —0,1%. Соответствующие значения для люминола составляют ±0,034 мл (±0,17%) и —0,2%).
Определение исков свинца и ртути(11)
Титрование раствора ЭДТА растворами меди (II) позволяет определять ионы, образующие комплексы с ЭДТА в аммиачной среде. Избыток ЭДТА можно титровать обратным методом в при
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
433
сутствии люцигенина или люминола как индикаторов стандартным раствором сульфата меди (II).
Методика определения. К раствору нитрата свинца или рту-ти(П), концентрация которого примерно 0,01 М, добавляют в избытке 0,01 М раствор ЭДТА. Нагревают до кипения, приливают 20 мл 4 М гидроокиси аммония, 1 мл 3%-ной перекиси водорода и наконец 3 мл индикаторного раствора люминола (или 1 мл люцигенина). Смесь титруют в темноте стандартным раствором сульфата меди(II). В точке эквивалентности растворы, содержащие люцигенин, прекращают свечение, а в случае люминола темный раствор начинает хемилюминесцировать.
Стандартное отклонение при определении свинца составляет ±0,05 мл (±0,23%), а отклонение от истинного значения —0,1%. Соответственно при определении ртути(II) получены ±0,05 мл (±0,21%) и—0,05%.
ТИТРОВАНИЕ ПО МЕТОДУ ОСАЖДЕНИЯ
С ИСПОЛЬЗОВАНИЕМ ХЕМИЛЮМИНЕСЦЕНТНЫХ ИНДИКАТОРОВ
В области титрования по методу осаждения разработаны методики хроматометрического определения ионов сзинца [43] и сульфата [44] и титрование ионов кадмия раствором гексацианоферрата (III) в присутствии силоксена как индикатора, а также аргентометрическое титрование иодид-ионов в присутствии люцигенина.
ХРОМАТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ИОНОВ СВИНЦА [43]
Этот метод основывается на том, что при добавлении стандартного раствора хромата калия к умеренно кислому раствору ионов свинца (pH около 1,9—3) выпадает в осадок хромат свинца. После достижения точки эквивалентности окислительно-восстановительный потенциал раствора так возрастает, что добавленный в раствор силоксен начинает испускать красный хемилюминесцентный свет. Очень важно, чтобы значение pH раствора было налажено точно, поскольку в растворах с рН>1,9 хромат свинца тоже растворяется, тогда как при рН>3 окислительно-восстановительный потенциал раствора не достигает такого высокого значения, при котором силоксен начнет испускать свет.
Приготовление 0,1 М стандартного раствора хромата калия. Растворяют 38,8410 г х. ч. хромата калия в воде, доливают до 2 л при 26 °C. Рекомендуется титровать при этой же температуре.
Приготовление 0,1 М раствора нитрата свинца. Растворяют 41,4426 г полированного металлического х. ч. свинца в азотной кислоте, выпаривают раствор досуха, растворяют остаток в воде и доливают до 2 л при 26 °C.
28—518
434
’’ЛАВА X
Методика определения
Доводят 30 мл 0,1 М раствора нитрата свинца до pH 1,9— 3,0 путем добавления гидроокиси аммония или азотной кислоты, вносят 20—80 мг силоксена и титруют в темноте стандартным 0,1 М хроматом калия. Красную хемилюминесценцию, которая появляется в конечной точке, можно регистрировать также фотовольтметром.
Ошибка определения находится в пределах 0,07—0,13%- Осадок, образовавшийся при реакции, мешает визуальному установлению конечной точки титрования. Определению не мешает присутствие ионов марганца (II), никеля, железа(III), цинка, кальция и магния. Метод пригоден также для определения сульфат-ионов. Если сульфат-ионы осадить избыточным количеством ионов свинца, то этот избыток можно определить обратным титрованием раствором хромата калия в присутствии силоксена.
АРГЕНТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ИОДИД-ИОНОВ [10]
Люцигенин можно использовать в качестве хемилюминесцентного индикатора для аргентометрического титрования иодид-ионов. В аммиачных растворах на отрицательно заряженной поверхности иодида серебра (осажденного в присутствии избытка иодид-ионов) катионы люцигенина адсорбируются и поэтому не дают свечения с перекисью водорода. Если, однако, ионы серебра добавить к раствору в избытке, знак заряда поверхности осадка иодида серебра меняется на положительный, катионы люцигенина десорбируются, и если в растворе содержится перекись водорода, то начинается свечение. Поэтому свечение люцигенина наблюдается лишь в том случае, если иодид-ионы полностью перешли в осадок, а ионы серебра находятся в избытке. Титрование можно проводить только в 0,1 М растворах, так как при концентрации 0,01 моль-л-1 получается слишком высокая индикаторная ошибка.
Методика определения. К 50 мл анализируемого раствора иодида добавляют 10 мл 4 М гидроокиси аммония, 1 мл 0,5%-ного индикаторного раствора люцигенина и 1 мл 0,3%-ной перекиси водорода, а затем титруют в темноте 0,1 М нитратом серебра до появления зеленой хемилюминесценции.
Относительное стандартное отклонение метода ±0,16%. Определение можно проводить в присутствии ионов хлорида и бромида. Ионы цианида титруются вместе с иодид-ионами. Определению не мешают роданид-ионы.
ХЕМИЛЮМИНЕСЦЕНТНЫЕ ИНДИКАТОРЫ
435
ТИТРОВАНИЕ КАДМИЯ РАСТВОРОМ ГЕКСАЦИАНОФЕРРАТА(Ш) ПРИ ИСПОЛЬЗОВАНИИ СИЛОКСЕНА В КАЧЕСТВЕ ХЕМИЛЮМИНЕСЦЕНТНОГО ИНДИКАТОРА
Имеется сообщение об определении ионов кадмия титрованием по методу осаждения стандартным раствором гексацианоферрата (III) калия [58]. При титровании в кислой среде (pH ~0,5) ионы кадмия образуют с ионами гексацианоферрата(III) осадок состава Cd3[Fe(CN)6]2. Вследствие связывания ионов гексацианоферрата (III) окислительно восстановительный потенциал раствора понижается вплоть до самой конечной точки. Однако, если эти ионы находятся в избытке, наблюдается быстрое повышение потенциала и в итоге инициируется хемилюминесценция силоксена. Реакция стехиометрична лишь при медленном титровании (4 мл в 1 мин) при энергичном перемешивании. За ходом титрования следят с помощью фотометра с умножителем на 520 А. Если раствор содержит только ионы кадмия, коэффициент вариаций довольно низкий (ниже 0,5%) и не превышает 1% даже в присутствии больших количеств ионов аммония, калия, натрия, магния, стронция, кальция, бария и мышьяка(III).
СПИСОК ЛИТЕРАТУРЫ
1. Moldvai R., Disseration, Budapest, 1941.
2. Steienann A., Photographische Ind., 35, 1365 (1937); Chem. and Ind., 60, 889 (1941); J. Soc. Chem. Ind., 61, 36 (1942).
3. Filemoni M., Siesta A. J., Boll. soc. ital. biol. sper., 26, 1096 (1950); C. A., 49, 11, 497 (1955).
4. Пономаренко А. А., ДАН СССР, 102, 539 (1955).
5. Ojima H., Iwaki R., Nippon Kagaku Zasshi, 78, 1632 (1957); C. A., 52, 5200 (1958).
6. Tsumaki T., Yoshino T., Hiro H., J. Chem. Soc. Japan, 72, 624 (1951); C. A., 46, 6986 (1952).
7. Kenny F., Kurtz R. B., Anal. Chem., 23, 382 (1951).
8. Erdey L., Buzas 1., Acta Chim. Hung., 6, 80 (1955).
9. Kurtz К. B., Trans. N. Y. Acad. Sci., 16, 399 (1954).
10. Erdey L., Buzas 1., Anal. Chim. Acta, 22, 524 (1960).
11. Kautsky H., Kaiser N. H., Naturwiss., 31, 505 (1943).
12. Van der Burg A. S., Rec. trav. chim., Pays-Bas, 69, 1525 (1950).
13. Drew H. D. K, Sci. J. Roy. Coll. Sci., 8, 33 (1938); Trans. Faraday Soc., 35,
207 (1939).
14. Свешников Б., Дикун П. П., Acta Physica USSR, 17, 173 (1942).
15. Erdey L., Acta Chim. Hung., 3, 95 (1953).
16. Свешников Б., Дикун П. ГЕ, Ж. физ. химии, 19, 289 (1945).
17. Рыжков Б. Д., Изв. АН СССР, сер. физ.. 20, 533 (1956).
18. Kroh J., Roczniki Chem., 28, 70, 511 (1954); 29, 1053 (1955); С.А., 48, 11, 195 (1954); 49, 8003 (1955); 50, 76, 19 (1956).
19. Kroh J., Czernik Z., Roczniki Chem., 31, 915 (1957); C. A., 52, 6928 (1958).
20. Erdey L., Acta Chim. Hung., 3, 81 (1953).
21. Kenny F., Kurtz R. B., Anal. Chem., 24, 1218 (1952).
22. Kenny F., Trans. N. Y. Acad. Sci., 16, 394 (1954).
28
436
ГЛАВА X
23. Пономаренко А. А., Маркарян Н. А., Комлев А. Е., ДАН СССР 86 115» (1952), 89, 1061 (1953).
24. Weiss J., Trans. Faraday Soc., 35, 219 (1939).
25. Bernanose A., Bull. soc. chim. France, 1951, 329.
26. Bremer Th., Bull. soc. chim. beiges, 62, 569 (1953).
27. Erdey L„ Buzds I., Anal. Chim. Acta, 15, 322 (1956).
28. Trantz M., Z. phys. Chem., 53, 86 (1905).
29. Cottman E. W., J. Chem. Educ., 14, 236 (1937).
30. Erdey L„ Pickering W. F., Wilson C. L., Taianta, 9, 371 (1962).
31. Wohler F., Ann. d. Chem. u. Pharm., 127, 257 (1863).
32. Kenny F., Kurtz R. B., Anal. Chem., 22, 693 (1950).
33. Kautsky H., Kolloid Z., 102, 1 (1943).
34. Erdey L., Buzds I., Polos L., Z. anal. Chem., 169, 187 (1959).
35. Erdey L., Buzds I., Polos L., Z. anal. Chem., 169, 263 (1959).
36. Erdey L., Buzds I., Polos L., Z. anal. Chem., 169, 267 (1959).
37. Buzds I., Erdey L., Taianta, 10, 467 (1963).
38. Erdey L., Buzds I., Acta Chim. Hung., 6, 77 (1955).
39. Erdey L., Buzds I., Acta Chim. Hung., 6, 127 (1955).
40. Erdey L., Buzds I., Acta Chim. Hung., 6, 93 (1955).
41. Erdey L., Buzds I., Acta Chim. Hung., 6, 115 (1955).
42. Erdey L., Buzds I., Acta Chim. Hung., 6, 123 (1955).
43. Kenny F., Kurtz R. B., Anal. Chem., 25, 1550 (1953).
44. Kenny F., Kurtz R. B., Beck J., Lukosevicius I., Anal. Chem., 29, 543 (1957).
45. Erdey L., The Industrial Chemist, 1957, 576.
46. Erdey L., Buzds I., Takacs J., Acta Chim. Hung., 41, 37 (1964).
47. Totter J. R., Rhotochem. Photobiol., 3, 231 (1964).
48. McCapra F„ Richardson D. G., Tetrahedron Letters, 1964, 43.
49. Яровенко Я-, Козелева Г. H., Завод, лаб., 27, 407 (1961).
50. Chrzaszczewska A., Bravn A., Lodz. Towarcz. Navk: Wydzial III. Acta Chim.,
9, 189 (1964); C. A., 62, 9103g (1965).
51. Michelski E„ Turowska M., Chem. Anal. (Warszawa), 3, 599 (1958).
52. Turowska M., Chem. Anal. (Warszawa), 5, 815 (1960).
53. Michelski E., Turowska M., Chem. Anal. (Warszawa), 4, 651 (1959).
54. Stauff J., Hartmann G., Ber. Bunsenges. physik, Chem., 69, 145 (1965).
55. Dorabialska A., Kalinowska A., Rcznicki Chem., 38, 457 (1964).
56. Philbrook G. E., Maxwell M. A., Tetrahedron Letters, 1964, 1111.
57. Bermejo F., Badrinas A., Prieto A., Inform, quim. analyt, 14, 151 (1960).
58. Kenny F., Kurtz R. B., Vandenoever A. C., Sanders C. J., Novarro C. A., Menzel L. E., Kukla R., McKenna К- M., Anal. Chem., 36, 529 (1964).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азины 2 71—81
определение рК 2 76
потенциалы 2 76
применение 2 79
Азоиндикаторы 1 106—126 влияние заместителей 1 118, 119 наиболее важные 1 120
Азокрасители 1 322—399
Азо-фиолетовый 1 267
Ализариновый бриллиантовый синий 2 93
Алкалоиды чилибухи 2 107
N-Алкилбензидины 2 199
Алкоксибензидины 2 206
Альфазурин 2 229
потенциалы перехода 2 297
Алюминия, гидроокись 1 233
Амарант 2 351
Аминоантрахиноны 2 117 4-Аминодифениламин 2 196 2-Аминодифениламин-4 -сульфокислота 2 196
Аминоиндофенолы 2 67
З-Аминофталевой кислоты гидразид 2 416
«Амфи-индикаторы» I 238
Анилинсульфофталеины I 168—173
Антрахиноны 2 118
Апосафранины 2 75
Ауксохромные группы 1 96
4-(Ацетиламино)дифениламин 2 196
Белковые вещества, влияние на индикаторы I 105
Безеины 1 173, 175
Бензидин
предшественники и гомологи и их производные 2 167—225
производные, спектральные характеристики 2 215
спектральные характеристики 2 215
Бензидиновая перегруппировка 2 172
Бензидины 2 167
N-алифатические 2 198—202
3,3'-дизамещенные 2 167
индикаторные свойства 2 203
N-производные 2 167
с заместителями в кольце 2 202— 207
Бензоилаурамин G 1 185
Бриллиантовый ализариновый синий
2 92
Бриллиантовый крезиловый синий 2 83, 84
Бриллиантовый пунцовый 5R 2 351
Бромбензидины 2 206
Бромиды, определение 1 282; 2 36
Бромкрезоловый зеленый I 145—148
Бромкрезоловый пурпуровый I 149—
151
Бромтимоловый синий 1 153—156
Бромфеноловый синий 1 151—153
Бруцин 2 107, 111—113
Бруцихинон 2 112
Буферные растворы I 213—216
Вариаминовый синий 2 53—55, 99, 345
применение 2 103
производные 2 98—107
Виологены 2 113—115
Выбор окраски 1 73
Галлофенин 2 82, 83
Галлоцианин 2 82, 83
Гидразобензол 2 171
Гипогалогениты, определение 2 426, 430
Голохиноид 2 168
Двуокись углерода, влияние на индикаторы I 103
438
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
2,4-Диаминодифениламин 2 196
4,4'-Диаминодифениламин 2 197 о-Дианизидин 2 207 Дигидрорезоруфин 2 86
Дииминожелезо(II) и хелатные комплексы 2 303—343
исторический обзор 2 305
Диметиламиноазобензол I 106, 107— 109
бис- (Диметилглиокси мат) железо (II)
2 115
З.З'-Диметилнафтидин 2 209, 213
Диметиловый желтый 1 106, 107—109
бис-3,3'-15,6-Дим етил-1,2,4-триазин)
2 311
Диметилфеносафранин 2 76
3,3'-Диметоксибензидин 2 207
Динитрофенолы 1 128—132
3,7-Диоксифенотиазин 2 92
Дипиридил 2 113—115, 306, 311, 313, 317
константа кислотной диссоциации 2 326
Дифенил, спектральные характеристики 2 215
Дифениламин 2 167, 168, 186
дикарбоновые кислоты 2 196
индикаторные свойства 2 185
монометилпроизводные 2 195
окисление 2 170, 174
спектральные характеристики 2 215
Дифениламина производные 2 167, 186
N-замещенные 2 197
индикаторные свойства 2 185
окисление 2 170
полизамещенные 2 198
спектральные свойства 2 215
Дифенилбензидин 2 167, 168, 178, 186
индикаторные свойства 2 185 окисление 2 177—198
Дифенилбензидина производные 2 167, 186
индикаторные свойства 2 185
окисление 2 177—198
Дифенилбензидиновый фиолетовый 2 177, 178
Дифенилгидразины 2 171
Дифенилкарбазид 1 273; 2 119
Дифенилкарбазиддисульфонат натрия
1 278
Дифенилкарбазон 1 273
Дифенилкарбазон+бром феноловый
синий 1 276
Дифенилкарбазон+ксиленцианоло-вый FF I 277
Дифенилкарбазон+2-нитрозо-1-нафтол 1 280
2,2'-Дихинолилметан 2 207
2,6-Дихлориндофенол 2 48—53, 64
система 2 48
Железо(П)/железо(Ш) обмен электроном 2 304 оксидиметрическое титрование 2 422
Заместители
влияние на кинетику реакции 2 327
— на растворимость 2 321
полярные 2 321
Зеленый Биндшедлера 2 68—71
Изорозиндулины 2 75, 76
Индамины 2 67—71
потенциалы и рД 2 68
Индиго 2 350
Индигокармин 2 55, 59, 60
Индигосульфокислоты 2 55—61
применение 2 58
Индикаторная бумага I 228
Индикаторы
абсорбаты красителей 1 235
адсорбционные 1 35; 2 5—43
• — исторический обзор 2 5
— классификация и характеристика 2 7—17
— методы исследования 2 17
— образующие осадки 2 17
— применение 2 34—41
— теория 2 25—34
анионные 1 45
на бром 2 346—350
броматометрии 2 240, 265, 351
визуальные 1 25—91
— природа и действие 1 25
внутренние 2 126
выбор 1 71, 202, 205, 209, 211; 2 164
для высоких pH I 230
двухцветные 1 43; 2 146
двухэлектронное окисление 2 181
деструкция 2 179, 350—357
идеальные 1 193
изменение окраски 1 37
для измерения pH I 218—219
на иод 2 343
исследование 1 81—91, 194, 195, 198
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
439
кислотно-основные 1 35, 92—246 — неорганические I 233 классификация I 35, 320; 2 166 критерии 2 165
металлофлуоресцентные 2 393— 403
металлохромные 1 35, 289—469 методы наблюдения за изменением 1 36
необратимые 2 150
необратимые и разрушающиеся 2 350—357
неорганические 1 17, 233; 2 360 одноцветные I 43; 2 150
окислительно-восстановительные
1 35, 39, 50, 239; 2 44—372
— вопросы теории 2 46
— с высоким потенциалом 2 125 -372
— деструкция 2 213—225
— • классификация 2 166
— с потенциалом ниже 0,76 В 2 44—124
— применение 2 331
— принцип действия 2 145
— свойства 2 331
— скорость разложения 2 217, 218, 219
особого действия 2 150
оценка 2 161
поверхностные 2 17
— кислотно-основные 2 7, 19, 27 — комплексометрические 2 7, 25, 31
— окислительно-восстановительные 2 7, 23, 31
— для титрования по методу осаждения 2 25
— флуоресцентные 2 7, 25, 32 полифункциональные 1 109 предложенные Гамметом 1 232 применение 2 185
с присоединением ионов 1 35, 41 производные антрацена 1 421 — бензола 1 421, 427—451 — нафталина 1 421, 452—455 различные 1 421, 456—469 смесь с инертным красителем I
191, 194
смешанные 1 109, 191, 280; 2 362, 419, 420
специфического действия 2 343—• 350
теоретический подход 1 26 для титрования бихроматом 2
194, 241
— ванадатом 2 194
— кислот в неводных растворителях 1 261
— кислот в смешанных растворителях 1 248
— оснований в неводных растворителях 1 250
флуоресцентные 2 361, 373—405 — кислотно-основные 2 375—391 — комплексометрические 2 393—•
403
— окислительно-восстановительные 2 392
— для титрования по методу осаждения 2 391
хемилюминесцентные 2 362, 406— 435
— - кислотно-основное титрование 2 410—421
— комплексометрическое титрование 2 430—433
— окислительно-восстановительное титрование 2 421—430
— титрование по методу осаждения 2 433
чувствителпность I 106; 2 185
Индоаналины 2 67—71
потенциалы и рК 2 68
Индофенолы I 187; 2 61—67
применение 2 64
Индулиновый ярко-красный 2 76, 77
Интервал перехода 1 88—91, 100—
101; 2 148
в грет-бутилов о и спирте 1 262
методы измерения I 253
в пиридине 1 267
Интермедиаты разложения индикаторов 2 179
Йодиды, определение I 282; 2 37
Ион «промежуточный», структура 1
107
Кадмий, определение 2 435
Какотелин 2 107—111
применение 2 109
Капри синий С 2 88
2-Карбоксидифениламин-4'-сульфо-кислота 2 197
2-Карбокси-2/-метоксидифениламин 2 197
2-Карбокси-2'-нитродифениламин 2
197
G-Кислота феноксазина 2 83, 86
R-Кислота феноксазина 2 83, 86
Кислотное число жиров и масел, оп-
ределение 2 415
Коллоиды в качестве кислотно-основных индикаторов 1 235
140
предметный указатель
Комплекс ванадия с 1,10-фенантро-лином 2119
Комплексы
железа (П) с замещенными лигандами 2 320—321
меди(1) с замещенными лигандами 2 320—321
металлов с незамещенными лигандами 2 319
осмия 2 337
применение в качестве индикаторов 2 343
рутения 2 337
смешанные 2 339—342
— хелатные 2 341
— цианидные 2 340
хелатные 2 311, 313
Конго красный 2 351
Константа индикатора 1 42; 2 146,
151
определение потенциометрическим методом 2 155
— спектрофотометрическим методом 2 153
Константа комплексообразования 2 315
Константа скорости
восстановления хелатов Fe(III) 2 329
обмена электронами системы Fe (II)/Fe (III) 2 329
окисления комплексов индикаторов 2 330
Коэффициент молярной экстинкции индикаторов 1 73
Красители
индикаторные свойства 2 241, 266—294
как индикаторы в броматометрии 2 238, 239
органические I 92
свойства при титровании церием (IV) 2 265, 292—297, 299, 301, 303
со слабыми индикаторными свойствами 2 241—265
технические как окислительно-восстановительные индикаторы 2 231—303
трифенилметановые 1 173—182, 400
— как окислительно-восстановительные индикаторы 2 226—303 — свойства 2 231—237, 238 фталоцианиновые 2 302
Крахмал 2 343, 344, 345
Крезоловый красный 1 158—160 .м-Крезоловый пурпуровый 1 157—158
Кремниймолибденовая кислота 2 118
Кристаллический фиолетовый I 257
Лакмоид 1 186
Лакмус I 17, 188
Лейкотионолин 2 95
Лиганды, содержащие «.а'-диимино-
группу, стерические эффекты 2 308
Лиссаминовый синий BF 2 76, 78
Лофин 2 417—419
Люминол 2 416—417, 426
Люцигенин 2 411—415, 424
Малахитовый зеленый 1 258
Меконовая кислота 2 116
Меркаптаны алифатические, определение I 283
Меркуриметрия 1 271—284
применение 1 281
Металлоиндикаторы 1 289—469 коэффициенты побочных реакций 1 470
названия и синонимы I 483
pMtrans I 474
пояснения к таблицам 1 321
применение I 312, 316
структура 1 318
теория 1 289
титрование ступенчатое I 307 2-Метилвариаминовый синий 2 55 2-Метилдифениламин-4'-сульфокис-лота 2 196
Метиленовый белый 2 90
Метиленовый голубой I 109; 2 89, 93, 94—96, 98, 345, 351
Метилкапри синий 2 83, 84
Метиловый желтый 1 107
Метиловый красный I 106, 114—116, 258; 2 351
Метиловый оранжевый 1 106, 107, 110—113, 258; 2 351
Метиловый фиолетовый I 259
М-Метил-2-оксифеназин 2 76, 78 Метод
Гольдмана 1 278
Джилеспи I 223
Джоба 1 86
Кларка 1 277
Мак-Клери 1 282
Михаэлиса 1 219
Робертса I 275
Методы титрования I 250
З-Метокси-4-аминодифениламин 2 196 2-Метоксивариаминовый синий 2 55 Мочевина+дифенилкарбазон 1 281 Мускарин DH 2 88
предметный указатель
44t
Нафтидин и его производные 2 208— 213
потенциалы перехода 2 210
спектральные характеристики
окисленных форм 2 212, 215
Нафтидины 2 167
3,3'-дизамещенные 2 167
1-Нафтолбензеин 1 259
а-Нафтоловый оранжевый 1 107
Нафтоловый сине-черный 2 351
а-Нафтолфлавон 2 349
а-Нафтол фталеин 1 134—135
Нейтрализация
многокислотных оснований 1 209
многоосновных кислот I 205
смесей кислот 1 205
смесей оснований 1 209
Нейтральный красный I 184; 2 73, 76
Нейтральный синий 2 75, 76
Никель, цианометрическое определение 1 284
Нильский синий А 1 259; 2 83, 84, 88
Нитразиновый желтый 1 107
п-Нитро-п'-аминоазобензол 1 267
о-Нитроанилин 1 267
4-Нитродифениламин 2 195
Нитроиндикаторы 1 126—132
Нитропруссид натрия 1 272
Нитрофенолы I 127
Новый метиленовый голубой 2 88, 93
Область перехода 1 43, 44
Окраска индикаторов, влияние условий на ее изменение 1 101—106
Оксазины 2 81—89
потенциалы и рК 2 83
Оксиновый синий 1 183
8-Оксихинолин, азопроизводные 1 280
Оранжевый IV 1 260
Ошибки
белковая 1 226
дискриминационная 1 78
индикатора 1 79—81, 303; 2 426, 427
колориметрического определения pH 1 224—227
титрования 1 74—81; 2 161, 354, 355
химическая 1 74—78, 202, 205, 208; 2 162
pH, определение
без буферных растворов 1 219
быстрый метод 1 227
колоричгетрический 1 210, 224
с помощью буферных растворов 1 212
Парарозанилинфуксин 2 346, 347
Пиоцианин 2 72, 76
1-(2-Пиридилазо)-2-нафтол 1 281
3- (2-Пиридил) -5,6-дифенил-1,2,4-
триазин 2 311
трнс-(2'-Пиридил)-1,3.5-триазин 2 310
2. 6-Пиридиндиамидоксим 2 310
Порфирексид 2 359
Порфиридин 2 359
Потенциал точки эквивалентности 2 135
Потенциалы
зависимость от pH 2 48—55
окислительно-восстановительные 2 129
стандартные комплексов ос-мий(П)/осмий(Ш) 2 342
формальные 2 46
— комплексов медь(1)/медь(П) 2 338
Продукты деструкции индикатора 2 179
Простой нейтральный красный 2 73, 76
Прочный синий В для хлопка 2 88
Прюн 2 83, 85
Равновесия
индикаторов 1 39
титриметрические 1 37
Реакции
асимметричные 1 67
индуцированные 2 144
нейтрализации, определение конечной точки 1 198—200
обмена электронами 2 327
окислительно-восстановительные
1 33, 37, 2 127—129, 329
— установление равновесия 2 132
побочные 1 294, 298
— коэффициенты 1 470—473
— pMtrans 1 474—482
присоединения ионов 1 34, 38
симметричные 1 64—67
типы 1 33
титриметрические, кинетика и механизм 1 30
— классификация 1 29
титрования и равновесия 2 133— 145
Рез азурин 2 86
Резоруфин 2 83, 86
Роданид, определение 2 39
Розанилин 2 343, 346
Розидон сульфированный 2 76, 77
Розиндулин 2G 2 72, 73, 76
442
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Ртуть (I), определение 2 40
Ртуть(II), определение 2 432
Сафранин Т (или О) 2 74, 76
Свинец, определение 2 432, 433
Серебро, определение 2 39
Силоксен 2 421, 422, 435
Смеси индикаторов 1 193, 196
Смолы ионообменные, применение в
качестве индикаторов 1 237
Соли Вурстера 2 170, 177
Соли ртути (II), определение 1 285
Сульфат, определение 2 40
Сульфат железа(III) основной, коллоидный 1 234
Сульфат меди+хлорид аммония 1 234
Сульфофталеины 1 137, 142—168,
400—412; 2 357, 360
Теория
Оствальда 1 92—95
Оствальда, объединенная с хромофорной теорией 1 97—98
резонанса 1 98—100
хромофорная I 95—97
Терпиридил 2 311, 313, 318
Тетрабромтионолин 2 95
Тетраметилфеносафранин 2 76
Тетрафенилгидразины 2 173
Тетраэтилфеносафранин 2 76
Тиазиновый синий 2 93
Тиазины 2 89—98
потенциалы и рК 2 93
применение 2 94
Тимоловый синий 1 162, 163, 168, 260, 268
Тимолсульфофталеин 1 260
Тимолфталеин 1 136—137
Гиоиндиосульфокислоты 2 61
Тиокол 2 92, 93, 95
Тионолин 2 95
Тионин 2 89, 93
Тиофлуоресцеин I 283
Титриметрический анализ 1 27
Титрование
влияние различных параметров 1 50—51
выбор условий 1 70; 2 138, 141 — 144
гипобромитом 2 427
гипохлоритом 2 429
с замещением 1 209
комплексометрическое меди (II) 2 432
кулонометрическое 1 68 оснований 2 417
сильных и слабых кислот 2 419
сильных кислот сильными основаниями 1 200
сильных оснований сильными кислотами 1 200
слабых кислот 1 261; 2 416, 417 слабых кислот сильными основаниями 1 203
слабых оснований сильными кислотами 1 207
в среде двух несмешивающихся растворителей 1 210
ступенчатое 1 307
фотометрическое и автоматическое 2 409
цианометрическое 1 284—287 N-Толилантраниловые кислоты 2 197 о-Толуидиновый синий 2 68, 93, 94 .и-Толуилендиамининдофенол 2 68 Толуиленовый синий 2 68, 69 Точка
перехода 1 42, 43, 71
эквивалентности 1 71
Трифенилгидразины 2 173 Трифенилкарбинол 1 261 4,4',4"-Трифенил-2,2/,2"-терпиридил 2 310
Тропеолины 1 116—117
Универсальные индикаторные растворы 1 227
Феназин 2 71
1,10-Фенантролин 2 306, 311, 313, 315
трис- (о-Фенантролин)железо(П) 2
118
1,10-Фенантролин и его производные, константы кислотной диссоциации 2 324—326
п-Фениламиноазобензол 1 268
Фениламины
вторичные 2 176
третичные 2 175
N-Фенилантраниловая кислота 2 118
Фенилгидразин, производные 1 188— 190
Фенил-2-пиридилкетоксим 2 311
1-Фенилтиосемикарбазидат меди 1 283
Феноксазин 2 82
Феноксазон 2 87
Феноловый красный 1 160—162
Фенолфталеин 1 135, 268
Феносафранин 2 74, 76
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
443
Фенотиазин 2 89
Фиолетовый Лаута 2 89, 90
Фирмы — производители красителей
2 263- 264
Флороглюцин 1 280
Флуоресцеин + люминол 2 420
Фталеины 1 133—137, 138—142, 412—
420
Фталоцианины 2 297
Функция Q 1 68; 2 136
Хинальдиновый красный 1 260
Хиноны 2116
Хлориды, определение 1 271—281; 2
34
Хлорорафин 2 76, 78
Хлорпромазингидрохлорид 2 96, 97
Хлорфеноловый красный 1 156—157
Цианид ртути (II)+роданпд хро-ма(Ш) 1 234
Цианиды, определение 1 284
Цинк, определение 1 39
Экстракты растений 1 190
Этил-б«с-[2,4-динитро]фенилацетат 1 186
Этилендиаминтетрауксусная кислота, определение 2 432
Этилкапри синий 2 83, 84
п-Этоксихризоидин 2 351
л-Этоксихризоидингидрохлорид 1
109—110
Эффекты стерические 2 309
СОДЕРЖАНИЕ
Глава VII. Адсорбционные индикаторы. Е. Пунгор, Е. Шулек .... 5
Исторический обзор........................................ 5
Классификация и краткая характеристика адсорбционных индикаторов 7
Методы исследования адсорбционных индикаторов . 17
Теория адсорбционных индикаторов ................................ 25
Применение адсорбционных индикаторов......................34
Применение адсорбционных индикаторов в других областях аналитической химии............................................... 40
Список литературы ............................................... 41
Глава VIII. Окислительно-восстановительные индикаторы . 44
Л. Окислительно-восстановительные индикаторы, имеющие потенциал £q<0,76 В. Дж. Оттоуэй.......................................44
1. Введение....................................................44
2. Вопросы теории ........................................... 46
3. Индигосульфокислоты.........................................55
4. Индофенолы................................................. 61
5. Иидоаиалииы и индамины......................................67
6. Азины.......................................................71
7. Оксазины....................................................81
8. Тиазины.....................................................89
9. Производные вариаминового синего........................... 98
10. Какотелин и бруцин....................................... 107
11. Виологены...................................................ИЗ
12. Разные индикаторы..........................................115
Слисок литературы................................................119
Б. Окислительно-восстановительные индикаторы с высоким формальным потенциалом. Е. Бишоп......................................... 125
Введение..................................................... 125
Окислительно-восстановительные реакции.........................127
Окислительно восстановительные потенциалы..................... 129
Установление равновесия........................................132
Реакции титрования и равновесия .............................. 133
Общие принципы действия окислительно-восстановительных индикаторов ..................................................... 145
I
СОДЕРЖАНИЕ
445
Константы индикатора............................................146
Интервал перехода (интервал изменения окраски) ................ 148
Необратимые индикаторы и индикаторы особого действия . . . 150
Определение константы индикатора .............................. 151
Ошибки титрования и поправки на индикатор.......................161
Выбор подходящего индикатора....................................164
Классификация окислительно-восстановительных индикаторов . . 166
1. Бензидин, его предшественники и гомологи и их производные 167
А. Окисление дифениламина и его производных.................170
Б. Окисление дифенилбензидина и его производных . . . . 177
В. N Алифатические бензидины................................198
Г. Бензидины с заместителями в кольце.............. . . 202
Д. Нафтидин и его производные . .... 208
Заключение.....................................213
2. Трифенилметановые и другие красители..........226
Характеристика красителей в качестве окислительно-восстанови-тельных индикаторов ........................................231
Выводы.........................................303
3. Дииминожелезо (II) и близкие ему хелатные комплексы . . . 303
Введение.......................................303
Исторический обзор.............................305
Лиганды, содержащие а.а'-дииминогруппу ..... 306
Образование комплексов . . ...... 311
Влияние заместителей .... 321
Окислительно-восстановительные свойства 327
Свойства индикаторов . 331
Применение.....................................331
Смешанные комплексы............................339
Другие случаи применения комплексов в качестве индикаторов 343
4. Индикаторы специфического действия............343
5. Необратимые и разрушающиеся индикаторы . 350
6. Различные соединения.......................................358
7. Смешанные индикаторы . . . . . .............362
Списоклитературы..................................................362
Глава IX. Флуоресцентные индикаторы. Г. Ф. Киркбрайт............373
Введение.....................................................373
Флуоресцентные кислотно-основные индикаторы..................375
Флуоресцентные индикаторы в титровании по методу осаждения . . 391
Флуоресцентные индикаторы для окислительно-восстановительного титрования...................................................392
Флуоресцентные индикаторы для комплексометрического титрования 393
Списоклитературы. 403
Глава X. Хемилюминесцентные индикаторы. Л. Эрдей................406
Введение.....................................................406
Применение хемилюминесцентных индикаторов в кислотно-основном титровании.......................................................410
Люцигенин (диметилдиакридилдинитрат) в качестве хемилюминесцентного кислотно-основного индикатора...............................411
Люминол (гидразид 3-аминофталевой кислоты) в качестве хемилюминесцентного кислотно основного индикатора........................416
Лофин (2,4,5-трифенилимидазол) в качестве хемилюминесцентного кислотно-основного индикатора . . ...........................417
446 СОДЕРЖАНИЕ
Хемилюминесцентный смешанный индикатор...........................419
Хемилюминесцентный индикатор в окислительно-восстановительном титровании . ..... ....................... 421
Силоксен в качестве хемилюминесцентного окислительно-восстановительного индикатора..............................................422
Люцигенин в качестве обратимого хемилюминесцентного окислительно-восстановительного индикатора ... .......................424
Люминол в качестве хемилюминесцентного окислительно-восстанови тельного индикатора ............................................ 426
Комплексометрическое титрование с использованием хемилюминесцентных индикаторов..................................................430
Титрование по методу осаждения с использованием хемилюминесцентных индикаторов..................................................433
Список литературы................................................435
Предметный указатель ............................................437