/













































































































































































































































































































































































































































































Text
ОСНОВЫ АНАЛИТИЧЕСКОЙ ХИМИИ
ИЗДАТЕЛЬСТВО «ХИМИЯ»
МОСКВА 1971
4Шк
А.П.КРЕШКОВ
ОСНОВЫ
теоретические
основы
т
количественный
анализ
АНАЛИТИЧЕСКОЙ
ХИМИИ
качественный
и количественный
анализ
издание третье (переработанное)
¦
ДОПУЩЕНО МИНИСТЕРСТВОМ ВЫСШЕГО
И СРЕДНЕГО СПЕЦИАЛЬНОГО ОБРАЗОВАНИЯ СССР
В КАЧЕСТВЕ УЧЕБНИКА
ДЛЯ СТУДЕНТОВ ХИМИКО-ТЕХНОЛОГИЧЕСКИХ
СПЕЦИАЛЬНОСТЕЙ ВУЗОВ
УДК 543.062 (075.8)
К79
Крешков А. П. Основы аналитической химии.
Теоретические основы. Количественный анализ.
Книга является второй частью курса «Основы
аналитической химии» и предназначена в качестве учебника для
студентов химико-технологических специальностей высших
учебных заведений.
В книге изложены теория, методика и техника
количественного анализа. Наряду с объемным и весовым
методами в книге описаны основы электрохимических,
спектральных, радиометрических и хроматографических
методов конечного определения, а также методы разделения,
выделения и концентрирования отдельных компонентов
анализируемых смесей.
Особое внимание в книге уделено описанию техники
химического эксперимента, методике количественных
определений, правилам работы, способам расчета и учету
ошибок анализа.
В книге содержится 456 стр., 30 таблиц и 97
рисунков.
2-5-5
16-70
Анатолий Павлович Крешков
ОСНОВЫ АНАЛИТИЧЕСКОЙ ХИМИИ
Книга вторая
Издательство «Химия», М., 1971 г.
456 с УДК 643.062 (075.8)
Редактор Л. Н. Овсянникова
Технический редактор 3. И. Яковлева
Корректор Р. Л. Вилкомирская
T-14863. Подписано к печати 21/Х 1970 г.
Формат бумаги 7UXl08x/ie« Печ. л. 28,5. Усл. печ. л» 39,9
изд. л. 38,52. Тираж 80 000 (2-й завод 30 001—80000) экз. Типогр. 6ум.№ 2
Цена 1 р. 53 к. Тем. пл. 1970 г., № 16 Зак. № 931
Ленинградская типография № 6 Главполиграфпрома
Комитета, по печати при Совете Министров СССР
Ленинград, С-144, ул* Моисеенко, 10
СОДЕРЖАНИЕ
Предисловие к первому изданию 11
Предисловие ко второму изданию 13
Предисловие к третьему изданию 15
Введение < 17
§ 1. Понятие о количественном анализе 17
§ 2. Классификация методов количественного анализа 20
§ 3. Характеристика методов количественного анализа 21
§ 4. Анализ больших и малых количеств вещества 24
§ 5. Отбор средней пробы 25
§ 6. Подготовка вещества для взвешивания 26
§ 7. Взвешивание 27
§ 8. Техника взвешивания на аналитических весах 30
§ 9. Правила обращения с аналитическими весами 35
§ 10. Приготовление раствора для анализа 36
§ 11. Запись результатов анализа 36
ЧАСТЬ ПЕРВАЯ
ОБЪЕМНЫЙ АНАЛИЗ
Глава I. Основы объемного анализа 38
A. Общая характеристика объемного анализа 38
§ 1. Сущность объемного анализа 38
§ 2. Общее уравнение реакции титрования и выводы из него 45
Б. Техника химического эксперимента в объемном анализе 49
§ 3. Измерение объемов растворов 49
§ 4. Посуда, применяемая для измерения объемов растворов 51
§ 5. Работа с мерными колбами 54
§ 6. Работа с пипетками 56
§ 7. Работа с бюретками 58
§ 8. Приготовление стандартных растворов 61
B. Вычисления в объемном анализе 65
§ 9. Концентрация растворов и способы ее выражения 65
§ 10. Способы вычисления в объемном анализе 68
§ 11. Связь между точностью измерений и точностью вычислении .... 79
§ 12. Краткие сведения о статистической обработке экспериментальных
данных 82
Г. Полу микрообъемный метод анализа 88
§ 13. Понятие о пол у микрообъемном анализе 88
§ 14. Особенности техники измерения объемов растворов в полумикро-
методе % 88
Д. Безбюреточные методы титрования • 88
§ 15. Понятие о безбюреточных методах титрования 88
§ 16. Классификация методов безбюреточного титрования 88
6
СОДЕРЖАНИЕ
Е. Автоматические методы 90
§ 17. Химико-аналитический контроль производства 90
§ 18. Автоматические методы титрования 90
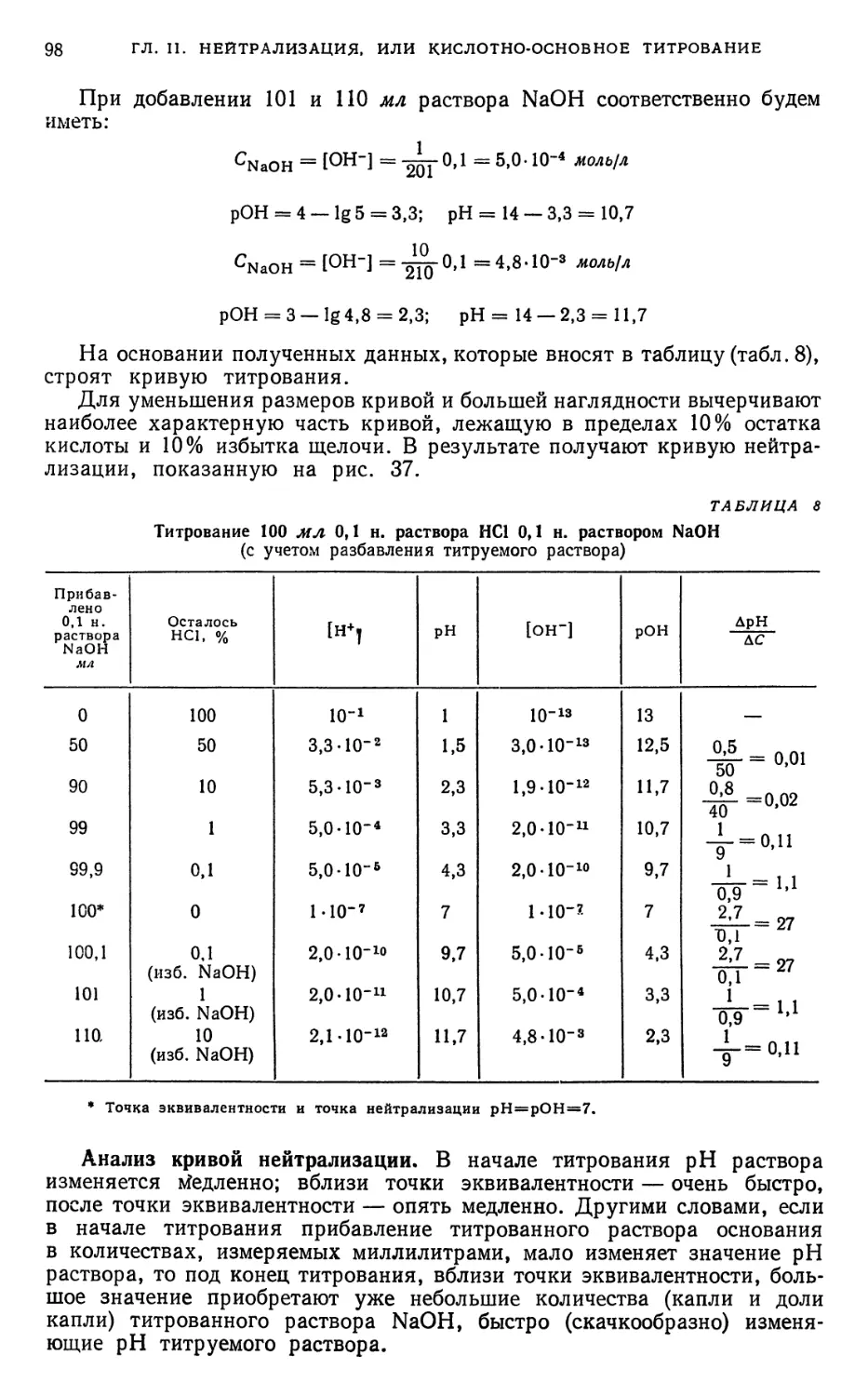
Глава II. Методы нейтрализации, или методы кислотно-основного титрования 93
A. Теоретическая часть 93
§ 1. Характеристика метода 93
§ 2. Установление точки эквивалентности 93
§ 3. Графический метод изображения процесса нейтрализации 94
§ 4. Вычисление концентрации ионов водорода в водных растворах
сильных кислот и оснований 94
§ 5. Вычисление активности ионов водорода в водных растворах сильных
кислот и оснований 96
§ 6. Титрование сильной кислоты сильным основанием 96
§ 7. Вычисление концентрации ионов водорода в растворах слабых кислот
и оснований 104
§ 8. Вычисление активности ионов водорода в водных растворах слабых
кислот и оснований 105
§ 9. Равновесия в водных буферных растворах слабых кислот в
присутствии солей этих кислот 106
§ 10. Равновесия в водных буферных растворах слабых оснований в
присутствии солей этих оснований 107
§ 11. Вычисление концентрации ионов водорода в водных буферных
растворах 109
§ 12. Вычисление активности ионов водорода в водных буферных рас*
творах , , , , . ПО
§ 13. Вычисление концентрации ионов водорода и степени гидролиза в
водных растворах гидролизующихся бинарных солей 111
§ 14. Вычисление активности ионов водорода в водных растворах
гидролизующихся бинарных солей 120
§ 15. Титрование слабой кислоты сильным основанием 121
§ 16. Титрование слабого основания сильной кислотой . . , 126
§ 17. Титрование многоосновных кислот 130
§ 18. Титрование солей, образованных катионами сильных оснований и
анионами слабых многоосновных кислот 133
§ 19. Изменение активности и показателя активности ионов водорода в
процессе титрования водных растворов кислот и оснований 136
§ 20. Выводы, вытекающие из рассмотрения кривых нейтрализации , % , 137
§ 21. Индикаторы 138
§ 22. Интервал перехода индикатора 147
§ 23. Выбор индикатора 150
§ 24. Ошибки титрования 153
Б. Практическая часть 158
§ 25. Организация рабочего места 458
§ 26. Приготовление стандартных (титрованных) растворов 158
§ 27. Приготовление 0,1 н. раствора хлористоводородной кислоты % % % , 160
§ 28. Установка титра 0,1 н. раствора хлористоводородной кислоты ... 160
§ 29. Приготовление 0,1 н. раствора едкого натра 170
§ 30. Установка титра 0,1 н. раствора едкого натра 170
§ 31. Определение карбонатов 174
§ 32. Определение содержания H2S04 в технической серной кислоте , , % . 176
§ 33. Определение содержания уксусной кислоты 177
§ 34. Определение содержания Na2C03 и NaOH при их совместном
присутствии 179
§ 35. Определение содержания Na2C03 и NaHC03 при их совместном
присутствии ; 181
§ 36. Определение жесткости воды 183
§ 37. Определение аммонийного азота в солях аммония 184
§ 38. Определение содержания фосфорной кислоты 187
B. Кислотно-основное титрование в неводных средах 188
§ 39. Неводные растворы , 188
§ 40. Современные представления о кислотах и основаниях 189
§ 41. Диссоциация электролитов в неводных растворах 192
§ 42. Влияние неводных растворителей на силу кислот и оснований , , % 193
§ 43. Применение закона действия масс к растворам сильных электролитов 194
§ 44. Титрование кислот и оснований в неводных растворах 198
СОДЕРЖАНИЕ
7
§ 45. Методы кислотно-основного титрования в неводных средах 202
§ 46. Примеры практических определений в неводных растворах 204
Глава III. Методы окисления — восстановления (оксидиметрия, оксредметрия,
ред-окс-методы) 211
Л. Теоретическая часть 211
§ 1. Значение окислительно-восстановительных потенциалов 211
§ 2. Реакции окисления—восстановления и комплексообразование , , . . 215
§ 3. Примеры окислительно-восстановительного титрования 217
§ 4. Константы равновесия окислительно-восстановительных реакций . . . 221
§ 5. Связь между константами равновесия
окислительно-восстановительных реакций и нормальными потенциалами 221
§ 6. Вычисление констант равновесия окислительно-восстановительных
реакций 222
§ 7. Вычисление окислительно-восстановительных потенциалов и
констант равновесия окислительно-восстановительных реакций с учетом
коэффициентов активностей 224
§ 8. Зависимость скорости реакций окисления—восстановления от
различных факторов 224
§ 9. Графический метод изображения процесса
окисления—восстановления 230
§ 10. Фиксирование точки эквивалентности в методах
окисления—восстановления 239
§ 11. Окислительно-восстановительные индикаторы (ред-окс-индикаторы) , . 239
Б. Перманганатометрия 244
§ 12. Основы перманганатометрии 244
§ 13. Титрование перманганатом в кислой среде 245
§ 14. Титрование перманганатом в щелочной среде ч • • 246
§ 15. Приготовление стандартного (титрованного) раствора перманганата
калия 247
§ 16. Установка титра стандартного раствора перманганата калия , . , , 248
§ 17. Установка титра и нормальности раствора перманганата калия по окса-
лату аммония 249
§ 18. Вещества, определяемые методом перманганатометрии 250
Определение восстановителей 251
§ 19. Определение щавелевой кислоты и оксалатов 251
§ 20. Определение соединений железа (II) , 252
§ 21. Определение содержания металлического железа в присутствии окислов
железа 253
§ 22. Определение азотистой кислоты и нитритов 254
§ 23. Определение содержания марганца (II) в рудах 254
Определение окислителей 256
§ 24. Определение соединений железа (III) 256
§ 25. Определение нитратов 257
§ 26. Определение бихроматов 258
§ 27. Определение содержания Мп02 в пиролюзите 259
Определение других веществ 260
§ 28. Определение ионов кальция . , 260
В. Иодометрия 262
§ 29. Основы иодометрии . , 262
§ 30. Методы иодометрического* титрования , , , 263
§ 31. Преимущества и недостатки иодометрического метода 265
§ 32. Приготовление стандартного (титрованного) раствора тиосульфата и
установка его титра 267
§ 33. Приготовление стандартного (титрованного) раствора иода и уста-*
новка его титра 270
Методы прямого титрования 270
§ 34. Определение мышьяка (III) 270
Методы обратного титрования 271
§ 35. Определение сульфита натрия 271
§ 36. Определение содержания формальдегида в формалине 272
Методы косвенного определения 273
§ 37. Определение ионов меди (II) 273
§ 38. Определение двуокиси свинца в сурике % , , , 275
Метод титрования заместителей 276
§ 39. Определение содержания двуокиси марганца в пиролюзите 276
Метод определения кислот 277
8
СОДЕРЖАНИЕ
§ 40. Определение хлористоводородной кислоты 277
§ 41. Определение воды по Фишеру 277
Г. Понятие о других методах окисления—восстановления 278
§ 42. Хроматометрия 278
§ 43. Определение содержания железа (II) 282
§ 44. Цериметрия 282
§ 45. Броматометрия 284
§ 46. Ванадатометрия 285
§ 47. Аскорбинометрия 286
§ 48. Титанометрия 288
Глава IV. Методы осаждения и комплексообразования 289
A. Теоретическая часть 289
§ 1. Общая характеристика методов 289
§ 2. Классификация методов осаждения и комплексообразования 291
§ 3. Применение теории осаждения к объемному анализу 292
§ 4. Вычисление растворимости электролитов в воде с учетом
коэффициентов активности 294
§ 5. Влияние одноименных ионов на растворимость малорастворимого
электролита 296
§ 6. Солевой эффект 300
§ 7. Влияние концентрации ионов водорода на растворимость
малорастворимых соединений 300
§ 8. Кривые титрования в методе осаждения 302
§ 9. Общие выводы, вытекающие из рассмотрения кривых осаждения . , , 311
§ 10. Адсорбционные явления, наблюдаемые при титровании по методу
осаждения 312
Б. Аргентометрия 318
§ 11. Характеристика метода 318
§ 12. Приготовление 0,1 н. раствора нитрата серебра 320
§ 13. Приготовление стандартного раствора хлорида натрия 320
§ 14. Установка титра 0,1 н. раствора нитрата серебра по точной навеске
хлорида натрия 320
§ 15. Определение ионов хлора в техническом хлориде натрия по методу Мора 322
§ 16. Определение хлоридов по методу Фаянса 322
B. Роданометрия 323
§ 17. Характеристика метода 323
§ 18. Приготовление 0,1 н. раствора роданида аммония 323
§ 19. Определение ионов хлора в растворимых хлоридах по методу Фоль-
гарда 324
§ 20. Определение серебра в сплавах 325
Г. Меркуриметрия * 326
§ 21. Характеристика метода 326
§ 22. Приготовление 0,1 н. раствора нитрата ртути (II) 327
§ 23. Установка титра раствора нитрата дтути (II) 327
§ 24. Определение ионов хлора в воде меркуриметрическим методом , , . 328
Д. Меркурометрия 328
§ 25. Краткая характеристика метода 328
Е. Комплексонометрия (хелатометрия) 329
§ 26. Характеристика метода 329
§ 27. Теоретические основы комплексонометрического титрования 332
§ 28. Классификация методов комплексонометрического титрования . . , 338
§ 29. Установка титра раствора комплексона III >% 339
§ 30. Определение содержания кальция 340
§ 31. Определение жесткости воды комплексонометрическим методом , . , 342
§ 32. Анализ смеси ионов кальция и магния 343
§ 33. Определение содержания алюминия 343
§ 34. Раздельное определение ионов кальция и алюминия 345
§ 35. Раздельное определение ионов алюминия и железа 345
СОДЕРЖАНИЕ
9
ЧАСТЬ ВТОРАЯ
ВЕСОВОЙ АНАЛИЗ
Глава V. Основы весового анализа „. 347
A. Общая характеристика весового анализа 347
§ 1. Сущность весового анализа 347
§ 2. Классификация методов весового анализа 347
§ 3. Расчеты в весовом анализе 349
Б. Техника весового анализа 353
§ 4. Взятие и растворение навески 353
§ 5. Техника осаждения 355
§ 6. Фильтрование и промывание осадков 356
§ 7. Получение весовой формы 359
§ 8. Взвешивание весовой формы 361
B. Теоретическая часть 361
§ 9. Теоретические основы выделения осадков из растворов с помощью
специфических неорганических и органических реактивов 361
§ 10. Требования, предъявляемые к осадкам ,_ . . . 363
§ 11. Методы повышения точности весовых определений 366
§ 12. Теоретические обоснования выбора оптимальных условий для весового
определения 367
Г. Практическая часть 369
§ 13. Определение кристаллизационной воды в ВаС12-2Н20 369
§ 14. Определение сульфат-ионов или серы 370
§ 15. Определение ионов железа (III) 373
§ 16. Определение содержания кальция в карбонате кальция . 375
§ 17. Определение содержания магния 376
§ 18. Определение ионов хлора в растворимых хлоридах или в
хлористоводородной кислоте 377
§ 19. Анализ силикатов 378
§ 20. Анализ доломита 388
§ 21. Анализ бронзы и латуни 390
Д. Методы весовых определений, основанные на применении органических
реактивов 392
§ 22. Определение никеля 392
§ 23. Определение алюминия 393
ЧАСТЬ ТРЕТЬЯ
ПОНЯТИЕ О ФИЗИЧЕСКИХ И ФИЗИКО-ХИМИЧЕСКИХ
(ИНСТРУМЕНТАЛЬНЫХ) МЕТОДАХ АНАЛИЗА
Глава VI. Классификация физических и физико-химических методов
количественного анализа 395
§ 1. Электрохимические методы 395
§ 2. Спектральные (оптические) методы 396
§ 3. Хроматографические методы 397
§ 4. Радиометрические методы 397
§ 5. Масс-спектрометрические методы 400
Глава VII. Электровесовые методы анализа 401
§ 1. Характеристика методов электроанализа 401
§ 2. Химические процессы, протекающие при электролизе 402
§ 3. Методы электроанализа 403
§ 4. Электровесовой анализ 404
§ 5. Метод внутреннего электролиза 406
§ 6. Определение меди в растворе сульфата меди с применением
платиновых сетчатых электродов 407
§ 7. Определение меди и свинца в латуни с применением платиновых
сетчатых электродов. 410
§ 8. Определение малых количеств меди методом внутреннего электролиза 411
10
СОДЕРЖАНИЕ
Глава VIII. Объемные электрохимические методы анализа 413
§ 1. Особенности объемных электрохимических методов анализа 413
§ 2. Кондуктометр ическое титрование 413
§ 3. Высокочастотное титрование 415
§ 4. Потенциометр ическое титрование 416
§ 5. Полярографический метод анализа 418
§ 6. Амперометрическое титрование 424
§ 7. Кулонометрическое титрование 425
Глава IX. Спектральные (оптические) методы анализа 428
A. Эмиссионный спектральный анализ 428
§ 1. Понятие об эмиссионном спектральном анализе 428
Б. Колориметрия 429
§ 2. Особенности колориметрических методов анализа 429
§ 3. Характеристика колориметрических методов анализа 430
B. Оптические методы установления точки эквивалентности 432
§ 4. Спектрофотометр ическое титрование 432
§ 5. Фототурбидиметрическое и фотонефелометр ическое титрование , . . . 434
Г. Лабораторные работы 434
§ 6. Определение содержания ионов железа методом колориметрического
титрования 434
§ 7. Определение содержания титана 435
Глава X. Методы разделения, выделения и концентрирования отдельных
компонентов анализируемых смесей 438
§ 1. Определение следов элементов (микропримесей) 438
§ 2. Метод осаждения малорастворимых соединений 439
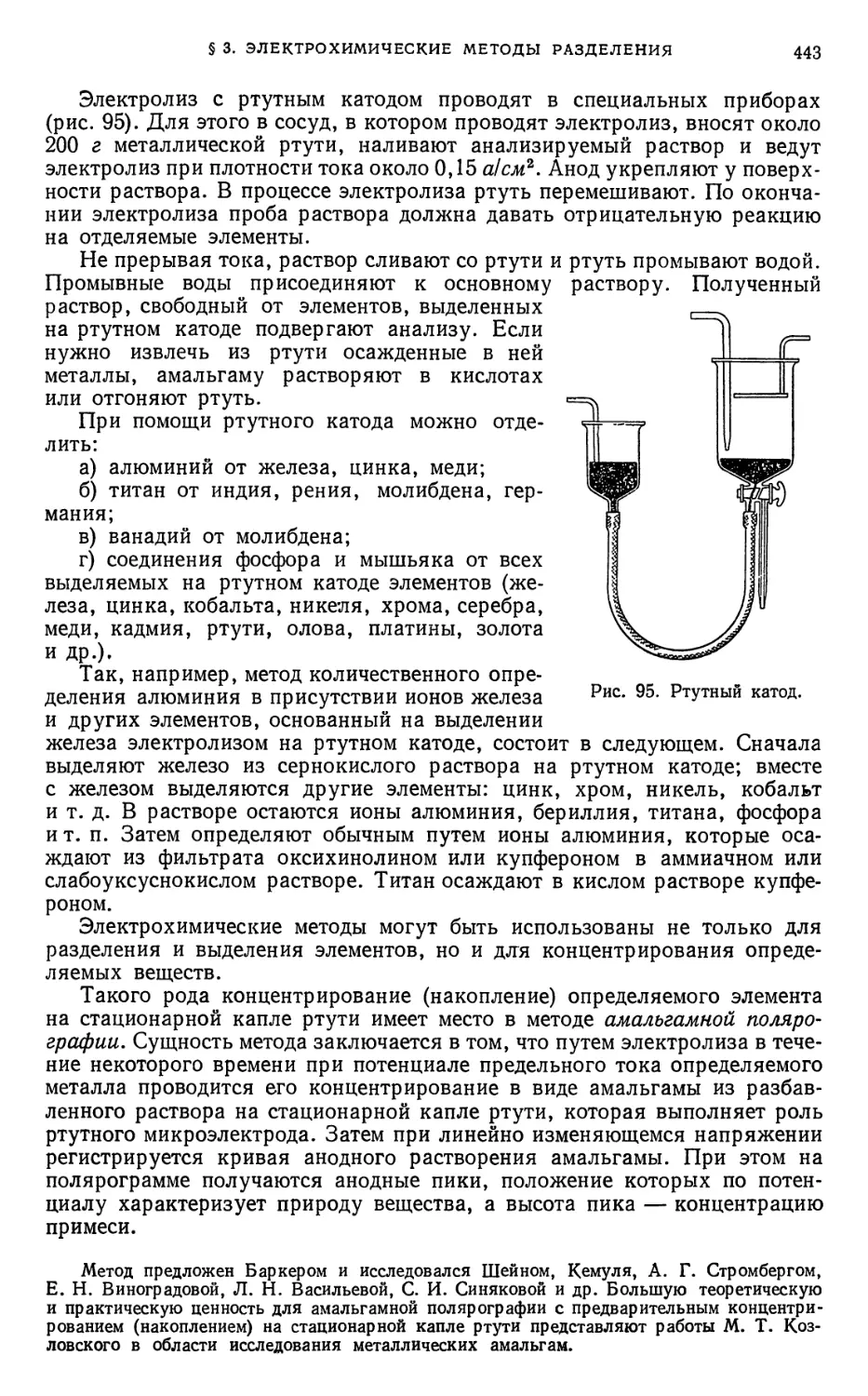
§ 3. Электрохимические методы разделения 442
§ 4. Метод экстрагирования 444
§ 5. Метод отгонки летучих соединений 444
§ 6. Хроматографические методы разделения 445
§ 7. Метод флотации 449
Литература * 449
Предметный указатель
450
ПРЕДИСЛОВИЕ К ПЕРВОМУ ИЗДАНИЮ
Настоящий учебник предназначен для студентов технологических
институтов и составлен применительно к программе по аналитической химии
для технологических специальностей высших учебных заведений,
утвержденной Министерством высшего и среднего специального
образования РСФСР. В методическом отношении он отражает многолетний опыт
постановки преподавания аналитической химии в Московском химико-
технологическом институте им. Д. И. Менделеева.
В книге кратко излагаются основы аналитической химии,
определяющие минимум знаний, который должен получить студент. Подробные
сведения из области аналитической химии, непрерывно развивающейся
в связи с прогрессом химической науки и производства, студент получит
из лекционного курса, излагаемого на основе опыта и достижений,
сложившихся в каждом втузе научных школ и направлений.
Учебник состоит из двух книг. Качественный анализ излагается в
первой книге, количественный — во второй. Общие вопросы теории,
являющиеся основой аналитической химии как науки, излагаются в первой
книге. Теоретические вопросы, относящиеся непосредственно к
качественному или к количественному анализу, рассматриваются при изложении
соответствующих разделов курса.
В первой книге описываются макро-, микро-, полумикрометоды, а также
хроматографические, люминесцентный и некоторые другие методы
анализа. Наряду с описанием реакций катионов и анионов, которые обычно
рассматриваются в учебниках по качественному анализу, приводится
описание реакций и методов разделения наиболее важных редких и
рассеянных элементов (лития, рубидия, цезия, бериллия, титана, циркония, тория,
урана, германия, ванадия, вольфрама, молибдена и др.), которые
изучаются студентами только некоторых специальностей. Однако материал
учебника расположен таким образом, что при необходимости описание
упомянутых элементов может быть выпущено без особого ущерба для
изложения основного курса.
Во второй книге излагаются основы весового и объемного химического
анализа, а также дается понятие о физических и физико-химических
методах анализа (электрохимических, спектроскопических, хроматографи-
ческих, радиометрических и др.), нашедших широкое применение в прак-
12
ПРЕДИСЛОВИЕ К ПЕРВОМУ ИЗДАНИЮ
тике научно-исследовательских и заводских лабораторий. При изучении
количественного анализа рассматриваются также основы теории и
практики методов титрования в неводных растворах, приобретших большое
значение в различных областях науки, промышленности и новой техники.
Особое внимание в учебнике уделено описанию техники химического
эксперимента, разбору условий проведения реакций, методикам
определения, правилам и способам расчетов.
Практические работы, помещенные в учебнике, являются примерными.
Тип и число заданий и последовательность их выполнения можно
варьировать в соответствии со специальностями данного втуза.
В конце каждой книги помещены списки использованной авторами и
рекомендуемой студентам литературы.
В составлении учебника приняли участие доценты, кандидаты
химических наук: С. С. Вильборг, написавшая главу III второй книги «Методы
окисления—восстановления», Ю. Я. Михайленко, написавший главу VIII
второй книги «Спектральные методы анализа», А. Н. Яровенко,
написавшая совместно с автором книги главу V второй книги «Основы весового
анализа».
Автор считает приятным долгом поблагодарить своих сотрудников,
принявших то или иное участие при подготовке рукописи к печати, и
выразить глубокую благодарность коллективу кафедры аналитической
химии Ленинградского технологического института им. Ленсовета и
профессору, доктору химических наук Н. П. Комарю, сделавшим
многоценных замечаний при просмотре рукописи.
А. П. КРЕШКОВ
ПРЕДИСЛОВИЕ КО ВТОРОМУ ИЗДАНИЮ
При подготовке второго издания учебника перед автором стояла
довольно трудная задача ответить на многочисленные пожелания
рецензентов. Основная причина возникавших затруднений обусловливалась резким
противоречием между возросшим за последнее время значением
аналитической химии в науке, промышленности и технике, вызывающим
обоснованные требования рецензентов расширить некоторые из разделов
учебника, с одной стороны, и резким снижением в учебных планах времени,
отводимого на этот предмет, — с другой стороны.
Стараясь не выходить за рамки учебной программы, автор подверг
учебник существенной переработке. Произведены некоторые сокращения
за счет исключения наименее важных сведений. Теоретические и
практические разделы учебника дополнены новыми литературными данными,
отражающими достижения современной химической науки. Впервые в
учебнике представлены безбюреточные методы титрования, приведены понятия
о кинетических методах анализа, описаны методы разделения,
концентрирования, обнаружения л определения ультрамалых количеств примесей
в особо чистых веществах и т. п.
Крупным шрифтом набран текст, отвечающий минимуму сведений,
требуемых для усвоения курса, предусмотренного программой. Мелким
шрифтом напечатан дополнительный материал, гармонически связывающий в
методическом отношении все разделы учебника.
В целях развития у студентов навыков самостоятельной работы со
справочной литературой из учебника изъяты все справочные таблицы
приложения, которые студенты должны научиться находить, пользуясь
рекомендуемой справочной литературой.
В составлении 2-го издания учебника приняли участие доцент
А. Н. Яровенко, написавшая совместно с автором главу V второй книги,
и старший преподаватель Е. К. Крешкова, написавшая совместно с
автором главу III второй книги.
В заключении автор считает своим приятным долгом выразить всем
коллективам кафедр аналитической химии и всем товарищам, приславшим
свои отзывы об учебнике, глубокую благодарность за весьма ценные
замечания и пожелания. Особую признательность автор выражает
коллективам кафедр аналитической химии Казанского химико-технологического
14
ПРЕДИСЛОВИЕ КО ВТОРОМУ ИЗДАНИЮ
института, Московского института тонкой химической технологии,
Московского технологического института мясо-молочной промышленности,
Ярославского технологического института, Харьковского и Горьковского
политехнических институтов, Ленинградского, Ростовского, Саратовского
и Уральского университетов; академику КазССР М. Т. Козловскому,
профессорам А. И. Бусеву, М. X. Карапетянцу, П. Н. Коваленко,
К. Н. Мочалову, И. С. Мустафину, К. М. Ольшановой, В. И. Тихомирову,
Ф. И. Тришину и доцентам Ф. К. Баеву, В. Ф. Барковскому, Д. В. Без-
углому, В. В. Васильеву, Н. И. Витальской, 3. М. Графовой, С. И. Дра-
кину, И. П. Ефимову, М. Н. Зверевой, Р. Н. Новикову, И. Л. Орестову,
А. А. Соболевой, Т. А. Худяковой, А. И. Черкесову.
Автор также выражает глубокую благодарность всем своим ученикам
и сотрудникам кафедры аналитической химии МХТИ им. Д. И.
Менделеева, принимавшим то или иное участие в подготовке рукописи к печати.
Все замечания, способствующие дальнейшему совершенствованию
учебника, автор примет с благодарностью.
АВТОР
ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
Аналитическая химия как наука о методах химического анализа
является одной из основных общехимических (а в ряде высших учебных
заведений и профилирующих) учебных дисциплин, изучаемых студентами
химических и химико-технологических вузов и факультетов. Она играет
огромную воспитательную роль в процессе подготовки молодых
специалистов. Будущий химик или инженер-технолог-химик начинает
формироваться при изучении аналитической химии, составляющей для него наряду
с другими химическими и общенаучными дисциплинами фундамент
материалистического мировоззрения и прочное основание для специальных
знаний.
С давних пор, по традиции, аналитической химии в учебных планах
отводилось место вслед за курсом неорганической химии. Поэтому
аналитическая химия являлась как бы естественным продолжением курса
неорганической химии. Это обстоятельство накладывало особый отпечаток
на программу аналитической химии, представлявшей собой теорию и
практику так называемых классических (качественного, весового и объемного)
методов анализа неорганических соединений. Все к этому привыкли, и
раньше это оправдывалось многими обстоятельствами.
В настоящее время теория и практика аналитической химии
претерпели существенное изменение. Современную аналитическую химию нельзя
изучать на основе только неорганической химии. В связи с широким
применением органических реагентов, используемых для обнаружения и
количественного определения многих неорганических и органических
веществ, а также применяемых в виде стандартных (титрованных) растворов
(например, комплексонов), индикаторов, экстрагентов, соосадителей,
ионообменных смол, органических растворителей и т. п., аналитическую химию
необходимо изучать не только на базе неорганической, но и органической
химии.
С другой стороны, современная аналитическая химия испытывает
сильное влияние экспериментальной физики и физической химии. Мощное
развитие этих наук, чрезвычайное разнообразие и точность их методов
изучения материи все больше изменяет основное направление развития
аналитической химии. Для решения задач химического анализа в различных
областях промышленности, науки и новой техники весьма широко исполь-
16
ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
зуются физические и физико-химические (инструментальные) методы,
именно вследстБие этого они составляют одну из неотъемлемых частей
аналитической химии и изучаются в курсе аналитической химии.
Вот почему подготовлен третий том настоящего учебника. В первой
книге излагаются общие теоретические основы аналитической химии и
качественный анализ; во второй — количественный анализ (объемный и
весовой); в третьей — физико-химические (инструментальные) методы
анализа (электрохимические, спектральные, хроматографические,
радиометрические и др.), а также методы определения редких элементов и
титрование неводных растворов.
При подготовке третьего издания учебника автор столкнулся с рядом
трудностей. Наиболее сложно было ответить на просьбы читателей,
высказавших пожелания ввести в учебник дополнения. Стремясь не выходить
за рамки учебной программы, автор сделал все от него зависящее и подверг
учебник существенной переработке. Первая книга «Качественный анализ»
сокращена. Вместе с тем в нее наряду с сероводородным методом анализа
катионов введен бессероводородный метод, разработанный и
апробированный на кафедре аналитической химии МХТИ им. Д. И. Менделеева. Вторая
книга «Количественный анализ» несколько расширена, в нее внесено много
новых материалов. Приведено описание некоторых новых методов
анализа; расширены вопросы теории; переработан раздел, посвященный
анализу силикатов; дано представление об автоматических методах
титрования; описаны способы статистической математической обработки
результатов анализа; рассмотрены некоторые вопросы теории строения вещества
и теории химической связи в их химико-аналитическом аспекте; особое
внимание уделено уточнению формулировок, определений и отдельных
положений.
В составлении 3-го издания учебника приняли участие старший
преподаватель Е. К. Крешкова, написавшая совместно с авторов главы III
и V, и доцент А. Н. Яровенко, написавшая также совместно с автором
главу V.
Пользуясь случаем, автор выражает глубокую благодарность за
консультативную помощь профессорам Е. М. Александровой, Н. А. Фи-
гуровскому, П. Я. Яковлеву и доценту Н. А. Каверину и всем своим
ученикам и сотрудникам за весьма ценные замечания.
АВТОР
ВВЕДЕНИЕ
§ 1. Понятие о количественном анализе
Количественный анализ предназначен для определения количественных
соотношений составных частей исследуемого вещества. Другими словами,
количественный анализ дает возможность установить количественный
элементный или молекулярный состав анализируемого вещества или
содержание отдельных его компонентов.
В ряде случаев требуется установить содержание всех элементов, ионов
или соединений, входящих в состав данного исследуемого вещества.
Например, при анализе медных сплавов (бронз и латуней) определяют
содержание меди, олова, свинца, цинка и других элементов. При анализе
растворов электролитных ванн, применяемых для никелирования металлов,
определяют содержание Ni++, Zn++, CN~, ОН" и т. п.
В других случаях требуется установить содержание некоторых
отдельных элементов, ионов или соединений, входящих в состав анализируемого
продукта. Так, при анализе металлического сплава химика-аналитика
может интересовать лишь содержание меди и олова, или ванадия и
вольфрама, или алюминия и магния, или только железа и т. д.
Иногда определяют не только общее содержание того или иного
элемента (иона), но и формы нахождения его в исследуемом веществе.
Например, при анализе руды определяют не только общее содержание серы, но
и содержание свободной (S0),. сульфидной (S~"~), пиритной ([S2]"~) и
сульфатной (S(V~) серы.
В задачу количественного анализа входит также определение
разнообразных реакционноспособных (активных) атомов и функциональных
групп в различных (преимущественно в органических) соединениях.
Совокупность химических, физических и физико-химических методов,
применяемых для решения этой задачи, называют функциональным анализом.
К такого рода методам относятся титриметрические, электрохимические
(потенциометрические, полярографические, хронокондуктометрические и
др.), спектроскопические [фотоколориметрические, спектрофотометр
ические, инфракрасная спектроскопия (ИКС), ультрафиолетовая
спектроскопия (УФС)], метод комбинационного рассеивания света (КРС), ядерный
магнитный резонанс (ЯМР), рентГеноспектроскопия, масс-спектроскопия,
хроматографические и другие методы (см. ниже).
Одним из важных разделов количественного анализа является так
называемый фазовый анализ, который имеет большое значение в цветной
и черной металлургии. Фазовый анализ представляет собой совокупность
разнообразных химических, физических и физико-химических методов
разделения и определения отдельных структурных (фазовых)
составляющих гетерогенных систем. К этим методам относятся: химические и
электрохимические методы избирательного растворения, рентгеноструктурный
18
ВВЕДЕНИЕ
петрографический, металлографический, кристаллооптический, электрон-
номикроскопический, термографический и др.
Фазовый анализ сталей и сплавов дает возможность судить о
содержании отдельных структурных (фазовых) составляющих исследуемого сплава
(карбидов, боридов, нитридов, карбонитридов, карбоборидов, интерметал-
лидов, свободного и связанного углерода и т. п.), т. е. о составе отдельных
фаз гетерогенных систем.
При исследовании вещества неизвестного состава (например, шлака,
руды, сплава и т. п.) количественному анализу предшествует
качественный, так как выбор метода количественного определения каждой составной
части анализируемого вещества зависит от результатов качественного
анализа.
Часто бывает известен качественный состав анализируемых веществ
(кислот, оснований, солей, сплавов и т. п.), а нередко известно и
приблизительное содержание в них отдельных компонентов. Поэтому при
исследовании известного вещества (например, соды, технической серной кислоты
и т. п.) в большинстве случаев не требуется предварительно проводить
качественный анализ этого вещества. В таких случаях определяют
содержание данного вещества в анализируемом образце или концентрацию его
раствора. Иногда определяют только содержание одного или нескольких
элементов, не являющихся основными компонентами данной сложной
смеси, т. е. определяют примеси, например серу и фосфор в чугуне и стали,
благородные металлы в отходах металлургического производства и т. д.
В последнее время в связи с развитием новых отраслей
промышленности стало необходимым определять содержание в анализируемом
веществе ничтожнейших количеств примесей (микропримесей). Определение
микропримесей имеет большое значение при анализе особо чистых
веществ.
Таким образом, количественный анализ позволяет установить:
1. Количественные соотношения составных частей неизвестного
индивидуального соединения, т. е. установить его формулу.
2. Содержание или концентрацию определяемого вещества в
исследуемом образце.
3. Содержание всех или некоторых элементов или ионов, входящих
в состав данного вещества.
4. Содержание всех или некоторых главных (основных) компонентов
анализируемой смеси (например, смеси солей, кислот, изомеров и т. п.).
5. Содержание определенных форм того или иного элемента или
простых и сложных веществ, образуемых им.
6. Содержание неглавных (неосновных) компонентов (примесей) в
данном известном веществе.
7. Содержание микропримесей в особо чистых веществах (металлах,
сплавах, полупроводниковых материалах, графите и т. п.).
8. Содержание определенных радикалов, активных атомов,
функциональных групп.
9. Состав отдельных фаз гетерогенных систем, в которых определяемые
вещества распределяются в зависимости от изменения рецептуры
получаемого технического объекта, способа его получения, термической и
механической обработки и т. д.
В широком смысле слова количественным анализом следует называть
совокупность химических, физических и физико-химических методов
исследования, позволяющих с требуемой точностью определять в образце
анализируемого вещества количественное содержание отдельных составных частей
или концентрацию их в растворе, а также устанавливать содержание
примесей в исследуемом техническом объекте.
§ 1. ПОНЯТИЕ О КОЛИЧЕСТВЕННОМ АНАЛИЗЕ
19
Основоположником современного количественного анализа является
М. В. Ломоносов, положивший начало систематическому применению весов
при химических исследованиях.В 1756 г. М. В. Ломоносов
экспериментальным путем доказал сформулированный им еще ранее (1748 г.) закон
сохранения массы вещества, являющийся основой количественного анализа.
М. В. Ломоносовым созданы основы физической химии, оказавшей
существенное влияние на развитие теории аналитической химии. В 1748 г.
М. В. Ломоносов организовал первую в России химическую лабораторию.
Его научные исследования имеют важное значение в истории развития
русской химической науки.
Примеры количественного анализа. Количественный анализ основан
на точном измерении массы и объема определяемых веществ или продуктов
их химических превращений, или расходуемых реактивов, вступающих
в реакции с определяемыми веществами.
Например, определяемую составную часть анализируемого вещества
выделяют в виде осадка, который отфильтровывают, промывают от
посторонних примесей, высушивают или прокаливают и взвешивают. Зная
массу выделившегося вещества (так называемой весовой формы) и его
формулу, можно вычислить содержание определяемого вещества. Так,
определяют НС1 по массе осадка AgCl, выделившегося при
взаимодействии Ag+ и С1".
Другим примером может служить измерение объема раствора AgN03
точно известной концентрации, израсходованного на реакцию с НС1. Зная
объем и концентрацию прибавленного раствора AgN03, можно вычислить
содержание НС1 в соляной кислоте.
Примером количественного анализа может также служить измерение
объема выделившейся двуокиси углерода, получаемой при взаимодействии
определенной навески мела с хлористоводородной кислотой. Зная объем
газа С02, можно вычислить содержание карбонатов в меле.
В ряде случаев в целях количественного анализа измеряют различные
показатели оптических, электрических и других физических свойств
исследуемых веществ. Данные измерений используют для вычисления
результатов анализа.
Результаты количественного анализа выражают различными
способами. Например, содержание в растворе хорошо растворимого соединения
выражают в граммах, в граммах на 100 г раствора, в процентах, в граммах
или в миллиграммах на 1 миллилитр;
содержание малорастворимых веществ в граммах
на 1 л и в молях на 1 л. Состав
металлических сплавов выражают в процентах
содержания элементов в сплаве и т. д.
Значение количественного анализа.
Количественный анализ является основным
методом контроля химических процессов, сырья,
промежуточных и готовых продуктов
производства, а также наряду с качественным
анализом служит важнейшим методом
исследования при выполнении химических научно-
исследовательских работ.
Количественный анализ играет большую
роль в науке, технике и промышленности,
в значительной степени способствуя
прогрессу химической промышленности и
связанных с ней отраслей производства, а также
развитию химии и других естественных М. В. Ломоносов (1711—1765).
20
ВВЕДЕНИЕ
наук, например геохимии, геологии, минералогии, агрохимии, биологии,
почвоведения, медицины и т. п.
Внедрение в производство и научно-исследовательскую работу
высокочувствительных и точных методов количественного определения
ультрамалых количеств примесей в значительной мере способствовало развитию
атомной и полупроводниковой техники, производству жаростойких
сплавов и высококачественных полимерных материалов.
§ 2. Классификация методов количественного анализа
Все методы количественного анализа в зависимости от характера
экспериментальной техники, применяемой для конечного определения
составных частей анализируемого вещества или смеси веществ, делят на три
группы: химические, физические и физико-химические (инструментальные)
методы анализа.
К химическим методам анализа относятся:
1. Весовой анализ — измерение массы определяемого вещества или его
составных частей, выделяемых в химически чистом состоянии или' в виде
соответствующих соединений.
2. Объемный анализ — измерение объема жидких, твердых и
газообразных продуктов или их водных и неводных растворов.
Известны разнообразные объемные методы:
1) объемный титриметрический — измерение объема
израсходованного на реакцию реактива точно известной концентрации;
2) газовый объемный — анализ газовых смесей, основанный на
избирательном поглощении из анализируемой газовой смеси определяемого
компонента подходящими поглотителями;
3) седиментационный объемный *, основанный на расслоении
дисперсных систем под действием силы тяжести, сопровождающемся отделением
дисперсной фазы в виде осадка и последующем измерении объема осадка
в калиброванной центрифужной пробирке. Например, в микро- и
ультрамикроанализе содержание серы находят путем окисления ее до сульфатной
и последующего осаждения в виде осадка сульфата бария, определяемого
указанным методом.
В более широком смысле седиментационным анализом называют метод определения
в дисперсных системах величины и относительного содержания частиц различных размеров
по скорости седиментации (оседания или всплывания).
Скорость седиментации сферических частиц при известных условиях описывается
уравнением Стокса:
2r«(tf-<f')g
9г)
где v — скорость седиментации;
г — радиус частицы;
d — плотность материала частицы;
d! — плотность дисперсной среды;
г) -— вязкость среды;
— ускорение силы тяжести.
чень часто в лабораторной практике применяют весовые методы седиментационного
анализа, основанные на гидростатическом взвешивании осадка в процессе его накопления
при помощи седиментационных стеклянных весов Н. А. Фигуровского.
В ряде случаев разделение методов анализа на химические и физико-
химические условно, так как иногда трудно или практически невозможно
решить вопрос о принадлежности того или иного метода анализа к какой-
либо из указанных групп.
* Название «седиментационный» происходит от латинского слова Sedimentum —
оседание.
§ 3. ХАРАКТЕРИСТИКА МЕТОДОВ КОЛИЧЕСТВЕННОГО АНАЛИЗА 21
Перечисленные методы являются лишь методами конечного определения
определяемого вещества или его составных частей и не отражают всех
особенностей химического анализа.
Существенной частью химического анализа, на выполнение которой
химику-аналитику иногда приходится расходовать больше времени и
труда, чем на конечное определение определяемого вещества, являются
методы разложения анализируемого вещества, а также методы разделения,
выделения и концентрирования определяемых элементов (или ионов).
§ 3. Характеристика методов количественного анализа
Весовой анализ. Весовой анализ основан на точном измерении массы
определяемого вещества или его составных частей, выделяемых в
химически чистом состоянии или в виде осадка точно известного постоянного
состава, в котором содержится определяемое соединение или ион.
Например, определение НС1 по количеству выделившегося осадка AgCl
относится к весовому методу анализа. Массу получаемого осадка определяют
при помощи аналитических весов.
Содержание определяемого компонента в исходном веществе
вычисляют исходя из весовых количеств вещества, взятого для анализа, и
вещества, полученного в результате химической реакции (после
соответствующей обработки). Так, если из а граммов исходного вещества получено
b граммов определяемого компонента, то процентное содержание этого
компонента в исходном веществе вычисляют по формуле:
х ^JLiooo/o (1)
Объемный (титриметрический) анализ. Объемный анализ основан
на измерении объема раствора реактива точно известной концентрации,
израсходованного на реакцию с данным количеством определяемого
вещества.
Например, определение содержания НС1 в соляной кислоте по объему
раствора AgN03 точно известной концентрации, пошедшему на реакцию
образования AgCl, относится к объемному методу анализа.
Прибавление к определяемому веществу А раствора реактива В
известной концентрации продолжают до завершения реакции, т. е. до
достижения так называемой точки эквивалентности. Объем израсходованного
раствора реактива VB, точно измеряют при помощи специальных приборов.
Весовое количество реактива В, пошедшего на реакцию, вычисляют,
умножая величину объема раствора реактива (VB) на содержание его в единице
объема (Тв). Величину Тв называют титром раствора. Так как 1 грамм-
эквивалент анализируемого вещества А (ЭА) вступает в реакцию с 1 грамм-
эквивалентом реактива В (Эв), то содержание анализируемого
вещества (#д) в граммах вычисляют из пропорции:
или в процентах
где а — навеска вещества,
ЭА-ЭВ
gA-^-TB
„ УВЭА
§А=-Эв~
Vb3a 100
г.
(2)
Отличие весового анализа от объемного (титриметрического). Для
определения одного и того же вещества в одном и том же объеме, например для
22
ВВЕДЕНИЕ
определения содержания НС1 в соляной кислоте, могут быть использованы
различные методы (весовой и титриметрический).
Весовой метод анализа отличается от титриметрического рядом
особенностей.
1. В основе весового метода анализа лежит точное измерение массы,
в основе объемного метода — измерение объема.
2. В весовом анализе измеряют массу определяемого вещества или
малорастворимого осадка, в котором содержится определяемое соединение или
ион; в объемном анализе измеряют объем реактива, использованного для
реакции с анализируемым веществом.
3. В весовом анализе используют растворы основных исходных
реактивов приблизительной концентрации; в объемном анализе применяют
реактивы точно известной концентрации.
4. В весовом анализе, как правило, растворы реактивов приливают
к анализируемому раствору в избытке; в объемном анализе — в строго
эквивалентных количествах.
5. В весовом анализе не имеет особого значения вопрос, когда наступил
момент эквивалентности; в объемном анализе решающее значение имеет
фиксирование точки эквивалентности, которую определяют с помощью
индикатора или другим способом.
6. В весовом анализе требуется много времени для осаждения
вещества, декантации, фильтрования, промывания осадка, подготовки его
к взвешиванию и т. д. Все операции в объемном анализе проводятся
быстрее, чем в весовом анализе, так как определение в объемном анализе
по существу начинается и заканчивается процессом постепенного прили-
вания раствора реактива к анализируемому веществу до завершения
реакции.
Так, для завершения определения весовым способом требуется от 2 до
24 ч\ определение объемным методом продолжается обычно от нескольких
минут до 1 ч.
7. Весовой анализ отличается большой точностью порядка 0,01—
0,005%. Объемный анализ менее точен, достигаемая точность составляет
0,1—0,05%.
Газовый объемный анализ *. Газовый анализ основан на поглощении
газов соответствующими поглотителями. Например, при анализе газовой
смеси, состоящей из СО2 +СО + 02 + СлН2л, двуокись углерода
поглощают раствором КОН или NaOH, окись углерода — аммиачным
раствором Си2С12, кислород — раствором пирогаллола, непредельные
углеводороды — серной кислотой. При этом содержание определяемого компонента
вычисляет на основании уменьшения объема оставшейся газовой смеси
или падения давления.
К химическим методам газового анализа относят также газоволюметри-
ческий метод, основанный на определении содержания того или иного
вещества по количеству выделившегося газа при взаимодействии
определяемого вещества с реактивом. Например, для того, чтобы установить
содержание карбонатов в соде, навеску анализируемого продукта
обрабатывают кислотой. Выделяющуюся при этом двуокись углерода поглощают
раствором едкого кали и взвешивают или измеряют объем выделившегося
газа. По привесу сосуда, содержащего раствор КОН, вычисляют
содержание С02, поглощенной щелочным раствором. Зная массу выделившейся
С02, легко вычислить содержание карбонатов в образце соды.
* Существующие химические, физические и физико-химические методы газового
анализа весьма многочисленны, но описание их не входит в нашу задачу.
§ 3. ХАРАКТЕРИСТИКА МЕТОДОВ КОЛИЧЕСТВЕННОГО АНАЛИЗА 23
Физические и физико-химические (инструментальные) методы анализа.
Весовой и объемный методы химического анализа дают возможность
определять количественный состав самых разнообразных веществ. Однако
выполнение определений этими методами иногда связано с большими
трудностями, возникающими главным образом в тех случаях, когда в ходе
анализа необходимо предварительно отделить определяемую составную
часть от примесей. Особенно трудно выделять индивидуальные вещества
из очень сложных смесей, компоненты которых обладают близкими
свойствами. Часто интересующая химика-аналитика составная часть
содержится в анализируемом веществе в столь малых количествах, что
выделение ее обычными химическими методами практически невозможно.
К недостаткам химических методов анализа относится и сравнительно
небольшой предел их чувствительности, несмотря на относительно
большую точность определений.
Наконец, для выполнения анализа объемным и в особенности весовым
методом, как правило, требуется много времени. Между тем на
производстве скорость аналитического определения часто играет решающую роль.
Поэтому наряду с химическими методами анализа все более широко
применяются физические и физико-химические методы, отличающиеся
многими преимуществами по сравнению с химическими методами. Иногда
эти методы анализа называют «инструментальными».
Обычные химические методы основаны на применении химических
реакций, протекающих с образованием осадков (в методах осаждения) или
с выделением газов (в газовом анализе), реакций окисления —
восстановления (в методах оксидиметрии) и т. п. Однако состав вещества иногда
можно определить и другими методами, не связанными с химическими
реакциями. В таких случаях для определения состава анализируемого
вещества оказывается достаточным измерить показатели каких-либо
физических свойств, например коэффициент лучепреломления, электро- или
теплопроводность, потенциал электрода, погруженного в исследуемый
раствор, и т. п. Так, определив плотность раствора кислоты или щёлочи,
можно найти по соответствующим справочным таблицам процентное
содержание их в данном растворе. Опустив в исследуемый раствор водородный
или другой подходящий электрод, можно очень быстро определить с
помощью потенциометра концентрацию ионов водорода (или рН) данного
раствора. Такие методы количественного анализа, позволяющие
определять состав анализируемого вещества, не
прибегая к использованию химических реакций,
называют физическими методами анализа.
Для количественного анализа вещества
можно использовать также химические
реакции, протекание которых сопровождается
изменением физических свойств
анализируемого раствора, например изменением его
цвета, интенсивности окраски, величины
электропроводности и т. п. Измеряя
электропроводность какого-либо электролита,
изменяющуюся в результате взаимодействия
его с другим веществом, можно определить
количество этого вещества в растворе.
Например, электропроводность баритовой воды
изменяется в процессе поглощения ею
двуокиси углерода. На этом свойстве основан
метод определения С02. Если через
баритовую воду пропускать газ, содержащий С02, Н. С. Курнаков (1860—1941).
24
ВВЕДЕНИЕ
и одновременно измерять ее электропроводность, то можно найти
количество С02, поглощенное баритовой водой, и рассчитать процентное
содержание двуокиси углерода в исследуемом газе.
Методы анализа, основанные на наблюдении изменений физических
свойств анализируемой системы, происходящих в результате
определенных химических реакций, называют физико-химическими методами.
Физические и физико-химические методы анализа отличаются большой
чувствительностью и быстротой выполнения аналитических определений.
Например, пользуясь радиоактивационным методом, можно определять
в анализируемых веществах 10"*% примесей, что во много раз
превосходит возможности весового, объемного, и других методов анализа. Время,
требуемое для завершения анализа физическими и физико-химическими
методами, иногда измеряется минутами.
Физико-химические методы количественного анализа не следует
смешивать с физико-химическим анализом по Н. С. Курнакову, с помощью
которого изучают физические свойства систем в зависимости от их
химического состава.
§ 4. Анализ больших и малых количеств вещества
В количественном анализе приходится иметь дело с анализом больших
и малых количеств исследуемого вещества. В соответствии с этим
различают микро-, полумикро- и макроколичественный анализ.
Микро- и полумикрометоды позволяют применять весовой и объемный
методы для анализа очень малых количеств (порядка 10 мг) определяемого
вещества, экономить реактивы и время, требующиеся для выполнения
анализов.
Отличие макро- от микрометода. При использовании макрометодов
анализа имеют дело с относительно большими количествами веществ.
В весовом анализе работают с навесками, превышающими 0,05—0,1 г,
которые взвешивают с точностью до десятитысячных долей грамма. В
объемном анализе измеряют объемы растворов и газообразных веществ,
превышающих 1 мл, и проводят отсчеты с точностью до 0,020—0,025 мл.
При использовании микро- и полумикрометодов работают со
значительно меньшими количествами веществ. Навески берут в пределах от
1 до 50 мг и применяют специальные микровесы. Объемы измеряют при
помощи микробюреток емкостью несколько миллилитров,
градуированных на 0,01 мл, и делают отсчеты с точностью до 0,0020—0,0025 мл.
В табл. 1 приведена сравнительная характеристика различных химико-
аналитических методов.
ТА БЛИЦА t
Сравнительная характеристика макро- и микроаналитических методов
Метод
Макро-
Полумикро-
Микро-
Ультрамикро-
Качественный анализ
масса
пробы, мг
>100
юо—ю
10—Ю-2
<ю-2
объем
раствора,
мл
>ю
10—1
1—Ю-1
ю-1-
—иг3
количество
определяемого вещества,
мг
>10"2
10-2—Ю-з
Ю-з__1о-б
10-е—ю-11
Количественный анализ
масса пробы,
мг
>100
100—10
10—1
1—Ю-1
объем
раствора, мл
>ю
10—1
1—Ю-1
до 10" 3—
—10-е
количество
определяемого вещества,
мг
>10"1
Ю-1—10-2
10~3
Ю- з—ю- е
^
§ 5. ОТБОР СРЕДНЕЙ ПРОБЫ
25
Микро-и полумикрометоды незаменимы в тех случаях, когда в
распоряжении аналитика имеется очень незначительное количество вещества.
Указанные методы очень полезны и тогда, когда для анализа есть достаточное
количество исследуемого вещества.
Теоретические основы микро-, полумикро- и макрометодов одни и те же.
В этом отношении между ними нет принципиального различия. Разница
состоит лишь в применяемой технике химического эксперимента, в
конструкции используемых приборов, аппаратов и химической посуды.
Следует иметь в виду, что при работе с маленькими навесками
вещества часто могут происходить ошибки, вызываемые неполнотой отделения
определяемого вещества, растворимостью осадков, значительными
потерями в процессе экспериментирования и т. д. Неизбежными являются
потери, наблюдаемые при отделении определяемого элемента от других
компонентов анализируемой смеси. Поэтому строго соблюдают все
указания, относящиеся к технике выполнения анализа. Используют по
возможности методы непосредственного (прямого) определения многих
элементов в присутствии посторонних веществ, а если это невозможно, то
применяют не более одной операции разделения.
Применяемые в микрометодах реактивы должны отличаться особенной
чистотой. Кроме того, как правило, с целью учета влияния небольших
количеств примесей, встречающихся даже в очень тщательно очищенных
реактивах, проводят параллельно «холостые» опыты.
Введение микрометодов в весовой анализ встречает некоторые
технические затруднения из-за необходимости пользоваться микровесами.
Методы анализа, позволяющие определять тысячные и миллионные доли
миллиграмма вещества и оперировать с объемами растворов, не
превышающими 0,001 мл, называют количественным ультрамикроанализом.
В микро- и ультрамикроанализе, как ц. в макроанализе, наряду с
весовым и объемным методами анализа широко применяют также
разнообразные физикр-химические методы анализа.
Большую роль в разработке и во внедрении в практику методов микро-
и ультрамикроанализа в СССР сыграли работы советских ученых И. П. Али-
марина, М. Н. Петриковой, И. М. Коренмана и др.
§ 5. Отбор средней пробы
Перед исследованием вещество предварительно подготавливают к
анализу. К числу, таких подготовительных операций относятся: 1) отбор
средней пробы; 2) приготовление вещества для взвешивания; 3)
взвешивание вещества; 4) приготовление раствора для анализа.
Массу пробы анализируемого вещества, отбираемой для
количественного определения макрометодом, измеряют долями грамма,
микрометодом — несколькими миллиграммами, а ультрамикрометодом —
несколькими микрограммами. Между тем на производстве обычно приходится
иметь дело с б©льшими количествами сырья, полупродуктов, готовой
продукции и других технических материалов, измеряемыми многими сотнями
тонн. Поэтому очень важно, чтобы химический состав отбираемой для
анализа пробы в точности соответствовал среднему химическому составу всей
партии анализируемого продукта.
Следовательно, прежде всего необходимо правильно отобрать большую
среднюю пробу от всей массы материала, а затем первоначальную пробу
отобранного материала разделить до получения проб, требуемых для
проведения химического анализа. Задача заключается в том, чтобы измельчить
и перемешать отобранную пробу, сократить ее массу и в то же время
сохранить в окончательной пробе такое содержание компонентов, которое
равно количественному содержанию их во всей массе начальной пробы.
26
ВВЕДЕНИЕ
Для того чтобы получить пробу, достаточную для полного и
всестороннего анализа, а также необходимую на случай повторного и арбитражного,
т. е. спорного, анализа, выполняемого в особом порядке, установленном
законом, обычно отбирают от нескольких десятков до нескольких сот
граммов вещества.
Отбор средней пробы подлежащего анализу вещества, не
представляющего собой простого вещества или индивидуального соединения, является
одной из важнейших подготовительных операций. Отбор средней пробы
преследует своей целью получить относительно небольшое количество
исходного вещества, в котором количественное содержание всех компонентов
должно быть равно количественному содержанию их во всей массе
анализируемого вещества.
Ошибка на один или несколько процентов, а иногда и долей процентов,
допускаемая при анализе, в результате неправильного отбора средней
пробы может привести к грубым просчетам.
Если средняя проба анализируемого вещества не соответствует составу
всей партии, то теряет смысл последующий за отбором средней пробы даже
самый тщательный анализ этого вещества. В этом случае полученные
результаты анализа будут расходиться с действительным составом
анализируемого продукта.
Методы отбора пробы различных материалов сильно отличаются друг
от друга. При отборе проб руководствуются правилами, подробно
описываемыми в технических руководствах, ГОСТ и в специальных
инструкциях, посвященных анализу этих материалов.
Особым случаем отбора пробы является метод без взятия стружки,
применяемый в анализе черных, цветных и драгоценных металлов и их
сплавов. Бесстружковый метод основан на растворении металлов
непосредственно на поверхности детали и удобен в тех случаях, когда взятие
стружки портит деталь или когда детали настолько малы, что невозможно
взять стружку.
Отбор средней пробы делают обычно тогда, когда требуется установить
средний состав большой партии каменного угля, минерального удобрения,
руды, сплавов и т. п.
§ 6. Подготовка вещества для взвешивания
В том случае, если формула индивидуального вещества неизвестна,
для изучения его состава и определения физических констант необходимо
сначала освободить его от примесей, т. е. получить в химически чистом
состоянии.
Для очисткл индивидуальных соединений применяют: растворение и
экстрагирование, -кристаллизацию и перекристаллизацию, осаждение и
переосаждение, возгонку, фракционную перегонку и т. п. Очистку веществ
выполняют также в тех случаях, когда требуется получить их в химически
чистом состоянии для анализа других веществ.
В том случае, когда формула вещества известна, подготовка его к
анализу состоит в извлечении и определении сопутствующих ему примесей.
Правильная подготовка вещества для взвешивания заключается в
удалении всех посторонних включений, попавших в пробу при отборе, упаковке,
транспортировке, хранении и т. д. Процентное содержание составных
частей в исследуемом образце меняется с изменением содержания воды в нем.
Поэтому в ряде случаев вещество предварительно высушивают при 105—
110° С до постоянной массы в сушильных шкафах (рис. 1).
Следует иметь в виду, что некоторые вещества при нагревании окис
ляются. Поэтому их сушат в атмосфере инертных газов или в вакууме
§ 7. ВЗВЕШИВАНИЕ
27
Пересчет на сухое вещество. Результаты анализа, полученные «в
расчете на сухое вещество», означают, что для анализа было взято вещество,
высушенное при определенной температуре, или же что результаты анализа
пробы пересчитаны на сухое вещество на основании одновременного
определения его влажности, проведенного в отдельной пробе.
Рис. 1. Сушильные шкафы:
а — с электрическим обогревом; б — с газовым обогревом; / — ручка
переключателя нагревающих спиралей; 2 — нижние вентиляционные
отверстия (приточные); 3 — верхние вентиляционные отверстия
(вытяжные); 4 — термометр.
Например, если воздушно-сухая проба вещества содержит: 60% А1203
30% Si02 и 10% Н20, «в расчете на сухое вещество» та же проба содержит
А12Оз = -^-=66,7о/о
Si02=i^- = 33,30/0
Таким образом, количественное содержание компонентов в пересчете
на сухое вещество отличается от их содержания в воздушно-сухой пробе.
Это необходимо учитывать, чтобы избежать грубых ошибок при
вычислении результатов анализа веществ, содержащих в своем составе воду.
§ 7. Взвешивание
Воздушно-сухие вещества взвешивают, не прибегая к особым
предосторожностям. При взвешивании высушенных проб и гигроскопических
веществ соблюдают особые меры предосторожности во избежание
поглощения этими веществами влаги из воздуха. В таких случаях вещество
высушивают в стеклянных стаканчиках — бюксах (рис. 2), закрывают
бюкс крышкой и помещают в эксикатор (рис. 3). Пришлифованную крышку
эксикатора через некоторое время на мгновение открывают для
выравнивания внешнего и внутреннего давления (рис. 4). Эксикатор из лаборатории
в весовую комнату переносят так, как показано на рис. 5. Взвешенное
вещество используют для анализа.
Лабораторные весы. Для измерения массы взвешиваемого тела в
химико-аналитических лабораториях применяют весы различного назначе-
28
ВВЕДЕНИЕ
ния, отличающиеся не только конструктивными особенностями, но и
допустимой предельной нагрузкой, точностью, чувствительностью, ценой
деления и т. п.
В связи с этим лабораторные весы подразделяют на:
аналитические, полумикроаналитические, микроаналитические и
пробирные;
Рис. 2. Бюксы. Рис. 3. Эксикаторы.
микровесы, отличающиеся от аналитических весов относительно
малыми значениями предельной нагрузки, цены деления и погрешности;
технические весы 1-го и 2-го классов, применяемые для менее точного
взвешивания;
специальные лабораторные весы, применяемые в особых условиях
лабораторной работы (например, в атмосфере агрессивных сред) или для
Рис. 4. Открывание эксикатора. Рис. 5. Переноска эксикатора.
особых видов анализа (термогравиметрические, седиментационные и т. п.).
К этому типу весов относятся электронные весы, в которых отсчет
показаний или уравновешивание нагрузки производится посредством
электронных приборов.
В студенческих химико-аналитических лабораториях применяют почти
исключительно рычажные аналитические весы, на которых сравнивают
массу взвешиваемого тела с массой разновесок (или гирь).
Устройство аналитических весов. Устройство аналитических весов
и их частей подробно описывается в курсах физики и изучается
студентами на первом курсе. Поэтому мы опускаем описание конструкции весов.
Схема аналитических весов дана на рис. 6.
Некоторые весы имеют приспособления для автоматического
подвешивания гирек (рис. 7).
§ 7. ВЗВЕШИВАНИЕ
29
Очень удобны в работе демпферные весы, при взвешивании на них
положение равновесия чашек определяют не по колебаниям стрелки, а по
ее положению после прекращения колебания. Демпфер (рис. 8) представ-
Рис. 6. Схема аналитических весов:
/ — коромысло весов; 2 — средняя опорная призма; 3 — стержень рейтерного приспособления;
4 — крайняя призма; 5 — крючок рейтера; 6 — горизонтальная балансирная гайка; 7 — стойка
арретира коромысла; 8 — планка сережки; 9 — крючок сережки; 10 — рычаг арретира коромысла;
11 — вертикальная балансирная гайка; 12 — колонка; 13 — дужка чашки; 14 — чашка; 15 —
стрелка; 16 — шкала стрелки; 17 -— ручка арретира; 18 — передняя ножка весов; 19 — подставка
ножки; 20 — задняя ножка весов; 21 — базисная (основная) доска весов; 22 — подвижная
подставка арретира чашек.
Рис. 7. Приспособление для
автоматического подвешивания гирек. В
данном случае на планку весов помещены
500 + 80 = 580 мг, или 0,580 г.
Рис. 8. Демпфер:
/ — подвижные цилиндры, прикрепленные
к дужкам весов; 2—неподвижные цилиндры.
Стрелки показывают движение воздуха.
ляет собой два легких полых металлических циливдра, подвешенных
к чашкам или дужкам весов.
В эти цилиндры свободно входят два других неподвижно укрепленных
на колонке или на основании весов цилиндра.
При колебании стрелки весов верхний цилиндр наклоняющегося плеча
коромысла сжимает находящийся в нижнем цилиндре эоздух, который
30
ВВЕДЕНИЕ
стремится выйти через узкую щель, образующуюся между цилиндрами.
В результате колебания коромысла тормозятся и чашки весов быстро
приходят в состояние покоя.
Разновес. Тела, служащие эталонами для измерения массы, называют гирями или
разновесками.
Мелкие гирьки (разновески) имеют форму квадратной пластинки с загнутым углом
или форму шестиугольника. Цифры, поставленные на мелких разновесках, показывают
миллиграммы (500, 200, 100 и т. д.) или доли грамма (0,5, 0,2, 0,1 и т. д.). Набор гирь,
необходимых для взвешивания на аналитических весах, называется аналитическим
разновесом (рис. 9). Этот набор составляется так, чтобы при наименьшем числе гирь можно
было получать любое число целых граммов и долей грамма. Гири помещают в определен-
Я
0,5 0,2 0,2 0,1 0,05 0,02 0,01
Рис. 9. Аналитический разновес и рейтер
ном порядке в футляре. Каждой гире предназначено особое место в виде углубления.
Расположение гирь систематическое (по убывающей массе), благодаря этому нетрудно
определить по пустому месту массу отсутствующей гири. Мелким разновескам отводится в
футляре особое отделение, которое покрывают стеклом. В этом же отделении помещают пинцет
или щипчики, при помощи которых берут гири. Щипчики снабжены костяными
наконечниками для того, чтобы они не царапали гири.
Обычно аналитический набор имеет в своем составе гири достоинством 100, 50, 20,
10, 10, 5, 2, 2, 1 г и 500, 200, 100, 100, 50, 20, 10, 10, 10 мг. Гири менее 10 мг не
употребляются: их заменяет рейтер, или гусарик.
Чувствительность весов. Чувствительность весов выражают числом,
делений шкалы, прикрепленной к основанию колонки, на которое
отклоняется стрелка весов при изменении нагрузки одной из чашек на 1 мг.
Пример. Если при добавочном грузе 2 мг стрелка весов отклонится на 8 делений,
это значит, что чувствительность весов равна -^- = 4 дел/мг.
Цена деления шкалы. Ценой деления шкалы называют груз,
необходимый для смещения стрелки шкалы на одно деление. Цену деления
выражают в миллиграммах.
Если цена деления шкалы весов равна, например, 0,5 мг/дел, то это
значит, что под влиянием гирьки массой 0,5 мг стрелка отклоняется на
1 деление.
Иногда цену деления шкалы называют чувствительностью весов, в
действительности цена деления обратно пропорциональна чувствительности
весов.
§ 8. Техника взвешивания на аналитических весах
Точка равновесия весов. Под точкой равновесия подразумевают то
деление шкалы, против которого должен бы остановиться конец стрелки
весов после свободного затухания колебаний коромысла. Точку равновесия
называют также центром колебаний стрелки.
§ 8. ТЕХНИКА ВЗВЕШИВАНИЯ НА АНАЛИТИЧЕСКИХ ВЕСАХ 31
Чувствительные аналитические весы обладают тем весьма важным
свойством, что трение опорной призмы в них доведено до минимума. Если
коромысло весов, не снабженных демпфером, вывести из положения
равновесия, то оно придет в затухающее колебательное движение, которое может
продолжаться довольно долго. Поэтому точку равновесия определяют из
колебаний. Шкала аналитических весов обычно разделена на 20 делений
(среднее деление обозначают цифрой 10, крайнее левое — 20 и крайнее
правое — 0, или соответственно 10—0—10).
Точку равновесия (или центр колебаний стрелки) непогруженных весов
называют нулевой точкой.
Взвешивание на аналитических весах, как правило, начинают с
определения нулевой точки.
Для определения точки равновесия ненагруженных весов (нулевой
точки) поступают следующим образом. Открыв арретир после того, как
весы сделают одно—два колебания, проводят три (один за другим) отсчета
показаний стрелки в тот момент, когда стрелка останавливается, меняя
направление своего движения. Положим, что 1± — крайнее левое
положение стрелки, /2 — крайнее правое положение, /3 — второе крайнее левое
положение стрелки.
Так как колебания стрелки затухающие, а затухающее колебательное
движение характерно тем, что при нем амплитуда колебания уменьшается
с течением времени, то /3 <3 1\. Точку равновесия (нулевую точку) п0
можно рассчитывать по формуле:
n° = T-\-2-+k)= 4
Пример. Остановка стрелки весов: слева справа
15,8 дел. 5,5 дел.
15,1 дел. —
Точка равновесия (нулевая точка) равна:
1 / 15,8+15,1 . -Л 1ЛК
по = -§- (——7?—— + 5»5 J = 10,5 дел.
Десятые доли делений шкалы отсчитывают на глаз. Чтобы получить
более надежные результаты, можно увеличить число наблюдений, но число
должно быть всегда нечетным. Начинать отсчеты делений можно либо
слева, либо справа, но всегда нужно придерживаться какого-либо одного
принятого за правило способа.
В весах с демпферами положение равновесия отсчитывают
непосредственно по шкале после полной остановки стрелки. Обычно делают три
отсчета, каждый раз арретируя, а затем освобождая весы. Из
полученных отсчетов берут среднее арифметическое.
Пример. По шкале стрелки демпферных весов сделаны следующие три отсчета: 10,20;
10,05; 10,11. Значение точки равновесия равно:
10,20+10,05 + ЮЛ1 ,Л1П
Следовательно, среднее значение точки равновесия соответствует
делению шкалы «10, 12».
Взвешивание с точностью до 0,001 г. Взвешиваемое тело иногда
предварительно взвешивают на технических весах (рис. 10) с точностью 0,01 г,
а потом — на аналитических. Это ускоряет процесс взвешивания и создает
гарантию, что масса взвешиваемого предмета не превышает предельной
32
ВВЕДЕНИЕ
нагрузки весов. Взвешиваемый предмет помещают на левую чашку весов,
пользуясь боковыми дверцами шкафа весов. Поднимать переднюю дверцу
не рекомендуется.
Взвешивание проводят в такой последовательности: вначале
определяют нулевую точку весов, как было указано выше. Затем на левую чашку
весов кладут взвешиваемое тело, а на правую при помощи пинцета ставят
какую-либо гирьку, по возможности минимальную по массе, но заведомо
большую, чем масса взвешиваемого тела. При этом весы должны быть
арретированы. Закрывают дверцу, осторожно опускают арретир и следят
за отклонением стрелки. Если стрелка наклонится влево, значит весы
перегружены. Тогда арретируют
весы, открывают правую дверцу,
снимают гирьку и вместо нее ставят
другую, следующую по порядку
в наборе разновесов. Закрывают
дверцу и опускают арретир. Если
стрелка опять отклонится влево, то
весы оказываются еще
перегруженными и, повторяя описанную выше
операцию, весы нагружают гирькой
меньшего веса. Когда стрелка будет
отклоняться вправо, т. е. весы
окажутся недогруженными, прибавляют
следующую по порядку гирьку и т. д.
до гирьки 10 мг. В конце концов
добиваются такого момента, когда
гирька в 10 мг (0,01 г),
прибавленная к гирькам, находящимся на правой чашке весов и не вызывающим
перегрузки, эызовет ее. Тогда снимают с чашки весов эту добавочную
гирьку, закрывают арретир, дверцу весов и футляр с разновесом и
дальнейшее уравновешивание взвешенного тела проводят при помощи рейтера.
Все операции рейтера провбдят, когда весы арретированы. Если шкала
коромысла двусторонняя, то рейтер помещают на правую часть коромысла,
например на 5-е деление, и наблюдают качание стрелки. Если стрелка
движется в пределах от середины шкалы до левого края (т. е. до 20), то
весы перегружены. Тогда передвигают рейтер на меньшее деление шкалы.
Если, наоборот, стрелка движется в пределах от середины шкалы до
правого края (т. е. до 10), тогда весы недогружены и рейтер передвигают на
большее деление шкалы. Эти операции проделывают до тех пор, пока
масса взвешиваемого тела не будет установлена с точностью ±0,001 г.
Взвешивание с точностью до 0,0001 г. Такое взвешивание проводят по методу
качаний. По этому методу взвешиваемое тело уравновешивают при помощи гирь и рейтера
до третьего знака, а четвертый знак, т. е. десятые доли миллиграмма, находят,
наблюдая за колебаниями стрелки. Для этого предварительно определяют цену деления шкалы
весов при данной нагрузке. Предположим, что весы недогружены в пределах 0,001 г.
Определяют точку равновесия недогруженных весов т, как было указано выше. Если за 0
считать крайнее правое деление шкалы, тогда щ < по (по — нулевая точка). Затем весы
перегружают на 1 мг, передвигая рейтер слева направо, и вновь определяют точку
равновесия яг. В этом случае пъ > по. Если считать, что отклонение стрелки весов прямо
пропорционально грузу, то можно сказать, что груз в 1 мг отклонит стрелку на п2—П\
делений. Отсюда находим цену одного деления (5), составляя пропорцию:
п2 — tii Дел» — 1 мг
1 дел. — S мг
10. Технические весы.
отсюда
S = -
1
мг/дел.
n2 — ni
где S — масса гирьки, которая отклоняет стрелку весов на одно деление шкалы.
§ 8. ТЕХНИКА ВЗВЕШИВАНИЯ НА АНАЛИТИЧЕСКИХ ВЕСАХ 33
Предположим, что весы недогружены массой Р граммов. К массе Р граммов нужно
добавить такую гирьку у граммов, чтобы сумма (Р + у) граммов равнялась массе тела.
В этом случае точка равновесия весов совпадает с нулевой точкой. Гирька у граммов
должна отклонять стрелку весов на по—п\ делений
1 дел. — S мг
отсюда
(п0 — щ) — у мг
y = (n0 — n{)S
При пользовании гирями найдена масса (Р) с точностью до третьего знака.
Найденная величина у выражается четвертым десятичным знаком, так как 1 мг = 0,001 г.
Таким образом, масса взвешиваемого тела (g) равна:
g = Р ± 0,000*/ = Р + 10~4*/ г
Величина у — положительная, когда весы недогружены, и отрицательная — при
перегрузке.
Все изложенное можно схематически изобразить таким образом:
Правая чашка Точка равновесия
0 /?0 (нулевая точка)
(гири) пг (весы недогружены)
гири+0,001 г л2 (весы перегружены)
Левая чашка
о
(тело)
(тело)
g=p.
по — п\
пг — пх
ю-
При точных взвешиваниях рекомендуется два раза определять нулевую точку: до и
после взвешивания и брать среднее значение.
Так как чувствительность весов обычно мало меняется, то при серийных
взвешиваниях ее определяют только один раз. Остается, таким образом, только определить точку
равновесия при небольшой недогрузке или перегрузке.
Пример. Нулевая точка весов по = 9,4 дел. Нагрузка, когда весы недогружены,
равна 20,233 г. Точка равновесия недогруженных весов щ = 8,9 дел. Рейтер был
передвинут слева направо на число делений, соответствующих 1 мг. Весы оказались
перегруженными (20,234 г). Точка равновесия перегруженных весов П2 = 11,3 дел. Отсюда цена
одного деления -rt-z «-q- = 0,4 мг/дел. Таким образом, масса взвешиваемого тела
g= 20,333 г+ (9,4 — 8,9) 0,4 мг = 20,3332 г (табл. 2).
Форма записи результатов взвешивания
К работе №
Взвешивание тигля с BaS04
ТАБЛИЦА 2
не на груженных весов
точка) п0
12,2
11,9
11,6
35,7 13,8
11,9+ 6,9
18,8
9,4
(нулевая
6,7
7,1
Точка равновесия
недогруженных весов (20,233 г)
«1
11,9 5,9
11,6 6,3
11,3
34,8 12,2
11,6+ 6,1
17,7
8,9
перегруженных весов
п2
13,8
13,5
13,2
40,5 18,2
13,5+ 9,1
22,6
11,3
(20,234 г)
8,8
9,4
Чувствительность: 11,3 — 8,9 = 2,4 дел.
Цена одного деления: -рг-^ 5-^ = 0,4 мг/дел.
11 ,о — о,У
Массу взвешиваемого тела (g) определяют по формуле:
g = 20,333 + (9,4 — 8,9) 0,4 мг = 20,333 + 0,0002 г = 20,3332 г
34
ВВЕДЕНИЕ
Вычисление можно сделать без промежуточных действий по формуле:
g = P+ "в"""1 Ю-4 = 20,333 + f/4~8Q'9Q Ю-4 г = 20,3332 г
П>2 ill 11 ,0 О,У
Метод взвешивания, основанный на передвижении рейтера. Вместо того
чтобы находить десятые доли миллиграмма путем вычислений, их можно
найти опытным путем, передвигая рейтер по коромыслу. Рейтер
передвигают до тех пор, пока точка равновесия не совпадет с нулевой точкой (при
передвижении рейтера весы должны быть арретированы), тогда четвертый
знак отсчитывают по мелким делениям коромысла. Этот метод особенно
удобен, когда шкала весов односторонняя и весы снабжены демпферами.
Рис. 11. Демпферные весы. Рис. 12. Одночашечные весы.
Взвешивание на демпферных весах. Наиболее широкое
распространение получили демпферные весы АДВ-200 (рис. 11), предназначенные для
точных и быстрых взвешиваний в научно-исследовательских и заводских
лабораториях. Для ускорения взвешивания весы снабжены демпферами
и специальным устройством для механического накладывания и снятия
мелких разновесок (от 10 до 990 мг) без открывания дверцы. Кроме того,
для удобства ведения отсчета весы снабжены оптическим приспособлением
со световым экраном (вейтографом), на котором видно увеличение
изображения микрошкалы, укрепленной на стрелке. Отсчет проводится по рискам
на экране.
Разновески достоинством меньше 1 г навешиваются посредством
рычагов. Опускание и подъем разновесок осуществляется поворотом двух
вращающихся дисков (внешнего и внутреннего), укрепленных на правой
стороне футляра весов. На дисках нанесены деления, указывающие массы
помещенных на планку весов разновесок. При повороте внешнего диска
против стрелки-указателя останавливаются цифры, показывающие массу
разновесок достоинством от 100 до 1000 мг; при повороте внутреннего
диска — от 10 до 100 мг. Например, если против стрелки-указателя на
внешнем диске стоит цифра 5 (500 мг), а на внутреннем 80, то это значит,
что на планку весов помещено 500 + 80 = 580 мг (см. рис. 7).
Миллиграммы (масса меньше 10 мг) и десятые доли миллиграмма на демпферных
весах отсчитывают по освещенной микрошкале. При взвешивании
освещение микрошкалы включается автоматически от сети через трансформатор.
Максимальная нагрузка весов АДВ-200 равна 200 г.
§ 9. ПРАВИЛА ОБРАЩЕНИЯ С АНАЛИТИЧЕСКИМИ ВЕСАМИ 35
Взвешивание на одночашечных весах. За последнее время в
лабораториях аналитической химии для взвешивания с точностью от 0,1 до
0,001 мг все чаще стали применять одночашечные весы (рис. 12).
Взвешивание на таких весах занимает очень мало времени. Взвешиваемый предмет
помещают на чашку весов. Разновески навешивают на планку весов
посредством вращения рукояток. Масса взвешиваемого тела указывается на табло.
Взвешивание на микровесах. Для более точного взвешивания
применяют микрохимические весы. На микровесах можно взвешивать с
точностью до 10 "6 г или 0,001 мг. При работе на микровесах подсчет
миллиграммов и десятых долей миллиграмма проводят по положению рейтера,
а сотые и тысячные доли миллиграмма отсчитывают по микрошкале.
§ 9. Правила обращения с аналитическими весами
1. Приступая к взвешиванию, следует проверить наличие всех
разновесок и рейтера. Все разновески должны быть расположены в футляре
в строгом порядке.
2. Перед взвешиванием необходимо сесть на табуретку против весов.
Все движения должны быть плавные, без толчков. Прежде всего следует
определить нулевую точку.
3. Взвешиваемый предмет нужно класть на левую чашку весов, а
разновески — на правую.
4. Категорически запрещается нагружать весы больше их предельной
нагрузки, превышение которой может вызвать остаточные деформации или
даже разрушение коромысла.
5. Тяжелые грузы следует класть посредине чашки. В противном
случае при опускании арретира чашка придет в колебательное движение
вокруг точки подвеса, вследствие чего появятся центробежные силы, которые
будут влиять на результаты взвешивания.
6. Накладывать и снимать взвешиваемое тело, разновески и рейтер
необходимо с арретированных весов. Несоблюдение этого правила может
повести к тому, что коромысло сдвинется со своего места и положение
нулевой точки изменится. Кроме того, при этом острия призм скорее
притупляются.
7. При вращении ручки арретира не следует допускать резких
движений, не давая слишком сильно раскачиваться чашкам весов. Открывать
и закрывать арретир необходимо осторожным плавным вращением ручки,
причем арретир следует закрывать в тот момент, когда стрелка проходит
мимо среднего деления шкалы.
8. Разновески нельзя брать руками, а только пинцетом. Нельзя
прикасаться руками к какой-либо части весов, кроме арретира и дверок
шкафчика.
9. Взвешивать тело можно только тогда, когда оно приняло
температуру окружающего весы пространства. С этой целью взвешиваемое тело
приносят за 30—40 мин до взвешивания в весовую комнату. В противном
случае токи воздуха, вызванные разностью температур, могут значительно
повлиять на результаты взвешивания. Если тело принесено из более
холодного помещения, то на нем осаждается влага, которая увеличивает массу
его.
10. Во время взвешивания, т. е. во время отсчетов колебаний стрелки
(при открытом арретире), дверцы весового ящика должны быть плотно
закрыты.
11. Каждый анализ или группа связанных между собой анализов
должны быть проведены ни одних и тех же весах и с одним и тем же
разновесом.
36
ВВЕДЕНИЕ
12. Взвешиваемое вещество ни в коем случае нельзя класть
непосредственно на чашку весов. Оно обязательно должно быть помещено в чистую
посуду (бюкс, тигель, часовое стекло и др.). Летучие вещества взвешивают
в специальных плотно закрывающихся сосудах. Запрещается взвешивать
вещество на бумажках и ставить взвешиваемый предмет непосредственно
на лабораторный стол.
13. Закончив взвешивание, нужно убедиться, что весы арретированы
(т. е. чашки весов находятся в неподвижном состоянии), дверцы футляра
весов закрыты, стол убран.
§ 10. Приготовление раствора для анализа
После взвешивания вещества его навеску растворяют в подходящем
растворителе. Растворение ведут на холоду или, если вещество плохо
растворяется, при нагревании.
Особым случаем приготовления раствора для анализа является
растворение металла или сплава непосредственно на поверхности детали. Для
этой цели на чистую обезжиренную поверхность металлической детали
помещают каплю кислоты, щелочи или другого реагента и после некоторой
выдержки снимают каплю для дальнейшего анализа. Такой метод
называют бесстружковым. Бесстружковый метод анализа разработан Н. А. Та-
нанаевым и имеет целый ряд преимуществ перед обычным химическим
методом анализа.
§11. Запись результатов анализа
Степень точности. Вычисление результатов анализа на основании
данных измерений является неотъемлемой частью любого количественного
определения. Поэтому важно не только тщательно выполнить само
определение, но и с требуемой точностью сделать соответствующие вычисления.
Неаккуратность в работе или неточность в вычислении приводят к
искажению результатов анализа и к необходимости повторить весь анализ
сначала.
Вычисления проводят либо приближенно, либо с точностью,
соответствующей степени точности применяемого метода определения и
измерительных приборов.
Окончательные результаты анализов записывают так, чтобы только
одна последняя цифра в них была не вполне достоверной, это позволяет
отразить степень точности полученных цифровых данных.
Например, запись «2,5 г» показывает, что масса определена с точностью
до 0,1 г. Запись «2,5324 г» указывает, что масса определена с точностью
до 0,0001 ?. Последняя цифра, лишь приближенно выражающая результат
определения, называется недостоверной.
Выражение результатов анализа. При вычислении результатов
анализа необходимо помнить, что точный результат вычислений получают
лишь в том случае, если имеют дело только с точными цифрами.
Так как при вычислении результатов анализа пользуются
результатами измерений, т. е. приближенными величинами, то и результаты этих
вычислений получают приближенными. Поэтому нет никакого смысла
вычислять результаты анализа, стремясь получить большое число цифр,
превышающих одну недостоверную цифру.
Если для анализа взята навеска вещества с точностью до 0,1 г, то
бессмысленно давать окончательный результат анализа в виде, например,
четырехзначного числа, так как лишь первая цифра является достоверной,
а все стоящие справа — недостоверны. Согласно сформулированному
§ 11. ЗАПИСЬ РЕЗУЛЬТАТОВ АНАЛИЗА
37
выше правилу, окончательный результат анализа должен быть записан
так, чтобы только одна последняя цифра была не вполне достоверной.
Поэтому в данном примере окончательный результат записывают с
точностью до 0,1 г.
Между тем студенты нередко пишут большое число цифр, полагая,
что это приводит к более точным результатам.
Искомый результат не может быть более точным, чем точность
применяемого метода анализа или наименее точное число, использованное в
процессе данного вычисления.
Для получения более точных результатов анализа необходимо
использовать более точные методы анализа и, тщательно выполняя само
определение, пользоваться наиболее точными измерительными приборами и
с требуемой точностью проводить соответствующие вычисления.
ЧАСТЬ ПЕРВАЯ
Объемный анализ
ГЛАВА I
ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
А. ОБЩАЯ ХАРАКТЕРИСТИКА ОБЪЕМНОГО АНАЛИЗА
§ 1. Сущность объемного анализа
Объемным (титриметрическим) методом анализа называют метод
количественного анализа, основанный на измерении количества реагента,
требующегося для завершения реакции с данным количеством определяемого
вещества.
Метод заключается в том, что к раствору определяемого вещества А
постепенно прибавляют раствор реактива В известной концентрации.
Добавление реактива В продолжают до тех пор, пока его количество не станет
эквивалентным количеству реагирующего с ним определяемого вещества А.
Количественные определения с помощью объемного метода
выполняются очень быстро. Время, требуемое для завершения определения
объемным методом, измеряется минутами. Это позволяет без особой затраты
труда проводить несколько последовательных и параллельных
определений.
Основоположником объемного анализа является французский ученый
Ж. Л. Гей-Люссак.
Пример. Предположим, требуется определить в данном растворе едкого натра
неизвестной концентрации содержание NaOH (определяемое вещество А). Для решения этой задачи
объемным методом анализа к исследуемому реактиву постепенно приливают по каплям
хлористоводородную кислоту точно известной
концентрации (реактив В).
При этом происходит реакция нейтрализации:
NaOH + HCl -* NaCl + H20
По мере прибавления НС1 постепенно
нейтрализуется NaOH; количество его непрерывно уменьшается,
и щелочность раствора падает. Наконец, введенное
количество НС1 становится равноценным
(эквивалентным количеству NaOH, содержащемуся в растворе
неизвестной концентрации. Зная концентрацию и объем
раствора реагента (НС1), израсходованного на реакцию
с определяемым веществом (NaOH), можно вычислить
содержание определяемого вещества в титруемом
растворе.
Определяемое вещество. Химический
элемент, простое или сложное вещество,
содержание которого определяют в данном образце
анализируемого продукта, называют
определяемым веществом и обозначают буквой А.
К определяемым веществам относят также
Ж. Л. Гей-Люссак (1778—1850).
§ 1. СУЩНОСТЬ ОБЪЕМНОГО АНАЛИЗА
39
атомы, ионы, связанные и свободные радикалы и функциональные группы.
Например, атомы водорода, непосредственно связанные с атомами
кремния (Si—Н), атомы галогенов, входящие в состав ядра или боковой цепи
ароматических углеводородов, и т. п.; ионы, входящие в состав растворов
кадмиевых, медных, никелевых и цинковых электролитов, ионы
алюминия в ваннах для анодирования, ионы магния в растворах для
оксидирования магниевых сплавов и т. д.
Реагент (реактив). Твердое, жидкое или газообразное вещество,
вступающее в реакцию с определяемым веществом А, называют реагентом
и обозначают буквой В.
Следует различать понятия «реагент» и «реактив». Реагентом называют
вещество, непосредственно вступающее в реакцию, а реактивом —
химический препарат, который может представлять собой сложную смесь
различных веществ, содержащую наряду с собственно реагентом
вспомогательные вещества и растворитель. Например, «реактив Чугаева» (для
определения № + +) представляет собой смесь диметилглиоксима со спиртом
и водным раствором аммиака (собственно реагентом в этом реактиве
является диметилглиоксим); «реактив Несслера» (для определения NH3) —
смесь Hgl2 + KI + КОН + Н20, реагентом в этом реактиве служит
[Hgl4]~ ~; «реактив Швейцера» — смесь: Си (ОН)2 + NH3 + H20,
соответствующий реагент [Си (NH3)41++. Поэтому термины «реактив» и
«реагент» не всегда можно считать синонимами.
Титрование. Количественное определение вещества А объемным (титри-
метрическим) методом, при котором к раствору исследуемого продукта
медленно приливают раствор реагента точно известной концентрации в
количестве, соответствующем содержанию определяемого вещества А,
называют титрованием. Слово «титрование» происходит от слова «титр». Иногда
к титруемому веществу прибавляют реагент в твердом, жидком или
газообразном состоянии.
Поэтому в широком смысле титрованием называют процесс непрерывно
контролируемого постепенного смешивания измеренного количества
твердого, жидкого или газообразного вещества, или чаще, точно измеренного
объема стандартного раствора реагента В с исследуемым веществом. При
этом количество реагента соответствует содержанию определяемого
компонента А, реагирующего с реагентом В в строго эквивалентных
количествах.
В так называемых методах безбюреточного титрования не измеряют
объем израсходованного на реакцию реактива, поэтому нет необходимости
знать точную концентрацию реагента. В этом случае при помощи счетчика
подсчитывают число капель израсходованного раствора реагента или
измеряют время, пошедшее на титрование данного определяемого вещества.
Стандартный, или титрованный, раствор (титрант). Раствор реагента В
точно известной концентрации, применяемый для титрования в методах
объемного (титриметрического) анализа, называют стандартным, или
титрованным, раствором, или титрантом.
Нормальность раствора Сн (или N). Нормальность раствора
выражают числом грамм-эквивалентов вещества, содержащегося в 1 л раствора
(см. стр. 66):
С = # = — = —
н у ЭУ
Титр (Тв). Точную концентрацию стандартного раствора реагента В
(титранта) называют титром и выражают числом граммов растворенного
вещества, содержащегося в 1 см* (мл) раствора (см. стр. 67).
40
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Правило (закон) эквивалентности. Химические элементы или их
соединения вступают в химические реакции друг с другом в строго
определенных весовых количествах, содтветствующих их химическим эквивалентам
(грамм-эквивалентам). Другими словами, грамм-эквивалент одного
вещества реагирует с одним грамм-эквивалентом другого вещества. Это правило
имеет большое значение в объемном анализе; им руководствуются при
расчете результатов анализа.
Согласно правилу эквивалентности:
а
NV а = ТУ
п юоо э ~ э
где пА, пв — число грамм-эквивалентов растворенного вещества А и В;
N — нормальность раствора, г-экв/л\
V — объем раствора, мл\
а — количество растворенного вещества, г;
Э — эквивалент растворенного вещества;
Т — титр, гУ'см3 (мл).
Точка эквивалентности и конечная точка титрования. Согласно
правилу эквивалентности, титрование необходимо продолжать до тех пор,
пока количество прибавленного реагента В не станет эквивалентным
содержанию определяемого вещества А. Наступающий в процессе титрования
момент, в котором количество стандартного раствора реагента В (титранта)
становится теоретически строго эквивалентным количеству определяемого
вещества А, реагирующего с прибавляемым реагентом В, согласно
определенному уравнению химической реакции, называют точкой
эквивалентности.
Точку эквивалентности устанавливают различными способами,
например по изменению окраски индикатора, прибавляемого в титруемый
раствор. Момент, при котором происходит наблюдаемое изменение цвета
индикатора, называют конечной точкой титрования. Очень часто конечная
точка титрования не совсем точно совпадает с точкой эквивалентности,
соответствующей теоретической точке конца титрования.
Точка эквивалентности наступает в тот момент, когда в титруемый
раствор прибавлено теоретически требующееся количество реагента В,
нацело реагирующего с определяемым веществом А. Следовательно,
теоретически в точке эквивалентности не должно быть ни вещества А, ни
реагента В, если реакция их взаимодействия протекает количественно.
Реакции, применяемые в объемном анализе, обратимы и в точке
эквивалентности практически не доходят до конца. Эта одна из причин того, почему
точка эквивалентности не всегда совпадает с конечной точкой титрования.
В тех случаях, когда точка эквивалентности полностью или почти
полностью совпадает с конечной точкой титрования, по количеству реагента,
израсходованного на реакцию с определяемым веществом (ТВУВ), согласно
правилу (закону) эквивалентности можно рассчитать количество
определяемого вещества в граммах (§д) или содержание его в процентах (*А).
Когда эти точки не совпадают, вводят поправочный коэффициент, который
вычисляют на основании данных, полученных при титровании в
аналогичных условиях растворов с известным (эталонным) содержанием
определяемого вещества или производят расчеты по методу, разработанному
румынским ученым Литеану.
Навеска анализируемого вещества. Небольшое количество средней
пробы вещества, которое берется для анализа, называют навеской. Для
получения наиболее точных результатов необходимо брать рассчитанную
§ 1, СУЩНОСТЬ ОБЪЕМНОГО АНАЛИЗА
41
навеску анализируемого вещества на аналитических весах. От точности
взвешивания навески зависит точность результатов анализа. Величину
навески выбирают в зависимости от относительного содержания
определяемых компонентов в исследуемом продукте и от избираемого метода
анализа (см. табл. 1, стр. 24). Зная приблизительное содержание в
анализируемом образце того или иного компонента, можно рассчитать
оптимальную величину навески (см. гл. V, § 3).
В анализируемом объекте различают основные компоненты,
составляющие от 1 до 99,99999%, и примеси, содержание которых составляет 0,01 —
1%. Эти составные части анализируемого образца называют
макрокомпонентами. Компоненты, содержащиеся в количествах меньше 0,01%,
называют микропримесями {микрокомпонентами).
Выбор навески, требуемой для проведения анализа и определения
макро- и микрокомпонентов, и выбор метода количественного анализа
зависят от содержания основных определяемых веществ и
микропримесей.
Реакции, используемые в объемном анализе. Титриметрические методы
анализа основаны на использовании самых разнообразных реакций:
нейтрализации, окисления—восстановления, осаждения, комплексообразо-
вания, ионного обмена, замещения, присоединения, конденсации
и т. п.
Реакции нейтрализации используют при титровании неорганических
и карбоновых кислот, фенолов, оксипроизводных гетероциклических
соединений, оснований, солей, гидрогалогенидов слабых органических
оснований, аминов, алкалоидов, оснований Шиффа, гетероциклических
оснований, аминокислот, фармацевтических и медицинских препаратов и т. п.
Например:
Н+ + ОН- -> НоО
Н+ + —NH2 ^ — ЫНз
H+ + = NH -> =NH2
Н+ + = N -> -ее NH
H+ + NH2 -> NH3
H+ + NH3 -> NH4
H+ + CH3COO- -+ CHgCOOH
NH3 + C2H50- -* -NH2 + C2H5OH
Основания обычно титруют стандартными растворами кислот, а
кислоты — растворами оснований, точку эквивалентности фиксируют с
помощью кислотно-основных индикаторов или физико-химическими
(инструментальными) методами. Так, содержание хлористоводородной или
уксусной кислоты определяют титрованием стандартным раствором едкого натра
в присутствии метилового красного (при титровании НС1) или
фенолфталеина (при титровании СН3СООН). Содержание оснований определяют
титрованием стандартным раствором серной кислоты.
Реакции окисления—восстановления используются при титрований
неорганических восстановителей, гидрохинона, аминофенолов, Сахаров,
щавелевой, малеиновой, аскорбиновой, лимонной и других оксикислот,
а также при титровании неорганических окислителей, ненасыщенных орга-
42
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
нических соединений, нитрозо-, нитро- и азосоединений, хинонов,
органических красителей, фармацевтических препаратов и т. п. Например:
12 + 2е -+ 21-
МпО; + 5е+8Н+ -* Мп+++4Н20
Сг207~ + бе + 14Н+ -> 2Сг+++ + 7Н20
Fe++ — e -> Fe+++
Tiul-e -+ TiIV
C6H8Oe — 2e -> СбН6Об + 2Н+
аскорбиновая дегидроаскорбиновая
кислота кислота
Восстановители обычно титруют стандартными растворами
окислителей, а окислители — растворами восстановителей. Точку эквивалентности
фиксируют с помощью окислительно-восстановительных (ред-окс)
индикаторов или другими способами.
Реакции осаждения используют при титровании веществ, образующих
в соответствующей среде нерастворимые продукты: соли бария, серебра,
свинца, ртути (II), меди (II), кадмия, таллия (I); галогениды, гексациано-
ферраты (II), тетрафенилбораты, пикраты, сульфаты, хроматы, оксалаты,
барбитураты и т. п. Например:
Ag+ + Cl" -^ |AgCl
Ba++ + SC>4" -> |BaS04
К+ + [(С6Нб)4В]- -* ^К[(СвНб)4В]
Hg++ + 2[(C8H5)4Br -* |Hg[C6H6)4B]2
В тех случаях, когда определяемое вещество находится в форме, не
пригодной для определения, его переводят в подходящую катионную или
анионную форму. Например, при определении в сплаве содержания
металлического серебра сплав предварительно растворяют в азотйой кислоте,
а затем катионы серебра титруют роданидом:
я HN03 л x SCN" , . 0___
Ag > Ag+ -» 4- AgSCN
При определении элементарной, сульфидной, пиритной и сульфитной
серы анализируемый продукт предварительно окисляют подходящим
окислителем. При этом сера окисляется в сульфатную форму, которую титруют
солями бария:
s°, s" [s2]", sor °кислител*
_» SO^'-?1—-> |BaS04
Реакции комплексообразования широко используют в объемном анализе.
К этому типу относятся реакции, протекающие между катионами
переходных элементов с аминокислотами, окислами, этилендиаминтетрауксусной
кислотой и другими реагентами. Например:
Hg*+ + 4I- ^ [HglJ-
H2NCSNH2+.Bi+++ -* [Bi (H2NCSNH2)]+++
Тиомочевина
Ag^-2CN" -* [Ag(CN)a]-
§1. СУЩНОСТЬ ОБЪЕМНОГО АНАЛИЗА 43
Реакции ионного обмена сопровождаются образованием малодиссоции-
рующих, малорастворимых и молекулярных соединений. Например:
Hg++ + Cl- -* HgCl+
HgCl+ + Cl- ;?? HgCl2
2R (Ar) COOMe + Hg (C104)2 -> I [R (Ar) COO]2 Hg + 2MeC104
(Ar, R — углеводородные радикалы: алкил, арил)
CHsCOOTl + CH3COCI -+ (СН3СО)2 О + i Т1С1
AgN03 4-Na [(СбНб)4 В] -> J Ag [(СбНб)4В] + NaN03
Реакции замещения. В качестве примеров использования реакций
замещения в объемном анализе можно привести следующие:
СбН6 + Вг2 -+ СбН5Вг + НВг
бензол бромбензол
С6Н5ОН + ЗВг2 -+ СвН2Вг3ОН + ЗНВг
Вг
I
I II I —^ I II I
| N Вг | N
НО НО
Реакции присоединения. Например:
= С = С = +Вг2 -» =СВг —СВг =
= С = С = + (СН3СОО)2 Hg + CH3OH ->
-> = С С = + СНзСООН
I I
ОСНз HgOCOCH3
= с — с = + нвг -> = с —с =
\/ I I
О ОН Вг
Реакции конденсации. Например:
C6H5NH2 + С6Н13СНО -+ C6H5N = СНС6Н13 + Н20
гептальдегид
C6H5NHNH2 + RR'CO -> C6H5NHN = CRR'+ Н20
фенилгидразин
Требования, предъявляемые к реакциям. Реакции, используемые
в объемном анализе, должны удовлетворять следующим условиям:
1) вещества, вступающие в реакцию, должны реагировать в строго
определенных количественных соотношениях (стехиометрических отношениях);
2) реакции, протекающие между определяемым веществом и
стандартным (титрованным) раствором реактива, должны протекать быстро и
практически до конца;
3) посторонние вещества, присутствующие в исследуемом продукте
и переходящие вместе с основным определяемым компонентом в раствор,
не должны мешать титрованию определяемого вещества;
4) точка эквивалентности должна фиксироваться тем или иным
способом резко и точно;
5) реакции должны по возможности протекать при комнатной
температуре;
6) титрование не должно сопровождаться побочными реакциями,
искажающими результаты анализа.
44
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Реагенты, применяемые при титровании. В качестве реагентов,
применяемых при титровании, используют не только растворы твердых,
жидких или газообразных веществ в жидких растворителях, но и газы,
жидкости, смеси жидкостей, твердые вещества и вещества, генерируемые
фотохимическим или электрохимическими методами.
Реагентами служат щелочи (основания), кислоты, окислители,
восстановители, осадители, комплексообразователи, разнообразные соли, спирты,
альдегиды, кетоны, ангидриды, металлорганические соединения,
красители, комплексоны, гексацианоферраты, гетерополикислоты, газообразный
водород, фтор, бром, иод, хлор, вода и многие другие вещества.
Условия титрования. В процессе титрования должны строго
соблюдаться условия, обеспечивающие достаточную быстроту выполнения
анализа и достижение требуемой точности количественного определения
(т. е. получение надежных результатов анализа).
Ниже указаны наиболее существенные условия титрования.
1. Место титрования должно быть тщательно подготовлено и хорошо
освещено дневным или электрическим светом.
2. Применяемые посуда и измерительные приборы должны быть
тщательно вымыты и точно откалиброваны перед началом титрования.
3. Используемые реактивы, побочные продукты, вводимые в титруемую
смесь, дистиллированная или деионизированная вода должны
удовлетворять предъявляемым к ним требованиям и не содержать посторонних
примесей, искажающих результаты анализа.
4. В тех случаях, когда на процесс титрования оказывают влияние
кислород или двуокись углерода для предотвращения побочных,
нежелательных реакций, титрование проводят в атмосфере достаточно
инертного газа (обычно в атмосфере газообразного азота).
5. Процесс титрования должен сопровождаться тщательным и
непрерывным перемешиванием реагирующих веществ (вручную или при помощи
механической или магнитной мешалки).
6. Титрование должно завершаться по возможности в течение 5—
10 мин и протекать в строго контролируемых условиях температуры,
рН раствора, в присутствии или отсутствие индикатора и т. п. Если
необходимо, титрование проводят при нагревании или охлаждении.
7. При титровании Следует, согласно методике определения,
поддерживать требуемую кислую или щелочную реакцию среды, добавлять в
титруемую смесь необходимые электролиты (фон), органические растворители
или экстрагенты, буферные смеси, комплексообразователи, катализаторы,
индукторы, индикаторы и т. п.
8. Выбранный индикатор или какой-либо инструментальный метод
определения конечной точки должны обеспечивать наиболее четкую и
резкую фиксацию этой точки титрования, по возможности совпадающей
с точкой эквивалентности.
9. Титры применяемых стандартных растворов перед титрованием
должны быть тщательно установлены или проверены по химически чистым
установочным веществам.
10. Все параллельные титриметрические определения необходимо
закончить в один рабочий день. Перенесение параллельных титрований на
следующий день возможно лишь в случае полной уверенности в
устойчивости титра стандартного раствора, применяемого для титрования. Если
такой уверенности нет, следует на другой день снова проверить титр
титр анта.
Значение титриметрических методов анализа. Титрование
представляет собой очень важный метод количественного анализа, широко
применяемый в практике заводских, учебных и научно-исследовательских
§ 2. ОБЩЕЕ УРАВНЕНИЕ РЕАКЦИИ ТИТРОВАНИЯ И ВЫВОДЫ ИЗ НЕГО 45
лабораторий для определения состава разнообразных неорганических,
органических и элементоорганических соединений. Кроме того, количестбен-
ные данные, получаемые методом титрования, используются для
установления строения исследуемых веществ, изучения кинетики химических
процессов, определения констант диссоциации кислот и оснований,
относительной силы электролитов, протяженности и положения шкалы
констант автопротолиза различных растворителей, определения
молекулярных весов, исследования механизма разнообразных химических
превращений, для разработки, модернизации и интенсификации методов синтеза,
получения особо чистых элементов и их соединений, обеспечения
оптимальности химико-технологических процессов и т. д.
Классификация методов объемного анализа. В зависимости от типа
используемых основных реакций объемные (титриметрические) методы
анализа классифицируют на следующие группы.
Методы нейтрализации или кислотно-основного титрования основаны
на использовании реакций нейтрализации кислот, оснований, солей
слабых кислот или слабых оснований, сильно гидролизующихся в водных
растворах, разнообразных неорганических и органических соединений,
проявляющих в неводных растворах кислые или основные свойства, и др.
Методы окисления — восстановления основаны на использовании
реакций окисления — восстановления элементов, способных переходить из
низших степеней окисления в высшие, и наоборот, а также ионов и
молекул, которые реагируют с окислителями или восстановителями, не
подвергаясь непосредственному окислению или восстановлению.
Методы осаждения основаны на использовании реакций осаждения.
Методы комплексообразования основаны на использовании реакций
комплексообразования, из которых наиболее широко применяют реакции
ионов металлов с так называемыми комплексонами (см. ниже).
§ 2. Общее уравнение реакции титрования и выводы из него
Методы установления точек эквивалентности. Установление
(фиксирование) конечной точки титрования или точки эквивалентности
представляет собой важнейшую операцию титриметрического метода
анализа, так как от точности определения точки эквивалентности зависит
точность результатов анализа. Обычно конец титрования устанавливают по
изменению окраски титруемого раствора или индикатора, вводимого в начале
или в процессе титрования. Применяют также безындикаторные методы,
основанные на использовании специальных приборов, позволяющих судить
об изменениях, которые происходят в титруемом растворе в процессе
титрования. Такие методы называют физико-химическими, или
инструментальными, методами определения точек эквивалентности. Они основаны
на измерении электропроводности, величин потенциалов, оптической
плотности и других физико-химических параметров титруемых растворов,
которые резко изменяются в точке эквивалентности.
Общее уравнение реакции титрования. Процесс титрования можно
представить в виде следующего общего уравнения:
а + аЧа11 Л ai + aii + d + e JH
-> AI + AII + D + E + BH36
где А, А1, А11 — определяемые вещества, находящиеся в титруемом растворе;
В — реагент (титрант);
D, Е — продукты реакции определяемого вещества А с реагентом (титран-
том) В;
ВЯзб — избыток реагента.
46
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
По мере титрования койцентрация определяемого вещества А
постепенно уменьшается до точки эквивалентности; в точке эквивалентности
она уменьшается до минимума; после точки эквивалентности — остается
практически равной нулю.
До точки эквивалентности концентрация реагента В остается равной
нулю, так как по мере приливания титрованного раствора реагент В
вступает в реакцию с определяемым веществом. В точке эквивалентности в
титруемом растворе появляются следы реагента В после точки
эквивалентности, разумеется, концентрация реагента начинает повышаться, так как
концентрация определяемого вещества в это время становится равной
нулю.
Концентрации продуктов реакции D и Е соответственно, по мере
вступления реагента В в реакцию с определяемым веществом, постепенно
повышаются, достигают максимума в точке эквивалентности и практически
остаются постоянными после точки эквивалентности.
ТАБЛИЦА 3
Изменение концентрации реагирующих и образующихся веществ
в процессе титрования
Вещество
А
В
D, Е
До точки эквивалентности
Сд постепенно
уменьшается
св = о
Оэ, Се постепенно
повышаются
В точке эквивалентности
Сд резко уменьшается
В растворе появляются
следы реагента В
Оэ» Се достигают
максимума
После точки
эквивалентности
СА#0
Св начинает повышаться
Оэ» Се практически
остаются постоянными
Точку эквивалентности можно определить следующими методами:
1) визуально — по изменению цвета раствора, если определяемое
вещество А или реагент В окрашены, так как в точке эквивалентности
концентрация определяемого вещества уменьшается до минимума, а
концентрация реагента В начинает повышаться;
2) визуально — по появлению помутнения или по изменению окраски
раствора, вызываемой образованием продуктов реакции, или индикатора,
если А и В бесцветны;
3) физико-химическими (инструментальными) методами с
последующим анализом кривых титрования, отражающих происходящие в процессе
титрования изменения физико-химических параметров титруемых
растворов (например, рН) независимо от окраски А и В. Точку
эквивалентности устанавливают по пересечению кривых или по скачку кривой
титрования.
Общие приемы титрования. Прямое титрование. Простейший
прием титрования в объемном анализе состоит в том, что к определенному
объему раствора или к определенной навеске вещества А, растворенного
в подходящем растворителе, по каплям приливают из точно
калиброванной бюретки стандартный (титрованный) раствор реагента В.
Точку эквивалентности устанавливают по резкому уменьшению СА,
сопровождающемуся, например, исчезновением окраски раствора (если
определяемое вещество А окрашено) или по началу повышения Св (по
появлению окраски раствора, если реагент В окрашен), или по
изменению окраски индикатора.
§ 2. ОБЩЕЕ УРАВНЕНИЕ РЕАКЦИИ ТИТРОВАНИЯ И ВЫВОДЫ ИЗ НЕГО 47
Если в распоряжении аналитика имеется титрованный раствор
требуемого реагента В, то весь процесс прямого титрования вещества А
занимает несколько минут.
Зная количество стандартного раствора реагента В, израсходованного
на реакцию с определяемым веществом А, можно легко вычислить
содержание вещества А.
Примером применения прямого метода титрования является
определение содержания НС1 в технической соляной кислоте с помощью NaOH.
Обратное титрование. Иногда по тем или иным причинам нельзя
применить метод прямого титрования. Тогда прибегают к так называемому
методу обратного титрования ( титрование по остатку). Этот прием
состоит в том, что к определенному объему раствора или к определенной
навеске вещества А, растворенного в подходящем растворителе,
приливают точно измеренный объем титрованного раствора реагента В, взятый
в избытке. Избыток не вошедшего в реакцию реагента В оттитровывают
стандартным раствором другого вспомогательного реагента Ъг точно
известной концентрации. На титрование избытка реагента В должно
идти не менее 15—20 мл реагента В г (макрометодом) или 1,5—2 мл
(микрометодом).
Например, для определения содержания НС1 в соляной кислоте
неизвестной концентрации можно прибавить к ней точно отмеренное
количество титрованного раствора AgN03, взятого с избытком, а затем непро-
реагировавший избыток AgN03 оттитровать стандартным раствором
подходящего реагента, реагирующего с ионами серебра, не вошедшими
в реакцию. Таким реагентом является роданид аммония, вступающий
в реакцию с ионами серебра с образованием малорастворимого AgSCN.
Если реакцию обратного титрования вести в присутствии ионов
железа (III), то лишняя капля титрованного раствора вспомогательного
реагента Вх вызывает окрашивание раствора в розово-красный цвет
вследствие образования Fe (SCN)3. Появление розовой окраски
свидетельствует об окончании реакции.
Как и при прямом титровании, зная количество стандартного раствора
реагента В, израсходованного на реакцию с определяемым веществом А,
можно легко вычислить содержание вещества А.
Косвенное титрование. Этот вид титрования имеет несколько
вариантов.
Титрование заместителя. Сущность этого варианта
заключается в следующем. К определяемому веществу А прибавляют
какой-либо вспомогательный реагент В19 реагирующий с ним с
выделением эквивалентного количества нового вещества Alf которое
оттитровывают стандартным раствором основного реагента В. Другими словами,
вместо непосредственного титрования определяемого вещества А титруют
его заместитель А±.
Поясним это на примере. Окислитель КМп04 можно оттитровать с
помощью стандартного раствора восстановителя. Но не всякий
восстановитель пригоден для этой цели. Например, Na2S203, являющийся хорошим
восстановителем, нельзя, по ряду причин непосредственно применить
для титрования таких сильных окислителей, какими являются КМп04,
К2Сг207, МпО 2 И Т. Д.
Однако раствор Na2S2Os с успехом применяют для титрования такого
слабого окислителя, как иод. Поэтому предварительно действуют на
КМп04 в кислой среде вспомогательным реагентом KI (BJ, который
реагирует с КМп04 с выделением иода (Aj).
Иод выделяется в эквивалентном количестве по отношению к КМп04.
Если оттитровать выделившийся иод [заместитель перманганата (Aj)]
48 ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
стандартным раствором основного реагента Na2S203 (В), то можно
вычислить количество КМп04 (А).
Конец титрования определяют с помощью индикатора — раствора
крахмала. В присутствии иода крахмал окрашивается в синий цвет.
По мере восстановления иода тиосульфатом синяя окраска крахмала
постепенно исчезает; в точке эквивалентности раствор полностью
обесцвечивается.
Количество израсходованного на реакцию основного реагента Na2S203
(В) эквивалентно количеству выделившегося иода (Aj). Количество
выделившегося иода (заместителя — Ах) в свою очередь эквивалентно
количеству КМп04 (А), содержащегося в образце.
Зная, сколько основного реагента Na2S203 израсходовано на
реакцию с 12, нетрудно вычислить содержание определяемого вещества —
КМп04.
Определение по содержанию продуктов
реакции титрования. Содержание продуктов реакции D или Е
определяют тем или иным способом после взаимодействия определяемого
вещества А с реагентом В.
Определение по разности. Если известно или
определено содержание всех компонентов и компонентов А1 и А11 в титруемом
растворе, легко можно вычислить содержание компонента А:
А = ? А, А1, А11-2 А1, А11
Например, при анализе смеси НС1 + NaCl сначала прямым или
обратным титрованием стандартным раствором AgN03 определяют
суммарное содержание HCI + NaCl
HCI + NaCi + п AgN03 -> 2AgCI + HN03 + NaN03 + (n — 2) AgN03
A Al B D E F Визб
Затем прямым или обратным титрованием стандартным раствором
в отдельной порции исследуемого раствора определяют содержание
в смеси НС1:
НС1 + NaCl + NaOH -> NaCl + NaCl + Н20
A A1 B D A1 E
Зная суммарное содержание HCI + NaCl и содержание НС1,
рассчитывают содержание NaCl.
Иногда для анализа сложной смеси комбинируют разные приемы
титрования. Например, анализ смеси бензоилхлорида и нафталина основан
на использовании следующей реакции с морфолином:
Н2С—СН2
Q^COCl+CwHg + HN^ \) -+
А А1 >> '
Н2С—СН2
в
НоС—СН2 Н2С—СН2
- C6H5CON^ Nd + HCI + CxoHs+HN^ \>
\ / Е А* N /
Н2С—СН2 Н2С—СН2
D визб
Избыток морфолина Визб обратно оттитровывают уксуснокислым
раствором хлорной кислоты. Хлористоводородную кислоту (Е),
образующуюся в эквивалентном количестве по отношению содержащегося
§ 3. ИЗМЕРЕНИЕ ОБЪЕМОВ РАСТВОРОВ
49
в растворе определяемого вещества А, оттитровывают стандартным
раствором едкого натра или солями серебра. Нафталин осаждают
пикриновой кислотой в виде пикрата, образовавшийся пикрат восстанавливают
соединениями титана (III) и затем избыток восстановителя обратно
оттитровывают солями железа (III).
Реверсивное титрование. Во многих случаях с целью получения более
надежных данных проводят так называемое реверсивное титрование.
При реверсивном титровании раствором определяемого вещества титруют
стандартный раствор реагента. Поясним это двумя примерами.
Определение азотистой кислоты и нитритов, основанное на
взаимодействии их с перманганатом калия в кислой среде, проводят не прямым
методом титрования анализируемого раствора стандартным раствором
перманганата, а предпочтительно реверсивным методом, титруя кислый
раствор перманганата калия анализируемым раствором. Такой способ
титрования дает возможность предотвратить разложение азотистой кислоты
и окисление ее кислородом воздуха. Благодаря этому результаты анализа
оказываются более точными, чем при использовании прямого метода
титрования.
Другим примером может служить реверсивный метод титрования
раствором определяемых солей ртути (II) стандартного раствора иодида
калия.
В начале титрования, когда в растворе находится много иодида и мало
ионов ртути, протекает реакция:
Hg++ + 4l- -* [Hgl4]~
а в конце титрования при эквивалентном содержании Hg++ и [Hgl4]"":
Hg++ + [HgI4]- -* 2HgI2
красный
осадок
Понятие «реверсивное» титрование не следует отождествлять с
понятием «обратное титрование», поскольку принципы их различны.
Б. ТЕХНИКА ХИМИЧЕСКОГО ЭКСПЕРИМЕНТА
В ОБЪЕМНОМ АНАЛИЗЕ
§ 3. Измерение объемов растворов
Объем и емкость (вместимость) измеряют в литрах (л) и кубических
метрах (м3) и их долях.
Литр (л) — объем одного килограмма химически чистой воды,
взвешенной при наибольшей ее плотности (т. е. при 3,98° С) и 1 атм\ 0,001
доля литра — миллилитр (мл).
Дольными единицами кубического метра являются: 0,001 доля м3 —
куб. дециметр (дм3); 0,001 доля дм3 — куб. сантиметр (см3), равный
0,000001 м3.
1 см3 не равен 1 мл. Поэтому нельзя отождествлять 1 л, равный 1000 мл,
с 1 дм3, равным 1000 см3. По этим же причинам не следует смешивать
понятия «миллилитр» (0,001 л) с кубическим сантиметром («кубиком» —
0,001 дм3).
1 л равен 1,000028 дм3\ 1 мл равен 1,000028 см3. На практике этой
разницей обычно пренебрегают и считают 1 мл равным 1 см3.
Точность измерения объемов. Измерение объемов растворов и емкости
сосудов необходимо проводить с достаточной точностью. Ошибки в
измерении объемов растворов и емкости посуды обусловливают ошибки
в окончательном расчете результатов анализа.
50
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
При несоблюдении точности измерений объемов нельзя получить
точные результаты анализа, если даже все другие аналитические операции
проводились с большой точностью.
Точность измерения (в процентах) вычисляют по формуле:
где Vi — объем, с точностью которого проводят отсчеты выливаемой из бюретки
жидкости, мл;
V — измеряемый объем раствора, мл.
Пример. Требуется вычислить точность измерения объема (в %), если известно, что
на титрование определяемого вещества пошло 25 мл раствора реактива, а отсчеты по
бюретке проведены с точностью до 0,02 мл.
Решение:
0,02-^-= 0,08%
Если в рассмотренном примере отсчеты по бюретке были проведены с точностью до
0,1 мл, то точность измерения составила бы только
о,, 4?-о,«
Следовательно, для получения точных результатов в объемном анализе
необходимо с большой точностью отсчитывать объемы титрованных
растворов, выливаемых из бюреток.
Вот почему измерение объемов растворов составляет наряду со
взвешиванием вещества очень важную аналитическую операцию в объемном
анализе.
Главными измерительными приборами в объемном анализе являются
бюретки, пипетки и мерные колбы.
Определение емкости измерительных приборов. Емкость
измерительных приборов при их калибровании обычно определяют взвешиванием
вмещаемой ими или выливаемой из них дистиллированной воды.
Поправка на температуру. Так как при взвешивании температура
воды отличается от температуры при ее наибольшей плотности (3,98° С), то вводят поправку,
учитывающую изменение плотности воды с температурой. При комнатной температуре
масса 1 л воды меньше 1 кг. Разница составляет 1000—\000d, где d — плотность воды
при данной температуре.
Поправка на взвешивание в воздухе. 1 л представляет собой
объем 1 кг химически чистой воды, взвешенной в безвоздушном пространстве.
По закону Архимеда, в воздухе вес тела уменьшается на величину, равную весу объема
вытесненного им воздуха. Например, 100 г воды вытесняют 100 мл воздуха, а вес 1 мг
воздуха равен 0,001293 gdun (g — ускорение силы тяжести). Следовательно, 100 г воды теряют
в весе 100-0,001293 5"= 0,1293 gdun.
100 г латуни (гирь) теряют в весе ^ ,_ -0,001293 = 0,0\53g дин, где 8,45 —
плотность литой латуни, т. е. вес 100 г воды меньше веса 100 г латунных гирь на 0,1293 g —
— 0,0153 g= 0,1140 g дин. Поэтому, отвесив 100 г воды, аналитик допускает ошибку
в определении массы на 0,1140 г.
Таким образом, взвешивая тело с относительно большим объемом на точных
аналитических весах с помощью латунных разновесов в воздухе, можно допустить существенную
неточность. Чтобы избежать этого, вводят поправку на взвешивание в воздухе.
Поправка на расширение стекла. Так как химику-аналитику
приходится работать главным образом со стеклянными измерительными ^приборами, то вводят
также поправку на расширение стекла, вызывающее изменение емкости сосудов при
колебаниях температуры.
Поправку находят по формуле:
^. = ^, + т^1(/2-<1)
где Vt — емкость сосуда при температуре W,
Vti — емкость сосуда при температуре h;
t2 — h — разность температур, где h^> h;
у\—коэффициент объемного расширения стекла (от 0,000012 до 0,00003).
§ 4. ПОСУДА, ПРИМЕНЯЕМАЯ ДЛЯ ИЗМЕРЕНИЯ ОБЪЕМОВ РАСТВОРОВ 51
Суммарная поправка. Таким образом, суммарная поправка складывается
из трех поправок:
1) поправки на температуру воды (Л);
2) поправки на потерю при взвешивании в воздухе (В);
3) поправки на расширение стекла (С).
Следовательно,
Ъп= А + В+С
Для нахождения истинной массы воды, взвешиваемой в стеклянном сосуде при
разных температурах в воздухе при помощи латунных гирь, пользуются специальной таблицей
(см. раздел «Литература»).
§ 4. Посуда, применяемая для измерения объемов растворов
Помимо обычной химической посуды, в объемном анализе широко
применяют мерную посуду, которая служит для точного измерения
объемов растворов.
Мерные колбы (рис. 13) представляют собой круглые плоскодонные
стеклянные сосуды с длинной узкой шейкой (горлом) с кольцевой меткой.
Мерные колбы служат для
измерения объемов растворов,
приготовления растворов определенной
концентрации. Объем жидкости,
вмещаемой колбой, выражают в
миллилитрах. На колбе указывают ее
емкость и температуру (обычно 20° С),
при которой эта емкость измерена.
Мерные колбы бывают различной f\
емкости: от 25 до 2000 мл.
Пипетки (рис. 14) представляют
собой длинные узкие стеклянные
трубки, расширенные в средней части; рИс. 13. Мерные колбы,
один конец трубки оттянут; трубка
снабжена кольцевой меткой. Некоторые пипетки имеют цилиндрическую
форму. Пипетки служат для отмеривания небольших объемов растворов
и перенесения определенного объема раствора из одного сосуда в другой.
Объем жидкости, вмещаемой пипеткой, выражают в миллилитрах. На
расширенной части пипетки указывают ее емкость и температуру (обычно
20° С), при которой эта емкость измерена.
Пипетки бывают различной емкости: от 1 до 100 мл.
Измерительные пипетки небольшой емкости не имеют расширения
и градуированы на 0,1—1 мл.
Бюретки (рис. 15) представляют собой узкие, градуированное по
длине цилиндрические стеклянные трубки. Один конец бюретки сужен
и снабжен стеклянным краном или резиновой трубкой, соединенной
с капилляром, через который из бюретки выливается раствор. Резиновая
трубка зажимается снаружи металлическим зажимом или закрывается
изнутри стеклянным шариком (рис. 16). При надавливании на зажим
указательным и большим пальцами или при-оттягивании резиновой
трубки в том месте, где помещается шарик, из бюретки выливается жидкость.
Обычно бюретки градуируют на миллилитры и доли миллилитра.
Для отсчета объемов растворов, расходуемых на титрование, на бюретке
нанесены деления и цифры. Нулевое деление помещено в верхней части
бюретки.
В макрометоде применяют бюретки емкостью 25—50 мл.
Такие бюретки градуируют с точностью до 0,1 мл. Расстояние между
двумя соседними делениями делят на глаз (рис. 17). Это дает возможность
52
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
проводить отсчет с точностью до сотых долей миллилитра; объемы
бесцветных растворов отсчитывают по нижней части мениска (рис. 18),
окрашенных — по верхней части (рис. 19). Для облегчения отсчета сотых
/S
25
МЛ
Ш
V
1
Illllllllll
Illllll
ТТЛ]
К'
§~
- L
1
- 1-
о\ |_
- 1-
Щ Ь
I
щ Ь
1 Е
0 0
1
- р-
- 1^
- !•-
щ Щ
1
- щ
i
|зо| |зр| |зо
1"
1
|_35l Is
I
1 1=
- ?
Ё
35 §35
|
«1 140
|45| |45| |45
(Ы |_
С
i \
к) ?т
1 '
Рис. 15. Б
50 рР
| ||
Т If
юретки.
i V
Рис. 14. Пипетки.
долей миллилитра применяют лупу. В лабораторный журнал результаты
отсчета записывают с двумя десятичными знаками (например, 23,05,
а не 23,1 или 23).
Для большей точности отсчета иногда противоположная от
наблюдателя стенка бюретки снабжается продольной узкой цветной полосой,
Ы—<*б
Щи <й-
ЬВ,51 *-
*L
^-^50
Рис. 16. Зажимы
для бюреток.
Рис. 17. Отсчеты по бюретке
при различных положениях
глаза.
нанесенной на молочно-белом стекле. В том месте, где устанавливается
мениск, наблюдается разрыв цветной полосы кажущийся
экспериментатору двумя сходящимися в одной точке углами (рис. 20). По месту
схождения углов отсчитывают объем вылитого из бюретки титрованного раствора.
S 4. ПОСУДА, ПРИМЕНЯЕМАЯ ДЛЯ ИЗМЕРЕНИЯ ОБЪЕМОВ РАСТВОРОВ 53
Бюретки, применяемые в микрометоде (рис. 21), градуированы на
0,01 мл, но, пользуясь ими, можно проводить отсчет и с большей
точностью — до тысячных долей миллилитра.
-=-24
-=-25
Рис. 18. Отсчеты по
бюретке при помощи белого
экрана с черной полосой.
В бюретку налит
прозрачный раствор. Отсчет
24,87 мл.
Рис. 19. Отсчет по
бюретке уровня
непрозрачного раствора. Источник
света находится сзади
наблюдателя. Отсчет
25,62 мл.
Бюретки заполняют титрованными растворами с помощью воронок.
Существуют бюретки с автоматически устанавливаемым уровнем; такие
бюретки заполняют при помощи специальных приспособлений (рис. 22).
Принцип действия
автоматической полу-
микробюретки (рис. 22, в).
Прежде всего устанавливают кран
бюретки 8 в положение /. Затем,
открыв пришлифованную стеклянную
Рис. 20. Отсчет по
бюретке, снабженной
цветной полосой.
Отсчет 48,21 мл.
Рис. 21. Микробюретка.
пробку /, через сухую стеклянную воронку наливают в баллон 2
стандартный (титрованный) раствор. Заполнив баллон 2 титрантом,
закрывают сосуд пробкой / и поворачивают кран 8 в положение //. При этом
жидкость под давлением собственной тяжести поступает по трубке 7
в бюретку 6. Избыток титранта через капилляр 10 переливается в
промежуточную емкость 5 и через капилляр 11 по трубке 3 автоматически сбра-
54
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
сывается в баллон 2. Благодаря такому устройству происходит
автоматическая установка уровня стандартного раствора на нулевой отметке.
Заполнив бюретку 6 стандартным раствором, кран 8 переводят в
положение /. При титровании кран 8 устанавливают в положение ///,
при котором титрант поступает в титруемый раствор.
Рис. 22. Бюретки:
а, б — с автоматически устанавливаемым уровнем; в — автоматическая полумикробюретка: / —
пришлифованная пробка; 2 — стеклянный баллон для стандартного раствора; 3 — трубка для сброса
избытка титранта; 4 — хлоркальциевая трубка, заполненная натронной известью и плавленным
хлоридом кальция; 5 — промежуточная емкость; 6 — бюретка емкостью 5 мл (цена деления 0,02 мл);
7 — подающая трубка, соединяющая баллон 2 с бюреткой 6; 8 — двухходовой кран; 9 — кончик
бюретки; 10 — капилляр автоматической установки нуля; // — капилляр трубки для сброса
избытка титранта.
Бюретки применяют для измерения объемов стандартных растворов,
расходуемых на титрование определяемых компонентов. Поэтому чем
тщательней проводят эти измерения, тем точнее результаты объемного
анализа. Следует помнить, что измерение объемов титрованных растворов
неточными измерительными приборами (бюретками) — ошибка
систематическая — или допущение небрежного отсчета показаний бюреток —
ошибка случайная — являются причинами неправильных результатов
объемного анализа.
§ 5. Работа с мерными колбами
Правила работы с мерными колбами. Колбу берут за верхнюю часть
горла, избегая прикасаться руками к ее выпуклой части. От тепла,
сообщаемого руками стенкам колбы, емкость колбы и, следовательно, объем
вмещаемой ею жидкости увеличиваются. Перед заполнением колбу ставят
на ровную, хорошо освещаемую поверхность стола.
Для растворения в мерной колбе твердого вещества его помещают
в колбу, которую заполняют растворителем не более чем на х/2 или 2/3.
Затем содержимое колбы взбалтывают плавными круговыми движениями
$ 5. РАБОТА С МЕРНЫМИ КОЛБАМИ
55
-J>
Рис. 23. Наблюдение за
установкой мениска в мерной, колбе.
до полного растворения вещества. Лишь после этого добавляют в колбу
новые количества растворителя.
Последние 1—2 мл растворителя прибавляют по каплям при помощи
пипетки, снабженной резиновым колпачком.
При добавлении последних капель жидкости глаза экспериментатора
и метка колбы должны находиться на одном уровне (рис. 23). Вогнутый
мениск поверхности жидкости своей нижней частью должен сливаться
с линией метки, а выпуклый мениск должен сливаться с линией метки
своей верхней частью.
Капли растворителя, удерживаемые на
внутренней поверхности шейки колбы выше
метки, осторожно удаляют при помощи
свернутой трубочкой фильтровальной бумаги.
Закрыв колбу хорошо пригнанной пробкой,
раствор очень тщательно перемешивают.
При отборе из мерной колбы части
раствора при помощи пипетки нижний конец ее
погружают в раствор почти до дна колбы.
Если пипетка опущена не до дна колбы, то
при засасывании раствора вместе с
жидкостью в пипетку будет проскакивать
воздух.
Не рекомендуется:
а) заполнять мерные колбы растворами трудно отмывающихся
веществ или образующих на стенках колбы трудно удаляемые гели;
б) хранить в мерных колбах в течение длительного времени
приготовленные растворы;
в) нагревать мерные колбы, сбивать в них остатки реактивов,
использовать их не по назначению.
Очистка мерных колб. Перед употреблением колба должна быть
тщательно вымыта.
Мерная колба (пипетка, бюретка) считается чистой только в том
случае, если на ней нет каких-либо видимых на глаз загрязнений и если
дистиллированная вода стекает с внутренних стенок сосуда, не оставляя
капель.
Если на стекле остаются капли воды, то это свидетельствует о
загрязнении его жировыми веществами. Жировые загрязнения сильно
искажают результаты измерения емкостей всех объемно-измерительных
сосудов, и поэтому их присутствие недопустимо.
Для удаления жировых загрязнений колбу моют хромовой смесью,
щелочным раствором перманганата, смесью спирта с эфиром, спиртовым
раствором едкого кали, горячим раствором тринатрийфосфата
(стиральным порошком) и т. п. Хромовую смесь готовят смешением равных объемов
насыщенного на холоду раствора бихромата калия и концентрированной
H2S04.
Помните, что работа с хромовой смесью требует соблюдения особой
осторожности. Хромовая смесь обжигает кожу, при попадании ее на
одежду происходит прожигание ткани или образование на ней
несмываемых пятен. Остатки хромовой смеси сливают обратно в склянку.
Для дополнительной очистки измерительных сосудов прибегают
к особому приему, называемому пропариванием с помощью водяного пара
(рис. 24). С этой целью колбу с водой укрепляют в штативе, зажигают
горелку и, когда вода закипит, пропускают струю пара в очищаемую
мерную колбу. Конденсирующаяся на стенках мерной колбы вода стекает
через воронку обратно в нагреваемую колбу. Пропаривание продолжают
56
ТЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
в течение 1 ч. После пропаривания на стенках колбы капли воды не
задерживаются и объем раствора, налитый в колбу, точно отвечает ее емкости.
Верным признаком тщательной очистки мерной колбы является хорр-
шая смачиваемость ее внутренней поверхности дистиллированной водой
и совершенно правильная сферически вогнутая
поверхность мениска раствора в шейке колбы.
Следует иметь в виду, что при пропаривании
мерной посуды может необратимо измениться ее емкость.
Поэтому после пропаривания посуды проверяют ее
емкость.
Проверка емкости мерной колбы. Действительная
емкость мерной колбы может значительно отличаться
от номинальной емкости, обозначенной на колбе.
Поэтому перед употреблением мерной колбы следует
проверить ее емкость. Мерные колбы калибруют
«на вливание», а бюретки и пипетки «на выливание».
Емкость мерной колбы проверяют путем
взвешивания на технических весах воды, вмещаемой колбой.
Для этого сначала взвешивают (или тарируют дробью)
пустую, предварительно тщательно очищенную и
высушенную горячим воздухом колбу, поместив ее на
левую чашку технических весов. Затем заполняют колбу
до метки дистиллированной водой, имеющей
температуру окружающего воздуха, и после этого вновь
взвешивают. Разность между массой колбы, заполненной
водой, и массой пустой колбы показывает массу воды,
вмещаемой колбой при данной температуре.
Массу воды делят на массу 1 мл воды при 20° С,
взвешенной при данной температуре с помощью
латунных разновесок.
Пример. Проверить емкость мерной колбы номинальной емкостью 250 ма, если масса
воды, вмещаемой указанной колбой при 15° С, равна 248,25 г.
Кажущаяся плотность воды при 15° С равна 0,99794 г/мл. Это значит, что 0,99794 г
воды, взвешенные при 15° С, занимают объем 1 мл при 20° С. Если разделить массу воды,
вмещаемой колбой, на массу 1 мл воды при той же температуре, то получим действительную
емкость колбы:
248,25 : 0,99794 = 248,8 мл
Разность между номинальной емкостью, обозначенной на колбе, и действительной
емкостью, установленной путем взвешивания воды, вмещаемой мерной колбой, составляет
ДУ = Уном — Действ = 250 — 248,8 = +1,2 м л
Рис. 24. Пропари-
вание мерной
колбы:
/—горелка; 2—колба
с водой; 3 — воронка;
4 — стеклянная
трубка; 5 — мерная колба.
§ 6. Работа с пипетками
Правила работы с пипетками. Во время работы с пипеткой избегают
прикосновения рукой к ее средней части. От тепла, сообщаемого рукой
стеклу, емкость пипетки и, следовательно, объем вмещаемой ею жидкости
увеличиваются.
При работе пипетку берут за верхнюю часть большим и средним
пальцами правой руки и глубоко (почти до самого дна сосуда) погружают
нижний конец пипетки в раствор. Придерживая левой рукой сосуд, из
которого берут жидкость, всасывают ртом жидкость в пипетку так, чтобы
уровень в ней поднялся на 2—3 см выше метки. Затем быстро закрывают
верхнее отверстие пипетки указательным пальцем (рис. 25), чтобы
жидкость не выливалась из пипетки. Избыток жидкости медленно сливают
из пипетки, ослабив слегка нажим пальца. Когда нижняя полоса мениска
§ 6. РАБОТА С ПИПЕТКАМИ
57
коснется метки, отверстие пипетки плотно закрывают, усилив нажим
пальца на верхнее отверстие, и переносят содержимое пипетки в колбу
для титрования. При этом пипетку держат так, чтобы метка находилась
на уровне глаза (рис. 26).
Выливать содержимое пипетки в другой
сосуд необходимо медленно, давая всей жидкости
равномерно тстечь из отверстия пипетки. При
быстром сливании раствора значительная часть
его в силу законов истечения жидкости
останется на стенках пипетки. Во время сливания
жидкости пипетку держат в вертикальном
положении.
К концу сливания, несмотря на все
предпринятые меры, в пипетке все же остается
некоторая часть жидкости. Поэтому по
окончании сливания прикасаются на мгновение
нижним концом пипетки к внутренней стенке
сосуда, в который сливают раствор.
Можно пользоваться и другими способами
наполнения пипетки и выливания из нее
жидкости. Но существует одно обязательное
правило: необходимо придерживаться во время
работы одного и того же способа наполнения
и выливания жидкости из пипетки.
Для наполнения пипетки легколетучими и
ядовитыми жидкостями (хромовая смесь,
растворы аммиака, органические растворители
и т. п.) пользуются специальными приспособлениями (рис. 27).
Засасывать такие жидкости ртом категорически запрещается.
Пипетки, промытые дистиллированной водой или соответствующим
растворителем, хранят в специальных штативах (рис. 28). Во избежание
Рис. 25. Положение пальцев
при отборе пробы с помощью
Рис. 26. Положение глаза
при измерении объема
с помощью пипетки.
Рис. 27. Прибор для заполнения пипетки
летучими или ядовитыми жидкостями:
/ — резиновая груша; 2 — резиновые
пробки; 3 — широкая стеклянная трубка;
4 — сосуд с раствором; 5 — пипетка.
Рис. 28. Штатив
для хранения
пипеток.
58
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
попадания в пипетку пыли верхнее отверстие пипеток накрывают
стеклянными или бумажными колпачками.
Очистка пипеток. Придерживаясь требуемых правил работы с
пипеткой, все же можно допустить очень большие ошибки при выполнении
объемно-аналитических определений. Это случается при работе с
недостаточно очищенной или загрязненной в процессе употребления пипеткой.
Ошибки, обусловливаемые загрязнением пипетки, могут быть
значительны, так как емкости пипеток сравнительно невелики, а искажения
вследствие загрязнения пипеток могут быть относительно большими.
Поэтому следует работать с абсолютно чистыми пипетками. Перед
употреблением пипетки должны быть тщательно вымыты и пропарены.
Проверка емкости пипетки. Действительная емкость пипетки может
значительно отличаться от номинальной емкости, обозначенной на
пипетке. Поэтому перед употреблением пипетки следует проверить ее емкость.
Емкость пипетки проверяют путем взвешивания воды, вмещаемой
пипеткой. Для этого предварительно взвешивают на технических весах
пустую колбу емкостью 100 мл. Потом выливают в нее воду из пипетки
и вновь взвешивают. Взвешивание воды проводится три раза. Далее
поступают так, как указано при проверке емкости мерной колбы.
Пример. Проверить емкость пипетки номинальной емкостью 25 мл, если масса воды,
вылитой из пипетки при 17° С, равна 24,35 а. Кажущаяся плотность воды при 17,0° С
равна 0,99767 г!мл. Следовательно, действительная емкость пипетки:
24,35 : 0,99767 = 24,41 мл
Д V = Уном - ^действ = 25 — 24,41 = +0,59 мл
§ 7. Работа с бюретками
Правила работы с бюретками. Бюретку укрепляют в штативе в строго
вертикальном положении. Перед каждым новым титрованием ее
заполняют до верхнего (нулевого) деления, предварительно заполнив
титрованным раствором нижний оттянутый конец бюретки или крана.
В момент отсчета показаний бюретки глаза экспериментатора должны
находиться на уровне мениска (см. рис. 17).
Отсчет проводят по нижней части вогнутого или верхней части
выпуклого мениска. Всякий раз установка уровня раствора на нулевое деление
должна соответствовать способу отсчета уровня оставшегося в бюретке
раствора.
Выливать жидкость из бюретки следует медленно, давая возможность
всей жидкости стечь со стенок бюретки, что имеет особое значение при
титровании неводными растворами. Под конец титрования раствор
выливают по каплям. Титрование нужно выполнять несколько раз. За
окончательный результат принимают среднюю величину, вычисляемую на
основании ряда параллельных определений. Объем расходуемого на
титрование стандартного раствора не должен превышать емкость бюретки.
Титрование считается оконченным, когда разность параллельных
определений це превышает ±0,1 мл.
По окончании титрования остающийся в бюретке раствор сливают;
после этого бюретку дважды ополаскивают дистиллированной водой,
а затем, заполнив ее доверху водой, накрывают верхний конец бюретки
колпачком для защиты от пыли. Перед употреблением сличают
дистиллированную воду до сужения бюретки. Затем бюретку дважды
ополаскивают тем раствором, которым будут титровать. Лишь после этого бюретку
наполняют стандартным раствором.
§ 7. РАБОТА С БЮРЕТКАМИ
59
Наполняют бюретки с помощью абсолютно чистой и сухой маленькой
стеклянной воронки. После употребления воронку немедленно вынимают
и ставят в штатив.
Очистка бюреток. Перед употреблением бюретки должны быть
тщательно вымыты (так же, как мерные колбы). Ошибки, обусловливаемые
загрязнением бюретки, могут достигать значительных величин.
Наибольшие ошибки вызываются жировыми загрязнениями. Вследствие этого
в процессе титрования на стенках бюретки остаются удерживаемые на
стекле капли жидкости. При этом, разумеется, искажаются результаты
измерений.
Во избежание попадания в бюретку маслянистых и жировых веществ
нельзя применять грязную посуду, обильно смазывать стеклянный кран
бюретки вазелином и закрывать при мытье посуды отверстие бюретки
пальцами.
Указанной степени чистоты бюретки нельзя достичь только мытьем.
Для окончательной очистки бюретки пропаривают водяным паром.
Проверка емкости бюретки. Действительная емкость бюретки и
отдельные ее деления могут значительно отличаться от номинальной емкости
и обозначений, нанесенных на бюретке. Поэтому перед употреблением
следует проверить емкость бюретки. Так как объемы, соответствующие
одинаковым делениям по всей длине бюретки, практически не могут быть
равны вследствие того, что трубка бюретки обычно не бывает строго
цилиндрической, то равным делениям бюретки в различных ее частях
соответствуют неравные объемы вмещаемого бюреткой раствора.
Для нахождения точного объема жидкости, вмещаемой между
определенными делениями бюретки, бюретку наполняют дистиллированной
водой и устанавливают мениск на нулевом делении. Затем под бюретку
подставляют бюкс, предварительно взвешенный с крышкой на
аналитических весах с точностью до 0,001 г. В бюкс медленно сливают из бюретки
определенный объем (5 мл) воды. После этого бюкс закрывают крышкой
и снова взвешивают. Разность между массой бюкса с водой и массой
пустого бюкса соответствует массе воды, вмещаемой в бюретке между
делениями 0 и 5 при данной температуре. После этого снова наполняют
бюретку дистиллированной водой до нулевого деления. Затем сливают
в бюкс 10 мл воды, которые взвешивают. Таким же способом взвешивают
15, 20, 25 и т. д. миллилитров воды.
С целью получения более точных результатов массу воды определяют
три раза и берут среднее арифметическое трех взвешиваний. При этом
пользуются следующей формой записи результатов взвешиваний:
Отсчет по шкале бюретки, мл 0—5 0—10 0—15
и т. д.
Масса бюкса, г
с водой
пустого
Масса воды, г
Результаты взвешиваний, г
первого
второго
третьего
Среднее арифметическое "трех взвешиваний
При выполнении точных ра0от вносят соответствующие поправки и
вычисляют точный объем вылитой жидкости.
60
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Если вычесть из величины истинного объема бюретки величину объема,
обозначенного на бюретке, то получим искомую поправку:
Уист — Уобозн = ДУ
т/ _ ?н2о
1000
где gHoQ — масса вылитой из бюретки воды, г\
-rfljk кажущаяся плотность воды, г!мл.
Например, если вода, вылитая из бюретки между делениями 0—5 мл,
при 15° С весит 5,052 г, то поправка равняется:
5Д52
0,9979
. 5 = +0,063 мл
Поправки вычисляют для каждых пяти миллилитров; данные
записывают в таблицу. На основании данных полученной таблицы
вычерчивают кривую поправок (рис. 29).
и, и о
0,0 h
ъ 0,02
^ о
-0,02
-П Пй
^0^"^ ,
10
20
t
30
4^\
50
t
60
Рис. 29. Кривая поправок емкости бюретки.
По оси ординат откладывают величины поправок, по оси абсцисс —
объемы, обозначенные на бюретке. По кривой находят поправки для
любого объема бюретки.
Примерная форма записи поправок, вычисляемых при калибровании
бюретки:
Номинальная
(Уобозн), МЛ
Масса воды (#н2о)> г • ¦
Температура опыта, °С
Истинная емкость бюретки
(и _ gH2Q \ иа
емкость бюретки
g20
Д1Л
1000 /
•'ист— ^обозн» мл
5
5,052
15
5,052
0,9979
5,052
0,9979
5=+0,063
10 и т. д.
10,074
16,2
10,074
0,9978
10,074
0,9978
-10=+0,09
Найдя нужную поправку, прибавляют (или вычитают, в зависимости
от ее знака), ее величину к обозначенной на бюретке величине емкости
и получают истинный объем раствора, помещающегося в бюретке или
части ее.
Например, если на титрование пошло 17,5 мл, истинный объем
раствора равен 17,53 мл; если на титрование пошло 45 мл, истинный объем
раствора составляет 44,99 мл и т. д.
§ 8. ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ РАСТВОРОВ
61
Определение объема капли растворителя. Очень полезно в начале
пользования бюреткой определить средний объем капли вытекающего
из нее растворителя. При употреблении водных растворов определяют
объем капли воды. Определение ведут следующим образом. Наливают
растворитель в бюретку. Устанавливают уровень жидкости на нуль.
Подставляют под бюретку колбу или стакан и очень медленно, ведя счет
каплям, сливают жидкость из бюретки. Отсчитав 100 капель, закрывают
кран бюретки и измеряют объем вытекшего растворителя. Отмеренный
объем жидкости делят на 10Q и вычисляют объем одной капли
применяемого растворителя.
Зная объем капли растворителя, можно вносить необходимые поправки
при вычислении результатов титрования.
§ 8. Приготовление стандартных растворов
Основным раствором в объемном анализе является титрованный, или
стандартный, раствор исходного реактива, при титровании которым
определяют содержание вещества в анализируемом веществе.
Приготовление растворов точно известной концентрации требует
соблюдения особых правил, исключительной точности и аккуратности
в работе. Несоблюдение требуемых условий впоследствии неизбежно
отражается на точности всех объемных определений, выполненных при
помощи приготовленного стандартного раствора, и очень часто приводит
не только к необходимости переделывать анализ, но и устанавливать вновь
титр исходного раствора.
Существуют различные способы приготовления титрованных растворов.
Грамм-эквивалент. Обычно готовят 0,1; 0,05 или 0,01 н. растворы,
содержащие соответственно 0,1; 0,05 и 0,01 грамм-эквивалента
растворенного вещества в 1 л раствора.
Грамм-эквивалентом (Э) называют количество вещества, выраженное
в граммах, которое в данной реакции соответствует (эквивалентно)
1,008 весовой части водорода (т. е. 1 грамм-атому водорода) или 8 весовым
частям кислорода (т. е. V2 грамм-атома кислорода).
Так как численное значение грамм-эквивалента связано с характером
реакции, в которую вступает данное вещество, то поэтому
грамм-эквивалент не является постоянным числом. В этом отношении
грамм-эквивалент отличается от грамм-молекулы, представляющей собой постоянное
число для данного вещества. Следовательно, для того чтобы вычислить
грамм-эквивалент, необходимо составить уравнение реакции, в которую
вступает данное вещество.
Для веществ, вступающих в реакции нейтрализации, грамм-эквивалент
равен:
П
где М — молекулярный вес данного вещества;
п — число ионов водорода или гидроксила, участвующих в данной реакции.
Например
^ _ ^NaOH ~ МВа (ОН)а
^NaOH — I ; ^Ва (ОН)2 — § '
. ^HsSQ4 Q __ ^СН»СООН
'H2S04 — 2 » ^CHaCOOH — |
62
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Для веществ, вступающих в реакции двойного обмена,
грамм-эквивалент равен:
П
где п — число зарядов ионов, обменивающихся в данной реакции.
Например
2AgN03 + ВаС12 -* 2AgCl + Ва (N03)2
а _ ^AgNQ8 Q ... МВаС12
^AgN08 — J *, ^BaCl, — 2
Для веществ, вступающих в реакции окисления—восстановления^
грамм-эквивалент равен:
П
где п — число электронов, отдаваемых восстановителем или принимаемых окислителем,
в данной реакции.
Например, КМп04 в зависимости от условий реакции может
приобретать 3 или 5 электронов и в соответствии с этим его эквивалент может
быть равен
^ ^КМп04
в щелочной среде ЭКМп04 = ^
^ ^КМп04
в кислой среде ЭКМп04 = g
Частное, получаемое при делении величины молекулярного веса
на величину эквивалента, выражает собой число грамм-эквивалентов
в 1 грамм-молекуле (1 грамм-атоме или 1 грамм-ионе) данного вещества.
Приготовление титрованного раствора по точной навеске исходного
вещества. Самым простым, на первый взгляд, способом приготовления
раствора точно известной концентрации, т. е. характеризующегося
определенным титром, является растворение точной навески исходного
химически чистого вещества в воде или другом растворителе и разбавление
полученного раствора до требуемого объема.
Зная массу (а) растворенного в воде химически чистого соединения
и объема (V) полученного раствора, легко вычислить титр (Тв)
приготовленного реактива:
Тв = у- г/мл
Этим способом готовят титрованные растворы таких веществ, которые
можно легко получить в чистом виде и состав которых отвечает точно
определенной формуле и не изменяется в процессе хранения.
Взвешивание вещества проводят в пробирке с притертой пробкой
(рис. 30), на часовом стекле или в бюксе. Ввиду того что некоторые
вещества очень трудно, а иногда практически невозможно получить в чистом
виде или трудно взвешивать на аналитических весах, прямой метод
приготовления титрованных растворов применяют лишь в отдельных
случаях. Таким путем нельзя приготовить титрованные растворы веществ,
которые отличаются большой гигроскопичностью, легко теряют
кристаллизационную воду, подвергаются действию двуокиси углерода воздуха
и т. д.
Установка титра раствора при помощи установочного вещества.
Второй способ установки титров основан4 на приготовлении раствора
§ 8. ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ РАСТВОРОВ 63
реактива приблизительно требуемой нормальности и последующем
точном определении концентрации полученного раствора.
Титр или нормальность приготовленного раствора определяют, титруя
им растворы так называемых установочных веществ.
Установочным веществом называют химически чистое соединение точно
известного состава, применяемое для установки титра раствора другого
вещества.
На основании данных титрования установочного вещества вычисляют
точный титр или нормальность приготовленного раствора.
Для приготовления титрованцого раствора растворяют в воде
взвешенную на технических весах навеску или смешивают с водой
определенный объем раствора данного вещества
приблизительно известной
концентрации; разбавляют полученный
раствор до требуемого объема и
устанавливают его концентрацию по
раствору другого (установочного) веще- Рис# 30. Пробирка для взвешивания твер-
ства, концентрация которого точно дых веществ,
известна. Раствор химически чистого
установочного вещества приготавливают растворением в воде
вычисленного его количества (взвешенного на аналитических весах) и
последующим доведением объема раствора до определенной величины в мерной
колбе. Отдельные (аликвотные) части приготовленного таким образом
раствора отбирают из мерной колбы пипеткой и титруют их раствором,
титр которого устанавливают. Титрование проводят несколько раз и
берут средний результат.
Иногда вместо отбора аликвотных частей раствора берут отдельные
точные навески установочного вещества, рассчитанные на одно титрование
(на 20—25 мл 0,1 н. раствора), растворяют их в воде и полученный раствор
титруют. Титрование аликвотных частей, отбираемых с помощью пипеток,
называют методом пипетирования; титрование точных навесок — методом
отдельных навесок.
Для вычислений пользуются соответствующими формулами (см. § 10).
Требования, предъявляемые к установочным веществам. Не всякое
вещество может быть применено в качестве установочного. Установочное
вещество должно удовлетворять следующим требованиям.
1. Иметь кристаллическую структуру и отвечать определенной
химической формуле.
2. Химический состав должен соответствовать его формуле.
3. Не содержать посторонних примесей выше допустимого предела,
устанавливаемого ГОСТ для данного вещества марки «х. ч.».
4. Способы очистки установочного вещества от сопутствующих
примесей (кристаллизация, экстракция, возгонка и др.) должны быть
доступными в аналитической лаборатории.
5. Химически чистое установочное вещество не должно быть
гигроскопичным, но должно сравнительно хорошо растворяться.
6. Растворы установочного вещества не должны изменять своего
титра при хранении и соприкосновении с воздухом.
7. Установочное вещество должно отличаться по возможности
наибольшим эквивалентным весом. Чем больше эквивалентный вес вещества,
тем больше точность установки титра раствора, так как при взвешивании
вещества с большим молекулярным весом ошибки взвешивания
оказываются незначительными.
Установка титра раствора с помощью другого титрованного раствора.
Очень часто титры устанавливают титрованием устанавливаемого раствора
64
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Рис. 31.
Устройство для
вскрытия ампулы фик-
санала.
другим титрованным раствором, приготовленным одним из указанных
выше способов.
Например, установить титр раствора едкого натра можно при помощи
титрованного раствора хлористоводородной кислоты, титр которого
установлен в свою очередь при помощи химически чистых карбоната или
тетрабората натрия.
Вычисления проводят по соответствующим формулам (см. § 10).
Приготовление титрованных растворов по «фиксаналу». Очень часто
на практике для приготовления титрованных растворов используют
приготовленные на химических заводах или в специальных лабораториях
точно отвешенные количества твердых химически чистых соединений или
точно отмеренные объемы их растворов, требуемые для
приготовления титрованных растворов определенной
нормальности.
Указанные вещества помещают в специальные
стеклянные ампулы и запаивают. Поступающие в продажу
ампулы с содержащимся в них определенным количеством
вещества называют фиксаналами.
Для приготовления требуемого титрованного раствора
ампулу разбивают над специальной воронкой,
снабженной пробивным устройством (рис. 31), содержимое ее
количественно переводят в мерную колбу и доводят объем
водой до метки.
Обычно в ампулах содержится 0,1 г-экв вещества, т. е.
столько, сколько требуется для приготовления 1 л точно
0,1 н. раствора.
Правила, соблюдаемые при приготовлении
титрованных растворов и определении их титров. При
установке титра стандартного раствора исходного реактива должны
соблюдаться следующие правила.
1. Исходное вещество, применяемое для приготовления стандартного
раствора, должно быть по возможности химически чистым.
2. Исходное вещество должно легко и быстро реагировать с
титруемыми веществами.
3. Раствор исходного вещества должен сохраняться долгое время без
изменения.
4. Реакции, протекающие между исходным и определяемым
веществами, должны проводиться по возможности методом прямого
титрования.
5. Процесс титрования должен заканчиваться быстро и четко.
Конечная точка титрования должна определяться легко и точно.
6. Устанавливать титры желательно либо путем растворения
рассчитанной навески исходного вещества в определенном объеме, либо при
помощи установочного вещества методом отдельных навесок. При этом
навеска должна быть по возможности большой (несколько сот
миллиграммов),
7. Для предурреждения ошибок при титровании необходимо
заботиться о том, чтобы объем стандартного раствора, расходуемого на
титрование определяемого вещества, был приблизительно равен 20 мл (при
пользовании бюреткой емкостью 25 мл). Лучше использовать для
титрования такие количества вещества, чтобы на титрование требовалось около
40 мл раствора (при пользовании бюреткой емкостью 50 мл). При
меньших объемах расходуемых реактивов и пользовании макробюретками
относительная ошибка будет превышать допустимую погрешность в
результате снижения точности измерения (см. § 3).
§9. КОНЦЕНТРАЦИЯ РАСТВОРОВ И СПОСОБЫ ЕЕ ВЫРАЖЕНИЯ 65
8. Не следует ограничиваться одним или двумя параллельными
определениями. Необходимо проводить титрование до тех пор, пока не
будет получено по крайней мере три сходящихся результата при
установке титра.
9. Приготовленные титрованные растворы должны сохраняться в
условиях, исключающих их загрязнение и изменение концентрации
вследствие поглощения влаги воздуха, а также испарение. Титры не должны
изменяться с течением времени.
10. Посуда и измерительные приборы, применяемые в объемном
анализе, должны быть тщательно вымыты, прокалиброваны, подготовлены
к титрованию и должны храниться в чистом месте.
11. Точность, с которой выполняют титрование, измерение объемов
и последующие вычисления, должна соответствовать точности
взвешивания. Поэтому совершенно недопустимо взвешивать навески исходных
или установочных веществ на технических весах с точностью до 0,1—
0,01 а и затем измерять объемы при помощи точных измерительных
приборов с точностью до сотых долей миллилитра или взвешивать навески
на аналитических весах с точностью, равной ±0,1%, и затем измерять
объемы растворов с точность^) ±0,5%.
В. ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
§ 9. Концентрация растворов и способы ее выражения
Обычно концентрацию раствора хорошо растворимых твердых или
жидких веществ выражают весовым количеством данного вещества,
содержащегося в определенном весоврм количестве (или объеме) раствора
или растворителя *. Концентрацию раствора можно выразить:
количеством граммов растворенного вещества в 100 а растворителя, или в 100 а
насыщенного при определенной температуре раствора, или же числом
молей вещества, содержащихся в 1000 мл раствора, или в 1000 а
растворителя, и т. д.
Существуют следующие способы выражения концентрации растворов.
1. В процентах (Со/о), т. е. числом граммов растворенного вещества,
находящегося в 100 а раствора:
Со/ = —Ц— 100 = -^ юо (1)
где ai — количество растворенного вещества, г;
а г — количество растворителя, г;
g — масса раствора, равная а\ + аг, г.
Пример 1. Вычислить концентрацию раствора карбоната натрия в процентах (Со/0),
если известно, что 25 г Na2COs растворены в 250 мл воды.
Решение. В данном случае а\ = 25 г; яг = К*р, где V — объем, ар — плотность **
растворителя, т. е. а2 = 250-1 = 250 г; g = 25 + 250 = 275 г.
Подставляем значения ах и 02 в формулу (1):
С 25 2500
С% ~ 25 + 250 Ш° ~ ^7Г "" 9'09 /о
* Числовые значения растворимости газов выражают преимущественно в
миллилитрах (при 20 и 100° С).
** Плотность р — отношение массы тела (g) к его объему (V) — выражают в г/см3.
66
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
2. В единицах молярности, т. е. числом грамм-молекул растворенного
вещества в 1 л раствора:
с - т - а (2)
где а — количество растворенного вещества, г;
т — число грамм-молекул растворенного вещества;
V — объем раствора, л;
Л1р.в — масса 1 моль растворенного вещества (численно равная молекулярному
весу), г<моль.
Если 1/=1, то формула (2) принимает вид:
См = —2— (2а)
м Мр.в
Очень часто См изображают просто индексом С. При числах моляр-
ность выражают буквой М; например — децимолярный раствор
обозначают 0,1 М.
Пример 2. Вычислить моляр ность раствора (См) серной кислоты, если известно,
что в 500 мл раствора содержится 49,04 г H2SO4.
а 49 04
Решение. В данном случае т= —г-я = nQ' = 0,5, где 98,08 — молекул яр-
mh2so4 98»08
ный вес H2S04; V = 0,5 л.
Подставляем значения т и V ъ формулу (2):
С 49,04 0,5
иЛ1~ 98,08-0,5 ~ 0,5 ~1М
Иногда концентрацию выражают числом молей вещества не на 1000 мл
раствора, а на 1000 в растворителя. Такие растворы, в отличие от
молярных, называют моляльными растворами:
г — т
("МОД —"~Г"
где т — число молей растворенного вещества;
/ — число тысяч граммов растворителя.
3. В единицах нормальности, т. е. числом грамм-эквивалентов (Э)
вещества, растворенного в I л раствора:
*.--f-sr (3>
где а — количество растворенного вещества, г;
п — число грамм-эквивалентов;
V — объем раствора, л;
Э — эквивалент растворенного вещества.
Иногда Сн обозначают просто N. При числах нормальность
выражают буквой «н». Например, децинормальный раствор обозначают 0,1 н.;
сантинормальный 0,01 н.
Если навеска исходного вещества (а) растворена в 1 л раствора (V =
= 1 л), то уравнение (3) примет вид:
откуда
#Э = аг/л (За)
Если навеска исходного вещества (а) растворена в V миллилитрах, то
где V — объем раствора, мл.
св^ = 4^ (Зб)
§9. КОНЦЕНТРАЦИЯ РАСТВОРОВ И СПОСОБЫ ЕЕ ВЫРАЖЕНИЯ 67
Так как нормальность (N) раствора означает число
грамм-эквивалентов вещества, содержащихся в 1 л (или число
миллиграмм-эквивалентов * в 1 мл) данного раствора, то, следовательно, произведение объема
раствора, выраженного в литрах, на его нормальность равно числу грамм-
эквивалентов (а выраженное в миллилитрах — числу
миллиграмм-эквивалентов) вещества.
Следовательно, если 1 л данного раствора содержит N
грамм-эквивалентов, то 1 мл его содержит Q грамм-эквивалентов или N
миллиграмм-эквивалентов. В V миллилитрах содержится 1QQQ V
грамм-эквивалентов, или ' * г, или А/"- Vмиллиграмм-эквивалентов, или N- V-Эмг.
Пример 3, Вычислить нормальность раствора (Сн) фосфорной кислоты
(реагирующей как трехосновная кислота), если известно, что в 250 мл раствора содержится 32,66 г
Н3Р04.
Решение. В данном случае
_ а _ 32,66 __
П~ ЭН,Р04 - 32,66 -1
где 32,66 — грамм-эквивалент ортофосфорной кислоты.
Подставляем значения п и V (250 мл) в формулу (36):
г _АТ_ 32,66-1000 ,
L*-N~ 32,66-250 "~4Н-
4. Точным числом граммов растворенного вещества в 1 мл раствора,
т. е. в виде титра (Т):
а _ N3 _ СММ^В
т-ТГ--щоб Гооо^г/И|Л (4)
где а — навеска растворенного вещества, г;
V — объем раствора, мл;
Э — эквивалент растворенного вещества.
Исходя из формул (3), (36) и (4), можно написать следующие основные
формулы:
1) Вычисление нормальности:
.. Т1000 а 1000 ,-ч
" = —э— =-Э1/- ®
где N — концентрация раствора, выраженная в единицах нормальности;
а — навеска вещества, г;
V — объем раствора, мл;
Э — эквивалент исходного вещества;
Т — титр раствора, г/мл.
2) Переход от нормальности к титру:
т=-щ-г/МЛ <6>
Пример 4. Вычислить титр (т. е. содержание вещества в граммах в единице объема)
1М раствора H2SO4 и 4 н. раствора НзР04 (см. примеры 2 и 3).
* Миллиграмм-эквивалентом называют эквивалентный вес (Э), выраженный в
миллиграммах, т. е. миллиграмм-эквивалент представляет собой величину в 1000 раз меньшую,
чем грамм-эквивалент.
68
ГЛЛ. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Решение. Подставляя в формулу (4) значения а и V для H2SO4 и НзР04,
получаем:
^ 49,04 1-98,08 плпопо .
TH2S04 = "500" = -1000- = 0j09808 г/МЛ
т 32,66 4-32,66 П10ПС/| .
Тн8Р04 = ~25Г = 1000- = °'13064 г1мл
§ 10. Способы вычисления в объемном анализе
В объемном анализе количество определяемого вещества или его
концентрацию рассчитывают несколькими способами:
1. По нормальности стандартного (титрованного) раствора (N).
2. По титру стандартного раствора (Тв) или по титру, выраженному
по определяемому веществу (ТВ/а).
3. С помощью поправочного коэффициента (К)-
Рассмотрим каждый из этих способов.
При вычислениях в объемном анализе приняты следующие
обозначения:
а — навеска образца анализируемого вещества, г;
Э — грамм-эквивалент;
N — нормальность (только в расчетных формулах; при цифрах
нормальность обозначается «н.»);
Т — титр, г/мл;
V — объем, мл;
g — содержание (масса) вещества, г.
Сочетание основных буквенных обозначений с соответствующими
индексами дает возможность построить любое обозначение.
Например, если необходимо обозначить грамм-эквивалент (Э)
определяемого вещества (А) или реактива (В), то соответственно пишут ЭА и Эв.
В соответствии с этим принято пользоваться следующими
обозначениями:
ТА — титр раствора определяемого вещества, г/мл;
Nк — нормальность раствора определяемого вещества;
УА — аликвотная часть раствора определяемого вещества, мл;
gA — содержание определяемого вещества в аликвотной (кратной)
части раствора, г;
gA — общее содержание определяемого вещества в исследуемом
продукте, г;
VK — общий объем определяемого раствора;
Тв — титр стандартного (титрованного) раствора реактива, г/мл\
Тв/а — титр стандартного (титрованного) раствора реактива по
определяемому веществу, выраженный в граммах определяемого
вещества А, эквивалентного количеству вещества В,
содержащегося в 1 мл стандартного раствора реактива В;
NB — нормальность стандартного (титрованного) раствора реактива;
/Св — поправочный коэффициент стандартного (титрованного)
раствора реактива;
VB — объем стандартного (титрованного) раствора реактива, мл;
gB — общее содержание реактива в стандартном (титрованном)
растворе, г.
Расчет содержания определяемого вещества по нормальности
исходного стандартного (титрованного) раствора реактива. Предположим, что
для количественного анализа было растворено в произвольном объеме
воды а граммов вещества А. Полученный раствор целиком был оттитрован
§10. СПОСОБЫ ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
69
стандартным (титрованным) раствором реактива В, нормальность
которого равна NB. На титрование всего раствора вещества А израсходовано
VB миллилитров стандартного раствора вещества В. Требуется вычислить
процентное содержание определяемого компонента в данном
анализируемом веществе.
Вычисления проводят исходя из правила эквивалентности (см. стр.40)
Прежде всего необходимо установить, сколько грамм-эквивалентов
реагента В прореагировало с определяемым веществом А. В 1000 мл
(в 1 л) стандартного (титрованного) раствора реактива В содержится NB
N V
грамм-эквивалентов В, а в Ув мл [fnjr = пв- Зная, сколько грамм-
эквивалентов В прореагировало с определяемым веществом А, можно
легко вычислить число грамм-эквивалентов определяемого вещества
в исследуемом продукте:
ЭА-ЭВ
- ^в
"А Ю00
где Яд — число грамм-эквивалентов определяемого вещества А.
Чтобы вычислить содержание определяемого вещества А (в граммах),
число грамм-эквивалентов его необходимо умножить на величину грамм-
эквивалента:
_AJB_B_ /7ч
ёА 1000 ч '
или в процентах:
gA100 3ANBVB юр
а 1000 'а к '
где а — навеска анализируемого вещества, г.
Пользуясь формулами (7) и (7а), можно рассчитать содержание
определяемого вещества в граммах или процентах по нормальности
стандартного (титрованного) раствора реактива В.
При определении концентрации раствора вычисления производят,
исходя из положения, что в точке эквивалентности произведение объемов
(в мл) реагирующих растворов на их нормальности равны между собой:
^в = ^А «)
Другими словами, в точке эквивалентности объемы прореагировавших
растворов реактива и определяемого вещества обратно пропорциональны
их нормальностям:
VA~ "в
Это правило называют правилом пропорциональности.
Предположим, что для анализа было использовано растворенное
в воде вещество А и объем раствора доведен до метки в мерной колбе
емкостью Укмл. На титрование ал иквотной-части раствора вещества А(УА)
потребовалось VB мл ^-нормального раствора реактива- В.
Требуется определить общее содержание вещества А (?д), титр (Тд)
и нормальность (N^ полученного раствора.
70
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
По уравнению (8) находим нормальность раствора определяемого
вещества А:
А ^А
Согласно формуле (6), титр МА-нормального раствора вещества А равен:
Т ^ЭА р,мл
Следовательно, общее содержание определяемого вещества А в объеме
VK мл, равное ТАУК, составит:
^А Ю00 к
ПОСКОЛЬКУ #А = ^-, ТО gд = ^^^" (9)
Растворы, характеризующиеся одинаковыми величинами нормальности,
реагируют друг с другом в равных объемах, или растворы, реагирующие
друг с другом в равных объемах, имеют одинаковую нормальность.
Если Л/в = NA, то VB = VA.
Пример /. Рассчитайте процентное содержание тетрабората натрия Na2B407* 10H2O
в образце технической буры, если известно, что на титрование навески буры, равной
0,2298 г, израсходовано 10,60 мл 0,1060 н. раствора НС1.
Решение. Прежде всего необходимо вычислить, сколько грамм-эквивалентов НС1
содержится в 10,60 мл (Кв) 0,1060 н. раствора хлористоводородной кислоты (пцс\)>
израсходованного на титрование.
В 1000 мл стандартного (титрованного) раствора соляной кислоты содержится
0,1060 г-экв НС1 (#НС1), а в 10,60 мл (VHCl):
0,1060-10,60 ™t
*HC1 = 1000 ' г-экв HCI
Согласно правилу эквивалентности, в оттитрованном растворе буры содержится
столько же грамм-эквивалентов Na2B407* ЮНгО. Чтобы установить, сколько граммов
ЫагВ407- IOH2O соответствует этому числу грамм-эквивалентов, его необходимо
умножить на 3Na2B4(V10H2O = 190,69 [см. уравнение (7)]:
_ 190,69-0,1060-10,60 _n01AQ
?Ыа2В4О7-10Н2О - [QUO -U,^14d 2
или в процентах [см. уравнение (7а)]:
190,69-0,1060-10,60 100
= 93,24о/0
1000 0,2298
Пример 2. Раствор технической соды неизвестной концентрации разбавлен водой
и доведен до метки в мерной колбе емкостью 250 мл. На титрование 25 мл полученного
раствора в присутствии индикатора метилового оранжевого израсходовано 22,45 мл
стандартного (титрованного) 0,1095 н. раствора хлористоводородной кислоты. Рассчитайте
общее содержание №гСОз в граммах в исходном растворе соды.
Решение. Согласно правилу пропорциональности:
^HCl^HCl = ^Na2COe^Na2C08
отсюда нормальность раствора ЫагСОз:
АЛ. ™ = Унарна 22,45-0,1095 ппшт
iVNa2C08 — -57 = or == 0.09835
^Na2CO, 25
§ 10. СПОСОБЫ ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
71
В 1000 мл титруемого раствора содержится 0,09835 г-экв, а в 250 мл:
0,09835-250 м „
fooo— г-экв Na2C03
Умножив эту величину на ЭКа2СОз = 52,99, получим gA:
0,09835-52,99-250 л опо м „
^а = — шоо = 1>ш г Na2C°3
Эту задачу можно решить, не делая промежуточных расчетов, т. е.
„ - jyHClvHCl9Na,co. VK 0,1095-22,45-52,99 250 _
*А iooo "W^7= looo гВ"-1'3038
Запись результатов и выполнение вычислений. При расчетах в
объемном анализе пользуются таблицей четырехзначных логарифмов.
Логарифмическая линейка дает возможность проводить вычисления до третьей
значащей цифры. Поэтому логарифмической линейкой можно
пользоваться только при ориентировочных вычислениях (например, при расчете
величины навески вещества, требуемой для проведения данного анализа).
Запись результатов вычислений должна отражать требуемую степень
точности, обусловливаемую точностью применяемого метода анализа
и точностью измерений массы и объема.
Для рассмотренного выше примера 1 соответствующая запись в лабораторном
журнале должна быть представлена следующим образом:
18ЭЫ.1В4О?-10Н,О = ^ 190'69 = 2'2803
+ 1Ш ^ на = Jg °»1060 = W253
1б^НС1 = 1б10»60 = 1.°253
2,3309
lg 1000 = 3,0000
1g^Na2B4O7.10H2O=1»3309
?Na2B4O7-10H2O = О»2143 г
lg *N..B4of .10H.O = lg0»2143 = 1,3309
+
lg 100 = 2,0000
1,3309
~~ lg a = lg 0,2298=7,3613
lg д: =1,9696
x = 93,24
Расчет содержания определяемого вещества по титру исходного
стандартного (титрованного) раствора реактива (Тв) или по титру,
выраженному по определяемому веществу (Тв/а). Предположим, что при
титровании методом отдельных навесок для анализа было растворено а граммов
вещества А. Полученный раствор был оттитрован стандартным раствором
реактива В, титр которого равен Тв. На титрование всего раствора пошло
Ув мл стандартного раотвора. Требуется вычислить процентное
содержание вещества А.
Вычисления проводят исходя из положения, что 1 г-экв вещества
А нацело реагирует с 1 г-экв вещества В.
72
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Количество граммов реактива В, пошедшего на реакцию с
определяемым компонентом, равно:
^в
Составляют пропорцию:
Эв-ЭА
gB-sA
8А Э,
еА-
в
(10)
эАтв^в
ЭВ
или в процентах:
При вычислении необходимо учитывать, что титрованию иногда
подвергают не весь раствор, объем которого равен VK, а лишь аликвотную
часть его (VA). Поэтому общее содержание определяемого компонента
в этом случае можно вычислить, установив предварительно титр
определяемого вещества по формуле:
т _ g'A
А~ Уа
Содержание (g'A) определяемого компонента (в г) в аликвотной части
(Уд) вычисляют по формуле:
• эатвув
*А=—Эв
По титру определяемого вещества вычисляют общее его содержание
(«а):
э.твквкк
«А = ТАУ«- АЭВ1/А <И>
Рассмотренный метод вычисления общего содержания определяемого
вещества по титру исходного раствора имеет значение при титровании
различных веществ одним и тем же стандартным раствором. При массовом
титровании одинаковых веществ удобнее выражать титр исходного
стандартного раствора в граммах определяемого вещества А (Тв/а),
количество которого соответствует 1 мл стандартного раствора реактива В.
Существенное отличие такого способа выражения концентрации
исходного стандартного раствора состоит в том, что титр стандартного
(титрованного) раствора реактива выражают в граммах определяемого вещества.
Тв/а показывает, сколько граммов определяемого вещества (А) реагирует
в процессе титрования с 1 мл раствора реактива (В).
Тв/а — титр по определяемому веществу выражают в граммах
определяемого вещества, количество которого эквивалентно количеству ее-
щества, содержащегося в 1 мл стандартного раствора, т. е. величина Тв/а
показывает, сколько граммов определяемого вещества эквивалентно
(равноценно) количеству вещества стандартного раствора реактива,
содержащемуся в I мл его раствора. Например, Та6ыо3 = 0,001699 г/мл
означает, что в I мл исходного стандартного раствора нитрата серебра
содержится 0,001699 г AgN03, TAgNo8/Hci = 0,0003646 г/мл, TAgNo8/cr =
= 0,0003546 г/мл, TAgNo8/Br- = 0,0007992 г/мл и TAgNo8/i- = 0,001269 г/мл
соответственно означают, что 1 мл исходного стандартного раствора,
§10. СПОСОБЫ ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
73
содержащего 0,001699 г AgN03, эквивалентен 0,0003646 г НС1, 0,0003546 г
хлорид-ионов, 0,0007992 г бромид-ионов и 0,001269 а иодид-ионов.
Поэтому, если на титрование хлористоводородной кислоты пошло
20,45 мл указанного стандартного раствора AgN03, то, следовательно,
в кислоте содержалось 20,45-0,0003646 =0,007458 г НС1 или 20,45 X
X 0,0003546 = 0,007251 г хлорид-ионов.
При массовых авдлизах способ выражения титра по определяемому
веществу значительно удобнее способа выражения концентрации по
титру исходного стандартного раствора. Для того чтобы вычислить этим
способом количество определяемого компонента, требуется только
умножить величину Тв/а на объем израсходованного стандартного раствора Ув
(в мл).
Для перехода от одного способа выражения титра к другому
прибегают к следующим рассуждениям. Предположим, что при титровании
раствора определяемого вещества А (например, КОН) неизвестной
концентрации было израсходовано VB миллилитров стандартного
(титрованного) раствора реактива В (например, НС1) с Тв, т. е. Thci = 0,003646 г/мл.
Требуется вычислить титр стандартного раствора реактива по
определяемому веществу, т. е. ТВ/а или THci/koh.
Расчет проводят исходя из основного положения эквивалентности:
В нашем случае
эв~~~ эа
ТВ"~ТВ/А
т
тсэ
В^А
ВМ--Э?
т 0,003646-56,11 ЛППСС11 ,
thci/koh = збДё =0,005611 г/мл
Это означает, что 0,003646 г реактива В (в нашем случае НС1),
содержащегося в 1 мл стандартного раствора, полностью нейтрализуют
0,005611 2 определяемого вещества А (в нашем случае КОН).
Другими словами, титры одного и того же раствора, выраженные
различными способами, зависят от величин грамм-эквивалентов
стандартного и определяемых веществ. Эту зависимость выражают следующей
формулой:
ТВ/А _ ЭА
ТВ ЭВ
Пользуясь этой формулой, можно вычислить титр раствора по
определяемому веществу:
Тв/а = -Р- (12)
•^в
Эд
где -^ величина постоянная; ее называют аналитическим множителем (фактором)
dB
объемного анализа и обозначают F.
Следовательно
тв/а = ^тв (13)
Для расчета содержания определяемого компонента необходимо титр
стандартного раствора умножить на аналитический объемный фактор
и на объем стандартного (титрованного) раствора, израсходованного на
титрование анализируемого образца:
?а = ^Тв1/в; gA = TB/AVB (W)
или в процентах:
* = tb/afb-^- (На)
74
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Таким образом, весь расчет сводится к умножению двух (если заранее
известен ТВ/а) или трех величин: F, Тв и VB-
Если определение велось из аликвотной части анализируемого
раствора, необходимо ввести в расчет сомножитель -тг-:
т т/ Ук т т/ Ук ЮО
SA = ^B/AVb-y-; ИЛИХА = ТВ/АУВ'ТГ--Т~
А А
Пример 3. На титрование навески а (0,2240 г) технической соды в присутствии
индикатора метилового оранжевого израсходовано 18,00 мл стандартного (титрованного)
раствора хлористоводородной кислоты (Vhci) c ^hci = 0,003646 г/мл.
Рассчитайте содержание №гСОз в исходной соде.
Решение. Для решения этой задачи можно воспользоваться формулой (10). Но те же
результаты можно получить, применив правило эквивалентности.
На титрование навески соды пошло:
ТНС1КНС1 = 0,003646-18,00 г НС1
По правилу эквивалентности (ЭНС1 = 36,46 и 3Na2COs = 52,99) содержание ЫагСОз
(в граммах) можно найти из пропорции:
36,46—52,99
или -в- процентах:
•0,003646-18,00 -gNa2COa
_ 0,003646-18,00-52,99
&Na2C08 ~ 36,46
0,003646-18,00-52,99 100 _ Ап enn7
*Na2C03 - 36Д6 0,2240 ~ АЬ /о
Пример 4. Азотная кислота неизвестной концентрации разбавлена водой и доведена
до метки в мерной колбе емкостью 250 мл. На титрование 25 мл полученного раствора
израсходовано 32 мл раствора едкого натра с TNaOH/HNOs = 0,06300 г/мл. Вычислите
содержание НШз (?hno8)-
Решение. Для решения этой задачи титр стандартного (титрованного) раствора
едкого натра, выраженный по определяемому веществу TNaOH/HNOa, умножают на его
* /1/ ч Ук 250 1Л
объем (yNaOH) и на отношение -тг"-— -of = *0:
?HNO, = ТНаОН/НЫОз^аОН ТГ~ = 0,06300-32-10 = 20,16 г
А
Расчет содержания определяемого вещества с помощью поправочного
коэффициента. Наряду с упомянутыми способами расчета содержания
определяемого вещества пользуются также способом расчета с помощью
так называемого поправочного коэффициента (7СВ), показывающего, во
сколько раз практическая нормальность или практический титр данного
стандартного (титрованного) раствора больше или меньше теоретической
нормальности или теоретического титра этого раствора определенной
концентрации:
Л^практ ТПракт
Ad = —
/vxeop TTeop
Например, готовят приблизительно 0,1 н. раствор хлористоводородной
кислоты и, титруя им установочное вещество (Na2B4O7-10H2O, Na2C03),
находят, что нормальность раствора НС1 равняется 0,1254. Значит,
приготовленный стандартный раствор более концентрированный, чем 0,1 н.
(теоретический) раствор НС1; концентрация его во столько раз больше,
§ 10. СПОСОБЫ ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
75
во сколько 0,1254 больше 0,1, т. е. в 1,254 раза. Число 1,254 и
представляет собой поправочный коэффициент 0,1254 н. раствора НС1 по
отношению к 0,1 н. раствору.
Следовательно, чтобы определить поправочный коэффициент данного
стандартного (титрованного) раствора, необходимо величину NnpaKt
разделить на величину NTeop. В данном примере:
^практ __ 0,1254
Поправка больше единицы указывает на то, что нормальность данного
раствора больше теоретической, а поправка меньше единицы указывает
на то, что нормальность данного раствора меньше теоретической.
Чтобы определить поправочный коэффициент данного раствора по
титру, необходимо величину Тпракт разделить на величину Ттеор.
Поправочный коэффициент можно также рассчитать, зная япракт
(навеску, взвешенную на аналитических весах) и атеор (теоретическую
навеску):
Япракт
в Отеор
Суммируя все изложенное, можно сделать вывод, что
^практ Тпракт Япракт е
Лд = —77 = -= = (15)
**теор Атеор атеор
Зная величину поправочного коэффициента, можно рассчитать общее
содержание определяемого компонента по теоретической нормальности
или теоретическому титру, так как произведение теоретической
нормальности или теоретического титра на поправочный коэффициент выражает
собой соответственно величину практической нормальности или
практического титра:
теор^В== практ' теор^В == практ
Предположим, что для титрования аликвотной части {Vp) раствора,
в. котором растворена навеска а вещества А, израсходовано Ув мл
Nq-нормального раствора вещества В, имеющего поправочный коэффициент
/(в. Требуется вычислить процентное содержание определяемого
компонента.
Содержание определяемого компонента (в г) вычисляют по
приведенной выше формуле (9):
„ W»A vK
gA ~ Ю00 VA
Но так как наш стандартный раствор не точно Л^в-нормальный, то для
вычисления NnpSLKT нужно NTeop умножить на 7(в. Следовательно
теор В В AJ^k_ /16ч
^А Ю00 VA К }
или в процентах
__^т€ор*ВУВЭА Ук ЮО п-
1000 VA a K }
При переходе к формуле определения вещества А методом отдельных
навесок в приведенной выше формуле опускают величину -^_.
* А
76
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Рассмотренный способ расчета с помощью поправочного коэффициента
широко применяют в практике лабораторий, в которых выполняют
массовые анализы.
С целью твердого усвоения студентами способов расчета
рекомендуется пользоваться одновременно всеми способами. В случае сходимости
результатов анализа, вычисленных тремя способами, можно быть
уверенным в правильности проведенных расчетов.
Пример 5. Вычислите содержание H2S04 в растворе объемом 250 мл\ установите
значения Тн so , NH so и Кн so » если на титрование 24,99 мл раствора серной кислоты
(УА) идет221,72 млЪУн. раствора NaOH (VB), KNaOH = 1,012.
Решение.
1) NA - -*-*-
# NaOH = ^ B = ^теор^В = 0,1 -1,012 = 0,1012
21,72-0,1012
NH2SO< — 24^9 ~~ °'087У4
2)TAeW» 3H2S04= 49,04
TH2so4 (практ) = °,°8^49'04 = 0.004313 г/мл
T _ ^теорЭд __ 0,1-49,04 __
1H2S04 (теор) - 1000 1000 ~~ U'UU4yU4 г/МЛ'
Л^практ _ Тпракт _ 0,08794 _ 0,004313
v-reop Ттеор
4) Расчет содержания определяемого компонента по NB:
«v „ _ " практ _ * практ __ и,ио/з* _ u,uu*oio __
з) *h2so4 - т^~ " "Т^Г ~ ~оХ~~ ~" 0,004904 - °*8794
„ УвЭАУк я „ 21,72-0,1012-49,04-250 _ , А„0 _
^А ~ 1000КД ' а *H,so4 ~ 1000-24,99 " 1,U™ 2
А *
5) Расчет содержания определяемого вещества по Т.:
#H2so< = 0,004313-250 = 1,078 г
6) Расчет содержания определяемого вещества по поправочному коэффициенту (Кв):
^А 10001/А
_ 0,1-1,012-21,72-49,04-250 _
gH2so4 ~ 1000-24,99 "~ 1,07в г
Расчет содержания вещества, определяемого методом обратного
титрования. В тех случаях, когда применяют метод обратного титрования
как, например, при определении НС1 с помощью AgN03 и NH4SCN
(см. § 2), расчеты проводят по формулам, имеющим несколько иной вид.
Обычно расчет сводится к определению:
1) числа грамм-эквивалентов (п'в) стандартного раствора вещества В,
затраченного с избытком на реакцию с определяемым веществом А:
• _^вув
*в~~"ТобсГ
§ 10. СПОСОБЫ ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
77
2) числа грамм-эквивалентов (nBt) стандартного раствора вещества Вь
пошедшего на титрование избытка вещества В:
Ув,
Лв1 " 1000
3) числа грамм-эквивалентов (яд) определяемого вещества А, равного
числу грамм-эквивалентов основного реактива В, прореагировавшего
с определяемым веществом А:
rtA==nB~ nBt
4) количества вещества А в граммах:
*а-яаэа
Подставляя значения п'в и nBi в формулу, выражающую значение ?д>
получаем:
wnvn — Nn vn \
*А-( им' 'Ь <'*>
Для титрования по методу отдельных навесок (в %):
х (NBVB-NBVB\ ЮО
ха ~ [ iooo ) эа ~Т~ (19)
Для метода пипетирования (в %):
х ("вУъ-УвЛ-
хь-\ 1000 / ^А
Расчет по титру (в %):
эв, )Эау.
Авкв
эв ^Bt / " "а
vK
VA
, V«
100
a
100
(20)
так как:
Т1000
N =—7?
Расчет содержания определяемого вещества методом обратного
титрования может быть проведен также исходя из следующих рассуждений.
Допустим, что к определяемому веществу А было добавлено в избытке
Ув миллилитров реактива В. По завершении реакции вещества А с
реактивом В избыток последнего (Ув мл) оттитровывают yBl миллилитрами
стандартного раствора реактива В1#
Следовательно, с определяемым веществом А прореагировало Ув— Ув=
5= Ув мл реактива В. Если Ув мл раствора реактива В эквивалентно
У^ мл реактива В1Э то можно вычислить, какой объем (в мл)
стандартного раствора реактива Вх эквивалентен содержанию определяемого
вещества А, прореагировавшего с Ув мл реактива В. Расчет проводят
по формуле:
vBt *% — KB<
где Vg —объем стандартного раствора реактива Bi (в мл), эквивалентного VB мл
реактива В, добавленного в избытке к определяемому веществу А;
VB — объем стандартного раствора реактива Вх (в мл), израсходованного на
титрование Vg мл избытка реактива В, не вошедшего в реакцию с определяемым
веществом А;
1/g1 — объем стандартного раствора реактива Вг (в мл), эквивалентного содержанию
определяемого вещества А, прореагировавшего с V^ мл реактива В.
78
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Таким образом, для того чтобы вычислить объем VBl стандартного
раствора реактива Bi (в мл), необходимо из Vb± мл стандартного раствора
реактива В1э эквивалентного общему количеству миллилитров VB
реактива В, добавленного в избытке к определяемому веществу А, вычесть
Ув! мл стандартного раствора реактива Вь расходуемого на титрование
избытка реактива В, не вошедшего в реакцию с определяемым веществом А.
Чтобы опытным путем определить, скольким миллилитрам
стандартного раствора реактива Вх эквивалентно Ув мл раствора реактива В,
предварительно в отдельной колбе оттитровывают Ув мл реактива В
стандартным раствором реактива В±.
Зная объем VlBi9 вычисляют содержание определяемого вещества А,
исходя из основного положения эквивалентности:
ЭА -~ ЭВх
ЭдУУ tr
gA= A *' <21>
А эв,
Если известна нормальность раствора Вх, то можно написать:
ЭАУВ ^В
^А^ ЮОО (22)
Если титрования проводились методом пипетирования, то полученный
результат нужно умножить на VK и разделить на VA (см. уравнение 15,
а также пример 6).
Нетрудно заметить, что при данном способе расчета нет необходимости
знать основные характеристики вспомогательного раствора реактива В:
титр Тв, нормальность NB и поправочный коэффициент Кв-
В табл. 4 приведены расчетные формулы, применяемые для
вычислений в объемном анализе.
Пример 6. Навеска пиролюзита (а) 0,0900 г восстановлена при соответствующих
условиях 50 мл tVB\ раствора оксалата (реактив В). По окончании восстановления Мп02
(определяемое вещество А) избыток восстановителя (Vg) оттитрован стандартным 0,1 н.
раствором КМп04 (реактив ВХ). На титрование избытка оксалата (Kg) израсходовано 15 мл
(VB±) раствора КМп04. На титрование 50 мл раствора оксалата (Vg) в «холостом» опыте
израсходовано 24 мл (VlB ) раствора КМп04. Вычислите, сколька процентов Мп02 содержал
анализируемый образец пиролюзита.
Решение. На титрование «холостого» опыта израсходовано 24 мл раствора КМп04,
а на титрование избытка оксалата —15 мл этого же раствора, т. е.
Vit - VBl = Vll = V1wtBOt - VmBOt = V»Mn0i - 24 - 15 = 9 мл
Следовательно, 9 мл — это объем стандартного раствора КМп04, эквивалентного
содержанию МпОг.
Исходя из правила эквивалентности, можем вычислить, сколько процентов МпОг
содержал анализируемый образец пиролюзита:
- _ ЭмпОа^кМпо/кМпО, 100 43,47-0,1-9 100 ..._
*Мп0*"" ЮОО Г" = —Гооо—"Шоо" = 43-47%
Пример 7. Навеску известняка, равную 0,7500 г, обработали 100 мл (Кв) 0,1 н.
раствора НС1; избыток кислоты оттитровали 20 мл (VBi) раствора NaOH (B^, титр которого
Тв, = 0,004216 г/мл. Вычислить, сколько процентов окиси кальция (А) было в известняке.
1) Определение числа грамм-эквивалентов (/ig) хлористоводородной кислоты (В),
взятой для обработки навески известняка:
' ^hci^hci ОД-ШО nm я а1ГО нп
лв = юоо = -щоо" = °'01 г'экв НС1
§ и. связь между точностью измерений и точностью вычислений 79
2) Определение числа грамм-эквивалентов (яВ1) едкого натра/В^, израсходованного
на титрование избытка хлористоводородной кислоты:
'TNaOH^NaOH 0,004216-20 n nnotno м пи
/iRj = = 77г = 0,002108 г-акв NaOH
1 3NaOH 40
3) Определение числа грамм-эквивалентов хлористоводородной кислоты,
прореагировавшей с известняком. Число грамм-эквивалентов хлористоводородной кислоты равно
числу грамм-эквивалентов (/гд) окиси кальция (А):
пА = пв — nBi = 0,01 — 0,002108 = 0,007892 г-же НС1 = 0,007892 г-экв СаО
4) Определение содержания окиси кальция в известняке (в %):
ЭСао=^= 28,04
28,04.0,007892-100
*СаО ~ QJ500 ~ У'5и/о
Вычисление по общей формуле для метода отдельных навесок (см. § 10).
Вычисление #NaOH по титру Тв:
А, TNaOH1000 0,004216-1000 nm-,
^NaOH = —а" = 40 = 0,Ю54
*Г*Г\ =
7NaOH
(^HCi^HCi-^NaOHl/NaOH) ЭСаО100
Ca° ~~ 1000 g
g — навеска
__ (0,1 ¦ 100 — 0,1054-20) 28,04-100 __ (10 — 2,108)28,04 __ 7,892-28,04 _ 0Q
*Ca° ~ 1000-0,7500 " ЪЬ ~~ 7^ " ^'&U/o
§ 11. Связь между точностью измерений и точностью вычислений
Все вычисления в объемном анализе проводят с требуемой точностью,
придерживаясь изложенных выше правил (см. Введение, §11).
Искомый результат не может быть более точным, чем точность
применяемого метода анализа (основанного на использовании измерительных
приборов, характеризующихся определенной точностью), поэтому
вычисления в объемном анализе необходимо проводить с той точностью,
6 какой проводится измерение с помощью бюретки объемов стандартных
(титрованных) растворов.
Обычные бюретки емкостью 25—50 мл калиброваны на 0,1 мл, и
точность отсчетов объемов по этим бюреткам до сотых долей миллилитра.
Микробюретки емкостью 1—2 мл градуированы на 0,01 мл\ точность
отсчетов по этим бюреткам до тысячных долей миллилитра.
Объем, отсчитываемый по бюретке, и числа, выражающие в объемном
методе анализа нормальность, титр и поправочный коэффициент, должны
содержать 4 значащие цифры.
Поэтому при вычислении результатов объемного анализа нельзя,
например, писать 0,1 н. вместо 0,1042 н. или 0,0034 г/мл вместо
0,003356 г/мл. В то же время бессмысленно выражать поправочный
коэффициент в виде числа 0,9350782, вместо того чтобы написать его в виде
четырехзначного числа 0,9351. Наконец, точность результата анализа не
повысится, если вместо 0,5634 г написать 0,5634428 г, так как в этом
случае просто увеличивается число недостоверных цифр.
Окончательный результат, вычисленный на основании данных,
полученных в процессе титрования с помощью макробюретки, выражают
четырехзначным числом, в котором только одна последняя цифра является
не вполне достоверной.
Расчетные формулы, применяемые для вычисления в объемном анализе'
ТАБЛИЦА 4 g
Исходные величины
Определение методом
отдельных навесок
пипетирования
Выражение нормальности, титра
и поправочного коэффициента
Определение содержания вещества А в граммах
Нормальность стандартного
(титрованного) раствора (N^
Титр стандартного раствора (Тв)
Титр стандартного раствора,
выраженный по определяемому веществу ТВ^А
Поправочный коэффициент (/Св)
?а =
*Wa
1000
а ЭАТВ^В
'В
8а = тв/а^
В/АКВ
gA Ю00
8а
1000- V.
вА
9B^A
#А = Тв/А^В-рг1-
8 =
10001/д
"в =
твюоо
а-1000 _ #bVb
*» А
Эв^в ' А"
В- ЮОО ' g_ Акк»
VA
эА эА
тв--Sf. -gj—ftTBIA-FT„
ЬВ/А
LB/A =riB
*B = "iv,
Л^практ Tr,paKT апракт
теор
i теор u теор
Нормальность стандартных растворов
(/VB и WBi) (метод обратного
титрования)
Титр стандартных растворов (Тв и TBJ
(метод обратного титрования)
Титр стандартного раствора В (Тв)
(метод обратного титрования)
Нормальность стандартного раствора
В (#в) (метод обратного титрования)
(УвГв-*в/в.)эа
gA ЮОО
„ /Ув тв/вЛ
8а~[ эв эв, J
8А
еА
Эа^в'Дв,
ЭдУв'Ав,
1000
«А =
(^вУв-^в,Ув1)ЭАУ»
1000V,
«А =
„,/Ув УвЛ
~ i эв Эв, }
"5вГ) А^
ЭА^в',Тв, VK
gA= Э
о эАкв>в, Кк
gA ЮОО Кя
ЕслиЛГв = ЛГВ(, то VB-KBi = l/y
Тв 1000
N — '
"В|— а
^в,
^', = ^,-4
Л'вЛ,
Тв' 1000
Определение содержания вещества А в процентах
100 о/
100 ..
* При переходе от формул, применяемых для вычисления содержания определяемого вещества А по методу отдельных навесок, к формулам определения
содержания вещества по методу пипетирования вводят коэффициент к-, где ^к — общий объем раствора, в котором растворена навеска; Уд — объем аликвотной
А А
части раствора определяемого вещества А.
82
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
§ 12. Краткие сведения о статистической обработке
экспериментальных данных
Даже точные наблюдения могут привести к неточным выводам, если
опытные данные обработаны неправильно. Поэтому обработка результатов
эксперимента является ответственной операцией при выполнении
лабораторных и исследовательских работ.
При обработке экспериментальных данных приходится: а) учитывать
и оценивать погрешности при вычислениях; б) статистически
обрабатывать полученную информацию (результатов измерения) и
анализировать ее.
Общие понятия теории ошибок. Аналитические операции и
измерения неизбежно сопровождаются ошибками. Ошибки принято делить
на систематические, случайные и грубые (нужно иметь в виду, что это
деление условно).
Грубые ошибки. При обработке экспериментальных данных
приходится считаться с возможностью возникновения ошибок при проведении
эксперимента или получения совершенно неверных результатов
вследствие внешних влияний. Очень серьезные ошибки могут возникнуть,
если, например, наблюдатель неправильно прочтет отсчет по бюретке,
потеряет вещество при пересыпании или перенесении из одного сосуда
в другой или допустит арифметические ошибки в вычислении. Наличие
ошибок проявляется в том, что среди сравнительно близких результатов
наблюдается одно или несколько значений, заметно выделяющихся по
величине из общего ряда. Если отличие настолько велико, что можно
говорить о грубой ошибке, то это измерение сразу отбрасывают. Однако
так просто дело обстоит редко. В большинстве случаев нельзя сразу
признать то или иное наблюдение неверным только по признаку
«выскакивания» из общего ряда и нужно провести дополнительные исследования.
Систематические ошибки. Обычные результаты измерений всегда
являются приближенными прежде всего вследствие ограниченной точности
измерительных приборов *. Поэтому к систематическим ошибкам
относятся в первую очередь ошибки, которые часто называют
инструментальными. Инструментальные ошибки не единственный вид систематических
ошибок. Например, при вычислении количества определяемого вещества
(см. § 10) ряд величин, входящих в соответствующие формулы, содержат
ошибки, которые систематически будут изменять определяемую по ним
величину.
К ошибкам этого рода можно отнести также ошибки, возникающие
вследствие использования недостаточно чистых реактивов, титрования
неправильно установленными стандартными растворами и т. д. Общим
признаком систематических ошибок можно считать принципиальную
возможность изучить их и исключить из результатов измерений.
Случайные ошибки. Многократные измерения одной и той же
величины, произведенные с возможной тщательностью, и после учета всех
систематических ошибок всегда дают различные числовые значения, т. е.
на результаты измерений влияют какие-то причины, не поддающиеся
учету. Например, проводится взвешивание на аналитических весах.
В момент измерения в результате внешнего воздействия стрелка весов
может отклониться в случайном направлении. На результаты измерения
могут влиять колебания температуры, атмосферные изменения или
индивидуальные особенности наблюдателя. Причиной случайных ошибок
* Точность измерительного прибора в большинстве случаев указывается в заводском
паспорте, прилагаемом к прибору.
G 12. СТАТИСТИЧЕСКАЯ ОБРАБОТКА ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ 83
в аналитической химии может быть недостаточно четкое проведение
титрования, осаждения, фильтрования и т. п. Случайных причин,
вызывающих отклонение от точного значения, много. Каждая из этих причин
дает мало заметное отклонение, ибо в противном случае оно было бы
отмечено и изучено. От случайных ошибок избавиться невозможно. Можно
лишь приближенно оценить их влияние на погрешность эксперимента.
Под теорией ошибок обычно подразумевается именно теория случайных
ошибок.
Абсолютная и относительная ошибки. Предположим, что некоторая
величина имеет определенное числовое значение «а», остающееся
неизменным во время процесса измерения. Допустим также, что при
измерении этой величины получено значение «х».
Точной ошибкой (Ал:') измерения называют разность между точным (а)
и приближенным (х) значениями:
а — х = А*' (23)
Это определение удобно тем, что понятие точной ошибки совпадает
с понятием поправки
а = х + Ах' (24)
т. е. точная ошибка есть число, которое нужно прибавить к приближенному
значению, чтобы получить точное.
«Точная ошибка» имеет только теоретическое значение, так как эта
ошибка не может быть определена. Во многих случаях легче определить
верхнюю границу абсолютного значения точной ошибки (абсолютной
погрешности Ал:). Предельная абсолютная погрешность Ал;
приближенного числа х является наименьшим числом, удовлетворяющим условию:
Ах ^ | Ах' | (25)
Абсолютная погрешность недостаточно характеризует качество
измерений, она позволяет только указать границы, в которых заключено
точное значение числа:
х — Дх^ а^х + Ьх (26)
В связи с этим вводится понятие предельной относительной
погрешности, которую называют относительной ошибкой. На практике
относительную ошибку выражают в процентах и рассчитывают по формуле:
Д*отн=-^-ЮОо/0 (27)
Математические критерии оценки результатов
эксперимента. Случайные ошибки в большинстве случаев подчиняются закону
нормального распределения с математическим ожиданием, равным 0.
Закон нормального распределения математически выражается
следующей формулой:
у = — *е^ (28)
сг К2я
где у — плотность распределения ошибок;
?—основание натуральных логарифмов, равное 2,72;
х = xi — х — ошибка результата единичного определения;
х — среднее арифметическое из п измерений;
Xi — результат единичного определения;
а2 — генеральная дисперсия.
84
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Графически закон нормального распределения может быть
представлен в виде кривой Гаусса. На рис. 32 представлены кривые Гаусса с
различными значениями дисперсии о\ <а| <аз. Сравнение этих кривых
показывает, что с уменьшением величины дисперсии улучшается
распределение и уменьшается предел, который практически могут достигнуть
ошибки. Например, ошибки, достигающие значений от ±10 до ±15%,
наблюдаются только при дисперсии ^о\ и ^о|; ошибки, составляющие
от ±5 до ±10%, при дисперсии ^о\ и ^af, а ошибки от 0 до ±5% встре-
'20 45
Рис. 32. Кривая Гаусса {о\<о\<(з^).
чаются только при дисперсии <;а?. Генеральная дисперсия является
понятием теоретическим. На практике обычно имеют дело с выборочной
дисперсией, обозначаемой S2.
Следовательно, одним из основных метрологических требований к
методу анализа является достаточно малая величина выборочной
дисперсии S2 или средней квадратичной ошибки отдельного определения,
равной KS2".
Обычно величину выборочной дисперсии рассчитывают по формуле:
52 =
i=n
t=i
/г—1
(29)
где Xi — результат единичного определения;
п — число измерений; п — 1 = k — число степеней свободы;
х — среднее арифметическое из п измерений.
Среднее арифметическое вычисляют по формуле:
- _ *i + *2 Л h xt H h xn _ _1_ уГ
п
п
*1
*=1
(30)
Если разность (xt — х) > 25, то такие результаты опыта
отбрасываются.
Другим математическим показателем, посредством которого
оцениваются полученные результаты опыта, является, наряду с дисперсией,
доверительный интервал — интервал, в котором, находится истинное
значение определяемой величины с заданной доверительной вероятностью.
Значение доверительного интервала определяется выражением:
(*-'°-ftw)<a<(*+^ftw) (31)
« 12. СТАТИСТИЧЕСКАЯ ОБРАБОТКА ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ 85
Дробь S/Vn обозначают через Sx и называют квадратичной ошибкой
среднего арифметического, a ta% k S~x = еа> t называют абсолютной ошибкой
среднего арифметического.
Тогда доверительный интервал выражается формулой:
или
(* ~ '«. kSx) < <* < (* + '«, А) (31а)
(х — ea,t) < а < (5 — еа, /) (316)
где а — истинное значение определяемой величины;
х — среднее арифметическое из п измерений;
S- — средняя квадратичная ошибка среднего арифметического;
tatk — критерий Стьюдента;
л — число измерений;
etti/ — абсолютная ошибка среднего арифметического.
Вероятность того, что доверительный интервал действительно
заключает в себе истинное значение величины а, называют надежностью
доверительного интервала а. В аналитической химии значение а принимают
равным 0,95. На практике надежность доверительного интервала может
быть отличная от 0,95 в зависимости от характера задачи.
Для того чтобы рассчитать доверительный интервал, задаются
величиной а и при известном количестве измерений п (или k = п — 1)
определяют по таблицам критерий Стьюдента tatk.
Пример статистической обработки экспериментальных данных. Определение фосфорной
кислоты объемным методом основано на титровании фосфорной кислоты стандартным
раствором NaOH или КОН в присутствии метилового оранжевого или фенолфталеина.
Содержание фосфорной кислоты в граммах рассчитывают по формуле (9):
или по формуле (11):
S-
g =
^ЫаОН1/1^аОНЭН8Р041/к
10001/А
9HsP04TNaOH^NaOH^K
^NaOH^A
где g — содержание фосфорной кислоты в титруемом растворе, г;
^NaOH' ^NaOH ~~ характеристики (нормальность и титр) стандартного раствора NaOH;
Эн РО — грамм-эквивалент фосфорной кислоты;
VK — общий объем анализируемого раствора, мл;
1Л — объем аликвотной части анализируемого раствора, мл;
3NaOH — грамм-эквивалент едкого натра;
VNaOH — объем стандартного раствора NaOH, израсходованного на
титрование, мл.
Согласно закону нормального распределения, суммарная ошибка
определения появляется в результате совместного действия ряда причин,
каждая из которых вносит малую долю в общую ошибку.
По закону сложения ошибок средняя квадратичная ошибка суммы
независимых величин равна корню квадратному из суммы дисперсий
отдельных слагаемых, т. е. ошибка определения содержания Н3Р04
в пробе — SXi равна:
S*l = VS*k + S*VA + ^ <T>NaOH + ^NaOH
Однако при условии, что ошибка установки параметров раствора
(Sn (T)NaOH) и ошибка калибровки мерной посуды (SvK> SvA) малы по
сравнению с ошибкой отсчета и ошибкой капли, в сумме обозначаемых
ГЛ.1. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
величиной SvNaOH, т. е. Svk + SyA + SN (T)NaOH C SvNaOH> поэтому
можно принять, что SXi = ySvNaOH.
Допустим, что в процессе экспериментальной работы получены
следующие результаты титрования:
№ определения 1 2 3 4 5 6 7
Объем раствора NaOH, мл . . 7,75 7,70 7,78 7,75 7,05 7,76 7,70
По данным титрования рассчитывают содержание Н3Р04 (**) в
граммах. Полученные результаты сводят в таблицу (табл. 5).
ТАБЛИЦА б
определения
1
2
3
4
5
6
7
п=7
Статистическая обработка результатов объемного определения
фосфорной кислоты
Объем
титранта
(^NaOH).
мл
7,75
7,70
7,78
7,75
7,05
7,76
7,70
Содержание
Н3Р°4(*/)-
г
0,7594
0,7543
0,7623
0,7594
0,6909
0,7605
0,7543
5=0,7487
Xf—X
+0,0107
+0,0056
+0,0136
+0,0107
—0,0578
+0,0118
+0,0056
Щ=—0,0002
(*Н0Я
0,00011449
0,00003136
0,00018496
0,00011449
0,00334084
0,00013924
0,00003136
2 = 0,00395672
С2 0,00395672
6 = 7-1 =
=0,00065945
5= V 0,00065945 =
=0,0256
Зная значение х{, вычисляют среднее арифметическое х по формуле:
- Е*<
Х = -=
п
В рассматриваемом случае
- = ?** = 0,7594 + 0,7543 + 0,7523 + 0,7594 + 0,6909 + 0,7605 + 0,7543 _
п 7 *
Определив х, приступают к расчету абсолютной ошибки результата
единичного определения xt — х. Результаты вносят в таблицу и
суммируют [2 (xt — х)—*0]. Затем вычисляют величину (xt — х)2,
результаты расчета также вносят в таблицу и суммируют.
На основании данных табл. 5 рассчитывают величину выборочной
дисперсии S2 и среднюю квадратичную ошибку отдельного
определения S по формулам:
2 (**-*)2
S2 =
/=1
л—1
s = V&
Затем проводят анализ полученных результатов, из табл. 5 следует,
что результат измерения № 5 имеет отклонение от среднего значения
(xt — х), превышающее величину 2S (2S = 0,0256 X 2 = 0,0512). Этот
$ 12. СТАТИСТИЧЕСКАЯ ОБРАБОТКА ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ 87
результат считают недостоверным, отбрасывают его и проводят
аналогичным путем вторую обработку данных, записывая результаты в таблицу
(табл. 6).
ТАБЛИЦА 6
Повторная статистическая обработка результатов
объемного определения фосфорной кислоты
определения
1
2
3
4
5
6
/1=6
Объем
титранта
(^NaOH).
мл
7,75
7,70
7,78
7,75
7,76
7,70
Содержание
Н3Р°4 (**)•
г
0,7594
0,7543
0,7623
0,7594
0,7605
0,7543
х = 0,7584
Xf— X
+0,0010
—0,0041
+0,0039
+0,0010
+0,0021
—0,0041
23-
= —0,0002
(Х;-Х)>
0,00000100
0,00001681
0,00001521
0,00000100
0,00000441
0,00001681
2 =0,00005524
С2 0,00005524
=0,00001104
S = ]/0,00001104=
= 0,0033
Исходя из вновь полученных результатов, рассчитывают квадратичную ошибку
среднего арифметического 5- по формуле:
* *-^-^?-^
Принимая надежность, равную 0,95, по справочным таблицам (для k = п — 1, т. е. k =
= 6 — 1 = 5) находят значения коэффициента ta% k-
Получив критерий tatk, вычисляют доверительный интервал, внутри которого
находится истинное значение 'определяемой величины:
0,7584 — 2,57-0,0014 < а < 0,7584 + 2,57-0,0014
или
0,7548 < а < 0,7620
и
а = х ± &a,t= 0,7584 ± 0,0036
Вычислив абсолютную ошибку среднего арифметического eaif, рассчитывают
относительную ошибку е0тн в процентах по фюрмуле:
-^1<Ю = е0ТН
х
Все результаты статистической обработки экспериментальных данных сводят в
заключительную таблицу (табл. 7).
ТАБЛИЦА 7
Результаты статистической обработки экспериментальных данных
объемного определения фосфорной кислоты
п
6
~х
0,7584
S*
0,00001104
S
0,003*3
8а=0,95
0,0036
еотн, %
0,45
88
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Г. ПОЛУМИКРООБЪЕМНЫЙ МЕТОД АНАЛИЗА
§ 13. Понятие о полумккрообъемном анализе
В настоящее время наряду с макроколичественными методами анализа широко
применяют разнообразные микроколичественные методы анализа (см. Введение, § 4).
Особое место среди микроколичественных методов анализа занимает объемный полу-
микрометод. Главное отличие полумикрометода от макрометода объемного анализа
заключается в технике химического эксперимента, доведенного до большой точности определений.
Титрование в пол у микрометоде ведется малыми объемами стандартных 0,1—0,01 н.
растворов, на отдельное титрование расходуется менее 10 мл раствора. Поэтому особое внимание
уделяется точности дозировки раствора, которая в данном случае играет решающую роль
для получения правильных результатов анализа. Применяемые в макрообъемном анализе
бюретки, при помощи которых проводят отсчет объемов с точностью до 0,02 мл, в
полумикроанализе не используют, для этих целей применяют специальные бюретки.
§ 14. Особенности техники измерения объемов растворов
в полумикрометоде
В полумикрообъемном анализе применяют бюретки емкостью от 1 до 10 мл,
позволяющие проводить отсчет объемов растворов с точностью до 0,002 мл.
Существуют разнообразные конструкции микробюреток. Очень удобна бюретка,
представляющая собой толстостенную стеклянную трубку длиной 20—50 см, с постоянным
внутренним диаметром около 2—4 мм (см. рис. 21). Цена деления шкалы такой бюретки
0,01 мл. Большая точность измерения объемов растворов достигается с помощью лупы
(на глаз) по положению мениска жидкости.
Одной из особенностей титрования по полумикрометоду является приливание
стандартного раствора к концу титрования (вблизи точки эквивалентности) не по каплям,
а долями капель. С этой целью бюретки снабжаются специальными приспособлениями.
Иногда к концу полумикробюретки присоединяют тонкий стеклянный капилляр диаметром
менее 0,5 мм.
В лабораторной практике широко применяют бескрановые микробюретки,
заполнение которых раствором и выливание последнего регулируются пневматическим
устройством. Принцип пневматического регулирования движения раствора основан на
применении мембраны, с помощью которой в верхней части бюретки создается разряжение.
Разряжение достигается при повороте винта. При вращении винта в противоположном
направлении раствор вытекает из бюретки. Раствор из такого рода бюретки подается по
1 капле в течение 5 сек. Отсчет уровня раствора проводят через 30 сек.
Полумикрообъемный анализ с успехом может быть применен в методах
нейтрализации, оксидиметрии, осаждения и комплексообразования.
Д. БЕЗБЮРЕТОЧНЫЕ МЕТОДЫ ТИТРОВАНИЯ
§ 15. Понятие о безбюреточных методах титрования
За последнее время в связи с возросшими требованиями промышленности в
отношении скорости и точности количественного определения и возможности автоматизации
процессов титрования все большее значение приобретают безбюреточные методы
титрования.
В то время как в основе обычных (бюреточных) методов титрования лежит измерение
объемов титранта при помощи бюреток самых разнообразных конструкций, в безбюреточных
методах титрования бюретки не применяются; расход титранта отсчитывается автоматически
при помощи приборов, фиксирующих, например, число капель стандартного раствора,
израсходованных на титрование определяемого вещества.
§ 16. Классификация методов безбюреточного титрования
Различают следующие методы безбюреточного титрования:
1) весовое (гравиметрическое) титрование',
2) капельное ^титрование, или титрование по числу капель',
3) титрование по времени.
Весовое (гравиметрическое) титрование. Принцип весового (гравиметрического)
титрования состоит во взвешивании стандартного раствора реактива В, содержание реагента
в котором выражается в весовых единицах: числом граммов растворенного вещества в 1 г
раствора, или числом грамм-эквивалентов растворенного вещества в 1 кг раствора.
§ 16. КЛАССИФИКАЦИЯ МЕТОДОВ БЕЗБЮРЕТОЧНОГО ТИТРОВАНИЯ 89
Весовой метод титрования применяют в микро- и ультрамикроанализе. Метод
позволяет избежать ошибок, происходящих при отсчете объемов при работе даже с
микробюретками, так как измерить объем в обыкновенной бюретке с точностью до 0,01 мл, а в
микробюретке — до 0,001 мл очень трудно, в то время как отвесить раствор с точностью до 0,01 г
и 0,001 г не составляет особого труда.
При весовом титриметрическом определении точность результатов анализа по
сравнению с бюреточными определениями может быть повышена в несколько раз.
Весовое титрование применяют в методах нейтрализации, окисления —
восстановления и осаждения. Наивысшая достижимая точность составляет 0,0015%.
Капельное титрование. Принцип метода состоит в том, что к титруемому раствору
определяемого вещества добавляют по каплям раствор реактива и автоматически
учитывают при помощи счетчика число
израсходованных на титрование капель.
Для последующего вычисления
необходимо знать содержание реактива
в одной капле раствора. При этом нет
необходимости измерять объем капли
или знать точный титр раствора
реактива.
Рис. 33. Прибор Г. Г. Кобяка для безбюреточного метода титрования:
/ — гидростатическая капиллярная трубка; 2 — электроды счетчика капель; 3 —
стакан с титрантом; 4 — колба с титруемым раствором; 5— подставка для стакана,
передвигающаяся при помощи кремальеры в вертикальном направлении; 6 — магнит;
7 — магнитная мешалка; 8 — электромагнитный импульсный счетчик капель.
Прибор для безбюреточного титрования состоит из капиллярной гидростатической
трубки, укрепляемой в штативе, который снабжен подставкой, передвигающейся при
помощи кремальеры в вертикальном направлении. Подставка служит для перемещения
стакана с раствором реактива вверх и вниз, чем регулируется нужное гидростатическое
давление в капиллярной трубке и осуществляется процесс титрования (рис. 33).
Капельное титрование применяют в методах нейтрализации и осаждения.
Титрование по времени (темпометрическое титрование). Принцип метода основан
на равномерной подаче титранта в анализируемый раствор. Благодаря этому измерение
объема титранта заменяется измерением времени титрования.
Титрование по времени отличается многими преимуществами по сравнению с
другими объемными методами: устраняется ряд ошибок, неизбежных при отсчете объема,
увеличивается скорость определения и осуществляется полная автоматизация процесса
титрования.
В МХТИ им. Д. И. Менделеева успешно применен метод капельно-темпометрического
титрования, основанный на применении поплавковой системы, снабженной
гидростатической капиллярной трубкой, обеспечивающей постоянство истечения титранта.
Титрование по этому способу применяется в методах нейтрализации, окисления —
восстановления, осаждения и комплексообразования.
Содержание определяемого вещества рассчитывается по формуле:
?а = Т
В/ А» сек
•t
где gA — масса определяемого вещества, г;
Тв/а-сек — ТИТР стандартного раствора по определяемому веществу, выраженный
в граммах определяемого вещества А, эквивалентного количеству
реактива В, расходуемого в 1 сек (г/сек);
t — время титрования, сек.
90
ГЛ. К ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
Е. АВТОМАТИЧЕСКИЕ МЕТОДЫ
§ 17. Химико-аналитический контроль производства
Автоматический контроль, регулирование и управление технологическими процессами.
В основе управления разнообразными химическими процессами и связанными с ними
различными отраслями производств лежит правильно построенная система химического
контроля как отдельных стадий технологических процессов, так и всего производства
в целом. Без надежного и систематического химического контроля, дающего яснее
представление о ходе тех или иных процессов, невозможно обеспечить нормальный ритм
производства, оптимальные технологические процессы и эффективные системы управления
химическими заводами. Высококачественный контроль обеспечивает максимальную
производительность, безаварийность, наибольшую рентабельность производства и высокое качество
выпускаемой продукции.
Автоматический контроль производства осуществляется с помощью специальных
приборов, выполняющих необходимые контрольно-измерительные функции без участия
человека. Для контроля и регулирования производственных процессов широко
используют новейшие достижения химии, физики, электроники, радиотехники, кибернетики и
других наук и применяют приборы, основанные на измерении различных
физико-химических величин. Для этой цели применяют радиоактивные изотопы, ультразвук,
фотоэлементы, спектральные приборы, управляющие вычислительные машины и т. д.
Автоматизация химического контроля производства имеет особо большое значение
в тех случаях, когда анализ связан с вредностью и опасностью для здоровья и жизни
обслуживающего персонала. Наконец, необходимость применения автоматического контроля
вызывается еще и тем, что некоторые сложные аналитические операции требуют для своего
осуществления много времени, в течение которого анализируемый объект или
контролируемая величина изменяются настолько, что результаты анализа запаздывают.
Современные аналитические лаборатории оснащены усовершенствованной
аппаратурой и точнейшими приборами. Для контроля производства широко применяются
контрольно-измерительные, автоматические, регулирующие и сигнализирующие устройства
и счетные решающие устройства. Аналитик, овладевший теорией аналитической химии
и современной техникой химического эксперимента, пользуясь приборами и аппаратурой,
может получать весьма точные результаты анализа.
§ 18. Автоматические методы титрования
Лабораторные автоматические приборы дают возможность проводить
массовые однотипные анализы; титровать жидкости, содержащие
радиоактивные изотопы, ядовитые или взрывоопасные вещества; проводить
анализ растворов, окраска которых или содержание в них твердых частиц
мешают применению индикаторов; титровать с помощью нестойких титран-
тов, которые реагируют с кислородом или двуокисью углерода,
находящимися в воздухе; изучать кинетику химических и биохимических
реакций. Титрующие анализаторы промышленного типа позволяют
контролировать почти все химико-технологические процессы, а в некоторых
случаях и регулировать их.
Методы автоматического титрования можно классифицировать по
следующим признакам:
по типу химической реакции, происходящей в процессе титрования
(нейтрализации, окисления — восстановления, осаждения, комплексо-
образования, вытеснения и замещения);
по способам дозирования титрующего раствора (объемный, весовой,
кулонометрический);
по способу определения точки эквивалентности (электрохимические
методы: потенциометрический, кондуктометрический, амперометричеедий,
высокочастотный; оптические: фотометрический, нефелометрический,
колориметрический; термохимический, манометрический).
Автоматические анализаторы. Целесообразность автоматизации
операций, связанных с титрованием, определяется условиями применения
тех или иных схем. Полностью автоматизированные установки
необходимы при многократном выполнении одного и того же анализа в поточных
§ 18. АВТОМАТИЧЕСКИЕ МЕТОДЫ ТИТРОВАНИЯ
91
производственных процессах. Например, широко применяется
автоматический анализатор непрерывного титрования с потенциометр ичес кой уста-
I
?
^
^
а
ь
Рис. 34. Принципиальная схема автоматического титратора БАТ-12-ЛМ:
/ — рН-метр; 2 — титратор; 3 — датчик конечной точки титрования; 4 — усилитель;
5 -— реле; 6 — магнитный клапан; 7 — сосуд с титрантом; 8 — аналитическая ячейка;
9 — резиновая тру6"ка.
новкой конечной точки при определении активного хлора в ваннах для
хлорирования. Для определения NaOH, Na2C03, AgN03 ц некоторых
кислот успешно используется автоматическая термометрическая
установка. Разработан автоматический
титратор типа АТ-2М для объемного кондукто-
метрического титрования. Точка
эквивалентности определяется по перегибу
кривой, соответствующему концу титрования.
Привязка точек кривой титрования к
количеству титранта осуществляется по
времени на стандартном регистрирующем
приборе типа ДС-1-01.
В заводских лабораториях широко
применяется автоматический титратор
БАТ-12-ЛМ для потенциометр ического
титрования до заданного значения рН или
потенциала (мв). Принципиальная схема
этого титратора представлена на рис. 34.
Напряжение (Ux), пропорциональноеэ. д.с.
электродной системы, с выхода рН-метра 1
подается на вход титратора 2 и
сопоставляется с напряжением (?/J,
установленным на датчике конечной точки
титрования 3. Разность этих напряжений
поступает на вход усилителя 4. На выходе
усилителя установлено реле 5,
управляющее работой магнитного клапана 6.
Магнитный клапан регулирует подачу
титрующего раствора из сосуда 7 в аналитическую
ячейку с испытуемым раствором 5. При
Ux = UK реле 5 отключает питание
магнитного клапана, который, зажимая
резиновую трубку 9, прекращает подачу
титрующего раствора в ячейку 8.
Общий вид установки для титрования представлен на рис. 35.
При исследовательских работах обычно стремятся получить
максимальную точность, достижимую в данном методе анализа. Кроме того,
Рис. 35. Общий вид установки для
титрования:
/ — сосуд с титрантом; 2 — бюретка;
3 — магнитный клапан; 4 —
аналитическая ячейка; 5 — магнитная мешалка;
6 — стакан для слива; 7 —
измерительный блок.
92
ГЛ. I. ОСНОВЫ ОБЪЕМНОГО АНАЛИЗА
приходится выполнять разнообразные анализы, а значит, необходимо,
чтобы установка была достаточно универсальной: допускала возможность
быстрой замены электродов, титранта, регулирование скорости
перемешивания раствора и т. д. Поэтому автоматизация подготовки и
дозирования исследуемого вещества здесь не нужна, также нецелесообразно авто»
матическое управление мешалкой, так как момент включения или
выключения мешалки при разных анализах может быть различным. Зато часто
возникает необходимость в регистрации кривых титрования для более
объективного проведения анализа. Для этой цели используют методы
титрования с автоматическим отсчетом количества израсходованного
Рис. 36. Принципиальная
схема работы
полуавтоматической установки:
/ — дозатор титранта;
2 — аналитическая ячейка;
3 — магнитная мешалка;
4—рН-метр ЛПУ-01; 5
—автоматический потенциометр
ЭПП-09.
титранта. Например, объем титранта находят по длине бумажной ленты
регистратора или резервуар с титрантом оборудуют системой с элементом,
следующим за поверхностью раствора, причем каждому положению
элемента соответствует строго определенное положение пера регистратора.
Указанный принцип использован при создании электронного
полуавтоматического потенциометрического титрометра.
На кафедре аналитической химии МХТИ им. Д. И. Менделеева создана
полуавтоматическая установка (рис. 36) для метода титрования по
времени. Характерной особенностью этого метода является постоянная
скорость подачи титранта в анализируемый раствор. Продолжительность
титрования фиксируют непосредственно при помощи секундомера или
использует автоматический регистратор. Постоянная скорость подачи
титранта делает возможной автоматическую запись кривых титрования
с электрометрической фиксацией точки конца титрования. Для этого
дозатор титранта соединяют с регистратором кривой титрования, ленточная
диаграмма которого протягивается синхронно с поступлением реагента.
При работе с этим титратором нет необходимости в применении бюретки
для стандартного раствора, а стандартной единицей может служить
«титр миллиметра» *. Эта установка может быть использована для
потенциометр ического титрования кислот и оснований, а также для
дифференцированного определения компонентов смесей кислот или оснований.
* Содержание реагента в стандартном растворе, введенном в титруемую смесь в течение
времени, за которое диаграммная лента передвинется на 1 мм.
ГЛАВА II
МЕТОДЫ НЕЙТРАЛИЗАЦИИ, ИЛИ МЕТОДЫ
КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
А. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
§ 1. Характеристика метода
Методы нейтрализации основаны на применении реакций
нейтрализации.
Основным уравнением процесса нейтрализации в водных растворах
является взаимодействие ионов гидроксония (или водорода) с ионами
гидроксила, сопровождающееся образованием слабодиссоциированных
молекул воды:
Н30+ + ОН" —> 2Н20
или
н+ + он--^н2о
Методы нейтрализации позволяют количественно определять кислоты
(с помощью титрованных растворов щелочей), основания (с помощью
титрованных растворов кислот) и другие вещестба, реагирующие в стехио-
метрических соотношениях с кислотами и основаниями в водных
растворах (например, соли аммония, реагирующие со щелочами; карбонаты,
реагирующие с кислотами, и т. п.).
Применяя специальные приемы титрования, методами нейтрализации
можно определять содержание многих солей, дифференцированно
(раздельно) титровать смеси сильных, слабых и очень слабых кислот, а также
смеси оснований и солей в неводных растворах.
Титрование кислотами и основаниями. Пользуясь каким-либо
титрованным раствором, содержащим ионы гидроксония (например, раствором
НС1), можно титровать основания; титрованными растворами оснований
титруют кислоты.
Техника определения состоит в том, что к определенному количеству
раствора основания (или кислоты) постепенно приливают из бюретки
титрованный раствор кислоты (или основания) до наступления точки
эквивалентности. Количество основания (или кислоты), содержащееся
в исследуемом растворе, вычисляют по объему титрованного раствора
кислоты (или основания), израсходованного на нейтрализацию
определенного объема раствора анализируемого образца или навески исследуемого
продукта (см. гл. I, § 10).
§ 2. Установление точки эквивалентности
Определение точки эквивалентности с помощью индикаторов. Точное
установление точки эквивалентности, т. е. того момента, когда количество
прибавленного реактива В станет эквивалентно количеству реагирующего
с ним определяемого вещества А, имеет очень важное значение в
объемном анализе вообще и в методе нейтрализации в особенности.
94 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Практически момент эквивалентности (точку эквивалентности)
устанавливают индикаторным методом по изменению окраски индикатора,
1—2 капли раствора которого добавляют в титруемый раствор.
Физико-химические, или инструментальные, методы установления
точки эквивалентности. В связи с тем, что цветные индикаторы
оказываются не пригодными для установления точки эквивалентности при
титровании сильно окрашенных или мутных растворов, разработаны и широко
применяются другие способы установления точки эквивалентности,
основанные на наблюдении свойств раствора, резко меняющихся в момент
эквивалентности. Большое значение приобрели так называемые физико-
химические, или инструментальные, методы установления точки
эквивалентности, основанные на измерении при помощи специальных
приборов некоторых величин, характеризующих какое-нибудь свойство
раствора (например, электропроводность), которое меняется в процессе
титрования постепенно, а в точке эквивалентности — резко. К этим
методам титрования относятся кондуктометрические, высокочастотные,
потенциометрические, амперометрические и некоторые другие (см. гл. VIII).
§ 3. Графический метод изображения процесса нейтрализации
Принцип построения кривых, изображающих ход изменения рН в
процессе нейтрализации. В ходе нейтрализации рН титруемого раствора
меняется в зависимости от объема прибавленного стандартного раствора
(VB) и от его титра (Тв). Следовательно, если на оси абсцисс откладывать
процентное содержание остающейся в растворе в разные моменты
титрования кислоты или щелочи или количество прибавленного стандартного
раствора в миллилитрах, а на оси ординат — отвечающие им значения
рН раствора, то получается ряд точек, соединив которые, можно
представить ход изменения рН в процессе нейтрализации.
На рис. 37—44 представлены кривые титрования различных кислот
и оснований.
Каждый процесс нейтрализации можно представить графически в виде
кривой титрования, изображающей изменение величины рН титруемого
раствора по мере приливания к нему стандартного (титрованного)
раствора кислоты или щелочи.
Построение кривой титрорания ведут с таким расчетом, чтобы
показать значения рН раствора, соответствующие главнейшим моментам
титрования.
Значения рН растворов, соответствующие различным моментам
титрования, вычисляют по формулам, выражающим значения
концентраций ионов водорода в воде, в водных растворах кислот, оснований, гидро-
лизующихся солей и, наконец, в буферных смесях.
Значение кривых нейтрализации. Кривые титрования дают
возможность проследить изменение рН раствора в различные моменты
титрования, изучить влияние температуры и концентрации реагирующих
веществ на процесс нейтрализации, установить конец титрования и, как
будет показано в дальнейшем, сделать правильный выбор индикатора.
§ 4. Вычисление концентрации ионов водорода
в водных растворах сильных кислот и оснований
Концентрация ионов водорода в предельно разбавленных водных
растворах сильных кислот (типа НО, HN03) практически равняется
концентрации этих кислот:
1н+]~снап и pHU—lg[H*] —igC-^ т
§ 4. КОНЦЕНТРАЦИЯ Н+ В РАСТВОРАХ КИСЛОТ И ОСНОВАНИЙ 95
Пример 1. Вычислить [Н+] и рН 0,01 н. раствора хлористоводородной кислоты.
Решение. [Н+ J ^ СНС1 = 0,01 = Ю^г-ион/л
рН = — lg [Н+] = — lg Ю-2 =2 lg 10 = 2
Пример 2. Вычислить [Н+ J в 0,05 М растворе хлористоводородной кислоты.
Решение. [Н+] = 0,05 = 5» 10~2 г-ион/л1 что можно представить так:
[Н+] = 10-=2.101г5 = Ю-МО0'7 = Ю-1'3 г-ион/л
рН = l,31g 10 = 1,3
или
рН = 2 — lg 5 = 2 — 0,7 = 1,3
Пример 3. Вычислить [Н+ ], [S04 "] и рН 0,01 М (а) и 0,01 н. (б) растворов серной
кислоты.
Р е ш е н и е. а) Серная кислота в водном растворе диссоциирует согласно уравнению:
h2so4:^2H+ + so;~
Следовательно, [Н+ ] в растворе H2SO4 превышает [S04 "] в два раза, т. е. в 0,01 М
растворе H2S04, в котором [S04 1 = Сн so
[Н+]=2 [SO"]=2.CHaS04 = 2-0,01 =2-10~2 г-ион/л
отсюда
рН = — lg [H+] = 2 lg 10 — lg 2 = 1,7
б) В 0,01 н. растворе H2S04 [SO"] = ^- = CH2SOa;
[Н+] = 2 [S04"~] = 2.Mi- = 0,01 = 10"2 г-ион/л
отсюда
рН = — lg [H+] = — lg Ю-2 = 2
Пример 4. Вычислить [Н+] и рН 0,5%-ного раствора H2SO4.
Решение.
100—0,5 ^=500_=52
юоо — х х юо
5 5
С„ «п =-т-л =т7^-т^г^5-Ю-2 моль/л
H*s°4 ^Haso4 98>08
[Н+] = 2-CHgS04 =2.5- Ю-2 = Ю-1 г-ион/л
рН = — lg [Н+] = — lg Ю-1 = 1
Пример б. Вычислить [ОН" ] в 0,01 М растворе NaOH.
Концентрация ионов гидроксила в предельно разбавленных водных растворах сильных
оснований (типа NaOH или КОН) практически равняется концентрации этих оснований:
[ОН-] = Скюн и рОН = - lg [ОН"] = -lg CKtOH (2)
Решение. [ОН" ] = Скаон = 0,01 = 10"2 г-ион/л.
Пример 6. Вычислить [ОН'], рОН и рН 0,02 н. раствора Ва (ОН)г.
Решение. Ва (ОН)2 ^ Ва+* + 20Н".
Следовательно, [ОН"] в растворе Ва (ОН)2 превышает [Ва++ J в два раза. В 0,02 н.
растворе Ва (ОН)2 [Ва++ ] = -~— = СВа (ОН)а = 0,01 г-ион/л.
[ОН"] = 2 [Ва++] = 2.10-2 г-ион/л; рОН = -lg [ОН"] = 2 lg 10 — lg 2 = 1,7.
Из уравнения рН + рОН = 14 находим значение рН = И — 1,7 = 12,3.
96 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
§ 5. Вычисление активности ионов водорода
в водных растворах сильных кислот и оснований
Показатель активности ионов водорода. При точных расчетах
следует помнить, что экспериментальные методы, применяемые для
определения [Н+] и рН, дают возможность измерять не концентрацию ионов,
а их активность и соответственно раН:
PaH = -lga H+ = lg—— (3)
paH, в отличие от рН, выражающего обратный логарифм KOH^HTpai
ции ионов водорода, представляет собой обратный логарифм активности
ионов водорода.
В тех случаях, когда ан+ и раН находят расчетным путем, а не
определяют экспериментально, необходимо величину [Н+] умножать на /н+-
Пример. Вычислить рН 0,1 н. раствора НС1 с учетом коэффициента активности.
Решение. Чтобы вычислить рН раствора с учетом коэффициента активности нужно
сначала вычислить ионную силу раствора jx, затем найти значение среднего коэффициента
активности /i, соответствующее найденной величине ионной силы (см. книга 1, табл. 1,
стр. 36), вычислить а„+ и, наконец, вычислить раН.
н
По уравнению (3) (см. книга I, стр. 36):
|x=-i-(0,l.l2 + 0,l.l2)=0,l
Ионная сила бинарных электролитов (типа Kt+An~) равна молярной концентрации
электролита [см. уравнение (5), книга 1, стр. 37)].
При [л = 0,1 коэффициент активности ионов водорода f + = 0,84.
н
Следовательно,
ян+ = [Н+] /н+ = 0,Ь0,84 = 0,084
pflH = — lg 0,084 = — lg 8,4 • Ю-2 = 2 — lg 8,4 = 2 — 0,92 = 1,08
Таким образом, для 0,1 н. раствора НС1 величина рН раствора без учета коэффициента
активности равна 1, а с учетом коэффициента активности равна 1,08, т. е. эти величины
отличаются друг от друга на 0,08.
§ 6. Титрование сильной кислоты сильным основанием
Вычисление рН раствора в различные моменты титрования кислоты
сильным основанием. Предположим, что для титрования взято 100 мл
0,1 н. раствора НС1, который титруется 0,1 н. раствором NaOH.
Пользуясь формулами 1, 2, можно вычислить рН раствора,
меняющийся в процессе нейтрализации сильной кислоты сильным основанием.
До начала титрования 0,1 н. раствора НС1:
pH = -lg[H+] = -lgCHCI =-lg io-i = i
Рассмотрим, что произойдет с 0,1 н. раствором НС1, если прилить
к нему 50; 90; 99; 99,9 мл 0,1 н. раствора NaOH. При приливании 50 мл
0,1 н. растцора NaOH останется не нейтрализованными 50% НС1; при
приливании 90 мл NaOH — 10% НС1; 99 мл NaOH — 1% НС1; 99,9 мл
NaOH —0,1% НС1.
Тогда, соответственно прибавленному количеству NaOH, объем
титруемого раствора увеличится при прибавлении 50 мл NaOH до 150 мл;
90 мл NaOH до 190 мл\ 99 мл NaOH до 199 мл и 99,9 мл NaOH до 199,9 мл.
Изменится также концентрация ионов водорода и рН раствора.
§ 6. ТИТРОВАНИЕ СИЛЬНОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ 97
Для вычисления концентрации СА определяемого вещества (Chci)
в процессе титрования можно пользоваться формулой:
VA — VB VHCl — VNa0H
Ca = TVM^a ИЛИ Сн«-Ина+^^ж*на
где VA — первоначальный объем (в мл) раствора определяемого вещества, нормальность
которого NA;
VB — объем (в мл) стандартного (титрованного) раствора реактива, прибавленный
в титруемый раствор вещества А. Нормальность стандартного раствора
реактива (NB) тоже равна NA.
При нейтрализации половины НС1 концентрация хлористоводородной
кислоты уменьшится во столько раз, во сколько 50 меньше 150, т. е.:
СП
chci = lH+] = -jgo °Л = 3»3'10'2 М0ЛЬ1Л
рН = - lg Снс, = - lg [H+] = 2 - lg 3,3 = 1,5
Для других случаев значение рН, предшествующее точке
эквивалентности, выражают следующим образом:
chci = [н+] = -j^-0,1 =5,3-10-3 моль/л
PH = -lgCHC1 = —lg[H+] = 3 —lg 5,3 = 2,3
chci = [Н+] =^-0,1 =5,0-Ю-4 моль/л
pH = -lgCHC1=-lg[H4=4-lg5 = 3,3
0,1
199,9
Chci = [H+] = -т^тг 0,1 = 5,0- Ц)-« моль/л
pH = -lgCHC1 = _lg[H4=5-lg5 = 4,3
Если к 100 мл 0,1 н. раствора НС1 прилить 100 мл 0,1 н. раствора
NaOH, то будет достигнута точка эквивалентности и рН = рОН раствора
станет равным 7. Заметим, что для перехода от рН = 4,3 к рН = 7 (т. е.
для изменения рН на 2,7 единицы) потребовалось всего 0,1 мл 0,1 н.
раствора NaOH. Между тем в начале титрования для перехода от рН = 1
до рН = 3,3 (2,3 единицы рН) потребовалось 99,0 мл 0,1 н. раствора
NaOH, т. е. в 1000 раз больше (99,9/0,1).
Если прилить к 100 мл 0,1 н. раствора НС1 100,1 мл, 101 мл, ПО мл
0,1 н. раствора NaOH, то раствор станет щелочным вследствие
прибавления избытка щелочи. Концентрация ионов гидроксила в этом случае
будет равна концентрации избытка едкого натра и может быть вычислена.
Прибавление 100,1 мл 0,1 н. раствора NaOH ведет к разбавлению
раствора до 200,1 мл. Такой раствор содержит 0,1 мл избытка 0,1 н.
раствора NaOH. Следовательно:
cNaOH =" [ОН"! = -ЗЩ-0Л = 5'0' 10~5 М0ЛЬ,Л
POH = 5-lg5 = 4,3
рН = 14 — рОН = 14 — 4,3 = 9,7
98 ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
При добавлении 101 и 110 мл раствора NaOH соответственно будем
иметь:
cNaOH = [°н~] = 25Г0'1 = 5>°-10"4 М0ЛЬ1Л
рОН = 4 — lg5 = 3,3; рН = 14 — 3,3 = 10,7
CNaOH = [ОН"] =^-0,1 =4,8-10-3 МОЛЫ Л
рОН = 3 — lg 4,8 = 2,3; рН = 14 — 2,3 = 11,7
На основании полученных данных, которые вносят в таблицу (табл. 8),
строят кривую титрования.
Для уменьшения размеров кривой и большей наглядности вычерчивают
наиболее характерную часть кривой, лежащую в пределах 10% остатка
кислоты и 10% избытка щелочи. В результате получают кривую
нейтрализации, показанную на рис. 37.
ТАБЛИЦА 8
Титрование 100 мл 0,1 н. раствора НС1 0,1 н. раствором NaOH
(с учетом разбавления титруемого раствора)
Прибавлено
0,1 н.
раствора
NaOH
мл
0
50
90
99
99,9
100*
100,1
101
па
Осталось
НС1. %
100
50
10
1
0,1
0
0,1
(изб. NaOH)
1
(изб. NaOH)
10
(изб. NaOH)
[Н+,
ю-1
3,3-Ю-2
5,3-Ю-3
5,0-Ю"4
5,0-10"6
МО"7
2,0- Ю"1*
2,0- 10-ц
2,1-10"12
РН
1
1,5
2,3
3,3
4,3
7
9,7
10,7
11,7
[он-]
ю-13
3,0-Ю-13
1,9-10~12
2,0-10-ц
2,0-10"10
МО"7-
5.010-5
5,0-Ю-4
4,8-Ю"3
рОН
13
12,5
11,7
10,7
9,7
7
4,3
3,3
2,3
АрН
ДС
¦59 "м
-^=27
тм
27
' = 27
0,1
0,9
4-м.
* Точка эквивалентности и точка нейтрализации рН=р0Н=7.
Анализ кривой нейтрализации. В начале титрования рН раствора
изменяется Медленно; вблизи точки эквивалентности — очень быстро,
после точки эквивалентности — опять медленно. Другими словами, если
в начале титрования прибавление титрованного раствора основания
в количествах, измеряемых миллилитрами, мало изменяет значение рН
раствора, то под конец титрования, вблизи точки эквивалентности,
большое значение приобретают уже небольшие количества (капли и доли
капли) титрованного раствора NaOH, быстро (скачкообразно)
изменяющие рН титруемого раствора.
§ 6. ТИТРОВАНИЕ СИЛЬНОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ 99
Изменение величины показателя иона водорода может быть выражено
величиной АрН/ДС, которая означает среднее изменение показателя
ионов водорода при добавлении 1 мл титрованного раствора (в данном
случае раствора NaOH). Здесь ДрН — приращение показателя иона
водорода, наблюдаемое на данном отрезке титрования; \С — количество
прибавленного титрованного раствора (в данном случае 0,1 н. раствора
NaOH в мл или в %).
Как видно из табл. 8, это отношение достигает максимума в точке
эквивалентности.
Следует иметь в виду, что чем больше величина ДрН/ДС на участке
вблизи точки эквивалентности, тем точнее титрование.
Линия
зквивалентшти
Интервал перехода
метилового оранжевого(3,1'-4,4)
Интервал перехода
метилового красного (4,ь.-6,2)
Точна эквивалентности Линия
^Точна нейтрализации моральности
1 Интервал перехода
г (ренолсрталеина {8,0-10,0}
jsraOH
100 ПО
Объем раствора NaOH ,мл
Рис. 37. Кривая титрования 0,1 н. раствора НС1 0,1 н.
раствором NaOH.
Скачок рН. Особенно резкое изменение рН наблюдается в интервале,
когда осталось 0,1% неоттитрованной НС1 или когда прилито 0,1%
избытка щелочи. В этом интервале рН быстро изменяется от 4,3 до 9,7,
а [Н+] — от 5,0-10 ~б до 5,0-10"10 г-ион/л. Резкое изменение рН раствора,
наблюдающееся вблизи точки эквивалентности, т. е. в конце титрования,
называют скачком рН. Величину скачка рН (или скачка титрования)
измеряют высотой вертикального участка кривой титрования. Чем больше
величина скачка рН, тем точнее можно оттитровать определяемое
вещество.
Влияние концентрации растворов кислот и оснований на характер
кривой титрования. Рассмотрим пример титрования 100 мл 1 н. раствора
НС1 1 н. раствором NaOH. В этом случае
PH = -lgCHC1 = — lg 1 = 0
Изменение рН в процессе титрования до и после точки
эквивалентности приведено в табл. 9.
В момент достижения точки эквивалентности рН = 7.
Для сравнения в таблице показаны изменения рН, наблюдающиеся
при нейтрализации 100 мл 1 н., 0,1 н. и 0,01 н. растворов НС1
растворами NaOH такой же концентрации, вычисленные с учетом разбавления
раствора.
100
ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
ТАБЛИЦА 9
Влияние концентрации раствора НС1 на изменение рН при титровании
(титрование 100 мл раствора различной нормальности
с учетом разбавления)
Прибавлено
раствора NaOH
такой же
концентрации, мл
0
50
90
99
99,9
100*
100,1
101
ПО
Осталось НО,
%
100
50
10
1
од
0
0,1 (изб. NaOH)
1 (изб. NaOH)
10 (изб. NaOH)
1 Н.
0
0,5 -
1,3
2,3
3,3
7,0
10,7
11,7
12,7
рН раствора
0,1 н*
1
1,5
2,3
3,3
4,3
7,0
9,7
10,7
11,7
0,01 н.
2
2,5
3,3
4,3
5,3
7,0
8,7
9,7
10,7
Точка эквивалентности и точка нейтрализации.
Данные этой таблицы представлены также графически на рис. 38.
Анализ кривой (рис. 38) и данных табл. 9 дает возможность
заключить, что чем выше концентрация титруемого и стандартного растворов,
тем больше скачок рН:
для 1 н. растворов он составляет
» 0,1 н. » » »
» 0,01 н. » » »
10,7—3,3=7,4 единицы рН
9,7—4,3=5,4 » »
8,7—5,3=3,4 » »
Следует, однако, заметить, что скачок рН достаточно велик и при
титровании 0,1 н. раствора. Поэтому без особой необходимости не следует
прибегать к титрованию растворов более высокой концентрации, чем 0,1 н.
Линия
зндивалентности
Интервал перехода
метилового оранжевого (3, / -4,4)
. Интервал перехода
_ J метилового красного (Ь,Ь-6,2)
Точка эквивалентности Линия
Точна нейтрализации нейтральности
* "1 Интервал перехода
К Г (ренолсрталеина (8,0-10,0)
NaOH
за
Рис. 38. Влияние
100
Объем раствора NaOH ,мл
по
/— 1 н.
концентрации растворов кислот и основании на
характер кривой титрования:
раствор HCI; 2 — 0,1 н. раствор HC1; 3 — 0,01 н. раствор НС1.
Ошибки титрования за счет неточности измерений, при пользовании более
концентрированными растворами значительно возрастают по сравнению
с ошибками при титровании менее концентрированных растворов.
§6, ТИТРОВАНИЕ СИЛЬНОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ Ю1
Влияние температуры водного раствора на изменения рН в процессе
титрования. Ионное произведение воды Kw сильно возрастает с
повышением температуры и при 100° С увеличивается почти в 100 раз по
сравнению с Kw при 18° С. При 100° С величина Kw становится приблизительно
равной Ю-12 (см. книга 1, гл. I, § 10). Поэтому повышение температуры
Линия
эквивалентности
Интервал перехода
. метилового краснс
-¦. Интервал перехода
_; метилового оранжевого (3,1-bfi)
. Точка эквивалентности
V ж, Линия нейтральности при /00 °С
-Точна нейтрализации
^ Линия нейтральности при 2h С
\ Интервал перехода
' фенолфталеина (8,0-10,0)>
NaOH
90
100
ПО
Объем раствора NaOH, мл
Рис. 39. Кривые нейтрализации 0,1 н. раствора НС1 0,1 н.
раствором NaOH:
/ — кривая титрования при 100° С; 2 — кривая титрования при 24° С.
оказывает сильное влияние на скачок рН. В точке нейтрализации
величина изменения рН зависит от значения Kw Если Kw == Ю""14, то точке
нейтрализации соответствует рН = 7; если Kw,= 10~12 (как это имеет
место при 100° С), то точке нейтрализации соответствует рН = 6, т. е.
скачок рН изменяется. Для сравнения приводим кривую титрования 0,1 н.
раствора НС1 0,1 н. раствором NaOH при 100° С (рис. 39). Данные,
соответствующие этой кривой, приведены в табл. 10.
ТАБЛИЦА 10
Изменение рН в процессе титрования 100 мл 0,1 н. раствора НС1 0,1 н.
раствором NaOH
(с учетом разбавления титруемого раствора)
Прибавлено
0,1 н.
раствора
NaOH, мл
0
50
90
99
99,9
Осталось
HC1,
%
100
50
10
1
0,1
РН
1
1,5
2,3
3,3
4,3
АрН
АС
ч—
?-»•»'
4—»¦»
таг-1'1
Прибавлено
0,1 н.
раствора
NaOH, мл
100*
| 100,1
101
по
Осталось НС1,
%
0
0,1
(изб. NaOH)
1
(изб. NaOH)
10
(изб. NaOH)
рН
6
7,7
8,7
9,7
АрН
АС
0,1 и
0,1 и
¦от-1'1
4-=0,11
* Точка эквивалентности и точка нейтрализации.
102 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Анализ кривой (рис. 39) и данных табл. 10 показывает, что при
титровании горячих растворов скачок рН уменьшается; нем выше температура
титруемого раствора, тем меньше скачок титрования.
Отсюда следует, что титрование по методу нейтрализации лучше
проводить на холоду (без нагревания).
Влияние разбавления раствора в процессе титрования на точность
расчета. При расчете рН мы учитывали разбавление титруемого раствора,
вызываемое постепенным прибавлением раствора щелочи к раствору
кислоты. Поэтому в конце титрования, когда вся кислота нейтрализована,
объем раствора увеличивается вдвое и становится равным 200 мл. Однако,
как показывает опыт, точность расчета мало уменьшается, если
пренебречь наблюдающимся разбавлением и считать объем титруемого раствора
постоянным. Ошибки в расчете, допускаемые в данном случае, небольшие
и не отражаются на принципиальных выводах, вытекающих из теории
нейтрализации, поэтому с ними можно не считаться.
Покажем на примере титрования 0,1 н. раствора НС1 0,1 н. раствором
NaOH, какая разница получается между значениями рН, вычисляемыми
с учетом и без учета разбавления.
До начала титрования рН = 1. Если прилить к 0,1 н. раствору НС1
50; 90; 99; 99,9 мл 0,1 н. раствора NaOH, то соответственно будет
нейтрализовано 50, 90, 99, 99,9% и останется не нейтрализованным 50, 10, 1,
0,1% НС1.
Изменится также концентрация ионов водорода и рН раствора:
50
при нейтрализации 50 мл НС1 (рН=1,3) . . [Н+ ]=-г^г-0,1=5,0»10"2 моль/л
при нейтрализации 90 мл НС1 (р№=2) . . . [Н+ ]=-г^тг-0,1=10~2 моль/л
при нейтрализации 99 мл НС1 (рН=3) . . . [Н+ ] =-т7л7г 0Л= Ю~3 моль/л
при нейтрализации 99,9 мл НС1 (рН=4) . . [Н+ ]= -^г-0,1=10"4 моль/л
Другими словами, если пренебречь изменением объема титруемого
раствора, то в первом случае не нейтрализованной НС1 останется в 2 раза
меньше, чем ее было до начала титрования. Следовательно, ее
концентрация станет в два раза меньше первоначальной:
—^— =5-10~2 моль/л
Когда к раствору НС1 прилито до 90 мл 0,1 н. раствора NaOH, ее
концентрация уменьшится в 10 раз и станет равной 0,1 : 10 = 10~2 моль/л.
Когда к титруемому раствору прибавят 99 мл NaOH, то концентрация
НС1 уменьшится еще в 10 раз (или в 100 раз по сравнению с
первоначальной) и будет равна 0,1 : 100 = 10"3 моль/л и т. д. В точке
эквивалентности количество прибавленного 0,1 н. раствора NaOH должно
составлять 100 мл, и при этом:
[Н+] = [ОН-] = 10-г г-ион/л; рН = рОН = 7
Именно в этот момент заканчивают титрование.
§6* ТИТРОВАНИЕ СИЛЬНОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ
103
Если прилить 0,1; 1 и 10 мл избытка 0,1 н. раствора NaOH, то
соответственно будет возрастать [ОН"1:
в первом случае (рОН=4; рН=
= 10) [ОН-] = М-0,1=10-4л/олб/>2
во втором случае (рОН=3; рН=
= 11) [ОН-] = -^0,\ = Ю-* моль/л
в третьем случае (рОН=2; рН=
10
= 12) [ОН-]= -^0,\=10-2 моль/л
Полученные результаты сведены в табл. 11.
ТАБЛИЦА 11
Титрование 100 мл 0,1 н. раствора НС1 0,1 н. раствором NaOH
(без учета разбавления титруемого раствора)
Прибавлено
раствора
NaOH,
мл
0
50
90
99
99,9
100*
100,1
101
110
Осталось HC1,
%
100
50
10
1
0,1
0
0,1
(изб. NaOH)
1
(изб. NaOH)
10
(изб. NaOH)
[н+].
г-ион/л
ю-1
5,0-Ю-2
ю-2
ю-8
10~4
ю-7
ю-u
10-i2
рН
1
1.3
2
3
4
7
10
И
12
[ОН"].
г-ион/л
ю-13
2,0-10"13
Ю-12
ю-"
Ю-ю
Ю-7
ю-4
ю-3
ю-2
рОН
13
12,7
12
11
10
7
4
3
2
ДрН
АС
___
4? = 0,006
^ = 0,0175
-J--*»
тог-1-1
1ГГ = 30
4—°'и
* Точка эквивалентности и точка нейтрализации.
Сопоставляя данные табл. 8 и 11, можно видеть, что:
во-первых, при титровании 0,1 н. раствора НС1 0,1 н. раствором NaOH
порядок и характер получаемых цифр одинаковы в обоих случаях расчета,
т. е. с учетом и без учета разбавления раствора во время титрования;
во-вторых, характер наблюдаемых кривых идентичен;
в-третьих, наибольшее расхождение в значении рН не превышает 0,3,
например значениям рН раствора, равным 2,3; 3,3; 4,3; 9,7; 10,7 и 11,7
(табл. 8), соответствуют следующие значения рН: 2; 3; 4; 10; 11 и 12
(табл. 11).Такое ничтожно малое расхождение не отражается на
Принципиальных выводах, которые можно сделать на основании данных
табл. 8 и анализа кривых.
Простота и удобство метода расчета рН без учета разбавления
титруемого раствора представляют некоторые преимущества по сравнению с
методом расчета с учетом разбавления раствора. В дальнейшем все приво-
104 ГЛ.И. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
— ^НАп
димые расчеты, применяемые для построения кривых титрования,
выполнены без учета изменения объемов растворов во время титрования. Это,
конечно, не соответствует действительности и приводит к известным
ошибкам опыта. Однако изменения рН в точке эквивалентности, т. е.
именно тогда, когда нужно заканчивать титрование, обычно настолько
значительны, что ошибки опыта при таком допущении оказываются
ничтожными.
Все приведенные рассуждения с одинаковым успехом применимы при
рассмотрении процесса титрования сильных оснований сильными
кислотами.
§ 7. Вычисление концентрации ионов водорода в растворах
слабых кислот и оснований
Зависимость (Н+) от Кнап и СНАп. В водном растворе слабая кислота
диссоциирует согласно следующему уравнению:
НАп^±Н+ + Ап-
Применим закон действия масс:
[Н+] [An-]
[HAn]
Так как [Н+] = [Агг ], то
[НЧ2 к
[HAn] ~"АНАп
или
[Н+] = //(HAn [HAn]
Подставляя в это уравнение значение [НАп] = Снап — Ш+],
получим:
[Н+] = У*НАп(СНАп-[НЧ)" (4)
Для слабо диссоциированной кислоты концентрация ионов водорода
величина сравнительно малая по отношению к Снап и ею можно
пренебречь. Уравнение при этом примет следующий вид:
ГН+]=У*напСиАп (5)
Аналогично можно вывести уравнение для расчета [ОН"] в слабых
основаниях:
[ОН-]=У*КЮНСкюн (6)
Пример* Вычислить концентрацию ионов водорода [Н+ ] в 0,1 М растворе уксусной
кислоты.
Решение. На основании уравнения (5) можно написать:
[Н+] = V 1,82-Ю-6- Ю-1 = 1,35.10-3 г-ион/л
Если величина [Н+ ] составляет более 5% от величины Снап, то нельзя
приравнивать [НАп] к Снап- В таких случаях пользуются более точным
уравнением, которое можно вывести из уравнения (4), проведя
соответствующие преобразования:
[Н+12 + * НАп [Н+] - * НАГНАН = 0
откуда
§ 8. ВЫЧИСЛЕНИЕ АКТИВНОСТИ Н+ В РАСТВОРАХ КИСЛОТ И ОСНОВАНИЙ Ю5
Аналогично для точных расчетов [ОН" ] в слабых основаниях:
т., *КЮН Т| / ^KtOH к г
*] = 2—± у —4 I-^KtoH'C]
[0Н-] = ^-± |/ _^ + ккюн.скюн (8)
§ 8. Вычисление активности ионов водорода
в водных растворах слабых кислот и оснований
Переход к активным концентрациям. Чтобы перейти от обычного выражения
концентрации к активностям, необходимо перейти от обычных, или концентрационных,
констант к истинным, или термодинамическим, константам.
Истинные константы К! определяются на основании активностей ионов в растворах:
их значения не изменяются в зависимости" от ионной силы раствора. Значения обычных,
или концентрационных, констант К определяются на основании равновесных концентраций
в растворах.
Истинные константы диссоциации кислот /СНАп и оснований K^tOH связаны с
обычными константами диссоциации /СнАп или ^кюн следующим образом.
Для кислот:
ГН+НАп-]
[НАп] ~лНАп
но
V - ^ V' V" - tAn'l 'а.- вНАп = 1НА"1 'нАп
Так как коэффициент активности недиссоциированных молекул слабого
электролита /Ндп равен единице, то
VflA„- ^ [Н+"н^АП-"А„-
"НАп
отсюда
- #НАп - [НАп]
#НАп ^НАп
V > *^НАп *хНАп /ЛЧ
*НАп = f ,f = 72 (9)
Подставляя значение ЯНАп, выраженное через —Н2Ап , в уравнение (5), получим:
h
[Н+]=-^]/^НАпСнАп (Ю)
вн+ = [Н+]/н+=^^С^ (11)
Здесь fi — среднее значение коэффициента активности для данного электролита.
Аналогично для оснований
%tOH = -1 } = —g (9a)
'Kt+/OH" /1
Подставляя значение /СКЮн» выраженное через —-=~—, в уравнение (6), получим:
106 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
В предельно разбавленных водных растворах химически чистых кислот или оснований
/н+ = /Ап- и /OH- = /Kt+ Равны 1. следовательно,
^НАп = ^НАп и ^KtOH = ^KtOH
Вычисление а + и р_Н. Пример, Вычислить (Н+), а ., рН и pflH в 0,01 н. растворе
н н
уксусной кислоты с учетом коэффициента активности ионов.
Решение.
|1 = 0,01, fH+ = 0,92
[H+]=-gi^l/"l,82-10-5.10-2^4,7.Ю-4 г-ион/л
рН = — Ig [Н+ ] = 4 — lg 4,7 = 4 — 0,7 = 3,3
я . = 1/1,82.10-б.10-2 = 4,2.10-4 г-ион/л
н
PflH = -lg«H+=4-lg4,2 = 4-0,6==3,4
§ 9. Равновесия в водных буферных растворах
слабых кислот в присутствии солей этих кислот
Если к раствору слабой кислоты НАп прибавлять малыми порциями
ее соль KtAn, то будет наблюдаться постепенное изменение концентрации
ионов водорода и можно получить растворы с различными значениями
[Н+] (или соответственно рН). Для вычисления [Н+] в смесях слабой
кислоты и ее соли можно воспользоваться следующими рассуждениями.
Для слабой кислоты
[Н+] [Atl-] # ГН+1-ГДп-1
[НАп] - *нап. tH+l = IAn I
При добавлении к этой кислоте сильного электролита KtAn
концентрация иона An" увеличится за счет анионов, образующихся при
диссоциации KtAn. Так как сильный электролит практически диссоциирует
полностью, то концентрация его аниона может быть приравнена к кон^
центрации соли, т. е.
[AnKtAn] = CKtAn
Таким образом, общая концентрация аниона равна сумме: [AnKtAn] +
+ [АпЙап], но так как [Н+] = [Ап?Ап] и [Ап^ап] = CKtAn, то можем
написать:
[An-]=CKtAn + [H+]
Равновесная концентрация недиссоциированных .молекул слабой
кислоты равна:
[НАп] = СНАп-[НЧ
Решив указанное выше уравнение в отношении [Н+ ] и подставив в него
значения [An"] и [НАп], получим:
ГН+1 - К СНАп — [Н* 1
[Н]~*НАп CKtAn + [H*]
Если принять во внимание, что [Н+] величина малая по сравнению
с Снап и CKtAn, то можно написать:
[Н^^/СнАп-^Т1- (15)
uKtAn
§ 10. РАВНОВЕСИЯ В БУФЕРНЫХ РАСТВОРАХ СЛАБЫХ ОСНОВАНИЙ JQ7
Из выведенной формулы следует, что концентрация ионов водорода
в буферных растворах, представляющих собой смесь слабой кислоты и ее
соли, зависит не только от концентрации самой кислоты и величины ее
константы электролитической диссоциации, но и от концентрации ее
соли. Чем больше концентрация соли, тем меньше [Н + ] в таких растворах.
Следовательно, [Н+] в таких растворах зависит не от абсолютных
количеств, а от соотношения концентраций кислоты и ее соли.
§ 10. Равновесия в водных буферных растворах слабых оснований
в присутствии солей этих оснований
Аналогично уравнению (15) выводится уравнение для вычисления
концентрации ионов гидроксила в смеси слабого основания KtOH и его
соли KtAn:
[ОН-]=*кюн-?М- (16)
Это означает, что [ОН"] в таких растворах зависит не от абсолютных
количеств, а от соотношения концентраций основания и его соли.
Из уравнения [Н + ] [ОН~] = Kw получим [Н + ] =-foffT и под"
ставляя в него значение [ОН"] из уравнения (16), можем написать
уравнение для вычисления концентрации ионов водорода в смеси слабого
основания KtOH и его соли:
[Н+] = /^°УА" (17)
AKtOHl'KtOH
Из формулы (17) следует, что концентрация ионов водорода буферных
растворов, представляющих собой смесь слабого основания и его соли зависит
не только от концентрации самого основания и величины его константы
электролитической диссоциации, но и от концентрации его соли. Чем
больше концентрация соли, тем больше [Н+] в таких смесях.
Значение рН в буферных растворах. Меняя соотношения ^НАп и
cKtAn
*юн, можно получить буферные растворы, отличающиеся плавным
CKtAn
изменением рН от их минимально до максимально возможных значений.
В водном растворе слабой кислоты
[Н+]=К*нАпСНАп
откуда
рН = -lg [Н+] = -1/2 lg tf нап -1/2 lg CHAn
Но так как Кнап представляет собой постоянную величину, то ее
лучше представить в формуле (подобно —lg [H+] = рН) в виде р/СнАп»
т. е. показателя константы электролитической диссоциации: р/СнАп =
= —lg /Снап
Тогда получим, что в водном растворе слабой) кислоты:
рН = - lg [Н*] = 1/2р/СНАп ~ W *g CHAn OS)
По мере прибавления к водному раствору слабой кислоты ее соли рН
раствора будет меняться.
108 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Так как, согласно уравнению (15), в растворе, содержащем смесь
слабой кислоты и ее соли
СНАп
cKtAn
ТО
PH = ~lg[H-]=-lg/CHAn-lgCHAn4-lgCK{An
или
рН = - lg [Н+] = р/СнАп - te CHAn + lg CKtAn (19)
Аналогично выводим формулы применительно к слабым основаниям:
[ОН-] = У КкЮНСкюн
рОН = - lg [ОН-] = -1/2 lg Ккюн - 1/2 lg CKtOH
или
рОН = - lg [ОН"] = 1/2 рЯКЮн - 1/2 lg CKiOH (20)
Концентрацию ионов водорода также выражают следующей формулой
[Н+]=ТШТ' П0ЭТ0МУ
РН = pKw - (1/2 р/СКЮн - 1/2 lg Скюн)
или
pH = 14-l/2p/CKtoH + l/21g °KtOH (21)
Согласно уравнению (17), в растворе, содержащем смесь слабого
основания и его соли
AKtOHcKtOH
т. е.
рН = - lg [Н+] = - lg Kw + lg ККЮн - lg CKtAn + lg скюн
или
рН = - lg [H+] = pKw - pKKtoH ~ !g CKtAn + Jg CKtOH
или
рН = 14 - pKKiOH - lg CKtAn + lg CKt0H (22)
Нет никакой необходимости запоминать выведенные формулы
значений рН, так как они очень легко выводятся путем логарифмирования
простых формул, выражающих значение [Н+ ].
Значение [Н+] и [ОН-] в буферных смесях, содержащих равные
концентрации кислоты и соли или основания и соли. Поскольку величина
[Н+ ] в буферной смеси зависит от концентрации соли, то, добавляя
различные количества этой соли, можно получить буферные смеси,
отличающиеся различными значениями концентрации ионов водорода.
При добавлении к буферной смеси HAn + KtAn (слабая кислота и ее
соль) небольшого количества основания и происходит незначительное
уменьшение концентрации кислоты и увеличение концентрации соли.
Например, если добавить 1 мл 0,1 М раствора сильного основания к 100 мл
смеси HAn + KtAn, в которой Снап = CKtAn = Ю"1 моль/л (пусть
Кнап = 1,8-10~б), то Снап уменьшится и станет равной 10"1 — 10~3 =
= 0,099 моль/л, a CKtAn увеличится и станет равной 10"1 + 10""3 =
= 0,101 моль/л. Тогда по уравнению (15)
О 0QQ
[Н+] = 1,8.10-б-^^= 1,76-Ю-5 г-ион/л
§11. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ Н+ В БУФЕРНЫХ РАСТВОРАХ Ю9
Эта величина мало отличается от Кнап = 1,8-10~5.
Таким образом, в тех случаях, когда концентрации кислоты и соли
в буферной смеси близки (отношение их равно приблизительно 1), величина
[Н+ ] численно приближается к величине Кнап даже при прибавлении
к ней небольшого количества сильной кислоты или основания
[Н+] = /СНАп -? — « КНАп
cKtAn
В равной мере это относится к величине концентрации ионов ОН""
для буферной смеси основание—соль [см. уравнение (16)]:
KtOH
[ОН-] = ККЮИ -= ^^KtOH
Если в рассмотренном примере к буферной смеси HAn + KtAn
добавить сильное основание в количестве, равном половине количества взятой
кислоты, то в результате реакции нейтрализации концентрация кислоты
станет равной 0,1 —0,05 = 0,05, a CKtAn увеличится до 0,1 + 0,05 =
= 0,15. Тогда по уравнению (15)
[H*j-l,8-10-g;;-g;g ~0,6.10- г-ион/л
т. е. и в этом случае величина [Н+ ] изменится незначительно.
То же самое получается, если к буферной смеси прибавить кислоту.
§ 11. Вычисление концентрации ионов водорода
в водных буферных растворах
Значения [Н+ ] и [ОН~ ] в водных буферных растворах вычисляют с помощью
формул (15—17).
Пример /. Вычислить [Н+] уксусно-ацетатного буферного раствора, являющегося
0,1 М в отношении СНзСООН и 0,01 М в отношении СНзСОСЖа.
Решение. Воспользуемся формулой (15), так как буферный раствор—смесь
слабой кислоты и ее соли:
[Н+] = 1,82.10-б-~щ- = 1,82-Ю-4 г-ион/л
Пример 2. Во сколько раз изменится величина [Н+] в буферном растворе,
рассмотренном в примере 1, если его разбавить в 10 раз?
Решение. При разбавлении буферного раствора в 10 раз концентрация СНзСООН
уменьшится и станет равной 0,01 М, также в 10 раз уменьшится концентрация CHsCOONa,
которая станет равной 0,001 М. Поэтому
[Н+] = 1,82.Ю-6 -^щ- = 1,82-Ю-4 г-ион/л
т. е. при разбавлении указанного буферного раствора в 10 раз [Н+ ] не изменится.
Пример 3. а) Вычислить значение [Н+ ] в уксусно-ацетатном буферном растворе,
который является 0,1 Мв отношении СНзСООН и 0,1 М ъ отношении СНзСООИа; б)
вычислить, во сколько раз концентрация ионов водорода в буферном растворе больше или
меньше концентрации ионов водорода в 0,1 М растворе СНзСООН.
Р е ш е н и е. а) Согласно формуле (15), концентрация Н+ в буферном растворе равна:
[Н+] = 1,82-10-*-giL = 1,82-Ю-6 г-ион/л
б) Концентрация Н+ [см. формулу (5)] в 0,1 М растворе СН3СООН равна:
[Н+]= ]/ 1,82- Ю-5-10-!= 1,35-Ю-3 г-ион/л
Следовательно, величина [Н+ ] в буферном растворе нримерно в 100 раз меньше
величины [Н+ J в растворе СНзСООН.
ПО ГЛ.И. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
§ 12. Вычисление активности ионов водорода
в водных буферных растворах
Для вычисления активности ионов водорода и гидроксила (а . и а \ в водных
V Н ОН /
буферных растворах пользуются формулами, полученными путем преобразования
формул (15—17).
Для вычисления а . буферного раствора, состоящего из кислоты и ее соли, выра-
н
жаем в формуле (15) КНАп через КНАп, так как
v ^НАп
дНАп = Л
/1
тогда
/2 t-KtAn
отсюда
^НАп СнАп
Для вычисления а + в буферном растворе, состоящем из слабого основания и его соли,
Н
выражаем в формуле (17) /CKtOH чеРез ^кюн и %w чеРез K\v:
%w o „ ^KtoH
Д^=-72~> а ДКЮН = 3
/1 /1
т. е.
ГИ+1 Kw?1 CKtAn *РГ CKtAn П7яч
/l^KtOH CKtOH /CRtoH CKtOH
Гтт+1 f f KW CKtAn
«H+ = [H+] /H+ = /,
H+ L J 'H+ 'H+ к' Стг+гм
AKtOH KtoH
Пример /. Вычислить рН буферного раствора 0,1 н. в отношении СНзСООН и 0,01 н.
в отношении СНзСОСЖа с учетом коэффициента активности.
Решение. Уксусная кислота является слабым электролитом. В присутствии
ацетата натрия уксусная кислота практически находится в недиссоциированном состоянии.
Поэтому ионная сила данного раствора определяется по ацетату натрия.
Ионная сила бинарного электролита (типа Kt+ An") равна его молярной концентрации
(см. § 8), т. е. 0,01. При [х = 0,01 / . = 0,92.
н
Согласно формуле (15а), для буферной смеси, состоящей из кислоты и ее соли
1,82-Ю-5 0,1 n 1Л ,
Vе —ода W~2*10 г'ион,л
paH = -lgffH+ = -lg2.10-* = 4-lg2 = 4-0,3 = 3,7
Пример 2. Вычислить рН аммиачно-аммонийной буферной смеси (0,01 н. NH4OH +
+ 0,1 н. NH4C1) с учетом коэффициента активности.
Решение. В данном случае \i = 0,1 (см. книга 1, табл. 1); /н+ = 0,81.
Для вычисления рН буферной смеси, состоящей из основания и его соли, пользуемся
формулой (156):
V = 0-81 i.'smo-' -W~4-10'9 г-тн/л
раН = — lgeH+ = —lg4-10-» = 9 — lg 4 = 9 -0,6^8,4
§ 13. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ Н+ И СТЕПЕНИ ГИДРОЛИЗА Щ
§ 13. Вычисление концентрации ионов водорода и степени
гидролиза в водных растворах гидролизующихся
бинарных солей
Вычисление [Н]+ и агидр в растворах гидролизующихся солей,
образованных катионами слабых оснований и анионами сильных кислот
Гидролиз по катиону. Соли типа NH4 C1 гидролизуются по уравнению:
NH4Cl+HOH ^± NH4OH + HCl
В ионной форме это можно записать:
NH^+HOH ^ NH4OH+H+
ИЛИ
NH+ + НОН т± NH3 + Н30+
Константа гидролиза. Применив к уравнению, выражающему в
ионной форме гидролиз соли типа NH4C1, закон действия масс, получим:
[NH4OH] [Н+]
[NH+]
= К [НаО]
Произведение (К [Н20]) называют константой гидролиза и
обозначают /Сгидр.
Поэтому в общем виде для солей типа NH4C1 можно написать:
IKtOH] [Н+]_
г^р-| = Д IH2UJ = Дгидр (23)
При математических расчетах, связанных с применением констант равновесия,
взаимодействие и образование воды не принимают в расчет, так как [Н20] является величиной
постоянной, равной ~55,5 моль/л. Поэтому равновесная концентрация воды в
разбавленных водных растворах практически не изменяется. Величина [Н20] входит в значения
констант равновесий, характеризующих процессы, протекающие в водной среде, как это
имеет место при выражении константы гидролиза.
Умножив и разделив левую часть уравнения (23) на [ОН""], можно
представить его в следующем виде:
ГКЮН] [Н+] [ОН-1 __
lKt+] [OH-] -А гиДр {ZV
Но
[KtOH] 1
[Kt+]{OH-] /CKtOH
[H+][OH-]=tfir
Подставляя эти значения в уравнение (24), получим:
#гидр = -т? (25)
дкюн
Например, для случая гидролиза NH4C1 можно написать:
*гадр-*^Г~ 1.81-10- -5'5'10
Следовательно, константа гидролиза бинарной соли, образованной
катионом слабого основания и анионом сильной кислоты, представляет
собой отношение ионного произведения воды к константе электролитической
диссоциации слабого основания, образующегося в процессе гидролиза.
112 ГЛ.П. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Вычисление концентрации гидролизованной части соли. Обозначим
через CKtAn концентрацию исходной соли и через хгидр концентрацию
гидролизованной части соли. Концентрация негидролизованной части соли,
равная концентрации катиона, не вступившего в реакцию гидролиза,
может быть представлена в виде разности CKtAn —<Wp.
Согласно уравнению реакции гидролиза:
Kt+ + HOH ^t KtOH + H+
^KtAn *гидр *гидр *гидр
[KtOH] = [H+ ] = л:гидР, так как при гидролизе соли образуется столько
молей слабого основания, сколько образуется грамм-ионов водорода и
сколько молей соли гидролизовалось.
Подставив в уравнение (23) значения [KtOH], [H+] и [Kt+],
выраженные соответственно через хгидр и через CKtAn — *гидр, можем написать:
X2
-7п — Г = Ятидр (26)
luKtAn *гидр)
Если концентрацию исходной соли принять за 0,01 М, то
[NH+] = CNH4 С1 -*гидр « 0,01 - *гидр
Поэтому, пользуясь уравнением (26), можем написать:
*гидр KW _., МО"14 с. 1П-1о
(0,01—^гидр) "/Сгидр~'^^Г-" 1,81-10-' -5'5'10
откуда
4ежо + б,б. 10-10*PMD - 5,5. Ю-12 = 0
"Гидр « ~>" iV "Гидр
Хп№ = т=_ M^L + yjM^pi+5>5.10.и ж1,он г.ион/л
Следовательно, концентрация гидролизованной части соли равна
~1 -Ю"6 г-ион/л.
Вычисление концентрации ионов водорода. Так как [Н+] = аггидР,
то для ее вычисления остается лишь вычислить, чему равняется л:гидр.
Значение хГИдР можно вычислить, решая квадратное уравнение (26).
В тех случаях, когда концентрация гидролизованной части соли л:гидР
представляет собой относительно малую величину по сравнению с большой
величиной CKtAn, можно знаменатель дроби уравнения (26) CKtAn — Ягидр
приравнять к CKtAn и вычислить [Н+ ] по формуле:
: *гидр — V^THflpCKtAn ~ Л/ J(
[НЧ -^ндр = У/СгидрСк4Ап=1/ ТСК*Л" (27)
KtOH
Уравнение (27) показывает, что концентрация ионов водорода в водном
растворе бинарных солей (типа NH4C1), образованных катионами слабых
оснований и анионами сильных кислоту пропорциональна корню
квадратному из произведения константы гидролиза на молярную концентрацию
* Уравнение (27), как и последующие аналогичные уравнения, является
приближенным, так как при выводе допущено, что
CKtAn "" *гидр = CKtAn
§ 13. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ Н+ И СТЕПЕНИ ГИДРОЛИЗА ЦЗ
Вычисление степени гидролиза. Степень гидролиза (для соли типа
NH4C1) выражает собой отношение гидролизованной части соли к общей
ее концентрации:
_ *гидр __1 / к 1 , / Kw (Ш
*гидр — г — \ / Агидр -р — I / 1? г ^°'
°KtAn I/ CKtAn I/ AKtOHCKtAn
Уравнение (28) показывает, что степень гидролиза солей, образованных
катионами слабых оснований и анионами сильных кислот пропорциональна
корню квадратному из константы гидролиза и обратно пропорциональна
корню квадратному из молярной концентрации соли.
Для двух сравниваемых солей указанного типа степень гидролиза
будет тем выше, чем меньше величина константы диссоциации слабого
основания (Ккюн).
При концентрации NH4C1, равной 0,01 М, получим:
Ы0"6
агидр = 0Q1 =1-10-*, или 0,01%
б) Вычисление [Н+] и аГИдР в растворах гидролизующихся солей,
образованных катионами сильных оснований и анионами слабых кислот
Гидролиз по аниону. Соли типа CH3COONa гидролизуются по
уравнению:
CH3COONa+HOH ^± CHgCOOH + NaOH
в ионной форме:
СН3СОО- + НОН 5± СНзСООН + ОН-
или в общем виде:
An- + НОН ^± НАп + ОН-
Раствор имеет щелочную реакцию.
Такой случай гидролиза называется гидролизом по аниону.
Константа гидролиза. Применив закон действия масс к уравнению,
выражающему в ионной форме гидролиз соли типа CHgCOONa, получим:
или в общем виде:
[СНзСООН] [ОН-] rrHOi-ff
[CHaC00.j = К [НаО] - КГЙДР
[HAn] [OH-] __ (т
[55=] ~^гидр к™>
Умножив и разделив левую часть уравнения (29) на [Н+], можно
представить это уравнение следующим образом
[НАп] [Н+] [ОН-] _„
[Н+] [An-] ~~ Агидр
но
[НАп] 1
[Н+] [An-] KHAn
[Н+][ОН-]=/Сц7
откуда
Ктиар=к*- (30)
114 гл. и. нейтрализация, или кислотно-основное титрование
Следовательно, константа гидролиза бинарной соли, образованной
катионом сильного основания и анионом слабой кислоты, представляет
собой отношение ионного произведения воды к константе электролитической
диссоциации слабой кислоты, образующейся в процессе гидролиза.
Например, для случая гидролиза KCN можно написать:
К* -к. 10"14 --М ю-»
KHCN - ДгиДР- 7,2.10-ю ~ м Ш
Вычисление концентрации гидролизованной части соли. Так как при
гидролизе соли KtAn (типа CHgCOONa) [HAn] = [ОН" ] = хгидр, то для
0,01 М раствора KCN можно считать, что
х2
IES5E = 1 4. Ю-5
(0,01-*гидр) '•*
откуда
^идр + 1,4.10-^гидр-1,4.10-7 = 0
Решая это квадратное уравнение, получим:
1 4-Ю-5 т Г(\ 4-\о~ъ)2
*гиДР = ^ + у [ 4 ) + 1,4. Ю-7 = 3,67-10-4 г-ион/л
Вычисление концентрации ионов гидроксила. Подставляя значения
[ОН"], [НАп] и [АгГ], выраженные через хгидр, в уравнение (29),
получим:
х2
гидр „
uKtAn *гидр
Так как хГИЛР по сравнению с CKtAn величина очень малая, то можно
написать:
[ОН"] = *гидр = |/^гидрСК1Ап = ]/ ^н^п
^KtAn (31)
Уравнение (31) показывает, что концентрация ионов гидроксила в
водном растворе бинарных солей (типа CHgCOONa), образованных катионами
сильных оснований и анионами слабых кислот, пропорциональна корню
квадратному из произведения константы гидролиза на молярную
концентрацию соли.
Чтобы найти [Н+ ], применяют следующее уравнение [Н+ ] = тт^тгу.
Подставляя в это уравнение значение [ОН~ ] из уравнения (31), получим:
_Kw Kw _ -I / Kw^HAn
V CKtAn
w=w=^fe=i/ -^
•I / KwCKtA
V #HAn
Например, если концентрация раствора KCN равна 0,01 М, то
концентрация ионов водорода в этом растворе будет равна
[Н+]= зУ°ш-4 = 2,7-10-" г-ион/л
Вычисление степени гидролиза. Из уравнения
§ 13. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ H+ И СТЕПЕНИ ГИДРОЛИЗА Ц§
следует, что степень гидролиза бинарных солей, образованных анионами
слабых кислот и катионами сильных оснований, пропорциональна корню
квадратному из константы гидролиза и обратно пропорциональна корню
квадратному из молярной концентрации соли.
Для двух сравниваемых солей указанного типа степень гидролиза
будет тем больше, чем меньше величина /(нал- В 0,01 М растворе KCN
степень гидролиза равна:
<*гиДр= 3,6Jo1°"4 =3.67» Ю-2, или 3,67о/0
Вычисление степени гидролиза солей, образованных катионами
сильных оснований и анионами слабых многоосновных кислот. Вычисление
степени гидролиза солей типа Na2C03 производят, не принимая в расчет
вторую ступень гидролиза таких солей. Другими словами, уравнения
остаются прежними [см. уравнение (33)], но в формулу подставляют
Янап — вторую константу электролитической диссоциации слабой
двухосновной кислоты, образующейся согласно уравнению первой ступени
гидролиза соли:
An"" + НОН ^± НАгг+ОН-
Лример. Вычислить степень гидролиза 0,1 М раствора ЫагСОз.
Решение. Гидролиз карбоната натрия протекает согласно следующим уравнениям:
СОд " + НОН :?± HCOg + ОН" (первая ступень)
НСОд + НОН ^± Н2С03 + ОН"" (вторая ступень)
Не принимая в расчет вторую ступень гидролиза, можно написать:
т Г 10"14
агидР= у 4,7-10-"-10-* ==4'6-1°-2» или 4>6%
в) Вычисление [Н+] и агидр в растворах гидролизующцхся солей,
образованных катионами слабых оснований и анионами слабых кислот
Гидролиз соли такого типа, как, например, CH3COONH4, можно
выразить уравнением:
CH3COONH4 + НОН ^± NH4OH + СНзСООН
в ионной форме:
NH4 + СН3СОО" + НОН ^± NH4OH + CH3COOH
или в общем виде:
Kt+ + An-+HOH ^± KtOH + HAn
Такой случай гидролиза называют гидролизом по катиону и аниону.
Водные растворы таких солей имеют нейтральную, кислую или
щелочную реакцию в зависимости от величины констант электролитической
диссоциации кислот и оснований (Ккюн и /Снап)» образующих эти соли.
Константа гидролиза. Применив закон действия масс к уравнению,
выражающему в ионной форме гидролиз соли типа CH3COONH4, получим:
[NH4OH] [СН3СООН]
[NH+] [СН3СОО-] ~ * 1Н2°] ~ *гидр
или в общем виде для солей типа CH3COONH4:
[КЮН] [HAn] _ m.
[Kt+] [An-] Лгвдр (а)
116 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Умножив числитель и знаменатель уравнения (34) на произведение
[Н+] [ОН-], получим:
[KtOH] [HAn] [Н+] [ОН-]
[Kt+] [ОН-] [Н+] [An-] "~ Лгидр
Однако
[KtOH] 1 [HAn] l
[Kt+] [ОН-] /CKt0H ' [Н+] [An-] Кнап
откуда
^гидр = J? ip
^KtOH^HAn
[Н+][ОН-]=/Ст^
Агидр — J? J7 (35)
Следовательно, константа гидролиза бинарной соли, образованной
катионом слабого основания и анионом слабой кислоты, представляет
собой отношение ионного произведения воды к произведению констант
электролитической диссоциации слабого основания и слабой кислоты,
образующихся в процессе гидролиза.
Например, для случая гидролиза CH3COONH4 можно написать:
^nh4oh^ch8cooh 1,81-Ю-6-1,82-Ю-6
^ГИДр 7? Ъ = 1 «1 in-R 1 оо 1Л.к = 3- 10"
Вычисление концентрации гидролизованной части соли. Обозначим
через Хгидр концентрацию гидролизованной части соли, тогда
концентрация негидролизованной части соли может быть представлена в виде
^KtAn — *гидр- Из уравнения гидролиза соли, гидролизующейся по катиону
и аниону
Kt+ + An- +HOH ^t KtOH + HAn
CKtAn *гидр ^KtAn *гидр *гидр *гидр
следует, что ^идР = [KtOH] = [HAn].
Равновесные концентрации катиона и аниона равны между собой т. е.
[Kt+] = [An-]=?KtAn-*w
Подставив значения равновесных концентраций в уравнение (34),
получим:
/С ^-х \% =^гндр (26а)
(cKtAn- *гидр)
Если концентрация гидролизованной части соли относительно
невелика, т. е. если данная соль гидролизуется относительно слабо, то
CKtAn — *гидр можно приравнять к CKtAn- Тогда, решая уравнение (26а),
получим:
*гидр
= [KtOH] = [HAn] - CKtAn V~K^ = CKtAn |/ KKtonKHAn <36>
Например, для 0,01 M раствора CH3COONH4
«гидр = [NH4OH] = [CHgCOQH] = CCHjCOONH4 VK^=-
-1 / IcR*
= 0-01У1,81.10-^.1,82.10-^5'4-10"3 тЛЬ'Л
§ 13. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ Н+ И СТЕПЕНИ ГИДРОЛИЗА Ц7
Вычисление концентрации ионов водорода и гидроксила. Подставив
в уравнение (36) значение х, выраженное через [КЮН ], из уравнений (23)
и (25) или, выраженное через [НАп], из уравнений (29) и (30), получим:
__ 1 f Ку/Кь
" V *KtC
'•КЮН
или
[н+] = 1/ ; НАп (37)
1Н+]=/и: -*НАп /ет^=«- ^^ (38)
Уравнение (38) показывает, что концентрация ионов водорода в водном
растворе бинарных солей (типа CH3COONH4), образованных катионами
слабых оснований и анионами слабых кислот, пропорциональна
произведению константы электролитической диссоциации кислоты на корень
квадратный из константы гидролиза.
Аналогично поступают при вычислении концентрации ионов
гидроксила:
[ОН-] = , К* = 1/ *ук'°н (39)
_ 1 / KwK
, Г KwKHAn V *н
V *Kt
НАп V ^НАп
КЮН
Умножим числитель и знаменатель на величину /Скюн» тогда
Г°н"] = V KWK\tOH = *кюн l/V^lC = ^кюн V^WT (40)
V ДНАпдКЮН V AKtOHAHAn V
Из уравнений (37) и (39) следует, что растворы бинарных солей слабых
кислот и слабых оснований, характеризующихся равными значениями /Снап
и Ккюн, имеют нейтральную реакцию даже в тех случаях, когда соль
в значительной степени гидролизовалась. Если Кнап не равна Ккюн,
то кислая, или щелочная, реакция зависит от отношения констант
диссоциации кислот и оснований.
Например, для 0,1 н. раствора NH4CN будем иметь
ги+1 т/" Ы0-14.7,2.10-10 л 1Л 1п
[Н+]= у if8bio-8 «6-Ю~10 г-ион/л
и, соответственно,
[ОН-] «±^«2,5-10-» г-ион/л
Следовательно, в растворе NH4CN концентрация ионов гидроксила
больше концентрации ионов водорода и раствор имеет слабощелочную
реакцию.
Как видно из уравнения (38), [Н+ ] в растворе солей, гидролизующихся
по катиону и аниону, не зависит от концентрации данной соли.
Вычисление степени гидролиза. Степень гидролиза представляет
собой отношение концентрации гидролизованной соли к ее общей
концентрации, поэтому, пользуясь приближенной формулой для вычисления
•*тидр> можно рассчитать агидР:
Н8 ГЛ. И. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Из этого уравнения следует, что степень гидролиза бинарных солей,
образованных катионами слабых оснований и анионами слабых кислот,
пропорциональна корню квадратному из константы гидролиза.
Например, для CH3COONH4.
«гидр = V Кгидр = у 1)81,lQ-6,1>82,10-6 = 5,4- Ю-з
или в процентах: 5,4-10"3-100 = 5,4-10"1 = 0,54%.
Все приближенные формулы для вычисления /СгидР, [Н+], [ОН"],
д:гидр и агидр приведены в табл. 12.
Вычисление степени гидролиза сильно гидролизующихся солей.
В случае сильно гидролизующейся соли концентрация ее гидролизованной
части д:гидР составляет относительно большую величину. В таком случае
уже нельзя C^tAn — *гиДР приравнивать к C^t An- Пользуясь
приближенными формулами при расчетах степени гидролиза сильно гидролизующихся
солей, можно допустить большие неточности. Поэтому они применимы
только для расчета степени гидролиза слабо гидролизующихся солей.
В аналитической практике имеет большое значение расчет степени
гидролиза ряда сильно гидролизующихся солей, образованных катионами
слабых оснований и анионами слабых кислот, например (NH4)2C03 и
(NH4)2S. Расчет степени гидролиза такого рода солей проводят исходя
из уравнения первой ступени гидролиза, так как гидролиз солей
многоосновных кислот протекает в основном по первой ступени, например:
2NH^ + СО" + НОН ^± NH4OH + HCO:f + NH^
*(NH4)2OV
или
NHf + CO^~ + НОН ^ NH4OH -f HCOif
Вычисление проводят по более точной формуле.
Так как степень гидролиза представляет собой отношение агпдр =
*гидр т,, А „
= g , то для соли типа KtAn, гидролизующейся по катиону и по
аниону:
*гидр ==агидр^К1Ап
Другие равновесные концентрации можно представить в следующем виде:
[КЮН] = [НАп[ =*гидр =агидрСк1Ап
[Kt+] = [An-] = CKtAn - *гидр = CKtAn - aCKtAn = CKtAn (1 - аГйдр)
Подставив эти значения равновесных концентраций в уравнение (34),
получим:
г,2 Г2
arHflpLKtAn ___ v
> — Агидр
CKtAn(1"~ агидр)
откуда
«гидр = (1 — «гидр) "1/ J? ^ (42)
Эту формулу и применяют для вычисления степени гидролиза
указанных выше солей с той лишь разницей, что вместо Кнап подставляют вторую
константу электролитической диссоциации слабой кислоты, образующейся
в результате полного гидролиза (NHJaAn.
ТАБЛИЦА 12
Формулы для вычисления А*Гидр» [Н+], [ОН~], лгГИДр и агидр в растворах гидролизирующихся бинарных солей
Электролит
гидр
[н+]
он
гидр
(концентрация гидролизо-
ванной части соли)
гидр
Соль, образованная
катионом слабого
основания и анионом
сильной кислоты (типа
NH4C1)
Соль, образованная
катионом сильного
основания и анионом
слабой кислоты (типа
CH3COONa)
Соль, образованная
катионом слабого
основания и анионом
слабой кислоты (типа
CHaCOONHJ
Kw
4tOH
Kw
К
HAn
Kw
К
KtOHAHAn
Ук,
y
гидр*-1 KtAn :
^WCKtAn
К
KtOH
V-
CKtAn
/
^KtOH
= ^HAnK^i
гидр
V-
KwKi^iow
CKtAn
уж,
гидр^Ап —
-/
KwCKtAn
К
HAn
^r^KtoH
К
HAn
KtOH
vx
гидр
уж,
-V
гидр^^Ап :
KwCKtAn
К
KtOH
V*r
-Y
идрСК1Ап :
KwCKtAn
К
HAn
'KtA
n V ^гидр ~~
'KXKnV «KtOH^HAn
J
KtAn
К
w
^KtOH^KtAn
уж,
гидр^МАп
-KtAn
=/:
К
w
К
HAnCKtAn
Т^гидр =
Yk
к
w
KtOH^HAn
120 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Пример. Вычислите степень гидролиза растворов (NH4)2C03 и (NH4)oS.
Решение. Для (МН4)гСОз:
*гидр
1/ Ю-14
= (1 — агидр) у 1}3l.i0-6-4,7-10-n ~ ^ ~~ агиДР^'3,46
3 46
«гидр = "4l6= °'77' ИЛИ 77%
Для (NH4)2S:
агидр = 0 — «гидр) у 1 8Ы0"б-10-15 = ^ ~"агиДР^'74^
740
«гидр = 74Г == 0,99, или 99%
§ 14. Вычисление активности ионов водорода
в водных растворах гидролизующихся бинарных солей
Вычисление ан+ для соли, гидролизующейся по катиону. Для соли типа NH4C1 (см.
табл. 12):
V дкюн
Подставив в эту формулу значения Kw и /CKtoH» выраженные с учетом
коэффициентов активности, получим:
V Kw. v — ^KtOH
'V—ir' дкюн
h h
ru+1 1 / %WCKtAnh 1 / *WCKtAi
11 = V 1? = I/ ~~7
f aAKtoH * дкюн
-fc./s
«н*- №1 ft,—fo> 1/ 'Г0""" <*»
KtOH
Пример. Вычислить ан+ в 0,01 н. водном растворе NH4C1.
Решение. [л = 0,01; /я+ = 0,92.
т /" Ы0"14-0 01
ан+ = 0,92 |/ ^ g110u:6U1^0,92»2,35-10-6^2,16-10-в г-ио«/л
Вычисление ан+ для соли, гидролизующейся по аниону. Для соли типа CH3COONa
(см. табл. 12)
[Н+] = l/ /v^AHAn
''KtAn
или, подставив в это выражение значения K,w и tfнап> выраженные с учетом активности,
получим:
V — ^НАп
дНАп — гГ"
§ 15. ТИТРОВАНИЕ СЛАБОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ 121
Уравнения (33) и (34) показывают, что значение ан+ растворов солей типа NH4CI
и СНзСОСЖа зависит не только от концентраций соли, но и от коэффициента активности,
величина которого меняется в зависимости от присутствия в растворе посторонних
электролитов.
Пример. Вычислить ан+ в 0,01 М водном растворе CH3COONa.
Решение, р, = 0,01; /н+ = 0,92
1 т/ Ы0-14.1,82-10-6 , *„ 1Л .
*н+=-оде V <Щ = 4,67-10- г-ит/л
Вычисление ан+ для соли, гидролизующейся по катиону и по аниону. Для соли типа
CHsCOONH4 (см. табл. 12)
-V-
1НЧ = 1/ ?^
AKtOH
или, подставляя в это уравнение значения Kw, #нап и ^кюн» можно написать:
ш+1 i/ J^w^haJi 1 1 / KWKY
1 1= V ~Шс = Т~ V ~F~~
r 'l'l^KtOH /1 ' AKtC
л/ K'WK
i+= V it
%* = [НЧ/Н*=|/ -7-^ <«>
4tOH
Уравнение (45) показывает, что значение ан+ растворов солей типа CH3COONH4
не зависит от концентрации соли.
Соотношение истинных констант диссоциации с обычными и значения ан+ в
растворах гидролизующихся солей сведены в табл. 13.
§ 15. Титрование слабой кислоты сильным основанием
Вычисление рН раствора в различные моменты титрования кислоты
щелочью. Предположим, что для титрования взято 100 мл 0,1 н. раствора
СН3СООН, титруемого 0,1 н. раствором NaOH.
В данном случае концентрацию ионов водорода слабой кислоты нельзя
приравнять к концентрации СН3СООН. Поэтому для вычисления рН
растворов в нулевой точке (до начала титрования) применяют выведенные
ранее формулы (5) и (18) для вычисления концентрации ионов водорода
и рН в растворах слабых кислот (см. § 7 и 10 и табл. 16);
^снзсоон =1,82-Ю-6
Р^снзсоон = -lg Ксн.соон =5 —lgl,82 = 5 — 0,26 = 4,74
Подставляя соответствующие значения р/Ссн8соон и lgCcH8cooH в
формулу (18), получим:
рН = V2*4,74 + V» IgO, 1 = 2,37 Н- 0,50 = 2,87
Таким образом, величина рН 0,1 н. раствора СН3СООН равняется 2,87.
Если прилить к титруемой уксусной кислоте 50, 90, 99, 99,8, 99,9 мл
0,1 н. раствора NaOH, то наряду со свободной СН3СООН в растворе
появится продукт нейтрализации уксусной кислоты — ацетат натрия.
Уксусная кислота с ее солью образует буферный раствор.
Концентрацию ионов водорода в водных буферных растворах слабых
кислот и их солей вычисляют по формулам (15) и (19) (см. § 9 и 10).
По формуле (19) вычисляют рН промежуточных точек титрования
предшествующих точке эквивалентности.
ТАБЛИЦА 13
Соотношения истинных констант (Я") с обычными константами (К) с учетом коэффициентов активности (/) и значения ан+
Электролит
К
Соотношение К и К'
Вода
Слабая кислота
Слабое основание
Соль типа NH4C1
Соль типа CHgCOONa
Соль типа CH8COONH4
*w = [H+][OH-]
_ [Н+] [An-]
ЛнАп ~~ [НАп]
__ [Kt+] [ОН-]
Ккюн ~ [КЮН]
*>гипп
Kw
гидр д-
KtOH
Кгидр — к
Kw
К,
НАп
Kw
ГИДР ~ J? J7
АКЮНАНАп
Kw =
К
W
'н+'он"
Kw
f2
Ан д п — "
тс
НАп
vHAn
^КЮН —"
К
KtOH
Кгидо —
К
w
К
кюн
Кг
К
w
гидр ¦
*1
НАп
Кгппъ —'
Kwfx
^КЮН^НАп
[H+)fH+
Укы*с>
Km
НАп
\W
a°U" УКппнС
KtOH^KtOH
с 1 / KwCKtAn
fl V иг—
J_l/ ^HAn
fl V CKfAn
V-
к
KtOH
§ 15, ТИТРОВАНИЕ СЛАБОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ
123
Подставляя соответствующие значения р/Ссн8соон, lg Ссн8соон и
lg CcH3cooNa в формулу (19), получим:
1. Когда прилито 50 мл NaOH
pH = 4.74-lg^0,l+lg^0.1=4.74
Таким образом, в тот момент, когда к титруемой кислоте прилита
половина (50 мл) требующейся для ее нейтрализации раствора основания
lg СнАп = lg CKtAn, PH = рКнАп-
2. Для случая, когда прилито 90 мл NaOH
pH = 4,74-lgl^0,l+lg^0,l =4,74 + 2-1,05 = 5,69
Аналогичным путем можно рассчитать значения рН для других
промежуточных точек. Данные этих расчетов сведены в табл. 14.
ТАБЛИЦА 14
Титрование 0,1 н. раствора СН3СООН 0,1 н. раствором
Прибавлено
ОД н. раствора
NaOH, мл
0
50
90
99
99,8
99,9
100*
100,1
100,2
101
110
Осталось СНзСООН, %
100
50
10
1
0,2
0,1
0
0,1 (изб. NaOH)
0,2 (изб. NaOH)
1 (изб. NaOH)
10 (изб. NaOH)
[Н+]
1,35.10-»
1,82-Ю-6
2,0-10" в
1,82.10~7
3,6-Ю-8
1,82.10-8
1,35-Ю-8
Ю-ю
5,0. Ю-11
ю-11
10-ia
РН
2,87
4,74
5,69
6,74
7,44
7,74
8,87
10,0
10,3
11,0
12,0
NaOH
АрН
АС
ж-0-037
3?-««
ip.0.H7
$-°*
0,3
"оТ-3
id?-из
од -11,3
i^-пз
со
&=»•'
4—°."
* Точка эквивалентности.
Следует отметить, что уже до достижения точки эквивалентности,
когда не вся уксусная кислота нейтрализована и прилито только 99,8 мл
0,1 н. раствора NaOH, рН раствора превышает 7 (7,44), т. е. среда раствора
становится щелочной.
Поэтому можно сказать, что точка эквиёалентности в случае
нейтрализации слабых кислот (или слабых оснований) сильными основаниями (или
сильными кислотами) не совпадает с точкой нейтрализации, т. е. с
моментом, при котором происходит изменение цвета индикатора, меняющего
свою окраску при рН = 7.
124 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Это указывает на то, что титрование кислот и оснований не всегда
следует заканчивать при рН = 7. В ряде случаев титрование
заканчивают при рН больше или меньше 7, так как при титровании кислот и
оснований необходимо точно установить момент эквивалентности, а не точку
нейтрализации.
В точке эквивалентности вся уксусная кислота будет нейтрализована
и в растворе будет находиться только продукт ее нейтрализации — ацетат
натрия, гидролизующийся под влиянием воды.
Концентрацию ионов водорода в водных растворах гидролизующихся
солей (типа CH3COONa) вычисляют по формуле (32) (см. § 13):
'-/
CKtAn
откуда
РН = -ig [НЧ = -V2 lg Kw - Vi lg *нAn + V2 lg CKtAn
Kw = Ю-1*; igKw = —14
Следовательно
рН = 7 + V.I*HAn + V, lg CKtAn (46)
По этой формуле и вычисляют рН в точке эквивалентности.
В нашем случае
РН = 7 + V2 P^CH.COOH + 2/г lg CCH8COONa =
= 7 + V2-4,74 + V2lg0,l =7 + 2,37-0,5 = 8,87
Это указывает на то, что при титровании уксусной кислоты едким
натром точка эквивалентности не совпадает с точкой нейтрализации
и лежит в области щелочной среды (рН = 8,87).
При добавлении избытка раствора едкого натра величина рН
определяется концентрацией присутствующей в титруемом растворе свободной
щелочи NaOH. При избытке 0,1 н. раствора NaOH в 0,1; 0,2; 1 и 10 мл
концентрация ионов гидроксила будет соответственно равна:
щ0,1 == 1(Г4 г-ион/л; ут^ОД =2-10-* г-ион/л
jJgO.l = Ю-з г.и0н/л; щОД = 10-2 г.ион/л
Концентрация ионов водорода и рН раствора, соответствующие
указанным значениям [ОН" ], равны:
[Н+] = Ю-10 г-ион/л; рН = 10
[Н+] = 5- Ю-11 г-ион/л; рН = 10,3
[Н+] = Ю-11 г-ион/л; рН = 11
[Н+] = Ю-12 г-ион/л; рН = 12
Результаты вычислений рН приведены в табл. 12. На основании
полученных данных построена кривая титрования (рис. 40).
Анализ кривой нейтрализации. Полученная кривая нейтрализации
показывает, что:
1. Точка эквивалентности при титровании слабой кислоты сильной
щелочью не совпадает с точкой нейтрализации, а находится в щелочной
области (при рН = 8,87).
2. Кривая нейтрализации слабой кислоты сильным основанием
несимметрична в отношении точки нейтрализации.
§ 15* ТИТРОВАНИЕ СЛАБОЙ КИСЛОТЫ СИЛЬНЫМ ОСНОВАНИЕМ
125
3. Скачок титрования, наблюдаемый у точки эквивалентности при
нейтрализации слабой кислоты сильным основанием (7,74—10), менее
резко выражен, чем при титровании сильной кислоты (НС1) (см. § 6).
Линия эквивалентности
Интервал перехода
[метилового красного &U-62)
Линия нейтральности
Ш \ Интервал перехода
У\иренол(рталеина (8,0-10,0)
\г^<[очка эквивалентности
(№=8,87)
NaOH
по
Объем раствора NaOH, мл
Рис. 40. Кривая нейтрализации 0,1 н. раствора СН3СООН
0,1 н. раствором NaOH.
4. Начальная и последующие точки титрования, предшествующие
точке эквивалентности, лежат в менее кислой области (при рН = 2,87),
чем при титровании сильной, например соляной кислоты (рН = 1)^
Линия эквивалентности
Объем раствора щелочи, мл
Рис. 41. Кривая нейтрализации 0,1 н. растворов кислот,
различающихся значениями рК:
/ — HC1; 2 — CH8COOH; 3 — кислоты слабее СН8СООН.
5. Кривая титрования слабой кислоты сильным основанием
характеризуется наличием треугольника, образующегося в результате
пересечения кривой титрования, линии нейтральности и линии эквивалентности.
126 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Вершины этого треугольника лежат в точке нейтрализации (а); в
начальной точке соприкосновения кривой с линией эквивалентности (Ь) и в точке
пересечения линий нейтральности и эквивалентности (с) (см. рис. 40).
Наличие указанного признака является характерной чертой всякой
кривой титрования слабой кислоты (типа СН3СООН) и, как увидим ниже,
слабого основания (типа NH4OH).
Титрование кислот более слабых, чем уксусная. При титровании
разбавленных растворов кислот более слабых, чем уксусная, скачок
титрования еще меньше. Определение точки эквивалентности при
титровании таких кислот с помощью индикаторов весьма затруднительно и
во многих случаях просто невозможно. Для количественного определения
очень слабых кислот прибегают к специальным методам титрования и
используют для этой цели неводные среды. Для вычисления рН раствора
вблизи точки эквивалентности при титровании очень слабых кислот
пользуются более сложными уравнениями.
На рис. 41 показаны кривые нейтрализации 0,1 н. растворов слабых
кислот, отличающихся друг от друга различными величинами р/СнАп-
Для сравнения также показана кривая нейтрализации
хлористоводородной кислоты.
§ 16. Титрование слабого основания сильной кислотой
Вычисление рН раствора в различные моменты титрования.
Предположим, что для титрования взято 100 мл 0,1 н. раствора аммиака (/Cnh4oh =
= 1,81-10"5), титруемого 0,1 н. раствором НС1. До начала титрования
согласно формуле (20) (см. § 10)
РОН = V, ptf KtOH - Vi lg CKtOH
или
рН = 14 - рОН = 14 - Vi Р*кюн + Vi lg CKt0H I47)
РКкн4он=-18*ын4он = 5-lg 1,81 =5-0,26 = 4,74
Подставляя соответствующие значения p/Cnh4oh и lg Cnh<oh =
= lg 0,1 = —1 в формулу (47), получим:
рН = 14 — V2-4,74 + V2 lg ОД = 14 — 2,37 — 0,50 = 11,13
Таким образом, величина рН 0,1 н. раствора NH4OH равна 11,13;
Смесь NH4OH с солями аммония образует буферные растворы.
Концентрацию ионов водорода и рН в водных буферных растворах
слабых оснований и их солей, вычисляют по формулам (см. § 10):
гы+1 KwCKtAn
I** J = "г? Г
AKtOHuKtOH
рН = 14 — р/Скюн — lg CKtAn + lg CKtoH
По формуле (22) вычисляют рН промежуточных точек титрования,
предшествующих точке эквивалентности.
Например, если прилито 50 мл NaOH, то
pH = 14-4,74-lg^0.1+lg^0,l=9,26
Таким образом, в тот момент, когда к титруемому основанию прилита
половина (50 мл) требующейся для его нейтрализации кислоты, lg CKtAn =
= lg СКюн и рН = pKw — рКкюн.
Аналогичным путем можно рассчитать значения рН для других
промежуточных точек. Данные этих расчетов сведены в табл. 15.
§ 16. ТИТРОВАНИЕ СЛАБОГО ОСНОВАНИЯ СИЛЬНОЙ КИСЛОТОЙ 127
ТАБЛИЦА 15
Титрование 0,1 н. раствора аммиака 0,1 н.
Прибавлено
0,1 н. раствора
НС1, мл
0
50
90
99
99,8
99,9
100*
100,1
100,2
101
ПО
Осталось аммиака, %
100
50
10
1
0,2
0,1
0
1,0 (изб. НС1)
0,2 (изб. НС1)
1 (изб. НС1)
10 (изб. НС1)
[Н+]
7,4-Ю'12
5,5-10-"
4,3-10"9
5,5-10" 8
2,8-Ю-7
5,5-Ю"7
7,4-10-6
ю-4
2-Ю-4
ю-3
ю-2
раствором
РН
11,13
9,26
8,36
7,26
6,56
6,26
5,13
4,0
3,7
3,0
2,0
НС1
ДрН
АС
-^ = 0,037
09
-g.- 0,023
Ц- = 0,122
-°^-=0,9
0,8 '
-РА-з
од -3
0,1 -ПА
-^-113
.М. = з
0,1
-°^-=09
0,8 *
-9- = °'И
Точка эквивалентности.
Следует отметить, что до достижения точки эквивалентности, т. е.
тогда, когда еще не весь NH4OH нейтрализован и прилито только 99,8 мл
0,1 н. раствора НС1, рН раствора меньше 7 (6,56), а среда раствора
становится кислой.
В точке эквивалентности весь NH4OH нейтрализован и в растворе
находится только продукт его нейтрализации — хлорид аммония, гидро-
лизующийся под влиянием ионов воды. Концентрацию ионов водорода
в водных растворах гидролизующихся солей (типа NH4C1) вычисляют
по формуле (27) (см. § 13):
[№
-V4
V А]
WCKtAn
KtOH
откуда
или
рН = -lg [Н+] = -V2 lg Kw- V, lg CKtAn + V. lg * кюн
pH = 7 — V2 P^KtOH — Vt Ig^KtAn
(48)
По этой формуле и вычисляют рН в точке эквивалентности. В нашем
случае
РН = 7 - Vb-4,74 - Va lg 0,1 = 7 — 2,37 + 0,5 = 5,13
Это указывает на то, что при титровании водного раствора аммиака
соляной кислотой точка эквивалентности не совпадает с точкой
нейтрализации и лежит в области кислой среды (рН = 5,13).
128 ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
При добавлении избытка раствора НС1 величина рН определяется
присутствующей в титруемом растворе свободной кислотой НС1 и
вычисляется по общей концентрации этой кислоты известным способом.
Результаты вычислений рН приведены в табл. 15. На основании
полученных данных построена кривая титрования (рис. 42).
Линия эквивалентности
HCI
! Интервал перехода
'метилового оранжевого (3,1-ty)
Интервал перехода
'метилового красного fat- -6,2)
Линия нейтральности
Интервал перехода
\сренолтталеина (8,0-10,0)
W0
Объем раствора НС1,л&7
110
Рис. 42. Кривая нейтрализации 0,1 н. раствора аммиака 0,1 н.
раствором НС1.
Анализ кривой нейтрализации. Полученная кривая нейтрализации
показывает, что:
1» Точка эквивалентности при титровании слабого основания сильной
кислотой не совпадает с точкой нейтрализации, а находится в области
кислой среды (при рН = 5,13). Скачок рН наблюдается в интервале
6,26—4.
2. Кривая нейтрализации слабого основания сильной кислотой
несимметрична в отношении точки нейтрализации.
3. Кривая титрования слабого основания сильной кислотой
характеризуется наличием треугольника, образующегося в результате
пересечения кривой титрования, линии нейтральности и линии эквивалентности.
Вершины этого треугольника лежат в точке нейтрализации (а); в
начальной точке соприкосновения кривой с линией эквивалентности (Ь) и в точке
пересечения линий нейтральности и эквивалентности (с).
Титрование оснований более слабых, чем водный раствор аммиака.
При титровании разбавленных растворов оснований более слабых, чем
раствор аммиака, скачок рН будет еще меньше. Определение точки
эквивалентности при титровании таких оснований с помощью индикаторов
весьма затруднительно и во многих случаях просто невозможно.
Для количественного определения очень слабых оснований
применяют специальные методы титрования в неводных средах (см. § 39).
Для вычисления рН раствора вблизи точки эквивалентности при
титровании очень слабых оснований пользуются более сложными уравнениями.
Формулы, применяемые для вычисления значений рН и рОН
растворов в процессе титрования, представлены в табл. 16.
ТАБЛИЦА J 6
Сводка формул, применяемых для вычисления значений рН и рОН растворов в процессе титрования кислот и оснований
Электролит
Значение рН среды
Значения рОН среды
Вода (при 25° С), Kw = Ю"*14
Вода (при 100° С), Kw ^ Ю"12
Сильная кислота
Сильное основание
Титравание слабой
кислоты сильным
основан и е м
а) слабая кислота (до
начала титрования)
б) буферная смесь слабой
кислоты и ее соли (до
точки эквивалентности)
в) соль, образованная
катионом сильного
основания и анионом слабой
кислоты (б точке
эквивалентности)
Титрование слабого
основания сильной
кислотой
а) слабое основание (до
начала титрования)
б) буферная смесь слабого
основания и его соли (до
точки эквивалентности)
в) соль, образованная
катионом слабого
основания и анионом сильной
кислоты (в точке
эквивалентности)
[Н+] = [ОН;[ = Kw = Ю-14
[Н+[ = [ОН-[ = VKw = 1Л0"14 = Ю-7 г-ион/л
рН = рОН = -Vt IgKw = ViPtfw = 7
pH = pOH = V2P^^6
PH = -lg[H4=-lgCHAn
рН = pKw - pOH = pKw+ lg CKi0H = 14+lg CKtOH
pH = V2pKHAn~1/2lgCHAn
рН = р/Сндп - lgCHAn + lgCKtAn
pH = V, pKw+ V. pKHAn + V. 1 g С KtAn =
= 7 + V2P^HAn + 1/2lgCKtAn
pH = pKw - ('AP^KtoH ~ Va ^ скюн)
PH = 14 - ViP^RtoH + Vi lg CKtOH
PH = 14 - p/CKtOH - lg CKtAn + lg CKt0H
PH = 7-V2p/CKt0H-1/2lgCKtAn
pOH = pH = 4tfKw = 7
pOH = 14 — pH
pOH = pH = V2p/Cw7^6
pOH=12 —pH
pOH = pKw - pH = pKw + lg CHAn = 14 + lg CHAn
pOH=-lgCKtOH
pOH=p/C*-pH=14 -V2 P^HAn+Vt Ig^HAn
POH = p/(^pH = 14~p^HAn+lgCHAn-lgCKtAn
pOH = p/Cu^ - pH = 7 - 1/2рУ(нAn -1/, lg CKtAn
pOH = 1/2 P^KtOH ~ V2 lg СКюн
pOH = pKw - pH = pKKt0H + lg CKtAn - lg CKtOH
pOH = p/Cv - pH = 7 + V2 P*KtOH + Vi lg CKtAn
130 ГЛЛ1. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
§ 17. Титрование мяогоосновных кислот
Ступенчатая нейтрализация многоосновных кислот. Диссоциация
многоосновных кислот совершается по ступеням. Соответственно ступенчатой
диссоциации многоосновных кислот нейтрализация их щелочами протекает
также ступенчато с образованием кислых и средних солей.
Каждой ступени нейтрализации соответствует своя точка
эквивалентности. Например, при нейтрализации трехосновной фосфорной
кислоты сильным основанием наблюдаются три точки эквивалентности
1-я линия 3-я линия
эквивалентности 2-я линия эквивалентности
эквивалентности
О
1
2
3
и
5
-6
¦ 7
8\
9
10
11
12
13
14
\- Интервал
перехода г-
метилового)
I красного |
Интервал
. перехода
тенолтталеина\
(8,040,0) ¦ 1
1-я точка эквивалентности
Интервал перехода метилового
оранжевого (3,)'-4,4)
H2P04,+HPQ4
нейтральности
2-я точка эквивалентности
Линия
+Р04"""
3-я точка эквивалентности
РОГ""
Число молей "$$д,0]1,приходтихся т один моль Н3Р04
Рис. 43. Кривая нейтрализации Н3РО4 раствором NaOH.
(рис. 43), соответствующие добавлению к одному молю кислоты одного,
двух и трех молей NaOH. При этом реакция сопровождается образованием
одно-, двух- и трехзамещенной соли.
Происходит как бы нейтрализация смеси трех кислот: Н3Р04 (НАпг),
Н2Р04-(НАпп) и НРО"—(НАпш). Сначала нейтрализуется более
сильная кислота Н3Р04, отличающаяся большей константой
электролитической диссоциации (Кг = 1,Ы0"2). Во вторую очередь нейтрализуется
Н2РО-(/С2 = 2,0-10—7). И, наконец, нейтрализуется НРО^—,
характеризующаяся наименьшей константой диссоциации (К3 = 3,6-10~13).
Вычисление рН раствора в различные моменты титрования
многоосновных кислот сильными основаниями. Так как диссоциация многоосновной
кислоты по второй и третьей ступеням ничтожно мала по сравнению с
диссоциацией по первой ступени, то при вычислении [Н+ ] и рН можем
допустить, что многоосновная кислота диссоциирует как одноосновная. В этом
случае для вычисления [Н+ ] можно пользоваться приближенной
формулой, выведенной для одноосновной кислоты.
[НЧ = КЯнАпСнАп. РН = ^ Р*НАп - гЬ lg С1
ЫАп
или в нашем случае
pH = V2P*HAnI--1/2lgCHAnI
Если нельзя пренебречь второй и третьей ступенями диссоциации,
то пользуются более точными уравнениями. Так как величина Кг фос-
§ 17. ТИТРОВАНИЕ МНОГООСНОВНЫХ КИСЛОТ
131
форной кислоты значительно превышает величины К2 и /С3> то второй и
третьей ступенями можно пренебречь:
*н.ро4 = 1-1 -10"2; Р*н3ро4 = 2-IgM = 1,96
Подставляя значение рКг и Сн3ро4 в приведенную выше формулу,
получим значение рН до начала титрования для 0,1 н. раствора Н3Р04:
рН = 1/2-1.96 — Va lg 0,1 = 0,98 + 0,5 = 1,48
Чтобы проследить, как изменяется рН раствора в первой* второй и
третьей точках эквивалентности, когда прибавлено 1, 2 и 3 моль NaOH,
приходящихся на 1 моль Н3Р04, рассмотрим сначала состояние смеси
двух кислот: НАпг (например, Н3Р04) и НАпп (например, Н2РО~),
которые характеризуются различными константами электролитической
диссоциации К± и К2-
Для первой кислоты
tH4 [АпГ] . „ г№1 [HAml
[НАщ] -*' IH]—|а^]-^ <49>
Для второй кислоты
tH+) [Апп] _ к . гн», _ [НАпп]
[НАп„] -** [Н1~ [Anj7] Кг (50)
При нейтрализации смеси этих кислот едким натром в начале
титрования практически нейтрализуется только более сильная кислота НАщ.
Когда величина [Н+] этой кислоты станет равной величине [Н+]
более слабой кислоты, тогда начнет нейтрализоваться вторая кислота
НАпп. В этот момент:
[HAml к [НАпп] v [НАпп] [НАщ] а „
[Ani J [AniiJ [Ann] [Ani J
Чем выше отношение Kt : /C2» тем полнее нейтрализуется только
кислота НАпг При этом [An-], соответствующая количеству частично
нейтрализованной НАпп, практически будет ничтожно малой.
Опыт показывает, что дифференцированное (раздельное) титрование
в водном растворе двух кислот может быть выполнено в случае, если
величина Кг в 10 000 или более раз превышает величину К2-
Для фосфорной кислоты
Кг ~ 2,0-Ю-7 - *'* Ш ' К3 ~~ 3,6.10-" ~ ^
Это значит, что фосфорную кислоту можно дифференцированно
титровать и как одноосновную и как двухосновную. Однако такие кислоты, как
щавелевая (/Сх : К2 = 1000) или винная (Ki: К2 — Ю)> оттитровать
в водном растворе как одноосновную и как двухосновную невозможно.
Предположим, что для титрования взята двухосновная кислота,
способная диссоциировать в две ступени, т. е. образующая в растворе
две кислоты, одна из которых HAri! более сильная, чем вторая НАпц.
Обе кислоты образуются в эквивалентных количествах; концентрации их
равны (Сх — первоначальная концентрация каждой из двух кислот).
Требуется вычислить рН раствора в первой точке эквивалентности,
наблюдающейся при титровании этой многоосновной кислоты.
Если к данной смеси кислот прибавить щелочь в количестве,
необходимом для полной нейтрализации НАп1э т. е. в количестве, эквивалент-
132 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
ном С19 то в первую очередь будет нейтрализоваться НАпь Однако как
только ее концентрация сильно понизится, то начнет нейтрализоваться
НАпц. Если для достижения первой точки эквивалентности будет
израсходовано х% щелочи, то многоосновная кислота перейдет в форму
соли в количестве (100 — х)%. При этом концентрация Ап~,
соответствующая частично нейтрализованной НАпп, будет равна:
[АпГ]=Шс1; [АпП] = :п^С1
_-, СхООО — х)
[HAnj] + [Anf] = Сг; [HAnj] = Cx — [Anf]
100
[HAn„] + [An" ] = Cl5 [HAnf ] = Cx - [An" ] = *g
Перемножив уравнения (49) и (50), получим:
[НАщ] [НАпц]
lH J = Гд -1 Га -1 KlK*
[Ап1 J [Ar4lJ
Подставляя в это уравнение значение [НАпЛ, [НАпП], [Anf]
и [An-], получим:
Откуда
d (100-*) 100 хСг }00
[Н] = ГОО^ 100 (100 — д;) Сх ^^ = ^^
[н+] = VbKt
рН = -lg [H+] = -V2 lg Кг - VJg^2
или
или
pH=V.(pKHAnI + P^HAnII) (51>
По этой формуле вычисляется рН в первой точке эквивалентности
при титровании многоосновной кислоты или смеси кислот. Например,
для фосфорной кислоты:
Кг = 1,1 • Ю-2; рКг = 2 — lg 1,1 =2 — 0,04 = 1,96
К2 = 2,0-10^; рК2 = 7 — lg 2 = 7 — 0,3 = 6,70
К3 = 3,6-10~13; рК3 = 13 — lg 3,6 = 13 — 0,56 = 12,44
В первой точке эквивалентности
pH=Va 0.96+ 6,70) =4,33
Следовательно, первая точка эквивалентности лежит вблизи области
изменения окраски индикатора метилового оранжевого (3,1—4,4).
Если для титрования взята трехосновная кислота, то как только
будет нейтрализована НАпь начнет нейтрализоваться смесь НАпп и
НАпш и, разумеется, в первую очередь более сильная кислота HAnIb
т. е. произойдет то же самое, что и при титровании двухосновной кислоты.
В рассматриваемом случае, т. е. во второй точке эквивалентности, рН
определяют по формуле:
pH = Vl(pKl+P*«) ИЛИ Р« = 1/2(Р^НАп11 + Р^НАпш)
§ 18, ТИТРОВАНИЕ СОЛЕЙ СИЛЬНЫХ ОСНОВАНИЙ И СЛАБЫХ КИСЛОТ 133
В нашем случае (при титровании НзРО^
рН = i/2 (6,70 + 12,44) = 9,57
Следовательно, вторая точка эквивалентности лежит вблизи области
изменения окраски фенолфталеина (8,0—10,0).
В третьей точке эквивалентности для трехосновных кислот, когда
останется не нейтрализованной только одна слабая кислота НАпш,
рН вычисляют так же, как при титровании слабых кислот (см. § 15):
рН = 7 + V, р/Снап + Vb lg CKtAn или РН = 7 + V.p/CHAnm + V. lg CKtАпш
В нашем случае
рН = 7 + V2-12.44 + i/з lg0,l = 12,72
Следовательно, третья точка эквивалентности лежит вне области
изменения метилового оранжевого и фенолфталеина, и поэтому ни с одним
из указанных индикаторов нельзя оттитровать Н3Р04 как трехосновную
кислоту. Константа электролитической диссоциации НРО", равная 3,6 X
X Ю-18, численно очень мала и соответственно рНнроГ/; равный 12,44,
больше 10, поэтому НРО~~ нельзя оттитровывать обычным способом.
Вследствие этого НРО"—-ионы осаждают в виде малорастворимых
солей, например, концентрированным раствором хлорида кальция:
2НРО; " + ЗСа+ +—> I Са3 (Р04)2 + 2Н+
Освобождающуюся при этом свободную кислоту титруют в
присутствии фенолфталеина или тимолового синего.
Таким образом:
1. При титровании многоосновных кислот или смесей кислот, равно
как и многоосновных оснований, каждая ступень титрования
характеризуется своей точкой эквивалентности. Если отношение величин констант
электролитической диссоциации велико (порядка 10 000), то
наблюдается достаточно резкий скачок рН в соответствующих точках
эквивалентности. Если Снапп значительно превышает Снапр то отношение
Ki : /С2 должно быть еще больше. Благодаря этому можно применять
индикаторы, меняющие свою окраску при значениях рН, соответствующих
этим точкам эквивалентности.
2. Кривые титрования многоосновных кислот или многоосновных
оснований, а также смеси кислот или смеси оснований различной силы
характеризуются несколькими скачками, число которых не превышает
суммы ступеней диссоциации этих кислот или оснований.
Обычно наблюдаемые перегибы кривых титрования многоосновных
кислот в водных растворах не столь резки, как перегибы кривых
титрования одноосновных кислот. Резкость скачков титрования можно
увеличить, титруя слабые электролиты в неводных растворах (см. § 39).
§18. Титрование солей, образованных катионами сильных
оснований и анионами слабых многоосновных кислот
Титрование тетрабората натрия. Водные растворы гидролизующихся
солей, образованных катионами сильных оснований и анионами слабых
многоосновных кислот, имеют сильнощелочную реакцию.
При титровании солей сильных оснований и слабых многоосновиых
кислот точка эквивалентности достигается в момент прибавления
теоретически рассчитанного эквивалентного количества сильной кислоты.
134 ГЛЛ1. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Например, хлористоводородной кислотой можно точно оттитровать тетра-
борат натрия.
Образующаяся при этом борная кислота относится к слабым кислотам,
поэтому рН в конце титрования достигает величины порядка 4—5. В
качестве индикатора при титровании тетрабората натрия
хлористоводородной кислотой применяют метиловый красный (4,2—6,2) или метиловый
оранжевый (3,1—4,4), меняющие свою окраску в указанных пределах.
Титрование карбоната натрия. При титровании карбоната натрия
хлористоводородной кислотой наблюдается два скачка рН:
1. При приливании половины кислоты, требующейся для полной
нейтрализации всего количества Na2C03 (т. е. 1 моль HC1 на \моль Na2C03),
образуется бикарбонат:
Na2C03 + HC1 —> NaHC03 + NaCl
или в ионной форме
СОз"+Н+->НСОз
2. При приливании теоретического количества хлористоводородной
кислоты (т. е. 2 моль НС1 на 1 моль Na2C03) получается угольная кислота:
НСОз + НС1 —> Н2С03 + NaCl
'Н20 + С02*
или в ионной форме
НСОз + Н+-+Н2С03
Поэтому карбонат натрия можно оттитровать хлористоводородной
кислотой двумя способами:
Способ 1. Хлористоводородной кислотой титруют карбонат
натрия до бикарбоната в присутствии фенолфталеина, меняющего окраску
в интервале 8,0—10,0. рН раствора бикарбоната равен 8,35. Затем NaHC03
титруют хлористоводородной кислотой до Н2С03 в присутствии метилового
оранжевого (3,1—4,4). Раствор Н2С03 имеет рН, равный 3,75.
Способ 2. Карбонат натрия может быть, подобно тетраборату,
полностью оттитрован хлористоводородной кислотой в присутствии
метилового оранжевого. Образующаяся при этом угольная кислота разлагается
и лишь небольшая ее часть остается в растворе, не влияя существенным
образом на рН раствора. Если С02 удалять кипячением, то можно для
титрования Na2C03 применять фенолфталеин и другие индикаторы.
Кривая титрования карбоната натрия хлористоводородной кислотой
показана на рис. 44. На кривой отчетливо видны два скачка: первый
соответствует титрованию Na2C03 до NaHC03, второй — до Н2С03 (Н20 +
+ С02).
Вычисление рН раствора в различные моменты титрования Na2C03
кислотой. Карбонат натрия в растворе гидролизуется согласно уравнениям
СОд " + НОН t=^ HCO3 + ОН" (I ступень)
НСОз+НОН^±Н2С03 + ОН' (И ступень)
Образующиеся HCOjT -ионы и молекулы Н2С03 способны реагировать
также согласно следующим уравнениям:
но); + н2о ^± н3о+ + со; -
Н2С03 + H20 ^t H30+ + НСОз-
§ 18. ТИТРОВАНИЕ СОЛЕЙ СИЛЬНЫХ ОСНОВАНИЙ И СЛАБЫХ КИСЛОТ 135
Первая ступень гидролиза сопровождается образованием очень слабой
кислоты HCOjT (НАпп), вторая — слабой кислоты H2C03 (НАпх).
НСО«Г — более слабый электролит, чем Н2С03.
[н+] [нСРз]
к.
Ш2СОз
= К
н2со,¦
[Н2С03]
: 4,13-Ю-з
ГнЧГсоП
Я2Н со = #нсо- = г 1 = 4,70-Ю-11
2Н2СОз НСОз [HCOJ-]
Сильнее подвергается гидролизу соль более слабой кислоты, т. е.
соль кислоты, константа диссоциации которой наименьшая. В данном
2-я линия эквивалентности
1-я линия эквивалентности
Н2СОз(Н20+С02)!
Интервал перехода[
метилового оранже6огб[
(3,1-М)
HCI
)2-я точка эквивалентности
Л Интервал перехода
]метилового красного (4,Ь-6,2)
?+ЩСР3 Линия нейтральности
•1-я точка эквивалентности
л "1 Интервал перехода
у/\ ^фенолфталеина/8,0-10,0)
1 Z 3
Число молей HCI, приходящихся на один мольШъСОъ
Рис. 44. Кривая нейтрализации Na2C03 раствором HQ.
случае Кнсо- <^н со в ^800 Раз- ОН "-ионы, образующиеся при
гидролизе Na2C03 по I ступени, тормозят гидролиз по II ступени.
Следовательно, щелочность раствора обусловливается
преимущественно первой ступенью гидролиза карбоната натрия, характеризующейся
выражением Кнсо-> т. е. второй константой электролитической
диссоциации К< ~
2Н2С03*
Поэтому при расчете рН водного раствора Na2C03
второй ступенью гидролиза можно пренебречь.
При вычислении рН водных растворов средних солей слабых много-
основных кислот в расчет принимают только последнюю константу
электролитической диссоциации данной кислоты.
Поэтому рН водных растворов средних солей слабых двухосновных
кислот до начала титрования вычисляют по формуле (46) (§ 15).
В нашем случае:
РН = 7 + v«pKH апп + Vt lg CKtAn (52)
Для 0,1 н. раствора
PH = 7 + V2P^HCor + V2lgO,l
«нсо—4'7-10-11
р/СнсоГ ^ ~~ lg 4,7°' 10"U = 11~~ lg 4,7° = 11 —0,7 = 10,3
136 ГЛ.П. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Отсюда рН первоначального раствора
рН = 7 + 5,15 — 0,5= 11,65
В первой точке эквивалентности
РН = 1/2(рКНАп1+Р^НАпп)
рН = V2 (6,4 + 10,3) = 8,35
Когда карбонат натрия превратится полностью в бикарбонат, раствор
будет слабощелочным. Этот момент может быть зафиксирован с помощью
фенолфталеина, переход окраски которого лежит в интервале рН = 8,0—
10,0.
Во второй точке эквивалентности значение рН вычисляют так же,
как для слабых одноосновных кислот:
pH=V2P^HAnI~1/2lgCHAni
сн2со,^5*10"2 (точнее 3,8-10-2)
рН = Vb-6,4 — Vs lg5- Ю-2 = 3,2 + 0,65 = 3,85
Этот момент может быть фиксирован с помощью метилового оранжевого
(3,1-4,4).
После достижения второй точки эквивалентности раствор будет
кислым. Значение рН этого раствора соответствует концентрации избыточной
хлористоводородной кислоты.
§ 19. Изменение активности и показателя активности ионов
водорода в процессе титрования водных растворов кислот
и оснований
Сравнение обычных (концентрационных) (К) и истинных (/С') констант равновесия
показывает, что в водных растворах значения К™, Кц&п и /CKtoH М0ГУТ изменяться в
зависимости от величины ионной силы раствора, так как с ее увеличением коэффициенты
активностей уменьшаются. Коэффициенты активностей могут значительно отличаться от единицы
в концентрированных растворах, а коэффициенты активностей многозарядных ионов даже
в разбавленных растворах много меньше единицы.
Поэтому применительно к водным растворам, в которых концентрации определяемых
ионов могут составлять большие величины, или в растворах, в которых концентрации
определяемых ионов составляют малые величины, а примесей — очень большие, следует
в расчетных формулах заменять концентрации ионов активностями.
Для того чтобы перейти к активностям, часто требуется сначала рассчитать величину
ионной силы раствора (р,), а затем найти значение среднего коэффициента активности,
соответствующее найденной величине ионной силы раствора (см. книга 1, гл. I, § 8).
Активности ионов водорода и показатели активностей ионов водорода вычисляют по
формулам, в которые входят истинные константы; значение этих констант не изменяется
в зависимости от величины ионной силы раствора (см. книга 1, гл. I, § 7, 8, 18). Для
удобства пользования эти формулы сведены в общую табл. 17.
Как видно из сводной табл. 17, формулы, применяемые для расчета показателей
активности ионов водорода (раН), аналогичны формулам, которые применяют для расчета
показателей ионов водорода (рН), или отличаются от них на величину ±lg/i, причем вместо
концентрационных констант в этих формулах значатся истинные константы.
Так как концентрационные константы определялись для сильно разбавленных
растворов химически чистых кислот и оснований, то их значения можно принять за истинные
константы. Вот почему в сильно разбавленных растворах, в которых значения
коэффициентов активностей равны единице, a lg /i = 0, можно пользоваться приближенными
формулами, применяемыми для вычисления рН и рОН.
Исходя из того, что значения коэффициентов активностей ионов в сильно
разбавленных растворах, с которыми приходится иметь дело химику-аналитику при титровании
кислот и оснований, близки к единице, пользуются обычными расчетными формулами.
20. ВЫВОДЫ, ВЫТЕКАЮЩИЕ ИЗ РАССМОТРЕНИЯ КРИВЫХ
137
ТАБЛИЦА 17
Формулы, применяемые для расчета активностей ионов водорода в процессе
титрования кислот и оснований (при 24° С)
Электролит
Активность водородных
ионов (ац+)
Показатель активности
водородных ионов
(РвН)
Сильная кислота
Сильное основание
Слабая кислота
до начала титро-
до точки
эквивалентности*
в момент
эквивалентности
Слабое основание
до начала
титрования
до точки
эквивалентности **
в момент
эквивалентности
СНАп/н+
К
W
К
W
а _ С f
OH KtOH'OH"
VTHA„c
HAn^HAn
^HAn CHAn
h CKtAn
— Л/ ^HAn
h V cKtAn
К
w
Vk'
KtOH^KtOH
%W CKtAn
^KtOH CKtOH
f 1 / ^UP^KtAn
h V ~P
r AKtOH
-lgaH+ = -lgCHAn-lg/H+
14 + lgC 4-lg/ -
^ * KtoHт ё/он
1/2P^HAn~1/2lgCHAn
P^HAn ~"~*g^HAn + 18 °KtAn
7 + V2p/<HAn + 1/2lgCKtAn+lg/1
14 — VsP^KtOH + Vs lgCKt0H
14 — p/C^toH — lgCKt An +
7 - ViP^toH - Vi Ig CKtAn - lg /t
* Слабая кислота титруется сильным основанием.
** Слабое основание титруется сильной кислотой.
Ошибки, допускаемые в этом случае, ничтожны и особого значения не имеют при
формулировании принципиальных основ теории кислотно-основного титрования. Например,
для 0,1 н. раствора НС1 величина рН раствора без учета коэффициента активности равна 1,
а с учетом — равна 1,08; для 0,01 н. раствора уксусной кислоты рН = 3,3, раН = 3,4
(см. книга 1, гл. I, § 18) и т. д.
§ 20. Выводы, вытекающие из рассмотрения кривых
нейтрализации
При построении кривых нейтрализации для любых водных растворов
кислот и оснований зависимость между рН и рОН выражают уравнением:
рН + рОН = pKw = И (при 25° С)
Для каждого растворителя существует специфичная для него шкала
рН (рНр). Индекс «р» в показателе кислотности указывает на различную
протяженность шкалы и различные значения рН, отвечающие точке
нейтрализации в невоДных растворах (см. § 39). Поэтому указанная
зависимость рН и рОН справедлива только для водных растворов.
Целью титрования является определение точки эквивалентности,
а не нахождение точки нейтрализации.
138 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Кривые титрования многоосновных кислот, многоосновных оснований,
смесей слабых и сильных кислот или слабых и сильных оснований
характеризуются наличием нескольких скачков рН, число которых
соответствует числу ступеней диссоциации этих кислот, оснований или числу
одноосновных кислот или одноосновных оснований, входящих в состав
данной анализируемой смеси.
Пользуясь представленными формулами, можно: 1) построить любую,
кривую нейтрализации; 2) с большой точностью рассчитать значение рН,
при котором необходимо закончить титрование.
Применяя ранее выведенные соотношения истинных констант с
обычными и формулы для перехода от [Н+]кан+ йот рН краН, можно перейти
от концентраций к активностям.
§ 21. Индикаторы
Понятие об индикаторах. Точку эквивалентности устанавливают
различными способами (см. гл. II, § 2). Одним из наиболее широко
применяемых способов фиксации точки эквивалентности является
индикаторный метод, основанный на взаимодействии титруемого раствора с
индикатором. При этом конечную точку титрования определяют по изменению
цвета, появлению или исчезновению осадка (или мути), излучению света
и т. п.
Индикаторами называют такие вещества, которые дают возможность
с известной степенью достоверности установить конечную точку
титрования. При правильном выборе индикатора точка эквивалентности
совпадает с конечной точкой титрования.
Внутренние и внешние индикаторы. Наиболее часто индикатор вводят
в титруемый раствор. В процессе титрования индикатор все время
находится в титруемом растворе. Такого рода индикаторы называют
внутренними индикаторами.
Иногда во время титрования отбирают каплю титруемого раствора и
помещают ее на индикаторную бумагу (фильтровальную бумагу,
предварительно пропитанную раствором индикатора) или смешивают ее с каплей
раствора индикатора на часовом стекле, фарфоровой пластинке или белой
бумаге. Такого рода индикаторы называют внешними. Титрование с
внешними индикаторами вызывает неизбежные потери анализируемого
раствора. Поэтому в настоящее время индикаторные методы, основанные на
использовании внешних индикаторов, вытесняются физико-химическими
(инструментальными) методами фиксации точки эквивалентности.
Обратимые и необратимые индикаторы. Индикатор может
представлять собой обратимую систему, изменяющуюся в ту или иную сторону
по мере изменения того или иного физико-химического параметра
(например, концентрации определяемого вещества, рН раствора,
окислительно-восстановительного потенциала и т. п.). Такого рода индикаторы
называют обратимыми. К ним, например, относятся кислотно-основные
индикаторы, применяемые в методе нейтрализации; эти индикаторы
способны менять свою окраску практически любое число раз по мере
изменения рН в зависимости от кислой или щелочной реакции среды.
Известны также и необратимые индикаторы, с помощью которых
возможно наблюдать конечную точку титрования только один раз, что
обусловливается необратимым изменением химического состава и
строения индикатора. К числу таких индикаторов относятся многие
окислительно-восстановительные индикаторы, которые в процессе окисления—
восстановления подвергаются химическому разрушению. Так ведут себя,
например, некоторые органические красители.
§ 21. ИНДИКАТОРЫ
139
Индикаторы, образующиеся в процессе титрования. В ряде случаев
индикаторного титрования роль индикатора выполняет одно из веществ,
принимающих участие в реакции или образующихся в процессе
титрования. Примерами такого рода титрования являются:
1) титрование восстановителей перманганатом, избыток которого
окрашивает титруемый раствор в малиново-красный цвет;
2) титрование броматом в кислом растворе, сопровождающееся при
избытке бромата образованием элементарного брома, окрашенного
в желто-бурый цвет;
3) титрование иодатом в концентрированном солянокислом растворе,
сопровождающееся выделением элементарного иода и последующим его
обесцвечиванием при избытке иодата.
Классификация индикаторов. В зависимости от типа используемой
реакции при титровании индикаторы подразделяют на следующие группы.
Кислотно-основные индикаторы, реагирующие на изменение рН
раствора. Эти индикаторы широко применяются в методах нейтрализации
и колориметрии для определения рН среды. К ним относятся
фенолфталеин, метиловый оранжевый и др. (см. ниже).
Окислительно-восстановительные (ред-окс) индикаторы, реагирующие
на изменение окислительно-восстановительного потенциала системы. Эти
индикаторы широко применяются в методах окисления—восстановления.
К ним относятся дифениламин, азокрасители и т. п. (см. гл. III, § 11).
Комплексометрические индикаторы, реагирующие на изменение pKt.
Эти индикаторы широко применяются в методах комплексообразования.
К ним относятся эриохром черный Т, ксиленоловый оранжевый и др.
(см. гл. IV, § 23).
Адсорбционные индикаторы, реагирующие на изменение концентрации
ионов, осаждаемых в виде малорастворимых соединений (например, гало-
генидов серебра). К ним относятся флуоресцеин и эозин (см. гл. IV, § 6).
Известны также другие типы индикаторов: радиоактивные, хеми-
люминесцентные, флуоресцентные {люминесцентные), флотационные и др.
Наибольшее значение из них имеют радиоактивные индикаторы,
представляющие собой радиоактивные изотопы некоторых элементов.
Радиоактивные изотопы применяют при изучении процессов осаждения,
экстракции, хроматографического разделения, дистилляции, растворения,
адсорбции, кинетики реакций и т. п.
Действие хемилюминесцентных индикаторов основано на
возникновении или исчезновении излучения видимого света в процессе
окислительно-восстановительных реакций при определенных значениях рН среды.
К их числу относятся: люминол, силоксен и др.
Флуоресцентные, или люминесцентные, индикаторы при освещении
ультрафиолетовыми лучами в процессе титрования при определенном
значении рН раствора вызывают не изменение окраски раствора, а изменение
цвета люминесценции, который не зависит от окраски и степени
прозрачности титруемого раствора. К их числу относятся родамин 6Ж, эозин,
резоруфин, хинин, риванол и др.
Кислотно-основные индикаторы. Индикаторы, применяемые в методе
нейтрализации, представляют собой органические вещества; это слабые
электролиты, обладающие кислыми или основными свойствами:
Hind zrt H+ + Ind-
кислота основание
(донор протона) (акцептор протона)
где Hind — молекулярная форма индикатора;
Ind" — ионная форма индикатора (анион).
140 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Та^им образом, индикаторы, применяемые в методе нейтрализации,
являются обратимыми кислотно-основными системами; поэтому они носят
общее название кислотно-основные индикаторы. Окраски кислотных форм
этих индикаторов отличаются от окрасок основных форм.
Кислотно-основные индикаторы меняют свою окраску при действии
кислот и оснований. Эти изменения окраски зависят от степени изменения
Увеличение рН
(щелочная область)
Повышение цвета
Уменьшение рН
(нислап область)"
углубление цвета
Желто- Оранжевый Пурпурный Синий Рес1инЮиит
-зеленый\Желтый\{расный\Фиолетовь1й / /Сине-зеленый
Рис. 45. Кривые поглощения метилового красного в
щелочной (/) и кислой (//) средах.
концентрации ионов водорода или рН раствора. В связи с этим
представляет большой практический и теоретический интерес исследование
поведения кислотно-основных индикаторов в процессе изменения рН растворов
и выяснение причин изменения окраски индикаторов в кислой и щелочной
средах.
Нелишне в связи с этим напомнить, что вещество представляется нам
окрашенным, если оно отражает часть лучей видимой области спектра
(с длинами волн от 400 до 750 нм). Другими словами, вещество принимает
цвет непоглощенных (отраженных) излучений белого цвета. Оно
представляется белым, когда вещество отражает все лучи видимой области спектра;
черным — когда их поглощает; цветным — когда избирательно поглощает
часть лучей.
Поглощение света веществом характеризуется кривой поглощения
(спектром), имеющей максимум поглощения (рис. 45). Под влиянием ряда
факторов (структура вещества, влияние заместителей, растворитель,
величина рН) максимум поглощения может сдвигаться в длинноволновую
область спектра (батохромный сдвиг) или в коротковолновую область
спектра (гипсохромный сдвиг).
Максимум поглощения на спектре метилового красного (рис. 45)
соответствует длине волны Ямакс (~435 нм) в щелочной среде и смещается
в кислой среде в сторону длинноволновой области спектра Л?акс (~530нм).
При переходе от кислой среды к щелочной максимум поглощения
индикатора смещается в коротковолновую область спектра к'макс, происходит
повышение цвета и окраска переходит от красной к желтой (гипсохромный
§ 21. ИНДИКАТОРЫ
141
эффект); переход от щелочной области к кислой сопровождается
углублением цвета индикатора и изменением его окраски от желтого к красному
(батохромный эффек^. Одновременно с углублением цвета увеличивается
и интенсивность поглощения е", с повышением цвета уменьшается
интенсивность поглощения е'.
Существуют различные теории индикаторов, каждая из которых по
своему объясняет поведение кислотно-основных индикаторов в кислых
и щелочных средах. Ниже приводятся некоторые из этих теорий.
Ионная теория индикаторов. В связи с тем что кислотно-основные
индикаторы представляют собой слабые кислоты или слабые основания,
любой такого рода индикатор диссоциирует в растворе согласно уравнению:
Hind ^± Н+ + Ind-
Окраска раствора, в котором индикатор находится в молекулярной
форме (Hind), отличается от окраски раствора, в котором индикатор
находится в ионной форме (Ind").
Если равновесие реакции сдвинуто в правую сторону, то в растворе
преобладает окраска, характерная для ионной формы индикатора. Если
же указанное равновесие смещено влево, то в растворе преобладает
окраска, характерная для молекулярной формы индикатора.
Например, нейтральный раствор метилового оранжевого, являющегося
относительно сильной кислотой (р/С = 3,7), имеет желтую окраску,
так как в растворе преобладает ионная форма:
Hind z^± H+ + Ind-
красная желтая
форма форма
В нейтральном растворе фенолфталеина — слабой кислоты (рК =
= 9,2) равновесие сдвинуто влево и молекулярная форма преобладает
над ионной
Hind ^± H+ + lnd-
бесцветная красная
форма форма
поэтому раствор бесцветен.
При известном соотношении количеств молекулярной и ионной форм
индикатора раствор может иметь промежуточную (переходную) окраску,
вызываемую смешиванием двух различных цветов молекулярной и ионной
форм индикатора.
Например, индикатор метиловый оранжевый в сильнокислом
растворе окрашен в красный цвет, так как в кислом растворе преобладает
его молекулярная форма (Hind). В щелочном растворе, в котором
превалирует ионная форма (Ind"), метиловый оранжевый окрашен в желтый
цвет. Смешение красного и желтого цветов [молекулярной (Hind) и
ионной (Ind"") форм индикатора метилового оранжевого] приводит к
появлению промежуточной оранжево-красной окраски, наблюдаемой при
рН я» 4.
Таким образом, согласно ионной теории индикаторов, предложенной
Оствальдом, все цветные ийдикаторы, применяемые в методе
нейтрализации, являются слабыми органическими кислотами или основаниями,
специфически реагирующими на ионы водорода и гидроксила.
Зависимость окраски кислотно-основных индикаторов от рН раствора.
Константу электролитической диссоциации индикатора выражают
формулой:
к _ [H+][Ind-]
ДинД — [Hind]
142 ГЛ.П. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Так как Ктд является величиной постоянной, то прибавление в
раствор кислоты ведет к увеличению [Н+] и [Hind] и уменьшению [Ind~ ],
т. е. к усилению окраски, свойственной молекулярной форме индикатора.
Прибавление в раствор щелочи вызывает обратное действие —
усиление окраски, свойственной ионной форме индикатора.
Таким образом, переход одной окраски, присущей молекулярной форме
кислотно-основного индикатора, в другую, свЬйственную его ионной форме,
происходит под влиянием Н+- или ОН"-ионов, т. е. зависит от рН
раствора.
Чем более слабой кислотой является индикатор, т. е. чем меньше его
^Hind (или чем больше его р/СникО» тем при более высоком значении рН
он изменяет свой цвет под влиянием прибавляемых в раствор оснований.
Например, p/CHind метилового оранжевого равен 3,7, а фенолфталеина —
9,2; поэтому метиловый оранжевый изменяет свой цвет при более низком
значении рН раствора (3,1—4,4), чем фенолфталеин, который изменяет
свою окраску при рН, значительно превышающем 3,1—4,4 (8,0—10,0).
Другими словами, окраска различных кислотно-основных индикаторов
зависит от их кислотной силы.
Хромофорная теория индикаторов. Поведение индикаторов,
объясняемое ионной теорией индикаторов, дополняется хромофорной теорией
индикаторов, согласно которой изменение окраски индикаторов связано
с изменением структуры их молекул, внутримолекулярной
перегруппировкой, вызываемой действием Н+- или ОН "-ионов. Согласно ионной
теории индикаторов, молекулы фенолфталеина диссоциируют по
уравнению:
Hlnd^±H+ + lnd-
бесцветная форма, окрашенная
форма в красный цвет
Чем выше значение рН, тем больше данное равновесие сдвинуто в
правую сторону. При рН = 10 молекулы фенолфталеина почти полностью
переходят в окрашенную ионную форму.
По хромофорной теории в процессе изменения рН раствора меняется
строение молекул кислотно-основных индикаторов. Это явление
обусловливается бензоидно-хиноидной таутомерией.
Простейшим случаем бензоидно-хиноидной таутомерии является
таутомерия монооксима хинона (I), который по элементарному составу
тождествен нитрозофенолу (II):
0=<^ \=NOH ^± НО-/ ^—N=0
i и
желтый зеленый
Обе формулы представляют одно и то же химическое соединение в
различных таутомерных формах. В растворе хиноксима устанавливается
подвижное химическое равновесие между обеими таутомерными формами,
зависящее от различных факторов. Подобные таутомерные превращения
наблюдаются и в растворах кислотно-основных индикаторов. Различные
таутомерные формы индикаторов имеют различную окраску растворов,
меняющуюся при действии кислот или оснований.
Например, п-нитрозодиметиланилин (III), обладающий зеленым
цветом, претерпевает при действии кислоты структурные изменения,
сопровождающиеся переходом зеленого цвета в желтый. При этом образуется
хлоргидрат (IV). Наблюдаемое изменение цвета объясняется присоедине-
§21. ИНДИКАТОРЫ
143
нием ионов водорода кислоты к атому кислорода нитрозогруппы, а не
Н3Сч
к атому азота „ ^ /N— -группы.
Н8Сх
Вследствие этого происходит смещение электронов во всей системе,
сопровождающееся образованием хиноидной структуры, что можно
представить следующей схемой:
Н3Сч.о(?-^ .. /-ГХ +HCI | Н3С +
vOy-V>,--/^x +hci нз^х+ /=\ .. ..
н r/N4=JXN=?- —> н ><3=n-o--h
"3^ I Н3С
Ш IV
СГ
Подобно я-нитрозодиметиланилину одни соли /г-аминоазобензола имеют
желтую окраску и бензоидное строение (V), а другие — синюю и хиноид-
ное строение (VI):
2й_(—)-n=n-<;
_> 1 а-^ [h^=<->=n-n-0] а"
VI
Аналогичное равновесие существует в растворах метилового
оранжевого, широко применяемого в качестве кислотно-основного индикатора
(см. ниже).
Таким образом, можно считать, что индикаторные свойства кислотно-
основных индикаторов обусловлены существованием таутомерных
превращений между бензоидной и хиноидной формами.
Возникновение хиноидной структуры и вызывает превращение одной
формы индикатора в другую. Исчезновение хиноидной структуры
обусловливает обратное превращение.
Цветность органических соединений, согласно хромофорной теории,
обусловливается не только хиноидной структурой молекул, но и
присутствием в них других атомных группировок, называемых хромофорными
группами (—N = N—, —N02, —NO, =С=С=, =С=0). При введении
в молекулы органических веществ, содержащих хромофорные группы,
некоторых других групп, называемых ауксохромами (—ОН, —NH2,
—NHR, —NR2), происходит углубление цвета окрашенного вещества.
Таким образом, изменение окраски индикатора происходит в том
случае, когда появляются или исчезают хромофорные группы и когда вместо
одних хромофорных групп появляются другие. Если хромофорные группы
превращаются в нехромофорные, окраска исчезает.
Ионно-хромофорная теория индикаторов. Таким образом, согласно
дополняющим друг друга ионной и хромофорной теориям в растворах
кислотно-основных индикаторов одновременно имеют место и равновесия,
обусловливаемые диссоциацией молекул, и равновесия, связанные с
внутримолекулярными перегруппировками одних форм индикатора в другие,
отличающиеся различным строением. Поэтому обе рассмотренные теории
слились в общую ионно-хромофорную теорию индикаторов, согласно
которой изменение цвета кислотно-основного индикатора, происходящее
вследствие последовательного присоединения органическими сбедине-
ниями ионов водорода при действии кислот или отнятия ионов водорода
при действии щелочей, связано со смещением ионных равновесий
индикаторов, сопровождающихся одновременным изменением их структуры.
Суммируя изложенное выше, можно сказать, что у бесцветных или
слабоокрашенных соединений появление или углубление окраски связано
144 ГЛ.И. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
с введением в их молекулы хромофоров и ауксохромов и увеличением
I I I I I I
числа сопряженных связей —С=С—С=С—С=С—, диссоциацией или
ассоциацией их молекул и, наконец, образованием внутрикомплексных
соединений (например, диметилглиоксимата никеля). Для
кислотно-основных индикаторов наиболее характерными факторами, вызывающими
изменение окраски, являются изменение соотношения количеств
молекулярной и ионной форм индикатора, происходящее под влиянием кислот
и щелочей, и появление или исчезновение хромофорных групп или
превращение одних хромофорных групп в другие.
Краткие сведения из теории цветности органических соединений. Цветность
органических соединений в настоящее время рассматривается как следствие их специфического
электронного строения; при этом особая роль отводится смещению валентных электронов
и перераспределению плотности электронного облака между хромофорами.
Поглощение световых волн данным соединением зависит от подвижности его
электронов. Чем более свободно электроны перемещаются в молекуле соединения, тем более
длинные волны поглощает данное вещество. «Зеленой улицей» для свободного перехода
электронов в молекулах индикаторов является наличие большого числа сопряженных связей.
Следовательно, меняя строение вещества или подбирая соответствующие вещества,
характеризующиеся определенным строением, можно повышать или понижать подвижность
электронов, которая сказывается на повышении или углублении цвета химического
соединения.
Для углубления цвета необходимо увеличивать свободу перемещения электронов,
что можно достичь увеличением числа сопряженных связей в молекуле индикатора
(удлинением системы конъюгации) или усилением ионизации молекул.
Значительное усиление поглощения при наличии системы конъюгации наблюдается
при введении в состав молекул индикаторов электронно-донорных [СНз, ОСНз, ОН, ЫНг,
NHR, N (R)2 ] и электронно-акцепторных (SO2NH2, SO2OH, СООН, COOR, CO, N02) групп.
Очень показательным в этом отношении является пример введения в хлористый трифе-
нилметил, характеризующийся желтой окраской, двух электронно-донорных (ауксохром-
Н3СЧ
ных) групп
НзС^
N, углубляющих его цвет до зеленого:
-.+
СГ
Изменения в строении молекулы индикатора, приводящие к уменьшению смещения
электронов в системе сопряженных связей, повышает окраску соединения.
Поляризующие заместители увеличивают подвижность л-электронов и интенсивность
поглощения света. В кислой среде электронофильность некоторых групп, например
=С=0:-групп, сильно возрастает благодаря присоединению ими ионов водорода:
>С=0: +Н+->С
:=он
Электронодонорность некоторых групп, например —ЫНг-группы, в кислой среде
сильно снижается вследствие перехода азота в новое валентное состояние,
характеризующееся отсутствием неподеленной пары электронов:
—NH2 + Н+ —* -NH3
Электронодонорность оксигруппы в щелочной среде усиливается вследствие того, что
кислород приобретает эффективный отрицательный заряд:
-он+он-
=,н2о
»-0:
§ 21 ИНДИКАТОРЫ
145
Наличие или отсутствие этих групп влияет на положение максимумов поглощения
света соответствующими соединениями. Например, максимум поглощения фенола СбН5ОН
находится в области 275 нм, а фенолят-ионов СвНбО~ — 289 нм; анилин C6H6NH2 — в обла-
+
сти 282 нм, а анилиний-ионов CeH5NHs — 253 нм.
Следовательно, на поглощение света оказывает сильное влияние среда, определяющая
ионизацию растворенных веществ. Примером такого влияния может служить трифенилме-
тановый краситель — бензауринсульфокислота, цвет которого углубляется в кислой и в
щелочной среде:
I I I I
с
/4/S03"
II 1
\//
красный
Ьмакс^503*-"
Н+
1! 1 1 1
II 1 1 1
с
II 1
II 1
\//
желтый
*MaKC = 430w*
:0
\/Ч
//\//®:
он-
—Н,0'
с
II I
\//
красный
^макс = 55бкл?
Углубление цвета бензауринсульфокислоты, связанное с увеличением интенсивности
поглощения, обусловливается протонизацией в кислой среде электронофильных групп
(>С=0:), а в щелочной среде потерей протонов электронодонорными группами (—ОН).
Уменьшение интенсивности поглощения, связанное с присоединением протона
электронодо норными заместителями, например аминогруппами (—NH2) в кислой среде (—NH3),
сопровождается повышением цвета соединения и сдвигом ^макс в коротковолновую область.
Этим также объясняется изменение окраски многих других индикаторов. Например, анилин-
су л ьфофта л ей на и фенолсульфофталеина.
Красители, содержащие несколько ауксохромных групп, в процессе изменения в
широких пределах рН могут периодически изменять свой цвет, т. е. проявляют батохромный
эффект, а затем гипсохромньш и снова батохромный.
Аналогично ведет себя в кислом растворе очень важный индикатор кристаллический
фиолетовый, широко применяемый в аналитической практике как индикатор и как реагент
для обнаружения, осаждения и отделения многих катионов (Hg+ + , Cd+ + , Sb+ + + и др.),
способных образовывать комплексные галогенидные анионы ([HgHal4]"~, [CdHal4]~",
[SbHal6]--- и т. п.):
(CH3)2N
II I I I
С
I
II I
\//
N (CN3)2
N (СНзЬ
+
G1-
Для кристаллического фиолетового наблюдается переход окраски от желто-зеленой до
синей в пределах рН от 0,5 до 2,0.
Изменение спектра кристаллического фиолетового в кислом растворе также
объясняется последовательным присоединением протонов к его —N (СНд)г-группам:
-N (СН3)2+Н+
-N (СН3)2
Н
Следовательно, между строением органических соединений и их цветом существует
определенная зависимость, обусловливаемая наличием большого числа сопряженных
связей и существованием специфического электронного распределения.
146 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Развитию современных взглядов на цветность органических соединений
способствовали работы русских (А. Е. Порай-Кошиц, В. А. Измаильский и др.) и зарубежных ученых
(Р. Вилынтеттер, Р. Вицингер и др.).
Фенолфталеин. Фенолфталеин представляет собой бесцветные
кристаллы, нерастворимые в воде, но растворимые в спирте. В лабораториях
поэтому применяют его 0,1%-ный раствор в 50%-ном спирте. Формула
индикатора:
0=С О
бесцветная форма
При действии разбавленных растворов едкого натра на фенолфталеин
получается двунатриевая соль, имеющая хиноидное строение,
окрашенная в красный цвет:
<_>-Ч/=\_0 +2№+
о=с-о-—
окрашенная форма
Реакция протекает согласно уравнению:
/=ч /Ч_/ ип 2он-
I I ч // 2Н
/=\_с/ч_//
г, I rs \—/
0=С— О—
-Ь2Н20
о=с-
бесцветная форма окрашенная форма
Эти структурные изменения в молекуле фенолфталеина происходят
при рН = 8—10. При действии кислот (при рН < 8) наблюдается обратная
картина: окрашенная форма переходит в бесцветную. ЕсЛи рН равен
13—14, фенолфталеин претерпевает новую структурную перегруппировку.
При этом образуются бесцветные анионы трехзамещенной натриевой
соли.
OH/=V-ONa
\—//~ C\/=\_ONa
0=С—ONa
Этим объясняется обесцвечивание фенолфталеина при действии
большого избытка щелочи.
Таким образом, интенсивно малиново-красная окраска фенолфталеина
обусловлена образованием хромофорной системы:
Метиловый оранжевый (гелиантин, натриевая соль я-диметиламино-
азобензолсульфокислоты) представляет собой оранжево-желтый порошок.
В лабораториях применяют его 0,05%-ный водный раствор. Формула
индикатора:
(CH3)2N~^\-N=N-^\-S03Na
§ 22. ИНТЕРВАЛ ПЕРЕХОДА ИНДИКАТОРА 147
В щелочной среде или в сильно разбавленном водном растворе
метиловый оранжевый имеет желто-оранжевую окраску (VII). При действии
кислот наблюдается переход желто-оранжевой окраски в
оранжево-красную и красную(VIII). Область перехода лежит в пределах рН = 3,1—4,4:
(CH3)2N-^~/""N=N- vZ/-S03 <=±<CH^)2N=^\=N~N-^/--S03
- - 4-он- - \н -
VII VIII
желтая форма красная форма
(^нейтральная или щелочная среда) (кислая среда)
При действии щелочей на растворы метилового оранжевого красного
цвета наблюдается изменение окраски раствора в желтую.
Таким образом, индикаторные свойства метилового оранжевого
обусловлены таутомерией между бензоидной и хиноидной формами.
§ 22. Интервал перехода индикатора
Область значений рН, в которой становится видимым изменение
цвета индикатора, называют интервалом перехода индикатора.
Оптимальное значение рН титруемого раствора, при котором
наблюдается наиболее резкое изменение окраски индикатора,
свидетельствующее об окончании титрования, называют показателем титрования
индикатора и обозначают символом рТ.
Зависимость цвета индикаторов от соотношения их молекулярной
и ионных форм.
Окраска раствора индикатора зависит от соотношения концентраций
его диссоциированной и недиссоциированнои форм, т. е. от отношения
[Ind-] _ *mnd итш [Hind] _ [НЧ
[Hind] - [НЧ [Ind-] - KHmd
Когда Kmnd = Ш+], то -щщ- = 1.
Другими словами, в этом случае концентрация диссоциированной
формы индикатора равна концентрации его недиссоциированнои формы.
Если "^ > 1, то в растворе превалирует диссоциированная форма,
а если rH+?d ^ *» то превалирует недиссоциированная форма.
При одной и той же концентрации ионов водорода отношение
#Hind/[H+] будет тем больше, чем больше /Снпнь
Для фенолфталеина
[H4[lnd4 ^10„
«Hind ~ [HInd] - Ш
Если условно для большей наглядности [Ind"] обозначить через
[Краен], a [Hind] через [Бесцв], то можно написать:
или
[Краен] "" [Ind-] ""Тб^
[Краен]
[ Бесцв]
[Бесцв]
[Ind4
~~ [Hind]
_ [Hind]
ю-9
[НЧ
= [НЧ
148 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Если рН = 7, т. е. [Н+] = 10~7 ?-ион/л, то
[Бесцв] _ [Hind] 10-»
[Краен] "" [Ind-] ~~ Ю-9 ~
Это означает, что при рН = 7 на каждые 100 бесцветных молекул
фенолфталеина приходится лишь 1 окрашенный ион, т. е. соотношение
между бесцветной молекулярной формой индикатора типа фенолфталеина
и его окрашенной ионной формой равно 100.
Так как нейтральные молекулы фенолфталеина бесцветны, то и
раствор при визуальном рассмотрении его тоже будет казаться бесцветным.
Если к раствору фенолфталеина прибавить щелочь и довести рН
раствора до 8, то соотношение * " j уменьшится по сравнению с
первым в десять раз и раствор станет бледно-розовым:
[Бесцв] _ [Hind] _ [Н+] _ Ю"8 _
[Краен] [Ind-] ~ /CHInd 10-«
сл о ы п [Бесцв] [Hind] 10"9
Если рН = p#Hind = 9, отношение [Красн] = [Ind-j = -^ станет
равным единице и в растворе окажутся равные количества (50%)
бесцветных молекул индикатора (Hind) и окрашенных в красный цвет ионов
(Ind"), при этом раствор окрасится в розовый цвет (переходная окраска).
При дальнейшем увеличении рН до 10, 11 и 12 раствор окрасится
в малиново-красный цвет. Однако при рН = 13,5—14 раствор снова
обесцветится вследствие образования тринатриевой соли индикатора.
Следовательно, переходная окраска индикатора становится заметной при рН
среды, равном pKnind, т. е. —lg/CHind- Но так как изменение цвета
индикатора происходит постепенно, то можно считать, что цвет недиссо-
циированных молекул индикатора начинает маскироваться цветом ионов
задолго до достижения соотношения * •" j == 1.
Таким образом, цвет водного раствора индикатора определяется
соотношением его молекулярной и ионной форм, отличающихся различной
окраской, и зависит от [Н+ ].
Расчет интервала перехода индикатора. Зная константу индикатора
(например, для фенолфталеина /Снта ^ Ю"9), можно рассчитать область
значений рН, в которой изменение окраски индикатора наблюдается
визуально.
Опытным путем найдено, что наблюдаемое изменение цвета появляется
в том случае, когда соотношение различных форм индикатора становится
равным 1 : 10.
Для фенолфталеина изменение окраски наблюдается в тот момент,
когда
-^—JQ = AHInd^ Ю 9
т. е.
[H+] = *Hlnd-10
рН = р/Снша — lg 10 = ptfmnd — I
или
[H+].1Q „ ,п_9
J = AHInd^ Ю 9
т. е.
[Н+] =
10 10
рН = ptfmnd + lg Ю = pKmnd + 1
§ 22. ИНТЕРВАЛ ПЕРЕХОДА ИНДИКАТОРА
149
Область значений рН, в которой визуально наблюдается изменение
окраски индикатора, можно рассчитать по формуле:
РН = p/tHind ± 1 (53)
Пользуясь этой формулой, можно вычислить интервал перехода
любого индикатора. Например, для индикатора, характеризующегося
Кнтд ^ Ю-4, интервал перехода будет соответствовать рН = 4 ± 1,
т. е. 3—5.
Область значений рН, при которой заметно изменяется цвет индика-*
тора, приблизительно равна двум единицам рН.
Поэтому для фенолфталеина интервал перехода окраски составляет
приблизительно:
рН^ — lgio-9 ± Ig 10^8 —10
Показатель титрования индикатора. Вместо интервала перехода
окраски индикатора в объемном анализе пользуются также показателем
титрования индикатора (рТ).
рТ определяет оптимальное значение рН, при котором индикатор
наиболее пригоден для титрования. Значения величины рТ
приблизительно совпадают со значениями величин р/Сшпа (табл. 18).
ТАБЛИЦА 18
Интервалы перехода окраски некоторых индикаторов *
Индикатор
Тимоловый голубой (1-й
переход)
Метиловый оранжевый
Метиловый красный
Лакмус
Феноловый красный
Тимоловый голубой (2-й
переход)
Фенолфталеин
Тимолфталеин
Ализариновый желтый
Интервал
перехода рН
1,2-2,8
3,1—4,4
4,4-6,2
5,0—8,0
6,4—8,0
8,0—9,6
8,0—10,0
9,4—1Q,6
10,0—12,0
Значение
рТ
2
4
5
7
7
8
9
10
11
Окр
в кислой
среде,
молекулярная
форма
Красный
Красный
Красный
Красный
Желтый
Желтый
Бесцветный
Бесцветный
Желтый
аска
в щелочной
среде,
ионная форма
Желтый
Желтый
Желтый
Синий
Красный
Голубой
Красный
Синий
Сиреневый
P^Hind
1,7
3,7
5,1
—
8,0
9,2
9,2
9,7
10,7
* Более полные сведения об индикаторах приведены в справочниках.
Индикаторы, меняющие окраску на всем протяжении шкалы рН.
Наряду с индивидуальными индикаторами иногда применяют
универсальные индикаторы. Универсальные индикаторы изменяют свою окраску
при различных значениях рН на всем протяжении шкалы рН (см. книга I,
гл. III, § 16).
Смешанные индикаторы. Иногда вместо одного индикатора добавляют
смесь двух индикаторов, или смеси индикатора и нейтрального красителя,
не изменяющего свою окраску при различных значениях рН. Например,
вместо метилового оранжевого применяют смесь 1 г метилового
оранжевого и 2,5 г индигокармина в 1 л воды. Эта смесь имеет в щелочном растворе
желто-зеленую окраску, которая в кислом растворе переходит в
фиолетовую. Смесь 1 г метилового оранжевого и 1,4 г ксиленолцианолового
в 500 мл 50%-ного спирта имеет в щелочном растворе зеленую, окраску и
красную в кислом. Такого рода индикаторы называют смешанными
индикаторами.
150 ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Смешанные индикаторы дают возможность сделать переход окраски
более контрастным и достаточно отчетливым в относительно узких
пределах рН, что увеличивает точность результатов титрования. Например,
смешанный индикатор, состоящий из 0,1%-ного водного раствора крезо-
лового красного и 0,1%-ного раствора тимолового голубого (1 : 3),
позволяет точно установить точку эквивалентности, соответствующую
нейтрализации карбоната до бикарбоната (рН = 8,3). При рН = 8,2 этот
смешанный индикатор имеет розоватый цвет, а при рН = 8,4 — фиолетовый.
Из других смешанных индикаторов укажем следующие:
а) метиловый красный — бромкрезоловый зеленый (рТ = 4,3)
изменяет окраску от оранжевой до сине-зеленой;
б) фенолфталеин — тимоловый синий (рТ = 9,0) изменяет окраску
от желтой до красно-фиолетовой;
в) тимолфталеин — ализариновый желтый (рТ = 10,2) изменяет
окраску от желтой до фиолетовой.
§ 23. Выбор индикатора
Самым важным условием, соблюдаемым при титровании, является
правильный выбор индикатора. При несоблюдении этого условия нельзя
получить точные результаты анализа, если даже все другие аналитические
операции проводились очень тщательно.
Интервал перехода окраски выбранного индикатора должен по
возможности совпадать со скачком рН (или скачком титрования),
наблюдаемым в данной системе, а показатель титрования индикатора (рТ) — с рН
в точке эквивалентности.
Поэтому при выборе индикатора сначала вычисляют область рН
раствора, в которой наблюдается скачок рН, а затем подбирают такой
индикатор, у которого интервал перехода окраски совпадал бы с вычисленным
скачком рН.
Например, если рН = 6 в точке эквивалентности, то подходящими
индикаторами могут быть: метиловый красный (4,4—6,2) и лакмус (5,0—
8,0), т. е. такие индикаторы, которые начинают менять окраску в
слабокислой среде.
Следует также учитывать влияние различных факторов на характер
кривых титрования, из которых можно видеть, например, что метиловый
оранжевый, успешно применяемый для титрования 1 н. и 0,1 н. растворов
НС1, не пригоден для титрования 0,01 н. растворов кислот (см. рис. 38,
§6).
Выбор индикатора в зависимости от типа титрования. Для титрования
определяемых веществ никогда не применяют слабых кислот или слабых
оснований, титруют сильные и слабые кислоты сильными основаниями или
сильные и слабые основания сильными кислотами.
Таким образом, все виды титрования в методе нейтрализации сводятся
к нескольким типам. Рассмотрим выбор индикатора применительно к
перечисленным случаям титрования.
а) Титрование сильной кислоты сильным
основанием или сильного основания сильной
кислотой. Титрование сильной кислоты сильным основанием
представлено кривой нейтрализации, показанной на рис. 37 (см. § 6). Анализ
кривой приводит к выводу, что резкий скачок титрования наблюдается
в интервале рН = 4,3—9,7. В этот скачок титрования полностью или
частично укладываются интервалы перехода окраски метилового
оранжевого, метилового красного, лакмуса, фенолового красного, фенолфталеина.
Поэтому указанные индикаторы пригодны как для данного случая ти-
§ 23. ВЫБОР ИНДИКАТОРА
151
трования, так и для случая титрования сильных оснований сильными
кислотами.
Последняя капля стандартного раствора вызывает изменение окраски
всех перечисленных индикаторов.
При титровании сильными кислотами отдают предпочтение метиловому
красному или метиловому оранжевому, так как на них не влияет двуокись
углерода, поглощаемая из воздуха. Фенолфталеин, в противоположность
метиловому красному или метиловому оранжевому, весьма чувствителен
к кислотам и на него влияет даже угольная кислота.
б) Титрование слабой кислоты сильным
основанием. Титрование слабой кислоты сильным основанием представлено
кривой нейтрализации, показанной на рис. 40 (см. § 15). Анализ кривой
приводит к выводу (см. § 15), что резкий скачок титрования
наблюдается в интервале рН = 7,74—10. В этот скачок титрования полностью
или частично укладываются интервалы перехода фенолового красного,
тимолового голубого, фенолфталеина, тимолфталеина. Поэтому
указанные индикаторы пригодны для данного случая титрования. Последняя
капля стандартного раствора вызывает изменение окраски всех
перечисленных индикаторов. Титрование с метиловым оранжевым приводит
к совершенно неверным результатам. Поэтому при титровании слабых
кислот нельзя применять индикаторы типа метилового оранжевого
(с интервалами перехода, лежащими в кислой области).
Фенолфталеин очень чувствителен к кислотам, поэтому при
титровании в его присутствии слабых кислот рекомендуется вблизи точки
эквивалентности растворы прокипятить для удаления С02. Так поступают,
когда титруют уксусную кислоту.
Для того чтобы легче и точнее определять конец титрования по
изменению окраски индикатора, рекомендуется сравнивать окраску
титруемого раствора с окраской индикатора, наблюдаемой, в точке
эквивалентности. Для этого применяют так называемый «свидетель»,
представляющий собой свободный от Н2С03 раствор, содержащий соль титруемой
кислоты.
Значение рН этого раствора соответствует рН в точке
эквивалентности. 3 такой раствор приливают столько же капель индикатора, сколько
прилито в титруемый раствор.
в) Титрование слабого основания сильной
кислотой. Титрование слабого основания сильной кислотой
представлено кривой нейтрализации, показанной на рис. 42 (см. § 16). Анализ
кривой приводит к выводу, что резкий скачок титрования наблюдается
в интервале рН = 6,26—4. В этот скачок титрования полностью или
частично укладываются интервалы перехода окраски метилового красного,
метилового оранжевого, поэтому указанные индикаторы пригодны для
данного случая титрования. Последняя капля стандартного раствора
вызывает изменение окраски всех перечисленных индикаторов. Титрование
с фенолфталеином приводит к совершенно неверным результатам. Поэтому
при титровании слабых оснований нельзя применять индикаторы типа
фенолфталеина с интервалами перехода, лежащими в щелочной области.
Титрование лучше выполнять со «свидетелем».
г) Титрование многоосновных кислот или
многоосновных оснований. Нейтрализация многоосновных
кислот (например, H2S04) и многоосновных оснований [например,
Ва(ОН)2] сразу приводит к образованию средних солей. Если величины
Ки К2 и К3 сильно отличаются друг от друга, то такие соединения при
условии подбора подходящего индикатора могут быть оттитрованы до
одно-, двух- и трехзамещенных солей.
152 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Для выбора соответствующих индикаторов нужно рассчитать
значение рН для каждой точки эквивалентности. Титрование фосфорной
кислоты представлено кривой нейтрализации, показанной на рис. 43 (см.
§ 17). Анализ кривой приводит к выводу, что первая точка
Эквивалентности соответствует значению рН = 4,33; вторая — 9,57; третья — 12,72.
Следовательно, для титрования до NaH2P04 можно применить метиловый
оранжевый, до Na2HP04 — фенолфталеин; до Na3P04 — тимоловый
голубой (8—9,6) в присутствии солей кальция.
Классификация кислотно-основных индикаторов. Все
кислотно-основные индикаторы классифицируют следующим образом:
1) индикаторы, чувствительные к кислотам (интервал перехода
рН >7), — фенолфталеин, тимолфталеин, ализарин;
2) индикаторы, чувствительные к основаниям (интервал перехода
рН < 7), — метиловый красный, метиловый оранжевый;
3) индикаторы, чувствительные и к кислотам и к основаниям, или
нейтральные индикаторы (интервал перехода рН = 7) — лакмус,
феноловый красный.
Индикаторы первой группы чувствительны даже к угольной кислоте.
Поэтому при титровании с этими индикаторами слабых кислот
угольная кислота и карбонаты, разлагающиеся при действии кислот с
образованием угольной кислоты, должны отсутствовать. Если все же
приходится титровать в присутствии угольной кислоты, то применяют
специальные способы (например, нагревание титруемого раствора до кипения).
При титровании сильных кислот сильными основаниями или сильный
оснований сильными кислотами практически можно применять любые
индикаторы. Однако следует иметь в виду, что конечная точка титрования
при пользовании различными индикаторами не соответствует одному
и тому же значению рН.
ТАБЛИЦА 19
Индикаторы, применяемые для титрования различных кислот и оснований
Разбавленные растворы
НС1, HN03, H2S04
HF
CHgCOOH
HCOOH
H3P04
до Н2Р04-
до НРО"
до РО"~
NaOH, КОН, Ва (ОН)2
NH3
Na2B407
Na2C03
до NaHC03
до H2C03
Na3P04
до Na2HP04
до NaH2P04
Рекомендуемые кислотно-основные индикаторы
Метиловый оранжевый, метиловый красный,
феноловый красный, фенолфталеин
Фенолфталеин, тимоловый голубой, феноловый
красный
Фенолфталеин, тимолфталеин, тимоловый голубой
(2-й переход)
Фенолфталеин, тимоловый голубой, феноловый
красный
Метиловый оранжевый
Фенолфталеин, тимолфталеин
Фенолфталеин, тимоловый голубой +СаС12
Метиловый оранжевый, метиловый красный,
феноловый красный, фенолфталеин
Метиловый оранжевый, метиловый красный
Метиловый оранжевый, метиловый красный
Фенолфталеин
Метиловый оранжевый
Фенолфталеин, тимолфталеин
Метиловый оранжевый
§ 24, ОШИБКИ ТИТРОВАНИЯ
153
Для титрования слабых кислот сильными основаниями применяют
индикаторы, чувствительные к кислотам; для титрования слабых
оснований сильными кислотами — чувствительные к основаниям.
В табл. 19 приведены индикаторы, применяемые при титровании
различных кислот и оснований.
Условия, соблюдаемые при титровйнии. Для получения точных и
воспроизводимых результатов анализа необходимо соблюдать
определенные условия при титровании.
1. Следует устанавливать титр стандартного раствора и применять
установленный для титрования раствор в присутствии одного и того же
индикатора.
2. Для титрования следует брать всегда одно и то же количество
индикатора и повторять титрование определяемого вещества несколько
раз до тех пор, пока не будут получены три близко сходящихся
результата.
3. Необходимо брать, как правило, не более 1—2 капель индикатора,
не забывая о том, что индикаторы, применяемые в методе нейтрализации,
сами являются кислотами или основаниями. На нейтрализацию их также
расходуется некоторое количество раствора.
4. Всегда следует титровать до появления одного и того же оттенка
окраски раствора, используя для титрования по возможности одинаковые
объемы титруемого раствора.
5. Необходимо выбирать такой индикатор, который изменяет свой цвет
вблизи точки эквивалентности.
§ 24. Ошибки титрования
Индикаторная ошибка. Изменение цвета индикатора происходит
не абсолютно точно в точке эквивалентности, а раньше или позже, поэтому
химик-аналитик допускает некоторую ошибку, прекращая титрование
раньше или позже требуемого момента. Такую ошибку называют
индикаторной ошибкой. Величина ее может колебаться в самых широких
пределах в зависимости от того, насколько удачно выбран примененный
индикатор.
Предположим, что для' титрования 0,1 н. раствора слабой кислоты
с К = Ю~б 0,1 н. раствором сильной щелочи мы использовали два
индикатора: метиловый оранжевый (3,1—4,4) и фенолфталеин (8,0—10,0).
Посмотрим, что произойдет в обоих случаях титрования. рН исходной
кислоты равен 2,87; в точке эквивалентности рН = 8,87 (см. § 16).
В случае прибавления к титруемой кислоте метилового оранжевого
раствор окрасится в красный цвет. Вскоре после прибавления первых
порций сильного основания рН раствора станет равным 3,1 и раствор
окрасится в оранжевый цвет, а при рН = 4,4 раствор окрасится в желтый
цвет, не изменяющийся при дальнейшем добавлении щелочи.
Следовательно, после прибавления нескольких миллилитров, а быть может и
капель щелочи придется прекратить титрование раньше достижения точки
эквивалентности. Рассчитанное количество титруемой кислоты может
оказаться в несколько раз ниже, чем действительное ее содержание, и
индикаторная ошибка может достичь 75—85%.
Если же к титруемой кислоте прибавить фенолфталеин, то раствор
будет оставаться бесцветным до тех пор, пока по мере прибавления к нему
щелочи рН раствора не достигнет 8,0. При рН = 8 раствор порозовеет,
а при рН = 10,0 раствор окрасится в красный цвет. Так как в точке
эквивалентности рН = 8,7, что укладывается в интервал 8—Ю, то рассчитан-
154 ГЛ.И. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
ное количество кислоты будет практически отвечать действительному ее
содержанию и индикаторная ошибка составит 0,1—1%.
Таким образом, если индикатор выбран правильно, то индикаторную
ошибку не принимают во внимание; если же индикатор выбран
неправильно, то индикаторная ошибка превышает допустимые погрешности и
может достигать очень большой величины. В случаях, требующих более
точных результатов, следует учитывать индикаторные ошибки.
Типы индикаторных ошибок. К индикаторным ошибкам относят такие,
которые вызываются недотитрованием или перетитрованием исследуемого
раствора. В методе нейтрализации различают несколько типов
индикаторных ошибок.
а) Водородная ошибка титрования, вызываемая наличием
в титруемом растворе по окончании титрования избытка ионов водорода,
остающихся в растворе в результате недотитрования сильной кислоты
сильной щелочью (обозначается Н^д-ошибка) или перетитрования
сильного основания сильной кислотой (обозначается Нпер-ошибка).
б)Гидроксильная ошибка титрования, вызываемая
наличием в титруемом растворе по окончании титрования избытка ионов
гидроксила, остающихся в растворе в результате недотитрования
сильного основания сильной кислотой (ОН^ед-ошибка) или перетитрования
сильной кислоты сильной щелочью (ОН^р-ошибка).
в) Кислотная ошибка титрования, вызываемая
присутствием в титруемом растворе по окончании титрования нейтральных
молекул недотитрованной слабой кислоты (НАп-ошибка).
г) Щелочная ошибка титрования, вызываемая
присутствием в титруемом растворе по окончании титрования нейтральных
молекул недотитрованного слабого основания (KtOH-ошибка).
Примеры вычисления ошибок титрования. Н*ед-о шибка. Предположим, что
для титрования сильной кислоты сильным основанием выбран индикатор с рТ = 4
(например, метиловый оранжевый). Титрование заканчивается при рН = рТ = 4, т. е. при [Н+ ]=
*=10~4 г-ион/л (в кислой среде). Следовательно, часть титруемой кислоты будет недоти-
трована и мы допустим Ннед-ошибку. Вычислим ее величину.
Пусть концентрация титруемой кислоты N = 0,1 н.; начальный объем кислоты Vi =
= 25,0 мл; объем раствора в конце титрования Уг «= 50,0 мл.
Каждый миллилитр 0,1 н. раствора кислоты или щелочи содержит г-экв. Для
N
титрования взято ш n Vi г-экв кислоты.
Неоттитрованных ионов водорода * останется 10"4 г-ион/л, или [H+J= 10-pt г-ион/л-
10~pT
в V2 мл раствора [H*J= n V2 г-экв кислоты-. Эта величина и составляет водородную
ошибку титрования, обусловливаемую недотитрованием Н+-ионов.
Величину Н -ошибки (в процентах) вычисляют согласно пропорции:
N
ух _ 100%
1000
Т00б-^2-Ннед-ошибка
В нашем примере
Н+нед-ошибка=^-М000/о ^
Н+ед-ошибка = ^^ 100 = 2-10-1 = 0>2%
* рТ = —lg [Н+ ] = рН, где [Н+ ] — концентрация ионов водорода определяемой
кислоты, при которой наблюдается наиболее резкое изменение окраски индикатора (см.
§22).
§ 24. ОШИБКИ ТИТРОВАНИЯ 155
ОН~р-о шибка. Предположим, что для титрования сильной кислоты сильным
основанием выбран индикатор с рТ = 9 (например, фенолфталеин). В этом случае
титрование заканчивается при рН = рТ = 9, т. е. при [Н+ ] = 10"9 г-ион/л-(в щелочной среде).
Следовательно, при титровании будет прилит некоторый избыток щелочи, что приведет
к ОН~ -ошибке. Вычислим ее величину.
Концентрация щелочи к концу титрования составит:
1П-14
[ОН-] = ш_9 = Ю-6 г-ион/л, так как [Н+] [ОН"] = Kw = Ю~14
или
[он-] = ю-(14-рТ)
ш-(14-рТ)
В У2 мл раствора [ОН" 1 = т^т V2 г-же щелочи.
Эта величина и составляет гидроксильную ошибку титрования, обусловливаемую
перетитрованием кислоты щелочью.
В процентах:
10-(14-рТ) у
ОН~р-ошибка = NVi V* lOOo/o (55)
В нашем примере (при Vi = 25 мл, V* = 50 мл, рТ = 9):
1О-6.^П
ОНпер-ошибка = ^ ^ 100 = 2-10"» = 0,02о/0
Это означает, что при титровании сильной кислоты сильным основанием в присутствии
метилового оранжевого изменение окраски индикатора наступает раньше, а при титровании
в присутствии фенолфталеина (при условии отсутствия в растворе Н2СО3) — после
достижения точки эквивалентности, причем ошибка титрования при использовании
фенолфталеина в 10 раз меньше ошибки титрования с индикатором метиловым оранжевым.
Так как величина концентрации титруемого раствора входит в знаменатель дроби,
то ошибка титрования будет тем больше, чем менее концентрированный раствор титруют.
Поэтому, чтобы избежать больших ошибок титрования, не следует
титровать слишком, разбавленные растворы очень разбавленными титран-
тами.
Влияние области интервала перехода индикатора на величину ошибки
титрования. При титровании оснований в присутствии индикатора
метилового оранжевого (рТ = 4) необходимо не только прибавить требуемое
по расчету количество сильной кислоты, но и добавить некоторый ее
избыток, для того чтобы окраска индикатора изменилась от желтой к
оранжево-красной.
Если Nhc\ =0,1, a V2 = 50 мл, то этот избыток (Vhci) вычисляют по
формуле:
Vuci= *НС1 =1о^ = о,05^
При рТ = 3 эта величина возрастает до 0,5 мл. Такая ошибка
совершенно недопустима. Поэтому, чтобы избежать слишком больших ошибок
титрования, не следует применять при титровании оснований
индикаторы, интервалы перехода которых лежат ниже 3—4 (рТ < 4).
При титровании кислоты в присутствии фенолфталеина (рТ = 10)
необходимо прибавить некоторый избыток щелочи, чтобы индикатор из
бесцветной формы перешел в окрашенную. Избыток VNa0n находят
по формуле:
При рТ, равном 11 и 12, эта величина возрастает до 0,5 и 5 мл. Такая
ошибка совершенно недопустима.
156 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Поэтому, чтобы избежать слишком больших ошибок титрования, не
следует применять при титровании кислот индикаторы, интервалы
перехода которых лежат выше 10—11 (рТ >> 10).
Гидроксильная ошибка титрования.
Предположим, что для титрования сильного основания сильной кислотой выбран
индикатор с рТ = 10; концентрация титруемого основания N = [ОН~] =
= 0,1 г-ион/л; начальный объем основания V х — 25 мл; объем в конце
титрования V2 = 50 мл.
Каждый миллилитр 0,1 н. раствора основания содержит QQQ г-экв.
Для титрования взято 1Q0Q V г-экв основания. Титрование заканчивают
при рН = рТ = 10, т. е. [Н+ ] = 10~10 г-ион/л (в щелочной среде);
следовательно, рОН = 14 — рТ, т. е. [ОН~ ] = 10~4 г-ион/л. Таким образом,
часть титруемой щелочи будет недотитрована, и мы допустим ОН^ед-
Ьшибку. Вычислим ее величину.
Концентрация ионов гидроксила в конце титрования составит
Ю-4 г-ион/л или ЮН"] = 10~<14-рт>.
10-(14-рТ)у,
В V" мл раствора [ОН" ] = г™ г-экв основания.
Эта величина и составляет гидроксильную ошибку титрования,
обусловленную недотитрованием ОН "-ионов.
Величину ОН "-ошибки (в процентах) вычисляют по формуле:
10-(14-рТ)у*
;ннед"ошиока = -
В нашем примере
ОН-д-ошибка = NV, 100% (56)
ОН-д-ошибка =|д=г^- ЮО = 0,2о/0
Если выполнять титрование в присутствии индикатора с рТ = 4, то
необходимо прибавить некоторый избыток кислоты.
Водородная ошибка титрования. Предположим, что
для титрования сильного основания сильной кислотой выбран
индикатор срТ = 4. Титрование заканчивают при [Н + ] = 10"4 = 10~рт г-ион/л.
до-—рТу*
[Н+ ] = —|щ—составляет водородную ошибку титрования,
обусловленную перетитрованием щелочи кислотой.
В процентах
Н+р -ошибка = Ш NV/ ЮОо/о (57)
Если V = 25 мл, V" — 50 мл9 то в этом случае получим:
1П~4.50
Н+р-ошибка = ?L^L 100 = 0,2о/0
Кислотная ошибка титрования. Предположим, что
для титрования дана слабая кислота НАп с /Снап = Ю"6, р/Сндп = 5.
Титрование ведут в присутствии индикатора с рТ = 10. В этом случае
[Н+] [An-]
А НАп ~ [НАп]
или
[НАп] [Н+]
[An-] KHAn
§ 24. ОШИБКИ ТИТРОВАНИЯ 157
Так как [НАп] для слабого электролита равна Снап, a [An"" ] = CKtAn ,
то можно написать:
[НАп-] С
[An-] ~ С
НАп
KtAn
Титрование заканчивается при Ш+] = 10^рт; Кнап = 10-р*,
следовательно, HAn-ошибка равна:
НАп-ошибка = т^Ап ^ HT^L = iqp*-pT (58)
Отношение Wjpf можно рассматривать как отношение концентраций
неоттитрованнои части кислоты к оттитрованной ее части и считать это
отношение критерием кислотной ошибки титрования.
В нашем примере НАп-ошибка = -г^р- = 10"5 или 10"3%.
При титровании той же самой кислоты в присутствии индикатора
с рТ = 4 соответствующая ошибка увеличилась бы в 1 млн. раз:
Ю-4
ю-
НАп-ошибка = -fTpg- • 100 = 103%
Пример. Вычислите ошибку титрования 0,1 н. раствора СНзСООН (р/С = 4,74) 0,1 н.
раствором NaOH в присутствии индикатора метилового оранжевого (рТ = 4):
НАп-ошибка = ЮР*^РТ = 104-74~4 = 100'74 = 5,5
Следовательно, на каждую оттитрованную молекулу СНзСООН приходится 5,5 неот-^
титрованных.
Отношение [СНзС00НД --*?-_ Б 5
0X110111611116 [сн3соо-] ~ НГ ~ 5*5
(1 + 5,5) — 100%
5,5 — х
Это значит, что 85% кислоты будет не оттитровано, если титровать СНзСООН
раствором NaOH в присутствии метилового оранжевого. Следовательно, титровать СНзСООН
с индикаторами, имеющими рТ < 7, нельзя.
Щелочная ошибка титрования. Предположив, что
для титрования дано слабое основание КЮН, щелочную ошибку
титрования вычисляют аналогично кислотной ошибке:
КЮН-ошибка = -$S!OH = [КЮН]_ = jOH^ ^ JO^^ = 1()Р«+рт-14
CKtAn lKt+] ККЮИ 10-P^
Пример. Вычислите ошибку титрования 0,1 н. раствора аммиака (р/С = 4,74) 0,1 н.
раствором НС1 в присутствии индикатора фенолфталеина (рТ = 9):
КЮН-ошибка = ю4»74+9-14 = 10~0'26 =0,55
Следовательно, на каждую оттитрованную молекулу ЫНз приходится 0,55
неоттитрованнои:
(1 + 0,55) — 100
0,55 — *
*=^й»=«
Это значит, что около 35% аммиака будет не оттитровано, если его титровать
раствором НС1 в присутствии фенолфталеина. Следовательно, титровать раствор NHs с
индикатором, имеющим рТ > 7, нельзя.
158 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Б. ПРАКТИЧЕСКАЯ ЧАСТЬ
§ 25. Организация рабочего места
Каждому студенту в лаборатории количественного анализа отводится
постоянное рабочее место и отдельный шкаф для хранения посуды и
реактивов. Перед началом экспериментальной работы в лаборатории
объемного анализа необходимо:
1) получить химическую посуду и измерительные приборы;
2) вымыть посуду и пропарить мерные колбы, пипетки и бюретки
(см. гл. I, § 4—7);
3) проверить емкость мерных колб и пипеток и правильность
калибрования бюреток (см. гл. I, § 4—7).
4) определить массу бюкса;
5) определить объем капли дистиллированной воды, вытекающей из
бюретки (см. гл. I, § 7);
6) приготовить и установить стандартные (титрованные) растворы
кислоты и щелочи (см. гл. I, § 8).
Взвешивание бюкса. Бюкс перед взвешиванием должен быть
тщательно вымыт, высушен в сушильном шкафу и охлажден до температуры
весовой комнаты. Предварительно следует определить приблизительную
массу бюкса, взвешивая его на технических весах, а затем определить его
точную массу на аналитических весах. Правила взвешивания на
аналитических весах изложены в «Введении» (§ 9). Результаты взвешивания
записывают в лабораторный журнал.
§ 26. Приготовление стандартных (титрованных) растворов
Исходные вещества, применяемые для приготовления стандартных
(титрованных) растворов кислот и оснований. Для титрования оснований
необходимо иметь стандартный раствор кислоты.
Чаще всего применяют, хлористоводородную кислоту. Ей отдают
предпочтение перед другими кислотами главным образом потому, что
хлористоводородная кислота является сильной кислотой и почти все ее
соли хорошо растворимы в воде. В тех случаях, когда приходится
нагревать или кипятить растворы, лучше брать серную и хлорную кислоты.
При титровании неводных растворов применяют преимущественно
хлорную кислоту.
Для титрования кислот необходимо иметь стандартный раствор
сильного основания. Наиболее широкое применение находят титрованные
водные растворы едкого натра или неводные растворы едкого кали, эти-
лата натрия, гидроокиси тетраэтиламмония.
Концентрации стандартных растворов могут изменяться от 0,01 н.
до 0,2 н. Иногда применяют 0,5 и 1 н. растворы.
Приготовление стандартных растворов. Техника приготовления
стандартного раствора сводится к следующему.
1. Если исходная кислота или щелочь представляет собой жидкость,
то измеряют ее плотность. Определив плотность жидкости, находят по
справочным таблицам соответствующее ей процентное содержание данного
вещества.
2. Концентрированные растворы предварительно разбавляют
приблизительно до требуемой нормальности, а затем устанавливают их
точную концентрацию при помощи установочного вещества или другого
титрованного раствора (см. гл. I, § 8).
§ 26 ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ (ТИТРОВАННЫХ) РАСТВОРОВ 159
3. Если исходная кислота или основание представляют собой
твердое вещество, то для приготовления стандартных растворов
соответствующие навески исходных веществ растворяют в требуемом объеме
дистиллированной воды.
Определение плотности раствора исходной кислоты или щелочи.
Плотность (р) — величина, численно равная массе, заключенной в единице
объема вещества, т. е. отношение массы тела (g) к его объему (V) —
выражается в г/смг или г1мл\ р = -|- . Плотность определяют при помощи
пикнометра или ареометра.
Определение плотности жидкости при помощи
ареометра. Ареометр (рис. 46) представляет собой полый,
герметически закрытый цилиндрический стеклянный
поплавок с нанесенными на нем метками. В нижней части
ареометра помещен груз (дробь), благодаря чему ареометр,
погруженный в жидкость, удерживается в вертикальном
положении. По глубине погружения ареометра судят
о плотности жидкости. Применение ареометра основано
на законе Архимеда.
Для определения плотности жидкости при помощи
ареометра исследуемую жидкость, выдержанную при
определенной температуре, наливают в сухой высокий
стеклянный цилиндр емкостью 250 мл. В жидкость погружают
сухой ареометр со шкалой, соответствующей плотности
данной жидкости. Как только ареометр установится
на определенном уровне, отмечают по мениску деление
шкалы, соответствующее уровню исследуемой жидкости
в цилиндре. Отсчет по шкале ареометра проводят сверху
вниз с точностью до третьего знака.
При пользовании ареометрами соблюдают следующие
правила:
1. Во избежание излишней потери времени на подбор
требуемого ареометра предварительно грубо определяют
плотность жидкости при помощи менее чувствительного
ареометра, отличающегося от других более широкой
шкалой (от 1 до 1,8 г/см3). Затем измеряют "плотность при
помощи ареометра, имеющего более узкую шкалу
(например, от 1,200 до 1,400 г/см3).
2. Жидкость не наливают до самого верха цилиндра,
чтобы избежать переливания ее из цилиндра на стол
при погружении ареометра. Одновременно наблюдают за тем, чтобы цилиндр был
наполнен жидкостью настолько, чтобы опущенный в нее ареометр не касался дна цилиндра.
3. Температура жидкости должна быть 20Q С, т. е. соответствовать указанной на
шкале ареометра.
4. Во время отсчета по шкале ареометр не должен вращаться и касаться стенок
цилиндра.
5. Отсчет показаний ареометра проводят по верхнему делению шкалы, выступающему
на уровне поверхности жидкости.
В СССР приняты ареометры со шкалой, непосредственно показывающей значения
плотности в г/см3 (денсиметры). Денсиметрами определяют плотности жидкостей легче
и тяжелее воды в пределах от 0,650 до 2 г/см3. Шкала денсиметров градуирована при 20Q С.
Рис. 46. Положение
ареометра в цилиндре с раствором
и отсчет по шкале ареометра.
Разбавление концентрированных растворов. Работа с
концентрированными растворами кислот и щелочей весьма неудобна, так как может
происходить разбрызгивание, вызывающее ожоги, порчу одежды
и т. д.
При необходимости разбавить концентрированную кислоту или
щелочь следует соблюдать все предосторожности, рекомендуемые в
подобных случаях. В первую очередь нужно помнить, что при разбавлении
концентрированных растворов кислот или щелочей их вливают в воду,
а не наоборот.
160 ГЛ.П. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОбНОВНОЕ ТИТРОВАНИЕ
§ 27. Приготовление 0,1 н. раствора
хлористоводородной кислоты
Обычно титрованные растворы хлористоводородной кислоты
приготавливают разбавлением рассчитанного количества исходной
концентрированной хлористоводородной кислоты известной концентрации в
дистиллированной воде. Затем устанавливают титр полученного раствора. Если
нужно приготовить точно 0,1 н. раствор, то его приготавливают сначала
несколько более концентрированным. После установки титра
приблизительно 0,1 н. раствора его разбавляют рассчитанным количеством воды и
снова устанавливают титр. Чаще всего готовят приблизительно 0,1 н.
раствор НС1 и ограничиваются точной установкой его нормальности.
Для выполнения предусмотренных планом учебных и контрольных работ по методу
нейтрализации требуется около 2 л стандартного раствора НС1. Поэтому все
вычисления необходимо произвести исходя из расчета приготовления 2 л 0,1 н. раствора НС1.
Количество хлористоводородной кислоты, необходимое для
приготовления 2 л 0,1 н. раствора НС1, рассчитывают следующим образом.
Предположим, что плотность исходного раствора НС1, определенная
при помощи ареометра, равна 1,179 г/см3. По справочной таблице находят,
что раствор НС1 такой плотности соответствует 36 весовым процентам НС1.
Сначала определяют, сколько граммов 100%-ного НС1 требуется для
/ мНС1 \
приготовления 2 л 0,1 н. раствора (Эна = —|— = 36,461.
Для приготовления 2 л 0,1 н. раствора нужно взять 36,46«0,1-2 =
= 7,292 г НС1.
Затем вычисляют, сколько граммов 36%-ной хлористоводородной
кислоты требуется для той же цели. Для этого составляют пропорцию:
100 г —36 г I 7,292-100
гнс1-7>292г| g™~ 36 г
Так как концентрированную хлористоводородную кислоту неудобно
взвешивать, то определяют, какой объем (V) занимают ga 36% -ной кислоты:
т/ S Т7 ?hci 7,292-100 t„10
Таким образом, для приготовления 2 л 0,1 н. раствора НС1 необходимо
отмерить при помощи мерного цилиндра (емкостью 20—25 мл) или бюретки
около 17,2 мл хлористоводородной кислоты (р = 1,179 г/см3) и разбавить
ее в дистиллированной воде до объема 2 л. Полученный раствор следует
тщательно перемешать, а затем установить титр кислоты.
При хранении растворов на стенках сосудов над поверхностью
жидкости конденсируются капельки воды. Конденсирующаяся вода должна быть
смешана с остальным раствором перед его употреблением.
§ 28. Установка титра 0,1 н. раствора
хлористоводородной кислоты
Способы установки титра раствора хлористоводородной кислоты.
Для установки титра раствора НС1 применяют весовой или объемный
способ.
Следует отметить, что установка титров весовым способом отнимает
больше труда и времени, чем установка объемным способом. Поэтому
применяют обычно объемный метод.
§ 28. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА НСГ
161
Объемный способ. Объемные способы установки титра
основаны:
1) на титровании определенного объема кислоты раствором щелочи
или
2) на титровании кислотой титрованных растворов установочных
веществ, обладающих основными свойствами (см. гл. I, § 8).
Объемные способы установки не уступают по точности весовому и
не требуют для своего выполнения много труда и времени. Поэтому
объемные способы установки титра предпочитают весовому способу.
Установочные вещества, применяемые для установки титров
стандартных растворов кислот. В качестве установочных веществ, применяемых
для установки титров стандартных растворов кислот, используют
разнообразные химически чистые соединения точно известного состава,
реагирующие с кислотами в строго определенных стехиометрических
соотношениях. Чаще всего для этой цели применяют:
1) безводный карбонат натрия *, Na2C03;
2) тетраборат натрия **, кристаллизующийся с 10 молекулами воды,
Na2B4O7-10H2O;
3) оксалат натрия, Na2C204, термически разлагаемый с образованием
карбоната натрия, который затем титруют кислотой;
4) окись ртути (II);
5) хлорид натрия, подвергающийся ионному обмену на катионитах
в Н-форме с образованием хлористоводородной кислоты;
6) дифенилгуанидин, HN= С (NHC6H5)2 и др.
Установка титров стандартных растворов кислот посредством
перечисленных выше установочных веществ основана на следующих реакциях:
По карбонату натрия. Водный раствор карбоната натрия
вследствие его гидролитического разложения под влиянием ионов воды
имеет сильно щелочную реакцию:
СО" ~ + НОН ^ HCOg" + ОН"
НСОз + НОЙ ^ Н2С03 + ОН"
При титровании карбоната натрия кислотой гидролиз протекает
полностью в сторону образования угольной кислоты, разлагающейся на
воду и двуокись углерода. Поэтому суммарное уравнение реакции
титрования карбоната натрия хлористоводородной кислотой можно
представить следующим уравнением:
Na2C03 + 2HC1 —> 2NaCl + НаС03
*H20 + (XV
Эквивалент карбоната натрия в этом случае равен половине его
молекулярного веса:
э Мыаасо._ Ю5,989
^Na2CO, ~ 2 ~~ 2 """ °*>*т
* Очень часто установку титра кислоты по карбонату натрия называют
«установкой рабочего титрованного раствора кислоты по соде». Употребление названия «рабочий
раствор» не оправдано, так как все употребляемые химиками растворы являются
«рабочими». Замена термина «карбонат натрия» словом «сода» недопустимо, так как под
названием «сода» подразумевают технический карбонат натрия, содержащий многие посторонние
примеси: хлориды, сульфаты, бикарбонаты, гидроокись натрия, воду и т. п.
** Замена термина «тетраборат натрия» термином «бура» недопустима, так как под
названием «бура» подразумевается технический тетраборат, содержащий примеси других
веществ.
162 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
По тетраборату натрия. Водный раствор тетрабората
натрия (подобно карбонату натрия) имеет сильно щелочную реакцию.
Титрование кислотой тетрабората натрия можно представить следующим-
суммарным уравнением реакции:
Na2B407.10Н2О + 2HC1 —* 2NaCl + 4Н3В03 + 5Н20
Эквивалент тетрабората натрия в этом случае равен половине его
молекулярного веса:
Q _ MNa2B4O7.10H2O _ 381,37 _ inn gn
^Na^Oj-lOHjiO — 2 ~~ 2 ~~ iyU'Dy
По оксалату натрия. Оксалат натрия непосредственно не
титруется кислотой, так как его водный раствор не обладает сильно
щелочной реакцией подобно карбонату или тетраборату натрия. Его
предварительно переводят в карбонат натрия путем нагревания в платиновом
тигле. Образующийся при этом карбонат натрия термически не
разлагается даже при температуре плавления и может быть использован для
установки титра кислоты. Уравнения протекающих при этом реакций
можно представить следующим образом:
Na2C204 -С Na2C03 + СО
СОз"+2Н+-^Н2С03
Эквивалент оксалата натрия в данном случае равен половине его
молекулярного веса:
q __ ^Na2C2Q4_ 134,000 *7QQQ
3Na2C204 = § 2 67ДЮ
По окиси ртути (II). Окись ртути (II) является очень подходящим
установочным веществом для стандартизации кислот, так как ее легко
получить в химически чистом виде, однако она практически не растворяется
в воде. Поэтому из нее нельзя приготовить установочный раствор путем
растворения ее в воде. Ее нельзя непосредственно использовать для
установки титра кислоты:
HgO + 2Н+ ^± Hg++ + Н20
так как реакция обратима. Для приготовления раствора окиси ртути ее
предварительно растворяют в нейтральном растворе йодида или бромида
калия. При этом раствор окиси приобретает сильно щелочную реакцию
вследствие связывания ионов ртути в комплекс, а ионов кислорода в гидро-
ксильные группы:
HgO + 21- + НОН —> Hgl2 + 20Н-
HgI2 + 2I-^[HgI4]"
Образующиеся при этом ионы гидроксила титруют кислотой.
Эквивалент окиси ртути (II) в данном случае равен половине ее
молекулярного веса:
а _ ^Hgo _ 216,59
dHgo = 2— —2~ '
Установка стандартных (титрованных) растворов кислот с помощью
катионитов. Наряду с описанными методами установки титров в
последнее время стали устанавливать стандартные растворы кислот с помощью
§ 28 УСТАНОВКА ТИТРА 0.1 н. РАСТВОРА HCI
163
катионитов. Метод основан на пропускании через колонку, заполненную
катионитом в Н-форме, раствора определенного количества химически
чистой соли (например, NaCl), которая подвергается ионному обмену
согласно уравнению:
R—Н + NaCl ^± R—Na + HC1
Образующуюся при этом в эквивалентном количестве по отношению
к взятой навеске соли (NaCl) кислоту вымывают из колонки, собирают
в мерную колбу и доводят соответствующим растворителем до метки.
Таким путем получают стандартный раствор кислоты, свободный от
посторонних примесей. При этом отпадает необходимость дополнительной
установки титра полученной кислоты с помощью установочных веществ.
Установка стандартных (титрованных) растворов кислот по карбонату
натрия. Безводный химически чистый карбонат натрия является наиболее
доступным установочным веществом для стандартизации растворов кислот.
Для приготовления безводного карбоната натрия применяют:
а) высушивание в сушильном шкафу в течение 1 ч при 250—270° С
исходного х. ч. препарата;
б) нагревание в платиновом тигле на песочной бане гидрокарбоната
(бикарбоната) при 250—270° С до постоянной массы:
2NaHC03 —> Na2C03 + H20 + С02
в) термическое разложение в платиновом тигле х. ч. оксалата натрия
Na2C204 —^ Na2C03 + СО
Водный раствор Na2C03 имеет щелочную реакцию.
Установка титра кислоты при помощи карбоната натрия основана
на реакции:
Na2C03 + 2Н+ -^ 2Na+ + Н20 + СОа
Приготовление 0,1 н. раствора карбоната
натрия. Для установки титра 0,1 н. раствора кислоты точно отвешивают
рассчитанное количество карбоната натрия (в г):
_3Na2C(V250 _^Na2co3-25Q_ 105,989 _ t 00,Q
flNa2co8- 10-1000 "" 2.10-1000 "" 2-10.4 "млв
Навеску растворяют в воде в мерной колбе емкостью 250 мл.
Следует иметь в виду, что нет никакой необходимости брать точно
рассчитанную навеску Na2C03. Вполне достаточно предварительно
взвесить на технических весах навеску, приближающуюся по своей массе
к 1,3248 г, и затем взвесить ее на аналитических весах. В зависимости от
взятой навески вводят соответствующий поправочный коэффициент:
v __ апракт
ЯТеор
Для установки стандартного раствора кислоты из мерной колбы
отбирают пипеткой аликвотные части приготовленного раствора карбоната
и титруют их приготовленным раствором кислоты.
Вместо приготовленного раствора можно титровать устанавливаемым
раствором кислоты уменьшенные в 10 раз навески сухого карбоната.
Титрование проводят в присутствии метилового оранжевого,
желательно в присутствии раствора — «свидетеля», содержащего NaCl и С02.
Карбонат натрия используют не только для установки титров водных
растворов кислот, но и для стандартизации неводных растворов кислот.
Для этого рассчитанную навеску химически чистого карбоната натрия
164 ГЛ. И. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
помещают в фарфоровую чашку, прибавляют несколько капель безводной
уксусной кислоты. Полученный раствор выпаривают досуха на водяной
бане, остаток образовавшегося ацетата натрия растворяют в нужном
растворителе, переносят в мерную колбу, доводят объем раствора до метки
и тщательно перемешивают.
Затем отбирают аликвотную часть раствора и титруют неводным
раствором кислоты. Чаще всего для этой цели применяют хлорную кислоту
НСЮ4. В качестве растворителей при установке титров обычно
применяют те же растворители, которые используют для последующих
количественных определений.
Установка титра 0,1 н. раствора хлористоводородной кислоты по
тетраборату натрия. Установка титра хлористоводородной кислоты по
тетраборату натрия имеет ряд преимуществ по сравнению с другими
способами:
1. Установка титра хлористоводородной кислоты по тетраборату
может быть выполнена с метиловым оранжевым или метиловым красным.
Установку же титра кислоты по карбонату проводят обычно в присутствии
метилового оранжевого.
2. Тетраборат натрия (х. ч.) при хранении в плотно закрытой банке
в течение долгого времени не изменяет своего состава. Карбонат натрия
легче изменяет свой состав под влиянием С02 и влаги воздуха.
3. Тетраборат натрия имеет более высокий эквивалентный вес (см.
стр. 162) по сравнению с эквивалентным весом карбоната натрия (см.
стр. 161).
Поэтому тетраборату натрия отдается предпочтение при установке
титров кислот.
К существенным недостаткам тетрабората натрия относится его плохая
растворимость в воде и легкость, с которой он теряет части
кристаллизационной воды при неправильном хранении, вследствие чего декагидрат
(десятиводная соль) Na2B407- 10H2O превращается в пентагидрат (пяти-
водную соль) Na2B407-5H20. Однако этот недостаток тетрабората натрия
легко устраним.
Если кристаллический тетраборат натрия не отвечает составу
Na2B407- 10H2O, то его тщательно перекристаллизовывают перед
употреблением.
Приготовление 0,1 н. раствора тетрабората
натрия. Установку титра кислоты по х. ч. тетраборату натрия можно
выполнить:
а) методом пипетирования, т. е. титрованием аликвотных частей
стандартного раствора тетрабората натрия приготовленным
приблизительно 0,1 н. раствором НС1;
б) титрованием кислотой отдельных точных навесок установочного
вещества, рассчитанных на одно титрование (см. гл. I, § 8).
Оба способа имеют свои преимущества и недостатки. Так как
тетраборат натрия плохо растворяется в воде, иногда предпочитают второй
способ титрования или растворяют тетраборат в горячей
дистиллированной воде. После полного растворения Na2B4O7-10H2O в горячей воде
раствор охлаждают и затем доводят водой до определенного объема.
а) Установка титра хлористоводородной
кислоты по тетраборату натрия методом
пипетирования. Величина навески тетрабората натрия, рассчитанная при
приготовлении 250 мл 0,1 н. раствора для многократного титрования
аликвотных (кратных) частей, должна быть равна (см. гл. I, § 8, 10):
__3NV 1П 190,69-0,1-250 _АПап
§ 28. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА НС1
165
Для приготовления 250 мл 0,1 н. раствора тетрабората натрия
необходимо взвесить около 4,767 г Na2B407' 10H2O. Отвесить на
аналитических весах в точности теоретически рассчитанное количество соли
очень трудно. Поэтому сначала взвешивают в бюксе на технических весах
с точностью до первого десятичного знака 4,5—5 г Na2B407-10Н2О.
Затем бюкс переносят на аналитические весы и взвешивают с точностью
до четвертого десятичного знака*. Взвешенный тетраборат осторожно
пересыпают через сухую воронку с широкой и короткой трубкой в мерную
колбу емкостью 250 мл с сухим горлом.
Кристаллы Na2B4O7-10H2O, оставшиеся в бюксе и на воронке,
тщательно смывают в колбу горячей водой из промывалки. В колбу через
ту же воронку добавляют около 150 мл горячей дистиллированной воды.
Затем содержимое колбы осторожно перемешивают плавными
вращательными движениями. После того как весь тетраборат растворится в воде,
колбу охлаждают до комнатной температуры и доводят объем полученного
раствора холодной дистиллированной водой до метки. Колбу закрывают
пробкой. Приготовленный раствор тщательно перемешивают. После
этого раствор тетрабората натрия готов для титрования.
Всю работу с мерной колбой выполняют, строго соблюдая все
правила обращения с нею (см. гл. I, § 5).
Отбор проб для титрования. Для титрования берут при помощи
пипетки 25 мл приготовленного раствора тетрабората натрия. Для этого
оттянутый конец пипетки опускают в мерную колбу почти до дна и
засасывают 10—15 мл раствора. Затем закрывают верхнее отверстие пипетки
указательным пальцем и вынимают ее из колбы. Слегка наклоняя
пипетку в разные стороны, ополаскивают ее раствором. Использованный
раствор отбрасывают. Снова набирают в пипетку 25 мл раствора и
выливают его в коническую колбу емкостью 200—250 мл. Пробы отбирают
параллельно в три колбы.
Всю работу с пипеткой выполняют, строго соблюдая все правила
обращения с нею (см. гл. I, § 6).
Титрование аликвотных частей раствора тетрабората натрия
соляной кислотой. Титрование тетрабората натрия как соли, образованной
катионом сильного основания и анионом слабой кислоты, ведут в
присутствии метилового красного, интервал перехода которого 4,4—6,2
(см. гл. II, § 22).
Для более точного титрования применяют «свидетель». Для
приготовления «свидетеля» используют 100 мл буферного раствора с рН =
= 5,1—5,2 или 100 мл сильно разбавленного (1 : 100) раствора борной
кислоты, к которой добавляют 2—2,5 мл 1 н. раствора NaCl и 1—2 капли
индикатора. Раствор «свидетеля» должен быть окрашен в оранжевый цвет.
Бюретку емкостью 25—50 мл, снабженную стеклянным краном,
укрепляют в штативе, заботясь о том, чтобы она приняла правильное
вертикальное положение. Если бюретка была перед употреблением
заполнена доверху водой, то сначала сливают воду. Во время работы с бюреткой
следят за тем, чтобы ее кончик оставался заполненным раствором. Сначала
2—3 раза бюретку ополаскивают 10—15 мл приготовленного раствора НС1,
который затем выливают. Потом бюретку наполняют раствором кислоты
несколько выше нулевого деления. Лишь перед самым началом
титрования устанавливают уровень кислоты на нулевое деление, осторожно
сливая избыток кислоты из бюретки в подставленный стакан.
* Навеску можно также брать в пробирке для взвешивания или в бюксе методом двух
взвешиваний (по разности).
166 ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Всю работу с бюреткой выполняют, строго соблюдая правила
обращения с ней (см. гл. I, § 7).
После окончания всех приготовлений приступают к титрованию.
Для этого в каждую приготовленную ранее коническую колбу,
содержащую раствор тетрабората натрия, приливают по 1—2 капли метилового
красного. Первую колбу подставляют под бюретку. Под колбу подкла-
дывают лист белой бумаги. Кончик бюретки опускают в горло колбы
на 1—2 см. Придерживая бюретку в нижней части левой рукой, осторожно
открывают правой рукой кран бюретки. При этом кислота начинает
понемногу выливаться из бюретки в колбу.
По мере вытекания раствора из бюретки содержимое колбы
непрерывно перемешивают плавными круговыми движениями. Титрование
прекращают в тот момент, когда прибавление очередной капли раствора
кислоты вызывает появление оранжевой окраски индикатора, похожей
на окраску «свидетеля».
В случае сомнения в правильности окончания титрования прибегают
к следующему приему. Записывают показания бюретки, добавляют в
коническую колбу еще каплю титрованного раствора и наблюдают за
изменением окраски. Если раствор покраснеет, значит тцтрование было
закончено. В противном случае продолжают титрование до момента изменения
окраски индикатора.
После первого титрования проводят второе и третье титрования.
Последующие титрования проводят быстрее первого. Во время титрования
следят за тем, чтобы не происходило разбрызгивания раствора. Нельзя
допускать появления на внутренних стенках колбы капель раствора.
В случае их появления капли смывают дистиллированной водой из про-
мывалки. Разбавление раствора не отражается на результатах
титрования. Необходимо следить также за тем, чтобы капли раствора не
оставались висеть на кончике бюретки, так как объем висящей капли войдет при
расчете в общий объем раствора, израсходованного на титрование, и это
поведет к ошибочным результатам.
Порле окончания каждого титрования отсчитывают по шкале бюретки
объем израсходованной на титрование кислоты и снова заполняют бюретку
раствором кислоты до нулевого деления. Результаты титрования
записывают в рабочий журнал.
Если результаты трех титрований не расходятся или отличаются на
несколько сотых долей миллилитра, титрование считают законченным.
Если расхождения превышают сотые доли миллилитра, то повторяют
титрование до тех пор, пока не получат по крайней мере три не
расходящихся результата. Сильно отличающиеся результаты титрования в расчет
не принимают. Окончательный расчет проводят исходя из среднего
арифметического близкосходящихся результатов титрований.
Вычисление Nhcu Кнс\ и Thci no данным титрования
хлористоводородной кислотой аликвотных частей раствора тетрабората натрия.
Расчеты проводят на основании уравнений реакций (см. гл. I, § 10) и
исходят из NA или ТА. Например, зная NA, т. е. нормальность раствора
тетрабората натрик, можно вычислить NB — нормальность раствора
кислоты. Предположим, что навеска Na2B407-10H20, взятая для
приготовления 250 мл раствора, равна 4,8524 г. Следовательно
Г Т(практ) Na2B4O7.10H2O Дпракт _ 4,8524 . mQ
ANa2B4O7.10H2O = "-т ~* "Z — 4 7fi7 ~~ '
х(теор)Ыа2В407 10H2O ^теор «,/Ь/
Г(практ) Na2B4O7.10H2O — #Na2B4O7.10H2O* ^(теор) Na2B4O7-10H2O — 1э018-0э
§ 28. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА HC1
167
Допустим, что на титрование 25 мл раствора тетрабората натрия
израсходовано 20,07; 20,08; 20,06 мл раствора НС1. Среднее
арифметическое составит:
20,07 + 20 08 + 20.06 _ ^ ^
О
Так как произведения объемов (в мл) реагирующих между собой
растворов на их нормальности равны между собой, т. е.
Vb = Va
то
В нашем примере
25-0,1018
20,07
^НС1= 3 = 0,1268
„ ^(практ) НС 1 ,0,1268 OCQ
НС1 ^(теор) HC1 <U
Зная нормальность, можно вычислить Thci:
т Л'НС1ЭНС1 ,0,1268.36,46 _.0001623 г1ш
1НС1~~ ГШ ~ Гооо - °'004623 г'мл
Проделайте самостоятельно расчет по значению ТА (см. гл. I, § 10).
Пользуясь найденными величинами, можно проводить любые
вычисления в процессе объемных определений при помощи приготовленного
раствора НС1.
б) Установка титра хлористоводородной
кислоты по тетраборату натрия методом
отдельных навесок. Расчет навески тетрабората натрия. Для
титрования методом отдельных навесок необходимо брать такие навески
установочного вещества, чтобы на их титрование расходовалось 20—25 мл
0,1 н. раствора (см. гл. I, § 8). Это означает, что величина отдельной
навески Na2B407-10Н2О, рассчитанная только на одно титрование, должна
быть эквивалентна 20—25 мл 0,1 н. раствора тетрабората натрия. Расчет
навески выполняют по формуле
а = TV =
1000
Поэтому навеска тетрабората должна быть равна:
_ 190,69 -о,ь 25
%а2в4о7.юн2о - [обо * ' г
где 190,69 — грамм-эквивалент Na2B407- 10H2O.
Взятие отдельной навески. В сухую пробирку с притертой пробкой
и проволочной петлей для подвешивания к коромыслу аналитических
весов помещают около 1,5 г Na2B407-10Н2О. Для взятия такой навески
пробирку предварительно тарируют на технических весах, всыпают в нее
примерно требуемое для трех титрований количество тетрабората натрия
и взвешивают сначала на технических весах, а затем на аналитических.
После этого отсыпают около V3 содержимого пробирки в коническую
колбу емкостью 100—250 мл и прббирку снова взвешивают. Разность
в массе после первого и второго взвешиваний на аналитических весах
168 ГЛ. II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
1
а
"1
2
<*i
Яг
3
<h
аз
указывает массу отсыпанного Na2B407- 10H2O. Таким же образом
отсыпают вторую и третью навески во вторую и третью колбы. Колбы
предварительно нумеруют. Все результаты взвешиваний записывают в
рабочий журнал по следующей форме:
Масса пробирки с Na2B4O7-10H2O . .
До отсыпания Na2B4O7-10H2O . . . .
После отсыпания Na2B4O7-10H2O . . .
Навеска, г а—ах ах—а2 а2—а3
Взятые навески растворяют приблизительно в 50—100 мл
дистиллированной воды и титруют приготовленным 0,1 н. раствором НС1.
Титрование растворов, приготовленных из отдельных навесок.
Титрование растворов, приготовленных из отдельных навесок тетрабората
натрия, ведут в присутствии метилового красного, интервал перехода
которого 4,4—6,2 (см. § 22). Для более точного титрования применяют
«свидетель».
При титровании необходимо принять во внимание следующее.
Если взятые навески Na2B407-10Н2О еще полностью не
растворились в воде, то колбы нагревают на песочной бане, затем дают им
охладиться и добавляют в них столько же капель индикатора, сколько было
прибавлено в «свидетель». Установив уровень кислоты в бюретке на нуль,
приступают к титрованию. Титрование начинают с раствора,
приготовленного из самой маленькой навески, и кончают раствором, приготовленным
из самой большой. Такой порядок титрования позволяет заранее знать,
что при каждом последующем титровании пойдет большее количество
кислоты.
Вычисление THci> A^hci и ^Chci на основании данных титрования
хлористоводородной кислотой отдельных навесок тетрабората натрия.
Когда определение ведут методом отдельных навесок, то вычисления
проводят иначе, чем при расчете THci> NKCX и ^hci на основании данных
титрования аликвотных частей раствора. В этом случае нельзя применять
уравнение VBNB = VANA, так как неизвестными являются и VA и NA.
Поэтому исходят из соответствия эквивалентов НС1 и Na2B407-10Н2О
(см. гл. I, § 10):
9Na2B4O7-10H2O — ЭНС1
aNa2B4O7.10H2O *~~ ^НС1ТНС1
откуда
^HClgNa2B4O710HgO
Т„„, =
HC1 3Na2B4O7-10H2O^HCl
ЭНС1 = р
9Na2B4O7.10H2O
где F — аналитический множитель (фактор) объемного анализа.
Следовательно
Т — р gNa2B4O7.10H2O
НС1' ^
Величину Thci вычисляют для каждой навески отдельно. Значения
Whci и /Chci вычисляют по среднему значению Thci- Расчет может быть
также проведен следующим путем:
так как
9Na„B4O7.10H2O ~~ ЭНС1
§28. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА HCI
169
ТО
лЫа2В4О7-10Н2О ~~ ПНС1
где nNa в о.-юНоО — число грамм-эквивалентов Na2B407- ЮНгО во взятой навеске;
пНС] — число грамм-эквивалентов НС1.
Отсюда
_ gNa2B4O7-10H2O _ ^HCl^HCl _ ,
^а2В4О7-10Н2О - Щб9 "" Г000 нс^
Л/ _ gNa2B4O7.10H2O'1000
НС! 190,691/
HCI
Если при расчете величины THci оказываются равными или близкими
по значению, то это указывает на правильность результатов отдельных
титрований. Если величины THci сильно отличаются друг от друга, то
повторяют титрование до тех пор? пока для трех титрований не получат
близко сходящихся результатов. Величины, значительно отличающиеся
друг от друга, в расчет не принимают.
Чтобы судить о порядке точности определений, нет необходимости
проводить все вычисления полностью. Достаточно вычислить отношение
а/V, так как F — величина постоянная. Допускается расхождение этих
отношений друг от друга на 3 единицы в четвертой значащей цифре.
Полученные данные рекомендуется записывать в рабочий журнал по
форме:
№ навески
Величина
навески (а), г
Объем раство-
НС1 fv), мл
Отношение: -тт-
ТНС!~Г*в""
На основании данных титрования вычисляют среднее значение Thci*
Зная среднее значение THci» легко вычислить NHCl и Кнсх-
Nuc\ = •
'HCI
1000
^hci —
Nk
yHCl
(практ) HCI
(теор) HCI
N.
Предположим, что для установки титра раствора НС1 была взята
навеска Na2B407« 10H2O, равная 0,4680 г. На титрование ее было
израсходовано 24,20 мл приготовленного приблизительно 0,1 н. раствора НС1.
Следовательно
gNa2B4O7-10H2O 0,4680
'HCI
В нашем примере
F
1НС1
HCI/Na2B4O7.10H2O
%а2В4О7.10Н2О
= 0,01934 г
= 0,1912
'на
%С1 =
190,69
F = 0,01934-0,1912 = 0,003699 г/мл
0,003699-1000
36,46
= 0,1015
Vi = q| = !.015
vHCf
170 ГЛ. II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
§ 29. Приготовление 0,1 н. раствора едкого натра
Едкий натр (гидроокись натрия) представляет собой белое
непрозрачное очень гигроскопичное кристаллическое вещество, которое
расплывается на воздухе и легко реагирует с С02 воздуха. Поэтому состав NaOH
непостоянен и это соединение не годится в качестве установочного вещества
для приготовления титрованных растворов.
Для приготовления раствора NaOH, свободного от Na2C03,
применяют специальные методы.
Обычно титрованные растворы едкого натра приготавливают путем
разбавления дистиллированной водой рассчитанного количества
концентрированного раствора NaOH известной концентрации. Сначала
готовят приблизительно 0,1 н. раствор едкого натра, а затем точно
устанавливают нормальность этого раствора.
Расчет ведут аналогично расчету количества хлористоводородной
кислоты (см. § 27).
Для приготовления 2 л 0,1 н. раствора NaOH требуется 100%-ного
NaOH:
3NaOH0,l -2 = 40,00-0,1 -2 = 8 г
Предположим, что плотность (р) исходного раствора NaOH равна
1,285 г/см3 (26%).
Для приготовления 2 л 0,1 н. раствора NaOH нужно взять 26%-ного
его раствора:
ЮОг — 26г
#NaOH ~ 8г
8-100
?NaOH = '
26
Объем, занимаемый g граммами 26%-ного раствора NaOH (p = 1,285):
Р
^NaOH =1^5- = ЖИГ = 23'94 * 24 М
Таким образом, для приготовления 2 л 0,1 н. раствора NaOH следует
отмерить около 24 мл исходного раствора NaOH и раэбавить его
дистиллированной водой до объема 2 л. Полученный раствор необходимо тщательно
перемешать.
Приготовленный раствор едкого натра предохраняют от поглощения
СО2 из воздуха.
§ 30. Установка титра 0,1 н. раствора едкого натра
Вещества, применяемые для установки стандартных растворов
оснований. В качестве веществ, применяемых для установки стандартных
водных растворов щелочей, используют:
1) щавелевую кислоту Н2С204-2Н20 (х. ч.);
2) бензойную кислоту С6Н6СООН (х. ч.);
A-COOK
I—(
3) бифталат калия | |_/-qqu (КНС8Н404) (х. ч.), водный раствор
которого имеет кислую реакцию.
Установка титра раствора сильного основания при помощи бифталата
калия основана на реакции:
КНС8Н404 + NaOH-->KNaC8H404 + Н20
§ 30. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА ЕДКОГО НАТРА 171
Кроме указанных веществ, для установки титров щелочей применяют
янтарную кислоту НООС (СН2)2СООН, бииодат калия КН (Ю3)2, битар-
трат калия КНС4Н406 и др.
Установку титров щелочей, предназначенных для титрования с
фенолфталеином, лучше выполнять с помощью органических кислот;
Предпочтение отдается щавелевой и янтарной кислотам и бифталату калия.
Следует заметить, что при неправильном хранении дигидрата
щавелевой кислоты наблюдается потеря некоторого количества
кристаллизационной воды. Поэтому, если щавелевая кислота не соответствует
формуле Н2С204-2Н20, ее перекристаллизовывают.
Установку титра раствора NaOH выполняют так же, как и установку
титра раствора НС1 (см. § 28). Для этого применяют метод пипетирования
и метод отдельных навесок. Располагая 0,1 н. раствором НС1, можно
установить титр NaOH по хлористоводородной кислоте; титрованный раствор
NaOH можно приготовить из фиксанала (см. гл. I, § 8).
Установка стандартных (титрованных) растворов оснований с
помощью анионитов. За последнее время подобно установке стандартных
растворов кислот с помощью катионитов (см. § 28) стали устанавливать
стандартные растворы оснований с помощью анионитов. Ддя этого через
колонку, заполненную анионитом в ОН-форме, пропускают раствор
определенного количества химически чистой соли (например, NaCl). При
этом в результате реакции
R __ ОН + NaCl ^± R — CI + NaOH
образуется эквивалентное количество по отношению к взятой навеске
соли основания, которое вымывают из колонки, собирают в мерную колбу
и доводят соответствующим растворителем до метки.
Установка титра раствора едкого натра по щавелевой кислоте.
Щавелевая кислота диссоциирует по двум ступеням:
H2C204 ^=t H+ + НС20; (РКн2с2о< = 1,2)
НС2О^Н+ + С2ОГ (р/СНс2о4- = 4,3)
Оба значения рК щавелевой кислоты относительно мало отличаются
друг от друга. Поэтому при титровании водных растворов щавелевой
кислоты едким натром сразу нейтрализуются обе ее карбоксильные группы.
Следовательно, ее эквивалент равен половине молекулярного веса:
~ Л*н2с2о4-2н2о 125,067 AQnQ.
dH2C204-2H20 = 2 = 2 = 63'034
В точке эквивалентности при титровании 0,1 н. раствора щавелевой
кислоты 0,1 н. раствором едкого натра рН = 8,55, а при титровании
0,01 н. раствора рН = 8,05. Следовательно, лучшими индикаторами для
данного случая являются тимоловый голубой (интервал перехода 8,0—
9,6) и фенолфталеин (интервал перехода 8—10).
Присутствие угольной кислоты, способной реагировать с одним или
двумя эквивалентами сильного основания с образованием бикарбонатов
и карбонатов, мешает титрованию щавелевой кислоты, так как р/Сн2со8 =
= 6,43 и р/СнсоГ = Ю,24 не очень сильно отличаются от р/С2 щавелевой
кислоты. Поэтому желательно при приготовлении растворов щавелевой
кислоты пользоваться свежепрокипяченной дистиллированной водой,
свободной от С02.
Установку титра стандартного раствора едкого натра по щавелевой
кислоте проводят методом пипетирования и методом отдельных навесок.
172 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Установка титра раствора едкого натра по
щавелевой кислоте методом пипетирования.
Величина навески щавелевой кислоты для приготовления 250 мл
0,1 н. раствора должна быть равна:
3NV
#теор :
2(теор) H2C204-2H20 ~ '
~ 1000
63,03- 0,1- 250
1000
= 1,575 г
Навеску Н2С204-2Н20 (величину
навески рассчитывают аналогично тому,
как описано выше—см. стр. 167)
растворяют в холодной воде, не
содержащей С02. Всю работу с мерной колбой
выполняют, строго соблюдая все
правила обращения с нею (см. гл. I, § 5).
Для титрования аликвотных частей
отбирают в конические колбы
пипеткой по 25 мл приготовленного раствора
щавелевой кислоты. Операцию отбора
проб выполняют, как описано выше.
Всю работу с пипеткой выполняют,
соблюдая все правила обращения с нею
(см. гл. I, § 6).
Титрование щавелевой кислоты,
являющейся относительно слабым
электролитом (/Сн2с2о< = 5,9-10"2; Кнс261=
= 6,4• 10~5), ведут в присутствии
фенолфталеина, интервал перехода
которого 8,0—10,0 (см. § 22 и 23). Для
более точного титрования применяют
«свидетель», для приготовления
которого к 100 мл буферного раствора
с рН = 9 или 100 мл дистиллированной воды, не содержащей С02,
добавляют 2—2,5 мл 1 н. раствора Na2C204, 1 каплю 0,1 н. раствора NaOH
и 1—2 капЛи индикатора. Раствор «свидетеля» должен быть окрашен
в розовый цвет. Установка для титрования щелочами показана на
рис. 47.
В коническую колбу наливают приготовленный раствор щавелевой
кислоты, в бюретку — раствор едкого натра. Титрование ведут до
появления неисчезающей при перемешивании в течение 10—15 сек розовой
окраски.
Выполняют три параллельных титрования.
Вычисление Л^каон, Кыаон и Тыаон на основании данных титрования.
Вычисления проводят исходя из среднего арифметического близко
сходящихся результатов трех титрований по формулам (гл. I, § 10),
аналогично вычислениям величин Nncu Кна и THci (см. § 28):
Рис. 47. Установка для титрования
сильными основаниями:
/ — бутыль со стандартным раствором
NaOH; 2 — бюретка; 3 — трубки с
натронной известью; 4 — зажим; 5 — трубка
для отсасывания воздуха.
М
NaOH
^H2C2Q4 -2H2Q ^H2C2Q4 -2H2Q
^NaOH
К,
Nt
NaOH
(практ) NaOH
^(теор) NaOH
1 NaOH
^NaOH^NaOH
1000
§ 30. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА ЕДКОГО НАТРА 173
Установка титра раствора едкого натра по
щавелевой кислоте методом отдельных навесок.
Навеска щавелевой кислоты должна быть равна
тт/ 3NV 63,03-0,Ь25 П1К.Г
e-TV~IB00-e ШОО =0Д575г
Навеску берут так, как описано в § 28.
Титрование растворов, приготовленных из отдельных навесок
щавелевой кислоты, ведут в присутствии индикатора фенолфталеина, интервал
перехода которого 8,0—10,0 (си, § 22).
Вычисление Tn3oh, Nn3oh и Кыаон- При титровании отдельных
навесок расчет ведут по формулам (см. гл. I, § 10).
т ЭЫаОН gH2C2Q4-2H2Q _ п gH2C2Q4-2H2Q
rfH2C204-2H20 ^NaOH KNaOH
., TNaOH^QO „ %2C2O4.2H2O'1Q0°
^NaOH = —9 ИЛИ ^NaOH = ar ' лГ,
^NaOH C,H2C204-2H20 KNaOH
к ^(практ) NaOH K gH2C2Q4-2H2Q-10Q0
ANaOH — "~й ИЛИ ANaOH — 5 7v M
/v(Teop) NaOH C,H2C204-2H20 KNaOHyvNaOH(Teop)
Установка титра раствора едкого натра по 0,1 н. раствору
хлористоводородной кислоты. Установка титра раствора едкого натра по 0,1 н.
раствору НС1, предварительно установленному по тетраборату натрия, может
быть выполнена в присутствии различных индикаторов: метилового
оранжевого (3,1—4,4), метилового красного (4,4—6,2), фенолфталеина (8,0—
10,0) и т. д. (см. § 22). Так как в растворе едкого натра присутствуют
карбонаты, которые разлагаются при титровании кислотой с
образованием двуокиси углерода, влияющей на фенолфталеин, титрование лучше
проводить в присутствии метилового оранжевого или метилового красного.
При этом в коническую колбу отбирают пипеткой 25 мл NaOH,
прибавляют 1—2 капли индикатора и титруют из бюретки 0,1 н. раствором НС1
до появления оранжевой окраски.
При титровании «от щелочи к кислоте» в присутствии индикатора
метилового оранжевого допускается некоторая ошибка (см. § 24). Эта
ошибка вызывается тем, что лишние 2—3 капли 0,1 н. раствора кислоты
расходуются на подавление электролитической диссоциации
индикатора, являющегося относительно сильной кислотой. Эту ошибку можно
корректировать с помощью «холостого» опыта.
Установка титра раствора NaOH no HC1 относится к наименее
точным способам. Это объясняется тем, что многократное пользование
различными измерительными приборами, неточности, допускаемые при
установке титра раствора НС1, неизбежное изменение условий титрования
и т. п. вызывают суммирование ошибок.
Вычисление результатов титрования. Результаты титрования
рассчитывают, пользуясь формулами (см. гл. I, § 10):
A/ ^HCl^HCl
^NaOH = JA~~T^
KNaOH
т , ^NaOH^NaOH
NaOH== looo
*i
_ ^(практ) NaOH Т(практ) NaOH
NaOH — л/ ~ T
^(теор) NaOH l (теор) NaOH
174 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Установка титра раствора едкого натра по фиксаналу. Метод
приготовления титрованных растворов при помощи фиксанала описан ранее
(см. гл. I, § 8).
Установка титра раствора едкого натра по бифталату калия. За
последнее время для установки титров стандартных растворов оснований все
чаще стали применять бифталат калия, КООССбН4СООН. Это объясняется
тем, что он вполне удовлетворяет требованиям, предъявляемым к
установочным веществам (см. гл. I, § 8).
В водном растворе бифталат калия проявляет себя как слабая
одноосновная кислота:
коосс6н4соон ^± кооссбн4соо- + н+
Образующийся при нейтрализации кислой соли средний фталат
(подобно ацетатам щелочных металлов) в водном растворе имеет слабо
щелочную реакцию, поэтому точка эквивалентности, наблюдаемая при
титровании бифталата раствором основания, лежит в щелочной области.
Следовательно, титрование заканчивают при рН, лежащем в области от 7, 8 до 10.
При визуальном титровании подходящим индикатором является
тимоловый голубой или фенолфталеин.
Расчеты ведут по следующим формулам:
~ мкоосс6н4соон 9fU 99Q
^кооссвн4соон = 1 = ™%*а>
3NV
^теор — "
1000
204,229-0,1.250
атеор KOOCC.H4COOH — [Щ = '
Для приготовления 250 мл 0,1 н. водного раствора бифталата
рассчитанную навеску (около 5,1 г) высушенного до постоянной массы
КО0ССвН4СООН растворяют в свежепрокипяченной дистиллированной
воде в мерной колбе емкостью 250 мл, доводят объем раствора до метки и
тщательно взбалтывают.
Далее приступают к титрованию аликвотных частей полученного
раствора бифталата калия устанавливаемым раствором едкого натра.
Все операции и вычисление нормальности титра и поправочного
коэффициента едкого натра проводят как указано при установке титра NaOH
по щавелевой кислоте.
Установку титра едкого натра по бифталату калия можно проводить
и методом отдельных навесок.
§ 31. Определение карбонатов
Определение содержания растворимых в воде карбонатов щелочных
металлов (например, Na2C03 в соде). Принцип метода. Определение
Na2C03 основано на титровании растворов соды стандартным раствором
хлористоводородной кислоты.
Если для анализа дан кристаллический продукт, то можно выполнять
титрование и методом пипетирования, и методом отдельных навесок.
Если же для анализа дан раствор соды, то определение ведут по методу
пипетирования. В качестве индикатора применяют метиловый оранжевый
или смешанный индикатор (1 г метилового оранжевого и 2,5 г индигокар-
мина в I л дистиллированной воды).
§ 31. ОПРЕДЕЛЕНИЕ КАРБОНАТОВ
I 175
Уравнения реакции:
СОз~ + Н+—>НСС?
НСОз+Н+ -^ H2C03 ^=t Н20 + С02
При добавлении половины кислоты, требующейся согласно
указанным уравнениям, Na2C03 нейтрализуется «наполовину» и превращается
в NaHC03. При дальнейшем добавлении кислоты NaHC03 реагирует с HC1.
Поэтому кривая нейтрализации Na2C03 кислотой характеризуется двумя
скачками титрования, соответствующими двум точкам эквивалентности
(см. § 8).
Методика определения. Если исходят из кристаллического (твердого)
продукта, то сначала рассчитывают соответствующую навеску (см. § 28).
При работе по методу отдельных навесок рассчитанную навеску переносят
в коническую колбу, добавляют 50—100 мл дистиллированной воды и
титруют хлористоводородной кислотой. При работе по методу пипетиро-
вания рассчитанную навеску переносят в мерную колбу, растворяют в
дистиллированной воде и доводят объем раствора до метки. Если для
анализа дан раствор соды, то его разбавляют в мерной колбе
дистиллированной водой до метки.
Содержимое мерной колбы тщательно перемешивают, с помощью
пипетки отбирают для титрования 25 мл приготовленного раствора в
коническую колбу и добавляют 1—2 капли индикатора. Титрование ведут
до перехода окраски из желтой в оранжевую (при пользовании метиловым
оранжевым) или из зеленой сначала в серую, а затем — от следующей
капли — в фиолетовую (при пользовании смешанным индикатором).
Техника титрования Na2C03 хлористоводородной кислотой такая же,
как при титровании кислотой раствора тетрабората натрия (см. § 28).
Если данный раствор окажется очень разбавленным, то на его
титрование расходуется небольшое количество 0,1 н. раствора НС1 (иногда
меньше 1 мл). В таком случае для титрования используют большую пробу
анализируемого раствора или титруют более разбавленной (0,01 н.)
кислотой, или раствор предварительно упаривают до нужного объема,
если он не содержит летучих веществ.
Расчет результатов анализа. В зависимости от тоцо, как проводилось
титрование, для вычисления содержания определяемого вещества
пользуются различными формулами (см. гл. I, § 10).
Определение нерастворимых в воде карбонатов. Принцип метода.
Определение содержания нерастворимых в воде карбонатов прямым
титрованием стандартным раствором кислоты представляет определенные
трудности:
1) вследствие нерастворимости в воде анализируемого карбоната
процесс прямого титрования навески его протекает медленно;
2) многие нерастворимые в воде карбонаты содержат примеси,
мешающие прямому титрованию;
3) выделяющаяся при титровании угольная кислота мешает
определению в присутствии индикаторов, меняющих свою окраску в щелочной
среде (например, в присутствии фенолфталеина).
Поэтому для определения нерастворимых в-воде карбонатов наиболее
рациональным является .метод обратного титрования, основанный на
растворении навески анализируемого карбоната в растворе НС1 и
последующем титровании избытка кислоты стандартным раствором щелочи.
Методика определения. Рассчитанную (см. § 28) и взвешенную сначала
на технических, а затем на аналитических весах навеску переносят в
коническую колбу, добавляют 25—30 мл дистиллированной воды и 40—50 мл
176 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
стандартного раствора хлористоводородной кислоты. Во избежание
разбрызгивания смеси и потерь анализируемого раствора вследствие
выделения С02 колбу ставят наклонно. Для окончательного завершения
растворения карбоната и удаления двуокиси углерода содержимое колбы
кипятят. По охлаждении колбы избыток кислоты оттитровывают
стандартным раствором едкого натра в присутствии метилового оранжевого.
Примечание. Вместо стандартного раствора НС1 можно применять и
нестандартный его раствор. Для этого 40—50 мл приблизительно 0,1 н. раствора НС1 предварительно
оттитровывают в отдельной конической колбе стандартным раствором едкого натра. Затем
навеску карбоната растворяют при нагревании в другой колбе в том же объеме кислоты.
По охлаждении колбы избыток кислоты оттитровывают едким натром. Зная сколько
миллилитров раствора NaOH израсходовано на титрование избытка кислоты (^иаон) и ^ мл
раствора HC1 (УМаОН), легко вычислить, какому объему стандартного раствора NaOH
(^Ыаон) соответствует (эквивалентна) навеска анализируемого карбоната.
Расчет результатов анализа. Для того чтобы вычислить Уыаон
стандартного раствора NaOH (в мл), эквивалентного содержанию
определяемого карбоната, прореагировавшего с V"nc\ мл хлористоводородной
кислоты, необходимо из FNaOH мл стандартного раствора NaOH,
эквивалентного общему количеству добавленного к пробе раствора НС1 (Vhci),
вычесть VNaOH мл стандартного раствора NaOH, расходуемого на
титрование избытка раствора НС1 (Vhci)
^NaOH — ^NaOH "~" ^NaOH
Зная объем Умаон, вычисляют содержание определяемого вещества
исходя из основного правила эквивалентности.
§ 32. Определение содержания H2S04
в технической серной кислоте
Определение H2S04 основано на титровании ее растворов стандартным
раствором едкого натра. Можно также титровать стандартные
(титрованные) растворы едкого натра раствором определяемой кислоты,
рассчитанную навеску которой предварительно разбавляют дистиллированной
водой до определенного объема. Желательно, как правило, титрование
вести тем же способом, какой применялся при установке титра раствора
основания. Очень часто прибегают к способу титрования от щелочи к
кислоте, хотя такой порядок титрования и менее точен по сравнению с
титрованием от кислоты к щелочи (см. § 24).
В качестве индикатора применяют метиловый оранжевый или
метиловый красный, но можно титровать и с фенолфталеином.
Уравнения реакций:
h2so4 + он" —> hso; + н2о
hsc? + он" —> so;" + н2о
Нейтрализация серной кислоты происходит в две стадии, но так как
H2SO4 и HSOI — довольно сильные кислоты, то при титровании водных
растворов серной кислоты наблюдается лишь один скачок титрования и
одна точка эквивалентности. Аналогично в водной среде почти невозможно
получить два изгиба кривой титрования смеси двух кислот до тех пор,
пока отношение KJK2 будет невысоким или величины р/С этих кислот
будут мало отличаться друг от друга. В среде неводных растворителей
возможно дифференцированное (раздельное) титрование смесей кислот,
§ 33. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ УКСУСНОЙ КИСЛОТЫ 177
которые нельзя раздельно оттитровать в водной среде (см. § 39). Например,
в среде метилизобутилкетона можно раздельно титровать H2S04 и HSOJ*.
При этом кривые титрования имеют два изгиба.
Методика определения. Прежде всего измеряют плотность (р)
раствора H2S04. Если исходная кислота очень концентрированная, то
навеску серной кислоты рассчитывают по формуле
3NV
а~ юоо
Если кислота разбавленная, то ее навеску увеличивают в
соответствии с ее концентрацией.
При взвешивании кислот соблюдают особые правила
предосторожности. Сначала взвешивают кислоту на технических весах в бюксе, в
который предварительно наливают 10 мл Н20, затем точно взвешивают на
аналитических весах, стараясь, чтобы серная кислота не попала ни на
весы, ни на внешнюю сторону бюкса. Если исходная кислота не
концентрированная, то воду в бюкс не добавляют. Для того чтобы взять требуемую
навеску, бюкс с водой ставят на левую чашку технических весов и
тарируют. На правую чашку весов помещают соответствующие гири, а затем
в бюкс из капельницы, снабженной резиновой грушей, осторожно
наливают серную кислоту до тех пор, пока чашки весов не
уравновесятся.
Иногда для взятия навесок жидкостей пользуются весовой
бюреткой.
Взвешенную кислоту переливают с помощью воронки в мерную колбу
емкостью 250 мл, в которую предварительно наливают 100—150 мл
дистиллированной воды. Бюкс и воронку несколько раз тщательно
ополаскивают из промывалки водой, чтобы на них не осталось кислоты. Затем
содержимое колбы разбавляют водой до метки и перемешивают.
Приготовленный таким путем раствор готов к употреблению.
Очень часто указанные выше операции опускают. Это происходит
в тех случаях, когда требуется определить содержание кисл'оты в данном
объеме. В таких случаях данный объем кислоты просто разбавляют водой
в мерной Колбе до метки и тщательно перемешивают. Приготовленный
раствор наливают в бюретку и титруют им 25 мл 0,1 н. раствора едкого
натра или 25 мл приготовленного раствора кислоты помещают в
коническую колбу и титруют 0,1 н. раствором NaOH.
Расчет результатов анализа. В зависимости от того, как
приготавливали раствор —из навески или разбавлением данного объема кислоты,
для вычисления результатов анализа пользуются различными формулами
(см. гл. I, § 10).
§ 33. Определение содержания уксусной кислоты
Уксусная кислота является слабой кислотой. Слабые кислоты титруют
в присутствии фенолфталеина, так как в этом случае точка
эквивалентности наступает тогда, когда титруемый раствор становится щелочным
(см. рис. 40, § 15).
Определение уксусной кислоты методом нейтрализации является
довольно трудной задачей. Во-первых, фенолфталеин весьма
чувствителен к действию угольной кислоты, образующейся при нейтрализации
карбонатов, содержащихся в растворах NaOH (см. § 23). Во-вторых,
титрование с фенолфталеином раствором щелочи, установленным с помощью
метилового оранжевого, неминуемо приводит к большим ошибкам.
178 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Для устранения указанных ошибок пользуются различными
способами:
1) для удаления Н2С03, накапливающейся в процессе титрования,
кислый титруемый раствор кипятят;
2) устанавливают титр едкого натра не по хлористоводородной
кислоте, а по щавелевой кислоте в присутствии фенолфталеина;
3) если титр раствора щелочи был установлен по хлористоводородной
кислоте с метиловым оранжевым, то рекомендуется перед определением
СН3СООН установить его по фенолфталеину.
Уравнения реакций:
СНдСООН + ОН- ^± Н20 + СН3СОО-
В присутствии карбонатов:
СНдСООН + СО3 ~ —> НСОд + СН3СОО"
НСОд + СНдСООН —> Н2С03 + СНдСОО" ^± Н20 + С02 + СНдСОО"
Устранение вредного влияния угольной кислоты на точность
титрования. При титровании слабых кислот, какой является СН3СООН, в
присутствии угольной кислоты, находящейся в их водных растворах и в
дистиллированной воде, а также образующейся при нейтрализации
карбонатов, содержащихся в растворах оснований, результаты анализа
искажаются.
Дистиллированная вода, не защищенная от действия двуокиси
углерода воздуха, имеет кислую реакцию рН = 5,7. Для нейтрализации
угольной кислоты в 100 мл такой воды требуется около 0,3—0,5 мл 0,1 н.
раствора едкого натра в зависимости от того, с каким индикатором (бромти-
моловым голубым или фенолфталеином) проводят титрование. Очевидно,
что такого рода ошибка титрования недопустима. Поэтому, чтобы
устранить вредное влияние угольной кислоты на точность титрования слабых
кислот, помимо кипячения кислого титруемого раствора принимают
особые меры предосторожности:
1) все используемые растворы и дистиллированную воду защищают
от действия атмосферной двуокиси углерода;
2) приготовление стандартных растворов оснований проводят в
условиях, исключающих попадание в них С02; раствор готовят из химически
чистых оснований и свежеприготовленной дистиллированной воды.
Методика определения. Взятие навески и приготовление раствора
СНдСООН для титрования выполняют так, как описано при определении
H2S04 (см. § 32).
С помощью пипетки 25 или 50 мл приготовленного раствора уксусной
кислоты переносят в коническую колбу, добавляют 1—2 капли
фенолфталеина и титруют 0,1 н. раствором NaOH до появления слабо-розовой
окраски. Так как образующаяся угольная кислота обесцвечивает
фенолфталеин, то желательно вблизи точки эквивалентности титруемый раствор
прокипятить для разрушения Н2С03 и затем охладить перед тем, как
продолжить титрование.
Для сравнения результатов анализа можно повторить титрование,
налив в бюретку приготовленный раствор уксусной кислоты, а в
коническую колбу поместить 25 мл 0,1 н. раствора NaOH и 2—3 капли
фенолфталеина. В этом случае титрование ведут до обесцвечивания. В конце
титрования титруемый раствор кипятят.
Расчет результатов анализа. Вычисления выполняют, пользуясь
соответствующими формулами (см. гл. I, § 10).
§ 34. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ Na2COs И NaOH
179
§ 34. Определение содержания Na2C03 и NaOH
при их совместном присутствии
Определение содержания Na2C03 и NaOH в их смеси может быть
выполнено двумя методами, основанными на различных принципах.
Метод 1. Сначала титруют анализируемую смесь 0,1 н. кислотой
в присутствии фенолфталеина. При этом нейтрализуется весь NaOH
и «наполовину» Na2C03, превращаемый в NaHC03. Этим путем можно
установить, сколько миллилитров кислоты (Vhci) идет на титрование
NaOH + 1/2Na2C03. Затем раствор дотитровывают кислотой в
присутствии метилового оранжевого. Так устанавливают, сколько миллилитров
кислоты требуется на титрование NaHC03, образовавшегося из Na2C03,
т. е. половины Na2C03.
Таким образом, пользуясь двумя индикаторами, удается наблюдать
две. точки эквивалентности: первую, когда полностью нейтрализован
NaOH и «наполовину» Na2C03, и вторую, когда нейтрализуется NaHC03.
В этот момент наступает полная нейтрализация смеси Na2C03 + NaOH.
В первой точке эквивалентности (см. § 7, 8):
РН = 1/2 (рКн2со8 + Р^нсо") = 1/2 (6'4 + 10'3) = 8'35
Следовательно, когда едкий натр будет полностью нейтрализован,
а карбонат натрия превратится в бикарбонат, раствор станет
слабощелочным. Этот момент фиксируют с помощью фенолфталеина, меняющего свой
цвет в интервале рН = 8,0—10,0.
Во второй точке эквивалентности (см. § 8):
РН = V2P* н2со8 - Vt lg Сн2сов = Vi- 6.4 - V« lg 5-10-2 e 3>85
Этот момент фиксируют с помощью метилового оранжевого,
меняющего свою окраску в интервале рН = 3,1—4,4.
Уравнения реакций:
ОН- + Н+ —> Н20
С03~ + Н+-^НС03
НСО~ + Н+ —> Н2С03 ^± Н20 + С02
Методика определения. К титруемому раствору прибавляют 3—*5
капель фенолфталеина и титруют 0,1 н. раствором НС1 до обесцвечивания.
По достижении первой точки эквивалентности отсчитывают количество
0,1 н. раствора кислоты, израсходованной на нейтрализацию NaOH и
«половины» Na2C03—Vhci- Затем к титруемому раствору прибавляют
1—2 капли метилового оранжевого и титруют до появления оранжевой
окраски. По окончании титрования отсчитывают израсходованное
количество кислоты Унсь
Титрование лучше проводить со «свидетелями».
Щелочные растворы легко поглощают С02 из воздуха. Поэтому
отмеренный объем анализируемого раствора титруют немедленно и быстро,
избегая излишнего взбалтывания.
Расчет результатов анализа. Зная V'HCl (первое показание по шкале
бюретки) и Vhci (второе показание по шкале бюретки), можно рассчитать
содержание в смеси Na2C03 и NaOH, воспользовавшись соответствующими
формулами (см. гл. II, § 10). Если на титрование NaOH + V2Na2C03
пошло Vhci мл, а на титрование смеси NaOH + №гСОз пошло Vhci мл,
180 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
то на титрование V2Na2C03 будет израсходовано Vhci = ^hci — Vhci-
На титрование всего Na2C03: Vhci = Vhci -2 = 2 (Vhci — Vhci)» на
титрование NaOH: Vhci' = Vhci — Vhci = Vhci — 2 (VHci — VHci) =
= 2 Vhci— Vhci.
1. Метод отдельных навесок:
^HCI '2 (^HCl - ^HCl) 3Na2COs 100
*Na2CO,- 1000 e aNa2c08+NaOH ^
^HCl (2I/HC1 - Уна) %aOH 100
*NaOH ~ Ю00 ' aNa,cOs+NaOH ^
2. Метод пипетирования:
_ ^HCl '2 {VWC\ — ^HCl) 9Na2CQ3 VK
eNa2C08~ 1000 " VA 2
_ ^HCl {2VHC\ — ^HCl) ^NaOH VK
^NaOH- 1000 V7 Z
A
3. Если известна навеска смеси Na2C03 + NaOH, растворенная в VK
(общий объем раствора), то можно вычислить процентное содержание
отдельных компонентов смеси, умножив gNa2co8 и gfaaOH на 100 и разделив
на величину навески.
Метод 2. Сначала титруют одну порцию анализируемого раствора
в присутствии метилового оранжевого. Этим путем устанавливают, сколько
миллилитров кислоты (Уна) идет на титрование смеси Na2C03+NaOH.
Затем к другой порции анализируемого раствора прибавляют ВаС12.
При этом выпадает осадок BaC03, a NaOH остается в растворе. Если,
не отфильтровывая осадка, оттитровать кислотой NaOH в присутствии
фенолфталеина, то можно определить, сколько миллилитров кислоты
(Уна) потребуется на титрование только NaOH.
Разность между объемом Vhci и объемом Vhci выражает объем 0,1 н.
раствора НС1, требующийся для нейтрализации Na2C03.
Уравнения реакций:
ОН" + С03 " + ЗН+ —> 2Н20 + С02
Ва+ + + СОз"~>ВаС03
ОН- + Н+ -+• НаО
При работе этим методом хорошие результаты получаются, если
анализируемая смесь содержит малые количества NaOH.
Методика определения. Для титрования берут 25 мл смеси.
Определение общей щелочности ведут в присутствии метилового оранжевого.
Для определения содержания NaOH к 25 мл новой пробы добавляют 20 мл
1 н. раствора ВаС12 и 2—Закапли фенолфталеина. ТитруютНС1 до
обесцвечивания фенолфталеина.
Необходимо помнить, что при поглощении титруемой смесью С02
из воздуха количество NaOH уменьшается, а количество Na2COs
увеличивается вследствие взаимодействия NaOH с угольной кислотой.
Для предупреждения поглощения С02 из воздуха анализируемый
раствор титруют немедленно и быстро, избегая излишнего взбалтывания.
§ 35. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ Na2COs И NaHCOa 181
Расчет результатов анализа. Зная Vhci (первое показание по шкале
бюретки) и V'hci (второе показание по шкале бюретки), можно легко
рассчитать содержание в смеси NaOH и Na2C03l пользуясь соответствующими
формулами (см. гл. I, § 10).
Если на титрование смеси Na2C03 + NaOH израсходовано Vhci мл,
а на титрование NaOH израсходовано Vhci мл, то на титрование Na2C03
будет израсходовано Vhci = Vhci — ^hci мл. Подставляя в формулы
значения Vhci и Vhci, рассчитывают содержание отдельных компонентов
анализируемой смеси (см. гл. I, § 10).
§ 35. Определение содержания Na2C03 и NaHC03
при их совместном присутствии
Определение Na2C03 и NaHC03 в их смеси может быть выполнено
двумя методами.
Метод 1. Сначала титруют анализируемую ciytecb кислотой в
присутствии фенолфталеина (Vhci)- При этом нейтрализуется «наполовину»
Na2C03, превращающийся, в NaHC03.
Затем раствор дотитровывают кислотой в присутствии метилового
оранжевого (Vhci)- При этом нейтрализуется NaHC03, находившийся
в начальной смеси и образовавшийся из Na2C03 (1/2Na2C03). Очевидно,
на титрование NaHC03 идет Vhci — Vhci — 2Vhci-
Используя два индикатора, удается, так же как и при определении
смеси Na2C03 + NaOH (см. § 34), наблюдать две точки эквивалентности:
первую, когда «наполовину» нейтрализован Na2C03, вторую, когда
нейтрализован NaHC03, содержащийся в исходной смеси и полученный в
результате нейтрализации Na2C03. В этот момент наступает полная
нейтрализация смеси Na2C03 + NaHC03.
Зная, сколько миллилитров хлористоводородной кислоты пошло на
нейтрализацию Na2C03 «наполовину», можно вычислить содержание
Na2C03.
Зная общую щелочность, легко вычислить содержание NaHCOg.
Интересно отметить, что при титровании по этому способу возможны
пять случаев:
1. Объемы 0,1 н. раствора кислоты, израсходованные на титрование
с фенолфталеином (V<j,) и метиловым4 оранжевым (VH.0), равны. В этом
случае в анализируемом растворе имеется только Na2C03. Это объясняется
тем, что в присутствии фенолфталеина Na2C03 оттитровывается только
«наполовину», т. е. до NaHC03, а с метиловым оранжевым оттитровывается
уже образовавшийся NaHC03 (вторая половина Na2COb, превращенного
в NaHC03).
2. Объем 0,1 н. раствора кислоты, пошедшей на титрование с
фенолфталеином, больше объема кислоты, израсходованной на титрование
с метиловым оранжевым. В этом случае в анализируемом растворе
содержится наряду с Na2C03 и NaOH. Это объясняется тем, что в присутствии
фенолфталеина оттитровывается не только половина Na2C03, но и NaOH.
3. Объем 0,1 н. раствора кислоты, израсходованной на титрование
с метиловым оранжевым, больше объема кислоты, расходуемой при
титровании с фенолфталеином. В этом случае в анализируемом растворе
находится смесь Na2C03 и NaHCOg. Это объясняется тем, что в присутствии
метилового оранжевого оттитровывается не только вторая половина
Na2C03, но и NaHC03, присутствовавший в исходном растворе.
182 ГЛ.П. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
4. Объем 0,1 н. раствора кислоты, израсходованной на титрование
с фенолфталеином, равен нулю, а с метиловым оранжевым он больше
нуля. В этом случае в анализируемом растворе имеется только NaHC03,
не титруемый в присутствии фенолфталеина.
5. Объем 0,1 н. раствора кислоты, израсходованной на титрование
с фенолфталеином, больше нуля, а с метиловым оранжевым равен нулю.
В этом случае анализируемый раствор содержит только NaOH, который
полностью оттитровывается в присутствии фенолфталеина.
Следовательно, при титровании щелочных растворов, содержащих
гидроокись, карбонат и бикарбонат натрия, в исследуемом растворе могут
быть: Na2C03, NaOH + Na2C03, Na2C03 + NaHC03, NaHC03 либо же
NaOH. Аналогичные варианты могут иметь место и для таких же
соединений калия. Случая титрования, соответствующего содержанию
тройной смеси NaOH + NaHC03 + Na2C03, практически не может быть, так
как сильные щелочи нейтрализуют бикарбонаты:
НСОз + ОН" —* Н20 + С03 "
Расчет результатов анализа.
1. Метод отдельных навесок:
_^hci Унс1эка2ау2 100 0,
*Na8COs- 1()00 • aNa2co8+NaHCo/°
^НС1 (Уна — 21/HCl) 3NaHCQ3 100 07
*NaHC°* - 1000 *NaaCO,+ NaHCO, *
2. Метод пипетирования:
_ ^HCl ^HCl 9Na,COj'2 Vk ,
^Na2CO, — Ю00 * VA
^HCl (^HCI — 2^HCl) 3NaHCO, VK
SNaHC03 ~ Щбб VI *
Если известны величина навески анализируемой смеси и общий объем
раствора (VK), то можно вычислить процентное содержание Na2C03
и NaHC03, умножив gNa2co8 ИЛИ ^NaHcOs на 100 и разделив на величину
навески.
Метод 2. Смесь Na2C03 + NaHC03 титруют 0,1 н. раствором HC1
в присутствии метилового оранжевого. Затем к другой пробе раствора
Na2C03 + NaHC03 прибавляют титрованный раствор щелочи с таким
расчетом, чтобы весь бикарбонат превратился в карбонат и, кроме того,
чтобы в растворе остался избыток NaOH. К полученной смеси приливают
избыток раствора ВаС12. При этом выпадает осадок ВаС03.
Не отфильтровывая осадка, оттитровывают избыток щелочи в
присутствии фенолфталеина.
Зная количество кислоты, пошедшей на титрование смеси Na2COs +
+ NaHC03, и количество едкого натра, потребовавшегося на превращение
бикарбоната в карбонат, можно рассчитать содержание каждого
компонента смеси.
Уравнения реакций:
НСОз" + он~~ —* со~ + н2°
СОз~ + Ва++-^ВаС03
§ 36. ОПРЕДЕЛЕНИЕ ЖЕСТКОСТИ ВОДЫ
183
Этот метод дает хорошие результаты, когда анализируемая смесь
содержит малые количества NaHC03 по сравнению с Na2C03.
Методика определения. Определение компонентов смеси Na2C03 +
+ NaHC03 проводят по методу пипетирования или по методу отдельных
навесок.
Для титрования берут 25 мл смеси. Определение общей щелочности
ведут в присутствии метилового оранжевого.
Для определения содержания бикарбоната берут 25 мл новой пробы
смеси, добавляют 50 мл 0,1 н. раствора NaOH, 20 мл 1 н. раствора ВаС12
и 2—3 капли фенолфталеина. Избыток NaOH оттитровывают до
обесцвечивания фенолфталеина.
Для достижения более точных результатов при тех же условиях
параллельно ставят «холостой» опыт. Разность между количеством 0,1 н.
раствора кислоты, пошедшей на титрование в «холостом» опыте, и при
титровании анализируемой пробы соответствует содержанию NaHC03.
Расчет результатов анализа. Если на титрование Na2COs + NaHC03
(с метиловым оранжевым) пошло Vhci мл, а на титрование избытка 0,1 н.
раствора NaOH (с фенолфталеином) Vhci мл 0,1 н. раствора НС1, то объем
0,1 н. раствора НС1, эквивалентный содержанию NaHC03, равен Vhci =
= 50 — Уна мл.
Зная Vhci и Унсь можно легко рассчитать содержание в смеси Na2C03
и NaHC03, воспользовавшись соответствующими формулами (см. гл. I,
§ 10 и выше «Метод 1»).
§ 36. Определение жесткости воды
Напомним, что жесткость воды зависит от наличия в ней солей
двухвалентных металлов, преимущественно кальция и магния. Жесткая вода
при кипячении образует накипь вследствие оседания некоторых солей
кальция, магния и железа (II). Мыло в жесткой воде не мылится (не
вспенивается), так как образуются нерастворимые в воде кальциевые и
магниевые соли жирных кислот. Жесткая вода не пригодна для питания
паровых котлов и применения в химической технологии, а также других
технических целей.
Определение жесткости воды имеет большое практическое значение
и очень широко применяется в технике и промышленности.
Различают жесткость временную (или устранимую) и постоянную.
Временная жесткость воды обусловлена присутствием в воде
бикарбонатов: Са (НС03)2, реже Mg (HC03)2 л иногда»также Fe (HC03)2. Ее можно
устранить кипячением воды.
При кипячении воды бикарбонаты разлагаются с образованием
нерастворимых в воде карбонатов, оксикарбонатов и гидроокисей:
Са (НСОз)а —> СаС03 + Н20 + С02
Mg (НС03)2 —> MgC03 + Н20 + С02
2Mg (НС03)2 —> Mg2 (ОН)2 С03 + Н20 + ЗС02
Fe (HC03)2 -^ Fe (ОН)2 + 2С02
Постоянная жесткость воды обусловлена присутствием в ней
преимущественно сульфатов и хлоридов кальция и магния и не устраняется
кипячением. Сумма временной (устранимой) и постоянной жесткости
составляет общую жесткость воды.
184 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Жесткость выражают в миллиграмм-эквивалентах Са++-ионов на 1 л
воды. 1 мг-экв жесткости соответствует содержанию —^— = 20,04 мг
24 31
Са+ + или —^— = 12,16 мг Mg+ + в 1 л воды, где 40,08 и 24,31 —
соответственно округленные атомные веса кальция и магния; 20,04 и 12,16
эквивалентны Са++ и Mg++.
Для выражения жесткости также применяют тысячную долю
миллиграмм-эквивалента — микрограмм-эквивалент в 1 л воды.
Существуют различные способы определения жесткости. Рассмотрим
два из них:
1) определение временной жесткости с помощью титрованного раствора
хлористоводородной кислоты и 2) комплексометрический метод (см. гл. IV)
определения общей жесткости.
При титровании образца воды хлористоводородной кислотой в
присутствии метилового оранжевого происходит разложение бикарбонатов,
обусловливающих временную жесткость:
Са (НС03)2 + 2Н+ —> Са++ + 2Н20 + 2С02
Методика определения. Отбирают в коническую колбу пипеткой или
мерным цилиндром 100 мл чисследуемой воды, добавляют 2—3 капли
метилового оранжевого и титруют 0,1 н. раствором НС1 до появления
оранжевой окраски.
Расчет результатов анализа. 1 мл 0,1 н. раствора НС1 соответствует
0,1/1000 г-экв или 0,1 мг-экв Са++. Vhci соответствует 0,1 Vhci/IOOO г-экв
или 0,1 Vhci мг-экв Са++. 0,1 Vhci мг-экв находится в объеме Va- Чтобы
выразить жесткость в миллиграмм-эквивалентах на 1 л воды, нужно
найденную величину разделить на VA и умножить на 1000, т. е. жесткость
исследуемой воды (х) равна:
^нс! Уна 1000
х = tz мг-экв/л
УА
§ 37. Определение аммонийного азота в солях аммония
Напомним, что азот входит в состав многочисленных неорганических
и органических соединений. Соединения азота имеют громадное значение
в природе и применяются в разнообразных отраслях промышленности
и народного хозяйства. Поэтому определение азота в его соединениях имеет
большое практическое значение.
Азот, входящий в состав различных соединений, очень часто
определяют в виде NH3, который образуется:
а) при кипячении солей аммония со щелочами;
б) при восстановлении нитратов и нитритов^ в щелочном растворе
при помощи сплава Деварда (45% А1, 5% Zn, 50% Си);
в) при нагревании с концентрированной H2S04 азотсодержащих
органических соединений и последующем разложении образовавшейся
соли аммония NH4HS04 щелочью (метод Кьельдаля). Метод основан на
том, что азотсодержащее вещество полностью разрушается при
нагревании с концентрированной H2S04 в присутствии добавок, действующих
как катализаторы.
Существует несколько способов определения NH3.
Определение аммиака в солях аммония методом
обратного титрования. Определение основано на титровании избытка ще-
§ 37. ОПРЕДЕЛЕНИЕ АММОНИЙНОГО АЗОТА В СОЛЯХ АММОНИЯ 185
лота, которую добавляют к соли аммония, разлагаемой при кипячении
щелочного раствора с образованием NH3:
NH? + (л + 1) NaOH —> NH3 + Н20 + п NaOHH36 + Na+
Рис. 48. Установка для отгонки аммиака:
/ — круглодонная перегонная колба; 2 — ловушка;
3 — тройник с краном; 4 — холодильник; 5 —
колба-приемник.
Зная количество NaOH, добавленного в раствор соли аммония до
кипячения и оставшегося после кипячений, можно по разности вычислить
содержание NH3 в исходной соли.
Методика определения.
Рассчитывают навеску исходной соли
аммония (см. § 28) для
приготовления 250 мл 0,1 н. раствора.
Взятую навеску растворяют
в воде или данный для анализа
готовый раствор переносят в
мерную колбу, точно нейтрализуют
свободную кислоту несколькими
каплями 0,1 н. раствора NaOH
в присутствии метилового
красного и доводят объем раствора
водой до метки. Три пробы по
25 мл приготовленного раствора
переносят при помощи пипетки
в три стакана. В каждый стакан
добавляют по 50 мл 0,1 н.
раствора NaOH. Стаканы ставят на
песочную баню и нагревают почти
до кипения, пока не улетучится
весь аммиак. Выпаривание обычно
прекращают тогда, когда испарится свыше Чь содержимого стакана.
Полноту удаления NH3 проверяют при помощи фильтровальной бумаги,
смоченной раствором Hg2 (N03)2. Если весь аммиак удален, бумага
не чернеет.
По окончании удаления NH3 стаканы снимают с бани, накрывают
часовыми стеклами и дают им остыть. После охлаждения содержимого
стаканов добавляют в них по 1—2 капли метилового красного и титруют 0,1 н.
р аствор ом кислоты.
Указанный метод определения NH3 весьма прост, но менее точен, чем
метод, описанный ниже.
Расчет результатов анализа. Содержание аммиака вычисляют, как
описано (в гл. I, § 10).
Определение аммиака в солях аммония методом отгонки.
Определение основано на отгонке аммиака, выделяющегося при
взаимодействии соли аммония со щелочью, улавливании его определенным
объемом стандартного раствора кислоты и последующем титровании
остатка неиспользованной кислоты щелочью в присутствии метилового
оранжевого или еще лучше в присутствии метилового красного.
Зная количество кислоты, пошедшей на реакцию с NH3, можно легко
вычислить содержание аммонийного азота в соли аммония.
Методика определения. Рассчитанную навеску соли аммония (взятую
из расчета нейтрализации около 30 мл 0,1 н. раствора кислоты) вводят
через воронку с укороченным носиком в круглодонную колбу / (рис. 48).
Воронку над перегонной колбой тщательно споласкивают
дистиллированной водой.
186 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
На дно колбы / опускают несколько кусочков неглазурованного
фарфора или пемзы величиной с конопляное зерно для равномерного кипения
жидкости при нагревании.
В некоторых руководствах для той же цели предлагается вместо
фарфора или пемзы поместить в колбу немного цинковой пыли. Мы
рекомендуем воздерживаться от введения цинка, восстанавливающего в
щелочном растворе при нагревании нитраты и нитриты (которые могут
содержаться в анализируемой смеси) до NH3. Это ведет к неизбежным ошибкам.
В колбу 5 вводят пипеткой 50 мл 0,1 н. раствора НС1 и 2—3 капли
метилового оранжевого или метилового красного. Нижний конец
холодильника 4 должен быть опущен в
кислоту на 1 см. Нижний отросток водяной
рубашки холодильника через
резиновую трубку присоединяют к
водопроводному крану, а верхний — к сточной
трубе и пускают в холодильник воду.
Затем с помощью воронки с длинной
трубкой быстро вливают в круглодон-
ную колбу 1 около 50 мл 50%-ного
раствора КОН и немедленно закрывают
колбу резиновой пробкой с вставленной
в нее насадкой. Пробка
заблаговременно должна быть хорошо пригнана
к горлу колбы.
При нагревании сначала из колбы
вытесняется воздух, затем начинает
выделяться аммиак. Как только воздух
будет вытеснен из колбы У, кислота
начинает засасываться в алонж
холодильника. На это не следует обращать
особого внимания.
Пламя горелки регулируют таким образом, чтобы кипение в колбе /
было равномерным и умеренным.
Если навеска соли аммония была рассчитана правильно, то жидкость
в колбе 5 до конца опыта остается красной. В противном случае раствор
начинает желтеть. Это указывает на то, что необходимо добавить еще
некоторое (отмеренное) количество 0,1 н. раствора хлористоводородной
кислоты.
Перегонку продолжают до тех пор, пока около 2/3 содержимого колбы /
не перегонится в колбу 5. По окончании отгонки, не прекращая
нагревания, вынимают из приемника алонж и ополаскивают его над колбой 5
дистиллированной водой из промывал ки. Затем содержимое колбы 5
титруют 0,1 н. раствором едкого натра до появления оранжево-желтой
окраски. Иногда для отгонки NH3 применяют упрощенную установку
(рис. 49) и вместо НС1 пользуются H2S04.
Расчет результатов анализа. Содержание аммиака рассчитывают, как
описано в гл. I, § 10.
Рис. 49.
Упрощенная установка для
отгонки аммиака.
*NHa ='
^НС1 ^НС1 ЭЫН8
1000
¦?«
где VHCl — объем хлористоводородной кислоты, израсходованный на нейтрализацию
выделившегося аммиака, мл:
^НС1 = ^НС1"
^NaOH ^NaOH
N
НС1
здесь Vhq — объем взятого раствора НС1, нормальность которого WHC1, мл.
§ 38. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ФОСФОРНОЙ КИСЛОТЫ 187
Определение аммиака в солях аммония формалиновым методом. При
взаимодействии солей аммония с формальдегидом СН20, содержащимся
в водном растворе в виде гидрата СН20-Н20, выделяется эквивалентное
количество свободной кислоты:
4NH4 + 6СН20- Н20 —*• (СН2)6 N4 + 4Н+ + 12Н20
уротропин
Содержание выделяющейся при этом свободной кислоты определяют
титрованием стандартным раствором едкого натра, установленным по
щавелевой или янтарной кислоте в присутствии фенолфталеина.
Методика определения. В коническую колбу емкостью 250 мл
наливают 15 мл 25%-ного водного раствора формальдегида (формалина),
добавляют 100 мл дистиллированной воды, 2—3 капли фенолфталеина и
точно нейтрализуют кислый раствор формалина (который может
содержать свободную муравьиную кислоту) 0,1 н. раствором NaOH до
слаборозовой окраски. В случае если исходный раствор формалина более
концентрирован или содержит менее 25% формальдегида, то берут его
соответственно меньше или больше 15 мл.
В нейтрализованный раствор вносят навеску около 0,2—0,3 а соли
аммония, взвешенной на аналитических весах с точностью до 0,0001 г.
Смесь тщательно перемешивают, дают постоять 3—5 мин и затем титруют
выделившуюся кислоту стандартным раствором NaOH.
Расчет результатов анализа. Содержание аммиака в исследуемой соли
рассчитывают исходя из правила эквивалентности, — см. гл. I, § 10.
§ 38. Определение содержания фосфорной кислоты
Титрование Н3Р04 (многоосновной кислоты) отличается некоторыми
особенностями (см. § 17).
Определение ее основано на титровании стандартным раствором едкого
натра. Первая константа диссоциации Н3Р04 достаточно большая (К\ =
= 1,1-10"2; р/С = 1,96), поэтому фосфорную кислоту можно титровать
в присутствии метилового оранжевого.
При титровании в присутствии метилового оранжевого фосфорная
кислота титруется как одноосновная кислота:
Н3Р04 + NaOH —> NaH2P04 + H20
или
н3ро4 + он"" -^ н2ро; + н2о
Эквивалент фосфорной кислоты в этом случае равен ее молекулярному
весу
Вторая константа диссоциации Н3Р04 мала, она в 100 раз меньше
константы диссоциации уксусной кислоты (К2 == 2,0-10"7; рК = 6,7).
Поэтому фосфорную кислоту как двухосновную кислоту можно титровать
в присутствии фенолфталеина (для подавления гидролиза в титруемый
раствор прибавляют хлорид натрия):
Н3Р04 + 2NaOH —* Na2HP04 + 2H20
или
Н2Р04 +ОН ->НР04 +Н20
188 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ THTPOBAHHF
В этом случае эквивалент фосфорной кислоты равен половине ее
молекулярного веса
э Л*Няро4 97,9953
с*Н8ро4 — 2 "" 2 '
Третья константа диссоциации Н3Р04 очень мала (К3 = 3,6-10~12;
р/С = 12,44). Поэтому оказывается невозможным оттитровать в водном
растворе Н3Р04 как трехосновную кислоту согласно уравнению
Н3Р04 + 3NaOH^Na3P04 + ЗН20
или
нро; " + он" ^± ро"" + н2о
Течению этой реакции слева направо мешает гидролиз Na3P04 — соли,
образованной катионом сильного основания и анионом очень слабой
кислоты. Гидролиз обусловливается взаимодействием РО™ с ионами
водорода. Если РО^~~ осадить в виде какой-либо малорастворимой соли
[например, Са3(Р04)2], то тогда оказывается возможным оттитровать
Н3Р04 как трехосновную кислоту:
1) Н3Р04 + ОН —> Н2Р04 + Н20 (метиловый оранжевый)
2) Н2Р04 +ОН~-^НР04~~+Н20 (фенолфталеин)
3) гНРО;" + ЗСа+ + + 20Н" -^ | Са3 (Р04)2 + 2Н20 (в присутствии
CaClo, фенолфталеин)
В этом случае эквивалент Н3Р04 равен ее молекулярному весу,
деленному на 3:
q ^Н8Р04 97,9953
ЭН8Р04 = § = 3 32'66
Методика определения. Прежде всего измеряют плотность раствора
Н3Р04. Если исходная кислота очень концентрированная, то навеску ее
рассчитывают по формуле (см. гл. I, § 9):
_ 3NV.
°"~~ 1000
Взятие навески и приготовление раствора кислоты выполняют, как
указано в § 32. Полученный раствор Н3Р04 доводят в мерной колбе
дистиллированной водой до метки, тщательно перемешивают, отбирают
аликвотную часть, приливают по 1—2 капли индикатора и титруют
стандартным раствором едкого натра.
Расчет результатов анализа. В зависимости от того, как
приготавливали раствор — из навески или разбавлением данного объема кислоты,
для вычисления результатов анализа пользуются различными формулами
(см. гл. I, § 10), подставляя в формулы Эн,ро4 в зависимости от характера
индикатора, который применялся в процессе титрования.
В. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ В НЕВОДНЫХ СРЕДАХ
§ 39. Неводные растворы
До самого последнего времени химики-аналитики использовали
преимущественно реакции, протекающие в водных средах. Поэтому
химические и физико-химические методы анализа основывались главным образом
на наблюдении явлений, протекающих в водных растворах.
§ 40. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О КИСЛОТАХ И ОСНОВАНИЯХ 189
За последнее время приобрели большое значение новые методы
анализа, основанные на исследовании явлений, наблюдаемых в неводных
растворах реагирующих веществ.
Успехи в аналитической химии неводных растворов обусловлены
развитием химической теории растворов, основы которой были заложены
русской школой ученых-химиков во главе с Д. И. Менделеевым.
С другой стороны, внедрению в практику методов анализа в неводных
средах в значительной степени способствовали экспериментальные
физико-химические исследования свойств растворов различных веществ
в жидких неорганических и органических растворителях и сжиженных
газах.
§ 40. Современные представления о кислотах и основаниях
Существование кислот и оснований в неводных растворах. В
настоящее время доказано, что кислоты и основания с присущими им
характерными свойствами существуют не только в водных растворах, в которых
имеются ионы водорода и гидроксила, но и в неводных растворах, где этих
ионов нет. Поэтому применительно к неводным растворам прежние
определения кислот и оснований как электролитов,- диссоциирующих с
образованием ионов водорода и ионов гидроксила, неточны.
Ионы лиония и ионы лиата. Доказано, что свободные ионы водорода
отсутствуют даже в водных растворах. Они гидратированы. При
электролитической диссоциации кислот в неводных растворах образуются
разнообразные сольватированные ионы водорода. В воде Н + -ионь1 образуют
Н30 + -ионы (гидроксония), в среде аммиака они образуют ЫН^-ионы
(аммония), в растворе пиридина — С5НбЫН + -ионы (пиридиния) и т. д.
Сольватированные молекулами растворителя ионы водорода
(протоны) называют ионами лиония, а анионы, образующиеся при ионизации
растворителя, — ионами лиата (табл. 20):
2Н20^± Н30+ + ОН-
гидроксоний гидроксил
(ион лиония) (ион лиата)
2NH3q=t NH4 + NH2
аммоний амид-ион
(ион лиония) (ион лиата)
Теория сольвосистем. В жидком аммиаке амид калия KNH2 ведет
себя так, как едкое кали КОН в водном растворе, т. е. является сильным
основанием. Это доказывается тем, что указанный раствор, по отношению
к фенолфталеину оказывается щелочным, нейтрализует кислоты и обладает
высокой электропроводностью.
Хлорид аммония в растворе жидкого аммиака ведет себя так,, как
хлористый водород в воде, т. е. является сильной кислотой. Это доказывается
тем, что указанный раствор нейтрализует основания и обладает высокой
электропроводностью.
Следовательно, основные и кислые свойства присущи не только
соединениям, характеризующимся наличием гидроксильных групп и свобод-
лых ионов водорода.
Проявление кислых и основных свойств, не являющихся кислотами
или основаниями в общепринятом смысле, наблюдается во многих
растворителях, как содержащих водород, так и не содержащих его, например
NaH4, жидких HF, H2S, COCl2, S02, S02C12, SOCl2, CH3COOH и др.
190 ГЛ.П. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
ТАБЛИЦА 20
Сольватированные протоны (ионы лиония) и соответствующие им анионы
(ионы лиата), образующиеся в процессе самоионизации растворителей
Растворитель
Фтористый водород жидкий
Уксусная кислота
Муравьиная кислота
Серная кислота (безводная)
Сероводород (жидкий)
Вода
Метиловый спирт
Этиловый спирт
Аммиак (жидкий)
Гидразин
Анилин
Ацетамид
Формамид
Этилендиамин
Формула
HF
СНдСООН
НСООН
H2S04
H2S
Н20
СН3ОН
С2Н5ОН
NH3
N2H4
C6H5NH2
CH3CONH2
HCONH2
H2NCH2CH2NH2
Ионы лиония
(кислота)
H2F+
CH3COOH2+
HCOOH2+
H3S04+
H3S+
н3о+
сн3он2+
С2Н5ОН2+
NH4+
N2H5+
C6N5NH3+
CH3CONH3+
HCONH3+
H2NCH2CH2NH3+
Ионы лиата
(основание)
HF2-
CHgCOO*
нсоо-
hso4-
sh-
он-
сн3о-
С^НбО-
NH2-
N2H3"
C6H6NH-
CH3CONH-
HCONH-
H2NCH2CH2NH"
Некоторые реакции нейтрализации кислот и оснований в среде
различных растворителей сопровождаются образованием молекул данного
растворителя:
а) в водном растворе:
НС1 + NaOH -^NaCl + H20
кислота основание соль растворитель
б) в среде жидкого аммиака:
NH4Cl + KNH2 —> КС1 + 2NH3
кислота основание соль растворитель
в) в среде безводной уксусной кислоты:
НСЮ4 + CH3COONa —> NaC104 + СН3СООН
кислота основание соль растворитель
И Т. Д.
Это общее начало — образование в процессе нейтрализации соли
и молекул растворителя — объединяет взгляды многих сторонников так
называемой теории сольвосистем. Согласно этой теории, между реакциями
аквосоединений в воде и соединениями в других растворителях имеется
большое сходство.
Кислоты и основания с точки зрения теории сольвосистем. Согласно
теории сольвосистем, кислотами и основаниями являются химические
соединения, образующие катионы и анионы, идентичные с катионами и анионами
данного растворителя.
Например, диссоциация аммиака происходит с образованием NHj
и NH2; NH4C1 в среде жидкого аммиака образует катионы аммония
(NH4), следовательно NH4C1 — кислота; KNH2 образует анионы амид-
ионов (NH2), значит KNH2 — основание; в среде безводной уксусной
кислоты НСЮ4 — кислота, CH5COONa — основание и т. д.
§ 40. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О КИСЛОТАХ И ОСНОВАНИЯХ 191
К недостаткам этой теории можно отнести то, что она не объясняет
существования типичных кислот и оснований в растворах неионизиру-
ющих растворителей.
Теория Бренстеда—Лоури. Согласно теории Бренстеда—Лоури,
придерживающихся того взгляда, что свойства кислот и оснований
объясняются их составом, кислотами являются химические соединения,
способные отдавать протоны {доноры протонов); основаниями — вещества,
способные присоединять протоны (акцепторы протонов).
С этой точки зрения общим для всех кислот является наличие в их
составе водорода. Кислотами могут быть электронейтральные молекулы,
катионы и анионы. Например:
Н20 —>Н++ ОН-
кислота основание
Н20 +Н+—> Н30+
основание кислота
СНзСООН —> Н+ + СНзСОО-
кислота основание
Сопряженные кислоты и основания. Каждая кислота имеет
сопряженное с ней основание и каждое основание имеет сопряженную с ним
кислоту.
Для того чтобы кислота (Ах), являющаяся донором протона и
образующая при этом основание (Вх), могла отдать протон, необходимо
присутствие основания (В2), акцептирующего (присоединяющего) протон
с образованием кислоты (А2):
Ах ^± Н+ + Вх; В2 + Н+ ^± А2
НС1 ^±Н++ СГ ; NH3 +H+^± NHj
кислотах основание! основаниеа кислота2
НС1 + NH3 ^± СГ + NHj
кислота! основаниеа основание! кислота2
Как видно из уравнений реакций, в приведенном выше равновесии
участвуют две пары кислот и оснований.
Состояние равновесия среди сопряженных пар кислот и оснований.
В общем виде состояние равновесия зависит от относительной силы
оснований и представляется следующим образом:
шт~к (60)
Процесс диссоциации слабой кислоты и слабого основания, согласно
этой теории, может быть представлен следующими схемами:
Слабая кислота:
СНзСООН + Н20 ^± СН3СОО- + Н30+
Ах Ва Вх А2
Слабое основание:
Н20 + NH3 ^± ОН" + NHj
Ai B2 Bt A2
Сила кислот и оснований зависит от природы растворителя. В
растворе жидкого аммиака, отличающемся сильноосновными свойствами,
все кислоты полностью диссоциированы. В среде других растворителей,
отличающихся менее выраженным акцепторным характером, т. е. не
отличающихся основным характером, диссоциация кислor неполная.
192 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Основным недостатком теории Бренстеда является то, что эта теория
исключает возможность проявления кислотного характера веществами,
не содержащими водорода.
Теория Льюиса. Согласно теории Льюиса, придерживающегося взгляда,
что свойства кислот и оснований объясняются их строением, основанием
является химическое соединение, обладающее неподеленной свободной
парой электронов, склонной к образованию устойчивой электронной
группировки (октета) другого атома. Согласно этой теории аммиак является
Н
основанием, потому что его молекула Н : N : обладает указанной парой
Н
электронов.
Кислотой является вещество, в молекуле которого не хватает пары
электронов до устойчивой электронной группировки и которое склонно
к обобществлению свободной пары электронов основания,
сопровождающемуся образованием устойчивого электронного октета. С этой точки
:С1:
зрения хлорид бора (III), строение которого : С1 : В, является кислотой
" :С1:
Следовательно, основания являются донорами, а кислоты акцепторами
пары электронов', реакция нейтрализации сопровождается образованием
ковалентной связи за счет общей свободной пары электронов основания:
Н :С1: H:Q:
H:N:+ В: CI:—>H:N: B:C1:
H :C1:" H:C1:**
Образование координационной ковалентной связи является
первичным процессом нейтрализации, за которым может следовать ионизация
и диссоциация, например:
Н Н
H:N: +H:C1: -+ H:N:H:Q:
Н Н
Г " Т
H:N:H + С1"
L й J
§ 41. Диссоциация электролитов в неводных растворах
Влияние растворителя на состояние ионов в растворе. Исследования
концентрированных водных и неводных растворов выявили ряд их
особенностей по сравнению с разбавленными водными растворами слабых и
сильных электролитов. В результате многочисленных исследований
в области растворов было отмечено влияние характера растворителя на
состояние ионов в растворах. Огромное значение химического
взаимодействия между молекулами растворенного вещества и растворителя
впервые было показано Д. И. Менделеевым. О сильном влиянии характера
растворителя свидетельствуют, например, данные по электропроводности
одинаково диссоциирующих в водных растворах солей, проявляющих
себя по-разному в различных по своей химической природе
растворителях.
В настоящее время, на основании исследований ряда ученых, со всей
определенностью можно утверждать, что одно и то же вещество в
зависимости от растворителя, в котором оно растворено, может быть отне-
§ 42. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ НА СИЛУ КИСЛОТ И ОСНОВАНИЙ 193
сено к классу сильных или слабых электролитов. Другими словами, сильные
электролиты в водном растворе могут оказаться слабыми в неводных
растворах и наоборот. Таким образом, полная или неполная
электролитическая диссоциация в водных растворах является только частным
случаем состояния электролитов.
Исследования показали, что диссоциация сильных электролитов
в неводных растворах подчиняется закону действия масс.
§ 42. Влияние неводных растворителей на силу кислот и оснований
Поведение электролитов в различных по характеру средах.
Многосторонние исследования показали, что некоторые вещества, которые ведут
себя как кислоты в среде одного растворителя, в другом проявляют себя
как основания. Соединения, проявляющие себя как основания в одних
средах, ведут себя как кислоты в других; нередко в неводных растворах
кислые или основные свойства проявляют также вещества, которые,
казалось бы, ничего общего не имеют с кислотами и основаниями в обычном
их представлении.
При переходе от амфотериых, или амфипротных. растворителей
(каким является вода) к основным или протофильным (какими являются
аммиак, этилендиамин и др.), отличающимся склонностью к
присоединению протонов, ионизация по типу кислот усиливается, а ионизация по
типу оснований уменьшается *.
При переходе от амфотерных, или амфипротных, растворителей к
кислым или протогенным (какими являются безводная уксусная кислота,
жидкий фтористый водород и др.), отличающимся склонностью отдавать
свои протоны, усиливается ионизация по типу оснований, а ионизация
по типу кислот уменьшается.
Одно и то же вещество может вести себя различно в зависимости от
растворителя, в котором оно растворено. Таково, например, поведение
мочевины, являющейся кислотой в растворе жидкого аммиака, сильным
* Встречающиеся здесь и далее понятия означают:
Амфотерный, или амфипротный, растворитель (НгО, СНзОН, СгНбОН и др.) —
вещество амфотерного характера, играющее роль основания по отношению к кислотам
и роль кислоты по отношению к основаниям. Молекулы амфипротных растворителей
отличаются способностью отдавать и присоединять протоны. Так вода отдает свои протоны
NH3, N2H4, CH3NH2 и принимает протоны от НС1, H2SO4, CH3COOH и др.
Кислый, или протогенный, растворитель (HF, H2SO4, HCOOH, СНзСООН и др.) —
вещество кислого характера, молекулы которого могут отдавать протоны. Молекулы про-
тогенного растворителя могут присоединять протоны лишь от сильных кислот. Например,
уксусная кислота присоединяет протоны от H2SO4, HC1, НСЮ4 и др.
Основной, или протофильный, растворитель (NH3, N2H4, этилендиамин, диоксан)—
вещество основного характера, молекулы которого обладают ясно выраженным
сродством к протону. Оторвать протоны от молекулы протофильного растворителя могут лишь
молекулы очень сильных оснований. Трудно, например, назвать такое основание, которое
способно отнять протоны от молекулы аммиака.
Апротонный, или апротный, растворитель (С6Нб, СвН5С1, СНзС1 и др.) —вещество
нейтрального характера, молекулы которого не способны ни отдавать, ни присоединять
протоны; молекулы апротного растворителя не ионизированы.
Нивелирующий растворитель — растворитель, в котором кислоты и основания
различной природы не изменяют соотношения в своей силе.
Дифференцирующий растворитель — растворитель, в котором кислоты и основания
различной природы заметно изменяют соотношения в своей силе.
Примечание. Не следует смешивать классификацию растворителей,
основанную на характере участия их в кислотно-основном процессе, — амфотер-
ные, кислые и основные, с классификацией по признаку их влияния на
относительную силу кислот, солей и оснований, проявляющегося в их способности изменять
соотношения в силе электролитов, — нивелирующие и дифференцирующие
растворители.
194 ГЛ.П. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
основанием в среде безводной уксусной кислоты и слабым основанием
в водном растворе.
Чем сильнее основные свойства растворителя, тем сильнее его
влияние на слабые кислоты.
Дифференцирующее действие некоторых растворителей на силу
растворенных в них электролитов. Исследования И. А. Каблукова, Нернста
и Томсона показали, что, как правило, чем больше величина
диэлектрической проницаемости (е) растворителя, тем лучше диссоциирует
растворенный в нем электролит. Однако наряду с значением е на
поведение электролита также оказывает
существенное влияние и химическая природа
растворителя. Известны растворители,
характеризующиеся приблизительно одинаковыми
значениями е, в которых одно и то же
растворенное вещество ведет себя по-разному.
Например, в нитрометане (е = 37),
нитробензоле (е = 34,5), метаноле (е = 31,5)
некоторые электролиты диссоциируют хорошо,
но многие диссоциируют плохо.
Исследования Вальдена показали, что
в воде и в спиртах (нивелирующих
растворителях) различные по своей природе соли
являются сильными электролитами, а в
нитробензоле, нитрометане и пиридине
(дифференцирующих растворителях) эти соли ведут
И. А. Каблуков (1857—1942) себя неодинаково. На основании этого все
растворители классифицируют как
нивелирующие (в которых электролиты хорошо и примерно одинаково
диссоциируют) и дифференцирующие растворители (в которых электролиты
диссоциируют по-разному, благодаря чему они отличаются друг от друга
в этих средах различными константами диссоциации К или соответственно
значениями р/С).
Кислотная сила растворенного вещества значительно увеличивается
в сильноосновном растворителе. Чем сильнее основные свойства
растворителя, тем сильнее его влияние на слабые кислоты. Все кислоты становятся
одинаково сильными в основных растворителях. Например, в среде
жидкого аммиака слабая синильная кислота становится столь же сильной, как
азотная кислота в водном растворе. Благодаря этому слабые кислоты
можно титровать в неводных растворителях.
Равным образом в кислых растворителях наблюдается сильное влияние
растворителя на основания. Чем сильнее кислотные свойства
растворителя, тем сильнее его влияние на слабые основания. В сильнокислом
растворителе основные свойства растворенного вещества значительно
увеличиваются. Все основания становятся одинаково сильными в кислых
растворителях. Этиловый спирт в среде жидкого фтористого водорода является
ясно выраженным основанием и отличается сильноосновными свойствами,
а уксусная и муравьиная кислоты, характеризующиеся кислыми
свойствами в врдных растворах, проявляют основные свойства в среде
жидкого HF.
§ 43. Применение закона действия масс
к растворам сильных электролитов
Н. А. Измайловым предложена новая схема диссоциации
электролитов, учитывающая все главнейшие процессы, протекающие в
растворах. На основании этой схемы и учета энергии взаимодействия ионов
§ 43. ПРИМЕНЕНИЕ ЗАКОНА ДЕЙСТВИЯ МАСС К ЭЛЕКТРОЛИТАМ 195
и молекул электролитов с растворителем выведены общие уравнения,
характеризующие зависимость силы кислот и оснований от физических и
химических свойств растворителей. Дифференцирующее действие
растворителей связано с различием в энергии сольватации ионов и молекул,
а также с различной ассоциацией ионов.
Диссоциация кислот, солей и оснований на ионы в водных или
неводных растворах зависит от ряда сопряженных динамических равновесий.
Для кислоты НАп эти равновесия можно представить в виде схемы:
1. Недиссоциированные молекулы кислоты НАп реагируют с
молекулами растворителя М, образуя продукты присоединения — сольваты
переменного состава:
НАп + лМ ^± НАпМл (а)
сольват
2. Образующиеся сольваты в результате последующего
взаимодействия с молекулами растворителя диссоциируют с образованием сольвати-
рованных ионов, ионов лиония (сольватированных ионов водорода) Нсол
и ионов лиата (сольватированных анионов кислот) Ап^оЛ:
НАпМл + тЖ ^ Н+ол + Ап;ол (б)
3. Сольватированные ионы в растворителях, особенно отличающихся
низкой диэлектрической проницаемостью, взаимодействуют между собой
с образованием ионных пар, или ионных двойников:
Кол + АЛсол ^ Нсол^сол + *М W
Схема непосредственного превращения продукта присоединения
НАпМл в ионную пару может быть представлена так:
НАпМл + уЖ ^± Н*0ЛАп;0Л + хЖ (г)
где х + у = т.
Соотношения между активными концентрациями продуктов
приведенных выше реакций (НАп, НАпМл, Нс0Л, Апёол и Нс0Л Апёол) зависят
от свойств растворенного электролита и растворителя, а также их
концентраций.
Указанными реакциями далеко не исчерпываются все процессы,
происходящие в неводных растворах электролитов.
В общем виде схема равновесий, устанавливающихся в неводном
растворе кислоты НАп, следующая:
НАп+(/г + т)М^±НАпМл^-тМ^±Н^0Л + Ап;0Л
+ + . w
уЖ ^± н:0ЛАпС0Л + *М
Константы равновесий этих реакций могут быть выражены
следующим образом:
1. Константа нестойкости продукта присоединения как комплексного
соединения [см. уравнение (а)]:
* * я
аНАпаМ
Анест — —^
аНАпМл
Индекс (*) показывает, что активности отнесены к бесконечно
разбавленному раствору НАп.
196 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
В разбавленных растворах активность растворителя ам является
величиной постоянной, поэтому константа нестойкости может быть
выражена в виде уравнения:
*
к» аНАп /лп
аНАпМ„
2. Константа диссоциации образовавшегося продукта присоединения
[см. уравнение (б)] соответственно может быть представлена в виде
уравнения:
* *
ан+ аА-
*Ап
аНАпМ,
„ СОЛ СОЛ /ЛОч
Адисс = ; \°г)
п
так как ам также является величиной постоянной.
3. Константа ассоциации сольватированных ионов в ионные пары
или ионные двойники [см. уравнение (в) ] может быть представлена
формулой:
UHT An
X сол сол
Аасс — *~
аП+ ' аАп"
сол сол
а обратная этой константе величина формулой:
аИ+ актГ
К—\ сил сил /Л9Ч
^асс = * (63)
аН+ An"
сол сол
4. Константа электролитической диссоциации кислоты НАп,
определяемая обычными методами (/(об)> может быть представлена следующим
уравнением:
аН+ актГ
т, СОЛ СОЛ
Лоб. = *
Величина а*- недиссоциированного вещества определяется суммой
активности свободных молекул кислоты андп, активности продукта
присоединения анАпм и активности ионных пар а*н+ ап" , т. е.
л сол сол
* * * •
снедис = аНАп "Г flHAnMn + аН+ An"
'* СОЛ СОЛ
Такое суммирование основано на том, что в предельно разбавленных
растворах а* = С и коэффициент активности у = 1.
Следовательно
аН+ аАп~
„ СОЛ СОЛ
flHAn + аНАпМя + аН+ An" „
«• СОЛ СОЛ
§ 43. ПРИМЕНЕНИЕ ЗАКОНА ДЕЙСТВИЯ МАСС К ЭЛЕКТРОЛИТАМ 197
Выразив в этом уравнении активности анлп, #напм и ан+ An"
П СОЛ СОЛ
через активность сольватированных ионов [уравнение (62)], получим:
аН+ дАп"
* сол сол
аНАпМп ~ F
П АдиСС
UH+ "Ап"
т, СОЛ. СОЛ
А об — * ; * *
"Н-1- иАп "Нт "An UHT "An
СОЛ СОЛ , СОЛ СОЛ . СОЛ СОЛ
К К*'1 Кдисс К~1
*хдисс*хнест м iVacc
Проведя соответствующие математические преобразования, можно
написать:
Коб = ? _ = ^ (64)
1 11 ^нест+1 1
Кдисс*Гест К*»сс <7с К*™с *Тсс
т. е.
А0б == ^дисс \ "•" ^нест/ "Г Аасс
Из уравнения (64) следует, что обратная величина обычной константы
диссоциации является суммой постоянных величин, характеризующих
превращение ионов в недиссоциированные молекулы НАп и НАпМ„ и
в ионные пары Н^лАп
сол»
В растворителях с высокой диэлектрической проницаемостью
ассоциация ионов отсутствует и уравнение принимает следующий вид:
^-^О + Ссг) (65)
В растворителях, отличающихся низкой диэлектрической
проницаемостью И ОСНОВНЫМ Характером, К7б = Касс-
Таким образом, можно заключить, что влияние растворителя на
константу диссоциации кислоты зависит от взаимодействия:
а) между Н+- и An "-ионами кислоты и молекулами растворителя;
б) между недиссоциированными молекулами НАп и молекулами
растворителя;
в) между Н+- и An "-ионами (сопровождается образованием молекул
НАп);
г) между сольватированными ионами (сопровождается
образованием ионных пар, и т. д.).
Уравнение (64) можно представить в следующем виде:
А-об = _*дисс
"г ^нест "г" ^дисс^асс
Обозначив произведение /СДИСс -Касс через /Спр (константа
превращения), получим константу, выражающую состояние равновесия (г):
*нт_ап:
н> сол сол
AjiD — 5
аНАпМи
198 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-OGHOBHOE ТИТРОВАНИЕ
В зависимости от свойств растворенного вещества и растворителя это
уравнение приобретает частные значения. Например, если /Сасс —* О,
Кпр-+0, то
^дисс
#об:
1+*„
если Кя
0; Кп
0; Кнест<1,тоЯ0
Кдисс и т- Д-
vacc r v» JVnp "" v' *хнест "*< х» iV Аоб '
В общем виде для электролита KtAn универсальная схема
рассматриваемых взаимосвязанных равновесий и соответствующих им констант,
указанных над знаками обратимости, по Н. А. Измайлову, представляется
следующим образом:
*и
*п
хнест *хдисс
KtAn+(Ai + m)M -* KtAnM„ + mM ^ Kt^+An^
+ It ^м
к,
*/М
пр
КС0ЛАп- + *М
§ 44. Титрование кислот и оснований в неводных растворах
Ионные произведения растворителей и константы ионизации кислот
и оснований в неводных растворах. Явления, наблюдающиеся в неводных
растворах, имеют исключительно важное значение в аналитической химии.
В неводных средах наблюдается неравномерное изменение отношений
К
НАп
^KtOH
^НАпц и ^КЮНП
^HAnx ^KtOHx
где /Cs—ионное произведение растворителя (табл. 21); /CHAn, ^HAni, *СнАпП'
^KtOH» ^KtOHi и ^KtOHn — соответственно константы электролитической
диссоциации кислот и оснований.
Точность кислотно-основного титрования определяется указанными
отношениями. Чем меньше эти отношения, тем выше точность титриметри-
ческого определения.
Значение ионных произведений некоторых растворителей
приведены в табл. 21.
ТАБЛИЦА 21
Ионные произведения (константы автопротолиза)
некоторых растворителей
Растворитель
Вода
Муравьиная кислота
Уксусная кислота
Серная кислота
*s
ыо-14
5-10-7
2,5-Ю-13
ыо-6
Растворитель
Метанол
Этанол
Гидразин
Аммиак
Ks
2.10-и
8-Ю-20
2-10"25
З-НГ33
Данные табл. 21 показывают, что ионные произведения таких
растворителей, какими являются безводные уксусная, муравьиная и серная
кислоты, больше ионного произведения воды. Особенно большой кон-
§ 44. ТИТРОВАНИЕ КИСЛОТ И ОСНОВАНИЙ В НЕВОДНЫХ РАСТВОРАХ 199
стантой автопротолиза отличается безводная серная кислота (Ы0~б).
Ионные произведения других растворителей меньше Kw> например
константа автопротолиза жидкого аммиака в 1019 раз меньше Kw
Из данных табл. 22 видно, что в среде безводной уксусной кислоты
наиболее сильной является хлорная кислота.
ТАБЛИЦА 22
Константы ионизации кислот в растворе безводной уксусной кислоты
Формула кислоты
НС104
НВг
H2S04
к
1,6-10-4
4-10-6
6-Ю-7
Формула кислоты
НС1
HN03
CCI3COOH
К
1,4-10-2
4,2-Ю-8
2,3-10"10
Такие сильные в водном растворе кислоты, какими являются серная,
хлористоводородная и азотная, становятся в среде безводной уксусной
кислоты слабыми.
Изменение относительной силы кислот и оснований в зависимости
от растворителя. Иллюстрацией изменения относительной силы кислот
могут служить данные для хлористоводородной и хлоруксусной кислот:
Растворитель
^НС1
^CH2ClCOOH
Вода Спирт Метилэтилкетон
631 150 000 10000000
Иллюстрацией изменения отношения KslКк^юн могут служить данные
для мочевины, растворенной в воде и в безводной муравьиной кислоте.
Для муравьиной кислоты
*s (нсоон) = [НСООН^-] [НСОО-] = б-Ю"7
Для мочевины СО (NH2)2 в среде безводной муравьиной кислоты
*СО (NH8)a/HCOOH = 5,6- Ю-2
Для мочевины в водной среде
^со (NH*)2/Hao = 1>5-Ш-14
Поэтому в воде
%w Ы0-14
^CO (NH2b/H20 1,5-10-14
В среде безводной муравьиной кислоты
^нсоон 5-1Q-7
^СО (NH2)2/HCOOH 5>6# 10"2
^1
ю-
Из этого следует, что в среде безводной муравьиной кислоты
происходит увеличение как Ks, так и /СКюн, но при этом отношение Ks/Ккюн,
становится меньше, чем в водной среде. Поэтому в среде НСООН сшга
слабых оснований (подобных мочевине) увеличивается настолько, что их
можно титровать с достаточной точностью как сильные основания.
200 ГЛ.II. НЕЙТРАЛИЗАЦИЯ. ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Аналогично в среде основных растворителей [подобных диметилформ-
амиду — HCON (СН3)21 сила слабых кислот (подобных фенолу)
увеличивается настолько, что их можно титровать как сильные кислоты.
Устранение помех, вызываемых сольволизом. Трудности,
возникающие при титровании слабых оснований и слабых кислот в водных
растворах, можно устранить, применяя соответствующий неводный
растворитель. Например, константа гидролиза (см. книга 1, гл. II, § 13)
к — ^w v — ^w v — ^w
Агидр — —7? ; Ащдр — -7? ; Агидр — ~77 7^
AKtOH Анап AKtOH-AHAn
(NH4C1) (CH3COONa) (CH3COONH4)
или, в общем виде, константа сольволиза равна:
к - Ks . к - Ks к - Ks
АсоЛ — J? , АсоЛ — р , А СОЛ — —т? 7?
AKtOH AHAn AKtoH-AHAn
(NH4C1) (CHgCOONa) (CHsCOONH4)
Сольволиз можно подавить или усилить, применяя растворитель,
отличающийся соответствующим значением Ks-
Например, заменяя воду абсолютным спиртом, Ks которого (8-Ю-20)
меньше Kw (Ю-14), и не изменяя сильно значение Ккюн, можно
замедлить процесс сольволиза и тем самым способствовать появлению более
резкого изгиба кривой титрования.
Таким образом, представляется возможность устранить многие
помехи, встречающиеся при анализе водных растворов, и использовать
неводные среды в тех случаях, когда применение водных растворов
невозможно.
Применение неводных растворов для кислотно-основного титрования.
В настоящее время неводные среды используют в аналитической практике
для титрования разнообразных неорганических и органических веществ
и для дифференцированного (раздельного) титрования
многокомпонентных смесей солей, кислот и основание Благодаря этому возможно
титровать:
не только сильные кислоты и основания, но и слабые и очень слабые
кислоты и основания;
смеси сильных кислот, смеси сильных оснований;
смеси сильных и слабых кислот, смеси сильных и слабых оснований;
смеси слабых и очень слабых кислот, смеси слабых и очень слабых
оснований;
смеси сильных, слабых и очень слабых кислот;
смеси сильных оснований и солей слабых кислот;
смеси сильных кислот и солей слабых оснований;
смеси свободных и связанных кислот;
соли неорганических и органических кислот;
вещества, не содержащие водорода и не являющиеся донорами или
акцепторами протонов, но являющиеся (по Льюису) кислотами или
основаниями и т. д.
Вещества, определяемые в неводных растворах. Быстрое развитие
методов неводного титрования привело к тому, что уже в настоящее время
можно количественно определять в неводных растворах гораздо больше
веществ, чем в водной среде.
Так, в соответствующих неводных средах можно титровать любые
кислоты и основания, амины, нитросоединения, фенолы, аминофенолы,
§ 44. ТИТРОВАНИЕ КИСЛОТ И ОСНОВАНИЙ В НЕВОДНЫХ РАСТВОРАХ 201
аминокислоты, алкалоиды, ангидриды и хлорангидриды, соли
органических и неорганических кислот и т. п.
В водной среде, вследствие «нивелирующего эффекта» воды,
невозможно получить два или несколько изгиба кривой титрования смеси
двух или нескольких кислот или оснований до тех пор, пока величины
их р/С не будут сильно отличаться друг от друга. Например, нельзя в
водной среде раздельно оттитровать смеси уксусной и серной кислот,
хлористоводородной и муравьиной кислот, серной и молочной и т. д.
ок
Рис. 50. Кривые потенциометрического титрования многокомпонентных смесей
кислот в среде метилэтилкетона.
При титровании смесей кислот в среде неводных растворителей
наблюдается несколько скачков титрования. Например, при титровании смеси
хлористоводородной и муравьиной кислот в среДе абсолютного спирта
на кривой титрования наблюдается два скачка. В среде ацетонитрила
можно дифференцированно (раздельно) оттитровать хлорную и уксусную
кислоты; в гликолевой среде раздельно титруются азотная и уксусная
кислоты; в пиридине — фенол и уксусная кислота и т. д.
Как показали наши исследования, в среде метилэтилкетона могут
быть оттитрованы бензольно-метаноловым раствором гидроокиси тетра-
этиламмония не только индивидуальные сильные, слабые и очень слабые
кислоты, но и их двух-, трех-, четырех-, пяти- и шестикомпонентные
смеси, которые не могут быть оттитрованы в водных растворах (рис. 50).
При титровании двухосновных кислот в среде метилэтилкетона
получается два скачка титрования даже у таких кислот, у которых первые
и вторые константы диссоциации в водных растворах мало отличаются
друг от друга, например у щавелевой (Кг = 5,9-10"2 и К2 = 6,4-10"5),
янтарной (Кг = 6,4-10"5 и К2 = 3,0-10"6), яблочной (Кг = 3,8-10~4 и
К2 = 7,80-10-в).
Все это говорит о значительных преимуществах методов титрования
в неводных средах по сравнению с титрованием в водных растворах.
Особенности титрования в неводных средах. Быстрое развитие
методов титрования в неводных средах объясняется многими их достоинствами:
202 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
1. Титрование в неводных средах представляет собой очень простой,
быстрый и удобный метод количественного анализа многих
неорганических, органических и элементорганических соединений.
2. Методы титрования в неводных растворах дают возможность с
большой аналитической точностью определять многочисленные вещества,
которые при титровании в водной среде не дают резких конечных точек
титрования.
3. Одним из важнейших преимуществ методов неводного титрования
является то, что они позволяют определять не только растворимые, но и
нерастворимые в воде соединения, а также вещества, разлагаемые водой
или образующие в водных растворах стойкие нерасслаиваемые эмульсии.
4. Методы титрования в неводных средах могут быть применены для
титрования как бесцветных, так и окрашенных растворов.
5. Титрование неводных растворов может проводиться индикаторным,
потенциометрическим, кондуктометрическим, амперометрическим и
другими физико-химическими методами.
6. При методах неводного титрования во многих случаях не нужно
предварительно разделять анализируемые вещества и отделять
сопутствующие им примеси или наполнители.
7. Вследствие небольшого, как правило, поверхностного натяжения
органических растворителей размеры капель неводных жидкостей меньше
размеров капель водных растворов, благодаря чему повышается точность
титрования по сравнению с точностью титрования водных растворов.
8. Использование неводных растворителей в аналитической практике
дает возможность расширить области их применения в других методах
анализа (осаждения, комплексообразования, окисления —
восстановления, хроматографии, электрометрических методах и т. д.) и увеличить
ассортимент веществ для приготовления титрованных растворов,
пригодных для титрования как мономерных, так и полимерных соединений.
§ 45. Методы кислотно-основного титрования в неводных
средах
Растворители. В качестве неводных растворителей для
кислотно-основного титроЕания применяют: кислые растворители в качестве сред для
титрования слабых оснований; основные растворители для титрования
слабых кислот; амфотерные, инертные и смешанные растворители—для
титрования слабых кислот и оснований.
Наиболее широко в практике неводного титрования применяются
безводные кислоты, гликоли и их смеси, амины, кетоны, спирты, диоксан,
ацетонитрил, диметилформамид, хлороформ, четыреххлористыи углерод
и др.
Кислые растворители. К числу кислых растворителей
относятся безводные кислоту (муравьиная, уксусная, хлоруксусные, про-
пионовая, серная), а также жидкий цианистый водород, жидкий
фтористый водород, жидкий сероводород, гликоли и их смеси и др.
Из кислых растворителей наиболее широко используются в качестве
среды для титрования оснований безводная уксусная кислота, гликоли и
их смеси с другими растворителями.
Основные растворители. К даслу основных
растворителей относятся: жидкий аммиак и его жидкие производные (среди них
в аналитических целях наиболее исследованы одно-, двух- и
многоатомные амины, являющиеся аналогами спиртов и гляколей), гидразин,
гидроксиламин, пиридин, формамид, диметилформамид и др.
§ 45. МЕТОДЫ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ 203
В качестве среды для титрования кислот широко применяются этилен-
диамин, пиридин, формамид, диметилформамид.
Амфотерные растворители. К числу неводных амфотер-
ных (амфипротных) растворителей относятся главным образом спирты
(метанол, этанол, пропанол, изопропанол, изобутанол) и их смеси с
инертными растворителями (например, с бензолом). К этой же группе можно
отнести и ряд других растворителей (например, ацетонитрил, ацетон,
метилэтилкетон, метилизопропилкетон, метилизобутилкетон и др.).
Из амфотерных растворителей наиболее широкое применение в
качестве сред для титрования нашли этанол, кетоны, ацетонитрил и др.
Инертные растворители. Инертные растворители не
отдают и не принимают протонов, т. е. не реагируют ни с кислотами,
являющимися донорами протонов, ни с основаниями, являющимися
акцепторами протонов. К числу инертных растворителей относятся жидкие
углеводороды (бензол, толуол, циклогексан и др.), уайт-спирит, петро-
лейный эфир, хлороформ, четыреххлористый углерод, дихлорэтан, тетра-
хлорэтан и др. Наибольшее значение из них приобрел бензол,
применяемый в смеси с другими органическими растворителями.
Смешанные растворители. Иногда индивидуальным
растворителям предпочитают смешанные растворители, отличающиеся
более высокой растворяющей способностью, определенным значением
диэлектрической проницаемости и другими нужными свойствами.
Например, при смешивании диоксана, характеризующегося хорошей
растворяющей способностью, но имеющего малую величину диэлектрической
проницаемости (е = 2), с водой (е = 81,5) можно получить растворитель,
отличающийся повышенной растворяющей способностью и любым
значением е в пределах от 2 до 80.
Из смешанных неводных растворителей наиболее широко применяются
в качестве среды для титрования разнообразных веществ гликоли в смеси
с алифатическими и ароматическими углеводородами, хлорпроизводными
углеводородов, спиртами, эфирами и кетонами, бензол в смеси с
метанолом и этанолом.
Специальные растворители. К числу специальных
растворителей, применяемых для исследования процессов
нейтрализации, относятся: жидкая двуокись серы, сульфурилхлорид, тионилхлорид,
оксихлорид селена, фосген, фторид бора и др.
Стандартные растворы. В качестве стандартного раствора
для титрования веществ осцовного характера применяют 0,1 н. раствор
НСЮ4 в безводной уксусной кислоте или б диоксане. Такой раствор
готовят смешением 8,5 мл 72%-ной НСЮ4 с 900 мл безводной уксусной
кислоты и 30 мл уксусного ангидрида или с 1 л диоксана.
Установку титра раствора хлорной кислоты ведут по гидрофталату
калия, который растворяют при нагревании в безводной уксусной кислоте
(0,5 г гидррфталата + 50 мл СН3СООН). Титрование проводят в
присутствии кристаллического фиолетового.
Кроме гидрофталата калия, применяют карбонат натрия,
растворяющийся в безводной уксусной кислоте с образованием ацетата натрия.
В качестве стандартного раствора для титрования веществ
кислотного характера применяют 0,1 н. растворы метилата калия или натрия
в смеси бензола с метанолом или 0,1 н. спиртовой раствор гидроокиси
тетраэтиламмония. Такие растворы готовят, растворяя рассчитанное
количество металлического натрия в смеси из 1 части метанола с 6 частями
бензола или рассчитанное количество металлического калия в смеси,
состоящей из 1 части метанола и 12 частей бензола. Для приготовления 0,1 н.
раствора гидроокиси тетрабутиламмония или гидроокиси тетраэтилам-
204 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
мония рассчитанное количество йодистого тетралкиламмония растворяют
в безводном метиловом спирте и встряхивают полученный раствор с окисью
серебра (см. ниже). Гидроокись тетралкиламмония получают также
методом ионного обмена при пропускании раствора галогентетралкиламмония
через ионит в ОН-форме.
Титрование в инертных и смешанных растворителях. Метод
титрования в инертных растворителях был применен для определения кислых
составных частей масел, жиров, смол, асфальта, каменноугольной смолы
и т. п., многих органических кислот (салициловой, ^-нитробензойной,
га-аминобензойной, стеариновой, бензойной, уксусной, щавелевой,
малоновой, янтарной, глутаровой, адипиновой, камфарной, фумаровой, ма-
леиновой, винной, яблочной, фталевой, антраниловой и др.), борной
кислоты, ангидридов и хлор ангидридов и целого ряда других веществ.
Титрование в среде кислых растворителей. В среде кислых
растворителей сильно возрастают (нивелируются) основные свойства слабых
оснований по сравнению с их водными растворами.
Метод использовался для количественного анализа первичных,
вторичных и третичных алифатических и ароматических аминов, жирных
аминов (с большим молекулярным весом) и их производных, аминокислот,
аминоспиртов, амидов кислот, сульфонамидов, оксазолинов, пиридин-
карбоновых кислот и их производных, пуринов, пиразолонов,
алкалоидов, витаминов, четвертичных аммониевых солей, комплексных
соединений, солей неорганических и органических кислот и т. д.
Титрование в среде основных растворителей. В среде основных
растворителей сильно возрастают кислые свойства слабых кислот по
сравнению с их водными растворами. Метод был применен для количественного
анализа карбоновых кислот, фенолов и их производных, сульфамидов,
енолов, имидов, нитросоединений, оксимов, аминокислот и т. д.
Титрование в среде амфотерных растворителей. Многие амфотерные
растворители обладают высокими дифференцирующими свойствами, что
дает возможность в их среде раздельно титровать многокомпонентные смеси
сильных, слабых и очень слабых кислот или оснований.
На кафедре аналитической химии МХТИ им. Д. И. Менделеева
разработаны методы кислотно-основного титрования в среде спиртов, кето-
нов и нитрилов. Эти методы позволяют количественно определять многие
моно- и дикарбоновые кислоты, их галоген-, нитро- и оксипроизвод-
ные, фенолы и их производные, а также смеси карбоновых кислот друг
с другом, с фенолами и минеральными кислотами; амины, диамины и их
производные, гетероциклические азотсодержащие соединения и их смеси,
кремнийорганические соединения, а также мономерные и полимерные
продукты.
§ 46. Примеры практических определений в неводных растворах
Определение воды в исходных органических растворителях по методу
Фишера. Определение воды по методу Фишера основано на реакции,
которую схематически можно представить следующим образом:
I2 + S02 + 2H20 ^± 2HI + H2S04
или
U + S02 + Н20 + СН3ОН + 3C6H5N -* C5H6NOS02OCH3 + 2C6H5NH + 21"
н
Для приготовления реактива Фишера растворяют йод и пиридин в'
метаноле, затем добавляют жидкий сернистый ангидрид. Титр реактива
Фишера определяют по стандартному раствору воды в метаноле.
§ 46. ПРИМЕРЫ ПРАКТИЧЕСКИХ ОПРЕДЕЛЕНИЙ
205
Для приготовления стандартного раствора в сухую мерную колбу с
притертой пробкой наливают 900 мл сухого метанола и помещают колбу
в водный термостат (25° С). В этот же термостат помещают меньшую
колбу, в которую налито 200 мл метанола. Затем отвешивают 15,0 г
дистиллированной воды и вливают в первую колбу. Когда содержимое
колбы примет температуру термостата, объем жидкости в колбе доводят
до метки, добавляя метанол из меньшей колбы.
Определение титра стандартного раствора воды в метаноле и титра
реактива Фишера. 2—3 пробы по 25,0 мл сухого метанола и 2—3 пробы
по 10,0 мл стандартного раствора пипеткой переносят в сухие колбы и
титруют реактивом Фишера. Титр устанавливают с предельной точностью.
Расчет проводят исходя из следующих данных:
V — средний объем реактива Фишера, требующийся на титрование
25 мл метанола (в миллилитрах);
V — объем (в миллилитрах) реактива Фишера, требующийся на
титрование 9,85 мл метанола, рассчитанный по величине V (10 мл
стандартного раствора, содержащего 0,15 мл воды в 9,85 мл
метанола);
V" — средний объем реактива Фишера, требующийся для титрования
10 мл стандартного раствора (в миллилитрах);
у " — V — объем реактива Фишера, требующийся на
титрование g мг воды, введенных в 10 мл стандартного
раствора (в миллилитрах);
v*f_y = Тф/н2о — масса воды, эквивалентная 1 мл реактива Фишера
(в миллиграммах);
g + У'Тф/н2о — масса воды, содержащейся в 10 мл стандартного
раствора (в миллиграммах).
В колбу для титрования пипеткой переносят 10,0 мл исследуемого
образца органического растворителя и титруют реактивом Фишера. При
приближении к конечной точке (о чем можно судить по уменьшению
скорости исчезновения бурой окраски и слабому изменению оттенка
«отработанного» реактива от канареечно-желтого до хроматно-желтого цвета)
реактив добавляют по 0,1—0,2 мл до тех пор, пока бурый цвет иода не
перестанет исчезать. Конечное изменение окраски от хроматно-желтого
до красновато-коричневого цвета иода отчетливо и воспроизводимо.
Влажность растворителя в объемных процентах (х) определяют по
формуле:
»_ тф/н2оУф 100
*- р тг
где Тф^На0 — масса воды, эквивалентная 1 мл реактива Фишера, мг\
Уф — объем реактива Фишера, израсходованный на титрование, мл\
р — плотность воды при температуре опыта;
УА — объем анализируемого растворителя, мл.
Приготовление титрованного раствора хлорной кислоты в диоксане.
Для титрования оснований в среде безводной уксусной кислоты иногда
применяют 0,1 н. раствор хлорной кислоты в диоксане. При этом конец
титрования наблюдается более четко, чем при использовании
титрованного раствора хлорной кислоты в среде безводной уксусной кислоты.
Конец титрования определяют либо по изменению окраски индикатора,
либо потенциометрически (см. гл. VII, § 5).
Для приготовления раствора исходной кислоты 8,5 мл 72%-ной
хлорной кислоты и 30 мл уксусного ангидрида растворяют в 1 л очищенного
диоксана. При наличии более разбавленной хлорной кислоты берут
206 ГЛ.II. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
соответственно большие количества кислоты и уксусного ангидрида. Титр
устанавливают по гидрофталату калия, растворенному в безводной
уксусной кислоте (см. выше).
Диоксан имеет почти такой же температурный коэффициент
расширения, как и уксусная кислота. При расчетах поэтому также необходимо
вводить соответствующую температурную поправку (см. ниже).
Для очистки диоксан в течение 1 ч встряхивают с асбестом, а затем
фильтруют, перегоняют над металлическим натрием и пропускают через
ионообменную колонку, заполненную ионитом в ОН-форме.
Приготовление титрованного раствора хлорной кислоты в ацетоне
или метилэтилкетоне. Для титрования смесей оснований в среде
дифференцирующих растворителей приготавливают раствор хлорной кислоты
в среде ацетона или метилэтилкетоне. Для этого растворяют 8,5 мл 75%-
ной хлорной кислоты в 1 л безводного кетона и насыщают раствор сухим
азотом в течение 5 мин. Раствор хранят в склянке-, снабженной трубкой,
наполненной хлоридом кальция.
Приготовление титрованного раствора хлорной кислоты в безводной
уксусной кислоте. Для титрования оснований в среде безводной уксусной
кислоты обычно применяют 0,1 н. раствор хлорной кислоты в безводной
уксусной кислоте или в диоксане.
Для этого к 900 мл ледяной уксусной кислоты постепенно прибавляют
при непрерывном перемешивании 8,5 мл 72%-ной хлорной кислоты и
затем рассчитанное количество (около 30 мл) уксусного ангидрида. При
наличии более разбавленной хлорной кислоты (например, 60%-ной)
берут соответственно большие количества кислоты и уксусного ангидрида
для связывания воды.
Титр раствора хлорной кислоты устанавливают по гидрофталату калия,
растворенному в безводной уксусной кислоте (см. выше). Для этого навеску
.около 0,5 г гидрофталата калия при нагревании растворяют в уксусной
кислоте и доводят объем раствора до 50 мл. Помимо гидрофталата калия,
применяют безводный карбонат натрия, который растворяется в
безводной уксусной кислоте с образованием ацетата натрия.
Титрование ведут в присутствии двух капель индикатора
кристаллического фиолетового (0,2%-ный хлорбензольный раствор). Конец
титрования соответствует переходу окраски от синей к сине-зеленой.
Реакция гидрофталата калия с хлорной кислотой протекает согласно
уравнению:
НООС-С6Н4-СООК + НСЮ4 —> НООС—СбН4—СООН + КС104
Для установки титра хлорной кислоты вместо индикаторного метода
применяют также потенциометрическое титрование (см. гл. VII, § 5).
Если титрованный раствор применяют при температурах, отличных
от тех, при которых был установлен титр кислоты, то необходимо вводить
температурную поправку. Для этого умножают величину объема
израсходованного раствора на разность (1 —0,0011), если нитруют при более
высокой температуре, и на сумму (1 + 0,0011), если титруют при более
низкой температуре. Здесь t — разность температур при установке титра
и при титровании.
Титрование анилина в среде неводных растворителей. Навеску
исходного анилина около 0,3—0,5 г, взятую на аналитических весах,
растворяют в конической колбе в 50 мл безводной уксусной кислоты и титруют
0,1 н. раствором хлорной кислоты. В качестве индикатора применяют
0,2%-ный хлорбензольный раствор кристаллического фиолетового. В точке
эквивалентности фиолетовая окраска индикатора меняется на синюю.
§ 46. ПРИМЕРЫ ПРАКТИЧЕСКИХ ОПРЕДЕЛЕНИЙ
207
Конечная точка титрования соответствует началу перехода окраски от
синей к сине-зеленой.
Конец титрования можно определить также потенциометрически.
Титрование анилина в безводной уксусной кислоте хлорной кислотой
протекает согласно уравнениям:
c6h5nh2 + сн3соон ^± c6h5nhg + ch3coo"
нс104 + сн3соон ^± сн3соон2 + сю4
сн3соон2 + сн3соо~ —* 2сн3соон
c6h5nh3+cio; —* c6h5nh2.hcio4
c6h5nh2 + нсю4 —> c6h5nh2-hc104
Расчет проводят хэбычным путем (см. гл. I, § 10).
Дифференцированное (раздельное) определение свободной щелочи
в присутствии солей одноосновных органических кислот (например,
NaOH + CH3COONa). Растворяют 0,2—0,5 г анализируемого вещества
в 25 мл гликолевой смеси — этиленгликоль + изопропиловый или я-
бутиловый спирт 1 : 1 (по объему), к которой прибавляют несколько капель
крезолового красного или фенолфталеина. Розовое окрашивание
указывает на присутствие щелочи. Анализируемую смесь титруют стандартным
0,1 н. гликолевым раствором НСЮ4. Когда вся свободная щелочь
нейтрализуется, розовая окраска исчезает. Объем стандартного раствора
кислоты, пошедший на титрование исследуемого вещества к моменту
изменения окраски, соответствует содержанию свободного основания в
анализируемом образце. Затем к этому же раствору прибавляют несколько
капель метилового оранжевого или метилового красного. При этом
раствор окрашивается в желтый цвет.
Раствор снова титруют 0,1 н. раствором НСЮ4 до тех пор, пока
индикатор не изменит окраску на розовую. Объем титрованного раствора
хлорной кислоты, пошедшей на титрование с момента изменения окраски
фенолфталеина до появления розовой окраски, соответствует содержанию соли
слабой кислоты.
Расчет проводят обычным путем (см. гл. I, § 10).
Вместо описанного двойного индикаторного метода можно применить
потенциометрический метод титрования. Кривые титрования дают два
резких изгиба; первый возникает при более высоком значении рН, что
соответствует содержанию свободного основания; второй — при более
низком значении рН, что соответствует содержанию соли органической
кислоты.
Приготовление стандартного (титрованного) спиртового раствора
едкого кали. Для титрования кислот в неводных растворах используют
0,1 н. спиртовые растворы едкого кали. Для их приготовления растворяют
навеску (5—6 г) кристаллического едкого кали в 1 л метилового,
этилового или изопропилового спирта, отфильтровывают осадок карбонатов и
полученный раствор хранят в склянке, снабженной хлоркальциевой
трубкой, которую наполняют натронной известью и хлоридом кальция.
Приготовление стандартного (титрованного) раствора гидроокиси те-
траэтиламмония. Обычно готовят бензольно-метаноловый раствор
гидроокиси тетраэти л аммония. Получение указанной гидроокиси основано на
реакции:
2(C2H5)4NI+Ag20 + HOH -> 2AgI + 2 (С2Н5)4 NOH
208 ГЛ.П. НЕЙТРАЛИЗАЦИЯ, ИЛИ КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Для приготовления 0,1 н. раствора гидроокиси тетраэтиламмония 30 г
перекристаллизованного и высушенного на воздухе йодистого
тетраэтиламмония растворяют в 300 мл абсолютного метилового спирта; к
полученному раствору прибавляют 30 г тонко измельченной чистой окиси
серебра и энергично перемешивают при комнатной температуре в течение 1 ч.
Центрифугированием отделяют несколько миллилитров жидкости и
испытывают на присутствие Г-ионов. При отсутствии их перемешивание
прекращают. В противном случае к смеси дополнительно прибавляют 2 г
свежей окиси серебра и снова перемешивают в течение 30 мин. По окончании
реакции осадок отфильтровывают, сосуд споласкивают тремя порциями
(по 50 мл) сухого бензола, бензольный раствор фильтруют через ту же
воронку и добавляют к первоначальному раствору (фильтрату).
Затем фильтрат разбавляют до 1 л сухим бензолом, насыщают в
течение 5 мин сухим азотом и сохраняют в склянке, снабженной хлоркальцие-
вой трубкой с натронной известью и плавленым СаС12 для предохранения
раствора от влаги и двуокиси углерода воздуха.
Установка титра стандартных растворов оснований. Титр спиртового
раствора едкого кали или бензольно-метанолового раствора гидроокиси
тетраэтиламмония устанавливают по х. ч. бензойной кислоте в
присутствии тимолового -синего до изменения окраски от желтой к синей или
потенциометрически (см. гл. VII, § 5) в среде того растворителя, в
котором проводят определение. Проводят «холостое» титрование для
нейтрализации кислых примесей в растворителе.
Дифференцированное (раздельное) титрование смеси фталевой и ма-
леиновой кислот в среде кетонов. В стакан для титрования емкостью
100 мл наливают 30 мл ацетона или метилэтилкетона, 2—3 капли
индикатора тимолового синего и точно нейтрализуют стандартным раствором
основания содержащиеся в растворе кислые примеси. Затем в стакан
помещают навеску анализируемой смеси или аликвотную часть исследуемого
раствора (содержащие приблизительно по 0,3 мг-экв каждой кислоты)
и титруют потенциометрическим методом стандартным спиртовым
раствором едкого кали. Во время титрования смесь перемешивают магнитной
мешалкой. На основании полученных данных строят кривую титрования
в координатах милливольты—миллилитры.
Первый скачок титрования соответствует совместной нейтрализации
одной карбоксильной группы фталевой и одной карбоксильной группы
малеиновой кислот; второй ч— нейтрализации второй карбоксильной
группы фталевой, а третий — нейтрализации второй карбоксильной группы
малеиновой кислоты.
Расчет результатов определения проводят, как указано в гл. I, § 10.
При этом из общего объема израсходовалного титранта вычитают
объем, пошедший на нейтрализацию кислых примесей в исходном
растворителе.
Дифференцированное титрование смеси салициловой и бензойной
кислот. Определение проводят так же, как при титровании смеси
фталевой и малеиновой кислот. В данном случае получают кривую с двумя
скачками титрования, из которых первый соответствует нейтрализации
салициловой кислоты, а второй — нейтрализации бензойной кислоты.
Дифференцированное (раздельное) определение свободной кислоты
и солей аммония при их совместном присутствии (например, HN03 +
+ NH4N03). Навеску анализируемого вещества около 0,1 г растворяют
в 2 каплях метанола и 20 мл ацетона, полученный раствор обрабатывают
5 мл стандартного бензольно-метанолового раствора гидроокиси
тетраэтиламмония. Гидроокись тетраэтиламмония нейтрализует азотную
кислоту и вступает в обменное взаимодействие с нитратом аммония, сопрово-
§ 46. ПРИМЕРЫ ПРАКТИЧЕСКИХ ОПРЕДЕЛЕНИЙ
209
ждающееся образованием эквивалентного количества свободного
основания (NH4OH). Этот процесс может быть представлен следующим
уравнением:
HN03 + NH4N03 + (m + 2) (С2Нб)4 NOH -+
—> Н20 + NH4OH + 2 (С2Нб)4 NN03 + т (С2Нб)4 NOH
Полученную смесь оснований (гидроокись тетраэтиламмония и
гидроокись аммония) титруют потенциометрическим методом стандартным метил-
этилкетоновым раствором хлорной кислоты. Это взаимодействие может
быть представлено следующим уравнением:
NH4OH + m(C2H5)4NOH + (m + 1)НСЮ4 —> NH4C104 + m(C2H5)4 NC104 + (m + 1) H20
-(C2H5)4NOH
^NH4OH
-J 1 I I I ! I L_
4hcio4
Объем стандартного
раствора HC104,/w
Рис. 51. Кривая титрования
метаноло-ацетонового
раствора гидроокиси аммония ме-
тилэтилкетоновым раствором
хлорной кислоты.
Объем стандартного
раствора ГЩ мл
Рис. 52. Кривая
титрования метаноло-метилэтил-
кетонового раствора
уксусной и хлорной кислот
бензольно-метаноловым
раствором гидроокиси
тетраэтиламмония .
На кривой титрования (рис. 51) наблюдается два скачка: первый
соответствует титрованию избыточного количества гидроокиси
тетраэтиламмония, не вступившего в реакцию взаимодействия с компонентами
анализируемой смеси; второй — титрованию гидроокиси аммония,
образовавшейся при взаимодействии соли с гидроокисью тетраэтиламмония.
Расчет содержания компонентов анализируемой смеси проводят по
следующим формулам:
Содержание нитрата аммония в смеси (в г):
__ (V' — ]r)N'3e
?nh4no, - iooo
Содержание азотной кислоты в смеси (в г):
(VN — V'N')BK
?hno, — iooo
где V — общий объем стандартного раствора хлорной кислоты, израсходованный на
титрование остаточного количества гидроокиси тетраэтиламмония и гидроокиси
аммония, мл;
V" — объем стандартного раствора хлорной кислоты, израсходованный на титрование
остаточного количества гидроокиси тетраэтиламмония, мл;
V — объем (5 мл) стандартного раствора гидроокиси, добавляемый в анализируемый
раствор перед началом определения, мл;
N — нормальность стандартного раствора гидроокиси тетраэтиламмония;
N' — нормальность стандартного раствора хлорной кислоты;
Эс — эквивалентный вес соли (нитрата аммония);
Эк — эквивалентный вес азотной кислоты.
210 ГЛ. II. НЕЙТРАЛИЗАЦИЯ ИЛИ КИСЛОTHO-OCHOBНОЕ ТИТРОВАНИЕ
Дифференцированное (раздельное) определение свободной кислоты
и соли одноосновной кислоты при их совместном присутствии (например:
CHgCOOH + CHgCOONa). Навеску анализируемого вещества 0,1 г
растворяют в 2 каплях метанола и 20 мл метилэтилкетона, полученный
раствор обрабатывают 5 мл стандартной хлорной кислоты в метилэтилкетоне.
Хлорная кислота вступает в обменное взаимодействие с ацетатом натрия,
сопровождающееся образованием эквивалентного количества уксусной
кислоты. Этот процесс может быть представлен следующим уравнением:
CHgCOOH + CHgCOONa + (т + 1) НС104 —> 2СН3СООН + NaC104 + mHCI04
Полученную смесь кислот (хлорная кислота и уксусная кислота)
титруют потенциометр ическим методом стандартным бензольно-метаноло-
вым раствором гидроокиси тетраэтил аммония. Это взаимодействие может
быть представлено следующим уравнением:
2СН3СООН + тНСЮ4 + (т + 2) (С2НБ)4 NOH —>
—> 2 (С2Нб)4 NCH3COO + т (С2Нб)4 NC104 + (т + 2) Н20
На кривой титрования (рис. 52) наблюдается два скачка: первый
соответствует титрованию избыточного количества хлорной кислоты, не
вступившей в реакцию взаимодействия с ацетатом натрия, второй —
титрованию суммарного количества уксусной кислоты (уксусной кислоты,
содержащейся в анализируемой смеси, и уксусной кислоты, выделившейся из
ацетата натрия при его взаимодействии с хлорной кислотой).
Расчет содержания компонентов анализируемой смеси проводят по
следующим формулам:
Содержание ацетата натрия в смеси (в г):
_ (VN' — VnN)3c
?cH8COONa ~~ ЮОО
Содержание уксусной кислоты в смеси (в а):
_ (ум — УМ')ЭК
?сн8соон — fooo
где V — общий объем стандартного раствора гидроокиси тетраэтиламмония,
израсходованный на титрование суммарного количества кислот (избыточного количества
хлорной кислоты, уксусной кислоты, содержащейся в смеси, и уксусной кислоты,
выделившейся из ацетата натрия при его взаимодействии с хлорной кислотой),
мл;
V" — объем стандартного раствора гидроокиси тетраэтиламмония, израсходованный
на титрование избыточного количества хлорной кислоты, мл;
V — объем (5 мл) стандартного раствора хлорной кислоты, добавляемого в
анализируемую смесь перед началом определения, мл;
N — нормальность стандартного раствора гидроокиси тетраэтиламмония;
W — нормальность стандартного раствора хлорной кислоты;
Эс — эквивалентный вес соли (ацетата натрия);
Эк — эквивалентный вес уксусной кислоты.
ГЛАВА III
МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
(ОКСИДИМЕТРИЯ, ОКСРЕДМЕТРИЯ, РЕД-О КС-МЕТОДЫ)
А. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
§ 1. Значение окислительно-восстановительных потенциалов
Понятие об окислительно-восстановительных методах титрования.
Методы окисления—восстановления основаны на использовании реакций
окисления—восстановления. Поэтому иногда эти методы называют ред-
окс-методами*. В качестве стандартных (титрованных) растворов в
методах окисления—восстановления применяют растворы самых
разнообразных окислителей и восстановителей:
окислители: перманганат калия, растворы элементарного иода, би-
хромат калия, гексанитратоцерат (IV) аммония (NH4)2 [Ce (N03)6j, бро-
мат калия, иодат калия, йодная кислота НбЮ6, периодат калия КЮ4,
ванадат аммония NH4V03, перкупраты (соединения Си111, например,
К7 [Си (Юв)2], К [Си (Те04)2Ь соединения свинца (IV) и многие другие;
восстановители: соль Мора (NH^ [Fe (SO^K сульфат железа (II),
хлорид титана (III), хлорид олова (II), сульфат хрома (II), аскорбиновая
кислота СвН806, соединения мышьяка (III), хлорид меди (I), гидрохинон
С6Н4 (ОН)2, тиосульфат натрия Na2S203, щавелевая кислота, перекись
водорода и многие другие.
Как правило, восстановители титруют растворами окислителей, а
окислители — растворами восстановителей.
Например, соединения низших степеней окисления многих элементов
(Fe11, Cr11, Mn11, Tim, VIV, UIV и т. п.) окисляют в соединения
высшей степени окисления (Fe111, Cr111, MnIV, TiIV, Vv, UVI и т. д.)
титрованием такими сильными окислителями, какими являются
перманганат, бихромат и др.:
мпо4
Fe++ ^ Fe+++
Соединения высших степеней окисления многих элементов
восстанавливают в их соединения низших степеней окисления титрованием такими
сильными восстановителями, какими являются Sn11, Cr11, Tim и т. п.:
Си++ -U- Си+
Очень часто определяемый окислитель предварительно
восстанавливают до низшей степени окисления, а затем титруют стандартным
раствором другого окислителя:
МпОГ
ре+ + + Zn-амалыама^ ре++ ^ ре+ + +
* Термин «ред-окс» часто встречается в химической литературе. Он происходит от
слияния начальных слогов двух английских слов: reduction — восстановление и oxidation —
окисление.
212
ГЛ. III. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
Иногда определяемый восстановитель предварительно окисляют до
высшей степени окисления, а затем титруют стандартным раствором
другого восстановителя:
«+ +
K2CraO, Fe*
Се+ + + -> Се+ + + + -> Се++ +
Н8Р04
Посредством стандартных растворов окислителей и восстановителей
можно определять и вещества, не обладающие окислительными или
восстановительными свойствами, но осаждающиеся в виде нерастворимых
соединений при действии окислителей или восстановителей. Определение
такого рода веществ основано на предварительном их осаждении и
последующем титровании ионов, связанных в осадок, или избытка окислителя,
или восстановителя, не вошедшего в реакцию:
Са++ + *С2С?*~ —> I СаС204 + (х — 1) С2С>4 " (изб)
М1Ю4
(*-DC2o4 —^—* со2
2 Ва++ + *Сг20~ ^? 2 |ВаСг04 + (* — 1) Сг207~ (изб)
Fe++
(* — 1)Сг207" -> 2Сг+++ + 7НоО
27 14Н+ 1
Изменение окислительно-восстановительных потенциалов в процессе
титрования. В процессе титрования по методу
окисления—восстановления наблюдается изменение окислительно-восстановительных
потенциалов взаимодействующих друг с другом систем. Поэтому, прослеживая за
изменением ред-окс-потенциалов систем во время титрования, можно
судить о протекании процесса окисления—восстановления на различных
его этапах.
Как было указано ранее (см. книга 1, гл. II, § 4), количественная
зависимость окислительно-восстановительного потенциала системы от
концентрации (активности) реагирующих веществ может быть представлена рядом
уравнений.
Для простейшей обратимой ред-окс-системы, выражаемой уравнением
а Окисл + пе ^± b Восст
реличина окислительно-восстановительного потенциала Е может быть
представлена уравнением (1):
f-fO .RT Тп [Окисл]°
* ~ ^Окисл/Въсст + -Jjjr 1П [Восст]& U)
Если а = Ь = I, то
^ - ^окисл/восст-г nF ш [Восст] (ia;
При замене концентраций активностями получаем:
р рО i RT \„ аОкисл
* - ^Окисл/Восст "I- -^Г ш flBoccT
где Е — окислительно-восстановительный потенциал системы, в;
Е° — нормальный окислительно-восстановительный потенциал, в;
R — газовая константа, равна 8,314 джоуля;
Т — абсолютная температура, °К;
п — число теряемых или приобретаемых электронов;
F — число Фарадея, равное 96 500 кулонам;
[Окисл] —концентрация окисленной формы;
[Восст] — концентрация восстановенной формы.
(16)
§ 1. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ 213
Если в реакции принимают участие ионы водорода, то величина Е
также зависит и от концентрации ионов водорода:
Е - Яокисл/Восст + -^Г 1П [Восст]6 [Н J (lB)
Если заменить константы их числовыми значениями и перейти от
натуральных логарифмов к десятичным (коэффициент перевода 2,303), то
при Т = 298° К (+25° С) уравнение (1) примет вид:
о 0,059 [Окисл]° г +]т
При замене равновесных концентраций активностями уравнение (1)
примет вид:
С ггО , 0,059 , ^ОКИСЛ m ,л ч
Е = ^Окисл/Восст + —7Г~ lg ~ aH+ (1Д)
°Восст
Для более сложной ред-окс-системы
аЛ + ЪВ + • • • пе ^± dD + eE
а Окисл! + b Восст2 + •• - пе ^zt d Bocctx + е Окисл2
потенциал выражают в виде уравнения:
о , 0,059 [А)а[В]ь
^Окисл/Восст "Г п 1& тщйхще
(2)
Исследуя уравнения (1—1д), можно прийти к следующим выводам.
1. Потенциал системы зависит от величины нормального окислительно-
восстановительного потенциала ?окисл/восст, концентрации окислителя
[Окисл ], восстановителя [Восст], ионов водорода [Н+] и от температуры Г.
По мере увеличения [Окисл ] или уменьшения [Восст ] Е увеличивается;
по мере уменьшения [Окисл] или увеличения [Восст] Е уменьшается.
Другими словами, изменение отношения концентраций (активности)
окислителя и восстановителя приводит к изменению величины
окислительно-восстановительного потенциала ред-окс-системы. В том случае,
когда йктивности всех веществ, участвующих в реакции, равны единице,
потенциал системы равен стандартному нормальному окислительно-
восстановительному потенциалу.
2. С повышением температуры (Т) потенциал ред-окс-системы увели-
RT
чивается, так как коэффициент б = -р- увеличивается по мере
возрастания температуры.
3. В тех случаях, когда в реакции принимают участие ионы водорода,
Е увеличивается по мере возрастания активности ионов водорода.
4. Введением в окислительно-восстановительную систему осадителя,
осаждающего в виде нерастворимого соединения окислитель или
восстановитель, можно изменить потенциал системы Е. Если при этом в осадок
выпадает окисленная форма, то Е° > Е; если выпадет в осадок
восстановленная форма, то Е° < Е; если в равной мере в осадок выпадают и
окисленная и восстановленная формы, то отношение концентраций обеих форм
равно единице, а Е° = Е.
Таким образом, осаждение, вызывая изменение концентрации ионов
окисленной или восстановленной форм, обусловливает изменение
окислительно-восстановительного потенциала системы.
214 Г Л .III. МЕТОДЫ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
Аналогичное явление наблюдается при образовании (вместо осадка)
слабодиссоциирующих соединений.
Например, нормальный окислительно-восстановительный потенциал
системы, состоящей из Fe+ и Fe++ (?pe+ + +/Fe++), равен 0,771 в. Если
изменить величину [Fe+++] (aFe+++) или [Fe++] (Яре++), то изменится
?Fe+ + +/Fe++-
Концентрацию (или активность) Fe+++ и Fe++ можно изменить
несколькими способами. Простейший способ — добавление в раствор Fe+++ или
Fe++. Другим способом является восстановление Fe+++ подходящим
восстановителем (например, SnCl2) или окисление Fe++ подходящим
окислителем (например, КМп04). Третьим способом может служить добавление
комплексующих агентов (например, фторида натрия, фосфорной кислоты,
тартратов и т. п.), которые с Fe+++ образуют стойкие комплексные ионы.
Существуют и другие способы изменения
окислительно-восстановительных потенциалов.
Изменение величины окислительно-восстановительных потенциалов
может привести к изменению направления реакции.
Поясним это на примере. Если к раствору FeCl3 прибавить KI, то
наблюдается выделение свободного иода:
2FeCl3 + 2KI ^± 2FeCl2 + 2КС1 + I h
или в ионной форме:
2Fe+ + + + 2I- -^t 2Fe+++|l2
Это означает, что ?ре+ + +д,е++>> Е°г /2Г:
?°Fe+ + +/Fe++ = +0,771 в; ?j2/2r = +0,5345 в;
э. д. с. =0,771 —0,5345 =Ю,2365 в.
Если предварительно к раствору FeCl3 добавить NaF, образующий
с Fe+++ комплексные ионы [FeFe] , то выделения иода не
наблюдается, потому что ?°реРвГ--/Ре++ (+0,4 в) меньше Е°и/2Г (+0,5345 в);
э. д. с. = 0,4 — 0,5345 = —0,1345 в.
При известных условиях (при соответствующем изменении потенциала
системы Fe+++/Fe++ см. табл. 23). Ре++-ионы окисляются элементарным
иодом. В этом случае реакцию представляют в виде следующего
уравнения:
2Fe++ + I2 ^± 2Fe+ + + + 2I~
т. е. реакция протекает справа налево. Это объясняется тем, что при
данных УСЛОВИЯХ ??2/2г > ^Fe+ + +/Fe++#
Таким образом, величины окислительно-восстановительных
потенциалов можно изменять в достаточно широких пределах, благодаря чему
оказывается возможным сдвигать течение реакции в нужном
направлении, причем если разность окислительно-восстановительных потенциалов
реагирующих между собой систем достаточно большая, то реакция
окисления — восстановления количественно протекает до конца и поэтому
возможно прямое титрование.
Частные случаи выражения величин окислительно-восстановительных
потенциалов ред-окс-систем. Для некоторых ред-окс-систем выражение
величин окислительно-восстановительных потенциалов могут быть
представлены частными уравнениями. Это возможно в тех случаях, когда в
процессе окисления—восстановления образуется или участвует растворитель
§ 2. РЕАКЦИИ ОКИСЛЕНИЯ— ВОССТАНОВЛЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ 215
(вода), а одно из реагирующих веществ выпадает в осадок, представляет
собой малорастворимый газ или полностью комплексуется, т. е.
практически не принимает участие в окислительно-восстановительной реакции.
Например, для ред-окс-системы, выражаемой уравнением:
МпО^ + Зе + 2НОН ^± J Mn02 + 40Н~
согласно уравнению (2):
? = ?о 0,059 [МпОГ] [НОН]2
сОкисл/Восст-Г з ё [Мп02][ОН-]4 {6)
Двуокись марганца Мп02 — твердое вещество и его концентрация
[Мп02] или активность aMno2 является величиной постоянной, так как
раствор всегда насыщен двуокисью марганца, характеризующейся
определенной растворимостью в данной среде. Концентрация воды также
является величиной постоянной. Поэтому уравнение (3) можно
представить в следующем виде:
о 0,059 [МпОГ]
? ^Окисл/Восст » з ** ГОН~14
Аналогично для реакции анодного окисления гидроксильных ионов,
выражаемой уравнением:
40Н- — 4е:^02 + 2Н20
согласно уравнению (2):
о . 0,059 [ОН-]4
с ~ ^Окисл/Восст "Т" 4 ё [02] [Н20]2 1 '
или, учитывая, что [02] является постоянной величиной (раствор насыщен
малорастворимым в воде кислородом), и концентрация воды также
представляет постоянную величину, можно уравнение (5) написать так:
* = *°Окисл/Восст + ^ 18 ЮН']4 (6)
Это означает, что потенциал указанной системы главным образом
зависит от концентрации гидроксильных ионов.
§ 2. Реакции окисления—восстановления и комплексообразования
Добавление в ред-окс-систему комплексующих агентов, которые
образуют с ионами окисленной или восстановленной формы комплексные
соединения, отличающиеся малыми константами нестойкости, обусловливает
уменьшение концентрации простых ионов либо окислителя, либо
восстановителя (например, комплексование Fe+ + + фторидом натрия).
Благодаря тому, что ионы окисленной и восстановленной форм ком-
плексуются добавляемыми реагентами в различной степени, отношение
[Окисл]/[Восст] либо увеличивается, либо уменьшается, а следовательно,
изменяется и величина потенциала системы. Допустим, что в системе:
Fe+++ + e^±Fe++
[Окисл] = [Восст], т. е. [Fe+++] = [Fe++]
При добавлении в указанную систему какого-либо лиганда
произойдет комплексообразование, например:
Fe+++ + 6X-^[FeXe]—
Fe++ + 6Х~ ^ [FeXef55
где X —F~, CN-, SCN- и т. п.
216
ГЛ. III. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
Константы нестойкости образующихся комплексных соединений
можно представить так:
[Fe+*+] [Х-]6
Кг-
*,=
[(FeXe)—]
[Fe++] [X-]6
Откуда
[Fe+++] =
[Fe++]:
[(FeX.rl
JCjKFeXe)-
[X-je
Xg[(FeXe)SB]
[Х-]в
(7)
(7a)
(8)
(8a)
Подставив эти значения из уравнений (8 и 8а) в уравнение (1),
получим:
,0 , RT ,, Кг [(FeXe)—]
К2 [(FeXe)==]
? = ?? + ^1п-
(9)
1. Если Ki = К2, то Е° = Е\, т. е. в этом случае ред-окс-потенциалы
пар не изменяются. Заметим, это мало вероятный случай.
2. Если Кг < К2> т. е. если ионы окисленной формы закомплексованы
сильнее, чем ионы восстановленной форм#, то ?J < ?°. Подобный случай
иллюстрирует комплексование Fem фторидом натрия:
^[FeF.] < ^[FeF.]==
3. Если Кг > К2, т. е. если ионы восстановленной формы
закомплексованы сильнее, чем ионы окисленной формы, то ?J > E°. Это явление
имеет место в тех случаях, когда Fe++ комплексуется фенантролином
или дипиридилом.
Изменение ред-окс-потенциала системы Fe+ + + +e-^±Fe++ под
действием комплексующих агентов весьма значительно (табл. 23).
ТАБЛИЦА 23
Изменение потенциала системы Fe+ + + +е ;zt Fe+ +
в зависимости от природы добавленного ли ганда
(?0Fe++w+ = 0.771e)
Комплексующий агент
НС1
н3ро4
HF
KCN
ЭДТА
(Этилендиаминтетрауксусная кислота)
НАО4
Фенантролин (ФА)
Дипиридил (ДП)
Преимущественно
образуемые
комплексные ионы
[Fed,]—
[Fe(P04)2]---
IFeF,]"--
[Fe(CN)e]--~
FeY"
IFetCOJ,]—
Ре(ДП)3+ +
Потенциал
системы, в
+0,53
+0,41
+0,40
+0,36
+0,12
—0,02
+ 1,12
+ 1,10
В большинстве случаев ионы железа высшей степени окисления
.сильнее комплексуются, чем ионы низшей степени окисления, что обычно
приводит к уменьшению потенциала ред-окс-системы. Лишь в отдельных
случаях комплексование Fe++ фенантролином и дипиридилом вызывает зна-
§ 3. ПРИМЕРЫ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯ 217
чительное увеличение потенциала указанной системы, оказывающейся
способной окислять многие вещества. С другой стороны, введение окса-
лата, легко комплексующего Fe+ + + и не оказывающего заметного
действия на Fe+ + , приводит к резкому возрастанию восстановительной силы
ионов Fe11. Используя эту особенность, индийский ученый Гопало Рао
предложил целый ряд оригинальных методов окисления—восстановления
(см. ниже).
Таким образом, комплексообразование, вызывая изменения
концентрации свободных ионов окисленной или восстановленной форм, избирательно
обусловливает изменение потенциалов ред-окс-систем.
§ 3. Примеры окислительно-восстановительного титрования
Количественное определение разнообразных веществ методами
окислительно-восстановительного титрования можно проводить следующими
способами.
1. Элементарные вещества (например, элементарный фосфор),
способные давать положительные или комплексные отрицательные ионы (РО~—),
окисляют стандартными растворами окислителей [КМп04, К2Сг207,
КВгОд, КЮ3, I2, Ce(S04)2 и др.].
Например, красный фосфор окисляют сульфатом церия (IV), заведомо
взятым в избытке (а), и затем оттитровывают (методом обратного
титрования) избыток ионов церия (IV) стандартным раствором соли Мора
(NH4)2Fe (S04)2 (б):
Р + (5 + п) Се*4"1"* + 4Н20 -* РО~" + 5Се+++ + 8Н+ + лСе++++ (а)
избыток
Р — Ъе + 4Н20 —> Р04 + 8Н+
Се++++ + е —* Се+++
/iFe++ + nCe++++ -> /iFe+++ + /iCe+++ (б)
избыток
Fe++ — e-+ Fe+++
дСе++++ + пе —> лСе+
2. Элементарные вещества, способные давать отрицательные ионы,
непосредственно титруют стандартными растворами восстановителей
[SnCl2, (NH4)2Fe(S04)2, TiCl3, СгС12 и др.].
Например, свободный кислород в природных водах можно определять
титрованием стандартным раствором хлорида хрома (II):
4Сг++ + 02 + 4Н+ -* 4Сг+++ + 2Н20
Сг++ — е —> Сг+++
02 + 4е + 4Н+ -+ 2Н20
3. Анионы (например, S20~) и катионы (например, Sn++), способные
окисляться в ионы высших степеней окисления, непосредственно титруют
стандартными растворами окислителей.
Например, в щелочном растворе тиосульфат-ионы окисляются перман-
ганатом до сульфат-ионов (см. стр. 267).
4. Ионы (типаСи+ + , MnOj-, СгО~, Сг20~ и др.), способные
относительно легко переходить в ионы низших степеней окисления,
непосредственно титруют стандартными растворами восстановителей.
Например, ионы меди (II) титруют стандартным раствором хлорида
титана в присутствии роданида и нескольких капель раствора
железа (III).
218 ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Во время титрования Си + + восстанавливается до Си+ (а), который
с SCN~ образует осадок роданида меди (б); Fe+ + + с SCN*" образуют
кроваво-красное окрашивание, вызываемое образованием роданида железа
(III) (в). По окончании титрования ионов меди начинают
восстанавливаться Ре+ + +-ионы и кроваво-красная окраска исчезает (г), что
указывает на окончание процесса восстановления Си + + .
Ti+++ + Си++ —> Ti++++ + Си+ (а)
Си+ + SCN- -» ^ CuSCN (б)
Fe+++ + 3SCN- ^ Fe (SCN)3 (в)
Fe (SCN)3 + Ti+++ —» Fe++ + 3SCN~ + Ti++++ (r)
5. Ионы, способные окисляться (например, Вг" и Fe++), переводят
с помощью окислителей в высшие степени окисления и затем оттитровы-
вают стандартными растворами восстановителей.
Например, бромиды окисляют гипохлоритом до броматов:
Вг" + ЗОСГ —¦> ВгОд + ЗСГ
ВГ —бе + ЗН20-> ВгОд + 6Н+ 11
ОС1-+2е + 2Н+—>Н20 + С1- |3
Образовавшийся бромат обрабатывают в кислом растворе иодидом:
6Г + ВгОд + 6Н+ -> * 312 + Вг" + ЗН20
2I- —2е—»12
ВгОд +6е + 6Н+ -* Вг" + ЗН20
Выделившийся иод оттитровывают стандартным раствором
тиосульфата:
2S20~ +12 — s40~- + 21-
6. Ионы, способные восстанавливаться (например, Fe+ + +), переводят
посредством восстановителей в их низшие степени окисления и затем
оттитровывают стандартными растворами окислителей.
Например, ионы железа (III) восстанавливают (а) хлоридом олова (II)
до железа (II), затем избыток ионов олова (II) окисляют (б) хлоридом
ртути (II); ионы железа (II) титруют стандартным раствором бихромата (в):
(1 + п) Sn++ + 2Fe+++ -> SnIV + 2Fe++ + nSn+* (a)
избыток
/zSn++ + 2/iHgCl2 —> /iSnlv + /iHg2Cl2 + 4/iCi" (6)
избыток
nSn++ — 2ne—>Snlv
2HgCl2 + 2e -» Hg2Cl2 + 2СГ
1
2л
6Fe++ + CrgOf" + 14H+ -> 6Fe+++ + 2Cr+++ + 7H20 (в)
Fe++— e—>Fe+++
Cr2°7 " + бе + 14H+ -> 2Cr+++ + 7H20
7. Ионы, количественно реагирующие с восстановителями с
образованием малорастворимых соединений (например, CaC204, Zn2 [Fe (CN)e]
и т. п.), обрабатывают стандартным раствором восстановителя, заведомо
взятым в избытке, и затем оттитровывают избыток восстановителя
стандартным раствором окислителя.
§ 3. ПРИМЕРЫ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯ 219
Например, ионы кальция осаждают в мерной колбе точно отмеренным
объемом титрованного раствора оксалата (восстановителя), заведомо
взятым в избытке:
Са++ + (1 + п) С?>1~ -» ф СаС204 + /iC20"
избыток
разбавляют содержимое колбы до метки, отбирают аликвотную часть
раствора и оттитровывают избыток оксалата стандартным раствором
перманганата (окислителя):
2/гМпО^ + 5/гС2С?~ + 16/гН+ —> 2/гМп++ + 8/гН20 + 10/гСО2
/гС204 — 2пе —> 2лС02
++
Мп04 + Ъе + StT —> Мп + 4Н20
5
2/г
8. Ионы, количественно реагирующие с осадителем с образованием
малорастворимых соединений (Zn3 [Fe (CN)6]2, обрабатывают осадителем,
взятым в избытке, и затем оттитровывают избыток осадителя (или его
заместителя) стандартным раствором восстановителя.
Например, ионы алюминия, магния, цинка и других металлов дают
с 8-оксихинолином малорастворимые кристаллические соединения —
оксихиноляты. Осадки оксихинолятов отделяют от раствора, промывают
и растворяют в хлористоводородной кислоте. К полученному раствору
прибавляют раствор бромида ибромата. При этом происходят следующие
реакции окисления—восстановления:
5Вг" + ВгОз + 6Н+ -> ЗВг2 + ЗН20
ОН ОН
N I N I вг
| || | + 2Вг2-»| || | +2НВг
I
Вг
К избытку бромата добавляют KI:
BrOg + бГ + 6Н+ —> Вг" + ЗН20 + 312
Выделившийся элементарный иод оттитровывают тиосульфатом, как
указано в пункте 5.
9. Ионы, реагирующие с окислителем или восстановителем с
образованием продуктов реакции, способных давать малорастворимые
соединения, обрабатывают при соответствующих условиях стандартным
раствором окислителя или восстановителя, а затем оттитровывают их избыток
стандартными растворами восстановителей или окислителей.
а) Растворы иодидов, содержащих ионы бария, окисляются в щелочной
среде избытком перманганата до периодатов. При этом образуется осадок
манганата бария:
Г + (8 + п) Мп04 + 8Ва++ + 80Н~ -> Ю4 + * 8ВаМп04 + 4Н20 + лМп04
избыток
б) Избыток перманганата оттитровывают стандартным раствором фор-
миата:
2лМп04 + ЗлНСОО" + лН20 —> * 2лМп02 + 3/iC02 + 5/iOH"
HCOO- — 2е + ОН- -» С02 + Н20
яМп04 + Зле + 2яН20 —> яМп02 + 4/гОН"
3л
2
220 ГЛ. III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
10. Элементарные вещества или ионы окисляют избытком окислителя
или восстанавливают избытком восстановителя и затем оттитровывают
избыток окислителя стандартным раствором восстановителя или избыток
восстановителя стандартным раствором окислителя, т. е. применяют также
и методы обратного титрования.
Таким образом, применяя стандартные растворы окислителей и
восстановителей, можно количественно определять не только разнообразные
элементарные вещества и ионы, способные окисляться или
восстанавливаться, но и вещества, не отличающиеся ярко выраженными
окислительными свойствами, но склонные реагировать с окислителями или
восстановителями в стехиометрических соотношениях с образованием
малорастворимых соединений (например, Са+ + , реагирующий с С20~" с
образованием малорастворимого осадка СаС204; Ва + + , образующий с СгО~~ мало-
растворимый ВаСг04, и т. п.).
Определение таких веществ проводят на основании расчета количества
прореагировавшего с ними окислителя или восстановителя.
Кроме того, окислительно-восстановительные методы дают возможность
количественно определять ионы, не реагирующие совсем с окислителями
и восстановителями, но способные, например, осаждаться в виде окси-
хинолятов (Al + + + , Mg++ и др.), глиоксиматов (Ni++), гексацианофер-
ратов (Zn++ и др.).
Определение таких веществ проводят на основании расчета количества
прореагировавшего с осадителем (вступившего в реакции с
определяемыми ионами) окислителя или восстановителя.
Наконец, применяя специальные приемы титрования, можно методами
окисления—восстановления дифференцированно (раздельно)
количественно определять содержание различных компонентов в
многокомпонентных смесях, сплавах, рудах, минералах и т. д.
Например, индийский ученый Гопало Рао с сотрудниками
разработал ряд методов дифференцированного окислительно-восстановительного
титрования стандартным раствором бихромата калия в среде
концентрированной фосфорной кислоты, действие которой, как комплексующего
агента, заключается в изменении ред-окс-потенциала ряда систем.
Некоторые из этих методов описаны ниже:
1) Определение VIV, основанное на титровании бихроматом калия
в среде более чем 12 М фосфорной кислоты при комнатной температуре.
Присутствие многих посторонних ионов не мешает определению.
Предложенный метод пригоден для дифференцированного титрования смесей:
Fe" и VIV; Mn11 и VIV; Fe11, Mn11 и VIV.
2) Определение Се111 титрованием бихроматом калия в среде 12—13,5 М
фосфорной кислоты. Метод применен для дифференцированного титрования
смесей: Fe11 и Се111; VIV и Се111; Fe11, VIV и Се111.
3) Определение UIV и его смесей с Fe11, Mn11, Се111 или VIV.
4) Определение UVI, основанное на предварительном
восстановлении его солями железа (II) и последующем титровании избытка
восстановителя дихроматом калия. Метод пригоден для дифференцированного
титрования смесей UIV с другими восстановителями (например, Fe11). При
этом наблюдается два скачка титрования. Первый соответствует
окислению Fe11, второй — UIV.
§ 4. Константы равновесия окислительно-восстановительных реакций
Реакции окисления—восстановления обратимы. Равновесие
наступает в момент, когда ?окисл становится равным jEbocct- В тех случаях,
когда реакции обратимы, к ним можно применять закон действия масс
§ 5. СВЯЗЬ МЕЖДУ КОНСТАНТАМИ РАВНОВЕСИЯ РЕАКЦИЙ И ПОТЕНЦИАЛАМИ 221
и вычислять изменения молярных концентраций реагирующих ионов,
что имеет особо важное значение в количественном анализе.
Например, для реакции
М1Ю4 +5Fe++ +8H+^lMn++ + 5Fe+++ + 4Н20
Окисл, Восст2 Восст! Окисл2
выражение закона действия масс имеет вид:
ГМп++] [Fe+++]5
[MnO;][H+]8 [Fe-]6 AMno;/Fe++ {Ш)
где /( _ . ++ — константа равновесия указанной реакции.
Mn04 /Fe
§ 5. Связь между константами равновесия
окислительно-восстановительных реакций и нормальными потенциалами
Общее уравнение, выражающее константу равновесия окислительно-
восстановительных реакций. Для реакции
а Окислх + Ь Восст2 ;~i d Восстх + е Окисл2
константу равновесия представляют формулой:
[Восстх]* [Окисл2]в
[Окисл^ [Восст2]* -докнсл/восст W
Если коэффициенты а = d и Ь = е, то можно написать:
( [Восстг] у ( [Окисл2] W
\ [Окислх] / \ [Восст2] | = ^Окисл/Восст (12)
Значение lg/Сокисл/восст. Рассмотрим следующие реакции:
а Окислх + пе ^ а Восстх (а)
b Восст2 — ne^lb Окисла (б)
Для реакции а:
. о 0,059 ( [Ошслг] \а
^Окислt/Восстх^ п ls\ [BOCCTJ /
Для реакции б:
,059 1 f [Окисл2] \ь
п g\ [Восст2] /
Е"=Е° « °'059 i~ f [Окисл2] \*
Окисл2/Восст2 '
При установившемся равновесии окислительные потенциалы обеих
систем равны Е' = Е", или
о 0,059 ( [Окислх] уа
^Окислг/Восст! -Г п i&\ [BOCCTJ /
о 0,059 . ( [Окисл2] )Ь
==^Окисла/Восста"Г п i§^ [В0ССТ2] J
откуда
о о 0,059 Г ( [Окисл,])ft . ( [Окислх] )<П
^ОКИСЛ^ВОССТ! ^Окисла/Восста ~" д [1§ \ [В0ССТ2] / ё \ [BOCCTJ / J
0,059 . / [Boccti] у ( [Окисл2] у
~" я g \ [Окислх] J # ( [Восст2] J
222
ГЛ. III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
На основании уравнения (12):
rt о _ 0,059
^ОкисЛх/Boccti ^ Окисл 2/Восст2 ~~ п #1б АОкисл/Восст
ИЛИ
п(Е°1-Е°и)_
QQ59 *?^Окисл/Восст {*• '
Значение отношений [Восст1]/[Окисл1] и [Окисл2]/[Восст2] в точке
эквивалентности.
Для реакций типа
Окислх + Восст2 ;=t Восстх + Окисл2
например, Се+ + + + +Fe++^Ce+ + + +Fe+ + + в момент
эквивалентности, т. е. в момент, когда количество прибавленного Восст2 эквивалентно
содержанию Окисл х (или количество прибавленного Окисл х
соответствует содержанию Восст2) устанавливаются следующие равновесия:
[Окислх] = [Восст2]; [Восстх] = [Окисл2]
Константа равновесия указанных реакций
к _ [BoccTt] [Окисл2]
дОкисл/Восст - [Окисл!] [Восст2]
Поэтому можно написать
К — [B°CCTi12 _ [Окисл2]2
ДОкисл/Восст- [0КИСЛ1]2 "~ [В0ССТ2]2
отсюда
[BocctJ __ [Окисл2] _ Г1г (Ш
[ОКИСЛх] ~~ [В0ССТ2] "" У ЛОкисл/Восст * >
Для общего случая, когда реакцию выражают уравнением
а Окисл! + Ь Восст2 "^L a Boccti + Ь Окисл2
аналогичным образом можно вывести, что
[Восст!] _ [Окисл2] __ a+*w п~
[ОкИСЛх] " [В0ССТ2] "~ У ЛОкисл/Восст ^°>
В точке эквивалентности отношения [ВосстхШОкислх] и [Окисл2]/[Восст2] не всегда
равны друг другу и зависят от первоначальных концентраций окислителя или
восстановителя. Это относится к реакциям, в которых коэффициенты у окисленных и восстановленных
форм реагирующих веществ неодинаковы (например, 12 7^- 21").
§ 6. Вычисление констант равновесия
окислительно-восстановительных реакций
Для вычисления константы равновесия какой-либо обратимой
окислительно-восстановительной реакции необходимо вычислить lg К по
формуле (13), а затем К:
(?°-?°)л
К = 1°К 0,059 <16>
Пример /. Вычислить Kpe+++/sn+* реакции:
2Fe+++ + Sn++ ^ 2Fe++ + SnIV
л= 2
§ б. КОНСТАНТЫ РАВНОВЕСИЯ РЕАКЦИЙ 223
Решение, ?°е+++/ре++ = 0,771; ?°nIV/Sn++ = 0,15.
2(0,771-0,15)
IgAFe+++/Sn++ 0,059
Следовательно,
Пример 2. Вычислить К -, ++ реакции:
MnOWFe
МпО; + 8Н+ + 5Fe++^Mn++ + 5Fe+++ + 4Н20
п = 5; [Н+1=1 г-ион/л
Решение.
\„К 5(1,52-0,77) _сос
lg KM„o;/Fe+1-= ОМ 63,6
Следовательно
Mn04/Fe
Сопоставление констант окислительно-восстановительных реакций
в примерах I и 2 дает основание полагать, что
1) обе реакции идут до конца слева направо и являются практически
необратимыми;
2) так как константа реакции выражает собой отношение
произведений концентраций конечных продуктов реакции к произведению
концентраций исходных продуктов, то:
а) равновесие первой реакции наступает тогда, когда [Fe++]2-
• [SnIV] станет больше [Fe+ + + ]. [Sn+ + ] в 1021 раз;
б) равновесие второй реакции наступает тогда, когда [Мп+ + ] • [ Fe+ + + ]б
станет больше [MnOj-]-[Н+]8-[Fe++]б в 1063 раза.
Любую реакцию окисления—восстановления можно представить в виде
суммы двух реакций: реакции окисления и реакции восстановления,
поэтому (согласно общему правилу) константу равновесия реакции
окисления—восстановления можно рассматривать как произведение констант
равновесия этих двух составляющих реакций:
^Окисл/Восст == ^ О кисл^В осст
Логарифмы констант равновесий реакций вычисляют по формуле
1?*Окисл=16^?°>
которая получается после преобразования уравнения
?0 = ^-1п#Окисл
В этом уравнении константы заменяют их числовыми значениями и натуральные
логарифмы десятичными.
Для каждой индивидуальной реакции окисления—восстановления
константа равновесия реакции окисления равна обратной величине
константы реакции восстановления, т. е.
*Ч)кисл == ^Восст» а *?Аокисл == ^Восст
Для вычисления логарифмов общих констант равновесия реакций
окисления—восстановления проводят алгебраическое сложение двух
величин: lg /Сокисл и lg /Свосст (см. книга 1, Приложение).
224
ГЛ. III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Пример 3. Пользуясь \gKOKRCJJ и \gKBoccT(cM. книга 1» Приложение), вычислить
константу равновесия реакции, протекающей между I" и Fe+ + + :
2I-+2Fe+ + + ?lI2 + 2Fe+ +
Решение. При составлении уравнения реакции окисления—восстановления
уравнение реакции восстановления умножают на коэффициент 2, поэтому логарифм
константы равновесия полной реакции составит:
lg /С п = lg К +++/1. = 2-13,04 — 18,07 = 8,01
6 Окисл/Восст s Fe+++/l ' *
К = К ""^ 10®
АОкисл/Восст AFe+++/l"
Для наглядности приводим подсобные уравнения
2I- —2е;П12 II
Fe+ + + + e^Fe+ + |2
По величине вычисленной константы равновесия можно считать, что реакция
предпочтительно протекает слева направо.
Пример 4. Вычислить константу равновесия реакции, протекающей согласно
уравнению
5Fe++ + МпО: + 8Н+ ^5Fe+++ + Мп++ + 4Н20
Решение.
lg/C -, ++= 128,5 — 5-13,04 = 63,3
6 Mn04/Fe
К -. ++«*1063
Mn04/Fe
§ 7. Вычисление окислительно-восстановительных потенциалов
и констант равновесия окислительно-восстановительных реакций
с учетом коэффициентов активностей
При более точных вычислениях окислительно-восстановительных потенциалов и
констант равновесия окислительно-восстановительных реакций [уравнение (6)] необходимо
учитывать коэффициент активности (см. книга 1, гл. II, § 6).
§ 8. Зависимость скорости реакций
окисления—восстановления от различных факторов
Реакции окисления—восстановления протекают с измеримой скоростью,
меняющейся в зависимости от условий их проведения. Наиболее важными
факторами, оказывающими сильное влияние на скорость реакции
окисления—восстановления, являются:
1) концентрации реагирующих веществ и [Н+];
2) температура, с изменением которой меняется величина окислительно-
восстановительного потенциала системы;
3) природа реагирующих веществ, характер среды, присутствие
катализаторов, комплексообразование, сопряженность реакций окисления—
восстановления и т. п. (см. ниже).
Влияние концентраций реагирующих веществ. Согласно закону
действия масс при постоянной температуре скорость данной химической
реакции прямо пропорциональна произведению концентраций реагирующих
веществ.
Для реакции:
Окислх + Восст2 ;zt Восст! + Окисл2
скорость прямой реакции (иг) равняется:
Vi = К [Окислх] [Восст2]
где К — коэффициент пропорциональности (константа скорости), численно равный
скорости реакции, когда молярные концентрации [Окислх1 = [Восст2] = 1.
§ 8. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИЙ ОТ РАЗЛИЧНЫХ ФАКТОРОВ 225
Поэтому, чем выше [Окислх], или [Восст2], тем больше скорость
прямой реакции, обычно выражающей процесс титрования в методах
окисления—восстановления.
Например, реакция
6Г + Сг20;Г + 14Н+ —> *312 + 2СГ"*4"1" + 7Н20
протекает практически до конца лишь в достаточно концентрированных
растворах. В разбавленных растворах при концентрациях менее 0,05 М
эта реакция протекает слишком медленно. Результаты, получаемые при
использовании указанной реакции, в особенности зависят от [Н+] и
[I"] в растворе и от продолжительности протекания реакции. Однако
при большой кислотности раствора (Ш+]>0,5 г-ион/л) наблюдается
окисление иодистоводородной кислоты кислородом воздуха:
41 + 02 + 4Н+ -> 212 + 2Н20
Следует иметь в виду, что многие реакции окисления—восстановления
протекают очень медленно, даже при высокой концентрации реагирующих
веществ. Это в особенности относится к реакциям, протекающим в
неводных растворах. Например, известно, что даже при высокой концентрации
такие окислители, как перманганат, персульфат, бихромат и некоторые
другие реагируют со многими восстановителями с очень малой скоростью.
В таких случаях прибегают преимущественно к методам обратного
титрования или добавляют в титруемый раствор катализаторы, повышающие
скорость реакции, или вводят индукторы, вызывающие реакции, которые
не протекают в их отсутствие.
Влияние рН среды. Скорость реакции в большинстве случаев является
функцией величины концентрации ионов водорода, поэтому реакции
проводят обычно при определенных значениях рН среды. Кроме того,
посредством увеличения концентрации кислот удается повысить окислительно-
восстановительные потенциалы некоторых систем. Например, путем
добавления очень концентрированной H2S04 можно повысить нормальные
потенциалы Е° системы МпО~/Мп++ от +1,52 до +1,9 в и системы IO^/IOg
от +1,50 до +2,0 в.
Влияние рН среды можно иллюстрировать на следующих реакциях
окисления—восстановления:
при рН < 7 ClOg + 3H2S03 ^1СГ + 3HSO; + 3H+ (a)
при рН > 7 ClOg + 3SOg " ^ СГ + 3SO" "
при рН < 7 Ag+ + Fe++ ^l Ag + Fe+ + + (б)
при рН > 7 Ag20 + 2Fe (OH)2 + НОН ^ 2Ag + 2Fe (OH)3
при рН < 7 НС10 + 2Fe++ + Н + ^ Н20 + 2Fe+++ + С1" (в)
при рН > 7 СЮ- + 2Fe (ОН)2 + НОН ^ С1~ + 2Fe (OH)3
при рН < 7 H3As04 + 2Г + 2Н+ -» HAs02 + I2 + 2Н20 (г)
при рН > 7 AsO 2 + 12 + 40Н" —> AsO™ + 21" + 2Н20
Необходимо учитывать влияние среды на состояние ионов (см. книга 1, гл. I, § 1).
Например, в зависимости от характера среды мышьяк (III) в водном растворе может
находиться в следующих формах: H3As03, HAs02, H2AsOg, AsOg, HAsOg"", AsO™,
As(OH)2, AsOH**, As"*4"*" и т. д.
226
ГЛ.1И. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
При составлении подсобных уравнений реакций окисления—восстановления, в
которых участвуют ионы водорода или ионы гидроксила, Н+-ионы, как правило, находятся по
одну сторону с окислителем, ОН~-ионы — с восстановителем. Например:
ВгОд + бе + 6Н+ ^ ВГ + ЗН20 (а)
Вг" — 6е + 60Н~" ^ ВгО~ + ЗН20 (б)
МпО~ + Бе + 8Н+ ^ Мп++ + 4Н20 (в)
МпО 4 + Ъе + 2Н20 ^1 Мп02 + 40Н~ (г)
Следует помнить, что на течение реакции оказывает действие не только добавление
кислоты или щелочи, учитываемых при составлении уравнений реакций, но и влияние
ионов водорода и гидроксила, которые образуются при диссоциации воды и участвуют
во многих реакциях окисления—восстановления.
Влияние температуры. При изменении температуры раствора
потенциал системы меняется. Из уравнения (1):
?_?о RT [Окисл]*
* ~ ^Окисл/Восст + -^Г in [Восст]6
следует, что чем выше температура, тем больше потенциал системы.
Коэффициент 0,059//г в уравнении (1) подставляют, если измерения потенциала
ведут при 25° С (Т = 298° К), если измерения делают при 30° С (Т =
= 303° К), то вместо величины 0,059//г подставляют величину 0,06//г, при
18° С (Г = 291° К) — 0,058/л и т. д.
От температуры также зависит скорость реакции, изменение которой
связано с энергией активации (см. книга 1, гл. 1, § 4).
Влияние катализаторов. Скорость реакции, схематически выражаемой
уравнением
Окислх + Восст2 ^ Восстх + Окисл2
может в сотни, тысячи и миллионы раз увеличиваться в присутствии
катализаторов (в частности, ионов водорода).
Многие реакции окисления—восстановления катализируются
кислотами.
Ионы водорода оказывают разлагающее действие на
кислородсодержащие окислители типа MnO~, MnO~~, AsO~""~ и др., направляя реакции
в сторону образования молекул Н20. Благодаря этому усиливаются
окислительные свойства указанных окислителей.
Действие катализатора на скорость прямой реакции объясняется
многими причинами. Одной из такого рода причин является образование
катализатором с Окислх или Восст2 комплексного соединения, реагирующего
с Восст2 или Окислх с образованием продуктов реакции Восстх и Окисл2.
При этом сам катализатор выделяется в свободном виде.
Например, перекись водорода образует с некоторыми веществами
(катализаторами): молибдатами, вольфраматами, ванадатами и другими
комплексные соединения:
(NH4)2 Мо04 + Н202 —> (ШЖ Мо04.Н202
Благодаря свойству образующихся при этом перекисных групп
растворы перкислот и их солей с щелочными металлами отличаются
исключительно сильным окислительным действием. Их активное окислительное
действие может быть продемонстрировано известным фактом окисления
ТЮН, не изменяющейся при действии перекиси водорода, в Т1 (ОН)3
в присутствии (NHJ 2Мо04 • Н 202:
ТЮН -рДОНЖ Мо04.Н202 -» * Т1 (ОН)8 + (NHJa Mo04
§ 8. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИЙ ОТ РАЗЛИЧНЫХ ФАКТОРОВ 227
Другим примером реакции каталитического окисления может служить
реакция иодидов с перекисью водорода в присутствии тех же
катализаторов:
H2W04.H202 + 2Н+ + 21- -> H2W04 + 2H20 + Ф 12
Иногда катализатор участвует в образовании чрезвычайно сильного
реакционноспособного промежуточного соединения. В присутствии такого
вещества Окисл! реагирует с Восст2 со значительно большей скоростью,
чем без катализатора.
Примерами подобных процессов могут служить многочисленные
каталитические реакции (иод-азидная, ферри-тиосульфатная и другие реакции),
рассмотренные в книге I «Качественный анализ».
Особый вид каталитических явлений состоит в образовании
катализатора во время самой реакции окисления—восстановления, которое
называют автокатализом.
Например, реакция окисления щавелевой кислоты перманганатом
протекает чрезвычайно медленно. При добавлении стандартного раствора
перманганата титруемый раствор долгое время остается окрашенным и~не
обесцвечивается, несмотря на присутствие в растворе достаточного
количества окислителя (МпО-) и восстановителя (С20~—), т. е. реакция идет
очень медленно, однако в результате ее постепенно накапливаются ионы
Мп11. Образующиеся Мп++-ионы оказывают заметное каталитическое
действие на процесс окисления—восстановления и вызываемая МпО~
окраска раствора быстро исчезает вследствие их восстановления оксалат-
ионами.
Окисление С2О4— перманганатом сопровождается образованием
промежуточных соединений, в которых важную роль играют ионы Мп11.
Поэтому иногда перед титрованием перманганатом в титруемый раствор
рекомендуется добавлять соли марганца (II).
Другим примером автокаталитической реакции может служить
окисление сернистой кислоты йодноватой кислотой:
5H2S03 + 2НЮ3 -> 5H2S04 + * I2 + Н20
Представленное уравнение выражает собой итоговое уравнение общей
реакции, протекающей на самом деле в несколько стадий.
Окисление сернистой кислоты йодноватой кислотой сначала
протекает очень медленно с образованием иодистоводородной кислоты (а);
иодистоводородная кислота быстро окисляется йодноватой кислотой до
элементарного иода (б), который мгновенно окисляет сернистую кислоту
до серной кислоты (в):
3H2S03 + HIO3 -» 3H2S04 + HI (a)
5HI + HI03 -» * 312 + ЗН20 (б)
H2S03 + I2 + Н20 -> H2S04 + 2HI (в)
Образующаяся HI вновь вступает во взаимодействие с НЮ3.
Таким образом, реакция катализируется иодистоводородной кислотой,
элементарный иод выделяется лишь тогда, когда окисляется вся сернистая
кислота.
Сопряженные реакции окисления—восстановления. Опыт показывает
(Н. А. Шилов), что иногда в одной и той же среде происходят две реакции
окисления—восстановления, из которых одна зависит от другой.
Например, хромовая кислота не окисляет винную кислоту, но окисляет мышья-
228 ГЛ.111. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
ковистую кислоту. При действии на смесь
винной и мышьяковистой кислот
хромовой кислотой наблюдается одновременное
окисление обеих кислот.
Следовательно, мышьяковистая
кислота, окисляясь хромовой кислотой,
способствует окислению винной кислоты.
Таким образом, реакция окисления винной
кислоты зависит от реакции окисления
мышьяковистой кислоты, так как обе
реакции протекают одновременно. Такие
реакции называют сопряженными
реакциями окисления—восстановления.
Поэтому реакцию окисления- хромовой
кислотой винной кислоты называют
сопряженной с реакцией окисления
мышьяковистой кислоты.
Н. А. Шилов (1872—1930). Известно много таких сопряженных
реакций. Укажем на следующие из них:
1) окисление С1"" перманганатом в присутствии железа (II):
2) окисление в кислой среде арсенитов перманганатом в присутствии
марганца (II);
3) окисление сульфитов кислородом воздуха в присутствии иода;
4) окисление арсенитов броматом в кислой среде в присутствии
сернистой кислоты;
5) окисление иодидов нитратами в кислой среде в присутствии цинка;
6) окисление оксалатов перманганатом в присутствии солей марганца
(II) и т. д.
Согласно Н. А. Шилову, вещество, участвующее в двух сопряженных
реакциях, называется актором (хромовая кислота); вещество,
непосредственно реагирующее с актором, называется индуктором
(мышьяковистая кислота): вещество, реагирующее с актором под влиянием индуктора,
называется акцептором (винная кислота).
В соответствии с этой терминологией вещества, принимающие участие
в приведенных выше реакциях, называют:
Акторы
Н2Сг207
КМп04
КМп04
02
НВгОз
HN03
KMn04
Индукторы
H3As03
Fe+ +
Mn+ +
I*
H2S03
Zn
Mn+ +
Акцепторы
C4H6Oe
ci-
As03
SO?"
As03
I-
ед-
Химизм протекающих сопряженных реакций весьма сложен и во
многих случаях окончательно не установлен. Так как скорости всех
протекающих одновременно реакций неодинаковы, то отсюда вытекает общее
правило: титрование во всех случаях надо вести с такой скоростью, чтобы все
реакции заканчивались во время титрования. В конце титрования
необходимо учитывать также уменьшение концентрации реагирующих веществ
и вести титрование медленнее.
Одновременное протекание нескольких реакций часто осложняет
объемно-аналитические окислительно-восстановительные определения. На-
§8. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИЙ ОТ РАЗЛИЧНЫХ ФАКТОРОВ 229
пример, при окислении перманганатом сульфитов в зависимости от
условий проведения реакции одновременно идет окисление SO~" до
сульфатов и дитионатов. Образование дитионатов объясняется появлением
промежуточного продукта — двуокиси марганца, которая окисляет
сульфиты только до дитионатов.
Изучение влияния катализаторов на течение реакций окисления—
восстановления интересно не только с теоретической точки зрения, оно
имеет большое значение в практической работе аналитиков. Например,
некоторые сильные окислители, обычно вполне устойчивые, могут во
время титрования разлагаться с выделением кислорода. Так иногда ведет
себя пермангаиат. Очень часто реакции, вполне удовлетворительные в
применении к индивидуальным реактивам, могут оказаться непригодными
при титровании смеси веществ и т. д. Сложность протекания реакций
окисления—восстановления требует тщательного изучения течения реакции
и выяснения влияния посторонних веществ. Только на основании
полученных данных возможно проведение реакции в наиболее благоприятных
условиях.
Влияние других факторов. На скорость реакций
окисления—восстановления, помимо указанных выше влияют и некоторые другие факторы.
1) Добавление в реакционную среду избытка окислителя (а) или
восстановителя (б) способствует сдвигу равновесия в нужном направлении
и обеспечивает полноту протекания реакции, например:
а) окисление сульфитов избытком иода, который затем обратно оттит-
ровывают тиосульфатом (см. § 35):
S03~ + «I2 + H20 -» 50Г + 2Г + 2Н+ + (/г-1)12
избыток
(n_l)I2-f2(/i--l)S203~ -* (/I_i)s4o;r + 2(/i-l)r
б) восстановление меди (II) до меди (I) иодистоводородной кислотой
или иодидами в кислой среде сопровождается образованием
элементарного иода, который оттитровывают тиосульфатом (см. § 37):
/zI- + 2Cu++ -* I2 + 2CuI + (п — 4) I-
избыток
I2+2S207 -* S407 + 2I"
2) Удаление из сферы реакции образующихся продуктов, например
элементарного иода, титрованием его тиосульфатом или отгонкой его, или
экстрагированием органическими растворителями способствует сдвигу
равновесия реакции в нужном направлении.
3) Введение в титруемый раствор (иногда к концу титрования)
посторонних веществ благоприятно влияет на точность результатов анализа.
Например, осадок иодида меди (I), выделяющийся в результате реакции (б),
адсорбирует на своей поверхности некоторое количество элементарного
иода. Это приводит к искажению результатов иодометрического
титрования. Для количественного выделения иода добавляют к концу титрования
роданид, адсорбирующийся осадком Cul сильнее иода и поэтому
вытесняющий адсорбированный иод.
4) Связывание в комплексные ионы посторонних веществ, мешающих
количественному определению окислителей или восстановителей, или
гасящих действие реагентов, обеспечивает полноту протекания аналитической
реакции. Например, с целью маскирования ионов Fem, мешающих
определению многих окислителей, в титруемый раствор предварительно
добавляют фосфат-, фторид- или оксалат-ионы, комплексующие железо (HI).
230 ГЛ.1И. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
5) Прибавление в титруемый раствор буферных смесей или реагентов
обеспечивает буферную среду, рН которой необходим для успешного
проведения данной реакции. Например, при титровании соединений
мышьяка (III) для поддержания рН 6—7 в титруемый раствор вводят
бикарбонат натрия (рН 8), который при частичной его нейтрализации кислотой
дает угольную кислоту. Бикарбонат с угольной кислотой образует
буферную смесь с требуемым значением рН.
Иногда для создания буферной среды с рН 3 в титруемый раствор
добавляют бифторид аммония NH4HF2, который одновременно выполняет
роль маскирующего комплексующего агента для Be11, Fem, TiIV и др.
§ 9. Графический метод изображения процесса
окисления—восстановления
Подобно тому, как процессы кислотно-основного титрования могут
быть представлены графически в виде кривой титрования,
изображающей изменение величины рН титруемого раствора по мере приливания
к нему стандартного (титрованного) раствора кислоты или щелочи (см.
гл. II, § 3), процессы окислительно-восстановительного титрования можно
также представить графически в виде кривой титрования, изображающей
изменение окислительно-восстановительного потенциала Е титруемого
раствора по мере приливания к нему стандартного (титрованного) раствора
окислителя или восстановителя.
Каждый процесс окисления—восстановления характеризуется своей
кривой титрования, изображающей изменение
окислительно-восстановительного потенциала в процессе титрования. Как видно из уравнения
о 0,059 [Окисл]
п ^Окисл/Восст-Г п хб [Восст]
величина потенциала Е зависит от величины нормального окислительно-
восстановительного потенциала данной системы Е° и от количественного
соотношения концентраций окислителя и восстановителя. Например,
потенциал раствора, содержащего одновременно соли Fe+ + + и Fe++,
будет тем выше, чем больше отношение
[ре+++] (окислитель)
[Fe++] (восстановитель)
При титровании железа (II) перманганатом в кислой среде, согласно
схеме Fe++-^Fe+ + + , [Fe+ + +] увеличивается по мере приливания
титранта и одновременно уменьшается [Fe+ + ]. Следовательно, изменяется
отношение [Fe+ + + ]/[Fe++ ]. По мере увеличения этого отношения
окислительно-восстановительный потенциал тоже увеличивается.
Это постепенное увеличение потенциала происходит неравномерно.
Вблизи точки эквивалентности наблюдается наиболее резкое
(скачкообразное) изменение потенциала, объясняемое более резким изменением
соотношения [Fe+ + + ]/[Fe++ ], которое достигает превышения
начального отношения в миллион раз.
По величине нормальных потенциалов можно рассчитать величину
константы равновесия данной окислительно-восстановительной реакции,
а зная ее величину, можно вычислить отношение равновесных
концентраций и рассчитать значение потенциалов в процессе титрования. В точке
эквивалентности раствор характеризуется определенным окислительно-
восстановительным потенциалом. Конец титрования может быть определен
по изменению окраски титруемого раствора, вызываемой избыточной
§ 9. МЕТОД ИЗОБРАЖЕНИЯ ПРОЦЕССА ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ 231
каплей стандартного раствора перманганата, или посредством индикатора,
изменяющего свою окраску при этом окислительно-восстановительном
потенциале, или инструментальными методами.
Изменение окислительно-восстановительного потенциала раствора в
процессе титрования. Если опустить в раствор, содержащий Fe+++ и Fe++,
пластинку из инертного металла (например, платины) и соединить такой
электрод с каким-либо другим окислительно-восстановительным (или
ред-окс) электродом, то образуется гальванический элемент. При этом
происходит окисление Fe+ + до Fe+ + + или восстановление Fe+ + + до Fe+ +:
Fe++ ^ Fe+++ + e
Окисление Fe+ + до Fe+ + + происходит в том случае, когда второй
электрод в гальваническом элементе является положительным, т. е. когда
в реакцию вступает более сильный окислитель, чем Fe+ + + .
Восстановление Fe+ + + до Fe+ + наблюдается тогда, когда указанный электрод является
отрицательным, т. е. когда в реакцию вступает более сильный
восстановитель, чем Fe+ + .
Таким образом, непременным условием протекания какой-либо
окислительно-восстановительной реакции является наличие системы двух пар
окислителей — Окисл х и Окисл2 и восстановителей — BoccTi и Восст2.
В начале титрования, когда имеется избыток восстановителя, величину
окислительно-восстановительного потенциала, меняющуюся по мере
изменения концентрации, вычисляют по формуле:
о 0,059 .[Окисл,] 7
с — ^ Окисл 2/Восст2 "Г п lg [B0CCT2] К '
где [Окисл2] и [Восст2] относятся к титруемому веществу (восстановителю).
В точке эквивалентности потенциал раствора вычисляют на
основании величины нормальных потенциалов восстановителя и окислителя
и константы равновесия данной реакции. После точки эквивалентности
величину потенциала определяют по избытку прибавляемого окислителя
по формуле:
о 0,059 [Окислу
с ~ ^Окисл^Восстч "Г —?- ^ [BoccTi] Vio;
где [Окислх] и [BoccTi] относятся к титрованному раствору (окислителю).
При титровании окислителя восстановителем вычисления проводят
аналогично.
Рассмотрим в качестве примеров титрование железа (II) сульфатом
церия (IV) и титрование железа (II) перманганатом калия в сернокислой
среде.
Титрование соли железа (II) сульфатом церия (IV). а) Расчет
величины потенциала в начале титрования.
Когда идет речь об окислительно-восстановительном потенциале, to имеется
в виду не отдельный элемент или его ионы, а система, характеризующаяся
определенным равновесием. Например:
Fe++++e ^± Fe++ (a)
Fe++ + 2e ^± Fe<> (б)
Fe+++ + 3e ^± Feo (в)
[Fe(CN)e]— + e ^ [Fe(CN).]M (г)
[FeFe]— + е -* Fe++ + 6F~ (д)
И т. Д,
232
ГЛЛН. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
Вследствие этого для систем, выражающих различное состояние
данного элемента, может быть не одно, а несколько значений нормального
потенциала. Так, для железа, в соответствии с уравнениями а, б, в, г, д,
существуют различные значения нормального потенциала (см. книга I,
гл. II, § 3, табл. 6).
При титровании солей железа (II) сульфатом церия (IV) ионы железа
(II) окисляются в ионы железа (III), в результате возникает система,
выражаемая следующим уравнением:
Fe++ — e ^± Fe+++
Если для титрования взято 100 мл 0,1 н. раствора соли железа (II),
то при прибавлении первой капли (~0,001 мл) 0,1 н. стандартного
раствора сульфата церия образуется эквивалентное количество Fe+ + +.
Поэтому в начале титрования отношение [Fe+ + + ]/ [Fe+ + ], определяющее
потенциал системы, составляет:
[Fe+++] 0,001
[Fe++] ~' 100 10~
Потенциал системы:
?Fe+++/pe++ = ?°Fe"*/Fe" + °'°59 Ig WT ~ °'771 + 0,°59 lg 10"6 ~
**0,771 —0,059-5^0,771 --0,295^0,476 в
Реакция окисления железа (II) церием (IV) идет согласно
уравнению:
Ce++++ + Fe++ ^± Ce+++ + Fe+++
?°Fe+++/Fe++ = 0,771 в; ?°Се++++/Се+++ = 1,45в
Реакция обратима, и во время титрования, после каждого
прибавления стандартного раствора сульфата церия, устанавливается равновесие:
Окисл! (Се++++) + Восст2 (Fe++) ^± Восстх (Се+++) + Окисл2 (Fe+++)
Окислительно-восстановительные потенциалы обеих систем
Fe+++ + e ^± Fe++ и Се+++++г т± Ce+++
при установившемся равновесии равны друг другу. Поэтому расчет
потенциала в каждый данный момент титрования после установления
равновесия может быть сделан по одному из следующих уравнений:
fFe+++l
?Fe+++/Fe++ = 0,771 + 0,059 lg ip^ (17a)
или
?ce++++/Ce+++ = 1.45 + 0,059 lg [^***j (18a)
б) Расчет величины потенциала в процессе
титрования до точки эквивалентности. При прили-
вании к 100 мл 0,1 н. раствора соли железа (II) 0,1 мл 0,1 н. раствора
сульфата церия (IV) происходит окисление Fe++ до Fe+ + + в количестве,
эквивалентном его содержанию в 0,1 мл 0,1 н. раствора. В этот момент,
если не учитывать разбавление раствора, отношение [Fe+ + + l/[Fe+ + l
составит:
[Fe+++] _ 0,1
[Fe++] - 100
5 9. МЕТОД ИЗОБРАЖЕНИЯ ПРОЦЕССА ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ 233
Следовательно:
?Fe++*/Fe++ = °»77l +0,059 lglgg^i^0,771 + 0,059 lg 10"» ~
^0,771—0,059-3^0,594 в
При приливании 1 мл стандартного раствора сульфата церия
образуется эквивалентное количество Fe+++ и
[Fe+"1 _ 1 _ 1ри
[Fe++J 100 ""
тогда
^Fe+++/Fe++ = О»77* + 0.059 lg [p***f ~ 0,771 + 0,059 lg 10~a =
= 0,771 — 0,059-2 = 0,653 в
При приливании к 100 мл 0,1 н. раствора соли железа (II) 10 мл 0,1 н.
раствора сульфата церия (IV)
[Fe+++] ю 1
= Ю-
lFe++] 90 10
Следовательно
TFe+++l
^Fe+++/Fe++ = О»77* + 0>059 lg Lp J ^0,771 + 0,059 lg 10"1 ^
^0,771—0,059^0,712 в
После добавления 50 мл раствора сульфата церия половина, т. е. 50 мл,
первоначально взятого объема раствора соли железа (II) будет окислена и
[Fe+ + + ] = [Fe++1; тогда
?Fe+++/Fe++ = 0,771 + 0,059 lg [ГрС7+7 = °»771 + °'059 ^ 1 = °'771 в
[re j
После прибавления 90 мл раствора сульфата церия неокисленными
останутся ионы железа (II) в количестве, соответствующем 10 мл 0,1 н.
раствора:
[Fe+++] 90
WT = -Io-~1()
EFe+++/Fe++ = 0,771 + 0,059 lg i^L ~
^0,771 + 0,059 lg 10^0,771 + 0,059 «* 0,830 e
После прибавления 99 мл раствора сульфата церия
[Fe++] l 1UU
?Fe+++/Fe+ + = О»771 + °>059 lg 1[Х**] ~ °'771 + °'°59 I8100w
«*0,771+0,118 «* 0,889 в
После прибавления 99,9 мл раствора сульфата церия
[Fe+++] 99,9
[Fe+Ч 0,1
>юоо
?Fe+++/Fe++ -0,771 +0,059 lg Jgj?L«0,771 +0,059lg 1000.
_ ^0,771+0,177=0,948 в
234
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
а) Расчет величины потенциала в точке
эквивалентности. В точке эквивалентности, т. е. в момент, когда 100 мл
0,1 н. раствора сульфата цёрия прибавлено к 100 мл 0,1 н. раствора соли
железа (II), в растворе устанавливаются следующие равновесия:
[Се+++] = [Fe+++]; [Ce++++] = [Fe++]
или
[Се+*+*]= [Fe++]
[Се+++] ~~ [Fe+++]
Для установившегося равновесия ?се++++/се+++ = Е?е+++/ге++ = Е
можно написать:
2? = 1,45 + 0,771+ 0,059 (igl^+Igl^L)
[Се+-""Ч [Fe+++]
2Е = 1,45+ 0,771+0,059 lg*
[Се+++] [Fe++]
Так как произведение взаимообратных чисел
[Се++++] [Fe++]
[Се+++] — [Fe+++]
равно единице, то выражение под знаком логарифма также равно
единице; lg 1 = 0. Поэтому
2? = 1,45+ 0,771
? = l,45 + 0.77,l=ul0g или ДмД? + ^1
Таким образом, в точке эквивалентности Е = 1,110 в. В точке
эквивалентности, согласно формуле (14) (см. § 5):
[Восстх] [Окисл2] Лг-гг
[ОкИСЛх] [В0ССТ2] У ЛОкисл/Восст
а согласно формуле (13):
1& лОкисл/Восст q 059
Для нашего случая титрования (п = 1)
- „ 1,45 — 0,771 11С
хь АОкисл/Восст ~~ о 059 ~~~
^Окисл/Восст ^ 4* ^
Поэтому в точке эквивалентности
[Се++++] [Fe++] v u ' и
Это означает, что указанная реакция будет идти практически до конца
слева направо и что в момент равновесия в точке эквивалентности [Fe+ + ]
§ 9. МЕТОД ИЗОБРАЖЕНИЯ ПРОЦЕССА ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ 235
в титруемом растворе будет более чем в миллион раз меньше [Fe+ + + ],
т.е. ионы железа(II) будут практически полностью оттитрованы (окислены),
г) Расчет величины потенциала после точки
эквивалентности. Окислительный потенциал после точки
эквивалентности вычисляют по уравнению (18):
?Се*+**/Се++* = ?Ce+++*/Ce*** + °'°59 lg [Се***]
Нормальный потенциал этой системы Е° . +++/ +++ равен 1,45 е.
Ов /Се
Прибавление избытка 0,1 мл 0,1 н. раствора соли церия (IV) создает
отношение:
[Се****] 0,1
[Се+++] 100
ю-3
ГГр+ +++1
ЯСе++++/Се+ ++ = 1,45 + 0,059 lg ig^i = 1,45 + 0,059 lg 10~з =
= 1,45 — 0,059.3 = 1,273 в
Прибавление избытка 1 мл 0,1 н. раствора сульфата церия создает
отношение:
[Се*++] 100
ЕСе****/Се+++ = 1АБ + °'°59 1*1№^] = !'45 + °'0591& 10'2 =
= 1,45 — 0,059-2=1,332 в
Прибавление 10 мл избытка 0,1 н. раствора соли церия (IV) создает
отношение:
[Се****] _ 10 _ 1 ,
[Се+*+] 100 ~~ 10
ЕСе****/Се+++ = 1ЛЪ "" °'°59 lg [С[С^\ = 1 >45 + °'°59 lg Ш"1 в
= 1,45 — 0,059 = 1,391 в
Другими словами, в начальный период после достижения точки
эквивалентности значение потенциала (подобно его значению в точке
эквивалентности) продолжает резко повышаться при добавлении первых капель
раствора сульфата церия, а затем значение потенциала изменяется в
меньшей степени.
Полученные данные сведены в табл. 24. При титровании применяют
индикатор ферроин.
Титрование солей железа (II) стандартным раствором перманганата
калия. Аналогичным образом можно рассчитывать потенциалы в различные
моменты титрования растворов солей железа (II) стандартным раствором
перманганата; ионов железа (III) ионами олова (II) или ионами титана
(III) и т. д.
Составьте самостоятельно таблицу титрования 100 мл 0,1 н. раствора
ионов железа (II) 0,1 н. стандартным раствором КМп04 в сернокислой среде
при рН = 1 (?0е+++/ре++ = о,771 в; ^по7+8Н+/Мп++ = 1,52 в).
236
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
ТАБЛИЦА 24
Титрование 100 мл 0,1 н. раствора ионов железа (II)
0,1 н. раствором ионов церия (IV)
Прибавлено
ОД н.
раствора сульфата
церия, мл
Осталось
Fe++-Ho-
нов, %
Соотношение концентраций ионов
Значение
потенциала, в
АС
До точки эквивалентности
1 капля
(~ 0,001 мм)
0,1
1
10
50
90
99
99,9
100
99,9 |
99
90
50
10
1
0,1
0,001
100
0,1
99,9
0,1
99
10
90
50
50
90
10
99
1
99,9
0,1
= ю-5
^10~3
• = 1
¦^10
«*102
**103
[Fe+++
IFeH
0,476
0,594
0,653
0,712
0,771
0,830
0,889
0,948
100
В точке эквивалентности
[Ce+++] = [Fe+++]; [Се++++] = [Fe++]
1,110
[Се+++] а [Ге+»+] =м.10.в
[Се++++] [Fe++]
0,118
0,1
0,059
0,9
0,059
10
0,059
40
0,059
40
0,059
9
0,059
0,9
0,162
0,1
= 1,18
= 0,065
= 0,006
= 0,0015
= 0,0015
= 0,0065
= 0,065
= 1,62
100,1
101,0
110
0,1
1
10
После точки эквивалентности
[Се*++Ч
[Се+++]
Избыток ионов церия (IV)
0,1
100
1
¦= 10-
100
10
100
ю-2
ю-1
1,273
1,332
1,391
0,163
0,1
0,059
0,9
0,059
= 1,63
= 0,065
= 0,0065
Расчет величины потенциала до точки эквивалентности проводится
точно так же, как при титровании Fe+ + сульфатом церия (IV), по формуле:
rpe+++i
? = 0,771+0,059 lgJg^J-
В точке эквивалентности, т. е. в момент, когда к 100 мл 0,1 н. раствора
ионов железа (II) прибавлено 100 мл 0,1 н. раствора КМп04,
устанавливается равновесие:
5Fe++ + МпО; + 8Н+ ^± 5Fe+++ + Мп++ + 4Н20
[Fe++]=5[MnO;]
[Fe+++] = 5 [Mn++]
« Q МЕТОД ИЗОБРАЖЕНИЯ ПРОЦЕССА ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ 237
Разделив второе уравнение на первое, получим:
[Fe-1""-] [Мп**]
[Fe**J ~ [мпо;]
B*~W = °'771 + °'059 lg WT (т
0059 [MnOTl ГнЧ8
Сложим уравнения (19) и (21):
?Fe"+/Fe" + 5?МпО:+8Н*/М„" = °'7Л + ^ + °'°59 '* W*f +
ГМпО;1 ГнЧ8 fFe-+1 [МпО;] [н+18
+ 0,059 lg [ [My+] J =0,771 + 1.52-5 + 0,059 lgi ^ [М^Ц J
Если в растворе [Н+] = 1, то
[Ре"*] [МпО;ПнЧ8 ,
так как
[Fe++J [Mn++]
[Fe+++] [MnOj]
= 1
[Fe++J [Mn++J
g [Fe++J [Mn++] lg ] ~ u
? +++/ +++5? - +/ ++ = 0,771 -f- 1,52-5
Fe+++/Fe++^ Mn04+8H7Mn++ ' ^ '
При установлении равновесия в точке эквивалентности
?Fe+++/Fe++ = ^МпОЦ-8Н+/Мп++ = ?
? + 5? = 6? = 0,771 + 5-1,52
?= 0,771 + 5.1,52 ==1395g (22)
Окислительный потенциал после точки эквивалентности вычисляют
по уравнению
Е _*• . 0,059 [Мп04-] [н+]»
?Мп07+8Н+/Мп++ — * Мп07-Ь8Н+/Мп++ ^ (Г" g [Mn++] { 6)
Построение кривых титрования. Если на оси абсцисс откладывать
содержание (в процентах) остающегося в растворе (в разные моменты
титрования) восстановителя или окислителя или* количество прибавленного
стандартного раствора (в миллилитрах), а на оси ординат — отвечающие
им значения Е (в милливольтах), то можно построить кривую,
представляющую ход изменения Е в процессе окисления—восстановления.
На рис. 53 показана наиболее характерная часть кривой титрования
0,1 н. раствора сульфата железа (II) 0,1 н. раствором сульфата церия (IV)
[10% остатка Ре++-ионов и 10% избытка стандартного раствора Се (SOJg].
238
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Как видно из рис. 53, кривая окислительно-восстановительного
титрования напоминает кривые, получаемые в методе нейтрализации. В начале
титрования, когда в титруемом растворе находится значительное
количество восстановителя, кривая изменяется плавно, даже при
добавлении значительных количеств реактива величина Е изменяется медленно.
Вблизи точки эквивалентности наблюдается резкое изменение потенциала
даже при прибавлении малых количеств реактива. По резкому скачку
кривой титрования устанавливают точку эквивалентности, которая не всегда
Линия эквивалентности
/4501
135о\
/250\
/Щ
? 950\-
^ 85о\
75и\
650\
550\-
I
Область изменения цвета
Т ^индикатора шерроина
fc (/?ри[Н+]=/)
Точка эквивалентности
100 НО
Объем 0,1н. стандартного раствора Ce(S04)2, мл
Рис. 53. Кривая титрования 0,1 н. раствора соли железа (II) 0,1 н.
раствором сульфата церия (IV).
лежит на середине скачка. Характер кривых в методах
окисления—восстановления не зависит от разбавления раствора, если стехиометрические
коэффициенты у окислителя и восстановителя одинаковы.
Например, в системе Fe+++ ^ Fe++ и Се++++ Z^L Ce+++ при
разбавлении раствора соотношения концентраций остаются те же. Если же
стехиометрические коэффициенты различны, то разбавление раствора
сказывается на отношениях окисленной и восстановленной форм, и потенциал
раствора несколько изменится*.
При более точных вычислениях, как уже было сказано раньше, применяют формулу:
Е = Е
Окисл/Восст '
0,059
lg
"Окисл
аВосст
При титровании ионов железа (II) сульфатом церия изменение потенциала раствора
может быть вычислено по уравнению:
0 [Fe+++] /Fe+++
Е == ^Окисл/Восст + °»059 *8 [Fe++] /F ++
где / — коэффициент активности.
* Заслуживающие большого внимания химиков-аналитиков методы расчета
окислительно-восстановительных процессов, применяющихся в объемном анализе, основанные
на теории ионных равновесий, подробно рассматриваются Н. П. Комарем [см. К о -
марь Н. П. Основы качественного химического анализа, изд. Харьковского
государственного университета, 1955; Зав. лаб., 28, 899 (1962) J.
§ 11» ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 239
1. Надо иметь в виду, что между окисленной и восстановленной формами образуются
нестойкие промежуточные соединения и потенциал определяется их активностью, а не
активностью полностью восстановленной формы. Так, например, обстоит дело при окислении
бихроматом и перманганатом.
2. Реакции окисления—восстановления обычно проводят в растворах, отличающихся
сравнительно большими ионными силами. Коэффициенты активности в таких растворах
не могут быть вычислены с достаточной степенью точности. Поэтому, учитывая указанные
обстоятельства, при обычных расчетах подставляют в формулы значения концентраций
вместо значений активностей.
§ 10. Фиксирование точки эквивалентности в методах
окисления—восстановления
В ряде случаев окислительно-восстановительного титрования точки
эквивалентности фиксируют по изменению окраски титруемого раствора,
вызываемой избытком окрашенного стандартного раствора (например,
перманганата).
В методах, основанных на титровании стандартным раствором иода,
или на выделении иода (иодометрия), точку эквивалентности
устанавливают при помощи индикатора — крахмала, специфически
реагирующего с иодом (в присутствии свободного иода бесцветный раствор крахмала
окрашивается в интенсивно синий цвет; в присутствии избытка
восстановителей, восстанавливающих нейтральный иод, интенсивно синее
окрашивание исчезает).
В некоторых случаях конец титрования определяется по
обесцвечиванию кроваво-красной окраски роданида железа в присутствии избытка
восстановителя (титанометрия).
Перечисленные вещества можно отнести к группе специфических
реактивов (индикаторов), действующих в пределах определенных методов
окисления—восстановления.
Помимо этой группы веществ (индикаторов), в методах окисления—
восстановления широко применяются индикаторы, называемые ред-окс-
индикаторами, которые изменяют окраску в зависимости от величины
окислительно-восстановительного потенциала (Е).
Иногда для более четкого фиксирования точки эквивалентности прямое
визуальное титрование проводят либо в неводных растворах, либо к
водному раствору добавляют небольшое количество органического
растворителя, не смешивающегося с водой (бензол, изоамиловый эфир, четырех-
хлористый углерод и т. п.). При этом органический растворитель
экстрагирует слабоокрашенное вещество, и окраска его усиливается. Так,
например, поступают при иодометрическом титровании, экстрагируя иод.
В ряде случаев используют внешние, флюоресцирующие, хемилюми-
несцентные и другие индикаторы (см. ниже).
Наиболее широко для фиксирования конечной точки титрования
применяют физико-химические (инструментальные) методы (потенциометри-
ческий, амперометрический, кулонометрический, кондуктометрический,
хронокондуктометрический, высокочастотный, фотометрический, спектро-
фотометрический и др.).
§ 11. Окислительно-восстановительные индикаторы
(ред-окс-индикаторы)
Окислительно-восстановительные индикаторы (ред-окс-индикаторы)
представляют собой органические соединения, окисленная и
восстановленная форма которых имеет различные окраски.
Изменение цвета ред-окс-индикатора обусловливается окислением
восстановленной формы индикатора и превращением ее в окисленную форму
240
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
или восстановлением окисленной формы и переходом ее в восстановленную
форму при определенном значении потенциала. Таким образом, две формы
индикатора представляют собой окислительно-восстановительную пару:
или
IndBoccT — ™
бесцветная форма
1пс1Окисл + пе
окрашенная форма
^±
^t
1п(1Окисл
окрашенная
IndBoccr
форма
бесцветная форма
Отношение концентраций окисленной формы к восстановленной
определяется уравнением (1):
f - ро л. RT in [1п(!окисл]
п? [IndBoccT]
Другими словами, ред-окс-индикаторы напоминают кислотно-основные
индикаторы, изменяющие свою окраску при определенных значениях рН
титруемого раствора.
Следует иметь в виду, что величина потенциала большинства
индикаторов ред-окс-систем сильно зависит от величины рН титруемого раствора,
поэтому применяемый ред-окс-индикатор может изменить свою окраску
не в точке эквивалентности, если резко изменился рН среды.
Появление в растворе избытка окислителя обусловливает окисление
молекул самого индикатора, сопровождающееся переходом одной формы
индикатора в другую. Избыток восстановителя вызывает восстановление
индикатора. Индикатор дает правильное показание, если момент
изменения его окраски совпадает с точкой эквивалентности, т. е. применяемый
индикатор должен вступать в реакцию окисления—восстановления вблизи
точки эквивалентности.
Ред-окс-индикаторы применяют в различных методах оксидиметри-
ческого титрования; поэтому они имеют большое значение в
аналитической практике.
Применяемые в оксидиметрии индикаторы должны удовлетворять
следующим требованиям:
1) индикатор должен быть чувствителен, т. е. реагировать в точке
эквивалентности с минимальным избытком окислителя или восстановителя;
2) окраски окисленной и восстановленной форм индикатора должны
резко отличаться друг от друга;
3) изменение окраски должно быть отчетливо заметно при применении
небольшого количества индикатора;
4) интервал индикатора должен быть невелик, т. е. изменение окраски
должно происходить в небольших пределах значений потенциала и
совпадать со скачком титрования;
5) индикатор должен быть устойчив по отношению к кислороду
воздуха, двуокиси углерода и свету.
Зависимость окраски индикатора от соотношения концентраций
окислителя и босстановителя. При титровании к раствору приливают очень
небольшое количество индикатора. В зависимости от потенциала раствора
меняется состояние равновесия ред-окс-индикаторов.
В качестве примера может быть приведена система 12 + 2е^21",
Свободный иод окрашен, иодид-ионы бесцветны. Иодид-ионы окисляются
до элементарного иода различными веществами с окислительными
потенциалами большими, чем потенциал указанной системы, а веществами
с меньшими окислительными потенциалами иод восстанавливается до иодид-
§11. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 241
ионов. Действие индикаторной системы иодид — иод основано на
соотношении окислительных потенциалов титруемой системы и индикатора.
Обычно ред-окс-индикаторами служат сложные органические
соединения. Как пример вещества, меняющего окраску при окислении и
восстановлении, может быть указан бензидин H2N—<f Ъ— <f Ъ— NH2.
При действии окислителей он окисляется, интенсивно окрашивая раствор
в синий цвет в слабощелочной, нейтральной и очень слабокислой (рН = 6)
средах и в желтый цвет в сильнокислой среде. Бензидин окисляется
хлором, бромом, хроматом, гексацианоферратом и т. д. Окисление бензидина
возможно только при определенном окислительном потенциале. Так,
водные растворы иода вызывают синюю окраску бензидина, растворы
иода, содержащие иодид-ионы, не реагируют с бензидином. Присутствие
иодид-ионов настолько снижает окислительный потенциал системы 12 +
+ 2е ^ 21", что бензидин не окисляется. Аналогичная картина
наблюдается при окислении бензидина ионами железа (III). В присутствии
большого количества Fe++ окисление бензидина железом (III) не
происходит.
Из рассмотренных примеров видно, что изменение окраски индикатора
зависит не от общего количества окислителя и восстановителя, а от
соотношения их концентраций, так как именно оно определяет величину
окислительного потенциала данной системы.
Интервал перехода окислительно-восстановительного (ред-окс)
индикатора. Появление или исчезновение интенсивной окраски ред-окс-инди-
катора связано с определенным количественным отношением двух форм
индикатора: 1пс!окисл + п^^Цпёвосст. Для этой системы можно написать:
? = ?ind +
о 0,059 [IndOKHcn]
md+ n lg[IndB0CCT]
где Е — окислительный потенциал системы индикатора;
?jnd — нормальный окислительный потенциал системы индикатора;
п — число приобретаемых или теряемых электронов.
Окислительный потенциал системы должен иметь определенную
величину, чтобы вызвать образование окрашенной формы индикатора. Окраска
индикатора и ее интенсивность, как указано выше, зависят от отношения
концентраций двух форм индикатора:
[1п(1Окисл1
[IndBoccT]
Из приведенного выше уравнения следует:
0,059 б [1п<*Восст]
Появление окраски связано с некоторой областью
окислительно-восстановительных потенциалов раствора, называемой интервалом перехода
индикатора.
Найдем интервал перехода индикатора, т. е. границы окислительно-
восстановительного потенциала раствора, при которых происходит
изменение окраски. Наш глаз замечает появление окраски, если в данной форме
242
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
индикатора (окисленной или восстановленной) присутствует другая форма
в отношении 10 : 1 или 1 : 10 ко взятой:
[1п(1Окисл] _ю и [МОкисл] _ 1
[^Восст] [IndBoccx] 10
[1пс30кисл] _ у [IndOKHCJI] n(E-E°lnd)
[IndBocCT] ~Aj lg [IndB0CCT] lgA 0,059
Если
[IndOKHCJI] п(Е-Е?ш)_
[внесет] °>059
TO
? = ^Ind
0,059
Если
то
[^Окисл] = 1 *(S-E?nd)
[lndBocCT] 10 > 0,059
о 0>O59
Поэтому интервал ред-окс-индикатора вычисляют по формуле:
'Ind
ПрИ А1= 1
*«*?.- ±-*^ (24)
Е = ??nd ± 0,059 (25)
Интервал перехода индикатора лежит между двумя окислительными
потенциалами, один из которых примерно на 59 мв больше, а другой на
59 мв меньше, чем нормальный окислительный потенциал индикатора.
Применение ред-окс-индикаторов. Окислительно-восстановительные
индикаторы, имеющие нормальные окислительные потенциалы меньше
+0,76 в, редко применяются в аналитической практике, но могут быть
использованы при титровании сильными окислителями солей олова (II),
хрома (II) и титана (III). Недостатками этих индикаторов является то,
что их окислительные потенциалы сильно зависят от рН и от ионной силы
раствора. Примерами таких индикаторов могут служить индигосульфоно-
вые кислоты, индофенолы и др.
Окислительно-восстановительные индикаторы, окислительные
потенциалы которых равны или превышают+0,76 в, широко применяются в
практике. Их используют главным образом для титрования солей железа
(Fe+++ + е^_ Fe++).
Наиболее распространены следующие индикаторы: дифениламин и его
производные, дифенилбензидин, дифениламинсульфокислота и некоторые
ее производные, фенилантраниловая кислота и др. При окислении
дифениламина сильными окислителями, например хроматом, бесцветный
дифениламин окисляется до бесцветного дифенилбензидина (процесс необратимый):
\_// \—// -Н20 \_// \—// \-.// \—//
дифениламин
(бесцветный)
дифенилбензи ди н
(бесцветный)
§11. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ИНДИКАТОРЫ 243
Бесцветная форма дифенилбензидина окисляется с изменением
структуры молекулы в форму дифенилбензидина фиолетового цвета (процесс
обратимый); в сильнокислых растворах возникает сине-фиолетовое
окрашивание:
дифенилбензидин
(бесцветный)
^~V-N =/~\=/_N>= N-/"\ + 2е + 2Н+
4_i"N =\=7=\=7= N~4_7 ¦
дифенилбензидин
(фиолетовый)
Интервал перехода индикатора лежит при окислительном потенциале
+0,76±0,1 в. Величина эта практически не зависит от концентрации ионов
водорода.
Дифениламин применяют при титровании бихроматом калия, вана-
датом аммония и очень разбавленными растворами перманганата калия.
Дифениламинсульфокислота C6H4NHC6H4S03H является одним из
лучших ред-окс-индикаторов. В восстановленной форме индикатор
бесцветен; окисленная форма окрашена в красно-фиолетовый цвет. Интервал
перехода лежит в области Е = 0,84 — 0,85 в. Химизм изменения окраски
аналогичен тому, какой наблюдается при окислении дифениламина.
Другой важной группой индикаторов является офенантролин и его
производные. Комплекс о-фенантролина с ионами железа (II) интенсивно
красного цвета; он называется «ферроин». Под действием сильных
окислителей ферроин переходит в бледно-голубой комплекс с железом (III):
[Fe(C12H8N2)3]++ ^± [Fe(C12H8N2)3]+++ + e
красный бледно-голубой
Реакция обратима.
Нормальный окислительный потенциал этого индикатора в I н.
растворе серной кислоты равен 1,06 в. Ферроин широко применяют при
титровании сильными окислителями: перманганатом, бихроматом, солями
церия (IV).
При использовании ред-окс-индикаторов рекомендуется вводить
индикаторные поправки. Например, если в титруемый раствор добавляют
две капли 1%-ного раствора дифениламина, то из объема 0,1 н.
стандартного раствора окислителя, израсходованного на титрование, вычитают
0,05 мл, а если добавляют четыре-пять капель дифениламинсульфокислоты,
то вычитают 0,1 мл.
Другие индикаторы. В ред-окс-методах титрования также применяют
необратимые индикаторы: метиловый оранжевый, метиловый красный,
конго красный и другие, которые в конечной точке титрования
необратимо окисляются избытком окислителя и при этом меняют свою окраску.
Например, метиловый оранжевый окисляется согласно уравнению:
(СН3)2 NCeH4N = NCeH4S03Na -^ (CH3)2NCeH4NO + ONCeH4S03Na
окрашенная форма бесцветная форма
Известно также применение флуоресцентных и хемилюминесцентных
индикаторов при титровании восстановителей сильными окислителями.
244
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
К числу флуоресцентных индикаторов относятся многие вещества
(акридин, эухризин, семикарбазон салицилового альдегида и др.), излучающие
видимый свет при определенных значениях рН раствора в процессе
облучения их ультрафиолетовыми лучами. Поглощая ультрафиолетовые лучи,
молекулы флуоресцентных индикаторов переходят в возбужденное
состояние, при котором электроны располагаются на более высоких
энергетических уровнях. При излучении энергии молекулы индикаторов переходят
в первоначальное (невозбужденное) энергетическое состояние.
Хемилюминесцентными индикаторами являются разнообразные
вещества (люминал, люцигенин, силоксен и др.), светящиеся в конечной
точке титрования вследствие экзотермических химических процессов.
Хемилюминесценция представляет собой один из видов люминесценции
и связана с образованием электронно-возбужденных частиц.
Хемилюминесценция наблюдается главным образом при реакциях окисления
перекисью водорода, гипохлоритами и некоторыми другими окислителями.
Преимуществом флуоресцентных и хемилюминесцентных индикаторов
является то, что их можно применять для титрования не только
прозрачных и бесцветных, но мутных или окрашенных растворов, для титрования
которых обычные ред-окс-индикаторы не пригодны.
Б. ПЕРМАНГАНАТОМЕТРИЯ
§ 12. Основы перманганатометрии
Основным веществом, применяемым в качестве стандартного раствора
окислителя в перманганатометрии, является КМп04.
Обычно титрование стандартным раствором перманганатом калия
проводят в кислой среде.
Принципиальную схему протекающей при этом реакции можно
представить следующим образом:
МпО; + Бе + 8Н+ 5± Мп44" + 4Н20
Окислительно-восстановительный потенциал данной системы
выражается уравнением:
В -, „-?» -, ,, + igg-l, fM"0№T (26)
Мп04/Мп++ Мп04/Мп++^ 5 ё [Мп++]
Е° -. ++ = + 1,52 6
Мп04/Мп
Из этого уравнения видно, что ?Mn0-/Mn++ B сильной степени зависит
от [Н + ].
Иногда титрование стандартным раствором перманганата калия
проводят в нейтральных или щелочных растворах.
Принципиальную схему протекающей при этом реакции можно
представить следующим образом:
Мп04 + Ъе + 2HOH ^± Мп02 + 40H"
р -р° . 0,059 [мпо;]
Мпо;/мпо2 мпо;/мпо2 "*" 3 щ [Мп02].[ОН-]4 '
Е° -, = + 0,57 в
Мп04/Мп02
Таким образом, ^Мп0-/Мп0 в сильной степени зависит от [ОН"]
или 1Н+].
§ 13. ТИТРОВАНИЕ ПЕРМАНГАНАТОМ В КИСЛОЙ СРЕДЕ 245
§ 13. Титрование перманганатом в кислой среде
Процесс окисления перманганатом в кислой среде различных
неорганических и органических веществ, несмотря на кажущуюся простоту
суммарных уравнений реакций, весьма сложен. Это объясняется тем, что ионы
марганца могут существовать в кислых растворах в различных степенях
окисления: Mn++, Mn+ + +, MnIV, MnVI и Mnvn.
Низкозарядные ионы марганца, образующиеся в процессе
восстановления М11О4, сами способны окисляться перманганатом в более высшие
степени окисления. Поэтому состояние динамического равновесия,
устанавливаемого между указанными ионами, зависит от концентрации
(активности) ионов водорода и избытка М11О4. Так как реакции окисления—
восстановления, совершаемые в присутствии КМп04, Протекают с
измеримой скоростью, то во избежание образования промежуточных форм
окисления ионов марганца титрование раствором перманганата рекомендуется
проводить относительно медленно, при определенных значениях [Н+] и
температуры.
Достаточно легко окисляются перманганатом в кислой среде:
щавелевая кислота, перекись водорода, мышьяковистая кислота, иодистоводород-
ная, сернистая, сероводородная, азотистая и другие кислоты, а также
гексацианоферраты (II), тиосульфаты, роданиды, фосфиты и
многочисленные ионы металлов в их низших степенях окисления: Fe++, V++, V02,
Ti + + +, Ce+ + +, Mn++, Tl + , Cr++ и др.
Например, реакция КМп04 с Н2С204 протекает согласно следующему
суммарному уравнению:
5С20~ + 2МпО; + 16Н+ —> 10СО2 + 2Мп++ + 8Н20
QfiT — 2е—>2С02
МпО; + 5е + 8Н+ —> Мп"*4" + 4Н20
Практически данная реакция протекает в несколько стадий:
а) Вначале перманганат очень медленно реагирует с щавелевой
кислотой, вследствие чего и раствор долго остается окрашенным в малиново-
красный цвет от нескольких капель раствора КМп04, прибавленных
к Н2С204.
б) Затем наступает период, в течение которого малиново-красная
окраска перманганата медленно исчезает. При этом образуются ионы
двух-, трех- и четырехвалентного марганца, слегка окрашивающие раствор
в бурый цвет:
Мпо; —* Мп02 —> Мп+++ —> Мп++
в) Наконец, ионы марганца с промежуточной валентностью быстро
окисляют щавелевую кислоту, превращаясь в Mn+ +.
Нагревание и повышение [Н + ] способствуют быстрому течению
указанной реакции окисления—восстановления. Ионы марганца низших
степеней окисления оказывают каталитическое действие на реакцию. При
этом перманганат очень быстро обесцвечивается.
246 ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
В общем схематически процессы взаимодействия КМп04 с Н2С204
можно представить следующим образом:
Очень медленная реакция Мп04+5е+8Н+-^Мп+ + +4Н20 (а)
Быстрая реакция 2Mn04+3Mn+ + 4-16H+-->5MnIV +
+8Н20 (б)
Быстрая реакция MnIV+Mn++—> 2Мп+++ (в)
Быстрая реакция 2Мп1У+С204 ~ —> 2Мп++++2С02 (г)
Более медленная, чем реакция (г) . . 2Мп+ + + +Q2OJ " —> 2Mn+ + Ч-2С02 (д)
Поэтому при окислении перманганатом медленно окисляющихся
восстановителей необходимо добавлять к титруемым растворам катализаторы
(Mn+ +, Fe+ + + и другие ионы), сильно ускоряющие течение реакций
окисления—восстановления.
Другим способом окисления в кислой среде медленно окисляющихся
веществ является прибавление к определяемому веществу избытка пер-
манганата и последующее обратное оттитрование избытка перманганата
стандартным раствором восстановителя (например, Н2С204).
О содержании вещества, определяемого методом обратного
титрования, судят по количеству прореагировавшего с ним КМп04 (Укмпо4)»
вычисляемого по формуле:
^КМп04 = ^КМп04 "" ^Н2С20<
где 1^кмп04 — объем первоначально взятого стандартного раствора КМп04, мл;
VH с 0 — объем стандартного раствора восстановителя (Н2С204), израсходованного
на обратное титрование КМп04 (той же концентрации, что и раствор
KMnOJ, мл.
§ 14. Титрование перманганатом в щелочной среде
Процесс окисления перманганатом в щелочной среде различных
неорганических и органических веществ не менее сложен, чем окисление в
кислой среде. Это объясняется тем, что в первой стадии реакции окисления—
восстановления образуется Сначала манганат:
Мп04+б^±Мп07~
Окислительно-восстановительный потенциал этой системы
выражают следующим образом:
? -, _ =?° ., _ + 0,059 lg-J =Уг-
Мп04/Мп04 Мп04/Мп04 Ь [Мп04 ]
Е° -, — = + 0,54 в
Мп04/Мп04
Из этого уравнения видно, что ?Мп0-/Мп0— практически не зависит
от [ОН"] или [Н+].
Если в процессе титрования перманганатом щелочного раствора
восстановителя добавить в титруемый раствор нитрат бария, то [MnOj]
снижается вследствие образования осадка ВаМп04 зеленого цвета
(ПР = 2,5-КГ10).
Благодаря этому ускоряется реакция восстановления перманганата.
§ 15. ПРИГОТОВЛЕНИЕ РАСТВОРА ПЕРМАНГАНАТА КАЛИЯ 247
Во второй стадии окисления—восстановления манганат
восстанавливается до соединений марганца (IV):
Мп04^ + 2е + 2НОН —> | Мп02 + 40Н"
Окислительно-восстановительный потенциал этой системы выражают
следующим образом:
Е __, =Е° ._ +О059,, [М"°Г]
Мп04 /Мп02 Мп04 /Мп02 2 б [Мп02][ОН"]4
Е° __ . = + 0,57 в
Мп04 /Мп02
Из этого уравнения видно, что ?Мп0--/Мп0 зависит от [ОН"] (или
Ш + ]).
Восстановление перманганата в манганат протекает быстрее, чем
восстановление манганата в манганит. Скорость реакции, протекающей во
второй стадии, возрастает по мере увеличения [ОН"" ] и практически
сводится к нулю в присутствии Ba(N03)2.
Концентрация твердого вещества Мп02 постоянна, поэтому
приведенное уравнение можно представить так:
о 0,059 [мпо;]
Мпо;/Мп02 Мпо;/Мп02 ^ 3 1ё [ОН-]4 ^
Следует иметь в виду, что при известных условиях манганат склонен
к реакциям диспропорционирования
(самоокисления—самовосстановления)
2Мп04~ + Мп04" + 2HOH ^ 2Мп04 + \ Мп02 + 40H"
При этом зеленая окраска манганата меняется и наблюдается
образование буро-черного осадка Мп02.
Относительно легко окисляются перманганатом в щелочной среде:
формиаты, иодиды, иодаты, цианиды, роданиды и многие органические
соединения. Интересно отметить, что в щелочной среде перманганат не
окисляет оксалаты. Поэтому многие органические вещества окисляются
перманганатом в щелочной среде до оксалатов, а не до карбонатов.
Так, например, реакция КМп04 с этиленгликолем в щелочном растворе
протекает согласно следующему уравнению:
ЗНОСН2СН2ОН + 8КМп04 -^ 3KOOC — COOK + 8Мп02 + 2КОН + 8Н20
§ 15. Приготовление стандартного (титрованного) раствора
перманганата калия
В качестве стандартного раствора в методе перманганатометрии
применяют 0,05 н. или 0,1 н. раствор КМп04. Использование перманганата калия
дает возможность фиксировать точку эквивалентности по появлению
розовой окраски раствора, вызываемой добавлением лишь одной капли избытка
стандартного раствора КМп04. Даже очень разбавленные растворы
перманганата калия интенсивно окрашены. Например, 100 мл
дистиллированной воды окрашиваются в розовый цвет от одной капли 0,01 й. раствора
КМп04. Поэтому фиксирование точки эквивалентности при титровании
перманганатом не представляет никаких трудностей.
Расчет навески. Титрованный раствор перманганата калия нельзя
приготовить путем взятия точной навески и растворения ее в определенном
248 ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
объеме воды, так как перманганат калия всегда содержит ряд трудно
удаляемых загрязнений; на перманганат действуют органические вещества,
содержащиеся в дистиллированной воде, ненасыщенные углеводороды
светильного газа и т. д. Поэтому обычно берут приблизительную навеску
КМп04 или применяют предварительно приготовленный его раствор,
который разбавляют до требуемой концентрации.
Для работы обычно готовят 1—2 л 0,05 н. или 0,1 н. раствора перман-
ганата калия. Если титрование проводят в кислой среде, то грамм-экви-
КМп04 158,04 01 сло и ,
валент его равен —=—- = —~- = 31,608 г. Для приготовления 1 л
0,1 н. раствора берут приблизительно 3,16 г КМп04.
Техника приготовления раствора. Навеску, рассчитанную для
приготовления нужного объема раствора КМп04 заданной нормальности,
отвешивают на технических весах в бюксе или на часовом стекле и
растворяют в требуемом количестве дистиллированной воды в конической
колбе с притертой стеклянной пробкой. Воду отмеривают цилиндром.
Перманганат калия растворяется медленно, поэтому рекомендуется его
предварительно измельчить. Приготовленный раствор взбалтывают и
оставляют стоять 7—8 дней. В течение этого времени имеющиеся в нем примеси
окисляются. Выделившийся осадок двуокиси марганца отфильтровывают
через воронку со стеклянной ватой или сливают раствор при помощи
сифона. Часто для ускорения приготовления раствора перманганата калия
рекомендуют нагреть его в конической колбе до кипения, охладить и после
этого отфильтровать осадок.
Иногда стандартный раствор перманганата готовят не из навески
КМп04, а из его раствора.
Титр раствора перманганата калия с течением времени несколько
изменяется, поэтому его необходимо периодически проверять. Может
происходить частичное разложение перманганата с образованием
двуокиси марганца и выделением кислорода. Особенно быстро этот процесс
идет в присутствии катализаторов (Мп++). Разложение ускоряется также
под действием света. Раствор, защищенный от света (в темной стеклянной
бутыли или в склянке, обернутой защитной черной бумагой), сохраняется
долго.
§ 16. Установка титра стандартного раствора перманганата калия
Установочные вещества. Титр раствора перманганата калия
устанавливают по щавелевой кислоте Н2С204-2Н20, оксалату аммония
(NH4)2«C204-H20, оксалату натрия Na2C204, мышьяковистому ангидриду
Аз2Оа,гексацианоферрату (II) калия 1^ [Fe(CN)6l, по металлическому
железу и некоторым другим веществам.
Наиболее удобны как установочные вещества щавелевая кислота и ее
соли, так как они легко могут быть очищены от примесей
перекристаллизацией из воды. Необходимо применять только свежеперекристаллизо-
ванную щавелевую кислоту. Более удобно пользоваться оксалатом
аммония или оксалатом натрия, которые не изменяются при хранении. Оксалат
натрия не содержит кристаллизационной воды и устойчив, но, к
сожалению, относительно трудно растворим.
В качестве установочного вещества наиболее часто употребляют
оксалат аммония. Соли железа (II) также являются хорошими
восстановителями, но неустойчивы, легко окисляются кислородом воздуха. Наиболее
устойчива соль Мора (NH4)2S04-FeS04-6H20. Она легко получается
в чистом виде перекристаллизацией, очень медленно выветривается
и медленно окисляется на воздухе.
§ 17. УСТАНОВКА ТИТРА РАСТВОРА КМп04 по (NH4)2C204 249
Титрование перманганатом калия обычно проводят в кислой среде.
Выбор кислоты. При титровании перманганатом в кислой среде
предпочитают использовать разбавленную серную кислоту. В тех случаях,
когда в растворе присутствуют ионы, которые образуют осадки с сульфат-
ионами, применяют азотную кислоту. Хлористоводородная кислота
окисляется перманганатом с образованием свободного хлора. Реакция сильно
ускоряется, в присутствии солей железа.
Количество прибавляемой кислоты должно быть достаточно, чтобы
обеспечить рН <7 в течение всего процесса титрования. Следует иметь
в виду, что в процессе титрования кислотность среды понижается
вследствие того, что восстановление М11О4 до Мп++ сопровождается
нейтрализацией 8 г-ионов Н+ на каждый г-ион МпО^ согласно уравнению:
МпО^ + Ъе + 8Н +—** Мп++ + 4Н20
Теоретическое количество серной кислоты вычисляют, исходя из
соотношения реагирующих веществ.
Зная концентрацию исходной кислоты, легко вычислить объем
требуемой кислоты. Обычно для титрования берут 15—20 мл 2 н. раствора H2S04.
§ 17. Установка титра и нормальности раствора перманганата калия
по оксалату аммония
Установка титра перманганата калия может быть проведена методом
отдельных навесок и методом пипетирования (см. гл. I, § 8).
Установка титра раствора КМп04 методом отдельных навесок. На
аналитических весах берут несколько навесок (3—5) оксалата аммония,
растворяют их в дистиллированной воде и каждый из полученных растворов
титруют перманганатом. Навески рассчитывают так, чтобы на титрование
каждой из них пошло около 20 мл приготовленного стандартного раствора
перманганата калия. Эквивалент оксалата аммония 3(nh4)2c2o<.h2o =
71ЛС7 п 0,Ь20-71,057
= 71,057. Следовательно, надо взять каждую навеску около -—^д~— г,
т. е. приблизительно 0,1421 г.
Навеску переносят в коническую колбу, растворяют в 20—25 мл воды,
прибавляют 15—20 мл 2 н. раствора серной кислоты и нагревают смесь
до 70—80° С. Кипятить раствор не рекомендуется, так как щавелевая
кислота довольно легко разлагается. Нагревание раствора следует проводить
на водяной бане.
Нагретый раствор титруют перманганатом до бледно-розовой окраски
раствора, не исчезающей в течение 2—3 мин. Первые капли перманганата
обесцвечиваются медленно. Затем обесцвечивание КМп04 происходит
быстро.
Стандартный раствор перманганата калия помещают в бюретку со
стеклянным краном. Каких-либо соединений с помощью резиновых
трубок следует избегать, так как перманганат калия окисляет резину и титр
раствора изменяется.
Установка титра раствора КМп04 методом пипетирования. На
аналитических весах берут одну навеску оксалата аммония и переносят ее
в мерную колбу емкостью 200—250 мл, растворяют в воде и объем раствора
доводят до метки. Затем пипеткой берут из мерной колбы аликвотную
часть (20—25 мл) оксалата аммония, подкисляют 15—20 мл 2 н. раствора
серной кислоты, нагревают смесь до 70—80° С и титруют перманганатом
до появления слабо-розового окрашивания.
250 ГЛ.111. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
При расчете навески в этом случае исходят из того, что на каждое
титрование, т. е. на 20—25 мл раствора установочного вещества, должно
расходоваться примерно 20 мл стандартного раствора перманганата калия.
Концентрации применяемых растворов оксалата аммония и
перманганата калия должны быть 0,05—0,1 н. Порядок расчетов — см. гл. I,
§ Ю.
§ 18. Вещества, определяемые методом
перманганатометрии
Титрование перманганатом является одним из самых распространенных
и доступных ред-окс-методов объемного анализа, широко применяемых
в химико-аналитической практике в заводских,
научно-исследовательских и учебных лабораториях. Методами перманганатометрии можно
определять разнообразные неорганические и органические вещества:
а) восстановители; металлы (Fe, Bi, Ag, Cd, Zn, редкоземельные
металлы и др.), неметаллы (Sb, As, P и др.), ионы низших степеней
окисления, способные давать соединения элементов высших степеней окисления
(Fe++, Сг++, Mn++, Sn++, Cu + , Ti + + +, V111, VIV, Mo111, UIV, WIV,
As111 и др.), заряженные ионы неметаллов типа CI", Br", I", S" и т. п.,
комплексные анионы восстановители (SOJT» БеОз", ТеО^ , SjAT»
SCN", CN", N07, [Fe (CN)e]==s, поли- и оксикарбоновые кислоты
(НООССООН, НООССН2СООН и др.), альдегиды, муравьиную, мочевую,
аскорбиновую, сульфиновые кислоты, сахара, полифенолы, ненасыщенные
соединения и многие другие продукты (см. ниже);
б) окислители: Fe+ + +L CeIV_, Vv, UVI, MoVI, WVI, CrVI, Mn02,
Pb02, NOi, ВгОз, CIOI, I03, S208"" и др. (см. ниже).
Титрование перманганатом может осуществляться прямым, обратным
и косвенным методами. Прямым методом титрования определяют многие
восстановители, которые легко без потерь окисляются перманганатом.
Методы обратного титрования обычно применяют для определения
восстановителей, которые окисляются перманганатом слишком медленно,
и окислителей, которые предварительно восстанавливаются
соответствующими восстановителями до соединений низших степеней окисления.
Для определения многих веществ, не окисляющихся перманганатом
в строго эквивалентных количественных отношениях и для которых
неприменимы методы обратного титрования, применяют косвенные методы
титрования перманганатом. Поясним это на примере. Многие металлы
(например, серебро), или соединения элементов низших степеней окисления
(например, Си1) непосредственно не могут быть оттитрованы перманганатом
ни прямым, ни обратным методом титрования. В этом случае можно
прибегнуть к предварительному их окислению Fe111. Для этой цели обычно
применяют раствор железных квасцов, не содержащий свободного
кислорода. Образующиеся при этом в эквивалентном количестве по отношению
определяемого металла ионы Fe11 затем титруют перманганатом.
Преимущества и недостатки перманганатометрического метода. По
сравнению с другими методами окисления—восстановления рассматриваемый
метод отличается рядом преимуществ.
1. Исходные стандартные растворы перманганата окрашены в малиново-
красный цвет. Поэтому конечную точку титрования можно определять
по покраснению титруемого раствора при титровании прямым методом
или по исчезновению окраски при титровании обратным методом, не
прибегая к использованию индикаторов.
2. Титрование перманганатом возможно осуществлять в кислой или
щелочной средах.
§ 19. ОПРЕДЕЛЕНИЕ ЩАВЕЛЕВОЙ КИСЛОТЫ И ОКСАЛАТОВ 251
3. Перманганат отличается высоким окислительно-восстановительным
потенциалом (?0 _ ++ = +1,52в). Поэтому многие вещества, которые
v Mn04/Mn '
невозможно оттитровать растворами более слабых окислителей, можно
определять перманганатометрическим методом.
4. Перманганат является дешевым и легко доступным реагентом.
5. Перманганат также применяют для определения веществ, не
обладающих окислительно-восстановительными свойствами. Такого рода
определения основаны на осаждении определяемых ионов в виде
малорастворимых соединений, получаемых при действии восстановителей (например,
оксалатов), и последующем титровании перманганатом избытка
восстановителя, или ионов осадителя, связанных в осадок.
Известны также определения, основанные на взаимодействии избытка
восстановителей с определяемыми веществами, которое не
сопровождается образованием осадка, и последующем титровании избытка
восстановителя перманганатом. К числу таких методов относится определение
уксусного ангидрида, основанное на его реакции с щавелевой кислотой (а)
и последующем титровании избытка ее перманганатом (б):
/гН2С204 + (СН3СО)20 -^ 2СН3СООН + С02 + СО + (п — 1) Н2С204 (а)
МпО^
Н2С204 -^ 2С02 + 2Н+ (б)
Перманганатометрическому методу свойственны и некоторые
недостатки.
1. Исходный реагент — перманганат калия, трудно получить в
химически чистом состоянии. Поэтому непосредственно, исходя из навески
кристаллического КМп04 или другой соли марганцевой кислоты, установить
титр перманганата нельзя. Поэтому титр раствора перманганата
устанавливают посредством установочных веществ или фиксанала.
2. Стандартные растворы перманганата не устойчивы, со временем
меняют свой титр, поэтому в процессе использования их необходимо
периодически проверять.
3. Титрование перманганатом, как правило, не рекомендуется
проводить в присутствии С1~-ионов, окисляющихся им до элементарного хлора.
Поэтому избегают применять хлористоводородную кислоту для подкисле-
ния титруемого раствора.
4. Некоторые реакции окисления перманганатом протекают при
комнатной температуре очень медленно. Поэтому обычно требуется некоторое
нагревание растворов. Однако в ряде случаев, в особенности при
титровании легко летучих и термически разлагаемых продуктов, нагревание не
допускается.
5. Состояние динамических равновесий, устанавливающихся в
процессе титрования перманганатом, зависит от различных факторов, поэтому
определения необходимо проводить при строго определенных условиях,
рекомендуемых методикой анализа.
ОПРЕДЕЛЕНИЕ ВОССТАНОВИТЕЛЕЙ
§ 19. Определение щавелевой кислоты и оксалатов
Определение щавелевой кислоты или ее солей основано на титровании
их растворов стандартным раствором перманганата калия в кислой среде.
По количеству перманганата, израсходованного на титрование известной
навески или известного объема исследуемого вещества, вычисляют
количество оксалата. Определение оксалат-ионов применяют при анализах
252 гл.ш. методы окисления-восстановления
технической щавелевой кислоты, ее солей и при перманганатометрическом
определении кальция.
Уравнение реакции:
5Ср7 + 2M11O4 + 16Н+ —> 10СО2 + 2Мп++ + 8Н20
Рассчитанную навеску (см. гл. I, § 10) щавелевой кислоты или окса-
лата взвешивают в- бюксе или на часовом стекле сначала на технических
весах, а затем — на аналитических. Величину навески определяют по
разности двух взвешиваний бюкса: до и после взятия навески. Взятую
навеску пересыпают в мерную колбу, растворяют, затем доводят объем
раствора до метки и перемешивают. Для титрования берут аликвотные
порции раствора в конические колбы и титруют, как указано выше.
Если определение оксалатов проводят методом отдельных навесок,
то поступают так же, как при установке титра перманганата калия этим
методом.
Выполнение расчетов см. гл. I § 10.
§ 20. Определение соединений железа (II)
Определение содержания железа в какой-либо соли железа, например
в железном купоросе FeS04-7H20 или соли Мора (NH4)2S04. FeS04-6H20,
обычно проводят прямым титрованием стандартным раствором
перманганата калия в сернокислой среде:
5Fe++ + МпО; + 8Н+ -> 5Fe+ + + + Мп+ + + 4Н20
Рассчитанную навеску сульфата железа (II) переносят в мерную колбу
и прибавляют туда 100—150 мл 2 н. серной кислоты, доводят объем
водой до метки и хорошо перемешивают. Прибавление избытка серной
кислоты необходимо для переведения в раствор примеси основных солей
окиси железа, обычно присутствующих в солях железа (II) и для
предотвращения гидролиза солей железа (III), образующихся в процессе
окисления ионов железа (II). Кроме того, серная кислота замедляет окисление
железа (II) кислородом воздуха.
При титровании берут аликвотные части раствора. Реакция окисления
железа (II) идет быстро, без нагревания раствора. Конец реакции
определяют по появлению розового окрашивания, вызываемого одной
избыточной каплей раствора перманганата калия. Окраска не должна изменяться
в течение 1 мин. Иногда в раствор вводят фосфорную кислоту,
связывающую окрашенные в желтоватый цвет ионы железа (III) в бесцветный
комплекс [Fe (P04)2] . Благодаря этому более отчетливо наблюдается
переход окраски от бесцветной к розовой. Чтобы ионы железа (II) не
окислялись кислородом, следует избегать длительного соприкосновения их
с воздухом. Все операции — взвешивание, растворение, титрование —
должны проводиться быстро.
При точных работах определения ведут в атмосфере двуокиси
углерода. Расчеты выполняют по формулам, приведенным ранее (см. гл. I,
§ Ю).
Определение содержания железа (II) в присутствии СГ-ионов. При
определении железа в присутствии СГ происходит сопряженное окисление
хлорид-ионов. Вследствие этого часть хлора выделяется из раствора в виде
газа, что приводит к повышенному расходу КМп04 и результаты
получаются неточными. Чтобы избежать этого, титрование проводят по методу
Циммермана—Рейнгардта с прибавлением защитной смеси, состоящей из
сульфата марганца, серной и фосфорной кислот. Сульфат марганца яв-
§ 21. ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА В ПРИСУТСТВИИ ОКИСЛОВ ЖЕЛЕЗА 253
ляется антикатализатором реакции окисления СГ-ионов и задерживает
их окисление, так как Мп++-ионы легче окисляются, чем СГ. Желтая
окраска хлорида железа (III) может маскировать розовую окраску пер-
манганата. Фосфорная кислота связывает железо (III) в бесцветный
комплекс [ Fe (P04) 2 ]""".
Определение железа (II) в присутствии С1" имеет большое практическое
значение при определении его в сплавах, металлах, рудах, которые обычно
растворяют в хлористоводородной кислоте.
К раствору, содержащему Fe++, прибавляют 10—15 мл раствора
Циммермана—Рейнгардта, затем смесь разбавляют водой приблизительно
до 100 мл и медленно титруют перманганатом до появления розовой
окраски, не исчезающей в течение 1 мин.
Расчеты проводят по формулам, приведенным в гл. I, § 10.
§ 21. Определение содержания металлического железа
в присутствии окислов железа
Определение металлического железа основано на растворении сначала
железа в разбавленной серной кислоте в колбе, снабженной клапаном
Бунзена *, и последующем титровании стандартным раствором КМп04
образовавшихся ионов железа (II).
Определение металлического железа в присутствии его окислов
основано на предварительной обработке смеси раствором, содержащим КО,
СцС12 и СНдСООН, отделении окислов железа и последующем титровании
стандартным раствором КМп04 ионов железа (II), образующихся согласно
следующему уравнению:
Fe + Cu++ -*¦ Fe++ + Си
Си + Си++ + 2С1- —> 2CuCi
Методика определения. Измельченную пробу, содержащую 0,1 г
металлического железа, помещают в коническую колбу емкостью 250 мл.
Затем в колбу наливают 30 мл насыщенного двуокисью углерода раствора
реактива, содержащего 1000 мл НаО, 54 г СиС12-2Н20, 47 г КС1, 0,5 мл
70%-ной СН3СООН.
В указанном реактиве окислы железа не растворяются. Смесь
перемешивают 30 мин. Осадок отфильтровывают. Фильтрат собирают во вторую
коническую колбу. Первую колбу и фильтр тщательно промывают 100 мл
дистиллированной воды. Промывные воды прибавляют к фильтрату.
Для восстановления избытка СиС12 в фильтрат добавляют 1 а
алюминиевых стружек и 6 мл разбавленной (1 : 1) хлористоводородной кислоты.
Смесь кипятят до обесцвечивания раствора:
2А1 + 3Cu++ —> 2А1+++ + ЗСи
Остаток отфильтровывают и промывают. Фильтрат и промывные воды
собирают в третью коническую колбу и титруют в присутствии реактива
Циммермана—Рейнгардта стандартным раствором перманганата до
слаборозового окрашивания.
Расчет результатов анализа производят по формулам, приведенным
в гл. I, § 10.
* Клапан Бунзена представляет собой короткую резиновую трубку, посредине
которой сделан продольный разрез. Верхний конец трубки закрыт стеклянной палочкой.
Благодаря такому устройству газы выходят из колбы, а воздух в колбу не попадает (см. рис. 54,
стр. 257).
254 ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
§ 22. Определение азотистой кислоты и нитритов
Определение азотистой кислоты и нитритов основано на
взаимодействии их с перманганатом калия в кислом растворе. При этом нитриты
количественно окисляются до нитратов:
5NC>2 + 2MnO; + 6Н+ —> 5NOg + 2Mn+ + + ЗН20
Азотистая кислота неустойчива, летуча, легко окисляется кислородом
воздуха. При медленном титровании перманганатом калия подкисленного
раствора нитрита получают пониженные результаты, поэтому метод
прямого титрования перманганатом нитритов не применяется.
Известно два способа определения нитритов.
1-й способ. Титрование определенного объема подкисленного раствора
перманганата калия анализируемым раствором нитрита до
обесцвечивания раствора (метод Лунге). Метод не очень точен, потому что нитриты
реагируют с перманганатом не мгновенно и возможна частичная потеря
азотистой кислоты вследствие ее разложения и улетучивания окислов
азота. Под конец титрования обесцвечивание идет очень медленно. Метод
применяется редко.
Для титрования переносят пипеткой в коническую колбу 25 мл 0,1 н.
раствора перманганата, добавляют 25 мл разбавленной серной кислоты и
250 мл воды, смесь нагревают до 40—50° С и медленно титруют из бюретки
заранее приготовленным приблизительно 0,1 н. раствором нитрита до
очень слабо-фиолетовой окраски. Точность титрования 0,5—1%.
2-й способ. Окисление нитрита проводят избытком перманганата,
затем определяют количество непрореагировавшего перманганата (метод
обратного титрования).
Для титрования рассчитанную навеску нитрита или соответствующий
объем его раствора помещают в мерную колбу емкостью 250 мл, приливают
дистиллированной воды, смесь перемешивают и доводят объем раствора
до метки. Затем 50 мл стандартного 0,1 н. раствора перманганата
переносят в коническую колбу или склянку с притертой пробкой, прибавляют
5 мл разбавленной серной кислоты и 25 мл приготовленного раствора
нитрита. Закрывают колбу пробкой, оставляют стоять на 10—15 мин,
потом перемешивают. К окрашенному перманганатом раствору приливают
избыток раствора оксалата аммония определенной нормальности, смесь
нагревают до 70—80° С и обесцвеченный раствор, в котором содержится
непрореагировавший остаток оксалата аммония, оттитровывают
перманганатом калия. На обратное титрование должно пойти около 15—20 мл
стандартного раствора перманганата калия. Рекомендуется провести
«холостой» опыт, чтобы установить соотношение объемов оксалата
аммония и перманганата калия.
Расчеты проводят по соответствующим формулам (см. гл. I, § 10).
§ 23. Определение содержания марганца (II) в рудах
Содержание марганца (II) может быть определено сначала окислением
его в марганец (VII) периодатом или висмутатом, а затем восстановлением
образовавшегося перманганата избытком восстановителя (Н2С204, солью
Мора и т. п.). Избыток восстановителя обратно оттитровывают
стандартным раствором перманганата.
Более простым способом является метод прямого титрования
соединений марганца (II) стандартным раствором перманганата калия. При
этом ионы марганца (II) окисляются в соединения марганца (IV):
ЗМп+ + + 2МпО; + 2Н20 —> \ 5Мп02 + 4Н+
§ 23. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МАРГАНЦА (II) В РУДАХ 255
В процессе титрования выпадает бурый осадок двуокиси марганца
и образуется свободная кислота.
Для полноты протекания реакции необходима нейтрализация ионов
водорода. Прибавление сильной щелочи недопустимо, так как она может
вызвать осаждение гидроокиси марганца и способствовать частичному
окислению ее кислородом воздуха. Для поддержания нужной
кислотности среды (рН = 5—6) прибавляют окись цинка, которая связывает
образующуюся кислоту:
ZnO + 2Н+ —> Zn++ + Н20
В процессе титрования наряду с Мп02 образуется Н2Мп03,
Мп(НМп03)2 и МпМп03 и окисление ионов марганца (II) замедляется.
При прибавлении окиси цинка выпадает осадок ZnMn03:
ZnO + H2Mn03 —> l ZnMnOs + H20
Поэтому окисление ионов марганца (II) идет беспрепятственно. Если
в растворе присутствуют ионы железа (III) или хрома, то они осаждаются
в присутствии окиси цинка в виде гидроокисей. При недостатке окиси
цинка образуется свободная кислота, окисление прекращается и раствор
принимает фиолетовую окраску до достижения точки эквивалентности.
Методика определения. Рассчитанную для приготовления 250 мл
раствора навеску руды помещают в стакан, обливают 15—20 мл
концентрированной хлористоводородной кислоты, добавляют 2—3 г хлората калия,
закрывают стакан часовым стеклом и нагревают под тягой до полного
разложения руды и окончания выделения хлора. Раствор количественно
переносят в мерную колбу, не отфильтровывая белого осадка кремневой
кислоты, объем раствора доводят до метки и тщательно перемешивают.
Отбирают пипеткой аликвотные части раствора в конические колбы
емкостью 400—500 мл, нейтрализуют их карбонатом натрия до слабокислой
реакции и прибавляют взмученную в воде окись цинка до тех пор, пока
после взбалтывания жидкости на дне не будет оставаться небольшой
осадок окиси цинка. Полученную смесь разбавляют 250—300 мл горячей воды
и горячий раствор титруют стандартным раствором перманганата.
При прибавлении из бюретки перманганата калия образуется осадок
двуокиси марганца, который затрудняет наблюдение за изменением
окраски раствора. Поэтому для ускорения анализа несколько изменяют-
обычную технику титрования. Прибавляют перманганат не по каплям,
а сразу по 1—2 мл. Смесь хорошо перемешивают и дают осадку в растворе
отстояться, опуская колбу в горячую воду или держа ее на бане. Двуокись
марганца быстрее коагулирует в горячем растворе. Если раствор не
окрашивается, то прибавляют еще перманганата калия. Незадолго до точки
эквивалентности раствор просветляется быстро, отстаивание идет хорошо
и при дотитровывании по каплям розовая окраска раствора становится
ясно заметной. Ко второй порции сразу приливают почти все необходимое
количество перманганата, дают осесть осадку и дотитровывают по каплям.
При расчетах необходимо помнить, что эквивалент КМп04 в проводимой
реакции равен МКмпо4/3, так как Mnvn переходит в Mniv. Если раствор
перманганата калия, как обычно, был установлен по щавелевой кислоте,
то расчет навески его был сделан на основании эквивалента, равного
Мкмпо4/5.
При восстановлении КМп04 до Мп02 0,1 н. раствор перманганата
калия должен содержать —^— = 52,6792 г/л, а при восстановлении
до Мп++ —^— = 31,6075 г/л. Следовательно, наш исходный стандартный
256
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
раствор перманганата слабее требуемого раствора во столько раз, во
сколько 31,608 меньше 52,68, т. е. 52' = 0,6.
Поэтому истинная нормальность ранее приготовленного нами 0,1 и.
раствора КМпр4 равна 0,1.0,6 = 0,06 н. Поэтому если пользуются
стандартными растворами, приготовленными путем растворения Мкмпо4/5 на
1 л, то при расчетах вводят вычислительную поправку, равную 0,6.
ОПРЕДЕЛЕНИЕ ОКИСЛИТЕЛЕЙ
§ 24. Определение соединений железа (III)
Определение соединений железа (III) перманганатометрическим
методом основано на предварительном восстановлении Fe+ + + до Fe+ +
соответствующим восстановителем и последующем титровании железа (II)
стандартным раствором перманганата.
Восстановление железа (III) до железа (II) может быть проведено
цинком, алюминием, висмутом, хлоридом олова (II), жидкими
амальгамами и др. Из металлов чаще всего пользуются гранулированным цинком,
цинковой пылью или амальгамированным цинком. Восстановление
металлическим цинком или амальгамированным цинком целесообразно
проводить при отсутствии в анализируемом растворе других ионов, способных
восстанавливаться (например, титана, ванадия, хрома, молибдена и т. п.).
Восстановление соединений железа (III) хлоридом олова (II). Иногда
Ре+ + + -ионы восстанавливают хлоридом олова (II):
2FeCl3 + SnCl2 —> 2FeCl2 + SnCl4
Избыток хлорида олова (II) окисляют прибавлением хлорида ртути (II):
SnCl2 + 2HgCl2 —> I Hg2Cl2 + SnCl4
Образующаяся каломель при титровании железа (II) перманганатом
окисляется очень медленно и не мешает определению.
Восстановление соединений железа (III) металлическим цинком. При
восстановлении цинком рассчитанную навеску соли железа (III)
помещают в круглодонную колбу, в которую прибавляют 50 мл разбавленной
(1 : 4) серной кислоты; затем в колбу высыпают рассчитанное (с большим
избытком) количество цинковой пыли или стружек. Колбу закрывают
пробкой, снабженной клапаном Бунзена (рис. 54) и нагревают на водяной
бане до полного растворения цинка. Колбу устанавливают косо во
избежание разбрызгивания. После растворения цинка раствор охлаждают
и количественно переносят в мерную колбу, перемешивают и титруют
перманганатом.
Восстановление соединений железа (III) амальгамированным цинком
(в редукторе Джонса). Редуктор Джонса (рис. 55) представляет собой
стеклянную трубку длиной 25—30 см и внутренним диаметром 1,8—2 см.
В верхней части трубка имеет расширение емкостью 25—30 мл. В нижней
части трубка сужена и снабжена стеклянным краном, как бюретка.
Приемником служит коническая колба. Если восстановленный раствор легко
окисляется воздухом, то колбу закрывают пробкой с двумя отверстиями,
через одно из которых проходит трубка редуктора (при титровании
заменяется бюреткой), через другое — трубка, через которую колбу можно
наполнить двуокисью углерода.
Примечание. Если восстановление вести при доступе воздуха, то образуется
перекись водорода, которая восстанавливает окислитель при последующем титровании
восстановителя.
§ 25. ОПРЕДЕЛЕНИЕ НИТРАТОВ
257
В нижнюю часть редуктора помещают дырчатую фарфоровую
пластинку, пористую стеклянную пластинку или несколько стеклянных
шариков, на которые положен слой стеклянной ваты. На фильтрующий слой
помещают кусочки (величиной 0,6—2 мм) амальгамированного цинка.
Цинк амальгамируют обработкой раствором хлорида окисной ртути
(яд!), для чего 200 г цинка высыпают в 200 мл 2%-ного раствора хлорида
окисной ртути, перемешивают 5—6 мин
и промывают декантацией горячей водой.
Для зарядки редуктор сначала наполняют
дистиллированной водой и затем
осторожно насыпают в него цинк, следя
Рис. 54. Колба с клапаном Бунзена.
Рис. 55. Редуктор Джонса.
за тем, чтобы в слой металла не попали пузырьки воздуха. Высота слоя
цинка 20—25 см. Верхний слой металла всегда должен быть покрыт
водой.
Рассчитанную навеску соли железа (III) растворяют в 100 мл 3 н.
раствора серной кислоты на водяной бане в конической колбе.
Полученный раствор пропускают через редуктор, скорость вытекания 3—
4 мл/мин. Полноту восстановления проверяют роданидом калия, который
в присутствии Ре+ + +-ионов образует кроваво-красное окрашивание.
После пропускания раствора редуктор промывают 3—4 раза 2 н.
раствором H2S04, не давая уровню жидкости опускаться до поверхности металла.
Промывные воды присоединяют к основному раствору. Затем редуктор
промывают водой.
Титрование. Содержимое колбы-приемника тотчас медленно титруют
0,1 н. раствором перманганата. Расчет результатов титрования
производят, как указано в гл. I, § 10.
§ 25. Определение нитратов
Определение нитратов основано на восстановлении их в кислой среде
ионами железа (II) и последующем титровании избытка ?е++ -ионов
стандартным раствором перманганата калия:
N03 + 3Fe+++4H+
NO+3Fe+ + + + 2H20
Рассчитанную навеску нитрата натрия или соответствующий объем
его раствора помещают в мерную колбу, добавляют до V3 объема колбы
дистиллированной воды, перемешивают и затем разбавляют водой до метки
258 ГЛ.1Н. МЕТОДЫ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
и снова тщательно перемешивают. Аликвотную часть полученного
раствора переносят пипеткой в коническую колбу.
Если в исследуемом растворе наряду с NaN03 присутствует NaN02, то добавляют
5 мл метилового спирта или 10 мл насыщенного раствора хлорида аммония и при
непрерывном помешивании приливают по каплям из пипетки 10 мл серной кислоты (пл. 1,14 г/см3).
Затем содержимое колбы кипятят 3—5 мин (тяга!). Нитриты с метиловым спиртом в кислой
среде образуют летучий метиловый эфир азотистой кислоты:
NOg + СН3ОН —> CH3ONO | + ОН"
ОН- + Н+ -*- Н20
При кипячении анализируемого раствора с СН3ОН образовавшийся эфир вместе с
избытком метилового спирта улетучивается. Нитриты можно также разрушить кипячением
с хлоридом аммония (см. книга 1, гл. XI, § 11):
NO; + NH4 —> N2 J + 2Н20
Если в растворе нитриты отсутствуют (качественная проба, см. книга 1, гл. XI, § 11),
то обработки метиловым спиртом или хлоридом аммония не делают.
Раствор нитрата, свободный от NaN02, нейтрализуют карбонатом натрия
(пофенолфталеину) и упаривают до объема 10—15 мл (для полного разложения нитритов и удаления
следов спирта).
К раствору приливают 25 мл 0,2 н. раствора сульфата железа (II),
5 мл раствора молибдата натрия (катализатор, ускоряющий
восстановление нитрата) и 5 мл серной кислоты (пл. 1,84 г/см3). Затем в колбу вносят
небольшими порциями 10 г нейтрализующей смеси *, каждый раз
закрывая колбу пробкой с газоотводной трубкой. По окончании реакции
приоткрывают пробку, вливают в колбу 20 мл концентрированной серной
кислоты. Повышение кислотности раствора и присутствие больших
количеств солей натрия сокращает время реакции.
Раствор нагревают в течение 5 мин, разбавляют водой и титруют
избыток сульфата железа (II) стандартным раствором перманганата калия
до появления розового окрашивания. Параллельно определяют в тех же
условиях объем стандартного раствора КМп04, необходимый для
титрования 25 мл 0,2 н. раствора сульфата железа (II).
Расчеты — см. гл. I, § 10.
При расчете содержания NaNOg следует учесть, что объем
стандартного раствора сульфата железа (II), израсходованного на восстановление
нитрата, равен разности:
^КМп04 — ^КМп04
где У^мпО ~~" °бъем 0,1 н. раствора КМп04, израсходованного на титрование 25 мл 0,2 н.
раствора сульфата железа (II), мл\
^КМпО — объем 0,1 н. раствора КМп04, израсходованного на титрование избытка
сульфата железа (II), мл.
§ 26. Определение бихроматов
Определение бихроматов перманганатометрияеским методом основано
на восстановлении их солью Мора (NH4)2S04-FeS04-6H20 и
последующем титровании избытка соли Мора стандартным раствором
перманганата:
Сг20; " + 6Fe+ + + 14Н+ —* 2Сг+ + + + 6Fe+ + + + 7Н20
Навеску соли Мора рассчитывают для приготовления 0,2 н. раствора.
Для этого около 80 г соли растворяют в 1 л воды, содержащей 50—60 мл
* Для приготовления нейтрализующей смеси 300 г NaHCOs нейтрализуют 100 мл
10% -ной H2SO4 и высушивают при 100Q С.
§ 27. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ MnQ2 B ПИРОЛЮЗИТЕ
259
концентрированной серной кислоты или соответствующее количество
2 н. раствора H2S04, и определяют титр и нормальность полученного
раствора титрованием перманганатом (см. § 7). Раствор соли Мора
неустойчив, поэтому установку титра его проводят в один день с определением
бихромата. Концентрация раствора соли Мора должна быть
приблизительно в два раза больше, чем концентрация раствора перманганата.
Навеску бихромата калия рассчитывают для приготовления 250 мл
приблизительно 0,1 н. его раствора. Раствор должен быть разбавленным,
так как в концентрированных растворах труднее уловить изменение
окраски в точке эквивалентности.
Пипеткой переносят в коническую колбу 25 мл исходного раствора
бихромата калия, прибавляют 20—25 мл концентрированной фосфорной
кислоты и затем 25 мл раствора соли Мора. При добавлении раствора
соли Мора наблюдается изменение оранжевой окраски раствора бихромата
в изумрудно-зеленую. Избыток соли Мора оттитровывают раствором
перманганата до изменения окраски в красно-фиолетовую или
сине-фиолетовую в зависимости от содержания хрома, зеленая окраска которого
несколько маскирует розовую окраску перманганата. На обратное
титрование должно быть израсходовано около 15—20 мл раствора КМп04.
Расчеты — см. гл. I, § 10.
§ 27. Определение содержания Мп02 в пиролюзите
Определение двуокиси марганца в пиролюзите представляет собой
пример обратного титрования, основанного на восстановлении двуокиси
марганца стандартным (титрованным) раствором восстановителя,
отмеренное количество которого предварительно было прилито к навеске
исходного пиролюзита, и последующем оттитровывании избытка
восстановителя стандартным раствором перманганата калия.
В качестве восстановителя применяют щавелевую кислоту, оксалаты,
соли железа (II) и др.:
Мп02 + С20; " + 4Н+ —+ Мп+ + + 2С02 + 2Н20
Мп02 + 2Fe+ + + 4Н+ —> Мп+ + + 2Fe+ + + + 2Н20
Методика определения. Сначала тщательно измельчают в агатовой
ступке образец пиролюзита. При анализе природных веществ типа
пиролюзита можно получить близкие результаты нескольких определений
лишь в том случае, когда точно взята средняя проба анализируемого
продукта. Исследуемый пиролюзит может быть дан в виде относительно
крупных кусков, поэтому его следует предварительно тщательно
перемешать и измельчить. Затем взвешивают на аналитических весах несколько
рассчитанных навесок измельченного пиролюзита. Каждая навеска
должна содержать такое количество пиролюзита, чтобы на восстановление
Мп02 расходовалось около 25 мл 0,1 н. раствора восстановителя
( июо'И) г'1 Рассчитанные навески помещают в конические колбы.
Затем прибавляют избыток 0,1 н. раствора оксалата аммония или
щавелевой кислоты (40—50 мл), 20 мл 20%-ной серной кислоты * и смесь
нагревают на водяной бане до 80° С.
Соотношение между содержанием Мп02 и применяемым
восстановителем должно быть таково, чтобы после окончания реакции в конической
колбе остался избыток восстановителя, для окисления которого потре-
* Вместо серной кислоты можно также применять 60%-ную хлорную кислоту. При
этом не образуются сульфаты Ва++ и РЬ++ (если они присутствуют в виде примесей),
которые в случае применения серной кислоты выпадают в осадок и мешают определению.
260 ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯ
буется около 15—20 мл 0,1 н. раствора перманганата калия. По
окончании восстановления Мп02 содержимое колбы разбавляют водой и избыток
восстановителя (С204~ -ионы) оттитровывают стандартным раствором
перманганата калия.
При определении содержания Мп02 иногда применяют растворы
восстановителей,, нормальность которых точно не установлена. В таких
случаях ставят параллельно «холостой» опыт, при помощи которого
устанавливают, сколько миллилитров раствора КМп04 расходуется на
определенный объем раствора Н2С204. В коническую колбу помещают щавелевую
кислоту или оксалат аммония и серную кислому в таких же количествах,
как в проводимом опыте, и оттитровывают перманганатом. Разность
между объемами КМп04, израсходованными на титрование всего взятого
оксалата в «холостом» опыте (Ккмпо4), и его избытка (Укмпо4) составляет
объем раствора КМп04 (Укмпо4), эквивалентный количеству щавелевой
кислоты, вошедшей в реакцию с пиролюзитом.
Содержание Мп02 вычисляют как указано в гл. I, § 10.
ОПРЕДЕЛЕНИЕ ДРУГИХ ВЕЩЕСТВ
§ 28. Определение иоков кальция
Определение катионов, осаждаемых в виде оксалатов, основано на
взаимодействии этих катионов с С2О4 " и последующем титровании оксалат-
ионов либо связанных с определяемым катионом, либо не
прореагировавших с ними.
Покажем это на примере определения Са++-ионов:
Са++ + С204~ —> \ СаС204
ЪС20~ " + 2МпО^ + 16Н+ —+ 10СО2 + 2Мп+ + + 8Н20
1-й способ. Содержание кальция может быть установлено путем
определения оксалат-ионов, связанных с кальцием (прямое титрование).
В этом случае ионы кальция осаждают оксалатом аммония или щавелевой
кислотой, осадок отфильтровывают, промывают, растворяют в серной
кислоте и титруют оксалат-ионы перманганатом. Количество перманганата
эквивалентно количеству кальция в осадке.
2-й способ. Ионы кальция осаждают определенным количеством
стандартного раствора оксалата аммония, взятого в избытке. Избыток
оксалат-ионов определяют титрованием перманганатом (обратное титрование).
По разности объемов взятого и оставшегося одсалата аммония
находят эквивалентное количество кальция.
Определение перманганатометрическим методом солей кальция, не
являющихся ни окислителями, ни восстановителями, может служить
примером проведения определения путем титрования заместителя *.
Осаждение оксалата кальция. Осаждение ионов кальция в виде
оксалата кальция может быть проведено из нейтральных, аммиачных или
кислых растворов. В первых двух случаях наряду с образованием осадка
СаС204 наблюдается значительное соосаждение посторонних веществ.
При осаждении ионов кальция в кислом растворе осадок получается более
* В настоящее время указанный метол определения солей кальция постепенно
вытесняется комплексонометрическим методом (см. гл. IV, § 26).
§ 28. ОПРЕДЕЛЕНИЕ ИОНОВ КАЛЬЦИЯ
261
чистым. При добавлении оксалата аммония к кислому раствору соли
кальция в зависимости от степени кислотности выпадает только часть
оксалата кальция или осадок может совсем не выпасть. Вследствие малого
перенасыщения раствора в отношении оксалата кальция осадок получается
крупнозернистым. При последующем медленном добавлении раствора
аммиака к горячему раствору оксалат кальция выпадает полностью.
При этом также получаются относительно крупные кристаллы СаС204.
Такой осадок хорошо осаждается, отделяется фильтрованием и
промывается. Поэтому обычно осаждение кадьция проводят в солянокислой
среде (рН = 3,5—4,5) и затем раствор нейтрализуют аммиаком.
Определение оксалат-ионов в осадке
оксалата кальция методом отдельных навесок.
Рассчитывают навеску соли кальция (например, СаС03), соответствующую
^СаСО *
25 мл 0,1 н. раствора оксалата аммония —iqqq!iq г.
Рассчитанную навеску исходного вещества помещают в химический
стакан и растворяют в 2 н. растворе хлористоводородной кислоты. Во
избежание потерь в результате разбрызгивания во время растворения
навески стакан накрывают часовым стеклом. После растворения
исходного вещества стенки стакана и часовое стекло обмывают дистиллированной
водой из промывалки. Затем к кислому раствору приливают 50 мл 0,1 н.
раствора щавелевой кислоты или оксалата аммония, смесь нагревают до
70—80° С и медленно прибавляют по каплям разбавленный (1 : 1)
раствор аммиака (при сильном перемешивании) до появления запаха аммиака.
При этом по мере нейтрализации раствора выпадает осадок оксалата
кальция. Для полного выделения и созревания осадка оксалата кальция
необходимо дать осадку отстояться в течение 30 мин. После этого его быстро
отфильтровывают через плотный фильтр и промывают на фильтре холодной
дистиллированной водой для удаления С1~ и избытка оксалата аммония
(проба с раствором AgN03 + HN03). Для промывания осадка следует
пользоваться возможно меньшим количеством воды, которую необходимо
прибавлять малыми порциями после того, как предыдущая порция воды
пройдет через фильтр. Общий объем промывных вод не должен превышать
100 мл.
Промытый осадок смывают горячей водой в стакан, в котором велось
осаждение, приливают 10 мл разбавленной (1 : 4) серной кислоты,
промывают фильтр разбавленной серной кислотой, подогревают раствор до
80° С и титруют перманганатом калия до появления розовой окраски.
Для получения точных результатов анализа необходимо взять 3—4
навески соли и найти среднее значение из полученных сходящихся данных.
Вместо отдельных маленьких навесок можно взять большую навеску,
рассчитанную для получения 250 мл 0,1 н. раствора, которую растворяют
в хлористоводородной кислоте (как было описано выше), переносят
раствор в мерную колбу и доводят объем водой до метки. Отбирают из мерной
колбы в стакан пипеткой аликвотную часть анализируемого раствора и
выполняют весь анализ, как было описано цри проведении анализа
методом отдельных навесок.
Расчеты — см. гл. I, § 10.
Определение избытка оксалат-ионов в
растворе (обратное титрование) методом пипети-
р о в а н и я. Рассчитанную навеску соли кальция для приготовления
250 мл 1 н. раствора растворяют в разбавленной азотной кислоте,
переносят раствор в мерную колбу емкостью 250 мл и доводят объем водой до
метки. Для анализа отбирают в стакан аликвотную часть полученного
раствора.
262
ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Рассчитывают навеску оксалата аммония для получения 250 мл 0,2 н.
раствора. Растворяют навеску (NH4)2C204-H20 в воде, раствор нагревают
и приливают к предварительно нагретой аликвотной части раствора соли
кальция. Вместо сухой навески оксалата аммония можно взять
соответствующее количество его раствора. Затем раствор нейтрализуют аммиаком,
как было указано выше. Когда осадок полностью осядет, его
отфильтровывают и тщательно промывают малыми порциями холодной воды.
Раствор и промывные воды собирают в мерную колбу емкостью 250 мл и
доводят объем водой до метки. Отбирают пипеткой часть раствора,
подкисляют 2 н. раствором H2S04, подогревают до 70—80° С и оттитровывают
перманганатом калия избыток оксалат-ионов.
Расчеты — см. гл. I, § 10.
В. ИОДОМЕТРИЯ
§ 29. Основы иодометрии
Основным веществом, применяемым в качестве окислителя в
иодометрии, является элементарный иод (Е° _ = +0,5345 б\. Иод окисляет
все восстановители (SO3", S2O3", S"", CN", HCHO, SCNT, N2H4,
ASO3 , Сг++ и др.), окислительно-восстановительный потенциал
систем которых меньше Е°и/2г-
Кристаллический иод малорастворим в воде. Поэтому обычно в
качестве стандартного раствора применяют его раствор в KI.
При растворении иода в растворе иодида калия образуются [131"~-
ионы:
12 + 1- ^[Id-
Нормальный окислительно-восстановительный потенциал системы три-
иодид—иодид равен Е° _ = +0,5355 в, т. е. окислительно-восста-
[Ц] /31
новительные потенциалы систем 12/21" и [13]~/31~ можно практически
считать равными.
Принципиальную схему реакции, протекающей при иодометрических
определениях, можно представить следующим образом:
I^+2e ^± 31"
Следовательно, окислительно-восстановительный потенциал этой
системы не зависит от концентрации ионов водорода, если реакции
протекают в кислых растворах.
Однако многие вещества, содержащие в своем составе атомы кислорода
и вступающие в реакции с [13]~, или I", в присутствии ионов водорода
реагируют с образованием нейтральных молекул воды. Например:
H2As04 + 2Г + ЗН+ —> HAs02 + 2Н20 + 12
Поэтому окислительно-восстановительные потенциалы таких систем
сильно зависят от [Н+] раствора.
Применительно к реакции восстановления мышьяковой кислоты
в мышьяковистую иодистоводородной кислотой (или иодидами в кислой
среде) влияние [Н+] на величину потенциала рассматриваемой системы
можно выразить уравнением:
Б = ?° 0,059 [H2AsQ4-][H+]3
H2As04~/HAs02 HjjAsO^HAsOg 2 ё [HAsOa]
§ 30. МЕТОДЫ ИОДОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
263
В кислых растворах вследствие того, что
окислительно-восстановительный потенциал системы H2AsOr ^ HAsCb сильно возрастает по
сравнению с ?fI ^зг, мышьяковая кислота окисляет I" до
элементарного иода, образующего с избытком 1~ трииодид-ионы. По мере
уменьшения [Н+ ] уже в слабокислых и нейтральных и, в особенности в
щелочных растворах
Е _ < Е
As04~"7As02 /3/3/
и поэтому 1з окисляет HAsCb в H3ASO4:
HAs02 + I2 + 2H20 ^ H3As04 + 2Г + 2Н+
Следовательно, приведенные выше уравнения представляют собой
систему динамического равновесия, сильно зависящего от рН раствора.
Основным веществом, применяемым в качестве восстановителя, в иодо-
метрии является тиосульфат натрия, который реагирует с иодом по
уравнению:
2S20~+I2 -^ S4op + 2r
Тиосульфат применяют для титрования избытка иода, добавляемого
в процессе титрования некоторых восстановителей, или иода,
образующегося при взаимодействии иодидов с окислителями, например:
С12 + 21- —> 2С1- + 12
§ 30. Методы иодометрического титрования
Метод прямого титрования. Вещества, легко окисляемые
элементарным иодом (т. е. такие, окислительно-восстановительные потенциалы
систем которых меньше Е[1а]-/зг), титруют непосредственно
стандартными растворами иода в иодиде калия, т. е. растворами К [181. Такие
методы определения называют методами прямого иодометрического
титрования. Этим путем легко определяют сульфиды, сульфиты, тиосульфаты
и другие сильные восстановители. Например, в слабощелочных растворах
иод количественно окисляет цианиды согласно уравнению:
CN- + I2 ^ ICN + I-
Аналогичным образом цианиды реагируют с бромом:
CN- + Br2 —> BrCN + Br-
Метод обратного титрования. Вещества, которые труднее окисляются
элементарным иодом (т. е. такие, окислительно-восстановительный
потенциал систем которых приближается по своему значению к Е^ ]-/3г)>
обрабатывают избытком раствора К [131 и затем, спустя некоторое время,
достаточное для окисления определяемого ве!цества, оттитровывают
избыток К [131 стандартным раствором тиосульфата натрия. Такие методы
определения называют методами обратного иодометрического титрования.
Этим путем определяют менее сильные восстановители, чем сульфиды и
тиосульфаты.
В щелочных растворах сульфиды частично окисляются иодом в
сульфаты:
S" " + 412 + 80Н" —* S01" + 8Г + 4Н20
Для устранения этой побочной реакции исследуемый раствор
сульфида вводят в кислый раствор иода, взятый в избытке, а затем
оттитровывают избыток иода стандартным раствором тиосульфата натрия.
264
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
При прямом титровании кислого раствора сульфида происходит
потеря сероводорода вследствие его улетучивания и результаты определения
получаются неточными. Применение метода обратного титрования дает
возможность получать более точные результаты анализа.
Косвенные методы. Вещества, которые относятся к группе
окислителей (окислительно-восстановительньщ потенциал систем которых больше
E[Ia]-/3r), обрабатывают иодидами калия или натрия, а затем оттитровы-
вают выделившийся при этом в эквивалентном количестве элементарный
иод стандартным раствором тиосульфата натрия. Такие методы
определения называют методами косвенного иодометрического определения. Этим
путем определяются перманганаты, хроматы, бихроматы, иодаты,
элементарные хлор и бром, ионы меди, двуокиси свинца и марганца, ионы FeIir
и другие окислители.
Например, в кислых растворах персульфаты количественно реагируют
в присутствии избытка иодида калия согласно уравнению:
S20g"+2r -* 2SO;-+l2
Выделившийся иод оттитровывают стандартным раствором тиосульфата.
К косвенным методам иодометрического титрования также относятся
определение брома, основанное на взаимодействии элементарного брома
с цианидами (см. выше) с последующим разложением образующегося бром-
циана иодидом:
BrCN + 2I- + H+ —> Br- + HCN + I2
Выделившийся иод титруют стандартным раствором тиосульфата.
На этом же принципе основано определение в нейтральных или
слабощелочных растворах формальдегида, что можно представить в виде
нескольких уравнений
№-Cf +(l + /i)KCN -^ H2C<" -MKCN
^Н ^CN избыток
nKCN + /iBra —> nBrCN + пК++ пВг
BrCN + 2r —> CN- + Br + I2
I2 + 2Na2S203 -^ 4Na+ + 2r-+S406~
Титрование заместителя. Известны методы иодометрического
определения иодидов, бромидов, хлоридов и других восстановителей,
основанные на окислении их соответствующими окислителями до ЮГ, ВгОГ,
ICN, BrCN и т. п., которые затем (после удаления избытка окислителя и
соответствующей обработки) титруют стандартным раствором
тиосульфата натрия, например:
ICN + 2S20~~ + H+ —> r+HCN + S40^'
Такие методы определения называют методами иодометрического
титрования заместителя-, они также относятся к косвенным методам.
Иодометрическое определение кислот. Иодиды с иодатами реагируют
в присутствии кислот согласно следующему уравнению:
БГ + Юз + 6Н+ -*» 312 + ЗН20
Используя эту реакцию, кислоты можно определять иодометрическим
методом. Такой метод определения называют методом иодометрического*
определения кислот.
§ 31. ПРЕИМУЩЕСТВА И НЕДОСТАТКИ ИОДОМЕТРИЧЕСКОГО МЕТОДА 265
§ 31. Преимущества и недостатки иодометрического метода
Вещества, определяемые иодометрическим методом. Наряду с пер-
манганатометрией иодометрический метод титрования также является
одним из наиболее широко применяемых ред-окс-методов. Окислительно-
восстановительный потенциал системы ?j /2l- меньше потенциала
^мпо"/мп++' °Днако» возможности иодометрического метода достаточно
велики. Поскольку иоД реагирует не только как окислитель, способный
окислять многие неорганические и органические вещества, но и проявляет
наряду с этим и другие свойства, используемые в объемном анализе.
Титрование иодом основано на следующих реакциях.
Реакция окисления—восстановления. Элементарный иод или его растворы
в KI или в органических растворителях способны окислять: Cr11, As111,
Sbm, Sn11, сульфиды, сульфиты, тиосульфаты, цианиды, роданиды,
гидразин, гидроксиламин, фосфористый водород, фосфористую кислоту,
полифенолы, аскорбиновую кислоту, меркаптаны, мочевую кислоту и др.
Используя восстановительные свойства иодистоводородной кислоты
или иодидов, возможно определять иодометрическим методом большое
число сильных окислителей, при взаимодействии с которыми иодиды,
окисляясь, образуют элементарный иод, титруемый затем тиосульфатом.
К таким окислителям относятся: нитриты, гипогалогениты, селениты,
перманганаты, бихроматы, иодаты, броматы, перекиси, Си11, FeIH,
Mn111, MnIV, PbIV, Vv и многие другие.
Реакции присоединения. Эти реакции широко применяются в
аналитической химии органических соединений для количественного определения
двойных связей в ненасыщенных соединениях:
>С = С< + 12 —> >С1 — С1<
Одним из таких способов определения ненасыщенности органических
соединений является метод Гюбля, основанный на способности спиртового
раствора иода в присутствии хлорида окисной ртути давать продукты
присоединения по месту двойной связи:
HgCl2 + 2I2 —> Hgl2 + 2JC1
>C = C< + IC1 —> >с —С<
I I
I С1
Стейень ненасыщенности определяется йодным числом, т. е.
количеством галогена (в процентах), перечисленном на иод, которое
присоединяется анализируемым продуктом, растворенным в индиферентном
растворителе (например, в хлороформе). Йодное число является одним из
важнейших критериев ненасыщенности органических соединений.
Реакции замещения. Иод способен замещать атомы водорода в
ароматических и гетероциклических кольцах органических соединений,
например, в ряде фенолов* дифенолов и ароматических диаминов.
Реакции образования периодидов. Многие органические основания и
красители способны реагировать с водными растворами иодида калия,
содержащего иод, с образованием нерастворимых в воде продуктов.
Например,
R«N+ + [I,]- -> R4NI3
Другие реакции. В анализе широко используются реакции иода,
например, с диазосоединениями:
CH2N2CONH2 + I2 -> CH2I2CONH2 + N*
266 ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Преимущества иодометрического метода. Иодометрические
определения отличаются большим разнообразием и этим объясняется широкое
применение метода в практике заводских, научно-исследовательских и
вузовских лабораторий.
1. Иодометрический метод применим для определения многих
соединений, не реагирующих непосредственно с иодом или иодидами. В
качестве примера приведем очень важный метод иодометрического
определения воды по Фишеру (см. § 41) и иодометрическое определение кислот
(см. § 40).
2. Иодометрический метод отличается большой точностью,
превосходящей точность других окислительно-восстановительных методов.
3. Растворы иода окрашены и поэтому иодометрическое титрование
можно осуществлять не прибегая к использованию индикаторов, так как
о конечной точке титрования можно судить по исчезновению или
появлению окраски иода. Желтая окраска комплексных ионов [13]~ наблюдается
при очень малой их концентрации, составляющей 5-10"5 н., при условии
отсутствия в растворе других окрашенных продуктов.
4. Иод хорошо растворим в органических растворителях, поэтому
широко применяют не только водные, но и неводные растворы иода для
всевозможных иодометрических определений.
Недостатки иодометрического метода. Главнейшие ошибки в иодо-
метрии возникают по ряду причин.
Потери иода вследствие его летучести.
Титрование следует проводить в условиях, исключающих потери иода в
результате его улетучивания, на холоду и по возможности быстро. Умеренный
избыток KI уменьшает эти ошибки титрования. В тех случаях, когда
титруемый раствор должен постоять некоторое время для завершения реакции,
титрование следует проводить в колбе, снабженной притертой пробкой.
Ни в коем случае не разрешается проводить титрование в химическом
стакане.
Окисление I-ионов кислородом воздуха:
4Г + 02 + 4Н+ ^± 212 + 2Н20
Окислению I" способствует уменьшение рН раствора и действие
солнечного света. Поэтому иодометрическое титрование следует проводить
при умеренной кислотности раствора. При хранении стандартных
(титрованных) растворов иода надо защищать их от действия прямого солнечного
света и хранить их в темных склянках или в закрытых шкафах.
Следует иметь в виду, что некоторые вещества индуцируют окисление I"
кислородом воздуха. Поэтому иодометрическое титрование необходимо
проводить в отсутствие таких веществ (Cu++, NO2, NO и др.). В тех
случаях, когда приходится вести определение в присутствии указанных
веществ, титрование следует заканчивать по возможности быстро.
Наличие свободных ионов гидроксила. Ионы
гидроксила вызывают реакцию диспропорционирования иода. Поэтому
иодометрическое титрование нельзя проводить в щелочной среде.
Недостаточность времени для завершения
реакции окисления иодидов окислителями или
восстановления иода восстановителями. Там,
где это требуется по условиям титрования (определение As03~", Cu++,
СГ2О7" и др.), необходимо давать достаточное время для достижения
полноты желаемой реакции окисления—восстановления.
Адсорбция элементарного иода.
Поверхностно-активные вещества и некоторые осадки, получающиеся в процессе иодометри-
§ 32. ПРИГОТОВЛЕНИЕ РАСТВОРА ТИОСУЛЬФАТА И УСТАНОВКА ТИТРА 267
ческого титрования, обладают способностью удерживать иод. Поэтому
к концу титрования следует тщательно взбалтывать титруемый раствор,
содержащий осадок, или вводить органический растворитель для
экстракции адсорбированного осадком иода.
Изменение титров стандартных растворов
иода и тиосульфата в процессе их хранения и
использования. Во избежание этих ошибок необходимо
периодически проверять титр тиосульфата по бихромату, а иода по тиосульфату.
Нарушение методики определения в
отношении концентраций применяемых растворов,
порядка приливания растворов и других
условий протекания реакций. Для получения точных и хорошо
воспроизводимых результатов необходимо строго придерживаться
рекомендуемых методик определения.
§ 32. Приготовление стандартного (титрованного) раствора
тиосульфата и установка его титра
Приготовление стандартного раствора тиосульфата натрия. Для
приготовления требуемого объема (обычно 1 л) стандартного раствора (обычно
0,1 н. или 0,05 н.) тиосульфата необходимо 0,1 или 0,05 2-5KeNa2S203-5H20
растворить в воде и довести объем раствора до 1 л. Грамм-эквивалент
тиосульфата не является постоянной величиной и зависит от реакции, в
которую он вступает.
В методе иодометрии используют реакцию тиосульфата с иодом,
протекающую по уравнению:
2S20-" + I2 -> S40e" + 2I" (a)
Из уравнения этой реакции видно, что тиосульфат реагирует с иодом
в отношении два S203~ -иона на два атома I, т. е. 2 молекулы тиосульфата,
окисляясь иодом, отдают 2 электрона.
Из этого делаем вывод, что величина грамм-эквивалента тиосульфата
равна 2 : 2, т. е. его молекулярному весу (MNa2s2o3-5H2o = 248,19).
В реакции
520з " + 4С12 + 5Н2° —* 2S04 " + 8С1~ + 10Н+ @)
молекула тиосульфата отдает 8 электронов. Следовательно,
грамм-эквивалент тиосульфата в этом случае равен MNa2s2oa-5H2o/8.
В реакции с перманганатом, протекающей в щелочной среде
3S203 " + 8МпО~ + Н20 —> 6S04 - + 8Мп02 + 20Н" (в)
3Na2S203.5H20 также Равен LX
^Na2S2Q8.5H20
Для приготовления 1 л 0,1 н. раствора тиосульфата согласно уравне-
248 19
нию (а) необходимо взять —^— = 24,819 г Na2S203-5H20.
Кристаллический Na2S203-5H20 легко теряет кристаллизационную
воду, поэтому его стандартный раствор по точной навеске приготовить
очень трудно. Для приготовления приблизительно 0,1 н. раствора
тиосульфата натрия взвешивают на технических весах около 25 г Na2S203 X
X 5Н20 и растворяют в 1 л свежепрокипяченной и охлажденной
дистиллированной воды. Воду кипятят для удаления С02, чтобы предотвратить
268
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
последующее изменение титра раствора тиосульфата, разлагаемого
угольной кислотой (см. ниже). К полученному раствору тиосульфата
прибавляют около 0,1 г карбоната натрия, связывающего попадающую в
стандартный раствор двуокись углерода:
СОз"+С02 + Н20 ^± 2НСО"
Стандартный раствор Na2S203 для защиты от двуокиси углерода
рекомендуется хранить в темной бутыли с пробкой, снабженной хлоркаль-
циевой трубкой с натронной известью. Перед установкой титра раствор
должен постоять не менее 1—2 дней. Титр раствора тиосульфата при
стоянии может изменяться под влиянием двуокиси углерода (г) и кислорода
воздуха (д):
Na2S203+C02 + H20 —> NaHCOa + NaHS08 + S (г)
2Na2S203 + 02 —> 2Na2S04 + 2S (д)
Под влиянием С02 нормальность раствора тиосульфата увеличивается,
ибо образующиеся ионы гидросульфита (бисульфита) реагируют с иодом
согласно следующему уравнению:
HSO3 + I2-f H20 _> HSO4+2HI
Под влиянием кислорода уменьшается нормальность раствора Na2S203,
и результаты титрования получаются неправильными.
При правильном приготовлении и, хорошем хранении титр раствора
тиосульфата не изменяется в течение 2—3 месяцев.
Крахмал как индикатор. Титрование иодом можно проводить без
какого-либо индикатора в растворах, концентрация которых выше 0,2 н.
Конечную точку титрования определяют по появлению или исчезновению
желтой окраски. Но обычно при титровании иодом применяют
специфический индикатор — раствор крахмала. Амилоза, входящая в состав
крахмала, образует с иодом адсорбционное соединение синего цвета.
На каждые 100 мл титруемого раствора прибавляют по 5 мл раствора
крахмала. При титровании иода тиосульфатом крахмал надо приливать
перед концом титрования, когда желтая окраска иода начинает ослабевать,
иначе большое количество иода будет адсорбировано крахмалом и реакция
иода с тиосульфатом замедлится. Чувствительность иодо-крахмалькой
реакции с повышением температуры понижается.
Установка титра стандартного раствора тиосульфата. На практике
в качестве установочного вещества для установки титра раствора
тиосульфата обычно применяют бихромат калия. Бихромат калия легко получить
в химически чистом виде перекристаллизацией его из водного раствора;
он не содержит кристаллизационной воды, растворы его очень устойчивы.
Бихромат калия — сильный окислитель (Е^ 0—/2Сг+ + + = 1,36 е\.
Прямое титрование тиосульфата бихроматом не применяют вследствие
того, что реакция протекает неоднозначно и не может быть выражена одним
определенным уравнением. При установке титра раствора тиосульфата
натрия по бихромату калия применяют метод замещения. К
определенному количеству бихромата прибавляют избыток иодида калия и кислоты.
При этом протекает реакция:
Сг20" + 6Г + 14Н+ ^ 312 + 2Сг+ + + + 7Н20
Указанная реакция в кислой среде проходит количественно.
Выделяющийся иод оттитровывают раствором тиосульфата, титр которого
§ 32. ПРИГОТОВЛЕНИЕ РАСТВОРА ТИОСУЛЬФАТА И УСТАНОВКА ТИТРА 269
устанавливают. Так как титр стандартного раствора тиосульфата
изменяется со временем, то его периодически следует проверять по бихромату
калия. Помимо бихромата для установки титра тиосульфата в качестве
установочных веществ применяют также иод, иодат калия КЮ3, бииодат
калия КН (Ю3)2, бромат калия КВг03, гексацианоферрат (III) калия,
персульфат калия K2S208 и стандартный раствор перманганата,
установленный по какому-нибудь восстановителю. (Напишите уравнения реакции
взаимодействия указанных веществ с иодидом калия.)
Установка титра раствора тиосульфата по бихромату методом
пипетирования. Удобнее устанавливать раствор тиосульфата натрия по
бихромату калия методом пипетирования. Раствор бихромата калия готовят
примерно 0,1 н. или 0,05 н. В более разбавленных растворах реакция
протекает очень медленно. Навеску рассчитывают для приготовления
250 мл 0,1 н. раствора бихромата калия. Реакция протекает в присутствии
иодида калия, который необходим и для окисления — восстановления и
для растворения выделяющегося иода. Для полноты протекания реакции
его берут обычно около 2—3 г.
Для увеличения скорости реакции необходим избыток кислоты. Опыт-^
ным путем найдено, что желательно брать около 20 мл 4 н. раствора
кислоты на 100 мл общего раствора. При меньшем количестве, например при
20 мл 2 н. серной кислоты, реакция идет медленно и раствор должен
постоять 5—10 мин для завершения реакции. Слишком большое количество
кислоты вредно, так как увеличивается возможность окисления иодида
калия кислородом воздуха.
В коническую колбу переносят пипеткой 25 мл приготовленного
раствора бихромата калия, прибавляют 2 г или соответствующий объем
раствора иодида калия, затем 10 мл 2 н. хлористоводородной или серной
кислоты и хорошо перемешивают. Колбу закрывают часовым стеклом, дают
постоять около 5 мин, разбавляют 200 мл воды и титруют выделившийся
иод тиосульфатом натрия. Когда бурая окраска выделившегося иода
перейдет в лимонно-желтую, прибавляют 1—2 мл раствора крахмала. Светло-
синяя окраска раствора в точке эквивалентности переходит в
светло-зеленую. Иногда наблюдается посинение раствора после окончания
титрования. Посинение через несколько минут может происходить вследствие
окисления иодида калия кислородом воздуха и на результатах титрования
не отражается. Посинение, происходящее тотчас же после окончания
титрования, указывает, что выделение иода бихроматом калия не прошло до
конца и разбавление было проведено слишком рано. Такое титрование
неправильно, опыт должен быть повторен.
При расчетах титра и нормальности тиосульфата натрия надо помнить,
что взятое число эквивалентов бихромата калия выделяет из иодида ка-
лия равное количество эквивалентов элементарного иода, а выделяющийся
иод для своего восстановления требует точно такого же количества
эквивалентов тиосульфата натрия:
^К2Сгй07^К2Сг20? _ NhVIt ^^Na^gOs.SHaO^NagSaOa.SHgO
1000 Ю00 1000
Установка титра раствора тиосульфата натрия по бихромату калия
методом отдельных навесок. Более точной является установка титра по
методу отдельных навесок. Величина навески должна быть такова, чтобы
на титрование выделенного иода расходовалось около 25 мл 0,1 н.
раствора тиосульфата. Необходимое количество бихромата (Эк^сг*о* = 49,032)
вычисляют по формуле:
0,1-25-49,03 п 10
aK*Cra07 — ]Ш\ = 0,12 г
270
ГЛ.III. МЕТОДЕ^ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
На аналитических весах взвешивают около 0,12 г чистого бихромата
калия и растворяют его в 50 мл воды. К полученному раствору прибавляют
2 г иодида калия и 15—20 мл 2 к. серной кислоты, закрывают колбу,
тщательно перемешивают, дают постоять 5 мин и затем титруют
выделившийся иод тиосульфатом, как указано выше.
§ 33. Приготовление стандартного (титрованного) раствора иода
to установка его титра
Приготовление стандартного раствора иода. Для приготовления
стандартных растворов применяют свежевозогнанный иод. Растворимость
иода в воде очень мала: насыщенный раствор содержит около 0,03%
иода. Поэтому обычно готовят растворы иода в водном растворе иодида
калия.
Для приготовления 1 л приблизительно 0,1 н. раствора иода в стакан
емкостью 250 мл помещают 40 г иодида калия и 25 мл воды, прибавляют
12,7 г возогнанного иода (навеску иода берут в бюксе и взвешивают на
технических весах), перемешивают до полного растворения иода, раствор
переливают в склянку и только тогда разбавляют водой до 1 л.
Концентрация раствора иода может изменяться вследствие летучести иода, поэтому
раствор необходимо хранить в сосудах с плотно пришлифованными
стеклянными пробками. При концентрации иодида калия не менее 4% раствор
иода устойчив.
Установка титра стандартного раствора иода по тиосульфату. Реакция
окисления — восстановления между иодом и тиосульфатом при
соблюдении определенных условий протекает строго количественно.
Окислительный потенциал системы 12 + 2е ^± 21" равен Е° = +0,5345 в,
а окислительный потенциал системы 2S20^ —2e^S406~ равен Е° =
= +0,15 в.
При титровании 0,1 н. раствором иода рН должен быть не больше
7,6; при рН = 8 иодометрический метод неприемлем, так как протекает
реакция диспропорционирования иода.
Нагревание недопустимо, так как иод очень летуч.
В коническую колбу переносят пипеткой 20—25 мл стандартного
раствора тиосульфата натрия, добавляют 1—2 мл раствора крахмала и
титруют иодом до появления синей окраски.
Иногда помещают тиосульфат натрия в бюретку, а в коническую колбу
пипеткой отбирают раствор иода. В этом случае крахмал прибавляют,
когда раствор приобретает бледно-желтую окраску, и титруют до
исчезновения синей окраски, появляющейся в результате прибавления
крахмала.
Расчеты — см. гл. I, § 10.
Для установки титра стандартного раствора иода, кроме тиосульфата
натрия, применяют тиосульфат бария (BaS203-H20), окись мышьяка (III)
(As203), антимонилтартрат калия (KSbOC^Oe-VjsHjjO), сернокислый
гидразин (N2H4-H2S04) и др. (Напишите уравнения реакций
взаимодействия указанных веществ с иодом).
МЕТОДЫ ПРЯМОГО ТИТРОВАНИЯ
§ 34. Определение мышьяка (III)
Определение соединений мышьяка (III) иодометрическим методом
основано на прямом титровании арсенитов стандартным раствором иода:
AsOg + 12 + 2Н20 ^ HASO4 + 21- + ЗН+
§ 35. ОПРЕДЕЛЕНИЕ СУЛЬФИТА НАТРИЯ
271
Реакция обратима. На состояние равновесия оказывает большое
влияние концентрация ионов водорода (см. книга 1). При титровании
мышьяковистой кислоты иодом величина рН раствора должна быть больше 5,
но не более 7—8. Если рН меньше 5, то протекает обратная реакция;
если рН > 8, то наступает реакция диспропорционирования иода.
Выделяющуюся при реакции кислоту нейтрализуют бикарбонатом
натрия (рН = 8). При нейтрализации NaHC03 устанавливается рН =
= 6—7 вследствие буферного действия NaHC03 + Н2С03.
Навеску определяемого вещества (например, As203) рассчитывают
для приготовления 250 мл 0,1 н. раствора. Рассчитанную навеску
переносят в мерную колбу и растворяют в концентрированном растворе едкой
щелочи, взятом в полуторном количестве против рассчитанного. Большой
избыток щелочи вреден. По окончании растворения As203 раствор
разбавляют до четверти емкости колбы и нейтрализуют 2 н. раствором
серной кислоты (индикатор—фенолфталеин). Затем прибавляют к
нейтральному раствору двойное количество по сравнению с рассчитанным
количеством бикарбоната натрия, т. е. 16—20 г/л. Нагревать раствор нельзя.
Если после растворения бикарбоната раствор окажется окрашенным,
то его обесцвечивают, прибавляя по каплям разбавленную серную
кислоту.
Если для анализа дан готовый раствор натриевой соли
мышьяковистой кислоты, то сначала приливают 2 н. раствор серной кислоты для
нейтрализации свободной щелочи, всегда сопутствующей натриевым солям
мышьяковистой кислоты, а затем бикарбонат натрия до рН = 6—7.
Приготовленный раствор разбавляют водой и доводят объем раствора
до метки, титруют стандартным 0,1 н. раствором иода. Для титрования
отбирают пипеткой, снабженной предохранительным шариком, 25 мл
анализируемого раствора, прибавляют индикатор — крахмал и титруют
иодом.
Следует иметь в виду, что под конец титрования, когда концентрация
арсенита понизится, реакция окисления — восстановления между иодом
и арсенитом замедляется. Поэтому синее окрашивание крахмала
появляется преждевременно. Но если дать раствору постоять некоторое время,
то синяя окраска исчезает. Когда появляется неисчезающее синее
окрашивание, титрование считают законченным и приступают к расчету
результатов анализа.
Порядок титрования может быть изменен: в бюретку можно поместить
раствор Na2HAs03 (рН ^ 6—7), а раствор иода отбирать пипеткой. Этот
порядок удобен при отсутствии пипеток с предохранительным шариком
и бюреток со стеклянным краном.
Расчеты — см. гл. I, § 10.
МЕТОДЫ ОБРАТНОГО ТИТРОВАНИЯ
§ 35. Определение сульфита натрия
Определение сульфитов основано на окислении их до сульфатов
титрованным раствором иода. Избыток иода оттитровывают стандартным
раствором тиосульфата:
S03~ + I2 + H20 ^± SO" + 2I~+2H+
I2+2S203~ ^± S40~"" + 2r
272
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
По количеству затраченного иода вычисляют количество сульфита.
Окисление сульфитов иодом может протекать в двух направлениях (см.
книга 1, гл. XII, § 3):
2S03~+I2 -^ S20g~ + 2r (a)
SO~~+I2 + H20 —+ SO" + 2I" + 2H+ (б)
Чтобы исключить реакцию образования дитионатов, сульфит вводят
в раствор иода, подкисленный хлористоводородной кислотой, и затем
оттитровывают избыток иода тиосульфатом натрия.
Рассчитанную навеску или соответствующий объем сульфита натрия
помещают в мерную колбу и разбавляют водой до метки. Затем при помощи
пипетки переносят в коническую колбу 50 мл 0,1 н. раствора иода,
подкисляют 2 н. раствором НС1 и прибавляют 25 мл сульфита. Непрореаги-
ровавший иод оттитровывают тиосульфатом. Индикатор — крахмал.
Расчет — см. гл. I, § 10.
Иногда для упрощения расчетов проводят «холостой опыт»: в
коническую колбу берут 50 мл 0,1 н. раствора иода и титруют тиосульфатом.
Если принять: V — объем раствора тиосульфата натрия,
израсходованного на «холостой опыт», V — объем раствора тиосульфата натрия,
израсходованного на титрование избытка иода, то V — V =
= ^Na2s2o3-5H2o —объем 0,1 н. раствора тиосульфата, эквивалентный
объему раствора иода, пошедшего на окисление сульфита.
Интересно отметить, что в то время как иод окисляет сульфит до сульфата, а тиосульфат
до тетратионата, йерманганат окисляет в щелочной среде и сульфит (а), и тиосульфат (б)
до сульфата:
2MnO; + 3S03~ + HOH —* 2Mn02 + 3S04" + 20H" (a)
8MnO; + 3S2C>3 " + НОН —> 8Mn02+6SO" + 20H~ (б)
На этом основан метод дифференцированного определения сульфита и тиосульфата.
Эквивалент сульфита в реакции с перманганатом и иодом равен его молекулярному
весу, деленному на 2:
"^NaaSOe ~ 2
Эквивалент тиосульфата в реакции с перманганатом (Э') и иодом (Э") отличаются друг
от друга и соответственно равны молекулярному весу, деленному на 8 и на 1:
ъ' _ MNa2S2Q, ^ _ MNa2S2Q8
^Na2S20, — g И "^NaaSaOa — J
Следовательно, на 1 моль сульфита требуется 2 г-экв перманганата или иода, а на
1 моль тиосульфата 8 г-экв перманганата и только 1 г-экв иода. Поэтому, если оттитровать
анализируемую смесь сульфита и тиосульфата сначала перманганатом, а затем такую же
аликвотную часть их смеси оттитровать раствором иода, то можно вычислить содержание
отдельных компонентов в анализируемой смеси.
Для этого из объема раствора перманганата, пошедшего на титрование смеси, вычитают
объем раствора иода, требующегося на окисление сульфита и тиосульфата. Разница в
объемах обоих окислителей, выраженная в грамм-эквивалентах, составляет семь восьмых
содержания тиосульфата.
§ 36. Определение содержания формальдегида в формалине
Определение содержания формальдегида в формалине иодометриче-
ским методом основано на окислении формальдегида иодом и последующем
оттитровывании тиосульфатом избытка иода.
Формальдегид количественно окисляется иодом в щелочном растворе
с образованием солей муравьиной кислоты.
§ 37. ОПРЕДЕЛЕНИЕ ИОНОВ МЕДИ (II)
273
Уравнение реакции:
НСНО + 12 + 3NaOH -+ HCOONa + 2NaI + 2Н20
По окончании реакции раствор подкисляют (рН = 6—6,5) и непрореаги-
ровавший иод оттитровывают тиосульфатом.
В мерной колбе 5 мл 40%-ного раствора формальдегида (формалина)
разбавляют до метки. К 5 мл полученного раствора прибавляют 5 мл
2 н. раствора NaOH и 50 мл 0,1 н. раствора иода. Полученную смесь
оставляют стоять 10—15 мин, чтобы реакция окисления формальдегида прошла
полностью. Затем прибавляют хлористоводородную кислоту до кислой
реакции. Непрореагировавший иод оттитровывают 0,1 н. раствором
тиосульфата.
Проводят «холостой» опыт, оттитровывая 50 мл раствора иода
тиосульфатом.
Если принять: V — объем раствора тиосульфата, израсходованный на
титрование 50 мл, раствора иода; V — объем раствора тиосульфата,
израсходованный на титрование' избытка иода, то V — V = VNa2s2os-su2o—
объем точно 0,1 н. раствора тиосульфата или, что то же, объем 0,1 н.
раствора иода, пошедшего на окисление формальдегида.
1 мл 0,1 н. раствора иода соответствует 0,0015008 г формальдегида.
Расчеты — см. гл. I, § 10.
МЕТОДЫ КОСВЕННОГО ОПРЕДЕЛЕНИЯ
§ 37. Определение ионов меди (II)
Ионы меди (II) не могут быть определены прямым или обратным иодо-
метрическими методами титрования, так как они находятся в максимально
окисленной форме. Поэтому иод, являющийся более сильным окислителем
(??2/2Г = +0,5345 «) по сравнению с ионами меди (II) (Е°Си++/Си+ =
= +0,167 в; ?cu++/cu = +0»3448 в], должен окислять Си++.
Определение ионов меди (II) представляет собой пример применения
косвенного иодометрического титрования и основано на взаимодействии
Cu+ + с I", сопровождающемся (вопреки ожиданиям) окислением ионов иода
в элементарное состояние, и последующем титровании образовавшегося
в эквивалентном количестве [13]~ стандартным раствором тиосульфата.
Реакцию можно представить следующим уравнением:
2е
2Г + 2Си++ + 2Г ^± |Ia+|2CuI; 12 + 4Г ^± 2 [18Г
При этом также получается малорастворимый осадок Cul. Реакция
обратима.
Чем же можно объяснить течение этой реакции слева направо,
которая, казалось бы, должна протекать справа налево, так как иод более
сильный окислитель, чем ионы меди? Это явление объясняется
образованием малорастворимого осадка Cul (Pcui = Ю"6 моль/л), благодаря
чему [Си+ ] = Pcui = Ю"6 г-ион/л в момент достижения состояния
равновесия незначительна по сравнению с [Си++ ], которая вследствие
хорошей растворимости некоторых солей меди может достигать больших
значений.
274
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Окислительно-восстановительный потенциал системы, как известно,
сильно зависит от концентрации окислителя и восстановителя:
? = ?cn-/c/ + 0'0591gW
Так как [Си++] значительно превышает [Си+], наблюдается резкое
увеличение потенциала системы Си++ + е^±Си+ до величин,
превышающих потенциал системы 12 + 2е ^± 21~~ Е°Си++/Си1 = +0,88 в.
Вследствие этого реакция взаимодействия ионов меди (II) с Г протекает
практически до конца слева направо.
Следовательно, теоретически для максимального сдвига
рассматриваемой реакции слева направо необходимо создавать такие условия реакции,
которые:
1) способствуют увеличению окислительно-восстановительного
потенциала системы Си++ + б^±Си+;
2) приводят к уменьшению окислительно-восстановительного
потенциала системы 12 + 2е^± 21"";
3) предупреждают понижение [Си++] вследствие, например, комплексо-
образования;
4) создают максимально благоприятные условия в отношении
кислотности раствора (рН = 4—5).
Повышение окислительно-восстановите л ь -
•ного потенциала системы Си+ + + е —> Си+ достигают
осаждением в ходе анализа ионов меди (I) в виде малорастворимого иодида
меди (I).
Понижение окислительного потенциала
системы I2 + 2e^t 21"может быть достигнуто различными методами:
уменьшением концентрации выделяющегося иода прибавлением
тиосульфата; отгонкой 12 и экстрагированием его органическими растворителями
или путем увеличения концентрации иодид-ионов, т. е. прибавлением
большого количества иодида калия. Обычно прибавляют около 5 г KI
на 100 мл раствора или соответствующий объем его концентрированного
раствора.
Предупреждение комплексообразования. При
проведении анализа необходимо избегать присутствия веществ,
образующих комплексные соединения с медью (II) и тем понижающих ее
концентрацию. Например, хлористоводородная кислота в больших
количествах вредна, так как образует комплекс [СиС14]"".
Соблюдение определенного значения
рНраствор а . Если значение рН раствора слишком высоко, реакция идет
очень медленно и не доходит до конца. Если значение рН раствора низко,
то иодид калия окисляется кислородом воздуха, причем медь (I)
каталитически ускоряет эту реакцию. Обычно для сохранения постоянного
значения рН раствора применяют буферные ймеси с рН = 4—5.
Для усовершенствования иодометрического определения ионов меди
было предложено заменить иодид калия смесью иодида калия и роданида
аммония. При добавлении этой смеси к раствору, содержащему ионы
меди (II), дротекают реакции:
2Си++ + 4Г ^± 2CuI + I2
2CuI + 2SCN~ —* Cu2 (SCN)2 + 2Г
B результате ионы меди (I) переходят в очень малорастворимый
роданид меди (I). Титрование проводят в сернокислой или солянокислой среде.
§ 38. ОПРЕДЕЛЕНИЕ ДВУОКИСИ СВИНЦА В СУРИКЕ 275
Иодометрический метод считается лучшим для определения меди.
Он достаточно точен. Присутствие посторонних веществ не оказывает
существенного влияния на точность результатов анализа. При анализах
сложных смесей, например медных руд, это обстоятельство имеет большое
значение. Мешают вещества, окисляющие иодид калия, например мышьяк
(V), окисляющиеся иодом, например мышьяк (III), сурьма (III), и
осаждающие иодид-ионы, например ионы висмута и серебра.
Определение меди (II) в солях. Рассчитанную навеску или
соответствующий объем раствора соли меди (II) помещают в мерную колбу,
растворяют в воде и доводят объем раствора до метки. Переносят пипеткой
25 мл полученного раствора в коническую колбу, добавляют 5 мл 2 н.
раствора серной кислоты. Прибавляют около 3—5 г иодида калия или
соответствующее количество раствора иодида.
Выделившийся иод оттитровывают тиосульфатом, не обращая
внимания на образующийся осадок иодида меди (I). Под конец титрования
прибавляют 1—2 мл раствора крахмала и дотитровывают до
обесцвечивания раствора.
Определение меди в рудах. При определении меди в рудах
(содержащих 10—30% меди) берут навеску около 1 г и растворяют ее в 20 мл
концентрированной азотной кислоты. Раствор в стакане выпаривают до
объема 5 мл, прибавляют 25 мл воды и нагревают до растворения солей и
удаления окислов азота. Остаток отфильтровывают и промывают.
Фильтрат выпаривают до объема 25 мл. Для нейтрализации кислоты к раствору
прибавляют раствор аммиака до появления слабого запаха; происходит
осаждение примеси железа в виде гидроокиси железа. Затем прибавляют
2 г бифторида аммония для растворения гидроокиси железа и переведения
его в комплекс, взбалтывают, добавляют 3 г иодида калия. Выделившийся
иод титруют тиосульфатом натрия.
Расчеты — см. гл. I, § 10.
§ 38. Определение двуокиси свинца в сурике
Определение содержания РЬ02 в сурике основано на способности РЬОа
в уксуснокислом растворе выделять в эквивалентном количестве иод
из иодида калия:
РЬ02 + 21- + 4СН3СООН —>- 12 + РЬ (СН3СОО)2 + 2СН3СОО" + 2Н20
I. + I- ^± [13Г
Выделившийся иод оттитровывают тиосульфатом.
Реакция хорошо протекает в присутствии большого избытка ацетата
натрия, при котором не происходит выпадения малорастворимого иодида
свинца.
Сурик тщательно растирают в агатовой ступке. В мерную колбу
помещают около 5 г иодида калия (или соответствующее количество его
раствора), 15 г ацетата натрия, 1—2 мл воды и 5 мл разбавленной (1 : 1)
уксусной кислоты. В полученную смесь вносят рассчитанную навеску
хорошо измельченной двуокиси свинца и смесь взбалтывают. Если
выпадает оранжевый осадок иодида свинца, то прибавляют еще ацетат натрия.
Затем пипеткой из мерной колбы в конические колбы берут аликвотные
части раствора и оттитровывают выделившийся иод тиосульфатом.
Лучшие результаты получаются при выполнении определения методом
отдельных навесок.
Расчеты — см. гл. I, § 10.
276
ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
МЕТОД ТИТРОВАНИЯ ЗАМЕСТИТЕЛЕЙ
§ 39. Определение содержания двуокиси марганца в пиролюзите
Определение двуокиси марганца в пиролюзите основано на окислении
НС1 до элементарного хлора и последующем его определении иодометри-
ческим методом:
Мп02 + 2С1- + 4Н+
Мп++ + С12 + 2Н20
Выделяющийся свободный хлор пропускают через газоотводную трубку
в приемник, содержащий раствор иодида калия:
СЦ + 2Г
12 + Г
2С1- +12
[lei-
Элементарный иод,
выделяющийся в эквивалентном
количестве, титруют тиосульфатом.
Прибор для разложения пиролюзита
показан на рис. 56. Аналогично
проводят анализ двуокиси свинца,
сурика, бертолетовой соли,
селеновой и теллуровой кислот
и т. д.
Навеску берут с таким
расчетом, чтобы количество
выделившегося иода соответствовало 250 мл 0,1 н. раствора. Согласно
приведенным выше уравнениям реакций:
Рис. 56. Прибор для определения двуокиси
марганца в пиролюзите.
М
откуда
аМп02 —
Мп02 "
Ммпо.у1,т1,
-М
h
12 *2
87-250-127
127-2.10-1000
1,087 г
если считать пиролюзит содержащим 100% Мп02. Обычно берут примерно
1,5 г.
В колбу, снабженную отводной трубкой (дистилляционную колбу),
помещают около 1,5 г пиролюзита, смачивают его небольшим количеством
воды и 20—25 мл концентрированной хлористоводородной кислоты. Затем
быстро закрывают колбу и опускают отводную трубку в приемник, куда
предварительно наливают раствор иодида калия. Конец трубки должен
быть опущен в раствор иодида калия. Колбу с анализируемым веществом
осторожно нагревают до кипения. В процессе нагревания колбы
приемник периодически поворачивают вокруг своей оси, чтобы выпустить
скапливающийся в ней воздух. В приемник перегоняют около 4/б объема
раствора. Нагревание продолжают до растворения темного осадка.
По окончании опыта содержимое приемника количественно переносят
в мерную колбу емкостью 250 мл и разбавляют раствор до метки. Для
титрования отбирают пипеткой 25 мл раствора. Выделившийся иод
титруют тиосульфатом. Индикатор — крахмал.
Содержание Мп02 вычисляют по формулам (см. гл. I, §' 10).
§ 41. ОПРЕДЕЛЕНИЕ ВОДЫ ПО ФИШЕРУ 277
МЕТОД ОПРЕДЕЛЕНИЯ КИСЛОТ
§ 40. Определение хлористоводородной кислоты
Иодометрическое определение кислот основано на использовании
реакции, протекающей в кислой среде между иодидом и иодатом с
выделением иода. В нейтральной среде выделение иода прекращается. По
количеству выделенного иода можно вычислить количество содержащейся
в растворе кислоты (см. § 30).
Взаимодействие между иодид- и иодат-ионами протекает быстро и
полно.
Иодометрическое определение кислот имеет большое значение, так
как дает возможность перейти от метода нейтрализации к методу иодо-
метрии. Слабые минеральные кислоты (например, борная кислота) и
органические кислоты иодометрически не определяются.
Для определения хлористоводородной кислоты готовят 250 мл
приблизительно 0,1 н. ее раствора.
Расчет количества иодида и иодата калия. Согласно реакции
КЮ3 + 5KI + 6НС1 —>- 6КС1 + 31а + ЗН20
312 + ЗГ ^± 3[13Г
6-36,46 г НС1 соответствует 5-166 г KI и 214 г КЮ3; 25 мл 0,1 н. НС1
36,46-25 ир1
содержит 1(М000 г НС1.
6-36,46 г НС1—5-166 a KI
36,46-25
10-1000
г НС1 — х гК1
__ 5-166-25
*ki- 6-10-юоо
Реакция обратима. Для полного протекания ее необходим избыток
реактива; нужно взять полуторное количество иодида калия. Но так
как KI требуется не только для реакции, но и для растворения
выделившегося иода, то рассчитанное количество иодида должно быть еще удвоено:
5-166-25-1,5-2 , п. ___
gKi= 6-10-1000 =Ь04аК1
Аналогичный расчет проводят для иодата калия и берут полуторное
его количество от теоретического:
214-25-1,5 nio. „?Г|
*КЮ,= 6-10-1000 =0'134гКЮз
Вместо взятия отдельных навесок удобнее пользоваться
соответствующими растворами иодида и иодата калия.
В коническую колбу наливают рассчитанные количества растворов
иодида и иодата калия. В полученную смесь пипеткой приливают 25 мл
определяемой кислоты. Содержимое колбы перемешивают в течение 1—
2 мин и титруют выделившийся иод тиосульфатом. Когда раствор примет
слабо-желтую окраску, приливают 2—3 мл раствора крахмала. Титруют
до обесцвечивания раствора.
Расчеты — см. гл. I, § 10.
§ 41. Определение воды по Фишеру
Иодометрическое определение кислот (уравнение реакции приведено
в § 40) представляют собой особый случай косвенного метода титрования,
отличающийся тем, что определяемое вещество непосредственно не всту-
278
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
пает в реакцию окисления — восстановления, т. е. не окисляется и не
восстанавливается.
Другим примером такого рода является иодометрическое определение
воды по Фишеру, широко применяемое в промышленных и
научно-исследовательских лабораториях.
Принцип метода. Иодометрическое определение воды по методу
Фишера основано на использовании реакции окисления—восстановления,
протекающей между двуокисью серы и иодом в присутствии воды в среде
метилового спирта. Для этого анализируемый объект (например,
органический растворитель, содержащий примеси воды) растворяют в безводном
метиловом спирте. Метаноловый раствор подвергают прямому титрованию
стандартным раствором реагента, представляющим собой смесь иода,
двуокиси серы и пиридина в безводном метиловом спирте. При этом
протекает следующая реакция:
Н
I
h + S02 + Н20 + СН3ОН + 3C5H6N —> C5H5NOS02OCH3 + 2C5H5NH + 2Г
Следовательно, 1 моль иода эквивалентен 1 моль Н20; вода
непосредственно не реагирует с иодом или с иодидами, а наряду с метанолом,
служит источником ионов кислорода, необходимых для образования группы
i
—OS02OCH3. Окисление серы происходит за счет элементарного иода,
который восстанавливается до I" -ионов.
Метод Фишера применим также для определения воды, поглощаемой
или выделяемой при многочисленных реакциях, например, для
определения воды, выделяемой при конденсации кремнийорганических
соединений, содержащих гидроксильные группы:
^SiOH+HOSi-^ —> "T^ — О —Si^ + H20
Конечную точку титрования фиксируют по появлению в титруемом
растворе избытка иода, обнаруживаемого по изменению желтой окраски
титруемого раствора в красновато-коричневую. Титрование рекомендуется
проводить в присутствии «свидетеля». Наряду с визуальным методом
определения конечной точки титрования применяют также физико-химические
(инструментальные) методы.
Реактив Фишера весьма чувствителен к действию воды, поэтому
хранение реактива и титрование им исследуемых растворов проводят в
условиях, исключающих влияние влаги воздуха. Приготовление стандартного
раствора Фишера и установка его титра, а также методика определения
воды в метаноле описаны выше (см. гл. II, § 46).
Г. ПОНЯТИЕ О ДРУГИХ МЕТОДАХ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
§ 42. Хроматометрия
Хроматометрией называют метод объемного анализа, основанный на
использовании в качестве стандартного водного раствора бихромата калия
или (реже) хромата калия. Иногда вместо указанных реагентов применяют
трехокись хрома (хромовый ангидрид), который растворяют в ледяной
уксусной или концентрированной серной кислоте.
Обычно титрование проводят в сернокислой, солянокислой или
фосфорнокислой средах.
§ 42. ХРОМАТОМЕТРИЯ 279
Применение хроматометрического метода основано на реакциях:
окисления—восстановления (см. ниже) и реакциях двойного обмена,
сопровождающихся образованием нерастворимых в воде соединений:
солей бария, серебра, свинца и т. д., а также производных оснований,
подобных алкалоидам.
Реакции окисления—восстановления. Бихромат калия в кислой среде
является довольно сильным окислителем:
Cr2°7" + 6e + 14H+ ^ 2Сг+ + + +7Н20
Нормальный окислительно-восстановительный потенциал этой
системы ?° -- + + + равен+1,36 в, т. е. соединения CrVI обладают боль-
шей окислительной способностью, чем элементарный иод (Е^ /21- =
= +0,5345 в), но меньшей, чем перманганат (Е° -. ++ = +1,52 в\.
Из приведенного выше уравнения реакции следует, что эквивалент
бихромата калия равен его молекулярному весу, деленному на 6:
^К2Сг20, — б
Бихромат калия способен окислять многие неорганические и
органические вещества: Fe11, Мпш, MnIV, VIV, W111, Mov, TeIV, Tim,
сульфит, дитионат, гексацианоферрат (II), арсенит, иодид, спирты,
гидрохинон, глицерин, аскорбиновую кислоту, тиомочевину и др.
Ниже приводятся некоторые типичные реакции окисления —
восстановления, протекающие с участием бихромата и используемые в объемном
анализе.
Окисление гексацианоферрата (II):
Сг2°7 " + 6 [Fe (CN)6] = = + 14Н+ -— 2Сг+ + + + б [Fe (CN)J + 7Н20
Окисление ванадия (IV):
няро4
Сг20; + 6VO+ + + 5Н20 —> 6VO3 + 2Сг+ + + + 10Н+
Окисление метилового спирта:
Сг2°7 " + СН3ОН + 8Н+ -^ 2Сг+ + + + С02 + 6Н20
Окисление гидрохинона:
Сг2°7 " + сбн4 (°Н)2 + 12Н+ -^ 2Сг+ + + + С6Н402 + 7Н20
Окисление ионов церия (III):
Н,РО<
Сг20" + 6Се+ + + + 14Н+ —> 6Се+ + + + + 2Сг+ + ++7Н20
Казалось бы, реакция окисления ионов церия (III) бихроматом
невозможна, поскольку нормальный окислительно-восстановительный
потенциал системы Се+ + + +/Се+ + + равен +1,44 в, т. е. больше потенциала
системы Сг20"/2Сг+ + +, который составляет + 1,36 в. Однако реакция
идет. Это обусловливается тем, что значения реальных потенциалов пар
CeIV/Cem и CrVI/CrIU сильно меняются в зависимости от концентрации
добавляемой фосфорной кислоты. В то время, как потенциал системы
280
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Се /Се постепенно уменьшается по мере прибавления фосфорной
кислоты, потенциал пары CrVI/Crm увеличивается. В 12 н. растворе
Н3Р04 реальный потенциал первой пары становится равным +1,24 в,
а второй — +1,66 в, т. е. разница потенциалов достигает 1,66—1,24 =
= +0,42 в, что составляет достаточно большую величину,
обеспечивающую течение реакции в нужном направлении (слева направо).
Аналогичное явление наблюдается при действии фосфорной кислоты
на системы UVI/UIV и FeHI/Fen. По мере прибавления фосфорной
кислоты потенциал первой системы увеличивается, а потенциал второй
системы уменьшается.
В рассмотренных случаях изменение потенциалов обусловливается
комплексообразованием (см. § 1).
Бихромат калия используется и для определения окислителей (UVI,
Vv, MoVI, Fe111, Co111, NOr, H2O2 и т. п.). Метод основан на
предварительном восстановлении окислителей до соединений низшей степени
окисления (например, в редукторе Джонса) и последующем окислении
восстановленных форм полученных соединений стандартным раствором бихро-
мата калия; или восстановление окислителей стандартным раствором
восстановителя (чаще всего Fe++ -ионами) и последующим титрованием
избытка восстановителя бихроматом.
Например, для определения нитратов к анализируемому раствору
прибавляют избыток раствора соли железа (II) и полученную смесь
кипятят. При этом протекает реакция окисления-восстановления:
t°
/iFe++ + NC>3 + 4H+ —> 3Fe+ + + + NO + 2H20 + (п — 3) Fe+ +
избыток
Затем избыток ионов железа (II) оттитровывают бихроматом:
6(л — 3)Fe+ + + (n — 3)Сг20"+ 14 (/г— 3) Н+ —> 6(п — 3)Fe+++ +
+ 2(/г-3)Сг+ + ++7 fr~3>H20
Реакция осаждения. Хромат и бихромат калия, как известно, вступают
в реакции двойного обмена со многими солями металлов (см. книга 1,
гл. XII, § 10), образуя осадки с Ва+ + , Ag+, Pb++ и другими ионами.
2) Бихромат образует нерастворимые соединения с алкалоидами и
некоторыми другими основаниями, реагирующими с ним в стехиометри-
ческом отношении.
Методы определения конечной точки титрования. Для фиксирования
конечной точки титрования в методе хроматометрии применяют
различные способы.
1) Безындикаторный метод основан на наблюдении за окраской
титруемого раствора, вызываемой избытком реагента. При этом зеленый цвет,
присущий ионам Сгш, переходит в желто-зеленый. Иногда изменение
окраски раствора вызывается окислением окрашенного определяемого
вещества, меняющего окраску при действии бихромата (например, синее
индиго меняет окраску на желто-коричневую).
2) Индикаторный метод основан на применении внутренних ред-
окс-индикаторов (дифениламин, дифениламинсульфокислота, фенилан-
траниловая кислота), хемилюминесцентных индикаторов (сщлоксен), а
также внешних индикаторов (лейкометиленовый голубой).
3) Инструментальные методы (потенциометрический, кондуктометри-
ческий, высокочастотный, амперометрический и др.).
§ 42. 'ХРОМАТОМЕТРИЯ
281
Преимущества хроматометрического метода и его недостатки. По
сравнению с другими окислительно-восстановительными методами, и в
частности, по сравнению с перманганатометрией хроматометрия отличается
рядом особенностей.
1. Бихромат калия легко получить в химически чистом состоянии.
Стандартный раствор бихромата калия готовят по точной его навеске.
Это позволяет использовать бихромат калия в качестве установочного
вещества при определении титров других стандартных растворов, например
для установки стандартных растворов тиосульфата (см. § 32), титана
(см. § 47) и т. п.
2. Растворы К2Сг207 очень устойчивы. Титр бихромата калия не
изменяется в течение долгого времени даже под влиянием различных факторов
(температуры, действия кислорода или двуокиси углерода воздуха
и т. д.).
3. Титрование бихроматом калия можно проводить в солянокислой
среде, тогда как титрование перманганатом в присутствии НС1 связано
с окислением С1~ в элементарный хлор.
4. Бихромат труднее, чем перманганат, восстанавливается
органическими веществами, попадающими в дистиллированную или деионизиро-
ванную воду.
5. В отличие от более слабых окислителей, каким является, например,
иод, бихромат способен окислять многие органические соединения.
6. В ряде случаев титрование бихроматом можно проводить на холоду,
в то время как окисление перманганатом или соединениями CeIV
происходит при повышенной температуре. Однако следует указать, что бывают
случаи, когда титрование бихроматом трудноокисляемых соединений
необходимо проводить также при повышенной температуре.
7. Помимо титрования восстановителей, бихромат широко применяют
для определения окислителей (MnVI1, CrVI, MoIV, UVI, Си11 и т. д.)
путем предварительного, восстановления их солями железа (II) и
последующего титрования избытка Fe++ стандартным раствором бихромата.
8. Титрование бихроматом в среде концентрированной фосфорной
кислоты, предложенное индийским ученым Гопало Рао, имеет еще и то
преимущество, что позволяет проводить дифференцированное титрование
двух- и трехкомпонентных смесей катионов, например, Fe11 + VIV,
Mn11 + VIV, Fe11 + Mn11 + VIV и т. п.
Наряду с указанными преимуществами, хроматометрическому методу
титрования присущи и некоторые недостатки.
1. Бихромат калия менее сильный окислитель, чем перманганат и
поэтому он использовался относительно меньше, чем перманганат. Однако
в настоящее время бихромат приобретает все более широкое
применение.
2. Ред-окс-реакции с участием бихромата протекают относительно
медленно. Скорость реакций заметно повышается в сильно кислых средах,
так как окислительно-восстановительный потенциал системы CrVI/Crm
возрастает по мере увеличения концентрации ионов водорода и в
присутствии некоторых лигандов.
3. Конечную точку титрованию по изменению окраски раствора,
вызываемой избытком реагента, относительно трудно установить, поэтому
титрование проводят предпочтительно индикаторным способом.
4. Вследствие того, что со многими восстановителями бихромат
реагирует слишком медленно, прямой метод титрования в хроматометрии
часто неосуществим; главным образом, применяют обратный метод
титрования.
282
ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
§ 43. Определение содержания железа (II)
Определение железа (II) хроматометрическим методом основано на
прямом титровании ионов железа (II) стандартным раствором бихромата
в сернокислой или солянокислой среде в присутствии фосфорной кислоты:
6Fe++ + Cr20~ " + 14Н+ —> 6Fe+ + + + 2Сг+ + + + 7Н20
Фосфорную кислоту прибавляют для того, чтобы связать в комплекс
образующиеся ионы железа (III), что благоприятствует титрованию Fe11:
6Fe+ + + + 12РО; " " -> 6 [Fe (P04)2]
Методика определения. Рассчитанную навеску соли железа (II)
растворяют в мерной колбе емкостью 100 мл в 40 мл дистиллированной воды,
к которой добавляют 40 мл 2 н. раствора серной или хлористоводородной
кислоты и около 20 мл концентрированной фосфорной кислоты.
Содержимое колбы тщательно взбалтывают, дают охладиться до комнатной
температуры и затем доводят фосфорной кислотой или дистиллированной
водой до метки. Аликвотную часть раствора (10 мл) переносят при помощи
пипетки в коническую колбу, добавляют несколько капель дифениламин-
сульфокислоты (или другого подходящего индикатора) и титруют при
перемешивании 0,1 н. раствором бихромата калия до изменения окраски
индикатора. В случае применения дифениламинсульфокислоты зеленая
окраска индикатора в конечной точке титрования переходит в фиолетово-
синюю.
Расчет — см. гл. I,, § 10.
§ 44. Цериметрия
Цериметрией называют метод объемного анализа, основанный на
использовании в качестве окислителя соединений церия (IV):
Се++++ + е ^± Се+++
В качестве исходных веществ для приготовления стандартных
растворов в цериметрии преимущественно применяют комплексные аммониевые
соли церия (IV): гексанитратоцерат (IV) (NH4)2[Ce(NO3)0], трисуль-
фатоцерат (IV) (NH4)2[Ce(S04)3], гексахлороцерат (IV) (NH4)2[CeCl6l,
гексаперхлоратоцерат (IV) (NH4)2[Се(СЮ4)6], этилендиаммонийтетра-
сульфатоцерат (IV) (H3NCH2CH2NH3)2[Ce(S04)4] • 7H20. Окислительно-
восстановительные потенциалы соответствующих систем имеют различные
значения в зависимости от состава титруемого вещества и среды:
солянокислые + 1,28 в, сернокислые + 1,45 в, азотнокислые + 1,60 в и хлорно-
кислые + 1,70 в.
В среде 6 М фосфорной кислоты окислительно-восстановительный
потенциал системы CeIV/CeI!I снижается до 1,25 в, а в более
концентрированной фосфорной кислоте — еще сильнее. Следовательно, реальные
окислительно-восстановительные потенциалы систем, содержащих
соединения церия (IV), колеблются в пределах от +1,3 до +1,7 в в
зависимости от среды и концентрации лиганда.
Высокий окислительно-восстановительный потенциал системы
CeIV/Celn позволяет проводить почти все определения, какие возможны
при участии перманганата калия.
§ 44. ЦЕРИМЕТРИЯ
283
Преимущества и недостатки цериметрического метода.
Цериметрическии метод отличается рядом преимуществ по сравнению с другими
методами окисления—восстановления.
1. Соли, используемые для приготовления стандартных растворов
CeIV, устойчивы, не расплываются на воздухе, не подвергаются
действию С02 и 02, не выветриваются и, самое главное, имеют относительно
высокие молекулярные веса. Стандартный раствор можно приготовить
по точной навеске исходной соли, не проверяя его по другим установочным
веществам.
2. Стандартные растворы долгое время сохраняют постоянство титра.
3. Титрование растворами солей церия (IV) можно проводить не
только в сернокислой, но и в солянокислой средах.
4. При титровании растворами солей церия (IV), как правило, не
образуются промежуточные и побочные продукты, снижающие точность
определения или замедляющие процесс титрования.
5. Широкий диапазон значений реальных потенциалов пары Се IV/CeIU,
зависящих от среды и концентрации лигандов, дает возможность
использовать цериметрическии метод для определения самых разнообразных
веществ, не определяемых другими ред-окс-методами.
6. Иногда применяют Се+ + + +-ионы, генерируемые
электрохимическим путем.
Цериметрии свойственны также и некоторые недостатки.
1. В цериметрии необходимы индикаторы, так как конечную точку
титрования трудно обнаружить rto изменению цвета раствора при
добавлении избытка титранта, хотя он и обладает желтой окраской. В случае
визуального метода определения конечной точки титрования вводят
поправки, устанавливаемые путем постановки холостых опытов.
2. При титровании соединениями церия (IV) растворов,
содержащих F~, образуются устойчивые комплексные ионы [CeF6]"~, а при
титровании в присутствии РО4 ~~ при недостаточной кислотности раствора
образуется нерастворимый фосфат церия (IV), вследствие этого могут быть
искажены результаты анализа.
3. В ряде случаев в растворах хлористоводородной кислоты церий (IV)
восстанавливается С1 и образуются комплексные ионы СеС1+++, СеСЬ
и другие в зависимости от концентрации НС1; в водных растворах,
содержащих хлорную кислоту, иногда наблюдается фотохимическое
восстановление церия (IV), сопровождающееся образованием свободного кислорода:
Ce+ + + ++/iv —> *Се+ + + +
*Се+ + + + + НОН —> Се+ + + + Н++.ОН
• ОН+.ОН —*» НОН+О
О + О —> 02
4. Титрование растворами соединений церия (IV) часто необходимо
проводить при нагревании до 50—75° С, что представляет большие
неудобства в случае определения легколетучих органических соединений.
Вследствие того, что многие реакции с соединениями церия (IV) протекают
медленно, титрование часто проводят в присутствии катализаторов —
иодхлорида, соединений марганца (II) и др.
Методы определения конечной точки титрования. В цериметрии
применяют различные способы -фиксации конечной точки титрования.
1. Безындикаторный метод основан на визуальном наблюдении за
изменением окраски титруемого раствора, который при избытке титранта
окрашивается в желтый цвет. Данный метод неприменим при титровании окра-
284
ГЛ.III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
шенных или мутных растворов, а также в тех случаях, когда появляется
окраска, вызываемая образованием цветных продуктов реакции.
2. Индикаторный метод, основанный на применении ред-окс-индика-
тороц (ферроина, 2,2'-дипиридила, 5,6-диметилферроина, сетополина С,
дифениламинсульфокислоты и др.) и необратимых индикаторов
(метилового оранжевого, метилового красного и т. п.).
3. Физико-химические (инструментальные) методы титрования (потен-
циометрический, фотометрический, амперометрический и др.).
Вещества, определяемые цериметрическим методом. Соединения
церия (IV) применяют для титрования восстановителей (As111, Sb111,
Sn++, Fe+++, Cr++, NO;, H2P02, H3P03, H202, H2C204, Р, S2OT,
I > [Hg2]++, Te03 , СбН4 (ОН)2, углеводы, ненасыщенные соединения,
аскорбиновая кислота).
§ 45. Броматометрия
Броматометрический метод объемного анализа основан на окислении
восстановителей броматом калия, который в кислой среде (при [Н+ ] = 1)
является сильным окислителем:
?° -. _ =+ 1,45 в
ВгОд/Вг
При восстановлении бромат переходит в бромид:
ВгОз + 6Н+ + бе —> Вг" + ЗН20
При титровании броматом первая лишняя капля бромата вступает
в реакцию с получающимся в растворе бромидом, выделяя свободный
бром, который может быть обнаружен по появлению желтой окраски
ВгО~ + 5ВГ + 6Н+ —> ЗВг2 + ЗН20
Обычно бромид прибавляют в раствор бромата. При титровании
применяют азоиндикаторы, например метиловый красный или метиловый
оранжевый. Эти индикаторы после окончания основной реакции
окисляются бромом и разрушаются: цвет раствора из красного переходит в
бледно-желтый. Так как переход окраски связан с разрушением индикатора,
процесс этот необратим; при титровании необходимо осторожное прили-
вание окислителя. Индикаторами могут служить также индигосульфоно-
вые кислоты.
Броматометрию применяют для определения мышьяка (III),
сурьмы (III), таллия (I) и гидразина в кислой среде; в инертной атмосфере
можно титровать, олово (II) и медь (I).
Для увеличения скорости реакции титрование проводят при
нагревании до 50—60° С.
Стандартным раствором служит 0,1 н. раствор бромата калия. Этот
раствор приготовляют по точной навеске чистого перекристаллизованного
бромата калия, высушенцого при 150° С. Раствор бромата калия может
быть проверен по титрованному раствору мышьяковистой кислоты.
Кроме того, броматометрию применяют для определения многих
других неорганических и органических соединений: фенолов и их
производных, аминов, аскорбиновой кислоты, 8-оксихинолина (и осаждаемых
этими соединениями ионов: Mg+ + , Al + + + , Bi+ + + , Fe+ + + , In+ + + и др.),
тиомочевины, меркаптанов и т. п.
§ 46. ВАНАДАТОМЕТРИЯ
285
Широкое применение броматометрии объясняется тем, что бромат-
бромидная смесь может участвовать не только в реакциях окисления—
восстановления, но и в реакциях замещения и присоединения:
реакции окисления—восстановления:
НС1
3As02 + Вг03 + ЗН20 —> Вг + 3H2As04
реакции замещения:
С6Н5ОН + ЗВг2 _*. 3HBr+CeH2Br3OH
реакции присоединения:
>С = С< + Вг2 —+ >СВг —СВг<
Преимущества и недостатки броматометрического метода. Бромато-
метрический метод отличается рядом преимуществ по сравнению с другими
методами.
1. Бромат-бродоидные растворы можно применять не только для
определения восстановителей и окислителей, но и для- анализа органических
ненасыщенных, ароматических и гетероциклических соединений, а также
для косвенного определения разнообразных ионов, осаждаемых в виде
нерастворимых в воде соединений, например в виде оксихинолятов.
2. В отличие от стандартных растворов иода или брома, применяемых
для анализа тех же соединений, растворы бромата калия устойчивы и не
меняют своего титра в течение продолжительного времени. Поэтому при
пользовании броматом получаются более надежные результаты анализа.
3. При введении в бромат-бромидную смесь ионов ртути (II)
увеличивается потенциал системы бром-бромид благодаря образованию
устойчивых комплексных ионов [HgBrJ". ?° ,„ -- в этом случае пре-
вышает ?°Вг0-/Вг-- При этом происходит окисление таких ионов и соеди-
з/
нений, которые в отсутствие ионов ртути не окисляются бромат-бромидной
смесью. Например, хром (III) легко окисляется до хрома (VI) в
присутствии Hg11.
Броматометрический метод имеет также ряд недостатков.
1. Вода, присутствующая в растворе или образующаяся в процессе
титрования неводных растворов, мешает определению многих
органических соединений.
2. Окисление некоторых органических соединений сопровождается
нежелательными побочными реакциями гидролиза, замещения и
присоединения, вызываемых действием ионов воды и элементарного брома.
3. В ряде случаев реакции бромата калия с органическими веществами
протекают не в строго стехиометрических отношениях, что приводит к
искажению конечных результатов анализа.
§ 46. Ванадатометрия
Ванадатометрия основана на окислении восстановителей солями
ванадиевой кислоты. Стандартным раствором является раствор ванадата
аммония в 6 н. растворе серной кислоты. Ванадий (V) восстанавливается
до ванадия (IV):
VOg-fe + 4H+ ^ VO++ + 2H20
286
ГЛ.111. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
Окислительный потенциал системы сильно зависит от рН среды и
может быть повышен путем увеличения кислотности раствора:
* ^ 1 g IVO++]
Е = +1,02 в (в 1 М растворе H2S04); Е = +1,1 в (в 5 М растворе H2S04).
Е = +1,30 в (в 8 М растворе H2S04).
Стандартный раствор ванадата аммония устанавливают по бихромату
калия и соли Мора или по оксалату натрия:
2VO; + H2C204.+ 6H+ —> 2VO++ + 2C02 + 4H20
Ванадатометрию широко применяют для определения железа (II),
молибдена после восстановления молибдена (VI) до молибдена (V), урана
после восстановления до урана (IV), сульфитов, тиосульфатов, гипофос-
фитов, гидрохинона, индиго и других органических соединений.
Особое значение имеют методы, основанные на осаждении
определяемых ионов органическими реактивами — купферроном, диметилглио-
ксимом, 8-оксихинолином и т. п.
/NO
Купферрон CeH5N\ применяют для осаждения ионов железа,
xONH4
ванадия, циркония, титана, олова, тантала, ниобия и др. Осаждение
купферроном проводят в сильнокислой среде, что позволяет отделять
перечисленные ионы от других элементов. Это обстоятельство является
большим преимуществом при анализах руд и сплавов, содержащих редкие
элементы. Осадки купферронатов отфильтровывают, промывают и
обрабатывают избытком титрованного раствора ванадата аммония для их
окисления. Избыток ванадата аммония оттитровывают солью железа (II)
в присутствии фенилантраниловой кислоты. Окраска переходит из бледно-
голубой в вишнево-красную.
Индикаторами в ванадатометрии, как и в хроматометрии, служат
фенилантраниловая кислота, дифениламин и его сульфокислоты. Ванадато-
метрические определения можно проводить в солянокислых растворах.
§ 47. Аскорбинометрия
Принцип метода. Ред-окс методы, описанные выше, основаны на
использовании в качестве главных реагентов окислителей, способных
окислять самые разнообразные восстановители. Аскорбинометрия основана на
применении в качестве главного стандартного раствора аскорбиновой
кислоты, обладающей восстановительными свойствами.
Аскорбиновая кислота (АК) применяется для прямого титрования
окислителей. При ее окислении образуется дегидроаскорбиновая
кислота (ДГАК):
О О
с—с
н+ / \
¦—> н—a Jc=o
-2е Z
он
НО
СН2ОН
§ 47. АСКОРБИНОМЕТРИЯ
287
Как видно из приводимой структурной формулы, кислый характер
аскорбиновой кислоты (витамин С, у"лактон 2,3-дегидро-/,-гулоновой
кислоты) обусловлен не карбоксильной группой, характерной для карбо-
новых кислот, а енольными гидроксильными группами (р/Сдк = 4,17;
Р^Сак = 11,57). Нормальный окислительно-восстановительный потенциал
системы АК/ДГАК (?ак/дгак = +0,18 в) при рН = 7 немного меньше,
чем системы SnIV/Sn++ (^snIV/sn++ == +0Л5 в). Она является более
слабым восстановителем по сравнению с солями хрома (II) (?'сг+++/сг++ =
= —0,41 в), ванадия (II) (?уш/уц = 0,2 в) и титана (III) (?°.iv/Tini =
= +0,1 в).
При действии восстановителей дегидроаскорбиновая кислота,
образующаяся при окислении аскорбиновой кислоты, вновь восстанавливается
в аскорбиновую кислоту, т. е. ред-окс система АК/ДГАК обратима.
Окислительно-восстановительный потенциал этой системы сильно зависит от рН
раствора, повышаясь по мере увеличения кислотности раствора.
При действии сильных окислителей аскорбиновая кислота может
окисляться в L-треоновую и щавелевую кислоты:
ОН Н
I I
сбн8о6 -^ сбнбов —> нооссоон + носн—с—с—соон
I I
н он
Поэтому эквивалент аскорбиновой кислоты может изменяться в
зависимости от силы окисляющего ее окислителя. При окислении
аскорбиновой до дегидроаскорбиновой кислоты под действием умеренных
окислителей типа элементарного иода, ее эквивалент равен молекулярному весу,
деленному на 2:
Вещества, восстанавливаемые аскорбиновой кислотой. Аскорбиновая
кислота восстанавливает многие неорганические и органические вещества:
FeIH, Hg11, Aum, PtIV, Ag1, элементарный иод, хлораты, броматы,
иодаты, ванадаты, цераты, кислород, растворенный в растворителях,
нитро-, нитрозо-, азо-, иминогруппы, индофенолы, перфориндины и другие:
СвН806 + 12 —> СвНбОв + 2Н+ + 21-
CeH6N02 + 3CeH8Oe —> C6H5NH2 + ЗС6Н6Ов + 2Н20
Поскольку аскорбиновая кислота способна восстанавливать
элементарный иод, она заменяет тиосульфат в тех случаях, когда по каким-либо
соображениям нельзя применять тиосульфат натрия в иодометрическом
методе. При этом вместо крахмала в качестве индикатора применяют ва-
риаминовый синий, обесцвечивающийся в конце титрования.
Преимущества и недостатки аскорбинометрического метода. Аскорбино-
метрия отличается рядом преимуществ по сравнению с другими методами,
основанными на использовании в качестве основного титранта растворов
восстановителей.
1. В отличие от других восстановителей — солей хрома (II),
титана (III) и т. п., растворы аскорбиновой кислоты являются более
устойчивыми, и поэтому титровать ими можно не прибегая к специальной
аппаратуре и использованию инертной атмосферы (Н2, С02, N2).
288 ГЛ.Ш. МЕТОДЫ ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ
2. Являясь более устойчивым реагентом по сравнению с другими
восстановителями, применяемыми в ред-окс методах, аскорбиновая кислота
применяется для большего числа определений неорганических и
органических соединений и служит заменителем тиосульфата в иодометрии.
Основные недостатки аскорбинометрии следующие: аскорбиновая
кислота подвержена действию энзим и ультрафиолетовых лучей,
каталитически ускоряющих ее разложение. Поэтому для приготовления
стандартных растворов рекомендуется применять особо чистые образцы
аскорбиновой кислоты и хранить их на холоду в темноте в склянках из
оранжевого стекла. Для стабилизации растворов аскорбиновой кислоты
рекомендуется добавлять к ним этилендиаминтетрауксусную кислоту (ЭДТА)
и муравьиную кислоту, благодаря чему ее стандартные растворы
сохраняются дольше.
Применение аскорбиновой кислоты для определения ионов железа (III).
Определение железа (III) относится к важнейшим определениям,
выполняемых титрованием стандартным раствором аскорбиновой кислоты.
В качестве титранта применяют 0,1 н. раствор аскорбиновой кислоты,
устанавливаемый иодометрическим методом. Титрование ведут в солянокислом
растворе при нагревании до 50—60° С в присутствии роданида калия,
образующего с ионами железа (III) кроваво-красное окрашивание.
Конечную точку титрования фиксируют по обесцвечиванию раствора.
§ 48. Титанометрия
Титанометрический метод объемного анализа основан на титровании
окислителей солями титана (III). Соли титана (III) являются очень
сильными восстановителями (E°Tiiv/Tim = +0,04 в).
В качестве стандартных растворов применяют растворы хлорида или
сульфата титана. Раствор сульфата титана (III) устойчивее, чем раствор
хлорида титана (III), очень быстро окисляющийся на воздухе; растворы
имеют фиолетовую окраску.
Титры растворов солей титана устанавливают по бихромату калия,
перманганату калия, сульфату железа (III) и некоторым другим
окислителям. Титрование ведут в атмосфере двуокиси углерода в
сильнокислой среде. Определение окислителей проводят прямым титрованием или
по остатку.
Титанометрическим методом можно определять бихроматы, хроматы,
перхлораты и другие окислители. Титанометрию применяют также для
определения нитросоединений и органических красителей, например
метиленового синего. Окислители титруют раствором хлорида титана (III)
в присутствии дифениламина. Например:
—N02 + 6TiCl8 + 6HCl —> — NH2 + 6TiCl4 + 2Н20
ГЛАВА IV
МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
А. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
§ 1. Общая характеристика методов
Метод осаждения. Методы осаждения основаны на применении
реакций осаждения. Рассмотренный ранее пример определения НС1 в соляной
кислоте по объему раствора AgN03 точно известной концентрации,
пошедшего на реакцию с НС1 (см. «Введение»), относится к методу осаждения.
Наибольшее значение приобрели те методы осаждения, которые,
связаны с образованием малорастворимых соединений серебра, бария,
ртути, свинца, цинка и некоторых других элементов. Например:
1) Титрование Ва++ сульфатом основано на реакции:
Ва++ + SO~ —+ BaS04
Точку эквивалентности устанавливают, применяя родизонат натрия.
При избытке Ва++ появляется красное окрашивание. В точке
эквивалентности красное окрашивание исчезает.
2) Титрование РЬ++ хроматом основано на реакции:
РЬ++ + СгО;""—> РЬСг04
Точку эквивалентности устанавливают при помощи Ag +-ионов
образующих с избытком СгО~~ осадок Ag2Cr04 красного цвета.
3) Титрование Zn++ гексацианоферратом (II) основано на реакции:
3Zn++ + 2 [Fe (CNJ-- + 2К+ -> Zn3K2 [Fe (CN)J,
Точку эквивалентности устанавливают с помощью нитрата уранила,
образующего с избытком К4 [Fe (CN)G] буро-коричневый осадок.
В отличие от индикаторов, вводимых в раствор и называемых поэтому
внутренними индикаторами, нитрат уранила относится к так
называемым внешним индикаторам, которые нельзя вводить в титруемый раствор,
так как они реагируют с образующимися осадками. При работе с внешними
индикаторами в конце титрования отбирают каплю прозрачной жидкости,
переносят ее на фарфоровую пластинку и прибавляют каплю цндикатора;
выпадение характерного осадка или изменение цвета раствора указывает
на окончание титрования.
Точку эквивалентности можно устанавливать и другими химическими
и физико-химическими методами. Например, титрование Zn++
гексацианоферратом (II) ведут в присутствии К3 [Fe (CN)6] и дифениламина.
При титровании Zn++ кроме Zn3K2 [Fe (CN)6]2 образуется также и
Zn3 [Fe (CN)6]2. Первая из этих солей менее растворима, чем вторая.
По мере прибавления стандартного раствора гексацианоферрата (II)
отношение [Fe(CN)6] /[Fe(CN)6]== в растворе медленно
увеличивается, так как [Fe (CNJe]" непрерывно связывается с Zn++, a
290
ГЛ IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
[Fe (CN)6] ионы постепенно освобождаются в,результате превращения
осадка Zn3 [Fe (CN)6]2 в Zn3K2 [Fe (CN)6]2. Это приводит к увеличению
окислительного потенциала:
?-?М 0 0501Г [Fe(CN>«]~"~
и, следовательно, [Fe (CN)6] окисляет дифениламин, при этом
появляется синее окрашивание. После достижения точки эквивалентности в
титруемом растворе появляется избыток [Fe (CN)6]==-hohob, отношение
[Fe (CN)6] /[Fe(CN)6]-= резко уменьшается и дифениламин
обесцвечивается. В случае приливания Zn++ к титруемому раствору в
присутствии системы, содержащей К3 [Fe (CN)6] + К4 [Fe (CN)6] + бензи-
дин, в точке эквивалентности, наоборот, появляется синее окрашивание,
как это бывает при комплексонометрическом титровании алюминия, в
присутствии индикаторной смеси Кз [Fe (CN)6] + К4 [Fe (CN)6] + бензи-
дин (см. ниже).
Методы осаждения дают возможность количественно определять
анионы, осаждаемые катионами серебра, бария, ртути, свинца, цинка и др.,
например хлориды, бромиды, иодиды, цианиды, роданиды, сульфаты,
хроматы, фосфаты, ферроцианиды и т. д., а также катионы, образующие
малорастворимые соединения с указанными выше анионами. Применяя
специальные приемы титрования (см. гл. I, § 2), можно этими методами
количественно определять не только отдельные катионы или анионы,
но и их смеси.
Метод комплексообразования. Методы комплексообразования
основаны на использовании реакций комплексообразования, например:
Ag+-j-2CN- ^± [Ag(CN)2]-
A1+ + + + 6F- ^t [AlFe]- —
Пользуясь методами комплексообразования, можно количественно
определять разнообразные катионы (Ag + , Hg++, Al+++ и др.) и анионы
(CN", F", С1~ и др.)» склонные вступать в реакции комплексообразования
(см. книга 1, гл. VII, § 12—14). Особое положение среди методов
комплексообразования занимает так называемая комплексонометрия (ком-
плексонометрическое титрование), основанная на применении реакций
образования прочных комплексных соединений с нитрилтриуксусной,
этилендиаминтетрауксусной и другими аминополикарбоновыми кислотами,
дающими комплексные соединения со многими катионами (см. ниже).
Связь между методами осаждения и комплексообразования. Методы
осаждения тесно связаны, с методами комплексообразования, так как
многие реакции осаждения сопровождаются комплексообразованием, а
образование комплексов сопровождается осаждением малорастворимых
соединений. Например:
AgCl — осадок; [AgCl2]~ — комплексный ион, образующийся при
действии избытка С1~ на AgCl.
AgCN — осадок; [Ag (CN)2]~ — комплексный ион, образующийся при
действии избытка CN" на AgCN.
AgSCN — осадок; [Ag (SCN),2]~ —комплексный ион, образующийся
при действии избытка SCN" на AgSCN.
Реакции комплексбобразования в свою очередь близко соприкасаются
с реакциями образования малодиссоциирующих солей [HgCl2, Hg (CN)2,
Hg(SCN)2 и т. п.].
Поэтому методы осаждения рассматриваются совместно с методами
комплексообразования и образования слабых электролитов.
§ 2. КЛАССИФИКАЦИЯ МЕТОДОВ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ 291
§ 2. Классификация методов осаждения и комплексообразования
Методы осаждения и комплексообразования классифицируют
следующим образом:
1) Аргентометрия — метод объемного анализа, основанный на
применении стандартного раствора нитрата серебра. Различают:
а) Метод, который был предложен еще Гей-Люссаком, основанный
на реакции, протекающей между ионами серебра и ионами галогенов и
выполняемой в отсутствие индикатора:
Ag+4-НаГ —* AgHal
При титровании бромидов определение ведется следующим образом.
К анализируемому раствору, содержащему, например, Вг~, приливают
из бюретки малыми порциями стандартный раствор AgN03. При этом
образуется творожистый осадок AgBr и раствор становится мутным.
Следующую порцию раствора нитрата серебра вводят лишь после того,
как раствор над осадком слегка посветлеет. Приливание из бюретки
стандартного раствора заканчивают в тот момент, когда последующая порция
приливаемого раствора AgN03 не вызывает образования новых количеств
осадка AgBr. В этот момент титруемый раствор полностью просветляется
и становится прозрачным вследствие коагуляции AgBr в точке
эквивалентности. Этот метод называется методом просветления.
При титровании безындикаторным методом хлоридов стандартным
раствором нитрата серебра титрование заканчивают по так называемому
методу равного помутнения. Точку эквивалентности по этому методу
устанавливают путем отбора двух проб титруемого раствора. На одну из них
действуют новой каплей стандартного раствора, на другую — каплей
стандартного раствора хлорида натрия той же концентрации. В случае
равного помутнения обеих проб титрование заканчивают.
Этот метод требует большого навыка работы. При известном навыке
работы метод приводит к точным результатам анализа. Раньше, до
разработки хороших индикаторных методов, способ Гей-Люссака имел большое
значение: в настоящее время он не применяется на практике.
б) Метод Мора, основанный на реакции, протекающий между Ag+
и С1~ и выполняемой в присутствии индикатора — раствора хромата
калия.
Определение ведется следующим образом. К анализируемому раствору,
содержащему С1~, приливают по каплям из бюретки стандартный рас-
створ AgN03. При этом образуется AgCl. Образование AgCl продолжается
до тех пор, пока к исследуемому раствору не будет прибавлена
достаточное количество раствора AgN03, эквивалентное содержанию C1" в
анализируемом растворе. Лишняя капля титрованного раствора AgN03
прибавленная после достижения точки эквивалентности, вызывает
выпадение красного осадка Ag2Cr04, образующегося в результате
взаимодействия Ag+ с ионами индикатора:
2Ag+ + CiOf& ^± Ag2Cr04
Таким образом, точку эквивалентности в методе Мора наблюдают
по образованию красного окрашивания, вызываемого Ag^Cr04.
в) Метод Фольгарда, основанный на реакции, протекающей между
Ag+ и SCN" и выполняемой в присутствии индикатора—ионов железа (III).
Определение ведется следующим образом. К анализируемому
раствору, содержащему, например Ag+, приливают по каплям из бюретки
292 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
стандартный раствор NH4SCN. При этом образуется малорастворимый
осадок AgSCN:
Ag+ + SCN~ —> AgSCN
Образование AgSCN продолжается до тех пор, пока к исследуемому
раствору не будет прибавлено достаточное количество раствора NH4SCN,
эквивалентное содержанию Ag+ в анализируемом растворе. Лишняя капля
раствора NH4SCN, прибавленная после достижения точки
эквивалентности, вызывает появление кроваво-красного окрашивания вследствие
взаимодействия SCN~ с ионами индикатора:
Fe+++ + 3SCN" ^± Fe(SCN)3
Таким образом, точку эквивалентности в методе Фольгарда
наблюдают по образованию красного окрашивания, вызываемого роданидом
железа.
г) Метод Фаянса, основанный на применении так называемых
адсорбционных индикаторов (см. ниже).
2) Меркуриметрия — метод объемного анализа основанный на
образовании слабодиссоциированных соединений ртути (II) HgCl2, Hg (CN)2,
Hg(SCN)2 и др.:
Hg++ + 2CN- ^ Hg(CN)2
В качестве индикатора в меркуриметрии применяют раствор нитро-
пруссида натрия Na2[Fe(CN)5 NO]-2H20, реагирующий с Hg++ с
образованием белого осадка Hg[Fe(CN)5 NO]-2H20, или дифенилкарбазон,
который с избытком ионов ртути образует комплексные соединения,
окрашенные в сине-фиолетовый цвет, и др.
3) Меркурометрия — метод объемного анализа, основанный на
образовании нерастворимых соединений ртути (I):
[Hg2]++ + 2C1- _> Hg2Cl2
В качестве индикатора в меркурометрии применяют кроваво-красный
раствор роданида железа, обесцвечивающийся в присутствии избытка
[Hga]++
3[Hg2]++ + 2Fe(SCN)3 —* 2Fe++++ 3Hg2 (SCN)2
или дифенилкарбазон, образующий с [Hg2]++ осадок синего цвета.
4) Комплексонометрия — метод объемного анализа, основанный на
использовании реакций комплексообразования с помощью комплексонов
Na2H2Y + Са++ -> Na2CaY + 2Н+
где Y — анион комплексона.
§ 3. Применение теории осаждения к объемному анализу
Требования, предъявляемые к реакциям, применяемым в методах
осаждения. Не все реакции осаждения могут быть использованы для
количественного определения. Основным требованием* предъявляемым к
реакциям, применяемым в методах осаждения, и комплексообразования,
является следующее:
выделяющиеся осадки должны по возможности обладать минимальной
растворимостью, а образующиеся комплексы наименьшей константой
нестойкости, являющейся мерой их устойчивости.
<j 3 ПРИМЕНЕНИЕ ТЕОРИИ ОСАЖДЕНИЯ К ОБЪЕМНОМУ АНАЛИЗУ 293
Основы теории образования осадков. Согласно закону действия масс
при установившемся равновесии в насыщенном водном растворе
малорастворимого электролита при данной температуре и давлении, независимо
от изменений концентраций отдельных ионов, величина произведения
концентраций ионов (ПРк;аАпь) остается постоянной и в общем виде
выражается формулой:
ПР^ А„ = fKtiafAnl6 = const (1)
'KtflAn, = [Kt]a[Anf = const
где nPKt An — произведение растворимости малорастворимого электролита;
[Kt], [Anj — концентрации катионов и анионов, образующихся при
электролитической диссоциации электролита KtaAn^;
a, b — коэффициенты, показывающие число соответствующих ионов,
образующихся при электролитической диссоциации одной молекулы
данного электролита (см. книга 1, гл. 1, § 24).
Если величина произведения [Kt]a [An]* в растворе становится больше
значения nPKtflAnft, то малорастворимое соединение выделяется в
осадок. Если [Kt]a [An]* < nPKtaAn6, осадок не выпадает или растворяется.
Это правило является важнейшим теоретическим положением,
лежащим в основе теории образования осадков. Из него следует, что
растворимость (Pi<;taAn6) электролита KtflAn& приобретает максимальное
значение в насыщенном растворе, свободном от посторонних ионов.
ТАБЛИЦА 25
Формулы, применяемые для вычисления растворимости
малорастворимых электролитов в воде
Электролит
ПР
Растворимость
ПА
Растворимость
с учетом
коэффициента активности
KtAn
Kt2An
Kt3An
KtAn,
KtAn3
Kt3An2
Kt2An3
[Kt+] [An-]
[Kt+]2[An--]
[Kt+]3[An---]
[Kt++] [An-]2
[Kt+++][An-]3
[Kt++]3[An---]2
[Kt+ + +]2[An--]3
уш
KtAn
V
V
V
4 Г
v
V'
f-
ПР
Kt2An
22
ПР
Kt8An
33
ПР
KtAn2
22
ПР
KtAn,
33
ПР
Kt8An2
33-22
ПР
Kt2An3
22-33
a .a
Kt+ AiT
o
Kt+ An
a3 +a
Kt+ An
Kt++ An
a .. .a
4 2
a2 j. j. ±a}
Kt+ + + An"
/nAKtAn
V
ПА
Kt2An
KtsAn
V
ПА
KtAn2
*A
ПА
KtAn8
5 Г
27//i
KtaAn2
\ lOS/f/f
1 / ПАК«гАп
v mm
Примечание. flt f2 и f9 — коэффициенты активности одно-, двух- и трехзарядных ионов.
294
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Добавление любого из ионов, образующих соединение KtaAn&
уменьшает растворимость. Добавление других электролитов, не имеющих с
соединением KtaAn& общих ионов, повышает растворимость (см. ниже).
Основные требования, предъявляемые к реакциям, используемым при
титровании в методах осаждения, сводятся к следующему.
1. Реакция, протекающая между определяемым веществом А и
стандартным (титрованным) раствором реагента В, должна проводиться в
условиях, обеспечивающих образование такого осадка, растворимость
которого (PKtflAn6) является наименьшей и, во всяком случае, не превышает
минимально допустимой растворимости 10"5 моль/л.
2. Так как добавление других электролитов, не имеющих с
осадком KtaAn6 общих ионов, повышает его растворимость, то реакция должна
протекать при таких условиях, когда посторонние вещества,
присутствующие в анализируемом веществе, не мешают титрованию определяемого
вещества, а реакция осаждения завершается быстро и практически идет
до конца.
Вычисление растворимости малорастворимого электролита. Как было
указано ранее (см. книга 1, гл. 1, § 26), для вычисления растворимости
малорастворимого электролита (табл. 25) пользуются общей формулой:
a+b f
.-У-
Kt An
PKtaAnb= Y aa^b (2>
§ 4. Вычисление растворимости электролитов в воде
с учетом коэффициентов активности
Для бинарного электролита (типа AgCl) имеем:
AKt
А
РЮАп = [Kt+] = [An-] = I/ 1— = TV 11AKtAn (3)
[Kt+] [An'] = -1^- [см. уравнение (36)]
Я
/nAKtAn 1 1/ТПГ-
—-p— = — УПА*
Следует иметь в виду, что при более точных расчетах растворимости необходимо
учитывать не только ионную силу раствора, но и гидролиз соли.
Пример 1. Вычислить растворимость BaCr04, SrCr04 и СаСг04 в г/л при 25° С.
Решение. Для ВаСг04:
рВаСго4 = [Ва++] = [CrO^~] = "J/ 2,3-10~10 = 1,5- 10~5 моль/л
или
1,5- Ю-5-253,33 ^ 3,8- 10~3 г/л (253,33 = МВаСг04)
Для SrCr04 и, соответственно, для СаСг04:
pSrCrO< = [Sr++] = [Сг0~] = ]/3,6-Ю-5 = 6-Ю-3 моль/л
или
6- Ю-3-203,61 « 1,22 г/л (203,61 = MSrCr04)
рсасг04 = [Са++] = [Сг0~] = "[/"2,3.10~2 = 1,5- Ю-1 моль/л
или
1,5- Ю-1-156,07 = 23,4 г/л (156,07 = МСаСг04)
Ионная сила [см. уравнение (3), § 8] равна:
для ВаСг04
М, = -у-(1,5-10-Б.22 + 1,5.10-Б.2а)=6.10-6 и /2 -н- 1
§ 4. ВЫЧИСЛЕНИЕ РАСТВОРИМОСТИ ЭЛЕКТРОЛИТОВ
295
для SrCr04
\i = -i- (6- Ю-3 -22 + 6- Ю-3 -22) = 2,4- Ю-2 и U = 0,57
для СаСг04
|i = -i-(l,5.10-1.22+l,5.10-1-2a) = 6.10-1 и /2<0,24
Для очень разбавленных растворов / = 1 и ПРК4Ап = ПАК1Ап. В этом случае
растворимость с учетом коэффициента активности
для ВаСг04
РВаСг04^ 1/"2,3-10-10« 1,5- Ю-6 моль/л
или
1,5- Ю-6-253,33 = 3,8-10-8 г/л
для SrCr04
Psrcro. ~"о^7~ ^3,6.Ю-5^ 1,05-10"2 моль/л
или
1,05-Ю-2-203,61 =2,14 г/л
для СаСг04
pcacr04^-g^4" 1^2,3. Ю-2 ^6,3-10-1 моль/л
или
6,3-КГ1-156,07 =98,3 г/л
Влияние ионной силы раствора на растворимость электролита. Чем
больше растворимость данного электролита, тем сильнее влияет на его
растворимость ионная сила раствора.
При низких концентрациях электролита в растворе, когда значение
PKtAn < Ю"6 моль/л (например, растворимость ВаСг04), коэффициент
активнрсти приближается к единице. Поэтбму результаты вычисления
растворимости электролитов в воде с учетом коэффициента активности
практически не отличаются от результатов, получаемых при расчете
по приближенным формулам — без учета /. Когда значение PKtAn больше
величины 10-Б моль!л, например растворимость (СаСг04), влияние ионной
силы раствора заметно увеличивается и коэффициент активности
уменьшается в несколько раз по сравнению с коэффициентом активности в сильно
разбавленных растворах.
Если бы мы не учли значение коэффициента активности при
вычислении растворимости ВаСг04 вводе, то получили бы РваСго4 = 3,8-10~8г/л,
т. е. такой же результат, как при вычислении с учетом коэффициента
активности.
Если бы мы не учли значение коэффициента активности при
вычислении растворимости СаСг04 в воде, то различие в величине
растворимости составило бы 98,3 — 23,4 = 74,9 г/л, т. е. очень большую величину.
Поэтому для относительно хорошо растворимых соединений вычисления
их растворимости следует проводить с учетом коэффициентов активности.
О связи произведения растворимости с растворимостью. Количественная связь между
произведением растворимости и растворимостью, выражаемых в равновесных
концентрациях ионов, образующих малорастворимое вещество, носит приближенный характер.
Как показали исследования, более точные результаты можно получить лишь с учетом всех
процессов (гидролиза, комплексообразования и т. п.), сопутствующих растворению данного
малор аствор имого соединения.
Наряду с простыми ионами, образующимися при растворении электролита типа KtaAn&,
в процессе гидролитического расщепления получаются сложные катионы [Kt (ОН)]6"1,
296
ГЛ.IV МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
[Kt (ОН)2]6 2 и т. д. и нейтральные молекулы Kt (ОН);,, а также сложные анионы НАп1 а,
Н2АгГ~а и т. д. и нейтральные молекулы НаАп:
KtaAn6 + &H+ + aOH- ^ a[KXOH]b-l + b[HAn]l~a
и т. д.
Kt~*~ и An-a могут образовывать и комплексные ионы, имеющие различный состав,
а также различный знак и величину заряда.
Вследствие этого расчеты растворимости значительно осложняются и требуют нового
подхода.
В связи с этим Н. Аксельрудом и В. Спиваковским предложено понятие «константа
диссоциации» /СДИСс для некоторых малорастворимых соединений. Это новое понятие
^Сдисс введено исходя из предположения о реальном существовании в растворе
нейтральных частиц типа KtAn. Если данное малорастворимое вещество не образует в растворе
таких частиц, то оно по-прежнему подчиняется правилу произведения растворимости,
а понятие /СДИсс Для него теряет свой смысл.
Соотношение между /Сдпсс и ПРк1Ап выражается формулой:
К — nPKtAn
Адисс- [К1Дп]
§ 5. Влияние одноименных ионов на растворимость
малорастворимого электролита
Уменьшение растворимости. Абсолютно нерастворимых веществ нет.
Все вещества растворимы в большей или меньшей степени. Добавление
избытка осаждающего реактива при осаждении является одним из
способов уменьшения растворимости осадков. Например, для осаждения С1~
в виде AgCl к раствору электролита приливают раствор AgN03. При
добавлении AgN03 в количестве, эквивалентном количеству хлорид-ионов.
[Ag+J = [Ci-] = PAgC1 = l/nP^
или (что то же):
PAeci = [a-] = -^p w
Таким образом, растворимость AgCl в этом случае будет равна
растворимости этого электролита в чистой воде.
При добавлении избытка AgN03 концентрация ионов серебра в
растворе над осадком будет больше концентрации ионов хлора. При этом
можно считать, что избыток ионов серебра прибавлен к насыщенному
водному раствору AgCl. Отсюда общая концентрация ионов серебра в
растворе будет равна [Ag+] + [Ag+]nPll6, где [Ag+]приб — концентрация
ионов серебра, прибавленных в избытке к насыщенному водному раствору
чистого электролита AgCl
Следовательно,
{[Ag+] + [Ag-b]npH6}[cr]=nPAgC1
и
^ AgCl
PAgci = IC1-] = [Ag4 + [Ag4nPH6 (4а)
При сравнении уравнений (4) и (4а) легко увидеть, что растворимость
малорастворимого электролита AgCl имеет наибольшее значение лишь в
насыщенном водном растворе этого электролита и уменьшается при
добавлении избытка AgN03, содержащего одноименные ионы.
Величиной [Ag+] в уравнении (4а) можно пренебречь, если [Ag+]np„6
значительно превышает концентрацию ионов серебра в насыщенном
растворе AgCl.
§ 5. ВЛИЯНИЕ ИОНОВ НА РАСТВОРИМОСТЬ ЭЛЕКТРОЛИТА 297
Тогда
PAgci = tC1"] = [Ag+bpLj
nPAgci
или в общем виде:
nPKtAn
Если же прибавляют избыток реактива, осаждающего катионы
данного электролита (например, Ag+ осаждают в виде AgCl избытком НС1),
то формула принимает вид
Р— = tKt+1 = W!^r (6)
Более точную формулу для вычисления растворимости
малорастворимого электролита типа KtAn в присутствии избытка осаждающего
реактива можно вывести следующим путем:
{[Ag+] + [Ag+]npH6}fCl-]= nPAgC1
Так как в насыщенном растворе
[Ag+] = [Cl-]=PAgC1
то при добавлении избытка AgN03, содержащего одноименный катион
с осадком, можно написать:
{PAgCl + [Аё+]приб} PAgCl = nPAgCl
или
PAgCl + PAgCl Мприб-ПРАбС1 =0
Решив это квадратное уравнение, получим:
[Ag+]npH6 ^ 1 /" [Ag+]^PH6
или в общем виде:
Р _ _ L ° J1^"" _j_ I / L ° .|прио , пр
FAgCl 9 ' У 4 Г llFAgCl
[К1+]Прнб i/ [Ю+]*риб
Соответственно для случая прибавления избытка реактива,
содержащего одноименные анионы:
_ [С1-]прнб ¦¦ / [С1 ]%ри6
FAgCl о h I/ 4 ^ UHAgCl
или в общем виде:
V-
[Ап-]приб , / [Ап"]2приб
FKtAn~ о г I/ 7 H"11FKtAn <б<
Из уравнений (7) и (8) видно, что растворимость осадка при
добавлении избытка осаждающего реактива уменьшается и зависит от избытка
прибавленного иона, общего с одним из ионов, образующих осадок, а также
от численного значения произведения растворимости данного
малорастворимого электролита.
Аналогичным образом можно доказать, что (если малорастворимое
вещество не растворяется с образованием комплексных соединений и не
относится к амфотерным соединениям) растворимость малорастворимого
298
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
электролита также понижается при добавлении не только осаждающего
реактива, но и любого другого хорошо растворимого в данной среде
электролита, имеющего общие ионы с малорастворимым веществом.
Оптимальные соотношения объемов при реакциях осаждения. При реакциях
осаждения, как это убедительно показал Л. П. Адамович, существуют определенные
соотношения объемов раствора осаждаемого иона и осадителя (реактива), при которых осаждение
для данной концентрации осадителя протекает наиболее полно. При определенных
условиях (если концентрация осадителя в 20 раз выше концентрации осаждаемого иона и ионные
силы растворов равны) для многих реакций эти соотношения равны отношениям
коэффициентов в уравнении реакций. Несоблюдение требуемых оптимальных отношений может
повести к серьезным ошибкам при выполнении качественных реакций и количественного
определения.
Вычисление растворимости. Пример 1. Вычислить растворимость AgCl, к
насыщенному раствору которого прибавлен 0,001 н. раствор AgN03 или 0,001 н. раствор NaCl.
Решение. Растворимость AgCl в воде составляет:
PAgCl = tA§+] = Iе1"] = l/*nPAgci = /1,7-10-*° = 1,3- Ю-6 моль/л
а) Растворимость AgCl при добавлении 0,001 н. раствора AgN03, т. е. при избытке
Ag+, будет равна:
РАвС1-[СИ- [Ag+]npH6
1 7-Ю"10
PAgCl = '10-з = 1>7' Ю"7 тль>Л
б) Растворимость AgCl при добавлении 0,001 н. раствора NaCl, т. е. при избытке СГ,
равна:
1 7-Ю"10
PAgci = ' 10-3 = 1.7-10-' моль/л
Таким образом, растворимость AgCl одинаково уменьшается от прибавления избытка
как ионов серебра, так и ионов хлора.
Пример 2. Вычислить растворимость Ag2Cr04 в воде; в 0,1 М растворе AgN03; в 0,1 М
растворе Na2Cr04.
Решение. Используя уравнение (1), можно написать:
а) Растворимость Ag2Cr04 в воде
р _ [Ag+] _ ГСЮ—1 _ У*/ nPAg2CrQ4
FAg2Cro4 - 2 ~ Г 4 J ~~ У 4
Подставив в уравнение величину ПРА^Сг04 = 2» 10~12, получим:
^— = 0,79.10-0 моль/л
б) Растворимость Ag2Cr04 в 0,1 М растворе AgN03.
Из уравнения
nPAg2Cr0l=[Ag+]2[Cr04--]
находим значение [Сг04 J:
ГГг0—1 - x"Ag2cro4
^ги4 J - [Ag+]2
Следовательно,
npAg2Cfp4
PAg2Cr04= ИГ] =-^?- = 2.10~10 моль/л
§ б. ВЛИЯНИЕ ИОНОВ НА РАСТВОРИМОСТЬ ЭЛЕКТРОЛИТА 299
Из этого следует, что растворимость Ag2Cr04 в 0,1 М растворе AgN03 (т. е. при избытке
Ag+) по сравнению с растворимостью в воде уменьшится от 0,79» 10"4 моль/л до 2Х
X Ю-10 моль/л.
в) Растворимость Ag2Cr04 в 0,1 М растворе Na2Cr04.
Из уравнения
npAg2crOl=[Ag+]2[Cr0ri
можно написать, что
/ пр
ГАе+1 = 1/ Ag'Cr0<
[Ag ] V [сю—]
Следовательно, растворимость равна:
PAgscro4 = -^f- = 4" УЩ^ = 2'25-10"6 мом/л
Из этого следует, что растворимость Ag2Cr04 в 0,1 М растворе Na2Cr04, т. е. при
наличии избытка Ст01 , уменьшилась до 2,25-10~"6 моль/л, т. е. избыток Ag~^~ и СтО^
по-разному влияет на растворимость осадка Ag2Cr04.
Из приведенных примеров следует, что растворимость малорастворимого осадка в воде
больше, чем в растворе электролита, имеющем общий ион с осадком, при условии, если
осадок не растворяется в избытке осадителя; избыток различных осадителей неодинаково
уменьшает растворимость электролита.
В свете современных научных положений расчеты изменения растворимости
электролитов, связанные с влиянием одноименных ионов, верны только для тех
концентраций и тех электролитов, для которых коэффициенты активностей ионов равны единице,
т. е. для случаев, когда концентрации равны активностям и / -> 1.
Когда коэффициенты активностей ионов значительно отличаются от единицы,
вычисления ведут с учетом влияния ионной силы раствора. Например:
Р«А. = [«Ч--^ (9)
или
Рк1А„ = [Ап-] = -^^- (10)
И Т. П.
Пример 3. Вычислить растворимость AgCl в 0,1 М растворе NaCl с учетом и без
учета коэффициента активности.
Решение, а) С учетом коэффициента активности. В 0,1 М растворе AgCl fx = 0,1
и fx = 0,76:
1 7-10"10
PAgci = [Ag+1 = ю-х.о.76* = 2>9'10"8 МАЬ1Л
б) Без учета коэффициента активности:
1 7-10-1°
PAgCl = [Аб+] = 'ю-! = 1.7-10-* моль/л
В воде:
PAgci = 1,3-10'5 моль!л
Следовательно, растворимость AgCl в 0,1 М растворе NaCl по сравнению с его
растворимостью в воде уменьшается более чем в 7500 раз, если не учитывать коэффициент актив-
/ 1 3-Ю"5 \
ности ( 17.1Q-9 = 7647J, и приблизительно в 4500 раз, если при расчете раствори-
(13-Ю"5 \
¦ ' д = 4483) .
Таким образом, прибавление одноименных ионов оказывает сильное
влияние на растворимость малорастворимого электролита и
способствует более полному осаждению твердой фазы.
300 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
§ 6. Солевой эффект
На величину растворимости малорастворимых солей оказывают
влияние не только электролиты, имеющие с ними общие ионы. Так,
растворимость некоторых малорастворимых солей увеличивается в присутствии
других растворимых солей, не имеющих с ними общих ионов.
Например, растворимость PbS04 увеличивается в присутствии NaN03;
растворимость BaS04, SrS04, CaS04 увеличивается от прибавления в
раствор КС1, NaN03, KN03 и. т. д.
Чем больше концентрация прибавляемой соли, не имеющей общих
ионов с малорастворимым электролитом, тем больше повышается его
растворимость.
Описываемое явление известно под названием солевого эффекта и
связано с увеличением ионной силы раствора вследствие введения в него
других ионов.
Увеличение растворимости электролита вследствие увеличения
ионной силы раствора вызывается, согласно теории сильных электролитов,
изменением подвижности ионов, которое обусловлено межионным
притяжением (см. §6) и связано с изменением величины коэффициента активностей.
Если ввести в насыщенный раствор любого малорастворимого
электролита какую-либо соль, не имеющую с ним общих ионов, то межионные
силы взаимодействия возрастут. Вследствие этого коэффициенты
активностей ионов понизятся, и если они были равны единице, то станут меньше
единицы. Это приведет к тому, что величина произведения растворимости
малорастворимого соединения возрастает по сравнению с величиной
произведения растворимости данного электролита в чистой воде. Увеличение
произведения растворимости электролита в солевом растворе, насыщенном
посторонними ионами, вызывает повышенную растворимость электролита.
Таким образом, увеличение растворимости малорастворимых
электролитов в присутствии других растворимых солей (солевой эффект)
обусловливается уменьшением коэффициентов активности в связи с увеличением
ионной силы раствора вследствие прибавления посторонних электролитов.
Прибавление различных электролитов по-разному влияет на
растворимость малорастврримого электролита, так как значение ионной силы
раствора зависит не только от концентрации солей, но и величин зарядов
ионов.
Пример. Вычислить растворимость CaS04 в воде и в 0,1 М растворе NaCl.
Решение, а) Растворимость в воде:
pCaSo4 = /6,26- Ю-6 = 7,9- Ю-3 моль/л
б) Растворимость в 0,1 М растворе NaCl (ц, = 0,1; f2— 0,33):
PCaso4 = "оЖ ,/"6>26'10"5 = 2'4*10"* тль,л
Следовательно, растворимость CaS04 в 0,1 М растворе NaCl возрастает по сравнению
(2 4-10~2 \
7Q 1П-з ^3) .
§ 7. Влияние концентрации ионов водорода на растворимость
малорастворимых соединений
Ионы водорода оказывают сильное влияние на растворимость
различных соединений.
Заметное растворяющее действие на некоторые гидроокиси, оксикарбо-
наты и карбонаты оказывают также аммонийные соли сильных кислот,
растворы которых имеют кислую реакцию.
§7. ВЛИЯНИЕ [Н+] НА РАСТВОРИМОСТЬ МАЛОРАСТВОРИМЫХ СОЕДИНЕНИЙ 301
В кислой с?>еде растворимость малорастворимой соли тем больше,
чем больше ее произведение растворимости и чем больше концентрация
ионов водорода.
Так, например, для Са++, Sr++ и Ва++ наибольшую величину ПР имеет
оксалат бария (ПРвас2о4 = 1>7-10"7), наименьшую — оксалат кальция
(ПРсас2о4 = 3,8-10"9), оксалат стронция занимает промежуточное
положение (nPsrc2o4 == 5,6-10"8). Поэтому в кислой среде лучше
растворяется ВаС204, хуже — SrC204 и еще хуже — СаС204. Следует отметить,
что растворимость этих солей в солянокислой среде больше, чем в
уксуснокислой.
Растворимость малорастворимой соли в кислоте тем больше, чем
меньше константа электролитической диссоциации кислоты, анионы
которой входят в состав данной соли. Другими словами, чем больше
растворимость данного электролита в воде и чем слабее кислота, образующая
его, тем больше этот электролит растворяется в кислоте.
Так, в уксусной кислоте оксалат кальция (Кж2с2о4 = 5,9-10~2)
практически не растворяется, а карбонат кальция (ПРсасо, = 1,7-10~8;
Кш,со, = 4,13-10"7) — растворяется.
Расчет растворимости электролита в кислой среде. Пусть дана
малорастворимая соль слабой кислоты KtAn.
В водном растворе:
KtAn ^=± Kt+ + An-
осадок раствор
Если в этот раствор прибавить сильную кислоту, то ионы An* будут
связываться с Н+ образованием слабой кислоты НАп.
При этом равновесие нарушается и осадок будет растворяться, так
как
[Kt+] [An"] = nPKtAn
Каждому аниону An", присутствующему в растворе и связанному1
в молекулы НАп, соответствует катион Kt+, следовательно,
[Kt+] = [НАп] + [An-]
Подставляя значение [Kt+] из уравнения, выражающего nPKtAm
получим:
KtAn. = [HAn] + [An-]
но
Следовательно,
1Ап-]
[НАп] - ^ ^
nPKtAn _ [Н+] [An-L + [Ап_]
[АЛ"] *НАп
Умножив левую и правую части этого уравнения на [Ап~ ], получим:
npM„-.An-,-(JSi + 1)
откуда
[An
/nPKtAn
\ ^KtAn
)
РК<А.-'К'*>--ПЙ^-У ПРкЦт^+') <">
302 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Здесь [Н+] = Снап — [НАп], т. е. концентрация ионов водорода
равна разнице между общей концентрацией кислоты и ее недиссоцииро-
ванной частью.
В частном случае, когда [Н+] = Кнап
P*tAn = 1/2ПРК1Ап = 1.41 УЩ^Гп
Физический смысл уравнения (11) заключается в следующем:
растворимость малорастворимых солей слабых кислот (в том числе и гидроокисей
и основных солей, которые можно рассматривать как кислые соли
двухосновной кислоты Н20) в водных растворах сильных кислот при данных
температуре и давлении повышается по сравнению с их растворимостью
в чистой воде. Причем эта растворимость тем выше, чем больше
произведение растворимости малорастворимого электролита, чем выше
концентрация ионов водорода и чем меньше константа электролитической
диссоциации слабой кислоты, образующей данную соль.
При вычислении растворимости малорастворимых солей в кислотах
с учетом коэффициентов активностей применяют формулу:
P*tAn = К] = h "]/nPKtAn (-jgL f\ + ij (12)
Значение теории осаждения и комплексообразования в объемном
анализе. Теории осаждения и комплексообразования имеют большое
значение для объяснения многих явлений и процессов, наблюдающихся
в объемных методах осаждения и комплексообразования. Они дают
возможность:
1) точно вычислить растворимость любого малордстворимого
электролита;
2) вычислить потери определяемых ионов в точке эквивалентности,
вызываемые растворимостью осаждаемых соединений;
3) представить течение процессов осаждения и комплексообразования
в различные моменты титрования;
4) построить кривые титрования;
5) правильно выбрать индикатор;
6) рассчитать концентрацию применяемого индикатора;
7) проследить влияние разбавления и температуры на точность
объемно-аналитического определения.
Все эти вопросы рассматриваются ниже при описании отдельных
методов осаждения и комплексообразования.
§ 8. Кривые титрования в методе осаждения
Осаждение хлорида серебра. При титровании раствора, содержащего
ионы хлора, стандартным раствором нитрата серебра образуется осадок
AgCl. Зная величину произведения растворимости AgCl и концентрации
ионов титруемого (С1~) и стандартного (Ag+) растворов, можно легко
вычислить изменения [Ag+] и [С1~ ] в любой момент титрования.
Предположим, что 100 мл 0,1 н. раствора NaCl титруют 0,1 н.
раствором AgN03.
До начала титрования 0,1 н. раствора NaCl, полностью
диссоциирующего на ионы:
[Cl"]=CNaC1
PCI = —lg CNaC1 = —lg 10-i = l
$ 8. КРИВЫЕ ТИТРОВАНИЯ В МЕТОДЕ ОСАЖДЕНИЯ 303
По мере приливания к 0,1 н. раствору NaCl 50; 90; 99; 99,9 мл 0,1 н.
раствора AgN03 концентрация С1~ будет уменьшаться, а рС1 —
увеличиваться.
При прибавлении 50 мл 0,1 н. раствора AgN03 останется не
осажденным 50% NaCl, т. е. величина [С1~ ] (без учета разбавления) уменьшится
в 2 раза:
[СГ] =-щ-0,1 =5-Ю-2 г-ион/л
рС1 = 2 lg 10 — lg 5 = 1,3
При прибавлении 90 мл 0,1 н. раствора AgN03 останутся не
осажденными 10% С1""-ионов, т. е. величина [С1~ ] уменьшится в 10 раз:
1СГ] = ~Ш~'0Д = 10~2 г'ион,л'
pCl =2lg ю = 2
Рассуждая аналогично, можно вычислить величину [С1~ ] и для
остальных случаев титрования.
При прибавлении 99 мл 0,1 н. раствора AgN03
[СГ] = -^-0,1 = 10-з г.тн,л
рС1 =3 lg 10 = 3
При прибавлении 99,9 мл 0,1 н. раствора AgNQ3
[СГ] =-Ь1_.0,1 = Ю-4 г-ион/л
рС1 = 4 lg 10 = 4
Так как [Ag+] [CL"] = nPAgci = 1,7-Ю"10
pAg + pCl = 10 — lg 1,7 = 9,77
Следовательно в процессе титрования по мере приливания 0,1 н.
раствора AgN03 получим следующие значения:
pCl 1,3 2 3 '4
pAg 8,47 7,77 6,77 5,77
[Ag+ ], г-ион/л 3,4-10-9 lJ-lO"8 lJ-lO"7 1,7-10-6
При добавлении 100 мл 0,1 н. раствора AgN03 наступает точка
эквивалентности. Однако в силу растворимости AgCl в растворе находятся
и ионы хлора и ионы серебра в количестве, соответствующем насыщенному
раствору AgCl. В этот момент
[Ag+] = [СГ] = ]/ПРАеС1 = 1Л.7-10-10 = 1,303-10-* г-ион/л
pAg = pCl =5 — lg 1,303 = 4,885
или в общем виде:
pKt = pAn = -V2lgnPKtAn
В процессе дальнейшего прибавления AgN03 в растворе появляется
избыток ионов серебра и вследствие этого растворимость AgCl
уменьшается.
При добавлении 0,1 мл избытка 0,1 н. раствора AgN03
[Ag+] =-^--0,1 = 10'* г-ион/л
pAg = 4; pCl = 5,77; [СГ] = 1,7-10~6 г-ион/л
304
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
При добавлении 1 мл избытка 0,1 н. раствора AgN03
1
[Ag+]
100
•0,1 = 10"3 г-ион/л
pAg = 3; pCl = 6,77; [CI"] = 1,7-10~7 г-ион/л
При добавлении 10 мл избытка 0,1 н. раствора AgN03
10
[Ag+]
100
•0,1 = 10~2 г-ион/л
pAg = 2; pCl = 7,77; [СГ] = 1,7-10"8 г-ион/л
Таким образом, от прибавления избытка AgNOg растворимость осадка
AgCl значительно и резко уменьшается. При прибавлении 0,1 мл 0,1 н.
раствора AgN03 после достижения точки эквивалентности концентрация
ионов хлора понижается от
[СГ] = [Ag+] = 1,303-10"5 г-ион/л
ДО
[C1-J--
ПР
AgCl
[Ag+
1,7-IP"10 1,7-Ю-10 , _ 1П_б
— 10-4—= 1,7-10 6 г-ион/л
0,1
100
0,1
1,303-Ю-5 пп
Т' е' В 1,7.10-е % 7»7 Раза-
При прибавлении 10 мл избытка 0,1 н. раствора AgN03 концентра-
1 303*10~5
ция ионов хлора понизится в ' Q_8 ^r 770 раз.
Полученные данные сведены в табл. 26.
ТАБЛИЦА 26
Титрование 100 мл 0,1 н. раствора NaCl 0,1 н. раствором AgN03
(без учета разбавления)
Прибавлено 0,1 н.
раствора
AgN03, мл
0
50
90
99
99,9
100*
100,1
101
ПО
Осталось
NaCl, %
100
50
10
1
0,1
0
0,1
(изб. AgN03)
1
(изб. AgN03)
10
(изб. AgN03)
[СГ[
ю-1
5-Ю-2
ю-2
ю-3
Ю"4
1,303-Ю-5
1,7-10-в
1,7-10"7
1,7-Ю-8
pCl
1
1,3
2
3
4
4,885
5,77
6,77
7,77
[Ag+]
—
3,4-Ю-8
1,7-10"8
1,7.10-7
1,7.10-6
1,303-10~б
10"4
10"3
ю-2
pAg
—
8,47
7,77
6,77
5,77
4,885
4
3
2
ДрС1
—
W-- 0,006
^=0,0,8
4-=о,11
тйг-w
^Г =8,865
^- = 8,85
w-M
-j—0.И
* Точка эквивалентности.
§8. КРИВЫЕ ТИТРОВАНИЯ В МЕТОДЕ ОСАЖДЕНИЯ 305
Построение кривой титрования на основании данных расчета рС1
и pAg. Строят систему координат, откладывая на оси абсцисс процентное
содержание хлорида натрия, оставшегося в растворе в разные моменты
титрования, и добавленного избытка нитрата серебра, а на оси ординат
отвечающие им значения рС1.
В целях уменьшения размеров кривой и для большей наглядности
строят наиболее характерную часть кривой, лежащую в пределах от 10%
остатка NaCl до 10% избытка AgN03.
В результате получают кривую осаждения, показанную на рис. 57.
Анализ кривой титрования в методе осаждения. Кривая титрования
NaCl раствором AgN03 симметрична в отношении точки эквивалентности.
Линия
о —
/
2
3
4
5
6
О 7
°- 8
9
10
11
12
13
14-
SO 100 НО
Объем раствора AgNCX,, мл
Рис. 57. Кривая титрования 0,1 н. раствора NaCl 0,1 н.
раствором AgN03.
Эта кривая дает возможность проследить изменения рС1 и pAgB различные
моменты процесса титрования. В начале титрования рС1 раствора
изменяется очень медленно, вблизи точки эквивалентности — очень быстро
(скачкообразно).
Другими словами, если в начале титрования прибавление
титрованного раствора AgN03 в количествах, измеряемых миллилитрами, мало
изменяет рС1 раствора, то под конец титрования, вблизи точки
эквивалентности, большое значение приобретают уже капли стандартного раствора
AgN03, быстро (скачкообразно) изменяющие рС1 титруемого раствора.
Изменение величины рС1 наглядно выражают в виде отношения
ДрС1/ДС, где ДрС1 — приращение показателя С1"-иона, наблюдаемое
на данном отрезке титрования; ДС — количество прибавленного
титрованного раствора в миллилитрах или процентах.
Это отношение показывает увеличение рС1 при добавлении 1 мл (1%)
раствора AgN03, требуемого для осаждения С1~ в виде AgCl. Как видно
из табл. 26, это отношение достигает максимума в точке эквивалентности.
Чем больше величина ДрС1/ДС в точке эквивалентности, тем больше
скачок титрования и тем точнее титрование.
Влияние концентрации титруемого и стандартного растворов на
характер получаемрй кривой титрования. Рассмотрим титрование 100 мл
0,01 н. раствора NaCl 0,01 н. раствором AgN03. В этом случае до начала
ТИТрОВаНИЯ рС1 = —lg СыаС! = 2.
Величины рС1 перед точкой эквивалентности представлены в табл. 27.
экоиоалентности
Точка эквивалентности
306
IVI.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
ТАБЛИЦА
Влияние концентрации титруемого и стандартного растворов на изменение
рС1 при титровании 100 мл раствора NaCl 100 мл раствора AgN03
27
Прибавлено
раствора AgNOs
мл
0
50
90
99
99,9
100*
100,1
101
110
Осталось NaCl
%
100
50
10
1
0,1
0
0,1 (изб. AgN03)
1 (изб. AgN03)
10 (изб. AejNO,)
1 н.
0
0,3
! 1
2
3
4,885
6,77
7,77
8,77
рС1
0,1 н.
1
1,3
2
3
4
4,885
5,77
6,77
1 7,77
0,01 н.
2
2,3
3
4
4,73
4,885
5,04
5,77
6,77
Точка эквивалентности.
Эти величины вычислены аналогично данным, приведенным в табл. 26,
с той лишь разницей, что в этом случае в силу большого разбавления
титруемого раствора (при титровании сильно разбавленных растворов
сильно разбавленными стандартными растворами) при расчете [С1 ]
и [Ag+] вблизи точки эквивалентности учитывают растворимость AgCl и
пользуются более точными уравнениями, позволяющими вычислить
растворимость малорастворимого электролита в присутствии избытка
осаждающего реактива:
PAgCl — ¦
PAgCl — '
[CI-
1приб.
+
/
[CI"
l2
*приб.
+ nPAgCi
[Ag+]n
[риб.
+
У
[Ag+]2
¦ПР
AgCl
Например, при расчете [С1~ ] в случае добавления 99,9 мл 0,01 н.
раствора AgN03 к 100 мл 0,01 н. раствора NaCl (т. е. когда в растворе
остается избыток в 0,1 мл 0,01 н. раствора NaCl) получают:
PAgCl —
0,1-0,01
100-2
+
// 0,1-0,01 \2
I "У > +1.7-10-»-
= -_о,5-Ю-6 + 1Л.95-10"10 = 0,896-Ю-5 г-ион/л
Следовательно, pAg = 5 — lg 0,896 = 5,04.
Так как pAg + pCl = 9,77, то pCl = 9,77 — 5,04 = 4,73.
В точке эквивалентности pAg = pCl = 4,885.
Вблизи точки эквивалентности, когда добавлено 0,1 мл избытка 0,01 н.
раствора AgN03, растворимость хлррида серебра составляет:
PAgCl — "
0,1-0,01
100-2
+
ft 0,1 0,01 \2
V [ 'Г } +1.7-
Ю-10 = 0,896-10-5 г-ион/л
Следовательно, pCl = 5 — lg 0,896 = 5,04, a pAg = 4,73.
Другие значения pCl после достижения точки эквивалентности
характеризуются значениями, приведенными в табл. 27
§ 8. КРИВЫЕ ТИТРОВАНИЯ В МЕТОДЕ ОСАЖДЕНИЯ
307
Для сравнения в табл. 27 показаны изменения рС1, наблюдаемые при
титровании 100 мл 1 н. 0,1 н. и 0,01 н., растворов NaCl растворами AgN03
такой же концентрации. Данные этой таблицы представлены также
графически на рис. 58.
Анализ кривой (рис. 58) и данных табл. 27 позволяет сделать
заключение, что чем выше концентрация титруемого и стандартного растворов,
тем больше скачок титрования:
для 1 н. растворов скачок
титрования составляет
для 0,1 н. растворов скачок
титрования составляет
для 0,01 н. растворов скачок
титрования составляет
6,77—3=3,77
5,77—4=1,77
5,04—4,73=0,31
Следует, однако, иметь в виду, что скачок титрования достаточно
велик и при титровании 0,1 н. раствора. Поэтому не следует прибегать
к титрованию растворов более высокой концентрации, чем 0,1 н. Ошибка
титрования в результате неточности измерений при пользовании более
концентрированными растворами значительно возрастает по сравнению
с титрованием менее концентрированными растворами.
Линия
эквивалентности
Точка эквивалентности
Объем раствора AdN03, мл
Рис. 58. Влияние концентрации титруемого (NaCl) и
стандартного (AgN03) растворов на ход изменения рС1.
Влияние растворимости выпадающего осадка на характер получаемой
кривой титрования. Соединения, образующиеся в процессе титрования
по методу осаждения, относятся к числу малорастворимых. Однако
ошибки, вызываемые их растворимостью, могут искажать результаты анализа.
Поэтому растворимостью все-таки нельзя пренебрегать.
Влияние величины растворимости выпадающего осадка на величину
скачка титрования показано в табл. 28.
На основании данных табл. 28 построены кривые осаждения (риге. 59).
Анализ кривой (рис. 59) и данных табл. 28 позволяет сделать
заключение, что чем меньше растворимость осаждаемого соединения, тем больше
скачок титрования. Например, при титровании 0,1 н. раствора:
NaCl скачок титрования составляет 5,77—4=1,77
NaBr скачок титрования составляет 8,42—4=4,42
Nal скачок титрования составляет 12,07—4=8,07
308
ГЛ.1У. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Скачок титрования достаточно велик во всех рассмотренных случаях
титрования.
Получаемые кривые симметричны в отношении точек
эквивалентности.
Титрование смеси галогенидов. В процессе титрования смеси гало-
генидов (I" + Вг-; I" + С1"; Вг" + С1"; Г + Вг" + С1~) стандарт-
Линия
эквивалентности
Точка эквивалентности
^ при титровании NaCl
ев
Скачок
тигррованияк
й:
Точка эквивалентности
' пРи титровании NaBr
Точка эквивален',., а —
ноет и при титр овании^Ш.
SO 100 ttO
Объем раствора AdNOj, мл
Рис. 59. Влияние величины растворимости выпадающего
осадка на величину скачка титрования:
/ — кривая титрования Nad; 2 — кривая титрования NaBr;
3 — кривая титрования Nal.
ным раствором нитрата серебра сначала осаждается Agl, отличающийся
наименьшей растворимостью, затем AgBr и, наконец, AgCl,
характеризующийся наибольшей растворимостью среди галогенидов серебра.
ТАБЛИЦА 28
Влияние растворимости выпадающего осадка на величину скачка
титрования при титровании 100 мл 0,1 н, раствора NaHal
100 мл 0,1 н. раствором AgN03
Прибавлено
раствора AgNOs
мл
0
50
90
99
99,9
100*
100,1
101
ПО
Осталось NaHal
%
100
50
10
1
0,1
0
0,1 (изб. AgN03)
1 (изб. AgNO,)
10 (изб. AgN03) .
ЫаС1(ПРАдС1 =
= 1,7.1(Г10)
1
1,3
2
3
4
4,885
5,77
6,77
7,77
pHal
при титровании
NaBr(nPAgBr =
= 3,3.10"13)
1
1,3
2
3
4
6,211
8,42
9,42
10,42
Nal (nPAgI =
= 8,5.10"^)
1
1.3
2
3
4
8,035
12,07
13,07
14,07
* Точка эквивалентности.
§ 8. КРИВЫЕ ТИТРОВАНИЯ В МЕТОДЕ ОСАЖДЕНИЯ
309
Однако до полного завершения процесса осаждения Agl, когда [I"]
сильно уменьшится, а [Вг~] останется прежней, произведение [Ag+] [Вг" ]
достигает величины произведения растворимости nPAgBr и начинает
выпадать также осадок AgBr. Это происходит вследствие того, что [I"]
достигает такой малой величины по сравнению с большой величиной [Вг" ],
когда создаются необходимые предпосылки для образования AgBr.
Чем меньше [1~] и чем больше [Вг" ] в смеси галогенидов, тем скорее
достигается IlPAgBr и начинается осаждение AgBr.
Покажем это на примере титрования смеси 0,1 н. растворов
галогенидов раствором нитрата серебра.
В случае прибавления эквивалентного количества раствора AgN03,
необходимого для осаждения иодида, концентрация оставшихся в растворе
ионов серебра может быть вычислена по формуле:
[Ag+] = [I-] = PAgI = 1/ПР^7 = 1^8,5-10"17 = 9,2- 10-э г-ион/л
При наличии бромида, концентрация которого в растворе 10"1 г-ион/л,
[Ag+] = 9,2-10~9 г-ион/л вполне достаточна, чтобы начал выпадать
осадок AgBr, произведение растворимости которого 3,3-10~13, т. е. меньше
произведения 9,2• 10~9• 10"1 = 9,2-10~10. Это означает, что осаждение
AgBr, по-видимому, начинается значительно ранее точки эквивалентности,
при которой практически завершается выпадение в осадок Agl.
При [Вг~ ] = 10"1 г-ион/л для осаждения AgBr концентрация серебра
(из уравнения [Ag+] [Br"] = nPAgBr) должна быть:
[Ag+) = J^rer. = 3^ _33.10.и г_ион/д
а эта [Ag+] будет достигнута, когда в растворе [I" ] (из уравнения [Ag+jx
[I~] = nPAgi) будет равна:
Таким образом, AgBr станет выделяться из смеси I" + Вг" раньше
достижения полноты осаждения Agl, т. е. до первой точки
эквивалентности, так как 2,6-10"5 > 9,2-10"9.
Осадок Agl в присутствии Вг" выпадает из раствора до тех пор, пака
отношение [Вг"]/[1"1 в растворе не станет равным отношению
произведений растворимости AgBr и Agl. При дальнейшем приливании
стандартного раствора AgN03 выпадают AgBr и Agl, причем отношение
[Вг" ]/ [I" ] остается постоянным:
[Br] nPAgBr 3,3-10-»
Ш ~~ ПРАе1 ~ 8,5.10-" ~4UUU
т. е. как только [I" ] станет в 4000 раз меньше, чем [Вг" ], начнет выпадать
AgBr.
Осадок AgBr в присутствии С1" выпадает из раствора до тех пор,
пока отношение [СГ~ ]/ [Вг" ] в титруемом растворе не станет равным
отношению произведений растворимости AgCl и AgBr:
[СГ] _ nPAgC} 1,7.10-ю
[Вг-] - nPAgBr 3,3-10-13 ~оии
т. е. как только величина [Вг" ] станет в 500 раз меньше величины [С1~ ],
начнет выпадать AgCl.
310
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Таким образом, AgCl начинает выпадать до полного завершения
процесса осаждения AgBr, а именно, в момент, когда [Ag+] [Cl"~] достигнет
величины, равной ПРа6сь
Отсюда следует, что дифференцированное (раздельное) титрование С1",
В" и I" при их совместном присутствии вполне осуществимо. Наиболее
точных результатов можно достичь при дифференцированном титровании
смеси I" и С1".
Линия
эквивалентности
„Точка эквивалентности
30 100 ПО
Объем раствори оксалата (хромата) натрияг мл
Рис. 60. Кривая титрования 0,1 н. раствора AgN03 0,1 н.
раствором оксалата {хромата) натрия.
Таким образом, при титровании смеси галогенидов (СГ, Вг", I"), равно
как и других смесей ионов, осаждаемых одним и тем же реактивом,
осаждение малорастворимых соединений происходит ступенчато: сначала
выпадает наименее растворимый осадок — продукт наиболее
чувствительной реакции, затем более растворимый и, наконец, наиболее
растворимый осадок. При известных сочетаниях концентрации осаждаемых ионов
достигают количественного их разделения.
Влияние температуры водного раствора на изменение рС1 и pAg в
процессе титрования NaCl раствором AgN03. С увеличением температуры
водного раствора растворимость галогенидов серебра значительно
повышается. Вследствие этого точность определения понижается. Чем больше
растворимость осаждаемого соединения, тем больше ионов остается в
растворе в точке эквивалентности. Так как в процессе
объемно-аналитического определения нельзя приливать избыток осадителя, уменьшающего
растворимость осадка, то концентрация остающегося в растворе
определяемого иона может достигать значительной величины. С целью уменьшения
растворимости понижают температуру титруемого раствора до 10—15° С.
Осаждение хромата, оксалата и других солей серебра типа Kt2An.
При титровании солей типа Kt2An получаются кривые не симметричные
в отношении точки эквивалентности (рис. 60). В этом случае вычисление
[Kt+] и [An"""] проводят при помощи формул:
при избытке Kt+.
при избытке An"
PKt2An = fAn""]=-
ПР
Kt2An
[Kt+]2
§9. ВЫВОДЫ, ВЫТЕКАЮЩИЕ ИЗ РАССМОТРЕНИЯ КРИВЫХ ОСАЖДЕНИЯ 311
Вблизи точки эквивалентности концентрации Kt+ или An""
рассчитывают по более сложным и точным формулам или вычисляют
графическим методом, при котором наносят значение растворимости Kt2An
в присутствии избытка Kt+ или Ап"~ на оси ординат, а известные
значения [Kt+] и [Ап~~]— на оси абсцисс.
Подробности подобных расчетов мы опускаем. В табл. 29 приведены
данные, иллюстрирующие процесс титрования 0,1 н. раствора нитрата
серебра 0,1 н. раствором электролита (оксалатом, хроматом и др.),
образующим с Ag+-HOHaMH осадки типа Ag2An с ПРАё2АП = 10~12.
На основании данных табл. 29 построена кривая осаждения (см. рис. 60).
Анализ кривой и данных табл. 29 показывает, что получаемая кривая
довольно пологая и не характеризуется столь резким скачком
титрования, какой наблюдается при осаждении солей типа AgHal при титровании
AgN03 солйми NaHal.
ТА ВЛИЦА 29
Титрование 0,1 н. раствором оксалата (хромата) 100 мл 0,1 н. раствора
нитрата серебра
Осаждено раствора
AgNOa, мл
0
90,0
99,0
99,9
100,0
Избыток с2о;р~~, %
0,1
1
Ag+
ю-1
ю-2
ю-3
1,69-10"4
1,26-10"4
1,0-Ю-4
4,4-10"6
pAg
1
2
3
3,77
3,90
4
4,36
рс2о4
8
6
4,46
4,20
4
3,28
ApAg
ДС
0,011
0,11
0,85
1,3
1
0,4
ДрС20*
ДС
0,022
0,22
1,70
2,6
2,0
0,8
Изменения pAg, наблюдаемые при каждом приливании очередной
порции раствора, содержащего осаждающий анион, в два раза меньше
изменений рАп.
Рассмотренный случай имеет большое значение для объяснения
явлений, наблюдаемых при осаждении методом Мора, основанным на
применении К2СЮ4.
§ 9. Общие выводы, вытекающие из рассмотрения кривых
осаждения
1. В начале титрования по мере приливания растворов, содержащих
осаждающие ионы, к титруемым растворам солей величины pKt и рАп
изменяются медленно. Незначительное изменение pKt и рАп в начале
титрования объсняется тем, что в растворе остается еще достаточно
большой избыток осаждаемых ирнов.
Под конец титрования вблизи точки эквивалентности наблюдается
резкий скачкообразный переход от одних значений рК и рАп к другим.
2. Величина скачка титрования, измеряемого высотой вертикального
участка кривой осаждения, как и в случае объемно-аналитических
определений по методу нейтрализации, играет решающую роль в процессе
титрования.
3. Скачкообразное изменение величин pKt и рАп уточки
эквивалентности выражают изменением величин ApKt/ДСи ДрАп/АС, показывающих
увеличение pKt или рАп при добавлении 1 мл (1 %) стандартного раствора,
312 ГЛ.IV. МЕТОДЫ ЬСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
требуемого для осаждения определяемого вещества. Чем больше величины
ApKt/AC и ApAn/ДС в точке эквивалентности, тем точнее титрование.
4. Резкость скачка титрования и, следовательно, точность определения
тем больше, чем больше концентрация титруемого и стандартного
растворов и чем ниже их температура.
5. Кривые осаждения электролитов типа KtAn симметричны
относительно точки эквивалентности. Кривые осаждения электролитов типа
Kt2An несимметричны.
6. При осаждении электролитов типа KtAn максимальное значение
ApKt/AC точно совпадает с точкой эквивалентности. При осаждении
осадков типа Kt2An отношение ApKt/AC приобретает максимальное
значение раньше достижения точки эквивалентности.
7. Чем меньше растворлмость осаждаемого соединения, тем точнее
титрование. Для бинарных соединений типа AgHal точность определения
возрастает в следующем порядке:
AgCl -^ AgSCN -> AgBr -> Agl
8. При титровании смесей ионов, осаждаемых одним и тем же
реактивом, изменение концентраций ионов у первой точки эквивалентности
определяется отношением произведений растворимости ПРК(АП1 и ПР^апц
и отношением концентраций осаждаемых ионов Агц и Апп.
9. Пользуясь формулами, представленными в § 8, можно:
1) построить кривую осаждения;
2) с большой точностью рассчитать значения pKt и рАп, при которых
необходимо закончить данный процесс титрования.
Пользуясь ранее выведенными соотношениями истинных констант
с обычными и формулами, позволяющими перейти от [Kt+] и [An"]
к aKt+ и #Ап-, можно перейти от концентраций к активностям.
§ 10. Адсорбционные явления, наблюдаемые
при титровании по методу осаждения
В процессе осаждения, фильтрования и промывания некоторых
осадков образуются коллоидные высокодисперсные микрогетерогенные
(неоднородные) системы (коллоидные растворы), сохраняющие поверхность
раздела между дисперсной фазой и дисперсионной средой, которые
отличаются от истинных растворов рядом характерных свойств (см. книга 1,
гл. VI, § 16).
Свойства коллоидных систем, встречающихся и используемых в
химическом анализе. Процессы образования и разрушения коллоидных систем,
а также их свойства всесторонне рассматриваются в коллоидной химии.
Здесь мы кратко рассмотрим лишь те стороны этого вопроса, которые
имеют непосредственное отношение к аналитической химии.
Характерные свойства коллоидных систем связаны в основном с
поверхностными явлениями на границе раздела фаз в этих системах.
Они определяются дисперсностью и характером физико-химического
взаимодействия, протекающего между диспергированным веществом
и дисперсионной средой на границе раздела фаз.
Коллоидным системам присущи электрические свойства,
обусловливаемые возникновением заряда на поверхности раздела — образование
двойного электрического слоя ионов, и связанные с ними адсорбционные
свойства; оптические свойства — рассеяние света, имеющие важную роль
в нефелометриче^ких и турбидиметрических методах анализа; структурно-
механические свойства, выражающиеся в возникновении пространствен-
§ 10. АДСОРБЦИОННЫЕ ЯВЛЕНИЯ ПРИ ТИТРОВАНИИ 313
ных сеток (структур) — каркасов с различной степенью заполнения
системы частицами дисперсной фазы (переход золь—гель в коллоидных
системах).
Адсорбционные свойства коллоидных частиц. Взвешедные частицы
коллоидных систем несут положительные или отрицательные заряды.
Зарядность коллоидных частиц обусловливается адсорбцией на их
поверхности анионов (отрицательный заряд) или катионов (положительный
заряд). Например, сульфиды адсорбируют S""- БН'-ионы и осаждаемые
катионы; галогениды серебра —Ag+- или Hal "-ионы; гидроокиси —ОН~-
ионы и осаждаемые катионы и т. д.
Явление адсорбции одних ионов сопровождается одновременным
притяжением других ионов, противоположных им по знаку. Например,
при медленном осаждении АГ++ в виде А1 (ОН)3 действием NaOH осадок
гидроокиси алюминия адсорбирует на своей сильно развитой поверхности
молекулы воды и положительно заряженные ионы: Al+ ++, [A1 (ОН)]+ + ,
[А1(ОН)2К
При этом коллоидная частица заряжается положительно.
Положительно заряженные частицы притягивают анионы (например,
ОН-, CI" и др.).
По мере прибавления избытка NaOH частицы А1 (ОН)3 начинают
адсорбировать гидроксильные ионы, заряжаясь отрицательно, и
образуют алюминат-ионы: [Al (OH)J-, [А1 (ОН)5]-_, [А1 (ОН)6] и т. д.
Эти частицы притягивают катионы, например Na + , Mg++, Ca*+ и др.
Таким образом, заряд взвешенной коллоидной частицы определяется
зарядом ионов, адсорбированных на ее поверхности и зависит от условий
образования золей. Коллоидные частицы, включающие непосредственно
связанные с ними молекулы и ионы, и находящиеся в растворе
противоположно заряженные ионы, образуют так называемые мицеллы.
Мицеллы представляют собой частицы дисперсных фаз в коллоидных
системах и являются сложными комплексами, состоящими из многих
тысяч атомов, молекул и ионов. Мицеллы состоят из нерастворимого
в данной среде ядра, окруженного двойным электрическим слоем ионов.
Один слой ионов, называемый адсорбционным, находится на поверхности
ядра, сообщая ему электрический заряд. В состав адсорбционного слоя
входит также часть ионов противоположного знака, осдовная масса которых
образует второй слой ионов. Мицеллы окружены сольватной оболочкой.
Алгебраическая сумма зарядов мицеллы равна нулю, т. е. мицелла
электронейтральна.
При изображении формул мицелл часть формулы, соответствующей
ядру мицеллы, заключают в квадратные скобки, а часть формулы,
соответствующей ядру и адсорбированному слою — в фигурные.
Например, строение мицелл, образуемых Al (OH)3; As2S3 и кремневой
кислотой изображают следующими формулами:
Мицелла А1 (ОН)3 в нейтральной среде
{[А1 (ОН)3]т-/гА1 (ОН)2+•[/?--*) С1~ }*+.*С1~
J - 5
Мицеллы А1 (ОН)з в щелочной среде:
{[А1 (ОН)3]т^А1 (ОН)^ (п — х) Na+}*-.*Na+
} 2 3
Мицеллы As2S3 в кислой среде
{[As2S3]m • nSH-. (ft — х) Н+ }х~ хН+
/ 2 Т
314 ГЛ.IV- МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Мицеллы кремневой кислоты:
{[Si02.jH20]m.nSiO;~ • 2(п — х)Н+}2х~.2хН+
? 2 ~5
где / — ядро мицеллы;
2 — ионы, определяющие положительный или отрицательный заряд золя;
3 — ионы противоположного знака, из которых х образует второй слой ионов.
Коагуляция. Коллоидные частицы могут соединяться (свертываться,
коагулировать) в более крупные агрегаты. Но самопроизвольное их
свертывание (старение) под влиянием физико-химических процессов,
протекающих в системе во времени (например, при стоянии), совершается
медленно.
Процесс образования более или менее крупных агрегатов приводит
в конце концов к образованию выпадающих в осадок или всплывающих
в дисперсионной среде хлопьев или к возникновению сплошной и рыхлой
массы геля (см. ниже), с которым соосаждаются ионы, не осаждаемые
в нормальных условиях данным осадителем. Увлеченные примеси с
трудом удаляются при промывании таких осадков.
Заряженные коллоидные частицы можно превратить в более крупные
частицы специальными приемами. Процесс соединения коллоидных частиц,
сопровождающийся укрупнением частиц, называют коагуляцией.
Коагуляции противодействуют электрические заряды коллоидных
частиц и сольватация их поверхности. Устойчивость коллоидных частиц
понижается в результате нейтрализации электрического заряда и
уменьшения сольватации, что происходит при добавлении в раствор
электролитов (коагуляторов, или коагулянтов). В химическом анализе для
образования гидрогелей применяют большей частью водные растворы кислот
или солей аммония. На степень коагуляции сильно влияет заряд катиона
или аниона прибавляемого электролита. Чем больше заряд ионов, тем
сильнее действие электролита. Например, при коагуляции отрицательно
заряженного золя — сернистого мышьяка — наиболее слабое действие
оказывает NH4C1, сильнее действует ВаС12 и еще лучше — А1С13. В
случае положительно заряженного золя (например, гидроокиси алюминия)
NH4C1 действует слабее, чем (NH4)2S04.
Таким образом, на положительно заряженные частицы золей
коагулирующее действие оказывают анионы, на отрицательно заряженные —
катионы, причем, чем выше заряд аниона или катиона, тем большее,
коагулирующее действие они оказывают на соответствующие золи.
Некоторые золи коагулируют лишь при добавлении концентрированных
растворов солей (высаливание) или при добавлении другого растворителя.
Повышение температуры раствора в ряде случаев способствует
коагуляции. Гели, образуемые из золей в процессе их коагуляции, представляют
собой дисперсные системы, отличающиеся развитой пространственной
структурой (сеткой) — каркасом. Пространственная сетка
характеризуется различной степенью объемного заполнения системы частицами
дисперсной фазы, .обусловливаемого силами молекулярного сцепления.
Коагуляция золей имеет большое значение в анализе. С целью
предотвращения образования золей при осаждении гидроокисей и сульфидов
катионов III аналитической группы сульфидом аммония к нему добавляют
хлорид аммония, способствующий коагуляции.
Коллоидные частицы, которые адсорбируют на своей поверхности
анионы слабых кислот, относительно легко коагулируют при действии
сильных кислот. Ионы водорода сильных кислот с анионами слабых
кислот образуют слабодиссоциирующие молекулы слабых кислот. В
результате этого заряд коллоидных частиц понижается, падая почти до
$ 10. АДСОРБЦИОННЫЕ ЯВЛЕНИЯ ПРИ ТИТРОВАНИИ 315
нуля, и коллоидные частицы коагулируют. Это правило в особенности
относится к золям типа сернистых соединений, кремневой кислоты
и т. п., например:
н+
{[As2S3]m-/*SH--(/i — х) H+}*--JtH+ —*• \ mAs2S3 + /*H2S t
Вследствие этого в сильнокислой среде происходит быстрая
коагуляция золей сернистого мышьяка, кремневой кислоты и т. п.,
сопровождающаяся образованием: As2S3, H2Si03 и т. п.
Коагуляцию можно также вызвать повышением температуры
(например, кипячением коллоидного раствора), действием света,
высокочастотных электромагнитных и ультразвуковых колебаний, путем
встряхивания, взбалтывания, перемешивания и пр.
Пептизация. При промывании и стоянии осадков или при действии
на осадки некоторых реагентов (пептизаторов) наблюдается явление,
обратное коагуляции. Это явление, называемое пептизацией, состоит
в том, что осадок переходит в золь. Примером пептизации под влиянием
пептизатора является расщепление агрегатов частиц гидроокиси железа
в водной среде ионами железа (III).
Пептизация, часто наблюдаемая при промывании осадков водой,
объясняется уменьшением концентрации коагулирующих ионов.
Например, пептизация наблюдается при промывании водой осадка сульфида
никеля. Для предупреждения пептизации осадки промывают не водой,
а растворами соответствующих электролитов. Так, осадок NiS промывают
раствором хлорида аммония, содержащим NH4OH + (NH4)2S. Однако
следует иметь в виду, что один и тот же электролит может в зависимости
от концентрации оказывать как стабилизирующее, так и коагулирующее
действие.
Коллоидные системы, применяемые в химическом анализе. Из
коллоидных систем наибольшее значение для химика-аналитика имеют
гидрозоли — двухфазные микрогетерогенные дисперсные системы,
характеризующиеся предельно высокой дисперсностью, в которых дисперсионной
средой является вода — наиболее часто применяемый в аналитической
практике растворитель. В связи с применением неводных растворов
встречаются также органозоли, в которых дисперсионной средой являются
неводные (органические) растворители.
В результате молекулярного сцепления частиц дисперсной фазы из
золей при их коагуляции образуются гели. При этом не происходит
разделения фаз; другими словами, переход золей в гель не является фазовым
превращением.
При образовании геля вся дисперсионная среда (например, вода
в гидрозоле) прочно связывается поверхностью частиц дисперсной фазы
и в ячейках пространственной структуры геля.
Гели способны обратимо восстанавливать свою пространственную
структуру во времени, но после высушивания наступает разрушение их
структуры, и они теряют указанную способность.
Коллоидные свойства галогенидов серебра. В процессе титрования
галогенид-ионов растворами солей серебра получаются галогениды
серебра, весьма склонные к образованию коллоидных растворов. В
присутствии избытка Hal"-ионов, т. е. до точки эквивалентности при
титровании галогенидов ионами серебра или после точки эквивалентности при
титровании ионов серебра галогенидами вследствие адсорбции Hal "-ионов
взвешенные частицы AgHal приобретают отрицательный заряд:
т AgHal + /zHai" -* [ AgHal ]mnHal"
316 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
В присутствии избытка Ag +-ионов (т. е. до точки эквивалентности при
титровании ионов серебра галогенидами или после точки эквивалентности
при титровании галогенидов ионами серебра) взвешенные частицы
приобретают доложительный заряд:
т AgHal + n Ag+ —> [AgHalJm./i Ag+
Таким образом, заряд взвешенной коллоидной частицы [AgHal ]m X
X п Hal" или [AgHal]m• п Ag+ определяется зарядом ионов,
адсорбированных на поверхности ядра мицеллы [AgHal ]m, и зависит от наличия
в системе избытка НаГ или Ag + , обусловливающих отрицательный или
положительный заряд взвешенной частицы золя.
Помимо адсорбционного слоя, находящегося на поверхности ядра
мицеллы и обусловливающего определенный электрический заряд, в
состав мицеллы входит также часть ионов противоположного знака,
образующих второй (внешний) слой ионов.
Например, в процессе титрования иодида калия раствором нитрата
серебра
Ag+ + NO" + К+ + Г-> | Agl + К+ + NO;
образуются мицеллы следующего строения:
а) мицеллы, образуемые Agl при избытке нитрата серебра
{[AgI]m.nAg+ • (n-x)NQ-3\x*.xNQ-
1 ' 2 3
б) мицеллы, образуемые Agl при избытке иодида калия:
{[AgI]m^I-v(/z-A:)K+^--A:K+
; 2 "3
Коллоидные частицы, несущие одноименные электрические зарядЬг,
отталкиваются друг от друга. Силы взаимного отталкивания мешают
частичкам сблизиться настолько, чтобы произошло взаимное притяжение.
В то же время заряженные частички обладают высокой адсорбционной
способностью, они притягивают к себе частицы, несущие обратные по знаку
электрические заряды, и образуют с ними малорастворимые соединения.
В первую очередь на поверхности заряженных коллоидных частиц
адсорбируются те из ионов, которые дают наименее растворимые осадки с
ионами, входящими в состав этих частиц. Кроме того, адсорбируются те
из ионов, концентрация которых наибольшая. Например, при осаждении
Agl могут соосаждаться вместе с ним Br", CI", SCN" и другие ионы.
При титровании галогенидов, не содержащих посторонних примесей,
осадком адсорбируются имеющиеся в растворе Hal "-ионы, сообщая
частичкам AgHal отрицательные заряды. И в том, и в другом случаях результаты
титрования искажаются. Поэтому требуется строго соблюдать условия
осаждения, рекомендуемые в методиках определения тех или иных
веществ.
Адсорбционные индикаторы. Образование коллоидов и
сопутствующие этому процессу явления используют в количественном анализе. На
использовании коллоидных свойств галогенидов серебра основано
применение так называемых адсорбционных индикаторов. Адсорбционные
индикаторы представляют собой органические соединения, являющиеся
слабыми кислотами, диссоциирующими согласно уравнению:
H2Ind^±2H+ + Ind--
§ 10, АДСОРБЦИОННЫЕ ЯВЛЕНИЯ ПРИ ТИТРОВАНИИ
317
Анионы этих индикаторов, адсорбируясь на поверхности положительно
заряженных коллоидных частиц, выпадающих в процессе титрования
осадков, вызывают изменение цвета поверхности этих осадков. Если
изменение цвета происходит вблизи точки эквивалентности, то можно
использовать такие адсорбционные индикаторы для установления конца
титрования.
Наиболее известными представителями этого класса органических
соединений являются флуоресцеин и эозин.
Водно-щелочные растворы двунатриевой соли флуоресцеина окрашены
в красновато-желтый цвет, отличающийся интенсивной флуоресценцией,
которая сохраняется даже при сильном разбавлении:
свн4соон г с6н4соо- -|
I II I I
флуоресцеин
(хиноидная форма желто-
коричневого цвета)
I II I I
2Na+
флуоресцеин
(двунатриевая соль красновато-
желтого цвета)
Эозин представляет собой тетрабромфлуоресцеин:
С6Н4СОО-
3\/\/Ч/Ч/В
I II I I
"o/YVY4°
Br
Br
2Na+
эозин
(двунатриевая соль красного цвета;
При титровании, например ионов хлора, стандартным раствором
нитрата серебра образуется осадок AgCl. Частицы осадка хлорида серебра
адсорбируют на своей поверхности ионы хлора с образованием [AgCl ]m X
X п С1"". Адсорбция ионов хлора наблюдается до тех пор, пока в
титруемом растворе находятся свободные СГ-ионы. Поэтому, если в титруемый
раствор добавить флуоресцеина, индикатор не будет изменять окраски
до тех пор, пока в растворе имеется избыток С1~-ионов. Это объясняется
тем, что окрашенные анионы индикатора, несущие отрицательные заряды,
не адсорбируются отрицательно заряженными коллоидными частицами
осадка.
При осаждении галогенидов серебра в момент достижения точки
эквивалентности при самом незначительном избытке AgN03 осадок AgHal
адсорбирует избыток Ag+'Ионов и вместе с ними NOif-ионы:
nAgCl + Ag+ + NO; -> [AgCl]w . Ag+NO^
Так как NO^-ионы не образуют ни с одним из ионов, входящих в
состав осадка, малорастворимое соединение, то они не очень прочно
удерживаются частицами [AgCl ]m • п Ag+ и склонны замещаться другими
анионами, способными сильнее адсорбироваться осадком. Поэтому
коллоидные частицы, теперь уже несущие положительные заряды,
адсорбируют интенсивно окрашенные анионы индикатора. При этом осадок
окрашивается в розово-красный цвет, что служит сигналом для прекращения
дальнейшего титрования раствора хлорида.
318 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
В случае прибавления избытка С1"-ионов к раствору, в котором
взмучен розово-красный осадок, образуемый флуоресцеином [AgC\]m-n Ag+,
ионы серебра, адсорбированные на поверхности осадка AgCl, образуют
дополнительное количество AgCl. Избыток С1"-ионов адсорбируется AgCl
с образованием [AgCl]m-nCl~\ При этом розово-красная окраска осадка
исчезает. Бромиды и иодиды титруют в присутствии эозина, который
нельзя применять в качестве адсорбционного индикатора при титровании
хлоридов, так как при титровании хлоридов нитратом серебра осадок
краснеет до достижения точки эквивалентности.
На первый взгляд может показаться, что адсорбционные индикаторы
по сути дела являются индикаторами на Ag + -HOHbi, адсорбированные на
поверхности заряженных коллоидных частиц. В действительности это
не так. Во-первых, адсорбционные индикаторы могут реагировать с
другими положительно заряженными ионами, адсорбированными на
поверхности AgHal; во-вторых, если к AgN03 прибавить флуоресцеин, то цвет
индикатора не изменится. Но если в присутствии флуоресцеина начать
титрование нитрата серебра хлоридом натрия, то выпадающий осадок
хлорида серебра адсорбирует ионы серебра и вместе с ними анионы
флуоресцеина. При этом белый осадок становится розово-красным.
Таким образом, адсорбционные индикаторы вызывают изменение
окраски^ не в растворе, а на поверхности коллоидного осадка, несущего
положительный заряд.
Б. АРГЕНТОМЕТРИЯ
§ 11. Характеристика метода
Аргентометрический метод объемного анализа (см. § 1) основан на
применении в качестве осадителя стандартного раствора, содержащего
ионы серебра:
Ag+ + An- —> AgAn
Стандартные растворы. В качестве стандартных растворов для
определения анионов применяют нитрат серебра; для определения катионов
серебра — хлорид натрия.
Аргентометрическим методом пользуются главным образом для
количественного определения галогенид-ионов и ионов серебра.
Определение конечной точки титрования. В методе аргентометрии
применяют как индикаторные, так и безындикаторные способы
определения точки эквивалентности.
Способы равного помутнения и способ просветления подробно описаны
в §2.
Индикаторный способ. В качестве индикатора
применяют раствор хромата калия (метод Мора, см. § 2).
Применяемый в методе осаждения индикатор не должен изменять
цвет до тех пор, пока определяемые ионы полностью не перейдут в осадок.
Очевидно, этому условию может удовлетворять такой индикатор, который
образует с ионами титрованного раствора окрашенный осадок,
отличающийся большей растворимостью, чем растворимость основного
осаждаемого вещества. Индикатор должен быть достаточно чувствительным по
отношению к ничтожно малому избытку AgN03. Таким условиям более
или менее удовлетворяют хроматы и арсенаты щелочных-металлов.
Наиболее широко в аналитической практике применяют хромат калия.
Требуемую концентрацию индикатора вычисляют следующим образом.
§11. ХАРАКТЕРИСТИКА МЕТОДА
319
Например, при титровании хлоридов стандартным раствором AgN03
в момент, когда начинает осаждаться хромат серебра (см. § 5):
rAg+1 = nPAgC1
LAg J [C1.j
гпр:
m-Vip^
}
Следовательно
npAgci = [C1-] ^ 1,7-10-iQ
/nPAg2Cr04 V\oGT] VTW^
1,2.10-*
Подставляя значение [CI"] = ]AlPAgci = V 1,7-10~10 в приведенное
выше уравнение, получим:
J/"1,7-10-10
= l,210-4
откуда
Зная, чему равняется [Ag+] в точке эквивалентности, можно также
вычислить [СгО"] по формуле:
[Ag+]2[Cr04"]=nPAg2Cr04
откуда
LCr°4 J"" [Ag+]2
Так как
[Ag+]2 = [Crp = nPAgC1
то, подставляя значение IIPAgci = 1,7-10~10, получим
г --т 2-Ю-12
[^ ]= 1>7.10-io^1Q-2g^^
Следовательно, теоретически осадок хромата серебра в точке
эквивалентности образуется только в том случае, если концентрация
индикатора будет равна 0,02 М. В действительности, для того чтобы заметить
окраску хромата серебра, необходим некоторый избыток индикатора
(«холостой» опыт!)
Метод Мора имеет ряд существенных недостатков:
1. Этот метод применим только для определения хлоридов и бромидов
и неприменим для определения иодидов и роданидов, титрование которых
сопровождается образованием коллоидных систем и адсорбцией,
затрудняющих установление конечной точки титрования.
2. Метод нельзя применять в кислых и сильнощелочных средах. В
кислых средах хромат переходит в бихромат, который образует с Ag+
красный осадок, растворимый в кислоте. В сильнощелочном растворе
образуется окись и гидроокись серебра. Потому рН раствора должен быть
не меньше 6,5 и не больше 10. В присутствии солей аммония рН раствора
должен быть равен 6,5—7,2.
3. Ионы, образующие с ионами индикатора осадки хроматов (Hg++,
Pb+ + , Ba++ и др.), мешают титрованию по методу Мора. Это можно
320
ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ и КОМПЛЕКСООБРАЗОВАНИЯ
заключить из сравнения растворимости соответствующих хроматов:
PAg2cro4 = 1,4-Ю""5 моль/л; Рвасю, = 1,5-10"6 моль1л\ Ррьсго, = 1,3 X
X 10~7 моль/л.
4. По методу Мора нельзя титровать окрашенные растворы,
маскирующие окраску хромата серебра в точке эквивалентности.
5. Титрованию по методу Мора мешают также многие анионы (S",
РО~~, AsO~"~, AsO~~~, СО~~, С20~" и др.), образующие с ионами
серебра малорастворимые соединения.
§ 12. Приготовление 0,1 н. раствора нитрата серебра
Способы приготовления и хранения раствора. 0,1 н. раствор нитрата
серебра получают растворением рассчитанного количества химически
чистого кристаллического AgN03 в определенном объеме воды (от 0,5
до 1 л) или растворением определенной навески химически чистого
серебра в химически чистой азотной кислоте.
Стандартные растворы нитрата серебра, приготовленные из
продажных препаратов AgN03, содержащих некоторое количество примесей,
устанавливают по химически чистому хлориду натрия. Приготовленный
раствор AgN03 изменяется при длительном хранении. Под влиянием
света разложение ускоряется. Поэтому раствор хранят в склянках
оранжевого стекла или в посуде, обернутой черной бумагой или покрытой
черным асфальтовым лаком.
Учитывая, что ЭА^кю, = 169,875, для приготовления 0,5 л 0,1 н.
раствора нитрата серебра необходимо взять
169.9-0,1-500
1000
= 8,495 г AgN03
Отвешивают на технических весах во взвешенном предварительно
бюксе рассчитанное количество нитрата серебра. Осторожно переносят
навеску через воронку в склянку емкостью 1 л из темного стекла, туда же
наливают мерным цилиндром определенный объем дистиллированной
воды, часть которой расходуют на промывание бюкса и воронки.
Содержимое склянки тщательно перемешивают и затем приступают к установке
титра полученного раствора.
§ 13. Приготовление стандартного раствора хлорида натрия
Стандартный раствор хлорида натрия получают растворением точно
известного количества химически чистого хлорида натрия в определенном
объеме воды.
Перед определением титра раствора нитрата серебра хлорид натрия
высушивают, рассчитанную навеску, предварительно взвешенную на
технических весах, взвешивают на аналитических весах и растворяют в
мерной колбе емкостью 0,5 или 1 л. Установка титра AgN03 no NaCl доожет
быть проведена различными методами.
§ 14. Установка титра 0,1 н. раствора нитрата серебра
по точной навеске хлорида натрия
Установка титра методом отдельных навесок. Для того чтобы на
титрование пошло не более 25 мл раствора, необходимо взять навеску:
_58,443-0,1»У
flNaCl- 1000 г
где V — объем титранта;
58,443 — грамм-эквивалент NaCl.
§ 14. УСТАНОВКА ТИТРА 0,1 н. РАСТВОРА НИТРАТА СЕРЕБРА 321
Высушенный хлорид натрия помещают во взвешенный бюкс;
отвешивают на технических весах рассчитанное количество этой соли, затем
навеску точно взвешивают на аналитических весах. Отвешенное
количество хлорида натрия осторожно ссыпают в коническую колбу и
обмывают бюкс 20—25 мл дистиллированной воды. Взвешивание NaCl можно
проводить в пробирке. Для титрования берут 4—5 навесок.
Титр раствора нитрата серебра по хлориду натрия устанавливают
методом прямого титрования в присутствии индикатора — хромата
калия.
Для этого приготовленный раствор AgN03 наливают в бюретку.
Предварительно бюретка должна быть тщательно промыта малым
количеством этого же раствора нитрата серебра. К навеске хлорида натрия,
находящейся в конической колбе и растворенной в малом объеме воды
(25 мл), прибавляют 1—2 мл 0,02 М раствора хромата калия и при
энергичном перемешивании титруют раствором AgN03, пока не появится
неисчезающее красное окрашивание, сходное с окраской «свидетеля».
Расчет результатов титрования проводят точно так же, как это делают
при установке титров других растворов.
Установка титра методом пипетирования. Для приготовления 0,5 л
0,1 н. раствора NaCl берут
*каС1=58,4423'0Д = 2*922gNaCl
Во взвешенном бюксе предварительно отвешивают на технических
весах рассчитанное количество хлорида натрия, затем точно взвешивают
на аналитических весах. Навеску осторожно через воронку переносят
в мерную колбу, смывают NaCl с воронки и бюкса дистиллированной
водой и растворяют в малом количестве воды. После растворения навески
раствор доливают водой до метки, закрывают мерную колбу пробкой и
содержимое ее тщательно перемешивают.
Для титрования отбирают из мерной колбы по 25 мл раствора NaCl,
переносят их в конические колбы емкостью по 250 мл, добавляют 1—2 мл
(всякий раз одинаковое количество) раствора индикатора К2СЮ4 и
медленно при непрерывном перемешивании титруют приблизительно 0,1 н.
раствором AgN03. Титрование продолжают до тех пор, пока не появится
неисчезающее красное окрашивание, сходное с окраской «свидетеля».
Определение повторяют до тех пор, пока результаты трех титрований
не будут расходиться больше чем на 0,05 мл.
Расчет результатов титрования проводят по формулам (см. гл. 1, § 10).
Поправка на индикатор. На практике расходуют на титрование
некоторый избыток AgN03 для того, чтобы стал заметен красный осадок
хромата серебра. При титровании сильно разбавленных растворов ошибка,
вызываемая перетитрованием, возрастает и достигает недопустимых
пределов. Поэтому при титровании сильно разбавленных растворов проводят
«холостой» опыт и находят поправку на индикатор. Для этого берут
такое же количество раствора индикатора, которое используется при
установке титра AgN03, разбавляют его 50 мл воды и титруют раствором AgN03
до тех пор, пока не появится неисчезающее красное окрашивание, сходное
с окраской, которую принял титруемый раствор определяемого вещества
в конце титрования.
Количество стандартного раствора, расходуемое на титрование
индикатора в «холостом» опыте, не должно превышать 0,03 мл. Найденную
поправку на индикатор вычитают из объема стандартдого раствора AgN03,
пошедшего на титрование галогенида.
322 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
§15. Определение ионов хлора в техническом хлориде натрия
по методу Мора
Определение ионов хлора в растворимых хлоридах основано на
прямом титровании навески анализируемого вещества или его раствора
стандартным раствором AgN03 в присутствии индикатора — хромата калия:
Ag+ + Cl--^AgCl
Рассчитанную навеску технического хлорида натрия или его раствор
переносят в мерную колбу, доводят объем водой до метки и тщательно
перемешивают. Отбирают пипеткой 25 мл полученного раствора и титруют
в присутствии К2Сг04 так же, как описано при установке титра AgN03.
Если исходят из кристаллического продукта, то вычисляют
процентное содержание NaCl или С1"-ионов в пробе. Если для анализа дан
раствор NaCl, то рассчитывают содержание в нем С1~ -ионов или NaCl в
граммах (см. гл. 1, § 10).
§ 16. Определение хлоридов по методу Фаянса
Метод Фаянса основан на прямом титровании галогенидов
стандартным раствором AgN03 в присутствии адсорбционных индикаторов (см. § 6):
Ag+ + Hal- —> AgHal
mAgHal + nAg+H36 -+ (AgHal)m Ag+
(AgHal)m Ag+ + Ind- -+ [(AgHal)m Ag+] IncT
Титрование хлоридов в присутствии флуоресцеина проводят в
нейтральной среде. При повышенной концентрации ионов водорода флуоре-
сцеин, являющийся кислотой (Hind), слабо диссоциирует вследствие
подавления его диссоциации кислотой. Поэтому концентрация Ind "-ионов
становится очень малой. В щелочном растворе осаждается Ag20. В
умеренно кислой среде обычно титруют в присутствии других индикаторов:
дихлорфлуоресцеина и эозина. Титрование в кислых средах выгодно
отличается от титрования в нейтральных растворах, так как дает возможность
вести определение в присутствии гидролизующихся солей,
разлагающихся водой с образованием осадков гидроокисей и оксихлоридов (А1+++,
Fe+++ и др.).
В условиях учебной аналитической лаборатории используют для
анализа нейтральные растворимые в воде галогениды щелочных металлов.
Рассчитанную навеску хлорида натрия количественно переносят в
мерную колбу и растворяют в небольшом объеме воды. После растворения
навески доводят объем раствора до метки и тщательно перемешивают.
Если анализируют готовый раствор хлорида, то операции расчета и
растворения навески опускают. В этом случае пробу анализируемого раствора
просто разбавляют дистиллированной водой в мерной колбе до метки.
Для определения хлорида отбирают аликвотные части исследуемого
раствора (по 25 мл)9 переносят их в конические колбы, прибавляют по 5
капель раствора флуоресцеина и титруют стандартным раствором AgN03,
непрерывно перемешивая.
По мере прибавления по каплям раствора AgN03 титруемая смесь
мутнеет. Вблизи точки эквивалентности наблюдается частичная
коагуляция коллоидного осадка AgCl. В этот момент титрование проводят еще
более внимательно и осторожно, сильно перемешивая содержимое колбы.
Титрование заканчивают, когда белый осадок AgCl окрашивается в
красный цвет. Титрование выполняют 3—4 раза и, получив три сходящихся
результата, вычисляют результаты анализа (см. гл. I, § 10).
§ 18. ПРИГОТОВЛЕНИЕ 0,1 н. РАСТВОРА РОДАНИДА АММОНИЯ 323
В. РОДАНОМЕТРИЯ
§ 17. Характеристика метода
Роданометрический метод (метод Фольгарда) объемного анализа
основан на применении в качестве осадителя титрованного раствора,
содержащего SCN~-HOHbi:
Ag+ + SCN- —> AgSCN
Более подробно — см. § 2.
В качестве стандартных растворов используют: для определения
Ag+-ионов — роданид аммония; для определения галогенидов и других
анионов — нитрат серебра и роданид аммония.
Роданометрическим методом пользуются для определения галоген-
ионов и серебра в серебряных сплавах.
В роданометрии в качестве индикатора для определения точки
эквивалентности применяют насыщенный раствор железо-аммонийных
квасцов (см. § 2).
В отличие от метода Мора, метод Фольгарда обладает рядом
преимуществ.
1. Роданометрический метод применим для определения хлоридов,
бромидов, иодидов, роданидов и ионов серебра.
2. Метод применим для титрования кислых растворов, так как осадок
AgSCN нерастворим в кислотах. Эта особенность метода делает его очень
удобным при анализе серебряных сплавов, которые растворяют в
кислотах, и количественном определении галогенидов в сильнокислых средах,
так как галогениды в указанных средах нельзя титровать по методу Мора
или в присутствии адсорбционных индикаторов.
3. Другие ионы (Ва++, РЬ++ и др.), мешающие определению по
методу Мора, в большинстве случаев не мешают определению по методу
Фольгарда.
§ 18. Приготовление 0,1 н. раствора роданида аммония
Раствор NH4SCN точной концентрации по навеске вещества
приготовить нельзя, так как NH4SCN гигроскопичен. Поэтому сначала готовят
раствор требуемой концентрации, а затем уже устанавливают титр
приблизительно 0,1 н. раствора NH4SCN по стандартному раствору AgN03.
Для приготовления 1 л 0,1 н. раствора роданида аммония необходима
(3nh4scn = 76,120) следующая навеска реактива:
%h4scn =76,120-0,1 =7,612 г
Отвешивают на технических весах во взвешенном бюксе рассчитанное
количество роданида аммония. Затем осторожно переносят навеску через
воронку в склянку емкостью 1 л, туда же наливают отмеренное мерным
цилиндром необходимое количество дистиллированной воды. Содержимое
склянки тщательно перемешивают и затем приступают к установке титра
полученного раствора по стандартному раствору AgN03.
Установку титра раствора роданида аммония по нитркту серебра
проводят в присутствии индикатора — железо-аммонийных квасцов
Fe2 (S04)3-(NH4)2S04-24H20, отличающихся от других солей железа (III)
наименьшей склонностью к гидролизу. При этом протекают следующие
реакции:
Ag+ + SCN" —> AgSCN
Fe+++ + 3SCN- ^± Fe(SCN)3
324
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Бюретку заполняют раствором роданида аммония. В коническую
колбу помещают точный объем (25 мл) ранее приготовленного
титрованного раствора нитрата серебра известной концентрации, к которому
прибавляют 2 капли концентрированной азотной кислоты и 1 мл
насыщенного раствора индикатора. Титрование ведут при энергичном
перемешивании. По мере приливания к титруемому раствору AgN03 стандартного
раствора NH4SCN в месте падения капель NH4SCN появляется красное
окрашивание, исчезающее при помешивании содержимого колбы.
Незадолго до достижения точки эквивалентности цвет раствора переходит
в оранжево-красный. В точке эквивалентности раствор приобретает
коричнево-красный оттенок. На этом титрование заканчивают и приступают
к расчету результатов анализа (см. гл. I, § 10).
Поправку на индикатор определяют «холостым» опытом.
§ 19. Определение ионов хлора в растворимых хлоридах
по методу Фольгарда
Определение С1"~-ионов по методу Фольгарда основано на применении
метода обратного титрования. Хлорид-ионы сначала осаждают
определенным объемом стандартного раствора AgN03, взятого с избытком. Затем
оттитровывают не вступивший в реакцию с хлоридом избыток AgN03
стандартным раствором NH4SCN в присутствии железо-аммонийных
квасцов в качестве индикатора. По разности результатов двух титрований
определяют объем раствора AgN03, израсходованного на осаждение C1".
Таким образом, последовательно протекают три реакции:
Ag+ + Cl- -> AgCl
Ag+ + SCN" —> AgSCN
3SCN" + Fe+++ ^± Fe (SCN)3
Однако в тот момент, когда избыток Ag+ будет оттитрован роданидом,
избыток SCN" вступает, кроме того, в реакцию с AgCl:
AgCl + SCN" 5=? AgSCN + СГ
Так как роданид серебра (nPAgscN = 10"12) менее растворим, чем
хлорид серебра (ПРдбс1 = 1,7-10"10), то указанное равновесие
сдвигается слева направо.
В момент равновесия отношение [C1"]/[SCN~] равно отношению
nPAgci/nPAgscN (см. § 8). Следовательнр
[С1-] . nPAgCI =1,7-Ю-10=17П
[SCN-] ~ nPAgSCN ю-12
т. е. равновесие устанавливается тогда, когда величина [SCN" ] станет
в 170 раз меньше величины [С1~ ]. В момент равновесия
[CI"] = УnPAgC1 = VlJ-lQ-io = 1,3-Ю-6 г-ион/л
Следовательно, в точке эквивалентности при избытке SCN" и
установившемся равновесии [SCN" ] = 1,3-10"5 : 170 = 8-Ю"8 г-ион/л.
Таким образом, равновесие установится тогда, когда практически
весь избыток SCN" вступит в реакцию двойного обмена с AgCl. Поэтому
конечную точку титрования трудно заметить, так как появившееся
розово-красное окрашивание, вызываемое образованием F§ (SCN)3, быстро
исчезает вследствие обменной реакции:
Fe (SCN)3 + 3AgCl -* Fe++++ 3C1" + 3AgSCN
§ 20. ОПРЕДЕЛЕНИЕ СЕРЕБРА В СПЛАВАХ
325
Для предупреждения этой реакции применяют различные способы.
Наиболее эффективно отделение осадка AgCl фильтрованием. При этом
С1~-ионы осаждают избытком раствора AgN03 в мерной колбе, доводят
объем раствора до метки, смесь взбалтывают 5—10 мин и
отфильтровывают по частям через сухой фильтр. Первые порции фильтрата
отбрасывают. Аликвотные части фильтрата (25 мл из 250 мл) титруют роданидом *.
Для проведения анализа по методу Фольгарда готовят 250 мл
приблизительное,! н. раствора NaCl, навеску вычисляют по формуле (Эыаа =
= 58,443):
58,4430,1 250 , _1П м г.
а== ШОО = !>4612aNaCl
Рассчитанную навеску NaCl осторожно переносят в мерную колбу
и тщательно смывают остатки NaCl с бюкса и воронки дистиллированной
водой. Сначала навеску растворяют в малом количестве воды, а затем
наполняют мерную колбу до метки, закрывают пробкой и тщательно
перемешивают содержимое колбы. Титрованные растворы нитрата серебра
и роданида аммония помещают в две различные бюретки. Отбирают из
мерной колбы в коническую колбу аликвотное количество раствора
хлорида натрия, добавляют 2 капли азотной кислоты, 1 мл раствора
индикатора и прибавляют отмеренный избыток раствора нитрата серебра (50 мл).
Затем приступают к титрованию полученной смеси титрованным раствором
роданида аммония до появления красного окрашивания раствора.
Определение повторяют до тех пор, пока результаты трех титрований не будут
расходиться более чем на 0,05 мл.
В случае необходимости выпавший осадок отфильтровывают или
добавляют в анализируемый раствор бензол и ведут определение, как
указано выше.
При известном навыке определение не занимает много времени и
приводит к достаточно точным результатам.
Расчеты — см. гл. I, § 10.
§ 20. Определение серебра в сплавах
Метод основан на растворении навески сплава серебра в азотной
кислоте и последующем титровании азотнокислого раствора 0,1 н.
раствором роданида аммония в присутствии железо-аммонийных квасцов
в качестве индикатора:
3Ag + 4HN03 —> 3Ag+ + 3NO:f + NO + 2H20
Ag+ + SCN- —> AgSCN
При избытке SCN~-hohob:
3SCN- 4- Fe+++ ^zt Fe (SCN)3
Рассчитанную навеску сплава (примерно 1—2 г) отвешивают на
часовом стекле сначала на технических, а затем на аналитических весах,
помещают ее в фарфоровую чашку и растворяют в 5 мл умеренно
разбавленной азотной кислоты. Во время растворения чашку накрывают часовым
стеклом, помещенным !на стеклянный треугольник. Растворение ведут
при нагревании на водяной бане в вытяжном шкафу. После растворения
сплава содержимое чашки выпаривают до полного удаления окислов
—^
* Предложено также добавлять перед титрованием в раствор галогенида 1 мл какого-
либо несмешивающегося с водой органического растворителя (эфир, амиловый спирт,
бензол, нитробензол, толуол и т. п.).
326 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
азота, затем разбавляют 20 мл дистиллированной воды и фильтруют в
мерную колбу емкостью 250 мл. Фильтр тщательно промывают водой. Объем
раствора доводят дистиллированной водой до метки и содержимое колбы
перемешивают.
Следует иметь в виду, что в присутствии продуктов восстановления
азотной кислоты (окислов азота и HN02) титрование приводит к
искаженным результатам вследствие окисления SCN" и образования
окрашенного в красный цвет продукта взаимодействия HN02 с NH4SCN. Поэтому
удаление окислов азота необходимо проводить очень тщательно.
Для титрования отбирают пипеткой из мерной колбы аликвотное
количество полученного раствора, помещают в коническую колбу,
добавляют 1 мл индикатора и титруют роданидом аммония до появления не-
исчезающего красного окрашивания. Определение заканчивают тогда,
когда получают три сходящихся результата. В присутствии в
анализируемом сплаве меди титруемый раствор окрашивается в синий цвет.
Поэтому для более точного определения серебра применяют «свидетель».
Вычисления проводят по формулам (см. гл. I, § 10).
Г. МЕРКУРИМЕТРИЯ
§ 21. Характеристика метода
Меркуриметрический метод объемного анализа основан на применении
в качестве титрованного раствора солей окисной ртути (меркури-ионов).
При взаимодействии Hg++ с хлоридами, бромидами, цианидами и
роданидами образуются слабодиссоциированные соединения, например:
Hg++ + cr ^± [HgCl]+
[HgCl]+ + Cl- ^± HgCl2
После достижения точки эквивалентности, когда титруемые ионы
галогенов полностью прореагируют с ионами ртути (II), в титруемом растворе
появляются избыточные Hg++-noHbi, которые обнаруживают при помощи
соответствующего индикатора, образующего с Hg++ характерные
соединения.
При титровании иодидов образуется устойчивый комплекс:
Hg++ + 4I- z^: [Hgl4]~
В этом случае конечную точку титрования устанавливают по
образованию неисчезающей мути красного осадка иодида окисной ртути:
Hg++ + [HgI4]- ^ Hg[Hgl4] (или2Нб12)
появляющейся вблизи точки эквивалентности.
В качестве стандартных растворов для определения галогенидрв,
цианидов и роданидов применяют нитрат или перхлорат окисной ртути,
а для определения ионов хорошо диссоциирующих солей ртути —
роданид аммония.
В меркуриметрии в качестве индикаторов применяют нитропруссид
натрия, дающий бесцветный осадок с Hg++, дифенилкарбазон,
образующий синий осадок, Р-нитрозо-а-нафтол, внутрикомплексное соединение
которого с Hg++ красного цвета. И. С. Мустафин и О. В. Сиванова в
1964 г. предложили для этой же цели применять нитрозооксин в смеси
с красителем кислотным синим антрахиноновым; последний прибавляется
в качестве светофильтра. Такой индикаторный раствор, названный
авторами гидрон III, при избытке галогенидов окрашен в зеленый цвет, пере-
§ 23. УСТАНОВКА ТИТРА РАСТВОРА НИТРАТА РТУТИ (II) 327
ходящий в красный при избытке Hg++. Индикатор позволяет работать
с 2,5• 10"3 н. раствором Hg++ и определять, например, 0,03 мг хлоридов
в 10 мл титруемого раствора.
Меркуриметрический метод широко применяется благодаря многим
преимуществам по сравнению с аргентометрическими методами.
1. Меркуриметрический метод позволяет вести прямое определение
анионов в кислой среде.
2. Этот метод применяется не только для определения галогенидов,
цианидов и роданидов, но и для определения ионов окисной ртути.
3. Многие ионы, мешающие определению по методу Мора и Фоль-
гарда, не оказывают влияния на точность определений с помощью нитрата
или перхлората окисной ртути.
4. Соединения ртути являются менее дефицитными, чем соли серебра,
и легко регенерируются.
Однако меркуриметрический метод, равно $ак и другие методы,
основанные на применении солей ртути, имеет весьма существенный
недостаток: соли phiymu ядовиты, работа с ними требует большой аккуратности
и применения необходимых мер предосторожности.
§ 22. Приготовление 0,1 н. раствора нитрата ртути (II)
Нитрат окисной ртути гигроскопичен, поэтому нельзя приготовить
стандартный раствор Hg (N03)2 растворением точной навески нитрата
ртути (II). Обычно сначала готовят раствор нитрата ртути приблизительно
требуемой концентрации, а затем устанавливают его титр по 0,1 н.
раствору NaCl.
Во взвешенном предварительно бюксе отвешивают на технических
весах около 17 г Hg (NOgb-VaHgO для приготовления 1 л 0,1 н. раствора.
Навеску переносят через воронку в склянку или большую мерную колбу.
Соль окисной ртути плохо растворима в воде, поэтому в склянку
прибавляют 20 мл 6 н. азотной кислоты. Когда навеска растворится, то доводят
раствор дистиллированной водой до требуемого объема и тщательно
перемешивают. Затем устанавливают титр раствора по установочному
веществу,
§ 23. Установка титра раствора нитрата ртути (II)
Установочными веществами для раствора нитрата окисной ртути
могут быть хлориды, роданиды и т. п. вещества, образующие с Hg++ мало-
диссоциированные соединения.
Приготовление растворов хлорида натрия и роданида аммония
описано ранее (см. § 8 и 13).
Установка титра по хлориду натрия. Титр раствора нитрата окисной
ртути по хлориду натрия устанавливают методом прямого титрования
в присутствии индикаторов — нитропруссида натрия или дифенилкар-
базона.
Для установки титра раствора нитрата окисной ртути отбирают
пипеткой определенный объем титрованного раствора хлорида натрия,
прибавляют 0,3 мл 10%-ного раствора нитропруссида натрия и титруют
раствором Hg (N03)2. Титрование заканчивают тогда, когда появляется
неисчезающая муть от образования нитропруссида ртути:
Hg++ + [Fe(CN)5NO]- -* Hg [Fe (CN). NO]
Если титрование ведут в присутствии дифенилкарбазона, то его
заканчивают тогда, когда в растворе появляется неисчезающее синее
окрашивание.
328 ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Установка титра по роданиду аммония. Установку титра нитрата
окиснои ртути по роданиду аммония можно проводить двумя способами.
а) Для установки титра раствора нитрата окисной ртути отбирают
пипеткой в коническую колбу определенный объем устанавливаемого
раствора, добавляют 5 мл разбавленного раствора соли железа (III) и
титруют раствором NH4SCN. Титрование заканчивают тогда, когда
появляется неисчезающее красное окрашивание роданида железа:
Hg++ + 2SCN" -+ Hg(SCN)2
При избытке SCN~-hohob:
3SCN- + Fe+++ ^± Fe(SCN)3
б) Для установки титра раствора нитрата окисной ртути отбирают
пипеткой определенный объем титрованного раствора роданида аммония,
прибавляют 5 мл раствора соли железа (III) и титруют раствором нитрата
окисной ртути до исчезновения красного окрашивания раствора:
Hg++ + 2SCN- _* Hg(SCN)2
При избытке Hg++-HOHOB:
3Hg++ + 2Fe (SCN)3 —> 3Hg (SCN)2 + 2Fe+++
§ 24. Определение ионов хлора в воде
меркуриметрическим методом
Определение ионов хлора меркуриметрическим методом основано на
том же принципе, что и установка титра раствора нитрата окисной ртути
по хлориду натрия (см. § 23). Титрование ведут в присутствии индикатора
нитропруссида натрия или дифенилкарбазона:
Hg++ + 2C1- ^ HgCl2
При избытке Hg++-HOHOB:
Hg+++[Fe(CN)5NO]~ -+ Hg [Fe (CN)5 NO]
Для выполнения определения отбирают в крническую колбу 100 мл
предназначенной для анализа воды, прибавляют 10 мл 2 н. раствора
химически чистой азотной кислоты и I мл 2%-ного раствора
дифенилкарбазона. Полученную смесь титруют раствором нитрата окисной ртути до
появления сине-фиолетового окрашивания.
Д. МЕРКУРОМЕТРИЯ
§ 25. Краткая характеристика метода
Меркурометрический метод объемного анализа основан на
применении титрованных растворов солей закисной ртути (меркуро-ионов).
При взаимодействии [Hg2]++-HOHOB с хлоридами^ бромидами, иоди-
дами и т. д. образуются осадки малорастворимых галогенидов Hg2Cl2,
Hg2Br2, Hg2I2, например:
[Hg2]++ + 2C1- -> Hg2ci2
Меркурометрический метод по сравнению с аргентометрическим имеет
некоторые преимущества.
1. При меркурометрическом методе не требуется ценных препаратов
серебра.
§ 26. ХАРАКТЕРИСТИКА МЕТОДА
329
2. Соли закисной ртути менее растворимы, чем соответствующие соли
серебра, и поэтому при титровании хлоридов нитратом закисной ртути
наблюдается резкий скачок титрования вблизи точки эквивалентности.
3. Определение меркурометрическим методом можно проводить в
кислых растворах методом прямого титрования.
Недостатком меркурометрического метода является ядовитость солей
ртути. Поэтому при работе с этими солями следует соблюдать большую
осторожность.
Применение меркурометрического метода при количественных
определениях растворимых хлоридов и бромидов пока ограничено.
В меркурометрическом методе титрования в качестве индикаторов
применяют:
1. Роданид железа Fe (SCN)S. При титровании (например, хлоридов)
растворами солей закисной ртути в точке эквивалентности раствор
обесцвечивается. Избыток [Hg2]++-HOHOB реагирует с Fe (SCN)3 по
уравнению:
3 [Hg2]++ + 2Fe (SCN)3 -+ 3Hg2 (SCN)2 + 2Fe+++
2. Дифенилкарбазон, образующий с [Hg2]++-noHaMH осадок сийего
цвета.
Е. КОМПЛЕКСОНОМЕТРИЯ (ХЕЛАТОМЕТРИЯ)
§ 26. Характеристика метода
В последнее время в заводских и научно-исследовательских
лабораториях широко применяют методы анализа, основанные на использовании
реакций, которые сопровождаются образованием комплексных
соединений катионов с органическими реактивами, называемыми комплексонами.
Эти реакции сопровождаются образованием внутрикомплексных
(клешневидных, хелатных) солей.
Понятие о комплексонах. Комплексонами обычно называют
органические соединения, представляющие собой производные аминополикарбо-
новых кислот. Простейший комплексон — нитрилотриуксусная кислота
(НТА), известная под названием комплексон 1 (сокращенно H3Y):
/ СН2СООН /СН2СООН
N—СН2СООН или Н—N—СН2СОО"
\ сн2соон чсн2соон
Наибольшее значение имеет четырехосновная эти денди аминтетраук-
сусная кислота (ЭДТА) — комплексон II (сокращенно H4Y):
нооссн2 сн2соон
\ /
N—СН2—СН2—N
/ \
нооссн2 сн2соон
или
нооссн2 сн2соо-
H~tf-CH2-CH2--N-H
-оосот2 сн2соон
Комплексоны наряду с карбоксильными группами (—СООН) содержат
аминный азот (=N). Благодаря такому строению эти соединения
отличаются мульти(поли)дентатностью, т. е. способностью образовывать
330
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
сразу несколько координационных связей с ионами металлов-комплексо-
образователей.
На практике обычно применяют двунатриевую соль этилендиамин-
тетрауксусной кислоты, которую называют комплексоном III или три-
лоном Б (сокращенно Na2H2Y):
нооссн2 сн2соон
\ /
N—СНо—СНо—N
/ \
NaOOCCH2 CH2COONa
Комплексон III образует со многими катионами устойчивые малодис-
социированные растворимые в воде внутрикомплексные соли,
построенные по типу известной комплексной соли диметилглиоксимата никеля.
С некоторыми ионами металлов-комплексообразователей
комплексны образуют настолько устойчивые слабые электролиты, что обычными
качественными реакциями невозможно доказать присутствие данного
катиона в растворе этого комплексного соединения.
Реакции между комплексоном и ионами металлов-комплексообразова-
телей протекают стехиометрически, т. е. в строго эквивалентных
отношениях, это обстоятельство открывает широкие возможности применения
комплексонов для количественного определения многих катионов, в том
числе кальция, цинка, меди, алюминия, индия и др.
Комплексонометрическое титрование. Комплексоны широко
используются для комплексонометрического титрования многих катионов и
анионов (косвенным методом). При этом используют метод прямого и
обратного титрования. В первом случае титрование ведут при определенном
значении рН стандартным раствором комплексона III. Точку
эквивалентности устанавливают с помощью индикаторов, представляющих собой
органические красители, образующие с катионами окрашенные
комплексные соединения (металлиндикаторы).
При титровании комплексоном III такого окрашенного комплексного
соединения оно постепенно разлагается вследствие образования нового
более прочного внутрикомплексного соединения катиона с
комплексоном. В точке эквивалентности первоначальный цвет комплексного
соединения, образуемого индикатором с определяемым катионом, исчезает и
появляется окраска, свойственная свободному индикатору.
При обратном методе титрования к анализируемому раствору
прибавляют отмеренный объем стандартного раствора комплексона III,
избыток которого оттитровывают стандартным раствором соли цинка (или
другого металла) в присутствии металл-индикатора, реагирующего на
ионы цинка (или ионы другого металла).
Таким образом, комплексонометрия (хелатометрия) представляет
собой титриметрический метод анализа, основанный на использовании
реакций ионов металлов-комплексообразователей с комплексонами,
сопровождающихся образованием устойчивых малодиссоциированных
растворимых в воде внутрикомплексных {клешневидных, хелатных) солей.
При титровании комплексоном III (Na2H2Y) солей металлов-комплек-
соббразователей протекают следующие реакции:
Na2H2Y ^± 2Na+ + H2Y~
Me++(Ca++) + H2Y-- —> MeY- + 2H+
Ме+++ (In+++) + H8Y~ —> MeY~ + 2H+
Me++++(Th++++) + H2Y- —> MeY + 2H+
§ 26. ХАРАКТЕРИСТИКА МЕТОДА
331
Согласно приведенным уравнениям, 1 г-ион реагирующих с
комплексном III катионов, независимо от их степени окисления, связывает 1 г-ион
комплексона III.
Типы реакций, используемых при комплексонометрическом титровании.
В комплексонометрии применяют реакции разнообразного типа.
1. Реакции непосредственного взаимодействия ионов металла ком-
плексообразователя с комплексоном; например, реакции, применяемые
для определения ионов цинка в аммиачной буферной среде или ионов
кальция в среде КОН и т. д. в присутствии индикатора, реагирующего на
изменение рМе аналогично тому, как кислотно-основной индикатор реагирует
на изменение рН раствора:
Zn-"- + H2Y" NH^N10H4C>ZnY- + 2№
2. Реакции взаимодействия избытка комплексона со стандартным
раствором сульфата цинка, магния, железа (III) и т. п.; например, реакции,
используемые для определения алюминия (при рН5 ацетатный буферный
раствор), при котором комплексон прибавляют в титруемый раствор
в избытке (а)у а затем избыток реагента оттитровывают стандартным
раствором (б):
M++++nH2Y-'E}tS Al Y" + 2Н+ + (п — 1) H2Y- (a)
избыток
(п — 1) Zn++ + (п — 1) H2Y—Е^% — 1) ZnY- + 2Н+ (б)
3. Реакции взаимодействия заместителей с определяемым элементом,
завершаемые титрованием выделившихся ионов заместителя стандартным
растворам ЭДТА.
4. Реакции кислотно-основного титрования, при которых замещенные
ионы водорода оттитровывают стандартным раствором сильного
основания в присутствии кислотно-основного индикатора (а) или иодометри-
ческим методом (б):
Me++ + H2Y- -^ MeY~ + 2H+ (a)
2Н-ь + 20Н-МетилрвыйкрасныД 2Н20
ЗМе++ + 3H2Y~~ + IOg + 51" -^
—> 3MeY— + ЗН20 + 312 (б)
Выделившийся элементарный иод оттитровывают тиосульфатом:
6S2C?" + 3I2 -> 3S40-- + 6r
Другие типы комплексонов. К числу новых комплексующих реагентов относятся
фосфор органические комплексоны, являющиеся производными аминоалкилфосфоновых кислот.
Например:
этилендиаминтетраметилфосфоновая кислота (ЭДТФ)
[(НО)2Р (О) CH2]2N-CH2-CH2-N [CH2P (О) (ОН)2]2
и
этилендиамин-N\N'-диуксусная-N,N''-диметилфосфоновая кислота (ЭДУФ)
НООССН2 СН2СООН
NH-CH2-CH2—ЙН
/ \
-но3рсн2 сн2ро3н-
Аминоалкилфосфоновые кислоты обладают комплексующими свойствами благодаря
протонно-донорным функциям кислотных групп и наличию аминных атомов азота, склонных
к образованию клешневидных соединений. Свойства этих комплексонов настолько
разнообразны, что они дают возможность проводить дифференцированные определения в смесях.
332
ГЛ.IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМШГЕКСООБРАЗОВАНИЯ
Например, этилендиаминдиизопропилфосфоновая кислота (ЭДДИФ)
не образует комплексов с ионами щелочноземельных металлов. Это
позволяет комплексонометрически определять катионы элементов первого
переходного периода в присутствии катионов щелочноземельных
металлов. ЭДДИФ и ЭДУФ проявляют уникальную способность по отношению
к ионам бериллия. Другие фосфорилированные амины отличаются
избирательностью по отношению к ионам магния. Некоторые фосфороргани-
ческие комплексоны образуют с ионами алюминия, железа, индия, галлия
и другими более прочные комплексные соединения, чем соответствующие
амииополикарбоновые кислоты.
Описаны комплексоны, содержащие конъюгированные системы
двойных связей. К их числу относятся производные бензола: I — фенилимино-
диуксусная (ФИДА), II —о-оксифенилиминодиуксусная (ОФИДА),
III — я-крезол-2-метилениминодиуксусная (КМИДА) кислоты:
/СН2сООН
I хСН2СООН
/ч
II I
\//
I
хт/СН2СООН
I хСН2СООН
А/он
II I
/ч/
/СНоСООН
\СН2СООН
он
Н3С
III
Варьирование конъюгированной системы двойных связей, дентат-
ности и строения хелатообразующих групп открывает большие
возможности создания новых реагентов, отличающихся особой
чувствительностью и избирательностью в отношении определенных катионов.
Существенную роль в химии новых комплексонов сыграли
исследования советских ученых.
§ 27. Теоретические основы комплексонометрического
титрования
Строение образуемых внутрикомплексных солей. Анион этилендиамин-
тетрауксусной кислоты, как четырехосновной кислоты, содержащей
в своем составе четыре подвижных иона водорода и 2 атома азота с
основными свойствами, обладает всего шестью атомами, которые участвуют
в качестве лигандов при построении пятичленных циклов
внутрикомплексных соединений. Классическими примерами такого типа соединений
являются:
гликоколят меди
0=С—О О—С=^0
\/
Си
. / \ .
Н2С-ЫН2 H2N-CH2
диметилглиоксимат никеля
О-.-Н-О
t I
Н3С—C=N N=C—СНз
\ /
\ *-
Ni
^ \
. / \
Н3С—C=N N=
I i
о—н..-o
=C-CH3
§ 27. ОСНОВЫ КОМПЛЕКСОНОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 333
Внутрикомплексные соли образуются во всех случаях, когда ионы
металла-комплексообразователя, с одной стороны, замещают активные
атомы водорода функциональных групп органического соединения, а
с другой стороны — взаимодействуют с какими-либо группами,
способными сочетаться с данным ионом координационной связью.
Примерами групп, содержащих ионы водорода, которые способны
замещаться на ионы металлов, и взаимодействующих с ионами металла-
комплексообразователя за счет главной валентности, служат:
карбоксильная— СООН, сульфоксильная—S03H, оксимная = NOH, гидроксиль-
ная — ОН и другие группы.
Примерами групп, соединяющихся с ионами
металла-комплексообразователя координационной (донорно-акцепторной) связью, служат:
аминогруппа— NH2, иминогруппа =NH, оксимная группа =NOH,
карбонильная =СО, тиоэфирная —S— и некоторые другие.
В комплексонах группами, взаимодействующими с ионами металла-
комплексообразователя за счет главной валентности, являются —СООН-
группы, а побочной валентности — третичные аминогруппы.
Внутрикомплексные соли с комплексоном III образуются, с одной
стороны, за счет замещения ионами металла-комплексообразователя
активных атомов водорода карбоксильных групп, с которыми он
соединяется главными валентностями, а с другой стороны — взаимодействия
с атомами азота, способными сочетаться с данным ионом
металла-комплексообразователя посредством побочной (координационной)
валентности.
Наиболее ценным свойством комплексонов, широко используемым
в анализе, является их способность давать внутрикомплексные соли даже
с ионами щелочноземельных металлов: магнием, кальцием и барием,
которые, как известно, трудно или невозможно перевести в комплексные
соединения другими средствами.
Строение внутрикомплексной соли кальция (Na2CaY) можно
схематически представить следующим образом:
о=с—о о-с=о
I \/ I
Н2С Са СН2
\/ \/
NaOOCCH2 CH2— CH2 CH2COONa
Способность карбоксильных групп образовывать соли металлов
усиливается эффектом возникновения внутрикомплексных солей. Поэтому
комплексоны образуют внутрикомплексные соединения с катионами
щелочноземельных, редкоземельных и других элементов. Образуемые
соединения отличаются достаточно малыми величинами констант
нестойкости комплексных ионов (например, для Са** около 10"10, Zn++ около
10"16) достаточно большими значениями рК (например, для Са++ рК ^ 10,
для Zn около 16).
Индикаторы комплексонометрии, подобно комплексонам, также
образуют с ионами металлов внутрикомплексные соли, которые по условиям
титрования должны быть менее устойчивы по сравнению с комплексом
ионов данного металла с комплексоном III.
Если представить себе комплексонометрический индикатор, в состав
молекул которого входит группа —ОН, занимающая орто-положение по
334 ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
отношению к группе —N=N—, то структурный фрагмент индикатора,
связанный с группами —ОН и —N=N, можно представить так:
ОН
При взаимодействии ионов металла-комплексообразователя с
индикатором, содержащим подобную группировку, образуется внутриком-
плексная соль следующего строения:
.О—Me
Эта внутрикомплексная соль о-оксиазокраситедя образуется в
результате замещения иона водорода фенольной гидроксильной группы
ионами металла-комплексообразователя и участия в комплексообразова-
нии неподеленных пар электронов одного из атомов азота азогруппы.
Наличие лишь одной гидроксильной группы в орто-положении в ряде
случаев недостаточно для образования устойчивого клешневидного (хелат-
ного) комплекса. Поэтому в молекулу комплексующего вещества должны
входить другие группы, реагирующие с металлом-комплексообразова-
телем, или в реакции должны участвовать две молекулы монооксиазо-
красителя.
По своему цвету такого рода соли отличаются от цвета индикатора.
При их разложении вновь появляется окраска, присущая свободному
индикатору. Поэтому, если на внутрикомплексную соль, образованную
металлом-комплексообразователем с индикатором, подействовать
комплексном, образующим с данным металлом-комплексообразователем
более устойчивую внутрикомплексную соль, то первоначальный комплекс
разрушается и наблюдается изменение окраски раствора.
Способы фиксирования точки эквивалентности. Определение точки
эквивалентности при комплексонометрическом титровании может быть
проведено различными способами.
Во-первых, используют обычные кислотно-основные индикаторы, так
как реакция комплексообразования -сопровождается выделением Н +-ионов
в количестве, эквивалентном количеству определяемого катиона:
Me++ + H2Y— —> MeY— + 2H+
Выделившуюся кислоту определяют методом нейтрализации с обычными
кислотно-основными индикаторами.
Во-вторых, применяют металл-индикаторы, представляющие собой
органические красители, которые образуют с катионами определяемых
элементов растворимые в воде окрашенные комплексные соединения.
Получаемые при этом комплексные соединения менее устойчивы, чем
внутрикомплексные соли, образуемые определяемыми катионами с ком-
плексонами. Поэтому в процессе титрования комплексоном раствора,
содержащего окрашенное комплексное соединение, образуемое одределяе-
мыми катионами с индикатором, в точке эквивалентности наблюдается
изменение окраски раствора. Это объясняется тем, что комплексное
соединение индикатора разрушается и индикатор выделяется в свободном
виде. Так как окраска комплексного соединения индикатора отличается
от окраски свободного индикатора, то происходит изменение титруемого
раствора.
§ 27. ОСНОВЫ КОМПЛЕКСОНОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 335
Схематично это можно представить следующим образом:
Kt++ + 2HInd
бесцветный окрашен
Ktlnd2 + H2Y— ^
окрашен
^ Ktlnd2 + 2Н+
окрашен в
иной цвет.
чем
свободный
индикатор.
KtY— + 2HInd
бесцветен окрашен
Таким образом, металл-индикатор реагирует на изменение
концентрации катиона аналогично тому, как кислотно-основной индикатор ведет
себя при изменении рН титруемого раствора.
В-третьих, для фиксирования точки эквивалентности применяют
окислительно-восстановительные (ред-окс) индикаторы и
инструментальные методы.
Индикаторы. В качестве индикаторов при комплексонометрических
титрованиях часто применяют красители: кислотный хром темно-синий,
кислотный хромоген черный специальный (иначе называемый эриохром
черный Т или хромоген специальный ЕТ 00) и др. Эти индикаторы в
щелочной среде имеют синюю окраску.
Ионы кальция, магния и ряда других металлов образуют с
индикаторами внутрикомплексные соединения, окрашенные в вишнево-красный
цвет. При титровании комплексоном III раствора, содержащего
определяемый катион и индикатор, ионы металла переходят от индикатора к ком-
плексону III, при этом выделяется свободный индикатор. В точке
эквивалентности красная окраска раствора переходит в синюю, свойственную
индикатору.
Эриохром черный Т. Эриохром черный Т является органическим азо-
красителем группы о,о'-диоксиазонафталина:
/он °н
H03S-^~~^-N= N—j^ Y^ (Halnd)
o2n/
диссоциирующий с образованием H + , H2Ind", Hind", Ind . В его
молекулу входят две фенольные группы и хромофорная азогруппа.
Поэтому эриохром черный Т способен реагировать с ионами металл а-ком-
плексообразователя с образованием комплексных соединений. Сам
индикатор окрашен в синий цвет. В нейтральной или щелочной среде при
рН=7—11 он образует с ионами металлов (Cu++, Mg++, Zn++, Mn++,
In+++, Al+++ и др.) соединения красного цвета.
Уравнение реакции можно представить в следующем виде:
заключительная стадия титрования комплексоном III протекает согласно
уравнению
Me++ + HInd~ ^± Melnd- + H+
синий красный
Melnd- + Na2H2Y+OH- —> Na2MeY + Hlnd~+H20
Для поддержания рН раствора на требуемом уровне обычно в
титруемый раствор добавляют аммиачную буферную смесь.
Ксиленоловый оранжевый. Ксиленоловый оранжевый является
органическим красителем трифени л метанового ряда. Сам индикатор окрашен
в желтый цвет. В кислой среде при рН даже значительно меньше 7 он
336
ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
дает с ионами металлов (Zn++, ThIV, ZrIVH др.) комплексные соединения
красного цвета. Переход окраски очень резкий.
К числу катионов, определяемых титрованием комплексоном III
в присутствии ксиленолового оранжевого, относятся Fe+++ (pH = l—1,5);
ZrIV(pH = l— 2); Bi+++ (рН=2—3,5); А1+++ (рН=2—4); 1п+++ (рН =
=3—4,5); Т1+++ (рН=4—5); Zn++, Cd++, Cu++, Hg++, РЗЭ (рН =
= 5—6).
По своему строению ксиленоловый оранжевый представляет собой
3,3'-бис (ди (карбоксиметил)-аминометил)-о-крезолсульфофталеин:
нооссн2\ /сн2соон
N—СН2 СН2—N
НООССН2/ I I \СН2СООН
но\/ч //\//°
I! I I I
A/SOsH
II II
\//
Кислотный хром темно-синий. Кислотный хром темно-синий является
органическим азокрасителем:
С \ /°Н ОН NHCOCH3
/~^_N-N I I
г,/"\г| I « I
I
S03Na
Сам индикатор окрашен в синий цвет. С катионами металлов в
щелочной среде он образует комплексные соединения вишнево-красного цвета.
Его применяют при титровании растворами комплексонов катионов цинка,
кадмия (рН=9—10), марганца (рН = 10), магния (рН = 10—11), кальция
(рН>12) и др.
Мурексид (пурпурат аммония). Мурексид представляет собой темно-
красный порошок. Водный раствор мурексида окрашен в фиолетово-
красный цвет, изменяющийся в зависимости от среды: при рН ^ 9 —
красно-фиолетовый, рН = 9—10 — фиолетовый, рН > И — синефиоле-
товый. С катионами кальция, никеля, кобальта, меди и другими мурексид
образует комплексные растворимые в воде соединения красного или
желтого цвета, разлагаемые комплексоном III с образованием более
устойчивых внутрикомплексных солей. В процессе титрования солей кальция
и других металлов в присутствии мурексида в точке эквивалентности,
наблюдается изменение красного цвета раствора в сине-фиолетовый цвет.
Структурную формулу мурексида можно представить следующим образом:
НО ОН
I II 1 I
N—С С—N
/ \ / \
0=С С—N=C С=О.Н20
\ // \ /
N—С С—N
I II I
ONH4 О Н
к
§27. ОСНОВЫ КОМПЛЕКСОНОМЕТРИЧЕСКОГО ТИТРОВАНИЯ 337
Гидроны. В ряде случаев в процессе комплексонометрического
титрования бывает очень трудно различить переход одной окраски к другой.
Поэтому в таких случаях прибегают к различным приемам,
обеспечивающим оптимальные условия наблюдения за наступлением конечной точки
титрования.
И. С. Мустафин и Е. С. Кручкова предложили добавлять к раствору
индикаторного вещества так называемый внутренний светофильтр,
значительно улучшающий контрастность перехода индикатора.
Рекомендованные ими индикаторные растворы гидрон I (для суммарного
определения кальция и магния) и гидрон II (для определения кальция в
присутствии магния) в присутствии избытка определяемых катионов имеют
вишнево-красный цвет, переходящий в зеленый при избытке комплексона.
У этих индикаторов внутренним светофильтром является краситель
нафтоловый желтый. Применяя гидроны, можно пользоваться 0,002 н.
растворами титранта и определять 0,003—0,005 мг-экв/л кальция и магния.
В настоящее время гидрон II является, вероятно, одним из наилучших
индикаторов для комплексонометрического определения кальция в
присутствий магния.
Условия комплексонометрического титрования. Основным условием
комплексонометрического титрования является требование,
предъявляемое к реакции, которая должна протекать таким образом, чтобы в точке
эквивалентности определяемые катионы были практически полностью
связаны в комплекс. Константа нестойкости таких комплексов должна быть
очень незначительной. При этом определяемые катионы должны
образовывать с металл-индикатором комплексы, отличающиеся меньшей
прочностью, чем их комплексы с комплексоном.
Титрование комплексоном III проводится при строго определенных
условиях, из которых наибольшее значение имеет соблюдение требуемого
значения рН титруемого раствора.
В сильно кислых растворах с рН <3 образуются менее устойчивые
кислые комплексные соединения. Комплексообразованию устойчивых
комплексных соединений способствует повышение значения рН
титруемого раствора. Однако в сильнощелочных растворах при рН > 10
наблюдается образование оксикомплексных соединений или осадков
гидроокисей определяемых катионов. Следует также иметь в виду, что при
образовании комплекса определяемого катиона с комплексоном освобождаются
ионы водорода, рН раствора понижается. Поэтому, если титруемые
растворы не защищены действием буферной смеси, понижение рН раствора
может достигнуть нескольких единиц рН и требуемые комплексные
соединения не образуются. Чтобы поддержать рН раствора на заданном уровне,
необходимо проводить титрование в буферных растворах, отвечающих
определенному значению рН.
Концентрация свободных анионов Y4" в кислых растворах
комплексона ЭДТА практически ничтожно мала. Численные значения обратных
логарифмов (рК) констант диссоциации H4Y характеризуются
следующими величинами:
H4Y
H3Y"
HgY~
HY—
Z~ H+ + H3Y_
Z~ Н++Н,у-~
^t H+ + HY—
^ H+ + Y4~
(P*i =
(pK«<
(PK3:
(P*4 =
= 2,0)
= 2,67)
= 6,16)
= 10,26)
Суммарная константа, являющаяся произведением отдельных
констант, равняется Ю-21'09, что соответствует р/С = 21,09. Эти данные
338 ГЛ, IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
говорят о том, что HY является очень слабой кислотой и поэтому
в растворе H4Y концентрация Y4~ очень мала.
Можно рассчитать [Y4~ ], применив закон действия масс:
[H4Yj ~ *h«y
rYM ^h4y[H4Y]
1 J ~ [H+]4
Это уравнение показывает, что [У4~] сильно зависит от
концентрации ионов водорода. По мере увеличения [Н+] уменьшается [Y4"].
Концентрация свободных У4~-ионов 0,1 н. раствора ЭДТА в 0,1 н.
растворе хлористоводородной кислоты (рН = 1) равна:
Ю—21. 09 in— 1
I x-i = '10 = in-18 >Юг-ион/л
1 J (10-i)4
Только катионы (Fe+++, In+++, Sc+++, ZrIV, ThIV), образующие
очень прочные комплексы с комплексном, титруются в кислой среде.
Катионы, образующие менее прочные комплексы с комплексоном, не
титруются в кислой среде и не мешают титрованию катионов, определяемых
в кислом растворе.
Вот почему титрование большинства катионов обычно проводят в
аммиачной буферной среде (NH4OH + NH4C1) при рН = 8—9. Во
избежание образования осадков гидроокисей и с целью маскирования отдельных
катионов наряду с буферными смесями добавляют вспомогательные
комплексообразующие вещества: тартраты или цитраты для Мп++,
Pb++, In+++; тайрон (динатриевая соль 1,2-диоксибензол-3,5-дисульфо-
кислоты) или фторид натрия для Ве++, Al+++, TiIV; цианид калия для
Zn44", Cu++, Cd++ и т. д. Благодаря этому удается успешно проводить
комплексонометрическое титрование большинства катионов и
косвенным путем анионов.
§ 28. Классификация методов комплексонометрического
титрования
Метод прямого титрования. Анализируемый раствор, содержащий
катионы определяемого металла, разбавляют в мерной колбе и берут для
титрования аликвотную часть раствора.
Титрование ведут стандартным раствором комплексона III в
щелочной среде с эриохром черным Т или в кислой среде с ксиленоловым
оранжевым. Для этого титруемый раствор предварительно перед титрованием
доводят до определенного значения рН при помощи буферного раствора.
Наряду с буферным раствором иногда добавляют еще вспомогательный
комплексующий агент (тартрат, цитрат и др.), связывающий некоторые
катионы и удерживающий их в растворимом состоянии во избежание
выпадения осадков гидроокисей в щелочном растворе.
В процессе прямого титрования концентрация определяемого катиона
сначала снижается постепенно, затем вблизи точки эквивалентности резко
падает. Этот момент замечается по изменению окраски введенного
индикатора, мгновенно реагирующего на изменение концентрации катионов
металла-комплексообразователя.
Методом прямого комплексонометрического титрования определяют
Cu++, Cd++, Pb++, Ni++, Co++, Fe+++, Zn++, ThIV, Al+++, Ba++,
Sr++, Ca++, Mg++ и некоторые другие катионы. Определению мешают
§ 29. УСТАНОВКА ТИТРА РАСТВОРА КОМПЛЕКСОНА III 339
другие комплексующие вещества, удерживающие ионы в виде
комплексных ионов, которые не разрушаются комплексонами.
Метод обратного титрования. В тех случаях, когда по тем или иным
причинам невозможно провести прямое титрование определяемого
катиона, пользуются методом обратного титрования. В подобных случаях
к анализируемому раствору прибавляют точно измеренный объем
стандартного раствора комплексона, нагревают до кипения для завершения
реакции комплексообразования и затем на холоду оттитровывают
избыток комплексона титрованным раствором MgS04 или ZnS04. Для
установления точки эквивалентности применяют металл-индикатор,
реагирующий на ионы .магния или цинка.
Метод обратного титрования применяют в тех случаях, когда нет
подходящего индикатора на катионы определяемого металла, когда в
буферном растворе катионы образуют осадок и когда реакция комплексооб-
разования протекает медленно. Методом обратного титрования также
определяют содержание катионов в нерастворимых в воде осадках (Са++
в CaC204, Mg++ в MgNH4P04, Pb++ в PbS04 и т. п.).
Метод титрования заместителя. В некоторых случаях вместо
описанных выше методов пользуются методом титрования заместителя. Метод
комплексонометрического титрования заместителя основан на том, что
Mg++-HOHbi дают с комплексоном менее устойчивое комплексное
соединение (р/С = 8,7), чем подавляющее большинство других катионов.
Поэтому, если смешать катионы определяемого металла с магниевым
комплексом комплексона III, то при этом произойдет реакция обмена.
Например, эта реакция используется для определения ионов тория,
когда в анализируемый раствор предварительно вводят комплексонат
магния MgY" (a), a затем оттитрбвывают выделившиеся Mg++-HOHbi
стандартным раствором ЭДТА (б):
рН=2
Th++++ + MgY— —> ThY + Mg++ (a)
рН=10
Afe++ + H2Y— -^ MgY—+2H+ (б)
Вследствие того что ThIV образует с комплексоном более устойчивое
комплексное соединение, чем Mg++, равновесие реакции (а) сдвигается
вправо.
Если по окончании реакции вытеснения оттитровать Mg++
стандартным раствором комплексона III в присутствии эриохрома черного Т,
то можно рассчитать содержание ТЬ1У-ионов в исследуемом растворе.
Метод кислотно-основного титрования. В процессе взаимодействия
комплексона с теми или иными катионами металлов выделяется
определенное количество эквивалентов ионов водорода.
Образующиеся при этом в эквивалентном количестве ионы водорода
оттитровывают обычным алкалиметрическим методом в присутствии
кислотно-основного индикатора или другими способами.
Существуют и другие методы комплексонометрического титрования,
описание которых не входит в нашу задачу.
§ 29. Установка титра раствора комплексона III
Для приготовления стандартного (титрованного) раствора
комплексона III применяют двунатриевую соль этилендиаминтетрауксусной
кислоты, кристаллизующуюся с двумя молекулами воды; ее состав
отвечает формуле Na2C10H14O8N2-2H2O.
340 ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Если двунатриевую соль, содержащую кристаллизационную воду,
высушить при 120—140° С, то получается безводная соль, состав которой
соответствует формуле Na2C10H14O8N'2.
Обе соли могут служить исходными веществами для приготовления
стандартного раствора комплексона III.
Для приготовления 1 л 0,1 н. раствора комплексона III надо взять:
м
Na2C10H14O8N2-2H2O 372,242
2^0 = 2-10 = 18'612 г
ИЛИ
MNa2C10H14O8N2 _ 336,211 1ЙЯ1.
2Л0 2-10 =16'811 2
Для установки титра комплексона III применяют х. ч. карбонат
кальция, х. ч. ZnO или х. ч. металлический цинк, рассчитанную
навеску которых растворяют в х. ч. хлористоводородной или серной кислоте,
нейтрализуют едким натром или аммиаком, разбавляют аммиачным
буферным раствором и титруют стандартным раствором комплексона III
в присутствии необходимого индикатора. Под конец титруют медленно.
Титр раствора комплексона III может быть установлен также по фикса-
налу соли магния (в продаже имеются 0,01 и 0,05 н. растворы сульфата
магния).
По результатам титрования рассчитывают Т, N и К комплексона III.
§ 30. Определение содержания кальция
Методы количественного определения кальция. Существуют
различные методы определения кальция.
а) Весовые методы.
1) Осаждение в виде оксалата СаС204 • Н20 и взвешивание в виде СаС03
или СаО (см. «Весовой анализ»).
2) Осаждение в виде сульфата CaS04 из спиртового раствора.
3) Осаждение в виде пикролоната Са (C10H7O5N4)2-8H2O.
б) Объемные методы.
1) Осаждение в виде оксалата кальция и последующее определение
связанного с кальцием оксалат-иона методом перманганатометрии или
цериметрии.
2) Осаждение в виде молибдата СаМо04, восстановление молибдена
и титрование его ванадатом аммония.
3) Комплексонометрический метод.
Весовой метод определения кальция имеет весьма существенные
недостатки.
1. Определение содержания кальция в различных технических
объектах весовым методом представляет собой весьма длительную операцию.
2. Осаждение ионов кальция в виде СаС204 связано с большими
трудностями, обусловливаемыми невозможностью достижения
количественного выделения оксалата кальция;
3. Осадок оксалата кальция бывает часто загрязнен посторонними
примесями и его трудно выделить в химически чистом виде.
4. Получение весовой формы (СаО) связано с применением
относительно высокой температуры, необходимой для термического разложения
оксалата кальция.
5. Получаемая весовая форма (СаО) нестабильна и подвергается
действию влаги и двуокиси углерода воздуха, вследствие чего ее масса
меняется в зависимости от условий получения, и хранения.
§ 30. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КАЛЬЦИЯ
341
Поэтому в настоящее время весовой метод определения кальция
утратил прежнее свое значение и вытеснен более прогрессивными
объемными методами анализа.
Один из этих методов описан выше (см. гл. II.I, § 28). Перманганато-
метрический метод определения кальция отличается рядом преимуществ
по сравнению с весовыми методами анализа. Одним из таких преимуществ
является более быстрое завершение операции
определения. Однако п.ерманганатометриче-
скому методу определения кальция,
основанному на осаждении ионов кальция в виде окса-
лата и последующем титровании оксалат-ионов
перманганатом, свойственны многие недостатки
весового анализа, связанные с невозможностью
полного количественного осаждения и
отделения оксалата кальция.
Из объемных методов анализа наиболее
точным и быстрым методом определения кальция
несомненно является комплексонометрическое
титрование ионов кальция комплексоном III.
Комплексонометрический метод определения
кальция. Комплексонометрическое определение
кальция основано на прямом методе титрования
его ионов стандартным раствором
комплексна III в присутствии мурексида или
кислотного хром темно-синего. Индикатор образует
с ионами кальция комплексное соединение
красного цвета. При титровании раствора
комплексоном III в точке эквивалентности красная
окраска переходит в окраску, характерную
для свободного индикатора.
В результате титрования солей кальция комплексоном III проис
ходит образование комплекса CaY~~ и кислоты:
Ca++ + H2Y~ ^± CaY—+2Н+
Образующийся комплекс CaY"" относительно неустойчив:
[Ca+1[Y°1
Обьем комплексона Ж, ил
Рис. 61. Кривые титрования
ионов кальция комплексоно-
метрическим методом при
различных значениях рН
раствора:
2 — рН = 8;
/ — рН = 6;
3 — РН = 10
рН = 12.
[CaY-]
= зю-11
Поэтому образование свободной кислоты во время реакции или
прибавление ее в титруемый раствор перед титрованием сдвигает указанное
равновесие влево, т. е. в сторону разрушения комплекса.
ЭДТА является четырехосновной кислотой, характеризуется
следующими константами: р/Сх = 2; р/Си = 2,7, рКт = 6,2; p/Civ = 10,3,
и представляет собой относительно слабую кислоту, поэтому рН раствора
ее комплекса с Са+ + не должен быть ниже 10,3. Если рН будет меньше,
то Y=== с Н+ образует соответствующие гидроанионы: HY , H2Y"",
H3Y" и кислоту H4Y. При этом комплекс CaY"" разрушается или не
образуется совсем.
Таким образом, устойчивость внутрикомплексной соли, образуемой
ионами кальция с комплексоном III зависит от величины рН раствора.
Поэтому для обеспечения оптимального течения реакции образования
комплекса CaY" " титрование солей кальция раствором ЭДТА нужно
проводить в сильнощелочной среде при рН > 12. В этом случае достигается
полная нейтрализация образующейся в процессе тгитрования свободной
кислоты и наблюдается максимальный скачок кривой титрования (рис. 61).
342 ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
Методика определения. В мерной колбе емкостью 250 мл готовят
приблизительно 0,1 н. раствор какой-либо растворимой в воде соли
кальция. Отбирают в коническую колбу пипеткой 25 мл приготовленного
раствора, добавляют 50 мл дважды перегнанной дистиллированной (или
деионизированной) воды, 25 мл 20%-ного раствора едкого натра, 2—3
капли индикатора и титруют при непрерывном перемешивании 0,1 н.
раствором комплексона III до перехода красной окраски в фиолетовую или
синюю. Под конец титрование проводят очень медленно.
Расчет ведут обычным образом (см. гл. 1, § 10).
Пр имечание. Если определяют процентное содержание окиси кальция в
силикатных материалах, то при расчете пользуются формулой:
_ ^эдтаТэдта/сзО WO-100-VK
Са°~ а (100— W)VA
г^е ^эдта — объем раствора комплексона III, израсходованного на титрование
навески анализируемого материала, мл;
^эдтА/СаО — ТИТР комплексона III, выраженный по определяемому веществу, г/мл
СаО;
а — навеска, г;
W — содержание в силикате воды, %.
§ 31. Определение жесткости воды
комплексонометрическим методом
Жесткость воды выражают числом миллиграмм-эквивалентов
кальция и магния в 1 л.
Определение общей жесткости проводят путем комплексонометриче-
ского титрования раствором комплексона III в присутствии эриохром
черного или хром темно-синего. Обычно определяют суммарное количество
кальция и магния. При необходимости раздельного определения кальция
и магния сначала определяют суммарное их количество. Затем в отдельной
пробе осаждают кальций в виде оксалата кальция и оттитровывают ионы
магния. Кальциевую жесткость определяют по разности. В присутствии
некоторых примесей ход анализа изменяется. Ионы меди и цинка
переводят в сульфиды, чтобы марганец не окислялся, прибавляют гидроксил-
амин.
Присутствие в воде Cl~, S04"~ и НС03" не мешает определению
общей жесткости. Так как рПРсасо3 <рККомпл.са+ +, осадок карбоната
кальция растворяется и кальций количественно оттитровывают комплек-
соном III.
Методика определения — см. § 29.
Расчет:
w 0,05Т/-/С-Ю00 50-1/ v
ж = = д
кн2о ^н2о
где Ж — жесткость воды, мг-экв/л;
V — объем израсходованного раствора комплексона III, мл;
К — поправочный коэффициент 0,05 н. раствора комплексона III;
VH о — объ^ем взятой для исследования воды, мл.
Пример. Для титрования взято 25 мл воды, израсходовано 2,20 мл 0,05 н. раствора
комплексона III. Поправочный коэффициент (К) этого раствора 0,9600.
w 0,05-2,20.0,9600* 1000
Ж = —^ = 4,22 мг-экв/л
Для перевода мг-экв/л в градусы жесткости полученную величину жесткости в мг-экв/л
умножают на 2,8: 4,22x2,8= 11,8°.
§ 33. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АЛЮМИНИЯ
343
Объем отбираемой пробы воды зависит от содержания солей кальция
и магния и колеблется от 10 до 100 мл.
Жесткость воды, градусы 1—15 15—30 30—60 60
Объем пробы, мл ... 100 50 25 10
Отмеряют требуемый объем анализируемой воды, добавляют 5 мл
аммиачного буферного раствора (рН ^ 9), 3—5 капель раствора эрио-
хрома черного Т или другого подходящего комплексонометрического
индикатора, имеющегося в лаборатории, доводят объем смеси до 100 мл
и титруют стандартным 0,05 н. раствором комплексона III до изменения
красной окраски раствора в синюю.
§ 32. Анализ смеси ионов кальция и магния
Раздельное определение ионов кальция и магния при их совместном
присутствии основано на предварительном определении общего
содержания ионов кальция и магния титрованием аликвотной части
анализируемого раствора стандартным раствором комплексона III в присутствии эрио-
хром черного Т (или кислотного темно-синего) и последующем
определении в отдельной пробе содержания ионов кальция титрованием
аликвотной части в присутствии мурексида.
Методика определения. Пробу анализируемой смеси помещают в
мерную колбу, разбавляют дважды перегнанной дистиллированной (или
деионизированной) водой, доводят до метки и тщательно перемешивают.
Из мерной колбы отбирают аликвотную часть полученного раствора,
добавляют 50 мл дважды перегнанной дистиллированной воды, 25 мл
аммиачной буферной смеси, 2—3 капли эриохром черного Т (или
кислотного хром темно-синего) и титруют стандартным раствором комплексона III
до перехода винно-красной окраски раствора в фиолетово-синюю. Под
конец титрование проводят очень медленно. На титрование смеси ионов
кальция и магния идет объем раствора комплексона III — Кэдта.
Для определения содержания ионов кальция вновь отбирают
аликвотную часть раствора, добавляют 5 мл 20%-ного раствора NaOH, доводят
общий объем до 100 мл> вносят на кончике шпателя 30—40 мг смеси
мурексида с хлоридом натрия и титруют стандартным раствором ЭДТА до
перехода красной окраски в фиолетовую. На титрование ионов кальция идет
объем раствора комплексона III — Уэдта.
Расчет. Содержание ионов кальция вычисляют по формуле:
_ ЭСа**УЭДТА^ЭДТАУк
^Са++~ 1000 VA
Содержание ионов магния вычисляют по формуле:
^Mg++ (^эдта "~ ^эдта) ^эдта^к
^Mg++"" 1000 VA
§ 33. Определение содержания алюминия
Методы количественного определения алюминия. Существуют
различные методы определения алюминия,
а) Весовые методы.
1) Осаждение в виде гидроокиси А1 (ОН)3 и взвешивание в виде А1208
(см. «Весовой анализ»).
2) Осаждение в виде оксихинолята Al (C9HeNO)3 и непосредственное
взвешивание полученного осадка или взвешивание после прокаливания
в виде А1203 (см. «Весовой анализ»).
344 ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
б) Объемные методы
1) Осаждение в виде оксихинолята алюминия, растворение
полученного осадка в хлористоводородной кислоте, обработка высвободившегося
оксихинолина смесью бромида и бромата калия и титрование избытка
окислителя иодометрическим методом.
2) Комплексонометрический метод.
Существуют и другие методы определения алюминия, основанные
на использовании цветных реакций ионов алюминия.
Среди весовых методов определения алюминия в сталях, сплавах
и других материалах металлургического производства, а также в
минеральном сырье, наиболее быстрым, точным и универсальным является
фторидный (криолитовый) метод, разработанный И. В. Тананаевым и
П. Я. Яковлевым. Метод основан на осаждении ионов алюминия в виде
криолита Na3 [A1F6] фторидом натрия при рН = 1 —3 в присутствии
цитрата и оксалата аммония, как комплексующих агентов, и
последующем переведении осадка криолита в оксихинолинат алюминия, который
прокаливают и взвешивают в виде А1203. Проведению весового анализа
мешают редкоземельные элементы и ионы щелочноземельных металлов.
Метод широко применяется. В настоящее время весовое окончание
определения заменено комплексонометрическим титрованием, получившим
наиболее широкое применение в практике определения алюминия.
Комплексонометрический метод определения алюминия. Комплексоно-
метрическое определение алюминия основано на образовании внутри-
комплексной соли алюминия с комплексоном III и последующем
оттитровании добавленного в анализируемый раствор избытка стандартного
раствора комплексона солями цинка, железа или тория. По количеству
вошедшего в реакцию с ионами алюминия комплексона III судят о
содержании алюминия в анализируемом растворе.
Определение алюминия в присутствии ксиленолового оранжевого.
Исходный раствор, содержащий ионы алюминия, разбавляют
дистиллированной водой в мерной колбе емкостью 250 мл, тщательно
перемешивают, доводят до метки и снова перемешивают. Берут пипеткой аликвот-
ную часть раствора (25 мл) и переносят в коническую колбу. Затем в
коническую колбу приливают избыток стандартного раствора комплексона
III, добавляют 20 мл ацетатного буферного раствора, кипятят 5 мин,
охлаждают, добавляют 7 капель ксиленолового оранжевого и титруют
избыток комплексона III стандартным раствором соли цинка до
перехода лимонно-желтой окраски раствора в розовую.
Характеристики стандартного раствора соли цинка предварительно
определяют прямым титрованием раствором комплексона III в присутствии
ксиленолового оранжевого в ацетатной буферной среде до перехода
розовой окраски раствора в лимонно-желтую.
Определение алюминия в присутствии сульфосалицилата натрия.
Анализируемый раствор готовят как указано выше, но титруют
стандартным раствором соли железа (III) в присутствии сульфосалицилата натрия
до возникновения красно-коричневого окрашивания, устойчивого в
течение I мин.
Характеристики стандартного раствора соли железа (III) определяют
прямым титрованием стандартного раствора комплексона III раствором
соли железа (III) также в присутствии сульфосалицилата натрия в
ацетатной буферной среде.
Расчеты — см. гл. I, § 10.
В тех случаях, когда алюминию сопутствуют мешающие его комплек-
сонометрическому определению ионы, принимают ряд необходимых мер
для устранения вредного влияния этих примесей.
§ 35. РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ ИОНОВ АЛЮМИНИЯ И ЖЕЛЕЗА 345
Один из вариантов комплексонометрического определения алюминия,
выполнению которого не мешают многие катионы и анионы, в том числе
катионы щелочноземельных металлов и железа и анионы серной,
фосфорной и других кислот, основан на образовании внутрикомплексной соли
алюминия с комплексоном III и последующем разложении ее фторидом
натрия. При этом образуется более устойчивое комплексное соединение
алюминия fAlF6]3-, а комплексон III, ранее связанный с ионами алюминия,
освобождается. Выделившийся комплексон III оттитровывают
стандартным раствором соли цинка в присутствии ксиленолового оранжевого.
Если в титруемом растворе не содержатся другие катионы,
мешающие определению алюмдния, то по количеству вступившего в реакцию
комплексона III легко вычислить содержание алюминия в анализируемом
продукте.
В том случае, когда в титруемом растворе содержатся также другие
катионы, например (Са++), реагирующие, подобно ионам алюминия,
с комплексоном III, то по количеству вступившего в реакцию
комплексона III нельзя судить о содержании алюминия. Необходимо установить,
сколько комплексона III непосредственно связано с ионами алюминия.
Для этого после того как весь избыток комплексона III будет связан
ионами цинка в виде внутрикомплексной соли, к раствору прибавляют
30 мл насыщенного раствора фторида натрия и кипятят 5 мин. В процессе
кипячения с NaF комплексное соединение алюминия с комплексоном III
разлагается. При этом освобождается эквивалентное количество
комплексона III, ранее связанного с ионами алюминия. Внутрикомплексное
соединение кальция при кипячении не разлагается. По охлаждении к раствору
вновь добавляют немного указанного выше индикатора и оттитровывают
выделившийся комплексон III стандартным раствором соли цинка.
Расчет результатов анализа проводят, исходя из того, что титр 0,1 н.
раствора соли цинка, выраженный через определяемое вещество,
соответствует 1,349 мг А1 или 2,548_жг А1203.
Другой вариант комплексонометрического определения алюминия
в присутствии ионов кальция основан на раздельном титровании их при
различном значении рН с разными индикаторами (см. § 34).
§ 34. Раздельное определение ионов кальция и алюминия
Подготавливают анализируемый раствор для титрования. Из одной
аликвотной части определяют содержание ионов кальция в сильно
щелочной среде в присутствии мурексида (см. § 29), а в другой содержание
ионов алюминия методом обратного титрования избытка комплексона III
стандартным раствором соли цинка в присутствии ксиленолового
оранжевого в ацетатной буферной среде (см. § 32). Ионы кальция не мешают
определению алюминия в этом случае.
§ 35. Раздельное определение ионов алюминия и железа
Исходный раствор подготавливают, как указано выше (см. § 32).
Аликвотную часть анализируемого раствора помещают в коническую
колбу для титрования и определяют в ней содержание ионов железа (III)
путем прямого титрования стандартным раствором комплексона III
в присутствии сульфосалициловой кислоты при рН = 2. В этих
условиях А1+ + +, Са++ и Mg++ не мешают определению Fe+ + +.
Оттитровав железо (III), приступают к определению ионов алюминия
в той же пробе анализируемого раствора. Ионы алюминия определяют
обратным титрованием предварительно введенного избытка раствора ком-
346 ГЛ. IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
плексона III стандартным раствором соли железа (III) при рН = 4,8—
5,0. При этом значении рН раствора ранее образовавшийся комплексонат
железа (III) не разрушается и не мешает определению ионов алюминия.
Расчеты. Если определяют содержание окиси железа в силикатных
материалах в процентах, то вычисляют по формуле:
_ УЭДТАТЭДТА/Ре203 ЮО'ЮО'Ук
*Fe203- а '(100— W)VA
где ^эдта— объем раствора комплексона III, пошедшего на титрование навески
анализируемого материала, мл;
^эдТА/Ре2о3 — ТИТР комплексона III, выраженный по определяемому веществу,
г/мл Fe203;
а — навеска, г;
W — содержание воды в силикате, %.
Содержание окиси алюминия рассчитывают по формуле:
(^эдта -~ ^эдта'^эдта) ТУдтА/АЬОз Ю0-100-Ук
*А1208~ а ' (100—W)VA
где ^эдта — объем стандартного раствора комплексона III, добавленного после
титрования FeIH, мл\
УЭдТА — объем стандартного раствора соли железа (III), пошедшего на обратное
титрование избытка комплексона III, мл;
^ЭДТА/А12о3 — ТИТР комплексона III, выраженный по определяемому веществу, г/мл
А1203;
а — навеска, г;
/Сэдта — поправочный коэффициент;
ЧАСТЬ ВТОРАЯ
Весовой анализ
ГЛАВА V
ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Л. ОБЩАЯ ХАРАКТЕРИСТИКА ВЕСОВОГО АНАЛИЗА
§ 1. Сущность весового анализа
Весовым анализом называют метод количественного химического
анализа, основанный на точном измерении массы определяемого вещества
или его составных частей, выделяемых в химически чистом состоянии или
в виде соответствующих соединений {точно известного постоянного
состава).
Непосредственное выделение из анализируемого продукта
определяемого вещества или его составных частей в химически чистом
состоянии представляет во многих случаях очень трудную, а порой и
неосуществимую задачу. Поэтому очень часто определяемое вещество выделяют
в осадок в виде соединения определенного состава. Для этого взвешенное
количество (навеску) анализируемого вещества переводят в раствор,
к полученному раствору прибавляют соответствующий реактив,
реагирующий с одним из компонентов анализируемой смеси с образованием
малорастворимого соединения. При этом определяемая составная часть
анализируемого вещества (катионы или анионы) выделяется из раствора
в виде практически нерастворимого осадка. Выделившийся осадок
отделяют от раствора фильтрованием или центрифугированием, промывают
с целью удаления всех растворимых в данном растворителе примесей,
высушивают или прокаливают до постоянной массы и взвешивают на
аналитических весах.
§ 2. Классификация методов весового анализа
Все многочисленные весовые определения можно разделить на три
большие группы методов, а именно: методы выделения, осаждения и
отгонки.
Методы выделения. В методах выделения определяемый компонент
количественно выделяют в свободном состоянии из анализируемого
вещества и взвешивают на аналитических весах. Так, например,
количественно определяют золото и медь в сплаве.
При растворении определенной навески сплава в царской водке
получают раствор, содержащий ионы золота и меди. При добавлении к
полученному раствору перекиси водорода, восстанавливающей ионы
золота до элементарного золота и не оказывающей влияния на ионы меди,
все золото выделяется в элементарном состоянии. Выделившееся золото
отфильтровывают, промывают разбавленным раствором
хлористоводородной кислоты от посторфнних примесей, помещают вместе с фильтром
в предварительно взвешенный фарфоровый тигель, высушивают, прока-
348
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
ливают для удаления летучих примесей и после охлаждения
взвешивают. По массе выделившегося золота судят о содержании его в
анализируемом сплаве.
Если через промывные воды и фильтрат, оставшийся после
отделения золота, пропустить при определенных условиях постоянный
электрический ток, то на предварительно взвешенном, инертном по отношению
к раствору платиновом катоде количественно выделяется металлическая
медь. По увеличению массы катода судят о содержании меди в сплаве.
Описанный метод определения золота в сплаве называют весовым,
а меди — электровесовым.
Другим примером подобного определения является определение
процентного содержания золы в твердом топливе, основанное на сжигании
и прокаливании до постоянной массы навески топлива в предварительно
взвешенном тигле. Оставшуюся в тигле золу взвешивают. По массе золы
вычисляют ее процентное содержание в данном образце твердого топлива.
Методы осаждения. В методах осаждения определяемый компонент
количественно осаждают химическими способами в виде малорастворимого
химического соединения строго определенного состава. Выделившийся
осадок промывают, высушивают или прокаливают. При этом осадок
большей частью превращается в новое вещество точно известного состава,
которое и взвешивают на аналитических весах. В весовом анализе
различают: форму осаждения, т. е. форму, в виде которой осаждают
определяемое вещество, и весовую форму, т. е. форму, в виде которой
определяемое вещество взвешивают. Весовая форма может иметь ту же формулу,
что и форма осаждения. Например, при определении сульфат-ионов
весовым методом, путем осаждения их ионами бария, формула формы
осаждения (осадка) и формула весовой формы при соблюдении всех требуемых
условий анализа одна и та же. Схема такого определения представляется
следующим образом:
Ba++ t
S04 —> BaS04 —> BaS04
определяемое форма весовая
вещество осаждения форма
В некоторых весовых методах определения путем осаждения формула
весовой формы отличается от формулы осадка. Например, при
определении ионов железа, осаждаемых в виде гидроокиси, схема определения:
6 (OH)" t
2Fe+++ —> ^2Fe(OH)3 —>¦ Fe203
-зн2о
определяемое форма весовая
вещество осаждения форма
Методы отгонки. В методах отгонки определяемый компонент
количественно отгоняют в виде летучего соединения. Отделение определяемой
части осуществляют путем нагревания анализируемого вещества или
действием соответствующих реагентов, сопровождающимся выделением
летучих продуктов. Методы отгонки бывают прямые и косвенные.
Прямые методы отгонки. Определяемый летучий
компонент поглощают специфическим поглотителем и по увеличению массы
последнего вычисляют, количество определяемого компонента.
Примером прямого весового определения летучего вещества является
определение С02 в карбонатных породах, основанное на разложении
карбонатов кислотами:
СаС03 + 2Н+ -^ С02| + Са++ + Н20
С02 + 2NaOH — Na2C03 + Н20
§ 3. РАСЧЕТЫ В ВЕСОВОМ АНАЛИЗЕ
349
Образец карбоната разлагают в специальных приборах,
позволяющих улавливать выделяющийся С02. Содержание С02 вычисляют по
увеличению массы поглотительной трубки, наполненной натронной известью
(CaO+NaOH).
Косвенные методы отгонки. В косвенных методах
определения узнают массу остатка вещества после полного удаления
определяемого вещества. Разность в массе до и после отгонки
определяемого вещества дает возможность вычислить количество определяемого
компонента. Схема этого определения:
ВаС12-2Н20 -U ВаС12 + 2Н20 f
Косвенные способы весовых определений применяют при определении
влажности материалов, кристаллизационной воды в кристаллогидратах,
потерь при прокаливании и т. п.
Преимущества и недостатки весового анализа. Весовые методы
анализа позволяют с относительно большой точностью определять в данном
образце анализируемого вещества количественное содержание
отдельных компонентов или (если дан раствор) концентрацию их в растворе.
Весовой анализ пригоден для определения очень многих металлов
(катионов) и неметаллов (анионов), составных частей сплавов, руд, силикатов,
органических соединений и т. д.
Существенным недостатком весового метода является большая
длительность определений, намного превосходящая длительность определений,
осуществляемых посредством объемных методов анализа.
По этой причине методы весового анализа утратили свое прежнее
значение; в практике их заменяют современными экспрессными химическими
и физико-химическими методами.
Однако весовые методы, отличающиеся большой точностью,
полностью сохраняют свое значение при арбитражных (спорных) анализах
и широко применяются при выполнении научно-исследовательских
работ для сравнения аналитических данных, полученных разными
-методами. При' помощи весового анализа ведут определения с точностью до
0,01—0,005%, что превосходит точность объемных методов.
§ 3. Расчеты в весовом анализе
Все вычисления в весовом анализе проводят согласно правилам,
изложенным ранее (см. «Введение», § 11 и гл. I, § 11).
При правильном выполнении всех аналитических операций весового
анализа ошибка опыта определяется точностью взвешивания. При
одинаковой абсолютной ошибке взвешивайия большая навеска исходного
вещества приводит к большей относительной точности результата анализа,
выражаемой в процентах.
Вместе с тем применение очень больших навесок тоже связано с
неудобствами, обусловливаемыми необходимостью затраты на их обработку
большого количества времени. Поэтому следует учитывать все
изложенные соображения при вычислении величин навесок, рассчитываемых
по формулам (1) и (2) (см. ниже).
Расчеты ведут с помощью четырехзначных таблиц логарифмов.
Расчет навески анализируемого вещества. Для расчета навески (а)
анализируемого вещества составляют пропорцию, исходя из уравнения
реакций. Следует также учитывать, что если анализируемое вещество
содержит значительное количество посторонних примесей, то навеска
должна соответствовать содержанию определяемого вещества в
исследуемом образце.
350
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
а) Для кристаллических осадков. Например, для
определения в СаС03 кальция в виде СаС204 навеску анализируемого
вещества вычисляют, исходя из следующих уравнений реакций:
СаСОз + 2Н+ —> Са++ + Н20 + С02 f
Са++ + С204-
СаСг^
СаС204 Л- СаО + СО | +С021
т. е.
СаСОз —> Са++ -
с0о
'2^4
-> СаСоО,—> СаО
Следовательно
Mt
СаСОз
-М
СаО
а —0,5 г
мСаСОз*0'5 100,1-0,5
М,
СаО
56,1
= 0,9 г
где 0,5 — рекомендуемая масса весовой формы определяемого вещества (в граммах) для
кристаллических осадков.
б) Для аморфных осадков. Например, если анализируют
Fe(N03)3-9H20, то навеску вычисляют, исходя из следующих
уравнений реакций:
-jFe(OH)*
¦ Fe203 + ЗН20 f
т. е.
Fe+++ + ЗОН-
2Fe(OH)3-^
вон
Следовательно
2Fe (N03)
ж,
Fe (N08)e-9H20 '
-А*,
Fe2Oa
^FelNOaJa-SH^"""0»1
2Fe(OH)3
2М,
•Fe203
a.
Fe(NOa)s.9H20 :
Fe (NOs)3»9H2Q,Q>1 808,0-0,1
M,
Fe208
*0,5 а
159,7
где 0,1 — рекомендуемая масса весовой формы (в граммах) определяемого вещества для
аморфных осадков.
В общем виде при расчете навесок анализируемых веществ
пользуются формулами:
а) при осаждении кристаллических осадков
тМА0,5
nMAt
б) при осаждении аморфных осадков
тМА0,1
a = ¦
пМй
(1)
(2)
где a — навеска образца анализируемого вещества, г;
Ма—молекулярный вес определяемого вещества;
МAi — молекулярный вес весовой формы;
0,5 — практически удобная масса весовой формы определяемого вещества для
кристаллических осадков (найдено опытным путем), г;
0,1 — практически удобная масса весовой формы определяемого вещества для
аморфных осадков (найдено опытным путем), г;
т, л — коэффициенты [в примере (а) т = 1 и п = 1, в примере (б) т = 2 и п = 1 ].
Расчет объема раствора осадителя. Для расчета объема раствора
осадителя составляют пропорцию, исходя из уравнения реакции.
Например:
AgN03 + HC1 -^ 4- AgCl + HNO3
3. РАСЧЕТЫ В ВЕСОВОМ АНАЛИЗЕ 351
Количество осадителя (хлористоводородной кислоты) в граммах,
необходимое для осаждения Ag +-ионов из образца нитрата серебра,
вычисляют на основании пропорции:
МНС1— MAgNOu
?НС1 ~-"aAgN03
MHC1aAgNo3
2HCi=^jr
aAgN03
Для определения объема раствора осадителя необходимо знать его
концентрацию и плотность.
Практически количество приливаемого осадителя должно превышать
теоретически рассчитанное в полтора раза.
В общем виде при расчете объема раствора осадителя пользуются
следующей формулой:
1,5тМваЮ0 „.
Ув= лМАСр <3>
где а — навеска образца анализируемого вещества, г;
Ма—молекулярный вес определяемого вещества;
Мв—молекулярный вес осадителя;
Vb — объем раствора осадителя, мл;
р — плотность раствора осадителя, г/мл;
С — концентрация раствора осадителя, %;
1,5 — практически найденный коэффициент;
т, п — коэффициенты, определяемые из уравнений реакций.
Расчет результатов определения при анализе по методу осаждения.
Содержание определяемого вещества обычно вычисляют или в граммах
или в процентах.
Расчет массы определяемого вещества. Массу
определяемого вещества (gjj в граммах находят по формуле:
Sa = *a/b W
где 0Ai — масса весовой формы определяемого вещества, г;
Fq — аналитический множитель (фактор) весового анализа.
Величину FB находят по справочным таблицам. Он представляет
собой отношение
р = тМ а
nMAi
где Ма — молекулярный вес определяемого вещества;
МAi — молекулярный вес весовой формы определяемого вещества;
m, n — коэффициенты.
Например, если нужно определить содержание железа в
анализируемом растворе по массе выделенного Fe203, то аналитический множитель
(^в) выражают отношением
2Л*Ре
MFea08
где т = 2 и п = 1.
Аналитический множитель (фактор) весового анализа обозначают
3 данном случае FFe/Fe2o8- Такое обозначение означает, что
аналитический множитель пригоден для расчета содержания железа по массе
весовой формы Fe203. Если аналитический множитель обозначить /*>е2о8/Ре,
то его можно использовать для расчета Fe2Od по массе выделенного
железа.
352
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
В первом случае:
во втором случае:
^Fe/Fe203 ~~
F Fe2Os/Fe =
. 2MFe
" MFe203
%е203
2MFe
= 0,6994
1,4297
Следовательно, при выражении аналитического множителя (фактора)
весового анализа в числителе указывают формулу определяемого вещества,
а в знаменателе — формулу его весовой формы.
Расчет процентного содержания
определяемого вещества. Для вычисления процентного содержания
анализируемого вещества исходят из соответствующих пропорций. Например,
если необходимо определить процентное содержание железа в
анализируемом образце Fe(N03)3'9H20 по массе выделенной весовой формы
Fe203, то по формуле (4):
gFe = ^FeaOa^Fe/FeaOa
Следовательно
aFe (N03)3-9H20 ~~^Fe I 100
100 —a: I flFe(N03)8-9H20
Подставив в данную формулу значение gpe, получим:
х = ^Oa^Fe/FesO,'100 %
aFe (N03)3-9H20
В общем виде при расчете процентного содержания определяемого
вещества применяют формулу:
где а — навеска образца анализируемого вещества, г;
адх — масса весовой формы определяемого вещества, г;
Fq — аналитический множитель (фактор) весового анализа.
Расчет результатов определения при анализе по методу выделения.
Процентное содержание определяемого вещества, выделенного в
свободном виде, вычисляют, исходя из соответствующих пропорций. Например,
если необходимо вычислить процентное содержание Si02 в анализируемом
образце известняка, то
^известняка"
100 — д:
х- gSi0'-100 «/о
^известняка
и так как формула весовой формы та же, что и формула определяемого
вещества, то F = 1 -и aSio2 = gsioa. Тогда
100
х = aSiQ %
2 ^известняка
В общем виде при расчете процентного содержания определяемого
вещества, выделенного в свободном виде, пользуются формулой:
100 100 п/
Расчет результатов анализа при прямом весовом определении по
методу отгонки. При прямых определениях летучие продукты обычно вы-
§ 4. ВЗЯТИЕ И РАСТВОРЕНИЕ НАВЕСКИ
353
деляют в форме летучих соединений определенного состава.
Кристаллогидраты разлагаются с выделением воды, карбонаты — с образованием С02
и т. п. Поэтому количество определяемого вещества вычисляют по
увеличению массы поглотительного прибора. Процентное содержание летучего
компонента вычисляют, исходя из соответствующих пропорций.
Например, если необходимо вычислить процентное содержание С02
в карбонате кальция по увеличению массы поглотительного прибора,
в котором улавливается выделяющаяся С02, то:
аСаСОа ~~аС02
100 — х
асп -100
*=-?^—%
аСаСОа
В общем виде при расчете процентного содержания определяемого
вещества прямым определением по методу отгонки пользуются формулой
* = «А,^% (7)
гДе tfAi — масса весовой формы вещества, определяемого по увеличению массы
поглотительного прибора, г;
а — навеска образца анализируемого вещества, г.
Расчет результатов анализа при косвенном определении по методу
отгонки. При косвенном способе определения летучие продукты удаляют
нагреванием или прокаливанием анализируемого образца.
Например, если необходимо определить процентное содержание влаги
в образце угля по потере массы после высушивания навески, то:
аугля — аН20 I аН2О*10° п/
\ х = — %
100 — a: J Яугля
гДе аи2о = аугля ~ а\ (а1 — масса остатка, г).
Процентное содержание летучего компонента при косвенном
определении по методу отгонки вычисляют по формуле:
* = (a-0i)-^-% (8)
где а — навеска образца анализируемого вещества, г;
ах — масса высушенного или прокаленного анализируемого вещества после удаления
летучих определяемых компонентов, г.
Расчет результатов анализа по разности косвенного определения.
Иногда определяемое вещество не выделяется, не осаждается и не
отгоняется. В таком случае о его содержании судят по разности между массой
суммы (в г или %) всех компонентов смеси (находят опытом) и найденной
опытом массой остальных компонентов.
Так обычно устанавливают содержание кислорода в органических
веществах, когда известно содержание в них углерода, водорода и азота
и других элементов.
Б. ТЕХНИКА ВЕСОВОГО АНАЛИЗА
§ 4. Взятие и растворение навески
Величина навески вещества, подвергаемого исследованию, влияет
на точность анализа. Чем больше величина навески анализируемого вещества,
тем выше относительная точность результатов анализа.
Для расчета навески необходимо знать приблизительное содержание
компонентов в исследуемой пробе анализируемого вещества или его
формулу.
354
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Кристаллические осадки обладают малым объемом, аморфные —
большим, следовательно, и величина навески вещества должна быть
различной. Получаемый осадок не должен быть большим, так как иначе
возрастают экспериментальные трудности, связанные с работой с большими
навесками, и требуется больше времени для анализа. В то же время
величина осадка должна быть достаточной для того, чтобы было удобно с ним
оперировать. Кроме того, применение слишком малых навесок может быть
причиной значительных относительных ошибок при взвешивании.
В весовом анализе допустимая ошибка при взвешивании не должна
превышать 0,1%. Предположим, что применяемые весы дают возможность
Рис. 62. Стеклянные ампулы для взятия навесок легколетучих
жидких веществ и раздавливание ампулы в стакане с
растворителем.
взвешивать с точностью до 0,0001 г, тогда минимальную навеску,
которую можно взвешивать, допуская ошибку в ±0,1%, рассчитывают
следующим образом.
Если взвесить 1 г с ошибкой ±0,0001 г, то допущенная ошибка
составит 0,0001-100 = ±0,01%; при взвешивании 0,1 г ошибка достигнет
предельной величины, т.е. ±0,1%.
Следовательно, минимальная навеска, которую можно взвешивать
на обычных аналитических весах, не должна быть меньше 0,1 а.
Чем меньше процентное содержание определяемого компонента в пробе,
тем больше должна быть навеска.
Взятие навески для анализа. Точное взятие навески играет решающую
роль в количественном анализе. От точности, с которой взята навеска
анализируемого вещества, зависит достоверность результатов анализа.
Гигроскопические и летучие вещества вызывают особые затруднения
при взятии навески и требуют в отдельных случаях специальных условий
при взвешивании и отборе навесок.
Для взятия навесок твердых веществ обычно применяют часовые стекла,
специальные пробирки или бюксы, а для жидких веществ — капельницы,
маленькие колбы емкостью 1—2 мл, желатиновые капсули или
подвешиваемые пипетки с пришлифованным краном. Для взятия навесок
легколетучих веществ применяют тонкостенные ампулы (рис. 62), из которых
перед их заполнением удаляют воздух.
Перенесение навески твердого вещества, а) Взвешенный бюкс
(часовое стекло или пробирку) с навеской осторожно берут с чашки весов
и содержимое его аккуратно высыпают в стакан. Затем постукивают
по стенкам бюкса стеклянной палочкой или указательным пальцем.
После перенесения навески в стакан бюкс с остатком навески снова
взвешивают на аналитических весах. В этом случае величину навески образца
для анализа определяют как разность между массой бюкса с навеской
и массой бюкса после высыпания навески.
§ 5. ТЕХНИКА ОСАЖДЕНИЯ
355
б) Взвешенный бюкс с навеской осторожно берут с чашки весов и
опрокидывают над стаканом. Затем остатки навески смывают
дистиллированной водой из промывалки. В этом случае величину навески образца
для анализа определяют как разность между массой бюкса с навеской и
массой чистого бюкса.
При перенесении навески в стакан следует тщательно следить, чтобы
не были потеряны даже незначительные количества вещества.
Перенесение навески жидкого вещества. Для взятия навески
жидкости применяют специальные пипетки с краном. При перенесении
навески жидких тел открывают кран пипетки и осторожно выливают
содержимое в стакан, после чего пипетку с остатками пробы снова
взвешивают. Величину навески жидкого вещества определяют как разность
между массой пипетки с навеской и массой пипетки с остатком вещества.
Растворение навески. Вещества, нерастворимые в холодной воде,
растворяют при слабом нагревании на водяной или воздушной банях
в химических стаканах или колбах. Стакан покрывают часовым стеклом,
которое кладут выпуклой стороной вниз, в колбу вставляют стеклянную
воронку для конденсирования паров.
Иногда растворение ведут в фарфоровых чашках. Чашки накрывают
часовым стеклом, которое кладут выпуклой стороной вниз, стекло
помещают не непосредственно на чашку, а на стеклянный треугольник.
Растворение в кислотах (разбавленных или концентрированных)
выполняют под тягой.
§ 5. Техника осаждения
Количество растворителя. Когда из раствора взятой пробы осаждают
кристаллический осадок, пробу разбавляют водой с таким расчетом,
чтобы получить примерно 0,1 н. раствор.
Так как объем раствора, содержащего 0,1 г-же основной части
анализируемого вещества, равен 1000 мл, то можно составить пропорцию:
0,1 Э — 1000 мл
а— V мл
где а — навеска пробы, г;
V— объем полученного 0,1 н. раствора вещества, мл.
Отсюда
Здесь не требуется большой точности, и объем воды, взятой для
растворения пробы, приравнивают к объему раствора, полученного после
растворения пробы.
Если из раствора осаждают аморфный осадок, то навеску растворяют
в минимальном количестве воды, чтобы получить концентрированный
раствор.
Количество осадителя. Для осаждения кристаллических и
аморфных осадков количество раствора осадителя следует брать примерно
в 1,5 раза больше рассчитанного.
Обычно в лабораториях имеются растворы осадителей определенной
нормальности. В этом случае рассчитанный объем раствора осадителя
отмеривают с точностью 0,1—0,2 мл. Если раствора осадителя нет, его
готовят, исходя из навески сухого вещества.
Для осаждения кристаллических осадков пользуются разбавленными
растворами осадителя, поэтому отмеренный объем или отвешенное ко-
356
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
личество осадителя разбавляют водой примерно до 50 мл. Для осаждения
аморфных осадков пользуются концентрированными растворами
осадителя, в этом случае отмеренный объем осадителя разбавляют водой только
до 5 мл.
Осаждение. Обычно осаждение ведут в том же сосуде, в котором
проводилось растворение пробы. Если же проба была растворена в
фарфоровой чашке, то содержимое чашки переливают в химический стакан
емкостью 200—300 мл и чашку тщательно обмывают водой, собирая воду
в тот же стакан. Осаждение ведут при нагревании. Техника осаждения
кристаллических и аморфных осадков несколько различна.
Осаждение кристаллических осадков.
Кристаллические осадки осаждают при нагревании на водяной или воздушной
бане. Как правило, не следует доводить раствор до кипения, так как
добавление реактива к кипящему раствору может вызвать резкое
выделение пара и разбрызгивание, что ведет к потерям. Осадитель добавляют
медленно по каплям при непрерывном помешивании раствора. Для
перемешивания пользуются стеклянной палочкой, но нужно следить, чтобы
палочка не прикасалась к стенкам и ко дну стакана и не царапала стекло.
Каждый раз, когда палочку вынимают из стакана, ее следует промывать
дистиллированной водой над стаканом. Осадитель необходимо приливать
по Стенке стакана. Когда осадок соберется на дне и жидкость над
осадком станет прозрачной, к ней приливают несколько капель реактива,
чтобы убедиться в полноте осаждения. Осажденный кристаллический
осадок не следует сразу отфильтровывать, а его необходимо оставить постоять
некоторое время (1—6 ч) на водяной бане в накрытом часовым стеклом
стакане. Эту операцию называют созреванием осадка.
Осаждение аморфных осадков. Аморфные осадки
осаждают из горячих концентрированных растворов и
концентрированными растворами осадителя. Осаждение проводят быстрым прибавлением
осадителя, что дает возможность получать осадки с минимальной
поверхностью.
Когда осаждение закончено, в стакан прибавляют 100—150 мл
горячей воды и быстро фильтруют, чтобы избежать пептизации аморфного
осадка.
Услбвия, при которых проводят осаждение, более подробно изложены
в методиках анализов.
§ 6. Фильтрование и промывание осадков
Фильтрование. В количественном анализе применяют беззольные
фильтры, которые сгорают почти полностью (без остатка). Масса
оставшейся золы составляет 0,00003—0,00007 г и обычно ею пренебрегают.
При проведении точных анализов массу золы, указанную на обертке пачки
фильтров, учитывают при расчетах.
Иногда для фильтрования применяют так называемую бумажную
пульпу (измельченную беззольную фильтровальную бумагу). Осадки,
легко восстанавливающиеся при обугливании фильтров и последующем
прокаливании, желательно фильтровать через пористые стеклянные и
фарфоровые тигли или через специальные воронки с пластинками из
пористого стекла.
В зависимости от размеров частиц полученного осадка применяют
бумажные фильтры разной степени пористости. В СССР выпускают три
сорта беззольных фильтров: 1) синяя лента — для мелкозернистых
осадков; 2) белая лента — для осадков средцей зернистости и 3) красная
лента — для крупнозернистых и аморфных осадков. Размер фильтра
§ G. ФИЛЬТРОВАНИЕ И ПРОМЫВАНИЕ ОСАДКбВ
357
определяется величиной осадка, а не объемом фильтруемой жидкости.
Только одна треть фильтра должна быть заполнена осадком. Размер
воронки подбирают так, чтобы края фильтра были на 0,5—1 см ниже края
воронки. ft
При сливании жидкости на фильтр поль- |
зуются стеклянной палочкой, служившей |
для перемешивания в процессе осаждения.
Палочку вынимают из стакана и держат
левой рукой в вертикальном положении над
воронкой. Нижний конец палочки должен
близко подходить к фильтру, но не касаться
бумаги. Правой рукой берут стакан с
фильтруемой жидкостью, прикладывают носик
стакана к палочке и осторожно сливают
жидкость на фильтр (рис. 63), пока он не
будет наполнен на 2/3. Каждый раз палочку
опускают обратно в стакан и ждут, пока
жидкость не стечет с фильтра, тогда снова
наливают жидкость в воронку. Когда
большая часть ее будет пропущена через фильтр,
осадок промывают декантацией (рис. 64).
Для этого к осадку приливают несколько
миллилитров промывной жидкости, смесь
перемешивают стеклянной палочкой, дают осадку осесть на дно стакана
и сливают жидкость по палочке на фильтр, стараясь не взмучивать осадка.
Когда промывание декантацией закончено, осадок количественно
переносят на фильтр. Для этого осадок смешивают с промывной
жидкостью и эту суспензию переливают по палочке на фильтр. Когда большая
Рис. 63. Фильтрование через
гладкий фильтр.
V
а 0 в г д
Рис. 64. Декантация:
а — положение перед началом сливания раствора; б, в, г — положение
во время сливания раствора; д — положение после сливания раствора.
часть осадка перенесена на фильтр, ополаскивают внутреннюю
поверхность стакана, направляя на нее струйку промывной жидкости из прот
мывалки. Частицы, плотно приставшие к стакану, удаляют со стенок
стакана протиранием стеклянной палочкой с резиновым наконечником
или палочкой и маленькими кусочками беззольной фильтровальной
бумаги, которые затем кладут в фильтр. При этом протирают всю
внутреннюю поверхность. Стакан и палочки ополаскивают промывной жидкостью
и выливают ее в воронку с осадком. После этого немедленно приступают
к промыванию осадка на фильтре.
358
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Выбор промывной жидкости. Жидкость для промывания выбирают
в зависимости от свойств осадка. Обычно применяют дистиллированную
воду, к которой прибавляют небольшое количество веществ,
понижающих растворимость осадка и препятствующих пептизации. Однако эти
вещества должны легко удаляться из осадка при подготовке его к
взвешиванию. При определении катионов, осаждаемых в виде
кристаллических осадков, промывание проводят разбавленными растворами солей
аммония, имеющих с осадком общий ион.
Аморфйые осадки промывают
разбавленными растворами электролитов,
препятствующими пептизации (например, NH4N03).
Соль аммония, оставшаяся в осадке после
промывания, улетучивается при
последующем прокаливании осадка.
Промывание осадка на фильтре. При
промывании осадка на фильтре струю
жидкости из промывалки направляют
в воронку, стараясь, чтобы жидкость
стекала сверху вниз и осадок заполнял
вершину конуса фильтра (рис. 65). Когда
фильтр будет наполнен примерно
наполовину, прекращают дальнейшее
промывание и дают жидкости полностью стечь
с фильтра. Затем на фильтр наливают новую порцию промывной
жидкости и операцию повторяют несколько раз, пока осадок полностью не
будет отмыт от растворимых примесей.
Если начальная концентрация примесей в 1 мл раствора равна х0 и после фильтрования,
на фильтре удержалось а миллилитров жидкости, то количество примесей на фильтре (А0)
будет: А0= ах0. Если для промывания осадка добавлено т миллилитров промывной
жидкости, а на фильтр ев этот момент будет (а + т) миллилитров жидкости, то после прохождения
жидкости через фильтр а миллилитров снова задержутся на фильтре, концентрация
примесей после первого промывания уменьшится и станет равной: хг = , ° ; количество
примесей на фильтре (Аг) окажется: Аг = ахг. Если вторично прилить т миллилитров
промывной жидкости, то
ахг ( а \2 Л
Ь=-^Г7Г = \-Ъ+^) *о; А2 = ах2
V
6
Рис.
65. Промывание осадка на
фильтре:
а — положение до промывания; б —
положение осадка после промывания.
а + т
аналогично
хз
ах2
,а + т
(-^г)3*о: Аз=ахз
а после п промываний
*«=( а + т ) Х°
Физический смысл полученного уравнения (уравнение Бунзена) состоит в том, что
лучше промывать малыми порциями много раз, чем два-три раза большими количествами
промывной жидкости.
Пример. Допустим, для промывания осадка на фильтре взято 100 мл промывной
жидкости. Это количество промывной жидкости может быть использовано различными способами
в зависимости от значений тип.
Например, если т = 50 мл, то п равно 2, т. е. если приливать к осадку каждый раз
по 50 мл жидкости, то ее хватит на два промывания. В данном случае, если а = 10 мл,
то после второго промывания
*2 = ( ю + 50 У Хо =°>02776*о~3.10-2*о
Следовательно, после второго промывания концентрация примесей, удерживаемых
осадком и фильтром, в 1 мл раствора составит 3-Ю"2 начальной концентрации примесей
в I мл раствора (*0).
$7. ПОЛУЧЕНИЕ ВЕСОВОЙ ФОРМЫ
359
Если же т = 10 мл, то п = 10, т. е. если приливать к осадку каждый раз по 10 мл
жидкости, то ее хватит на 10 промываний. В этом случае после десятого промывания
^=(lO+To) *<> = 9,7.10-%
Следовательно, после десятого промывания концентрация примесей в I мл раствора
3-10~2
составит 9,7* 10~4 х0, т. е. уменьшится в 4 я^ 30 раз по сравнению с
концентрацией после двукратного промывания.
Нужно иметь в виду, что нельзя прерывать процесс промывания осадка.
Если непромытый осадок оставить на некоторое время на фильтре, то
масса осадка быстро затвердеет, растрескается на куски и тогда будет
почти невозможно промыть его полностью. Во время фильтрования
полезно накрывать стакан с осадком стеклянной пластинкой, имеющей на
краю вырез для стеклянной палочки.
Для проверки полноты промывания помещают несколько капель
промывной жидкости в пробирку или на часовое стекло и проделывают
чувствительную качественную реакцию на один из тех ионов, которые должны
быть отмыты от осадка. Промывание заканчивают только в том случае,
когда проверочная реакция даст отрицательные результаты.
§ 7. Получение весовой формы
Для получения весовой формы осадок должен быть высушен в
сушильном шкафу или прокален до постоянной массы. Осадки прокаливают
(иногда без предварительного высушивания) в фарфоровых, кварцевых
Рис. 66. Тигли:
а — низкие фарфоровые; б — высокие фарфоровые; в — платиновый тигель
с крышкой.
и платиновых тиглях. Тигли (рис. 66) выпускаются различных размеров
под несколькими номерами. Размер тигля подбирают в зависимости от
объема осадка.
Тигель тщательно промывают, сушат и прокаливают на газовой
горелке, в муфельной или тигельной печи (рис. 67). Прокаливать тигель
следует в тех же условиях, в которых будут прокаливать осадок.
Например, осадок сульфата бария прокаливают на пламени газовой горелки,
поэтому при подготовке тигля для этого осадка его следует прокалить
также на газовой горелке. Если осадок должен быть прокален в
электрической печи при 1000—1200° С, то подготовка тигля также ведется в элек-
360
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
трической печи при той же температуре. Тигель прокаливают 10—15 мин,
вынимают его из печи подогретыми тигельными щипцами и осторожно
ставят в эксикатор.
Эксикатор закрывают пришлифованной стеклянной крышкой и
переносят в весовую комнату.
Когда тигель совершенно остынет и примет температуру воздуха
весовой комнаты (для этого требуется примерно 30—40 мин), его выни-
Рис. 67. Электрические печи:
а — муфельная; б — тигельная; в — высокотемпературная муфельная.
мают холодными щипцами из эксикатора и взвешивают на аналитических
весах с точностью до 0,0002 г. Прокаливание тигля с осадком проводят
не менее двух раз. Если разность результатов двух параллельных
взвешиваний будет не более 0,0002 г, то прокаливание считают оконченным.
В противном случае прокаливание тигля повторяют до тех пор, пока масса
тигля не станет постоянной, т. е. разность между параллельными
взвешиваниями не будет превышать ±0,0002 г.
Рис. 68. Свертывание фильтра с осадком:
а, б, в — сгибание краев фильтра; г — свертывание в спираль,
д — свернутый фильтр с осадком.
Сжигание влажного фильтра с осадком. Обычно сжигают
подсушенный фильтр с осадком, но иногда сжигают влажный фильтр. В этих
случаях его вынимают из воронки, осторожно заворачивают края (рис. 68)
и кладут в тигель. Зятем тигель накрывают крышкой так, чтобы он с
одного края был немного приоткрыт (для выхода паров воды), и ставят
вертикально в фарфоровый треугольник, который помещен на кольцо
штатива (рис. 69). Снизу подставляют газовую горелку. Тигель должен
находиться примерно на 8—10 см выше пламени горелки. Нагревание
следует проводить очень медленно, не давая жидкости в тигле кипеть и
разбрызгиваться. Когда влага будет удалена, тигель постепенно наклоняют
к горелке и увеличивают пламя. Когда бумага фильтра начнет
обугливаться, нужно следить, чтобы она не загоралась. Если все же фильтр на-
§ 9. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ВЫДЕЛЕНИЯ ОСАДКОВ 361
чал гореть, то тигель временно закрывают крышкой. Когда бумага
полностью почернеет и выделение газа прекратится, тигель наклоняют под
углом 45° и нагревают до темно-красного каления до тех пор, пока
углерод полностью не сгорит. Затем тигель опять ставят вертикально и
прокаливают в течение положенного времени. Если осадок требует
прокаливания при высокой температуре (порядка 900—1200° С), то его обычно
заканчивают в электрической печи или на паяльной горелке.
Прокаливание осадка во влажном состоянии
рекомендуется проводить в
платиновом или кварцевом тиглях
(фарфоровые тигли часто лопаются).
Сжигание сухого фильтра с
осадком. Воронку с осадком накрываю!
сверху листом бумаги, проколотым
в нескольких местах булавкой,
помещают в сушильный шкаф и
подсушивают при температуре около 100° С.
Затем воронку С фильтром ВЫНИ- Рис- 69' Прокаливание тигля на горелке,
мают из сушильного шкафа. Фильтр
заворачивают, как показано на рис. 68, и переносят во взвешенный
тигель. Тигель помещают в фарфоровый треугольник над горелкой и
обугливают фильтр, регулируя пламя горелки так, чтобы бумага фильтра
не загоралась, а тлела. При горении фильтра частицы осадка могут быть
подхвачены током горячих газов и вынесены из тигля, что поведет к
потерям. После обугливания фильтра осадок прокаливают при
определенной температуре до постоянной массы.
Если при обугливании фильтра осадок может восстанавливаться
углеродом фильтра, то большую часть осадка высыпают на глянцевую
бумагу, фильтр озоляют, а затем в том же тигле прокаливают осадок.
Этот метод в настоящее время применяется редко. В таких случаях
гораздо рациональнее пользоваться фильтровальными тиглями.
Прокаленный тигель с осадком помещают в эксикатор, дают ему остыть и
взвешивают. Затем прокаливание повторяют в тех же условиях до достижения
постоянной массы осадка, для чего обычно требуется 2—3 прокаливания.
Точные данные о продолжительности и температуре прокаливания
указываются в методиках анализов.
§ 8. Взвешивание весовой формы
Высушенную или прокаленную весовую форму взвешивают на
аналитических весах, как указано в «Введении», § 8. Высушивание или
прокаливание продолжают до тех пор, пока взвешиваемое вещество не
достигнет постоянной массы.
В. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
§ 9. Теоретические основы выделения осадков из растворов
с помощью специфических неорганических
и органических реактивов
Одной из наиболее важных операций весового анализа является
операция образования осадка.
От качества, формы, структуры и степени чистоты получаемых
осадков в значительной степени зависит точность и надежность результатов
весового анализа.
362
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Форма, структура и степень чистоты образующегося осадка зависят
от условий осаждения и главным образом от скорости осаждения, поэтому
обоснование оптимальных условий осаждения является важнейшим
элементом теории осаждения малорастворимых соединений с помощью
специфических органических и неогранических реактивов.
Структура и свойства образующихся осадков зависят от
концентрации осаждаемого вещества, осадителя, продолжительности осаждения,
температуры, перемешивания, растворимости образующихся осадков,
рН среды и т. д. Рассмотрим подробнее влияние некоторых из этих
факторов.
Влияние скорости осаждения. По мере прибавления в анализируемый
раствор реактива-осадителя, когда произведение концентраций ионов
осаждаемого соединения превысит величину произведения
растворимости (nPKtAn), начинает образовываться осадок малорастворимого
соединения. Процесс образования частиц осадка называют агрегацией.
Расположение частиц осадка в процессе агрегации в строго определенном
порядке называют ориентацией. В начале процесса агрегации образуются
частицы осадка очень малого размера, идентичные тем, которые
характерны для коллоидных растворов. Затем по мере образования осадка
его частицы увеличиваются за счет мелких частиц.
При медленном осаждении образующиеся первичные кристаллы
успевают правильно ориентироваться по отношению друг к другу. В
результате получаются правильной формы крупные кристаллы. Чем менее
растворимо вещество, тем быстрее образуются осадки и мельче кристаллы.
Чем больше скорость образования осадка, тем быстрее нарушается
правильная ориентация при кристаллизации. При быстром осаждении сразу
появляется много центров, кристаллизации. В результате получается
много мелких кристаллов. Мелкие кристаллы могут собираться в хлопья,
в которых кристаллическую структуру можно различить лишь под
микроскопом или посредством рентгеноструктурного анализа.
Наконец, при еще большей скорости осаждения первичные центры
кристаллизации не успевают правильно ориентироваться и
располагаются хаотично. В результате получаются аморфные (студенистые, жела-
тинообразные и т. п.) осадки, в которых кристаллическая структура не
различима даже при рентгеноструктурном исследовании.
Влияние концентрации. Выпадению осадка предшествует пересыщение
раствора. В пересыщенном растворе образуются зародыши кристаллов
осаждаемого вещества, которые по мере созревания осадка увеличиваются
в размерах. Скорость образования этих зародышей и их количество
зависят от степени пересыщения. Чем больше пересыщение, тем больше
зародышей. Поэтому при осаждении из концентрированных растворов
получаются мелкокристаллические или аморфные осадки.
Осаждение из разбавленных растворов вследствие малого количества
зародышей кристаллов протекает медленнее и в процессе осаждения
мелкие кристаллы растут. Таким образом, при осаждении из разбавленных
растворов выпадают крупнокристаллические осадки.
Влияние перемешивания раствора. Перемешивание способствует
снижению высоких местных концентраций и увеличению скорости
растворения; количество зародышей кристаллов при этом уменьшается и
создаются условия для роста крупных кристаллов. Поэтому осаждение
необходимо вести при перемешивании раствора.
Влияние температуры. При нагревании растворимость осадков, как
правило, увеличивается и крупные кристаллы образуются быстрее.
Следовательно, осаждение необходимо вести из горячих растворов
горячими растворами осадителя.
$ 10. ТРЕБОВАНИЯ, ПРЕДЪЯВЛЯЕМЫЕ К ОСАДКАМ
363
При нагревании раствора (или при стоянии осадка в соприкосновении
с маточным раствором) в процессе созревания осадка крупные кристаллы
растут за счет мелких. При этом происходит уменьшение общей и удельной
поверхности кристаллов, так как чем мельче частицы вещества,
занимающего определенный объем, тем больше их суммарная поверхность.
Покажем это на примере. Удельная поверхность 5Уд равна:
для куба
с _ 6fl2 _ 6
для шара
_ 4яг2-3 _ 3
Ьуд- 4яг3 "" г
где а — ребро куба;
г — радиус шара.
Удельная поверхность зависит от линейных размеров геометрических тел, т.че. чем
больше линейные размеры, а следовательно, чем меньше число кристаллов, тем меньше
общая поверхность.
Объем кристаллического тела, имеющего форму куба с длиной ребра 1 см, равен 1 смг,
а его поверхность — 6 см2.
Если мысленно раздробить этот куб на более мелкие кубы с ребром, равным 10~6 см,
то получится 1018 кубов с поверхностью
5= 6 (Ю-6)2 1018= б-Ю6 см2
т. е. поверхность тела увеличится.
Схема образования кристаллических осадков. Схематически процесс
осаждения может быть представлен следующим образом:
Осаждаемое вещество + Осадитель —>• Первичный осадок
Первичный осадок —>• Вторичный (созревающий) осадок
Мелкие частички осадка —> Крупные частички осадка
При соприкосновении осадка и раствора протекают обратные друг
другу процессы — образование осадка и его растворение. Первичный
осадок непрерывно растворяется и по достижении границы пересыщения
вновь выпадает. Этот динамический процесс ведет к растворению мелких
и росту крупных зерен осадка.
В результате между осадком и частью вещества, находящегося в
растворе, наступает состояние динамического равновесия, при котором
скорость растворения осадка равняется скорости его образования.
§ 10. Требования, предъявляемые к осадкам
Не всякий осадок удовлетворяет требованиям, предъявляемым к
осадкам в весовом анализе.
Растворимость. Прежде всего соединение, в виде которого осаждают
одну из составных частей анализируемого раствора, должно быть
практически нерастворимо в среде, из которой ведется осаждение. Лишь при
соблюдении этого условия возможно количественное осаждение.
Обычно в количественном анализе считают практически
нерастворимым такое соединение, растворимость которого не превышает 10 "5 моль/л.
Но и в этом случае потери вещества при весовом анализе могут
превышать допустимые. Благодаря специальным приемам ошибки анализа,
вызываемые повышенной растворимостью осаждаемого вещества, можно
свести к минимуму. Например, можно понизить растворимость осадка,
действуя избытком осаждающего реактива.
Состав. Чтобы вычислить по массе осадка или по его весовой форме
содержание определяемого элемента, выделяющегося в осадок, весовая
364
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
форма осадка должна отвечать определенному постоянному химическому
составу. Соединения переменного состава, даже если они мало растворимы,
непригодны для весового анализа.
Структура. Образование осадка сопровождается несколькими
сопутствующими процессами: пересыщение, формирование центров
кристаллизации, рост кристаллов, коллоидообразование, коагуляция,
адсорбция. Осаждение ведут в условиях, обеспечивающих получение осадка,
обладающего (по возможности) кристаллической структурой.
Осадок кристаллической структуры должен отличаться минимальной
склонностью к загрязнению и не увлекать посторонних примесей
из анализируемого раствора, т. е. осадок должен быть химически
чистым.
Осаждение малорастворимого соединения должно быть по
возможности специфичным и не сопровождаться образованием осадков других
сопутствующих элементов. Равным образом присутствующие в
анализируемом растворе другие компоненты не должны препятствовать
выделению осаждаемого малорастворимого соединения.
Таким образом, осадок должен быть практически нерастворимым,
иметь постоянный состав и структуру, обеспечивающую успешное
выполнение с ним дальнейших операций фильтрования, промывания и
превращения его в определенную весовую форму.
Причины загрязнения осадков. Причиной загрязнения осадков
является сопряженное осаждение.
Сопряженное осаждение (соосаждение) обусловливается склонностью
некоторых соединений, хорошо растворимых в данном растворителе,
осаждаться во время образования осадка совместно с выпадающими
в осадок малорастворимыми соединениями. Например, относительно
хорошо растворимая соль MgC204 осаждается вместе с мало^астворимой
солью CaC204; Na2S04 с BaS04; Ba(N03)2 с BaS04; Ва(Мп04)2 с BaS04
и т. д.
Соосаждение может быть объяснено различными явлениями:
1. Образованием химических соединений между осаждаемым
веществом и присутствующими в растворе примесями.
2. Образованием смешанных кристаллов, состоящих из основного
соединения и примесей, кристаллизующихся в одной и той же
кристаллической форме.
3. Окклюзией, т. е. захватом образующимся осадком растворимых
в данной среде примесей, что наблюдается при быстром росте
кристаллических осадков. При окклюзии в отличие от поверхностной адсорбции
загрязнение осадков происходит не только на поверхности, но и по всей
массе осадка, внутри его кристаллов.
4. Переходом примесей в осадок после его образования при стоянии
(адсорбцией).
Адсорбция, или поглощение примесей (ионов или молекул) из
раствора поверхностью частиц осадка, проявляется у всех твердых веществ,
но больше всего она присуща веществам, обладающим относительно
большой удельной поверхностью.
Чем больше поверхность, тем сильнее адсорбция примесей, тем больше
загрязнение осадка. Поэтому к адсорбции более склонны осадки с
развитой поверхностью, т. е. аморфные осадки.
Переосаждение. Надежным средством получения чистых осадков
является их промывание дистиллированной водой или соответствующими
растворителями, а также повторное осаждение (переосаждение), которое
заключается в растворении полученного осадка и последующем его
вторичном осаждении.
§ 10. ТРЕБОВАНИЯ. ПРЕДЪЯВЛЯЕМЫЕ К ОСАДКАМ 365
При растворении осадка раствор будет содержать лишь те примеси,
которые были увлечены при первом осаждении, т. е. незначительные их
количества, и после второго осаждения получаются уже относительно
чистые осадки.
Оптимальные условия осаждения кристаллических осадков. В связи
с тем, что точность весового анализа во многом зависит от того, насколько
хорошо прошло образование осаждаемого соединения, удовлетворяющего
предъявляемым к нему требованиям, необходимо снизить до минимума
влияние различных факторов, нарушающих нормальный процесс
выделения твердой фазы.
Одним из таких факторов, как известно, является сопряженное
осаждение, обусловливаемое образованием химических соединений между
осаждаемым веществом и присутствующими в анализируемом растворе
посторонними примесями, выделением смешанных кристаллов
(состоящих из примесей и основного соединения), кристаллизующихся в одной
и той же кристаллической форме, адсорбцией примесей поверхностным
слоем осаждаемого вещества и окклюзией (см выше).
Для осаждения кристаллических осадков необходимо соблюдать
следующие условия.
1. Вести осаждение из достаточно разбавленных растворов
разбавленными растворами осадителя. При осаждении из разбавленных
растворов выпадают крупнокристаллические осадки.
2. Прибавлять осадитель медленно, по каплям. При быстром
осаждении сразу появляется много центров кристаллизации. В результате
получается много мелких кристаллов.
3. Перемешивать раствор стеклянной палочкой, чтобы избежать
сильных местных перенасыщений при прибавлении осадителя.
Перемешивание способствует созданию условий для роста крупных кристаллов.
4. Вести осаждение из горячего раствора горячим же раствором
осадителя. При нагревании увеличивается растворимость мелких кристаллов
и за их счет образуются крупные кристаллы.
5. В некоторых случаях на отдельных стадиях процесса осаждения
необходимо добавлять вещества, повышающие растворимость
формирующегося осадка.
Соблюдение перечисленных условий осаждения дает возможность
получать кристаллические осадки высокого качества.
Для уменьшения явления окклюзии следует прибавлять осадитель
медленно, по каплям, так чтобы в момент образования кристаллов не
было избытка посторонних ионов в растворе. Избыток осаждающего иона
следует прибавлять по окончании осаждения. При образовании
кристаллических осадков для полного выделения веществ из раствора требуется
некоторое время. После прибавления избытка реактива осадку дают
возможность созреть.
Оптимальные условия осаждения аморфных осадков. Аморфные осадки
склонны к адсорбции и к образованию коллоидных растворов, поэтому
при осаждении аморфных осадков необходимо вести осаждение из
горячего раствора и в присутствии коагулирующего электролита. Для
предупреждения явления адсорбции осаждение аморфных осадков следует
вести из концентрированных растворЬв. Образующиеся при этом осадки
хорошо свертываются, их легче отфильтровывать и промывать. К
раствору с осадком тотчас же по окончании осаждения приливают большой
объем (до 100 мл) воды и смесь перемешивают. При этом происходит
нарушение адсорбционного равновесия и часть адсорбированных ионов
переходит в раствор. Осадок не рекомендуется оставлять в соприкосновении
с раствором длительное время.
366
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
§11. Методы повышения точности весовых определений
При осаждении даже самых малорастворимых веществ все же остается
некоторое количество осаждаемых ионов, так как абсолютно
нерастворимых веществ в природе нет. Чем больше растворимость осаждаемого
соединения, тем менее точен результат весового определения. Поэтому
основная задача аналитика сводится не только к получению осадка,
отличающегося определенной формой и структурой, но и к тому, чтобы
потери вещества, обусловливаемые неполнотой осаждения, были сведены
к допускаемому в этих случаях минимуму.
Понижение растворимости осаждаемого соединения. Одним из способов
понижения растворимости осадка является действие одноименного иона
(подробно см. гл. IV, § 5).
Для бинарного электролита
nPKtAn
D гл«-.-т мап
PKtAn-[An]=rKF];^
или
Следовательно, растворимость осадка в случае прибавления избытка
осаждающего реактива уменьшается и зависит от избытка прибавленного
иона, общего с ионами, образующими осадок, а также от численного
значения произведения растворимости малорастворимого электролита
(nPKtAn).
Например, для уменьшения растворимости BaS04 необходимо
добавить в раствор либо Ва+ +, либо SOi"""- Если осаждают Ва+ +, то
прибавляют избыток БОГ" и, наоборот, при осаждении SOr~~ добавляют
избыток Ва+ +.
Другим способом понижения растворимости является осаждение
в неводных растворах. Например, растворимость сульфата свинца сильно
уменьшается, если вести осаждение ионов свинца SOr~ в среде этилового
спирта.
Повышение специфичности реакций. Повышения специфичности
отдельных реакций можно добиться: а) удалением посторонних ионов,
осаждающихся данным осадителем; б) созданием определенного значения
рН раствора; в) связыванием мешающих ионов в прочные комплексные
соединения. Отделение мешающих ионов сильно замедляет выполнение
анализа и поэтому подбор специфических осадителей имеет большое
практическое значение. Очень удобны для этого органические реактивы.
Для определения той или иной составной части анализируемого ве-
щества> применяют различные способы анализа. Следует помнить, что
выбор того или иного метода анализа определяется главным образом
целью, которую ставит аналитик. В тех случаях, когда большая
точность не требуется или когда надо провести анализ возможно быстрее,
выбирают простые и быстрые способы анализа взамен очень точных
методов, требующих для своего выполнения много времени.
Параллельные определения. Подавляющее число аналитических
операций весового анализа (осаждение, фильтрование, промывание,
прокаливание, взвешивание) сопровождается потерями вещества, приводящими
к ошибочным результатам анализа. Некоторые ошибки можно учесть.
Другие ошибки учесть и устранить нельзя. Для проверки правильности
проделанной работы обычно проводят параллельные определения*
«> 12. ВЫБОР УСЛОВИЙ ДЛЯ ВЕСОВОГО ОПРЕДЕЛЕНИЯ Зб7
Результаты параллельных определений должны достаточно хорошо
сходиться друг с другом. Проведение параллельных определений
исключает случайные грубые ошибки в работе.
При обсуждении результатов химического анализа пользуются
терминами — точность и воспроизводимость. Воспроизводимость является
мерой того, как повторяются результаты при многократном проведении
определения одним способом, а точность говорит о правильности
полученных результатов. Точность химического определения зависит от
избранного метода анализа, способа взвешивания пробы и осадка и т. п.
Точность полученного результата можно иногда проверить, повторив
анализ, но совершенно другим методом. Если два метода, сильно
отличающиеся друг от друга, приводят к одинаковым результатам, считают
вполне вероятным, что полученные результаты мало отличаются от
истинного значения.
Поправки на ошибки, возникающие при выполнении определения,
находят, выполняя «холостой» опыт. При выполнении «холостого» опыта
точно следуют тому ходу работы, какой применяют при анализе пробы.
Лучше «холостой» опыт проводить одновременно с анализом пробы.
§ 12. Теоретические обоснования выбора оптимальных
условий для весового определения
Главной целью аналитика при выполнении того или иного определения
является получение точных результатов анализа, поэтому выбор
оптимальных условий определения очень важен.
Выбор осадителя. При выборе осадителя решающее значение имеет
величина растворимости образующегося осадка. Обычно выбор
останавливают на наименее растворимом осадке. Например, наиболее пригодными
для количественных определений ионов бария (из его форм ВаС03, ВаС204,
ВаСЮ4 или BaS04) являются BaS04 и ВаСЮ4. Это видно из
сопоставления величин растворимости:
Осадок ВаС03 ВаС204 ВаСг04 BaS04
Растворимость, моль/л . . 7,015-10"6 4,123-1СГ4 1,517- 1СГб 0,9949-1(Гб
Однако не всякое малорастворимое вещество может быть
использовано в количественном методе анализа. Малорастворимое вещество может
быть пригодно Для этих целей лишь в том случае, если оно отвечает целому
ряду требований, изложенных в § 10.
Количество осадителя. Согласно правилу произведения
растворимости образование осадков происходит лишь при условии, если
произведение концентраций соответствующих ионов в растворе превышает
величину произведения растворимости осаждаемого соединения при данной
температуре. Так как абсолютно нерастворимых в воде веществ не
существует, то ни одно осаждение не бывает совершенно полным. Полнота
осаждения в первую очередь зависит от количества прибавленного
осадителя.
Осложнения, вызываемые избытком осадителя. Применение избытка
осадителя, помимо полезного действия, может повести и к нежелательным
результатам (вызвать явления комплексообразования, сопровождающиеся
растворением осадка).
Наличие в растворе сильных электролитов также увеличивает
растворимость осадков вследствие солевого эффекта (см. гл. IV, § 6).
Влияние концентрации ионов водорода на полноту осаждения. Если
осаждаемое вещество представляет собой малорастворимую соль
сильной кислоты, то оно практически не растворяется в кислотах. В этом слу-
368
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
чае растворимость осадка, являющегося солью сильной кислоты,
практически не зависит от [Н+] в растворе. Если же вещество представляет
собой соль слабой кислоты, растворимость в кислой среде сильно
увеличивается, так как анионы реагируют с ионами водорода, образуя
растворимую в воде слабую кислоту. Например С2ОГ~-ионы в кислой среде
образуют НС2ОГ или молекулы щавелевой кислоты.
Растворимость осадков тем больше, чем больше концентрация ионов
водорода и величина произведения растворимости и чем меньше константа
диссоциации кислоты, анион которой входит в состав осадка.
Следовательно, влияние рН на растворимость различных осадков различно (см.
гл. IV, § 7).
Влияние явления комплексообразования. Действие избытка осадителя
приводит к уменьшению растворимости осадка. В ряде случаев
образование комплексных ионов приводит к увеличению растворимости осадка.
Поэтому не следует применять при осаждении большой избыток
осадителя, если для этого нет особой необходимости. Следует вести осаждение
преимущественно в виде таких соединений, которые не склонны к ком-
плексообразованию.
Выбор промывной жидкости. При фильтровании и промывании осадка
происходит частичное удаление адсорбированных им примесей, а также
пропитывающего осадок маточного раствора.
Промывание осадков необходимо для удаления таких солей, которые
не улетучиваются при прокаливании.
В редких случаях осадки промывают чистой водой, так как обычно
это может привести к нежелательным явлениям: увеличению
растворимости осадка, образованию коллоидных растворов или гидролизу. Кроме
того, пользуясь водой, практически невозможно полностью отмыть
посторонние электролиты. Поэтому необходимо подбирать состав промывной
жидкости в соответствии с составом промываемого осадка.
Для уменьшения растворимости осадка в состав промывной
жидкости вводят электролиты, имеющие общие ионы с ионами осадка. Для
предупреждения коллоидообразования в состав промывной жидкости
вводят электролиты (чаще всего применяют соли аммония), способные
вступать в реакции двойного обмена с адсорбированными ионами и легко
улетучиваться при прокаливании. В состав промывной жидкости
прибавляют также электролиты, подавляющие гидролиз.
Весовая форма. Осадок может иметь неопределенный состав, поэтому
его высушивают и в большинстве случаев прокаливают. Получаемый
при этом остаток, состав которого выражается определенной формулой,
называют весовой формой.
Весовая форма, получаемая путем прокаливания, должна отличаться
устойчивостью при высоких температурах.
Примеры перевода осадков в весовую форму прокаливанием:
2 [Fe (Н20)*] (ОН)з —> Fe203 + (3 + 2x) H20 f
t
CaC204 • Н20 —* СаО + CO f + C02 f + H20 f
t
2MgNH4P04.6H20 —> Mg2P207 + 2NH3 f + 7H20 f
Схема анализа. Приступая к анализу неизвестного вещества или
к определению составных частей сложной смеси нескольких веществ,
химик-аналитик должен обстоятельно продумать ход анализа. Метод,
дающий вполне удовлетворительные результаты при определении того
или иного вещества в одном случае, может оказаться совершенно неудов-
§ 13. ОПРЕДЕЛЕНИЕ КРИСТАЛЛИЗАЦИОННОЙ ВОДЫ В ВаС12 2Н,0 369
летворительным в другом. Особенно сильно искажаются результаты
определений при анализе сложных смесей. Примеры несостоятельности хорошо
известных методов весьма многочисленны. Например, метод определения
кремневой кислоты путем выпаривания досуха солянокислого раствора
анализируемого вещества и последующего обезвоживания сухого остатка
дает хорошие результаты, если кремневой кислоте не сопутствуют примеси,
выпадающие вместе с нею в осадок. Но этот метод нельзя применять
в присутствии таких элементов, как бор, фтор, сурьма, титан, висмут и др.
Осаждением смесью едкого натра и карбоната натрия можно хорошо
отделить ионы алюминия от ионов железа и кальция, выпадающих в осадок
в виде Fe(OH)3 и СаС03. Но тот же метод непригоден для отделения
ионов алюминия от ионов железа и цинка. Оксалатный метод, который
обычно применяют для определения кальция в присутствии магния,
неприменим, если ионы кальция содержатся в незначительном количестве,
а ионы магния — в большом количестве. Определение свинца в виде
сульфата дает вполне хорошие результаты, если это определение проводят
в отсутствие ионов бария, кальция, серебра и сурьмы.
Анализ смесей. Методы исследования простых соединений не могут
быть с уверенностью применены к анализу смесей. Много затруднений
вызывают вещества, которые препятствуют осаждению или замедляют
его. Вещества, вызывающие нежелательные побочные явления и реакции,
должны быть предварительно удалены. Наибольшие затруднения
возникают потому, что многие из применяемых реакций не приводят к полному
разделению ионов. Так, кремневая кислота не может быть полностью
отделена от бора выпариванием досуха кислых растворов и последующим
обезвоживанием; ионы цинка при осаждении сероводородом частично
осаждаются с ионами меди, ионы кобальта — вместе с ионами олова (IV)
и т. д. Осадок гидроокиси алюминия удерживает ионы меди и цинка даже
после повторного переосаждения аммиаком. Фосфат магния увлекает в
осадок ионы щелочных металлов. Успех зависит в значительной степени от
того, с какой тщательностью был подготовлен раствор к анализу. Эту
подготовку нельзя выполнить по определенным заранее сделанным
предписаниям. Каждый случай должен быть обдуман и рассмотрен особо.
Г. ПРАКТИЧЕСКАЯ ЧАСТЬ
§ 13. Определение кристаллизационной воды в ВаС12 2Н20
Определение кристаллизационной воды в разлагаемых при
нагревании кристаллогидратах основано на высушивании или прокаливании
навески исходного кристаллогидрата при определенной температуре до
постоянной массы. При этом кристаллогидрат разлагается с выделением
воды. Например, высушивание кристаллогидрата хлорида бария
достигается при 105—125° С; для высушивания CuS04-5H20 необходима teM-
пература 210—220° С и т. д. Разность в массе вещества до и после
высушивания или прокаливания показывает массу кристаллизационной воды,
содержащейся в навеске исходного кристаллогидрата.
Уравнение реакции:
ВаС12-2Н20 -^ ВаСЦ -f 2H20 f
Предварительно необходимо подготовить бюкс. Для этого бюкс
тщательно моют и помещают на 30 мин в сушильный шкаф, в котором
поддерживают температуру 105—125° С (в процессе высушивания бюкс не
закрывают крышкой). После этого бюкс закрывают, вынимают из сушиль-
370
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
ного шкафа и помещают в эксикатор. Эксикатор переносят в весовую
комнату и когда бюкс охладится (примерно через 30 мин) до температуры
весовой комнаты, его взвешивают. Взвешивание и высушивание
продолжают до постоянной массы. Рассчитанную навеску анализируемого
вещества сначала взвешивают на технических весах в бюксе, а затем снова
взвешивают на аналитических весах. Бюкс с навеской помещают в
сушильный шкаф на 2 ч. Высушивание проводят в открытом бюксе, крышку бюкса
помещают поперек его отверстий. Бюкс с высушенной навеской
вынимают из сушильного шкафа, помещают в эксикатор, охлаждают в
помещении весовой комнаты и взвешивают на аналитических весах. Затем
бюкс с навеской снова помещают в сушильный шкаф на 1 ч и по
охлаждении взвешивают. Образец высушивают и взвешивают до постоянной массы,
т. е. до тех пор, пока кристаллизационная вода полностью не испарится.
Результаты всех взвешиваний должны быть обязательно записаны в
журнал.
Расчет результатов анализа проводят, как указано в § 3.
§ 14. Определение сульфат-ионов или серы
Метод основан на реакции взаимодействия сульфат-ионов с ионами
бария, сопровождающейся образованием малорастворимого
мелкокристаллического осадка сульфата бария. Осадок сульфата бария
отфильтровывают, промывают, прокаливают, взвешивают и рассчитывают в нем
содержание SO" или серы. На этом принципе основано определение свободной,
пиритнои и сульфидной серы в каменном угле, рудах и минералах с той
лишь разницей, что серу предварительно окисляют до сульфатной серы.
Уравнение реакции:
Ba++ + S07~>;BaS04
Весовая форма при этом методе определения идентична форме
осаждения. При обработке осадка при переведении его в весовую форму могут
произойти следующие нежелательные- процессы:
а) при озолении фильтра может произойти восстановление сульфата
бария:
BaS04 + 2С —> 2С02 + BaS
Обратный переход сульфида бария в сульфат происходит в процессе
длительного нагревания осадка на воздухе:
BaS + 202 —> BaS04
б) при прокаливании при слишком высокой температуре наблюдается
термическое разложение сульфата бария:
BaS04—>BaO + S03
Превращение весовой формы BaS04 в другую форму приводит к
неверным результатам, и работу приходится переделывать. Иногда
образовавшийся BaS или ВаО обрабатывают 2—3 каплями H2S04 и затем
осторожно нагревают под тягой до полного разложения избытка кислоты,
что узнают по прекращению выделения паров S03.
При этом происходят реакции:
Ва§ + H2S04 —> BaS04 + H2S
t
ВаО + H2S04 -+ BaS04 + H20
H2S04—>H20-f S03
§ 14. ОПРЕДЕЛЕНИЕ СУЛЬФАТ-ИОНОВ ИЛИ СЕРЫ
371
Однако следует иметь в виду, что обработка осадка в тигле серной
кислотой может привести к неверным результатам, если работа
выполняется недостаточно тщательно. Поэтому к обработке осадка серной
кислотой прибегают только в исключительных случаях.
Условия проведения реакций осаждения. 1. Осаждение ведут из
кислого раствора при рН <2и при температуре, близкой к температуре
кипения.
2. Осаждению мешают некоторые анионы (SiO", SnOr~~, WOr~
и др.), выпадающие в осадок в виде соответствующих кислот (H2Si03,
H2Sn03, H2W04) при подкислении раствора, поэтому мешающие анионы
должны быть предварительно удалены из анализируемого раствора.
3. Неудовлетворительные результаты анализа получаются также
в том случае, если в анализируемом растворе присутствуют в большом
количестве ионы, не осаждаемые ионами Ва++, но соосаждаемые вместе
с BaS04 (Fe+ + +, Al+ + +, МпОГ, С1" и др.). Поэтому осаждение нужно
вести в отсутствие больших количеств соосаждаемых ионов.
Расчет навески. MNa2so4 = 142,041; MBaso4 = 233,40. Так как осадок
BaS04 имеет кристаллическую структуру, то масса его весовой формы
должна быть около 0,5 г. Следовательно, для анализа необходимо взять
навеску исходного вещества (в пересчете на Na2S04), рассчитываемую
по формуле:
0,5-142,0 П9
%a2S04= 233,4 ~0'32
Если исходят из готового раствора Na2S04, то его разбавляют в
мерной колбе до метки и для анализа берут аликвотные части.
Расчет количества осадителя. Ионы SOi""- осаждаются 1 н. раствором
ВаС12-2Н20. Для расчета требуемого количества ВаС12-2Н20 составляют
уравнение реакции:
ВаС12-2Н20 + Na2S04 —* l BaS04 + 2NaCl + 2H20
Из уравнения реакции следует, что
242,2 — 142
л: —0,3
0,3-242,2 пс
Х= 142 ~°'5г
Так как в 1000 мл 1 н. раствора хлорида бария содержится 121,1 г
ВаС12-2Н20, то 0,5 г его содержится в V мл раствора:
т/ 0,5-1000
v= 121 ~4 мл
Практически берут в 1,5 раза больше: 4-1,5 = 6 мл.
Методика определения. Рассчитанную и взвешенную на аналитических
весах навеску анализируемого сульфата переносят в стакан емкостью
200—250 мл и растворяют в 100 мл дистиллированной воды. К
полученному раствору приливают 2 мл 6 н. раствора хлористоводородной кислоты
и нагревают содержимое стакана почти до кипения. Исходный 1 н. раствор
хлорида бария (6 мл) предварительно разбавляют водой до 50 мл,
нагревают почти до кипения и осторожно по каплям при перемешивании
(стеклянная палочка не должна касаться стенок стакана) приливают к
нагретому раствору осаждаемого сульфата. Затем стакан ставят на кипящую
водяную баню. Как только осадок осядет и жидкость над ним
просветлеет, приступают к прббейа полноту осаждения. Для этого к раствору над
осадком осторожно по палочке приливают 2—3 капли горячего раствора
372
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
хлорида бария. Если при этом не наблюдается помутнения раствора, то
полное осаждение достигнуто. Если раствор мутнеет, то добавляют еще
О 5—1 мл раствора осадителя и снова проверяют полноту осаждения.
Приливание осадителя продолжают до тех пор, пока не будет достигнута
полнота осаждения.
После достижения полноты осаждения стакан ставят на кипящую
баню на 2 ч или для созревания осадка оставляют стоять в рабочем шкафу
Рис. 70. Смывание на фильтр частиц осадка со дна
и стенок стакана.
до следующего занятия. Необходимо помнить, что перед отделением
раствора, находящегося над осадком, раствор следует охладить. На
следующем занятии приступают к декантации и фильтрованию. Для
фильтрования применяют фильтр «синяя лента». Фильтрование проводят, сливая
раствор с осадка на фильтр по палочке. Осадок от раствора отделяют
декантацией, при этом раствор сливают, стараясь не взмутить осадок.
Когда из стакана будет слит почти весь раствор, приступают к
промыванию осадка. В стакан наливают 20 мл промывной жидкости
(дистиллированную воду, подкисленную НС1), взмучивают осадок палочкой, дают
отстояться и сливают жидкость с осадка на фильтр. В стакан наливают
снова промывную жидкость.
Декантирование проводят не менее трех раз. В стакане легче отмыть
примеси от осадка, чем промывать осадок на фильтре. После окончания
промывания декантацией осадок количественно переносят на фильтр.
Для этого осадок взмучивают и осторожно по палочке переносят на фильтр.
Частицы, оставшиеся на дне и стенках стакана, удаляют при прмощи
стеклянной палочки. Затем стеклянную палочку вынимают правой рукой из
стакана, кладут ее поперек стакана (рис. 70) так, чтобы она выдавалась
наружу у его носила на 2—3 см. Прижимая палочку указательным пальцем
левой руки, наклоняют стакан над воронкой; жидкость должна стекать
в воронку по лалочке, не разбрызгиваясь. В правую руку берут промы-
валку и направляют из нее струю дистиллированной воды на дно и
внутренние стенки стакана, смывая ею частицы осадка на фильтр.
Частицы, приставшие прочно к стенке стакана, удаляют смачиванием
стенки стакана водой и протиранием с помощью стеклянной палочки
кусочками фильтра или палочкой с резиновым наконечником. Затем стакан
§ 15. ОПРЕДЕЛЕНИЕ ИОНОВ ЖЕЛЕЗА (III)
373
и стеклянную палочку тщательно просматривают на свет и если
обнаруживают на них частицы осадка, операцию повторяют.
Когда весь осадок перенесен на фильтр, приступают к его
промыванию на фильтре. Для этого струю промывной жидкости из промывалки
направляют в воронку, обводя ею края фильтра. Осадок осторожно
смывают вниз. Когда фильтр будет наполнен водой примерно наполовину
своего объема, прекращают добавление промывной жидкости и дают
ей полностью стечь с фильтра. Промывание фильтра начинают с
верхнего края фильтровальной бумаги. Осадок сульфата бария обладает
достаточно большой растворимостью, поэтому объем промывных вод следует
ограничить (не более 200 мл).
Промывание осадка необходимо для удаления таких примесей,
которые при последующем прокаливании осадка не будут улетучиваться.
Промывание заканчивают тогда, когда проба на полноту промывания
(каплей раствора AgN03) даст отрицательные результаты на С1"-ионы.
После того как стекут с воронки последние капли жидкости, воронку
с фильтром накрывают фильтровальной бумагой и помещают в
сушильный шкаф для высушивания. После высушивания фильтр осторожно
вынимают из воронки, складывают и помещают в чистый и доведенный до
постоянной массы тигель. Тигель с осадком ставят в фарфоровый
треугольник и на малом пламени горелки озоляют фильтр, избегая его
воспламенения. После того как фильтр озолен, приступают к прокаливанию
осадка на пламени горелки при температуре примерно 600—800° С.
Прокаливают тигель с осадком около 40 мин, затем при помощи тигельных
щипцов переносят его в эксикатор, осторожно закрывают эксикатор крышкой.
По охлаждении тигля с осадком (в весовой комнате) приступают
к взвешиванию.
Прокаливание осадка и его взвешивание повторяют до тех пор, пока
не получат постоянной массы, т. е. пока результаты взвешиваний не будут
расходиться более чем на 0,0002 г.
Расчет результатов анализа проводят, как указано в § 3.
§ 15. Определение ионов железа (III)
Метод основан на осаждении гидроокисью аммония ионов железа (III)
в виде Fe(OH)3, получении весовой формы Fe203 прокаливанием Fe(OH)3
и пересчете весовой формы на железо.
Уравнение реакции:
Fe+"H"+3NH4OH —> i Fe (OH)3 + 3NH+
При прокаливании гидроокись железа превращается в безводную
окись железа (III), которую и взвешивают:
2Fe(OH)3 —* Fe203-b3H20f
Длительного прокаливания избегают, так как это приводит к
частичному восстановлению окиси железа:
6Fe203 -^ 4Fe304-b02t
Но если к охлажденной Fe304 прибавить 1 каплю концентрированной
азотной кислоты и осторожно нагреть тигель на пламени горелки, то
восстановленное железо окисляется:
2Fe304 + 2HN03 —> 3Fe203 -f Н20 + 2N02 f
Гидроокись железа осаждают из нагретых растворов; при этом соли
железа гидролизуются, например:
FeCl3 + 2НОН -> Fe (ОН)2 CI + 2HC1
374
ГЛ-V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Поэтому в процессе анализа для подавления гидролиза и для
окисления железа (II) в железо (III) прибавляют азотную кислоту:
Fe (ОН)2 С1 + 2Н+ —> 2H20 + Fe++++Cl-
3Fe++ + 4HN03 —у 3Fe+++ + NO f + 2H20 + 3NO~
Условия проведения реакции осаждения. 1. Осаждение ведут из
кислого раствора при рН = 2—3 и при 75—90° С. Заканчивают осаждение
в нейтральной или слабощелочной среде при рН = 7—9.
2. Катионы железа (II), возможно присутствующие в растворе, должны
быть предварительно окислены до Fe+ + +.
3. Для предупреждения образования коллоидной системы и для
быстрой коагуляции образующегося аморфного осадка в анализируемый
раствор предварительно добавляют коагулянт—нитрат аммония (или,
что менее желательно, NH4C1).
4. Осаждению мешают многие другие катионы, осаждаемые NH4OH
в виде гидроокисей вместе с Fe(OH)3. Число катионов металлов,
осаждаемых аммиаком, очень велико. К ним относятся катионы бериллия,
алюминия, хрома, циркония, титана, урана, ниобия, тантала, висмута,
таллия, галлия, индия, редкоземельных металлов и др. Поэтому
передосаждением Fe(OH)3 мешающие катионы должны быть удалены из
анализируемого раствора.
5. Осаждению мешают многие анионы, которые осаждаются в
аммиачной среде в виде нерастворимых солей катионов, присутствующих в
анализируемом растворе. К ним относятся силикаты, фосфаты, бораты,
арсенаты и др. Поэтому осаждению Fe(OH)3 должно предшествовать
выделение кремневой кислоты и отделение мешающих анионов.
6. Неудовлетворительные результаты анализа получаются даже в том
случае, если в анализируемом растворе находятся в большом количестве
ионы, не осаждаемые в присутствии избытка аммиака, но соосаждаемые
вместе с Fe(OH)3. К ним относятся катионы меди, кальция, цинка, никеля,
кобальта и др. Поэтому осаждение нужно вести в отсутствие больших
количеств. соосаждаемых катионов.
7. Ионы железа (III) весьма склонны к образованию прочных
комплексных соединений с органическими и неорганическими соединениями
(оксисоединениями, фторидами, фосфатами и др.). Поэтому в
анализируемом растворе не должны содержаться комплексообразующие вещества,
маскирующие ионы железа.
8. При последующем прокаливании Fe(OH)3, которую осаждали
в присутствии хлоридов или промывали жидкостью, содержащей NH4C1,
может произойти потеря железа вследствие улетучивания FeCl3,
образующегося по уравнению:
Fe (ОН)з + 3NH4C1 —> FeCl3t +3H20+3NH3t
Поэтому, чтобы избежать возможных потерь FeCl3, необходимо Fe(OH)3
промывать смесью NH4N03+NH4OH, а в конце промыть осадок 5—10 мл
горячей дистиллированной воды.
Методика определения. Если исходят из твердого вещества, то,
предварительно рассчитав навеску исходного вещества, приготовляют его
раствор. Для анализа должно быть взято такое количество соли железа,
чтобы масса получаемой весовой формы при данном определении не
превышала 0,1 г.
Если для анализа дан готовый раствор (например, FeCl3), то в
зависимости от концентрации его либо разбавляют в мерной колбе и для
анализа берут аликвотную часть, либо используют для осаждения весь
§ 16. ОПРЕДЕЛЕНИЕ С ОДЕРЖАНИЕ КАЛЬЦИЯ В КАРБОНАТЕ КАЛЬЦИЯ 375
раствор. В первое случае для работы применяют мерную колбу; во
втором — раствор помещают в химический стакан емкостью 150—200 мл.
Перед, осаждением ионов железа раствор, содержащий соль железа
и не содержащий мешающих осаждению ионов, подкисляют азотной
кислотой и осторожно нагревают до 75—90° С. К горячему раствору при
перемешивании прибавляют 10%-ный раствор NH3 до появления слабого
запаха аммиака. Убеждаются в полноте осаждения. После окончания
осаждения к раствору с осадком прибавляют 100 мл горячей дистиллированной
воды и фильтруют через беззольный фильтр «красная лента».
Промывают горячим 2%-ным раствором нитрата аммония, к которому
прибавляют до слабого запаха раствор аммиака. Заканчивают промывание
тогда, когда будут полностью отмыты ионы хлора. Промытый осадок
подсушивают в сушильном шкафу и еще влажным помещают во взвешенный
тигель. Сначала нагревают его на слабом пламени для озоления фильтра,
а затем прокаливают при 1000—1100° С и взвешивают.
Расчет результатов анализа — см. § 3.
§ 16. Определение содержания кальция в карбонате кальция
Определение кальция в карбонатных породах основано на
растворении в кислоте определенной навески исходного карбоната, осаждении
ионов кальция в виде СаС204-Н20 и последующем прокаливании окса-
лата кальция до постоянной массы (СаО).
Уравнения реакций:
СаС03 + 2НС1 —> СаС12-{-С02| +Н20
СаС12 + 2НС1+2(Ш4)2С204 —> Са (НС204)2 + 4NH4C1
Са (НС204)2 + 2NH4OH —> СаС204 • Н20 + (NH4)2 С204 + Н20
t
СаС204.Н20 —> СаО + С02 f +CO f + H20 f
Условия проведения реакции осаждения СаС204— см. книга 1, гл. V,
§з.
Методика определения. Навеску карбоната кальция осторожно
пересыпают в стакан, смачивают 5 мл дистиллированной воды, закрывают
стакан часовым стеклом и затем осторожно из мензурки через носик
стакана (не снимая стекла) приливают небольшими порциями 6 н.
раствор соляной кислоты, непрерывно помешивая раствор вращением
стакана. Когда вся навеска растворится, разбавляют содержимое стакана
100 мл дистиллированной воды, нагревают до 70—80° С, прибавляют 2
капли метилового оранжевого и затем медленно, по каплям, при непрерывном
перемешивании, вводят нагретый 0,5 н. раствор оксалата аммония.
Количество оксалата аммония рассчитывают, как указано выше (см.
§ 3). Затем по каплям прибавляют раствор аммиака до перехода
розовой окраски раствора, вызываемой индикатором, в желтую. По
окончании осаждения раствор с осадком оставляют стоять на кипящей водяной
бане не менее 1 ч. После отстаивания проводят пробу на полноту
осаждения и далее прозрачную жидкость декантируют на фильтр. Для
промывания используют тот же раствор оксалата аммония, но разбавленный в 6
раз. Промывание заканчивают, когда полностью будут отмыты все ионы
хлора (проба с AgN03+HN03).
Подсушенный осадок вместе с фильтром переносят во взвешенный
тигель. Тигель помещают в фарфоровый треугольник на кольце штатива
и осторожно озоляют фильтр на маленьком пламени горелки. Когда весь
фильтр сгорит, тигель с осадком помещают в муфельную печь и прокали-
376
ГЛ.У ОСНОВЫ ВЕСОВОГО АНАЛИЗА
вают при 1200° С до постоянной массы. Взвешивание весовой формы СаО
во избежание поглощения влаги и двуокиси углерода воздуха проводят,
помещая охлажденный тигель в предварительно взвешенный большой
бюкс с крышкой, и взвешивают тигель с осадком в бюксе.
Результаты анализа рассчитывают по приведенным ранее формулам
(см. § 3).
§ 17. Определение содержания магния
Метод определения основан на осаждении ионов магния в виде
MgNH4P04-6H20:
Mg++ + (nh4)2 нро4 + nh3 + бн2о -+ ф MgNH4Po4. бн2о + 2NHf
При прокаливании MgNH4P04-6H20 образуется пирофосфат магния
(весовая форма):
2MgNH4POr6H20 -U Mg2P207 + 2NH8r + 7H80 t
Для предупреждения образования Mg(OH)2 осаждение MgNH4P04
ведут в присутствии солей аммония. Но большого их избытка следует
избегать, так как он приводит к выпадению осадка, отвечающего составу
Mg (NH4)4 (P04)2; этот осадок после прокаливания образует не только
Mg2P207, но и Р205:
Mg (NH4)4 (P04)2 Л Mg(P03)2 + 4NH3f +2H20f
2Mg(P03)2 Л Mg2P207 + P206
Осадок MgNH4P04 при промывании водой частично подвергается
гидролизу:
MgNH4P04 + H20 ^± MgHP04 + NH4OH
Для подавления гидролиза осадок промывают разбавленным
раствором аммиака.
Условия проведения реакции осаждения MgNH4P04 — см. книга 1,
гл. IV, § 10.
Методика определения. Рассчитанную навеску соли магния
растворяют в 100 мл воды, прибавляют 5 мл 6 н. раствора соляной кислоты,
2 капли индикатора метилового красного и рассчитанное количество
10%-ного раствора гидрофосфата аммония. Затем медленно, по каплям,
при перемешивании приливают концентрированный раствор аммиака
до перехода окраски индикатора в желтый цвет. Добавляют избыток
в 5 мл и оставляют стоять в рабочем шкафу доследующего занятия. Перед
фильтрованием делают пробу на полноту осаждения. Осадок
отфильтровывают через фильтр «синяя лента» и промывают 2,5%-ным раствором
аммиака. При очень точных определениях магния в виде фосфата магния-
аммония осадок переосаждают. Необходимость двойного осаждения
вызывается тем, что анализируемый раствор всегда содержит большое
количество солей аммония и поэтому при первом осаждении нельзя получить
осадок, точно соответствующий формуле MgNH4P04. При повторном
осаждении избыток гидрофосфата аммония вводят в том минимуме,
который необходим для уменьшения растворимости; при этом также
уменьшается соосаждение.
Промывание заканчивают тогда, когда проба на полноту осаждения
показывает отсутствие ионов хлора (проба с AgN03+HN03). Воронку
с промытым осадком помещают для высушивания в сушильный шкаф.
« 18. ОПРЕДЕЛЕНИЕ ИОНОВ ХЛОРА
377
Высушенный осадок высыпают из фильтра на глянцевую бумагу и
накрывают его перевернутой воронкой. Фильтр помещают в доведенный до
постоянной массы тигель и озоляют, затем ссыпают туда же осадок с бумаги
и осторожно прокаливают его в муфельной печи при 1000—1100° С до
постоянной массы. Взвешивание и прокаливание повторяют до тех пор,
пока масса не станет постоянной.
Если осадок не отделять от фильтра, то он приобретает темный цвет.
Образования темного, содержащего углерод осадка можно избежать если,
вести обугливание фильтра при низкой температуре. Обрабатывать
осадок фосфата магния-аммония, содержащего темные частички угля,
азотной кислотой не рекомендуется, так как это не дает хороших
результатов.
Вычисление результатов анализа проводят по ранее приведенным
формулам (см. § 3).
§ 18. Определение ионов хлора в растворимых хлоридах
или в хлористоводородной кислоте
Метод определения хлоридов основан на взаимодействии ионов
серебра с ионами хлора, сопровождающемся образованием
малорастворимого осадка AgCl. Осадок хлорида серебра легко разлагается на свету
с выделением металлического серебра, что придает белому осадку хлорида
серебра фиолетовый, а иногда и черный оттенок. Для получения точных
результатов не следует осажДать хлорид серебра при ярком солнечном
освещении. При нагревании AgCl легко разлагается, поэтому перевод
в весовую форму требует определенных предосторожностей.
Наилучшим методом получения весовой формы является
высушивание осадка при 110° С. Это возможно при отделении осадка через
стеклянный фильтрующий тигель.
Уравнение реакции:
Ag+ + Cl-~-> jAgCl
Условия проведения реакции осаждения AgCl"— см. книга I, гл. VIII,
§3.
Методика определения. Рассчитанную навеску хлорида или
определенный объем соляной кислоты количественно переносят в химический
стакан и растворяют в дистиллированной воде; в стакан доливают
дистиллированную воду до объема 100 мл, в случае определения хлоридов раствор
подкисляют 1 мл концентрированной азотной кислоты и нагревают смесь
почти до кипения. Подкисление и нагревание обеспечивают коагуляцию
осадка. В случае определения НС1 операция подкисления раствора
опускается.
В другом стакане нагревают до 75—90° С 0,1 н. раствор нитрата
серебра. Затем осторожно по каплям при энергичном перемешивании
приливают раствор осадителя к анализируемому раствору до тех пор, пока
не прекратится образование осадка AgCl. Убедившись в полноте
осаждения, обертывают стакан темной бумагой и оставляют стоять на 1—2 ч.
Отделение осадка можно осуществить фильтрованием через бумажный
или стеклянный фильтр.
Фильтрование через бумажный фильтр. Если
нет стеклянного фильтра, то отделяют осадок от фильтра, как было
указано ранее (см. § 17). После окончания фильтрования осадок промывают
водой, к которой прибавлена 2 н. азотная кислота в количестве 5 мл на
100 мл воды. Проба на полноту промывания осадка: отсутствие ионов
серебра в промывных водах. В дальнейшем осадок обрабатывают анало-
378
ГЛ/V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
гично тому, как это делают при определении магния в его солях. Надо
помнить, что при нагревании осадка AgCl для получения весовой формы
всегда происходит частичное восстановление его до металлического
серебра. В этом случае необходимо к осадку добавить 1—2 капли
концентрированной азотной кислоты для растворения металлического серебра
и затем несколько капель 6 н. раствора соляной кислоты для образования
хлорида серебра и осторожно подогреть содержимое тигля.
Фильтрование через стеклянный фильтр. Для
количественного отделения от раствора осадков, изменяющих состав
своей весовой формы при высокой температуре, применяют
фильтровальные тигли или стеклянные фильтры. Фильтровальный тигель
промывают водой, затем разбавленной кислотой и высушивают до постоянной
массы. Тигель вставляют при помощи пробки в коническую колбу для
отсасывания, колбу соединяют с насосом. Переносят осадок в стеклянный
фильтр и промывают.
После отделения осадка его высушивают в тигле в сушильном шкафу
до постоянной массы и взвешивают.
На этом же принципе основан метод определения ионов серебра с той
лишь разницей, что AgCl осаждают из предварительно нагретого до 75° С
раствора, содержащего ионы серебра, 0,2 н. раствором НС1.
При пробе на полноту промывания осадка стекающий из воронки
фильтрат испытывают на присутствие в нем ионов хлора, а не ионов серебра,
как делают при определении С1~-ионов.
Результаты определения вычисляют, как указано в § 3.
§ 19. Анализ силикатов
Краткие сведения о силикатах. В химии силикатами называют соли
кремниевых кислот. В технике силикатами называют природные
соединения и технические материалы, в состав которых входих двуокись
кремния (кремнезем).
Химический состав силикатов изображают как сочетание различных
окислов металлов и неметаллов с Si02.
Например, формулу силиката натрия можно написать как
натриевую соль кремневой кислоты Na2Si03 или как сочетание Na20-Si02.
Формула полевого шпата, выраженная как сочетание различных окислов,
может быть представлена следующим образом:
K2O.Al203.6Si02 или K2AlSie016
Все силикаты по их отношению к минеральным кислотам и к воде
делят на три группы: растворимые в воде; разлагаемые кислотами; не
разлагаемые кислотами.
1. К силикатам, растворимым в воде, относят: силикаты щелочных
металлов, отвечающие общей формуле /nKt20-nSi02 (где Kt — ион
щелочного металла), растворимость которых в воде определяется их
кремнеземистым модулем и дисперсностью.
2. К силикатам, разлагаемым кислотами, относят: нефелины,
портландцемент, романцемент, гидравлическую известь и некоторые виды
шлаков и силикатных минералов.
3. К силикатам, не разлагаемым кислотами, относят:
Естественное сырье. Глины и каолины — Al203-2Si02-3H20;
полевые шпаты: ортоклаз — K20-Al203-6Si02, альбит — Na20-Al203X
X 6Si02, анортит —CaO-Al203-2Si02; слюда — 2K20-3Al203-6Si02 X
хН20; асбест — CaO-MgO-2SiOa; тальк — 3MgO-4Si02-H20; минералы
§ 19. АНАЛИЗ СИЛИКАТОВ
379
силикатной группы — Al203-Si02 (андалузит, силиманит, кианит),
муллит— 3Al203-2Si02 и ряд других силикатных горных пород и
минералов.
Искусственное сырье. Металлургические и котельные шлаки,
отвечающие общим формулам х KtO-#Kt203-2Si02; m KtO-/z Si02, а также
различные золы и др.
Силикатные материалы. Стекло, эмали, глазури, керамика,
фарфор, фаянс, шамот, высокоглиноземистые, динасовые изделия,
глиняный (красный) строительный кирпич, клинкерный кирпич, облицовочные
плиты.
Силикаты, силикатное сырье и силикатные материалы анализируют
на содержание в них: 1) гигроскопической влаги; 2) потерь при
прокаливании (сокращенно «п. п. п.»); 3) Si02; 4) А1203; 5) Fe203; 6) Ti02; 7) CaO;
8) MgO; 9) S03; 10) Na20; 11) К20 и др. Реже определяют Р205, MnO, FeO
(в шлаках, эмалях, глазурях и некоторых стеклах).
Порядок определений методом весового анализа может быть
следующим:
1-я навеска — определение гигроскопической влаги;
2-я навеска — определение п. п. п.;
3-я навеска — определение: Si02; Kt203+Ti02; CaO и MgO;
4-я навеска — определение содержания сульфатной серы;
5-я навеска — определение содержания К20 и Na20.
Определение гигроскопической влаги. Исследуемую среднюю пробу
силиката измельчают в агатовой или фарфоровой ступке. Отвешивают на
аналитических весах в бюксе с притертой крышкой около 1 г
измельченного силиката и сушат его при 100—125° С в сушильном шкафу в
течение 2 ч. По истечении этого времени бюкс. закрывают крышкой, ставят
в эксикатор на 30 мин для охлаждения и взвешивают. Затем повторно
бюкс с навеской ставят в сушильный шкаф, сушат еще 1 ч, охлаждают
в эксикаторе и снова взвешивают.
Высушивание и взвешивание повторяют до постоянной массы.
Отклонение должно составлять —0,0002 а.
Расчет. Относительную влажность или влажность, отнесенную к
влажной навеске, в процентах рассчитывают по формуле:
r = iL^.ioo (9)
а
где W — содержание воды, %;
а — исходная навеска, г;
Ь — масса остатка после высушивания, г.
Содержание всех составных частей анализируемого силиката (Si02,
Fe203, A1203, CaO, MgO и т. д.) пересчитывают на сухую навеску.
Для этого пользуются следующей формулой:
% (Ю)
_ g-100-100
*А~ а (100 — W)
где g — содержание определяемого вещества, г;
хА—масса определяемого компонента, %;
а — исходная навеска анализируемого силиката, г;
W — содержание в силикате воды, %.
Определение потерь при прокаливании («п. п. п.»). При
прокаливании исходной навески силикатов улетучиваются гигроскопическая
влага, двуокись углерода, образующаяся при термической диссоциации
карбонатов и сгорании органических примесей и другие летучие продукты.
380
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Для определения п. п. п. берут либо навеску исходного воздушно-
сухого силиката или навеску, в которой предварительно была определена
гигроскопическая влага, т. е. сухую навеску.
В первом случае около 2—3 г средней пробы исследуемого силиката
тонко измельчают в агатовой ступке; приблизительно 1 а измельченной
пробы помещают в предварительно прокаленный и взвешенный
фарфоровый (или) лучше платиновый тигель и нагревают в муфельной печи
при 1000° С в течение 30 мин. Прокаленный тигель ставят в эксикатор
для охлаждения, после охлаждения взвешивают jh затем повторно
прокаливают до постоянной массы.
Расчет. Содержание потерь при прокаливании вычисляют в
процентах по формуле:
гДе gn. п. п.— потеря при прокаливании, г;
а — масса исходной навески, г;
W — содержание в силикате воды, %.
Определение кремневой кислоты (или двуокиси кремния). Если
силикат, например портландцемент, разлагается минеральными кислотами,
его обрабатывают хлористоводородной кислотой:
ЗСаО- Si02 + 6HC1 -^ ЗСаС12 + 4- H2Si03 + 2Н20
при этом образуется осадок кремневой кислоты.
Около 1 г — из воздушно-сухой лабораторной пробы, или около 0,5 г —
из остатка после определения потерь при прокаливании силиката
помещают в фарфоровую чашку диаметром 12 см, а бюкс (или тигель) снова
взвешивают, массу взятого вещества определяют по разности
взвешиваний.
Навеску силиката смачивают дистиллированной водой и приливают
к ней по каплям 50 мл концентрированной хлористоводородной кислоты.
Содержимое чашки выпаривают досуха на водяной бане, снова
приливают 30 мл хлористоводородной кислоты (1 : 1) и опять выпаривают
досуха. Осадок смачивают концентрированной хлористоводородной кислотой,
приливают 50—75 мл горячей воды и оставляют на водяной бане на 15 мин.
Осадок кремневой кислоты без потерь переносят на фильтр (красная лента)
и промывают горячей водой до полного удаления СГ -ионов. Фильтрат
вместе с промывными водами собирают в мерную колбу емкостью 250 мл,
доводят водой до метки, тщательно перемешивают и сохраняют для
определения суммы (Kt203+Ti02), CaO и MgO весовым или объемным
методами.
Фильтр с осадком кремневой кислоты помещают в тигель,
подсушивают, осторожно сжигают и прокаливают до постоянной массы.
Если силикат не разлагается минеральными кислотами, то его
сплавляют с карбонатом натрия или смесью Na2C03 и К2С03
Si02 + Na2C03 -> Na2SiOa + C021
при этом неразлагаемые кислотами силикаты переходят в растворимую
в воде форму.
Для сплавления навеску силиката после определения потерь при
прокаливании помещают во взвешенный платиновый тигель, смешивают
с шести-, восьмикратным количеством безводного Na2C03 и нагревают
на газовой горелке сначала осторожно, на маленьком пламени, а затем
постепенно увеличивают пламя.
« 19. АНАЛИЗ СИЛИКАТОВ
381
Разложение следует считать законченным после прекращения
выделения пузырьков СО2, о чем судят по образованию сплошной однородной
массы. Затем тигель нагревают еще 10 мин. Весь процесс можно закончить
за 30—40 мин. Тигель с осадком помещают в фарфоровую чашку.
Полученный плав, содержащий только силикаты, разлагаемые минеральными
кислотами, выщелачивают водой в фарфоровой чашке и затем
обрабатывают хлористоводородной кислотой (1 : 1), прибавляя ее осторожно,
сначала до прекращения выделения С02, а затем еще 20—30 мл избытка.
Платиновый тигель вынимают щипцами и тщательно обмывают из про-
мывалки дистиллированной водой. Кислый раствор разложенной пробы
силиката выпаривают на водяной бане для выделения из него геля
кремневой кислоты в виде п Si02-/n H20.
Кремневая кислота при прокаливании превращается в двуокись
кремния:
п Si02m Н20 —> n SiOs + т Н20
Истинное содержание Si02 определяют, обрабатывая прокаленный
и взвешенный осадок кремневой кислоты смесью фтористоводородной
и серной кислот (1—2 капли концентрированной серной кислоты на
10 мл 40%-ной фтористоводородной кислоты). При этом кремний
улетучивается в виде SiF4.
Нагревание содержимого платинового тигля со смесью H2S04 + H2F2
проводят сначала на водяной бане. Затем для полного превращения
сульфатов в соответствующие окислы металлов остаток прокаливают:
A12(S04)3 —> A1203+3S03
Fe2(S04)3 —> Fe203 + 3S03
Ti(S04)2 —> Ti02 + 2S03
После этого тигель снова взвешивают, по разнице результатов
взвешивания тигля с прокаленным Si02 и тигля с осадком окислов Fe203,
А1203 и Ti02 находят массу Si02 (gsio2).
Расчет. Содержание Si02 в процентах находят по формуле:
gsio2-100
xsio2 — a
При пересчете на абсолютно сухую навеску содержание Si02 в
процентах вычисляют по формуле:
_ gsuv100'100
*Si°* a (100— W)
гДе SsiOi — масса Si02» 2.
В случае обработки прокаленного осадка Si02 фтористоводородной
кислотой содержание Si02 высчитывают по потере в массе после
химической обработки.
Определение полуторных окислов и двуокиси титана. К полуторным
окислам относят окись железа и окись алюминия. Для их определения
используют фильтрат, полученный после отделения кремневой кислоты.
В присутствии кремневой кислоты определение полуторных окислов
не проводят, так как кремневая кислота мешает их количественному
определению. К солянокислому анализируемому раствору сначала приливают
азотную кислоту для перевода железа (II) в железо (III), затем осаждают
Fe+ + + и А1+ + + аммиаком. При этом выпадают осадки гидроокисей
железа, алюминия и титана, которые прокаливают и взвешивают (весовая
форма Fe203, A1203 и Ti02).
Получаемые осаДки гидроокисей представляют собой аморфные
вещества и склонны к адсорбции и образованию коллоидных растворов,
382
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
поэтому при осаждении необходимо соблюдать следующие условия:
осаждение вести из горячего раствора в присутствии коагулирующего
электролита (NH4N03), разлагающегося без остатка в процессе
последующего прокаливания полуторных окислов. Для предупреждения
адсорбции осаждение аморфных осадков ведут из концентрированных растворов.
После осаждения сразу приливают 100 мл горячей воды и выдерживают
несколько минут на водяной бане. Осадки при этом хорошо коагулируют,
раствор над осадком становится прозрачным. Осадки отфильтровывают
в горячем состоянии и промывают горячей промывной жидкостью (2%-ным
раствором NH4N03). Промывание осадков промывной жидкостью
препятствует пептизации аморфных осадков.
Рекомендуется проводить два параллельных определения. Для этого
из мерной колбы берут две пробы по 50 мл, переносят в стаканы емкостью
500 мл; приливают в каждый по несколько капель концентрированной
HN03 и нагревают до кипения (для окисления двухвалентного железа
в трехвалентное).
К кипящим растворам приливают несколько капель метилового
красного (0,2%-ный спиртовый раствор), 0,3 г сухого хлорида аммония,
осаждают АГ++-, Fe+++- и TiIV -ионы в виде Al (OH)3, Fe (OH)3,
Ti (OH)4 10%-ным водным раствором аммиака, прибавляя: его по каплям
при постоянном перемешивании жидкости до пожелтения раствора и
появления запаха аммиака. При этом благодаря избытку NH4C1 ионы
щелочноземельных металлов остаются в растворе.
При наличии в исследуемом силикате примесей фосфорной кислоты в аммиачном
растворе выпадают А1Р04 и FeP04:
Fe+++ + РО~ -> I FeP04
Al+++ + Р0~ -* I А1Р04
что может привести к неточным результатам определения полуторных окислов.
Растворы с осадками нагревают 3—5 мин на водяной бане для
коагуляции (раствор над осадком должен быть прозрачным).
Затем осадки отфильтровывают в горячем состоянии через
беззольные фильтры (красная лента). Фильтрат и промывные воды
собирают в мерные колбы емкостью 250 мл. Осадок промывают 3 раза
2%-ным раствором NH4N03. Затем для получения более точных
результатов проводят повторное осаждение полуторных окислов. Для этого осадки
вместе с фильтрами переносят в химические стаканы, прибавляют по
15 мл HN03 (1 : 1), добавляют по 50 мл дистиллированной воды и повторно
осаждают гидроокиси 10%-ным раствором NH4OH в присутствии
метилового красного. Выпавшие осадки гидроокисей снова otфильтpoвывaют
через беззольный фильтр (красная лента) в те же. мерные колбы емкостью
250 мл и промывают 2%-ным раствором NH4N03.
Содержимое мерных колб используют для определения СаО и MgO
весовым или комплексонометрическим способами (см. «Анализ
силикатов объемным комплексонометрическим методом», определение СаО и MgO).
Один из осадков подсушивают на воронке в сушильном шкафу,
прокаливают до постоянной массы и вычисляют содержание суммы
полуторных окислов и Ti02 в процентах по формуле;
gA-100-100-5
*KteO,+TIO. ^ а (ЮО— W)
где gA — масса прокаленного осадка (сумма полуторных окислов + Ti02), г;
5 — коэффициент разбавления.
§ 19. АНАЛИЗ СИЛИКАТОВ
383
Воронку с другим осадком помещают над колбой емкостью 250 мл
и на фильтре растворяют в 50 мл 2 н. раствором H2S04. Фильтр промывают
водой, полученный раствор используют для определения железа
колориметрическим (см. гл. IX, § 6), перманганатометрическим (см. гл. III,
§ 24) или иодометрическим (см. гл. III, § 30) методами.
По 25 мл раствора из мерной колбы объемом 250 мл используют для
количественного определения двуокиси титана колориметрическим,
перманганатометрическим или иодометрическим методами (см. главу IX,
§7).
Содержание Fe203(Ti02) вычисляют в процентах по формулам:
при определении иодометрическим методом
_ ^.0,^ Na.S.o/W.O, ¦ 100 - Ю0 - Ук
*Fe2o8 (ТЮ2) 10оо а (100 — W) VA
при определении перманганатометрическим методом
TKMnO4/Fe2O3^KMnO4-1Q0'10Q^K
*Fe203 (TiOz) - а (100 _ щ уА
Определение железа в полуторных окислах. Определение содержания
железа в прокаленном осадке полуторных окислов основано на осаждении
сероводородом ионов железа в присутствии солей винной кислоты. Тар-
тратные комплексные ионы железа в отличие от тартратных комплексных
ионов алюминия разрушаются при действии сероводорода. Дальнейшее
определение железа проводят обычным методом — путем растворения FeS
в НС1 + HN03, осаждения Fe+ + + в виде гидроокиси, прокаливания
Fe (ОН)3 до Fe203 и взвешивания *. Перевод полуторных окислов в
растворимые соли осуществляется сплавлением их с K2S207.
Прокаленный осадок полуторных окислов после его взвешивания
сплавляют в платиновом тигле с пиросульфатом калия. К осадку прибавляют
5 г чистого безводного пиросульфата калия и, закрыв тигель крышкой,
нагревают его. Сплавление продолжают до тех пор, пока в плаве нельзя
будет обнаружить частичек окислов. Когда сплавление закончено, дают
тиглю остыть, прибавляют 50 мл горячей воды, 3—5 мл
концентрированной серной кислоты и нагревают до полного растворения плава.
К раствору прибавляют винную кислоту в некотором избытке по
отношению к массе полуторных окислов, раствор аммиака и пропускают
сероводород. Осадок сульфида железа отфильтровывают, растворяют на
фильтре в горячей 2 н. хлористоводородной кислоте, прибавляют к
раствору 1 мл концентрированной азотной кислоты, кипятят для удаления
сероводорода и окисления железа (II) до ионов железа (III); ионы железа
осаждают в виде гидроокиси (см. § 23).
Определение алюминия. Для определения массы окиси алюминия
из массы прокаленного осадка полуторных окислов вычитают общее
содержание железа * в пересчете на Fe203.
Определение окиси кальция. Окись кальция и окись магния
определяют в фильтрате, полученном после отделения полуторных окислов, так
как ионы железа и алюминия мешают определению кальция и магния,
они дают с реактивами, применяемыми при определении окиси кальция и
* В настоящее время весовые определения этих и других элементов вытесняются
комплексонометрическими методами титрования (см. гл. IV, § 22—28).
384
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
магния, малорастворимые осадки. По тем же соображениям сначала
определяют окись кальция, а затем окись магния.
Ионы кальция определяют в виде оксалата кальция, а ионы магния —
в виде аммоний-магний фосфата. Эти осадки кристаллические, поэтому
при их осаждении необходимо соблюдать соответствующие условия
осаждения: 1) осаждение вести из разбавленных растворов разбавленными
растворами осадителей; 2) прибавлять осадитель медленно, по каплям.
При быстром осаждении появляется много центров кристаллизации и
образуются мелкие кристаллы; 3) перемешивать раствор стеклянной
палочкой, чтобы избежать сильных местных пересыщений; 4) осаждение вести
из горячего раствора горячим раствором осадителя; при нагревании
увеличивается растворимость мелких кристаллов и образуются крупные
кристаллы; 5) в некоторых случаях добавляют вещества, повышающие
растворимость кристаллов.
После прибавления осадителя осадку дают созреть, для чего
анализируемую смесь оставляют стоять на несколько часов. При созревании
образуются крупные кристаллы. Крупные кристаллы легко
отфильтровываются и быстро отмываются от примесей.
Промыван"ие осадков ведут разбавленным раствором, содержащим
одноименный ион с осадителем, а именно 2%-ным раствором (NH4)2C204,
это способствует понижению растворимости осадков.
Нейтральный или слабоаммиачный фильтрат с промывными водами,
оставшийся после определения Kt203 и ТЮ2, собирают в мерную колбу
емкостью 250 мл, доводят водой до метки и отбирают для анализа пипеткой
50 мл две параллельные пробы. Пробы подкисляют концентрированной
уксусной кислотой до розовой окраски в присутствии метилового красного,
нагревают до кипения и прибавляют 70—80 мл 4%-ного раствора оксалата
аммония. В осадок выпадает СаС204, а ионы магния остаются в растворе.
После 4 ч стояния осадок СаС204 отфильтровывают и промывают 2%-ным
раствором (NH4)2C204 до удаления ионов хлора. Фильтраты с
промывными водами собирают раздельно в химические стаканы емкостью 500 мл
для определения содержания магния.
Фильтр с осадком помещают в тигель, предварительно доведенный
до постоянной массы, подсушивают, озоляют на маленьком пламени
и прокаливают до постоянной массы.
Расчет. Содержание окиси кальция вычисляют в процентах по
формуле:
gCaQ.100-100.5.5
*CaO- а (100— W)
где gcaO — масса СаО, г;
5 и 5 — коэффициенты разбавления.
Осаждение ионов кальция можно вести из солянокислого раствора,
добавляя при нагревании кристаллическую Н2С204-2Н20 и за^ем
нейтрализуя раствор NH3 по метиловому красному-.
Определение окиси магния. Содержание окиси магния можно
определить несколькими способами. Ниже описываемый весовой метод основан
на осаждении ионов магния в виде MgNH4P04, последующем прокаливании
полученного осадка и взвешивании весовой формы — Mg2P207.
К фильтратам с промывными водами после осаждения окиси кальция
приливают 40—50 мл I и: раствора Na2HP04 и нагревают до кипения.
К горячим растворам в присутствии фенолфталеина добавляют 10%-ный
водный раствор NH3 до щелочной реакции:
Mg++ + HPOT" + NH3 —* ^MgNH4P04
§ 19. АНАЛИЗ СИЛИКАТОВ
385
Осадки оставляют для созревания на несколько часов.
После этого осадки отфильтровывают, промывают холодной
дистиллированной водой до удаления ионов хлора и прокаливают до постоянной
массы:
2MgNH4P04 -1> Mg2P207 + 2NH3 t + Н20 t
Расчет. Содержание MgO вычисляют в йроцентах по формуле:
_ gMgaPao/-МО-100-5-5
*MgO~ a (100— W)
гДе ?Mg2p2o7 — масса прокаленного осадка пирофосфата магния, г;
F — фактор пересчета, равный 2iWMg0/MMg2p2o7;
5 и 5 — коэффициенты разбавления.
Определение сульфатной серы. Сульфаты определяют при анализе
многих природных силикатов, а также готовых силикатных изделий.
В природных силикатах сульфатная сера находится иногда в
значительных количествах, в готовых силикатных изделиях содержание сульфатной
серы обычно не превышает десятых долей процента.
В природных силикатах встречается также и сульфидная сера.
Для определения сульфатной серы анализируемый образец силиката
разлагают кислотами или сплавляют с карбонатом натрия, не
содержащим сульфатов, полученный плав выщелачивают. Разложение плава
ведут горячей водой, при этом выпадает осадок основной углекислой
соли железа, титановой кислоты и гидроокиси алюминия. Осадок
отфильтровывают, промывают разбавленным раствором карбоната натрия.
В фильтрате с промывными водами осаждают сульфатную серу раствором
хлорида бария. В случае разложения силиката кислотой в раствор
переходят все компоненты; мешающие примеси осаждают раствором аммиака.
При этом выпадает объемистый аморфный осадок, состоящий из
кремневой кислоты, полуторных окислов и гидроокиси титана. Отфильтровав
осадок и отмыв его до отрицательной реакции на ионы хлора, в фильтрате
с промывными водами осаждают сульфатную серу раствором хлорида
бария (см. § 14).
Определение фосфора. Пробу обрабатывают концентрированной
азотной кислотой при кипячении. При этом соединения фосфора переходят
в раствор в виде фосфорной кислоты, которую можно определять
различными методами.
Фосфат-ионы осаждают молибдатом аммония в виде аммониевой
соли фосфорномолибденовой кислоты (NH4)3H4 [P (Мо207)6] или
(NH4)3P04-12Мо03-2Н20. Этот осадок высушивают и взвешивают в виде
(NH4)3H4 [P (Мо207)6] или (NH4)3P04-12Mo03 или прокаливают до
образования осадка Р2Об-24Мо03:
Н3Р04 + 12 (NH4)2 Mo04 + 21HN08 —*
—> (NH4)3 H4 [P (Mo207)e) + 21NH4N03 + ЮНаО
Можно также полученный осадок (NH4)3H4 [P (Мо207)6] растворить
в растворе аммиака и осадить Р04~~-ионы в виде MgNH4P04:
(NH4)3 Н4 [Р (Мо207)61 + 24NH3 + 10Н2О —>- (NH4)3 P04 + 12 (NHja Mo04
(NH4)3 Р04 + MgCl2 -> J MgNH4P04 + 2NH4C1
2MgNH4P04 -Л Mg2P207 + 2NH3 f + H20 f
386
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Определение фосфора в виде Р205 -24Мо03. Навеску
около 0,5 г вещества помещают в платиновую чашку, прибавляют 5 мл
воды, 3 мл концентрированной азотной кислоты и выпаривают досуха на
водяной бане. Остаток снойа обрабатывают 2 мл азотной кислоты и кислоту
выпаривают. Затем вновь полученный остаток обрабатывают 10 мл
разбавленной (1:3) азотной кислоты, смесь кипятят и раствор
отфильтровывают через фильтр «белая лента» в стакан. Общий объем жидкости не
должен быть больше 40 мл. К раствору прибавляют рассчитанное
количество 6 н. раствора молибдата аммония. (Для полноты осаждения
необходим десятикратный избыток осадителя). Полученную смесь сильно
перемешивают и оставляют на ночь. Осадок отфильтровывают через
взвешенный фильтровальный тигель и промывают 5%-ным раствором нитрата
аммония, затем высушивают и нагревают при 400° С в течение 1 ч. Остаток
должен иметь сине-черный цвет. Весовая форма — Р205-24Мо03. Осадок
гигроскопичен, его взвешивают так же, как осадок СаО при определении
кальция (см. § 16).
Определение фосфора в виде Mg2P207. Осадок фосфоро-
молибдата аммония растворяют в 20 мл раствора цитрата (25 г лимонной
кислоты в 700 мл воды и 350 мл концентрированного раствора аммиака).
Полученный раствор собирают в стакан емкостью 250 мл, фильтр
последовательно промывают разбавленным (1 : 20) раствором аммиака, горячей
водой и разбавленной (1 : 20) хлористоводородной кислотой. Общий
объем жидкости не должен превышать 150 мл. Прибавляют 2 капли
метилового красного и хлористоводородную кислоту до появления красной
окраски раствора, затем добавляют смесь NH4C1 и MgCl2.
К охлажденному раствору фосфата медленно прибавляют по каплям
при перемешивании концентрированный раствор аммиака до появления
желтой окраски индикатора. Осадок с раствором оставляют стоять в
рабочем шкафу до следующего занятия и далее обрабатывают, как при
определении магния (см. § 17).
Определение фосфора в виде Mg2P207 может быть осуществлено и без
образования осадка фосфоромолибдата аммония. Осаждение проводят
из раствора, содержащего фосфат-ионы, действием магнезиальной смеси;
далее поступают так же, как и при определении магния (см. § 17).
Расчет результатов анализа — см. § 3.
Определение двуокиси углерода проводится так же, как при анализе
доломита (см. § 20).
Помимо описанных выше весовых методов определения составных
частей силикатов, в настоящее время широко применяют и другие
ускоренные химические и физико-химические методы анализа. Для удобства
сопоставления весового и объемного методов анализа силикатов они
рассматриваются в одном разделе, что в известной мере нарушает
принятый принцип изложения (объемные методы анализа описаны выше — см.
гл. I). Ниже приводится описание комплексонометрического метода
анализа силикатов.
Анализ силикатов объемным комплексонометрическим методом. Комплексонометри-
ческий метод по точности уступает весовым методам, как и все методы объемного анализа,
но отличается экспрессностью. В основе этого метода лежит способность комплексона III
давать с катионами, растворимыми в воде, комплексные соединения.
Устойчивость комплексных соединений зависит от рН среды. Комплексные соединения
кальция и магния устойчивы в щелочных средах. Комплексные соединения железа (III)
и алюминия устойчивы в кислых средах при рН = 1—2.
При определении отдельных срставных частей строительных материалов применяют
прямое и обратное комплексонометрическое титрование (см. гл. IV, § 1 и ел.).
Определение Fe203 (в цементе). Для определения Fe203 из мерной колбы емкостью
550 мл отбирают пипеткой две пробы по 50 мл солянокислого раствора, оставшегося после
-отделения кремневой кислоты, помещая их в конические колбы емкостью 250 мл. Приливают
§ 19. АНАЛИЗ СИЛИКАТОВ
387
для окисления Fe++ до Fe+ + + концентрированной HN03, прибавляют 10 мл
хлористоводородной кислоты (1 : 1), нагревают до 60—70° С, приливают 1—2 мл индикатора —
сульфосалицилата натрия (до появления фиолетовой окраски раствора) и титруют комплек-
соном III до перехода фиолетового окрашивания в светло-желтое.
Раствор сохраняют для определения А1203.
Расчет. Содержание Fe203 в процентах рассчитывают по формуле:
^ДТА/РегОз
1/ЭДТА. 100-100-5
*Fe203- a (100— W)
где ^эдта — объем раствора комплексона III, израсходованный на титрование, мл;
ТэдтА/Fe Ор — ТИТР раствора ЭДТА, выраженный через определяемое вещество (Fe203),
г/мл;
5 — коэффициент разбавления.
Определение А1203 (в цементе). В коническую колбу, в которой определяли окись
железа, прибавляют 10 мл комплексона III. Нагревают до кипения, добавляют 10 мл
ацетатного буферного раствора, 1—2 мл индикатора — сульфосалицилата натрия. Охлаждают
до комнатной температуры и титруют раствором соли трехвалентного железа (железным»
квасцами) до перехода светло-желтой окраски в золотисто-оранжевую.
Расчет. Содержание А1203 в процентах вычисляют по формуле:
_ (Ю - ^эдта) ТЭДтА/А12су100- ЮР"5
*А1«оч a(\00—W)
где (10—Уэдта) — объем раствора комплексона III, израсходованный на реакцию-
с ионами алюминия, мл\
Тэдта/аьОя — ТИТР раствора ЭДТА, выраженный через определяемое вещество
(А1203), г/мл.
Определение СаО (в цементе). Кальций определяют титрованием комплексоном III
в присутствии мурексида при рН = 12. Для определения СаО отбирают пипеткой две пробы
по 50 мл раствора, получившегося после определения полуторных окислов, в конические
колбы емкостью 250 мл. Добавляют 10 мл 20%-ного раствора щелочи и мурексида (50—
ПО мг) до появления розовато-сиреневого окрашивания, затем титруют комплексоном III
до перехода розовато-сиреневого окрашивания в устойчивое сине-голубое.
Расчет. Содержание СаО в процентах вычисляют по формуле:
ТэДТА/СаО^ЭДТА'ЮО-ЮО^-б
Са° а (100— W)
где ^эдта — объем раствора комплексона III, израсходованного на титрование, мл;
ТэдтА/СаО — тИТР раствора ЭДТА, выраженный через определяемое вещество (СаО),.
г/мл;
5 и 5 — коэффициенты разбавления.
Определение суммы СаО и MgO (в цементе). Содержание СаО и MgO определяют в
фильтрате после отделения осадка полуторных окислов. Из мерной колбы емкостью 250 мл
отбирают пипеткой две пробы по 50 мл раствора и переносят в конические колбы емкостью»
250 мл. Затем прибавляют 7—10 мл аммиачной буферной смеси, 7—8 капель кислотного
хром темно-синего и титруют сумму Са++ и Mg++ 0,1 н. раствором комплексона III до
перехода окраски из винно-красной в сиренево-фиолетовую. Титрование повторяют несколько
раз. Затем вычисляют содержание MgO.
Расчет. Содержание MgO в процентах вычисляют по формуле:
х тэдта^о (Уэдта - Уэдта) 10°-100'5'5
м*° а (100— W)
где ТЭдТА/Мб0 — титр раствора комплексона III no MgO, гТмл;
^эдта — объем раствора комплексона III, израсходованный на титрование ионов-
кальция и магния, мл;
^эдта—объем раствора комплексона III, израсходованный на титрование-
ионов кальция, мл;
5 и 5 — коэффициенты разбавления.
388
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
Пламеннофотометрическое определение натрия и калия. Для определения в силикатах
натрия и калия широко применяют пламеннофотометрический метод, основанный на
предварительном фотометрировании серии стандартных растворов образцов с известным
содержанием натрия и калия и последующем фотометр ировании на пламенном фотометре
растворов исследуемого образца, содержащего калий и натрий (см. книга III, гл. VII, § 16 ел.).
§ 20. Анализ доломита
Доломит представляет собой горную породу, состоящую в основном
из карбонатов кальция и магния. В состав доломитов входят, кроме того,
карбонаты, силикаты и сульфаты железа (II), марганца, алюминия, титана
и т. д., а также двуокись кремния, пириты, фосфаты, углеродистые
соединения, вода и др. Для того чтобы судить о пригодности доломита для
данной технической цели, обычно проводят краткий анализ, при котором
определяют:
а) потери при прокаливании (сокращенно «п. п. п.»);
б) нерастворимые вещества;
в) сумму полуторных окислов Kt203, состоящих из Fe203, A1203 и др.;
г) окись кальция;
д) окись магния;
е) двуокись углерода.
Определение потери при прокаливании. При высокой температуре
доломит теряет влагу, двуокись углерода и органические вещества.
Часто анализ начинают с определения гигроскопической воды, для чего
навеску вещества перед прокаливанием сначала высушивают в сушильном
шкафу при 105—125° С до постоянной массы. Затем эту навеску
прокаливают до постоянной массы в муфельной печи. Разность в массе вещества
до прокаливания и после прокаливания дает массу потери при
прокаливании.
Определение и расчет проводят так, как указано при анализе силикатов
(см. § 19).
Определение веществ, нерастворимых в кислотах. При обработке
навески доломита хлористоводородной кислотой в осадке остается
нерастворимая кремневая кислота и некоторые другие соединения.
Полученный кислый раствор выпаривают досуха. Обработку кислотой и
выпаривание повторяют. Осадок снова обрабатывают хлористоводородной
кислотой, отделяют от раствора, прокаливают до постоянной массы, охлаждают
и взвешивают. При этом кремневая кислота переходит в Si02.
Определение ведут по той же методике, которая рекомендуется при
анализе силикатов (см. § 19).
Содержание нерастворимого остатка рассчитывают по ранее
приведенным формулам (см. § 3).
Определение полуторных окислов. Определение суммы полуторных
окислов основано на выделении в один общий осадок гидроокисей железа,
алюминия и других катионов, осаждающихся при действии раствора
аммиака. Весовая форма, получаемая при прокаливании гидроокисей,
отвечает формуле Kt203.
Методика определения описана в § 19.
Расчет содержания суммы полуторных окислов проводят по формулам,
приведенным в § 3.
Определение окиси кальция. Фильтрат и промывные воды после
определения полуторных окислов выпаривают до объема 200 мл, подкисляют
5 мл 6 н. хлористоводородной кислоты и определяют кальций, как было
указано ранее (см. § 16).
Определение окиси магния. В фильтрате и промывных водах после
определения Са++ определяют содержание магния. Для этого прибав-
§ 20. АНАЛИЗ ДОЛОМИТА
389
ляют к раствору 5—7 мл концентрированной азотной кислоты и
выпаривают на водяной бане досуха (под тягой). Сухой остаток растворяют
в 2 мл концентрированной хлористоводородной кислоты и 25 мл воды и
определяют магний, как было описано ранее (см. § 17).
Рис. 71. Схема установки для поглощения двуокиси углерода:
/ — натронный асбест; 2
сульфата меди;
- Mg(Cl04)2; 3 — пемза, пропитанная раствором
4 — концентрированная серная кислота.
Определение двуокиси углерода. Двуокись углерода, находящуюся
в доломитах в виде карбонатов, определяют обработкой отдельной навески
кислотой и поглощением выделяющейся при этом двуокиси углерода
натронным асбестом:
СаС03 + 2Н+ —+ СО* f + Са++ + Н20
C02 + 2NaOH -*- Na2C03 + HaO
Количество С02 определяют по увеличению массы поглотительного
прибора. Схема установки для поглощения С02 представлена на рис. 71.
Установка состоит из реакционной колбы, снабженной небольшим
холодильником для конденсации воды и хлористоводородной кислоты.
Холодильник соединен с промывалкой, содержащей концентрированную
серную кислоту для обезвоживания С02, а также с трубкой, заполненной
безводным сульфатом меди, и трубкой, заполненной Mg (С104)2 для
окончательного высушивания газа. Для поглощения С02 служат две
поглотительные склянкц, заполненные натронным асбестом. Протягивание
воздуха и анализируемого газа через установку осуществляют при помощи
аспиратора или водоструйного насоса.- В колбу, в которой проводят
разложение пробы, вставляют также капельную воронку; конец воронки,
находящийся внутри колбы, должен быть загнут кверху, чтобы не было
утечки СО2 через воронку. Верхний конец воронки закрывают хлоркаль-
циевой трубкой.
Навеску анализируемого вещества (около 1 г) помещают в
реакционную колбу прибора и приливают 50 мл воды. Затем колбу закрывают
пробкой, э которой укреплены холодильник и капельная воронка.
Холодильник соединяют с осушительными сосудами и протягивают через всю
систему воздух, очищенный от С02, до полного удаления из системы
двуокиси углерода. Затем закрывают кран капельной воронки и включают
390
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
в установку предварительно взвешенные поглотительные сосуды.
Наполняют капельную воронку раствором разбавленной (1 : 1)
хлористоводородной кислоты, открывают кран капельной воронки и медленно (не более
двух пузырьков выделяющегося газа в секунду) приливают кислоту
в колбу. Когда выделение пузырьков ослабевает, пускают в холодильник
воду и нагревают колбу до слабого кипения. По окончании нагревания
пропускают через систему воздух еще 20 мин для полного удаления С02
из раствора. Отделяют поглотительные сосуды, закрывают вводной и
отводной краны поглотительных трубок, помещают трубки в шкафчик
аналитических весов и по охлаждении взвешивают. По разности в массе
поглотительных сосудов до и после поглощения двуокиси углерода
определяют массу выделившейся С02.
Расчет результатов анализа — см. § 3.
§ 21. Анализ бронзы и латуни
Бронзы и латуни представляют собой сплавы цветных металлов,
в состав которых входят медь, цинк, олово, свинец, железо и т. д.
Латуни отличаются от бронз тем, что они в основном состоят из сплава
меди и цинка, а бронзы — из сплава меди и олова.
При растворении сплава в концентрированной азотной кислоте
в осадке остается лишь метаоловянная кислота, которую отфильтровывают.
Фильтрат обрабатывают серной кислотой, при этом в осадок выпадает
сульфат свинца.
Фильтрат, полученный после отделения свинца, подвергают
электролизу. Металлическая медь выделяется на катоде. Железо определяют
осаждением в виде гидроокиси, цинк — в виде двойной соли — фосфата
цинка-аммония.
Медь можно также определить и весовым методом — действием
роданид-ионов (см. книга 1, гл. VH, § 4).
Определение олова. Метод основан на осаждении нерастворимого
осадка метаоловянной кислоты, получающегося при действии на
металлическое олово концентрированной HNOs. При прокаливании
метаоловянной кислоты получается Sn02:
Sn + 4HN03 -Л* l H2Sn03 + 4N02 f + H20
H2Sn03 -^ Sn02 + H20f
Навеску латуни (около 1 г) помещают в стакан емкостью 50 мл,
накрывают часовым стеклом и осторожно через носик стакана приливают 15 мл
концентрированной азотной кислоты и 10 мл воды. По окончании
растворения раствор упаривают при 80—90° С до объема 5—8 мл. Упаривание
ведут медленно, около 1 ч\ такое медленное упаривание способствует
количественному выделению H2Sn03. Горячий раствор фильтруют через фильтр
«красная лента», осадок на фильтре промывают несколько раз горячей
разбавленной (1 : 20) азотной кислотой, высушивают, озоляют при
возможно низкой температуре. Для окисления восстановленных форм олова
осадок после сжигания фильтра обрабатывают 1—2 каплями
концентрированной HN03; избыток кислоты выпаривают и осадок прокаливают
при хорошем доступе воздуха при 1100° С до постоянной массы. Весовая
форма ^п02.
Расчет результатов анализа — см. § 3.
Определение свинца. Определение основано на осаждении ионов
свинца в виде PbS04. Весовую форму получают высушиванием осадка.
Сульфат свинца довольно хорошо растворим, поэтому определение еле-
§ 21. АНАЛИЗ БРОНЗЫ И ЛАТУНИ
391
дует вести в присутствии избытка сульфат-ионов. В качестве промывной
жидкости применяют вначале разбавленную серную кислоту, а затем —
спирт. Прибавление спирта понижает растворимость осадка сульфата
свинца.
К фильтрату и промывным водам, собранным после отделения олова,
прибавляют 5 мл концентрированной серной кислоты, выпаривают на
электрической плитке до появления паров серной кислоты, охлаждают,
осторожно обмывают водой стенки сосуда и снова выпаривают до
появления паров серной кислоты.
Смесь охлаждают, добавляют 40 мл дистиллированной воды, снова
нагревают около 1 ч почти до кипения и перемешивают. Когда содержимое
стакана примет комнатную температуру, приступают к отделению осадка
сульфата свинца через фильтровальный тигель. Промывают осадок
разбавленной серной кислотой. Фильтрат и промывные воды количественно
собирают для проведения следующего определения.
Недостатком определения свинца в виде сульфата является
необходимость промывать осадок серной кислотой, вследствие этого приходится
прокаливать его. Отделение осадка через беззольные бумажные фильтры
проводят так же, как это было указано при определении магния в его
солях (см. § 17). Восстановившийся при этом свинец окисляют
прибавлением 1 капли азотной кислоты, как это делают при определении AgCl
(см. § 18).
Тигель с осадком прокаливают при 500° С до постоянной массы. При
пользовании горелкой фильтровальный тигель надо вставлять в
фарфоровый тигель.
Вычисление результатов анализа проводят по ранее приведенным
формулам (см. § 3).
Определение меди. Определение основано на осаждении в слабокислом
растворе ионов меди в виде Cu2 (SCN)2. Осадок высушивают в сушильном
шкафу и взвешивают.
Уравнения реакций:
CuS04 + 2NH4SCN —> i Cu (SCN)2 + (NH4)2 S04
2Cu (SCN)2 + (NH4)2 S03 + H20 —> 4, Cu2 (SCN)2 + (NH4)2 S04 + 2HSCN
К фильтрату, полученному после определения свинца, прибавляют
раствор аммиака до появления неисчезающего осадка, затем 10 мл 2 н.
раствора серной кислоты, разбавляют дистиллированной водой до 300 мл
и добавляют 25 мл 5%-ного раствора сульфита аммония. Фильтрат
нагревают до кипения, медленно прибавляют к нему при перемешивании
рассчитанное количество 10%-ного раствора роданида аммония и 2 н.
раствора (NH4)2S03. Осадку дают постоять 1—72 ч, затем отфильтровывают его
через фильтровальный тигель и промывают несколько" раз промывной
жидкостью, состоящей из смеси 0,1%-ного раствора роданида аммония и
0,01%-ного раствора сульфита аммония. Осадок высушивают в
сушильном шкафу при 105—110° С до постоянной массы, фильтрат сохраняют
для последующих определений.
Расчет результатов анализа — см. § 3.
Электролитическое определение меди описано в гл. VII (§ 6, 7).
Определение железа. К фильтрату, полученному после отделения
роданида меди, добавляют 40 мл концентрированной азотной кислоты и
20 мл концентрированной хлористоводородной кислоты и выпаривают
почти досуха. К остатку прибавляют 4 мл той же кислоты, нагревают до
растворения осадка и разбавляют раствор 75 мл дистиллированной воды.
Раствор, содержащий ионы железа и ионы цинка, нагревают почти до кипе-
392
ГЛ.У. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
ния и осаждают Fe+++ концентрированным раствором аммиака. Далее
поступают так, как указано в § 15.
Фильтрат и промывные воды сохраняют для определения цинка.
Определение цинка. Определение основано на осаждении ионов цинка
в виде двойной соли—фосфата цинка-аммония. Наиболее полно его
осаждение происходит при рН = 5,5—7. В начале осаждения образуется
фосфат цинка, очень быстро переходящий в кристаллический осадок
двойного фосфата. При прокаливании выделившегося осадка образуется
Zn2P207.
Уравнение реакции:
Zn++ + (NH4)2HP04-f NH4OH —> | ZnNH4P04 + H20 + 2NHJ
2ZnNH4P04 \ \ Zn,P207 + 2NH3 t + H20 f
Весовую форму взвешивают и пересчитывают на содержание цинка
в сплаве. Очень часто в латуни цинк не определяют аналитически, а
содержание его вычисляют по разности.
Объем раствора, в котором будут вести определение цинка, доводят
водой до 200 мл, прибавляют 2 капли метилового красного и 2 н. раствор
аммиака до исчезновения красной окраски. Затем содержимое стакана
нагревают до кипения и медленно, по каплям, при перемешивании
прибавляют рассчитанное количество горячего 7%-ного раствора
гидрофосфата аммония. Смесь оставляют стоять на холоду 2 ч, фильтруют через
фильтровальный тигель и промывают остаток сначала 1%-ным раствором
гидрофосфата аммония, потом дистиллированной водой. Пробу на полноту
промывания проводят до отрицательной реакции на Р04 -ионы. Осадок
прокаливают до постоянной массы в муфельной печи при 1000° С.
Вычисление результатов анализа — см. § 3.
Содержание цинка определяют также комплексонометрическим
методом (см. гл. IV, § 27).
Общая сумма результатов определения всех составных частей сплава
должна быть близкой к 100%.
Д. МЕТОДЫ ВЕСОВЫХ ОПРЕДЕЛЕНИЙ, ОСНОВАННЫЕ
НА ПРИМЕНЕНИИ ОРГАНИЧЕСКИХ РЕАКТИВОВ
§ 22. Определение никеля
Методы количественного определения. Определение никеля может
быть осуществлено различными методами:
а) осаждением в виде диметилглиоксимата никеля;
б) осаждением в виде никель-а-бензилдиоксима;
в) осаждением в виде гидроокиси никеля (III) едким кали и бромной
водой (весовая форма Ni203);
г) осаждением в виде сульфида (весовая форма №0);
д) электролитическим методом (см. гл. VII);
е) объемным методом — титрованием цианидом калия до образования
комплексного цианида K2lNi(CN)4];
ж) колориметрическим методом (см. гл. IX), основанным на
измерении интенсивности синего окрашивания иона [Ni (NH3)6]++или красного
окрашивания растворимого комплексного соединения, образующегося
при взаимодействии ионов никеля (III) с диметилглиоксимом в щелочном
растворе в присутствии окислителя;
з) комплексонометрическим методом и др.
§ 23. ОПРЕДЕЛЕНИЕ АЛЮМИНИЯ
393
Определение никеля в виде диметилглиоксимата никеля. Определение
никеля основано на осаждении ярко-красной внутрикомплексной соли—
диметилглиоксимата никеля,, образующейся при взаимодействии диметил-
глиоксима с ионами никеля.
Условия проведения реакции — см. книга I, гл. VI, §11.
Образующийся осадок диметилглиоксимата никеля очень объемистый,
поэтому исходный раствор должен содержать около 0,02 г никеля. Осадок
практически нерастворим в разбавленных растворах
аммиака, солях аммония, избытке осадителя и в аце-
татно-буферных смесях. Он растворим в минеральных
кислотах, в растворах цианидов и в спирте.
Осаждению мешают не только осаждаемые
реактивом катионы (палладий, золото), но и ионы железа,
алюминия, титана и др., осаждающиеся аммиаком
в виде гидроокисей, так как осаждение ионов никеля
завершается в аммиачной среде. Реакции мешают
также фосфаты, арсенаты, бораты, оксалаты и
некоторые другие анионы.
Осаждение начинают в слабокислой среде. Для
этого к холодному раствору, содержащему
рассчитанное количество никеля и разбавленному до 200 мл,
прибавляют 5 мл 6 н. раствора хлористоводородной
кислоты. Слабокислый раствор нагревают. Как только
раствор нагреется почти до кипения, его снимают
с бани и прибавляют к нему по каплям при
непрерывном перемешивании спиртовый раствор диметилглиок-
сима, взятый в количестве, превышающем
рассчитанное не более чем в 1,5 раза. Затем, продолжая
тщательно перемешивать содержимое стакана, прибавляют пб каплям 2 н.
раствор аммиака до слабого запаха аммиака. Стакан с осадком ставят
на водяную баню на 2—3 ч, а затем снимают и оставляют стоять до
следующего дня.
Осадок отфильтровывают под вакуумом через мелкопористый
стеклянный фильтр (рис. 72), высушивают в сушильном шкафу при 120—
125° С до достоянной массы, взвешивают и вычисляют содержание никеля
(см. § 3). Формула весовой формы NiC8Hu04N4.
Рис. 72.
Приспособление для
фильтрования с
отсасыванием через
фильтровальный тигель
(стеклянный или
фарфоровый).
§ 23. Определение алюминия
Определение ионов алюминия в его растворимых солях можно
проводить, аналогично определению ионов железа, осаждением А1+++
в виде гидроокиси и взвешиванием весовой формы А1203. Но этот метод
определения осложняется тем, что А1 (ОН)3 является амфртерным
соединением, а А1203 обладает достаточной гигроскопичностью. Поэтому для
весового определения ионов, алюминия применяют 8-оксихинолин (см.
книга I, гл. VI, § 4). При этом в качестве весовой формы можно
применять А1203 или оксихинолят алюминия.
Уравнение реакции:
А1+++ + 3C9H6NOH + 3CH3COONH4 —> | Al (C9H6NO)3 + 3CH3COOH + 3NH4+
Условия проведения реакции. 1. Осаждение начинают вести из кислого
раствора при рН = 4 и 75—90° С. Заканчивают осаждение в
слабощелочной среде при рН = 8—9.
2. Осаждению мешают катионы магния, серебра, меди, свинца,
висмута, кадмия, цинка, молибдена, урана, железа, титана, циркония, мар-
394
ГЛ. V. ОСНОВЫ ВЕСОВОГО АНАЛИЗА
ганца, никеля, кобальта, бериллия, кальция, стронция, бария, олова,
тантала, ниобия и др., осаждаемые оксихинолином из ацетатных
буферных или аммиачных и сильнощелочных растворов.
3. Осаждению мешают многие анионы, образующие осадки с А1+ + +
в условиях среды, при которой идет осаждение оксихинолином алюминия,
а именно: фосфат, борат, арсенат, фторид и т. п. Поэтому указанные
анионы должны отсутствовать в анализируемом растворе.
4. В присутствии минеральных кислот осаждение оксихинолята
алюминия не происходит.
5. Используя подходящую среду, можно отделить ионы алюминия от
многих других ионов:
а) в ацетатном буферном растворе катионы алюминия отделяются от
ионов магния и бериллия;
б) в аммиачной среде в присутствии перекиси водорода — от
молибдена, ванадия и титана;
в) в аммиачно-карбонатной среде — от урана.
6. В присутствии тартратов и других органических оксисоединений
в щелочной среде оксихинолин не осаждает алюминий. Поэтому
органические соединения должны быть предварительно удалены.
Методика определения, Навеску соли алюминия, например алюмо-
калиевых квасцов (около 0,1 а в пересчете на алюминий), растворяют
в 10 мл 6 н. хлористоводородной кислоты, раствор разбавляют водой до
200 мл, прибавляют 15 мл 2 н. раствора ацетата аммония, 8 капель
0,04%-ного раствора бромкрезолового пурпурного или метилового
красного. Раствор нагревают до кипения. К полученному раствору добавляют по
каплям при перемешивании 2,5%-ный уксуснокислый раствор оксихино-
лина в количестве в 1,5 раза больше, чем это требуется по расчету. Затем
медленно при перемешивании добавляют разбавленный раствор аммиака
до изменения окраски индикатора. Смесь кипятят в течение 1 мин,
охлаждают и фильтруют через стеклянный фильтр № 3.
По окончании фильтрования осадок промывают несколько раз
горячей водой (порциями по 10 мл) и затем холодной водой до тех пор, пока
промывные воды не станут бесцветными. Объем промывных вод не должен
превышать 100 мл. После того как осадок промыт, отключают колбу от
водоструйного насоса, закрывают водопроводный кран и вынимают тигель.
Тигель высушивают в сушильном шкафу при 120—140° С. Высушивание
и взвешивание тигля повторяют до постоянной массы. По разцости масс
пустого тигля и тигля с осадком находят массу весовой формы, т. е. массу
оксихинолята алюминия, содержание в котором А1203 составляет 11,10%.
Предварительно стеклянный фильтровальный тигель вставляют при
помощи резинового кольца в горло колбы для отсасывания, тщательно
промывают при слабом отсасывании горячей разбавленной уксусной
кислотой и горячей водой, затем помещают в сушильный шкаф,
высушивают при 120—140° С и взвешивают на аналитических весах.
Высушивание и взвешивание повторяют до постоянной массы. После этого
приступают собственно к отфильтровыванию осадка.
Расчет проводят обычным образом.
ЧАСТЬ ТРЕТЬЯ
Понятые о физических
и физико-химических
(инструментальных)
методах анализа
ГЛАВА VI
КЛАССИФИКАЦИЯ ФИЗИЧЕСКИХ И
ФИЗИКО-ХИМИЧЕСКИХ МЕТОДОВ
КОЛИЧЕСТВЕННОГО АНАЛИЗА
В настоящее время в научно-исследовательских и заводских
лабораториях широко применяется большое число разнообразных физических
и физико-химических методов количественного анализа.
Все физические и физико-химические методы анализа обычно делят
на следующие группы:
1) электрохимические;
2) спектральные (оптические);
3) хроматографические;
4) радиометрические;
5) масс-спектрометрические.
§ 1. Электрохимические методы
К электрохимической группе методов анализа относятся:
1. Электровесовой анализ, основанный на выделении из растворов
электролитов веществ, осаждающихся на электродах при прохождении
через раствор постоянного электрического тока. Выделившийся при
электролизе металл (окисел) взвешивают и по массе осадка судят о
содержании вещества в растворе.
Разновидностью электровесового анализа является метод
внутреннего электролиза, основанный на использовании электрического тока,
возникающего при погружении в анализируемый раствор двух
электродов, составляющих гальваническую пару, например Zn и Pt.
Выделившееся на электродах вещество взвешивают и по массе осадка
судят о содержании его в растворе.
2. КондуктометриЯу основанная на измерении электропроводности
анализируемых растворов, изменяющейся в результате химических
реакций и зависящей от свойств электролита, его температуры и
концентрации растворенного вещества.
3. Потенциометрия, основанная на измерении изменяющегося в
результате химической реакции потенциала электрода, погруженного
в анализируемый раствор. Величина потенциала электрода зависит от
концентрации соответствующих ионов в растворе при других постоянных
условиях измерения. Потенциалы измеряют при помощи специальных
приборов — потенциометров.
4. Полярография, основанная на измерении силы тока, изменяющейся
в зависимости от напряжения в процессе электролиза, в условиях, когда
один из электродов имеет очень малую поверхность.
396 ГЛ. VI. ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
При полярографических измерениях таким электродом являются
капли ртути, вытекающие из очень тонкого отверстия капиллярной
трубки, а также платиновый (вращающийся), графитовый, серебряный
и другие электроды.
5. Кулонометрия, основанная на измерении количества
электричества, израсходованного на электролиз определенного количества вещества.
В основе этого метода лежит закон Фарадея.
С перечисленными электрохимическими методами анализа тесно
связаны методы амперометрического и высокочастотного титрования (см.
гл. VII).
§ 2. Спектральные (оптические) методы
Спектральные методы анализа основаны на изучении спектров
излучения, поглощения и рассеивания. К этой группе относятся:
1. Эмиссионный спектральный анализ, основанный на изучении
эмиссионных спектров (спектров испускания или излучения) элементов
анализируемого вещества; этот метод дает возможность определять
элементарный состав вещества.
2. Абсорбционная спектроскопия, основанная на изучении спектров
поглощения исследуемого вещества. Различают исследования в
ультрафиолетовой, в видимой и в инфракрасной областях спектра.
Абсорбционный спектральный анализ включает:
а) спектрофотометрический и б) колориметрический методы.
Спектрофотометрия основана на определении спектра поглощения
или измерении светопоглощения при строго определенной длине волны,
которая соответствует максимуму кривой поглощения данного
исследуемого вещества.
Колориметрия основана на сравнении интенсивностей окрасок
исследуемого окрашенного раствора и стандартного окрашенного раствора
строго определенной концентрации.
3. Анализ по спектрам комбинационного рассеяния света,
основанный на явлении, открытом одновременно советскими физиками
Г. С. Ландсбергом и Л. И. Мандельштамом и индийским физиком
Ч. В. Раманом. Это явление, обусловливаемое молекулярной структурой
исследуемого вещества, сопровождается изменением длины волны
рассеиваемого данной- средой света.
К оптическим методам анализа также относятся:
Турбидиметрия, основанная на измерении количества света,
поглощаемого неокрашенной суспензией. В турбидиметрии свет, поглощенный
раствором или прошедший через него, измеряют так же, как и при
колориметрии окрашенных растворов.
Нефелометрия, основанная йа использовании явлений отражения
или рассеивания света окрашенными или неокрашенными частицами
взвешенного в растворе осадка.
Этот способ дает возможность определять очень малые количества
вещества, находящегося в растворе в виде взвеси.
Люминесцентный, или флуоресцентный, анализ, основанный на
флуоресценции веществ, облученных ультрафиолетовым светом, и измерении
интенсивности излучаемого ими видимого света. Флуоресцентные методы
нашли широкое применение для определения следов различных примесей
в неорганических и органических соединениях.
Фотометрия пламени (пламенная фотометрия), основанная на
распылении анализируемого раствора, в пламени, выделении характерной
для данного элемента световой волны и измерении интенсивности
излучения.
4. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ
397
§ 3. Хроматографические методы
Хроматографические методы количественного анализа основаны на
избирательном поглощении (адсорбции) отдельных компонентов
анализируемой смеси различными адсорбентами. Они широко применяются для
разделения близких по составу и свойствам неорганических и
органических веществ. Хроматографию применяют, в частности, для разделения
редкоземельных, а также радиоактивных элементов.
Хроматографические методы анализа подробнее описаны в гл. IX,
§ 6, см. также книга I, гл. III, § 10.
§ 4. Радиометрические методы
Радиометрические методы анализа основаны на измерении излучений,
испускаемых радиоактивными элементами. Для регистрации излучений
применяют специальные установки с использованием счетчиков Гейгера—
Мюллера (рис. 73). При действии н$ приемник радиоактивных излучений
в нем возникает электрический ток в виде кратковременных импульсов,
которые специальной радиотехнической аппаратурой усиливаются,
выравниваются по величине и поступают на регистрирующее счетное устройство.
Радиометрические методы анализа отличаются рядом преимуществ
по сравнению с химическими методами. Прежде всего следует отметить
их высокую чувствительность, которая значительно выше
чувствительности химических и других физических и физико-химических методов
анализа.
Чувствительность этих методов характеризуется, например,
следующими данными:
ИЗОТОП 65Ni 60Q, 64QJ 127Те 76AS
Чувствительность, г 10~8 10"8 3,4-Ю"10 1,2-КГ8 3,4-10~10
Эти методы анализа применяются для количественного определения
микропримесей различных элементов- в металлах и неметаллах высокой
степени чистоты.
Следует отметить, однако, что точность радиометрических методов
невысока и составляет ±5—20% (относительных). Химические методы
анализа отличаются более высокой точностью. Но там, где обычные
весовой и объемный методы анализа дают большие ошибки, как, например,
при определении ничтожно малых примесей, радиометрические методы
являются незаменимыми.
Различают следующие радиометрические методы:
1. Метод изотопного разбавления. Метод изотопного разбавления,
как указывает само название, основан на разбавлении соединения,
меченного радиоактивным изотопом, неактивным компонентом смеси. Для этого
к анализируемой смеси добавляют некоторое количество соединения,
меченного одним из радиоизотопов и по своему составу совпадающего
с определяемым компонентом.
При этом удельная активность соединения, меченного
радиоактивным изотопом, уменьшится. Если выделить часть анализируемого
вещества, то можно определить конечную удельную активность. Зная
начальную и конечную удельные активности, легко вычислить содержание
определяемого вещества.
Преимущество этого метода анализа заключается в том, что отпадает
необходимость в количественном выделении определяемого вещества при
условии полного смешения изотопов. Достаточно выделить лишь часть
его в химически чистом виде.
Рис. 73. Установка для регистрации радиоактивных излучений:
а _ счетчики Гейгера—Мюллера; б — внешний вид счетной установки; в — схема счетной установки^
/ — газовый счетчик; 2 — высоковольтный выпрямитель; 3 — усилитель; 4 — пересчетное
устройство; 5 — электромеханический счетчик.
§ 4. РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ
399
Если некоторое количество вещества, отвечающее по составу
определяемому соединению, имеет массу Wx и радиоактивность Л, то его
удельная активность 1г равна:
1г = имП'мин/мг
Wi
При добавлении точного количества такого вещества к определенной
навеске W2 анализируемого неактивного соединения удельная
активность смеси /2 будет равна:
72 = w/ . w/ имп-мин/мг
wx -J- w2
Решая систему приведенных выше уравнений, получим:
но так как W = VC, то
VA = ViCi(-tJ--i) 0)
где Vx — объем радиоактивного раствора с известной концентрацией Сг;
V2 — объем исследуемого раствора;
С2 — концентрация анализируемого раствора.
В случае если V± = V2, формула (1) принимает вид:
с--*(¦?-0 (2)
Метод изотопного разбавления имеет преимущество перед другими
радиометрическими методами в тех случаях, когда полное выделение
исследуемого вещества из анализируемой смеси затруднительно или
невозможно.
2. Радиоактивационный анализ. Принцип этого метода заключается
в переводе стабильных изотопов элемента в радиоактивные, измерение
радиоактивности которых служит критерием содержания данного
элемента в анализируемом объекте. Для этого анализируемые образцы
подвергают облучению, например, в атомном реакторе.
Активность измеряют при помощи специальных счетных устройств.
Период полураспада и энергия излучения являются специфичными
для индивидуальных радиоизотопов, т. е. применяя
радиоактивационный анализ, можно контролировать чистоту получаемых веществ.
Измерив радиоактивность и зная время облучения, интенсивность
потока облучающих частиц, соответствующие ядерно-физические
данные определяемого элемента, можно вычислить его весовое количество.
Одновременно с исследуемым веществом облучают стандартные
образцы, содержащие точно известные количества определяемых элементов.
Сравнивая в одинаковых условиях активности определяемого вещества
и стандартных образцов, можно вычислить содержание определяемого
элемента.
Радиоактивационный метод отличается многими преимуществами
по сравнению с другими методами анализа. Метод обладает высокой
чувствительностью. Основным недостатком его является то, что не все
элементы можно определять этим методом. Образующийся после облучения
радиоактивный элемент должен иметь сравнительно большой период
полураспада, достаточный для того, чтобы можно было успеть провести
химическое разделение и измерение активности выделенного элемента.
400 ГЛ. VI. ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Применяя радиоактивационный метод анализа, можно определять
микроколичества различных элементов в морской воде; редкоземельных
металлов в рудах; золото, платину, палладий и иридий в серебре и никеле;
никель, кобальт, медь, мышьяк, теллур в сурьме и т. д.
К числу радиометрических методов анализа также относятся:
радиохроматография, нейтронная абсорбциометрия, радиометрическое
титрование и др.
§ 5. Масс-спектрометрические методы
Масс-спектрометрические методы анализа основаны на определении
отдельных ионизированных атомов, молекул и радикалов посредством
разделения потоков ионов, содержащих частицы с разным отношением
массы (т) к заряду (е), в результате комбинированного действия электри-
Рис. 74. Схема масс-спектрографа.
ческого и магнитного полей. Регистрация разделенных частиц достигается
электрическим (масс-спектрометрия) или фотографическим (масс-спек-
трография) способами.
Определения проводят с помощью специальных приборов — масс-
спектрометров или масс-спектрографов (рис. 74), описываемых в курсах
физики.
Масс-спектрометрические методы применяют в химическом анализе
для определения изотопного состава элементов, содержания различных
радикалов, отдельных компонентов в сложных смесях или примесей.
Масс-спектрометрия позволяет выполнять количественные
определения различных элементов методом изотопного разбавления с
использованием стабильных изотопов.
i
1
ГЛАВА VII
ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
§ 1. Характеристика методов электроанализа
Методы электровесового анализа основаны на точном измерении
массы определяемого вещества или его составных частей, выделяемых
в химически чистом состоянии или в виде осадка на погруженных в
анализируемый раствор электродах при прохождении через раствор
постоянного электрического тока. На электродах могут выделяться металлы
(например, Си) или образовываться окислы (например, РЬ02).
Образовавшийся при электролизе металл (окисел) взвешивают и
по массе выделившегося осадка вычисляют содержание вещества в
анализируемом растворе.
Законы Фарадея. Зависимость, существующая между количеством
вещества, выделившегося при электролизе, и количеством прошедшего
через данный раствор электричества, определяется первым законом
Фарадея, согласно которому масса вещества, выделенного на электроде,
пропорциональна количеству прошедшего через исследуемый раствор
электричества.
Согласно второму закону Фарадея, количество вещества, выделяющегося
в разных электролитах при пропускании через раствор одинакового
количества электричества, пропорционально их эквивалентам (Э):
э = А (1)
где А — атомный вес;
z — валентность.
Таким образом, количества веществ, выделяющихся при электролизе
на электродах в случае постоянного тока силой /, протекающего в течение
времени /, можно вычислить по формуле:
вА_Л/А «,.,_*_ (2)
гДе ?д — масса определяемого вещества, выделенного на электроде, г\
I — сила тока, а;
t —время, сек;
F — число Фарадея, равное 96 500 кулонам;
Э — грамм-эквивалент определяемого вещества, г.
Количество вещества, которое выделяется на электроде при
прохождении через электролит единицы количества электричества (одного
кулона), называют электрохимическим эквивалентом Э/F.
Так, например, электрохимический эквивалент серебра в реакции
Ag+ +e—»Ag равняется 1,118 мг/к.
402
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
Когда на электродах выделяется 1 грамм-эквивалент вещества,
gA = Э и F = It. Следовательно, величина F (число Фарадея) представляет
заряд, переносимый 1 грамм-эквивалентом какого-либо вещества.
Другими словами, прохождение через электролит 1 фарадея
электричества сопровождается выделением на электродах 1 грамм-эквивалента
определяемого вещества.
§ 2. Химические процессы, протекающие при электролизе
Анодное окисление и катодное восстановление. Схематически процесс
электролиза может быть представлен следующим образом.
При пропускании через раствор постоянного электрического тока
катионы электролита под влиянием электронов источника тока
восстанавливаются на катоде в ионы низшей валентности, нейтральные атомы или
атомные группы. Продукты восстановления отлагаются на катоде,
вступают в реакции с молекулами растворенных веществ или растворителя
или, наконец, реагируют с материалом электрода. В то же время анионы
окисляются на аноде.
Поэтому основные химические процессы, протекающие при электролизе,
можно рассматривать как реакции окисления — восстановления.
Процессы восстановления, протекающие на катоде, называют
катодным восстановлением; процессы окисления, протекающие на аноде,
называют анодным окислением.
Применительно к целям электровесового анализа процесс электролиза
водного раствора CuS04 можно изобразить следующей схемой:
На катоде выделяется медь:
Си++ + 2е —> Си
На аноде выделяется кислород:
4Н20 z^: 4H+ + 40H-
40Н-— 4е —> 02 + 2Н20
При этом в растворе накапливается серная кислота:
4Н+ + 2SO^— ^± 2H2S04
Образование на аноде газообразного кислорода объясняется тем, что
ОН--ионы окисляются легче S04~-hohob.
В тех случаях, когда Н + -ионы восстанавливаются легче других
катионов, на катоде выделяется газообразный водород. Например, при
электролизе Na2S04 на катоде вместо металлического натрия выделяется
газообразный водород.
При надлежащих условиях электролиза анодное окисление Pb+ +
сопровождается переходом в PbIV, Mn++ — в MnIV и т. п.
Например, электролиз водного раствора, содержащего ионы свинца
и меди, сопровождается:
на катоде выделением металлической меди:
Си++ 4- 2е —> Си
на аноде образованием РЮ2:
РЪ++ — 2е —> PblV
PbIV + 2H20 —> РЬ024-4Н+
а в растворе накапливается серная кислота:
4Н+ + 2SO— ^± 2H2S04
§ 3. МЕТОДЫ ЭЛЕКТРОАНАЛИЗА
403
§ 3. Методы электроанализа
Электролиз с наложением и без наложения тока извне. Существует
два варианта электровесового метода. По первому из них, наиболее
широко распространенному, который собственно и называют электровесовым
методом, выделение веществ на электродах происходит при действии
постоянного тока, полученного от внешнего источника (аккумулятора,
выпрямителя и т. п.). По второму варианту постоянный ток возникает
при погружении в анализируемый раствор так называемой гальванической
пары и в этом случае внешнего источника тока не требуется.
Этот вариант электровесового анализа называют методом
внутреннего электролиза.
Гальваническая пара представляет собой два соединенных
металлическим проводником электрода, отличающихся величинами нормальных
электродных потенциалов. Чем больше разность электродных потенциалов
металлов, образующих гальваническую пару, тем больше
электродвижущая сила данной пары.
Электровесовым анализом (с наложением тока извне) можно
пользоваться для определения состава растворов, содержащих большие
количества определяемых веществ. Однако для выполнения определений этим
методом требуется специальное оборудование (источник постоянного
тока, электроизмерительные и регулирующие приборы и т. д.). При
выполнении анализов методом внутреннего электролиза сложного
оборудования и приборов не требуется, но этот метод пригоден только для
определения малых количеств металлов, а потому находит ограниченное
применение.
Особым видом электролиза является метод электролиза с ртутным
катодом.
Потенциал разложения. Электролиз данного электролита начинается
при определенном напряжении, называемом потенциалом разложения.
Для того чтобы электролиз проходил быстро, напряжение тока в цепи
поддерживают выше потенциала разложения. При этом часть
электроэнергии превращается в тепло.
Если исследуемый раствор содержит несколько компонентов,
отличающихся различными потенциалами разложения, то можно выделять их из
смеси в определенной последовательности, строго регулируя напряжение.
При этом в первую очередь выделяются те металлы, которые имеют
меньший потенциал разложения.
Преимущества и недостатки электровесовых методов анализа.
Электровесовые методы анализа широко применяются в аналитической
практике, особенно при исследовании цветных металлов и их сплавов. Они
весьма просты, удобны и достаточно точны. Электровесовые определения
выполняются сравнительно быстро. Особым их преимуществом является
то, что они не требуют введения в анализируемый раствор посторонних
веществ, как это делается в обычном весовом анализе.
Так как каждый элемент электролитически осаждается при
определенном напряжении разложения, то, подбирая и регулируя
электродный потенциал, можно электролитически разделять многие
элементы.
Например, при пропускании тока через раствор, содержащий ионы
свинца и кадмия в равных концентрациях (аРь++ = #cd++ = 1), ионы
свинца восстанавливаются легче (?рь++/рь = —0,126 в), чем ионы кадмия
(Есд++/сб = —0,4020 в). Если во время электролиза поддерживать
потенциал катода равным, например, —0,35 в, то будет выделяться только
свинец, а ионы кадмия будут оставаться в растворе.
404
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
Применяя электролитическое осаждение, можно отделять
посторонние примеси от главных компонентов анализируемой смеси. При
электроанализе отпадает необходимость в отфильтровывании выделенного осадка.
Одним из существенных недостатков электровесовых методов анализа
является то, что не во всех случаях можно достигнуть количественного
осаждения определяемого вещества.
Когда в анализируемом растворе находятся катионы только одного
металла, то применение электровесового анализа, как правило, не
встречает особых затруднений. Присутствие в растворе катионов нескольких
элементов иногда создает большие трудности для их раздельного
осаждения.
Многие элементы невозможно определить электровесовым способом.
В тех случаях, когда в анализируемой смеси присутствует два или
несколько близких по своим электрохимическим свойствам элементов, может
происходить их одновременное осаждение на электроде, что
затрудняет проведение электровесового анализа. Тем не менее обычно удается
подобрать подходящий электролит или соответствующий потенциал
электрода, при котором определяемый компонент количественно осаждается.
§ 4. Электровесовой анализ
Источники постоянного тока. Для количественного выделения какого-
либо катиона в виде металла или окисла металла электролиз следует
проводить при строго определенных условиях. Электролиз проводят при
определенном напряжении. Следует, однако, иметь в виду, что количество
выделяющегося при электролизе вещества остается неизменным независимо
от источника постоянного тока, от температуры, от плотности тока, от
формы электродов.
На практике потенциал электрода, на котором проводят осаждение
определяемого вещества, может быть изменен в некоторых пределах.
Иногда электролиз проводят при контролируемой плотности тока.
Для поддержания постоянной разности потенциалов в процессе
электролиза применяют реостаты различных конструкций или специальные
автоматические приборы. Разность потенциалов обычно измеряют при
помощи вольтметров, включенных параллельно электродам в цепь
источника постоянного тока.
В качестве источников постоянного тока в электроанализе
используют свинцовые или щелочные аккумуляторы. Для преобразования
переменного тока в постоянный применяют выпрямители. Силу тока,
протекающего через раствор, измеряют при помощи амперметров,
включенных в цепь последовательно.
Электролиз сильно ускоряется при перемешивании и подогревании
раствора.
Перемешивание раствора можно проводить, пропуская через него
воздух или азот, а также с помощью мешалок различных конструкций.
Платиновые электроды. При электровесовых определениях обычно
применяют платиновые электроды. Для увеличения скорости
выполнения анализа им придают возможно более развитую поверхность. В
большинстве случаев электроды изготовляют в виде сетчатых цилиндров,
вставляемых один в другой. Иногда применяют сетчатый катод и.
свернутый в спираль анод, а также некоторые другие типы электродов
(рис. 75).
Правила работы с платиновыми
электродами. При работе с платиновыми электродами необходимо строго
выполнять следующие привила:
§ 4. ЭЛЕКТРОВЕСОВОЙ АНАЛИЗ
405
1. При электровесовом определении металлов, образующих при
электроосаждении сплавы с платийой (например, индия, цинка, олова),
платиновые катоды предварительно покрывают медью или кадмием, легко
удаляемыми с поверхности катода по окончании электролиза.
2. Электровесовое определение нельзя проводить в присутствии ионов
хлора, так как последние вызывают заметное разрушение платинового
электрода, происходящее ^ процессе электролиза.
3. Для удаления продуктов электролиза с поверхности платинового
катода следует применять азотную кислоту или смесь азотной и серной
кислот без примеси соляной кислоты, чтобы исключить возможность
образования царской водки, энергично разрушающей платину. Для удаления
Рис. 75. Типы электродов:
а — сетчатый катод и спиральный анод; б — конический катод и
спиральный анод; в — катод в виде чашки, анод — дисковый.
двуокиси свинца с поверхности анода применяют разбавленную (1 : 1)
соляную кислоту, которая не должна содержать примеси азотной кислоты
Очистку анода в этом случае проводят на холоду.
4. При электровесовом определении ртути нельзя удалять
выделившуюся ртуть путем обработки электрода азотной кислотой, так как часть
платины теряется, а поверхность катода становится рыхлой и
непригодной для электровесовых определений. Для удаления ртути с поверхности
катода рекомендуется нагревать его длительное время в муфельной печи.
При этом ртуть с поверхности платины испаряется, а катод остается
чистым, не рыхлым и пригодным для дальнейших электровесовых
определений. Эту операцию необходимо проводить в вытяжном шкафу с мощной
вентиляцией, чтобы избежать отравления парами ртути.
Неплатиновые электроды. Вследствие высокой стоимости
платиновых электродов их заменяют танталовыми, вольфрамовыми, серебряными,
никелевыми, графитовыми и другими. Можно применять катоды из
нержавеющей стали и аноды из свинца, железа и никеля.
Условия электроосаждения. Для выделения различных элементов
требуются разные условия (определенные температура, рН, состав
электролита, разность потенциалов между анодом и катодом и т. д.). Например,
количественное выделение металлической меди на катоде и осаждение
свинца в виде двуокиси на аноде хорошо протекают в азотнокислой среде,
в то время как никель в этой среде не выделяется. Такое различие
условий объясняется тем, что различные ионы с неодинаковой легкостью
принимают и отдают заряды на электродах. Например, для того чтобы ионы
серебра восстановить на катоде в металлическое серебро, требуется
меньшая разность потенциалов, чем для восстановления ионов меди; ионы
иода легче отдают свои электроны на аноде, чем ионы хлора.
406
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
Практически напряжение при электролизе приходится поддерживать
выще напряжения разложения.
Избыточное напряжение, которое необходимо создать на данном
электроде, чтобы нормально протекал электролиз, называют
перенапряжением.
Перенапряжение объясняется протеканием на поверхности
электродов и в непосредственной близости к ним сложных физико-химических
процессов.
Условия электролиза должны быть выбраны так, чтобы происходило
выделение только одного металла, а не смеси каких-либо металлов.
При электроанализе большое значение имеет характер выделяющегося
осадка. Для получения правильных результатов анализа осадки,
выделенные в результате электролиза, должны удовлетворять следующим
требованиям:
1. Осадок должен быть чистым и не содержать посторонних примесей.
Наиболее чистыми осадками являются мелкокристаллические.
2. Осадок должен обладать хорошим сцеплением с поверхностью
электрода. Если осадок плохо сцепляется с поверхностью электрода, то при
последующей обработке, промывании, высушивании и взвешивании
электрода часть осадка может быть потеряна и результаты анализа будут
неточными.
3. Состав отложившегося на электроде вещества не должен
изменяться под действием кислорода воздуха. При некоторых условиях
электролиза (высокой плотности тока) получаются мелкокристаллические, но
пористые осадки с весьма развитой поверхностью. Такие осадки легко
окисляются. Например, при электролитическом осаждении меди из
аммиачных растворов при высокой плотности тока металл образует на
поверхности катода губчатый порошкообразный осадок, легко окисляющийся
на воздухе в процессе сушки. Результаты определения в этом случае
получаются повышенными.
§ 5. Метод внутреннего электролиза
Если в раствор соли анализируемого металла опустить две пластинки
(электроды) — одну платиновую, а другую из какого-либо более
электроотрицательного металла, чем определяемый, и
замкнуть их вне раствора металлическим
проводником, то по цепи начнет протекать электрический
ток. Находящиеся в растворе ионы металла
разряжаются на катоде (платине), образуя осадок
металла. Процесс начинается с выделения более
электроположительного из находящихся в
растворе металлов.
При наличии в растворе катионов нескольких
металлов, подобрав соответствующие аноды
(алюминий, цинк, кадмий, железо, свинец), удается
точно отделить некоторые элементы друг от друга.
Например, отделение меди от цинка можно
проводить в нейтральной среде, применив в
качестве анода никель, так как в этой среде ?zn <
<?ni <?cu, или в аммиачной среде в
присутствии тартрата аммония, используя в качестве
Рис. 76. Прибор для ана- анода алюминий, так как и в этом случае ?zn<
лиза методом внутреннего ^ Ь ^ ?
электролиза: гт *
F Приборы для анализа методом внутреннего
/ — анод: 2 — катод: 3 — о
держатель; 4 - стакан. ЭЛеКТрОЛИЗа МОГуТ ИМеТЬ раЗЛИЧНОе уСТрОИСТВО.
§ б. ОПРЕДЕЛЕНИЕ МЕДИ В РАСТВОРЕ СУЛЬФАТА МЕДИ 407
В приборах одного типа оба электрода непосредственно погружены
в анализируемый раствор (рис. 76). В приборах другого типа имеется
пористая диафрагма, отделяющая анодное пространство от катодного.
Катодное пространство заполняют анализируемым раствором, а
анодное — каким-либо другим подходящим электролитом.
Метод внутреннего электролиза применяют главным образом при
анализе цветных металлов, преимущественно для определения малых
количеств посторонних примесей.
§ 6. Определение меди в растворе сульфата меди
с применением платиновых сетчатых электродов
Электровесовой метод является одним из наиболее точных методов
определения меди.
Медь осаждают на поверхности предварительно взвешенного
индифферентного катода; после осаждения высушенный катод взвешивают.
Осаждение осуществляют при помощи постоянного электрического тока,
пропускаемого через сернокислый или аммиачный растворы соли меди.
Поэтому ионы металлов, стоящих в ряду напряжений ниже меди
(ионы золота, серебра, ртути и др.), а также соединения мышьяка, сурьмы,
олова, молибдена, селена, теллура мешают этому определению.
Поляризация. При прохождении через раствор электрического тока
на катоде выделяется металлическая медь, на аноде — газообразный
кислород. В присутствии H2S04 на катоде также выделяется газообразный
водород.
Таким образом, на катоде протекают химические реакции
восстановления ионов меди и ионов водорода, а на аноде — реакция окисления
ионов гидроксила, сопровождающиеся выделением газообразного
кислорода.
В момент пропускания тока противоположные системы соединены
между собой проводником, поэтому они образуют гальванический
элемент, при разрядке которого наблюдаются обратные реакции. Э. д. с.
этого гальванического элемента направлена против э. д. с, прилагаемой
от источника тока. Вследствие электролиза возникает некоторое
напряжение между электродами, направленное противоположно направлению,
прилагаемому от источника тока; это явление называют поляризацией.
Различают химическую и концентрационную поляризацию.
Химическая поляризация. Рассматриваемый вид
поляризации, возникающей вследствие того, что выделение продуктов
электролиза приводит к возникновению гальванического элемента,
называют химической или электрохимической поляризацией. Химическая
поляризация наблюдается при любом случае электролиза. Следовательно,
электрический ток проходит через раствор электролита лишь в том
случае, если приложенное извне напряжение превышает встречное
напряжение возникающего при электролизе гальванического элемента; это
встречное напряжение обусловлено образованием выделяющихся на электродах
продуктов разложения.
Таким образом, электролиз данного электролита начинается при
напряжении, превышающем электродвижущую силу возникающего при
электролизе гальванического элемента.
Химическую поляризацию ослабляют прибавлением соответствующих
реагентов, реагирующих с веществами, ее вызывающими.
Поляризацию, обусловливаемую выделением на катоде водорода, подавляют при
помощи окислителей. Для ослабления поляризации, вызываемой
образованием на аноде кислорода, в качестве деполяризаторов применяют
восстановители.
408
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
Концентрационная поляризация возникает вследствие изменения
концентрации ионов в анодном и катодном пространствах вблизи электродов
по сравнению с их концентрацией в основной массе раствора.
Таким образом, одинаковые индифферентные электроды, опущенные
вначале в электролит одинаковой концентрации, в результате
электролиза оказываются опущенными в растворы разной концентрации.
Вследствие этого возникает концентрационный гальванический элемент,
электродвижущая сила которого направлена навстречу приложенному извне
постоянному электрическому току. Э. д. с. возникшего концентрационного
элемента называют э. д. с. концентрационной поляризации. Это явление
приводит к уменьшению приложенной э. д. с. и силы тока.
Концентрационная поляризация возрастает по мере увеличения плотности тока.
Величина концентрационной поляризации зависит от размеров электродов.
При одинаковой силе тока, протекающего через раствор, на малых
электродах поляризация значительно больше, чем на больших, так как
плотность тока в этом случае больше.
Влияние среды. Точные результаты получаются при осаждении меди
в сернокислой среде в присутствии свободной азотной кислоты,
действующей как деполяризатор и препятствующей выделению на катоде
газообразного водорода. Но если электролиз протекает только в азотнокислой
среде, то процесс электроосаждения проходит медленно и осадок
выделяется не полностью.
Осаждение меди из аммиачных растворов приводит к менее точным
результатам, так как в процессе электролиза вместе с металлической медью
осаждаются окислы и гидроокись меди и других металлов.
В качестве источников тока применяют аккумуляторы, батареи,
мотор генераторы и различного рода выпрямители, присоединенные к
питающей сети переменного тока.
Для перемешивания электролита применяют электромагнитные мег
шалки. Электромагнитная мешалка (рис. 77) представляет собой
электромотор, на валу которого закреплен мощный постоянный магнит. Над
магнитом мотора расположена медная плитка, на которой устанавливают
сосуд с анализируемым раствором. Внутри стакана помещают
маленький постоянный магнит, запаянный в стекло, который при вращении
закрепленного на валу магнита также вращается и перемешивает
электролит.
Медная плитка снабжена электронагревательной спиралью и
змеевиком для охлаждения, что позволяет в процессе электролиза поддерживать
нужную температуру электролита.
Для крепления электрододержателя мешалка снабжена специальным
штативом. Схема установки представлена на рис. 78.
Подготовка установки для электролиза. Прежде всего погружают
электроды на некоторое время в горячую разбавленную азотную кислоту.
Эта операция необходима для раствбрения меди и других отложений,
оставшихся на электродах от предыдущего определения. Затем электроды
промывают водой, а под конец споласкивают дистиллированной водой.
Подготовка анода на этом заканчивается. Катод необходимо высушить
и взвесить.
Для ускорения высушивания катод промывают спиртом и затем
высушивают в сушильном шкафу при 80—90° С. Высушенный катод хранят
в эксикаторе. По охлаждении катод взвешивают и приступают к монтажу
установки. Сначала закрепляют анод, затем катод. При этом электродо-
держатель должен быть установлен так, чтобы магнитная мешалка и
стоящий на плитке сосуд для электролиза не мешали укреплению
электродов.
§ 6. ОПРЕДЕЛЕНИЕ МЕДИ В РАСТВОРЕ СУЛЬФАТА МЕДИ 409
В процессе работы нельзя касаться руками тех частей электродов,
которые погружают в раствор электролита, так как на загрязненной жиром
поверхности электродов не осаждаются продукты электролиза. Поэтому
электроды берут руками за их верхние части на расстоянии 1—2 см от
конца. Концы электродов закрепляют в зажиме электрододержателя:
анод — к положительному полюсу, обозначенному знаком плюс (+),
катод — к отрицательному полюсу, обозначенному знаком минус (—).
При установке катода его подводят снизу и закрепляют в зажиме
держателя так, чтобы анод проходил точно через центр сетчатого цилиндра
и ни в коем случае не касался его поверхности. Кончик спирали должен
немного выступать из-под сетчатого
катода.
На этом можно считать подготовку
установки для электроанализа
законченной.
^
в
-лл/wv
'ш
д
Рис. 77. Магнитная мешалка.
Рис. 78. Схема установки для
электроанализа:
/ — источник постоянного тока; 2 —
амперметр; 3 — вольтметр; 4 — катод; 5 — анод;
6 — реостат; 7 — предохранитель; 8—
выключатель.
Методика определения. Разбавьте анализируемый раствор до 175—
200 мл. Прибавьте к нему 5 мл 2 н. раствора серной кислоты и 3 мл 2 н.
раствора азотной кислоты. Установите стакан на плитку магнитной
мешалки и погрузите в него магнит. Включите ток. Установите на клеммах
электродов напряжение в пределах 2—2,5 в. Плавно опустите электроды
в электролит так, чтобы они не соприкасались ни с дном, ни со стенками
стакана. Уровень жидкости в стакане должен быть на 1—1,5 см ниже
верхнего края катода. Установив электроды в электролите, снова
отрегулируйте напряжение.
Желательно накрыть стакан для электролиза двумя половинками
часового стекла с вырезанными в них отверстиями для электродов, чтобы
избежать потерь, от разбрызгивания или загрязнения извне.
Наиболее точные результаты прлучаются при медленном осаждении
меди на холоду без перемешивания раствора. Поэтому к нагреванию и
перемешиванию анализируемого раствора прибегают после того, как основная
масса меди выделится из электролита.
Температура нагретого электролита не должна превышать 50—70° С.
В процессе электролиза поддерживают напряжение 2 в и силу тока 0,5 а.
По мере осаждения меди раствор обесцвечивается, а катод
окрашивается в оранжево-красный цвет. Тусклый цвет меди указывает на ее
окисление или присутствие посторонних примесей. Выделившаяся медь
должна плотно держаться на катоде.
Электролиз продолжается около 1 ч (до полного обесцвечивания
раствора). Для проверки полноты осаждения обмойте часовые стекла
дистиллированной водой, долейте в стакан 20—25 мл дистиллированной воды и
410
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
продолжайте электролиз еще 10 мин. Если на свежепогруженной части
катода не будет наблюдаться нового выделения меди, электролиз можно
считать законченным.
По окончании электролиза выключите плитку. Не выключая ток
(во избежание растворения в кислоте выделенной меди), поднимите
Электр о до держатель, промойте электроды погружением несколько раз в
дистиллированную воду и только после этого выключите ток. Возьмите катод
за его верхнюю часть на расстоянии 1—2 см от края, отверните винт
зажима и снимите его. Промойте катод спиртом, высушите в течение
непродолжительного времени в сушильном шкафу при 80° С, охладите
в эксикаторе и взвесьте на аналитических весах.
Расчет. Содержание в растворе меди (gcu) в граммах вычисляют по
следующей формуле:
?cu =g2 — gi (3)
гДе &i — масса катода до электролиза, г;
g2 — масса катода после электролиза, г.
По окончании определения очистите электроды, опустив их в горячую
азотную кислоту. После этого промойте их сначала водопроводной,
а затем дистиллированной водой.
§ 7. Определение меди и свинца в латуни с применением
платиновых сетчатых электродов
При электровесовом определении меди и свинца в латунях применяют
платиновые сетчатые электроды. Ионы меди выделяются в виде металла
на катоде, а ионы свинца — в виде двуокиси свинца на аноде.
Если раствор, подвергаемый электролизу, содержит недостаточное
количество азотной кислоты, то одновременно с выделением на аноде
двуокиси свинца возможно частичное осаждение металлического свинца
на катоде (вместе с медью). Для предотвращения этого явления к
электролиту добавляют концентрированную азотную кислоту (пл. 1,4 г/см3)
в количестве 10 мл на 100 мл электролита.
В растворах, не содержащих примеси серной кислоты, выделение
меди в конце электролиза несколько замедляется. Для полного выделения
меди необходимо в конце электролиза добавлять к анализируемому
раствору небольшое количество серной кислоты.
Навеску латуни (в виде стружки или опилок) около 1 а, взвешенную
на аналитических весах, помещают в стакан емкостью 150 мл и осторожно
обрабатывают 15 мл концентрированной азотной кислоты (пл. 1,4 г/см3).
и 10 мл дистиллированной воды. Чтобы не было потерь вследствие
разбрызгивания раствора, стакан накрывают часовым стеклом, а кислоту
приливают пипеткой по стенке через носик стакана.
После полного растворения навески часовое стекло снимают и
раствор осторожно упаривают при 80—100° С на песочной или водяной бане
до объема 5—10 мл.
Если при этом на дне стакана появляется осадок метаоловянной или
кремневой кислоты, то раствор разбавляют водой, осадок отделяют
фильтрованием и промывают 8—10 раз разбавленной (1 : 20) горячей азотной
кислотой.
Фильтрат и промывные воды собирают в стакан для электролиза и
упаривают до объема 100 мл. Если при этом осадок больше не образуется,
то раствор разбавляют дистиллированной водой до 200 мл и нагревают
до 90° С.
§ 8. ОПРЕДЕЛЕНИЕ МАЛЫХ КОЛИЧЕСТВ МЕДИ
411
Высушенные и взвешенные электроды закрепляют в специальных
держателях. При этом цилиндрический сетчатый анод устанавливают
внутри сетчатого катода (см. § 6).
Во избежание короткого замыкания между электродами к наружной
поверхности анода обычно припаяны специальные стеклянные
предохранительные палочки.
Во время взвешивания, сушки и закрепления электродов в держателе
электролизной установки стараются не прикасаться к обезжиренной
поверхности электродов руками.
Закрепленные в держателе электроды погружают в подготовленный
электролит и включают ток (см. § 6). При помощи реостата регулируют
разность потенциалов между анодом и катодом так, чтобы она составляла
2,5—2,7 в при силе тока 2—3 а. При этих условиях выделение свинца
заканчивается в течение 10—15 мин.
Через 20—25 мин от начала электролиза по стенке стакана
добавляют 1—4 мл разбавленной (1 : 1) серной кислоты и продолжают процесс
осаждения меди до полного ее выделения. Для проверки полноты
выделения меди к раствору добавляют 20—25 мл дистиллированной воды и
продолжают электролиз еще 10 мин. Если в течение этого времени на
погруженной в жидкость части электрода не будет выделяться медь
(выделение меди обнаруживают по покраснению поверхности катода),
электролиз считают законченным. Процесс электролиза ведут при энергичном
перемешивании раствора с помощью электромагнитной мешалки (см. § 6).
По окончании процесса электроосаждения, не выключая ток,
держатель с электродами вынимают из раствора и электроды быстро
ополаскивают, погружая их несколько раз в дистиллированную воду. После
этого выключают ток, снимают электроды и промывают катод спиртом,
а анод вторично промывают дистиллированной водой.
Если ток будет выключен в то время, когда электроды еще находятся
в растворе, медь, осевшая на поверхности катода, начнет растворяться
в кислом электролите.
Промытые электроды сушит в сушильном шкафу: катод — при 80° С,
а анод — при 180° С. Высушенные электроды взвешивают на
аналитических весах и по увеличению массы катода и анода рассчитывают
процентное содержание меди (a:Cu) и свинца (хрЬ) в латуни по формулам:
Хси=(Е^Ш> (4)
где gi — масса катода до электролиза, г;
g2 — масса катода после электролиза, г;
а — навеска латуни, г.
(g2A-gf) ЮО-0,86
хръ = - (О)
где gf — масса анода до электролиза, г;
g$ — масса анода после электролиза, г;
а — навеска латуни, г;
0,86 — коэффициент пересчета двуокиси свинца на металлический свинец (при условии
высушивания анодного осадка при 180° С).
§ 8. Определение малых количеств меди методом
внутреннего электролиза
Медь выделяют из кислого раствора на платиновом катоде,
применяемом в паре с алюминиевым анодом, без наложения тока извне.
Известно три варианта внутреннего электролиза. Первый из них
основан на отделении катода и анализируемого раствора от анода, погру-
412
ГЛ. VII. ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
женного в раствор какой-либо соли, не содержащей определяемого
элемента. Отделения катода от анода достигают при помощи коллодиевой
диафрагмы (перегородки) с целью предотвращения разряжения части ионов
определяемого элемента непосредственно на самой анодной пластинке
(в нашем случае на алюминиевой пластинке):
2А1 + 3Cu++ -> 2Ai+++ + 3Cu
Этот процесс носит название цементации на аноде.
Поэтому для полного устранения процесса цементации в
анализируемый раствор погружают только катод.
По другому варианту, разработанному советским ученым Ю. Ю. Лурье,
электролиз ведут без диафрагмы и без перемешивания раствора
электролита. В этом случае электроды погружают непосредственно в
анализируемый раствор. Этим способом можно определять малые количества меди
и других металлов (не более 20 мг), так как при повышенной
концентрации осаждаемого элемента в исходном растворе возникают явления
цементации на аноде.
Метод очень прост и не требует сложной аппаратуры.
Прибор для определения меди состоит из платинового сетчатого
катода и тщательно очищенной наждачной бумагой алюминиевой пластинки
(длина 130 мм, ширина 6—7 мм, толщина 1—2 мм). Электроды
соединяются между собой металлической муфтой. В качестве сосуда для
электролиза служит обычный химический стакан емкостью 400 мл.
Третий вариант, разработанный советским ученым Ю. А. Чернихо-
вым, основан на непосредственном покрытии анода коллодиевой пленкой,
что дает возможность практически полностью выделять металл при
относительно высокой концентрации его в исходном растворе.
Рассчитанную навеску (2—3 г) исходного сплава помещают в
химический стакан и растворяют в серной или азотной кислоте. Нагревание
способствует растворению металла.
Полученный раствор разбавляют до 200 мл и подвергают внутреннему
электролизу. Если для анализа дан нейтральный исходный раствор, то
его сначала подкисляют 4 мл разбавленной (1:1) серной кислоты или
5—10 мл концентрированной уксусной кислоты, разбавляют до 200 мл,
нагревают до 85—90° С и затем подвергают внутреннему электролизу.
Для этого в нагретый раствор опускают соединенную пару электродов.
Платиновый катод должен быть предварительно очищен, высушен и
взвешен. Электролиз, ведут в течение 40 мин при нагревании. По окончании
электролиза электроды вынимают и обмывают водой. Катод промывают
дистиллированной водой, затем спиртом, высушивают и взвешивают.
Расчет ведут обычным способом.
Следует иметь в виду, что совместно с медью на катоде осаждаются
серебро, ртуть, золото, платина, поэтому анализу подвергают сплавы, не
содержащие указанных металлов. В случае содержания их в исходном
растворе выделенные металлы обрабатывают азотной кислотой и в
полученном растворе определяют медь другими методами.
Таким образом, осажденный на катоде методом внутреннего
электролиза металл определяют весовым способом путем взвешивания катода
с выделившимся на нем металлом или путем растворения металла в кислоте
и последующего определения его в растворе фотоколориметрическим или
другим физико-химическим методом.
ГЛАВА VIII
ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
АНАЛИЗА
§ 1. Особенности объемных электрохимических
методов анализа
Физико-химические методы определения точки эквивалентности.
Способы определения точек эквивалентности с помощью индикаторов не
являются единственными. Существуют и другие, так называемые физико-
химические методы определения точки эквивалентности. Во многих
случаях применение индикаторов в процессе объемно-аналитических
определений оказывается невозможным по разным причинам. Трудно или
практически невозможно наблюдать изменение окраски индикатора в
окрашенных или мутных растворах. В других случаях, например при
титровании слабых и очень слабых кислот и оснований, индикаторы
оказываются бесполезными, так как с их помощью невозможно достичь
требуемой точности определения. Наконец, для многих объемно-аналитических
определений еще не найдены требуемые индикаторы, йто в особенности
относится к случаям дифференцированного (раздельного) титрования
смесей разнообразных кислот или оснований.
Помимо методов титрования в присутствии индикаторов, применяются
и электрохимические методы определения точки эквивалентности. В
процессе электрохимического титрования наблюдение ведут не за изменением
окраски раствора (так как в этом случае индикаторы не применяют),
а за изменением электрохимических показателей титруемого раствора:
электропроводности (кондуктометрическое титрование), окислительно-
восстановительного потенциала (потенциометрическое титрование),
диффузионного тока (амперометрическое титрование) и т. п. При этом
титрование выполняют обычным способом, но вместо визуального наблюдения
за изменением окраски индикаторов пользуются приборами, показания
которых не зависят от субъективных наблюдений экспериментатора.
Электрохимические методы определения точек эквивалентности имеют
ряд преимуществ перед обыкновенными индикаторными методами
титрования. Они отличаются высокой чувствительностью, быстротой
выполнения и объективностью получаемых результатов анализа.
Электрохимические методы дают возможность организовать непрерывный контроль
производства.
Ниже приводится краткое описание некоторых электрохимических
методов титрования.
§ 2. Кондуктометрическое титрование
Электропроводность раствора зависит от характера электролита, его
температуры и концентрации растворенного вещества.
С повышением температуры электропроводность увеличивается. При
данной температуре удельная электропроводность разбавленных рас-
414 ГЛ. Vni. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
творов приблизительно пропорциональна концентрации электролита.
Это позволяет пользоваться кондуктометрическими измерениями для
аналитических определений. Практическое применение этого метода
ограничено тем, что электропроводность зависит не только от концентрации
определяемого вещества, но и от концентрации ионов всех веществ,
присутствующих в исследуемом растворе.
Определение точки эквивалентности по электропроводности.
Наибольший интерес представляет измерение электропроводности в объемном
анализе для определения точки эквивалентности при титровании.
При титровании определяемого вещества А стандартным раствором
реактива В в случае получения нерастворимого или слабодиссоцииру-
!
Шъем pac/nffo/pa Яисмош/
Рис. 79. Кривая кондуктометр и-
ческого титрования NaOH
раствором НС1.
Одъел/ pacmffqpa BaCl2
Рис. 80. Кривая кондуктометри-
ческого титрования AgNOs
раствором ВаС12.
ющего соединений (без учета разбавления) электропроводность раствора
титруемого электролита уменьшается. Минимум на кривой титрования
наблюдается к концу титрования (в точке эквивалентности). При избытке
реактива электропроводность возрастает. Это служит признаком конца
титрования.
Метод титрования, при котором точку эквивалентности фиксируют
по резкому изменению электропроводности исследуемого раствора, носит
название кондуктометрического титрования.
Кондуктометрическое титрование имеет большое значение при
анализе окрашенных и мутных растворов, когда определение точки
эквивалентности с помощью индикаторов затруднено. Обязательным условием
применимости метода кондуктометрического титрования является
заметное изменение электропроводности раствора в момент достижения точки
эквивалентности.
Так, например, в процессе титрования едкого натра
хлористоводородной кислотой
Na+ + OH- + H+ + Cl-
Na+ + Cl- + H20
концентрация щелочи уменьшается. Так как электропроводность
раствора NaCl значительно ниже электропроводности раствора NaOH и
хлористоводородной кислоты, то по мере прибавления кислоты
электропроводность раствора уменьшается и в точке эквивалентности будет
наименьшей. При дальнейшем прибавлении хлористоводородной кислоты
электропроводность вновь возрастает *.
* Уменьшение или увеличение электропроводности титруемого раствора объясняется
изменением подвижности ионов (X). Так в реакции нейтрализации подвижность ионов
водорода (А, + = 350) больше подвижности ионов гидроксила (А, „- = 200) и ионов натрия
н он
(К + = 50). Поэтому кривая кондуктометрического титрования несимметрична.
§ 3. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
415
Точна эн0и0а-
\ /леятяоспи
]/
Кривые кондуктометрического титрования. Если отложить по оси
абсцисс значения объемов прибавляемого титрованного раствора кислоты
(в миллилитрах), а по оси ординат — величины электропроводности,
получится характерная кривая
кондуктометрического титрования (рис. 79). Точка эквивалент- I
ности при этом титровании совпадает с точкой §
минимума электропроводности. |
Другой вид имеет кривая кондуктометриче- §
ского титрования нитрата серебра хлоридом |^
бария (рис. 80). В этом случае в процессе титро- §,.
вания не наблюдается заметного изменения |
электропроводности, но после достижения точки с^
эквивалентности прибавление даже незначи-
тельного избытка хлорида бария вызывает Объем раствора NaOH
повышение электропроводности. Кривые треть-
его типа получаются при титровании слабых ^рическог^и^овТи^а:
кислот сильными основаниями или слабых бой кислоты сильным осно-
оснований сильными кислотами (рис. 81). Здесь ванием.
электропроводность до точки эквивалентности
возрастает менее резко, чем после ее достижения.
Таким образом, форма получаемой кривой кондуктометрического
титрования зависит от типа титрования, а изменения электропроводности
обусловлены различной подвижностью ионов (в см/сек) в одинаковых
условиях опыта.
§ 3. Высокочастотное титрование
Недостатки, свойственные обычным кондуктометрическим методам
титрования, которые вызываются поляризацией электродов или
изменением их электропроводности вследствие осаждения твердых частиц при
титровании по методу осаждения, в значительной мере устраняются при
так называемом методе высокочастотного титрования.
При обычных измерениях электропроводности электроды погружают
в анализируемый раствор. В случае высокочастотного титрования
электроды помещают вне анализируемого раствора непосредственно у стенок
ячейки и повышают частоту переменного тока до нескольких мегагерц
(1 мегагерц равен 10е герц).
Форма кривых высокочастотного кондуктометрического титрования
определяется совокупностью всех факторов, влияющих на
электропроводность: температуры, концентрации электролита, подвижности ионов
и т. п.
Работа с металлическими электродами, находящимися вне ячейки,
не требует дорогостоящих платиновых электродов. Метод оказывается
также полезным в тех случаях, когда нежелательно по тем или иным
причинам помещение электрода в анализируемый раствор.
Высокочастотный метод титрования широко применяется для
определения кислот, оснований, смесей кислот и смесей оснований в неводных
средах.
На практике при высокочастотном титровании пользуются
различными конструкциями ячеек. Простейшим видом ячейки конденсаторного
типа является стеклянный сосуд с прижатыми вплотную к наружным
стенкам металлическими пластинками (электродами). В ячейках
индукционного типа стеклянный сосуд плотно вставляется внутрь индукционной
катушки.
416 ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
§ 4. Потенциометрическое титрование
Потенциометрический метод титрования основан на измерении
потенциала электрода, погруженного в раствор. Величина потенциала зависит
от концентрации соответствующих ионов в растворе. Например,
величина потенциала серебряного электрода,
погруженного в раствор соли серебра, изменяется с
изменением концентрации Ag+ в растворе. Поэтому,
измерив потенциал электрода, погруженного в раствор
данной соли неизвестной концентрации, можно
определить содержание соответствующих ионов
в растворе.
Электрод, по потенциалу которого судят о
концентрации определяемых ионов в растворе,
называют индикаторным электродом.
Величину потенциала индикаторного электрода
определяют, сравнивая ее с величиной потенциала
другого электрода, который принято называть
электродом сравнения. В качестве электрода сравнения
может быть применен только такой электрод,
величина потенциала которого остается неизменной при
изменении концентрации определяемых ионов.
Обычно в качестве электрода сравнения применяют
нормальный водородный электрод (см. книга 1,
гл. II, § 3).
Потенциал нормального водородного электрода
условно приравнивают нулю, поэтому э. д. с. (Ех)
гальванического элемента, составленного из
индикаторного электрода и нормального водородного электрода сравнения,
приравнивают потенциалу индикаторного электрода:
Рис. 82. Каломельный
электрод:
У — слой ртути;
2—платиновая проволока для
контакта; 3—стеклянная
трубка; 4— паста из
каломели; 5 — насыщенный
раствор КО; 6 — сифон;
7 — боковая трубка;
8 — медная проволока
для включения электрода
б цепь.
при ?сэ = О
ЕХ = Е,
инд"
-ср
-инд
(1)
(2)
На практике часто в качестве электрода сравнения с известным
значением электродного потенциала пользуются не водородным, а
каломельным электродом (рис. 82).
~ ТАБЛИЦА 30
Потенциал каломельного электрода
(по отношению к нормальному водородному
электроду)
Потенциал каломельного
электрода во время работы
практически не изменяется.
Зависимость величины
потенциала каломельного
электрода (по сравнению с
потенциалом нормального
водородного электрода) от условий
анализа приведена в табл. 30.
Электрод, потенциал
которого хотят измерить
(например, серебряный), соединяют
с электродом сравнения
(каломельным, водородным
и т. д.) и получившуюся ячейку включают в схему, измеряющую
напряжение (потенциометр, ламповый вольтметр). Во время измерения
напряжения поддерживают достаточно низкой силу тока (от 10"в до 10"14 а),
проходящего через ячейку. При малой силе тока характеристики ячейки
не изменяются.
Концентрация раствора
KC1
Насыщенный
1 н.
0,1 н.
Потенциал каломельного
электрода, в
при 25 °С
0,2458
0,2819
0,3369
при 20 °С
0,2490
0,2859
0,3379
4. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
417
Потенциал электрода является функцией активности веществ.
Например, потенциал серебряного электрода в растворе ионов серебра
выражают уравнением:
Е = Е°+ 0,059 lgaAg+
где Е° = 0,7995 в — нормальный потенциал для процесса, выражаемого
уравнением
Ag+ + е —> Ag
Следовательно, активность ионов серебра (равно как и активность
других ионов) можно определять по величине потенциала Е.
Для определения концентрации ионов серебра (CAg+) необходимо
знать коэффициент активности (fAg+)'
CAg+ = ^Ag+//Ag+
Определение точки эквивалентности по потенциалу индикаторного
электрода. Зависимость величины потенциала индикаторного электрода
от концентрации раствора может быть использована дЛя определения
точки эквивалентности при титровании. В точке эквивалентности, когда
концентрация определяемого иона сильно уменьшается и становится
практически ничтожной, происходит резкое изменение потенциала,
регистрируемое чувствительным потенциометром.
Например, если в раствор, содержащий Ag+, опустить серебряный
индикаторный электрод и электрод сравнения, то серебряный электрод
приобретает положительный заряд. При титровании раствора,
содержащего Ag+, раствором NaCl концентрация ионов серебра уменьшается
и потенциал индикаторного электрода изменяется. Потенциал же
электрода сравнения является постоянным. Поэтому во время титрования по
мере взаимодействия определяемого вещества (Ag+) со стандартным
раствором прибавляемого реактива (NaCl) наблюдается постепенное и
нарастающее изменение потенциала индикаторного электрода. Вблизи точки
эквивалентности происходит сильное изменение концентрации ионов
серебра, сопровождающееся резким изменением потенциала
индикаторного электрода, что и является признаком конца титрования.
Титрование, при котором точка эквивалентности определяется по
скачку потенциала электрода, погруженного в раствор, называют
потенциометр ическим титрованием. Потенциометрическое титрование
применяют при методах нейтрализации, оксидиметрии, осаждения, комплек-
сообразования.
Кривые потенциометрического титрования. Характерная кривая
потенциометр ического титрования приведена на рис. 83, где по оси ординат
отложены значения потенциала индикаторного электрода, а по оси
абсцисс — объем раствора хлорида, добавляемого к раствору.
Потенциометрическое титрование может быть с успехом применено
не только для титрования растворов, к которым применим индикаторный
метод, но и для титрования окрашенных и мутных растворов, когда
индикаторные методы титрования применить нельзя. Потенциометр ическое
титрование широко применяют для определения слабых и очень слабых
кислот и оснований, смесей кислот или оснований, смесей окислителей
или восстановителей в неводных средах. Наконец, потенциометрический
метод применяют для определения рН исследуемых растворов.
Если на оси ординат откладывать не величину потенциала, а отношение
Д?/ДС, т. е. величину изменения потенциала при добавлении 1 мл
раствора хлорида, то получается кривая (рис. 84), на которой еще более
отчетливо видно положение точки эквивалентности.
418
ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Определение рН. Потенциал индикаторного электрода функционально
связан с концентрацией ионов водорода. Водородный электрод,
получаемый погружением платиновой пластинки, покрытой платиновой чернью,
в насыщенный газообразным водородом кислый раствор, находится в
равновесии с ионами водорода:
К-
[HJ
или К = •
Ям+
где [Н+ ] — концентрация ионов водорода в растворе;
[Н] — концентрация водорода в платине при постоянном давлении
(величина постоянная):
5
4
V
< 2
In aH+
(10)
Осаждено
ЮО%А$+-иопод
о г 4- s в ю 1 г
Раствор хлорида, мл
Рис. 83. Кривая потенциометрического
титрования AgN03 раствором NaCl.
JK
9 W //
С,пл
Рис. 84. Кривая потенциометри-
ческого титрования AgN03
раствором NaCl, построенная в
системе координат Д?/ДС.
Так как потенциал нормального водородного электрода (Е°) равен
нулю, то потенциал водородного электрода выражают формулой:
*н = -§Г1п[Н+] «ли ?H = -|?-lnV
(11)
или
?н = 0,059 lg[H+] или Ен = 0,059 lg aR+ (12)
При пользовании каломельным электродом, потенциал которого
(Екал) известен, значения Ех исследуемого раствора вычисляют по
формуле:
или
Так как Ен = 0,059 lg [H+ ], то
-,,[№,-pH-^gffiL
(15)
где Ех — э. д. с. исследуемого элемента.
§ 5. Полярографический метод анализа
Полярографический метод анализа основан на использовании
концентрационной поляризации (см. гл. VII, § 6), возникающей в процессе
электролиза, на электроде с малой поверхностью. По характерной кри-
§ 5. ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
419
вой, показывающей изменение силы тока в процессе электролиза в
зависимости от приложенного напряжения, можно с достаточной точностью
определить качественный и количественный состав анализируемого,
вещества. Кривая силы тока в момент восстановления анализируемого иона
поднимается резко вверх, образуя так называемую полярографическую
волну. По расположению этой волны мощно судить о качественном составе
электролита', по высоте волны — о концентрации восстанавливающегося
иона.
Полярографический метод анализа впервые был предложен в 1922 г.
чешским ученым Я. Гейровским.
Полярографический метод дает возможность определять примеси
металлов, содержащиеся в технических образцах в количестве порядка
0,001%, с точностью в среднем до 1%.
Отличительными особенностями полярографического метода анализа
являются:
1. Быстрота аналитического определения, не превышающая нескольких
минут.
2. Большая чувствительность, позволяющая вести аналитические
определения очень малых количеств исследуемого вещества (до
10"5 моль/л).
3. Независимость результатов определений от индивидуальных
особенностей экспериментатора, так как о них судят по объективным
показаниям чувствительного гальванометра.
4. Возможность одновременно вести определение нескольких
элементов, не прибегая к предварительному их разделению.
Основы метода. Если приложить разность потенциалов к электродам,
опущенным в раствор электролита, и постепенно увеличивать эту разность
потенциалов, то вначале ток через раствор протекать почти не будет.
При увеличении разности потенциалов до величины, достаточной для
разложения электролита, сила тока резко возрастает. Эту величину
разности потенциалов называют потенциалом разложения (см. гл. VII, § 3).
Если взять один из электродов с малой поверхностью (обычно
применяют капельный ртутный катод), другой — с большой поверхностью, то
при пропускании через раствор постоянного электрического тока
основное изменение концентрации будет наблюдаться у электрода с малой
поверхностью. Такое явление обусловлено большой силой тока,
приходящейся в процессе электролиза на единицу поверхности малого электрода,
т. е. высокой плотностью тока на электроде.
По мере повышения разности потенциалов между электродами
увеличивается сила тока, протекающего через раствор, и плотность тока на
малом электроде. При этом скорость обеднения раствора
восстанавливающимися ионами в непосредственной близости к поверхности малого
электрода также возрастает.
Постепенное повышение напряжения и связанное с этим возрастание
плотности тока на малом электроде приводит в конечном итоге к такому
моменту, когда все движущиеся к катоду ионы успевают разрядиться.
Приэлектродный слой пополняется ионами из раствора медленнее, чем
протекает процесс разрядки на поверхности электрода. В этом случае
дальнейшее повышение разности потенциалов не вызывает заметного
возрастания силы тока, протекающего через раствор.
Диффузионный ток. При установившемся подвижном равновесии,
когда количество восстановленных ионов становится равным количеству
ионов, продиффундировавших к ртутному катоду, сила тока становится
постоянной. Такую силу тока, при которой достигается полный разряд
всех ионов анализируемого вещества, поступающих в приэлектродное
420 ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
пространство за счет диффузии, называют предельным или диффузионным
током.
Скорость диффузии вещества из раствора более высокой концентрации
в раствор более низкой концентрации пропорциональна разности
концентраций обоих растворов. Поэтому диффузионный ток пропорционален
концентрации определяемого иона в растворе (концентрация ионов у
поверхности катода приближается к нулю, когда ток достигает
предельного значения).
Уравнение Ильковича. Зависимость силы диффузионного тока от
концентрации выражается уравнением, выведенным Ильковичем:
I=605nDl/2m2'4'6C (16)
где / — сила тока, мка (1 мка — 10~6 а);
п — число электронов, принимаемых ионом при восстановлении;
D — коэффициент диффузии иона, см2'сек'1;
т — масса ртути, вытекающей из капилляра в 1 сек, мг\
т — период капания, сек (время «жизни» одной капли);
С — концентрация определяемого иона, ммоль/л.
Если полярографирование проводят с каким-нибудь одним типом
ионов, то для них п и D являются величинами постоянными; Ь случае,
если работают с одним и тем же капилляром и с одной и той же скоростью
вытекания ртути, то и произведение т2/НУ6 будет величиной постоянной
и тогда
1 = КС (17)
Полярографическая кривая. Кривая зависимости силы тока от
напряжения показана на рис. 85. По оси абсцисс отложена разность
потенциалов между электродами, по оси ординат — значения силы тока,
протекающего через раствор. На участке кривой от нуля до А электролиз не
протекает. Соответственно по мере возрастания напряжения не происходит
практически изменения силы тока в цепи. Сила протекающего тока весьма
невелика. Ток в этом месте вызывается заряжением ртутной капли и
восстановлением примесей. На этом участке кривой потенциал разложения
анализируемого вещества не достигается.
На участке от Л до В незначительное повышение разйости потенциалов
вызывает резкое возрастание силы протекающего через раствор тока.
Этот участок характеризует собой нормальное прохождение процесса
электролиза, сопровождающегося все более интенсивным обеднением
приэлектродного слоя.
Участок от В до С характеризует собой процесс, когда все
находящиеся в приэлектродном слое ионы анализируемого вещества успевают
разрядиться. Скорость диффузии меньше скорости выделения ионов.
Этот участок характеризуется неизменностью тока в цепи при
постепенном повышении разности потенциалов между электродами.
Изображенную на рис. 85 кривую зависимости силы тока,
протекающего через раствор, от напряжения между электродами называют вольт-
амперной, или полярографической кривой, или полярографической волной.
Полярографическая волна характеризует собой как количество
анализируемого вещества, так и его характер.
Высота волны Н {рис. 86) характеризует предельный ток и дает,
таким образом, возможность определить концентрацию анализируемого
вещества.
Если расстояние от точки А до точки В разделить пополам и из
полученной точки опустить перпендикуляр на ось абсцисс, то он отсечет на ней
определенный отрезок, характеризующий потенциал, необходимый для
§5. ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
421
достижения половины предельного тока. Потенциал середины
полярографической волны называют потенциалом полуволны Е\/2.
Потенциал полуволны не зависит от концентрации растворенного
вещества, а только от природы восстанавливаемого иона. Поэтому
потенциал полуволны служит качественной характеристикой
присутствующего в растворе иона.
Капельный ртутный катод. Рассмотренный выше процесс является
идеальным процессом. Практически, когда поверхность микроэлектрода,
служащего катодом, покрывается продуктами восстановления, его
химический состав меняется и получить воспроизводимые результаты
невозможно. Поэтому указанный выше
случай может быть осуществлен q
только при условии постоянства
размеров и химической природы
катода.
^
Рис. 85. Вольт-амперная кри- Рис. 86. Определение потенциала полу-
вая. волны и высоты полярографической
волны:
Е — потенциал полуволны; Н— высота
полярографической волны.
Ближе всего соответствует требованиям, предъявляемым к
микроэлектроду для полярографии, капельный (непрерывно обновляющийся)
ртутный катод. Такой электрод (рис. 87) представляет собой ртуть,
вытекающую по каплям из капилляра с определенной скоростью.
Поверхность капельного ртутного катода по составу и размерам
остается постоянной. Ртуть непрерывно обновляется. Вследствие этого
потенциал восстановления каждого иона сохраняется на определенном
уровне. Образование амальгамы при выделении катионов металла на
поверхности ртутного капельного катода и стекание ее на дно электролизера,
а также постоянное обновление поверхности его за счет вытекания ртути
и замены одной капли другой способствует приближению условий
проведения анализа на капельном электроде к идеальным.
Полярографический фон. При рассмотрении процесса нами было
принято, что число ионов металла в приэлектродном слое пополняется
в результате диффузии. В действительности, если в растворе присутствуют
только ионы анализируемой соли, то перенос тока от одного электрода
к другому осуществляется иоязми растворенной соли. Эти ионы под
действием электрического тока передвигаются: к катоду — катионы и
к аноду — анионы. Скорость движения ионов под действием
электрического тока зависит от многих факторов: разности потенциалов между
электродами, диаметра и заряда анализируемого иона, его концентрации
в растворе и ряда других причин. Поэтому перенос ионов под действием
электрического тока искажает процесс полярографического определения
и сильно затрудняет проведение анализа.
Однако если в анализируемый раствор добавлена другая соль,
разложение которой наступает при большей разности потенциалов, чем это тре-
422 ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
буется для определяемого вещества, то катионы фона также движутся
к катоду, но они не разряжаются при заданном потенциале. Таким
образом, доля участия ионов определяемого вещества в переносе тока
уменьшается.
Если концентрация добавленной соли велика по сравнению с
концентрацией анализируемого вещества, то практически весь ток переносится
ионами добавленной соли. В этом случае скорость
движения ионов анализируемого вещества под
действием электрического тока приближается
к нулю и им можно пренебречь.
Индифферентную соль, добавляемую для
устранения движения ионов анализируемого вещества
под действием электрического тока, называют
полярографическим фоном или просто фоном.
Для того чтобы постоянный электрический
ток, протекающий через раствор, не вызывал
заметного движения ионов анализируемого
вещества, концентрация фона должна в 100—1000 раз
превышать концентрацию определяемой соли.
Поэтому полярографические определения
применимы для анализа незначительных примесей
вещества в растворе.
Область применения полярографического
метода анализа. Полярографическим методом можно
определять как неорганические, так и
органические вещества, способные восстанавливаться или
окисляться на поверхности электродов при
прохождении постоянного электрического тока. Поэтому
полярографические определения широко
применяются в работе заводских и
научно-исследовательских химико-аналитических лабораторий.
За последнее время все более широкое
распространение приобретает метод
полярографического анализа с твердым вращающимся
микроэлектродом. Использование твердого
вращающегося микроэлектрода дает возможность применять
методы полярографического анализа при
определениях в расплавленных средах.
Методика полярографического анализа. Для
анализа какого-либо вещества
полярографическим методом его прежде всего переводят в
раствор. Затем создают необходимую среду и удаляют мешающие
полярографическому определению примеси. Большие неудобства для полярографи-
рования создают вещества с потенциалами, близкими к потенциалу
восстановления определяемого элемента или более низкими. Заметно мешает
определению и растворенный кислород. Для удаления мешающих веществ
широко применяют осаждение, койшлексообразование, окисление —
восстановление и т. п. Для удаления растворенного кислорода,
восстанавливающегося цг катоде, через раствор пропускают водород. В щелочные
растворы добавляют Na2S03.
Большое значение имеет правильный выбор и создание фона для поля-
рографирования. Обычно это решается опытным путем.
Подготовленный к анализу раствор, свободный от посторонних
примесей, помещают в электролизер (см. рис. 87). Включают постоянный
электрический ток и постепенно повышают напряжение. По мере непре-
Рис. 87. Схема
простейшего капельного ртутного
катода:
/ — трубка с ртутью и
платиновым контактом; 2 —
резервуар (груша) со ртутью,
питающей капельный
ртутный катод; 3 — резиновая
трубка, соединяющая
резервуар со стеклянным
капилляром; 4—стеклянный
капилляр, через который
поступает ртуть (ртутный катод);
5 — электролизер; 6 — слой
ртути, соединенный с
платиновым контактом (ртутный
анод); 7 — боковая трубка
для пропускания водорода
или азота с целью
вытеснения кислорода.
$ 5. ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
423
рывного увеличения напряжения регистрируют происходящее при этом
изменение силы тока в зависимости от напряжения. На основании
полученных данных строят полярографическую кривую.
Для количественных определений в полярографии применяют метод
калибровочных кривых, метод стандарта или метод добавок.
При применении метода калибровочных кривых снимают несколько
полярограмм стандартных образцов. По высоте волны h и концентрации С
строят калибровочную кривую. По высоте волны неизвестного образца
определяют его концентрацию по
калибровочной кривой.
В методе стандарта снимают поля-
рограмму только одного стандартного
раствора и рассчитывают концентрацию
неизвестного раствора по формуле:
С* ?- (18)
Для анализа методом добавок в
электролизер наливают определенный объем
анализируемого вещества и получают
волну определяемого вещества hA. Затем
добавляют к раствору 1—3 мл
стандартного раствора определяемого вещества
и снова снимают полярограмму.
Получают суммарную волну Л0бщ-
Неизвестную концентрацию (мг/мл) вычисляют
по формуле:
|Aaaa/wwwv\Aaaa/wwww\ww^J
к/)
ГТ-7
— "ЯГ (ЛСг = Лобщ-АА) <19>
Рис. 88. Схема полярографической
установки:
/ — батарея аккумуляторов; 2 — реохорд;
3 — вольтметр; 4 — подвижной контакт;
5, 6 — электроды; 7 — электролизер; 8 —
гальванометр; 9 — сопротивление (шунт).
где
^общ — высота волны испытуемого вещества после добавления стандартного раствора;
ССт—концентрация добавляемого иона в электролизере:
Сст_ у
Сетует
г+Уд
(20)
Уст
количество стандартного раствора, добавленное в электролизер, мл;
объем испытуемого раствора в электролизере, мл;
Сст — первоначальная концентрация стандартного раствора.
Полярографы. Для полярографического анализа применяют приборы,
называемые полярографами. Они бывают визуальные и автоматические.
Последние позволяют отсчитывать силу тока и напряжение не только
визуально, но и по кривой зависимости силы тока от напряжения,
автоматически записываемой специальным прибором.
Схема полярографической установки показана на рис. 88.
При помощи внешнего источника тока / (батареи аккумуляторов на
4—6 в) напряжение подается на реохорд 2. Напряжение на реохорде
измеряется вольтметром 3.
Постепенным передвижением подвижного контакта 4 (вправо по шкале
реохорда) дают все увеличивающееся напряжение на электроды 5, 6
(5 — капельный ртутный катод, опущенный в анализируемый раствор,
6 — ртуть, налитая на дно электролизера 7). Сила тока, возникающего
при этом в цепи, измеряется посредством чувствительного (обычно
зеркального) гальванометра S. Так как при одной и той же чувствительности
гальванометра нельзя снимать полярограммы в растворах различных
концентраций, то параллельно гальванометру подключают градуированное
424 ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
сопротивление (шунт) 9. При небольшой концентрации анализируемого
раствора возникает также и небольшой ток. В этом случае шунт включают
полностью, чтобы практически весь ток проходил через гальванометр.
При больших концентрациях растворов сопротивление шунта уменьшают
и через гальванометр проходит только часть тока.
Определение цинка в растворе методом стандарта. В мерную колбу
емкостью 50 мл помещают точно 10 мл раствора ZnS04 ([Zn+ + ] = Ct =
= 0,5 мг/мл), 20 капель раствора желатина и доводят до метки 1 н.
раствором КО. Полученный раствор тщательно перемешивают. Около 20—
25 мл этого раствора помещают в сухой (или сполоснутый этим же
раствором) электролизер и пропускают через раствор водород в течение
20 мин. Затем снимают полярограмму (при соответствующем положении
шунта), строят график и определяют высоту волны h±.
К раствору ZnS04 неизвестной концентрации, помещенному в колбу
емкостью 50 мл, добавляют 20 капель раствора желатина и доводят объем
до метки 1 н. раствором КС1. Затем раствор перемешивают.
Подготовленный раствор наливают в электролизер, пропускают через него
водород в течение 20 мин, снимают полярограмму, строят график и
определяют высоту волны (hx). Находят концентрацию Zn+ +-ионов (Сх) по
формуле (в мг1мл):
где Сг = 0,5 -эд- = 0,1 мг/мл.
Содержание цинка в миллиграммах в данном растворе вычисляют,
умножая Сх на емкость мерной колбы (50 мл).
§ 6. Амперометрическое титрование
Полярографией пользуются не только для непосредственного
определения концентрации анализируемого вещества, но и для определения
точки эквивалентности в процессе титрования. Сущность метода амперо-
метрического титрования заключается в следующем. Аликвотную часть
исследуемого раствора помещают в электролизер, снабженный
капельным ртутным катодом и большим ртутным анодом. Между электродами
устанавливают заданное напряжение, требуемое для выделения на катоде
того или иного металла, и приступают к титрованию. В процессе
титрования отмечают показания гальванометра. На основании результатов
титрования строят кривую амперометрическогр титрования, откладывая
на оси ординат показания гальванометра, а на оси абсцисс — объем
стандартного раствора (в мл). По этой кривой находят точку
эквивалентности. Для амперометрического титрования применяют
полярографическую установку.
Количество определяемого вещества вычисляют по объему стандартного
раствора реактива, израсходованного до достижения точки
эквивалентности.
Методом амперометрического титрования можно определять многие
катионы и анионы, а также органические вещества. Метод
амперометрического титрования отличается рядом преимуществ перед методом
полярографии. Одним из таких преимуществ является то, что с помощью
метода амперометрического титрования можно определять вещества,
которые не восстанавливаются и не окисляются на индикаторном
электроде. При этом применяют титрант, который восстанавливается или
окисляется.
§ 7. КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
425
Метод амперометрического титрования широко применяют в
аналитической практике. Хотя он отличается относительно небольшой точностью
(до 2%), но относится к группе очень чувствительных методов по
сравнению с обычными методами титрования. Этот метод применяют для
определения веществ при концентрации 10 ~3—10~4 моль/л и титрования
окрашенных и мутных растворов.
Кривые амперометрического титрования.
При амперометрическом титровании
наблюдаются кривые различной формы.
1. По мере прибавления реактива
диффузионный ток уменьшается, пока не
достигнет минимального значения (рис. 89, /).
Такого рода кривая наблюдается при
титровании восстанавливающегося вещества
посредством невосстанавливающегося реагента,
например при титровании ионов свинца
раствором сульфата. В этом случае
первоначально сила диффузионного тока
сравнительно высока; затем она уменьшается
по мере того, как РЬ++-ионы реагируют
с SO" "-ионами с образованием PbS04. После
достижения точки эквивалентности
дальнейшее прибавление стандартного раствора
сульфата не вызывает изменение силы тока.
2. По мере добавления в анализируемый
раствор реактива диффузионный ток
уменьшается, а затем, достигнув минимального значения в точке
эквивалентности, вновь увеличивается (см. рис. 89, II). Такого рода
кривая наблюдается при титровании восстанавливающегося вещества
восстанавливающимся реагентом, например при титровании ионов свинца би-
хроматом.
3. По мере прибавления реактива диффузионный ток остается
постоянным и минимальным до тех пор, пока не будет добавлен избыток реактива.
При избытке реактива диффузионный ток возрастает (см. рис. 89, ///).
Такого рода кривая наблюдается при титровании невосстанавливающегося
вещества восстанавливающимся реагентом, например при титровании
сульфат-ионов нитратом свинца.
Точка перегиба соответствует точке эквивалентности.
§ 7. Кулонометрическое титрование
Количество вещества, превращаемого при электролизе, прямо
пропорционально силе тока и времени его прохождения. Поэтому измерение
количества электричества, израсходованного при электролизе, может
служить мерой превращения вещества, подвергшегося электролизу, и
используется в аналитических целях.
Соответствующие аналитические методы объединяют под общим
названием ку лоно метрических методов.
Если при электролизе через раствор протекает Q кулонов, то по
закону Фарадея количество прореагировавшего вещества (gA в г) равно:
^а= 96 500л (22)
где М — молекулярный вес окисленного или восстановленного вещества;
п — количество принимаемых или отдаваемых веществом электронов;
96 500 кулонов — число Фарадея.
Рис. 89. Форма кривых,
получаемых при амперометрическом
титровании:
/ — кривая титрования по
спадающей волне (титрование ионов евин4-
да сульфатом); // — кривая
титрования по спадающе-Еозрастающей
волне (титрование ионов свинца
бихроматом); /// — кривая
титрования по возрастающей волне
(титрование сульфат-ионов нитратом
свинца).
426 ГЛ. VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Одним из важнейших преимуществ кулонометрии является то, что
этот метод анализа применяют в тех случаях, когда в результате
электролиза образуются не только нерастворимые, как при весовом
электроанализе, но и растворимые соединения.
Принцип метода поясним на примере кулонометрического титрования
окислителя (например, бихромата) ионами восстановителя (например,
двухвалентного железа). Ионы железа (II) получают методом
электрохимического восстановления ионов железа (III), добавляемых в электролизер
вместе с определяемым окислителем. Раствор бихромата помещают в
электролизер, подкисляют серной кислотой, добавляют фосфорной кислоты
и раствор железо-аммонийных
квасцов. Полученную смесь
разбавляют водой до требуемого
объема и подвергают электролизу,
предварительно удалив из
раствора свободный кислород при
помощи газообразного азота. Конец
реакции (точку эквивалентности),
когда Сг20~~-ионы количественно
восстановятся Fe+ + -ионами в ионы
хрома (III), устанавливают
потенциометр ическим или
индикаторным методами. Зная время,
израсходованное на кулонометрическое
титрование, рассчитывают
содержание определяемого вещества
по приведенной выше формуле.
Аналогичным образом кулоно-
метрическим методом титрования
можно окислять восстановители генерируемыми ионами окислителя.
Обычно кулонометрия осуществляется при заданном потенциале или
заданной силе тока.
Для кулонометрических определений может быть использована
установка, применяемая для электровесового анализа при контролируемом
потенциале. Для этого в электрическую сеть достаточно включить прибор,
измеряющий количество прошедшего электричества, потребовавшегося
для полного превращения анализируемого вещества.
Если в анализируемый раствор, содержащий ионы определяемого
элемента, добавить в большом избытке окисляемые в процессе электролиза
другие ионы, которые в свою очередь способны окислить ионы
определяемого элемента, то по времени, израсходованному на электролиз при
заданной силе тока, можно судить о количестве определяемого вещества
(восстановителя).
Избыток ионов, образующихся в процессе анодного окисления, также
обнаруживают индикаторным или электрохимическим методом.
Таким образом, описанный выше метод кулонометрического
определения при постоянной силе тока весьма напоминает обычное титрование и
называется кулонометрическим титрованием с внутренним генерированием
реагента.
Пряем генерирования реагента позволяет поддерживать на
необходимом уровне потенциал электрода при заданной силе тока. В противном
случае, если в течение всего электролиза поддерживать силу тока
постоянной,— обычно не удается контролировать потенциал электрода, на
котором происходит превращение анализируемого вещества. В результате
возможно снижение эффективности тока ниже 100%.
Рис. 90. Схема прибора для
кулонометрического титрования при постоянной силе тока:
/ — сосуд для титрования; 2, 3 — электроды;
4 — стеклянная трубка; 5 — источник
постоянного тока; 6 — ключ; 7 — амперметр; 8 —
мешалка; 9 — электрические часы.
§ 7. КУЛОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
427
Предположим, что требуется определить содержание ионов железа (II)
в растворе. Для окисления можно применить ионы церия (IV), которые
в свою очередь могут быть получены электроокислением ионов церия (III).
Для кулонометрического титрования собирают установку, показанную
на рис. 90. Анализируемое вещество помещают в сосуд 1, в который
опущено два электрода 2 и 3. Рабочий электрод 2, индифферентный по
отношению к данному раствору, погружают непосредственно в раствор,
вспомогательный электрод 3 помещают в стеклянную трубку 4. Внизу в трубку
впаивают стеклянный фильтр. Трубку заполняют раствором нитрата
калия или другого электролита, не содержащим ионы железа.
Электроды присоединяют к источнику постоянного тока 5,
поддерживающему при электролизе заданную силу тока независимо от процессов,
возникающих у электродов. Электролиз начинают, замкнув цепь ключом 6У
который одновременно приводит в действие электрические часы 9,
присоединенные к сети переменного тока. Сила тока контролируется при помощи
амперметра 7. В процессе электролиза раствор перемешивают
механической мешалкой 8.
В анализируемый раствор предварительно добавляют раствор,
содержащий Се+++ в стократном избытке по отношению к содержанию Fe++.
В процессе электролиза протекают следующие реакции:
на аноде
Се+++-е—>CeIV
на катоде
2Н+ + 2е—>Н2
По мере появления CeIV немедленно происходит окисление Fe++:
Fe++ + CeIV->Fe+++ + Се+++
Как только ионы железа (II) полностью окислятся, появляется избыток
ионов церия (IV). Появление в растворе CeIV обнаруживают индикаторным
или электрохимическим способом. На этом электролиз прекращают.
Зная величины Q и t, легко вычислить содержание ионов железа (II).
Аналогичным путем осуществляют кулонометрическое титрование
различных окислителей (CeIV, Vv, CrVI, Mnvn и др.) Ре++-ионами,
образующимися в процессе электролиза из Ре+++-ионов, а также
нейтрализацию кислот гидроксильными ионами, образующимися на катоде в процессе
электролиза водных растворов кислот, и другие реакции.
ГЛАВА IX
СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
А. ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
§ 1. Понятие об эмиссионном спектральном анализе
В эмиссионном анализе определение основано на измерении интенсив-
ностей характерных спектральных линий спектров элементов, входящих
в состав данного анализируемого вещества.
Между интенсивностью спектральных линий определяемого элемента
и его концентрацией в исследуемом веществе существует
пропорциональная зависимость, т. е. чем выше концентрация определяемого элемента,
тем больше интенсивность спектральных линий.
Определение ведут следующим образом. Сначала измеряют
интенсивности спектральной линии определяемого элемента и близкой к ней линии
другого элемента (линии сравнения), содержание которого в исследуемой
пробе постоянно *. Этот элемент называют внутренним стандартом.
Между отношением интенсивности линии определяемого элемента /0.э
к интенсивности линии внутреннего стандарта /в. с и концентрацией
определяемого элемента существует пропорциональная зависимость, которую
устанавливают экспериментальным путем на основании измерения
интенсивности линий эталонных образцов.
Так как /в. с постоянно, то /0. JIB. с зависит только от содержания
определяемого элемента. Благодаря этому легко определить его
содержание.
В зависимости от степени точности измерений различают
полуколичественный и количественный спектральный анализ. Полуколичественный
анализ выполняют с помощью приборов, называемых стилоскопами. Для
количественного анализа применяют более совершенные приборы,
называемые стилометрами. Эти приборы предназначены для визуального
наблюдения спектров. Для получения фотографий спектров применяют
спектрографы, например ИСП-22, ИСП-28, ИСП-30 и др.
В этом случае для измерения интенсивности спектральных линий
спектр исследуемого вещества снимают-на фотографическую пластинку.
На пластинке видны линии, степень почернения которых зависит от
интенсивности снятых спектральных линий, которая в свою очередь
пропорциональна концентрации определяемого элемента. Количественно
почернение фотопластинки (плотность почернения) измеряют при помощи
специальных оптических приборов микрофотометров.
Получив экспериментальные данные измерений интенсивностей линий,
находят концентрацию определяемого элемента по градуировочной кривой,
* Линия, используемая в качестве сравнения, должна по длине волны и по
интенсивности приближаться к линии определяемого элемента, тогда изменения их
интенсивностей при изменениях в режиме источника возбуждения будут близкими и не вызовут
больших ошибок. Такие две подобранные линии называют гомологической парой линий.
§ 2. ОСОБЕННОСТИ КОЛОРИМЕТРИЧЕСКИХ МЕТОДОВ АНАЛИЗА 429
вычерчиваемой на основании измерения интенсивности линий эталонных
образцов.
Этот метод дает возможность определять элементарный качественный
и количественный состав вещества.
Эмиссионный спектральный анализ широко применяют в различных
областях науки и промышленности, особенно в металлургии, геологии,
астрофизике и т. п.
Спектральный количественный анализ отличается по сравнению с
химическими методами анализа высокой чувствительностью и быстротой
выполнения. С его помощью определяют многие элементы, содержание
которых не превышает тысячные и десятитысячные доли процента. Одним
из преимуществ эмиссионного спектрального анализа является то, что
для его осуществления требуется ничтожно малое количество
анализируемого вещества (иногда несколько миллиграммов).
Б. КОЛОРИМЕТРИЯ
§ 2. Особенности колориметрических методов анализа
Из оптических методов анализа в практике аналитических лабораторий
наиболее широко применяются колориметрические методы.
Колориметрические методы основаны на измерении интенсивности светового потока,
прошедшего через окрашенный раствор.
Колориметрический метод анализа был предложен русским ученым
В. М. Севергиным еще в 1795 г.
В колориметрическом методе используются химические реакции,
сопровождающиеся изменением цвета анализируемого раствора. Измеряя
светопоглощение такого окрашенного раствора или сравнивая
полученную окраску с окраской раствора известной концентрации, определяют
содержание окрашенного вещества в испытуемом растворе.
Существует зависимость между интенсивностью окраски раствора
и содержанием в этом растворе окрашенного вещества.
Эта зависимость, называемая законом Бугера—Ламберта—Бера,
выражается уравнением:
/=:/0.10-еС/ (23)
где / — интенсивность света, прошедшего через раствор;
/0 — интенсивность падающего на раствор света;
е — коэффициент поглощения света — постоянная величина, характерная для
каждого окрашенного вещества и зависящая от его природы;
С — концентрация окрашенного вещества в растворе;
/ — толщина слоя светопоглощающего раствора, см.
Физический смысл этого закона можно выразить следующим образом.
Растворы одного и того же окрашенного вещества при одинаковой
концентрации этого вещества и толщине слоя раствора поглощают равное
количество световой энергии, т. е. светопоглощение таких растворов
одинаковое.
Для окрашенного раствора, заключенного в стеклянную кювету
с параллельными стенками, можно сказать, что по мере увеличения
концентрации и толщины слоя раствора его окраска увеличивается, а
интенсивность света /, прошедшего через поглощающий раствор, уменьшается
по сравнению с интенсивностью падающего света 10.
Оптическая плотность раствора. Если прологарифмировать уравнение
закона Бера и изменить знаки на обратные, то уравнение принимает вид:
lgA. = 8C/ (24)
430 ГЛ. IX. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Величина lg -у- является очень важной характеристикой окрашенного
раствора; ее называют оптической плотностью раствора и обозначают
буквой D:
D = \g-^=eCl (25)
Из этого уравнения вытекает, что оптическая плотность раствора
прямо пропорциональна концентрации окрашенного вещества и толщине
слоя раствора.
Другими словами, при одинаковой толщине слоя раствора данного
вещества оптическая плотность этого раствора будет тем больше, чем
больше в нем содержится окрашенного вещества. Или, наоборот, при
одной и той же концентрации данного окрашенного вещества оптическая
плотность раствора зависит только от толщины его слоя. Отсюда может
быть сделан следующий вывод: если два раствора одного и того же
окрашенного вещества имеют различную концентрацию, одинаковая интенсивность
окраски этих растворов будет достигнута при толщинах их слоев, обратно
пропорциональных концентрациям растворов. Этот вывод очень важен,
так как на нем основаны некоторые методы колориметрического анализа.
Таким образом, чтобы определить концентрацию (С) окрашенного
раствора, необходимо измерить его оптическую плотность (D). Чтобы
измерить оптическую плотность, следует измерить интенсивность светового
потока.
§ 3. Характеристика колориметрических методов анализа
Интенсивность окраски растворов можно измерять различными
методами. Различают субъективные (или визуальные) методы колориметрии
и объективные (или фотоколориметрические).
Визуальными называются такие методы, при которых оценку
интенсивности окраски испытуемого раствора делают невооруженным глазом.
При объективных методах колориметрического определения для
измерения интенсивности окраски испытуемого раствора вместо
непосредственного наблюдения пользуются фотоэлементами. Определение в ,этом случае
проводят в специальных приборах — фотоколориметрах, откуда и метод
получил название фотоколориметрического.
Визуальные методы. К визуальным методам относятся:
1) метод стандартных серий;
2) метод дублирования (колориметрическое титрование);
3) метод уравнивания.
Метод стандартных серий. При выполнении анализа
методом стандартных серий интенсивность окраски анализируемого
окрашенного раствора сравнивают с окрасками серии специально
приготовленных стандартных растворов (при одинаковой толщине слоя).
Колориметрическое титрование (метод
дублирования). Этот метод основан на сравнении окраски
анализируемого раствора с окраской другого раствора — контрольного. Для
приготовления контрольного раствора готовят раствор, содержащий все
компоненты исследуемого раствора, за исключением определяемого вещества,
и все употреблявшиеся при подготовке пробы реактивы, и к нему
добавляют из бюретки стандартный раствор определяемого вещества. Когда
этого раствора будет добавлено столько, что интенсивности окраски
контрольного и анализируемого раствора уравняются, считают, что в
анализируемом растворе содержится столько же определяемого вещества, сколько
его было введено в контрольный раствор.
§ 3. ХАРАКТЕРИСТИКА КОЛОРИМЕТРИЧЕСКИХ МЕТОДОВ АНАЛИЗА 431
Метод уравнивания. Этот метод основан на уравнивании
окрасок анализируемого раствора и раствора с известной концентрацией
определяемого вещества — стандартного раствора.
Существуют два варианта выполнения колориметрического
определения этим методом.
По первому варианту уравнивание окрасок двух растворов с разной
концентрацией окрашенного вещества проводят путем изменения толщины
слоев этих растворов при одинаковой силе проходящего через растворы
светового потока. При этом, несмотря на различие концентраций
анализируемого и стандартного растворов, интенсивность светового потока,
проходящего через оба слоя этих растворов, будет одинакова.
Соотношение между толщинами слоев и концентрациями
окрашенного вещества в растворах в момент уравнивания окрасок будет
выражаться уравнением:
-?—-?l. (26)
где 1Х — толщина слоя раствора с концентрацией окрашенного вещества Сх;
/2 — толщина слоя раствора с концентрацией окрашенного вещества С2.
В момент равенства окрасок отношение толщин слоев двух
сравниваемых растворов обратно пропорционально отношению их концентраций.
На основании приведенного уравнения, измерив толщину слоев двух
одинаково окрашенных растворов и зная концентрацию одного из этих
растворов, легко можно рассчитать неизвестную концентрацию
окрашенного вещества в другом растворе.
Для измерения толщины слоя, через который проходит световой
поток, можно применять стеклянные цилиндры или пробирки, а при более
точных определениях специальные приборы — колориметры.
По второму варианту, для уравнивания окрасок двух растворов с
различной концентрацией окрашенного вещества, через слои растворов
одинаковой толщины пропускают световые потоки различной
интенсивности.
В этом случае оба раствора имеют одинаковую окраску, когда
отношение логарифмов интенсивностей падающих световых потоков равно
отношению концентраций.
В момент достижения одинаковой окраски двух сравниваемых
растворов, при равной толщине их слоев, концентрации растворов прямо
пропорциональны логарифмам интенсивностей падающего на них света.
По второму варианту определение может быть выполнено только
с помощью колориметра.
Фотоколориметрические методы. Все фотоколориметрические методы
определения основаны на одном общем принципе. Световой поток
проходит через кювету или пробирку, наполненную испытуемым окрашенным
раствором. Прошедший через раствор световой поток воспринимается
фотоэлементом, в котором световая энергия превращается в
электрическую. Возникающий при этом электрический ток измеряют при помощи
чувствительного гальванометра. Как показал А. Г. Столетов, сила
электрического тока, возникающего при действии световой энергии на
фотоэлемент, прямо пропорциональна интенсивности освещения.
Для определения этим методом концентрации исследуемого вещества
измеряют оптическую плотность испытуемого раствора (DHCn) и
эталонного раствора, концентрация которого известна (?>ЭТал). Расчет проводят
по формуле:
Сисп = "гч Сэтал (27)
ь'этал
432 ГЛ. IX. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Основными преимуществами фотоколориметрических методов
измерения интенсивности окраски являются быстрота и легкость определений
при высокой их точности.
Нашей промышленностью выпускаются фотоколориметры марки
ФЭК-М, в которых интенсивности световых потоков измеряются с помощью
селеновых фотоэлементов. Действие прибора основано на принципе
уравнивания интенсивности двух световых потоков при помощи щелевой
диафрагмы. Подробное изложение конструкции прибора и порядок
работы на нем даются в прилагаемых к прибору паспорте и инструкции.
Построение калибровочной кривой. При массовых
фотоколориметрических анализах, определяя концентрацию испытуемого раствора, не
сравнивают каждый раз его светопоглощение
со светопоглощением эталонного раствора, а
предварительно строят так называемую калибровочную
кривую. Для этого пользуются серией эталонных
растворов различной концентрации. Имея такую
кривую, при определении концентрации
испытуемого раствора достаточно измерить его
светопоглощение и по калибровочной кривой найти вели-
Ионцентрсщияс чину концентрации, соответствующую найденному
Рис. 91. Зависимость светопоглощению.
оптической плогности рас- Для построения калибровочной кривой нужно
твора от концентрации приготовить серию эталонных растворов, содер-
(калибровочная кривая). жащих разные количества определяемого
вещества. Сначала приготовляют стандартный раствор,
содержащий строго определенное количество исследуемого вещества.
С помощью бюретки отбирают в мерные колбы емкостью 100 мл
различные, точно измеренные объемы этого стандартного раствора и
соответствующих реактивов, вызывающих окраску анализируемого раствора.
Затем содержимое каждой мерной колбы разбавляют дистиллированной
водой, доводя объем раствора до метки.
С помощью фотоколориметра измеряют оптические плотности
приготовленных эталонных растворов и результаты измерений записывают
в виде таблицы.
Пример такой записи приведен ниже:
Эталонный раствор 1 2 3 4 5
Содержание определяемого
вещества, мг/100 мл раствора 1 4 6 8 10
Оптическая плотность .... 0,1 0,4 0,6 0,8 1,0
На основании полученных результатов строят кривую зависимости
оптической плотности раствора от его концентрации. Это и есть
калибровочная кривая (рис. 91).
Для ее построения на миллиметровой бумаге откладывают по оси
абсцисс значения концентраций эталонных растворов, а по оси ординат —
величины их оптических плотностей. Затем из точек, найденных на осях,
восстанавливают перпендикуляры, и точки их пересечения соединяют
одной линией.
В. ОПТИЧЕСКИЕ МЕТОДЫ УСТАНОВЛЕНИЯ ТОЧКИ
ЭКВИВАЛЕНТНОСТИ
§ 4. Спектрофотометрическое титрование
Оптические методы установления точки эквивалентности применимы
лишь в том случае, если существует линейная зависимость между
величиной светопоглощения и концентрацией определяемого вещества в иссле-
§ 4. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
433
дуемом растворе. Метод применим для определения точки
эквивалентности при кисдотно-основиом, окислительно-восстановительном, комплексо-
метрическом и других методах титрования.
Оптические методы установления точки эквивалентности в объемном
анализе основаны на измерении светопоглощения исследуемой системы,
изменяющегося в процессе титрования.
График строят в координатах: оптическая плотность (D) — объем
стандартного реактива (V). Титрование проводят при определенной длине
волны. Графически процесс титрования представляется в виде пересека-
¦6
Рис. 92. Схема установки для фотометрического титрования:
/ — источник света; 2 — линза; 3 — светофильтр; 4 — кювета;
5 — фотоэлемент; 6 — бюретка; 7 — магнит; 8 — магнитная мешалка;
9 — гальванометр.
ющихся прямых линий, напоминающих собой кривые амперометриче-
ского титрования (см. рис. S9, стр. 425). Пересечение прямых (излом)
соответствует точке эквивалентности.
Для уменьшения влияния разбавления на светопоглощение
применяют концентрированные растворы реагентов.
Фотоэлектрическое титрование с цветным индикатором.
Фотоэлектрическое титрование с цветным индикатором основано на наблюдении
изменения поглощения лучей определенной длины волны цветным
индикатором в точке эквивалентности.
Схема установки для фотоэлектрического титрования приведеца на
рис. 92. Лучи от источника света / (лампа накаливания) направляют
линзой 2 через светофильтр 3 и кювету 4 на фотоэлемент 5. Над кюветой
установлена бюретка 6. Магнитная мешалка 8 дает возможность
перемешивать жидкость в кювете 4. Фотоэлемент связан с гальванометром 9.
Примером фотоэлектрического титрования может служить титрование
щелочи кислотой с /г-нитрофенолом в качестве индикатора. При рН > 7
нитрофенол имеет желтый цвет. При рН <;7 он бесцветен. В этом случае
применяют синий светофильтр. Раствор щелочи и индикатор наливают
в кювету, а кислоту — в бюретку. Титрование ведут обычным способом,
перемешивая раствор при помощи мешалки. Записывают объем
прибавленного раствора кислоты и регистрируют при этом силу фототока
гальванометром. Пока щелочь не будет вся нейтрализована, титруемый
раствор окрашен в желтый цвет. Так как синие лучи сильно поглощаются
желтым раствором, то гальванометр будет регистрировать слабые
фототоки, но как только будет достигнута точка эквивалентности, индикатор
обесцветится, лучи начнут проходить через раствор без поглощения и
сила фототока в фотоэлементе резко увеличится. На графике сила тока —
объем прилитой кислоты получают кривую с сильным перегибом. Этот
перегиб соответствует точке эквивалентности.
^ f
434
ГЛ.IX, СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Обычно в установках для фотоэлектрического титрования применяют
вакуумные фотоэлементы и фотоумножители; фототоки усиливаются
ламповыми усилителями. Это дает возможность применять в качестве
измерительного прибора миллиамперметр. Титрование можно
автоматизировать, если применять реле, которое приводит в действие механизм,
закрывающий и открывающий кран бюретки.
§ 5. Фототурбидиметрическое и фотонефелометрическое
титрование
Метод применяется в тех случаях, когда титруемое вещество дает
муть с титрующим веществом. Прибавление каждой новой порции
титрующего вещества (осадителя) ведет к образованию некоторого количества
осадка. При этом мутность раствора повышается, что ведет к увеличению
поглощения света раствором до достижения точки эквивалентности. При
дальнейшем прибавлении титрующего вещества образование мути
прекращается, мутность понижается вследствие
разбавления, и поглощение света раствором
будет понижено. Максимальная мутность и
максимальное поглощение световых лучей
будут соответствовать точке эквивалентности.
Наблюдение за изменением степени мутности
проводят при помощи специальных
аппаратов — турбидиметров (визуальных или
фотоэлектрических).
'объемстандартногорастбора,мл Схема установки для фототурбидиметри-
ческого титрования напоминает схему, пока-
Рис. 93. Кривая фототурбиди- занную на рис. 92.
метрического титрования. Титруемый раствор ставят между
источником света 1 и фотоэлементом 5. По мере
образования мути освещенность фотоэлемента ослабляется и
соответственно jio мере ослабления силы фототока уменьшается отклонение
стрелки гальванометра. В точке эквивалентности изменение силы
фототока не наблюдается.
Кривая фототурбидиметрического титрования имеет вид, показанный
на рис. 93.
Если фотоэлемент установить над кюветой (перпендикулярно к лучу
света, падающему от источника света), то до начала титрования
гальванометр не реагирует, что указывает на отсутствие фототока. По мере
появления мути частички образующегося осадка рассеивают свет, который
падает на фотоэлемент, и гальванометр начинает реагировать на
воздействие фотоэлектрического тока. В точке эквивалентности наблюдается
максимальное отклонение стрелки гальванометра. Такой способ иногда
называют фотонефелометрическим титрованием.
Г. ЛАБОРАТОРНЫЕ РАБОТЫ
§ 6. Определение содержания ионов железа методом
колориметрического титрования
Колориметрическое определение ионов железа (III) основано на
взаимодействии их с SCN", сопровождающемся образованием железо-
роданидных комплексов, окрашивающих раствор в кроваво-красный
цвет, тем более интенсивный^ чем больше Fe+++ содержалось в растворе.
К недостаткам этого метода относится то, что в зависимости от содержа-
§ 7. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ТИТАНА
435
ния SCN" в растворе состав комплексов может быть различным (см.
книга I, гл. IV, § 25).
Эта реакция весьма чувствительна и позволяет определять железо
при содержании его в растворе 10 "7 г/мл.
Если к раствору, содержащему роданид железа, прибавить немного
изоамилового спирта и смесь тщательно взболтать, роданид железа
растворится в спирте. Так как изоамиловый спирт с водой не смешивается,
окрашенный слой всплывает на поверхность. Ввиду малого количества
прибавленного изоамилового спирта концентрация роданида железа
в нем значительно выше, чем во всем водном растворе, и поэтому при коло-
риметрировании спиртового раствора чувствительность метода
увеличивается.
Необходимые реактивы. Для выполнения определения требуются следующие реактивы:
Стандартный раствор соли железа, содержащий 0,1 мг ?е+++/мл.
Роданид аммония, 25%-ный раствор.
Азотная кислота (пл. 1,15 г/см3),
Изоамиловый спирт.
Серная кислота (пл. 1,84 г/см3).
Приготовление стандартного раствора соли железа.
Навеску 0,864 г химически чистых железо-аммонийных квасцов растворяют в воде,
подкисляют 4—5 мл серной кислоты, дистиллированной водой доводят объем раствора до 1 л
и тщательно взбалтывают. Из полученного раствора отбирают 10 мл и в мерной колбе
емкостью 100 мл разбавляют дистиллированной водой. В 1 мл приготовленного таким
образом раствора содержится 0,1 мг железа. Раствор пригоден только в день изготовления.
Ход определения. В стакан емкостью 100 мл отбирают пипеткой
3—5 мл анализируемого раствора, прибавляют 50 мл дистиллированной
воды и 0,5 мл азотной кислоты, кипятят 2 мин и затем охлаждают.
Полученный раствор переносят в мерный цилиндр емкостью 100 мл с притертой
пробкой, прибавляют 5 мл 25%-ного раствора NH4SCN и 10 мл
изоамилового спирта и тщательно взбалтывают.
В другом таком же цилиндре готовят контрольный раствор. Для этого
в цилиндр наливают 50 мл дистиллированной воды, прибавляют 5 мл
25%-ного раствора NH4SCN и 10 мл изоамилового спирта и также
тщательно взбалтывают. К этому раствору, содержащему все употребляемые
реактивы, но не содержащему железа, добавляют из микробюретки по
каплям, при энергичном перемешивании, стандартный раствор соли
железа. Титрование ведут до тех пор, пока окраска спиртового слоя
контрольного раствора не сравняется с окраской спиртового слоя в цилиндре
с анализируемым раствором.
Расчет. Зная количество добавленного из микробюретки стандартного
раствора соли железа, можно вычислить содержание железа в
анализируемом растворе. Расчет проводят по формуле:
cFe=-ppiooo
где V — объем стандартного раствора соли железа, добавленного из микробк>ретки, мл;
TFe — титр стандартного раствора соли железа, г/мл;
Уг — объем испытуемого раствора, взятого для колориметрирования, мл.
§ 7. Определение содержания титана
Колориметрическое определение малых количеств титана основано
на реакции ионов титана с перекисью водорода, сопровождающейся
образованием комплексного катиона ШО (Н202)2]++» окрашенного в желтый
цвет.
436 ГЛ. IX. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
Эта реакция очень чувствительна и позволяет определять титан при
его концентрации до 10 '5 г/мл.
Определению мешают фториды, фосфаты и окрашенные ионы,
например железа, меди, кобальта, никеля и некоторых других элементов.
Необходимые реактивы. Для выполнения определения требуются следующие реактивы:
Стандартный раствор сульфата титана, содержащий 0,1 мг/мл ТЮ2.
Серная кислота, 2 н. и разбавленный (1 : 1) растворы.
Перекись водорода, 3%-ный раствор.
Пиросульфат калия.
Фосфорная кислота (пл. 1,7 г/см3).
Приготовление стандартного раствора сульфата
титана. Навеску 0,1 г двуокиси титана х. ч. сплавляют в тигле с 4—5 г пиросульфата
калия. Полученный плав растворяют в серной кислоте (1 : 1) и затем раствор разбавляют
дистиллированной водой до 1 л с таким расчетом, чтобы получить 2 н. раствор в отношении
серной кислоты. Полученный раствор тщательно взбалтывают. 1 мл приготовленного таким
образом раствора соответствует 0,1 мг Ti02.
Методика определения. В мерной колбе емкостью 25 мл к исследуемому
раствору, содержащему ионы титана, прибавляют несколько капель
3%-ного раствора перекиси водорода и доводят объем содержимого колбы
Рис. 94. Общий вид фотоэлектрического колориметра ФЭК-М:
/ — гнезда; 2 — светофильтры; 3, 6 — винты для фокусировки лампы накаливания; 4 —- крышка
отделения для светофильтров; 5 — лампа накаливания; 7 — крышка отделения для кювет; 8 —
рукоятка для закрывания шторок; 9, 16 — линзы; 10 — держатель кювет; // — корректор; 12 —
арретир гальванометра; 13 — барабан; 14 — шкала для отсчетов; 15 — ручка для смены
светофильтров; 17 — ручка для тонкой настройки; 18 — ручка для грубой настройки; 19 —
переключатель; 20 — гнезда для включения гальванометра.
до метки 2 н. раствором серной кислоты. Затем измеряют оптическую
плотность (D) раствора при помощи фотоколориметра ФЭК-М (рис. 94),
применяя светофильтр с областью пропускания 400—500 ммк. Можно
также проводить определение путем сравнения окраски полученного
раствора с окраской эталонного раствора в колориметре погружения или
пользоваться методом колориметрического титрования.
Рекомендуется избегать большого избытка перекиси водорода,
разлагающейся с образованием кислорода; при этом пузырьки газообразного
кислорода оседают на стенках кювет, что приводит к искажению
оптической плотности раствора.
§ 7. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ТИТАНА 437
В присутствии ионов железа (III) к исследуемому и эталонному
растворам добавляют по 5 капель фосфорной кислоты с целью маскирования
ионов железа, мешающих определению титана.
Содержание титана находят по калибровочной кривой, построенной
по серии эталонных растворов, приготовленных из исходного стандартного
раствора.
Расчет. Зная количество двуокиси титана А, найденное по
калибровочной кривой, можно вычислить в процентах содержание титана в
пересчете на Ti02:
А-100
где а — навеска, мг.
ГЛАВА X
МЕТОДЫ РАЗДЕЛЕНИЯ, ВЫДЕЛЕНИЯ И
КОНЦЕНТРИРОВАНИЯ ОТДЕЛЬНЫХ КОМПОНЕНТОВ
АНАЛИЗИРУЕМЫХ СМЕСЕЙ
§ 1. Определение следов элементов (микропримесей)
В последнее время с развитием современных отраслей
промышленности и новой техники, потребляющих материалы высокой чистоты,
большое значение приобрели методы определения следов элементов
(микропримесей). Поэтому возросли требования в отношении точности и
чувствительности методов, применяемых для анализа особо чистых веществ.
Современная инструментальная техника позволяет обнаруживать
или определять небольшие количества веществ. Например, с помощью
камеры Вильсона и счетчика Гейгера—Мюллера можно обнаружить
около 10~10 г вещества. Масс-спектрометрия дает возможность определять
10~8—10~6 мкг примесей. Кинетическими методами можно открывать
в 1 мл раствора 10"19 моль, или 60 000 молекул, а в случае использования
техники ультрамикроанализа — определять еще меньшие их количества.
Область концентраций микропримесей (в %), определяемых другими
методами, составляет:
Эмиссионная спектрография .... 10_б—10~4 (например, В—10"6)
Фотометрия и колориметрия .... 4-10"7—10"4 (например, А1—10"6)
Фотометрия пламени 10"6—10"б (например, Са—10~в)
Люминесцентный анализ 10"6—10"б (например, Ga—10"в)
Полярография 10"8—10"6 (например, In—10"6)
Активационный анализ 10"9—10"8 (например, Аи—10"9)
Химико-аналитическая работа с веществами особо высокой чистоты
связана с неизбежными потерями анализируемого вещества и его
загрязнением, что в конечном счете приводит к неминуемым ошибкам
аналитического определения. Важнейшими источниками загрязнений являются
дистиллированная вода, лабораторный воздух и реактивы.
Дистиллированная вода содержит примеси соединений mhofhx химических элементов
до 10"4—10~2%. Поэтому обычная дистиллированная вода совершенно
непригодна для определения следов элементов. Для получения воды,
содержащей допустимые количества примесей (10'8—10~7%), применяют
специальные методы ее очистки: многократную перегонку в кварцевой
или полиэтиленовой посуде, деионирование с помощью ионитов (катио-
нитов и анионитов) и др. Работа с особо чистыми веществами в атмосфере
загрязненного лабораторного воздуха также недопустима, так как в
воздухе находятся соединения различных химических элементов (Na, Mg,
Са, Fe, Си, Zn, Al, S, С и др.). Поэтому при анализе особо чистых веществ
химик-аналитик должен работать в особо чистых лабораториях с
нагнетаемым в них очищенным от посторонних примесей воздухом. Реактивы
также должны подвергаться специальным методам очистки.
§ 2. МЕТОД ОСАЖДЕНИЯ МАЛОРАСТВОРИМЫХ СОЕДИНЕНИЙ 439
При химико-аналитической работе с металлами и неметаллами особой
чистоты требуется применение специальных устройств, позволяющих по
возможности избежать потерь и загрязнений анализируемых веществ.
Для определения микропримесей, а также макроколичеств, очень
часто прибегают к разнообразным методам выделения и
концентрирования следов определяемых элементов. Для этого применяют следующие
методы:
1) осаждение малорастворимых соединений;
2) электрохимическое разделение;
3) экстрагирование;
4) отгонка летучих соединений;
5) хроматографическое разделение;
6) флотация и другие методы.
§ 2. Метод осаждения малорастворимых соединений
Теория и практика метода осаждения подробно изложены при
изучении качественного анализа (см. книга 1, гл. I и в гл. IV и V
настоящей книги).
В химическом анализе осаждение малорастворимых соединений
применяют:
а) для разделения отдельных компонентов сложных смесей;
б) для обнаружения катионов и анионов;
в) для количественного определения составных частей
индивидуального вещества или смеси веществ.
Осадители. В качестве осадителей для разделения и выделения
отдельных компонентов анализируемых смесей применяют разнообразные
химические соединения. Главнейшими из них являются: сероводород,
осаждающий в виде сульфидов ионы V, IV и частично III аналитических
групп (см. книга 1, гл. VI—VIII), а также разлагающий при
определенных значениях рН анионы AsOg , AsO™, VO~, MoO^", WO^" и др.
(см. книга 1, гл. XII); водный раствор аммиака, осаждающий катионы
бериллия, железа (III), алюминия, таллия, галлия, индия, ниобия,
тантала, урана, редкоземельных металлов и др.; фосфаты щелочных
металлов и аммония; ацетат натрия; едкие щелочи; сульфид аммония и т. д.
Коллекторы. Особым случаем осаждения является использование
коллекторов (см. книга 1, гл. III, § 14), применяемых для отделения малых
количеств (следов) определяемого вещества от основного компонента.
Действие коллектора основано на использовании явления соосаждения
определяемых примесей, осаждающихся вместе с коллектором.
При осаждении наблюдается очень интересное явление,
заключающееся в том, что вместе с осаждаемым веществом в осадок выпадают
некоторые соединения, относящиеся к группе хорошо растворимых
веществ и сами по себе не осаждающиеся данным реактивом.
После того как осадок такого рода образовался, соосажденные примеси
не удается отмыть дистиллированной водой от осадка нерастворимого
в воде вещества.
Явление соосаждения очень часто мешает химику-аналитику получить
осадок требуемой чистоты. Однако это явление оказывает большую пользу
там, где им пользуются для отделения и определения микроэлементов,
содержащихся в ничтожно малых количествах в составе анализируемых
природных или технических объектов.
С целью выделения в осадок ионов микроэлементов, в анализируемом
растворе создают требуемые условия для образования осадка другого
соединения, с которым во время его формирования соосаждаются микро-
440 гл- X. РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ КОМПОНЕНТОВ СМЕСЕЙ
элементы. Осадок, соосаждающий следы определяемых элементов из их
разбавленных растворов, называют коллектором.
Примерами осаждения на коллекторе могут служить методы отделения
примесей от Al + + + , Cu++ и др. Содержание примесей часто столь мало,
что обычные методы осаждения при помощи наиболее чувствительных
реагентов не приводят к желаемым результатам. Применяя коллектор,
можно количественно осадить эти примеси. Например при осаждении
из раствора примесей, содержащихся в металлической меди, в качестве
коллектора применяют гидроокись и оксикарбонат железа (III),
образующийся при действии Na2C03 на раствор, содержащий Fe+++.
В процессе образования гидроокиси железа все примеси
неблагородных металлов, входящих в состав анализируемого образца меди, соосаж-
даются на коллекторе. Осаждение ведут из азотнокислого раствора,
получаемого растворением рассчитанной навески исходной меди в HN03,
к которому предварительно прибавляют немного раствора соли железа
(III). Затем в полученном осадке определяют содержание примесей
обычными химическими, физическими и физико-химическими методами.
При соосаждении следов катионов в процессе осаждения коллектора
[Fe (ОН)3, А1 (ОН)3 и т. п. ] они количественно увлекаются в осадок с
гидроокисью, если следовые количества катионов не связаны в виде
комплексных соединений и если рН раствора на 2—3 единицы больше рН
осаждения их гидроокисей. При этом наблюдается количественное сооса-
ждение посторонних катионов независимо от того, достигнута или не
достигнута величина ПР их гидроокисей. В качестве коллектора, как
правило, можно брать гидроокись любого другого металла. То же самое
относится и к сульфидам металлов, используемых в качестве коллекторов.
Микрокомпонент с коллектором образует твердый раствор или
замещает катионы макрокомпонента в структуре осадка на катионы
микрокомпонента. При этом микрокомпонент, по-видимому, распределяется
по всему объему осадка.
Органические осадители. За последние годы в количественном
анализе широко применяются органические реагенты (см. книга I, гл. VI,
§ 18). Они используются для приготовления стандартных (титрованных)
растворов, как индикаторы, маскировочные средства, осадители и сооса-
дители и т. д.
Преимущества органических осадителей. Органические осадители
отличаются рядом преимуществ по сравнению с обычными неорганическими
осадителями.
1. Пользуясь органическими осадителями, можно осаждать и
разделять различные элементы из очень сложных смесей. Например, при помощи
диметилглиоксима возможно количественное осаждение катионов никеля
в присутствии многих других катионов.
2. Осадки, получающиеся с органическими осадителями, хорошо
отфильтровываются и промываются (например, осадки комплексных
соединений катионов, содержащих в качестве лигандов пиридин или
другие органические соединения). Это дает возможность легко отмывать от
осадков примеси, содержащиеся в анализируемом растворе.
3. Осадки, получающиеся при действии на катионы или анионы
органических осадителей, отличаются большим молекулярным весом.
Вследствие этого точность анализа повышается. Например, определение магния,
алюминия и других катионов проводится с большой точностью осаждением
их в виде оксихинолятов, обладающих большим молекулярным весом.
4. В составе осадков, являющихся соединениями неорганических
веществ с органическими компонентами, обычно содержится мало соосаж-
дающихся иримесей.
2. МЕТОД ОСАЖДЕНИЯ МАЛОРАСТВОРИМЫХ СОЕДИНЕНИЙ 441
Применение органических осадителей в количественном анализе.
В количественном анализе для осаждения и разделения многих катионов
применяют дитизон, купферон, пиридин, 8-оксихинолин, фениларсо-
новую кислоту как высокоизбирательный осадитель для ниобия и тантала;
диэтилдитиофосфорную кислоту и ее аналоги для осаждения и отделения
ряда элементов; 6- или 8-метилхинальдин для осаждения вольфрама;
1,8-аминонафталинсульфонат магния, осаждающий ионы натрия в
присутствии ионов калия, и многие другие.
Опыт показывает, ч^го изменяя рН раствора, очень часто можно
провести последовательное осаждение и разделение различных катионов при
помощи одного и того же органического осадителя. Так, например,
купферон из сильнокислых растворов осаждает только ионы ниобия, тантала,
титана, циркония, ванадия, железа (III), олова и позволяет отделять их
от неосаждающихся в тех же условиях ионов алюминия, хрома, урана
(VI), бериллия, марганца, никеля, кобальта, цинка, фосфора, бора.
Пользуясь растворимостью в органических растворителях некоторых
осадков, получаемых с помощью органических осадителей, можно
экстрагировать их (см. § 4).
Соосаждбние — один из способов концентрирования элементов. За
последнее время предложен ряд новых органических реагентов —
органических соосадителей, соосаждающих [подобно указанным выше
неорганическим соосадителям: гидроокиси и оксикарбонату железа (III)] следы
элементов при их содержании до 10~б—10~8%.
Органические соосадители представляют собой осадки
малорастворимых органических соединений, образующихся при взаимодействии
органических катионов или анионов (которые находятся в анализируемом
растворе) с солями тяжелых органических ионов противоположного
знака (которые вводятся в анализируемый раствор в процессе соосаждения).
С этой целью применяют метиловый фиолетовый, родамины и другие
соединения (поставляющие катионы) и нафталин-(3-сульфокислоту,
метиловый оранжевый и другие соединения (поставляющие анионы).
Органические соосадители отличаются от неорганических и
смешанных коллекторов (образованных ионами металлов при действии
органических осадителей и комплексообразователей) некоторыми преимуществами.
Прежде всего следует отметить легкость разложения органической части
коллектора при сжигании и озолении осадка, что позволяет получить
соосажденные элементы в концентрированном состоянии и в чистом виде.
С другой стороны, органические соосадители дают возможность сооса-
ждать следы элементов в присутствии больших количеств других
элементов, что имеет особое значение при анализе объектов сложного
состава.
Таким образом, соосаждение с неорганическими и органическими сооса-
дителями представляет собой один из эффективных способов
концентрирования элементов.
Природа доминирующих процессов при соосаждении. За последнее
время в связи с возрастающим практическим значением соосаждения для
концентрирования элементов уделяется должное внимание разработке
теории соосаждения.
Единой теории по этому вопросу не существует.
Различают несколько видов процессов соосаждения: окклюзия,
адсорбция ионов или соединений соосаждаемого элемента на поверхности сооса-
дителя, образование твердых растворов между компонентами, образование
соосаждаемым соединением элемента центров кристаллизации, на которых
происходит отложение выпадающего соосадителя, ионообменные
процессы и т. п.
442 ГЛ. X. РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ КОМПОНЕНТОВ СМЕСЕЙ
По-видимому, природа доминирующих процессов, которыми
обусловливается захватывание микроэлементов осадками, зависит от многих
факторов и способа концентрирования.
За последнее время расширен ассортимент органических соосадителей.
Найдены соосадители, пригодные для растворов, характеризующихся
высокой кислотностью; предложены индифферентные соосадители, к их
числу относятся органические вещества, не вызывающие непосредственное
осаждение элементов и не содержащие в своих молекулах активных
атомных группировок, реагирующих с элементами. Примерами могут
служить <х-х (3-нафтолы, 2,4-динитроанилин, дифениламин и др. Указанные
соосадители позволяют полностью извлекать ультрамалые количества
элементов из чрезвычайно разбавленных растворов.
Комбинирование такого рода соосадителей с осаждением элементов
в присутствии обычных органических соосадителей позволяет полностью
извлекать уран при соосаждении его в виде 8-оксихинолята 2,4-динитро-
анилином. Кроме 8-оксихинолятов соосаждаемыми формами могут
служить также комплексные и внутрикомплексные соединения. Соосаждае-
мый элемент отделяют от органической части озолением.
Особенный интерес представляют неокрашенные органические
соосадители, при использовании которых отпадает надобность в озолении
осадков. Соосаждение может быть комбинировано с последующим
спектральным, полярографическим или иным методом определения.
§ 3. Электрохимические методы разделения
Электрохимические методы разделения основаны на выделении на
инертном электроде определяемого вещества в элементарном состоянии
или в виде нерастворимого осадка, выпадающего при пропускании через
анализируемый раствор постоянного электрического тока.
Теория и практика электрохимических методов разделения подробно
изложены в гл. VI.
В настоящее время электрохимические методы применяются для
разделения соединений большинства химических элементов и оказались
очень удобными вследствие того, что они не требуют введения в
анализируемый раствор посторонних веществ. Используя различные способы
электрохимического осаждения с применением платиновых или других
электродов и ртутного катода, а также внутреннего электролиза (см.
гл. VI, § 5), можно разделять катионы алюминия, титана, циркония,
ванадия, урана от катионов хрома, железа, кобальта, никеля, цинка,
меди, серебра, кадмия, германия, молибдена, олова, висмута и других
элементов. Можно также отделять примеси от основных компонентов при
анализе цветных металлов, их сплавов и руд.
Разделение сложной смеси, состоящей из различных
электролитически осаждаемых ионов, достигают либо подбором соответствующего
электролита, либо проведением электролиза с автоматическим контролем
электродного потенциала, при котором ведется электроосаждение.
Осаждение на ртутном, катоде. Особым видо& электролитического
разделения является осаждение на ртутном катоде. При этом металлы
под действием электрического тока выделяются на ртути с образованием
амальгам. Вследствие этого выделение и разделение многих металлов
происходит быстро и количественно. Некоторые металлы (например, Fe),
не выделяющиеся на твердом катоде, количественно выделяются при
электролизе на ртутном катоде. Однако ряд металлов (например, А1) не
осаждается на ртутном катоде.
§ 3. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ РАЗДЕЛЕНИЯ
443
Электролиз с ртутным катодом проводят в специальных приборах
(рис. 95). Для этого в сосуд, в котором проводят электролиз, вносят около
200 г металлической ртути, наливают анализируемый раствор и ведут
электролиз при плотности тока около 0,15 а/см2. Анод укрепляют у
поверхности раствора. В процессе электролиза ртуть перемешивают. По
окончании электролиза проба раствора должна давать отрицательную реакцию
на отделяемые элементы.
Не прерывая тока, раствор сливают со ртути и ртуть промывают водой.
Промывные воды присоединяют к основному раствору. Полученный
раствор, свободный от элементов, выделенных
на ртутном катоде подвергают анализу. Если
нужно извлечь из ртути осажденные в ней
металлы, амальгаму растворяют в кислотах
или отгоняют ртуть.
При помощи ртутного катода можно
отделить:
а) алюминий от железа, цинка, меди;
б) титан от индия, рения, молибдена,
германия;
в) ванадий от молибдена;
г) соединения фосфора и мышьяка от всех
выделяемых на ртутном катоде элементов
(железа, цинка, кобальта, никеля, хрома, серебра,
меди, кадмия, ртути, олова, платины, золота
и др.).
Так, например, метод количественного
определения алюминия в присутствии ионов железа
и других элементов, основанный на выделении
железа электролизом на ртутном катоде, состоит в следующем. Сначала
выделяют железо из сернокислого раствора на ртутном катоде; вместе
с железом выделяются другие элементы: цинк, хром, никель, кобальт
и т. д. В растворе остаются ионы алюминия, бериллия, титана, фосфора
и т. п. Затем определяют обычным путем ионы алюминия, которые
осаждают из фильтрата оксихинолином или купфероном в аммиачном или
слабоуксуснокислом растворе. Титан осаждают в кислом растворе
купфероном.
Электрохимические методы могут быть использованы не только для
разделения и выделения элементов, но и для концентрирования
определяемых веществ.
Такого рода концентрирование (накопление) определяемого элемента
на стационарной капле ртути имеет место в методе амальгамной
полярографии. Сущность метода заключается в том, что путем электролиза в
течение некоторого времени при потенциале предельного тока определяемого
металла проводится его концентрирование в виде амальгамы из
разбавленного раствора на стационарной капле ртути, которая выполняет роль
ртутного микроэлектрода. Затем при линейно изменяющемся напряжении
регистрируется кривая анодного растворения амальгамы. При этом на
полярограмме получаются анодные пики, положение которых по
потенциалу характеризует природу вещества, а высота пика — концентрацию
примеси.
Рис. 95. Ртутный катод.
Метод предложен Баркером и исследовался Шейном, Кемуля, А. Г. Стромбергом,
Е. Н. Виноградовой, Л. Н. Васильевой, С. И. Синяковой и др. Большую теоретическую
и практическую ценность для амальгамной полярографии с предварительным
концентрированием (накоплением) на стационарной капле ртути представляют работы М. Т.
Козловского в области исследования металлических амальгам.
444 ГЛ. X РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ КОМПОНЕНТОВ СМЕСЕЙ
Сочетание электрохимического концентрирования определяемых
элементов на стационарной капле ртути с последующей анодной
поляризацией полученной амальгамы дает возможность на несколько порядков
повысить чувствительность метода по сравнению с обычным методом
полярографии. Это имеет важное практическое значение для анализа особо
чистых металлов, химических реактивов и полупроводниковых материалов.
Существует несколько вариантов концентрирования микропримесей
порядка 10"6—10"8% электрохимическим (полярографическим) методом.
Общим принципом их является накопление определяемых элементов
путем предварительного электролиза на стационарном электроде.
С целью электрохимического концентрирования разработаны методики
амальгамной полярографии для определения 10 ~3—10"7% примесей
в цинке, алюминии, индии, олове, мышьяке, галлии, в урановых солях,
в химически чистых реактивах, в биологических объектах, пищевых
продуктах и т. п.
§ 4. Метод экстрагирования
Метод экстрагирования основан на извлечении веществ при помощи
органических растворителей. Главным условием успешного разделения
анализируемых веществ методом экстрагирования являете^
растворимость извлекаемого вещества в органическом растворителе. При этом не
имеет значения, растворяется экстрагируемое вещество в воде или нет.
Очень часто в аналитической практике экстрагируемое вещество
образуется в результате реакции, протекающей в водной среде, при
взаимодействии определяемых ионов преимущественно с органическими
реагентами. В этом случае органический растворитель не должен смешиваться
с водой. Метод экстрагирования дает возможность выделять очень малые
количества веществ из больших количеств анализируемых продуктов.
Экстракция органическими растворителями широко используется
с целью не только разделения, но и концентрирования элементов.
Соединения концентрируемых элементов экстрагируются органическими
растворителями; иногда экстракционное концентрирование проводят путем
экстракции соединений основного элемента, а примеси остаются в водной
фазе.
В ряде случаев концентрирование примесей методом экстракции имеет
преимущества перед другими методами концентрирования. Так,
например, сравнение чувствительности метода экстракции и соосаждения
показало, что экстракция является более эффективной.
Особый интерес представляет сочетание метода экстракции с
последующими методами определения (спектральными, радиометрическими,
полярографическими и др.). Среди них наибольшее значение представляют
экстракционно-фотометрические методы анализа, отличающиеся большой
чувствительностью и избирательностью. В этих методах процесс
получения окрашенного раствора сочетается с одновременно происходящим
экстракционным отделением определяемого вещества, благодаря чему
весь ход определения оказывается несложным.
§ 5. Методы отгонки летучих соединений
В тех случаях, когда определяемая составная часть анализируемой
смеси отгоняется при нагревании, широко применяют метод отгонки.
Анализируемую смесь нагревают и улавливают или поглощают
отгоняемый летучий компонент.
В некоторых случаях отгоняемый компонент не улавливают и не
поглощают, а, закончив отгонку, взвешивают остаток. В тех случаях,
§ 6. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ РАЗДЕЛЕНИЯ 445
когда определяемое вещество нелетуче, его превращают в летучее
соединение. Для разгонки смесей органических соединений применяют
лабораторные ректификационные колонки (рис. 96).
Простейшим случаем метода отгонки является выделение воды из
твердого вещества, происходящее при нагревании в сушильном шкафу.
Выделяющиеся пары воды
улавливают прокаленным или плавленым
хлоридом кальция или безводным
перхлоратом магния.
Методом отгонки отделяют:
а) бор в виде летучих эфиров
борной кислоты, например В (ОСН3)3;
б) мышьяк в виде AsCl3; в)
германий в виде GeCl4; г) железо в
виде FeCl3; д) ртуть в виде Hg и т. д.
Метод отгонки также
применяют при определении ванадия,
вольфрама, молибдена, олова,
сурьмы, иода, фтора, осмия, серы,
селена, теллура, кремния и
других элементов.
Отгонка SiF4. В качестве
примера рассмотрим метод
отгонки SiF4. Для химика-аналитика
представляет особый интерес
отделение кремневой кислоты от
других сопутствующих ей соединений
(ТЮ2 и др.). Количественное
определение содержания Si02 в
силикатах основано на выделении
в нерастворимом состоянии
кремневой кислоты.
В процессе осаждения
кремневой кислоты при анализе
природных и искусственных силикатов
в осадок увлекаются примеси
соединений титана, железа,
щелочных и щелочноземельных
металлов и др.
После прокаливания H2Si03
до Si02 осадок обрабатывают
в платиновом тигле фтористоводородной кислотой в присутствии
нескольких капель серной кислоты. При выпаривании смеси досуха Si02
превращается в SiF4 и улетучивается. Окислы других элементов образуют
нелетучие соединения. По разности массы до обработки осадка
фтористоводородной кислотой и после обработки и прокаливания вычисляют
содержание кремневой кислоты в силикатах.
§ 6. Хроматографические методы разделения
Принцип и классификация хроматографических методов анализа
описаны ранее (см. книга 1, гл. III, § 10).
Хроматографические методы анализа широко применяют в
количественном анализе для разделения и концентрирования отдельных
компонентов сложных смесей неорганических и органических соединений.
Рис. 96. Схема лабораторной
ректификационной колонки, применяемой для разгонки
смесей органических веществ:
/ — конденсатор; 2 — кран для отбора
дистиллята; 3 — холодильник; 4— специальный алонж;
5 — хлоркальциевая трубка; 6 — приемник;
7 — трубка для термометра; 8 — соединительная
трубка; 9 — верхний счетчик капель (счетчик
орошения); 10 — центральная трубка с насадкой;
// — обогревающая трубка-рубашка; 12 —
изолирующая трубка-рубашка; 13 — расширение
центральной трубки с сетчатым колпачком;
14—нижний счетчик капель; 15 — хвостовик; 16 — колба
(куб); 17 — колбонагреватель; 18, 19 — реостаты;
20, 21 — амперметры.
446 ГЛ. X. РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ КОМПОНЕНТОВ СМЕСЕЙ
Выделенные компоненты определяют обычными химическими,
физическими и физико-химическими методами анализа. Наиболее широко в
количественном анализе применяют ионообменную хроматографию для
разделения составных частей анализируемых веществ:
а) отделения катионов от некоторых анионов;
б) разделения катионов;
в) разделения анионов;
г) выделения примесей и получения химически чистых препаратов;
д) определения общей концентрации электролитов в растворе;
е) концентрирования ионов микропримесей из растворов и т. д.
Рис. 97. Адсорбционные колонки:
/ — воронка; 2 — конечный уровень жидкости; 3 —
адсорбент; 4 — пористый фильтр; 5 — напорный сосуд; 6 —
раствор; 7 — стеклянная вата.
Примеры отделения, разделения и идентификации катионов и анионов
рассмотрены при изучении качественного анализа. Ниже приведено
несколько других примеров применения ионообменной хроматографии
в количественном анализе.
Определение общей концентрации электролитов в растворе. Это
определение основано на том, что при пропускании анализируемого раствора
через катионит в Н-форме выделяется кислота в количестве,
эквивалентном содержанию соли в растворе, по схеме:
R — Н + KtAn -+ R - Kt + HAn
Количество выделившейся кислоты определяют титрованием щелочью.
Определение концентрации соли в растворе.
Катионит марки СБС (около 15 г), помещенный в стеклянную колонку
диаметром 2 см и высотой 30 см (рис. 97), переводят в Н-форму
пропусканием через него 100 мл 2 н. хлористоводородной кислоты со скоростью
2 капли в секунду. Скорость вытекания жидкости из колонки регулируют
с помощью крана. Кислоту вливают в колонку отдельными порциями по
10—15 мл. Затем катионит тщательно отмывают от кислоты
дистиллированной водой до нейтральной реакции фильтрата по метиловому
оранжевому. Отмытый катионит готов для определения концентрации соли.
Определение проводят следующим образом. Анализируемый
приблизительно 0,1 н. раствор соли (20—25 мл, отобранные пипеткой) помещают
в колонку и пропускают раствор через катионит. Скорость пропускания
$ 6. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ РАЗДЕЛЕНИЯ 447
жидкости через катионит — 2 капли в 1 сек. Фильтрат собирают в
коническую колбу. Затем через катионит пропускают дистиллированную воду,
вливая новую порцию воды тогда, когда уровень жидкости в колонке
достигнет поверхности катионита. Воду пропускают через катионит до
тех пор, пока капля фильтрата, взятая на часовое стекло, не будет иметь
нейтральную реакцию по метиловому оранжевому. Все промывные воды
тщательно собирают в ту же коническую колбу, что и фильтрат.
Содержимое конической колбы оттитровывают 0,1 н. раствором NaOH с
индикатором метиловым оранжевым. Определение проводят три раза.
После окончания определения катионит регенерируют. Для этого
через колонку с катионитом пропускают 100 ж2н. хлористоводородной
кислоты для извлечения поглощенных катионов, затем отмывают катионит
от кислоты дистиллированной водой.
Количество соли (g) в анализируемом объеме раствора вычисляют по
формуле:
^ЫаОН^ЫаОНЭсоли
8~ 1000
где V'jsjaOH — объем щелочи, пошедшей на титрование, мл;
NNaOH — нормальность раствора NaOH;
Эсоли — грамм-эквивалент соли.
Определение свободной кислоты в растворах различных солей.
Свободную кислоту в растворе соли вычисляют по разности между
суммарной кислотностью, которую определяют титрованием элюата, йолучен-
ного при пропускании раствора соли через катионит в Н-форме, и
кислотностью, соответствующей по расчету содержанию катиона в растворе
соли, определяемому заранее весовым или объемным методом.
Концентрирование ионов из разбавленных растворов. При
пропускании через катионит большого количества разбавленного раствора
катионит поглощает катионы из раствора. При промывании катионита
разбавленной соляной кислотой наблюдается вытеснение поглощенных
катионов в фильтрат. При этом получается небольшой объем раствора,
содержащего все катионы. Таким образом, можно провести
концентрирование катионов из разбавленных растворов. На этом основан метод контроля
качества конденсата пара при помощи катионита.
Разделение катионов, основанное на использовании амфотерных
свойств. Для разделения катионов раствор, содержащий смесь
амфотерных и неамфотерных катионов, пропускают через катионит в Н-форме.
Затем промывают катионит раствором щелочи (NaOH или КОЙ). В
результате обменной реакции поглощенные катионы вытесняются из катионита.
При этом катионы неамфотерных соединений образуют со щелочью
гидроокиси, осаждающиеся на зернах катионита, а катионы амфотерных
соединений образуют в избытке щелочи анионы и проходят в фильтрат.
Так, например, можно отделить алюминий, цинк, молибден, сурьму,
вольфрам от железа, меди и др. Метод отделения амфотерных катионов
имеет существенное преимущество вследствие устранения соосаждения
амфотерных катионов с осадком гидроокисей катионов.
Разделение катионов, основанное на использовании комплексообра-
зования. Возможны два варианта разделения.
1. Раствор, содержащий смесь катионов, пропускают через катионит.
В результате обменной реакции катионы, содержащиеся в растворе,
поглощаются катионитом. Через катионит пропускают раствор
электролита, который образует комплексное отрицательно заряженные ионы
с одними из катионов. Другие катионы могут образовывать с раствором
448 ГЛ X- РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ КОМПОНЕНТОВ СМЕСЕЙ
электролита положительно заряженные комплексные ионы или же
оставаться в виде простых ионов.
Катионы, образующие с раствором электролита отрицательно
заряженные комплексные ионы, проходят в фильтрат. Катионы, не
образующие таких комплексных ионов, остаются на катионите.
2. К раствору, содержащему смесь катионов, предварительно
прибавляют вещество, образующее комплексные, отрицательно заряженные
ионы с одними из катионов. Другие катионы при этом не образуют
отрицательных комплексных ионов. При пропускании полученного раствора
через катионит поглощаться будут только ионы, которые находятся
в растворе в виде простых либо в виде комплексных катионов. Ионы,
находящиеся в растворе в виде комплексных анионов, пройдут в фильтрат.
Оба варианта разделения катионов широко применяются в
аналитической химии. В качестве веществ, образующих с катионами комплексные
ионы, используют лимонную, винную, сульфосалициловую кислоты,
пирофосфат натрия, оксалат аммония, глицерин и др.
Для разделения катионов методом комплексообразования используют
как катиониты, так и аниониты.
Методом ионообменной хроматографии можно разделить
редкоземельные элементы, используя различия констант нестойкости их комплексных
соединений.
Разделение катионов можно проводить на анионитах, насыщенных
анионами, способными образовывать с некоторыми катионами
комплексные ионы. Так, при пропускании через анионит в цитратной форме
раствора, содержащего смесь катионов, происходит поглощение анионитом
тех катионов, которые образуют с цитрат-ионами комплексные анионы.
Катионы, не образующие комплексных анионов с цитрат-ионами,
проходят в фильтрат. Таким путем отделяются: Mg++, Ca++, Sr++, Ba++ от
Fe+++, Cu++, Ni++. Катионы Mg++, Ca++, Sr++, Ba++ проходят в
фильтрат, a Fe+++, Cu++, Ni++ образуют с цитрат-ионами комплексные
анионы и прочно удерживаются анионитом.
Определение фтора во фториде алюминия, криолите и плавиковом
шпате. Исходный нерастворимый в воде фторид превращают в
растворимый фторид щелочного металла сплавлением пробы со смесью
карбоната калия и Si02. При выщелачивании плава водой фторид калия
переходит в раствор, а гидроокись и оксикарбонат алюминия и карбонат
кальция остаются в нерастворимом остатке. Раствор фильтруют и пропускают
через катионообменную колонку. Вытекающая из колонки жидкость
содержит HF, которую оттитровывают стандартным раствором щелочи.
Уравнения реакций:
а) реакции сплавления1
A1F3 + 3Si02 + ЗК2СО3—>КзА1 (Si03)3 + 3KF + 3C02
CaF2 + 3Si02 + 3K2C03—>KdCa (Si03)3 + 2KF + ЗСОа
CaF2 + K2C03—>CaC03 + 2KF
Si02 + K2C03—>K2Si03 + C02
б) реакции ионного обмена:
R — H + KF—*R -к + HF
2R — H + K2C03-^2R — К + С02 + Н20
2R — Н + K2Si03—*2R — К + H2Si03
в) реакция титрования:
HF + OH- ^± H20 + F-
§ 7. МЕТОД ФЛОТАЦИИ
449
Сплавляют в платиновом тигле 0,5 г пробы с 5—10 г карбоната калий-
натрия и 1,5 г Si02 и далее поступают так, как описано в гл. V, § 20.
После выщелачивания раствор фильтруют. Фильтрат, содержащий
растворимые фториды, карбонаты и силикаты, пропускают через
ионообменную колонку, наполненную ионитом в Н-форме. Содержимому
колонки дают постоять 15 мин для удаления двуокиси углерода,
образующейся при взаимодействии растворимых карбонатов с ионитом. После
удаления С02 раствору дают стечь и затем титруют его 0,1 н. раствором
NaOH в присутствии бромкрезолового фиолетового. При титровании
желтая окраска раствора становится зеленой, затем голубой и, наконец,
фиолетовой. В этот момент титрование прекращают.
§ 7. Метод флотации
Флотацией называют метод разделения веществ, основанный на
использовании различий физико-химических свойств частиц разделяемых
компонентов. Например, частицы сульфидов металлов гидрофобны, они не
смачиваются водой. Поэтому если поместить в воду тонко измельченную
горную породу, содержащую даже небольшой процент сульфида металла,
то смачиваемые водой частички силикатной породы, имеющие большую
плотность, тонут, а частички извлекаемого (или обогащаемого) сульфида
металла, не смачиваемого водой, не тонут. Если сильно вспенить
разделяемую смесь струей воздуха или другим способом, то частички обогащаемой
минеральной суспензии увлекаются пеной и собираются на поверхности
воды, образуя минерализованную пену. Флотация происходит при
перемешивании мелко измельченной смеси твердых- веществ не только с водой,
но и с маслами, специальными реагентами и др.
Метод флотации дает возможность концентрировать соединения
различных элементов путем отделения сопутствующих им примесей. Методы
селективной (избирательной) флотации позволяют не только отделять
извлекаемые соединения от пустой породы, но и успешно разделять
отдельные соединения. Методом флотации разделяют индивидуальные минералы
полиметаллических руд, обогащают вольфрамовые, оловянные, титановые
и другие концентраты, отделяют друг от друга сульфидные и окисленные
свинцовые, цинковые, медные, молибденовые и другие руды. Все большее
значение приобретают флотационные методы разделения неорганических
солей и органических соединений.
Флотационные реагенты. С целью изменения физико-химических
свойств поверхности минеральных частиц применяют особые органичен
ские и неорганические вещества — флотационные реагенты:
органические кислоты и их соли, растворимые в воде ксантогенаты, дитиофосфаты
и др. С помощью флотационных реагентов удается разделять самые
разнообразные минералы и соли, часто не разделяемые другими способами.
Помимо рассмотренных выше методов, для разделения веществ
используют также диализ, электрофорез, газовую диффузию,
центрифугирование, электромагнитные свойства и т. п., но описание этих методов не
входит в нашу задачу.
* * *
ЛИТЕРАТУРА
Основные справочники, реферативные журналы, основная периодическая литература
и задачники по аналитической химии указаны в списке литературы I тома.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбциометрия нейтронная 440
Автокатализ 227
Автопротолиза константа 59, 198
Агрегация 362
Адсорбция 364, 441
при титровании по методу осаждения 312
Азот, определение 184 ел
Азотистая кислота, определение 254
Активационный анализ 438
Активность
ионов водорода 9С ел., 136
коэффициенты 224
показатель 96, 136
Актор 228
Акцептор 228
Ализарин 152
Ализариновый желтый 149
Алюминий
определение в силикатах 383, 387
— весовое 393
— комплексонометрическое 344 ел.
— объемное 343
отделение 443
Аминоалкилфосфоновые кислоты 331
1,8-Аминонафталинсульфонат магния 441
Аммиак
ионное произведение 198
кривая нейтрализации 126
определение 184 ел.
применение в весовом анализе 373
Аммоний
ванадат 286
оксалат 248
пурпурат 336
роданид 323, 328
соли, определение азота 184 ел.
в смеси с кислотой 208
Амперометрическое титрование 424 ел.
Ампулы для взятия навесок 354
Анализ
активационный 438
бесстружковый метод 26, 36
больших количеств вещества 24
весовой, см. Весовой анализ
воспроизводимость 367
вычисление результатов в весовом
анализе 249 ел.
в объемном анализе 65 ел.
газоволюметри чески й 22
газовый 20, 22
инструментальные ме-годы 23, 94, 395 ел.
кинетические методы 438
колориметрические методы 429 ел.
кулонометрические методы 425 ел.
люминесцентный 3?6, 438
макроколичественный 24
малых количеств вещества 24
масс-спектрометрический 18, 400
методы внутреннего электролиза 395, 403,
406
— изотопного разбавления 397
микроколичественный 24
объемный, см. Объемный анализ
оптические методы 395, 428 ел., 432 ел.
ошибки 26, 54
пересчет на сухое вещество 27
полумикроколичественный 24
Анализ
полумикрообъемный метод 88
полярографический метод 18, 418 ел.
приготовление растворов 36
радиоактивационный 399 ел
радиометрический 397
разделение и выделение компонентов 438 ел,
седиментационный 20
спектральный, см. Спектральный анализ
титриметрический 21
точность 36, 367
ультрамикрометод 25, 438
фазовый 17
физико-химические методы 20, 23, 94,
295 ел.
физико-химический по Н. С. Курнакову 24
физические методы 20, 23, 395
флуоресцентный 396
фотоколориметрический 430 ел.
функциональный 17
химические методы 20
хроматографические методы 19, 395, 397,
445 ел.
электровесовой 348, 395, 401 ел.
электрохимические методы 19, 395, 413 ел.,
442 ел.
эмиссионный 396, 429
Анализаторы автоматические 90
Аналитический множитель 73
в весовом анализе 351
в объемном анализе 73, 168
Ареометры 159
Ауксохромные группы 144
Бария хлорид, определение
кристаллизационной воды 369
Батохромный сдвиг 140
Бензидин 241
Бензойная кислота, титрование 170, 208
Бертолетова соль, анализ 276
Бихроматы, определение 258, 288
Бор, отгонка 445
Бренстеда — Лоури теория 191
Броматометрия 284 ел.
Бромиды, определение 323
Бронза, анализ 17, 390
Бугера — Ламберта — Бера закон 429
Бунзена
клапан 253, 257
уравнение 358
Буферные растворы 106 ел., 126
активность Н+-ионов ПО
концентрация Н+-ионов 109 ел.
Бюксы 27, 158
Бюретки 51 ел., 79
для полу микрометода 88
правила работы 58 ел.
с автоматически устанавливаемым
уровнем 53
Ванадатометрия 285 ел.
Ванадий, осаждение 441, 443
Весовая форма 348, 361, 368
получение 359
Весовой анализ 20, 347 ел.
аналитические множители 351
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
451
Весовой анализ
вычисление результатов 349 ел
классификация методов 347
методы вычисления 347
неизвестного вещества 368
обработка осадков 356 ел.
осаждения методы 348 ел.
отгонки методы 348
реактивы 361 ел., 392
теоретические основы 361
техника работы 353 ел.
точность 366
требования к осадкам 363 ел.
условия 367 ел.
Весы 27 ел.
аналитические 28 ел.
— правила обращения 35
демпферные 29, 34
лабораторные 27
микроаналитические 427
нулевая точка 31
одночашечные 35
полумикроаналитические 27
пробирные 27
равновесия точка 30 ел.
седиментационные 20
специальные 28
технические 28
чувствительность 30
цена деления шкалы 30
центр колебания стрелки 31
Взвешивание 27 ел., 31 ел., 50
метод качаний 32
подготовка вещества 26
Вильсона камера 438
Вместимость 49
Внутреннего электролиза метод 359, 403, 406
Внутренний стандарт 428
Вода
ионное произведение 198
определение в органических
растворителях 204
— в силикатах 379
— жесткости 183, 342
— кристаллизационной 369
— по Фишеру 204, 277
— хлора 328
очистка 438
Водорода ионы
активность 96 ел.
концентрация, влияние на полноту
осаждения 367
— — на растворимость электролитов 300
Водородная ошибка 154, 156
Волна полярографическая 419 ел.
Воспроизводимость анализа 367
Восстановители 250 ел.
Восстановление катодное 402
Выделения методы 347, 352
Высокочастотное титрование 396, 415
Вычисления
в весовом анализе 349 ел.
в объемном анализе 65 ел.
степень точности 36, 367
Денсиметры 159
Джонса редуктор 256
Диализ 449
я-Диметиламиноазобензолсульфокислота,
натриевая соль 146
Диметилглиоксим 286
Дисперсия выборочная 84
Дитизон 441
Дифениламин 242 ел., 286
Дифениламинсульфокислота 242
Дифенилбензидин 242 ел.
Дифенилгуанидин 161
Дифенилкарбазон 292, 326, 328 ел.
Диффузия газовая 449
Диэтилдитиофосфорная кислота 441
Добавок метод 423
Доломит, анализ 388 ел.
Дублирования метод 430
Едкий натр
кривая кондуктометрического титрования
414
определение при совместном присутствии
с Na2C03 179
приготовление растворов 170 ел.
установка титра 170 ел.
Едкое кали, спиртовой раствор, титрованный
207
Емкость 49 ел.
Железо
двухвалентное
определение ванадатометрическое 286
— кулонометрическое 427
— перманганатометрическое 235 ел.,
252
— хроматометрическое 282
— цериметрическое 231
металлическое, определение 253
трехвалентное
восстановление 256
определение весовое 373
— в бронзах и латунях 391
— в силикатах 383, 386
—• бихроматометрическое 282
— колориметрическое 434
— перманганатометрическое 256
осаждение 441
отгонка 445
Жесткость воды
определение комплексонометрическое 342
— методом нейтрализации 183 ел.
Зажимы для бюреток 52
Закон(ы)
Бугера—Ламберта—Бера 429
действия масс 194 ел.
нормального распределения 83
Фарадея 401
эквивалентности 40
Заместителя титрование 47, 264, 276, 339
Галогениды, титрование смеси 308
Гаусса кривая 108
Гейгера — Мюллера счетчик 397, 399, 438
Гелиантин 146
Германий, отгонка 445
Гидразин
ионное произведение 198
определение 284
Гидроксил-ионы, концентрация 114, 117, 119,
129
Гидроксильная ошибка 154, 156
Гидролиз солей
по аниону 113 ел.
по катиону 111
степень 111
Гидроны, индикаторы 326
Гидрохинон, определение 286
Гипофосфиты, определение 286
Гипсохромный сдвиг 140
Грамм-эквивалент 61
Гусарик 30
Двойники ионные 195
Деварда сплав 184
Декантация 357
Демпфер 29
Излучения радиоактивные, установка для
регистрации 388
Изотопного разбавления метод 397
Илъковича уравнение 420
Индиго, определение 286
Индикаторная ошибка 153
Индикаторы 94, 138 ел., 335 ел.
адсорбционные 139, 316
броматометрии 284
внешние 138, 289
внутренние 138, 289
выбор 150
интервал перехода 147 ел., 155, 241
ионная теория 141
ионно-хромофорная теория 143
кислотно-основные 93, 139, 141, 152
классификация 139
комплексонометрии 139, 289, 330, 335
люминесцентные 139
меркуриметрии 326
меркурометрии 329
нейтрализации 139
необратимые 138
образующиеся в процессе титрования 130
обратимые 138
окислительно-восстановительные 139, 239ел.
452
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Индикаторы
показатель титрования 147, 149
радиоактивные 139
ред-окс 139, 239 ел.
роданометрии 324
смешанные 149
универсальные 149
флуоресцентные 139
хемилюминесцентные 139
хроматометрии 280
хромофорная теория 142
цветные 433
Индуктор, 228, 433
Интервал доверительный 84
надежность 85
Иод
диспропорционирования реакция 266
стандартный раствор 270
Иодиды
окисление кислородом воздуха 266
определение 323
Иодометрия 262 ел.
Ионные пары 195
Калибрование измерительных приборов 50
Калия
бииодат 1.71
битартрат' 171
бифталат 170, 174
определение в силикатах 388
перманганат 235, 244, 247 ел.
Кальций
окись, определение в доломите 388
в силикате 383, 387
определение в СаСОа 375
— в сумме с магнием 343, 387
— весовое 388
— комплексонометрическое 341 ел., 345
— перманганатометрическое 260 ел.
Камера Вильсона 438
Капля, определение объема 61
Карбонаты, определение 174
Катализаторы 226
Катиониты 162
Катод ртутный 421 ел., 443
Кислотная ошибка титрования 154, 156
Кислотный хром темно-синий 336
Кислотный хромоген черный специальный 335
Кислоты 189 ел.
в неводных средах 189
диссоциация 195
изменение активности Н+-ионов в
процессе титрования 136 ел.
константы ионизации в неводных
растворах 198
концентрация Н+-ионов 96, 104
— влияние на кривую титрования 99
— многоосновные, титрование 130 ел., 151
определение в неводных растворах 201, 210
— иодометрическое 264, 277
— методом нейтрализации 93, 176 ел., 187
— хроматографическое 447
плотность 159
сила 191, 193, 199
сильные, титрование 96 ел., 150
слабые, титрование 121 ел., 151
сопряженные основания 191
установка титра 161 ел.
Клапан Бунзена 253, 257
Коагуляция 314
Колбы мерные 51, 54 ел.
Коллекторы 439
Коллоидные системы 312 ел.
Колонки
адсорбционные 446
ректификационные 445
Колориметрический анализ 429 ел.
Колориметрия 396, 429, 438
Комплексон III 330, 338 ел.
Комплексонометрическое титрование 290,
336 ел.
классификация методов 338
Комплексонометрия 290, 292, 329 ел.
Комплексоны 329
Комплексообразование 215
влияние на осаждение 215
методы 289 ел., 302
Комплексы, константа нестойкости 195, 216
Компоненты анализируемого объекта 51
Конго красный 243
Кондуктометрическое титрование 413 ел.
Кондуктометрия 395
Константа(ы)
автопротолиза 198
ассоциации 196
гидролиза 111, 113, 115, 119
ионизации кислот и оснований в неводных
растворах 198
истинные 122
концентрационные 136
нестойкости комплексов 195, 216
объемные 122
превращения 197
равновесия реакции 220 ел
скорости реакции 224
термодинамические 136
Концентрация растворов 65 ел
активная 105
Концентрирование определяемых веществ 21,
438, 443, 447
Коэффициент
активности 136, 238
поглощения света 429
поправочный, при расчетах 75
Крахмал, индикатор 268
я-Крезол-2-метиленаминодиуксусна
кислота 332
Кремневая кислота, определение 380 ел.
Кремний
двуокись, определение 380 ел.
фторид, отгонка 445
Кривые
вольтамперные 420
калибровочные 423, 432
осаждения 302 ел., 311
поглощения света 140
полярографические 420
титрования амперометрического 425
— кислотно-основного 94 ел., 124 ел.,
128 ел., 135, 137
— кондуктометрического 415
— окислительно-восстановительного
230 ел., 237 ел.
— по методу осаждения 302 ел.
— построение 237
— потенциометрического 417
— фототурбидиметрического 434
Криолит, определение фтора 448
Критерий Стьюдента ПО
Кристаллизация 441
Ксиленоловый оранжевый 335, 338
Кулонометрический анализ 425
Кулонометрическое титрование 425
Кулонометрия 396
Купферон 286, 441
Лакмус 152
Латунь, анализ 17, 390 ел., 410 ел.
Лиата ионы 189
Лиония ионы 189
Литр 49
Лунге метод 254
Льюиса теория 192
Люминесцентный анализ 396, 438
Магний
определение в доломите 388
— в силикатах 384, 387
— в сумме с кальцием 343, 345
— весовое 376
— комплексонометрическое 343, 345
— окиси 384, 387 ел.
Макрокомпоненты 51
Малеиновая кислота, титрование смеси с фта-
левой кислотой 208
Марганец
определение 254
—двуокиси 259, 276
Масс-спектрографы 300
Масс-спектрометрические методы 400
Масс-спектрометры 400
Медь
Гликоколят 332
определение броматометрическое 284
— в рудах 275
— в солях 275
— в CuS04 407
-— в сплавах 391, 410
— иодометрическое 273
— методом внутреннего электролиза 411
— электровесовое 407, 410
Меркуриметрия 292, 326 ел.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
453
Меркурометрия 292, 328
Металл индикаторы 330
Метанол, ионное произведение 198
Метиленовый синий, определение 288
Метиленовый красный 152, 243, 284
Метиловый оранжевый 146, 150, 152 ел.,
243, 284, 441
Метиловый фиолетовый 441
8-Метилхинальдин 441
Метод просветления 291
Метод равного помутнения 291
Метод стандарта 423
Мешалка электромагнитная 409
Микробюретки 53, 88
Микровесы 35
Микропримеси 17, 41, 438
Микрокомпоненты 57
Микрофотометры 428
Микроэлектроды 422
Миллиграмм-эквивалент 67
Миллилитр 49
Мицеллы 313
Молибден, определение 286
Молярность 66
Мора метод 291, 318, 323
Муравьиная кислота, ионное произведение 198
Мурексид 336
Мышьяк
определение 270 сл.,у 284
отгонка 445
отделение 443
Навеска 40
взятие и растворение 36, 353 ел.
расчет 349
Натрий
бикарбонат и карбонат, определение при
совместном присутствии 181
карбонат 161, 163
— титрование 133
карбонат и едкий натр, определение при
совместном присутствии 179
нитропруссид 292, 326
оксалат 161 ел.
определение в силикатах 388
сульфит, определение 271
тетраборат 161 ел., 164
— титрование 133, 162
тиосульфат, стандартный раствор 267
хлорид 161
— стандартный раствор 267
— технический, определение хлора 322
Нафталин-у-сульфокислота 441
Нафтоловый желтый 337
Неводные растворы 158, 188 ел.
Нейтрализации методы 89, 93 ел.
Нефелометрия 396
Никель
диметилглиоксимат 392
определение весовое 392
Ниобий, осаждение 286, 441
Нитраты, определение 257
Нитрилтриуксусная кислота 329
Нитриты, определение 254
Р-Нитрозо-а-нафтол 326
Нитрозооксин 326
Нитросоединения, определение 288
Нормальность раствора 39, 63, 67, 80 ел.
Объемный анализ 23, 25 ел., 38 ел.
аналитический множитель 73, 168
вычисления 65 ел.
газоволюметрический 22
газовый 20, 22
измерительные приборы 51
классификация методов 45
методы 38 ел.
реакции 41
седиментационный 23
техника эксперимента 49 ел.
титриметрический 20, 25 ел., 38 ел.
точность 49
характеристика 38
электрохимические методы 413 ел.
Объемы, точность измерения 49 ел.
Окисление
анодное 402
каталитическое 62, 227
Окисление—восстановление 62
реакции 41, 215 ел., 279
— скорость 224
— сопряженные 227
Окисления—восстановления методы 89, 211 ел.
фиксирование точки эквивалентности 239
Окислители, определение 250, 256
Окислы полуторные
определение в доломите 388
— в силикатах 381
Окклюзия 251, 364, 441
Оксалаты, определение 261, 310
Оксидиметрия 211 ел., 417
Оксифенилиминодиуксусная кислота 332
8-Оксихинолин 286, 441
Оксредметрия 211 ел.
Олово
определение броматометрическое 284
— в сплавах 390
осаждение 441
хлорид, восстановление железа (III) 256
Определяемое вещество 38
Оптическая плотность 429 ел.
Оптические методы анализа 395, 428 ел.,
432 ел.
Осадители 439
в весовом анализе 350, 355, 367
органические 440 ел.
Осадки
аморфные 350, 356, 365
в весовом анализе 350, 356 ел.
выделение из растворов 361 ел
высушивание 359
загрязнение 364
кристаллические 350, 356, 363, 365
обработка 356
образование 293
прокаливание 359 ел.
промывание 356 ел.
растворимость 293 ел., 363, 366
созревание 356
состав 363
структура 364
Осаждение 89, 289 ел., 348 ел., 356, 439
классификация методов 291
на ртутном катоде 442
оптимальные соотношения объемов 298
скорость 362
сопряженное 364
теория 292
техника 355
Основания
активность 105
вычисление концентрации Н+-ионов 96, 104
диссоциация 195
изменение активности Н+-ионов в
процессе титрования 137 ел.
константа ионизации в неводных
растворах 198
концентрация, влияние на кривую
титрования 99
многоосновные, титрование 151
нейтрализация 93
плотность 159
сила 191, 193, 199
сильные, титрование 126 ел., 150
слабые, титрование 151
сопряженные кислоты 191
титрование в неводных растворах 198
установка титра растворов 171, 208
Отгонка летучих' веществ 348, 352,
444
Отдельных навесок метод 63
Очистка
бюреток 55, 59
вещества анализируемого 26
воды 438
мерных колб 55
пипеток 55, 58
Ошибка(иу
абсолютная 83
анализа 26, 54
водородная 154, 156
гидроксильная 154, 156
грубые 82
индикаторная 153
инструментальные 82
кислотная 154, 156
относительная 83
систематические 54, 82
случайные 54, 82, 105
средняя 85
титрования 153 ел.
точная 83
щелочная 154, 156
454
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Пептизация 315
Перенапряжение 406
Переосаждение 364
Пересчет на сухое вещество 27
Перманганатометрия 235 ел.
Перхлораты, определение 288
Печи электрические 360
Пикнометр 159
Пипетирования метод 63
Пипетки 51, 56
Пиридин 441
Пиролюзит, определение двуокиси марганца
259, 277
Плотность растворов
определение 159
оптическая 429 ел.
Показатель активности 136
Полумикробюретка автоматическая 53
Полумикрообъемный метод анализа 88
Поляризация 407, 418
Полярографическая волна 419
Полярографический анализ 18, 418 ел.
Полярография 395, 418 ел., 438
амальгамная 443
Полярографы 423
Помутнения равного метод 291
Поправка на индикатор 321
Посуда мерная 51
Потенциал(ы)
нормальные 220
окислительно-восстановительные 212, 224,
231 ел.
полуволны 421
разложения 403, 419
Потенциометрическое титрование 413, 416 ел.
Потенциометрия 395
Потенциометры 395
Потери при прокаливании
доломитоз 388
силикатов 379
Правила пропорциональности 69
Проба средняя, отбор 25 ел.
Пробирка для взвешивания 63
Промывная жидкость 358, 368
Пропаривание колб 55
Просветления метод 291
РН
в буферных растворах 107
влияние на скорость реакции 224
— температуры 101
изменение в процессе нейтрализации 96 ел.,
121 ел., 126 ел., 129
определение потенциометрическое 418
скачок 99
рОН, вычисление 129
Радиоактивационный анализ 399 ел.
Радиометрический анализ 397
Радиометрическое титрование 400
Радиохроматография 400
Разбавление
влияние на точность титрования 102
изотопное, метод 397
Разделения методы 438 ел,
электрохимические 442
Разновес аналитический 30
РастворимЬсть осадков 293 ел., 363, 366
влияние одноименных ионов 296 ел.
вычисление 298
Растворители
ионные произведения 198
неводные 193 ел., 202 ел.
определение воды 204
Растворы
измерение объемов 49
концентрация 65 ел.
молярные 66
неводные 158, 188 ел.
нормальность 39
разбавление 159
стандартные (титрованные) 39, 62, 158,
188
— приготовление 61 ел., 64, 158 ел.
твердые 441
титр 21, 39
— установка 62, 71, 160
Реагент 39
Реактив(ы) 39, 49
флотационные 449
Циммермана—Рейнгардта 253
Реакции
в методах осаждения и комплексообразо-
вания 292 ел.
двойного ©бмена 62
замещения 43
ионного обмена 43
комплексообразования 42, 215, 331
конденсации 43
нейтрализации 41, 93 ел.
окисления—восстановления 41, 215 ел.,
279
осаждения 42, 292
присоединения 43
сопряженные 227
специфичность 366
Ред-окс-индикаторы 139, 239 ел.
Ред-окс-методы 211 ел.
Ред-окс-системы 214
Ред-окс-электрод 231
Редуктор Джонса 256
Рейтер 30
Родамины 441
Роданиды, применение 323
Роданометрия 323 ел.
Ртути нитрат, стандартный раствор 327
Ртуть
окись 161 ел.
отгонка 445
Руда, анализ 254, 275
Салициловая кислота, дифференцированное
титрование с бензойной кислотой
208
Свинец
определение в бронзах и латунях 390,
410 ел.
— двуокиси 275
— электровесовое 410
Селеновая кислота, анализ 276
Сера
определение 370
— в силикатах 385
Серебро
галогениды 315
нитрат, стандартный раствор 320 ел.
— титрование кондуктометрическое 413
— — потенциометрическое 418
определение 323, 325
соли, осаждение 310
хлорид 302
Серная кислота
ионное произведение 198
техническая, определение H2S04 176
Силикаты
анализ весовой 378 ел.
— объемный 387 ел.
Скачок титрования 99
Солевой эффект 300
Соли
внутрикомплексные 330, 332
диссоциация 195
органических кислот, определение в
присутствии щелочи 207
титрование 133 ел.
Сольваты 197
Сольволиз 200
Сольвосистемы 189
Соосаждение 361, 441 ел.
Спектр поглощения 140
Спектральный анализ 396, 428
абсорбционный 396 ел.
по спектрам комбинационного рассеяния
света 396
эмиссионный 396, 428 ел.
Спектрография эмиссионная 438
Спектрографы 428
Спектроскопия 18
Спектрофотометрическое титрование 433 ел.
Спектрофотометрия 396
Сплав(ы)
анализ 286, 325
Деварда 184
Средняя арифметическая 84
Стандартов метод 423
Стандартные серии, метод 430
Статистическая обработка данных 82 ел.
Стилометры 428
Стилоскопы 428
Стокса уравнение 20
Стьюдента критерий НО
Сульфаты, определение 370
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
455
Сульфиты, определение 286
Сурьма, определение 284
Счетчик Гейгера—Мюллера 399, 438
Тайрон 338
Таллий, определение 284
Тананаева бесстружковый метод анализа 36
Тантал, осаждение 286, 441
Теллуровая кислота, анализ 276
Температура, влияния на скорость реакций 226
Теория ошибок 104
Тетраэтиламмония гидроокись 207
Тигли 359
Тимоловый4 голубой 152
Ти молфталеи н 152
Тиосульфаты, определение 286
Титан
осаждение 441, 443
определение колориметрическое 435
— двуокиси 381
Титанометрия 239, 288
Титр 21, 39, 67, 80 ел.
установка 62 ел., 161 ел.
Титрант 39
Титрование 39, 44
автоматическое 90
амперометрическое 424 ел.,
безбюреточные методы 39, 88
в методе нейтрализации 89, 93 ел.
в неводных средах 188, 201 ел.
весовое 88
высокочастотное 396, 415
вычисление результатов 65 ел.
капельное 88 ел.
капельно-темпометрическое 89
кислотами 93, 96 ел.
кислотно-основное 93, 339
комплексонометрическое 290, 330 ел.
кондуктометрическое 413 ел.
конечная точка 40
косвенное 47
кривые, см. Кривые титрования
кулонометрическое 425
методом отдельных навесок 63
обратное 47
окислительно-восстановительное 211 ел.
основаниями 93
ошибки 153 ел.
перманганатом 244 ел.
по времени 88 ел.
по методу Циммермана—Рейнгардта 253
по остатку 47
по разности 48
по содержанию продуктов реакции 48
по числу капель 88
показатель 147 ел.
потенциометрическое 413, 416 ел.
приемы 64
прямое 46
радиометрическое 400
реверсивное 49
скачок 99
спектрофотометрическое 433 ел.
темпометрическое 89
точка конечная 40
— эквивалентности 40
точность 102
условия 57
фотонефелометрическое 434
фототурбидиметрическое 434
фотоэлектрическое 433
Ток
диффузионный 419 ел.
постоянный, источники 404
предельный 420
Точка эквивалентности, см. Эквивалентности
точка
Точность анализа 36, 367
вычисление 79
Трилон Б 330
Турбидиметрия 396, 434
Углерода двуокись, определение в доломите
389
Угольная кислота, влияние на точность
титрования 178
Уксусная кислота
ионное произведение 198
определение 177
титрование едким натром 121 ел.
Ультрамикроанализ 438
Уравнение
Бунзена 358
Ильковича 420
Уравнивания метод 431
Уран, определение 286
Установочные вещества 63 ел.
для растворов кислот 161 ел.
— — для оснований 170 ел.
Факторы аналитические
весового анализа 351
объемного анализа 73, 168
Фарадея законы 401 ел.
Фаянса метод 292, 322
о-Фенантролин 243
Фенилантраниловая кислота 242, 286
Фениларсоновая кислота 441
Фенилиминодиуксусная кислота 332
Феноловый красный 152
Фенолфталеин 146 ел., 152 ел.
Ферроин 243
Фиксаналы 64, 174
Фильтрование 356 ел.
Фильтры
сжигание с осадком 360 ел.
стеклянные 393
Фишера метод определения воды 204 ел.
Флотация 449
Флуоресцеин 317
Флуоресцентный анализ 396
Фольгарда метод 291, 323 ел.
Фон полярографический 421
Формалин, определение формальдегида 272
Фосфор
определение в силикатах 385
осаждение 443
Фосфорная кислота
кривая нейтрализации 130
определение 187
Фотоколориметрический метод 430 ел.
Фотоколориметры 430
Фотометрия пламени 396, 438
Фотонефелометрическое титрование 434
Фототурбидиметрические титрование 433
Фотоэлектрическое титрование 433
Фталевая кислота, титрование в смеси с ма-
леиновой кислотой 208
Фтор, определение 448
Хелатометрия 329
Хлор, определение в воде 328
Хлориды
определение весовое 377
— по Мору 322
— по Фаянсу 322
— по Фольгарду 324
Хлористоводородная кислота
кривая нейтрализации 96 ел.
определение 277
— ионов хлора 377
приготовление растворов 160
установка титра 160 ел., 164 ел.
Хлорная кислота, растворы 205 ел.
«Холостой» опыт 367
Хроматография 395, 397, 445 ел.
Хроматометрия 278 ел.
Хроматы, определение 288, 310
Хромовая смесь 55
Хромоген специальный ЕТ 00 335
Хромофорные группы 143
Цветность органических соединений 144
Цемент, анализ 386 ел.
Цементация 412
Центрифугирование 449
Цериметрия 282
Циммермана—Рейнгарда раствор 253
Цинк
амальгамированный 256
определение в сплавах 392
— полярографическое 424
Цирконий, определение 286, 441
Шкафы сушильные 26
Шпат плавиковый, определение фтора 448
456
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Щавелевая кислота 170 ел.
определение перманганатометрическое 251
Щелочи, см. Основания
Щелочная ошибка 154, 156
Эквивалент электрохимический 401
Эквивалентности правило (закон) 40, 90
Эквивалентности, точка 40, 45
методы установления 60
определение в комплексонометрическом
титровании 334
в методе аргентометрии 318
нейтрализации 93 ел.
окисления—восстановления 239
осаждения 389
хроматометрии 280
цериметрии 283
оптическое 432 ел.
физико-химическими методами 94,
413 ел.
Эксикаторы 27
Экстрагирование 444
Электроанализ 401 ел.
Электровесовой анализ 348, 395, 401, 403 ел
Электрод(ы) 405
вращающийся 422
индикаторный 416 ел.
каломельный 416
неплатиновые 405
платиновые 404, 410
Электроды
ред-окс 231
сравнения 416
Электролиз 402 ел.
внутренний 395, 403, 406
потенциал разложения 403
с ртутным катодом 403
Электролиты
диссоциация в неводиых растворах 192 ел.
концентрация в растворе 446
растворимость 300
— вычисление 293 ел.
сильные 194
Электроосаждение 405
Электрохимические методы 395 ел., 413, 442 ел
Электрофорез 449
Эмиссионный анализ 396, 429
Эозин 317
Эриохром черный Т 335, 338
Этанол, ионное произведение 198
Этилендиаминдиизопропилфосфоновая кислота
332
Этилендиаминдиуксусная диметилфосфоновая
кислота 331
Этилендиаминтетраметилфосфоновая кислота
Этилендиаминтетрауксусная кислота 329
Эухризин 244
Янтарная кислота 171