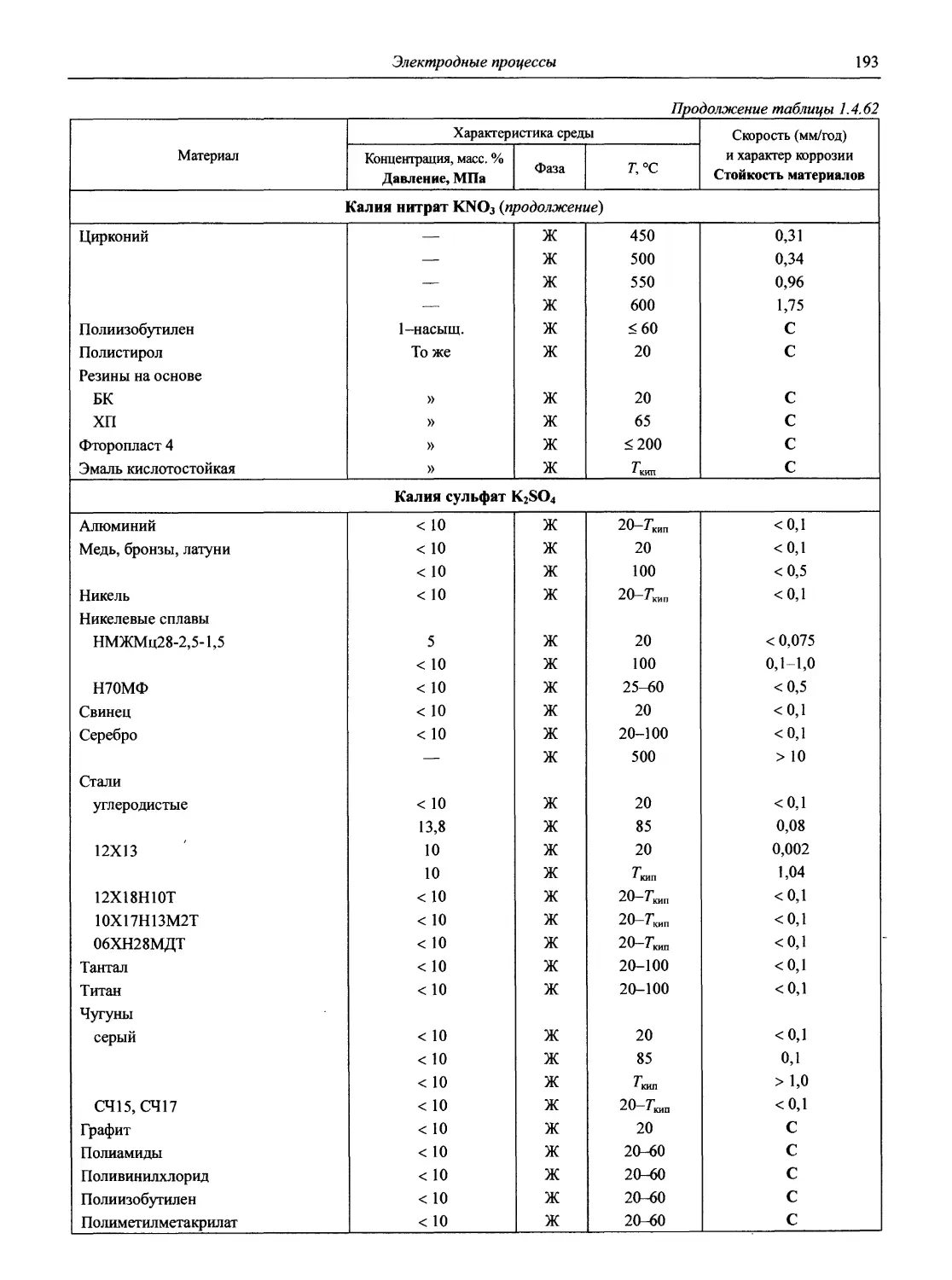
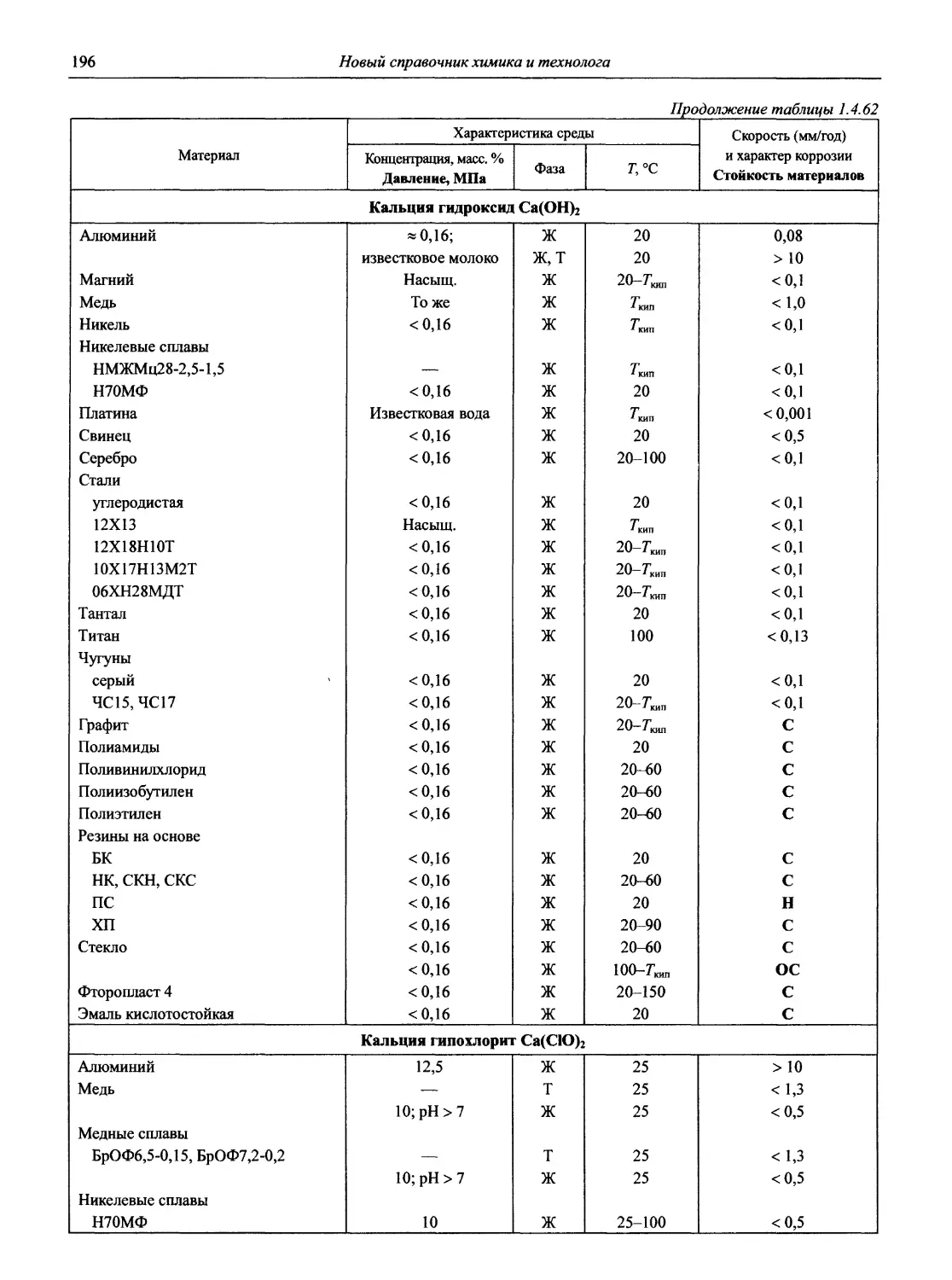
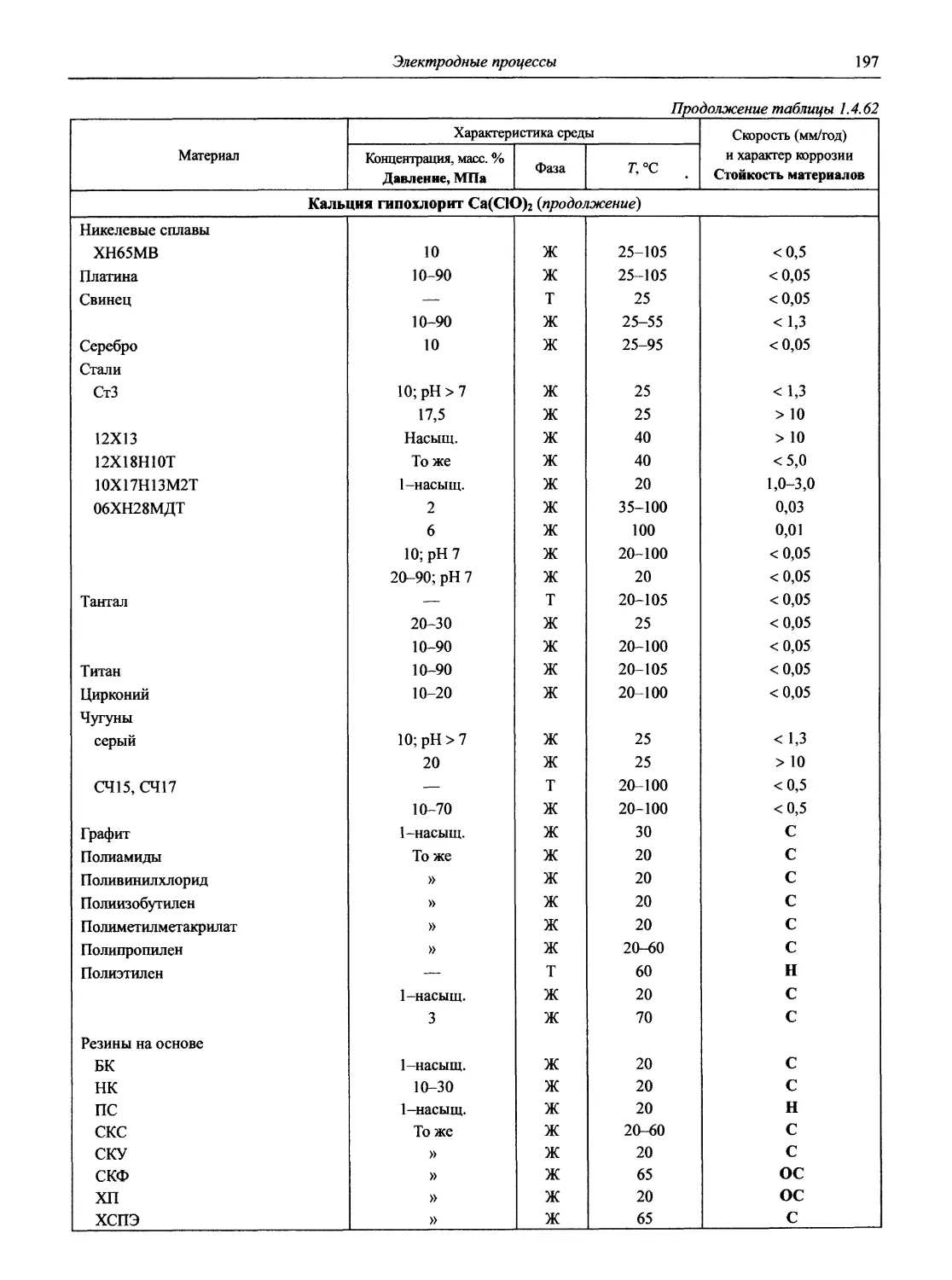
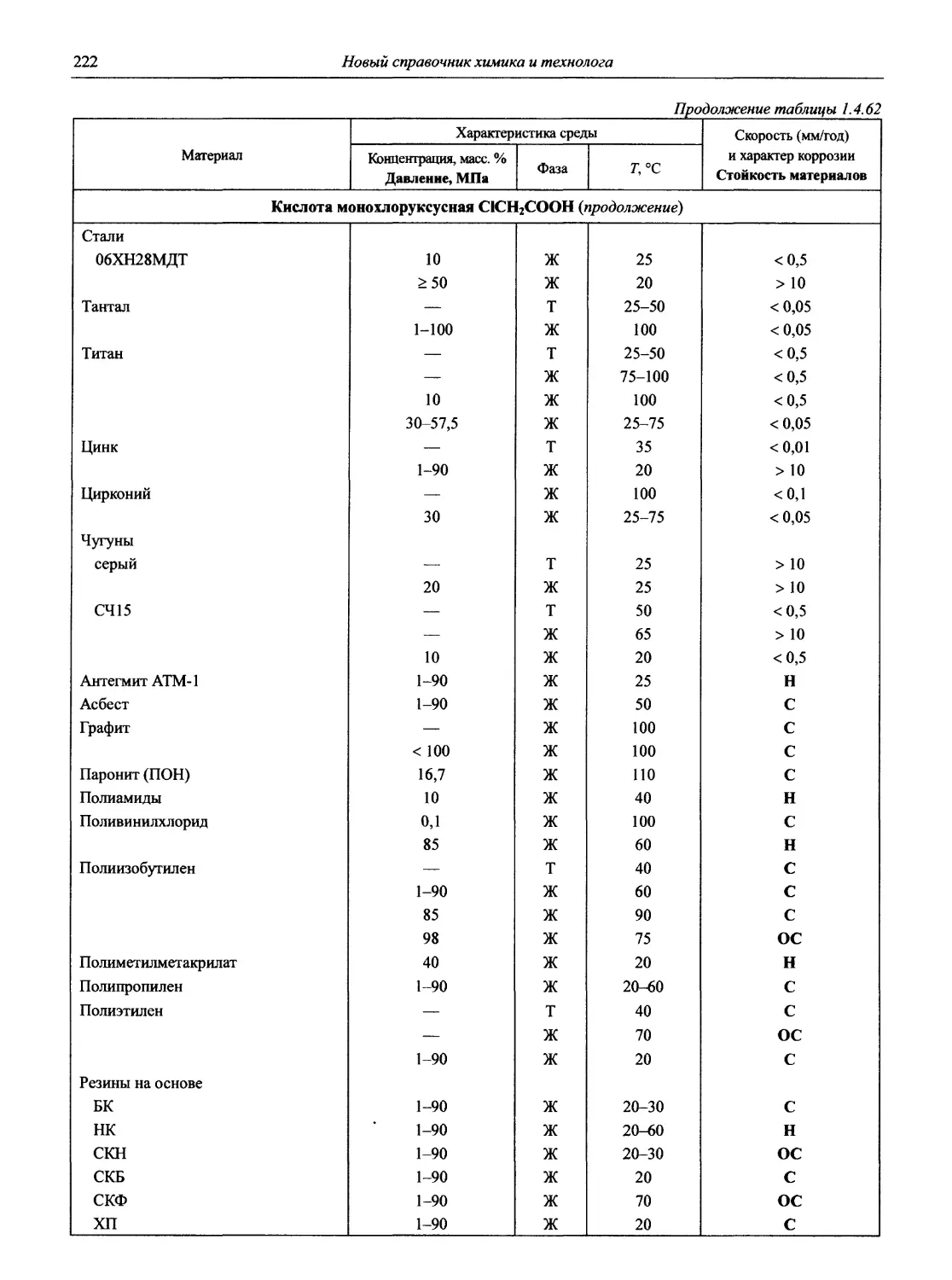
/













































































































































































































































































































































































































































































































































































































































































































































































































































Text
М&ВЬГЙ
СПРАВОЧНИК ХИМИКА и ТЕХНОЛОГА
Электродные процессы Химическая кинетика и диффузия Коллоидная химия
MMIV Санкт-Петербург 2004
ББК 24.4 24.46
Н72
Авторы:
д.т.н., проф. Абиев Руфат Шофкетович
д.х.н., проф. Бибик Ефим Ефимович
д.т.н., проф. Власов Евгений Александрович
д.т.н., проф. Ермаков Борис Сергеевич
д.т.н., проф. Зотиков Владимир Степанович к.т.н., доц. Иванов Валентин Анатольевич
акад. РАЕН, д.х.н., проф. Симанова Светлана
Александровна
к.т.н., доц. Суворов Константин Александрович к.х.н., доц. Хохряков Константин Анатольевич д.т.н., проф. Яблокова Марина Александровна
Редактор тома: акад. РАЕН, д.х.н., проф. Симанова Светлана Александровна
Н 72 Новый справочник химика и технолога. Электродные процессы. Химическая кинетика и диффузия. Коллоидная химия — С.-Пб.: АНО НПО «Профессионал», 2004. — 838 с.
Справочник содержит сведения об электродных процессах, кинетике и диффузии сложных систем (растворов, сплавов), о физике, химии и технологии дисперсных систем (коллоидная химия).
Предназначен для научного и инженерно-технического персонала отраслевых химических лабораторий, инженеров-химиков, химиков-технологов, преподавателей и студентов университетов, химико-технологических и химико-фармацевтических вузов, а также для специалистов смежных профессий.
Все права защищены и принадлежат издателю.
Любое использование материала данной книги, полностью или частично, без разрешения АНО НПО «Профессионал» запрещено и будет преследоваться по закону.
ISBN 5-98371-016-8
© АНО НПО «Профессионал», 2004
ОТ ИЗДАТЕЛЬСТВА
Петербургское издательство «Профессионал» предлагает специалистам-химикам и всем заинтересованным читателям 7-томный «Новый справочник химика и технолога» (Справочник). («Справочник химика» в 7 томах, под общей редакцией чл.-корр. АН СССР Б.П. Никольского, последний раз был переиздан в 1962— 1966 г.г.)
Название Справочника отражает основную задачу издателей и авторов: помимо базовых, «академических», публиковавшихся в научной и справочной литературе (например, в «Справочнике химика») сведений представить в максимально возможном объеме новую информацию, данные, полученные за последние 40 лет в области не только химической науки, но и химических технологий, — и таким образом сделать Справочник интересным, нужным, необходимым широкой «химической» аудитории — как ученым и студентам, так и производственникам-практикам.
В создании Справочника приняли участие крупные ученые и ведущие специалисты — химики из Москвы, Санкт-Петербурга, других городов страны (всего около 150 авторов); материалы являются оригинальными авторскими разработками либо подготовлены на основе современной литературы. Справочник обобщает опыт работы российских и зарубежных ученых и технологов второй половины XX в. и показывает перспективу развития химической и смежных областей науки и производства.
В процессе работы неоднократно возникала необходимость привлечения дополнительных материалов,
разработки новых тем, более расширенного и углубленного изложения уже включенных в Справочник разделов. В связи с этим значительно увеличился, по сравнению с первоначально запланированным, объем издания (некоторые тома выйдут в двух книгах), сроки выпуска данного и следующих томов отодвинулись. Однако авторско-издательский коллектив считает эти производственные потери и трудности оправданными в связи с необходимостью оптимального решения заявленной глобальной задачи.
Продолжает издание том «Электродные процессы. Химическая кинетика и диффузия. Коллоидная химия». Настоящий том дает представление о современном состоянии, достижениях, возможностях этих областей химической науки.
Основные темы изданных и готовящихся к изданию томов:
♦ основные свойства неорганических, органических и элементоорганических веществ;
♦ свойства растворов, химическое равновесие;
♦ сырье и продукты промышленности;
♦ процессы и аппараты химических технологий;
♦ аналитическая химия;
♦ вредные химические вещества.
Руководители издательства выражают благодарность за самоотверженную работу всем принявшим участие в создании данного тома.
ОТ АВТОРОВ ТОМА
Технический прогресс в области химии и химической технологии на рубеже XX-XXI столетий привел к возникновению информационного «взрыва», отражающего современное состояние науки и техники.
Почти 40 лет прошло после выхода в свет в нашей стране «Справочника химика». За это время появилось огромное количество новых фундаментальных данных, касающихся теоретических и практических проблем современной химии. Создание «Нового справочника химика и технолога» (Справочника), включающего в себя научные и практические достижения последних десятилетий, для издательства «Профессионал» представлялось весьма актуальным.
В данном томе рассмотрена информация, относящаяся к важнейшим разделам физической химии, электрохимии, коллоидной химии. Предлагаемые материалы касаются не только фундаментальных данных по этим важнейшим разделам химической науки, но и за
трагивают многие практические проблемы, представляющие интерес для инженеров-химиков, химиков-технологов разных специальностей, то есть производственников-практиков. Так, в настоящем томе в разделе «Электродные процессы» большое внимание уделено различным видам коррозии и коррозионной стойкости различных материалов в разных средах, способам защиты от коррозии. Рассмотрены кинетика гомогенных химических реакций, топохимические реакции, диффузия в газах, жидкостях и различных твердых материалах. К интенсивно развивающимся наукам относится коллоидная химия. Она изучает дисперсные системы и поверхностные явления на границе раздела фаз. Благодаря успехам коллоидной химии разрабатываются научные основы многочисленных технологических процессов, в частности создаются современные композиционные и строительные материалы, пластмассы, синтетические волокна и т. д.
Справочник содержит современную терминологию и номенклатуру. Оценка физико-химических констант приведена в Международной системе единиц (СИ). Номенклатура химических соединений в основном дана в соответствии с правилами Международного союза теоретической и прикладной химии — IUPAC (1979 и 1993 гт.). В качестве синонимов в ряде случаев сохранены и традиционные названия соединений, широко используемые в инженерной практике. При характеристике химических веществ наряду с единицами СИ иногда используются разрешенные к применению внесистемные единицы, которые химики и технологи продолжают активно употреблять.
Справочник дополнен современной библиографией по всем разделам. В списках рекомендуемой литературы предпочтение отдается монографиям, в которых можно найти дополнительные сведения по каждому разделу.
Материалы, представленные в данном томе Справочника, заинтересуют химиков-исследователей, преподавателей вузов и техникумов, инженеров-технологов многих специальностей, аспирантов, студентов.
Авторы будут признательны читателям за конструктивную критику и пожелания.
Академик РАЕН, доктор химических наук, профессор
С.А. Симанова
К ЧИТАТЕЛЯМ
Издательство с благодарностью примет и учтет при подготовке последующих изданий все ваши замечания, предложения и пожелания
Раздел 1 ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ
Значения коэффициента 2,3026—
т,°с RT 2,3026— F RT 1g 2,3026-у- Г, °C RT 2,3026— F RT 1g 2,3026^- Г, °C RT 2,3026— F RT 1g 2,3026— F
0 0,054197 -1,26602 23 0,058760 -1,23092 46 0,063323 -1,19844
1 0,054395 -1,26444 24 0,058958 -1,22946 47 0,063522 -1,19708
2 0,054593 -1,26287 25 0,059157 -1,22799 48 0,063720 -1,19572
3 0,054792 -1,26128 26 0,059355 -1,22654 49 0,063918 -1,19437
4 0,054990 -1,25972 27 0,059554 -1,22509 50 0,064117 -1,19303
5 0,055189 -1,25815 28 0,059752 -1,22365 51 0,064315 -1,19168
6 0,055387 -1,25659 29 0,059950 -1,22221 52 0,064514 -1,19035
7 0,055585 -1,25504 30 0,060149 -1,22077 53 0,064712 -1,18902
8 0,055784 -1,25349 31 0,060347 -1,21934 54 0,064910 -1,18769
9 0,055982 -1,25195 32 0,060546 -1,21792 55 0,065109 -1,18636
10 0,056181 -1,25041 33 0,060744 -1,21650 56 0,065307 -1,18504
11 0,056379 -1,24888 34 0,060942 -1,21509 57 0,065506 -1,18372
12 0,056577 -1,24736 35 0,061141 -1,21366 58 0,065704 -1,18240
13 0,056776 -1,24583 36 0,061339 -1,21227 59 0,065902 -1,18110
14 0,056974 -1,24432 37 0,061538 -1,21085 60 0,066101 -1,17979
15 0,057173 -1,24281 38 0,061736 -1,20946 65 0,067093 -1,17332
16 0,057371 -1,24131 39 0,061934 -1,20807 70 0,068085 -1,16695
17 0,057569 -1,23981 40 0,062133 -1,20669 75 0,069077 -1,16067
18 0,057768 -1,23831 41 0,062331 -1,20529 80 0,070069 -1,15448
19 0,057966 -1,23682 42 0,062530 -1,20391 85 0,071061 -1,14836
20 0,058165 -1,23534 43 0,062728 -1,20253 90 0,072053 -1,14235
21 0,058363 -1,23386 44 0,062926 -1,20117 95 0,073045 -1,13641
22 0,058561 -1,23239 45 0,063125 -1,19979 100 0,074037 -1,13055
6
Новый справочник химика и технолога
1.1. Электродные процессы в растворах
(доц., к.х.н. К.А. Хохряков, проф., д.х.н. С.А. Симанова)
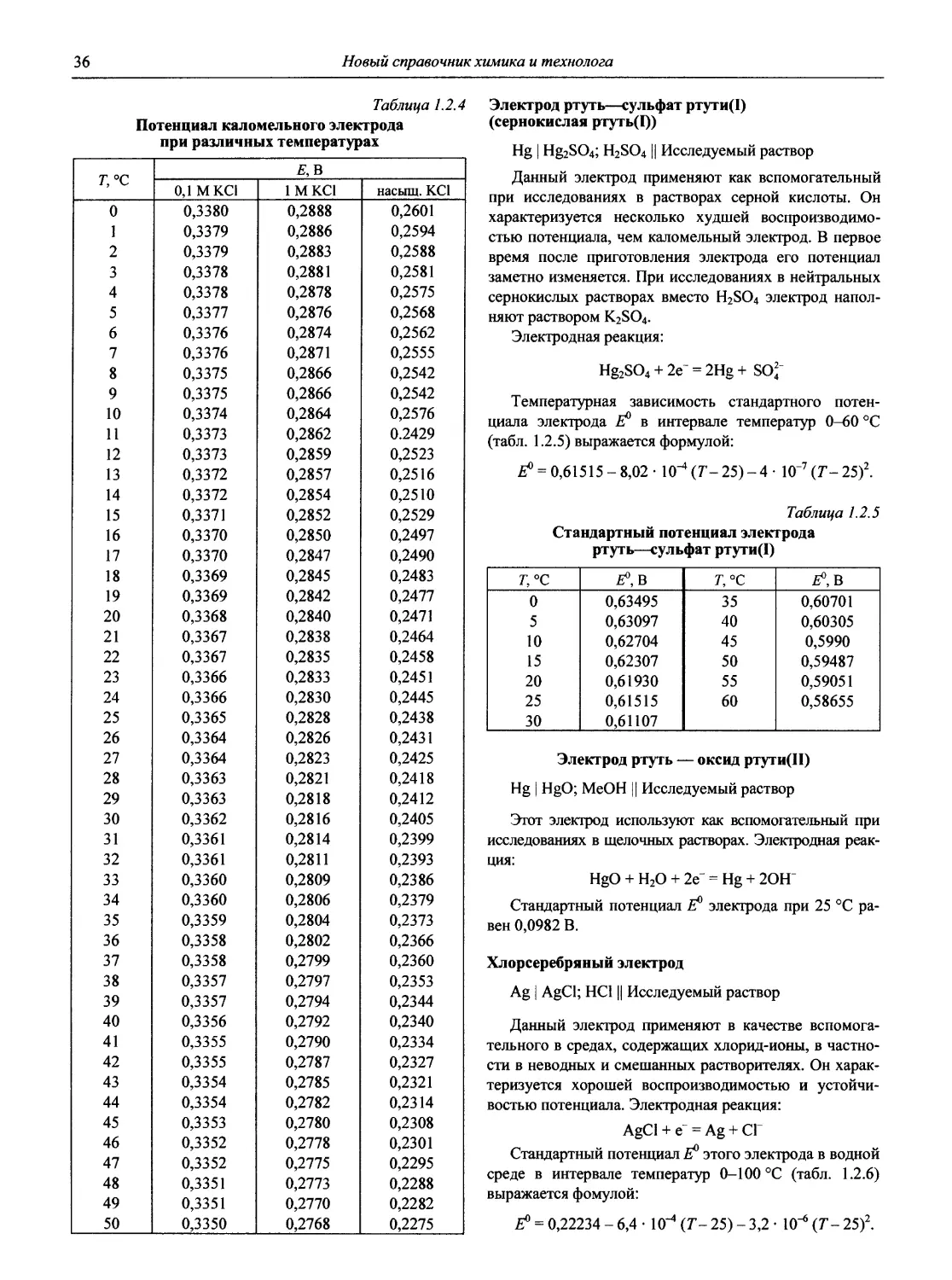
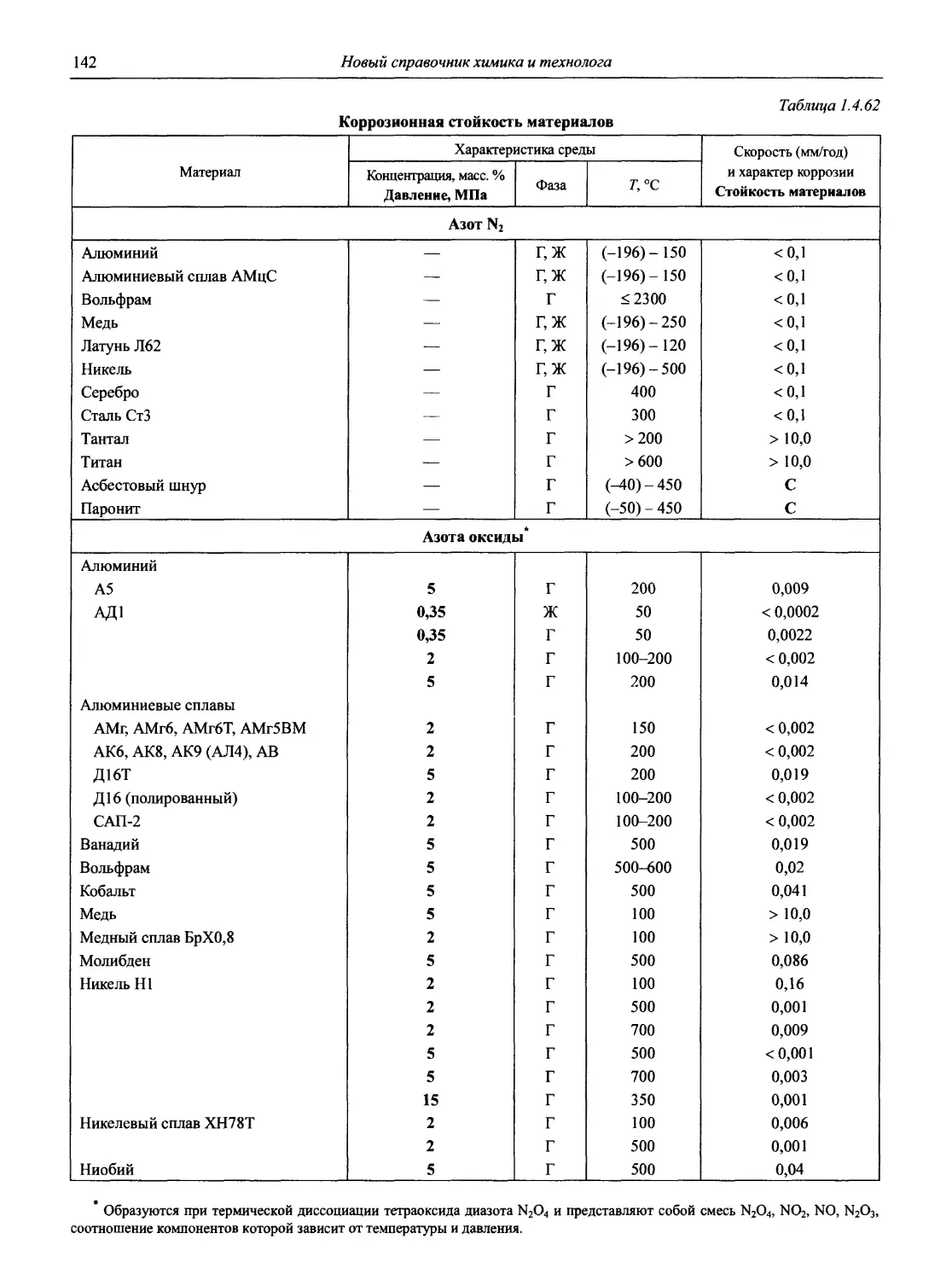
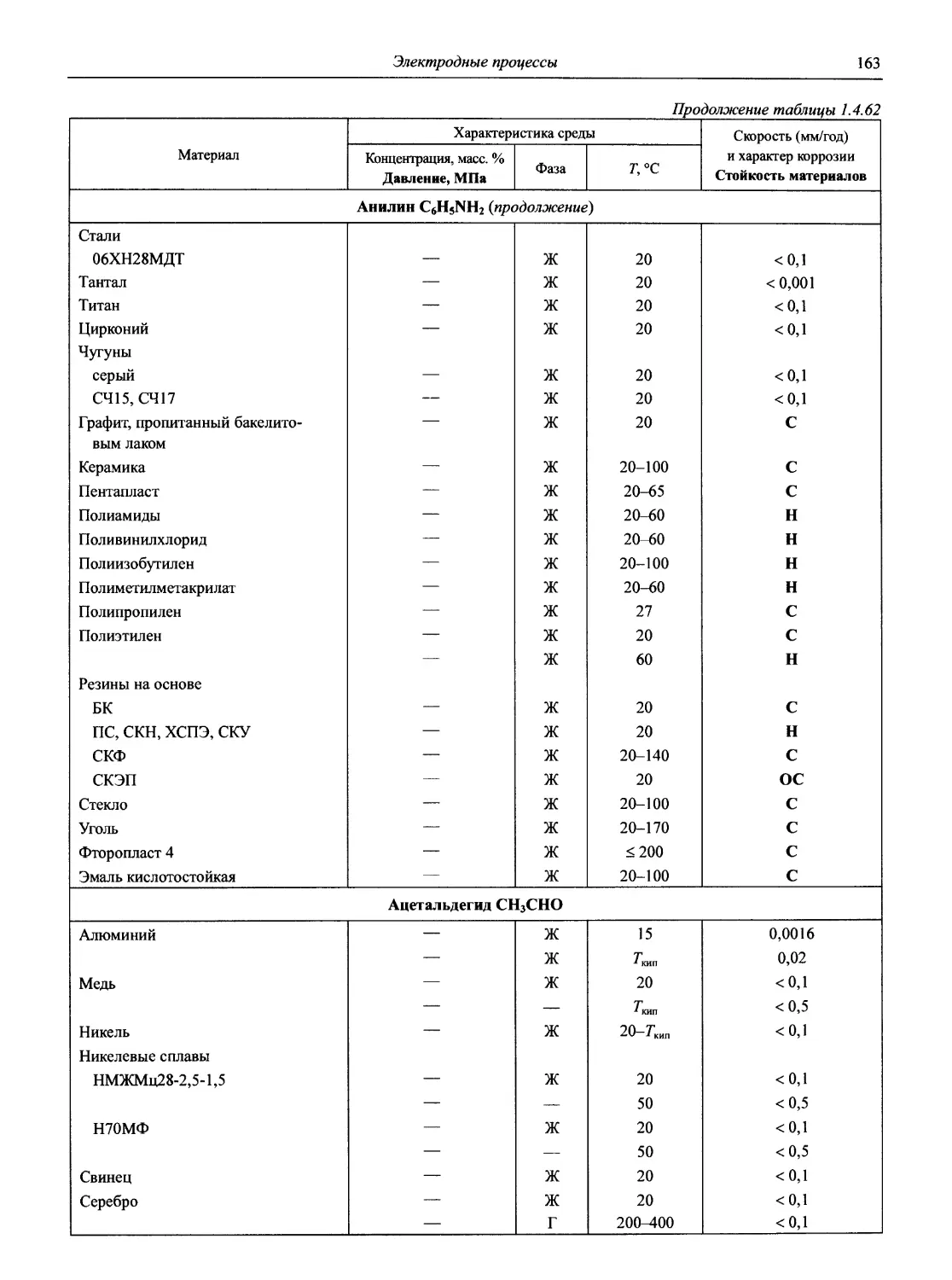
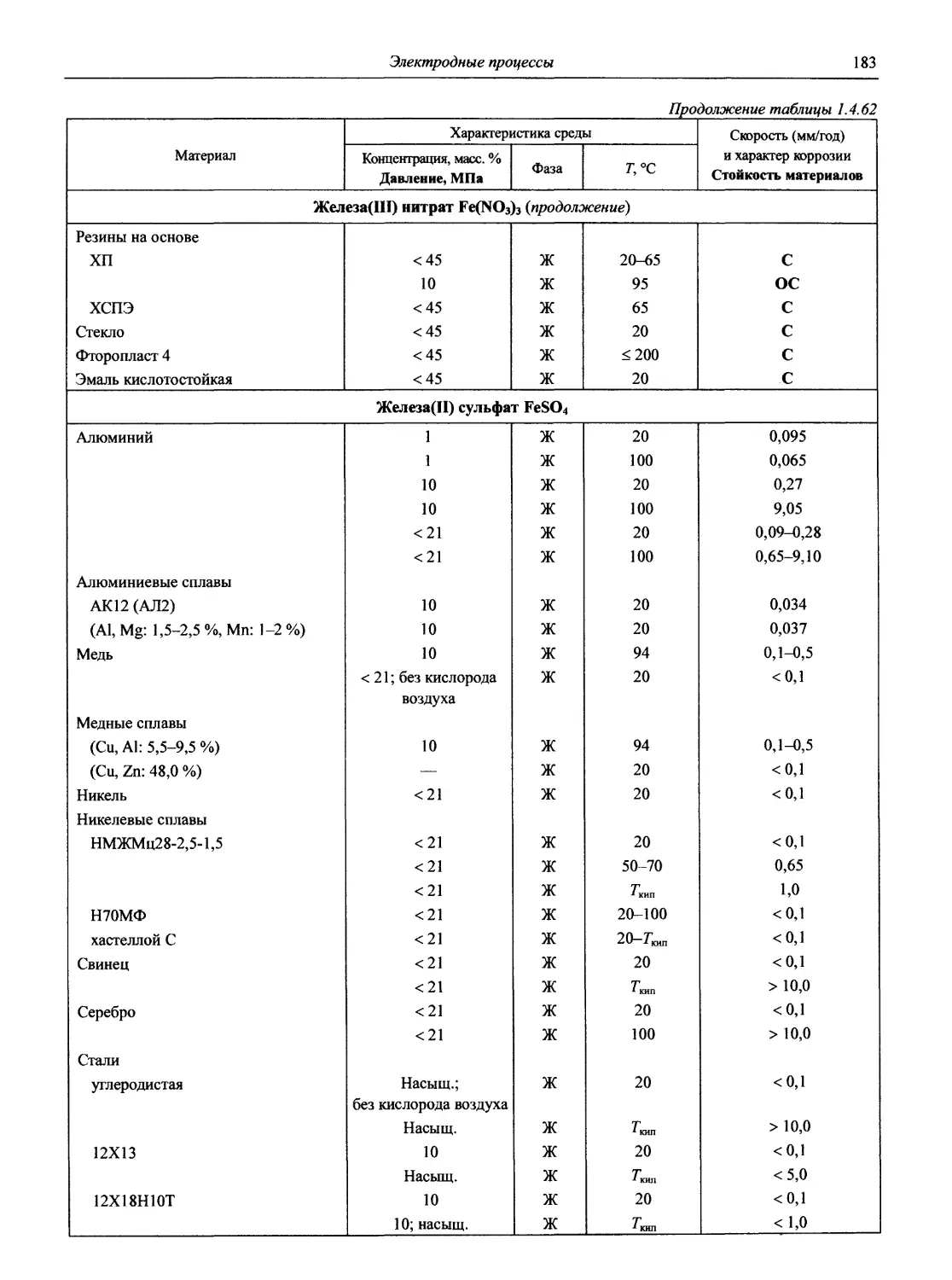
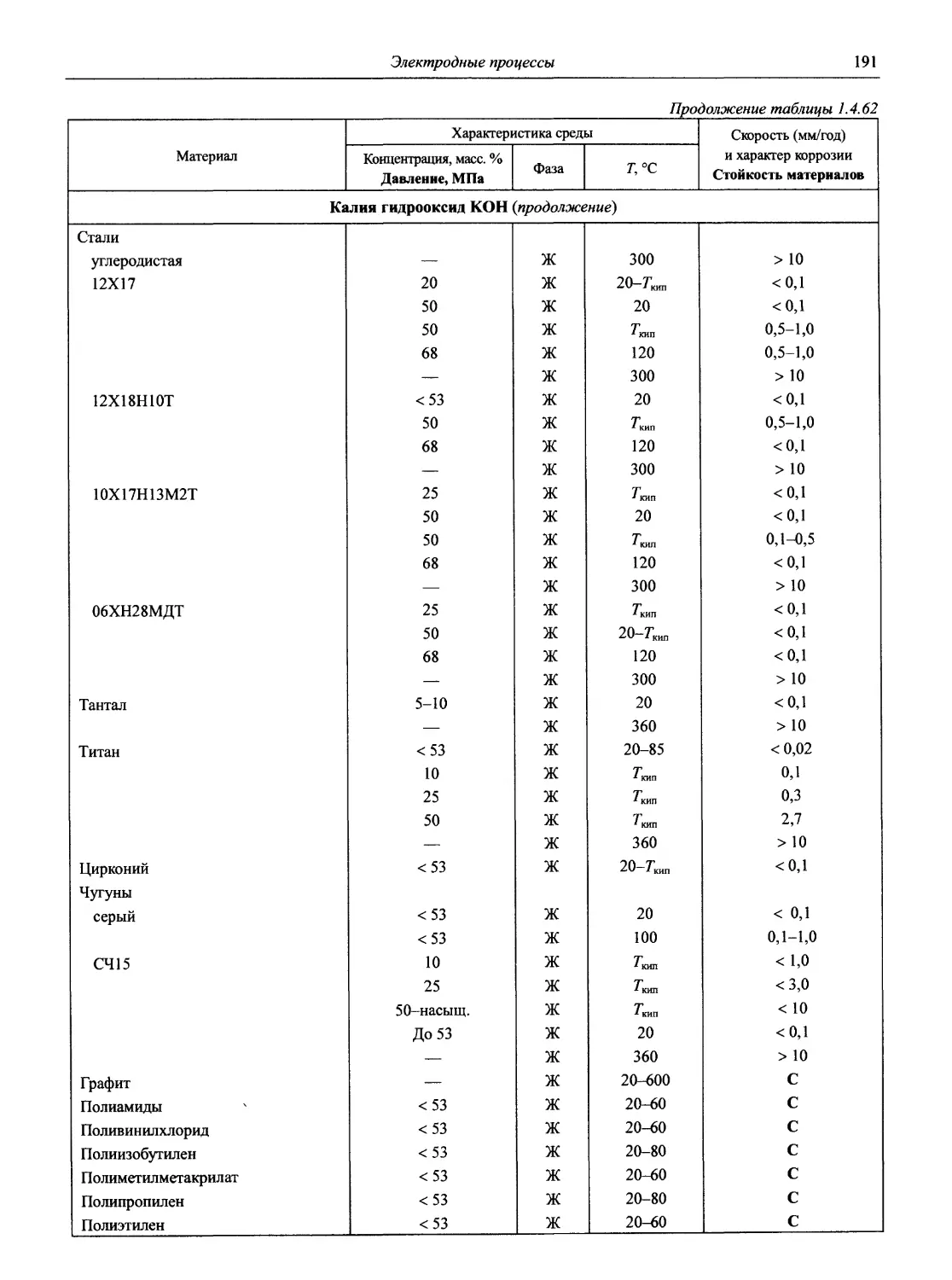
Таблица 1.1
Стандартные электродные потенциалы в водной среде
В таблице приведены величины стандартных электродных потенциалов в водных растворах при температуре 25 °C и давлении 1 атм. Величины потенциалов выражены в вольтах по отношению к стандартному потенциалу водородного электрода, принятому при всех температурах за нуль.
В некоторых случаях представлены данные других авторов, в скобках приведены результаты более ранних работ.
Принятые сокращения: aq — акватированный; бел. — белый, ж. — желтый, красн. — красный, кор. — коричневый; г. — газ, кр. — кристаллический, т. — твердый; Ьру — 2,2' -бипиридин, dien — диэтилендиамин, dip — 4,4' -бипиридин, edta — этилендиаминтетраацетат, еп — этилентриамин, Gly — глицин (аминоуксусная кислота), phen — 1,10-фенантролин, ру — пиридин, trpy — терпиридин
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Азот
N2H4 + 4Н2О + 2е" = 2NH3 • Н2О + 2ОН 0,11
N2H; + ЗН* + 2е” = 2NH; 1,275
NH2OH + 2Н2О + 2е" = NH3 ♦ Н2О + 2ОН" 0,42
2NH2OH + 2е = N2H4 + 2ОН" 0,73
NH3OH++ 2ГГ + 2е~ = NH; + Н2О 1,35
2NH3OH+ + Н+ + 2е" = N2H^ + 2Н2О 1,41
NH2C1+2Н+ + 2е“ = NH/ + СГ -1,48
NH2C1 + Н2О + 2е" - NH3(r)+ СГ + ОН -0,81
NH2 Cl + NH4+ + 2е“ = 2NH3(r) + СГ -1,4
HN3 + ЗН+ + 2е~ = NH? + N2 1,96
HN3+11Н++ 8е“ = 3NH; 0,695
N' + 7H2O + 6e" = NH3(aq) + + 7OH" -0,62
1^2(г.) + СО2(г) + 6НГ + 6e = CO(NH2)2 + 0,1
+ H2O
N2(r.) +2H2O + 4H" + 2e~ = 2NH3OH" -1,87
N2(r.) +4H2O + 2e = 2NH2OH+ 2OH" -3,04
N2(r) + 5lT + 4e“ = N2H, -0,23
N2(r) + 4H2O + 4e“ - N2H4 + 4OH" -1,16
N2(r) + 8lT + 6e~ = 2NH, 0,275
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
N2(r.) + 6Н1 + бе = 2NH3(r) 0,057
Мад +2Н2О + 6Н+ + 6е“ = 2NH3 • Н2О -0,092
N2(r.) +8Н2О + бе" = 2NH3 • Н2О + 6ОН" -0,74
3N2(r.) + 2е" = 2N; -3,40
NHC12 + ЗН+ + 4е“ = NH; + 2СГ -1,39
H2N2O2 + 2Н+ + 2е~ = N2(r) + 2Н2О 2,65
H2N2O2 + 6Н+ + 4е“ = 2NH3OH+ 0,378
N2O2" + 2Н2О + 2е” = N2(r) + 4ОН" 1,52
N2O^’ + 6Н2О + 4е" = 2NH2OH + 6ОН" -0,76
C6H5NO + ЗН+ + 2е~ = C6H5NH2OH+ 0,60
N2O(r.) + 2Н+ + 2е" - N^.) + Н2О 1,766(1,77)
N2O(r) + 10Н+ + 8е~ = 2NH4+ + Н2О 0,647
N2O(r) + 6Н+ + Н2О + 4е“ = 2NH3OFr -0,05
N2O(r) + 5Н2О + 4е" - 2NH2OH + 4ОН" -1,05
N2O(r) + 8Н1 + Н2О + 8е“ = 2NH3 • Н2О 0,510
2NO(r) + 4Н * + 4е" = N2 + 2Н2О 1,678
NO(r) + 6Н+ + 5е” = NH; + Н2О 0,836
NO(r) + 5Н+ + 5е~ = NH3 • Н2О 0,727
2NO(n) + 2Н2О + 4е“ = N2 (n) + 4ОН" 0,85
2NO(r.) + 2Н+ + 2е" = N2O(r) + Н2О 1,59
HNO3 + Н+ + е" = NO2 (г) + Н2О 0,775
NO3" + 2Н+ + е“ = NO2(r) + Н2О 0,775
2HNO3 +2Н+ + 2е" = NjO^.j + 2Н2О 0,803
2NO; + 4Н+ + 2е" = N2O4(r) + 2Н2О 0,803
NO; + 2Н+ + 2е" = NO; + Н2О 0,835
HNO2 + 7Н * + бе" = NH/ + 2Н2О 0,864
HNO2 + 6lT + 6е“ = NH3(r) + 2Н2О 0,753 (0,755)
2HNO2 + 6Н+ + бе" = N2(r) + 4Н2О 1,44(1,454)
2HNO2 +4Н" + 4е“ = N2O(r) + ЗН2О 1,297
HNO2 + Н" + е" = NO(r) + Н2О 1,004(0,98)
2HNO2 + 4Н+ + 4е“ = H2N2O2 + 2Н2О 0,86
NO; + 2Н+ + е“ = NO(r) + Н2О 1,202
NO2 + Н2О + е“ = NO(r) + 2ОН" 0,46
NO; + 8lT + бе" = NH; + 2Н2О 0,897
Электродные процессы
7
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Азот
NO’ + 7Н+ + бе’ = NH3 • Н2О + Н2О 0,806
NO’ + 6Н2О + бе’ = NH3 • Н2О + 7OJT -0,15
2NO’ + 6Н++ 4е’ = N2O(e) + ЗН2О 1,396
2NO’ + ЗН2О + 4е” = N2O(r) + 6OJT 0,15
2NO” + 2Н2О + 4е‘ = N2O3’ +4ОН -0,14
2NO’ + 8Н+ + бе’ = N2(r) + 4Н2О 1,520
2NO’ + 4Н2О + бе’ = N2(r) + 8OJT 0,41
NO++ е’= NO(r) 1,45
N2O<(r.) + 2е = 2NO2 0,867
N^.) + 4H' + 2e~ = 2NO+ + 2H2O 0,77
N2O4(r) + 2H++ 2e’ = 2HNO2 1,07
ЬЬО^.) + 4H+ + 4e" = 2NO(r.) + 2H2O 1,039
N^.) + 8H++ 8e’ = N2(r) + 4H2O 1,357
N2O4(r) + 4H2O+ 8e = N2(r) + 8OH~ 0,53
N2O4(r.) + I6FF +14e= 2NH4+ + 4H2O 0,89
NO2(r) + 8H+ + 7e" = NH4 + 2H2O 0,897
2NO2(r) + 8FF f 8e’ = N2(r) + 4H2O 1,363
NO2(e) + 2H' + 2e~ = NO(r) + H2O 1,049 (1,045)
NO2(r) + H+ + e’ = HNO2 1,093
NO’ + 2H+ + e’ = NO2(r) + H2O 0,80
NO’ + H2O + e’ = NO2(r) + 2ОВГ -1,688
NO; + 2H* + 2e" = no; + H2O 0,835
no; + 3H* + 2e" = HNO2 + H2O 0,94
no; + H2O + 2e“ = no; + 2ОВГ 0,01
no; + 4H+ + 3e~ = NO(r) + 2H2O 0,96
no; + 2H2O + 3e' = NO(r) + 4ОВГ -0,14
no; + 8H+ + 6e’ = NH3OH+ + 2H2O 0,73
NO; + 5H2O + 6e’ = NH2OH + 7OH -0,30
NO; + ЮН* + 8e’ = nh; + 3H2O 0,87
no; + 7H2O + 8e“ = NH3 • H2O + 9OH -0,12
2NO; + 4H+ + 2e“ = N^) + 2H2O 0,803
NO; + NO + 2H+ + e" = 2HNO2 0,517
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
2NO; + 2Н2О + 2е’ = N2O4(r) + 4ОН —0,86
2NO; + 4Н2О + 8е’ = N,O3’ + 8ОН -0,18
2NO; + 10Н+ + 8е’ = N2O(r) + 5Н2О 1,116
2NO; + 12Н* + 10е" = N^j + 6Н2О 1,246
Актиний
Ас3+ + Зе = Ас -2,13
Ас(ОН)з + Зе' = Ас + ЗОН’ -2,60
Алюминий
[A1F6]3’ + Зе’ = Al + 6F -2,069
А13+Зе =А1 -1,663
А1(ОН)3(т) + ЗН' + Зе’ = А1 + ЗН2О -1,471
АЮ2- + 4Н* + Зе’ = А1 + 2Н2О -1,262
[А1(ОН)4]’ + Зе’ = Al + 4ОН’ -2,35
А1(ОН)3(т) + Зе’ = А1 + ЗОЬГ -2,30
Америций
Ат3+ + Зе= Ат -2,07 (-2,320)
Ат(ОН)3(т) + ЗН' + Зе- Ат + ЗН2О -1,872
Ат2О3(т) + 6Н+ + бе’ = 2Ат + ЗН2О -1,676
2Ат(ОН)4(Т) + 2Н+ + 2е’ = Ат2О3(т.) + + 5Н2О -0,185
2АтО2(т) + 2Н+ + 2е“ = Ат2О3(т) + Н2О -0,072
Ат(ОН)4(т) + Н* + е’ = Ат(ОН)3(т) + + Н2О 0,420
АтО2(Т) + Н2О + Г + е — Am(OH)3(Tj 0,533
АтО2 + 4Н+ + е" = Ат4+ + 2Н2О 1,261 (0,82)
Ат2О5(т) + 2Н+ + 2е’ = 2АтО2(Т) + Н2О 1,418
Ат2О5/т) + ЗН2О + 2Н* + 2е = = 2Ат(ОН)ад 1,530
Ат2О5(т) + 10Н+ + 4е" = 2Ат3+ + 5Н2О 1,639
АтО2+ + 4Н+ + Зе’ = Ат3+ + 2Н2О 1,694
АтО; + 4Н+ + 2е’ = Ат3+ + 2Н2О 1,721
АтСОН)^.) + 4Н+ + е~=Ат3+ + 4Н2О 1,746
2АтО2+ + Н2О + 2е — Ат2О3(Т) + 2Н+ 1,804
АтО^т.) + 4Н+ + е’ = Ат3+ + 2Н2О 1,856
2АтО3(т) + + 2е’ = Ат2О5(т) + Н2О 1,930
Ат4+ + е“ = Ат3+ 2,181 (2,62)
8
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Астат
At2 + 2е" = 2АГ 0,2
2НАЮ + 2ЬГ + 2е’ = At2 + 2Н2О 0,7
2АЮ“ + 2Н2О + 2е" = At2 + 4OFT 0,0
HAtO3 + 4Н+ + 4е’ = HAtO + 2Н2О 1,4
AtO; + 2Н2О + 4е” = AtO’ + 4ОН’ 0,5
Барий
Ва2+ + 2е’ = Ва -2,905
Ва(ОН)2(т) + 2е’ = Ва + 2ОН~ -2,81
Ва(ОН)2 • 8Н2О(т) + 2Н+ + 4е = Ва + -2,99
+ 2ОН’ + 8Н2О
ВаО(ач) + 2Н+ + 2е = Ва + Н2О -2,166
ВаО(т) + 2Н+ + 2е = Ва + Н2О -1,509
ВаО2(т) + 2Н+ + 2е — ВаО<Т) + Н2О -1,023
ВаО2(т) + 2Н+ + 2е — ВаО(а<1) + Н2О 1,679
ВаО2(т) + 4Н+ + 2е' - Ва2+ + 2Н2О 2,419(2,365)
BaO2(aq) + 2Н+ + 2е — ВаО + Н2О 1,047
BaO2(aq) + 2Н+ + 2е = BaO(aq) + Н2О 1,626
BaO2(aq) + 4Н+ + 2е = Ва2+ + 2Н2О 2,365
Бериллий
Ве2+ + 2е = Be -1,847 (-1,97)
Ве(ОН)2(т) + 2Н+ + 2е’ = Be + 2Н2О -1,820
Ве2О3" + 6Н+ + 4е’ = 2Ве + ЗН2О -1,387
ВеО2’ + 4Н++ 2е’= Be + 2Н2О -0,909
Ве(ОН)2(т) + 2е’ = Be + 2OFT -2,599
ВеО + Н2О + 2е = Be + 2ОН" -2,613
Ве2О3" + ЗН2О + 4е“ = 2Ве + 6ОН’ -2,62
[Ве(ОН)4]2’ + 2е = Be + 4ОН’ -2,52
[BeF4]2 +4Н+ + 2е = Be + 4HF -2,042
Берклий
Вк3+ + Зе = Вк -2,01
Вк4+ + е" = Вк3+ 1,6
Вк4+ + 4е = Вк -1,05
Бор
[BF4]’ + Зе" = В + 4F" -1,284 (-1,06)
H3BO3(aq) + ЗН+ + Зе = В + ЗН2О -0,890 (-0,90)
Н2ВО3’ + Н2О + Зе" = В + 4ОН" -1,79
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
ВО3’ + 6Н+ + Зе" = В + ЗН2О -0,165
[В(ОН)4] +Зе=В + 4ОН~ -1,811
[В(ОН)4]“ + 4Н2О + 8е“ = [ВН4] + 8ОН" -1,240
Бром
Вг3“ + 2е’ = ЗВг“ 1,05
Вг2(ж) + 2е“ = 2Вг“ 1,065
ЗВг2(ж) + 2е“ = 2Вг‘ 1,11
Br2(aq.) + 2е = 2Вг “ 1,087
НВгО + Н+ + 2е“ = Вг’ + Н2О 1,341
2НВгО + 2Н+ + 2е" = Вг2 + 2Н2О 1,59(1,609)
ВгО’ + Н2О + 2е’ = Вг’ + 2ОН" 0,766
2ВгО + 2Н2О + 2е = Вг2(г) + 4ОН 0,45
ВгС1 + 2е’= Вг “+СГ 1,2
ВгО3 + ЗН2О + бе" = Вг " + 6ОН" 0,61 (0,584)
ВгО’ + 6Н+ + бе’ = Вг" + ЗН2О 1,44
ВгО, + 2Н2О + 4е“ = ВгО’ + 4ОН" 0,492 (0,54)
ВгО3 + 5Н+ + 4е" - НВгО + 2Н2О 1,49(1,447)
2ВгО“ + 6Н2О + 10е" = Br2 + 12ОН" 0,50
2BrO3 + 12Н+ + 10е’ = Вг2 + 6Н2О 1,52,(1,478)
ВгО’ + Н2О + 2е“ = ВгО3’ + 2ОН" 1,025
ВгО’ + 2Н+ + 2е" = ВгО“ + Н2О 1,853 (1,763)
Ванадий
V2+ + 2е" = V -1,175 (-1,186)
V2O2 + 4Н4 + 4е’ = 2V + 2Н2О -0,820
V3+ + е“ = V2+ -0,255
V3+ + Зе’ = V -0,87 (-0,835)
V2O3(T.) + 2Н+ + 2е’ = 2VO(T) + Н2О -0,486
V2O3(T) + 6Н4 + 2е’ = 2V2+ + ЗН2О 0,135
VO2+ + е’ = VO+ -0,044
VO2+ + 2Н+ + е’ = V3+ + Н2О 0,337
HV2O’ + 9Н + 4е’ = 2V2+ + 5Н2О 0,338
[V(CN)s]3- + е" = [V(CN)„14 0,51
H3V2O; + ЗН4 + 2е’ = V2O4 + ЗН2О 0,806
V2O5 + 6Н+ + 2е“ = 2VO2+ + ЗН2О 0,958
Электродные процессы
9
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Ванадий
hvo3+ зн+ + е = vo2+ + 2н2о 1,1
VO2+ + 2Н+ + е - = VO2+ + Н2О 1,004
VO2+ + 4Н+ + 2е" = V3+ + 2Н2О 0,668
VO2+ + 4Н+ + Зе' = V2+ + 2Н2О 0,361
VO2 + 4Н+ + 5е" = V + 2Н2О 0,25
H3V2O7’ + 7Н+ + 2е’ = 2VO2+ + 5Н2О 1,096
HV2O7’ + 4Н+ + 2е' - HV2O5' + 2Н2О 0,991
2HV2O7’ + 8Н+ + 4е = V4Oj’ + 5Н2О 0,997
VO4 + 6Н+ + 2е" = VO+ + ЗН2О 1,256
VO4" + 4Н+ + 2е = [V(OH)4]+ 1,13
V(OH)3 + е" = V(OH)2 + ОН -1,313
H2VO; + 4Н+ + е = VO2+ + ЗН2О 1,314
HV6OJ7 + 16Н2О + ЗОе" = 6V + ЗЗОН' -1,154
VO4 + 6Н+ + е" = VO2+ + ЗН2О 1,031
VO34 + 6Н+ + 2е" = VO+ + ЗН2О 1,26
Висмут
BiO + 2Н+ + 2е" = Bi + Н2О 0,285
Bi3+ + Зе" = Bi 0,215(0,317)
Bi2O3 + 6FT + бе’ = 2Bi + ЗН2О 0,376
Bi2O3 + ЗН2О + бе = 2Bi + 6ОН' -0,452 (-0,46)
Bi(OH)3 + Зе’ = Bi + ЗОН“ -0,46 (-0,383)
BiOH2+ + Зе’ = Bi + ОН’ 0,072
BiOH2+ + Н+ + Зе’ = Bi + Н2О 0,254
[Bi6(OH)i2]6+ + 18е’ = 6Bi + 12ОН’ -0,234
[Bi9(OH)22]5+ + 27е’ = 9Bi + 22ОН' -0,336
BiO+ + e“ = BiO 0,39
BiO+ + 2H+ + 3e“ = Bi + H2O 0,320
BiO(OH) + H2O + 3e‘ = Bi + ЗОН’ -0,46
BiOCl + 2H+ + 3e’ = Bi + H2O + СГ 0,160 (0,170)
BiOBr + 2ЬГ + Зе’ = Bi + H2O + Br’ 0,152
[BiCl]2+ + 3e’ = Bi + СГ 0,271
[BiCl2]+ + 3e" = Bi + 2СГ 0,249
[BiCU]’ + 3e’ = Bi + 4СГ 0,168 (0,199)
[BiCl6]3’ + 3e" = Bi + 6СГ 0,190
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
[BiBr]2+ + Зе’ = Bi + Br’ 0,273
[BiBr2]+ + Зе’ = Bi + 2Br’ 0,231
[BiBr4]’ + Зе’ = Bi + 4 Br“ 0,168
[BiBr6]3’ + 3e~ = Bi + 6 Br’ 0,135
[Bil4]" + 3e" = Bi + 4Г 0,027
[Bil6]3’ + 3e’ = Bi + 6Г -0,047
Bi4O7 + 2H’ + 2e’ = 2Bi2O3 + H2O 1,338
ВцО7 + H2O + 2e = 2Bi2O3 + 2OH 0,51
2Bi2O4 + 2H* + 2e — Bi4O7 + H2O 1,541
Bi2O4 + 4H4 + 2e’ = 2BiO+ + 2H2O 1,593
Bi2O4 + H2O + 2e’ = Bi2O3 + 2OH" 0,56
2Bi2O4 + H2O + 2e — Bi4O7 + 2OH 0,62
Bi5+ + 2e~ - Bi34 2,0
NaBiO3 + 4H+ + 2e~ = BiO+ +Na+ + 2H2O >1,8
Bi2O5 + 21^ + 2e ~ Bi2O4 + H2O 1,607
Bi2O5 + 10H+ + 4e’ = 2Bi3+ + 5H2O 1,759
Bi2S3 + 6e = 2Bi + 3S2 -0,709
Bi2S3 + 6H+ + 6e = 2Bi + 3H2S(aq) -0,12
Bi2S3 + 6H+ + 6e" = 2Bi + 3H2S(r) -0,091
Водород, дейтерий
H2 + 2e" = 2H’ -2,251
ЬГ + е” = Н(г) -2,106
Н+ + 2е’ = Н“ -1,125
2Н’ + 2е’ “ Н2 0,000
2D+ + 2е~ = D2 -0,0034
Н2О + е’ = Н(г) + ОН’ -2,9345
2Н2О + 2е" = Н2(г) + 2ОН“ -0,828
2D2O + 2е = D2(r.) + 2OD -0,87
Вольфрам
WO2 + 4Н+ + 4е’ = W + 2Н2О -0,119
W2O5 + 2Н+ + 2е" = 2WO2 + Н2О -0,031
2WO3 + 2Н+ + 2е" = W2O5 + Н2О -0,029
WOJ’ + 8Н+ + бе' = W + 4Н2О 0,049 (0,05)
[W(CN)8]3" + е'= [W^N^]4" 0,457
[W(CN)e]’- + е’ = [W(CN)6]4 0,50
2WO4" + 6Н+ + 2е“ = W2O5 + ЗН2О 0,801
WO2’ + 2Н2О + 2е’ = WO2(T) + 4ОН’ -1,259
10
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Вольфрам
WO42 + 4Н2О + бе’ = W + 8ОН’ -1,074 (-1,05)
WO3(T) + 6ЬГ + бе’ = W + ЗН2О -0,09
WO3(T) + 4Н* + е" = WO3+ + 2Н2О 0,26
2WO3(t) + 2РГ + 2е“ = W2O5 + Н2О -0,025 (0,03)
Гадолиний
Gd3+ + Зе" = Gd -2,397 (-2,28)
Gd(OH)3 + ЗН* + Зе’ = Gd + ЗН2О -1,994
Gd(OH)3 + Зе" = Gd + ЗОН -2,82
Галлий
Ga2O + 2Н+ + 2е = 2Ga + Н2О -0,4
Ga2' + 2е" = Ga -0,45
Ga3+ + е" = Ga2+ -0,67
Ga3+ + Зе’ = Ga -0,529
Ga(OH)3 + 3H+ + Зе’ = Ga + ЗН2О -0,419
GaO3’ + 6Н+ + Зе’ = Ga + ЗН2О 0,319
Ga2O3 + 4Н+ + 4е" - Ga2O + 2Н2О -0,5
H2GaO3 + Н2О + Зе’ = Ga + 4ОН’ -1,22
GaOH2+ + Н+ + Зе" = Ga + Н2О -0,505
Ga(OH)2+ + 2Н+ + Зе’ = Ga + 2Н2О -0,435
Ga(OH)3 + Зе" = Ga + ЗОН -1,258
[Ga(OH)4]’+ Зе’ = Ga + 4OFT -1,326
Гафний
Hf4t + 4е" = Hf -1,700 (-1,474)
НЮ2 • 2Н2О + 4Н+ + 4е" = Hf + 4Н2О -1,685
НГО2 + 4Н+ + 4е" = Hf + 2Н2О -1,505 (-1,570)
НЮ2+ + 2Н* + 4е" = Hf + Н2О -1,70
Hf(OH)2+ + 2Н+ + 4е" = Hf + 2Н2О -1,680
НГО(ОН)2(Т) + Н2О + 4е" = Hf + 4ОН -2,50 (-2,60)
Германий
Ge2+ + 2е = Ge 0,000 (0,247)
GeO(aq юр) + 2Н+ + 2е Ge + Н2О -0,286 (0,29)
GeO(aq; ж) + 2ЕГ + 2е = Ge + Н2О -0,130
H2GeO3 + 4Н+ + 2е" = Ge2+ + ЗН2О -0,363
GeO2(T) + 2Н+ + 2е = GeO + Н2О -0,12
GeO2(T) + 4Н+ + 2е" = Ge2+ + 2Н2О -о,з
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
GeO2(T) + 4Н+ + 4е — Ge + 2Н2О -0,246, (-0,15, -0,069)
H2GeO3 + 4Н+ + 2е" = Ge2+ + ЗН2О -0,363
H2GeO3 + 4Н+ + 4е" = Ge + ЗН2О -0,182 (-0,13)
HGeO3’ + 5Н+ + 4е’ = Ge + ЗН2О -0,14
HGeO3 + 2Н2О + 4е" = Ge + 5ОН -0,89
GeO3’ + 6Н+ + 4е" = Ge + ЗН2О 0,328
[Ge(OH)6]2’ +4е" = Ge + 6ОН’ -0,972
Гольмий
Но3+ + Зе’ = Но -2,319 (-2,33)
Но(ОН)3 + ЗН+ + Зе’ = Но + ЗН2О -1,937
Но(ОН)3 + Зе’ = Но + ЗОН’ -2,77 (-2,85)
Диспрозий
Dy2 + + 2е” = Dy -2,2
Dy3+ + е" = Dy2+ -2,5
Dy3+ + Зе’ = Dy -2,353 (2,29)
Dy(OH)3 + 3H+ + Зе’ = Dy + ЗН2О -1,956
Dy(OH)3 + Зе" = Dy + ЗОН" -2,80 (-2,78)
DyO2 + 2Н2О + е“ = Dy(OH)3 + ОН" 3,50
Европий
Еи2+ + 2е" = Ей -3,395 (-2,80)
Еи3+ + Зе“ = Ей -2,406 (-1,99)
Eu3+ + е" = Еи2+ -0,550 (-0,35; -0,429)
Eu(OH)3 + ЗН+ + Зе’ = Ей + ЗН2О -2,002
Еи(ОН)3 + Зе’ = Ей + ЗОН" -2,51
Железо
Fe2+ + 2е" = Fe -0,441 (-0,473)
FeO(T) + 21-Г + 2е — Fe + Н2О -0,052
FeS(T) + 2е = Fe + S2 -0,97 (-0,95; -0,965)
FeS(T.) + Н+ + 2е = Fe + HS -0,997
FeCO3(T) + 2е = Fe + СО3 -0,756
F e2S3(T.) + 2е = 2FeS(T) + S2 -0,7 (-0,715)
Fe2O3(T.) + Н2О + 2Н+ + 2е — 2Fe(OH)2(T) -0,057
Fe2O3(T.) + бН4^ + бе — 2Fe + ЗН2О -0,051
у-РегОз^.) + 6Н4 + 2е’ = 2Fe2+ + ЗН2О 0,74 (0,85)
Электродные процессы
11
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Железо
y-Fe2O3(T) + 6Н+ + бе = 2Fe + ЗН2О -0,007
2y-Fe2O3(T.) + 4Н+ + 2е — Fe2+ + Fe3O4(T) + + 2Н2О 0,71
3y-Fe2O3(T) + 2Н + 2е — 2Fe3O4(T)+ Н2О 0,58
3a-Fe2O3(T) + 2Н+ + 2е = 2Fe3O4(T) + Н2О 0,22
Fe(OH)2(T) + 2Н+ + 2е" = Fe + 2Н2О -0,047
Fe(OH)2(T) + 2е‘ = Fe + 2ОН -0,877
FeOH+ + Н+ + 2е“ = Fe + Н2О -0,279
HFeO2 + Н2О + 2е’ = Fe + ЗОН -0,740
FeOj + Н2О + е“ = HFeO’ + ОН- 0,55
Fe3+ + Зе’ = Fe -0,037
Fe(OH)3(T) + ЗН+ + Зе’ = Fe + ЗН2О 0,059
Fe(OH)3 + Н+ + е’ = Fe(OH)2(T) + Н2О 0,271
Fe(OH)3(T) + е’ = Fe(OH)2(T) + OFF -0,56
[Fe(OH)6]4’ + 2е’ = Fe + 6ОН -0,762
[Fe(CN)6]4' + 2e“ = Fe + 6CN“ -1,728 (-1,50)
[Fe(CN)6]3 *e=[Fe(CN)f,]4- 0,361 (0,543)
(Fe(CN)5(NH3)]2 + e-=[Fe(CN)s(NH3)]3' 0,374
[Fe(phen)3]3+ + e" = [Fe(phen)3]2+ 1,140
[Fe(C2O4)3]3’ + e’ = [Fe(C2O4)2]2’ + + C2OJ’ 0,020
[FeF6]3’ + e" = Fe2+ + 6F 0,400
Fe3+ + H2O + e’ = FeO + 2H+ -0,035
Fe3+ + e’ = Fe2+ 0,771
FeOH2+ + H+ + e’ = Fe2+ + H2O 0,914
3Fe3+ + 4H2O + e~ = Fe3O4(T.) + 8H+ 0,35
Fe3O^T) + 81-Г + 8e = 3Fe + 4H2O -0,085
Fe3O4(T) + 8H+ + 2e’ = 3Fe2+ + 4H2O 0,980 (1,21)
Fe3O4(T.) + 2H+ + 2e = 3FeO(Tj+ H2O -0,173
Fe3O^T) + 6H+ + 2e = FeO(T.) + 2Fe2+ + + 3H2O 0,593
FeO3’ + 5H+ + 4e” = HFeO’ + 2H2O 1,001
FeO3’ + 8H+ + 3e‘ = Fe3+ + 4H2O 1,700(1,9)
FeO3’ + 4H2O + 3e" = Fe(OH)3(T) + 5OH’ 0,72
FeO3’ + 2H2O + 3e" = FeO’ + 4OH’ 0,55
Золото
Au+ + e = Au 1,692(1,83)
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
АиС1(т.) + е’ = Au + СГ 1,17(1,157)
[АиС12]’ + е’ = Аи + 2СГ 1,154
[AuBr2]’ + е’ = Au + 2Br“ 0,960
AuI(T) + е” = Au + Г 0,50 (0,530)
[Aul2]’ + е“ = Au + 2Г 0,578
[Au(CN)2]’ + е’ = Au + 2CN“ -0,61 (-0,764)
[Au(SCN)2]“ + e’ = Au + 2SCN“ 0,69 (0,662)
AuOH(T) + H+ + e“ = Au + H2O 2,33
Au2+ + e“ = Au+ > 1,29
AuO + 2H+ +2e“ = Au + H2O 1,37
Au(OH)2(t) + 2FT +2e’ = Au + 2H2O 2,01
2Au(OH)2(t) + 2H* + 2e’ = Au2O + 3H2O 2,75
Au3+ + e" = Au2+ 1,29
Au3+ + 2e’ = Au+ 1,401 (1,36)
Au3+ + 3e“ = Au 1,498(1,52)
[AuBr4]“ + 2e” = AuBr2“ + 2Br ’ 0,82 (0,802)
[AuBr4]“ + 3e’ = Au + 4 Br ’ 0,854 (0,87)
[AuCU]’ + 2e’ = [AuCl2]’ + 2СГ 0,926
[AuC14]’ + 2e" = AuCl(T) + ЗСГ 0,924
[AuCl4]” + 3e “ = Au + 4СГ 1,00(1,002)
[AuI4]“ + 2e’ = [AuI2] + 2Г 0,55
[AuI4] ’ + 3e = Au + 4Г 0,56
[Au(SCN)4]“ + 2e’ = [Au(SCN)2]’ + + 2SCN’ 0,623
[Au(SCN)4]’ + 3e’ = Au + 4SCN" 0,636 (0,655)
[AuCl3(OH)]’ + H+ + 3e’ = Au + ЗСГ + + H2O 1,123
[Au(OH)4]’ + 3e’ = Au + 4OH“ 0,490
AuO(OH) + 3H+ + 3e" = Au + 2H2O 1,253
Au2O3(t.) + 6H+ + 6e’ = 2 Au + 3H2O 1,457
Au2O3(T) + 2FT + 2e’ = 2AuO(T) + H2O 1,79
Au2O3(t) + 4FF + 4e = Au2O(T) + 2H2O 1,64
Au(OH)3(T) + H+ + e — AuO(T) + 2H2O 1,95
Au(OH)3(T) + 3H+ + 2e" = Au+ + 3H2O 1,50
Au(OH)3(T) + 3H+ + 3e’ = Au + 3H2O 1,45
2Au(OH)3(T) + 4НГ + 4e = Au2O<T) + + 5H2O 1,71
AuO’ + 4H* + 3e’ = Au + 2H2O 1,60
H2 AuO’ + 4H+ + 2e’ = Au+ + 3H2O 1,85
12
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Золото
Н2АиО3" + 4Н+ + Зе = Аи + ЗН2О 1,80
H2AuOJ + Н2О + Зе = Au + 4ОН" 0,7
HAuO3 + 5Н+ + Зе" = Аи + ЗН2О 2,059 (2,06)
HAuO3" + 5Н+ + 2е" = Аи+ + ЗН2О 2,24
AuO3" + 6Н1 + Зе’ = Аи + ЗН2О 2,33
H3AuO3 + ЗН* + 2е" = Аи+ + ЗН2О 1,502
НзАиОз + ЗЬГ + Зе" = Аи + ЗН2О 1,565
АиО2(Т) + 4Н + е = Аи3+ + 2Н2О 2,507 (2,51)
АиО2(т) + Н2О + е’ = НАиО3" + Н+ 0,822 (0,82)
АиО2(т.) + Н2О + е" = H2AuOJ 1,61
AuO2(t.) + Н2О + Н+ + е" = Н3АиО3 2,30
АиО2(т) + 2Н++ 2е“ = АиО + Н2О 2,14
АиО2(т) + 4Н Ч 4е" = Аи + 2Н2О 1,75
2АиО2(т.) + 2Н++ 2е = Аи2Оз(Т) + Н2О 2,46
2АиО2(Т) + 6Н + бе = Аи2О(т) + ЗН2О 1,91
Индий
In+ + е" = In -0,139 (-0,126; -0,25)
1пС1(Т) + е“ = In + СГ -0,34
In(OH)3(T) + ЗН+ + Зе" = In + ЗН2О -0,172
1п(ОН)з(т) + Зе’ = In + ЗОН’ -1,0 (-1,066)
1п2О3(т) + ЗН2О+ бе— = 21п + 6ОН -1,18
In2+ + е’ = 1п+ -0,35
In3+ + е" = 1п2+ -0,45
In3+ + 2е" = 1п+ -0,443 (-0,444)
1п3+ + Зе- = In -0,338
InO" + 4Н+ + Зе’ - In + 2Н2О 0,146
1п28з(Т) + 6НЧ бе — 2In + 3H2S(aq) -0,568
1п28з(т) + 6Н++ бе = 2In + 3H2S(rj -0,539
Иод
1з(т.) + 2е = 2Г 0,536
l2(aq) + 2е" = 2Г 0,621
I; + 2е~ = ЗГ 0,536 (0,545; 0,535)
10’ + Н2О + 2е" = Г + 2ОН" 0,49 (0,472)
210" + 2Н2О + 2е" = I2(r) + 4ОН" 0,433 (0,45)
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
НЮ + Н+ + 2е" = Г + Н2О 0,99 (0,985)
2IBr(aq)+ 2е" = 12(т.) + 2Вг" 1,02(1,026)
2IBr(aq) + 2е" - 12Вг'+ Вг’ 0,973
21Вг + 2е" = 12(т) + 2Вг" 0,874
21Вг2" + 2е" = 12(т) + 4Вг” 0,87
21Вг" + 2е“ = 12Вг" + ЗВг’ 0,821
21С1Вг" + 2е" = 12(т) + 2СГ + 2Вг" 0,928
2ICN + 2е" - Г + CN" 0,30
2ICN + 2Н+ + 2е’ - 12(т) + 2HCN(aq) 0,695 (0,609)
2ICN + 2Н4 + 2е’ = I2(aq) + 2HCN 0,63
2IC1’ + 2е" “ 12(т) + 4СГ 1,06(1,07)
2IC1' + 2е" = 12СГ + ЗСГ 0,995
1С13(Т) + 2е" = 12С1(Т.) + 2СГ 1,312
21С13(Т) + бе — I2(T) + 6С1 1,281
2ICl(aq) + 2е = I2(T) + 2С1 1,19(1,20)
21СЦ) + 2е’ = 12СГ + СГ 1,12
НЮ3 + 4Н+ + 4е" - HIO(aq) + 2Н2О 1,098(1,128)
НЮ3 + 5Н+ + 2СГ + 4е" = 1С12" + ЗН2О 1,214
10; + 5Н+ + 4е" = HIO(aq) + 2Н2О 1,14
210; + 12Н+ + 10е’ = 12(т) + 6Н2О 1,195
210’ + 6Н2О + 10е - I2(T) + 12ОН’ 0,21
ю; + ЗН2О + бе’ = Г + 6ОН’ 0,26 (0,257)
Ю3" + 2Н2О + 4е" = Ю" + 4ОН’ 0,56 (0,14)
ю; + 6Н ‘ + бе" = Г + ЗН2О 1,085
ю; + 2Н* + 2е" = ю; + 6Н2О 1,653
ю; + 8Н+ + 8е" = Г + 4Н2О 1,4
Н5Ю6 + 2Н+ + 2е" = НЮз + ЗН2О 1,626
Н5Ю6 + Н+ + 2е" = ю; + ЗН2О 1,603
Н5Ю6 + 7Н+ + 8е" - Г + 6Н2О 1,24
Н4Ю; + 2Н+ + 2е" = Ю3 + ЗН2О 1,70
Н3Ю2" + 2е" = Ю3" + ЗОН’ 0,656
Н3Ю62" + ЗН2О + 8е" = Г + 9ОН" 0,37
H3IO2 +2е’= IO3" +ЗОН" 0,7
Н2Ю3’ + Н2О + 2е" = IO" + 4ОН" 0,603
Электродные процессы
13
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Иридий
1г3+ + Зе = 1г 1,15 (1,156; 1,000)
[IrCtf +Зе Мг + бСГ 0,77 (0,86; 0,720)
[IrCtf- + е" = [IrCUJ3 1,017(0,867)
[IrCU]2 + 4е~ = Ir + 6СГ 0,86
[IrBr6]2'4-e = [1гВг6]3 0,947, (0,99)
[1гВг6]3 + е’= [1гВг6]4 0,99
1г20з(Т) + 6Н+ 4- бе — 21г 4- ЗН2О 0,926
1г2О3(т) 4- ЗН2О 4- бе = 21г 4- 6OFT 0,1
[Irl6|2 +e- = [lrl6f- 0,48
IrO2(T) + 4Н+ 4- 4е~ = 1г 4- 2Н2О 0,93
1гО2(т) + 4Н+ 4- е" = 1г3+ 4- 2Н2О 0,720
1гО2(т) 4- 2Н2О 4- 4е" = 1г 4- 4ОН 0,1
21гО2(Т.) 4" 2Н 4- 2е = 1г2О3(Т) 4" Н2О 0,926
IrO2 4- 2Н2О 4- 2е" = 1гО2(т) 4- 4ОН' 0,4
Иттербий
УЪ2+ 4- 2е = УЪ -2,8
Yb3' 4- е' = Yb2+ -1,205 (-1,05)
Yb3+ 4-Зе'=Yb -2,22 (-2,267)
Yb(OH)3(T) 4- Зе’ = Yb2+ + ЗОН -2,74
Иттрий
Y3+ 4- Зе“ = Y -2,372 (-2,37)
Y2O3 4- 6Н' 4- бе’ = 2Y 4- ЗН2О -1,676
Y(OH)3(T) 4- Зе' = Y 4- ЗОН“ -2,81 (-2,85)
Кадмий
Cd 4- ЬГ 4- е = CdH -2,417
Cd2+ + 2е" = Cd -0,403
Cd2+ 4-2e’ = 2Cd >W)
2Cd2+4-2e'= Cd22^ <(-0,6)
CdO(T) 4- 21-Г + 2e~ = Cd + H2O 0,063
CdO(T) 4- H2O 4- 2e~ - Cd 4- 2OH’ -0,783
Cd(OH)2(T) 4- 2H+ 4- 2e~ = Cd + 2H2O 0,005
Cd(OH)2(T) 4- 2e' = Cd 4- 2OH' -0,824
[Cd(OH)3]' 4- 2e" = Cd 4- 3OH“ -0,668
[Cd(OH)4]2' 4- 2e" = Cd 4- 4OH’ -0,670
CdCO3(T) 4- 2e = Cd 4" CO2 -0,734
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
[Cd(NH3)4]2+ + 2е' = Cd + 4NH3 -0,61 (-0,622)
[Cd(CN)4]2' 4- 2e' = Cd + 4CN' -1,09 (-1,028; -0,943)
[CdCl]+ + 2e’ = Cd 4- CT -0,483
CdCl2 + 2e'-Cd + 2СГ -0,502
[CdCl3]' + 2e' = Cd 4- ЗСГ -0,483
[CdCl4]2 4- 2e" = Cd 4- 4СГ -0,453
[CdBr]+ 4- 2e" = Cd 4- Br’ -0,466
CdBr2 + 2e' = Cd 4- 2Br" -0,488
[CdBr3]' 4- 2e’ - Cd + 3Br’ -0,495
[CdBr4]2' 4- 2e’ = Cd 4- 4Br’ -0,488
[Cdl]+ 4- 2e’ = Cd 4- Г -0,467
Cdl2 + 2e" = Cd 4- 2Г -0,508
[Cdl3]“ 4- 2e" = Cd 4- ЗГ -0,542
[Cdl4]2'4-2e" = Cd 4-4Г -0,568
CdS(T) 4- 2e = Cd 4- S2 -1,24 (-1,208; -1,255)
CdS(T) + H2O 4- 2e' = Cd 4- HS’ 4- OH' -1,239
CdS(T) 4- 2H+ 4- 2e — Cd 4- H2S(aqj -0,619
CdS(T ) 4- 2H+ 4- 2e = Cd 4- H2S(r.) -0,589
CdSe(T) 4" 2e — Cd 4- Se2 -1,38
CdTe(T) 4- 2e" = Cd 4- Те2' -1,62
Калий
K+ + e' = К -2,925 (-2,924)
Кальций
Са2+ 4- 2е" = Са -2,866 (-2,84)
Са 4" 2Н* 4- 2е = СаН2 0,776
СаО^ 4- 2Н* 4" 2е = Са 4- Н2О -2,189
СаО 4- 2Н* 4- 2е — Са 4- Н2О -1,902
Са(ОН)2(Т.) 4- 2е = Са 4- 2ОН -3,03
СаО2 4-4^4- 2е" = Са2+ 4- 2Н2О 2,224
СаОг 4- 2Н+ 4- 2е = СаО^ 4- Н2О 1,547
СаО2 4- 2Н+ 4- 2е = СаО 4- Н2О 1,260
Кислород
Н2О2 4- 2Н+ 4- 2е" = 2Н2О 1,776 (1,77)
Н2О2 4- 2е' = 2ОНГ 0,78
НО; + 2Н' + 2е' = ОН' 4- Н2О 1,706
О + Н2О4- 2е' = 2ОН' 1,602
14
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Кислород
О + 2Н" + 2е- Н2О 2,34(2,421)
О2 + 2Н+ + 2е= О + Н2О 0,037
О2 + 21-Г + 2е’= Н2О2 0,682
O2 + W + 2e = НО’ 0,338
О2 + Н2О + 2е’ = НО’ + ОН’ -0,076
О2 + 2Н2О + 2е" = Н2О2 + 2ОН’ -0,076
О2 + 4Н+ + 4е" = 2Н2О 1,229(1,228)
О2 + 2Н2О + 4е" = 4ОН’ 0,401
О3 + Н2О + 2е = О2+2ОН 1,24(1,246)
O3 + 2tf + 2e=O2 + H2O 2,076 (2,07)
О3 + 6Н* + бе’ = ЗН2О 1,511
OF2(r) + 2Н+ + 4е’ = Н2О + 2F’ 2,153
OF2(r) + 4Н+ + 4е“ = Н2О + 2HF(aq) 2,246
ОРад + ЗН+ + 4е- = Н2О + HF“(aq) 2,209
Кобальт
Со2+ + 2е" = Со(г) -0,277
СоСО3(т) + 2е" = Со + СО2’ -0,64 (-0,98)
Со(ОН)2(т) + 2е = Со + 2ОН" -0,733
Со(ОН)2(т) + 21Г + 2е = Со + 2Н2О 0,095 (-0,277)
Со3+ + е’ = Со2+ 1,808 (1,95)
Со3+ + Зе — Со(Т.) 0,46, (0,40)
[Co(NH3)6]2+ + 2е = Со + 6NH3 -0,42
[Со(СО)4]2+ + 2е“ = [Со(СО)4] -0,4
p-CoS(T.) + 2е — Со + S2 -1,07 (-1,02)
a-CoS(T) + 2е = Со + S2 -0,90 (-0,89)
СоО(т) + 2Н+ + 2е" = Со + Н2О 0,166
Со3О4(Т) + 2HF + 2е — ЗСоО(Т.) + Н2О 0,777
СозО^.) + 2Н2О+ 2Н% 2е = ЗСо(ОН)2 0,993
Со3О4(т.) + 8НЧ 2е’ = ЗСо2+ + 4Н2О 1,746
Со(ОН)з(т) + е" = Со(ОН)2(т) + ОН 0,17
[Co(NH3)6]3t + е‘ = [Co(NH3)6]2+ 0,108 (0,10)
[Co(NH3)s]3+ + е- = [Co(NH3)5]2+ 0,37
[Co(dien)2]3+ + е’ = [Co(dien)2]2+ -0,233
[Cofdip)3]3h + е- = [Cofdip)3]2+ 0,34
[Со(рЬепЬГ + e- = [Co(phen)3f+ 0,40
[Co(CN)6f “ + e- = [Co(CN)6]“- -0,83
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Со2О3(т) + Н2О + 2е’ = 2НСоО2 -0,128
ЗСо2О3(т)+ 2Н+ + 2е“ = 2Со3О4(т) + Н2О 1,018
[Со(Н2О)6]3+ + е’ = НСоО’ +4Н2О+ЗН+ -0,0065
a-[Co(Gly)3 ] + е“ = a-[Co(Gly)3]’ 0,20
[Co(edta)]’ + е’ = [Co(edta)]2’ 0,38 (0,60)
НСоО’ + ЗН+ + 2е = Со + 2Н2О 0,659
СоО2(т) + 4Н+ + е= Со3+ + 2Н2О 1,416
СоО2(т) + 4Н+ + 2е = Со2+ + 2Н2О 1,612(1,746)
СоО2(Т) + Н2О + 2е = СоО(Т) + 2ОН 0,7
2СоО2(Т) + 2Н+ + 2е — Со2О3(Т) + Н2О 1,477
Кремний
Si + 4H+ + 4e’ = SiH4 0,102
SiO + 2Н+ + 2е~ = Si + Н2О -0,809
SiO2(K„pi„ + 4fT + 4е~ = Si + 2Н2О -0,857 (-0,86)
FUSiO4 + 2Н+ + 2е" = SiO + ЗН2О -0,887
FUSKX + 4Н* + 4е’ = Si + 4Н2О -0,848
H3SiO; + ЗН’ + 2е’ = SiO + ЗН2О -0,615
H3SiO; + 5Н+ + 4е’ = Si + 4Н2О -0,712
H2SiO2’ + 4Н+ + 2е’ = SiO + ЗН2О -0,248
H2SiO3(T) + 4Н+ь 4е’ = Si + ЗН2О -0,807 (-0,84)
H2SiO3(aq) + 4Н+ + 4е’ = Si + ЗН2О -0,79 (-0,789)
SiO3’ + ЗН2О + 4е’ = Si + 6ОН’ -1,69
SiO3’ + 6Н++ 4е" = Si + ЗН2О -0,455
SiF62’ +4e = Si + 6F" -1,2 (-1,24;-1,37)
Ксенон
XeF + e’ = Xe + F” 3,4
XeF2 + е" - XeF + F 0,90
XeF2 + 2tf+2e= Xe + 2HF 2,64
XeO3 + 61Г+ 6e’ = Xe + 3H2O 2,10(1,80)
HXeO: + 3H2O + 6e’ = Xe + 7OH 1,24
H4XeO6 + 2tf+ 2e~ = XeO3 + 3H2O 3,0(2,42)
H4XeO6 + 8H*+ 8e’ = Xe + 6H2O 3,18
HXeO3’ + 2H2O + 2e’ = HXeO; + 4OH 0,99
HXeO3’ + 5H2O + 8e“ = Xe + 11 OH 1,18
Электродные процессы
15
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Лантан
La3+ + Зе = La(T.) -2,38 (-2,522)
La2O3(r) + 6Н+ + бе — 2La + ЗН2О -1,856
La(OH)3(T.) + Зе"= La + ЗОН" -2,80 (-2,72)
Литий
Li+ + е = Li -3,040 (-3,045)
Лютеций
Lu3+ + 3e =Lu -2,255
Lu2O3(t) + 6IT" + бе — 2Lu + ЗН2О -1,892
Lu(OH)3 + Зе" = Lu + ЗОН" -2,83 (-2,72)
Магний
Mg2+ + 2е" = Mg -2,356 (-2,363)
Mg(OH)2(T) + 2е"= Mg + 2OFT -2,69
Mg(OH)2(T) + 21Г + 2e" = Mg + 2H2O -1,862
MgOH+ + H4 + 2e" = Mg + H2O -2,023
Марганец
Mn2+ + 2e = Mn -1,179 (-1,18;-1,192)
MnOH4 + H+ + 2e" = Mn + H2O -0,878
Mn(OH)2(T) + 2H* + 2e" = Mn + 2H2O -0,727
Mn(OH)2(T) + 2e" = Mn + 2OH" -1,56
MnS(T) + 2H + 2e" = Mn + H2S -0,986
MnS(T.) + 2e“ = Mn + S2~ -1,575
MnCO3(T.) + 2e~ = Mn + CO2" -1,48
[Mn(CN)6]4" + e" = [Mn(CN)6]5" -1,06
Mn3+ + e" = Mn2+ 1,509 (1,51; 1,499)
Mn(OH)3(T) + e" = Mn(OH)2(T) + OH 0,1 (0,15)
Mn(OH)3W + 3H++ e" = Mn2+ + 3H2O 1,84
Mn2O3(T.) + 3H2O + 2e“ = 2Mn(OH)2(T) + + 2OH" -0,25 (-0,225)
Mn2O3(T) + 6H+ + 2e" = 2Mn2+ + 3H2O 1,443(1,48)
[Mn(CN)6]’- + e = [Mn(CN)6]4- -0,22 (-0,244)
Mn3O4(T.) + 8H4 + 2e~ = 3Mn2+ + 4H2O 1,75
Mn3O4(T) + 4H2O + 2e“ = 3Mn(OH)2(T) + + 2OH" -0,324
MnO2(T) + 41~T + e" = Mn3+ + 2H2O 0,95
Mn4+ + 2e“ = Mn2+ 1,84
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
МпО2(тэ + 4Н+ + 2е" = Мп2+ + 2Н2О 1,228 (1^3)
МпО2(Т) + 2Н2О + 2е~ = Мп(ОН)2(т.) + + 2ОН" -0,05
2МпО2(Т) + 2Н4 + 2е" = Мп 2О3(т) + Н2О 0,98
2МпО2(т) + Н2О + 2е = Мп 203<Т) + + 2ОН" 0,15
2уМпО2(т) + Н2О + 2е~ = Мп 2О3(т.) + + 2ОН" 0,32
МпО2(т) + Н2О + е" = у-МпО(ОН)(Т) + + ОН" 0,19
у-МпО2(т) + Н2О + е” = у-МпО(ОН)(т.) + + О1Г 0,36
у-МпО2(Т) + Н2О + е“ = а-МпО(ОН)(Т) + + ОН" 0,30
МпО4 + 8Н+ + 2е" = Мп3" + 4Н2О 0,95
МпО4 + 2Н2О + е" = МпО2(т) + 4ОН" 0,96
МпО2" + 4Н + 2е" = МпО2(т) + 2Н2О 2,27 (2,257)
МпО2" + 2Н2О + 2е" = МпО2(т) + 4ОН" 0,62
МпО4 + е“ = МпО4“ 0,564 (0,558)
МпО; + 4Н+ + Зе" = МпО2(т) + 2Н2О 1,692 (1,69; 1,725)
МпО4" + 2Н2О + Зе" = МпО2(т) + 4ОН" 0,60
МпО4" + 8Н" + 5е" = Мп2+ + 4Н2О 1,507(1,51)
МпО; + 4Н2О + 5е" = Мп(ОН)2(Т) + + 6ОН" 0,34
Медь
Си + Н+ + е" = СиН -2,775
Си+ + е" = Си 0,520
[Cu(NH3)2]+ + е" = Си + 2NH3(aq) -0,100 (-0,12)
СиС1(т) + е” = Си + СГ 0,137(0,121)
СиВГ(Т) + е" = Си + Вг" 0,03
Си!(Т) + е" = Си + Г -0,185 (-0,182)
CuSCN(r) + е - Си + SCN" -0,27 (-0,31)
[Cu(CN)2]~ + е’ = Си + 2CN" -0,43 (-0,44)
[СиС12]" + е" = Си + 2СГ 0,19
[СиВг2]" + е" = Си + 2ВГ 0,033
[Си12]“ + е" = Си + 2Г 0,00
16
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Медь
Си2О(Т) + 2Н4 + 2е — 2Cu + Н2О 0,471
Cu2O(T) + Н2О + 2е" = 2Си + 2ОН -0,365
Cu2S(T) + 2е = 2Cu + S2 -0,898 (-0,93)
Cu2S(T) + Н2О + 2е" = 2Cu + HS” + ОЬГ -0,923
Cu2S(T.) + 2Н’ + 2е = 2Cu + H2S(aq) -0,302
Cu2S(T.) + 2Н4 + 2е = 2Cu + H2S(r) -0,273
Cu2+ + Q = Cu+ 0,153 (0,153)
Cu2+ + 2e” = Cu 0,34 (0,337; 0,338)
Cu2+ + СГ + e = CuCl(T.) 0,538 (0,559)
Cu2+ + Br~ + e“ = CuBr(T) 0,654 (0,640)
Cu2+ +1 + e = CuI(T) 0,861 (0,86)
Cu2+ + SCN” + q = CuSCN(r) 0,96
Cu2+ + 2SCN“ + e = [Cu(SCN)2]“ 1,12
Cu2+ + CN’ + e‘ = CuCN 1,12
2Cu2+ + H2O + 2e = Cu2O + 2tT 0,203
CuCO3(T) + 2e — Cu + CO2 0,053
[Cu(NH3)4]2+ + e ’ = [Cu(NH3)2]+ + 2NH3 0,100
[Cu(NH3)4]2+ + 2e" = Cu + 4NHW -0,05 (-0,07)
2Cu(OH)2(t) + 2e = Cu2O(t) + 2OH + + H2O -0,08
Cu(OH)2(T.) + 2H+ + 2e = Cu + 2H2O 0,609
Cu(OH)2(T) + 2e’ -Cu + 2OH -0,22
CuO(T) + 2H4 + 2e — Cu + H2O 0,570
2CuO(T) + 2H4 + 2e = Cu2O + H2O 0,669
2CuO(T) + H2O + 2e = Cu2O + 2OH -0,22 (-0,226)
CuO(T) + H2O + 2e = Cu + 2OFF -0,29
HCuO” + 3H+ + 2e" = Cu + 2H2O 1,127
CuO2’ + 4fC + 2e" = Cu + 2H2O 1,515
[Cu(OH)4]2 + 2e’ = Cu + 4ОНГ -0,132
2CuO2’ + 6H+ + 2e" = Cu2O + 3H2O 2,560
[Cu(SCN)]+ + e" = CuSCN -0,854
CuS(T) + 2e — Cu + S2 -0,70 (-0,722; -0,79)
CuS(T) + H2O + 2e" = Cu + HS’ + OFF -0,754
CuS(T.) + 2H4 + 2e — Cu + H2S(aq) -0,133 (-0,259)
CuS(T) + 2H+ + 2e — Cu + H2S(r) 0,104
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Си3+ -е-Си2' 1,8
СиО2 + 2Н2О + е" = Си(ОН)2(т) + 2ОН 0,8
Менделевий
Md2+ + 2е’ = Md -2,4
Md3+ + е = Md2+ -0,15
Md3+ + Зе’ = Md -1,7
Молибден
Мо3+ + Зе’ = Мо -0,200
[Mo(CN)6]3’ + е = [McXCNM4- 0,725
[Mo(CN)7]4’ + е‘ = [Mo(CN)7]5’ 0,730
[Mo(CN)8]3- + е = [Mo(CN),]4- 0,725
[Mo(CN)8]2’ + 2e’ = [Mo(CN)8]4’ 0,73
MoO2(T) + 4H+ + e' = Mo3+ + 2H2O -0,008
MoO2(T) + 4H+ + 4e“ = Mo + 2H2O -0,072 (-0,152)
MoO2(T) + 2H2O + 4e’ = Mo + 4OH’ -0,983
MoO2(T) + 4tT + 2e’ = Mo2+ + 2H2O = 0,0
MoO2 + e ’ = MoO2(T) -0,08
Moo* + 4H" + 2e" = Mo3+ + 2H2O 0,089 при 30 °C
MoO2 + e" = MoO24 0,48
MoO*+ + 2H* + e’ = MoO3+ + H2O 0,483 при 30 °C
H2MoO4(aq) + 2J-C + 2e” = MoO2(T) + 2H2O 0,39
H2MoC>4(T.) + 2Ff + 2e” - MoO2(T) + 2H2O -1,091
H2MoO4 + 6H+ + 3e’ = Mo3+ + 4H2O 0,428
H2MoO4 + 6H+ + 6e’ = Mo + 4H2O 0,0
H2MoO4 + 2H4 + e~ = MoO* + 2H2O = 0,4
3H2MoO4 + 2H+ + 2e’ = (MoO2)2MoO4 + + 4H2O = 0,6
4H2MoO4 + 2H* + 2e“ = Мо4Оц(т) + + 5H2O 0,865
HMoO; + 3H4 - 2e - MoO2(T) + 2H2O 0,429
HMoO; + 7H+ + 6e’ = Mo + 4H2O 0,095
MoO2’ + 4H" + 2e" = MoO2(T) + 2H2O 0,606
MoO^’ + 2H2O + 2e’ = MoO2(T) + 4OH’ -0,780
MoO2’ + 4H2O + 6e" = Mo + 8OFT -0,913
Электродные процессы
17
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Молибден
МоО4 + 8Н+6е -Мо + 4Н2О 0,154
МоО3 + 4Н* + е’ = МоО3* + 2Н2О 0,5
МоО3(Т.) + 2НГ + 2е’ = МоО2(Т.) + Н2О 0,320 (0,34)
МоО3(т) + бН’ + Зе’ = Мо3* + ЗН2О 0,317
МоО3(Т) + 6Н* + бе’ = Мо + ЗН2О 0,06
МоО4(т) + 2Н* + 2е’ = Н2МоО4 1,567
МоО4(т) + 2Н* + 2е” = МоО3(Т.) + Н2О 1,402
МоО<цТ) + 4Н* •+ 4е = МоО2(Т) + 2Н2О 0,861
МоО^) + 8Н* + 8е’ = Мо + 4Н2О 0,394
Мышьяк
As + ЗН2О + Зе’ = AsH3 + ЗОН’ -1,370
As + ЗНГ + 3e-AsH3 -0,608 (-0,238)
As3+ + Зе’ = As 0,3
AsO* + 2Н* + Зе’ = As + Н2О 0,254
As2O3(T.) + 6Н* + 6e“ = 2As + 3 H2O 0,234
HAsO2 + 3H* + 3e“ = As + 2H2O 0,248
AsO’ + 2H2O + 3e’ = As + 4OH -0,68
AsO3’ + 3H2O + 3e~ = As + 6OH’ -1,572
As2S3(T) + 6e“ = 2 As + 3S2~ -0,609
As2S3(t) + 3H2O + 6e — 2As + 3HS + -0,641
+ ЗО1Г
As2S3(T) + 6H* + 6e — 2 As + 3H2S(aq) -0,020
As2S3(t.) + 6H*+ 6e — 2 As + 3H2S(rj 0,009
AsS2 + 3e’ = As + 2S2’ -0,8 (-0,75)
AsO2(T.j + 4H* + 4e = As + 2H2O 0,429
As2Os(T) + 4H* + 4e — Аз20з(Т) + 2H2O 0,721
As2O5(T) + I OH* + 10e“ = 2As + 5H2O 0,429
H3AsO4 + 3H* + 3e’ = As + 3H2O 0,24
H3AsO4 + 3H* + 2e” = AsO* + 3H2O 0,550
H3AsO4 + 2H* + 2e’ = H3AsO3 + H2O 0,559
H3AsO4 + 2H’ + 2e’ = HAsO2 + 2H2O 0,560
2H3AsO4 + 4НГ + 4e“ = Аз20з(Г) + 5H2O 0,580
H2AsO4 + 2H* + 2e = AsO2 + 2H2O 0,666
HAsO2’ + 3H* + 2e’ = AsO2 + 2H2O 0,609
HAsO2’ + 4H* + 2e~ = HAsO2 + 2H2O 0,881
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
2HAsO42’ + 8Н’ + бе’ = As2O3(t) + 5Н2О 0,901
AsO4“ +4Н’ + 2е - AsO’ +2Н2О 0,977
AsO* + 2Н2О + 2е" = AsO’ + 4ОН’ -0,67
AsO| + 8Н’ + 5е = As + 4Н2О 0,648
2AsO|’ + ЮН* + 4е’ = As2O3(T.) + 5Н2О 0,687
AsS3’ + 2е’ = AsS3’ + S2’ -0,600
Натрий
Na+ + e =Na -2,714 (-2,713)
Неодим
Nd2* + 2е’ = Nd -2,2
Nd3* + е“ = Nd2* -2,6
Nd3* + Зе’ = Nd -2,431
Nd2O3(T) + 6H* + 6e’ = 2Nd + 3H2O -1,811
Nd(OH)3(T) + 3e’ = Nd + ЗОН’ -2,78 (-2,54)
Nd4* + e’ = Nd3* 4,9
NdO2(T) + 2H2O + e’ = Nd(OH)3(T.) + OH’ 2,5
Нептуний
Np3* + 3e’ = Np -1,79 (-1,856)
Np2O3(T.) + 6H* + 6e’ = 2Np + 3H2O -1,420
Np(OH)3(T) + 3e’ = Np + ЗОН’ -2,2
Np4* + e’ = Np3* 0,152(0,15)
Np4* + 4e” = Np -1,3
NpO2(T) + 4H* + e’ = Np3* + 2H2O 0,337
NpO2(T) + 2H2O + e’ = Np(OH)3(T) + OH -2,2
2NpO2(T.) + 2H* + 2e’ = Np2O3(T) + H2O -0,962
Np(OH)4(T) + 4H* + e’ = Np3* + 4H2O 0,391
2Np(OH)4(T) + 2H* + 2e — Np2O3(T.) + + 5H2O -0,928
NpO* + 4H* + e’ = Np4* + 2H2O 0,55
NpO* +e=NpO2 0,564
NpO* + 2H2O + e“ = Np(OH)4(T.) 0,530
NpO* + 4H* + 2e" - Np3* + 2H2O 0,451
Np2O5(T.) + 2H* + 2e" = гИрО^х) + H2O 1,253
Np2Os(t) + 3H2O + 2FT + 2e = = 2Np(OH)4<t) 1,219
NpO2OH(r) + e“ = NpO2(T) + OH“ 0,3
18
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Нептуний
NpO^ + е = NpO* 1,149(1,24)
NpO3(T.) + 2Н+ + е” = NpO2+ + Н2О 1,998
2NpO3(T) + 2Н* + 2е — Np2O3(T) + Н2О 1,310
NpO2(OH)2(T) + е" = NpO2(OH)(T) + ОН 0,6
NpO3+ + 2Н+ + е~ = NpO2 + Н2О 2,04
NpO' + 4Н” + е’ = NpO2(OH)2(T) + Н2О 0,58
Никель
Ni2+ + 2е” = Ni -0,250 (-0,25)
NiO(T) + 2Н+ + 2е” = Ni + Н2О 0,116
NiOH2O + 2Н+ + 2е” = Ni + 2Н2О 0,117
Ni(OH)2(T) + 2Н + 2е" = Ni + 2 Н2О 0,110
Ni(OH)2(T) + 2е” = Ni + 2ОН -0,72
HNiO’ + ЗЬГ + 2е” - Ni + 2Н2О 0,648
[Ni(NH3)6]2+ + 2е" = Ni + 6NH3(aq) -0,49 (-0,476)
NiCO3(T) + 2е" ~ Ni + СО3 -0,45
a-NiS(T.) + 2е” = Ni + S2“ -0,83 (-0,814)
p-NiS(T.) + 2e” = Ni + S2” -0,960
y-NiS(T) + 2e” = Ni + S2’ -1,04 (-1,07)
NiS2O3(T) + 2e — Ni + S2O3” -0,372
[Ni(CN)4]2 + e = [Ni(CN)3]2 + CN’ -0,401
[Ni(CN)4]2 + e” = [Ni(CN)4]3’ -0,82
Ni3O4(T) + 2H2O + 2e” = 3HNiO” + H+ -0,718
Ni3O4(T.) + 2H+-+ 2e" = 3NiO(T) + H2O 0,897
Ni3O4(T) + 8H+ + 2e” = 3Ni2+ + 4H2O 1,977
Ni3O4(T) + 6FT + 6e = 3Ni + ‘/2O2 + 3H2O 0,0
Ni3O4(T) + 8H* + 8e” = 3Ni + 4H2O 0,307
Ni(OH)3(T) + H + e = Ni(OH)2(T) + H2O 1,020
Ni(OH)3(T) + 3H' + e- Ni2+ + 3H2O 2,08
Ni(OH)3(T) + ЗНГ + Зе” = Ni + 3H2O 0,586
Ni(OH)3(T) + e” = Ni(OH)2(T) + OH” 0,48
[Ni(CN)6]3~ + e’ = [Ni(CN)6]*- 0,82
Ni2O3(T) + 2FT + 2e” = 2NiO + H2O 1,020
Ni2O3(T) + 2H" + H2O + 2e” = 2Ni(OH)2(T) 1,032
Ni2O3(T) + 6H+ + 2e” = 2Ni2+ + 3H2O 1,753
Ni2O3(T) + H2O + 2e — 2HNiO2 -0,044
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
3Ni2O3(T) + 2Н” + 2е” = 2Ni3O4 + Н2О 1,305
Ni2O3(T) + ЗН2О + бе” = 2Ni + 6ОН” -0,450
3NiO(OH)(T) + Н” + е” = Ni3O2(OH)4(T.) -0,478
NiO2(T) + 4ЬГ + 2е” = Ni2+ + 2Н2О 1,65 (1,593)
NiO2(T) + 2Н2О + 2е” = Ni(OH)2(T) + + 2ОН” 0,490
NiO2(T) + 2Н+ + 2е” - Ni2+ + 2ОН” 1,75
NiO2(T) + 4Н+ + 4е” = Ni + 2Н2О 1,678
2NiO2(T) + 2Н+ + 2е — Ni2O3(T) + Н2О 1,434
Ni(OH)4(T) + е” = Ni(OH)3(T) + ОН” 0,60
NiOj + 8Н+ + 4е” = Ni2+ + 4Н2О 1,60
Ниобий
NbO(T) + 2Н+ + 2е” = Nb + Н2О -0,733
Nb3+ + Зе’ = Nb -1,1
NbO2(T) + 2FT + 2е” = NbO(T) + Н2О -0,625
Nb5+ + 5е” = Nb -0,96
NbO3+ + 2ЬГ + 2e” = Nb3+ + H2O -0,34
Nb2O3(T) + 2I-f + 2e = 2NbO2<T) + H2O -0,289
Nb2O5(T) + ЮН* + 4e” = 2Nb3+ + 5H2O -0,1
Nb2O5(T.) + 10H+ + lOe” - 2Nb + 5H2O -0,65
[NbO(SO4)2]~ + 2H+ + 2e” - Nb3+ + + H2O + 2SO2’ -o,i
[NbO(SO4)2]’ + 2H” + 5e” = Nb + H2O + + 2SO2’ -0,63
Олово
Sn + 4I-T + 4e” = SnH4 -1,074
Sn2+ + 2e” = Sn -0,136 (-0,14)
[SntOHM4” + 2e” = Sn + 6OH’ -0,790
Sn(OH)2(T) + 21-Г + 2e” = Sn + 2H2O -0,091
HSnO’ + ЗЬГ + 2e” = Sn + 2H2O 0,333
HSnO’ + H2O + 2e” = Sn + ЗОН” -0,91
SnO(T.) + 2H* + 2e = Sn + H2O -0,104
Sn3(PO4)2(T) + 6e" = 3Sn + 2PO3” -0,865
SnS(T.) + 2H* + 2e = Sn + H2S(aq) -0,366
SnS(T.) + 2H^ + 2e — Sn + H2S(r.) -0,337
SnS(T.) + 2e — Sn + S2 -0,94
SnCl(OH) ♦ H2O(T)+ 2e” = Sn + OH” + + СГ + H2O -0,631
Электродные процессы
19
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Олово
[SnCl3]“ + 2е“ = Sn + ЗСГ -0,201
[SnCl4]2’ + 2е~ = Sn + 4СГ -0,19
Sn4+ + 2е" = Sn2+ 0,151 (0,152)
Sn4+ + 4е" = Sn 0,010
Sn(OH)4(T.) + H’ + 2e = HSnO’ + 2H2O -0,349
Sn(OH)4<T.) + 4H+ + 4e’ = Sn + 4H2O -0,008
[Sn(OH)6]2‘ + 2e" = [Sn(OH)6]4‘ -0,93 (-0,96)
SnO2(T) + 2НГ + 2e = SnO(T) + H2O 0,088 (-0,108)
SnO2(T) + 41Г + 2e“ = Sn2+ + 2H2O 0,125
SnO2(T) + H+ + 2e~ = HSnO, -0,35
SnO2(T) + 4H + 4e — Sn + 2H2O -0,106
SnO32’ + 6H+ + 2e“ = Sn2+ + 3H2O 0,849
SnO2’ + 31Г + 2e” = HSnO’ + H2O 0,374 (0,375)
SnO2' + 4H+ + 2e" = SnO + 2H2O 0,810
SnF62’ + 4e" = Sn + 6F -0,200 (-0,250)
SnS2’ + 2e = SnS (T.) + 2S2 -0,6
[SnCl6]2’ + 2e’ - [SnCl3]’ + ЗСГ 0,139
Осмий
Os2+ + 2e = Os 0,85
[OsCl6]3’ + e = Os2+ + 6СГ 0,40
[OsCl6]3’ + 3e" = Os + 6СГ 0,71
[Os(CN)6]3’ + e' = [Os(CN)6]4’ 0,634
[Os(bpy)3]3++ e" = [Os(bpy)3]2+ 0,885
[Os(trpy)2]3+ + e" = [Os(trpy)2]2+ 0,987
[Os(bpy)(py)4]3+ + e~ = [Os(bpy)(py)4]2+ 0,805
[Os(bpy)2(py)2]3+ + e~ = [Os(bpy)2(py)2]2+ 0,834
[Os(py)3(trpy)]3+ + e“ = [Os(py)2(trpy)]2+ 0,80
[Os(bpy)(py)(trpy)]3+ + e” = = [Os(bpy)(py)(trpy)]2+ 0,871
[Os(bpy)2(py)Cl]2++ e’= = [Os(bpy)2(py)Cl]+ 0,484
[Os(bpy)2(py)Br]2+ + e” = = [Os(bpy)2(py)Br]+ 0,487
[Os(bipy)2(py)I]2+ + e’ = [Os(bipy)2(py)I]+ 0,489
[Os(bpyXtrpy)Cl]2+ + e" = = [Os(bpy)(trpy)Cl]+ 0,563
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
[Os(bpy)(trpy)Br]2+ + е’ = = [Os(bpy)(trpy)Br]+ 0,567
[Os(bpy)(trpy)I]2+ + е’ = [Os(bpyXtrpy)I]+ 0,567
[Os(bpy)(py)3Cl]2+ + е- = = [Os(bipy)(py)3Cir 0,426
[Os(bpy)(py)3Br]2+ + e = = [Os(bpy)(py)3Br]+ 0,444
[Os(bpy)(py)3I]2+ + e" = [Os(bpy)(py)3I]+ 0,451
[Os(bpy)(3-CH3pyXtrpy)]3+ + e’ = = [Os(bpy)(3-CH3py)(trpy)]2+ 0,867
[Os(bpy)(4-CH3py)(trpy)]3+ + e’ = = [Os(bpy)(4-CH3py)(trpy)]2+ 0,851
[Os(bpyX4-C2H5py)(trpy)]3+ + e’ = = [Os(bpyX4-C2H5py)(trpy)]2+ 0,859
[Os(bpy)(4-C3H7py)(trpy)]3+ + e" = = [Os(bpy)(4-C3H7py)(trpy)]2+ 0,861
OsO2(T) + 4H+ + 4e — Os + 2H2O 0,687
OsO2(T) + 2H2O + 4e = Os + 4OH -0,15
OsO2 -2H2O(t) + 4bT + 4e” = Os + 4H2O 0,72
[OsCl6]2“ + e = [OsCl6]3’ 0,85
[OsBr6]2- + e" = [OsBr6]3’ 0,45
Os(OH)4(T) + 4e” = Os + 4OH“ -0,15
OsO2’ + 2H2O + 2e’ - OsO2 + 4OH’ 0,1
[OsO2 (OH)4]2’ + 2H2O + 2e" = = Os(OH)4 + 4OH’ 0,1
OsO4 + 4H+ + 4e’ = OsO2(T) + 2H2O 1,005 (0,96)
OsO4()K) + 8H' + 8e” = Os + 4H2O 0,85
OsO4(aq) + 8H + 8e” = Os + 4H2O 0,78
OsO4 +6СГ + 8H+ + 4e“ = [OsCl6]2’ + 4H2O 1,00
OsO4 + СГ + 8H+ + 9e = OsCl2’ + 4H2O 1,00
HOsO; + 2e’= OsO42 + OH“ 0,3
HOsO; + 4H2O + 8e~ = Os + 9OH’ 0,02
HOsOj + 4H2O + 4e = OsO2 • 2H2O(T) + + 5OH” 0,1
[OsO4 (OH)2]2’ + 2H2O + 2e’ = = [OsO2 (OH)412’ + 2OH“ 0,30
Палладий
Pd2+ + 2e" = Pd 0,915(0,987)
PdO(T) + 2H* + 2e’ = Pd + H2O 0,85 (0,896)
Pd(OH)2(T) + 2РГ + 2e’ = Pd + 2H2O 0,897
20
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Палладий
Pd(OH)2(T) + 2е“ = Pd + 2ОН 0,07
[PdCI4]2’ + 2е" = Pd + 4СГ 0,623
[PdBr4]2 + 2е" = Pd + 4ВГ 0,6
[Pdl4]2 + 2e“ - Pd + 41 0,18
PdO2(T) + 2H+ + 2e“ = PdO(T) + H2O 1,263(1,283)
PdO2(T) + 4H* + 2e" = Pd2+ + 2H2O 1,194
PdO2(T) + 2H* + 2e" = Pd(OH)2(T) 1,283
Pd(OH)4(T) + 2H* + 2e" = Pd(OH)2(T) + 1,258
+ 2H2O
Pd(OH)4(T) + 2e" = Pd(OH)2(T) + 2О1Г 0,73
Pd(OH)4(T) + 4H+ + 2e" = Pd2+ + 4H2O 1,128
Pd(OH)4(T) + 4H+ + 4e“ = Pd + 4H2O 1,228
[PdCI6]2 + 2e~ - [PdCU]2’ + 2СГ 1,288(1,470)
[PdBr6]2 + 2e~ = [PdBr4]2’ + 2Br 0,993
[Pdl6]2 + 2e' = [Pdl4]2 + 2Г 0,482
[PdCI6]2 + 4e" - Pd + 6СГ 0,96
PdO3(T) + 2H+ + 2e = PdO2(T) + H2O 2,030
PdO3(T) + H2O + 2e = PdO2(l) + 2OH 1,2
Платина
Pt2+ + 2e - Pt 1,188
PtO(T) + 2H* + 2e’ = Pt + H2O 0,980
Pt(OH)2(T) + 2e = Pt + 2OH“ 0,15
Pt(OH)2(T) + 2H* + 2e~ = Pt + 2H2O 0,980
PtS(T) + 2e = Pt + S2’ -0,95
PtS(Tj + 2H+ + 2e = Pt + H2S(aq) -0,327
PtS(T) + 2H + 2e = Pt + H2S(r) -0,297
[PtCl4]2 + 2e’ = Pt + 4СГ 0,73 (0,758)
[ PtBr4]2 + 2e" = Pt + 4Br“ 0,698 (0,58)
Pt3O4(T) + 8РГ + 8e = 3Pt + 4H2O 1,Н
PtO2(T.) + 2H* + 2e = PtO(T) + H2O 1,045
PtO^) + 2H+ + 2e — Pt(OH)2(T) 1,045
PtO2(T)+ 4H* + 2e = Pt2* + 2H2O 0,837
[PtCU]2" + 2e~ = [PtCl4]2 + 2СГ 0,726
[PtBr6]2 + 2e" = [PtBr4]2- + 2Br“ 0,59
[РАб^+ге^рч^+гг 0,393
[Pt(SCN)6]2~ + 2e = [Pt(SCN)4]2- + 2SChT 0,468
[PtCU]2" + 4e~ = Pt + 6СГ 0,744
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
[Pt(OH)6]2 + 2е = Pt(OH)2(T) + 4ОН“ 0,1-0,4
PtS2(T) + 2е — PtS(T)+ S2 -0,64
z/wc-[PtBr2(NH3)2(NO2)2](aq) + 2е = = ^wc-[Pt(NH3)2(NO2)2](aq) + 2ВГ 0,782
wpa«c-[PtBr2(NH3)2(NO2)2](aq) + 2е = = m/?<7HC-[Pt(NH3)2(NO2)2](aq) + 2Br “ 0,786
[Pt(CN)4Cl2]2’ + 2e~ = [Pt(CN)4]2’ + 2СГ 0,89
PtO3(Tj + 2H* + 2e — PtO2(T) + H2O 2,000
PtO2' + 4H2O + 2e“ = [Pt(OH)6]2 + 2OH 0,4
Плутоний
Pu3++3e“ = Pu -2,031 (-2,03)
Pu2O3(T) + 6H+ + 6e” = 2Pu + 3H2O -1,592
Pu(OH)3(T) + 3e“ = Pu + ЗОН" -2,46
Pu4+ + e- = Pu3+ 0,967 (0,97)
PuO2(T) + H2O + H* + e" = Pu(OH)3(t) -1,4
PuO2(T) + 4H+ + e" = Pu3+ + 2H2O 0,862
2PuO2(T) + 2H+ + 2e ~ Pu2O3(T) + H2O -0,455
Pu(OH)4(T) + e’ = Pu(OH)3(T) + OH" -0,95
Pu(OH)4(t) + 4H+ + e = Pu3+ + 4H2O 1,182
гР^ОН)^) + 2H* + 2e" = PuA^) + 5H2O -0,135
PuO2+ + 4H* + e“ - Pu4+ + 2H2O 1,15(1,157)
Pu2O5(T) + 2H* + 2e — 2PuO2(t) + H2O 1,908
Pu2O5(tj + 3H2O + 2H+ + 2e — = 2Pu(OH)4(t) 1,588
PuOj+ + e~ = PuOj 0,916 (0,928)
PuO2+ + 2H2O + 2e’ = Pu(OH)4(t) 0,935
PuO2+ + 4H* + 2e = Pu4+ + 2H2O 1,042(1,04)
PuO2+ + 2H2O + 2e“ = Pu(OH)4(T) 0,935
PuO2+ + 4H* + 3e = Pu3+ + 2H2O 1,017
PuO2+ + 2e = PuO2(T) 1,095
PuO3(T) + 2H* + 2e = PuO2 + H2O 1,485
PuO3(T) + H2O + 2H* + 2e = Pi^OH)^) 1,325
2PuO3(T.) + 2H* + 2q — Pu2O5(t) + H2O 1,062
PuO2(OH)2(T) + H* + e’ = PuO2(OH)(T) + + H2O 0,3
PuO2(OH)2(T) + e = PuO2(OH)(T) + OH 0,234
PuO3- + 4H* + e = PuO2(OH)2(T) + H2O 0,95
Электродные процессы
21
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Полоний
Ро + 2е“ = Ро2' -1,4
Ро2+ + 2е" = Ро 0,368 (0,65)
Ро3+ + е‘ = Ро2+ 0,330
Ро3+ + Зе' = Ро 0,651
Ро4+ + 4е“ = Ро 0,775
РоО2(т.) + 4Н* + 4е = Ро + 2Н2О 0,775
РоО2(т) + 4Н" + е = Ро3+ + 2Н2О 1,5
РоО2(т) + 4Н+ + 2е" = Ро2+ + 2Н2О 1,095 (® 0,8)
РоОСОН)2(т) + Н2О + 4е" = Ро + 4ОН -0,098
РоО2 + ЗН2О + 4е - Ро + 6ОН -0,5 (-0,494)
РоО2' + 6Н+ + 2е" = Ро2+ + ЗН2О 0,847
РоО2' + 6ЬГ + 4е" = Ро + ЗН2О 0,748
РоО3(Т) + 2Н + 2е — РоО2(т) + Н2О 1,509
РоОз(Т) + 2е' = РоО2' 1,477
РоО3(т) + 61Г + 4е' = Ро2+ + ЗН2О 1,161
Празеодим
Рг3+ + Зе~ = Рг -2,462 (-2,35)
Рг2О3(т) + 6Н+ + бе' = 2Рг + ЗН2О -1,829
Рг(ОН)3(т) + Зе' = Рг + ЗОН' -2,79
Рг4+ + е' = Рг3+ 3,2
РгО2(Т) + Н2О + Н+ + е = Рг(ОН)3(т) 1,431
РгО2(т.) + 2Н2О + е" = Рг(ОН)3(т) + ОН' 0,8
2РгО2(т.) + 2Н+ + 2е = Рг2О3(т) + Н2О 0,863
Прометий
Рт3+ + Зе' = Рт -2,423 (-2,29)
Pm(OH)3(T) + ЗН* + Зе' = Рт + ЗН2О -2,008
Рт(ОН)3(т) + Зе' = Рт + ЗОН' -2,76
Протактиний
Ра4+ + 4е' = Ра -1,46
РаО; + 4Н+ + 5е’ = Ра + 2Н2О -1,0
РаО(ОН)2+ + ЗИ! + е - Ра4+ + 2Н2О -0,1
РаО(ОН)2+ + ЗРГ + 5е' = Ра + 2Н2О -1,19
Радий
Ra2+ + 2е“ = Ra -2,92 (-2,916)
RaO(T) + 2Н+ + 2е — Ra + Н2О -1,319
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Рений
Re + е' = Re' -0,10
Re+ + е' = Re -0,324
Re2+ + е" = Re+ 0,02
Re3+ + e'= Re2+ -0,23
Re3+ + 3e“ = Re -0,18(0,300)
Re3 + 4e' = Re' 0,125
Re(OH)3(T) + 3e' = Re + ЗОН' -0,58
Re2O3(T) + 6H + 6e = 2Re + 3H2O 0,227
Re2O3(T) + 6H+ + 8e = 2Re + 3H2O 0,07
Re2O3(T.) + 3H2O + 6e' = 2Re + 6OH' -0,333
ReO2(T.) + 4H + e = Re3++ 2H2O 0,157
ReO2(T.) + 4H* + 4e — Re + 2H2O 0,26
2ReO2(T) + 2H++ 2e — Re2O3(T) + H2O 0,375
2(ReO2 2H2O) + 2e = Re2O3(T) + 3H2O + + 2OH~ -1,25
[ReCl6]2' + 4e' = Re + 6СГ 0,51 (0,414)
2[ReCl6]2' + 2e' =[Re2Cl8]2' + 4СГ 0,756
ReO3(T) + 2H++ 2e — ReO2(T) + H2O 0,4
ReO3(T) + 6H"+ 3e' = Re3+ + 3H2O 0,318
ReO3(T) + 3H2O + 2e = Re(OH)4(T) + 2OH -0,429
ReO2' + 4H+ + 2e — ReO2(T.) + 2H2O -0,50
ReO42' + 4H2O + 2e' = Re(OH)4 + 4OH' -0,541
ReO2' + 8FT + 7e' = Re' + 4H2O 0,412
ReOj' + 8FT + 3e' = Re3+ + 4H2O 0,795
ReO4 + 2H+ + e — ReO3(j) + H2O 0,768
ReO' + e' = ReO2' 0,700
ReO4 + 8H+ + 4e' = Re3+ + 4H2O 0,422
ReO4 + 8H* + 8e' = Re' + 4H2O 0,273
ReO4 + H2O + e = ReO3(T) + 2OH 0,890
ReO4 + 4H* + 3e = ReO2(T) + 2H2O 0,510
ReO' + 4H2O + 3e' = ReO2 • 2H2O + 4OH' -0,594
ReO4 + 8НГ + 7e" = Re + 4H2O 0,34
ReO' + 4H2O + 7e" = Re + 8OH' -0,604
ReO4' + 8H" + 6СГ + 2e' = [ReCl6F + 4H2O 0,12
22
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Родий
Rh2O(T) + 2Н+ + 2е“ = 2Rh + Н2О 0,796
Rh+ + е“ = Rh 0,6
Rh2 + е = Rh+ 0,6
2RhO(T) + 2Н+ + 2e — Rh2O(T) + H2O 0,882
RhO(T) + 2H+ + 2e" = Rh + H2O 0,81
Rh3+ + 3e“ = Rh 0,76
Rh(OH)3(r) + 3e~ =Rh + ЗОН 0,00
Rh2O3(T) + 6H+ + 2e" = 2Rh2+ + 3H2O 1,349
Rh2O3 + 6FT + 4e" = 2Rh+ + 3H2O 0,975
Rh2O3(T) + 4H+ + 4e“ = Rh2O(T) + 2H2O 0,877
Rh2O3(r) + 6H+ + 6e = 2Rh + 3H2O 0,87 (0,88)
[RhCl6]3 + 3e" = Rh + 6СГ 0,44 (0,5)
[Rh(CN)6]’ + e“ = [Rh(CN)„]4 0,9
RhO2+ + 2Ff + e = Rh3+ + H2O 1,40
RhO2(T) + 4H + e = Rh3+ + 2H2O 1,881
RhO2(T) + 4IH + 6СГ + e = [RhCl6]3’ + + 2H2O 1,4
2RhO2(T) + 2H + 2e — Rh2O3(T) + H2O 1,73
[RhCl6]2 +e =[RhCl6]3 1,2
RhO2" + 4H2O + 3e = Rh(OH)3(T) + 5OH" -0,1
RhO2 + 8НГ + 3e =Rh3+ + 4H2O 1,5
RhO2" + 8H+ + 6e“ =Rh + 4H2O 1,1
RhO2' + 6НГ + 2e“ =RhO2+ + 3H2O 1,46
Ртуть
Hg + H+ + e=HgH -2,281
Hg2+ + 2e‘ = 2Hg 0,789 (0,796)
Hg2F2(T) + 2e“ = 2Hg + 2F" 0,656
Hg2Cl2(T) + 2e“ = 2Hg + 2СГ 0,268 (0,2676)
Hg2Br2(T) + 2e“ = 2Hg + 2ВГ 0,139 (0,140)
Hg2I2(T.) + 2e" = 2Hg + 2Г -0,04 (0,405)
Hg2O(T j + H2O + 2e" = 2Hg + 2OH" 0,123
Hg2(SCN)2 + 2e~ = 2Hg + 2NCS" 0,22
Hg2CrO4(T) + 2e" = 2Hg + CrO2 0,51
HgzHPO^.) + НГ + 2e’ = 2Hg + H2PO; 0,638
Hg2CO3(T) + 2e — 2Hg + CO2 0,309
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Hg2SO4(T) + 2е — 2Hg + SO2- 0,613(0,615)
Hg2(IO3)2(T) + 2e-=2Hg+ 2IO; 0,395
Hg2(OOCH)2(T.) + 2e“ = 2Hg + 2HCOO 0,567
Hg2(OOCCH3)2(T) + 2e" = 2Hg + 2CH3COO 0,511
Hg2(OOCC2H5)2(T) + 2e’ = 2Hg + + 2C2H5COO 0,51
Н&(С2О4)(Т) + 2e- = 2Hg + 2C20j- 0,416
Н&(ООСС6Н5)2(Т) + 2e’ = 2Hg + + 2C6H,COO 0,426
Hg2(OC6H2(NO2)3)2(T) + 2e’ = 2Hg + + 2(О^)3СбН2О- 0,492
2Hg2+ + 2e“= Hg2’ 0,920 (0,911)
Hg2* + 2e = Hg 0,853
HgO(T) + 2H+ + 2e“ = Hg + H2O 0,926
HgO(T) + H2O + 2e' = Hg + 2OH 0,0977
Hg(OH)2(T) + 2H+ + 2e‘ = Hg + 2H2O 1,034
HgS(T) + 2НЧ 2e‘ = Hg + H2S -0,096
HgS(T) + 2e” = Hg + S2’ -0,75 (-0,67)
Hg(IO3)2(T) + 2e=Hg+ 2IOj 0,460
[Hg(CN)4]2’ + 2q = Hg + 4CN -0,37
2HgCl2(T) + 2e~ = Hg2Cl2(T) + 2СГ 0,62
[HgCl4]2 + 2e = Hg + 4СГ 0,48 (0,38)
[HgBr4]2” + 2e“ = Hg + 4Br” 0,21 (0,223)
[HgU]2- + 2e = Hg + 4Г -0,038
Рубидий
Rb+ + e“ = Rb -2,925
Рутений
Ru2+ + 2e~ = Ru 0,45
Ru3+ + e - Ru2+ 0,249
Ru3++ 3e = Ru 0,38
[Ru(CN)6]3- + e = [Ru(CN)6]4~ 0,86
[Ru(NHj)6]34 + e- = [Ru(NH3)6]24 0,10
[Ru(en),]54 + e = [Ru(en)3]24 0,210
RuCl3 + 3e — Ru + 3C1 0,68
Ru(OH)3(T) + 3e“ = Ru + ЗОН" -2,42
Ru(OH)4(t) + 3e" = Ru(OH)3(T) + ЗОН" -0,963
RuO2(T.) +2H2O + 4e = Ru + 4OH -0,04
RuO2(T) + 4H+ + 4e = Ru + 2H2O 0,79 (0,68)
Электродные процессы
23
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Рутений
[RuCI5(OH)]2~ + Н' + е = [RuC15]2 + Н2О 1,3
[RuCl5] +2e -Ru2, + 5C1 0,3
RuCl2’ + 4Н+ + 2е = RuO2(t) + 2Н2О 2,005
RuO2+ + 2Н2О + 2е" = Ru(OH)4(t) 0,935
RuO3(T) + ЗН2О + 2е’ - Ri^OH)^ + 2ОН -0,331
R11O4 + е“ = RuO2’ 0,593 (0,6)
R11O4 + 4Н' + Зе = RuO2(t) + 2Н2О 1,533
RuO4(t) + е = RuO4 0,99
RuO4(t) + 4Н+ + 4е’ = RuO2(t ) + 2Н2О 1,387
RuO4(t) + 8Ff + 8е = Ru + 4Н2О 1,04
H2RuO5 + 4ЬГ + 4е = RuO2(T) + ЗН2О 1,40
Самарий
Sm2+ + 2е“ = Sm -2,67
Sm3+ + е“ = Sm2+ -1,000 (-1,55)
Sm3+ + Зе = Sm -2,414 (-2,30)
Sm(OH)3(T) + Зе’ = Sm + ЗОН" -2,80 (-2,83)
Свинец
Pb + 2Н+ + 2е" = РЬН2 -1,507
Pb2+ + 2е" = РЬ -0,125 (-0,126)
Pb(OH)2(T) + 2е’ = Pb + 2ОН” -0,714
РЬО(т) + 2Н+ + 2е" = РЬ + Н2О 0,25 (0,248)
РЬО(Т) + Н2О + 2е” = Pb + 2ОН' -0,580
РЬ(ОН)2(Т) + 2ЬГ + 2е~ = РЬ + 2Н2О 0,277
НРЬО’ + Н2О + 2е” = РЬ + ЗОН’ -0,502
НРЬО’ + ЗН++ 2е’ = РЬ + 2Н2О 0,702
PbF2(T) + 2е’ = Pb + 2F’ -0,350
РЬС12(Т) + 2е — Pb + 2С1 -0,226 (-0,268)
РЬВг2(т) + 2е“ = РЬ + 2Вг“ -0,280
РЫ2(Т) + 2е = РЬ + 21 -0,365
РЬНРО4(Т) + 2е" = Pb + НРО2’ -0,465 при 30 °C
[Pb(OH)4]2’ + 2е’ = Pb + 4OFT -0,538
РЬ3(РО4)2(Т) + 12е~ = ЗРЬ + 4РОд’ -0,54 при 30 °C
PbSO4(T) + 2е = Pb + SO2 -0,350 (-0,356)
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
РЬСО3(Т) + 2е” = Pb + СО2 -0,506 (-0,509)
PbS(T) + 2е’ = Pb + S2’ -0,98 (-0,93)
РЬ3О4(Т) + Н2О + 2е" = ЗРЬО(Т) + 2ОН’ 0,25
РЬ3О4 (т) + 2Н + 2е — ЗРЬО(Т) + Н2О 0,972
РЬ4+ + 2е’ = РЬ2+ 1,694(1,69)
РЬ4+ + 4е" = РЬ 0,77
РЬО2(Т) + 4Н' + 2е’ = РЬ2+ + 2Н2О 1,449 (1,468)
РЬО2(Т.) + 4Н+ + 4е’ = РЬ + 2Н2О 0,666
РЬО2(Т) + Н2О + 2е — РЬО(Т) + 2ОН 0,248
РЬО2(Т.) + SO4 + 4Н+ + 2е = PbSO4(T) + + 2Н2О 1,685 (1,698)
РЬО2(Т) + СО3 + 4Н+ + 4е’ = РЬСО2’ + + 2Н2О 1,83
ЗРЬО2(Т) + 4Н+ + 4е — РЬ3О4(Т) + 2Н2О 1,127 (0,972)
Р-РЬО2(Т) + Н2О + 2е" = НРЬО’ + ОН’ 0,208
РЬО2’ + Н2О + 2е’ = РЬО2’ + 2ОН’ 0,2
РЬО2’ + ЗН+ + 2е" = НРЬО’ + Н2О 1,547
РЬО2’ + 4Н+ + 2е’ — РЬО(Т)(кр.) + 2Н2О 2,001
РЬО2’ + ЗН2О + 4е" = Pb + 6ОН" -0,127
РЬО32’ + SO2’ + 6Н+ + 2е" = PbSO4(T) + + ЗН2О 2,34
ЗРЬО32’ + 10Н+ + 4е’ = РЬ3О4(Т) + 5Н2О 2,515
PbOj’ + ЗН2О + 2е’ = НРЬО’ + 5ОН’ 0,683
РЬ3О4(Т) + 4Н2О + 4ОН’ + 2е’ = = 3[РЬ(ОН)4]2’ 0,076
РЬ3О4(Т) + 4Н2О + 2е’ - ЗРЬ(ОН)2(Т) + + 2ОН’ 0,587
[РЬ(ОН)6]2’ + 2е" = [Pb(OH)4]2’ + 2ОН” 0,305
Селен
Se + 2е = Se2 -0,670 (-0,92)
Se + 2Н+ + 2е — H2Se(r) -0,082
Se + 2Н+ + 2е = H2Se(aq) -0,115 (-0,399; -0,40)
Se + Н+ + 2е’ = HSe’ -0,227
Se2Cl2 + 2е’ = 2Se + 2СГ 1,1 (1,Ю8)
H2SeO3 + 6Н' + бе’ = H2Se + ЗН2О 0,360
24
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Селен
H2SeO3 + 4Н+ + 4е’ = Se + ЗН2О 0,741 (0,739)
HSeO3’ + 7Н+ + бе’ = H2Se + ЗН2О 0,386
HSeO3 + 5Н+ + 4е’ = Se + Н2О 0,777
HSeO3’ + 6Н+ + бе’ = HSe’ + ЗН2О 0,349
SeO3’ + 7Н+ + бе’ = HSe’ + ЗН2О 0,414
SeO2’ + 6Н+ + 4е" = Se + ЗН2О 0,885
SeO2’ + 8Н+ + бе’ = H2Se + ЗН2О 0,276
SeO2’ + ЗН2О + 4е’ = Se + 6ОН’ -0,357 (-0,366)
HSeO; + ЗН+ + 2е’ = H2SeO3 + Н2О 1,094
SeO2 + 4Н' + 2е~ = H2SeO3 + Н2О 1,151
SeO2 + 3FT + 2е" = HSeO; + Н2О 1,074
SeO4 + Н2О + 2е" = SeO2’ + 2ОН’ 0,031
Сера
S2’ + 2е" = 2S2’ -0,483 (-0,524)
S22’ + 2Н+ + 2е" = 2HS" 0,287 (0,298)
(SCN)2(r) + 2е’ = 2SCN’ 0,77
S2’ + 2е’ - S2’ + S2’ -0,49
S2’ + ЗН+ + 4е’ = 3HS’ 0,097
2S2’ + 2е" = 3S2’ -0,473 (-0,506)
S2’ + 2е’ = S2’ + S2’ -0,52
S2’ + 4Н+ + бе’ - 4HS’ 0,033 (0,036)
3S2’ + 2е’ = 4S2’ -0,453 (-0,478)
S52’ + 10Н+ + 8е’ = 5H2S(e) 0,303
S2’ + 5Н+ + 8е’ = 5HS" 0,003 (0,007)
4S2’ + 2е’ = 5S2’ -0,426
S + Н* + 2e“ = HS“ -0,062 (-0,065)
S + Н2О + 2е" = HS’ + ОН’ -0,52
S + 2FT + 2е“ = H2S(e) 0,171 (0,174)
S + 2Н+ + 2е ~ H2S(aq) 0,144
S + FT + 2е’ = HS’ -0,065
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
S + 2е" = S2’ -0,447
2S + 2е" = S2’ -0,476
4S + 2е" = S2’ -0,23
5S + 2е" = S2’ -0,340
S2Cl2(r) + 2е = 2S + 2С1 1,19
SF(r) + е = S + F 3,36
SO(E) + 2Н+ + 2е = S + н2о 1,507
H2SO2 + 2Н+ + 2е" = S + 2Н2О 0,5
SF2(e) + 2е" = S + 2F 2,12
H2SO3 + 2Н+ + 2е" = H2SO2 + Н2О 0,4
H2SO3 + 4Н+ + 4е’ = S + ЗН2О 0,449 (0,50)
2H2SO3 + Н+ + 2е~ = HS2O; + 2Н2О -0,068
2H2SO3 + 2Н+ + 4е” - S2O2’ + ЗН2О 0,40
3H2SO3 + 2е’ = S3O2 + ЗН2О 0,290
4H2SO3 + 4Н+ + бе’ = S4O2’ + 6Н2О 0,507
5H2SO3 + 8FT + 10е’ = S5O2’ + 9Н2О 0,416
HSO” + 5Н+ + 4е~ = S + ЗН2О 0,0
2HSO; + ЗН+ + 2е~ = HS2O; + 2Н2О 0,173
2HSO3’ + 2Н+ + 2е" - S2O2 + 2Н2О 0,099
2HSO3’ + 4Н+ + 4е’ = S2O2 + ЗН2О 0,453
4HSO; + 8Н+ + бе’ = S4O2’ + 6Н2О 0,577
SO2’ + ЗН2О + 4е’ = S + 6OFF -0,659
SO2’ + 61-Г + бе’= S2’+ ЗН2О 0,231
2SO2’ + 4Н+ + 2е’ = S2O2’ + 2Н2О 0,416(0,526)
2SO2’ + 2Н2О + 2е’ = S2O2’ + 4ОН’ -1,13
2SO2’ + 6Н+ + 4е" - S2O3’ + ЗН2О 0,666
2SO2’ + ЗН2О + 4е’ = S2O2’ + 6ОН’ -0,576
4SO2’ + 12Н+ + бе’ = S4O2’ + 6Н2О i 4 0^ 0,862
2SO2’ + 6Н+ + 4е’ = S2O2’ + ЗН2О 0,705
SO2(E) + 4Н* + 4е" = S + 2Н2О -0,659
4SO20.) + 4Н* + бе’ = S4O2’ + 2Н2О 0,511
SF4(r) + 4е = S + 4F 0,97
Электродные процессы
25
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Сера
S2O2’ + 6Н+ + 8е" = 2S2' + ЗН2О -0,006
S2O2' + бЬГ + 4е" = 2S + ЗН2О 0,465
S2O2' + 8Н+ + 8е = 2HS' + ЗН2О 0,200
5S2O2’ + 30Н+ + 24е' = 2S2’ + 15Н2О 0,331
S4O2 + 2е" = 2S20j- 0,080
S4O2' + 12Н+ + 1 Ое~ = 4S + 6Н2О 0,416
S,02' + 12FT + 1 Ое’ = 5S + 6Н2О Э 0 х 0,484
S2O2' + 4Н* + 2е“ = 2H2SO3 0,569
S2O2’ + 2Н+ + 2е = 2HSO3 0,468
S2O2' + 2e" = 2SO2’ 0,026 (0,037)
SO2’ + 4H4 + 2e = S02(r) + 2H2O 0,138
SO42' + 4H+ + 2e" = H2SO3 + H20 0,158(0,172)
SO2 + H20 + 2e" = SO2' + 20H“ -0,936
SOJ' + 8H+ + 6e = S + 4H2O 0,357
SO42’ + 8H4 + 8e" = S2 + 4H2O 0,149
SO2' + 9Ff + 8e" - HS' + 4H2O 4 z 0,252
SO2 + 10H+ + 8e' = H2S(r) + 4H2O 0,311
S04 + 10H+ + 8e' = H2S(aq) + 4H2O 0,303
2SO2' + 10H4 + 8e' = S2O2' + 5H2O 0,29
2SO2' + 4H+ + 2e" = S20?' + 2H2O 4 2 о z -0,253
HSO4 + 9H4 + 8e’ = H2S(aq) + 4H2O 0,289 (0,275)
HSO; + 7H+ + 6e~ = S + 4H2O 0,339
SF^r) + 6e" = S + 6F 0,97
S2O2’ + 2e" = 2SO2’ 2,010(1,961)
S2O2' + 2H+ + 2e' = 2HSO4 2,123
Серебро
Ag+ + e’ = Ag 0,799
AgF(T.) + e" = Ag + F“ 0,779
AgCl(T.) + e~ = Ag + СГ 0,222
AgBr(T) + e' = Ag + Br' 0,03 (0,0711)
Agl(1) + e'= Ag + Г -0,152
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
AgCN(T.) + е' = Ag + CN“ -0,017
AgSCN(T.) + е' = Ag + SCN' 0,09 (0,0895)
AgCNO(T) + e" = Ag + CNO' 0,41
AgClO4(T) + e' = Ag + C1O4 0,787
a-Ag2S(T) + 2e“ = 2Ag + S2“ -0,691 (-0,66)
Ag2S(T.) + H++ 2e‘ = 2Ag + HS' -0,272
AgN3(T.) + e~ = Ag + N' 0,292 (0,293)
AgBrO3(T) + e' = Ag + BrOj 0,55 (0,546; 0,588)
AgIO3(T.) + e’ = Ag + IO; 0,35 (0,354)
Ag3PO4(T) + 3e' - 3Ag + PO4' 0,452
Ag2CrO4(T) + 2e“ = 2Ag + CrO4' 0,446 (0,449)
Ag2SeO3(T.) + 2e“ = 2Ag + SeO2' 0,363
Ag2SeO4(T) + 2e" = 2Ag + SeO2' 0,55
Ag2CO3(T) + 2e' = 2Ag + CO2' 0,47
Ag2C2O4(T.} + 2e' = 2Ag + C2O2' 0,472 (0,465)
Ag3AsO4(T) + 3e" = 3Ag + AsO4' 0,401
Ag2MoO4(T) + 2e' = 2Ag + MoO2' 0,486
Ag2WO4(T) + 2e' = 2Ag + WO2' 0,466
AgNO2(T.) + e~ = Ag + NO’ 0,562 (0,564)
AgOOCCH3 (T) + e' = Ag + CH3COO' 0,643
Ag2SO4(T.) + 2e' = 2Ag + SO2' 0,654
Ag3[Co(CN)6] + 3e’ = 3Ag + [Co(CN)J3- 0,298
Ag4[Fe(CN)6] + 4e' = 4Ag + [Fe(CN)6]4' 0,194(0,148)
Ag2O(T) + 2H++ 2e — 2Ag + H2O 1,173
Ag2O(T) + H2O + 2e" = 2Ag + 2OH 0,342
[Ag(IO3)2]- + e- = Ag+ 210;- 0,725
[Ag(CN)2]' + e' = Ag + 2CN' -0,31 (-0,43)
[Ag(CN)3]2 +e~ = Ag + 3CN- -0,51
[Ag(S,O3)2]1 +e- = Ag+ 2S;0j- 0,01 (0,017; 0,004)
[Ag(SO3)J3 +e" = Ag+ 2S0J- 0,43
[Ag(SCN)2]' + e' = Ag + 2SCN' 0,304
[Ag(SCN)3]2' + e’ = Ag + 3SCN' 0,231
26
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Серебро
[Ag(SCN)4]2’ + е‘ = Ag + 4SCNT 0,214
[Ag(S2O8)2]3-+ е“ = Ag + 2S2OJ~ -0,01
[Ag(NH3)2]* + 2H2O + e"=Ag + 2NH3 • H2O 0,373 (0,367)
[Ag(NH3)2]* + e- = Ag + 2NH3(r) 0,164
[Ag(OH)2] + e" = Ag + 2OFF 0,563
Ag2* + e = Ag* 1,98
AgO(T) + 2H+ + e = Ag+ + H2O 1,772
2AgO(T.) + 2H* + 2e — Ag2O(T) + H2O 1,398
2AgO(T) + H2O + 2e” = Ag2O(T_> + 2OFF 0,604
Ag2O3(T.) + 6H* + 2e"= 2Ag2*+ 3H2O 1,360
Ag2O3(T.) + H2O + 2e“ = 2AgO(T)+ 2OFF 0,739
Ag2O3(T) + H2O + 2e = Ag2O2(T j+ 2OH 1,711
Ag2O3(T) + 2H+ + 2e = 2AgO(T) + H2O 1,569
Ag2O3(T) + 4H* + 4e = Ag2O(T) + 2H2O 0,960
Ag2O3(T) + 6H* + 4e — 2Ag* + 3H2O 1,670
Ag2O3(T) + 3H2O + 6e — 2Ag + 6OH 1,757
AgO* + e’ = AgO(T) 1,288
AgO* + 2H* + e’ = Ag2* + H2O 2,016
AgO+ + 2H+ + 2e“ = Ag+ + H2O 1,998
Скандий
Sc3* + 3e" = Sc -2,077 (-2,03)
Sc2O3(T) + 6H* + 6e =2Sc + 3H2O -1,591
Sc2O3(T) + 3H2O + 6e —2Sc + 6OH -2,74
Sc(OH)3(T) + 3e = Sc + ЗОН -2,663
Sc(OH)3(T) + 3H* + 3e = Sc + 3H2O -1,784
ScF3(T) + 3e = Sc + 3F -2,37
ScF* + 3e’ = Sc + 2F -2,28
Стронций
Sr2* + 2e~ = Sr ^-2,888 (-2,89)
Sr(OH)2 + 2e“ = Sr + 2OFF -2,88
Sr(OH)2 • 8H2O + 2e" = Sr + 2OFT + 8H2O -2,99
SrO(8q) + 2H* + 2e — Sr + H2O -2,047
SrO2(aq) + 2H* + = SrO + H2O 1,492
SrO2(aq) + 4H* + 2e~ = Sr2* + 2H2O 2,333
Сурьма
Sb + 3H*+3e=SbH3(r) -0,510
Sb3* + 3e" = Sb 0,24
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Sb2O3(T) + 6Н* + 6е = 2Sb + ЗН2О 0,152
Sb4O6(T) + 12Н* + 12е’ = 4Sb + 6Н2О 0,150
Sb4O6(T) + 6Н2О ч- 12е“ = 4Sb + 12ОН’ -0,683
HSbO2 + ЗН+ + Зе"= Sb + 2Н2О 0,230
SbO* + 2Н* + Зе = Sb + Н2О 0,212 (0,204)
SbO2 + 2Н2О + Зе = Sb + 4ОН -0,639
SbO2 + 4Н* + Зе = Sb + 2Н2О 0,446
[SbCUp + Зе’ = Sb + 4СГ 0,17
Sb2S2 +2e=Sb + 2S2- -0,85
Sb2S24’ + бе- - 2Sb + 4S2- -0,763
SbSf +4e=Sb + 3S2- 0,45
SbSO + H2O + 3e = Sb + 2OH + S2 -0,350
SbO2(T) + 4H* + 4e = Sb + 2H2O 0,446
Sb2O4(T) + 4H* + 2e- = 2SbO* + 2H2O 0,68
Sb2O4(T) + 2H* + 2e = Sb2O3(T) + H2O 0,863
2Sb2O4(T) + 4H* + 4e" = Sb4O6(r) + 2H2O 0,342
H3SbO4 + 2H* + 2e" = H3SbO3 + H2O 0,75
Sb2O5(T) + 2FT* + 2e = Sb2O4(T> + H2O 1,055
Sb2O5(T) + 6H* + 4e“ = 2SbO* + 3H2O 0,605
Sb2O5(T) + 4H* + 4e = Sb2O3(T) + 2H2O 0,671
2Sb2O5(T) + 8H* + 8e = Sb4O6(T.) + 4H2O 0,699
SbO" + 3H* + 2e" = HSbO2 + H2O 0,678
SbO3- + H2o + 2e- = SbO2 + 2OH~ -0,43
SbO3- + 4H+ + 2e- = SbO* + 2H2O 0,720 (0,704)
SbO3- + 2ЬГ + 2e’ = SbO2 + H2O 0,353
2SbO3’ + 6H* + 4e- = Sb2O3(T) + 3H2O 0,794
SbO* + 2H* + 2e" = SbO* + H2O 0,720
[Sb(OH)6]- + 2H* + 2e = [Sb(OH)4f + + 2H2O 0,363
SbS3- + 2e" = SbS2 + 2S2- -0,6
[SbCUf + 2e~ = [SbCUf + 2СГ 0,746
Таллий
Tl* + e' = T1 -0,336
Т1ОН(т) + H* + e“ = Tl + H2O 0,778
T1OH(I) + e“ = Tl + OFF -0,343
Электродные процессы
27
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Таллий
Т1С1(т3 + е“ = Т1 + СГ -0,557
Т1Вг(Т) + е = Т1 + Вг -0,658
ТП(т) + е” = Т1 + Г -0,753 (-0,752)
T1SCN(T) + е“ = Т1 + SCN -0,56
Т110з(т) + е = Т1 + Ю3 -0,666
T1N3(T.) + е- = Т1 + N3- -0,554
T12S(T.) + 2е" = 2Т1 + S2 -0,93 (-0,90)
ТЬСгО^т.) + 2е = 2Т1 + СгО42' -1,056
Т13+ + 2е = Т1+ 1,252 (1,25)
Т13+ + Зе' = Т1 0,72
Т120з(т.) + ЗН2О + 4е' = 2ТГ + 6ОГГ 0,02
Т1(ОН)з(т) + 2е" = Т1ОН(т) + 2ОН“ -0,05 (0,05)
Т1С13(т) + 2е“ = TIC^.) + 2СГ 0,89
Тантал
Та5+ + 5е" = Та -1,12
Та2О5(т) + 10Н+ + 1 Ое = 2Та + 5Н2 -0,750 (-0,812)
[TaF7]2' + 5е' -Та + -0,45
Теллур
Те2' + 2е-2Те2' -1,445
Те + 2е' = Те2' -1,143
Те + 2Н++ 2е'= Н2Те(г) -0,72
2Те + 2е~ = Те|’ -0,84
ЗТе2 + 8е' = 2Те2' + 2Те2' -0,92
Te2S + 2е = 2Те + S2 -0,90
Те2+ + 2е" - Те 0,40
Те4+ + 4е' = Те 0,568
2Те4++ Юе'= Те2' 0,286
ТеО2(т) + 4Н" + 4е“ = Те + 2Н2О 0,521 (0,529)
ТеО2(у aq) + 4ГГ + 4е = Те + 2Н2О 0,604
НгТеОзоц) + 4ГГ + 4е" = Те + ЗН2О 0,589
ТеСЦ + 4е' = Те + 4СГ 0,745
[ТеСЦ]2' + 4е' = Те + 6СГ 0,55
ТеО(ОН)+ + 3FT + 4е' = Те + 2Н2О 0,559
2ТеО(ОН)+ + 6Н+ + 1 Ое' = Те2' + 4Н2О 0,273
НТеО; + 5Н+ + 4е' = Те + ЗН2О 0,713
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
2НТеО3 + 10Н++ 10е“ = Те2' + 6Н2О 0,402
ТеО2’ + 6ГТ + 4е“ = Те + ЗН2О 0,827
2ТеО2' + 12Н+ + 10е' = Те2’ + 6Н2О 0,493
ТеО2' + ЗН2О + 4е' = Те(т) + 6ОН’ -0,415 (-0,57)
[Те(ОН)6]2' + 4е" = Те + 6ОН' -0,412
ТеО3(Т.) + 2Н + 2е = ТеО2<Т) + Н2О 0,975
TeO3(T,aq)+ 2Н+ + 2е' = ТеО2(Т) + Н2О 1,02
ТеО3(Та<1) + 2Н+ + 2е' = ТеО2(Т.>аЧ) + Н2О 0,850
Н2ТеО4 + 61Г + 2е" = Те4+ + 4Н2О 0,926
Н2ТеО4 + 2ЬГ + 2е" = ТеО2(т) + 2Н2О 1,004
Н2ТеО4 + 2Н+ + 2е = ТеО2(Т + 2Н2О 0,854
Н2ТеО4 + ЗН+ + 2е' = ТеО(ОН)+ + 2Н2О 0,959
Н2ТеО4 + Н+ + 2е‘ = НТеО3 + Н2О 0,637
Н6ТеО6(Т) + 2Н+ + 2е = ТеО2(Т.) + 4Н2О 1,02
ТеС16 + бе' = Те + 6СГ 0,55
TeF6 + бе' = Те + 6F' 0,755
НТеО4 + Н+ + 2е' = ТеО2' + Н2О 0,590
НТеО4' + 2Н+ + 2е' = НТеО' + Н2О 0,819
НТеО; + ЗН+ + 2е' = ТеО2(т) + 2Н2О 1,186
НТеО4 + ЗН‘ + 2е~ TeO2(T>aq) + 2Н2О 1,036
ТеО2 + 2Н+ + 2е‘ = ТеО32 + Н2О 0,892 (0,897)
ТеО2' + 4FT + 2е' = ТеО2(т) + 2Н2О 1,494
ТеО2' + 4Н+ + 2е" = TeO2(T.>aq) + 2Н2О 1,343
ТеО4" + Н2О + 2е' = ТеО32' + 2ОН“ 0,4
Тербий
ТЬ3* +Зе-ТЬ -2,391 (-2,31)
ТЬ(ОН)з(т) + ЗН+ + Зе' = ТЬ + ЗН2О -1,999
ТЬ(ОН)з(т) + Зе' = ТЬ + ЗОН' -2,82 (-2,79)
ТЬ4+ + е’ = ТЬ3+ 3,1
ТЬО2(Т) + 2Н2О + е' = ТЬ(ОН)3(т)+ ОН 0,9
Технеций
ТсОН(Т) + FT + е' = Тс + Н2О 0,031
Тс2+ + 2е' = Тс 0,400
Тс(ОН)2(Т) + Н + е' — ТсОН(Т) + Н2О 0,113
28
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Технеций
Тс(ОН)2(т) + 2Н+ + 2е" = Тс + 2Н2О 0,072
Тс3О4(Т.) + 2Н2О + 2Н+ + 2е" = ЗТс(ОН)2(т) 0,234
Тс3О4(т) + 5Н+ + 5е" = ЗТс(ОН)(т) + Н2О 0,161
Тс(ОН)з(т.) + Н‘ + е“ = Тс(ОН)2(т) + Н2О 0,412
Тс(ОН)3(т) + 2ЬГ + 2е" = ТсОН(т) + 2Н2О 0,262
Тс(ОН)з(т) t- 3HJ + Зе" = Тс + ЗН2О 0,185
ЗТс(ОН)3(т) + Н+ + е" = Тс3О4(т) + 5Н2О 0,768
Тс4О7(Т) + 10Н+ + Юе = 4ТсОН(т) + ЗН2О 0,338
ТсО2(т) + 4FT + 4е" = Тс + 2Н2О 0,272
ТсСОН)^.) + Н‘ + е" = Тс(ОН)з(т) + Н2О 0,620
Тс(ОН)4(т) + 2Н+ + 2е" = Тс(ОН)2(т) + + 2Н2О 0,518
Тс(ОН)4(т) + ЗН+ + Зе’ = ТсОН(т) + ЗН2О 0,382
Тс(ОН)4(т) + 4Н+ + 4е" = Тс + 4Н2О 0,294
3Tc(OH)4(T) + 4Н‘ + 4е” - ТсзО^.) + 8Н2О 0,657
ТсОз(Т) *" 2Н+ + 2е = ТсО2(Т> + Н2О 0,757
Тео; + 2Н+ + е" = ТсО3(т) + Н2О 0,700
ТсО4 + 4Н‘ + Зе = ТсО2(т) + 2Н2О 0,738 (0,757)
ТсО4" + 8ЬГ + 5е" = Тс2+ + 4Н2О 0,500
ТсО4 + 8Н‘ + 7е" = Тс + 4Н2О 0,472
ТсО4 + 4Н2О + Зе" = ТсСОН)^) + 4ОН" -0,366
Титан
Ti2+ + 2е" = Ti -1,630
TiO + 2Н+ + 2е = Ti + Н2О -1,306
Ti3+ + е" = Ti2+ -0,369 (0,368)
Ti3+ + Зе’ = Ti -1,209
Ti2O3(T) + 6FT + 2е = 2Ti2+ + ЗН2О -0,478
Ti2O3(T) + 2Н+ + 2е — 2TiO(T) + Н2О -1,123
Ti2O3(T.,a<1) + 6Н+ + 2е" = 2Ti2+ + ЗН2О -0,248
TizOscr.aq) + 2Н+ + 2е = 2TiO(T) + Н2О -0,894
Ti2O4(T) + 2Н+ + 2е = Ti2O3(T) + Н2О -0,490
Ti2O4(r) + 2Н+ + 2е“ = Ti2O3(T aq) + Н2О -1,178
Ti4+ + е" = Ti3+ -0,04
Ti4+ + 4е" = Ti -0,88
Ti(OH)3+ + H+ + e’ = Ti3+ + H2O -0,098
Ti(OH)2+ + 2H+ + e" = Ti3+ + 2H2O -0,012
TiO2+ + 2H+ + e" = Ti3+ + H2O 0,100
TiO2+ + 2H+ + 2e" = Ti2+ + H2O -0,135
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
ТЮ2т + 2Н+ + 4е" = Ti + Н2О -0,892
ТЮ2(Рутил) + 4Н+ + е“ = Ti3+ + 2Н2О -0,666
ТЮ2(ругил) + 4Н+ + 2е - Ti2+ + 2Н2О -0,502
TiO2(T) + 4Н‘ + 4е" = Ti + 2Н2О -0,86
2TiO2(T) + 2Н+ + 2е = Ti2O3(T) + Н2О -0,556
2ТЮ2(т) + 2Н+ + 2е“ = Ti2O3(Taq) + Н2О -0,786
3TiO2(T) + 2Н+ + 2е = Ti3O5(T) + Н2О -0,589
TiO2(T, aq) + 4Н‘-е- Ti3+ + 2Н2О 0,029
TiO2(r + 4Н+ + 2е“ - Ti2+ + 2Н2О -0,169
2TiO2(r aq) + 21-Г + 2е“ = Ti2O3(T) + Н2О -0,139
2TiO2(T aq) + 2Н + 2е — Т120з(Т aq) + Н2О -0,091
3TiO2(T, aq) + 2Н+ + 2е" = Ti3O5(T) + Н2О -0,493
TiF4+ 4е” = Ti + 4F” -0,89
TiCl4+ 4е" = Ti + 4СГ -0,39
TiBr4+ 4е" = Ti + 4Br" -0,52
Til4+ 4е“ = Ti + 41“ -0,58
[TiF6]2" + 4е" = Ti + 6F" -1,19 (-1,191)
HTiO; + 5H+ + 2e" = Ti2+ + 3H2O 0,362
TiO2+ - 2H- 2e" - TiO2+ + H2O 1,800
TiO2+ + H2O + 2e" = HTiO3" + H+ 1,303
TiO2+ + 4H++ 6e" = Ti + 2H2O 0,012
TiO^ + 2e" = TiO2(T) 2,182
Торий
Th4+ + 4e" = Th -1,899 (-1,83;-1,875)
ThO2(T.) + 4H+ + 4e" = Th + 2H2O -1,789
ThO2(T) + 2H2O + 4e" = Th + 4OH" -2,56
Th(OH)4(T) + 4e" = Th + 4OH" -2,48 (-2,513)
Th(OH)4(T) + 4H+ + 4e" = Th + 4H2O -1,650
ThOH3+ + 1Г + 4e" = Th + H2O -1,818
Th(OH)2+ + 21Г + 4e" = Th + 2H2O -1,755
П1(ОН)з(т) + 3H+ + 4e" = Th + 3H2O -1,663
Тулий
Tm2+ + 2e = Tm -2,3
Tm3+ + e’ = Tm2+ -2,3
Tm3+ + 3e" = Tm -2,278 (-2,32)
Тт(ОН)з(т.) + 31Г + 3e" = Tm + 3H2O -1,913
Tm(OH)3(T) + 3e" = Tm + ЗОН" -2,83 (-2,74)
Электродные процессы
29
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Углерод
ОгН^) + 2Н* + 2е - С2Н6<1.') 0,52
CH3OH(aq) + 2Н+ + 2е" = СН^ Н2О 0,588
CH3OH(aq) + Н2О + 2е" = СН4(Г)+ 2ОН -0,25
C2H5OH(aq) + 2FT + 2е" = + Н2О 0,46
С2Н2(г) + 2Н+ + 2е = 0,731
C4H4S(rj + 4Н+ + 4е — C4H8S(r.) 0,209
С + 4Н+ + 4е’ = СН^ 0,132
CH2O(aq) + 2Н+ + 2е’ = СН3ОН(а(1) 0,232 (0,24)
СН2О (aq) + 2Н2О + 2е” = CH3OH(aq) + + 2ОН -0,59
CH3CHO(aq) + 2ЬГ + 2е" = C2H5OH(aq) 0,192
СО(Г) + 2Н+ + 2е — С + Н2О 0,517(0,518)
СО(Г) + 6Н+ + бе = СИцг)+ Н2О 0,497 (0,260)
HCOOH^aq) + 2Н+ + 2е = С + 2Н2О 0,523
HCOOH(aq) + 2Н" + 2е = CH2O(aq) + + Н2О 0,034 (-0,056)
HCOOH(aq) + 4Н+ + 4е“ = CH3OH(aq)+ + Н2О 0,100 (0,145)
НСОО’ + ЗН+ + 2е" = СН2О (aq) + Н2О 0,167 (0,082)
НСОО’ + 2Н2О + 2е’ = СН2О (aq) + ЗОН’ -1,14
НСОО" + 5Н+ + 4е’ = CH3OH(aq) + Н2О 0,199 (0,157)
QHjO^aq) + 2Н+ + 2е” = C6H4(OH)2(aq) 0,6994
CH3COOH(aq) + 2bT + 2е’ = = CH3CHO(aq) + H2O -0,118
C2H2O4(aq) + 2Н+ + 2е = 2HCOOH(aq) 0,254
СгНгО^ац) + 6Н+ + бе — CHjCOOH^aq) + + 2Н2О 0,31
С2О^ + 2Н+ + 2е" = 2НСОО’ 0,145
C6H5COOH(aq) + 6Н+ + бе’ = = C6H5CH3(aq) + 2H2O 0,21
CH3COCOOH(aq) + 2Н+ + 2е’ = = СНзСНОНСООН 0,20
(CN)2(r) + 2Н+ + 2е" = 2HCN 0,37
СН3ОН(ад) + СОг(Г) + 2Н+ + 2е = = СНзСООН^ + НгО 0,35
2HOCN + 2Н+ + 2е” = 2HCN + Н2О 0,35
2HOCN + 2Н+ + 2е’ = (CN)2(r) + 2Н2О -0,33
NCO’ + Н2О + 2е‘ = CN + 2ОН’ -0,97
CO2(r) + 2Н* + 2е’ = HCOOH(aq) -0,196
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
СО2(Г) + 2Н+ + 2е = СО(Г) + Н2О -0,106 (-0,12)
СО2(Г) + 4Н+ + 4е’ = С +2Н2О 0,207 (0,169)
СО2(г) + 8Н* + 8е = СН^г) + 2Н2О 0,1
2СО2(г) + 2Н+ + 2е’ = C2H2O4(aq) -0,481
2СО2(г) + С2Н6(Г) + 2Н+ + 2е = = 2CH3COOH(aq) 0,12
Н2СО3 + 2Н+ + 2е’ = HCOOH(aq) + Н2О -0,391 (-0,156)
Н2СО3 + 4Н+ + 4е = CH2O(aq) + 2Н2О 0,228
Н2СО3 + 4Н+ + 4е’ = С + ЗН2О 0,044
Н2СО3 + 6Н+ + бе’ = CH3OH(aq) + 2Н2О 0,311
2Н2СО3 + 2Н+ + 2е’ = C2H2O4(aq) + 2Н2О -0,050
СО?’ + ЗН* + 2е" = НСОО’ + Н2О 0,478
СО; + 6Н+ + 4е" = СН2О (aq) + 2Н2О 0,197
СО? + 6Н+ + 4е“ = С + ЗН2О 0,475
COj’ + 8Н+ + бе’ = CH3OH(aq) + 2Н2О 0,209
2СО; + 4Н+ + 2е" = С2О42 + 2Н2О 0,441
СО? + 2Н2О + 2е’ = НСОО’ + ЗОН’ -0,95
СО? + 6Н* + 4е’ = С + ЗН2О 0,475
СС14 + 4е’ = С + 4СГ 1,18
Уран
UO + 2ЬГ + 2е’ = U + Н2О -1,438
UO + 5Н+ + 5е’ = UH3 + Н2О -0,422
U3+ + Зе’ = U -1,798, (-1,66; -1,780)
U2O3(T.) + 2Н+ + 2е’ = 2UO + Н2О -1,163
U2O3(T.) + 6Н+ + бе = 2U + ЗН2О -1,346
U2O3(T) + 12Н+ + 12е“ = 2UH3 + ЗН2О -0,545
U(OH)3(T) + Зе’ = и + ЗОН -2,17
U3+ + ЗН+ + бе’ = UH3 0,772
и4+ + е" = и3+ -0,52 (-0,607)
U4+ + 4е” = U -1,38
ион3+ + Н+ + е’ = и3+ + Н2О -0,538
UO2(T) + 4Н+ + е’ = U3+ + 2Н2О -0,382
UO2(T) + 4Н+ + 4е’ = U + 2Н2О -1,444
UO2(T) + 2Н2О + 4е’ = U + 4ОН’ -2,39
UO2(T) + 7Н* + 7е’ = ин3 + 2Н2О -0,716
30
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Уран
2UO2(T.) + 2Н+ + 2е = и2Оз(Т) + Н2О -1,738
U(OH)4<T)+ 4Н+ + е" = и3+ + 4Н2О -0,019
U(OH)4(r) + е’ = U(OH)3 + ОН" -2,39 (-2,20)
U(OH)4(T.) + 4Н+ + 4е- U + 4Н2О -1,353
U(OH)4(T) + 7Н+ + 7е" = UH3 + 4Н2О -0,664
2U(OH)4(t) + 2ЬГ + 2е" = U2O3(T.) + 5Н2О -1,375
UO2+ + ЗН++ е = UOH3+ + Н2О 0,546
UO2 + 4Н + е“ = U4+ + 2Н2О 0,38 (0,62; 0,612)
изО8(т) + 4ЬГ + 4е“ = 3UO2(T.) + 2Н2О 0,533
U6+ + 2е’ = U4+ 0,4
UO2+ + е" = ио2+ 0,16 (0,052; 0,05)
UO2+ + 2е = ио2(т) 0,45 (0,221)
UO2+ + 4Н+ + 2е" = и4+ + 2Н2О 0,27 (0,33; 0,333)
UO2+ 4- 2SO42“ + 4ЬГ + 2е -= U(SO4)2(aq) + 2Н2О 0,42
UO2+ + ЗН+ + 2е" - ион3+ + Н2О 0,299
UO2+ + 2Н2О + 2е" = 11(011)^.) -0,040
3UO2+ + 2Н2О + 2е" = U3O8(t) + 4Н+ -0,538
UO2+ + 4Н+ + 6е - U 4 2Н2О -0,82
3UO22+ + 2Н2О + 2е’ = U3O8(T.) + 4Н+ -0,538
UO3(T.) 4- 2Н+ + 2е" = UO2(T) + Н2О 0,657
UO3(T) + Н2О + 2Н+ + 2е‘ = U(OH)4(T) 0,475
3UO3(T) + 2FT + 2е" - U3O8(t) + Н2О 0,904
UO3 • Н2О + 2Н+ + 2е" = U(OH)4(r) 0,186
UO3 • Н2О + 2РГ + 2е" = UO2(t) + 2Н2О 0,368
Зио3 • Н2О + 2Н* + 2е" = U3O8(T) + 4Н2О 0,038
Фосфор
Р2Н4 + 2Н+ + 2е’ = 2РН3(г) 0,006
Р4Н2 + 6Н+ 4- бе = 2P2H4(aq) -0,014
Р4Н2 4- ЮН* + 10е’ = 4РН3(г) -0,006
4Р(красн.)+ 2Н* 4- 2е~ = Р4Н2 -0,633
4Р(бел.)4-2Н+4-2е- = Р4Н2 -0,347
4Р(бел 0+ 2Н2О + 2е“ = Р4Н2 + 2ОН~ -1,18
Р(красн.) + ЗН+ + Зе“ = РНз -0,111
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Р(бел.) + ЗН+ 4- Зе’ = РН3 -0,063 (-0,06)
Р(бел.) + ЗН2О + Зе- = РН3(Г) + ЗОН- -0,89
2Р(красн.) + 4Н+ 4- 4е~ = Р2Н4 -0,169
2P(tol,+ 4Н' + 4е =Р,Н„ -0,100
Н3РО2 4- Н+ 4- е — Р(красн.) + 2Н2О -0,365
Н3РО2 4- Н+ 4- е = Р(бел) + 2Н2О -0,508 (-0,51)
Н3РО2 + 4Н+ + 4е" = РН3(Г) 4- 2Н2О -0,174
4Н3РО2 + 6Н+ + бе- = Р4Н2+ 8Н2О -0,455
Н2РО2 + 2Н+ 4- е" = Р(красн.) 4- 2Н2О -0,248
Н2РО2 + 2Н+ + е- = Р(6ел) + 2Н2О -0,391
Н2РО2 +е- = Р(бея.) + 2ОН- -2,05
Н2РО2 4- ЗН2О + 4е’ = РН3(Г) + 5ОН -1,18
Н2РО2 + 5Н+ + 4е = РН3(Г) + 2Н2О -0,145
4Н2РО2 + 10Н+ + бе- = Р4Н2 + 8Н2О -0,376
Н3РОз + 2Н+ + 2е = Н3РО2 + Н2О -0,499
Н3РОз + ЗНЧ + Зе" = Р(красн) + ЗН2О -0,454
Н3РО3 + ЗН+ + Зе’ = Р(бел) + ЗН2О -0,502
Н3РОз + 6ЬГ + бе’ = РН3(Г) + ЗН2О -0,282
4Н3РО3 + 14Н + 14е’ = Р4Н2 + 12Н2О -0,480
Н2РО; + ЗЬГ + 2е’ = Н3РО2 + Н2О -0,446
Н2РО3 4-4Н+4-Зе ~ Р(красн) + ЗН2О -0,419
Н2РО3- + 4Н+ + Зе- = Р(бел) + ЗН2О -0,467
Н2РО3’ + 7Н+ + бе’ = РНз(г) + ЗН2О -0,265
4Н2РО3 + 18Н+ + 14е’ = Р4Н2 + 12Н2О -0,450
НРО3 + 2Н+ + 2е’ = НРО2’ + Н2О -0,323
НРО3- + ЗН+ + 2е’ = Н2РО2 + Н2О -0,504
ПРО2- + 2Н2О + 2е’ = Н2РО2 + ЗОН’ -1,57
НРО3 4- 5Н+ 4- Зе = Р(красн.) + ЗН2О -0,346
НРО3’ + 5РГ + Зе- = Р(бел) + ЗН2О -0,298
НРО2- + 8FT + бе’ = РН3(Г) + ЗН2О -0,205
4НРО2- + 22РГ + 14е = Р4Н2 4- 12Н2О -0,346
Н,Р2О6 4- 2е- = 2Н2РО; 0,275
РЦРгОб + 2РГ + 2е’ = 2Н3РО3 0,380
Электродные процессы
31
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Фосфор
Нзр,о; + Н+ + 2е' = 2Н,РО; 5 2. 0 2 3 0,340
Н2Р2О62’ + 2е" = 2НРО2’ 2 Z О 3 -0,061
Н2Р,О2' + 2Н+ + 2е' = 2Н2РО; 2 Z О 2 J 0,423
НР2О3’ + Н+ + 2е" = 2НРО2 2 о 3 0,275
Р,СГ + 2Н+ + 2е" = 2НРО2' 0,570
HPO3(aq) + 5Н4 + 5е = Р(бел ) + ЗН2О -0,346
Н3РО4 + FT + 2е" = Н2РО’ + Н2О -0,329
Н3РО4 + 2Н+ + 2е = Н3РО3 + Н2О -0,276
Н3РО4 + 4Н+ + 4е = Н3РО2 + 2Н2О 0,39
Н3РО4 + 5Н+ + 5е’ = Р(красн.) + 4Н2О -0,383
Н3РО4 + 5ЬГ + 5е' = Р(бел э + 4Н2О -0,411
Н3РО4 + 8Н+ + 8е = РН3(Г) + 4Н2О -0,281
2Н3РО4 + 2Н+ + 2е" = Н4Р2О6 + 2Н2О -0,94 (-0,933)
4Н3РО4 + 22Н+ + 22е = Р4Н2 + 16Н2О -0,406
н2ро; + Н+ + 2е = НРО2 + Н2О -0,447
Н2РО4 + 2Н+ + 2е" = Н2РО’ + Н2О -0,260
Н2РО; + 6Н+ + 5е" = Р(красн) + 4Н2О -0,358
Н2РО; + 6Н+ + 5е" - Р(бел) + 4Н2О -0,386
Н2РО; + 9Н+ + 8е = РН3(Г) + 4Н2О -0,265
2Н,РО: + 2Н* + 2е' = Н2Р2О2' +2Н2О -0,955
2Н2РО; + ЗН+ + 2е’ = Н3Р2О; +2Н2О -0,872
2Н2РО; + 4FT + 2е’ = ЩРгОб + 2Н2О -0,807
4Н2РО; + 26Н+ + 22е’ = Р4Н2 + 16Н2О -0,383
НРО2 + 2РГ + 2е" = НРО2 + Н2О -0,234
НРО’ + 7Н+ + 5е~ = Р(красн0 + 4Н2О -0,288
НРО2 + 7FT + 5е" = Р(беЛ.) + 4Н2О -0,316
НРО2 + 10Н+ + 8е' = РН3(Г) + 4Н2О -0,212
2НРО4' + 2FT + 2е = Р?О* + 2Н2О -1,039
2НРО2 + ЗН4 + 2е' = НР2О3~ + 2Н2О -0,744
2НРО2' + 4Н+ + 2е' = Н2Р2О2’ + 2Н2О -0,551
4НРО4” + 30Н+ +22е' = Р4Н2 + 16Н2О -0,305
РО3’ + ЗН* + 2е~ = НРО2" + Н2О -0,121
РО3’ + 2Н2О + 2е" = НРО2" + ЗОН’ -1,12
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
РО4 + 8Н+ + 5е" = Р(красн.) + 4Н2О -0,128
POJ" + 8Н’ + 5е" = Р(бел) + 4Н2О -0,156
POJ + 11ЬГ + 8е" = РН3(Г) + 4Н2О -0,123
2РО3’ + 4Н* + 2е" = Р2О*" + 2Н2О -0,328
4РО3’ + 34Н+ + 22е" = Р4Н2 + 16Н2О -0,176
Фтор
F(r) + е" = F' 2,850
F(r.) + Н+ + е’ = HF(aq) 3,030
F2(r) + 2е" = 2F" 2,87
F2(r) + 2Н4- + 2е“ = 2HF(aq) 3,06 (3,090)
F2(r.) + Н+ + 2е" = HF2 3,013 (2,979)
Хлор
С1; + 2е" = ЗСГ 1,415
С12(г) + 2е = 2С1 1,359(1,359)
С12(Г)+ 2Н+ + 2е" = 2НС1(г) 0,987
C12(aq) + 2е = 2С1 1,396(1,395)
НС1О + Н+ + 2е" = СГ + Н2О 1,50; (1,494)
2НС1О + 2Н+ + 2е" = Cl2(aq) + 2Н2О 1,594
2НСЮ + 2Н+ + 2е" = С12(г) + 2Н2О 1,630
СЮ’ + Н2О + 2е" = СЮ" + 2OFT 0,890
2СЮ’ + 2Н2О + 2е" = С12(г) + 4ОН’ 0,421 (0,49)
С12О(Г.) + 4Н+ + 4е’ = 2НС1(Г) + Н2О 1,351
Cl2O(r) + 2Н" + 2е’ = Cl2(aq) + Н2О 1,679
С12О(Г) + 2Н+ + 2е" - С12(г) + Н2О 1,714
С12О(Г) + 2FT + 4е’ = 2СГ + Н2О 2,152
НСЮ2 + 2FT + 2е" = НСЮ + Н2О 1,674(1,645)
НСЮ2 + ЗН+ + 4е’ = СГ + 2Н2О 1,570 (1,584; 1,56)
2НСЮ2 + 6Н+ + бе' = Cl2(aq) + 4Н2О 1,628
2НСЮ2 + 6Н4 + бе’ = С12(г) + 4Н2О 1,640(1,639)
СЮ’ + Н2О + 2е' = СЮ’ + 2ОН' 0,681 (0,66)
СЮ' + 2Н2О + 4е" = СГ + 4ОН" 0,77
СЮ2(Г) + е' = СЮ2 1,071
С1О2(Г) + Н+ + е = НСЮ2 1,188(1^75)
СЮ2(Г) + 4Н+ + 5е’ = СГ + 2Н2О 1,511 (1,50)
32
Новый справочник химика и технолога
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Хлор
2С10г(г.)+ 8Н* + 8е — Cl2(aq) + 4Н2О 1,540
2C102(r) + 8Н* + 8е’ = С12(г) + 4Н2О 1,549
С1О2(Г) + 5Н* + 5е’ = НС1(Г) + 2Н2О 1,436
СЮ2(И + 2Н2О + 5е = СГ + 4ОН 0,85
СЮ; + 2Н* + е’ = С1О2(Г) + Н2О 1,175(1,15)
сю; + Н2О + е’ = СЮ2(Г) + 2ОН 0,481
сю; + ЗН* + 2е’ = НСЮ2 + Н2О 1,181 (1,21)
сю; + Н2О + 2е’ = сю; + 2ОН’ 0,295
СЮ; + Н2О + 4е = СГ + 4ОН 0,488
сю; + 5Н* + 4е’ = НОС1 + 2Н2О 1,41
сю; + 6Н* + бе’ = СГ + ЗН2О 1,451 (1,45)
сю; + ЗН2О + бе’ = СГ + 6ОН" 0,622
2СЮ; + 12Н* + 1 Ое’ = Cl2(aq) + 6Н2О 1,463
2СЮ; + 12Н* + 1 Ое’ = С12(г) + 6Н2О 1,470(1,470)
сю; + 2Н* + 2е" = сю; + Н2О 1,189 (1,201; 1,19)
сю; + Н2О + 2е’ = сю; + 2ОН’ 0,374 (0,36)
СЮ; + 8Н* + 8е“ = СГ + 4Н2О 1,389(1,388)
СЮ4 + 4Н2О + 8е“ = СГ + 8ОН” 0,56
2СЮ; + 16Н* + 14е’ = Ci2(r) + 8Н2О 1,389 (1,392; 1,34)
2СЮ4 + 16Н+ + 14е" = Cl2(aq) + 8Н2О 1,385
Хром
Ст2* + 2е = Сг -0,90 (-0,913;-0,852)
Сг(ОН)2(т) + 2е’ = Сг + 2ОН’ -1,41
СгО(т) + 2Н* + 2е’ = Сг + Н2О -0,588
Сг3* + е" = Сг2* -0,424 (-0,407;-0,408)
Сг^+Зе’^Сг -0,744
Cr(OH)3(aq) + е” = Сг(ОН)2 + ОН" -1,175
Cr(OH)3(aq) + Зе’ = Сг + ЗОН" -1,34
Сг(ОН)3(т) + Зе’ = Сг + ЗОН- -1,48
Сг(ОН)з(Т.) + ЗН* + Зе’ = Сг + ЗН2О -1,654
[Cr(OH)6]3’ + е” = Cr(OH)2 + 4ОН -1,057
(Cr(CN)6f+ e~ = [Cr(CN)6]4 -1,043 (-1,28)
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
[Cr(edta)(H2O)]“ + е’ = = [Cr(edta)(H2O)]2’ -0,99
CrO3” + 6Н* + Зе’ = Сг + ЗН2О 0,374
СгО; + 4Н* + е’ = Сг2* + 2Н2О -1,188
CrO; + 4Н* + Зе" = Сг + 2Н2О -0,213
СгО; + 2Н2О + Зе’ = Сг + 4ОН’ -1,27
CrCi; + е’ = Сг2* + 2СГ -0,403
CrCi; + Зе = Сг + 2С1 -0,74
СгО2(т.) + 4Н* + е" = Сг3* + 2Н2О 1,556
Н2СгО4 + 6Н* + Зе’ = Сг3* + 4Н2О 1,335
Н2СгО4 + 6Н* + бе’ = Сг + 4Н2О 0,295
HCrO; + 7Н* + Зе’ = Ст3* + 4Н2О 1,38
СгО4‘ + 4Н* + 2е" = СгО2(т) + 2Н2О 1,437
CrOj’ + 2Н* + Зе’ = СгО3 + Н2О 0,359
СгО4" + 8Н* + Зе” “ Сг3* + 4Н2О 1,477
CrO2’ + 8Н* + бе" - Сг + 4Н2О 0,366
CrO2- + 4Н* + Зе’ = СгО; + 2Н2О 0,945
СгО2’ + 4Н2О + Зе” = [Cr(OH)4]’ + 4ОН” -0,13
СгО2 + 4Н2О + Зе’ = [Сг(ОН)6]3’ + 2ОН” -0,165
СгО2’ + 4Н2О + Зе’ = Сг(ОН)3(т) + 5ОН” -0,11 (-0,13;-0,125)
СгО4’ + Н2О + Зе’ = СгО3’ + 2ОЕГ -0,20
Cr2O2’ + 14Н* + бе’ = 2Сг3* + 7Н2О 1,36(1,333)
Сг2О2” + 14Н* + 12е’ = 2Сг + 7Н2О 0,294
Цезий
Cs* + е” = Cs -2,923
Церий
Се3* + Зе’ = Се -2,34 (-2,483)
Се(ОН)3(т) + Зе’ = Се + ЗОН” -2,78
Се(ОН)22* + 2Н* + е" = Се3* + 2Н2О 1,731 (1,70)
Се4* + е" = Се3* 1,72(1,743)
Се4* + 4е” = Се 1,68
СеО2 + 4Н* + е’ = Се3+ + 2Н2О 1,4(1,293)
СеО2(т) + 2Н2О + е’ = Се(ОН)3 + ОН’ -0,70
Электродные процессы
33
Продолжение таблицы 1.1
Окислительно-восстановительная полуреакция Стандартный электродный потенциал, В
Церий
[CeCU]2’ + e" = Ce3+ + 6СГ 1,28
[Ce(C104)6]2- + e’ = Ce3+ + 6CIO4 1,70
[Ce(NO3)6]2' + e = Ce3+ + 6NO3- 1,61
[Ce(SO4)3]2 + e" = Ce3+ + 3SO42- 1,44
Цинк
Zn2+ + 2e- = Zn -0,763 (-0,76)
ZnO(T) + H2O + 2e — Zn + 2OH -1,248
ZnS(BIopuHT) + 2e — Zn + S -1,44 (-1,405)
Zn(OH)2(T) + 2e = Zn + 2OH -1,246
Zn(OH)2(T) + 2H+ + 2e = Zn + 2H2O -0,439
Zn(OH)2(aq) + 2H+ + 2e- = Zn + 2H2O -0,400
HZnO2 + 31Г + 2e = Zn + 2H2O 0,054
HZnO- + H2O + 2e = Zn + ЗОН -1,2
ZnO2" + 4H++ 2e~ = Zn + 2H2O 0,441
ZnO2" + 2H2O + 2e" = Zn + 4OH -1,216
[Zn(OH)4]2” + 2e" = Zn + 4OH“ -1,285
[Zn(NH3)4]2+ + 2e“ = Zn + 4NH3(aq) -1,04
[Zn(CN)4]2“ + 2e = Zn + 4CN- -1,34 (-1,26)
ZnCO3(T) + 2e = Zn + CO2 -1,06
[Zn(C2O4)2]2 + 2e = Zn + 2C2O4- -0,99
[гп^ЩОб),]6- + 2e" = Zn + 4C4H4O2- -1,15
Цирконий
Zr4+ + 4e" = Zr -1,55 (-1,539; -1,529)
ZrO2(T) + 4FT + 4e" = Zr + 2H2O -1,45 (-1,553;-1,473)
ZrO2+ + 2ЕГ + 4e" = Zr + H2O -1,570
HZrO; + 5FT + 4e = Zr + 3H2O -1,276
[ZrF6]2- + 6H+ + 4e" = Zr + 6HF(aq) -1,608
[Zr(OH)2]2+ + 2H+ + 4e = Zr + 2H2O -1,355
ZrO(OH)2(T) + H2O + 4e = Zr + 4OH -2,225
Эрбий
Er3+ + 3e" = Er -2,32 (-2,296)
Er(OH)3(T) + 3e~ = Er + ЗОН- -2,84
Литература
1. Справочник химика. Т. III Химическое равновесие и кинетика. Свойства растворов. Электродные процессы / Гл. ред. Б.П. Никольский. М.-Л.: Химия, 1965. 1005 с.
2. Латимер В.М. Окислительные состояния элементов и их потенциалы в водных растворах. М.:ИЛ, 1954. 235 с.
3. Турьян Я.И. Окислительно-восстановительные потенциалы в аналитической химии. М.: Химия, 1989. 248 с.
4. Лидин Р.А., Андреева Л.Л., Молочков В.М. Справочник по неорганической химии. М.: Химия, 1987. 320 с.
5. Лидин Р.А. Справочник по общей и неорганической химии. М.: Просвещение, 1997. 253 с.
6. Краткий химический справочник / Под ред. А.А. Потехина и А.И. Ефимова. СПб.: Химия, 1994.432 с.
7. Гончаров А.И., Корнилов М.Ю. Справочник по химии. Киев: В. школа, 1977. 303 с.
8. Химический энциклопедический словарь / Гл. ред. И.Л. Кнунянц. М.: Сов. Энциклопедия, 1983. 792 с.
9. Свойства неорганических соединений: Справочник / А.И. Ефимов, Л.П. Белокурова, И.В. Васильева, В.П. Чечев. Л.: Химия, 1983. 392 с.
10. Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1989. 448 с.
11. Добош Д. Электрохимические константы. Справочник для электрохимиков / Пер. с англ, и венг. Под ред. Я.М. Колотыркина. М.: Мир, 1980. 36 с.
12. Справочник по электрохимии / Под ред. А.М. Сухотина. Л.: Химия, Лен. отд., 1981. 486 с.
13. Bard A.I., Parsons R., Iordan I. Standart Potentials in Aqueous Solutions. NY: Marcel Dekker, 1985.
34
Новый справочник химика и технолога
1.2. Дополнительные сведения
(проф., д.х.н. С.А. Симанова)
В этом разделе приведена информация о стандартных и вспомогательных электродах, применяемых для измерений pH и исследований в различных средах.
1.2.1. Стандартные электродные потенциалы в неводных средах
В табл. 1.2.1 приведены значения стандартных потенциалов электродных процессов в неводных средах.
Таблица 1.2.1
Стандартный электродный потенциал
Значения стандартных потенциалов Е0 даны при температуре 25 °C и давлении 1 атм. Потенциалы выражены в вольтах и отнесены к потенциалу стандартного водородного электрода в данном растворителе, принятому за нуль.
Электродный процесс E °, В, в растворителе
CH3OH C2H5OH CH3CN НСООН NH3 N2H4
Li+ + е = Li -3,095 -3,042 -3,23 -3,48 -2,24 -2,20
Na+ + е- = Na -2,727 -2,656 -2,87 -3,42 -2,01 -1,82
К+ + е' -К -3,16 -3,36 -1,98 -2,02
Rb+ + е“ = Rb -3,17 -3,45 -1,93 -2,01
Cs+ + е~ = Cs -3,16 -3,44 -1,95
Са2+ + 2е = Са -2,75 -3,2 -1,64 -1,91
Zn2+ + 2е“ = Zn -0,74 -0,64 -0,74 -1,05 -0,53 -0,41
Cd2+ + 2е" = Cd -0,43 -0,38 -0,47 -0,75 -0,20 -0,10
Tl+ + е" = Т1 -0,379 -0,343
РЬ2’ + 2е = РЬ -0,20 -0,15 -0,12 -0,72 +0,32 +0,35
2Н+ + 2е = Н2 0,000 0,000 0,000 0,000 0,000 0,000
Сп+ + е“ = Си -0,38 +0,41 +0,22
Си2++2е“= Си +0,34 +0,21 -0,28 -0,14 +0,43
Hg2+ + 2e” = 2Hg +0,74 +0,76 +0,18
Hg2+ + 2e“ = Hg +0,25 +0,75
Ag+ + e = Ag +0,764 +0,749 +0,23 +0,17 +0,83 +0,77
I2 + 2e" = 2Г +0,357 +0,305 +1,45
Br2 + 2e = 2ВГ +0,887 +0,777 +1,83
Cl2 + 2e" = 2СГ +1,116 +1,048 +2,03
1.2.2. Вспомогательные электроды и электроды для измерения pH
Все значения потенциалов выражены в вольтах и отнесены к потенциалу стандартного водородного электрода при соответствующей температуре.
Водородный электрод
Pt (платинированная) | H2SO4 || Исследуемый раствор
Этот электрод применяют для точных определений pH. Электродная реакция:
2Н+ + 2е" = Н2
Стандартная свободная энергия и стандартный электродный потенциал этой реакции при любой температуре приняты равными нулю.
Потенциал водородного электрода Е зависит от активности ионов водорода:
£ = 2,3—Ig-^-
? Рн2 или, при рН2 = 1 атм ,
Для приведения потенциала водородного электрода, найденного при измерениях, к нормальному давлению (1 атм) следует вносить поправку А, учитывающую истинное давление водорода над раствором. Эта поправка вычисляется по формуле А£:
_ 2,3/?7\ 760
£ = —-1g---------(табл. 1.2.2),
F Р-р
где Р — барометрическое давление, ар — суммарное давление насыщенных паров над раствором.
Электродные процессы
35
Таблица 1.2.2
Поправка ЛЕ для разбавленных водных растворов при различных давлениях (мм рт. ст.)
7, °C 780 770 760 750 740
0 -0,00024 -0,00008 -0,00007 0,00023 0,00039
5 —0,00021 -0,00006 0,00010 0,00026 0,00043
10 —0,00017 -0,00001 0,00015 0,00031 0,00048
15 —0,00012 0,00004 0,00021 0,00038 0,00055
20 -0,00004 0,00012 0,00029 0,00046 0,00064
25 0,00007 0,00024 0,00041 0,00059 0,00076
30 0,00020 0,00038 0,00056 0,00074 0,00092
35 0,00040 0,00058 0,00076 0,00095 0,00113
40 0,00064 0,00082 0,00101 0,00121 0,00141
45 0,00098 0,00115 0,00136 0,00156 0,00176
50 0,00140 0,00160 0,00181 0,00202 0,00223
55 0,00195 0,00217 0,00239 0,00261
60 0,00269 0,00291 0,00314 0,00338
Хингидронный электрод
Pt (гладкая) | Хингидрон (насыщ.) || Исследуемый раствор
Каломельный электрод
Hg | Hg2Cl2; КС11| Исследуемый раствор
Данный электрод используют для измерений pH в интервале от 0 до 6, а в буферных растворах в отсутствие сильных окислителей — от 0 до 8,5. Электродная реакция:
С6Н4О2 + 2Н+ + 2е’ = С6Н4(ОН)2
Зависимость потенциала электрода Е от активности ионов водорода в интервале температур 0-50 °C выражается формулой:
£ = 0,6992-7,4- 10^(7-25) +
+ [0,0591 + 2- Ю4 (Т-25)] lgaH+
Таблица 1.2.3
Стандартный потенциал хингидронного электрода
7, °C Е°, В 7, °C 7е, В 7, °C Е°, В
0 0,6807 17 0,6933 34 0,7059
1 0,6814 18 0,6940 35 0,7066
2 0,6822 19 0,6948 36 0,7073
3 0,6829 20 0,6955 37 0,7081
4 0,6837 21 0,6962 38 0,7088
5 0,6844 22 0,6970 39 0,7096
6 0,6851 23 0,6977 40 0,7103
7 0,6859 24 0,6985 41 0,7110
8 0,6866 25 0,6992 42 0,7118
9 0,6874 26 0,6999 43 0,7125
10 0,6881 27 0,7007 44 0,7133
11 0,6888 28 0,7014 45 0,7140
12 0,6896 29 0,7022 46 0,7147
13 0,6903 30 0,7029 47 0,7155
14 0,6911 31 0,7036 48 0,7162
15 0,6918 32 0,7044 49 0,7169
16 0,6925 33 0,7051 50 0,7177
Этот электрод применяют в качестве вспомогательного. Он характеризуется хорошей воспроизводимостью потенциала. Электродная реакция:
Hg2Cl2 + 2е" = 2Hg + 2СГ
Стандартный электродный потенциал Е? этой реакции в интервале температур 0-50 °C выражается формулой:
7ю = 0,26647 - 3,465 • 10 4 (7- 30) -- 2,87 • 10 6 (7- 30)2 - 8,5 • 10 9 (7- 10)3.
Электрод имеет следующие разновидности:
1. Насыщенный каломельный электрод, заполняемый насыщенным раствором КС1. Этот электрод отличается наилучшей воспроизводимостью потенциала, но имеет больший, чем другие разновидности, температурный коэффициент. Потенциал электрода Е:
£ = 0,2438-6,5- 10^(7-25).
2. Нормальный каломельный электрод, заполняемый насыщенным раствором КС1. Потенциал электрода Е:
£ = 0,2828-2,4-10^(7-25).
3. Децимолярный каломельный электрод, заполняемый 0,1 М раствором КС1. Потенциал электрода £:
£=0,3665-6 - 10^(7-25).
36
Новый справочник химика и технолога
Таблица 1.2.4
Потенциал каломельного электрода при различных температурах
Г, °C Е,В
0,1 МКС1 1 МКС1 насыщ. КС1
0 0,3380 0,2888 0,2601
1 0,3379 0,2886 0,2594
2 0,3379 0,2883 0,2588
3 0,3378 0,2881 0,2581
4 0,3378 0,2878 0,2575
5 0,3377 0,2876 0,2568
6 0,3376 0,2874 0,2562
7 0,3376 0,2871 0,2555
8 0,3375 0,2866 0,2542
9 0,3375 0,2866 0,2542
10 0,3374 0,2864 0,2576
11 0,3373 0,2862 0.2429
12 0,3373 0,2859 0,2523
13 0,3372 0,2857 0,2516
14 0,3372 0,2854 0,2510
15 0,3371 0,2852 0,2529
16 0,3370 0,2850 0,2497
17 0,3370 0,2847 0,2490
18 0,3369 0,2845 0,2483
19 0,3369 0,2842 0,2477
20 0,3368 0,2840 0,2471
21 0,3367 0,2838 0,2464
22 0,3367 0,2835 0,2458
23 0,3366 0,2833 0,2451
24 0,3366 0,2830 0,2445
25 0,3365 0,2828 0,2438
26 0,3364 0,2826 0,2431
27 0,3364 0,2823 0,2425
28 0,3363 0,2821 0,2418
29 0,3363 0,2818 0,2412
30 0,3362 0,2816 0,2405
31 0,3361 0,2814 0,2399
32 0,3361 0,2811 0,2393
33 0,3360 0,2809 0,2386
34 0,3360 0,2806 0,2379
35 0,3359 0,2804 0,2373
36 0,3358 0,2802 0,2366
37 0,3358 0,2799 0,2360
38 0,3357 0,2797 0,2353
39 0,3357 0,2794 0,2344
40 0,3356 0,2792 0,2340
41 0,3355 0,2790 0,2334
42 0,3355 0,2787 0,2327
43 0,3354 0,2785 0,2321
44 0,3354 0,2782 0,2314
45 0,3353 0,2780 0,2308
46 0,3352 0,2778 0,2301
47 0,3352 0,2775 0,2295
48 0,3351 0,2773 0,2288
49 0,3351 0,2770 0,2282
50 0,3350 0,2768 0,2275
Электрод ртуть—сульфат ртути(1) (сернокислая ртуть(1))
Hg | Hg2SO4; H2SO4 || Исследуемый раствор
Данный электрод применяют как вспомогательный при исследованиях в растворах серной кислоты. Он характеризуется несколько худшей воспроизводимостью потенциала, чем каломельный электрод. В первое время после приготовления электрода его потенциал заметно изменяется. При исследованиях в нейтральных сернокислых растворах вместо H2SO4 электрод наполняют раствором K2SO4.
Электродная реакция:
Hg2SO4 + 2е= 2Hg + SO2
Температурная зависимость стандартного потенциала электрода Е? в интервале температур 0-60 °C (табл. 1.2.5) выражается формулой:
Е? = 0,61515 - 8,02 • 10-4 (Г-25)-4 • 10’7 (Т-25)2.
Таблица 1.2.5
Стандартный потенциал электрода ртуть—сульфат ртути(1)
Г, °C £°, В Г, °C £°,В
0 0,63495 35 0,60701
5 0,63097 40 0,60305
10 0,62704 45 0,5990
15 0,62307 50 0,59487
20 0,61930 55 0,59051
25 0,61515 60 0,58655
30 0,61107
Электрод ртуть — оксид ртути(П)
Hg | HgO; МеОН || Исследуемый раствор
Этот электрод используют как вспомогательный при исследованиях в щелочных растворах. Электродная реакция:
HgO + Н2О + 2е = Hg + 2ОН”
Стандартный потенциал £° электрода при 25 °C равен 0,0982 В.
Хлорсеребряный электрод
Ag | AgCl; НС11| Исследуемый раствор
Данный электрод применяют в качестве вспомогательного в средах, содержащих хлорид-ионы, в частности в неводных и смешанных растворителях. Он характеризуется хорошей воспроизводимостью и устойчивостью потенциала. Электродная реакция:
AgCl + е = Ag + СГ
Стандартный потенциал Е? этого электрода в водной среде в интервале температур 0-100 °C (табл. 1.2.6) выражается фомулой:
5ю = 0,22234 - 6,4 • Ю'4 (Т- 25) - 3,2 • 10-6 (Г-25)2.
Электродные процессы
37
Таблица 1.2.6
Стандартный потенциал хлорсеребряного электрода
Вода
Т,°С £°, В Г, °C £°, В Т,°С £°, В Г, °C £°, В
0 5 10 15 20 0,23655 0,23413 0,23142 0,22857 0,22557 25 30 35 40 45 0,22234 0,21904 0,21565 0,21208 0,20835 50 55 60 70 80 0,20449 0,20056 0,19640 0,18782 0,1787 90 95 0,1695 0,1651
Метанол—вода
Г, °C £°, В при концентрации СН3ОН
10 масс. % 20 масс. % 43,3 масс. % 64 масс. % 84,2 масс. % 94,2 масс. % 100 масс. %
5 10 15 20 25 30 35 40 45 50 0,22762 0,22547 0,22328 0,22085 0,21821 0,21535 0,21220 0,20892 0,20550 0,22022 0,21837 0,21631 0,21405 0,21155 0,20881 0,20567 0,20246 0,19910 0,2010 0,1975 0,1939 0,1901 0,1860 0,1818 0,1771 0,1864 0,1813 0,1765 0,1717 0,1668 0,1620 0,1563 0,1452 0,1384 0,1319 0,1252 0,1184 0,1114 0,1039 0,0979 0,0908 0,0838 0,0768 0,0693 0,0618 0,0539 0,0014 -0,0044 -0,0103 -0,0164 -0,0228
Этанол—вода Диоксан—вода
Г, °C Е?, в, при концентрации С2Н5ОН Г, °C Е°, В, при концентрации С4Н8О2
10 масс. % 20 масс. % 20 масс. % 45 масс. % 70 масс. % 82 масс. %
0 10 20 25 30 40 0,22726 0,22328 0,21901 0,21467 0,21383 0,20783 0,21606 0,21367 0,21013 0,20757 0,20587 0,19962 0 5 10 15 20 25 30 35 40 0,21975 0,21677 0,21362 0,21025 0,20674 0,20303 0,19914 0,19505 0,19080 0,18938 0,18468 0,17972 0,17454 0,16916 0,16358 0,15778 0,15182 0,14560 0,10584 0,09784 0,08970 0,08123 0,07267 0,06395 0,05500 0,04587 0,03661 -0,0130 -0,246 -0,0370 -0,0487 -0,0614 -0,0738 -0,0871 -0,1012
Изопропанол— -вода
Г, °C £°,В, при концентрации СН3СНОНСН3 45 50 0,18634 0,18171 0,13925 0,13282 0,02705 0,01746 -0,1172
5 масс. % 10 масс. % 20 масс. % Бромсеребряный электрод
0 5 10 15 20 25 30 35 40 0,23106 0,22892 0,22654 0,22390 0,22107 0,21807 0,21494 0,21164 0,20809 0,22543 0,22365 0,22158 0,21922 0,21667 0,21383 0,21081 0,20754 0,20410 0,21612 0,21492 0,21336 0,21138 0,20906 0,20637 0,20341 0,20009 0,19652 Ag | AgBr; НВг || Исследуемый раствор Этот электрод применяют как вспомогательный электрод в средах, содержащих бромид-ионы. Электродная реакция: AgBr + е“ = Ag + Вг Стандартный потенциал электрода Е° в водной среде при 25 °C равен 0,07131 В (табл. 1.2.7), в метаноле при 25 °C равен -0,1375 В. Потенциал бромсеребряного электрода плохо воспроизводится в присутствии растворенного кислорода.
38
Новый справочник химика и технолога
Таблица 1.2.7
Стандартный потенциал бромсеребряного электрода в водной среде
Т,°С £°, В Т,°С В
0 0,08163 30 0,06872
5 0,07991 35 0,06597
10 0,07802 40 0,06304
15 0,07595 45 0,05995
20 0,07372 50 0,05667
24 0,07131
Иодсеребряный электрод
Ag | Agl; HI || Исследуемый раствор
Данный электрод используют в качестве вспомогательного электрода в средах, содержащих иодид-ионы, в отсутствие растворенного кислорода и других окислителей. Электродная реакция:
Agl + е = Ag + I
В водной среде при 25 °C стандартный потенциал данного электрода Л° равен -0,15225 В (табл. 1.2.8).
Таблица 1.2.8
Стандартный потенциал иодсеребряного электрода
Т,°С £°, В Г, °C В
5 -0,14712 25 -0,15225
10 -0,14805 30 -0,15396
15 -0,14920 35 -0,15586
20 -0,15062 40 -0,15787
Электрод диоксид свинца—сульфат свинца(П) (сернокислый свинец)
РЬО21 PbSO4; H2SO4 || Исследуемый раствор
Этот электрод применяют при исследованиях в растворах серной кислоты и ее солей, особенно при исследованиях свинцовых аккумуляторов. Электродная реакция:
РЬО2 + SOJ’ + 4Н+ + 2е - PbSO4 + 2Н2О
При 25 °C стандартный потенциал электрода £° равен 1,68488 В (табл. 1.2.9).
Таблица 1.2.9
Стандартный потенциал электрода с диоксидом свинца
Т,°С Ер, в Т,°С Е?,в
0 1,67694 35 1,68847
5 1,67846 40 1,69036
10 1,67998 45 1,69231
15 1,68159 50 1,69436
20 1,68322 55 1,69649
25 1,68488 60 1,69861
30 1,68671
1.2.3. Нормальные элементы
Нормальный элемент Вестона
Cd(Hg) | CdSO4 • 8/3Н2О, CdSO4, Hg2SO4 | Hg
(12,5%) (насыщ. р-р)
Нормальный элемент Вестона отличается долговечностью, хорошей воспроизводимостью и постоянством напряжения. Потенциал Е в интервале температур 0-40 °C воспроизводится с точностью ± 0,02 мВ:
Е = 1,017954-4,06- 10-5 (Т-20) -
- 9,5 • 10’7 (Т -20)2 + 1 • Ю 8 (Т -20)3.
Внутреннее сопротивление элемента 500-1000 Ом.
Для технических целей, когда не требуется высокая воспроизводимость, можно применять вторичный элемент Вестона:
Cd(Hg) | CdSO4; Hg2SO41 Hg
(12,5%) (насыщ. р-р при 4 °C)
В интервале температур 0-30 °C этот элемент имеет ничтожно малый температурный коэффициент. Потенциал в указанном интервале температур воспроизводится с точностью ± 0,1 мВ:
Е = 1,0184 ±0,0001.
Нормальный элемент Кларка
Zn(Hg) | ZnSO4 • 7Н2О; ZnSO4; Hg2SO4 ] Hg
(10%) (насыщ. р-р)
Потенциал Е этого элемента в интервале температур 0-30 °C воспроизводится с точностью ±0,1 мВ:
Е = 1,4320- 1,19 • 10 3(Т- 15)-7 • 10^(7- 15)2.
1.2.4. Перенапряжение выделения водорода
При восстановлении водорода на различных катодах зависимость перенапряжения т| (В) от плотности тока j (А/см2) выражается уравнением Тафеля:
П = а + b Igy,
где а и b — табличные константы (табл. 1.2.10).
Таблица 1.2.10
Константа а и Ь для уравнения Тафеля (20 °C)
Материал катода Раствор а Ь
Pt (гладкая) 1,0МНС1 l,0MNaOH± + 3,0 М Na2SO4 0,10 0,31 0,13 0,097
W 1,0МНС1 5,0 М НС1 0,23 0,55 0,040 0,11
Pd 1,1 м КОН 0,53 0,130
Со 1,0МНС1 0,62 0,140
Ni 0,11 MNaOH 0,64 0,100
Fe 1,0МНС1 2,0 М NaOH 0,70, 0,76* 0,125 0,112
Электродные процессы
39
Продолжение таблицы 1.2.10 Продолжение таблицы 1.2.10
Материал катода Раствор а b Материал катода Раствор а b
Ag 1,0МНС1 10,0MH2SO4 0,95 0,95 0,116 0,13 Hg 1,0МНС1 2,0 М H2SO4 1,0 М КОН 1,406 1,415 1,51 0,116 0,113 0,105
Си 2,0 М H2SO4 0,80 0,115
Sn 1,0МНС1 1,24 0,116 Т1 3,4 М H2SO4 1,55 0,140
Zn 2,0 М H2SO4 1,24 0,118 РЬ 2,0 М H2SO4 1,56 0,110
Cd 2,6 М H2SO4 1,40 0,120 * Экстраполировано оту =10 5 А/см2.
L2.5. Потенциалы нулевого заряда металлов и амальгам
В табл. (1.2.11-1.2.13) приведены значения потенциалов нулевого заряда £пнз (в вольтах по водородной шкале) некоторых металлов и амальгам при 20 °C. Потенциалы металлов восьмой группы определены по измерениям емкости двойного электрического слоя в водных растворах, содержавших различные количества Na2SO4 (0,04 М), H2SO4 (0,04 М) и NaOH (0,02 М). Измерения проводились после предварительной катодной поляризации в течение 1 ч при плотности тока 0,25 мА/см2. Потенциалы амальгам при различных концентрациях металла измерены по положению максимума электрокапиллярной кривой. N— мольная доля металла в амальгаме.
Таблица 1.2.11
Потенциалы нулевого заряда металлов
Металл Раствор Е В нз ? и Метод измерения
Кадмий 0,04 М H2SO4 (-0,7Н-0,9) Емкостный
Медь 0,04 М H2SO4 -0,03 То же
Платина, насыщенная 2 М Na2SO4 + 0,02 М H2SO4 0,4-1,0 Адсорбционный
кислородом
Ртуть 0,04 М H2SO4 -0,21 Емкостный
Свинец 0,002 М H2SO4 -0,67 То же
Серебро 0,04 М H2SO4 0,05 »
Таллий 2 М Na2SO4 -0,69 По твердости
Теллур 2 М Na2SO4 0,6 То же
Цинк, монокристалл грань [0101] 0,04 М H2SO4 -0,68 Емкостный
Цинк, монокристалл грань [0101] 0,04 М H2SO4 -0,60 То же
Таблица 1.2.12
Потенциалы нулевого заряда металлов восьмой группы периодической системы элементов
Металл Еп н з, В, при pH раствора
1 2 3 4 5 6 7 8 8,8 10 11
Железо -0,30 -0,35 -0,38 -0,40
Кобальт -0,33 -0,40 -0,45 -0,47 -0,48 -0,48
Никель -0,23 -0,29 -0,32 -0,34 -0,35 -0,36
Палладий +0,19 -0,06 -0,19 -0,28 -0,31 -0,34 -0,37 -0,39 -0,39 -0,40
Платина +0,48 +0,30 +0,13 +0,01 -0,13 -0,22 -0,28 -0,31 -0,34 -0,35
Платина после 5 ч предварительной катодной поляризации +0,48 +0,30 +0,19 +0,12 +0,08 +0,06 +0,03 +0,02 +0,01
40
Новый справочник химика и технолога
Таблица 1.2.13
Потенциалы нулевого заряда амальгам металлов
А £пнз, В, 1 MNaOH
Натрий
0,000 -0,21
0,000126 -1,69
0,000783 -1,73
0,00587 -1,78
0,0174 -1,81
0,0558 -1,82
N £п.н 3., В, 2 М H2SO4
Цинк
0,000 -0,21
0,000594 -0,35
0,00587 -0,40
0,0558 -0,45
Кадмий
0,000 -0,21
0,00118 -0,24
0,00587 -0,32
0,0558 -0,42
Таллий
0,000 -0,21
0,00927 -0,39
0,033 -0,45
0,102 -0,52
0,118 -0,55
0,332 -0,64
0,411 (насыщ.) -0,65
1.2.6. Стандартные токи обмена металлов
Значения стандартных токов обмена металлов /*, (т. е. токов обмена при а = 1, где а — активность ионов металла в растворе) в водных растворах при 20 °C и значения параметра 0 приведены в табл. 1.2.14.
Зависимость плотности катодного тока j от потенциала электрода Е выражается формулой:
fizFEj
Таблица 1.2.14
Стандартные токи обмена металлов
Металл Раствор 7°, А/см2 P Метод определения
Железо FeSO4 1 • IO’7 0,5 Осцилограммы включения
Кобальт CONO3 6,3 • 10 8 0,27 Радиоактивные индикаторы
COSO4 1,3 • 10~5 0,35 Осцилограммы включения
СоС12 8,0 • 107 0,5 То же
СоС12 2,0 • 10 7 Радиоактивные индикаторы
Медь CuSO4 1 • 104 0,5 Осцилограммы включения
Никель NiCl2 io-7 -108 То же
Серебро AgNO3 3,2 • 10’5 0,5 Радиоактивные индикаторы
Цинк ZnSO4 4 • 105 0,5 Осцилограммы включения
1.2.7. Дифференциальная емкость двойного электрического слоя на ртути
Значения дифференциальной емкости двойного электрического слоя на ртутном электроде в водных и спиртовых растворах серной кислоты приведены при 20 °C.
М — молярность раствора серной кислоты, Ск и Са — емкости двойного слоя, образованного соответственно катионами или анионами.
Растворитель м, моль/л с мкФ/см2 с мкФ/см2
Н2О 4,0 35-40 18
С2Н5ОН 4,0 7,2
С3Н7ОН 4,0 5,1
С4Н9ОН 4,0 3,75
С5НнОН 2,9*
С6Н13ОН 0,8 2,2
С7Н15ОН 1,65*
С8Н17ОН 1,30*
С9Н19ОН 0,18 1,05
* Получено интерполяцией.
1.3. Электродные процессы в расплавах
{проф., д.х.н. С.А. Симанова)
Таблица 1.3.1
Равновесные напряжения электрохимических систем с твердыми или расплавленными хлоридами металлов
В таблице приведены вычисленные из термохимических данных значения напряжения электрохимических систем типа:
Me | МеС1„ | С12, р - 1 атм, где Me — металл; МеС1„ — хлорид этого металла.
Температура плавления (7^) дана для давления 760 мм рт. ст. Если после температуры плавления стоит слово «разл.», это означает, что вещество плавится при данной температуре с разложением; если температура стоит после «разл.», это означает, что при данной температуре вещество разлагается без плавления.
В тех случаях, когда значения напряжения относятся к температуре кипения, возгонки или разложения хлорида, соответствующая температура приводится в скобках с добавлением сокращений «кип.», «возг.», «разл.».
МеС1„ Напряжение, В, при температуре
100 °C 200 °C 300 °C 350 °C 400 °C 450 СС 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
AgCl 1,093 1,037 0,984 0,959 0,935 0,911 0,896 0,883 0,870 0,826 0,784 0,665 455
А12С16 2,150 2,097 (180 возг.)
AsC13 0,986 0,973 (130 кип.) -18; (-16)
AuCl 0,138 0,082 0,037 (287 разл.)
AuCl3 0,162 0,101
ВаС12 4,139 4,056 3,975 3,935 3,888 3,848 3,808 3,768 3,728 3,568 3,412 3,079 960
ВеС12 2,382 2,315 2,252 2,222 2,192 2,167 2,144 2,122 (547 кип.) 404
BiCl3 1,051 0,986 0,926 0,896 0,867 0,844 (441 кип.) 320
СаС12 3,830 3,754 3,680 3,643 3,607 3,570 3,534 3,498 3,462 3,323 3,208 2,926 772 (782)
CdCl2 1,715 1,637 1,560 1,521 1,481 1,442 1,403 1,364 1,331 1,193 1,002 (980 кип.) 564 (568)
Электродные процессы
Продолжение таблицы 1.3.1
МеС1„ Напряжение, В, при температуре 7k, °C
100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
СоС12 1,408 1,337 1,269 1,236 1,203 1,171 1,140 1,109 1,079 0,977 0,900 0,881 (1050 кип.) 724 («740)
СгС12 1,795 1,729 1,664 1,632 1,600 1,568 1,537 1,505 1,474 1,352 1,262 1,137 (1302 кип.) 815 (824)
СгС13 1,646 1,567 1,489 1,451 1,412 1,374 1,336 1,299 1,261 1,113 1152
CsCl 4,109 4,002 3,896 3,843 3,791 3,739 3,692 3,645 3,599 3,362 3,078 2,667 (1300 кип.) 638 (642)
CuCl 1,191 1,140 1,093 1,071 1,050 1,035 1,024 1,013 1,003 0,970 0,943 430
CuCl2 0,791 0,721 0,654 0,621 0,589 0,558 0,528 630
DyCl3 3,149 3,075 3,004 2,970 2,935 2,901 2,868 2,835 2,802 2,690 2,599 2,359 655 (680)
ErCl3 3,062 2,989 2,918 2,883 2,849 2,815 2,781 2,748 2,715 2,589 2,488 2,257 (1497 кип.) 774
EuCl3 3,283 3,210 3,139 3,104 3,070 3,036 3,002 2,969 2,936 2,828 2,815 (827 разд.) 623 ±21
FeCl2 1,516 1,451 1,388 1,357 1,327 1,297 1,267 1,237 1,207 1,118 1,050 1,041 (1026 кип.) 672 (677)
FeCl3 1,147 1,084 1,023 1,016 (319 кип.) 304 (309)
GaCl2 1,659 170,5
GaCl3 1,572 1,528 (200 кип.) 78
GdCl 3,260 3,187 3,116 3,081 3,047 3,013 2,979 2,946 2,913 2,807 2,709 2,483 612 (628)
GeCLt 1,190 (83 кип.) -49,6
2,481 2,409 2,340 2,328 (317 возг.)
HgCl2 0,892 0,814 0,743 0,741 (304 кип.) 277
Hg2Cl2 1,022 0,930 0,840 0,800 0,730 возг.« 400
HoCl3 3,077 3,003 2,932 2,897 2,863 2,829 2,796 2,762 2,729 2,610 2,511 2,283 718(821)
InCi 1,654 1,581 1,520 1,492 1,465 1,439 1,414 1,389 1,364 1,360 (609 кип.) 225 ± 1
Новый справочник химика и технолога
Продолжение таблицы 1.3.1
МеС1„ Напряжение, В, при температуре Тт,°С
100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
1пС12 1,601 1,528 1,464 1,435 1,407 1,380 1,361 (485 кип.) 235
1пС13 1,588 1,519 1,431 1,417 1,384 1,352 1,321 (498 возг.)
IrCl 0,751 0,699 0,650 0,626 0,603 0,580 0,558 0,536 0,515 0,433 (799 разл.)
1гС12 0,685 0,625 0,567 0,539 0,512 0,485 0,458 0,432 0,406 0,320 (771 разл.)
IrCl3 1,610 1,539 1,472 0,439 0,406 0,374 0,342 0,311 0,280 0,180 (765 разл.)
КС1 4,158 4,056 3,954 3,904 3,854 3,805 3,755 3,707 3,658 3,441 3,155 768-770
LaCl3 3,504 3,426 3,350 3,313 3,277 3,241 3,205 3,170 3,134 2,997 2,876 2,607 855 (872)
LiCl 3,955 3,881 3,800 3,761 3,722 3,684 3,646 3,508 3,671 3,457 3,352 3,122 (1382 кип.) 613
LuCl3 2,988 2,909 2,834 2,796 2,760 2,723 2,687 2,652 2,616 2,478 2,356 2,108 (1477 кип.) 890-895
MgCl2 3,006 2,922 2,840 2,800 2,760 2,720 2,680 2,641 2,602 2,460 2,346 1,974 (1418 кип.) 708(714)
MnCl2 2,235 2,166 2,098 2,065 2,032 1,999 1,967 1,935 1,902 1,807 1,725 1,649 (1190 кип.) 650
MoCl2 0,711 0,651 0,593 0,566 0,538 0,511 0,485 0,471 (527 разл.)
MoCl3 0,669 0,602 0,536 0,519 (327 разл.)
M0CI4 0,600 0,582 (127 разл.)
M0CI5 0,550 0,491 0,463 (268 кип.) 194
NaCl 3,910 3,810 3,712 3,663 3,615 3,566 3,519 3,471 3,424 3,240 3,019 2,366 (1465 кип.) 801
NdCl3 3,369 3,291 3,215 3,177 3,140 3,103 3,067 3,030 2,994 2,856 2,736 2,455 760 (784)
NiCl2 1,355 1,282 1,210 1,174 1,139 1,104 1,070 1,036 1,003 0,875 0,763 (987 возг.)
Электродные процессы
Продолжение таблицы 1.3.1
МеС1„ Напряжение, В, при температуре 7™ , °C
100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
РЬС12 1,569 1,493 1,420 1,383 1,345 1,307 1,271 1,243 1,215 1,112 1,039 (954 кип.) 501 ±5
PdCl2 0,714 0,646 0,581 0,549 0,518 0,487 0,457
PmCl3 3,353 3,279 3,208 3,173 3,139 3,105 3,072 3,038 3,006 2,884 2,784 2,554
PrCl3 3,420 3,342 3,266 3,229 3,192 3,156 3,120 3,085 3,049 2,911 2,795 2,523 818(823)
PtCl 0,525 0,465 0,407 0,380 0,353 0,326 0,300 0,274 0,257 (583 разд.)
PtCl2 0,513 0,449 0,387 0,357 0,328 0,299 0,270 0,242 0,225 (581 разд.)
PtCl3 0,426 0,351 0,279 0,243 0,209 0,185 (435 разд.)
Ptcu 0,412 0,344 0,278 (370 разд.)
RaCl2 4,272 4,189 4,108 4,068 4,029 3,990 3,952 3,913 3,876 3,723 2,569 3,098 900
RbCl 4,101 3,998 3,897 3,846 3,795 3,745 3,695 3,645 3,595 3,314 0,001 2,428 (1381 кип.) 715
RhCl2 0,524 0,460 0,398 0,368 0,339 0,310 0,281 0,253 0,225 0,142 0,093 (958 разд.)
RhCl3 0,539 0,471 0,406 0,374 0,343 0,312 Разд. 450-500
RhCU 0,478 0,425 0,376 0,352 0,328 0,305 0,282 0,260 0,238 0,153 0,084 (965 разд.)
RuCl3 0,494 0,428 0,364 0,333 0,303 0,273 0,243 0,214 0,185 0,170 (627 разд.)
SbCl3 1,077 1,028 1,019 (221 кип.) 73,4
SbCl5 0,701 2,8 (4,0)
ScCl3 2,885 2,807 2,731 2,694 2,657 2,621 2,585 2,549 2,514 2,375 939 (960)
SiCl4 1,466 (57 кип.) -70 (под давлением)
Si2Cl6 1,736 1,713 (145 кип.)
SmCl2 4,147 4,071 3,998 3,962 3,926 3,891 3,856 3,822 3,787 3,661 3,559 3,317 740
Новый справочник химика и технолога
Продолжение таблицы 1.3.1
МеС1„ Напряжение, В, при температуре ТПЯ,°С
100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
SmCl3 3,322 3,249 3,178 3,143 3,109 3,075 3,041 3,008 2,975 2,861 2,763 2,712 (1107 кип.) 678 ±2
SnCl2 1,556 1,490 1,428 1,400 1,373 1,346 1,320 1,295 1,270 1,259 (623 кип.) 247
SnCU 1,184 1,176 (113 кип.) -33 (-30,2)
SrCl2 3,987 3,909 3,832 3,794 3,757 3,720 3,684 3,648 3,612 3,469 3,333 873
Т1С1 1,865 1,798 1,732 1,696 1,660 1,629 1,606 1,583 1,561 1,473 1,470 (806 кип.) 427 (430)
ТЬС13 3,204 3,130 3,059 3,025 2,990 2,956 2,923 2,890 2,858 2,754 2,657 2,433 588 (591)
TbCU 2,779 2,699 2,622 2,584 2,546 2,509 2,472 2,435 2,399 2,264 2,208 (921 кип.) 770
TiCl2 2,202 2,134 2,069 2,037 2,006 1,975 1,945 1,914 1,885 (разл.)
TiCl3 2,097 2,024 1,954 1,919 1,886 1,852 1,836 (475 разл.)
TiCl4 1,700 1,678 (136 кип.) -23,0
TuCl3 3,029 2,956 2,884 2,850 2,815 2,781 2,748 2,715 2,682 2,553 2,447 2,221 (1487 кип.) 824
UC13 2,788 2,713 2,639 2,602 2,566 2,530 2,494 2,458 2,423 2,280 2,162 1,886 835 (842)
UCI4 2,436 2,362 2,289 2,252 2,217 2,181 2,146 2,111 2,078 1,974 590
VC12 2,044 1,970 1,898 1,863 1,828 1,794 1,761 1,727 1,695 1,566 1,441 1325- 1375
VC13 1,674 1,596 1,576 (227 разл.)
VC14 1,281 1,253 (152 кип.) -25,7
WC12 0,581 0,521 0,463 0,435 0,408 0,393 (427 разл.)
WC14 0,513 0,449 0,432 (227 разл.)
Электродные процессы
Продолжение таблицы 1.3.1
МеС1„ Напряжение, В, при температуре Тт_, °C
100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C 550 °C 600 °C 800 °C 1000 °C 1500 °C
WC15 0,491 0,431 0,392 (276 кип.) 248 (253)
WCU 0,464 0,401 0,343 0,325 (337 кип.) 275 (284)
YC13 3,106 3,032 2,961 2,926 2,892 2,858 2,824 2,791 2,758 2,643 2,548 2,329 680 (703)
YbCl3 3,017 2,944 2,873 2,838 2,804 2,770 2,736 2,703 2,670 2,542 2,434 2,449 (1027 разя.) 857
ZnCl2 1,854 1,776 1,706 1,680 1,655 1,629 1,603 1,577 1,552 1,476 (756 кип.) 320
ZrCl2 2,847 2,772 2,699 2,664 2,629 2,594 2,560 2,526 2,508 (577 разл.)
ZrCl3 2,736 2,668 2,603 2,571 2,540 2,509 3,492 (477 разл.)
ZrCl4 2,209 2,135 2,063 2,041 (331 возг.)
Новый справочник химика и технолога
Электродные процессы
47
Таблица 1.3.2
Электродные потенциалы металлов в расплаве КС1—NaCI
В таблице приведены величины электродных потенциалов некоторых металлов относительно электродного потенциала серебра в растворах хлоридов этих металлов в эквимолярном расплаве КС1—NaCI. Значения электродных потенциалов определены по напряжению электрохимических систем с серебряным электродом сравнения при 660-900 °C:
Me | МеС1„ (А2), КО—NaCI || AgCl (ty), КО—NaCI | Ag,
где и N2 —мольные доли AgCl и МеС1„ в соответствующих
расплавах.
Величины электродных потенциалов, приведенные в таблице, есть напряжения электрохимических систем, для кото-
N2 , рых —- = 1 .
А,"
Для перевода потенциалов в шкалу относительно хлорного электрода в конце таблицы приведены потенциалы хлорного электрода.
Электродная реакция Электродный потенциал, В, при температуре
700 °C 800 °C 900 °C
Мп2* + 2е" = Мп -1,206 -1,190 -1,172
Zn2+ + 2е“ = Zn -0,860 -0,835 -0,810
Сг2* + 2е" = Сг -0,758 -0,740 -0,728
Т1+ + е“ = Т1 -0,665
Cd2* + 2е = Cd -0,620 -0,580
Fe2* + 2е’ = Fe -0,520 -0,510 -0,498
Сг3* + Зе = Сг -0,425 -0,385 -0,345
Pb2+ + 2е’ = РЬ -0,390 -0,376 -0,355
Sn2+ + 2е“ = Sn -0,370 -0,354 -0,340
Со2* + 2е" = Со -0,324 -0,300 -0,275
Ni2* + 2e’ = Ni -0,140
Ag* + e’ = Ag 0,0 0,0 0,0
Cu2*+2e’ = Cu 0,170 0,180 0,192
Cl2 + 2е” = 2СГ +0,845 +0,820 +0,795
Таблица 1.3.3
Электродные потенциалы металлов в эвтектическом расплаве LiCl—КС1
В таблице приведены величины электродных потенциалов некоторых металлов относительно электродного потенциала платины в растворах хлоридов этих металлов в эвтектическом расплаве LiCl—КС1. Значения электродных потенциалов определены при 450 °C по напряжению электрохимических систем с платиновым электродом сравнения:
Me | МеС1„ (А2), LiCl—KCI || PtCl2 (АД LiCl—КС11 Pt,
где N\ и N2 —мольные доли PtCl2 и МеС1„ в соответствующих расплавах.
Величины электродных потенциалов, приведенные в таблице, есть напряжения электрохимических систем, для кото-
N2 1
рых —- = 1 .
Af
Для перехода к хлорной шкале потенциалов в конце таблицы приведен потенциал хлорного электрода.
Электродная реакция Электродный потенциал, В Электродная реакция Электродный потенциал, В
Li+ + е“ = Li -3,410 Т1+ + е’ = Т1 -1,370
Mg2+ + 2е’ = Mg -2,580 Cd2* + 2е’ = Cd -1,316
Мп2* + 2е" = Мп -1,849 Pb2+ + 2е” = РЬ -1,101
А13* + Зе-А1 -1,797 Sn2* + 2е" = Sn -1,082
Fe2* + 2е = Fe -1,711 Со2+ + 2е“ = Со -0,991
Zn2+ + 2е“ = Zn -1,566 Ga3* + Зе’ = Ga -0,88
Сг2* + 2е = Сг -1,425 Cu+ + е“ = Cu -0,851
In3* + Зе’ = In -0,835 Hg2, + 2e~ = Hg -0,5
Ni2* + 2е’ = Ni -0,795 Pd2* + 2e" = Pd -0,214
Sb3+ + Зе’ = Sb -0,670 Pt2* + 2e" = Pt 0,0
Ag* + e’ = Ag -0,637 Cu2* + e” = Cu* 0,045
Cr3* + e’ = Cr2* -0,631 Au* + e’ = Au 0,311
Bi3* + 3e“ = Bi -0,588 Cl2 + 2e’ = 2СГ +0,216
Таблица 1.3.4
Электродные потенциалы металлов в расплавах индивидуальных галогенидов металлов
В таблице приведены величины электродных потенциалов некоторых металлов в расплавленных галогенидах этих металлов при температуре 700 °C по отношению к электродному потенциалу натрия. Величины электродных потенциалов определены на основании измерений потенциалов разложения, а также по напряжению электрохимических систем со стеклянно-натриевым электродом сравнения.
Электродная реакция Электродный потенциал, В, в расплаве
MeF„ МеС1„ МеВг„ Ме1„
Al3* + Зе’ = Al 1,78 1,78 1,72
Ag* + е" = Ag 2,55 2,25 1,74
Ва2* + 2е" = Ва -0,47 -0,23 -0,27 -0,01
Be2* + 2е’ = Be 1,47
Bi3* + Зе’ = Bi 2,75 2,54 2,14
Са2* + 2е" = Са -0,29 0,01 0,10 0,18
Cd2* + 2е’ = Cd 2,11 1,89 1,62
Со2* + 2е" = Со 2,42 2,30 2,24
Cs* + е“ = Cs -0,24
Си* + е” = Си 2,65 2,29 1,98
Hg2, + 2e~ = Hg 2,53 2,44 2,18
К* + е’ = К 0,22 -0,14 -0,18 -0,17
La3* + Зе’ = La 0,22
Mg2* + 2е“ - Mg 0,56 0,78 0,77 0,80
Мп2* + 2е“ = Мп 1,51 1,52 1,37
Ni2* + 2е’ = Ni 2,36
РЬ2* + е’ = РЬ 2,27 2,07 1,82
48
Новый справочник химика и технолога
Продолжение таблицы 1.3.4
Электродная реакция Электродный потенциал, В, в расплаве
MeF„ МеС1„ MeBr„ Mel„
Rb+ + е" = Rb -0,30
Sb3+ + Зе’ = Sb 2,90 2,56 2,30
Sn2+ + 2е“ = Sn 2,31 2,22 1,78
Sr2* + 2e' = Sr -0,40 -0,15 -0,06 -0,13
Tl+ + e’ = T1 1,92 1,66 1,40
Th4+ + 4e~ = Th 1,17
Zn2+ + 2e“ = Zn 1,96 1,85 1,54
Данные по Электродным потенциалам и pH гид-ратообразования в системах элемент—вода (диаграмма Пурбе) см. Справочник химика. Изд. 2-е, пере-раб. и дополн. Т. 3. М.-Л.: Химия, 1964. С. 755-825.
1.4. Коррозия. Виды коррозии, методы испытаний и способы предотвращения коррозионных повреждений
(проф., д.т.н. Б.С. Ермаков)
1.4.1. Понятие коррозии, основные виды коррозионных повреждений металлов и сплавов
Коррозия (от лат. corrosio — разъедание и corrodo — грызу) — разрушение материалов под влиянием окружающей среды в результате ее химического или электрохимического воздействия.
Обычно рассматривают коррозию металлических материалов. Однако это явление характерно не только для металлов и сплавов, аналогичные процессы происходят и в неметаллических системах, например пластмассах или керамике. Примером такого коррозионного воздействия на неметаллические материалы может служить износ футеровки плавильных печей под действием жидкого, химически активного шлака.
Ущерб, причиняемый коррозией, может быть разделен на две категории. Первая — прямые потери: затраты при ремонте оборудования, замене поврежденных элементов и т. п.; вторая — косвенные потери, связанные с простоем ремонтируемого и сопряженного с ним оборудования, ухудшением качества выпускаемой продукции в результате ее загрязнения, увеличением расхода топлива, материалов, энергии. Средние ежегодные потери, связанные с коррозионными повреждениями оборудования, для промышленно развитых стран достигают 3-10 % валового продукта. Влияние коррозионных повреждений на надежность и безопасную эксплуатацию оборудования можно проиллюстрировать следующим примером. По данным фирмы «Du Pont» (США) в 247 случаях выхода из строя оборудования этой фирмы из 313, или 79 % аварий, произошли по причине коррозионных повреждений. При этом на долю общей коррозии пришлось 31,5% случаев, на коррозионное растрескивание (стресс-коррозию) — 21,6 %
случаев, питтинговая коррозия стала причиной 15,7% выходов оборудования из строя, а межкристаллитная коррозия явилась причиной аварий в 10,2 % случаев.
1.4.1.1. Способы классификации коррозии
Коррозию относят к поверхностным явлениям и классифицируют по тем изменениям, которые происходят с поверхностью материала в результате протекания процесса коррозии. При взаимодействии всей поверхности материала с окружающей средой наблюдается общая или сплошная коррозия, при взаимодействии части поверхности — местная или локальная коррозия. Принято различать два вида общей коррозии. При равномерной коррозии вся поверхность металла равномерно разъедается внешней средой без изменений в топографии поверхности. К такой коррозии, например, относится коррозия углеродистой стали в растворах серной кислоты (рис. 1.4.1, а). Второй тип общей коррозии — неравномерная коррозия, когда поверхность металла под слоем продуктов коррозии носит «изрытый» характер, т. е. на поверхности возникают места более глубоких повреждений — коррозионные каверны (например, коррозия углеродистой стали в морской воде — рис. 1.4.1, б). К неравномерной коррозии относится структурно-избирательная коррозия, когда одна из фаз или структурных составляющих сплава растворяется с большей скоростью, чем остальные, например процесс обесцинкивания латуней (рис. 1.4.1, в).
Рис. 1.4.1. Виды коррозионных повреждений металлов и сплавов: a-в) общая коррозия: а) равномерная коррозия, б) неравномерная коррозия, в) избирательная коррозия; г-м) местная коррозия: г) коррозия пятнами, д) язвенная коррозия, е) питтинговая коррозия, ж) сквозная коррозия, з) нитевидная коррозия, и) подповерхностная коррозия, к) межкристаллитная коррозия, л) ножевая коррозия, м) транскристаллитное коррозионное растрескивание
Местная, или локальная, коррозия характеризуется разрушением отдельных участков поверхности материала. Местная коррозия более опасна, чем общая, т. к. при сравнительно малых потерях массы материала возможен выход из строя дорогостоящей конструкции. Местная коррозия обычно разделяется на несколько групп: коррозия пятнами, когда поверхностные разме-
Электродные процессы
49
ры пятна значительно превышают глубину проникновения коррозионного дефекта (например, коррозия латуни в морской воде — рис. 1.4.1, г); язвенная коррозия, когда размеры коррозионного повреждения на поверхности соизмеримы с глубиной повреждения (коррозия углеродистых сталей в грунте — рис. 1.4.1, д). Точечной или питтинговой коррозией называется такой вид коррозии, при котором глубина коррозионного дефекта значительно больше его поверхностных размеров, например коррозия аустенитной хромоникелевой стали в морской воде (рис. 1.4.1, е\ в частности, к этому виду коррозии относится сквозное разъедание материала (рис. 1.4.1, ж). К местной коррозии также относятся нитевидная коррозия, распространенная в основном в материалах с неметаллическими защитными пленками (рис. 1.4.1, з) и подповерхностная коррозия (рис. 1.4.1, и). Этот вид коррозии начинается с поверхности материала, а затем распространяется под его поверхностью таким образом, что продукты коррозии оказываются сосредоточенными в некоторых зонах материала. Этот вид коррозии вызывает местное вспучивание материала, приводит к его расслоению. Типичным примером такого вида коррозии может служить коррозия при травлении некачественного проката.
К локальной коррозии относится также межкристаллитная коррозия (рис. 1.4.1, к), когда коррозионные разрушения локализуются в границах зерен материала. К типичным видам этого типа коррозии относится коррозия в некоторых средах нержавеющих хромоникелевых сталей после нагревов в диапазоне температур 550-700 °C. Этот тип коррозии может быть признан наиболее опасным. Так, при одинаковой потере массы, например, в 5 мг/см2 снижение временного сопротивления материала при общей коррозии составит примерно 10-15 %, при язвенной коррозии — до 40 %, а при межкристаллитной коррозии снижение прочности материала может достигать 80 %.
Еще одним типом местной коррозии является ножевая коррозия — локализованная коррозия металла, имеющая вид надреза острым предметом (рис. 1.4.1, л). Обычно очаги такой коррозии обнаруживаются в зонах сплавления сварного шва в сильно агрессивных средах (например, ножевая коррозия стали 12Х18Н10 в азотной кислоте).
В условиях эксплуатации оборудования достаточно часто возникает ситуация, когда кроме коррозионных сред на материал воздействуют внешние механические нагрузки или внутренние напряжения, что приводит к повышению скорости протекания коррозионных процессов. Такой вид коррозии получил название коррозионного растрескивания или стресс-коррозии и коррозионной усталости. Такая коррозия может распространяться как по телу зерна (рис. 1.4.1, м) — транскристал-литная трещина, так и по его границам — межкристаллитная трещина.
Коррозию принято классифицировать также по условиям контакта металла с агрессивной коррозионной
средой. В зависимости от условий контакта различают коррозию при полном погружении, когда объект полностью погружен в агрессивный коррозионный раствор, и коррозию при неполном погружении — одним из примеров такой коррозии является коррозия по ватерлинии кораблей. Отмечают также такие виды коррозии, как коррозия при периодическом погружении и струйная коррозия.
В зависимости от того, как распространяются коррозионные трещины, коррозионные разрушения принято делить на транскристаллитные (разрушение протекает по телу зерен) и интеркристаллитные, или межкристаллитные (разрушение происходит по границам зерен).
Одним из главнейших способов классификации коррозии, который позволяет наиболее полно охарактеризовать процессы, протекающие при взаимодействии материалов и коррозионных сред, является классификация по механизму коррозионного процесса. По этому методу классификации коррозию принято делить на следующие виды: коррозия химическая, электрохимическая и биохимическая.
Химическая коррозия металлов — это процесс взаимодействия металла с коррозионной средой, при котором окисление металла и восстановление окислительного компонента коррозионной среды протекают одновременно. Продукты коррозии при этом процессе возникают непосредственно на корродирующих участках. К химической коррозии относятся газовая коррозия (окисление металла в процессе высокотемпературных нагревов, например при термической обработке) и коррозия в неэлектролитах, например в нефтепродуктах.
Электрохимическая коррозия — это процесс взаимодействия металла с коррозионной средой (раствором электролита), при котором ионизация атомов металла и восстановление окислительного компонента коррозионной среды протекают в две различные стадии, а скорости процессов на этих стадиях могут быть различны и зависимы от электродного потенциала. При этом виде коррозии одновременно протекают две реакции — анодная и катодная, локализованные на определенных участках поверхности корродирующего металла, причем участки протекания таких реакций могут меняться в процессе коррозии. К видам электрохимической коррозии относятся: атмосферная коррозия во влажной газовой или. воздушной атмосфере; коррозия в жидких средах или электролитах; коррозия в расплавах солей; почвенная и подземная коррозии; электрокоррозия под действием внешнего источника тока и т. п.
Биохимическая коррозия — это процесс, связанный с воздействием на материал микроорганизмов. При этом металл может разрушаться из-за того, что он служит для этих микроорганизмов питательной средой, или из-за воздействия на металл продуктов жизнедеятельности этих микроорганизмов. В чистом виде биохимическая коррозия встречается достаточно редко, поскольку в присутствии влаги происходит также процесс электрохимической коррозии материала. Поэтому
50
Новый справочник химика и технолога
при анализе причин и экспертизе разрушения различных конструкций, поврежденных коррозией, разрушения, связанные с биохимической коррозией, обычно относят к разрушениям, связанным с электрохимической коррозией.
1.4.1.2. Химическая коррозия
Одним из наиболее распространенных видов химической коррозии является газовая коррозия. Она возникает на поверхности металлов в отсутствие влаги, т. е. в условиях, когда электрохимические процессы развиться не могут. Это либо коррозия металлов в ходе их термической обработки, либо коррозия в сухих газах при нормальной температуре.
Поведение металлов при высоких температурах имеет важное практическое значение. Повреждения поверхности металлов и сплавов при термической обработке могут достигать глубин до нескольких миллиметров и приводить к значительному увеличению объема последующей механической обработки. Основным показателем, определяющим стойкость металла и спла
ва против окисления при высокотемпературном нагреве, является жаростойкость, т. е. способность металла сопротивляться коррозионному воздействию газа.
Первопричиной химической коррозии металлов и сплавов является их термодинамическая неустойчи
вость в различных средах при данных внешних условиях, т. е. самопроизвольный переход металла в более устойчивое — окисленное — состояние, которое дости
гается в результате реакции окислительный металл + компонент среды
продукт реакции
за счет соответствующего уменьшения термодинамического потенциала этой системы.
Основными факторами, определяющими скорость газовой коррозии, являются температура и состав газовой среды. Влияние температуры на скорость газовой коррозии приближенно описывается уравнением Аррениуса:
1п# = Я-Я/7;
где К — скорость реакции (коррозии), А и В — константы, Т—температура (К).
При химическом взаимодействии углеродистых сталей с кислородом воздуха на поверхности образуется окалина — оксидная пленка, в состав которой при умеренно высоких температурах входят гематит — Fe2O3 и магнетит — Fe3O4, при более высоких температурах нагрева (более 575 °C) на поверхности раздела окалина—металл возникает еще один оксид железа — вюс-тит — (FeO). Одновременно с процессом окисления железа идет процесс обезуглероживания поверхности стали — Fe3C + О2 —» 3Fe + СО2, в результате чего цемен-титная фаза «вымывается» с поверхности стали. При увеличении времени нагрева глубина обезуглероженного слоя увеличивается и может достигнуть нескольких
миллиметров, что приведет к снижению прочности и твердости поверхности обрабатываемого изделия.
Кроме изменения температуры на скорость химической коррозии влияет давление газовой среды. С повышением давления скорость коррозии резко возрастает вследствие наличия в газовой среде водорода, который при повышенном давлении вызывает водородное охрупчивание стали.
Большинство металлов при взаимодействии с кислородом воздуха или другими окислителями покрываются пленкой химического соединения. Первой стадией этого процесса является адсорбция окислительного компонента среды (О2, Н2О, СО2, SO2 и т. п.) на поверхности металла. В табл. 1.4.1 приведена стандартная энтальпия образования оксидов и энтальпия адсорбции кислорода на ряде металлов. Эти данные указывают на химическую природу связи между адсорбатом и адсорбентом — хемосорбцию атомов кислорода на поверхности металла. Связь, возникающая между кислородом и поверхностными атомами металла, — ионная. Она оказывается значительно сильнее, чем связь, возникающая между этими элементами в оксиде, т. к. за счет энергии поляризации на атом кислорода оказывает воздействие поле, создаваемое нижележащими атомами металла.
Таблица 1.4.1
Энтальпии образования оксидов и адсорбции кислорода на металлах
Металл Оксид Стандартная энтальпия образования оксида Д/^98’ кДж/моль О2 Энтальпия адсорбции О2 Ад ^298’ кДж/моль О2
Серебро Ag2O -61,9 -225,7
Палладий PdO -175,6 -100,3
Медь Cu2O -334,4 -501,6
Кобальт СоО -476,5 -501,6
Никель NiO -480,7 -627,0
Вольфрам WO2 -585,2 <(-627,0)
После насыщения поверхности металла хемосорбированным окислителем, происходящего практически мгновенно, на поверхности металла возникает монослой окислителя. При наличии химического сродства между металлом и окислителем (термодинамической стабильности оксида) хемосорбированная пленка быстро переходит в состояние оксидной пленки в результате протекания химической реакции:
wMe (т) + 1/2 тп О2 (аде) = = ?иМе+ + 1/2 тп О2 = МетОтя/2 (т)
Таким образом, при химическом взаимодействии окислительный компонент среды вступает с металлом во взаимодействие и образует химическое соединение,
Электродные процессы
51
которое в большинстве случаев располагается на поверхности металла в виде пленки. Образование пленки продуктов коррозии на поверхности металла приводит к эффекту самоторможения процесса коррозии во времени в том случае, если пленка образует достаточно плотный слой, замедляющий транспортировку окислителя к поверхности металла. Толщина таких пленок может изменяться в широких пределах и (в зависимости от толщины слоя) подразделяется на три группы:
- тонкие (невидимые) пленки толщиной от 0,1 до 40 нм;
- средние (дающие цвета побежалости — см. табл. 1.4.2), толщина которых от 40 до 500 нм;
- толстые (видимые) пленки, толщина которых свыше 500 нм и во многих случаях, как например в случае окисления углеродистых сталей, может достигать нескольких миллиметров.
Жаростойкость металлов, а также закономерности роста толщины пленки на металлах и сплавах во времени в значительной степени зависят от свойств образующихся пленок. Защитные свойства пленки оценивают по скорости окисления металла, которая устанавливается при возникновении пленки, и характеру изменения этой скорости во времени.
Таблица 1.4.2
Цвета побежалости материалов
Углеродистые стали Коррозионностойкие стали и жаропрочные сплавы
Г, °C Цвета побежалости Г, °C Цвета побежалости
12Х18Н10Т ХН756БТЮ
220 Светло-желтый 300 Светло-соломенный —
240 Темно-желтый 400 Соломенный Светло-желтый
255 Коричнево-желтый 500 Красновато-коричневый Желтый
265 Красно-коричневый 600 Фиолетово-синий Коричневый
275 Пурпурно-красный 700 — Синий
285 Фиолетовый 800 — Голубой
295 Васильково-синий
315 Светло-синий
330-350 Серый
Защитными свойствами могут обладать только сплошные пленки. Такие пленки должны покрывать сплошным слоем всю поверхность материала. Возможность образования такой пленки определяется условием сплошности, при соблюдении которого молекулярный объем Гок соединения, возникающего из металла и окислителя, должен быть больше объема металла ГМе, израсходованного на образование пленки. В других случаях образующегося соединения не хватит на то, чтобы покрыть слоем весь металл, в результате чего пленка продукта коррозии получается рыхлой и пористой. Таким образом, если Иок/ ИМе < 1, то пленка не может быть сплошной, если Гок/ ГМе - 1, то пленка может быть сплошной. Отношение объемов соединения металла с окислителем и самого металла может быть рассчитано по формуле:
Гок/ ^Ме М>к ' РМе ! ’ Рок " ^Ме,
где Мок — молекулярная масса соединения, АМе — атомная масса металла, рок и рме — плотность оксидного соединения и металла соответственно, т — число атомов металла в соединении. К металлам, не удовлетворяющим условию сплошности, относятся все щелочные и щелочноземельные металлы (кроме бериллия). Данные по отношениям Гок / ГМе для этих и ряда других металлов при-
приведены в табл. 1.4.3. Однако защитные свойства пленки определяются не только условием сплошности.
Таблица 1.4.3
Отношение объемов оксида и металла
Металл Оксид ГОк/ ГМе Металл Оксид F0K/ FMe
Li Li2O 0,57 Pb PbO 1,15
К K2O 0,48 Nb NbO 1,57
Mg MgO 0,79 Nb2O5 2,81
Са CaO 0,63 Ta Ta2O3 2,32
Sr SrO 0,66 Cr Cr2O3 2,02
Си Cu2O 1,67 Mo MoO2 2,18
CuO 1,74 MoO3 3,45
Ag Ag2O 1,58 W WO2 1,86
Be BeO 1,67 wo3 3,36
Zn ZnO 1,58 Fe FeO 1,77
Cd CdO 1,27 Fe3O4 2,09
и UO2 1,96 Fe2O3 2,14
U3O8 3,12 Co CoO 1,75
Al A12O3 1,31 Co3O4 2,00
Ti TiO2 1,76 Co2O3 2,42
Zr ZrO2 1,60 Ni NiO 1,52
Sn SnO2 1,33 Pt PtO 1,56
52
Новый справочник химика и технолога
В реальных условиях роста пленки в ней могут возникать значительные внутренние напряжения, которые могут приводить к частичному или полному нарушению сплошности, т. е. разрушению защитной пленки. Опытным путем было показано, что подобного разрушения защитных пленок не наступает при выполнении условия 2,5 > Кок/ Кме> 1.
Для защиты от газовой коррозии используют в основном жаростойкие сплавы. Так, например, чтобы уменьшить скорость окисления углеродистой стали при 900 °C в три раза, достаточно ввести в нее 3,5 % алюминия; в четыре раза — 5,5 % алюминия. Кроме жаростойкого легирования используется метод, заключающийся в применении защитных атмосфер. Газовая среда не должна содержать окислителей, находящихся в контакте со сталью, и восстановителей в контакте с медью. В качестве защитной атмосферы при термической обработке и сварке применяют инертные газы — аргон и азот. Также можно осуществлять термическую обработку сталей в атмосфере, содержащей азот, водород и оксид углерода. Сварка титановых и алюминиево-магниевых сплавов должна осуществляться в защитной среде аргона.
Возможно покрытие поверхности материала различными защитными пленками — термодиффузионными железо-алюминиевыми или железо-хромовыми — методами химико-термической обработки (хромирование и алитирование), нанесение металлокерамических покрытий, керметов, металлооксидных покрытий, для получения которых в качестве неметаллических ингредиентов применяют тугоплавкие оксиды (например, А12О3, MgO), карбиды и нитриды различных металлов. Металлическими составляющими таких покрытий могут служить тугоплавкие металлы — вольфрам, молибден, хром и т. п.
К химической коррозии также относится коррозия в среде неэлектролитов. Органические жидкости, не обладающие электропроводимостью, исключают возможность протекания электрохимических реакций. К таким жидкостям относятся органические растворители (бензол, толуол, тетрахлорид углерода), жидкое топливо (мазут, бензин, керосин) и некоторые неорганические вещества (бром, расплав серы, жидкий фтороводород). В этих средах коррозию вызывает реакция между металлом и коррозионной средой. Наибольшее практическое значение имеет коррозия металлов в нефти и нефтепродуктах. Коррозионноактивными составляющими нефти являются сера, сероводород, сероуглерод, тиофены, тиолы и т. п. Сероводород образует сульфиды с железом, свинцом, медью и их сплавами. При взаимодействии меркаптанов с никелем, серебром, медью и свинцом получаются производные тиолов — тиолаты. Сера взаимодействует с медью и серебром с образованием сульфидов. Повышение температуры ускоряет коррозию металлов в нефти; наличие воды в нефти резко ускоряет процесс, вызывая электрохимическую коррозию.
Основными путями борьбы с коррозией в неэлектролитах является использование коррозионностойких материалов, химико-термическая обработка (хромирование, алитирование, хромоалитирование) поверхности применяемых изделий.
1.4.1.3. Электрохимическая коррозия
Электрохимическая коррозия металлов и сплавов возникает на границе раздела фаз металл—электролит. Этот вид коррозии не зависит от типа электролита, будь то химически чистая вода, раствор соли или кислота. Существенной роли не играет и количество электролита — коррозию может вызвать слой влаги толщиной несколько десятков нанометров. Единственное необходимое условие для протекания коррозионного процесса — это возможность совместного возникновения и развития анодной реакции ионизации металлов и катодной реакции восстановления тех или иных ионов и молекул на поверхности металла. Такое условие реализуется в случае, когда равновесный анодный потенциал более отрицателен, чем потенциал хотя бы одной из возможных катодных реакций.
Анодный процесс электрохимической коррозии всегда сопровождается ионизацией металла. В катодном процессе могут участвовать различные ионы или молекулы, окисляющие металл. Возможны следующие основные типы катодных реакций: восстановление катионов:
Н++е“—* 1/2 Н2,
Ag++e’-> Ag;
восстановление анионов:
S2O82 + 2е—► 2SO2 , МпО4“ + е“ —> МпО2 ;
восстановление оксидных и гидроокисных пленок:
Fe3O4 + Н2О + 2е“ 3FeO + 2ОН ’,
Fe(OH)3 + е Fe(OH)2 + ОН '
Возможны также реакции восстановления молекул газов, растворенных в электролите, восстановления органических соединений и т. п.
В электролитах всегда содержится более одного окислителя. В водных растворах кроме водородных ионов постоянно присутствует некоторое количество растворенного кислорода, а часто и различные органические примеси. Их совместное влияние на скорость анодного процесса зависит от эффективности отдельных катодных процессов.
Технически чистые металлы всегда загрязнены примесями, а сплавы содержат еще большее количество инородных атомов (атомов легирующих элементов). Поверхность таких металлов и сплавов структурно и термодинамически неоднородна, поэтому в реальных условиях происходит коррозия многокомпонентного металлического материала с неравновесным состояли-
Электродные процессы
53
ем поверхности. Ток, протекающий в системе металл— электролит—металл, называется локальным током, а сама система представляет собой своеобразный коротко замкнутый гальванический элемент. Теория, объясняющая механизм электрохимической коррозии как реакции внутри гальванического элемента, была создана в 1830 г. де ля Ривом, а позднее доработана Акимовым и Эвансом. Теория локальных элементов достаточно проста, хорошо описывает процессы, происходящие при электрохимической коррозии. Полученные таким образом результаты в виде коррозионных диаграмм «потенциал—ток», называемых диаграммами Эванса, или поляризационных коррозионных диаграмм Шульгина в координатах «потенциал—плотность тока» очень наглядны и хорошо описывают процесс развития коррозии. Однако теория локальных элементов имеет и ряд недостатков. Так, в рамках этой теории оказывается достаточно сложно провести расчеты процесса, сложно определить суммарную поверхность всех катодных и анодных участков металла. Не учитывается тот факт, что часто некоторые участки являются катодными в одной системе и анодными в другой. Согласно теории локальных элементов, все окислители, способствующие протеканию катодной реакции, называются деполяризаторами, а катодный процесс — деполяризацией. Эти термины общеприняты независимо от того, отвечают ли они по физическому смыслу современной теории электродных процессов, протекающих в гальванических элементах.
Известно, что все металлы можно расположить в так называемый ряд напряжений: Zn, Fe, Си, Ag, Аи. При погружении любой пары этих металлов в раствор по внешней цепи проходит ток, т. е. появляется электродвижущая сила, поскольку металл, расположенный левее, более электроотрицателен, чем стоящий за ним в
ряду. Электрохимические эквиваленты ряда металлов и газов приведены в табл. 1.4.4.
На эффективную работу гальванических элементов, в том числе и коррозионных элементов, основное влияние оказывают три фактора — природа металла и его структура; природа коррозионной среды; физические условия, в которых протекает коррозионный процесс: температура, давление и т. п. Причины, вызывающие образование коррозионных гальванических элементов, приведены в табл. 1.4.5.
Таблица 1.4.4 Электрохимические эквиваленты металлов и газов
Элемент Степень окисления Атомная масса Количество вещества (г), перенесенного 1 Кл Количество вещества (г), перенесенного 1 Ач
Алюминий +3 26,97 0,0932 0,3356
Водород +1 1,008 0,0104 0,0376
Железо +2 55,85 0,2894 1,0424
Железо +3 55,85 0,1929 0,6949
Кислород -2 16,00 0,0829 0,2986
Магний +2 24,23 0,1260 0,4539
Медь +1 63,57 0,6588 2,3729
Медь +2 63,57 0,3294 1,1864
Натрий +1 23,00 0,2383 0,8585
Никель +2 58,69 0,3041 1,0954
Свинец +2 207,21 1,0376 3,8673
Серебро +1 107,88 1,1179 4,0269
Хлор -1 35,46 0,3675 1,3236
Цинк +2 65,38 0,3388 1,2202
Таблица 1.4.5
Классификация факторов, вызывающих электрохимическую гетерогенность металлической поверхности
Основной фактор возникновения неоднородности Причина возникновения электрохимической гетерогенности Практические примеры коррозии
Структурные неоднородности в металле А) Наличие металлических или электропроводных неметаллических включений. Включения с более положительным электропотенциалом работают как катоды Б) Неравномерность концентрации твердого раствора (ликвация). Участки сплавов, обогащенные положительным компонентом, работают как катоды В) Наличие границ зерен. Обычно граница работает как катод А) Контактная коррозия при контакте с более электроположительным металлом. Растворение цинка, загрязненного примесями, в кислоте. Ускоряющее влияние включений графита на коррозию чугунов в кислых средах Б) Усиление коррозионного процесса твердого раствора сплава А1—Zn в водных растворах вследствие ликвации В) Межкристаллитная коррозия нержавеющих сталей типа XI8Н (после нагревов при 600-700 °C); коррозия алюминиевых и магниевых сплавов
54
Новый справочник химика и технолога
Продолжение таблицы 1.4.5
Основной фактор возникновения неоднородности Причина возникновения электрохимической гетерогенности Практические примеры коррозии
Структурные неоднородности в металле Г) Анизотропность металлического кристалла Г) Избирательное травление отдельных кристаллов на металлографических шлифах; неодинаковая скорость растворения различных граней монокристаллов магния, цинка и меди
Структурные неоднородности в металле А) Наличие разнородных атомов в твердом растворе Б) Наличие энергетических флуктуаций атомов на поверхности жидкого или твердого металла А) Избирательное растворение атомов цинка из латуней Б) Возможность протекания периодически чередующихся (флуктуирующих) анодных и катодных процессов на поверхности даже самых чистых металлов, помещенных в электролит
Неоднородности в продуктах коррозии А) Микропоры в пленке; участки, не покрытые пленкой, — аноды Б) Неравномерность распределения продуктов коррозии; участки под ржавчиной, как правило, являются анодами В) Микропоры в защитной пленке. Металл в поре — анод А) Ускоренная коррозия железа вследствие несплошности слоя окалины Б) Ускоренная коррозия под действием окалины В) Растворение металла в момент активирования пассивного состояния железа, алюминия, нержавеющих сталей
Неоднородные деформации и внутренние напряжения в металле А) Наличие неодинаковых по величине внутренних напряжений в различных точках металлического тела в результате неоднородной (локальной) деформации или приложения внешних нагрузок. Более напряженные участки — анод А) Многочисленные случаи коррозии паровых котлов. Коррозионное растрескивание латуни; ускоренная коррозия корпусов судов в местах концентрации напряжений; коррозия в местах изгиба листов и труб. Коррозия головок заклепок
Неоднородность жидкой фазы А) Различие в концентрации собственных ионов металла в электролите; участки с меньшей концентрацией собственных ионов — аноды Б) Различная концентрация нейтральных солей в растворе; участки, омываемые электролитом с большей концентрацией солей (например NaCl), — аноды; в растворах солей пассивирующего действия (Na2Cr2O7) эти участки являются катодами В) Различная концентрация водородных ионов (pH); участки с более высокими значениями pH — аноды; при активировании пассивного металла участки с более высоким pH могут работать катодами Г) Различная концентрация кислорода или других окислителей; участки, омываемые растворами с меньшей концентрацией кислорода или окислителя, служат анодами А) Коррозия меди в растворах; более сильное разрушение в местах протекания свежих растворов Б) Коррозия химической аппаратуры. Усиление местной коррозии морских судов и сооружений при наличии пресного слоя воды вблизи устья реки В) Коррозия химической аппаратуры и почвенная коррозия Г) Коррозия в зонах с неравномерной аэрацией раствора — узких щелях; интенсивная коррозия конструкций и сооружений из-за деполяризующего действия ионов Fe+3 или Си 2
Электродные процессы
55
Продолжение таблицы 1.4.5
Основной фактор возникновения неоднородности Причина возникновения электрохимической гетерогенности Практические примеры коррозии
Неоднородность жидкой фазы Д) Периодически изменяющаяся субмикроскопическая неоднородность раствора из-за теплового движения молекул Д) Протекание чередующихся анодных и катодных процессов на идеально однородных поверхностях металла
Неоднородность физических условий А) Влияние температуры различных участков корродирующей поверхности; более нагретые участки поверхности — аноды Б) Неравномерное распределение лучистой энергии по корродирующей поверхности; более интенсивно облучаемые участки — аноды В) Неравномерное наложение внешнего электрического поля; участки, где положительные электрические заряды выходят из металла в электролит, — аноды А) Коррозия паровых котлов, теплообменников, холодильных установок Б) Усиленная коррозия сооружений в морской воде с южной стороны; усиленная коррозия установок под действием мощных электромагнитных излучений В) Образование катодных и анодных участков под влиянием блуждающих токов при почвенной коррозии
На скорость, вид и характер развития электрохимической коррозии влияет ряд внешних и внутренних факторов. К внешним факторам можно отнести такие, как pH среды и температура среды, состав и концентрация растворов, концентрация растворенного кислорода, скорость относительного движения среды. Внутренними факторами, оказывающими существенное влияние на скорость коррозии металлов и сплавов, являются их термодинамическая неустойчивость, положение металлов в таблице Менделеева, тип и структура сплава и механический фактор. Под механическим фактором понимается воздействие на материал механических нагрузок — постоянных или периодических, внешних или внутренних напряжений. Механический фактор, усиливая термодинамическую нестабильность металла и сплава, может привести к разрушению сплошности защитных пленок на его поверхности. К таким видам коррозии относится коррозия под напряжением, которая возникает при совместном действии на металл постоянных растягивающих напряжений и коррозионной среды; коррозионная усталость, возникающая при одновременном воздействии среды и периодического или знакопеременного механического воздействия. На устойчивость металла к коррозионно-механическим повреждениям оказывает влияние ряд дополнительных факторов. Это технологические и конструкционные особенности деталей и изделий, условия их эксплуатации, такие факторы, как температура и перемешивание коррозионной среды и аэрация.
1.4.1.4. Основные виды общей электрохимической коррозии металлов и сплавов
Число видов электрохимической коррозии крайне велико. Однако наиболее распространенными и опасными (с точки зрения их влияния на работоспособность
конструкций различного назначения) среди этих видов можно считать атмосферную коррозию, коррозию металлов и сплавов в грунте, коррозию в воде. К числу наиболее опасных видов местной коррозии металлов и сплавов следует отнести питтинговую коррозию, межкристаллитную коррозию и коррозию под напряжением или коррозионное растрескивание.
Атмосферная коррозия металлов и сплавов
Атмосферная коррозия считается самым распространенным видом коррозии металлов и сплавов. До 80 % металлических конструкций эксплуатируется в условиях атмосферы. Это сельскохозяйственные и горнодобывающие машины, конструкции электропередач, оборудование промышленных предприятий и транспорт, мосты, здания и сооружения. Основным фактором, определяющим механизм и скорость атмосферной коррозии, является степень увлажненности корродирующей поверхности материалов оборудования. По степени увлажненности поверхности металлов и сплавов коррозию принято разделять на:
- мокрую атмосферную коррозию — коррозию при наличии на поверхности металла видимой пленки влаги толщиной от 0,1 мкм до 0,1 мм. Этот тип коррозии наблюдается в атмосфере со 100 % влажностью и при наличии капельной конденсации влаги, в условиях дождя;
- влажную атмосферную коррозию, когда на поверхности металла образуется тончайшая, невидимая пленка влаги. Такая пленка, толщиной от 10 нм до 0,1 мкм, возникает в результате капиллярной, адсорбционной конденсации влаги при относительной влажности воздуха около 100 %;
- сухую атмосферную коррозию — коррозию при полном отсутствии влаги на поверхности металла.
56
Новый справочник химика и технолога
В этом случае толщина пленки обычно не превышает 10 нм.
По механизму своего воздействия мокрая и влажная коррозия относятся к электрохимической коррозии, а сухая — к химической коррозии (см. раздел 1.4.1.2).
Мокрая атмосферная коррозия металлов по своему механизму приближается к электрохимической коррозии при полном погружении металла в электролит. Видимая пленка влаги на поверхности металла возникает при мокрой коррозии в результате непосредственного попадания электролита на поверхность металла (дождь, обливание водой и т. п.). Еще одной причиной возникновения видимых пленок может быть капельная конденсация влаги, которая происходит при относительной влажности воздуха около 100 %. Пленка влаги, при влажной атмосферной коррозии, также возникает при относительной влажности воздуха около 100 %. Причинами появления пленки могут являться несколько процессов. Это капиллярная конденсация влаги, связанная с зависимостью давления паров, насыщающих пространство, от формы поверхности и степени кривизны мениска жидкости, над которым устанавливается такое давление. Равновесное давление пара будет максимальным над выпуклым мениском, а минимальным — над вогнутым, причем зависимость равновесного давления в этом случае будет описываться уравнением Томсона:
Р\ =ро ехр (2оГ„ / RTr), где рх и ро — давление насыщенного пара над вогнутым и плоским мениском соответственно, о — поверхностное натяжение жидкости, Vn — молярный объем жидкости, R — универсальная газовая постоянная, Т — абсолютная температура, г — радиус кривизны вогнутого мениска. С уменьшением радиуса кривизны вогнутого мениска уменьшается давление насыщенного водяного пара над этим мениском (табл. 1.4.6). Таким образом, наличие капилляров со смачиваемыми стенками приводит к конденсации водяного пара, не насыщенного по отношению к плоскому мениску жидкости.
Такими капиллярами на поверхности изделий, подвергающихся коррозии, могут быть микротрещины, щели между соединяемыми деталями, поры в оксидной пленке, риски, царапины, частицы пыли и грязи, осевшей на поверхность металла.
Следующим процессом, приводящим к формированию пленки, может быть адсорбционная конденсация влаги, обусловленная проявлением адсорбционных сил на поверхности металла и способная создавать слои влаги толщиной до нескольких десятков молекулярных слоев. Наибольший эффект образования пленок методом адсорбционной конденсации возникает при наличии в металле тонких капилляров.
Кроме того, возможен процесс химической конденсации в виде химического взаимодействия продуктов коррозии с водой, приводящего к образованию гидратированных соединений. Таким соединениям соответ
ствует пониженное давление насыщенного водяного пара (табл. 1.4.7). Конденсацию влаги облегчает и наличие на поверхности металла пленки раствора соли, которая также способствует понижению давления насыщенного водяного пара (табл. 1.4.8).
Таблица 1.4.6
Зависимость давления насыщенного пара от радиуса кривизны вогнутого мениска при 25 °C
Радиус кривизны вогнутого мениска, мм Давление насыщенного пара над вогнутым мениском, мм рт. ст.* Отношение давлений над вогнутым и плоским менисками Относительная влажность, %
00 КГ4 10’5 5 • 10 6 2 • 10 6 10 6 23,8 23,5 21,4 19,3 14,1 8,3 1,0 0,99 0,90 0,81 0,59 0,35 100 99 90 81 59 35
*1 мм рт. ст. = 133 Па.
Таблица 1.4.7
Давление водяного пара, находящегося в равновесии с кристаллогидратами CuSO4 при 25 °C
Среда Давление водяного пара, мм рт. ст.
Чистая Н2О 23,8
Насыщенный раствор CuSO4 23,1
CuSO4 • 5Н2О + CuSO4 • ЗН2О 7,4
CuSO4 • ЗН2О + CuSO4 • Н2О 5,6
CuSO4 • Н2О + CuSO4 0,8
Таблица 1.4.8
Давление водяного пара над насыщенными растворами солей при 20 °C
Насыщенный раствор соли Давление водяного пара, мм рт. ст. Относительная влажность, % Насыщенный раствор соли Давление водяного пара, мм рт. ст. Относительная влажность, %
ZnCl2 0,23 10 Na2SO4 1,89 81
СаС12 0,82 35 КС1 2,00 86
Zn(NO3)2 0,98 42 ZnSO4 2,12 91
NH4NO3 1,56 67 KNO3 2,17 93
NaCl 1,82 78 K2SO4 2,31 99
Электродные процессы
57
Особенности атмосферной коррозии связаны с малой толщиной пленок электролита на поверхности корродирующего металла. В данном случае электролитом является как сама влага, так и увлажненный слой продуктов коррозии металла. Для атмосферной коррозии типична легкость проникновения кислорода к поверхности металла. Это приводит к тому, что даже под тонкой кислой пленкой влаги коррозия в основном протекает с кислородной деполяризацией. Одновременно с этим легкость доступа кислорода к поверхности металла облегчает наступление пассивного состояния металла т. е. при уменьшении толщины пленки электролита при атмосферной коррозии катодный процесс облегчается, а анодный — затрудняется. При сверхмалой толщине пленки влаги (адсорбционных пленках) возможно совместное торможение катодной деполяризационной
реакции и анодной реакции из-за недостатка воды для их осуществления. Малая толщина слоев электролитов приводит к заметному увеличению омического сопротивления электролита при работе коррозионных микропар.
Таким образом, атмосферная коррозия металлов и сплавов, в частности на основе железа, протекает со смешанным катодно-анодным омическим контролем. Такой контроль в зависимости от толщины, состава и электропроводности электролита и природы корродирующего металла может переходить преимущественно в катодный контроль — мокрая атмосферная коррозия, или преимущественно в анодный контроль — влажная коррозия легко пассивирующихся металлов при отсутствии депассиваторов, или преимущественно в омический контроль (рис. 1.4.2).
Рис. 1.4.2. Поляризационные коррозионные диаграммы для основных практических случаев контроля атмосферной коррозии металлов:
а) смешанный катодно-анодный омический контроль; б) преимущественно катодный контроль; в) преимущественно анодный контроль; г) преимущественно омический контроль
Скорость атмосферной коррозии связана с протеканием нескольких процессов. Одним из основных процессов, определяющих скорость коррозии, является увеличение влажности воздуха, способствующей ускоренному образованию на поверхности металла пленки электролита (рис. 1.4.3). Величина критической влажности воздуха, определяющей начало интенсивного развития коррозионных процессов, зависит от состояния поверхности металла и состава атмосферы (табл. 1.4.9). Крайне велико влияние примесей воздуха на увеличение скорости атмосферной коррозии.
Среди примесей можно выделить посторонние, т. е. не входящие в постоянный состав воздуха. Это такие газы, как SO2, SO3, H2S, NH3, Cl2, НС1. Эти газы, попадая в пленку влаги, увеличивают ее электропроводимость и гигроскопичность продуктов коррозии, действуют как депассиваторы, как катодные деполяризаторы. Твердые частицы, попадающие из воздуха на корродирующую поверхность, могут сами быть коррозионными, например NaCl, Na2SC>4; могут увеличивать электропроводность, депассивирующую способность среды, выступать адсорбентами, облегчающими адсорбцию на поверхности металла различных газов и влаги.
влажность, %
Рис. 1.4.3. Влияние относительной влажности воздуха на скорость атмосферной коррозии железа в воздухе, содержащем 0,01% SO2, в течение 55 суток
58
Новый справочник химика и технолога
Таблица 1.4.9
Критическое значение относительной влажности воздуха при различном состоянии поверхности металлов
Металл Состояние поверхности металла (состав атмосферы) Критическая влажность, %
Железо Чистая (в чистом воздухе) Чистая (в воздухе с 0,01% SO3) Со следами слабой предварительной коррозии в Н2О Со следами коррозии в 3% NaCI Около 100 70 65 55
Медь Со следами слабой коррозии в чистом воздухе Предварительно обработанная газом SO2 Чистая (в воздухе с добавкой пара 12) 87 80 30-40
Подземная коррозия металлов и сплавов
Подземное расположение различных транспортных, производственных и коммуникационных объектов стало неотъемлемым показателем современной цивилизации. Это нефтепроводы, канализационные и кабельные сети, системы метро, сваи и другие строительные конструкции, которые эксплуатируются в подземных условиях, соприкасаясь с поверхностью земли (почвой) или нижележащими породами (грунтом). В этих условиях эксплуатации также наблюдается коррозионное разрушение металлов и сплавов, особенно интенсивное у тех подземных сооружений, которые находятся в зоне действия блуждающих токов. Приблизительный объем коррозионных повреждений и связанный с ним объем замен оборудования, поврежденного подземной коррозией, оценивается примерно в 2-3 % от общего объема металла подземных сооружений. Почва и грунт содержат различные химические реагенты, влагу и обладают ионной электропроводимостью, т. е. они являются коррозионноактивными электролитами по отношению к материалам подземных объектов и сооружений. Таким образом, можно прогнозировать электрохимическое коррозионное повреждение подземных объектов при их контакте с почвой и грунтом.
Почва и грунт представляют собой коллоидные капиллярно-пористые системы, поры которых заполнены воздухом и влагой, т. е. их можно рассматривать как микропористые электролиты с большой неоднородностью строения и свойств. Наиболее характерным катодным процессом в подземных условиях является кислородная деполяризация с преобладанием торможения транспортировки кислорода к металлу. В сильнокислых средах может происходить водородная деполяризация, в ряде случаев возможно также электрохимическое вос
становление продуктов жизнедеятельности различных грунтовых микроорганизмов.
Подземную коррозию принято подразделять на грунтовую, обусловленную электрохимическим взаимодействием подземных металлических сооружений с коррозионноактивным грунтом, и электрокоррозию, связанную с наличием подземных металлических сооружений в зоне действия блуждающих токов, что приводит к дополнительному усилению разрушения этих конструкций. Электрокоррозия как процесс ускоренного разрушения конструкций является во много раз более опасной, чем грунтовая коррозия.
Для грунтовой коррозии металлов характерен преимущественно язвенный характер разрушения. Скорость коррозии металлов в грунте зависит от состава грунта, его влагоемкости, воздухопроницаемости. Основным фактором, определяющим скорость коррозии, является наличие влаги, которая делает грунт электролитом и вызывает электрохимическую коррозию находящихся в нем металлических конструкций. Увеличения влажности грунта облегчает протекание анодного процесса, уменьшает электросопротивление грунта, но затрудняет протекание катодного процесса при значительном насыщении водой пор грунта, уменьшая скорость диффузии кислорода. Поэтому зависимость скорости коррозии металлов от влажности грунта имеет вид кривой с экстремумом (рис. 1.4.4). Следующим фактором, влияющим на скорость коррозии в грунте, является его воздухопроницаемость, которая зависит от влажности, особенностей состава и плотности грунта. Повышение воздухопроницаемости ускоряет коррозионное разрушение металлов, облегчая катодный процесс. В случае неравномерной воздухопроницаемости грунта различного состава на более воздухопроницаемых участках (песках) локализуется катодный процесс, на более плотных (глинистых) — анодный процесс. Еще одним фактором является удельное электросопротивление грунтов, которое может изменяться от нескольких единиц до сотен Ом • метр. Электросопротивление зависит от влажности грунта, его состава и структуры. Во многих случаях показатель электросопротивления грунта с достаточной достоверностью может дать информацию о коррозионной агрессивности грунта и часто используется для этих целей (табл. 1.4.10а).
Рис. 1.4.4. Влияние влажности на скорость коррозии стали:
1 — в песке; 2 — в глине
Электродные процессы
59
К факторам, определяющим скорость коррозии металлов в грунте, также относятся кислотность грунта, pH которого колеблется в пределах от 3 до 9, неоднородность грунта по структуре, плотности, составу,
влажности, кислотности и т. п. (табл. 1.4.106), наличие в грунте микроорганизмов, температура грунта. Сводные данные о влиянии различных факторов на скорость грунтовой коррозии приведены в табл. 1.4.11.
Таблица 1.4.10а
Характеристика коррозионной активности грунтов
Оценка агрессивности грунта Особо высокая Высокая Повышенная Средняя Низкая
Удельное электросопротивление грунта, Ом • м <5 5-10 10-20 10-100 > 100
Таблица 1.4.106
Классификация коррозионной активности осадочных и насыпных пород
Таблица 1.4.11
Влияние изменений условий на основные стадии и скорость коррозии металлов
Характер изменения условий грунтовой коррозии Анодный процесс Катодный процесс Протекание тока Общая скорость коррозии
Увеличение влажности грунта Облегчается Затрудняется Облегчается Проходит через максимум
Увеличение воздухопроницаемости грунта Затрудняется Облегчается Затрудняется Проходит через максимум
Увеличение засоленности грунта Облегчается Незначительно затрудняется Облегчается Увеличивается
Увеличение удельного электросопротивления грунта Непосредственно не влияет Непосредственно не влияет Затрудняется Мало изменяется
Увеличение температуры грунта Облегчается Облегчается Облегчается Увеличивается
Повышение кислотности грунта Облегчается Облегчается Облегчается Увеличивается
Повышение содержания H2S в грунте Облегчается Облегчается Облегчается Увеличивается
Жизнедеятельность бактерий, восстанавливающих SO*- Облегчается Облегчается Мало изменяется Увеличивается
Жизнедеятельность бактерий, окисляющих H2S Облегчается Облегчается Облегчается Увеличивается
60
Новый справочник химика и технолога
Электрокоррозия представляет собой электрохимическую коррозию, протекающую под действием внешнего источника постоянного тока, т. е. так называемых блуждающих токов, возникающих вблизи электрифицированных транспортных путей (железнодорожных, трамвайных, кабельных и т. п.) и сооружений (цехов, оснащенных электросварочным оборудованием, силовыми электрическими установками и т. п.). Источники блуждающих токов возникают при плохой изоляции рельсов от поверхности земли или силовых шин от пола, при наличии солевых электролитных мостов в электролизных цехах, образующихся при центральном подводе или отводе электролита.
Если в зоне действия блуждающих токов оказываются металлические трубопроводы, кабели, длинномерная металлическая арматура, то они играют роль параллельной цепи тока, т. к. обладают гораздо более высокой электропроводимостью, чем почва или грунт. Зона входа электрического тока становится катодной зоной, зона выхода — анодной зоной, а средняя часть конструкции — нейтральной зоной.
Разрушение металлических конструкций под действием электрокоррозии происходит со значительной скоростью. Так, общая сила блуждающих токов может находиться в пределах от 2,1-20 А до 200 А. При хорошей электропроводимости почвы и грунта и повреждениях в изоляции металлического объекта плотность тока в отдельных точках анодной зоны может достигать очень высоких значений.
Следует иметь в виду, что стальные конструкции под действием электрокоррозии обычно разрушаются только в анодных зонах, а такие металлы, как алюминий или свинец и их сплавы, разрушаются на катодных участках из-за подщелачивания среды при протекании коррозионного процесса с кислородной деполяризацией.
Морская коррозия металлов и сплавов
Коррозии в морской воде или морской атмосфере подвержены металлические части морских судов, различные портовые и нефтедобывающие устройства и сооружения, механизмы и трубопроводы, морская авиация, оборудование рыбоперерабатывающих цехов и предприятий и многие другие объекты, расположенные как в море, так и на его побережье.
Наиболее широко применяемыми материалами «морского» оборудования являются стали; в морской авиации и при изготовлении маломерных быстроходных судов кроме сталей достаточно широко применяются легкие сплавы (гребные валы и винты); ряд других судовых механизмов изготавливается из сплавов меди.
Морская вода представляет собой хорошо аэрированный нейтральный электролит. Такой электролит отличается высокой электропроводимостью, обусловленной наличием в воде растворов ряда солей (в первую очередь хлоридов и сульфатов натрия, магния, кальция и калия, содержание которых в морской воде
может доходить до 3,5-4,5 %), высокой депассивирующей способностью благодаря большому содержанию растворенных в воде хлоридов. Количество кислорода, растворенного в морской воде, достигает 8 мг/л, ее pH находится в пределах 7,2-8,6, электропроводимость напрямую зависит от объема растворенных солей и в различных водоемах колеблется от 0,025 до 0,03 Ом-1 • см1.
Морская коррозия протекает по механизму электрохимической коррозии с кислородной деполяризацией. Особенностями морской коррозии металлов при этом являются как высокая агрессивность морской воды и морской атмосферы, так и наличие дополнительных механических факторов воздействия на материал — эрозии и кавитации. Не менее важна роль биологического фактора — обрастания подводной части металлических конструкций морскими организмами.
Для разрушения металлов в морской воде характерно как протекание процессов равномерной коррозии, так и наличие на корродирующей поверхности глубоких местных повреждений язвенного типа. При этом коррозионная активность различных водоемов колеблется в широких пределах. Так, средняя скорость равномерной коррозии сталей составляет от 0,08 до 0,20 мм/г, а максимальная глубина язвин может изменяться в пределах от 0,4 до 1,0 мм/г. Разрушение металлов и сплавов в морской атмосфере в основном протекает по механизму равномерной коррозии, язвенное повреждение металлов, как правило, отсутствует, а сама морская атмосфера оказывается значительно менее агрессивной, чем, например, индустриальная.
Одним из наиболее опасных видов морской коррозии является коррозия по ватерлинии, т. е. в зоне периодического смачивания металла морской водой; обычно эта зона возвышается над уровнем воды на 0,4-1,0 метра. Это связано с облегченным доступом кислорода к поверхности металла, ухудшением условий для возникновения и сохранения защитных пленок на металле при периодическом смачивании и энергичным коррозионным воздействием брызг морской воды. При быстром испарении брызг на поверхности металла образуются микрокристаллы солей, смоченные насыщенным раствором, которые еще более затрудняют появление и сохранение защитных пленок. Дополнительное отрицательное воздействие оказывает солнце, лучи которого нагревают металл, ускоряя коррозионный процесс в условиях усиленной аэрации.
Особое внимание в морских конструкциях следует уделять наличию в них щелей и зазоров. Эти дефекты оказывают крайне неблагоприятное влияние на сохранность металлических конструкций в морской воде, т. к. в них из-за плохой аэрации ускоряется анодный процесс растворения металла. Не менее важную роль играет механическое воздействие среды на корродирующую поверхность, вызывающее явления коррозионной усталости, коррозионной эрозии и коррозионной кавитации.
Электрокоррозия морских систем и сооружений является достаточно распространенным явлением и вы
Электродные процессы
61
зывается прохождением электрического тока через подводную часть устройств. Этот вид коррозии обычно связан с неправильными схемами питания потребителей электрического тока находящегося на плаву достраиваемого судна (например, при однопроводной схеме питания сварочных работ и других потребителей тока, высоком сопротивлении обратного провода и т. п.), наличием вблизи стоянки судов подводных конструкций, в которых возникли блуждающие токи, и т. п.
Весьма важную роль в регулировании коррозионной стойкости морских судов играет биологический фактор: обрастание днищ и бортов кораблей различными микроорганизмами растительного и животного происхождения — кораллами, диатомеями, мшанками и т. д. Появление некоторых из них, например болянусов, разрушает защитные покрытия, приводит к неравномерной аэрации корродирующей поверхности и возникновению щелевой коррозии. Некоторые микроорганизмы (например, диатомеи) в процессе фотосинтеза выделяют кислород, что ускоряет и облегчает процесс коррозии. Однако в ряде случаев наличие на поверхности металла биологических организмов может тормозить коррозионный процесс. Так, обрастание стали мидиями снижает скорость коррозии, что связано со значительным потреблением кислорода этими моллюсками и, как следствие, снижением его концентрации у поверхности корродирующего металла.
Кроме прямого ущерба, наносимого поверхности конструкции микроорганизмами, существует еще один фактор, требующий борьбы с этим явлением. Обрастание днищ судов, систем водоснабжения, водоводов биологическими объектами приводит к уменьшению скорости и увеличению потребляемой мощности судна, уменьшению пропускной способности водотока и т. п. Наиболее подвержены обрастанию морскими организмами алюминий и его сплавы, все виды сталей, сплавы на никелевой основе, олово, свинец и их сплавы. Наименее обрастающими материалами являются магний, цинк, медь и их сплавы.
1.4.2. Питтинговая коррозия
Питтинговая или точечная коррозия (рис. 1.4.1, е), наблюдаемая у металлов и сплавов в пассивном состоянии, связана с разрушением защитной пленки и часто возникает на совершенно гладкой поверхности. Она появляется там, где защитная оксидная пленка на металле подвержена только местному разрушению, а в остальной своей части устойчива к раствору, который воздействует на металл. Область вероятных значений pH при этом, согласно диаграммам Пурбэ, соответствует нейтральной — слабощелочной области.
Различают два основных типа образования питтингов. Во-первых, питтинг, вызываемый работой элементов дифференциальной аэрации (подобной щелевой коррозии), которому подвержены многие металлы. Во-вторых, питтинг, поражающий пассивированные или покрытые оксидами высокого сопротивления металлы,
при котором любые повреждения пленок в незащищенном состоянии становятся местами интенсивного разъедания.
Точечные повреждения — питтинги — зарождаются в дефектах (несплошностях) оксидной пленки в тех местах конструкций, где имеются различные переходы, выступы и т. п., и при наиболее подходящих для этого локальных условиях внешней агрессивной среды. Такими местами конструкций могут быть выступающие болты и головки заклепок, а также направляющие и острые ребра. Кроме того, питтинг может вызываться внешними элементами, оседающими на конструкцию, например, когда какие-либо вещества оседают на ее поверхность. На возникновение питтингов оказывает влияние также структура металлической поверхности, даже защищенной оксидной пленкой. Так, они могут возникать в местах выхода на поверхность гетерогенных составляющих сплава, в границах его фаз и зерен. Твердые неметаллические включения, возникающие в сталях и сплавах при выплавке, литье и термической обработке, также могут оказаться местами зарождения питтингов.
Питтинг часто затягивается мембраной из продуктов коррозии, и если часть этих продуктов отпадает от вертикально расположенной поверхности корродирующего материала, то это может способствовать интенсификации коррозионного процесса. Такой же эффект может быть вызван другими причинами, например, морскими организмами, илом и т. п.
В стальных трубах, которые часто покрыты прокатной окалиной, состоящей из магнетита — Fe3O4, питтинги часто затягиваются мембраной из гидроксида железа Fe(OH)3. В содержимом такой точки при pH ~ 6 устанавливается равновесие.
На алюминии реакция вызывает утолщение пленки, иногда до толщины, отвечающей интерференционным цветам. Образующаяся при этом щелочь стремится растворить пленку и не обязательно способствует локализации разъедания. Однако, если щелочь в значительной степени расходуется (например, в реакции с гидрокарбонатом кальция) и местные анодные продукты не нарушаются, то может начаться питтинг, причем такая реакция будет поставлять ионы водорода:
А1 + ЗН2О = А1(ОН)3 + ЗН+ + Зе
Если ионы П не будут расходоваться на восстановление или уноситься из зоны реакции, то их накопление вызовет постепенное понижение pH. При этом разъедание будет усиливаться, приобретая автокаталитический характер. В конечном итоге пленкообразование прекратится и на дне питтинга единственной протекающей реакцией останется:
А1 ~ А13 +Зе
Питтинг алюминия вызывается водами, содержащими гидрокарбонат кальция, кислород, следы меди, способные образовывать катоды значительно более эффективные, чем глинозем и хлорид-ионы, которые
62
Новый справочник химика и технолога
препятствуют пленкообразованию. Величина pH внутри питтинга, образовавшегося на алюминиевой поверхности в хлоридных растворах, составляет 3,5 при pH 7 в объеме раствора.
Медь не склонна к питтингообразованию, хотя осаждающиеся на ее поверхности частицы и морские организмы могут способствовать его зарождению за счет эффекта дифференциальной аэрации. Питтинг в меди также может возникать в местах разрывов слоев углеродистых веществ с низким сопротивлением, остающихся в медных изделиях после прокатки.
Питтинг может усиливаться ионами металлов, окислительно-восстановительные потенциалы которых находятся в области пассивного состояния основного металла, поскольку добавляются объекты катодного восстановления. Так, если в присутствии кислорода все хлориды вызывают питтинг у нержавеющих сталей, то в отсутствие кислорода лишь некоторые из них, а именно только СпС12 и FeCl3, будут вызывать образование язвин.
Питтинговое коррозионное разрушение может возникнуть у самых разнообразных материалов — меди, алюминия и их сплавов, никеля, циркония, титана, нержавеющих хромистых и хромоникелевых сталей; в средах, в которых наряду с пассиватором-окислителем (в том числе растворенным кислородом) присутствуют активирующие анионы, такие как СГ, Вг или Г-ионы (например, в морской воде, растворах NaCl, FeCl3, в смесях соляной и азотной кислот и т. п.).
Питтинг может возникать на поверхности всех металлов. На практике он чаще всего обнаруживается на пассивных сплавах, у которых благодаря весьма стойкой пленке разъедание приобретает местный характер. Это также относится к металлам, которые могут быть пассивированы ингибиторной добавкой с недостаточно высокой концентрацией, причем наиболее интенсивно питтинг развивается в хлоридных средах.
Зарождение питтинга может потребовать довольно значительного времени, поэтому необходимо контролировать процесс образования питтингов с помощью анодной поляризации. Для этого испытуемые образцы анодно поляризуются, и пробой пассивной пленки определяется по повышению тока при потенциале пробоя Еь — потенциале питгингообразования (рис. 1.4.5). Чем больше потенциал Еь, тем выше стойкость сплава к питтинговой коррозии.
Существуют три стадии образования питтингов.
1. Питтинг возникает в слабых местах пассивной пленки по достижении определенного потенциала Еь (потенциала питгингообразования) за счет окислителя или анодной поляризации в растворе в присутствии активирующих ионов, которые вытесняют адсорбиро
ванный кислород или, взаимодействуя, разрушают оксидную пленку. Местное ослабление пассивности может быть обусловлено неоднородностью структуры металла (неметаллические и интерметаллидные включения, границы фаз и зерен и т. п.), случайными механическими повреждениями защитной пленки или др. причинами.
2. Рост питтинга происходит из-за интенсивного растворения защитной пленки, что приводит к сильному возрастанию скорости анодного процесса (активационный режим роста питтинга), которая со временем падает в связи с расширением поверхности питтинга и возникающими диффузионными ограничениями (диффузионный режим роста питтинга).
3. Возможная репассивация (прекращение роста) значительного числа питтингов в начальный период коррозии, вследствие чего число питтингов во времени поддерживается примерно на одном уровне. Реализация этой стадии коррозии необязательна. Причин репассивации питтингов в преддиффузионном режиме может быть несколько. В частности, сдвиг потенциала всей поверхности или самого питтинга в отрицательную сторону — в область пассивности. Растущий питтинг, перешедший в диффузионный режим роста, является стабильным питтингом, неспособным к репассивации. При значительном углублении растущего питтинга возможно некоторое снижение скорости его роста, определяемое диффузионным торможением.
Потенциал пробоя (потенциал питтингообразова-ния) Еь находится в хорошем соответствии с очаговым показателем коррозии (число язвин на 1 см2) и пригоден для исследования влияния различных эксплуатационных факторов на склонность металлов и сплавов к питтинговой коррозии. В табл. 1.4.12. приведены значения Еь в 0,1 М NaCl при 25 °C. Как видно из таблицы, наименьшей стойкостью к питтинговой коррозии обладает алюминий, наибольшей — титан и хром.
1g'
Рис. 1.4.5. Анодная поляризационная кривая пассивирующего сплава, у которого нарушение пассивности (пробой) происходит при Еь
Таблица 1.4.12
Потенциал питгингообразования некоторых металлов и сплавов
Материал А1 Fe Ni Zr Cr Ti 08X18H9
Потенциал Еь, В -0,45 + 0,23 + 0,28 + 0,46 + 1,0 + 12,0 + 0,26
Электродные процессы
63
В табл. 1.4.13 приведены результаты сравнительных коррозионных испытаний рада широко применяемых сталей. Как видно, увеличение концентрации хрома, никеля и молибдена в сталях повышает их стойкость к питтинговой коррозии (рис. 1.4.6). Кроме хрома и молибдена стойкость хромоникелевых сталей к питтинговой коррозии повышают кремний и рений, препятствуя зарождению и способствуя репассивации питтингов. В то же время углерод, титан и ниобий снижают стойкость сталей к питтинговой коррозии, что, вероятно, связано с образованием в структуре этих сталей новой фазы — карбидов титана и ниобия, выступающих как инициаторы зарождения и развития язвин.
На склонность хромоникелевых сталей к питтинговой коррозии значительное влияние оказывает качество состояния ее поверхности. Механическая полировка понижает склонность к питтингообразованию при обычных температурах, в то время как электролитическое полирование повышает ее. Предварительная пассивация металлов, например в (HNO3 + К2Сг2О7), увеличивает стойкость хромоникелевых сталей к питтинговой
коррозии.
Питтинговая коррозия на металлах возникает в рас
творах, содержащих анионы галогенов, из которых наиболее агрессивны хлорид- и бромид-ионы, в то время как фторид-ионы точечную коррозию вообще не вызывают, обеспечивая значительное и равномерное растравление всей поверхности металла. Некоторые кислородсодержащие анионы ингибируют питтинговую коррозию, вытесняя хлорид-ионы с поверхности. При определенном соотношении SO^/СГ питтинговая коррозия сталей не возникает.
Зависимость потенциала пробоя от pH среды для стали 12X18Н9Т в растворах NaCI при сСг = const имеет сложный характер (рис. 1.4.7). Наиболее опасная область— кислые растворы. С повышением температуры число питтингов на стали 12X18Н9Т возрастает, а глубина питтингов остается практически неизменной (рис. 1.4.8).
10 20 30 40
Сг, %
Рис. 1.4.6. Влияние содержания хрома и никеля в сталях на потенциал питтиногообразования:
а) хром в сплаве Fe—Сг в 0,5 М NaCI при 25 °C;
1 — экспериментальные плавки, 2 —20X13, 3 —30X13,4 — Х28;
б) никель в стали Х22Т в 0,1 М NaCI при pH 2 и 25 °C
Рис. 1.4.7. Зависимость потенциала питтингообразования от pH среды в стали 12Х18Н9Т в растворе NaCI при сСг = const
Таблица 1.4.13
Потенциал питтингообразования и очаговый показатель коррозии хромистых и хромоникелевых сталей По результатам 15-дневных испытаний.
Марка стали Потенциал питтингообразования, В, в 0,5 М NaCI Очаговый показатель коррозии, число питтингов/см2
При переменном погружении При постоянном погружении
0,17MFeCl3 0,49 М NaCI + + 0,01 МНС1 0,17MFeCl3
20X13 0,204 НО 50 100
08X17 0,343 30 12 18
08X28 0,543 8 5 6
10Х13Н4Г9 0,270 55 15 25
12Х18Н9Т 0,410 50 15 35
12Х18Н9 0,447 20 7 10
08Х18Н12М2Т 0,468 15 8 8
0,529 10 5 6
0,951 3 1 2
64
Новый справочник химика и технолога
Рис. 1.4.8. Влияние температуры электролита на число возникающих питтингов и скорость развития коррозии настали 12Х18Н9Т:
1, 1а — число питтингов, 2,2а — средняя глубина питтингов, 3, За — максимальная глубина питтингов;
1-3 — в электролите 2% FeNH4(SO4)2 • 2Н2О + 3% NH4C1, 1а-3а — в электролите, содержащем
11,5 г/л FeCU • 6Н2О + 28,5 г/л NaCl
1.4.3. Коррозионное растрескивание металлов и сплавов
Коррозионное растрескивание (стресс-коррозия) — один из наиболее опасных видов коррозии, т. к. имеет остро локализованный характер. Коррозионное растрескивание возникает при одновременном воздействии растягивающих напряжений или пластической деформации и агрессивной среды, содержащей активирующие добавки (хлорид-ион, окислители и т. п.)
Проблема коррозионного растрескивания металлов и сплавов стала наиболее актуальной в последние полвека, когда широкое распространение получили такие виды масштабного оборудования, как магистральные и промысловые нефте-, газо- и продуктопроводы, системы синтеза углеводородов, энергетические и энерготехнологические системы большой единичной мощности, атомные энергетические системы.
1.4.3.1. Механизм коррозионного растрескивания
Видимое проявление коррозионного растрескивания состоит в появлении трещин, которые напоминают хрупкое разрушение, поскольку их возникновение не сопровождается значительной пластической деформацией. Коррозионное растрескивание, вызывающее хрупкое разрушение, в пластичном материале обычно обуслов
лено воздействием ряда факторов — определенной внешней среды, растягивающих напряжений, действующих в материале, его химического и фазового состава и ряда эксплуатационных особенностей: температурных, временных и т. п. Энергетический баланс коррозионного растрескивания можно представить следующим образом:
изменение поверхностной энергии + работа пластической деформации = изменение начального запаса энергии + выделение энергии при электрохимической реакции.
Величиной поверхностной энергии в этом уравнении можно пренебречь, т. к. она весьма мала по сравнению с работой пластической деформации при стресс-коррозии пластичных материалов. С учетом этого данное уравнение приобретает вид:
Р = К' -(1-Ц2) + £^р А (1.4.3.1) Е f| М
где Р — работа пластической деформации, К\ — коэффициент интенсивности напряжений, ц — коэффициент Пуассона, Е — модуль Юнга, z — формальный заряд сольватированных ионов, F — постоянная Фарадея, р— плотность, 5 — высота надвигающегося фронта трещины, М — молекулярная масса металла, ц — перенапряжение анодной реакции. При пороговом значении коэффициента интенсивности напряжений Кт, т. е. при минимальном значении К\, при коррозионном растрескивании уравнение (1.4.3.1) преобразуется к следующему виду:
see = F Р 5—)тп • (1 -4.3.2)
у 1-ц М
Таким образом, видно, что переменными, которые могут оказывать влияние на значение К\$сс, являются Р и ц , т. е.
(1.4.3.3)
Понижение значения работы пластической деформации Р Ъур&х происходить в результате увеличения или предела текучести, или скорости механического упрочнения в вершине трещины. В результате каждый из этих факторов при постоянном значении ц будет понижать величину К\^сс и, следовательно, понижать степень сопротивления материала коррозионному растрескиванию. Увеличение перенапряжения анодной реакции f| (потенциал металла становится более электроположительным) при определенном значении работы пластической деформации Р, согласно уравнению (1.4.3.3), будет приводить к понижению сопротивления коррозионному растрескиванию. Величина анодного перенапряжения является функцией электрохимических условий внутри трещины, контролирующих активно-пассивные переходы, от которых в свою очередь зависит, будет ли происходить растрескивание. Следовательно, коррозионное растрескивание
Электродные процессы
65
является в определенной степени функцией от pH, активности анионов, химического состава сплава и электродного потенциала.
1.4.3.2. Основные теории, описывающие природу возникновения склонности материалов к коррозионному растрескиванию
К настоящему времени сформировалось несколько основных теорий, описывающих природу возникновения и развития процесса коррозионного растрескивания. Выдвигаемые различными научными школами предложения можно обобщить в несколько основных моделей, которые с большим или меньшим уровнем приближения, с большей или меньшей степенью достоверности описывают коррозионные процессы в сталях и сплавах при одновременном воздействии агрессивной среды и деформаций (напряжений), возникающих в материалах.
Адсорбционная теория
Теория основана на открытом в начале двадцатого века эффекте понижения поверхностной энергии в результате адсорбции (эффекте Ребиндера). Согласно этой теории адсорбция типичных поверхностно-активных веществ из окружающей среды вызывает облегчение деформации и разрушения твердых тел, часто в значительно большей степени, чем при химическом воздействии. Эффект адсорбционного понижения прочности, согласно этой теории, обусловлен тем, что поверхностно-активные вещества, понижая поверхностную энергию металлов, способствуют зарождению пластических сдвигов. При этом процесс коррозионного растрескивания протекает не путем химического или электрохимического растворения металла в вершине трещины, а вследствие ослабления межатомных связей в напряженном сплаве при адсорбции специфических компонентов раствора. Благодаря адсорбции снижается поверхностная энергия, что облегчает разрыв межатомных связей металла, находящегося под растягивающим напряжением. Уменьшение сродства между атомами на поверхности металла происходит при наличии одного адсорбционного монослоя, при этом наиболее эффективно действуют частицы, проявляющие специфическую адсорбцию. Инициирование трещины стресс-кор-розии вызывается адсорбционным снижением сил взаимодействия между смежными атомами в вершине надреза материала, подвергающегося действию высоких растягивающих напряжений.
При коррозионном растрескивании материалов анионы адсорбируются преимущественно на подвижных дислокациях или других несовершенствах структуры, выходящих на подвергающуюся агрессивному воздействию поверхность. Т. к. адсорбция протекает с конечной скоростью, дефекты кристаллической решетки на поверхности, которые могли бы служить зародышем стресс-коррозионной трещины, должны существовать в течение определенного времени. Примеси в
сплаве могут оказывать сильное влияние на время жизни поверхностных дефектов. Атомы примесей, скапливаясь вокруг дислокаций, образуют облака Коттрелла, тормозящие движение дефекта. Такое торможение увеличивает время жизни дислокации, которое становится достаточным для хемосорбции нитрат- или гидроксидиона на активных плоскостях скольжения сталей. Если же поверхностные дефекты успевают мигрировать с поверхности сплава до того, как завершится процесс адсорбции аниона, склонность к коррозионному растрескиванию не наступает.
Водородная теория
В процессе наводороживания аустенитных сталей наблюдается увеличение параметров решетки аустенита, а, следовательно, дополнительно возрастает растворимость водорода в ГЦК решетке. В сплавах, подверженных коррозионному растрескиванию в процессе наводороживания, возникает ферритная фаза, которая получила название «деформированный феррит», или ОЦК мартенситная фаза. После насыщения сталей водородом их структура представляет собой смесь фаз — исходный стабильный аустенит, и метастабильные фазы — аустенит с повышенным параметром решетки и деформированный водородом феррит. Изменение параметров кристаллических решеток исходной — аустенитной — фазы и вновь образующихся метастабильных фаз вызывает появление в сталях значительных внутренних напряжений, провоцирующих стресс-кор-розионные процессы. После непродолжительного старения при комнатной температуре происходит распад метастабильных фаз на исходный аустенит и гексагональную гидридную фазу.
Таким образом, наводороживание способствует объемным изменениям и возникновению внутренних напряжений в сталях, приводит к увеличению дефектов структуры и зарождению микротрещин на вновь образованной межфазной границе.
Дислокационная теория
Коррозионное растрескивание с точки зрения дислокационного строения металла хорошо объясняет взаимосвязь между величиной приложенного напряжения и склонностью материала к стресс-коррозионному растрескиванию. Увеличение приложенных растягивающих напряжений до определенных значений повышает скорость образования и развития стресс-коррозионной трещины в материале, что связано с возникновением дислокаций в процессе деформирования, их движением и образованием плоских скоплений. Если упорядочение выражено не слишком сильно, приложенные высокие напряжения могут вызвать поперечное скольжение и снизить восприимчивость металла к стресс-коррозии. Поэтому в ряде случаев склонность материалов к коррозионному растрескиванию после значительных степеней пластической деформации оказывается ниже, чем у слабодеформированных сталей и сплавов. Это не относится к аустенитным сталям, где
66
Новый справочник химика и технолога
при значительных степенях пластической деформации возможно протекание структурных (у —» а и у —» б) мартенситных превращений, резко повышающих склонность этих материалов к коррозионному растрескиванию, что объясняется перестройкой кристаллической решетки сплава, в результате которой на границе раздела фаз возникает скопление дислокаций, которые становятся зародышами микротрещин.
Механохимическая теория
В этой теории предпринята попытка количественно связать напряжения, возникающие в материалах, и скорость развития стресс-коррозионного дефекта с помощью термодинамики необратимых процессов. Деформация металла рассматривается на стадии линейного упрочнения, когда дислокации выстраиваются и двигаются в системе параллельных плоскостей скольжения при отсутствии поперечного скольжения. Из математических построений этой теории вытекает ряд феноменологических уравнений, указывающих на взаимосвязь процессов пластической деформации и стресс-коррозии в металлах, которые при одновременном процессе деформирования и электрохимической коррозии принимают следующий вид:
N=La -A + Lb-f\; и l = Lc + Ld-x\,
где N— плотность потока дислокаций (дислокационный ток); I— плотность тока при коррозии; А — обобщенная сила — химическое сродство процесса образования и движения дислокаций; f| — перенапряжение; феноменологические коэффициенты: La— коэффициент, регулирующий проводимость; Ld — характеризует поляризуе
мость электрода; Lc = Lb регулируют степень взаимного влияния деформации и электрохимической коррозии и выражают количественно явление механохимического эффекта. Физический смысл этого коэффициента заключается в изменении химического потенциала металла в результате его пластического деформирования, которое связано с ослаблением межатомных связей в местах скопления разрежающих дислокаций.
Пленочная теория
Согласно этой теории при пластической деформации металла у концентратора напряжения разрушается защитная оксидная пленка — значительно более хрупкая, чем основной металл. При этом поверхность металла — анода по отношению к покрытой пленкой поверхности — обнажается, а процесс коррозионного растрескивания ускоряется. На основании этой теории был сформулирован ряд условий, необходимых для развития стресс-коррозии. Для образования трещины стресс-коррозии необходимо, чтобы в материале, в ходе пластической деформации, образовывались широкие ступени скольжения; металл должен обладать способностью образовывать на поверхности защитную пленку (для того, чтобы образовалась пара металл—пленка); необходима среда, в которой участки поверхности металла с дефектами кристаллической решетки не смогли бы полностью репассивироваться. Было также показано наличие взаимосвязи между толщиной защитной пленки и склонностью материала к коррозионному растрескиванию, которая по данным В.В. Герасимова может быть выражена соотношениями, приведенными в табл. 1.4.14.
Таблица 1.4.14
Толщина продуктов коррозии, образовавшихся при выдержке в различных растворах при 560 К
Материал Раствор (pH) Толщина слоя, см Восприимчивость к коррозионному растрескиванию
Fe-10% Ni-8% Cr 10’2MNa2SO4 H2SO4 (2,5) 1 • ю4 1 • 10’3 Низкая Высокая
Ni-8% Fe-5% Cr 102MNa2SO4 H2SO4 (2,5) 1 • Ю5 3 • 10’3 Очень низкая Высокая
Сталь 304 (холодно-деформированная) 102MNa2SO4 H2SO4 (2,5) 1 • 104 3 • 10"3 Низкая Высокая
Сталь 304 10”2MNa2SO4 H2SO4 (2,5) 2 • 10’5 3 • 104 Очень низкая Низкая
Инконель 600 H2SO4 (2,5) 3 • 104 Очень низкая
Сплав AL326 10’2MNa2SO4 H2SO4 (2,5) 3 • IO4’ 1 • 10~5 Очень низкая Очень низкая
Электрохимическая теория
Идея, положенная в основу этой теории, заключается в признании того, что существует разница в электродных потенциалах в пластически деформированном — напряженном — металле (вершина трещины) и
ненапряженной боковой поверхности трещины. Роль растягивающих напряжений при этом сводится главным образом к созданию разности потенциалов между отдельными участками поверхности и образованию электрохимических пар, являющихся ответственными
Электродные процессы
67
за процесс зарождения коррозионной трещины. Таким образом, в деформированном металле создается активно-пассивное состояние, причем в активном состоянии находится деформированный металл, в пассивном — недеформированный. Разница, возникающая в скорости растворения деформированного и недеформированного металлов, приводит к возникновению локальных процессов, способствующих возникновению коррозионной трещины.
1.4.3.3. Коррозионное растрескивание углеродистых и низколегированных сталей
Влияние химического состава стали на склонность к коррозионному растрескиванию
В большинстве работ, проведенных на малопрочных (ферритных) сталях, в качестве агрессивной коррозионной среды использовались растворы нитратов. Поэтому данные, приведенные в этом разделе, в основном касаются поведения ферритных сталей именно в этих средах.
Основным фактором, определяющим коррозионную стойкость низкоуглеродистых сталей, применяемых в нормализованном или отожженном состояниях, является содержание углерода. На рис. 1.4.9. показано изменение значений пороговых напряжений, при которых не происходит разрушения в этих сталях, в зависимости от содержания углерода. В качестве коррозионной среды был использован 4 М NH4NO3. Влияние углерода на сопротивляемость стали коррозионному растрескиванию может быть объяснено с двух позиций. С одной стороны, углерод — основной элемент, обеспечивающий в сталях повышение прочности, с другой — он, сегрегируя в границы зерен стали, или образует там насыщенные углеродом двухмерные твердые растворы, или выделяется в виде карбидных фаз. Такое распределение изменяет электрохимические характеристики стали, ее сопротивляемость стресс-коррозии.
О 0,04 0,08 0,12 0,16
Содержание углерода, масс. %
Рис. 1.4.9. Влияние углерода в отожженных углеродистых сталях на величину пороговых напряжений при испытаниях на коррозионное растрескивание в кипящем 4MNH4NO3
При повышении содержания углерода увеличивается доля перлитной фазы и соответственно повышается доля карбидных частиц в границах зерен сталей, что в конечном итоге приводит к изменению места протекания избирательной коррозии и развитию коррозионного растрескивания.
На рис. 1.4.10. приведены данные о влиянии пластической деформации на стойкость сталей к коррозионному растрескиванию, которые также можно объяснить углеродным влиянием. Как известно, после значительных пластических деформаций сегрегаций атомов углерода в границах ферритных зерен низкоуглеродистых сталей не обнаруживается.
Рис. 1.4.10. Влияние степени пластической деформации на чувствительность низкоуглеродистой стали (С = 0,017 %) к коррозионному растрескиванию в 4 М NH4NO3 (цифры у кривых — степень пластической деформации образцов)
Таким образом, можно сделать заключение, что распределение углерода в сталях определяет место активных участков, вдоль которых происходит избирательная коррозия, которая и способствует коррозионному растрескиванию. Подобным действием, приводящим к возникновению многочисленных электрохимически активных участков в сталях, обладает еще целый ряд легирующих и примесных элементов, которые так же, как и углерод, образуют в сталях зернограничные сегрегации. В табл. 1.4.15 приведены данные о распределении примесных элементов в границах стали 20 после различных режимов ее термической обработки (Н — нормализация стали: 1173 К — 2 часа + изотермические выдержки в диапазоне температур 573-1073 К — 1 час). Данные по распределению элементов были получены двумя методами. Метод Оже-эмиссионной спектроскопии (ОЭС) при толщине исследуемого слоя 1 нм — т. е. при толщине, сопоставимой с толщиной границы зерна, — позволяет точно оценить сегрегацию того или иного элемента в границе зерна, когда вид сегрегации приближается к равновесной. Дополнительно был проведен анализ методом эмиссионного спектрального микроанализа (ЭСМА) при толщине анали
68
Новый справочник химика и технолога
зируемого слоя 100 нм. Этот метод особенно ценен в том случае, когда элемент сегрегируется по неравновесному механизму и повышенное содержание примеси растянуто глубоко в тело зерна. Оценить степень склон-
ности легирующего или примесного элемента к перераспределению в границы зерен позволяет энергия взаимосвязи атома примеси с границей зерна ЕЛ (табл. 1.4.16).
Таблица 1.4.15
Распределения атомов (атом. %) примесных элементов в границах зерен стали 20, полученные методами ОЭС, ЭСМА
Элемент Режим термической обработки 20кп 20пс 20сп
ЭСМА ОЭС ЭСМА ОЭС ЭСМА ОЭС
N Н — 0,008 — 0,007 — 0,008
Н + 573 К 0,75 8,53 0,31 2,47 0,22 2,1
Н + 673 К 1,33 19,22 0,38 3,62 0,44 3,34
Н + 573 К 1,19 16,71 0,20 2,09 0,18 1,43
С Н — 0,24 — 0,23 — 0,21
Н + 673 К 0,21 0,32 0,22 0,41 0,20 0,33
Н + 773 К 0,27 0,53 0,29 0,63 0,26 0,57
Н + 823 К 1,53 17,32 2,63 19,22 1,41 16,22
Н + 873 К 3,87 33,67 3,91 32,77 3,31 29,43
Н + 923 К 3,15 27,33 3,10 24,39 2,61 19,31
Р Н 0,09 0,47 0,11 0,50 0,09 0,44
Н + 773 К 0,62 5,52 0,43 3,18 0,50 3,63
Н + 823 К 1,31 9,37 1,21 7,57 1,16 8,04
Н + 873 К 1,26 9,33 1,24 8,42 1,25 8,43
Н + 923 К 0,57 3,78 0,61 4,23 0,63 4,71
S Н 3,46 31,85 0,11 0,83 0,10 0,74
Н + 873 К 0,73 5,45 0,09 0,31 0,09 0,26
Н + 973 К 1,37 11,42 0,14 0,61 0,12 0,44
Н + 1073К 1,91 16,57 0,18 0,79 0,19 0,77
I РЬ + Sn Н — 0,019 — 0,021 0,017
Н + 773 К 0,41 3,21 0,21 1,62 0,20 1,54
Н + 823 К 0,48 3,97 0,36 2,73 0,32 2,33
Н + 873 К 0,61 4,82 0,58 4,11 0,59 4,55
Н + 923 К 0,20 1,53 0,21 1,62 0,29 2,03
Мп Н — — 0,44 0,51 0,48 0,63
Н + 473 К — — 0,49 0,67 0,48 0,57
Н + 573 К — — 0,51 0,82 0,52 0,71
Н + 873 К — — 1,73 1,63 1,77 1,93
Н + 973 К — — 1,02 1,02 1,0 1,24
Н + 1073К — — 0,81 0,91 0,83 0,99
Сг Н 0,21 0,30 0,22 0,28 0,73 1,20
Н + 773 К 0,16 0,27 0,14 0,37 0,97 1,53
Н + 873 К 0,33 0,96 0,41 0,92 1,39 3,27
Н + 973 К 0,15 0,31 0,19 0,40 0,88 1,02
Таблица 1.4.16
Энергии взаимодействия (эВ) атомов примесных элементов с границами зерен в промышленных плавках стали 20 при различных температурах
Элемент 673 К 873 К 973 К
20кп 20-1 20-2 20кп 20-1 20-2 20кп 20-1 20-2
N 0,81 0,52 0,46 0,31 0,14 0,16 0,11 0,09 0,07
Р 0,31 0,27 0,24 1,12 1,07 1,06 0,31 0,29 0,30
РЬ 0,12 0,11 0,14 0,47 0,46 0,52 0,16 0,17 0,17
Sn 0,09 0,09 0,10 0,48 0,47 0,44 0,14 0,11 0,13
S 0,07 0,04 0,03 0,24 0,16 0,12 0,61 0,38 0,41
Электродные процессы
69
Как следует из табл. 1.4.16, наибольшие величины достигающие 1,07 и 1,12 эВ, отмечены у самой опасной с точки зрения снижения межатомных сил сцепления в зернограничном a-твердом растворе примеси — атомов фосфора — при температуре 873 К. У атомов азота и серы энергии взаимодействия с границей зерна в кипящей стали 20 достигают максимальных величин 0,81 эВ при 673 К (азот) и 0,61 эВ при 973 К (сера). В сталях 20пс и 20сп эти примеси находятся в основном в связанном состоянии и не оказывают столь резкого воздействия на величину Еа, а, следовательно, их роль в снижении когезии границ и изменении величины их электрохимической активности не так заметна.
В табл. 1.4.17 приведены данные о влиянии зернограничных сегрегаций на интенсивность процесса избирательной коррозии в стали 20, при анализе которой отчетливо прослеживается взаимосвязь между коррозионной стойкостью и распределением примесей в стали 20. Максимальная скорость проникновения коррозионного дефекта наблюдается в стали при максимальной активности сегрегированной примеси — атомов фосфора, сегрегация которых при температурах 823-923 К по форме приблизилась к равновесной.
В легированных сталях равновесные сегрегации могут образовываться также легирующими элементами. При этом их влияние на коррозионные свойства сталей
может быть как положительным, так и отрицательным. В ряде случаев склонность к образованию сегрегаций определенных элементов увеличивается или уменьшается в присутствии второго растворенного элемента, т. е. в присутствии легирующих элементов склонность углерода, фосфора и др. примесных атомов к образованию зернограничных сегрегаций, снижающих когезию границ зерен, может как понижаться, так и возрастать. Эти эффекты сильных взаимодействий между двумя растворенными элементами часто не учитываются в модели, в основу которой заложена только совместимость напряжений, вызванных растворенными атомами, и искаженной сегрегациями структуры границ зерен. Таким образом, присутствие дополнительного легирующего элемента может оказаться как полезным, так и вредным — повышающим склонность стали к коррозионному растрескиванию, т. е. существует целый ряд факторов, которые могут оказывать влияние на скорость коррозии границ зерен за счет образования электрохимической неоднородности между сегрегированными элементами и матрицей стали. Легирующие элементы также оказывают влияние на чувствительность сталей к растрескиванию. Это происходит вследствие влияния легирующих элементов на способность стали к образованию защитной пленки в определенных коррозионных средах и за счет их влияния на механические свойства сталей.
Таблица 1.4.17
Влияние температуры изотермической выдержки на глубину проникновения коррозионного дефекта
Сталь Глубина проникновения коррозионного дефекта, мкм/ч, после изотермической выдержки
473 К 573 К 673 К 773 К 823 К 873 К 923 К 973 К 1073 К
20 кп 173,6 292,0 415,3 444,3 477,0 590,2 473,0 348,0 332,0
20-1 144,0 167,3 175,0 200,5 280,2 340,5 300,0 210,5 197,3
20-2 129,2 158,0 165,0 200,0 265,0 340,0 290,5 200,0 195,0
Влияние термической обработки на склонность сталей к коррозионному растрескиванию
Для низкоуглеродистых сталей в отожженном или нормализованном состояниях важнейшими структурными параметрами, определяющими их склонность к коррозионному растрескиванию, являются размер зерна и последствия фазовых превращений в сварном шве и око-лошовной зоне. Влияние размера зерен на склонность сталей к коррозионному растрескиванию приведено на рис. 4.1.11. Из приведенных данных видно, что снижение размеров зерен приводит к повышению уровня напряжений, при которых сталь становится чувствительной к коррозионному растрескиванию. Это связано, в первую очередь, с числом сегрегированных в границах зерен примесных атомов — фосфора, цветных металлов, серы, углерода. В табл. 1.4.18. приведены данные, позволяющие проанализировать эту взаимосвязь. Как видно из представленной таблицы, наблюдается пропорциональная зависимость между размерами зерен и суммарным содержанием сегрегированных атомов на их границах. Такая особенность может быть объяснена тем, что при уменьшении размеров зерен суммарная площадь их гра-
ниц повышается, а суммарный объем сегрегированных атомов остается практически неизменным. Таким образом, число сегрегированных атомов на единицу площади
Рис. 1.4.11. Влияние размеров зерен в низкоуглеродистой стали (0,08 % С) при испытаниях в 4 М Ca(NO3)2 на механические свойства: 1 — предел текучести;
2 — напряжения, вызывающие 5 % пластическую деформацию;
3 — напряжения, вызывающие разрушения при коррозионном растрескивании
70
Новый справочник химика и технолога
Таблица 1.4.18
Влияние размеров зерен на химический состав зернограничного твердого раствора
Балл (средний диаметр зерна) Содержание элементов, атом. %, в слое толщиной 1 нм
Fe Р Есегр
3 (111 мкм) 5 (55,3 мкм) 7 (26,7 мкм) 9 (13,8 мкм) 61,2 67,3 75,2 80,4 14,2 11,6 7,4 3,8 38,3 32,7 24,8 19,6
Основными методами, снижающими размеры зерен в углеродистых и низколегированных сталях, являются термическая обработка и микролегирование одним из элементов группы титана, ванадия, ниобия и циркония (в количестве, не превышающем их предельную растворимость в твердом растворе стали). В табл. 1.4.19 приведены данные о возможных путях измельчения зерен стали 20. Наиболее заметные результаты достигаются при замене режима нормализации стали, принятого для этой марки на сегодняшний день, на термоциклирование. Влияние микролегирования дает по сравнению с заменой режима термической обработки менее значимый результат в измельчении зерен, однако одновременно с ним у сталей, легированных ванадием, ниобием, цирконием или титаном, наблюдается рост трещиностойкости, т. е. происходит повышение сопротивляемости стали действию растягивающих напряжений.
1.4.3.4. Коррозионное растрескивание в аустенитных сталях
Влияние растягивающих напряжений на стойкость аустенитных сталей
Одним из основных факторов, оказывающих воздействие на стойкость сталей и сплавов против коррозии под напряжением, является влияние растягивающих напряжений на электрохимические процессы, протекающие в них. Оценка такого влияния показана на примере одной из самых распространенных марок аустенитных сталей 12Х18Н(1О-12)Т. Эти марки сталей широко применяются в качестве материалов оборудования углеводородного синтеза, в криогенной технике, пищевой и др. отраслях промышленности. Для проведения таких исследований аустенитизированные при 1320 К (выдержка 1 час, охлаждение в воде) образцы были подвергнуты холодной дробной прокатке, а их склонность к возникновению трещин коррозии под напряжением оценивалась по изменению скорости анодного процесса. Испытания были проведены на кольцевых образцах, вырезанных из трубы 108 х 8 мм, шириной 15 мм. С целью оценки влияния локальных напряжений на изменение склонности к коррозионному разрушению (КР) стали, на ряде образцов наносили треугольные риски — надрезы с углом при вершине 90° и радиусом скругления в вершине 0,2 мм. Глубина надрезов составляла 25 % от толщины стенки трубного образца.
Таблица 1.4.19
Механические свойства, балл зерна и содержание вредных примесей в границах зерен углеродистых сталей
Есегр — содержание примесных атомов в слое толщиной 1 нм.
Содержание элементов в сталях:
20БЛ: Nb — 0,06 %, Мо — 0,15 %; 20ФЛ-1: V — 0,15 %; 20ФЛ-2: V — 0,12 %; 20ТЛ: Ti — 0,03 %.
Механические свойства Хсегр, атом. %
№ Режим термической обработки Марка стали Ов ст0,2 £5 rz-293 К ^1с Балл аустенита
МПа % МПа • м,/2
1 Нагрев 1173 К, выдержка 2 ч, 20Л 485 270 19 48 5 24,3
охлаждение на воздухе 20МЛ 552 318 29 50 6 22,0
20ФЛ-1 610 410 28 51 8 16,0
20ФЛ-2 590 360 33 54 7-8 16,2
20БЛ 610 375 31 54 7 16,8
20ТЛ 600 370 30 55 7-8 16,6
2 Нагрев 1213 К, выдержка 2 ч, 20Л 473 251 14 60 — 19,4
охлаждение в воде + + отпуск при 873 К в течение 1 ч 20МЛ 615 503 21 67 — 17,9
3 Гомогенизация при 1373 К в течение 20Л 525 280 23 54 — 17,0
2 ч + режим 2 20МЛ 620 408 25 69 — 14,9
4 Гомогенизация при 1373 К в течение 20Л 563 351 24 70 9-10 11,6
2 ч + четырехкратная ТЦО в меж- 20МЛ 635 440 25 70 10-11 10,3
критическом интервале с охлажде- 20ФЛ-2 640 460 28 74 12 8,8
нием на воздухе — 1, 3 и 4-го цик- 20БЛ 625 430 29 76 11-12 9,1
лов и в воде после 2-го цикла 20ФЛ-1 640 470 26 69 11 9,0
20ТЛ 655 480 28 74 12 9,1
Электродные процессы
71
Влияние напряженного состояния на электрохимические характеристики было исследовано при непрерывной деформации образцов со скоростью 1 мм/мин. Кинетика анодного процесса изучалась в 1 М Na2SO4 при электрохимическом потенциале 0,6 В, что соответствует пассивному, т. е. не склонному к КР состоянию этих сталей. Результаты исследований приведены в табл. 1.4.20. Было обнаружено, что резкий скачок скорости анодного процесса на гладких (без надреза) образцах соответствует напряжениям, примерно равным пределу текучести хромоникелевых сталей — 250-270 МПа.
Вероятность возникновения склонности к КР у сталей 12Х18Н(10-12)Т крайне мала, т. к. в области упругих напряжений скорость анодного процесса практически не изменяется,. Возникновение пластических течений в образцах приводит к ускорению анодного процесса и, как следствие, к появлению опасности повреждений по механизму КР.
С целью оценки влияния локальных напряжений, возникающих вокруг концентраторов напряжений, были испытаны образцы с заранее нанесенными дефектами. В этом случае изменение скорости анодного процесса, указывающего на опасность возникновения КР, наступает значительно раньше, до достижения напряжений, соизмеримых с пределом текучести металла (табл. 1.4.20). Это связано с возникновением зоны локальных напряжений в зоне надреза, величина которых значительно превышает средние значения.
Изменение электрохимических характеристик анодного процесса следует связывать с тем, что при превышении предела текучести начинается пластическое формоизменение образца с образованием линейных дефектов кристаллической решетки — дислокаций. Потенциальная энергия атомов в ядре таких дислокаций увеличивается в десятки и сотни раз по отношению к недеформированному состоянию, а возникающие ступеньки скольжения металла значительно увеличивают площадь поверхности контакта с коррозионной средой.
При переходе от кольцевых трубных образцов, испытанных при постоянной скорости деформации, к листовым прокатанным пробам, которые затем подвер-
Таблица 1.4.20
Скорость анодного процесса в гладких (1) и надрезанных (2) образцах
Растягивающие напряжения, МПа Скорость анодного процесса, мкА/см2
1 2
0 -1,4 -1,4
100 -1,4 -1,4
200 -1,4 -0,4
250 -1,3 +0,3
270 -0,1 +0,4
300 +0,2 +0,4
350 +0,4 —
гались коррозионному воздействию, было установлено, что после пластической деформации в 15 % скорость анодного процесса достигает 0,2 мкА/см2, после 40 % — 0,4 мкА/см2.
Следует также отметить, что растягивающие напряжения, резко изменяя электрохимию анодного процесса, не оказывают заметного воздействия на скорость катодного процесса ионизации кислорода в аустенитных сталях. Видимо, это следует объяснять тем, что под действием растягивающих напряжений пассивирующая пленка разрушается лишь на отдельных, незначительных по величине участках поверхности образца.
Влияние способа задания пластической деформации на скорость анодного процесса в аустенитных сталях
Процесс растворения металлов и сплавов в кислых средах принято описывать формулой Me - Me (z+) + Ze. Однако электролитическое окисление металлов может принимать иные формы, когда при окислении материала образуется стойкий в данном электролите оксид. В этом случае окисляемый материал становится пассивным, т. е. покрывается слоем пассивирующей пленки. Если для такого материала построить анодную поляризационную кривую, то она примет вид, показанный на рис. 1.4.12. Когда плотность тока, приложенного извне, превысит порог критической плотности тока, произойдет скачок потенциала, и кислород начнет выделяться на поверхности материала. При потенциале, превышающем точку А (рис. 1.4.12), металл начнет покрываться слоем оксидной пленки — пассивироваться. В интервале потенциалов между точками А и В гальва-ностатический анализ, используемый при оценке коррозионной стойкости сталей и сплавов, становится неприменим, и для анализа состояния материалов принято использовать потенциостатический метод, т. е. при анализе в этой области принято задавать не ток, а потенциал и наблюдать изменение плотности тока в образце.
На рис. 1.4.13 приведена схема потенциостата, используемая при проведении подобных исследований. С целью определения влияния непрерывной деформации на скорость анодного процесса на ряде аустенитизированных образцов в процессе снятия поляризационной кривой проводилось непрерывное деформирование со скоростью 1,5 мм/мин.
Рис. 1.4.12. Поляризационные кривые пассивирующегося образца
72
Новый справочник химика и технолога
Рис. 1.4.13. Принципиальная схема потенциостата:
1 — сопротивление (малое сопротивление); 2 — электрод сравнения;
3 — милливольтметр; 4 — образец; 5 — зонд; 6 — электрод
Возникающие на поверхности стали пассивирующие пленки не являются электрическими изоляторами. Такие пленки обладают некоторой электронной проводимостью, и поэтому в них может устанавливаться малая (порядка 1 В) разность потенциалов. Если к стали прикладывать более высокие анодные потенциалы, то на обращенной в сторону электролита стороне пленки начнутся анодные реакции, свойственные более благородным металлам. При этом произойдет значительное снижение потенциала пассивации, и пленка станет утолщаться — возникнет эффект анодирования. В этом случае оксид пассивного металла или сплава будет иметь постоянную толщину.
В присутствии агрессивных для аустенитных сталей соединений, и в первую очередь хлорид-ионов, поддержание пассивности в стали становится весьма сложной задачей — возникает опасность появления в материале склонности к коррозионному растрескиванию под напряжением. При возрастании концентрации хлоридов в электролите критическая плотность тока возрастает, потенциал первичной пассивности повышается, плотность тока в условиях пассивности возрастает, и интервал потенциалов пассивной облас
ти сокращается. Это явление следует объяснять особенностями хлорид-иона — высокой плотностью заряда на ионе и его легкой миграцией. В интервале потенциалов пассивности этот ион конкурирует с ионами молекулы окислителя и встраивается в пассивирующую пленку, что вызывает дефекты ее кристаллической решетки и, как следствие, снижение электросопротивления оксида.
Учитывая то, что главенствующую роль в формировании структуры пассивирующей пленки играет количественное содержание в электролите хлорид-иона, такие исследования необходимо проводить на электролитах различных (приближенных к реальным) составов. На рис. 1.4.14 приведены анодные поляризационные кривые сталей 12Х18Н(10-12)Т в двух видах электролитов. Первый электролит содержал 5,5 М H2SO4 и 0,5 М NaCl на 1 литр раствора, второй — 5,5 М H2SO4 и 1 М NaCl на 1 литр раствора. Испытания проводились в кипящих растворах, скорость изменения потенциала задавалась равной 0,75 В/мин. Из приведенных данных видно, что с ростом концентрации хлорид-иона скорость анодного процесса как в аустенитизированном, так и в деформированном состоянии резко возрастает. Напряженное состояние также приводит к повышению скорости анодного процесса и уменьшению величины интервала потенциалов пассивации, причем наиболее интенсивно влияние напряженного состояния проявляется, когда коррозионная среда и пластическая деформация воздействуют на сталь одновременно — кривая 3 (рис. 1.4.14, а). Причиной максимального роста плотности анодного тока в этом случае, видимо, следует считать суперпозицию двух одновременно протекающих процессов:
1) количественный и качественный рост ступеней скольжения в ходе пластического деформирования, когда возникают участки металла, не защищенные пасси-вационной пленкой; 2) резкий рост потенциальной энергии атомов в ядрах возникающих в процессе деформации дислокаций.
-0,5 О 0,5 1 1,5 2 -0,5 0 0,5 1 1,5 2
Потенциал пассивации, В 11отенциал пассивации, В
Рис. 1.4.14. Влияние напряженного состояния в аустенитных хромоникелевых сталях на скорость анодного процесса в электролитах: а) 5,5 М H2SO4 и 0,5 М NaCl; б) 5,5 М H2SO4 и 1 М NaCl; / — аустенитизированное состояние; 2 — предварительная холодная прокатка со степенью пластической деформации 10 %;
3 — непрерывное деформирование со скоростью 1,5 мм/мин
Электродные процессы
73
Примерное значение энергии деформации на один атомный промежуток в ядре дислокации достигает 5,9 • 1019 Дж. В то же время вне ядра дислокации изменение потенциальной энергии вследствие деформационных искажений не превышает 0,4-0,8 кДж/моль или около 0,63 • 10'21 Дж/ат., т. е. примерно в тысячу раз меньше. Плотность линий дислокаций в деформированной стали 12Х18Н10Т можно оценить примерно как 1012 см1. Условно принимая диаметр трубки дислокации в 10 межатомных расстояний, а число атомов на поверхности за 5 • 1015 наем2, легко определить, что поверхность, занимаемая дислоцированными атомами, может достигать примерно 1 % от общей площади образца.
Таким образом, в аустенитных хромоникелевых сталях возникает дополнительный термодинамический потенциал дислоцированных атомов, снижающий энергию активации анодного процесса и, как следствие, повышающий его скорость. Этот тезис подтверждается результатами следующего эксперимента. На образцы, помещенные в потенциостат, одноразово давалась нагрузка (ниже или выше предела текучести стали), а затем измерялось изменение скорости анодного процесса во времени. Установлено, что в активной зоне
(рис. 1.4.15, а) скорость анодного процесса прямо пропорциональна величине приложении) напряжения: чем выше действующее напряжение, тем выше скорость анодного процесса в активной области (области потенциалов до 0,25 В). В области потенциалов, соответствующих началу пассивационного процесса, тип деформационно-временной зависимости более неоднозначен. При приложении постоянной нагрузки скорость анодного процесса уменьшается с течением времени, проходит через минимальное значение и вновь возрастает, не достигая, однако, значений плотности тока для не-деформированного состояния (рис. 1.4.15, б). В области пассивации тип зависимости вновь изменяется (рис. 1.4.15, в). В ходе нагружения образцов в пассивной области наблюдается резкий рост значений плотности тока в первые минуты испытаний и падение до величин, близких к недеформированному состоянию, в последующие несколько минут. Это связано с влиянием энергии атомов, подвергающихся пластической деформации, на кинетику процесса пассивации. Увеличение энергии атомов эквивалентно увеличению потенциала и снижению величины энергии активации процесса. Чем выше приложенные напряжения, а, следовательно, величина пластической деформации, тем больше число
Рис. 1.4.15. Влияние времени выдержки в электролите, состоящем из 2,5 М серной кислоты и 0,5 М хлорида натрия, на скорость анодного процесса в аустенитной хромоникелевой стали 12Х18Н12Т, находящейся под действием постоянных напряжений
74
Новый справочник химика и технолога
дислокаций, больше ступеней скольжения — т. е. участков металла, не защищенного пассивирующей пленкой, больше пик скорости анодного процесса. С увеличением времени весь металл вновь покрывается пассивирующей пленкой, и скорость анодного процесса уменьшается. Это позволяет объяснить, почему предварительная пластическая деформация оказывает значительно меньшее влияние на ускорение анодного процесса в аустенитных сталях (о - 100 4-250 МПа, см. рис. 1.4.15, а). В случае предварительной пластической деформации значительно ослаблен фактор «чистого», т. е. не покрытого пассивирующей пленкой металла, участки которого возникают при непосредственном воздействии деформации образца в электролите. За время, прошедшее с момента деформации до момента испытаний, эти участки успевают закрыться слоем пассивирующих оксидов и не участвуют в процессе анодного растворения.
В зоне, близкой к потенциалу пробоя (рис. 1.4.15, г), отмечается следующая зависимость. В случае, если приложенное напряжение выше предела текучести, то чем оно больше, тем больше степень пластической деформации, которая смещает потенциал пробоя в сторону меньших значений. Таким образом, скорость анодного процесса увеличивается. Если приложенное напряжение ниже предела текучести, влияние напряжения на скорость анодного процесса не отмечается.
Особое внимание следует обратить на роль никеля в изменении скорости анодного процесса в хромоникелевых сталях. При повышении его концентрации даже внутри марочного диапазона легирования стали 12Х18Н12Т (с 11 до 13 %) при одном и том же растягивающем напряжениии рост скорости анодного процесса постепенно снижается. Еще заметнее влияние никеля проявляется при значительном повышении его концентрации в хромоникелевых аустенитных сталях (рис. 1.4.16). Благоприятную роль никеля на формирование стойкости стали против коррозионного растрескивания можно объяснить тем, что никель, практически
не изменяя механических свойств хромоникелевых сталей, изменяет механизм их пластического деформирования, облегчая в них поперечное скольжение. Тем самым часть вновь образованных при пластическом деформировании дислокаций уходит в другие плоскости скольжения, уменьшая количество атомов с повышенной потенциальной энергией в поверхности образца.
Влияние скорости нагружения на стойкость к коррозионному растрескиванию хромоникелевых аустенитных сталей
К числу факторов, влияющих на стабильность поведения трещин при растрескивании в коррозионной среде в ходе эксплуатации и испытаний, относится скорость деформирования материала. Особо важным становится влияние этого фактора, когда скорость деформирования крайне мала и составляет десятые и сотые доли процента в час. В этом случае возникновение стресс-коррозион-ных трещин может смещаться в зону пластической деформации значительно меньшей, чем принято считать опасной для данной группы материалов, что и приводит к неожиданным повреждениям конструкций.
Испытания с целью определения влияния скорости деформирования на механические свойства сталей или испытания на длительную пластичность достаточно широко используются при аттестации материалов энергомашиностроения. В данном случае к стандартным испытаниям длительной пластичности был добавлен дополнительный фактор — в момент достижения образцами предела текучести, который определяется как перегиб на диаграмме растяжения при достижении площадки текучести, образец помещался в ванну с коррозионной средой: кипящим водным раствором 2,5 М H2SO4 и 0,5 М NaCl. При испытаниях были использованы десятикратные разрывные образцы с длиной рабочей части 60 и диаметром 6 мм. Скорость деформации изменилась от 314 % в час до 0,01 % в час. Результаты испытаний приведены на рис. 1.4.17 и в табл. 1.4.21.
Рис. 1.4.16. Влияние никеля на скорость анодного процесса в аустенитных хромоникелевых сталях при однократном растяжении (напряжения равны 350 МПа), время нахождения в среде — 2,5 мин, соответствующее минимальной скорости процесса (см. рис. 1.4.17)
260-1
230 т-0
100 200 300
Скорость деформирования:
-4-1-10% -ж-2-1%
-л- 3-0,1 % 4-0,01 %
—।
400
Рис. 1.4.17. Влияние скорости деформирования и времени воздействия коррозионной среды при определенном уровне деформации образцов аустенитной хромоникелевой стали 12Х18Н12Т на поддерживающее деформацию усилие
Электродные процессы
75
Таблица 1.4.21
Механические свойства стали 12Х18Н12Т при испытаниях в воздушной и коррозионной среде
Скорость Испытания в воздушной среде Испытания в коррозионной среде
деформирования, ст0,2 5К £5 ст0,2 £5
% в час МПа % МПа %
314 265 585 1170 41 265 585 1170 41
100 265 585 1170 41 265 585 1165 40
10 255 575 1140 39 255 500 865 29
1 250 565 1100 37 250 — 610 3,7
0,1 245 565 1070 34 245 — 505 1,9
0,01 242 555 995 29 242 — 415 1,0
В зависимости от скорости деформирования в коррозионной среде результаты, приведенные в табл. 1.4.21, можно разделить на две группы:
1. Образцы, испытанные при высоких скоростях нагружения (314 и 100 % в час), которые в коррозионной среде не изменяют свойств, полученных при испытаниях на воздухе. Эти изменения могут проявляться крайне незначительно, т. к. разрушение образца протекает за время меньшее, чем необходимо для возникновения в образце трещины стресс-коррозии.
2. При малых скоростях деформирования влияние коррозионной среды на механические свойства стали становится превалирующим.
Полученные результаты резко отличаются от данных по свойствам образцов, испытанных в воздушной среде. Такое влияние коррозионной среды может быть объяснено только возникновением и ростом в образце коррозионной трещины, которая принципиально изменяет всю картину разрушения материала.
На рис. 1.4.17 приведены диаграммы растяжения образцов в координатах напряжение—время, которые позволяют выделить два участка кривой. На первом — требуемый для деформирования образца уровень напряжений остается неизменным, на втором — уровень напряжений снижается, и тем значительнее, чем больше время нахождения образца в коррозионной среде, что может быть объяснено только уменьшением реальной площади поперечного сечения образца, т. е. возникновением и развитием в нем коррозионного дефекта. Точка перегиба — время инкубационного периода коррозионного процесса, т. е. время, необходимое для зарождения в образце трещины КР. Со снижением скорости деформации время инкубационного периода увеличивается, хотя и не так значительно, как снижается скорость деформирования, а степень деформации, при которой возникает трещина КР, уменьшается (табл. 1.4.22).
Таким образом, вид разрушения образца при одновременном воздействии пластической деформации и коррозионной среды напрямую будет зависеть от скорости деформирования. Если разрушение протекает ускоренно (за время, меньшее инкубационного периода
зарождения трещины КР), роль среды крайне мала. Однако она повышается с уменьшением скорости деформирования; при малых скоростях (0,1 и 0,01 % в час) , когда трещина КР развивается в практически аустенитизированной матрице, ее роль становится главенствующей, а возникающие напряжения локализуются только в зоне трещины.
Таблица 1.4.22 Время инкубационного периода и степень
пластической деформации образца в момент образования стресс-коррозионной трещины
Скорость деформирования, % в час 10 1 о,1 0,01
Время инкубационного периода — точка А, мин 132 198 240 252
Степень пластической деформации, % 22 з,з 0,4 0,04
Увеличение времени инкубационного периода до зарождения трещины КР в аустенитных хромоникелевых сталях может быть достигнуто путем дополнительной поверхностной обработки материала, создающей в его поверхности сжимающие напряжения, например при обкатке роликом, дробеструйной или пескоструйной обработке. На рис. 1.4.18 приведены данные об изменении времени до зарождения трещины КР в образцах стали 12Х18Н12Т, деформируемой со скоростью 1 % в час. Как видно из рис. 1.4.18, максимальное время инкубационного периода отмечается у аустенитизированного образца после его пескоструйной обработки песком с максимальным диаметром частиц не более 0,1 мм. Даже нанесение на рабочую часть такого образца (до его пескоструйной обработки) кольцевого надреза глубиной в 1 мм и радиусом закругления при вершине в 0,2 мм, хотя и приводит к снижению инкубационного периода по отношению к гладкому образцу, но все же остается выше, чем у гладкого аустенитизированного образца без пескоструйной обработки. Минимальное время до зарождения трещины КР отмечается у образца с надрезом, подвергающегося непрерывному деформированию с напряжениями выше
76
Новый справочник химика и технолога
предела текучести стали — 300 МПа (рис. 1.4.18). Скорость роста коррозионной трещины зависит также от способа обработки образца и степени его пластического деформирования. На рис. 1.4.19 приведены данные о влиянии состояния образца на скорость роста стресс-коррозионной трещины в образцах стали 12Х18Н12Т. Максимальная скорость роста трещины отмечается при непрерывном пластическом деформировании образца, т. е. когда растягивающие напряжения в образце превышают его предел текучести. В случае аустенитизированного образца и образца после аустенитизации и пескоструйной обработки скорость роста трещины в аустенитизированном образце вначале оказывается большей,
а затем их величины выравниваются. Выравнивание скоростей роста трещин наступает в тот момент, когда трещиной преодолен слой металла со сжимающими напряжениями, вызванными пескоструйной обработкой, и далее ее распространение протекает в единой для обоих образцов аустенитизированной матрице (рис. 1.4.19, а). Повышение степени предварительной пластической деформации в образцах аустенитных сталей также приводит к повышению скорости роста стресс-коррозионной трещины (рис. 1.4.19, б), хотя и в этом случае пескоструйная обработка приводит к снижению скорости ее роста на первом этапе, когда трещина распространяется в поверхностном слое металла.
на «время до зарождения» (инкубационный период) трещины коррозионного растрескивания
/—Аустенизация + пескоструйная обработка
2—Аустенизация + пескоструйная обработка + надрез
3 —Аустенизация
4—Аустенизация + надрез
5—Аустенизация + прокат е = 10 %
6—Аустенизация + прокат g = 10 % + надрез
/—Прокат е = 1,0 % 2—Прокат £ = 2,5 % 3—Прокат е = 5,0%
4—Прокат е = 10% 5 — Прокат е - 20 % 6 — Прокат е = 30 %
Рис. 1.4.19. Влияние состояния поверхности (а) и степени пластической деформации, наведенной холодной прокаткой (б), на скорость роста трещины коррозионного растрескивания в аустенитной хромоникелевой стали 12Х18Н12Т
Электродные процессы
77
1.4.3.5. Коррозионное растрескивание в высокопрочных сталях
В конце XX века резко возросли требования к надежности и долговечности высокопрочных сталей, прочность которых ов > 1500 МПа обеспечивается за счет упрочняющей термической обработки. Это связано с их широким использованием в газо- и нефтепереработке, химической промышленности, авиации и ядер-ной физике. К основным недостаткам таких сталей относится их чувствительность к коррозионному растрескиванию.
Коррозионное растрескивание высокопрочных сталей (предел прочности более 1500 МПа) наблюдается в большинстве сред, включая влажный H2S, аэрированные растворы NaCl, Na2SO4, NaNO3, растворы аммиака, морскую и промышленную атмосферу. Предлагается два основных механизма для объяснения коррозионного растрескивания высокопрочных сталей: 1) механизм, предполагающий существование активных участков, при котором коррозионные реакции приводят к высокой скорости растворения вдоль этих участков в процессе коррозионного растрескивания (электрохимическая теория); 2) теория водородного растрескивания, при котором коррозионные реакции способствуют внедрению в сталь водорода с последующим ослаблением зернограничной когезии материала за счет образования его сегрегаций в границах зерен.
Водородное охрупчивание высокопрочных сталей включает в себя зарождение трещины и ее слабый рост — основные фазы разрушения. Как правило, чувствительность сталей к водородному охрупчиванию увеличивается при повышении предела текучести материала. Пластичность охрупченного материала уменьшается до минимума и практически не изменяется при повышении температуры. Однако теория водородного охрупчивания не позволяет достаточно достоверно связать влияние температуры и скорости деформации материала. Среди наиболее распространенных объяснений такого процесса можно выделить следующие:
- образование микротрещины коррозионного растрескивания происходит в результате скопления дислокаций на границах зерен, а адсорбция водорода на внутренних поверхностях этих трещин понижает поверхностную энергию и напряжения, необходимые для ее распространения;
- активизированная напряжениями диффузия водорода в области с высокими локальными напряжениями приводит к понижению межзеренной когезии или понижению разрушающих напряжений в сталях. В этом случае инкубационный период зарождения стресс-коррозионной трещины определяется активизированной напряжениями диффузией водорода в области с максимальными объемными напряжениями. Длительность инкубационного периода зависит от скорости достижения в стали критической суперпозиции концентрации водорода и напряжений. Слабый рост трещины на первом этапе его развития связывается с диффузией, инициированной напряжениями непосредственно в области фронта трещины, где наблюдаются максимальные напряжения. При достижении критической концентрации водорода начинается интенсивный рост трещины, которая будет проходить через области, обогащенные водородом.
Наряду с теорией, объясняющей коррозионное охрупчивание высокопрочных сталей только с точки зрения водородного охрупчивания, существует теория, где используются элементы электрохимической теории растрескивания. Согласно этой теории зависимость электрохимической поляризации от времени до разрушения материала может являться критерием того, происходит ли разрушение за счет водородного охрупчивания или за счет растворения активных участков. На рис. 1.4.20 представлены различные типы кривых (в осях «время до растрескивания—поляризация»), где на основании этих представлений указаны области, в которых разрушение протекает или по механизму водородного охрупчивания, или за счет растворения актив-
Рис. 1.4.20. Влияние анодной (А) и катодной (С) поляризации на «время до разрушения» высокопрочных сталей АУК — активные участки коррозии, ВР — время до разрушения, Л и С — анодная и катодная поляризация соответственно, Н — водородное охрупчивание, О — образцы не разрушаются
78
Новый справочник химика и технолога
1.4.3.6. Коррозионное растрескивание цветных сплавов
Коррозионное растрескивание титановых сплавов
Число разрушений конструкций из титана и его сплавов, произошедших по вине коррозионного растрескивания, к настоящему времени достаточно мало. Однако в ряде сред и условий эксплуатации титановые сплавы оказываются склонны к коррозионному растрескиванию. К основным механизмам коррозионного растрескивания титановых сплавов относятся солевое высокотемпературное растрескивание и растрескивание при комнатной температуре. Растрескивание при комнатной температуре в основном происходит в водных и метанольных средах, содержащих хлориды; при прямом контакте сплава с рядом жидких и твердых металлов, газов; в ряде других сред, например, тетраоксиде диазота — N2O4, дымящей азотной кислоте и т. п. Солевое растрескивание происходит под действием внешних или внутренних напряжений при непосредственном контакте материала с твердыми хлоридами в присутствии кислорода и водяного пара при температурах выше 250 °C. Такое растрескивание носит преимущественно межкристаллитный характер. В зависимости от степени коррозионного воздействия на титановые сплавы, хлориды по степени интенсивности воздействия можно распределить следующим образом:
MgCl2 < SrCl2 < СаС12< КС1 < ВаС12 < NaCl < LiCl
Бромиды и иодиды этих же металлов также могут вызывать коррозионное растрескивание титановых сплавов; фториды не оказывают значительного воздействия на снижение сопротивляемости титановых сплавов коррозионному растрескиванию.
По степени подверженности коррозионному растрескиванию титановые сплавы можно разделить на три основные группы:
- сплавы, высокочувствительные к коррозионному растрескиванию (химические составы, масс. %): Ti—5 Al—2,5Sn; Ti—12Zr—7А1; Ti—5 Al—5Sn—5Zr; Ti—8A1— IMo— IV; Ti—8Mn;
- сплавы средней чувствительности:
Ti—5Al—5Sn—5Zr— IMo— 1V; Ti—6A1^V;
Ti—6A1—4 V—2Sn; Ti—13 V—1 ICr—3A1;
- сплавы с высоким сопротивлением к коррозионному растрескиванию:
Ti—4А1—ЗМо—IV; Ti—HSn—5Zr—2,25A1— IMo—0,025Si; Ti—4Mo—4Zr—2A1.
К элементам, резко понижающим стойкость сплавов против коррозионного растрескивания, относятся алюминий, олово, медь, ванадий, хром, марганец, железо и никель; к элементам, слабо влияющим на понижение коррозионной стойкости, — цирконий, тантал и молибден. Сплавы со структурой а-титана более чувствительны к коррозионному растрескиванию, чем сплавы с 0-титаном. Термическая обработка приводит к некоторому повышению чувствительности а-сплавов к корро
зионному растрескиванию и изменению характера разрушения; холодная пластическая деформация, наоборот, снижает чувствительность сплавов к коррозионной повреждаемости.
Высокотемпературное растрескивание титановых сплавов может быть заторможено или даже предотвращено за счет наклепа поверхности изделия, например методами дробеструйной или пескоструйной обработки. Повысить сопротивляемость можно также нанесением на изделия гальванических защитных покрытий (алюминия и цинка или никеля).
Многие титановые сплавы подвержены коррозионному растрескиванию при комнатной температуре в водных или метанольных средах, содержащих хлориды.
В нейтральных (водных) растворах хлоридов титановые сплавы не подвергаются коррозии при комнатной температуре, если поверхностная защитная оксидная пленка материала не нарушена. Если же нарушение оксидной пленки происходит, то у сплавов, склонных к коррозионному растрескиванию, следует ожидать развития коррозионного дефекта. Впервые коррозионное растрескивание в водных растворах было изучено на примере сплава Ti—7Al—2Nb, испытанного в растворе поваренной соли на образцах с предварительно нанесенной усталостной трещиной при консольном нагружении в условиях плоской деформации. Испытания были проведены при напряжениях ниже К^с- Развитие трещины в образцах продолжалось до тех пор, пока задаваемый коэффициент интенсивности напряжений К(а. • о с,/2) не достигнет К\с. График зависимости коэффициента К от времени до разрушения Ткр приведен на рис. 1.4.21. Значение К, ниже которого растрескивания не происходит, обозначено на рис. 1.4.21 как параметр /fiscc- Отношение K\JK\SCc показывает чувствительность сплавов к коррозионному растрескиванию. Для сплавов, чувствительных к растрескиванию, эта величина находится на уровне 0,2.
Рис. 1.4.21. Зависимость коэффициента интенсивности напряжений от «времени до разрушения» для титановых сплавов, чувствительных к коррозионному растрескиванию при испытании в нейтральном водном растворе в условиях плоской деформации
Коррозионное растрескивание в метанольных средах также типично для титановых сплавов. В а-сплавах развитие коррозионной трещины происходит в широком диапазоне скоростей развития трещины. В присут
Электродные процессы
79
ствии НС1 к исходному механизму разрушения добавляется эффект межкристаллитного разрушения сплава за счет диффузионного насыщения его атомами водорода, Добавление воды приводит к торможению коррозионного растрескивания, причем ее концентрация, необходимая для эффективного торможения процесса, зависит от содержания хлоридов в растворе.
Высокочистый тетраоксид диазота N2O4 вызывает растрескивание сосудов высокого давления, изготовленных из сплава Ti—6А1—4V. Растрескивание может быть предотвращено путем добавок NO или Н2О. Величина K\scc сплава понижается при повышении температуры испытания. Считается, что коррозионное растрескивание титановых сплавов в тетраоксиде диазота связано с разрушением оксидной пленки TiO(NO3)2, возникающей в процессе контакта тетраоксид диазота—сплав и не обладающей защитными свойствами:
Ti + 2N2O4 TiO(NO3)2 + NO + NO2 + e ',
2TiO(NO3)2-----> 2TiO2 + 2N2O4 + O2
Промышленные титановые сплавы подвержены коррозионному растрескиванию в дымящейся азотной кислоте, содержащей 20%NO2. При исключении NO2 коррозионное растрескивание наблюдается только у некоторых сплавов, а добавление в эту среду 2% Н2О устраняет коррозионное растрескивание полностью.
Коррозионное растрескивание магниевых сплавов
Коррозионное растрескивание магниевых сплавов происходит в водных средах при комнатной температуре. В основном оно наблюдается в деформируемых сплавах. Данных о коррозионном растрескивании литейных сплавов крайне мало, и они носят достаточно противоречивый характер. Основным легирующим элементом, определяющим склонность магниевых сплавов к коррозионному растрескиванию, является алюминий. Основным деформационным механизмом, ответственным за коррозионное растрескивание магниевых сплавов, является действие внутренних остаточных напряжений в материале. В качестве примера, подтверждающего объективность этих тезисов, можно рассмотреть проблему коррозионного растрескивания промышленных сплавов системы Mg—Al—Zn. Склонность этих сплавов к стресс-коррозии наблюдается при содержании в них алюминия в диапазоне концентраций 3-10 % и отношении Al / Zn > 2. Чувствительность к коррозионному растрескиванию увеличивается с повышением в сплаве содержания алюминия. Введение в эти сплавы железа или меди еще более повышает склонность сплавов к стресс-коррозии. Магниевые сплавы, не содержащие алюминия, по-видимому, не склонны к коррозионному растрескиванию в большинстве коррозионноактивных сред. Однако в ряде безалюминиевых сплавов склонность к коррозионному растрескиванию может наблюдаться в определенных средах. Так, сплавы Mg—Мп, легированные Се (при его содержании не ме
нее 0,5 %), не чувствительны к коррозионному растрескиванию в растворах (NaCI + К2Сг2О7), однако они растрескиваются в средах с KHF2. Магний-литиевые сплавы, имеющие объемно-центрированную кристаллическую решетку, разрушаются во влажном воздухе; сплавы магния с цинком и цирконием растрескиваются в дистиллированной воде и KHF2.
В качестве основного метода предохранения магниевых сплавов от коррозионного растрескивания можно предложить виды термической или термомеханической обработки, снижающие уровень остаточных напряжений в сплавах. Например, для сплавов Mg—6,5Al—IZn—0,2Мп рекомендуется отжиг для снятия напряжений при температуре 125 °C в течение 8 ч. Такой же отжиг рекомендуются при обработке сварных соединений магниевых сплавов. Отмечается положительный эффект от дробеструйной и пескоструйной обработки, обкатки поверхности деталей роликом и шлифования, применения защитных покрытий, например оксидирования поверхностей деталей.
Коррозионное растрескивание алюминиевых сплавов
Коррозионное растрескивание в деталях и изделиях, изготовленных из чистого алюминия, не наблюдается. Также крайне редко отмечаются случаи коррозионного растрескивания литейных алюминиевых сплавов. Однако в ряде деформируемых алюминиевых сплавов высокой прочности за счет изменения их химического состава, холодной деформации и термической обработки возникают повреждения, связанные со стресс-корро-зией. К таким материалам относятся, в первую очередь, сплавы на основе систем А1—Mg, Al—Си. Системы сплавов Al—Ag, Al—Cu—Mg, Al—Mg—Si, Al—Zn, Al—Zn—Mg—Cu также подвержены коррозионному растрескиванию, однако в меньшей степени, чем системы алюминий—магний или алюминий—медь. Следует отметить, что во всех этих сплавах склонность к коррозионному растрескиванию повышается с повышением концентрации легирующих элементов. Введение в сплавы алюминия, хрома, марганца, циркония, титана, ванадия, никеля и лития может понижать склонность алюминиевых сплавов к коррозионному растрескиванию. Большинство разрушений изделий из алюминиевых сплавов, связанных с коррозионным растрескиванием, происходит в водных средах, однако были отмечены случаи коррозионного растрескивания в тетраоксиде диазота (N2O4), минеральных маслах, спиртах, ртути, гексане.
Меры предохранения алюминиевых сплавов от коррозионного растрескивания такие же, как и для сплавов магния — рациональный выбор формы изделия и его термической обработки, наклеп поверхности изделия, защита лакокрасочными и гальваническими противокоррозионными покрытиями. Также применяется металлизация поверхности или одно- или двухсторонняя плакировка деталей. Возможна катодная защита деталей, хотя ее применение весьма ограниченно.
80
Новый справочник химика и технолога
1.4.4. Межкристаллитная коррозия в металлах и сплавах
Межкристаллитная коррозия является одним из наиболее опасных видов местной коррозии (рис. 1.4.1, к), приводящей к избирательному разрушению границ зерен, что сопровождается потерей прочности и пластичности металлов и сплавов. Опасность заключается в том, что зачастую изменений во внешнем виде изделий, поврежденных межкристаллитной коррозией, не происходит. Коррозия этого вида наблюдается у многих материалов — хромистых и хромоникелевых нержавеющих сталей, никелевых и алюминиевых сплавов и т. п.
Причиной межкристаллитной коррозии чаще всего являются структурные изменения на границах зерен, которые превращают зернограничный твердый раствор в малополяризующийся анод, подвергающийся усиленному коррозионному разрушению. Сложность этого процесса и зависимость его от многих факторов затрудняет описание межкристаллитной коррозии даже для отдельной группы сплавов.
1.4.4.1. Влияние основных легирующих и примесных элементов на стойкость к межкристаллитной коррозии (МКК) аустенитных хромоникелевых сталей
Аустенитные хромоникелевые стали являются одним из основных материалов оборудования нефтехимической, химической и пищевой промышленности, они широко применяются в низкотемпературной технике и физике высоких энергий. Одним из основных факторов, снижающих надежность и долговечность такого оборудования, являются коррозионные повреждения, развивающиеся по механизму межкристаллитной коррозии. Межкристаллитным коррозионным повреждениям подвержено оборудование, в эксплуатационный цикл которого включаются высокотемпературные технологические разогревы в интервале температур 773-973 К, ремонтные сварочные операции. Этот температурный интервал нагрева характеризуется возникновением и ростом карбидной сетки Ме23С6 в границах зерен стали 12Х18Н12Т (рис. 1.4.22), определяющей возникновение склонности к МКК.
Возникновение склонности к МКК и скорость роста коррозионных трещин у хромистых и хромоникелевых сталей следует связывать с образованием в границах зерен карбидной сетки Ме2зСб—(Cr,Fe)23C6 и обезлеги-рованием по хрому приграничных областей твердого раствора, по которым развивается коррозионная трещина. Скорость роста карбидных частиц, в свою очередь, связана с температурой разогрева и химическим составом стали, в основном с содержанием углерода, никеля, кремния и др. примесей.
При фиксированном содержании углерода в хромоникелевых аустенитных сталях главную роль в изменении скорости диффузии атомов хрома и углерода в границы зерен играет никель, ускоряющий и облегчающий процесс карбидообразования. Кроме никеля, на свойства приграничных зон оказывают влияние атомы кремния, замещающие в твердом растворе уходящие во вновь образуемую карбидную фазу атомы хрома, а также фосфор, сегрегирующий в границах зерен при высокотемпературных нагревах. В табл. 1.4.23 приведены химические составы опытных плавок стали 12Х18Н12Т с различным содержанием никеля, кремния и фосфора.
Рис. 1.4.22. Карбидные включения в границах зерен стали 12Х18Н12Т после отпуска при температуре 873 К в течение 3-х часов. х200
Таблица 1.4.23
Химический состав (масс. %) сплавов на основе стали 12Х18Н12Т
Плавка № С Сг Si Ni Mn Р S Ti
1 0,11 18,1 0,09 Н,1 1,05 0,009 0,011 0,30
2 0,10 17,9 0,11 12,0 1,09 0,011 0,012 0,23
3 0,12 17,9 0,12 10,2 1,02 0,010 0,014 0,27
4 0,11 18,1 0,09 11,1 1,05 0,021 0,011 0,30
5 0,11 18,1 0,37 Н,1 1,05 0,021 0,011 0,30
6 0,11 18,1 0,93 П,1 1,05 0,021 0,011 0,30
7 0,11 18,1 1,03 11,1 1,05 0,021 0,011 0,30
8 0,11 18,1 0,64 11,1 1,05 0,009 0,011 0,30
9 0,11 18,1 0,64 11,1 1,05 0,012 0,011 0,30
10 0,11 18,1 0,64 П,1 1,05 0,030 0,011 0,30
Электродные процессы
81
Для получения наиболее достоверных данных о влиянии этих элементов был применен метод последовательной дошихтовки сплава, когда плавки №№ 4-10 были получены путем дополнительного легирования тем или иным химическим элементом плавки 1. Испытания коррозионной стойкости опытных плавок проводили по методу АМУ (ГОСТ 6032-84) после провоцирующих нагревов, имитирующих высокотемпературные технологические разогревы металла оборудования в диапазоне от 723 до 1023 К во временном интервале от 0,5 до 100 часов.
Влияние никеля и кремния. Данные о влиянии никеля и кремния на стойкость хромоникелевых аустенитных сталей к межкристаллитной коррозии приведены на рис. 1.4.23. и 1.4.24. Установлено, что температура минимальной устойчивости сталей к МКК не зависит от содержания никеля и для плавок с 0,64 % кремния составляет 923 К (рис. 1.4.24). При изменении со
„ ог, Плавка 1 (никель — 11 масс. %)
О - не склонна к МКК
• - склонна к МКК
Рис. 1.4.23. Влияние никеля на температурно-временные области склонности к межкристаллитной коррозии в стали 12Х18Н12Т
держания кремния от 0,09 до 1,03 % эта температура плавно возрастает от 873 до 973 К (рис. 1.4.24).
Анализ результатов исследования влияния никеля на коррозионную стойкость стали 12Х18Н12Т к МКК показал, что с ростом его концентрации (плавка 1) до 13 масс. % (плавка 3) температурно-временная зона склонности к МКК увеличивается. С-образные кривые областей склонности хромоникелевых сталей к МКК с повышением в них содержания никеля смещаются влево в сторону меньших времен выдержек — от 4 часов у плавки 1 до 3 часов у плавки 3 (рис. 1.4.23). Причиной такого воздействия никеля является его влияние на увеличение скорости диффузионных процессов в хромоникелевых аустенитных сталях.
Если влияние никеля на коррозионную стойкость хромоникелевых сталей явно отрицательно, то воздействие кремния носит далеко не однозначный характер. Кремний способствует повышению пассивации хромоникелевых сталей наряду с такими металлами, как молибден, титан, тантал и алюминий. В хромоникелевых сталях кремний образует зернограничные плены — сегрегации, наличие которых подтверждается как замерами микротвердости по телу зерна (рис. 1.4.25), так и методом эмиссионного спектрального микроанализа (табл. 1.4.24). В объемах зерна, удаленных от границы более чем на Юмкм (при среднем размере зерен в исследованных сталях 60-80 мкм), микротвердость твердого раствора практически неизменна. При удалении зерна от границы на расстояние менее 10 мкм микротвердость резко возрастает, причем суммарное повышение микротвердости зависит от концентрации кремния в стали (рис. 1.4.25). Результатами эмиссионного спектрального анализа (табл. 1.4.24) было подтверждено, что ответственность за повышение микротвердости несут неравновесные (растянутые на значительные расстояния в глубь зерна) сегрегации кремния.
Кремний образует в границах зерен неравновесные сегрегации, толщина слоя которых соизмерима с размерами карбидных включений. Эти сегрегации образуются в ходе высокотемпературных нагревов под аустенитизацию стали, а затем в процессе провоцирующих нагревов стремятся принять более равновесную (т. е. сосредоточенную в тонком, близком к моноатомному, слое границы) форму. В ходе провоцирующих нагревов наблюдается ускоренное перераспределение атомов кремния в сторону межзеренной границы. Было обнаружено, что при нагревах до температур 873-923 К это перераспределение носит равномерный характер, а при более высоких температурах
82
Новый справочник химика и технолога
происходит постепенное расслоение сегрегации с образованием в границах зерен зон с пониженной и повышенной концентрацией кремния (табл. 1.4.25), т. е. в границах зерен возникают кластеры предвыделений кремнистых фаз. Именно этой особенностью поведения кремния в границах зерен объясняется его неоднозначное влияние на стойкость стали 12Х18Н12Т к МКК.
При температурах нагревов ниже 873-923 К кремний насыщает обедненный по хрому твердый раствор границ зерен, повышая его пассивацию, а следовательно, и стойкость к МКК. При более высоких температурах нагревов в границах зерен возникают кластеры предвыделений кремнистых фаз, образуются участки зернограничного твердого раствора, обедненного по хрому и кремнию, стойкость которого к МКК резко понижается. Так, содержание хрома в твердом растворе приграничных зон зерен стали 12Х18Н12Т после нагрева при 973 К в течение 5 часов для плавки, содержащей 0,37 % кремния, составило 11,8 масс. %, а содержание кремния в обедненных по этому элементу зонах — 1,41 масс. %, в обогащенных — 5,46 масс. %
(табл. 1.4.25).
Рис. 1.4.24. Влияние кремния на температуру минимальной устойчивости хромоникелевых сталей к межкристаллитной коррозии
Рис. 1.4.25. Изменение микротвердости аустенита в теле зерна стали 12Х18Н12Т в аустенитизированном состоянии в зависимости от концентрации кремния
Следует подчеркнуть, что с повышением концентрации кремния в хромоникелевых сталях уход хрома из твердого раствора в карбидную фазу ускоряется. Это связано с повышением конкуренции за места в зернограничном твердом растворе между этими элементами. Так, если у сплава 4 (0,09 % Si) после провоцирующего нагрева при 923 К в течение 5 часов содержание хрома в твердом растворе границ зерен составило 13,1 %, у сплава 5 (0,37 % Si) — 12,2 %, то у сплава 6 (0,93 % Si) — 11,7 и у сплава 7 (1,03% Si) — 11,4 масс. % хрома. В плавках 6 и 7 содержание хрома в границах зерен становится менее 12 масс. %, что сразу же отражается на величине времени минимальной устойчивости стали к МКК (т кр), приведенной на рис. 1.4.26.
Таким образом, кремний, конкурируя с хромом за места в границах зерен, уменьшает приток атомов хрома взамен ушедших в карбидную фазу. Одновременно с этим при температурах выше 923 К изменяется характер распределения атомов кремния в границах зерен. Если до 873 К это распределение носит равномерный характер и оказывает благоприятное воздействие на стойкость стали к МКК, то при более высокой температуре концентрационное расслоение зернограничных зон на низко- и высококремнистые приводит к снижению стойкости стали к МКК.
Таблица 1.4.24
Содержание элементов в границах и объемах зерна стали 12Х18Н12Т в аустенитизированном состоянии
Плавка № (Si, масс. %) Элемент Среднее содержание элемента, масс. %, в слое толщиной 100 нм на расстоянии от поверхности (границы) зерна
0-0,1 мкм 0,1-0,2 мкм 10,0-10,1 мкм
4 (0,09) Сг 17,9 18,0 17,3
Ni 11,2 И,4 12,0
С 0,12 0,11 0,12
Si 1,14 1,13 0,36
5 (0,37) Сг 18,1 18,0 17,3
Ni Н,7 11,7 11,8
С 0,11 0,11 0,12
Si 2,91 2,73 0,52
7(1,03) Сг 17,9 18,2 18,0
Ni 11,6 11,8 11,7
С 0,11 0,12 0,12
Si 4,70 4,21 1,94
Таблица 1.4.25
Содержание хрома и кремния в твердом растворе приграничных зон стали 12Х18Н12Т (0,37 масс. % Si)
Температура провоцирую-щего нагрева, К Распределение элементов, масс. %, в слое толщиной 100 нм после провоцирующего нагрева при 973 К в течение времени
1 ч 5 ч
Сг Si Сг Si
823 18,1 2,91 16,1 3,22
873 18,0 2,96 14,2 3,39
923 17,0 2,99 12,2 3,68/3,41
973 16,7 3,03 11,8 5,46/1,41
Электродные процессы
83
Рис. 1.4.26. Влияние концентрации кремния на время минимальной устойчивости стали 12Х18Н12Т в зависимости от температуры провоцирующего нагрева
Влияние фосфора. Фосфор напрямую не оказывает воздействия на вид С-образных кривых температурновременных зон склонности стали 12Х18Н12Т к МКК,
хотя его роль в изменении надежности металла оборудования, изготовленного из аустенитных хромоникелевых сталей, достаточно велика.
Фосфор во время нагревов под аустенитизацию образует сегрегации в границах зерен сталей. Концентрация атомов фосфора в этих зонах зернограничного твердого раствора резко возрастает в процессе карби-дообразования. Карбидная фаза, обладающая жесткой кристаллической решеткой химического соединения, вытесняет фосфор из объема, который она занимает, во вновь образующиеся межфазные границы. При этом содержание фосфора в границах зерен может возрастать в десятки и сотни раз (рис. 1.4.27). Когезия границ зерен с повышенным содержанием атомов фосфора резко уменьшается, тем самым снижается механическая прочность границ, облегчается распространение трещины по границам зерен. Вблизи равновесного состояния температура области максимального роста сегрегации атомов фосфора совпадает с температурой образования непрерывной карбидной сетки Ме23С6 в границах зерен, т. е. с температурой состояния минимальной устойчивости к МКК.
Рис. 1.4.27. Изменение содержания фосфора в слое толщиной 100 нм от границы зерна в зависимости от интенсивности провоцирующего нагрева: 1 — 973 К, 2 — 923 К, 3 — 773 К; Сгр — концентрация фосфора в граничном слое, сс? — концентрация фосфора в зерне
84
Новый справочник химика и технолога
На рис. 1.4.28 приведены данные о влиянии никеля, кремния и фосфора на скорость проникновения коррозионного дефекта в глубь образца из стали 12Х18Н12Т. Установлено, что изменение концентрации никеля в исследуемых пределах практически не изменяет скорости коррозионного процесса. В то же время кремний и фосфор резко повышают скорость движения трещины МКК в глубь металла.
Скорость проникновения коррозионного дефекта, мкм/ч
Рис. 1.4.28. Влияние никеля, кремния и фосфора на скорость роста коррозионного дефекта в границах зерен стали 12Х18Н12Т в кипящем растворе 25% HNO3
1.4.4.2. Влияние пластической деформации на стойкость стали 12Х18Н12Т к МКК
Возникновение зон локальной пластической деформации в материале оборудования представляет серьезную проблему при прогнозировании запаса пластичности и надежности, необходимого для оценки остаточного ресурса работоспособности оборудования в процессе эксплуатации. В тех же случаях, когда развитие возможного очага пластической деформации может происходить в местах, обладающих пониженной стой
костью к МКК, оба процесса — МКК и пластическая деформация, — накладываясь друг на друга, взаимо-усиливают свое влияние, и опасность преждевременного исчерпания запаса надежности оборудования резко возрастает.
С целью оценки влияния пластической деформации на стойкость к МКК и коррозионным повреждениям и на пластичность аустенитных хромоникелевых сталей 12Х18Н10Т с содержанием никеля 10,1 и 11,0 % и 12Х18Н12Т с содержанием никеля 12,0 и 12,9 % были изготовлены образцы, которые после аустенитизации и провоцирующих нагревов в диапазоне 773-1023 К в течение от 0,5 до 10 ч подвергали пластической деформации — холодной дробной прокатке при 293 К до степени деформации е = 5, 10, 12, 14, 16, 18 и 20 %. После пластической деформации образцы испытывались по методу АМУ на определение стойкости сталей к МКК. Результаты исследований приведены на рис. 1.4.29. Установлено, что пластическая деформация понижает стойкость хромоникелевых сталей к МКК. Время т,ф до появления коррозионных повреждений уменьшается, вследствие чего действительное время до наступления коррозионных повреждений тдейств = Ткр - Ат (где Ат — уменьшение времени провоцирующего нагрева до появления МКК повреждений под действием пластической деформации) снижается. С увеличением степени пластической деформации стойкость хромоникелевых сталей к МКК плавно понижается (Ат растет) до определенного критического значения пластической деформации Екр. При достижении гкр происходит резкое падение стойкости к МКК хромоникелевых сталей. Критическая степень деформации гкр зависит от содержания в хромоникелевых сталях никеля. Так, при увеличении содержания никеля с 11 % до 13 % критическая степень деформации уменьшается с 16 до 10 %, т. е. больше, чем в полтора раза.
Время до возникновения склонности к МКК, ч
Рис. 1.4.29. Влияние пластической деформации на стойкость против межкристаллитной коррозии хромоникелевых сталей: а) температурно-временные области склонности к межкристаллитной коррозии хромоникелевой стали с 11,0 % Ni в аустенитизированном и деформированном состояниях; б) время минимальной устойчивости до наступления склонности к межкристаллитной коррозии у хромоникелевых сталей, содержащих: 11 % Ni, 12 %Ni и 13 % Ni (кривые 1,2,3 соответственно)
Электродные процессы
85
Резкое падение стойкости аустенитных хромоникелевых сталей к МКК при £ = (рис. 1.4.29) необходимо связывать с несколькими факторами. Это наличие неоднородных микронапряжений на границах зерен, возникающих в процессе провоцирующего нагрева из-за образования в межграничном пространстве карбидных включений — фазы с большим объемом, а также с образованием сегрегаций примесных атомов, в частности фосфора, уменьшающих когезию границ и отрицательно влияющих на пластичность. Кроме того, в ходе пластической деформации при достижении е^, в границах зерен возникают микроповреждения типа микротрещин, на развитие и качественное состояние которых оказывают большое влияние как внутренние межграничные напряжения, так и проявление эффекта П.А. Ребиндера. Суммарное воздействие всех этих факторов приводит к активизации коррозионных процессов, к резкому падению пластичности.
На рис. 1.4.30 представлена микроструктура образцов стали с 13 % Ni, деформированных на 16% (е > £кр), где отчетливо видны повреждения границ зерен, вызванных пластической деформацией. Появление зернограничных повреждений — микротрещин — резко усиливает воздействие на сталь коррозионной среды, увеличивает глубину проникновения коррозионных трещин МКК. В табл. 1.4.26 приведены данные о влиянии пластической деформации на глубину проникновения трещины МКК от поверхности в глубь образца в слабокислых (например, 0,5 М H2SO4) и сильнокислых (например, кипящий раствор 25 % HNO3) средах.
Из анализа табл. 1.4.26 следует, что при превышении критической степени пластической деформации Екр,
при которой возможно возникновение микротрещины деформации, наблюдается резкое увеличение глубины проникновения коррозионного дефекта. Наибольшее его увеличение происходит при испытании в слабокислой среде. Так, если в слабокислой среде скорость увеличения глубины коррозионного дефекта составила 490 мкм/час при е > и 160 мкм/ч при деформации меньше критической, т. е. возросла более, чем в 3 раза, то в сильнокислой среде при скорости 540 и 280 мкм/ч соответственно наблюдается рост при е > £кр примерно в 1,9 раза.
Развитие микроповреждений оказывает влияние на запас пластичности стали, который постепенно понижается при возрастании степени деформации до Екр.
При превышении критической степени пластической деформации происходит резкое, вплоть до нуля, падение запаса пластичности (рис. 1.4.31). Таким образом, можно считать установленным, что пластическая деформация повышает склонность хромоникелевых сталей к МКК. Причем влияние пластической деформации по величине степени деформирования можно разделить на два этапа. На первом этапе (е < е^,) время провоцирующего нагрева до появления склонности к МКК в аустенитных хромоникелевых сталях плавно уменьшается, на втором (е > е^) склонность сталей к МКК резко возрастает за счет возникновения в структуре стали дефектов деформации— зернограничных микротрещин. Эти трещины не только понижают пластичность, но, являясь каналами-проводниками коррозионной среды, способствуют развитию межкристаллитной коррозии в глубь изделия. Никель, увеличивая склонность стали к МКК, уменьшает величину критической степени деформации £кр.
Рис. 1.4.30. Микротрещины (показаны стрелками) в границах зерен хромоникелевой стали с 13 %Ni после провоцирующего нагрева при 923 К в течение 5 часов и пластической деформации е > = 16 %
Рис. 1.4.31. Влияние пластической деформации на пластичность хромоникелевой стали, содержащей 11 % Ni
86
Новый справочник химика и технолога
Таблица 1.4.26
Глубина развития коррозионных трещин при МКК в слабо- и сильнокислой среде
Марка стали (содержание Ni в стали) Термическая обработка Слабокислая среда Сильнокислая среда
Пластическая деформация, %
5 10 16 20 5 10 16 20
Глубина проникновения МКК в границу зерна, мкм/ч
12Х18Н10Т (11,0 %) Аустенитизация + + нагрев 923 К, 3 часа ПО 135 160 490 230 250 280 540
12Х18Н12Т (12,9 %) 150 170 495 525 285 310 635 710
1.4.4.3. Пути повышения стойкости хромоникелевых сталей к межкристаллитной коррозии
К числу наиболее перспективных путей повышения коррозионной стойкости аустенитных хромоникелевых сталей к межкристаллитной коррозии относится способ дополнительного легирования этих сталей такими элементами, как кремний, азот и молибден. В табл. 1.4.27 собраны результаты исследований влияния этих элементов на коррозионную стойкость хромоникелевых сталей. Пределы изменения концентрации каждого из элементов в таблице определялись тремя уровнями, которые обозначены знаками «-» — минимальное содержание, «о» — среднее содержание и «+» — максимальное содержание. Абсолютные интервалы легирования составили (масс. %): для молибдена — 1,0-4,0; азота — 0,18-0,45; кремния — 0,58-5,0. Содержание углерода варьировалось в пределах от 0,03 до 0,11 %, хрома — 17,9-18,1 %; серы — 0,011-0,012 %; фосфора
0,018-0,023 %; титана от 0,27 до 0,31 %. Известно, что молибден и кремний относятся к группе ферритообразующих элементов. Это может привести к частичному распаду аустенитного твердого раствора и, как следствие, к снижению коррозионной стойкости стали. Для стабилизации аустенитной матрицы в сталях такого типа содержание никеля обычно увеличивают от 11 до 16,5 масс. %. В таблице приведены также данные по Ткр— минимальному времени провоцирующего нагрева, приводящего к возникновению склонности к МКК. Анализ таблицы 1.4.27 позволяет сделать несколько выводов о влиянии легирующего комплекса на коррозионную стойкость аустенитных хромоникелевых сталей к МКК. Для удобства анализа стали таблицы разбиты на группы и обозначены номерами в зависимости от содержания никеля: первая группа — стали с 11 % Ni, вторая — с 13 % Ni, третья — с 14 % Ni, четвертая — с 15 % Ni, пятая — с 16,5 % Ni, шестая — прочие.
Таблица 1.4.27
Составы хромоникелевых сталей и минимальное время провоцирующего нагрева ткр при 923 К до наступления склонности к МКК
№№ сталей Химический состав, масс. %* Содержание элемента Ъср
С Ni Мо Si N Мо Si N
1.1 0,11 — 0,64 — 3,9
2 0,11 Н,1 1,0 0,90 0,18 — — — 5,2
3 0,11 11,0 2,0 2,50 0,38 о о о 6,6
4 0,11 11,0 4,0 5,0 0,45 + + + 6,9
5 0,05 Н,1 2,0 2,5 0,38 о о о 8,3
2.1 0,11 13,0 — 0,58 — 2,9
2 0,11 12,9 1,0 0,9 0,18 — — — 4,7
3 0,11 13,1 2,0 2,5 0,37 о О о 5,5
4 0,11 13,0 4,0 5,0 0,45 + + + 5,8
5 0,05 13,1 2,0 2,5 0,37 о о о 8,0
3.1 0,11 13,9 2,0 2,5 0,38 о о о 5,0
2 0,11 13,8 1,0 0,9 0,18 — — — 4,2
3 0,10 13,9 2,0 0,9 0,18 о — — 4,7
4 0,11 14,1 1,0 2,5 0,38 — о о 4,5
5 0,11 13,9 1,0 2,5 0,18 — о — 4,2
6 0,11 14,0 4,0 5,0 0,45 + + 4- 5,8
7 0,05 13,8 2,0 2,5 0,38 о о О 6,2
4.1 0,11 15,0 1,0 0,9 0,18 — — — 3,8
2 0,11 15,3 2,0 2,5 0,38 о о о 4,2
Электродные процессы
87
Продолжение таблицы 1.4.27
№№ сталей Химический состав, масс. %* Содержание элемента Ткр
С Ni Мо Si N Мо Si N
4.3 0,11 15,1 3,0 4,5 0,45 4,5
4 0,11 15,2 4,0 5,0 0,45 + + + 4,2
5 0,11 15,0 4,6 5,5 0,50 2,7
6 0,05 15,0 2,0 2,5 0,38 о о О 5,3
5.1 0,11 16,5 1,0 0,9 0,18 — — — 2,3
2 0,11 16,5 1,0 2,5 0,38 — О О 2,8
3 0,11 16,4 2,0 2,5 0,38 о О О 3,5
4 0,11 16,5 4,0 5,0 0,45 + + + 3,7
5 0,05 16,7 2,0 2,5 0,38 О О о 4,0
6.1 0,03 11,0 2,0 2,5 0,30 10,6
2 0,05 13,0 3,0 3,8 0,38 11,5
3 0,06 15,0 4,0 5,0 0,45 11,0
4 0,03 11,0 4,0 3,8 0,38 12,3
* Содержание хрома в сталях 18 %.
С повышением содержания кремния при одновременном введении молибдена и азота, даже при относительно высоком содержании углерода (С > 0,1 %), с позиции коррозионной стойкости сталей, образцы приобретают повышенную устойчивость против МКК. Кривые температурно-временной области склонности сталей к МКК (кривые Ролассона) (рис. 1.4.31 и 1.4.32) при этом смещаются вправо, в сторону увеличения времени, и вверх, в сторону повышения температуры провоцирующего нагрева. В этом случае молибден, находясь в твердом растворе у-железа, по-видимому, замедляет диффузию к границам зерен атомов легирующих и примесных элементов. Причем это свойство молибдена проявляется как в сталях аустенитизированных, так и после провоцирующего нагрева.
В табл. 1.4.28 приведены средние данные по распределению легирующих и примесных элементов в зернограничном твердом растворе толщиной 100 нм и в твердом у-растворе различных сталей, определенные методом эмиссионного спектрального микроанализа. В этой же таблице под чертой приведены сведения по содержанию фосфора, определенного методом Оже-эмиссионной спектроскопии, в моноатомном слое границ на образцах, подвергнутых провоцирующему нагреву в течение 5 ч при 923 К.
Сравнительный анализ образцов сталей 1.1 и 1.3 (содержание Ni 11 %) и сталей 2.1 и 2.3 (содержание Ni 13 %) показывает, что после провоцирующего нагрева в течение 5 ч при 923 К дополнительное легирование (стали 1.3 и 2.3 — молибденом 2 %, кремнием 2,5 %) способствует понижению содержания фосфора в границах зерен с 3,56 % (сталь 1.1) до 2,10 % (сталь 1.3) и с 4,34 % (сталь 2.1) до 2,21 % (сталь 2.3). Также наблюдается уменьшение сегрегации кремния в границах зерен. Если без дополнительного легирования превышение кремния в границах зерен аустенитизированных образцов над его средним содержанием для стали 1.1
составляет 3,11 : 0,64 = 4,86 раза, для стали 2.1 — 3,17 : 0,58 = 5,47 раза, а после провоцирующего нагрева это превышение соответственно достигает для стали 1.1 — 6,53 и стали 2.1 — 7,44 раза, то при дополнительном легировании (стали 1.3 и 2.3, табл 1.4.28) это превышение для аустенитизированных образцов в среднем составляет 1,7 раза, а с провоцирующим нагревом — 2,5 раза.
Обращает на себя внимание способность молибдена, сдерживая процессы диффузии, замедлять скорость карбидообразования в хромоникелевых сталях. Таким образом, комплексное легирование хромоникелевых сталей кремнием, никелем и азотом приводит к уменьшению видимой площади шлифа, занятой карбидами, примерно в 1,5 раза (табл. 1.4.28).
Повышение содержания азота до 0,38 % в сталях с 11-13 % никеля, а в сталях с большей концентрацией никеля (стали 3.1-4.4, табл. 1.4.27) до 0,45 % практически не уменьшает их стойкость к МКК. Повышение содержания никеля до 16,5 % заметно снижает стойкость к МКК, что, видимо, обусловлено понижением растворимости углерода в у-твердом растворе, увеличением скорости диффузии, вызванным ростом содержания никеля. Однако и в этом случае добавка кремния, молибдена и азота сдвигает кривые Ролассона вправо — в сторону увеличения т^, что, видимо, объясняется уменьшением вероятности образования зародышей карбидов Ме2зСб по границам аустенитных зерен при одновременном повышении устойчивости пассивного состояния стали.
Уменьшение содержания углерода в два раза — до 0,05 % — резко повышает стойкость хромоникелевых сталей к МКК, однако отрицательно сказывается на их прочностных свойствах, что не позволяет понижать содержание углерода в этих материалах ниже 0,05 % из-за ограничения по предъявляемой конструктивной прочности.
88
Новый справочник химика и технолога
Температура, К Температура, К Температура, К Температура, К
1100-]
1050-
8004--------------------.---------------------.
1 10 100
Рис. 1.4.32. Влияние никеля на температурно-временные области склонности к межкристаллитной коррозии хромоникелевых сталей: а) 08Х18Н11МС, б) 08Х18Н11М4АС, в) 12Х18Н13,г) 12Х18Н13М4С5, д) I2X18H14M, е) 12Х18Н15М, ж) 12Х18Н17М2АСЗ, з) 03XI8H11M4C4
1100-,
1050-
Н 850-800 -----------------------------------------------
1 10 100
Электродные процессы
89
Таблица 1.4.28
Распределение легирующих и примесных элементов в границах зерен хромоникелевых сталей
Среднее содержание элемента в границе, масс. % Содержание Ме23С6,
Сталь Состояние* в слое излома в твердом растворе
С Сг Si Р С Сг Si Р масс. %
1.1 А 0,11 18,0 3,11 0,032 0,09 17,2 3,11 0,032 —
ПН 0,16 33,2 4,18 0,061 0,06 14,2 4,10 0,060 0,39
3,56
1.3 А 0,11 17,7 4,15 0,027 0,08 17,5 2,10 0,026 —
ПН 0,13 22,7 6,15 0,041 0,08 16,5 3,10 0,040 0,22
2,10
2.1 А 0,12 18,1 3,17 0,032 0,10 17,7 3,15 0,032 —
ПН 0,19 39,7 4,32 0,073 0,05 12,3 4,30 0,070 0,48
4,34
2.3 А 0,11 18,2 4,28 0,022 0,10 17,8 2,25 0,022 —
ПН 0,14 25,4 6,23 0,039 0,08 15,6 3,20 0,039 0,31
2,21
3.2 А 0,11 18,1 4,36 0,023 0,10 17,9 2,36 0,022 —
ПН 0,15 26,4 6,48 0,041 0,07 15,0 3,42 0,041 0,33
4.2 А 0,11 18,3 4,34 0,026 0,09 17,9 2,30 0,026 —
ПН 0,17 32,1 6,51 0,045 0,06 14,4 3,50 0,045 0,36
* А — аустенитизированный образец; ПН — провоцирующий нагрев.
1.4.4.4. Контроль качества оборудования из аустенитных коррозионностойких сталей после расчетного срока службы с целью обнаружения поврежденных или потенциально склонных к межкристаллитной коррозии зон
Взаимосвязь между коррозионной стойкостью аустенитных хромоникелевых сталей и их магнитной проницаемостью
Одной из наиболее острых проблем, стоящих перед промышленностью, является резкое старение основных производственных фондов. Это связано, с одной стороны, с событиями, произошедшими в нашей стране в 1980-1990 гг., когда практически полностью были сорваны сроки плановых замен оборудования предприятий. С другой стороны, — со значительным удорожанием как отдельных агрегатов, так и крупных производственных комплексов в целом, произошедшим в последние десять-пятнадцать лет. Все это привело к необходимости переосмысления понятия «ресурс оборудования». В настоящее время даже самые крупные и успешные предприятия ищут пути продления срока службы оборудования за пределы расчетного, что невозможно без создания принципиально новых методов контроля состояния металла такого оборудования. Одной из причин ускоренного выхода из строя оборудования нефте- и газоперерабатывающих предприятий, физики высоких энергий, криоэнергетики, пищевой и целого ряда других отраслей промышленности является развитие межкристаллитной коррозии в трубопроводах,
емкостях, сосудах, колоннах и др. крупномасштабных агрегатах. Обнаружить в этих системах места, поврежденные или потенциально склонные к МКК, необычайно трудно, что связано как с размерами самих агрегатов, так и с наличием в них значительного числа труднодоступных для прямого контроля металла зон. Особо усложняется задача при анализе криогенного оборудования, в котором практически все поверхности металла закрыты теплоизоляционной защитой, стоимость которой сопоставима со стоимостью самого оборудования.
Анализ полученных ранее данных о влиянии различных факторов на формирование структуры, физико-химических и механических свойств термически обработанных и деформированных образцов из аустенитостабильных хромоникелевых сталей показал, что между их стойкостью к МКК, степенью пластической деформации и магнитной проницаемостью существуют закономерные связи. Эти связи обусловлены химическим составом и структурой стали, а также процессами, протекающими в них при провоцирующих нагревах, пластической деформации и эксплуатации. Однако большое число трудно учитываемых переменных факторов, воздействующих на металл оборудования в процессе эксплуатации, затрудняет создание обобщенной математической модели его поведения в условиях коррозионного воздействия среды.
Поэтому была предложена полуэмпирическая модель, включающая данные непосредственно проведенных исследований и набора статистических данных, полученных на основе метода математического плани
90
Новый справочник химика и технолога
рования эксперимента. Это позволило построить функции отклика системы при учете наиболее значимых воздействий: химического состава (содержания никеля и кремния), пластической деформации и условий провоцирующего нагрева. Построенные по этим данным графики уточнялись прямыми наблюдениями. В табл. 1.4.29 приведены данные по магнитной проницаемости хромоникелевых сталей с различным содер
жанием никеля в зависимости от режима провоцирующего нагрева и температуры ее измерения. Из таблицы следует, что основное влияние на величину магнитной проницаемости оказывают содержание никеля в стали и интенсивность провоцирующего нагрева. Температура измерения (в диапазоне 4,2-293 К), хотя и оказывает влияние на рост ее значения, но в значительно меньшей
проницаемости связывается с образованием в процессе нагрева сильномагнитной фазы — карбидов — и происходит тем значительнее, чем больше температура и время нагрева и чем выше содержание никеля в стали. На первой стадии процесса карбидообразования (рис. 1.4.33, а), когда объем карбидной фазы относительно невелик (табл. 1.4.29), рост магнитной проницаемости незначителен. Резкое повышение магнитной
проницаемости соответствует формированию в границах зерен карбидной фазы в виде зернограничной сетки (рис. 1.4.33, б), увеличению объема карбидной фазы, возникновению в сталях склонности к МКК (рис. 1.4.34, табл. 1.4.30). Повышение магнитной проницаемости при воздействии коррозионной среды связывается также с образованием продуктов коррозии с повышенной
степени. магнитной проницаемостью.
При провоцирующих нагревах, не вызывающих в
стали проявления склонности к МКК, рост магнитной
Таблица 1.4.29
Влияние температуры и времени провоцирующего нагрева на магнитную проницаемость ц и склонность плавок стали 12Х18Н12Т к МКК
Содержание Ni в хромоникелевой стали, % Нагрев Склонность к МКК
Г, К т, ч В поле Земли В поле с напряженностью 320 кА/м
Г, К
293 293 77 50 20 4,2
Н,1 973 3 Нет 1,351 1,162 1,383 1,415 1,400 1,400
5 Да 2,150 2,100 2,200 2,295 2,310 2,344
10 Нет 2,210 2,356 2,388 2,488 2,510 2,600
923 3 Нет 1,133 1,150 1,193 1,205 1,200 1,200
5 Да 1,917 1,855 2,150 2,160 2,215 2,230
873 3 Нет 1,063 1,035 1,088 1,093 1,089 1,090
5 Да 1,815 1,800 1,855 1,973 1,870 1,863
11,8 973 3 Нет 1,415 1,380 1,415 1,500 1,500 1,493
5 Да 2,195 2,400 2,456 2,459 2,510 2,560
923 3 Нет 1,200 1,193 1,215 1,311 1,310 1,310
5 Да 1,999 2,000 2,105 2,122 2,120 2,120
873 3 Нет 1,084 1,100 1,115 1,200 1,195 1,190
5 Да 1,890 1,900 1,995 2,050 2,020 2,020
13,0 973 3 Нет 1,530 1,545 1,570 1,600 1,600 1,560
5 Да 2,450 2,680 2,690 2,700 2,740 2,800
923 3 Нет 1,210 1,195 1,200 1,215 1,210 1,210
5 Да 2,200 2,152 2,200 2,270 2,270 2,300
873 3 Нет 1,096 1,090 1,106 1,120 1,120 1,120
5 Да 2,015 2,025 2,020 2,100 2,200 2,250
Электродные процессы
91
Рис. 1.4.33. Микроструктуры хромоникелевой стали 12Х18Н12Т после провоцирующих нагревов при 923 К: а) в течение 1 часа; б) в течение 10 часов. х650
Таблица 1.4.30
Влияние температуры и времени провоцирующего нагрева на химический состав и объемное содержание карбидных межграничных включений
Режимы провоцирующего нагрева Содержание Ni в плавке, % Химический состав карбидной фазы, об. % Содержание карбидов в стали, об. % Содержание Сг в твердом растворе границы, масс. %
Т,К т, ч С Fe Сг Ti
720 10 Н,1 20,4 31,3 47,0 1,3 0,25 16,9
920 10 Н,1 21,0 25,6 52,0 1,7 0,47 13,7
970 10 И,1 20,7 21,3 56,2 1,5 0,70 11,6
920 10 11,9 20,6 24,8 53,2 1,4 0,54 12,5
920 10 13,0 21,2 23,0 54,3 1,5 0,58 11,9
Рис. 1.4.34. Влияние степени пластической деформации при растяжении на магнитную проницаемость в магнитном поле напряженностью 320 кА/м стали 12Х18Н12Т в аустенитизированном состоянии:
а) при температурах 4,2 К (кривая 1) и 293 К (кривая 2);
б) и в) влияние степени пластической деформации на магнитную проницаемость стали 12Х18Н12Т в магнитном поле напряженностью 320 кА/м (кривые 2,4,6) и в магнитном поле Земли (кривые 1,3, 5, 7) после провоцирующих нагревов при 923 К в течение Тц, - 60 мин (кривые 1-4) и при + 60 мин (кривые 5-7) при температурах 4,2 К (кривые 3,4, 6, 7) и при 293 К (кривые 1,2, 5)
92
Новый справочник химика и технолога
На рис. 1.4.34, а показано влияние пластической де-фомации при растяжении образцов из стали 12Х18Н12Т в аустенитизированном состоянии на магнитную проницаемость во внешнем магнитном поле напряженностью 320 кА/м: кривые (1) — при температуре 4,2 К и (2) — при 293 К. На рис. 1.4.34, б, в представлено влияние степени деформации на магнитную проницаемость тех же образцов после провоцирующего нагрева при 920 К. При нагреве в течение времени (Ткр + 60 мин) (кривые 1-4) в структуре стали происходят изменения, не приводящие к проявлению МКК. При нагреве в течение времени (т^, + 60 мин) (кривые 5-7) при воздействии коррозионной среды в стали проявляются коррозионные повреждения.
Анализ магнитной проницаемости в магнитном поле Земли и во внешнем магнитном поле напряженностью 320 кА/м показал, что с увеличением степени пластической деформации магнитная проницаемость повышается. Характер ее повышения в зависимости от деформации аналогичен как для аустенитизированных образцов, так и для образцов после провоцирующего нагрева — плавное медленное нарастание до степени деформации 8 = 8^-10 % и ускоренное нарастание магнитной проницаемости при дальнейшем увеличении пластической
деформации. Криогенные температуры 4,2 К, не изменяя общего характера зависимости магнитной проницаемости от пластической деформации, несколько повышают ее величину. Выдержки при провоцирующем нагреве (рис. 1.4.34, б и 1.4.34, в) за счет процессов, происходящих в металле при повышении температуры, приводят к росту магнитной проницаемости, особенно значительному при превышении т^, (рис. 1.4.34, в: Тдейст ^кр + 60 МИН).
В табл. 1.4.31 приведены данные о влиянии содержания в хромоникелевых сталях никеля, времени провоцирующего нагрева (от (т^, - 60 мин) до (т^, + 60 мин)) при температуре 920 К и коррозионной среды на величину магнитной проницаемости. Анализ табл. 1.4.31 и рис. 1.4.34, в убедительно доказывает, что увеличение степени пластической деформации и содержания никеля, наличие коррозионной среды понижают стойкость хромоникелевых сталей к коррозионным повреждениям, повышают их магнитную проницаемость. Из данных табл. 1.4.31 также видно, что даже в отсутствие коррозионного воздействия при постоянной температуре провоцирующего нагрева в течение времени (Ткр--60 мин) увеличение степени пластической деформации и содержания никеля в стали повышает магнитную
Таблица 1.4.31
Влияние содержания никеля и продолжительности провоцирующего нагрева при 920 К на магнитную проницаемость хромоникелевых сталей до и после испытания на стойкость против МКК
Содержание Ni в стали, % Г, К Магнитная проницаемость в поле Земли при провоцирующем нагреве продолжительностью, мин Нкр
Ткр 60 Ткр Ткр 4-60
До испытаний После испытаний До испытаний После испытаний До испытаний После испытаний В поле Земли В поле с напряженностью 320 кА/м
11,0 293 1,063 1,089 1,315 1,393 1,615 1,876 1,310 1,118
4,2 1,143 1,170 1,420 1,515 1,940 1,995 1,400 1,372
11,8 293 1,084 1,110 1,400 1,450 1,765 1,907 1,395 1,250
4,2 1,158 1,255 1,600 1,730 1,975 2,050 1,585 1,485
13,0 293 1,092 1,115 1,550 1,690 1,915 2,015 1,550 1,360
4,2 1,193 1,290 1,715 1,833 2,140 2,310 1,895 1,575
13,8 293 1,114 1,170 1,640 1,870 2,090 2,235 1,600 1,385
4,2 — — — — — — — 1,630
15,0 293 1,156 1,190 1,740 1,900 2,160 2,300 1,700 1,470
4,2 — — — — — — — 1,720
16,1 293 1,195 1,200 1,855 1,960 2,210 2,430 1,840 1,525
4,2 1,286 1,400 1,995 2,150 2,390 2,595 — 1,790
17,1 293 1,210 1,230 1,895 2,040 2,240 2,570 1,890 1,585
4,2 — — — — — — — 1,830
18,2 293 1,240 1,270 1,930 2,060 2,290 2,650 1,910 1,620
4,2 1,365 1,415 2,110 2,245 2,490 2,700 — 1,915
20,4 293 1,270 1,360 1,980 2,090 2,305 2,800 1,980 1,695
4,2 — — — — — — — 2,010
22,6 293 1,320 1,415 2,040 2,150 2,340 2,315 2,000 1,720
4,2 1,440 1,565 2,230 2,390 2,600 3,015 — 2,065
Электродные процессы
93
проницаемость и, следовательно, способствует проявлению в хромоникелевых сталях склонности к межкристаллитной коррозии. При увеличении выдержки при провоцирующем нагреве до (т^, + 60 мин) наблюдается более резкое возрастание магнитной проницаемости и интенсификация процесса МКК.
Установленная взаимосвязь величины магнитной проницаемости, определяемой параметрами провоцирующего нагрева, с химическим и фазовым составом хромоникелевых сталей аустенитного класса, их механическими свойствами, степенью пластической деформации, стойкостью к МКК, напряженностью магнитного поля и температурой исследования позволила ввести универсальный параметр, величина которого зависит от всех вышеприведенных факторов, воздействующих на металл оборудования. В качестве такого универсального параметра было предложено критическое значение магнитной проницаемости превышение которого сопряжено с опасностью возникновения аварийных ситуаций в оборудовании, связанных с возникновением и развитием коррозионных дефектов по механизму МКК.
В табл. 1.4.31 приведены критические значения магнитной проницаемости Цкр хромоникелевых сталей с различным содержанием никеля, а на рис. 1.4.35 значения параметра цкр представлены в виде графической зависимости от содержания никеля в хромоникелевых сталях при температуре 293 и 4,2 К в магнитных полях различной напряженности (Н) и магнитном поле Земли. Значения параметра цкр были получены на основе непосредственных измерений магнитной проницаемости на образцах и действующем оборудовании, а также данных, определенных методом математического планирования эксперимента. Как следует из табл. 1.4.31 и рис. 1.4.35, величина критического значения магнитной проницаемости зависит от содержания никеля в стали, напряженности магнитного поля, температуры ее измерения. С увеличением содержания в сталях никеля, как в магнитном поле Земли, так и во внешнем магнитном поле (независимо от температуры измерения), критическая величина магнитной проницаемости возрастает. Увеличение напряженности магнитного поля, наоборот, уменьшает ее значение. Однако определяющими факторами критического значения магнитной проницаемости являются структурное состояние стали, зависящее от ее химического состава, режима провоцирующего нагрева и, главным образом, склонности стали к МКК как основного фактора, определяющего работоспособность оборудования. В связи с этим использование подобных таблиц (табл. 1.4.31) и графиков, подобных приведенным на рис. 1.4.35, в сочетании с металлографическим анализом позволяет непосредственно на контролируемом объекте оборудования в короткое время выявить состояние металла и опасные зоны в нем.
На основании данных исследований был предложен метод неразрушающего контроля крупномасштабного оборудования, изготовленного из аустенитных хромо-
никелевых сталей, предназначенный для обнаружения поврежденных зон или зон, потенциально склонных к межкристаллитной коррозии.
Степень деформации, %
Рис. 1.4.35. Параметр цкр в зависимости от содержания никеля в хромоникелевых сталях: а) при температуре 293 К в магнитном поле напряженностью 40, 80, 160, 320 кА/M (кривые 2, 3, 4,5); б) при температуре 4,2 К в магнитном поле Земли (кривая 1);
в) при температуре 293 К в магнитном поле напряженностью 40, 80, 160, 320 кА/M (кривые 2,3, 4 ,5)
94
Новый справочник химика и технолога
1.4.4.5. Межкристаллитная коррозия хромистых нержавеющих сталей
Склонность к межкристаллитной коррозии у высоко-хромистых нержавеющих сталей (Сг > 17 %, С > 0,025 %) проявляется после ускоренного охлаждения с высоких температур (1000-1100 °C) и обусловлена выделением в границах зерен сталей карбидов хрома, приводящим к обеднению по этому элементу зернограничного твердого раствора. Протекающая в ряде сред, например, в растворах (H2SO4 + CuSO4) или (Н3РО4 + CuSO4), межкристаллитная коррозия этих сталей является следствием резкого снижения анодной поляризации границ зерен и сопровождается переходом в раствор только железа. Склонность к межкристаллитной коррозии у хромистых сталей можно ликвидировать повторным нагревом до 600-800 °C. Такой нагрев приводит к завершению выпадения карбидов и коагуляции выпавших ранее карбидных частиц, к обогащению границ зерен хромом в результате его диффузии и снятию внутренних напряжений, возникших в процессе выделения карбидных включений из твердого раствора стали при ускоренном охлаждении от 1000 °C и более.
Дополнительным средством повышения стойкости высокохромистых сталей к межкристаллитной коррозии может служить их легирование титаном, связывающим углерод в труднорастворимые специальные карбиды и препятствующим образованию хромистых карбидных включений в границах зерен.
1.4.4.6. Межкристаллитная коррозия алюминиевых сплавов
Явление межкристаллитной коррозии в деформируемых алюминиевых сплавах встречается достаточно часто. Известно несколько путей повышения стойкости алюминиевых сплавов к межкристаллитной коррозии. Здесь могут использоваться как механические методы, повышающие качество поверхности материалов, так и термическая обработка сплавов.
Типичным представителем этой группы сплавов являются сплавы системы А1—Си—Mg—Si—Fe. Межкристаллитная коррозия этой системы сплавов связана с распадом зернограничного твердого раствора, образованием и последующим разрушением в границах зерен непрерывной сетки интерметаллидных соединений СиА12 в том случае, когда коррозионные процессы сопровождаются выделением водорода. При этом на включениях интерметаллидного соединения не образуется защитная пленка, которая обычно, например при кислородной деполяризации, предохраняет включения СиА12 от коррозии. Первоначальными очагами выделения водорода и возникновения межкристаллитной коррозии в данном случае служат микропоры на поверхности сплава. Поэтому основным механизмом, повышающим стойкость дуралюминов к межкристаллитной коррозии, является поверхностное упрочнение и повышение сплошности наружных поверхностей изделий.
Склонность к межкристаллитной коррозии еще одной группы алюминиевых сплавов — магналоев (сплавы алюминия с магнием: обычное содержание магния в сплавах от 5 до 10 %, сплавы дополнительно могут легироваться марганцем в концентрации до 1 %) — может быть устранена или значительно снижена путем проведения правильно подобранного режима термической обработки. В основном таким режимом является высокотемпературный отпуск магналоев при температурах 250-400 °C. При такой термической обработке в сплавах происходит коагуляция анодных частиц Mg2Al, выделяющихся в границах зерен, что нарушает непрерывность анодной зоны.
1.4.5. Коррозионностойкие стали и сплавы
Стойкость сталей против различных видов коррозии определяется, в первую очередь, их химическим составом — содержанием таких элементов, как хром, углерод, никель, алюминий, кремний. Не менее важную роль играют технологические свойства сталей — способ получения (выплавки) стали, ее деформируемость и свариваемость, способность воспринимать тот или иной вид термической обработки, а также эксплуатационные особенности конструкций, в которых применены эти стали — качество поверхности изделия, возможность возникновения застойных зон и зон локальных пластических деформаций.
В зависимости от основной структуры, полученной при охлаждении на воздухе после высокотемпературного нагрева, коррозионностойкие стали принято разделять на следующие классы:
- ферритный класс — стали, имеющие структуру феррита и не претерпевающие превращения а—*у. Эти стали содержат от 13 до 30 % хрома и отличаются низким содержанием углерода — менее 0,15 %, например 08X13. В ряде случаев они дополнительно легируются титаном, ниобием, кремнием, молибденом и алюминием;
- мартенситно-ферритный класс — стали, структура которых представляет собой смесь двух фаз: мартенсита и не менее 10% феррита. Это хромистые стали с 13-18% хрома, содержание углерода в них обычно не превышает 0,15 %. К ним относятся стали типа 12X13, 15X13. В ряде случаев эти стали могут быть дополнительно легированы алюминием, титаном, никелем и кремнием;
- мартенситный класс — стали, основной структурой которых является мартенсит. Это хромистые стали с 12-17 % хрома, содержащие более 0,15% углерода (например 20X13) и некоторое количество ванадия, вольфрама, молибдена, никеля;
- аустенитно-мартенситный класс — стали, имеющие двухфазную аустенитно-мартенситную структуру. Соотношение аустенита и мартенсита в этих сталях может изменяться в широких пределах. Это стали, содержащие 12-18 % хрома, 4-9 % никеля, добавки алюминия и молибдена, а также ряд сталей на основе системы железо—углерод—хром—марганец;
Электродные процессы
95
- аустенитно-ферритный класс — стали, имеющие структуру аустенита и феррита (в количестве не менее 10 %): хромоникелевые, хромоникельмарганцевые и хромомарганцевые стали с добавками молибдена, титана и др. легирующих элементов;
- аустенитный класс — стали, имеющие структуру аустенита и содержащие 17-22% хрома, более 10% никеля, стабилизированные или нестабилизированные титаном, ниобием, цирконием или молибденом, а также хромоникельмарганцевые стали.
1.4.5.1. Стали ферритного, мартенситно-ферритного и мартенситного классов
В основном к этим классам относятся хромистые стали, которые можно разделить на три основные группы:
1) стали, не претерпевающие превращений при охлаждении, — стали ферритного класса;
2) претерпевающие частичное превращение — стали ферритно-мартенситного класса;
3) стали с полным превращением — стали мартенситного класса.
Химические составы наиболее распространенных сталей этого класса приведены в табл. 1.4.32 и 1.4.33.
Таблица 1.4.32
Химические составы хромистых нержавеющих сталей
Сталь Химический состав (содержание элемента не более), масс. % *
С Сг Ni Si Мп Ti Прочие
Стали ферритного класса
08X13 <0,08 12-14 — 0,8 0,8 — —
12X17 <0,12 16-18 — 0,8 0,8 — —
08X17Т <0,08 16-18 — 0,8 0,8 5 • С-0,8 —
15Х25Т <0,15 24-27 — 1,0 0,8 5 • С-0,9 —
15X28 <0,15 27-30 — 1,0 0,8 — —
Стали ферритно-мартенситного класса
12X13 0,09-0,15 12-13 — 0,8 0,8 — —
14Х17Н2 0,14-0,17 16-18 1,5-2,5 0,8 0,8 — —
Стали мартенситного класса
20X13 0,16-0,25 12-14 — 0,8 0,8 — —
30X13 0,26-0,35 12-14 — 0,8 0,8 — —
40X13 0,36-0,45 12-14 — 0,8 0,8 — —
25Х13Н2 0,20-0,30 12-14 1,5-2,0 0,8 0,8-1,2 — —
20X17Н2 0,17-0,25 16-18 1,5-2,5 0,8 0,8 — —
95X18 0,90-1,00 17-19 — 0,8 0,8 — —
09X16Н4Б 0,05-0,13 15-18 3,5—4,5 0,6 0,5 — 0,05-0,20 Nb
* Сера и фосфор (не более): S — 0,025 % и Р — 0,035 %.
Таблица 1.4.33
Некоторые зарубежные хромистые нержавеющие стали
Марка стали Номер USN Химический состав, масс. %
С Сг Ni Мо N Ti Прочие
Стали ферритного класса
405 S40500 <0,15 13,0 — — — — 0,2 А1
406 — <0,15 13,0 — — — — 4,0 А1
409 S40900 <0,08 11,0 0,5 — — б' Cmin —
430 S43000 <0,12 16,0 — — — — —
18SR — 0,05 18,0 0,5 — — 0,40 2,0
18Сг-2Мо — — 18,0 — 2,0 — — А1
Стали ферритного класса
446 S44600 <0,20 25,0 — — 0,25 — —
E-Brite 26-1 — <0,01 26,0 — 1,0 <0,015 — 0,1 Nb
29Сг-4Мо — <0,01 29,0 — 4,0 <0,2 — —
Стали мартенситного класса
403 S40300 <0,15 12,0 — — — — —
410 S41000 <0,15 12,5 — — — — —
416 S41600 <0,15 13,0 — 0,6 — — —
96
Новый справочник химика и технолога
Продолжение таблицы 1.4.33
Марка стали Номер Химический состав, масс. %
USN С Cr Ni Mo N Ti Прочие
Стали мартенситного класса
422 Н-46 Moly Ascoloy Greek Ascoloy Jethe M-152 431 S42200 S43100 0,20 0,12 0,14 0,15 0,12 <0,20 12,5 10,75 12,0 13,0 12,0 16,0 0,75 0,50 2,4 2,0 2,5 2,0 1,0 0,85 1,8 1,7 0,07 0,05 — l,0W 0,22V 0,2 V 0,3 Nb 0,35 V 3,0 W 0,3 V
Дисперсионно-твердеющие мартенситные стали
Custom 450 — <0,05 15,5 6,0 0,75 — — 1,5 Cu 8 • Cmin
Custom 455 — 0,03 11,75 8,5 — — 1,2 Nb 2,5 Cu
15-5PH SI5500 0,07 15,0 4,5 — — — 0,3 Nb 3,5 Cu 0,3 Nb
Термическая обработка большинства из перечисленных сталей (мартенситного и ферритно-мартенситного классов) состоит в закалке в масло при 975-1050 °C и отпуске, температура которого зависит от химического состава стали. Стали ферритного класса используются в отожженном состоянии (отжиг 680-780 °C, охлаждение на воздухе или в воде).
1.4.5.2. Высокопрочные хромоникелевые стали аустенитно-мартенситного класса
Стали этого класса применяют в тех отраслях техники, где требуется сочетание высокой прочности, надежности и трещиностойкости материала с хорошими технологическими параметрами, в первую очередь свариваемостью. Механические свойства этих сталей зависят от содержания в них аустенита и мар
тенсита, вторичных интерметаллидных и карбидных фаз, оказывающих значительное влияние на упрочнение материала.
Высокие прочностные свойства стали аустенитномартенситного класса достигаются после комплексной термической обработки, состоящей из закалки или нормализации при температурах 925-1050 °C, обработки холодом при минус 70 °C или высокого отпуска при 745-775 °C и старения при 350-500 °C с охлаждением на воздухе. Основные марки аустенитно-мартенситных отечественных коррозионностойких сталей приведены в табл. 1.4.34. В табл. 1.4.35 дана информация о составах некоторых зарубежных сталей, называемых в американских источниках полуаустенитными, дисперсион-но-твердеющими, а в табл. 1.4.36 приведены основные области использования этих материалов.
Таблица 1.4.34
Химические составы коррозионностойких сталей аустенитно-мартенситного класса
Сталь Химический состав, масс. %
С Сг Ni Si* Мп* Прочие
20Х13Н4Г9 0,15-0,30 12,0-14,0 3,7-4, 7 0,8 8-10 —
09X15Н8Ю <0,09 14,0-16,0 7,0-9,4 0,8 0,8 А1 0,7-1,3
07X16H6 0,05-0,09 15,5-17,5 5,0-8,0 0,8 0,8 —
08X17H5M3 0,06-0,10 16,0-17,5 4,5-5,5 0,8 0,8 Мо 3,0-3,5
09X17Н7Ю <0,09 16,0-17,5 7,0-8,0 0,8 0,8 А10,5-0,8
09Х17Н7Ю1 <0,09 16,0-18,0 6,5-7,5 0,8 0,8 А10,5-0,8
06Х16Н7М2Ю <0,09 15,0-16,5 6,5-7,5 0,7 0,7 А1 0,5-1,0
Мо 1,0-2,0
03X14H7B <0,09 13,5-15,0 6,0-7,2 0,7 0,7 W 0,4-0,8
* Содержание элемента не более.
Электродные процессы
97
Дисперсионно-твердеющие полуаустенитные нержавеющие стали
Таблица 1.4.35
Марка стали Номер USN Химический состав, масс. %
С Сг Ni Мо N Ti Прочие
АМ-350 S35000 0,10 16,5 4,25 2,75 0,10 — —
АМ-355 S35OOO 0,13 15,5 4,25 2,75 0,10 — —
17-7РН S17700 0,07 17,0 7,0 — — — 1,15 А1
РН15-7Мо S15700 0,07 15,0 7,0 2,25 — — 1,15 А1
Таблица 1.4.36
Области применения коррозионностойких сталей аустенитно-мартенситного класса
Сталь Область применения
20Х13Н4Г9 Заменитель холоднокатаной стали 12Х18Н9 и 17Х18Н9 для прочных и легких конструкций, соединенных точечной электросваркой
09X15Н8Ю Для изделий, работающих в атмосферных условиях, уксуснокислой и солевых средах; для упругих элементов
07X16Н6 Не имеет 6-феррита. Используется как высокопрочная сталь для изделий, работающих в атмосферных условиях, уксуснокислой и солевых средах; для упругих элементов
08Х17Н5МЗ Для изделий, работающих в условиях сернокислых сред и для изделий, работающих в атмосферных условиях, уксуснокислой и солевых средах; для упругих элементов
09X17Н7Ю Для корпусных, рулевых и крыльевых устройств изделий, работающих в морской воде
09Х17Н7Ю1 Для судовых валов, работающих в морской воде
06Х16Н7МЮ Для дисков распыливающих сушилок при сушке двойного суперфосфата, клапанных пластин в компрессорах конвертированного газа, плунжеров и пружин карбоматных насосов
03X14Н7В Для валов погружных центробежных насосов нефтедобывающих скважин
1.4.5.3. Стали аустенитного и аустенитно-ферритного классов
Стали аустенитно-ферритного класса характеризуются высоким содержанием хрома (до 18-22 %) и пониженным содержанием никеля (4-6 %).
Эти стали имеют двухфазную структуру, состоящую из феррита и аустенита. Основными дополнительными легирующими элементами в них являются молибден, медь, титан и ниобий. Оптимальным считается такое соотношение легирующих элементов, при котором после термической обработки содержание ферритной и аустенитной фаз в сталях составляет 1:1.
К достоинствам сталей аустенитно-ферритного класса относится их большая, примерно в полтора-два раза, прочность, чем у аустенитных сталей при сохранении запасов пластичности и вязкости на удовлетворительном уровне. У этих сталей отмечается повышенная сопротивляемость к воздействию ударных нагрузок и трещиностойкость, большая коррозионная стойкость к межкристаллитной коррозии и коррозионному растрескиванию. К недостаткам этих сталей следует отнести склонность к охрупчиванию после нагревов в диапазоне температур 400-750 °C.
Аустенитные коррозионностойкие стали по объему своего производства в классе коррозионностойких и кислотоупорных материалов по праву занимают первое
место. Эти стали имеют высокую коррозионную стойкость в широком диапазоне агрессивных сред, прекрасную обрабатываемость и технологичность. Они нашли широкое применение в различных отраслях техники — от криогенной до высокотемпературных энергетических систем, от пищевой промышленности до оборудования по переработке нефтепродуктов и производства различных кислот. Наиболее высокую коррозионную стойкость при высоком запасе пластичности и вязкости эти стали приобретают после термической обработки, состоящей из нагрева при температуре 1000—1050 °C и последующего ускоренного охлаждения на воздухе или в воде (режима аустенитизации). Структура сталей после такого режима термической обработки — аустенит.
Химические составы наиболее широко применяемых сталей отечественных аустенитно-ферритного и аустенитного классов приведены в табл. 1.4.37. В табл. 1.4.38 представлены некоторые зарубежные марки нержавеющих сталей аустенитного класса.
Наряду со сталями хромоникелевого класса достаточно широкое применение нашли безникелевые и хромоникельмарганцевые коррозионностойкие стали аустенитного класса. Появление этих групп материалов связано, в первую очередь, с экономическими проблемами и попытками снизить стоимость одних из наиболее дорогостоящих конструкционных материалов —
98
Новый справочник химика и технолога
сталей аустенитного класса. Это привело к созданию сталей с полным или частичным замещением никеля в сталях марганцем, стоимость и дефицитность которого на мировом рынке значительно меньше, чем у никеля. Второй причиной такой замещающей технологии явилась необходимость повышения прочностных свойств аустенитных сталей — у хромоникелевых сталей аустенитного класса прочность, особенно при комнатной температуре, невелика: например, для стали 12Х18Н10Т временное сопротивление составляет 520 МПа, для стали 08X18Н10 — 500 МПа.
Аустенитная структура у безникелевых и малони-келевых сталей обеспечивается за счет дополнительного введения марганца или совместного легирования материала марганцем и азотом вместо никеля. Следует обратить внимание на несколько пониженное, по сравнению с хромоникелевыми сталями, содержание хрома, например сталь 10Х14АГ15 по сравнению со сталью 08Х18Н10Т. Это приводит к снижению коррозионной стойкости безникелевых или хромоникельмарганцевых сталей.
Таблица 1.4.37
Химические составы коррозионностойких сталей аустенитно-ферритного и аустенитного классов
Сталь Химический состав, масс. %
С Сг Ni Ti Si Мп Прочие
Стали аустенитно-ферритного класса
08Х22Н6Т <0,08 21-23 5,3-6,3 5 • С-0,65 <0,8 <0,8 —
12X21Н5Т 0,09-0,14 20-22 4,8-5,8 0,25-0,50 <0,8 <0,8 —
08Х21Н6М2Т <0,08 20-22 5,5-6,5 0,20-0,40 <0,8 <0,8 Мо 1,8-2,5
08Х18Г8Н2Т <0,08 17-19 1,8-2,8 0,20-0,50 <0,8 7-9
15Х18Н12С4ТЮ 0,12-0,17 17-19 11-13 0,4-0,7 3,8-4,5 0,5-1,0 А1 0,13-0,35
Стали аустенитного класса
08Х10Н20Т2 <0,08 10-12 18-20 1,5-2,5 <0,8 <2,0 А1< 1,0
10Х14Г14НЗ 0,09-0,14 12,5-14 2,8-3,5 — <0,8 13-15
10Х14Г14НЗТ <0,10 13-15 2,8-4,5 5(С-0,02)-0,6 <0,8 13-15
10Х14АГ15 <0,10 13-15 — — <0,8 14-16 N0,15-0,25
12Х17Г9АН4 <0,12 16-18 3,5-4,5 — <0,8 8-10,5 N 0,15-0,25
08Х17Н13М2Т <0,08 16-18 12-14 5 • С-0,7 <0,8 <2,0 Мо 2-3
10Х17Н13М2Т <0,10 16-18 12-14 5 • С-0,7 <0,8 <2,0 Мо 2-3
12X18Н9 <0,12 17-19 8-10 — <0,8 <2,0
12Х18Н9Т <0,12 17-19 8-10 5 • С-0,8 <0,8 <2,0
04Х18Н10 <0,04 17-19 8-10 — <0,8 <2,0
08Х18Н10Т <0,08 17-19 8-10 5 • С-0,7 <0,8 <2,0
12Х18Н10Т <0,12 17-19 8-10 5 • С-0,8 <0,8 <2,0
03Х18Н12 <0,03 17-19 11,5-13 — <0,8 <2,0
12Х18Н12Т <0,12 17-19 11-13 5 • С-0,7 <0,8 <2,0
03Х21Н21М4ГБ <0,03 20-22 20-22 — <0,6 1,8-2,5 Мо 3,4-3,7 Nb 15 • С-0,8
Химические составы некоторых зарубежных хромоникелевых и хромоникельмарганцевых нержавеющих сталей
Таблица 1.4.38
Марка стали Номер USN Химический состав, масс. %
С Сг Ni Мо N Ti Прочие
304 S30400 <0,08 19,0 10,0 — — — —
304L S30403 <0,03 19,0 10,0 — — — —
304N S30451 <0,08 19,0 9,25 — 0,13 — —
309 S30900 <0,20 23,0 13,0 — — — —
310 S31000 <0,25 25,0 20,0 — — — —
Электродные процессы
99
Продолжение таблицы 1.4.38
Марка стали Номер USN Химический состав, масс. %
С Сг Ni Мо N Ti Прочие
316 S31600 <0,08 17,0 12,0 2,5 — — —
316L S31603 <0,03 17,0 12,0 2,5 — — —
316N S31651 <0,08 17,0 12,0 2,5 0,13 — —
317 S31700 <0,08 19,0 13,0 3,5 — — —
321 S32100 <0,08 18,0 10,0 — — 5 • С ^vm —
347 S34700 <0,08 18,0 11,0 — — — 10 • Cvin
Nb
19-9DL К63198 0,30 19,0 9,0 1,25 — 0,3 0,4 Nb, 1,25 W
19-9DX К63199 0,30 19,2 9,0 1,5 — 0,55 1,2 W
17-14CuMn — 0,12 16,0 14,0 2,5 — 0,3 3,0 Cu
202 S20200 0,09 18,0 5,0 — 0,10 — 8,0 Mn
216 S21600 0,05 20,0 6,0 2,5 0,35 — 8,5 Mn
21-6-9 S21900 <0,04 20,25 6,5 — 0,30 — 9,0 Mn
Nitronic 32 — 0,10 18,0 1,6 — 0,24 — 12,0 Mn
Nitronic 33 — <0,08 18,0 3,0 — 0,30 — 13,0 Mn
Nitronic 50 — <0,06 21,0 12,0 2,0 0,30 — 0,2 Nb, 5,0 Mn
1.4.5.4. Кислотоупорные сплавы
Кислотоупорные (кислотостойкие) стали и сплавы в основном изготавливаются на железоникелевой или никелевой основе. К числу наиболее широко применяемых марок относятся такие сплавы, как 06ХН28МДТ, 03ХН28МДТ — железо-никелевые сплавы, дополнительно легированные хромом, молибденом, медью и титаном. Эти сплавы нашли широкое применение в кислотном и целлюлозно-бумажном производ-
Химические составы сплавов на »
ствах, производствах минеральных удобрений. Основным недостатком этих сплавов является их относительно низкая стойкость против межкристаллитной коррозии, возникающей в зоне термического влияния сварного шва или после технологических нагревов. В особо агрессивных средах применяются высоконикелевые сплавы типа Н70МФ или ХН65МВ. В табл. 1.4.39, 1.4.40 и 1.4.41 приведены химические составы и свойства кислотоупорных материалов.
Таблица 1.4.39 езо-никелевой и никелевой основе
Элемент Химический состав, масс. %
06Х28МДТ 03ХН28МДТ Н70МФ ХН65МВ
Углерод <0,06 <0,03 <0,05 <0,03
Кремний <0,8 <0,8 <0,2 <0,15
Марганец <0,8 <0,8 <0,5 < 1,0
Хром 22,0-25,0 22,0-25,0 <0,3 14,5-16,5
Никель 26,0-29,0 26,0-29,0 Основа Основа
Титан 0,5-0,9 0,5-0,9 — —
Вольфрам — — — 3,0-4,5
Молибден 2,5-3,0 2,5-3,0 25,0-29,0 15,0-17,0
Ванадий — — 1,4-1,7 —
Железо Основа Основа <4,0 < 1,0
Сера < 0,020 < 0,020 < 0,020 < 0,020
Фосфор <0,035 < 0,035 < 0,020 < 0,020
Медь 2,5-3,5 2,5-3,5 — —
Таблица 1.4.40
Некоторые зарубежные нержавеющие сплавы на основе железа, никеля и кобальта
Марка сплава Номер USN Химический состав, масс. %
С Fe Сг Ni Со Мо W Nb Ti Прочие
Сплавы на основе железа
16-25-6 — 0,06 50,7 16,0 25,0 — 6,0 — — — l,35Mn, 0,7Si, 0,15N
17-14СиМо — 0,12 62,4 16,0 14,0 — 2,5 — 0,4 0,3 0,75Mn, 0,5Si, 3Cu
19-9DL K63198 0,30 66,8 19,0 9,0 — 1,25 1,25 0,4 0,3 1,1 Mn, 0,6Si
Incoloy 800 N08800 0,05 45,7 21,0 32,5 — — — — 0,38 —
Incoloy 801 N08801 0,05 46,3 20,5 32,0 — — — — 1,13 —
Incoloy 802 — 0,35 44,8 21,0 32,5 — — — — 0,75 —
А-286 K66286 0,04 55,2 15,0 26,0 — 1,25 — — 2,0 O,2A1, 0,005B, 0,3V
Discaloy K66220 0,06 55,0 14,0 26,0 — 3,0 — — 1,7 O,25A1
Incoloy 903 — 0,04 41,0 <0,1 26,0 15,0 0,1 — 3,о 1,4 O,7A1
W-545 K66545 0,06 55,8 13,5 38,0 — 1,5 — — 2,85 O,2A1, 0,05B
Сплавы на основе никеля
Hastelloy В N10001 <0,05 5,0 < 1,0 63,0 <2,5 28,0 — — — 0,03V
Hastelloy В-2 N10665 <0,02 <2,0 < 1,0 69,0 < 1,0 18,0 — — — —
Hastelloy С N10002 <0,15 6,0 16,5 56,0 — 17,0 4,5 — — —
Hastelloy С4 N06455 <0,015 <3,0 16,0 63,0 <2,0 15,5 — — <0,7 —
Hastelloy С-276 N10276 <0,02 5,0 15,5 59,0 — 16,0 3,7 — — —
Hastelloy N N10003 0,06 <5,0 7,0 72,0 — 16,0 — — <0,5 —
Incjnel 600 N06600 0,08 8,0 15,5 76,0 — — — — — < 0,25Cu
Incjnel 601 N06601 0,05 14,1 23,0 60,5 — — — — — 1,35A1, 0,5Cu
Nimonic 75 — 0,12 2,5 19,5 75,0 — — — — — 0,15Al, < 0,25Cu
Nimonic 90 N07090 0,06 1,5 19,5 55,5 18,0 — — — 2,4 1,4A1,
Nimonic 95 — <0,15 <5,0 19,5 53,5 18,0 — — — 2,9 2,0А1, +B, +Zr
Nimonic 100 — <0,30 <2,0 11,0 56,0 20,0 5,0 — — 1,5 5,0А1, +B, +Zr
Nimonic 105 — 0,08 — 15,0 54,0 20,0 5,0 — — 1,2 4,7 Al, 0,005B
Nimonic 115 — 0,20 1,0 15,0 55,0 15,0 4,0 — — 4,0 5,0Al, 0,04Zr
Nimonic 263 — 0,06 <0,7 20,0 51,0 20,0 5,9 — — 2,1 O,45A1
Новый справочник химика и технолога
Продолжение таблицы 1.4.40
Марка сплава Номер USN Химический состав, масс. %
С Fe Сг Ni Со Мо W Nb Ti Прочие
Udimtt 500 N07500 0,08 <4,0 19,0 48,0 19,0 4,0 — — 3,0 3,0А1, 0,005Zr
Udimtt 520 — 0,08 — 19,0 57,0 12,0 6,0 1,0 — 3,0 2,0А1, 0,005Zr
Сплавы на основе кобальта
Haynes 188 R30188 0,10 <3,0 22,0 22,0 37,0 — 14,5 — — —
S-816 R30816 0,38 4,0 20,0 20,0 42,0 4,0 4,0 4,0 — 0,90La
Stelite 60 — 1,0 1,0 30,0 1,0 61,5 — 4,5 — — —
UMCo-50 — <0,12 21,0 28,0 — 49,0 — — — — —
AR-213 — 0,17 <0,05 19,0 <0,51 65,0 — 4,5 — — 3,5А1, 6,5Та, 0,15Zr, 0,lY
MP-35N R30035 — — 20,0 35,0 35,0 10,0 — — — —
MP-159 — — 0,9 19,0 25,0 36,0 7,0 — 0,6 3,0 0,2А1
Электродные процессы
102
Новый справочник химика и технолога
Таблица 1.4.41
Режимы горячей обработки давлением, термической обработки и механические свойства кислотоупорных сплавов
Сплав Режимы обработки Механические свойства (не менее)
Горячая обработка давлением Термическая обработка
Начало, °C Конец, °C ов, МПа о0,2, МПа е5, %
06ХН28МДТ 03ХН28МДТ 1100-1150 850 Закалка при 1050-1080 °C в воду, масло или воздух 550 250 10
Н70МФ ХН65МВ 1200-1220 950 Закалка при 1050-1100 °C в воду 900 400 37
1.4.6. Коррозия и коррозионная стойкость неметаллических материалов
Неметаллические материалы также испытывают на себе разрушающее воздействие агрессивных коррозионных сред. В целом неметаллические материалы можно подразделить на две основные группы — неорганические и органические соединения. К первой группе — группе неорганических соединений — относятся главным образом строительные материалы. По данным различных источников, до 90-95 % всей массы неорганических материалов, используемых человеком, применяются для сооружения жилых и производственных зданий, коммуникаций, труб, бассейнов, печей, других конструкций. Одни из этих материалов используются как самостоятельные конструкционные материалы, обладающие достаточной прочностью и надежностью, другие применяются в качестве покрытий различного назначения.
Самым привычным видом разрушения неорганических соединений является эрозия — разрушение конструкций под действием дождей, ветра, изменений температуры. Однако кроме эрозии, т. е. разрушения, связанного с механическими воздействиями на материал, выделяют еще одну причину разрушения неметаллических конструкций, а именно разрушение под действием различных химических и физико-химических факторов. Чаще всего при разрушении неметаллических материалов наблюдается совместное воздействие эрозионной и коррозионной сред. Поэтому, говоря о коррозии строительных материалов, обычно имеют в виду одновременное протекание обоих процессов.
Одной из основных причин коррозионно-эрозионного разрушения конструкций является солнечная активность. Резкие колебания температуры в дневные и ночные часы в сочетании с низкой теплопроводностью строительных материалов вызывают значительный градиент температур в объеме конструкции. При этом объем нагретых элементов увеличивается, а охлажденных уменьшается. В результате появляются капиллярные трещины. В такие трещины проникает вода, которая при замерзании увеличивается в объеме, что со временем приводит к разрушению материала. Аналогичный
эффект вызывает кристаллизация солей. Находящаяся на поверхности строительной конструкции соль поглощает конденсирующуюся ночью влагу, а образующийся солевой раствор постепенно заполняет все трещины и поры материала. Днем под воздействием солнца влага испаряется, и в трещинах начинается кристаллизация солей, приводящая к их дополнительному раскрытию.
В промышленных и жилых районах городов воздух дополнительно загрязнен химическими соединениями, находящимися в нем в виде пыли, частицы которой становятся ядрами промышленного тумана. Химически активные пылинки, растворяясь в каплях тумана, образуют агрессивные растворы. Обычно такие соединения значительно тяжелее воздуха и быстро оседают на поверхности конструкций, угрожая в первую очередь нижним ярусам зданий и сооружений.
1.4.6.1. Коррозионная стойкость горных пород
Сопротивление коррозии в кислых средах у горных пород определяется содержанием в них SiO2. Природные материалы, содержащие 55 % и более SiO2, относятся к кислым и, следовательно, к кислотоупорным. Считается, что SiO2 не растворим в кислотах. Структура SiO2 может быть как кристаллической — структура зерна песка, так и аморфной — диатомит. Кристаллический диоксид кремния обладает более высокой стойкостью в кислотах, чем аморфный. К числу кислот, растворяющих диоксид кремния, относятся фтороводородная кислота — HF, ортофосфорная — Н3РО4 (при температуре 300 °C), вольфрамовая — H2WO4 и молибденовая — Н2МоО4 — кислоты. Основными неорганическими материалами, применяемыми в строительстве, являются андезит, асбест, базальт, диабаз, гранит, кварц, сиенит и известняковые породы.
Андезит (бештаунит, липарит) — излившаяся горная порода, состоящая главным образом из полевого шпата и темноцветных минералов, представляет собой вулканическую породу с высоким содержанием SiO2. Этот материал чрезвычайно стоек в серной, соляной и азотной кислотах, часто применяется как наполнитель кислотоупорных бетонов и растворов.
Электродные процессы
103
Асбест — минерал волокнистого строения, используемый как наполнитель химически стойких растворов и замазок, широко применяется в футеровках нагревательных устройств и печей. Асбесты, в основном хри-зотил-асбесты, прекрасно работают в щелочных средах, однако нестойки в средах разбавленных кислот, особенно при повышенных температурах. В этом случае рекомендуется применение антофиллит-асбеста.
Базальты — магматические горные породы, состоящие из темноцветных минералов и вулканического стекла. Особо стойки при воздействии атмосферы и кислот. Отливки из базальтов широко применяются в виде плит кислотоупорных полов и труб. Новая группа материалов — базальтопласты — нашла применение в качестве материалов защитных и теплоизоляционных покрытий в химической промышленности и энергетике. Аналогично базальтам используется еще одна группа горных материалов — диабазы. Диабазы — глубинные горные породы, состоят в основном из плагиоклаза и авгита, традиционное применение диабазов — мощение улиц, декоративная отделка зданий и сооружений.
Граниты — магматические породы серого или красно-коричневого цвета. Состоят из кварца, полевых шпатов, слюды, цветных минералов. Граниты разделяют по составу и структуре (мелко- и грубозернистые граниты), что и определяет их свойства. Наибольшее применение получили мелкозернистые граниты. Граниты стойки в атмосферных условиях и широко применяются в строительстве, благодаря высокому содержанию кварца являются кислыми породами, стойкими к воздействию кислот.
Доломиты, известняки, мраморы. Доломит — осадочная горная порода, состоящая в основном из карбонатов кальция и магния. Известняки — осадочные горные породы, состоящие в основном из кальцита — СаСО3. Мраморы — метаморфические породы, образованные в результате перекристаллизации известняков и доломитов. Эти минералы, состоящие в основном из карбонатов и оксидов, считаются основными. Они нестойки в кислых средах, но отличаются высокой стойкостью в щелочных средах. Их применяют в виде отдельных плит и в качестве наполнителей щелочестойких бетонов и растворов.
Кварц (минерал) или диоксид кремния (SiO2), относится к кислым материалам, стойким к воздействию сильных и слабых кислот (кроме плавиковой и орто-фосфорной). Кварц нестоек в щелочных средах, особенно в растворах едких щелочей.
Сиениты — глубинная изверженная порода, состоящая из калиевого полевого пшата — ортоклаза и незначительного количества плагиоклаза и темных минералов — биотита и пироксена. Эти материалы обладают улучшенной обрабатываемостью, отлично полируются и широко применяются в качестве отделочных и декоративных покрытий. По свойствам напоминают граниты, однако обладают пониженной, по сравнению с этим материалом, стойкостью в кислотах.
1.4.6.2. Коррозионная стойкость керамических и других неметаллических материалов
Стойкость керамических изделий в кислых и щелочных средах определяется их химическим составом, а также объемом и типом пор. Чем выше доля открытой пористости керамики, тем меньше ее коррозионная стойкость. Закрытые (изолированные) поры снижают агрессивное воздействие внешней среды. Количество видов керамики, стойкой к коррозионному воздействию среды, достаточно велико. Исследование кислотоупорных свойств керамических материалов определяют по их стойкости в кипящей концентрированной серной кислоте. Изделия, предназначенные для эксплуатации в условиях щелочных сред, обрабатывают 10%-м раствором гидроксидов натрия и калия.
Терракотовые (керамические) плитки применяются для футеровки полов и стен в помещениях, где ведутся работы с кислотами (за исключением фтороводородной — плавиковой кислоты), с растворами солей и щелочей, а также для облицовки сборников и хранилищ агрессивных отходов. Терракотовая плитка нестойка к истиранию, поэтому ее укладка в местах интенсивного движения не рекомендуется.
Кислотоупорные плитки из керамической глины — глазурованные или неглазурованные светлые плитки, применяемые для облицовки полов и стен, подверженных воздействию кислот, щелочей, солей. Иногда в керамическое сырье добавляется фарфоровый лом, шамот и ряд флюсов. Обожженные изделия из такой массы применяются для футеровки колонн и емкостей, предназначенных для концентрированных и разбавленных кислот (кроме фтороводородной), для изготовления травильных и гальванических ванн.
Клинкерные плитки и строительный клинкерный кирпич — глиняные изделия, обожженные до состояния полного спекания (имеющие темно-вишневый цвет) — клинкер или частичного спекания — полуклинкер. Применяется для защитной облицовки стен, каналов для отвода агрессивных отходов, для полов, подверженных интенсивному истиранию. Стоек в среде горячих и холодных кислот (кроме фтороводородной), слабых (до 10 %) растворах щелочей, солей. Дорожный клинкер производится из низкоспекающихся глин и обжигается при температуре, обеспечивающей полное спекание массы. Используется для облицовки химиче-скистойких полов, каналов и сборников для отвода агрессивных жидкостей.
Каменное литье. Изделия из каменного литья выпускаются в виде плиток, желобов, труб, колен. Имеют серо-черный цвет. Отличаются повышенной хрупкостью, низкой термостойкостью, колебания температуры при эксплуатации изделий не должны превышать 90°. Используются при изготовлении деталей, подверженных воздействию сильных кислых и щелочных сред.
104
Новый справочник химика и технолога
Углеграфитовые материалы производятся из облагороженного угольного сырья, смешанного со связующим материалом (каменноугольным пеком, дегтем и т. п.). Масса формуется и спекается при температуре 2500 °C. Полученные изделия могут быть пропитаны феноло-формальдегидными олигомерами. Полученные изделия стойки при воздействии большинства кислот, солей, различных органических соединений, нестойки в среде концентрированных азотной и хромовой кислот, кислорода. Основное применение — футеровка химической аппаратуры, настилы безис-кровых полов. Пропитанные изделия разрушаются в щелочах.
Непропитанные угольные изделия в условиях отсутствия доступа воздуха могут использоваться до температуры 2500 °C, при нагревах в воздушной среде — до 350 °C.
Силикатные соединения. Силикатные соединения применяются в виде силикатных замазок, получаемых смешением двух компонентов — твердого кислотоупорного наполнителя и жидкого связующего вещества (силиката натрия). Силикатные замазки стойки в среде концентрированных неорганических кислот, разбавленные кислоты и соли уменьшают их стойкость. Нестойки в щелочных средах, плохо переносят вибрации, резкие перепады температур.
Силикатные замазки применяются для соединения кислотоупорных кирпичей и плиток при футеровке емкостей. Иногда могут применяться в сочетании с другими замазками, например с битумной или синтетическими смолами.
1.4.6.3. Коррозионная стойкость бетонов
Основными ингредиентами обычных портландцементов, применяемых при замешивании бетонов, являются кальций-силикатные соединения. Бетоны содержат от 47 до 60 % алита (ЗСаО • SiO2) и 11-26 % — белита (2СаО • SiO2).
Принято различать несколько видов коррозии бетонов.
К первому виду можно отнести все виды коррозии, происходящие при воздействии на бетон воды с небольшой жесткостью. При этом твердеющие составляющие бетона растворяются и уносятся протекающей водой. Наибольшую интенсивность эти процессы получили при просачивании воды сквозь массу бетона.
Ко второму виду относится коррозия, связанная с воздействием вод, в которых растворены химические соединения, вступающие в обменные реакции с массой бетона. Образовавшиеся при этом соединения оказываются либо хорошо растворимы в воде и вымываются ею, либо не обладают вяжущими свойствами и в виде аморфной массы остаются в зоне реакции. К этому типу коррозии относится также коррозия бетона в кислотах и растворах солей.
К третьему типу коррозии относятся процессы, вызванные накопившимися в порах, капиллярах и трещи
нах бетона малорастворимыми солями, кристаллизация которых приводит к увеличению объема бетона. К этим видам разрушений относятся разрушения, связанные с накоплением в бетоне гипса, сульфата-алюмината кальция и т. п.
Угольная кислота, содержащаяся в природных водах, чаще других соединений вызывает коррозию бетона. Природные воды обогащаются угольной кислотой благодаря биохимическим процессам, идущим как в самой воде, так и в грунтах, с которыми вода контактирует. Раствор угольной кислоты агрессивно действует на бетон, реагируя с карбонатом и гидроксидом кальция и образуя при этом легкорастворимый гидрокарбонат кальция:
СаСОз + СО2 + Н2О -» Са(НСО3)2
или Са(ОН)2 + 2СО2—> Са(НСО3)2
После вымывания водой растворимых соединений в бетоне остается только нерастворимый кремнезем.
Проблема коррозии бетона в морской воде, в частности, связана с растворением в ней достаточного количества солей магния (15-18 % от объема растворенных солей). Основной магниевой солью в морской воде является сульфат магния, а идущую при контакте морской воды и бетона реакцию можно описать следующим уравнением:
MgSO4 + Са(ОН)2—> CaSO41 + Mg(OH)2J,
Наибольшие разрушения морских бетонных сооружений наблюдаются в зоне переменного уровня воды при одновременном факторе замерзания и таяния. В бетоне, постоянно погруженном в воду, коррозия протекает гораздо медленнее.
Концентрированные растворы щелочей также могут вызвать интенсивную коррозию бетона. В щелочной среде в бетоне идут реакции, ведущие к разрушению кислых компонентов массы:
ЗСаО • SiO2 + 6NaOH 3Na2SiO3 + ЗСа(ОН)2
Образующиеся в ходе этой реакции продукты — гидроксид кальция и силикат натрия — вымываются водой.
Еще один вид коррозии бетонов — сульфатная коррозия. Известно, что при взаимодействии сульфатов с известью образуется гипс — вещество, резко увеличивающее объем бетона. При дальнейшем течении реакции возможно образование сложного соединения ЗСаО • А120з • 3CaSO4 • 31Н2О, которое разрывает бетонную массу. Для предотвращения возникновения данной соли возможно введение в раствор бетона ряда химических соединений, например NaCl, NaBr.
В табл. 1.4.42 пердствлена классификация газов по степени агрессивности воздействия на бетон.
Электродные процессы
105
Таблица 1.4.42
Классификация газов по их агрессивному воздействию на бетон
Группа агрессивных газов Название газов Формула газов Концентрация, мг/л
Группа А Диоксид серы Фтороводород Оксиды азота Сероводород Тетрафторид кремния SO2 HF NO, NO2 H2S SiF4 <0,02 <0,01 < 0,005 <0,01 < 0,001
Группа Б Диоксид серы Фтороводород Оксиды азота Сероводород Хлороводород Хлор so2 HF NO, NO2 H2S HC1 Cl2 0,02-0,1 0,01-0,05 0,005-0,025 >0,01 <0,01 > 0,001
Группа В Диоксид серы Фтороводород Оксиды азота Хлороводород Хлор so2 HF NO, NO2 HC1 ci2 0,1-0,5 0,05-0,2 0,025-0,125 0,01-0,05 0,001-0,005
Бетонные соединения, находящиеся в чистом и влажном воздухе, не подвергаются коррозии. Однако наличие в воздухе некоторых газов (сероводорода, оксидов азота, хлороводорода, фтороводорода и т. п.) может привести к разрушению бетона. Воздействие на бетон агрессивных сред можно определять по специальной шкале (табл. 1.4.43).
Классификация воздушно-газовых смесей по степени активности их воздействия на бетон, применяемый в строительных конструкциях, приведена в табл. 1.4.44. Степень агрессивности среды была определена в условиях постоянных температур в диапазоне 25-230 °C. При увеличении температуры среды до 70-80 °C степень агрессивности увеличивается на один класс. Уровень защиты бетона от воздействия воздушно-газовых сред зависит от ее агрессивности. Для сред I-П классов достаточно применить непроницаемый бетон или цементный раствор, подвергнутый флюатированию или гидрофобизации, для сред III—IV классов необходимо выполнять изоляцию бетона.
Таблица 1.4.43
Шкала агрессивности сред
Степень агрессивности среды (класс агрессивности) Понижение прочности в зоне коррозии, % Внешние признаки
Неагрессивная (I) 0 —
Слабоагрессивная (II) <5 Слабое поверхностное разрушение материала
Среднеагрессивная (III) 5-20 Повреждение углов и граней, капиллярные царапины на поверхности
Сильноагрессивная (IV) >20 Явные признаки разрушения материала, растрескивание, выпадение кусочков
Таблица 1.4.44
Агрессивность воздушно-газовых смесей при различной влажности воздуха
Степень агрессивности среды (класс агрессивности) Относительная влажность воздуха, % Характеристика газов
Неагрессивная (I) <75 <60 Без агрессивных газов Агрессивные газы группы А
Слабоагрессивная (II) >75 61-75 <60 Без агрессивных газов Агрессивные газы группы А Агрессивные газы группы Б
Среднеагрессивная (III) >75 61-75 <60 Агрессивные газы группы А Агрессивные газы группы Б Агрессивные газы группы В
Сильноагрессивная (IV) >75 >60 Агрессивные газы группы Б Агрессивные газы группы В
106
Новый справочник химика и технолога
1.4.6.4. Коррозионная стойкость древесных материалов
Коррозионная стойкость древесины в воде и на воздухе велика, однако на нее разрушающе влияют влага, ряд химических соединений, грибки и насекомые.
В условиях воздушной среды коррозионная стойкость древесины может колебаться от десятка до нескольких тысяч лет. В зависимости от коррозионной стойкости древесные породы принято разделять на очень стойкие породы (дуб, лиственница), среднестойкие (бук, пихта, ель) и малостойкие (ольха, береза). В воде стойкость древесины также велика. Такие породы, как дуб и лиственница, могут сохраняться в воде (на глубине более 50 см) на протяжении сотен лет. Это объясняется малым содержанием кислорода в воде, что тормозит развитие в материале древесных грибков. По долговечности в воде древесину также принято делить на несколько групп. К первой группе (очень стойкие — более 500 лет) относятся такие породы, как дуб, лиственница, ольха. Ко второй группе — среднестойкие (50-100 лет) — ель и сосна. К третьей группе — малостойкие (менее 20 лет) — береза, липа, тополь, каштан. У поверхности воды скорость разрушения древесины резко возрастает. Максимальная скорость разрушения достигается в условиях периодического изменения уровня воды, когда в ходе эксплуатации происходит периодическое замачивание и высыхание древесины. Поведение древесины в почве зависит в первую очередь от степени влажности почвы. Так, в болотистых почвах поведение древесины соответствует ее поведению в воде, когда в ней развиваются гнилостные бактерии и споры грибов. Однако в кислых (торфяных) почвах гниения древесины не происходит. Гниение древесины в плотных грунтах замедляется, ее долговечность, по сравнению с долговечностью в легких грунтах, возрастает в несколько раз.
Влияние газов на коррозионную стойкость древесины можно описать на ряде примеров. Хлор проникает до наиболее глубоких слоев древесины, окисляет и хлорирует лигнин, отбеливает и разлагает целлюлозу. Во влажной древесине при абсорбции возникает соляная кислота, которая разрушает целлюлозу. Аммиак сильно абсорбируется древесиной, при комнатной температуре он разлагает смолу и жиры. Оксид cepw(VI) — серный ангидрид — хорошо абсорбируется древесиной, отбеливает волокна, частично подвергает древесину гидролизу. Фтороводород не вызывает видимых разрушений древесины. Хлороводород во влажном дереве, соединяясь с водой, образует соляную кислоту, разрушающую древесину.
Древесина в среде кислот обладает высокой стойкостью. Если стали и бетон разрушаются при pH < 5, то древесина начинает разрушаться при pH < 2. В области 2 < pH < 7 коррозия древесины в кислотах невелика. Действие кислот на древесину можно описать следующим образом: азотная кислота окисляет лигнин до муравьиной, уксусной и щавелевой кислот. Целлюлоза
при этом остается неповрежденной. Холодный 5%-й раствор азотной кислоты слабо действует на древесину хвойных пород, холодный 25%-й раствор азотной кислоты разрушает все виды древесины. Уксусная кислота вызывает сильное разбухание древесины, 15%-й раствор уксусной кислоты приводит к разрушению ее волокон. 32%-я соляная кислота на первом этапе вызывает разбухание древесины, а затем и растворение содержащейся в ней целлюлозы, которая превращается в глюкозу. Так же воздействует на древесину 70-80%-й раствор серной кислоты, а 90-96%-я серная кислота приводит к обугливанию древесины.
Разбавленные растворы щелочей (pH 8-10) при обычной температуре вызывают интенсивное разбухание древесины, растворяют содержащиеся в ней углеводы (пентозу и гексозу), омыляют смолу. При повышении концентрации щелочи при обычной температуре начинается частичное растворение лигнина, при одновременном повышении давления и температуры происходит полное растворение лигнина. На этом эффекте основан процесс получения целлюлозы. Действие щелочей тем интенсивнее, чем выше степень измельчения древесины. В случае крупных кусков разбухание наружных слоев прекращает доступ щелочей к внутренним зонам, и растворение лигнина прекращается.
1.4.6.5. Коррозионная стойкость синтетических изоляционных покрытий
Наибольшее распространение в промышленности получили изоляционные материалы на основе битума. Эти материалы являются продуктами переработки нефти и каменного угля. В зависимости от вида агрессивной среды для изготовления изоляционных битумных материалов используются различные наполнители — картоны, ткани, сетки и т. п. Наполнители могут быть как органического, так и неорганического происхождения. Ограничением применения битумных материалов является их низкая термоустойчивость. Температура защищаемых битумными материалами объектов не должна превышать 30-50 °C. Битумные материалы нестойки в органических растворителях, жирах и маслах. Битумные материалы принято делить на нефтяные битумы и каменноугольные дегти и пеки.
Нефтяные битумы — это вещества, имеющие темный цвет, твердую или полутвердую консистенцию, значительную вязкость. В их состав входят углеводороды и различные органические соединения, содержащие серу, кислород и азот. При использовании этого материала следует учитывать эффект старения битумов, связанный с испарением легких фракций в процессе эксплуатации материала, полимеризацией и карбонизацией битума. У каменноугольных пеков и дегтей процесс старения выражен еще значительнее, чем у нефтяных битумов. Старение приводит к увеличению доли свободного углерода в объеме пеков и дегтей, что приводит к снижению их вязкости и пластичности.
Электродные процессы
107
Основными изоляционными материалами на основе битумов, пеков и дегтей являются битумные грунтовки (смесь битума и органического растворителя); горячие битумные мастики (представляют собой окисленный битум или смесь специально обработанных нефтяных битумов с добавками пластификаторов и соединений, повышающих клеящую способность материала); холодные битумные мастики (смесь нефтяных битумов, пылеволокнистых наполнителей, пластификаторов, жировых пеков и органических растворителей); битумные замазки и различные битумные рулонные материалы и ленты.
Кроме битумных материалов в качестве изоляционных материалов получили распространение феноло-формальдегидные, эпоксидные, полиэфирные замазки и покрытия на их основе.
Феноло-формальдегидные замазки получают смешением двух веществ — феноло-формальдегидного олигомера и сухой мелко размолотой коксовой муки с кислым катализатором. После смешения образуется вязкая масса со временем затвердевания при комнатной температуре 7-8 часов. Она обладает высокой стойкостью в кислотах, достаточной механической прочностью. В сильных окислителях, щелочах и органических растворителях стойкость замазок ограничена. Такие замазки применяют при кладке и соединении кислотоупорных плиток и кирпичей, максимальная температура эксплуатации замазок составляет 150 °C.
Эпоксйдные замазки образуются при смешении двух компонентов — эпоксидного олигомера с минеральным наполнителем и отвердителя. Время затвердевания при комнатной температуре — 4 часа. Затвердевшая замазка обладает высокой стойкостью в разбавленных и концентрированных щелочах при обычных и повышенных температурах, в воде, растворах солей и разбавленных кислот. Имеет хорошее сцепление с керамикой и бетоном, обладает достаточно высокой механической прочностью, минимальной усадкой и влагопоглощением. В среде органических кислот, разбавленных неорганических кислот при повышенных температурах и растворителей стойкость замазки невысока. Замазка применяется для кладки и соединения коррозионностойкой футеровки, бетонных и кирпичных конструкций.
Полиэфирные замазки — продукт смешения полиэфирных олигомеров, отвердителей, минерального наполнителя и коллоидного кремнезема. Часто вместо минерального наполнителя используется угольный. Продолжительность затвердевания при комнатной температуре — 3 часа. Отвердевшие замазки являются кислотоупорными материалами, имеют хорошую сцеп-ляемость с бетоном и керамикой. Полиэфирные замазки обладают высокой стойкостью в воде, растворах солей, неорганических кислот при комнатной температуре, бензине, минеральных маслах. Стойкость замазок в щелочах и ароматических углеводородах невысока. Полиэфирные замазки не пригодны для работы в жидких средах при температуре выше 30 °C.
Эпоксидные и полиэфирные покрытия, армированные стекловолокном, получаются при укладывании на бетонное основание нескольких слоев эпоксидных или полиэфирных смесей, между которыми закладываются ткани, волокна, маты. Такие покрытия обладают хорошей сцепляемостью с основой, большой механической прочностью, сопротивлением к истиранию и отсутствием поглощения жидкостей даже под большим давлением.
Изоляционный поливинилхлоридный пластикат производится вальцеванием смеси поливинилхлорида с пластификаторами и стабилизаторами. При комнатной температуре он имеет хорошую стойкость в разбавленных растворах органических и неорганических кислот и щелочей, дистиллированной воде, 3%-м растворе пероксида водорода, 20%-м растворе хлорида натрия, 10%-м растворе хлората натрия. Нестоек в концентрированной серной кислоте, 5%-м растворе фенола, ацетоне, бензине, бензоле, толуоле, этаноле, керосине, маслах. Температура эксплуатации не должна превышать 40 °C. К основе приклеивается с помощью клеев. В частности, может быть использован перхлорвиниловый клей, получаемый растворением перхлорвиниловой смолы в смеси ацетона, дихлорэтана, уксусноэтилового и уксуснобутилового эфиров. Этот клей применяется как для склеивания листов изоляционного материала между собой, так и для приклеивания его на стальную, бетонную или виниловую основы. Расход клея при приклеивании поливинилхлоридного пластиката на бетонное основание составляет около 1 кг/м2.
1.4.6.6. Коррозионная стойкость полимерных материалов
По происхождению все полимерные материалы делятся на природные (биополимеры), например белковые соединения, нуклеиновые кислоты, смолы, и синтетические — полученные искусственным путем: полиэтилен, полипропилен, фторопласты и многие другие. Атомы или атомные группы в макромолекуле полимера могут располагаться в виде открытой цепи или вытянутой в линию последовательности циклов (линейные полимеры, например, каучук натуральный), цепи с разветвлением (разветвленные полимеры, например, амилопектин), могут обладать более сложными структурами типа лестничных структур или трехмерной сетки (пространственно-сетчатые структуры) (рис. 1.4.36.)
Рис. 1.4.36. Различные типы структур полимеров: а) линейная, б) линейно-разветвленная, в) лестничная, г) пространственно-сетчатая
108
Новый справочник химика и технолога
В зависимости от состава основной цепи полимеры делятся на гетероцепные, в основной цепи которых содержатся атомы различных элементов (чаще всего углерода, водорода, азота, кремния и фосфора), и гомоцеп-ные, основные цепи которых построены из однотипных атомов. Из гомоцепных полимеров наиболее распространены карбоцепные полимеры — полиэтилен, полиметилметакрилат, политетрафторэтилен. Отдельную группу полимеров образуют неорганические полимеры — пластическая сера, полифосфорнистый хлорид и т. п.
Линейные полимеры обладают специфическим комплексом свойств, а именно: способностью образовывать высокопрочные анизотропные высокоориентированные волокна; способностью к большим, длительно развивающимся обратимым деформациям; способностью в высокоэластическом состоянии набухать перед растворением, иметь высокую вязкость самих растворов. Этот комплекс свойств связан со значительной молекулярной массой, цепным строением, а также гибкостью макромолекул. При переходе от линейных цепей к разветвленным, к редким трехмерным сеткам и, наконец, к густым сетчатым структурам этот комплекс свойств становится все менее ярко выражен.
Полимеры могут существовать в аморфном или кристаллическом состояниях. Необходимое условие кристаллизации — регулярность достаточно длинных участков макромолекулы. В кристаллических полимерах возможно образование более сложных надмолекулярных структур, тип которых будет определять весь комплекс физико-механических и химических свойств полимера. Незакристаллизованные полимеры могут находиться в одном из трех физических состояний — стеклообразном, высокоэластическом и вязкотекучем, причем в зависимости от ряда факторов возможен переход полимера из одного состояния в другое. Полимеры с низкой (ниже комнатной) температурой перехода из стеклообразного состояния в высокоэластическое называются эластомерами, с высокой — пластиками.
Важнейшими свойствами полимеров являются химический состав, молекулярная масса и молекулярномассовое распределение, степень разветвленности и гибкости макромолекулы. Полимеры обладают высокой стойкостью в щелочных и кислых средах. В отличие от металлов они не подвержены электрохимической коррозии. Однако во времени происходит постепенное изменение структуры и свойств полимеров — так называемая деструкция полимера.
Деструкцией (старением) полимера называют самопроизвольное необратимое изменение важнейших технических характеристик, происходящее в результате сложных химических и физических процессов, развивающихся в материале при эксплуатации и хранении. Этот процесс может ускоряться под действием света, при частой смене циклов нагрев—охлаждение, под воздействием среды — кислородной или озонной и т. п. Деструкция также может ускоряться под действием многократной деформации материала.
Повышение живучести полимера — его стабилизация — может достигаться как физическими, так и химическими методами. Физические методы стабилизации заключаются в изменении скорости транспорта (диффузии) реагирующих частиц. Так, например, для замедления процесса гидролитической деструкции полимеров необходимо снизить скорость диффузии агрессивной среды (воды, растворов кислот, оснований, солей) в полимерную матрицу. Химические методы стабилизации, как правило, связаны с добавками в полимер различных веществ, которые перехватывают активные частицы (в первую очередь осколки молекул — атомы, радикалы, ионы), ответственные за деструкцию полимеров.
Существует два основных типа распада полимерной цепи. Это деполимеризация — процесс, обратный полимеризации — и распад полимерной цепи по закону случая, когда разрыв любой связи в полимерной цепи равновероятен. К основным видам такого распада — деструкции полимеров — относятся: термическая и термоокислительная деструкции, фотодеструкция, радиационная деструкция, гидролитическая деструкция, механодеструкция, биологическая деструкция, озонное разрушение полимеров и т. п.
Термическая деструкция полимеров
Термическая деструкция — это распад полимера под действием повышенных температур. Общий механизм термораспада полимеров по цепному механизму можно описать на примере карбоцепного полимера. Обобщенная формула карбоцепного полимера может быть изображена в виде: -СНг-СНХ-СНг-СНХ-, где X — некий гетероатом или некая группа атомов. Распад полимера начинается со стадии инициирования, причем наиболее вероятен распад по закону случая. Тогда в результате разрыва макромолекулы будет получено два радикала (осколки молекул). Такие макрорадикалы могут в дальнейшем подвергаться деполимеризации с образованием мономеров. Распад будет проходить с развитием двух стадий — внутримолекулярной и межмолекулярной передачи цепи, а затем деструкции самой макромолекулы. Такой механизм распада — радикальный — встречается наиболее часто, однако существует еще несколько видов распада — ионный (так распадаются полиформальдегиды и др. гетероцепные молекулы) и молекулярный распады.
Если в формуле карбоцепного полимера X — это хлор, то представленный полимер — поливинилхлорид (ПВХ). Распад такого полимера можно представить следующим образом. При повышении температуры из ПВХ выделяется молекула НС1 и образуется двойная связь, которая является катализатором дальнейшего дегидрохлорирования, ускоряющим распад поливинилхлорида примерно в 10 раз:
~СН2-СНС1-СН=СН~; ~СН=СН-СН=СН~ + НС1, при этом группировка ~СН=СН-СН=СН~ вновь приводит к инициированию процесса распада, ускоряя его
Электродные процессы
109
уже примерно в тысячу раз по сравнению с группировкой ~СН=СН~, в результате чего возникает новая последовательность в ПВХ.
Таким образом, термический распад (или пиролиз) полимеров может начинаться с концов макромолекулы (деполимеризация) или с ее середины (распад по закону случая). Чтобы предотвратить распад полимера, необходимо блокировать концы макромолекул. Распад по закону случая предотвратить невозможно, т. к. он определяется только энергиями связи в молекуле, однако, связав свободные радикалы ингибиторами свободнорадикальных реакций, представляется возможным замедлить процесс термической деструкции полимера.
Термоокислительная деструкция полимеров
Механизм реакции окисления органических соединений RH (включая полимеры) в жидкой фазе на неглубоких стадиях окисления (т. е. когда в системе образуются только гидропероксиды) может быть записан в следующей последовательности:
зарожде- вырожденное _
продолжение обрыв
ние —► > разветвление —> „
цепей w цепей
цепей цепей
Для торможения процессов термоокислительного разложения полимеров необходимо применение ингибиторов (стабилизаторов, антиоксидантов). По механизму действия эти ингибиторы могут быть отнесены к одной из следующих групп:
- ингибиторы, обрывающие цепи по реакции с пе-роксидными радикалами. К ним относятся фенолы, ароматические амины, ароматические многоядерные углеводороды;
- ингибиторы, обрывающие цепи по реакции со свободными радикалами. К таким ингибиторам относятся хиноны, молекулы иода и т. п.;
- ингибиторы, разрушающие гидропероксиды. В реакциях автоокисления главный инициатор процесса — гидропероксид (ROOH), поэтому автоокисление также тормозят вещества, разрушающие гидропероксиды без образования свободных радикалов — это сульфиды, эфиры фосфористой кислоты.
Озонное разрушение каучуков, резин и пластиков
Скорость реакции озона с высокомолекулярными соединениями, содержащими двойную связь С=С, в 105 раз выше, чем скорость реакции озона с соединениями, имеющими связь С-С. Поэтому от озона, в первую очередь, страдают каучуки и их производные — резины. Реакция озона со связью С=С интенсивно протекает при достаточно низких температурах, и уже ниже 0 °C ее влияние достаточно ощутимо.
В технической практике наибольший защитный эффект достигается применением антиозонаторов в сочетании с восками (предельными углеводородами). Для защиты используют воски, парафины, церизины. Эти вещества на поверхности резинового изделия создают прочный эластичный защитный слой. Наилучшие результаты достигаются при нанесении защитных слоев
из изопарафинов — эти вещества не кристаллизуются и дают механически прочные покрытия.
Фотодеструкция, радиационная деструкция и с вето стабилизация полимеров
Солнечный свет несет кванты света с длиной волны более 200 нм, коротковолновая граница спектра у поверхности Земли соответствует 290 нм. Часть света с длиной волны от 200 до 290 нм рассеивается атмосферой, а видимая и инфракрасная часть спектра достигает земной поверхности.
Насыщенные углеводородные молекулы в значительной части ультрафиолетового спектра излучения прозрачны. Свет с длиной волны более 290 нм инициирует поглощение макромолекулой кислород- и азотсодержащих групп, примесей металлов переменной валентности (остатков катализаторов полимеризации) и образование двойных связей. Поглощение света приводит к образованию радикалов и сопровождается деструкцией полимера. Под действием света в полимерах могут инициироваться различные превращения, которые в конечном счете приводят к его разрушению.
В качестве светозащиты полимеров могут быть использованы ультрафиолетовые абсорберы света, применение которых основано на принципе — если невозможно отразить свет, то его необходимо поглотить. В качестве таких абсорберов используют, например, гидроксибензофенон. Возможно применение эффекта отражения света. Так, например, сажа в ультрафиолете отражает свет, представляясь в ультрафиолетовом излучении абсолютно белым телом. Таким образом, сажа является простейшим и весьма действенным фотостабилизатором резин различного назначения.
Если свет не был отражен или поглощен и он попал на полимер, то последний переходит из нормального состояния в возбужденное, предшествующее началу распада. В этом случае необходимо использование специальной группы веществ, так называемых тушителей возбужденного состояния полимеров. К этим веществам относится, например, бензотиазол.
В то время как свет поглощается полимером только когда его частота соответствует частоте поглощения молекулы, радиационное излучение поглощается всеми молекулами, вызывая акты ионизации и переводя молекулы в возбужденное состояние. Ионизирующее излучение делят на корпускулярное (электронное, протонное, нейтронное) и электромагнитное (рентгеновское излучение, у-излучение). Под действием ионизирующего облучения происходит не только обрыв, но и сшивание молекул. В качестве стабилизаторов-антирадов могут быть предложены вторичные амины.
Гидролитическая деструкция, механодеструкция и биологическая деструкция полимеров
Гидролиз полимеров протекает не по радикальному, а по ионному механизму. Это лишает возможности применить для его предотвращения антиоксиданты.
но
Новый справочник химика и технолога
Основным методом предотвращения или снижения скорости гидролиза может быть признан способ, затрудняющий доставку агрессивной среды (воды, кислоты, соли) в полимерную матрицу.
Механизм механодеструкции полимеров состоит в снижении энергии активации процесса деструкции под действием внешних напряжений. Образующиеся макрорадикалы на воздухе превращаются в пероксидные радикалы, а затем идет процесс окислительной деструкции, инициированной механохимической реакцией.
Под биологической деструкцией полимеров понимают взаимодействие полимеров с бактериями, грибами и т. п. При этом взаимодействии, как правило, возникает гидролитическое ферментативное разложение полимеров. Поскольку размеры подобных ферментов весьма велики, их проникновение в матрицу полимера маловероятно, и обычно процесс биологической деструкции идет от поверхности в глубь полимерного материала. Защита от биодеструкции состоит в нанесении на поверхность полимера защитных покрытий и топографической стабилизации. Под топографической стабилизацией понимается процесс диффузии в подповерхностный слой полимера специальных добавок. Задача таких добавок — создать на поверхности изделия охранную зону, не вступающую в реакции с ферментами.
1.4.7. Промышленная безопасность оборудования. Способы контроля качества деталей с целью выявления коррозионных дефектов различных типов
1.4.7.1. Проблемы промышленной безопасности оборудования на современном этапе
Эксплуатационная надежность оборудования, отработавшего расчетный срок службы в условиях агрессивного воздействия внешних
и рабочих сред
Вопросы повышения надежности и безопасной эксплуатации оборудования опасных производств решаются в рамках Федерального закона «О промышленной безопасности опасных производственных объектов» от 20.06.1997 № 116-ФЗ. На основании этого закона Российская Федерация приняла на себя обязательства по совершенствованию системы промышленной безопасности, ее интеграции в европейские и общемировые нормативы в рамках международного стандарта ИСО 9001.
Проблемы промышленной безопасности, охраны труда и снижения аварийности и травмоопасное™ производства особо остро встали в последнее десятилетие XX века. Это связано с резким старением парка оборудования, отсутствием его плановых реноваций, повторным введением отдельных агрегатов и производств в целом в эксплуатацию после длительных — иногда многолетних — простоев. Такая ситуация не могла не сказаться на безопасности оборудования. И действи
тельно, только на химических предприятиях Северо-Западного округа, по данным Главной Государственной Инспекции по охране труда, за период с 1999 по 2001 г. число аварий превысило 9000, при этом погибли 340 человек и еще 963 получили инвалидность или профессиональные заболевания. В целом по стране число аварий в промышленности в 2001 г. выросло по сравнению с 1999 г. более, чем в полтора раза, причем около 70 % всех аварий совершаются по причине изношенного оборудования, снижения надежности и работоспособное™ материалов и их коррозионных и коррозионноусталостных повреждений.
Столь тяжелая картина по аварийности и необходимость приведения нормативно-технической документации по промышленной безопасности в Российской Федерации в соответствие с международными стандартами не могли не сказаться на изменении отношения к проблемам промышленной безопасности. В нормативных документах по вопросам промышленной безопасности, принимаемых в последние годы, отмечается явная тенденция к значительному ужесточению требований к надежности, долговечности, ресурсу и параметрам безопасной эксплуатации оборудования. В дополнение к Федеральному закону о промышленной безопасности принят ряд подзаконных и ведомственных актов, касающихся регулирования отношений в сфере промышленной безопасности: «Правила применения технических устройств на опасных производственных объектах», введенные Постановлением Правительства Российской Федерации № 1540 от 25.12.1998 г., распоряжение Правительства Российской Федерации № 854-р от 20.07.2000 г. об утверждении «Перечня технических устройств, применяемых на опасных производственных объектах» и др. правительственные документы. На основании этих документов ответственность за безопасность эксплуатации оборудования и охрану труда на опасных предприятиях была возложена на МЧС, Министерства труда и здравоохранения, профсоюзы, Государственный горный и технический надзор (И TH РФ) и Госстандарт РФ. В результате совместной деятельности ГГТН и Госстандарта РФ был издан согласованный перечень оборудования опасных производственных объектов, подлежащих обязательной сертафикации документов, в частности: Сборник документов ГГТН РФ №25 «Перечень оборудования опасных производств, подлежащих обязательной сертификации»; приказ ГГТН РФ № 115 от 27.08.2001 г.; РД 03-484-02. Положение о порядке продления срока безопасной эксплуатации технических устройств, оборудования и сооружений на опасных производственных объектах; СП 111-10-58-01. «Организация и проведение производственного контроля охраны труда и промышленной безопасности».
Новейшими среди документов, регулирующих правила безопасной эксплуатации оборудования опасных производств, является подготовленный к изданию и введению нормативный документ «Общие требования к
Электродные процессы
111
охране труда в организации», гармонизирующий отношения в этой области между отечественными актами и общеевропейской системой безопасности труда УНСАС 18.01.99 и ГОСТ Р 51.901-2002 «Управление надежностью. Анализ технического риска технологических систем», который введен в действие с 2003 года.
ГОСТ Р 27.002-2002 расшифровывает термин «надежность» и дает основные направления развития системы экспертизы промышленной безопасности и технического диагностирования опасного оборудования. Согласно ГОСТ Р 27.002-2002 термин «надежность» включает в себя такие понятия, как «долговечность» — срок службы оборудования до списания или капитального ремонта. Долговечность определяется как время (число лет) от изготовления оборудования до его списания. «Ресурс» или средний ресурс до списания оборудования или его капитального ремонта — часть долговечности, которая определяется как время нахождения оборудования под эксплуатационной нагрузкой. Ресурс может определяться в часах, циклах работы или пуско-остановах оборудования. Следующей составляющей долговечности является «безотказность» или вероятность безотказности работы оборудования, которая согласно требованиям ГОСТ Р 27.002-2002 должна находиться в пределах Р = 0,95-гО,99. Для получения точного значения безотказности величину Р следует умножить на коэффициент оперативной готовности, который, например, для арматуры различного оборудования опасных производств, составляет 0,99999-0,999999. В понятие «надежность» входит также ремонтируемость или ремонтопригодность оборудования — т. е. время или стоимость ремонтных работ, необходимых для поддержания его безотказности в период всего срока эксплуатации (ресурса), и «сохраняемость» — неизменность формы, размеров и эксплуатационных параметров оборудования вплоть до исчерпания его ресурса.
Оценить комплекс проблем промышленной безопасности оборудования, учесть все факторы риска, возникающие в ходе проектирования, изготовления, монтажа, эксплуатации и ремонта «вручную», без применения современных систем АСУТП, оказывается практически невозможно. Поэтому требования всех международных и отечественных стандартов по вопросам промышленной безопасности сведены в программные комплексы моделирования надежности и безопасности промышленных систем. Самыми известными и широко применяемыми программными комплексами являются: «Риск спектрум» (Швеция) (этот комплекс получил наибольшее признание, принят и широко используется в 37 странах мира, в том числе в России); «Сапфир» (США); отечественные программные системы типа ПК АСМ-201, ПК АСМ СЭМА и др. Эти комплексы включают в себя нормирование всех стадий проектирования оборудования и основных операций по его монтажу и эксплуатации. Построение отечественных и зарубежных программных комплексов ведется по единой логи
ческой схеме и заключается в следующих операциях (в области обеспечения надежности оборудования):
- разработка структурной схемы ошибок;
- построение деревьев событий, деревьев отказов и т. п.;
- создание исходных параметров надежности;
- постановка задачи и создание схемы функциональной надежности;
- ввод структурных схем и создание математической модели надежности;
- расчет математической модели надежности и выдача заключения и параметров надежности для проектирования нового оборудования.
Влияние эксплуатации, в частности коррозионные повреждения оборудования, учитывается только в ролевых функциях элементов, что связано с практически полным отсутствием информации о структурных и коррозионных изменениях в сталях и сплавах в ходе длительной эксплуатации. Отсутствует информация о влиянии термоциклической эксплуатации низкотемпературного оборудования. Не учитываются воздействия коррозионных сред и климатического холода на металл вспомогательного оборудования различного назначения.
Первой попыткой учета влияния эксплуатационных факторов на надежность и долговечность оборудования в нефте- и газоперерабатывающей промышленности и низкотемпературной технике можно считать программные продукты, вышедшие под эгидой ГГТН РФ — Методические указания «Автоматизированная система управления надежностью и безопасностью» и Методические указания «Анализ и оценка риска опасных производственных объектов нефтехимических производств». Эти программные комплексы призваны учесть ролевые вклады элементов, в число которых впервые включен элемент «качество и надежность материалов». Однако в настоящее время оба методических указания введены в действие без оценки действительного состояния материала оборудования. Учет качества материалов ведется по так называемым средневзвешенным свойствам, и данные программные продукты (до особого указания) используются без единого блока «фактического состояния надежности и свойств материалов», что связано с недостаточным объемом статистически достоверной информации по данному вопросу.
В основном аналитическая цепочка «срок службы — изменение коррозионного состояния материалов» сводится только к двум точкам контроля: исходное состояние оборудования и состояние оборудования по исчерпанию гарантированного производителем и подтверждаемого ГГТН РФ ресурса. После исчерпания гарантированного ресурса оборудование подлежит автоматическому изъятию из производственного цикла и обязательной замене. Однако во всем мире в целом и в России в частности старение оборудования носит столь интенсивный характер, что его автоматическое списание привело бы к остановке как отдельных производств, так и целых отраслей промышленности. Поэтому во всех странах мира разработаны и утверждены
112
Новый справочник химика и технолога
правила и нормы продления срока службы оборудования за пределы его гарантированного ресурса. В странах ЕС эти нормы были приняты на основании правил, разработанных системой TUV (объединение технического надзора Германии) и согласованных с системой стандартизации Германии (DIN), в нашей стране — это Правила ГГТН РФ. В качестве обобщенного положения по проведению оценки промышленной безопасности оборудования в Российской Федерации используется РД 10-369-00, утвержденный ГГТН РФ.
Особенности разрушения крупногабаритных конструкций
Одной из главных трудностей, с которыми приходится сталкиваться при контроле состояния и оценке надежности аппаратов химической, нефтехимической, газоперерабатывающей промышленности и оборудования криогенной техники, являются их размеры. Такое оборудование, стационарно размещенное на производственных площадках на открытом воздухе, из-за своих размеров и весовых характеристик практически невозможно транспортировать в специально оборудованные ремонтные зоны для проведения их планового контроля и ремонта. Это необходимо учитывать при назначении методов контроля (в случае работы с подобным оборудованием возможно использование только мобильных контролирующих систем), при проведении последующих расчетов этого оборудования на прочность и оценке критериальных соотношений механики разрушения.
Предельное состояние материала в наиболее опасной зоне элемента конструкции описывается уравнением:
F[o,; 77(а, Р); 7; М; S; Т] = С, (1.4.7.1)
где С — критерий наступления предельного состояния; Xi — характеристика материала; П(а, Р) — функция, описывающая вид напряженного состояния (oi > о2 > о3; а = о2 / си; Р = оз / Oi); I — размер дефекта; М — геометрический фактор; 5 — фактор условий эксплуатации; Т — технологический фактор.
Уравнение (1.4.7.1) характеризует, в зависимости от природы материала, начало пластического течения материала (критерий течения), либо момент его разрушения (критерий разрушения). В последнем случае в рамках классических теорий прочности в качестве расчетных параметров могут быть использованы характеристики механических свойств: о0,2 — предел текучести, ов — временное сопротивление и SK — сопротивление разрыву, а в области механики разрушения — характеристики трещиностойкости материала: критические значения коэффициентов интенсивности напряжений — Кс, раскрытия трещины — 5С, J-интеграла — Jc, коэффициента интенсивности деформаций в упруго-пластической области — Kiec и т. п.
Особенности расчетов крупногабаритных систем связаны с моделированием аварийных ситуаций в экстремальных условиях эксплуатации, что является обя
зательным при создании крупногабаритных машин и конструкций, отличающихся повышенной степенью опасности. Наиболее сложной является расчетноэкспериментальная оценка трещиностойкости в условиях агрессивного воздействия среды и живучести технической системы.
Целью расчетов на трещиностойкость является количественная оценка и анализ работоспособности металлоконструкций, оценка их способности сопротивляться развитию дефектов (трещин) в ходе штатных условий эксплуатации. Однако в данные для расчета могут закладываться различные параметры состояния системы (исходные — до эксплуатации или реальные — на момент проведения расчетов). Такая «свобода» операций ввода исходных данных приводит к тому, что либо не будет выполнено одно из основных условий достоверности расчета (учет изменений, произошедших в металле в ходе его эксплуатации), либо до проведения расчетов необходимо выполнить прямые механические и коррозионные испытания металла оборудования.
Следует отметить, что в области оценки производственных и эксплуатационных рисков оборудования опасных производств разработан ряд новых алгоритмов и принципов обеспечения безопасности производственных систем, выделяемых в новое научное направление — механику катастроф. На рис. 1.4.37а и 1.4.376 приведен ряд алгоритмов расчета безопасной эксплуатации оборудования опасных производств, решаемых в рамках механики катастроф.
Предметом механики катастроф являются основные аварии, связанные с механическими и коррозионномеханическими разрушениями, последствия которых имеют принципиальное, с точки зрения безопасности, значение. Методы механики катастроф включают в себя совокупность моделей, теоретических положений и принципов науки о прочности, в том числе с учетом образования трещин, зон локальных пластических деформаций, влияния коррозионных сред. Во многих странах мира ведутся интенсивные разработки обобщенной концепции максимальной гипотетической аварии. Эта концепция позволила сформулировать первоочередные задачи в развитии промышленной безопасности оборудования опасных производств и основные направления в изучении технических систем в рамках механики катастроф, в частности:
- установление внешних нагрузок, действующих на элементы системы, исходя из реальных режимов ее эксплуатации как при нормальных, так и при аварийных условиях;
- исследование напряженно-деформированного состояния высоконагруженных несущих элементов системы с учетом внешних и внутренних динамических нагрузок;
- определение прочности, повреждаемости и масштабов возможных разрушений элементов конструкции технических систем;
- оценка последствий подобных разрушений;
Электродные процессы
113
- выбор мер и рекомендаций по исключению или снижению возможного ущерба от катастрофических и опасных разрушений.
Аварии и катастрофы, как правило, сопровождаются возникновением и развитием трещин в зонах повышенной концентрации напряжений, в местах локального изменения механических свойств — охрупчивания, которое может быть связано с межкристаллитными коррозионными повреждениями, коррозионным растрескиванием, усталостно-коррозионными процессами. Таким образом, проблема анализа и обеспечения безопасной эксплуатации оборудования опасных производств оказывается тесно связанной с решением задач анализа кинетики трещинообразования, причин возникновения в материале конструкций раз
личного вида дефектов, изменения структурного состояния сталей, их охрупчивания в процессе эксплуатации.
До настоящего времени наблюдается определенный разрыв между новейшими научными достижениями в области механики разрушений и механики катастроф и общепринятыми, нормированными расчетами прочности и остаточного ресурса оборудования. Используемые в таких расчетах механические свойства сталей рассматриваются как константы материала, не учитываются изменения локальной прочности в условиях существенной неоднородности химического состава стали, агрессивного влияния эксплуатационных сред и поля напряжений.
Рис. 1.4.37а. Концепция обеспечения безопасности оборудования опасных производств
Рис. 1.4.376. Принципы анализа и обеспечения инженерной безопасности оборудования опасных производств
114
Новый справочник химика и технолога
1.4.7.2. Способы испытаний коррозионной стойкости сталей и сплавов
При выборе материала при конструировании и изготовлении деталей машин и аппаратов стали и сплавы подвергаются целому ряду испытаний. Они включают в себя стандартные методики, объединяющие оценку физических (теплопроводность, электропроводимость, плотность и т. п.), механических (прочностные свойства, пластичность, вязкость, трещиностойкость и т. п.), технологических (свариваемость, литейные свойства и способность к формоизменению) и химических свойств. К числу испытаний химических свойств материалов относятся испытания на коррозионную стойкость материала в тех или иных агрессивных средах при различных условиях нагружения: при воздействии высоких температур (оценка окалиностойкости материала), при совместном воздействии растягивающих напряжений и агрессивных коррозионных сред (стресс-коррозия или коррозия под напряжением) и т. п. Способы испытаний на коррозионную стойкость разнообразны, а их методики зависят от условий эксплуатации того или иного изделия.
Основными задачами, решаемыми при коррозионных испытаниях сталей и сплавов, являются:
- определение принципиальной возможности возникновения коррозионных повреждений в том типе оборудования, в котором предполагается использование материала;
- выявление тех коррозионных механизмов, действие которых может привести к разрушению данного материала от возникновения и развития коррозионных дефектов;
- определение возможности применения стали или сплава в качестве материала той или иной конструкции, вероятности появления в нем коррозионных, коррозионно-механических или коррозионно-усталостных дефектов и интенсивности развития этих дефектов в ходе эксплуатации или при простое оборудования;
- проведение сравнительной оценки коррозионной склонности этого материала в различных средах, воздействию которых может подвергаться оборудование в ходе эксплуатации, сопоставление полученных данных с информацией о стойкости ранее применявшихся материалов и прогноз целесообразности использования этого материала; оценка изменения коррозионной стойкости материала в ходе длительной эксплуатации оборудования и влияния этих изменений на ресурс оборудования в целом. Такой прогноз должен включать не только оценку повышения коррозионной стойкости конструкции при применении нового материала, но также оценку экономической эффективности при замене, определение возможности возникновения технологических и эксплуатационных сложностей при изготовлении и работе оборудования, где будет применяться данный материал.
Первые три группы испытаний можно провести в лабораторных условиях. Они позволяют ускоренными
методами и за короткий срок оценить коррозионные свойства материала в заданных условиях. Лабораторные исследования в зависимости от методики их проведения условно можно подразделить на собственно коррозионные испытания, а также электрохимические и физические методы исследования.
Четвертая группа исследований предполагает полупромышленные испытания материала, сопоставимые по длительности со сроком службы оборудования, в котором предполагается применение материала, анализ условий монтажа, влияние эксплуатационных и технологических особенностей этого оборудования на коррозионные свойства материала.
1.4.7.3. Испытания на общую коррозию
Испытания на общую коррозию проводят на образцах с большим отношением поверхности к объему. Среда испытания зависит от условий эксплуатации конструкции, изготовленной из исследуемого материала, и должна быть максимально приближена к реальной. Испытания проводятся в жидкой среде при постоянном или многократно повторяемом (циклическом) погружении образцов, в средах кипящих солевых растворов, в парах жидкостей, в окружающей атмосфере или почве.
В Российской Федерации и др. развитых странах принята единая пятибалльная шкала оценки общей коррозии материала (табл. 1.4.45). Критерием коррозионной стойкости при данном типе испытания является скорость коррозии материала vKop, мм/год — т. е. глубина проникновения коррозионного дефекта в глубь материала за единицу времени.
Кроме глубины проникновения коррозионного дефекта, оценка коррозионной стойкости материалов может производиться по анализу потери массы образца определенной площади за единицу времени. Оценка выполняется по десятибалльной шкале потери массы образца ТС (табл. 1.4.46).
Пересчет показателей vKOp и К производится по формуле vKop = К! у, где у — плотность материала, г/см3.
Для оценки коррозионной стойкости полимерных материалов применяется трехбалльная шкала, построенная по принципу учета изменений механической прочности материала и изменения его массы под воздействием агрессивной среды (табл. 1.4.47).
Таблица 1.4.45
Шкала общей коррозионной стойкости материалов
Балл коррозионной стойкости Скорость коррозии, мм/год Категория стойкости
1 2 3 4 5 Не более 0,10 0,10-1,00 1,10-3,00 3,10-10,0 Более 10,0 Сильностойкие Стойкие Пониженностойкие Малостойкие Нестойкие
Электродные процессы
115
Таблица 1.4.46
Шкала коррозионной стойкости материалов
Балл корроз. стойкости Категория стойкости Потеря массы К, г/(м 2 • ч ')
Железо и сплавы Медь и сплавы Никель и сплавы Алюминий и сплавы
1 Совершенно стойкие < 0,0009 <0,001 < 0,001 < 0,0003
2 Весьма 0,0009-0,0045 0,001-0,0051 0,001-0,005 0,0003-0,0015
3 стойкие (> 0,0045)-0,009 (> 0,0051 >-0,01 (> 0,005)-0,01 (> 0,0015>-0,003
4 Стойкие 0,009-0,045 0,001-0,051 0,01-0,05 0,003-0,015
5 (> 0,045)^0,09 (>0,051)-0,1 (> 0,05>-0,1 (> 0,015)-0,03
6 Пониженостойкие (> 0,09)-0,45 (> 0,1 >-0,5 (> 0,1 >-0,5 (> 0,031)-0,154
7 (> 0,45)-0,9 (>0,5)-1,02 (> 0,5)—1,0 (> 0,154)—0,31
8 Малостойкие (> 0,9Н,5 (> 1,020)—5,1 (> 1,0)—5,0 (> 0,31)-1,54
9 (>4,5)-9,1 (>5,1)-10,2 (> 5,0)—10,0 (> 1,54)-3,1
10 Нестойкие >9,1 > 10,2 > 10,0 >3,1
Шкала коррозионной стойкости полимерных материалов
Таблица 1.4.47
Тип пластмассы Изменение механических показателей Изменение массы (за 42 сут.)
Оценка стойкости Изменение прочностных свойств, % Изменение деформационных свойств, % Оценка стойкости Увеличение массы, % Уменьшение массы, %
Термо- 1 < 10 <10 1 <5 <3
пласты 2 (> 10)—15 (> 10)—20 2 (>5М5 (> 3)-20
3 > 15 >20 3 > 15 >20
Реакто- 1 < 15 — 1 <5 <5
пласты 2 (> 15)—25 — 2 (>5)-8 (>5)-8
3 >25 — 3 >8 >8
1.4.7.4. Определение склонности сталей и сплавов к питтинговой коррозии
Возникновение питтинговой коррозии в металлах и сплавах в процессе эксплуатации оборудования представляет собой большую опасность и требует строгого контроля, выполнение которого в производственных условиях весьма затруднено. Поэтому особое внимание уделяется лабораторным испытаниям на склонность материалов к питтингообразованию. Эти испытания основаны на подверженности легко пассивирующихся металлов к местному разрушению в окислительных средах в присутствии галоген-ионов. В результате такого разрушения коррозия развивается в отдельных центрах в виде глубоких поражений (язвин), называемых питтингами. Этому виду коррозии широко подвержены стали, титановые, алюминиевые и некоторые др. сплавы.
Наиболее полно изучен процесс питтингообразова-ния в коррозионностойких сталях. При наличии в среде хлорид-ионов возникает процесс активирования поверхности металла в некоторых участках, пассивное состояние которых по каким-либо причинам менее устойчиво, чем на остальной поверхности. Такими уча
стками могут быть неметаллические включения, структурные дефекты, границы зерен и т. п. В этих участках хлорид-ионы вытесняют кислород с поверхности материала и способствуют возникновению коррозионных повреждений. Коррозионный процесс, начавшийся в отдельной точке, резко уменьшает вероятность его появления на остальной поверхности материала. Поскольку основная часть поверхности остается в пассивном состоянии, возникает коррозионный электрохимический элемент, в котором анодом является питтинг, а катодом — остальная (неповрежденная) часть поверхности. Разность потенциалов между анодом и катодом может достигать величин 0,5-0,6 В. Преимущественное развитие в питтингах анодной реакции, протекающей с большой скоростью, приводит к значительному подкислению коррозионной среды, pH в питтингах понижается до 3-4.
Таким образом, исходя из типов и особенностей химических и электрохимических процессов, протекающих при питтинговой коррозии, методы исследования стойкости сталей и сплавов к питтинговой коррозии были разделены на химические и электрохимические.
116
Новый справочник химика и технолога
Химические методы исследования
При использовании химических методов исследования стойкости материалов к питтинговой коррозии образцы помещают в электролит, содержащий активатор и окислитель. Составы электролитов подбирают таким образом, чтобы потенциал металла сместился в сторону положительных значений. Так, для коррозионностойких сталей чаще всего применяют 10%-й раствор FeCl3 (pH = 2,2). Однако этот электролит не позволяет варьировать содержание компонента окислителя (Fe3+) или активатора (СГ), что бывает необходимо при испытаниях сталей с различным содержанием легирующих элементов. Для устранения этого недостатка иногда используется более сложный электролит, состоящий из 3 % NHjCl + 2% FeNHj (SO4)2 при 30 °C и длительности испытаний 30-650 минут. Образцы при таких испытаниях рекомендуется вращать со скоростью 100 об/мин.
В некоторых случаях используется смесь 1 г/л гексацианоферрата калия с хлоридом натрия, концентрация которого может составлять 5; 1; 0,1; и 0,001 моль/л. В этом электролите образуется химическое соединение FeK[Fe(CN)6] • 6Н2О, имеющее ярко-голубую окраску, поэтому места появления питтингов легко выявляются.
Способы измерения глубины при анализе питтинговой коррозии
Не меньшую (чем способ травления) важность в процессе определения склонности металлов и сплавов к питтинговой коррозии имеет способ оценки. При ускоренных испытаниях необходимо определять скорость проникновения коррозии в наиболее активных центрах, а также коэффициент неравномерности, который характеризует отношение глубины 2-3 наиболее глубоких питтингов к средней глубине питтингов. Глубина питтингов может определяться либо прямым — индикаторным — методом, либо косвенным — по потере массы образца.
Существует ряд приборов для измерения глубины коррозии. Наиболее точные из них — оптические. При таких замерах могут применяться обычные металлографические микроскопы — в этом случае замер глубины питтинга идет методом расфокусировки прибора. Сначала резкость фокусируется на поверхности образца, затем на дне язвины, а глубина определяется по микрометрическому лимбу на рукоятке тонкой настройки прибора. Кроме обычных микроскопов применяют еще целый ряд оптических приборов, например профилографов. Преимуществом профилографа является возможность измерения язвины и получение в увеличенном масштабе фотографической или компьютерной записи микрогеометрии поверхности образца, поврежденного коррозией. По такой профилограмме представляется возможным судить не только о глубине питтингов, но и об их форме и типе распределения по исследуемой поверхности.
При определении глубины проникновения питтингов возникает ряд сложностей. Так, например, невозможно измерить глубину большого числа питтингов, т. е. необходим произвольный отбор дефектов при контроле глубины питтинга, что может привести к занижению реальных результатов. В этом случае, для уменьшения вероятности ошибки, рекомендуется разделение контрольного образца на несколько зон (обычно квадратов, равных по площади) и проведение на каждой из них выборочного контроля глубины питтингов с последующим усреднением полученных данных. Таким образом определяется средняя глубина коррозии. Кроме того, на каждом из участков фиксируется максимальная глубина питтингов для определения коэффициента неравномерности коррозии.
Важнейшей характеристикой склонности к питтин-гообразованию является коэффициент питтингообразо-вания. Он представляет собой отношение средней глубины всех питтингов к условной глубине, вычисленной по потере массы образца, при допущении, что коррозия носит равномерный характер. Например, если коэффициент питтингообразования, вычисленный из данного соотношения, оказывается равным 30, то это значит, что глубина коррозии в отдельных зонах в 30 раз больше по сравнению с ее средней величиной, вычисленной по потере массы.
Коррозионная стойкость сплавов в растворах ингибиторов при образовании питтингов приведена в табл. 1.4.48. При оценке коррозионной склонности по этой шкале не учитывают очаги, расположенные на расстоянии до 3 мм от краев образца и на его торцах. Величина очагов подсчитывается с помощью трафарета, представляющего собой сетку из 100 одинаковых ячеек. Трафарет должен закрывать всю поверхность образца. Для испытаний рекомендуется использовать образцы размерами 100 х 50 х 2 или 50 х 25 х 2 мм.
Таблица 1.4.48
Шкала оценки склонности к питтинговой коррозии
Балл Максимальная глубина питтинга, мм/год Интенсивность распространения питтингов по поверхности, %
1 >0,4 >50
2 >0,4 <50
3 0,25-0,4 >50
4 0,25-0,4 <50
5 0,15-0,25 >50
6 0,15-0,25 <50
7 0,05-0,15 >50
8 0,05-0,15 <50
9 > 0,005 >50
10 > 0,005 <50
Электродные процессы
117
Электрохимические методы исследования
Склонность сталей и сплавов к питтинговой коррозии электрохимическими методами определяют по потенциалу питтингообразования, вычисляемому с помощью поляризационных кривых. При достижении для данного материала определенного значения потенциала наступает разрушение защитной пленки на образце в одной или нескольких точках. При этом наблюдается падение потенциала. Максимальное значение потенциала, при котором начинается его резкое изменение, называют потенциалом питтингообразования или потенциалом пробоя.
Поляризационные кривые снимают потенциометрическим или гальваностатическим методами. Кроме того, изучают кривые заряжения, снятые при постоянном потенциале или при постоянной плотности тока. Получаемые такими методами кривые приведены на рис. 1.4.38. На рис. 1.4.38, а, кроме потенциала питтингообразования фпо, можно также определить потенциал репассивации — фрп. В практических целях потенциал репассивации даже более значим, чем потенциал питтингообразования, т. к. он показывает, при каких отрицательных потенциалах питтинги не возникают. Для определения потенциала репассивации потенциометрическим методом дополнительно снимается поляризационная кривая обратного хода. При определении потенциала питтингообразования потенциометрическим методом следует иметь в виду, что его величина, хотя и не зависит от скорости снятия кривой в широких пределах от 0,1 до 10 В/ч, при высоких скоростях (> 10 В/ч) может увеличиваться, а при более малых (< 0,1 В/ч) — уменьшаться. При снятии потенциометрических кривых важную роль играет расположение образца в электрохимической ячейке. Ранее было принято располагать образец горизонтально между двумя платиновыми электродами, позднее наибольшее распространение получил метод вертикального закрепления образца. При этом платиновые электроды, между которыми создается электрическое поле, помещаются в пористые сосуды. Достаточно широко применяется вращение образца в процессе испытания.
В основном электрохимические методы контроля питтингообразования, как не требующие значительного времени для своего проведения, относятся к ускорен-
Рис. 1.4.38. Схематические потенциометрические (а) и гальваностатические (б) анодные кривые
1.4.7.5. Испытания на межкристаллитную коррозию
Испытания на этот вид коррозии нормированы государственным стандартом ГОСТ 6032-84.
Основной причиной возникновения межкристаллитной коррозии являются нагревы сталей при пластическом деформировании, термической обработке, сварке или технологические разогревы оборудования, приводящие к возникновению электрохимической гетерогенности между объемом зерен и приграничными участками в материале. В основном такая гетерогенность проявляется в образовании и развитии карбидных частиц, выделяющихся в границах зерен сталей при высокотемпературных нагревах. При этом происходит резкое обеднение по хрому зернограничного твердого раствора стали и изменение его электродного потенциала в местах фазовых превращений. Температурновременная область выделения зернограничных карбидных фаз в коррозионностойкой стали представлена на рис. 1.4.39. Внутри очерченной на рисунке области сталь обладает повышенной склонностью к межкристаллитной коррозии — сенсибилизацией. Область сенсибилизации может быть описана с помощью температурно-временных параметров: Гт1П и Ттах — температурный интервал сенсибилизации, Tmjn — минимальное время, необходимое для развития сенсибилизации.
Рис. 1.4.39. Температурно-временная область склонности коррозионностойкой аустенитной стали к межкристаллитной коррозии (МКК), связанной с обеднением границ зерен по хрому:
Гр — температура растворения карбидов; у — аустенит; К — карбиды
Для построения температурно-временных зон сенсибилизации сталей их подвергают специальному виду термической обработки — провоцирующему нагреву, температуры которого определяются химическим составом стали. Для хромистых сталей эти температуры находятся в области 950-1100 °C. Для хромоникелевых сталей (например 08Х18Н10Т) эта температурная область находится в пределах 500-750 °C. Этот темпера
118
Новый справочник химика и технолога
турный интервал гарантированно перекрывает интервал сенсибилизации аустенитных хромоникелевых сталей. Время нагревов обычно варьируется от десятых долей часа до 30 ч у аустенитных хромоникелевых сталей и до 60 ч у хромистых сталей. Термически обработанные образцы подвергаются испытаниям на коррозионную стойкость в различных средах, например в кипящих водных растворах азотной и серной кислот. Выбор вида коррозионной среды и длительности испытаний зависит от марки стали и ее назначения.
Контроль сталей с целью обнаружения склонности материала к межкристаллитной коррозии проводится путем изгиба на угол 90° образца, выдержанного в коррозионной среде. Отсутствие видимых невооруженным глазом трещин на растянутой поверхности образца говорит об отсутствии у стали склонности к межкристаллитной коррозии, наличие трещин указывает на то, что в данной температурно-временной области провоцирующих нагревов возможна сенсибилизация стали. При контроле растянутой зоны изогнутого образца допускается использовать увеличительные приборы (лупы). Кроме анализа состояния поверхности изогнутого образца допускается проводить контроль склонности стали к МКК с помощью травления образцов специальными травителями, позволяющими выявить межкристаллитные трещины.
1.4.7.6. Испытания на стойкость сталей и сплавов к коррозионному растрескиванию
Склонность стали к коррозионному растрескиванию может быть оценена по электрохимическим характеристикам напряженного и ненапряженного металла, а также путем физических исследований и прямых коррозионных испытаний. К физическим методам контроля относятся акустический и ультразвуковой методы, рентгеноструктурный анализ, оценка электросопротивления материала, магнитометрические методы. Общим во всех этих методах является то, что в их основу положен поиск поверхностной трещины, причина возникновения которой может быть как следствием коррози
онной агрессии среды, так и следствием технологических ошибок при производстве стали и детали из нее (микродефекты литья и сварки, термические и шлифовочные трещины). Таким образом, поиск дефектов стресс-коррозии сводится к поиску поверхностной трещины в исследуемом материале, а все существующие методы могут быть оценены по ряду формальных признаков. К ним относятся (вне зависимости от причины возникновения) надежность обнаружения дефекта, простота метода и возможность его применения в промышленных условиях.
Все методы контроля стойкости металлов против коррозионного растрескивания можно разделить на три группы в зависимости от условий задания напряжений, возникающих в образце при испытаниях. Это испытания при постоянной общей деформации, постоянной нагрузке и постоянной скорости деформации. В первом случае происходит имитация напряжений, возникающих в конструкции при изготовлении или под воздействием монтажных или эксплуатационных дефектов — т. е. остаточных напряжений. Так как коррозионное растрескивание большинства деталей оборудования различного назначения связано именно с остаточными напряжениями в конструкции, то такие испытания можно считать наиболее реалистичными. Испытания при постоянной нагрузке имитируют разрушения под действием рабочих нагрузок в оборудовании, например в условиях внутреннего (рабочего) давления в сосуде или трубопроводе. Анализ повреждений при постоянной скорости деформации относится к группе методов, не имеющих непосредственного производственного значения, так как вероятность стресс-коррозионного разрушения материала при таком виде нагружения конструкции мала. Однако эта группа методов позволяет глубже понять процессы, происходящие в материале при коррозионном растрескивании, и незаменима при лабораторных исследованиях.
В табл. 1.4.49 приведены основные составы электролитов, принятых при испытаниях сплавов на склонность к коррозионному растрескиванию.
Таблица 1.4.49
Составы электролитов для испытаний сплавов на склонность к коррозионному растрескиванию
Испытуемый материал Электролит Режим
Алюминиевые сплавы 3% NaCI Периодическое погружение при 18-25 °C*
3% NaCI + 0,1% Н2О2; 3% NaCI Погружение в электролит
, 3%NaCl Распыление в камере
Магниевые сплавы 0,001% NaCI; дистиллированная вода Периодическое погружение при 18-25 °C; распыление в камере (влажность 95-98 %)
3% NaCI Распыление в камере
Электродные процессы
119
Продолжение таблицы 1.4.49
Испытуемый материал Электролит Режим
Медные сплавы 20% NH3 • Н2О + 1% Си(ОН)2 5% NaNO2 + 4% CuSO4 • 5Н2О 1% Hg(NO3)2 + 1% HNO3 Погружение в электролит
Низколегированные стали 3% NH4NO3 + (50-57)% Ca(NO3)2 42% MgCl2 Погружение при кипячении
3%NaCl Распыление в камере
3% NaCl Периодическое погружение
Коррозионностойкие стали 3% NaCl Периодическое погружение
3% NaCl Распыление в камере
42% MgCl2 Погружение при кипячении
65% HNO3 Погружение при кипячении
Титановые сплавы 3% NaCl; CH3OH + 1,5% HC1 + 1,0% H2O CH3OH + 0,01 M NaCl; + 0,2% H2O Погружение в электролит
♦ Периодическое погружение проводят по режиму: 10 мин в электролите, 50 мин на воздухе.
Склонность к коррозионному растрескиванию принято определять по нескольким показателям. Это может быть время, необходимое для появления первой трещины или полного разрушения образца. Также может быть применен показатель сравнения механических свойств образцов в напряженном и ненапряженном состояниях при их разрушении в коррозионной среде. При испытаниях с постоянной скоростью деформации может быть применен показатель максимально достигаемой нагрузки или показатели изменения пластичности материала (длительная пластичность образцов и ее изменение в зависимости от условий испытания или изменение относительного сужения разрушенных образцов). Формы и типы образцов при испытаниях на стойкость против коррозионного растрескивания достаточно разнообразны и зависят от метода испытания, формы изделия, типа внешних нагрузок, которые может испытывать оборудование в процессе эксплуатации. На рис. 1.4.40 приведено одно из приспособлений для испытаний образцов при постоянной нагрузке. В настоящее время достаточно широко распространены так называемые С-образные образцы, некоторые виды которых представлены на рис. 1.4.41. При испытаниях могут применяться гладкие или ступенчатые образцы, а также образцы с предварительно нанесенной усталостной трещиной.
Испытания при постоянной общей деформации, хотя и являются наиболее простыми и распространенными, при исследовании стойкости сталей и сплавов к коррозионному растрескиванию имеют один общий недостаток. Фиксация на образцах заданной нагрузки достигается с помощью копирующего устройства (например, струбцин, в которых зажимаются образцы). При этом предполагается, что заданная в начале испытаний деформация остается неизменной в течение всего времени испытаний, т. е. не учитывается явление релак-
Рис. 1.4.40. Приспособление в виде скобы для испытаний образцов в напряженном состоянии:
7 — скоба; 2 — образец; 3 — вкладыш; 4 — винт
Рис. 1.4.41. Кольцевой образец для испытаний алюминиевых сплавов в напряженном состоянии: а) при постоянной деформации; б, в) при постоянной нагрузке;
1 — винт; 2 — образец; 3 — пружина
120
Новый справочник химика и технолога
сации напряжений в материале, величина которой может достигать 30-35 % от исходной величины. В качестве задаваемых вариантов нагрузки рассматриваются либо растягивающие усилия, либо изгиб, где применяются образцы в форме петли, либо U-образный образец и скобы, позволяющие менять стрелу прогиба испытуемого образца, тем самым изменяя величину его деформации. Образцы в виде скоб и петель обычно используются для листовых материалов толщиной до 3 мм. Растягивающие напряжения можно создать, используя кольцевые образцы, что делает этот вид испытаний особенно привлекательным при оценке стойкости труб. В этом случае растягивающие напряжения на внутренней поверхности кольца создаются путем введения клина определенной толщины между концами разрезного кольцевого образца. Для испытаний труб применяются образцы шириной 15 мм.
Испытания при постоянной нагрузке также имеют ряд недостатков. При их проведении следует учитывать тот факт, что по мере зарождения и развития коррозионной трещины «живое» сечение металла в образце уменьшается, а напряжения в процессе испытаний возрастают. Следует иметь в виду, что в ряде случаев при таком методе испытаний причина разрушения образца может быть иная, не связанная с коррозионным растрескиванием. Поэтому на разрушенных образцах факт коррозионного растрескивания следует подтверждать дополнительными методами контроля, например металлографическими исследованиями разрушенных образцов.
Испытания при постоянной скорости деформации позволяют максимально ускорить наступление эффекта коррозионного растрескивания в испытуемом образце. Это наиболее жесткий из всех методов испытаний, т. к. при его проведении удается достигнуть того же состояния образца, что и в случае применения образцов с предварительно нанесенной трещиной. Поэтому такие испытания значительно ускоряют процесс выявления склонности материалов к коррозионному растрескиванию и могут быть рекомендованы как метод предварительного контроля.
Физические методы исследования позволяют оценить возможность появления трещин стресс-коррозии по ряду косвенных показателей. К числу таких методов относятся рентгеноструктурный метод, методы, осно
ванные на измерении внутреннего трения, замере электросопротивления.
Рентгеноструктурный метод позволяет установить связь между степенью искажения кристаллической решетки сплава и стойкостью сталей к коррозионному растрескиванию. Эти опыты были впервые проведены на аустенитных сталях типа 08X18НЮТ, 08Х23Н18, ОЗХ23Н28МЗДЗТ, сплаве ХН38ВТ. Все сплавы были деформированы методом холодной прокатки на степень обжатия в 20 %. Время до разрушения образцов определяли в среде 42%-го хлористого магния и 5%-го хлорного железа при температуре 154 °C (табл. 1.4.50). Анализ табл. 1.4.50 подтверждает сделанный вывод о взаимосвязи искажений в кристаллической решетке материалов, определяемых по увеличению полуширины линий (311) сплавов, с их стойкостью к коррозионному растрескиванию.
Анализ образцов, склонных к коррозионному растрескиванию, проведенный методом внутреннего трения, показал, что у образцов, склонных к развитию трещины, наблюдается значительное изменение внутреннего трения по параметру «декремент затухания»; у образцов, не склонных к развитию трещины, никаких изменений в декременте затухания не происходит.
Метод электросопротивления нашел свое применение при изучении процесса развития коррозионной трещины в ходе испытаний. Однако точность этого метода невелика, а на изменение величины электросопротивления влияет не только эффект развития трещины, но и еще целый ряд факторов, например температура испытаний и т. п. Кроме того, метод достаточно трудоемок, поэтому в основном используется при научных и научно-практических исследованиях. Достаточно широкое распространение при контроле коррозионного растрескивания сталей и сплавов получили методы оптической, электронной и растровой электронной микроскопии, методы эмиссионного анализа.
Однако большинство из перечисленных способов контроля представляют собой методы, применимость которых ограничена базой лабораторных исследований. Их применение в производственных условиях или крайне ограниченно, или просто невозможно из-за сложности и громоздкости применяемых установок.
Таблица 1.4.50
Относительное увеличение полуширины линии (311) и стойкость деформированных сталей к коррозионному растрескиванию
Показатели Марка стали
08Х18Н10Т 08Х23Н18 ОЗХ23Н28МЗДЗТ 06Х20Н14С2 ХН38ВТ
Время до разрушения образца, ч 4,5 10 35 60 100*
Относительное увеличение полуширины линии (311) после деформации 20 % 2,3 2,0 1,7 1,8 1,4
* После испытаний трещин не обнаружено.
Электродные процессы
121
1.4.7.7. Методы неразрушающего контроля крупногабаритного оборудования, отработавшего расчетный срок службы, с целью обнаружения зон, поврежденных различными видами коррозии
Анализ нормативно-технических документов по вопросам промышленной безопасности, выпущенных ГГТН и Госстандартом РФ, позволяет утверждать, что ни один из документов, регламентирующих правила и нормы продления срока службы оборудования, законодательно не предписывает проведения прямых металлографических, механических и коррозионных испытаний металла на вырезках из элементов котлов, сосудов, паро- или продуктопроводов, реакционных колонн или нефтехранилищ, отработавших расчетный срок службы. Поэтому подобные испытания ограничиваются, как правило, неразрушающими методами контроля — визуально-измерительного и акусто-эмиссионного, ультразвуковой, магнитопорошковой или цветной дефектоскопией, замерами толщины стенок и их твердости.
К числу методов, получивших наибольшее распространение при оценке состояния металла с целью обнаружения коррозионных дефектов, относятся:
1. Оптико-визуальный метод, основанный на различном отражении света от различных участков контролируемой поверхности. Поверхностные дефекты можно наблюдать невооруженным глазом и с помощью оптических устройств. Достоинствами метода являются простота контроля и применяемых приборов, относительно небольшая трудоемкость. К недостаткам — малая чувствительность и, как следствие, недостаточная надежность метода, необходимость применения других методов контроля для подтверждения полученных результатов.
2. Акустические методы, основанные на сравнении различий в упругих колебаниях, возбуждаемых в материале, при наличии или отсутствии в нем дефектов. Наибольшее распространение в этой группе методов получили метод звукового (свободных колебаний) и ультразвукового диапазонов. Применение ультразвука дает возможность фактически неограниченного проникновения в глубину металла и обнаружения дефектов при любом их расположении.
3. Акустическая эмиссия, которая возникает в диапазоне звуковых колебаний вследствие освобождения энергии в твердых телах, подвергнутых пластической деформации или разрушению. Часть этой энергии преобразуется в упругие волны, которые распространяются в материале и могут быть обнаружены на его поверхности с помощью соответствующих преобразователей. Основные методы измерения акустической эмиссии — свободных колебаний и импедансный (импеданс — комплексное сопротивление, вводимое при рассмотрении колебаний акустических систем). Импедансный метод, основанный на использовании зависимости полного механического сопротивления (импеданса) контролируемого оборудования от качества соединения его отдельных элементов между собой, как и метод свобод
ных колебаний, используется для обнаружения зон нарушения жесткой связи (зарождения и роста трещины) между элементами системы. В соответствии с Правилами ГГТН РФ метод акустической эмиссии, как один из методов контроля, широко применяется при контроле сосудов, трубопроводов, емкостей, теплообменников, обязательно используется при контроле оборудования, подвергаемого пневматическим испытаниям.
4. Метод вихревых токов, основанный на регистрации распределения вихревых токов, наводимых электромагнитным преобразователем в контролируемом объекте. Применяется для обнаружения поверхностных дефектов в магнитных и маломагнитных деталях. Этот метод позволяет выявить нарушения сплошности, в основном трещины на различных по конфигурации деталях, имеющих защитные покрытия. На основе метода вихревых токов разработаны приборы для контроля лопаток турбин, сварных соединений и т. п.
5. Радиационные методы обнаружения дефектов и несплошностей в контролируемом объекте. В этом случае используется эффект изменения параметров проникающего ионизирующего излучения, взаимодействующего с контролируемым объектом. Наиболее распространенным среди них является рентгеноструктурный метод, широко применяемый, например, при контроле основного металла и сварных соединений трубопроводных систем.
6. Методы проникающих жидкостей, нашедшие широкое применение в производственных условиях благодаря своей простоте, высокой скорости и технологичности контроля. Эти методы, которые иногда называют методами химического контроля, основаны на использовании эффектов капиллярности, диффузии, сорбции светового и цветового контраста. При анализе состояния поверхности изделий этими методами на подготовленный участок оборудования наносят проникающую жидкость, которая заполняет полости дефектов (язв, раковин, волосовин, трещин). Затем проникающая жидкость удаляется с поверхности контролируемой зоны, оставаясь только в полостях, расположенных ниже общей поверхности. Таким образом, все поверхностные дефекты оказываются заполненными проникающей жидкостью, а неповрежденные участки освобождены от нее. Последующее проявление поврежденных зон возможно либо с помощью специального освещения (люминофорные методы), либо с помощью наносимых на контролируемую поверхность проявителей. Проявитель адсорбирует оставшуюся в дефектах проникающую жидкость, образуя индикаторный рисунок. Последний метод получил наибольшее распространение и вошел в обязательные способы контроля металла оборудования, перечисленные в соответствующих разделах нормативных требований ГГТН, под названием капиллярной или цветной дефектоскопии. Благодаря своей простоте и высокой чувствительности этот метод используется не только для обнаружения дефектов в поверхности мс-
122
Новый справочник химика и технолога
талла оборудования, но и как метод, подтверждающий результаты контроля состояния поверхности оборудования, полученные другими способами. Кроме цветной дефектоскопии, к этой группе методов относятся методы люминесцентной и люминесцентно-цветной дефектоскопии, фильтрующихся жидкостей, радиоактивных жидкостей.
7. Магнитные методы контроля металла оборудования основаны на изменении состояния и регистрации локальных магнитных полей над зоной дефектов или единичным повреждением металла. С помощью магнитных методов удается выявить как поверхностные дефекты, так и участки с подповерхностными изменениями структуры материала, различными видами повреждений. Дефект выявляется с помощью изменения магнитного сопротивления, намагниченности, магнитной проницаемости или магнитной индукции в зоне повреждения. К числу таких методов относятся магнитопорошковый, магнитографический и феррозондовый методы.
Полностью решить поставленную перед исследователями задачу — провести контроль фактического состояния оборудования в сжатые сроки ремонтного останова производства, соблюдая при этом максимально возможную точность и достоверность контроля, — не позволяет ни один из существующих методов. Поэтому в настоящее время разработаны и проходят опытнопромышленные испытания новые методы контроля, основанные на иных физических константах материалов. Одним из таких методов является метод магнитометрической оценки состояния металла оборудования, изготовленного из маломагнитных сталей. В основу метода заложено сравнение фактической магнитной проницаемости ЦфаКт, измеренной с помощью магнитометрических датчиков, установленных на элементах оборудования, в магнитном поле Земли или во внешнем магнитном поле определенной напряженности, с величиной критического значения магнитной проницаемости Цкр.
Цкр представляет собой предельно допустимое значение магнитной проницаемости, превышение которой приводит к проявлению в металле МКК. Цкр является универсальной величиной, при определении которой учитывается химический и фазовый состав стали, ее локальные пластические деформации, условия эксплуатации оборудования.
Метод включает четыре основных этапа.
1. Анализ химического состава стали (в основном содержания никеля в случае хромоникелевых сталей), изучение нормативно-технической документации на изготовление и монтаж оборудования, мест и технологии сварки, термической обработки, эксплуатационных и ремонтных журналов с целью выявления мест, потенциально склонных к проявлению МКК и других дефектов, составление карты-схемы исследования и определение объема исследования.
2. Измерение фактического значения магнитной проницаемости (согласно разработанной на основании
п. 1 карты-схемы) в магнитном поле Земли или во внешнем магнитном поле определенной напряженности. Сравнение фактического значения магнитной проницаемости с величинами Цкр, приведенными на графиках, подобных графикам рис. 1.4.35. Для определения магнитной проницаемости в процессе эксплуатации оборудования магнитометрические датчики устанавливаются на анализируемых поверхностях оборудования (во время ремонтных остановов оборудования) до монтажа теплоизоляции. Такая установка магнитометрических датчиков позволяет организовать непрерывные наблюдения за происходящими изменениями в металле в течение всего срока его работы. При технологических остановках определение магнитной проницаемости может производиться как с помощью датчиков, установленных на поверхности оборудования, так и с помощью приборов, вводимых внутрь оборудования через контрольные люки (рис. 1.4.42). В этом случае магнитометрический датчик закрепляется на конце волоконно-оптического эндоскопа. Введение такого комбинированного прибора внутрь агрегата позволяет не только получить данные о величине магнитной проницаемости, но и за счет визуального осмотра внутренней поверхности изучить ее состояние и более точно привязать места контроля к карте-схеме, установить координаты поврежденных зон и произвести вскрытие теплоизоляции только в местах, требующих проведения ремонтных работ.
3. При обнаружении зон, в которых Цфакт> Цкр, рекомендуется вскрыть теплоизоляцию над поврежденной зоной и провести ультразвуковую дефектоскопию и металлографический анализ металла с помощью реплик. При обнаружении зон, поврежденных МКК, необходим обязательный ремонт конструкции. Эксплуатация оборудования с трещинами МКК, особенно при низких и сверхнизких температурах, недопустима, т. к. приводит к снижению надежности и резкой потере запасов пластичности металла (табл. 1.4.51).
Рис. 1.4.42. Принципиальная схема прибора для неразрушающего контроля:
7 — постоянный магнит Mi (сильный — около 500 Т); 2 — мембрана;
3 — постоянный магнит М2 (слабый Mi: М2 = 10 : 1);
4 — катушка индуктивности; 5 — реостат; 6 — амперметр;
7 — генератор переменного тока и индикатор индуктивности;
8 — магнитный датчик; 9 — компенсатор; 10 — корпус
Электродные процессы
123
4. Выдача рекомендаций по характеру ремонтных работ, обеспечивающих необходимую прочность, надежность и безопасность дальнейшей эксплуатации оборудования.
Предложенный метод неразрушающего контроля аустенитных хромоникелевых сталей как в эксплуатационных условиях, так и при исследовании в лабораториях резко сокращает сроки испытаний стойкости металла к МКК. Снижается срок регламентных работ при оценке фактического состояния металла оборудования очистки и сжижения газов, их транспортировки, а также оборудования прецизионной техники, аэрокосмических систем.
В качестве примера использования метода неразрушающего контроля могут служить данные, полученные при исследовании трубопровода системы сжижения гелия диаметром 108 мм с толщиной стенок 8 мм, изготовленного из стали 12Х8Н12Т, содержащей 0,11 % углерода, 18 % хрома, 0,3 % титана и 12,2 % никеля.
Время работы трубопровода при температуре 4,2 К на момент контроля составляло 72108 ч. Время технологических разогревов металла трубопровода до температур 925 К — 82,5 ч, время простоя в процессе межэксплуатационных остановок криосистемы при температуре ~ 300 К — около 12000 ч.
Таблица 1.4.51
Механические свойства образцов хромоникелевых сталей без коррозионных повреждений и с трещинами межкристаллитной коррозии глубиной не менее 100 мкм в поверхностном слое рабочей части образца
K.CV — ударная вязкость образцов с V-образным надрезом; б5 — относительное удлинение образца, длина рабочей части которого равна 5 диаметрам; ств — временное сопротивление (предел прочности при одноосном статическом растяжении).
Провоцирующий 12Х18Н10Т 12Х18Н12Т
нагрев 293 К 4,2 К 293 К 4,2 К
Г, К т, ч ов, МПа б5, % KCV, МДж/м2 ств, МПа б5, % KCV, МДж/м2 ств, МПа б5, % KCV, МДж/м2 ств, МПа б5, % KCV, МДж/м2
Без трещин МКК на рабочей поверхности образца
— 570 40 3,0 1620 24 2,2 625 41 3,1 1650 24 2,2
2 590 39 3,0 1800 21 2,0 645 37 3,0 1850 19 2,0
873 3 595 36 3,0 2100 19 1,7 645 35 3,0 2000 19 1,8
4 615 34 2,3 2150 15 1,5 670 24 2,6 2150 16 1,4
5 660 24 1,9 2150 11 1,0 695 22 2,1 2250 9 0,7
8 710 11 1,2 2300 7 0,6 710 9 1,0 2700 6 0,4
2 615 37 3,0 1900 19 1,7 665 35 2,7 1950 18 1,9
3 645 34 2,7 1950 19 1,3 680 33 2,5 2100 18 1,9
923 4 670 31 2,0 1950 16 1,1 685 22 1,9 2250 12 1,2
5 695 17 1,6 2100 9 0,8 700 17 1,0 2400 7 0,6
8 710 9 1,0 2150 6 0,5 725 8 1,0 2400 7 0,3
2 645 34 2,6 1950 18 1,5 690 33 2,1 2100 15 1,6
3 665 31 2,2 2100 14 1,5 695 31 2,0 2150 15 1,2
973 4 690 22 1,4 2200 11 0,7 710 20 1,1 2400 11 1,0
5 715 14 1,1 2250 7 0,4 710 14 0,8 2450 6 0,3
8 720 8 0,7 2400 2 0,1 745 6 0,5 2600 4 0,2
Трещины МКК глубиной не менее 100 мкм на поверхности рабочей части образца
2 660 27 2,2 2570 15 1,7 715 26 2,0 2200 15 1,8
3 710 24 2,0 2610 15 1,4 725 24 1,6 2250 16 1,2
1023 4 745 22 2,0 2700 14 1,2 740 20 1,4 2400 15 1,0
5 795 19 1,7 2710 11 1,0 760 18 1,0 2650 14 0,8
8 810 16 1,5 2800 11 0,5 800 15 1,0 2800 12 0,4
873 5 8 * 690 8 0,7 2100 2 0 695 700 14 6 1,2 0,6 2000 2150 1 0 0 0
923 5 700 6 0,5 1950 2 0 700 9 0,4 1900 0 0
8 700 4 0,3 1740 0 0 710 3 0,1 1700 0 0
973 5 690 5 0,5 1710 1 0 710 12 0,6 2100 4 0,1
8 700 3 0,2 1670 0 0 — — — — — —
* Получить трещины на рабочей поверхности образца не удалось.
124
Новый справочник химика и технолога
Контроль состояния металла трубопровода проводился во время планово-ремонтной остановки оборудования. На основании анализа документальных данных была составлена карта-схема контроля гиба (рис. 1.4.43).
При исследовании внутренней поверхности гиба трубопровода датчик крепился на конце волоконно-оптического эндоскопа, который вводился внутрь трубопровода на заданное расстояние через расположенный рядом с гибом вентиль. Замер магнитной проницаемости совмещался с одновременным визуальным осмотром внутренней поверхности гиба, что позволяло четко определить и зафиксировать координаты контролируемых зон.
Рис. 1.4.43. Карта-схема контроля гиба криотрубопровода (108 х 8 мм) из стали 12Х18Н12Т
Определение величины магнитной проницаемости в контролируемых зонах проводилось в магнитном поле Земли и во внешнем магнитном поле напряженностью 40 кА/м. Исходная величина магнитной проницаемости стали до эксплуатации составляла: в магнитном поле Земли — 1,023, во внешнем магнитном поле — 1,029. Критическое значение магнитной проницаемости для стали с 12,2% никеля, согласно данным рис. 1.4.35, составляет цкр^ 1,435 в поле Земли и цКр= 1,470 во внешнем поле напряженностью 40 кА/м. Данные по определению магнитной проницаемости в различных зонах гиба приведены в табл. 1.4.52.
Анализ табл. 1.4.52 показывает, что в зонах 2, 3,4, 8, 9, 10 величина фактической магнитной проницаемости отличается небольшим ее увеличением по сравнению с исходной магнитной проницаемостью. Более отчетливо ее повышение наблюдается в зонах 1, 5, 6, 7 и 11.
В зоне 6 фактическое значение магнитной проницаемости значительно превышает ее критическое значение » Цкр и составляет в магнитном поле Земли Цфакт = 1,926; во внешнем магнитном поле напряженностью 40 кА/м Цфакт = 1,985. Следовательно, согласно сделанным ранее выводам, зона 6 является наиболее опасной, склонной к коррозионным повреждениям, понижающим запас прочности металла. С целью под
тверждения полученных данных гиб был вырезан из трубопровода для проведения ультразвуковой дефектоскопии, микроанализа и определения глубины коррозионных трещин. При исследовании в зоне 6 были обнаружены коррозионные трещины глубиной более 20 % от толщины стенки гиба. При металлографическом анализе внутренней поверхности гиба с использованием реплик в структуре металла зоны 6 наблюдались многочисленные трещины, разъедание межзеренных граничных слоев (рис. 1.4.44, а). В зонах 5 и 7 наблюдались лишь единичные точечные коррозионные дефекты (рис. 1.4.44, б).
Широкое применение неразрушающих методов контроля, повышенный интерес к их внедрению в практику и к разработке новых методов неразрушающего анализа обусловлены ситуацией, сложившейся в промышленности на современном этапе. Все более заметное старение основных фондов требует повышенного внимания к обеспечению надежной и безопасной эксплуатации оборудования, невозможной без проведения периодического контроля состояния металла с целью обнаружения поврежденных, в частности поврежденных различными видами коррозии, зон, их ремонта или замены отдельных поврежденных деталей или узлов.
В табл. 1.4.53 приводятся результаты сравнительной оценки возможностей применения различных методов неразрушающего контроля для обнаружения того или иного вида коррозии или других дефектов металла. К числу основных показателей возможности применения того или иного метода относится его чувствительность к определению различных видов дефектов (табл. 1.4.54).
Таблица 1.4.52
Величина магнитной проницаемости в зонах гиба трубопровода жидкого гелия
№ зоны Магнитная проницаемость цфакг (средняя из 5 измерений)
В поле Земли Во внешнем магнитном поле напряженностью 40 кА/м
Мфает Мфаю
1 1,126 1,163
2 1,099 1,099
3 1,099 1,105
4 1,098 1,100
5 1,236 1,277
6 1,926 1,985
7 1,262 1,283
8 1,097 1,090
9 1,099 1,110
10 1,097 1,102
11 1,119 1,133
* В поле Земли = 1,435, во внешнем магнитном поле напряженностью 40 кА/м цкр = 1,470.
Электродные процессы
125
Рис. 1.4.44. Микроструктура внутренней поверхности металла гиба криотрубопровода жидкого гелия из стали 12Х18Н12Т: а) зона 6 (рис. 1.4.43); б) зона 5. х200
Таблица 1.4.53
Условия применимости (+) или неприменимости (-) методов неразрушающего контроля
Принятые обозначения. А — металлы магнитные, Б — металлы немагнитные, В — неметаллы; Г — простая форма (параллелепипед, труба, лист и т. п.), Д — сложная форма; Е — поверхность с высокой степенью шероховатости (сварка, литье и т. п.), Ж — высокое качество подготовки поверхности; 3 — на поверхности, И — в подповерхностном слое, К — в глубине металла, Л — под слоем защитного покрытия; М — в условиях производства, Н — в условиях эксплуатации, О — при ремонте.
Метод контроля Материал контролируемой детали Форма детали Чистота обработки поверхности Место расположения дефекта Контроль
А Б В Г д Е Ж 3 И К л М н О
Визуальный + + + + + + + + — — — + + +
Рентгенографический + + + + + + + + + + + + + +
у-Графический + + + + + + + + + + + + + +
Магнитопорошковый + — — + — — + + + — + + + +
Магнитографический + — — + — + + + + — — + + +
Феррозондовый + — — + — — + + + + — + + —
Вихревых токов + + — + + — + + + — + + + +
Цветной + + + + + — + + — — — + + +
Люминесцентный + + + + + — + + — — — + — +
Ультразвуковой теневой + + + + — + + + + + — + — —
Ультразвуковой резонансный + + + + — — + — + + — + — —
Ультразвуковой эхо-импульсный + + + + + + + + + + + + + +
Акустический импедансный + + + + — — + — — — — + + —
Свободных колебаний + + + + - + + - - + - + - -
Таблица 1.4.54
Чувствительность различных методов неразрушающего контроля
Метод Чувствительность
Визуальный Дефекты протяженностью около 0,1 мм с помощью оптических приборов (до сотых долей мм)
Рентгенографический Дефекты шириной 0,1 мм и глубиной 1-2,5 % от толщины изделия
у-Графический Дефекты шириной 0,1-0,2 мм и глубиной 1-4 % от толщины изделия
Магнитопорошковый Трещины шириной раскрытия 0,0005-0,001 мм и глубиной 0,01 мм и более
126
Новый справочник химика и технолога
Продолжение таблицы 1.4.54
Метод Чувствительность
Магнитографический Трещины и непровары протяженностью 10 % от толщины сварного соединения
Феррозондовый Трещины шириной раскрытия 0,0005-0,001 мм и глубиной 0,01 мм и более
Вихревых токов Трещины глубиной 0,25 мм и более
Цветной Трещины шириной раскрытия 0,001-0,01 мм и глубиной 0,05 мм
Люминесцентный Трещины шириной раскрытия 0,001-0,01 мм и глубиной 0,05 мм
Ультразвуковой теневой Трещины протяженностью от 1 мм, глубиной от 0,01-0,05 мм и более
Ультразвуковой эхоимпульсный Поверхностные трещины глубиной 0,01 мм, внутренние дефекты площадью 0,5 мм2
Свободных колебаний Непроклей, непропай общей площадью 15 мм2
1.4.8. Способы защиты металлических и неметаллических материалов от коррозии
Основные методы борьбы с коррозией могут быть классифицированы на основе закономерностей протекания коррозионных процессов. Скорость коррозии может быть снижена следующими способами:
- выбором соответствующего для данных условий эксплуатации коррозионностойкого сплава или новых конструкционных решений и методов;
- введением в материал или во внешнюю коррозионную среду химических веществ, замедляющих протекание катодных или анодных процессов;
- изоляцией поверхности материала от воздействия внешней агрессивной среды путем нанесения на его поверхность металлических покрытий, органических или неорганических пленок;
- электрохимической защитой.
Первая группа методов реализуется на стадии формирования технических условий на изготовление устройства и его конструирования, на стадии металлургического производства применяемого при его изготовлении материала — стали или сплава — введением в них соответствующих легирующих добавок. При разработке изделий, работающих в агрессивных коррозионных средах, должны применяться такие конструктивные решения, которые не допустят образования в изделии застойных зон, узких щелей. При конструировании корпусных изделий должны быть предусмотрены вентиляционные отверстия и т. п.
Вторая группа методов — снижение эффективности катодного или анодного процесса — может быть реализована несколькими способами. К их числу относятся мероприятия, замедляющие катодный процесс, для чего необходимо уменьшение площади макрокатодов, например путем закалки для углеродистых сталей или путем улучшения химической чистоты применяемых материалов; повышение перенапряжения катодного процесса, например путем легирования сталей мышьяком, сурьмой или висмутом. Эффективность анодного процесса можно понизить введением в применяемый материал легирующих добавок, повышающих термодинамическую устойчивость анодной фазы: легированием сталей никелем, никеля — медью, меди — золотом. Возможно также дополнительное легирование сплавов
элементами, способствующими пассивированию анодной фазы: легирование сталей хромом, кремнием или алюминием, никеля — хромом. Благотворно влияет на снижение эффективности анодного процесса введение в хромистые и хромоникелевые стали стабилизирующих добавок — титана, ниобия, ванадия, тантала и т. п. Наконец, снижения эффективности анодного процесса также можно добиться уменьшением площади анодной фазы на поверхности материала путем проведения термической обработки, снижающей внутренние напряжения в материалах, и легированием сталей и сплавов элементами, препятствующими образованию зернограничных сегрегаций примесных атомов, например легированием сталей такими элементами, как вольфрам и молибден.
Эта же группа методов борьбы с коррозией предусматривает модифицирование агрессивной внешней среды путем введения в нее ингибирующих добавок, снижающих ее агрессивность.
Третья группа методов подразумевает использование для борьбы с коррозией защитных покрытий. Основное назначение защитного покрытия, с одной стороны, состоит в создании барьерного слоя, препятствующего прониканию агрессивной среды к поверхности материала; с другой — в ограничении или предотвращении образования новой фазы (продуктов коррозии) на поверхности раздела «материал — покрытие», т. е. защитные покрытия должны обладать высокой химической устойчивостью, слабой проницаемостью для жидкостей и газов, хорошей адгезией к металлу или неметаллическому материалу, высокой стабильностью структуры и относительно высокой механической прочностью и долговечностью.
Защитные покрытия разделяются на неметаллические и металлические. Неметаллические покрытия, в свою очередь, подразделяются на органические и неорганические покрытия. К органическим покрытиям относятся лаковые, битумные, пластмассовые, эпоксидные, резиновые и т. п. покрытия, к неорганическим прикрытиям — оксидные, бетонные, фосфатные, силикатные и т. п.
Для повышения эффективности защиты изделия, работающего в агрессивных средах, часто последовательно наносится несколько разнотипных покрытий,
Электродные процессы
127
например цинковое покрытие с последующим фосфатированием или нанесением нескольких слоев лакокрасочного материала.
Четвертая группа защитных методов предусматривает электрохимическую защиту, т. е. наведение в материале защищаемого изделия токов, препятствующих протеканию катодных или анодных процессов.
1.4.8.1. Электрохимическая защита
Электрохимическая защита наиболее широко применяется для каркасов зданий и сооружений химической промышленности, транспортных объектов, в том числе трубопроводов наземного и подземного расположения, эксплуатационных колонн буровых скважин, станций подземного хранения газов и т. п.
Катодная и протекторная защиты
Катодная защита, открытая Деви, известна с 1824 г. Она заключается в уменьшении скорости электрохимической коррозии путем катодной поляризации или с помощью вспомогательных электродов (протекторов), являющихся анодами по отношению к корродирующей системе. Катодная защита применяется в основном для подводных или подземных сооружений — морских конструкций, пирсов, трубопроводов. Она может быть осуществлена с помощью внешних источников тока или с помощью «жертвенных» анодов — протекторов.
Принцип катодной защиты заключается в том, что потенциал поверхности металла (относительно электролита, грунта и т. п.) сдвигается в сторону отрицательных значений за счет подвода электронов (рис. 1.4.45). В результате этого атомы железа не переходят в раствор в виде положительно заряженных ионов, а pH электролита, контактирующего непосредственно с металлом, смещается в щелочную область. Благодаря высокому pH на защищаемую поверхность осаждаются гидроксид магния, карбонаты кальция и магния, образуя пленку, подобную накипи. Эта пленка экранирует металлическую поверхность и затрудняет диффузию кислорода. Таким образом, в металлическую поверхность извне должен подводиться постоянный электрический ток. Этот ток может идти от гальванического элемента или выпрямителя, отрицательный полюс которых связан с защищаемым элементом, а положительный полюс — с анодом. Плотность защитного тока зависит от толщины осаждаемой пленки и может уменьшаться по мере ее роста.
Катодная защита широко распространена для подземных и гидротехнических сооружений, реакторов. В химической промышленности она используется весьма ограниченно, в основном для защиты конструкций в технической воде, сточных водах предприятий и в ряде сред, содержащих ионы хлора. Однако в агрессивных средах ее применение затруднено, т. к. для достижения защитного катодного потенциала необходимо прилагать высокую плотность тока, при которой на защищаемой поверхности происходит интенсивное выделение водорода.
При использовании катодной защиты от коррозии трубопроводов и подземных складских помещений и сооружений необходимо предусмотреть несколько обязательных условий.
1. Защищаемый трубопровод должен иметь высокую электропроводимость в продольном направлении, что легко достигается при сварных соединениях труб. При использовании других типов соединений, например гуммированных, винтовых или вставных муфт, следует предусмотреть электропроводящие перемычки.
2. Защищаемый трубопровод должен быть электрически изолирован от всех других, проложенных ранее металлических систем — трубопроводов, кабелей, заземлений и т. п.
3. Стальные конструкции должны иметь изолирующую оболочку.
Для обеспечения указанных требований необходимо выполнение ряда мероприятий.
1. Для экономически оправданного применения катодной защиты необходимо оборудовать защищаемый трубопровод во всех концевых точках системами, изолирующими его от посторонних конструкций.
2. При использовании оболочковых труб на перекрестках или пересечениях дорог нужно предусмотреть изолирующие вставки для предотвращения контакта между трубопроводом и оболочкой.
3. Для предотвращения металлического контакта защищаемого трубопровода с другими металлическими объектами его прокладка должная вестись таким образом, чтобы минимальное расстояние между ними было не менее 0,4 м. Если по техническим причинам расстояние между объектами оказывается менее 0,1 м, то должна быть предусмотрена механическая защита в виде изолирующих плит между трубопроводом с катодной защитой и другими объектами. При параллельной прокладке трубопроводов расстояние между ними должно быть не менее 1 м.
Катод Электролит (грунт) Анод
Рис. 1.4.45. Принцип действия катодной защиты
128
Новый справочник химика и технолога
4. Арматура, расположенная на бетонном фундаменте, должна быть изолирована от бетона и железобетонных конструкций изолирующими прокладками и плитами.
5. Конструкция крепления бетона, стенок и дюкеров должна быть выполнена таким образом, чтобы исключить электрический контакт с бетоном или железобетоном.
6. Если прокладка трубопровода с катодной защитой осуществляется по методу мостовых конструкций, подвесные устройства этих мостов не должны иметь электрического контакта с металлом трубопровода, т. е. должны быть предусмотрены изолирующие системы в местах соединения трубопровода и мостовой конструкции, а электрические соединения надземных (мостовых) и подземных частей трубопровода должны быть выполнены с помощью специальных электрических кабелей.
7. Управляющие элементы трубопровода с электрическим приводом — задвижки, заслонки, клапаны и т. п. — не должны быть заземлены. Электрическая защита этих элементов осуществляется с помощью выключателей защиты.
8. Через каждые 1-2 км на защищаемом трубопроводе выделяются измерительные участки для определения потенциала труба/грунт, а через каждые 5 км — участки для измерения тока. Минимальная защитная разность потенциалов (по отечественным нормативам) должна составлять (-1,1)-(-0,72) В по отношению к медносульфатному электроду сравнения. Превышение этих величин до значений > (-1,22) В может нарушить адгезию изоляции трубопровода, поэтому максимально допустимая разность потенциалов в изолированных трубопроводах должна быть < (-1,22) В.
Для морских гидротехнических сооружений применяют двухступенчатую схему катодной защиты. На первом этапе поддерживается высокая плотность тока, при которой образование и отложение защитной пленки протекает ускоренно. После того, как на защищаемой поверхности образуется достаточно толстая пленка из труднорастворимых солей, приступают ко второму этапу защиты. При этом плотность тока делают более низкой, чем первоначальная. Двухступенчатая защита может быть также обеспечена монтированием двух типов протекторов — основных и вспомогательных. Основные протекторы рассчитаны на весь период действия защиты, а вспомогательные — только на начальный этап повышенной плотности тока. Поэтому для основных протекторов отношение массы к поверхности уменьшено, зачастую они выполняются в виде сфер, в то время как вспомогательные имеют вид дисков или лент.
Чтобы обеспечить эффективную защиту, протекторы должны обеспечивать:
- максимальное количество электрической энергии, соответствующей единице массы протектора;
- минимальную потерю мощности в результате коррозии самого протектора;
- максимальную первоначальную электродвижущую силу в системе протектор—сооружение;
- минимальное омическое сопротивление системы протектор—сооружение;
- максимальный срок службы протектора;
- минимальную анодную поляризацию.
Протекторы в основном изготавливаются из магниевых, цинковых или алюминиевых сплавов, реже — из углеродистых сталей. Эффективность протекторной защиты подземных сооружений может быть повышена, если поместить протектор в специальную смесь солей, называемую активатором или наполнителем. Наполнитель служит для понижения собственной коррозии протектора, уменьшения анодной поляризации, уменьшения сопротивления протекающему к защищаемой поверхности току и для устранения причин, вызывающих образование плотных пленок продуктов коррозии на поверхности протектора. Применение наполнителя обеспечивает стабильную силу тока в цепи протектор— сооружение и высокий коэффициент полезного действия системы защиты. В случае магниевых сплавов основными компонентами наполнителя служат гипс, глина, сульфаты магния и натрия. Возможно применение ряда минералов, в частности астраханита, мирабилита, эпсомита и т. п. Наполнители приготавливаются путем смешивания сухих солей и глины с водой до получения сметанообразной пасты.
В зависимости от вида защищаемого объекта и эксплуатационных условий защищаемой системы можно использовать одиночные, групповые и сплошные протекторные устройства, обеспечивающие защиту на участках большой протяженности.
Некоторые виды катодной защиты
А. Катодная защита с гальваническим анодом. Гальванический анод может быть использован для катодной защиты труб с хорошо изолирующим покрытием или коротких трубопроводов, например трубопроводов с пластмассовым покрытием, складских емкостей, защитных труб, дюкеров или ограниченных участков трубопроводов, находящихся под активным коррозионным воздействием, несмотря на незначительную отдачу ими тока в землю. Металлом гальванического элемента обычно служит магний. В грунте с удельным электросопротивлением не более 20 Ом • м можно использовать цинк, имеющий более высокую эксплуатационную стойкость. Напряжение между цинком и сталью с катодной защитой составляет Д(7=0,2В, а между магнием и сталью---0,6 В.
В табл. 1.4.55 приведены основные свойства анодных материалов.
Электродные процессы
129
Таблица 1.4.55
Некоторые свойства основных анодных материалов
Свойство Материал
Цинк Магний Алюминий
Стандартный потенциал UH, В Годовые потери, кг/(А • г) Фактическая емкость тока, А • ч/кг Эффективная емкость тока, А • ч/дм3 Коэффициент использования, % Масса электрода, кг, при 10-летней эксплуатации для 0,1 А Равновесный потенциал грунта относительно Cu/CuSO4, В Напряжение относительно стали с катодной защитой (tfcuzcuso, =-0,85 В), В -0,8/-0,85 11 820 5800 95-99 12 (—0,9)—(—1,1) -0,2 -1,0/-1,3 8 2200 3840 50-55 8 (—1,4)—(—1,6) -0,6 -0,7/-1,1 3-5 2900 5300-7600 50-95 3,7 (-0,9)-(-1,2) -0,3
Для сооружений и трубопроводов, испытывающих сильное воздействие токов утечки, гальванические аноды непригодны, поскольку большие изменения потенциалов вследствие колебаний токов утечки не могут быть выровнены их относительно низкими потенциалами. Кроме того, при изменении полярности может возникнуть дополнительный ток утечки от гальваниче
ского анода.
Б. Катодная защита с посторонним (внешним) источником тока. Катодную защиту с использованием тока питающей сети, пропущенного через выпрямитель, целесообразнее всего использовать для протяженных или разветвленных систем, например питательных трубопроводов, магистральных трубопроводов и т. п., а также для оборудования с износившимся защитным покрытием.
Необходимость в защитном токе и удельное электросопротивление грунта являются решающими факторами для выбора того или иного способа защиты. В частности, в системах с пластмассовым защитным покрытием рационально использовать катодную защиту с гальваническими анодами из магния или цинка. Для защиты протяженных систем с высокой величиной потребляемого защитного тока повсеместно используется катодная защита с внешним источником тока. Схема использования такой защиты приведена на рис. 1.4.46.
Величину требуемого защитного тока / = А £7//?а можно регулировать изменением исходного напряжения выпрямителя At/ (до 50 В), устанавливаемого между анодом и защищаемым объектом. Величина /?А является сопротивлением рассеянию в грунте горизонтальных или вертикальных анодов внешнего источника тока, служащих для ввода защитного тока в грунт. Они могут быть выполнены в виде группы отдельных анодов или в виде непрерывных продольных анодов. Для получения необходимого низкого сопротивления рассеянию в земле и длительной стойкости аноды укладывают в коксовую подушку. Преимущественно используются графитовые электроды, элек
троды из ферросилиция, а при высоком удельном электросопротивлении грунта — электроды из железного скрапа. В табл. 1.4.56 приведены основные свойства таких электродов.
Аноды постороннего источника тока обычно используются в зонах с низким удельным электросопротивлением грунта. Это позволяет снизить требуемое анодное
напряжение и связанные с ним затраты на энергию для катодной защиты. Используемые для этих целей выпрямители должны быть рассчитаны на определенное сопротивление и длительный срок службы. Напряжение обычно устанавливается на вторичной обмотке трансформатора или через регулировочный трансформатор на первичной обмотке. Система должна быть оснащена амперметром и высокоомным вольтметром для измерения потенциала с целью контроля основных параметров. При влиянии тока утечки рекомендуются выпрямители с регулированием потенциала.
Рис. 1.4.46. Структура катодной защиты с внешним источником тока:
1 — шкаф защитного тока; 2 — корпус шкафа;
3 — соединительная муфта; 4 — анодный кабель;
5 — подсоединение низкого напряжения; 6 — подсоединение к вольтметру; 7 — подсоединение защитного тока; 8 — электрод сравнения; 9 — защищаемый трубопровод; 10 — анод внешнего источника; 11 — коксовая подушка
130
Новый справочник химика и технолога
Таблица 1.4.56
Основные свойства анодов постороннего (внешнего) источника тока
Характеристики анода Материал анода
Сталь Чугун Ферросилиций Графит
Длина анода, м
1 (стальные консоли) I (стальные рельсы) 1 0,5 1 1,5 1 1,2 1,5
Диаметр, м 0,3 0,14 0,28 0,04 0,06 0,075 0,06 0,06 0,08
Высота, м 0,13 0,13
Масса, кг 56 43 35 3 28 43 5 6 8
Плотность, кг/дм3 7,8 7,8 7,6 7 7 7 2,1 2,1 2,1
Износ без коксовой подушки, кг/(А-г) 10 10 10 0,2-0,3 0,2-0,3 0,2-0,3 1 1 1
Износ с коксовой подушкой, кг/(А-г) 5 5 8 Около 0,1 Около 0,1 Около 0,1 Около 0,5 Около 0,5 Около 0,5
Стойкость для 1 А/г без коксовой подушки, г 5 4 3 15 80 140 5 6 8
Стойкость для 1 А/г с коксовой подушкой, г 1 8 8 30 260 430 10 12 16
Возможность разрушения Нет Нет Малая Малая Малая Малая Большая Большая Большая
Сфера использования Только с коксовой подушкой в грунтах с низкой электропроводимостью (р> 100 Ом • м) Там, где требуется длительная стойкость даже без коксовой подушки В коррозионноактивных грунтах даже без коксовой подушки
В. Катодная защита от электрокоррозии. Этот вид коррозии может возникнуть в случае нахождения защищаемого оборудования в зоне действия сильных внешних источников тока, например вблизи от высоковольтных линий электропередачи, трамвайных путей и т. п. Если в таких системах возникают токи утечки, то они могут послужить причиной появления в защищаемой системе электрокоррозии. При этом виде коррозии (рис. 1.4.47) ток утечки возвращается через кабель к рельсам. В этом случае электродренаж через металлический кабель к конструкциям, вызывающим ток утечки, может предотвратить электрокоррозию. В ряде случаев такой защиты оказывается достаточно, однако иногда требуется надежно отвести ток и обеспечить эффективную катодную защиту объектов. Так, в непосредственной близости от выпрямителя на соседних с ним кабелях или трубопроводах часто наблюдается коррозия, обусловленная током утечки. В этом случае, если через дренаж нельзя отвести весь ток утечки, то катодная защита достигается с помощью принудительного отвода тока утечки (рис. 1.4.47, в). При этом в систему отвода тока утечки дополнительно включается выпрямитель, связанный с сетью питания. При сильных колебаниях потенциала отводимого тока утечки применяют защитный выпрямитель, ограничивающий ток. Перегрузка катодного защитного выпрямителя в результате короткого замыкания контактных проводов, разрыва рельсов или влияния кабеля и трубопровода при слишком высоких напряжениях может быть предотвращена с помощью соответствующих предохранителей.
Рис. 1.4.47. Ток утечки транспорта, работающего на постоянном токе: а) вблизи тягового выпрямителя;
б) катодная защита с помощью поляризованного электродренажа; в) катодная защита путем отвода тока утечки;
1 — тяговый выпрямитель; 2 — контактный провод; 3 — рельсовый ток; 4 — кабель и защищаемый трубопровод; 5 — коррозионный ток;
б — анодная зона; 7 — переходная зона; 8 — катодная зона
Электродные процессы
131
Г. Катодная защита оборудования в промышленных системах. Для промышленных установок и систем, например электростанций, химических и нефтехимических предприятий, требуется значительное число трубопроводных систем различных диаметров, прокладываемых в грунте; установленных на поверхности земли или заглубленных емкостей, сосудов и т. п. Для оборудования этих технологических конструкций катодной защитой требуется большое число изолирующих блоков. Еще большие проблемы возникают при переоборудовании производства, когда приходится заново определять все исходные параметры для катодной защиты. Даже один неправильно рассчитанный блок может привести к потере эффективности катодной защиты всего оборудования.
Расположенные на промышленных предприятиях защищаемые системы — трубопроводы, емкости, сосуды, колонны и др. промышленные агрегаты — все чаще сооружаются таким образом, чтобы их системы были металлически связаны с конструкциями из бетона и стали (фундаменты, стены зданий, опоры и т. п.). Так как сталь в бетоне имеет более высокий положительный потенциал, чем сталь в грунте (примерно на 0,2-0,5 В), то объекты, контактирующие с бетоном, подвержены интенсивному коррозионному воздействию. При создании новых конструкций из бетона и стали необходимо предусмотреть электрическую изоляцию бетонных поверхностей. Опасность коррозии металл—бетон может быть устранена созданием локальной катодной защиты. С помощью катодной поляризации постоянным током стремятся выровнять различные потенциалы металлов. Хотя сталь в бетоне сама по себе не корродирует, однако ее катодно поляризуют, чтобы не было коррозионного воздействия на проложенные в земле металлические системы (трубопроводы, кабели, складские емкости газов и т. п.). Для этого требуется защитный ток поверхности бетона плотностью 2-5 А/м2. Защитный ток защищаемых объектов должен быть в пределах 10-50 мА/м2, что в сравнении с защитным током бетона представляется весьма незначительной величиной. Это связано с тем, что из-за больших площадей бетонных конструкций (фундаментов и т. п.) в грунт надо вводить большие токи.
Значительные затраты на оборудование и эксплуатацию защитной системы компенсируются экономией изолирующих материалов, повышением эксплуатаци
онной надежности и долговечности защитной антикоррозионной системы. Такая защита должна быть произведена сразу после монтажа защищаемого объекта и сразу же введена в действие, т. к. с развитием процесса схватывания бетона сталь внутри железобетонных конструкций пассивируется, в результате чего возрастает опасность коррозионных повреждений проложенных в земле защищаемых объектов.
Д. Катодная защита внутренних поверхностей труб, емкостей и сосудов. В трубопроводах, по которым транспортируется агрессивная среда, в емкостях, где хранятся агрессивные жидкости (например, золы, химические воды, загрязненная сливная вода и т. п.), опасность коррозии устраняется с помощью покрытия на цементной основе. Катодная защита применяется, в основном, для небольших по размеру объектов — светлых труб, дюкеров и т. п. В этом случае важен расчет анодов и их расположение из-за относительно высокого электросопротивления, небольшого объема электролита и большой плотности защитного тока. В качестве анодного материала хорошо зарекомендовали себя ферросилиций и платинированный титан. Также достаточно широко применяются кремниевые аноды, имеющие преимущество по отношению к платинированным титановым, состоящее в том, что кремниевые аноды не ограничивают анодное напряжение, в то время как в анодах из платинированного титана напряжение анод—электролит должно быть не менее 12 В, иначе пробивается нерастворимый слой диоксида титана и электрод интенсивно корродирует. Преимущество платинотитановых электродов заключается в их большей технологичности. Такие аноды можно изготавливать в виде проволоки, благодаря чему достигается необходимое распределение токов и потенциалов внутри защищаемого объекта. Состав и свойства анодов при катодной внутренней защите с посторонним источником тока приведены в табл. 1.4.57. Пределы катодной защиты внутренних поверхностей зависят, прежде всего, от требуемой плотности защитного тока, т. е. от внутреннего покрытия. Для защиты светлых поверхностей (т. е. поверхностей без специальной защиты) требуется плотность защитного тока 50-220 мА/м2 в зависимости от скорости истечения среды. Для поверхностей с покрытиями требуется плотность тока в пределах 0,2-0,5 мА/м2.
Таблица 1.4.57
Свойства анодов при катодной защите с посторонним источником тока (агрессивная среда — морская вода)
Материал анода Состав анода, масс. % Плотность анода, г/см3 Плотность анодного тока, А/дм2 Расход анода, г/(А • год)
максимальная минимальная
Сложные по составу аноды
Графит 100 С 1,6 0,5-1,5 0,1-0,5 30-450
Ферросилиций 14 Si, 1 С, ост, —Fe (возможно 5 Сг, 1 Мп, 1 Мо) 7,0-7,2 3 0,1-0,5 90-250
Свинец/серебро 1 Ag, 6 Sb, ост. — РЬ 11,0-11,2 3 0,5-2 45-90
Свинец/серебро 1 Ag, 5 Sb, 1 Sn, ост. — Pb 11,0-11,2 5 1-2,5 30-80
Свинец/платина Карандаши 11,0-11,2 5 1-2,5 2-60
132
Новый справочник химика и технолога
Продолжение таблицы 1.4.57
Металлические аноды
Материал Плотность, 103 кг/м3 Материал поверхности Толщина слоя, мкм Плотность анодного тока, AIjw? Предельное напряжение, В Расход анода, мг/(А • г)
Максимальная Средняя
Платина 21,45 Платина Мала >100 — — >2
Титан 4,5 Платина 2,5-10 > 10 6-8 12-14 4-10
Ниобий 8,4 Платина 2,5-10 > 10 6-8 40 4-10
Тантал 16,6 Платина 2,5-10 > 10 6-8 80 4-10
Титан 4,5 RuO2 — — 7,5-11 12-14 5
Анодная защита
Анодная электрохимическая защита металлов от коррозии — сравнительно новый и достаточно специфический метод. Он основан на переходе металла из активного состояния в пассивное вследствие смещения его потенциала при анодной поляризации от (постороннего) внешнего источника тока.
В отличие от катодной защиты в этом случае процесс коррозии связывается не полностью. Для выполнения анодной защиты защищаемая поверхность должна быть поляризована посторонним источником так, чтобы точечная коррозия не могла иметь места, а общая коррозия металла была настолько мала, что с технической точки зрения ею можно было бы пренебречь. Системы, в которых можно использовать анодную защиту, приведены в табл. 1.4.58.
В настоящее время анодная защита сформировалась как самостоятельное направление электрохимической защиты. С ее появлением началось активное использование электрохимических защитных процессов в химической промышленности.
Применение анодной защиты позволяет использовать в качестве конструкционного материала для оборудования химической промышленности различные нержавеющие стали и титан, хорошо пассивирующиеся в различных средах. Приложенный анодный ток ускоряет наступление пассивности, способствует ее сохранению в течение длительного времени, позволяет подобрать условия оптимального пассивирования, а в ряде случаев заменить более легированные стали на менее легированные.
Так, в 0,65 н H2SO4 защитная плотность тока для углеродистой стали при катодной защите составит примерно 3,5 А/м2, а при анодной поляризации плотность тока пассивации металла ниже 0,1 А/м2.
Анодная защита применяется как для предохранения оборудования от коррозионных повреждений, так и для сохранения чистоты химического продукта.
В случае подземных и гидротехнических сооружений анодная защита не может конкурировать с катодной, хотя в ряде случаев удается получить достаточно значимый положительный эффект. Так, при анодной защите некоторых алюминиевых сплавов и нержавеющих сталей в морской воде наблюдается высокий защитный эффект, необходимая при этом плотность тока составляет всего 0,02 А/м2, но для обеспечения доста
точной эффективности защиты необходимо постоянно поддерживать потенциал, близкий к напряжению пробоя пассивной пленки, что неудобно с практической точки зрения.
Таблица 1.4.58
Некоторые среды и металлы, для которых возможно применение анодной защиты
Материал Агрессивная среда
Углеродистые стали Серная кислота Олеум Очищенная серная кислота, содержащая органические вещества Целлюлозные щелочные среды Водные растворы аммиака Аммиачные удобрения Фосфорная кислота Фосфорная и серная кислоты Щавелевая кислота Растворы алюминатов 5% NaOH и 15% Na2CO3
Хромоникелевые и хромоникельмолибденовые стали Серная кислота Сульфат гидроксиламмония (NH3OH)2SO4 Серная кислота—акрилонитрил Азотная кислота Фосфорная кислота Удобрения с хлоридом калия Серная кислота, содержащая НС1, НВг, HF Изобутилсерная кислота Сульфатные (хромоаммониевые) растворы Щелочи NaOH и LiOH Щелочь и кислота в процессе нейтрализации Сульфокислоты RSO3H Серная и азотная кислоты Соляная кислота Муравьиная кислота Щавелевая кислота Сульфаминовая кислота Фосфорная кислота Растворы хлорида хрома Сг(Ш) Серная кислота, H2S, CS2
Электродные процессы
133
Правильный подбор интервала защитного потенциала позволяет не только защитить металл от общей коррозии, но и предотвратить развитие местных видов коррозионных повреждений. Так, анодная защита сдерживает развитие избирательной коррозии феррита, межкристаллитной коррозии и коррозионное растрескивание стали 12Х18Н9 в растворе 20%-й серной кислоты, содержащей 1% NaCl, независимо от условий ее сварки или термической обработки. Она позволяет избежать развития питтинговой коррозии стали 12Х18Н9Т в разбавленной азотной кислоте, содержащей ионы хлора, предотвращает растрескивание вследствие наводороживания металла и т. п. В табл. 1.4.59 приведены некоторые сведения о пределах защиты систем анодной защиты металлов.
Основными факторами, характеризующими зависимость скорости растворения металла от потенциала (рис. 1.4.48) и существенно влияющими на эксплуатационные условия и эффективность анодной защиты, являются следующие параметры: критическая плотность пассивного тока; критический интервал пассивации; интервал потенциалов, в котором сохраняется пассивное состояние, и ток растворения в пассивном состоянии.
При эксплуатации производственного оборудования с анодной защитой установлено, что наличие сварных швов (включая швы, соединяющие разнородные материалы) не вызывает ускоренной коррозии. Однако необходим тщательный контроль сварных соединений: не допускается к эксплуатации оборудование с дефектами типа горячих и холодных сварочных трещин, непроваров, пор, шлаковых включений. Такие дефекты могут служить инициаторами возникновения и ускоренного развития коррозионных дефектов. Интенсивное пере
мешивание электролита также не оказывает влияния на эффективность анодной защиты.
При анодной электрохимической защите необходимо контролировать потенциал конструкции, что требует применения электрода сравнения, простого в изготовлении и пригодного для длительной эксплуатации в условиях реального производства. В некоторых случаях для облегчения первоначального пассивирования рекомендуется вводить окислители или ингибиторы.
Рис. 1.4.48. Зависимость стационарной скорости растворения металла от потенциала, возникающего в конструкции
Таблица 1.4.59
Пределы защиты и плотность тока систем анодной защиты
Система Пределы защиты, В Плотность защитного тока после достигнутой пассивации, мА/м2 Примечание
Fe в H2SO4 (+0,7Н+1,7) 70
Сталь с 17 % Сг в соляных растворах удобрений, содержащих КС1 и KNO3 (+0,4)—(+1,2) 1000 Из-за наличия иона NO3 возможно применение анодной защиты
Нержавеющая сталь 4401 (18 % Сг, 10 % Ni, 2 % Мо) в 67 % H2SO4 (+0,2Н+0,8) 1 Сопротивление образующейся оксидной пленки 16500 кОм/м2
Нержавеющая сталь 4401 в 15 % Н3РО4 (+0,25Н+0,5) 0,2
Нержавеющие стали (18-25 % Сг, 8-14 %Ni) в 67 % H2SO4 (+0,4Н+1,0) 1-110 Сопротивление образующейся оксидной пленки 230-1800 кОм/м2
Нержавеющая сталь в 20%NaOH (+0,ЗН+0,6) 100
Котельная сталь HIYL или 13СгМо44 в Na[Al(OH)4] (-0,4) 10 Анодная защита применяется для устранения коррозионного растрескивания
134
Новый справочник химика и технолога
1.4.8.2. Ингибиторы коррозии
Ингибитор — это химическое вещество, при добавлении которого в небольших количествах в данную коррозионную среду значительно уменьшается скорость коррозии металлов, находящихся в контакте с этой средой. Ингибиторы широко используются для защиты от разрушений внешних и внутренних поверхностей труб и аппаратов в циркуляционных охладительных системах, реакторах, емкостях для хранения и транспортировки химических продуктов, коммуникационных и энергетических системах. Их основное преимущество состоит в том, что они пригодны для защиты пораженных коррозией систем без замены поврежденных деталей и узлов. В зависимости от способа действия ингибиторы делятся на пленкообразующие (пассиваторы) и адсорбирующиеся.
А. Пленкообразующие ингибиторы. В качестве пассиваторов могут быть использованы все те вещества, которые образуют с ионами металлов нерастворимые продукты и формируют пленку. В общем случае — это пассиваторы металла, определяющую роль в ингибирующей способности которых играет величина pH раствора.
Некоторые пассиваторы образуют на поверхности металла оксидную пленку, толщина которой доходит до 0,01 мкм и может быть значительно больше толщины пленки, образованной на поверхности металла под действием воздуха. Так, хроматы создают оксидную пленку на сплавах железа, алюминия и цинка. К пассиваторам могут относиться кислород, гидроксид-, нитрат-, нитрит-, фосфат-, молибдат-, бензоат-ионы. Они непосредственно или в виде продуктов реакции блокируют анодные и катодные участки поверхности металла, повышая ее потенциал. Их большое сродство к металлу сочетается с высокой энергией активации образования веществ с новой кристаллической решеткой (хемосорбцией). Так, например, кислород при хемосорбции пассивирует поверхность, в то время как хлорид-ионы из-за низкой энергии активации образования хлоридов вытесняют адсорбированные на поверхности металла атомы пассиватора.
Фосфаты, силикаты и бензоаты щелочных металлов являются анодными пассиваторами. Так, Na3PO4 образует на анодных участках стали фосфат железа, который ингибирует коррозию стали, погруженной в водный раствор хлорида натрия. Катионы пассиватора могут образовывать нерастворимый гидроксид на катодных участках корродирующего металла. Если погрузить стальную пластину в морскую воду, содержащую хлорид магния, то на катодных участках поверхности образуется пленка Mg(OH)2, т. е. хлорид магния служит катодным ингибитором коррозии стали в растворе хлорида натрия. Некоторые вещества обладают одновременно анодным и катодным действием. К ним относятся атмосферные пассиваторы, например Са(НСО3)2. Ион HCOj образует карбонат железа(П) на анодных участках, а ион Са2+ — пленку Са(ОН)2 на катодных участках.
Часто в качестве неокислительных пассиваторов используют водорастворимые силикаты. К ним относятся продукты состава R2O • «SiO2, где R2O — К2О или Na2O, а п — число молекул SiO2. Отношение между числом молекул диоксида кремния и числом молекул оксида щелочного металла называется силикатным модулем. Модуль является основной характеристикой силикатов — их способности растворяться в воде, образовывать коллоидные системы, взаимодействовать с электролитами и т. п. Для защиты металлов от коррозии рекомендуется использовать силикаты с высоким модулем. Оптимальная концентрация силиката натрия в пересчете на SiO2 находится в пределах от 0,0008 до 0,0012%. Силикат натрия пригоден для защиты цветных металлов и сплавов и входит в состав ряда моющих средств.
Б. Адсорбирующие ингибиторы. Действие адсорбирующих ингибиторов характеризуется главным образом их адсорбцией. Известны два типа адсорбции — физическая и химическая. Физическая адсорбция обусловлена действием ван-дер-ваальсовых сил между ингибитором и металлом. Десорбцию таких адсорбентов легко осуществить промывкой или протиркой поверхности. Химическая адсорбция, или хемосорбция, возникает в результате химического сродства между металлом и ингибитором, приводящего, в ряде случаев, к возникновению на поверхности металла химических соединений.
Адсорбционные ингибиторы — это в основном органические соединения, в молекулах которых содержатся определенные функциональные группы, хотя иногда применяются и неорганические вещества. К органическим ингибиторам относятся амины и их производные, аминоэтанолы, альдегиды, спирты, мочевины, тиолы и т. п.
Все существующие типы ингибиторов можно разделить на несколько основных групп по возможности их применения в тех или иных средах. Ингибиторы для травления — серосодержащие соединения, производные ацетилена. Слабые органические азотсодержащие основания эффективны в кислых растворах и не проявляют защитного действия в некислых растворах. Еще одной группой ингибиторов являются органические производные гидроксиламина, восстанавливающие растворенный кислород.
Известно, что низкомолекулярные органические кислоты вызывают коррозию металлов, однако их натриевые соли обладают защитным действием при условии, что константа диссоциации кислоты имеет величину порядка 10 4—10 8. При недостаточной концентрации ингибитора такого типа будет наблюдаться равномерная коррозия. При этерификации карбоксильной группы кислоты их защитное действие прекращается.
Фенолы оказывают ингибирующее воздействие только в виде фенолятов при условии, что их константа диссоциации выше, чем 10-8. Исключение составляет о-нитрофенол, обладающий хорошим защитным действием.
Электродные процессы
135
Действие низкомолекулярных аминов зависит от концентрации гидроксид-ионов в растворе. Защитное действие нерастворимых в воде аминов с длинной цепью и кислот металлического ряда и их производных с концевой полярной группой связано с гидрофобными свойствами этих соединений. Амины с длинной цепью способствуют образованию защитных пленок. Они оказывают эффективное защитное действие, причем у ок-тилдециламина оно значительно сильнее, чем у цикло-гексиламина.
Натриевые соли двухосновных карбоновых кислот оказывают такое же действие, как нитрит натрия, гидрокарбонат калия или дициклогексилпентилнитрит. Бензоат натрия также известен как хороший ингибитор. В концентрации 1-2 % он хорошо защищает сталь, а в водных растворах блокирует действие хлорид-ионов. В сочетании с нитритом натрия бензоат натрия защищает системы с этиленгликолевыми антифризами.
1.4.8.3. Антикоррозионные защитные покрытия
К защитным материалам, используемым для покрытия металлических материалов, предъявляется ряд требований, гарантирующих эффективную стойкость защитного слоя. Если для защиты от атмосферной коррозии путем окраски прежде всего необходимо высокое качество подготавливаемой к окраске поверхности, то при создании внешней оболочки или внутреннего защитного слоя (в трубах или емкостях) следует добиваться высокой химической и механической стойкости покрытия, низкой влаго- и газопоглощающей способности и кислородопроницаемости, высокого электросопротивления, термической стабильности и устойчивости против старения и т. п.
Для внешних покрытий особо важны высокие пределы значений прочности при сжатии и ударной нагрузке, сохранение защитных свойств при нанесении их тонкими и сверхтонкими слоями.
Особое внимание следует уделять целостности наносимого защитного (пассивного) покрытия — отсутствия в нем пор, язвин, трещин и т. п., т. к. такие покрытия достаточно эффективны только в случае отсутствия в них участков, где агрессивная среда может соприкасаться с защищаемой поверхностью. Иногда, преимущественно в трубопроводах с катодной защитой, под действием эксплуатационных нагрузок происходит частичное отслаивание или растрескивание защитного покрытия, и щелочные среды проникают к поверхности труб. Полевые испытания такого трубопровода не выявят изменения защитного тока, что может привести к непредсказуемым разрушениям конструкции.
Металлические покрытия
А. Горячее цинкование. Одним из наиболее распространенных способов защиты металлических поверхностей от коррозии является горячее цинкование. С этой целью обрабатываемые изделия предварительно подвергается травлению для получения поверхности,
имеющей хорошее сцепление с цинком. После этого поверхность обрабатывается специальным флюсом и погружается в расплавленный цинк. Когда изделие нагревается до температуры цинковой ванны (~ 450 °C), флюс испаряется и образуется сплав железной основы и расплава цинка. Слой полученного сплава обеспечивает последующее хорошее сцепление обрабатываемой поверхности с чистым цинком, слой которого образуется в процессе извлечения изделия из цинковой ванны.
Высокая стойкость цинка в условиях атмосферной коррозии объясняется образованием плотного защитного слоя гидроксида цинка. Эффективность антикоррозионной защиты зависит от климатических условий, прежде всего от количества осадков и содержания в атмосфере диоксида серы. Годовое уменьшение защитного цинкового покрытия в среднем составляет примерно 1 мкм для морского и материкового климата и 5 мкм для промышленных условий.
Высокая коррозионная стойкость может быть получена при комбинировании цинкования с окраской (дуплекс-система). Для обеспечения работоспособности дуплекс-системы очень важна подготовка грунтовочного слоя, поскольку лак и краска имеют слабое сцепление с гладким слоем цинка.
Детали, которые нельзя подвергать горячему цинкованию, могут быть покрыты слоем цинка методом напыления. Цинкование напылением дает шероховатую поверхность, наилучшую с точки зрения надежности работы дуплекс-системы.
Б. Электролитические покрытия. Особо тонкие металлические покрытия лучше всего наносить электролитическим способом. Кроме антикоррозионной защиты, они часто служат декоративными украшениями деталей. Чаще всего этот способ используется для покрытия небольших по размеру деталей специального назначения в автомобильной, мебельной и судостроительной промышленности, при изготовлении бытовой техники. Эти покрытия с химической точки зрения должны быть более благородны, чем защищаемый металл, не должны иметь трещин, вздутий, пор и других дефектов, что достигается нанесением нескольких разнородных слоев. Так, например, при нанесении хрома на стальную деталь на нее предварительно наносят слой меди, затем слой никеля и только после этого — слой хрома.
В. Плакирование. В некоторых случаях, прежде всего в химической промышленности, особо тонкие слои металла наносят на поверхность сталей путем плакирования с использованием прокатки или сварки. В плакированном изделии получается комбинированный слой, состоящий из отдельных компонентов, имеющих различную прочность и различные антикоррозионные свойства. Плакированные трубы применяют в тех случаях, когда применение толстостенных труб из дорогостоящих металлов и сплавов экономически нецелесообразно. Плакированные изделия могут соединяться между собой посредством сварочных операций.
136
Новый справочник химика и технолога
Г. Эмалирование. В химической промышленности широко используются трубы и емкости, внутренние поверхности которых покрыты эмалью. В этом случае соединение труб или их присоединение к емкостям возможно только с помощью соответствующих фланцев. Механическая прочность эмалированного покрытия невелика, поэтому при транспортировке, монтаже и эксплуатации эмалированных изделий необходимо избегать дополнительных механических нагрузок.
Как правило, эмалирование включает в себя нанесение грунтовой эмали и обладающей высоким сопротивлением к агрессивному воздействию среды покровной эмали. В ряде случаев наносится несколько слоев эмали, что часто используется в приборостроении, например два грунтовочных слоя и от трех до шести покровных слоев. При нанесении одного слоя толщиной до 300 мкм невозможно получить беспористое покрытие, чем и объясняется многослойное нанесение. Однако, если по эмалированным трубам не транспортируются вещества с кислотными свойствами, то поры в поверхности покрытия допускаются. Это связано с тем, что поры при однослойном эмалировании оказываются закрыты для продуктов коррозии. Образование электрического элемента между основанием поры и окружающей средой невозможно (в отличие от всех других тонкослойных покрытий), так как эмаль неэлектропроводна.
Перед нанесением эмали защищаемое изделие нужно предварительно обезжирить и протравить, после чего на поверхность наносят тестообразный эмалевый шликер, который подвергается сушке, а затем обжигу при температуре 600-850 °C. После охлаждения детали этот процесс повторяется еще несколько раз.
Неметаллические покрытия
Независимо от типа используемого антикоррозионного покрытия для достижения хорошего сцепления с металлической поверхностью защищаемого изделия необходимо тщательно зачистить ее поверхность и полностью удалить оксидные отложения (окалину). Перед нанесением битумных или пластмассовых защитных покрытий окалину и ржавчину удаляют до тех пор, пока невооруженным глазом виден рельеф вследствие наличия пор. При нанесении реакционноспособных лаков окалина и ржавчина удаляются полностью. Прокатная окалина с поверхности стали может быть удалена путем термического воздействия, пескоструйной обработки или путем травления. Механические способы удаления ржавчины включают ручную обработку, например, проволочными щетками, шпателем, шабером, молотком для отстукивания и другими ручными инструментами, вращающимися проволочными щетками, шлифовальными кругами и лентами и т. п. При термической обработке окалина сбивается пламенем ацетиленовой горелки. При такой обработке можно использовать Т-образные горелки гребенного типа со скоростью подачи 3-5 м/мин, наводимые на обрабатываемую поверхность либо за один ход, либо за несколько ходов с промежуточными охлаждениями.
А. Фосфатирование. Фосфатирование, в строгом понимании этого процесса, не является антикоррозионной обработкой. Это способ нанесения грунтовочного слоя для последующего защитного слоя. Фосфатирование заключается в погружении предварительно протравленного изделия в растворы фосфатов марганца, железа, аммония. В результате реакции между сталями и этими растворами образуются фосфатные пленки, которые часто содержат ионы других компонентов (цинка, марганца и т. п.). Поставка фосфатированных изделий от производителей потребителям практически не производится, так как пленки фосфата легко могут быть повреждены при транспортировке и при промежуточном складировании. Операция фосфатирования должна выполняться непосредственно перед нанесением на поверхность изделия защитного слоя.
Б. Нанесение защитных покрытий путем окраски и лакирования. При монтаже стальных конструкций на открытом воздухе используются преимущественно покрытия, в которых образование пленки происходит либо благодаря физическому испарению растворителя, либо в результате химического отверждения с участием кислорода воздуха. Для грунтовочных слоев чаще всего используют свинцовый сурик, а для покрывных слоев — железную слюдку и цинковый пигмент, либо смесь железной слюдки со свинцовыми белилами и цинковым пигментом. При качественном исполнении такие покрытия обеспечивают надежную длительную защиту. При нанесении алкидного олигомера получают более тонкие слои покрытий (порядка 40 мкм) по сравнению с масляной краской (50 мкм).
Антикоррозионная защита, обеспечиваемая покрытиями, и ее долговечность напрямую зависят от общей толщины слоя и прочности сцепления. В зависимости от условий эксплуатации изделия и сил сцепления покрытий и основного металла используют покрытия, состоящие из одного или двух грунтовых или покровных слоев. В качестве грунтовочного слоя при высоких нагрузках используют слои из цинковой пыли, которую необходимо наносить на чистые металлические поверхности. Грунтовка из цинковой пыли включает в себя саму цинковую пыль и связующий компонент — реакционноспособный лак, эпоксидный эфир, эпоксидный олигомер или этилсиликат. Следует иметь в виду, что такая грунтовка может использоваться только совместно с покрывным защитным слоем.
Для конструкций морского базирования используют специальные покрытия большой толщины, на которые часто сверху наносят ядовитые покрывные слои (проти-вообрастающая краска). Это специальное покрытие представляет собой слои каменноугольного пека и эпоксидного олигомера, наносимые несколько раз. Общая толщина покрытия в среднем составляет около 500 мкм.
В системах с высокой влажностью используют грунтовку из цинковой пыли с покрывным слоем из эпоксидных материалов. При высоких температурах (более 120 °C) эпоксидные олигомеры заменяются полиуретаном, стойким до температуры 150 °C, или жаростойки
Электродные процессы
137
ми покрывными материалами, изготовленными из силиката цинка, сохраняющими свои свойства до 400 °C.
Также используются защитные покрытия (грунтовки), изготовленные на основе хроматов цинка, из-за их улучшенной свариваемости, в то время как грунтовки на основе цинковой пыли обладают невысокой свариваемостью, что ограничивает области их применения.
В. Изоляция труб. Защитные оболочки прокладываемых в земле трубопроводов должны обеспечить работоспособность труб без капитального ремонта в течение 30-50 лет. Это требование может быть выполнено с большей степенью гарантии путем нанесения защитных покровных слоев. Структура обычных оболочек стальных труб, прокладываемых в земле, приведена в табл. 1.4.60.
Коррозионностойкие чугунные трубы покрывают слоем толщиной около 0,07 мм, получаемым методом погружения. Если прокладка труб ведется в грунтах с высокой коррозионной активностью, то применяют усиленные покрытия, толщина которых может составлять от 0,1 до 0,15 мм. Для защиты чугунных труб, прокладываемых в особо агрессивных грунтах, применяют также цинкование и покрытия из пластмасс.
Битумная защитная оболочка стальных труб выполняется как покрытие большой толщины, усиленное стеклохолстом. Минимальная толщина слоя составляет 4 мм. Недостатком битумных оболочек является их невысокая механическая стойкость. Это приводит к повреждениям труб при транспортировке, которые при монтаже системы приходится устранять вручную. Из-за низкой прочности битумных оболочек такие трубы не укладывают в каменистых грунтах. Пластмассовые оболочки преимущественно выполняют из термопластичных материалов и прежде всего из полиэтилена. Основными преимуществами полиэтиленовых оболочек, по сравнению с битумными, являются:
- высокая механическая прочность, стойкость и сцепление, что имеет большое значение при транспортировке, складировании и монтаже труб;
- высокое электрическое сопротивление оболочки. Потребность в защитном токе таких труб в 100 раз меньше, чем у труб с оболочками из каменноугольных пеков и с битумными оболочками. В течение длительного времени защитный ток остается низким;
- высокая химическая стойкость и устойчивость против старения при использовании полностью стабилизированного полиэтилена;
- слабая восприимчивость к кислороду и водяному пару;
- широкий интервал рабочих температур;
- высокая механическая стойкость против прорастающих корней деревьев и кустарников.
Полиэтиленовые оболочки в основном получают одним из двух способов. Это наплавление порошков и экструзия со скошенной головкой. При экструзии обработанную пескоструйным методом заготовку нагревают до 200 °C. На нагретую поверхность подается двухслойное покрытие, состоящее из слоя клеящего вещества и слоя полиэтилена, выходящее из экструдера с кольцевым зазором. Внутренний — клеевой слой — обеспечивает сцепление между металлом и полиэтиленовой оболочкой. При порошковом наплавлении полиэтиленовый порошок или выстреливается снизу, или распыляется сверху на предварительно обработанную и нагретую до 320 °C защищаемую поверхность. С помощью этого метода удается изготовить защитные оболочки для колен, гибов, тройников и др. сложных по форме конструкций.
Г. Защита внутренних поверхностей труб, емкостей, сосудов. Для защиты внутренних поверхностей в основном применяют битумные, цементные и эпоксидные покрытия. Эпоксидные покрытия наибольшее распространение нашли в газопроводных системах, где покрытия толщиной до 50 мкм наносят для уменьшения коэффициента трения. Битумные покрытия применяют для антикоррозионной защиты внутренних поверхностей водопроводов для питьевой воды, поэтому они должны удовлетворять не только техническим и механическим параметрам, но и требованиям санитарно-гигиенических правил. Толщина битумных покрытий, применяемых для защиты внутренних поверхностей, зависит от условий эксплуатации объекта, свойств агрессивной среды. Особые требования выдвигаются при пневматических испытаниях систем с защитным внутренним покрытием, выполненным из битума. При пневматических испытаниях трубопроводов с толщиной битумного покрытия в 1 и более мм не допускается превышение давления испытания над рабочим более чем на 1 бар при общей длительности испытаний более 6 ч, максимальная температура испытаний не должна превышать 25 °C. При ремонтах и длительных остановах таких трубопроводов не допускается осушка защитного битумного покрытия, что может привести к его повреждению и растрескиванию.
Таблица 1.4.60
Структура защитных оболочек стальных труб
Параметры оболочки Из полиэтилена Из битума
Толщина слоя, мм 2-4 4
Структура Экструзия: клей и полиэтилен. Наплавленный полиэтиленовый порошок Битумная грунтовка. Битумная масса. Стеклохолст
Интервал температур эксплуатации, °C (—40)—(65) (-5Н30)
138
Новый справочник химика и технолога
Продолжение таблицы 1.4.60
Параметры оболочки Из полиэтилена Из битума
Катодный защитный ток, мА/м2 1 30-100
Окончательная изоляция сварных швов Антикоррозионные связующие вещества, сжимающиеся шланги из полиэтилена Антикоррозионные связующие вещества из битума или пластмасс
Основные сферы использования Для грунтов всех типов (при наличии тока утечки требуются защитные мероприятия), в том числе каменистых и проходящих в черте города, для горячих трубопроводов (до 65 °C) Для грунтов всех типов (при наличии тока утечки требуются защитные мероприятия) возможно применение катодной защиты
Для защиты внутренних поверхностей объектов в системах водопроводов и транспортировки соленой воды используют покрытия на основе цементного раствора. С этой целью используется цемент с повышенной сульфатной стойкостью. В качестве добавок применяется песок, который по размеру зерен и гранулометрическому составу соответствует определенной толщине слоя. Существуют различные способы нанесения покрытий. В трубопроводах широко применяется центробежный способ, благодаря которому достигается высокая степень уплотнения. Плотная структура защитного слоя является основным требованием к данному виду покрытия, т. к. в этом случае обеспечивается высокое сопротивление химическому воздействию среды.
Покрытие на цементной основе представляет собой пассивный слой, изолирующий внутреннюю поверхность оборудования от воздействия протекающей воды. Следует иметь в виду, что такой слой водопроницаем и на поверхности раздела цемент—сталь могут протекать различные химические реакции, что отличает цементное покрытие от битумного или эпоксидного. Проникающая в цементное покрытие вода, особенно мягкая, постепенно растворяет кальциевую основу цемента, хотя это явление почти не оказывает влияния на прочность покрытия в том случае, если обедненный кальцием поверхностный слой постоянно находится во влажном состоянии. Это нужно учитывать при трассировке транспортных систем, т. к. высыхающий, лишенный кальция цемент растрескивается и крошится. Однако даже подобное повреждение покрытия не уменьшает его антикоррозионного значения — гидроксид железа образует с цементом прочное соединение. Не опасны цементным покрытиям и упругие напряжения, возникающие при укладке или подвижке проложенных труб. Поверхность стали в зоне трещины оказывается прикрыта образующимся при взаимодействии металла и цемента покрывным слоем. Этот эффект известен под названием «самолечения». Параметры воздействия воды на бетон приведены в табл. 1.4.61.
Д. Временная защита от коррозии. Для защиты изделий из углеродистой и низколегированной стали от коррозионного воздействия атмосферы при транспортировке, складировании и строительстве могут быть использованы следующие способы.
1. Применение защитного масла (в основном минерального масла с ингибиторами или добавкой раствори
теля). Масляное покрытие пригодно для защиты от дождя, снега и других атмосферных осадков в течение 1-5 месяцев. Его можно легко удалить механическим путем. Полное удаление масляной пленки достигается при применении бензола, уайт-спирита, хлороуглеводородов.
2. Применение светлых лаков или аналогичных продуктов, которые обеспечивают прочное покрытие, не взаимодействующее с пылью. Стойкость покрытия при складировании на открытом воздухе составляет от 1 до 5 месяцев. Светлый лак удаляется растворителем или с помощью пескоструйной обработки.
3. Применение воска или воскообразных покрытий, которые позволяют получать разнообразные пленки — от мягких до твердых с малой степенью взаимодействия с пылью. Такое покрытие пригодно для защиты изделий при хранении на открытом воздухе от 6 до 12 месяцев. Удаление покрытия возможно с помощью горячих очистителей.
4. Покрытия на основе ланолина, аналогичные восковым покрытиям. Они образуют мягкие пленки, слабо взаимодействующие с пылью. Применяются для складирования изделий под навесом или на открытом воздухе в течение 6 месяцев. Удаление покрытия осуществляется с помощью щелочных средств очистки или органических растворителей.
Таблица 1.4.61
Оценка воздействия воды на покрытия из цементного раствора
Кислотность и примеси Степень воздействия
Слабая Сильная Очень сильная
Величина pH 6,5-5,5 5,5-4,5 4,5
Диоксид углерода (СО2), растворяющий известь (по Тейеру), мг/л 15-30 30-60 60
NH*, мг/л 15-30 30-60 60
Mg2+, мг/л 100-300 300-1500 1500
Сульфат( SO2-), мг/л 200-600 600-3000 300
5. Лаки с битумным или каменноугольным пеком при толщине покрытия порядка 20 мкм. Подобные покрытия предназначены для защиты изделий, хранящихся под навесом в течение 12 месяцев или на открытом воздухе в течение 1-2 месяцев. При морской транспор
Электродные процессы
139
тировке изделий рекомендуется применять вязкие битумные или каменноугольные лаки, позволяющие получать слои большей толщины. Удаление битумных и каменноугольных лаков осуществляется с помощью бензина или других углеводородов.
6. Применение грунтовочных покрытий с пассивирующими пигментами (в виде окончательного покрытия), преимущественно на основе свинцового сурика и хромата цинка. Рекомендуется в том случае, когда эти покрытия используются в качестве грунтовки под последующие покрывные защитные слои. Нанесение внешних покрывных слоев на слой свинцового сурика следует осуществлять не позднее 3 месяцев со дня нанесения грунтовочного слоя. Если внешнее покрытие будет нанесено в более поздние сроки, необходимо двухслойное грунтовочное покрытие.
1.4.8.4. Защита неметаллических конструкций
Все строительные объекты общественного назначения и промышленные здания содержат конструкционные элементы неорганического происхождения, в основном выполненные из кирпича и бетона. Изделия из горных пород также применяются при строительстве зданий, печей, емкостей, химической аппаратуры. Если изделия из горных пород, имея достаточно высокую химическую стойкость, практически не нуждаются в коррозионной защите, то бетоны, являющиеся одним из основных строительных материалов, требуют дополнительной изоляции. Защита бетонных конструкций основывается, главным образом, на улучшении их структурных характеристик, а именно: плотности, непроницаемости, химической стойкости. Поэтому основные мероприятия по защите бетонных конструкций можно разделить на две группы: первичную — защиту путем структурных изменений, и вторичную — защиту с помощью изоляционных материалов. Если структурная защита оказывается недостаточно действенной с точки зрения химической стойкости бетонов, то она дополняется применением защитных изоляционных материалов.
Техника структурной защиты бетонов известна, а ее возможности по созданию непроницаемых бетонов изучены достаточно подробно. Структурная защита может выполняться химическими или механическими методами.
А. Механическое уплотнение бетонов. Самым простым способом механического уплотнения является трамбование бетонов, однако изготовить этим методом непроницаемые бетоны удается далеко не всегда. Основным дефектом трамбовки является расслаивание бетона, вызванное оседанием более крупных по размеру частиц. Более совершенным является метод виброуплотнения. Виброуплотненные бетоны, имеющие то же водоцементное отношение и консистенцию, что и утрамбованные, являются значительно более водонепроницаемыми. Виброуплотнение дополнительно пластифицирует бетон и не создает значительной разницы в проницаемости в горизонтальном и вертикальном направлении, что присуще бетонам утрамбованным.
Еще одним способом уплотнения бетонов является торкретирование, при котором сухая смесь цементного раствора, нагнетаемая при помощи сжатого воздуха, перемешивается с распыленной водой, которая вводится в форсунку в нескольких сантиметрах от ее сопла. Образующаяся бетонная смесь выбрасывается наружу и покрывает торкретируюмую поверхность. Торкретирование позволяет получать непроницаемые бетоны.
Б. Химическое уплотнение бетонов. Применяемая для уплотнения бетона силикатизация может быть достигнута при помощи пропитки верхних слоев изделий из бетона или горных пород раствором силиката натрия, а после его высыхания — раствором хлората или хлорида кальция. При этом протекают следующие реакции:
Na2SiO3 + Са(С1О3)2 -> CaSiO3>k + 21ЧаС1О3
и Na2SiO3 + СаС12 —> CaSiO34< + 2NaCl, в результате которых образуются нерастворимый силикат кальция, заполняющий поры, и растворимые хлорат и хлорид натрия, которые вымываются водой. Первый способ силикатизации (верхняя реакция) применяется для уплотнения поверхности природных строительных материалов, второй — нижняя реакция — для уплотнения бетонов. Кроме того, в результате описанных процессов возрастает кислотоупорность материалов, т. к. в ходе реакций выделяется некоторое количество кремневой кислоты.
Еще одним способом химического уплотнения бетонов является флюатирование. Оно применяется для стабилизации и уплотнения поверхности конструкций из горных пород и бетона. Флюатами называются водные растворы солей гексафторокремневой кислоты, например, цинковая (ZnSiF6 • 6Н2О) или магниевая (MgSiF6) соли. Водные растворы этих солей имеют кислую реакцию. Они вызывают коррозию стали и стекла, являются токсичными материалами.
Кроме флюатирования достаточно широкое применение нашли тартратизация и гидрофобизация. Тартра-тизация заключается в обработке поверхности бетона винной кислотой, образующей тартрат кальция при реакции с находящейся в бетоне несвязанной известью. Тартратизацию можно применять для стабилизации любой бетонной поверхности. Например, этим способом защищают винные бетонные бочки от действия разбавленных органических кислот.
Гидрофобизация предназначена для создания не-смачиваемых поверхностей и применяется для защиты поверхности изделий из бетона и горных пород от увлажнения. Основным средством, применяемым для гид-рофобизации, является спиртовый раствор калийного мыла. Нанесенное покрытие из калийного мыла следует стабилизировать уксуснокислым алюминием. Достаточно широко для гидрофобизации применяются растворы метилсиликонов. Штукатурку можно гидрофоби-зировать введением в раствор стеарата кальция в объеме 3-5 % от объема применяемого цемента. Защищенная таким образом штукатурка отличается повышенной стойкостью в промышленной атмосфере.
140
Новый справочник химика и технолога
В. Изоляционная защита бетонов. Изоляционные покрытия наносятся на поверхность бетонных конструкций, на цементные покрытия и на другие прочные основы. Поверхности стен, выполненные из кирпичей или блоков, пригодны для нанесения покрытий только после сглаживания всех неровностей и заполнения раствором щелей и повреждений в этих стенах. Поверхность может быть шероховатой, но размер впадин не должен мешать нанесению грунта и последующих слоев мастики или изоляционной массы.
Нанесение горячей мастики на бетонную поверхность осуществляется следующим образом. Разогретые битумные и дегтевые мастики наносятся щеткой на покрытую битумной грунтовкой поверхность. При одноразовом покрытии получают слой толщиной около 1 мм. Для получения более толстых слоев операцию повторяют.
Холодные мастики и изоляционные массы обычно поставляются в готовом виде. В зимнее время рекомендуется их подогрев до температур 30-40 °C. Расход мастики при однослойном наложении находится в пределах 0,7-1,1 кг/м2. Толщина наносимых на загрунтованную поверхность слоев определяется консистенцией мастики. Наносят мастику при помощи кистей и щеток.
Изоляционные массы густо наносятся шпателем, при этом толщина слоя составляет 1,0-1,5 мм. Многослойные покрытия наносятся одно на другое после частичного высыхания предыдущего слоя. Стойкость такого покрытия зависит от вида наносимого материала и основы, а также от сцепления их друг с другом. Сушка покрытий осуществляется с помощью паяльной лампы или потока горячего воздуха.
Нанесение рулонных изоляционных материалов на битумной или любой другой основе без наклеивания возможно только при наличии выровненных защищаемых поверхностей и при условии, что они поджаты балластом или уложены между двумя поджимными досками. В этом случае не приклеивается только контактирующий с защищаемой поверхностью слой — остальные слои битумных рулонных материалов приклеиваются друг к другу.
Для приклеивания слоев битумного рулонного материала к бетонной основе поверхность последней грунтуется мастикой или битумом и, не дожидаясь их засыхания, закрывается рулонным материалом с нанесенной на него мастикой или любой другой битумной массой. Такой способ приклеивания «мокрое к мокрому» обеспечивает наилучшее сцепление. Рулонные материалы при прокладке следует разрезать на куски длиной 3-5 метров. Закладки при однослойной изоляции должны иметь длину не менее 100 мм, при многослойной — не менее 50 мм.
Г. Защита фундаментов и свай. В строениях без подвалов и строениях с подвалами, фундаменты которых не подвержены воздействию грунтовых вод, достаточно наличия в стенах и под полом горизонтальной двухслойной рулонной изоляции — двух слоев битумных рулонных материалов, склеенных между собой при помощи битумных мастик.
Встречающиеся в грунтах природные воды обычно не столь агрессивны, чтобы потребовалась какая-то специальная защита фундаментных конструкций. Для гидроизоляции фундаментов обычно применяются битумные материалы. В случае загрязнения грунтовых вод промышленными стоками агрессивность воды может резко повышаться, что потребует специальных методов защиты конструкций. В этих условиях рекомендуется применение максимально непроницаемых бетонов, специальных изоляционных покрытий.
При высокой агрессивности грунтов производят изоляцию бетона от самого грунта, для чего на слой насыпанного до насыщения битумом гравия укладывается литой слой асфальта. На рис. 1.4.49 приведен пример использования изоляции, которую в свою очередь защищает кирпичная кладка. Защита этого типа полезна, если возможны повреждения изоляции в ходе дальнейших строительных работ или во время засыпки котлована.
Рис. 1.4.49. Изоляция фундамента в агрессивных средах: I — бетонная стена и плита фундамента подвала;
2 — сплошная изоляция из рулонного битумного материала;
3 — слой литого асфальта толщиной около 20 мм;
4 — защитная кирпичная кладка; 5 — смесь песка и гравия, пролитая битумом; 6 — грунт
Особую сложность представляет защита железобетонных свай в химически агрессивных грунтах. Фундаментные сваи делятся на сборные, забиваемые в грунт сваи и сваи, формируемые в самом грунте. Во всех случаях необходимо, чтобы бетон свай был непроницаемым и стойким в данном типе агрессивной среды. Повышенной непроницаемостью отличаются сваи, формируемые непосредственно в грунте, при правильном выборе бетона и технологии производства. Получить такую же непроницаемость в забиваемых сваях крайне сложно, поэтому перед забивкой их в грунт на поверхность свай следует нанести защитное эпоксидное покрытие или пропитать их под давлением битумом.
Электродные процессы
141
1.4.9. Коррозионная стойкость материалов
(проф., д.х.н. В.С. Зотиков)
В таблице 1.4.62 представлены данные по коррозионной стойкости наиболее распространенных металлических и неметаллических материалов в различных жидких и газовых средах. Для каждой среды сначала дается характеристика коррозионной стойкости металлов и сплавов, а затем неметаллических материалов. Данные для металлов относятся к материалам, содержащим не менее 99 % основного вещества. Для наиболее распространенных сплавов указаны конкретные марки, для опытных сплавов приводится содержание основных компонентов.
Названия химических сред расположены в алфавитном порядке, причем в отличие от принятой в последние годы в химии номенклатуры сложных соединений название начинается с катиона.
В графе «Концентрация. Давление» приведены массовая концентрация основного вещества либо примеси с указанием ее наименования и давления в МПа. Дополнительно сообщается о факторах, влияющих на коррозионную стойкость материалов (аэрация среды, pH, присутствие кислорода воздуха и т. д.). В случае, когда не указан растворитель агрессивного вещества, имеется в виду водный раствор. Прочерк означает,
что сведения относятся к чистому продукту. Термин «фаза» характеризует агрегатное состояние химической среды.
В графе «Скорость и характер коррозии. Стойкость материалов» представлены скорость коррозии (мм/год), характер коррозии и химическая стойкость материалов.
Условные обозначения и сокращения
• характер коррозии: т — точечная коррозия, я — язвенная коррозия, н — неравномерная коррозия, мк — межкристаллитная коррозия, кр — коррозионное растрескивание;
• химическая стойкость материалов: С — стоек, ОС — относительно стоек, Н — нестоек;
• агрегатное состояние вещества: Т — твердая фаза; Ж — жидкая фаза; Г — газообразная фаза;
• физические параметры: Ткии — температура кипения; Тпд — температура плавления; р — давление;
• количественная характеристика среды: на-сыщ. — насыщенная (ый);
• резины на основе каучуков: БК — бутилкаучук, НК— натуральный, ПС — полисульфидный, СКБ — бутадиеновый, СКИ — изопреновый, СКН — бутадиен-нитриль-ный, СКС — бутадиен-стирольный, СКТ — кремнийоргани-ческий, СКУ — уретановый, СКФ — фторкаучук, СКЭП — этилен-пропиленовый, ХП — хлоропреновый, ХСПЭ — хлорсульфированный полиэтилен.
Перечень химических сред
A3otN2
Азота оксиды
Аллилхлорид СН2=СНСН2С1
Алюминия сульфат A12(SO4)3
Алюминия хлорид А1С13
Аммиак NH3
Аммиак NH3 водные растворы Аммония гидродифторид NH4HF; Аммония нитрат NH4N()3
Аммония ортофосфат (NH4)3PO4
Аммония сульфат (NH4)2SO4
Аммония фторид NH4F Аммония хлорид МЬЦС!
Ангидрид уксусный (СН3СО)2О
Анилин C6H5NH2
Ацетальдегид СН3СНО Ацетилен СН=СН
Ацетон СН3СОСН3
Бария хлорид ВаС12 Бензин
Бензол С6Н6
Бром Вг2
Бромоводород НВг (газ)
Вода Н2О
Вода морская
Водород Н2
Водорода пероксид Н2О2
Глицерин СНОН(СН2ОН)2 1,2-Дихлорэтан С1СН2СН2С1 1,2-Дихлорэтилен С1СН=СНС1
Железа(Ш) нитрат Fe(NO3)3 Железа(П) сульфат FeSO4
Железа(Ш) сульфат Fe2(SO4)3
Железа(Ш) хлорид FeCl3
Иод 12
Иодоводород HI (газ)
Калия гидроксид КОН
Калия нитрат KNO3
Калия сульфат K2SO4
Калия хлорид КС1
Кальция гидроксид Са(ОН)2
Кальция гипохлорит Са(С1О)2
Кальция хлорид СаС12 Кислота
азотная HNO3
борная Н3ВО3
бромоводородная НВг
винная НООССН(ОН)СН(ОН)СООН гексафторокремневая H2SiF6 иодоводородная HI
лимонная НОС(СН2СООН)2СООН малеиновая СООНСН=СНСООН масляная СН3(СН2)2СООН молочная СН3СН(ОН)СООН монохлоруксусная С1СН2СООН муравьиная НСООН ортофосфорная Н3РО4 пропионовая СН3СН2СООН серная H2SO4
трихлоруксусная СС13СООН уксусная СН3СООН фтороводородная HF хлорная НС1О4 хлороводородная НО хлорсульфоновая SO2C1(OH) щавелевая НООССООН
Лития хлорид LiCl
Магния хлорид MgCl2
Меди(П) сульфат CuSO4 Метиленхлорид СН2С12 Метилхлорид СН3С1 Натрия гидроксид NaOH
Натрия гипохлорит NaClO
Натрия карбонат Na2CO3
Натрия нитрат NaNO3
Натрия ортофосфат Na3PO4
Натрия перхлорат NaC104
Натрия сульфат Na2SO4
Натрия хлорат NaC103
Натрия хлорид NaCI
Никеля(П) сульфат NiSO4
Никеля(П) хлорид NiCl2
Серы диоксид SO2
Сероводород H2S
Спирт
метиловый СН3ОН
этиловый С2Н5ОН
Тетрахлорметан СС14
1,1,2-Трихлорэтан СН2С1СНС12
Трихлорэтилен СНС1=СС12
Фенол С6Н5ОН
Формальдегид НСНО
Фтор F2
Фтороводород HF
Хладоны:
дифтордихлорметан CF2C12 (R12)
дифторметан CF2H2 (R32)
дифторхлорметан CF2C1H (R22) октафторпропан CF3CF2CF3 (R218) 1,1,1,2-тетрафторэтан CF3CFH2 (R134a) 1,1,2-трифгоргрихлорэтан CF^lCFCk (R113) фтортрихлорметан CFC13 (R11)
Хлор Cl2
Хлороводород HC1 (газ)
Хлороформ СНС13
Цинка хлорид ZnCl2
Этилен Н2С=СН2
Этилхлорид С2Н5С1
142
Новый справочник химика и технолога
Таблица 1.4.62
Коррозионная стойкость материалов
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Азот N2
Алюминий —- Г, Ж (-196)-150 <0,1
Алюминиевый сплав АМцС — Г, Ж (-196)- 150 <0,1
Вольфрам — Г <2300 <0,1
Медь — Г, Ж (-196)-250 <0,1
Латунь Л62 -— г, ж (-196)-120 <0,1
Никель — г, ж (-196)-500 <0,1
Серебро — г 400 <0,1
Сталь СтЗ — г 300 <0,1
Тантал — г >200 > 10,0
Титан — г >600 > 10,0
Асбестовый шнур — г (-40)-450 с
Паронит — г (-50) - 450 с
Азота оксиды*
Алюминий
А5 5 г 200 0,009
АД1 0,35 ж 50 < 0,0002
0,35 г 50 0,0022
2 г 100-200 < 0,002
5 г 200 0,014
Алюминиевые сплавы
АМг, АМгб, АМгбТ, АМг5ВМ 2 г 150 < 0,002
АК6, АК8, АК9 (АЛ4), АВ 2 г 200 < 0,002
Д16Т 5 г 200 0,019
Д16 (полированный) 2 г 100-200 < 0,002
САП-2 2 г 100-200 < 0,002
Ванадий 5 г 500 0,019
Вольфрам 5 г 500-600 0,02
Кобальт 5 г 500 0,041
Медь 5 г 100 > 10,0
Медный сплав БрХ0,8 2 г 100 > 10,0
Молибден 5 г 500 0,086
Никель Н1 2 г 100 0,16
2 г 500 0,001
2 г 700 0,009
5 г 500 <0,001
5 г 700 0,003
15 г 350 0,001
Никелевый сплав ХН78Т 2 г 100 0,006
2 г 500 0,001
Ниобий 5 г 500 0,04
* Образуются при термической диссоциации тетраоксида диазота N2O4 и представляют собой смесь N2O4, NO2, NO, N2O3, соотношение компонентов которой зависит от температуры и давления.
Электродные процессы
143
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Азота оксиды (продолжение)
Свинец — Г 100 > 10,0
Стали
СтЗ 2 Г 100 0,22
2 Г 700 0,18
20X13 2 Г 100 0,003
2 Г 500 < 0,001
2 Г 700 0,001
5 Г 500 0,001
5 Г 700 0,001
15 Г 350 0,004
15 Г 500 0,001
12Х18Н10Т 0,35 Г 50 0,0025
0,35 Ж 50 0,0002
2 г 100 0,002
2 г 500 0,001
5 г 500 0,001
5 г 600 < 0,001
15 г 350 0,006
15 г 600 0,004
06ХН28МДТ 2 г 100 0,001
2 г 500 <0,001
2 г 700 0,001
5 г 500 <0,001
15 г 350 0,004
15 г 600 0,003
Тантал — г 500 > 10,0
Титан ВТ1-1 2 г 200 < 0,001
Титановые сплавы
ТСЗ, ТС5, ВТ5-1, АТ6 2 г 200 <0,001
ТС4 2 г 200 0,001
ОТ4 2 г 600 0,008
48-ОТЗ 2 г 200 0,004
Цирконий 5 г 100 <0,001
5 г 500 0,011
Циркониевые сплавы
110 5 г 600 0,063
125 5 г 500 0,16
Чугун серый СЧ15 2 г 100 1,14
Графитопласт 7В-2А — г 30 С
Полиизобутилен — г 20 ОС
11 г 60-100 н
Стекло — г 20 С
144
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Азота оксиды {продолжение)
Фторопласт 4 — Г, Ж 50 С
Эмаль кислотостойкая — Г 20 С
Аллилхлорид СН2=СНСН2С1
Алюминий Безводный Ж <45 <0,1
Кобальтовый сплав То же Ж <45 <0,1
(Со 55 %, Сг 33 %, W 6 %)
Медь » Ж <45 <0,1
Сухой Г 70 <0,5
Медные сплавы
БрОФ6,5-0,15 Безводный ж 20 <0,5
БрОФ7,2-0,2 Сухой г 70 <0,5
Никель Безводный ж <45 <0,1
НМЖМц28-2,5-1,5 То же ж <45 <0,1
ХН78Т Вода 0,07 ж 45 0,001
То же г 45 0,002
Вода 2,7 ж 45 0,039
То же г 45 0,033
Н70МФ — ж 25 <0,05
— г 50 <0,05
хастеллой А Безводный ж <45 <0,1
хастеллой В То же ж <45 <0,1
хастеллой С » ж <45 <0,1
инконель (Ni; Сг: 13-17 %, » ж <45 <0,1
Fe: 6-8 %)
Осмий » ж <45 0,1
Палладий, платина, родий » ж <45 0,1
Свинец » ж <45 <0,1
Серебро » ж <45 0,1
Стали
СтЗ Вода 0,03 ж 45 0,02
Вода 0,1 г, ж 45 0,11
Вода > 1 ж 20 0,88
То же г 45 0,22
12Х18Н10Т Вода 0,07 ж 45 0,003
Вода > 1 г, ж 20 0,35
То же г 45 0,13
10Х17Н13М2Т Вода > 0,07 г 45 0,014
То же ж 45 0,01
Вода > 2,7 г 45 0,16
То же ж 45 0,03
06ХН28МДТ » г 45 0,24
» ж 45 0,11
Электродные процессы
145
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аллилхлорид СН2=СНСН2С1 (продолжение)
Титан ВТ1 Вода < 2,7 Г 45 0,027
То же Ж 45 0,002
Цирконий — Ж 25 <0,05
— Г 50 <0,05
Чугуны
серый Безводный Ж 20 <0,1
СЧ15, СЧ17 — Ж 20-50 <0,05
Антегмит АТМ-1 — Ж 45 С
Паронит — Ж 45 с
Полиамиды — Ж 25 с
Поливинилхлорид — Ж 45 н
Полиизобутилен — Ж 45 н
Полиметилметакрилат — Ж 25 н
Полипропилен — Ж 45 н
Полиэтилен — ж 45 н
Резины на основе —
НК + СКВ (2566) — ж 20 н
СКФ (ИРП-1287, ИРП-1345) — ж 20 ОС
Стекло — ж 45 с
Фторопласт 4 — ж 45 с
Эмаль кислотостойкая — ж 45 с
Алюминия сульфат A12(SO4)3
Алюминий Безводный т 20 > 10,0
и влажный
<26 ж 20 0,09
<26 ж Ткип > 10,0
Алюминиевый сплав АК12 (АЛ2) 25 ж 20 0,018
Медь Ml 1-насыщ. ж 20-100 <0,1
Никель 10-насыщ. ж 20 0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 <26 ж 20 <0,1
<26 ж 35 0,05
<26 ж 115 0,41
Н55Х15М16В <26 ж 20 <0,1
хастеллой С 1-насыщ. ж Лин <0,1
хастеллой D То же ж Лип <0,1
Свинец » ж 20—Ткип <0,1
Серебро <26 ж 20 <0,1
<26 ж 100 <0,5
Стали
СтЗ 1-насыщ. ж 20 > 10,0
12X13 <26 ж 20 3
<26 ж Лин > 10,0
146
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза г. °с
Алюминия сульфат AI2(SO4)3 (продолжение)
Стали
12Х18Н10Т 5-насыщ. Ж 20-65 <0,1
10Х17Н13М2Т То же Ж 20-65 <0,1
06ХН28МДТ <26 Ж 20-Ткнп <0,1
Тантал 1-насыщ. Ж 20-Ткип <0,1
Титан 50 Ж 100 <0,05
Чугуны
СЧ15, СЧ17 10-насыщ. Ж 20 0,1-1,0
10 Ж Гкип 3,0
хромистый (Сг 32,9 %) 10-насыщ. Ж 20 <0,1
Антегмит АТМ-1 1-насыщ. Ж Т 1 кип С
Графит, пропитанный бакелите- То же Ж т 1 кип С
вым лаком
Полиамиды <26 Ж 20-60 С
Поливинилхлорид <26 Ж 20-40 С
<26 Ж 60 ОС
Полиизобутилен <26 Ж 20-60 С
<26 Ж 70-100 ОС
Полиметилметакрилат <26 Ж 20 С
<26 Ж 60 ОС
Полипропилен <26 Ж 20-60 С
Полиэтилен <26 Ж 20-60 С
Резины на основе
БК <26 ж 20-80 С
НК, СКС <26 ж 20-70 С
ХП <26 ж 20-90 С
Стекло <26 ж 20- С
Фторопласт 4 <26 ж 20-100 С
Эмаль кислотостойкая <26 ж 20-ТКИ11 С
Алюминия хлорид А1С13
Алюминий Безводный т 20 <0,1
10 ж 20 > 10,0
Медь Безводный, т 20 <0,1
без воздуха
10 ж 20 > 10,0
Медный сплав БрА7 10 ж 25 > 10,0
Никель 10 ж 25 <1,3
<31,4 ж 100 > 10,0
Никелевые сплавы
Н70МФ <31,4 ж 20-Ткнп <0,1
хастеллой А 1-насыщ. ж <50 <0,1
хастеллой В 20-80 ж 20-Ткип <0,1
хастеллой С 25 ж 20 <0,1
Электродные процессы
147
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Алюминия хлорид А1С13 (продолжение)
Платина — Т 50-100 0,05
1-насыщ. Ж Ткип <0,1
Свинец 10 Ж 25 > 10,0
Серебро 10-30 ж 20-100 0,05
50 ж 20 <0,5
Стали
СтЗ Безводный т 20 0,2
— т 75 > 10,0
Безводный ж 200 > 10,0
Сухой г 200 > 10,0
<31,4 ж 20-7 1 кип > 10,0
12X13 <31,4 ж 20 Гкип > 10,0
12Х18Н10Т 10 ж 20 < 1,0 т
< 10 ж Т’кип 1,0-3,0
50 ж 50 < 5,0 т
— ж 200 > 10,0
10Х17Н13М2Т 10 ж 20 < 1,1 т
06ХН28МДТ — т 75 < 0,5 т
12,5 ж 30 < 0,5 т
<31,4 ж 100 1,0-3,0
Тантал <31,4 ж 20-150 <0,001
Титан ВТ 1-0 10-25 ж ТКип <0,1
30 ж 95 0,16
Титановый сплав ОТ4 25 ж ТКип <0,1
30 ж 95 0,16
Цирконий 10-30 ж 25-100 <0,5
Чугуны
серый Безводный т 20 <0,1
— т 75 > 10,0
<31,4 ж 20 > 10,0
СЧ15 <31,4 ж 20-70 <0,1
Антегмит АТМ-1 <31,4 ж Т 1 кип С
Графит, пропитанный бакелито- <31,4 ж Ткип с
вым лаком
Полиамиды <31,4 ж 20-60 С
Поливинилхлорид 25 ж 40 с
25 ж 60 ОС
Поливинилхлорид 25 ж 80 н
Полиизобутилен <31,4 ж 20-60 С
<31,4 ж 100 ОС
Полиметилметакрилат <31,4 ж 20 с
<31,4 ж 60 ОС
Полистирол <31,4 ж 20-60 с
148
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Д °C
Алюминия хлорид А1С13 (продолжение)
Полипропилен <31,4 Ж 20-60 С
Полиэтилен <31,4 Ж 20-60 С
Резины на основе
БК <31,4 Ж 20-100 С
НК, СКС, ХП <31,4 Ж 20-70 с
С КН <31,4 Ж 20-60 с
ХСПЭ <31,4 Ж 20-80 с
Стекло <31,4 Ж 20-100 С
Фторопласт 4 1-насыщ. Ж <200 с
Эмаль кислотостойкая <31,4 Ж 20-150 с
Аммиак NH3 с содержанием воды < 0,2 масс. %
Алюминий
А7 (А1 > 99,7 %) 0,6-1,0 Г, Ж 16-20 <0,001
— Г, Ж 50 <0,001
А5 (AI > 99,5 %) ГОСТ 11069 0,6-1,0 ж 16-20 0,026 т
0,6-1,0 г 16-20 0,013 т
— ж 50 0,028 т
— г 50 0,012 т
АО (А1 > 99,0 %) 0,6-1,0 ж 16-20 0,026 т
0,6-1,0 г 16-20 0,012 т
— ж 50 0, 022 т
— г 50 0,011 т
АД00, АДО, АД1, Сухой г <200 <0,1
Алюминиевые сплавы
АВ, АМг, АМгЗМ, Д16, АК9 0,6-1,0 ж 16-20 < 0,016 т
(АЛ4), АМгбН, АМгбТ, АМг7
0,6-1,0 г 16-20 < 0,013 т
АВ, АМг, АМг7 — ж 50 < 0,028 т
— г 50 < 0,013 т
АМг5В, АМгбН, АМгбТ, АМц 0,1-0,3 г, ж -30 < 0,003 т
АМгб — г 50 0,007 т
0,1-0,3 г, ж -30 < 0,019 т
АМгбМ, АЛ4, Д16 0,1-0,3 г, ж -30 < 0,003
АМцС Сухой г <200 <0,1
Вольфрам — г 650 <0,1
Медь
Ml 0,1-03 г, ж -30 < 0,006
0,1-0,3 ж -30 0,075
М2 — г 50 0,05
М3 0,1-0,3 ж -30 0,075
0,1-0,3 г -30 <0,001
0,6-1,0 ж 16-20 0,01
Электродные процессы
149
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммиак NH3 с содержанием воды < 0,2 масс. % {продолжение)
Медь
М3 0,6-1,0 Г 16-20 0,004
— Г, Ж 50 <0,11
Медные сплавы
БрОФ, БрА5 0,1-0,3 Г, Ж -30 0,001
БрОФ7-0,2, БрАЖМц 10-3-1,5, БрОС 5-25 0,6-1,0 Г, Ж 16-20 <0,1
БрОФ7-0,2, БрАЖМц 10-3-1,5 — Г, Ж 50 <0,1
БрОС 5-25 — Ж 50 <0,3
— Г 50 <0,2
Л90 0,1-0,3 Ж -30 0,05
Л68, Л90, ЛС59-1 0,1-0,3 Г -30 0,001
Л62, Л68, Л90, ЛС59-1 0,6-1,0 Ж 16-20 <0,013
Л62, Л90 0,6-1,0 Г 16-20 0,005
Л62, ЛС59-1 — Г, Ж 50 <0,1*
Никель 0,1-0,3 Ж -30 0,001
0,1-0,3 Г -30 0,001 т
0,6-1,0 Г, Ж 16-20 < 0,002
— Г, Ж 50 <0,001
— Г 400-500 0,05
Никелевые сплавы
НМЖМц28-2,5-1,5 0,1-0,3 г, ж -30 < 0,002
0,6-1,0 г, ж 16-20 < 0,002
— г, ж 50 <0,001
Н70МФ, Н55Х15М16В — г 500 0,01
Свинец 0,1-0,3 г, ж -30 <0,02
0,6-1,0 г, ж 16-20 < 0,004
— г 300 <0,1
Стали
СтЗ, 10 0,1-0,3 г, ж -30 < 0,003
0,6-1,0 г, ж 16-20 < 0,012 т
25 — г, ж 50 <0,001
12X13, 12Х18Н10Т, 10Х17Н13М2Т 0,1-0,3 г, ж -30 <0,001
— г, ж < 100 <01
06ХН28МДТ 0,6-1,0 г, ж 16-20 <0,001
— г, ж 50 <0,001
Титан ВТ1,ВТ1-1 0,1-0,3 г -30 <0,001
0,6-1,0 ж 16-20 <0,001
Сухой г <40 <0,1
То же г 450 < 0,001
Чугун серый 0,1-03 ж -30 0,03
0,1-03 г -30 <0,001
Обесцинкивание.
150
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммиак NH3 с содержанием воды < 0,2 % масс, {продолжение)
Чугун серый 0,6-1,0 Ж 16-20 0,023
0,6-1,0 Г 16-20 0,004
Антегмит АТМ-1 — Г 20 С
Полиамиды — Г 20-60 с
Поливинилхлорид — Г 20-60 с
— Ж 20 ОС
Полиметилметакрилат — г 20 н
— ж 15-20 с
Полипропилен — г 20-60 с
Полиэтилен — г 20-60 с
— ж -33 с
Резины на основе
БК, НК, СКН, СКС — г 20 с
СКТ — г 60 ОС
СКФ — г 20 н
Резины
НО-68-1, 1751,4327,4770 — ж 15-20 С
518,6138 — ж 15-20 ОС
Стекло — г 20-150 С
Фторопласт 4 — ж -33 С
— г 20-100 С
Эмаль кислотостойкая — г 20 С
Аммиак NH3 водные растворы
Алюминий
(AI > 99,99 %) 0,5 ж 20 5,8
2,0 ж 30 9,2
5,0-10,0 ж 20 > 10,0
21,8 ж 20 2,2
А5 (А1 > 99,5 %) 15 ж 22-27 0,001
25 г, ж 22-27 < 0,006
Алюминиевые сплавы
(Al; Mg 3,8-10,3 %) 24 ж 20 0,01
(Al, Mg: 1,5-2,5 %; Mn: 1,0-2,0 %; Fe: 24 ж 20 0,173
< 0,8 %)
Медь — ж 20 > 10,0
Никель 3,5 ж 20 0,03
10 ж 20 <0,1
10 ж 100 > 10,0
Никелевый сплав — ж 20 0,06
НМЖМц28-2,5-1,5
Свинец 35 ж 20 0,04
Свинец сурьмянистый CCyl Насыщ. ж 20 0,01
Электродные процессы
151
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
АммиакХНз водные растворы (продолжение)
Стали
СтЗ, 10, 20 5-25 Ж 20-Гкнп <0,1
12X13 1-30 Г, Ж 20-100 <0,1
12Х18Н10Т 1-30 Г, Ж 20-100 <0,1
10Х17Н13М2Т 1-30 Г, Ж 20-100 <0,1
06ХН28МДТ 1-30 Г, Ж 20-100 <0,1
Тантал 10 Ж 100 <0,001
Титан 28 Ж 20— Ттп <0,1
Чугуны СЧ15, СЧ17 25 Ж 20 <0,1
Антегмит АТМ-1 1-30 Ж т * кип с
Графит, пропитанный бакелито- 1-30 Ж Ткип с
вым лаком
Полиамиды 30 ж 20-60 с
Поливинилхлорид 25 ж 20-60 с
Полиизобутилен ПСГ 25 ж 20 с
Полиметилметакрилат 25 ж 20 с
Полистирол 25 ж 20 с
Полиэтилен 25 ж 20 с
Фторопласт 4 30 ж 20-60 с
Аммония гидродифторид NH4HF2
Алюминий 10 ж 100 > 10,0
Золото — т 25 <0,05
Медь 12,5; без кислорода ж 30 > 10,0
воздуха
Медные сплавы
БрА5, БрА7 12,5 ж 25 > 10,0
Л59, Л63, Л68, Л80 12,5-15 ж 25 > 10,0
Никель 10 ж 25 <0,5
Никелевые сплавы
НМЖМц28-2,5-1,5 — т 30 <0,5
12,5 ж 25 <0,5
12,5 ж 95 > 10,0
ХН78Т 12,5 ж 25 <0,5
ХН65МВ — т 30-105 <0,5
— ж 130-220 <0,5
12,5 ж 30 <0,5
Платина — т 25 <0,05
Свинец — т 30 <0,5
10 ж 24 <0,5
10 ж 100 > 10,0
Серебро — т 25 <0,05
Стали
СтЗ 10 ж 100 0,26
152
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония гидродифторид NH4HF2 (продолжение)
Стали
СтЗ 20 Ж 100 0,21
40 Ж 100 0,52
50 Ж 50 0,8
50 Г 50 0,7
50 Ж 100 1,2
60 Ж 100 8,3
80 Ж 90 4,7
80 Г 90 5,5
80 Ж 100 9,4
12Х18Н10Т 10 Ж 100 2,11
30 Ж 100 0,4
50 Ж 50 0,8
50 Г 50 0,27
50 Ж 100 1,6
60 Ж 100 3,2
80 Ж 90 0,85 я
10Х17Н13М2Т 10 Ж 100 2,05
30 Ж 100 0,44
50 Ж 100 0,51
60 Ж 100 0,27
80 Ж 100 0,12
06ХН28МДТ — Т 20-100 <0,5
10 Ж 20-75 <0,5
Титан 10 Ж 25 > 10,0
Чугуны
серый 12,5 Ж 30 > 10,0
СЧ15, СЧ17 — Т 25 <0,5
Кварц <20 Ж 25 Н
Поливинилхлорид <20 ж 20 С
Полиэтилен <20 ж 20 с
Стекло <20 ж 20 н
Фарфор <20 ж 20 н
Эмаль кислотостойкая <20 ж 20 н
Аммония нитрат NH4NO3
Алюминий 8 ж 20 <0,01
Насыщ. ж 20—Тюш <0,1
— ж 180 <0,1
Алюминиевый сплав 10 ж 20 0,006
(Al, Mg: 1,5-2,5 %; Мп: 1,0-2,0 %)
Медь и медные сплавы <64 ж 20 > 10,0
Никель 10 ж 20 <0,1
10 ж 100 <3,0 .
Электродные процессы
153
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония нитрат NH4NO3 (продолжение)
Никелевые сплавы
НМЖМц28-2,5-1,5 <64 Ж 20 > 10,0
Н70МФ <64 Ж 20 <0,1
Свинец <64 Ж 20 > 10,0
Серебро <64 Ж 20 0,1
<64 Ж Ткип 3,0
Стали
СтЗ 10 Ж 20 0,54
10 Ж 50 0,63
20 Ж 20 1,0
20 Ж 50 1,0
40 Ж 20 1,9
40 Ж 50 0,44
60 ж 20 1,0
60 ж 50 0,26
12X13 10 ж <90 <0,1
50 ж ткип <1,0
65 ж < 125 <0,1
75 ж <90 <0,1
12Х18Н10Т 10 ж <90 <0,1
10Х17Н13М2Т 50-60 ж Ткип <0,1
75 ж <90 <0,1
85 ж 80-120 <0,1
Насыщ. ж Т 1 кип < 1,0
06ХН28МДТ <64 ж 20-Т 1 кип <0,1
Насыщ. ж т 1 кип < 1,0
Тантал <64 ж 20-Г 1 кип <0,001
Титан 64 ж Гкип <0,05
ЧугунСЧ15, СЧ17 «65 ж 20 0,003
Графит, пропитанный бакелите- <64 ж 20 с
вым лаком
Полиамиды <64 ж 20-60 с
Поливинилхлорид 25 ж 40 с
25 ж 60 ОС
Полиизобутилен Насыщ. ж 20-100 с
Полиметилметакрилат <64 ж 20-60 с
Полипропилен <64 ж 20-100 с
Полиэтилен <64 ж 20-60 с
Резины на основе
БК, НК, СКС, СКУ <64 ж 20-60 с
СКН <64 ж 20 с
ХП <64 ж 20-90 с
ХСПЭ <64 ж 20-80 с
154
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония нитрат NH4NO3 (продолжение)
Стекло <64 Ж 20-Гкип С
Фторопласт 4 <64 Ж <200 С
Эмаль кислотостойкая <64 Ж 20-Ткнп С
Аммония ортофосфат (NH^PC^
Алюминий 3 Ж 20 0,027
3 Ж 60 0,7
10 Ж 20 0,23
10 Ж 60 25,5
Медь <40 Ж 20 1,0-3,0
Никель <40 Ж 20 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 <40 Ж 20 <0,1
Н70МФ <40 Ж 20 <0,1
Свинец <40 Ж 20 <0,5
Серебро <40 Ж 20 0,1-1,0
<40 Ж 100 1,0-3,0
Стали
углеродистая < 10 Ж 20 <0,5
30 Ж 20 > 10,0
12X13 < 40; pH 7 Ж 20 <0,1
<40 Ж Ткип > 10,0
12Х18Н10Т <40 Ж 20 <0,1
<40 Ж Гкип 1,0-10,0
10Х17Н13М2Т <40 Ж 20 <0,1
<40 ж Ттп 0,1-3,0
06ХН28МДТ <40 ж 20 <0,1
<40 ж 100 0,1-1,0
Тантал <40 ж 20-100 <0,1
Титан <40 ж 20 <0,1
<40 ж Гкип > 10,0
Чугуны
серый <40 ж 20 0,1-1,0
СЧ15, СЧ17 <40 ж 20 > 10,0
Антегмит ATM-1 <40 ж 20 С
Графит, пропитанный бакелито- <40 ж 20-120 С
вым лаком
Полиамиды <40 ж 20-60 С
Поливинилхлорид <40 ж 20-60 С
Полиизобутилен <40 ж 20-60 С
Полиметилметакрилат <40 ж 20-60 С
Полипропилен <40 ж 20-60 С
Полиэтилен <40 ж 20-100 С
Электродные процессы
155
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Аммония ортофосфат (NH^PC^ (продолжение)
Резины на основе
БК <40 Ж 20 С
НК, СКС <40 Ж 20-70 С
СКН <40 Ж 20-65 С
ХП <40 Ж 20-90 С
Стекло <40 Ж 20-100 С
Фторопласт 4 1-насыщ. Ж <200 С
Эмаль кислотостойкая <40 Ж 20-100 С
Аммония сульфат (NH4)2SO4
Алюминий 10 Ж 20 0,01 т
Алюминиевый сплав 10 Ж 20 0,016 т
(Al, Mg: 1,5-2,5 %, Мп: 1,0-2,0 %)
Медь 10 Ж 20 0,26
10 Ж 40 0,56
>30 Ж 100 > 10,0
Медные сплавы
БрАМц9-2 10 Ж 40 0,054
БрАЖ9-4 10 Ж 40 0,07
БрКМцЗ-1 10 Ж 40 0,59
БрОЦ4-3 10 Ж 40 0,65
БрОЦС4-4-2,5 10 Ж 40 0,75
Никель 10-насыщ. Ж 20 <0,1
То же Ж 100 <1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 10 Ж 40 0,05
<43 Ж 20 <0,1
<43 Ж 100 <1,0
Н70МФ <43 Ж 20 <0,1
<43 Ж 100 < 1,0
Свинец <43 Ж 20 <0,01
43 Ж 50 0,07
43 Ж ПО 0,9
Серебро <43 ж 20 <0,1
Стали
СтЗ 5 ж 20 0,25
30-50 ж 20 0,05
<43 ж 100 > 10,0
12X13 <43 ж 20 <1,0
<43 ж Ткип > 10,0
12Х18Н10Т 1-насыщ. ж 20-Г 1 кил <0,1
10Х17Н13М2Т То же ж 20— ТКИП <0,1
06ХН28МДТ <43 ж 20-100 <0,1
Тантал <43 ж 20-100 <0,001
156
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Аммония сульфат (NH4)2SO4 (продолжение)
Титан <43 Ж 20-Ткип <0,13
Полиамиды <43 Ж 20-60 С
Поливинилхлорид <43 Ж 20-40 с
<43 Ж 60 ОС
Полиизобутилен <43 Ж 20-100 с
Полиметилметакрилат <43 Ж 20-60 с
Полипропилен <43 Ж 20-100 с
Полиэтилен <43 Ж 20-60 с
Резины на основе
БК, НК, СКС, СКН, СКУ, ХП <43 Ж 20-60 с
ХСПЭ <43 Ж 20-80 с
Стекло <43 ж 20-Ткип с
Фторопласт 4 1-насыщ. ж <200 с
Эмаль кислотостойкая <43 ж 20-Ткип с
Аммония фторид NH4F
Алюминий 7,5 ж 100 > 10,0
10-12 ж 20 0,003
10-12 г 20 <0,01
10-12 ж 50 0.08
10-12 г 50 <0,01
10 ж 75 <0,5
50 ж 50 0,045
— г 50 0,042
80 ж 115 0,54
— г 115 0,019
Алюминиевые сплавы
АМгЗ 50 ж 50 0,02
— 50 0,02
АМгб 10-12 ж 20 0,03
10-12 г 20 <0,01
10-12 ж 50 0,05
10-12 г 50 <0,01
Д16 50 ж 50 0,06
— г 50 0,06
Золото — т 25 <0,05
10-50 ж 20-100 <0,05
Магний Мг1 50 ж 70 0,43
— г 70 0,43
50 ж 90 1,2
— г 90 0,12
80 ж 115 2,96
— г 115 0,08 я
Электродные процессы
157
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония фторид NH4F (продолжение)
Медь Без кислорода Т 50 > 10,0
воздуха 50 Ж 50 0,60
— Г 50 1,8 т
Медные сплавы
БрА5, БрА7 — Т 55 > 10,0
12,5 Ж 25 > 10,0
БрОФ6,5-0,15 Без кислорода Т 50 > 10,0
Л59, Л63, Л68, Л80 воздуха Т 45-55 > 10,0
12,5 Ж 25 > 10,0
ЛО68-1 12,5 Ж 25 10,0
ЛС59-1 50 Ж 50 0,58
— Г 50 1,3
Никель 20 Ж 20-95 <0,05
50 Ж 70 0,009
— Г 70 0,045
50 Ж 90 0,013
— Г 90 0,009
80 Ж 50 0,02
— Г 50 0,07
Никелевый сплав 10-20 ж 20-70 >0,5
НМЖМц28-2,5-1,5
20 ж 95 < 10,0
50 ж 90 0,06
— г 90 0,002
80 ж 115 0,23
— г 115 0,014
Свинец С2 50 ж 70 0,05
— г 70 0,25
50 ж 90 0,08
— г 90 0,3
80 ж 115 0,2
— г 115 0,14
Стали
СтЗ — т 25 <0,5
— т 100 > 10,0
0,7 ж 20 < 1,0
10 ж 20 0,16
— г 20 <0,001
10 ж 50 1,06
— г 50 0,003
10 ж 70 > 10,0
ЗОХГСА 50 ж 50 0,81
— г 50 0,07 т
158
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония фторид NH4F (продолжение)
Стали
12X13 50 Ж 50 0,19
— Г 50 0,18
12Х18Н10Т 10 Ж 50 < 0,003
50 Ж 50 0,025
— Г 50 0,028
50 Ж 90 0,25
— г 90 0,11
80 ж 115 0,12 т
— г 115 0,025 т
10Х17Н13М2Т 20 ж 25 <0,5
20 ж 75 > 10,0
50 ж 90 0,23
— г 90 0,1
80 ж 115 0,035
— г 115 0,051 я
06ХН28МДТ 50 ж 70 0,1
— г 70 0,18
50 ж 90 0,013
— г 90 0,08
80 ж 115 0,12
— г 115 0,015
Тантал 5 ж 25 > 10,0
Титан 7,2-33,0 ж 20 0,2-0,5
Цинк 5 ж 20 1,0
10 ж 20 1,9
20 ж 20 2,1
Чугуны
серый 20 ж 100 > 10,0
СЧ15, СЧ17 10 ж 25 > 10,0
Поливинилхлорид 20 ж 20 С
<42 ж 60 ОС
20 ж 100 н
Полиизобутилен <42 ж 20-60 с
<42 ж 100 ОС
Полиметилметакрилат <42 ж 20-40 с
Полипропилен 20-42 ж 20-60 с
Полиэтилен <42 ж 20-60 с
Резины на основе
БК, НК, СКН, СКС, ХП <42 ж 20 с
Стекло <42 ж 20 н
Уголь <42 ж 20 с
Фторопласт 4 <42 ж 20-100 с
Эмаль кислотостойкая <42 ж 20 н
Электродные процессы
159
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Аммония хлорид 1ЧН4С1
Алюминий 1 Ж 20-50 <0,06
1 Ж 100 0,1
10 Ж 20 < 0,09 т
10 Ж 50-100 0,2 т
Влажный Т 20 > 10,0
Безводный Т 20 < 1,0
Алюминиевые сплавы
АК12 (АЛ2) 10 Ж 20 0,03 я
(Al, Mg: 9,5-11,5%) 10 Ж 20 0,03
Медь 5 Ж 20 6,0
<27 Ж 20 > 10,0
Медные сплавы
(Си, Sn: 4,3-5,0 %) 5 Ж 20 0,36
10 Ж 40 3,9
БрА5 10 Ж 25 > 10,0
Л62 5 Ж 20 <0,1
Л59, Л63, Л68 10 Ж 35 0,005
10 Ж 100 0,06
Молибден 10 ж 25 10,0
Никель 10 40 ж 25-100 <0,5
50 ж 20-100 <1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 10 ж 20-40 0,5
20-50 ж 20-100 < 1,3
ХН78Т 1-насыщ. ж Т’кип <0,1
Н70МФ 10 ж 20-100 < 0,5 т
ХН65МВ 25 ж Т’кип <0,01
хастеллой В 1-насыщ. ж 140-144 <0,1
хастеллой С Насыщ. ж Т 1 кип <0,1
Платина 1-насыщ. ж <0,04
Свинец 10 ж 25 <0,5
10 ж 55 > 10,0
25 ж 20 > 10,0
Серебро 1 -насыщ. ж ?kn <0,08
Стали
СтЗ 1 ж 18-20 0,062
— г 18-20 0,02 т
5 ж 18-20 0,064 т
— г 18-20 0,016 т
10 ж 18-20 0,1
10 ж 55 > 10,0
12X13 1 ж 20 < 0,05 т
5-10 ж 90 <5,0т
160
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Т,°С и характер коррозии Стойкость материалов
Аммония хлорид NH4CI (продолжение')
Стали
12X13 5 Ж Гкип < 10,0
10 Ж 20 <0,5т
10 Ж Гкип > 10,0
12Х18Н10Т 5-10 Ж 20-90 <0,1
10-25 Ж Гкип < 5 т
30 Ж 50 < 0,1 т
40-50 Ж Гкип < 10,0
10Х17Н13М2Т 1 Ж 20 <0,01 т
5-10 Ж 90 < 0,05 т
5 Ж т 1 кип <0,1 т
10 Ж Гкип < 0,5 т
25 ж 90 < 0,1 т
30 ж 50 0,1 т
40-50 ж Ткип <5,0т
06ХН28МДТ 1, 10; аэрированные ж 35-100 <0,1
25-50 ж Ткип <0,5т
Тантал 10-50 ж 20-105 0,05
Титан 0,6 ж 160 0,02
10 ж 100 <0,01
4,5 ж 115 0,001
1-насыщ. ж 100 <0,13
Цирконий 1 ж 35 0,0008
1 ж 60 0,0013
1 ж 100 0,0033
10 ж 35 0,0003
10 ж 60 0,0008
Насыщ. ж Гкип 0,0005
Чугуны
серый 5 ж 93 4,8
20 ж 93 5,8
28-40 ж 25 > 10,0
СЧ15, СЧ17 10 ж 30-105 <0,05
Х28Л 28-40 ж 25-100 0,3
ЧН15Д7Х2 5 ж 20 0,08
5-20 ж 93 <0,3
Антегмит АТМ-1 <27 ж 20-Т 1 кип С
Графит, пропитанный бакелито- <27 ж 20—Ткип с
вым лаком
Полиамиды <27 ж 20-60 с
Поливинилхлорид <27 ж 20-40 с
10 ж 60 ОС
Электродные процессы
161
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Аммония хлорид NH4C1 (продолжение)
Полиизобутилен <27 Ж 20-100 С
Полиметилметакрилат <27 Ж 20-100 С
Полипропилен <27 Ж 20-60 С
Полиэтилен <27 Ж 20-60 с
Резины на основе
БК, СКН <27 Ж 20-60 с
НК, СКС, ХСПЭ <27 Ж 20-80 с
СКФ (ИРП-1225, ИРП-1345) 30 Ж 70 н
СКФ (ИРП-1287) 30 Ж 70 ОС
ХП <27 Ж 20-90 с
Стекло <27 Ж 20—Ткип с
Фторопласт 4 <27 Ж 20-100 с
Эмаль кислотостойкая <27 Ж 20-Т^п с
Ангидрид уксусный (СН3СО)2О
Алюминий — Ж 25 0,005
— Г, Ж 60 0,12
Медь — Г, Ж 25 <0,1
— Ж 75 1,16
— Ж Ткип >3,0
Медные сплавы
(Cu, А1: 4-8 %), (Си, Sn: 7-9 %) — Ж 20 <0,1
— Ж ТКип >3,0
Л90 — Ж 20 <0,1
— Ж т 1 кип >3,0
Никель НП2 — ж 20-7 1 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж Т’кип <0,1
хастеллой В — ж < 100 <0,1
Свинец С1 — ж 20 <0,1
— ж т 1 кип >3,0
Серебро — ж 20-Ткип <0,1
Стали
СтЗ — ж 20 < 1,0
— ж Ткип >3,0
12X13 -— ж 20 <0,1
12X17 — ж <80 <0,1
— ж т 1 кип >3,0
12Х18Н10Т — ж 20-7 Z'V 1 кип <0,1
10Х17Н13М2Т — ж 20-7 Z'V 1 кип <0,1
06ХН28МДТ 99,5 ж 7 1 кип <0,1
Тантал — ж 7 1 кип <0,1
Титан ВТ-00, ВТ 1-0 99,5 ж Ткип <0,1
Цинк — ж 20 >3,0
162
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Ангидрид уксусный (СН3СО)2О (продолжение)
Цирконий 99,5 Ж ^кип 0,001
Чугун серый — Ж 25 0,72
— Ж Т 1 кип 3,95
Антегмит АТМ-1 — Ж Гкип С
Асбовинил — Ж 20 С
Графит и уголь, пропитанные — Ж Гкип с
фенольными олигомерами
Графит, пропитанный бакелите- — Ж < 100 с
вым лаком
Полиамиды — Ж 20 н
Поливинилхлорид — Ж 20-60 н
Полиизобутилен — Ж 20 с
— Ж 60 ОС
Полиметилметакрилат — Ж 20-60 н
Полипропилен — Ж 20 с
Полиэтилен — Ж 20 с
— Ж 60 ОС
Резины на основе
БК, ХП — Ж 20-50 с
НК, СКС — Ж 20 с
СКФ — Ж 20 н
Стекло — Ж 20-7 •4'V * кип с
Фторопласт 4 — Ж 20-Т Z'V кип с
Эмаль кислотостойкая — Ж 20-7 1 кнп с
Анилин C6HsNH2
Алюминий — Ж 20 <0,1
— Ж 180 > 10,0
Алюминиевый сплав АК12 (СИЛ-1) — Ж 20 <0,1
Медь — Ж 20 > 10,0
Медные сплавы
(Си, Sn: 3,4 %) — Ж 20 > 10,0
Л62 — Ж 20 > 10,0
Никель — Ж 20 0,1-1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20 <0,1
Н70МФ — ж 20 <0,1
Свинец — ж 20 <0,1
Серебро — ж 20 <0,1
Стали
углеродистая — ж 20 <0,1
12X13 — ж 20 <0,1
12Х18Н10Т — ж 20 <0,1
10Х17Н13М2Т — ж 20 <0,1
Электродные процессы
163
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Анилин C6H5NH2 (продолжение)
Стали
06ХН28МДТ — Ж 20 <0,1
Тантал — Ж 20 <0,001
Титан — Ж 20 <0,1
Цирконий — Ж 20 <0,1
Чугуны
серый — Ж 20 <0,1
СЧ15, СЧ17 — Ж 20 <0,1
Графит, пропитанный бакелито- — Ж 20 с
вым лаком
Керамика — Ж 20-100 с
Пентапласт — Ж 20-65 с
Полиамиды — Ж 20-60 н
Поливинилхлорид — Ж 20-60 н
Полиизобутилен — Ж 20-100 н
Полиметилметакрилат — Ж 20-60 н
Полипропилен — Ж 27 с
Полиэтилен — Ж 20 с
— Ж 60 н
Резины на основе
БК — Ж 20 с
ПС, СКН, ХСПЭ, СКУ — Ж 20 н
СКФ — Ж 20-140 с
СКЭП — Ж 20 ОС
Стекло — Ж 20-100 с
Уголь — Ж 20-170 с
Фторопласт 4 — Ж <200 с
Эмаль кислотостойкая — Ж 20-100 с
Ацетальдегид СН3СНО
Алюминий — Ж 15 0,0016
— Ж Ткип 0,02
Медь — Ж 20 <0,1
— — Т 1 кип <0,5
Никель — ж 20-Ткип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20 <0,1
— — 50 <0,5
Н70МФ — ж 20 <0,1
— — 50 <0,5
Свинец — ж 20 <0,1
Серебро — ж 20 <0,1
— г 200-400 <0,1
164
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Ацетальдегид СН3СНО (продолжение)
Стали
СтЗ 5-7 Ж 20 0,38
— Ж 20 <0,001
— Ж 50-60 0,095
12X13 — Ж 20 1,0-3,0
12X17 — Ж 20 <0,1
12Х18Н10Т 5-7 Ж 20 0,0005
40 Ж Лсип <0,1
10Х17Н13М2Т — Ж 20 <0,1
06ХН28МДТ — Ж 20 <0,1
Тантал — 20-100 <0,1
Титан 99 Ж 20 <0,1
Чугуны
серый — Ж 20 1,0-3,0
кремнистый — Ж 20 <0,1
Антегмит АТМ-1 5-7 Ж 65-75 с
100 Ж 20 с
Поливинилхлорид = 40 Ж 40 с
= 99 Ж 20 н
Полиизобутилен = 99 Ж 20 ОС
Полиметилметакрилат = 99 Ж 10 н
Полипропилен = 99 Ж 10 с
Полиэтилен = 99 Ж 10 с
Резины на основе
БК, ХСПЭ — Ж 20 ОС
СКН, СКС, СКФ — Ж 20 н
ХП — Ж 20-30 ОС
Стекло — Ж 20 с
Фторопласт 4 — Ж 20-60 с
Эмаль кислотостойкая — Ж 20 с
Ацетилен СН= СП
Алюминий АО, А1 Сухой и влажный г 20 <0,1
Алюминиевые сплавы АДО, АД1, То же г 20 <0,1
АМцС, АМг, Д16
Медь » г 20 Неприменима
Медные сплавы
(Си, Sn: 3,42-6,5 %) Влажный г 20 То же
(Си, Sn: 9,58 %, Zn: 1,76 %) — г 20 <0,1
Л62 — г 20 <0,1
Никель — г 20-60 <0,1
— г <200 <0,1
Возможно образование ацетиленида меди, взрывающегося при нагреве и ударе.
Электродные процессы
165
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Ацетилен CHsCH (продолжение)
Никелевые сплавы
НМЖМц28-2,5-1,5 — Г 20 <0,1
Н70МФ — Г 20-100 <0,1
Олово Сухой и влажный Г 20 <0,1
Свинец С2 То же Г 20 <0,1
Серебро — Г 20 Неприменимо
Стали
СтЗ — Г 20 <0,1
12X17 — Г 20 <0,1
12Х18Н10Т — Г 20 <0,1
10Х17Н13М2Т — Г 20-100 <0,1
06ХН28МДТ — Г 20 <0,1
Тантал — г 20 <0,1
Титан — г 20 <0,1
Цинк Сухой г 20 <0,1
Цирконий — г 20 <0,1
Чугун серый — г 20-80 <0,1
Графит, пропитанный бакелито- — г 20 С
вым лаком
Асбест Сухой и влажный г 100 С
Керамика — г 20-400 С
Полиамиды — г 20 С
Поливинилхлорид — г 20 ОС
Полиизобутилен — г 20 с
— г 60 ОС
Полиметилметакрилат — г 20 с
Полиэтилен — г 20 с
Резины на основе
НК, ПС — г 20 ОС
БК — г 20-100 с
СКН, СКС. ХП — г 20-30 с
Стекло — г 20-100 с
Фторопласт 4 — г 20-100 с
Эмаль кислотостойкая — г 20-450 с
Ацетон СН3СОСН3
Алюминий — ж 20 < 0,001
— ж Ткип <0,1
Алюминиевый сплав — ж 20 <0,1
(Al, Mg: 3,8-10,3 %)
Медь — ж 20-Ткип <0,1
Возможен взрыв.
166
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Ацетон СН3СОСН3 (продолжение)
Медные сплавы
(Си, А1: 4-10 %) — Ж Гкип <0,1
(Си, Zn: 1(М0 %) — Ж 20 <0,1
Никель — Ж Т 1 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж т 1 кип <0,1
Н70МФ — Ж т 1 кип <0,1
Олово — Ж т * кип <0,1
Платина — Ж /’кип <0,1
Свинец — Ж 20 <0,1
Серебро — Ж 20 <0,1
Стали
углеродистая — Ж 20-Г 1 кип <0,1
12X17 — Ж т 7 кип <0,1
12Х18Н10Т — Ж т • кип <0,1
10Х17Н13М2Т — Ж т 1 кип <0,1
06ХН28МДТ — Ж т 1 кип <0,1
Тантал — Ж Ткип <0,1
Титан — — 100 0,02
Чугуны
серый — Ж 20 >3,0
СЧ15, СЧ17 — ж т 1 кип <0,1
(Fe, Сг: 25-30 %) — ж /’кип <0,1
Антегмит АТМ-1 — ж 20 С
Керамика — ж 20-Г Z.V 1 кип С
Полиамиды — ж 20— TKim с
Поливинилхлорид — ж 20-60 н
Полиметилметакрилат — ж 20-60 н
Полипропилен — ж 20 ОС
— ж Ткип н
Полиэтилен — ж 20 ОС
— ж 60 н
Резины на основе
БК, НК — ж 20 с
— ж /’кип ОС
СКФ, ХП, ХСПЭ — ж 20 н
Стекло — ж 20-Г z'v 1 кип с
Фторопласт 4 — ж 20—Ткип с
Эмаль кислотостойкая — ж 20— 7^ с
Бария хлорид ВаС12
Алюминий 10 ж 20 0,006 т
5-50 ж 25-105 <0,5
Алюминиевые сплавы
(Al, Si: 12 %) 5-10 ж 20-50 0,03
Электродные процессы
167
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бария хлорид ВаС12 (продолжение)
Алюминиевые сплавы
(Al, Mg: 1,5-2,5 %, Мп: 1,0-2,0 %) 10 Ж 20 0,003
Медь <26 Ж 20 <0,1
10-50 Ж 25-105 <0,5
Медные сплавы
БрА5, БрА7 10 Ж 20-100 <0,5
БрОФ 6,5-0,15 10-50 Ж 25-105 <0,5
Л59, Л63, Л68, Л80, Л068-1 10 Ж 20-100 <0,5
Никель <26 Ж 20-100 <0,1
Безводный Ж 850 0,3
Никелевые сплавы
НМЖМц28-2,5-1,5 <26 Ж 20 <0,1
Насыщ.; Ж Т’кип <0,1
pH 7,1-7,3
ХН78Т
Н70МФ <26 Ж 20-100 <0,1
ХН65МВ 10 Ж 25-105 <0,5
хастеллой С 20-25 Ж 60-100 <0,1
Платина — Ж 20-100 <0,05
Свинец 10-40 Ж 20 <0,5
Серебро 10-50 Ж 20 <0,05
10-30 Ж 50 <0,05
20 Ж 100 > 10,0
Стали
углеродистая < 26; pH > 7,0 Ж 20 <1,0
12X13 <26 Ж 20—Ткип <3,0
12Х18Н10Т <26 Ж 20 <0,1
<26 Ж ^кип 0,1-1,0
10Х17Н13М2Т <26 Ж 20—Ткип <0,1
06ХН28МДТ <26 Ж 20-Ткип <0,1
Тантал 10-20 Ж 100 <0,5
Титан 20 Ж 20-100 <0,1
25 Ж 20 <0,1
Цирконий 5 ж 35-100 <0,01
20 ж 35 0,001
20 ж 100 0,0018
Чугуны
серый < 26; pH > 7,0 ж 20 < 1,0
10 ж 100 > 10,0
СЧ15, СЧ17 10-30 ж 20-100 <0,5
40-50 ж 20-100 <0,5
Графит <26 ж 20-100 С
168
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бария хлорид ВаС12 (продолжение)
Графит, пропитанный бакелите- <26 Ж 20-100 С
вым лаком
Керамика <26 Ж 20-Ткип С
Пентапласт 1-насыщ. Ж Ткип С
Полиамиды <26 Ж 20-60 С
Поливинилхлорид <26 Ж 20-60 С
Полиизобутилен <26 Ж 20-100 С
Полиметилметакрилат <26 Ж 20-60 С
Полипропилен <26 Ж 20-60 С
Полистирол <26 Ж 60 С
Полиэтилен <26 Ж 20-60 С
Резины на основе
БК <26 Ж 20-100 С
НК <26 Ж 20-70 С
СКБ, СКН, ХП <26 Ж 20-60 С
Стекло <26 Ж 20-100 С
Фторопласт 4 — Ж <200 С
Эмаль кислотостойкая <26 Ж 20-Ткип С
Бензин
Алюминий — Ж 20-ТкИП <0,1
Алюминиевые сплавы — Ж 20 <0,1
АЛ2, АМцС, АМг5
Медь, бронзы, латуни Ж 20-Г 1 кип <0,1
Никель — Ж 20-Г 1 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20-Г 1 кип <0,001
Н70МФ — Ж 20-Г ZrV 1 кип <0,1
Стали
углеродистая — Ж 20-Г 1 кип <0,1
12X13 — Ж 20-Т 1 кип <0,1
12Х18Н10Т — Ж 20-Г <0,1
10Х17Н13М2Т — Ж 20-Г 1 кип <0,1
06ХН28МДТ — Ж 20-Г z'v 1 кип <0,1
Тантал — Ж 20-Г 1 кип <0,1
Титан — ж 20-Г 1 кип <0,01
Чугуны
серый — ж 20-Ткип <0,1
СЧ15, СЧ17 — ж 20 <0,1
Антегмит — ж 20—Ткип С
Асбест — ж <200 С
Графит, пропитанный бакелите- — ж 20—Ткип С
вым лаком
Полиамиды — ж 20 С
Электродные процессы
169
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Бензин (продолжение)
Поливинилхлорид — Ж 20-60 С
Полиметилметакрилат — Ж 20-40 С
— Ж 60 н
Полипропилен — Ж 20 н
Полиэтилен — Ж 20 ОС
— Ж 60 н
Резины на основе
БК, НК, ХСПЭ — Ж 20 н
СКН — Ж 20 с
СКС — Ж 20-60 н
С КУ — Ж 20-40 с
СКФ — Ж 20-145 с
ХП — Ж 20 ОС
Стекло — Ж 20— Ткип с
Фторопласт 4 — Ж 20-95 с
Эмаль кислотостойкая — Ж 20-Ткип с
Бензол С6Н 6
Алюминий — Ж 20 <0,1
Алюминиевые сплавы
АЛ2 — ж 20 <0,01
(Al, Mg: 3,8-10,3 %) — ж 20 0,002
(Al, Mg: 1,5-2,5 %, Mn: 1,0-2,0 %) — ж 20 <0,01
Медь — ж 20-7^ <0,1
Медные сплавы
БрОФ6,5-0,25 — ж 20 <0,01
Л62, Л68, Л90 — ж 20—Ткип <0,1
Никель — ж 20 <0,1
— ж 60—Ткип <0,5
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20 <0,01
Н70МФ — ж 20 <0,01
— ж 60—Ткип <0,5
Свинец — ж 20— Ткип <0,1
Серебро — ж 20-Ткип <0,1
Стали
углеродистая — ж 20 <0,1
— ж Ткип 0,1-1,0
12X13 — ж 20-Ткип <0,1
12Х18Н10Т — ж 20—Ткип <0,1
10Х17Н13М2Т — ж 20-Т,™ <0,1
06ХН28МДТ — ж 20-Ткип <0,1
Тантал — ж 20—Ткип <0,1
Титан — ж Ткип <0,05
170
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Г, °C и характер коррозии Стойкость материалов
Бензол С6Н6 (продолжение)
Чугуны
серый — Ж 20 <0,1
— Ж Т’кип <3,0
кремнистый — Ж 20-Т 1 кип <0,1
(Fe; Сг 32 %) — Ж Гкип <0,1
Асбест — Ж <90 с
Анте гм иг АТМ-1 — Ж Т 1 кип с
Графит, пропитанный бакелито- — Ж т 1 кип с
вым лаком
Графит, пропитанный феноло- — Ж Гкип с
формальдегидным олигомером Паронит — Ж 250 с
Полиамиды — Ж 20 с
Поливинилхлорид — Ж 20 с
Полиизобутилен — Ж 20-60 н
Полиметилметакрилат — Ж 20 н
Полипропилен — Ж 20 с
Полиэтилен Резины на основе — Ж 20 с
БК, НК, СКВ, СКН, СКС, ПС, ХП — Ж 20 н
СКФ — Ж 20-70 ОС
Стекло — Ж 20-Г с
Фторопласт 4 — Ж т 1 кип с
Эмаль кислотостойкая — ж т 1 кип с
Бром Вг2
Алюминий Безводный ж 20-30 <0,5
Влажный ж 20 > 10,0
Сухой г <250 <0,1
Вольфрам То же г 250 <0,1
Золото Безводный ж 20 3,5
Влажный ж 20 > 10,0
Иридий Безводный ж 20 < 0,001
Влажный ж 20 <0,001
Кобальт То же ж 20 > 10,0
Медь Безводный, без воздуха ж 20 < 1,0
Влажный ж 20 > 10,0
Медные сплавы
БрА5, БрА7, БрОФ6,5-0,15 То же ж 25-30 > 10,0
Л59, Л63, Л68, Л80, ЛО68-1 » ж 25-30 > 10,0
Молибден » ж 20 > 10,0
Сухой г <250 0,001
Электродные процессы
171
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бром Вг2 {продолжение)
Никель Безводный, без воздуха Ж 20 0,02 т
Вода 0,05, без воздуха Ж 20 0,03 т
То же Г 20 < 0,06 т
Вода 0,05 Ж 20 0,2 т
То же Г 20 0,8 т
Сухой Г 20-50 < 0,04 т
Под слоем воды Ж 20 0,8 т
То же Г 20 3,0 н
Никелевые сплавы
НМЖМц28-2,5-1,5 Безводный Ж 20 <0,1
Вода 0,05, без воздуха Ж 20 0,08 т
Вода 0,05 Г, Ж 20 1,3
Сухой Г 20 0,2
Под слоем воды Г, Ж 20 2,0 н
ХН78Т Безводный Ж 20 < 0,05 т
Вода 0,05, без воздуха Ж 20 0,07 т
То же Г 20 0,1 т
Вода 0,05 Ж 20 0,2 т
Под слоем воды ж 20 0,6 н
То же г 20 2,6 н
Н70МФ Безводный ж 20-50 <0,05
Влажный ж 25 <0,05
Сухой г 75 <0,05
То же г 95-338 <0,5
ХН65МВ Безводный и влажный ж 25-50 <0,05
Сухой и влажный г 75 <0,05
Сухой г 100-350 <0,5
хастеллой В Сухой и влажный г 20 <0,1
хастеллой С То же г 20 <0,1
Ниобий Безводный, без воздуха ж 20-50 <0,001
Вода 0,05, без воздуха г, ж 20 <0,001
Вода 0,05 г, ж 20 < 0,001
— г 100 < 0,001
Ниобиевый сплав с 5 % Та Безводный, без воздуха ж 20 <0,01
Вода 0,05, без воздуха г, ж 20 <0,001
Вода 0,05 г, ж 20 <0,001
172
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бром Вг2 (продолжение)
Олово — Ж 20 > 10,0
Осмий Безводный Ж 20 > 10,0
Палладий Безводный и влажный Ж 20 > 10,0
Платина То же Ж 20 > 10,0
Родий » Ж 20 < 0,001
Рутений » Ж 20 <0,001
Свинец Безводный Ж 44 > 10,0
Влажный Ж 25 > 10,0
Сухой Г 20 <0,1
Сухой и влажный Г 75 > 10,0
Серебро Безводный Ж 20 0,04
То же Ж 58 > 10,0
Стали Влажный Ж 20 > 10,0
СтЗ Безводный, без воздуха Ж 20 0,13 н
То же г 20 0,5 н
» ж 50 0,46 н
» г 50 0,6 н
Вода 0,05, без воздуха ж 20 0,6
То же г 20 1,5 н
Под слоем воды ж 20 7,0 н
То же г 20 12,0 н
20X13 Безводный, без воздуха г, ж 20 0,1 т
То же ж 50 0,04
» г 50 0,1 т
Вода 0,05, без воздуха ж 20 0,3 т
То же г 20 1,4 н
Под слоем воды ж 20 7,0 н
— То же г 20 6,5 н
12Х18Н10Т Безводный, без воздуха ж 50 0,02 т
То же г 50 0,2 т
Вода 0,05, без воздуха ж 20 0,2 т
То же г 20 0,8 т
Под слоем воды ж 20 4,0 н
10Х17Н13М2Т Безводный, без воздуха г, ж 20 0,05 т
Вода 0,05, без воздуха ж 20 0,2 т
Электродные процессы
173
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Г, °C и характер коррозии Стойкость материалов
Бром Вг2 (продолжение)
Стали
10Х17Н13М2Т Вода 0,05, без воздуха Г 20 0,47 т
Под слоем воды Г, Ж 20 < 5,0 н
06ХН28МДТ Безводный, без воздуха Ж 50 0,02
То же Г 50 0,13 т
Вода 0,05, без воздуха Ж 20 0,13
То же Г 20 0,5 н
Под слоем воды Ж 20 5,0 н
То же Г 20 6,0 н
Тантал Безводный и влажный Ж 20 <0,1
Сухой Г < 150 < 0,001
То же Г 244 <0,05
» Г 315 > 10,0
Влажный Г < 150 <0,001
То же Г 250 <0,05
» г 306 > 10,0
Титан Безводный и влажный г, ж 20 > 10,0
Титановые сплавы АТЗ, ВТ14, — ж 0 > 10,0
ВТ5-1, ОТ4 Цирконий Чугуны Безводный ж 20 <0,1
серый Безводный и влажный ж 20 > 10,0
То же г 75 > 10,0
СЧ15, СЩ7 » ж 20 > 10,0
Сухой и влажный г 75 > 10,0
Асбест — ж 50 с
Антегмит АТМ-1 Безводный и влажный ж 20 н
— г 100 н
Графит, пропитанный олиго-
мерами феноло-формальдегидным — ж 57 н
фенольным Безводный ж 20 н
эпоксидным — ж 20 н
Керамика кислотостойкая Влажный ж 20 с
Полиамиды Безводный ж 20 н
Поливинилхлорид Безводный и влажный ж 20 н
Полиизобутилен То же ж 20 н
174
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бром Вг2 (продолжение)
Полиметилметакрилат Безводный Ж 20 Н
и влажный
Полипропилен То же Ж 20 Н
Полиэтилен » Ж 20 Н
Резины на основе
БК, НК, СКИ, СКС, СКТ, ХП, Безводный Ж 20 Н
ХСПЭ и влажный
СКФ То же Ж 20 С
Стекло » ж 20 С
Фторопласт 4 Влажный ж 20 С
Сухой г 100 С
Влажный г 150 С
Эмаль кислотостойкая Безводный и влажный ж 20 С
Сухой и влажный г 100 С
Бромоводород НВг (газ)
Алюминий Сухой г 20 <0,1
— г 100 > 10,0
Медь Без воздуха г 20 <0,1
Никель Сухой г 20 <0,1
Никелевые сплавы
Н70МФ Сухой г 20 <0,1
ХН65МВ То же г 80-105 <0,5
Свинец » г 100 > 10,0
Серебро » г 20 > 10,0
Стали
СтЗ Сухой г 25 <0,5
То же г 105 > 10,0
12X17 » г 20 <0,1
12Х18Н10Т » г 20 <0,1
» г 100 < 1,3
10Х17Н13М2Т » г 20 <0,1
06ХН28МДТ » г 20 <0,1
Тантал » г 25-371 <0,05
Чугуны
серый » г 20 <0,1
СЧ15, СЧ17 » г 20 <0,5
» г 100 > 10,0
Антегмит АТМ-1 » г 20 С
Полиамиды — г 20-60 н
Поливинилхлорид — г 20 С
Полиизобутилен — г 20 С
Полиметилметакрилат — г 20 С
Электродные процессы
175
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Бромоводород НВг (газ) (продолжение)
Резины на основе
БК, НК, СКИ, СКС, — Г 20 С
ХП — Г 20 Н
Стекло — Г 20 С
Фторопласт 4 — Г 20 С
Эмаль кислотостойкая Сухой г 20 С
Вода Н2О
Алюминий — ж 25-80 <0,1
— г 200 <0,1
Алюминиевый сплав АК12 (АЛ2) — ж 20 <0,01
Медь — ж 20 <0,1
— г 250 <0,1
Без кислорода г 535 <0,1
Медные сплавы
(Си, А1: 9,0-10,0 %) — ж 20-100 <0,1
(Си, Sn: 3,42 %) — ж 20-100 <0,01
Л62, Л68, ЛО68-1, ЛО70-1, Л90 — ж 20—Ткип <0,1
Никель — ж 20 <0,01
Никелевый сплав — ж 20— Ттп <0,1
НМЖМц28-2,5-1,5
— г 250 <0,1
Свинец — ж 20-7’кип <0,1
Серебро — ж 20 <0,1
Стали
СтЗ — ж 20 <0,2
— ж 100 0,1-1,0
12X13 — ж 20-100 <0,1
— г 300 <0,1
12Х18Н10Т — ж 20 <0,001
— ж Ткип <0,1
— г 300 <0,1
10Х17Н13М2Т — ж 35 < 0,001
— ж т 1 кип <0,1
06ХН28МДТ — ж /’кип <0,1
Тантал — ж 20—Тит <0,1
— г 400 <0,1
Титан — ж 100 <0,001
Чугуны
серый — ж 20 <0,1
СЧ15, СЧ17 — ж 20-100 <0,01
Антегмит АТМ-1 — ж 20-100 С
Полиамиды — ж 20 С
176
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Вода Н2О (продолжение)
Поливинилхлорид — Ж 20-40 С
— Ж 60 ОС
Полиизобутилен — Ж 20-60 с
— Ж 100 н
Полиметилметакрилат — Ж 20-60 С
Полипропилен — Ж 20-100 с
Полиэтилен — Ж 20-60 С
Резины на основе
БК — Ж 20-100 с
НК — Ж 20-65 с
— Ж 100 н
СКН — Ж 20 С
— Г 100 н
СКС — Ж 20-60 с
— Г 100 н
СКФ — Г 175 с
С КУ — Ж 20 с
— Ж 65 ОС
— Ж 90 н
ХП — Ж 20-70 с
— Г 100 н
Стекло — Ж 20-100 с
Фторопласт 4 — Ж 20— ТКИП с
Эмаль кислотостойкая — Ж 20-100 с
Вода морская
Алюминий — Ж 20 Неприменим*
Алюминиевые сплавы АМгЗ, — Ж 20 <0,1
АМг5В, АМгб, АМц
Медь — Ж 20-80 <0,1
Медные сплавы
бронза алюминиевая — Ж 20-50 <0,05
бронза оловянистая — ж 20-40 <0,1
ЛА77-2 — ж 20-90 0,01
Никель — ж 20 <0,1
Никелевый сплав НМЖМц28-2,5-1,5 — ж 20 0,02
Олово — ж 20 <0,01
Свинец — ж 20 <0,1
Стали
углеродистая — ж 20-80 0,1-1,0
12X17 — ж 20 <3,0
2Х18Н10Т — ж 20 0,1-1,От
Подвергается интенсивной точечной коррозии.
Электродные процессы
YT1
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Вода морская (продолжение)
Стали
10Х17Н13М2Т — Ж 20 <0,1
Титан — Ж 20 <0,001
— Ж Т’кип <0,05
Чугуны
серый — Ж 20 <0,1
СЧ15, СЧ17 — Ж 20-100 0,1
Асбовинил — Ж 20 С
Поливинилхлорид — Ж 20 с
Полиметилметакрилат — Ж 20-60 с
Полипропилен — Ж 20-100 с
Полиэтилен — Ж 20-60 с
Резины на основе СКУ, ХП — Ж 20 с
Фторопласт 4 — Ж 20-60 с
Эмаль кислотостойкая — Ж Т’кип с
Водород Н2*‘
Водорода пероксид Н2О2
Алюминий <90 ж 20-50 0,001-0,060*2
Алюминиевые сплавы
(Al, Mg: 3,8-10,3 %) 6 ж 20 0,012
(Al, Mg: 1,5-2,5 %; Мп: 1-2 %) 6 ж 20 0,0006
Медь <30 ж 20 Неприменима*3
Медный сплав (Cu, Si: 6,5 %) <30 ж 20 <0,1
Никель <30 ж 20 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20 0,1-1,0
Н70МФ <30 ж 20-60 <0,1
Платина <30 ж 20 Неприменима*3
Свинец <30 ж 20 То же
Серебро <30 ж 20 »
Стали
углеродистая <30 ж 20 »
12X13 20 ж 20 <0,1
12Х18Н10Т 30 ж 20 <0,1
10Х17Н13М2Т 30 ж 20 <0,1
06ХН28МДТ 30 ж 20-60 <0,1
Тантал <30 ж 20-100 <0,001
Титан 3-30 ж 20 <0,13
*' Сведения о нем приведены в конце таблицы.
*2 В 90 %-м растворе скорость коррозии больше, чем в менее концентрированных.
*3 Вызывает каталитическое разложение пероксида водорода.
178
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Водорода пероксид Н2О2 (продолжение)
Чугуны
(Fe, Сг: 28 %) 30 Ж 20 <0,1
(Fe, Сг: 6,0 %; Ni: 14,0 %) 20 Ж 20 0,03
Керамика <30 Ж 100 с
Поливинилхлорид <30 Ж 20-60 с
Полиизобутилен <30 Ж 20 с
<30 Ж 60 н
Полиметилметакрилат 3 Ж 25 с
Полистирол 3 Ж 25 с
Полипропилен <30 Ж 20 с
<30 Ж 100 н
Полиэтилен <30 ж 20 с
Резины на основе
НК, СКС, ПС <30 ж 20 н
БК, СКФ, ХСПЭ <30 ж 20 с
ХП <30 ж 20 с
<30 ж 60 н
Стекло <30 ж 20-60 с
Фторопласт 4 <30 ж 20-60 с
Эмаль кислотостойкая <30 ж 20-150 с
Глицерин СНОН(СН2ОН)2
Алюминий — ж Т’кип <0,01
Алюминиевый сплав АК12 (АЛ2) — ж 20 <0,01
Медь — ж 20-60 <0,1
Никель — ж 20-100 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20-100 <0,1
Н70МФ — ж 20-100 <0,1
Свинец — ж 20 <0,1
— ж 100 <0,5
Серебро — ж 20—Гкип <0,1
Стали
углеродистая — ж 20 <0,1
12X13 — ж 20-Т 1 кип <0,1
12Х18Н10Т — ж 20—7’кип <0,1
10Х17Н13М2Т — ж 20-100 <0,1
06ХН28МДТ — ж 20-100 <0,1
Тантал — ж 20—Гют <0,1
Титан — ж 20-100 <0,1
Чугуны
серый — ж 20 <0,1
СЧ15, СЧ17 .— ж 20-100 0,1
Электродные процессы
179
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Глицерин СНОН(СН2ОН)2 (продолжение)
Графит — Ж 20-170 С
Поливинилхлорид — Ж 20-60 С
Полиизобугилен — Ж 20-100 С
Полиметилметакрилат — Ж 20-60 С
Полипропилен — Ж 20-60 С
Полиэтилен — Ж 20-60 С
Резины на основе
БК, НК, СКН, СКС — Ж 20-60 С
С КУ — Ж 20 С
ХП — Ж 20-70 С
ХСПЭ — Ж 20-90 С
Стекло — Ж 20-100 С
Фторопласт 4 — Ж 20-200 С
Эмаль кислотостойкая — Ж 20-Ткип С
1,2-Дихлорэтан С1СН2СН2С1
Алюминий Безводный Г, Ж 20— Ткип <0,1
Влажный Г, Ж 20 0,3
То же Ж Т’кип <0,5
Золото Безводный Ж 20-80 <0,05
Медь Безводный, Г, Ж 20 <0,1
без воздуха
То же ж 25-75 <1,3
» г 100 <0,5
Влажный ж 20 0,65
То же г 20 0,19
Медные сплавы
БрА5, БрА7 — ж 50 < 1,3
БрОФ6,5-0,15 Без воздуха ж 25-50 <1,3
Л59, Л63, Л68, Л80 — ж 25-50 < 1,3
ЛО68-1 — ж 25 < 1,3
Никель Безводный г, ж Ткип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20-80 <0,1
— г 100 <0,5
Н70МФ Безводный ж 20-80 <0,1
ХН78Т — ж 20-80 <0,1
— г 95 <0,5
Платина Безводный ж 20-80 <0,04
Свинец — ж 20-75 <0,5
— г 100 <0,5
Серебро — ж 50 <0,5
180
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
1,2-Дихлорэтан CICH2CH2CI (продолжение)
Стали
СтЗ Безводный, Г, Ж 30— Ткип <0,1
без воздуха
Насыщенный Ж 70 0,04
водой
То же Г 70 0,002
12X13 Безводный Ж 20 <0,1
Насыщенный Г, Ж 70 <0,01
водой
12Х18Н10Т То же Г, Ж 70 <0,01
Вода 0,1-1,0 Ж 80 0,004 т
Вода 50 Ж 80 0,05 т
10Х17Н13М2Т — ж 20 <0,1
Вода 0,1-1,0 г, ж 80 0,004 т
Вода 50 ж 80 0,05 т
То же г 80 <0,01 т
06ХН28МДТ — ж 20-80 <0,1
Тантал Безводный ж 20-80 <0,05
Влажный ж 83 <0,1
То же г 100 <0,05
Титан Безводный ж ^КИП <0,1
и влажный
Вода 50 ж 80 0,002
Цирконий — ж 25-75 <0,05
— г 100 <0,05
Чугуны
серый Безводный ж 20 <0,1
СЧ15, СЧ17 Безводный ж 20-80 <0,1
и влажный
Антегмит АТМ-1 — ж <80 С
Асбест — ж 20-80 с
Графит, пропитанный — ж <80 с
феноло-формальдегидным
олигомером
Паронит — ж <80 с
Полиамиды — ж 20 н
Поливинилхлорид — ж 20 н
Полиизобутилен — ж 20 н
Полиметилметакрилат — ж 20 н
Полипропилен — ж 20 н
Полиэтилен — ж 20 н
Резины на основе
БК, НК, ПС, СКН, СКС, СКТ, ХП — ж 20 н
СКФ — ж 20 ОС
Стекло — ж 20-80 с
Фторопласт 4 — ж 20-80 с
Эмаль кислотостойкая — ж 25 с
Электродные процессы
181
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
1,2-Дихлорэтилен С1СН=СНС1
Алюминий Безводный Ж Т 1 кил <0,1
Золото Безводный Ж 20- Т <0,05
и влажный
Медь Безводный Ж 20—Ткип <0,1
Влажный Ж 45 <0,3
Сухой Г 80-105 <0,5
Медные сплавы
БрА5, БрА7 Безводный Ж 20—7КИП <0,1
БрОФ6,5-0,15 То же Ж 25 <0,5
Л68 — Ж 20-Г 1 кип <0,1
Никель — Ж 20-Г 1 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20-7 1 кип <0,1
ХН78Т — Г 75-100 <0,5
Н70МФ — Ж 20 <0,1
— Г 100 <0,5
Олово Безводный; pH 7 Ж 20-Ткип <0,1
Платина Безводный Ж 20-7 ZrV— 1 кип <0,04
и влажный
Свинец То же Ж 20— Ткип <0,08
— Г 70-95 <0,05
Серебро Безводный ж 20-7 <0,08
и влажный
Стали
СтЗ Безводный ж 20— Ткип <0,1
Влажный ж 20 < 1,0
12X13 — ж <60 <0,1
12X17 — ж 20-7 ZrV— 1 кип <0,1
12Х18Н10Т — ж <60 <0,1
— г 70-95 <0,5
10Х17Н13М2Т — ж 20-7кип <0,1
— г 70-95 <0,5
06ХН28МДТ — ж 60 <0,1
— г 80-105 <0,5
Тантал Безводный ж 20-Ткип <0,05
Титан — ж 20 <0,05
— г 70-95 <0,05
Чугуны
серый Безводный ж 20 <0,1
СЧ15, СЧ17 — ж 25 <0,5
— г 75 <0,5
Асбест — ж 20-Ткип С
Графит — ж 20—T^n С
Полиамиды Безводный ж 20 с
182
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
1,2-Дихлорэтилен С1СН=СНС1 (продолжение)
Поливинилхлорид — Ж 20 Н
Полиизобугилен — Ж 20 Н
Полиметилметакрилат — Ж 20 Н
Полипропилен — Ж 20 Н
Полиэтилен — Ж 20 Н
Резины на основе
НК, ПС, СКИ, СКН, СКС, ХП, — Ж 20 Н
ХСПЭ
СКФ — Ж 20 С
— Ж 50 ОС
Стекло — Ж 20-Ткнл С
Фторопласт 4 — Ж 20-7 1 кип С
Эмаль кислотостойкая — Ж 20-7 X'V 1 кип С
Железа(ПГ) нитрат Fe(NO3)3
Алюминий <45 Ж 20 > 10,0
Медь и ее сплавы <45 Ж 20—Гкип > 10,0
Никель <45 Ж 20 > 10,0
НМЖМц28-2,5-1,5 <45 Ж 20 > 10,0
Н70МФ <45 Ж 20 <0,1
Свинец <45 Ж 20 <0,1
<45 Ж 100 > 10,0
Серебро <45 Ж 20 > 10,0
Стали
углеродистая <45 Ж 20 > 10,0
12X13 <45 Ж 20- Гкип <0,1
12Х18Н10Т <45 Ж 20—Гкип <0,1
10Х17Н13М2Т <45 Ж 20-7 1 кип <0,1
06ХН28МДТ <45 Ж 20-7^ <0,1
Тантал <45 Ж 20-150 <0,001
Чугуны
серый <45 Ж 20 > 10,0
кремнистый <45 Ж 20 <0,1
Графит <45 ж 20-Гкип с
Полиамиды <45 ж 20-60 с
Поливинилхлорид <45 ж 20-60 с
Полиизобутилен <45 ж 20-60 с
Полиметилметакрилат <45 ж 20-60 с
Полипропилен <45 ж 20-60 с
Полиэтилен <45 ж 20-60 с
Резины на основе
БК <45 ж 20-65 с
НК, СКН, СКС <45 ж 20 с
Электродные процессы
183
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Железа(Ш) нитрат Fe(NO3)3 (продолжение)
Резины на основе
ХП <45 Ж 20-65 С
10 Ж 95 ОС
ХСПЭ <45 Ж 65 С
Стекло <45 Ж 20 С
Фторопласт 4 <45 Ж <200 С
Эмаль кислотостойкая <45 Ж 20 С
Железа(П) сульфат FeSO4
Алюминий 1 Ж 20 0,095
1 Ж 100 0,065
10 Ж 20 0,27
10 Ж 100 9,05
<21 Ж 20 0,09-0,28
<21 Ж 100 0,65-9,10
Алюминиевые сплавы
АК12 (АЛ2) 10 Ж 20 0,034
(Al, Mg: 1,5-2,5 %, Мп: 1-2 %) 10 Ж 20 0,037
Медь 10 Ж 94 0,1-0,5
<21; без кислорода Ж 20 <0,1
воздуха
Медные сплавы
(Cu, А1: 5,5-9,5 %) 10 Ж 94 0,1-0,5
(Си, Zn: 48,0 %) — Ж 20 <0,1
Никель <21 Ж 20 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 <21 Ж 20 <0,1
<21 Ж 50-70 0,65
<21 Ж Ткип 1,0
Н70МФ <21 Ж 20-100 <0,1
хастеллой С <21 Ж 20— Ткип <0,1
Свинец <21 Ж 20 <0,1
<21 Ж Т’кип > 10,0
Серебро <21 Ж 20 <0,1
<21 ж 100 > 10,0
Стали
углеродистая Насыщ.; ж 20 <0,1
без кислорода воздуха
Насыщ. ж Ткип > 10,0
12X13 10 ж 20 <0,1
Насыщ. ж Ткип <5,0
12Х18Н10Т 10 ж 20 <0,1
10; насыщ. ж Ткип < 1,0
184
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Железа(П) сульфат FeSO4 (продолжение)
Стали
10Х17Н13М2Т 10; насыщ. Ж Т 7 кип <0,1
06ХН28МДТ То же Ж Ткип <0,1
Тантал <21 Ж 60 0,001
<21 Ж 100 <0,1
Титан 50 Ж 100 <0,05
Чугуны
серый ®21 Ж 20 <0,1
кремнистый <21 Ж 20-Т^п <0,1
Антегмит АТМ-1 1-насыщ. Ж 100 с
Графит, пропитанный бакелито- То же Ж Ткип с
вым лаком
Полиамиды <21 Ж 20-60 с
Поливинилхлорид <21 Ж 20-60 с
Полиизобутилен <21 Ж 20-100 с
Полиметилметакрилат 5 Ж 60 с
Полипропилен <21 Ж 20-60 с
Полиэтилен <21 Ж 20-60 с
Резины на основе
БК <21 Ж 20-95 с
НК, СКС, ХП <21 Ж 20 с
<21 Ж 60 ОС
СКН <21 Ж 20 с
Стекло <21 Ж 20—Ткип с
Фторопласт 4 1-насыщ. Ж <200 с
Эмаль кислотостойкая <21 Ж 20 с
Железа(Ш) сульфат Fe2(SO4)3
Алюминий <81 ж 20 > 10,0
Золото 1-насыщ. ж Ткип <0,1
Медь То же ж 20 > 10,0
Медные сплавы
(Си, Si: 2,6 %) » ж 20-Ткип <0,1
БрА5 <81 ж 20 > 10,0
Л68 <81 ж 20 > 10,0
Никель <81 ж 20 <0,5
Никелевые сплавы
НМЖМц28-2,5-1,5 <81 ж 50-100 0,5-1,27
Н70МФ <81 ж 20 <0,1
<81 ж 100 <0,5
хастеллой С 1-насыщ. ж <40 <0,1
Свинец То же ж 20—Ткип <0,1
Серебро <81 ж 20 3,0-10,0
Электродные процессы
185
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Железа(Ш) сульфат FetCSO^ (продолжение)
Стали
углеродистая 3,95 Ж 45-85 > 10,0
12X13 5-насыщ. Ж 20 <0,1
20 Ж ^кип <1,0
12Х18Н10Т <81 Ж 20-7 2 кип <0,1
10Х17Н13М2Т <81 Ж 20-7 <0,1
06ХН28МДТ <81 Ж 20—Ткип <0,1
Тантал <81 Ж 20-150 <0,001
Титан Насыщ. Ж 20 <0,01
Чугуны
серый <81 ж 20—Ткип > 10,0
(Fe, Сг: 30-32 %) 10 ж 20-100 <0,1
Антегмит <81 ж 20-7’кип С
Полиамиды <81 ж 20-60 С
Поливинилхлорид 1-насыщ. ж <65 С
Полиизобутилен <81 ж 20-60 С
Полиметилметакрилат <81 ж 20-60 С
Полипропилен 1-насыщ. ж 20-60 С
Полиэтилен То же ж 20-60 С
Резины на основе
БК <81 ж 20-100 С
НК, СКС <81 ж 20 С
<81 ж 60 ОС
ПС, СКН <81 ж 20-60 С
ХП <81 ж 20-90 ОС
Стекло <81 ж 20-100 С
Фторопласт 4 1-насыщ. ж Ткип С
Эмаль кислотостойкая <81 ж 20-100 С
Железа(Ш) хлорид FeCI3
Алюминий <48 ж 20 8,7
Алюминиевые сплавы АК12 (АЛ2), 20 ж 20— 7’кип > 10,0
АМц
Медь 10 ж 25 > 10,0
Медные сплавы
БрА5, БрА7, БрОФ7,5-0,15 10 ж 25 > 10,0
Л59, Л63, Л69, Л80, ЛО68-1 10 ж 25 > 10,0
Молибден 20 ж 35 > 10,0
Никель 5 ж 20 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 10 ж 25 > 10,0
Сухой т 340 0,7
ХН78Т 10 ж 25 < 1,3
10 ж 55 > 10,0
Сухой т 340 0,17-0,71
186
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Железа(Ш) хлорид FeCl3 (продолжение)
Никелевые сплавы
ХН78Т Влажный Т 340 3,7
Н70МФ 30 Ж 20 <0,1
ХН65МВ — Т 25 <0,5
—- Т 50 > 10,0
10 Ж 30-60 <0,05
10-30 Ж 75 <0,5
40-50 Ж 25 <0,5
60 Ж 80 3,0
80 Ж 95 4,2
хастеллой С 5-10 Ж 20 0,007
10 Ж 65 0,17
45 Ж 30 0,24
45 Ж 65 1,2
Ниобий 10 Ж 25 <0,001
Олово — ж 20 > 10,0
Палладий 12 ж 20 0,9
Платина <48 ж 20 <0,1
Свинец 10 ж 20 0,2
<48 ж 20 < 1,0
<48 ж 100 > 10,0
Серебро 5 ж Ткип <1,0
Стали
СтЗ Сухой т 20 <0,1
Влажный т 20 > 10,0
Сухой и влажный т 340 > 10,0
<48 ж 20 >3
12X13 5-75 ж 20 > 10,0
12Х18Н10Т Сухой т 20 <1,0
Влажный т 120 > 10,0
1 ж 20 < 1,0
5 ж 20 > 10,0
10Х17Н13М2Т 5-75 ж 20 < 5,0 т
5 ж 60 > 10,0
06ХН28МДТ 5-75 ж 20 < 10,0 т
Тантал —- ж 25 <0,001
10-80 ж 100 <0,05
Титан 1-5 ж 30-100 <0,01
5-50 ж 20-80 < 0,002
60 ж 20-80 0,01
80 ж 80 3,4
Титановые сплавы ВТЗ-1, ВТ5-1, 40 ж 95 <0,01
ВТ5, ВТ6, ОТД
Электродные процессы
187
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Железа(Ш) хлорид FeCl3 {продолжение}
Цирконий 1-10 Ж 35 <0,1
1-5 Ж 60 <0,1
15-20 Ж 35-60 < 1,3
25-30 Ж 35 < 1,3
Чугуны
серый Сухой Т 20 <0,1
Влажный Т 20 > 10,0
<48 Ж 20 >3,0
СЧ15, СЧ17 50 Ж 50 > 10,0
Антегмит АТМ-1 — Ж Ткип с
Графит — Ж Т’кип с
Полиамиды 10 Ж 20 с
Поливинилхлорид <48 Ж 20 с
<48 Ж 60 ос
Полиметилметакрилат <48 Ж 60 с
Полипропилен <48 Ж 60 с
Полиэтилен <48 Ж 60 с
Резины на основе
БК, НК, СКН, СКС <48 Ж 20-65 с
СКУ, СКФ <48 Ж 20 с
ХП <48 Ж 20-90 с
ХСПЭ <48 Ж 20-95 с
Стекло <48 Ж 20—Гкип с
Фторопласт 4 <48 Ж 20-150 с
Эмаль кислотостойкая <48 Ж 20-Ткип с
Иод 12
Алюминий Безводный т 20 <0,1
То же т 100 > 10,0
— г 115 15я
Медь Безводный; без т 20 <0,5
кислорода воздуха
— т 50 2,4
Безводный; без т 100 > 10,0
кислорода воздуха
Сухой г 200 > 10,0
Медные сплавы
БрОФ6,5-0,15, БрОФ7,2-0,2 Безводный; без т 20 <0,5
кислорода воздуха
МН20, МНЗО Безводный т 20 <0,09
ЛО62-1 — т 50 8,7
Никель Безводный т 30 < 0,05 т
То же т 100 < 1,3 т
— г 115 0,7
188
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Иод 12 (продолжение)
Никель Безводный Ж 170 0,8 т
Вода 0,5 Ж 170 1,9 т
— Г 200 0,2 т
— Г 490 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 Безводный Т 25 <0,05
— Г 220 2,6
Н70МФ — Т 20-100 <0,1
Безводный Ж 170 0,3 т
Вода 0,5 Ж 170 1,1 т,я
— Г 450 1,2
ХН65МВ — Т 25-105 <0,05
— Ж 125-175 <0,05
— Г 450 > 10,0
Свинец — Т 25 <0,08
Влажный Т 20 > 10,0
— Г 115 0,15
— Г 200 > 10
Стали
углеродистая Безводный Т 20 <0,1
Влажный Т 20 > 10
— Т 50 14
— Г 20 0,5 т
— г 200 9,4 я
12X13 Безводный т 20 <0,1
Влажный т 20 > 10
— г 200 1,3
12Х18Н10Т Безводный т 25 <0,05
— т 50 12 т, я
Влажный т 20 > 10
— г 20 0,2 т
— г 175 5
— г 200-250 < 4,8 т, я
— г 450 > 10
10Х17Н13М2Т Безводный т 25 < 0,05 т
— т 55 < 0,5 т
Влажный т 20 > 10
— г 20 0,2 т
— г 175 2,3 т
— г 450 4,4 т, я
06ХН28МДТ — т 50 13 т, я
Безводный ж 170 0,6 т
Вода 0,5 ж 170 3,9 т
— г 450 3,5 т
Электродные процессы
189
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Иод 12 (продолжение)
Титан — Т 60 > 10
Влажный Т 30 <0,1
То же Т 115 0,0001
Чугуны
серый Безводный Т 20 <0,1
Сухой Г 20 <0,1
СЧ15 — Г 175 0,07
— Г 200 2,0 т
Графит Безводный Т 25 С
Графит, пропитанный
олигомером
феноло-формальдегидным — Т 25 н
фурфурольным — Т 25 н
Полиамиды — Т 20 н
Поливинилхлорид — Т 20 н
Полиметилметакрилат — Т 20 н
Полипропилен — Т 20 н
Полиэтилен — Т 20 ОС
—- Т 60 н
Резины на основе НК, СКН, СКС, Безводный Т 20 н
ХП и влажный
Стекло — Т < 113 с
— Ж < 189 с
Фторопласт 4 — Т 20-60 с
Эмаль кислотостойкая — Т 20 с
Иодоводород HI (газ)
Алюминий Сухой, без воздуха Г 20 <0,1
Золото — Г <300 <0,05
Медь Сухой, без воздуха Г 20-100 <0,08
Медные сплавы МН20, МНЗО То же Г 20-100 <0,09
Никель » Г 20-100 <0,1
Сухой г 200-230 0,08
То же г 370-400 1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 Сухой, без воздуха г 20-100 <0,1
Н70МФ Сухой г 200-230 0,013
То же г 370-400 0,08
Серебро » г 20 <0,1
Стали
СтЗ Сухой, без воздуха г 20-100 <0,1
12X17 То же г 20-100 <0,1
12Х18Н10Т » г 20-100 <0,1
10Х17Н13М2Т » г 20-100 <0,1
06ХН28МДТ » г 20-100 <0,1
190
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Иодоводород HI (газ) (продолжение)
Тантал — Г 150 <0,1
Титан — Г 20 <0,1
Чугуны
серый Сухой, без воздуха Г 20-100 <0,1
ЧС15 Сухой Г 20 <0,1
Графит, уголь То же Г 20 с
Полиамиды » Г 20 н
Поливинилхлорид » Г 20 с
Полиизобутилен — Г 20 с
Полиэтилен Сухой Г 20 с
Резины на основе БК, НК, СКИ, — Г 20 н
СКН, СКС, ХП
Стекло Сухой Г 20-150 с
Фторопласт 4 — Г 20 с
Эмаль кислотостойкая Сухой Г 20-150 с
Калия гидроксид КОН
Алюминий 0,1-53 Ж 20 > 10
Вольфрам <53 Ж 20 <0,1
— Ж 360 > 10
Медь <53 Ж 20 <0,1
Медные сплавы
(Си, А1 8,7 %; Fe 0,2 %) 20 Ж 20 0,013-0,02
20 Ж 100 0,45
бронза оловянистая До 53 Ж 20—Ткип <0,1
Молибден — ж 360 > 10
Никель <53 ж 20— Ткип <0,1
— ж 500-750 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 <53 ж 20-100 <0,1
Н70МФ <53 ж 20-100 <0,75
Олово <53 ж 20 > 10
Платина <53 ж 20— Ткип <0,1
Аэрированная ж 410 > 10
Свинец 5-10 ж 20 <0,1
<53 ж 100 > 10
Серебро <53 ж 20-Ткип < 0,001
Без кислорода ж 400 0,1
воздуха
Стали
углеродистая <53 ж 20 <0,1
40 ж 70 0,13 т
- <53 ж 100 < 1,0
68 ж 120 <5,0
Электродные процессы
191
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Калия гидрооксид КОН (продолжение)
Стали
углеродистая — Ж 300 > 10
12X17 20 Ж 20— Ткип <0,1
50 Ж 20 <0,1
50 Ж Ткип 0,5-1,0
68 Ж 120 0,5-1,0
— ж 300 > 10
12Х18Н10Т <53 ж 20 <0,1
50 ж Ткип 0,5-1,0
68 ж 120 <0,1
— ж 300 > 10
10Х17Н13М2Т 25 ж Ткип <0,1
50 ж 20 <0,1
50 ж Ткип 0,1-0,5
68 ж 120 <0,1
— ж 300 > 10
06ХН28МДТ 25 ж Ткип <0,1
50 ж 20-Ткип <0,1
68 ж 120 <0,1
— ж 300 > 10
Тантал 5-10 ж 20 <0,1
— ж 360 > 10
Титан <53 ж 20-85 <0,02
10 ж Т 1 кип 0,1
25 ж т 1 кип 0,3
50 ж Ткип 2,7
— ж 360 > 10
Цирконий <53 ж 20-Ткип <0,1
Чугуны
серый <53 ж 20 < 0,1
<53 ж 100 0,1-1,0
СЧ15 10 ж т 1 кип <1,0
25 ж т 1 кип <3,0
50-насыщ. ж Ткип < 10
До 53 ж 20 <0,1
— ж 360 > 10
Графит — ж 20-600 с
Полиамиды <53 ж 20-60 с
Поливинилхлорид <53 ж 20-60 с
Полиизобутилен <53 ж 20-80 с
Полиметилметакрилат <53 ж 20-60 с
Полипропилен <53 ж 20-80 с
Полиэтилен <53 ж 20-60 с
192
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Калия гидроксид КОН (продолжение)
Резины на основе
БК, НК <53 Ж 20-70 С
СКН, СКС <53 Ж 20 С
ХП <53 Ж 20-100 С
СКФ, СКУ <53 Ж 20 ОС
ХСПЭ <53 Ж 20-95 С
Стекло <53 Ж 20 С
Фторопласт 4 <53 Ж 20-150 С
Эмаль кислотостойкая 1-20 Ж 20 С
<53 Ж 100 Н
Калия нитрат KNO3
Алюминий 10 Ж 20-98 0,47
Алюминиевые сплавы
АК12(АЛ2) 5 Ж 20 0,07
(Al, Mg: 3,8-10,3 %) 5 Ж 20 0,03
Золото — Ж 550 <0,1
Медь Насыщ. Ж 20 <0,1
— Ж 400 2,0
— Ж 450 4,4
— Ж 500 7,0
Медноникелевый сплав МН 19 10- ж 15-20 <0,001
Молибден — ж 350 > 10
Никель 10 ж 20-100 <0,1
— ж 400 0,47
— ж 450 0,17
— ж 500 0,12
Платина — ж 550 > 10
Свинец 0,5-10 ж 8 0,08
Серебро — ж 550 > 10
Стали
углеродистые Насыщ. ж 115 5,5
— ж 400-500 1,45
12X13 1-насыщ. ж 20- Гкип <0,1
— ж 550 > 10
12Х18Н10Т 1-насыщ. ж 20—Гкип <0,1
— ж 400 0,33
— ж 450 0,41
— ж 500 0,53
10Х17Н13М2Т 1-насыщ. ж 20— Гкип <0,1
Титан Насыщ. ж 100 <0,05
80 ж 100 <0,05
Чугун (Fe, Сг: 30-32 %) 25 ж Ткип <0,1
Цирконий — ж 400 0,24
Электродные процессы
193
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Калия нитрат KNO3 (продолжение)
Цирконий — Ж 450 0,31
— Ж 500 0,34
— Ж 550 0,96
— Ж 600 1,75
Полиизобутилен 1-насыщ. Ж <60 С
Полистирол То же Ж 20 С
Резины на основе
БК » Ж 20 С
ХП » Ж 65 С
Фторопласт 4 » Ж <200 С
Эмаль кислотостойкая » Ж Гкип С
Калия сульфат K2SO4
Алюминий < 10 Ж 20—Ткип <0,1
Медь, бронзы, латуни < 10 Ж 20 <0,1
< 10 Ж 100 <0,5
Никель < 10 ж 20—Ттп <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 5 ж 20 < 0,075
< 10 ж 100 0,1-1,0
Н70МФ < 10 ж 25-60 <0,5
Свинец < 10 ж 20 <0,1
Серебро < 10 ж 20-100 <0,1
— ж 500 > 10
Стали
углеродистые < 10 ж 20 <0,1
13,8 ж 85 0,08
12X13 10 ж 20 0,002
10 ж т 1 кип 1,04
12Х18Н10Т < 10 ж 20-7 1 кип <0,1
10Х17Н13М2Т < 10 ж 20-7 1 кип <0,1
06ХН28МДТ < 10 ж 20—Гют <0,1
Тантал < 10 ж 20-100 <0,1
Титан <10 ж 20-100 <0,1
Чугуны
серый < 10 ж 20 <0,1
< 10 ж 85 0,1
< 10 ж 7 1 кип >1,0
СЧ15, СЧ17 < 10 ж 20—Ткип <0,1
Графит < 10 ж 20 С
Полиамиды < 10 ж 20-60 С
Поливинилхлорид < 10 ж 20-60 С
Полиизобутилен < 10 ж 20-60 С
Полиметилметакрилат < 10 ж 20-60 С
194
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Калия сульфат K2SO4 (продолжение)
Полипропилен < 10 Ж 20-60 С
Полиэтилен < 10 Ж 20-60 С
Резины на основе
БК, ХСПЭ < 10 Ж 20-80 С
НК, СКН < 10 Ж 20-70 С
СКС < 10 Ж 20-60 С
ХП < 10 Ж 20-90 С
Стекло < 10 Ж 20-100 С
Фторопласт 4 < 10 Ж 20-100 С
Эмаль кислотостойкая < 10 Ж 20-100 С
Калия хлорид KCI
Алюминий <25 Ж 20 > 10
— Ж 770 > 10
Медь — Т 25 < 0,5 кр
Безводный Ж 800-950 <6,6
10-20 Ж 25-100 <0,05
Медные сплавы
БрА5, БрА7 10 Ж 25 <1,3
БрОФ6,5-0,15 10-20 Ж 25-100 <0,05
Л59, Л63, Л69 10 Ж 25 <1,3
Л80, Л068-1 — Т 25 <0,5
10 Ж 30-55 <0,5
Никель — т 175 <0,05
Безводный ж 800-810 0,8
— ж 860-900 <3,0
10-30 ж 20-100 <0,5
Никелевые сплавы
НМЖМц28-2,5~1,5 — т 175 <0,05
10-30 ж 25-105 <0,5
ХН78Т — т 175 <0,05
Безводный ж 800 1,2
Н70МФ — т 175 <0,05
7,5-30 ж 25-105 <0,05
ХН65МВ — т 175 <0,05
10 ж 25 <0,05
10-27,5 ж 50-100 <0,5
Свинец 10 ж 25-75 <0,5
Серебро 10-20 ж 20-75 <0,05
Стали
СтЗ 5 ж 20-25 1,5 т
10; без кислорода ж 25 <0,5
воздуха
Электродные процессы
195
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Калия хлорид КС1 {продолжение)
Стали
СтЗ 40; без кислорода воз- Ж 25-50 <1,3
духа
12X13 35 Ж 50 < 0,1 т
12Х18Н10Т Безводный Ж 775-900 <3,7
35 Ж 50 <0,1 т
35 Ж Гкип < 1,0 т
10Х17Н13М2Т Безводный Ж 800-850 < 1,5
35 Ж 50 <0,1 т
35 Ж Гкип < 1,0 т
06ХН28МДТ 10-20 Ж 25-100 < 0,05 т
30 Ж 50-105 <0,5
35 Ж 50 <0,1 т
Тантал 10-40 Ж 25-100 <0,05
Титан 25 Ж 120 <0,001
10-40 Ж 30-105 <0,05
Цирконий Безводный Ж 700 1,5
— Ж 800 6,1
Чугуны
серый 10 Ж 25 <0,5
25 Ж 25-50 < 1,3
СЧ15, СЧ17 — Т 25-100 <0,05
10-20 Ж 30-105 <0,05
Графит <25 ж 20 С
Поливинилхлорид 25 ж 40 С
25 ж 60 ОС
Полиизобутилен Насыщ. ж 60 с
То же ж 100 ОС
Полиметилметакрилат <25 ж 20-60 с
Полипропилен <25 ж 20-60 с
Полиэтилен — т 60 с
<25 ж 20 с
Резины на основе
БК 35 ж 100 с
НК 10-20 ж 25 с
СКН, СКС <25 ж 20-60 с
СКФ <25 ж 20—Гкип с
хп <25 ж 20-100 с
Стекло 10-20 ж 100 с
Фторопласт 4 1-насыщ. ж <200 с
Эмаль кислотостойкая <25 ж 20-60 с
196
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кальция гидроксид Са(ОН)2
Алюминий -0,16; Ж 20 0,08
известковое молоко Ж,Т 20 > 10
Магний Насыщ. Ж 20-Ткип <0,1
Медь То же Ж Т 1 кип <1,0
Никель <0,16 Ж т 2 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж Ткип <0,1
Н70МФ <0,16 Ж 20 <0,1
Платина Известковая вода Ж Ткип <0,001
Свинец <0,16 Ж 20 <0,5
Серебро <0,16 Ж 20-100 <0,1
Стали
углеродистая <0,16 Ж 20 <0,1
12X13 Насыщ. Ж т 1 кип <0,1
12Х18Н10Т <0,16 Ж 20-Т *^v 1 кип <0,1
10Х17Н13М2Т <0,16 Ж 20-Т 1 кип <0,1
06ХН28МДТ <0,16 Ж 20—Ткип <0,1
Тантал <0,16 Ж 20 <0,1
Титан <0,16 Ж 100 <0,13
Чугуны
серый <0,16 Ж 20 <0,1
ЧС15, ЧС17 <0,16 Ж 20-Т z'v 1 кип <0,1
Графит <0,16 Ж 20-Ткип с
Полиамиды <0,16 Ж 20 с
Поливинилхлорид <0,16 Ж 20-60 с
Полиизобутилен <0,16 Ж 20-60 с
Полиэтилен <0,16 Ж 20-60 с
Резины на основе
БК <0,16 Ж 20 с
НК, СКН, СКС <0,16 Ж 20-60 с
ПС <0,16 ж 20 н
хп <0,16 ж 20-90 с
Стекло <0,16 ж 20-60 с
<0,16 ж 100-Ткип ОС
Фторопласт 4 <0,16 ж 20-150 с
Эмаль кислотостойкая <0,16 ж 20 с
Кальция гипохлорит Са(С1О)2
Алюминий 12,5 ж 25 > 10
Медь — т 25 < 1,3
10; pH >7 ж 25 <0,5
Медные сплавы
БрОФ6,5-0,15, БрОФ7,2-0,2 — т 25 < 1,3
10; pH > 7 ж 25 <0,5
Никелевые сплавы
Н70МФ 10 ж 25-100 <0,5
Электродные процессы
197
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кальция гипохлорит Са(С1О)2 (продолжение)
Никелевые сплавы
ХН65МВ 10 Ж 25-105 <0,5
Платина 10-90 Ж 25-105 <0,05
Свинец — Т 25 <0,05
10-90 Ж 25-55 <1,3
Серебро 10 Ж 25-95 <0,05
Стали
СтЗ 10; pH > 7 Ж 25 <1,3
17,5 ж 25 > 10
12X13 Насыщ. ж 40 > 10
12Х18Н10Т То же ж 40 <5,0
10Х17Н13М2Т 1-насыщ. ж 20 1,0-3,0
06ХН28МДТ 2 ж 35-100 0,03
6 ж 100 0,01
10; pH 7 ж 20-100 <0,05
20-90; pH 7 ж 20 <0,05
Тантал — т 20-105 <0,05
20-30 ж 25 <0,05
10-90 ж 20-100 <0,05
Титан 10-90 ж 20-105 <0,05
Цирконий 10-20 ж 20-100 <0,05
Чугуны
серый 10; pH > 7 ж 25 < 1,3
20 ж 25 > 10
СЧ15, СЧ17 — т 20-100 <0,5
10-70 ж 20-100 <0,5
Графит 1-насыщ. ж 30 С
Полиамиды То же ж 20 с
Поливинилхлорид » ж 20 с
Полиизобутилен » ж 20 с
Полиметилметакрилат » ж 20 с
Полипропилен » ж 20-60 с
Полиэтилен — т 60 н
1-насыщ. ж 20 с
3 ж 70 с
Резины на основе
БК 1-насыщ. ж 20 с
НК 10-30 ж 20 с
ПС 1-насыщ. ж 20 н
СКС То же ж 20-60 с
СКУ » ж 20 с
СКФ » ж 65 ОС
хп » ж 20 ОС
хспэ » ж 65 с
198
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кальция гипохлорит Са(С1О)2 (продолжение)
Стекло 1-насыщ. Ж 20-100 С
Фторопласт 4 То же Ж 20-60 С
Эмаль кислотостойкая » Ж 20-100 С
Кальция хлорид СаС12
Алюминий — Т 25 <0,05
Влажный Ж 167 9,5
Безводный ж 840 > 10
1 ж 20 0,2
1 ж 50 <0,5
10 ж 50-100 <0,5
25 ж 20 <0,1 т
30 ж 120 < 1,3
Алюминиевые сплавы АМгМ, 30 ж 25-30 <0,1
АМг2М, АМгЗМ, АМг5ВМ,
АМгбМ, АВМ, АМцМ
Медь Влажный ж 167 1,4
— ж 260 > 10
38 ж 95 1,1
Медные сплавы
БрА5 38 ж 95 0,7
БрАЖМц 10-3-1,5 30 ж 90 0,01
ЛН65-5 38; без кислорода ж 95 1,1
Никель 10-35; без кислорода ж 25-150 < 0,05 т
воздуха
18 ж 120 0,006
<43 ж 20 <0,1
<43 ж 100 0,1-1,0
50; без кислорода воз- ж 25-150 <0,5т
духа
55 ж 112 0,42
62 ж 154 > 1,0
62 ж 310 0,4
— ж 310 0,4
— ж 815 > 10
Никелевые сплавы
НМЖМц28-2,5-1,5 1-насыщ. ж 7’кип <0,1
ХН78Т — ж 816 > 10
10-60 ж 20-100 < 0,05 т
27,5 ж 125 < 0,5 т
Н70МФ — т 20-100 <0,05
10-80 ж 25-105 <0,05
ХН65МВ — т 95-100 < 0,05 т
— ж 816 > 10
Электродные процессы
199
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Кальция хлорид СаС12 (продолжение)
Никелевые сплавы
ХН65МВ 10-80 Ж 25-105 < 0,05 т
хастеллой С Насыщ. Ж ^кип <0,1
Платина — Т 100 <0,05
Свинец Влажный Ж 167 5,6
60 Ж 75 <1,3
80 Ж 40 0,8
Серебро — Т 132 <0,05
10-60 Ж 20-100 <0,05
Стали
СтЗ 10; pH 6 Ж 20 0,03
10; pH 6 Ж 70 0,09
12,5; pH >7 Ж 125 <0,5
20; pH 5,6 Ж 20 0,02
30; pH 4,9 Ж 70 0,05
<43 Ж 20 0,2
55 Ж 104 0,62
55 Ж 112 1,0
62 Ж 164 >2,0
62 Ж 310 1,8
73 Ж 350 2,57
12X13 Сухой Т 20 <0,1 т
5 Ж 20 <0,1 т
12Х18Н10Т Сухой т 20 <0,1 т
5 ж 20 <0,1 т
30 ж 90 0,013 кр
58 ж 100 0,04 кр
10Х17Н13М2Т Сухой т 20 <0,1 т
5 ж 20 <0,1 т
10-50 ж 25-55 < 0,5 т
Насыщ. ж 100 <1,0
06ХН28МДТ 30 ж 90 0,007
Тантал 10-30 ж 20-100 <0,05
40 ж 20 <0,05
Титан 30 ж 90 0,001
40; pH 7 ж 120-140 <0,001
Цирконий Безводный, ж 850 0,8
над расплавом Аг
— ж 900 2,6
— ж 950 5,5
Чугуны
серый 38 ж 95 0,14
40-58 ж 120-140 <0,8
(Fe; Ni 20 %) 10-40 ж 25-50 <0,5
200
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кальция хлорид СаС12 (продолжение)
Асбовинил <43 Ж 20 С
Графит 1-насыщ. Ж 100 С
Графит, пропитанный бакелито- Насыщ. Ж 100 С
вым лаком
Графит, пропитанный феноло- 1-насыщ. Ж < Ткип с
формальдегидным олигомером (МНГ-Г-ФФ, ЭГ-ФФ)
Полиамиды <43 Ж 20-100 с
Поливинилхлорид <43 Ж 20 с
Полиизобутилен Насыщ. Ж 70 с
Полиметилметакрилат То же Ж 60 с
Полипропилен <43 Ж 20-60 с
Полиэтилен 25 Ж 70 с
Резины на основе
БК, ХП <43 Ж 20-100 с
НК 1-насыщ. Ж 20 с
ПС, СКН, СКС <43 Ж 20-60 с
ХСПЭ <43 Ж 20-80 с
Стекло <43 Ж 20-100 с
Фторопласт 4 1-насыщ. Ж < 150 с
Эмаль кислотостойкая <43 Ж 20-100 с
Кислота азотная HNO3
Алюминий 4,5 Ж 20 0,48
12,5 ж 20 0,88
23,5 ж 20 1,05
31 ж 20 0,96
35 ж 20 0,88
40 ж 20 0,79
40 ж 60 10,0
43 ж 20 0,7
49 ж 20 0,58
55 ж 20 0,45
61 ж 20 0,41
65 ж 20 0,13
71 ж 20 0,13
77 ж 20 0,09
84,5 ж 20 0,044
94 ж 20 0,035
95 ж 57 0,025
97 ж 20 0,017
100 ж 20 0,008
Алюминиевые сплавы
АМц 1 ж 20 0,33
Электродные процессы
201
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Кислота азотная HNO3 (продолжение)
Алюминиевые сплавы
АМц 1 Ж 50 4,40
1 Ж 98 > 10
10 Ж 20 1,57
10 Ж 50 > 10
25 Ж 20 1,7
25 Ж 50 > 10
45 Ж 20 1,66
45 Ж 50 > 10
АК7ч(АЛ9) 1 ж 20 0,5
1 ж 50 3,99
1 ж 98 > 10
10 ж 20 1,65
10 ж 50 > 10
25 ж 20 1,74
25 ж 50 > 10
45 ж 20 2,1
45 ж 50 > 10
65 ж 20 1,89
65 ж 50 > 10
85 ж 20 0,2
85 ж 50 1,41
85 ж 98 8,88
АМг2 0,5 ж 20 0,25
5 ж 20 1,68
20 ж 20 3,56
65 ж 20 1,4
95 ж 20 0,025
Вольфрам 1-100 ж 20-100 <0,1
Золото 1-100 ж 20 <0,1
Кадмий 1-100 ж 20 > 10
Кобальт 1-94 ж 20 > 10
Магний 4,5-21,0 ж 20 > 10
Медь 4,5-70 ж 20 > 10
71 ж 20 9,63
84,5 ж 20 <0,001
97-100 ж 20 <0,05
Медные сплавы
БрАЮ 30 ж 20 2,28
30 ж Лип > 10
50 ж Лип > 10
66 ж 20 > 10
(Си; Sn: 3,4 %) 6 ж 15 1,8
>90 ж 15 > 10
202
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота азотная HNO3 {продолжение)
Медные сплавы Л70 4,5 Ж 20 > 10
12,5 Ж 20 > 10
77 Ж 20 1,59
84,5-94,0 Ж 2 <2,0
97-100 Ж 20 > 10
Молибден 12,5-61,0 Ж 20 > 10
65-94 Ж 20 <0,1
70 Ж 50 > 10
97 Ж 20 0,22
100 Ж 20 1,27
Никель 4,5 Ж 20 0,87
10 Ж 100 > 10
12,5 Ж 20 4,38
23,5—43,5 Ж 20 > 10
49 Ж 20 0,87
55 Ж 20 1,31
61 Ж 20 2,63
65 Ж 20 4,03
77 Ж 20 0,61
100 Ж 20 0,35
Никелевые сплавы НМЖМц28-2,5-1,5 1 Ж 20 0,07
1 Ж Т’кип 6,4
10 Ж 20 0,11
10 Ж ГКИП > 10
Н70МФ <70 Ж 20 0,1-1,0
<70 ж Ткп 1,0-3,0
хастеллой С 10 ж 20 0,29
10 ж 70 0,96
10 ж Т 1 кип 1,09
20 ж ттп 4,57
50 ж 20 0,23
50 ж 70 3,5
70 ж 70 3,0
70 ж Ткп > 10
Ниобий 12,3 ж 20-100 <0,001
70 ж 100 3,5
Олово 4,5 ж 20 15,8
77-100 ж 20 <0,001
Осмий 70-95 ж 20 > 10
Палладий 1 ж 18 <0,1
Электродные процессы
203
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Кислота азотная HNO3 (продолжение)
Палладий 6 Ж 18 <1,0
12 Ж 18 <3,0
70 Ж 18 > 10
Платина 1-100 Ж 20— Гкип <0,1
Свинец 4,5-23,5 Ж 20 > 10
31 Ж 20 3,07
35 Ж 20 2,23
40 Ж 20 0,7
61 Ж 20 0,26
65 ж 20 2,01
71 ж 20 1,84
77 ж 20 0,61
94 ж 20 0,39
97 ж 20 0,96
100 ж 20 1,48
Серебро 4,5 ж 20 <0,001
12,5-55,0 ж 20 > 10
61 ж 20 0,45
71 ж 20 0,13
77 ж 20 0,08
84,5 ж 20 <0,001
94 ж 20 0,052
100 ж 20 0,17
Стали
углеродистая 6-50 ж > 10
66 ж 20 0,28
66 ж ^кип > 10
98 ж 18 0,44
12X13 7 ж 20 0,004
20 ж Ткип 2,6 кр
30-66 ж 20 <0,1
30 ж Гкип 1,43
35 ж 80 0,34
35 ж Тккп 3,05
65 ж 15 0,08
65 ж 40 0,2
65 ж 90 < 1,15
65 ж ПО <3,45
80 ж 65 < 1,15
90 ж 20 <0,12
12Х18Н10Т 0,5-99,0 ж 20 <0,1
6 ж 100 <0,1
30 ж Ткип 0,11
204
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Кислота азотная HNO3 {продолжение)
Стали
12Х18Н10Т 50 Ж Т’кип 0,29
65 Ж 100 0,2*
65 Ж Ткип 0,64 мк
70 Ж 60 0,003
70 Ж 80 0,03*
70 Ж Ткип 1,2 мк
98 Ж 20 0,02 мк
10Х17Н13М2Т 1-5 Ж 20- Т 1 кип <0,1
8 ж 7'кип 0,11
10 ж 20-85 <0,1
10-20 ж Ткип 0,1-0,5
25 ж 20-90 <0,1
25 ж Ткип 0,1-0,5
30 ж 85—ТКИП <0,1
40 ж 60-90 <0,1
40 ж Ткип <0,5
50 ж 20-80 <0,1
50 ж Ткип <1,0
60 ж 60 <0,1
60 ж Ткип < 1,0
65 ж 20-85 <0,1
65 ж Ткип <5,0
80 ж 30-65 <0,1
80 ж Ткип <5,0
85 ж 85 5,0-10,0
90 ж 20 <0,1
90 ж 80 <5,0
90 ж 7'кип > 10
99 ж 20 0,5-1,0
06ХН28МДТ 10 ж т л кип 0,1-0,5
20 ж т * кип 0,1-0,5
25 ж т Л кип 0,1-0,5
30 ж т * кип <0,1
37 ж Ткип 0,1-0,5
40 ж Ткип 0,1-0,5
50 ж Ткип 0,5-1,0
60 ж 60 <0,1
65 ж 50 <0,1
65 ж т * кип 0,5-5,0
Тантал 0,5-6,5 ж Ткип <0,01
>90 ж 19-26 <0,001
На сварных образцах наблюдается межкристаллитная коррозия.
Электродные процессы
205
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Кислота азотная HNO3 (продолжение)
Титан 5 Ж 35 0,0025
5 Ж 100 0,015
10 Ж 35 0,005
10 Ж 100 0,033
20 Ж 35 0,005
50 Ж 35-100 <0,01
65 Ж 35 <0,001
65 Ж 100 0,1
70 Ж 35-100 <0,1
98 Ж 20 0,0025
Цинк 4,5-100 Ж 20 > 10
Цирконий 10 Ж 35 0,003
10 Ж 100 <0,001
30 Ж 60-100 0,002-0,006
50 Ж 100 0,004
65 Ж 100 0,003
Чугуны
серый 6,8-66,0 Ж 15-ТКШ1 > 10
СЧ15, СЧ17 30-66 Ж 20 <0,01
Х34 7 Ж Ткип <0,12
37 Ж 20 <0,12
37 Ж Ткип < 1,18
66 Ж 20 <0,12
66 Ж ТКИП <3,54
99,5 Ж 20 0,12
99,5 Ж 40 > 10
Антегмит <70 Ж 20 С
<70 ж 50-100 н
90 ж 20 н
Графитопласт < 10 ж 20 с
10-70 ж 20 ОС
Полиамиды < 10 ж 20 с
< 10 ж 60-80 ОС
20-40 ж 20 ОС
20-40 ж 60-80 н
50 ж 20 н
>90 ж 20 н
Поливинилхлорид < 10 ж 20 с
< 10 ж 60-80 ОС
20-40 ж 20 с
30 ж 60 ОС
50 ж 20 с
80 ж 60 с
>90 ж 20 н
206
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота азотная HNO3 (продолжение)
Полиизобутилен < 10 Ж 20 С
< 10 Ж 60-80 С
20-40 Ж 10 С
20-40 Ж 60-80 С
50 Ж 20 ОС
50 Ж 60-80 Н
>90 Ж 20-80 Н
Полиметилметакрилат < 10 Ж 20 С
20-40 Ж 20-80 Н
50 Ж 20-80 н
>90 Ж 20 н
Полипропилен < 10 Ж 20 с
< 10 Ж 60-80 с
20-40 Ж 20 с
20-40 Ж 60-80 с
50 Ж 20 с
50 Ж 60-80 н
>90 Ж 20 н
Полиэтилен < 10 Ж 20 с
< 10 Ж 60-80 с
20-40 Ж 20 с
20-40 Ж 60-80 с
50 Ж 20 ОС
50 Ж 60-80 ОС
>90 Ж 20 н
Резины на основе
БК <5,0 Ж 20-70 с
5-10 Ж 70 с
20-35 Ж 20 с
20-35 ж 70 ОС
56 ж 20 с
НК, СКИ <5,0 ж 20 с
5-10 ж 20 с
5-10 ж <70 н
20-35 ж 20 н
СКН <5,0 ж 20 с
5-10 ж 70 н
20-35 ж 70 н
50-70 ж 10 н
СКС <5,0 ж 20 ОС
5-10 ж 70 ОС
20-35 ж 70 ОС
Электродные процессы
207
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Т, °C и характер коррозии Стойкость материалов
Кислота азотная HNO3 (продолжение)
Резины на основе
СКТ 50-70 Ж 20 Н
СКФ 5-10 Ж 60 С
20-35 Ж 20 ОС
20-35 Ж 70 Н
56 Ж 20 Н
ХП <5 Ж 20 С
5-10 Ж 20 Н
ХСПЭ 5-10 Ж 20 С
<40 Ж 20-70 С
70 Ж 20 С
>90 Ж 20 Н
Стекло <70 Ж 20—Ткип С
>90 Ж 20-Т™ С
Фторопласт 4 10-50 Ж 20-150 С
>90 Ж 20-150 С
Эмаль кислотостойкая <40 Ж 20-Ткип С
>90 Ж 20 с
Кислота борная Н3ВО3
Алюминий 5 Ж 20 0,003
<5 ж 60 0,1-1,0
Алюминиевые сплавы
АК12 (АЛ2) 5 ж 20 0,004
(Al, Mg: 3,8-10,3 %) 5 ж 20 0,013
Медь <5 ж 20-100 <0,1
Никель Никелевые сплавы Насыщ. ж 20—Тки, <0,1
НМЖМц28-2,5-1,5 <5 ж 20-100 0,1-1,0
Н70МФ <5 ж 20—Ткип <0,1
Свинец <5 ж 20 <0,1
<5 ж 100 0,1-1,0
Серебро Стали <5 ж 20 <0,1
СтЗ <5 ж 20 <3,0
<5 ж 100—Гкип > 10
12X13 <5 ж Ткип < 0,004
12Х18Н10Т <5 ж 20-150 0,002
10Х17Н13М2Т <5 ж 20-7 •4*v * кип <0,1
06ХН28МДТ <5 ж 20-7 1 кип <0,1
Тантал <5 ж 20—Ткип <0,001
Титан Насыщ. ж 20 <0,01
10 ж Ткип <0,01
208
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота борная Н3ВО3 (продолжение)
Чугуны
серый <5 Ж 20-Т 1 кип <3,0
СЧ15, СЧ17 <5 Ж 20—Ткип <0,1
Антегмит АТМ-1 Насыщ. Ж 100 с
Полиамиды <5 Ж 20 с
<5 Ж 60 н
Поливинилхлорид <5 Ж 20-40 с
<5 Ж 60 ОС
Полиизобутилен <5 Ж 20-60 с
Полиметилметакрилат <5 Ж 20-60 с
Полипропилен <5 Ж 20-60 с
Полиэтилен <5 Ж 20-60 с
Резины на основе
БК, НК, СКН, СКС <5 Ж 20-65 с
СКФ <5 Ж 20 с
ХП <5 Ж 20-95 с
ХСПЭ <5 Ж 20-93 с
Стекло <5 Ж 20-ТКИ11 с
Фторопласт 4 <5 Ж 20-100 с
Эмаль кислотостойкая <5 Ж 20—Ткип с
Кислота бромоводородная НВг
Алюминий <40 Ж 20 > 10
Медь 10; без кислорода воз- Ж 25 > 10
духа
Медные сплавы
БрА5, БрА7 10 Ж 25 > 10
БрОФ6,5-0,15 10; без кислорода воз- Ж 25 > 10
духа
Л59, Л63, Л68, Л80 10 Ж 25 > 10
Никель <40 Ж 20-Ткип > 10
Никелевые сплавы
НМЖМц28-2,5-1,5 10 Ж 25 > 10
Н70МФ 10-40 Ж 25 < 1,3
хастеллой А 5 Ж 50-70 0,3
10-20 Ж 50 0,34
10-20 ж 85 0,64
10 ж 100 0,91
20 ж 100 1,3
30-40 ж 100 0,88
Олово <40 ж 20 > 10
Платина 10-40; без кислорода ж 25-105 <0,5
воздуха
Свинец < 15 ж 20 > 10
Электродные процессы
209
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота бромоводородная НВг (продолжение)
Серебро 10 Ж 25 > 10
Стали
СтЗ 10 Ж 25 > 10
12X13 <40 Ж 20 > 10
12Х18Н10Т 10 Ж 25 > 10
10Х17Н13М2Т 10 Ж 25 > 10
06ХН28МДТ 20 Ж 25 > 10
Тантал 10—40 Ж 25-105 <0,5
Титан 0,5 Ж 100 <0,1
5 Ж 25 <0,1
10-20 Ж 25 <0,05
Цирконий 10 Ж 25 <0,5
20 Ж 25 > 10
Чугуны
серый 10 Ж 25 > 10
СЧ15, СЧ17 <40 Ж 20 <0,1
<40 Ж 100 > 10
Антегмит АТМ-1 <40 Ж Ткип н
Графит <40 Ж 20-Ткип С
Полиамиды 40 Ж 20 н
Поливинилхлорид 20-40 Ж 20 С
Полиизобутилен 10 Ж 40-100 С
Полиметилметакрилат <48 Ж 20-60 С
Полипропилен <40 Ж 20-60 С
Полиэтилен <40 Ж 20-60 С
Резины на основе
БК <40 Ж 20-60 С
НК <40 Ж 60 ОС
СКС,ХП <40 Ж 20 ОС
ХСПЭ <40 Ж 20 с
Стекло <40 ж 100 с
Фторопласт 4 <40 ж Т 1 кип с
Эмаль кислотостойкая <40 ж 20-Г 1 кип с
Кислота винная НООССН(ОН)СН(ОН)СООН
Алюминий — т 20 <0,1
10 ж 20 0,002
Насыщ. ж 100 > 10
Алюминиевые сплавы
АК12 (АЛ2) 10 ж 20 0,064
(Al, Mg: 1,5-2,5 %, Мп: 1,0-2,0 %) 10 ж 20 0,0085
Медь < 58; без кислорода ж 20 <0,2
воздуха
Никель 0,7 ж 20 0,01
210
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Кислота винная НООССН(ОН)СН(ОН)СООН (продолжение)
Никель <58 Ж 60-100 > 10
Никелевый сплав
НМЖМц28-2,5-1,5 30 Ж 20 0,03
Свинец <58 Ж 20 > 10
Серебро <58 Ж 75 <0,1
Стали
СтЗ <58 Ж 20 <0,1
<58 Ж Гкип <1,0
12X13 10-50 Ж 20 <0,1
10-50 Ж т 1 кип <1,0
12Х18Н10Т 10-50 Ж 20-Т ^v кип <0,1
10Х17Н13М2Т 10-50 Ж 20-Т 1 кип <0,1
06ХН28МДТ <58 Ж 20-Т^ <0,1
Тантал <58 Ж 20 <0,001
Титан <58 Ж 20-100 <0,01
Чугун СЧ17 10-50 Ж 20-Т 1 КИП <0,1
Антегмит <58 Ж 20-Ткип С
Дерево <58 Ж 20 с
Полиамиды <58 Ж 20-60 С
Поливинилхлорид <58 Ж 20-40 С
<58 Ж 60 ОС
Полиизобутилен <58 Ж 20-60 с
Полиметилметакрилат <58 Ж 20 с
Полипропилен <58 Ж 20-60 с
Полиэтилен <58 Ж 20-60 С
Резины на основе
БК, НК, СКН, СКС, ХП, ХСПЭ <58 Ж 20-65 С
Стекло <58 Ж 20—Ткип с
Фторопласт 4 <58 Ж 20-60 С
Эмаль кислотостойкая <58 Ж 20—Ткип с
Кислота гексафторокремневая H2SiF6*
Алюминий <35 Ж 20-70 > 10
Ванадий 10 ж 70 > 10
Вольфрам 5-20 ж 20 <0,1
5-40 ж 50-80 <0,1
Магний 2 ж 20 > 10
Медь 6 ж 20 0,2
6 ж 50 0,45
6 ж 80 0,69
10; без кислорода ж 100 > 10
воздуха
20 ж 20 0,26
Водный раствор гексафторокремневой кислоты (H2SiF6) содержит H2SiF6, HSiF5 • Н2О, SiF4 • 2Н2О, Si(OH)F3 • 2Н2О и Hf.
Электродные процессы
211
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота гексафторокремневая H2SiF6 (продолжение')
Медь 20 Ж 50 0,62
20 Ж 80 0,95
<35 Ж 20 0,75
<35 Ж 70 2,8
40 Ж 20 0,43
40 Ж 50 0,87
40 Ж 80 1,37
60 Ж 20 0,19
60 Ж 50 0,37
60 Ж 80 0,85
Медные сплавы
БрОФ6,5-0,15; БрОФ7,2-0,2 10-30 ж 25 < 1,3
10; без кислорода ж 100 > 10
воздуха
ЛО68-1, Л80 То же ж 25 > 10
Молибден 10 ж 70 0,17
<60 ж 100 0,05-0,10
Никель 6 ж 20 0,25
6 ж 50 0,49
6 ж 80 0,69
10-30; без кислорода ж 25 < 0,05 кр
воздуха
То же ж 50 < 1,3 кр
10 ж 70 2,2
<35 ж 20 <0,1
<35 ж 100 > 10
40 ж 20 0,35
40 ж 50 1,38
40 ж 80 4,47
60 ж 20 0,5
60 ж 50 0,7
60 ж 80 1,79
Никелевые сплавы
НМЖМц28-2,5-1,5 14,9 ж 70 1,3
<35 ж 20 <0,1
40 ж 20 <0,1
Н70МФ <35 ж 20 <0,1
<35 ж 50-70 1,0-3,0
ХН65МВ 9,7 ж 70 2,2
1-15 ж 40-50 0,05-0,1
10-20 ж 80 1,0-5,0
Ниобий 10 ж 70 4,5
212
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота гексафторокремневая H2SiF6 (продолжение)
Олово 10 Ж 70 3,7
Платина <60 Ж 20—Тит <0,04
Свинец 10^10 Ж 25 < 1,3
10 Ж 70 3,0
Серебро 10 Ж 85 <0,04
<35 Ж 20-60 <0,1
Стали
СтЗ 2 Ж 20 2,4
5-10 Ж 20 4,0
15-20 Ж 20 5,0
12X13 2 Ж 50 > 10
<35 Ж 20 > 10
12Х18Н10Т 2 Ж 50 1,0
<35 Ж 50-60 1,2-1,8
10Х17Н13М2Т 0,5-3,0 Ж 40-60 0,1
4 Ж 40 0,7
4 Ж 80 2,7
5 Ж 40 0,9
5 ж 80 5,3
06ХН28МДТ 5 ж 40 0,2
5 ж 60 0,4
5 ж 80 0,6
10 ж 40 0,3
10 ж 60 0,5
10 ж 80 0,8
15 ж 40 0,5
15 ж 80 5,5
20 ж 40 0,9
20 ж 60 3,0
Тантал 5 ж 30 0,13-1,30
10 ж 70 0,6
<35 ж 20-100 > 10
Титан 1-10 ж 20 > 10
Чугуны
серый <35 ж 20 > 10
СЧ15 10 ж 20 > 10
(Fe; Ni 20 %) 10 ж 25 <0,5
Антегмит <60 ж 20—Гют С
Графит <60 ж 20—Ткип С
Полиамиды <35 ж 20 С
<35 ж 60 н
Поливинилхлорид 30 ж 20-60 С
Полиизобутилен <35 ж 20 С
Электродные процессы
213
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Кислота гексафторокремневая H2SiF6 (продолжение)
Полиметилметакрилат <35 Ж 20-60 С
Полипропилен <35 Ж 20-60 С
Полиэтилен 30-32 Ж 20-60 С
Резины на основе
БК 5-20 Ж 60 С
НК, СКН, СКС <35 Ж 20-70 С
ПС <35 Ж 20 ОС
СКФ 10 Ж 70 Н
ХП <35 Ж 20-100 С
ХСПЭ <35 Ж <65 С
Стекло <60 Ж 20 С
<35 Ж 100 Н
Фторопласт 4 32-40 Ж ^О-Т'кип С
Эмаль кислотостойкая <60 ж 20 С
<35 ж 100 Н
Кислота иодоводородная HI
Алюминий 0,13 ж 20 > 10
Золото <57 ж 20 <0,1
Иридий <57 ж 20-Г„п <0,1
Медь ^1,0 ж 100 > 10
< 10; без кислорода ж 20 < 0,008
воздуха
Никель То же ж 20 <0,1
Никелевый сплав
НМЖМц28-2,5-1,5 » ж 20 <0,1
Олово <57 ж 20 > 10
Платина <57 ж 20-Т„„ <0,1
Свинец < 10 ж 20 < 1,0
Стали
СтЗ <57 ж 20 > 10
12X13 < 1,0 ж 20 <0,1
> 1-57 ж 20 > 10
12Х18Н10Т < 1,0 ж 20 <0,1
<10; без кислорода ж 20 <0,1
воздуха
> 1-57 ж 20 > 10
10Х17Н13М2Т ^1,0 ж 20 <0,1
< 10; без кислорода ж 20 <0,1
воздуха
> 1-57 ж 20 > 10
06ХН28МДТ <1,0 ж Т’кип > 10
< 10 ж 20 <0,1
Титан <57 ж 20-Г„ <0,1
Чугун серый <57 ж 20 > 10
214
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота иодоводородная HI (продолжение)
Графит <57 Ж 20—Гкип С
Полиамиды <1,0 Ж 20 Н
Поливинилхлорид 40 Ж 20-40 С
Полиизобугилен 40 Ж 20-40 С
Полиэтилен <1,0 Ж 20 С
Стекло «Пирекс» <57 Ж 20—Ткип С
Фторопласт 4 <1,0 Ж 20 С
Эмаль кислотостойкая < 1,0 Ж 20 С
Кислота лимонная НОС(СН2СООН)2СООН
Алюминий 1-10 Ж 20 <0,01
5 Ж 60-70 0,01
25 Ж 65 0,13
50 Ж 93 0,13-0,50
<59 Ж 100 0,1-1,0
Алюминиевый сплав АК12 (АЛ2) 10 ж 20 0,006
Медь 0,2 ж 20 0,06
50; в присутствии ж 20 0,10
водорода
50; в присутствии ж 20 0,57
кислорода
<59 ж 100 1,0-3,0
Медные сплавы
(Си, А1: 9-10 %) 5 ж 20 0,04
БрОФ6,5-0,25 5 ж 20 0,04
Никель 5 ж 20 <0,1
1-50 ж 20-100 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 30 ж 20 0,03
30 ж 60 0,18
<59 ж Гкип <1,0
Н70МФ <59 ж 20—Гкип < 0,075
Олово < 59; без кислорода ж 20 <0,1
воздуха
Платина <59 ж 20-Т <0,05
Серебро 1-20 ж 20-Т 1 КИП <0,05
30 ж Гкип <0,5
Стали
СтЗ 5 ж 20 1,0-3,0
5 ж >40 > 10
12X13 1 ж 20 <0,1
25 ж 20 < 1,0
67 ж Гкип > 10
1-50 ж 20 <0,1
25 ж 80 < 1,0
Электродные процессы
215
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота лимонная НОС(СН2СООН)2СООН (продолжение)
Стали
12X13 25 Ж Т’кип > 10
58 Ж 54 0,2 т
12Х18Н10Т 67 Ж Т 1 кип > 10
10Х17Н13М2Т 1-50 Ж 20-Г <0,1
06ХН28МДТ 10; 25; 50; Ж 35-100 <0,1
аэрированные
Титан То же Ж 100 < 0,001
50 Ж Т’кип 0,35
85 Ж 80 0,001
Цирконий <59 Ж 20— Ткт <0,05
Чугун СЧ15 5; 25-50 Ж 20 <0,1
25-50 Ж т 1 кип <1,0
Графит <59 Ж 20—Т^п С
Полиамиды <59 Ж 20-60 с
Поливинилхлорид <59 Ж 20 С
<59 Ж 60 ОС
Полиизобутилен <59 Ж 20-60 С
Полиметилметакрилат <59 Ж 20 С
<59 Ж 60 ОС
Полипропилен <59 Ж 20-100 с
Полиэтилен <59 Ж 20-60 с
Резины на основе
БК, НК, СКН, СКС, СКФ, ХП, <59 Ж 20-65 с
ХСПЭ
ПС <59 Ж 20 н
Стекло х <59 Ж 20-100 С
Фторопласт 4 <59 Ж 20-60 С
Кислота малеиновая НООССН=СНСООН
Алюминий 20 Ж 25 0,01 я
20 г 25 0,003 я
20 ж 100 5,99 я
45 ж 100 4,86 я
Медь 20 ж 25 0,25 я
20 г 25 0,002 я
20 ж 100 0,29 я
Никель 20 ж 100 1,69
45 100 5,34
Свинец 45 ж 100 > 10
Стали
СтЗ 20 ж 25 0,42
20 ж 100 > 10
20 г 25 0,06
216
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среда Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°с
Кислота малеиновая НООССН=СНСООН (продолжение)
Стали
СтЗ 45 Ж 100 > 10
— Ж 140 3,06
— Г 140 0,54 я
12X13 1 Ж 20 <0,1
50 ж 100 > 10
12Х18Н10Т 1 ж 20 <0,05
20 г, ж 25 <0,01
20 ж 100 > 10 я
25 ж 60 0,21 я
25 г 60 < 0,001 я
25 ж 100 4,8
25 г 100 0,23
45 г, ж 40 0,04
45 ж 60 0,21
45 г 60 <0,001
45 ж 80 0,68
45 г 80 0,02
45 ж 100 > 10
45 ж 90-105 2,95
70 ж 90 0,008
70 г 90 0,006
90 ж 90 0,082
90 г 90 0,008
10Х17Н13М2Т 1 ж 20 <0,01
20 г, ж 25 <0,001
25 г, ж 100 0,01
45 ж 40 0,02
45 г 40 <0,001
45 г, ж 60 <0,001
45 г, ж 100 0,01
45 ж 95-105 0,2
70 ж 90 0,005 я
70 г 90 0,003 я
90 ж 90 0,009 я
90 г 90 0,016 я
— ж 140 <0,001 я
— г 140 0,01 я
06ХН28МДТ 1 ж 20 <0,01
70 г, ж 90 0,002
90 г, ж 90 0,011
— г, ж 140 <0,001
Титан 70 ж 90 0,008
70 г 90 0,004
Электродные процессы
217
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота малеиновая НООССН=СНСООН (продолжение)
Титан — Ж 140 0,001
Чугун Х28 20 Ж 100 > 10
20 Г 100 6,72
60 Ж 100 10,6
60 Г 100 6,02
Антегмит АТМ-1 20-45 Ж 90 С
Каучук натуральный Насыщ. Ж 20 с
Каучук хлоропреновый 1-насыщ. Ж 40 с
Керамика То же Ж Т'кии с
Полиамиды — Ж 20-60 с
Поливинилхлорид Насыщ. Ж 40 с
— Ж 60 ОС
Полиизобутилен Насыщ. Ж 40 с
— Ж 100 ОС
Полиметилметакрилат — Ж 20 с
— Ж 60 ОС
Полипропилен — Ж 20-60 с
Полиэтилен — Ж 20-60 с
Резины на основе
БК, НК, СКН — ж 20-60 с
ХП — ж 20-50 с
Фторопласт 4 1-насыщ. ж Т 1 кип с
Эмаль кислотостойкая То же ж т 1 кип с
Кислота масляная СН3(СН2)СООН
Алюминий 5-98 ж 20 <0,05
5-50 ж 50 <0,07
5 ж Т’кип 0,2 т, я
10 ж т 1 кип 0,5 т, я
15 ж т 1 кип 0,9 т, я
25 ж т 1 кип 1,0 т, я
50 ж Гкип 1,5 т, я
75 ж 50 0,2
75 ж Ткп 5,0 т, я
98 ж 50 <0,01
Медь 5-98 ж 20 <0,05
5 ж Гкип 0,01
10-98 ж 50 <0,1
10 ж т 1 кип 0,1
15-98 ж Гкип 0,2-0,5
Медные сплавы
БрАМц5-1 5-98 ж 20 <0,02
5-98 ж 50 <0,1
5-25 ж Т’кип <0,1
218
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота масляная СН3(СН2)СООН {продолжение)
Медные сплавы
БрАМц5-1 50 Ж Т 1 кип 0,4
75 Ж т 1 кип 0,7
98 Ж Т’кип 0,1
БрА5 5-98 Ж 20 <0,04
5-98 Ж 50 <0,1
5-15 Ж т 1 кип <0,1
25-75 Ж т 1 кип 0,3-0,4
98 Ж т 1 кип 0,1
БрОФ6,5-0,4 5-98 Ж 20 <0,06
5-25 Ж 50 0,01-0,05
50-98 Ж 50 0,1-0,2
5-15 Ж ^КИП <0,1
25-50; 98 Ж т 1 кип 0,4-0,5
75 Ж ^кип 0,9
Л62, ЛО62-1, Л68, ЛС59-1 5-98 Ж 20 0,01-0,08
5-98 Ж 50 0,01-0,20
5-25 Ж т 1 кип 0,01-0,30
50-98 Ж ^КИП 0,3-0,5
Никель 0,5 Ж 20 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20-100 0,1-1,0
ХН78Т 5-98 Ж 20-50 0,004-0,040
Н70МФ 15-98 Ж Т’кип 0,4-1,3
Олово 1 Ж 60 0,02
Серебро — Ж 20-Гкип <0,1
Стали
20 5 Ж 20 0,05
10 Ж 20 0,11
5-25 Ж 50 0,3-1,0
5-10 Ж ^кип 5,0-6,0
15 ж 20 0,20
25 ж 20 0,61
50 ж 20 1,0
75 ж 20 0,8
50-75 ж 50 1,2-2,2
98 ж 20 0,1
98 ж 50 0,1
15-98 ж Гкип > 10я
12X13 5-98 ж 20 <0,01
5-25 ж 50 <0,1
5-15 ж Гкнп < 1,0 т
Электродные процессы
219
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота масляная СНз(СНг)СООН (продолжение)
Стали 12X13 25 Ж Т’кип 2,5 т
50-75 Ж 50 0,4-0,5
50-75 Ж ткнп 1,5 т
98 Ж 50 < 0,001
98 Ж Ткип 0,3 т
12Х18Н10Т 5-98 Ж 20-50 <0,001
5-15 Ж Т 1 кип < 0,01 т
25-98 Ж Ткип 0,1-0,2 т
10Х17Н13М2Т 5-98 Ж 20—Ткип <0,001
06ХН28МДТ 5-98 Ж 20—Ткип <0,001
Тантал 1-98 Ж 20-100 <0,1
Титан 5-98 Ж 20-Ткип <0,001
Чугун (Fe, Сг: 30-32 %) 80 Ж 20 <0,1
Графит — Ж 20-Г -^-v 1 кип С
Керамика — Ж 20-Ткип с
Полиамиды — Ж 20 с
Поливинилхлорид — Ж 20 с
— Ж 60 н
Полиизобутилен — Ж 20-100 н
Полиметилметакрилат — Ж 20 ОС
— Ж 60 н
Полипропилен — Ж 20 с
Полиэтилен — Ж 20 с
— Ж 60 ОС
Резины на основе БК, СКН — Ж 20 ОС
С КУ — Ж 20 с
СКФ,ХСПЭ — Ж 20 н
ХП — ж 20 ОС
— ж 60 н
Стекло — ж 20-100 с
Фторопласт 4 — ж 20-100 с
Эмаль кислотостойкая — ж 20-Ткип с
Кислота молочная СН3СН(ОН)СООН
Алюминий 0,5 ж 20 0,019
80 ж 20 0,018
Алюминиевый сплав АК12 (АЛ2) 0,5 ж 20 0,016
80 ж 20 0,014
Медь 1 ж 20 0,06
1 ж 65 0,30
— ж Ткип 1,0-3,0
Никель 10 ж 20-100 <1,0
220
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Г, °C и характер коррозии Стойкость материалов
Кислота молочная СН3СН(ОН)СООН (продолжение)
Никелевые сплавы
НМЖМц28-2,5-1,5 5 Ж 20 0,03
Н70МФ — Ж 20-60 <0,1
Олово 1 Ж 20 0,09
Свинец — Ж 20 > 10
Серебро Стали — Ж 20 <0,1
углеродистая — Ж 20-100 > 10
12X13 10 Ж 20 0,4
10 Ж ?кип > 10
12Х18Н10Т 1,5 Ж 20-Ткип <0,1
1-20 Ж 20 <0,1
10 Ж Т 1 кип <1,0
10Х17Н13М2Т 1,5-10 Ж т 1 кип <0,1
Тантал — Ж 20-Т 1 кнп <0,001
Титан 10-85 Ж 20-Т 1 кип <0,05
Цирконий 10-85 Ж 35-Т < 0,003
Чугуны СЧ15, СЧ17 — Ж 20-Ткип <0,1
Графит — Ж 20—Ткип С
Поливинилхлорид 90 Ж 20 н
Полиизобутилен — Ж 20-100 с
Полиметилметакрилат 90 Ж 20 н
Полиэтилен Резины на основе -— Ж 20-60 с
БК, НК, СКФ, ХП — Ж 20-60 с
ПС — Ж 20-60 н
СКУ, ХСПЭ — Ж 20 с
Стекло — Ж 20—Ткип с
Фторопласт 4 — Ж 20-100 с
Эмаль кислотостойкая — ж 20—Ткип с
Кислота монохлоруксусная С1СН2СООН
Алюминий — т 25 > 10
1 ж 25 4,4
10 ж 25 > 10
Ванадий Ванадиевые сплавы 10 ж 100 0,08
(V, Ti: 2 %) 10 ж 100 0,07
(V, Ti: 30-50 %) 10 ж 100 0,02
Медь — т 25 > 10
10 ж 25 > 10
95 ж Ткип 7,0
100 ж 100 6,8
Электродные процессы
221
Т/родаъж-ение таблицы 7.2.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Кислота монохлоруксусная С1СН2СООН (продолжение)
Медные сплавы
БрА5, БрА7, БОФ6,5-0,15, — Т 25 > 10
БрОФ7,2-0,2
10 Ж 25 > 10
Л59, Л63, Л68, Л80, ЛО68-1 — т 25 > 10
10 ж 25 > 10
Никель — т 25-50 < 0,5 т
— ж 80 < 0,5 т
10 ж 20-100 < 0,5 т
90 ж 95 < 1,3 т
Никелевый сплавы
НМЖМц28-2,5-1,5 — т 25 < 1,3
— ж 75 < 1,3
10 ж 20-75 <0,5
ХН78Т — ж 75 <1,3
— ж 125 > 10
90 ж 25 <0,5
Н70МФ — т 25-50 <0,05
— ж 75 <0,05
— ж 100-125 <0,5
10-70 ж 20-100 <0,5
90 ж 25 <0,05
— г 210 <0,05
ХН65МВ — т 25-50 <0,05
— ж 75 <0,05
— ж 105 <0,5
10-70 ж 20-100 <0,5
90 ж 25 <0,05
— г 210 <0,05
Платина 1-100 ж Т’кип <0,04
— г 210 <0,05
Свинец — т 25 > 10
10 ж 25 > 10
Серебро 1-100 ж Т’кип <0,1
Стали
СтЗ — т 25 > 10
10 ж 25 > 10
100 ж 120 8,5 т
12X13 1-90 ж 20 > 10
12Х18Н10Т — т 25 > 10
10 ж 25 > 10
10Х17Н13М2Т — т 25 > 10
> 10 ж 25 > 10
222
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Кислота монохлоруксусная С1СН2СООН (продолжение)
Стали
06ХН28МДТ 10 Ж 25 <0,5
>50 Ж 20 > 10
Тантал — Т 25-50 <0,05
1-100 Ж 100 <0,05
Титан — Т 25-50 <0,5
— Ж 75-100 <0,5
10 Ж 100 <0,5
30-57,5 Ж 25-75 <0,05
Цинк — Т 35 <0,01
1-90 Ж 20 > 10
Цирконий — Ж 100 <0,1
30 Ж 25-75 <0,05
Чугуны
серый — Т 25 > 10
20 Ж 25 > 10
СЧ15 — Т 50 <0,5
— Ж 65 > 10
10 Ж 20 <0,5
Антегмит АТМ-1 1-90 Ж 25 н
Асбест 1-90 Ж 50 с
Графит — Ж 100 с
< 100 Ж 100 с
Паронит (ПОН) 16,7 Ж ПО с
Полиамиды 10 Ж 40 н
Поливинилхлорид 0,1 Ж 100 с
85 Ж 60 н
Полиизобутилен — т 40 с
1-90 ж 60 с
85 ж 90 с
98 ж 75 ОС
Полиметилметакрилат 40 ж 20 н
Полипропилен 1-90 ж 20-60 с
Полиэтилен — т 40 с
— ж 70 ОС
1-90 ж 20 с
Резины на основе
БК 1-90 ж 20-30 с
НК 1-90 ж 20-60 н
СКН 1-90 ж 20-30 ОС
СКБ 1-90 ж 20 с
СКФ 1-90 ж 70 ОС
ХП 1-90 ж 20 с
Электродные процессы
223
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота монохлоруксусная С1СН2СООН {продолжение)
Резины на основе
ХП 1-90 Ж 60 ОС
ХСПЭ 1-90 Ж 30 С
Стекло 1-100 Ж Т’кип С
Фторопласт 4 1-100 Ж Т 1 кип С
Эмаль кислотостойкая 1-100 Ж т 1 кип С
Кислота муравьиная НСООН
Алюминий 5 Ж 20 0,05
5 Ж 50 0,43
5 Ж 70 2,03
5 Ж 100 4,7
25 Ж 20 0,06
25 Ж 50 0,07
25 Ж 70 2,84
25 Ж 100 8,1
50 Ж 20 0,04
50 Ж 50 0,59
50 Ж 70 1,72
50 Ж 100 5,85
86 ж 20 0,2
86 ж 50 1,2
86 ж 70 3,16
86 ж 100 10,0
Алюминиевые сплавы
АМц 10 ж 20 0,006
100 ж 20 0,06
АМгЗ 5 ж 20 0,07
5 ж 50 1,3
5 ж 70 2,03
5 ж 100 8,3
25 ж 20 0,09
25 ж 50 1,29
25 ж 70 4,7
25 ж 100 9,41
50 ж 20 0,08
50 ж 50 1,34
50 ж 70 3,9
50 ж 100 7,55
86 ж 20 0,76
86 ж 50 2,91
86 ж 70 4,8
86 ж 100 > 10
АК12 (АЛ2) 10 ж 20 0,05
224
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота муравьиная НСООН (продолжение)
Алюминиевые сплавы
АК12 (АЛ2) 100 Ж 20 0,02
Молибден 10 Ж 20 <0,001
20 Ж 20 0,025
Медь 1 Ж 20 <0,1
1 Ж 80 0,37
1 Г 80 0,21
5 Ж 80 0,91
5 Г 80 1,5
20-50 Ж 20 <0,1
20-50 Ж Т’кип 0,18-0,37
90-100 Ж 20-100 0,1-1,0
Медные сплавы
БрА7 40 Ж 30 0,07
40 Ж 100 1,29
(Cu, Sn: 4,3-5,0 %) — Ж 20 > 10
ЛО68-1 50 Ж 80 0,66
Л62 1 Ж 80 0,24
1 Г 80 0,78
5 Ж 80 0,41
5 Г 80 1,5
Никель 10 Ж 20 <0,1
20 Ж 20 <1,0
20 Ж 100 <3,0
85 Ж 100 0,45
90-100 Ж 20-60 0,1-1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 < 10 Ж 20-100 <0,5
20-50 Ж 60—Ткип 0,58-0,75
90-100 ж 20-100 0,25-0,33
Н70МФ < 10 ж 20-7кИ11 <0,1
20-50 ж 60—Ткип <0,1
90-100 ж 60—Ткип <0,1
хастеллой В 40 ж 65-Ткип <1,0
хастеллой С 10 ж <70 <0,1
20 ж Ткип <0,1
40 ж 65—Ткип < 1,0
Ниобий 13-25 ж 25-100 <0,001
Платина 1-100 ж 100 <0,01
Свинец 3,5 ж 20 0,05
Серебро < 10; 20-50 ж 20 <0,01
Стали
углеродистая <10; 20-50; 90-100 ж 20-100 > 10
Электродные процессы
225
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота муравьиная НСООН (продолжение)
Стали
12X13 5 Ж 20 2,25
5 Ж 50 4,2
5 Ж 70 > 10
25 Ж 20 2,5
25 Ж 50 5,0
25 Ж 70 > 10
50 Ж 20 2,3
50 Ж 50 5,9
50 Ж 70 > 10
86 Ж 20 0,67
86 Ж 50 3,3
86 Ж 70 5,07
86 Ж 100 8,65
12Х18Н10Т 5 Ж 20-100 <0,001
5 Ж 140 1,09
25 Ж 20-50 <0,001
25 Ж 70 1,0
25 Ж 100 2,3
25 Ж 140 > 10
50 Ж 20-50 <0,001
50 ж 70 1,1
50 ж 100 2,8
50 ж 140 10,0
86 ж 20-50 <0,001
86 ж 70 0,5
86 ж 100 1,3
86 ж 140 > 10
10Х17Н13М2Т 5 ж 50-100 <0,001
5 ж 140 0,27
25 ж 20-70 <0,001
25 ж 100 0,2
25 ж 140 1,18
50 ж 20-50 <0,001
50 ж 70 0,1
50 ж 100 0,3
50 ж 140 1,35
86 ж 20-50 <0,001
86 ж 70 0,1
86 ж 100 0,4
86 ж 140 > 10
06ХН28МДТ 5 ж 20-140 0,001
25 ж 20-100 <0,001
226
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота муравьиная НСООН (продолжение)
Стали
06ХН28МДТ 25 Ж 140 0,5
50 Ж 20-100 <0,001
50 Ж 140 0,6
86 Ж 20-70 <0,001
86 Ж 100 0,1
86 Ж 140 0,75
Тантал 10 Ж 100 0,01
Титан 5 Ж 20-140 <0,001
25 Ж 20-100 <0,001
25 Ж 140 0,06
50 Ж 20-50 <0,001
50 Ж 70 0,6
50 Ж 100 1,3
50 Ж 140 4,65
86 Ж 20-50 <0,001
86 Ж 70 0,3
86 Ж 100 0,7
86 Ж 140 6,64
Титановые сплавы
ОТ4 30 Ж 95 0,98
ВТЗ 5-60 Ж 200 <0,1
ВТ15 5-25 Ж 200 <0,1
60 Ж 200 < 1,0
Цирконий 10 Ж 60-100 <0,01
Чугуны
серый 5 Ж 20 > 10
СЧ15 10 Ж 20-70 <0,1
10 Ж 100 3,0
50 Ж 20-70 <0,1
50 ж 100 <1,0
(Fe, Сг: 30-33 %) 10-50 ж 20 <0,1
10 ж Гкип <0,1
50 ж Т * кип < 10,0
100 ж ^кип < 1,0
Антегмит АТМ-1 50 ж 50 с
Графит 1-100 ж Гкип с
Дерево <50 ж 20 с
Керамика — ж 20-100 с
Полиамиды <10; 20-50; 90-100 ж 20 н
Поливинилхлорид <10; 20-50; 90-100 ж 20-40 с
<10; 20-50 ж 60 ОС
90-100 ж 60 н
Электродные процессы
227
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота муравьиная НСООН (продолжение)
Полиизобутилен < 10; 20-50 Ж 20-40 С
< 10 Ж 60-100 ОС
90-100 Ж 20-60 ОС
Полиметилметакрилат < 10; 20-50 Ж 20 С
20-50 Ж 60 ОС
90-100 Ж 20 Н
Полипропилен < 10; 20-50; 90-100 Ж 20-60 С
Полиэтилен < 10; 20-50; 90-100 Ж 20-60 С
Резины на основе
БК 1-100 Ж 20 С
НК 20 Ж 60 Н
ПС 10 Ж 60 Н
С КУ < 10 Ж 20 Н
СКФ < 10 Ж 70 н
СКС, ХП, ХСПЭ < 10 Ж 20 с
Стекло 1-100 Ж Гкип с
Фторопласт 4 1-100 Ж Т 1 кип с
Эмаль кислотостойкая 1-100 Ж т 1 кип с
Кислота ортофосфорная Н3РО 4
Алюминий 1 Ж 20 0,05
1 Ж 60-70 <0,12
5 Ж 20 0,26
5 Ж 60-70 0,56-0,62
10 Ж 20 0,49
10 Ж 60-70 1,24
20 Ж 20 0,9
20 Ж 60-70 1,34
40 Ж 20 10,3
80 Ж 20 5,8
80 Ж ГКИП > 10,0
Алюминиевые сплавы
АК12 (АЛ2) 2,5 ж 20 1,43
(Al, Mg: 1,2-2,5 %; Мп: 1,0-2,0 %) 2,5 ж 20 0,64
Золото 1-94 ж 20-100 <0,1
Медь 25 ж 95 0,42
25; аэрированная ж 95 9,0
40 ж Гкип 3,2
42;аэрированная ж 21 1,22
76 ж 21 0,2
80 ж 21 0,5
85 ж 95 0,12
Медные сплавы
БрА7 40 ж Гкип 0,01
228
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота ортофосфорная Н3РО4 {продолжение)
Медные сплавы
БрА7 80 Ж ^КИП 0,23
(Cu; Sn 9,58 %; Zn 4,0 %) 40 Ж 20 0,05
40 Ж Т’кип 0,11
80 Ж 20 < 0,001
80 Ж Т’кип 0,36
ЛС59-1В 40 Ж 20 0,04
40 Ж Т’кип 0,48
80 Ж 20 <0,001
80 Ж Т’кип 0,8
Никель 5-15 Ж 65 <0,13
10 Ж 20 0,015
28 Ж 80 > 10,0
40 Ж 26-28 0,025
57 Ж 26-28 0,013
75-85 Ж 20 <0,01
78-85 Ж 50 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 1-90 Ж 20-80 <0,2
Н70МФ 76 Ж 125 0,25
80 Ж НО 0,07
80 Ж 145 0,33
94 Ж 145 0,16
94 Ж 150 0,34
хастеллой В 10 Ж 20— 7КИП <0,04
30 Ж 20 0,022
80-85 Ж 20 0,005
85 Ж Т’кип 0,06
хастеллой С 10 Ж 20 0,002
* 10 Ж 80 0,2
10 ж Т’кип 0,004
25 ж 80 0,17
50 ж 80 0,07
85 ж Гкип > 10,0
хастеллой D 10 ж 20 0,037
30 ж 20 0,013
50 ж 20 0,1
80 ж 20 0,007
Олово 5 ж 20 3,1
Платина 1-94 ж 20-100 <0,1
85 ж 160-200 <0,001
Свинец 40 ж 20 0,08
40 ж ^кип > 10,0
Электродные процессы
229
Продолжение таблицы 1.4.62
Материал s Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота ортофосфорная Н3РО4 (продолжение)
Свинец 80 Ж 20 0,08
80 Ж Ткип > 10,0
Серебро 5 Ж 102 0,003
45 Ж 20-60 < 0,001
45 Ж ПО 0,007
67 Ж 20 <0,001
67 Ж 60 0,004
67 Ж 125 0,02
85 Ж 20 0,1
85 Ж 60 0,002
85 Ж 140 0,048
Стали
СтЗ 10-90 Ж 20 > 10,0
12X17 10 Ж 20 <0,1
24 Ж 20— Tf^n 0,1-3,0
80 Ж 20 <0,1
80 Ж 110-120 > 10,0
12Х18Н10Т 28 Ж 80 0,67
34 Ж 80 7,9
60 Ж 60 1,7
80 Ж 60 <0,1
80 Ж ПО > 10,0
10Х17Н13М2Т 1-80 Ж 20 <0,1
1-45 Ж Ткип <0,1
70-78 Ж 130 <3,0
80 Ж 60 <0,1
80 Ж ЮО-Ткип 1,0-3,0
06ХН28МДТ 1-94 Ж по <3,0
76 Ж 120 0,4
85 ж 120-140 0,33-0,60
Тантал 1-94 ж 20-100 <0,1
Титан 1 ж 20 0,008
1 ж 50 0,096
5 ж 50 1,15
10 ж 20 0,008
10 ж 50 10,8
20 ж 20 0,012
5-30; аэрированная ж 35 <0,03
20-80 ж 112 > 10,0
50; аэрированная ж 35 0,45
87,4 ж 20 0,86
87,5; аэрированная ж 35 0,74
230
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота ортофосфорная Н3РО4 (продолжение)
Чугуны
СЧ15, СЧ17 1-94 Ж 20 <0,1
10-50 Ж 97 <4,0
Х23, Х32 10 Ж 20 0,12
45 Ж Т’кип <0,1
73 Ж ^кип 2,35
80 Ж 60 0,02
80 Ж по 1,76
80 Ж 120 6,5
80-85 Ж Т 1 кип > 10,0
Антегмит ATM-1 10-90 Ж 20-Г 1 кип С
Графит, пропитанный бакелите- 85 Ж Гкип С
вым лаком
Полиамиды 10-90 Ж 20 н
Поливинилхлорид <30 Ж 20-60 ОС
Полиизобутилен 10-90 Ж 20-100 с
Полиметилметакрилат <30 Ж 20-60 с
Полипропилен 10-90 Ж 20-100 с
Полиэтилен 10-90 Ж 20 с
80-90 Ж 60 ОС
Резины на основе
БК, СКФ 10-90 Ж 20-65 с
НК 1-85 Ж <65 с
С КУ 10-90 Ж 20 с
ХСПЭ 10-90 Ж 20-95 с
Стекло 10-90 Ж 20-200 с
Фторопласт 4 10-90 Ж 200 с
Эмаль кислотостойкая 10-90 Ж 20-160 с
Кислота пропионовая СН3СН2СООН
Алюминий 1-100 Ж 20 <0,11
80 ж Ткип 3,6
100 ж 30 0,04
100 ж 80 0,13
100 ж 120 1,8
Алюминиевый сплав АМгЗ 5 ж 50 0,26
25 ж 50 0,66
50 ж 50 0,56
98 ж 100 0,09
Золото 1-100 ж 20— Ткип <0,1
Медь »100; без кислорода ж < 100 <0,1
воздуха
Медные сплавы
БрА5, БрА7 То же ж < 100 <0,1
Электродные процессы
231
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота пропионовая СН3СН2СООН (продолжение)
Никель « 100 Ж 20 0,1
~ 100 Ж 30 0,34
« 100 Ж 80 0,62
~ 100 Ж 120 1,24 я
Никелевые сплавы
НМЖМц28-2,5-1,5 99,9 Ж Т’кип 4,0
ХН78Т 100 Ж 30 0,27
100 Ж 80 0,68 я
100 Ж 120 1,2 я
хастеллой В 1-100 Ж 20-Т 1 кип <0,1
хастеллой С 1-100 Ж 20-Т 1 кип <0,1
Платина 1-100 Ж 20-7’кип <0,1
Свинец 1-100 Ж 20 >2,34
100 Ж 120 12,0 я
Серебро 1-100 Ж 20— Ткт <0,1
Стали
СтЗ 100 Ж 30 1,2 я
100 Ж 120 5,3 я
1-98 Ж 20 > 10,0
12X13 5 Ж 20 0,11
5 Ж 50 0,48
5 Ж 100 2,36
25 Ж 20 0,25
25 Ж 50 1,8
25 Ж 70 3,52
25 Ж 100 > 10,0
50 Ж 20 0,32
50 Ж 50 2,0
50 Ж 70 4,0
50 Ж 100 > 10,0
99 ж 20 0,13
99 ж 50 1,0
99 ж 100 2,45
99 ж 140 4,68
12Х18Н10Т 5-50 ж 20-100 <0,001
5 ж 160 0,04
25 ж 160 0,34
50 ж 140 1,05
50 ж 160 1,64
99 ж 20-70 <0,001
99 ж 100 0,05
99 ж 140 0,68
99 ж 160 1,86
232
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота пропионовая СН3СН2СООН (продолжение)
Стали
10Х17Н13М2Т 5-99 Ж 20-100 <0,001
25 Ж 160 0,07
50 Ж 140 0,08
50 Ж 160 0,42
99 Ж 140 0,14
99 Ж 160 0,34
06ХН28МДТ 5-99 Ж 20-140 < 0,001
50 Ж 160 0,12
99 Ж 160 0,04
Тантал 1-100 Ж 20-Ткип <0,1
Титан 5-25 Ж 20-160 <0,001
50 Ж 20-100 < 0,001
50 Ж 140 0,01
50 Ж 160 0,04
99 Ж 20-160 < 0,001
100 Ж 30 0,001
100 Ж 80-120 0,012-0,080
Цирконий 1-100 Ж 20-Т 1 кип <0,1
Чугуны
СЧ15, СЧ17 1-100 Ж 20-Г z-v 1 кип <0,1
(Fe, Сг: 25-30 %) 100 Ж Т 1 кип <0,1
Асбест 1-100 Ж 20—Ткип С
Графит, пропитанный феноло- — Ж < 120 с
формальдегидным олигомером
Полиамиды 1-98 Ж 20 н
Поливинилхлорид <80 Ж 20-50 с
Полиизобутилен — Ж 20 ОС
Полипропилен — Ж 20 с
Полиэтилен <60 ж 20-50 с
Резины на основе
БК <50 ж 20 с
НК, СКН — ж 20 н
Стекло 1-100 ж 20—Гют с
Фторопласт 4 1-100 ж <200 с
Эмаль кислотостойкая — ж 20 с
Кислота серная H2SO4
Алюминий 1 ж 20 0,14
1 ж 50 1,79
1 ж 80 5,8
10 ж 20 0,28
10 ж 50 3,9
10 ж 80 > 10,0
20 ж 20 0,31
Электродные процессы
233
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Алюминий 20 Ж 50 4,2
20 Ж 80 > 10,0
50 Ж 20 2,4
78 Ж 20 > 10,0
96 Ж 20 3,5
Алюминиевые сплавы
АК12 (АЛ2) 25 Ж 20 4,9
95,6 Ж 20 0,62
АМц 1 ж 20 0,16
1 ж 80 7,3
10 ж 20 0,33
20 ж 20 0,65
20 ж 80 > 10,0
62,5 ж 20 2,5
62,5 ж 50 > 10,0
78 ж 20 6,25
78 ж 50 > 10,0
96 ж 20 3,72
АМгЗ 1 ж 20 1,0
1 ж 50 2,66
1 ж 98 > 10,0
10 ж 20 1,73
10 ж 50 > 10,0
25 ж 20 2,76
25 ж 50 > 10,0
62,5 ж 20 3,86
62,5 ж 50 > 10,0
78 ж 20 8,49
78 ж 50 > 10,0
96 ж 20 4,73
96 ж 98 > 10,0
Вольфрам 1-98 ж 20 <0,1
Золото 1-100 ж 20—Гкип <0,1
Магний 1-96 ж 20 > 10,0
Медь <5 ж 20 0,1-1,0
<5 ж 50 > 10,0
10-40 ж 20 <0,01
10 ж 40-60 3,36-3,73
25 ж 40-60 1,60-2,09
35 ж 40 0,14
40 ж 40-60 1,33
45 ж 115 2,06
62 ж 95 2,35
234
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Медь 90-98 Ж 20 0,07-1,01
90-98 Ж 50 <2,1
Медные сплавы
БрА5 10 Ж 20 0,24
10 Ж 40 0,53
10 Ж 80 1,3
35 Ж 20 0,15
35 Ж 40 0,37
35 Ж 80 1,5
50 Ж 20 0,1
50 Ж 40 0,23
50 Ж 80 0,5
55 Ж 20 0,07
55 Ж 40 0,11
55 Ж 80 0,36
БрА7 10-40 Ж 20 < 0,002
10-25 Ж 40-60 4,6-5,2
40 Ж 60 2,4
БрАЮ 10-55 Ж 20 <0,1
10 Ж 40 0,17
10 ж 80 0,55
35-50 ж 40 <0,1
35-50 ж 80 <0,5
92 ж 20 0,58
92 ж 40 0,86
БрАЖМц 10-3-1,5 10 ж 20-80 <0,2
35-55 ж 20-40 <0,1
35 ж 80 0,45
55 ж 80 0,06
92 ж 20 0,029
92 ж 40 0,16
БрОФ6,5-0,25 10 ж 20-40 0,21-0,31
10 ж 80 0,94
35 ж 20-40 0,11-0,13
35 ж 80 0,42
55 ж 20 0,019
55 ж 40-80 <0,23
БрОЦС6-6-3 10 ж 20 0,2
10 ж 40 0,4
10 ж 90 0,73
35 ж 20 0,058
35 ж 40 0,1
35 ж 90 0,29
Электродные процессы
235
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Кислота серная H2SO4 (продолжение)
Медные сплавы БрОЦС6-6-3 55 Ж 20 0,066
55 Ж 40 0,086
55 Ж 90 0,17
92 Ж 20 0,078
92 Ж 40 0,26
(Си, Zn: 10-40 %) 1-98 Ж 20 > 10,0
(Си, Zn: 45,78 %, Pb: 0,54 %, 10-40 Ж 20 0,09-0,12
Mn: 2,85 %, Fe: 0,21 %) 10 Ж 40 2,98
10 Ж 60 3,85
25 Ж 40-60 3,5
40 Ж 40-60 1,2-1,5
Молибден 10 Ж 20 0,02
10 Ж 71 0,036
40 Ж 71 0,023
40 Ж Т’кип 0,039
65 Ж 152 0,125
75 Ж 71 0,004
75 Ж Т’кип 0,86
95 Ж 71 0,006
95 Ж 7кп > 10,0
Никель 1 Ж 20 0,1
5 Ж 30 0,23
10 Ж 20 U2
10 Ж 100 3,0
30 Ж 20 0,73
50 Ж 20 0,6
50 Ж 100 10,0
60 Ж 20 0,1
93 ж 65 3,7
95 ж 20 1,8
Никелевые сплавы НМЖМц28-2,5-1,5 1 ж Т’кип 1,0
<5 ж 20 0,02-0,07
10 ж 20 0,06
10 ж 7™п 0,95
65 ж 105 5,7
78 ж 105 3,6
Н70МФ <5 ж 20 0,03
<5 ж 70 <0,15
<5 ж Т’кип 0,025
93 ж 120 0,25-0,30
236
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Никелевые сплавы
Н70МФ 93 Ж 140 1,7-1,9
98 Ж 120 5,4-5,6
хастеллой А 60 Ж 20 0,01
60 Ж 70 0,3
60 Ж Ткип > 10,0
80 Ж 20 0,023
80 Ж 70 0,72
80 Ж 7"кип > 10,0
96 Ж 20 0,008
96 Ж 70 0,2
96 Ж 150 9,0
96 Ж Ткип > 10,0
хастеллой В 2 Ж 20 0,025
2 Ж ТКИП 0,025
10 Ж 20 0,045
10 Ж 70 0,54
10 Ж 7"кип 0,06
25 Ж 20 0,03
25 Ж 70 0,23
25 Ж 7"кип 0,045
60 Ж 20 0,009
60 Ж 70 0,054
60 Ж Т’кип 0,93
80 Ж 20 0,012
80 Ж 70 0,033
80 Ж 7"кип > 10,0
93 Ж 120 0,13-0,18
93 ж 140 0,35-0,40
93 ж 160 0,85-1,0
93 ж 180 2,3-2,4
96 ж 20 0,001
96 ж 70 0,03
96 ж 150 1,2
96 ж 7"кип > 10,0
98 ж 120 6,5-6,7
хастеллой С 2 ж 20 0,008
2 ж Лип 0,375
10 ж 70 0,096
10 ж 95 0,23
10 ж 7"кип 1,26
20 ж 95 0,25
20 ж ЛиП 0,9
Электродные процессы
237
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Г, °C и характер коррозии Стойкость материалов
Кислота серная H2SO4 (продолжение)
Никелевые сплавы
хастеллой С 25 Ж 20 0,004
25 Ж 70 0,19
25 Ж Ткип 1,2
30 Ж 95 0,31
30 Ж Т’кип U
40 Ж 95 0,34
40 Ж Т’кип 4,2
50 Ж 95 0,36
50 Ж Ткип > 10,0
60 Ж 20 0,003
60 Ж 70 0,29
60 Ж Ткип >10,0
65 Ж 95 1,8
65 Ж Ткип > 10,0
78 Ж 95 0,93
78 Ж Ткип > 10,0
80 Ж 20 0,002
80 Ж 70 0,75
80 Ж Ткип > 10,0
93 Ж 95 0,14
93 Ж 120 1,9
93 Ж 140 2,5-4,0
93 Ж 160 3,0-5,3
93 Ж 180 2,7-7,3
93 Ж Ткип 4,3
96 Ж 70 0,063
96 Ж 150 7,8
96 Ж 150 7,8
96 ж Ткип 7,8
Ниобий 10 ж 100 0,006
20 ж 21 < 0,001
98 ж 21 0,002
Олово 1 ж 20 0,08
Платина 1-98 ж <200 <0,1
Свинец 5 ж 20-90 0,05-0,07
10 ж 90 0,07
0,5-10 ж Т 1 КИП 0,05
60 ж т 1 КИП 0,08
78 ж т 1 КИП 0,16
98 ж т 1 КИП 0,29
Серебро 10 ж т 2 КИП 0,003
20 ж т 1 КИП 0,004
238
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Серебро 50 Ж Т 1 кип 0,034
60 Ж Т’кип 0,88
96 Ж 20 0,14
Стали
углеродистая 1 Ж 20 3,3
2 Ж 90 6,2
5 Ж 20 4,4
50 Ж 90 2,04
60 Ж 20 1,5
65-98 Ж 25 0,3-0,5
75 Ж 50 1,3
90 Ж 20 0,09
90 Ж 90 0,97
98 Ж 80 2,8
98 Ж 120 > 10,0
99,5 Ж 20 0,06
12X13 0,4 Ж 36^10 3,0
2-50 Ж 20-Гкип > 10,0
52 Ж 15 2,11
52 Ж 60 8,6
63,4 Ж 15 2,1
63,4 Ж 40 5,27
65 Ж 20 0,03
65 Ж 50-100 > 10,0
80 Ж 60 > 10,0
85 Ж 20 <5,0
85 Ж 120 > 10,0
98 Ж 20 <0,5
98 Ж 100 <5,0
12Х18Н10Т <5 ж 20 0,1-3,0
<5 ж 50 1,0-10,0
10 ж 20 1,0-3,0
40 ж 20 <5,0
40 ж 100 > 10,0
60 ж 20 <0,05
60 ж 100 > 10,0
80 ж 20 <0,1
80 ж 60 < 10,0
85 ж 20 <0,05
93 ж 70 > 10,0
98 ж 20 <0,05
98 ж 100 > 10,0
Электродные процессы
239
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Кислота серная H2SO4 (продолжение)
Стали
10Х17Н13М2Т 5-10 Ж 50 < 1,0
20 Ж 50 <2,0
40-80 Ж 70 > 10,0
50 Ж 30 <2,0
70 Ж 30 <6,0
85-90 Ж 50 <1,0
94 Ж 70 1,П
96 Ж 70 <3,0
98 ж 80 0,15
06ХН28МДТ 10 ж 95 0,02
10 ж 105 0,41
10 ж ^кип 0,51
20 ж 95 0,2 кр
30 ж 95 0,45 кр
30 ж Т’кип 1,5
40 ж 95 0,42 кр
40 ж Гкип 1,09 кр
50 ж 95 0,37 кр
65 ж 95 > 10,0
78 ж 95 0,39
78 ж 105 1,04
78 ж Т’кип > 10,0
93 ж 95 0,4
93 ж 130 6,9
93 ж 150 > 10,0
98 ж 80 0,05
98 ж 100 0,16
98 ж 120 0,51
Тантал 10 ж 250 0,02
20 ж 93 <0,001
50 ж 23 < 0,001
90 ж 250 0,04
95 ж 174 0,003
95 ж 250 0,73
Титан 1,5 ж 95 > 10,0
2 ж 80 0,016
2 ж 95 > 10,0
3 ж 65 1,0
3 ж т J кип > 10,0
5 ж Ткип > 10,0
10-20 ж 20 <0,001
15-50 ж ^кип > 10,0
240
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Титановые сплавы
4200 (Ti, Pd: 0,2 %) 3 Ж ТКИП 0,19
5 Ж 100 1,35
5 Ж 150 0,3
5 Ж Ткип 0,51
10 Ж 25 0,02
10 Ж 60 0,097
15 Ж 100 10,4
15 Ж 150 2,8
20 Ж 100 > 10,0
40 Ж 20 0,22
40 Ж 60 0,84
50 Ж 100 > 10,0
4201 (Ti, Мо: 33 %) 5 Ж 150 0,25
5 Ж 175 1,4
15 Ж 125 0,16
15 Ж 150 6,0
40 Ж 125 0,3
85 Ж 120 > 10,0
Цирконий 10 Ж 20 0,005
30 Ж 35 0,009
60 Ж 35 0,012
75 Ж 35 0,03
80 Ж 35 1,4
90 Ж 35 > 10,0
Чугуны
серый 5-20 Ж 20 > 10,0
СЧ15 5 Ж 20 0,04
5 Ж 50 0,09
5 Ж 100 0,32
20 ж 20 0,01
20 ж 50 0,05
20 ж 100 0,24
25 ж 195 0,76
45 ж 120 0,1
50 ж 20 0,01
50 ж 50 0,03
50 ж 100 0,25
65 ж 20 0,01
65 ж 100 0,05
80 ж 100 0,16
95 ж 100 0,03
97 ж Ткип 0,12
Электродные процессы
241
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Чугуны Х34 5 Ж 20 6,7
5 Ж 50 > 10,0
15-50 Ж 20 > 10,0
65 Ж 20 1,2
65 Ж 50 > 10,0
80 Ж 50 1,8
95 Ж 50 <0,01
Антегмит АТМ-1 40 Ж 100 С
<70 Ж 120 с
75 Ж < 120 с
Асбест 3-10 Ж 60 с
30 Ж <600 с
40 Ж <300 с
80-96 Ж 30-75 с
Графит, пропитанный бакелите- 25 Ж Т’кип с
вым лаком 25-75 Ж < 135 с
Полиамиды < 10 Ж 20-80 н
70-95 Ж 20 н
Поливинилхлорид < 10 Ж 20-80 с
20-60 Ж 20 с
20-60 Ж 60-80 н
70-95 Ж 20 ОС
70-95 Ж 60-80 н
Полиизобутилен < 10 Ж 20 с
< 10 Ж 60-80 с
Полиметилметакрилат < 10 Ж 20-80 с
20-60 ж 60-80 с
70-95 ж 20-80 н
Полипропилен < 10 ж 20-80 с
20-95 ж 20-60 с
Полиэтилен < 10 ж 20-60 с
20-60 ж 20 с
20-60 ж 60-80 ОС
70-95 ж 20 ОС
70-95 ж 60-80 н
Резины на основе БК < 10 ж 20 с
20-50 ж 20 с
60-75 ж 20-70 ОС
>93 ж 70 н
242
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота серная H2SO4 (продолжение)
Резины на основе НК, СКИ < 10 Ж 20-70 С
20-50 Ж 70 ОС
60-75 Ж 20-70 С
>93 Ж 20 н
СКС < 10 Ж 20-70 С
60-75 Ж 20-70 с
>93 Ж 20 н
СКН < 10 Ж 20-70 с
20-50 Ж 20-70 с
60-75 Ж 20 ос
60-75 Ж 70 н
>93 Ж 20 н
СКФ < 10 Ж 20-70 с
20-50 Ж 70 ос
60-75 Ж 20-70 ос
>93 Ж 20 н
ХП < 10 Ж 20-70 с
20-50 Ж 20 с
20-50 Ж 70 ос
60-75 Ж 70 н
>93 Ж 20 н
ХСПЭ < 10 Ж 20-70 с
20-50 Ж 70 с
60-75 Ж 20 н
>93 Ж 20 н
Стекло <98 Ж 20— Ткип с
Фторопласт 4 <98 Ж 20-150 с
Эмаль кислотостойкая <98 ж 20-100 с
Кислота трихлоруксусная СС13СООН
Алюминий — т 25 > 10,0
— ж 120 2,1
10-90 ж 25 > 10,0
Алюминиевый сплав АМц 1,6 ж 35 > 10,0
Медь — т 25 > 10,0
10—90 ж 25 > 10,0
Медные сплавы БрА5, БрА7, БрОФ7,2-0,2 — т 25 > 10,0
10—90 ж 25 > 10,0
Л59, Л63, Л68, Л80 — т 25 > 10,0
10—90 ж 25 > 10,0
Никель — т 25 <0,5
90 ж 25 <0,5
Электродные процессы
243
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота трихлоруксусная СС13СООН (продолжение)
Никелевые сплавы
НМЖМц28-2,5-1,5 10-90 Ж 20-50 <0,5
<55 Ж 100 > 10,0
ХН78Т — Ж 120 < 1,2
Н70МФ 10-90 Ж 25-75 <0,5
20 Ж 100 <0,5
ХН65МВ — Т 25-50 <0,5
10-90 Ж 25-100 <0,5
хастеллой С 1-100 Ж 20 <0,1
Свинец — Т 25 > 10,0
10-90 Ж 25 > 10,0
Стали
12X13 55 Ж 20 > 10,0
12Х18Н10Т — Т 25 > 10,0
10-90 Ж 25 > 10,0
10Х17Н13М2Т — Т 25 > 10,0
10-90 Ж 25 > 10,0
Тантал 50 ж Т’кип <0,001
Титан — т 25 > 10,0
20-50 ж 25-100 <0,05
80 ж 105 > 10,0
Цинк <55 ж 20 > 10,0
Цирконий — т 25 > 10,0
20 ж 25 > 10,0
Чугуны
серый — т 25 > 10,0
10-90 ж 25 > 10,0
(Fe; Ni 20 %) — т 25 > 10,0
10-90 ж 25 > 10,0
Графит 10-30 ж 100 с
Поливинилхлорид 40 ж 20 с
40 ж 60 н
Полиизобутилен <55 ж 20 с
Полиметилметакрилат 30 ж 20 н
Полиэтилен 10 ж 20 с
<55 ж 60 н
Резины на основе
БК, СКН <55 ж 20 н
СКФ <55 ж 70 с
ХП 10 ж 30 ОС
Стекло 10 ж 20 с
Фторопласт 4 30-100 ж 20-100 с
Эмаль кислотостойкая <55 ж 20— Ткип с
244
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота уксусная СН3СООН
Алюминий <0,25 Ж 20 <0,05
0,25 Ж ЛиП 10,2
1-30 Ж 50 <0,11
1 Ж Т 1 кип > 10,0
10 Ж Лип 7,8
40 Ж 20-50 <0,08
40 Ж Лип 6,4
50-80 Ж 20 <0,01
50-70 Ж 50 0,11
60 Ж Лип 5,7
80 Ж 50 0,08
80 Ж Лип 4,0
90-95 Ж 20 0,004
90 Ж Лип 2,5
95 Ж 50 0,01
95 Ж Лип 1,4
98-99,8 Ж 50 0,007
98-99,8 Ж Лип 0,17
Алюминиевые сплавы
АК12(АЛ2) 5-10 Ж 20 0,018
15 Ж Лип 8,32
АМгЗ 5 Ж 20 0,11
5 Ж 100 1,36
25 Ж 20 0,06
25 Ж 70 1,11
50 Ж 20 <0,001
50 Ж 50 0,58
98 Ж 20-50 <0,01
98 Ж 70 0,07
98 ж 100 0,18
98 ж 140 0,8
АМцС, АМг2, АМг5 99,5 ж Лип <0,1
ДШ 5 ж 20 0,12
(Al, Zn: 1,0-1,6 %) 5 ж 20 0,22
Медь 1-5 ж 20 0,07-0,09
6; аэрированная ж 20 0,48
1-30 ж 40 0,21-0,25
7,6 ж т 1 кип 1,2
7,6 г Лип 0,96
20 ж Лип 1,7
65,5 ж т 1 кип 4,9
65,5 г т 1 кип 1,8
79,2 ж т 1 кип 6,2
Электродные процессы
245
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота уксусная СН3СООН (продолжение)
Медь 79,2 Г Т’кип 2,3
Медные сплавы
БрА5 1 Ж 40 0,22
5 Ж 40 0,12
10 Ж 40 0,32
30 Ж 40 0,25
БрАЖМцб-1-1 30 Ж 40 0,12
(Си, А1: 9-10 %) 1-5 Ж 40 0,28
30 Ж 20 0,21
30 Ж 40 0,48
(Си, Sn: 3,5-5,0 %) 33 Ж Т’кип <0,09
Л90 100 Ж 80 2,49
Никель 1-99,8 Ж 20 0,04-0,20
1-99,8; Ж 20 < 1,6
аэрированные
Никелевые сплавы
НМЖМц28-2,5-1,5 1-99,8 Ж 20 <0,1
10-90 Ж Т 1 кип 0,36-0,62
Н70МФ 50-80 Ж Т’кип <0,1
98 Ж 165 <0,1
хастеллой С 10-99 Ж 20—Гкип <0,01
Олово 1 Ж 20 0,36
5 Ж 20 0,45
10 Ж 40 0,62
60 Ж т 1 кил 0,40
Платина 1-99,8 Ж 20— 7’кнп <0,08
Свинец 1-2 Ж 20 0,50
10 Ж 20 0,47
60 Ж 20 0,72
98 Ж 20 2,13
98 ж т 1 кип 6,0
Серебро 1-99,8 ж 20— Ткип <0,1
Стали
СтЗ 3 ж 38 1,21
5 ж 20 0,79
15 ж 20 1,23
33 ж 20 1,34
33 ж Т’кип 2,33
12X13 1 ж 90 <0,1
5 ж 20 0,20
5 ж 50 0,64
5 ж 100 3,32
25 ж 20 0,20
25 ж 50 1,2
246
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Г, °C и характер коррозии Стойкость материалов
Кислота уксусная СН3СООН (продолжение)
Стали
12X13 25 Ж 70 4,3
50 Ж 20 1,2
50 Ж 50 2,8
50 Ж 70 4,4
98 Ж 20 0,12
98 Ж 50 1,42
98 Ж 100 3,4
98 Ж 140 5,4
12Х18Н10Т 5-25 Ж 20-140 <0,001
5-25 Ж 165 0,08-0,42
50 Ж 20-70 <0,001
50 Ж 100 0,12
50 Ж 140 1,52
50 ж 165 3,46
80 ж 20-80 <0,1
80 ж Ткип 1,0-3,0
98 ж 20-70 <0,001
98 ж 100 0,18
98 ж 140 1,04
98 ж 165 2,22
10Х17Н13М2Т 5 ж 20-165 <0,001
25 ж 20-140 <0,001
25 ж 165 0,12
50 ж 20-100 < 0,001
50 ж 140 0,80
50 ж 165 1,24
80 ж 20-80 <0,1
80 ж Ткип 0,1-1,0
98 ж 20-100 <0,001
98 ж 140 0,12
98 ж 165 0,44
06ХН28МДТ 5-25 ж 20-165 <0,001
50 ж 20-100 <0,001
50 ж 140 0,28
50 ж 165 0,62
80 ж Ткип 0,1-1,0
90 ж 95 <0,1
98 ж 20-100 <0,001
98 ж 140 0,04
98 ж 165 0,12
Тантал 1-99,8 ж 20—Ткип <0,1
Электродные процессы
247
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота уксусная СН3СООН (продолжение)
Титан 5-25 Ж 20-165 < 0,001
50 Ж 20-100 < 0,001
50 Ж 140 0,06
50 Ж 165 0,07
98 Ж 20-165 <0,001
Цирконий 5 Ж 35-60 0,0007
25 Ж 60 0,0002
50 Ж 100 0,0005
75 Ж 35 0,001
99,5 Ж 35 0,001
Чугуны
СЧ15 10-99,8 Ж 20 <0,1
10-80 Ж ^кип 0,1-1,0
(Fe, Сг: 22,5 и 36,0 %) 80 Ж 20 0,0025
Антегмит АТМ-1 98 Ж 102 С
Графит 1-99,8 Ж Ттп с
Полиамиды 1 Ж 20 н
Поливинилхлорид 6 Ж 60 ос
<80 Ж 20 с
<80 ж 50 ОС
99,8 ж 40 н
Полиизобутилен 25-60 ж 60 с
80 ж 40 с
80 ж 100 ОС
85 ж 100 н
Полиметилметакрилат 30 ж 20 н
Полипропилен 1-99,8 ж 20-80 с
Полиэтилен 10 ж 80 с
85 ж 60 с
99,8 ж 20 с
99,8 ж 60 н
Резины на основе
БК 50 ж 40 с
НК, ПС, ХП 10 ж 20 ОС
СКУ, СКФ 1-99,8 ж 20 н
Стекло 1-99,8 ж т 1 кип с
Фторопласт 4 1-99,8 ж т 1 кип с
Эмаль кислотостойкая 1-99,8 ж т 1 кип с
Кислота фтороводородная HF
Алюминий 1-70 ж 20 > 10,0
80 ж -10 0,17
85 ж -10 0,01
90-95 ж -10 0,007
99,5 ж -10 0,004
248
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т. °C
Кислота фтороводородная HF (продолжение)
Алюминий 97,0-99,5 Ж 15 0,28
Алюминиевые сплавы
АМц 40-55 Ж 20 > 10,0
70 Ж 20 7,0
80 Ж -10 0,036
85 Ж -10 0,013
95 Ж -10 0,007
99,6 Ж -10 0,004
АМг5В 80 Ж -10 0,88
85 Ж -10 0,55
90 Ж -10 0,058
95 Ж -10 0,025
99,5 Ж -10 0,002
Ванадий 30 Ж 20 > 10,0
Вольфрам 7 Ж 20 0,017
7 Ж 50 0,03
7 Ж 80 0,1
13 Ж 20 0,028
13 Ж 50 0,052
13 Ж 80 0,24
20 Ж 20 0,01
20 Ж 50 0,06
20 Ж 80 0,29
25 Ж 20 0,015
25 ж 50 0,045
25 ж 80 0,024
35 ж 20 0,01
35 ж 50 0,043
35 ж 80 0,14
50 ж 20 0,01
50 ж 50 0,04
50 ж 80 0,1
Золото 10;аэрированная ж 25-75 <0,05
Иридий 40 ж 20 0,001
Кобальт 5-14 ж 20 <0,09
27-37 ж 20 0,1
Магний 18; без кислорода воздуха ж 90 <0,01
97,0-99,5 ж 15 0,28
Медь 40-60 ж 20 0,08
38 ж НО 1,2
70 ж 20 0,89
93 ж 20 0,66
97,0-99,5 ж 15 0,28
Медные сплавы
БрА7 18 ж 20 0,07
38 ж НО 1,1
40 ж 25 0,1
Электродные процессы
249
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота фтороводородная HF (продолжение)
Медные сплавы БрОФ6,5-0,15 10; без кислорода воз- Ж 100 <0,5
БрАЖ9-4 духа 38 Ж по 1,3
40 Ж 20 0,05
Л068-1 10-30; без кислорода Ж 25 <0,5
воздуха 40; без кислорода воз- Ж 100 > 10,0
Л80 духа 10; без кислорода Ж 25 > 10,0
Молибден воздуха 1 Ж 20 0,005
Никель 8-24 Ж 20 <0,09
10;аэрированная Ж 75 > 10,0
20; без кислорода Ж 75 < 1,3 кр
воздуха 20; без кислорода Ж 100 > 10,0
воздуха 48 Ж 20 0,23
93 Ж 20 0,15
98 Ж 115 0,08
97,0-99,5 Ж 15 0,55
Никелевые сплавы НМЖМц28-2,5-1,5 8-14 ж 20 0,09
10 ж 50 < 0,5 кр
30-37 ж 20 <0,2
48 ж 20 < 0,002
48 ж 115 0,022
70 ж 20 < 0,002
70 ж 115 0,43
98 ж 20 0,05
Н70МФ 5-10 ж 20 <0,05
10 ж 70 0,2
10 ж 95 0,9
20-30; без кислорода ж 25 <0,5
воздуха 30 ж 70 0,8
30 ж 95 1,7
40; без кислорода ж 25 <0,5
воздуха 40; аэрированная ж 100 > 10,0
ХН65МВ 5 ж 25 <0,1
10 ж 70 0,2
10 ж 95 1,2
20 ж 25 <0,5
30 ж 70 0,8
40 ж 44 1,4
40;аэрированная ж 100 > 10,0
250
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота фтороводородная HF {продолжение)
Никелевые сплавы
хастеллой В 24 Ж 20 0,07
40 Ж 20 0,06
40 Ж 44 0,14
хастеллой С 24 Ж 20 0,06
40 Ж 20 0,06
40 Ж 44 1,4
Ниобий 20 Ж 20 1,7
Осмий 40 ж 20 0,001
Палладий 40 ж 20 0,001
Платина 40 ж 20 0,001
Родий 40 ж 20 0,001
Рутений 40 ж 20 0,001
Свинец 10-30; без кислорода воздуха ж 25 <0,05
То же ж 50-75 <0,5
10; без кислорода воздуха ж 100 <0,5
30; аэрированная ж 75 > 10,0
30; без кислорода воздуха ж 100 > 10,0
40; без кислорода воздуха ж 25 <0,5
Серебро 30; аэрированная ж 100 <0,5
40 ж 115 <0,05
<50 ж 20-Ткип <0,08
Стали
углеродистая 0,05 ж 20 1,2
0,1 ж 20 1,9
1 ж 20 > 10,0
48 ж 20 > 10,0
70 ж 20 5,8
80-95 ж 25-35 0,29
12X13 10 ж 20 > 10,0
40 ж 20 > 10,0
97,0-99,5 ж 15 0,12
12Х18Н10Т 0,05 ж 20 <0,001
0,1 ж 20 0,04
0,5 ж 20 0,4
1 ж 20 0,7
10 ж 100 > 10,0
20;аэрированная ж 25 > 10,0
40 ж 20 > 10,0
97,0-99,5 ж 15 0,19
10Х17Н13М2Т 0,05 ж 20 < 0,001
Электродные процессы
251
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота фтороводородная HF (продолжение)
Стали
10Х17Н13М2Т 0,1 Ж 20 0,02
0,5 Ж 20 0,08
1 Ж 20 0,2
5 Ж 20 0,5
20; аэрированная Ж 25 > 10,0
40 Ж 20 > 10,0
06ХН28МДТ 0,05 Ж 20 <0,001
1 Ж 20 0,01
5 Ж 20 0,1
< 20;аэрированная Ж 20 < 1,0
25 Ж 65 > 10,0
40 Ж 20 <3,0
Тантал 20 Ж 20 2,3
Титан 1 Ж 25 > 10,0
Титановый сплав ВТ-15 10 Ж 25 > 10,0
Цирконий 1 Ж 25 > 10,0
Чугуны
серый 10 Ж 25 > 10,0
ЧС15 10; без кислорода воз- Ж 25 > 10,0
духа
97,0-99,5 ж 15 0,61
Антегмит АТМ-1 <48 ж < Т’кип С
Асбест 5 ж 20 ОС
Графит <40 ж 20 С
Полиамиды 40 ж 20 С
Поливинилхлорид 40 ж 20 с
Полиизобутилен <40 ж 20-60 с
Полиметилметакрилат <20 ж 25 с
40 ж 20 н
Полипропилен <50 ж 20-50 с
Полиэтилен <48 ж 60 с
Резины на основе
БК <50 ж 50-70 с
НК <40 ж 20 ос
ПС 10 ж 20 н
СКИ 10 ж 20 ОС
СКН 10 ж 20 н
СКС <50 ж 20 ОС
СКТ 10 ж 20 н
СКФ 34 ж 20 н
хп 30 ж 20 с
ХСПЭ <50 ж 20-60 с
Стекло < 100 ж 20 н
Фторопласт 4 < 100 ж 20-60 с
Эмаль кислотостойкая < 100 ж 20 н
252
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлорная нсю4
Алюминий <72 ж 20 > 10,0
Медь 10 ж 20 > 10,0
Медные сплавы
БрА5, БрА7, БрОФ6,5-1,5, 10 ж 25 > 10,0
БрОФ7,2-0,2
Л59, Л63, Л68, Л80, ЛО68-1 7,5 ж 25 > 10,0
Никель 12,5 ж 30 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 10 ж 25 > 10,0
Н70МФ 1-10 ж 20 < 0,005
1-2 ж 70 <0,01
1-2 ж Гкип <0,5
5 ж 70 <0,5
5-10 ж Т’кип < 1,0
15-20 ж 20 <0,01
15 ж 70 <0,5
15 ж Т'кип <1,0
20-37 ж 70 <1,0
20 ж Т'кип <5,0
25-37 ж 20 <0,05
ХН65МВ 1-10 ж 20 <0,01
1-2 ж 70 <0,1
5 ж 70 <0,5
1-10 ж 7"кип <5,0
15 ж 20 <0,05
15 ж 70 <1,0
15 ж Гкип > 10,0
20 ж 20 <0,1
20-25 ж 70 <5,0
Ниобий <72 ж 20-100 <0,08
Олово <72 ж 20 > 10,0
Платина 10-70 ж 25-100 <0,05
Свинец 10 ж 25 > 10,0
Стали
СтЗ <72 ж 20 > 10,0
12X13 <72 ж 20 <3,0
12Х18Н10Т <72 ж 20 <3,0
10Х17Н13М2Т <72 ж 20 <3,2
06ХН28МДТ <72 ж 20 <1,0
Тантал 10-70 ж 25 <0,05
Титан <72 ж < 130 <0,08
Титановые сплавы
АТЗ 51-52 ж 20 0,0016
Электродные процессы
253
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Кислота хлорная НС1О4 (продолжение)
Титановые сплавы
АТЗ 75 Ж 20 < 0,001
АТ4 51-52 Ж 20 0,0019
75 Ж 20 <0,001
АТ6 51-52 Ж 20 < 0,0035
75 Ж 20 <0,001
АТ9 51-52 Ж 20 0,011
75 Ж 20 <0,001
ОТ4 75 Ж 20 0,0006
Цирконий 10-72 Ж 25-100 <0,05
Чугуны
серый 10 Ж 25 > 10,0
(Fe, Ni: 20 %) 10 Ж 30 > 10,0
Антегмит АТМ-1 10 Ж 80 с
<72 Ж 20 с
Графит <72 Ж 20 н
Графит, пропитанный бакелито- 10 Ж 20 ОС
вым лаком
Поливинилхлорид 10-40 Ж <23 с
Полиизобутилен <70 Ж <50 с
Полипропилен 20 Ж 20 с
<70 Ж 60 н
Полиэтилен <72 Ж 20 с
<72 ж 60 ОС
Резины на основе
СКН, СКС <72 ж 45 н
СКФ 10 ж 20-60 с
<72 ж 20 с
ХП 10 ж 60 с
ХСПЭ <72 ж 20 с
Стекло <72 ж 20-100 с
Фторопласт 4 <72 ж 20— ТКИ11 с
Эмаль кислотостойкая <72 ж Ткип с
Кислота хлороводородная НС1
Алюминий 1 ж 20 0,26
1 ж 50 2,02
1 ж 98 > 10,0
3,5-35 ж 20-Ткип > 10,0
Алюминиевые сплавы
АМц 1 ж 20 3,33
1 ж 98 > 10,0
АК12 (АЛ2) 1 ж 20 1,14
1 ж 50 > 10,0
254
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Кислота хлороводородная НС1 (продолжение)
Алюминиевые сплавы
АМг2 1 Ж 20 5,92
1 Ж 50 10,4
Ванадий 0,5 Ж 25 0,0013
0,5 Ж 100 0,068
10 Ж 25 0,068
10 Ж 100 4,17
20 Ж 100 6,8
30 Ж 25 0,57
36 Ж 25 1,79
36 Ж 100 6,8
Вольфрам 1-37 Ж 20 <0,0001
1-37 Ж 100 0,11
Золото 5 Ж 20 0,004
36 Ж 20 <0,1
Индий 0,4 Ж 20 0,3
1 Ж 20 0,51
Иридий 36 Ж 20 0,06
Иттрий 0,4-37 Ж 25 5,1
Кадмий 1-37 Ж 20 > 10,0
Кобальт 1-10 Ж 25 <0,06
20 Ж 25 0,2
36 Ж 25 0,6
Магний 1-37 Ж 20 > 10,0
Медь 4 Ж 20 0,04
10 Ж 20 4,74
10; без кислорода воз- Ж 20 0,08
духа
10 Ж 40 > 10,0
20-30 ж 20 > 10,0
Медные сплавы
БрА7 3 ж 30 0,7
10 ж 100 > 10,0
20 ж 40 3,5
30 ж 20 2,5
30 ж 40 5,0
БрАЖ9-4 10 ж 20 3,9
БрАЖМцЮ-3-1,5 10 ж 20 1,5
БрАМц9-2 10 ж 20 2,2
БрБ2 3,6 ж 15 1,0
БрМц5 4 ж 20 0,6
БрОб 10 ж 40 5,9
БрОФ4-0,25 3,6 ж 15 1,0
БрОЦ4-3 10 ж 20 2,6
Электродные процессы
255
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлороводородная HCI (продолжение)
Медные сплавы БрХ0,8 2 Ж 20 0,5
4 Ж 20 1,0
11 Ж 20 1,6
Л62 6—11 Ж 20 <2,8
11,7 Ж 10 2,8
ЛЦ59-1 2 Ж 20 1,5
6 Ж 20 3,6
11 Ж 20 4,5
ЛО62-1 6 Ж 20 2,8
11 Ж 20 4,0
ЛО70-1 0,1-0,3 Ж 30-90 0,6
Л80 4 Ж 20 1,2
Молибден 1,5 Ж 100 0,03
10-30 Ж 100 0,04
Никель 0,07 Ж 20 0,1
0,5 Ж 100 7,6
3,6 Ж 60 2,2
5; насыщ. азотом Ж 30 0,09
5; аэрированная Ж 30 1,6
10-30 Ж 80 <3,0
18 Ж 30 1,2
30; аэрированная Ж 30 1,6
Никелевые сплавы НМЖМц28-2,5-1,5 0,5; насыщ. воздухом Ж 80 2,75
5 Ж 70 1,3
5; насыщ. азотом Ж 70 1,2
5;аэрированная Ж 30 2,1
10; без воздуха Ж 80 0,5
10; насыщ. воздухом Ж 80 3,25
20; без воздуха ж 80 2,88
20; насыщ. воздухом ж 80 4,0
30; без воздуха ж 80 7,2
30; насыщ. воздухом ж 80 9,3
Н70МФ 1-35 ж 20 <0,05
1-35 ж 40 <0,1
1-35 ж 60-80 <0,3
10 ж 100 <0,4
37 ж 70 0,5
ХН65МВ 1,5 ж 20-70 0,1
1-2 ж Т’кип 3,0
5 ж 70 2,4
256
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза Т,°С и характер коррозии Стойкость материалов
Кислота хлороводородная НС1 (продолжение)
Никелевые сплавы
ХН65МВ 10-25 Ж 20 <0,1
10 Ж 70 <1,0
10;аэрированная Ж 70 0,3
10 Ж Т’кип > 10,0
20-25 Ж 70 <5,0
хастеллой А 5 Ж 20 0,2
5 Ж т 1 кип 3,3
10 Ж Т’кип 4,5
хастеллой В 10 Ж 20 0,17
37 Ж 70 0,51
37 Ж т 1 кип 0,44
хастеллой С 1 Ж т 1 кип 2,51
4 Ж т J кип > 10,0
10 Ж Т’кип > 10,0
37 Ж 20 0,44
Ниобий 18-37 Ж 10-26 < 0,003
37 Ж ПО 0,1
Олово 0,05 Ж 20 0,05
1,5 Ж 20 0,2
6; насыщ. кислородом Ж 20 > 10,0
10 Ж 20 0,4
Осмий 36 Ж 20 <0,1
Палладий 5 Ж Т’кип 0,01
36 Ж 20 0,07
Платина 36 Ж 20 0,01
Рений 10-20 Ж 100 < 0,004
Родий 30 Ж 100 0,1
Рутений 36 Ж 20 0,02
Свинец 4; насыщ. водородом ж 20 0,43
4; насыщ. кислородом ж 20 4,1
5 ж 20 < 1,0
10 ж 20 2,0
10 ж 100 3,9
20 ж 20 > 10,0
Серебро 5 ж 20 0,1
5;аэрированная ж 20 0,04
15 ж 20 0,007
15;аэрированная ж 20 0,085
25; аэрированная ж 20 0,36
36 ж 20 0,07
36;аэрированная ж 100 2,5
Электродные процессы
257
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлороводородная НС1 (продолжение)
Серебро 35-37 Ж Т’кип > 10,0
Стали
СтЗ 0,02 Ж 30 2,6
0,04 Ж 30 4,2
>0,4 Ж 30 > 10,0
12X13 0,5 Ж 20 2,3
0,5 Ж 70 > 10,0
12Х18Н10Т 1 Ж 20 0,1
3 Ж 20 1,63
3,6 Ж 20 <3,0
10 Ж 60 > 10,0
10Х17Н13М2Т 0,2-1,0 Ж 20 <0,1
0,2 Ж 50 < 1,0
1,5 Ж 60 <3,0
20 Ж 20 <3,0
20 Ж 60 10,0
06ХН28МДТ 0,5 Ж Тюш <5,0
1-2 Ж 20 <0,1
1-2 Ж 70 3,0
1 Ж Ткип > 10,0
2,5-5,0 Ж 20 1,0
5 Ж 70 > 10,0
10-20 Ж 20 3,0
25 ж 20 > 10,0
Тантал 10 ж 100 0,001
20 ж 60 0,002
20-30 ж 100 <0,01
35-37 ж Ткип <0,1
Титан 0,5 ж 60-100 0,009
1 ж 60-95 0,003
1 ж 100 0,5
1,5 ж 100 4,4
2 ж 50-60 <0,02
2 ж 95-100 1,0-10,0
3 ж 50 1,02
4 ж 60 1,08
4 ж 100 > 10,0
5 ж 20 0,004
5 ж 60 1,39
5 ж 90 9,21
10 ж 20 0,1
10 ж 60 8,9
15 ж 20 0,14
258
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлороводородная HCI (продолжение)
Титан 15 Ж 60 > 10,0
20 Ж 20 0,58
20 Ж 60 > 10,0
Титановые сплавы
ОТ4-1 0,4 Ж 30-90 0,08
2 Ж 90 2,9
4 ж 90 8,7
5 ж 90 > 10,0
10 ж 30 0,7
10 ж 60 7,0
15-20 ж 60 > 10,0
ВТ5-1 0,5-1,0 ж 95 <0,012
5-15 ж 30 < 1,1
20-30 ж 30 2,4-2,5
ВТ14 5 ж 40 0,2
5 ж 60 2,7
10 ж 60 8,2
30 ж 40 > 10,0
Цинк 0,4 ж 20 > 10,0
Цирконий 1-5 ж 60-100 <0,001
5;аэрированная ж 60-100 <0,02
10 ж 40 0,003
10 ж 60 0,02
10; аэрированная ж 60-100 <0,01
20 ж 40 0,02
20 ж 60 0,07
20 ж 100 0,2
20;аэрированная ж 100 0,02
Чугуны
серый 1,8 ж 20 > 10,0
СЧ15, СЧ17 30 ж 80 1,4
(Fe, Ni: 14 %) 0,5-25,0 ж 20 0,21-0,83
Х28Л 10 ж 20 > 10,0
Х27, Х34 10-37 ж 20 12,0
Антегмит АТМ-1 <37 ж < Т’кип С
Асбест 20 ж 100 н
30 ж 20 с
Графит <37 ж 20—Гют с
Полиамиды 2 ж 25 ОС
5 ж 60 н
10 ж 20 н
Поливинилхлорид <30 ж <50 с
35 ж 20 с
Электродные процессы
259
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлороводородная НС1 (продолжение)
Поливинилхлорид 35 Ж 60 ОС
Полиизобутилен <30 Ж <50 С
35 Ж 40 С
Полиметилметакрилат < 10 Ж 60 С
<30 Ж <50 С
35 Ж 20 Н
Полипропилен 10 Ж 60 С
20 Ж 60 ОС
<30 Ж <50 С
35 Ж 20-40 С
35 Ж 66 Н
Полиэтилен 10 Ж 100 С
20 Ж 60 С
<30 Ж <50 С
<36 Ж 25 С
Резины на основе
БК 8 Ж 52 С
20 Ж 20 С
33 Ж 32 ОС
34 Ж 70 н
НК < 10 Ж 60 с
<20 Ж 20 с
35 Ж 20 н
ПС < 10 Ж 20 с
20 Ж 20 ОС
35 Ж 20 н
СКИ < 10 Ж 60 с
<20 Ж 20 с
35 ж 20 н
СКС < 10 ж 60 с
20 ж 60 ОС
<25 ж 20 с
35 ж 25 н
СКТ < 10 ж 20 с
20 ж 20 н
СКУ 10-35 ж 20 н
СКФ 10-35 ж 20-70 с
ХП <20 ж 20-60 с
ХСПЭ 20 ж 20-60 с
35-37 ж 20 с
35-37 ж 70 ОС
Стекло <37 ж ^кип с
Фторопласт 4 <37 ж 20-7 1 КИП с
Эмаль кислотостойкая 10-35 ж 20-7 с
260
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлорсульфоновая SO2C1(OH)
Алюминий 87,5 Ж 25 > 10,0
99,5; безводная Ж 20-75 0,05-0,5
Алюминиевые сплавы Тоже Ж 20-75 0,05-0,5
(Al, Mg: 1,3 %), (Al, Мп: 1,2 %),
(Al, Si: 5-12 %),
Золото Безводная Ж < 100 <0,1
Магний То же Ж 20 >3,0
Медь 10; без кислорода Ж 25 > 10,0
воздуха
87; без кислорода Ж 25 > 10,0
воздуха
Безводная Ж Т’кип <0,1
Медные сплавы
БрА5, БрА7, БрКЗ То же Ж Т 1 кип <0,1
Л90 » Ж Ткип <0,1
Молибден » Ж < 100 <0,1
Никель 10 Ж 20 <0,5
Безводная Ж 20 <0,5
Никелевые сплавы
НМЖМц28-2,5-1,5 12,5 Ж 20 <0,5
Безводная Ж 20 <0,5
Н70МФ 10-50 Ж 25 <0,05
87,5 Ж 20-105 <0,05
ХН65МВ 10-50 Ж 25 <0,05
хастеллой В Безводная Ж < 100 <0,1
хастеллой С То же Ж < 100 <0,1
Ниобий » Ж < 100 <0,1
Платина » Ж < 100 <0,1
Свинец 10 Ж 25 <0,05
90 Ж 20 < 1,3
Серебро Безводная Ж 20 <0,1
Стали
СтЗ 10 Ж 25 > 10,0
87,5 Ж 25 > 10,0
Безводная Ж 20 <0,1
То же ж Ткип >3,0
12X13 10 ж 20 > 10,0
12Х18Н10Т 0,5 ж 20 <0,1
30 ж 25 > 10,0
90 ж 25 > 10,0
Безводная ж 20 <0,12
То же ж Ткип >3,0
10Х17Н13М2Т 0,5 ж 20 <0,1
10 ж 20 > 10,0
Электродные процессы
261
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота хлорсульфоноваям SO2CI(OH) {продолжение)
Стали
10Х17Н13М2Т Безводная Ж 20 <0,1
То же Ж Ткип >3,0
06ХН28МДТ » Ж 20 <0,1
Ж Ткип >3,0
Тантал 12,5 Ж 30 <0,05
Титан Безводная Ж < 100 <0,1
Титановые сплавы
Цинк То же Ж 20 >3,0
Чугуны
серый » Ж 20 0,1-1,0
СЧ15 » Ж 20 <0,1
Асбест 97 Ж 20-Ткип ОС
Графит Безводная Ж 20 с
Полиамиды То же Ж <60 н
Поливинилхлорид » Ж 20 н
Полиизобутилен » Ж <60 н
Полипропилен » Ж <60 н
Полиэтилен » Ж <60 н
Резины на основе БК, НК, СКН, » Ж <60 н
СКС, СКУ, СКФ, ХП, ХСПЭ
Стекло » Ж 100 с
Фторопласт 4 97 Ж 20-Ткип с
Эмаль кислотостойкая Безводная Ж 100 с
Кислота щавелевая НООССООН
Алюминий 0,5 Ж 20 0,05
0,5 Ж 70-80 0,073
2 Ж 20 0,1
2 Ж 70-80 0,41
5 Ж 20 0,116
5 Ж 70-80 1,П
10 ж 20 0,11
10 ж 70-80 1,49
Насыщ. ж 20 0,019
Алюминиевые сплавы
АК12 (АЛ2) 2 ж 20 0,355
Насыщ. ж 20 0,026
(Al, Mg: 1,5-2,5 %, Мп: 1,0-2,0 %) 2 ж 20 1,19
Насыщ. ж 20 0,019
Золото < 10 ж 20—Ткип <0,1
Медь < 10; без кислорода ж 20 <0,1
воздуха
Никель Насыщ. ж 20 <0,1
То же ж 100 < 1,0
262
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Кислота щавелевая НООССООН (продолжение)
Никелевые сплавы
хастеллой В 10 Ж 20-70 <0,1
хастеллой С 5-50 Ж 20- Т’кип <0,1
Ниобий < 10 Ж 20— Т’кип <0,1
Олово 0,75; без кислорода Ж 20 0,001
воздуха
Платина < 10 Ж 20-Т’кип <0,1
Свинец 0,6 Ж 20 0,01
1-10 Ж 20 <0,1
Серебро < 10 Ж 20-Т’кИП <0,1
Стали
углеродистая 6,3 Ж 20 0,41
12X13 10 Ж 20 < 1,0
10-50 Ж Т’кип > 10,0
12Х18Н10Т 10 Ж 20 <0,1
10 Ж 102 2,0
10-50 Ж Т’кип > 10,0
10Х17Н13М2Т 3-6 Ж 20 <0,11
10 Ж 102 1,4
Тантал < 10 Ж 20-Т’кИП <0,1
Насыщ. ж 20 <0,001
Титан 1 ж 35 <0,01
1 ж 60 0,49
0,5-10,0 ж 60 >1,3
0,5-10,0; ж 35 <0,13
аэрированная
Цинк 0,01-0,10 ж 20 > 10,0
Цирконий 0,5-10,0; ж 35-100 <0,02
аэрированная
25;аэрированная ж 60-100 0,10-0,31
Чугун СЧ15 Насыщ. ж 20 <0,1
То же ж 100 < 1,0
Антегмит < 10 ж 20-7 1 кип С
Графит < 10 ж 20-Т’кИП С
Полиамиды 10 ж 20 н
Поливинилхлорид < 10 ж 20-50 С
Полиизобутилен < 10 ж 20-100 С
Полиметилметакрилат < 10 ж 20-60 С
Полипропилен <10 ж 20 С
< 10 ж 60 ОС
Полиэтилен < 10 ж 20-60 С
Резины на основе
БК < 10 ж 20-80 С
Электродные процессы
263
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Кислота щавелевая НООССООН (продолжение)
Резины на основе
СКУ < 10 Ж 20-60 Н
СКФ <10 Ж 20-70 С
ХП < 10 Ж 20-95 С
ХСПЭ < 10 Ж 65 С
Стекло < 10 Ж 20—Ткип С
Фторопласт 4 < 10 Ж 20-100 С
Эмаль кислотостойкая < 10 Ж 20—Ткип С
Лития хлорид LiCl
Алюминий — Т 25 > 10,0
10 Ж 25 > 10,0
Медь Без кислорода Т 25-100 < 1,3 т
воздуха
Безводный Ж 650 5,3
То же Ж 700 > 10,0
10-90; без кислорода Ж 25-100 < 1,3 т
воздуха
Медные сплавы
БрОФ6,5-0,15, БрОФ7,2-0,2 10-90; без кислорода Ж 25-100 < 1,3 т
воздуха
Л80, ЛО68-1 10-40; без кислорода Ж 25-105 < 1,3
воздуха
Никель Безводный Ж 650 4,0
То же Ж 700 9,0
10-40; без кислорода Ж 25-100 <0,05
воздуха
Никелевые сплавы
НМЖМц28-2,5-1,5 То же Ж 25-105 <0,05
ХН78Т — Т 50-105 < 0,05 т, кр
10-40 Ж 25-105 < 0,05 т, кр
Вода: 0,5-1,5 Т 150-200 <0,01
10-40 Ж 25-105 <0,05
Н70МФ Вода: 0,5-1,5 Т 150-200 0,01
10-40 Ж 25-100 <0,05
Платина — Т 125-450 <0,05
10-40 Ж 25-105 <0,05
Свинец — т 25 <0,05
— т 55 <0,5
— т 75 < 1,3
10-40 ж 25 <0,05
10-40 ж 55 <0,5
10-40 ж 75 < 1,3
Серебро 10-40 ж 25-105 < 1,3
264
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Лития хлорид LiCl (продолжение)
Стали
углеродистая Безводный Ж 650 > 10,0
10 Ж 21 <0,5
10 Ж Ткип > 10,0
40 Ж 140 10,0
12X13 Вода: 0,5-1,5 Т 150-200 <0,1 т
12Х18Н10Т То же Т 150-200 <0,1 т
10 Ж Ткип < 0,1 т
10Х17Н13М2Т — Т 25 < 0,05 т, кр
Вода: 0,5-1,5 Т 150-200 < 0,01 т, кр
10 Ж Ткип <0,1 т
06ХН28МДТ Вода: 0,5-1,5 Т 150-200 <0,01
10-40 Ж 25-100 <0,05
Тантал 10-40 Ж 25-105 <0,05
Титан Вода: 0,5-1,5 Т 150-200 <0,01
— Ж 625 > 10,0
40; pH 6 ж 120 <0,001
10-40 ж 25-105 <0,05
50 ж 150 <0,01
Цинк 30 ж 115 > 10,0
Цирконий 10-40 ж 25-100 <0,05
Чугуны
серый <45 ж 20-Ткип 1,0-(> 10,0)
СЧ15, СЧ17 10-40 ж 25-100 <0,5
(Ni: 20 %) 30 ж 20-75 <0,05
30 ж 100 < 1,3
Графит 1-насыщ. ж 20-Г„п С
Графит, пропитанный феноло- То же ж 20-Ткип С
формальдегидным олигомером
Полиамиды <45 ж 20 С
Поливинилхлорид <45 ж 20 С
Полиметилметакрилат <45 ж 20 н
Полипропилен 1-насыщ. ж 20-60 С
Полиэтилен <45 ж 20 С
Резины на основе <45 ж 20 С
БК, НК, СКН,СКС, ХП
Стекло 1-насыщ. ж 20-Т 1 кип С
Фторопласт 4 То же ж 20-Ткип С
Эмаль кислотостойкая <45 ж 20 С
Магния хлорид MgCI2
Алюминий Сухой т 20 <0,001
Влажный т 20 0,002 я
10 ж 20 0,0038
10 ж 50 0,1 я
10 ж 100 0,3 я
Алюминиевые сплавы
АК12(АЛ2) 10 ж 20 0,0076 я
Электродные процессы
265
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т. °C
Магния хлорид MgCl2 (продолжение)
Алюминиевые сплавы
(Al, Mg: 1,2-2,5 %; Мп: 1,0-2,0 %) 10 Ж 20 0,0054
Золото — Ж 710 > 10,0
1-насыщ. Ж 20-ТКЙП <0,1
Медь — Ж 705 > 10,0
5 Ж 183 > 10,0
10-40; без кислорода воздуха Ж 25-95 <0,5
42 Ж 120 0,012
Медные сплавы
БрА5, БрА7, БрАЖН 10-4-4 10-30; без кислорода воздуха Ж 25 < 0,5 кр
БрОФ6,5-0,15, БрОФ7,2-0,2 То же Ж 25-95 <0,5
Л59, Л63, Л68 » Ж 25 < 0,5 кр
Никель — Ж 710 > 10,0
0,6 Ж 20 0,15
10—40; без кислорода воздуха Ж 25-105 <0,05
42 Ж 120 0,095
Никелевые сплавы
НМЖМц28-2,5-1,5 Без кислорода воздуха Т 25 < 0,5 кр
10—30; без кислорода воздуха Ж 25-105 < 0,5 кр
42 Ж 120 0,11 я
ХН78Т 10—40; без кислорода воздуха Ж 25-125 < 0,05 кр
Н70МФ — Т 25-100 <0,05
10-40 Ж 30-105 <0,05
30 Ж 125 <0,05
42 Ж 120 <0,001
ХН65МВ — Т 25-100 <0,05
10-32,5 Ж 30-125 <0,5
40 Ж 30-105 <0,05
Олово 1 ж 20 2,8
Платина — ж 710 > 10,0
1-насыщ. ж 20-Гкип <0,1
Свинец <35 ж 20 <0,5
<35 ж 100 > 10,0
Серебро 10-40 ж 20-105 <0,05
Стали
углеродистая Безводный ж 800 > 10,0
42 ж 120 0,35 я
266
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Магния хлорид MgCl2 (продолжение)
Стали
углеродистая Насыщ. Ж 85 0,15
12X13 2,5-10 Ж 20 < 0,1 т
10-30 Ж 20 < 10,0
42 Ж 135 < 0,03 кр
Насыщ. Ж 85 0,052
12Х18Н10Т 2,5; 10-30 Ж 20 <0,1 т
14 Ж 50 0,09
36 Ж 70 <0,01
48 Ж 165 0,1 кр
10Х17Н13М2Т 2,5; 10-30 Ж 20 <0,1 т
42 Ж 120 0,02 кр
06ХН28МДТ 2,5 Ж 20 <0,1 т
42 Ж 120-Ткип 0,01 кр
Тантал 10-40 Ж 25-105 <0,05
42 Ж 120 0,001
Титан 5^42 Ж Ткип <0,13
Титановые сплавы
4200 30 ж 120-160 <0,001
4207 30 ж 120 <0,001
Цинк 1,5 ж 20 0,2
Цирконий 1-насыщ. ж 20-Т^ <0,08
Чугуны
серый 42 ж 120 0,12
СЧ15 1-насыщ. ж 20—Ткип <0,1
Антегмит <35 ж 20-60 С
Графит — ж 720 с
1-насыщ. ж 20-100 С
Графит, пропитанный феноло- То же ж 20—Ткип с
формальдегидным олигомером
Полиамиды » ж 20-60 С
Поливинилхлорид 10 ж 40 С
<35 ж 60 ОС
Полиизобутилен 1-насыщ. ж 20-60 С
<35 ж 100 ОС
Полиметилметакрилат 1-насыщ. ж 20-60 с
Полипропилен <35 ж 20-100 с
Полиэтилен 1-насыщ. ж 20-60 с
Резины на основе
БК, ХП <35 ж 20-100 с
НК, СКС <35 ж 20-70 с
ПС <35 ж 20 ОС
СКН <35 ж 20-60 с
Стекло 1-насыщ. ж 20-Ткип с
Фторопласт 4 То же ж <200 с
Эмаль кислотостойкая <35 ж 20—Ткип с
Электродные процессы
267
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Меди сульфат CuSO4
Алюминий < 17,5 Ж 20 > 10,0
Алюминиевый сплав
(Al, Si: 4,5-6,0 %) 1-10 Ж 20 0,28
Иридий — Ж 100 < 0,005
Медь < 17,5 Ж 20 <0,1
Никель < 17,5 Ж 20 <0,5
< 17,5 Ж юо-ткип > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 < 17,5 Ж 20 <0,1
Н70МФ < 17,5 Ж 20-100 <0,1
Палладий — Ж 100 <0,01
Платина — Ж 100 <0,05
Родий — Ж 100 < 0,005
Рутений — Ж 100 < 0,005
Свинец < 17,5 Ж 20-100 <0,1
Серебро < 17,5 Ж 20-100 <0,1
Стали
углеродистая < 17,5 Ж 20 > 10,0
12X13 1-насыщ. Ж 20 <0,01
То же Ж Т’кип <0,1
12Х18Н10Т » ж 20 <0,001
» ж Т 1 кип <0,01
10Х17Н13М2Т » ж 20-Г * кип <0,01
06ХН28МДТ » ж т * кип <0,001
Тантал < 17,5 ж 20-Г 1 кип <0,001
Титан < 17,5 ж 20-Ткип <0,1
Цинк < 17,5 ж 20 > 10,0
Чугуны
серый < 17,5 ж 20 > 10,0
СЧ15 25 ж 20 0,02
Насыщ. ж 100 <0,1
Антегмит < 17,5 ж 20-60 с
< 17,5 ж 100—Ткип ОС
Поливинилхлорид < 17,5 ж 20 с
< 17,5 ж 60 ОС
Полиизобутилен < 17,5 ж 20-100 с
Полиметилметакрилат < 17,5 ж 20-60 с
Полипропилен < 17,5 ж 20-60 с
Полиэтилен < 17,5 ж 20-60 с
Резины на основе
БК, СКН, СКС < 17,5 ж 20-60 с
НК, ХСПЭ < 17,5 ж 20-80 с
ПС, СКФ, хп < 17,5 ж 20 с
Стекло < 17,5 ж 20—Ткип с
Фторопласт 4 < 17,5 ж 20-60 с
Эмаль кислотостойкая < 17,5 ж 20—Ткип с
268
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Метиленхлорид СН2С12
Алюминий Безводный Ж 25 0,1
Вода: 0,2 Ж 25 0,1
Алюминиевый сплав АМгб Безводный Ж 25 0,1
Вода: 0,2 Ж 25 0,1
Медь Безводный Ж 25 <1,0
— Г 50 <1,3
Медные сплавы
БрА7 Безводный Ж 25 < 1,0
— Г 50 < 1,3
БрОФ6,5-0,15, БрОФ7,2-0,2 — Г 50 <1,3
(Си, Si: 6,5 %) Влажный Ж 25 < 1,0
Л59, Л63, Л68 — Г 50 <1,3
Л80 Безводный Ж 25 < 1,0
Никель Безводный Ж 25-40 <0,1
и влажный
— Г 55-100 <0,5т
Никелевые сплавы
НМЖМц28-2,5-1,5 Безводный Ж 25 <0,1
и влажный
ХН78Т — Ж 25 <0,05
— Г 50 <0,05
Н70МФ — Ж 25 < 0,05 т
— Г 50-100 < 0,05 т
ХН65МВ — Г 50-100 < 0,05 т
Платина Безводный ж 25-40 <0,04
Свинец Безводный ж 25 <0,08
и влажный
Серебро Безводный ж 25-40 <0,08
Стали
СтЗ Безводный ж 25 <0,01
Вода: < 0,2 г 50-75 <0,5
08X13 — ж 40 <0,1
12ХГ8Н10Т Безводный ж 20-40 <0,001
Вода: 0,2 ж 25 <0,1
— г 75 < 0,5 т, кр
— г 120 > 10,0
10Х17Н13М2Т
06ХН28МДТ Безводный ж 25-40 <0,1
и влажный
Тантал Безводный ж 25-40 <0,05
Титан Тоже ж 25-40 <0,2
— г 50-100 <0,05
Чугуны
серый Безводный ж 25 < 1,1
Электродные процессы
269
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Метиленхлорид СН2С12 (продолжение)
Чугуны
серый Сухой Г 55-105 <0,5
СЧ15 Безводный Ж 25-40 <0,1
Влажный Ж 25 <0,1
(Ni: 20 %) Сухой Г 95 <0,5
Х28Л Безводный Ж 25 <0,1
и влажный
Антегмит ATM-1 — Ж <40 С
Асбест — ж 25-40 с
Графит, пропитанный феноло- — ж <40 с
формальдегидным олигомером
Полиамиды — ж 20 н
Поливинилхлорид — ж 20 н
Полиизобутилен — ж 20 н
Полиметилметакрилат — ж 20 н
Полипропилен — ж 25 н
Полиэтилен — ж 20 н
Резины на основе
БК, СКН, СКС, СКУ, СКФ, СКЭП, — ж 25 н
ХП, ХСПЭ
НК, СКИ — ж 20 н
Стекло — ж 20 с
Фторопласт 4 — ж 20 с
— г 150 с
Эмаль кислотостойкая — ж <40 с
Метилхлорид СН3С1
Алюминий Сухой и влажный г 20 <0,001
Алюминиевый сплав АМгб Сухой г 20-60 <0,1
Золото — ж 20-300 <0,05
Медь Сухой и влажный г < 100 <0,08
Медные сплавы
БрА5, БрА7 То же г < 150 <0,08
(Cu, Si: 6,5 %) » г < 100 <0,08
Л59, Л63, Л68 Влажный г 25 <3,0
Л80 Сухой г < 150 <0,1
Влажный г Ткип <0,1
Никель Сухой и влажный г <300 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 То же г <300 <0,1
ХН78Т » г <300 <0,1
Н70МФ — г 20-60 <0,1
Платина — г <300 <0,04
Свинец Сухой и влажный г < 150 <0,08
270
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Метилхлорид СН3С1 (продолжение)
Серебро Сухой и влажный Г <300 <0,08
Стали
углеродистая Сухой Г < 100 <0,1
Вода: 0,02 Г 15-50 0,0001
Вода: до насыщения Г 15-50 0,04-0,08
То же Г 60 0,11
» Г 70 0,36
12X17 Сухой г 150 <0,1
12Х18Н10Т Безводный ж -30 <0,05
Сухой г < 150 <0,1
Вода: г 15-80 0,001-0,07
до насыщения
10Х17Н13М2Т Сухой г < 100 <0,1
06ХН28МДТ То же г < 150 <0,05
Влажный г 40 <0,1
Тантал — г 55-105 <0,05
Титан — г 20-100 <0,1
Влажный г Ткип <0,001
Цинк Сухой г 20 <0,1
Чугуны
Серый То же г 100 <0,1
Вода: г 15 0,03
до насыщения
То же г 70 0,3
СЧ15 Сухой г < 150 <0,1
Влажный г < ткип <0,1
Асбест — г 20-100 С
Графит Сухой г < 120 С
Графит, пропитанный феноло- — г 20 с
формальдегидным олигомером
Полиамиды — г 20 с
Поливинилхлорид — г 25 н
Полиизобутилен — г 20 н
Полиметилметакрилат — г 20 н
Полистирол — г 20 н
Полиэтилен — г 20 н
Резины на основе
БК — г 20 с
НК, ПС, СКН, СКС, ХП — г 25 н
СКТ, СКФ — г 20 н
Стекло — г 20 с
Фторопласт 4 — г 20 С
Эмаль кислотостойкая — г 20 с
Электродные процессы
271
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия гидроксид NaOH
Алюминий <52 Ж 20 > 10,0
— Ж 320 1,08
Алюминиевый сплав АК12 (АЛ2) 5 Ж 20 > 10,0
Вольфрам <52 Ж < 100 <0,1
Золото — Ж 500 < 0,005
Иридий — Ж 500 <0,5
Медь 4 Ж 20 0,06
50 Ж 35 <0,001
<50 Ж 82 <0,06
— Ж 400 20,0
Медные сплавы
(Си, А1: 9 %) 0,2-20 Ж 95 0,83-1,92
(Си, Si: 15 %) 35 Ж 100 <0,001
(Си, Sn: 5 %) 50 Ж 80 0,5
(Си, Zn: 15 %) 4 Ж 20 0,05
30 Ж 60 0,4
(Си, Zn: 29 %; Sn: 1 %) 4 Ж 20 0,15
Молибден <52 Ж < 100 <0,001
Никель 14 Ж 88 0,005
20 Ж 20 <0,01
30-50 Ж 82 0,002
— Ж 500 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 <52 Ж 30-110 < 0,005
— Ж 320 1,37
Н70МФ <52 Ж 20-100 <0,1
хастеллой В 10 Ж 121 0,5
50 Ж Т’кип 0,05
хастеллой С 30; 70 Ж Т’кип 0,5
Олово 0,8 ж 20 0,22
Осмий — ж 500 <5,0
Платина <90 ж < Т’кип <0,1
Родий — ж 500 <0,5
Свинец 20 ж 20 1,9
Серебро 20 ж 40 2,0
<95 ж < Т’кип <0,1
— ж 400 0,015
— ж 800 3,0
Стали
СтЗ 1 ж 70 0,01
10 ж 70 0,01
40 ж 70 0,29 кр
12X13 20 ж 20 <0,1
12Х18Н10Т 1 ж 70 <0,01
272
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия гидроксид NaOH (продолжение)
Стали
12Х18Н10Т 10 Ж 70 0,01
40 Ж 70 0,01
~90 Ж 280 0,94 кр
— Ж 320 < 1,0 кр
10Х17Н13М2Т 1 Ж 70 <0,01
10 Ж 70 0,01
40 Ж 70 0,01
40 Ж 200 0,2
— Ж 320 < 1,0
06ХН28МДТ <52 Ж 20—Гкип <0,1
Тантал 40 Ж 20 0,25
— Ж 350 >0,3
Титан 10-40 Ж 100 <0,05
20 Ж Гкип <0,05
50-70 Ж 25-100 <0,5
— Ж 350 >0,3
Цирконий 1 Ж 60 <0,001
5-10 Ж Гкип <0,013
10-73 Ж < 100 < 0,025
Чугуны
серый 14 Ж 100 0,05
35-50 Ж 88 0,17
50 Ж Гкип 0,45
СЧ15 2 Ж 100 0,01
12 Ж 100 < 1,0
20 Ж Гкип < 10,0
50 Ж 80 1,3
— Ж 318 > 10,0
Антегмит ATM-1 <52 Ж 20 н
Графит, пропитанный бакелито- <52 ж 20 н
вым лаком
Графит, пропитанный феноло- <52 ж 20 н
формальдегидным олигомером
Полиамиды 52 ж <60 с
Поливинилхлорид <52 ж 20-50 с
Полиизобутилен <50 ж 20-60 с
Полиметилметакрилат <52 ж 20-60 с
Полипропилен <52 ж 20-60 с
Полиэтилен <52 ж 20-60 с
Резины на основе
БК, НК, СКС <52 ж 20-70 с
СКУ 10 ж 20 н
Электродные процессы
273
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия гидроксид NaOH (продолжение)
Резины на основе
СКФ <52 Ж 20-90 С
хп <52 Ж 20-100 С
хспэ <52 Ж 20-116 С
Стекло «Пирекс» <52 Ж 20 с
Фторопласт 4 <52 Ж 20-Ткип с
Эмаль кислотостойкая <52 Ж 20 с
Натрия гипохлорит NaClO
Алюминий Влажный Т 25 > 10,0
10; pH >7 Ж 25 > 10,0
Медь 2 Ж 20 <0,08
20 Ж 20 > 10,0
Медные сплавы
БрА5, БрА7 10 Ж 20 > 10,0
Л59, Л63, Л68, Л80, ЛО68-1 10 Ж 20 > 10,0
Никель <34 Ж 20 0,1-3,0
Никелевые сплавы
НМЖМц28-2,5-1,5 <34; Ж 20 0,007
активный хлор: 3
Н70МФ <34 Ж 35-100 < 0,004
Платина <34 Ж < 100 <0,1
Свинец <34; Ж 20 0,54
активный хлор: 1
<34; Ж 40 1,4
активный хлор: 1
Серебро <34 Ж 20 <0,1
Стали
СтЗ Безводный Т 25-30 <0,05
0,1; pH > 10 Ж 20 <0,1
>0,1 Ж 25 > 10,0
12X17 5 Ж 20 > 10,0
12Х18Н10Т 5 Ж 20 > 10,0
10Х17Н13М2Т <34; Ж 40 <0,001
активный хлор: 2
<34; Ж 7™п 1,0-3,0
активный хлор: 2
06ХН28МДТ <34 Ж 20—Гют <0,1
Тантал <34 Ж 20 <0,05
Титан 10-20 ж 25-105 <0,05
40 ж 25 <0,05
Цирконий 10 ж 30-110 <0,05
20 ж 30 <0,05
Чугуны
серый <0,1; pH >7 ж 25 <0,05
274
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия гипохлорит NaCIO (продолжение)
Чугуны
серый >0,1 Ж 25 > 10,0
СЧ15, СЧ17 <34 Ж 25-105 < 1,3
Асбест 14 Ж 20-100 с
Графит, пропитанный феноло- <25 Ж Гкип с
формальдегидным олигомером
Полиамиды <34 Ж 20-60 с
Поливинилхлорид <34 Ж 20 с
<34 Ж 65 ОС
Полиизобутилен <34 Ж 20 с
<34 Ж 60 ОС
<34 Ж 100 н
Полиметилметакрилат <34 Ж 20 с
Полипропилен <34 Ж 20-60 с
Полиэтилен <34 Ж 20-60 с
Резины на основе
БК 10 Ж 20-65 с
Насыщ. Ж 65 с
НК 10-30 Ж 65 с
СКТ 1-насыщ. Ж 20-100 с
СКФ <34 Ж 20-93 с
ХП 20 Ж 24 ОС
Насыщ. Ж 65 н
ХСПЭ <34 Ж 20-60 с
Стекло <34 Ж 20-60 с
Фторопласт 4 1-насыщ. Ж 20-100 с
Эмаль кислотостойкая То же Ж < 100 с
» Ж ткип ОС
Натрия карбонат Na2CO3
Алюминий Влажный Т 20 <0,1
~0,05 Ж 20 0,48
= 0,25 ж 20 4,54
= 0,5 ж 20 5,7
= 2,5 ж 20 > 10,0
Медь < 17 ж 20-Ткнп <0,1
Латунь Л68 = 10 ж 20 <0,001
Никель 1-20 ж 20-100 <0,1
— ж 900 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 < 17 ж 20— Т’кип <0,1
< 17 ж 135 0,04
Н70МФ < 17 ж 20—Гкип <0,1
Электродные процессы
275
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия карбонат Na2CO3 (продолжение)
Свинец < 17 Ж 20 <0,1
< 17 Ж 60-100 > 10,0
Серебро < 17 Ж 20-100 <0,1
Стали
СтЗ 10 Ж 70 0,04
Насыщ. Ж 104 0,066
12X13 = 22 ж 80 <0,01
31 ж Ткип <0,1
— ж Т’пл > 10,0
12Х18Н10Т 10 ж 70 <0,01
< 17 ж Т 1 кип <0,1
31 ж т 1 кип <0,1
— ж Ткл > 10,0
10Х17Н13М2Т 10 ж 70 <0,01
< 17 ж 20—Ткип <0,1
31 ж т 1 кип <0,1
— ж 900 > 10,0
06ХН28МДТ < 17 ж 20-Ткип <0,1
— ж 900 <0,1
Тантал < 17 ж 20-100 <0,1
Титан 20 ж Ткип <0,13
40 ж 100 <0,05
Чугуны
серый < 17 ж 20—Ткип <0,1
10,6 ж 104 0,13
Насыщ. ж 104 0,50
СЧ15, СЧ17 < 17 ж 20 <0,1
< 17 ж 100—Ткип 0,1-1,0
— ж 900 > 10,0
Антегмит АТМ-1 1-насыщ. ж 20—Ткип С
Полиамиды < 17 ж 20-60 С
Поливинилхлорид 1-насыщ. ж 20-60 С
Полиизобутилен < 17 ж 20-60 С
Полиметилметакрилат < 17 ж 20-60 С
Полипропилен < 17 ж 20-60 с
Полиэтилен 1-насыщ. ж 20-60 С
Резины на основе
БК < 17 ж 20-80 С
НК, СКН, СКС < 17 ж 20-60 С
ХП < 17 ж 20-90 С
ХСПЭ < 17 ж 20 С
Стекло < 17 ж 20 С
Фторопласт 4 1-насыщ. ж < 100 С
Эмаль кислотостойкая < 17 ж 20 С
Tib
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия нитрат NaNO3
Алюминий 5 Ж 20 0,03
<47 Ж 20-100 <0,001
Алюминиевый сплав АК12 (АЛ2) 5 Ж 20 0,01
Золото — Ж Тпл <0,01
Иридий — Ж Tki < 0,005
Медь <47 Ж 100 0,5-1,27
— Ж 400 8,0
— Ж 450 5,3
— Ж 500 3,2
Молибден — Ж 350 > 10,0
Никель <47 Ж 20-100 <0,1
— Ж 400 0,08
— Ж 450 0,27
— Ж 500 0,40
Никелевые сплавы
НМЖМц28-2,5-1,5 1-насыщ. Ж 20 <0,1
<47 Ж 60-100 0,1-1,0
Н70МФ <47 Ж 20-100 <0,1
<47 Ж 130 <0,5
Ниобий — Ж 350-600 > 10,0
Осмий — Ж Т 1 Ш1 > 10,0
Палладий — Ж т 1 пл <0,5
Платина — Ж т 1 пл <0,001
Родий — Ж т 1 пл <0,5
Рутений — Ж т 1 пл <0,5
Серебро — Ж Т„л > 10,0
Стали
углеродистая <47 Ж 20 <0,1
— Ж 400 1,2
— ж 450 2,1
— ж 500 3,45
12X13 1-насыщ. ж 20-Ткип <0,1
— ж 360 <0,1
12Х18Н10Т 1-насыщ. ж 20—Ткип <0,1
— ж 360 <0,1
— ж 400 0,25
— ж 450 0,46
— ж 500 0,60
10Х17Н13М2Т 1-насыщ. ж 20-Ткип <0,1
06ХН28МДТ То же ж 20—Ткип <0,1
Тантал <47 ж 20-Г 1 кип <0,001
— ж Тпл < 0,001
Титан Насыщ. ж 100 <0,05
Электродные процессы
277
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия нитрат NaNO3 (продолжение)
Цирконий — Ж 350 0,1
— Ж 400 0,08
— Ж 450 0,07
— Ж 500 0,05
— Ж 550 0,8
— Ж 600 1,8
Чугун серый <47 Ж 20-Т’кИП <0,1
Графит, пропитанный бакелита- 1-насыщ. Ж т 1 кип С
вым лаком
Полиамиды <47 Ж 20-60 С
Поливинилхлорид 1-насыщ. Ж <60 с
Полиизобутилен <47 Ж 20-60 с
Полиметилметакрилат <47 Ж 20-60 с
Полипропилен <47 Ж 20-60 с
Полиэтилен <47 Ж 20-60 с
Резины на основе
БК, НК, СКН, СКС <47 Ж 20-60 с
ХП <47 Ж 20-80 с
ХСПЭ <47 Ж 20 с
Стекло <47 Ж 20-100 с
Фторопласт 4 1-насыщ. ж <200 с
Эмаль кислотостойкая <47 ж 20-100 с
Натрия ортофосфат Na3PO4
Алюминий 1-10 ж 20-98 > 10,0
Алюминиевый сплав АК12 (АЛ2) 1-10 ж 20-98 > 10,0
Медь < 10,8 ж 20 <0,1
< 10,8 ж 100 0,1-1,0
Никель < 10,8 ж 20 <0,02
8 ж 100 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 8 ж 18 <0,1
Н70МФ < 10,8 ж 20-50 <0,05
< 10,8 ж 100 <0,1
< 10,8 ж Т’кип <0,5
Свинец < 10,8 ж 20 <0,1
Серебро < 10,8 ж 20 <0,1
— ж тт > 10,0
Стали
углеродистая < 10,8 ж 20 0,0016
< 10,8 ж 60— Т’кип <1,0
12X13 1-насыщ. ж 20-Т’кИП <0,1
12Х18Н10Т То же ж 20-Т’кИП <0,1
10Х17Н13М2Т » ж 20-Т’кИП <0,1
278
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия ортофосфат Na3PO4 (продолжение)
Стали
06ХН28МДТ 1-насыщ. Ж 20—Гкип <0,1
Титан < 10,8 Ж 20 <0,001
Насыщ. Ж 100 <0,05
Чугуны
серый < 10,8; pH > 7,2 Ж 20-Т * кип <0,1
СЧ15, СЧ17 1-насыщ. Ж Ткип <0,1
Антегмит < 10,8 Ж 60 С
Полиамиды < 10,8 Ж 20-80 С
Полиизобутилен < 10,8 Ж 100 С
Полиметилметакрилат < 10,8 Ж 20-50 с
Полипропилен < 10,8 Ж 20-60 с
Полиэтилен < 10,8 Ж 20-60 с
Резины на основе
БК, ХП < 10,8 Ж 20-100 с
НК, СКН, СКС < 10,8 Ж 20-65 с
СКУ < 10,8 Ж 20 н
ХСПЭ < 10,8 Ж 20-65 с
Стекло < 10,8 Ж 20 с
Фторопласт 4 < 10,8 Ж 20-100 С
Эмаль кислотостойкая < 10,8 Ж 20-60 с
Натрия перхлорат NaCIO4
Алюминий — Т 25-75 <0,05
10-40 Ж 25 < 1,3
Вольфрам — ж 487 > 10,0
Золото — ж 482 > 10,0
Медь 10-70 ж 105 <0,05
Медные сплавы
БрОФ6,5-0,15, БрОФ7,2-0,2 10-70 ж 105 <0,05
Молибден — ж 487 > 10,0
Никель — т 25-100 <0,05
10-70 ж 105 <0,05
Никелевые сплавы
НМЖМц28-2,5-1,5 10-70 ж 100 <0,05
ХН78Т 10-70 ж 100 <0,05
ХН65МВ 1-насыщ. ж Ткип <0,1
Палладий — ж 482 > 10,0
Платина — ж 482 > 10,0
10-70 ж 105 <0,05
Свинец 10-70 ж 105 <0,05
Серебро — ж 550 > 10,0
10-70 ж 100 <0,05
Электродные процессы
279
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия перхлорат NaClO4 (продолжение)
Стали
СтЗ 10 Ж 25 > 10,0
12X13 1-насыщ. Ж 20-Ткип <0,1
12Х18Н10Т — Т 30 <0,5
1-насыщ. Ж 20-7 кип <0,1
10Х17Н13М2Т То же Ж 20-Т 1 кип <0,1
06ХН28МДТ » Ж 20-Ткип <0,1
Тантал — Т 100 <0,05
— Ж 482 > 10,0
Титан — ж 482 > 10,0
10-70 ж 100 <0,05
Насыщ. ж 100 <0,05
Цирконий — ж 487 > 10,0
Чугуны
серый 10 ж 25 > 10,0
(Ni: 20 %) 10 ж 100 <0,05
Асбест 1-насыщ. ж 20-Ткин С
Графит 0,5 ж 20-100 С
Поливинилхлорид 1—насыщ. ж 50 С
Резины на основе
БК, НК, СКС, СКФ То же ж 65 С
ХП » ж 90 с
Стекло «Пирекс» » ж 482 С
Фторопласт 4 » ж Т’кип С
Натрия сульфат Na2SO4
Алюминий 10 ж 20 0,011
10 ж Т’кип <0,1
Алюминиевый сплав АК12 (АЛ2) 20 ж 20 <0,01
Золото — ж 884 <0,1
Иридий — ж 884 < 0,005
Медь < 19,4 ж 20-Т’кИП <0,1
Медные сплавы
(Си, Sn: 3,42 %) 10 ж 20 <0,001
(Си, AI: 9 %; Fe 8 %) 10 ж 20 0,001
(Си, Zn: 35,67 %) 10 ж 20 0,0014
Никель < 19,4 ж 20 <0,1
< 19,4 ж 100 0,1-1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 < 19,4 ж 20-100 <0,1
ХН78Т < 19,4 ж 20-100 <0,1
Осмий — ж 884 <0,5
Палладий — ж 884 <5,0
Платина — ж 884 > 10,0
280
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия сульфат NA2SO4 (продолжение)
Родий — Ж 884 <5,0
Рутений — Ж 884 <0,5
Свинец < 19,4 Ж 20 <0,01
< 19,4 Ж 100 0,1-1,0
Серебро — Ж 884 > 10,0
Стали
углеродистая 10 Ж 20 0,05
14,2 Ж 85 0,11
12X13 < 19,4 Ж 20 <0,1
15X28 1-насыщ. Ж Ткип <0,1
12Х18Н10Т То же Ж Т л кип <0,1
10Х17Н13М2Т » Ж Ткип <0,1
Тантал < 19,4 Ж 20-100 < 0,001
Титан Насыщ. Ж Ткип <0,05
Цинк 1 Ж 20 0,08
Чугуны
серый 10 Ж 20 0,04
14,2 Ж 85 0,16
СЧ15 < 19,4 Ж 100—Ткип <0,1
Антегмит 10 Ж 100 С
Графит, пропитанный бакелите- 10 Ж 100 с
вым лаком
Полиамиды < 19,4 Ж 20-60 с
Поливинилхлорид 10 Ж 60 с
Полиизобутилен 10 Ж 20-100 с
Полиметилметакрилат 10 Ж 20-60 с
Полипропилен < 19,4 Ж 20-60 с
Полиэтилен 10 Ж 20-60 с
Резины на основе
БК < 19,4 Ж 20-80 с
НК < 19,4 Ж 20-70 с
СКН, СКС < 19,4 ж 20-60 с
хп < 19,4 ж 20-100 с
хспэ < 19,4 ж 20 с
Стекло < 19,4 ж 20-100 с
Фторопласт 4 < 19,4 ж 20-60 с
Эмаль кислотостойкая < 19,4 ж 20-60 с
Натрия хлорат NaCIO3
Алюминий <50 ж 20 <0,1
<50 ж 110 < 0,009
Медь — т 20 <0,5
<50 ж ПО < 0,041
10-70 ж 25-105 <0,05
Электродные процессы
281
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия хлорат NaCIO3 (продолжение)
Медные сплавы
БрА5, БрА7 < 50; pH 6, 0 6, 5 Ж 110 < 0,026
БрАЖ9-1 < 50; pH 6, Об, 5 Ж но < 0,036
БрОФ6,5-0,15, БрОФ7,2-0,2 10-70 Ж 25-105 <0,05
Л62, ЛН65-5, ЛО70-1 < 50; pH 6, Об, 5 Ж 110 < 0,045
Никель < 50; pH 6,0 б,5 Ж по < 0,008
Никелевый сплав Н70МФ < 50; pH 6,0 6,5 Ж но <0,001
Свинец <50 Ж 20 0,1
<50 Ж 100 0,10-0,16
Серебро <50 Ж 20-Т^ <0,1
Стали
СтЗ <50 Ж <40 < 0,002
<50 Г <40 0,034 я
<50 Ж 90 0,001-0,034 я
<50 Г 90 0,96-1,10 я
12X13 <50 ж 20 0,1-1,0
12Х18Н10Т <50 ж Ткип < 1,0
10Х17Н13М2Т — т 20 <0,5
10-40 ж 25-100 <0,5
60-70 ж 100 <0,5
06ХН28МДТ 10-40 ж 20-100 <0,5
60-70 ж 100 <0,5
Тантал <50 ж 95 < 0,002
Титан 10 ж 25-100 <0,05
40 ж 25 <0,05
— ж Тпл <0,1
Графит 1-насыщ. ж 100 С
Графит, пропитанный феноло- <50 ж 100 с
формальдегидным олигомером
Полиамиды <50 ж 20-60 с
Поливинилхлорид <50 ж 20-40 с
Полиизобутилен <50 ж 20-100 с
Полиметилметакрилат <50 ж 20-60 с
Полипропилен <50 ж 20-60 с
Полиэтилен <50 ж 20-60 с
Резины на основе
БК 50 ж 88 с
СКН <50 ж 20 ОС
СКС <50 ж 60 с
ХП <50 ж 20 с
ХСПЭ <50 ж 20 с
Стекло <50 ж 20-100 с
Фторопласт 4 <50 ж 20-100 с
Эмаль кислотостойкая <50 ж 100 с
282
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия хлорид NaCI
Алюминий Влажный Т 220 0,021
— Ж 800 > 10,0
3 Ж 20 0,02
3 Ж 98 0,26 я
10 Ж 20 0,02
10 Ж 98 0,22 я
25 Ж 20 0,01
25 Ж 98 > 10,0
Насыщ. Ж 20 0,037
Алюминиевые сплавы
АМг2 0,1 Ж 45 0,031
Насыщ. Ж 20 0,043
Амг5 3 Ж 80 0,02
Ванадий 3 Ж 25 0,05
Вольфрам 1-насыщ. Ж 20-Ткип <0,1
Золото То же Ж 20-Ткип <0,1
Медь — Ж 800-850 > 10,0
1-насыщ. Ж 20-7^ <0,08
Медные сплавы
БрА5, БрА7 <26,4 Ж 20 <0,1
БрАЖМц 2 Ж 80 0,2
5 Ж 80 0,4
Л70 <26,4 Ж 20 <0,1
Молибден 1-насыщ. Ж 20-Ткип <0,1
Никель — Ж 800-950 1,0-3,5
26 Ж 100 <0,01
Никелевые сплавы
НМЖМц28-2,5-1,5 23 Ж 20-70 <0,02
Насыщ., Ж 75 2,25
pH 2,0-4,5
Н70МФ <26,4 Ж 20 <0,001
ХН65МВ 26 Ж 20-70 <0,001
хастеллой С Насыщ. ж 20-70 <0,001
Ниобий — ж 950 > 10,0
3 ж 25 <0,001
Олово 1-насыщ. ж 20 <0,1
Платина — ж 801 <0,05
1-насыщ. ж 20-Ткип <0,05
Свинец 6 ж 20 0,1
Серебро — ж 801 > 10,0
1-насыщ. ж 20-Ткип <0,08
Стали
СтЗ — ж 880 4,1
4 ж 90 <0,4
Электродные процессы
283
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Натрия хлорид NaCl (продолжение)
Стали
СтЗ 26 Ж 96-98 0,047
Насыщ. Ж 25 0,08
12X13 — Ж 850 7,3
10 Ж 20 0,034
12Х18Н10Т — Т 90 0,001 кр
— Ж 880 5,4
20 Ж 20 0,002 т
20 Ж Т'кип < 0,1 т, кр
26 Ж 75 0,2 т, кр
Насыщ. Ж 35 0,03 т, кр
10Х17Н13М2Т 3 ж 20 < 0,05 т
20 ж 20 < 0,1 т
20 ж Т 1 кип <0,1 т
06ХН28МДТ 3;аэрированная ж 35-Г <0,1
Насыщ., ж 35-7 <0,1
аэрированная
Тантал 1-насыщ. ж 20-7 1 кип <0,1
Титан 20 ж 7 1 кип <0,001
Насыщ. ж Т’кип 0,0013
Цирконий Над расплавом Аг ж 850 5,0
1-насыщ. ж 20—Ткип <0,1
Чугуны
серый 3 ж 50 0,2
Насыщ. ж 93 1,8
СЧ15 1-насыщ. ж 7 1 кип <0,1
Антегмит АТМ-1 То же ж < 7 1 кип С
Графит » ж Ткип с
Графит, пропитанный бакелито- Насыщ. ж 20-60 с
вым лаком
Графит, пропитанный феноло- 1-насыщ. ж < 170 с
формальдегидным олигомером
Полиамиды <26,4 ж 20-60 с
Поливинилхлорид 1-насыщ. ж 60 ОС
Полиизобутилен <26,4 ж 20-60 с
Полиметилметакрилат 25 ж 60 с
Полипропилен <26,4 ж 20-60 с
Полиэтилен 26 ж 60 с
Резины на основе
БК 25 ж 100 с
30 ж 90 с
НК 1-насыщ. ж 70 с
СКС, ХСПЭ <26,4 ж 20-80 с
284
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Натрия хлорид NaCI (продолжение)
Резины на основе
СКФ 30 Ж 90 С
ХП 30 Ж 90 ОС
Стекло 1-насыщ. Ж ттп с
Фторопласт 4 <26,4 Ж 20-100 с
Эмаль кислотостойкая 1-насыщ. Ж 20 с
Никеля(П) сульфат NiSO4
Алюминий <28 Ж 20 > 10,0
Медь <28 Ж 20 > 10,0
Никель < 28; без кислорода Ж 20-100 0,1-1,0
воздуха
Никелевые сплавы
НМЖМц28-2,5-1,5 <28 Ж 20-100 <0,05
Н70МФ <28 Ж 20-70 <0,05
Свинец <28 Ж 20-100 <0,1
Серебро <28 Ж 20 <0,1
Стали
углеродистая Насыщ. Ж Ткип > 10,0
12X13 <28 Ж 100 > 10,0
12X17 Насыщ. Ж т * кип <0,1
12Х18Н10Т То же Ж Ткип <0,1
10Х17Н13М2Т <28 Ж 88 0,0025
Насыщ. Ж т 1 кип <0,1
06ХН28МДТ То же Ж Ткип <0,05
Тантал <28 Ж 20-150 < 0,001
Титан 28 Ж 130 <0,05
Чугуны
серый <28 Ж 20 > 10,0
СЧ15, СЧ17 <28 Ж 20-Т Z'V 1 кип <0,1
Графит, пропитанный феноло- 1-насыщ. Ж 20—Ткип С
формальдегидным олигомером
Полиамиды <28 Ж 20-60 с
Поливинилхлорид <28 Ж 20-65 с
Полиизобутилен <28 Ж 20-100 с
Полиметилметакрилат <28 Ж 20-65 с
Полипропилен <28 ж 20-100 с
Полиэтилен <28 ж 20-80 с
Резины на основе
БК, ХП <28 ж 20-100 с
НК, СКН, СКС <28 ж 20-65 с
ХСПЭ <28 ж 20-95 с
Стекло <28 ж 20 с
Фторопласт 4 <28 ж 20-120 с
Эмаль кислотостойкая <28 ж 20 с
Электродные процессы
285
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Никеля(11) хлорид NiCl2
Алюминий — Т 25 > 10,0
10 Ж 25 > 10,0
Медь 10 Ж 25 > 10,0
Медные сплавы
БрА5, БрА7 — т 25 > 10,0
10 ж 25 > 10,0
БрОФ6,5-0,15, БрОФ7,2-0,2 10 ж 25 > 10,0
Л59, Л63, Л68 10 ж 25 > 10,0
Никель <37,5 ж <70 <0,05
<37,5 ж 100 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 <37,5 ж <70 <0,05
<37,5 ж 100 > 10,0
ХН78Т — т 25-105 <0,5
Н70МФ — т 55 <0,05
10-30 ж 20-105 <0,05
Свинец <37,5 ж 20 > 10,0
Серебро <37,5 ж 20— 7КИП <0,1
Стали
СтЗ 10 ж 25 > 10,0
12X13 <37,5 ж 20 > 10,0
12Х18Н10Т <37,5 ж 20 < 0,1 т
10Х17Н13М2Т <37,5 ж 93 < 0,05 т
06ХН28МДТ <37,5 ж 20-80 < 0,05 т
Тантал <37,5 ж 20-150 <0,001
Титан 30; pH 3,5 ж 120 <0,001
37,5 ж 100 0,003
Титановый сплав 4200 30 ж 120 <0,001
Цирконий 5 ж 100 0,0003
20 ж 35-100 <0,001
Чугуны
серый 7,5 ж 25 > 10,0
СЧ15, СЧ17 10-30 ж 20-100 <0,5
Графит 1-насыщ. ж Гкип С
Полиамиды <37,5 ж 20-60 с
Поливинилхлорид <37,5 ж 20-60 с
Полиизобутилен <37,5 ж 20-70 с
Полиметилметакрилат <37,5 ж 20-60 с
Полипропилен <37,5 ж 20-100 с
Полиэтилен <37,5 ж 20-60 с
Резины на основе
БК, НК, СКН, СКС <37,5 ж 20-70 с
ХП, ХСПЭ <37,5 ж 20-95 с
286
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
х Никеля(П) хлорид NiCl2 (продолжение)
Стекло <37,5 Ж 20-Т’кИП С
Фторопласт 4 <37,5 Ж 20-100 С
Эмаль кислотостойкая <37,5 Ж 20 С
Серы диоксид so2
Алюминий — г 20 0,0014
— ж 20 < 0,001
— г 100-400 < 0,001
Вольфрам — г 700 > 10,0
Золото — г 600 <0,1
Медь — г 20-100 <0,1
— ж Ткип <0,1
Молибден — г 600 <0,1
— г 1160 > 10,0
Никель — г 20-300 <0,1
— г 700 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 Сухой и влажный г Ткип-300 <0,1
— г 700 > 10,0
Н70МФ Сухой и влажный г 20-750 <0,1
Олово — г Ткип-20 <0,1
Платина Сухой и влажный г 1000 <0,1
Свинец То же г Ткип-200 <0,1
Серебро » г <300 <0,1
Стали
СтЗ — г 70 <0,05
Влажный г 150 0,25-0,40
— г 500 7,5
12X13 — г 700 0,19
— г 900 0,34
12Х18Н10Т Сухой и влажный г Гкип-100 <0,11
— г 300 <0,1
Влажный г 500 < 1,0
— г 900 < 10,0
10Х17Н13М2Т Сухой и влажный г Ткип-ЮО <0,11
— г 200 <0,11
— г 300 <0,1
— г 500 < 1,0
— г 900 <3,0
06ХН28МДТ Сухой и влажный г Т’Кип~420 <0,1
Тантал — г <350 <0,1
Титан Сухой и влажный г 70 <0,1
Чугуны
серый — г 20 0,12
Электродные процессы
287
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Фаза Т,°С и характер коррозии
Давление, МПа Стойкость материалов
Серы диоксид SO2 (продолжение)
Чугуны
серый Вода: 0,1 Г 20 < 1,0
Вода: 0,3 Г 20 <3,0
— Г 700 > 10,0
СЧ15, СЧ17 — Г <750 <0,1
Влажный Г 20 <1,0
Антегмит АТМ-1 — Г < 170 с
Полиамиды — Г 20-50 ОС
Поливинилхлорид — Г <60 с
Влажный Г 40 с
Полиизобутилен — Г <80 с
Полиметилметакрилат — Г 20-100 с
Полипропилен — Г 20-80 с
Полиэтилен — Г 20-60 с
Резины на основе
БК — Г 20 с
НК — г 20 с
— г 50-80 ОС
СКИ, СКН — г 20-60 ОС
СКС — г 20 ОС
СКУ — г 20 с
— г 80 н
СКФ — г 20-100 с
ХП — г 20-60 с
ХСПЭ — г 20 н
Фторопласт 4 Сухой и влажный г < 100 с
Эмаль кислотостойкая — г < 150 с
Сероводород H2S
Алюминий Сухой и влажный г 20 <0,1
То же г <250 <0,1
Иридий Влажный г 18 <0,001
Медь Сухой г 20 <0,1
Влажный г 20 > 10,0
Медные сплавы
(Cu, А1: 5-9,5 %) Сухой г 20 <0,1
ЛЖМц59-1-1, ЛО60-1, Л62, ЛО68-1, Л68, ЛО70-1, Л80 То же г 20 <0,1
Никель » г 20 <0,1
Влажный г 20 0,1-1,0
— г 100 10,0
Влажный г 100 > 10,0
Никелевый сплав Сухой и влажный г 20 <0,1
НМЖМц28-2,5-1,5
288
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Сероводород H2S (продолжение)
Никелевый сплав Сухой Г 100 10,0
НМЖМц28-2,5-1,5 Влажный Г 100 > 10,0
Осмий То же Г 18 <0,001
Палладий » Г 18 < 0,005
Платина — Г < 1000 <0,1
Рутений Влажный Г 18 <0,001
Свинец — Г 20 <0,1
Серебро Влажный Г 18 <0,01
Стали
углеродистая Сухой Г 20 0,1-1,0
— Ж 20 <0,1
12X13 — Г 100 <0,1
— Г 200 < 10,0
Влажный г < 100 0,1-1,0
12Х18Н10Т — г <200 <0,1
— г >200 > 10,0
Влажный г 20 <0,1
10Х17Н13М2Т — г <200 <0,1
— г >200 > 10,0
Влажный г 20 <0,1
06ХН28МДТ — г 20-100 <0,1
Влажный г 20 <0,1
Титан Сухой и влажный г 100 <0,05
Чугуны
серый Влажный г 20 0,5-1,0
СЧ15, СЧ17 — г <900 <0,1
Влажный г 20-100 <0,1
Антегмит Сухой г 150 с
Графит, пропитанный бакелито- » г 100 с
вым лаком
Полиамиды » г 20 н
Поливинилхлорид » г 20-60 с
Сухой и влажный г 20-40 с
Влажный г 60 ОС
Полиизобутилен Сухой и влажный г 20-60 с
Полиметилметакрилат Сухой г 20-60 с
Полипропилен То же г 20-60 с
Полиэтилен Сухой и влажный г 20-60 с
Резины на основе
БК Сухой г 20-80 с
Влажный г 20-80 ОС
НК, СКФ, ХСПЭ Сухой г 80 с
Электродные процессы
289
Продолжение таблицы 1.4.62
Характеристика среды Скорость (мм/год)
Материал Концентрация, масс. % Давление, МПа Фаза т,°с и характер коррозии Стойкость материалов
Сероводород H2S (продолжение)
Резины на основе
СКИ, СКН Сухой и влажный Г 20 ОС
Влажный Г 60 Н
СКС Сухой Г 20 С
Влажный Г 20 ОС
То же Г 60 Н
СКУ Сухой Г 20 ОС
хп Влажный Г 20-70 С
Стекло Сухой и влажный Г 20-100 С
Фторопласт 4 То же Г < 100 С
Эмаль кислотостойкая » Г 20-100 С
Спирт метиловый (метанол) СН3ОН
Алюминий 2-20 Ж 20 <0,001
5 40 Ж 20 0,0004
75-99 Ж 20 0,002
Медь Медные сплавы 1-99 Ж 20-Т’кИП <0,1
БрОФ6,5-0,25 — Ж 20 <0,1
Л62, Л80, ЛО60-1, ЛМц58-2 1-99 Ж 20—Ткип <0,1
Никель — Ж Т’кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20 <0,001
Н70МФ — Ж 20—7^ <0,1
Свинец — Ж 20 <0,1
— Ж Т 1 кип > 10,0
Серебро Стали — Ж Т'кип <0,1
СтЗ — Ж 20 <0,1
— ж Т’кип <5,0
12X13 — ж 20 <0,01
1-99 ж 20—Ткип <0,1
12Х18Н10Т 1-99 ж 20—Ткип <0,1
10Х17Н13М2Т 1-99 ж 20—Ткип <0,1
06ХН28МДТ 1-99 ж 20—Ткип <0,1
Тантал — ж Т’кип <0,001
Титан — ж 35 0,1-1,0 мк, кр
Цинк Чугуны — ж 20—Ткип > 10,0
серый — ж 20—Ткип 0,1-3,0
СЧ15, СЧ17 — ж 20—Ткип <0,1
Антегмит АТМ-1 — ж < т 1 кип с
Графит, пропитанный феноло- — ж < т 1 кип ОС
формальдегидным олигомером
290
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Спирт метиловый (метанол) СН3ОН (продолжение)
Полиамиды — Ж 20 С
Поливинилхлорид — Ж 20-40 С
— Ж 60 ОС
Полиизобугилен — Ж 2 0455 С
Полиметилметакрилат — Ж 20 Н
Полипропилен — Ж 20-100 С
Полиэтилен — Ж 20 ОС
— Ж 52 Н
Резины на основе
БК, НК, СКН, СКС, ХП — Ж 20450 С
С КУ — Ж 20450 н
СКФ, ХСПЭ — Ж 20 с
Стекло — Ж 20-Ткип с
Фторопласт 4 — Ж Т 1 кип с
Эмаль кислотостойкая — Ж 20-Т 1 кип с
Спирт этиловый (этанол) С2Н5ОН
Алюминий 1-99 Ж 20-7 1 кип <0,1
— Ж 7 1 кип <0,01
Медь 1-99 Ж 20-7 ZrV— 1 кип <0,1
Ж 7 1 кип <0,1
Медные сплавы Л62, ЛС59-1, 1-99 Ж 20-Ткип <0,1
ЛЖМц59-1-1
Никель — Ж 20 < 0,005
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20450 <0,1
Н70МФ — Ж 20450 <0,1
Свинец — Ж 20 <0,1
Серебро — Ж 20 <0,1
Стали
углеродистая — Ж 20 <0,17
— Ж ТКИП <0,5
12X13 1-99 Ж 20—ТкИП <0,1
— ж 20-Ткип <0,1
12Х18Н10Т 1-99 ж 20-Ткип <0,1
— ж 20-7 1 КПП <0,1
10Х17Н13М2Т 1-99 ж 20—Ткип <0,1
— ж 20-Ткип <0,1
06ХН28МДТ — ж 20—Ткип <0,1
Тантал — ж 20 <0,001
Титан — ж 7 1 кип <0,05
Цинк — ж Ткип <0,1
Чугуны
серый — ж 20 <0,1
Электродные процессы
291
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Спирт этиловый (этанол) С2Н5ОН (продолжение)
Чугуны
СЧ15, СЧ17 — Ж 20 <0,1
Антегмит АТМ-1 1-99 Ж 20- Ткип С
Графит, пропитанный феноло- 1-99 Ж < Т’кип с
формальдегидным олигомером
Поливинилхлорид 1-99 Ж <40 с
Полиизобутилен — Ж <60 с
Полиметилметакрилат — Ж 20-Т 1 кип н
Полипропилен — Ж 20-Ткип с
Полиэтилен 1-99 Ж <40 с
96 Ж 60 с
Резины на основе
БК, ХП — Ж 20-70 с
НК, СКН, СКФ — Ж 20-65 с
СКС — Ж 20-60 с
ХСПЭ — Ж 20-95 с
Стекло — Ж 20-Т X’V 1 кип с
Фторопласт 4 — Ж 20-Т 1 кип с
Эмаль кислотостойкая — Ж 20-Т 1 кип с
Тетрахлорметан СС14
Алюминий Безводный Ж 20 <0,001
Вода: 1 Ж 20 0,009 т
Безводный Ж Гкип <5,0
и влажный
Медь Безводный Ж 20 <0,05
То же Ж 75 <0,08
Вода: 1 Ж 20 0,002 т
То же Ж 76-78 0,02
Медные сплавы
БрОЦ4-3 Безводный Ж 20 <0,001
и влажный
Л90 Безводный Ж 20-77 <0,1
Никель То же Ж 20-77 < 0,001
Влажный Ж 20 0,001
То же Ж т 1 кип 0,5
Никелевые сплавы
НМЖМц28-2,5-1,5 Безводный ж 20-Т < 0,003
Влажный ж Гкип 0,1-0,35
Н70МФ — ж 25-50 <0,05
Олово Безводный ж 20 0,001
Олово То же ж Гкип 0,1-1,0
Влажный ж 20 0,0035
То же ж 67 0,027
292
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Тетрахлорметан СС14 (продолжение)
Платина Безводный Ж 75 <0,04
Свинец Безводный Ж 25-75 <0,08
и влажный
Серебро То же Ж 25-75 <0,08
Стали
СтЗ Безводный Ж Т'кип <0,1
Вода: 1 Ж 20 0,2 т
То же Ж 76-78 0,4 я
Вода: 2 Ж 40 5,7
12X13 Безводный Ж 20-Т 7 кил <0,1
12Х18Н10Т То же Ж 20-7-кк1| <0,001
Вода: 1 Ж 20-25 < 0,001 т
Влажный Ж Т'кип 0,34 т
10Х17Н13М2Т Безводный Ж 20-78 <0,001
Вода: 1 Ж 20-25 <0,001 т
Влажный Ж Т'кип 0,1-1,0
06ХН28МДТ Безводный ж 25-75 <0,1
Тантал Безводный ж < Т 1 кип 0,001
и влажный
Титан То же ж т 1 кип 0,003
Цирконий Влажный ж Т'кип < 0,005
Чугуны
серый Безводный ж 20 <0,1
СЧ15, СЧ17 Безводный ж 20-7кип <0,1
и влажный
Антегмит АТМ-1 — ж 20-75 с
Графит, пропитанный феноло- — ж <75 с
формальдегидным олигомером
Полиамиды — ж 20-60 с
Поливинилхлорид — ж 25 н
Полиизобутилен — ж 20 н
Полиметилметакрилат — ж 20 н
Полипропилен — ж 25 н
Полиэтилен — ж 20 н
Резины на основе
БК — ж 15 н
НК, СКИ. СКС, ХП, СКУ — ж 25 н
СКН, ХСПЭ — ж 20 н
СКФ — ж 20-70 ОС
Стекло — ж 20 с
Фторопласт 4 — ж 25-75 с
Эмаль кислотостойкая — ж < 150 с
Электродные процессы
293
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
1,1,2-Трихлорэтан СН2С1СНС1 2
Алюминий — Ж 25 <0,05
— Ж Ттп <0,1
Медь — Ж 25 <0,05
Медные сплавы
БрА5, БрА7, — Ж 30 <0,05
БрОФ6,5-0,15, БрОФ7,2-0,2 — Ж 25 <0,05
Л59, Л63, Л68 — Ж 30 <0,05
Никель — Ж 30-110 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20-100 <0,1
ХН78Т — Ж 30 <0,05
Н70МФ — Ж 25 <0,05
ХН65МВ — Ж 25 <0,05
Свинец — Ж 20 <0,1
Стали
СтЗ — Ж 20 <0,1
— Ж НО <1,0
12X13 — Ж ПО < 1,0
12Х18Н10Т — Ж НО 0,1
10Х17Н13М2Т — Ж НО <0,1
06ХН28МДТ Безводный Ж 25-50 <0,05
Титан — Ж 100 <0,05
Цинк — Ж 20 3,0
Цирконий — Ж 25 <0,05
Чугуны
серый — Ж 25 <0,05
СЧ15, СЧ17 — Ж 50 <0,05
Асбест — Ж 12 С
Поливинилхлорид — Ж 20 н
Полимстилметакрилат — Ж 20 н
Полиэтилен — Ж 25 н
Резины на основе БК, НК, СКН, — Ж 20 н
СКС, СКИ, ХП
Фторопласт 4 — ж 20-100 с
Эмаль кислотостойкая — ж 20-Ткип с
Трихлорэтилен СНС1=СС12
Алюминий — ж 20-7 1 кип <0,1
Медь — ж 20—Ткип <0,1
Медные сплавы
БрА5, БрА7, БрОФ6,5-0,15, Безводный ж 75 <0,5
БрОФ7,2-0,2
Л59, Л63, Л68, Л80, ЛО68-1 То же ж 20 <0,1
Никель » ж Ткип <0,001
294
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Трихлорэтилен СНС1=СС12 (продолжение)
Никель Вода: 0,1 Ж 25-30 0,01
То же Ж Ткип 0,03
Никелевые сплавы
НМЖМц28-2,5-1,5 Безводный Ж 20-Ткип 0,002
Вода: 0,1 Ж 25-30 0,018
То же Ж Ткип 0,28
ХН78Т — Ж 30 <0,05
Вода: 0,1 Ж Ткип <0,001
Н70МФ — Ж 25-75 < 0,005
— Ж Ткип <0,1
ХН65МВ — Ж 25-75 <0,05
Свинец Безводный Ж Ткип <0,001
Вода: 0,1 Ж 20 0,02
То же Ж Ткип 0,1
Серебро — Ж 25 <0,05
Стали
СтЗ Безводный Ж Ткип <0,001
Вода: 0,1 Ж 20 0,02
То же Ж Т 1 кип <0,1
12X13 Безводный Ж 20-Т 1 кип <0,1
12Х18Н10Т Вода: 0,1 Ж т 1 кип <0,001
10Х17Н13М2Т То же Ж т ' кип <0,001
06ХН28МДТ » Ж Ткип <0,001
Титан — Ж 25-75 <0,05
Вода: < 0,1 Ж т 1 кип <0,001
Цинк Безводный Ж т 1 кип <0,1
Влажный Ж Ткип >3,0
Цирконий — Ж 20-70 <0,05
Чугуны
серый Безводный Ж 20 <0,5
(Ni: 20 %) — Ж 25-75 <0,5
Антегмит ATM-1 — ж 30 С
Графит — ж 20-60 с
Графит, пропитанный феноло- — ж <80 с
формальдегидным олигомером
Полиамиды — ж 20 с
Поливинилхлорид — ж 20 н
Полиизобутилен — ж 20 н
Полиметилметакрилат — ж 20 н
Полипропилен — ж 20 н
Полиэтилен — ж 20 н
Резины на основе
БК, НК, СКИ, СКН, СКС, СКУ, ХП, — ж 20 н
ХСПЭ
Электродные процессы
295
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Трихлорэтилен СНС1=СС12 (продолжение)
СКФ — Ж 20 С
Стекло — Г, Ж 66-100 С
Фторопласт 4 — Ж Т’кип С
Эмаль кислотостойкая — Ж 20 с
Фенол С6Н5ОН
Алюминий 1-50 Ж 20 <0,001
1 Ж 60-70 0,004
3-10 Ж 60-70 0,02
50-75 Ж 60-70 0,013
Безводный Ж Т’кип > 10,0
Алюминиевый сплав АМц — Ж 70 <0,001
Медь — Ж 20 0,008
Безводный Ж <200 <0,1
Никель — Ж 20 <0,1
— Ж Т’кип <0,05
Никелевые сплавы
НМЖМц28-2,5-1,5 — Ж 20 <0,1
— Ж Т 1 кип <0,05
Н70МФ — Ж Т’кип 0,05
Олово — Ж 20 <0,01
Серебро — Ж Т’кип <0,1
Стали
углеродистая — Ж 20 0,002
— Ж 50 0,02
— Ж 175 0,03
— Г 220 0,0052
— Ж 260 0,4
— Ж 340 1,17
12X13 — Ж 20 0,1
Вода: 10 Ж Т’кип <0,1
12Х18Н10Т — ж 20— Т’кип <0,1
Вода: 10 ж Т’кип <0,1
10Х17Н13М2Т — ж 20-Т’кИП <0,1
Вода: 10 ж Т’кип <0,05
— ж 240 <0,001
— ж <425 <0,5
06ХН28МДТ — ж 20-Т’кИП <0,1
Вода: 10 ж Т’кип <0,05
— ж 240 < 0,001
— ж <425 <0,5
Тантал Насыщ. ж 20 <0,001
Титан 1-насыщ. г, ж 70 <0,001
— ж 20 <0,13
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс, % Давление, МПа Фаза Г, °C
Фенол С6Н5ОН (продолжение')
Чугуны
серый — Ж 40 <0,5
СЧ15, СЧ17 — Ж 20-100 <0,1
Антегмит — Ж 20— ТКИп С
Полиамиды — Ж 20 н
Поливинилхлорид — Ж 20 с
— Ж 40 ОС
Полиизобутилен — Ж 40-60 ОС
— Ж 100 н
Полиметилметакрилат — Ж 20-60 ОС
Полипропилен — Ж 20-60 с
Полиэтилен — Ж 20-40 с
Резины на основе
С КУ — Ж 20 с
СКФ — Ж 20-100 с
ХСПЭ — Ж 20 н
Стекло — Ж 20-Т XrV— 1 КИП с
Фторопласт 4 — Ж 20-Г 1 кип с
Эмаль кислотостойкая — Ж 20-Т Z'V 1 кип с
Формальдегид НСНО
Алюминий 5 Ж 20 <0,13
5 Ж 60-70 0,92-1,95
10 Ж 20 0,18-0,22
10 Ж 60-70 2,61-4,56
40 Ж 20 0,28
40 Ж 60-70 3,2
Алюминиевые сплавы
АК12(АЛ2) 10 Ж 20 0,21
(Al, Mg: 3,8-10,3 %) 10 Ж 20 0,0006
Медь = 40 Ж 20— Ттп <0,1
= 40 Ж 104 <0,013
Медные сплавы
БрА5 12-15; муравьиная ж 135 0,018
кислота: 2
(Cu, Si: 1,5 %) То же ж 135 0,03
Никель 10 ж 104 0,023
= 40 ж 20— Ттп <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 10 ж 104 0,013
= 40 ж 20— 7'кип <0,1
= 40 ж 135 0,2
Н70МФ = 40 ж 20—Гют <0,1
Электродные процессы
297
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Формальдегид НСНО (продолжение)
Никелевые сплавы
хастеллой С 10 Ж 104 <0,1
Олово 10-40 Ж 20 <0,1
Свинец 40 Ж Гкип > 10,0
Серебро 40 Ж Ткип <0,1
Стали
СтЗ 40 Ж 20 <0,1
40 Ж Ткип > 10,0
12X13 40 Ж 20 <0,01
40 Ж Ткип <0,1
12Х18Н10Т 12-15; муравьиная Ж 135 > 10,0
кислота: 2
40 Ж 20 <0,01
40 Ж Ткип <0,1
40 Ж 135 <0,1
10Х17Н13М2Т 12-15; муравьиная Ж 135 1,84
кислота: 2
40 Ж 20 <0,05
40 Ж Ткип <0,1
06ХН28МДТ 40 Ж 20 <0,05
40 Ж Ткип <0,1
Тантал 40 Ж 100 <0,001
Титан 10-70 Ж 100 <0,05
37 Ж Ткип <0,01
Чугуны
серый ~40 Ж 20 0,07
СЧ15, СЧ17 ~40 Ж 20 <0,1
Х34 40 Ж 20 0,1
Антегмит ATM-1 40 Ж т * кип С
Графит 1^10 Ж т 1 кип С
Графит, пропитанный бакелито- 1^10 Ж Ткип С
вым лаком
Поливинилхлорид 30 Ж 20 ОС
Полиизобутилен 40 ж 60 С
Полиметилметакрилат 40 ж 25 н
Полиэтилен 37 ж 50 С
Резины на основе
БК ~ 40 ж 20-60 С
СКН, ХП, ХСПЭ ~40 ж 20 С
СКУ ~40 ж 20 ОС
Стекло 1^10 ж т 1 кип С
Фторопласт 4 40 ж Ткип С
Эмаль кислотостойкая 40 ж 150 С
298
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Фтор F2
Алюминий Сухой Г <400 0,003
Влажный Г 20 > 10,0
Вольфрам Сухой Г 20 > 10,0
Золото То же Г 120 0,15
» Г 150 0,3
» Г 204 > 10,0
Магний » Г 200-300 <0,01
Медь — Г 25-150 <0,05
— Г 175 <0,5
— г 200-250 <1,3
— г 500 > 10,0
Медные сплавы
БрА5, БрА7 — ж -200 <0,05
Сухой г 60 <0,1
— г 250-300 <0,5
БрОФ6,5-0,15, БрОФ7,2-0,2 — г 25-150 <0,05
— г 175 <0,5
— г 200-250 <1,3
— г 500 > 10,0
Молибден — г 20 > 10,0
Никель — г 200 <0,1
— г 300 0,002
— г 400 0,013
— г 450 0,6
— г 500-650 <5,0
— г 700 > 10,0
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж -200 <0,05
Сухой г 400^450 <0,5
— г 500 0,6
— г 600 > 10,0
ХН78Т — ж -200 <0,05
— г 300 0,4
— г 400 0,3
Н70МФ Сухой г 25 <0,1
— г 300 2,18
ХН65МВ — г 480 0,8
— г 538-565 <3,0
— г 650 > 10,0
Ниобий Сухой г 300 > 10,0
Олово То же г 20 > 10,0
Платина — г 25^400 <0,05
Сухой г 480 0,8
Электродные процессы
299
Продолжение таблицы L4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Фтор F2 (продолжение)
Платина Сухой Г 510-535 <3,1
То же Г 565 > 10,0
Свинец — Ж -195 <0,05
Сухой Г 20 <0,1
— Г 25-100 < 1,3
Серебро Сухой Г 20 <0,1
То же Г 40 0,8
» Г 65-120 <3,1
» Г ' 260 > 10,0
Стали
углеродистая » Ж -196 <0,1
— Г 20-100 <0,1
Сухой Г 250 4,9
12X13 То же Г 200 0,21
12X17 » Г 300 > 10,0
12Х18Н10Т » Ж -200 <0,1
» Г 25 <0,05
Влажный Г 20 > 10,0
— Г 200 8,8
— Г 250 > 10,0
10Х17Н13М2Т Сухой Г 20 <0,1
— Г 200 0,2
— Г 350 1,8
— Г 400 > 10,0
06ХН28МДТ Сухой Г 20 <0,1
— Г 200 0,05
— Г 300 0,12
— Г 350 0,85
— Г 400 3,4
Тантал — г 20 > 10,0
Титан — г 20 > 10,0
Цинк Сухой г 25 > 10,0
Цирконий — г 150 > 10,0
Антегмит АТМ-1 — г 25 н
Асбест — г 100 н
Графит — г 20 н
Графит, пропитанный феноло- — г 20 н
формальдегидным олигомером
Полиамиды — г 25 н
Поливинилхлорид — г 20 ОС
Полиизобутилен — г 20 н
Полиметилметакрилат — г 20 н
Полипропилен Сухой и влажный г 20 н
300
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Фтор F2 (продолжение)
Полиэтилен — Г 20 Н
Резины на основе
БК Сухой Г 20 ОС
НК Влажный Г 20 н
СКИ, СКН, СКС Сухой Г 20 н
Фторопласт 4 — Ж < Т’кип н
Сухой Г 20 С
— Г 150 ОС
— Г 175 н
Влажный Г 20 н
Эмаль кислотостойкая — Г 20 н
Фтороводород HF
Алюминий 99,5 Ж -10 0,004
97,0-99,5 Ж 15 0,28
— Г 40 0,03
— Г 500 4,88
— Г 600 > 10,0
Алюминиевые сплавы
АМц 99,6 Ж -10 0,004
АМг5В 99,5 Ж -10 0,002
Вольфрам Вода: 0,6 Г 300 0,007
То же Г 400-600 <0,05
» Г 700 2,1
Магний 97,0-99,5 Ж 15 0,28
— Г 500 2,5
Медь — Ж 15 0,5
— Г 100 0,44
— Г 300 0,98
— Г 500 1,5
Медные сплавы
БрОФ4-0,25 — Ж 15 0,5
— Г 25 0,5
— г 80 1,5
Л80 — г 65 > 10,0
Молибден Вода: 0,6 г 300 0,004
То же г 400-600 0,03
» г 700 0,2
» г 800 1,5
Никель — ж 20 <0,1
— г 40 0,04
— г 100 0,01
— г 300 0,04
Электродные процессы
301
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Фтороводород HF (продолжение)
Никель — Г 600 <0,1
— Г 650 0,23
Никелевые сплавы
НМЖМц28-2,5-1,5 — Г 100-500 <0,1
— Г 650 0,13
ХН78Т — Г 100-550 <0,1
Н70МФ Сухой Ж 20 <0,1
Олово — Г 20 > 10,0
Платина — Г 25-150 <0,05
Свинец Сухой Г 20 <0,05
То же Г 100 > 10,0
Серебро » Г 20 <0,1
Стали
СтЗ — Г 20 0,004
— Г 50 0,03
— Г 500 > 10,0
12X13 97,0-99,5 Ж 15 0,12
— Г 100 0,05
— Г 300 0,14
— Г 500 8,40
— Г 600 > 10,0
12Х18Н10Т 97,0-99,5 Ж 15 0,19
— г 40 0,15
— г 100 0,05
— г 300-500 > 10,0
10Х17Н13М2Т — г 20 0,004
— г 200 <0,1
— г 500 > 10,0
06ХН28МДТ — г 50 0,001
— г 500 > 10,0
Тантал Сухой г 25 > 10,0
Титан То же г 25 > 10,0
Цинк — г 20 > 10,0
Цирконий Сухой г 25 > 10,0
Чугуны
серый 97,0-99,5 ж -15 0,55
— г 20 <0,1
— г 40 0,35
— г 300 3,5
— г 600 6,3
СЧ15 97,0-99,5 ж 15 0,61
— г 40 0,19
— г 100 0,18
302
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Фтороводород HF (продолжение)
Чугуны
СЧ15 — Г 300 0,48
— Г 500 > 10,0
Антегмит АТМ-1 — Г 20 Н
Графит — Г < 100 С
Поливинилхлорид — Г 20 с
Полиизобутилен — Г 20-60 с
Полиэтилен — Г 60 н
Резина на основе ХП — Г 20 с
Стекло Сухой Г 20 с
Влажный Г 20 н
Фторопласт 4 — Г 60 с
Эмаль кислотостойкая — Г 20 н
Дифтордихлорметан CC12F2 (R12)
Алюминий — Г, Ж 50 < 0,006
— Г 100 0,002
— Г 200 0,069 н
Алюминиевый сплав АМц — Г 20 <0,03
Медь — Ж -30 <0,05
— Г, Ж 50 <0,012
— Г 200 0,01
Вода: > 0,01 Ж 20 0,02-0,05
Медные сплавы
БрА5 — Г, Ж 50 0,002
— Г, Ж 100 0,008
— Г 200 0,048
БрАЖМц 10-3-1,5 — г 50-100 0,003
— г 150-200 «0,1
— г 250 0,42
ЛО62-1 — г, ж 50-100 <0,01
— г 150 0,004
Никель — г, ж 50-100 < 0,002
Никелевый сплав
НМЖМц28-2,5-1,5 — г, ж 50 < 0,003
— г 200 < 0,003
Олово — ж -30 <0,1
Платина — г 20-150 <0,05
Свинец — г 50 0,015
— г 100 0,11
Серебро — г 25 <0,05
Стали
СтЗ — ж -30 <0,05
— г, ж 50 < 0,05 т
Электродные процессы
303
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т, °C
Дифтордихлорметан CC12F2 (R12) (продолжение)
Стали
СтЗ — Г, Ж 100 < 0,025 н
—- Г 150 0,007 т
— Г 250 0,023
Вода: > 0,01 Ж 20 0,02-0,05
12X13 — Г, Ж 50 0,001
— г 100-200 < 0,004
12Х18Н10Т — г, ж 50 0,002-0,003
— г 100-250 < 0,003
06ХН28МДТ — г 250 0,001
Титан — г, ж 50-100 < 0,004
— г 200 0,027
— г 250 0,2
Титановый сплав ОТ4 — г 100-150 <0,001
Цирконий — г 30 <0,05
Чугуны
серый — ж -30 <0,05
СЧ15, СЧ17 — г 25 <0,05
(Ni: 20%) — г 20-105 <0,05
Асбест — ж -30 С
Паронит ПОН — ж -30 с
— г 100 с
Полиамиды — г 25 с
Поливинилхлорид — г <40 с
П олиизобутилен — г 20 н
Полипропилен — г 20 н
Полиэтилен — г 20 н
Резины на основе
БК, НК, СКИ, СКН, СКС, СКТ, — г 20 н
СКЭП, ХП
СКФ — г -20 с
— г 20 н
Стекло — г 20 с
Фторопласт 4 — ж 15-20 с
Дифторметан CH2F2 (R32)
Алюминий — г < 150 <0,001
Алюминиевые сплавы
АМгЗ — г < 150 <0,001
АМц — г < 150 <0,001
Д16 — г < 150 <0,001
Медь — г 150 0,02-0,03
Бронза БрАМц9-2 — г 150 0,02-0,03
Никелевый сплав ХН78Т — г < 150 <0,001
304
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Дифторметан CH2F2 (R32) (продолжение)
Стали
СтЗ — Г < 150 <0,001
20X13 — Г < 150 <0,001
12Х18Н10Т — Г < 150 <0,001
10Х17Н13М2Т — Г < 150 <0,001
06ХН28МДТ — Г < 150 <0,001
Титан — Г 150 0,02-0,03
Парониты ПОН, ПМБ-1 — Ж 50 с
Фторопласт 4 — Ж 50 С
Дифторхлорметан CHC1F2 (R22)
Алюминий — Г, Ж 50 0,016-0,023
— Г, Ж 100 < 0,002
— Г 150 <0,001
Алюминиевые сплавы
АМгЗ — Г, Ж 50 0,002-0,003
— Г 100 0,002
— Г 150 0,012
АМгб — Г, Ж 50 0,001-0,002
— Г 100 0,002
— Г 150 0,012
АМц — Г 20 0,03
Медь — Ж 50 < 0,005
— Г 100 0,004
— Г 200 0.33
Медные сплавы
БрА5 — Г, Ж 50 0,034
— Г 100 0,001-0,005
— г 150 0,013
Л59, ЛО62-1, Л63 — г, ж 50 0,002
— ж 100 0,004
— г 100 0,022 н
— г 150 0,012 н
— г 200 0,20 н
Л90 — ж (—15)—50 0,002
Никель — г, ж 50 < 0,003
— г 100-150 <0,001
Никелевые сплавы
НМЖМц28-2,5-1,5 — г, ж 50 < 0,003
— г 100-150 <0,001
ХН78Т — г 25 <0,05
ХН65МВ — г 25 <0,05
Свинец — г 25 <0,05
— ж 50-100 0,13
Электродные процессы
305
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Дифторхлорметан CHC1F2 (R22) (продолжение)
Свинец — Г 100 <0,001
— Г 150 0,046
Серебро — Г 20 <0,05
Стали
СтЗ — Г, Ж 50 0,040-0,049
— Г 100 0,003 т
— Г 150 0,002 т
12X13 — Г, Ж 50 0,002-0,003
— Г 100 <0,001
— Г 150 0,002
12XI8H10T — Г, Ж 50 <0,001
— Г 100 <0,001
Титан — Г, Ж 50 <0,001
— Г 100-150 <0,001
Цирконий — Г 25 <0,05
Паронит ПОН — Ж 15-30 С
Поливинилхлорид — Ж 15-30 с
Полиизобутилен — Ж 15-30 с
Полиэтилен — Ж 15-30 с
Резина на основе БК, СКИ, СКН, — Ж 15-20 н
СКС, СКФ, СКЭП, ХСПЭ
Фторопласт 4 — Ж 15-30 с
Октафторпропан CF3CF2CF3 (R218)
Алюминий — Г 150 < 0,005
Медь — Г 150 <0,05
Медные сплавы
БрАМц9-2 — Г 150 <0,05
Л63 — Г 150 <0,05
Никель — г 150 < 0,005
Никелевый сплав — г 150 < 0,005
НМЖМц28-2,5-1,5
Стали
СтЗ — г 150 <0,05
20X13 — г 150 < 0,005
12Х18Н10Т — г 150 < 0,005
10Х17Н13М2Т — г 150 < 0,005
06ХН28МДТ — г 150 < 0,005
Резины на основе СКЭП — г 100 С
Паронит ПМБ-1 — г 100 с
Фторопласт 4 — г 100 с
1,1,1,2-Тетрафторэтан CF3CFH2 (R134a)
Алюминий — г, ж 50-150 <0,001
Медь — г, ж 50-150 0,020-0,005
306
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
1,1,1,2-Тетрафторэтан CF3CFH2 (R134a) {продолжение)
Медные сплавы
БрАМц9-2 — Г, Ж 50-150 0,020-0,005
Л62 — Г, Ж 50-150 0,020-0,005
Никель — Г, Ж 50-150 <0,001
Никелевые сплавы
НМЖМц28-2,5-1,5 — Г, Ж 50-150 <0,001
ХН78Т — Г, Ж 50-150 <0,001
Стали
СтЗ — Г, Ж 50-150 0,020-0,005
20X13 — Г, Ж 50-150 <0,001
12Х18Н10Т — Г, Ж 50-150 <0,001
10Х17Н13М2Т — Г, Ж 50-150 <0,001
06ХН28МДТ — Г, Ж 50-150 <0,001
Титан — Г, Ж 50-150 <0,001
Парониты ПМБ-1, ТИИР — ж 50 с
Полиамиды — ж 50 с
Полипропилен — ж 50 с
Полиэтилен — ж 50 с
Резины на основе СКН, СКЭП — ж 50 с
Фторопласт 4 — ж 50 с
1,1,2-Трифтортрихлорэтан CCIF2CCI2F (R113)
Алюминий — г, ж 50 0,002
Алюминиевые сплавы
АМц — г, ж 50 0,002
Д16 — г, ж 50 <0,01
Медь — г, ж 50 <0,01
Медные сплавы
БрАМц9-2 — г, ж 50 <0,01
БрБ2 — г, ж 20 0,001
Л62, Л68 — г, ж 50 <0,01
Никель — г, ж 50 < 0,005
Никелевые сплавы
НМЖМц28-2,5-1,5 — г, ж 50 < 0,005
Н70МФ — ж 25 <0,05
Свинец — ж 30 <0,05
Стали
СтЗ — г, ж 50 < 0,003
12X13 — г, ж 50 < 0,005
12Х18Н10Т — г, ж 50 <0,001
10Х17Н13М2Т — г, ж 50 < 0,005
06ХН28МДТ
Титан — г, ж 50 <0,001
Электродные процессы
307
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
1,1,2-Трифтортрихлорэтан CC1F2CC12F (R113) (продолжение)
Пентапласт — Г 105 С
Полиамиды — Ж 50 С
Поливинилхлорид — Ж 50 С
Полиизобутилен — Ж 25-30 н
Полиметилметакрилат — Ж 50 ОС
Полипропилен — Ж 50 н
Полиэтилен — Ж 50 н
Резины на основе БК, СКИ, СКФ, — Ж 25 н
СКЭП, ХСПЭ
Фторопласт 4 — Ж 50 с
Фтортрихлорметан CCI3F (R11)
Алюминий — Г, Ж 50 0,001-0,002
— Г, Ж 100 0,004-0,007
— Г, Ж 150 0,06-0,08
— Г 200 0,24 т
Медь — Г, Ж 50 0,002
— Г, Ж 100 0,02-0,14
— Г, Ж 150 5,0
— Г 200 > 10,0
Медные сплавы
БрА5 — Г, Ж 50 0,001
— г, ж 100 0,02-0,045
— г, ж 150 0,14
БрБ2 — г, ж 50 0,001
БрОФ6,5-1 — г, ж 50 <0,001
ЛО62-1 — г, ж 50-100 0,002-0,003
— г, ж 150 0,60-0,70
ЛС59-1 — г, ж 50 <0,001
Никель — г, ж 50 0,001-0,002
— г, ж 100 < 0,003
— г, ж 150 0,005
Никелевый сплав
НМЖМц28-2,5-1,5 — г, ж 50 0,001
— г, ж 100 < 0,002
— г, ж 150 0,003-0,004
Свинец — г, ж 50-100 0,04-0,05 т
— г, ж 150 0,19-0,22 т
Стали
СтЗ — г, ж 50 0,004-0,008 т
— г, ж 100 0,003-0,010 т
— г, ж 150 0,012-0,013 т
— г 200 0,15 т
12X13 г, ж 50 0,001
308
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Фтортрихлорметан CC13F (R11) (продолжение)
Стали
12X13 — Г, Ж 100 0,002-0,003 т
— Г, Ж 150 0,003
— Г 200 0,012 т
12Х18Н10Т — Г, Ж 50 0,001
— Г, Ж 100 <0,001 т
— Г, Ж 150 0,004-0,005
— г 200 0,002-0,014 т
Титан — г, ж 50 0,001-0,004 т
— г, ж 100 0,001 т
— г, ж 150 0,017-0,050 т
Полиамиды — ж 50 С
Поливинилхлорид — ж 20 с
Полиметилметакрилат — ж 20 н
Полипропилен — ж 20 н
Полиэтилен — ж 20 н
Хлор С12
Алюминий — ж 20 <0,1
Сухой г 20 < 0,001
То же г < 100 < 1,0
» г 120 > 10,0*
Вода: 0,04 г 20 0,065
Вода: 0,10-0,03 г 20 0,26-0,93
Вода: 0,4-0,6 г 20 2,6-19,0
Вода: 0,06 г 130 < 0,001
То же г 160 > 10,0
Вода: 1,5 г 130-200 < 0,001
То же г 290-320 1,0
» г 350 > 10,0
Вода: 30 г 140 <0,001
То же г 170 0,3
» г 630 > 10,0
Бериллий Сухой г 60 > 10,0
Ванадий То же г 20 > 10,0
Вольфрам » г 200 > 10,0
Гафний » г 50 0,4
» г 100 8,4
» г 150 > 10,0
Иридий Сухой и влажный г 20 <0,001
Воспламеняется.
Электродные процессы
309
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлор С12 {продолжение)
Золото — Ж 20 > 10,0
Сухой Г 20 0,2
То же Г 150 <5,0
Влажный Г 20 < 1,7
Медь Сухой Г 20 0,1
То же Г 120 1,8
» Г 200 > 10,0
Вода: 0,06 Г 150-170 1,0-5,6
Вода: 0,5 Г 150-200 8,5-15,0
Вода: 0,6 Г 20 10,0
Вода: 1,5 Г 130 4,8
Вода: 0,06-1,50 г 220 > 10,0
Медные сплавы
БрА5, БрА7 — ж 20 <0,1
Сухой г 20 <0,1
Влажный г 100 1,5
То же г 300 > 10,0
БрО4 — ж 20 <0,1
Сухой г 20 <0,1
Влажный г 20 > 10,0
Л80 — ж 20 <0,1
Сухой г 20 <0,1
Влажный г 20 > 10,0
Молибден Сухой г 200 > 10,0
Влажный г 95 0,6
Никель Сухой г 20-250 <0,018
То же г 400 <0,16
» г 550 < 1,0
Вода: 0,06 г 600 > 10,0
Вода: 0,1-0,3 г 20 <0,06
Вода: 0,4 г 20 0,3
То же г 400 <0,1
» г 550 0,7
Вода: 4-15 г 20 <5,6
То же г 100 0,12-1,70
» г 150-400 <0,05
» г 550 2,0-2,4
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 20 <0,1
Сухой г 20-30 <0,09
Вода: 0,1-0,2 г 20 0,03
Вода: 0,3 г 20 0,16
Вода: 0,4 г 20 0,3
310
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлор С12 (продолжение)
Никелевые сплавы
НМЖМц28-2,5-1,5 Вода: 0,4 Г 100 0,005
Вода: 4-36 Г 150-200 0,034
ХН78Т Сухой Г 20-300 <0,013
То же Г 400 0,12-0,13
» Г 550 1,1-1,2
Вода: 0,1-0,2 Г 20 0,04-0,07
Вода: 0,3 Г 20 0,14
Вода: 0,4 Г 20 0,6
То же Г 50-250 <0,02
Вода: 4 Г 100 0,1
То же Г 150-400 <0,3
» Г 550 1,4
Вода: 15 Г 100 1,5
То же Г 550 1,9
Н70МФ — Ж 20 <0,1
Сухой Г 20-250 <0,02
То же Г 300 0,08
» г 400 0,12-0,13
» г 550 <2,0
Вода: 0,4 г 100 <0,01
Вода: 4 г 100 <0,05
То же г 550 2,3
Вода: 15 г 100 6,9
То же г 150-400 <0,05
Вода: 22 г 100 8,3
ХН65МВ — г 200-300 < 1,2
— г 400-500 <5,4
Ниобий Сухой г 50 0,1
То же г 100 5,0
» г 150 > 10,0
Олово » г 20 > 10,0
Осмий » г 20 0,005
Палладий » г 20 0,3
Платина » г 20 0,008
» г 320-370 <1,4
» г 430 2,4
» г 480 5,5
» г 630 > 10,0
Рений » г 100 0,2
» г 150 0,8
» г 200 > 10,0
Родий Сухой и влажный г 20 <0,001
Электродные процессы
311
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Хлор Ch {продолжение)
Родий Сухой Г 500 > 10,0
Рутений То же Г 20 0,001
Влажный Г 20 0,09
Свинец Сухой Г 20 <0,001
То же Г 200 0,6
» Г 250 1,2
» Г 300 > 10,0
Вода: 0,5 Г 150 1,2
То же Г 250 4,5
Вода: 4 Г 170 0,38
Вода: 1,5-30,0 Г 130 0,8
То же Г 155 1,2
» Г 250 4,5
» Г 275 9,0
Серебро — Ж 20 <0,001
Сухой Г 20-60 <0,8
То же Г 425 > 10,0
Стали
СтЗ — Г, Ж 20 <0,05
Вода: 0,03 Г, Ж 20 0,16-0,27
Вода: 0,3 Г, Ж 20 0,4-1,1
Сухой Г 100 0,026
То же Г 150 0,19
» Г 200 6,6
» Г 250 > 10,0
Вода: 0,04 Г 250 0,36
То же г 300 > 10,0*
Вода: 0,4 г 150 <0,1
То же г 400 3,5
» г 550 > 10,0*
Вода: 4-36 г 100 > 10,0
То же г 200-300 <0,1
» г 400 0,55
» г 550 > 10,0
08X17Т Сухой г 20-200 <0,015
То же г 300 0,77
» г 400 > 10,0
Вода: 0,01-0,02 г 20 0,3
То же г 100 <0,03
Вода: 0,5 г 440 0,33
12Х18Н10Т — ж 20 <0,1
Воспламеняется.
312
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Хлор С12 (продолжение)
Стали
12Х18Н10Т, 10Х17Н13М2Т Сухой Г 20-200 <0,1
То же Г 300 0,77
» Г 400 10,6
Вода: 0,01 Г 20-150 <0,06
Вода: 0,02 Г 20 0,2
Вода: 0,03 Г 20 0,46
Вода: 0,04 Г 20 <0,77
То же Г 100-250 <0,22
» Г 300-550 0,9-8,2
Вода: 0,4 Г 20 1,7-5,0
То же Г 100 1,0
» Г 120 0,015
» Г 150-400 <0,13
» Г 550 > 10,0
Вода: 4-15 Г 100 > 10,0
То же г 150 <3,1
» г 170-400 <0,12
» г 550 <0,78
06ХН28МДТ — ж 20 <0,1
Сухой г 200 0,08
То же г 300 0,3
» г 400 0,6
» г 500 > 10,0
Вода: 0,04 г 20-250 <0,09
То же г 300-400 0,22-0,40
» г 550 1,7
Вода: 0,4 г 20 2,0
То же г 100—400 <0,05
Вода: 4-15 г 100 2,8-6,3
То же г 150 <0,67
» г 160-400 <0,02
» г 550 <0,22
Тантал — ж 20 <0,1
Сухой г 20 <0,01
То же г 150 <0,1
Вода: 0,06 г 150 <0,001
Тоже г 200 0,05
» г 300 1,7
» г 350 > 10,0
Вода: 0,5 г 300-400 0,55
Вода: 30 г 150 <0,001
То же г 300 400 0,05
Электродные процессы
313
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлор С12 (продолжение)
Тантал Вода: 30 Г 500 2,1
Титан — Ж 40 > 10,0*
Сухой Г 20 > 10,0*
Вода: 0,4 Г <200 <0,1
Вода: 4 Г <350 <0,1
Вода: 15 Г <400 <0,1
Титановые сплавы ВТ5-1, ВТ14, Сухой Г 30 > 10,0
ОТ4-0, ОТ4-1,4200
Цинк То же Г 20 <0,001
Влажный Г 20 > 10,0
Цирконий Сухой Г 100 0,08
То же Г 150 0,5
» Г 200 > 10,0
Влажный Г 30 1,3
То же Г 90 3,6
Чугуны
серый — Г 20-25 <0,1
Вода: 1,5 Г 130 2,0
То же Г 280 0,8
» Г 440 > 10,0
Вода: 30 Г 170 0,8
То же Г 440 3,0
» Г 550 > 10,0
ЧС15 Сухой г 20 < 1,0
Антегмит — ж 20 С
Сухой г 20 С
— г 150 ОС
Влажный г 20 ОС
Графит Сухой г 50 С
Графит, пропитанный феноло- То же г 20 С
формальдегидным олигомером
Полиамиды » г 20 С
Влажный г 20 н
Поливинилхлорид Сухой г 20 С
Влажный г 95 ОС
Полиизобутилен Сухой г 20 ОС
То же г 40 н
Влажный г 20 н
Полиметилметакрилат — ж 20 н
Сухой г 20 С
Влажный г 20 н
Воспламеняется.
314
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлор С12 {продолжение)
Полипропилен — Ж 20 Н
Сухой Г 20 ОС
То же Г 60 Н
Влажный Г 20 ОС
Полиэтилен — Ж 20 Н
Сухой Г 25 С
То же Г 60 Н
Влажный Г 25 ОС
Резины на основе
БК — Ж 60 Н
Сухой Г 40 ОС
Влажный Г 45 ОС
То же Г 95 н
НК — Ж 60 н
Сухой Г 65 н
Влажный Г <40 н
СКИ — Ж 60 н
Влажный г 85 н
СКН — ж 20 н
Влажный г <40 н
СКС То же г 20 н
СКУ » г <40 н
СКФ — ж 20 С
Сухой и влажный г < 100 С
ХП — ж 20 н
Сухой г 20 н
Влажный г 20 н
ХСПЭ — ж 20 ОС
Сухой г 20 ОС
Влажный г 20 н
Стекло — ж 20 С
Сухой г 20-100 С
Влажный г 20-60 С
Фторопласт 4 — ж 20 С
Сухой г <200 С
Влажный г < 100 С
Эмаль кислотостойкая — ж 20-100 С
Сухой г 20-100 С
Влажный г 20-60 С
Хлороводород НС1 (газ)
Алюминий Сухой г -15 4- 40 0,032-0,098
То же г 154 0,13
» г 250 1,0
Электродные процессы
315
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлороводород НС1 (газ) (продолжение)
Алюминий Влажный Г 20 > 10,0
Золото Сухой Г 982 0,76
Медь То же Г -15 4- 40 <0,01
» Г 95 0,8
» Г 150 1,5
» Г 205 3,0
» Г 315 > 10,0
Медные сплавы
БрА5, БрА7, БрО4 » Г 20 <0,1
Л68 » Г (—15)—40 0,29-0,53
» Г 100-250 2,3-3,0
Никель » Г 20-100 <0,001
» Г 250 0,023
Никель » Г 400 0,28
» Г 450-550 0,19-0,65
Никелевые сплавы
НМЖМц28-2,5-1,5 » Г 20-100 <0,1
» Г 232 0,16
» Г 250 0,76
» Г 260 1,5
» Г 343 3,0
» Г 425 > 10,0
ХН78Т » Г 400 <0,1
» Г 600 0,96
хастеллой А » Г 340 0,84
хастеллой С » Г 370 0,76
» г 427 1,5
» г 482 3,0
» г 620 > 10,0
Олово » г 20 > 10,0
Платина » г 1260 0,76
Свинец » г -15 0,24
» г 40 0,35
Серебро » г 232 0,76
» г 260 1,5
» г 343 3,08
» г 454 > 10,0
Стали
СтЗ » г 20-250 <0,05
» г 280 0,1
» г 400 1,4
» г 450 2,0
» г 585 10,0
316
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г. °C
Хлороводород HCI (газ) (продолжение)
Стали Сухой
12X13 То же Г 250 0,05
» Г 300 0,25
» Г 400 0,67
» Г 500 1,67
» г 600 7,9
12Х18Н10Т » г 20 0,002
» г 250 0,02
» г 340 0,1
» г 400-450 0,46
» г 500-550 2,2
10Х17Н13М2Т » г 250 0,016
» г 400-450 0,26-0,47
» г 500-550 2,3
06ХН28МДТ » г 20-250 0,002-0,007
» г 400^450 0,17-0,31
» г 500-550 0,63-1,20
Тантал » г 20-100 0,1
Титан » г 100 0,01
» г 150 0,1
» г 200 4,5
Титановый сплав 4200 » г 200 0,002
» г 250 3,4
Цинк » г 20 > 10,0
Чугуны
серый » г -15 0,084
» г 100 0,1
» г 250 1,04
» г 315 3,0
» г 330 5,4
» г 360 6,9
» г 455 > 10,0
СЧ15, СЧ17 » г 100 <1,0
» г 150 > 10,0
Антегмит АТМ-1 — г 20-100 С
Графит Сухой г 20 С
Графит, пропитанный феноло- То же г 25 С
формальдегидным олигомером
Полиамиды — г 20 н
Поливинилхлорид — г <60 с
Полиизобутилен — г 20-60 С
Полипропилен — г 20-60 С
Полиэтилен — г 20-60 С
Электродные процессы
317
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлороводород HCI (газ) (продолжение)
Резины на основе
БК, НК — Г 20 С
ХП — Г <70 С
Стекло — Г < 100 С
Фторопласт 4 — Г <200 С
Эмаль кислотостойкая — Г <300 С
Хлороформ СНС13
Алюминий Ж 20 0,007
Безводный Ж 60 <0,05
Влажный ж 20 0,04 т
То же ж 50 > 10,0
Алюминиевый сплав АМгб Безводный ж 20 <0,1
Медь То же ж 20 0,02
» ж 61 0,2
Сухой г 100 <0,5
Влажный ж 20 0,003
То же ж 61 0,3
Медные сплавы
БрА5 — ж 20 <0,05
•— г 75 <0,5
Влажный ж 61 0,15
БрО4, БрОб — ж 20 <0,1
БрОФ6,5-0,15, БрОФ7,2-0,2 Безводный; без ки- ж 25 <0,05
слорода воздуха
Сухой; без кислорода г 100 <0,5
воздуха
Л59, Л63, Л68, Л80 Сухой г 75 <0,5
ЛО68-1 Безводный ж 20 <0,5
Сухой г 75 <0,5
Никель Безводный ж 20-60 < 0,003
Влажный ж 20-60 < 0,005
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 25 <0,05
Влажный ж 20-60 <0,1
ХН78Т — ж 20-60 0,1
Н70МФ — ж 25 <0,05
— г 75-105 <0,5
ХН65МВ — ж 25 <0,05
— г 75-105 <0,5
Платина — ж 20-60 <0,04
Свинец — ж 20-50 <0,5
— г 75-100 <0,5
Влажный ж 60 <0,08
Серебро Безводный ж 20-60 <0,08
318
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлороформ СНС13 (продолжение)
Серебро — Г 75-100 <0,05
Влажный Ж 20-60 <0,08
Стали
углеродистая Безводный Ж 61 0,22
Вода: 0,03 Ж 20 0,02
Вода: 0,05 Ж 20 0,06
12X13 Безводный Ж Т’кип 0,01
Влажный Ж 20 0,1-1,0
12Х18Н10Т Безводный Ж 20-50 0,1 кр
Влажный Ж 20 0,1 т, кр
То же Ж 60 3,3 т, кр
10Х17Н13М2Т Безводный Ж 20 <0,001
То же Ж Гкип <0,01
06ХН28МДТ » ж 20 <0,001
» ж Гкип <0,01
Влажный ж 20 60 0,1
Тантал Безводный и ж 20-60 <0,05
влажный
Титан Безводный ж Гкип <0,01
— г 80-105 <0,05
Влажный ж 20-60 <0,2
Цинк Безводный ж 20 <0,001
— ж Гкип > 10,0
Влажный ж 20 > 10,0
Цирконий — ж 25-50 <0,05
— г 75-100 <0,05
Чугуны
серый Вода: 0,03-0,05 ж 20 0,1
Вода: < 0,03 ж 60 1,0
Вода: 0,05 ж 60 3,0
СЧ15, СЧ17 -— ж 25 <0,5
(Ni: 20 %) — ж 25 <0,05
Асбест — ж 20-60 С
Графит — ж 60 с
Графит, пропитанный феноло- — ж 60 с
формальдегидным олигомером
Полиамиды — ж 25 н
Поливинилхлорид — ж 25 н
Полиизобутилен — ж 20 н
Полиметилметакрилат — ж 20 н
Полипропилен — ж 25 н
Полиэтилен — ж 20 н
Резины на основе БК, НК, СКИ, — ж 20 н
СКН, СКС, XII
Электродные процессы
319
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Хлороформ CHCI3 (продолжение)
СКФ — Ж 20 ОС
хспэ — Ж 25 н
Стекло — Ж 60 с
Фторопласт 4 — Ж 20—Т'кип с
Эмаль кислотостойкая — Ж 20 с
Цинка хлорид ZnCl2
Алюминий 1 Ж 20-75 0,013-0,034
5 Ж 20-75 0,022-0,40
10 Ж 25-50 < 0,05 т
10 Ж 100 < 0,5 т
10 Ж 125 > 1,3 т
60-90 Ж 105 > 1,3 т
Алюминиевые сплавы
АК12(АЛ2) 10 Ж 20 0,012
(Al, Mg: 1,5-2,5 %; Мп: 1,0-2,0 %) 10 Ж 20 0,011
Золото 10-90 Ж 25-105 <0,05
Магний 3 Ж 35 3,6
Медь 10-30; без кислорода Ж 25-50 < 0,05 т
воздуха
То же Ж 75-100 < 0,5 т
40-90; без кислорода Ж 25-105 < 0,5 т
воздуха
Медные сплавы
БрА5, БрА7 1-насыщ. Ж 20-140 < 1,0 т
(Си; Zn 38 %; Мп 2 %) 50 Ж Т'кип 0,32
Никель — Ж 370 <1,0
10 Ж 20 <0,1
10 Ж 100 <1,0
8-20 ж 40 <0,13
20-70 ж 115 1,0
Никелевые сплавы
НМЖМц28-2,5-1,5 — ж 370 < 1,0
1-насыщ. ж 20— Т'кип <0,5
Н70МФ 10-50 ж 20-100 <0,5
60-90 ж 100 <0,5
ХН65МВ — ж 370 < 1,3 т
10-90 ж 25-75 < 0,5 т
Ниобий 40 ж Т * кип <0,001
Платина 10-90 ж 25-100 <0,05
Свинец — т 25 <0,5
10-30 ж 25 <0,05
10 ж 50-100 <0,5
20-80 ж 30-100 <1,3
Серебро — т 25 <0,5
320
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Т,°С
Цинка хлорид ZnCl2 (продолжение)
Серебро 10-30 Ж 25 <0,05
1-насыщ. Ж < 100 <0,1
Стали
СтЗ — Т 25-155 <0,05
— Ж 350 > 10,0
10-70 Ж 25 >1,3
12X13 20 Ж 20 <0,1
20 Ж 90 <1,0
70 Ж Гкип 3,5
Насыщ. Ж 20 < 1,1
12Х18Н10Т — Ж 350 0,03 т
1-насыщ. Ж 20 <0,1
50 Ж т 1 кип < 1,0 т
60 Ж Гкип < 1,0 кр
10Х17Н13М2Т — Ж 350 0,04 т
20 Ж 20 <0,1
40 Ж 20 0,007
06ХН28МДТ 10-90 Ж 25-105 <0,5
Тантал 1-20 Ж < т 1 кип <0,12
Титан 1-20 Ж гкип <0,12
30 Ж 130 <0,04
80-90 Ж 100 < 1,3
Цирконий 1-20 Ж 20-Гкип <0,12
75-90 Ж 200 >1,3
Чугуны
серый — Т 150 <0,5
— Ж 350 > 10,0
30-70 Ж 20-50 > 10,0
СЧ15,СЧ17 50 Ж Гкип <0,1
Антегмит АТМ-1 <40 Ж <95 С
Асбест 1-насыщ. ж 20-120 с
Графит — ж 500 с
1-насыщ. ж т 1 кип с
Графит, пропитанный феноло- То же ж Гкип с
формальдегидным олигомером
Полиамиды <79 ж 20-60 с
Поливинилхлорид 1-насыщ. ж 60 с
Полиизобутилен <79 ж 20-100 с
Полиметилметакрилат <79 ж 20-60 с
Полипропилен <79 ж 20-60 с
Полиэтилен — т 70 с
<79 ж 20-60 с
Электродные процессы
321
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Цинка хлорид ZnCI2 (продолжение)
Резины на основе
БК <79 Ж 20-100 С
НК 1-насыщ. Ж 20-60 С
ПС <79 Ж 20 С
<79 Ж 60 ОС
СКН, СКС 75 Ж 65 с
СКУ, СКФ <79 Ж 20 с
ХП <79 Ж 20-60 с
ХСПЭ <79 Ж 95 с
Стекло <79 Ж 20-Ткип с
Фторопласт 4 <79 Ж 20-100 с
Эмаль кислотостойкая 1-насыщ. Ж Гкип с
Этилен Н2С=СН2
Алюминий — Г 20 <0,1
Медь — Г 20 <0,1
Никель — Г 20 <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — Г 20-100 <0,1
Н70МФ — Г 20 <0,1
Свинец — Г 20 <0,1
Серебро — Г 20 <0,1
Стали
углеродистая — Г 0-60 <о,Г
12X13 — Г' (-7)-60 <0,001
12Х18Н10Т — г (-7)^60 < 0,001
10Х17Н13М2Т — г (—95)—20 <0,1
06ХН28МДТ — г (-95)-20 <0,1
Тантал — г 20 <0,1
Титан — г 100 <0,05
Антегмит — г -20 с
Графит, пропитанный феноло- — г 20 с
формальдегидным олигомером
Полиамиды — г 20 с
Поливинилхлорид — г (-15)-60 с
Полиизобутилен — г (-15)-60 н
Полиметилметакрилат — г -15 н
Полиэтилен — г -15 с
— г 60 н
Резины на основе
БК, НК, СКС — г 20 н
СКН, СКФ — г 20 с
Фторопласт 4 — г 20-60 с
Эмаль кислотостойкая — г 20 с
* Ускоряет процесс полимеризации этилена.
322
Новый справочник химика и технолога
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза Г, °C
Этилхлорид С2Н5С1
Алюминий Сухой Г 20 <0,1
То же Г 100 <0,5
Влажный Г 20 > 10,0
Вольфрам Сухой Ж Т’кип <0,1
Золото — Ж Т 1 кип <0,1
Иридий — Ж Ткип <0,1
Медь — Г 20 <0,1
Медные сплавы
БрА5, БрА7, БрО4 — Г 20 <0,1
Л80 — Г 20 <0,1
Никель — Г 20 <0,1
Влажный Г, Ж Т -20 1 кип <0,1
Никелевые сплавы
НМЖМц28-2,5-1,5 — г, ж Гкип-20 <0,1
— г 100 <0,5
ХН78Т Сухой г 25-300 <0,05
Н70МФ — г 20 <0,1
хастеллой В — г 20 <0,1
хастеллой С Сухой и влажный ж т 1 кип <0,1
Олово Сухой ж т 1 кип <0,1
Осмий — ж Т’кип <0,1
Свинец — г 50 <0,05
Серебро Сухой ж ^кип <0,1
— г 20 <0,1
Стали
СтЗ Сухой г 20-100 <0,1
Влажный г 20 > 10,0
12X13 — г 20 <0,1
12Х18Н10Т Сухой г 20-100 <0,1
Влажный г 20-100 <0,1 т, кр
10Х17Н13М2Т Сухой г 20-100 <0,1
Влажный г 20-100 < 0,1 т, кр
06ХН28МДТ — г 25-150 <0,05
Тантал — г 20-100 <0,1
Сухой и влажный ж т 1 кип <0,1
Титан — ж Т’кип 0,1
— г 25-100 <0,05
Цинк — г 20 > 10,0
Цирконий — г 30-105 <0,05
Чугуны
серый Сухой г 20 <0,1
Влажный г 20 > 10,0
СЧ15, СЧ17 Сухой и влажный г 20 <0,1
Антегмит — ж ^кип С
Электродные процессы
323
Продолжение таблицы 1.4.62
Материал Характеристика среды Скорость (мм/год) и характер коррозии Стойкость материалов
Концентрация, масс. % Давление, МПа Фаза т,°с
Этилхлорид С2Н5С1 (продолжение)
Графит, пропитанный феноло- — Г <80 С
формальдегидным олигомером Полиамиды —_ Г 25 С
Поливинилхлорид — Г 25 н
Полиизобутилен — Г 20 н
Полиметилметакрилат — Г 20 н
Полипропилен — Г 20 н
Полиэтилен Резины на основе — Г 20 н
НК — Г 25 н
СКИ, СКС, СКУ, ХСПЭ — г 20 н
Стекло ( — г 150 с
Фторопласт 4 — г <70 с
Эмаль кислотостойкая — г 25 с
Растворимость водорода в металлах
Данные приведены для газообразной среды (фаза Г) при давлении р = 0,1 МПа.
Материал 7. °C Растворимость водорода в металлах, см3/100 г
Алюминий 0 <0,0001
20 0,0015
300 0,0010
350 0,0012
400 0,0032
470 0,004
500 0,012
530 0,011
560 0,018
590 0,032
660 0,36
Вольфрам 110-1200 <0,01
1700 0,08
1800 0,11
2000 0,17
2200 0,27
2400 0,33
2600 0,41
2800 0,5
Железо 409 0,39
514 0,83
620 1,31
724 1,98
827 2,69
Таблица 1.4.63
Материал T,°C Растворимость водорода в металлах, см3/100 г
Железо 904 4,25
1033 5,85
1250 8,64
Кобальт 600 0,9
700 1,22
800 1,85
900 2,52
1000 3,32
1100 4,34
1200 5,46
Магний 650 19
Магниевые сплавы: (Mg, Al: 3 %) 650 22
(Mg, Al: 6 %) 650 23
(Mg, Al: 10%) 650 20,5
(Mg, Al: 20 %) 650 18
(Mg, Al: 30 %) 650 15,3
Медь 432 0,15
500 0,30
558 0,20
600 0,70
630 0,26
700 1,1
800 1,6
324
Новый справочник химика и технолога
Продолжение таблицы 1.4.63
Материал Г, °C Растворимость водорода в металлах, см3/100 г
Медь 900 2,4
1000 3,2
Молибден 400 0,4
500 0,9
600 1,3
700 1,8
800 2,2
900 1,9
1000 1,2
1100 1,0
1200 . 0,6
Никель 300 1 2,34
400 3,30
500 4,35
600 5,60
700 6,67
800 7,74
1555 40,6
1690 48,6
Никелевые сплавы: (Ni, Fe: 20 %) 1000 4,7
(Ni, Fe: 50 %) 1000 3,0
(Ni, Fe: 60 %) 1000 3,0
Ниобий (98,5 %) 20 10400
200 9330
300 8800
400 7680
500 4740
600 1850
700 970
800 610
900 400
Олово 20-rM <0,01
400 0,10
500 0,33
Палладий 138 439,7
183 278,6
221 210,9
315 146,5
Продолжение таблицы 1.4.63
Материал Т, °C Растворимость водорода в металлах, см3/100 г
Палладий 418 115,4
519 98,8
620 89,9
822 82,7
1022 81,0
Платина 409 0,07
827 0,1
1033 0,2
1136 0,4
1239 0,6
1342 0,9
Свинец 0-800 0,01
Серебро 400 0,006
600 0,019
700 0,025
800 0,036
900 0,046
Тантал 170 470
268 3660
400 2400
500 1300
600 630
800 320
1000 230
1100 210
Хром 400 0,4
600 0,5
800 1,0
900 2,0
1000 3,0
1100 4,2
1150 5,0
Цирконий 300 27400
400 24000
600 18400
800 16500
1000 7800
1200 3200
Таблица 1.4.64
Коррозия при контактах между металлами и сплавами
Цифровые обозначения: 0 — при соприкосновении указанных металлов и сплавов коррозия не возникает; 1 — небольшая коррозия, однако контакт допустим, если подвижные детали покрыты смазкой, а неподвижные — лаком; 2 — сильная коррозия, металлы и сплавы необходимо разделять защитными покрытиями.
Условия эксплуатации: П — эксплуатация в отапливаемых и вентилируемых помещениях, температура воздуха 10-25 °C, относительная влажность 15-65 %; Н — эксплуатация под навесами и в неотапливаемых помещениях, среда характеризуется отсутствием воздействия атмосферных осадков и солнечной радиации, атмосфера загрязнена небольшим количеством промышленных газов, температура среды от -60 до +60 °C, средняя относительная влажность 95 ± 3 %; А — эксплуатация на открытом воздухе, среда характеризуется воздействием атмосферных осадков, морских туманов, солнечной радиации, атмосфера загрязнена промышленными газами, пылью, температура среды от -60 до +80 °C, средняя относительная влажность 95 ± 3 %.
Соприкасающиеся металлы и сплавы Серебряное, золотое, палладиевое и родиевое покрытия Медь, латунь, бронза Никелевое покрытие Хромовое покрытие (многослойное) Цинковое покрытие (хроматированное) >ытие ное) Оловянное и оловянно-свинцовое покрытие Алюминий и его сплавы Титан его сплавы Азотированная сталь h S?
Кадмиевое покр S э X 5 S э < оксидированные неоксидированные Нержавеющая с О 5 к S X о S а X,
П Н А П н А П Н А П Н А П Н А П н А П Н А П Н А П н А П Н А П Н А П Н А
Серебряное, золотое, палладиевое и родиевое покрытия 0 0 0 0 0 0 0 0 0 0 0 0 2 2 2 2 2 2 0 0 0 1 1 2 2 2 2 0 0 0 1 2 2 0 0 0
Медь, латунь, бронза 0 0 0 0 0 0 0 0 0 0 0 0 2 2 2 2 2 2 0 0 0 0 1 2 1 1 2 0 0 0 1 1 2 0 0 0
Никелевое покрытие 0 0 0 0 0 0 0 0 0 0 0 0 0 1 2 0 1 2 0 0 0 0 0 1 0 1 2 0 0 0 0 0 0 0 0 0
Хромовое покрытие (многослойное) 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 1 1 0 0 0 0 0 0 0 1 2 0 0 0 0 0 0 0 0 0
Цинковое покрытие (хроматированно е) 2 2 2 2 2 2 0 1 2 0 1 1 0 0 0 0 0 0 0 1 2 0 1 1 0 0 0 1 1 2 0 0 0 0 1 2
Кадмиевое покрытие (хроматированное) 2 2 2 2 2 2 0 1 2 0 1 1 0 0 0 0 0 0 0 1 1 0 1 1 0 0 0 1 1 2 0 0 1 0 1 1
Оловянное и оловянно-свинцовое покрытие 0 0 0 0 0 0 0 0 0 0 0 0 0 1 2 0 1 1 0 0 0 0 0 0 0 1 1 0 0 0 0 0 1 0 0 0
Электродные процессы
Новый справочник химика и технолога
Электродные процессы
327
Литература
1. Рекомендации по выбору химически стойких материалов для прокладок: В 2-х кн. / Под ред. А.В. Горяйновой. М.: Машиностроение, 1980. Кн. 1. 368 с.
2. Мигай Л.Л., Тарицына Т.А. Коррозионная стойкость материалов в галогенах и их соединениях: Справочник. М.: Металлургия, 1988. 304 с.
3. Рускол Ю.С. Титановые конструкционные сплавы в химических производствах: Справочное издание. М.: Химия, 1989. 288 с.
4. Туфанов Д.Г. Коррозионная стойкость нержавеющих сталей, сплавов и чистых металлов. М.: Металлургия, 1982. 352 с.
5. Повышение эксплуатационной надежности оборудования, работающего в агрессивных средах: Сб. научн. тр. / Под ред. В.С. Зотикова, М.Н. Игнатова. Пермь: Изд-во Пермского политехи, ин-та. 1990. 256 с.
6. Рачев X., Стефанова С. Справочник по коррозии / Пер с болг. Под ред. С.И. Нейковского. М.: Мир, 1982. 520 с.
7. Натай И.Н., Томашов Н.Д., Селедцов Д.К., Кочергина В.М. // Защита металлов. 1981. Т. 17, № 5. С. 530— 537.
8. Барабанов В.Г., Зотиков В.С., Лимонова Л.П. и др. Коррозия оборудования в производстве галогенсодержащих веществ: Справочное издание / Под ред. В.С. Зотикова. СПб.: ТЕЗА, 1998. 252 с.
9. Коррозионная стойкость оборудования химических производств. Коррозия под действием теплоносителей, хладагентов и рабочих тел: Справочное издание / Под ред. А.М. Сухотина, В.М. Беренблит. Л.: Химия, 1988. 360 с.
10. Коррозия конструкционных материалов. Газы и неорганические кислоты: Справочник: В 2-х кн. / В.В. Батраков, В.П. Батраков и др. Неорганические кислоты. М.: Металлургия, 1990. Кн. 2. 320 с.
11. Мамулова Н.С. и др. Все о коррозии: Справочник. СПб.: Химиздат, 2000. 577 с.
Раздел 2 ХИМИЧЕСКАЯ КИНЕТИКА И ДИФФУЗИЯ
2.1. Кинетика
2.1.1. Основные понятия в химической кинетике гомогенных химических реакций
(доц., к.т.н. В.А. Иванов)
Настоящий раздел справочника посвящен рассмотрению общих закономерностей и количественных соотношений, необходимых для решения кинетических задач, возникающих при исследовании химических реакций и при их практической реализации.
Химическая кинетика изучает химические превращения веществ как процесс, протекающий по определенному механизму с характерными для него закономерностями во времени. Объектами химической кинетики служат реакции, в которых принимают участие различные реакционные частицы, в том числе — молекулы, ионы, радикалы, комплексы и т. д. Кинетические данные дают возможность уточнить механизм реакции, выявить корреляционные зависимости между реакционной способностью молекул и их строением; они необходимы при проектировании реакторов для химического синтеза, при разработке новых и модернизации старых технологических процессов.
Корректное использование кинетического анализа возможно только в том случае, если обо всех реагентах и продуктах существует качественная и количественная информация. Именно на основе материального баланса составляется стехиометрическое уравнение реакции, где количество израсходованных веществ соответствует количеству образовавшихся соединений. Кинетические исследования обычно начинают с выявления влияния на состав продуктов реакции времени реакции в периодических реакторах или времени контакта в проточных реакторах.
Классификация кинетических кривых
Первичные данные кинетических экспериментов представляют собой набор концентраций химических компонентов реагирующей системы (или некоторых пропорциональных им величин) при разных значениях времени реакции. Кинетическая кривая — изменение
концентрации реагента или продукта, или связанного с ними свойства системы во времени в результате протекания химического процесса. Эта зависимость концентрации вещества от времени протекания реакции может быть выражена в графической, табличной или аналитической форме. Одним из основных кинетических параметров, характеризующих химическую реакцию, является скорость химической реакции — количество реакционных частиц определенного вида, реагирующих в единицу времени (dc/dt).
Форма полученных кинетических кривых позволяет выявить особенности кинетики изучаемой реакции. Возможные формы кинетических кривых приведены на рис. 2.1.1. Эти кривые характеризуют 4 основных типа расходования и накопления соединений в реакционной смеси:
1. Реагент расходуется практически нацело. Его концентрация уменьшается во времени: с^) > c(Z2) при /1 < /2, или dc/dt < 0, и, если реакция идет до конца, то c(t) —> 0 при / —> оо.
2. Реагент расходуется до конечной степени превращения, меньшей 100 %, что характерно для химических процессов, основу которых составляют: обратимые реакции; каталитические реакции, в ходе которых катализатор отравляется; сложные реакции, протекающие с образованием ингибитора. Кроме того, не расходуются до конца реагенты в многокомпонентной реакции, если реагенты взяты в нестехиометрическом соотношении. В этих случаях при t оо сда ± 0.
3. Концентрация продукта возрастает монотонно. Если вещество — конечный продукт, то его концентрация увеличивается во времени: c(ti) < c(t2) при Ц < t2, или dc/dt > 0.
4. Концентрация продукта проходит через максимум. Подобный тип зависимости характерен для промежуточного продукта, его концентрация увеличивается, проходит через максимум и уменьшается, т. е. какое-то время dc/dt > 0 для Л < tm^x, а затем dc/dt < О Для t > /щах (где /щах — время достижения максимальной концентрации промежуточного продукта).
330
Новый справочник химика и технолога
Для каждого из указанных типов возможны кривые четырех-шести видов, которые классифицируют по знаку второй производной концентрации по времени (см. на рис. 2.1.1 сверху вниз):
- скорость расходования реагента уменьшается в ходе химической реакции: dc/dt < 0 и cfc/dt2 > 0 . Этот вид кинетической кривой характерен для простых реакций;
- реакция протекает с постоянной скоростью, т. е. dc/dt = const и cfc/dt2 = 0;
- скорость расходования реагента увеличивается и, в некоторых случаях, сложная реакция переходит в режим взрыва. При этом \dc/dt\ нарастает во времени, ctc/dt2 < 0;
- скорость реакции сначала увеличивается, cfc/dt2 < 0 при t < /тах, проходит через максимум, когда \dc/dt\ = const при / = Zmax, а затем уменьшается и при t > /щах d* c/dt2 > 0. Подобный вид кинетических кривых характерен для автокаталитических реакций;
- в начальный период реакция протекает с низкой скоростью, затем ускоряется {dc/dt увеличивается во времени и fc/dt2 < 0) и переходит в стационарный режим dc/dt = const и (fc/dt2 = 0;
- скорость процесса периодически проходит через минимум и максимум, и cfc/dt2 в ходе реакции несколько раз меняет свой знак. Подобный вид кинетических кривых характерен для осциллирующих реакций.
Рис. 2.1.1. Типы кинетических кривых «концентрация—время». Знаки у кривых характеризуют производную d/cldt2
Форма и тип полученных кинетических кривых позволяют выявить особенности в кинетике исследуемой реакции и осуществить кинетическую классификацию химических реакций.
Кинетическая классификация химических реакций
Согласно кинетической классификации, реакции делят на простые и сложные. В простой реакции реагенты
непосредственно превращаются в конечные продукты без образования промежуточных, в реакционной системе отсутствуют катализаторы и ингибиторы, а химический процесс состоит из однотипных элементарных актов. Сложная (многостадийная) реакция состоит из элементарных стадий, связанных друг с другом через реагенты или продукты. Суммарные признаки кинетически простой и сложной реакций приведены в табл. 2.1.1.
Химическая кинетика и диффузия
331
Таблица 2.1.1
Кинетические признаки, лежащие в основе различий простой и сложной реакций
Простая реакция Сложная реакция
Число реагирующих веществ невелико, а количественный состав продуктов реакции сравнительно прост. Количественные соотношения продуктов не зависят от условий протекания реакции (времени, температуры, состава реагирующей исходной смеси, давления) Скорость реакции монотонно увеличивается с ростом температуры; энергия активации всегда положительна и иногда может зависеть от температуры эксперимента Скорость реакции монотонно меняется при изменении условий проведения реакции и не зависит от материала и размеров реакционного сосуда Число реагирующих веществ значительно. Состав продуктов реакции определяется временем, температурой, исходным составом реагентов и давлением реакционной смеси Скорость реакции с ростом температуры может увеличиваться, уменьшаться или проходить через экстремум и зависит от материала и размеров реакционного сосуда. Энергия активации, определенная в широком интервале температур, может быть положительной, отрицательной или близкой к нулю При варьировании условий проведения реакции возможно скачкообразное изменение скорости реакции на несколько порядков, обусловленное критическими явлениями катализа или ингибирования химической реакции
Другой тип кинетической классификации химических реакций основан на делении простых реакций по числу частиц, участвующих в превращениях: мономо-лекулярная реакция — реакция, в элементарном акте которой превращается одна частица (молекула, радикал, ион); бимолекулярная реакция — реакция, в которой претерпевают превращения две частицы (молекула, радикал, ион); тримолекулярная реакция — реакция, протекающая с участием трех частиц при их одновременном столкновении.
Основные типы простых мономолекулярных и бимолекулярных реакций приведены в табл. 2.1.2.
Тримолекулярные реакции характерны для каталитических и ингибированных процессов, а также процессов, протекающих с выделением значительного количества энергии, когда стабилизация образовавшейся в ходе бимолекулярной химической реакции молекулы достигается путем передачи энергии третьей частице.
Кинетические и термодинамические параметры химических реакций
Основная цель анализа кинетических кривых с -fif) состоит в том, чтобы найти математическое уравнение,
которое описывает их форму. Раздел химической кинетики, в котором рассматривается метод нахождения зависимости скорости химической реакции от концентраций реагирующих веществ, получил название формальной кинетики. В основу способа нахождения скорости реакции положен закон действующих масс, согласно которому скорость химической реакции пропорциональна концентрациям реагирующих веществ, возведенным в некоторые степени.
Для реакции
v,ai+v2a2+...+v)(a)(->v;ai'+v'2a;+...+vX (2.1.1.1) скорость химической реакции v равна
=Пса, , (2.1.1.2)
I
где k — константа скорости химической реакции', при условии равенства концентраций реагентов единице скорость реакции совпадает с величиной константы скорости; сА — концентрация реагента А(; п, — показатель степени при реагенте, который называют порядком реакции по реагенту А.,.
Таблица 2.1.2
Типы простых мономолекулярных и бимолекулярных реакций
Простые мономолекулярные реакции Простые бимолекулярные реакции
Реакция изомеризации — превращение одного реагента (частицы) в один продукт: СН3С(О)— СН2С(О)ОС2Н5—► СН3С(ОН)= СНС(О)ОС2Н5 (СН3)2СНСН2—>• (СН3)3С СНз(СН2)5СН2—► СН3СН2СН(СН2)3СН3 Диссоциация — распад молекулы, связанный с разрывом одной связи: С6Н5С(СН3)2ООН-» С6Н5С(СН3)26+ОН Реакция присоединения — взаимодействие двух реагентов друг с другом, из которых, по крайней мере, один является молекулой: (СН3)3С + СН2= С(СН3)2—► (СН3)3ССН2С(СН3)2 сн3 + сн2= СНСН3—► СН3СН2СНСН3 Реакции электрофильного и нуклеофильного замещения — реакции замены активной группы в молекуле на другую группу: СН3СН2Вг + ОН"-*- СН3СН2ОН + Вг"
332
Новый справочник химика и технолога
Продолжение таблицы 2.1.2
Простые мономолекулярные реакции Простые бимолекулярные реакции
Реакция распада — превращение одной молекулы (частицы) в две или несколько частиц (молекула, радикал, ион): RN= NR—► 2R + N2 СН2СН2(СН2)4СН3—► СН2= СН2 + СН2(СН2)3СН3 сн3— сн— сн2— сн3—► сн3сн= сн2 + сн3 Реакция элиминирования — распад многоатомной молекулы на несколько молекул: —> СН3СН= С(СН3)2 + НВг СН3СН2СВг(СН3)2— —> СН3СН2С(СН3)= СН2 + НВг Реакция отрыва — взаимодействие молекулы с атомом, радикалом или ионом, приводящее к образованию молекулярного продукта и новой активной частицы: СН3СН3 + С1—► СН3СН2 + НС1 СН3СНСН2СН3 + СН3СН(СН3)2—► ► СН3СН2СН2СН3 + С(СН3)3 Реакция рекомбинации — взаимодействие двух атомов, радикалов, ионов, приводящее к образованию молекулярного продукта: СН3СН2 + Вг—► СН3СН2Вг СН3 + С1-► СН3С1 Реакция диспропорционирования — перераспределение атомов или их группировок между двумя одинаковыми молекулами, радикалами в результате их взаимодействия: 2СН3СН2—► СН3СН3 + СН2= СН2 2С6Н5СНО + КОН->- С6Н5С(О)ОК + СбНзСЩОН
Параметры (к, п,) в кинетическом уравнении химической реакцииг^определяют расчетным методом при обработке кинетического эксперимента (кинетических кривых расходования реагентов и накопления промежуточных и конечных продуктов). Для простых реакций, когда реакция протекает в одну стадию (элементарный химический процесс), порядок по реагенту совпадает по величине со стехиометрическим коэффициентом при реагенте в уравнении (2.1.1.1). Для сложных реакций порядок реакции по реагенту, как правило, не равен стехиометрическому коэффициенту (и, v,) и может быть целочисленным, дробным или отрицательным. Общий порядок реакции равен сумме показателей степени по всем реагентам: п = В сложных реакциях, когда химический процесс протекает через ряд промежуточных стадий, уравнение (2.1.1.2) является формальной записью скорости химического процесса, при этом порядок реакции может быть дробным и отрицательным. Если сложная реакция состоит из нескольких последовательных стадий, из которых медленная определяет скорость всего процесса, то порядок суммарной реакции обычно равен порядку этой определяющей скорость реакции.
Под скоростью реакции (v) понимают скорость превращения вещества в единице объема (И). Со скоро-стью превращения реагента ---принимающего уча-
dt
стие в химической реакции, скорость реакции связана через стехиометрические коэффициенты vA :
1 ^А, ___1 dCA2 ______1
vA| dt vA2 dt vAk dt
1 de*. _ 1 dc^ 1 JcA-vA] dt v'A dt vA dt
(2.1.1.3)
Скорость гомогенных химических реакций по реагенту измеряют по изменению количества реагента или продукта реакции за единицу времени (рис. 2.1.2).
Рис. 2.1.2. Расчет скорости расходования реагента А с использованием графика зависимости концентрации реагента А от времени
Количественная аналитическая запись этого соотношения зависит от условий проведения реакции, в том числе от типа реактора и режима, в котором осуществляется синтез целевых продуктов.
Химическая кинетика и диффузия
333
Для периодического реактора идеального перемешивания скорость простой химической реакции по реагенту Ак или по продукту реакции А* составляет
±v=——(2.1.1.4) к Vdt
где знак «минус» — для реагента, знак «плюс» — для продукта, п — число молей реагента или продукта, V— объем реактора.
Если реакция проводится в проточном реакторе идеального вытеснения, то число молей компонентов пк, проходящих через объем dV за единицу времени (мольный поток), изменяется на dnK и скорость реакции по веществу равна
В проточном реакторе идеального перемешивания скорость реакции по компоненту связана с изменением мольного потока Длк в объеме реактора
±V,=^. <2.1.16)
Кинетическое уравнение для простой реакции выполняется при определенных условиях:
1. При протекании реакции не нарушается распределение частиц по энергиям Максвелла — Больцмана (по степеням свободы реагирующих частиц). Это возможно, когда скорость передачи энергии от частицы к частице больше скорости их химического превращения.
2. Изменение концентраций реагентов не изменяет свойства среды и физического состояния реагентов.
3. Химическая реакция осуществляется только за счет тепловой энергии.
4. Константа скорости реакции и порядки реакции по реагентам остаются постоянными в течение всей реакции, а также при вариации начальных концентраций реагентов (при неизменных условиях эксперимента).
В кинетике простых реакций наиболее важны реакции первого, второго и третьего порядков. Простые реакции более высокого порядка практически неосуществимы из-за чрезвычайно малой вероятности одновременного соударения четырех или более молекул.
Скорость химической реакции, как правило, с ростом температуры резко увеличивается. Для большинства химических реакций влияние температуры на скорость реакции отражается на типе кривых, описывающих зависимость скорости реакции от температуры (рис. 2.1.3).
Особо следует рассмотреть тип кривых д и е. Вид кривой д связан с возможностью активации побочного процесса расходования реагента, как правило, на катализаторе. Подобные зависимости получены для некоторых процессов окисления и полимеризации. Кривые
типа е характерны для сложной химической реакции, в число элементарных стадий которых входит стадия образования промежуточного интермедиата по обратимой реакции, причем вероятность его образования с ростом температуры уменьшается.
Согласно эмпирическому правилу повышение температуры на 10 °C приводит к росту скорости реакции в 2-4 раза. В качестве характеристики подобной зависимости используют температурный коэффициент скорости реакции уг, который представляет отношение констант скоростей химических реакций при температуре Т и Т+ 10: у7’= kT+io / kT. Однако температурный коэффициент сам является функцией температуры, т. к. скорость взаимодействия полярных молекул (ионов) во многом определяется степенью их сольватации, диэлектрической проницаемостью раствора, т. е. параметрами, зависящими от температуры, что уменьшает возможности его практического применения.
Превращение исходных реагентов в продукты реакции связано с преодолением потенциального барьера химической реакции Е (рис. 2.1.4), обусловленного затратой энергии на внутреннюю перестройку молекулы (иона, радикала), и сопровождается разрывом или ослаблением некоторых химических связей в активированной частице, превращающейся в продукт реакции.
Рис. 2.1.3. Основные типы кинетических кривых «скорость реакции—температура», наблюдаемые в лабораторных и промышленных реакторах: а) простые некаталитические реакции; б) гетерогенные реакции; в) реакции с автоускорением, сопровождающиеся взрывом; г) каталитические и ферментативные реакции (повышение температуры приводит к разрушению или дезактивации катализатора); д) многостадийные сложные реакции;
е) реакции с термодинамическими ограничениями Количество энергии (кинетическая энергия молекул, энергия света и др.), которое должно быть преобразовано в потенцальную (внутреннюю) энергию реагирующих молекул, чтобы протекание химической реакции стало возможным, называется энергией активации.
334
Новый справочник химика и технолога
Рис. 2.1.4. Зависимость потенциальной энергии системы от типа реакции: Еа — энергия активации экзотермической реакции, Еа — энергия активации эндотермической реакции
Доля частиц с энергией больше энергии активации составляет e~EalRT, и зависимость константы скорости к от температуры может быть представлена в экспоненциальной форме:
k = Ae~E°IRT (2.1.1.7)
или логарифмической форме:
/ 1п£ = 1пЯ-Е IRT, (2.1.1.8)
/
где Еа — эффективная (эмпирическая) энергия активации; А — константа, называемая предэкспоненциаль-ным множителем (предэкспонентой), частотным фактором или аррениусовским множителем.
Уравнение Аррениуса, связывающее константу скорости химической реакции с ее энергией активации, выведено теоретически на основе равновесной термодинамики при следующих предположениях:
1. В химические реакции вступает активированная частица, образование которой требует затраты определенной энергии Еа.
2. Активация частицы протекает по обратимой реакции, характеризующейся определенной константой равновесия.
3. Концентрация активных частиц во много раз меньше концентрации исходных частиц.
4. Превращение активированной частицы протекает со скоростью, не зависящей от температуры, при которой осуществляется химический процесс.
Для объяснения физического смысла параметра А используют две основные теории — теорию столкновений и теорию активированного комплекса.
В первом приближении, по теории столкновений, реагирующие частицы рассматриваются как жесткие шарики, не обладающие силами притяжения друг к другу. Вероятность протекания реакции между двумя частицами определяется частотой их столкновения Z, причем кинетическая энергия соударяющихся частиц должна быть равна или больше энергии активации.
В соответствии с законом распределения Максвелла — Больцмана, доля частиц п , обладающих необходимой кинетической энергией для осуществления химической реакции, определяется как
* / -EJRT п / п = е °
Согласно кинетической теории газов, число столкновений двух частиц может быть описано с помощью уравнения:
2 = [(ЯЛ + ЛВ)2^Г(^ + ^)
«А «в =^«А«в- (2.1.1.9)
где Z — число соударений молекул А и В в единице объема за единицу времени; R, т и п — соответственно радиус, масса и концентрация (число молекул А и В); к — постоянная Больцмана.
Для реакций, происходящих в газовой фазе, величина Z зависит от температуры, массы и размеров молекул и меняется в диапазоне 1010—1012 л / (моль- с).
Расчет константы скорости по частоте соударений и энергии активации
Уравнение
к = Ze~EJRr
(2.1.1.10)
удовлетворительно совпадает с экспериментальными данными для ряда газофазных реакций. Однако в большинстве случаев, и особенно для жидкофазных реакций, опытные значения величин констант скорости были меньше вычисленных в 10-108 раз. Это обусловлено тем, что для того, чтобы столкновение частиц могло привести к химической реакции, частицы в момент удара должны быть определенным образом ориентированы в пространстве. Поэтому для учета вероятности пространственной ориентации молекул при столкновении, благоприятной для протекания реакции, был введен стерический фактор р < 1:
k=pZe~Ea'RT. (2.1.1.11)
Значения величин стерического фактора некоторых газофазных бимолекулярных реакций представлены в табл. 2.1.3.
Таблица 2.1.3
Значения стерического фактора р газофазных реакций
Реакция Р
Н + Н2->Н2 + Н СН3 + Н2 -> СН4 + Н СН3 + СН3СН3 СН4 + СН3СН2 сн3 + с6н5сн3 -> сн4 + С6Н5СН2 СНз + сн2=сн2 -> СН3СН2СН2 СНз + СНз -> СН3СН3 С2Н4 + бутадиен -> цикло-С6Ню 2 молекулы циклопентадиена -> Димер 1 10 3 7- Ю'4 з • ю-4 10’3 0,5 4 • 10’5 3 • 10’7
Химическая кинетика и диффузия
335
Согласно теории активированного комплекса, элементарная реакция протекает непрерывно от начального до конечного состояния и проходит через образование активированного комплекса, характеризующегося максимальной энергией. Для элементарных реакций, протекающих в газовой фазе, скорость реакции равна
рассмотрении реакции в растворах, где разность энергий между активированным комплексом и исходными частицами измеряется при постоянном давлении, используют энтальпию, величина которой составляет АН* = Еа~ RT. Скорость реакции частиц в растворе описывается уравнением
v ~
h h
kT Q" h QA
„-EJRT „ e a c,c
(2.1.1.12)
кТ _ кТ ^ц^
v = c —=—К с, с, =—е с. с. =
h h Ai h * А2
=^^/^/лгсАсА гГ/яе-£“/лгсАс h 2 h 1
(2.1.1.13)
где c* — концентрация активированного комплекса, t — среднее время прохождения активированного комплекса через энергетический барьер t = hlkT, к -— константа Больцмана, К* — константа равновесия реакции образования активированного комплекса К*
А1 + А2 < >[А, • А2]*, 2а , 2Aj , 2* — статистические
суммы состояний реагирующих частиц Аь А2, [А] -А2]*.
Независимые расчеты предэкспоненциального мно-кТ О*
жителя А=-------— показали хорошее совпадение ре-
зультатов с экспериментально определенными величинами для реакций, протекающих в газовой фазе.
В жидкофазных реакциях в растворе трудно рассчитать суммы состояний реагирующих частиц. При
где AG'*, AS* — изменение энергии Гиббса и изменение энтропии соответственно при переходе системы из исходного состояния в активированный комплекс.
Величина AS* характеризует меру упорядоченности системы и для бимолекулярных реакций составляет отрицательную величину; AS* = (-168)4-(-63) Дж/(моль-К), для мономолекулярных реакций AS* > 0.
Полученное уравнение (2.1.1.13) позволяет рассчитать параметры AG*, AS*, AHZ на основе экспериментально определенных величин А и Еа.
В табл. 2.1.4, 2.1.5 приведены коэффициенты пересчета для энергий активации и предэкспонент, рассчитанные в соответствии с теориями столкновений и активированного комплекса.
Таблица 2.1.4
Коэффициенты пересчета для энергий активации бимолекулярных реакций, вычисленные с использованием уравнения Аррениуса, теорий столкновений, активированного комплекса и энтальпии активации
Пояснение, а = 0,5, если один из реагентов атом, а = 1,5, если один из реагентов двухатомная молекула, а = 2, если реаги-
руют две многоатомные молекулы.
Е Уравнение Аррениуса Теория столкновений Теория активированного комплекса Энтальпия активации
Ея Е' Ео ЫТ Ея E.d-0,5RT Е.Л + aRT EZ~2RT £' + О,57?Г £' £' + (« + О,5)7?Г £' - 1,5/?Г £0 - aRT EQ-(a + 0,5)RT Ео E0-(2 + a)RT АН* +2RT МГ+ l,5RT ЬН* + (2 +a)RT МГ
Таблица 2.1.5
Коэффициенты пересчета для предэкспонент бимолекулярных реакций, вычисленные с использованием уравнения Аррениуса, теорий столкновений и активированного комплекса
Пояснение, а = 0,5, если один из реагентов атом, а = 1,5, если один из реагентов двухатомная молекула, а = 2, если реагируют две многоатомные молекулы. — постоянная Авогадро.
Предэкспонента А, л/ (моль • с) pZ, см3 / (молекула • с) (Г 3 , , см / молекула qa2
А А UT3NApZje RTe~a Q" c_a Ю3А 0A|0A2 6
pZ 103 А (аа^)" pZ EE c-(a+0,5) Q* N.h QAxQA2
О' 103Л . а Ае PZNJl O'
0,,0,, RT RT Qk2
336
Новый справочник химика и технолога
2.1.1.1. Формальная кинетика химических реакций, протекающих в реакторах периодического и непрерывного действия
Кинетика гомогенных химических реакций в статических условиях
Химико-технологические процессы с применением реакторов периодического действия обычно используют в малотоннажной химии, а также при невысокой скорости химических реакций или при затруднениях в транспортировке исходных реагентов или продуктов реакции. Реакторы периодического или полупериоди-ческого действия представляют собой емкостные аппараты, снабженные механическими перемешивающими устройствами и элементами поверхности теплообмена, необходимыми для поддержания определенного температурного режима. В таком реакторе для жидкостей с вязкостью 10310! Па • с при идеальном перемешивании температура реакционной массы постоянна по всему объему, а концентрации компонентов в каждый момент времени во всех точках одинаковы и меняются лишь со временем. Именно для подобных реакторов наиболее полно развиты теоретические и практические положения формальной химической кинетики.
Кинетика гомогенных химических реакций в реакторах идеального перемешивания
Для реакции (2.1.1.1) скорость расходования реагента может быть представлена в виде:
а скорость накопления продукта реакции:
Исходя из определения мольно-объемной концентрации скорость расходования реагента, например соединения А, составляет
где dn^ — разность числа молей вещества в начальный и конечный момент времени, V— объем рассматриваемой системы.
Если объем реакционной массы не изменяется в ходе химической реакции, то его можно ввести под знак дифференциала
(Ос
vA=~ НА • (2.1.1.17)
к СТ )у
Для простых мономолекулярных реакций скорость реакции совпадает со скоростью расходования реагента.
В основе способов определения порядка и константы скорости реакции лежит определение характера за
висимости скорости расходования реагента от его концентрации. Для упрощения кинетического расчета разработаны два основных метода — метод разбавления и метод использования реагентов в эквимолекулярных количествах.
Наиболее широко распространен прием определения порядка по всем реагентам би- и тримолекулярных реакций, основанный на последовательном определении порядка (метод разбавления). Согласно этому методу, исходные концентрации всех реагентов реакционной смеси, кроме одного, например Аод, взяты в таком избытке >сАо ,...,сАо> >cAoJ, что позволяет пренебречь их изменением в ходе опыта. В этом случае для бимолекулярной реакции уравнение (2.1.1.1) может быть преобразовано к виду уравнения для моно-молекулярной реакции «-порядка:
Ч=*Ч=ЬХсл.’ (2.1.1.18)
где п = п\.
Если все реагенты взяты в стехиометрическом соотношении, то в бимолекулярной реакции вещества А) и А2 расходуются в постоянной пропорции, соотношение реагентов сохраняется в течение всей реакции и скорость реакции равна
v = k”c\ =^v1’("2+1)4, (2.1.1.19)
где п = п\ + «г-
Сочетанием методов разбавления и использования реагентов в эквимолекулярных количествах можно определить константу скорости реакции, порядок по реагенту и суммарный порядок реакции.
При определении кинетических параметров химической реакции, базирующихся на вышеприведенных приемах, используют либо одну полную кинетическую кривую, либо начальные участки серии кинетических кривых.
При использовании одной кинетической кривой наиболее широкое применение нашли следующие методы:
1. Метод, основанный на исследовании зависимости изменяющейся во времени скорости реакции от концентрации реагента. Скорость реакции при данной текущей концентрации реагента при исследовании процесса методом разбавления определяют дифференцированием кинетической кривой. Используя логарифмическую форму уравнения (2.1.1.18)
(dn. А
1пуА] =1п1 1 = In &, + иА11п сА|, (2.1.1.20)
графическим (рис. 2.1.5) или иным методом можно рассчитать порядок реакции. Зависимость в координатах lnvA -lncA) в случае простых реакций должна быть прямолинейной, отклонение от линейности связано с протеканием в системе сложных реакций.
Химическая кинетика и диффузия
337
Рис. 2.1.5. Графическое определение порядка реакции по заданному веществу
2. Метод, базирующийся на трансформации кинетической кривой расходования исходного вещества. Кинетическую кривую расходования реагента трансформируют в координатах fla)—t, где вид функции соответствует интегральной форме кинетического уравнения (табл. 2.1.6). Спрямление данных по одной из этих зависимостей в широком интервале исследованных концентраций дает возможность сделать вывод о порядке реакции, обосновать вид интегрального
ния, с высокой степенью вероятности описывающего кинетическую кривую расходования реагента, и рассчитать константу скорости.
3. Метод, основанный на определении времени превращения реагента на 1/р часть. Измерив текущую концентрацию реагента при разных значениях t, используя табличные данные (табл. 2.1.7), можно определить порядок и константу скорости реакции по времени превращения исходного вещества на определенную долю, например на одну четверть г1/4, на одну треть /1/3 ит. д.
4. Метод, в основе которого лежит зависимость глубины превращения реагента от времени. Соотношение, связывающее относительную глубину превращения у = l-cA/cA0 со временем, необходимым для ее достижения, имеет вид:
t _ 1 п
7~Л(ЧГ1+2Г
(2.1.1.21)
Кинетические параметры реакции определяют путем трансформации кинетической кривой расходования реагента в зависимость с координатами t—(y-t).
уравне-
Интегральные и дифференциальные формы уравнений скорости для моно- и бимолекулярных реакций
Таблица 2.1.6
Дифференциальная форма de, кинетических уравнении — % Интегральная форма кинетических уравнений
к 1 - х = кс' t Ао
К2
К -Inx = kt
кс^ x'l2-\ = -kc^t 2 0
кс\ х1 -1 = кс, t Ао
кс'«
[(х + х)1/2 + Х,/2 J 2
>0 1п< (х + х),/2+ х'/2 х1/2[(1 + хГ + х"\
ксКсх£ 0 * 1 1 II м | л-о <5
<0 z д!/2 z \1/2 , .и 1 + х 1 . -11 x + х 1 к, 4i/2 1/2 th1 - -th =-(-%) Сд/ 1 -X ) 1 -X ) 2^ А“
^/24/2 ^0 / \i/2 1 ( % + X 1 \1/2 к 1/2 t X J + =iXCA-r г
338
Новый справочник химика и технолога
Дифференциальная форма - dcA кинетических уравнении — dt X Интегральная форма кинетических уравнений
кс3/2сш А,еА СВ 0 X
Лсв <*+х;
*<4 >0 / \l/2 z х1/2 л-' 1 -th4 - =-x''2d”rt IxJ IxJ 2Л 4"
0 XV2_1=* И ; 2
<0 In- (1 + х)'/2(*1/2+|хГ2) (* + x)1/2(l + |xf/2)
/0 i x + TL i In 5 ——^t^Xca rt b(i+x)j
^АСВ 0 x"1 -1 = kc. rt Ao
(х + хГ-С + х)1'2
7^0 (1 + xf‘/2- xm (x + x)-1/2 = ^A//2t
^4/2 0 x4 -1 = kc.P^t
(х + хГ’-О + х)-1 =^a/2'
Таблица 2.1.7
Зависимость периода превращения на \!р часть от порядка реакции п
t}/p n =1 n =2 n =3 n
1 1 4 1 1 f-Г-*
t\H k} 3 ЗЛ2 Co 18£3Со
(«-DW
t\/3 1 , 3 —In— Ь 2 1 2fc2c0 5 8fc3c02 । K" СП 1 СЧ
(rt-wr
—In2 К 1 3 2"-I-l
t\!2 ^2C0 2A3C0
—In4 К 3 15 4n’*-l
*3/4 2^c0
Л/2 2"“’ -1
2,4 3 3,86 PY-1 .
Л/4 Ijj
Л/2 A/3 г"-1-!
1,7 2 2,4 7 en I
Л/2 ^3/4 0,5 0,33 0,20 2й'1-1
Продолжение таблицы 2.1.6
Для установления величин п и к с использованием начальных скоростей реакции предложены методы:
1. Метод, основанный на использовании зависимости начальной скорости реакции от исходной концентрации реагента. В основе методики определения ки
нетических параметров реакции лежат методы разбавления и использования реагентов в эквимолеку
лярных количествах. Проводится серия опытов с определением величин начальных скоростей реакции с последующей обработкой экспериментального материала на основе уравнений (2.1.1.18) и (2.1.1.19), представленных в логарифмической форме. Для надежного определения порядка и константы реакции необходимо
менять концентрацию реагента Ai в широких пределах.
Данный метод находит широкое применение при
исследовании химических реакций, когда нельзя пренебречь влиянием продуктов на их кинетику.
2. Метод, использующий величины отношения логарифмов начальных скоростей реакции. Относительно точно порядок реакции (и) рассчитывается аналитиче-ским методом с использова- 1g ~
нием начальных скоростей w =---------р-. (2.1.1.22)
реакции vj, v2, соответст- 1g
вующих начальным концен- соа, трациям реагентов и с2^ :
Химическая кинетика и диффузия
339
Как следует из вышеприведенного материала, при расчете константы и определении порядка реакции широко используются дифференциальные и интегральные уравнения скорости.
Главное преимущество дифференциального метода состоит в том, что не требуется никаких предположений по поводу механизма реакции, так как дифференциальное уравнение скорости выводится непосредственно из эксперимента. Основной недостаток метода заключается в том, что исходные данные сА—t нужно сначала перевести в данные v— сА («второе поколение» экспериментальных данных). Как правило, экспериментальные кривые с—t не записываются непрерывно, а строятся по отдельным точкам. Повысить точность метода можно с помощью графических редакторов на ЭВМ путем аппроксимации точек какой-либо кривой, описываемой/ известной функцией, с последующим ее дифференцированием.
Интегральный метод основан на использовании экспериментальной зависимости сА—/, при этом кривые расходования (накопления) вещества обрабатываются без предварительного преобразования. Глав
ный недостаток интегрального метода состоит в том, что нужно априори предположить вид интегрального уравнения скорости, а затем проверить правомочность его использования. При квалифицированном использовании данный метод широко применяется при исследовании простых реакций, а также реакций псевдопервого порядка.
В основу расчета кинетических параметров сложных химических реакций положен принцип независимости различных реакций, протекающих в одной системе. Согласно этому принципу, скорость каждой реакции в системе прямо пропорциональна концентрациям реагирующих веществ и независима от других реакций. Полученные с его использованием уравнения для некоторых обратимых, параллельных и последовательных реакций хорошо согласуются с опытом и представлены в табл. 2.1.8,2.1.9.
Численные значения констант зависят от единиц, выбранных для измерения времени и концентрации. Коэффициенты пересчета приведены в табл. 2.1.10, 2.1.11.
Интегральные и дифференциальные формы уравнений скорости для обратимых реакций первого и второго порядка
Таблица 2.1.8
(при /—► 00, САО —► с дос, Сро —► Сроо, к = kx!k-d
Реакция Дифференциальное уравнение Интегральное уравнение
dcK dcp . . = —- - k}c. - к ,ср dt dt 1 А '* Р СК =САос+(СА0-САоо> ' к, к. ~ 1 1 L- См ’ Ср°° ~ 1 / СА0 к} + к2 к}+ к2
А,+А:<^=±Р,+Р2 dt dt ~ ~ к-\СРСРг При условии сА = сА2: г — Or р с Or г 1П СА0 ZCAoo_ + b А_ = ^АрСд^ СМ СК ~ СЛоо СА0~ СА°О При условии сА # сАз: + -к_у, Q (СА, + где (са2о сЛ(о) + 4cAi0cAj0K
А^=^+Р2 ^-^-к}с^+к^(см-с^ |п СА0 (СА ~ САсо ) _ СА0 + СА°О ft t СМ~СКСЬ<х> СМ>~ СЛоо
д Л. >в_^р dcA 1 , i = -«.с. + к dt 1 А в — = с dt 2 в П Ci п -о W > II II II Ci > М t-J — ' > о h—* О 1 z ч 1 1 О >-> J. 1 ~ 1 Л (Ъ % 5 । ~ "2. I >t+ 1 К) * 1 -Л 1 £ 'сь' \ s д
340
Новый справочник химика и технолога
Продолжение таблицы 2.1.8
Реакция Дифференциальное уравнение Интегральное уравнение
А<==>В—^->Р где = i (k} + + k2) + + k_} + fc2 )2 - 4^
А<==±В^=>Р *-2 c _ c ( r2 k-2 ~ r\ c-rxt _ r\ ^-2 ~ r2 c-r2t + k_2 P\'i-''2 k2 Г\~Г2 k2 k2 ) cp =<tJ 1+ e 1 e 2 , 1 'i~'2 'i ~Г2 ) где cPo = J , , . , K]<2 + k}k2 + k }k 2 где r12 = ^^- + ~kik2[ 1+—+—1 u 2 2 J 1 k_2 k, J
Таблица 2.1.9
Интегральные и дифференциальные формы уравнений скорости для необратимых реакций первого и второго порядка
Реакция Дифференциальное уравнение Интегральное уравнение
kl Ap’ A \k2 U>P2 dcv ~Г = ^1CA dt dc„ h. - k c dt 2 A c -c e-^+k2)> VA сА0с c =—^—c h-e_(*,+*2H ” ’ c =—!b c h-e*Q) P1 kl+k, 1
«.Л dCK —-L = -^iCa dt 1 A‘ dcx dt 2 A2 CA, =CA,0e c = c e k?1 CA2 eA20e
ki k-> A B—► P dCk 7 —7^ = -^^ dt de» , , & ~ ~ k2CB При условии ki ~ k2'. c — c e ‘'A cA0c Св~ k -k См^в e ) я2 Л1 c- = c« L k2~kl J При условии ki » k2: с ~ с e~^' c — c e~^ CA CA0e CP ^A0c
k\ Al + a2 В -4>P dcA dcA de» , , dt ~^1CAiCA2 ^2CB at При условии cAj0 > CA)O: c c ca2o ~ca,o CA2O eXP^i (CA20 ~ CA]O “ CA]0
Химическая кинетика и диффузия
341
Таблица 2.1.10
Коэффициенты пересчета для констант скорости химической реакции второго порядка
Размерность к см3 • моль 1 • с 1 л • моль 1 • с 1 см3 • молекула ’ • с 1 атм 1 • с 1
1 см3 • моль-1 • с’1 1 10‘3 1,66 ♦ 10’24 1,64- 10’7Т
1 л • моль”1 • с”1 103 1 1,66- 10’21 1,64- 10 4Г
1 см3 • молекула’1 • с”1 6,022 • 1023 6,022 • Ю20 1 9,85 • Ю16/7
1 атм”1 • с”1 6,115 • 1067”’ 6,115 • Ю3/7’1 1,015 • Ю’17/7’1 1
Таблица 2.1.11
Коэффициенты пересчета для констант скорости химической реакции третьего порядка
Размерность к см6 • моль 2 • с 1 л • моль 2 • с 1 см6 • молекула 2 • с 1 атм 2 • с 1
1 cs/’ • моль’2 • с’1 1 л • моль’2 • с”1 1 см6 • молекула’2 • с”1 1 атм’2 • с’1 1 106 3,626 • 1047 3,47 • 10137”2 106 1 3,626 • 104' 3,74 • Ю7?7’2 2,76 • 10"48 2,76 • 10"42 1 1,02 • 10’347”2 2,67 • Ю’14/72 2,67 • Ю’8/72 9,69 • Ю33/72 1
Для определения величины энергии активации предложены способы, основанные на использовании функции In^-l/r, представленной в координатах соответствующих переменных:
1. Способ, основанный на построении зависимости константы скорости реакции от температуры в ар-рениусовских координатах IgA-(1/7). Величины констант скорости реакции определяют в независимых экспериментах при разных температурах (рис. 2.1.6).
Рис. 2.1.6. Зависимость lg£ от МТ
Значение величины энергии активации рассчитывают с помощью соотношения Еа - -2,303 R [AlgA / А(7) ’*].
2. Способ, основанный на построении зависимости логарифма коэффициента трансформации (%) от обратной температуры lg% = fil/T). Величину коэффициента % получают путем трансформации кинетических кривых расходования (накопления) вещества, полученных при разных температурах на одну кривую. Коэффициент трансформации рассчитывают с использованием соотношения: х/%1,2= h/ti, где Л и t2 — время достижения одной и той же степени превращения в опытах при температурах Т} и Т2; величина энергии активации Еа = -2,303 R [Alg%/A(7)”’].
Для простых некаталитических реакций с участием молекул значения энергии активации лежат в интервале 80-250 кДж/моль. Необходимо отметить, что значение энергии активации характеризует всегда определенный температурный интервал. Вне этого интервала у реакций может наблюдаться отсутствие линейной полулогарифмической зависимости, вызванное изменением механизма химической реакции и сопровождающееся изменением вида кинетического уравнения или его кинетических параметров.
В табл. 2.1.12 приведены соотношения между различными единицами энергии.
Таблица 2.1.12
Соотношение между различными единицами энергии
Энергия кДж/моль ккал/моль эВ/молекула
1 кДж/моль 1 0,239 1,036 • 10’2
1 ккал/моль 4,184 1 4,336 • 10’2
эВ/молекула 96,48 23,06 1
Кинетика гомогенных химических реакций в непрерывных процессах
Характерная особенность непрерывных процессов — постоянное движение реакционных масс с заданной температурой в струе жидкости или газа через реакционное устройство. Примерами реакций, осуществляемых в потоке в широких технических масштабах, являются большинство процессов нефтепереработки, основного органического и нефтехимического синтеза. Проведение процесса в потоке обеспечивает высокую эффективность использования реакторов, создает возможность для регулирования и автоматизации процесса.
342
Новый справочник химика и технолога
Реакции в потоке можно классифицировать по режимам проведения. Предельными режимами являются режим идеального вытеснения и режим идеального перемешивания. На практике могут существовать и промежуточные режимы.
В реакторах идеального вытеснения реализуется непрерывный поток реагентов через реакционную зону с постоянной линейной скоростью потока в любой точке поперечного сечения реактора, т. е. для данного режима характерно отсутствие продольного и поперечного перемешивания.
Для реакторов идеального перемешивания, работающих в непрерывном режиме, характерно равенство концентраций всех реагирующих веществ в любой точке объема реактора и на выходе из реактора, что достигается интенсивным перевешиванием реакционной массы.
Изменение концентрации одного из реагирующих веществ вдоль потока в реакторе при двух идеальных режимах процесса показано на рис. 2.1.7.
Рис. 2.1.7. Изменение числа молей иА( реагирующего вещества вдоль реактора длины /:
1 — режим идеального вытеснения;
2 — режим идеального перемешивания
Структура потока при транспорте реакционной массы описывается уравнениями гидродинамики, которые послужили основой при расчете кинетических параметров реакций, протекающих в потоке.
Кинетика гомогенных химических реакций в режиме идеального вытеснения
При обработке кинетических закономерностей, полученных при исследовании химических процессов в режиме идеального вытеснения для одномерного потока, необходимо учитывать перемещение веществ вдоль оси реактора. Поэтому в уравнение, устанавливающее взаимосвязь между скоростью реакции и химическими превращениями сореагентов, помимо времени необходимо вводить дополнительный параметр — координату пространства. Как правило, реактор идеального вытеснения имеет цилиндрическую форму, и в реакторе отсутствует перемешивание слоев при их движении по реактору. Если длина реактора /, то общее уравнение динамики гомогенного химического процесса для вещества А, может быть представлено в виде:
мса( ) dcM
---—=VA +—
dl А' dt
(2.1.1.23)
где и — линейная скорость потока; vA — скорость расходования вещества А.
В условиях стационарного состояния проведения реакции, которое достигается вводом постоянного количества реагентов во времени
5са
-^ = 0, dt
уравнение (2.1.1.23) приобретает вид:
(2.1.1.24)
-------- = v dl
С учетом того, что мольный поток вещества А, (яА — число молей вещества А„ проходящих через заданное сечение в единицу времени) есть произведение объемной скорости (v) на концентрацию компонента «Л =vcA, а линейная скорость потока связана с его объемной скоростью и сечением реактора (р) известным соотношением и = v/p, уравнение (2.1.1.24) может быть представлено в виде
1 = ^а, = <&А,
р dl dV d(V/v)
de.
-^- = vA , (2.1.1.25) at
где t — время пребывания потока в аппарате.
Выразив величину мольного потока через степень превращения (£) nAj = иоа,(1 - О» преобразуем уравнение (2.1.1.25) и получим уравнение зависимости скорости реакции от концентрации реагента для любой химической реакции в режиме идеального вытеснения в общем виде:
\->А. (2.1.1.26)
pdl
Используя математическое выражение постулата химической кинетики (2.1.1.2), уравнение (2.1.1.26) можно представить в следующем виде:
для необратимых реакций:
«».A = <2.1.1.27)
'pdl 2
для обратимых реакций:
А.Л,-^ = *.С*Л • (2.1 -1 -28)
’ pdl 12 " 12
где к\ и Л_1 — константы прямой и обратной реакций.
Необходимо отметить, что формы кинетических уравнений (2.1.1.25) и (2.1.1.27), выведенных для реактора, работающего в режиме идеального вытеснения, и формы уравнений, предложенных для описания процесса в периодическом реакторе идеального перемешивания, совпадают. Различие состоит в том, что величина t для периодического реактора — время реакции, для
Химическая кинетика и диффузия
343
проточного — время пребывания потока в реакторе. Поэтому для реакций в потоке в жидкой или газовой фазе, протекающих без изменения числа молей, можно для расчета кинетических параметров пользоваться методами, разработанными для реактора, работающего в режиме идеального перемешивания.
Для процессов, реализуемых в газовой фазе, для веществ, близких по свойствам к идеальным газам, концентрация вещества А, будет равна
. Р }~^ Р
А‘ ^nRT a + ^RT'
(2.1.1.29)
где р — общее давление смеси всех газов, £ — количество прореагировавшего реагента в долях от исходного количества, = (а + Р£),
р=Л+21+...+2к_Л_Х2.--------21.
V, v. V' vt V, V,
При условии, что перепад давления по длине реактора невелик, а реагирующий газ сильно разбавлен инертным газом, интегрированием уравнений (2.1.1.27) и (2.1.1.29) получены некоторые аналитические выражения для констант обратимых и необратимых реакций гомогенных реакций, приведенных в табл. 2.1.13.
Методика расчета величин константы скорости и порядка реакции по реагенту с использованием интегральной формы уравнения скорости не отличается от методики, изложенной выше для процессов в реакторе идеального перемешивания.
Используя соотношения (2.1.1.25) и (2.1.1.29), можно рассчитать объем проточного реактора идеального вытеснения:
--------!---------। п-----------
П0А, П0А, W0A,
(2.1.1.30)
Таблица 2.1.13
Кинетические параметры гомогенных реакций, протекающих в режиме идеального вытеснения
Обозначения переменных уравнений приведены в тексте.
Реакция Кинетическое уравнение в дифференциальной форме Аналитическое выражение к из интегрального уравнения
А -> ViAt + v2A2 + -+ vkXk (1-^)р ()Л рс// (1<ж рУ при условии: А —> А] . 1 * = ^А—Ч рУ 1-х
А,^ф=>А2 Л-| р<7/ л0А1 RT <А, гДе "ооа. = > «ОА, отвечает t —> оо "ооА . RT 1-Иооа, Л = И0А In ! , ' РУ где к = к\ + A_i
A+B^C + D dx = . р2 (1-^)(и0В-и0А^) dl 9R2T2 (иоа+иов)2 к = (»oa+wqb)2 ^21п ”ов(1-0 . Л0А — Л0В VP (W0B ~ W0A ^э) , r2t2 при условии с А0 - Сво - с А. к = 4п0А 2 Ур 1-S
А + В< *Ь»С + Р dx р2 (1-5)(”OB-”OA5) _ pdl ' Л!Т2 (»0А+»„в)2 t Р‘ 1 R2T2 (п0А + «ов)2 7 7Z ч2 Р2Р2 1 к, - к , = (л0А + ) — In —-—1 , 1 -1 k0A ов; р2У(тх-тг) тх(т2-^ П(}. -nm±4z где т. 2 = , ’ 2и0а(/С-1) z = (»0А - nJ К1 - 4пЛК(К -1) , К
344
Новый справочник химика и технолога
На основе анализа уравнения (2.1.1.30) предложено несколько универсальных способов определения порядков реакции:
1. Проведение реакции при постоянном количественном составе исходной реакционной смеси и одинаковой степени превращения, но при различном мольном потоке и давлении в системе, что достигается
выбором объема смеси V = const Графическое решение данной зависимости позволяет рассчитать порядок реакции по реагенту А, (рис. 2.1.8), где угол наклона прямой равен порядку реакции.
Рис. 2.1.8. Зависимость lgn0 от Igp при постоянных объемной скорости и температуре
2. Проведение реакции при постоянном количественном составе исходной реакционной смеси в реакторах разного объема и одинаковой степени превращения А„ что достигается выбором давления, при котором Vpm = const. На основе линейной зависимости lg V-Igp рассчитывается порядок реакции по А, (рис. 2.1.9).
Рис. 2.1.9. Зависимость 1g И от Igp при постоянных конверсии и температуре
Энергию активации реакции, протекающей в режиме идеального вытеснения, определяют, используя аррениусовскую зависимость константы скорости или скорости реакции от температуры, в полулогарифмических координатах 1пЛ (lnv)-l/T.
При исследовании кинетики реакций в газовой фазе и условии, что реагенты подчиняются законам идеального газа, скорость реакции может быть выражена
< d^ va, =—71—’ Pl Ча,
где £ - доля превратившегося вещества А,.
(2.1.1.31)
Величины —— определяют из экспериментальных Ча,
зависимостей ^-«0А, полученных при различных температурах. Вычисленные значения скорости подставляют в уравнение (2.1.1.32):
v7,
Я1п — ут.
(2.1.1.32)
Ъ Т2
и рассчитывают эффективную энергию активации.
Кинетика гомогенных химических реакций в потоке в режиме идеального перемешивания
При проведении синтеза органических веществ в жидкой фазе в реакторе идеального перемешивания, в ходе которого происходит увеличение объема реакционной смеси, скорость расходования реагента A (vA) равна
(С0А, СА, )Vl dCA
^0+(Vl~V2)Z dt
(2.1.1.33)
где Го — исходный объем реагирующей смеси; vb v2 — объемные скорости ввода и вывода смеси реагирующих веществ соответственно.
При стационарном режиме работы реактора V] и v2 равны v ~ V] = v2, объем реакционной смеси постоянен Го = Гр, a dc/Jdt = 0, что приводит к упрощению уравнения (2.1.1.33):
(С0А, СА, )V
(2.1.1.34)
Для реакции, протекающей в газовой фазе в режиме идеального перемешивания, газ во время процесса будет занимать весь объем реактора Гр. Если отбор газовой смеси будет меньше, чем алгебраическая сумма, равная поступающей газовой смеси (или изменение объема газовой смеси происходит в связи с химическими превращениями), то давление в реакторе не будет постоянным, а скорость расходования реагента А будет составлять
Cqa,VCa,V2
(2.1.1.35)
Осуществление процесса в стационарном режиме обеспечивает независимость концентраций реагентов, промежуточных и конечных продуктов реакции от времени (dc/Jdt = 0). Это приводит к упрощению уравнения (2.1.1.35):
C0A,Vl CA,V2
(2.1.1.36)
Химическая кинетика и диффузия
345
С учетом кинетического уравнения (2.1.1.2) можно установить взаимосвязь между кинетическими параметрами реакции (к, п, Еа) и параметрами процесса (с„ ph 7) (табл. 2.1.14), что позволяет оптимизировать условия проведения реакции и осуществить расчет объема реак-
Расчет эффективной энергии активации реакций, проводимых в потоке в реакторах типа идеального перемешивания, осуществляется по методикам, приведенным выше.
Таблица 2.1.14
тора.
Кинетические параметры гомогенных реакций, протекающих в потоке в режиме идеального перемешивания
Реакция Кинетическое уравнение в дифференциальной форме Аналитическое выражение к из интегрального уравнения
А-> ViA1 + v2A2+...+ vkAk Al + А2 —► В + С „ (>-ч>р 0АГ (1 + р^)/?Г’ гдер = У! + v2 + 1 "олЛ ;.(1-^)(г-4) рг V (1 + у)2 R2T2 ’ «оа2 где у = —- W0A| 0-Ж ’ при условии: А —> А! (1+г)Чл0А|/?2г (1-^Xy-^)/^
Мономолекулярные реакции в газовой фазе
Известно большое количество гомогенных газовых реакций, удовлетворяющих кинетическим уравнениям первого порядка. Причем мономолекулярный распад претерпевают, как правило, сложные молекулы, состоящие из большого количества атомов, а энергии активации большинства реакций относительно высоки.
Источником активации в химической реакции являются процессы с участием двух частиц: молекулярные столкновения при термических превращениях, поглощение кванта энергии в фотохимических реакциях, передача энергии молекуле вещества при корпускулярном столкновении частиц. Поэтому в реальных условиях мономолекулярные реакции всегда включают бимолекулярные стадии обмена энергией, и энергию молекула А может накопить путем соударений как за счет соударения с другой молекулой А, так и за счет соударения с молекулой постороннего вещества, не участвующего в химической реакции. Процесс активации (дезактивации) молекулы реагента, претерпевающего мономолекулярный распад, можно записать в виде:
А+М(А)«==>А‘ +М(А). (2.1.1.37)
*-i
Превращение активной молекулы А* возможно по обратимой реакции (2.1.1.37) или по реакции (2.1.1.38):
А* —> Продукты реакции . (2.1.1.38)
При малых степенях превращения (или низких концентрациях продуктов превращения) их рекомбинацию (реакцию, обратную 2.1.1.38) можно не учитывать. В стационарных условиях скорость активации равна сумме скоростей дезактивации и скорости реакции
ZTjCaCm ^-1 Сд СМ "I" ^2 Сд , а скорость реакции вычисляют по формуле:
При высоких концентрациях (давлениях) частиц в реакторе, когда вероятность столкновения частиц велика и большинство активированных молекул А дезактивируется, не успевая претерпеть превращение, к2 Сд <к Л_1 с* См, скорость реакции равна
z/z» lr k
= (2.1.1.40)
dt кЛ
что соответствует процессу первого порядка.
При низких концентрациях (давлениях) реагента и/или инертного вещества вероятность столкновения реакционных частиц уменьшается, и в так называемой «линдема-новской» области суммарный процесс распада молекулы А определяется стадией активации молекулы А к2 с‘ » к_х Сд см, скорость реакции равна
de
-^=^сАсм. (2.1.1.41)
dt
346
Новый справочник химика и технолога
Такил^образом, реакция мономолекулярного распада соединения А имеет общий порядок п = 2. Между этими двумя крайними случаями существует промежуточная область, где величина п меняется от 1 до 2.
С увеличением температуры величина к2 растет быстрее, чем величина к\, и поэтому, при прочих равных условиях, вероятность протекания реакции, описываемой кинетическим уравнением 2-го порядка, больше.
При кинетическом моделировании реакций мономолекулярного распада используют эмпирические соотношения, описывающие зависимость константы скорости распада от давления. Так, при расчете кинетического параметра для реакции пиролиза используется поправка Линдемана—Хиншелвуда (а):
k = kwa = kw/(\ +ро,5/рэф), (2.1.1.42)
где ро 5 — давление, при котором к\ = 0,51^; р^ = асА + + Ьсм; а и b — экспериментально установленные коэффициенты, характерные для каждого органического соединения.
При высоких давлениях для расчета к*, может использоваться соотношение, полученное с помощью метода переходного состояния:
* (2.1.1.43)
h
где Еа — экспериментально установленная энергия активации.
Очевидно, что не всегда молекула, обладающая энергией, равной или большей энергии активации, способна к мономолекулярному распаду. Активными молекулами будут только те, у которых избыточная энергия за счет внутри- и межмолекулярных процессов передачи колебательного возбуждения концентрируется на определенных связях.
В настоящее время наибольшее распространение получила квантовая статистическая теория Райса — Рамспергера — Касселя — Маркуса (РРКМ), позволяющая с наименьшей погрешностью предсказывать зависимости скорости реакции от давления, а также аррениусовские параметры как при высоких, так и при низких давлениях реакционной смеси.
Колебательная энергия многоатомной молекулы в гармоническом приближении является функцией колебательных квантовых чисел vk:
5 ( d \
Ev(yvv2^.v\ vk +^- , (2.1.1.44)
*=i \ J
где dk - степень вырождения колебательного состояния.
Согласно теории Касселя, молекула рассматривается как совокупность 5 гармонических осцилляторов, обладающих одинаковой постоянной частотой колебания и воспринимающих и отдающих энергию, кратную энергии кванта hv. Число гармонических осцилляторов
принимается равным числу колебательных степеней свободы:
s = 3N-6 (для нелинейной молекулы), s = 3N- 5 (для линейной молекулы).
В процессе активации участвует лишь часть из общего числа 5 колебательных степеней свободы, и в активной молекуле А подведенная энергия сосредоточивается на определенных степенях свободы. В теории РРКМ сделано предположение об активации молекулы за счет «сильных» столкновений, когда при столкновении частиц молекуле А передается сравнительно большая порция энергии, и процесс активации и дезактивации частицы рассматривается как одностадийный процесс. В результате активная молекула А переходит в активированный комплекс А#, который претерпевает превращение (мономолекулярный распад или изомеризацию):
А* —> А* —> Продукты реакции.
Для расчета концентраций А и А" используется аппарат статистической механики, применяя который можно получить общее выражение для константы скорости мономолекулярной реакции:
_ ГГ,* ехр(-Е0 /RT) % Вехр(-£* /RT^dE*
(2.1.1.45) где Fi и F* — статистические суммы вращений молекул А и А*, энергия которых не принимает участия в активации А и A*; F2 — статистическая сумма активных степеней свободы молекулы А, которая принимает участие в активации; Е* — полная текущая энергия активированного комплекса A*; L* — статистический множитель, учитывающий кинетически эквивалентные пути образования А^ из A*; Eq — пороговая энергия, необходимая для образования активированного комплекса А^; Е* — полная энергия активированного комплекса Е* = Eq + Е\
При высоких давлениях константа мономолекулярного превращения молекулы А равна:
кТ F*
k„ = L'——exp(-E,,/RT), (2.1.1.46)
h F
где F — статистическая сумма всех активных степеней свободы молекулы А.
При низких давлениях
* = (2.1.1.47)
F
где F* — статистическая сумма молекул, энергия которых превышает Eq.
Химическая кинетика и диффузия
347
В табл. 2.1.15 и 2.1.16 приведены кинетические параметры химических реакций, протекающих в жидкой фазе. Расположение материала в таблицах основано на принципах классификации реакций в соответствии с их кинетической классификацией.
Пояснения к таблицам 2.1.15 и 2.1.16
В первой колонке приведена формальная запись химической реакции, причем ключевые реагенты располагаются по системе Хилла в порядке возрастания числа атомов углерода, внутри каждой из получающихся групп — в порядке возрастания числа атомов водорода, а остальные элементы — в алфавитном порядке символов.
Во второй колонке представлены данные по типу приведенной химической реакции: гомолитические (гм) и гетеролитические (гт).
В третьей колонке указан растворитель, в котором осуществляется химическая реакция. Список растворителей, их формулы и обозначения в сокращенном виде приведены ниже:
Название Формула Сокращение
Вода Н2О
Ароматические углеводороды
Бензол с6н6 PhH
Толуол СбН5СН3 PhMe
Этилбензол С6Н5СН2СН3 PhEt
Кумол С6Н5СН(СН3)2
Алифатические углеводороды
Бутан С4Н10 BuH
Изобутан (СН3)2СНСН3 z-BuH
Пентан с5н12 PentH
Изопентан (СН3)2СНСН2СН3 z-PentH
Гептан СН3(СН2)5СН3 HeptH
Октан с8н18 OctH
Нонан С9Н2О NonH
Циклопентан c-PentH
Циклогексан c-Hex
Спирты
Метанол СН3ОН MeOH
Этанол С2Н5ОН EtOH
Пропан-2-ол (СН3)2СНОН z-PrOH
2-Метил пропан- (СН3)3СОН Z-BuOH
2-ол
Циклогексанол c-HexOH
Фенол С5Н5ОН PhOH
Продолжение
Название Формула Сокращение
Галогенпроизводные углеводородов
Трихлорметан Дихлорметан Хлорбензол СНС13 СН2С12 С6Н5С1 PhCl
Кетоны
Ацетон Циклогексанон (СН3)2СО An
Кислоты
Трифторуксусная кислота Уксусная кислота Пропионовая кислота Изомасляная кислота CF3COOH СН3С(О)ОН СН3СН2С(О)ОН (СН3)2СНС(0)ОН AcOH
Амиды
А',А'-диметил-формамид А',А-диметил-ацетамид HCON(CH3)2 CH3C(O)N(CH3)2 DMFA DMAA
Нитрилы
Ацетонитрил CH3CN MeCN
Простые эфиры
Диэтиловый эфир 1,4-Диоксан (СН3СН2)2О (/ \) Et2O D
Амины
Пиридин c5h5n Py
Нитросоединения
Нитрометан Нитробензол ch3no2 c6h5no2 MeNO2 PhNO2
Серосодержащие соединения
Диметилсульфоксид (CH3)2SO DMSO
В четвертой колонке приведены условия эксперимента:
- молярные концентрации веществ с — в моль/л, молярные доли х — в %, массовые доли w — в %, объемные доли ф — в %, ионная сила растворов / — в моль/л.;
- формула катализатора и концентрации реагентов, выраженные в моль/л;
- pH раствора;
- выход целевого продукта (G);
348
Новый справочник химика и технолога
- температура опыта, при которой были определены кинетические параметры реакции.
В пятой колонке таблицы 2.1.15 представлено уравнение константы равновесия, а в таблице 2.1.16 — кинетическое уравнение реакции, где:
- А, В, С— реагенты, взаимодействующие друг с другом;
- Kt — катализатор реакции;
- М, N, О — продукты реакции.
В шестой, седьмой и девятой колонках приведены кинетические параметры химической реакции, где предэкспонента и константа скорости имеют размерности, отвечающие типу кинетического уравнения (пятая колонка): с’1, л/(моль • с), л2/(моль2 • с) соответственно.
В восьмой колонке указана температура в °C, при которой рассчитана константа химической реакции, приведенной в девятой колонке; комн. — комнатная температура.
В десятой колонке приводится погрешность (воспроизводимость) табулируемой величины, выраженная либо в процентах, либо в логарифмических единицах.
В одиннадцатой колонке указывается экспериментальная методика с использованием сокращений, приведенных ниже:
Агт Аргентометрическое титрование
Алт Алкалиметрическое титрование
Ацт Ацидометрическое титрование
В Весовой метод
гжх Газо-жидкостная хроматография
Дл Дилатометрия
Диэ Метод диэлькометрии
Е.р. Импульсный радиолиз под воздействием
электронов
Ит Иодометрия
Им.р. Импульсный радиолиз
К Кондуктометрия
Кал Калориметрия
Кин Кинетический метод
Кокл Кинетика образования конечного продукта
Кол Колориметрия
Кот Кислотно-основное титрование
Кт Кондуктометрическое титрование
Мкр Метод конкурирующих реакций
Мрт Меркуриметрическое титрование
Нра Кинетика расходования акцептора свобод-
ных радикалов
Об Объемный метод
Ост Метод остановленной струи
ПО Метод прерывистого освещения
Пол Полярография
Пт Потециометрия
Раз Метод разбавления
Рив Кинетика расходования исходного вещества
pH ст pH-статический метод
Сп Спектрофотометрия
Тот Ториметрическое титрование
Хл Хемилюминесцентный метод
Х.р. Импульсный радиолиз под воздействием
рентгеновских лучей
Фот Метод фотолиза
ЭПР Электронный парамагнитный резонанс
ЯМР Ядерный магнитный резонанс
у.р. Импульсный у-радиолиз под воздействием
у-лучей
Константы равновесия химических реакций, протекающих в жидкой фазе
Таблица 2.1.15
Реакция Тип реакции Растворитель Условия проведения реакции Константа равновесия Энергия активации кДж/моль , A — постоянная из E_ авнения k-Ae ЯТ 0 0 1g К, К — константа равновесия Г Ошибка Метод
£
Реакции таутомерии
CH3C(S)NH2 CH3C(SH)= NH n Н2О К ^М-^А 25,0 8,6 2 Пт
CsXnh2 H cu ГТ Н2О К ~ С^/С\ 21,0 1,99 Сп
CH3C(S)NHCH3 - <=► CH3C(SH) —NCH3 ГТ Н2О К ~~ См-^А 25,0 9,6 2 Пт
CH3C(S)CH2C(O)CH3 CH3C(SH)=СНС(О)СН3 Q -Q kN^'NH, 'N^NH h ГТ ГТ ЕЮН z-PrOH DMFA, DMSO ССЦ H2O < < < < < < ..Jj s s s s s s II II II II II II 1^ 1^ 1^ 25,0 25,0 25,0 25,0 25,0 21 35 0,01 0,15 0,16 0,20 0,28 5,30 6,16 Сп Сп Сп Сп Сп Пт Сп
CH3C(O)CH2C(O)CH3 ^=*- CH3C(OH) = CHC(O)CH3 ГТ ГТ H2O Н2О-МеОН H2O-EtOH Н2О ссн,он = 15,1 мол. % сСн}он ~ мол- % Сс2н2он — 6,46 % сс2н5он ~ % Сс о он = 77,9 % 11 11 11 11 11 11 11 > > >> > > > x 25,0 25,0 25,0 25,0 25,0 25,0 35,0 -0,73 0,08 -0,46 0,35 -0,39 -0.62 0,0 ЯМР ЯМР ЯМР ЯМР ЯМР ЯМР Сп
^n^nh2 1 "N^NH
Химическая кинетика и диффузия
СД4 nhso2ch3 nso2ch3 H C^nhso2ch3"*~” 0^nso2ch3 A CH3C(O)CH2C(O)C2H5 .«=► CH3C(OH) = CHC(O)C2H5 CH3C(O)CH(CH3)C(O)CH3^=*T ^=*r CH3C(OH)=C(CH3)C(O)CH3 C6H5C(S)NH2 ^=*r C6H5C(SH)= NH C^NHC(O)CH3"*~~ NC(O)CH3 I1! СД- NHC(O)CH3 NC(O)CH3 A CH3C(O)CH2C(O)C3H7 ^=*r CH3C(OH)= CHC(O)C3H7 Реакция
3 3 3334 3 3 Тип реакции
я я яяяя я я NJ NJ NJ NJ NJ NJ NJ NJ о о о о о о о о PeaKi Растворитель
7 о N> дии таутомерии Условия проведения реакции
Рч Рч Рч Рч II II II II II II II II о о О О О О О О 2 2 2 S 2 2 2 2 О' О' О' О' О' О' О' .О' > > >>>> > > Константа равновесия
Энергия активации Еа, кДж/моль
1g Л, Л- постоянная из £_ уравнения к = Ае НТ
N> § § N> N> N> § § Ml 5* » Mi Ml Ml Ъ S 5 о о Ъ S S Т,°С
-7,5 -0,95 -0,83 -1,54 8,3 5,11 -1,5 -0,72 1g К, К - константа равновесия
N> Ошибка
n n nano n n д я я ч и a a a Метод
Продолжение таблицы 2.i.~l5
vzouoHXdui n vxnwnx ytnHhosvduo rmsoii
OSE
СН3С(О)СН2С(О)СН(СНз)2 -й=^ ^=*г СН3С(ОН)= СНС(О)СН(СН3)2 CH3C(O)CH(CH2CH3)C(O)CH3^z»r -й=^ СН3С(ОН)= С(СН2СН3)С(О)СН3 C6H5C(S)NHCH3 ^=*г СбН5С(8Н)= NCH3 СН3С(О)СН(СН2СН2СН3)С(О)СН3 ^=»гСН3С(ОН)= С(СН2СН2СН3)С(О)СН3 СН3С(О)СН2С(О)СН(СН3)2 -й=^ ^=*г СН3С(ОН)= СНС(О)СН(СН3)2 Cj^NHC6H5 н ЫН 1W;” /r-NH » /т-N NH Н5Сб Ът—N s xZAsh о о Реакция
3 3 3 3 3 3 3 Тип реакции
Пи Пи Пи Пи Пи Пи Пи Пи Пи tQ Ь4 Ь4 Ю Ю М Ь4 bJ О о о о о о о о о PeaKi Растворитель
дии таутомерии Условия проведения реакции
>5 >: и и и и и и и и и CJ CJ CJ С5С5С5П CJC5 2 3 2 2 2 2 2 22 О' О' О' n'n' > > > > > > > > > Константа равновесия
Энергия активации Еа, кДж/моль
1g А, А- постоянная из Е. уравнения к^Ае т
N> N> N> gbJKJbJ Kits) у» ел ел g tz> u» tz> ел и» О О О Ей О © О О О Т,°С
X б. 6» Л А о® Л Л \о V1 >— os Os 40 -~J Os © и» -р». Ji. и» 1g К, К- константа равновесия
Ошибка
S’ Р 5 29^5 19 Метод
нпк^ффпд n шпшэнт кп^ээнпгтх
Продолжение таблицы 2.1.15
ISE
HC(O)OH HC(O)O’+ н+ С13СС(О)ОН + СН3ОН ^2* С13СС(О)О"+ сн3бн2 Реакции диссоциации /FNH л * //“N H5c/<S^S h5c6XsAsh H5C6\ NH H5C6\ N v*s у- H H //— и— n H5c/^s H5C/<J<SH H H СН3С(О)СН2С(О)СбН5 ^2* ^*CH3C(OH)= СНС(О)СбН5 CH3(CH2)2C(O)CH2C(O)CH2CH(CH3)2 ^2* ^2*CH3(CH2)2C(OH)=CHC(O)CH2CH(CH3)2 (CH3)2CHCH2C(O)CH2C(O)CH2CH(CH3)2^2* ^2* (CH3)2CHCH2C(OH)= CHC(O)CH2CH(CH3)2 Реакции таутомерии Реакция
3 3 3 3 3 3 3 3 Тип реакции
? к Л) к, $ О XXX X XX ООО 0 00 Растворитель
7 7 и 7 р р р р о о о о •I- -1- -1- о о о о м м м м Условия проведения реакции
Il II II II II 2 2 2 2 2 Г» Ci Ci Ci Ci z z z z z o' o^ o' o^ o' > > > > > >5 II II II II II II ООО о о о 2 2 2 2 2^ 2^ Константа равновесия
Энергия активации Еа, кДж/моль
1g А, А- постоянная из Е_ уравнения к^Ае gT
u> u> 0 У* >° 0 0 0 0 0 М N> N> М ЮМ у> у> U> и> у» у» О о о о о о Т,°С
LA OS Ю 00 0 xj is) is) a -5,4 -0,29 -0,58 -0,48 1g К, К- константа равновесия
Ошибка
Я ДДД Д ООО о о о я я я я я я Метод
Продолжение таблицы 2.1.15
пгоконхэш n r»tnwnx xnuhosvduo nndofj
Продолжение таблицы 2.1.15
Реакция Тип реакции Растворитель Условия проведения реакции Константа равновесия Энергия активации Еа, кДж/моль 1g А, А- постоянная из £ уравнения k = Ae R1 О о 1g К, К - константа равновесия Ошибка Метод
Реакции диссоциации
НС(О)С(О)ОН НС(О)С(О)О" + Н+ НОС(О)С(О)ОН ^=*гНОС(О)С(О)О- + н+ ВгСН2С(О)ОН + сн3он^=»- ВгСН2С(О)О-+ сн3бн2 С1СН2С(О)ОН С1СН2С(О)О" + н+ СН2С1С(О)ОН + СН3ОН СН2С1 С(О)СГ + СН3ОН2 FCH2C(O)OH FCH2C(O)O" + н+ FCH2C(O)OH + СН3ОН fch2C(O)o-+ сн36н2 NH2C(O)C(O)OH^*NH2C(O)C(O)O“ + н+ СН3С(О)ОН*^ СН3С(О)О" + н+ СН3С(О)ОН + СН3ОН^=»Г СН3С(О)О" + сн3дн2 СН3С(О)ОН + СН3СН2ОН^=* СН3С(О)О- + сн3сн2бн2 ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ Н2О Н2О МеОН Н2О МеОН Н2О МеОН Н2О Н2О МеОН EtOH / = 0,05 / = 0,0002:0,001 /=0,0002-5-0,03 / = 0 1=0 /=0,02-0,02 /=0.02: 0,02 / = 0.02-0,02 / = 0 1 = 0 / = 0,01 4-0,2 / = 0,01-0,2 / = 0,01-0,2 / = 0,01-0,2 1 = 0 1=0 < <<<< <<<< <<<<< < <<<< < < zzzzz zzzz zzzzz z zzzz z z ч и у у у сэ S S I !S SSSS SSSS2- s II II II II II II II II II II II II II II II II II II II II II 25,0 20,0 25,0 30,0 25,0 0,0 10,0 18,0 25,0 15,0 20,0 25,0 30,0 25,0 25,0 0,0 5,0 10,0 15,0 25,0 25,0 3,46 1,250 1,270 1,31 8,06 2,816 2,827 2,834 7,96 2,554 2,572 2,586 2,604 7,99 2,10 4,782 4,770 4,762 4,758 9,52 10,41 0,06 0,03 0,0003 0,01 0,04 0.005 0,005 0,005 0,04 0,001 0,001 0,001 0,001 0,04 0,0005 0,0005 0,0005 0,0005 0,02 Пт Пт К Пт Пт Пт Пт Пт Пт К К К К Пт К Пт Пт Пт Пт Пт Пт
Химическая кинетика и диффузия
О к 11 о X о о о + о х о х о+ д о X N> II о X о о о X + о Д о Д о X О X к> II о X о о о + о X о+ X о X к> о X о о о X + о X о X О X 11 о X о 6 о + X о X Ti о X о о о X О X III о X о о о X о X III о X о 6 о 2! О О X NJ О о о 1 + о X о+ X 2! о о X о 6 о X + о X о X 1 X о о о + о X о X о+ X X § X о о о X + о X о X о X X 8 г? о § + «я S3 о-,зс § I Зч + п JX 3 Ъ X X О О X Ъ о q + о X О+ X X о п X о о о X + о X о X П X О о о о X о X о о 6 о 1 + + X СЛ О X о о + п X СИ X HSCH2C(O)OH + СН3ОН Реакция
+ X +
3 3 3 3 3 3 3 3 3 3 Тип реакции
EtOH МеОН X о X о МеОН EtOH EtOH МеОН X о МеОН Реакщ Растворитель
7=0 II о 7 о 7 о 7=0 7 о и © ии диссоциации Условия проведения реакции
* *
2 п п Z 2 2 z^ 2 Z^ (?м /са Vj/ Nj Wj _ п 2 п 2^ 2 о z^ п 2 2^ Константа равновесия
Энергия активации Еа,
IgA, А-постоянная из £_ уравнения к = Ае *г
К) LA о N> la О К) LA © N> LA © 25±2 N> LZ, О N> LZ> О 25±3 ю у> о N> LA О Г, °C
ъ» 00 9,271 к) ел 1,887 ъ» о 11,95 © 7,504 К) Os о 00 \gK,K~ константа равновесия
о о р о О О 00 р © LZ> © © LA Ошибка
5> н Я 5 о я О л О л П л ? Метод
Продолжение таблицы 2.1.15
огоконхэш n vxnwnx >tnHhO9VdUD nrwojj
^=»г СбН5С(О)СГ + сн3сн2бн2 СбН5С(О)ОН + СН3СН2ОН с6н5с(О)он + сн3он^гс6н5с(о)о + сн3бн2 О Я О О о о я о % о о о о + я + СН3СН2СН(СН3)СН2С(О)О~ + Н+ СН3СН2СН(СН3)СН2С(О)ОН ;<=► ;*=► СН3(СН2)2С(О)О- + сн3сн2бн2 СН3(СН2)2С(О)ОН + СН3СН2ОН ;<=► -<=► СН3(СН2)2С(О)О~+ СН3ОН2 СН3(СН2)2С(О)ОН + СН3ОН^=*г СН3СН2С(О)О + сн3бн2 о я о я bJ о о о я + о я о я П Я О я о о о я о я о я о о о 1 + я 4- СН3СН2С(О)ОН СН3СН2С(О)О- + Н+ Реакция
3 3 3 3 3 3 3 3 Тип реакции
m б I о О Я я N) О Я о m 5 я о о я о о я я о я о -та 0> р я Е Растворитель
и о О 7 7 о о ш 7 о 7 7 7 II о о II о о 7 = 0,01 н 7 = 0,01 н II о Ъ II о о 7 7 о о о Ъ ии диссоциац Условия проведения
о о V1 о о ш о о 40 р о 40 о о о о 40 40 о о 40 о о 40 о о о о 40 40 S S реакции
>4 II 2 >4 II 2 Z /С см /Сд К - См cN /сд и 2 Z II 2 п Z /С СмСц/Сд Л - См Cn/Сд >! II 2 Z /С См Cjsj /Сд /С — См Cjsj /Сд и и 2 2 > > >! >! II II 2 2 Константа равновесия
Энергия активации Еа, кДж/моль
1g А, А- постоянная из Е_ уравнения к-Ае Л7
ьо V1 о ьо V1 о ьо V1 ъ ьо Ъ ьо о ЬО V1 ъ ЬО V1 ъ V1 о о ъ У1 ° о о V1 о р о V1 о о Ъ Г, °C
о о 00 Ъ 00 ЬО ю 04 сь 00 ъ о р 40 р 00 00 00 00 00 00 40 -J V1 00 00 00 00 00 00 40 -J V1 1g К, К - константа равновесия
р о о О о о р о ьо V1 о о о р ъ ЬО р о ЬО о о о ш Р Ъ О о о 8 8 о о о ш о о о ш 0,003 0,003 Ошибка
я я Я н Я я н я Я Я Я Я н н н н Я Я Я Я н н н н Метод
кпеАффпр n пхпшэнпх иэхээыжпх
Продолжение таблицы 2.1.15
о.
Кинетические параметры химических реакций, протекающих в жидкой фазе
Таблица 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Ea, кДж/моль 1g А, А- постоянная из k~ Ае уравнения л О о 1g k, k - константа скорости реакции Ошибка Метод
Перегруппировка Гофмана
CH3C(O)NHBr + "ОН —►CH3NH2 + СО2 + Вг " ГТ Н2О Н2О Скон _ 0,25 Скон = 0,25 < < и и 30,0 40,0 -5,65 -4,42 Ит Ит
Н2О — v = fcA 50,9 -3,80 Ит
C2H5C(O)NHBr + "ОН—*>C2H5NH2 + СО2 + Вг~ ГТ Н2О Н2О скон = 0,25 Скон 0,25 £ II II 30,0 40,0 -3,51 -2,81 Ит Ит
Н2О скон ~ 0,25 V = fcA 50,0 -2,32 Ит
Н3С-^2)>— C(°)NHC1 +"он —► ГТ Н2О Н2О cNaOH ~ 0,5 cNaOH = 0,5 II II 0,0 5,0 -4,734 4,352 0,2 0,2 Ит Ит
Н2О cNaOH = 0,5 v = Ata 10,0 -3,982 0,2 Ит
—► Н3С<)-NH2 + СО2 + СГ Н2О ^NaOH = 0,5 v = £ca 15,0 -3,626 0,2 Ит
СбН5С(О)КНВг + "ОН —►C6H5NH2 + СО2 + Вг" ГТ Н2О сА = 0,025 v = fcA 20,0 -4,602 0,1 Ит
cNaOH = 0,5; сА = 0,025 cNaOH = 0,5 v = fcA 25,0 -3,504 0,2 Ит
C6H5C(O)NHCI + ОН ►c6h5nh2 + со2 + сг ГТ Н2О сА = 0,05 CNaOH = 0,5 v = fcA 15,0 -4,106 0,1 Ит
Cl^~^>— C(O)NHBr + "ОН —* ГТ Н2О н2о ^NaOH = 0,5 ^NaOH = 0,5 £ II II 20,0 30,0 -4,392 -3,692 0,2 0,1 Ит Ит
н20 ^NaOH ~ 0,5 v = kcK 35,0 -3,362 0,2 Ит
—► c^}-NH2 + СО2 + Вг-
Н3С^- C(O)NHBr +"ОН—* ГТ Н2О Н2О cNaOH = 0,5 cNaOH ~ 0,5 £ £ Il II 10,0 14,0 -4,086 -3,789 0,1 0,1 Ит Ит
Н2О CNaOH 0,5 v = ZrA 18,0 -3,502 0,1 Ит
—*Н3С—Q-NH2 + СО2 + Вг- н2о CNaOH = 0,5 v = fcA 20,0 -2,64 3 Ит
Новый справочник химика и технолога
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Ea, кДж/моль 1g А, А- постоянная из E. уравнения л 71 е О О 1g k, k - константа скорости реакции Ошибка Метод
Перегруппировка Клайзена
H3C-Z~Y- О —СН2СН = СН2 —► сн2сн=сн2 —►н3с^2^—он ГТ носн2сн2он НОСН2СН2ОН НОСН2СН2ОН PhOH PhOH PhOH PhOMe и и и и и и и 170,3 184,9 185,3 150,3 165,3 180,3 184,9 -4,17 -3,74 -3,69 4,62 -4,16 -3,69 -4,62 2 6 1 5 Сп Раз Сп Сп Сп Сп Раз
Перегруппировка Курциуса
СбН5СОН3 —► C6H5NCO + n2 ГТ H2O-AcOH AcOH AcOH AcOH AcOH AcOH AcOH - H2SO4 AcOH - H2SO4 AcOH - H2SO4 PhMe PhMe wacoh _ 55,8 Фн2вО4 ~ 20 Фн2во4 — 20 Фн2во4 — 20 '<<<<<<< £JJ££££ £ £ £ £ II II II II II II II II II II II 65,2 55,6 62,0 65,2 70,0 74,5 34,8 44,7 54,8 65,2 70,1 -3,65 -4,39 -4,06 -3,89 -3,68 -3,44 -4,74 -4,18 -3,66 -4,44 -4,18 2 2 2 2 2 2 2 2 2 0,3 0,3 Об Об Об Об Об Об Об Об Об Об Об
Реакции распада и фрагментации
НОСН2ОО—* С>27 + нсно + н+ ООСН(ОН)СН2ОН—► б]7 + онсн2сно + н+ (СН3)2С(ОН)ОО —*О17- СН3СОСН3 + н+ (СН3)2С(ОСН3)ОО —► О17+СН3£(ОСН3)2 СН3С(О)СН(ООН)СН3—► СН3С(О)СН(СН3)д + 6н гм гм гм гм гм H2O H2O H2O H2O PhCl pH = 6,7 pH = 5,5 pH = 5,5 pH = 5,0 (30 °C)-(70 °C) V = fcA V = fcA V = fcA V = fcA 33,0 56,0 114,8 8,3 12,8 13,53 комн. 22 22 20 120,0 < 1 2,28 2,81 4,81 -1,89 Фот Фот Фот Им.р Хл
Химическая кинетика и диффузия
HOCH2CH(OH)CH(OH)CH(OH)OO —►012”+ НОСН2СН(ОН)СН(ОН)СНО + н+ (СН3)3СООН—► (СН3)3СО + Он НОСН2СН(ОН)СН(ОН)СН(ОН)СН(ОН)ОО—► —►О12“+ НОСН2СН(ОН)СН(ОН)СН(ОН)СНО + н+ сн3 сн3 сн3 СН3С— N=N—ССН3 —► 2СН3О +N2 C=N C=N C=N (CH3)3CN=NC(CH3)3 —► 2(CH3)3C + N2 (CH3)3COOC(CH3)3 —^2(CH3)3CO C6H5-N=N— C(CH3)2CN —► -*C6H15‘ + C(CH3)2CN + N2 C6H5CH2C(CH3)2OOH —► -*► C6H5CH2C(CH3)2d + OH CH3(C2H5XCN)C -N=N— c(cn)(c2h5)ch3—► — 2CH3(C2H5)C(CN) + N2 (C2H5)2C(CN)-N=N-C(CN)(C2H5)2—► — 2(C2H5)2C(CN) + N2 Реакция
ГМ ГМ ГМ ГМ ГМ ГМ ГМ ГМ ГМ ГМ Тип реакции
b—1 W 1 2 Я o> = ir n > 5 « Q i-q -a P "° -21 о я § я "о а ж и д 2 Js 2 Ж 2 8 о о ° S 2 ® a* sr о й*Д д x 2 ° 2= ° “ £ Й P 3 s | = | 3 д Реакции p Растворитель
2? О О ~-4 4^ -Й. Ji Ui Ui >rt Д LA £ !4T“r> f ? x?t s g ti ti 8 3 53 g 8 m “ « -ooo 0s О О о о о о чИ* _ (-л П П О О о 0 оОООП Л ° la <J ПО о с J о аспада и фрагментации Условия проведения реакции
<<<< < <<<<<<< << < II II II II II II II II II II II II II II II > > > > > >>>>>>> >> > Кинетическое уравнение скорости реакции
171,0 138,3 52,0 127,6 146,7 142,5 127,4 183,5 146,7 163,0 125,7 123,2 133,5 133,0 143,3 Энергия активации Еа, кДж/моль
16,03 12,56 11,4 15,0 17,70 17,30 14,60 17,48 14,86 15,2 10,85 14,15 15,96 15,85 17,10 1g А, А- постоянная из Е. 1- ^ Др в? уравнения Л
22 120,0 120,0 22 80,0 80,0 80,0 80,0 80,0 80,0 120,0 80,0 80,0 80,0 80,0 Г, °C
UJ X- X. Ja 00 3° LA Jh J4* \O 00 w o 00 S© 'J LU о 00 V? 00 « О la О la N> о N> о <O N> 'J -J 1g к, к - константа скорости реакции
Ошибка
£ £ £ £ £ £ a £ £ я £ Л3 s £ Метод
Продолжение таблицы 2.1.16
пгоионхэш п vxnwnx xnHhOsvduz nvisojj
8S£
но+ -(oto to •*— (hoo)hd-to Гидропероксид полиэтилена с6н5(с4н9)2с— схс^ьСбЩ —► гс^с^с о о Ж “г о в Ж i о о Ж О’ (СбН5)2(С2Н5)С-С(С2Н5)(С6Н5)2— 2(СбН5)2(С2Н5)С (С6Н5)2(СН3)С - С(СН3)(С6Н5)2—2(С6Н5)2(СН3)С о э z: b ж о S3 К) 1 Ю О £ о-ж + О £ % p ж кГ о ж X- 1 Ю О £ Р’ о ж + X ю СбВДСНз);,— N=N-C(CH3)2C6H5 — СбНзСССНзЬООСССНзЪСбНз—►2С6Н5С(СН3)2О С6Н5С(О)ООС(О)С6Н5—^2 СбН5С(О)д Х=/ х=, | СН(СН3)2 СН(СН3)2 Н0+ ОНЭ—< H00HD < > . 1 1 СН(СН3)2 СН(СН3)2 —►2СН3СН2СН2С(СН3)2 + n2 CH3CH2CH2C(CH3)2N=NC(CH3)2CH2CH2CH3—► Реакция
2 § 2 2 2 2 2 2 2 3 3 Тип реакции
”0 Q Q о о PhCH3 ”0 о PhMe PhH о-С6Н4С12 -is ж Аром. утл. Аром. утл. PhC(0)CH3 с-Нех ж H”CiooH2o2 Реакции р Растворитель
110-130 °C 111-126 °C 20-100 °C 20-50 °C 60-80 °C Dofr9-fr£ 60-85 °C 75-100 °C 40-69 °C 60-80 °C 70-95 °C 80-90 °C 134-174 °C 160-200 °C аспада и фрагментации Условия проведения реакции
II ?г > и ?Г > II ?Г > ¥ду = Л II ?г > II ?Г > II II ?г?г > > и ?г > II ?г > II II ?г?г > > и ?г > и ?г > Кинетическое уравнение скорости реакции
fe 159,22 оо о Ъ» fe Ml fe ж СЛ so <1 о fe J4> 00 fe 129,05 126,5 118,2 1—* ЬО ьо ъ» Энергия активации Еа, кДж/моль
о К> SO <> 16,125 13,755 13,76 15,04 О к> Os о 14,48 14,34 13,49 о Os к> 00 1g А, А- постоянная из к=Ае w уравнения Л
о о Os о Ъ Os О О о> о о os о о 00 о о 120,0 120,0 00 о о 00 00 о о ъ ъ fe о ъ 00 о о Т,°С
fe о о fe fe fe Os „fe -2,04 -1,66 4 -4,34 -3,97 । оо £ о к> 1g к, к - константа скорости реакции
Ошибка
£ ж 45 Р ж 45 Р Ж 45 Р Ж 43 Р S’ w я ЖЖ 45 45 $ а ж 45 Р •45 *0 Щ S s -о И И р а? св S’ § Метод
хпсбффщ) п пмпшэнпя кохээыытх
Продолжение таблицы 2.1.16
6££
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль из Е. Эо А 1g к, к - константа скорости реакции Ошибка Метод
1g А, А- постоянная Jr := Ар уравнения л 71 е
Реакция элиминирования
транс-С1СН= СНС1 + CH3ONa —► ГТ МеОН v = Ar Асв
—►Cite CH + СН3ОН + NaCl
цис-С1СП= СНС1 + CH3ONa—► ГТ МеОН v = kcAcn 95,8 -6,46 2 Пт
МеОН v= ксАсъ 108,9 -5,84 2 Пт
—►Cite CH + CH3OH + NaCl МеОН v = ксксь 121,7 -5,22 16 Пт
МеОН v = ксКсц 133,4 -4,79 11 Пт
z/z/c-OC(O)CH= CClC(O)O’+NaOH —► ГТ Н2О NaNO3; I =0,10 v = Аг Асв 71,0 -4,04 Агт
Н2О NaNO3; / =0,10 v = Аг Асв 80,5 -3,61 Агт
-► "OC(O)- te C— C(O)O" + H2O + NaCl Н2О NaNO3; / =0,10 v = ксКсъ 90,6 -3,16 Агт
H2O-EtOH xc2h5oh “3L6; NaNO3; I =0,11 v = Аг Асв 69,5 -4,09 Агт
H2O-EtOH xc2h5oh “31,6; NaNO3; / =0,11 V = Аг Асв 72,3 -3,96 Агт
H2O-EtOH xc2h5oh = 31,6 NaNO3; / = 0,11 v = Аг Асв 80,1 -3,61 Агт
H2O-EtOH xc2h5oh 31,6: NaNO3; / = 0,11 v = Аг Асв 88,3 -3,23 Агт
C1CH2CH2C1 + NaOH—► CHC1=CH2 + H2O + NaCl ГТ H2O G(C2H3CI)= 100% v = ксАсъ 120,0 -1,82 Агт
CICH2CH2C1 + CH3COONa—► ГТ н2о G(C2H3CI) = 18,7 % v = кс^Сь 120,0 -4,37 Агт
-► C1CH = CH2 + CH3COOH + NaCl
CICH2CH2C1 + C6H5ONa—► ГТ Н2О G(C2H3CI) = 57,3 % v =ксАсв 120,0 -2,76 Агт
-► C1CH = CH2 + СбН5ОН + NaCl
CH3CH2C1 + CH3COONa—-► ГТ Н2О G(C2H4) = 35,8 % v = Аг Асв 100,0 -3,57 Агт
G(C2H4)) = 37,7 % v = ЛсАсв 110,0 -3,14 Агт
—►CH2= CH2 + CH3COOH + NaCl G(C2H4) = 38,7 % v = ксАсв 120,0 -2,83 Агт
Новый справочник химика и технолога
lu Ch о
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из Е. к^Ает уравнения л 71 е О О f-4 1g к, к - константа скорости реакции Ошибка Метод
Реакция элиминирования
CH2(CN)CH2C1 + NaOH—► - ► CH(CN) = СН2 + Н2О + NaCl z/wc-CH3CH= СНС1 + CH3ONa—► - ► СН3С= CH + СН3ОН + NaCl wpa«c-CH3CH— СНС1 + CH3ONa—► — ► СН3С= CH + CH3OH + NaCl CH3OCH2CH2C1 + CH3COONa—► -► CH3OCH= CH2 + CH3COOH + NaCl 2/wc-OC(O)CH= CC1C(O)O'+ NaOH —► -► ’OC(O)- C= C— C(O)O"+ H2O + NaCl ™рш/с-"ОС(О)СН= CC1COO + NaOH— —►-OC(O)C= CC(O)O" + H2O + NaCl ГТ ГТ ГТ ГТ ГТ Н2О МеОН МеОН Н2О Н2О Н2О Н2О Н2О - EtOH Н2О - EtOH Н2О - EtOH Н2О Н2О Н2О Н2О - EtOH Н2О - EtOH Н2О - EtOH G(C3H3N)= 100% G(C3H3N) - 100 % G(C3H3N) = 100 % G(C3H3N) = 100 % G(C3H6O) = 59,2 % NaNO3; 7=0,10 NaNO3; 7=0,10 NaNO3; 7=0,10 xc2h5oh _31,6 NaNO3; 7=0,11 •*c2h,oh — 31,6 NaNO3; 7=0,11 xc2h5oh ~ 31,6; NaNO3; 7=0,11 NaNO3; 7 = 0,10 7 = 0,10 7=0,10 xc2h5oh = 31,6; NaNO3; 7 = 0,1 xc2h5oh “31,6; NaNO3; 7 = 0,11 xc2hsoh “ 31,6 NaNO3; 7 = 0,11 v = kc Асв v = kc Асв v = kc Асв v = kc Асв v = kc Асв v = Агасв v = kc Асв v = kc Асв v = kc Асв v = kc Асв v = kc Асв v = kc Асв v = Агасв v = кс Асв v = ЛсАсв V = /ссАсв v = ксу?в v = ксАсв v = кс Асв 10,0 14,0 17,8 25,0 125,3 120,0 71,0 80,5 90,6 69,5 72,3 80,1 59,5 71,0 80,5 61,5 73,0 81,1 -0,34 -0,17 -0,01 0,28 -6,71 -3,59 -4.04 -3,61 -3,16 -4,09 -3,96 -3,61 -3,53 -3,04 -2,69 -3,41 -2,86 -2,50 7 Мрт Мрт Мрт Мрт Кот Агт Агт Агт Агт Агт Агт Агт Агт Агт Агт Агт Агт Агт Агт
Химическая кинетика и диффу
-► С6Н5С= СН + (СН3)3СОН + NaCl транс-С${5С[к= СНС1 + (CH3)3CONa—► -► СбН5С= СН + (СН3)3СОН + NaCl г/мс-СбН5СН= СНС1 + (CH3)3CONa—* -► С6н5с= СН + С2Н5ОН + NaCl ^анс-СбН5СН= CHCI + C2H5ONa —* -► СбН5С= СН + С2Н5ОН + NaCl -с к о П £ П К II о к о + о к> к о 2! 9 -► СН3С(О)СН= С(СН3)2 + (CH3)3N + НОСНз СН3С(О)СН2С(СН3)2ОСНз + (CH3)3N—► СН3С(О)СН2С(СНз)2ОСН3 + NaOH —► -► СН3С(О)СН= С(СН3)2 + Н2О + CH3ONa -► СН3С(О)СН= СН2 + Н2О + СН3С(О)ОК СН3С(О)СН2СН2ОС(О)СНз + кон—► СН3С(О)СН2СН2ОСН3 + NaOH—► —► СН3С(О)СН= СН2 + Н2О + CH3ONa -► C2H5CH= CH2 + CH3COOH + NaCl C2H5CH2CH2C1 + CH3COONa — Реакция
3 3 3 3 3 3 3 3 Тип реакции
СО С О Ж W с О ж tn О X tn О X X м о X О X м о X м о X 0 n a> P ss 9 Растворитель
7? Q 7 © -о X II X© 00 о КС1;7 = 1,0; pH = 12,07 pH = 11,99-5-12,69; КС1; 7=1,0; pH =13,07 КС1;7 = 1,0; pH = 12,60 КС1;7 = 1,0; pH =10,55:11,32 КС1;7 = 1,0; pH = 11,98: 12,68 Я О 7 о КС1;/= 1,0; pH = 14,61 КС1;/= 1,0; pH = 12,38 КС1; 1= 1,0; pH = 12,20 P?P К X X oo oo II 00 XO 0 0 Q X© X© qX Ф4 Ф4 ия элиминирования Условия проведения реакции
II и II В? II о? II В? II ?г п стз II ст? II II II f f f п Ъ n СТЗ СТЗ СТЗ II ст? < < II II II II СТЗ .ю . СО СТЗ II B? II II F F s? 0? Кинетическое уравнение скорости реакции
Энергия активации кДж/моль
1g А, А- постоянная из k = Ae уравнения л
ъ» 1—^ о Н-сл lo © © Lk> LA © Lk> bJ bJ © LA © © о © Lk> © © Я © bJ bJ — Ui © Ui © © © LA © © 120,0 130,0 Г, °C
1 Ох 00 00 © х© Д* хо bJ b bJ "н- LM Ох 00 ч о © X© 1 00 ХО jb jb jL 7— V) 1л rb x© bJ 1 1 p w LA 00 bo bo 1g к, к - константа скорости реакции
Ошибка
О Я О а ОПП 9 9 9 П 9 ПОПП 9 9 9 9 Метод
Продолжение таблицы 2.1.16
огоионхэш п oxnwnx xnuhoavdiio ппэоц
Z9£
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из _£-k- Ар уравнения л 71 е О о 1g к, к- константа скорости реакции Ошибка Метод
Реакция элиминирования
NCCH2CH2OC6H5 + CH3CH2ONa_► - ► NCCH= СН2 + СН3СН2ОН + C6H5ONa ОСНСН2СН2ОС6Н5 + C2H5ONa —► - ► ОСНСН= СН2 + С2Н5ОН + C6H5ONa СН3ОСН2СН2ОСбН5 + CH3CH2ONa—► - ► СН3ОСН= СН2 + СН3СН2ОН + CeH5ONa СН3С(О)СН2СН2ОС(О)С6Н5 + кон—► - ► СН3С(О)СН= СН2 + Н2О + С6Н5С(О)ОК СН3С(О)СН2СН2ОС(О)С6Н5 + ch3nh2—*► - * СН3С(О)СЕЬ= СН2 + СН3ЙН3Г)С(О)С6Н5 сн3с(О)т2сн2ос(О)с6Н5 + c2h5och2ch2nh2—► -► СН3С(О)СН= СН2 + С2Н5ОСН2Н2ЙН3 "ОС(О)С6Н5 СН3С(О)СН2СН2ОС(О)С6Н5 + HOCH2CH2NH2—► -► СН3С(О)СЕЬ= СН2 + НОСН2СН2Лн3~ОС(О)С6Н5 СН3С(О)СН2СН2ОС(О)С6Н5 + (CH3)2NCH2CH2NH2—► -► СН3С(О)СН= СН2 + (CH3)2NCH2CH2tfH3~OC(O)C6H5 (CH3)2CHCH(CH3)OS(O)2—Q^CH3 + (C4H9)4N Вг’ — (СН3)2С= СНСН3 + НВг + Н3С-^2^- S(O)2O~ N(C4H9) C^Oi^OSO^s + (СН3)3СОК —► С6Н5СН= СН2 + (СН3)3СОН + C6H5SO2OK ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ ГТ EtOH EtOH EtOH Н2О Н2О Н2О Н2О Н2О Ап Z-BuOH КС1;/ = 1,0 КС1;/ = 1,0; pH = 10,66 КС1;7= 1,0; pH = 9,06 КС1;7= 1,0; pH = 8,51 КС1;/ = 1,0 Р % = 44,6м v = ксАсв v= ксАсв v = ксАсв v = ксАсв v = ксАсв v = ксАсв v = ксАсв v = ксАсв v = ксАсв v = ксАсв 25,4 25,4 25,4 30,0 30,0 30,0 30 30 75,0 40,0 -1,03 1,42 1,50 0,97 -0,99 -1,75 -1,142 -0,84 -2,51 -2,210 3 3 1 0,2 0,7 Сп Сп Сп Сп Сп Сп Сп Сп Кот Сп
Химическая кинетика и диффу
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, 1g А, А- постоянная из Е. 1г = Ае^ уравнения Л 71 с О 0 1g к, к- константа скорости реакции Ошибка Метод
Реакция элиминирования
CH2CH2OS(O)2-^}>- СН3 + CH3CH2ONa —► —► СН= СН2 + СН3СН2ОН + Н3С-£}- S(O)2ONa CH2CH2OS(O)2-^2^- СНз + (СН3)3СОК —► —^2)- СН= СН2 + (СНз)зСОН + н3с—<Q>- S(O)2ok ^-CH(CH3)CH2OS(O)2-^2}— СНз + СС^Й Br" —► —* ^У~С(СН3)= СН2 + НВг + Н3С—8(О)гО' МС^) Qh CH2CH(CH3)OS(O)2-hQ>- СН3 + CH3CH2ONa—* —► СН= СНСНз + СН3СН2ОН + H3C-<Q>—S(O)2ONa ГТ ГТ ГТ ГТ EtOH t-BuOH An EtOH _о чО хО э э □ xC j хЭ U о4 э ®х 5 ° с э с II II » и н II *7 II *7 KJ <—> - - 00 О 0 „ 00 11 00 и 'Л о О О II о И о л л » л л О »О < << <<<< < < и II и и и и и и II ?г ?г > >> >>>> > > п п п П П П П П 2? ш оз оз оз оз оз оз оз ” , 30,0 40,5 50,0 30,0 40,0 50,0 75,0 75,0 30,0 -4,40 -3,80 -3,42 -2,71 -2,40 -1,96 -4,5 -3,28 -4.38 2 1 4 Кот Кот Сп Кот Кот Кот Сп
Реакции сольволиза
СН3С(О)С1 + Н2О —► СН3СООН + НС1 СН3С(О)С1 + СН3ОН —► СН3СООСН3 + НС1 СН3С(О)С1 + С2Н5ОН —►СН3С(О)ОС2Н5 + НС1 СН3СОС1 + СН3ОН^-ОН —► —► СН3СОО-^2^—ОСН3 + НС1 ГТ ГТ ГТ ГТ H2O H2O Н2О Н2О МеОН МеОН МеОН EtOH MeCN <?А = 0,70 < <<<<<<<< II II II II II II II II II ?г ?г ?г ?г ?г ?г ?г ?г ?г > >>>>>>>> 15,0 20,0 35,0 45,0 15,0 25,0 35,0 20,0 25,1 -0,32 -0,13 0,45 0,78 -4,18 -3,79 -3,44 -0,97 -3,71 5 5 5 5 1 3 Кал Кот Кот Кот Агт Сп
364 Новый справочник химика и технолога
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из Е. к = Ае уравнения л Эо А 1g к, к - константа скорости реакции Ошибка Метод
Реакции сольволиза
ch3coci + (Q(^) он—► ГТ MeCN сА= 0,42 сА= 0,42; и и со ио 25,1 25,1 -4,58 -5,50 3 3 Сп Сп
kJw + нс1 C(C2H,)4NC1 -0,13
СН3ОС(О)С1 + Н2О —► СН3ОС(О)ОН + НС1 ГТ Н20 V = fcA 19,6 -3,48 < 1 К
НС(О)ОСН3 + Н+ —► НС(О)ОН + СН3ОН ГТ Н20 Orel ~ 1,0 v = fcA 25,0 -2,50 2 Сп
НС(О)ОСН3 + ОН - —*40(0)0 - + СН3ОН ГТ Н20 1= 1,0; pH = 8,0-9,9 V = kcAcB 46,0 2,17 9 рНст
НС(О)ОСН3 + Н20 —►НС(О)ОН + СН3ОН ГТ Н20 II II II II 23,6 29,0 35,1 40,7 -8,04 -7,70 -7,36 -7,04 9 9 9 9 Алт Алт Алт Алт
НС(О)ОСН2СН3 + Н+ —► НС(О)ОН + СН3СН2ОН Н20 О С5 О О <Э o' о" o' о о" II II II II II U О U О U я я я я я и U и u U ^4^44 и и и и и 20 25 30 35 40 -4,75 -4,57 -4,39 -4,22 -4,04 Кот
НС(О)ОСН2СН3 + Н20 —►НС(О)ОН + сн3сн2он ГТ Н20 Н20 - EtOH •^c2h5oh 23 v = ЛсА v = fcA 20,0 40,5 -7,70 -9,18 9 Алт
СС13С(О)ОСН3 + Н20 —► СС13С(0)0Н + СН3ОН ГТ Н20 Н2О Н20 Н20 - EtOH •^c2h5oh — 23 II II II II 15,0 25,0 35,0 40,5 -3,36 -3,10 -2,88 -7,21 Алт Алт Алт Алт
CF3C(O)OCH3 + Н20 —► CF3C(O)OH + СН3ОН ГТ Н20 Н2О-Ап X = 36,4 ЛС2Н5ОН V = ксА v = kcA 15,0 24,9 -2,33 -3,89 1 10 К Кт
Н2О-Ап •^c2hsoh “ 36,4 v = kcA 34,9 -3,71 3 Кт
Н2О-Ап Л'С2Н5ОН “ 36,4 v = kcA 44,9 -3,53 1 Кт
С2Н5С(О)С1 + Н20 —► С2Н5С(О)ОН + НС1 ГТ Н20 v = kcA 14,8 -3,89 < 1 К
Химическая кинетика и диффузия
СЛ
-► НС(О)ОН + (СН3)2СНОН НС(О)ОСН(СН3)2 + н2о — -► O2NCH2C(O)OH + СН3СН2ОН O2NCH2C(O)OCH2CH3 + Н2О —► -► F3CC(O)OH + СН3СН2ОН F3CC(O)OCH2CH3 + Н2О —► -► С13СС(О)ОН + СН3СН2ОН С13СС(О)ОСН2СН3 + Н2О —► —► H2NCH2C(O)O-+ СН30Н H2NCH2C(O)OCH3 + ОН - -► CH3OC(O)C(O)OH + СН30Н СН3ОС(О)С(О)ОСН3 + Н2О—► СН3С(О)ОСН3 + ОН" —►СН3С(О)О -+ СН3ОН*3 СН3С(О)ОСН3 + Н+—*► СН3С(О)ОН + СН3ОН*2 • ?— • — сн3сн(он)оо + нро; —► сн3сн(о-)оо + н2ро4 QHsOQOP + СН3ОН —► С2Н5ОС(О)ОСН3 + НС1 Реакция
3 3 3 3 3 3 3 3 3 3 Тип реакции
X о X О X м о 1 > X о X о X О X о X О X о X о МеОН Растворитель
Реакции сольволиза
п I Q II © © X я о я II Ох 7 © 7= 0,1 7= 0,1 cNaCI ~ 0,1 С =01 сКС1О4 X Q II © я Q II © pH = 6,7 Условия проведения реакции
1! II II II II II ?г > со и II f f и“ t? II < < < II II II f f f t? с? с? II ?г II II и Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из _ Е‘ к-Ае^Т уравнения л
00 © ох © © к> У* © NJ У* © © © о 25,0 37 nj У* © У о 30,0 35,0 О © © § 2 Я У о Т, °C
\О 00 1 У1 ui 40 1 Ох NJ л Ох м м, © Ох 00 0,11 0,38 1 Л А> i Ох 1 и» 1g к, к - константа скорости реакции
— — nj NJ NJ Ошибка
г О я о я ТЗ X К К Z? 3 3 3 г X 2 “У § Метод
Продолжение таблицы 2.1.16
огоионхэш п oxnwnx nnuhoonduo тяэоц
99£
-► КОС(О)СН2С(О)ОН + СН3СН2ОН КОС(О)СН2С(О)ОСН2СН3 + н+ —► -► F3CC(O)OH + СН3СН2СН2ОН F3CC(O)OCH2CH2CH3 + Н2О—► -► H2NCH2CH(NH2)C(O)O" + СН3ОН H2NCH2CH(NH2)C(O)OCH3 + ОН - —► —► HOCH2CH(NH2)C(O)O- + СН30Н HOCH2CH(NH2)C(O)OCH3 + ОН~—► Н3СС(О)ОСН2СН3 + Н2О —► -► Н3СС(О)ОН + СН3СН2ОН сн3с(о)осн2сн3 + н2о 2!^ -► СН3С(О)Н + СН3СН2ОН Реакция
3 3 3 3 3 3 Тип реакции
а ю о а ю о а о а о а о а о -d о 05 Растворитель
п X п II р о 1= 0,1; pH = 10,5 /= 0,1; pH = 10,5 7 7 р р 45 45 а а и и О 00 7 р 1—* Ci к II ъ снс1~0,05; п 2 0Q п II UJ О Я £ V" о 5м ил о X II р о У $ я я я II 00 с> я II о Ъ У ! X я II 3? "|| р о У £ “и р о и> а с с я я я Q Q 2 “и “и II о р р о о о IZl Wl кции сольволиза Условия проведения реакции
II II и > > ri < II II = Л Tl > II II f II II II f II II II II ?г ?г ?г > > > Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из k = А ~JT уравнения Л
о ил о <-»J N> -4 U> О о К> К> У У о о м о м У о ил о ил ъ ил о к> У о tQ О ил о и w ю и> р у> о о о Г, °C
1 ил 1 UM >-* Л Л ЬО Й оо к> о о 1—* 1 р 1 ил 1 им 4 UJ о 1 У1 им 1 ил ил Л У У оо К> ьэ 00 VO 00 1g Л, к - константа скорости реакции
>—* Ошибка
О О О О я д я д 45 а 3 1 $ § о н « « « 3 S 3 Метод
Продолжение таблицы 2.1.16
L9£
юиЖффпр n хмпшэнпм нпхээнпгтх
НОС(О)СН2С(О)ОСН2СН3 + Н20 -► НОС(О)СН2С(О)ОН + СН3СН2ОН СН3СН2С(О)ОСН2СН3 + Н20—► -► СН3СН2С(О)ОН + СН3СН2ОН СН3С(О)ОСН(СН3)2 + Н20 -► СН3С(О)ОН + (СН3)2СНОН СН3С(О)ОСН2СН2СН3 + Н20 X* -► СН3С(О)ОН + СН3СН2СН2ОН F3CC(O)OC(CH3)3 + Н20 —► -► F3CC(O)OH + (СН3)3СОН СН3СН2СН2С(О)ОСН2СН3 + Н20 —► -► СН3СН2СН2С(О)ОН + СН3СН2ОН Н3СС(О)ОС(СН3)3 + Н20 —► -► Н3СС(О)ОН + (СН3)3СОН СбН5С(О)С1 + СН3ОН —► С6Н5С(О)ОСН3 + НС1 С6Н5С(О)С1 + С2Н5ОН —► С6Н5С(О)ОС2Н5 + НС1 С1СН2С(О)ОС6Н5 + Н20 —► -► С1СН2С(0)0Н + СбН5ОН Реакция
333333 3 33 3 Тип реакции
я я - и 222 о „ _ _ О _ .?= 5 «« «о * । 5е 5е 5е । г? Oxggglomo о ост о я я rt 0Э Растворитель
О Е - = Р к = к я -• z s § 2 £ 2 § R 2 2 2 R 2 Q = 2 2 2 2 - g и и ni и и "и 11 и 111 11 Д § 11 Н II .. о СТ о СТ - w Я А Я н- Я я .. Я Я Я Я II II V О V о О -Г О »_ О Я II о о о о Н- ~ N) *.СТ,. ° ° 1Л о 1Л ° Ui 'л bJUiUiUiUi .1. Я <*> w кции сольволиза Условия проведения реакции
ri < t t t < ri Ti Ti Ti Ti Ti Ti Ti Ti Ti t! t! t! t! II II II II II II II II II II II II II II II II II II II II II iTiT ?T ?T ?T ?T ?T > >>>> >> > > > > > > > > > >>>>> Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из Ар уравнения Л
N) N) - n SJ H- -P- -P- -P- -P- -P- -й-4*. -Й- _ Ui -Й- W <-Л О О О CT Ui о <-Л О о о 00 00 00 00 OgJUiUiUi Ъ © о о ° Ъ© © © ©© © © ©Ъ 1л о оо Т,°С
AAAjLA A A A A w w A A A A A A A A 1л CTi \о V> Vi V> ui К> V> o\ os os oo oo os 4*. N) W bJ OS 00 00 W 00 00 W © '-J 00 1>J 00 W О \O — 1g к, к- константа скорости реакции
— bJ — Ошибка
£> 75ЯИЯ ” ПИ И? Метод
Продолжение таблицы 2.1.16
пгоконхэш п vxnwnx xnHhoavduo птявоц
89£
X п п 6 о о о I п X + I м О I1 X О о о О I I + о X о I О 2! О О д + о д о III о о д о X о X о о о о д о III о о Д + д о 1 •i о о о д + о д 4i о и о д о д о д о о о о о д о д II о II о д + д о I1- —► C6H5C(O)NHCH2C(O)O + СН3ОН СбН5С(О)КНСН2С(О)ОСН3 + ОН" —► __^O2N—С(О)ОН + сн2=снсн2он о X ф о о о о д о д II о д к> + д о И —► НОС(О)СН2С(О)ОН + С6Н5ОН НОС(О)СН2С(О)ОС6Н5 + Н2О —► O2N—с(°)°н + сн= СНСН2ОН °2N—С(°)°СН2С= СН + Н2О СбН5С(О)ОСН3 + Н2О —► СбН5С(О)ОН + СН3ОН СН3С(О)ОС6Н5 + Н2О —СН3С(О)ОН + С6Н5ОН Реакция
3 3 3 3 3 3 3 3 3 Тип реакции
X К) X К)
Н20 NJ О I bJ о X о X К) о X о X NJ О Н2О 0 - EtOH 0 - EtOH о Растворитель
1= 1,0 хэ X II о •I- 7 = 0,1 NaCl; 7 = 0,1; 7 о "я о II X — II о к> хэ X II 00 о И 'll О X II 1кции сольволиза Условия проведения реакции
II гь си II со II со < xi II II t? с? хС II Ci CD Xi II гь CD II гь CD II II Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из -Л Лр уравнения л 71 е
39,6 <-Л р о Ui о bO ЬО 0» О» 0 0 о» р о р Ох о» р о 00 ьо р у» ьо о р о Г, °C
1 00 Ui 1 о» ЧО 1 У1 p p £j й 1 У 1 00 1 У 1 1 00 р хо Ci ох 4^ 1 У OS Ui 1g к, к - константа скорости реакции
и» и» UJ и» Ошибка
о я о о я я О я П я о я о н а Метод
Продолжение таблицы 2.1.16
69£
кпЕЛффпр п пмпшэнпх мэмээкпппх
О
КОС(О)СН2С1 + NH3 —*KOC(O)CH2NH3 + Cl- СН3С(О)Вг + СбН5ОН —► СН3С(О)ОС6Н5 + НВг Я О о д о о-+ о Г д Чз о + о п д о о- К О п д о О’ + О' к д о + d о д о о- гН + ОНЭ Н + ОНЭН • • . к г-► Снсь + нс1 chci3 + н-LJ 1—► £ci3 + н2 6Н5С(О)ОС6Н5 + Н2О СбН5С(О)ОН + СбНзОН —► O2N-^hC(O)OH + СбН5ОН °2Ь[“ОНС(О)ОСбН5 + Н2° —►НОС(О)С(СН3)2С(О)ОН + С6Н5ОН НОС(О)С(СН3)2С(О)ОСбН5 + Н2О —► O2N-^y.C(O)OH + СН3СН=СНСН2ОН о о о о о Е о д II о д о д + д о н Реакция
3 3 3 3 2 2 3 3 3 3 Тип реакции
ж о MeCN Н2О Е о Е О Е О П5 о CD Н2О Н2О Н2О Н2О о Q3 Растворитель
сА = 0,7934 св = 0,2492 сА = 0,270 с А = 0,270 Е и •1-чО pH = 1,0 pH = 1,0 Цецин замещения 1= 1,0 кции сольволиза Условия проведения реакции
и и 7i оо со a3v^y = Л = Л II 0? a^v^y = Л II гь a3v^y = Л a^V^y = л 'll гь 0 Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g Л, Л- постоянная из _А уравнения л
25,0 0,0 25,1 22,0 22,0 § 2 я § 2 я 49,5 50,0 39,6 Ml о Г, °C
-5,20 -3,14 -2,35 -6,30 10,23 6,56 7,04 -5,22 -5,20 -5,48 -5,09 1g к, к - константа скорости реакции
'j — SO Ошибка
Алт Сп Сп Им.р. Им.р. X W W О я О я О я Метод
Продолжение таблицы 2.1.16
пгоконхэш п vxnwnx хпньоэпдиэ птдоц
ОДЕ
СН3С1 + KF —► CH3F + КС1 СН3С1 + KCN —► CH3CN + КС1 CH3CI + AgNO3 —►CH3ONO2 + AgCI CH3Br + KCN —► CH3CN + КВг НС(О)ОН + ОН —*- С(О)ОН + Н2О гн + но(о)э*—H + но(о)эн • CH2C12 + (CH3)3N —►С1СН2Й(СН3)3 + CI“ CH2C12 + OH —►CHC12 + H2O 1ЭН + ЮгНЭ 1 Н-н + Who . t 1—► CHC12 + H2 CH3CH(0H)00+X)H —► CH3CH(O~)OO + H2O -► H0CH2C(0)0Na + NaCl KOC(O)CH2C1 + CH3NH2—► -► KOC(O)CH2NH2CH3 + CI-KOC(O)CH2C1 + (CH3)3N —► -► KOC(O)CH2N(CH3)3 + Cl" CICH2C(O)ONa + NaOH —► Реакция
3 3 3 3 3 2 3 3 3 3 3 3 3 Тип реакции
Ж о X о Н20 - EtOH Ж о д о I О Ж о 1 r о X о о I О Ж О о cP Растворитель
г> СО II р о *11 р о сс2н5он 41,9 мол. % о X II о Q I II p •1- О CB = 0,1428 xch3c(O)ch3 68,9; cA= 1,435 о ж и о о Ж II о о » и о с a =0,3195 св = 0,0446 cA = 0,819 cB = 0,1697 1кции замещения Условия проведения реакции
v = ксЛсв ri II W v = ксКсв Ti II ? п со Ti II f » Ti II f n CD Ti II О » 11 W Ti II f £ Ti II C0 Ti II f n C0 и f » Ti II f w Кинетическое уравнение скорости реакции
0‘09 Энергия активации Еа, кДж/моль
1g А, А- постоянная из Е. уравнения л
50,2 04 Q4 О к> s § u» о § § K> О K> о о N> K> о Т,°С
-5,31 40 1 ьэ к> о 1 к> 40 p 04 1 40 40 04 P 04 О p 40 О i 04 1 U) U) 40 U) 1g к, к- константа скорости реакции
Ошибка
1 г 5 s p s 2 p rn “p S 2 p rn “p S 2 p > 3 r Метод
Продолжение таблицы 2.1.16
U£
КП£&ффщ> п пятиэнпх XVXZdhnwnX
СН3С1 + KSCN—►CH3SCN + КС1 CH3F + KI —►СНз! + KF CH3F + KCN —►CH3CN + KF CH3C1 + NaOH —► CH3OH + NaCI CH3C1 + Na2S2O3 —► CH3S2O3Na + NaCI CH3SO2C1 + H —► CH3s6]’ + HC1 НзС n°2 + 2cH3NH2~* cr N _ НзС-N-^^, CH3NH2.HCt CH3NH N CH3I + AgNO3 —► CH3ONO2 + Agl CH3I + NaNO2 —► CH3NO2 + Nal CH3I + NaN3—*-CH3N3 + Nal CH3I + KCN —► CH3CN + KI CH3I + KSeCN —*• CH3SeCN + KI Реакция
3 3 333 3^33333 Тип реакции
ж X Ж X N) N) N) N) 2 2 ~ О 2 2 2 - о О О О g Л) CD 2м CD CD CD X 1 1 1 1 X I X ЗС X ст> LX <ч ю LX. s LX. ьо to n> to to to LX. $ $ 2 2 о 2222 00000$ hL *Jm о О CD О о о о о X X X X с? ел Растворитель
СА ~ СВ ~ = 0,005 - 0,03 •*ch3oh - 5,0 *ch3oh — 5,0 *ch3oh — 5,0 *ch,oh — 40 св = 0,010 св = 0,02 4- 0,08 СА = СВ = = 0,03 4- 0,05 (C6H5)4As са = св = = 0,05 - 0,15 са = 0,0182; св = 0,0310 СА = СВ = = 0,03 - 0,05 1кции замещения Условия проведения реакции
<<<<<<< <<<< <<<<< < II II и II II и и и Il II II II II II f fff f f f f F F f F F f f f f CoCOCDCOCDtO CD CO CO CD CO 00 coco co co co Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из _ Е> уравнения л
ЬЭЬЭЬЭ— bJNJNJbJ N> -t*. U0 M § x -J OS LA LA О LA LA LA LA LA О LA jyi g 7“ JsO О О JD о о X ° О ~ О О О О О О Я UJ М Г, °C
—4,09 -5,94 -3,57 -3,84 -3,31 ~ 7,45 —4,18 -3,86 -3,70 -4.32 -2,328 -4,53 -4,11 -4,11 -0,86 -3,48 -3,19 -2,04 1g к, к - константа скорости реакции
CZ* ЬЭ UJ UJ •— UJ Ошибка
3 J 3 3 3 3 f 3 г г Г Метод
Продолжение таблицы 2.1.16
югоионхэш п пхпюпх xriHhovvduo птэоц
ш
—► NaOCH2CH2OC(O)CH3 0 Н2С—СН2 + CH3C(O)ONa —► (д о X ю + п о ; о о X ю о X ю о о р NCN=C(NH2)2 + ОН—►NCN=C(NH)(NH2) + Н2О О О X ю о X ю о 6 о F О X ю о X ю о + о ВгСН2СН2ОН + Н —*- НВг + СН2СН2ОН СН3С(О)СГ + Н—*- Н2 + СН2С(О)О- -► NCC(O)OCH2CH = СН2 + Н2О NCC(O)OH + сн2= СНСН2ОН —► 1 о о X ю о о о о X ю о X II о X + X ю о С1СН2С(О)ОН + СН2СНСН2ОН—► BrCH2C(O)OH + Н—►НВг + СН2С(О)ОН 1 П О о о о о X ю п X II о X ю + X ю о о о о о о X + п X ю II о X о X ю о X 1 СН3ОН + н—*-н2 + СН2ОН CH3I + C6H5SNa—►C6H5SCH3 + Nal Реакция
3 3 2 3 2 2 3 3 2 3 2 3 Тип реакции
X о АсОН X о н20 X о X о сн2 = СНСН2ОН сн2 = СНСН2ОН X о сн2 = СНСН2ОН X о H2O- MeOH а Растворитель
7? 7? 7? я я s
Я >|| II А ° Р о 8 2 а? II о о <Л Ch X II <Л X II 40 X II -1-00 X о ф X о ф X II о •I- X о ф X) 13 X X II II n* я s II p i замещения Условия проведения реакции
о X р X о >о3н
= л = л II F ’Эу = л = л II II F = л = л II II "и II Кинетическое уравнение
и? и? с? W CJ со и? 5s со W с? t? скорости реакции
Энергия активации кДж/моль
1g А, А- постоянная из к-Ае уравнения л 21К
30,0 60,0 комн. 110,0 комн. комн. 50,0 30,0 40,0 30,0 комн. 40,0 о © Т,°С
-5,33 -3,42 6,86 1 w ЧО 8,40 5,54 -3,02 -3,72 -3,34 -2,70 8,52 -3,82 Ch Ch k> UJ о -0,66 1g к, к- константа скорости реакции
Ml Ml Ml Ml сл Ошибка
Алт СО 4з со 4з р и 4з X X X Е.р. Y-P- X Мкр Мкр Метод
Продолжение таблицы 2.1.16
£L£
кшЛффпр n пхпшэнпя колээнпгтх
SC О о О О I Z—Ч д О о rs О
2NCH2C(O)NH2 + OH-^NH2CHC(O)NH2+ H2O H3CH2I + KCN —►CH3CH2CN + KI 2H5C1 + CH3C(O)ONa —►C2H5OC(O)CH3 + NaCI Н3СН2С1 + (CH3)3N-^CH3CH2^(CH3)3 + сг 2H5Br + NaSCN -^C2H5SCN + NaBr )С(О)СН2С(О)ОН + Н—► НОС(О)СНС(О)ОН + н2 (СН3)2С(О-)ОО + Н2РО4 :н3)2с(он)оо + НРО4 — ОСН2С(О)ОН + Н —НОСНС(О)ОН - н2 ► СН3С(О)ОСН2СН=СН2 + Н2О Н3С(О)ОН + СН2=СНСН2ОН—► ► СН3С(О)ОСН2СбН5 + Н2О Н3С(О)ОН + СбН5СН2ОН—-► ► СН3С(О)ОСН2СН2СН3 + Н2О :н3с(о)он + сн3сн2сн2он—► Н3С(О)ОН + Н —* Н2 + СН2С(О)ОН <т> Р 71 а §
3 3 3 3 3 2 3 3 Тип реакции
I w о I о 1 tn О X Д О I Ю о 1 > э Д О 1 ш о л к К) о д К) о д К) о сн2 =СНСН20Н о о д о 45 CD ео Растворитель
pH = 10,0 CA “ CB “ = 0,05-0,15 X о X II bJ CM Os o? II Q> О СЛ *11 о Ъ bJ Os сА= 1,434; св = 0,1405 И X § х II Os 00 S© 0? II о ьм > II о о S© *х о X II so сл д II о ХЗ д II см д II о 5 Kt: Н3С-^~Л-8О3Н £ ? II II О 6J о 0 д tsj CZ) р X II о сА = 0,10; Kt: H2SO4 Х5 д II жции замещения Условия проведения реакции
II a? II w , II ?г w II ?г > из II ?г СЬ и II t? II СЬ W II F t? < и 3* 7? II ;> X < II $ ;> X < II ?г ;> X Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из уравнения Л
KOMH. 1 о о см сл сл о ЬМ СЛ О § 2 X ЬМ bJ Q> § 2 X СМ О О OS сл о о о Ъ о о OS 0 0 § § Т, °C
9,45 CM Л см ю А см л 00 сл OS О 0 к) см Л so см сл л сл SO so 1g к, к - константа скорости реакции
CM м сл кма* Ошибка
Им.р. д 1 Д 2 W д 2 Ъ и д £ н н ? m 4з Метод
Продолжение таблицы 2.1.16
пгоионхэш n tmnwnx xnuhodvduo ппэоц
о п ж । о 1 о I о з xxn Q Q д я ▼ S * * 1 £ 2 о g я «5е s 2 •? 2 ’ll я ч * Il Mr 2 2 Q 2? II 2 й 11 S 1 2 S ® 2 2 о 1 Я о Q 11 £ Д Я Я P P я 2 ж я о 2 6 + о о + + + 5 S S о £ -8 о So о- ® ® S’ $• I. и s - ч ? s: е. j. J JL । Реакции замещения Реакция
к- я* ?’ к £ JF я S $ £| 1 1 о. . 1 1 1 J 5= ,? +я£Я§«-^яяя + '2 S S х S §1 || 2’ | | Ш | о- 7 + + s' о I 2 я Q 2 1 S °* 2 £? ® g 2‘ ” I ’ +l'6o;z:c:£®S о € | ' 1 £ f S ® § “ 1 о ж .я °i + + чЬ "Y я £ о к я + + y + о о о “ я ® ± о о о ° □ яд °
гм гм гм гм гм гм ГТ ГТ ГТ гм гм гм Тип реакции
Я 1-fH NJ ,? m О Я Я Я О g 1 дядя яя ООО ' У я оооо оо ел b р Растворитель
Г) Г) .я п 5 с\ м о io тз г. п> Q тзтз ж я я я« >„ "82° я я 7 ’£ ’£ ц и и II 11 Я А Я II || и я я <—> ° м и >?- И 11 11 О и II — — — -° г- Г1 и 11 W ю чо — 7 , о о Ъ О Я Р л Я1 о •'• -1 - * J Т. оо |“‘ й S© О4 Условия проведения реакции
II II II II И И II II II II II II ?Г £ f ?г?г ъ 00 00 00 00 р р 00 00 00 00 00 00 Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из _ Д. k- Ае^ уравнения Л
23 комн, комн, комн, комн, комн. 19,5 30,0 40,0 60,0 70,0 80,0 90,0 35,0 комн, комн, комн. Т,°С
ps оо у_> X I 11 111 5 Т-1 7° 7° 5> -1 S Сл О оо '— ъ Оч оо О Ъ» N> Я R мл ° к> -N <-Л U1 ОО UJ ЧО t-Л О t-Л О ООО 1g к, к- константа скорости реакции
К> t-Л и» и» Ошибка
3 з ? шгг Ш Ш?{ f ‘ 1 Метод
Продолжение таблицы 2.1.16
кшХффпд n пмпшэнпх южэдыгитх
CH3CH2CH2C1 + KNO3—►CH3CH2CH2ONO2 + KC1 -► CH3CH2CH2NH(C2H5)2 + Br- CH3CH2CH2Br + (C2H5)2NH—► CH3CH(OH)C(O)OH + H —► CH3C(OH)C(O)OH + H2 CH3CH(SH)C(O)OH + H—► CH3CH(S)C(O)OH + H2 —►CH3CH2C(O)OCH2(CH2)2CH3 + H2O CH3CH2C(O)OH + CH3(CH2)2CH2OH—► -► C2H5C(O)OCH2CH=CH2 + H2O C2H5C(O)OH + CH2=CHCH2OH—► CH3CH2C(O)OH + H —► CH3CHC(O)OH + H2 —► CH3CH2C(O)OCH2CH2CH3 + H2O CH3CH2C(O)OH + CH3CH2CH2OH—► CH3C(O)CH3 + H —► H2 + CH2C(O)CH3 C1CH2CH2C(O)OH + H—►CH2CH2C(O)OH + HC1 °r C1CH2HG— CH2 + KSCN —► C1CH2CHCH2SCN + K+ Реакция
3 3 3 3 3 3 2 3 2 3 3 Тип реакции
H2O - EtOH X о 1 m s X X о X о CH3CH2C(O)O H CH2=CHCH2OH X о о X w о X о X о a Растворитель
Св= 0,50 I и © © c =419- cC2H5OH -6 n a о a II so X II © X II © Kt: H3C<V SO3H ( Kt: Н3С-ЛЛ-8О3Н X II © Kt: H2SO4; XH2SO4 “ 0’ Ci JI о II M pp X II •1- UJ X II © 1кции замещения Условия проведения реакции
II B? II B? II w II B? II £ II II в? II £ II I? II w II B? Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из уравнения л 71 с
40 © © © © § 2 a s 2 a © k> UJ © © § Os © О § § © о Г, °C
1 M 00 Lj о 00 SO J- OS 1 UJ LA LA p UJ LA © 1g к, к - константа скорости реакции
N> LA -J Ошибка
x Prt b % Prt Prt § Метод
Продолжение таблицы 2.1.16
игоионхэш n vxnwnx xnuhoanduo niQsojj
9L£
CH3CH2CH2SO2C1 + Н—► -► CH3CH2CH2SO2 + НС1 CH3CH2CH2I + C6H5SNa —► -► C6H5SCH2CH2CH3 + Nal HC(O)N(CH3)2 + OH—►HC(O)N(CH3)CH2 + H2O CH3CH2CH2OH + Н—► СН3СН2СНОН + н2 (СН3)2СНОН + Н—►(СН3)2СОН + н2 СН3СН2СНОН (54,3 %) + Н2О СН3СН2СН2ОН +он Сн2сн2сн2он + н2о СН3СНСН2ОН (46 %) + Н2О НОСН2СН2СН2ОН + Н—►НОСНСН2СН2ОН + н2 (СН3О)3РО + ОН —*> СН2ОР(О)(ОСН3)2 + Н2О + . К г—► CH2NH(CH3)2 + Н2О (CH3)3NH + ОН ——— + 1—► (CH3)3N‘+ Н2О (CH3)3CNH3 + ОН—► CH2C(CH3)2NH3 + Н2О НОС(О)СН=СНС(О)ОН + н —*--** НОС(О)ССНС(О)ОН + н2 Реакция
гм ГТ гм гм гм гм гм гм гм гм гм Тип реакции
Ди Ж Д- Ди Ди Ди Ди Ди Ди еГ ЬО ЬО bJbObO bJbObJbJ /А bJ О О ООО о о о о $ о Ди о a co Растворитель
тзтз тз I _ та » > II I II I I II II II II II -j Т 11 11 <=> Я — -~J .|. — Ui о ° Ъ к> Ъ> - “ о м р; жции замещения Условия проведения реакции
<< <<< <<<< < < и II II II II и и и и и и ?Г ?Г ?г ?г ?г ?г ?г ?г 00 00 00 00 00 00 00 00 00 00 00 Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
IgA, А- постоянная из k= уравнения Л 71 е
8 S S S S 8 5 3 5 о 5 II = = = i I i S § g Г, °C
00 00 00 -J \O --J 4O --q M 00 O> O bJ t>J oo Lj %. 00 OOLh О 00 cs Cs --J oo UJ 22 <-Zl 1g к, к - константа скорости реакции
to Ошибка
₽ ? I ₽ f I S *0*0 00*0 00**0 • о Метод
Продолжение таблицы 2.1.16
LLt
ишАффпр n ОХПШЭНПХ ИПХЭЭЪПМПХ
( > + он—► < >. + Н2О о о огн + ох О *— но + о^ о —►(СН3)2СНС(О)ОСН2(СН2)2СН3 + Н2О (СН3)2СНС(О)ОН + СН3(СН2)2СН2ОН—► о X о X к> о я к> о о о о X о X о X + X ю о СН3СН2СН2С(О)ОН + СН3СН2СН2ОН—► -► H2 + CH3CH2CHC(O)OH о X n X О X к> n о g + X* CH3OCH2OCH3 + OH —► CH2OCH2OCH3 + H2O CH3C(O)OCH2CH3 + H—► CH3C(O)OCHCH3 + H2 cQ) + о* X cCj • + X K> о гн +_o(o)dq2(£hd) H + -O(O)3H34EH3) -► HOC(O)CHCH2C(O)OH + H2 HOC(O)CH2CH2C(O)OH + H —► о X о о о о X + о-X о X к> о о о о о ,х + X к> о + н —- О + Н2 Реакция
3 2 3 3 2 2 2 3 3 2 3 2 Тип реакции
X о X о (СН3)2СНС(О)ОН о X о X о X о X о X о X о X о X о €? CD Растворитель
X II © Kt: H3C<ySO3H * 55 II © сА=ол; Kt: H,SO4; X II © X II о X II X II © X II to © 1кции замещения Условия проведения реакции
II Д W II Д п? II Д' II S' > £ ii Д c? II S' > co II д t? II Д' II S' > w II Д' w II Д' W II S' > со Кинетическое уравнение скорости реакции
Энергия активации кДж/моль
IgA, А- постоянная из уравнения Л ле
§ % § % LA о к> OS © © § s 2 a s § § § § § § § Г, °C
so os с so 1 N> о 00 00 OS 00 so о 00 ДЛ О SO Qs © os tso OS ж so bJ so so LA 1g к, к - константа скорости реакции
Ошибка
X 2 р X 2 р % r p (Tf p X 2 p 2 £ X 2 p -< m p p X 2 p X 2 р X 2 p Метод
Продолжение таблицы 2.1.16
игоионхэш n tninwnx xnHhosvduD пкюоц
8Z,£
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из _Я, к = Ае уравнения о о к 1g к, к - константа скорости реакции Ошибка Метод
Реакции замещения
СК СН СН СН СН (С1 (С1 СН (С1 СН к [3(СН2)2СН2С1 + AgNO3 —> - CH3(CH2)2CH2ONO2 + AgCl [3(СН2)2СН2С1 + CH3OLi —► ► СН3(СН2)2СН2ОСН3 + LiCl 3(CH2)3I + NaN3—►CH3(CH2)3N3 + Nal NH + (CH3)3COO —► |^N* + (CH3)3COOH [3c(O)N(ch3)2 + Он — - »► CH3C(O)N(CH3)CH2 + H2O . k r— CH3(CH2)2CHOH + H2 I3(CH2)3OH + h£J CH3CH2CHCH2OH + H2 43)3COH + H—►(CH3)2C(CH2)OH + H2 43CH2)2O + H —► CH3CH2OCHCH3 + H2 3CH2CH(OH)CH3 + H—»-H2 + CH3CH2C(OH)CH3 Щ)2СНСН2ОН + H—*-(CH3)2CHCHOH + H2 • к ш [3CH2CH2CH2OH + QH— * —► СН3СН2СН20НОН (41 %) + H2O — ► 0h2ch2ch2ch2oh + H2O — ► CH3CHCH2CH2OH + H2O — ► СН3СН20НСН2ОН (58,5 %) + H2O ГТ ГТ ГТ гм гм гм гм гм гм гм гм Ру МеОН МеОН Н2О Н2О Н2О Н2О Н2О Н2О Н2О сА = 0,0464 св = 0,0493 сА = св = 0,025 сА= 0,0185 св = 0,0450 pH = 6 - 7 pH = 1,0 pH = 2 pH = 1,0 pH = 1,0 pH = 1 v = Асасв v = Асасв v = Асасв v = Асасв v = Асасв v = ЛсАсв v = ЛсАсв v = ЛсАсв v = Асасв v = ЛсАсв v = Асасв 27,6 36,0 60,4 30 комн. комн. комн, комн, комн. комн, комн. -3,67 -6,73 -3,07 2,58 9,54 7,54 5,23 7,63 8,04 7,72 9,62 3 2 5 Ацт Пт Фот Им.р. Е.р. у.р. Им.р. Е.р. Е.р. у.р. Е.р. у.р. Им.р.
Химическая кинетика и диффузия 379
О о 1 ас о п ас 1 г> ас 1 о *г J с о ас
NH + (СН3)3С(О)О —► ^/Г+ (СН3)3С(О)ОН + *сн3 —► Q> • + сн4 НОС(О)(СН2)3С(О)ОСН3 + Н20 С(О)(СН2)3С(О)ОН + СН30Н —► CH2C(CH3)2NH2 + Н20 . К г-*- (CH3)3CNH + Н20 :3)3cnh9 + он^н . CH3CH2CH2CHNH2 + Н20 .ch2ch2ch2nh2 + он—► CH3CH2SSCH2CH3 +0Н- I3CH2SSCH2CH3 + ОН —► —►ён2СН(ОН)СН(ОН)СН3 + Н20 —►СН3С(ОН)СН(ОН)СН3 + Н20 • /с 1 (СН(0Н)СН(0Н)СН3 + ОН-^ ПЗ о> £ я §
2 2 3 2 2 2 2 Тип реакции
I £ I H2O-HNO3 I о к м о I о I о ПЗ о> ео Растворитель
О S о о О 5* О II о £ р о II о г* i р Z о II о я г* i р § о II о i р I II о I и о .кции замещения Условия проведения реакции
= л II F с? II II II |Г II |Г с? II |Г С? II |Г С? II ?г > ас II с? Кинетическое уравнение скорости реакции
54,1 Энергия активации Еа, кДж/моль
11,265 ! 1g А, А— постоянная из уравнения Л 71 е
о о сл о о и! ъ о о м и> о S 2 X § % § % § % Г, °C
1,32 1 00 о 00 о м ъ <L 40 М 1 о SO 00 00 SO р сл 1g к, к - константа скорости реакции
о о О о Ошибка
Фот -3 £ 1 £ г S S р S г Р S г Метод
Продолжение таблицы 2.1.16
пгоконхэш п vyinwnx xnuhosvduo пмэоц
ОЗЕ
+ HOC(O)CH2NHC(O)CH2NH2. НС1 о z: ГД 1 Z д п д п о Z д п д о о о д + O2N-^2/~C1 + 2NH2CH2C(O)NHCH2C(O)OH ► no2 О no2 —► 02N-Д2^-ОС2Н5 + НС1 O2N—Д- С1 + С2Н5ОН —► n°2 —»-O2N-(>-NHCH2C(O)NH2 + NH2C(O)CH2NH2-HC1 1 О /N02 02N-£)-Cl + 2NH2CH2C(O)NH2 —»• —►ChN— NHNH2 + NH2NH2. HC1 z: 0 O2N-4}-Cl + 2NH2NH2 —► z: 0 /=\/NC>2 —►O2N—4 )—ONa + NaCl + H2O ZN°2 02N-0-Cl +2NaOH —► Реакция
ц 3 3 3 3 Тип реакции
д м о Н20 - EtOH д о Д tQ 0 H20 - EtOH Д О <? £0 Растворитель
7 о X ъ X II О X о I II la Lj X о X II la II © II © Cl 2: № О X II О О * P X 0 X II Cl z О X II $ 'Z $ V to to ООО XXX II II II M — © LA ООО 1КЦИИ замещения Условия проведения реакции
xi II S' > со II S' W II S' W S' > со II S' > СО и w II > И II < < ci II II II ?r > > > Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из к = Ае уравнения л
р о р © м la © р о © о © О LA © LA © w W Ю LA LA LA © © © Т,°С
1 LU ъ I М М О 1 N> 00 “о о tL 5 © jb 00 -4,00 -3,67 -3,14 1g А, к- константа скорости реакции
Ошибка
О я Д Д Д О я 0 a n a r> a Метод
Продолжение таблицы 2.1.16
ISE
ишЛффпр n плпшэнпл хояээьпппх
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации кДж/моль 1g А, А- постоянная из L__ П5 уравнения л и 0 1g Л, к- константа скорости реакции Ошибка Метод
Реакции замещения
_ no2 O2N-£^c| + 2C4I5CH2NH2 — ,n°2 —► ОгТ^ЛИНСНгСбНз + СбЩСН^Щ- НС1 NO2 °2mC~ci + 2 CO ~ no2 —► O2N-£> + O~ NH2’hC1 Z=KN°2 Cl + 2 —*" no2 /=\ _ nh2-hci —nh-qqq+Q-^Y Cl-АУ-F + NaOC,H5 —». —► Cl^V OC2H5 + NaF no2 no2 (jCl + 2NH3 —^-Т4Н2 + NH4CI ГТ ГТ ГТ ГТ ГТ Н2О - МеОН Н2О- МеОН Н2О - МеОН Н2О - EtOH н2о А —! £ £ * п5 П п п5 “ •• 3 ? 3 2 || II = О О II II о „ О = = = = р р о = Я oo S ~ О II II II II Ъ ~ II II 11 X 5 “ ^ 00 000000 4^^40 <£? о ,lj — •I- см 5? и, о " W • \© чэ р О V = fa?ACB V = fa?ACB V = fa?ACB V = fa?ACB V = fa?ACB V = kcAc3 v = kc AcB v = fcAcKt 25,0 30,0 35,0 100,0 100,0 76 150,0 160,0 170,0 180,0 140,0 -3,38 -3,17 -2,99 -3,56 -3,48 -4,39 -5,22 -4,58 -4,77 -4,52 -3,99 6 3 4 4 Диэ Диэ Диэ Диэ Диэ Алт Пт Пт Пт Пт Агт
Новый справочник химика и технолога
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из lf = Де КТ уравнения л 71 с О о Е4 1g к, к - константа скорости реакции L .. Ошибка Метод
Реакции замещения
no2 no2 (j^-Cl + сн3он —*- <^-ОСН3 + НС1 ГТ Н2О - МеОН хсн3он 49,9 •^сн.он =91,5 V = ЛсдСв V = ЛсАСв 80,0 80,0 -2,48 -2,38 Кот Кот
no2 no2 ГТ Н2О - EtOH хс2н5он “ 78, 1 V = ЛсдСв 70,0 -2,85 Кот
+ с2н5он —{^-ос2н5 + нс1 хс2н5он = 78 Л хс2н5он ~ 78,1 V = АсдСв V = ЛсАсв 80,0 90,0 -2,45 -2,06 Кот Кот
О2Н-^-С1 + СН3ОН—► ГТ Н2О - МеОН хсн3он = 49,9 V = ЛсдСв 80,0 -1,95 Кот
—► O2N-QhOCH3+ НС1 хсн3он = 84,3 V = АсдСв 80,0 1,97 Кот
o2K^yci+ С2Н5ОН —► Н2О - EtOH хс2н5он = 40,8 V = ЛсдСв 80,0 -2,54 Кот
—► O2N-Q- ОС2Н5 +НС1 xc2hsoh = 94,1 V = АсдСв 80,0 -1,93 Кот
СбН5С1 + 2NH3—*C6H5NH2 + NH4CI ГТ Н2О Kt: CuCl; cNH, = 15 - 30 % Kt: CuCl2; wnh3 = 15-30% 5 5 2 2 2 о о и о о £ £ II II II II II >. >. > > > 180,0 190,0 200,0 210,0 180,0 -3,26 -3,07 -2,91 -2,77 -3,65 Агт Агт Агт Агт Агт
С6Н5С1 + Na2S2O3 —*- C6H5S2O3Na + NaCl ГТ Н2О - МеОН xch3oh = 24,4; V = ЛсдСв 15,0 -2,26 Ит
СбН58О2С1 + C6H5NH2 —► QHjSQjNHC^ + HC1 ГТ МеОН cA = cB = 0,02 V = ЛсАсв 35,0 -0,94 5 К
,CH3 C6H5SO2C1 + O^nh2 ^CH3 ГТ МеОН V = ЛсАсв 35,0 -2,73 5 К
H3C. —^C6H5SO2NH-Zj> + HC1 НзС^
Химическая кинетика и диффузия
—► NHC(O)-^J>- Cl + HCI СГ * \ // Qnh2 + C1-Q-COC1 —► сг H2N-^~~^- ci + 2NH3—► HoN-Z^VNH, + NH.CI C6H6 + СбН5СН2СН2С1 —>С6Н5СН2СН2С6Н5 + НС1 С6Н6 + С6Н5СН2С1—^С6Н5СН2С6Н5 + НС1 С6Н6 + (С6Н5)2СНОН —СбНзСНССдаг + Н2О С6Нб + (СН3)2СНВг —► С6Н5СН(СН3)2 + НВг о о, X q + о X о о-; п X о о + о % о* О o2n o2n —* Ху-МНС(О)СбН5 + HCI Z^-NH2 + СбН5С(О)С1 — О Реакция
3 3 3 3 3 3 Тип реакции
1Э ЕГ К X о 2 о Z о N> z: р > о X 2 z: р X о ЕГ z о to 1Э с? ьэ Растворитель
Я и
7^ О ca = 1,12; св = 0,4 Kt: CuCl; cnh3 =15-^30% сА = 1,75 ; СВ = 0,15 caici,ch,no2 - 0,2; п ь 3 'z о II р п> X о II р и! 2 § Хр о4 сА = 1,75 ; св = 0,15 CH2SO4 _ ^,75 п ь £ 'z о II р Х5 X II ?5 о £ о г> Р X X о о X ,:(СН3(СН2)2СН2)2Р(О)ОН [ии замещения Условия проведения реакции
II 0? п 5 п Я? и & II ? с? II £ II £ II ? с? II > <> 00 <> £ II f 0? <> II f * Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из 1г= Ао уравнения л
к> о «о p о о о К) У О к> о к> о § к> О к> сл О к> о Т,°С
р 1 К) i К) UJ Os Os сл л OS о р й с* 1g к, к- константа скорости реакции
Ошибка
□ Г % О я X 2 хз X X X Метод
Продолжение таблицы 2.1.16
пгоионхэш п tmnwnx xnuhosoduo птвоц
ж
o2n _ —► ^2^~NHC(°)C6H5 + НС1 o2n z + о X о о о О z:. § 0 I о p о % LA + К w Cl £>NH2 + C6H5C(O)Br 1ЭН+еНЭОН( у-(о)энм-\ ) *— Cl r>NH2 + Н3сЦ^СОС1-► cr —* < \-NHC(O)—( У-СНз+HCl (~Vnh2 + H3C-Z~)-COC1 — cr 1ЭН + £н9э(о)энм-{ /-Ю Cl С1~СУ” NH2 + СбН5С(О)С1 —► Д о p i z p + я Q 6pNII2 + O2N-^-CO€l —► Cf Реакция
3 3 3 3 3 3 Тип реакции
PhH AcOEt PhH PhH ТЭ § p 1 PhH о Растворитель
СбН5ч Kt: P(O)OH CH3' Kt: (CH3(CH2)2CH2)2P(O)OHj Kt: N<^N^CH3 Я о я о я £) Я €) :кции замещения Условия проведения реакции
v = kcKcB cKt II > И £ v = ксАсв cKt II > 09 2? II 09 2 v = ксдСв cKt Кинетическое уравнение скорости реакции
- Энергия активации Еа, кДж/моль
IgA, А- постоянная из 1г = Ле уравнения л 71 с
to СЛ © 25,0 1 25,0 25,0 25,0 25,0 I 25,0 Г, °C
s i, О U) 0,16 1,68 8l‘l 1g к, к - константа скорости реакции
Ошибка
3 о я a H a Метод
Продолжение таблицы 2.1.16
S8£
кп&Сффпр n гмпшэнпх юхээкпмпх
C6H5NH2 + С6Н5С(О)Вг—►C6H5NHC(O)C6H5 + НВт О 1 Ъ 1 NH2 + (СН3)3С(О)б —► (CH3CH2)3N + ОН—►CH3CHN(CH2CH3)2 + Н2О СбН58Н + (СН3)3С(О)6—► C6H5S + (СН3)3С(О)О1 С6Н5ОН + (СН3)3С(О)О—► (СН3)3С(О)ОН + СбН5д ЮН + 0(0)0—«<— о 2! о
NH2 + СбН5С(О)Р —►C6H5NHC(O)C6H5 + HF О X о о о X + X —► <Г~У- NHC(O)C6H5 + НС1 1 п Z X + о £ о о о
о о + X О *7
3 3 § 3 3 § 3 3
М3 М3 М3 X мз М3
а* а* <т <т а а*
X X а о я в о X
X X X z:
75 Т Т 1 Kt: 7^
SO SO
о о к о о О X d, о о X о о X to <0 п / о С7 X X О О' X MS
о [1 1 о о О о о О О о Q X о X О О
X
II и и II II
(Г II II II II II
из W Ci *Ci со со со со
со со со
?! ?! г. г. ?! ?!
ю to
о
SO Os 00
Os
OS U1 ю
Ю ю 1 § 1 1 to to ю Ю Ю
о о я о о о о о
1 to © is 1 1 р р
го N) U1 о> о о О U1 ,00 ,01 1л
X Пт Фот Мкр Фот Фот Кот X X
в ?! Я I § <Т> В <т> я Я
в Я я §
Тип реакции
Растворитель
Условия проведения реакции
Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из
уравнения Л
Г, °C
lg к, к - константа скорости реакции
Ошибка
Метод
Продолжение таблицы 2.7.16
огоионхэш п oxnwnx xnuhosoduo ттаоц
98 £
о Tl a n
Н3С(О)ОСбН5 + Н+ —► СН3С(О)ОН + С6Н5 £ О + о % О' Г) ?j _^F3c-<^j>_nh2 + nh4ci 2: О 3С-( />-С1 +2NH3 — 1 •z О (CH3)2CHSSCH(CH3)2 + ОН" НО + z(EH3)H3SSH3z:(EHC 2>+сн3 —► 0. + сщ * НО-£У NHCXOJCfjHs + HCI 0-(Qh NH2 + C6H5C(O)C1 —► sH5NH2 + СбН5С(О)С1—►C6H5NHC(O)C6H5 + HCI <? co Й §
3 2 3 2 2 3 3 Тип реакции
Н20 I м о I о I м о I 8 S 0 I 0 ar I s p <? ea Растворитель
п ас 5 II о тз I II о п 1 и 00 тз I и (Эо ОбНЭо 09) Kt: (CH3CH2)4NC1 Kt: (CH3CH2)3NHCr 75 Ф Kt: C6H5NO2 Kt: CH3CN Kt: CH3COC2H5 Kt: CH3COC6H5 cKt = 0,03 - 0,08 cB= 0,01 ; Kt: CH3COCH3; *11 p 0 0 01 75 0 1кции замещения Условия проведения реакции
II о? II |Г С? II |Г t? II |Г 0? и |Г W 11 0? v = fe?AcB cKt II 1 2? 11 £ £ 11 0? £ 11 0? 11 0? v = kcAcB cKt II 0? £ Кинетическое уравнение скорости реакции
47,3 Энергия активации Еа, кДж/моль
10,06 1g А, А- постоянная из к^Ает уравнения ле
50,0 о 2 ас р 0 § 2 ас СЛ N> LA О N> LA О N> LA О N> Oi О N> Oi О N> Oi О N> Oi О N> LA О LA О Г, °C
1 00 00 1 bJ 0 р о N> О О О О 04 Й л i Л чО 00 N> 00 04 N> О 1g к, к - константа скорости реакции
±13% Ошибка
о д S 2 *ХЗ a н 2 $ s 2 ТЗ О Д О Д $ $ H H § Метод
Продолжение таблицы 2.1.16
Z.8E
хшЛффпд п пхпшэнпх ктмэдыжпх
С(0)СН2С1 + <Qn —► - Вк —► -^>+ С1 - 0-C(O)H2CI+ Q4^ — <Qrt-cH2c(O)HQ>+С1- 0^ОСН2С1 + Ск —СЙ-СН2С&О+СГ С6Н5СНО + Н —►С6Н6СНО1’ С6Н5С(О)ОН + Н—►С6Н6С(О)ОН1’ СбН5СН2С1 + н—► ir + сг+ С6Н5СН2 СбН5СН2С1 + CH3ONa СбН5СН2ОСН3 + NaCI С6Н5СН2С1 + C^NH^ C6H5CH2N(C6H5)H2 + СГ СбН5СН2С1 + N(CH3)3 —► СбН5СН2й(СН3)3 + СГ Реакция
ГТ ГТ ГТ гм гм гм ГТ ГТ ГТ Тип реакции
2 2 2 ~ 2 2 2 <т> <т> 1 <т> гг" гг1 <т> <т> <т> ОО'рООО о о о = 1 2 1 * х сл О О Растворитель
° п >.. ,, > 3 п II II II 1 II П и « И — п —, о —о о о = £ > " r II "О "О -g > Р > £ > % © II II и О © Е ® ® II ® II ® II о§ i Sg i 11 - - - ъ d 2 w о и © и © и Й и S © © © 1кции замещения Условия проведения реакции
<<<<<<< < < < II II II II II II II II II II ?Г ?Г ?Г ?Г ?Г ?Г $ >>>>>>> > > > о о Ш Ш Ш Ш Ш Ш Ш и и и Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из _ Е-к=Ае^ уравнения Л 71 v
bJ <>J bJ N> § § § bJ bJ bJ -lb. (» (» -|b. g g g о © © OS © © OS я я я © © © Т, °C
Jb Jb Л P® « Jb ik Jb — bJ uj r; S© Lj os Ui bJ Ui -J bJ so 1g к, к - константа скорости реакции
UJ Ошибка
5 f f £ *ч H *P *p Метод
Продолжение таблицы 2.1.16
пгои'онхэш п vytnwnx xnuhosvduo тязоц
88£
О Z q О О ‘ 35 д 1 ''т? 1 О д о О\ д о о 1 о о О\ д о о О О\ Д о д 3х 0 5 О Д
Д + О’ к л к о О <£-О Jr О' *-?> к л о А К о К о-Sd ,5е ’ + Д о Н + СбН5СОС1—► NC(O)C6H5 + НС1 С2Н5Х Н00(сНЭ)НЗ +снэнэ^( Н2СН3 + ^2)- СН(СН3)ОО —► 5OCH2CH2NH(CH2CH2CH3)2 + Br" Н2СН2Вг + (CH3CH2CH2)2NH —► ОСН2С(О)ОСН3 + Н2О Н2С(О)ОН + СН3ОН —► сн3 ,ОН + Н—►С6Н6СН2ОН>- —► / J—NHC(O)C6H5 + НС1 NH2 + СбН5С(О)С1 —► —► (CH3)3C(O)OH + NCH3 HCH3 +(CH3)3C(O)O’—► —► CH3-^y-OC2H5 + NaF Ti + Z sa О О Д Реакция
2 3 2 3 3 2 3 2 3 Тип реакции
X Д
X О PhH PhEt ,0 - EtOH МеОН X о PhH HeptH ,O - EtOH *0 G> Растворитель
X
pH = 9 я о 25-65 °C Фс2н5он 95 % Kt: НС1 pH = 0,4 -ь 1,0 Kt: N<S/N'CH, (-75 °CH-43 °C) cA= 0,2162 cB = 0,2149 я о я II LZi (ни замещения Условия проведения реакции
II II и и II II II и II Кинетическое
/Г Ci /Г уравнение
£ п я сь W "сь св =£ £ Ci 5 & "n СВ скорости реакции
35,62 10,5 Энергия активации Еа, кДж/моль
6,51 t/l 1g А, А- постоянная из _£-k— Ар уравнения л 71 е
комн. 25,0 о 70,0 30,0 40,0 комн. 25,0 Os Г, °C
10,15 -0,20 0,16 ю L/I J- J-К> UJ 9,04 3,38 2,76 -5,49 1g к, к - константа скорости реакции
к> Ошибка
Им.р. О я 5 у.р. Алт Алт Е.р. a Фот Алт Метод
Продолжение таблицы 2.1.16
68£
кпЕ^ффпд п охпшэнпх кпхээнпппх
(CH3)3CSSC(CH3)3 + 6н—► -► (CH3)3CSSC(CH3)3 + ОН-(CH3CH2CH2CH2)2NH + он—► -► ch3ch2ch2chnhch2ch2ch2ch3 + Н2О СбН5СН=СНС(О)ОН + СН3ОН—► -► С6Н5СН= СНС(О)ОСН3 + Н2О СбН5ОСН2СН2Вг + (CH-^NH —► -► C6H5OCH2CH2NH(CH3)2 + Br" ^усн(сн3)2 + с(сн3)2оо —► -^<QhC(CH3)2 + <QhC(CH3)2OOH QQ"OH +(CH3)3COO —► —► QQ°-+(CH3)3COOH OH 6 (^Q^+(ch3)3coo —► (0[^1 + (ch3)3cooh nh2 nh (0[^)+ (сн3)3сод —► [^j[^)+(ch3)3cooh + (СНз)зСо6 —► +(CH3)3COOH Реакция
ГМ ГМ ГТ ГТ ГМ ГМ ГМ ГМ ГМ 1 Тип реакции
о O' X k> 7' 7' 7’ 7' О Ж 75 Ж Ж О , Ж Ж о о го го ж' 2 25= m О О О к х х х Q о 1 У X о Растворитель
Т Т 1 т « чо оо 2Z чо ° ° о ° У х я !-?> ж _ О О о О У о и " п X х ; т I & д ? 1Й i: i В О s gpd -о * О О о О П П о о 1кции замещения Условия проведения реакции
<<<< << << II II II II II II II II F f ? F F ?г F f Ci CiCSCS CSCS>CJCS 05 CD Q5 Q5 05 CD CD CD Кинетическое уравнение скорости реакции
о N 04 M — 4^ Энергия активации Еа, кДж/моль
У У У У -°'' Ъ к> 4^ 4^ У* 1g А, А- постоянная из к^Ае^ уравнения Л Z1V
комн, комн. 25,0 35,0 45,0 55 70,0 30,0 -73 -73 -73 -73 Г, °C
У У Л У Я <L II I о Я fe X У лй Я w th OS W tz. М ®2 43 00 ~ О >— 40 40 4Л 04 1g к, к - константа скорости реакции
Ошибка
е е е е я 3 § 3 3 о Метод
Продолжение таблицы 2.1.16
огоионхэш п nwiwnx unuhoaoduo тчиощ
06£
о О
—► 02N—s —+ 02N——ONa ^Z О Ю ] /N02 °2N^C_/~°~С~/N°2+v#SNa BKO—^--y_N3O+ £N N.3O -« z p 0 Ю /NO2 °2N—C_/— ° V_Z N°2 + NaN3 Mo2 1 О z ф о 1 п 'и> X °+ р Z О' р w р Z о Г J Z р + X п б ь сл О Г Z О 1 § 9 1 о б + о 2: ф о 75 о К) о К) б & б д о ю + о о 75 Z о К) j —►(СН3)3С-/2У °’ + (СН3)3СООН :снз)зс-<уон +(СН3)3СОО —► Реакция
3 3 3 3 3 Тип реакции
0> 0 X 0> 0 X X ? о X ? □ X о X б? Растворитель
cA= 1.64 10 2; cB = 3,9 • 10 2 X р II tzi X р II Vi X о II V с? о II V (-64 °С)-(-32 °C) кции замещения Условия проведения реакции
< < ri < II II II II > > > > ъ ъ ъ ъ w w w « 11 0? < t < II II II Гь ГЬ Ci ф ш ш II v = ксАсв v = ксксв Т1 II £ Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
tj 1g А, А- постоянная из к^Ае^ уравнения л
N) LZi м о £ 0 © О N> LZl О 20,0 1 30,0 р о о о ю о> ф © © Г, °C
О LZi р © к> 0,19 0,62 jb Qs Os L 2- QS 00 О — л о д* £ ю 2-© Os к> Os 1g k, к - константа скорости реакции
<-Л к> ю — Ошибка
о о о о я я я я о я ООО я я я О я О я О я е о Метод
ЬПЕ&ффЩ) П ПХПШЭНПЯ KDXOdhnwnX
Продолжение таблицы 2.1. J б
I6£
|i о к —►NC-Zy-O* + (СНз)зСООН jn о к Ji о к NC-Zy-OH + (СНз)зСОО—► jQ о к Ji о к —►/ уо* + (СНз)зСООН 1 О о к о о к (/ У ОН + (СНз)зСОО’ —► о о к о к —► С1-7 У О’ + (СНз)зСООН о к о о к /_<С(СНз)з Cl-f Уон + (СНз)зСОО—► 1 О n 1 ’Т) о> ё ё §
о о Л 7 z о О о о к + д о \/ Z к + о о X о о о т
§ 3 2 3 Тип реакции
о г;
3 X Аз 3 ^т- Аз 3 £ 1 1 •тз £ ’Т) Растворитель
ж д
т чо 00 0 о X о о т SO 00 о О X 1 OS о о SO 0 о X 00 о о ✓ (И а II so SO 00 * а II SO so Os я II p> II LA О SO (И JS II ьо ьо о JS II о os 7s о X 2! > II II ° II О II ьо ' £ II 2 W I о> в о> X я Условия проведения реакции
II II II < II II II II II Кинетическое
/г /г /г д /г уравнение
& п? Ci СО с? n а £ 'Ci w "п а п а скорости реакции
ЬО ЬО ьо Энергия активации Еа, кДж/моль
ъ 4^ 4^ 1g А, А- постоянная из Е. к = Ае^ уравнения л 71 е
ъ ьо LA О Т, °C
ьо LM LM X )—* 1— >— 1g к, к - константа ско-
о О и! UJ ьо Ml bi OS с> рости реакции
Ошибка
е о е о н е о § $ я г Метод
Продолжение таблицы 2.1.16
огоионхэш n nxnwnx unuhosoduo tUQSOJJ
S6£
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из k^Ae^ уравнения л 71 е О о 1g к, к- константа скорости реакции Ошибка Метод
Реакции замещения
С(СН3)э гм z-PentH (-100 ОСН-32 °C) v = Асасв 3,3 4,6 -73 3,73 Фот
Н3сУ”Л-ОН + (СН3)3СОб—► V4C(CH3)3
С(СН3)3 —►H3cV~Vo- + (СН3)3СООН х:(сн3)3
/С(СН3)3 гм z-PentH (-98 ОСН-36 °C) v = Асасв 1,7 4,7 -73 4,26 Фот
СН3О-0-ОН + (СН3)3СОО —► ^€<СНз)з С(СН3)3 —► СН3оУЧ-О- + (СН3)3СООН ^С(СН3)3
CVI5CH(COC6H5)Br + C«H5NH2—>. С6Н5СН2ЛН2СН(С6Н5ХСОС6Н5) + Вг- ГТ Н2О - EtOH ^CjHjOh “ 23,6; сА= 0,025; св =0,25 v = Аса 50,0 -3,19 3 Агт
Реакции рекомбинации и диспропорционирования радикалов
Н + Н —► Н2 гм Н2О pH = 0,4 -е- 3,0 v = Аса2 25,0 10,42 Им.р.
СС13ОО’+ СС13ОО’ —► Продукты гм Вода - z-PrOH ^(СНз^СНОН ~ 48 v = Аса2 комн. 8,30 Им.р.
СН3 + Сн3 —► Продукты НОСН2ОО + НОСН2ОО —► Продукты гм гм Н2О с-Нех Н2О pH = 3,0 ££ £ II II II > > > 25,0 25,0 22,85 7,34 9,95 8,48 ЭПР ПО Им.р.
СН3СН2 + СН3СН2 —► Продукты гм СН3СН3 v = Аса2 3,48 10,11 30,0 9,51 Им.р.
СН3СН(ОН)ОО + СН3СН(ОН)ОО —► Продукты гм Н2О pH = 6,8 v = Аса2 19,85 8,85 Им.р.
Химическая кинетика и диффузия
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Ea, кДж/моль IgA, А- постоянная из E. k = Aew уравнения л О О 1g к, к - константа скорости реакции Ошибка Метод
Реакции рекомбинации и диспропорционирования радикалов
(СН3)2СНОО + (СН3)2СНОО —► Продукты гм PentH CF2CC12 (-63 °C)-(27 °C) (-63 °C)-(27 °C) v = kc/? v = kc/? 9,22 8,08 7,68 7,33 24,85 24,85 6,08 5,91 Фот Фот
cf2ci2 (-63 °C)-(27 °C) v = ЛсА2 26,0 10,68 -73,15 3,89 Фот
(СН3)2С(ОН)ОО + (СН3)2С(ОН)ОО —► Продукты гм Вода pH = 3,0 v = ЛсА2 22,85 7,04 Им.р.
(СН3)3С + (СН3)3С —► Продукты гм c-Hex v = ЛсА2 25,0 9,32 ПО
(СН3)3СОО + (СН3)3СОО —► Продукты ж HeptH г-BuH (-64 °C)-(-12 °C) (-80 °CH-16°C) II II 26,8 32,6 9,1 9,2 -73,15 -73,15 2,11 0,69 Фот Фот
МеОН v = kc/C 21,85 4,00
PhH v = fcA2 21,85 3,60
PentH (-40 °C)-(37 °C) V = kc\ 10,9 24,85 6,60 Им.р.
СН3СН2СН(СН3)ОО + СН3СН2СН(СН3)ОО —► гм BuH CF2C12 (-80 0CH-16 °C) (-98 0CH-73 °C) •4 r! II II 11,3 4,6 9,0 7,1 -73,15 -73,15 6,04 5,90 Фот
—► Продукты
(СН3)2С(ОН)СН2ОО + (СН3)2С(ОН)СН2ОО —► гм H2O pH = 9,4 v = kc/? 19,85 8,90 Им.р.
—► Продукты
|^>—ОО + [^>—ОО —► Продукты гм CF2C12 c-PentH (-98 °C)-(-73 °C) (-80 °CH-16 °C) v = ЛсА2 V = kCf? 10,9 13 9,6 10,0 -73,15 -73,15 6,76 6,60 Фот Фот
—ОО + —ОО —► Продукты гм c-Hex PhH 12-60 °C v = kc/? v = kc/? 5,4 7,29 24,85 26,85 6,34 6,58 Фот Фот
С6Н13ОО + СбН13ОО —► Продукты гм Hex 10-47 °C v = fcA2 8,4 7,48 24,85 6,00 Фот
С6Н5СН2 + С6Н5СН2 —► Продукты гм PhH v = kc^ 25,0 9,915 ПО
С8Н17ОО + С8Н17ОО —► Продукты гм OctH 10-85 °C v = kc/f 8,4 7,46 24,85 5,99 Фот
С9Н19ОО + С9Н19ОО —► Продукты гм NonH 10-51 °C v = kc^ 8,4 7,46 24,85 5,99 Фот
394 Новый справочник химика и технолога
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из Е, к = Jz, ЕТ уравнения л О О 1g к, к - константа скорости реакции Ошибка Метод
Реакции рекомбинации и диспропорционирования радикалов
С16Н33ОО + С]бН33о6 —► Продукты гм Гексадекан 20-78 °C v = ксА2 8,4 7,46 24,85 5,99 Фот
СС14 + Н —► Н+ + СГ + СС13 гм Н2О pH = 1,0 v =ЛсАсв комн. 7,58 Е.р.
Им.р.
C(NO2)4 + Н —* C(NO2)3" + no2 + Н+ гм Н2О pH - 2,0 v =ЛсАсв комн. 8,74 Им.р.
cs2 + н—>Н+ + CS1/ гм Н2О pH = 1,0 v = ЛсАсв комн. 10,30 Им.р.
Реакции присоединения
CS2 + Он —►SC(OH)§ гм Н2О pH = 7,6 v = ксАс3 комн. 9,90 Им.р.
СНС12 + О2—►С12СНОО гм СНС13 v = ЛсАсв комн. 9,67 Им.р.
СН2С1 + О2 —► С1СН2ОО гм СН2С12 v - ЛсАсв комн. 9,80 Им.р.
Сн3 + о2—►СН3ОО гм Н2О v = kcfiCB комн. 9,67 Им.р.
СН3СС12 + О2 —*- СН3С(С1)2ОО гм Н2О pH = 1,0 v = £сасв комн. 9,18 Им.р.
СН2=СНС1 + ОН —►СНС1СН2ОН гм н2о pH ~ 6,5 v = ЛсАсв комн. 10,08 Им.р.
СН2ОН + 02 —► НОСН2ОО гм н2о pH = 7,0 v = ЛсАсв комн. 9,69 Им.р.
pH = 10,7 v = ЛсАсв комн. 9,62 Им.р.
СНС1=СС12 + ОН—►С1СН(ОН)СС12 гм Н2О pH ~ 6,5 v = ксАсв комн. 9,62 Мкр
СН2=СС12 + ОН —► СН2(ОН)СС12 гм Н2О pH » 6,5 v = ЛсАсв комн. 9,83 Мкр
С1СН =СНС1 + С12—► НСС12СНС12 ГТ Н2О-АсОН хсн3с(о)он = 55,8 v = ЛсАсв 24,0 -3,66 Ит
носн=’сн + о2—►носн=сноо гм н2о v = ЛсАсв комн. 9,00 Им.р.
Химическая кинетика и диффузия 395
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из _ Е‘ к-Ае уравнения л О о Е-ч 1g к, к - константа скорости реакции Ошибка Метод
Реакции присоединения
h2nch2cn + н—►h2nch2Cnh сн3Сн2 + О2—► СН3СН2ОО СН3СНОН + О2 —► СН3СН(ОН)ОО Сн2сн2он + О2—► НОСН2СН2ОО 0Н(ОН)СН2ОН + О2—*НОСН2СН(ОН)Об НС=СС(О)ОН + НВг—► ВгПС=СНС(О)ОН С1СН2Са=СН + НС1 —► С1СН2СС1=СН2 CH2=CHCN + II—* CH3CHCN НС=СНС(О)ОН + НС1 —► С1НС=СН2С(О)ОН Н2С = СНСНО + он —*НОСН2СНСНО СН3ОС^СН + Н2О—►СН3ОС(О)СН3 СН2= СНС(О)ОН + Н—*СН3СНС(О)ОН НОС(О)СП=СП2 + С12 —► НОС(О)СНС1СН2С1 гм гм гм гм гм ГТ ГТ гм ГТ гм ГТ гм ГТ Н2О Н2О Н2О Н2О Н2О Н2О АсОН Н2О Н2О Н2О Н2О Н2О АсОН pH = 7 pH = 7,0 pH = 1,0 pH = 7,0 св = 2,0 cNaCl ~ 3,0 cHgci2 ”0,185 pH = 1 св = 1,84 NaCI;/=0,25; pH = 5,92; скн2ро4 = 0’03 рН= 1 V = &САСВ V = ЛсАСв V = Асасв V = kcAcs V = ксАсв v = kcK V = ЛсАсв V = kcKc3 v = ЛсАсв V = fcACB V = fcAcB v = ЛсА V = fccAcB V = ксАсв комн, комн. комн. комн. комн. 75,0 75,0 50,0 комн. 75,0 75,0 комн. 25,0 комн. 25,0 7,72 9,46 9,66 9,82 9,51 -3,43 (транс-) -4,53 (цис-) -5,37 9,34 -4,51 (транс-) -5,22 (цис-) 9,85 -3,72 9,52 -3,52 ±5% ±5% 5 5 Е.р. Им.р. Им.р. Им.р. Им.р. ГЖХ ГЖХ ГЖХ Мкр ЯМР ЯМР Им.р. Дл Им.р. Им.р.
396 Новый справочник химика и технолога
О п X Г) о о X О О о X
2С=СНС(О)СН3 + ОН—► НОСН2СНС(О)СН3 Н3СН=СНСНО + ОН—► СН3СН(ОН)СНСНО ► СН3СС1=СН2С(О)ОН Н3С =СНС(О)ОН + НС1—* ► HO(O)CCH(OH)CHC(O)OH O(O)CCH=CHC(O)OH + OH —► Г О X о X о X n X о X Н2= СНСНоОН + ОН -Ч г (-►НОСН2СНСН2ОН Н2С(О)ОСН3 + 02 —* ООСН2С(О)ОСН3 Н3С(О)ОСН2 + 02—► СН3С(О)ОСН2ОО 2C=CHC(O)NH2 + ОН—►HOCH2CHC(O)NH2 1СН2СН=СН2 + Н0С1 —► С1СН2СНС1СН2ОН ICH2C=CCH3+ НС1 —► С1СН2СН= С(С1)СН3 Н3С = СС(0)0Н + НВг —► СН3СВг— СНС(О)ОН 2С =СНСН0 + ОН—► Н0СН2СНСН0 о g я «0
2 2 3 2 2 2 2 2 3 3 3 2 Тип реакции
X о X о X о X о X о X о X о X о X о > о о X X О X о о м X Растворитель
pH = 7 о и II 04 pH = 4 - 10,5 "О X II © "О X II © X II 04 pH = 7-12 CJ £ о II © © bJ CJ я 5 II © © UJ са=0,02; св = 0,0005 CJ II © 00 п и II 04 [ни присоединения Условия проведения реакции
II £ II £ II £ II 'n co II £ II & = д II 2 и СО II 2 II 1! S' > II £ Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из k— Ар уравнения л 71 е
1 комн. 1 S я я LA о LA О § g я § 2 я § я я § ж я § ж я bJ © LZ1 © © LA О LA ъ § р Г, °C
9,93 40 04 -4,39 (транс-) -4,97 (цис-) 40 00 40 00 40 ьо О' р LA 40 bJ 04 л? LZ1 С> © slz xi в 40 S w о 1 40 00 40 00 1g к, к - константа скорости реакции
LA LA ОА LA Ошибка
Им.р. X S •р I X S •p X я •р X 2: •р X 2: •р X ж •р X в 1 X 2: •р Метод
хпЕбффщ) n пятиэнпх uvxoawiwnx
Продолжение таблицы 2.1.16
L6£
-► СН3СН2СС1=СНСН2СН3 СН3СН2С S ССН2СН3 + НС1 —► -► СН3ОС(О)СН2СНС(О)ОСН3 СН3ОС(О)СН =СНС(О)ОСН3 + н —► СбН5>Ш2 + OH—►HOC6H5NHp СбН5ЫН2 + Н—»C6H6NH1‘ + он—►С6Н6ОНТ СбН5МО2 + Й—►C6H6NO?‘ (CH3)3CCN + Н —► (CH3)3CCNH —► СН3СН2ОС(О)СНС1СН2С1 о Е О Е ю о о о о X II о X ю + О ю- -► F(CH2)3C(OOCCF3) = сн2 F(CH2)3C =СН + CF3COOH—► (CH3)3t + О2 —► (СН3)3СОО (СН3)2С=СН2 + НС1—► (СН3)2СС1СН3 СН3СН2ОСН = СН2 + Н2О—►СН3СН2ОСН(ОН)СН3 (СН3)2С=СН2 + СН3ОН—►(СН3)2С(ОСН3)СН3 Реакция
3 2 2 2 2 2 2 3 3 2 3 3 3 Тип реакции
АсОН X о X К) о X о X о X о X о АсОН о о О о X X о (Et)2O MeN02 X К) о МеОН Peai Растворитель
С
о? II II © о ъ» к> О О pH = 0,7 pH = 8,1 - 9,5 pH = 7,8 - 8,4 pH = 7 PH = 1,0 pH = 1 = 0,125 о о Z II св = 0,57; св = 0,005 I 5 II р © 7^ X о 1 0 сл О Фа = 20; ;ии присоединения Условия проведения реакции
X
с II и и II II II и II и и и Кинетическое
II > 00 0? 0? 00 Г и II а? а? II уравнение скорости реакции
Энергия активации Еа, кДж/моль
1g А, А- постоянная из к = Ар уравнения л
25,0 комн. комн. комн. комн. комн. комн. 25,0 Os О 25,0 N> N) Ui Ui о Ъ 26,7 25,0 Г, °C
-7,85 9,95 10,15 9,38 9,89 9,36 7,15 -3,03 1 00 о 9,69 1 1 JQ у» оо О сл -1,68 19 V 1g k, k - константа скорости реакции
© Ошибка
ГЖХ Им.р. Им.р. Им.р. Им.р. Им.р. Е.р. X ГЖХ Кин Пт Кот О я & Метод
Продолжение таблицы 2.1.16
огоионхэш п vxnwnx xriHhosvduD ппэоц
86£
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из к = уравнения л О 0 К 1g k, к - константа скорости реакции Ошибка Метод
Реакции присоединения
СН3(СН2)3С ^СН + Н20 —► СН3(СН2)3С(О)СН3 СН3(СН2)3С=СН + F3CC(O)OH—► - * CH3(CH2)3C(OC(O)CF3) =сн2 СН3О(СН2)3С=СН + F3CC(O)OH—► - * CH3(CH2)3C(OC(O)CF3) =сн2 СН3(СН2)3СН = СН2 + С12 —► - * СН3(СН2)3СНС1СН2С1 СН3(СН2)3СН= СН2 + Н20—► - * СН3(СН2)3СН(ОН)СН3 СбН5ОН + Н—►С6Н6ОНП’ C6H5CN + бн —► HOQHsCN1’ С^С^ОН + ОН—►НОС6Н5С(О)ОЯ1-с6н5Сн2 + о2—►с6н5сн2об С^зСНрН + ОН—* НОСбНзСНгОН1’ С6Н5Сз=СН + С12 ►С6Н5СС1 = СНС1 . с6н5Снсн3 с6н5сн= сн2 + н—U2 L-4>C6H6CH=CH?’ ГТ ГТ ГТ ГТ ГТ ГМ гм гм гм гм ГТ гм Н20 CF3COOH CF3COOH АсОН Н20 Н20 Н20 Н20 Н20 Hex PhH MeCN г-PrOH Н20 S Н20 CH2SO4 6’9 CCF3C(O)ONa -= 0,125 CCF3C(O)ONa = = 0,125 ch2so„ = рН = 2-7 рН<3 pH = 7,0 АсОН pH = 1,0 м - < <<<<<< < < < < II II II II II II II II II II II II " II „ П п ££ Ъ Ъ £ ££ я я я со со w я я я оа 09 03 25,0 60,0 60,0 25,0 25,0 комн. комн. комн. комн, комн, комн, комн, комн. комн. 25,0 комн. -4,27 -3,57 -3,97 2,89 -3,71 9,23 9,64 9,63 9,30 9,45 9,46 9,53 9,40 9,92 1,00 9,76 9,81 Сп ГЖХ ГЖХ Сп Сп Им.р. Им.р. Им.р. Им.р. Фот Фот Фот Фот Им.р. Сп Им.р.
Химическая кинетика и диффузия
чо 40
C6H5CH= CH2 + СН3С(О)ОН—* ^СбН5СН(ОС(О)СН3)СН3 С6Н5СН= СН2 + С12—► С6Н5СНС1СН2С1 С6Н5СОСН3 + н—*- СбНбСОСНр СбН5СН2С(О)ОН + Н—*- С6Н6СН2С(О)ОН~’-c6h5nhc:och3 + н—*c6h5nhcoch1-(CH3CH2CH2CH2)2SO + он—*- -► (CH3CH2CH2CH2)2S(O)OH СбН5СН=СНС(О)О-+ ОН k р^НОС6Н5СН=СНС(О)О^ • СбН5СНСН(ОН)С(О)О-СбН5С = ССН3 + НС1—► С6Н5СС1= снсн3 СбН3(сн3)3 + н—► С6Н4(СН3)7 СбН5СН(СН3)2 + ОН —* (СН3)2СНС6Н5ОНП’ (СН3)3СС6Н3(ОН)2+ ОН (СН^ССбН^ОН)1/ C6H5CH2CH(NHC(O)CH3)CONH2 + н —* -► C6H6CH2CH(NHC(O)CH3)CONbn • Реакция
ГТ ГТ гм гм гм гм гм ГТ гм гм гм гм Тип реакции
Ж Ж Ж Ж £ ЕЕ х х х х £ О О О О g о о о о о g g Реакг Растворитель
Ci та тз тз тз "2. тз тз Ti х а ее а к х а о „ и и и и н и и ‘ 11 -~J — Ox *-* — — " О О О О О о Г- чо м о щи присоединения Условия проведения реакции
< -е -е -е -е -е и и и и и И и и и и и и 'j: >>>>•>•> > >>>>> п fi ОГъООСЬ > W СО СО W ч О 00 00 00 00 00 00 Кинетическое уравнение скорости реакции
Энергия активации Еа, кДж/моль
1е А, А- постоянная из х,_ Я7 уравнения л
§ § § § к> N> § - 8 3 S S У У § 8 8 8 8 У У ядяяоо я яяяяо о Г, °C
У> I s' | 1 х© so >о u> -с У У О 00 00 04 £ V g SO SO О О О 00 KJ -р- оо оо о ® ~ ° ° ° 04 OS 1g к, к - константа скорости реакции
Ошибка
SSSS7171 s s , m s s _ _ 8 8 8 2 « 2 3 F 2 2 Q Q Метод
VZOlfOHXdUl n minwnx UnHhOdVdUO ПП90Ц
Продолжение таблицы 2.1.16
OOP
Продолжение таблицы 2.1.16
Реакция Тип реакции Растворитель Условия проведения реакции Кинетическое уравнение скорости реакции Энергия активации Еа, кДж/моль 1g А, А- постоянная из £„ A уравнения л и о 1g к, к - константа скорости реакции Ошибка Метод
Реакции присоединения
СбН5СН2СН(ННС(О)СН3)СО2Н + Н -► C6H6CH2CH(NHC(O)CH3)CO2H1 • С6Н(СН3)5 + Н-^СбН2(СН3)1’ одад + Н ^С6НбС6Н? СбН5С6Н5 + ОН —►С6Н5С6Н5ОН1-(СбН5)2СО + ОН —-► НОС6Н5С(О)С6Нр (С6Н5)2СН + О2 —► (С6Н5)2СНОО (С6Н5)2СНЙН3 + dH-^HOC6H5CH(C6H5)NHl' (С6Н5)2С= СН2 + Н—► (С6Н5)2ССН3 гм гм гм гм гм гм гм гм Н2О Н2О Н2О н2о Н2О Н2О - г-РгОН н2о н2о pH = 2,0 pH = 1,0 рН = 3 ^(СН^СНОН _ 10% pH = 6,5 pH = 7 рН= 1 v = /ссАсв v = кс^св V = ЛсАсв v = Агасв v = ЛсАсв v = £сасв v = ксАсв v = /ссАсв комн. комн, комн. комн. комн, комн. комн. комн. 9,04 9,46 9,70 9,95 9,94 8,88 9,84 9,83 Им.р. Им.р. Им.р. Им.р. Им.р. Им.р. Им.р. Им.р.
Химическая кинетика и диффу:
* кп
1 Р % обозначает относительный процент реакции отщепления отсуммарного бимолекулярного процесса: Р % = -г—тпг— 100 %, где к01, к^ — константы скорости реакции лОт ' ^зам
отщепления и защемления соответственно.
*2 Механизм реакции:
о он
II К । + ь +
R1—с—OR2 + Н+ + Н2О ^=^Rl ~C—О—R2 ---------► R1—С(ОН)2 + R2OH----► R’COOH + R2OH + Н+
ОН Н
*3 Механизм реакции:
о-
^11 ^2
R1C—OR2 + ОН- R1 С—OR2--------► R1__C—ОН + R2O< R’COO- + R2OH
Таблица 2.1.17
Константы скорости гомолитических химических реакций, протекающих в газовой фазе
В таблице приведены кинетические параметры гомолитических химических реакций, протекающих при повышенных температурах в газовой фазе. Расположение материала в таблице основано на принципах кинетической классификации химических реакций.
В первой колонке приведена формальная запись химической реакции, причем ключевые реагенты располагаются по системе Хилла в порядке возрастания числа атомов углерода, внутри каждой из получающихся групп — в порядке возрастания числа атомов водорода, а остальные элементы — в алфавитном порядке символов. *СН2 - синглетный метилен, 3СН2 - триплетный метилен.
Во второй колонке приведен диапазон температур, при котором определена форма представления константы химической реакции. В третьей колонке представлено кинетическое уравнение скорости реакции, где А, В — реагенты молекулярной и радикальной природы, принимающие участие в химических превращениях: М — нейтральный газ, в среде которого протекает химическая реакция. В четвертой колонке приведены, как правило, усредненные экспериментальные или рекомендован-
ные рассчитанные значения констант скоростей химических реакций, имеющие размерность согласно виду кинетического уравнения (третья колонка) — с-1, см3 • молекула'1 • с'1, см6 • молекула'2 • с'1, соответственно. Если невозможно оценить однозначно надежность приведенных данных или они рассчитаны для различных интервалов температур, то приводятся два значения константы скорости реакции. В выражение константы может входить значение температуры (Т — температура, К) и величина газовой постоянной (R). В некоторых случаях реакции проводятся в присутствии инертных в данных условиях газов. Их давления также приводятся здесь. В пятой колонке приводится величина погрешности (воспроизводимости) табулируемой величины, выраженная либо в процентах %, либо в логарифмических единицах 0,... Если величина погрешности величины константы зависит от температуры эксперимента, то в скобках приведены области температур, которым соответствуют приведенные величины погрешности. В шестой колонке указана температура Тр, при которой рассчитана константа химической реакции, приведенная в седьмой колонке.
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, к IgA при Тр
Реакции распада
Н2 + Аг—* ► 2Н + Аг 2500-8000 v = kCfiC^ 3,71О~10 ехр(-48350/Г) 0,3 2500 -17,83
н2 + н2— ►2Н + Н2 2500-8000 v = 1,5-10'9 ехр(-48350/7’) 0,3 2500 -17,22
Н2 + М — ► 2Н + М 600-2000 v = ксксм 7,6-Ю'5?'1’4 ехр(-52530/Г) 3 1000 -31,13
О2 + М - -►20 + М 300-1500 v = кс дСм 3,0-10 67"1 ехр(-59380/Г) 2 1000 -34,31
НО2 + N2 —► н + о2 + n2 200-2200 v = ксКсщ 2,0-10"5/"1’8 ехр(-24363/7’) 3 1000 -18,82
Н2О + N2 - —►h + oh+n2 2000-6000 v = кс*сщ 5,8-10"9 ехр(-52900/Г) 1,5 2000 -19,72
Н2О2 -► 2 OH 1000-1500 v = кс& 3-1014 ехр(-24400/Т) 1200 5,65
Н2О2+ Аг —►2ОН + АГ 1000-1500 v = kc^Cbr З Ю'8 ехр(-21600/7") 0,2 1200 -15,34
Н2О2+ n2 —►2oh + n2 700-1500 300-1500 V = acn2 v = ксАс^ 2-10'7 ехр(-22900/Г) I-IO9 33?”4’86 ехр(-26795/Г) 0,2 2 (> 500 К) 900 900 -17,62 -17,96
NCO + Аг —►CO + -N- + Ar 1450-2600 v = кс^с Аг 1,7-10'9 ехр(-23500/Г) 0,4 2000 -13,87
402 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое Константа скорости Ошибка То, к Igfc
уравнение реакции к при Гр
Реакции распада
сно+м —►со+н+м 300-1500 V = fa?ACM 8,5-10~3Г~214 ехр(-10278/Г) 5 900 -13,35
к} . . 1500-2500 V1 = ^1САСМ кх - 2,110“8 ехр(-39200/Г) 0,3 2000 -16,19
[—► н + сно + м СН2О + М —\к2 1500-2500 v2 = к2САСм к\ /)12 = 0,5 (2200 К)
L-4>H2+CO + M
сн3 + м —►•сн2 + н + м 1500-3000 V= АГдСад 1,710“8 ехр(—45600/Г) 0,5 2000 -16,28
СН3б +м—►СН2О + Н + М 300-1000 V = ^САСМ 3,16102Г"2’7 ехр(-15400/Т) 1,0 700 -14,73
СН3ООН + М —►СНзО +ЙН + М 400-1000 V = fa?ACM 4-1015 ехр(-21600/Г) 0,5 (600 К); 1,0(400; 1000 К) 700 -12,68
СН4 —►СНз+Н 1000-3000 V=kcA 2,4- Ю16 ехр(-52800/Г) 0,5 2000 4,92
СНд + Аг —► СНз + Н + Аг 1000-3000 v = ксКсм L2- 10-6 ехр(—47000/Г) 0,3 2000 -16,13
СН4 + СН4 —► СН3 + Н + СН4 1000-2000 v = ^acch4 1,4- Ю 5ехр(-48100/Г) 0,3 1500 -18,78
QH3—►QA + H 500-2500 V = fa?A 2 • 1014 ехр(-20000/Г) 0,5 1500 8,51
<^Н3 + М —► С2Н2+Н + М 500-2500 v = kcAcM 6,9 • 10,7Г"7’5 ехр(-22900/Г) 0,5 1500 -12,61
СН3СН3 —► СН3 + СН3 v = kcK 5,01 • 10,6ехр(-372900/ЯГ) 700 -11,1
СН3СН2СН3 —►СНз + С2Н5 v = kcA 7,94- 10’6 ехр(-356600/ЯГ) 700 -9,7
CH3CH=CH2 —►H + C3H5 v = kcA 6,31 • 1014 ехр(-373300//?Г) 700 -13,1
к} Vi = k}CA кх = 3,16 1017 ехр(-359000//?Г) 700 ^ = -9,3
1 СНз + С3Н7 сн3сн2сн2сн3 v2 = k2cA к2= 2,51 • 1016ехр(-344000/7?Г) 700 к2 = -9,2
СН3СН(СН3)СН3 СНз+ Сзн7 v=kcA 2,00 • 1017 ехр(-349400/ЯГ) 700 -8,8
СН2-СНСН2СН3 —► СН3 + СН2=СНСН2 v=kcA 1.26 • 1016 ехр(-307500//?Г) 700 -6,8
Vi = kxcA ki = 6,31 • 1016 ехр(-357800/ЯГ) 700 Л1 = -9,9
СН3СН2СН2СН2СН3 —р^Г v2 = k2cA Л2=6,31 • 1016 ехр(-343200/7?Г) 700 Л2 = -8,8
Д-СНз + Сн2сн2сн2сн3 lXcH3£H2+ tH2CH2CH3
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Гр
Реакции распада
*1 • л (Т-ед+СзН, *э • СН3СН(СН3)СН2СНз —►СНз +СН2СН(СН3)СН3 Л3 . L-*- сн3 + СН3СНСН2СН3 сн2=снсн2сн2сн3 —►С2Н5 + £3Н5 СН3СН=СНСН2СН3 —► —► ён3+сн2сн=снсн3 СН2=СН(СН2)3СН3—►С3Н7 + сн2сн=сн2 г-^СНз + QHh СН3(СН2)4СН3 —4^ С2н5 +С4Н9 ЬХс3Н7 + СзН7 С’Н3СН=СН(С’Н2)2С’Нз—► С2Н5 + СН3СН=СНСН2 СН3СН2СН=СНСН2СН3 —► -► сн3+СН3СН2СН=СНСН2 СН3СН(СН3)СН2 СН2СН3 сн3 +СН3СНСН2СН2СН3 _-^СзН7+СзН7 -V ед + (сн3)зС LS- йн3 + £н2сн2сн(СНз)2 сн2=снсн2сн2сн=сн2 —► -► ch2=ch<Jh2 + сн2=сн(*:н2 СН3СН=СН(СН2)зСНз —► -► СН3СН=СНСН2 + СН3СН2СН2 СН2-СН(СН2)2СН-СНСН2СН3 —► сн2=снсн2 + СН3СН2СН=СНСН2 Ш .0-0 II II II II II II II II II II II II II II II II II II - - - > > > - - £> > > Р? > к, = 3,98 • 10'6 ехр(-331800//?7) к2 = 1,26 • 1017 ехр(-347400/ЛГ) £з=5,01 • 1016ехр(-356200/ЯГ) 1,00 • 1016 схр(-299200//?7) 5,01 • 1016 ехр( 305900/7?Г) 2,51 • 1016 ехр(-300400/ЯГ) кх = 6,31 • 1016 схр(-357800/7?7) к2 = 6,31 • 1016 ехр(-343200/7?Г) к3 = 3,16 • 1016 ехр(-343200//?Т) 1,59- 10,6ехр(-290400//?Г) 1,00- 1017ехр(-305900/ЯГ) ki = 1,26 • 1017 ехр(-347400/ЛГ) * 2 = 3,98- 1016ехр(-331800/ЯГ) * з = 6,31 • 1016 ехр(-339400//?7’) * 4 = 5,01 • 1016ехр(-356200//?Г) 2,51 • 1014 ехр(-248900//?Г) 1,59- 1016 ехр(-290400/7?7 ) 5,01 • Ю15 ехр(-246000/ЛГ) 700 700 700 700 700 700 700 700 700 700 700 700 700 700 700 700 700 700 еч ое о oo >— ootNvyos op 00 оС СП —' 00 сК °₽ с*4 «Н 00 00 00 00 о сч ГГ \О ’ ' ' чсГ V"? V"? 1111 id сч II II II 7 7 1 II II И Т 7 II II II И III -S?
404 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К IgA; при Тр
Реакции распада
СН3СН=СН(СН2)4СН3 —► —► С4Н9 + СН3СН=СНСН2 v = fcA 1,59 • 1016 ехр(-290000//?Г) 700 -5,4
СН3СН=СН(СН2)2СН-СНСН3 —► -► СН3СН=СНСН2 + СН3СН=СНСН2 V = fcA 6,31 • 1015 ехр(-236800//?Г) 700 -1,9
СН3СН-СН(СН2)5СН3 —* —► QHj J + СН3СН=СНСН2 V = fcA 1,59- 1016ехр( 290000//?7) 700 -5,4
сн3сн<:нсн2сн(сн3)(сн2ьсн3 —► —► C5H! i + СН3СН=СНСН2 V = fcA 1,00 • 10’7 ехр(-283300/ЛТ) 700 -4,2
сн3сн-снсн2сн(сн2сн3х:н2сн3 —► -► СН3СН2СНСН2СН3 + СН3СН=СНСН2 V = fcA 1,00- 1017 ехр(-283300//?Г) 700 -4,2
Реакции изомеризации
’CH2+Ar ► 3СН2 + Аг 300-2000 V = AcaCb 6,0 • 10'12 0,3
1ch2+n2- ► 3ch2+n2 300-2000 V = AcaCb 1,0- 10'" 0,3
1СН2 + СН4 ->- 3СН2 + СНд 300-2000 V = AcaCb 1,2- 10'" ±0,4
Реакции замещения
н + О2—♦ .КПД МП _ к id + 6* —►n2o+oh + Л. 300-2500 300-1250 270-380 V = AcaCb V = AcaCb V = AcaCb 3,3 • 1О'10 ехр(-8460/Г) 2,8- 10'7Г°’9 ехр(-8750/Г) к = 5,0- 10'" 0,1 (300 К); 0,2 (2000 К) 2 (300 К); 1,3 (1500 К) 0,2 900 700 -13,56 -9,42
_^n2 + 0h
•NH + OH - ОН +о2 - к ► no + h2 1—► h2o+*n* d‘+HO2 300-1000 300-1500 V = AcaCb V = AcaCb к = 8,0 • 10'" 3,7 -10'" ехр(-26500/Г) 0,5 3 700 -26,87
н2 + дн - н2 + но2 - ►Н2О + Н -н+н202 300-2500 240-2400 300-1500 V = fcACB V = AcaCb V = fcACB 1,7- 10'16Г16 ехр(-1660/Г) 1,06 • 10'17Г2 ехр(-1480/Г) 5,0- 10'" ехр(-13100/Г) 0,1 (300 К); 0,3 (2500 К) 1,4 (240 К); 3 (2400 К) 3 1000 1000 700 -11,69 -11,62 -18,43
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, к IgA при Гр
Реакции замещения
• кг~►•<М + н 300-1000 v = £сасв к = 2,4- 1О~10 ехр(-1760/Г) 0,3 700 -10,71
Н2 + *СН-^Ц . 2
1—сн3
н2+ СНО—►СН2О + Н 300-1500 v = ксАсв 3,0 • 1О’|8Г2 ехр(-8972/Г) 5 900 -15,94
Но + ’о-Ь, ► £н3 + Н 300-1000 v = ксАсв 1,2- Ю’10 ±0,1 (300К); ± 0,3 (1000 К)
н2+3сн2—► Сн3 + н 300-1500 v = ксАсв <5,0- 10 15
Н2 + СН3—►СН4 + Н 300-2500 v = £сАсв 1,14 • Ю-20Г2’74 ехр(-4740/Г) 0,15 (300-700 К); 1000 -13,72
700-2500 v = &сАсв 4,8- 10’22Г3'2 ехр(—4384/Г) 1,5 (700 К); 3 (2500 К) 1000 -13,86
Н2 + С2Н—►С2Н2 + Й 300-2500 v = £сАсв 2,5 • 10’" ехр(-1560/Г) 0,3 (300 К); 0,7 (2500 К) 1000 -11,28
300-2500 v = £сАсв 1,9 • 10 11 ехр(-1450/Г) 1,3 (300 К); 3 (2500 К) 1000 -11,35
Н2+ С2Н3 —► су-ц + н 300-1500 v = ксАсв 5,0 • ю 20Г2 63 ехр(-4298/Г) 5 700 -14,49
н2 + сн2он —► СН3ОН+н 300-1500 v = ксАсв 1.12 • 1О’18Г20 ехр(-6722/Г) 4 900 -15,29
н2 + СН3СО —► СН3СНО+н 300-1500 v = ксАсв 6,8- 10”18Г182 ехр(-8862/Г) 3 900 -16,07
Н2+С2Н5—►С2Н6 + Н 700-1200 v = ксАсв 5,1 • 10”24Г3,6 ехр(-4253/Г) 1,5 (700 К); <4 (1200 К) 900 -14,71
Н2 + С2Н5СН2 — н + С3Н8 v = ксАсв 2,63 • 10“12 ехр(-64430//?Г) 700 -16,39
н2 + СН3СНСН3 —-*-н + С3Н8 v - £сАсв 6,61 • 1012 ехр(-69870//?Г) 700 -16,39
Н2+ СН2СН=СН2—«-н +С3Нб v = £сасв 5,25 10 " ехр(-82420//?Г) 700 -16,43
н2 + сн3сн2сн2дн2 —►н +с4н10 V = £сдсв 5,25 • 10“12 ехр(-65690//?Г) 700 -16,38
н2 + сн3снсн2сн3 —► н*+С4Н10 v - £сдсв 8,32 • 10”12 ехр(-71960//?Г) 700 -16,45
Н2+(СН3)2СНСН2 —►Н + (СН3)2СНСН3 v - fccAcB 5,25 • 1012 ехр(-65690/ЯГ) 700 -16,18
н2 + сн3снсн==сн2 — v = £сасв 6,61 • 10’" ехр(-105400//?Г) 700 -18,05
-►н +СН3СН2СН=СН2 1
406 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое Константа скорости Ошибка гР, к IgA:
уравнение реакции к. при Гр
Реакции замещения
Н2 + СН3СН= =СНСН2—► V = Асасв 5 • 25 • 10“н ехр(-93300/ЯГ) 700 -17,24
-►н+сн3сн=снсн3
FL + 6.—►ОН +Н 300-2500 V = ЛсАсв 8,5 • 10~2ОГ2’67 ехр(-3160/Г) 0,5 (300 К); 0,2 (> 500 К) 1000 -9,68
300-1500 v =Асасв 1,8- 10’2°Г2,8 ехр(-2980/Г) 1,6 (400-1600 К); 2 (1600 К) 1000 -9,97
н2+ 02 —* • н +но2 300-1500 v = ксАсв 2,4- 10’10 ехр(-28500/Г) 5 700 -27,30
Н2О + н —► он+н2 300-2500 v = ксАсв 7,5 • 10"16Г1’6ехр(-9270/Г) 0,2 1000 -14,35
300-1500 v = ксАсв 1,03 • 10~16Г1,9 ехр(-5290/Г) 2,5 900 -12,58
Н2о + ‘СН - -► продукты реакции 300-1000 V = Асасв 9,5- 10’12 ехр(-380/Г) 1,0 700 -11,26
Н2О+ сно - —► СН2О + ОН 300-1500 v = ксАсв 3,9- 10_16r’’35 ехр(-13146/Г) 5 900 -17,76
н2о+ сн3— СН4+ОН 300-1500 v = ксАсИ 8,0 • 10’22Г2-9 ехр(-7480/Г) 1,6 900 -16,14
Н2О+ СгН3 —► СгНд + ОН 300-1500 v = ксАсв 8,0 • 10’22Г2’9 ехр(-7480/Г) 5 (> 1000 К) 700 -17,49
Н2О + б > •2 ОН 300-1500 v =ксАсИ 7,6 • 10~15Г’-3 ехр(-8605/Г) 2,5 1000 -6,48
300-1000 V1 = &1САСВ kx = 2,8- 1012 ехр(-1890/7 ) 0,3 700 -12,73
Н2О2 + Н — —4 Н2+Н02 Л? • 300-1000 v2 = А2сасв Л2= 1,7- 10’” ехр(-1800/Г) 0,3 700 -11,89
-4- он +н2о
н2о2+6н - -*»н2о+но2 300-1000 V = ксАсв 1,3 • 10’11 ехр(-670/Г) 0,2 700 -11,30
300-1500 v = ксАсИ 2,9 • 10’12 ехр(—160/Г) 1,25 (300-460 К); 700 -11,64
3(Г> 1000 К)
Н2О2 + о- — ► но2+он 300-500 v =ксАсв 1,1 • 10’12 ехр(-2000/Г) 0,3 400 -14,13
300-1500 v = ксАсв 1,6- 10’17Г2 ехр(-2000/Г) 3 900 -6,74
Н2О2 + о2 — -►2 но; 300-1500 v = ксАсв 9,0 • 1О’П ехр(-20000/Г) 5 700 -22,45
NH3 + б ► nh2 + он 500-2500 v = ксАсИ 1,6- 10’” ехр(-3670/Г) 0,5 1500 -11,86
—►•NH + CO 1400-1500 v = ксАсИ k = 8,7- 10’11 0,5
NCO + H
—► HCN + O*
HCN + OH - -*h2o+cn 1500-2500 v = Асасв 1,5 10 11 ехр(-5400/Г) 0,5 2000 -12,00
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция Г, К Кинетическое уравнение Константа скорости реакции к Ошибка Гр, к IgA: при Гр
Реакции замещения
|—► NCO + Н 450-2500 v = ЛсАсв к = 2,3 • 10~18Г2’’ ехр(-3075/Г) 0,2 (450 К); 0,3(2500 К) 1000 -12,67
• 1с UCNJ + П • Г^Гх 4- UNI •
LAJ 1 111N •
L-» он +CN
HNCO + H — |—► nh2 + co -Й2 L-4- h2+nco 500-1000 v = ксАсв к2 = 3,4 • Ю‘,0Г0-27 ехр(-10190/Г) 1,0 700 -13,08
СО+ НО — ►н +со2 300-2000 v = ксАсв 1,05 • 10"17Г1’5 ехр(-250/Г) 0,2 (300 К); 0,5 (2000 К) 1000 -12,59
СО+ НО2 —► СО2 + он 300-1500 v = кеАсв 2,5 • Ю’10 ехр(-П900/Г) 3 900 -15,34
СО + -СН — ► продукты реакции 300-1000 v = ксАсв 4,6 • 10’13 ехр(-860/Г) 1,0 700 -12,87
СО+СН3О —► СН3 + СО2 300-1500 v = ксАсв 2,6 • 10’11 ехр(-5940/Г) 5 700 -14,27
СО + ОД, - -► Q>H + СНО 300-1500 v = ксАсв 8,0- 1О“10 ехр(-53700/Г) 10 700 -42,41
со+о2 —* со2 + о- 300-1500 v = ксАсв 4,2 • 10“12 ехр(-24000/Г) 2 700 -26,27
СО2 + Н —» ►со+он 300-1500 v = ксАсв 2,5- 1О“10 ехр(1330/Г) 1,2 900 -11,64
СО2 + -сн - -► продукты реакции 300-1000 v = ксАсв 5,7- 10“'2 ехр(-345/Г) 1,0 700 -11,46
СО2 + 3СН2 - СН2О + СО 300-1500 v = ксАсв < 1,0- 10~15
со2 + о ► СО + О2 300-1500 v =ксАсв 2,8 • 10‘" ехр(-26500/Г) 2 900 -23,34
Н2О + *СН2 - -► СН3ОН 300-1500 v ~= ксАсв 3,0- 10’” 3
Н2О + 3СН2 - —► СН3 + ОН 300-1500 v = ксАсв < 1 • 10’16
H2O + CN - £ |—»• HCN + OH 1—► HOCN + H 500-3000 v = ксАсв к = 1,3 • 10"11 ехр(-3750/Г) 0,3 (500К); 0,5 (3000 К) 1000 -12,52
Н2О+ ОД- -►од+он 300-1500 v =ксАсв 5,6- lO ^r’^expHOlSO/r) 2 900 -17,90
Н2О2 + Q2H3 —C2H4 + HO2 300-1500 v = ксАсв 2,0- 10’14ехр(300/Г) 10 700 -13,89
408 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое Константа скорости Ошибка гр, К IgA:
уравнение реакции k при Гр
Реакции замещения
а • * 300-1500 v = ксАсв < 1,0- 10~14
СН2О + '’СН^ ► СН3 + СН1
СН2О + ОН —►Н2О1 СНО 300-3000 v = /ссАсв 5,7- IO-15/’1,18 ехр(225/Г) 0,1 (300 К); 0,7 (3000 К) 1000 -10,80
СН2О + НО2 —► Н2О2 + СНО 600-1000 v = Асасв 5,0- 10“12 exp(-6580/T) 0,5 700 -15,38
300-1500 v =Асасв 3,3 • 10” exp(-5870/T) 3 700 -15,12
СН2О + СН3 —► СН4 + СНО 300-1000 v = Асасв 6,8- 10“12 exp(—4450/T) 0,3 700 -13,93
СН2О + СН3О —► СН3ОН + СНО 300-1500 v = Асасв 1,7- 1013exp(-1500/T) 3 700 -13,70
СН2О + С2Н3 —► С2Н4 + СНО 300-1500 V = Асасв 9,0- 10’21r2’81 exp(-2950/7) 5 (> 1000 К) 700 -13,88
СН2О + СН3СО —► СН3СНО1 СНО 300-1500 v = Асасв 3,0- 10“!3 exp(-6500/7) 10 900 -15,66
СН2О+ С2Н5—► С2Н61 СНО 1000-2500 v = ксАсв 9,12- Ю”Г2’81 exp(-2950/T) 2(1000 К); 5 (2500 К) 1000 -12,89
СН2О + О ► ОН + СНО 250-2200 v = kcAcB 6,9- 10”Г°’57ехр(-1390/Г) 0,1 (250 К); 0,3(2200 К) 1200 -10,91
300-1500 v = kcAcB 3,0- 10’" ехр(-1550/Г) 1,5(750 К) 900 -11,27
СН2О + О2 —►НОо + СНО 700-1000 v = kcAcB 1,0- 10“10exp(-20460/T) 0,5 850 -20,18
300-1500 v =kcAcB 3,4- 10” ехр(-19600/Т) 2 700 -22,63
СНд + Н —►н2 + сн3 300-2500 v = keAcB 2,2- 10~20Г3’° ехр(-4045/Г) 0,2 1000 -12,41
300-1500 v = kcAcB 3,73 • Ю’20Г3 ехр(-4406/7) 1,3 (500-600 К); 3 (2500 К) 1000 -12,34
СЩ + ОН —► Н2О + СН3 250-2500 v =kcAcB 2,6- 10-17Т’1’83 ехр(-1400/Т) 0,7 1000 -11,70
300-1500 v = kcAcB 3,2- 10’19Т2-4 ехр(-ЮбООТ) 1,4 1000 -15,90
СН4 + НО2 —► Н2О2 + СНз 600-1000 v = keAcB 1,5 - 10” ехр(-12400/Т) 0,2 (600 К); 0,3 (1000 К) 700 -18,52
300-1500 v -= kcAcB 3,0- 10~13ехр(-9350/Г) 5 700 -18,32
СН4 + *СН —► продукты реакции • 200-700 v = kcAcB 5,0 • 10” ехр(-200/Г) 1,0 500 -10,48
СН4 + СНО —► СН3+СН2О 300-1500 v=keAcB 1,21 • Ю-2ОГ2’85 ехр(-11330/Т) 5 900 -16,96
300-1500 v = kcAcB <3,0- 10”
СН4 + сн2 —► сн3 + сн3
СН4 + СН2ОН —►СН3ОН1СН3 300-1500 v = kcAcB 3,68 • Ю’23?3’1 ехр(-8166/Т) 3 700 -18,68
CH4 + CH3O —► CH3OH + CH3 300-1500 v = kcAcB 2,6- 10”ехрК450/Г) 3 700 -15,35
СН4 + С2Н —► q,h2 + сн3 300-2500 v = kcAcB 3,0 • 10"12 ехр(-250/Т) 1,3 (300 К); 3 (2500 К) 1000 -11,63
Химическая кинетика и диффузия 409
Продолжение таблицы 2.1.17
Реакция г к Кинетическое Константа скорости Ошибка гр, К IgA
уравнение реакции к при Гр
Реакции замещения
СН4 + C,H3 —►С2Н4 + СН3 300-1500 v = Асасв 2,4 • 1(Г24Г4’02 ехр(-2754/Г) 5 (>1000 К) 700 -13,89
СН4 + СН3СО —►СН3СНО + СН3 300-1500 v = ксАсв 3,6- 10“21Г2’88 ехр(-10800/Г) 5 900 -17,15
СН4 + С2Н5 —► с2н6 + СН3 300-1500 v = ксАсв 1,43- 10”25Г4,14 ехр(-6322/Г) 2 900 -15,67
СН4 + О« —► ОН + СН3 300-2500 v = ксАсв 1,5- 10_15r1,56 ехр(-4270/Г) 0,3 (300 К); 0,15(2500 К) 1400 -11,24
300-1500 v = ксАсв 1,7- Ю15/4,5 ехр(—4330/Г) 1,25 900 -12,43
СН4 + О2 —► НО2 + СН3 500-2000 v = ксАсв 6,6 • Ю" ехр(-28630/Г) 0,5(500 К); 1,0 (2000 К) 1000 -22,61
300-1500 v = ксАсв 6,7 • 10’11 ехр(-28640/Г) 5 700 -27,94
600-2000 V = кх + к2 = 3,25 • 1017Г21 ехр(-2450/Г) 1,3 (600 К); 3 (2500 К) 1000 -11,26
• г-^СНоОН + Н, CH3OH+H—\к2 . 2 = (^ + к2)с Асв кх / к2 = 4
1——► CH3O + H2
• Н*- СН2ОН + Н2О CH3OH + OH —\к2 . 300-2000 V, = Л1САСВ кх + к2 = 1,1 • 10 19Г2 5 ехр(483/Т) 1,1 (300 К); 3 (2000 К) 1000 -11,67
v2 = к2САСВ к} / к2 = 3,7 ехр(-1020/Г) 1,6 (500 К)
ЬЛ- СН3О + Н2О
СН3ОН+НО2 —► СН2ОН + Н2О2 300-1500 v = ксАсв 1,6- 10 13 ехр( 6330/Т) 10 900 -15,85
СН3ОН + ’СНО —► -СН2ОН + СН2О 300-1500 v = ксАсв 1,6- 10~20Г2’9 ехр(-6596/Г) 10 900 -14,41
300-1500 v = ксАсв 2,5 • 10~12 3
СН3ОН + ^Нз —► *сн3 + *СН2ОН
к\ • • 300-1500 V) = Л]САсв кх = 5,3 • 10“23Г3’2 ехр(-3609/Г) 3 900 -14,56
, 1—► СН2ОН + сн3 СНзОН + 3СН2 —\к2 . . 300-1500 V2 = А2СаСВ к2 = 2,4 • 10~23Г3’’ ехр(-3490/Г) 3 900 -15,15
3 2 1-4. СН3О + СН3
к\ 600-2000 V, = Л1САСВ кх = 5,3 • 10~23Г3-2 ехр(-3609/Г) 1,4 (600 К); 3 (2000 К) 1000 -14,24
• I—СН2ОН + СНд СН3ОН+СН3 — 600-2000 v2 = к2сАсв к2 = 2,4 • 10’23r"31 ехр(-3490/7’) 1,4 (600 К); 3(2000 К) 1000 -14,84
СН3О + СН4
СН3ОН + СН2ОН —► СН3ОН + СН3О 300-1500 v = ксАсв 1,3 • 10’14 ехр(-6070/Г) 10 700 -17,65
СН3ОН + СН3О —► СН2ОН + СН3ОН 300-1500 v = ксАсв 5 • 10~13 ехр(-2050/Г) 10 700 -13,57
СН3ОН + СН3ОО —► СН2ОН + СН3ООН 600-1000 v = ксАсв 3 - 1013 ехр(-6900/Г) 1,3 (600 К); 2 (1000 К) 800 -16,27
кх , 300-1500 v = Л]САсв кх = 1 • Ю”11 5
Г—► СН2ОН + С2Н2 СНзОН + СзнЦ^.^ 300-1500 V = А2сасв А2 = 2- 10’12 5
410 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция 7, К Кинетическое уравнение Константа скорости реакции к Ошибка гр, к IgA: при 7р
Реакции замещения
СН3ОН + С2Н3 кх л, г-► дн^он+од 1^2 • СН3О + С2Н4 300-1500 300-1500 V1 = &1САСВ v2 = к2сКсв ki кг = 5,3 • 10’2V3 2 ехр(-3609/Т) = 2,4 • 10’23r3J ехр(-3490/Т) 10 10 700 700 -15,41 -15,97
CU3OH + CU3CO —► си2он+сн3сно 300-1500 v = ксАсв 8,06- 10‘21Г3 ехр(-6210/Г) 3 900 -14,23
СН3ОН + С2Н5 кх . Г-* СН.ОН + ОЯб СНзО + СгНб 300-1500 300-1500 V] - к}сАсв v2 = к2сАсв к\ кг = 5,3 • 10~23T3,2 ехр(-4610/Т) = 2,4 • 10’23Г31 ехр(-4500/Т) 3 5 900 900 -15,05 -15,63
CH3OH+O— *1 *- сн2он+дн * CH3O + OH 750-1100 vi = к}сАсв v2 = к2сАсв к]^г = 6,44 • 10’19Т2 5 ехр(-1550/Г) к\ > к2 1,3 (750 К); 2 (1100 К) 900 -11,55
CH3OOH+OH кх г—► Н2О СН3ОО -\к2 Н2ОСН2ООН 300-1000 300-1000 vi = к\СА.св v2 = к2сАсв к} = 1,2- 10‘12 ехр(-130/Г) к2= 1,8- 10’12 ехр(-220/Г) 0,2 (300 К); 0,4 (3000 К) 0,1 (300 К); 0,3 (3000 К) 700 700 -12,00 -11,88
ен3оон+д«- ь- -►дн + сн2оон —► дн+сн3од 250-2200 v = ке^Съ к = 6,9- 1О“137 057 ехр(-1390/7’) 0,1 (250 К); 0,3(2200 К) 1000 -11,1
С2Н2 + н —►н + С2Н 1000-3000 v= кедов 1,0- Ю’10 ехр(-14000/Г) 1,0 2000 -16,08
• k г СгНг + ОН-^Ч ► Н2О + СНО ► н2 + снсо 1000-2000 v = ксКсв к= 1,0- 10~‘° ехр(-6500/Г) 1,0 1500 -11,88
С2Н2НО2 — сн2=с=о + он 300-1500 v = ксАсв 1,0- 10“|4ехр(-4000/Т) 10 700 -16,48
QHj+’CH — продукты реакции 200-700 v = кс^св 3,5 • 10 10ехр(-61/7) 1,0 500 -9,51
С2Н2 + !сн2 - к Я 3СН2+С2Н2 + с,и4 300-1500 300-1500 V] = ^1САСВ v2 = к2сАсв 3,0- 10"11 1,0- Ю’10 3 10
С2Н2 + QH — С4Н2+Н 300-1500 v = ксАсв 4,0- 10’” 1,2
С2Н2 i &H, - СгН + СзНб 300-1500 v - ксАсв 4,5 • 10“13 ехр(-11800/7) 5 700 -19,67
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция лк Кинетическое уравнение Константа скорости реакции к Ошибка гр, к IgA: при Тр
Реакции замещения
СгН2 + б. — *1 - СО+3СН2 ► СНСО4Н 300-2500 v = ксКсв V! = ЛсАсв к = 3,6 • Ю’20Г28 ехр(-250/Г) кх /к = 0,5±0,3 0,2 1400 -10,71
сн2со+ н - к Г —*► сн3 + СО —► сн2сно 200-2000 v = ксАсв к = 3,0- 10“” ехр(-1700/Г) 0,5 (200 К); 1,0 (2000 К) 1000 -11,26
СгЩ + Н—► н2 +QH3 700-2000 v = ксАсв 9,0- 10~10 ехр(-7500/Г) 0,5 1000 -12,30
С2Н4 + ОН — ►H2O + Q2H3 650-1500 v =ксАсв 3,4 • 10’" ехр(-2990/Г) 0,5 1000 -11,77
СгЩ + НОг - ►OH+CH3CHO 600-900 v = ксАсв 1,0- 1014 ехр(—4000/Г) 3 (773 К); 10 (500 К, 1500 К) 700 -16,48
cyV-CH - продукты реакции 200-700 v = ксКсв 2,2- 1О“10 ехр(-173/Г) 1,0 500 -9,81
С2Н4+ ^2 *1 3 3СН2+С2Н4 Ьл. с3н6 300 -2000 300-1500 vi = ЛсАсв v2 = к2сКс в 2,3 10’" 1,5 • Ю’10 0,4 2
С2Н4 + СН3 - СНд + СгНз 400-3000 V = ксКсв 6,9 • 10’12 ехр(-5600/Т) 0,3 1000 -13,59
C^ + QH- СдНд+Н 300-1500 v = ксАсв 2,5 • 10’" 3
СгЩ + б* — к\ ► СН2СНО + Н ► СНО + бНз ►СН2О + СН2 ► СН2СО + Н2 300-2000 V = Асасв V] = к}сАсв v2 = к2сАсв А =5,75 • 1О’18Г2’08 к} / к = 0,35 ± 0,05 (р > 400 Па) к2 /к = 0,6±0,10 (300 К) ±0,1 (< 1000 К); ±0,3 (2000 К)
СН3СНО + Н к I—► Н2 + СН3СО н2+сн2сно 300-2000 V =Асасв к =6,8- 10’15Г116ехр(-1210/Г) 0,1 (300 К); 0,4 (2000 К) 1000 -11,21
сн3сно+бн £г— Н2О + СН3&> 1—► н2о+Сн2сно 250-1200 v = кс^св к = 3,9 • Ю 14Г0’73 ехр(-560/Г) 0,1 (250 К); 0,3 (1200 К) 700 -11,68
СН3СНО + НО2 —► Н2О2 + СН3СО 900-1200 v = Асасв 5,0- 10’12 ехрС-бООО/Г) 0,7 1000 -13,91
СН3СНО + б« к 1—► CH3CO + OH 1—► сн2сно+дн 300-1500 v =Асасв А: = 9,7- 10“12 ехр(-910/Г) 0,05(1000 К); 0,5 (1500 К) 1000 -11,41
412 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К lg& при Гр
Реакции замещения
сн3сно+о2 —► нд2+сн3со 600-1100 v= ксксв 5,0- 10” ехр(-19700/Г) 0,5 (600 К); 1,0 (1100 К) 700 -22,52
_тт а сдюон + он QH5OOH + O«—4 4 . . 250-2200 v = &сАсв к~ 6,9 • 10”r°’57 ехр(-1390/Г) 0,1 (150 К); 0,3 (2200 К) 1000 -11,06
1—► QH-jOO + OH
(^Нб + Н—►Н2 + С2Н5 300-2000 V = &сАсв 2,4 • 10’15Г*5 ехр(-3730/Г) 0,15(300 К); 0,3 (2000 К) 1000 -11,74
300-2500 v = kcAcB 9,2 • 10‘22Г3 5 ехр(-2600/Г) 1,5 (< 1000 К); 0,3 (2500 К) 1000 -11,66
СгНб + ОН —* Н2О + QHs 250-2000 v = ксдся 1,2 • Ю-17?'2’0 ехрК35/Г) 0,07 (250 К); 0,15 (2000 К) 1000 -11,11
300-2000 v = ксАсв 1,47 • Ю"14!1’04 ехр(-913/Г) 1,25 (300 К); 2 (2000 К) 1000 -11,11
QHe + Нб2 —► Н2О2 + С2Н5 500-1000 v = ксАсв 3,7- 1012 ехр(-10300/Т) 0,2 (500 К); 0,3 (1000 К) 700 -17,82
300-1500 v = &сАсв 4,9 • 10” ехр(-7520/Г) 3 700 16,98
С^Нб + *СН —► продукты реакции 200-700 v = ксАс3 1,8- IO10 ехр(-132/Г) 1,0 500 -9,86
ДД + ОНО —► С>Н5 + СН2О 300-1500 V = &сАсв 7,8- 10“20Г2,72 ехр(-9176/Г) 5 900 -15,53
300-2000 V] = &1САСВ 3,6- 10” 0,4
300-1500 v2 = к2сАсв 1,9- Ю10 2
СгНб + СН2 |_Д. сн3 + QH5
СЛЬ + СНз ~►СН4+С2Н5 300-1500 V = fcACB 2,5- 10“31Г6’°ехр(-3043/Г) 0,1 (300К); 0,2 (1500 К) 900 -14,35
300-2500 V = ксдСя 9,1 • 10 257'4 ехр( 4169/Г) 1,3 (300 К); 3 (2500 К) 900 -14,24
СгНб + СНзО —►СН3ОН + С2Н5 300-1500 v =ксАсв 4,0 • 10” ехр(-3570/Г) 3 700 -14,61
ДД + ён2ОН—►СН3ОН + £2Н5 300-1500 v =кс^сь 3,3 • 10’22Г3 ехр(-7033/Г) 5
0,0,+&н —►с2н2+с\н5 300-2500 v = &сАсв 6,0 • 1012 1,3 (300 К); 3 (2500 К)
с2н6 + он3—►с2н4+й2н5 300-1500 v = &сАсв 1,0- 10”21Г3’3 ехр(-5285/Г) 5 (> 1000 К) 700 -17,74
QHe + СН3СО —* СН3СНО + QHs 300-1500 v = kcAcB 3,0- Ю 20Г2-75 ехр( -8820/Г) 5 900 -15,66
С2Нб + СН2СН=СН2—►С2Н5 + СН3СН=СН2 V = kcACB 1,31 • 10-12 ехр(-85770//?Г) 700 -18,28
С2Нб-СН3СН2СН2 —►С2Н5 + С3Н8 v = kcKcB 5.25 • 10” ехр(-51460/ЯГ) 700 -16,12
(^Нб + СН3СНСН3 —►СН3СН2 + С3Н8 v = fcAcB 1,66 • 10” ехр(-53970//?Г) 700 -16,81
QHe + СН3СНСН=СН2—►QHs + С4Н8 v = kcAcB 1,66- 1012 ехр(-100800//?Г) 700 -19,31
Химическая кинетика и диффузия 413
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К IgA: при Тр
Реакции замещения
Сущ + СН3СН=СНСН2—►СуЩ + С4Н8 v = ЛсАсв 1,05 • IO’12ехр(-96230/ЛГ) 700 -19,61
Сущ + СН3СН2СН2СН2 —►СУЩ + С4Н10 v = ЛсАсв 5,25 • 10-13 ехр(-51460/Я7Э 700 -16,12
сущ + сн3снсн2сн3 —►сущ + С4Н10 v = ЛсАсв 1,66 • 1013 ехр(-53970//?Т) 700 -16,81
С2Н6 + (СЩЬСН^Щ—►СУЩ + (СЩУСНСЩ v = ЛсАсв 5,25 • 1013 ехр(-51460/Л7 ) 700 -16,12
сущ + б—► он+СН3СН2 300-1200 v = ЛсАсв 1,66- 10"15Г1’5 ехр(-2920/Г) 0,3 (300 К); 0,15(1200 К) 750 -12,16
СН3СН=СН2 + н—►СН2СН=СН2 + щ v = ксксй 1,66 - Ю1оехр(-1464О/Л7’) 700 -10,87
СН3СН=СН2 + сн3 —►(’щен =сн2 + сн4 v = ЛсАсв 2,63 • 10“13 ехр(-36820//?Г) 700 -15,33
СН3СН=СН2 + С2Н5-^СН2СН=СН2 + СуЩ v = ЛсАсв 1,66- 1013 ехр( 41000/Я7 ) 700 -15,84
СН3СН=СН2 + СН3СН2СЩ—► v = ЛсАсв 1,66- 10”13 ехр(—41000/ЛГ) 700 -15,84
—► сн2сн=сн2 + сущ
л'тл л'тл—-гчи -4- Лтд mm — v = ЛсАсв 6,61 • Ю'14 ехрН10580/ЯГ) 700 -16,21
СНзСг!—<хГ12 ' СПзСлСПз £н2сн=сн2+С3Н8
сн3сн =сн2 + сн3сн2сн2ён2—► v = ксАсв 1,66- 10,3ехр(-41000/7?Г) 700 -15,84
► СН2СН— сн2 + С4Н10
СН3СН=СН2 + СН3СН2СНСН3 —► v = ЛсАсв 6,61 • 10"'4ехр(^Ю580//?Г) 700 -16,21
СН2СН =сн2 + С4Н10
СН3СН=СЩ + (СНзУСНёЩ v = ZrAcB 1,66 • 1013 ехр(-41ООО/Л 7 ) 700 -16,21
-► СН2СН =СН2 + С4Ню
к\ v =ксАсв 2,09- Ю’10 ехр(-40580//?Г) 700 -12,71
р— СЩСЩСЩ + Щ сущ + н —L СЩСНСЩ + Щ 1,05 • 10“'° ехр(-32220//?Г) 700 -12,38
к\ 1—► СН3СЩСЩ + сн4 сущ + сщ-Hfo 1—► сн3днсн3 + сщ v=ксАсв 1,66- 1012ехр(-48120/ЯГ) 1,05 • 10~12ехрК3930//?Г) 700 700 -15,37 -15,26
414 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Гр
Реакции замещения
к\ . I—СН3СН2СН2 + 0?Н8 + С2Н5 —р . L-*► СН3СНСН3 + С2Н6 k\ , , Г—► СН3СН2СН2 +С3Нб С3Н8 + СН2СН=СН2 —\к2 1—►СН3СНСН3+С3Н6 С3Н8 + СН3СНСН3—►СН3СНСН3 + С3Н8 С3Н8 + СН3СН2СН2—►СН3СНСН3 + С3Н8 *1 сн3сн2сн3 + сн3снсн==сн2—рь/ к\ р* СН3СН2СН2 + С4Н8 сн3ёнсн3 + С4Н8 к\ СН3СН2СН3 + СН3СН=СНСН2—\к^ к\ . L—*" г—► СН3СН2СН2 + С4Н8 \^2 1—СН3СНСН3 + С4Н8 к\ СзН8 + сн3сн2сн2^н2 ~~|Ар р* СН3СН2СН2 + С4Н10 \кг *—-*• СН3СНСН3 + С4Н10 v = *сАсв v = *сАсв v = *сАсв v = *сАсв v = kcAcQ v = *сАсв v = £сАсв 5,25 1013 ехр(-51460/ЯГ) 8.32-10 14ехр(М3510/7?7') 1,32 • 10“12 ехр(-85770//?Г) 3,31 • 10-13 ехр(-67360/ЛГ) 1,66 • 1 (Г13 ехр(-53970//?Г) 8,32 • 10”14 ехр(—43510/ЛГ) 1,66 • 1012 ехр(-100800/ЛГ) 1,05 • 10-13 ехр(-77820/ЯГ) 1,05 • 10”12 ехр(-96230/7?Г) 1,05 • 1О13ехрК3510/7?Г) 5,25- 10 13 ехр(- 51460/7?7’) 8,32- 1014 ехр(-43510/7?7) 700 700 700 700 700 700 700 700 700 700 700 700 -16,12 -16,33 -18,28 -17,51 -16,81 -16,32 -19,31 -18,79 -19,16 -18,92 -16,12 -16,33
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция лк Кинетическое уравнение Константа скорости реакции к Ошибка Тр, к IgA: при Тр
Реакции замещения
*1 С3Н8 + СН3СНСН2СН3 — П* СН3СН2СН2 +С4Н10 —и2 СН3СНСН3 + С4Н10 Д* ci i3ci i2ch2+с4н10 QH8 + (CH3)2CHCH2 -к L-k СН3СНСН3 + С4Н10 СН3СН2СН =СН2 + Н—►СНзСН =снсн2 + н2 СН3СН=СНСН3 + й -►СН3СН=СНСН2 + н2 СН3СН2СН=СН2.+ сн3 ► СН3СН=СНСН2+сн4 СН3СН=СНСН3 + сн3 ► -► СН3СН=СНСН2 + сн4 СН3СН=СНСН3 + С2Н5 ► СН3СН=СНСН2 + QHe СН3СН2СН=СН2 + ► -*СН3СН=СНСН2 + СзНб сн3сн2сн=сн2+сн2сн=сн2—► СН2СН=СНСН2 + С3Нб СН3СН=СНСН3 + СН2СН=СН2 —► -► сн3сн=снсн2+с3н6 сн3сн2сн=сн2 + сн3сн2ён2 —► —► сн3сн=снён2 + с3н8 сн3сн2сн=сн2 + сн3£нсн3 —► —► сн3сн=снён2 + с3н8 V = fcACB V = АсаСв V - Асасв V = ксксъ V = fcACB V = fcACB V = fcAcB V = fcAcB v = fcAcB v = ксАсъ v = fcAcB v = Ar AcB 1,66 10“13 ехр(-53970/ЯГ) 1,32 • IO1’ exp(-51460//?Г) 5,25 • 10’13 ехр(-51460//?Г) 8,32 • 10 14 ехр(-43510/ЯГ) 1,66- 1О’оехр(-1632О//?Г) 3,31 • Ю’10 ехр(-14640//?7) 1,66 • 10 ’3 ехр(-30540/ЯГ) 4,17- 10 13 ехр(-34310/2?Г) 3,31 • 10“13 ехр(-41000/ЯГ) 1,66 • 10“13 ехр(-34730//?Г) 1,32 • 10"13 ехр(-51880//?Г) 4.17 • 1013 ехр(-56070/ЯГ) 1,66- 10|3ехр(-34730/ЯГ) 6,61 • 1014 ехр(-40580//?Г) 700 700 700 700 700 700 700 700 700 700 700 700 700 700 -16,81 -16,72 -16,12 -16,37 -11,00 -10,57 -15,06 -14,94 -15,54 -15,37 -16,75 -16,56 -15,37 -16,21
416 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции k Ошибка гр, К 1g# при Тр
Реакции замещения
сн3сн=снсн3 + сн3сн2Сн2 ► —► сн3сн=сн£н2 + С3Н8 сн3сн=снсн3 + сн3снсн3—► ► СН3СН=СНСН2 + С3Н8 сн3сн=снсн3 + сн3сн=снён2 —► > СН3СН=СНСН2 + СН3СН2СН=СН2 СН3СН=СНСН3 4- СН3СН=СНСН2 — > СН3СН=СНСН2 4- СН3СН=СНСН3 СН3СН2СН=СН2 + СН3СН2СН2СН2 ► —► сн3сн=снён24-с4н10 СН3СН=СНСН3 4- сн3сн2сн2сн2 ► —► сн3сн=снсн2 + с4н10 СН3СН2СН=СН2 4- сн3сн2снсн3 ► ► СН3СН=СНСН2 4-С4Ню СН3СН=СНСН3 4- сн3сн2снсн3 ► ► СН3СН=СНСН2 4-с4н10 СН3СН2СН=СН24-(СН3)2СНСН2 ► ► СН3СН=СН(^Н24-(СН3)2СНСН3 СН3СН=СНСН3 4- (СН3)2СНСН2 ► ► СН3СН=СНСН2 4- (СН3)2СНСН3 к\ . |—> СН3(СН2)2СН2 4- Н2 С4Н10 + Н — к 3\ СН3СНСН2СН3+н2 V = ЛсАСв v = Асасв v =#сАсв v = #сАсв v = #сАсв v = #сАсв v =ЛсАсв V = ЛсАСВ v=#сАсв V = ЛсАсв V =#сАсв 3,31 • Ю'13 ехр(—41000/ЛГ) 1,32 • 1013 ехр(-40580//?Г) 1,66 • Ю13 ехр(-62760//?Г) 6,61 • 10-14 ехр(-51880//?Г) 1,66 • 10~13 ехр(-34730//?Т) 3,31 • 10~13 ехр(-41000/7?Г) 6,61 • 10-14 ехр(—40580/ЛГ) 1,32 • 1013 ехр(-40580//?Г) 1,66 • IO-13 ехр(-34730//?Г) 3,31 • 1(Г13 ехр(-41000//?Г) 2,09 • IO40 ехр(-40580//?Г) 2,09 • Ю"10 ехр(-32220//?Г) 700 700 700 700 700 700 700 700 700 700 700 700 -15,54 -15,91 -17,46 -17,05 -15,37 -15,54 -16,21 -15,91 -15,37 -15,54 -12,71 -12,08
Химическая кинетика и диффузия 417
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К IgA при Гр
Реакции замещения
С4Н10 + СНз — Al г—► СН3(СН2)2СН2 + сн4 -Л- СН3СНСН2СН3 + сн4 V = Асасв 6,61 • 1013 ехр(-47700/ЯТ) 6,61 • 1013 ехр(-40170/RT) 700 700 -15,74 -15,77
С4Н10 + С2Н5- к *СН3(СН2),СН2 + С2Н6 ♦ СН3СНСН2СН3 + С2Н6 V = Асасв 5,25 • 10’13 ехр(-51460/ЯТ) 1,66- 1013ехр(-43510/ЯТ) 700 700 -16,2 -16,03
С4Н10 + СН2=СНСН2 г-* СНзССНг^СНз + СзНб ^2 >—► СН3СНСН2СН3 + С3Н6 v = кс^св 1,32 • 10’12 ехр(-85770/ЯГ) 5,25 • 10’13 ехр(-68620/ЯГ) 700 700 -18,28 -17,40
С4Н10 + СН3СН2СН2 к\ г-^снзсснзьснз + ед |А2 *—► СН3СНСН2СН3 + С3Н8 v = Асасв 5,25 • Ю13 ехр(-51460/ЯГ) 1,66 • I013 ехр(-43510/ЯТ) 700 700 -16,12 -16,03
С4Н10 + СН3СНСН3 - кх р-^СНзССНгЬСНг + СзЩ 4*2 !—► СН3СНСН2СН3 + С3Н8 v = Асдсв 1,66- 10 13 ехр(-53970/ЛГ) 1,66- 10 " ехр(-51460//?Г) 700 700 -16,81 -16,62
с4н10 + сн3сн=сна-1 к\ Г—► СН3СН2СН2СН2 + С4Н8 2 \к1 *—► СН3СН2СНСН3 + С4Н8 v = Асасв 1,66- 10*12 ехр(-84100/7?Г) 2,63 • 10’13 ехр(-79500/ЯГ) 700 700 -18,06 -18,51
С4Н10 + СН3СНСН2 сн3 V = Асасв 1,66- 10’13 ехр(-53970/ЯГ) 700 -16,81
—► СН3(СН2)2СН2 + С4Н10
С4Н10 + СН3СН2СН2СН2 —► V = ЛсАсв 1,66- 10~13ехр(-43510/ЯГ) 700 -16,03
СН3СНСН2СН3 + С4Н10
С4Н10 + (СНзЬСНСНз кх . Д^СНзССНг^СНг+С.Ню \ki * СН3СНСН2СН3 + С4Н10 v = fcAcB 5,24 • 10’13 ехр(-51460/7?7 ) 1,66- 10~13 ехр(-43510/ЯГ) 700 700 -16,12 -16,03
к СбНб + ОН—[I 1 2 Н2О + С6Н5 СбНбОН 400-1500 1000-1150 V] = к^с^св v2 = kiC^C-Q к} = 2,7 • Ю-16Г’’42 ехр(-730/Т) к2 = 2,2- 10"” ехр(-5330/Г) 0,3 0,3 1000 1000 -11,62 -12,97
418 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция Г, К Кинетическое уравнение Константа скорости реакции к Ошибка Гр, К IgA при Гр
Реакции замещения
• к QHs + OH QH6+ 0.^4 6 5 QH6O к\ • I—* QH5O + H2 QH5OH + H—^2 • 65 L^QHe + OH . к г-* и2о+с6н5д СбН5он+ он -4 2 . 5 I—► н2о+одон с6н5сн3 + н —► н2+С6Н5СН2 с6н5сн3+он —*Н2О+С6Н5СН2 одод+дн *Г* "2о+с6н5сн2сн2 1—►н20^с6н4с2н5 Н3С —СН3 + он —► Н3С —СН2 + Н20 300-1000 300-1500 1000-1150 1000-1150 600-2800 400-1200 773 500-960 300-600 v = ЛсАсв v = ЛсАсв V1 = Л]САсв v2 = £2ca<?b V = ЛсАсв V = ксКсъ v = ЛсАсв V = АсаСв V = АгдСв V = ЛСдСв к=\,2- 10’22Г3’7 ехр(-570/Г) = 2,0 • 1О"12Г0’6 ехр(-3680/Г) кх = 1,9 • Ю10 ехр(-6240/Г) к2 = 3,7 • Ю11 ехр(-3990/Г) к= 1,0- 101’ 6,6- 10 227 3'44ехр(-1570/7) 8,6 • 10“15 ехр(-1440/Г) к = 8,7- 10'12 6,46 • 1O’U ехр(-1440/Г) к= 1,7- 1011 ехр(-200/Г) 0,5 2 0,3 0,3 0,5 0,3 (600 К); 0,5 (2800 К) 0,5 (400 К); 0,3 (1200 К) 0,1 0,1 0,5 700 700 1000 1000 1000 700 700 450 -11,75 -12,28 -12,43 -12,17 -11,39 -14,96 -11,04 -10,96
к Ъ. ХГ 4- С€\~
Q2 + CN— О2 + СНО О^СНг к —►n2+co+o« <со+д« —►со+нд2 ь.С|Ц 4- ccl 300-2500 300-2500 300-1000 V = ксКсъ V = ЛсдСв V = АгдСв 1,1 • 10~п ехр(-205/Г) 5,0 • 10~12 к = 5,2- 10“11 0,25 (300 К); 0,5 (2500 К) 0,3 ±0,3 (300 К); ±0,5 (1000 К) 1000 -11,05
к ► Un т VV>2 —►сндз —►со+н + дн — СОг + Н2 —►СО + Н2О —^СНз + Ог
Химическая кинетика и диффузия 419
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр,к 1g* при Тр
Реакции замещения
Oj + QH — *-к4 * СОг + *СН ►2СО + Н. -►сую+д* ^►со + сно 300 300-2500 300-2500 V = *сАсв V = *сАсв v = *сАсв к = 3,0- 10'11 к3= 1 • 10~12 к4 = 4- Ю12 0,5 1,3 (300 К); 10 (2500 К) 1,3 (300 К); 10 (2500 К)
о2 + снсо к —► сно + со2 2СО + ОН 300-550 V =*сАсв к = 2,7- 10“12 ехр(-430/Г), р(Не) = 270 Па ±0,7 450 -11,98
—►суз + н‘о2
О2 + СН3 — ку tl ► сн3д2 *• сн3д+д« 700-2500 300-1500 Vi = *]САСВ V2 = ^2сАсВ к} = 1,5 • 10"35Г’,501ехр(-8567/Т) *2 = 3,3 • 10 V 157ехр(14710/Т) 3 3 1000 1000
о2 + ен3д— сн2о + нб2 300-1200 V = *сАсв 6,7 • 10’14 ехр(-1070/Г) 0,2 (500 К); 0,3 (300 К, 1000 К) 700 -17,31
о2 + сн2он *сн2о + нд2 300-1200 V = *сАсв 2,6- 1О-9Г“'’0 + + 1,2- Ю10 ехр(-1800/Г) 0,1 (300 К); 0,3 (1200 К)
о2 + С2Н3 — к Л 1 • -► СН2О + СНО 2 • С2Н2 + НО2 300-2000 300-1500 V1 = *]CacB v2 = 9,0 • 1012 2,0 • 1013 0,3 (300 К); 0,5 (2000 К) 5
О2+СН3СО ►сн3сод2 300-1500 V = *cAcB 3,0 • 10“12 2
OyWM ►(ед+ндз 600-1200 V =*cAcB 1,7- 1014ехр(-1100/Г) 0,3 700 -14,45
о2 + едд- >сн3сно+нд2 300-1000 V = *cAcB 1,07 • 1013 ехр(-830/Г) 0,3 (300 К); 0,5(1000 К) 700 -13,49
Реакции обмена
СН3ОН + О2- ►сн2он + нд2 300-1500 V = *CACB 3,4 • 10‘” ехр(-22600/Т) 10 1000 -20,28
С,Н4 + СО - с>н3 + сно 300-1500 V = *cAcB 2,5 • Ю’10 ехр(-45600/Г) 5 900 -31,61
С>Н2 + С>Н2 -^ед + с^н 300-1500 V = *cAcB 1,6 • 10-11 ехр(^2500/Г) 5 700 -37,16
ед + ед -►20,113 300-1500 V = кс^сц 4,0 • 10’11 ехр(-34400/Г) 10 700 -31,74
420 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция Г, К Кинетическое уравнение Константа скорости реакции к Ошибка Гр, К IgA: при Гр
Реакция присоединения
СО + С2Н —►С2НСО ед+сн3—-ед С2Н2 + С2Н5 —► С4Н7 С2Н4 + Н—►С2Н5 к\ , [-^СН2СН2СН3 С2Н4 + СН3 1с2 1—► СН3СНСН3 к\ . |-^СН3СН2СН2 СзНб + Н—L2 1—► СН3СНСН3 к\ I—► СН3СН(СН3)СН2 С3Нб + СН3—\к2 *—► СН3СНСН2СН3 к\ г-► сн3сн2сн2снсн3 ед + ед—U2 . ед (СН3)2СНСН2СН2 300-1500 <900 > 1000 > 1000 > 1000 > 1000 > 1000 > 1000 > 1000 V = fcAcB V = fcAcB V = АсаСв V = Асдсв v = A]CAcb V = А2СдСВ v = А,СдСв V = А2СдСв V = Л]САСв V = Л2СдСв V = £1СдСв V = к2сКсъ 2,5 • Ю-13 ехр(-2420/Г) 4,17- 10’14 ехр(-32200 / RT) 4,17- 10~’4 ехр(-29300 / RT) 2,09 • 1011 ехр(-6300 / RT) 5,25 • 1013ехр(-32600//?Г) 5,25 • 10’13ехр(-12100/ЯГ) 1,32 • 10" ехр(-12100 / RT) 11,32- Ю'" ехр(-5000 / RT) 5,25 • 10’13 ехр(-38100 / RT) 5,25- 10’13 ехр(-31000/ RT) 2,09- 10~14 ехр(-31400//?Г) 2,09 • 10”14 ехр(-38500//?Г) 10 900 1100 1100 900 1100 1100 1100 1100 1100 1100 1100 1100 -13,77 -14,91 -14,77 -11,04 -13,83 -13,75 -11,45 -11,12 -14,09 -13,75 -15,21 -15,51
Реакции рекомбинации и диспропорционирования
•NH + H—> •NH + OH - •NH + O2 — Н2 + -N- ц— no + h2 “еднзО+.к. —►NO + ОН —►NC^ + H ^-►HNO + 6- -h2+-nh 1500-2500 300-1000 270-550 2000-3000 V = АсдСВ V =АсдСв V = Асд<?в V = АсдСв 1,7- 10п к = 8,0 • Ю"11 к= 1,62 • 10’13 ехр(-770/Г) 1,0- 10"11 1,0 0,5 0,2 (270 К); 0,5 (550 К) 1,0 350 -13,75
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция Г, К Кинетическое уравнение Константа скорости реакции А Ошибка гр, К IgA при Гр
Реакции рекомбинации и диспропорционирования
nh2+oh - кх . г—*-«O + NH3 1—► h2o + -nh 500-2500 V! = А1СдСВ кх = 3,3 • 1О14/°’405 ехр(-250/Г) 0,5 1000 -11,34
ОН + Н—> •д+н2 300-2500 V = Асасв 8,1 • 1021/2 ехр(-1950/7 ) 2 1000 -14,94
он+он — ► Н2О+б- 250-2500 300-1500 V =Асасв V = Ас-дСв 2,5 • 1О’,5Г114 ехр(-50/Г) 3,5 • 10“1бГ1,4 ехр(-200/Г) 0,2 2 1000 1000 -11,20 -11,34
он+о ► о2+н 220-500 1000-2000 V = Ас-дСв V = Асасв 2,0 • 10’п ехр(-112/Г) 2,4 Ю” ехр(-353/Г) 0,2 0,2 350,0 1500 -11,14 -10,72
он + но2 - —► Н2О + о2 300-2000 300-1500 V = Ас-дСв V = Ас-дСв 4,8 • 1011 ехр(-250/Г) 2,4 • Ю^Г’1 0,2 (300 К); 0,5 (2000 К) 2 1000 1000 -10,42 10,62
к\ 300-1000 V! = AjCaCb А] = 7,1 • 10"11 ехр(-710/Г) 0,3 700 -10,59
НО2 + Н — т** н2 + о2 л2 • —►гон А3 . —►Н2О + О- 300-1000 300-1000 V2 = А2СдСв V3 = А3СаСв А2 = 2,8- 10-10 ехр(-440/Г) А3 = 5,0 • 1О‘П ехр(-866/Г) 0,3 0,3 700 700 -9,82 -10,84
Ai 300-1500 V] = А,сасв кх = 2,8 • Ю10 ехр(-440// ) 2 900 -9,77
НО2 + Н — 2ОН 1^. н2+о2 300-1500 V2 = А2СдСв к2= 1,1 • Ю10ехр(-1070/7) 2 900 -10,48
НО2 + НО2- —►Н2О2 + О2 550-1250 300-1500 V = Ас-дСв V = Ас-дСв 3,1 • 10’12 ехр(-775/Г) 3,0 • ю '2 0,15 (550 К); 0,3 (1250 К) 2 900 -11,88
но2+’д — * • А CN + OH - • . 1 NCO + O-- сн+.д—> ►о2 + он И—*-6+HCN "1—► NCO + H j—► NO + CO 1—► О2+ CN ►со+н 300-1500 1500-3000 1450-2600 300-2000 V = Ас-дСв V = АСдСв V = Ас-дСв V = Ас-дСв 2,9 10 ” ехр(+200/Г) А= 1,0 Ю10 А = 7,0- 1011 6,6 • IO’11 1,2 0,5 0,8 0,5 1000, 0 -10,45
•сн +.д - -►СНО + е 300-2500 V = Ас-дСв 4,2 • 1013 ехр(-850/7) 0,5 1250 -12,67
422 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Тр
Реакции рекомбинации и диспропорционирования
. k I—► СНО + О • •сн+о2-н 1—►со + он СНО + Н—Э-Н2 + СО сно+он —►Н2О+СО СНО + НО2 —>ОН + НгСО2 СНО + Зсн2 —* сн3 + со *1 г—->-2СО + Н2 СНО + СНО —\к2 ► СН2О + СО СНО + сн3 —*• сн4 + со кх + СО CHO + Qttj —Н2 1—-►С2Н3СНО Д^сн3сно+со сно+сн3со Н^Годсосно *1 г-*СН3ОН + СО СНО + СН2ОН —К 3 *—►СНгО + СНзО кХ . г—►он+со СНО + О • —*2 L—*-СО2 + Н СНО + О2—►НОз + СО 'сНг + Н—►•СН + Н2 300-2000 300-1500 300-2500 300-1500 300-1500 300-1500 300-1500 300-1599 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-2500 300-2500 300-1500 300-1500 v =Асасв v = Асасв v =*сАсв V = *СдСв V = АсдСв V] = fciC'AC'B v2 = *2сАсв v = ксАсв V! = к}сАсв v2 = к2сАсв V1 = ^СдСв v2 = *2cAcB V] = кхсАсв v2 = к2сАсв v = ксАсв v = ксАсв v = ксАсв v = *сАсв к=5,5- 10’” 2,0- Ю’10 1,7- Ю’10 ~ 5,0 • 10’11 3,0- Ю"’1 к} = 5,0 • 10’12 * 2 = 3,0- 10"11 2,0 • Ю10 к{ = 1,5 • 1О“10 * 2 = 3,0- 10’11 1,5- 10’11 3,0- 1011 * ! = 2,0 • Ю’10 * 2 = 3,0 • 1О“10 5,0 10“” 5,0- 10“” 8,5 • 10 ” ехр(-850/Т) 5,0- 10"” 0,3 (300 К); 0,5 (2000 К) 2 о,з 5 3 1,5 1,5 2 3 5 3 3 3 3 0,3 0,3 1,5 (300 К); 5 (1500 К) 3 1000 -10,44
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Гр
Реакции рекомбинации и диспропорционирования
]СН2 + НО2 —► сн2о + ОН ’сед сно —►СН3+СО ’СН2 + 1СН2 —► ед ’СНз + СНз—►С2Н4 + Н ’сн2+СН2ОН —► СН3СНО+н ’сн2 + сн3О —сн2о + бн3 ’сн2+сед—► <ед+сн3 ’сн2 + сн3Со _► сн2со + Сн3 к] (-^(ед+снз 1 . [-^СО + 2Н ед+о. _\k 1-4>со + н2 Зсн2+н —►•снед *1 гЛ.едед Зсн2 + Зсн2—*2 2 2 1-^(ед + 2н Зсн2+сн3—►сед + н к\ 3 . г—►С2Н4 + ОН 3 СН2 + бН2ОН —\кГ. !—►СН3 + СН2О 300-1500 300-1500 300-1500 300-1500 3OO-J5OO 300—1500 300-1500 300-1500 300-1500 300-1500 300-1500 ЗОО-ЗООО ЗОО-ЗООО 300-1500 300-1500 V = *СдСВ V = *СдСВ V = *СдСВ V = *СдСВ V = *СдСВ V = *СдСВ V = *СдСв V = *СдСВ V] = *1СдСв v2 = *2Ca<?b V] = *1СдСВ v2 = кгС^Съ V = *СдСв V =*СдСв V = *2СдСВ V = *СдСВ V, =*1СдСВ V2 = *2СдСВ 5,0 • 10"” 3,0 • 10” 5,0- 10"” 3,0- 10"” 3,0- 10"” 3,0- 10"” 8,0- 10” 3,0- 10"” 1,5 • 10"” 1,5 • 10"” к} =2,5- 10"” Л2 = 2,5- 10"” 2,7- Ю"10 * = 2,0- 1О",оехр(-4ОО/Г) к2 / к = 0,9 ± 0,1 (ЗОО-ЗООО К) 7,0- 10"” *! = 4- 10’” *2 = 2 • 10’12 3 3 5 3 2 3 3 3 3 3 3 2 0,5 0,3 (300 К); 0,5 (3000 К) 3 3 1000 -9,87
424 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Гр
Реакции рекомбинации и диспропорционирования
^Н2 + О2 k к\ . . г-—*- со + н + он -Т-+ СО2 + 2Н \з w- го + НоО 300-1000 300-1500 300-1500 v = *сАсв V1 = ^1САСВ V3 = *3САСВ * = 4,1-10’” ехр(-750/Г) кх = 1,0 • Ю"10 *з = 4,0- 10’13 0,3 (300 К); 0,5 (1000 К) 2 2 700 -10,85
—► СО2 + Н2 _► СН2О + О-
СН2О + Н — СН2ОН + О- ►Н2 + СНО -*-сн2о+он 300-2200 500-2500 300-1500 V = *сАсв V = *сАсв v = ксАсв 3,8- 1О'14Г105 ехр(-1650/Г) 3,64 • 10 16Т'77 ехр(1510/Г) 7 • 10"” 0,1 (300 К); 0,5 (2200 К) 1,3 (500 К); 3 (2500 К) 2 1000 1000 -10,99 -10,79
сн3+н—► ги 4- Ац k Н2 + 'СН2 ►Н + СН2ОН 300-2500 300-2000 v = *сАсв v = ксАсв 1,0- 1О~10 ехр(-7600/Г) * = 6,0- 10” 1,0 0,7 1000 -14,30
1—►Н2О + 1СН2
СНз + НОг - к\ г—-►СН3О + ОН -|*2 1-^СН4 + О2 300-1500 300-1500 V] = *iCAeB v2 = к2сАсв *! =3,3 • 10"” *2 = 6,0- Ю"12 3 5
СН3+СН3 — ►С2Н5+Н 1300-2500 v = *сАсв 5 -10"” ехр(-6800/Т) 0,6 1900 -11,86
СН3 + С2Н5- -►СН4 + С2Н4 300-800 V = *САСВ 1,9- 10"12 0,4
CH3 + CH3O к\ г-Л-око+сНд -]к2 1—► СН3ОСН3 300-1500 300-1500 V! = *1САСВ v2 = *2сАсв *! =4,0- 10"” *2 = 2,0- 10’” 5 5
СН3+.0 —» ► СН2О + Н 300-2500 V = *сАсв 1,4- Ю’10 0,2
CH3O + H — ► Н2+СН2О 300-1000 V = кеЛсв 3,0- 10’” 0,5
*1 . 1—► СН3ОН + СН2О СН3О+СН3О -\к2 S 3 L_СН3ООСН3 300-1500 300-1500 vi = *1САсв v2 = *2сАсв 1,0- Ю’10 3,0 • 10"12 5
Химическая кинетика и диффузия
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К IgA: при Гр
Реакции рекомбинации и диспропорционирования
СН3О + СН3О2 —► СН2О + СН3ООН СН3О + С2Н3 —► СН2О + QH4 h 1—СН3ОН + СН2СО СН3О + СН3СО— 1—СН2О + СН3СНО Л1 • • ।—► Оо + СН3 СН3О + О.—\кГ . L-^OH + CH2O L-CH2O + H2 СН2ОН + Н —\к2 . I-^CH3+OH СН2ОН + он —► СН2О + Н2О СН2ОН + НО2 —* СН2О + Н2О2 СН2ОН + СН2О —►СН3ОН + СНО НОСН2СН2ОН сн2он+сн2он—к2 1—-► СН3ОН + СН2О • • |—► С9Н4 + СН2О СН2ОН + С^Нз — kj • • СзН5+ОН ^тт ^тт ^1Т 300-1500 300-1500 300-1500 300-1500 300-1000 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 v = Асасв v = Асасв V 1 = кхсАсъ v2 = £2сАсв v = Асдсв v2 = £2сАсв V 1 = к}сАсъ v2 = к2сАсъ v = ксАСц v = ксАсъ v ~-= kcAcQ V 1 = А,сасв v2 = Л2сАсв V] = £1САСВ v2 = к2сАсъ vi =Л]САсв v2 = £2сасв v3 = £3сасв 5,0 • 1013 4,0- 10"11 1,0- 10" 1,0 • 10’11 к = 2,5 • 10-11 к2/к = 0,12 ±0,1 (300 К) кх = 1 • 10’11 к2= 1,6- IO40 4- 10” 2,0 • 10-11 9,1 • 10'21Т2’6 ехр(-2950/Г) кх = 1,6- 10” * 2 = 8- 1012 кх = 5 • 10’" к2 = 2- 10” кх = 2- 10” * 2 = 4- Ю12 * 3 = 4- 10“12 10 3 10 10 0,3 (300 К); 0,7(1000 К) 2 2 2 2 3 2 2 2 2,5 2 5 5 700 -14,47
(Чъ.ГИД 4- с н п? —C\I-L + CFLO
СН2ОН + О2 —► л3 L-^QHj + CH^H ен2о+но2 300-1500 V = ксАсв 2,0 • 1012 2
426 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К lg£ при Гр
Реакции рекомбинации и диспропорционирования
С2Н + ОН —к, СН2 + СО С2Н2+О2 QH + HCb —\k2 . L^QHO + OH QH + CHO —► C2H2 + CO СзН + СНз —Э-С3Н3 + Н 300-1500 300-1500 300-1500 300-1500 300-1500 300-1500 700-2500 V1 = AqCACB v2 = А^аСв V] = АлСдСв v2 = А:2СдСв V = ЛСдСв V = ЛсдСв V1 = /чСаСв V2 = £2СдСв ^ = 3,0- 10п fc2 = 3,0 • 10 n 3,0- 1011 3,0 • 10“n 1,0 • 10 '° 4,0 • 10 41 kx +fc2 = 3,0- 10’11 kxl k2 = 8,3 • 10-17Г5’25 ехр(1611/Г), p(N2) = 1 атм 10 10 10 10 3 3 3
С и + с 14 tf.4"2 . —ш. С,14 + 14
+ - 14 + CL14-, ™ Ч-/Д£ а. * Хс^+с, -*С4Н2 + Н к\ ГГ*С4Н4 л2 • • w. Г.14 . + 14 300-2700 700-2500 300-1500 300-1500 300-1500 300-2500 300-1500 300-2500 Ъ =А^СдСв V = ЛСдСв V] = А^СдСв V2 = &2САСВ Vj = ЛзСдСв V! = кхсАсв v2 = к2сАсв Vj = £3САСВ Vi = £]CACB v2 = k2cAcB V = kcAcB v = kcAcB .? 11 ??• , ? x + Il w — II II У II Z о .? У И У о -~j uj uj 0 1— il> и 1=1 +• 171 - - 00 _ bs и w Д © . . 0 0 0 1—• J* 0 © "11 — — I — b> 1 —* ©_= 00 x © S *" ’ - 0 - 2.1. 2- 5 n •— Д ° — ю rj^X© k> OS c?' 0 Э 0,3 3 3 3 3 1,0 3 0,4
V2n т </2Пз СЛ + СНзСО QH + QHs - QH + O ► • • k СЧЛС'Сь 4-14 \удП1 П *3 L-4-+ С2Н2 —►С2НСО^СН3 £1 \к2 L-^СзНз + СНз со+.сн |—► сн2+со 14- + С-.О
\м/£ £\^ЛмХ 1 1 £ —► нссон
Химическая кинетика и диффузия 427
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка гр, К 1g* при Гр
Реакции рекомбинации и диспропорционирования
СНСО + О- -►2СО + Н 300-2500 v = ксАсв 1,6- Ю10 0,3
СН2СО + ОН Jq—► СН2ОН + СО сн2о+сно 300-2000 v = *сАсв к = 1,7- 10” 1,0
СоН, + Н — ’Н2 + С2Н2 300-2500 300-1500 v = ксАсв v = ксАсв 2,0- 10” 2,0- 10” 0,5 2,5
С2Н3 + ОН *С>Н2 + Н2О 300-1500 v = ксАсв 5,0 • 10” 3
С2Н3+НО2 -*сн3 + он+со 300-1500 v = ксАсв 5,0 • 10” 3
С2Н3 + СН3СО —► СН3 + С^СО 300-1500 v = ксАсв 3,0- 10” 3
л —► OH + QjH, 300-2000 v =ксАсв к = 5,0 • 10” 0,5
си + Г) . СО 4- С14,
L-► сно+сн2
СН3СО + Н -^сн3 + сио 300-1500 v = ксАсв 1,6 • 10~10 3
СН3СО + ОН к\ г-—*- СН2СО + Н2О — L-^CH3+CO + OH 300-1500 300-1500 V] = £iCACb v2 = *2сА^в кх =2,0- 10” к2 = 5,0 • 10” 3 3
СН3СО + СН3 СО —►СН3СОСОСН3 300-1500 v = ксАсв 2,0- 10” 2
СН3СО + С2Н5 —► С2Н5СОСН3 300-1500 v = ксАсв 5,19- Ю’10/”0’5 2 900 -10,79
СН3СО + О • —► СН3 + СО2 300-1500 v = *сАсв 1,6 • 10” 3
СЛ-К + н к к *С2Н6 2 • • CFL + СН> 700-2500 Vi =кхсАсв v2 = к2сАсъ +£2 = 6,0- 10” \о%(к2/ к\) = -1,915 + + 2,69- 10’3Г -2,35 • 10-7Г2, p(N2) = 1 атм *з = 3,0- 10’12 3
к ^с2н4 + н2 300-1500 v3 = ^3САСВ 3
QHs + Он *1 Р^С^Нд + НоО ~\к2 . сн3 + сн2о + н 300-1500 300-1500 V, =кхсАсв V2 = к2сАсв II II © Ъ о © । । 4 4
428 Новый справочник химика и технолога
Продолжение таблицы 2.1.17
Реакция г, к Кинетическое уравнение Константа скорости реакции к Ошибка Гр, к 1g* при Гр
Реакции рекомбинации и диспропорционирования
С2Н5 + НО2 — С2Н5 + С2Н5 — С2Н5 + 6.-Ц* ед+сь-*] C6H5CH2 + к\ г—♦ СН3 + СН2О + ОН k-i *3 L2>C2H4 + H2O2 ► СгНб + СгЩ -►СН3СНО + Н ^СН2О + СН3 т62 + С2Н5 Ь—*СбН6+СНО 1—►с6н5сн + бн 300-1500 300-1200 300-2500 500-2000 300-1500 300 V] = *1САСВ v2 = кгСьСъ Vi = к3сАсв v =ксАсв v = ксАсв v2 = *2сАсв V = ксАсв v=ксКсв v = *сАсв к3 = 4,0 • 10'11 *2 = 5,0- 1013 *з = 5,0- 1013 2,4 • IO'12 *= 1,1 • 1О~10 *2/* = 0,17 ±0,2 (300 К) 1,0- Ю’10 ехр(-26100/7 ) 6,7 • 10’11 ехр(-25600/Г) *=5,5 • 10’’° 5 0,4 0,3 (300-1000 К); 0,5 (1000-2500 К) 0,5 (500 К); 1,0 (2000 К) 5 0,3 1000 700 -21,34 -26,06
Литература
1. Денисов Е.Т. Константы скорости гомолитических жидкофазных реакций. М.: Наука, 1971.712 с.
2. Денисов Е.Т., Саркисов О.М., Лихтенштейн Г.И. Химическая кинетика. М.: Химия, 2000. 568 с.
3. Ямпольский Ю.П. Элементарные реакции и механизм пиролиза углеводородов. М.: Химия, 1990. 216 с.
3. Жоров Ю.М. Кинетика промышленных органических реакций: Справ, изд. М.: Химия, 1989. 384 с.
4. Денисов Е.Т. Кинетика гомогенных химических реакций. М.: Высш, шк., 1988. 391 с.
5. Шмид Р., Сапунов В.Н. Неформальная кинетика. В поисках путей химических реакций. М.: Мир, 1985. 264 с.
6. Панченков Г.М., Лебедев В.П. Химическая кинетика и катализ. М.: Химия, 1974. 592 с.
7. Таблицы констант скорости и равновесия гетеролитических органических реакций / Под ред. В.А. Пальма. Тарту: ТГУ, 1985-1990. Т. 1-5.
Химическая кинетика и диффузия 429
430
Новый справочник химика и технолога
2.1.2. Гетерогенные реакции
Раздел содержит сведения о кинетике двух типов реакций с участием твердых фаз: 1) топохимические реакции — превращения твердых тел, протекающие на границе раздела фаз; 2) диффузионная кинетика — кинетика реакций, протекающих при диффузии реагирующего вещества сквозь слой твердых продуктов реакции (пористых или непористых) или сквозь их смесь с исходным веществом.
2.1.2.1. Топохимические реакции
(доц., к.х.н. К.А. Суворов)
В традиционном понимании топохимические реакции — твердофазные реакции, протекающие локально в определенных участках твердого тела.
Понятия «топохимия» и «топохимические реакции» предложены Колшюттером [26]. К топохимическим он относит, по крайней мере, реакции трех типов: 1) химические и псевдоморфные превращения минералов (выветривание, дегидратация); 2) электрохимические реакции; 3) локализованные реакции в животных и растительных организмах. В настоящее время вторая группа составляет один из разделов химии твердого тела. Третья группа развилась в самостоятельное направление и заслуживает отдельного рассмотрения в рамках органической, биооргани-ческой или бионеорганической топохимии. В биологии и медицине топохимия развивается как один из разделов гистохимии [19].
Существуют представления, в соответствии с которыми топохимическими (или топотактическими) называют твердофазные процессы, при которых исходная конфигурация частиц в решетке твердого тела однозначно определяет конфигурацию продукта. Подобные реакции имеют место при фотоциклодимеризации диенов, полимеризации кристаллических диацетиленов и т. п. [1].
Таким образом, в широком смысле термины «топохимия» и «топохимические реакции» используются для обозначения различных явлений: локализации химических и др. процессов, химических соединений, химических связей, молекул, атомов.
В узком смысле к топохимическим относят реакции типа Атв Втв + Сгаз, у которых химическое взаимодействие не осложнено диффузионными процессами и локализовано на границе раздела «твердое исходное вещество—твердый продукт реакции» [12]. Такое сужение понятия приводит к тому, что из класса топохимических реакций выпадает большая группа твердофазных процессов, для которых характерен эффект локализации, но которые протекают без образования границы раздела твердых фаз. К ним, в частности, относятся реакции термического разложения твердых веществ, протекающие без образования твердого продукта реакции.
Материал, изложенный ниже, основан на представлениях [12, 14], в соответствии с которыми к топохи
мическим относятся гетерогенные химические процессы, локализованные в определенных местах твердого тела, которое расходуется или образуется в ходе реакции. В некоторых случаях подобное разграничение условно и требует внимательного подхода к интерпретации пограничных явлений между топохимией, с одной стороны, и катализом, кристаллохимией, электрохимией, коллоидной химией и физикой твердого тела — с другой.
На основании вышеизложенного к топохимическим не причисляются реакции, в которых твердое вещество не расходуется и не накапливается, а играет только роль неизменного субстрата (подложки), носителя, катализатора. Такие процессы локализованы на стационарных поверхностях раздела, тогда как топохимические реакции локализованы на постоянно возобновляющихся поверхностях. В топохимических реакциях твердая фаза выступает в роли либо исходного вещества, которое подвергается превращению с образованием новых химических соединений, либо продукта реакции, который образуется в результате химического превращения исходного твердого, жидкого или газообразного вещества. Многие случаи роста кристаллов из растворов, расплавов или газовой фазы, связанные с одновременным протеканием химических процессов (химическая кристаллизация), могут быть отнесены к топохимическим реакциям [14].
В большинстве случаев топохимические реакции начинаются в области протяженных дефектов кристаллической решетки. В результате образуются зародыши новой фазы продукта и формируются межфазные границы между исходной твердой матрицей и твердой фазой продукта. Дальнейший рост фазы продукта происходит вследствие реакции на этих границах, а скорость процесса пропорциональна поверхности раздела фаз.
Типичные топохимические реакции: обжиг, выщелачивание, химическое травление. Топохимический характер имеют реакции окисления металлов и разложения многих твердых неорганических и органических веществ.
На топохимических реакциях основано восстановление металлов из руд, промышленное получение строительных и полупроводниковых материалов.
Вероятно, к классу топохимических реакций могут быть отнесены некоторые процессы самораспростра-няющегося высокотемпературного синтеза [20-22].
Формально-кинетический анализ топохимических реакций
Характерной особенностью топохимических реакций является сигмоидный (или 5-образный) вид зависимости степени превращения а от времени t (рис. 2.1.10).
В случае, когда реакция протекает чисто в кинетической области, топохимическую реакцию можно представить как последовательность следующих стадий [2]:
Химическая кинетика и диффузия
431
Рис. 2.1.10. Типичные кинетические кривые топохимических реакций: а — степень превращения; v — скорость реакции; t — время от начала реакции; — время индукции;
Vmax, «шах, /max — максимальная скорость, соответствующие ей степень превращения и время от начала реакции
1) образование отдельных молекул или элементарных ячеек продукта реакции на поверхности исходного вещества;
2) возникновение отдельных зародышей (ядер) фазы продукта реакции (рис. 2.1.11,12)',
3) рост ядер вплоть до смыкания их (рис. 2.1.11, /3), образование на поверхности исходного вещества сплошного продукта реакции;
4) рост сплошного слоя продукта реакции за счет сокращения объема и поверхности еще не прореагиро-
Рис. 2.1.11. Схема развития реакционной зоны на зернах порошка
Кинетика зародышеобразования
Предполагается, что скорость образования зародышей зависит от числа энергетически доступных мест Nq (в основном определяемых дефектами кристалла или примесями). Если в момент времени t число зародышей равно N, то для кинетики изотермического зародышеобразования справедливо уравнение:
(2.1.2.1)
где kN — константа скорости зародышеобразования.
Несложные преобразования уравнения (2.1.2.1) приводят к экспоненциальной зависимости:
N = kNNoexp(-kNt).
(2.1.2.2)
Если kN мало, уравнение (2.1.2.2) можно аппроксимировать линейной зависимостью N - kNNot. Границы применимости уравнения (2.1.2.1) расширяются, если его дополнить либо степенной функцией вида (No - N)n, что увеличивает или уменьшает наклон кривой либо множителем ехр(//70), учитывающим возможную задержку процесса зародышеобразования (рис. 2.1.12) [3].
Рис. 2.1.12. Кинетические кривые зародышеобразования (а) и зависимости скорости зародышеобразования от времени (б): 0 соответствует N = 0 (мгновенное зародышеобразование);
/— N = kN(N — No)', 2 — N = k,,N0-
3 — N = k,,(N - 2V0)” для n > 1;
7— 2V = ^(y-y,,)exp/1//0
(/« — инкубационный период зародышеобразования);
5 — N = kt, (N - No)exp t2/t0 при ц < /2;
kx — константа скорости зародышеобразования;
No — число мест, доступных для зародышеобразования
Формы локализации
Формы локализации топохимических реакций обычно рассматриваются на примере монокристалла в предположении, что полученные результаты в последующем
432
Новый справочник химика и технолога
с определенными оговорками могут быть перенесены на поликристаллические и массивные образцы.
Среди типичных форм топохимических реакций локализации можно выделить следующие [14]:
1) сплошная локализация на всех гранях (в реакцию мгновенно вступает вся поверхность граней);
2) сплошная локализация на активных гранях (неактивные грани либо не участвуют в реакции, либо вступают в реакцию через определенные промежутки времени);
3) очаговая локализация на гранях (последние вступают в реакцию одновременно или последовательно);
4) очаговая локализация в объеме кристалла (реакция начинается на изолированных центрах внутри кристалла);
5) сплошная локализация в объеме кристалла (реакция протекает гомогенно по всему кристаллу).
За исключением последнего случая, реакция распространяется в разных направлениях изотропно или анизотропно, в виде плоского или волнистого фронта, послойно, нитевидно, по границам между зернами и т. д.
Вступление в реакцию всей поверхности отличают от делокализованного ее протекания по признаку: в частично прореагировавшем кристалле, если его расколоть, обычно видно прозрачное ядро, которое отделено от непрозрачной прореагировавшей части кристалла резкой или диффузной поверхностью раздела. Подобная поверхность образуется также при очаговой локализации, когда реакция распространяется от начального центра, образуя характерные фигуры правильной или неправильной формы.
Сплошная локализация реакции на поверхности кристаллов. Одновременное вступление в реакцию всех частиц поверхности, скорее всего, означает, что превращение происходит не в кинетическом режиме и что звеном, определяющим скорость, является не химический акт, а процессы тепло- и массообмена подвижных составных частей реагентов и образующихся продуктов. Для реализации кинетического режима реакцию проводят в мягких условиях, соответствующих низким скоростям химического превращения, при которых улавливается разница между реакционной способностью активных и неактивных участков поверхности.
Сплошная форма поверхностной локализации при химическом превращении кристалла, протекающем в кинетическом режиме, связана не столько с одновременным вступлением в реакцию всех частиц поверхности, сколько с тем, что поверхность быстро покрывается большим числом очагов, либо на ней появляется незначительное количество очагов, которые быстро расширяются в плоскости грани до образования сплошного фронта реакции. Другими словами, «мгновенное вступление в реакцию всей поверхности» следует понимать не буквально, а лишь в том смысле, что на ранних стадиях превращения кристалла на его поверхности образуется сплошной фронт реакции. Его дальнейшее перемещение рассматривают уже независимо от того, каким образом он сформировался.
На рис. 2.1.13 представлена модель гомотетично (с сохранением исходных пропорций) сокращающегося прямоугольного ядра кристалла. Величины va, v<>, vc означают линейные скорости присоединения элементарных прореагировавших блоков или линейные скорости перемещения фронтов реакции в направлениях соответственно [100], [010], [001], a v(100), v(010), v(001) — те же линейные скорости присоединения или линейные скорости перемещения по поверхности граней соответственно (100), (010) и (001). Половинки сторон исходного кристалла в направлениях обозначены а0, со, половинки сторон непрореагировавшего ядра кристалла в момент времени t — а, b, с.
г(1°°)
v V(100) ,,(100) bf ’
Рис. 2.1.13. Модель гомотетично сокращающегося прямоугольного ядра кристалла
Для гомотетично сокращающегося прямоугольного кристалла протекание реакции в кинетическом или диффузионном режиме отражается на характеристиках формы кинетических кривых а — t, в том числе на величине кинетического параметра п в уравнении:
а = 1-ехр(-Л7"),
где а — доля прореагировавшего вещества; К — постоянная, связанная с константой скорости равенством k = nKlln; t — время разложения, п — кинетический параметр. Для кинетического режима п > 1, для диффузионного п = 0,5.
Сплошная локализация на всех гранях. В реакцию одновременно вступает вся поверхность образца с последующим развитием процесса по геометрической модели движущихся с постоянной линейной скоростью плоских поверхностей. Отличительная особенность: интегральные кинетические кривые а—t или а—//Ах = const (где ///а = const — приведенное время) не имеют участков нарастания скорости во времени, а соответствующие дифференциальные кинетические кривые не проходят через максимум. Возможное изменение формы кинетических кривых ограничивается пределами, соответствующими второму (п = 2) и нулевому (п = 0) формальному порядку реакции:
Химическая кинетика и диффузия
433
в случае реагирования полидисперсных фракции кристаллов. При реагировании монокристаллов формальный порядок имеет значение п = 0, п = 0,5 (уравнение диффузионно-контролируемых процессов) и п = 2/3 (закон топохимических реакций).
Для модели равномерно сжимающегося шара или изотропно реагирующего куба (рис. 2.1.14) п = 2/3. Аналогичная зависимость наблюдается для кристаллов, имеющих другую форму, например форму прямоугольного параллелепипеда при условии гомотетичности (если а0 = 2/>о = 4с0, то va = 2v0 = 4vc).
Рис. 2.1.14. Случай равномерно сжимающегося шара или изотропно реагирующего куба (п = 2/3)
Сказанное относится к кристаллам любой формы, исключая пластинчатые и игольчатые. По мере того, как форма кристалла приближается к форме тонкой пластинки или иголки, интегральная кинетическая кривая постепенно выпрямляется, приближаясь к прямой, а п стремится к нулю (рис. 2.1.15).
Рис. 2.1.15. Случай негомотетично сжимающегося куба (и = 0):
/ — Vc » Va, Vb = Va,2 — Va» Vb, Vc = Vb
Сплошная локализация на активных гранях. Разновременное вступление граней кристалла в реакцию может приводить к усложнению формы кинетической кривой.
Примером реакции, локализованной на активных гранях (010) и (001) и не протекающей на (100), служит термическое разложение кристаллов троны [23]
Na2CO3 • NaHCO3 • 2Н2О. При сравнительно быстром термическом разложении вытянутых в направлении [010] кристаллов (длина 3-15 мм, ширина 0,1-0,4 мм) в вакууме реализуется модель параллелепипеда, который сокращается в направлениях [010] и [001], но не сокращается в направлении [100] (рис. 2.1.16).
С учетом индукционных периодов, разных для различных граней, зависимость степени превращения от времени будет выглядеть так:
ан [q,-v.('-g)] [».-4-<У)]
(2.1.2.3)
где — индукционные периоды вступ-
ления в реакцию соответственно граней (100), (010), (001); остальные обозначения даны ранее.
Рис. 2.1.16. Термическое разложение кристалла троны Na2CO3 • NaHCO3 • 2Н2О в вакууме (Т= 80 °C, р = 10’5 гПа, /05 =10 мин, = оо, va = 0)
Своеобразно протекает термическое разложение кристаллов оксида ртути(П), имеющих форму правильной шестигранной призмы [23]. В определенных условиях реакция может зарождаться по центру шестиугольной грани, быстро проникать в кристалл вдоль a-оси, а затем перемещаться плоским фронтом по модели «отрицательно» растущего кристалла, который воспроизводит огранку исходного кристалла (рис. 2.1.17).
Рис. 2.1.17. Термическое разложение кристалла HgO в вакууме (Т= 460 °С,р = 10“3 гПа, = оо):
а) а = 0; б) а = 0,5; в) а = 0,9
в
Очаговая локализация реакции на поверхности граней. Очаговая локализация реакции может быть связана с дефектами на поверхности кристалла и с определенными элементами его симметрии (плоскостями, осями, ребрами), приводящими к наличию более реакционноспособных участков.
434
Новый справочник химика и технолога
Типичный пример — химические превращения кристаллов гидрокарбоната натрия при комбинированном циклическом воздействии температуры и влаги (рис. 2.1.18) [24].
Рис. 2.1.18. Схема термического разложения кристалла NaHCO3 в условиях циклического изменения температуры и парциального давления водяного пара
У призматического кристалла оксида ртути(П) плоскости двойникования выходят на ребра, параллельные a-оси. При термическом разложении, наряду со схемой «отрицательно» растущего кристалла (см. рис. 2.1.14), равновероятно реализуется схема, когда реакция зарождается на боковых ребрах шестигранной призмы и проникает в кристалл по плоскостям, соединяющим боковые ребра с a-осью (рис. 2.1.19).
Рис. 2.1.19. Термическое разложение кристалла HgO в вакууме (Т= 460 °C, р = 10’3 гПа, t^0) = С'0’ = oo):
а) а = 0; б) а = 0,14-0,2; в) а = 0,440,5
Повышенная реакционная способность характерна для мест выхода на поверхность плоскостей раскалывания кристаллов или плоскостей, совпадающих с определенными слоями в кристаллах слоистого строения. Высокая активность обычно характерна для ребер, идущих по контуру наиболее быстро растущих граней кристалла.
Математические модели механизмов топохимических реакций
Чтобы получить уравнение кинетики топохимической реакции, нужно знать или предположить, по крайней мере, следующие основные параметры:
- закон образования ядер новой фазы;
- закон роста ядер;
- форму ядер.
Возможны различные сочетания законов образования, роста и формы ядер продукта, поэтому существует большое количество уравнений, описывающих кинетику топохимических реакций. Но наиболее широко применяются лишь несколько из них.
Задачей эмпирической кинетики является нахождение функции /(а), содержащей наименьшее возможное
число констант, которые можно было бы интерпретировать и соотносить с другими физическими параметрами системы и процесса. Кроме того, функция f (а) должна наилучшим образом аппроксимировать экспериментальные результаты.
Эмпирический подход в кинетических исследованиях преобладает, поскольку [3]:
- в большинстве случаев практически невозможно подобрать такую физико-математическую модель, которая соответствовала бы характеру топохимического процесса на всем протяжении реакции или температурного интервала;
- при протекании параллельных процессов в поли-дисперсных системах предпочтительнее использовать физико-математическое описание суммарного процесса;
- в технологии детальное исследование механизма реакции является менее важной задачей, чем оптимизация процесса или определение влияния различных факторов на его скорость;
- использование эмпирических зависимостей позволяет достаточно легко и быстро получать численные характеристики.
В ряде случаев описание кинетики топохимических реакций на основе простых приближенных моделей приводит к достаточно надежным результатам.
Наиболее часто уравнение кинетики топохимических реакций выражается в виде формального соотношения:
/(а) = ат(1 -а)” (2.1.2.4)
где показатели степени т и п — некоторые постоянные, определяемые механизмом реакции и имеющие смысл кинетических параметров процесса. Довольно часто считают т = 0, а показателю п приписывают смысл порядка реакции.
Другое эмпирическое приближение для описания механизма реакции использует функцию g(a) (табл. 2.1.18).
Описательные свойства уравнения (2.1.2.4) улучшают путем добавления корректирующих сомножителей, содержащих фактор времени:
a = kptp~'(l-a). (2.1.2.5)
Множитель If нельзя рассматривать как константу скорости, так как уравнение (2.1.2.5) содержит две переменные а и t. Оценка параметров кинетики с помощью уравнения (2.1.2.5) не позволяет дать реальные кинетические характеристики, пока оно не преобразовано к виду уравнения с одной переменной.
Кинетические уравнения, имеющие вид функциональной зависимости a = f(t) или daJdt = f (/), можно рассматривать как разновидности уравнения (2.1.2.3). В таком виде уравнения используются для описания кинетики реакций термического разложения, процессов кристаллизации, реакций твердофазного синтеза между твердыми и между твердыми и жидкими веществами и т. д. (табл. 2.1.19).
Химическая кинетика и диффузия
435
Уравнения в интегральной g(a) = J—, дифференциальной /(а) формах о a
и формальное соотношение /(а) = аж(1-а)'’ [3]
Таблица 2.1.18
Характер образования и роста зародышей Стадии, определяющие скорость процесса
Тип и симметрия процесса Скорость диффузии реагента через слой продукта реакции
g(a) /(a) m n
Зародышеобразование по степенному закону; скорость зародышеобразования определяет скорость процесса в целом Линейный Квадратичный Кубический Степенной Экспоненциальный Автокаталитический (уравнение Праута — Томпкинса) Автокаталитический обобщенный Автокаталитический (уравнение Рогинского — Шульца) 2a2 a-1 -1 0
Постоянная скорость зародышеобразования Двухмерный рост зародышей Трехмерный рост зародышей l/2[-ln(l - a)]1/2 2/5[-ln(l - a)]2/5 (l-a)Hn(l-a)]1'2 (l-a)[-ln(l-a)]3'5 1/2 3/5 0,77 0,63
Нулевая скорость зародышеобразования при постоянном числе зародышей Двухмерный рост зародышей Трехмерный рост зародышей [-ln(l-a)] 2/3[-ln(l -a)]273 1 - a (1 — <x)[—ln(l — <x)]1/s 0 2/3 1 0,7
Зародышеобразование по степенному закону; скорость зародышеобразования определяет скорость процесса в целом Линейный Квадратичный Кубический Степенной Экспоненциальный Автокаталитический (уравнение Праута — Томпкинса) Автокаталитический обобщенный Автокаталитический (уравнение Рогинского — Шульца) a 2a2 2/3a2/3 rnam In a ln[a/(l - a)] + C 1 a1/2 a’/3 a1-17'” a a(l - a) (J a2/3(l -a)273 0 1/2 1/3 1 - Mm 1 1 ~~k 2/3 0 0 0 0 0 1 ~k 2/3
Постоянная скорость зародышеобразования Двухмерный рост зародышей Трехмерный рост зародышей l/3[—ln(l - a)]1/3 l/4[-ln(l - a)]1/4 (1-a)[-ln(l-a)]273 (l-a)[-ln(l-a)]3'4 2/3 3/4 0,7 0,66
Нулевая скорость зародышеобразования при постоянном числе зародышей Двухмерный рост зародышей Трехмерный рост зародышей l/2[-ln(l - a)]1/2 l/3[-ln(l - a)]1/3 (1 -a)[-ln(l -a)]172 (1 -a)[-ln(l -a)]273 1/2 2/3 0,77 0,7
36
Новый справочник химика и технолога
Продолжение таблицы 2.1.18
Характер образования и роста зародышей Тип и симметрия процесса Стадии, определяющие скорость процесса
Скорость диффузии реагента через слой продукта реакции
g(a) Ла) т п
Применение уравнения Иохансона — Меля — Аврами — Ерофеева — Колмогорова без выделения возможных стадий - (kt »1) (&<1) 1/5[-1п(1 - а)]1/5 (kt~ 1) (1-а)[-1п(1-а)]4'5 2/3 3/4 4/5 0,73 2/3 3/4 0,64 0,68
Мгновенное зародышеобразование. Реагирующая частица изменяется гомотетично Двухмерная симметрия Простая трехмерная симметрия Трехмерная симметрия (уравнение Гистлинга — Браунштейна) Трехмерная симметрия Противодиффузия (уравнение анти-Яндера) Диффузия с уменьшением активности реагента (1 -а)1п(1 -а) + а 3/2[1 - (1 - а)1/3]2 2/3[1 —2/За — (1—<х)2/3] к In? 2/3 [(1 + а)1/3-1]2 2/3 [(1 - а) 1/3 - I]2 ~[-1п(1-а)Г’ (l-ani-d-a)173]-1 [(1 -а)1/3- I]’’ [(1 + а)2/3[(1 +а)ю-1]’1 [(1 — а)4/3[(1 — а)13]’
Применение уравнения Иохансона — Меля — Аврами — Ерофеева — Колмогорова без выделения возможных стадий
Мгновенное зародышеобразование. Реагирующая частица изменяется гомотетично Двухмерная симметрия Простая трехмерная симметрия Трехмерная симметрия (уравнение Гистлинга -— Браунштейна) Трехмерная симметрия Противодиффузия (уравнение анти-Яндера) Диффузия с уменьшением активности реагента 1/2[1 - (1 - а),/2] 1/3[1 - (1 - а)1/3] (1-а)1/2 (1-а)2/3 0 0 1/2 2/3
Таблица 2.1.19
Уравнения кинетики топохимических реакций и области их применения [4]
Уравнение* Области применения уравнения
l-(l-a)1/3 = kt Описание кинетики реакций разложения перманганатов, карбонатов, оксалатов, гидроксидов
-ln(l-a) = ktn Описание кинетики реакций термического разложения и взаимодействия твердого тела с газом. Описание кинетики процессов кристаллизации
[1-(1-а),/3 ]2 = kt Уравнение Яндера. Описание кинетики реакции твердофазного синтеза равновеликих сферических зерен при параболическом законе роста слоя продукта
Химическая кинетика и диффузия
437
Продолжение таблицы 2.1.19
Уравнение* Области применения уравнения
1--|а-(1-а)2//3 = kt Уравнение Гистлинга — Браунштейна
z-1 V 7 z-1 Уравнение Картера — Валенси. Описание кинетики реакций взаимодействия в порошках между твердыми и между твердыми и жидкими веществами
|jl- a) 1/3 -ij =kt Уравнение Журавлева
[l-(l-a)'l/3]2 = ki Уравнение Кригера — Циглера
1 2 (1 + az)273 —+—a-- — = kt z 3 z Уравнение анти-Валенси — Картера
—+|-a-(l + az)273 = kt Уравнение анти-Гистлинга — Браунштейна
1 l-(l-a) - — = kt am \-m Универсальное уравнение Акулова
’ z — отношение объемов продукта реакции и реагента; т — фактор гетерогенности: т = 1 для гомогенной реакции и т < 1 для гетерогенной.
Влияние температуры и давления на кинетику топохимических реакций
Общие законы химической кинетики, отражающие влияние температуры и давления на скорость реакций, своеобразно преломляются в топохимических процессах. Здесь обнаруживаются эффекты, которые не встречаются в гомогенной кинетике и природа которых до конца не раскрыта. В случае обратимых реакций ряд эффектов (Смита — Топли, Завадского — Бретшнай-дера, некоторые случаи компенсационного эффекта) могут быть интерпретированы с единых позиций, принимая во внимание степень удаления изучаемой системы от равновесного состояния.
Закономерность Завадского — Бретшнайдера
Если обратимое термическое разложение твердого вещества проводить в вакууме в области АГ, затем в атмосфере газообразного продукта реакции в соответствующей области АГ при одном давлении р = const, потом при более высоком постоянном давлении р = const и т. д., то эффективная энергия активации Еа закономерно возрастает при условии, что реакция протекает в кинетическом режиме и не осложнена побочными процессами (табл. 2.1.20). Эта закономерность известна как правило Завадского — Бретшнайдера [14, 27]. Увеличение энергии активации Е& как правило, компенсируется изменением А и сравнительно слабо зависит от изменения Tcv.
Аналогичная закономерность обнаружена для многих неорганических веществ: карбонатов серебра и кадмия; гидроксидов магния и кадмия; пероксида лития; гидроксосульфатов алюминия и аммония; кристал
логидратов хлорида лития; сульфатов меди, кальция и бериллия; гидрофосфата кальция; карбоната натрия; оксалата цинка [12].
Таблица 2.1.20
Термическое разложение СаСО3 в атмосфере углекислого газа [27]
Гсо2> мм Рт- ст-* 1 5 10 20 30 45
Еа, кДж/моль 186 289 390,0 576 629 1534
* 1 мм рт. ст. - 133,3 Па.
Закономерность Завадского — Бретшнайдера связана с проявлением одного из вариантов так называемого компенсационного эффекта химической кинетики. Его сущность заключается в том, что в уравнениях, выражающих зависимость константы скорости к или скорости v химической реакции от температуры:
( Е ) ( Е \
к = А. ехр---— или v = Av exp---- ,
к ( rt) I rt)
изменение аррениусовской энергии активации Ек и Ev, вызванное воздействием различных факторов на реакционную способность веществ, компенсируется изменением предэкспоненциального множителя Ак и Av. Взаимосвязь между этими параметрами выражается эмпирическим уравнением типа:
1g А = а + ЬЕ,
где anb — эмпирические константы, а А и Е могут быть представлены как Ак и Ек, так и Av и Ev.
438
Новый справочник химика и технолога
Компенсационный эффект иногда называют также изокинетической зависимостью, изопараметрическим соотношением, 0-правилом [14].
Компенсационный эффект проявляется не всегда. Обычно он наблюдается при разложении карбонатов, гидроксидов, комплексных соединений, дегидратации кристаллогидратов, при различных термических превращениях твердых веществ в изо- и неизотермических условиях [27].
Отчетливо компенсационный эффект проявляется при термораспаде в изобарических условиях, когда переход от низкого давления газообразного продукта реакции к высокому вызывает увеличение Е. Однако и в этом случае имеются исключения. В процессе термического разложения карбоната железа в атмосфере диоксида углерода вследствие протекания побочных процессов взаимосвязанного изменения Ей Л не наблюдается [27].
Эффект Смита — Топли
Если удаление воды из кристаллогидратов проводить при постоянной температуре, то с ростом давления водяного пара скорость реакции проходит последовательно через минимум и максимум (рис. 2.1.20) вместо того, чтобы монотонно уменьшаться.
Рис. 2.1.20. Схематическое изображение эффекта Смита — Топли, наблюдаемого при дегидратации кристаллогидратов
Вероятная причина этого эффекта заключается в образовании в ходе реакции плотного слоя твердого продукта реакции, сохраняющего структуру кристаллической решетки исходной фазы и оказывающего сопротивление удалению водяного пара из зоны реакции. На участке АБ (рис. 2.1.20) наблюдается увеличение толщины тормозящего слоя и соответствующее уменьшение скорости реакции. На участке БВ происходит разрыхление плотного слоя и уменьшение его толщины вследствие кристаллизации твердого продукта реакции. Процесс сопровождается увеличением скорости реакции. На участке ВГ толщина тормозящего слоя стабилизируется, и дальнейшее изменение скорости реакции от давления приходит в соответствие с общими правилами химического равновесия.
Кроме дегидратации кристаллогидратов эффект Смита — Топли обнаружен для термического разложения карбонатов и гидрокарбонатов в атмосфере диоксида углерода и паров воды [24].
Реакции топохимического разложения твердых веществ
Формально-кинетический анализ процессов топохимического разложения осуществляется на основании теоретических зависимостей (степенных, экспоненциальных и др.), построенных на предположениях о различных законах образования и роста ядер [5].
Наиболее часто для анализа процессов разложения используется уравнение Ерофеева:
ot = l-exp(-fc"), (2.1.2.6)
где a — доля прореагировавшего вещества; к — константа скорости; t — время разложения, п — кинетический параметр.
Константа скорости реакции к (размерность [Г1]) в уравнении (2.1.2.6) может быть вычислена по формуле Саковича:
к = пК'/п. (2.1.2.7)
Значение показателя степени п позволяет судить об условиях образования и роста зародышей и механизме реакции:
Скорость пропорциональна массе непрореагировавшего вещества п = 1
Реакция находится в кинетической области п > 1
Скорость реакции мало зависит от скорости зародышеобразования и определяется ростом существующих ядер п »1
Реакция лимитируется диффузией. Чем больше отклонения п от единицы, тем значительнее влияние диффузионных процессов п < 1
Дифференциальная форма уравнения (2.1.2.6) имеет вид:
^Lzz^^-lnfl-ay^jfl-a). (2.1.2.8)
Из уравнения (2.1.2.8) можно получить приближенное уравнение:
— = каа(\-а)ь, (2.1.2.9)
dt
где а и b — постоянные, соответствующие определенным значениям л [5]:
п а b
2 1/2 0,774
3 2/3 0,700
4 3/4 0,664
5 4/5 0,642
оо 1 0,556
Химическая кинетика и диффузия
439
Для описания автокаталитических реакций топохимического разложения применяется уравнение Праута — Томпкинса:
= kt + const.
(2.1.2.10)
Уравнение (2.1.2.10) получено в предположении о цепном механизме реакции в твердом веществе за счет растрескивания кристаллов на границе с продуктом разложения. В дифференциальном виде уравнение (2.1.2.10) выглядит следующим образом:
^ = Аа(1-а). (2.1.2.11)
Уравнение (2.1.2.10) является частным случаем уравнения Ерофеева и пригодно для описания кинетики периода ускорения реакции. В период спада может быть применено уравнение сокращающейся сферы:
а = 1-
R-krf Я J
(2.1.2.12)
где R — начальный радиус сферической частицы; к — константа скорости.
Для описания автокаталитического разложения твердых веществ (в частности, перхлората аммония) можно применять общее уравнение вида:
— = А] (1-а)" + А2а(1-а)". (2.1.2.13)
dt
Уравнение (2.1.2.6) в виде:
-ln(l -a) = kf
(2.1.2.14)
известно как уравнение Иохансона — Меля — Аврами — Ерофеева — Колмогорова [6] (в отечественной литературе его называют уравнением Аврами — Колмогорова или Аврами — Ерофеева). В уравнении (2.1.2.14) к — обобщенная константа скорости, п — показатель степени.
Практически все уравнения изотермической кинетики можно выразить единым соотношением [6]:
где к — константа скорости; р — безразмерный фактор, обычно близкий к 1; z — величина, определяемая механизмом взаимодействия; т — величина, зависящая как от механизма, так и от формы реагирующих частиц.
Величина т, формально сходная с порядком реакции, называется индексом реакции. Различным кинетическим моделям соответствуют различные комбинации параметров z, р и т [6]:
Z ₽ т Физическая модель
1 1 2/3 Реакция лимитируется процессами на границе раздела фаз, реагенты состоят из сферических частиц
1 1 1/2 То же, но реагенты состоят из игл
1 1 0 То же, но реагенты — очень тонкие диски
1/2 1 0 Диффузионные модели
1/2 1 0,29 То же
1/2 1 2/3 »
1/2 1 0,43 »
> 1 1 1 Механизм зародышеообра-зования
Среди реакций термического разложения твердых веществ принято различать эндотермические (разложение гидратов, аммиакатов, гидроксидов, оксидов, карбонатов и т. п.) и экзотермические (разложение азидов, оксалатов, формиатов, перманганатов, перхлоратов, стифнатов, фульминатов и т. п.).
Для реакций разложения часто наблюдаются кинетические кривые сигмоидного типа (рис. 2.1.21) [7].
Рис. 2.1.21. Типичные кривые а(0, встречающиеся в реакциях термического разложения твердых веществ:
а) сигмоидная кривая с периодом индукции; б) сигмоидная кривая без периода индукции; в) период ускорения значительно короче периода замедления; г) вторая часть кривой имеет сигмоидную форму
Анализ экспериментальных данных [5, 7] свидетельствует о том, что в реакциях разложения часть кинетической кривой а(/), примыкающая к началу координат t - 0 в отсутствие периода индукции или к точке t = Zo> отвечающей концу периода индукции, описывается двумя различными законами: степенным и экспоненциальным (табл. 2.1.21).
На обобщенной кривой термического разложения для случая нулевой начальной скорости (рис. 2.1.22) можно выделить следующие участки [7]:
440
Новый справочник химика и технолога
1. Начальный период I — относится к значениям степени превращения 0,01 < а < 0,05. Изменения на этом участке могут быть обусловлены либо десорбцией газа, либо термическим разложением нескольких атомных слоев, прилегающих к поверхности твердого реагента.
2. Участок II — относится к периоду индукции, в течение которого протекание реакции весьма ограниченно.
3. Участок III — относится к периоду ускорения. По достижении критического времени /0 скорость реакции быстро возрастает, достигая максимального значения в точке перегиба (а„ 7,).
4. Период спада IV — характеризует быстрое уменьшение скорости реакции, стремящейся к нулю в конце реакции. На этой стадии прореагировавшая часть а/ может соответствовать (или нет) полному превращению исходного твердого реагента.
Таблица 2.1.21
Классификация топохимических реакций разложения твердых веществ [7]
Твердый реагент Тип кривых (рис. 2.1.21) Закон разложения в начале периода ускорения
Азиды Ме(П) (N3)2: Ba(N3)2 Sr(N3)2 Ca(N3)2 Азид меди(1) CuN3 Оксалат серебра(1) Ag2C2O4 (состаренный) Стифнат бария(П) моногидрат BaC6HN3O8 • Н2О (мелкие кристаллы) Стифнат свинца(П) PbC6HN3O8 (дегидратированный) Фульминат ртути(П) Hg(OCN)2 (состаренный) а Степенной закон а = l^(t-t^n и = 6-^8 п = 3 п = 3 п = 3 п = 3 -4 п = 2 п = 2 п - 3
Азид свинца(П) Pb(N3)2 (a-модификация, мелкие кристал лы 250 х 5 мкм) Оксалаты: свинца(П) РЬС2О4 (состаренный) серебра(1) Ag2C2O4 (свежеприготовленный) тория(1У) Th(C2O4)2 Перманганаты щелочных металлов и серебра(1) Фульминат ртути(П) Hg(OCN)2 (свежеприготовленный) б Экспоненциальный закон а = Сек‘
Азид свинца(П) Pb(N3)2 (a-модификация, кристаллы 2-4 мг, 7 -200-300 °C) Азид серебра(1) AgN3, кристаллизованный при 230 °C Оксалат лантана(Ш) Ьа2(С2О4)3 Кривые для степени превращения имеют небольшой период индукции и трансформируются друг в друга при изменении масштаба по времени в интервале температур 360-410 °C Оксалат ртути(П) HgC2O4 в Очень короткий период ускорения по экспоненциальному закону, затем действует закон сжимающейся сферы Очень короткий период ускорения А затем действует закон сжимающегося куба Экспоненциальный период ускорения (до а = 0,15), затем действует период замедления Короткий период ускорения (а - 0,15), описывающийся моделью расширяющихся дисков при постоянном числе потенциальных центров зародышеобразования; затем действует закон сжимающейся сферы
Азид калия KN3 Алюмогидрид лития ЫА1Н4 (тетрагидридоалюминат(Ш) лития) Фульминат ртути(П) Hg(OCN)2 (состаренный) г Быстрое начальное газовыделение (1 % разложения, 50-100 монослоев) Начальный участок отвечает разложению на глубину 20-30 монослоев Начальное газовыделение соответствует а = 0,004
Химическая кинетика и диффузия
441
Рис. 2.1.22. Обобщенная форма кривой термического разложения твердых веществ
Вещества, разлагающиеся топохимически [5, 7, 8, 10, 13]:
- перхлораты: NH4CIO4, NOC1O4, NO2C1O4;
- карбонаты: СаСОз, MgCO3, S1CO3, ВаСОз, CdCO3, РЬСОз, ZnCO3, CaMg(CO3)2, La2(CO3)3, Y2(CO3)3, EuCO3;
- оксиды: Ag2O, HgO, PbO2, Pb3O4, MnO2, Li2O2, CaO2, SrO2, BaO2;
- гидроксиды: Mg(OH)2, Cd(OH)2, Zn(OH)2;
- оксалаты: Ag2C2O4, NiC2O4, HgC2O4, Y2(C2O4)3, Th(C2O4)2, La2(C2O4)3, UO2C2O4, PbC2C>4;
- перманганаты: LiMnO4, NaMnO4, KMnO4, RbMnO4, CsMnO4, AgMnO4, Ba(MnO4)2;
- нитраты, в частности, Pb(NO3)2;
- азиды: KN3, NaN3, RbN3, CsN3, AgN3, CuN3, Ca(N3)2, Ba(N3)2, Sr(N3)2, Pb(N3)2;
- дихроматы, в частности, (NH4)2Cr2O7;
- сульфиды: Ag2S, FeS2, Сщ 75S, Cu)<7gS, Cu1>8S;
- аммиакаты: LiCl • NH3, LiCl • 4NH3, LiBr • NH3, CuBr • NH3, CuBr • 3NH3;
- врывчатые вещества: Hg(OCN)2, 1,3,5-триазидо-2,4,6-тринитробензол (C6N]2O6), стифнат свинца(П) моногидрат (PbC6HN30g • Н2О), стифнат бария моногидрат (BaC6HN30g • Н2О), стифниновая кислота (C6H3N3O8), пикриновая кислота (C6H3N3O7), пикраты калия (KC6H2N3O7) и аммония (NH4C6H2N3O7).
Топохимический характер имеют процессы дегидратации веществ:
КНС2О4 • 0,5Н2О, NiC2O4 • 2Н2О, МпС2О4 • 2Н2О,
Си(НСОО)2 • 4Н2О, CaSO4 • 2Н2О, CuSO4 • 5Н2О,
ZnSO4 • 7Н2О, MnSO4 • 4Н2О, NiSO4 • 7Н2О,
MgSO4 • 7Н2О, La2(SO4)3 • 8Н2О, Ce2(SO4)3 • 8Н2О,
СаСОз • 6Н2О, КНСО3, NaHCO3, Na2CO3 • 3NaHCO3, CoCl2 • 6H2O, Na2CO3 • NaHCO3 • 2H2O, La2(CO3)3 • 8H2O, Y2(CO3)3 • 4H2O, Na5P3O9 • 6H2O, Na3CuP3O10 • 12H2O, Na3MnP3Oio • 12H2O, MnHPO4 • 3H2O, ThF4-2,5H2O, UF4 • 2,5H2O, цеолитов NaA и NaX.
При введении добавок, например МпО2, топохимический характер приобретает разложение таких солей, как КС1О3, NaC103, КС1О4, NaC104 и др.
Разложение перхлората аммония
Разложение перхлората аммония (ПХА) в зависимости от условий, в которых оно протекает, относится к типу Ата Браз + Враз + Дгаз +... ИЛИ Ата Бгв + Враз "Ь Дгаз + • • •
Существует три основных температурных области разложения ПХА: низкотемпературное разложение (200-300 °C), высокотемпературное разложение (350-400 °C) и дефлаграция (начинается при 450 °C) [8].
Для описания кинетики разложения ПХА в каждой из температурных областей применяются различные кинетические уравнения (табл. 2.1.22).
Таблица 2.1.22
Уравнения, используемые для описания кинетики разложения перхлората аммония
Название уравнения Вид уравнения Область применения
Низкотемпературное разложение (200-300 °C)
Праута — Томпкинса Мономолекулярного разложения Степенного вида Аврами — Ерофеева 1g—-— = А, + с и 1g—-— = k2 + с Pf-P Pf-P -^>(pj - p) = k3t + с p = ktn Г 11 -ln(l-a)" - kt Период ускорения (&i) и замедления (£2) Период замедления Период ускорения
Высокотемпературное разложение (350-400 °C)
l-(l-a)« = kt da — — k dt я - 3 при описании кинетики разложения кристаллов и таблеток а = 0,1 4-0,3
442
Новый справочник химика и технолога
Низкотемпературное разложение ПХА. Результаты анализа продуктов разложения ПХА сведены в табл.
2.1.23.
Таблица 2.1.23
Состав продуктов низкотемпературного разложения перхлората аммония [8]
Г, °C о2 n2 N2O Cl2 + С1О2 1Г HNO3 НС1 NO
225 0,51 0,126 0,362 0,426 0,155
230 0,48 0,12 0,328 0,427 0,155
235 0,51 0,09 0,348 0,43 0,143
245 0,553 0,061 0,387 0,423 0,141
250 0,61 0,055 0,37 0,39 — 0,14 0,15 0,006
255 0,567 0,063 0,373 0,421 0,137
260 0,567 0,063 0,391 0,420 0,152
265 0,567 0,063 0,391 0,387 0,143
270 0,556 0,062 0,453 0,408 0,141
275 0,571 0,062 0,437 0,39 0,135
300 0,53 0,052 0,37 0,37 — 0,12 0,21 0,018
325 0,57 0,063 0,365 0,285 — 0,25 0,28 0,032
Примечание. Состав продуктов дан в числах молей, отнесенных к молю разложенного перхлората аммония. Предполагается, что HNO3 и НС1О4 образуются в сравнимых количествах.
В табл. 2.1.24 приведены значения констант скорости разложении ПХА, полученные в результате применения уравнения Праута — Томпкинса и уравнения мономолекулярного разложения. Значения энергии активации для kb k2 и k3, полученные с применением всех перечисленных формул, находятся в пределах:
Кристаллическая форма ПХА Г, °C Энергия активации, кДж/моль
Орторомбическая до 240 112,1-120,5
Кубическая 240-275 71,55-96,23
Таблица 2.1.24
Значения констант скорости разложения перхлората аммония [8]
Т,°С 105/Г, К~' Значение констант в уравнении Праута — Томпкинса*1 Константа мономолекулярного разложения 104£з*2
103 103А2
215 204,9 1,18 3,20 3,03
220 202,8 1,47 4,59 3,99
225 200,8 1,79 7,54 5,99
— 2,07 7,38 5,5
230 198,8 2,46 9,48 —
— 2,66 8,97 8,71
235 196,9 3,27 10,74 8,29
Продолжение таблицы 2.1.24
Г, °C 105/Г, К’1 Значение констант в уравнении Праута — Томпкинса*1 Константа мономолекулярного разложения 1оЧ’2
103 юЧ
235 — 3,10 10,5 9,91
240 194,9 4,92 12,8 11,5
— 5,03 13,2 11,4
245 193,1 3,84 5,45 4,74
— 3,10 5,33 5,15
250 191,2 5,05 8,61 7,42
— 5,76 6,34 8,71
255 189,4 6,64 9,14 7,98
— 6,98 8,2 7,01
260 187,6 7,68 10,2 8,71
— 8,23 10,1 8,71
265 185,9 10,23 10,1 8,8
— 10,23 11,46 8,98
270 184,2 12,8 10,44 11,7
— 10,8 14,0 11,4
275 182,5 15,44 11,2 10,4
— 14,92 11,84 10,9
*’ 1g—-— = ^+с и 1g—-— = k2+c, где и k2 — Р/-Р Pf~P
константы скорости периодов ускорения и замедления реакции соответственно; pj — полное давление, достигающееся при разложении вещества на 28-30 %; р — текущее давление.
*2 -lg(P/-p) = ^ + c-
Химическая кинетика и диффузия
443
На практике кинетические данные могут быть описаны уравнением Аврами — Ерофеева в виде:
[-ln(l -a)]lz" = kt
вплоть до степеней превращения 90 % в области температур до 470 °C. Величина п может принимать значе
ния 4, 3, 2, за исключением ранних стадий разложения монокристаллов.
При разложении кубической формы ПХА показатель степени п близок к единице (табл. 2.1.25), что может быть объяснено мономолекулярной реакцией распада и переходом ее в диффузионную область.
Таблица 2.1.25
Кинетические характеристики термического разложения перхлората аммония [9]
Температура разложения, °C Пределы применимости уравнения Аврами— Ерофеева, а, % п Предэкспоненциальный множитель*1, мин1 Энергия активации*1
кДж/моль ккал/моль
214-336 0-12 4,5 1,6 • 1016 167,8 40,1
250-300 0-20 1,2 1,3 • 108 105,0 25,1
330-450*2 0-40 0,6 4,5 • Ю10 163,6 39,1
330-450 40-80 1,0 5,9 • 109 148,5 35,5
400-470 10-50 0,8 2,0 • 107 118,4 28,3
400-470 50-90 1,1 1,1 • 106 99,2 23,7
*’ Пересчитано по уравнению Саковича Z = nkxln.
*2 Остаток от низкотемпературной реакции.
Высокотемпературное разложение ПХА [8]. Для высокотемпературного разложения предложено уравнение реакции
2NH4C1O4 = С12 + 4Н2О + 2NO + О2.
В диапазоне температур 380-450 °C разложение подчиняется степенному закону: р = kf, где величина п < 1 и в зависимости от температуры принимает следующие значения:
Т,°С 380 400 420 440 450
п 0,5 0,5 0,66-0,67 0,86-0,98 0,78-0,82
Энергия активации в диапазоне температур 400-440 °C составляет 306 кДж/моль.
Для описания высокотемпературного разложения может быть использовано уравнение:
1 — (1 — a)» — kt, в котором значение п = 3 хорошо описывает результаты по кинетике разложения кристаллов и таблеток.
При разложении на воздухе порошка ПХА с размером частиц 50-100 мкм формально скорость реакции при a = 0,14-0,3 может быть описана уравнением нулевого порядка.
Энергия активации высокотемпературного разложения Еа, рассчитанная по уравнению первого порядка, зависит от размера частиц ПХА:
Размер частиц, мкм 28 56 80
Еа, кДж/моль 129,7 149,4 206,7
Влияние добавок на термическое разложение ПХА. Многие оксиды переходных металлов являются эффективными катализаторами разложения ПХА (табл. 2.1.26).
Таблица 2.1.26
Кинетические характеристики термического разложения перхлората аммония в присутствии оксидов металлов по уравнению Аврами — Ерофеева [9]
Добавка, (5 масс. %) 0 0 Предэкспоненциальный множитель, мин-1 Энергия активации
кДж/моль ккал/моль
Со2О3 214-245 1,4 7,41014 160,3 38,3
Сг2О3 260-309 1,1 8,0-109 125,9 30,1
CuO 239-270 2,2 51O20 223,8 53,5
Cu2O 250-270 1,6 l,4-1017 190,4 45,5
Fe2O3 250-296 1,2 2,6-1014 171,5 41,0
MnO2 208-229 1,6 2,41017 201,7 48,2
NiO 270-296 1,2 6,31017 205,9 49,2
V2O5 250-303 1,2 l,6-1015 182,8 43,7
Значения энергии активации разложения ПХА в присутствии ZnO, CuO, Fe2O3 (в том числе допирован-ных различными присадками) приведены в табл. 2.1.27.
Эффекты влияния оксидов на разложение ПХА можно оценить следующим образом [27]:
а) Cu2O, CuO, ZnO ускоряют как низко-, так и высокотемпературные реакции;
б) NiO и Сг2О3 ускоряют в основном низкотемпературное разложение;
в) МпО2 ускоряет главным образом высокотемпературное разложение;
г) А12О3, TiO2, Fe2O3 и V2O5 не оказывают влияния на разложение ПХА.
444
Новый справочник химика и технолога
Таблица 2.1.27
Энергия активации разложения перхлората аммония в присутствии оксидов металлов [8]
Катализатор Массовое соотношение Присадка Количество присадки, масс. % Энергия активации, кДж/моль, рассчитанная по уравнениям Г, °C
ПХА и катализатора (1) (2) (3) (4) (5) (6)
СиО 5 : 1 — — — — 125,5 — 162,8 —
1 : 1 — — — — 136,8 — 137,7 — 180-200
5 : 1 Сг2О3 1 — — 134,3 — 125,5 —
5 : 1 Li2O I — — 122,2 — 132,6 —
5 : 1 — — — — 179,5 — 171,1 —
СиО 1 : 1 5 : 1 Сг2О3 1 — — 168,2 172,4 — 176,6 178,2 — 200-320
5 : 1 Ы2О I — — 155,2 — 154,8 —
1 : 1 — — — 124,7 149 129,3 — 128
1 :5 — — — 115,5 119,2 121,3 — 129,3 240-270
Fe2O3 50: 1 — — — — — — — 129,3
20 : I 8: 1 — — — — — — 107,5 92 320-380
100: 1 — — — — — — — 116,7
50: 1 — — 135,1 135,1 131 94,1 — 123,4
16: I — — 128,5 86,2 124,3 141 — 139,3
1 : 1 — — 126,8 123 129,7 128 — —
ZnO 1 : 8 — — 121,3 142,3 103,3 — — — 215-235
16 : 1 А12О3 1 135,1 126,8 123 131 — 135,6
1 : 1 А12О3 1 129,7 — — — — —
16 : 1 Li2O 1 136,8 142,3 141,4 133,5 —- —
1 : 1 Li2O 1 142,7 — — — — —
* Расчетные уравнения:
(1) lg/ = EJRT + с; / — время начала реакции.
(2) lg/max = EJRT + с', tmaK — время достижения максимальной скорости реакции.
(3) lg[a/( 1 - a)] = k}t + с — уравнение Праута — Томпкинса, период ускорения реакции.
(4) lg[a/( 1 - a)] = k2t + с — уравнение Праута — Томпкинса, период замедления реакции.
(5) lg(l - a) = kf — уравнение Аврами — Ерофеева, п = 3.
(6) 1 - (1 - а) 1/3 = kt — уравнение стягивающегося куба.
Разложение нитрата свинцаЩ)
При разложении твердого Pb(NO3)2 образуется твердый оксид РЮ:
Pb(NO3)2 -► РЬО + 2NO2 + 0,5О2
Образование налета оксида наблюдается вначале на вершинах и ребрах кристаллов. Затем поверхность кристалла покрывается оболочкой оксида. В дальнейшем реакция распространяется в глубь кристалла.
При разложении нитрата свинца(П) в запаянной трубке при 360 °C и последующем охлаждении вновь образуется нитрат свинца(П), что свидетельствует об обратимости реакции. После плавления разложение резко интенсифицируется (табл. 2.1.28a).
Температура начала заметного разложения нитрата свинца(П) на воздухе равна 380 °C. В изотермических условиях при температуре несколько выше 380 °C процесс, хотя и очень медленно, протекает до полного раз
ложения нитрат-иона. Данные о продуктах термического разложения приведены в табл. 2.1.286.
При термическом разложении нитрата свинца(П) идут одновременно два процесса — диссоциация и автоокисление [25]. На первой стадии разложения, соответствующей степени денитрации 2/3, образуется оксонитрат состава 2РЬ0 • Pb(NO3)2. На второй и третьей стадиях разложения, соответствующих степеням денитрации 3/4 и 4/5, образуются соответственно оксонитраты состава ЗРЬО • Pb(NO3)2 и 4РЬО • Pb(NO3)2. Тетраоксонитрат разлагается с образованием РЬО, который является единственной твердой фазой в конечном продукте. Наличие в продуктах разложения РЬ4+ (табл. 2.1.286) свидетельствует об образовании в процессе реакции пероксонитрата и пероксидных соединений, образующихся в результате окисления РЬО газообразными продуктами разложения нитрат-иона.
Химическая кинетика и диффузия
445
Таблица 2.1.28а
Скорость разложения Xm/Xt нитрата свинца(П) при различных температурах, определенная по убыли массы (% разложившегося нитрата в минуту) [8]
350 °C 360 °C 370 °C 450 °C 550 °C 600 °C
t, мин Am/AZ t, мин t, мин t, мин kml&t t, мин t, мин
0 — 0 — 0 — 0 — 0 — 0 —
20 0,139 20 0,124 21 0,205 10 2,5 2 12 3 28
40 0,030 40 0,035 42 0,021 15 4,2 5 21,5 5 14
60 0 65 0,038 62 0,015 20 4,0 8,5 6 7 3,5
— — 105 0,007 102 0,013 25 2,7 13 1,5 10 0
— — 165 0,014 122 0,022 30 1,2 15 0 — —
Таблица 2.1.286
Продукты термического разложения нитрата свинца(П) [8]
Продукты разложения (по стадиям) — 100% Aw0 Массовая доля, % Цвет продукта
РЬобщ РЬ4+ no;
Исходный 0 62,5 0,08 37,3 Белый
1-я стадия 20,4 77,4 5,04 15,9 Желтовато-белый
2-я стадия 23,3 82,0 5,32 13,4 Желтовато-белый с очагами красной фазы
3-я стадия 27,8 88,0 2,80 7,1 Красновато-желтый
4-я стадия 32,8 91,0 Нет Нет Желтый
Разложение хлоратов и перхлоратов калия и натрия в присутствии катализаторов
Термическое разложение хлоратов и перхлоратов калия и натрия приобретает топокинетический характер при введении в их состав катализаторов. В присутствии катализаторов снижается температура начала разложения, увеличивается скорость и изменяется сам характер разложения. В качестве катализаторов разложения могут выступать соединения Fe, Мп, Cu, А1, РЬ и др.
Оксиды металлов по снижению каталитической активности по отношению к хлорату калия могут быть расположены в следующий ряд: Со2О3 > Сг2О3 > > MnO2 > Fe2O3 > Ni2O3 > CuO > TiO2. При нагревании хлоратов и перхлоратов калия и натрия с добавкой МпО2 (около 1 %) разложение солей происходит в твердом состоянии и подчиняется уравнению: а -1 - ехр(-Лт") (табл. 2.1.29).
Таблица 2.1.29
Параметры уравнения а = 1 - ехр(-Лг") топокинетического разложения хлоратов и перхлоратов в присутствии 1% МпО2 [10]
Вещество п Условия
а Г, °C
КС1О3 0,5 1,0 0,68-0,85 0,2 315-330 335
КС1О4 0,66 0,66-0,85 425-465
NaClO3 0,66 1,05 — 240 255
NaClO4 0,63 0,7-0,81 350-380
Экзотермические реакции разложения
Особенностью экзотермических реакций разложения является саморазогрев исходного вещества. Дополнительные трудности связаны с отсутствием достаточно полных характеристик микрокристаллических препаратов, используемых в кинетических исследованиях.
Основные параметры кинетики процессов экзотермического разложения некоторых веществ приведены в табл. 2.1.30.
446
Новый справочник химика и технолога
Таблица 2.1.30
Параметры экзотермических процессов разложения [5]
Вещество Условия и параметры кинетики разложения
Область а Область температур, °C Уравнение разложения Ea, кДж/моль Примечание
Фульминат ртути(П) Hg(OCN)2 <0,2 <0,35 — In da (da\ . — - kt + const dt dt 70J a-a0 = k(t-t^ ln(a-a0) = kt + const 127,1-134,6 106,2 112,9
Оксалат никеля(П) NiC2O4 < 0,058 <0,01 0,05 < а < 0,85 280-320 i S' ~ P p ~ P JI • । J2 p и X > 1 1 .1 I*2 - kxt 1/2 = k2t 199,0 152,2 137,5 137,5 В азоте То же В вакууме То же
Оксалат серебра Ag2C2O4 0,03 < а < 0,35 a = £(z-/0 3,2 < п < 3,5
Кинетика окисления металлов
Для характеристики процессов окисления металлов во времени используются: Aw — изменение массы образца, отнесенное к единице площади его поверхности (г/см2, мг/см2), 5 — толщина оксидной пленки (мкм), dm/dt или d&dt — скорость окисления в момент времени t.
Основные законы изотермического роста оксидной пленки во времени:
1. Линейный закон:
Aw = ^/ + ^j. (2.1.2.15)
2. Параболический закон:
(&m)2 = k2t + A2. (2.1.2.16)
3. Кубический закон:
(Дт)3 = k3t + А3. (2.1.2.17)
4. Логарифмический закон:
(Aw) = k4]g(at + b), (2.1.2.18)
или обратный логарифмический закон:
(Aw)’1 = Л 5 - k5\gt. (2.1.2.19)
В выражениях (2.1.2.15)-(2.1.2.19) Aw может обозначать количество кислорода, расходуемого на единичную площадь поверхности окисляемого металла; количество металла, перешедшего в оксид; толщину
оксидной пленки (при условии, что пленка однородна и плоскопараллельна поверхности металла); к{-к5 — константы скорости окисления; Ау-А5 — постоянные.
Общая кривая окисления может сочетать в себе две или несколько зависимостей. Металл или сплав, например, может начинать окисляться по параболическому закону с дальнейшим переходом окисления по линейному закону. Подобная зависимость называется пара-линейной.
Применимость той или иной временной зависимости к конкретному металлу или сплаву во многом зависит от толщины образовавшейся пленки, то есть от времени и температуры. При низких температурах для тонких оксидных пленок преобладают логарифмические и обратно-логарифмические закономерности (табл. 2.1.31).
Сопоставление экспериментальных и расчетных данных свидетельствует о возможности применения уравнений (2.1.2.15-2.1.2.19) для прогнозирования образования поверхностной оксидной пленки на металлах (табл. 2.1.32).
Константы скорости окисления (табл. 2.1.33, 2.1.34) растут с повышением температуры, подчиняясь уравнению Аррениуса:
(Е к = &пехр—— , °
где Еа — энергия активации, R — газовая постоянная, Т — абсолютная температура.
Химическая кинетика и диффузия
447
Таблица 2.1.31
Законы кинетики окисления металлов*[15]
Металл Температура, °C 100 200 300 400 500 600 700 800 900 1000 1100 1200
Mg 1'1 1 г 1 Г ' 1 Г 1 1 1 1 лог. пар. паралин. лин.
Са лог. пар. лин. лин.
Се лог лог-лог лин уск
Th пар. лин. лин.
и пар. паралин. лин.-уск
Ti лог куб. куб. паралин. паралин.
Zr лог куб куб. куб. куб. лин
Nb пар. пар. паралин. лин. лин. уск. асимпт.
Та лог обр.-лог. пар. паралин лин. лин. замедл.
Мо пар. паралин. паралин. лин. лин.
W пар пар паралин паралин паралин.
Fe лог лог. пар. пар. пар. пар. пар. пар.
Ni лог лог. куб. пар. пар. пар
Си лог. куб. пар. пар. пар. пар.
Zn лог. лог. пар. пар.
Al лог. обр.-лог (лог), пар (асимпт )(лин )
Ge пар. паралин
* Обозначения: асимпт. — асимптотический, куб. — кубический, лин. — линейный, лог.— логарифмический, обр.-лог. — обратно-логарифмический, пар. — параболический, паралин. — паралинейный, уск. — ускоренный, замедл. — замедленный, лог-лог. — двойной логарифмический, лин.-уск. — линейно-ускоренный.
Таблица 2.1.32
Вычисленные и измеренные константы скорости образования поверхностной пленки на металлах [16]
Металл Газовая фаза Давление рх, мм рт. ст. Состав поверхностной пленки Г, °C Значение к, г см 1 • с 1
эксперимент расчет
о2 3,0-10 4 Си2О 1000 2,2 • 10’9 2,1 • 109
о2 2,3 103 Си2О 1000 3,1 • ю9 3,4 • 10’9
Медь О2 1,5-102 Си2О 1000 4,5 • 10“9 4,8 • 10’9
О2 8,3-10~2 Си2О 1000 6,2 • 10’9 6,6 • ю 9
ь 0,06 Cui 195 3,4 • 10~10 3,8 • 10~10
Серебро S Ag2S 220 1,6-10“* 2,4 • 10 6
Вг2 0,23 AgBr 200 3,8- 10'” 2,7 • 10'11
Таблица 2.1.33
Параметры кинетики окисления вольфрама [15]
Г, °C Константа скорости окисления Максимальная толщина внутреннего слоя, см Период образования внутреннего слоя половинной толщины, с
на параболическом участке, г • см"4 • с-1 на линейном участке, г • см'2 • с'1
700 2,5 • Ю 9 4,0 • IO'7 2,0 • 10'3 1490
800 3,0- 10~8 3,2 • 10^ 3,1 • 10“3 280
900 ю-7 6,0 • 10 6 5,5 • 10~3 264
1000 7,3 • 10’7 1,8- 10"5 1,35 • 10'2 214
448
Новый справочник химика и технолога
Таблица 2.1.34
Экспериментальные данные по кинетике окисления металлов [15, 16, 17]
Металл (сплав) Параметры кинетики окисления Условия эксперимента
Закон окисления Энергия активации Е кДж/моль Константа скорости к окисли-тельная среда Г, °C давление, МПа
значение размерность
Be (А/и)2 = kt 35 500 210 300 313 500 1,8 • 1012 3,5 • 10'3 23,6 г2 • см 4 • с'1 о2 350-700 750-900 0,01
N2 725-925
Cd (Ат)2 = kt 1,0 Ю'9 г2 • см 4 • с'1 Воздух 500 0,1
Со (Ат)2 = kt 20 000 65 000 4,1 • 10^ 6,4- 104 г2 • см 4 • с'1 400-700 700-1200
Сг (Ат)2 - kt 156 700 3 • 106 г2 • см 4 • с"1 о2 700-1100 0,01
Си (Ат)2 = kt 20 140 37 700 1,5 • 10'5 0,266 г2 • см-4 • с'1 Воздух 300-550 550-900 0,1
Fe (Ат)2 = kt 33 000 36 600 0,37 0,11 г2 • см-4 • с'1 Воздух 500-1100 400-600 0,1
Mg (МА-1) Am ~ kt 126 300 3,369- 105 мг • см'2 • мин'1 Воздух 400-550
Mg (МЛ-4) 179 200 1,063 • Ю10
Mg (МЛ-5) 176 800 1,469- Ю10
Mg 211 100 1,7- 106 2 „ -2 -1 г •см -с о2 475-575
Мп (Ат)2 = kt 28 300 1,95 • 10 3 г2 • см 4 • с'1 Воздух 400-1200 0,1
Nb (Ат)2 = kt 114 500 106 200 2,6 • 10'5 8 • 10'8 г2 • см 4 • с'1 о2 200-375 500-800 0,01
Ni (Ат)2 = kt 45 000 41 200 68 300 0,032 8 • 10"4 1,2 • 102 г2 • см 4 • с'1 Воздух 750-1240 400-850 900-1050 о,1
Si (Ат)2 = kt 43 000 1,13 • 10 4 г2 • см 4 • с'1 Воздух 1200-1360 0,1
Sn (Ат)2 = kt 20 000 1,7- 10'5 г2 • см 4 • с'1 Воздух 325-450 0,1
Ta (Ат)2 = kt 114 500 164 700 3,5 • Ю'4 1,4- 10-4 г2 • см-4 • с'1 о2 250-450 600-850 0,01
Ti (Ат)2 = kt 188 100 151 700 0,16 3,8 ’ 10 5 г2 • см-4 • с'1 о2 n2 550-850
Th (Ат)2 = kt 101 600 0,92 • 10'5 г2 • см4 • с'1 n2 670-1250
U (Ат)2 = kt 106 600 64 800 3,2 • 10 4 6,2 • 10^ г2 • см-4 • с'1 n2 550-750 775-900 0,1
V (Ат)2 = kt 128 300 131 300 1,3 • Ю3 0,94- 10'5 г2 • см 4 • с'1 О2 N2 400-600 600-900 0,01
Zn (Ат)2 = kt 2,3 • 10'13 1,9- 10'” г2 • см4 • с'1 О2 400 430
Zr (Ат)3 = kt 47 200 0,012 г3 • см 6 • с'1 О2 575-950
38 000 42 700 2,8- 10'5 0,001 350-650
(Ат)2 = kt 39 200 52 000 48 000 5 • 10'5 3,2 • 10'3 7,8 • 10'3 г2 • см-4 • с'1 n2 400-825 860-1045 975-1640 0,01 0,001-0,04 0,1
Химическая кинетика и диффузия
449
Литература
1. Герасимов Г.Н. Топохимические реакции. Химическая энциклопедия: в 5 т. Т. 4. М.: Большая Российская энциклопедия, 1995. С. 1216-1218.
2. Кузнецова Т.В., Кудряшов И.В., Тимашев В.В. Физическая химия вяжущих материалов: Учебник для хим.-технол. спец, вузов. М.: Высш, шк., 1989. 384 с.
3. Шестак Я. Теория термического анализа: Физико-химические свойства твердых неорганических веществ / Пер. с англ. М.: Мир, 1987. 456 с.
4. Кононюк Ф.В. Модели реакций твердофазного синтеза в смесях порошков И Гетерогенные химические реакции и реакционная способность. Минск: Наука и техника, 1975. С. 93-115.
5. Янг Д. Кинетика разложения твердых веществ/ Пер. с англ. К.А. Печерской. Под ред. Б.В. Ерофеева. М.: Мир, 1969. 263 с.
6. Третьяков Ю.Д. Твердофазные реакции. М.: Химия, 1978. С. 192.
7. Барре П. Кинетика гетерогенных процессов. / Пер. с франц. Н.З. Ляхова. Под ред. В.В. Болдырева. М.: Мир, 1976. С. 91.
8. Джекобс П.У.М., Уайтехид Х.А. Разложение и горение перхлората аммония И Механизм, кинетика и катализ термического разложения перхлората аммония (переводы с англ.). Новосибирск: Наука, Сиб. отд., 1970. С. 5-141.
9. Шидловский А.А., Шмагин Л.Ф., Буланова В.В. Влияние некоторых добавок на термическое разложение перхлората аммония // Изв. высш. учеб, заведений. Сер. химия и хим. технология. 1965. Т. 8, вып. 4. С. 533-538.
10. Хорунжий Б.И., Ильин К.Г. Твердофазное разложение хлоратов и перхлоратов калия и натрия в присутствии двуокиси марганца И Известия высш, учеб, заведений. Сер. химия и хим. технология. 1972. Т. 15, вып. 12. С. 1886-1888.
11. Розовский А.Я. Кинетика топохимических реакций. М.: Химия, 1974. 224 с.
12. Продан Е.А., Павлюченко М.М., Продан С.А. Закономерности топохимических реакций. Минск: Наука и техника, 1976. 264 с.
13. Андреев К.К. Термическое разложение и горение взрывчатых веществ. М.: Наука, 1966. С. 76.
14. Продан Е.А. Неорганическая топохимия. Минск: Наука и техника, 1986. 134 с.
15. Кубашевский О., Гопкинс Б. Окисление металлов и сплавов / Пер с англ. 2-е изд. М.: Металлургия, 1965. С. 64.
16. Ванюков А.В., Зайцев В.Я. Теория пирометаллургических процессов. М.: Металлургия, 1973. 504 с.
17. Маколкин И.А. Кинетика газовой коррозии магниевых сплавов // Журн. прикл. химии. 1957. Т. 30, вып. 10. С. 1542-1547.
18. Гетерогенные химические реакции / Под ред. Е.А. Продана. Минск: Наука и техника, 1979. 184 с.
19. Чеботин В.Н., Перфильев М.В. Электрохимия твердых электролитов. М.: Химия, 1978. 312 с.
20. Самораспространяющийся высокотемпературный синтез: теория и практика. Черноголовка: Территория, 2001. 432 с.
21. Мержанов А.Г. Твердопламенное горение. Черноголовка: ИСМАН, 2000. 224 с.
22. Мержанов А.Г. Процессы горения и синтез материалов / Под ред. В.Т. Телепы, А.В. Хачояна. Черноголовка: ИСМАН, 1998. 512 с.
23. Продан Е.А. Топохимия кристаллов. Минск: Наука и техника, 1990. 245 с.
24. Продан Е.А. Общие закономерности термического разложения кристаллогидратов неорганических соединений. Плоцк: Ин-т химии, 1979. 21 с.
25. Маргулис Е.В. О термическом разложении нитрата свинца // Журн. неорган. химии. 1972. Т. 17, вып. 1.С. 43 ^7.19.
26. Kohlschiitter V.Z. // Anorg. Allg. Chem. 1919. Bd. 105, № 1/2. S. 1-25.
27. Zavadski J., Bretsznajder S.Z. // Elektrochem. 1935. Bd. 41, №4. S. 215-223.
2.1.2.2. Диффузионная кинетика
(проф., д.х.н. Е.А.Власов)
В данном разделе приводятся краткие сведения о диффузионной кинетике гетерогенных каталитических процессов с участием твердой фазы (катализатора и газовой фазы) реакционной смеси. Каталитический процесс реализуется путем пропускания исходной газовой смеси через слой гранул катализатора различной формы (сферической, цилиндрической и т. д.).
Гетерогенный каталитический процесс можно представить состоящим из пяти последовательных этапов, четыре из которых (I, И, IV, V) имеют физическую природу, а один (III) — химическую (рис. 2.1.23). Первый этап включает диффузию молекул исходных компонентов к наружной поверхности гранулы катализатора через пленку газа (диффузионный слой, сформировавшийся у твердой поверхности), осуществляемую за счет молекулярной (1М) и конвективной (1К) диффузии. На втором этапе происходит диффузия молекул исходных компонентов в порах гранул катализатора, которая реализуется путем молекулярной (2М) и кнудсеновской (2Кн) диффузии. Третий этап состоит из адсорбции молекул (атомов) исходных компонентов на поверхности пор катализатора, сопровождающейся разрывом связей и перегруппировкой атомов в молекулах исходных компонентов с образованием молекул продуктов реакции. Четвертый этап включает диффузию молекул продуктов реакции в порах к наружной поверхности гранул катализатора, которая реализуется путем молекулярной (4М) и кнудсеновской (4Кн) диффузии. Пя
450
Новый справочник химика и технолога
тый этап — диффузия молекул продукта через пленку газа в ядро газового потока, осуществляемая за счет молекулярной (5М) и конвективной (5К) диффузии. Одновременно с диффузионными потоками реагирующих молекул и продуктов реакции происходит тепло-перенос внутри гранул и теплообмен между поверхностью гранул и газовым потоком [1-5]. При условии стационарности, т. е. при отсутствии накопления вещества (с) и теплоты (Q) — dc/dt = 0 и dQ/dt = 0, — скорость гетерогенного каталитического процесса будет
равна наименьшей скорости одного из пяти этапов. Если наименьшей является скорость первого или пятого этапа, то гетерогенный каталитический процесс протекает во внешнедиффузионной области. Если наименьшей является скорость второго или четвертого этапа, то гетерогенный каталитический процесс протекает во внутридиффузионной области. Если наименьшей является скорость третьего этапа, то гетерогенный каталитический процесс протекает в кинетической области.
Внешнедиффузионная область гетерогенного каталитического процесса
Скорость процесса во внешнедиффузионной области (скорость этапов I и V — Vi 5) равна
V1.5 = Vm + Vb
где vm — скорость молекулярного переноса, который происходит вследствие теплового движения молекул, кмоль/(м2 • с); vk — скорость конвективного массопере-носа, который реализуется за счет принудительного перемещения газа, осуществляемого работой насосов, вентиляторов и т. д., кмоль/(м2 • с).
Скорость молекулярного переноса vm определяется уравнением
Vm — В • (со — сп)/8,
где D — коэффициент молекулярной диффузии, м2/с; с0 — концентрация реагирующего вещества в объеме потока, кмоль/м3; сП — концентрация реагирующего вещества на наружной поверхности гранулы катализатора, кмоль/м3; 8 — толщина диффузионного слоя, м.
Для необратимой реакции сп = 0, для обратимой сп = ср, где ср — равновесная концентрация.
Скорость конвективного массопереноса определяется выражением
vk = Р • (с0 - сп),
где 3 — коэффициент массопередачи, м/с.
Перенос молекул реагирующих веществ из газового потока к наружной поверхности гранул катализатора зависит от характера движения газа в каналах, образующихся в слое катализатора. При ламинарном движении (критерий Рейнольдса Re < 60) массоперенос осуществляется по всему потоку только за счет молекулярной диффузии, при турбулентном (Re > 60) молеку
лярный перенос преобладает только в пленке газа, так называемом диффузионном слое, у поверхности гранулы катализатора. Доля молекулярной диффузии в общем массопереносе (при Re > 60) составляет ~ 10 %. Поэтому в технологических расчетах промышленных процессов, в которых часто движение газа имеет турбулентный характер, принимают Vi 5 ® vk.
Коэффициент массопередачи (при Re > 60) определяют из критериального уравнения:
Nim = 0,43 • Re0’7 • Prd0’33,
где Nud и Prd — диффузионные критерии Нуссельта и Прандтля соответственно. В более широком интервале значений Re справедливо следующее уравнение:
Niid =2,0 + 1,8 • Re0’5 • Prd0’33.
Диффузионные критерии определяются формулами:
Re = то • d/v,
Kv^-ddD,
Prd = v/Z),
где w — линейная скорость газа в каналах между гранулами, м/с; d — определяющий размер, м (для слоя катализатора определяющим размером является эквивалентный диаметр канала между гранулами d = dK); v — кинематическая вязкость, м2/с; dQ — приведенный диаметр гранулы, не зависящий от плотности упаковки слоя, м.
Диаметр канала dK = 4e/s, где е — доля свободного объема слоя катализатора; 5 — наружная поверхность гранул в единице объема, м2/м3, определяемая размером, формой и плотностью упаковки гранул.
Химическая кинетика и диффузия
451
Приведенный диаметр гранулы do =4 • Vq/sq, где Ко — объем, м3, a So — поверхность одной гранулы, м2. Тогда s = (1 - е) • sq/Fo = 4(1 - e)/do, adK = doe/(l - е).
Если ввести фиктивную линейную скорость газа (ыф), приходящуюся на полное сечение реактора, П5ф = £05, ТО Re = О5ф • dyjy- (1-е)].
Скорость гетерогенного каталитического процесса во внешнедиффузионной области определяется уравнением:
V1,5 = NUd • D/d0 • (с0 - сп).
Характерные черты гетерогенного каталитического процесса во внешнедиффузионной области:
1) для необратимых реакций зависимость скорости реакции от концентрации исходных веществ соответствует первому порядку, независимо от вида кинетического уравнения химического превращения;
2) скорость процесса возрастает с увеличением линейной скорости газа и уменьшением размера гранул катализатора;
3) высокая разность температур между поверхностью гранулы катализатора и газовым потоком.
Внутридиффузионная область гетерогенного каталитического процесса
Протекание процесса во внутридиффузионной области характерно для большинства промышленных катализаторов, при этом диффузия сопровождается протеканием химической реакции. Для крупнопористых катализаторов, у которых диаметр пор больше средней длины свободного пробега молекул (при 0,1 мПа — 100 нм; при 30 мПа — 1 нм), перенос массы осуществляется за счет молекулярной диффузии. Для тонкопористых катализаторов при расчете скорости используют эффективный коэффициент диффузии (£>э), определяемый, как правило, экспериментально:
1/£>э =!/£> + 1/£>кл, где £>Кн — коэффициент диффузии Кнудсена.
Для каталитической реакции первого порядка при стационарном протекании процесса дифференциальное уравнение баланса в поре имеет следующий вид (для катализатора в виде пластины):
D3d2ddl2 - kc
с граничными условиями: с = сп при / = L (на внешней поверхности гранулы) и dddl = 0 при I = 0 (в центре гранулы). Здесь к — константа скорости реакции первого порядка (с1); L — радиус сферической гранулы катализатора или половина внешнего размера гранулы другой формы, м; / — текущая координата.
При введении переменной у = UL уравнение преобразуется к виду:
d2c/dy2 = \|/2с
с граничными условиями: с - сп при у =1 и dddy = 0 при у = 0; у — параметр Тиле.
Степень использования внутренней поверхности катализатора г|, распределение концентрации по глубине гранулы у и скорость процесса во внутридиффузионной области определяются безразмерным параметром Тиле:
= Lk^5/D3'5 (для реакции первого порядка).
Решения дифференциальных уравнений для гранул различных форм имеют вид гиперболических функций:
г| = th(\|/)/\|/,
у = ch(\|/ • y)/ch(\|/) — для гранулы в виде пластины;
г| =3/\|/ • [cth(\|/) - 1 А|/],
у = Му • sh(\|/ • y)/sh(\|/) — для сферической гранулы. При \|/ < 0,5, что отвечает кинетической области протекания процесса, r| « 1; при \|/ > 2,5 процесс протекает во внутридиффузионной области.
Параметр Тиле для любого вида кинетического уравнения:
V = L^5/(D,c,)0-5
Если кинетическое уравнение имеет вид:
v = kc"'
где Ci — концентрация ключевого реагента или вещества, взятого в недостатке по сравнению со стехиометрией реакции, концентрация которого в глубине гранулы стремится к нулю; с2 и с3 — концентрации остальных исходных реагентов; и, — порядки в кинетическом уравнении; k = Аоехр(-Е(//КГ) — уравнение Аррениуса, ко — предэкспонента, Еа — энергия активации, R — газовая постоянная, Т — температура, то параметр Тиле примет следующий вид:
D, “
Скорость во внутридиффузионной области (скорость этапов II и IV — v24) равна: v24 = r|v. Используя данную зависимость, а также параметр Тиле для любого порядка реакции, получим:
v2,4 ^koD^expi-E^RT)- с,(”1+1)/2 • с?12 • с.?72.
Характерные черты гетерогенного каталитического процесса во внутридиффузионной области:
1) наличие дробных порядков в кинетическом уравнении. Порядок реакции по ключевому компоненту становится средним между порядком реакции в кинетической области и первым порядком; порядки по остальным компонентам снижаются в 2 раза;
2) наблюдаемая энергия активации уменьшается в 2 раза по сравнению с энергией в кинетической области.
452
Новый справочник химика и технолога
Таблица 2.1.35
Кинетические параметры некоторых гетерогенных каталитических реакций
Принятые обозначения: р — текущее парциальное давление; р'— равновесное парциальное давление; Кр — константа равновесия; вп — водяной пар.
Катализатор Кинетическое уравнение Предэкс-понента, ^0 Энергия активации, кДж/моль
Конверсия метана водяным паром
ГИАП-3 (никель на оксиде алюминия) %н„ v = z— 0 ~ РсоРнг) Рн2 ’ КрРсн^вп 2,03 • 106 91,8
Конверсия оксида углерода водяным паром
СТК-1 (железохромовый) у _ ^Рсо Р Со)Рвп Рн2 4,92 • 105 78,0
Окисление сероводорода
Магнийхромоксидный на у-А12О3 — 35,0
Окисление оксида углерода кислородом воздуха
Медьхроммарганцевый на алюмосиликате v ~ *Рсо ! Рсо2 2,2 • 105 39,9
Окисление диоксида серы
Ванадиевый на алюмосиликате kPo-, Pso2 v = 1 2 3- (Pso, +0,75PsOiXl Рю2р6гкр 1,6- 106 85,0
Хромоловооксидный А?о2 Pso2 V = i £ 0,5 zi PSO3 ч PsO30 0,5 r ) Pso2Po2 2,0 • 102 125,0
Восстановление оксида азота водородом
Хроммедный v_ 0,75 + 0,25/0 Рыо 3,6 - 106 56,4
Литература
1. Боресков Г.К. Гетерогенный катализ. М.: Наука, 1986. 304 с.
2. Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964. 607 с.
3. Киперман С.Л. Основы химической кинетики в гетерогенном катализе. М.: Химия, 1979. 349 с.
4. Франк-Каменецкий Д.А. Диффузия и теплопередача в химической кинетике. М.: Наука, 1967. 491 с.
5. Саттерфильд Ч.Н. Массопередача в гетерогенном катализе. М.: Химия, 1976. 240 с.
Химическая кинетика и диффузия
453
2.1.3. Реакции изотопного обмена
(проф., д.х.н. С.А. Симанова)
Материал представлен в виде таблиц (2.1.36 и 2.1.37), в которых приведены реакции изотопного обмена в гомогенной и гетерогенной средах.
В табл. 2.1.36 элементы расположены по алфавиту русских названий; внутри каждого элемента изотопы даны в порядке возрастания массового числа; в скобках указан период полураспада данного изотопа. В графе «Реагирующие вещества» радиоактивные изотопы выделены жирным шрифтом. Для каждого изотопа сначала приведены реакции неорганических веществ по алфавиту элементов, а затем — органических, как правило, в порядке усложнения реагирующих веществ.
В графе «Концентрация реагирующих веществ» указывается также среда, в которой изучалась реакция; если среда не указана, имеется в виду водный раствор. В этой же графе иногда приводятся pH среды и ионная сила раствора ц.
Если не указана температура реакции, это означает, что реакция проводится при комнатной температуре.
В графе «Период полуобмена и другие кинетические данные» иногда приводятся значения констант скоростей реакции первого к' (с1) и второго к” (л • моль1 • с4) порядков, а также энергии активации Еа (кДж/моль). Единицы измерения, отличные от указанных, приведены при соответствующих значениях констант.
Условные обозначения
абс. — абсолютный мол. отн. — молярное отношение
а. с. — атомные слои насыщ. — насыщенный
бв. — безводный орг. соед. — органическое соединение
бзл. — бензол осажд. — осаждение
вак. — вакуум охл. — охлажденный
водн. — водный разб. — разбавленный
г. — газ, в газообразном состоянии Р-Р — раствор
ж. — жидкость, жидкий СП. — спирт (этанол)
карб. к-та — карбоновая кислота ТВ. — твердый, в твердом состоянии
кис л. — кислый щел. — щелочной
к-та — кислота экстр. — экстракция
крист. — кристаллизация эф. — эфир
к. с. — комплексная соль Ей — этилендиамин
масс. отн. — массовое отношение Ру — пиридин
Таблица 2.1.36
Кинетика реакций изотопного обмена в гомогенной среде
Пояснение'.
*’ — Обмен водорода в ароматическом кольце.
*2 — Обмен водорода в метильной группе.
*3 — Обмен водорода в метиленовой группе.
*4 — Процент обмена вычислялся от величины, рассчитанной для водорода в а-положении.
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Азот
№3 (10 мин) N2O5 + NO2 0,1 MN2O5, 0,05 М NO2; СС14 10 2 мин Осажд. AgNO2
Бром
Вг82 (36 ч) ВгО3 + Вг2 0,1 MNaBrO3, 2,59 • 10м М Вг2; 0,1 М HNO3 25 220 ± 20 ч Экстр. Вг2 при помощи СС14
l,13MNaBrO3, 0,012 М Br2; 1,01 MHNO3 или НС1О4 25 40 мин То же
K2[PtBr4] + Вг" 0,0055 М к. с., 0,022 М КВг 20 120 мин Осажд. [Pt(NH3)4] • [PtBr4]
454
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Бром
Вг82 (36 ч) СгВг3 • Н2О + Вг (зеленый) 0,33 М СгВгз • Н2О, КВг 25 150 ч Осажд. Вг с помощью AgNO3 при 0 °C
0,33 М СгВгз • Н2О, КВг; 2МНВг 25 163 ч То же
СС13Вг + Вг2 0,207 М СС13Вг, 0,0028 М Вг2; СС14 135 ~3ч Экстр. Вг2 при помощи Г (водн.)
0,194 МСС13Вг, 0,00274 М Вг2; СС14 170 ~ 10 мин То же
0,0042 М СС13Вг, 3,7 • 10 4 М Вг2; г. 170 = 5 ч »
0,0335 М СС13Вг, 0,0034 М Вг2; г. 146 18,7ч Экстр. Вг2 при помощи NO'(водн.)
0,0325 М СС13Вг, 0,0033 М Вг2; г. 183 46 мин То же
С2Н5СН2Вг + ВГ 0,1244 МС3Н7Вг, 0,0375 М LiBr; ацетон (90 об. %) + Н2О 25 33,1 ± 1,7 ч Экстр, бзл. (С3Н7Вг) и водой (ВГ)
СНзСНВгСНз + Вг" 0,1504 МС3Н7Вг, 0,0433 М LiBr; ацетон (90 об. %) + Н2О 25 810 ± 40 ч То же
СНзСНВгСООН + Вг" 0,333 М СНзСНВгСООН, 0,333 М LiBr; ацетон 22 33,7 ± 10 мин Экстр. СНзСНВгСООН при помощи эф.
С4Н9Вг + Вг- 0,25 М С4Н9Вг, 0,10 М LiBr 24,4 23,9 ± 1,3 ч Экстр, бзл. (С4Н9Вг) и водой (ВГ)
0,905 М С4Н9Вг, 0,037 М LiBr; ацетон (90 об. %) + Н2О 59,6 120 ± 5 мин То же
0,0993 М С4Н9Вг, 0,0119 М LiBr; бв. ацетон 25 96 ± 3 мин »
изо-С4Н9Вг + Вг- 0,1163 М С4Н9Вг, 0,0501 М LiBr; ацетон (90 об. %) + Н2О 24,9 830 ±50 ч »
mpem-C4H9Br + Вг” 0,079 М С4Н9Вг, 0,033 М LiBr; диацетат этиленгликоля 100 48 мин Экстр. ВГ водой
С6Н5СНВгСН3 + Вг” 0,20 М С6Н5СНВгСН3, 0,20 М LiBr; бв. ацетон 30,2 33 ± 4 мин То же
а-СюН7Вг + Вг- 0,21 МС10Н7Вг, 0,20 М LiBr; диэтиленгликоль 216 17ч
СН2=СНСН2Вг + ВГ 0,01 моль СН2-СНСН2Вг в95млС2Н5ОН (95%), 0,01 моль NaBr в 5 мл С2Н5ОН 10 к" -0,6- 10 4
То же 20 Г=2,6 • 10'4
Химическая кинетика и диффузия
455
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Бром
Вг82 (36 ч) СН2=СНСН2Вг + Вг" 0,01 моль СН2=СНСН2Вг в 95 мл С2Н5ОН (95 %), 0,01 моль NaBr в 5 мл С2Н5ОН 40 Г =12,5 • 10’4
То же 61 Г =46- 10"4
o-NO2C6H4CH2Br ± Вг" 0,0106 М NO2C6H4CH2Br, 0,0290 М LiBr; ацетон (90 об. %) + Н2О 30,0 81 ±0,3 мин Экстр, бзл. (NO2C6H4CH2Br) и водой (Вг)
о-ЫОгСбНдВг + LiBr 0,21 М <?-NO2C6H4Br, 0,21 М LiBr; диэтиленгликоль 216 46 ч
2,4-C6H3(NO2)2Br ± + Br" 0,0766 М C6H3(NO2)2Br, 0,0577 М LiBr; диацетат этиленгликоля 100 62,6 ± 3,3 ч Экстр, бзл. [C6H3(NO2)2Br] и водой (Вг )
Водород
D Нафталин + KND2 0,02 М KND2; ND3; ж. 25 к' -5.5 • 10"4 KND2 связывается NiSO4 (бв), углеводород сжигается
Дифенил + KND2 0,02 М KND2; ND3; ж. 25 к' =2,8- 10’4 То же
Бензол + KND2 То же 25 к' =8,2- 10’5 »
Толуол*1 + KND2 » 25 Г =2,1 • 10’5 »
Тетралин*1 + KND2 » 25 А'=4- 10 6 »
Бензол ± DBr Среда DBr 25 к' = 4- 10’8 Углеводород сжигается
Толуол*1 + DBr То же 25 Г = 3 • 10"4 То же
Пропилен*2 + KND2 0,02 М KND2; ND3; ж. 25 к'=3 • 10’3 KND2 связывается NiSO4 (бв), углеводород сжигается
Изобутилен*2 + KND2 То же 25 к'=1- 10’4 То же
2-Метилбутен-2*2 + + knd2 » 25 к'=5 • 10’5 »
2,3-Диметилбутен-2*2 + + knd2 » 25 к'= 1 • 10’5 »
Пропилен*3 + KND2 » 25 к' = 4- 10~4 »
Изобутилен*3 + + knd2 » 25 к' =9- 10"5 »
ИЗО-С3Н7СООН + ± D2SO4 Мол. отн. к-т 1 : 2,1 20-25 82 % за 116 ч*4 Отделение карб. к-ты перегонкой в вак.
с2н5х CHC(X)H+D2SO4 C2H5Z Мол. отн. к-т 0 : 0,9 20-25 100 % за 120 ч*4 То же
CH3 XCHCH2OOOH + D2SO4 Ch/ Мол. отн. к-т 1 : 3,1 20-25 94 % за 189 ч*4 »
CH, XCH(CH2)3COOH. D2SO4 ch/ Мол. отн. к-т 1 : 2,7 20-25 42 % за 240 ч*4 »
456
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Т,°С Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Водород
D СН3 СН3— ССООН+ D2SO4 сн/ Мол. отн. к-т 1 : 1 20-25 Обмен не обнаружен за 460 ч*4 Отделение карб. к-ты перегонкой в вак.
НСООН + d2so4 0,082 М НСООН, 0,008 М D2SO4 20-25 1 % за 307 ч*4 То же
0,066 М НСООН, 0,006 М D2SO4 20-25 1,4 % за 216 ч*4 »
СН3СООН + D2SO4 0,057 М СН3СООН, 0,058 М D2SO4 20-25 87 % за 95 ч*4 »
0,068 М СН3СООН, 0,064 М D2SO4 20-25 89 % за 286 ч*4 »
С2Н5СООН + D2SO4 0,031 МС2Н5СООН, 0,024 М D2SO4 20-25 38 % за 56 ч*4 »
0,040 М С2Н5СООН, 0,041 М D2SO4 20-25 92 % за 118 ч*4 »
С3Н7СООН + D2SO4 0,043 М С3Н7СООН, 0,043 М D2SO4 20-25 40 % за 48 ч*4 »
0,045 М С3Н7СООН, 0,039 М D2SO4 20-25 100 % за 110 ч*4 »
ИЗО-СЗН7СООН + + D2SO4 0,042 М С3Н7СООН, 0,087 М D2SO4 20-25 82 % за 116 ч*4 »
0,038 М С3Н7СООН, 0,031 MD2SO4 20-25 98 % за 185 ч*4 »
С5НцСООН + D2SO4 0,030 м С5НПСООН, 0,020 М D2SO4 20-25 102 % за 121 ч*4 »
НСООН + D3PO4 0,068 М НСООН, 0,035 М D3PO4 20-25 0,5 % за 408 ч*4 »
CH3COOH + D3PO4 0,054 М СН3СООН, 0,034 М D3PO4 20-25 56 % за 216 ч*4 »
П-С3Н7СООН + D3PO4 0,069 М С3Н7СООН, 0,057 М D3PO4 20-25 27 % за 93 ч*4 »
СНСООН+р3Ю4 qh5z 0,024 М С5НцСООН, 0,030 М D3PO4 20-25 88 % за 165 ч*4 »
C5H11COOH + D3PO4 0,021 МС5НнСООН, 0,022 М D3PO4 20-25 86 % за 42 ч*4 »
HOOC(CH2)2COOH + + D2SO4 0,011 МС4НбО4, 0,041 MD2SO4 20-25 3 % за 1560 ч*4 Крист, дикарбоновой к-ты из охл. абс. эф. или нейтрализация Na2CO3
HOOC(CH2)4COOH + + D2SO4 0,022 М С6Н10О4, 0,101 MD2SO4 20-25 85 % за 170 ч*4 То же
То же 20-25 100 % за 650 ч*4 »
Химическая кинетика и диффузия
457
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Водород
D НООС(СН2)7СООН + + D2SO4 0,008 М C9Hi6O4, 0,053 М D2SO4 20-25 83 % за 262 ч*4 Крист, дикарбоновой к-ты из охл. абс. эф. или нейтрализация Na2CO3
НООС(СН2)8СООН + + D2SO4 0,010 МС10Н18О4, 0,057 М D2SO4 20-25 54 % за 98 ч*4 То же
0,007 М С10Н18О4, 0,032 М D2SO4 20-25 97 % за 312 ч*4 Крист, дикарбоновой к-ты из охл. эф. или нейтрализация 10 % Na2CO3
СНз(СН2)5СНО + d2o 0,1 М альдегид, 0,05 М D2O3,0,1-0,2 г катализатора (диметилпиридо-нилимина); диоксан 100 90 % за 50 ч*4 Отгонка воды из диоксана
Цикламеновый альдегид + D2O То же 100 92 % за 50 ч*4 То же
С6Н5СНО + D2O » 100 1 % за 50 ч*4 »
Жасминовый альдегид + D2O » 100 5 % за 50 ч*4 »
С6Н5СН=СНСНО +D2O » 100 2 % за 50 ч »
Цитраль + D2O » 100 70 % за 50 ч »
СН3СНО + CH3COOD 9,6807 г СН3СНО, 9,3252 г CH3COOD 150 Обменное равновесие за 24 ч
СН3СООК + + CH3COOD 8,0455 г СН3СНО, 9,6443 г CH3COOD 150 Обменное равновесие за 30 ч
РЬ(СН3СОО)2 + + CH3COOD 11,8619 г (СН3СОО)2РЬ, 9,3325 г CH3COOD 150 Обменное равновесие за 170-200ч Отток воды и диоксана
СН3СООС2Н5 + + CH3COOD 8,1023 г СН3СООС2Н5, 10,5335 г CH3COOD 150 20 % за 100 ч То же
Европий
Ей152 (5,2 года) Eu(II) + Eu(III) 0,0129 М Eu(II), 0,015 М Eu(III); 1,88 M HCI; ц - 2,0 39,4 59 мин Осажд. Еи(Ш) разб. А1(ОН)3
0,0135 М Eu(II), 0,0398 М Eu(III); 1,51 МНС1; р = 2,0 39,4 29 мин То же
Иод
j 128 (25 мин) 12 + Г < 15с Экстр, эф. (12)
сн212 + г 0,48 М СН212, 0,06 М Nal; бв. С2Н5ОН 60 20,6 ± 0,6 мин Экстр, эф. (СН212) и водой (12)
с2н51 + г 0,91 МС2Н51, 0,135 М Nal; бв. С2Н5ОН 25 63,5 ± 3 мин То же
0,0199 МС2Н51, 0,0174 М Nal; С2Н5ОН 50 100 ± 2 мин Отгонка С2Н51
То же 70,1 16,0 ± 3 мин То же
458
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Иод
|128 (25 мин) С3Н71 + г 0,75 М С3Н71, 0,135 MNal; С2Н5ОН 35 35 мин Экстр, эф. (С3Н71) и водой (Г)
0,01976 МС3Н71, 0,01635 MNal; С2Н5ОН 49,8 164 ± 3 мин Отгонка С3Н71
0,01959 МС3Н71, 0,01733 MNal; С2Н5ОН 70,1 24,5 ± 4 мин То же
изо-С3Н71 + Г 1,62МС3Н71, 0,142 MNal; С2Н5ОН 50 66 ± 3 мин Экстр, эф. (С3Н71) и водой (Г)
0,01988 МС3Н71, 0,01742 MNal; С2Н5ОН 60 16,3 ± 0,2 ч Отгонка С3Н71
С4Н91 + Г 0,64МС4Н91, 0,135 MNal; С2Н5ОН 50 14 ± 1 мин Экстр, эф. (С3Н71) и водой (Г)
ИЗО-С4Н91 + Г 1,42МС4Н91, 0,142 MNal; С2Н5ОН 50 49 ± 5 мин То же
втор-С^)\ + Г То же 50 61 ± 4 мин »
wpew-C4H9I + Г 1,24МС4Н91, 0,37 MNal; С2Н5ОН 40 7,6 ± 0,6 мин »
С5НП1 + Г 0,56МС5Нп1, 0,135 MNal; С2Н5ОН 50 12,2 ± 0,2 мин Экстр, эф. (С5Нц1) и водой (Г)
изо-С5Нн1 + Г 0,715 МС5НП1, 0,137 MNal; С2Н51 50 23,6 ± 1 мин То же
K2[PtI4] + 4KI 0,103 MK2[PtI4] Обменное равновесие за 25 мин Осажд. к. с. Еп в виде [PtI4][Ni(En)3]
I131 (8 дней) Ю3 + Г Нейтральный р-р 50,0 > 30 лет Р-р подкисляется экстр. 12 бзл.
То же 100 Г-0,27- 10’4 Осажд. Г в виде Agl
» 200 Г = 0,68- 10’4 То же
» 250 к "-1,6- 10’4 »
» 300 Г-27 • 10~4 »
ю3-+12 0,099 М НЮ3, 7 • 104 М 12 25,0 280 ± 4 ч Экстр. 12 при помощи СНС13 или CCI4
То же 50,0 22,4 ± 0,6 ч Экстр. 12 при ПОМОЩИ СС14
С2н51 + г 0,1 МС2Н51, 0,1 MNal; С2Н5ОН (90 масс. %) 10 Г =0,23 • 10’4 Экстр, циклогексаном (С2Н51)и(Г)
То же 20 Г = 0,92- 10’4 То же
» 30 Г-2,15 • 10 4 »
0,1 МС2Н51, 0,1 MNal; С2Н5ОН (90 масс. %) 40 Г -4,2- 10’4 »
То же 80 Г = 1,53 • 10’4 »
С3Н71 + г 0,05 М С3Н71, 0,05 М Nal; С2Н5ОН (90 масс. %) 35,9 Г = 46,3 • Ю 5 Экстр, циклогексаном (С3Н71) и водой (Г)
То же 55 Г-304 • 10“5 То же
» 80,9 Г = 2460- 10’5 »
Химическая кинетика и диффузия
459
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Т,°С Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Иод
J131 (8 дней) изо-С3Н71 + Г 0,1 МС3Н71, 0,1 MNal; С2Н5ОН (90 масс. %) 40 Г =5,52 • 10’5 Экстр, циклогексаном (С3Н71) и водой (Г)
То же 60 50,3 • 10 5 То же
» 80 208 • 10’5 »
Иридий
1г192 (74 дня) Na3 [1гС16] + СГ 0,092 М к.с., 1 М НС1 50 Полный объем за 9 мин Осажд. (NH4)2 [1гС16]
Na2 [IrCl6] + СГ 0,097 М к.с., 1 М НС1 1 93 % за 2 мин То же
Кислород
о18 NaC104 + Н2О18 НС1О4 (70 %), 1 М NaOH 160 Обмен не обнаружен за 5 ч
NaC103 + Н2О18 l,7rNaC103, 1 г Н2О18; 0,5 М H2SO4 100 8 ч
КС1О3 + Н2О18 0,47 г КС1О3, 1 г Н2О; 0,5 М H2SO4 100 6,6 ч
LiBrO3 + Н2О18 1,3 гЫВгО3, 1 гН2О 25 280 ч
То же 120 1,3 ч
» 140 0,3 ч
Н3РО2 + Н2О18 Н2О обогащена О18 в 3-4 раза 40 <0,2ч Отгонка воды в вак.
Н3РО3 + Н2О18 То же 63 6,5 ч То же
Н3РО4 + Н2О18 » 100 70 ч »
NaH2PO2 + Н2О18 Мол. отн. соли к воде 1 : 2 и 1 :3 100 10ч »
Na2HPO3 + Н2О18 То же 100 Обмен не обнаружен »
Na3PO4 + Н2О18 » 100 То же »
Na2HPO4+ Н2О18 » 100 » »
NaH2PO4 + Н2О18 » 100 » »
К2НРО4 + Н2О18 » 100 » »
КН2РО4 + Н2О18 » 100 » »
С6Н5СООН + Н2О18 Н2О, содержащая 0,6 % О18; диоксан (33 масс. %), 0,1 М НС1 80 Г= 1,61 • ю-4 Крист, к-ты при охл.
и-СН3С6Н4СООН + + Н2О18 То же 80 к' = 1,5 • 10 4 То же
я-СН3ОС6Н4СООН + + Н2О18 » 80 к' = 1,00- 10-4 »
л/-С1СбР4СООН + + Н2О18 » 80 к'= 1,77- 10"4 »
л-С1СбН4СООН + + Н2О18 » 80 к'= 1,1 • 10"4 »
Кобальт
Со56 (72 дня) [Co(NH3)6]3+ + Со(П) 0,011 MCo(NH3)6+, 0,011 МСо(П) > 50 дней Экстр. Co(SCN)2 смесью эф.— изопентанол
460
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Кобальт
Со56 (72 дня) [CoCI(NH3),]2*+ + Со(П) 0,0048 М Со(Ш), 0,0048 М Со(П); в темноте 23 > 7,3 дня Экстр. Co(SCN)2 смесью эф.— изопентанол
[CoC1(NH3)3](NO2)2 + + Со(П) То же 23 > 2,8 дня То же
[Co(NH3)3](NO2)3 + + Со(П) » 99 > 1 дня »
[Co(CN)6]3 + Со2+ » > 57 дней »
[Со(Еп)3]2* + + [Со(Еп)3]3+ 0,090 М [Со(Еп)3]С13, 0,090 М [Со(Еп)3]С12; 0,39 М ВаС12, 0,55 М Еп 25 10,5 ч »
Со60 (5,3года) [Со(ЫН3)б]3+ + Со(1П) 0,005 М к.с., 0,005 М Co2(SO4)3; 0,5 М H2SO4 20 > 100 ч
[Co(NH3)6]2* + Со(11) 0,011 Мк.с, 0,011 МСоС12 50 > 410 дней Экстр. Co(SCN)2 смесью эф.—изопентанол
Магний
Mg27 (Ю,2 мин) Хлорофилл «а» + Mg2+ Водн. р-р ацетона; pH - 7,8 > 2,3 дня Экстр. петролейным эф.
Хлорофилл «б» + Mg2+ То же То же То же
Марганец
Мп54 (291 день) МпО4 +МпО; 0,43 -Ю4 М МпО4“, 0,48 • 10 4 М МпО4’; 0,16 М NaOH (LiOH, КОН) 0,1 10,6 с; Г=710±30; Е = 44,0 Соосаждение КМпО4 с тетрафенил-арсониумперренатом
Мп56 (2,59 ч) Мпо; + Мп2+ 0,0086 ММпО 4, 0,00087 М Мп2+; 0,043 М НС1О4 > 147 ч Осажд. МпО2
Мп(С2О4)з +Мп2+ 0,00049 ММп(С2О4)' , 0,00508 М Мп2+; 0,101 МН2С2О4 < 5 с То же
Мп(С2О4)з’ +МпО4 0,00053 М Мп(С2О4) ' , 0,0109 М МпО4; 0,106 М Н2С2О4 > 34 мин »
Мп-ацетилацетонат + + Мп2+ < 5 мин Осажд. МпСО3
Медь
Си64 (12,8 ч) Cu-салицилальдегид + + Си2+ 0,015 М к. с., 0,015 М Си(С2Н3О2)2Ру; Ру 25 < 15с Экстр, к. с. СН3С1
Си-салицилальдегид-метилимин + Си2+ То же 25 То же То же
Си-салицилальдегид-1,8-нафтилендиимин + + Си2+ » 25 1 мин »
Химическая кинетика и диффузия
461
Продолжение таблицы 2.1,36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Медь
Си64 (12,8 ч) Cu-сал ицилальдегид-этилендиимин + Си2+ 0,015 М к. с., 0,015 М Си(С2Н3О2)2Ру; Ру 0 90 ч Экстр, к. с. СН3С1
То же 25 2,1 ч То же
Cu-сали цилальдегнд-о-фенилендиимин + + Си2+ 0,015 М к. с. Си, 0,015 М Си(С2Н2О2)2Ру; Ру 25 > 4 ч »
То же 50 9 ч »
Си-ацетилацетон-этилендиимин + Си2+ 0,0076 к. с. Си, 0,0076 М Си(С2Н3О2)2Ру; Ру 250 37 ч »
Мышьяк
As76 (26,8 ч) H3AsO4 + H3AsO3 0,105 М H3AsO3, 0,098 М H3AsO4; 0,107 М НС1, 0,100 М NaCl, 0,100 М KI 25,2 6,8 ч Осажд. AsO^ в виде MgNH4AsO4
Нептуний
Np Np(V) + Np(VI) 1,41 • 10’5MNp(V), 1,41 • 105MNp(VI); 0,106 М НС1О4 в водн. этиленгликоле 0 к" = 20,4 ±3,7 Экстр, трибутилфосфатом
Np(IV) + Np(VI) 0,00133 MNp(IV), 0,00115 MNp(VI); 0,9 М НС1О4 в этиленгликоле 25 Г = 0,158 ± ± 0,009(1) То же
Олово
Sn113 (105 дней) Sn(IV) + Sn(II) 0,1 М SnCl4, 0,1 MSnCl2; 9М НС1 7 мин Осажд. Cs2SnCl6
Плутоний
Ри238 (86,6 года) Pu(III) + Pu(IV) Свежие р-ры 0,33 • 10-6 М Ри(Ш), 0,2- 10"5MPu(IV); НС1О4 (водн.) 0 1-5 мин Экстр. Pu(IV) трибутилфосфатом из 6 М НС1
Pu(III) + Pu(IV) 5 • Ю^МРи(Ш), 5 • 10 6 М Pu(IV); 1 М НС1О4 25 0,68 мин; Е = 32,2 То же
Ртуть
Hg203 (48 дней) Hg2+ + Hg2+ 10 5 М Hg(C104)2, 10 3 М Hg2(C104)2; ЗМ NaClO4, 0,5 М НС1О4 < 15с Осажд. Hg 2+ в виде Hg2Cl2
Hg2+ + Hg7+ 0,0044 М Hg(C104)2, 0,0069 М Hg2(C104)2; 0,1 МНС1О4 < 15с Осажд. Hg7+ в виде Hg2CrO4
HgCl2 + + w-ClHgC6H4COOH 0,001 М HgCl2, 0,001 М w-ClHgC6H4COOH 25 Г= 5,4 ± 0,2; Е = 50,2 ± 2,5 Осажд. w-ClHgC6H4COOH из 0,5 М H2SO4
C6H6HgCl + HgCl2 0,00314 MC6H5HgCI, 0,00338 М HgCl2 25 61,7 ± 0,6 мин Образование HgC12[H2NC6H4C6H4NH2]
То же 40 46,0 ± 0,5 мин То же
^•моль-^-с-1.
462
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Сера
S35 (87,1 дня) NH4SCN + S 0,016 моль S, 0,016 моль NH4SCN; 5 мл ксилола, 12 мл изопентанола 115 Обмен не обнаружен за 5 ч Крист. S
NH4C(S)NH2 + s 0,004 моль NH2C(S)NH2, 0,004 моль S; 5 мл ксилола, 12 мл изопентанола 118 51 % за 70 мин NH4C(S)NH2 отмывается сп. после совместной крист, с S
S+ S (С2Н5О)2Р / S— С2Н5 0,387 г тиосоединения, 0,064 г S; 1 мл С6Н5СН3 130 Обмен не обнаружен за 12 ч Перегонка в вак.
(изо-С4Н9О)3Р=8 + S 0,304 г тиосоединения, 0,064 г S; 1 мл С6Н5СН3 130 То же То же
(изо-С5НцО)3Р-8 + S 0,321 г тиосоединения, 0,064 г S; 1 мл СбН5СН3 130 » »
S (CzHsO^P CH2S—С2Н5 xs—сн2+ s 0,296 г тиосоединения, 0,064 г S 130 » »
(C2H5O)2PS(SK) + S 0,449 г орг. соед., 0,064 г S; 2 мл С2Н5ОН, 6 мл С6Н6 50 Обмен не обнаружен за 13 ч Разделение в вак. при 30 °C
То же 85 Обмен не обнаружен за 6 ч То же
» 100 Обмен не обнаружен за 18 ч; Е = 167 »
Этилксантогенат Ni + S 20 мг этилксантогената Ni, 8 мг S; 2 мл С6Рб 139 10 ч; к' = 0,07 ч 1 Экстр, этилксантогената Ni 7 % NH3 • Н2О
То же 156 2 ч; к' = 0,35 ч’1 То же
Ксантогенат К + S 128 мг S, 324 мг ксантогената К; 30 мл смеси толуол + сп. 18,3 27 % за 3 мин; к' = 0,1045 мин-1 Экстр, водой; водноспиртовый слой содержит ксантогенат К
128 мг S, 324 мг ксантогената К; 10 мл смеси CS2 + сп. 20 66 % за 10 мин; А' = 0,117 мин-1 То же
Cr2(SO4)3 (зеленый) + + Na2SO4 0,163 М Cr2(SO4)3 20 1200 мин; к' = 0,00058 мин-1 Разделение хлороводородным бензидином; осажд. 8Од“ в виде BaSO4
То же 30 1200 мин; к = 0,00026 мин1; Е= ПО То же
2-Аминобензотиазол + + S Масс. отн. реагентов 1 : 1 140- 150 Обмен не обнаружен за 75 мин Выделение аминобензотиазола растворением в воде и упариванием
Химическая кинетика и диффузия
463
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Сера
S35 (87,1 дня) 2-Метилбензотиазол + + S Масс. отн. реагентов 1 : 1 137 Обмен не обнаружен за 15 мин Отгонка метилбензотиазола в вак.
2-Меркаптобензими-дазол + S 0,1 г 2-меркаптобензими-дазола, 0,2 г S 172 Обмен не обнаружен за 30 мин Сплавление меркаптобензимидазола с NaOH
NH2C(S)NH2+ S 0,25 г NH2C(S)NH2, 0,11 г S 183 Интенсивный обмен за 30 мин Сплавление NH2C(S)NH2 с NaOH
SOj + SO2’ Кисл. или щел. р-р 100 > 40 дней SO2 (г.): осажд. SO2 в виде SrSO3
S2O32’ + so2 0,0650 М Na2S2O3, 0,0719 MNa2SO3; 1,737 М NaCl, pH = 5,0 62 137 ± 14 мин То же
CS2 + H2S 0,108 MCS2, 0,0042 M H2S; C6H6 120 275 дней H2S (г.)
SOC12 + so2 Избыток SOC12; катализатор (CH3)4NC1 0 Л" =0,06 2 -2 -1 л • моль • ч ; £ = 61,5
Избыток SOC12; катализатор RbCl 0 к” =0,22 2 -2 -1 л • моль • ч ; £ = 56,9
Свинец
РЬ210 (22 года) PbO2 + PbO2’ ЮМ NaOH 25 > 1,3 года Осажд. ВаРЬО3
Pb(C2H5)4 + + Pb(C2H5)3Cl 0,0063 М Pb(C2H5)4, 0,0063 М Pb(C2H5)3Cl; С6Н6 1,7 ч Осажд. РЬ(С2Н5)3С1 при помощи СбНи
РЬ212 (10,6 ч) PbCl2 + Pb(NO3)2 0,027 М РЬС12, 0,027 М Pb(NO3)2; Ру <15 мин Осажд. РЬС12 при охлаждении
Pb(C2H3O2)4 + + Pb(C2H3O2)2 0,099 М РЬ(С2Н3О2)2 • ЗН2О, 0,099 М РЬ(С2Н3О2)4; СН3СООН 80 < 10 мин Осажд. РЬО2 или РЬ(С2Н3О2)4
Pb(C6H5)4+PbCl2 0,035 М РЬ(С6Н5)4, 0,034 М РЬС12; Ру > 3,4 ч Осажд. РЬС12 при охлаждении
РЬ(СбН5)4 + + Pb(C2H3O2)2 0,026 М РЬ(С2Н3О2)2 • ЗН2О, 0,028 М РЬ(С6Н5)4; пентанол > 3,4 ч Осажд. РЬ(С6Н5)4 при охлаждении
Сурьма
Sb124 (60 дней) Sb(III) + Sb(V) 0,0200 М Sb(III), 0,0200 М Sb(V); 6МНС1 25,0 53 ±5 ч Экстр. SbCl5 изопропиловым эф.
То же 9,8 610±60ч То же
Sb(III) + Sb(V) 0,0235 М Sb(III), 0,0198 MSb(V); 12,0 МНС1 25,0 36 ± 5 мин »
464
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Таллий
»р|204 (3,5 года) Tl(III) + T1(V) 0,0001 М Т1(С1О4)3, 0,01 М Т1С1О4; 0,40 М НС1О4 49,5 31,5 Осажд. Т12СгО4
0,00847 М Т1(С1О4)3, 0,0245 М Т1С1О4; 6 М НС1О4 32,2 302 ч То же
0,0067 М Т1(С1О4)3, 0,0010 МТ1С1О4; 5,6 М НС1О4, 0,4 М НС1 31,8 32,5 мин »
0,010 МТ1(С1О4)3, 0,010 МТ1С1О4; 2,5 М НС1О4, 1,0 М NaClO4 37,3 60,0 ч »
0,010 МТ1(С1О4)3, 0,01 МТ1С1О4; 2,5 М НС1О4, 0,6 М NaNO3 37,3 5,6 ч »
0,0244 М T1(NO3)3, 0,0244 М T1(NO3); 1,5MHNO3 24,8 1,7 ч Осажд. Т1Вг или Т1(ОН)3
0,0244 М Т1(С1О4)3, 0,0244 М Т1С1О4; 1,5 М НС1О4 24,8 33 ч То же
Углерод
С11 (20,5 мин) С2О2 + СО2’ 2М NaOH в соприкосновении со свежим МпО2 100 > 290 дней
К3[А1(С2О4)3] + + нс2о4 0,06 М к. с., 0,012 М К2С2О4; pH “ 4,5 + 6,0 35 < 6 с Осажд. СаС2О4
К3[Сг(С2О4)з] + + с2о42- 0,06 М к. с., 0,012 М К2С2О4 35 > 5,7 ч Осажд. Ag2C2O4
К3[Со(С2О4)3] + + с2о2- То же 35-50 > 5,7 ч То же
K3[Fe(C2O4)3] + + нс2о4 0,06 М к. с., 0,012 М К2С2О4; pH = 4,5 4- 6,0 35-50 < 6 с Осажд. СаС2О4
K2[Pt(CN)4] + CN 0,037 М к. с., KCN 95% за 5 мин
с14 (5600 лет) [Cr(CN6)]3’ + CN ’ pH = 3 4- 4 25 ~200ч
pH >7 25 ~ 2000 ч
K4[Mn(CN)6] + CN’ 0,0025 М к. с., 0,0571 М KCN; pH = 9,0-И 0,8 0 Г= 2,64 х х 10”2 мин’1; £ = 35,6
MMoCChOd + KCN 0,0491 М к. с., 0,0439 М KCN; pH = 4,3 25 Обмен не обнаружен за 138 ч Осажд. AgCN
K4Fe(CN)6] + KCN 0,0852 М к. с., 0,0568 М KCN; pH = 11,8 25 Обмен не обнаружен за 118ч То же
K3[Fe(CN)6] + KCN 0,0560 М к. с., 0,0500 М KCN; pH =10 25 Обмен не обнаружен за 115 ч »
K3[Co(CN)6] + KCN 0,017 М к. с., 0,0500 М KCN; pH = 10,8 25 Обмен не обнаружен за »
190 ч
Химическая кинетика и диффузия
465
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Углерод
С14 (5600 лет) K2[Ni(CN)4] +KCN 0,0500 М к. с., 0,0500 MKCN; pH = 10,1 25 < 3 мин Осажд. AgCN
K2[Pd(CN)4] + KCN 0,500 М к. с., 0,500 М KCN; рН= 10 25 То же То же
K2[Hg(CN)4] + KCN 0,025 М к. с., 0,024 М KCN 25 < 5 мин »
CH3CN + CN" 1 М CH3CN, 0,175 М NaCN; 0,5 М NaOH 75 >275ч »
(CH2)2(CN)2 + CN" 0,25 М (CH3)2(CN)2, 0,05 М NaCN; 0,1 М NaOH 100 >230ч »
HOCH2CH2CN + CN" 0,9 М C2H4CNOH, 0,05 М NaCN; 0,1 М HNO3 или 0,1 М NaOH 100 >7ч »
C6H5CN + CN" 0,2 М C6H5CN, 0,05 М NaCN; 0,1 М NaOH 75 > 700 ч »
Уран
и233 (1,63 X х ^лег) UO2+ + U(IV) 0,0274 М UO2C12, 0,025 М UCU; pH = 1,5 25 15 мин Осажд. UF4
0,0274 М UO2C12, 0,025 М UCU; pH = 0,96 31 72 мин То же
0,0274 М UO2C12, 0,025 М UCU; pH = 0,90 25 150 мин »
0,0274 М UO2C12, 0,056 MUCU; pH = 1,22 25 16 мин »
0,0274 М UO2C12, 0,0135 М иСЦ; pH = 1,22 25 360 мин »
0,0274 М UO2C12, 0,025 М UOC12; pH = 0,96 25 31 мин »
0,0206 М U(V1), 0,0205 М U(IV); 0,886 М Н+, 0,95 М SOJ-, на свету 29,6 35,7 мин Осажд. купфероната U(IV)
0,0206 М U(VI), 0,0205 М U(IV); 2,79 М Н+, 0,95 М SO; 29,6 17 ч Тоже
То же 29,6 51 мин »
и237 (6,8 дня) UO2+ + U(IV) 0,043 М U(SO4)2, 0,060 М UO2SO4; 0,186 MH2SO4 20-25 43 ч Осажд. UF4
Фосфор
р32 (14,3 дня) hpo;- + h2po- 0,05 М NaNH4HPO4, 0,05 MNaH2PO2 100 > 34 дней Осажд. Са3РО4
h2po;+(hpo3)„ 0,0182 М Н3РО4, 0,091 МНРО3; pH = 2,0 29 > 17ч Осажд. Ва(РО3)2
hpo42- + (HPO3)„ 0,0047 М Н3РО4, 0,0047 М НРО3; pH = 12,5 100 >2ч То же
466
Новый справочник химика и технолога
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Фосфор
р32 (14,3 дня) н2ро4 + н2р2о?~ 0,0167 М Н3РО4, 0,0083 М Н4Р2О7; pH = 2,2 100 > 34 ч Осажд. Cd2P2O7
poj- + НРО2 0,46 М РО34~, 0,43 М НРО2~; 0,42 М NaOH 100 >35 дней Осажд. Mg(NH4)PO4
н3ро4 + Н3РО3 0,036 М Н3РО4, 0,035 М Н3РО3; 5,63 М НС1 25 > 53 лет То же
РС13 + РС15 0,0037-0,0232 М РС15, 0,0051-0,0709 М РС13; ССЦ 10 £ = (0,90 ± 0,08) х xioV Е = 66,5 Осажд. P(V) из водн. р-ра в виде MgNH4PO4
То же 25 Г=(10,6± 1,4) х х 10-3ч-1 То же
» 50 Г= (82 ± 6) х х 103ч-1 »
Фтор
р!8 (112 мин) f2 + hf 38 мл F2, 36 мл HF; в темноте или при освещении ртутной лампой; г. 24 > 5 ч
38 мл F2, 35 мл HF; объемы газов измерены при 24 °C; г. 190 > 6 ч
Хлор
С136 (106 лет) КЬ[АиСЦ] + NaCl 0,0124 М [АиСЦГ, 0,0760 М СГ; ц = 0,088 0 1,64 ± 0,02 мин Осажд. (C6H5)4As[AuC14]
0,0119 М [АиСЦ] , 0,0469 М СГ; ц = 0,088 20 0,30 ± 0,1 мин То же
K2[PtC14] + СГ 0,268 М КС1, 0,0166 М K2[PtC14]; 0,0010 М [PtCl3 • Н2О]; (1 = 0,318 25 15,3 ч Осажд. [Pt(NH3)4] [PtCl4]
0,134 МКС1, 0,0083 М K2[PtC14]; 0,0010 М [PtCl3 ♦ Н2О]; ц = 0,318 25 15,2ч То же
0,938 М КС1, 0,0166 М K2[PtCl4]; 0,0025 М [PtCl3 • Н2О]“; ц = 0,318 25 13,2 ч »
0,638 М КС1, 0,0042 М K2[PtCl4]; 0,0016 M[PtCl3H2Or; (1 = 0,318 25 13,8ч »
H2[PtCl6] + СГ 0,039 М к. с., 0,087 М СГ; в темноте 20 % за 20 мин Осажд. Cs2[PtCle]
0,039 М к. с., 0,087 М СГ; на свету 100 % за 20 мин То же
Химическая кинетика и диффузия
467
Продолжение таблицы 2.1.36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Т,°С Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Хлор
С138 (38 мин) С12 + СГ 0,012 М С12, 0,06 М ацетанилид; 0,15 М NaCI, 0,5 М H2SO4 < 10-3 с Осажд. хлорацетанилида
СЮ2 + С12 0,043 М С1О2, 0,28 М С12, 0,06 М НС1 25 > 4 ч Экстр. СЮ2 при помощи СС14
С1О2 + сг 0,035 МС1О2, 0,1 М NaCI; 0,05 М НСЮ4, pH-7,2 25 То же То же
СЮ 2 + СГ+СЮ 0,048 М С1О2, 0,017 М СЮ’, 0,017 М СГ; 0,1 МОН" 25 7 ± 2,5 ч
С1О3 +С12 0,75MNaC103, 0,015 МС12; 4,5 М H2SO4 25 > 18ч Экстр. С12 при помощи СС14
1 М NaClO3, 0,026 М С12; 3,0 М H2SO4 93 > 4 ч То же
wpew-C4H9Cl + СГ 0,255 М С4Н9С1, 0,0256 М LiCl; бв. НСООН 15 4,1 ± 0,2 мин Экстр, эф. и водой
a?-NO2C6H4CH2C1 + СГ 0,05771 М NO2C6H4CH2C1; ацетон (90 об. %) + Н2О 30 150± 10ч Экстр, бзл., (NO2C6H4CH2C1) и водой (LiCl)
Хром
Сг51 (26,5 дня) Сг2О2 +Сг(Ш) 0,0358 М Cr(NO3)3, 0,0179 М К2Сг2О7, 8,45 М HNO3; pH = 2 18-20 > 160 дней Осажд. РЬСгО4
0,0358 М Cr(NO3)3, 0,0179 МК2Сг2О7, pH = 1,5 18-20 > 92 дней То же
0,010 МСг(СЮ4)3, 0,0050 М К2Сг2О7; 2,0 М НСЮ4 18-20 > 100 дней »
0,010 М Сг(СЮ4)3, 0,0050 М К2Сг2О7; 0,05 М НСЮ4 18-20 > 430 дней »
То же 45 60 ± 6 дней »
0,002 М Cr2(SO4)3 (зеленый), 0,002 М K2Cr2O7; H2SO4; pH = 2 + 3 25 > 208 дней Осажд. Сг(ОН)3
СгО42 +Сг(Ш) 0,0358 М Cr(NO3)3, 0,0358 М К2СгО4; 2,38 М NaOH; pH = 2 25 > 160 дней Осажд. РЬСгО4
0,010 МСг(СЮ4)3, 0,010 М К2СгО4; 3,1 М NaOH 25 > 68 дней Осажд. Сг(ОН)3
K3[Cr(CN)6] + Сг(Ш) 0,0049 М СгС13 (зеленый), 0,0049 М к. с.; 0,050 М НС1 25 > 100 дней Осажд. Ag3[Cr(CN)6] и AgCl
468
Новый справочник химика и технолога
Продолжение таблицы 2.1,36
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Период полуобмена и др. кинетические данные Метод разделения реагирующих веществ
Хром
Сг51 (26,5 дня) (СгССзОЛ^ + СгОП) 0,010 М Сг(СЮ4)3, 0,010 М К3[Сг(С2О4)з]; 0,025 М НС1О4 > 210 дней Осажд. Ва3[Сг(С2О4)3]2
0,0050 М СгС1з (зеленый), 0,0050 М К3[Сг(С2О4)з]; 0,050 М НС1 25 > 14 дней То же
[Cr(SCN)6]3’ + Сг(Ш) 0,010 МСг(С1О4)з, 0,010 М K3[Cr(scN)6]; 0,05 М или 2,0 М НС1О4 18-20 > 210 дней Осажд. Ag3[Cr(SCN)6]
0,0067 М СгС1з (зеленый), 0,0067 М K3[Cr(SCN)6]; 0,067 М НС1 25 > 69 дней То же
(У5 (2 ч) Сг2О2 +Сг(1П) pH = 3 в присутствии KI 25 > 13 ч Осажд. ВаСгО4
Церий
Се141 (30 дней) Ce(III) + Ce(IV) 0,05 М Се(С1О4)3, 0,05 М Се(С1О4)4; 1,0 или 1,8 М НС1О4; на свету или в темноте 4 < 1 ч
0,005 М Ce2(SO4)3, 0,01 М Ce(SO4)2; l,0MH2SO4 15 < 20 мин
Се144 (275 дней) Ce(III) + Ce(IV) 0,00198 М Ce(NO3)3, 0,00187 MC1(NO3)4; 6,18 М HNO3 25 11,2 ± 0,3 мин Экстр. Ce(NO3)4 диэтиловым эф.
То же 0 88 ± 4 мин То же
Цинк
Zn65 (250 дней) Zn-ацетилацетонат + + Zn2+ 0,01 М Zn(C2H3O2)2(Py)2, 0,01 М к.с.; Ру 25 <30 с Экстр. СНС13 (Zn-ацетилацетонат) и водой (Zn2+)
Zn-бензоил ацетон-аммиакат + Zn2+ То же 25 То же То же
Zn-никотинилацетон + + Zn2+ 0,0034 MZn(C2H3O2)2(Py)2, 0,0034 М к. с.; Ру 25 То же »
Zn(SCN)2 + Zn2+ 0,01 М Zn(C2H3O2)2(Py)2, 0,01 М Zn(SCN)2(Py)2 25 То же Экстр, хлороформом Zn(SCN)? и водой (Zn2 )
Таблица 2.1.37
Кинетика реакций изотопного обмена в гетерогенной среде
Пояснение, т — изотоп в возбужденном состоянии с измеримым временем распада.
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Степень обмена (по массе)
Бром
Вг80 (18 мин) CuBr (тв.) + Вг 0,00135 моль CuBr, 10 мл 0,008 М NaBr 18-20 8 % за 30 мин
Вг80/" (4,54 ч) Вг2 (ж.) + Вт" 0,18 моль Вг2, 100 мл 1 М NaBr 18-100 75 % за 30 мин
Химическая кинетика и диффузия
469
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Степень обмена (по массе)
Бром
Вг80/” (4,54 ч) AgBr2 (тв.) + ВГ 0,5 ммоль свежего AgBr2, 20 мл 0,0015 МКВг 18-20 86 % за 15 мин
То же 18-20 98 % за 1 ч
10,6 ммоль старого AgBr, 14 мл 0,001 МКВг 18-20 75 % за 1 ч
Вг80'” (4,54 ч) AgBr (тв.) + Вг2 (в С2Н5Вг) AgBr, выдержанный различное время в С2Н5Вг 18-20 68-100 % за 70 мин
AgBr (тв.) + Вг2 (г.) 5,3 ммоль свежего воздушносухого AgBr 18-20 64,2 % за 100 мин
AsBr3 (тв.) + С2Н5Вг (ж.) ~ 15 < 5 % за 10 мин
АиВг3 (тв.) + С2Н5Вг (ж.) ~ 15 5 % за 30 мин
СаВг2 (тв.) + mpem-C4H9Br (ж.) ~ 15 < 5 % за 1 ч
СиВг2 (тв.) + mpem-C4H9Br (ж.) ~ 15 > 1 % за 1,5 ч
1пВг3 (тв.) + wpew-C2H5Br (ж.) * 15 > 1 % за 30 мин
Вг2 (тв.) + С2Н5Вг (ж.) ~ 15 < 5 % за 5 мин
РВг5 (тв.) + С2Н5Вг (ж.) ~ 15 < 5 % за 2 ч
SbBr3 (тв.) + С2Н5Вг (ж.) 15 < 5 % за 100 мин
SbBr3 (тв.) + + СН2ВгСН2Вг (ж.) * 15 < 5 % за 2 ч
SbBr3 (тв.) + m/?ew-C4H9Br (ж.) ~ 15 > 95 % за 30 мин
Вг82(36 ч), Вг80,"(4,54 ч) AgBr (тв.) + ВГ 5,3 ммоль свежего AgBr, 0,0059 М КВг 20 ~ 10 % за 1 мин
То же 20 > 95 % за 30 мин
5,3 ммоль старого AgBr, 0,0059 М КВг 20 = 50 % за 48 ч
5,3 ммоль плавленого AgBr, 0,0059 М КВг 20 < 5 % за 48 ч
Свежий AgBr, NaBr в Н2О + С2Н5ОН 20 % за 3 мин
Старый AgBr 100 ~ 50 % за 30 мин
А12Вг6 (тв.) + Вг2 (г.) 95 % за 10 мин
А12Вг6 (ж.) + Вг2 (г.) То же
А12Вг6 (тв.) + НВг (г.) То же
АиВг (тв.) + Вг2 (ж.) <60 ~ 40 % за 1 ч
АиВг3 (тв.) + Вг2 (ж.) = 37 % за 48 ч
CuBr (тв.) + Вг2 (ж.) <60 50 % за 1 ч
СиВг2 (тв.) + Вг2 (г.) 16 % за 48 ч
Hg2Br2 (тв.) + НВг НВг в CCI4 < 5 % за 5 мин
Вг82 (36 ч) AgBr (тв.) + ВГ 1,0 ммоль свежего AgBr, 40 мл 0,005 М КВг 25 48 % за 5 мин
То же 25 90 % за 30 мин
470
Новый справочник химика и технолога
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Степень обмена (по массе)
Бром
Вг82 (36 ч) AgBr (та.) + Вг’ 0,76 ммоль AgBr в фотографической пластине, 100 мл 0,0076 М Вг’ 18-20 < 5 % за 31 ч
А12Вг6 (те.) + СН3Вг (г.) 6,5 мл плавленого, просеянного (через сито с шириной отверстия 210 мкм) А1Вг3; 298 мл СНзВг 25 66 % за 33 мин
ВаВг2 (та.) + СН3Вг (г.) 6,5 мл плавленого, просеянного (через сито с шириной отверстия 210 мкм) ВаВг2; 368 мл СН3Вг 95 25 % за 160 мин
КВг (та.) + СН3Вг (г.) 18-20 <5%
NaBrO3 (тв.) + СН3Вг (г.) 18-20 <5%
СбН5Вг (ж.) + Вг- 18-20 < 5 % за 1 мин
Ванадий
у52 (3,74 мин) V-8-оксихинолят (тв.) + + VO, 18-20 < 5 % за 5 мин
V-8-оксихинолят (тв.) + VO2+ 18-20 То же
V-купферонат (та.) + VO’ 18-20 »
V-купферонат (тв.) + VO2+ 18-20 »
V-купферонат + VO’ Купферонат в бзл. 18-20 10 % за 5 мин
V-купферонат + VO2+ То же 18-20 24 % за 2,6 мин
» 18-20 24 % за 12 мин
Висмут
Bi210 (5 дней) Bi (тв.) + Bi(III) 0,03 М BiCl3; 1 МНС1 19 40 а. с. за 30 с
0,03 М Bi(NO3)3; 1 MHNO3 17 15 а. с. за 30 с
0,003 М Bi (III); 0,05 М H2SO4 18-20 160 а. с. за 30 мин
Bi212 (60,5 мин) Bi (тв.) + Bi(III) 0,001 М BiCl3; ЗМНС1 18-20 5 • 104 а. с. за 2 ч
Водород
D Гептан + D2SO4 0,023 ммоль углеводорода, 0,085 М D2SO4 22-25 2,1 % за 1 ч
З-Метилгексан + D2SO4 0,022 ммоль углеводорода, 0,052 М D2SO4 20-25 96 % за 11 ч
Октан + D2SO4 2,00 г углеводорода, 4,63 г D2SO4 18-20 1,8 % за 10,5ч
2,03 г углеводорода, 4,71 rD2SO4 18-20 2 % за 50 ч
З-Метилгептан + D2SO4 1,42 г углеводорода, 3,28 г D2SO4 18-20 100 % за 10 ч
2,2-Диметилгексан + D2SO4 0,029 моль углеводорода, 0,051 М D2SO4 20-25 2,3 % за 4 ч
2,2,4-Триметилпентан + + D2SO4 0,015 моль углеводорода, 0,104 М D2SO4 20-25 100 % за 2,5 ч
Химическая кинетика и диффузия
471
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Степень обмена (по массе)
Водород
D 2,3,4-Триметилпентан + + D2SO4 1,45 г углеводорода, 3,75 г D2SO4 18-20 98 % за 5 ч
1,28 г углеводорода, 5,09 г D2SO4 18-20 98 % за 10 ч
Додекан + D2SO4 0,008 моль углеводорода, 0,037 М D2SO4 20-25 1,2 % за 4 ч
Циклопентан + D2SO4 0,022 моль углеводорода, 0,042 М D2SO4 20-25 2,2 % за 3 ч
Метилциклопентан + + D2SO4 0,027 моль углеводорода, 0,041 М D2SO4 20-25 83 % за 4 ч
1,1-Диметилциклопентан + + D2SO4 0,016 моль углеводорода, 0,051 М D2SO4 20-25 1,8 % за 4 ч
Циклогексан + D2SO4 0,033 моль углеводорода, 0,050 М D2SO4 20-25 1,4 % за 3 ч
Метилциклогексан + + D2SO4 0,029 моль углеводорода, 0,058 М D2SO4 20-25 97 % за 10,5 ч
/иранс-Декалин + D2SO4 0,020 моль углеводорода, 0,053 М D2SO4 20-25 2,4 % за 32 ч
Золото
Au198 (2,7 дня) Au (тв.) + Au(III) 0,1 М АиС13; pH = 1,0 19 20 а. с. за 30 с
То же 68 60 а. с. за 30 с
Иод
|128 (25,0 мин) Cui (тв.) + 1 1,05 ммоль Cui, 10 мл 0,008 М Nal 8 % за 30 мин
Agl (тв.) + Г Свежий Agl 100 > 95 % за 30 мин
I130 (13,6 ч) Agl (тв.) + Г То же 18-20 ~ 80 % за 36 ч
Старый Agl 18-20 ~ 20 % за 24 ч
Иридий
1г194 (19,0 ч) [1г(Ру)2СЬ] (тв.) + [1гС16]2- 0,0286 ммоль [1г(Ру)2С14], 30 мл 0,005 М (NH4)2 [1гС16] 100 < 5 % за ~ 3 мин
Кобальт
Со60 (5,3 года) Со(ОН)3 (тв.) + Со(П) 0,76 ммоль Со(ОН)3, 35 мл 0,02 М COSO4; 0,22 М NaOH, 0,32 М Н2О2 18-20 < 13 % за 24 ч
Лантан
La140 (40 ч) La (тв.) + La(III) 0,1 моль La(NO3)2; рН~5 19 ~ 40 а. с. за 30 с
То же 60 300 а. с. за 30 с
Магний
Mg27 (10,2 мин) Mg-8-оксихинолят (тв.) + + Mg2+ Свежий осадок 18-20 > 95 % за 30 мин
Марганец
Мп56 (2,59 мин) Мп (тв.) + Мп(И) 0,05 моль MnSO4; 5 • 10 4 М H2SO4 17 ~ 10 а. с. за 30 с
То же 66 95 а. с. за 30 с
472
Новый справочник химика и технолога
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Г, °C Степень обмена (по массе)
Марганец
Мп56 (2,59 мин) МпО2 (тв.) + Мп(П) 2,3 ммоль МпО2, полученного взаимодействием МпО4 и Мп2+, 25 мл 0,091 М Мп(П) 18-20 39 % за 15 мин
То же 100 95 % за 30 мин
2,3 ммоль МпО2 из прокаленного Mn(NO3)2, 25 мл 0,091 М Мп(П) 100 15 % за 15 мин
То же 18-20 2 % за 15 мин
МпО2 (тв.) + МпО4“ 23 ммоль МпО2, 5 мл 0,1 М МпО2'; 0,5 М NaOH 18-20 < 5 % за 30 мин
Медь
Си64 (12,8 ч) Си (тв.) + Си(П) 0,05 М Cu(NO3)2; pH = 3,5 17 ~ 20 а. с. за 30 с
То же 68 ~ 80 а. с. за 80 с
0,1 М CuBr2;pH-3,7 20 ~ 700 а. с. за 135 мин
То же 67 ~ 20 000 а. с. за 135 мин
CuBr (тв.) + Си(П) 1,35 ммоль CuBr, 10 мл 0,16 М CuSO4 18-20 56 % за 30 мин
Си! (тв.) + Си(П) 1,05 ммоль Cui, 10 мл 0,16 М CuSO4 18-20 52 % за 30 мин
CuSO4 • 5Н2О + Си(П) Кристаллы CuSO4 0,5-1,0 мм, эквивалентное количество насыщ. р-ра 18-28 90 % за 5 мин
Кристаллы CuSO4 2,0-2,5 мм 18-20 45 % за 5 мин
Мышьяк
As76 (26,8 ч) (NH,)3 AsO4 (тв.) + + As2S5 (ж.) As2S5 в NH3 ~ 50 % за 40 мин
(NH4)3 AsO4 (тв.) + + As2S3 (ж.) As2S3+NH3 То же
As2S5 (тв.) + AsS3' 5 % за 114 ч
As2S3 (тв.) + AsS3 10 % за 112 ч
Cd3 (AsS4)2 (tb.) + AsS3- > 95 % за 2 мин
Cd3 (AsS4)2 (tb.) + AsS3/ То же
Ag3AsO3 (tb.) + AsO3- < 9 % за 2 ч
Ag4As2O5 (tb.) + AsO3 18-20 9 % за 2 ч
Ртуть
Hg203 (48 дней) Hg (ж.) + Hg(I) 0,05 М Hg2(ClO4)2 или 0,05 М Hg2(NO3)2; pH =1,5 4-1,6 18 50 % за 32 мин
Hg(>.) + Hg(II) 0,1 М Hg2(NO3)2; pH =1,7 18-20 50 % за ~ 30 мин
Hg2Cl2 (TB.) + Hg(II) Свежий Hg2Cl2 18-20 30 % за 2,5 мин
Hg2Cl2, выдержанный 7,5 мин 18-20 10 % за 2,5 мин
Химическая кинетика и диффузия
473
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда т,°с Степень обмена (по массе)
Ртуть
Hg203 (48 дней) Hg2CrO4 (тв.) + Hg(II) Свежий Hg2CrO4 10-20 > 95 % за 2 мин
Hg2CrO4, выдержанный 6 мин 18-20 < 5 % за 2 мин
(CH3OC6H4)2Hg + Hg 0,026 ммоль ртутьорг. соед., 2,6 моль Hg 60 87 % за 2 ч
(C^QH^Hg + Hg 0,028 ммоль ртутьорг. соед., 1,4 моль Hg 60 56 % за 2 ч
(C^Hg + Hg То же 60 11 %за2ч
(ClC6H4)2Hg + Hg 60 8 % за 2 ч
(O2NC6H4)2Hg + Hg 60 4 % за 2 ч
Свинец
РЬ212 (Ю,6ч) Pb (тв.) + Pb(II) 0,1 М РЬ (С2Н3О2)2; pH = 5 300 а. с. за 30 с
PbO2 (тв.) + Pb(II) 1,25 ммоль РЬО2, 25 мл 0,05 М РЬ(Н) 15 % за 15 мин
То же 100 50 % за 30 мин
PbCrO4 (тв.) + Pb(II) 1,55 ммоль старого РЬСгО4, 20 мл 0,001 М (PbNO3)2 35 % за 5 ч
Свежий РЬСгО4, 20 мл 0,001 М Pb(NO3)2 95 % за 4 ч
PbCrO4 (тв.) + Pb(II) 0,5 ммоль свежего РЬСгО4, 20 мл 0,001 М РЬ(С2Н3О2)2 в С2Н5ОН 50 % за 4 ч
Pbl2 (тв.) + РЬ(П) 13 ммоль РЫ2, 1,5- 104MHNO3 0 25 % за 30 мин
PbSO4 (тв.) + Pb(II) 4,0 ммоль старого PbSO4, 34 мл насыщ. р-ра PbSO4 23 39 % за 1 мин
То же 24 93 % за 6 ч
1,0 ммоль свежего PbSO4, 30 мл 0,0024 М Pb(NO3)2 26 100 % за 3 мин
Серебро
Ag106 (8,2 дня) AgCl (тв.) + Ag+ Свежий AgCl 25 90 % за 10 мин
AgCl, перекристаллизованный из NH3 • Н2О 25 35 % за 10 мин
Ag110 (225 дней) Ag (тв.) + Ag+ Травленое Ag, 0,1 М AgNO3 18-20 100 а. с. за несколько секунд
Полированное Ag, 0,1 М AgNO3 18-20 100 а. с. за 24 ч
AgBr (тв.) + Ag+ 187 мл свежего AgBr, 40 мл 0,0055 М AgNO3 25 78 % за 3 мин
187 мг перекристаллизованного AgBr, 40 мл 0,0055 М AgNO3 25 56 % за 15 мин
Сурьма
Sb122 (2,8 дня) Sb (тв.) + Sb(III) 0,1 М SbCl3; 4 н. НС1 19 ~ 15 а. с. за 30 с
То же 67 к 40 а. с. за 30 с
Таллий
у |204 (3,5 года) T1(OH)3 (тв.) + T1(I) Свежий Т1 (ОН)3, TI (I) в NH3 • Н2О 18-20 20 % за 10 мин
474
Новый справочник химика и технолога
Продолжение таблицы 2.1.37
Изотоп Реагирующие вещества Концентрация реагирующих веществ; среда Т,°С Степень обмена (по массе)
Таллий
гр|204 (3,5 года) Т12СгО4 (тв.) + [T1(CN)6]3’ Свежий Т12СгО4, [Tl (CN)6]’“; NH3- Н2О, CN 18-20 1 % за 210 мин
Теллур
Те’27 (9,3 ч) Те (тв.) + Te(IV) 0,1 М ТеСЦ 19 95 а. с. за 30 с
То же 91 280 а. с. за 30 с
Торий
Th234 (24,5 дня) ThP2O6 (тв.) + Th(I V) 0,09 ммоль ThP2O6, 5 мл 2,2 • 10^ М ThCl4 18-20 75 % за 10 мин
Th(C2O4)2 + Th(IV) 0,12 ммоль Th(C2O4)2, 5 мл 1,9 • 10~5 М ThCl4 18-20 45 % за 10 мин
Th(C2O4)2 + Th(IV) 0,10 ммоль Th(C2O4)2, выдержанного 2,5 ч в горячем р-ре, 5 мл 1,3 • 10’5М ТЬСЦ 18-20 12 % за 10 мин
Углерод
С14 (5600 лет) Fe2C (тв.) + CO (r.) Рсо = 160 -Н 90 мм рт. ст.*1 250 5 ± 2 % за 30 мин
Рсо = 140 мм рт. ст. 322 29 ± 4 % за 7 мин
Fe2C (тв.) + CH4 (r.) Рсн4 = 60 мм рт. ст. 253 0 ± 1 % за 16 ч
Fe2C(TB.) + C2H2 (r.) РС = 113 -5- 149 мм рт. ст. 250 0 ± 3 % за 33 мин
Fe2C (тв.) + н-С4Н10 (r.) Рс н ~ 40 мм рт. ст. 250 0 ± 3 % за 26 мин
Fe2C (тв.) + (r.) Рс4н8 = 37 мм рт. ст. 250 2 ± 1 % за 480 мин
BaCO3 (тв.) + CO2 (г.) 178 мг ВаСО3, 2 л СО2, насыщ. парами Н2О; /?со2 = 760 мм рт. ст. 22 ~ 37 % за 65 ч
CH3COONa (ТВ.) + + (C2H3O2)2O (ж.) 2,0 ммоль NaC2H3O2, 48 ммоль (С2Н3О2)2О 18-20 55 % за 20 мин
Уран
и237 (6,8 дня) UF4 (тв.) + UO2+ 0,66 ммоль UF4, 90 мл 0,1 М UO2SO4; 0,16 М H2SO4 20-25 21 % за 70 ч
UF4 (тв.) + UF6 (ж.) 2,1 ммоль UF4, 1,5 ммоль UF6 98 ~ 18 % за 98 ч
*’ 1 мм рт. ст. = 133,3 Па.
*2 Бут-1-ен.
Литература
1. Несмеянов А.Н. Радиохимия. 2-е изд. М. 1978.
2. Радиоактивные индикаторы в химии. Основы метода / Под ред. В.Б. Лукьянова. 3-е изд. М., 1985.
Химическая кинетика и диффузия
475
2.2. Диффузия
(проф., д.т.н. М.А. Яблокова)
Перенос вещества внутри фазы может происходить либо только путем молекулярной диффузии, либо путем конвекции и молекулярной диффузии одновременно. Посредством одной молекулярной диффузии вещество перемещается, строго говоря, лишь в неподвижной среде. В движущейся среде перенос вещества осуществляется как молекулярной диффузией, так и самой средой в направлении ее движения или отдельными ее частицами в разнообразных направлениях.
В турбулентном потоке перенос молекулярной диффузией преобладает только вблизи границы фазы. При турбулентном течении возникают нерегулярные пульсации скорости, под действием которых, наряду с общим движением потока, происходит перемещение частиц во всех направлениях, в том числе и в поперечном. Конвективный перенос вещества, осуществляемый под действием турбулентных пульсаций, часто называют турбулентной диффузией. Роль молекулярной диффузии в турбулентных потоках незначительна, и ею обычно можно пренебречь, за исключением области вблизи поверхности раздела фаз.
2.2.1. Молекулярная диффузия
Молекулярной диффузией называется перенос распределяемого вещества, обусловленный беспорядочным тепловым движением молекул, атомов, ионов, коллоидных частиц. Молекулярная диффузия описывается Первым законом Фика, согласно которому масса вещества dm, диффундирующего за время dt через элементарную поверхность dS (нормальную к направлению диффузии), пропорциональна градиенту концентрации dc/dx этого вещества:
— = -DdS— (2.2.1.1)
dt dx
или
m = -DSt—. (2.2.1.2)
dx
Из выражения (2.2.1.2) следует, что удельный поток вещества, переносимого молекулярной диффузией через единицу поверхности S' в единицу времени t, или скорость молекулярной диффузии, составляет:
q. = — = ~D—. (2.2.1.3)
4 St dx 7
Градиент концентрации dc/dx представляет собой изменение концентрации диффундирующего вещества, приходящееся на единицу длины нормали между двумя поверхностями постоянных, но различных концентраций.
Коэффициент пропорциональности D в выражении закона Фика называется коэффициентом молекулярной диффузии, или просто коэффициентом диффузии. Знак
«минус» в правой части уравнения, описывающего закон Фика, указывает на то, что молекулярная диффузия всегда протекает в направлении уменьшения концентрации распределяемого компонента.
Согласно уравнению (2.2.1.1), коэффициент диффузии выражается следующим образом:
r -|_ mdx _ кг-м _ м2
*• J" ~dcSt ' кг 2 ”Т’
L J —г • М -С м
откуда вытекает физический смысл коэффициента D (до сокращения одноименных величин). Коэффициент диффузии показывает, какая масса вещества диффундирует в единицу времени через единицу поверхности при градиенте концентрации, равном единице.
Коэффициент молекулярной диффузии представляет собой физическую константу, характеризующую способность данного вещества проникать вследствие диффузии в неподвижную среду. Величина D, таким образом, не зависит от гидродинамических условий, в которых протекает процесс.
Значения коэффициентов диффузии D являются функцией свойств распределяемого вещества, свойств среды, через которую оно диффундирует, температуры и давления. Обычно величины D возрастают с увеличением температуры и понижением давления (для газов). В каждом конкретном случае значение коэффициента молекулярной диффузии определяют по опытным данным или по теоретическим и полуэмпирическим уравнениям [1-3] с учетом температуры и давления, при которых протекает процесс диффузии.
2.2.1.1. Диффузия в газах
Молекулярная диффузия в газовых системах является предметом теоретических и экспериментальных исследований на протяжении уже более 100 лет [2, 4-6]. Например, в табл. 2.2.1 приведены полученные экспериментально [2, 4-6] значения коэффициентов само-диффузии некоторых газов при различных температурах и давлении 0,1 МПа. Табл. 2.2.2 содержит определенные опытным путем [2, 5] значения коэффициентов взаимной молекулярной диффузии в газах и парах при тех же условиях (р = 0,1 МПа). Однако надежные значения коэффициентов диффузии можно получить и с помощью полуэмпирических уравнений, коррелирующих опытные данные.
Возможно, наилучшей из многочисленных, предложенных различными авторами расчетных зависимостей является корреляция Фуллера, Шеттлера и Гиддингса [6, 7], которая была получена с помощью ЭВМ с целью выявления наиболее точного соответствия между уравнением и приблизительно 340 экспериментальными точками. Это уравнение имеет вид:
ОАВ =____0.00010»r-”_.._pZnr (2.2.1-4)
/>[(ХОаш + (£Пв''1] и‘
476
Новый справочник химика и технолога
ще Т — температура, К; р — абсолютное давление, МПа; (£И)а и (ЕИ)В — молярные объемы веществ А и В в жидком состоянии при нормальной температуре кипения; Л/д и Л/в— молярные массы веществ А и В.
Величины (SF)a и (2И)в получаются суммированием атомных диффузионных объемов для каждого компонента бинарной системы. Значения V и SK для простых молекул приводятся в табл. 2.2.3. Среднее расхождение между расчетными и опытными значениями для бинарных газовых систем при низких давлениях составляет 4-7 %.
Современная математическая теория неоднородных газов [7] расширяет пределы применимости классической кинетической теории путем введения допущения о взаимодействии молекул газа. Молекулы отталкиваются друг от друга, когда они располагаются в непосредственной близости, и притягиваются, находясь друг от друга на некотором расстоянии. Межмолекулярные потенциалы взаимодействия выражаются эмпирически любой из ряда «потенциальных функций», самой употребительной среди которых является [1-4, 8] потенциал Леннарда — Джонса:
Ф(г) = 4е
(2.2.1.5)
где Ф(г) — потенциальная энергия; г — расстояние между центрами молекул; 8, ст — параметры потенциала Леннарда — Джонса.
Основываясь на этой зависимости как на исходной, Чэпмен и Энског [2, 3, 8] развили теорию диффузии, приводящую к следующему уравнению для расчета коэффициента взаимной диффузии в бинарной газовой смеси при давлениях до 2 МПа:
. 3/7Г(л/А+л/в)/(л/Ал/)”|1/2
Пдв = 1,858 • 10'8Г3/2 J , (2.2.1.6)
AD ' I 7 X Z
где Т— температура, К; р — абсолютное давление, МПа; Л/А и — молярные массы веществ А и В; стАВ — характеристическое расстояние, A; £1D — интеграл столкновений для диффузии.
Для вычисления параметров стАВ и часто используют следующие соотношения [3]:
«лв=(а*+ав)/2. (2-2.1.7)
Пв = 4-+—2— + —£_ + _®— (2.2.1.8) Т* expDT. exp ft; exp НТ.
где Т, = Шедв; А = 1,06036; В = 0,15610; С = 0,19300; D= 0,47635; Е = 1,03587; F = 1,52996; G = 1,76474;
77 = 3,89411; к — постоянная Больцмана; sAB — константа взаимодействия Леннарда — Джонса для бинарной смеси:
е =(е -8 )' 2.
АВ X А В /
(2.2.1.9)
Коэффициенты 8 и ст, необходимые для расчетов по формулам (2.2.1.7)-(2.2.1.9), представлены в [7, 8].
Согласно уравнению (2.2.1.6), коэффициент диффузии в газах обратно пропорционален давлению, что соответствует экспериментальным данным при низких и умеренных давлениях. Температурная зависимость коэффициента диффузии при р - const определяется соотношением [3]
j»3/2
> .
“ Ц>(г)
(2.2.1.10)
Анализ функции Qd(7) показывает, что величина Qo пропорциональна Т в степени от 0 до -0,5; следовательно, можно записать
(2.2.1.11)
где п =1,75^2; £)Аво— коэффициент молекулярной диффузии при температуре То = 273,15 К. Если известно значение коэффициента диффузии Z)AB0 при нормальных условиях (температуре То и давлении Ро = 0,1 МПа), то значение его DAB при температуре Т и давлении р можно найти из уравнения
D =d A.IZ.
АВ ABO rr
(2.2.1.12)
Для систем, где имеются экспериментальные данные в сравнительно широком диапазоне температур, проведено [2] обобщение этих данных по удобному для практических расчетов соотношению (2.2.1.11). Значения коэффициента Z)2o (А\во при температуре То = 293 К) и показателя степени п приведены в табл. 2.2.4. При температурах до 1200 К они получены в непосредственных опытах по диффузии, а при температурах от 1200 до 3000 К потенциал межмолекулярного взаимодействия определен из опытов по рассеянию газового пучка в среде второго газа [2, 5].
Диффузия при высоких давлениях изучена еще не достаточно. Приведенные выше уравнения применимы при давлениях до 2 МПа [6]. При более высоких показателях в правую часть уравнения (2.2.1.12) следует вводить в качестве сомножителя коэффициент сжимаемости газовой смеси. Зависимость коэффициентов молекулярной диффузии газов от давления представлена в табл. 2.2.5. Данные [5] о коэффициентах диффузии паров некоторых металлов приведены в табл. 2.2.6.
Химическая кинетика и диффузия
477
Таблица 2.2.1
Коэффициенты самодиффузии газов D при различных температурах и давлении р = 0,1 МПа
В скобках указано давление р в МПА.
Газ Т,К D • 104, м^/с Газ Г, К D- 104, м^/с Газ Г, К D • 104, м^/с
Азот (диазот), n2 77,5 194,5 273 293 298 353 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500 6000 6500 7000 7500 8000 8500 9000 9500 10000 1,0168 0,104 0,179 0,212 0,221 0,287 1,56 3,08 5,05 7,48 10,4 13,8 17,6 21,9 26,6 31,8 37,3 43,2 49,6 56,5 63,5 71,0 78,9 87,0 95,5 Аргон, Аг 4500 5000 5500 6000 6500 7000 7500 8000 8500 9000 9500 10000 11000 12000 13000 14000 15000 322,4 22,1 26,8 31,9 37,4 43,3 49,5 56,1 62,8 69,9 77,4 85 93,2 111 129 148 169 190 0,00272 (6,80) 0,00194 (8,88) 0,00145 (15,12) 0,000875 (24,3) 0,00270 Гелий, Не 64,8 76,1 192 296 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500 6000 6500 7000 8000 9000 10000 11000 12000 13000 14000 15000 0,147 0,187 0,843 1,68 13,3 26,9 46,7 70,3 98,6 130 167 207 250 299 350 407 467 595 738 898 1070 1260 1460 1670 1890
11000 12000 13000 14000 15000 113 133 153 174 197 (9,50) 0,00161 (11,77) 0,00095 (19,40) Диоксид углерода, СО2 194,5 194,6 296 298 312,6 0,0500 0,0516 0,109 0,111 0,1248
Аргон, Аг 77,5 90 194 194,5 273 0,0134 0,0180 0,0830 0,0833 0,157 0,179 0,212 0,249 1,51 3,00 287 0,000659 (29,10) 0,00115 (15,65) 0,152 318 353 362,4 273 0,129 0,153 0,1644 0,00598 (1,43)
295 326,5 353 1000 1500 Водород (диводород), н2 20,4 85 273 288,2 293,2 0,00816 0,172 1,285 1,43 1,40 273,2 0,01501 (0,606) 0,002902 (2,55) 0,01126
2000 2500 5,03 7,50 Вода (пар), Н2О 273 0,277 307,9 (0,78) 0,01016
3000 3500 4000 10,4 13,9 17,8 Гелий, Не 273 14,4 19,6 1,945 0,0124 0,0199 308 (1,12) 0,002347 (4,098)
478
Новый справочник химика и технолога
Продолжение таблицы 2.2.1
Газ Г, К D • 104, iZ/c Газ Г, К D • 104, mVc Газ Г, К D- 104, м*/с
Диоксид углерода, СО2 373,0 296,2 296,9 0,001378 (5,845) 0,004285 (2,479) 0,000942 (6,995) 0,0002869 (8,928) 0,000649 (7,859) 0,000594 (7,757) 0,000457 (7,934) 0,0002732 (9,805) 0,000309 (8,601) 0,000932 (6,968) 0,0002973 (8,826) 0,000926 (6,981) 0,00768 (2,161) 0,00390 (4,129) 0,00529 (3,098) 0,002088 (7,Ш) 0,001316 (10,22) 0,000888 (13,57) 0,000497 (20,48) 0,000638 (17,17) 0,01121 (1,454) 0,0847 (0,1243) 0,0424 (0,249) Диоксид углерода, СО2 296,1 296,6 296,4 296,5 295,7 296,2 296,5 269,0 296,0 297,1 296,0 295,7 295,9 295,6 296,0 317,7 317,6 297,9 297,9 317,8 318,2 317,8 298,3 0,0320 (0,277) 0,0150 (0,638) 0,0104 (0,933) 0,0071 (1,849) 0,0163 (0,634) 0,0101 (0,924) 0,00494 (1,859) 0,00476 (2,14) 0,00384 (2,81) 0,2060 (0,0506) 0,1030 (0,098) 0,0910 (0,118) 0,0500 (0,218) 0,0437 (0,246) 0,0065 (1,605) 0,0111 (0,970) 0,0131 (0,920) 0,0110 (0,950) 0,00577 (0,167) 0,00529 (1,630) 0,00519 (1,630) 0,00415 (2,720) 0,00325 (3,420) Диоксид углерода, СО2 318,3 298,2 298,1 273,3 273,4 286,6 294 295 303,2 317,7 303,2 296,8 273,75 286,4 316,8 317,6 317,5 298 295,8 295,5 294,5 296,3 303,2 0,00340 (3,430) 0,00271 (3,430) 0,00179 (4,760) 0,00131 (3,430) 0,00183 (3,430) 0,00119 (4,780) 0,00260 (3,640) 0,00197 (3,840) 0,00166 (4,720) 0,00197 (4,770) 0,00158 (4,770) 0,00410 (1,670) 0,00130 (3,430) 0,00114 (4,750) 0,00149 (6,120) 0,00105 (6,810) 0,00130 (6,810) 0,00106 (6,120) 0,00228 (3,640) 0,00250 (3,640) 0,00306 (3,060) 0,00150 (4,770) 0,00126 (5,950)
Химическая кинетика и диффузия
479
Продолжение таблицы 2.2.1
Газ Г, К D- 104, м2/© Газ Г, К D- lOW/c Газ Г, К D • 104, Л
Диоксид углерода, СО2 303,3 303,3 313,4 296,8 303,8 313,6 313,6 314,5 313,5 313,4 305,06 305,06 313,25 313,25 313,08 298,3 305,06 313,26 313,26 313,26 298,1 298,1 0,00105 (6,120) 0,000822 (6,550) 0,000638 (8,080) 0,00269 (7,060) 0,00192 (4,560) 0,00154 (5,760) 0,00113 (6,800) 0,000924 (7,50) 0,000804 (7,950) 0,000530 (8,560) 0,000563 (7,430) 0,000538 (7,465) 0,000507 (8,605) 0,000550 (8,605) 0,000293 (9,630) 0,000105 (12,490) 0,000345 (7,360) 0,000300 (8,605) 0,000182 (10,850) 0,000149 (12,990) 0,000216 (6,510) 0,000104 (9,020) Диоксид углерода, СО2 298,1 273,2 273,3 273,2 313,21 298,1 298,2 298,2 313,1 313,2 298,4 298,0 302,1 300,6 298,1 297,9 298,1 323,2 323,2 323,1 2987,0 298,2 0,0000822 (9,020) 0,0000456 (7,107) 0,0000599 (3,840) 0,0000404 (13,790) 0,000104 (14,490) 0,000103 (8,905) 0,0000702 (14,990) 0,000133 (6,510) 0,000166 (10,790) 0,000130 (11,980) 0,000111 (6,510) 9,05 • 10~5 (11,32) 13,7-105 (10,31) 7,02 • 10’5 (17,89) 9,34- 10’5 (10,31) 7,64 • 10’5 (13,03) 5,12 • 10 5 (56,20) 9,16- Ю 5 (23,23) 8,84 10 5 (25,00) 7,04- 105 (47,05) 4,09 • 10’5 (40,25) 4,34- 10~5 (40,25) Диоксид углерода, СО2 298,0 298,2 298,2 272,9 273,2 273,2 273,1 323,0 322,9 322,9 322,9 298,2 298,1 296 1103 1180 1204 1213 1218 1250 1278 1302 1321 1330 1338 1372 1416 1434 1445 1450 1451 1471 3,54 • 105 (56,20) 3,18- IO 5 (75,2) 2,53 • 10’5 (102,30) 4,04 • 10’5 (13,71) 3,2 • 105 (30,71) 2,46 • 10’5 (51,20) 1,98- Ю 5 (80,25) 4,98 • 10’5 (47,03) 4,53 • 10"5 (59,65) 3,92 • Ю 5 (83,70) 3,48- IO 5 (102,90) 3,51 • 10’5 (56,10) 2,45 • 10 5 (103,05) 0,109 1,63 1,78 1,78 1,94 2,04 2,12 2,19 2,26 2,19 2,38 1,99 2,30 2,34 2,56 2,83 2,56 2,47 2,62
480
Новый справочник химика и технолога
Продолжение таблицы 2.2.1
Газ Г, К D • 104, м^/с Газ Г, К D • 104, м^/с Газ г, к D • 104, м^/с
Диоксид углерода, СО2 1478 1487 1490 1498 1576 1580 1580 1607 1610 1654 1665 1671 1680 1703 1719 1745 1782 1796 1944 2,69 2,88 2,98 2,49 3,12 2,78 3,33 3,12 2,95 3,21 3,29 3,13 3,50 3,33 3,45 3,31 3,67 3,59 4,30 Криптон, Кг 5500 6000 6500 7000 8000 9000 10000 11000 12000 13000 14000 15000 194,5 16,4 19,2 22,2 25,5 32,6 40,7 49,6 59,6 70,2 81,5 93,7 107 0,0257 Метан, СН4 90 194,5 273 298 353 0,0266 0,0992 0,206 0,240 0,318
Неон, Ne 77,5 194,5 273,0 298,0 353,0 293 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500 6000 6500 7000 8000 9000 10000 11000 12000 13000 14000 15000 0,0492 0,255 0,452 0,516 0,703 0,473 3,93 7,76 12,7 18,5 25,2 32,6 41,1 50,4 60,7 72,0 83,8 96,5 НО 139 172 207 244 284 325 368 414
Ксенон, Хе 273 329,7 377,8 300,3 293 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500 6000 6500 7000 8000 9000 10000 11000 12000 13000 14000 15000 0,0480 0,0684 0,0900 0,058 0,044 0,502 1,01 1,65 2,43 3,32 5,32 5,42 6,64 7,97 9,43 10,9 12,6 14,4 18,2 22,5 27,2 32,4 37,7 43,4 49,4 55,7
Кислород (дикислород), о2 77,5 194,5 318 273 298 353 0,0153 0,104 0,230 0,181 0,232 0,301
Криптон, Кг 199 273 292,7 293 373 474 1000 1500 2000 2500 3000 3500 4000 4500 5000 0,045 0,083 0,090 0,093 0,140 0,214 0,853 1,72 2,83 4,14 5,67 7,38 9,32 11,4 13,8
Оксид азота(1) (оксид диазота), N2O 273 0,0890
Оксид углеро-Да(П), СО 273 194,7 273,2 319,6 373,0 0,175 0,109 0,190 0,247 0,323
Химическая кинетика и диффузия
481
Таблица 2.2.2
Коэффициенты взаимной диффузии в газах и парах Длв? полученные различными исследователями при давлении р = 0,1 МПа [2]
Газ Г, К D- 104, м2/с
I. Азот, N2
Аммиак, NH3 273 0,214
Аргон, Аг 244,2 0,1348
274,6 0,1689
293 0,199
303,5 0,2433
Бензол, С6Н6 273 0,0823
311,3 0,1022
Бутан, С4Н10 298 0,0960
(2)-Бут-2-ен, С4Н8 298 0,095
Вода (пар), 273 0,232
Н2О 297,93 0,256
307,4 0,260
312,79 0,268
327,95 0,290
328,4 0,303
328,8 0,313
349 0,354
352 0,359
Водород (диводород), 137 0,173
н2 153 0,208
193 0,368
253 0,600
273 0,699
288 0,743
289,2 0,737
293 0,780
298 0,780
300 0,80
303,5 0,852
315,8 0,849
335,5 1,001
347,6 0,991
373 1,20
400 1,270
401,0 1,30
435 1,45
436 1,61
471 h? 1
473 1,86
483 1,92
573 2,56
583 2,64
673 3,36
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Водород (диводород), н2 705 773 873 973 1000 1083 3,54 4,15 5,12 6,06 6,30 7,20
Гекса-1,5-диен, СбНю 288,1 0,0772
Гексан, 263 0,0630
СбН14 273,0 0,0684
283,0 0,0734
288,6 0,0753
293,0 0,0784
Гексафторид серы 289,8 0,103
(шестифтористая сера), 328,0 0,115
SF6 348,0 0,130
373,0 0,147
410,0 0,179
455,0 0,218
473,0 0,231
Гелий*1, 243,2 0,477
Не 275,0 0,596
289,4 0,651
293 0,705
298 0,71
300 0,743
600 2,40
900 4,76
1200 7,74
1300 8,67
1500 11,41
1800 15,80
2100 20,79
2400 26,39
2700 32,56
3000 39,28
289,4 0,651
298,2 0,687
303,5 0,719
315,6 0,788
323,2 0,766
332,5 0,811
348,4 0,915
353,2 0,893
374,5 1,07
383,2 1,08
400,0 1,15
413,2 1,20
433,0 1,31
482
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D- 104, м^с
Гелий ♦*, 443,2 1,29
Не 470,0 1,53
473,2 1,569
498,2 1,65
Гептан, 263 0,0561
с7н16 273 0,0600
283 0,0653
293 0,0702
303 0,0750
313 0,0807
Декан, CioH22 363,9 0,0763
2,3 -Диметилбута-1,3-диен, СбНю 288,1 0,0748
2,3-Диметилбутан, С$Н14 288,7 0,0751
2,3-Диметилбут-2-ен, СбН12 288,3 0,0705
2,4-Диметилпентан, с7н16 303,1 0,0744
Диоксид углерода, 288 0,158
СО2 293 0,159
298 0,165
300 0,173
315,4 0,184
348 0,223
373 0,257
410,0 0,307
435 0,323
455,0 0,361
473 0,371
483 0,387
573 0,509
583 0,55
600 0,605
673 0,683
705 0,75
773 0,898
873 1,067
900 1,217
973 0,253
1000 1,33
1083 1,46
1200 1,976
Дициан, 273 0,0895 •
c2n2 1073 1,000
1273 1,346
Додекан, С]2Н26 399,4 0,0829
Продолжение таблицы 2.2.2
Газ т,к D • 104, м^с
Изобутан, С4Ню 298 0,0908
Изооктан, 253 0,0484
с8н18 263 0,0546
273 0,0587
283 0,0630
293 0,0682
303 0,0729
313 0,0776
Иод (дииод), 273 0,070
ь 583 0,275
705 0,377
773 0,445
873 0,558
Кислород (дикислород), О2 293 0,219
Ксенон, 242,2 0,0854
Хе 274,6 0,1070
303,45 0,1301
334,2 0,1549
Метилциклопентан, с6н12 285,9 0,0758
Натрий, 526,5 0,68
Na 655 0,91
Нонан, С<>Н2о 339,8 0,0737
Оксид азота(П), 273 0,1379
NO 1073 1,532
1273 2,073
Оксид углерода(П), 194,7 0,105
СО 273 0,189
288 0,211
319,6 0,242
373,0 0,318
Октан, С8Н18 303,1 0,0726
Пиперидин, c5HnN 314,9 0,0953
Пиридин, C5H5N 317,9 0,1068
Ртуть, Hg 273 0,01190
Тиофен, C4H4S 302,1 0,0992
2,2,4-Триметилпентан, с8н18 303,1 0,0713
2,3,3 -Триметилпе нтан 363,8 0,0681
Циановодород 273 0,1290
(синильная кислота), 1073 1,435
HCN 1273 1,940
Циклогексан, СбН12 288,5 0,0746
Химическая кинетика и диффузия
483
Продолжение таблицы 2.2.2
Газ Г, К D- 104,м2/с
Четыреххлористый углерод (тетрахлорид углерода), СС14 273 0,0737
Этан, с2н6 298 0,148
Этанол, С2Н5ОН 273 0,111
Этилен, с2н4 273 298 0,144 0,163
II. Аргон, Аг
Аммиак, NH3 254,7 275,1 308,1 333,1 0,150 0,175 0,222 0,253
Ацетилен, с2н2 287,0 0,152
Водород (диводород), Н2 118 154 197 231 242,2 273 274,2 287,9 291,0 293 295,0 296 303,9 313,0 341,2 344,8 373,0 404,0 418 436,0 448,0 473,0 476 523 573,0 628,0 692,5 806,0 958,0 1069,0 0,151 0,247 0,391 0,510 0,562 0,674 0,698 0,828 0,764 0,772 0,819 0,821 0,830 0,874 1,010 1,05 1,26 1,46 1,632 1,68 1,76 1,96 1,918 2,275 2,619 3,21 3,684 4,86 6,81 8,10
Гексан, с6н14 288,6 0,0662
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Гексафторид серы 328,0 0,100
348,0 0,112
373 0,127
410,0 0,151
447,0 0,183
472,0 0,200
Гелий *2, 118 0,153
Не 143 0,212
169,2 0,285
177 0,304
181,2 0313
194,7 0,347
202,2 0,384
214,7 0,390
223,2 0,414
225 0,463
251 0,544
259 0,583
269 0,639
273,0 0,637
273,2 0,645
276,2 0,646
280 0,703
287,0 0,684
287,9 0,697
288 0,701
289 0,731
291,0 0,715
292,2 0,736
296 0,740
300 0,76
313,0 0,815
315,2 0,833
317,2 0,797
318 0,825
323 0,832
328 0,851
333,2 0,932
337 0,889
343,0 0,961
346,2 0,924
353,2 0,982
353,3 1,005
354 1,000
354,2 0,979
366 1,060
373,0 1,105
383,2 1,120
387 1,165
484
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D- Ю4,^
Гелий *2, 393,9 1,203
Не 401,0 1,26
405 1,288
413,2 1,24
418,0 1,371
434,0 1,45
443,2 1,40
464,0 1,61
473,2 1,61
479,5 1,652
498,2 1,73
500 1,86
1000 6,06
1100 7,68
1500 12,29
2000 20,40
2500 30,22
3000 41,70
3500 54,3
4000 68,6
4500 84,2
5000 101
5500 120
6000 139
7000 182
7500 205
8000 230
8500 256
9000 284
9500 313
10000 343
11000 406
12000 474
13000 546
14000 624
15000 705
Гептан, с7н16 303,2 0,0658
2,3-Диметилбутан, C6Hi4 288,9 0,0652
2,4-Диметилпентан, с7н16 303,2 0,0655
Диоксид углерода, 273 0,124
СО2 288,6 0,135
293 0,139
328,0 0,185
348,0 0,208
373,0 0,235
410,0 0,280
Продолжение таблицы 2.2.2
Газ Г, К D- 104, mVc
Диоксид углерода, 455,0 0,336
СО2 473,0 0,363
Изооктан, 253 0,0407
С8Н18 263 0,0473
273 0,0500
283 0,0509
293 0,0575
303 0,0614
313 0,0657
Калий, К 723,0 1,03
Кислород (дикислород), 243,2 0,135
О2 273 0,181
293 0,200
304,5 0,202
334,0 0,239
Криптон, 199,5 0,072
Кг 273 0,123
288 0,128
303 0,140
318 0,153
343 0,179
353 0,197
373 0,216
381 0,227
407 0,254
473 0,327
Ксенон, 194,7 0,0518
Хе 273 0,0951
288 0,102
303 0,114
315,0 0,123
318 0,128
329,9 0,137
354,1 0,1620
378,0 0,178
394,1 0,1920
Метилциклопентан, СбН12 288,6 0,0715
Натрий, Na 655 0,88
Неон, 90 0,036
Ne 118 0,0606
154 0,0984
194,5 0,159
197 0,155
231 0,207
273 0,281
288 0,300
Химическая кинетика и диффузия
485
Продолжение таблицы 2.2.2
Газ Г, К £)• 104, м^с
Неон, 296 0,317
Ne 300 0,325
303 0,327
318 0,357
353 0,442
374 0,474
473 0,738
573 0,973
680 1,302
Октан, 273 0,0497
с8н18 383 0,0516
293 0,0561
303 0,0610
313 0,0639
323 0,0667
333 0,0735
Толуол, 273 0,0714
с7н8 283 0,0783
283 0,0826
293 0,087
301 0,087
303 0,0885
313 0,0942
323 0,0998
333 0,1062
2,2,4-Триметилпентан, 296 0,0662
с8н18
Цезий, 299 0,19
Cs 723 0,67
Циклогексан, 287,2 0,0791
с6н)2
Этилен, 298 0,170
С2Н4 317 0,184
340 0,210
379 0,258
407 0,288
III. Вода (пар), Н2О
Воздух * 273 0,251
289 0,244
297,8 0,257
298 0,260
312,4 0,277
315 0,288
332 0,305
365,4 0,345
373 0,353
381 0,376
382 0,382
420 0,52
425 0,472
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м^с
Воздух *3 476 0,586
483 0,623
489 0,630
519 0,690
528,5 0,709
542 0,754
573 0,849
584,5 0,861
618,5 0,905
634 0,961
675 1,08
677,5 1,10
773 1,556
923 2,191
1073 2,650
1093 2,808
1273 3,253
1373 3,938
1463 4,480
1493 4,490
Гелий, 298 0,908
Не 307 0,932
328,3 1,011
352,3 1,121
Диоксид углерода, 297,7 0,164
СО2 307,3 0,181
322,5 0,202
328,5 0,205
352,2 0,245
365,4 0,254
372,4 0,2594
432,5 0,3399
433,0 0,3418
433,7 0,3431
434,0 0,3488
Кислород (дикислород), 298,0 0,270
О2 307,9 0,292
328,5 0,328
352,2 0,367
500 0,691
1000 2,21
1500 4,32
2000 6,96
2500 10,09
3000 13,65
Метан, СН4 297,8 307,0 313,0 323,0 328,6 0,251 0,278 0,292 0,297 0,313
486
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Метан, 333,0 0,331
СН4 352,1 0,356
Пропан, с3н8 297,8 0,156
Фреон-12, CF2C12 298 0,105
Этан, с2н6 297,8 0,191
Этилен, 297,8 0,178
С2Н4 307,6 0,204
328,3 0,233
352,4 0,247
IV. Водород (диводород), Н2
Аммиак, NH3 273 0,736
Ацетилен, с2н2 273 0,458
Ацетон, С3Н6О 273 0,361
Бензол, 253 0,2451
С6Н6 263 0,2722
273 0,3062
283 0,3161
293 0,3390
303 0,3576
318 0,3993
Бром (дибром), Вг2 273 0,402
Бутан, 287,9 0,361
с4н10 354,2 0,507
430 0,763
(7)-Буг-2-ен, с4н8 298 0,378
Вода (пар), 292,95 0,850
Н2О 297,93 0,802
307,0 0,915
312,79 0,937
322,5 1,000
324,5 1,012
328 1,06
352,2 1,200
365,4 1,215
372,34 1,28
Воздух 273 0,66
289 0,596 •
291,5 0,700
Гексан, 253 0,2225
СбН14 263 0,2415
273 0,2597
283 0,2795
Продолжение таблицы 2.2.2
Газ Г, К D 104, м2/с
Гексан, 288,6 0,2914
СбН]4 293 0,3008
Гекса-1,5-диен, СбНю 288,1 0,298
Гексафторид серы 286,2 0,396
(шестифтористая сера), 290,0 0,418
SF6 298 0,434
306,9 0,458
313,0 0,488
344,4 0,570
370,8 0,647
376,0 0,662
401,0 0,750
418 0,838
429,0 0,855
473,0 1,03
273 1,13
288 1,24
295 1,28
295,5 1,289
378 1,952
485 2,938
Гелий, 52 0,076
Не 58 0,093
118 0,318
137 0,344
143 0,443
153 0,521
177 0,657
225 0,985
273 1,21
292,4 1,383
295 1,550
296 1,550
380 2,401
476 3,554
573 4,89
692 6,76
Гептан, 263 0,2122
с7н16 273 0,2263
283 0,2433
293 0,2848
313 0,3008
Дейтерии (дидейтерий), 65,1 0,081
d2 76,6 0,116
115 0,253
155 0,418
192 0,583
197 0,649
232 0,857
Химическая кинетика и диффузия
487
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м^/с
Дейтерий (дидейтерий), 273 1,13
d2 288 1,24
295 1,28
295,5 1,289
378 1,952
485 2,938
Декан, С10Н22 363,8 0,293
2,3-Диметилбута-1,3-диен, 288,1 0,312
СбНю 303,3 0,297
2,3-Диметилбут-2-ен, СбН12 288,8 0,299
2,3-Диметилбутан, СбН14 288,1 0,296
2,4-Диметилпентан, с7н16 273 0,407
Диоксид углерода, 250 0,481
СО2 257 0,488
259 0,50
267 0,548
273 0,565
279 0,606
293 0,629
296,5 0,635
298 0,646
332 0,885
348 0,893
368 0,991
373 0,994
404,0 1,15
435 1,35
473 1,51
483 1,66
573 2,18
583 2,29
673 2,82
705 3,06
773 3,57
873 4,47
973 5,24
1000 5,56
1083 5,70
Дициан, C2N2 292,9 0,354
Диэтиловый эфир, С4Н ioO 399,6 0,311
Додекан, С12Н26 273 0,535
Изоокган, 263 0,2251
с8н18 273 0,2400
283 0,2580
293 0,2760
303 0,2945
313 0,3162
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Кислород (дикислород), 142 0,174
о2 153 0,215
273 0,692
300 0,824
500 2,07
1000 7,02
1500 13,7
2000 22,2
2500 32,1
3000 43,4
Ксенон, 242,2 0,410
Хе 274,2 0,508
303,9 0,612
341,2 0,751
Метан, 273 0,625
СН4 298 0,726
Меганол, 263 0,4690
СН3ОН 273 0,5033
283 0,5402
293 0,5786
303 0,6229
313 0,6656
Метилциклопентан, с6н12 288,5 0,312
Натрий, Na 655 3,14
Неон, 115 0,225
Ne 155 0,392
197 0,588
232 0,778
242,2 0,792
274,2 0,974
295 1,170
341,2 1,405
379 1,7687
484 2,688
575 3,585
680,5 4,68
Нонан, С9Н20 339,8 0,284
Оксид азота(1) (оксид диазота), 253 0,2045
n2o
Оксид азота(П), NO 273 0,643
Оксид углерода(П), 273 0,651
СО 295,6 0,743
315,0 0,856
345,0 0,979
488
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Оксид углерода(П), 374,5 1,14
СО 401,0 1,27
436,0 1,49
471,0 1,68
Октан, 273 0,2060
с8н18 283 0,2370
293 0,2533
303 0,2718
303,2 0,277
313 0,2908
323 0,3100
333 0,3330
Пиперидин, C5HnN 314,7 0,403
Пиридин, c5h5n 317,9 0,437
Пропан, С3Н8 273 0,385
Сернистый ангидрид (диоксид серы, оксид cepbi(IV)), 273 0,480
so2
Серный ангидрид (оксид серы(У1)), 273 0,379
so3
Сероуглерод 292,9 0,4255
(дисульфид углерода), CS2 305,8 0,4626
Тиофен, C4H4S 302,2 0,400
Толуол, 273 0,466
с7н8 283 0,5000
293 0,5327
301,0 0,570
303 0,5765
313 0,6044
323 0,6428
333 0,6844
2,3,3-Триметилпентан, с8н18 363,9 0,271
2,2,4-Триметилпентан, с8н18 303,0 0,292
Уксусная кислота, 273 0,4096
СН3СООН 283 0,4441
293 0,4851 .
303 0,5142
313 0,5455
323 0,5827
333 0,6161
343 0,6654
Продолжение таблицы 2.2.2
Газ Г, К D- 104, м*/с
Хлор (дихлор), С12 273 0,438
Хлорметан, СН3С1 273 0,453
Циановодород (синильная кислота), 273 0,565
HCN
Циклогексан, 253 0,2459
с6н12 263 0,2671
273 0,2861
283 0,3070
288,5 0,319
293 0,3265
303 0,3477
313 0,3731
Четыреххлористый
углерод (тетрахлорид углерода), 274 0,293
СС14
Этан, 298 0,537
с2н6
Этанол, 263 0,3229
С2Н5ОН 273 0,377
283 0,4016
293 0,4346
303 0,4673
313 0,5046
323 0,5465
339,9 0,5430
340 0,586
Этилацетат, 253 0,2432
С4Н8О2 263 0,2542
273 0,2730
283 0,2979
293 0,3161
303 0,3400
Этилен, 273 0,625
С2Н4
Этилпропионат, 273 0,326
CsHio02 300,8 0,372
300,9 0,409
V. Воздух
Авиационный бензин 283 0,0845
293 0,091
303 0,097
313 0,102
Аммиак, 293 0,227
NH3
Анилин, 298 0,0726
c6h5nh2 298,9 0,074
312,4 0,079
332 0,090
Химическая кинетика и диффузия
489
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Антрацен, С14Н16 372,2 0,0783
Ацетилен, 287,6 0,191
с2н2 289 0,206
Ацетон, С3Н6О 273 0,109
Бензол, 263 0,0711
С6Н6 273 0,0781
283 0,0831
293 0,0886
298 0,0930
303 0,0962
313 0,0993
315 0,1011
318 0,102
323 0,1060
340 0,117
343 0,124
363,8 0,135
377,8 0,147
418,0 0,179
440,2 0,187
451,5 0,199
473,0 0,222
526,8 0,272
530,2 0,269
530,8 0,271
573,8 0,318
579,2 0,332
617,5 0,361
Бензидин, C]2H]2N2 372 0,0555
Бифенил, 298 0,0727
С12Ню 491 0,160
Бром (дибром), Вг2 293 0,091
Бутан-1-о л, 298,9 0,087
С4Н10О 312,4 0,092
332 0,104
Бутан-2-ол, 298,9 0,089
С4Н10О 312,4 0,096
332 0,108
Бутилстеарат, С22Н44О2 293 0,030
Гексан, 263 0,0608
СбНи 273 0,0658
283 0,0709
293 0,0755
303 0,0793
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Гексан, 361,9 0,102
СбНи 399,0 0,120
415,4 0,125
453,9 0,143
487,3 0,161
526,7 0,180
571,2 0,210
575,5 0,217
Гексадекан, 287,97 0,0384
С1бН34 293,01 0,0392
297,98 0,0402
302,98 0,0425
307,88 0,0435
Гексафторид серы (шестифтористая сера), 289,8 0,096
SF6
Гелий, 273 0,63
Не 287 0,689
315,6 0,785
348,4 0,920
374,5 1,04
400,0 1,17
430,0 1,32
469,0 1,52
Гептадекан, 287,97 0,0403
С17Н36 293,01 0,0422
297,98 0,0423
302,98 0,0436
307,88 0,0453
312,94 0,0463
Гептан, 377,5 0,101
с7н16 410,2 0,111
414,2 0,119
434,5 0,126
445,8 0,127
462,5 0,133
469,2 0,138
474,8 0,148
498,2 0,151
527,2 0,168
564,5 0,190
Декан, 453,8 0,105
CioH22 464,5 0,110
467,0 0,111
467,8 0,112
478 0,113
482,2 0,1135
485 0,114
490,8 0,115
490
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D • 104, Л
Декан, 492,2 0,116
С10Н22 507,7 0,122
514,8 0,128
527,2 0,131
536,8 0,137
Дибромметан, 273 0,0708
С2Н4ВГ2 280,1 0,0736
288 0,0778
293 0,0813
Дибутил фталат, 288 0,034
С16Н22О4 292,9 0,0386
298 0,0415
303 0,0426
Дибутилфталат, 308 0,0442
С16Н22О4 313 0,0473
Диоксид углерода, 273 0,138
со2 290 0,152
293 0,158
373 0,257
473 0,321
573 0,507
673 0,670
773 0,921
873 1,062
973 1,270
1073 1,447
1273 1,835
1388 2,148
1473 2,432
1533 2,446
Дифенил, 298 0,0727
С12Н10 491 0,160
Дициан, 273 0,0970
C2N2 1073 1,080
1273 1,458
Диэтиловый эфир, 283,4 0,0835
С4Н10О 292,9 0,0893
Изоокган, CgHjg 323 0,060
Изопентан, 288 0,0774
С5Н,2 293 0,0782
303 0,0843
Иод (дииод), 273 0,0692
ь 298 0,1108
Кислород (дикислород), О2 273 0,178
Метан, СН4 273 0,196
Продолжение таблицы 2.2.2
Газ Г, К D- 104, м2/с
Метанол, 263 0,1249
СН4 273 0,1342
283 0,1424
293 0,1533
303 0,1627
313 0,1725
323 0,1808
Метилциклогексан, 283 0,0571
с7н14 293 0,0615
303 0,0660
313 0,0706
Нафталин, 298 0,0611
CioHg
Нитробензол, 298 0,0855
C6H5N(J2
Нитротрихлорметан
(хлорпикрин), 298 0,088
CCI3NO2
Нонан, 424,3 0,101
С9Н20 439 0,103
474,8 0,119
496,5 0,128
525,5 0,139
Октадекан, 287,97 0,0388
С18Н38 293,01 0,0396
297,98 0,0408
302,98 0,0418
307,88 0,0421
312,94 0,0445
Октан, 273 0,0497
CgHjg 283 0,0534
293 0,0576
298 0,0598
303 0,0622
313 0,0671
323 0,0718
405,5 0,103
424,6 0,112
465,2 0,127
495,2 0,141
510,8 0,150
528,4 0,162
Пентан-2-ол, 298,9 0,071
С5Н]2О 312,4 0,076
332 0,086
Пероксид водорода, Н2О2 333 0,188
Пропан-2-ол, 298,9 0,099
с3н8о 312,4 0,107
332,0 0,121
Химическая кинетика и диффузия
491
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Пропилацетат, с5н10о2 315 340 0,092 0,101
Ртуть, Hg 614 0,473
Сернистый ангидрид (оксид серы(1У)), SO2 293 0,122
Сероуглерод 253 0,0785
(дисульфид углерода), 263 0,0836
CS2 273 0,0887
283 0,0939
293 0,0983
303 0,1022
305,8 0,1120
1,2,3,4-Тегращдронафталин 484,2 0,159
(тетралин), 488,4 0,162
СюН22 493,8 0,166
501,2 0,170
503,6 0,172
507,0 0,175
Толуол, 273 0,0709
с7н8 293 0,080
198 0,0844
298,9 0,086
312,4 0,092
332 0,104
Топливо Т-5 523 0,103
573 0,123
623 0,145
673 0,168
Тяжелая вода (пар), 297,8 0,247
D2O 313,0 0,277
318,0 0,288
333 0,314
Уксусная кислота, 273 0,1064
СНзСООН 283 0,1141
293 0,1215
303 0,1297
313 0,1379
323 0,1477
333 0,1574
343 0,1615
Фосген, СОС12 273 0,095
Хлор (дихлор), С12 289 293 0,102 0,124
Хлорбензол, С6Н5С1 298,9 312,4 315,0 0,074 0,079 0,090
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Хлорбензол, 332 0,094
С6Н5С1 340 0,100
Хлороформ, СНС13 273 0,091
Хлорциан (цианид хлора), 273 0,111
C1CN
Циановодород (синильная кислота), 273 0,173
HCN
Циклогексан, 263 0,0600
с6н]2 273 0,0646
283 0,0696
293 0,0745
303 0,0787
313 0,0832
318 0,086
358,5 0,108
385 0,125
387,3 0,126
414,5 0,146
441 0,162
465,3 0,177
500,3 0,205
540,8 0,231
Четыреххлористый
углерод (тетрахлорид углерода), 273 0,07
СС14
Этанол, 273 0,105
С2Н5ОН 298 0,135
313,4 0,1372
339,9 0,1475
340 0,1534
Этилацетат, 298,9 0,087
с4н8о2 312,4 0,094
332 0,106
Этилпропионат, 273 0,069
CsHio02 300,8 0,082
300,9 0,0852
VI. Гелий, Не
Аммиак, 274,2 0,668
NH3 308,2 0,783
333,1 0,0881
Ацетилен, С2Н2 298,8 0,585
Бензол, с6н6 298 0,384
492
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Гексафторид серы 290,5 0,388
(шестифтористая сера), 313,0 0,430
SF6 344,4 0,517
376,3 0,601
399,4 0,669
430,0 0,755
464,6 0,865
Гептан, 263 0,1967
с7н16 273 0,2169
283 0,2328
293 0,2508
303 0,2689
313 0,2887
2,4-Диметилпентан, с7н16 303,2 0,263
Диоксид углерода, 250 0,392
СО2 (Данные от 250 К до 404 К описываются урав- 259 268 0,433 0,469
нением 280 0,515
£>=1,6- 10 5 - 7'184) 288 0,576
298,4 0,600
321 0,626
342 0,752
365 0821
382 0,867
404 1,005
Диоксид углерода, 260 0,050
СО2 273 0,54
283 0,57
287,0 0,592
298,2 0,612
318 0,684
323,2 0,69
344,8 0,782
353,2 0,80
358 0,81
373,0 0,877
383,2 0,884
400,0 1,02
413,2 1,04
443,2 1,13
473,2 1,28
498,2 1,4.1
Калий, К 723,0 2,22
Кислород 244,2 0,533
(дикислород), 274,0 0,646
О2 287,0 0,685
Продолжение таблицы 2.2.2
Газ Г, К D- 104, м2/с
Кислород 298,2 0,73
(дикислород), 304,4 0,771
о2 323,2 0,809
334,0 0,901
353,2 0,987
383,2 1,12
413,2 1,24
443,2 1,42
473,2 1,60
498,2 1,68
Криптон, 273 0,556
Кг 288 0,605
303 0,659
318 0,720
Ксенон, 273 0,501
Хе 288 0,550
303 0,604
318 0,655
354,2 0,721
394 0,882
Натрий, Na 655 2,17
Неон, 118 0,232
Ne 154 0,361
197 0,557
231 0,716
273 0,906
288 0,986
296 1,080
299 1,116
303 1,125
318 1,158
374 1,614
479,5 2,433
573,5 3,290
683 4,45
Нитробензол, C6H5NO2 298 0,372
Оксид углерода(П), 295,6 0,702
СО 314,8 0,788
345,0 0,914
374,5 1,05
400,0 1,17
436,0 1,35
470,0 1,54
Октан, 273 0,1993
С8Н18 283 0,2112
293 0,2336
Химическая кинетика и диффузия
493
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Октан, 303 0,2496
с8н18 313 0,2742
323 0,2867
333 0,3081
2,2,4-Триметилпентан, 253 0,1780
С8Н18 263 0,1938
273 0,2062
283 0,2220
293 0,2379
303 0,2534
313 0,2732
Цезий, 299 0,37
Cs 723 2,00
Этанол, С2Н5ОН 298 0,494
VII. Диоксид углерода, СО 2
Аммиак, 273 0,151
NH3
Ацетилен, 273 0,098
с2н2
Бензол, 263 0,0492
с6н6 273 0,0525
283 0,0561
293 0,0600
303 0,0640
313 0,0682
318 0,0715
Бром (дибром), 273 0,0664
Вг2
Гексафторид серы 290,8 0,0647
(шестифтористая сера), 328,0 0,0774
SF6 348,0 0,0872
373,0 0,0980
410,0 0,118
447,0 0,143
472,0 0,151
Дейтерий (дидейтерий), 250 0,337
d2 256 0,370
(данные описываются 267 0,383
уравнением D= 1,35 • 10~5- Г1’84) 297 0,494
346 0,594
372 0,660
Дибромбензол, 273 0,032
С6Н4Вг2
Дициан, 273 0,0788
C2N2
Диэтиловый эфир, 273 0,0541
С4Н10О 292,9 0,0632
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Иод (диод), 554 0,176
Ь 583 0,189
705 0,264
773 0,320
873 0,407
1000 0,522
1083 0,585
1136 0,63
1223 0,723
1275 0,79
Кислород 273 0,139
(дикислород), 287,8 0,154
02 293 0,157
300 0,163
500 0,415
1000 1,43
1500 2,98
2000 4,84
2500 7,17
3000 9,88
Метан, 273 0,153
СН4 298 0,180
Метанол, 263 0,0824
СН3ОН 273 0,0884
283 0,0937
293 0,0996
303 0,1060
313 0,1125
323 0,1185
Оксид азота(1) 194 0,0531
(оксид диазота), 273 0,098
n2o 288,6 0,107
298 0,117
312,6 0,128
362,4 0,168
Оксид азота(П), NO 273 0,129
Оксид углерода(П), 273 0,137
СО 296,1 0,152
298 0,160
315,4 0,185
348,0 0,222
373,0 0,253
410,0 0,295
455,0 0,359
473,0 0,381
494
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Оксиран, С2Н4О 298 0,0918
Пропан, с3н8 298 0,0863
Сернистый ангидрид
(оксид серы(1У)), 273 0,087
SO2
Серный ангидрид (оксид серы(У1)), 273 0,0708
so3
Сероуглерод 292,9 0,0726
(сульфид углерода(1У)), CS2 305,8 0,0789
Уксусная кислота, 273 0,0716
СНзСООН 283 0,0767
293 0,0831
303 0,0879
313 0,0938
323 0,1001
333 0,1066
343 0,1135
Хлор (дихлор), С12 273 0,078
Хлорметан, СН3С1 273 0,085
Циановодород (синильная кислота), 273 0,117
HCN
Этанол, 273 0,0686
С2Н5ОН 313,4 0,0898
339,9 0,1026
340 0,1064
Этилацетат, 253 0,0339
с4н8о2 263 0,0447
273 0,0483
283 0,0525
293 0,0564
303 0,0606
313 0,0647
Этилен, 298 0,15
с2н4
VIII. Дейтерий (дидейтерий), d2
Гептан, 263 0,1667
с7н16 273 0,1757
283 0,1901
293 0,2047
303 0,2181
313 0,2342
2,4-Диметилпентан, с7н16 303,2 0,224
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Октан, 273 0,1607
с8н18 283 0,1793
293 0,1957
303 0,2068
303,2 0,208
313 0,2295
323 0,2392
333 0,2555
2,2,4-Триметилпентан, 253 0,1482
с8н18 263 0,1587
273 0,1730
283 0,1858
293 0,1987
303 0,2118
303,2 0,212
313 0,289
IX. Оксид азота(1) (оксид диазота), N2O
Оксиран, С2Н4О 298 0,0914
Пропан, С3Н4 298 0,0860
X. Кислород (дикислород), о2
Аммиак, NH3 273 0,224
Ацетилен, с2н2 287,0 0,202
Бензол, 273 0,067
с6н6 298 0,073
311,3 0,1010
Гексан, с6н14 288,6 0,0754
Гексафторид серы 287,0 0,099
(шестифтористая сера), 317 0,109
SF6 340 0,129
379 0,160
408 0,193
2,3-Диметилбут-2-ен, с6н14 288,4 0,0742
Дициан, 273 0,0580
c2n2 283 0,0605
293 0,0664
303 0,0702
313 0,0748
1073 1,177
1273 1,592
Криптон, 298 0,146
Кг 343 0,198
377 0,244
408 0,268
Химическая кинетика и диффузия
495
Продолжение таблицы 2.2.2
Газ Г, К D-104, м2/с
Ксенон, 242,2 0,084
Хе 274,75 0,100
303,55 0,126
333,6 0,149
Метан, 300 0,229
СН4 500 0,580
1000 1,98
1500 4,03
2000 6,65
2500 9,82
3000 13,51
Метилциклопентан, СбН12 287,1 0,0744
Оксид азота(И), 273 0,1754
NO 1073 1,951
1273 2,636
Оксид углерода(П), 273 0,191
СО 298 0,21
300 0,224
500 0,540
1000 1,75
1500 3,47
2000 5,65
2500 8,24
3000 11,22
Октан, 263 0,0486
с8н18 273 0,0566
283 0,0606
293 0,0654
303 0,0705
313 0,0761
323 0,0816
Пиперидин, c5hun 315,0 0,0953
Пиридин, C5H5N 318,3 0,1050
Циановодород 273 0,1563
(синильная кислота), 1073 1,740
HCN 1273 2,35
Циклогексан, 253 0,0574
СбН12 263 0,0615
273 0,0663
283 0,0718
288,6 0,0731
293 0,0761
303 0,0810
Четыреххлористый
углерод (тетрахлорид углерода), 273 0,0651
ССЦ
Продолжение таблицы 2.2.2
Газ Г, К D • 104, м2/с
Этанол, С2Н5ОН 273 0,096
Этилен, с2н4 273 0,148
XI. Криптон, Кг
Аммиак, NH3 254,7 275,1 308,1 333,0 0,121 0,142 0,178 0,208
Неон, Ne 273 288 303 318 0,233 0,240 0,266 0,244
XII. Ксенон, Хе
Аммиак, NH3 274,2 308,4 331,1 0,114 0,145 0,173
Неон, Ne 273 288 303 318 0,186 0,202 0,221 0,244
XIII. Метан, СН4
Аммиак, NH3 273 1073 1273 0,1574 1,752 2,368
Монотритийметан, сн3т 273 0,0053 (2,0)*4 0,00248 (4,02) 0,00151 (6,0) 0,00109 (8,1) 0,00075 (10,0) 0,000575(12,5) 0,00040(15,0) 0,000248 (20,0) 0,000150 (30,0)
298 0,00291 (4,05) 0,00198 (6,0) 0,00138(8,0) 0,00098(10,0) 0,00087(10,0) 0,00069(12,5) 0,00071 (12,5) 0,00057(15,0) 0,00038 (20,0) 0,00021 (30,0)
323 0,00365 (4,0) 0,00246 (6,0) 0,00160 (8,03) 0,00113(10,0) 0,00094(12,48) 0,000664(15,0)
496
Новый справочник химика и технолога
Продолжение таблицы 2.2.2
Газ Г, К D- 104, м^с
Этилен, С2Н4 273 0,136
XIV. Неон, Ne
Аммиак, 274,2 0,298
NH3 308,4 0,378
333,1 0,419
Дейтерий 115 0,163
(дидейтерий), 155 0,286
d2 197 0,434
232 0,569
295 0,875
379 1,308
484,5 2,013
575,5 2,688
667 3,444
XV. Оксид углерода(П), СО
Аммиак, 233 0,108
NH3 273 0,151
313 0,209
353 0,269
Гексафторид серы 296,8 0,0887
(шестифтористая сера), 315,4 0,105
SF6 348,0 0,125
Продолжение таблицы 2.2.2
Газ Г, К D- 104. м2/с
Гексафторид серы 373,4 0,144
(шестифтористая сера), 410,0 0,174
SF6 455,0 0,212
473,0 0,222
Этилен, с2н4 273 0,116
XVI. Пентан, С5Н)2
Натрий, Na 655 0,23
XVII. Четыреххлористый углерод
(хлорид углерода(IV)), ССЦ
Трифторид бора, BF3 303 0,045
Трихлорид бора, ВС13 303 0.034
XVIII. Этан, С2Н6
Пропан, с3н8 273 0,074
XIX. Этилен, С2Н4
Аммиак, NH3 273 0,177
* ’ При 300 К < Т< 1200 К справедливо уравнение D = 4,81 • 10~5 • Г1,691.
При 1300 К < Т< 3000 К справедливо уравнение Dab = 2,48 • 10-5 • Г1,783.
* 2 При 300 К < Т< 1100 К справедливо уравнение DAB = 3,61 • 10“5 • Тх,746.
При 1000 К < Т< 3000 К справедливо уравнение Dab = 2,81 • 10~5 • Г1’776.
* 3 Экспериментальные данные описываются уравнением вида D = Do(77273)", где Do и п имеют следующие значения:
- в интервале 273 К < Т< 677,5 К Do = 0,2106 и п - 1,82;
- в интервале 273 К < Т< 370 К Do = 0,2232 и п = 1,81;
- в интервале 378 К < Т< 575 К Do = 0,2165 и п = 1,80.
*4 Здесь и далее в таблице в скобках указано давление р в МПа.
Таблица 2.2.3
Атомные и молекулярные диффузионные объемы V для определения £>ЛВ по уравнению (2.2.1.4)
Наименование V Наименование V
А. Атомные и структурные составляющие диффузионных объемов
С Н О N 16,5 1,98 5,48 5,69 С1 S Ароматическое кольцо Гетероциклическое кольцо 19,5 17,5 -20,2 -20,2
Б. Диффузионные объемы для простых молекул
н2 d2 7,07 6,70 СО СО2 18,9 26,9
Наименование Наименование V
Б. Диффузионные объемы для простых молекул
Не 2,88 N2O 35,9
n2 17,9 NH3 14,9
02 16,6 Н2О 12,7
Воздух 20,1 SF6 69,7
Аг 16,1 Cl2 37,7
Кг 22,8 Вг2 67,2
Хе 37,9 SO2 41,1
Химическая кинетика и диффузия
497
Таблица 2.2.4
Сводная таблица значений коэффициентов Р20 = Даво (при Т = 293 К) и показателя степени п в уравнении (2.2.1.11) прир = 0,1 МПа
Газы Яаво • Ю4, кг^/с п Область температур (Т, К), для которых имеются экспериментальные данные
Аг—Аг 0,157 1,92 77-353
Аг—Н2 0,715 1,89 273-418
Аг—Не 0,638 1,75 250-3000
СН4—СН4 0,200 1,69 90-353
СО2—СО2 0,0965 1,90 273-362
со2—о2 0,138 1,80 273-1000
н2—со2 0,575 1,76 250-1083
Н2—Н2О 0,734 1,82 290-370
н2—02 0,661 1,89 142-1000
н2о—со2 0,146 1,84 298-434
Н2О—02 0,240 1,73 298-1000
Н2О—воздух 0,216 1,80 273-1493
Не—СО2 0,494 1,80 250-404
Не—Не 1,62 1,71 14-296
N2—СО2 0,144 1,73 288-1200
N2—н2 0,689 1,72 137-1083
N2—Не 0,621 1,73 293-3000
n2—n2 0,178 1,90 77-353
Воздух—СО2 0,124 1,70 273-1533
Воздух—бензол 0,0783 1,89 273-617
Воздух—гексан 0,0646 1,60 273-575
Воздух—гептан 0,0594 1,60 373-573
О,—СО 0,188 1,68 273-100
02—О2 0,186 1,92 77-353
Воздух—декан 0,0461 1,60 545-537
Воздух—нонан 0,0490 1,60 425-525
Воздух—октан 0,0544 1,60 298-528
Воздух—тетралин 0,0536 1,90 484-507
Воздух—толуол 0,0709 1,90 273-332
Воздух—топливо Т5 0,0287 1,96 523-673
Воздух—этанол 0,105 1,77 273-340
Таблица 2.2.5
Зависимость коэффициентов диффузии газов от давления
Диффунди- Диффузион- Г, К Р, D • 104,
рующий газ ная среда МПа м^с
Аргон, Аргон, 322,4 6,8 0,00272
Аг Аг 322,4 11,77 0,00161
322,4 19,40 0,000950
322,4 29,10 0,000659
Водород Кислород 288,8 0,066 1,718
(диводород), (дикисло- 284,4 0,096 0,8012
н2 род), 287 0,097 0,778
О2
288,4 0,065 0,9184
Диоксид Водород 291 0,097 0,606
углерода, (диводо- 285,5 0,098 0,6142
СО2 род), Н2 288,7 0,039 0,4139
Воздух 288,2 0,047 0,3376
290,6 0,096 0,1653
273 0,606 0,01501
Диоксид 273,2 0,780 0,01126
углерода, 273 1,430 0,00598
со2 273 2,550 0,002902
273,3 3,43 0,00131
273,4 3,43 0,00183
273,75 3,43 0,00130
273,3 3,84 0,04599
273,2 7,107 0,04456
272,9 13,71 0,04404
273,2 13,79 0,04404
273,2 30,71 0,0432
273,1 80,25 0,04198
297,9 0,950 0,0110
298,3 3,420 0,00325
298,2 3,430 0,00271
298,1 4,760 0,00179
298 6,120 0,00106
298,1 6,510 0,000216
298,2 6,510 0,000133
298,4 6,510 0,000111
298,1 8,905 0,000103
298,1 9,020 0,000104
298,1 9,020 0,04822
298,1 10,31 0,04934
298,0 11,32 0,04905
298,3 12,49 0,000105
498
Новый справочник химика и технолога
Продолжение таблицы 2.2.5
Диффундирующий газ Диффузионная среда Г, К А МПа D • 104, м2/с
Диоксид Диоксид 297,9 13,03 0,04764
углерода, углерода, 298,2 14,99 0,04702
СО2 СО2 298,0 40,25 0,04409
298,2 50,25 0,04434
298,2 56,10 0,04351
298,1 56,20 0,04512
298,0 56,20 0,04354
298,2 75,2 0,04318
298,2 102,30 0,04253
289,1 103,05 0,04245
Монотритий- Метан, 273 2,0 0,0053
метан, СН4 273 4,02 0,00248
СН3Т 273 6,0 0,00151
273 8,1 0,00109
273 10,0 0,00075
273 12,5 0,000575
273 15,0 0,00040
273 20,0 0,000248
273 30,0 0,000150
298 4,05 0,00291
298 6,0 0,00198
298 8,0 0,00138
298 10,0 0,00098
298 10,0 0,00087
298 12,5 0,00069
298 12,5 0,00071
298 15,0 0,00057
298 20,0 0,000368
298 30,0 0,00021
323 4,0 0,00365
323 6,0 0,00246
323 8,03 0,00160
323 10,0 0,00113
323 12,48 0,00094
323 15,0 0,000664
Таблица 2.2.6
Коэффициенты диффузии паров некоторых металлов при р = 0,1 МПа
Диффундирующий пар Диффузионная среда Т, к D 104, м2/с
Cd n2 273 0,17
Na n2 288 20,4
Hg Н2 273 0,53
N2 273 0,14
2.2.1.2. Термическая диффузия в газах
Процесс молекулярного переноса массы, вызванный неоднородностью температуры внутри смеси, называется термической диффузией. В результате термической диффузии система приходит в равновесное состояние, при этом эффекты разделения и перемешивания взаимно уравновешиваются. Эффект разделения вызывается разностью температур, эффект перемешивания — возникшей при этом разностью концентраций. Эффект термической диффузии оценивается величиной разделения ДХ или термодиффузионным отношением КТ, которые связаны между собой следующим образом:
ДХ = К7.1п^-, (2.2.1.13)
^2
ДХ = С]-с2, (2.2.1.14)
где С] — объемная концентрация более легкого компонента в области, где температура Ть и с2 — объемная концентрация того же компонента в области, где температура Т2;
(2.2.1.15)
где D — коэффициент обычной (концентрационной) диффузии, Dr — коэффициент термической диффузии. В табл. 2.2.7 приведены значения дХ и Кт для ряда газовых смесей в широком диапазоне температур, полученные по методу Усманова [2]. Величины С) даны для более легкого компонента.
Таблица 2.2.7
Данные по термодиффузии в газовых смесях Данные разных авторов отделены чертой.
С], об. % 7], К Л, К ДА, об. % Кт
Аг—Кг
40 373 288 0,29 0,0112
50 0,36 0,0139
60 0,40 0,0155
70 0,37 0,0143
80 0,30 0,0116
58 0,40 0,0155
53,5 369 293 0,52 0,0226
464 1,23 0,0265
585 2,20 0,0319
293 233 0,38 0,0165
185 0,65 0,0141
147 0,84 0,0122
117 0,95 0,0105
197 0,54 0,0135
179 0,66 0,0130
144 0,82 0,0116
134 0,86 0,0110
Химическая кинетика и диффузия
499
Продолжение таблицы 2.2.7
С], об. % т„к т2,к АХ, об. % КТ
Аг—Кг
53,5 293 122 0,86 0,0098
111 0,93 0,0092
634 293 2,65 0,0338
577 2,15 0,0323
507 1,55 0,0199
470 1,35 0,0286
442 1,06 0,0258
397 0,69 0,0227
Аг—Хе
30 373 288 0,33 0,0128
40 0,41 0,0159
50 0,46 0,0178
60 0,48 0,0186
70 0,43 0,0167
58 0,47 0,0182
56,4 369 293 0,62 0,0269
464 1,50 0,0326
585 2,60 0,0377
293 233 0,47 0,0204
185 0,83 0,0180
СО—СО2
11,8 373 283 0,13 0,0047
26,4 0,27 0,0098
32,1 0,33 0,0120
39,9 0,40 0,0145
44,6 0,40 0,0145
50,3 0,42 0,0152
51,2 0,43 0,0156
59,8 0,40 0,0145
67,8 0,34 0,0123
77,0 0,26 0,0094
60,71 588 283 0,96 0,0137
773 1,35 0,0139
1073 1,82 0,0140
1273 2,20 1,0150
10 373 283 0,14 .0,0051
650 0,44 0,0053
1200 0,84 0,0058
1900 1,17 0,0062
20 373 283 0,24 0,0087
650 0,77 0,0093
1200 1,49 0,0103
1900 2,06 0,0108
30 373 283 0,32 0,0116
650 0,03 0,0124
1200 0,95 0,0135
1900 2,57 0,0140
Продолжение таблицы 2.2.7
С\, об. % Г,,К Г2,К ДХ, об. % Кт
50 373 283 0,41 0,0148
650 1,33 0,0160
1200 2,45 0,0170
2000 3,51 0,0180
70 373 283 0,31 0,0112
650 0,90 0,0118
1200 1,78 0,0124
1900 2,45 0,0129
90 378 0,11 0,0040
650 0,33 0,0041
1200 0,64 0,0043
1900 0,83 0,0044
СО—n2o
11,4 373 283 0,11 0,0040
23,0 0,21 0,0076
33,3 0,32 0,0116
40,8 0,36 0,0130
47,8 0,39 0,0141
51,7 0,40 0,0145
55,3 0,40 0,0145
62,0 0,35 0,0127
68,0 0,31 0,0112
80,7 0,20 0,0072
d2-n2
28,1 283 234 0,97 0,0513
205 1,62 0,0503
184 1,97 0,0459
162 2,52 0,0453
135 3,06 0,0418
88 4,47 0,0384
81 4,62 0,0370
42,6 225 1,67 0,0726
204 2,24 0,0685
189 2,82 0,070
166 3,28 0,0614
151 3,89 0,0619
132 4,54 0,0596
83 6,21 0,0508
80 6,36 0,0505
D2—Ne
82,2 293 20 5,60 0,0147
81,3 90 4,70 0,0399
81,65 69 5,40 0,0374
70,55 20 7,30 0,0191
62,55 90 6,10 0,0518
69,9 69 6,80 0,0471
500
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
Cl, об. % Т1,К г2,к ДА, об. % КТ
D2—Ne
58,3 293 90 6,40 0,0544
58,9 69 7,60 0,0526
59,25 20 8,30 0,0218
46,1 90 6,80 0,0578
46,5 69 7,60 0,0526
34,6 20 7,60 0,0529
33,5 90 5,80 0,0493
34,0 69 7,00 0,0485
Н2—Аг
47 286 108 5,19 0,0533
126 4,40 0,0538
130 4,24 0,0539
137 4,09 0,0556
142 3,91 0,0559
149 3,67 0,0565
165 3,42 0,0623
172 3,11 0,0611
190 2,47 0,0603
197 2,43 0,0652
218 1,79 0,0651
231 1,40 0,0654
55,6 290 103 5,78 0,0559
112 5,43 0,0572
126 4,96 0,0595
130 4,81 0,0601
143 4,18 0,0593
161 3,72 0,0634
178 3,30 0,0676
193 2,80 0,0688
205 2,54 0,0732
216 2,01 0,0681
231 1,57 0,0853
6 369 289 0,27 0,0111
428 0,47 0,0120
493 0,65 0,0122
11 369 290 0,46 0,0193'
448 0,87 0,0200
507 1,17 0,0209
547 1,39 0,0219
16,1 376 289 0,84 0,0320
447 1,40 0,0320
506 1,88 0,0336
73,5 330 288 0,57 0,0420
375 1,07 0,0408
431 1,52 0,0377
499 2,15 0,0392
Продолжение таблицы 2.2.7
С[, об. % ТьК г2,к ДА, об. % Кт
73,5 541 288 2,50 0,0397
85 324 286 0,72 0,0581
365 1,46 0,0599
402 2,09 0,0614
441 2,62 0,0607
442 2,74 0,0630
518 3,64 0,0613
Нг—СН4 (данные по термодиффузии при высоких температурах, рассчитанные по методу А.Г. Усманова)
59,10 483 293 2,80 0,0541
628 5,04 0,0662
773 6,57 0,0690
Н2—СО
24 284 81 4,13 0,0330
131 3,14 0,0409
149 2,71 0,0405
171 2,29 0,0450
183 1,92 0,0437
197 1,62 0,0442
210 1,38 0,0457
233 0,92 0,0465
32,5 285 81 6,12 0,0487
103 5,19 0,0510
115 4,80 0,0529
133 4,13 0,0543
147 3,78 0,0572
156 3,54 0,0587
170 3,08 0,0597
185 2,60 0,0600
208 1,94 0,0616
53 286 81 7,32 0,0581
106 6,33 0,0640
128 5,22 0,0650
143 4,55 0,0643
166 3,60 0,0652
184 3,34 0,0755
190 3,46 0,0844
210 2,29 0,0744
5,5 373 283 0,28 0,0102
8,0 0,41 0,0129
15,7 0,84 0,0304
24,4 1,21 0,0439
30,1 1,49 0,0540
35,7 1,69 0,0613
42,4 1,95 0,0707
Химическая кинетика и диффузия
501
Продолжение таблицы 2.2.7
сь об. % т„к Т2,К ДА, об. % КТ
н2-со
48,5 373 283 2,18 0,0790
55,0 2,45 0,0921
61,7 2,53 0,0917
63,3 2,64 0,0957
65,3 2,62 0,0950
70,4 2,46 0,0892
77,8 2,19 0,0794
81,9 1,65 0,0598
83,4 1,57 0,0569
39,9 293 90 5,60 0,0476
40,2 66 6,20 0,0416
51,7 90 6,60 0,0561
52,3 66 7,00 0,0470
62,6 90 6,60 0,0561
63,1 66 7,00 0,0470
73,7 90 5,70 0,0484
74,1 66 6,20 0,0416
81,7 90 4,50 0,0382
82,1 293 66 4,90 0,0329
88,7 90 3,20 0,0272
89,1 66 3,90 0,0262
Н2—СО2
5,8 320 290 0,12 0,0121
370 0,29 0,0119
414 0,43 0,0121
452 0,58 0,0132
499 0,70 0,0129
548 0,82 0,0129
11,8 317 289 0,23 0,0250
365 0,53 0,0228
373 0,64 0„250
417 0,82 0,0224
459 1,07 0,0233
502 1,30 0,0235
530 1,47 0,0243
557 1,60 0,0244
24,3 317 288 0,32 0,0232
368 1,00 0,0244
378 1,12 0,0414
405 1,38 0,0406
443 1,71 0,0398
488 2,22 0,0422
518 2,45 0,0418
570 2,91 0,0427
Продолжение таблицы 2.2.7
С\, об. % Ti, К г2,к ДА, об. % Кт
65 318 291 0,59 0,0679
370 1,80 0,0754
372 1,89 0,0775
410 2,57 0,0775
449 3,21 0,0742
488 3,72 0,0721
514 4,14 0,0729
547 4,53 0,0719
53 305 288 0,34 0,0618
314 0,62 0,0713
321 0,75 0,0695
330 0,99 0,0713
343 1,26 0,0720
350 1,37 0,0710
367 1,69 0,0693
388 2,07 0,0693
398 2,24 0,0697
414 2,50 0,0693
430 2,81 0,0702
445 3,20 0,0739
457 3,36 0,0731
472 3,63 0,0705
488 3,98 0,0755
516 4,44 0,0760
538 4,78 0,0765
561 5,14 0,0773
54,3 353 300 1,27 0,0780
371 1,33 0,0658
404 2,00 0,0669
415 2,27 0,0701
419 2,50 0,0749
435 2,64 0,0708
454 3,14 0,0759
492 4,00 0,0808
561 5,20 0,0832
597 5,82 0,0845
612 6,00 0,0841
717 7,66 0,0881
836 9,66 0,0922
863 9,67 0,0934
882 10,00 0,0929
42,9 759 273 6,94 0,1681
50,0 753 8,01 0,0987
54,4 759 8,68 0,0851
59,9 748 8,60 0,0855
66,9 767 8,40 0,0815
502
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
сь об. % ГЬК г2,к ДА, об. % Kf
Hr-СО2
72,3 753 273 7,198 0,0806
86,2 766 5,45 0,0529
48,1 393 293 1,61 0,0552
443 2,32 0,0560
37,3 393 1,37 0,0470
443 2,02 0,0488
88,6 393 0,85 0,0291
443 1,23 0,0297
18,4 284 197 1,18 0,0322
202 1,06 0,0311
206 1,00 0,0311
212 0,91 0,0312
221 0,78 0,0315
234 0,63 0,0326
239 0,54 0,0314
256 0,34 0,0327
34 285 197 1,74 0,0473
201 1,69 0,0488
208 1,53 0,0486
213 1,42 0,0490
219 1,29 0,0492
225 1,14 0,0485
234 0,96 0,0485
45,2 286 197 2,34 0,0628
198 2,30 0,2625
202 2,09 0,0603
208 1,97 0,0620
213 1,82 0,0620
223 1,55 0,0625
229 1,41 0,0613
235 1,28 0,0657
3,5 373 283 0,16 0,0058
8,4 0,43 0,0156
13,6 0,68 0,0246
15,7 0,83 0,0301
19,2 0,95 0,0344
22,2 1,15 0,0417
23,0 1,18 0,0428
24,4 1,21 0,0439
28,3 1,43 0,0518
32,8 1,56 0,0565
34,0 1,54 0,0558
37,1 1,74 0,0631
42,1 1,94 0,0703
46,7 1,99 0,0722
Продолжение таблицы 2.2.7
сь об. % Л, К г2,к ДА, об. % КТ
63,5 373 283 2,18 0,0790
71,4 2,10 0,0762
78,0 1,095 0,0707
80,0 1,83 0,0663
54,3 670 293 6,57 0,0795
604 5,54 0,0766
525 4,51 0,0775
490 3,97 0,0773
478 3,77 0,0768
431 2,88 0,0746
385 1,95 0,0712
362 1,51 0,0712
347 1,15 0,0677
316 0,51 0,0671
293 250 0,91 0,0573
224 1,53 0,0573
217 0,56 0,0522
197 2,16 0,0546
50,89 676 299 7,04 0,0865
53,22 373 302 1,52 0,0717
477 3,29 0,0723
487 3,51 0,0738
487 3,53 0,0752
531 4,63 0,0822
583 5,46 0,0831
629 6,37 0,0871
681 7,22 0,0884
737 8,05 0,0904
10 400 293 0,76 0,0245
700 2,17 0,0250
1200 3,72 0,0264
2000 5,34 0,0278
20 400 293 1,32 0,0425
700 3,77 0,0434
1200 6,53 0,0464
2000 9,43 0,0492
30 400 293 1,80 0,0580
700 5,22 0,0601
1200 8,49 0,0603
2000 12,82 0,0668
40 400 293 2,24 0,0722
700 6,36 0,0732
1200 10,89 0,0773
2000 15,45 0,0805
400 2,56 0,0824
700 7,21 0,0829
Химическая кинетика и диффузия
503
Продолжение таблицы 2.2.7
Cl, об. % Г,. К Т2,К АХ, об. % КТ
Н2—СО2
50 1200 293 12,25 0,0870
2000 17,37 0,0908
60 400 293 2,64 0,0818
700 7,47 0,0859
1200 12,54 0,0891
2000 17,82 0,0929
70 400 293 2,46 0,0792
700 7,02 0,0807
1200 11,69 0,0830
1900 16,51 0,0861
80 400 293 2,03 0,0654
700 5,79 0,0656
1200 9,57 0,0680
2000 13,54 0,0706
90 400 293 1,34 0,0432
700 3,75 0,0433
1200 6,13 0,0436
2000 8,64 0,0450
59,95 473 293 3,83 0,0800
473 4,02 0,0840
580 5,79 0,0850
580 5,87 0,0862
698 7,29 0,0840
698 7,55 0,0871
798 8,42 0,0841
798 8,61 0,0860
н2—С2Н4
17 288 183 1,35 0,0298
187 1,23 0,0288
199 1,08 0,0294
208 0,99 0,0303
215 0,90 0,0310
228 0,68 0,0291
239 0,54 0,0290
28 287 175 2,14 0,0435
180 0,07 0,0446
189 1,90 0,0457
202 1,61 0,0460
213 1,36 0,0458
223 1,15 0,0458
242 0,82 0,0483
58,5 175 3,27 0,0663
180 3,10 0,0667
176 3,19 0,0654
192 2,70 0,0675
Продолжение таблицы 2.2.7
Ci, об. % ТЬК г2,к АХ, об. % Кт
58,5 287 201 2,38 0,0669
222 1,74 0,0680
225 1,66 0,0680
5,4 373 283 0,24 0,0087
10,5 0,47 0,0170
17,7 0,79 0,0286
25,1 1,И 0,0402
29,0 1,28 0,0464
39,6 1,60 0,0580
48,2 1,73 0,0627
58,3 2,16 0,0783
64,4 2,40 0,0870
74,5 2,34 0,0848
74,8 2,28 0,0826
82,0 2,32 0,0808
84,6 1,69 0,0613
85,1 1,52 0,0551
89,6 1,37 0,0497
Н2—С12
62,4 473 293 3,68 0,0768
473 3,82 0,0798
613 5,31 0,0718
613 5,75 0,0778
673 6,31 0,0759
673 6,81 0,0820
773 7,07 0,0730
773 7,35 0,0759
923 8,50 0,0741
923 8,61 0,0751
Н2—d2
56,9 417 293 1,73 0,0489
480 2,27 0,0460
585 3,06 0,0443
690 3,85 0,0450
10 373 288 0,37 0,0143
20 0,68 0,0264
30 0,92 0,0337
50 1,11 0,0430
60 1,07 0,0415
70 0,94 0,0364
80 0,73 0,0283
90 0,42 0,0163
65,5 293 234 0,81 0,0360
194 1,61 0,0391
92 4,30 0,0372
88,2 20 4,20 0,0110
86,6 90 1,60 0,0136
504
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
сь об. % Г„К Г2,К ДА, об. % КТ
Н2—d2
76,0 293 90 3,00 0,0255
76,7 69 4,40 0,0305
66,0 20 8,30 0,0217
54,3 20 8,70 0,0228
52,4 90 5,60 0,0476
53,1 69 6,80 0,0471
41,9 20 7,10 0,0186
38,4 90 5,60 0,0476
38,6 69 6,00 0,0416
Н2—H2S
45,95 433 293 2,51 0,0646
538 4,19 0,0690
658 5,68 0,0705
738 6,18 0,0669
963 7,73 0,0651
1075 8,59 0,0662
1085 8,90 0,0680
1158 9,21 0,0671
Н2—Не
19,4 292 90 2,50 0,0213
19,8 65 3,50 0,0233
21,3 20 5,90 0,0220
32,3 90 3,40 0,0290
32,8 65 4,60 0,0307
35,0 20 8,20 0,0306
44,8 90 4,00 0,0341
44,8 65 5,40 0,0360
50,5 90 4,60 0,0392
51,1 65 5,80 0,0387
53,7 20 9,90 0,0370
65,4 90 3,70 0,0317
65,0 65 5,00 0,0334
68,4 20 9,20 0,0344
81,8 90 2,50 0,0213
81,9 65 3,20 0,0213
83,3 20 5,40 0,0202
78,8 290 228 0,50 0,0209
66,2 0,90 0,0377
52,0 290 228 1,20 0,0503
46,6 1,10 0,0461
26,0 1,00 0,0419
89,0 291 90 1,60 0,0136
67,4 4,10 0,0349
65,3 4,10 0,0349
45,4 4,70 0,0400
Продолжение таблицы 2.2.7
Су, об. % ГЬК г2,к ДА, об. % Кт
32,8 290 90 3,90 0,0334
17,8 288 2,30 0,0198
86,4 291 77 2,90 0,0219
65,5 290 4,40 0,0332
47,7 291 5,20 0,0392
43,4 288 4,80 0,0364
41,4 289 4,80 0,0364
18,6 288 3,40 0,0258
54,0 290 288 1,20 0,0502
291 90 4,44 0,0379
290 77 4,99 0,0377
Н2—n2
32,7 328 284 0,76 0,0535
379 1,71 0,0616
418 2,26 0,0592
456 2,50 0,0530
479 3,03 0,0580
510 3,46 0,0591
547 3,67 0,0560
4,7 324 285 0,12 0,0079
369 0,23 0,0089
412 0,30 0,0082
450 0,43 0,0095
500 0,52 0,0092
543 0,60 0,0093
10,7 325 283 0,28 0,0202
366 0,53 0,0228
411 0,76 0,0206
448 0,97 0,0211
499 1,23 0,0218
538 1,32 0,0206
25,7 321 284 0,44 0,0355
373 1,21 0,0446
415 1,72 0,0456
450 2,01 0,0437
500 2,67 0,0474
543 2,92 0,0452
37,4 326 0,85 0,0616
328 0,92 0,0648
374 1,70 0,0616
411 2,22 0,0603
445 2,79 0,0621
492 3,45 0,0611
539 4,25 0,0664
50,5 324 282 1,03 0,0737
369 1,95 0,0731
Химическая кинетика и диффузия
505
Продолжение таблицы 2.2.7
С\, об. % ТьК т2,к ДХ, об. % КТ
Н2—n2
50,5 418 282 2,81 0,0713
468 3,98 0,0782
506 4,56 0,0780
540 4,77 0,0734
29,4 289 81 5,02 0,0396
99 4,47 0,0418
116 4,04 0,0444
123 3,84 0,04512
138 3,22 0,436
148 3,05 0,0457
174 2,48 0,0491
187 2,23 0,0513
193 2,04 0,0506
203 1,79 0,0509
242 0,95 0,0537
42 81 6,62 0,0521
97 5,84 0,0539
130 4,74 0,0594
139 4,48 0,0613
149 4,17 0,0632
159 3,77 0,0633
179 3,06 0,0639
186 2,85 0,0671
198 2,61 0,0691
203 2,41 0,0685
224 1,82 0,0720
240 1,30 0,0699
77,5 288 81 6,15 0,0486
99 5,89 0,0552
114 5,42 0,0587
129 4,79 0,0588
136 4,67 0,0622
143 4,38 0,0627
152 4,14 0,0647
166 3,77 0,0685
191 2,69 0,0722
244 1,15 0,0693
5,2 373 283 0,26 0,0094
9,6 0,45 0,0163
18,9 0,97 0,0352
25,4 1,34 0,0486
36,9 1,80 0,0652
46,6 2,25 0,0815
54,7 2,54 0,0920
55,8 2,60 0,0942
Продолжение таблицы 2.2.7
С], об. % ТЬК г2,к ДХ, об. % Кт
61,0 373 283 2,63 0,0953
62,1 2,58 0,0935
66,4 2,62 0,0950
68,2 2,54 0,0920
76,4 1,98 0,0718
81,6 2,03 0,0736
85,0 1,45 0,0525
87,2 1,21 0,0438
39,8 293 254 0,73 0,0515
238 1,41 0,0681
202 2,47 0,0740
191 2,62 0,0612
182 3,03 0,0639
163 3,56 0,0609
153 3,89 0,0599
135 4,55 0,0588
123 4,85 0,0561
ПО 5,38 0,0549
98 5,60 0,0512
90 6,32 0,0536
28 283 230 1,17 0,0566
214 1,598 0,0572
195 1,19 0,0512
178 2,42 0,0525
151 3,05 0,0486
133 3,67 0,0486
89 4,79 0,0415
42,7 283 82 5,04 0,0407
217 2,09 0,0793
188 2,94 0,0718
166 3,62 0,0678
147 4,14 0,0633
137 4,46 0,0616
134 4,76 0,0639
82 6,83 0,0552
39,8 655 293 5,89 0,0734
513 4,08 0,0731
410 2,46 0,0741
357 1,66 0,0839
329 0,93 0,0810
46 293 93 10,10 0,1882
193 3,64 0,0876
79,9 93 7,80 0,0681
193 3,10 0,0746
46 393 293 2,53 0,0867
443 3,23 0,0784
79,9 393 2,02 0,0692
506
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
СЬ об. % Ть К г2,к ДА, об. % Кт
Нг—N2
79,9 443 293 2,77 0,0672
40,2 292 90 6,30 0,0537
40,9 64 7,60 0,0501
50,0 90 7,20 0,0614
50,4 77 8,00 0,0447
50,8 64 8,40 0,0555
51,0 55 8,30 0,0465
57,4 90 7,40 0,0631
58,1 64 8,50 0,0561
62,8 90 7,40 0,0631
63,1 77 7,50 0,0419
63,5 64 8,10 0,0535
63,8 55 8,10 0,0483
70,6 90 6,50 0,0554
70,9 77 7,00 0,0391
71,5 55 7,50 0,0447
77,4 90 5,60 0,0477
77,7 77 5,90 0,0329
78,0 64 6,30 0,0416
78,1 55 6,50 0,0387
82,4 77 5,40 0,0301
82,9 55 5,70 0,0340
81,3 90 5,00 0,0426
82,3 64 5,70 0,0376
90,3 90 3,40 0,0290
90,5 77 3,70 0,0206
90,7 64 3,90 0,0257
90,8 55 3,90 0,0232
90,3 90 3,40 0,0290
90,5 77 . 3,70 0,0206
90,7 54 3,90 0,0231
90,8 55 3,90 0,0232
89,9 291 90 3,20 0,0273
81,0 5,00 0,0427
66,7 6,40 0,0546
57,1 6,20 0,0529
49,4 6,10 0,0521
40,1 5,50 0,0469
24,5 4,00 0,0341
н2—n2o
55,3 312 288 0,55 0,0618
327 0,88 0,0691
339 1,18 0,0724
368 1,69 0,0693
379 1,89 0,0690
Продолжение таблицы 2.2.7
ch об. % т}, к г2,к ДА, об. % Кт
н2—n2o
55,3 398 288 2,25 0,0758
401 2,38 0,0723
414 2,54 0,0698
415 2,67 0,0729
431 2,88 0,0715
447 3,18 0,0723
461 3,45 0,0703
493 4,08 0,0762
503 4,25 0,0763
520 4,47 0,0760
35 288 195 1,68 0,0432
207 1,43 0,0435
220 1,18 0,0442
228 0,98 0,0419
244 0,72 0,0434
42,5 286 187 2,02 0,0478
193 1,87 0,0475
198 1,81 0,0492
213 1,49 0,0507
230 1,08 0,0495
235 0,98 0,0503
243 0,84 0,0522
59 287 187 2,31 0,0540
199 2,03 0,0555
207 1,81 0,0559
220 1,47 0,0534
234 1,13 0,0560
2,0 377 283 0,12 0,0044
9,4 0,45 0,0163
11,1 0,57 0,0206
12,5 0,61 0,0221
18,7 0,93 0,0337
25,6 1,24 0,0449
31,0 1,44 0,0522
36,4 1,58 0,0573
38,1 1,63 0,0491
40,8 1,71 0,0620
43,9 1,83 0,0663
50,1 1,93 0,0700
53,2 1,99 0,0721
61,0 2,07 0,0751
69,0 2,02 0,0732
72,7 1,97 0,0714
78,6 1,88 0,0681
83,5 1,52 0,0551
Химическая кинетика и диффузия
507
Продолжение таблицы 2.2.7
С], об. % К г2,к ДХ, об. % КТ
Hz—Ne
27,8 283 82 7,84 0,0696
128 5,43 0,0752
147 4,46 0,0682
162 3,70 0,0664
173 3,23 0,0657
197 2,47 0,0679
228 1,41 0,0653
40,6 285 82 9,44 0,0760
138 6,66 0,0919
147 5,40 0,0814
163 4,63 0,0829
176 3,97 0,0822
200 3,10 0,0877
228 1,90 0,0852
49,6 282 81 10,00 0,0803
129 7,06 0,0902
144 5,87 0,0873
161 5,26 0,0941
171 4,40 0,0879
196 3,30 0,0908
205 2,85 0,0896
211 2,04 0,0704
84 290 20 6,40 0,0242
76,8 90 7,70 0,0660
77,3 69 8,20 0,0573
66,1 90 8,40 0,0720
66,9 69 9,80 0,0685
65,0 20 10,70 0,0404
57,8 90 8,80 0,0754
58,6 69 10,00 0,0698
45,8 20 11,30 0,0426
41,8 90 7,30 0,0625
42,6 69 9,00 0,0629
28,2 90 5,70 0,0488
28,9 69 7,00 0,0489
24,9 20 7,50 0,0283
53,6 615 293 2,93 0,0396
549 2,02 0,0324
432 1,52 0,0390
54 293 90 4,20 0,0355
20 10,10 0,0265
20 9,90 0,0260
66 20 9,40 0,0246
50 373 288 1,24 0,0481
60 1,14 0,0442
Продолжение таблицы 2.2.7
сь об. % 7ЬК Тъ К ДХ, об. % Кт
70 373 288 0,90 0,0349
80 0,64 0,0248
90 373 288 0,34 0,0132
32,78 769 273 2,50 0,0244
40,33 753 3,21 0,0317
47,82 758 3,22 0,0319
60,30 763 3,02 0,0295
53,6 293 139 2,52 0,0338
115 3,40 0,0365
93 4,00 0,0355
93 4,55 0,0398
67 5,75 0,0391
21,6 290 90 5,50 0,0471
22,0 60 6,20 0,0419
23,4 20 7,20 0,0270
35,6 90 6,90 0,0591
36,3 66 8,40 0,0568
38,3 20 10,00 0,0375
50,9 90 8,20 0,0702
51,7 66 9,20 0,0622
53,8 20 10,90 0,0409
66,7 90 7,70 0,0659
67,5 66 8,90 0,0602
69,2 18 9,80 0,0353
69,3 20 10,10 0,0378
84,8 90 4,70 0,0402
85,2 66 5,40 0,0365
86,2 17 5,70 0,0201
86,3 20 5,90 0,0221
Нг—СЬ
29,8 290 102 4,07 0,0390
115 3,74 0,0404
134 3,43 0,0448
147 3,15 0,0464
153 2,96 0,0463
167 2,62 0,0476
178 2,35 0,0480
187 2,09 0,0478
199 1,81 0,0481
213 1,45 0,0471
223 1,26 0,0481
242 0,88 0,0483
33,8 294 90 4,00 0,0339
34,3 66 4,90 0,0329
48,2 294 90 5,50 0,0465
48,9 66 6,60 0,0443
508
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
С], об. % Ть К г2,к ДХ, об. % КТ
Н2—О2
63,1 294 90 6,00 0,0508
63,6 66 6,80 0,0456
73,7 90 5,30 0,0449
74,1 66 5,70 0,0382
84,6 90 3,70 0,0313
84,8 66 3,80 0,0255
Не—Аг
з,з 329 290 0,11 0,0087
373 0,20 0,0079
445 0,33 0,0078
500 0,42 0,0078
579 0,53 0,0077
11,2 322 292 0,29 0,0299
383 0,70 0,0257
430 1,04 0,0269
508 1,23 0,0222
567 1,73 0,0260
18,5 329 290 0,51 0,0404
384 1,25 0,0445
453 1,99 0,0447
523 2,56 0,0435
543 2,78 0,0438
38,1 377 291 1,90 0,0737
450 3,38 0,0778
480 3,80 0,0762
553 4,88 0,0762
70,8 387 290 2,85 0,0994
412 3,43 0,0975
485 5,00 0,0975
10 . 373 288 0,65 0,0252
20 1,23 0,0477
30 1,70 0,0659
40 2,09 0,0810
50 2,40 0,0930
65 2,60 0,1009
51,2 369 293 2,18 0,0949
464 4,40 0,0957
585 6,70 0,0972
293 233 2,18 0,0949
185 4,40 0,0957
147 6,35 0,0921
117 8,35 0,0912
93 10,20 0,0889
20,85 283 81 6,79 0,0542
121 4,34 0,0512
Продолжение таблицы 2.2.7
съ об. % ТьК г2,к ДХ, об. % Кт
20,85 283 145 3,76 0,0564
161 3,22 0,0571
176 2,74 0,0578
182 2,25 0,0511
206 1,85 0,0585
223 1,43 0,0604
241 0,95 0,0590
38,1 282 80 10,60 0,0845
122 6,86 0,0819
150 5,71 0,0905
166 4,84 0,0916
184 4,00 0,0940
195 3,41 0,0927
213 2,67 0,0950
234 1,80 0,0990
51,1 283 81 11,80 0,0948
130 7,99 0,1028
147 6,72 0,1027
162 5,62 0,1009
176 4,82 0,1014
187 4,19 0,1011
207 3,34 0,1067
229 2,27 0,1070
57,2 402 3,15 0,1079
463 4,65 0,1074
567 6,39 0,1005
638 8,00 0,1060
44,4 402 2,93 0,1002
463 4,10 0,0948
567 6,15 0,0969
638 7,42 0,0983
23,2 402 1,72 0,0590
463 3,31 0,0765
567 4,43 0,0698
638 5,40 0,0715
11,5 402 0,95 0,0326
463 1,28 0,0296
567 2,67 0,0421
638 2,75 0,0364
74,4 402 2,29 0,0785
463 3,54 0,0818
567 5,15 0,0811
638 6,00 0,0795
90,6 402 1,31 0,0449
463 1,64 0,0379
567 2,83 0,0446
638 3,03 0,0401
Химическая кинетика и диффузия
509
Продолжение таблицы 2.2.7
Су об. % Л, К Л, К АХ, об. % КТ
Не—СО2
38,0 764 273 8,41 0,0818
52,2 763 9,80 0,0956
62,3 758 10,23 0,1001
66,6 750 10,30 0,1020
75,4 760 9,51 0,0930
78,8 765 8,90 0,0863
Не—Не
20 373 288 1,37 0,0531
30 1,87 0,0725
40 2,23 0,0864
50 2,50 0,0969
60 2,59 0,1003
77,9 293 90 8,00 0,068
78,7 69 10,10 0,070
79,8 20 11,70 0,031
69,6 90 8,50 0,072
7U 69 11,30 0,078
71,5 20 14,40 0,038
68,6 20 17,20 0,045
59,5 90 9,70 0,082
60,4 69 11,40 0,079
47,9 20 16,00 0,042
44,5 90 9,50 0,081
45,0 69 10,50 0,073
53,8 369 293 1,80 0,0783
464 3,60 0,0783
585 5,42 0,0788
293 233 1,80 0,0783
185 3,60 0,0783
147 5,30 0,0770
117 7,02 0,0763
93 8,65 0,0752
39,9 288 81 10,00 0,0792
128 6,64 0,0820
147 5,43 0,0808
163 4,54 0,0799
189 3,41 0,0811
226 2,29 0,0946
53,6 81 11,2 0,0885
133 7,30 0,0945
151 5,98 0,0925
171 4,95 0,0952
191 3,79 0,0925
214 2,67 0,0900
25,5 287 81 7,71 0,0609
Продолжение таблицы 2.2.7
Су об. % Л, К Л, к АХ, об. % Кт
25,5 287 129 5,02 0,0629
144 4,22 0,0612
158 3,61 0,0606
176 2,98 0,06112
200 2,21 0,0616
226 1,45 0,0607
53,7 402 300 2,93 0,100
463 4,00 0,0924
567 5,82 0,0918
638 7,36 0,0976
43,3 410 2,69 0,0860
470 3,60 0,0803
567 5,44 0,0857
638 6,47 0,0857
30,8 402 2,25 0,0771
463 3,23 0,0747
567 4,57 0,0720
638 5,52 0,0732
19,6 402 1,80 0,0617
463 2,44 0,0563
567 3,52 0,0555
638 4,25 0,0563
75,7 402 2,53 0,0867
463 3,48 0,0804
567 4,91 0,0773
638 5,85 0,0775
89,9 402 1,62 0,0555
463 2,52 0,0582
567 3,40 0,0536
638 3,95 0,0524
53,8 293 192 3,45 0,0814
146 5,28 0,0761
122 6,74 0,0750
112 7,38 0,0767
100 8,08 0,0753
92 8,87 0,0766
609 293 5,87 0,0807
457 3,58 0,0807
385 2,35 0,0948
Не—Кг
30 373 288 1,75 0,0678
40 2,20 0,0853
50 2,58 0,1000
60 2,79 0,1081
70 2,76 0,1070
63 2,80 0,1084
510
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
С\, об. % ГЬК Л, к ДХ, об. % КТ
Не—Кг
55,0 369 293 2,52 0,1097
464 5,10 0,1109
585 7,64 0,1108
293 233 2,60 0,1131
185 5,10 0,1109
147 7,50 0,1087
117 9,70 0,1054
Не—N2
34,5 284 81 9,35 0,1176
122 5,99 0,0709
146 5,15 0,0775
170 4,05 0,0789
185 3,30 0,0771
198 2,78 0,0775
218 2,16 0,0815
240 1,40 0,0833
81 11,86 0,1493
133 7,77 0,1028
146 6,78 0,1020
165 5,59 0,1028
180 4,72 0,1038
192 4,05 0,1037
204 3,32 0,1040
227 2,44 0,1093
Не—Ne
61,0 295 90 6,20 0,0522
62,1 66 8,50 0,0568
61,0 90 6,20 0,0522
62,1 66 8,50 0,0568
Не—Хе
10 373 288 0,53 0,0206
20 1,06 0,0411
30 1,55 0,0601
40 2,03 0,0787
50 2,53 0,0981
70 3,15 0,1222
53,6 369 293 2,52 0,1097
464 5,02 0,1001
585 7,53 0,1091
293 233 2,50 0,1088
185 4,90 0,1066
45 373 288 0,10 0,0388
N2—Аг
46 289 94 1,14 0,0102
128 0,92 0,0113
154 0,83 0,0119
Продолжение таблицы 2.2.7
С], об. % Ть К г2,к ДХ, об. % Кт
46 289 177 0,77 0,0157
195 0,65 0,0165
215 0,55 0,0186
222 0,50 0,0189
232 0,40 0,0184
62,5 94 0,95 0,0085
139 0,87 0,0119
181 0,73 0,0156
201 0,59 0,0163
221 0,46 0,0172
70 290 114 0,92 0,0099
148 0,77 0,0114
166 0,72 0,0129
169 0,72 0,0134
184 0,67 0,0147
191 0,59 0,0142
203 0,54 0,0151
217 0,42 0,0145
229 0,39 0,0164
N2—СО2
76,5 367 284 0,25 0,0980
405 0,40 0,0113
458 0,53 0,0111
521 0,73 0,0120
59,4 326 282 0,24 0,0166
371 0,58 0,0212
412 0,68 0,0181
442 0,94 0,0209
493 U9 0,0214
500 1,13 0,0197
88,6 378 284 0,17 0,0597
418 026 0,0672
473 0,33 0,0947
475 0,38 0,0741
531 0,48 0,0769
39,3 309 288 0,08 0,0116
322 0,11 0,0102
338 0,20 0,0124
354 0,23 0,0111
390 0,37 0,0123
402 0,42 0,0126
422 0,49 0,0128
3,7 0,04 0,0015
12,5 0,13 0,0047
17,6 0,19 0,0069
26,9 0,28 0,0101
35,3 0,37 0,0134
Химическая кинетика и диффузия
511
Продолжение таблицы 2.2.7
сь об. % ГЬК г2,к ДА, об. % КТ
N2-CO2
40,6 373 283 0,39 0,0141
44,9 0,41 0,0146
45,1 0,40 0,0145
49,8 0,42 0,0152
57,0 0,41 0,0149
67,2 0,36 0,0130
80,8 0,20 0,0072
92,9 0,09 0,0033
10 400 283 0,17 0,0049
700 0,47 0,0052
1200 0,80 0,0055
2000 1,17 0,0060
20 400 0,29 0,0084
700 0,85 0,0094
1200 1,40 0,0097
2000 2,00 0,0102
30 400 283 0,42 0,0122
700 1,15 0,0127
1200 1,98 0,0138
2000 2,78 0,0142
50 400 0,52 0,0151
700 1,46 0,0161
1200 2,50 0,0173
2000 3,55 0,0182
70 400 0,44 0,0128
700 1,18 0,0130
1200 1,91 0,0133
2000 2,73 0,0140
90 400 283 0,16 0,0046
.700 0,45 0,0050
1200 0,74 0,0051
2000 1,05 0,0054
n2—n2o
45 307 288 0,09 0,0141
318 0,11 0,0111
335 0,18 0,0118
353 0,23 0,0114
373 0,33 0,0119
389 0,37 0,0124
16,3 373 283 0,16 0,0058
24,4 0,22 0,0080
34,7 0,31 0,0112
39,3 0,34 0,0123
41,0 0,35 0,0127
41,0 0,36 0,0130
45,6 0,39 0,0141
Продолжение таблицы 2.2.7
С], об. % ГьК г2,к ДА, об. % Кт
49,0 373 283 0,39 0,0141
54,8 0,38 0,0138
56,6 0,38 0,0138
59,6 0,37 0,0134
72,6 0,27 0,0098
84,1 0,16 0,0058
Ne—Аг
20 373 288 0,60 0,0233
30 0,85 0,0330
40 1,05 0,0407
50 1,18 0,0457
60 1,21 0,0469
58 1,20 0,0465
51,2 369 293 1,09 0,0474
464 2,20 0,0478
585 3,30 0,0478
293 233 0,95 0,0413
185 1,83 0,0398
147 2,63 0,0381
117 3,21 0,0349
93 3,68 0,0320
19,1 283 79 2,22 0,0174
135 1,52 0,0206
168 1,21 0,0233
190 0,96 0,0241
205 0,79 0,0245
233 0,51 0,0270
36,1 282 81 3,40 0,0273
133 2,44 0,0325
141 2,16 0,0312
168 1,78 0,0345
184 1,53 0,0360
209 1,10 0,0368
226 0,82 0,0371
51,7 283 81 3,71 0,0297
129 2,80 0,0357
152 2,30 0,0371
171 1,93 0,0386
194 1,52 0,0403
227 0,98 0,0444
51,2 293 264 0,63 0,0612
234 0,89 0,0399
196 1,63 0,0407
174 2,05 0,0394
145 2,61 0,0371
125 3,00 0,0207
512
Новый справочник химика и технолога
Продолжение таблицы 2.2.7
С], об. % ГЬК Г2,к ДА, об. % КТ
Ne—Аг
51,2 293 116 3,26 0,0237
105 3,26 0,0237
91 3,65 0,0322
635 293 3,80 0,0492
600 3,54 0,0494
522 2,78 0,0484
471 2,17 0,0458
408 1,59 0,0481
348 0,90 0,0530
Ne—Кг
20 373 288 0,84 0,0326
30 1,15 0,0446
40 1,44 0,0559
50 1,72 0,0667
60 1,90 0,0757
63 1,80 0,0698
53,0 369 293 1,75 0,0761
464 3,58 0,0778
585 5,47 0,0793
293 233 1,62 0,0704
185 2,93 0,0637
147 4,02 0,0583
117 5,00 0,0547
Ne—Хе
20 373 288 0,67 0,0260
30 0,99 0,0384
40 1,26 0,0489
50 1,49 0,0578
60 1,69 0,0656
70 1,70 0,0660
54,2 369 293 1,85 0,0805
464 3,88 0,0844
585 6,15 0,0892
233 0,0172 0,0748
185 0,330 0,0718
О2—СО2
20,0 373 283 0,20 0,0073
32,9 0,32 0,0116
39,8 0,36 0,0130
43,3 0,38 0,0138
45,5 0,40 0,0145
47,0 0,38 0,0138
49,4 0,40 0,0145
53,8 0,39 0,0141
55,8 0,39 0,0141
Продолжение таблицы 2.2.7
сь об. % Тх, К г2,к ДА, об. % Кт
57,3 373 283 0,39 0,0141
58,5 0,39 0,0141
61,1 0,37 0,0134
68,7 0,32 0,0116
76,6 0,28 0,0101
79,6 0,29 0,0105
87,5 0,20 0,0073
90,4 0,17 0,0062
10 373 283 0,10 0,0036
650 0,30 0,0039
1200 0,63 0,0044
1900 0,88 0,0047
20 373 283 0,20 0,0072
650 0,66 0,0079
1273 1,25 0,0087
1900 1,74 0,0092
30 373 283 0,30 0,0100
650 0,89 0,0117
1200 1,84 0,0128
1900 2,54 0,0134
50 373 283 0,41 0,0148
650 1,32 0,0159
1200 2,49 0,0173
1900 3,44 0,0181
70 373 283 0,35 0,0127
650 1,17 0,0141
1200 2,07 0,0144
1900 2,86 0,01050
90 373 283 0,19 0,0069
650 0,57 0,0070
1200 1,05 0,073
1900 1,43 0,075
60,80 515 293 0,84 0,0149
515 1,01 0,0179
515 1,01 1,0179
701 1,40 0,0161
879 1,75 0,0160
1034 2,02 0,0160
1034 2,14 0,0170
1250 2,32 0,0160
о2—no2
17,1 373 283 0,16 0,0058
23,1 0,24 0,0087
27,0 0,28 0,0101
33,3 0,35 0,0127
35,0 0,38 0,0138
Химическая кинетика и диффузия
513
Продолжение таблицы 2.2.7
С], об. % ТЬК т2,к ДА, об. % Кт
О2—no2
38,6 373 283 0,38 0,0138
42,4 0,39 0,0141
46,1 0,39 0,0141
47,9 0,39 0,0141
50,2 0,41 0,0149
52,9 0,40 0,0145
57,9 0,40 0,0145
64,5 0,37 0,0134
80,0 0,25 0,0091
85,7 0,20 0,0073
93,8 0,13 0,0047
2.2.1.3. Диффузия в жидкостях
В жидкостях коэффициент диффузии на несколько порядков ниже, чем в газах при атмосферном давлении: в невязких жидкостях при 20 °C он имеет порядок 10 9 м2/с, а в газах — (1-10) • 105 м2/с. Однако отсюда не следует, что плотность потоков в жидких средах меньше, чем в газовых, так как плотность жидкостей и градиенты концентрации в них обычно значительно выше [3, 7].
В жидкостях коэффициент диффузии существенно зависит от концентрации распределяемого вещества. Это обусловлено более плотной упаковкой молекул и, как следствие, более сильным молекулярным взаимодействием.
В табл. 2.2.8-2.2.17 приведены экспериментальные данные [5, 7, 8] по коэффициентам диффузии в бинарных жидких смесях, большая часть которых относится к коэффициентам взаимодействия в бесконечно разбавленных растворах. Для таких растворов коэффициент взаимной диффузии равен коэффициенту самодиффу-зии в бесконечно разбавленной смеси.
Современный уровень развития молекулярной теории жидкого состояния не позволяет дать определения коэффициентов диффузии с такой степенью достоверности, которая возможна для разбавленных газовых систем.
В бинарных жидких смесях коэффициент взаимной диффузии £>дв зависит от температуры, размера и формы диффундирующих молекул, их способности к ассоциации и диссоциации, а также от свойств растворителя (вязкость, способность молекул диффузионной среды к ассоциации). При анализе экспериментальных данных и построении корреляций обычно выделяют следующие случаи: диффузия в растворах неэлектролитов, диффузия в растворах электролитов, диффузия в растворах полимеров [7-9].
На основе модели, согласно которой частица растворенного вещества рассматривается как сфера, движущаяся через сплошную среду растворителя, получены [3]: уравнение Стокса — Эйнштейна
£>ЛВ=——, (2.2.1.16)
'’"''лПв
эмпирическое уравнение Ауэрбаха
^ab=W> (2.2.1.17)
где к — постоянная Больцмана; гА — радиус диффундирующей сферической молекулы; т]в — вязкость растворителя; Л/А — молярная масса диффундирующих молекул; К, b — опытные константы.
Коэффициенты К и b в уравнении (2.2.1.17) зависят от химической природы диффузанта; в частности, коэффициент b для различных гомологических рядов углеводородов изменяется от 0,5 до 2,7 [3, 8].
Для практических расчетов коэффициентов диффузии в жидких смесях неэлектролитов используют корреляцию Уилки и Ченга [3, 7, 8], полученную на основе уравнения (2.2.1.16):
/4=7,410-’
(фМ'2
Т
Пв ИА°’6
(2.2.1.18)
где £)дВ — коэффициент взаимной диффузии растворенного вещества А при очень низких его концентрациях в растворителе В, см2/с; Л/в — молярная масса вещества В; Т — температура, К; т]в — вязкость В, мПа-c; ИЛ — молярный объем растворенного вещества при нормальной температуре кипения, см3/моль; <р — параметр ассоциации растворителя В, имеющий следующие значения: 2,6 — для воды; 1,9 — для метанола; 1,5 — для этанола; 1,0 — для неассоциированных растворителей. Значения ИЛ находят по нормальной температуре кипения или по аддитивным объемам по Ле Ба (табл. 2.2.18) [7, 8].
Для приближенных расчетов коэффициента диффузии в жидкостях рекомендуют разные уравнения [1-3, 6, 10, 11]. Например, коэффициент диффузии £>АВ вещества А в жидкости В можно вычислить по приближенной формуле [6, 10]:
ЬЮ"6^372 I 1 1
(2.2.1.19)
где ИА и ИВ — молярные объемы растворенного вещества и растворителя; МА и Мв — молярные массы растворенного вещества и растворителя; А и В — коэффициенты, зависящие от свойств растворенного вещества и растворителя.
По данным Касаткина [11], при диффузии газов или капельных жидкостей
_8,2-10-127’ ^Ав~ и И1/3 ЧВ ' А
(2.2.1.20)
где т]В — динамическая вязкость растворителя, мПа • с.
514
Новый справочник химика и технолога
Таблица 2.2.8
Значения коэффициентов А и В уравнения (2.2.1.19) для некоторых веществ, растворенных в воде
Вещества Коэффициент А Коэффициент В
Ацетон — 1,15
Вода — 4,7
Газы 1 —
Метанол 1,19 2,0
Неассоциированные — 1,0
жидкости
Уксусная кислота 1,27 —
Этанол 1,24 2,0
Таблица 2.2.9
Коэффициенты диффузии веществ в водных растворах
Коэффициенты диффузии соответствуют нулевым концентрациям и получены экстраполяцией.
Диффундирующее вещество Концентрация, моль/л Г, °C D- 104, м2/сут.
I. Неорганические вещества
Вг2 0,1 12 0,8
0,00105 25 1,078
0,00173 25 1,067
0,00183 25 1,064
0,00193 25 1,061
0,00230 25 1,052
0,00309 25 1,036
0,00429 25 1,030
0,00501 25 1,019
со2 18 1,26
СаС12 0,29 9 0,68
0,37 9 0,94
1,5 9 0,72
CdSO4 0 16,8 0,304
0,5 16,8 0,292
1,0 16,8 0,282
1,5 16,8 0,376
2,0 16,8 0,374
3,0 16,8 0,383
3,5 16,8 0,292
4,5 16,8 0,314
5,5 16,8 0,346
6,5 16,8 0,386
7,0 16,8 0,410
С12 0,1 12 1,22
0,1 16,3 1,10
СоС12 0,0062 18 0,600
0,0127 18 0,629
СиС12 1,5 10 0,43
Продолжение таблицы 2.2.9
Диффундирующее вещество Концентрация, моль/л Г, °C D 104, м2/сут.
CuSO4 0,10 17 0,39
0,50 17 0,29
1,95 17 0,23
d2o — 25 2,16
HCI 0 10 1,87
0 25 2,71
0,02 10 1,77
0,02 25 2,56
0,05 10 1,74
0,05 25 2,53
0,20 10 1,77
0,20 25 2,57
0,35 10 1,82
0,35 25 2,64
0,43 0 1,39
1,0 25 3,09
2,0 0 1,52
2,0 11 2,12
5,0 0 1,86
6,5 11 2,67
8,0 0 2,31
9,0 11,5 2,93
HNO3 0,84 5,5 1,50
3,0 6,0 1,54
3,0 7 2,08
20 9 1,94
Н2 — 18 3,10
Н2О2 0,011 20 0,7635
0,099 20 0,9039
0,103 20 0,8542
0,05*’ 18 1,60
0,35*' 18 1,90
0,85*’ 18 2,36
КВг 1,0 10 1,13
КС1 0,00125 25 1,694
0,00194 25 1,688
0,00325 25 1,679
0,00585 25 1,668
0,00704 25 1,662
0,00980 25 1,657
0,02 25 1,685
0,1 25 1,931
0,5 25 1,573
0 18,5 1,464
0,05 18,5 1,348
Химическая кинетика и диффузия
515
Диффундирующее вещество Концентрация, моль/л Г, °C D • 104, м2/сут.
КС1 0,10 18,5 1,326
0,20 18,5 1,315
0,50 18,5 1,331
1,00 18,5 1,386
1,50 18,5 1,439
2,00 18,5 1,489
КОН 0,1 13,5 1,72
0,9 13,5 1,86
3,9 13,5 2,43
KI 0,01 10 1,117
0,01 18 1,460
0,10 10 1,064
0,10 18 1,391
1,0 10 1,045
1,0 18 1,366
5,5 10 1,185
5,5 18 1,549
KNO3 0,05 18 1,26
0,20 18 1,20
0,40 18 0,16
0,80 18 1,10
1,0 18 1,07
2,0 18 0,99
2,5 18 1,01
К2СО3 3,0 10 0,60
K2SO4 0,02 19,6 1,01
0,05 19,6 0,97
0,28 19,6 0,86
0,95 19,6 0,79
LiBr 2,3 10 0,80
4,4 10 0,90
LiCl 0,01 9 0,757
0,01 18 1,000
1,0 9 0,697
1,0 18 0,920
4,2 9 0,724
Lil 1,3 10 0,80
MgSO4 0,5 15,5 0,461
1,0 15,5 0,453
3,0 15,5 0,509
4,5 15,5 0,627
NH3 0,686 4 1,06
3,55 4,5 1,06
n2 — 18 1,40
Продолжение таблицы 2.2.9 Продолжение таблицы 2.2.9
Диффундирующее вещество Концентрация, моль/л Г, °C D • 104, м2/сут.
NaBr 2,9 10 0,86
NaC2H3O2 0,2 12 0,67
NaCl 0,05 18,5 1,08
0,40 18,5 1,03
1,00 18,5 1,07
2,0 18,5 1,11
3,0 18,5 1,17
4,0 18,5 1,23
5,0 18,5 1,28
NaOH 0,02 12 1,12
0,10 12 1,11
0,90 12 1,045
3,9 12 0,985
Nal 1,0 10 0,80
2,0 10 0,90
NaNO3 0,6 13 0,90
3,0 10,5 0,76
5,0 10,5 0,83
6,0 13 0,77
Na2CO3 2,4 10 0,39
Na2SO4 1,4 10 0,66
Ni(NO3)2 0,0088 18 0,57
0,0226 18 0,711
0,068 18 0,802
Pb(NO3)2 0,22 12 0,71
0,82 12 0,66
4,2 18 0,956
Zn(C2H3O2)2 2,0 0 0,120
2,0 18 0,210
ZnSO4 0,025 19,5 0,50
0,050 19,5 0,47
0,55 19,5 0,36
2,95 19,5 0,33
II. Органические вещества
Бутан-1-ол 0,25*2 18 0,76
Глицерин 0,125*3 20 0,72
6,25 18 0,962
15,0 18 0,762
25,0 18 0,744
Глюкоза 0,25*2 18 0,49
2,5 18 0,478
6,0 18 0,419
12,0 18 0,296
516
Новый справочник химика и технолога
Продолжение таблицы 2.2.9
Диффундирующее вещество Концентрация, моль/л Г, °C D • 104, м2/сут.
Метанол 0,25*2 18 1,18
2,901 20 1,414
11,032 20 1,240
16,102 20 1,170
Мочевина 0,25*2 20 1,02
2,0 18 1,437
5,0 18 1,205
10,5 18 1,178
Муравьиная кислота 1,0*2 12 0,97
Пентан-1-ол 0,25*2 18 0,76
Пирогаллол 0,25*2 18 0,57
Пропан-1-ол 0,25*2 18 0,85
4,160 20 0,660
8,764 20 0,671
13,450 20 0,544
Уксусная кислота 0,2*3 13,5 0,77
1,0*3 12 0,74
2,О*3 12 0,69
з,о‘3 12 0,68
4,О*3 12 0,66
Уретан 0,25*2 18 0,75
Фенол 0,25*2 18 0,69
Формамид 9,2 18 1,808
18,4 18 1,706
27,6 18 1,214
Этанол 0,25 *2 18 0,95
3,448 20 0,873
11,908 20 0,881
17,113 20 0,905
*' Н2О2 с 1 % CH3CONH2 в качестве стабилизатора. *2
Концентрация выражена в масс. %.
*3 Молярная концентрация, моль/л.
Таблица 2.2.10
Коэффициенты диффузии в неводных растворителях
Диффундирующее вещество Растворитель Г, °C D- 10 4, м2/сут.
Бензол Бромбензол 7,5 0,885
Бромбензол Бензол 7,3 1,22
Гексан 7,3 1,22
Декалин 7,3 1,224
л/-Ксилол 7,3 1,31
Тетралин 7,3 0,416
Продолжение таблицы 2.2.10
Диффундирующее вещество Растворитель Г, °C D- 10 4, м2/сут.
Бромбензол Толуол 7,3 1,37
Циклогексан 7,3 0,989
Метанол 20 1,67
Этанол 20 2,93
1-Бромнафталин Дибензиловый эфир 7,3 0,129
Декалин 7,3 0,293
Тетралин 7,3 0,310
Бромоформ Ацетон 20 2,32
Бензол 20 1,46
Пентан-Гол 20 0,52
Гексахлорбензол Бензол 7,6 0,883
о-Дихлорбензол Бензол 7,6 1,40
м-Дихлорбензол Бензол 7,6 1,36
л-Дихлорбензол Бензол 7,6 1,23
Иод Бензол 20 1,670
(0,1 М раствор) Бромбензол 20 1,038
Гептан 20 2,386
Метанол 20 1,572
Сульфид водорода 20 2,697
Хлороформ 20 1,831
Этанол 7,3 3,2
Тетрахлорид углерода 20 1,177
Этилацетат 20 1,859
Иодбензол Бензол 7,3 1,17
Подметан Бензол 7,3 1,78
Йодоформ Бензол 7,3 0,968
1 -Иодпентан Бензол 7,3 1,22
Нафталин Бензол 7,6 1,03
Нитробензол Диэтиловый эфир 7,6 2,80
1,2,4,5-Тетра-хлорбензол Бензол 7,6 1,07
Тетрахлорид углерода Бензол 7,3 1,30
1,2,4-Три- Бензол 7,6 1,16
хлорбензол
Фенантрен Бензол 7,6 0,82
Хлорэтилен Бензол 7,3 1,53
Химическая кинетика и диффузия
517
Таблица 2.2.11
Коэффициенты взаимной диффузии в жидких двухкомпонентных органических системах
Приводятся значения D • 104 (м2/сут.) для систем, содержащих 50 мол. % каждого компонента.
Система Т, °C
25 45 65
Бензол—гексадекан 0,83 — —
Бензол—гептан 2,14 2,94 3,73
Бензол—декан 1,49 2,17 2,68
Бензол—додекан 1,21 — —
Бензол—октадекан 0,75 — —
Гептан—гексадекан 0,864 1,24 1,77
Гептан—додекан 1,36 1,85 2,32
Гептан—октадекан 0,795 — —
Гептан—тетрадекан 1,11 — —
Таблица 2.2.12
Диффузия в расплавленных металлах
Диффундирующий металл Диффузионная среда r,°c D- 109, m2/c
Ag Sn 500 4,8
Au Bi Pb Sn 500 500 500 5,22 3,7 5,37
Mg Al 700 7,54
Pb Sn 500 3,68
Pb212 (самодиффузия) Pb 343 2,55
Pt Pb 490 1,95
Rh Pb 500 3,52
Si Fe 1480 1560 2,4 10,8
Таблица 2.2.13
Диффузия в амальгамах
В таблице приводятся данные различных авторов.
Диффундирующий металл T,°C D • 109, m2/c Диффундирующий металл T,°C D- 109, m2/c
Ag 16 1,H К 10,5 0,61
Au 11 0,83 25 0,71
25 0,73 Li 8,2 0,76
Ba 7,8 0,60 25 0,93
Bi 25 1,5 Na 9,6 0,74
Ca 10,2 0,62 25 0,86
Cd 8,7 1,68 Pb 9,4 1,74
99,1 3,43 15,6 1,59
15 1,8 25 2,1
20 1,51 Rb 7,3 0,53
(при 0,1 МПа) Sn 10,7 25 1,77 2,1
Sr 9,4 0,54
20 1,45 Tl 11,5 0,99
(при 150 МПа) 25 1,18
Zn 11,5 2,52
99,2 3,35
25 2,0 15 2,42
Cs 7,3 0,52 20 1,5
25 0,64 25 2,4
Диффузия в расплавленных солях
Диффундирующая соль Диффузионная среда r,°c D • 109, m2/c
AgBr КВг 780 4,92
AgCl KC1, LiCl 480 520 600 740 4,61 4,63 5,30 6,63
Agl KI 720 780 4,63 5,09
AgNO3 KNO3 360 390 4,56 4,86
NaNO3 330 360 4,57 5,06
Ba(NO3)2 KNO3 NaNO3 370 360 2,06 3,72
Таблица 2.2.14
Диффундирующая соль Диффузионная среда T,°C D- 109, m2/c
КВг KNO3 360 3,01
KI KNO3 360 2,96
NaNO3 KNO3 360 5,22
PbCl2 KC1, LiCl 530 720 2,03 4,40
Sr(NO3)2 KNO3 360 370 2,81 3,06
NaNO3 345 360 4,17 4,40
TIBr KBr 770 4,28
T1C1 KC1, LiCl 520 580 3,10 3,47
518
Новый справочник химика и технолога
Продолжение таблицы 2.2.14
Диффундирующая соль Диффузионная среда Т,°С D- 109, М2/с
ТП KI 720 780 3,14 3,30
T1NO3 KNO3 345 3,17
Продолжение таблицы 2.2.14
Диффундирующая соль Диффузионная среда Г, °C D • 109, М2/с
T1NO3 KNO3 380 3,38
NaNO3 320 360 3,94 4,31
Таблица 2.2.15
Экспериментальные значения коэффициентов диффузии различных газов и неэлектролитов в сильноразбавленных водных растворах
Растворенное вещество Г, К м2/с Растворенное вещество Г, К O^'lO9. м2/с Растворенное вещество Г, К ^ю9, м2/с
Азот (диазот) 293 2,6 Диоксид углерода 298 1,92 Пропан-1,2-диол 293 0,88
Аммиак 285 1,64 Диэтиламин 293 0,97 Пропан-1-ол 288 0,87
Анилин 293 0,92 Кислород (дикислород) 298 2,10 Пропан-2-ол 288 0,87
Аргон 293 2,00
Пропен 298 1,1
Аргон 298 2,5 Криптон 293 1,68
Салициловая кислота 298 1,06
Ацетон 293 1,16 Ксенон 293 0,60
Ацетонитрил 288 1,26 Метан 293 1,49
Уксусная кислота 293 1,19
Бензиловый спирт 293 0,82 Метанол 283 0,84
З-Метилбутан- 1-ол 288 0,69 Уретаны 288 0,80
Бензойная кислота 298 1,00
2-Метилпро-пан-1-ол 288 0,77 2-Фуральдегид 293 1,04
Бензол 293 1,02
Хлор (дихлор) 298 1,25
Бутан 293 0,89 Мочевина 293 1,20
Бутан-1-ол 288 0,77 Неон 293 3,00 Щавелевая кислота 298 1,53
Вода 298 2,44 Оксид азота(1) 298 1,69
Этан 293 1,20
Водород (диводород) 293 5,0
298 4,50 Оксид азота(И) 293 2,07 Этан-1,2-диол 298 1,16
Воздух 293 2,5 Этанол 283 0,84
Гелий 293 6,8 Оксид углерода(П) 293 2,03 Этилацетат 293 1,00
Гелий 298 6,28
Этилен 298 1,87
Глицерин 293 0,82 Пиридин 288 0,58
293 1,06 Пропан 293 0,97
Таблица 2.2.16
Коэффициенты взаимной диффузии для Н2, СН4 и СО2 в водных растворах солей
Газ Электролит Концентрация электролита, моль/л -IO9, м2/с
25 °C 45 °C 65 °C
СН4 КС1 2,4 — — 3,86
СН4 КС1 3,65 — 2,52 3,29
СН, MgCl2 1,03 1,60 2,44 3,36
СН) MgCl2 2,95 0,79 1,42 1,70
сщ MgSO4 0,5 1,88 2,58 3,56
СН) MgSO4 2,0 0,73 1,27 1,97
Газ Электролит Концентрация электролита, моль/л JO9. м2/с
25 °C 45 °C 65 °C
СО2 NaCl 1,041 1,73
СО2 NaCl 3,776 1,30
СО2 NaNO3 1,076 1,76
СО2 NaNO3 3,602 1,34
СО2 Na2SO4 0,318 1,74
СО2 Na2SO4 0,898 1,50
Химическая кинетика и диффузия
519
Продолжение таблицы 2.2.16
Газ Электролит Концентрация электролита, моль/л D°Mi • 109, м2/с
25 °C 45 °C 65 °C
СО2 MgCl2 0,377 1,80
СО2 MgCl2 1,262 1,43
СО2 MgSO4 0,195 1,89
СО2 MgSO4 0,969 1,28
СО2 Mg(NO3)2 0,215 1,85
СО2 Mg(NO3)2 1,219 1,64
н2 KC1 1,0 3,79 5,97 —
Н2 KC1 3,0 3,34 5,27 8,30
Н2 MgCl2 1,5 3,09 4,13 6,04
Н2 MgCl2 3,0 1,45 2,71 3,47
Н2 MgSO4 0,5 3,90 5,29 7,18
Н2 MgSO4 2,0 1,57 2,90 4,27
Таблица 2.2.17
Экспериментальные значения коэффициентов диффузии в неводных растворителях при бесконечном разбавлении
Растворенное вещество A Растворитель В Д К м2/с
Азобензол Этанол 293 0,74
Ацетон Толуол 293 2,93
Ацетон Хлороформ 288 2,36
Ацетон Четыреххлористый углерод 293 1,86
Бензойная кислота Ацетон 298 2,62
Бензойная кислота Толуол 293 1,74
Бензол Хлорбензол 293 1,25
Бензол Хлороформ 288 2,51
Вода Анилин 293 0,70
Вода Глицерин 293 0,0083
Вода Пропан-1,2-диол 293 0,075
Вода Пропан-1-ол 288 0,87
Вода Этан-1,2-диол 293 0,18
Вода Этанол 298 1,132
Глицерин Этанол 293 0,51
Диоксид углерода Газойль*1 298 1,95
Диоксид углерода Газойль*2 298 0,73
Диоксид углерода Гептан 298 6,03
Диоксид углерода Керосин 298 2,50
Диоксид углерода 2-Метил-пропан-1-ол 298 2,20
Продолжение таблицы 2.2.17
Растворенное вещество А Растворитель В ДК ^в-Ю9, м2/с
Диоксид углерода Пентан-1-ол 298 1,91
Диоксид углерода Уайт-спирит 298 2,11
Диоксид углерода Этанол 290 3,20
Диоксид углерода Этанол 298 3,42
Карбамид Этанол 285 0,54
Камфора Этанол 293 0,70
Коричная кислота Бензол 298 1,12
Метанол Бензол 298 3,82
Мочевина Этанол 285 0,54
Нафталин Бензол 280,6 1,19
Олово Ртуть 303 1,60
Пиридин Этанол 293 1,10
Тетрахлорид Бензол 298 1,92
углерода
Тетрахлорид Гексан 298 3,70
углерода
Тетрахлорид Циклогексан 298 1,49
углерода
Толуол Гексан 298 4,21
Уксусная кислота Ацетон 298 3,31
Уксусная кислота Бензол 298 2,09
Уксусная кислота Толуол 298 2,26
Хлорбензол Толуол 293 2,06
Хлорэтилен Бензол 280,6 1,77
Этанол Бензол 288 2,25
Этанол Толуол 288 3,00
Этанол Хлороформ 288 2,20
** ГКИ11 = 200 4-300 °C.
’2 Дш = 300 ч- 400 °C.
Таблица 2.2.18
Аддитивные приращения объемов для расчета молярных объемов растворенных веществ при нормальной температуре кипения
Атом (структура) Приращение объема, см3/моль
Азот
образующий связи X=N 15,6
в первичных аминах 10,5
во вторичных аминах 12,0
Бром 27
Водород 3,7
Иод 37
Кислород (кроме случаев, 7,4
указанных в примечании)
в сложных и простых эфирах
метиловых 9,1
520
Новый справочник химика и технолога
Продолжение таблицы 2.2.18
Атом (структура) Приращение объема, см3/моль
этиловых 9,9
высших 11,0
в кислотах 12,0
в соединениях с S, Р, N 8,3
Сера 25,6
Углерод 14,8
Фтор 8,7
Хлор 24,6
Кольца
трехчленные -6,0
четырехчленные -8,5
пятичленные -11,5
шестичленные -15,0
в нафталине -30,0
в антрацене -47,5
Процедуру аддитивного сложения объемов не следует применять для простых молекул. При оценке коэффициентов диффузии используют следующие приближенные значения: Н2 — 14,3; О2 — 25,6; N2 — 31,2; воздух — 29,9; СО — 30,7; СО2 — 34,0; SO2 — 44,8; NO — 23,6; N2O — 36,4; NH3 — 25,8; H2O — 18,9; H2S — 32,9; COS — 51,5; Cl2 — 48,4; Br2 — 53,2; I2 — 71,5 (данные Ле Ба).
2.2.1.4. Диффузия в растворах электролитов
При диссоциации соли в растворе диффундируют не молекулы, а ионы. Без использования электрического потенциала диффузию простой соли можно рассматривать как молекулярную диффузию.
Коэффициент диффузии для простой соли при бесконечном разбавлении вычисляется по уравнению Нернста — Хескелла [7]:
-RT (1/z++1/z-)
>АВ ~ F2 (l/Xj+l/X®)’
(2.2.1.21)
где — коэффициент диффузии, являющийся коэффициентом пропорциональности между молекулярным потоком и градиентом молекулярной концентрации, см2/с; Т — температура, К; 1°, V — предельные (при нулевой концентрации) молярные ионные проводимости соответственно катиона и аниона при температуре Т, См • см2/моль; F— постоянная Фарадея; R — универсальная газовая постоянная; z+, z_ — числа эквивалентности соответственно катиона и аниона.
В табл. 2.2.19 приведены значения и V при 25 °C для наиболее распространенных ионов. В табл. 2.2.20 представлены экспериментальные значения для некоторых водных растворов солей.
Теория диффузии солей в разбавленных водных растворах разработана достаточно хорошо, но она позволяет получить лишь ограниченную информацию о поведении более концентрированных растворов, представляющих интерес для техники [7].
Данных о коэффициентах диффузии газов в растворах солей опубликовано недостаточно. В табл. 2.2.21 приведены типичные значения коэффициентов диффузии водорода и метана в различных электролитах.
Таблица 2.2.19
Предельные ионные проводимости в воде при 25 °C
Катион См • см2/моль Анион х°, См • см2/моль
Ag+ 61,9 Вг’ 78,3
1/2Ва2+ 63,6 HCO' 54,6
1/2Са2+ 59,5 нсо; 44,5
l/3[Co(NH,)6]3t 102 i/2C2o; 74,2
1/2Cu2+ 54 нс2о4 40,2
Н+ 349,8 С1СН2С0‘ 39,8
К+ 73,5 СН3СО2 40,9
1/ЗЬа3+ 69,5 C(N)CH2CO’ 41,8
Li+ 38,7 СН3СН2СО" 35,8
l/2Mg2+ 53,1 СН3(СН2)2СО- 32,6
NH? 73,4 с6н5со- 32,3
Na+ 50,1 сг 76,3
1/2SF2" 50,5 сю; 68,0
Tl+ 74,7 l/3[Fe(CN)6]3' 101
l/2Zn2+ 53 l/4[Fe(CN)6]4- 111
Г 76,8
no; 71,4
ОН* 197,6
i/2so; 80
Химическая кинетика и диффузия
521
Таблица 2.2.20
Коэффициенты взаимной диффузии неорганических соединений в водных растворах
Растворенное вещество Т, °C Концентрация, моль/л Рав ‘ Ю5, см2/с
AgNO3 15 0,17 1,28
Са(ОН)2 20 0,2 1,6
Ca(NO3)2 14 0,14 0,85
СоС12 20 0,1 1,0
CuSO4 14 0,4 0,39
НС1 12 0,1 2,29
HNO3 20 0,05 2,62
20 0,25 2,59
H2SO4 20 0,25 1,63
Н3РО4 20 0,25 0,89
HgCl2 18 0,25 0,92
КВг 15 0,046 1,49
КС1 18 0,4 1,46
Растворенное вещество T, °C Концентрация, моль/л Рав ' 10s, см2/с
КС! 18 0,8 1,49
18 2,0 1,58
KNO3 18 0,2 1,43
КОН 18 0,01 2,20
18 0,1 2,15
18 1,8 2,19
LiCl 18 0,05 1,12
MgSO4 10 0,4 0,39
NLUCl 20 1,0 1,64
NaCl 18 0,4 1,17
18 0,8 1,19
18 2,0 1,23
NaOH 15 0,05 1,49
Таблица 2.2.21
Коэффициенты взаимной диффузии для водорода и метана в водных растворах солей
Газ Электролит 109, м^с Газ Электролит • 109, м2/с
формула концентрация, моль/л 25 °C 45 °C 65 °C формула концентрация, моль/л 25 °C 45 °C 65 °C
н2 н2о — 4,10 6,30 9,44 CH4 н2о — 1,81 2,78 3,84
н2 КС1 1,0 3,79 5,97 — CH4 КС1 2,4 — — 3,86
Н2 КС1 3,0 3,34 5,27 8,30 СН4 КС1 3,65 — 2,52 3,29
Н2 MgCl2 1,5 3,09 4,13 6,04 сн4 MgCl2 1,03 1,60 2,44 3,36
Н2 MgCI2 3,0 1,45 2,71 3,47 сн4 MgCl2 2,95 0,79 1,42 1,70
Н2 Mgso4 0,5 3,90 5,29 7,18 сн4 MgSO4 0,5 1,88 2,58 3,56
Н2 MgSO4 2,0 1,57 2,90 4,27 сн4 MgSO4 2,0 0,73 1,27 1,97
Литература
1. Бретшнайдер С. Свойства газов и жидкостей. Инженерные методы расчета / Пер. с польск. Под ред. П.Г. Романкова. М.; Л.: Химия, 1966. 535 с.
2. Варгафтик Н.Б. Справочник по теплофизическим свойствам газов и жидкостей. 2-е изд., перераб., доп. М.: Наука, 1972. 720 с.
3. Рудобашта С.П., Карташов Э.М. Диффузия в химико-технологических процессах. М.: Химия, 1993. 208 с.
4. Рид Р., Шервуд Т. Свойства газов и жидкостей (определение и корреляция) / Пер. с англ.; Под ред. В.Б. Когана. Л.: Химия, 1971. 704 с.
5. Справочник химика. М.: Химия, 1966. Т. 5. С. 658.
6. Рамм В.М. Абсорбция газов. 2-е изд., переработ. и доп. М.: Химия, 1976. 655 с.
7. Шервуд Т., Пигфорд Р., Уилки Ч. Массопередача / Пер. с англ.; Под ред. В.А. Малюсовой. М.: Химия, 1982. 695 с.
8. Рид Р., Праусниц Дж., Шервуд Т. Свойства газов и жидкостей. 3-е изд., перераб. и доп. / Пер. с англ. Под ред. В.И. Соколова. Л.: Химия, 1982. 591 с.
9. Чалых А.Е. Диффузия в полимерных системах. М.: Химия, 1987.312 с.
10. Павлов К.Ф., Романков П.Г., Носков А.А. Примеры и задачи по курсу процессов и аппаратов химической технологии. Л.: Химия, 1987. 576 с.
11. Касаткин А.Г. Основные процессы и аппараты химической технологии. М.: Химия, 1973. 752 с.
522
Новый справочник химика и технолога
2.2.2. Диффузия в твердой фазе
{проф., д.т.н. Р.Ш. Абиев)
2.2.2.1. Общие понятия
Уравнения, формально описывающие диффузионное перемещение атомов в твердых телах, были предложены Фиком в 1855 г. без экспериментальных оснований, поскольку первоначально были сформулированы применительно к молекулярной диффузии в сильно разбавленных жидких растворах. Первый закон Фика имеет вид:
/,=-Z)Vc,, (2.2.2.1)
где /, — плотность потока z-ro компонента, моль/(м2 • с); D, — коэффициент диффузии z-ro компонента, м2/с; Vc, — градиент его концентрации, моль/м4.
Второй закон Фика, описывающий зависимость концентрации от времени, непосредственно вытекает из первого при учете закона сохранения вещества. Увеличение концентрации частиц в некотором выделенном элементе объема при отсутствии посторонних источников возможно только за счет разности потоков этих частиц, входящих и выходящих из элемента объема, и описывается формулой:
дс, /dt = -divZ, = -VI,. {22.2.2)
Подставляя поток из (2.2.1.22) в (2.2.1.23), получим выражение второго закона Фика’.
dcJdt = V(DyCiY {2.2.2.3)
В большинстве практических задач коэффициент диффузии считается не зависящим от концентрации, а следовательно, и координат. В этом случае второй закон Фика выражается уравнением
дс, Idt = D,V2c, , {22.2.4)
где V2 = div (grad) — оператор Лапласа.
В простейшем случае одномерной диффузии, когда градиенты концентрации и потоки направлены вдоль оси х, уравнение второго закона Фика принимает вид
dc,/dt = D,d2c,/dx2. {22.2.5)
Первый закон Фика является сугубо эмпирическим и не вскрывает природы диффузионных явлений. Более того, в некоторых случаях он вообще приводит к неправильным результатам. Например, при диффузии в поле механических напряжений частицы могут двигаться в сторону увеличения их концентрации (восходящая диффузия), что соответствует физически бессмысленному результату D, < 0. В термодинамике необратимых процессов четко доказывается, что поток частиц в химическом (концентрационном) поле определяется не градиентом их концентрации, а градиентом
химического потенциала [1]. Вагнером сформулировано уравнение для потока заряженных частиц в химическом и электрических полях:
I,н^х^+^ф,), (2-2'2<5>
где х, — парциальная удельная ионная проводимость z-ro компонента, q, — заряд частицы z-ro компонента (алгебраическое значение), ц, — химический потенциал частиц z-ro компонента, отнесенный к одной частице, (pz — электростатический потенциал.
Физический смысл уравнения Вагнера заключается в следующем: Vp, + q,Vcp, есть градиент электрохимического потенциала частиц z-ro компонента Vp(, поток частиц пропорционален этому градиенту, который и рассматривается как движущая сила диффузии заряженных частиц. При отсутствии градиента химического потенциала Vp, уравнение (2.2.1.27) превращается в уравнение закона Ома.
2.2.2.2. Диффузия в непористых материалах
Диффузия при хаотических блужданиях. Можно показать, что между уравнениями (2.2.2.1) и {2.22.6) существует связь. Если диффузионная среда представляет собой идеальную термодинамическую систему, то справедливо соотношение Нернста — Эйнштейна
I, =-kT%,/^q2ci^Vcj, {22.2.7)
представляющее собой уравнение первого закона Фика, в котором роль коэффициента диффузии играет величина
D, = kT%,/(q2c,). (2.2.2.8)
Здесь к — постоянная Больцмана, Т — абсолютная температура.
Коэффициент диффузии £), является фундаментальной характеристикой диффузионной среды и играет чрезвычайно важную роль в теории всех диффузионных процессов. Действительно, идеальная термодинамическая система представляет собой ансамбль невзаимодействующих частиц, которые диффундируют в результате хаотических «блужданий». Теория хаотических «блужданий» опирается на строгие законы статистической механики и достаточно хорошо разработана. Поэтому коэффициент диффузии в данном случае может быть вычислен достаточно точно, что создает условия для разработки теории более сложных диффузионных процессов.
В научно-технической литературе оперируют несколькими диффузионными параметрами, которые при различных диффузионных процессах, формально удовлетворяющих уравнениям Фика, играют роль эффективных коэффициентов диффузии. Все они так или иначе связаны с вышеназванным коэффициентом £)„ однако могут существенно отличаться от него как по величине (зачастую на несколько порядков), так и по характеру зависимости от состава диффузионной среды.
Химическая кинетика и диффузия
523
В современной литературе до сих пор не разработана единая терминология для обозначения различных коэффициентов диффузии. Так, £), иногда называют просто «коэффициент диффузии», иногда «коэффициент самодиффузии» или «коэффициент диффузии компонента». Все эти термины нельзя признать удачными, поскольку они не отражают главную физическую особенность коэффициента £>„ отличающую его от других коэффициентов. В дальнейшем в данной главе для его обозначения будем в основном придерживаться термина «коэффициент хаотической диффузии z-частиц». Термин «коэффициент самодиффузии» будем использовать как для Dj, так и для рассматриваемого ниже коэффициента диффузии радиоактивных изотопов D, в тех случаях, когда речь идет о взаимном перемещении атомов в химически однородной среде (чистом соединении или твердом растворе постоянного состава) и когда нужно подчеркнуть его отличие от направленного потока атомов в поле химических сил, связанных с переменным составом среды.
С помощью соотношения Нернста — Эйнштейна (2.2.2.7) уравнение Вагнера (2.2.2.6) можно представить в виде
7 =----------------Г, (22.2.9)
*r(Vn,+9,V<p,)
при этом коэффициент пропорциональности выражен через коэффициент хаотической диффузии. Эта форма записи эквивалентна уравнению (2.2.2.6), но имеет по сравнению с ним следующее преимущество. Уравнение (2.2.2.6) применимо только к заряженным частицам, а уравнение (2.2.2.9) не содержит заряда частиц перед выражением в скобках и поэтому применимо к любым частицам, в том числе и к нейтральным атомам.
Термодиффузия. Уравнения Вагнера (2.2.2.6) и (2.2.2.9) обобщаются и в более сложном случае, когда к химической и. электрохимической движущим силам добавляется тепловая, если кристалл находится в температурном поле. Тогда при независимом движении частиц разных сортов для потока частиц z-ro компонента записывают так называемое основное уравнение переноса
7V ,
кТ
(2.2.2.10)
где м — энергия переноса частиц z-ro компонента, характеризующая энергию, переносимую через кристалл единичным потоком частиц z-ro компонента при изотермических условиях.
Диффузия радиоизотопов. Коэффициент хаотической диффузии является величиной гипотетической и не поддается прямому экспериментальному измерению, поэтому для его определения используются косвенные методы. Нахождение его с помощью соотношения (2.2.2.8) требует надежного измерения парциальной электрической проводимости которое достигается
только в тех случаях, когда ионный перенос осуществляется исключительно ионами z-ro сорта.
При смешанном переносе наиболее надежную информацию о хаотической диффузии дают измерения коэффициентов диффузии радиоактивных изотопов того химического элемента, диффузия которого изучается. Поскольку по химическим свойствам радиоактивный изотоп не отличим от обычного, его добавление в кристалл равносильно образованию идеального твердого раствора. Однако в большинстве случаев коэффициент диффузии радиоизотопа D' не совпадает с коэффициентом хаотической диффузии соответствующего атома Di, эти коэффициенты связаны соотношением Хэвена
D*=f.D^ (2.2.2.11)
в котором f носит название корреляционного множителя. Природа отличия этого множителя от единицы связана со взаимным влиянием встречных потоков обычного и радиоактивного изотопов, в результате которого нарушается постулат Вагнера о независимом движении частиц разных сортов. В большинстве случаев значение / лежит в пределах от 0,5 до 1.
Диффузия в поле напряжений. Еще один случай отклонения от первого закона Фика связан с влиянием градиента упругих напряжений на диффузию. Градиент потенциала создает поток атомов, который добавляется к концентрационному потоку в уравнении сохранения. Эмпирически установлено, что градиент потенциала V приводит к диффузионному перемещению атомов с некоторой скоростью в соответствии с соотношением
V--BVT, (2.2.2.12)
где В — подвижность, м/(Н с), которая может быть вычислена по формуле
B = DlkT. (2.2.2.13)
Целесообразно отметить, что, в отличие от второго закона Ньютона, в уравнении (2.2.2.12) сила, вызванная градиентом потенциала и равная (-VF), приводит к возникновению постоянной скорости вместо постоянного ускорения, так как атомы все время хаотически меняют направление движения и поэтому не могут ускоряться под действием силы, как свободные частицы [2].
Диффузия в неизотропных телах. Выше коэффициент диффузии выражался скалярной величиной, не зависящей от направления диффузии. В действительности существует много примеров того, что диффузия описывается тензором второго ранга. Так, если кристаллическая решетка вещества кубическая, то направления диффузии неразличимы, т. е. ненулевыми являются компоненты тензора диффузии только на главной диагонали, при этом они одинаковы. В веществах с тетрагональной, гексагональной или орторомбической решетками главные диагональные компоненты отличаются друг от друга, а остальные равны нулю [2-4].
524
Новый справочник химика и технолога
Эмпирические выражения для коэффициентов самодиффузии. [1] Основной специфической чертой коэффициентов самодиффузии (хаотической диффузии и диффузии радиоизотопов) является их экспоненциальная зависимость от температуры (соотношение Аррениуса):
D = Do ехр(-Еа /RT). (2.2.2.14)
Здесь Do — фактор диффузии, Еа — энергия активации, R — универсальная газовая постоянная. Экспоненциальная зависимость коэффициентов самодиффу
зии от температуры наблюдается в широких температурных интервалах для подавляющего большинства твердых тел. Это хорошо видно на рис. 2.2.1-2.2.3, где она приведена в координатах Аррениуса для металлов, их галогенидов и оксидов. Большинство приведенных данных получено методом меченых атомов, меньшая часть относится к хаотической диффузии; однако в используемом масштабе графиков различие между D и D несущественно.
Параметры выражения (2.2.2.14) для различных твердых тел представлены в табл. 2.2.22-2.2.46 [22,23].
Рис. 2.2.1. Температурная зависимость коэффициента самодиффузии металлов (а) и галогенов (б) в галогенидах
Рис. 2.2.2. Температурная зависимость коэффициента самодиффузии металлов (а) и кислорода (б) в оксидах
Рис. 2.2.3. Температурная зависимость коэффициента самодиффузии в металлах
Химическая кинетика и диффузия
525
Таблица 2.2.22
Параметры соотношения (2.2.2.14) для коэффициента взаимной диффузии металлов и сплавов
Прочерки означают, что концентрация примеси исчезающе мала.
Диффундирующий элемент Диффузионная среда Концентрация диффундирующего элемента, ат. % Т,К Dq, см2/с Еа, кДж/моль
Al 1,26 740-850 1,1 137
Ag Au 9 1120-1270 2,9 • Ю2 159
Cu 3 1000-1070 2,9 • Ю 2 156
Pb 0,12 500-600 7,4 • 10"2 64
Al Cu 15-21 770-1120 7,1 • 10’2 164
Ag 100 500-870 5,3 • 10“2 125
Au 18,4 1040-1200 1,1 • Ю 4 112
Cu 2,4-3,5 670-1250 6,8 • 10^ 94
Pb 0,03-0,09 370-570 0,35 59,5
В a-Fe — < 1800 105 260
Bi Pb 2,0 500-560 1,8 • Ю3 77
Карбонизация 1100-1400 1,67 • 10 2 120
Fe 1,1 масс. % 1200-1500 0,49 153
С 0,1-1,1 масс. % 1000-1500 0,12 137
a-Fe — < 1800 7,9 • Ю3 76
— < 1800 2 • Ж2 84
Ag 2,0 920-1170 4,9 • 10 5 94
Cd — < 1200 0,44 175
Cu 3 1000-1130 3,04 • 10 4 99
Pb 1,6 440-520 1,83 • 10’3 65
a-Fe — < 1800 0,4 226
Со y-Fe — < 1800 1,2 • 10“5 435
Сталь — < 1800 90 335
a-Fe — < 1800 3 • 104 343
Cr y-Fe — < 1800 1,8- 104 406
Сталь — < 1800 10 314
С<? W 1-й адсорбционный слой 300-700 0,2 58,6
2-й адсорбционный слой 300-700 1,64 • 10“2 96
Ag 2,0 920-1170 5,9 • 10’5 104
Al Эвтектика 700-800 2,3 146
0,085-0,17 730-840 8,4 • 10’2 137
Au 100 570-890 1,06 • Ю 3 115
Cu 25,6 700-1000 5,8 • 10 4 115
Ge — < 1200 1,9 • 10-4 172
Pt 13,9 1310-1730 4,8 • 10‘2 233
(3-латунь (45 % Zn) — < 1100 3,8 • Ж2 104
Au 18,3 1030-1280 1,16 - 10 4 102
Fe-20% — < 1800 18 • 1(Г°’92х[С]* 314 х [С]*
Fe Ni-C — < 1800 71'10-0,65 х 314 х [С]*
Ni — < 1800 8,4 • 103 214
W 0,04 2200-2800 11,5 586
Ge Ge Самодиффузия < 1200 87 308
Hg Cd 4 430-475 2,6 82
In Ag 20 920-1170 7,3 • Ю 5 102
526
Новый справочник химика и технолога
Продолжение таблицы 2.2.22
Диффундирующий элемент Диффузионная среда Концентрация диффундирующего элемента, ат. % т,к £>о, см2/с Еа, кДж/моль
Li Ge — < 1200 2,5 • 10"3 49,4
Si — < 1700 2,3 • 10’3 63,6
Mg A1 Эвтектика 640-710 1,5 • 10’2 161
Al 5,5-11,0 670-850 0,1 • 10’1 119
Mn Cu 8-11,4 670-1120 7,2 • Ж6 96
Мо w Монокристалл 1800-2530 6,3 • ю 4 33,7
Поликристалл 1800-2530 5 • 10~3 33,7
Fe — < 1800 3,1 • Ж3 75
— < 1800 3 • 10’3 76
N Th — < 1800 6,6 • 10’3 78
a-Ti — < 1970 2,1 • 10’3 94
P-Ti — < 1940 1,2- Ю 2 190
Au 15,0 1070-1280 1,74 • 10~3 131
Ni Cu 7,5-11,8 820-1220 6,5 • 10’5 125
Pt 14,9 1320-1670 7,8 • Ю 4 181
О P-Ti — < 1940 1,6 202
Ag 20,2 720-1200 6,4 • Ж6 84,5
Pd Au 17,1 1000-1250 1,13 • Ж3 91,5
Cu 4,3-6,2 760-1240 1,6 • 10 6 163
Pt Au 20,1 1000-1250 1,24 • Ж3 163
Cu 2,4-3,5 760-1230 1,0- 10^ 91,5
Sb Ag 2,0 920-1170 5,3 • 10’5 91
Si Al 0,5 740-870 0,9 128
Sn Ag 20 920-1170 7,8 • Ж6 89,5
Cu 3,9-5,6 670-1120 4,1 • 10~3 131
p-Sn Pb 2,0 510-560 4,0 ПО
Диффузия на границе 2050-2500 1,13 394
Th w между зернами
Объемная диффузия 2400 1,0 503
Поверхностная диффузия 1650 0,47 278
Tl Pb 2,0 500-550 2,5 • 10"2 81
U w — 2000 1,14 420
a-Fe — < 1800 380 293
W y~Fe — < 1800 103 377
Сталь — < 1800 13 314
Y W — 2000 0,11 260
Самодиффузия в Zn <690 0,22 60
Zn чистотой 99,999 % Самодиффузия в Zn
<690 0,38 61
чистотой 99,99 %
Zn Al 0,84 12 116
6,8-9,7 630-1150 3 • 10^ 8,5
Cu 3 1000-1130 3,7- 10^ 92
0-28,6 910-1150 3,2 • Ж3 176
Р-латунь (45 % Zn) — <1100 0,024 95,5
Zr W — 2000 1,1 326
* [С] — концентрация углерода, ат. %.
Химическая кинетика и диффузия
527
Таблица 2.2.23
Параметры соотношения (2.2.2.14) для коэффициента самодиффузии металлов
Металл r,K Do, CM^/C Ea, кДж/моль
Ag 700-1200 0,81 191
Al 600-700 0,10 128
700-900 1,7 143
Au 900-1200 0,031 165
1100-1300 0,11 177
Вех 800-1300 0,52 158
Бец 800-1300 0,62 165
Cd_L 380-490 0,10 80
Cd| 380-490 0,05 76
Со 1050-1300 0,5 274
1300-1600 0,2 260
Г'-. 1200-1500 10"* 221
СГ 1500-1900 0,28 309
Cs 1100-1300 47 25,7
1000-1200 11 23,9
Си 800-1300 70 235
1000-1200 120 278
a-Fe 1200-1500 3,6 298
y-Fe 1350-1600 1,1 284
1700-1800 6,8 259
S-Fe 1000-1400 0,48 276
Hf 2070-2300 0,0012 162
I ILL 320-420 3,7 78,2
Inl 320-420 2,7 78,2
к 220-230 0,16 3,92
La 930-1120 1,5 189
Li • 208-520 0,24 55,2
MgL 740-900 1,5 136
Mg || 740-900 1,0 135
Mo 2000-2200 2,8 465
2100-2600 0,3 398
Na 208-520 0,2 41,9
Nb 1150-2700 1,1 402
Ni 1300-1700 1,9 285
Pb 450-600 0,28 102
Pd 1300-1800 0,20 267
Pr 1100-1200 0,087 123
Pt 1500-2000 0,22 279
8-Pu 310-700 0,0045 100
8-Pu 800-900 0,027 77,4
Rb 208-520 0,23 39,4
Sbx 770-900 0,10 150
Sbj 770-900 56 201
Продолжение таблицы 2.2.23
Металл Г, К Dq, cm2/c Ea, кДж/моль
P-Snx 430-500 10,7 110
P-Sn|| 430-500 7,7 107
Ta 1500-2400 0,12 413
2100-2800 2 462
a-Ti 960-1100 6,4 • 108 123
P-Ti 1170-2850 0,0016 146
a-Th 1400-1700 700 350
P-Th 1700-1800 105 417
a-TL 420-560 0,4 94,5
470-690 0,39 104
a-Tlj 470-690 0,08 92
a-U 850-920 0,002 168
p-u 970-1020 0,014 176
y-U 1050-1300 0,002 113
V 1150-1600 0,36 308
1600-2100 210 394
w 1560-1730 6,3 • 107 570
2300-3000 0,54 502
2900-3600 43 640
a-Zr 600-1000 5 • 108 92
900-1100 5 • 10’2 218
P-Zr 1200-1800 0,0024 159
Примечание. 1 — поперечное направление диффузионного потока [001], || — продольное направление [010].
Таблица 2.2.24
Концентрационная зависимость констант Z)o и Еа для диффузии металлов в меди при 800 °C
Данные, соответствующие нулевой концентрации, получены экстраполяцией.
Диффундирующий металл Концентрация диффундирующего металла, ат. % Do, см27с Еа
кДж моль ккал моль
Al 0 0,0175 157,8 37,7
4 0,0454 165,4 39,5
8 0,375 180 43
12 6,75 201 48
16 252 226 54
Be 0 2,3 ♦ Ю 4 117,4 28
4 7,1 • 10 4 126 30
8 0,033 155 37
Са 0 2,0- 109 34 8
4 2 • 108* 42* 10*
Si 0 0,037 167,5 40,0
4 0,405 201,8 48,2
8 18,7 225,2 53,8
528
Новый справочник химика и технолога
Продолжение таблицы 2.2.24
Диффундирующий металл Концентрация диффундирующего металла, ат. % £>о, см2/с Еа
кДж моль ккал моль
Sn 0 1,13 188,4 45,0
4 0,0032 127,7 30,5
Zn 0 0,0166 157,8 37,7
4 0,0525 167 40
8 0,0776 167 40
12 0,0046 130 31
16 0,0063 130 31
* Для концентрации 0,5 ат. %.
Таблица 2.2.25
Константы диффузионного соотношения (2.2.2.14) для диффузии металлов в солях
Диффундирующий металл Диффузионная среда см2/с Еа
кДж моль ккал моль
Au КС1 11,0 125,6 30,0
NaCI 0,2 103,0 24,6
Си КС1 55,0 127,3 30,4
NaCI 0,5 105,5 25,2
Ni NaCI 0,2 106,3 25,4
Таблица 2.2.26
Коэффициент диффузии ионов в кристаллах при различных температурах
Диффундирующий ион Диффузионн ая среда Г, К Do- 10Л CM2/c
Ag‘ CuBr 518 51
Cu+ a-Cu2S 503 1,85
603 4,6
693 9,4
a-Cu2Te 603 0,359
749 1,98
794 4,66
Cu3Sb 723 0,139
Li+ AgCl 511 0,24
a-Agl 753 37-41
a-Ag2S 443 0,4
503 4,1
603 6,3
693 16,5
Na+ AgBr 573 0,023
AgCl 723 0,116
Pb2+ PbCl2 473 4,5
523 11
578 15
653 23
Продолжение таблицы 2.2.26
Диффундирующий ион Диффузионная среда T,K Do-ЮЛ CM2/c
Pb2+ Pbl2 387 7,3 - IO’11
420 1,35 • 10’9
438 7,35 • 10’9
cr Agl 453 1,1 • 10 4
Se2“ Cu2S 844 1,1 • 10’3
967 4,9 • 10’3
Таблица 2.2.27
Параметры соотношения (2.2.2.14) для коэффициента диффузии иона Ag+ в кристаллах
Диффузионная среда T,K Do, cm2/c E кДж/моль
a-Agl 454-744 — 9,45
a-Agl 451-701 1,6 - 10 4 9,45
a-Ag2I 473-653 5,8 • 10’3 19,1
a-Ag2S 443-693 4,6 • 10 4 13,3
a-Ag2Se — 6,7 • 10 4 85,5
a-Ag2Se 421-673 1,6 • 1 O'4 12,3
a-Ag2Te 428-678 3,8 • 10~5 11,1
a-CuI 685-753 4,5 • 10’3 28,3
a-Cu2S 586-1192 3,3 • 10 4 19,1
a-Cu2Te 603-794 2,4 87
NaBr — 50 199
NaCI <550 1,6 75
>550 3,1 174
PbCl2 439-743 7,7 154
Pbl2 387-438 7,8 126
528-588 10,6 127
PbS 733-1043 1,3 176
Таблица 2.2.28
Коэффициент взаимной диффузии твердых солей
Диффундирующее вещество Диффузионная среда Г.К D- 10”, CM2/c
Ag (из AgNO3) Na-стекло 627 25,2
Na-пермутит 627 14,8
Ag+Sn Cu4Sn 723 231
BaMoO4 BaWO4 1223 3,47
MgMoO4 MgWO4 1073 2,31
MgWO4 MgMoO4 1073 1,39
SrMoO4 SrWO4 1223 9,14
SrWO4 SrMoO4 1223 3,01
ZnMoO4 ZnWO4 1073 3,94
1123 6,36
ZnWO4 ZnMoO4 1073 1,00
1123 2,89
Химическая кинетика и диффузия
529
Таблица 2.2.29
Самодиффузия в твердом водороде
Г, К D Ю20, см2/с Еа
Дж/моль кал/моль
11,3 11,8 13,2 13,6 <0,017 0,035 1,2 ±0,25 >2 >3302 ±544 790±130
Таблица 2.2.30
Параметры соотношения (2.2.2.14) для коэффициента диффузии газов в твердых телах
Диффундирующее вещество Диффузионная среда Г, К D 106, см2/с Еа, кДж/моль
н2 SiO2 473 773 8,5 11 42,7 42,7
Не SiO2 293 773 5,7 2,9 23 23
NH3 Анальцим* Пирекс 773 293 5,5 163 36,4
* Природный алюмосиликат.
Таблица 2.2.31
Параметры соотношения (2.2.2.14) для коэффициента диффузии малой примеси в серебре
Примесь T,K Do, см2/с Ea, кДж/моль
Ag* 450-900 0,81 191
Au 650-950 0,26 191
Cd 592-937 0,44 175
Со — 104 251
Cu 700-950 1,23 193
Fe 718-927 2,42 205
Ge — 0,084 153
Hg 650-950 0,08 160
In 592-937 0,41 170
Ni 750-950 21,9 230
Pb 700-825 0,22 160
Pd 735-939 9,57 238
Ru 794-945 180 276
Sb 468-942 0,17 160
Sn 592-937 0,25 164
Tl 650-800 0,15 159
Zn 640-925 0,54 174
‘ Самодиффузия.
Таблица 2.2.32
Параметры соотношения (2.2.2.14) для коэффициента диффузии малой примеси в меди
Примесь T,K Do, cm2/c Ea, кДж/моль
Ag < 1356 0,63 195
As < 1356 0,12 176
Au 700-1000 0,15 192
Cd < 1356 0,93 189
Co 701-1077 1,93 226
Cu* 500-1000 70 234
Fe 716-1074 1,4 217
Ga < 1356 0,55 192
Hg < 1356 0,35 184
Ni 743-1076 2,7 237
Pd 807-1056 1,71 228
Sb 600-1000 0,34 176
Tl 785-996 0,71 181
Zn < 1356 0,34 191
Самодиффузия.
Таблица 2.2.33
Параметры соотношения (2.2.2.14) для коэффициента диффузии малой примеси в золоте
Примесь T,K Do, CM2/c Ea, кДж/моль
Ag 700-1000 0,072 168
Au* 700-1050 0,091 175
Co 700-950 0,068 174
Fe 700-950 0,082 174
Hg 500-1030 0,116 157
Ni 700-950 0,034 176
Pt 800-1050 7,6 255
* Самодиффузия.
Таблица 2.2.34
Параметры соотношения (2.2.2.14) для коэффициента диффузии малой примеси в никеле
Примесь T,K Dq, CM2/c Ea, кДж/моль
Al 1070-1250 1,1 249
1400-1600 1,87 268
Au 1200-1400 2 272
Co 1020-1460 0,75 271
1430-1650 1,11 272
Cr 940-1170 0,03 171
1400-1600 1,1 273
Cu 1320-1620 0,57 258
Fe 1200-1800 0,8 255
Mg 1070-1250 2,3 • IO5 131
1400-1600 0,44 235
Mn 1400-1600 7,5 281
Mo 1400-1600 3 288
530
Новый справочник химика и технолога
Продолжение таблицы 2.2.34
Примесь Т,К Do, см2/с Еа, кДж/моль
Si 1070-1250 10,6 271
1400-1600 1,5 258
Ti 1400-1600 0,86 257
W 1400-1600 11,1 322
Продолжение таблицы 2.2.35
Металл Примесь T,K Do, CM2/c Ea, кДж/моль
Sn Zn 360-510 5,9 • 10 3 49,4
TI Ag Au 360-500 390-500 0,033 5,3 • 104 48,2 21,8
* В квадратных скобках указаны индексы Миллера, характеризующие направление диффузионного потока.
Таблица 2.2.35
Параметры соотношения (2.2.2.14) для коэффициента диффузии различных примесей в металлах Ш и IV групп
Металл Примесь* T,K Do, CM^/c Ea, кДж/моль
Р-А1 Ag+ 470-670 1,65 • 104 16,8
К+ 470-670 0,78 • 10 4 22,4
Li+ 470-670 14,5 • 10 4 36,4
Na+ 470-670 2- 10 4 15,9
Rb+ 470-670 0,34 • 10 4 30,1
Tl+ 470-670 0,65 • 10 4 34,4
In Ag 300-420 0,11 48,2
Au 300-400 9 • 103 281
Pb Ag >400 4,6 - 1 O’2 60,3
390-570 7,5 • 10 2 63,6
Au 460-570 4,1 • 10’3 39,1
460-600 8,7 • 10~3 41,9
Cd >420 0,41 88,9
Cu >500 7,9 • Ю 3 33,5
Hg 470-570 1,05 95
Na 520-490 6,3 119
Ni 480-870 9,4 ♦ 10 3 44,4
Pd 480-860 3,8 ♦ 10"3 36
Sn 510-560 0,16 96,5
TI 480-600 0,5 104
Zn 450-570 1,6 • 10’2 47,3
Sn Ag [001] 400-510 7,1 • IO3 51,5
Ag[100] 400-510 0,18 77
Au [001] 400-510 5,8 • 10’3 46
Au [100] 400-510 0,16 74,1
Cd [001] 460-500 220 118
Cd [100] 460-500 120 113
Cu 410-500 2,4 • 10 3 33,1
Hg [001] 450-500 7,5 106
Hg [110] 450-500 30 112
In [001] 450-500 12 107
In [100] 450-500 34 108
Sb [001] 470-500 79 122
Sb [100] 470-500 77 123
Таблица 2.2.36
Параметры соотношения (2.2.2.14) для коэффициента диффузии атомов примесей в щелочных металлах
Металл Примесь У. К Do, cw^/c E ‘-•a, кДж/моль
К Au 280-326 1300 12,4
Li Ag 340-430 0,12 52,8
Au 320-420 0,21 46,1
Cu 320-450 0,47 38,6
Na 330-450 0,41 52,8
Zn 330-450 0,57 54,5
Na Au 270-350 3,3 9,2
Таблица 2.2.37
Параметры соотношения (2.2.2.14) для коэффициента диффузии атомов примеси в лантанидах и актинидах
Металл Примесь T, К Do, cw^/c E L~,a, кДж/моль
La Au 980-1070 0,022 75,8
Pr Ag 880-1040 0,14 106
1070-1190 0,032 90,0
Au 880-1020 0,043 82,5
1070-1180 0,033 84,1
Co 880-1040 0,047 68,7
Cu 930-1060 0,084 75,8
1090-1190 0,057 74,5
Zn 880-1040 0,18 104
1100-1200 0,63 113
U Co 1050-1350 3,5 • Ю 4 52,8
Cr 1050-1350 5,4 • 10’3 103
Cu 1050-1350 2 • IO-3 101
Fe 1050-1350 2,7 • 10 4 50,2
Mn 1050-1350 1,8-10^ 58,2
Nb 1050-1350 0,049 166
Ni 1050-1350 5,4 • 10^ 65,8
Химическая кинетика и диффузия
531
Таблица 2.2.38
Параметры температурной зависимости коэффициента диффузии малой примеси в P-Ti
D = D01 ехр (-£а1 / RT) + D02 exp (-Еа2 / RT)
При месь Т,К А • 104, CM2/c Ea\, кДж/моль АгХ cm^/c £д2, кДж/моль
Со 1200-1900 130 130 16 257
Сг 1220-1900 74 154 14 274
Fe 1200-1900 80 126 15 254
Мп 1200-1900 76 144 12 270
Мо 1170-1900 7 155 3,6 272
Nb 1270-1900 13 146 9,5 291
Ni 1200-1900 170 133 20 252
Р 1220-1900 36,2 101 5 237
Sc 1200-1560 21 137 34 —
Sn 1230-1870 3,8 132 9,5 291
Та 1300-1900 7,2 151 50 335
Ti 1170-1800 2 125 1,0 250
V 1200-1820 10 146 3,4 258
Таблица 2.2.39
Параметры соотношения (2.2.2.14) для коэффициента диффузии ионов в кристаллах солей
Ион Соль T,K Dq, CM^/c F L-‘ai кДж/моль
Cs+ CsCl 630-730 1,1 130
760-880 0,1 134
Cs+ CsI 570-770 80 158
K+ KBr 730-1000 0,01 122
K+ KC1 800-950 137 207
860-1020 1,8 • IO 5 76
K+ KF 870-1100 2 172
K+ KI 730-950 10’5 62
Li+ LiF 850-1000 0,8 192
1000-1200 5,6 289
Na+ NaBr 770-950 0,67 147
Na+ NaCl 550-820 3,5 • Ю 6 81,2
860-1070 76,9 196
Rb+ RbCl 840-970 33,3 193
Br KBr 670-970 103 196
Br~ NaBr 920-1020 1,0 164
СГ CsCl 550-730 1,51 122
760-880 0,7 152
СГ KC1 770-1010 178 216
СГ NaCl 770-1020 60,7 206
СГ RbCl 870-970 33,3 193
Г CsI 680-830 0,39 122
Г KI 730-950 1,2 ♦ 10“3 108
Таблица 2.2.40
Параметры соотношения (2.2.2.14) для коэффициента диффузии примесей в кристаллах солей
Кристалл Примесь T,K Do, cm2/c F кДж/моль
KBr Pb 550-670 1,5 • 10~3 87,5
Tl 410-480 0,091 96,5
620-770 50 192
KC1 Ag 470-920 — 40,6
Bi 670-950 5,6 • 10’3 94
Cd 620-770 4,7- 1 O’5 52,5
Ce 770-970 1,1 ♦ 10’3 100
Co 470-920 — 19,3
Cs 840-1020 0,7 169
Cu 620-920 10,6 118
Eu 700-820 0,064 124
Li 770-1000 20 142
Na 860-1020 2,2 169
Pb 500-750 10’3 96,5
Rb 880-1040 26,8 195
Tl 500-730 5,8 • Ю11 23
730-1000 1,3 • 10’3 126
СГ 770-920 1,5 • 10’3 109
Г 770-920 50 193
OH 670-890 1,2 • 103 193
NaCl Ag 850-1000 380 192
Au — 0,2 103
Ca 700-1020 6- 104 87
Cd 800-930 3,9 • 10’3 251
Co 880-1030 8 • 10’3 106
Cs 870-970 1,62 192
Cu 620-920 33,8 138
Hg 720-820 8,2 • 10 5 55
К 670-930 — 48,3
Mn 670-970 10 4 65
Ni 900-1020 0,02 125
Pb 600-800 0,015 94,5
Rb 870-1020 28,5 162
Sr 390-1070 1,7- Ж3 126
Zn 810-1000 0,04 94,5
Br“ 770-920 20 187
Г 800-970 500 216
NaF Li 850-1000 0,8 192
1000-1100 5,6 290
Nal Tl 670-910 5 • 10“3 113
532
Новый справочник химика и технолога
Таблица 2.2.41
Параметры соотношения (2.2.2.14) для коэффициента диффузии атомов водорода и его изотопов в металлах
Атом Металл T,K Do, CM2/c Ea, кР&Змоль
Н Си 600-1100 3,5 • Ю 4 39,5
Н a-Fe < 1800 7,5 ♦ 10^ 10
Н Nb >273 5,0 • 10^ 10,2
>223 0,9 • Ю 4 6,5
Н Ni >631 6,9 • 10’3 40,5
Н Ni <631 4,8 • 10’3 39,4
Н Pd <900 2,9 • 103 22,2
Н Ta >273 4,4 • 10^ 13,5
<200 2 • 10 6 3,85
Н V <600 3,1 • 104 4,36
D Cu 800-1100 2,5 • Ю 4 39,2
D Nb < 1000 5,2 • 10 4 12,3
D Ta <600 4,6- 104 15,5
D V <600 3,8 • 10 4 7,12
Т Cu 800-1100 2- 104 39,2
т Nb < 1000 4,5 • 10 4 13
Таблица 2.2.42
Параметры соотношения (2.2.2.14) для коэффициента диффузии атомов инертных газов в кристаллах солей
Кри- сталл Газ T,K Do, cm2/c Ea, кДж/моль
CsBr Xe 530-850 100 140
CsCl Xe 620-740 740-920 0,1 0,1 86,6 83,5
CsF Xe 720-870 102 19,3
CsI Xe 420-770 0,57 96
KBr Ar Xe 290-570 290-570 105 10 145 135
KC1 Ar Kr >670 450-520 520-770 7,9 • 10^ 8 • 10-5 106 36,7 107 203
KI Xe 420-770 1,5 98
RbBr Kr 460-600 >600 4- 107 5- Ю4 163 28,9
RbCl Kr 630-970 7,9 • 104 43
RbF Kr 570-1000 2,5 132
Rbl Kr Xe 450-610 >610 420-770 1,3 • 106 1,6 • 10’3 0,082 136 29,4 89,5
Таблица 2.2.43
Параметры соотношения (2.2.2.14) для коэффициента диффузии атомов примеси в полупроводниках
Полупроводник Диффундирующее вещество Г, К Do, cm2/c Ea, кДж/моль
AlSb Al — — 174
Cu 420-770 0,0035 34,5
Sb — — 145
Zn 930-1130 0,33 186
CdS Ag 570-770 0,24 77,2
Au 770-1070 200 174
Cd 670-1000 1,1 • 105 60
Cd 1000-1420 3 193
Cu 420-670 2000 92,7
Cu 670-1020 0,0015 73,4
Li 880-1230 3 • 10 6 65,6
CdSe P 1070-1270 — 203
Se 1220-1700 1,3 • 105 42,8
CdTe Au 600-1000 67 193
Cu 370-570 3,7 • 10-4 64,7
In 720-1300 0,041 154
Se 950-1200 1,7- Ю4 130
GaAs As 1400-1500 4- 104 980
Au 860-1330 103 106
Cd 1070-1370 0,0013 212
Ga 1400-1500 107 540
Ge 1320-1410 7,5 348
Li 520-770 0,53 96,5
Mg 1070-1270 4 • 10’5
Mn 1000-1300 0,65 240
S 110-1500 180 251
Sn 1300-1500 6 - 104 241
Те 1270-1370 2,6- 10’5 139
Zn 1070-1370 3 • IO 7 96,5
GaP S 1400-1700 3200 453
Ge Ag 900-1200 0,044 96,5
Al 1000-1200 0,05 261
Au 1000-1200 0,055 242
As 900-1200 4,5 232
В 1000-1200 0,084 444
Be 900-1200 0,5 242
Bi 900-1200 — 234
Cu 900-1200 0,0033 17,4
Co 900-1200 0,16 106
Fe 1000-1200 0,13 106
Ga 1000-1200 20 320
Химическая кинетика и диффузия
533
Продолжение таблицы 2.2.43
Полупроводник Диффундирующее вещество T,K £>0, CM2/c Ea, кДж/моль
Ge Ge 1000-1200 49 209
Н 1000-1200 0,0027 36,7
Не 900-1200 0,0061 68
In 1000-1200 0,048 231
Li 900-1200 0,0012 49,2
N 900-1200 — 249
Ni 900-1200 0,42 87
0 900-1200 0,17 195
P 1000-1200 1,3 240
Pb 1000-1200 — 348
Sb 1000-1200 3,6 232
Sn 1000-1200 0,017 183
Ta 900-1200 2,5 • 10* 112
Zn 900-1200 3,3 247
HgTe Cd 520-620 3,1 • io4 66,7
InAs Ag 730-1200 7,3 • 104 25
As 1000-1200 3 • 107 432
Au 900-1200 0,0058 62,8
Cd 770-900 1,5 • 10* 69,5
900-1200 6- 104 112
Cu 1200 0,036 51
Ga 930-970 3200 304
Ge 900-1200 3,7 • 10* 113
In 1000-1200 6 - 105 386
Li 1070 2,3 • 104 184
Mg 900-1200 2- 10* 113
P 920-1170 130 261
S 900-1200 6,8 212
Sb 600-920 0,0087 109
920-970 34000 333
Se 900-1200 13 212
Sn 600-920 900-1200 2,4 ♦ 10’5 1,5 • 10^ 77,4 113
Те 600-920 900-1200 3,8 • 104 3,4 • 10 5 116 124
Zn 900-1200 0,0037 104
InP Ag 800-1200 3,6 • 104 57
Au 870-1100 1,3 • 10’5 463
Cd 1000-1200 1,1 • 10’7 70
Cu 900-1200 30 66,5
In 1120-1270 IO5 373
P 1120-1270 7 • IO10 545
Продолжение таблицы 2.2.43
Полупроводник Диффундирующее вещество T, К Do, CM2/c Ea, кДж/моль
InSb Ag 700-800 io7 24,2
Au 400-800 7 • 104 30,9
Cd 520-770 105 106
Cu 500-760 3,5 • 10’5 35,7
Co 770-770 2,7 • 10n 37,7
Fe 700-780 107 24,2
Ge 600-770 5 • 10^ 91,5
Hg 700-770 4- 10^ 113
In 600-800 2 • 10 9 27
Li 770 7 • 104 27
S 470-720 4- IO5 101
Sb 600-800 1,4 - IO4 363
Se 470-720 0,016 125
Sn 570-770 1,3 • 10^ 62,7
Те 630-770 6,6 • Ю 5 115
Zn 620-770 1,6 - 106 222
Se Ge 490 9,4-104’ 38,6
Fe 490 10’5 37,5
In 490 5,2-10* 30,9
S 490 4,9 26
Se* 490 2,8 • 10’8 28
Sn 490 4,8 • 10’8 37,6
Те 490 5,4 • 10 * 39,6
TI 490 1,4 • 10* 33,8
Zn 490 3,8 • 10’7 28
Si Ag 1400-1600 0,002 154
Al 1400-1700 4,8 323
As 1300-1600 34 376
Au 1100-1500 0,0011 106
В 1000-1400 6 • 10’7 163
Bi 1300-1600 IO3 444
C 1350-1700 0,33 282
Cr 1200-1500 0,01 96,5
Cu 670-970 0,0047 41,5
1100-1400 0,4 96,5
Fe 1300-1600 0,0062 83
Ga 1300-1600 90 376
Ge 1400-1700 6,3 • 105 510
H 1240-1480 0,0094 46,3
He 1300-1600 0,11 121
In 1300-1600 18 374
К 800-1060 0,0011 72,5
Li 300-400 0,0025 63,6
700-1100 0,0023 62,7
534
Новый справочник химика и технолога
Продолжение таблицы 2.2.43
Полупроводник Диффундирующее вещество Г К CM2/c Ea, кДж/моль
Si Na 800-110 0,0016 69
О 1300-1600 0,21 292
Р 1300-1600 20 364
S 1300-1600 0,92 212
Sb 1300-1600 6,2 380
Si* 1400-1700 5,4- 103 483
Те 1300-1600 16,5 375
Zn 1250-1550 0,1 135
Те Hg 640-700 7,9 • 10’5 78
Se 590-700 260 120
Те* 600-700 3,5 • 10"1 96
Ti 630-700 320 172
ZnS S 970-1160 — 328
Zn 1170-1200 3 • IO4 147
Zn 1210-1300 1,5 • 104 314
ZnSe Cu 470-840 1,7 • 10 5 64
Zn 1000-1100 1000 332
ZnTe Те 1000-1270 2 • 10 4 367
Zn 1060-1220 0,01 183
* Самодиффузия.
Таблица 2.2.44
Параметры соотношения (2.2.2.14) для коэффициента самодиффузии в некоторых кристаллах
Кристалл 7', К Dq, cm2/c p L-ai кДж/моль
Ar 78 350 17,4
С (графит) 2270-2620 10 680
Ge 1000-1200 10,8 261
a-P (белый) 273-303 1,1 • 10~3 39,3
Xe 121-158 7,3 34,9
Таблица 2.2.45
Коэффициенты диффузии атомов металлов в амальгамах
Металл ГК D • 10 s, cm2/c Металл Г, К D- IO’5, cm2/c
Ag 289 1,11 Li 298 0,93
Au 298 0,73 Na 298 0,86
Ba 281 0,60 Pb 298 2,1
Bi 298 1,5 Rb 280 0,53
Ca 283 0,62 Sn 298 2,1
Cd 298 2,0 Sr 283 0,54
Cs 298 0,64 TI 298 1,18
К 298 0,71 Zn 298 2,4
Таблица 2.2.46
Коэффициент диффузии в расплавленных металлах
Диффундирующий металл Диффузионная среда 7, К По • 10’5, cm2/c
Ag Sn 800 4,8
Au Bi 800 5,22
Au Sn 800 5,37
Mg Al 1000 7,54
Pb Sn 800 3,68
Pt Pb 760 1,95
Rh Pb 800 3,52
Si Fe 1750 2,4
Si Fe 1830 10,8
2.2.2.3. Диффузия в капиллярно-пористых материалах
Массоперенос в капиллярно-пористых телах — сложный процесс, который обусловлен рядом причин, зависящих от вида технологического процесса (сушка, десорбция, экстрагирование, гетерогенный катализ на пористых катализаторах и т. д.), характеристик пористой среды (величина и конфигурация пор, распределение пор по размерам, характер соединения их между собой), энергетического состояния поверхности стенок пор, физико-химического сродства молекул извлекаемого вещества (диффузант) и «скелета» твердого тела, температуры, давления, степени заполнения пор извлекаемым веществом.
Диффузия при сушке. При сушке движение влаги в капиллярно-пористом материале происходит как в виде жидкости, так и в виде пара. Миграция жидкости может осуществляться за счет массопереноса под действием разности капиллярных потенциалов, пленочного течения, обусловленного градиентом расклинивающего давления пленки, поверхностной диффузии в микропорах (г < 10 9 м) и переходных порах (г = 10”9 4-10 7 м), термокапиллярного течения жидкости во всем объеме поры, термокапиллярного пленочного движения вдоль стенок пор, фильтрационного переноса жидкости под действием градиента общего давления в материале и т. д. Движение пара происходит за счет молекулярной диффузии пара, кнудсеновской диффузии, стефанов-ского потока, термодиффузии пара, теплового скольжения в микро- и макропорах (г > 10 7 м), циркуляции парогазовой смеси в порах, конвективно-фильтрационного переноса под действием градиента общего давления, бародиффузии (молекулярного переноса компонента с большей массой в область повышенного давления) и т. д. [5]. При большом влагосодержании материала преобладает капиллярный поток, с уменьшением влагосодержания материала возрастает вклад парового и пленочного потоков, а также поверхностной диффузии.
Влияние твердой фазы на массоперенос в капиллярно-пористых материалах проявляется различным образом: скелет твердого тела загромождает часть сечения,
Химическая кинетика и диффузия
535
по которому движется диффузионный поток, удлиняет путь диффузии, изменяет свойства граничной фазы. При этом возникают особые виды массопереноса — поверхностная диффузия, пленочное течение, капиллярный поток и т. д.
Диффузию пара в порах материала подразделяют на свободную, когда длина свободного пробега Л молекулы много меньше диаметра поры: Л <к 2г, и кнудсенов-скую — при Л » 2г. При Л ~ 2г механизм массопереноса смешанный. В свою очередь свободную диффузию пара подразделяют на нормальную диффузию, происходящую в открытых с обоих концов порах, и одностороннюю (стефановскую) — в порах, открытых только с одного конца.
Свободная и кнудсеновская диффузии описываются уравнением типа:
/г =-Z)rVcr, (2.2.2.15)
где JT — плотность диффузионного потока пара, сг — концентрация пара в парогазовой смеси в порах материала, DT — коэффициент диффузии пара, учитывающий диффузионное сопротивление пористой среды.
Коэффициент нормальной диффузии DT в капиллярно-пористом материале связан с коэффициентом диффузии в гомогенной парогазовой среде D соотношением
Dr = De/Цд, (2.2.2.16)
где £ — пористость материала, цд — диффузионное сопротивление со стороны материала.
Коэффициент стефановской диффузии связан с коэффициентом нормальной диффузии уравнением, учитывающим поправку на возникающий в тупиковых порах конвективный перенос парогазовой смеси:
£)стг = DTP/(P-p), (2.2.2.17)
где Р — полное давление, Па; р — парциальное давление пара, Па.
При кнудсеновской диффузии молекулы, достигая стенок пор, адсорбируются на них некоторое время, а после десорбции движутся в произвольном направлении (отраженная диффузия). Отраженная диффузия и время задержки молекул на стенках пор являются дополнительными факторами, снижающими плотность диффузионного потока (по сравнению со свободной диффузией в порах). В этом случае роль межмолекулярных соударений незначительна, поэтому понятия «поток» и «диффузия» при кнудсеновской диффузии совпадают, и каждый компонент ведет себя так, как если бы в смеси присутствовал он один.
Коэффициент кнудсеновской диффузии можно рассчитать по уравнению
где М — молярная масса диффузанта, кг/кмоль; при этом коэффициент кнудсеновской диффузии уменьшается с уменьшением радиуса пор.
Пленочное течение обусловлено градиентом «расклинивающего» давления пленки, его поток равен
, (2.2.2.19)
где т| — динамическая вязкость жидкости, Па с; р — плотность жидкости, кг/м3; р — расклинивающее давление, Па; h, ho — толщина пленки и неподвижной граничной фазы соответственно, м; цпл — коэффициент сопротивления пленочному переносу.
Поверхностная диффузия описывается уравнением
/n = -Z>npMVa, (2.2.2.20)
где Dn — коэффициент поверхностной диффузии; рм — плотность абсолютно сухого материала, кг/м3; а — величина сорбции, кг/кг.
Диффузия при адсорбции. При адсорбции обычно рассматривают четыре вида диффузии в порах: свободную, кнудсеновскую, поверхностную и «твердотельную». Доминирование одного из названных видов диффузии зависит от пористой структуры зерна и степени его заполнения сорбатом. «Твердотельная» диффузия наблюдается в порах диаметром в несколько ангстрем (d ~ 1О”10 м), при этом потенциальные поля противоположных стенок перекрываются. Поверхностная диффузия происходит практически на любой поверхности — как в широких, так и в узких порах. Однако заметную роль она начинает играть только в достаточно узких порах, когда кнудсеновская диффузия мала.
При нормальных физических условиях коэффициент диффузии газов и паров в капиллярно-пористых материалах имеет ориентировочно следующий порядок: при свободной диффузии 10 5—10 4 м2/с, при кнудсеновской < 10-6 м2/с, при поверхностной < 107 м2/с, при твердотельной < 10-9 м2/с.
Дифференциальное уравнение диффузии (аналог второго закона Фика) записывают для суммы плотностей потоков сорбата в зерне адсорбента в газовой и конденсированной фазах
^-+^ = div(Z)' grade,.), (2.2.2.21)
где D' — коэффициент внутренней диффузии, равный D'~ Dr + Dn (да/dcdi-
Для линейной изотермы адсорбции (да/дсД = = Г = const и, следовательно, выражение (2.2.2.21) можно представить в виде
дс
= div(Z)egradcr), (2.2.2.22)
dt
где De = D7(l + Г) - эффективный коэффициент диффузии при адсорбции, который при (da/dcT)t » 1 совпада
536
Новый справочник химика и технолога
ет с коэффициентом массопроводности при адсорбции [5].
Значения коэффициентов диффузии D' и De при адсорбции приведены в работах [6, 7]. Коэффициенты D* и De в значительной степени зависят как от типа адсорбента, так и от природы адсорбата. Особенно сильным избирательным свойством обладают цеолиты, что объясняется молекулярно-ситовым действием этих адсорбентов. Зависимость коэффициентов D’ и De от определяющих факторов (температуры, величины адсорбции) имеет сложный характер, поэтому при расчетах кинетики и динамики адсорбции часто используют усредненные по концентрации значения коэффициентов диффузии.
Диффузия при экстрагировании. При экстрагировании из капиллярно-пористого материала миграция распределяемого вещества в твердой фазе обычно осуществляется посредством молекулярной диффузии. Плотность диффузионного потока в материале, отнесенную к единице его поверхности, описывают уравнением Фика с использованием эффективного коэффициента диффузии (коэффициента массопроводности) De [8]
/ = -DeVc, (2.2.2.23)
где I — вектор плотности диффузионного потока в расчете на полное сечение материала; De — эффективный коэффициент диффузии, зависящий от структурных параметров материала; с — концентрация извлекаемого вещества.
Влияние пористой структуры материала на эффективный коэффициент диффузии проявляется в следующей последовательности: 1) удлиняется путь диффузионного потока вследствие извилистости капилляров; 2) элементы «скелета» твердого тела уменьшают свободное сечение потока; 3) потенциальное поле стенок пор воздействует на прилегающие слои жидкости, что в ряде случаев приводит к образованию граничной фазы и адсорбционного слоя молекул извлекаемого вещества. В последнем случае перенос извлекаемого вещества в капиллярно-пористом материале происходит в основном за счет молекулярной диффузии в объеме пор, а поверхностной диффузией в слое зачастую можно пренебречь.
Зависимость эффективного коэффициента диффузии De от параметров пористой структуры характеризуется соотношением
De = ПеП, (2.2.2.24)
где D — коэффициент диффузии в гомогенной среде (в чистой жидкости); П — коэффициент диффузионной проницаемости; т = сП — параметр, зависящий от пористости структуры [9].
При использовании переменного давления в жидкости перенос распределяемого (извлекаемого) вещества определяется в существенной мере конвекцией, обусловленной колебательным движением жидкости в макро
порах, что приводит к увеличению эффективного коэффициента диффузии на несколько порядков. Это позволяет сократить продолжительность процесса экстрагирования в десятки и сотни раз.
Диффузия при гетерогенном катализе. Диффузионный массоперенос играет большую роль в гетерогенном катализе, в котором обычно используют твердые пористые катализаторы. Во многих случаях диффузия реагентов (или продуктов реакции) через поры в гранулах катализатора (внутренняя диффузия) значительно влияет на скорость процесса. Промышленные катализаторы имеют активную поверхность порядка нескольких сотен квадратных метров на 1 г, что обусловлено их тонкопористой структурой, т. е. они фактически являются капиллярно-пористыми материалами.
При анализе массопереноса в гранулах пористых катализаторов выделяют следующие виды диффузии: свободную, кнудсеновскую и поверхностную. Если поры катализатора заполнены жидкостью, кнудсеновская диффузия отсутствует.
Для описания диффузии в пористых катализаторах используют уравнение (2.2.2.23). Влияние пористой структуры материала на эффективный коэффициент диффузии можно рассматривать на основе различных моделей, из которых наиболее широко применяют модель извилистых капилляров, модель со «случайным» пересечением пор и серийную модель [12]. Согласно модели извилистых капилляров, эффективный коэффициент диффузии для изотропных однородно-пористых материалов выражается соотношением типа (2.2.2.24) [13]:
De = Dsn = De /у2, (2.2.2.25)
где П = 1 /у2; у — коэффициент извилистости.
Для модели со «случайным» пересечением пор можно принять П = е. Анализ опытных данных показывает, что зависимости вида De-f (s) применимы для описания диффузии в материалах с однотипным морфологическим строением, для которых в уравнении (2.2.2.25) можно с некоторым допущением принять у =Л£)' В общем случае пористые материалы различаются не только пористостью, но и целым рядом других характеристик (функция распределения пор по размерам, форма боковой поверхности поры, конфигурация ее поперечного сечения, профиль поры в продольном разрезе, извилистость порового канала, взаимное соединение и расположение пор, микрорельеф поверхности стенок пор и т. д.) [14], влияние которых на коэффициент De необходимо учитывать.
В частности, в так называемой серийной модели предусмотрено влияние функции распределения пор по размерам. Согласно серийной модели эффективный коэффициент молекулярной или кнудсеновской диффузии можно выразить соотношением
De = Dsn = De х/у2, (2.2.2.26)
где П = %/у2; х — фактор формы.
Химическая кинетика и диффузия
537
Для молекулярной диффузии фактор формы определяется выражением
а для кнудсеновской — формулой
00 00 1
X = Хкн = \^f№r J— f(r)dr ’ (2.2.2.28)
о о'
где fir) — дифференциальная функция распределения радиусов элементарного диффузионного канала по размерам, м1; г и Гер — текущий и средний радиусы пор соответственно, м.
Уравнение (2.2.2.26) учитывает гофрировку пор, т. е. более детально описывает влияние пористой структуры на эффективный коэффициент диффузии.
Исследования показали, что значение коэффициента извилистости у реальных однородно-пористых материалов, не имеющих бидисперсной структуры, лежит в пределах от 1 до 1,57 и с увеличением пористости уменьшается, приближаясь к 1. Экспериментальная зависимость у = Де) аппроксимирована следующими уравнениями [5]:
у = 1,57- 8,03 • 10 3ехр(9,5Е), б < 0,37; (2.2.2.29)
у= 1 + 10ехр(-9,5Е), 8 >0,37. (2.2.2.30)
Определение фактора формы по уравнению (2.2.2.27) связано с целым рядом проблем экспериментального характера. Для однородно-пористых материалов функции распределения пор, по размерам которых хорошо описываются логарифмически нормальным законом распределения, предложены следующие аппроксимирующие зависимости для расчета Хм [5]:
Хм = ехр( -19,5olg(r)), с lg(r) < 0,25; (2.2.2.31)
Хм = 2,08 ехр( -4,76 olg(r)),
0,25 < ст lg(r) < 0,7,
(2.2.2.32)
где ст lg(r) = X A [1g “ (1g r )ср ] стандартное
\ i=l J
отклонение логарифмов радиусов пор;
п
(1gг) = ]Гд Igr — средний логарифм радиусов пор;
i=l
Pi = Ае; /е — доля объема пор z-й фракции.
2.2.2.4. Диффузия в коллоидных капиллярно-пористых материалах
К коллоидным капиллярно-пористым телам относятся материалы растительного и животного происхождения, которые частично применяют в химической технологии, а также пористые полимеры. В последних перенос вещества через стенки пор осуществляется за счет молекулярной диффузии и может либо подчиняться закону Фика (первая подгруппа коллоидных капиллярно-пористых материалов), либо не подчиняться (вторая подгруппа) — в зависимости от масштаба и скорости происходящих в материале структурных изменений.
В коллоидных капиллярно-пористых материалах характер диффузии зависит от того, являются ли поры замкнутыми или образуют сквозные, сообщающиеся между собой каналы. В материалах с замкнутыми порами диффузия молекул через стенки пор (наиболее медленная стадия процесса) лимитирует общую скорость диффузии, при этом коэффициент диффузии имеет тот же порядок величины, что и в материале, составляющем стенки пор.
Кинетические исследования и результаты математического моделирования показывают, что диффузия воды в гранулах полистирола при сушке в интервале температур 30-80 °C подчиняется закону молекулярной диффузии Фика, т. е. данный материал относится к первой подгруппе класса коллоидных капиллярно-пористых материалов. Возникновение в полистироле некоторого количества пор объясняется значительным локальным уменьшением объема кристаллических областей при охлаждении расплава.
Другой полимерный материал — целлулоид — относится ко второй подгруппе класса коллоидных капиллярно-пористых материалов, для которой характерна аномальная диффузия. Данные сорбционных измерений показывают, что целлулоид поглощает пар этилового спирта в количестве, существенно превышающем пористость материала, т. е. поглощение пара происходит как порами, так и матрицей полимера. Кривые распределения массовых долей этилового спирта по толщине пластины из целлулоида имеют обычный для таких распределений вид, необычным для них является медленное понижение поверхностной концентрации этилового спирта в образце в течение всего времени сушки, что не характерно для процесса, контролируемого внутренней диффузией.
Медленное установление десорбционного равновесия на поверхности раздела фаз объясняется напряженным состоянием полимера: под действием набухшего центрального ядра поверхностные слои растягиваются; растягивающие усилия препятствуют десорбции находящихся в материале молекул этилового спирта [5]. По мере сушки перепад концентраций по толщине материала уменьшается, механические напряжения релаксируют, и поверхностная концентрация снижается, приближаясь к равновесному значению.
538
Новый справочник химика и технолога
В отдельных случаях аномальную диффузию в полимерах с достаточной точностью можно описать на основе нелинейного дифференциального уравнения Фика, однако в целом для ее описания требуются специальные математические модели.
2.2.3. Диффузионная проницаемость через мембраны (проф., д.т.н. Р.Ш. Абиев)
Проникновение газа, пара или жидкости через мембрану осуществляется в зависимости от ее структуры. Если материал обладает капиллярно-пористой структурой, перенос вещества в нем обусловлен явлениями, происходящими в порах; если же он в предельном случае является непористым, распределяемое вещество мигрирует путем молекулярной диффузии [16-18].
Для описания плотности потока газа (пара) через мембрану используют уравнение проницаемости:
i = K(pl-p2)/d, (2.2.3.1)
где К — коэффициент проницаемости, м2/(с-Па); Р\,Рг— парциальное давление газа (пара) в средах по обе стороны мембраны (вдали от нее), Па; d — толщина мембраны, м.
Диффузия газов и паров через пористую мембрану в зависимости от размеров пор может происходить по типу свободной, кнудсеновской, поверхностной или твердотельной диффузии. Кроме того, определенную роль играют капиллярная конденсация пара и пленочное течение. Общее сопротивление массопередаче складывается из диффузионных сопротивлений пограничных слоев и диффузионного сопротивления собственно мембраны, которое, как правило, является определяющим.
Проницаемость газов и паров через непористые полимерные мембраны складывается из последовательности элементарных актов: диффузии (молекулярной или турбулентной) распределяемого вещества из ядра первой среды к поверхности мембраны, абсорбции его мембраной, диффузии в ней, десорбции и диффузии его от поверхности мембраны в ядро потока второй среды. При этом, в силу большого диффузионного сопротивления мембраны, диффузионными сопротивлениями пограничных слоев обычно можно пренебречь и считать концентрацию газа (пара) у поверхности мембраны равной концентрации в ядре потоков фаз. В этих условиях параметрами, определяющими процесс, являются характеристики изотермы сорбции—десорбции распределяемого вещества и коэффициент молекулярной диффузии его в полимере. Если коэффициент диффузии газа в мембране D = const, изотерма сорбции—десорбции линейная, то коэффициент проницаемости можно выразить соотношением
К = (КТ^)АД), (2.2.3.2)
где R — газовая постоянная диффузанта, Дж/(кг-К); Tq = 273 К; ро = 101325 Па; А? — коэффициент рас
пределения, вычисляемый как тангенс угла наклона линии равновесия к оси абсцисс при рабочих условиях процесса, кг/м3 • Па.
Температурная зависимость коэффициента проницаемости газов обычно описывается уравнением Аррениуса. Проницаемость паров и жидкостей через непористые полимерные мембраны в значительной степени зависит от их растворимости в полимере. При соприкосновении с жидкостями или их парами полимеры в большей или меньшей степени набухают. При этом межмолекулярные силы полимера ослабевают, энергия активации обычно снижается, что приводит к увеличению коэффициента диффузии. В общем случае коэффициент диффузии зависит от концентрации распределяемого вещества в полимере [5]. При сорбции водяного пара полимерными пленками наблюдается сильное отклонение от закона Генри уже при относительной влажности воздуха 40-60% [19]. Решение задачи стационарной диффузии газов и паров через плоскую мембрану при условии зависимости коэффициента диффузии от концентрации дано в работе [5].
2.2.4. Диффузия в ионообменных смолах
(проф., д.т.н. Р.Ш. Абиев)
Ионный обмен можно отнести к одному из самых сложных явлений переноса, связанных с диффузией [18,19]. Следовательно, процесс ионного обмена осложнен влиянием внутренней и внешней диффузии, скоростью химической реакции, а также электродиффузион-ным потенциалом. Учет одного или нескольких доминирующих факторов возможен в рамках упрощенной кинетической модели [20,21].
Для расчета скорости ионного обмена требуется знание коэффициентов переноса ионов в ионите, в качестве которых могут использоваться коэффициенты самодиффузии индивидуальных ионов или эффективные коэффициенты взаимодиффузии [20]. Коэффициенты диффузии обычно рассчитывают по данным экспериментальных измерений, используя три способа исследований: 1) метод постоянно обновляемого раствора; 2) метод ограниченного объема раствора; 3) измерение электрической проводимости ионитов.
По первым двум методам для определения коэффициентов самодиффузии изучают кинетику изотопного обмена с использованием радиоактивных изотопов. Расчет коэффициентов диффузии на основании кинетических данных производят по следующим уравнениям:
1. При постоянно изменяющемся объеме раствора в случае внутридиффузионной кинетики степень использования ионита/[21] дается бесконечным рядом:
/=i-4^-Vexp(-Fv2«2)’ <2-2-4-1)
где Род = Dtlr$ — число Фурье или диффузионный критерий Фурье (обобщенное время); D— коэффициент диффузии в ионите, м^с; t — время, с; г0 — радиус зерна ионита, м.
Химическая кинетика и диффузия
539
Для малых степеней заполнения ионита, практически до /и ~ 0,3 4- 0,4, можно использовать приближенное уравнение
„ lFon
(2.2.42)
V 7t
Для степени заполнения / = 0,50 так называемое время полуобмена /0,5 может быть выражено уравнением [21]
Го,5 = O,O3Oro2/Z>. (2.2.4.3)
2. При ограниченном объеме раствора и неселективном обмене получено решение для степени отработки ионита в следующей форме [21]:
2 exp(-Sn2Fofl)
3w 1 + Sn2/9 w(w +1) ’
(22.4.4)
где w — параметр ограниченности раствора, Sn — корни трансцендентного уравнения SnctgSn = 1 + Sn2/(3w).
3. Метод определения коэффициента диффузии по измерению электрической проводимости ионитов основан на уравнении, связывающем коэффициент самодиффузии /-го иона с удельной электрической проводимостью ионита х:
1О37?7хУ,
^2cr
(22.4.5)
где N, — число единиц переноса /-го иона; cR — емкость ионита в расчете на 1 см3; F — постоянная Фарадея; Z, — формальный заряд иона.
В табл. 2.2.47 приведены значения коэффициентов самодиффузии ионитов, рассчитанные по уравнениям (2.2.4.1)-(2.2.4.5) [19, 21].
Таблица 2.2.47
Значение коэффициентов самодиффузии ионитов
Марка ионита Содержание ДВЕ, масс. % Противоион T, °C Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии D • 106, см2/с
Катиониты*1
КБ-4П-2 2 Н+ — — 0,02
2 Li+ — — 1,30
2 Na+ — — 2,30
2 К+ — — 4,30
2 Cu2+ 21 — 0,00187
КУ-2 — Н+ 19 — 18,7
— н1 — — 11,0
— к+ 19 — 7,9
— к+ — — 1,70
— Na+ 19 — 4,1
4 Cu2+ 21 — 1,87
4 FT 25 0,25 37,81
4 Na+ 25 0,25 5,82
4 Ni2+ 21 — 1,40
4 UO2+ 21 — 1,35
8 Ca2+ 25 0,25 0,57
8 FT 25 0,05 16,81
8 H+ 25 0,10 20,23
8 H+ 25 0,25 23,56
8 FT 25 0,50 25,68
8 FT 25 1,0 27,21
8 K+ 25 0,25 5,74
8 Li+ 25 0,25 2,36
8 Mg2+ 25 0,25 0,51
8 Na+ 25 0,05 2,01
8 Na+ 25 0,10 2,32
540
Новый справочник химика и технолога
Продолжение таблицы 2.2.47
Марка ионита Содержание ДВБ, масс. % Противоион Т,°с Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии D • 106, см2/с
Катиониты^
КУ-2 8 Na+ 25 0,25 3,44
8 Na+ 25 0,50 3,99
8 Na+ 25 1,0 3,99
12 IT 25 0,25 12,63
12 Na+ 25 0,25 1,84
16 H* 25 0,25 10,19
16 Na+ 25 0,25 1,29
КФ-1-4 4 Cu2+ 21 — 0,0084
4 Ni2+ 21 — 0,0152
СДВ-3 — H‘ — — 1,87
— K+ — — 0,02
— Na+ — — 4,10
— Sr2+ — — 0,86
Nepton CR-51 — Cu2+ 27 — 0,53
— Na+ 23 — 2,15
— Zn2+ 27 — 0,43
Аниониты *1
АВ-17 6 СГ — — 3,15
6 СГ 20 — 4,2
6 СГ 25 — 5,6
6 СГ 30 — 6,5
6 СГ 35 — 7,5
6 СГ 40 — 8,7
6 C9H8N3O2S’ Норсульфазол 20 — 0,00013
(анион)
6 То же 25 — 0,00027
6 » 30 — 0,00056
6 » 35 — 0,001
6 » 40 0,00178
6 C12H13N4O2S Сульфадимезин 20 — 0,00012
(анион)
6 То же 25 — 0,00024
6 » 30 — 0,00036
6 » 35 — 0,00065
6 » 40 — 0,001
6 C12H13N4O4S-Сульфадиметоксин 20 — 0,000036
(анион)
6 Тоже 25 — 0,000065
6 » 30 — 0,00012
6 » 35 — 0,00027
6 » 40 — 0,00042
Химическая кинетика и диффузия
541
Продолжение таблицы 2.2.47
Марка ионита Содержание ДВБ, масс. % Противоион Г, °C Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии D • 10б, см2/с
Аниониты ♦1
АВ-17 6 C10H9N4O2S Сульфазин (анион) 20 — 0,0018
6 То же 25 — 0,0032
6 » 30 —• 0,0049
6 » 35 — 0,0075
6 » 40 — 0,0115
6 ChHhN4O3S-Сульфапиридазин 20 — 0,00065
(анион)
6 То же 25 — 0,0012
6 » 30 — 0,002
6 » 35 — 0,0027
6 » 40 — 0,0049
6 C8H9N2O3S~ Сульфацил (анион) 20 — 0,015
6 То же 25 — 0,024
6 » 30 — 0,032
6 » 35 — 0,042
6 » 40 — 0,056
6 NO3 — — 2,08
6 ОН — — 1,58
6 so^ — — 1,27
8 C6H7N2O2S Сульфаниламид 20 — 0,033
(анион)
8 То же 25 — 0,042
8 » 30 — 0,056
8 » 35 — 0,075
8 » 40 — 0,087
— Г — — 0,77
АВ-27 4 СГ 25 — 3,08
8 сг 25 — 1,00
16 сг 25 — 0,64
4 сг 50 — 6,25
8 сг 50 — 2,28
16 сг 50 — 1,45
4 сг 75 — 10,43
8 сг 75 — 4,43
16 сг 75 — 2,95
АН-31 — сг — — 0,012
Катиониты*2
КБ-4П-2 2 Na+ — — 3,40
КУ-2 — н+ — — 13,0
— к+ — — 1,6
542
Новый справочник химика и технолога
Продолжение таблицы 2.2.47
Марка ионита Содержание ДВБ, масс. % Противоион T,°c Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии D • 106, см2/с
Катиониты*2
КУ-2 8 Ва2+ 20 0,1 0,091
8 Ва2+ 20 0,2 0,103
8 Ва2+ 20 0,4 0,112
8 Ва2+ 20 1,0 0,124
8 Са2+ — — 0,16
8 FT — — 16,50
8 Na+ — — 2,58
8 Na+ — — 2,00
8 Na+ 25 1,0 4,10
8 Zn2+ 20 0,1 0,30
8 Zn2+ 20 0,25 0,38
8 Zn2+ 20 0,5 0,45
12 Cu2+ 20 IO-4 0,062
12 Cu2+ 43 10-4 0,12
12 Cu2+ 60 104 0,20
12 Cu2+ 20 10’3 0,072
12 Cu2+ 43 102 0,14
12 Cu2+ 60 10’3 0,22
12 Cu2+ 20 102 0,096
12 Cu2+ 43 10’2 0,19
12 Cu2+ 60 102 0,29
12 Cu2+ 20 0,1 0,11
12 Cu2+ 43 0,1 0,23
12 Cu2+ 60 0,1 0,34
12 Cu2+ 20 1,0 0,12
12 Cu2+ 43 1,0 0,24
12 Cu2+ 60 1,0 0,36
12 K+ — — 4,70
16 Na+ 20 1,0 1,50
КУ-23 (60) 8 Na+ 65 — 2,35
СДВ-3 — Na+ — — 3,50
Цеокарб 225 8 FT — — 5,60
8 Na+ — — 1,20
Dowex 50 — Na+ 0,3 — 0,12
Na+ 25 —- 0,29
4 FT 0,3 — 4,51
4 H+ 25 — 9,14
4 Na+ — — 1,60
4 Na+ 0,3 — 0,67
4 Na+ 25. — 1,41*3
4 Na+ 25 — 5,25*3
4 Zn2+ 25 — 0,40
8 Ag* 25 — 0,38
Химическая кинетика и диффузия
543
Продолжение таблицы 2.2.47
Марка ионита Содержание ДВБ, масс. % Противоион T,°C Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии D • 106, см2/с
Катиониты*2
Dowex 50 8 А13+ 25 — 0,0645
8 Ва2+ — — 0,0792
8 Ы 0,3 — 2,70
8 IT 25 — 5,40
8 Na+ 25 — 2,03
8 Zn2+ 25 — 0,23
8,6 Ag+ 0,3 —• 0,26
8,6 Ag+ 25 — 0,64
8,6 Cs+ 0,3 — 0,66
8,6 Cs+ 25 — 1,37
8,6 K+ 0,3 — 0,63
8,6 K+ 25 — 1,34
8,6 Na+ 0,3 — 0,35
8,6 Na+ 25 — 0,94
8,6 Rb+ 0,3 — 0,64
8,6 Rb+ 25 — 1,38
12 Na+ 25 — 0,73
12 Zn2+ 25 — 0,21
16 Ag+ 0,3 — 0,10
16 Ag+ 25 — 0,28
16 IT 0,3 — 1,10
16 H+ 25 — 2,20
16 Na+ 0,3 — 0,0375
16 Na+ 25 — 0,11
16 Sr2+ 0,3 — 0,0006
16 Sr2+ 25 — 0,00298
24 Ag+ 0,3 — 0,0336
24 Ag+ 25 — 0,0113
24 Cs+ 0,3 — 0,0332
24 Cs+ 25 — 0,073
24 Na+ 0,3 — 0,0269
24 Na+ 25 — 0,10
24 Rb+ 0,3 — 0,0967
24 Rb+ 25 — 0,23
24 Y+ 0,3 — 0,000059
24 Y+ 25 — 0,000218
24 Zn2+ 25 — 0,000263
4 Br’ 3,4 — 3,2
8 Br" 3,4 — 1,11
16 Br’ 0,3 — 0,000153
16 Br’ 3,4 — 0,24
16 Br 25 — 0,000510
DowexA-1 — Co2+ 29 0,4 0,0061
544
Новый справочник химика и технолога
Продолжение таблицы 2.2.47
Марка ионита Содержание ДВБ, масс. % Противоион T,°C Молярная концентрация эквивалента, ммоль/мл Коэффициент диффузии/) • 106, см1 2 3 4/с
Катиониты*2
Dowex А-1 — Со2+ 29 — 0,0074
— Со2+ 29 — 0,0075
Na+ 29 0,08 0,12
(pH = 3,2)
Na+ 29 0,1 и
(pH = 7,6)
Na+ 29 о,1 0,67
(рн= 12,6)
— Zn2+ 29 0,1 0,0051
— Zn2+ 29 0,2 0,0091
— Zn2+ 29 0,2 0,0074
Nepton CR-51 — Ca2+ 27 — 0,31
— Na+ 23 — 1,95
— Zn2+ 27 — 0,41
Аниониты*2
АВ-17 — Г 25 — 0,77
6 cr — — 2,18
6 г 20 — 2,18
6 г 40 — 0,82
6 г 60 — 1,86
АВ-27 4 cr 25 — 1,85
4 cr 50 — 2,70
4 cr 75 — 3,40
8 cr 25 — 0,76
8 cr 50 — 1,30
8 cr 75 — 1,80
16 cr 25 — 0,40
16 cr 50 — 0,90
16 cr 75 — 1,40
1 Значения D определены на основе измерения электрической проводимости по уравнению (2.2.4.5).
*2 Значения D определены на основе измерения радиоактивности.
*3 Данные разных авторов.
Литература
1. Чеботин В.Н. Химическая диффузия в твердых телах. М.: Наука, 1989. 208 с.
2. Шьюмон П. Диффузия в твердых телах. М.: Металлургия, 1966. 195 с.
3. Старк Дж. П. Диффузия в твердых телах / Пер. с англ.; Под. ред. Л.И. Трусова. М.: Энергия, 1980. 240 с.
4. Бокштейн Б.С., Бокштейн С.З., Жуховицкий А.А. Термодинамика и кинетика диффузии в твердых телах. М.: Металлургия, 1974. 280 с.
5. Рудобашта С.П., Карташов Э.М. Диффузия в химико-технологических процессах. М.: Химия, 1993.208 с.
6. Романков П.Г., Рашковская Н.Б., Фролов В.Ф. Массообменные процессы химической технологии. Л.: Химия, 1975. 336 с.
7. Тимофеев Д.П. Кинетика адсорбции. М.: Изд. АН СССР, 1962.211 с.
8. Аксельруд Г.А., Альтшулер М.А. Введение в капиллярно-химическую технологию. М.: Химия, 1983.264 с.
Химическая кинетика и диффузия
545
9. Аксельруд Г.А. Массообмен в системе твердое тело—жидкость. Львов: Изд. Львовского ун-та, 1970.186 с.
10. Абиев Р.Ш., Островский Г.М. Моделирование процесса экстрагирования из капиллярно-пористой частицы с бидисперсной структурой // Теор. основы хим. технол. 2001. Т. 35, № 3. С. 270-275.
11. Абиев Р.Ш. Исследование процесса экстрагирования из капиллярно-пористой частицы с бидисперсной структурой // Журн. прикл. химии. 2001. Т. 74, № 5. С. 754-761.
12. Решетов В.А. Диффузия газов в пористых катализаторах // Теор. основы хим. технол. 1983. Т. 17, №2. С. 194.
13. Саттерфилд Ч.Н. Массопередача в гетерогенном катализе. М.: Химия, 1976. 240 с.
14. Рудобашта С.П. Массоперенос в системах с твердой фазой. М.: Химия, 1980. 248 с.
15. Рейтлингер С.А. Проницаемость полимерных материалов. М.: Химия, 1974. 269 с.
16. Дытнерский Ю.И. Обратный осмос и ультрафильтрация. М.: Химия, 1978. 352 с.
17. Рудобашта С.П., Дмитриев В.М., Планов-скийА.Н. Исследования паропроницаемости полимерных мембран // Высокомол. соединения. Сер. А. 1978. Т. 20, № 3. С. 572.
18. Кокотов Ю.А., Пасечник В.А. Равновесие и кинетика ионного обмена. Л.: Химия, 1970. 336 с.
19. Кокотов Ю.А., Золотарев П.П., Елькин Г.Э. Теоретические основы ионного обмена: Сложные ионообменные системы. Л.: Химия, 1986. 280 с.
20. Смирнов Н.Н., Волжинский А.И., Константинов В.А. Расчет и моделирование ионообменных реакторов. Л.: Химия, 1984. 224 с.
21. Волжинский А.И., Константинов В.А. Регенерация ионитов. Теория процесса и расчет аппаратов. Л.: Химия, 1990. 240 с.
22. Физические величины: Справочник / А.П. Бабичев, Н.А. Бабушкина, А.М. Братковский и др.; Под. ред. И.С. Григорьева, Е.З. Мейлихова. М.: Энергоатомиздат, 1991. 1232 с.
23. Справочник химика. М.; Л.: Химия, 1965. Т. 3. 1005 с.
Раздел 3
ФИЗИКА, ХИМИЯ И ТЕХНОЛОГИЯ ДИСПЕРСНЫХ СИСТЕМ (КОЛЛОИДНАЯ ХИМИЯ)_______________________________
Автор-составитель: проф., д.х.н. Е.Е. Бибик
ЗЛ. Поверхность. Фундаментальные свойства
3.1.1. Геометрия дисперсной системы
Дисперсными принято называть гетерогенные системы, состоящие как минимум из двух фаз, причем одна из них, например твердая (фаза <«»), находится в дисперсном, т. е. измельченном состоянии. Другая фаза— сплошная, например жидкая (фаза «/»), является при этом средой (дисперсионной), несущей в себе дисперсную фазу (рис. 3.1). Фаза — часть системы, которая по химическому составу или физическому (агрегатному) состоянию отличается от остальной части системы и отделена от нее поверхностью раздела. Наряду с термином «поверхность раздела фаз» иногда будут использоваться эквивалентные термины «поверхность контакта фаз», «межфазная поверхность», именуемая также «поверхностью» или «границей раздела».
Если дисперсная система содержит более двух фаз, то обычно только одна из них может быть средой, а остальные, следовательно, должны быть дисперсными. Примером являются краски, составленные для придания им нужного цвета из нескольких пигментов (ZnO, РЬ3О4 и др.) и общей среды (олифы, раствора полимера, воды). О таких системах можно говорить, что они имеют двухкомпонентную (ZnO, РЬ3О4), трехкомпонентную (ZnO, Pb3O4, CdS) и т. д. дисперсную фазу (хотя правильнее их называть «двухфазная» или «трехфазная дисперсная фаза»).
Каждая из фаз дисперсной системы может быть в любом агрегатном состоянии — газообразном, жидком или твердом — и иметь иную химическую природу (или ту же самую), чем другие фазы. В соответствии с этим принята следующая классификация: системы с газообразной средой называются аэрозолями (дым, пыль, порошок, туман), дисперсная смесь двух нерастворимых жидкостей — это эмульсия, а к системам с
жидкой средой и твердой дисперсной фазой относятся суспензии и собственно коллоидные растворы или золи.
Последние отличаются от суспензий меньшим размером частиц. Весьма разнообразны системы с твердой средой — это подавляющее большинство конструкционных материалов, включая сплавы металлов и природных минералов.
Общей геометрической особенностью и фундаментальным свойством любой дисперсной системы является большая величина удельной (на единицу массы или объема системы) поверхности контакта фаз. Если дисперсная фаза системы состоит из частиц одинакового размера и формы (например, из сферических частиц радиусом г), то она называется монодисперсной, и ее удельная поверхность Ay = s/m может быть легко вычислена по формуле (3.1.1) при известном размере частиц как отношение поверхности 5 = 4лг2 одной частицы к ее массе т = vp, где р — плотность дисперсной фазы и v = 4ттг3/3 — объем одной частицы:
Рис. 3.1. Дисперсная система:
s — частицы дисперсной фазы;/— дисперсионная среда; d — адсорбционный слой
548
Новый справочник химика и технолога
Удельная поверхность единицы объема дисперсной фазы Ао — это отношение поверхности к объему частиц Ао = s / И. По порядку величины и размерности она обратна размеру частиц и называется дисперсностью и описывается формулой
Ао = 3/г.
(3.1.2)
Но эти формулы не находят применения по двум основным причинам: 1) реальные системы полидис-персны и полиморфны, т. е. содержат частицы разного размера и формы, и поэтому величина а не имеет определенного значения; 2) не всякую дисперсную систему можно однозначно разделить на дисперсную фазу, состоящую из частиц, и дисперсионную среду. Примером могут служить пористые материалы с открытыми порами (активированный уголь, губка, пемза и др.) и волокнистые материалы. В них обе фазы непрерывны, т. е. нет отдельных частиц, разделенных прослойками другой фазы. Хотя пористые материалы иногда характеризуются некоторым размером пор, но геометрически он имеет иной смысл, чем размер частиц, — это скорее диаметр каналов неопределенной длины (или волокон для волокнистых материалов). В противоположность этому к пористым материалам с закрытыми порами (их можно назвать твердыми пенами) применимы понятие «размер частицы» и приведенные выше формулы (в той же мере, как и к реальным суспензиям).
Более универсальной геометрической характеристикой дисперсной системы является ее удельная поверхность, а не размер частиц. Удельная поверхность однозначно характеризует любую дисперсную систему независимо от формы и размера частиц и ее топологических особенностей (прерывности или непрерывности фаз, характера их взаимного проникновения).
Удельная поверхность обычно более доступна для экспериментального определения, чем размер частиц. Универсальный метод измерения удельной поверхности основан на явлении адсорбции, при которой происходит налипание на поверхность частиц молекул некоторого вещества, присутствующего в дисперсионной среде (в одной из непрерывных фаз) в качестве одного из компонентов среды. Это может быть растворенное в жидкой среде вещество или сама газообразная фаза, если средой является газ (для единообразия газообразное вещество можно считать «растворенным» в вакууме и характеризовать его концентрацией с, как и «настоящий» раствор). Термин «адсорбция» употребляется для обозначения явления и количества адсорбированного вещества. Адсорбированное на поверхности частицы вещество увеличивает ее размер. На рис. 3.1 слой адсорбированного вещества (адсорбционная оболочка) показан в виде прозрачного ореола вокруг более темного тела самой частицы. Роль оболочки не сводится к изменению основной геометрической характеристики системы — размера частиц. Самым важным свойством таких оболочек является их способность предотвращать слипание частиц при их столкновениях.
Символом «Г» обозначим удельную (на единицу площади) величину адсорбции, выраженную числом киломоль на 1 м2 (кмоль/м2). В связи с этим предпочтения отдаются базовым единицам системы СИ: килограмм, метр, секунда (кг, м, с). Основная химическая единица количества вещества моль приравнена числу граммов в одном моле. По этой причине за химическую единицу количества вещества примем киломоль (кмоль) — количество килограммов, численно равное молярной массе вещества.
В свою очередь это требует соответствующего изменения значений и размерностей химических констант: постоянной Авогадро NA = 6,023 • 1026 1/кмоль, газовой постоянной R = 8310 Дж/(кмоль • К), постоянной Фарадея F - 9,65 • 107Кл/кмоль (кулон/киломоль).
Для измерения удельной поверхности адсорбционным методом экспериментально определяется величина X (кмоль/кг) — удельное (на единицу массы дисперсного материала) количество адсорбированного вещества. Оно связано с адсорбцией Г уравнением
JT=r-Jy, (3.1.3)
которое позволяет легко вычислить удельную поверхность Ау = X / Г. Однако это предполагает, что адсорбция Г либо доступна теоретическому вычислению, либо она предварительно определена экспериментально на материале той же природы, что и исследуемый, но с известной поверхностью. Эта проблема упрощается с помощью модели мономолекулярной адсорбции. Согласно ей молекулы могут налипать на поверхность только в один слой, и его полное заполнение соответствует достижению предельно возможной величины адсорбции Гот (предельной адсорбции) — образованию насыщенного мономолекулярного адсорбционного слоя (рис. 3.2).
Предельная адсорбция выражается через площадь So, занимаемую в насыщенном слое одной молекулой (посадочную площадку молекулы). В случае простой геометрии адсорбированных молекул s0 выражается через их размер. Если считать, что молекула — это сфера радиусом г, то 50 ~ лг2, и тогда справедливы соотношения
r.= 4-"4=W> (3-1.4)
Здесь Хт — предельная адсорбция на единицу массы сорбента.
Рис. 3.2. Насыщенный мономолекулярный слой адсорбированных молекул (светлые кружки) на поверхности частицы
Физика, химия и технология дисперсных систем
549
Условия эксперимента не всегда позволяют довести адсорбцию до ее предельной величины Хт, и поэтому последняя должна находиться экстраполяцией серии измеренных значений адсорбции к предельной величине. Обычно для этого измеряется адсорбция при различных концентрациях адсорбируемого компонента в дисперсионной среде. Чаще всего используется адсорбция газов с простой геометрией молекул (аргон, азот) при различных давлениях газа. В силу малой адсорбционной активности простых молекул опыты приходится проводить при низких температурах (порядка температуры кипения жидкого азота). Можно использовать адсорбцию более активных веществ (пары воды, спирта, растворенных веществ), но тогда снижается достоверность вычисленного значения посадочной площадки молекулы сорбированного вещества (сорбата), а главное — теряется уверенность в независимости этой величины от химической природы адсорбента (материала, на поверхности которого адсорбируются химически активные молекулы).
Зависимость адсорбции от концентрации сорбата называется изотермой адсорбции (при постоянстве температуры). Эксперимент дает некоторый участок этой изотермы. Априори можно считать, что адсорбция должна увеличиваться и стремиться к пределу при увеличении концентрации сорбата, поэтому для нахождения предельной адсорбции необходимо экстраполировать изотерму к бесконечно большой концентрации. Корректно эту процедуру можно выполнить только при наличии уравнения изотермы адсорбции — аналитического описания зависимости адсорбции от концентрации адсорбирующегося вещества, причем уравнение должно включать в себя предельную величину адсорбции в качестве одного из параметров. Существует ряд подходящих уравнений и отработанных алгоритмов их применения для адсорбционного определения удельной поверхности [39]. Но изучение адсорбции не сводится только к измерению удельной поверхности дисперсных и пористых материалов, а имеет прикладное значение. Адсорбция лежит в основе улавливания и концентрирования редких элементов, очистки газов и жидкостей от нежелательных примесей. Адсорбенты предназначены для поглощения (адсорбции) различных веществ из растворов или газов. Адсорбция также является частью термодинамического цикла холодильных установок. Существует отдельная индустрия по производству как универсальных, так и специализированных адсорбентов.
3.L2. Поверхностный слой
В понятие поверхности, или границы раздела фаз, прежде всего вкладывается геометрический смысл, т. е. нечто нематериальное, что разграничивает две соприкасающиеся материальные фазы. В математическом смысле это — двухмерный объект, не имеющий третьего измерения, т. е. толщины, массы и т. п. При таком
понимании поверхность характеризуется величиной площади и кривизной (если отбросить несущественные в данном случае характеристики ее положения в пространстве). Именно такое понимание поверхности лежит в основе наших представлений о размере, форме, пространственном положении тел. Оно необходимо и не может быть заменено никакими другими представлениями о границе фаз, но сильно идеализирует и упрощает представление о той части гетерогенной системы, которая лежит на границе между двумя соприкасающимися фазами. Гетерогенные системы, которые описываются с помощью такого упрощенного представления о границе раздела фаз, являются гипотетическими, т. е. в действительности не существуют. Реальная граница фаз устроена сложнее, прежде всего с геометрической точки зрения. Дело в том, что реальная поверхность практически никогда не бывает абсолютно гладкой: одни атомы и молекулы возвышаются над некоторой плоскостью (разделяющей поверхностью), которую условно можно принять за границу раздела фаз, другие атомы или молекулы отсутствуют, т. е. имеются углубления на поверхности вещества (рис. 3.3). Иначе говоря, на молекулярном уровне поверхность шероховата, и, следовательно, не существует никакой единственной и однозначно выбираемой границы между фазами. Реально они разделены не математической плоскостью, а слоем, в котором в переменном соотношении присутствуют молекулы обеих соприкасающихся фаз [26].
Толщина поверхностного слоя неопределенная, несколько молекулярных диаметров, но в некоторых случаях она может достигать гигантских величин, например двойной электрический слой.
Рассмотрим гипотетическую двухфазную систему с заданным общим объемом V, занимающую сосуд такого же объема. Плоская граница делит систему на две фазы: 7 и 2, объемы которых Ии К2 в сумме равны объему всей системы V. Координата х направлена по нормали к этой плоскости, причем площадь сечения сосуда А задана вдоль оси сосуда х и неизменна. Состав каждой из фаз также известен, т. е. заданы концентрации су и с2, каждого /-го компонента в обеих фазах. Задача состоит в том, чтобы вычислить количество п, i-ro компонента в системе.
Рис. 3.3. Поверхностный слой между фазами:
7 — вакуум; 2 — кристаллическое вещество; светлые ячейки — незанятые узлы кристаллической решетки; хо — разделяющая поверхность;
S — толщина поверхностного слоя
550
Новый справочник химика и технолога
Пусть 0, %о и В — координаты на оси х нижней границы сосуда (дна), плоской межфазной границы и верхней границы сосуда соответственно (рис. 3.4), тогда V=А(В - 0) Vi = А(х0 - 0) и У2 = А(В - х0). Если считать, что составы каждой из фаз неизменны вплоть до межфазной границы х0, т. е. концентрации с, каждого компонента скачком изменяются при переходе через границу, то количество z-ro компонента в гипотетической системе вычисляется по формуле (3.1.5) как сумма его содержания в первой (сцИ) и второй (с2(И2) фазах:
и/г = А [си (х0 - 0) + с2, (В - х0)]. (3.1.5)
В действительности, как уже отмечалось, плоскости, однозначно делящей гетерогенную систему на две фазы, не существует, и плоскость х0 может быть проведена произвольно. Следовательно, общий объем системы У не может быть однозначно разделен на две части И и У2, известно лишь, что их сумма равна общему объему. В реальной системе фазы разделены слоем переменного состава, в котором концентрации всех компонентов меняются постепенно, каждый компонент по своему закону с, = с,(х), поэтому проделанные вычисления относятся не к реальной, а к некоторой гипотетической гетерогенной системе. Для вычисления количества /-го компонента в реальной системе сумму (3.1.5) необходимо заменить интегрированием по координате х концентрации /-го компонента CiAdx в тонком слое толщиной dx, параллельном гипотетической межфазной границе:
в
Рис. 3.4. Реальное и гипотетическое распределение компонента в двухфазной системе:
кривая с(х) — реальное, а, с2— гипотетическое распределение компонента в системе (/ — газ, 2 — жидкость);
ct и сг — концентрации компонента в этих фазах соответственно; области серого цвета М и N2 — поверхностные избытки компонента
в тех же фазах, Xi и Х2 — границы поверхностного слоя;
Хо — координата разделяющей поверхности;
3 — толщина поверхностного слоя;
отрезок 0S — протяженность системы вдоль оси х, нормальной к границе раздела фаз
Если распределение некоторого компонента в системе представить графически (рис. 3.4) как функцию координаты х, то объемы У\, У2и У будут соответствовать длине отрезков 0х0, х0В и 0В. Геометрическим образом распределения компонента в гипотетической системе являются площади прямоугольников с основаниями по линиям Ci и с2, а в реальной системе — площадь фигуры под кривой с,(х). Эти образы наглядно демонстрируют, что суммарная площадь двух прямоугольников и площадь под кривой могут не совпадать по величине и что количество компонента в гипотетической системе (площадь двух прямоугольников) зависит от положения межфазной границы хо и поэтому не может быть объективной величиной, в то время как в реальной системе количество компонента является объективным параметром системы, не зависящим от выбора положения границы. Несмотря на произвольность одной величины и объективность другой, нельзя отдать предпочтение только последней, так как невозможно отказаться от понятия гипотетической межфазной поверхности с нулевой толщиной.
Согласование разных методов описания гетерогенных систем достигается путем введения поверхностных избытков — величин, представляющих собой разность количеств /-го компонента в реальной и гипотетической системах. В частности, поверхностный избыток /-го компонента N, = п^- п1Т. Обычно интерес представляет удельная (на единицу площади межфазных границ) величина избытка. Поверхностный избыток компонента, приходящийся на единицу площади межфазной границы, называется адсорбцией данного компонента. Графическим образом избытка (и адсорбции) на рис. 3.4 является разница площадей под кривой с((х) и под прямоугольниками (область серого цвета). Поверхностный избыток компонента, приходящийся на единицу площади межфазных границ, называется адсорбцией по Гиббсу, или гиббсовским избытком компонента.
Адсорбция как поверхностный избыток и количество вещества в гипотетической системе являются произвольными величинами, зависящими от положения межфазной границы хо. В теории поверхностных явлений гипотетическую межфазную границу принято называть разделяющей поверхностью, что подчеркивает различие между реальной поверхностью раздела двух фаз и гипотетической (плоской), положение которой в пределах поверхностного слоя не определено и может выбираться произвольно. Необходимо, однако, помнить, что соседние фазы в реальности разделены поверхностным слоем, а не плоской границей.
Общепринятая терминология не всегда удачна. Следует избегать применения термина «граница (или поверхность) раздела двух фаз» в силу его многословности и избыточности. Граница (в смысле поверхность) может возникнуть только между двумя фазами (три фазы могут иметь только общую линию соприкосновения, а четыре — только общую точку), поэтому указание на причастность именно двух фаз к созданию гра
Физика, химия и технология дисперсных систем
551
ницы явно излишне. Понятие «граница раздела фаз» абсолютно эквивалентно терминам «граница фаз» или «межфазная граница», которым и будет отдаваться предпочтение.
Понятие адсорбции многогранно. Говорят, например, что адсорбция компонента — это изменение его концентрации в поверхностном слое по сравнению с концентрацией в объеме. При этом может иметься в виду изменение во времени, т. е. процесс установления равновесной концентрации компонента в объемной фазе (растворе или газе), и, следовательно, за эталон сравнения принимается система, состояние которой первоначально было неравновесным, система с равномерным распределением компонентов в пространстве вплоть до разделяющей поверхности или гипотетическая система. На практике за эталонное (исходное) состояние часто принимается состояние объемной фазы до ее соприкосновения с адсорбентом — твердым веществом с большой удельной поверхностью. Под изменением концентрации можно понимать и разницу концентраций компонента в поверхностном слое и в объеме фазы. По размерности эта величина (кмоль/м3) не соответствует адсорбции (кмоль/м2), но наглядно отражает суть адсорбции как величины, характеризующей гетерогенную систему, и поэтому полезна при качественном обсуждении явления. В технической литературе термином «сорбат» («адсорбат») обозначают ту часть компонента, которая перешла в адсорбированное состояние, а термином «сорбтив» — компонент, участвующий в процессе адсорбции (десорбции). Адсорбция может быть обратимой, т. е. сорбат при изменении условий может десорбироваться — переходить из поверхностного слоя в объемную фазу без каких-либо остаточных явлений.
Адсорбция как явление и как физико-химический параметр гетерогенной системы не имеет практического смысла в однокомпонентной двухфазной системе, например в системе вода—водяной пар, хотя формально определение адсорбции как поверхностного избытка полностью сохраняет смысл и в этом случае. Так как величина избытка зависит от положения разделяющей поверхности, которое может быть произвольным, то нулевое значение адсорбции в однокомпонентной системе обеспечивается надлежащим выбором положения разделяющей поверхности. Разделяющая поверхность, выбранная указанным способом, называется эквимолекулярной. Таким образом, признание того, что адсорбция в однокомпонентной системе равна нулю, равносильно закреплению разделяющей поверхности в определенном — эквимолекулярном — положении. Это можно сделать и в многокомпонентной системе, но всегда только по отношению к одному, хотя и любому компоненту системы. Разделяющая поверхность, расположенная таким образом, что избыток (адсорбция) какого-либо компонента оказывается равным нулю, называется эквимолекулярной по отношению к упомянутому компоненту. По отношению к другим компо
нентам системы эта поверхность, скорее всего, не будет эквимолекулярной, и невозможно выбрать такое положение разделяющей поверхности, чтобы она стала эквимолекулярной по отношению ко всем или хотя бы к двум компонентам многокомпонентной гетерогенной системы. Это обстоятельство используется для того, чтобы придать определенность величине адсорбции, которая остается неопределенной до тех пор, пока не будет достигнута какая-либо договоренность относительно положения разделяющей поверхности. Следуя Гиббсу, в качестве разделяющей поверхности, относительно которой вычисляются поверхностные избытки, т. е. адсорбции компонентов системы, принята поверхность, являющаяся эквимолекулярной по отношению к растворителю. Соответствующие этому выбору избытки и адсорбции называются относительными [26]. При большом числе компонентов в системе (растворе) следует указывать, относительно какого компонента вычисляются избытки. При небольшом числе компонентов в системе (растворе) нет надобности делать это, поскольку указание значений избытков всех компонентов кроме одного из них означает, что это избытки относительно последнего. Если перечисляются избытки всех компонентов, то это означает, что они указываются относительно некоторого произвольного положения разделяющей поверхности, которая не является эквимолекулярной ни для одного из компонентов системы.
В общем распределения компонентов с,(х) около границы неизвестны, и поэтому прямые вычисления их избытков и адсорбций по Гиббсу невозможны. Фактически приведенные выше соотношения представляют интерес только как определения понятий «поверхностный избыток» и «адсорбция» по Гиббсу и ряда других избыточных экстенсивных (зависящих от размера системы) функций гетерогенной системы. К числу таковых, наравне с адсорбцией, относятся избыток энергии Гиббса и избыток энтропии. Прикладное значение величин гиббсовской адсорбции и других избытков состоит в том, что они могут быть связаны между собой и с экспериментально определяемыми величинами с помощью термодинамических уравнений, и, таким образом, искомая термодинамическая функция, например изотерма адсорбции, может быть выражена через другую, доступную экспериментальному определению термодинамическую функцию, например изотерму поверхностного натяжения.
3.1.3. Поверхностное натяжение
Поверхностное натяжение — это работа образования единицы поверхности в обратимом изотермическом процессе. Более привычно определение поверхностного натяжения как силы, действующей вдоль поверхности и направленной на ее сокращение. Эти два определения эквивалентны, так как работа образования единицы площади в точности равна касательной к поверхности силе, действующей на единицу длины любого контура, выделяющего часть поверхности. Натяжение поверхно
552
Новый справочник химика и технолога
сти проявляет себя в стремлении капли жидкости принять сферическую форму, поскольку она имеет минимальную площадь при фиксированном объеме капли. Другое хорошо известное проявление натяжения — поднятие жидкости (воды) в стеклянном капилляре или ее впитывание в пористый материал, соприкасающийся с жидкостью.
В качестве более значимого, чем натяжение, свойства вещества можно рассмотреть силу или энергию взаимодействия молекул этого вещества между собой (силу Ван-дер-Ваальса) и попытаться представить натяжение как результат взаимодействия молекул.
Рассмотрим механизм возникновения натяжения на примере границы между жидкостью и газом (рис. 3.5). Асимметрия окружения молекул жидкой фазы, расположенных на межфазной границе, приводит к тому, что на них действует отличная от нуля суммарная сила ван-дер-ваальсова притяжения. Эта сила, отнесенная к единице площади межфазной границы, называется внутренним давлением жидкости. Оно измеряется сотнями и тысячами атмосфер (107-108Па).
Внутреннее давление является составляющей в уравнениях состояния реальных газов, например в уравнении Ван-дер-Ваальса:
(P + Pin)(V-Vn) = RT,
где Р — давление, Pin — внутреннее давление, V — объем газа, Vn — молярный объем вещества в конденсированном состоянии, R — газовая постоянная, Т — температура.
Поверхностное натяжение о определяется как работа W внешних сил над системой, необходимая для увеличения площади А поверхности межфазной границы на единицу в обратимом изотермическом процессе, т. е. о = W / АА. Увеличение площади границы фаз требует перемещения этих молекул из объемной фазы в поверхностный слой, что совершается против силы их втягивания Fm в глубь жидкости, на что и тратится энергия внешних сил.
Внутреннее давление в жидкости Рт нарастает в поверхностном слое по мере продвижения из одной фазы (газа) в другую (жидкость), так как оно создается в слое конечной толщины 8 (рис. 3.5). Это означает, что в поверхностном слое существует градиент внутреннего давления dPin/ dx.
Рис. 3.5. Силы, действующие на молекулу возле границы фаз:
1 — газ; 2 — жидкость; Fin — суммарная сила взаимодействия молекулы с жидкой фазой 2
Из условия механического равновесия элементарного объема dV жидкости внутри слоя 8 легко установить, что dPin / dx = nFim где п — локальная концентрация молекул, в том числе в слое 8 и за его пределами, F(n — сила, действующая на одну молекулу со стороны всех остальных молекул гетерогенной системы, nFm — объемная плотность межмолекулярных сил в поверхностном слое, обусловленная расстоянием х от некоторой поверхности, принимаемой за разделяющую межфазную поверхность.
Важным качеством рассмотренного метода доказательства существования натяжения поверхности, который может быть назван молекулярно-кинетическим, является физическая наглядность, возможность объяснения адсорбции как следствия градиента внутреннего давления в поверхностном слое. Недостаток метода заключается в том, что он оперирует формальным параметром — внутренним давлением, которое не может быть измерено каким-либо прибором.
Предварительно необходимо вычислить работу деформирования dW, совершаемую внешними силами над плоским слоем в виде прямоугольного параллелепипеда с большой гранью площадью А, толщиной X и длиной двух больших ребер L (рис. 3.6), так что А = А2, а объем слоя V = ХА. Допустим, что внешние силы, действующие на параллелепипед, заданы давлениями Рп и Ph действующими на его грани, причем давление Рп, нормальное по отношению к большим граням площадью А, не равно тангенциальному давлению Pt, действующему на четыре узкие грани площадью XL каждая.
Согласно фундаментальному определению работы dW ~Y.FI ds„ где Ft — силы, s, — перемещения в направлении действия этих сил. Перемещение граней будем отсчитывать от одной из вершин параллелепипеда (точка 0 на рис. 3.6), так что для верхней грани ds = -dX, а для правой и лицевой граней ds = -dL.
Знак «-» указывает, что действующие на образец внешние силы уменьшают длины ребер X и L. Сопряженные с этими перемещениями силы равны РпА и P(XL соответственно, поэтому:
dW--PnAdX-2PtXLdL или dW =-PnAdX-PtXdA,
Рис. 3.6. Перемещение граней:
X— толщина слоя вещества (в виде прямоугольного параллелепипеда); L — длина ребер; Рп и Pt— давления, действующие на слой вещества
Физика, химия и технология дисперсных систем
553
Если прибавить к полученному выражению величину PnXdA и вычесть ее, а также учесть, что
-PnAdX- PnXdA = -Рп dV, то работу деформации слоя вещества толщиной dX можно описать уравнением
dW=-Р„ dV + (Рп - Р,) XdA. (3.1.7)
Таким образом, в общем случае работа деформирования включает работу сжатия -Рп dV, которая может быть совершена, если деформирующие силы вызывают изменение объема слоя V, и работу изменения поверхности (Рп - Pt)XdA, которая совершается при условии, когда (Рп - Pt) не равно нулю. В частном случае несжимаемой среды (жидкости) dV = 0, и тогда работа деформирования тонкого слоя (dW = (Рп - Pt) XdA) — это только работа по изменению площади А слоя вещества.
Неравенство Рп и Р, означает нарушение закона Паскаля. По этому закону величина давления не зависит от ориентации площади, на которую оно действует. В объемной изотропной среде (обычные жидкости и газы) этот закон, как известно, выполняется. В поверхностном слое он нарушается из-за анизотропности его структуры (рис. 3.3). Действительно, в направлении нормали к межфазной границе каждая молекула имеет контакт с соседними молекулами, и поэтому может воспринимать и передавать механические воздействия в этом направлении. В тангенциальных направлениях многие молекулы не имеют контактов с соседями и потому лишены возможности передавать механические импульсы вдоль этих направлений, что и приводит к нарушению закона Паскаля.
Поверхностный слой между двумя фазами может быть представлен как набор тонких слоев вещества (рис. 3.7), растяжение или сжатие которых сопровождается работой dW = (Рп - Pt)dxdA, где dx — толщина слоя. Работа по изменению величины межфазной поверхности А будет в этом случае равна сумме работ dW по всем тонким слоям dx поверхностного слоя или:
W = d4j(Pn-Pl)dx.
Приращение площади dA слоя здесь вынесено за знак интеграла, так как это не меняющаяся от слоя к слою величина. Интегрирование в бесконечных пределах вместо конечных не влияет на результат интегрирования, так как слои, расположенные вне поверхностного слоя толщиной 5, дают нулевой вклад в работу W, поскольку здесь Рп = Pt. Если поместить начало координат в середину межфазного слоя, то можно указать и конечные пределы интегрирования: от -5/2 до +5/2.
По определению о = W/dA, поэтому предыдущий результат можно записать в виде уравнения
+8/2
а= -/;)<*. (3.1.8)
-8/2
Это выражение известно как формула Баккера. Величина (Рп - P^dx имеет смысл натяжения dcsL слоя толщиной <&, а (Рп - Pt) — локального удельного натяжения csL, т. е. натяжения тонкого слоя вещества, отнесенного к его толщине dx, так что (Рп - Pt) ~ и, следовательно, выражение (3.1.8) может быть записано в виде уравнения:
о= JdaL . (3.1.9)
Формулы (3.1.8) и (3.1.9) определяют аддитивность натяжения отдельных частей межфазного слоя вещества как прием в описании (вычислении) межфазного натяжения. Но это не означает физической аддитивности— независимости локального натяжения некоторой тонкой части межфазного слоя от наличия, состава, натяжения других частей этого слоя.
Зависимость локального натяжения (разницы давлений Рп - Pt) от нормальной к поверхности координаты х может быть произвольной, однако она должна удовлетворять следующим требованиям:
1) о > 0, т. е. некоторая часть слоев (рис. 3.7) может давать отрицательную величину вклада в общее натяжение, если (Рп - Pt) < 0, но вклад (Рп - Pt) остальных слоев поверхностной фазы в общее натяжение должен быть положительным и большим;
2) за границами слоя Рп = Р,\
3) по условиям механического равновесия каждого слоя dPn Idy = 0, т. е. Рп не зависит от нормальной координаты х.
Условие (3) вместе с условием (2) означает, что в случае плоской границы раздела фаз Рп = Pf = Pv~ Р. Здесь Pj Vi.Pv — давления в соприкасающихся фазах «/» (жидкой) и «V» (газообразной или второй жидкой фазой, выступающей в качестве среды), равные давлению Р в гетерогенной системе. Отсюда следует, что различие нормальной и тангенциальной составляющих давления в поверхностном слое может возникать только благодаря отличию тангенциальной компоненты давления от ее значения в объемных фазах. Этот вывод важен для правильной трактовки механизма действия ПАВ. Существенное отличие рассмотренной трактовки поверхностного натяжения от молекулярно-кинетического состоит в том, что в этой макроскопической трактовке фигурируют термодинамические (манометрические) давления, которые поддаются прямым измерениям.
Рис. 3.7. Поверхностный слой толщиной 5 как совокупность тонких анизотропных слоев толщиной dy (ось у нормальна к межфазной границе)
554 Новый справочник химика и технолога
В частности, интегральная (по всему поверхностному слою) величина Pf= Pvn есть измеряемая в опыте сила, равная поверхностному натяжению о, а Рп может быть измерено по давлению Р в любой из фаз системы.
Плоская граница раздела фаз не может существовать в отсутствие внешних сил, обеспечивающих натяжение поверхности с силой, равной силе поверхностного натяжения. Отсюда следует еще одно определение поверхностного натяжения как внешней силы, действующей на единицу длины вдоль межфазной границы фаз и обеспечивающей ее равновесие. Поверхностное натяжение создается в слое толщиной 5, разделяющем объемные фазы, однако в духе метода поверхностных избытков натяжение а можно приписать не поверхностному слою толщиной 8, а некоторой поверхности нулевой толщины, которая называется поверхностью натяжения. В случае плоской границы координаты этой поверхности на оси х не имеют значения; в частности, можно считать, что поверхность натяжения совпадает с эквимолекулярной разделяющей поверхностью какого-либо компонента, например растворителя.
Поверхностное натяжение границы, разделяющей жидкую и газообразную фазы или две разные жидкости, проявляет себя как сила, которая стягивает (сокращает) поверхность, и поэтому может быть измерена. Натяжение поверхности твердого тела не может быть измерено, но, тем не менее, и в этом случае оно является фундаментальной характеристикой межфазной границы, поэтому одной из задач является нахождение соотношений, позволяющих вычислить натяжение границы твердого вещества с каким-либо другим, в том числе и твердым, веществом.
3.1.4. Адсорбция газов на поверхности твердых тел
Адсорбция газов играет самостоятельную и очень важную роль в технологии как процесс улавливания и концентрирования различных летучих веществ, как процесс очистки газов от вредных примесей, как основа ряда аналитических методов (хроматография, измерение удельной поверхности). Несмотря на многочисленные и разнообразные, в том числе прикладные, аспекты адсорбции газов [2], нас это явление и разные подходы к его описанию будут интересовать лишь в той мере, в какой это позволяет понять и описать фундаментальные свойства поверхности, главным образом, твердых веществ. В этом отношении непреходящую ценность имеет теория мономолекулярной адсорбции Ленгмюра.
В теории Ленгмюра адсорбционные силы по природе близки к химическим, для которых характерна их насыщаемость: молекула, образовавшая связь с другой молекулой или с поверхностью твердого вещества (адсорбента), теряет способность к дальнейшему взаимодействию. Это относится и к адсорбенту — он не может удерживать на своей поверхности более одного слоя молекул, т. е. адсорбция будет мономолекулярной. Другая типичная черта химического взаимодействия — избирательность: во взаимодействие при определенных
условиях могут вступать только атомы или молекулы определенного вида. Адсорбционным силам, в отличие от химических, избирательность не присуща. Иначе говоря, это силы универсальные, и любой газ может адсорбироваться на поверхности любого твердого вещества, хотя различие в их сродстве существенно влияет на константы уравнения адсорбции. В некоторых случаях не исключается и избирательное (химическое) взаимодействие компонентов среды с поверхностью адсорбента.
Адсорбция происходит благодаря столкновениям молекул газа с поверхностью, точнее — с ее незанятой частью. Если при определенных условиях адсорбированными молекулами занята 0-я доля поверхности, то доля свободной поверхности будет равна (1-0). Из молекулярно-кинетической теории газов известно, что число столкновений I молекул газа за единицу времени, приходящееся на единицу площади, пропорционально давлению газа Р и описывается уравнением
I = PNAi(2nMRT)m. (3.1.10)
В идеальном газе Р = cRT, и тогда эту величину можно выразить уравнением (3.1.10а) через концентрацию газа с:
I=cNA(RT/2nM)l/2. (3.1.10а)
Здесь М— молярная масса сорбата, Т — температура.
Произведение /(1-0) представляет собой число попаданий молекул на свободную часть поверхности за единицу времени и отождествляется со скоростью адсорбции газа <?а (обычно считается, что каждое столкновение молекулы со свободной поверхностью ведет к адсорбции). Целесообразно представить скорость адсорбции в форме уравнения:
^а = Л.с(1-0). (3.1.11)
Здесь ka — коэффициент при концентрации в формуле (3.1.10а) или при давлении в формуле (3.1.10), если скорость выразить через давление. В том и другом случае этот коэффициент имеет значение константы скорости адсорбции.
Каждая адсорбированная молекула с некоторой вероятностью кД может отрываться от поверхности адсорбента, т. е. десорбироваться. Скорость десорбции с единицы площади адсорбента пропорциональна количеству находящихся на поверхности молекул:
<7д — кдГНА.
При данной степени заполнения поверхности 0-адсорбция составляет 0-ю долю от предельной адсорбции, наступающей при 0=1, поэтому Г = 0Гт, и тогда скорость десорбции описывается формулой
qa = karmNA®. (3.1.12)
В состоянии адсорбционного равновесия скорости адсорбции и десорбции равны, поэтому, приравняв
Физика, химия и технология дисперсных систем
555
правые части этих скоростей, найдем равновесную степень заполнения поверхности при данной концентрации (давлении) газа:
0 = Кс / (Кс + KNArm).
После объединения всех констант в одну константу адсорбционного равновесия (3.1.13) степень заполнения поверхности сорбатом определяется уравнением (3.1.14):
K=K/Wm, (3.1.13)
® = Кс/(1 + Кс). (3.1.14)
Используя связь степени заполнения с адсорбцией, получим искомое уравнение изотермы мономолекуляр-ной адсорбции газа — уравнение Ленгмюра:
Г = Г^с/(1 + Кс). (3.1.15)
Графически эта изотерма представлена на рис. 3.8. Ее вид (уравнение (3.1.16)) не меняется при переходе к адсорбции, отнесенной к единице массы адсорбента: Хт = ЛуГт, где Ау — удельная поверхность адсорбента:
X=Х„Кс / (1 + Кс). (3.1.16)
Это же уравнение в форме линейной функции концентрации (3.1.16а) используется для экстраполяции адсорбции к ее предельной величине (рис. 3.9):
с/Х=\/КХт+с/Хт. (3.1.16а)
Предельная адсорбция Хт определяется из этого уравнения как величина, обратная угловому коэффициенту уравнения прямой. Из экспериментальных данных также может быть вычислена величина Гт, если адсорбция отнесена к единице площади поверхности сорбента.
Найденное указанным способом значение предельной адсорбции (емкости монослоя) используется не только для вычисления удельной поверхности адсорбентов, но и для проверки самой теории адсорбции, поскольку для молекул простой формы величина Гт вычисляется независимо по формуле:
Предельные адсорбции, вычисленные по изотерме и по величине посадочной площадки молекулы, часто не совпадают: теоретическое значение Гт= l/A^^o обычно больше, чем экспериментальное. Следовательно, не вся поверхность способна участвовать в адсорбции, а только некоторые ее участки, получившие название «активных центров» поверхности твердого вещества. Открытие активных центров является одним из важнейших достижений теории мономолекулярной адсорбции Ленгмюра. Понятие активных центров играет фундаментальную роль в физике твердого тела и в современной физико-химии поверхности.
Тот факт, что адсорбция протекает не на всей поверхности, а только на ее активных центрах (дефектах кристаллической решетки), никак не влияет на вид уравнения адсорбции, но определяет его константы. Величина предельной адсорбции связана с числом центров Nc на единице площади сорбента соотношением (рис. 3.10, а):
(3.1.18)
rm = Nc/NA.
Рис. 3.8. Зависимость адсорбции газа: Г или X — адсорбции газа; Р — давление; Хт и Гт— предельные значения адсорбции
Рис. 3.9. Линейная форма уравнения изотермы мономолекулярной адсорбции: светлые кружки — экспериментальные данные; сплошная прямая — аппроксимация экспериментальных данных; предельная адсорбция Гт = AD / BD, константа равновесия К = 1/(0А • Гт)
OQQOQQQOQQQQQQQ
Рис. 3.10. Предельная адсорбция молекул малого (а) и большого (б) размера на активных центрах поверхности. Активные центры обозначены вертикальными штрихами
556
Новый справочник химика и технолога
Выбор между формулами (3.1.17) и (3.1.18) при оценке емкости монослоя диктуется следующим соображением: применять следует ту формулу, которая дает меньшее значение Гт. ПриNclNA> 11s§NA вся поверхность участвует в адсорбции, так как размер молекул слишком велик и центры расположены слишком плотно, чтобы каждый из них мог присоединить отдельную молекулу (рис. 3.10, б).
При адсорбции на центрах параметр ® имеет смысл доли занятых центров. Вычисление скорости адсорбции должно учитывать лишь попадания молекул газа на свободные центры, которым, следовательно, необходимо приписать некоторую площадь захвата sa. Тогда скорость адсорбции описывается уравнением (3.1.19), в котором произведение N<sa представляет собой площадь активной поверхности на единице площади сорбента:
^а = Л.с(1-в)^а. (3.1.19)
Соответственно этому изменится выражение для константы (3.1.13) адсорбционного равновесия: К = kj^^l kJ^AVm. Поскольку при адсорбции на центрах Гт = Nc / NA, то, в отличие от уравнения (3.1.13), константа адсорбционного равновесия в уравнении (3.1.20) не включает в себя предельной адсорбции, но содержит площадь центра захвата sa:
£ = тД (3.1.20)
КД
При sa = s0 обе формулы имеют одинаковое решение (3.1.17) s0 = 1/ЛлГт- Таким образом, способы вычисления констант скорости адсорбции несколько отличаются при адсорбции на центрах и на всей поверхности.
Вопреки классической теории молекулярной физики, необходимо различать просто столкновение молекулы с поверхностью (или центром) и акт адсорбции. Выше было подчеркнуто, что классическая теория мо-номолекулярной адсорбции эти два события не различает. Первое из них для краткости будем называть упругим столкновением, а второе — адсорбцией. Количественное различие этих понятий можно определить на основе описания поведения адсорбированной молекулы на поверхности адсорбента. Очевидно, что она находится в состоянии колебательного теплового движения, в том числе такие колебания совершаются в направлении нормали к поверхности адсорбента. Средняя кинетическая энергия тепловых колебаний равна, как известно, RT72Na на каждую степень свободы. Приближенно указанные колебания можно считать гармоническими с некоторой частотой f Фаза колебательного движения молекулы, в которой оно направлено от поверхности сорбента, может рассматриваться как попытка молекулы разорвать адсорбционную связь и десорбироваться. Вероятность разрыва w связи за одну попытку (т. е. за один цикл колебаний) известным образом выражается через энергию этой связи W (работу адсорбции):
w = exp(~W/RT),
а вероятность десорбции одной молекулы за единицу времени, т. е. константа скорости десорбции ка, будет в f раз больше:
ka=fexp(-W/RT). (3.1.21)
С этих позиций упругое столкновение — это такое взаимодействие молекулы с поверхностью, при котором она десорбируется с первой попытки, т. е. ее единственный цикл колебаний будет состоять из фазы приближения молекулы к поверхности, которая затем сменится удалением от поверхности за пределы радиуса действия адсорбционных сил. Следовательно, вероятность упругого отскока молекулы от поверхности совпадает с вероятностью ее отрыва за один цикл колебаний:
w = exp (-W/ RT). (3.1.22)
Тогда по правилу сложения вероятностей двух взаимоисключающих событий вероятность адсорбции будет равна (1 - w), и формулу (3.1.12) следует заменить уравнением
^ = ЛДГИ№®(1-^). (3.1.23)
При этом вместо уравнения (3.1.10) с учетом формул (3.1.17) и (3.1.18) получим:
K=ka(\-w)/fivNArm
или
К = (ka/fNATm)[exp (WIRT) - 1].
Необходимо также помнить, что, согласно формуле (3.1.11), ka-NA (RTI 2лЛ/)1/2, а частота колебаний может быть оценена по формуле f = (2RT / Л/)1/2 / га, где га— радиус действия адсорбционных сил, который, вероятно, близок к радиусу молекулы. При объединении всех физико-химических констант с точностью до числового коэффициента порядка единицы получается следующее выражение: К - га[ехр(И// RT) - 1] / Гт. Поскольку величина Гт / га имеет порядок молярного объема сорбата Vm, то для константы адсорбционного равновесия можно записать уравнение [13]:
К = Vm[exp(W/RT)- 1]. (3.1.24)
Эта формула корректно описывает предельный случай адсорбции, когда W = 0.
В соответствии с физическим смыслом этого явления, адсорбция отсутствует при нулевом сродстве адсорбата к поверхности адсорбента. Взаимодействие молекул с поверхностью адсорбента сводится в этом случае к упругим столкновениям. Классическая теория мономолекулярной адсорбции включает и упругие столкновения в число актов адсорбции, что дает неправильное описание явления. На практике формулы типа (3.1.24) в классическом виде (без вычитания 1 из экспоненциального сомножителя) применяются для вычис
Физика, химия и технология дисперсных систем
557
ления работы адсорбции по экспериментально найденной константе адсорбционного равновесия К. Такие расчеты содержат, следовательно, ошибку, и тем большую, чем слабее сродство адсорбата к адсорбенту.
Следует предостеречь еще от одной распространенной ошибки, встречающейся при интерпретации теории Ленгмюра. Обычно она возникает при попытке теоретически вычислить константу адсорбционного равновесия на основе предположения, что количество молекул в адсорбционном слое Ленгмюра можно найти путем умножения концентрации молекул возле поверхности на толщину мономолекулярного слоя (или интегрированием концентрации по толщине, что не меняет сути дела). Ошибка здесь в том, что адсорбционный слой Ленгмюра как операнд математических выражений не имеет толщины: это двухмерный объект — геометрическое место точек, представляющих центры закрепленных на поверхности молекул. Расстояние между этими центрами в силу конечного и определенного размера молекул сорбата равно некоторому эффективному радиусу молекулы (длина адсорбционной связи) — они могут располагаться только в одной плоскости с нулевой толщиной. Монослой имеет физическую толщину, равную размеру адсорбированных молекул, но это не значит, что число молекул в этом слое можно вычислять умножением концентрации молекул возле поверхности на физическую толщину слоя, что означало бы, что центры адсорбированных молекул могут находиться на любом расстоянии от поверхности, в том числе равном нулю. Следовательно, молекулам приписывается неопределенный размер. Это является чисто математической стороной ошибки. Кроме того, упомянутый выше способ вычисления количества адсорбированного вещества неявным образом подменяет адсорбцию по Ленгмюру на адсорбцию по Гиббсу (поверхностный избыток).
Изотерма мономолекулярной адсорбции в графическом представлении имеет узнаваемый вид (рис. 3.9): с увеличением концентрации сорбента адсорбция сначала растет пропорционально концентрации вещества в растворе, а затем, при больших его концентрациях, сменяется асимптотическим приближением адсорбции к ее предельной величине Гт. Приближенное значение Гто можно определить из графика, однако точное вычисление предельной адсорбции требует представления изотермы в форме уравнения прямой (3.1.16а).
Экспериментально полученная изотерма адсорбции может заметно отличаться от рассчитанной теоретически по уравнению (3.1.16) или (3.1.15) даже при самом благоприятном подборе констант Гто и К. Одна из причин этого — наличие активных центров разной силы (неоднородность поверхности по величине работы адсорбции W). Таким образом, более полной характеристикой поверхности является функция распределения центров по их силе или по иным признакам, например по их кислотности (метод РЦА — распределение центров активности).
В теории полимолекулярной адсорбции газов (известной также под названием «потенциальная теория») изначально заложено предположение о возможности адсорбции одинаковых молекул с разной силой. Это происходит, если поверхность способна удерживать слой адсорбата толщиной в несколько молекулярных диаметров (несколько монослоев). Графики экспериментальных изотерм адсорбции могут быть при этом довольно разнообразны. Классический же вид графика (рис. 3.11) первоначально (при относительно малых давлениях газа) подобен изотерме мономолекулярной адсорбции с тенденцией к насыщению мономолекулярного адсорбционного слоя, которая затем сменяется прогрессивным увеличением адсорбции с последующим ростом давления газа.
Превышение количества адсорбированного вещества той величины, которая соответствует емкости монослоя, свидетельствует о полимолекулярном характере адсорбции. В таком случае ближайший к поверхности слой сорбата имеет наибольшую энергию адсорбции, а последующие слои — меньшую. Следовательно, емкость адсорбента определяется не только его удельной поверхностью Ау, но и радиусом действия га адсорбционных сил.
Адсорбент с этой точки зрения характеризуется величиной адсорбционного объема Иа = Луга, приходящегося на единицу массы адсорбента (или объема Иа = га, приходящегося на единицу площади адсорбента), и интенсивностью поля адсорбционных сил, действующих в пределах адсорбционного объема. Это поле количественно описывается величиной адсорбционного потенциала 8 — работой переноса одного моля (киломоля) сорбата из бесконечности в адсорбционный объем. Зависимость адсорбционного потенциала от степени заполнения адсорбционного объема сорбатом является основной характеристикой сродства сорбента к конкретному сорбируемому компоненту гетерогенной системы. При увеличении концентрации сорбата сначала заполняются те части адсорбционного объема, которые имеют наибольший потенциал, т. е. ближайший к поверхности слой, а затем и удаленные слои. Адсорбция продолжается до заполнения адсорбатом всего адсорбционного объема. Если принять, что при адсорбции газа происходит его конденсация в адсорбционном объеме, то можно конкретизировать значения некоторых параметров.
Рис. 3.11. Изотерма полимолекулярной адсорбции: Г„ — предельная величина мономолекулярной адсорбции
558
Новый справочник химика и технолога
Во-первых, плотность конденсата рс обычно известна (порядка 103 кг/м3 для обычных жидких веществ), что позволяет вычислить толщину пленки конденсата гс= ГЛ// рс, находящуюся в равновесии с газом при данной его концентрации (давлении Р). Во-вторых, если газ конденсируется, то адсорбционный потенциал £, соответствующий расстоянию гс от поверхности, можно вычислить как работу сжатия газа от равновесного давления Р в объеме газообразной фазы до давления насыщенного пара Ps данного вещества при температуре адсорбции Т. В случае идеального газа £ = RT In (Ps / Р).
В совокупности с полученной экспериментально изотермой адсорбции Г - Г(с) два уравнения (3.1.25) задают в параметрической форме зависимость адсорбционного потенциала £ от степени заполнения поверхности сорбатом (рис. 3.12), которая в данном случае характеризуется толщиной гс пленки конденсата на поверхности:
ГЛ/ гс=-----, £ = ЯГ1п(Л/ Р). (3.1.25)
Рс
Если причиной адсорбции являются силы молекулярного притяжения (силы Ван-дер-Ваальса), что и предполагается в этой теории, то полученная указанным образом зависимость будет отображать распределение потенциала молекулярных сил возле поверхности твердого тела.
Вместо толщины пленки гс можно использовать иные параметры х заполненности адсорбционного слоя, в частности — саму адсорбцию Г или величину 0 и др. При разных условиях полученная зависимость £ от х представляет собой изотерму адсорбции в других координатах: аргументом является адсорбция, выраженная величиной х, а функцией — адсорбционный потенциал £, связанный при данной температуре с равновесной концентрацией (давлением) сорбата. Графическое выражение изотермы адсорбции в указанных координатах называется характеристической кривой данной пары веществ — адсорбата и адсорбента.
Рис. 3.12. Адсорбционный потенциал (работа адсорбции) е в зависимости от толщины х слоя адсорбированного вещества (от величины адсорбции Г):
при повышении температуры Т точка, изображающая состояние адсорбционного слоя, перемещается из позиции 1 в позицию 2; е — адсорбционный потенциал (работа адсорбции); х — толщина слоя адсорбированного вещества; Г — величина адсорбции
Ее ценность заключается в инвариантности по отношению к температуре опыта, т. е. точки этого графика, полученные из изотерм, относящихся к разным температурам, ложатся на одну общую кривую. Иначе говоря, характеристическая кривая, построенная по одной изотерме (при одной фиксированной температуре), позволяет рассчитывать изотермы при других температурах. Теория полимолекулярной адсорбции, хотя и не дает аналитического описания изотермы, имеет большое прикладное значение в отличие от теории Ленгмюра, представляющей удобное для технологических расчетов уравнение изотермы адсорбции.
Введение в научный обиход понятия адсорбционного потенциала является наиболее важным достижением теории полимолекулярной адсорбции. Понятие адсорбционного потенциала применимо и к описанию мономолекулярной адсорбции, в том числе на активных центрах и при адсорбции из растворов. Характеристическая кривая в этом случае отражает распределения центров по энергиям адсорбции. Разумеется, что при этом формальная толщина заполненного сорбатом слоя не должна превышать размера молекул сорбата, поэтому при мономолекулярной адсорбции более содержательным аргументом характеристической кривой будет сама адсорбция Г или степень заполнения монослоя. В случае полимолекулярной адсорбции степень заполнения Г/ Гт может превышать единицу.
Отсутствие аналитического описания изотермы адсорбции является с прикладной точки зрения недостатком теории полимолекулярной адсорбции. Этот недостаток устранен в теории БЭТ (Брунауэра, Эммета, Теллера), которая использует кинетический вывод уравнения изотермы адсорбции теории Ленгмюра, но дополняет его положением о том, что каждая адсорбированная молекула также является активным центром адсорбции. В связи с этим положением энергия адсорбции включает в себя энергию конденсации газа (энергию адсорбции молекул газа самого на себе), что придает сходство этой теории с потенциальной теорией и обеспечивает полимолекулярность адсорбции. Уравнение изотермы адсорбции получается путем суммирования изотерм для первого, второго и т. д. адсорбционных слоев, причем величина адсорбции при данном давлении в предыдущем слое играет роль адсорбционной емкости текущего слоя. Результатом такого суммирования является уравнение
Г = ГтХ(Р/Л)/[(1 -P/Ps) + (К-\)P/PS]. (3.1.26)
При малом давлении газа (Р / Ps 1) и большом значении константы адсорбционного равновесия (К » 1) конденсация не имеет решающего значения, и это уравнение становится эквивалентным уравнению изотермы Ленгмюра. При произвольном значении этих параметров адсорбция может превышать предельную величину Гт (рис. 3.11), которую в данном случае уместнее называть емкостью монослоя. Предполагается, что она оп
Физика, химия и технология дисперсных систем
559
ределяется размером молекул адсорбата по формуле (3.17), поскольку нельзя безоговорочно согласиться с тем, что конденсация газа может успешно развиваться при незаполненном первом адсорбционном монослое, т. е. если предельная адсорбция по Ленгмюру не соответствует сплошному монослою. Впрочем, с некоторыми оговорками это можно допустить. Они относятся к взаимосвязи смачиваемости поверхности жидкостью (конденсатом) и адсорбции паров той же жидкости. Этот вопрос затрагивается в следующей главе.
3.2. Капиллярные явления
3.2.1. Основные понятия
Поверхностное натяжение направлено на уменьшение площади межфазной границы, поэтому в отсутствие внешних по отношению к межфазному слою сил плоская граница не может существовать. В сосуде с жидкостью внешней силой, обеспечивающей существование плоской поверхности жидкости, является реакция стенок сосуда. В концентрированной пене или эмульсии такой силой является давление со стороны соседних пузырьков или капель, которое деформирует их в многогранники с плоскими гранями.
В двухфазных системах в отсутствие указанных сил, а также в трехфазных системах, имеющих общую линию соприкосновения трех фаз, межфазные границы искривлены. В частности, свободно висящая капля жидкости имеет сферическую форму, так как ей соответствует минимальная площадь межфазной границы 4лг2, где г — радиус капли.
Рассмотрим шаровой сегмент сферической капли (рис. 3.13). В соответствии с принципами механики (статики) действие отброшенной части сферы заменим силами, с которыми она действовала на сегмент. Это сила поверхностного натяжения границы двух фаз и разность давлений Р\ - Р2 во внутренней (7) и наружной (2) фазе.
Сила натяжения действует на линию разреза длиной 27trsin<p по касательной к сферической поверхности сегмента, а разность давлений действует на основание сегмента площадью 7t(rsin<p)2. По условию равновесия сегмента векторная сумма этих сил равна нулю, т. е.
27trsin(pcos(7t/2 - (р)ст12- Tt(rsin(p)2(Pi - Р2) = 0.
Отсюда следует формула Лапласа, в которой (Рк = Р[ - Р2) — капиллярное давление есть разность давлений двух фаз, разделенных искривленной поверхностью:
Рк = 2п12/г. (3.2.1)
Положительный знак означает, что давление внутри капли выше, чем в сфере. Знак и величина Рк не зависят от того, какая фаза находится внутри капли, а какая — снаружи. Давление всегда повышено в той фазе, которая охватывается поверхностью.
Следует обратить внимание на аналогию, а точнее на идентичность (с точки зрения механического описания) капиллярного давления и натяжения поверхности, с одной стороны, и избыточного давления внутри сферического сосуда и напряжения (на единицу длины) в его оболочке — с другой. Аналогия сохраняется и при несферической форме капли и сосуда, например эллипсоидной. Это утверждение требует пояснения и развития, так как из него вытекают важные следствия. Прежде всего необходимо обобщить понятие радиуса в формуле Лапласа на случай несферической капли (сосуда). Можно показать, усложнив тригонометрическую часть задачи, что при произвольной форме капли (оболочки) формула Лапласа (3.2.1) имеет вид:
Рк = ^012,
где К = 1/Г] + 1/г2 — кривизна поверхности, п и г2 — радиусы кривизны двух ортогональных линий на поверхности (обычно используются главные радиусы, один из которых максимален, а другой — минимален). В частном случае сферы ri = г2 - г и К = 2/г.
Очевидно, что в сосуде с газом и в капле давление одинаково во всех областях, охваченных поверхностью раздела фаз или оболочке сосуда. В этом состоит условие отсутствия потоков внутри капли и сосуда. Из равенства давления в разных частях сосуда и формулы (3.2.1) следует хорошо известный в механике результат: напряжения максимальны в тех частях оболочки, которые имеют минимальную кривизну, т. е. в случае вытянутого эллипсоида напряжения максимальны в экваториальной области оболочки и минимальны на полюсах эллипсоида. Применительно к поверхностному натяжению разных участков капли эллиптической формы такой результат может показаться неправильным, так как до сих пор предполагалось, что натяжение при данной температуре зависит только от состава фаз. Различие натяжений на экваторе и полюсах эллипсоида означало бы, что оно зависит от ориентации поверхности. Разумеется, что в изотропной жидкости в отсутствие градиента давления в капле и вне ее капля может быть только сферической, т. е. натяжение не зависит от ориентации межфазной границы.
Рис. 3.13. Шаровой сегмент сферической капли.
Силы, действующие на сегмент капли: о — поверхностное натяжение; Р— капиллярное давление; г — радиус капли
560
Новый справочник химика и технолога
Несферичность означает анизотропию свойств жидкости или наличие градиента давления в изотропной среде, наличие градиента температуры или состава вдоль поверхности капли. Практически эллиптичность капель или пузырей газа можно создать вращением капли (или жидкости с пузырьком газа) вокруг некоторой оси. Под воздействием центробежной силы возникают разные давления на полюсах и экваторе вращающейся капли, а натяжение не зависит от ориентации поверхности. Можно сферическую каплю вытянуть в эллипсоид действием достаточно сильного электрического поля (или магнитного поля, если это капля магнитной жидкости). Поле создает анизотропию внутренней структуры жидкости (ориентацию или поляризацию молекул); тонкая структура поверхности зависит от ориентации молекул относительно поверхности, следовательно, и натяжение зависит от ориентации поверхности относительно осей анизотропии вещества.
Многим твердым веществам анизотропия присуща в силу их кристаллического строения, поэтому различные грани кристалла имеют разное по величине натяжение. Постоянство капиллярного давления внутри равновесного по форме кристалла означает в этом случае, что давление может быть выражено через натяжение ст, любой грани и расстояние L, от этой грани до некоторого центра кристалла. Величина Z, заменяет радиус кривизны в формуле Лапласа, поэтому:
Ст] / L\ = СТ2 / L2= Стз / Lj =...
Это соотношение известно как правило Вульфа.
Также важной причиной нарушения сферичности капель является соприкосновение капли одновременно с двумя различными фазами. Новизна этой ситуации в том, что она возможна только в трехфазных гетерогенных системах. Простейший пример такой системы — капля жидкости (фаза 2) на поверхности твердого вещества (фаза 7), окруженная газом (фаза 3).
Общая линия соприкосновения трех фаз называется периметром смачивания. На схеме (рис. 3.14) она представлена точкой пересечения трех линий, изображающих три межфазные границы. На единицу длины периметра смачивания (точку на схеме) действуют силы натяжения каждой из трех границ. Периметр может перемещаться только по — поверхности твердого тела, поэтому поведение капли будет определяться суммарной силой, действующей на единицу длины периметра в этой плоскости, т. е. суммой проекций всех сил натяжения F на направление возможного движения:
F = Ст]з - Ст12 - eicosa. (3.2.2)
Если F отличается от нуля, то периметр будет двигаться. При F > 0 капля растекается. В обратном случае (F < 0) капля собирается под действием поверхностных сил. F > 0 является условием растекания. Процесс заканчивается при достижении равновесного значения угла а, который называется краевым углом смачивания
и обозначается буквой 0. При этом F = 0, т. е. выполняется соотношение:
СТ13 - 012 - CT23COS0 = 0. (3.2.3)
Это соотношение известно как формула Юнга. Условие растекания F > 0 может быть заменено эквивалентным условием а > 0, где а — произвольный первоначальный угол соприкосновения фаз на периметре смачивания.
В капле, лежащей на подложке, как и в свободной капле, давление повышено на величину Рк = 2стгз / г = = 2ст2з sin0 / г0, где г — радиус сферической поверхности капли, а г0 — радиус пятна контакта жидкости с субстратом (подложкой). Капиллярное давление в данном случае не нужно учитывать как возможную причину движения капли. Оно обусловлено общими принципами механики: некоторая обобщенная сила (в данном случае Рк) участвует в процессе в качестве движущей силы (со знаком «+» или «-»), что возможно при изменении сопряженной с данной силой обобщенной координатой. По отношению к давлению сопряженной координатой является объем (капли). При растекании или собирании он не меняется, поэтому Рк «не работает». Но меняются координаты, сопряженные с натяжениями <Т]2, <Т]з и ст23, площади соответствующих межфазных поверхностей Л12, Л13, Л2з, что и учтено в выражении силы F через параметры трехфазной системы.
Указанный принцип позволяет во всех и сложных и простых случаях выделить необходимый и достаточный набор параметров состояния системы, определяющих ее поведение. Число таких параметров называется числом степеней свободы системы.
Равновесная форма капли, т. е. величина угла 0, отражает взаимодействие жидкости с субстратом (подложкой). Если 0 < тг/2, то считается, что поверхность твердого вещества смачивается данной жидкостью; при 0 = 0 происходит полное смачивание, при 0 > л/2 поверхность считается несмачиваемой.
Смачивание, или взаимодействие жидкости с субстратом (прилипание), оказывает более сильное влияние на форму капли, чем взаимодействие молекул жидкости между собой. Последнее проявляется в величине поверхностного натяжения жидкости. Чем оно выше, тем ниже при прочих равных условиях смачивающая способность жидкости. Ртутью, например (ст = 0,47 Дж/м2), почти никакие вещества не смачиваются (кроме ряда металлов).
Рис. 3.14. Силы, действующие на периметр смачивания (линию соприкосновения трех фаз: 1.2 и 3): а — угол контакта фаз;
on. он, с?2з — натяжения (раниц фаз / 2, ! 3 и 2 3
Физика, химия и технология дисперсных систем
561
Вещества, которые смачиваются водой, называются гидрофильными. Это вещества с ионным типом связи атомов, т. е. полярные с точки зрения поляризованно-сти межатомной связи (водонерастворимые соли, стекло, оксиды металлов и т. д.). Вещества с неполяризо-ванной связью атомов (ковалентная, молекулярная) являются гидрофобными, т. е. водой не смачиваются (графит, твердые парафины и др.).
Неполярными жидкостями с низким поверхностным натяжением (углеводороды, фреоны) хорошо смачиваются все вещества, в том числе полярные. Тем не менее некоторые твердые вещества лучше смачиваются водой, чем углеводородом, а некоторые хуже. Мерой относительного сродства двух несмешивающихся жидкостей к поверхности является избирательная смачиваемость, которая определяется краевым углом капли одной из жидкостей, нанесенной на поверхность твердого вещества под слоем другой жидкости. При этом наблюдаются все те же соотношения, что и при смачивании в газовой среде, но о23 и — натяжения границ жидкости и твердого вещества с другой жидкостью.
Характеристикой взаимодействия двух конденсированных фаз 1 и 2 между собой является адгезия 1Г12 — удельная (на единицу поверхности контакта фаз) работа по их разъединению с образованием двух новых межфазных границ (рис. 3.15).
Из определения натяжения как работы образования межфазной поверхности непосредственно следует формула:
JFi2 = Oi + о2 - Oi2. (3.2.4)
Если одна из разъединяемых фаз, например 1, твердая, то возникает необходимость выразить в формуле (3.2.4) неизмеряемые величины натяжений си и о12 с помощью уравнения (3.2.3) через измеряемые величины о2 и 0. Это приводит к формуле Дюпре
1Г12 = о2(1+cos0). (3.2.5)
При полном смачивании (0 = 0, cos0 = 1) адгезия достигает своей максимально возможной величины, равной 2о2. Эта величина называется когезией жидкости. Согласно определению (3.2.4), когезия (FF22 = 2о2) — частный случай адгезии, когда однородная фаза (oi - о2, Oi2 - 0) разделяется по сечению с единичной площадью.
При полном отсутствии смачивания (0 = л, cos0 = -1) формально 1Г12 = 0, однако практически взаимодействие веществ любой природы между собой имеет конечную величину И7]? > 0. Это означает, что случай 0 = п является абстракцией.
Наглядно капиллярные силы проявляют себя поднятием жидкости в капилляре (рис. 3.16), что и дало название всей группе аналогичных явлений. Равновесную высоту поднятия h в капилляре радиуса гк при краевом угле смачивания 0 обычно находят из равен
ства капиллярного давления Рк = 2о / гк гидростатическому давлению столба жидкости в капилляре РГ = pgh, где р — плотность жидкости, g — ускорение силы тяжести, по формуле
. 2о я =---
Pgr
(3.2.6)
Здесь г = rK / cos0 — радиус поверхности мениска. Величина Иг представляет собой кривизну поверхности мениска. Более детально рассмотрим причину капиллярного поднятия.
Согласно уравнению (3.2.3), при равновесном значении угла 0 результирующая сила межфазных натяжений F = 0 и не может быть причиной капиллярного давления и поднятия. Какие-либо иные движущие силы капиллярного поднятия отсутствуют.
Рис. 3.15. Адгезия И7]? — работа разъединения двух фаз по площади контакта, равной единице
Рис. 3.16. Поднятие жидкости в капилляре:
/ — стенки смачиваемого капилляра;
2 — равновесное положение мениска жидкости;
г — радиус кривизны мениска; гк — радиус капилляра; h — высота капиллярного поднятия жидкости, 0 — угол смачивания
562
Новый справочник химика и технолога
При капиллярном поднятии, в отличие от рассмотренного ранее растекания капли, не меняется площадь и форма границы раздела жидкости с газовой фазой, поэтому силы натяжения этой границы не могут совершать работу, т. е. участвовать в рассматриваемом процессе. Меняется площадь контакта твердого тела с газом и твердого тела с жидкостью, поэтому только сопряженные с ними натяжения ст12 и ст13 будут «работать» при капиллярном поднятии (здесь и далее используется индексация фаз, принятая выше при рассмотрении равновесия и движения капли на поверхности твердго субстрата). Таким образом, движущей силой в этом случае будет разница натяжений - Ст12, но согласно уравнению (3.2.3) она равна с обратным знаком величине o23cos©, и тогда Рк =2лго23со50/лгк2 =2су23/г. Это приводит к указанному выше результату. Однако причиной капиллярного давления и поднятия является не поверхностное натяжение жидкости ст23, а разность натяжений Ст13 - ст 12 несмоченной и смоченной поверхностей капилляра, которая выражается через ст23. В данном случае неправильное толкование причины капиллярного поднятия и давления не привело к ошибке, но в некоторых случаях такого рода неточности ведут к неверным результатам. Примером может служить ошибочная формулировка условия растекания, выражаемого соотношением Wa > WK между величинами адгезии жидкости к субстрату Wa и когезии жидкости WK, которая довольно часто встречается. Символы Wa и WK будут и далее использоваться наряду с символами типа и W22 в тех случаях, когда не возникает сомнений, к каким именно фазам относятся величины адгезии и когезии соответственно. Числовые индексы будут применяться тогда, когда необходимо явно указывать, какие фазы имеются в виду.
Согласно уравнению (3.2.5), адгезия Wa не может быть больше когезии жидкости WK. Причина упомянутой ошибки — перенос понятий (адгезия, когезия, краевой угол, натяжение), относящихся к макроскопическим фазам, на тонкие слои вещества, в которые превращается неограниченно растекающаяся жидкость. Указанное соотношение обычно и выводится из условия неограниченного растекания жидкости по субстрату. Средняя толщина тонких слоев жидкости может при этом стать соизмеримой с молекулярными размерами, а состояние слоя — близким к состоянию двухмерного газа. Слой жидкости на субстрате перестанет существовать как конденсированная фаза, а поэтому утрачивают смысл и понятия, относящиеся к жидкой фазе.
Кроме того, растекание, как любой самопроизвольный процесс, возможно лишь в том случае, когда исходное состояние системы (капли до начала ее растекания) отличается от равновесного (краевой угол отличается от равновесного значения угла смачивания). Направление процесса (растекание или собирание) также целиком зависит от того, в какую сторону от равновесия отклоняется первоначальное состояние системы (капли): если в исходном состоянии краевой угол окажется больше рав
новесного, то он самопроизвольно начнет уменьшаться, т. е. капля будет растекаться; в обратном случае капля будет собираться. Эти соображения здесь приведены с единственной целью: подчеркнуть, что ни при каких условиях нельзя предсказать направление процесса, если не указано, в какую сторону от равновесия отклоняется исходное состояние системы.
В формулировках условия растекания наряду со значениями равновесных параметров системы (угла смачивания или однозначно связанных с ним величин типа адгезии) используют величины, относящиеся к текущему неравновесному состоянию (фактическое значение краевого угла в этом состоянии).
Работа переноса сферической частицы через межфазную границу. Равновесное положение частицы (фаза 7) на плоской межфазной границе таково, что углы соприкосновения трех фаз задаются смачиваемостью частицы одной из фаз, например 2. Граница флюидных фаз 2 и 3 не должна быть при этом искривлена (рис. 3.17), а глубина погружения А3 частицы в фазу 3 определяется формулой
А3 = г (1-cos0). (3.2.7)
Работа отрыва частицы от этой фазы, т. е. работа ее перехода с границы в фазу 2 в соответствии с определением натяжения и адгезии, будет:
Аз2 = Аз (<^12 ~ CFi3) + т423О23,
где Л13 = 2гсг2(1 - cos©) — площадь поверхности сферы в фазе 3 (исчезающая при переходе частицы в фазу 2), Аз = ro^sin2© — площадь поверхности между фазами 2 и 3, которая возникает при уходе частицы с поверхности.
Если использовать условие равновесия частицы на границе, которое полностью совпадает с условием равновесия капли на субстрате (3.2.3), то получаются соотношения:
^32 = (><23 - A3cos0)ct23 ИЛИ FK32 = лст23 Л,2. (3.2.8)
Рис. 3.17. Граница флюидных фаз:
1 — твердое тело; 2 — жидкость; 3 — газ; © — угол смачивания; он, он, 023 — натяжения межфазных границ 12,13 и 23
соответственно
* В отсутствие силы тяжести.
Физика, химия и технология дисперсных систем
563
Работа перехода W22 частицы с границы в фазу 3, выраженная через тот же параметр, т. е. глубину Л3 погружения в фазу 3, описывается формулой
fP23 = ло23 (2г - Л3)2. (3.2.8а)
Работа переноса частицы из фазы 3 в фазу 2 через границу этих фаз может быть представлена как разность работ переноса:
^23 - ^32 = 71023 [(2г - Лз)2 " ] =
= яо23 (4г2 - 4гй) = 4лго23 (г - й).
Однако это выражение, как и два предыдущих, нельзя применять для вычисления работы изменения глубины погружения h частицы в одну из сред, поскольку здесь h — это равновесное положение частицы относительно границы, которое нельзя изменять произвольно, не подразумевая под этим изменение природы фаз или адсорбционного изменения натяжения границ. Уравнение (3.2.86) описывает работу переноса тела из одной фазы в другую:
^=4лго23(г-й). (3.2.86)
Оно дает правильный результат при любом h, являющемся мерой смачиваемости переносимой частицы той средой, в которую она переносится, в том числе при h = 0 и h = 2г.
Если величины >Р23 и fF32 сопоставимы с энергией теплового движения частиц кТ, то становится возможным флуктуационный отрыв частиц от границы в обе фазы. В этом случае соотношение равновесных концентраций с3 и с2 частиц в этих фазах определяется законом Больцмана, в котором с3мс2 — коэффициент распределения частиц между фазами:
с3/с2 = ехр(-^/Щ (3.2.9)
Уравнение (3.2.9) справедливо и в отношении молекул ПАВ, но тогда величина JV имеет смысл преимущественного сродства ПАВ к одному из растворителей. Формально оно применимо и к границе между жидкостью и газом, хотя говорить о равновесной концентрации частиц в газовой фазе не принято. Фактически лиофобные частицы (или молекулы нелетучего вещества) при этом скапливаются на поверхности жидкости, «плавают» по ней. Технологическая практика широко использует регулирование смачиваемости частиц с помощью явления адсорбции ПАВ, в том числе при флотационном обогащении ископаемых, в производстве красок, феррожидкостей и т. д.
3.2.2. Равновесие поверхности твердого вещества с летучей жидкостью
Если фаза 3 представляет собой газ, выражения (3.2.3)-(3.2.6) являются гипотетическими: поверхность
твердой фазы 1 взаимодействует с остальной частью гетерогенной системы 2 только по пятну контакта с жидкостью. Тем самым игнорируется взаимодействие с газовой фазой, которая в состоянии равновесия насыщена парами той же жидкости (или другого летучего компонента). Взаимодействие газа и твердого вещества выражается в адсорбции паров поверхностью твердого вещества. Согласно уравнению БЭТ адсорбции газа (3.1.26), в насыщенном паре на поверхности должна образовываться пленка конденсированного пара, т. е. пленка жидкости. Ее толщина при этом не описывается никакими известными уравнениями, а смачиваемость поверхности конденсатом может входить некоторым образом в константу адсорбционного равновесия. Наличие такой пленки отвечает равновесию в системе твердая фаза—насыщенный пар, и, следовательно, эта пленка образуется даже в случае практически нелетучей жидкости, скорее всего путем растекания жидкости по поверхности, даже если поверхность плохо ею смачивается.
Таким образом, в условиях полного равновесия трехфазной системы 7, 2, 3 (рис. 3.18) вместо границы твердой фазы 1 с газом 3 и ее натяжения Ов должна фигурировать пленка и ее натяжение оп, включающее в себя натяжения границы твердого вещества со слоем жидкости о'12 и натяжение границы слоя жидкости с паром <У23.
В многофазных системах любая из фаз может находиться в пленочном состоянии, и в соответствии с этим обозначение натяжения пленки должно содержать в себе указание, о пленке какой фазы идет речь. В подобных случаях вместо общего обозначения натяжения пленки оп будут использоваться альтернативные обозначения с помощью символа поверхностного натяжения cxyz с тремя нижними индексами. Средний индекс относится к фазе, образующей пленку, а первый и третий — к ограничивающим ее фазам. Например, в системе, показанной на рис. 3.18, Oi23 — это натяжение пленки фазы 2 (жидкости), ограниченной снизу твердой фазой 7 и сверху газовой фазой 3.
При наличии пленки условие равновесия капли по форме останется таким же, что и при ее отсутствии (формула (3.2.3)), причем о123 = о'12 + о'23:
СГ 123 - °12 - O23cos0 = 0. (3.2.10)
Рис. 3.18. Многофазная система:
1 — субстрат (подложка пленки конденсата);
2 — капля; 3 — пар; h — толщина пленки конденсата; ст12, <т23 — натяжение границ пленки с субстратом и паром
564
Новый справочник химика и технолога
Величины со штрихом указывают, что они отличаются от натяжений и ст23 границ между фазами 1 и 2, 2 и 3 соответственно.
Как уже отмечалось, межфазные натяжения формируются в поверхностном слое конечной толщины и достигают своей номинальной величины Ст12 или о23 при условии, что протяженность соприкасающихся фаз не ограничена в направлении нормали к границе между ними. Поэтому если толщина пленки h меньше, чем сумма толщин SJ2 и 523 поверхностных слоев, в которых создаются межфазные натяжения границ 12 и 23 соответственно, то поверхностные слои на обеих границах пленки не получают полного развития, а пленка считается тонкой. Это и создает различие величин межфазных натяжений ст12 и о23 и натяжений границ пленки ct'i2 и ст'23. При h > St2 + 623 пленка считается толстой, и натяжения границ совпадают с межфазными натяжениями.
В дальнейшем удобно использовать также разность натяжений толстой и тонкой пленок:
Л<7123 = (<712 + <7гз) - (<7'12 + <7'2з)-
Внутри толстой пленки имеется вещество с теми же свойствами, что и у объемной фазы, из которой она образована. Величина Acti23 может быть и положительной, и отрицательной, так как локальное натяжение отдельных микрослоев всего поверхностного слоя (рис. 3.18) может иметь разные знаки и переход к тонкой пленке может сопровождаться удалением слоев, вносящих или положительный, или отрицательный вклад в поверхностное натяжение.
В ряде случаев толщина пленки h = 3i2 + З23. Такую пленку можно назвать предельной. Пленка, с обеих сторон ограниченная идентичными фазами, называется симметричной.
Если исключить величину (о']2 + о'23) из определения До123 с помощью условия равновесия капли (3.2.10), то получится формула
До123 = о23 (1 - cos©). (3.2.11)
Если далее допустить, что o'i2 = о12, т. е. если натяжение границы субстрат—пленка останется таким же, как натяжение границы субстрата—неограниченная жидкая фаза, то получим связь между натяжением границы пленка—пар и углом смачивания субстрата жидкостью:
0*23 = o23cos0.
Из уравнения (3.2.11) вытекает также, что До)23 > О, т. е. самопроизвольно образующаяся и равновесная с объемной жидкой фазой тонкая пленка всегда имеет меньшее натяжение, чем толстая. Толстая и предельная пленка (Д<Т123 = 0), согласно (3.2.11), всегда полностью смачивается (cos© = 1) той жидкостью, из которой она образована. Поскольку значение cos© не может превышать единицы, формула (3.2.11) не охватывает случаев,
когда Дст123 < 0, т. е. когда натяжение пленки больше суммы натяжений межфазных границ. Повышенное натяжение пленки относится к случаям, когда условие равновесия (3.2.10) не выполняется, т. е. когда она создается под действием некоторых дополнительных сил, например при расплющивании капли жидкости между двумя поверхностями.
Формула (3.2.11) выявляет проблему взаимосвязи смачиваемости (cos0) и адсорбции паров. Насыщенный пар некоторой жидкости образует на поверхности твердого вещества адсорбционную пленку, толщина которой в состоянии полного равновесия в соответствии с уравнением полимолекулярной адсорбции паров (3.1.17) ничем не ограничена. Тогда при равновесии системы твердое вещество—жидкость—пар адсорбционная пленка должна быть, по крайней мере, предельной, а жидкость — полностью смачивать поверхность твердой фазы. В действительности равновесие капли на поверхности чаще соответствует неполному смачиванию. Для устранения возникающего противоречия необходимо принять во внимание, что плохо смачивающие жидкости не могут образовывать предельную пленку ни путем адсорбции ее насыщенного пара, ни путем растекания капли. Последнее общеизвестно, но в теории адсорбции паров какие-либо ограничения, связанные со смачиваемостью, отсутствуют.
Таким образом, конечная смачиваемость (cos0 < 0) в условиях полного равновесия означает, что адсорбция паров плохо смачивающей жидкости не может завершиться образованием толстой или предельной пленки.
Формула (3.2.11) позволяет внести уточнения и в некоторые другие известные положения. Гипотетический случай 0 = п означает полное отсутствие взаимодействия жидкости с субстратом, а значит, и отсутствие адсорбционной пленки на границе субстрат—пар. Для сохранения общности описания удобно, однако, считать, что и в этом случае имеется пленка нулевой толщины и натяжение такой пленки тождественно натяжению о13, т. е. Qi23 = о13. Тогда, по общему определению, Ао123 = о12 + о23 - О]3. В то же время из уравнения (3.2.11) следует, что при дефицит натяжения пленки До123 = 2о23. Приравнивая правые части последних выражений, получим о12 = + о23, т. е. натяжение
границы между одной фазой (7) и полностью несмачивающей ее другой фазой (2) равно сумме натяжения границ этих фаз с газовой фазой (3). Такой результат является правильным в той мере, в какой случай 0 = тс (отсутствие взаимодействия между фазами 7 и 2) соответствует действительности. В самом деле, если взаимодействие между фазами 7 и 2 отсутствует, то толщина газовой прослойки между ними не влияет на натяжения Ст13 и о23 границ этой прослойки с твердой и жидкой фазами. Тогда при уменьшении толщины прослойки натяжение границ остается неизменным вплоть до ее исчезновения, т. е. до образования общей границы этих фаз, натяжение которой в таком случае будет равно сумме натяжений границ толстой газовой пленки.
Физика, химия и технология дисперсных систем
565
Эмпирическое правило Антонова, связывающее межфазное натяжение с натяжениями границ конденсированных фаз с газовой средой (воздухом): <у12 = ов - <з2з, при котором происходит конечное (0 < л) или возможно полное (© = 0) смачивание, можно заменить обобщающей расширенной формулой:
012 = сПз - O23COS®. (3.2.12)
Она идентична условию равновесия капли в форме уравнения (3.2.3). Ее правая часть является суммой или разностью натяжений при 0 = п и 0 = 0 соответственно. Таким образом, правило Антонова относится к случаю полного смачивания и является частным видом выражения (3.2.3) или (3.2.12). Следует, однако, заметить, что натяжение о13 поверхности твердого вещества обычно вычисляется из экспериментальных данных о его смачиваемости жидкостью. Если эксперимент проводится в условиях равновесия системы твердое вещество—жидкость—пар, то тогда вычисляется величина Ops, а не Ов, и в формуле (3.2.12) следует сделать соответствующую замену, что характерно для формул, где фигурирует натяжение Ов «сухой» границы. В трехфазных системах граница всегда «мокрая», т. е. имеет слой адсорбированных паров жидкой фазы или эквивалентный ему слой, образующийся путем поверхностной диффузии нелетучей жидкости. Даже с оговорками такой способ распространения вещества жидкой фазы по поверхности субстрата нельзя назвать «растеканием жидкости».
Наличие равновесной пленки фазы 2 на поверхности фазы 7, которая образуется независимо от того, находится капля жидкости 2 в контакте с поверхностью фазы 7 или нет, требует пересмотра связи адгезии фаз 7 и 2 с поверхностными натяжениями этих фаз и ревизии формулы Дюпре. С учетом образования пленки в обратимом процессе разделения фаз, согласно данному ранее определению адгезии фаз 7 и 2, работу адгезии можно описать уравнением
^12 = <7123 + ^23 _ ^12- (3.2.13)
Если в этом уравнении учесть условие равновесия капли (3.2.10) или (3.2.11), то снова пролучается формула Дюпре (3.2.5). Формула (3.2.6) для капиллярного поднятия также не изменяется.
3.2.3. Равновесие поверхности твердой фазы с двумя жидкими
В адгезионном взаимодействии участвуют три фазы: две взаимодействующие и третья — среда, в которой они взаимодействуют. Взаимодействующие фазы могут быть твердыми или жидкими, или одна из них может быть твердой, а другая — жидкой, среда — газообразной или жидкой. К сожалению, придется отказаться от преемственности в индексации фаз, принятой в предыдущем параграфе. Если предусмотреть возможность
образования пленки любой фазы на границе между двумя остальными, то потребуется четыре индекса для обозначения конфигурации трехфазной системы, что делает обозначения неприемлемо громоздкими. Чтение формул облегчается, если индексы несут в себе некоторый смысл: поэтому индексом «5» (solid) будет обозначаться твердая фаза, индексом «/» (fluid) — жидкая фаза и индексом «v» (vapour) — газообразная фаза. Последний может обозначать и вторую жидкую фазу, если она выступит в качестве среды взамен газовой фазы. Твердым фазам, в отличие от жидкости и газа, мы не будем придавать статуса среды. Это позволит несколько упростить индексацию величин, и исключит рассмотрение с общих позиций пористых тел, у которых средой служит твердая фаза. При наличии во флюидной среде (жидкости или газе) двух разных твердых веществ они могут обозначаться цифровыми индексами 7 и 2. Среда при этом также может быть двухфазной — смесью жидкости и газа или двух жидкостей, т. е. гетерогенная система оказывается четырехфазной.
Примеры известных и практически важных четырехфазных гетерогенных систем: нефтеносная геологическая порода (кварц, глина, нефть, вода), ванны жидкостной и пенной флотации смеси минералов, смазочно-охлаждающие эмульсии и абразивный инструмент в процессе шлифовки или резания металлов и т. д.
При наличии двух объемных жидких фаз/и v и одной твердой фазы 5 сохраняют силу приведенные выше формулы при замене в них индексов 7 на s, 2 на/и 3 на v. Вместе с тем возникают особенности, связанные с ненулевой адгезией фазы v (среды) к поверхности твердого вещества.
Рассмотрим предварительно процесс погружения пластины твердого вещества s из одной жидкой среды v в другую f через их границу (рис. 3.19).
Работа погружения твердой фазы из одной среды в другую, отнесенная к единице площади поверхности, сменившей при погружении среду, описывается уравнениями
lFv/= -Wfv = asf- = -oypcos©^ = O/,cos0sv7. (3.2.14)
Рис. 3.19. Процесс погружения пластины твердого вещества из одной жидкой среды в другую: v,f— фазы погружения; 0Г./, ®vfi— межфазные углы;
Ojv, Ov/, — натяжения; sv, vf, sf— межфазные границы
566
Новый справочник химика и технолога
Средний индекс при угле смачивания ® относится к той фазе, которая лежит внутри этого угла. Очевидно, работа погружения из фазы v в фазу f равна с обратным знаком работе погружения из фазы f в фазу у. Отметим, что работа погружения на величину од отличается от адгезии Ид фазы s к фазе f в среде у, так как погружение из фазы f в фазу v не сопровождается образованием дополнительной поверхности контакта фаз f и у между собой. По этой причине справедливы соотношения:
= Wyf+
Wsf=Wp + <^ (3.2.15)
Вместе с формулами (3.2.14) они указывают прямой метод измерения адгезии, так как величина одсоз®д непосредственно измеряется (метод пластинки Виль-гельми). Такой путь надежнее, чем раздельное измерение од и ®д, необходимое в соответствии с формулой Дюпре (3.2.5).
Из формул (3.2.15) и (3.2.14) получаются уравнения:
Wsv = Од (1 + COS®.пу), (3.2.16)
Wsf - од (1 - cos®svy), (3.2.17)
Wsf = од (1 + соз®5д). (3.2.18)
Из формул (3.2.16) и (3.2.17) следует уравнение (3.2.19), которое означает, что величина Wsv не равна величине WSf, за исключением случая ® = л/2:
JTvv - Wrf = 2ОдС08®,д. (3.2.19)
Формула (3.2.19) не нарушает закон сохранения энергии. Работа цикла равна сумме работ погружения Иди Ид (3.2.14), т. е. нулю. Из формулы (3.2.17) вытекает, что адгезия ни в какой среде не может быть отрицательной.
Соотношения (3.2.14) и (3.2.15) не учитывают возможности образования пленок одной жидкости под слоем другой жидкости. Не существует каких-либо аргументов, позволяющих допускать такую возможность в системе твердое вещество—жидкость—пар и отвергать ее в системе твердое вещество—двухфазная жидкость. При этом обе жидкости равноправны в отношении такой возможности, следовательно, на поверхности фазы s, частично погруженной в одну жидкость f и частично в другую v (рис. 3.20), одновременно может существовать пленка жидкости f между фазами лиги пленка жидкости v между фазами 5 и/ Натяжение этих пленок будем обозначать символами Од и оуд, где средний индекс указывает пленочную фазу.
Хорошо известно явление, которое можно считать подтверждением склонности одной жидкости накапливаться под слоем другой, и известна симметрия этой способности в смеси двух жидкостей и твердого вещества. Это явление — смена знака адсорбции компонен
тов бинарного раствора из двух жидких веществ. В растворе, который состоит в основном из компонента 1, на поверхности твердого адсорбента может накапливаться компонент 2. Из раствора, состоящего в основном из компонента 2, на поверхности накапливается компонент 1. Наличие пленки означает наличие границы этой пленки с сопредельной жидкостью. Возможные конфигурации границ в зоне трехфазного контакта (края капли фазы f в среде v на поверхности твердого вещества s) показаны на рисунке (рис. 3.20). Различные варианты топологически несовместимы, поэтому возникает неясность с действительным строением зоны трехфазного контакта, которая ранее представлялась как линия (периметр смачивания). Если предположить, что условие равновесия капли при наличии пленок сохранит ту же форму, что и уравнение Юнга, можно записать уравнение:
<ysfi> - ^svf - одсо8®д = 0. (3.2.20)
Если одна из пленок или обе пленки метастабильны, т. е. могут отсутствовать, тогда в формуле (3.2.20) од или Gsyfi или обе эти величины нужно заменить при исчезновении пленок на величины оп, и Од. Отсюда следует, что в одной и той же системе могут в некоторых случаях реализовываться четыре разных состояния с различными углами смачивания, которые описываются формулами:
cos® д = (о д - о, д) / од, jv, (3.2.21 а)
COS®д = (osv - Оуд) I Од, v, (3.2.216)
СО8®Д = (Од - Од) / од,/ (3.2.21 в)
COS®д = (osv - Од) / Од. (3.2.21 г)
Справа от формул указаны индексы фаз, которые присутствуют и в обычном, и в пленочном состоянии. Экспериментальным подтверждением того, что одна и та же трехфазная система может иметь несколько углов смачивания, является гистерезис смачивания — зависимость величины угла смачивания от того, из какого состояния капля пришла к равновесию — от угла большего, чем равновесный, или меньшего (угол натекания и угол оттекания). Другая известная причина гистерезиса смачивания — шероховатость поверхности.
Если принять во внимание наличие пленок на межфазных границах для адгезии жидкости f к поверхности 5 в среде v, то справедливо уравнение
— ^sfv "I" ^fv &syf'
(3.2.22)
Рис. 3.20. Конфигурации границ в зоне трехфазного контакта: а) непрерывная фаза/; б) — непрерывная фаза v;
5 — твердое вещество; v и/ — жидкости
Физика, химия и технология дисперсных систем
567
Здесь средний индекс по-прежнему относится к пленочной фазе. Формула (3.2.22) вместе с условием (3.2.20) сводится к уравнению (3.2.18). Работа погружения Wfv = csSyf -Qsjv связана с адгезией соотношением (3.2.15). В силу гистерезиса смачивания адгезия также окажется неоднозначной величиной при любом способе ее нахождения: по формулам типа (3.2.15) или (3.2.16)-(3.2.18). Следует иметь в виду и влияние кинетики образования равновесных пленок. По порядку величины время установления равновесной формы капли t - т]г / о, где т] — вязкость жидкости иг — радиус капли.
Адгезия двух твердых фаз в жидкой среде в присутствии небольшого количества другой жидкости. Пусть адгезия двух различных твердых фаз 7 и 2 в жидкой среде v происходит в присутствии небольшого количества другой жидкости f которая обеспечивает образование пленки на границах фаз 7v и 2v. Контакт твердых фаз 7 и 2 между собой в общем случае осуществляется через пленку фазы f Тогда адгезия в среде v через пленку f описывается уравнением:
= ~ СТУ2- (3.2.23)
Здесь средний индекс относится к пленочной фазе, c?i/2 — натяжение равновесной пленки, сохраняющейся в зазоре между фазами 7 и 2 в отсутствие каких-либо сторонних сил, искусственно поддерживающих существование пленки. В отличие от предыдущих случаев здесь нет натяжения Од. границы подвижных фаз, которое можно непосредственно измерить и через которое есть смысл выражать адгезию, подобно формуле Дюпре (3.2.5). Не исключено, что есть возможность выразить W\2 через углы смачивания 0^ и 02> но это ограничит применимость полученного результата двумя условиями: наличием объемной жидкой фазы f и равновесностью пленок на поверхности обеих твердых фаз. Имеется в виду равновесие, которое установится в отсутствие каких-либо иных сил, кроме адгезионных. В общем случае (взаимодействие твердых фаз в жидкости) интерес представляет и иной случай равновесия — при действии некоторых сторонних сил, например внешней силы сдавливающей пленки. Очевидно, что искусственно сдавленная (растянутая) пленка отличается от равновесной, и ее параметры нельзя выразить только через адгезионно-равновесные показатели типа о и 0.
Если фазы 7 и 2 идентичны (каждая из них является частью одной фазы 5), их взаимодействие характеризуется величиной когезии
= 2ст,Л - asfs. (3.2.24)
Если пленка, разделяющая две части одной фазы s, устранена путем сближения этих частей до их соприкосновения, то ее натяжение = 0. Тогда выражение (3.2.24) приобретает известную форму Wss = 2asjv с той разницей, что вместо межфазного натяжения asv в ней фигурирует натяжение пленки возникающей на вновь образованной поверхности после когезионного разъединения фазы s на две части.
Так как в формулах (3.2.23) и (3.2.24) не используются условия равновесия двух объемных жидких фаз, находящихся в контакте с третьей (твердой) фазой, то они справедливы и тогда, когда пленка возникает и в отсутствие второй жидкой фазы f, т. е. когда компонент, способный в принципе образовывать пленку на границе фаз 7v, 2v, присутствует в среде v только в растворенном виде (раствор пленкообразующего компонента не насыщен). В этом случае понятия пленкообразующего компонента и пленки становятся тождественными понятиями поверхностно-активного вещества и адсорбционного слоя этого вещества соответственно. Возможность существования пленки, разделяющей две разные (или идентичные) фазы при их адгезионном (когезионном) взаимодействии, приобретает тогда статус экспериментально установленного факта, так как адсорбционные слои могут сохраняться и в зазоре между слипшимися частицами.
Понятие адгезии и когезии возникло из рассмотрения работы разъединения двух фаз s nf или двух частей одной фазы в некоторой среде v как работы перехода от пленки нулевой толщины между поверхностями этих фаз к толстой пленке, образованной самой средой. По существу формулы (3.2.23), (3.2.24) расширяют это понятие на конечную толщину исходной равновесной пленки и иной ее состав, чем состав среды. Пленкообразующее вещество f может присутствовать в среде v только в растворенном состоянии, т. е. отдельная фаза f не образуется. В этом случае состояние межфазных поверхностей характеризуется не наличием или отсутствием на поверхностях растворенного вещества, а величиной его адсорбции и соответствующим ей натяжением, являющимся функцией концентрации раствора. Поверхностный слой в зазоре между фазами 7 и 2 следует в этом случае считать состоящим из фазы v, обогащенной адсорбированными молекулами на межфазных границах 7v и 2v. Таким образом, формулу (3.2.23) можно сейчас записать иначе:
^12 ~ CTlv + &2v~ Cflv2-
Если в последней формуле заменить натяжение пленки OiV2 суммой натяжений ее границ o'iv + o'2v, то получаются соотношения:
= (oi v + o2v) - (o'iv + o'2v), (3.2.25)
^12 = Cfiv2(oo) - o1v2(/7o). (3.2.25a)
В формуле (2.2.25a) Oiv2(oo) = (<*iv + <*2v) и OiV2(/?o) = = (cfiv + o2v) — натяжения неопределенно толстой (h = 00) и равновесной (h = h0) пленок, сохраняющихся между фазами 7 и 2 в отсутствие каких-либо сил, кроме сил молекулярного взаимодействия всех трех фаз между собой.
Термин «пленка» общеупотребим и означает тонкий слой вещества или материала (кинопленка, полиэтиленовая пленка, мыльная пленка и т. д.). Ранее он использовался для обозначения слоев конденсата и адсорби
568
Новый справочник химика и технолога
рованных веществ на поверхности субстрата. Вещества и материалы в виде тонких пленок являются основой микроэлектроники. Микросхемы (чипы) изготавливаются, в частности, путем напыления (конденсации) на субстрат проводящих, полупроводниковых, диэлектрических и др. материалов. В коллоидной химии пленка — это часть фазы, ограниченная с двух сторон границами с другой фазой или с двумя разными фазами (рис. 3.21). В симметричной пленке обе ограничивающие ее фазы одинаковы по природе, т. е. являются двумя частями одной и той же фазы. Различают автономные и неавтономные пленки. Первые могут существовать отдельно — без ограничивающих и формирующих их твердых или жидких фаз с обеих сторон. В то же время они не могут существовать без внешней силы, препятствующей сокращению поверхности пленки.
Мыльная пленка не может существовать без жесткого замкнутого каркаса, на который она натянута. Существование стенок мыльного пузыря также поддерживается принудительно, за счет избыточного давления внутри пузыря. Упоминавшийся ранее адсорбционный слой паров на смачиваемом субстрате формально не подпадает под данное выше определение пленки, так как между ним и средой, из которой он образовался, по определению понятия «адсорбция» границы не существует. В то же время с точки зрения теории полимолекулярной адсорбции слой адсорбата — это пленка конденсата. Так, слой конденсированных паров алюминия на пластине кремния — вполне осязаемая и реально существующая деталь электронных чипов. Таким образом, принятые в настоящее время определение пленки и классификацию пленок не следует считать универсальными и исчерпывающими.
Деление пленок на толстые и тонкие (см. пояснения к формуле 3.2.10) является более однозначным. Толстая пленка имеет толщину h большую, чем сумма толщин S поверхностных слоев на каждой из границ пленки при их бесконечном удалении одна от другой.
Рис. 3.21. Пленка фазы в зазоре между двумя частями фазы: s, v — фазы пленки; h — толщина пленки; ст™ — натяжение границы фаз s и v; ст'™ — натяжение границы пленки;
8 — толщина поверхностного слоя на границе фаз s и v; П — расклинивающее давление пленки
Толщина тонкой пленки меньше, чем указанная сумма толщин поверхностных слоев. Это могут быть адсорбционные слои ПАВ, слои структурно измененной среды и др. В симметричных толстых пленках h > 2S, а в тонких пленках h < 2S (рис. 3.21).
Особый случай представляют тонкие пленки равновесной толщины й0. Такая пленка не нуждается в наличии каких-либо внешних сил для сохранения неизменной ее толщины. Напротив, произвольная (не равновесная) толщина пленки может поддерживаться только внешней силой, предотвращающей самопроизвольное увеличение или уменьшение толщины пленки. Удельная (на единицу площади) граница пленки и нормальная к ней сила, необходимая для сохранения неизменной толщины пленки, называется расклинивающим давлением пленки (П). Оно может быть положительным, отрицательным или равным нулю. Пленка, которая стремится к самопроизвольному увеличению толщины, создает положительное расклинивающее давление (П > 0) на ограничивающие ее тела. Пленка, которая стремится к самопроизвольному уменьшению своей толщины, создает отрицательное расклинивающее давление (П < 0). У равновесной пленки П = 0.
Состояние пленки задается тремя ее параметрами: толщиной h, натяжением оп и расклинивающим давлением П.
При увеличении толщины пленки от равновесной величины ho до произвольной h необходимо совершить работу, которая определяется формулой
^12(Л) - О1/2(Л) - а1/2(М- (3-2.26)
При изменении толщины от h до h + dh работа адгезии равна Wx2 = (fifoi/2 / dh) dh.
Производная <7oiy2 I dh с обратным знаком и есть сила П, которую необходимо приложить извне для поддержания системы в равновесии при произвольной величине зазора h, разделяющего фазы 7 и 2 во флюидной среде v:
П(Л, с) = -d<3xf2(h, с) / dh. (3.2.27)
Обозначение (Л, с) подчеркивает, что расклинивающее давление, адгезия и натяжение пленки в зазоре между двумя фазами 1 и 2 являются не только функцией величины зазора h, но также и концентрации ПАВ в растворе с.
Поскольку самопроизвольный процесс возможен только при уменьшении энергии Гиббса, то натяжение пленки 01^2, как и адгезия Wy2, должны уменьшаться при любом самопроизвольном изменении толщины пленки, что и обеспечивается отрицательным знаком в правой части формулы (3.2.27).
Взаимодействие поверхностей или коллоидных частиц через разделяющую их прослойку чаще характеризуют энергией U(h) расклинивающего давления. Отсчет энергии принято вести от состояния, соответствующего бесконечно большой толщине прослойки. В таком случае U(h) = -W(h) и, следовательно, справедливо уравнение
ад = о1/2(Л)-о1/2(со). (3.2.28)
Физика, химия и технология дисперсных систем
569
Отыскание функции П(Л, с) или С/(Л) является одной из основных задач теории поверхностных явлений и практической коллоидной химии. Для этого необходимо располагать уравнением состояния пленки, которое обычно получается на основе той или иной модели строения поверхностного слоя и пленки. Двойной электрический слой является в этом отношении наиболее благодатным объектом, поскольку его строение хорошо изучено теоретически и экспериментально. Как ни парадоксально, но наименее доступен исследованию самый простой случай, когда растворенный компонент f вообще отсутствует. Формулы (3.2.27) и (3.2.28) полностью сохраняют свою силу и в этом случае. Разность натяжений толстой и тонкой пленок в этом случае может быть целиком обусловлена различием в структуре слоев среды, расположенных на разных расстояниях от межфазной границы. Понятно, что структурную неоднородность поверхностного слоя самой среды исследовать значительно труднее, чем неоднородность ее состава, обусловленную адсорбцией ПАВ или ионов.
3.3. Термодинамика поверхности
3.3.1. Фундаментальное уравнение и термодинамические потенциалы
Фундаментальное уравнение термодинамики (3.3.1) выражает закон сохранения энергии: изменение энергии системы равно сумме энергий, переданных ей различными воздействиями:
dU = j^Xidxi. (3.3.1)
I
Здесь X, и х, — параметры состояния системы, причем Xt — интенсивные параметры, или обобщенные силы, действующие на систему, х, — экстенсивные параметры (обобщенные координаты), характеризующие состояние системы.
Уравнения термодинамики можно рассматривать как обобщение некоторых уравнений и законов механики, а также связанных с ними понятий. Прежде всего это относится к понятиям силы, координаты, энергии, работы. Работа определяется как произведение Fdl силы F на путь dl (изменение dl координаты /-той точки, к которой приложена сила). Совершенная силой работа может быть преобразована в потенциальную (внутреннюю) энергию U тела, над которым совершена работа. При изменении координаты сила может оставаться постоянной (например сила веса тела F, если совершается работа U = Fh его подъема на некоторую высоту Л), а может изменяться (например сила растяжения пружины). В последнем случае актуальна задача нахождения зависимости силы от координаты (в данном случае от длины пружины), которая в общем случае может оказаться не столь простой и очевидной, как в случае пружины, например зависеть не только от собственной сопряженной координаты, но и от других координат или сил. Упомянутая зависимость есть не что иное, как
уравнение состояния объекта, причем оно может быть представлено и в форме зависимости энергии от координаты, и в других формах. Примерами немеханических сил и связанных с ними (сопряженных) координат могут служить электрический потенциал Ue и заряд конденсатора q, напряженность магнитного поля Нт и индуцированный им магнитный момент тела <р. Еще одним важным примером механической и одновременно типично термодинамической пары сопряженных параметров сила—координата являются давление Р и объем газа V.
Природа и физический смысл некоторых термодинамических параметров интуитивно или по аналогии с механикой известен и понятен. Таково, например, давление и объем тела (газа). Смысл же других параметров не столь очевиден. К их числу относится энтропия 5 и химический потенциал ц. Дальнейшее изложение направлено не столько на выяснение физического смысла этих понятий, сколько на обоснование их необходимости и выяснение эмпирического смысла. Эмпиризм в науке не всегда считается ее позитивной стороной, но термодинамика — это наука эмпирическая по своей сути. Эмпирический, теоретический или какой-то иной подход к проблеме требует в первую очередь введения однозначного определения тех величин и понятий, которые используются при обсуждении проблемы. Сложность в том, что в одни и те же понятия иногда вкладывается разный смысл. Во избежание недоразумений далее будем исходить из того, что определить смысл некоторой величины — указать, как ее выразить через другие величины, смысл которых известен. В частности, это значит указать закон (уравнение, формулу), в который определяемая величина входит наряду с известными величинами. Примером может служить второй закон механики, служащий определением понятия «масса тела», закон Ома, служащий определением понятия «электрическое сопротивление», и т. д. Этот же принцип должен быть положен в основу определения смысла упомянутых выше понятий: энтропии и химического потенциала (/-го компонента системы). Уравнение (3.3.1) как раз для этого и предназначено.
Каждой силе Xt соответствует одна, сопряженная с ней координата х„ так что в уравнении (3.3.1) к — это число пар таких сопряженных параметров. Любая из обобщенных сил X, изменяет энергию системы на величину dUt = Xjdxt в том и только в том случае, если ее действие вызывает изменение сопряженной координаты х,. В простейшей термодинамической системе — газе — это его давление Р и объем V, а также температура Т и некоторый сопряженный с ней экстенсивный параметр (координата) S, который получил название «энтропия». Таким образом, энтропия вводится как сопряженный с температурой параметр, предназначенный для того, чтобы можно было отразить те изменения состояния системы (газа), которые возникают при передаче системе некоторого количества энергии 8Q = TdS в форме теплоты Q. Этого определения энтропии доста
570
Новый справочник химика и технолога
точно, чтобы, не вдаваясь в физический смысл величины S, корректно использовать ее в уравнениях, описывающих равновесие и обратимые изменения состояния термодинамической системы.
Подобным же образом может быть определен смысл химического потенциала ц, некоторого z-ro компонента термодинамической системы. Он является параметром (обобщенной силой), сопряженным с количеством nt данного компонента в системе. Энергия системы U может быть изменена на величину dUt = \^!dnl путем увеличения (или уменьшения) количества п, содержащегося в ней z-ro компонента или на величину dU='^\jLldni при изменении содержания каждого из к компонентов системы. Физический смысл параметра ц, при этом достаточно очевиден — это удельная энергия компонента при тех условиях, при которых он уже присутствует в системе. Здесь и далее символ суммирования «Е» без указания под ним и над ним пределов суммирования означает, что оно проводится по всем к компонентам системы. Важно, что изменить содержание одного компонента можно только его подводом (или отводом) извне, тогда как изменение содержания нескольких компонентов может произойти за счет их взаимных превращений (химической реакции) внутри системы.
В общем случае силы X, зависят от координат и при наличии уравнения состояния системы могут быть через них однозначно выражены, после чего в уравнении (3.3.1) останутся только к координат. Тогда оно в принципе может быть проинтегрировано, поэтому полная энергия системы (или внутренняя энергия системы, если не учитывать ее энергию во внешних полях и кинетическую энергию движения всей системы в целом) может быть представлена как однозначная функция координат:
U= U(xx,x2,... хк). (3.3.2)
Согласно уравнению (3.3.2), полному дифференциалу энергии соответствует формула
(3.3.3)
Из сравнения уравнений (3.3.1) и (3.3.3) следует выражение
(3.3.4)
дх,
Это формула выражает важное свойство внутренней или полной энергии: ее частные производные по координатам равны сопряженным с этими координатами обобщенным силам, т. е. силы могут быть вычислены, если известна зависимость полной энергии от экстенсивных параметров х,. Если при изменении размера (массы) системы в 1 раз в то же число раз изменяются ее энергия U и координаты х„ то это означает постоянство X, при изменении хь что позволяет проинтегрировать выражение (3.3.1) и получить формулу
и = £ Х,х,. (3.3.5)
I
Если изменить энергию на величину dU уравнения (3.3.1), но при этом не фиксировать величину Xh т. е. не принимать мер по стабилизации температуры, давления и т. д. в ходе процесса, вызывающего указанное изменение энергии, то, согласно формуле (3.3.5), полному дифференциалу энергии отвечает уравнение
к к
dU = £ Xtdx, + £ xidXi. (3.3.6)
1 1
Из сравнения формул (3.3.1) и (3.3.6) следует, что в процессах, когда обобщенные силы не остаются постоянными, они не могут изменяться произвольно — между их изменениями имеется связь в виде уравнения Г иббса — Дюгема:
£*А,=0. (3.3.7)
1
Внутренняя энергия U может быть представлена как функция интенсивных (X) или смешанных (Xt и х,) параметров. При этом она теряет свойства, выражаемые формулой (3.3.4).
Функции состояния, частные производные от которых по параметрам, интенсивным или экстенсивным, позволяют вычислить сопряженные с ними параметры состояния, называются характеристическими функциями. Ценное свойство характеристических функций состоит в том, что работа обратимого процесса выражается через изменение этих функций и не зависит от пути, по которому совершается процесс. Функции с такими свойствами называются термодинамическими потенциалами. В состоянии равновесия термодинамический потенциал имеет экстремальную величину.
Вид характеристической функции Т определяется условиями, которые наиболее удобно контролировать и регулировать в том или ином процессе. В общем случае среди них будет f обобщенных сил Xh где z = 1,2, ... /и к -/координат х7, где/ =f+ 1,/+2 ... к, т. е.
Т = ОД,Х2,... Xfi xf+ b х/+ 2... хк). (3.3.8)
Полный дифференциал функции состояния выражается через ее частные производные по всем независимым переменным:
^ат
1 sxi
д^ат
м Sr,
(3.3.9)
f
Введем дополнительную функцию ^Хр^ и най-1
дем ее полный дифференциал:
d(£xix.)=£xidxl+ftx,dX,. 1 1 1
Физика, химия и технология дисперсных систем
571
Если добавить к левой и правой частям этого урав-k
нения сумму Xidxj, которой не хватает для 7+>
образования полного дифференциала внутренней энергии (3.3.1), то получается выражение:
7 k f
d(^Xixi) + YxidXi=du + LxidXr i y+i 1
Его перегруппировка приводит к формуле:
d(U - £ ХЛ) = *4, + £ X,dx,. (3.3.10)
1 1 7+1
Левая часть уравнения (3.3.10) есть полный дифференциал, т. е. величина под знаком дифференциала представляет собой некоторую функцию состояния. Поскольку правая часть уравнения содержит дифференциалы именно тех параметров, которые были выбраны в уравнении (3.3.8) как независимые переменные функции Т, то она и есть искомый термодинамический потенциал:
V.J,,.../;,»,.,,!/.,,.. X,) = U-^X,X,. (3.3.11) 1
Правая часть уравнения (3.3.10) — его полный дифференциал:
7 к
-2>А,+2>А- (3.3.12)
1 7+1
Из сравнения формул (3.3.9) и (3.3.12) получаются соотношения:
ат х ат дХ, ’ дх, ‘
(3.3.13)
В частном случае при f- 0 формула (3.3.12) переходит в уравнение (3.3.1), и Т = £7.
3.3.2. Уравнения термодинамики гетерогенных систем
При отсутствии электромагнитных полей и пренебрежении действием силы тяжести состояние гетерогенной системы определяется объемами фаз И,, энтропией St, составом (числом молей п, компонентов), величиной межфазной поверхности А и сопряженными с ними интенсивными параметрами — давлениями Р} в фазах, температурой Т, химическими потенциалами ц, компонентов и поверхностным натяжением ст на межфазных границах. В дальнейшем будут рассматриваться двухфазные гетерогенные системы.
В системах с плоской границей между фазами давления Pj в фазах одинаковы, а в случае искривленной межфазной границы давление Л во внутренней (дисперсной) фазе больше давления Р2 во внешней фазе (среде) на величину капиллярного давления Рк. Фунда
ментальное уравнение (3.3.1) при наличии искривленной межфазной границы принимает вид
dU = -PxdVx -P2dV2 + TdS + adA + ^dn*. (3.3.14)
В случае плоской границы:
-PxdVx-P2dV2 = ~PdV,
Г= И + V2, dV= dVx + dV2, РХ = Р2 = Р.
При отсутствии межфазных границ справедливо уравнение:
dU = -PdV+ TdS + ^dnh (3.3.15)
Внутренняя энергия в этих двух случаях будет равна и описывается уравнениями:
U = -PXVX-P2V2 + TS+ аА + Еца, (3.3.16)
V = -PV + TdS + ^n,. (3.3.16а)
Уравнение Гиббса — Дюгема (3.3.7) для гетерогенной системы с искривленными границами принимает вид:
-VxdPx - V2dP2 + SdT + Ada + 0. (3.3.17)
В случае плоской границы справеливо уравнение:
- VdP + SdT + Ada + Wp, = 0. (3.3.18)
В гомогенной системе (А = 0) оно сводится к формуле:
-VdP + SdT + = 0. (3.3.18а)
В идеализированной (гипотетической) гетерогенной системе поверхностное натяжение как работа деформации поверхностного слоя с особыми механическими свойствами (т. е. с нарушением закона Паскаля) отсутствует. Для такой системы приведенные выше уравнения не будут содержать слагаемых аА, adA, Ada. В частности, вместо уравнения (3.3.18) следует записать уравнение, в котором верхний индекс h указывает на принадлежность величины к гипотетической системе:
-VdP + tfldT+ Yn-d^ =0. (3.3.19)
Вычитанием уравнения (3.3.19) из уравнения (3.3.18) получается уравнение:
tfdT + Ada + = 0. (3.3.20)
Здесь<S° = 5'-SAHHa = H-«A — поверхностные избытки энтропии и компонента в гетерогенной системе. Идеализированная система имеет тот же объем, что и реальная, поэтому в уравнении (3.3.20) слагаемое типа VdP отсутствует.
В случае искривленной межфазной границы уравнение Гиббса — Дюгема для идеализированной системы
* Здесь и далее для краткости в знаке суммирования пределы суммирования опущены, если, как в данном случае, оно распространяется на все к компонентов системы.
572
Новый справочник химика и технолога
совпадает с уравнением (3.3.19), поэтому при вычитании его из уравнения (3.3.17) получается уравнение:
-V.dP^^dT+A da + Z<^ = 0. (3.3.21)
Вид первого слагаемого (~VidPK) определяется условием сопряжения по давлению реальной системы с окружающей средой. В данном случае принято, что контролируется давление Р2 в фазе 2, тогда давление Рх в фазе 1 повышено, вследствие кривизны поверхности, на величину капиллярного давления Рк. Соответственно этому V\dP\ + V2dP2 = VdPi + V\dPK, где V= Vx + V2, что и приводит к уравнению (3.3.21). Вопрос о сопряжении по давлению идеализированной системы или реальной системы с плоскими межфазными границами не возникает в силу равенства давлений во всех фазах.
Если поделить все слагаемые уравнения (3.3.20) на площадь межфазной границы А, то величины 5 = S’/А и Г\ = п° / А представляют собой удельные (на единицу поверхности) избыточную энтропию гетерогенной системы и адсорбцию /-го компонента этой системы. Тогда уравнение Гиббса — Дюгема (3.3.20) преобразуется в уравнение
do + SdT + 2Г, 4i, = 0. (3.3.22)
Оно называется адсорбционным уравнением Гиббса и при постоянной температуре сводится к уравнению, которое называется уравнением изотермы адсорбции Гиббса:
dc = -YTidpl. (3.3.23)
Для двухкомпонентного раствора можно записать:
da = -Г !ф1 - Г2412.
Это уравнение можно решить, если адсорбцию одного из компонентов Г2 относить к такому состоянию поверхности, разделяющей фазы, которое является эквимолекулярным по отношению к другому компоненту (растворителю), например, что Г] = 0. Если обозначить в этом случае Г = Г2 и ц = ц2, а также считать смесь (раствор) компонентов 1 и 2 идеальной, т. е. что ц = ц° + 7? Tin с, где с — концентрация растворенного о
вещества и ц — его стандартный потенциал, то получается уравнение изотермы адсорбции Гиббса в наиболее пригодном для практических вычислений виде:
Г = -—. (3.3.24)
R Т de
Уравнение изотермы адсорбции Гиббса (3.3.24), как и другие термодинамические уравнения, используется только для преобразования одной зависимости, получаемой, как правило, экспериментально, в другую. В данном случае конкретный вид изотермы адсорбции Г(с) определяется зависимостью натяжения от концентрации раствора.
Проведем анализу капиллярных явлений термодинамическим методом. Для наглядности будем иметь в виду уже рассмотренную ранее систему капель однокомпонентной жидкости в среде ее насыщенного пара.
Каждую каплю или их совокупность можно считать самостоятельной однофазной термодинамической системой. Наличие сферической границы в этом случае отражает условие сопряжения жидкой фазы 7 с окружающей средой, каковой является фаза 2. Действие поверхностного натяжения на жидкую фазу в таком случае сводится только к увеличению давления в жидкой фазе на величину Рк = 2о / г по сравнению с равновесным давлением Р в фазе 2. Уравнение Гиббса — Дюгема (3.3.15) для жидкой фазы будет таким же, что и для гомогенной системы. При постоянстве температуры VdP = nxdp или dp = VmdP, так как V! пх есть молярный объем Vm вещества жидкой фазы. При изменении радиуса капли г давление в капле изменится на величину, равную изменению капиллярного давления dP ~~ 2о {dr I г2). При интегрировании уравнение dp--2<jVm(dr / г2) в пределах от г = <ю (плоская граница фаз) до некоторой конечной величины г можно найти приращение химического потенциала жидкого вещества при равновесном переходе жидкости из сплошного состояния в капельное:
p-ps = 2<sVml г (3.3.25)
Здесь ц — химический потенциал в дисперсном (капельном) СОСТОЯНИИ, Us — в сплошном состоянии. Если газовую фазу считать идеальной, то в ней ц = ц° + RT\vtP.
В состоянии равновесия жидкости и пара Р есть давление насыщенного пара Рг над искривленной поверхностью капель, а химические потенциалы в обеих фазах равны. Следовательно, в формуле (3.3.25) ц = ц°+ 7?71пРг и = ц° + 7?71п/’5, где Ps — давление насыщенного пара Ps над плоской поверхностью жидкости. Из равенства химических потенциалов в паре и жидкости вытекают соотношения между упругостями насыщенных паров над плоской и искривленной поверхностями (уравнение Кельвина):
ЯТ(1пРг-1пР,) = К.Р, (3.3.26) или
С = Р, = Р, e^VjRTr).
В случае вогнутой поверхности жидкости (газовые пузыри в жидкости) кривизна поверхности К отрицательна (К = -2 / г), и тогда упругость пара в газовых полостях понижена до величины
Ps ехр(-2оГ„Л / RTr).
В общем случае соотношение между упругостью насыщенных паров над плоской и искривленной поверхностями можно предствить уравнением
Pr = Ps exp (Ко Vm I RT). (3.3.27)
Здесь К и Рк в уравнении (3.3.26) могут принимать как положительные, так и отрицательные значения.
Физика, химия и технология дисперсных систем
573
3.3.3. Работа образования поверхности
В термодинамике работа — это форма обмена энергией между термодинамической системой и внешней средой. Энергия является в этом смысле внутренним параметром и не входит в число обобщенных координат, учитываемых при вычислении работы. В связи с этим уравнение (3.3.1) может быть представлено в форме tZ17=8IK+8g, где 81К— элементарная работа процесса, 8g — энергия, переданная в форме теплоты. Знак 8 означает, что 8 W и 8g не являются полными дифференциалами, т. е. приращения функций зависят от условий, при которых совершается процесс. Условия протекания процесса, или набор параметров состояния, которые при этом контролируются или регулируются, определяют вид характеристической функции состояния, через которую может быть выражена работа обратимого процесса. С математической точки зрения, контролируемые параметры — это параметры, выбранные в качестве независимых переменных, характеризующих состояние системы и изменение ее состояния на всех этапах процесса. Число независимых переменных равно числу степеней свободы системы, которое определяется правилом фаз Гиббса. Это число меньше, чем количество параметров состояния, через которые можно описать состояние системы, на число дополнительных связей, налагаемых на систему, т. е. на величину, равную числу уравнений, связывающих между собой параметры состояния.
Подавляющее большинство процессов в термохимических системах (системах, которые могут обмениваться энергией только через механическую работу, массообмен, теплообмен) контролируется составом, температурой и объемом или составом, температурой и давлением. Соответственно этому характеристическими для гомогенных систем будут функции F (Т, V, п,) и G (Т, Р, п,), а для гетерогенных систем — те же функции F (Т, Vj, nh A), G (Т, Pj, nh А), но с расширенным набором параметров. Здесь и, — число молей z-x компонентов в системе, Vj, Pj — объемы и давления j-х фаз, А — площадь межфазных границ.
Согласно уравнениям (3.3.11), (3.3.12) и (3.3.16), для гомогенных систем справедливы соотношения:
F(T, V, ni) = U-TS = - V+Zm, nh
dF (T, V, ni) = SdT-PdV + Ym, dnh
G (T, V, n,)=U-TS + PV=-PV+ Ym, щ, dG (T, V, n.) = SdT + VdP + Em, dn,.
(3.3.28)
Для гетерогенной двухфазной системы можно запи-ать уравнения:
F(Т, Vj, nb А)= U-TS = -PiKi -P2V2 + aA + Ym.n,, dF (T, Vj, n„ A) = SdT-P}dV} - P2dV2 + adA + Xm, dnh
G(T, Pj, щ, A)=U-TS + PlVi+P2V2 = -PKVt + oA+ dG (T, Pj, щ, A) = SdT-PKdVx + VdP2 + adA+ bmt dnb
(3.3.29)
В отличие от потенциала Гельмгольца F (энергии Гельмгольца) потенциал Гиббса G = F + PjV} (энергия Гиббса) определяется для гетерогенной системы неоднозначно. При наличии искривленных границ давления Р} отличаются в разных фазах на величину капиллярного давления Рк, и тогда в качестве независимой переменной следует брать давление в одной из фаз. Легче контролировать давление Р2 во внешней фазе 2 (дисперсионной среде) и, следовательно, целесообразно его использовать как фактическую независимую переменную характеристической функции, что и наблюдается для dG в серии уравнениий (3.3.29). Сопряженной с этим давлением координатой выбран общий объем системы V= V\ + V2. Внутренняя фаза 1 имеет при этом избыточный запас механической энергии благодаря повышенному давлению в этой фазе, что и отражает слагаемое P*dV\. В формуле (3.3.16) присутствуют слагаемые -P\V\ и -P2V2. При вычислении G по правилу (3.3.11) из них следует вычесть величину -P2(V\ + V2), чтобы получить в уравнениях (3.3.29) -PKVj, т. к. Р\= Р2 + Рк. Процедура вычисления dG по правилу (3.3.12) с учетом тех же соотношений между объемами фаз и давлениями в фазах дает указанный в уравнениях (3.3.29) результат. В случае гетерогенных систем с плоской межфазной границей из уравнений (3.3.29) исключаются величины, содержащие Рк.
Величины Рк и А не являются независимыми. Каждая определяется при фиксированном объеме V} дисперсной фазы 1 с размером частиц г в соответствии с известными формулами. Это пример упомянутого выше снижения числа степеней свободы по сравнению с числом независимых переменных гетерогенной системы. Разумеется, что в термодинамических формулах, предназначенных для конкретных расчетов, параметры Рк и А должны быть выражены через размер частиц г.
Формулы (3.3.29) позволяют уточнить определение натяжения о межфазной плоской границы как работы образования поверхности в обратимых процессах с помощью формул:
o-(dF/dA)TVn-(dG/dA)TP2^n (3.3.30) или
a = (dG/dA)Tpn . (3.3.30а)
Фактически в разнообразных реальных процессах, связанных с изменением площади границ, не всегда удается или невозможно поддерживать постоянство указанных в формулах (3.3.30) и (3.3.30а) параметров состояния, поэтому работа упомянутого процесса не
574
Новый справочник химика и технолога
обязательно совпадет с работой изменения поверхности adA. Типичными примерами таких процессов являются диспергирование жидкости и конденсация пара. Рассмотрим их на примере простейшей гетерогенной однокомпонентной системы жидкость—пар.
Диспергирование сплошной жидкой фазы 7 на капли радиусом г сопровождается ростом межфазной поверхности А и, согласно формуле Лапласа, увеличением давления Pi в жидкой фазе, т. е. изменением состояния объемной фазы 7, которое может быть выражено через изменение ее химического потенциала ц. При наличии равновесия жидкости с газовой фазой на ту же величину изменится и химический потенциал в газовой фазе. Последний в случае идеальной газовой фазы легко связать с давлением Р2 в этой фазе (3.3.27).
Если в качестве независимых параметров принять А и V (при постоянстве Т и и), то трудно будет выбрать какую-либо предпочтительную величину конечного объема, который должна занять система после увеличения давления в газовой фазе при диспергировании, т. е. работа диспергирования при переменном объеме может быть произвольной и поэтому не представляет интереса. При контроле параметров А и Р2 и постоянстве Т и п работа диспергирования Wd равна изменению энергии Гиббса гетерогенной системы AG. Если предположить, что в исходном монолитном состоянии жидкой фазы можно пренебречь ее поверхностной энергией аА и капиллярным давлением, то из уравнений (3.3.29) при V= const получается следующее соотношение:
Wd =&G = -PKV\ + <зА + иАц.
Согласно уравнению (3.3.25), Ац = РкРт. Если п = п\ (в случае малолетучей жидкости), то иАц = Рк^ъ и при постоянстве Т, V и п приведенное выше соотношение сводится к формуле
Wd=aA. (3.3.31)
При получении капель из газовой фазы необходимо перевести пар в метастабильное состояние (перенасыщенный пар), из которого система самопроизвольно может перейти в гетерогенное состояние, которое не обязательно окажется устойчивым. Представляет интерес работа Wz этого перехода, так как именно от нее зависят параметры образующейся гетерогенной системы при заданном перенасыщении гомогенной системы. Например, число зародышей новой фазы q, возникающих путем флуктуации плотности пара, связано с работой образования зародыша Wz законом Больцмана:
q = qoexp(-Wz/kT). (3.3.32)
Здесь qQ — константа, имеющая смысл вероятности образования зародыша того же размера при Wz = 0, т. е. за счет случайного попадания нужного числа частиц в элемент объема, численно равный объему будущего зародыша. Объем и работа образования зародыша опре
деляются при прочих равных условиях размером зародыша, поэтому в формуле (3.3.32) константы qQ и Wz не являются независимыми величинами.
Если выбрать в качестве независимой переменной давление (давление в газовой фазе при переходе системы в гетерогенное состояние), то работа образования капли Wz равна изменению энергии Гиббса AG. Гомогенная перенасыщенная система с фиксированным количеством вещества п в ней характеризуется, согласно уравнениям (3.3.28), химическим потенциалом ц, а гетерогенная — величиной потенциалов обеих фаз щ и ц2, а также, согласно уравнениям (3.3.29), величинами Рк, И и А. При п - const и после вычитания уравнений (3.3.28) из уравнений (3.3.29) получается следующее выражение:
AG = -PKV\ + оА + ц1и1 + р2п2 - рп.
При равновесии Ц1 = р2, И = Аг / 3 и, согласно уравнению (3.3.25), РкИ = 2сА / 3. Тогда справедливо уравнение, в котором индексы 1 и 2 относятся к конденсированной и газовой фазам соответственно:
AG = аА / 3 + (ц2 - р)и. (3.3.33)
Здесь [1 — химический потенциал, п — число молей газовой фазы исходной гомогенной системы.
В самом начале процесса конденсации, когда в большом объеме перенасыщенного пара образуется небольшое число капель новой фазы, химический потенциал ц (давление пара) остается практически неизменным, т. е. Ц- Из уравнения (3.3.33) получается классическая формула Гиббса для работы образования новой фазы:
W* = gA!3. (3.3.34)
Количество вещества в новом агрегатном состоянии (жидкости, в данном случае) здесь представлено только площадью А поверхности новой фазы. Так как механическая работа, необходимая для увеличения поверхности при неизменности других параметров состояния, равна <5А, то при конденсации пара часть этой работы 2оА / 3 совершается за счет внутренних ресурсов — энергии конденсации части перенасыщенного пара. Остальная часть работы аА / 3 приходится на работу внешних сил над системой.
Формула (3.3.34) не дает ответа на вопрос о размере и устойчивости образующихся капель по отношению к возможному самопроизвольному их росту или испарению. В поисках ответа на этот вопрос рассмотрим эволюцию системы из капель и пара, которая может быть неравновесной в условиях постоянства объема, температуры и количества вещества в системе. Предполагается, что в результате флуктуаций концентрации, которые по мере приближения системы к условиям сосуществования двух разных фаз очень сильно возрастают, возникают капли разного размера. Их летучесть Рг
Физика, химия и технология дисперсных систем
575
(давление насыщенного пара над поверхностью капель) определяется формулой Кельвина (3.3.26). Мелкие капли с упругостью большей, чем давление перенасыщенного пара в системе, будут испаряться. Крупные капли окажутся в перенасыщенном по отношению к ним паре и, следовательно, будут увеличиваться в размере за счет конденсации на них пара, и только капли некоторого критического радиуса гс с упругостью пара, равной давлению пара рассматриваемой неравновесной гетерогенной системы, будут находиться в состоянии неустойчивого равновесия. Равновесие неустойчиво, так как небольшие флуктуации размера капель в ту или иную сторону приведут к их испарению или росту, что сопровождается уменьшением энергии Гиббса. Таким образом, образование капель критического размера отвечает максимуму приращения энергии Гиббса, или максимуму работы образования зародышей новой фазы (капель жидкости). Размер этих капель определяется формулой Кельвина (3.3.26), где Рг = Р2 — давление пара в гетерогенной системе. Проблема в том, что его невозможно предсказать на основе оговоренных выше условий.
На самом начальном этапе конденсации число образовавшихся капель настолько мало, что можно пренебречь изменением давления перенасыщенного пара, и тогда Рг- Р2 = Р, так что на основе данного выше описания свойств критического зародыша его размер вычисляется по формуле Кельвина (3.3.26). В общем случае давление Р2 определяется количеством вещества и2, оставшегося в газообразном состоянии после образования капель. Поэтому необходимо определить долю общего объема исходной системы, приходящегося на одну каплю новой фазы или, что то же самое, число таких капель во всей системе. Эта задача пока не имеет решения, по крайней мере в приемлемой для технологии форме, однако можно ориентироваться на достаточно очевидные качественные критерии при определении условий, способствующих получению нужного результата.
При постоянстве объема системы образование, испарение и увеличение размера капель будут сопровождаться повышением или снижением давления пара в газовой фазе. Величина этого эффекта существенно зависит от соотношения между объемом пара V2 и жидкости И в системе. Очевидно, что при малом объеме пара, приходящемся на одну каплю, ее испарение или рост могут сильно повысить или, соответственно, понизить давление в газовой фазе, что прервет процесс испарения или конденсации пара на каплях. В таких условиях размер капель стабилизируется, равновесие в системе капли—пар становится устойчивым. В обратном случае (Г2 » И) испарение и конденсация практически не меняют давление в газовой фазе, и система будет неустойчивой в отношении неограниченного увеличения размера капель. Таким образом, возникновение большого числа зародышей новой фазы в перенасыщенной гомогенной системе благоприятствует образованию
высокодисперсной гетерогенной системы. Практически это можно осуществить путем создания очень сильного перенасыщения. Оно сравнительно легко достигается при химическом синтезе труднорастворимых веществ из концентрированных растворов исходных реагентов.
В природе и технике широко распространены процессы гетерогенного образования новой фазы. Они, например, играют основную роль в получении тонкопленочных изделий микроэлектроники методом вакуумного напыления, электрохимическими и другими методами, в метеорологии и т. д. Формула Гиббса (3.3.34), если она относится и к работе образования зародыша новой фазы Wz на поверхности твердого тела (на субстрате), может быть записана в виде уравнения
И7=оР7г. (3.3.35)
Здесь V — объем линзы, г — радиус кривизны ее поверхности на границе с паром, а — натяжение границы жидкость—пар. Так как при одинаковом радиусе г сферической капли и линзы, лежащей на плоской поверхности, объем последней меньше, то соответственно снижается и работа образования зародыша на поверхности. При постоянстве объема линзы конденсата работа конденсации тем меньше, чем меньше краевой угол смачивания 0. Приняв во внимание геометрические соотношения, связывающие V, г и 0, можно получить формулу Фольмера:
Wzs = Wz° (1 / 2 - 3 cos 914 + cos3 9 / 4). (3.3.36)
При 0 0 Wzs - 0 и при 0 - тт Г -> Wz°. В дей-
ствительности при 0 —» 0 существенную роль начинает играть линия соприкосновения трех фаз (длина L периметра смачивания). По аналогии с плоской границей раздела фаз она характеризуется линейным натяжением X. По этой причине при 0 = 0 (полное смачивание) Wz = KL /2. Роль линейного натяжения периметра может быть существенной в высококонцентрированных пенах и эмульсиях, где общая длина линий контакта фаз (ребер двухгранных углов) достигает большой величины. Следует иметь в виду, что при наличии неограниченной поверхности и полного смачивания растекание идет действительно неограниченно, т. е. равновесие в системе недостижимо.
3.3.4. Термодинамика тонких пленок
Если одна из подвижных (флюидных) фаз (газ, жидкость) или ее часть находится в состоянии тонкой пленки, то для термодинамического описания такой системы необходимо ввести еще одну пару сопряженных параметров, с помощью которых можно выразить работу изменения толщины пленки dW=-V\.dh. Один из этих параметров h — толщина пленки, а второй — П — ее расклинивающее давление. Расклинивающее давление — это удельная (на единицу площади одной из сторон пленки), нормальная к границе пленки внешняя (по отношению к системе) сила, которую необходимо при-
576
Новый справочник химика и технолога
дожить для обеспечения неизменности толщины пленки. Сила считается положительной, если она противодействует самопроизвольному увеличению толщины пленки, и отрицательной — в обратном случае.
Сила, стабилизирующая толщину пленки, часто возникает за счет взаимодействия фаз в данной термодинамической системе, т. е. не является по отношению к ней внешней. Примером могут служить равновесные пленки, рассмотренные в подразделах 3.2.2 и 3.2.3. Тогда расклинивающее давление термодинамической системы равно нулю. Иногда возникает необходимость рассматривать часть системы — только саму пленку, образующую ее флюидную фазу, и поверхности, ограничивающие пленку. В этом смысле часто употребляют понятие «некоторая составляющая расклинивающего давления пленки», которое является одним из слагаемых полного расклинивающего давления гетерогенной системы.
Гидростатическая интерпретация расклинивающего давления пленки П = Ph - Р представляет его как разницу гидростатических давлений в пленке (в щели) Ph и давления Р в той фазе, продолжением которой является пленка.
Необходимо обратить внимание на то, что расклинивающее давление пленки формируется с участием той субстанции, из которой она состоит, и ограничивающих ее фаз, поэтому даже мысленное их отбрасывание для выделения одной из составляющих давления пленки не всегда корректно. В дальнейшем будут рассмотрены примеры, в которых одну из сил можно считать внешней по отношению к рассматриваемой системе, или будет оговорено разделение общего расклинивающего давления на якобы независимые слагаемые (теория ДЛФО).
Рассмотрим термодинамическую систему, состоящую из однокомпонентной флюидной фазы, имеющей объем V при давлении Р, в которую погружены две пластины, разделенные плоскопараллельным зазором толщиной h. Площадь одной стороны пластины равна А. На пластины, кроме всестороннего давления Р, действует внешняя сила F = ГЫ, обеспечивающая постоянство указанной толщины пленки.
Фундаментальное уравнение (3.3.1) для пленки имеет вид:
dU- -PdV+ TdS + pdn + 2adA - llAdh. (3.3.37)
Для той же системы, но с гипотетическими пластинами, не имеющими натяжения поверхности и избытка компонента на ней, в уравнении (3.3.37) отсутствуют два последних слагаемых. Количество подвижного компонента п = п - 2ГЛ отличается на величину поверхностного избытка п = 2ГА от значения п для системы с реальными пластинами, поэтому избыток энергии будет:
dlf = TdS1 + pdn + 2odA - ndh.
Интегрирование этого уравнения при постоянстве интенсивных параметров дает:
Если независимыми переменными являются параметры Т, ц, о, А, а их функцией является соответствующий термодинамический потенциал Т = Т(Г, ц, су, h) (уравнение (3.3.11)), то получается уравнение равновесия системы, означающее равенство нулю полного дифференциала (3.3.12) этого потенциала:
SdT - ndp + 2Ad^ + HAdh = Q. (3.3.38)
При постоянстве температуры и после деления на 2Л уравнения (3.3.38) получается формула
tZo = -Гф - П dh/2. (3.3.39)
Здесь, согласно уравнению (3.3.13), а также ранее оговоренному знаку обобщенной силы X, = П, сопряженной с координатой h,
Г = -{d<31 dp)Th, (3.3.40)
П = -2(с/о/ dh)T>Vi. (3.3.41)
Этот же результат можно получить, сравнивая уравнение (3.3.39) с выражением для полного дифференциала поверхностного натяжения, которое при постоянстве температуры следует считать в данном случае функцией двух переменных ц и А:
des = (tZo I dp)h dp + (<Zo / tZA)g dh.
Следует обратить внимание на то, что уравнение (3.3.40) указывает на применимость уравнений изотермы адсорбции Гиббса (3.3.23), (3.3.24), полученных при бесконечной толщине пленки, и к случаям адсорбции в тонких пленках при h - const, а также, следовательно, и к тонкопористым адсорбентам. Однако адсорбционная емкость тонкопористых адсорбентов уменьшается с увеличением размера молекул адсорбируемого компонента. Это следует из соотношения (3.3.40), так как при прочих равных условиях (tZo/tZp) зависит от А. Очевидно, что чем меньше А/8, где 8 — размер молекул, тем меньше (tZo/tZp) и, следовательно, Г, по крайней мере при А < 8. При дифференцировании Г по А при ц = const иПпоц при А = const уравнений (3.3.40) и (3.3.41) и сравнении результатов получаются следующие равенства:
(<Г / dh) T^ = (dn/ dp\ h /2 = -(tZo / ф) dh.
Из них следуют выражения для расклинивающего давления:
<1 = 2 (бГ / dh)T^ dp (3.3.42)
или
П = 2 [f—"I dp.
Kdh) -оо х 'т
Физика, химия и технология дисперсных систем
577
С помощью формул (3.3.42) через адсорбцию и химический потенциал адсорбирующегося компонента можно выразить энергию расклинивающего давления пленки (7:
А Н / jt-A и
1/(А) = -2рй J — U = 2 |[Г(оо)-Г(Л)]</ц. (3.3.43) оо -ОО \ ) -оо
Эту же величину, используя формулу (3.3.41), можно выразить и через поверхностное натяжение границ пленки с помощью уравнения
ОД = 2[ОД - а(оо)]. (3.3.44)
Величины 2о(Л) и 2о(оо) представляют собой натяжения тонкой и толстой пленок, соответственно. Формула (3.3.44) легко обобщается на случай несимметричной пленки, после чего она совпадает с формулой (3.3.29), получившейся при рассмотрении адгезии фаз через пленку. Формула (3.3.44) более универсальна, чем (3.3.43), так как не связана с предполагавшейся одно-компонентностью фазы, образующей пленку. В то же время адсорбция более доступна экспериментальному определению (по адсорбции в тонких порах) и теоретическому вычислению на основе нетермодинамических (модельных) методов, чем натяжение тонкой пленки. Обе формулы (3.3.43) и (3.3.44) находят применение для получения конкретной зависимости энергии расклинивающего давления пленок, которая чаще именуется энергией взаимодействия плоских поверхностей (тел). Для этого необходимы данные о строении поверхностного слоя в разных случаях.
3.4. Поверхностное натяжение и адсорбция
3.4.1. Механизм адсорбции из растворов
Рассмотрим двухкомпонентную двухфазную систему — раствор и его пар, причем для простоты обе жидкости будем считать практически нелетучими. В первую очередь представляет интерес состав поверхностной фазы этой системы.
Из молекулярно-кинетической природы поверхностного натяжения жидкости следует, что в поверхностном слое возникает градиент внутреннего давления Р вдоль нормали х к границе двух фаз (dP/dx) = nF, где п— локальная концентрация молекул растворителя (1/м3), F — сила внутреннего давления, отнесенная к одной молекуле растворителя.
По законам гидростатики на всякое тело объемом И, погруженное в среду с градиентом давления (dP/dx), действует гидростатическая «архимедова» сила V(dPldx), выталкивающая тело в сторону меньшей величины давления Р, т. е. к межфазной границе. Исключений из этого закона нет, он актуален для молекул, рассматриваемых как «погруженные тела», и для градиента внутреннего давления (разумеется, с учетом флуктуационного характера силы, действующей на тела молекулярных размеров). Это утверждение не противо
речит тому, что внутреннее давление отличается от обычного (гидростатического) давления недоступностью первого для измерения с помощью манометра. Недоступность обусловлена не невозможностью поместить манометр внутрь поверхностного слоя, а тем, что поверхность любого датчика давления сама порождает такое же, но противоположно направленное давление. Если находящаяся в поверхностном слое молекула — это молекула растворителя, то действующая на нее «архимедова» сила в точности равна суммарной силе F= V(dP!dx) ее втягивания в глубь жидкой фазы, поэтому полная сила равна нулю. В случае посторонних молекул объемом Vx она отлична от нуля, что и приводит к изменению среднестатистического распределения молекул растворенного вещества относительно межфазной границы, т. е. к адсорбции.
Ограничимся случаем, когда концентрация посторонних молекул в слое мала в сравнении с концентрацией молекул растворителя. Тогда наличие посторонних молекул в растворе и в поверхностном слое практически не изменит внутреннее давление жидкой фазы и его градиент, т. е. действующая на молекулы примеси гидростатическая сила Fa = Vx(dP!dx) и сила ее втягивания в глубь раствора F, не будут зависеть от концентрации примесных молекул в поверхностном слое и в объеме. Полная сила Fx = Fj-Fa в этом случае будет определяться как профилем внутреннего давления в поверхностном слое Р(х) или его градиента (dPldx\ так и профилем силы F,(x) (зависимостью F, от расстояния х до межфазной границы):
Fx = F, (х) - (dPIdx). (3.4.1)
С учетом того, что (dP!dx) = FIV, можно записать уравнение:
Fx = F(x)-F(VxIV). (3.4.2)
Таким образом, адсорбционная сила Fx может отличаться от нуля не только из-за различия сил взаимодействия F и Fj разных молекул со всеми остальными молекулами гетерогенной системы, но и просто из-за различия их размеров (объемов И и Vx). Однако если растворитель и растворенное вещество близки по химической природе (например, это смесь октана и гексадекана, молекулы которых отличаются по размеру примерно вдвое), то есть основания считать, что обе составляющие адсорбционной силы взаимно компенсируются, т. е. F,(x) = F(VXIV), и тогда адсорбция будет отсутствовать. Проблема здесь в возможности выполнения последнего равенства на любом расстоянии от границы фаз. Поскольку F также является функцией расстояния х, то последнее предположение эквивалентно предположению о полном подобии функций F,(x) и F(x), т. е. предположению, что на любом расстоянии от поверхности они отличаются только постоянным множителем (VXIV). Это заведомо невозможно для молекул разного размера, когда они приближаются к межфазной
578
Новый справочник химика и технолога
границе на расстояние, меньшее размера большой молекулы. Ведь при этом большая молекула будет частично внедряться в соседнюю фазу (в газ), тогда как меньшая молекула может сохранить прежнее окружение, что неизбежно нарушит подобие двух упомянутых функций взаимодействия молекул с межфазной границей. Иначе говоря, в смеси химически близких, но разных по размеру молекул адсорбция неизбежна.
Чисто статическое рассмотрение условий адсорбции подразумевает, что адсорбированная молекула займет в поверхностном слое положение, при котором действующая на нее полная сила равна нулю, и тогда приведенное выше равенство обязательно должно выполняться, т. е. справедливо уравнение
FM = F(x^VxIV\ (3.4.3)
Важно отметить, что это равенство выполняется только на одном определенном равновесном расстоянии %о адсорбированной молекулы от поверхности. Иначе говоря, формула (3.4.3) определяет положение х0 адсорбированных молекул относительно межфазной границы. Разумеется, что в действительности оно более или менее размыто, как и сама межфазная граница. Вместе с тем хорошо известны случаи очень четкого позиционирования адсорбированных молекул на границе между раствором и газом. Это, например, слои Ленгмюра — Блоджет из практически нерастворимых ПАВ [1].
Согласно формуле (3.4.2), движущая сила адсорбции, равная с обратным знаком полной силе воздействия межфазной границы на молекулу Fx, максимальна при F,(x) = 0, т. е. при нулевой величине силы взаимодействия молекулы растворенного вещества с растворителем. Это возможно, если «растворенным веществом» являются пустоты, не занятые молекулами растворителя (вакансии). Можно думать, что адсорбция данных вакансий не изменит состав поверхностного слоя и, следовательно, поверхностного натяжения. Предоставляем возможность читателю самостоятельно разрешить некоторые несоответствия, возникающие при таком предположении. При понижении температуры растворителя концентрация вакансий в нем понижается (растет плотность). Натяжение при этом растет. Можно ли такое изменение трактовать как результат неизотермической адсорбции вакансий?
Статическое (как и статистическое) описание адсорбции требует принятия той или иной модели строения молекул, раствора, поверхностного слоя и т. д., и поэтому получаемые этими методами результаты в значительной мере зависят от выбранной модели. В отличие от этого законы феноменологической термодинамики, целиком основанные на опыте, базирующемся на ряде постулатов, не требуют навязывания каких-либо представлений о строении вещества и механизмах явления. Основой термодинамического метода описания адсорбции является уравнение изотермы адсорбции Гиббса (3.3.24). Особенность его в том, что для сужде
ния о величине, знаке и зависимости адсорбции Г от концентрации растворенного вещества с (об изотерме адсорбции) необходимо располагать зависимостью поверхностного натяжения о от концентрации раствора (изотермой натяжения). Обычно изотерма натяжения о(с) устанавливается экспериментально. Здесь и далее обсуждаются бинарные растворы, которые характеризуются концентрацией и адсорбцией растворенного вещества.
Согласно формуле (3.3.24), положительно адсорбируются (Г > 0) вещества, которые понижают натяжение раствора (<7о Ide < 0), и отрицательно адсорбируются (Г < 0) вещества, повышающие натяжение. В этом суть правила адсорбции Гиббса. Очевидно, что понижать натяжение раствора могут вещества с меньшим, чем у растворителя, натяжением, поэтому правило адсорбции можно формулировать иначе: из раствора на границе с газовой фазой положительно адсорбируется менее полярный компонент.
Для понимания механизма адсорбции и других поверхностных явлений полезно иметь в виду, что адсорбция является по существу формой выделения вещества из раствора (и из газовой фазы). При отсутствии межфазных границ выделение растворенного вещества в виде новой фазы требует определенного перенасыщения раствора, тогда как при наличии межфазной границы (как и зародышей кристаллизации) выделение растворенного вещества сильно облегчается и происходит в виде адсорбционного слоя на межфазной границе при любой концентрации раствора. По этой причине чем слабее растворимость вещества (летучесть для газов), тем сильнее оно адсорбируется. Для водных растворов эта зависимость в ряде случаев известна количественно (см. ниже правило Траубе).
3.4.2. Поверхностно-активные вещества (ПАВ)
Вещества, изменяющие поверхностное натяжение раствора при изменении его концентрации, называются поверхностно-активными веществами (ПАВ). Положительно поверхностно-активные вещества (ППАВ) уменьшают поверхностное натяжение, отрицательно поверхностно-активные вещества (ОПАВ) увеличивают натяжение. Инактивное вещество (ИНАВ) не изменяет натяжение (рис. 3.22).
Практическое применение ПАВ ориентировано в основном на положительно поверхностно-активные вещества, т. е. все другие не представляют интереса и поэтому названы инактивными. В соответствии с этим выстраивается терминология, принятая в технической литературе. При этом утрачивается необходимость подчеркивать, что имеются в виду только положительно активные вещества и за ними закрепляется общий термин «поверхностно-активное вещество», или ПАВ. В дальнейшем мы будем придерживаться этой традиции, тем более что действительно обсуждаться будут в основном проблемы, связанные с положительно активными веществами. Следует учитывать, что многие
Физика, химия и технология дисперсных систем
579
представления, в том числе зафиксированные в терминологии, сложились в процессе изучения важнейшей, но не единственной системы — водных растворов ПАВ, а также и водных коллоидных растворов. Перенос этих представлений на иные гетерогенные системы часто неоправдан. При рассмотрении роли межкристаллитных границ отмечено, что поверхностную активность, и притом необычайно сильную, способны проявлять вещества, весьма далекие от традиционных поверхностно-активных веществ. Это машинные масла и смазки, где средой (растворителем) являются углеводороды, — ряд так называемых минеральных масел, получаемых из нефтепродуктов. В роли среды выступают многочисленные и весьма разнообразные крем-нийорганические и фторорганические жидкости, в том числе ряд синтетических масел. Некоторые фторорганические жидкости по уровню сродства к другим веществам близки к газовым средам, но тем не менее найдены вещества, способные и в этих средах выполнять некоторые функции ПАВ водных и углеводородных сред, обусловленные положительной адсорбцией этих веществ из кремний- или фторорганических жидкостей на межфазных границах. При этом нет оснований считать, что здесь сохранят силу механизмы и закономерности, установленные для традиционных ПАВ, например непременная для водных сред дифильная структура молекул ПАВ.
По отношению к воде положительно поверхностноактивными являются вещества с дифильным строением молекул. Дифильность буквально означает двойное сродство. Оно обеспечивается наличием в молекуле полярной группы (-ОН, -СООН, -COONa, -NH3X, -OSO3H и т. д.), обеспечивающей сродство к воде (т. е. более или менее приемлемую растворимость), и неполярного радикала (алифатического, ароматического и др.), обеспечивающего меньшую, чем у воды, полярность вещества, а следовательно, и меньшую величину натяжения границы этого вещества с газовой средой, что необходимо для того, чтобы адсорбция этого вещества на границе раствор—газ привела к снижению натяжения раствора по сравнению с натяжением растворителя. В символическом изображении таких молекул на разного рода схемах кружок обозначает полярную группу, а черта — неполярный радикал.
Отрицательную поверхностную активность обычно проявляют неорганические электролиты. Натяжение разбавленных растворов электролитов увеличивается очень слабо с увеличением концентрации электролита. При высоких концентрациях оно должно возрастать очень круто, так как само растворяемое вещество (например поваренная соль, серная кислота) имеет большое поверхностное натяжение, которое не может быть достигнуто экстраполяцией плавной зависимости к составу раствора, эквивалентному растворенному веществу.
Аналитически изотерма натяжения раствора ПАВ (положительно активного) в ряде случаев описывается эмпирической формулой Шишковского:
с = с0 - В In (1 + Кс). (3.4.4)
Здесь о0 — натяжение растворителя, В — общая для всех ПАВ одного гомологического ряда константа и К— индивидуальная для каждого вещества константа. Точное совпадение с экспериментальными результатами дает формула В.А. Малова:
со - о = В Кс / (1 + Кс). (3.4.4а)
Основной характеристикой ПАВ является его поверхностная активность: G = lim (-da I dc)c - 0, т. e. крутизна начального участка графика изотермы натяжения — зависимости натяжения от концентрации раствора (рис. 3.22). Эмпирическое правило Траубе для водных растворов устанавливает, что в гомологическом ряду ПАВ одной химической природы при комнатной температуре поверхностная активность любого последующего члена ряда больше, чем у предыдущего, в среднем в 3,2 раза:
G„+1 / (7„ = 3,2. (3.4.5)
Индексы п + 1 и п означают число атомов углерода в неполярной части молекулы ПАВ. Вид семейства изотерм натяжения для веществ одного гомологического ряда схематично показан на рис. 3.23. Из формулы (3.4.5) следует, что, согласно уравнению (3.3.24), при одинаковых и достаточно малых концентрациях растворов адсорбции соседних членов ряда ПАВ различаются во столько же раз, во сколько и активности, т. е. Г„41/Г„ = 3,2.
Рис. 3.22. Изотермы натяжения растворов положительно поверхностно-активного вещества (ПЛАВ), отрицательно поверхностно-активного вещества (ОПАВ) и инактивного вещества (ИНАВ)
Рис. 3.23. Семейство изотерм натяжения ПАВ одного гомологического ряда: п — число атомов углерода в молекуле типа СяН(2л + i>F;
F — полярная группа
580
Новый справочник химика и технолога
Следовательно, различие в поверхностной активности целиком обусловлено различием в количестве адсорбированного ПАВ при равной концентрации молекул в растворе. Это очевидный вывод из правила Траубе, и уравнение изотермы адсорбции приводится здесь лишь потому, что существуют противоречащие ему объяснения правила Траубе. До сих пор можно встретить объяснение повышенной активности последующих членов ряда тем, что адсорбированные молекулы большего размера, располагаясь на поверхности плашмя, занимают («экранируют») большую поверхность, чем молекулы меньших размеров, и именно поэтому сильнее снижают натяжение. Если бы это было так, то благодаря тому, что и адсорбции соседних членов ряда обязаны отличаться в 3,2 раза. Тогда отношение активностей превышало бы отношение адсорбций соседних членов ряда, что противоречит уравнению адсорбции Гиббса.
При дифференцировании формулы Шишковского получается уравнение:
des I de = -ВК / (1 + Кс). (3.4.6)
Подстановка этого выражения в уравнение изотермы адсорбции Гиббса приводит к зависимости Г от с:
Г = ГДКс/(1+Кс)]. (3.4.7)
Здесь постоянная Гт = В / RT объединяет совокупность констант, появившихся при указанной подстановке. По форме это выражение идентично уравнению изотермы адсорбции Ленгмюра, а константа К совпадает с одноименной константой уравнения Ленгмюра. Это побуждает к поискам сходства этих двух по существу разных уравнений. Графически полученная зависимость представлена на рис. 3.24 кривой 7. Она ничем не отличается от аналогичной зависимости для мономолекулярной адсорбции газов и, следовательно, характеризуется наличием предельной величины адсорбции Гт. На границе жидкость—газ и жидкость—жидкость активные центры заведомо отсутствуют, поэтому предельная адсорбция ПАВ в данном случае определяется исключительно размером молекул и их ориентацией относительно плоскости адсорбции. Поскольку молекулы ПАВ практически любого типа имеют ярко выраженную анизо-диаметричность, то их длина намного больше поперечного размера. Величина предельной адсорбции связана с площадью сечения молекулы соотношением:
rm=l/A0NA. (3.4.8)
Здесь Ао — площадь сечения молекулы ПАВ плоскостью, параллельной плоскости адсорбции т. е «посадочная площадка молекулы».
Экспериментальные данные о значениях предельных адсорбций Гт ПАВ одного ряда показывают, что они оказываются практически одинаковыми при изменении размера молекулы (ее длины) в несколько раз.
Такое постоянство объясняется с позиций формулы (3.4.8) равенством посадочных площадок Ао молекул одного ряда, что возможно только при их ориентации в насыщенном монослое длинными осями перпендикулярно поверхности. Иначе говоря, величина Ао в формуле (3.4.8) — это площадь поперечного сечения длинных молекул ПАВ.
Из сравнения полученной зависимости с уравнением Ленгмюра выясняется смысл констант уравнения Шишковского. Константа В с точностью до размерной константы 1 / RT совпадает с предельной адсорбцией:
В-ГтВТ.
(3.4.9)
При использовании выражения (3.4.6) для вычисления поверхностной активности G как предела отрицательного значения производной des / de при стремлении концентрации с к нулю получается соотношение
G = BK.
(3.4.10)
Поскольку константа В является общей для членов одного гомологического ряда ПАВ, то отсюда следует, что эмпирическая константа К уравнения Шишковского, а также равная ей константа адсорбционного равновесия уравнения Ленгмюра с точностью до постоянного множителя В совпадают с поверхностной активностью веществ G и их отношение должно подчиняться правилу Траубе: К„ <. । / Кп = 3,2.
Обнаружившееся соответствие между теориями адсорбции Гиббса и Ленгмюра на самом деле является чисто внешним и порождает множество вопросов. Прежде всего, адсорбция как поверхностный избыток не может оставаться постоянной после достижения ею некоторой величины Гт, которую формула (3.4.8) связывает с полным заполнением мономолекулярного адсорбционного слоя плотно упакованными молекулами ПАВ. Образование такого слоя означает, что далее он уже не может изменяться при увеличении концентрации раствора.
Рис. 3.24. Изотермы:
1 — адсорбции Ленгмюра; 2 — поверхностного избытка;
3 — адсорбции Гиббса; х — мольная доля адсорбируемого вещества в растворе; Гт — его предельная адсорбция;
4 — прямая, описываемая уравнением Г = Гт- Vjc (Иа — адсорбционный объем (толщина адсорбционного слоя))
Физика, химия и технология дисперсных систем
581
Однако постоянство состава поверхностного слоя не соответствует постоянству адсорбции по Гиббсу:
Г = Гт-сй. (3.4.11)
По определению, адсорбция представляет собой разность между полным количеством растворенного вещества в поверхностном слое, которое в заполненном монослое равно Гт, и тем количеством вещества с<5, которое способен вместить тот же по толщине слой в отсутствие адсорбции, т. е. при равенстве концентраций в поверхностном слое и в объеме раствора. Величина непрерывно и пропорционально растет с увеличением концентрации и становится равной предельной величине адсорбции Гт при 100 % содержании ПАВ в объемной фазе. Следовательно, поверхностный избыток (адсорбция Г) должен линейно снижаться с увеличением концентрации после формирования на поверхности насыщенного мономолекулярного слоя ПАВ и достигать нулевой величины при 100 % содержании ПАВ в растворе (рис. 3.24; кривая 2). После заполнения монослоя (рис. 3.25) постоянства может достигать полное количество вещества в нем, но не адсорбция. На этом основании можно полагать, что уравнение (3.4.7) дает не адсорбцию (по Гиббсу), а полное количество растворенного вещества в поверхностном слое (адсорбцию по Ленгмюру).
Преобразование уравнения Гиббса в уравнение Ленгмюра с помощью формулы Шишковского означает, в сущности, подмену одного понятия другим. Принципиальное различие адсорбций по Гиббсу и по Ленгмюру состоит в том, что понятие гиббсовской адсорбции базируется на анализе пространственного распределения компонента возле межфазной границы, тогда как понятие адсорбции по Ленгмюру основано на анализе избыточного времени пребывания молекул (их геометрических центров) в плоскости адсорбции. Адсорбция по Ленгмюру ни при каких условиях не может быть представлена в виде произведения концентрации в поверхностном слое на его толщину, поскольку в математическом смысле этот слой является плоскостью, не имеющей толщины.
Зависимость Г (с) чаще находят графическим дифференцированием экспериментальной кривой а(с), минуя нахождение констант уравнения Шишковского. Из графика о(с) можно сразу найти величину cdcs / de как отрезок на оси ординат между касательной к любой точке кривой и ординатой этой точки. Подстановка этой величины в уравнение изотермы Гиббса Г = -{c/RTfcks /de) обычно дает иную зависимость Г(с), чем в уравнении Ленгмюра (3.4.7). Как правило, после достижения максимума адсорбция начинает снижаться с увеличением концентрации раствора (рис. 3.24; кривая 3). Точность измерения натяжения ограничена величиной порядка 0,1 мДж/м2, и поэтому, начиная с некоторой концентрации раствора ст, пологий участок изотермы натяжения в пределах точности измерений оказывается практически горизонтальным, а производная do!de равна нулю.
Соответственно и адсорбция на этом участке становится равной нулю. Это явно не согласуется ни с какими представлениями о ней (случай смены знака адсорбции в точке ст рассматривается ниже при обсуждении адсорбции из раствора на поверхности твердых веществ), в том числе противоречит тому, что минимум натяжения должен иметь место при адсорбции, близкой к ее предельной величине.
Рис. 3.25. Строение мономолекулярного насыщенного адсорбционного слоя ПАВ на границе X жидкость—газ: Ло — посадочная площадь молекулы ПАВ
Таким образом, имеющиеся данные о зависимости натяжения от состава растворов в области значительных концентраций ПАВ приводят к неудовлетворительным результатам и требуют дальнейшего анализа проблемы.
Обсуждавшиеся выше детали не имеют решающего значения при описании адсорбции и натяжения растворов технических ПАВ, как правило, полуколлоидных, классическим примером которых можно считать многочисленный отряд моющих средств, включая обычное мыло (олеат натрия или калия). В этих случаях используются вещества с очень высокой поверхностной активностью, благодаря чему они насыщают адсорбционный монослой при весьма низких концентрациях раствора. К тому же они ограниченно растворимы, и поэтому в интервале тех концентраций ПАВ, которые представляют интерес, количественное различие между поверхностным избытком (адсорбцией по Гиббсу) и полным количеством компонента в поверхностном слое (адсорбцией по Ленгмюру) становится, согласно уравнению (3.4.11), незначительным. По этой причине ход трех кривых на рис. 3.24 практически совпадают. Вместе с тем и для теории, и для ряда нетрадиционных областей применения ПАВ актуальной задачей остается устранение обнаружившейся неоднозначности изотерм натяжения и адсорбции. Для ее решения можно воспользоваться рядом соображений:
1. Перейти от концентрации с растворенного компонента к его активности а - ус в растворе, где у — коэффициент активности. Таким способом, как известно, формально учитывается неидеальность раствора. Химический потенциал вещества в растворе при этом выражается через активность так же, как через концентрацию: ц = ц° + 7? Лп<х Дифференцирование химического потенциала по концентрации дает:
d\Ji = RT (ydc + cdy) /ус,
582
Новый справочник химика и технолога
а подстановка полученного выражения в уравнение (3.3.23) приводит к зависимости
Г = (3.4.12)
RT de ydc + cdy
Это уравнение сводится к уравнению (3.3.24), если у не зависит от с. Решение проблемы данным способом оправдано, если существуют независимые данные о коэффициентах активности раствора, например полученные изучением упругости паров смесей, электрической проводимости растворов и т. п. При этом коэффициенты, характеризующие поверхностную активность, летучесть, электрическую проводимость и другие свойства растворов, должны совпадать. Обобщенные данные по методу активностей, которые могли бы дать ответы на поставленные выше вопросы, отсутствуют.
2. Предположение, выражаемое формулой (3.4.11), носит частный характер, и вытекающие из него следствия не представляют интереса для всего диапазона концентраций.
3. В поисках приемлемого вида изотерм адсорбции и натяжения при концентрациях, превышающих величину ст, можно обратиться к данным о натяжении растворов отрицательно поверхностно-активных веществ (ОПАВ). Изотермы натяжения ПАВ и ОПАВ, в сущности, однотипны. Их видимое различие (рис. 3.22) исчезнет, если растворителем является ОПАВ, а растворенным веществом — растворитель, который в силу меньшей полярности будет играть роль положительно активного вещества. Пологая часть изотермы этой смеси, соответствующая малой концентрации ОПАВ, изучена полнее, чем пологая часть изотермы натяжения ПАВ (большая концентрация ПАВ). Считается, например, что натяжение раствора ОПАВ линейно зависит от состава раствора. В силу общности законов поведения растворов та же зависимость должна описывать пологую часть изотермы натяжения для обычных ПАВ. Тогда на этом участке da / de = -Gm, где Gm — некоторая положительная постоянная, и в соответствии с уравнением Гиббса адсорбция должна линейно расти с увеличением концентрации. Такой результат ничем не подтверждается.
4. Если обратиться, наконец, к альтернативному способу решения общего уравнения изотермы адсорбции (подраздел 3.3.2), то его математическая сторона сводится к наложению условия (3.4.13) на величины поверхностных избытков компонентов раствора:
Г1К1+Г2К2 = 0. (3.4.13)
Здесь Ki и V2 — молярные объемы компонентов 7 и 2. Условие (3.4.13) просто означает, что поверхностный избыток одного компонента в точности равен недостатку другого, т. е. замещение одного компонента другим в поверхностном слое идет путем вытеснения эквивалентного по объему количества другого вещества. Это условие заменяет прежнее, по которому предполагалось
равенство нулю адсорбции растворителя Гь Условие (3.4.13) позволяет выразить адсорбцию растворителя через адсорбцию растворенного вещества:
Г1=-(^/И)Г2.
Содержание компонентов в объемной фазе связано очевидным соотношением:
c\V\+c2V2=\.
Из него находим следующие соотношения:
С1=(1-с2К2)/К1, dc}=AV2tV^dc2.
Они дают возможность выразить изменение химического потенциала растворителя через концентрацию растворенного вещества следующим образом:
= RT^- = -RT-У- dc2. г 1 1 jy L
С] 1 —c2K2
В уравнении изотермы сорбции двухкомпонентного раствора, являющегося частным случаем общего уравнения (3.3.23),
da = -Г1ф1 - Г2ф2 (3.4.14)
первое слагаемое можно выразить через величины, относящиеся к растворенному веществу:
V V de
da = -~r2RT—dc2 -T2RT^.
К, 1-с2И2 с2
Второе слагаемое имеет обычный вид. Решением этого уравнения относительно адсорбции растворенного вещества является выражение:
| 2 2 2
И 1-^2
Здесь Г = Г(с2) — адсорбция, вычисляемая рассмотренным выше классическим способом по уравнению Г = -(с/RT)(da/ de) (3.3.24), а Г2 — адсорбция, предполагающая одновременное отличие от нуля адсорбций обоих компонентов раствора. Для согласования обозначений при концентрации с2 сохранен индекс второго компонента (ПАВ). Легко заметить, что это уравнение дает еще более резкое уменьшение адсорбции с увеличением концентрации раствора.
3.4.3. Пленки нерастворимых ПАВ
На поверхности жидкости (обычно воды) можно искусственно создать слой ПАВ вполне определенного состава. Для этого нерастворимое в воде и нелетучее ПАВ (высшие жирные кислоты, некоторые их соли и т. д.) растворяется в неполярном летучем растворителе, и капля этого раствора наносится на выгороженную часть поверхности воды с известной площадью А. Капля полностью растекается, оставляя после испарения
Физика, химия и технология дисперсных систем
583
растворителя определенное количество ns вещества, равномерно распределенного на выгороженном участке поверхности. Таким образом, в этом методе однозначно задается адсорбция Г = ns /А, которая из-за нерастворимости ПАВ совпадает с полным количеством вещества на единицу межфазной поверхности.
В устройстве (рис. 3.26), известном под названием «весы Ленгмюра», площадь, по которой растекается капля раствора ПАВ, ограничена бортами кюветы с водой и двумя барьерами. Борта кюветы водой не смачиваются (парафинированы), так что поверхность воды возвышается над бортами кюветы. Один из барьеров (тяжелый 3) лежит на бортах кюветы, а второй (легкий 2) подвешен к силоизмерительному устройству (весам 4). Площадь А выгороженной части поверхности воды регулируется путем перемещения тяжелого барьера, а сила, действующая на легкий барьер в плоскости поверхности воды, регистрируется весами.
Опыты на весах Ленгмюра заключаются в измерении зависимости силы, действующей на подвешенный к весам барьер, от площади, занимаемой определенным количеством вещества. Интерес представляет удельная (на единицу длины барьера) величина р этой силы, равная разности натяжений До - о0 - о по обе стороны подвижного барьера. Типичные изотермы р(А), полученные этим путем для ряда ПАВ, напоминают своим видом (рис. 3.27) изотермы зависимости давления от объема газов с различной степенью отклонения от идеальности.
Результаты опытов Ленгмюра допускают двойственную интерпретацию. С одной стороны, измеряемая сила р является разностью натяжения о0 поверхности чистой воды и натяжения о поверхности, покрытой слоем молекул ПАВ. Следовательно, измеряемая величина есть не что иное, как эффект понижения поверхностного натяжения, создаваемый адсорбционным слоем ПАВ. С другой стороны, измеряемую величину р можно также интерпретировать как силу, с которой молекулы поверхностно-активного вещества действуют на барьер, т. е. как давление слоя двухмерного вещества на ограничивающий его контур.
Двухмерная форма существования вещества от обычной трехмерной отличается тем, что молекулы вещества могут располагаться и двигаться только в двух измерениях в пределах некоторой площади. Близкое по геометрическим признакам понятие мы использовали при описании поверхностного слоя гетерогенной системы. Его вполне можно было воспринимать как теоретическую абстракцию, необходимую для решения уравнений термодинамики поверхностного слоя. В опытах Ленгмюра оно возникает как рукотворный объект. Если считать, что слой вещества является идеальным двухмерным газом, то зависимость его давления р на стенки контура от площади А, занимаемой газом, можно описать уравнением состояния идеального газа: pA = nsRT, которое после деления на площадь дает р = VRT. Уравнение состояния двухмерного газа являет
ся полным аналогом уравнения состояния PV=nRT обычного трехмерного газа, п молей которого занимают некоторый объем V.
Равноправность двух разных способов интерпретации одной и той же величины означает, что можно приравнять правые части соответствующих формул и получить уравнение:
Ао = Г/?Г. (3.4.16)
Это означает, что действие ПАВ на натяжение раствора имеет ту же физическую природу, что и давление вещества на стенки сосуда, ограничивающие занимаемое им пространство, в том числе если оно имеет только два измерения. Иначе говоря, натяжение снижается ровно на ту величину, с которой молекулы адсорбционного слоя действует на единицу длины контура, охватывающего произвольный участок поверхности.
Рис. 3.26. Весы Ленгмюра:
1 — кювета; 2 — легкий барьер; 3 — тяжелый барьер; 4 — весы; А — площадь, занятая слоем ПАВ
Рис. 3.27. Изотермы давления разных веществ в двухмерном состоянии;
р — давление; А — площадь на 1 моль вещества; цифры возле кривых — число атомов углерода в молекулярной цепи ПАВ
584
Новый справочник химика и технолога
Формула (3.4.16), основанная на аналогии законов идеальных трехмерных и двухмерных газов и представляющая в сущности «механическую» интерпретацию поверхностной активности веществ, следует и из уравнения изотермы адсорбции Гиббса. При малых степенях заполнения поверхности do / de = (о- аоХс - 0) = -Аа/с, поэтому уравнение Гиббса (3.3.24) может быть сразу записано в форме уравнения (3.4.16): Г = AgRT. Отсюда в частности следует, что разреженный (Г Гт) слой ПАВ является по своему состоянию двухмерным газом.
При значительном заполнении поверхности слои нерастворимых ПАВ, как и обычные газы, обнаруживают заметные отклонения от свойств идеального газа. На экспериментальных зависимостях это проявляется в усложнении вида кривых при сжатии слоев и в усилении их различия для разных веществ. Общие закономерности в отклонениях от идеальности те же, что и для трехмерного состояния вещества: при достаточно сильном сжатии двухмерной фазы наблюдается ее конденсация, проявляющаяся в появлении горизонтальных участков на графиках р(Л). После завершения конденсации и превращения всего вещества в конденсированное (жидкое или твердое) состояние возникает очень сильное сопротивление дальнейшему сжатию, выражающееся в появлении почти вертикальных участков графиков. Как и в обычных газах, отклонения от идеальности усиливаются с увеличением размера молекул. Результаты измерений с различными веществами сводятся к следующим закономерностям.
1. В конденсированном состоянии площадь Ло, занимаемая одной молекулой ПАВ на поверхности, не зависит от длины ее углеводородной цепи. Это означает, что молекулы ПАВ в конденсированном слое плотно упакованы и ориентированы перпендикулярно поверхности, т. е. Ао — это площадь поперечного сечения молекул (рис. 3.25). Для нормальных одноосновных органических кислот, их солей и спиртов А0~2 • 10“19м2. Ориентация длинных осей молекул ПАВ по нормали к поверхности означает, что толщина слоя вещества, как и адсорбционного слоя растворимых ПАВ, равна длине молекул ПАВ.
2. Поверхностное давление р слоя ПАВ, как и разность натяжений Ао, существенно зависит от сродства полярной группы ПАВ к растворителю (воде): чем выше сродство к растворителю, тем больше р при одинаковой степени заполнения поверхности. Например, в ряду ПАВ — олеат серебра, олеиновая кислота, олеат натрия — нарастает растворимость этих веществ в воде и соответственно этому р составляет 1, 6 и 14 Дж/м при одном и том же числе молекул на единицу площади [1].
3. Измерение поверхностного давления р проводится в отсутствие полного равновесия в системе слой ПАВ—вода—чистая водная поверхность. Как бы ни мала была растворимость ПАВ, полному равновесию отвечает одинаковое заполнение поверхности воды молекулами ПАВ по обе стороны барьера, и, следователь
но, р = 0. Быстрое сжатие двухмерного вещества создает давление р большее, чем равновесная разность натяжений Аа, так как поверхностный слой будет перенасыщен по отношению к равновесному раствору ПАВ. Таким образом, в зависимости от условий опыта р может колебаться от 0 до величины большей, чем Аа.
Приведенный выше пример различия в действии на натяжение (поверхностное давление р) олеиновой кислоты и ее солей при одинаковых степенях заполнения поверхности, а также другие факты указывают на зависимость активности ПАВ от индивидуальных свойств их молекул, в частности от свойств полярных групп дифильных молекул. Следует подчеркнуть, что речь идет не о правиле Траубе, которое обусловлено различием в адсорбции ПАВ, а об эффекте, относящемся к одинаковой величине адсорбции.
Индивидуальность молекул ПАВ, конечно, не учитывается уравнением состояния адсорбционного слоя рА = nsRT. Простейший способ введения характеристик молекул (их размера) в это уравнение, как и в уравнения состояния трехмерных фаз, состоит в замене общей площади А, занятой адсорбированным веществом, на свободную площадь (А - Ао). Тогда можно получить уравнение состояния:
р (А - Ао) = nsRT. (3.4.16а)
Увеличение гидрофильности полярной группы дифильных молекул эквивалентно увеличению эффективной площади Ао поперечного сечения этих молекул. Введение этой площади в уравнение состояния является простейшим способом учесть отталкивание адсорбированных молекул между собой. Чем сильнее отталкивание, тем больше эта площадь и двухмерное давление и тем выше поверхностная активность вещества. Очень наглядно влияние взаимодействия между адсорбированными молекулами на натяжение поверхности видно на примере электрокапиллярного эффекта (подраздел 3.4.8).
3.4.4. Мицеллярные растворы ПАВ
Поверхностно-активные вещества практического назначения, имеющие высокую поверхностную активность, должны, согласно правилу Траубе (подраздел 3.4.2), иметь достаточно большой размер гидрофобной части молекул и, следовательно, слабую растворимость в воде. Высокая поверхностная активность в неполярном растворителе также связана с ограниченной растворимостью ПАВ в этом растворителе.
Выделение ПАВ из раствора при достижении предела растворимости происходит путем перехода вещества в состояние коллоидного раствора. Молекулы ПАВ объединяются при этом в более или менее крупные агрегаты — мицеллы. Переход осуществляется при определенной для каждого ПАВ концентрации, которая называется критической концентрацией мицеллообра-зования (ККМ).
Физика, химия и технология дисперсных систем
585
Несмотря на большое разнообразие химического строения молекул технических ПАВ, структура их мицелл одинакова (рис. 3.28) — это или сферические (7), или цилиндрические (2), или пластинчатые (3) агрегаты.
Рис. 3.28. Структура мицелл ПАВ в водной среде:
1 — сферическая; 2 — цилиндрическая; 3 — пластинчатая
Форма мицелл изменяется от сферической к пластинчатой по мере увеличения концентрации раствора. В водных растворах внешняя поверхность мицелл состоит из полярных групп дифильных молекул ПАВ, а в неполярных растворителях — из углеводородных концов этих молекул. При переходе к пластинчатой форме мицелл в концентрированных растворах понятие внешней поверхности мицеллы теряет смысл: раствор превращается в регулярную слоистую структуру. Каждый слой такой структуры — это дифильный лист, одна сторона его гидрофильная, а другая гидрофобна. Соседние слои обращены один к другому одинаковыми по гидрофильности сторонами. Растворитель, в зависимости от его полярности, находится или в гидрофильных, или в гидрофобных промежутках между слоями.
Аналогичную структуру имеют некоторые чистые жидкости, причем в мицеллярной форме находится все вещество. В сущности, такое состояние вещества по степени упорядоченности молекул ближе к кристаллическому, чем к жидкому, но по механическим свойствам оно остается жидкостью и называется жидкокристаллическим. Молекулы этой жидкости также имеют дифильное, или отчетливо выраженное стержневидное строение.
Основной термодинамической характеристикой жидкокристаллических веществ (ЖК) является температура фазового (жидкокристаллического) перехода. При температуре выше температуры фазового перехода они имеют структуру обычных жидкостей, а при понижении температуры переходят в ЖК состояние. В силу хорошо выраженных электрооптических эффектов используются для изготовления дисплеев.
Концентрированные растворы ПАВ с пластинчатыми или цилиндрическими мицеллами также находятся в жидкокристаллическом состоянии. Их термодинамической характеристикой является концентрация жидкокристаллического перехода. Они называются лиотропными ЖК.
Технические ПАВ практически применяются в основном при концентрациях, несколько превышающих ККМ, так как: 1) ряд применений основан именно на мицеллярном состоянии и 2) при валовой концентрации раствора, превышающей критическую, концентрация вещества в молекулярной форме остается равной ККМ. Иначе говоря, концентрация истинно растворенного
вещества все равно не может превышать ККМ, поэтому в ряде случаев нет смысла существенно превышать эту концентрацию. Ее повышение увеличивает только концентрацию мицелл.
Многие свойства растворов ПАВ определяются концентрацией вещества в молекулярной форме. К их числу относится, в первую очередь, поверхностное натяжение раствора. Другие свойства (прозрачность, электрическая проводимость, вязкость) ощутимо меняют характер зависимости от концентрации при достижении ее критической величины — на графиках зависимости свойства от концентрации в точке ККМ наблюдается излом. На этом основано измерение критической концентрации мицеллообразования.
Отличительной особенностью мицеллярных растворов ПАВ является их солюбилизирующая способность, т. е. способность растворять вещества, которые в чистом растворителе нерастворимы. Например, водный мицеллярный раствор ПАВ растворяет углеводороды, углеводородные мицеллярные растворы ПАВ становятся растворителями воды. Достаточно эффективное ПАВ делает несмешивающиеся жидкости неограниченно растворимыми. Разумеется, это не истинные, а коллоидные растворы. Солюбилизация происходит в результате проникновения молекул слабо растворимого вещества внутрь мицелл. Именно там оно накапливается в больших количествах, обеспечивая растворимость нерастворимого вещества. Мицеллы ПАВ при этом увеличиваются в размере. Типичным представителем водорастворимых мицеллообразующих ПАВ являются мыла щелочных металлов.
Важным классом технических ПАВ и составов на их основе являются моющие средства. Отмывание жидких загрязнений в значительной мере основано на солюбилизации. Механические воздействия на слой жидких или твердых загрязнений вызывают дробление (диспергирование) слоя загрязнения на капли (частицы), их поверхность покрывается адсорбционным слоем ПАВ, играющм роль эффективного стабилизатора коллоидно растворяемых загрязняющих веществ.
Разрыхление осадка, образованного из твердых частиц, и перевод его в состояние устойчивого коллоидного раствора при действии на осадок стабилизатора (и механических усилий) называется пептизацией осадка. На пептизации основано моющее действие ПАВ, приготовление технических суспензий из готовых высокодисперсных порошков, получение практически важных концентрированных коллоидных растворов.
Эмульгирование, солюбилизация, отмывание поверхности твердых веществ от слоя углеводородов очень широко применяются в производстве порошкообразных полимеров, добыче нефти. Эти же явления играют важную роль в усвоении организмом пищи, жиров в частности. Разнообразные функции масел в двигателях внутреннего сгорания и смазок также обусловлены указанными явлениями. Производство ПАВ — одно из основных в современной химической индустрии.
586
Новый справочник химика и технолога
3.4.5. Градиент поверхностного натяжения
Если каким-либо образом вызвать быстрое локальное растяжение границы раздела фаз, то это приведет к локальному уменьшению адсорбции и повышению поверхностного натяжения. Соседние участки поверхности при этом сохранят в большей или меньшей мере прежнее значение натяжения. Это приведет к возникновению разницы натяжений вдоль поверхности, которая количественно характеризуется величиной градиента натяжения des / dy вдоль направления у растяжения (или сжатия) поверхности. Такое состояние границы является неравновесным и будет переходить в к равновесное за счет ряда параллельно идущих процессов: механической деформации жидкости под действием сил поверхностного натяжения, диффузии адсорбированного вещества вдоль поверхности, обмена веществом между адсорбционным слоем и прилегающими слоями раствора, т. е. за счет процессов адсорбции и десорбции. В растворах мыла, например, требуется около 0,1 с для восстановления равновесия растянутого поверхностного слоя.
Наиболее интересная сторона поведения деформируемой поверхности заключается в появлении у нее динамической упругости. Пусть поверхность раствора ПАВ площадью 2А перегорожена барьером (рис. 3.26), разделяющим ее на две равные части А\ иЛ2, т. е. площадь каждой равна А. Пусть при быстром смещении барьера площадь первой половины увеличилась на величину dA, а площадь второй уменьшилась на ту же величину. Если допустить, что поверхностная диффузия происходит очень быстро, а обмен поверхности с раствором очень медленно по сравнению со скоростью перемещения барьера, то количество вещества на обеих частях поверхности останется неизменным. Оно равномерно перераспределится на новые величины поверхности, понизив адсорбцию на первой части поверхности и увеличив на второй на одну и ту же величину dT. Легко доказать, что при этом dT =-Гdy, где dy = dA/ А — величина деформации поверхности. Соответственно, натяжение первой части поверхности возрастет, а второй снизится на da - (da I tZT)tZT. В итоге возникает сила, равная разности натяжений двух частей поверхности, т. е. 2 da, которая будет действовать на барьер, возвращая его в исходное положение.
Важно, что появление силы вызвано такой деформацией поверхности, при которой ее общая поверхность 2А остается неизменной, и, следовательно, она не эквивалентна обычному поверхностному натяжению раствора. Задача состоит в том, чтобы найти модуль динамической поверхностной упругости Н = da / dy, который определяет возвращающее усилие 2da = 2Hdy, действующее на единицу длины барьера при быстрой деформации этим барьером прилегающих к нему поверхностей на величину dy = dA/ А.
Для нахождения производной da / tZT можно воспользоваться формулой Шишковского. Для этого урав
нение адсорбции Ленгмюра необходимо записать в следующем виде: 0 = Кс / (1 + Кс), где 0 = Г / Гт, и подставить величину Кс - 1 / (1 - 0) в формулу Шишковского. Тогда получается формула:
сг0-сг = -Гт/?Пп(1-0). (3.4.17)
Дифференцирование сг по 0 приводит к уравнению, которое выражает локальное изменение натяжения da через первоначально равновесную степень заполнения поверхности 0 и ее отклонение <70 от равновесной величины:
da = VmRTd® / (1 - 0). (3.4.18)
Эта форма уравнения применима к любому моменту времени после растяжения (или сокращения) поверхности, но в ней локальное отклонение от равновесного состояния есть некоторая функция времени.
При оговоренных выше соотношениях скоростей деформации поверхности и релаксации адсорбционного слоя dT = -ГсЬ{ или d® = -0tfy. Подстановка этой величины в уравнение (3.4.18) позволяет найти модуль динамической поверхностной упругости Н= da / dy с помощью уравнения:
H=rj?70/(1-0). (3.4.19)
Эта формула справедлива только в момент растяжения (сокращения) поверхности — в той мере, в какой выполняется соотношение d® = -0afy. С течением времени перепад d® степеней заполнения поверхности по обе стороны барьера уменьшается и поверхностная упругость снижается вплоть до нуля при медленном, т. е. равновесном перемещении барьера. Известные опыты Ленгмюра с поверхностными слоями нерастворимых ПАВ соответствуют очень медленной релаксации искусственно созданной неоднородной по натяжению поверхности, вследствии чего перепад адсорбций d® оставался неизменным во времени.
Само поверхностное натяжение нельзя считать проявлением упругости. Упругость — это способность противодействовать деформации. Упругая сила равна нулю, если деформация отсутствует. Поверхностное натяжение действует всегда, когда есть поверхность, и не зависит ни от величины, ни от скорости деформации поверхности, если при этом не изменяется величина адсорбции.
Для проявления динамической поверхностной упругости наличие барьера необязательно: разность поверхностных натяжений соседних участков, т. е. градиент натяжения, может быть получен и другими способами, например распространяющимися по поверхности волнами. Это могут быть видимые поперечные волны рельефа поверхности (капиллярные волны), а также продольные невидимые волны сгущения и разрежения адсорбционных слоев. В любом случае под действием разности натяжений площадь участков с повышенным
Физика, химия и технология дисперсных систем
587
натяжением будет самопроизвольно уменьшаться за счет участков с пониженным натяжением. Такое поведение неоднородной по величине натяжения поверхности называется эффектом Марангони.
Режимы деформации адсорбционного слоя могут быть весьма разнообразны, и соответственно режиму будет изменяться и модуль динамической упругости. Предельным можно считать режим очень быстрой деформации, при котором за двигающимся барьером остается зона полностью очищенной от ПАВ поверхности, а перед ним — зона предельно сгустившегося слоя ПАВ, в котором адсорбция равна ее предельной величине. Скорость движения барьера должна быть в этом случае много больше скорости диффузионного выравнивания концентраций перед фронтом и за фронтом движущегося барьера. Кажущийся очевидным ответ, что в этом режиме сила противодействия движению барьера равна (о0 - от), скорее всего будет ошибочным: о0 — натяжение растворителя, ат — натяжение раствора при насыщении его поверхности поверхностноактивным веществом. Сгустившийся перед барьером слой ПАВ в механическом смысле (в силу несжимаемости) можно считать продолжением барьера, т. е. единственным эффектом появления уплотненной зоны будет увеличение толщины барьера. Действующим на эту сторону барьера натяжением будет натяжение о еще не возмущенной поверхности перед фронтом барьера. Таким образом, в указанном режиме противодействие равно (о0 - о).
Режим, промежуточный между двумя рассмотренными, — это режим стационарного движения барьера. К нему можно перейти от предыдущего режима, уменьшив скорость движения барьера до величины, при которой зона высокой поверхностной концентрации ПАВ перед барьером будет заметно размываться поверхностной диффузией. Соответственно будет размываться (наполняться молекулами ПАВ) и зона за барьером.
Для решения задачи о стационарном движении барьера удобно считать, что он неподвижен, а жидкая фаза вместе с адсорбционным слоем набегает на барьер. Набегающее вещество адсорбционного слоя соскребается с поверхности раствора барьером, и некоторая часть его диффундирует навстречу набегающему потоку. Под стационарным подразумевается режим, когда поток набегающего на барьер вещества пи уравнивается его диффузионным потоком Ddn / dy, т. е. пи = -Ddn/dy. Здесь п — поверхностная концентрация (адсорбция по Ленгмюру), являющаяся искомой функцией расстояния у от поверхности барьера, и — линейная скорость потока и D — коэффициент поверхностной диффузии ПАВ. Решение этого уравнения дает формулу
п - nb exp(-uy / D). (3.4.20)
Здесь пь — поверхностная концентрация непосредственно перед барьером (при у = 0), которая в ряде слу
чаев и определяет поверхностную упругость. Поверхностная концентрация п и адсорбция (по Ленгмюру) — это одно и то же. Здесь используется другое обозначение и название адсорбции, для того чтобы различать адсорбции по Ленгмюру и по Гиббсу.
Все многообразие вариантов, которое может дать уравнение (3.4.20), определяется дополнительными условиями, необходимыми для нахождения неизвестной константы распределения пь. Можно, например, исходить из того, что количество вещества должно остаться неизменным и равным его первоначальному количеству Hq£0 на всем двухмерном пространстве перед барьером, протяженность которого в направлении движения обозначена символом L. Здесь п^ и Lq — концентрация ПАВ и протяженность поверхности до начала деформации. Интегрирование ndy с помощью уравнения (3.4.20) в пределах от у = 0 до у = L приводит к выражению общего количества вещества перед барьером, которое затем можно приравнять количеству вещества до начала деформации поверхностного слоя ПоЬо‘.
nh(D/ w )[1 - ехр (-и L / £>)] = hqLq.
Отсюда получается формула
nb = n0(L0 / Ld) [1 - exp (-Z / Ш (3.4.21)
Здесь Ld- DI и — диффузионная длина, характерная для данного соотношения скорости движения барьера и коэффициента диффузии.
При типичных для молекул значениях коэффициента диффузии порядка 10-10м2/с путем варьирования скорости движения барьера можно реализовать следующие условия: L Ld и L » Ld. В первом случае константа распределения (концентрация непосредственно перед барьером) пь = n$Lo / L нарастает пропорционально сокращению площади, а во втором пь = noLo / Ld остается постоянной. В забарьерной области диффузионной длине следует приписать обратный знак, тогда в ней пь = 0 при L » Ld. Первый вариант реализуется в условиях опытов Ленгмюра, а второй — при движении барьера по неограниченной по величине поверхности. Первый вариант иллюстрирует случай нарастающего по мере деформации сопротивления, а второй — постоянного.
Двухсторонняя мыльная пленка, в отличие от адсорбционного слоя, находящегося в равновесии с большим количеством раствора, обладает и равновесной (статической) упругостью на растяжение. Она обусловлена обеднением внутренней части пленки поверхностно-активным веществом за счет роста количества ПАВ в поверхностном слое при увеличении площади пленки. Адсорбция при этом снижается, а натяжение растет довольно сложным образом, поскольку убыль концентрации ПАВ внутри пленки зависит и от ее толщины, которую следует выразить через первоначальную толщину и приращение площади пленки. Рав
588
Новый справочник химика и технолога
новесная упругость пленки называется упругостью Гиббса. В сущности, она ничем не отличается от обычного поверхностного натяжения, и только в силу особых геометрических условий натяжение пленки растет при увеличении ее поверхности. Равновесная и неравновесная упругости пленок действуют одновременно, поэтому суммарный эффект увеличения натяжения при увеличении площади называют эффектом Маранго-ни — Гиббса.
3.4.6. Адсорбция из раствора на поверхности твердого вещества
Свойства растворов и компонентов раствора, которые приведены выше как важные для адсорбции на границе раствор—газ (полярность, дифильность, растворимость), сохраняют значения и при адсорбции растворенных веществ поверхностью твердого адсорбента.
Кроме того неоходимо учитывать особенности поверхности твердых веществ, в частности их химическую природу, свойства активных центров поверхности и конкурентоспособность компонентов раствора за эти центры. Основой количественного описания могут тогда служить теории Ленгмюра, БЭТ, а также потенциальная теория Поляни, оперирующая усредненным адсорбционным потенциалом центров.
Если к границе твердое вещество—жидкость относиться так же, как к любой другой однородной межфазной границе, то подходящей основой для анализа явления адсорбции и построения количественных зависимостей будет уравнение изотермы адсорбции Гиббса. В этом случае важно понять, каким компонентом должен обогащаться поверхностный слой и как это скажется на величине межфазного натяжения. Анализ явления на этой основе может опираться на различные характеристики веществ, участвующих в адсорбции (натяжения на границе с газом, диэлектрическая проницаемость, строение молекул, смачиваемость и др.), поэтому и правила адсорбции формулируются по-разному.
1. Адсорбируется (имеется в виду положительно) тот компонент раствора, который имеет промежуточную полярность в ряду полярностей всех участников процесса: растворителя, растворенного вещества и адсорбента. Мерой полярности может служить диэлектрическая проницаемость компонентов, их натяжение на границе с газом, а также дипольные моменты и поляризуемости молекул и их строение (в том числе дифильность). Это правило известно как правило Ребиндера, или правило уравнивания полярностей.
2. Растворенное вещество способно адсорбироваться, если адсорбент плохо смачивается растворителем, и адсорбция будет тем лучше, чем хуже смачиваемость. Такая формулировка с равным успехом может быть обоснована как уравнением Гиббса, так и теорией Ленгмюра.
Использование этих правил не вызывает трудностей только в случае типичных ПАВ, типичных полярных
(вода) или неполярных (углеводороды) растворителей, отчетливо гидрофильных или гидрофобных твердых веществ и заведомо физических адсорбционных сил. Правило 2 тогда можно конкретизировать: из водных растворов ПАВ адсорбируется на гидрофобных веществах, а из неполярных растворителей — на гидрофильных. Дифильные молекулы ориентируются при адсорбции полярной частью к полярной фазе, а неполярной частью — к неполярной фазе. В результате адсорбции улучшается смачиваемость поверхности адсорбента растворителем в силу благоприятной для этого ориентации молекул ПАВ.
Правило Траубе при адсорбции на поверхности твердых веществ претерпевает ряд изменений. При адсорбции из водных растворов оно имеет вид:
br±L>i. (3.4.22а)
При адсорбции из неполярных растворителей это соотношение обращается в неравенство
bj±L<i. (3.4.226)
Уменьшение кратности адсорбции гомологов обусловлено непроницаемостью твердой фазы для углеводородных радикалов адсорбированных молекул, следовательно, и уменьшением выигрыша в свободной энергии при переходе этих радикалов из водной фазы на поверхность твердого вещества в сравнении с поверхностью раствор—газ.
В неполярных растворителях сродство ПАВ к среде усиливается по мере роста длины гидрофобной части молекулы, поэтому адсорбция высших гомологов снижается. Обращение правила Траубе может быть обусловлено недоступностью внутренней поверхности пористых адсорбентов для крупных молекул.
В технологии часто применяются смешанные растворители, а многие из жидких технических продуктов, например бензин, представляют собой трудно разделяемую смесь близких по свойствам соединений. Эти обстоятельства требуют обратить внимание на нетипичные поверхностно-активные вещества, когда любой из компонентов смешанного растворителя можно рассматривать как слабое поверхностно-активное вещество. В литературе этот случай классифицируется как адсорбция из раствора неограниченно смешивающихся компонентов. Характерные особенности адсорбции в этом случае иллюстрируются серией изотерм (рис. 3.29), когда состав раствора выражен в мольных долях х второго компонента (растворенного вещества), который изменяется от 0 до 1. Решающее влияние на вид изотермы оказывают два параметра адсорбционной системы: неидеальность раствора, которую можно характеризовать энергией Гиббса сольватации молекул в растворе Gs и избирательностью взаимодействия ком
Физика, химия и технология дисперсных систем
589
понента 2 с адсорбентом (энергией Ga). Неполным аналогом величины Gs для несмешивающихся жидкостей 1 и 2 является их адгезия 7Г]2, а величины Ga — адгезия к твердому веществу второго компонента под слоем первого. Аналогия неполная, так как Gs и Ga могут быть как отрицательными, так и положительными. Такого рода характеристики предложены в решеточной модели растворов.
Положительное отклонение от идеальности (Gs > 0) обусловлено тем, что компоненты смешиваются хуже, чем в идеальном растворе (Gs = 0). При этом раствор имеет некоторую тенденцию к самопроизвольному уменьшению концентрации смеси, что и произойдет при выходе на поверхность того компонента, которого меньше в растворе. Следовательно, при х < 0,5 растворенное вещество адсорбируется положительно, а при х > 0,5 — отрицательно (рис. 3.29; кривая 7).
Отрицательная величина Gs означает, что растворимость лучше, чем у идеальной смеси. Компоненты имеют тенденцию к наиболее полному смешиванию, т. е. к поддержанию мольной доли компонента в растворе равной 0,5, поэтому из раствора на поверхность «выжимается» компонент, доля которого выше 0,5. Это ведет к отрицательной адсорбции второго компонента при х < 0,5 и к положительной при х > 0,5 (рис. 3.29; кривая 2). В идеальном растворе (Gs = 0) адсорбция равна нулю при любом составе смеси. Эти закономерности выполняются и на границе растворов с газовой средой.
Состав идеального раствора х = 0,5 наиболее выгоден, если компоненты имеют равное сродство к поверхности адсорбента. Если компонент 2 имеет преимущественное сродство к поверхности (Ga < 0), то адсорбционный слой обогащается этим компонентом, смещая состав раствора, при котором адсорбция равна нулю, в сторону повышенного содержания растворенного компонента (рис. 3.30; кривая 7). Такая тенденция сохраняется при отклонении от идеальности раствора (параметра Gs) в любую сторону. Адсорбционным азеотропом называется раствор, который повторяет по составу адсорбционный слой, т. е. дает нулевую величину адсорбции. При Gs < 0 точка азеотропа смещается в область повышенных концентраций раствора. В обратном случае точка азеотропа лежит левее эквимолекулярной смеси (рис. 3.30; кривая 2). В идеальном растворе (Gs = 0) при Ga < 0 адсорбция второго компонента положительна (рис. 3.30; кривая 3) и при Ga > 0 отрицательна при любом составе раствора, что, в основном, и предполагалось при рассмотрении адсорбции ПАВ на границе с газовой средой.
Рассмотренные закономерности подтверждают возможность выделения тонкой пленки одной жидкости под слоем другой при избирательном смачивании. Необходимая для этого слабая взаимная растворимость (Gs > 0) естественна для несмешивающихся жидкостей.
Наиболее сильные поправки в рассмотренные выше простые случаи адсорбции ПАВ из растворов вносит
химическое взаимодействие ПАВ с адсорбентом. Достаточно характерным в этом отношении является пример взаимодействия высших жирных кислот или их солей с оксидными твердыми адсорбентами, которые в силу плохой растворимости остаются на поверхности в химически связанном состоянии. Первым следствием этого, более сильного взаимодействия растворенного вещества с поверхностью, чем физические адсорбционные силы, является нарушение правил адсорбции и ориентации адсорбированных молекул: адсорбция идет в любой среде, на любой (в смысле гидрофильности) поверхности, имеющей активные центры подходящей химической природы. Молекулы при хемосорбции всегда ориентированы углеводородным радикалом к растворителю независимо от его полярности. При этом нарушается монотонная зависимость адсорбции от температуры, вытекающая из уравнения изотермы Гиббса.
Химическая адсорбция инициируется нагреванием смеси. Поверхность твердого вещества после химической адсорбции дифильных молекул гидрофобизуется. Это обстоятельство широко используется для подготовки природных смесей твердых минералов к разделению по признаку смачиваемости — путем пенной или масляной флотации.
Рис. 3.29. Адсорбция Г одного из компонентов раствора неограниченно смешивающихся жидкостей при положительном (/) и при отрицательном (2) отклонении свойств раствора от идеальности:
х — мольная доля компонента в растворе
Рис. 3.30. Адсорбция Г одного из компонентов раствора с отрицательным отклонением от идеальности при положительном (7) и отрицательном (2) сродстве компонента к адсорбенту. Адсорбция из идеальной смеси при положительном сродстве к адсорбенту положительна (3) при любом составе смеси:
х — мольная доля компонента в растворе
590
Новый справочник химика и технолога
Хемосорбция, как правило, преобладает, если ПАВ является ионогенным, независимо от природы растворителя. В неполярных средах, в которых диссоциация ПАВ сильно затруднена или невозможна, важную роль начинает играть адсорбированная на поверхности твердого вещества вода. В этом слое адсорбированной воды и разыгрываются основные этапы хемосорбционного процесса — диссоциация физически сорбированного ПАВ и обменная химическая реакция. Выделяющаяся при кислотно-основном взаимодействии вода в неполярной среде остается в основном на поверхности адсорбента вместе с молекулами ПАВ, увеличивая толщину водного слоя.
Хемосорбция ПАВ из водных растворов и, следовательно, гидрофобизация поверхности адсорбента подготавливают следующий этап адсорбции — обычную физическую сорбцию ПАВ на гидрофобной поверхности. При этом образуется бимолекулярный адсорбционный слой, при условии наличия в растворе достаточного количества ПАВ (рис. 3.31), а на изотермах адсорбции могут возникать хорошо различимые ступеньки (рис. 3.32) — одна соответствует образованию насыщенного гидрофобизующего монослоя, а вторая — второму, гидрофилизующему насыщенному слою ПАВ.
Если энергии химического взаимодействия молекул с поверхностью и физического взаимодействия их гидрофобных концов между собой близки, т. е. близки константы хемосорбции и физической сорбции, то ступенчатость изотермы сглаживается.
Хемосорбция в большей или меньшей мере необратима по отношению к изменению концентрации ПАВ, температуры, растворителя. Кроме ступенчатости изотермы (необязательной), для хемосорбции характерна неоднозначность зависимости величины адсорбции от равновесной концентрации ПАВ — на начальном участке изотермы из-за полного хемосорбционного связывания первых порций ПАВ адсорбентом начальный участок кривой совпадает с осью ординаты (рис. 3.32).
Рис. 3.31. Хемосорбция из водного раствора
Рис. 3.32. Зависимость хемосорбции ПАВ от его концентрации
Таким образом, имеется достаточное число признаков, позволяющих обнаружить хемосорбционное взаимодействие в системе раствор—твердое вещество. Появление упомянутых признаков должно в первую очередь служить поводом для более внимательного изучения химического состояния поверхности твердого вещества (часто это гидроксильный покров твердого вещества), а также химических свойств полярных групп поверхностно-активных веществ, прежде всего кислотно-основных. Следует иметь в виду, что в подобных случаях решающее влияние могут оказывать pH среды и наличие нейтральных неорганических солей в растворе и растворимых примесей в твердом веществе. Предметом особого внимания должны быть при этом многовалентные ионы, которые, как правило, переводят ионогенные ПАВ в нерастворимые соли.
3.4.7. Адсорбция на внутренней поверхности поликристаллических веществ
Наряду с коллоидными растворами и водной дисперсионной средой коллоидная химия изучает поверхностно-активные вещества. Это твердые поликристал-лические вещества, т. е. практически все те вещества, которые используются в качестве конструкционных материалов. В первую очередь к ним относятся металлы и металлические сплавы. ПАВ играют решающую роль в формировании структуры сплавов, но по строению компоненты этих веществ не имеют ничего общего с дифильными молекулами.
Следует помнить, что фактическая прочность металлов в сотни раз меньше теоретической. Основная причина потери прочности — дефектность кристаллической решетки. Самым распространенным видом дефектов является поликристалличность структуры, а также примеси посторонних атомов. Поликристалличность — неотъемлемая черта строения массовых конструкционных металлов. Уже отмечалось, что межкристаллитная граница имеет поверхностное натяжение и способна адсорбировать на себе всевозможные примеси. По признаку способности к адсорбции примесные компоненты сплавов относятся к поверхностно-активным веществам. Ограниченная растворимость примесей делает их сходными с мицеллообразующими веществами, а плохая совместимость (низкая растворимость) других примесей придает гетерогенность многим металлическим сплавам.
В железе (стали) в силу известных особенностей технологии его получения (восстановление оксидов углеродом) основными примесными компонентами являются углерод, карбиды, ферриты примесных металлов, оксиды двух- и трехвалентного железа. Во многих металлах содержится примесь фосфора, серы, кремния. «Атомом пустоты» является вакансия — один незанятый узел кристаллической решетки. В соответствии с формулой (3.4.2) универсальная «примесь», т. е. пустоты, наиболее склонна к адсорбции.
Физика, химия и технология дисперсных систем
591
Характерная особенность твердой среды и твердых растворов с точки зрения протекания в них процессов адсорбции, мицелл ©образования, коагуляции, капиллярных эффектов, состояния пленочных фаз и других поверхностных явлений заключается в неравновесности состояния этих систем, что обусловлено очень малой (практически нулевой) скоростью диффузии примесей в кристаллической решетке, в том числе к поверхности. По этой причине состояние и свойства металлов решающим образом зависят от того, в какой мере будет позволено пройти в них процессам перераспределения примесей (включая вакансии) на стадии кристаллизации сплава. Мощным средством регулирования этих процессов является термообработка — закалка (резкое охлаждение металла), отпуск (выдержка при повышенной температуре) и другие операции термообработки.
Не менее важна способность металлов и любой другой среды к самоочищению при кристаллизации. Фронт кристаллизации вытесняет любые примеси — посторонние атомы, вакантные узлы, коллоидные частицы и т. д. Именно поэтому льдины в соленых морях состоят из пресной воды, а перекристаллизация является эффективным способом очистки веществ. Примеси, в том числе коллоидные, при кристаллизации выталкиваются на межкристаллитные границы. Аналогичные явления происходят при переходе жидкокристаллических веществ из нормального в ЖК состояние, а в некоторой мере и при полимеризации мономеров. Это ведет к коагуляции коллоидных растворов в этих средах.
Время вытеснения примеси из растущего кристалла зависит от размера кристалликов. По этой и другим причинам резкое переохлаждение сплава, т. е. быстрая кристаллизация, образует мелкокристаллическую структуру, медленное охлаждение — крупнозернистую структуру. Размер кристаллов обычным образом влияет на величину удельной межкристаллитной поверхности — следовательно, и на ее насыщенность примесными компонентами сплава. При постоянстве содержания примеси в материале ее состояние на межфазной границе может изменяться от состояния разреженного или насыщенного адсорбционного монослоя до состояния пленки (тонкой или толстой) и дисперсных частиц более или менее округлой формы. Выделение примеси в виде частиц можно рассматривать как результат собирания пленки в капли при плохом смачивании кристаллов металла выделившейся примесной фазой.
На поверхности твердого вещества, в том числе внутренней, диффузия должна быть выражена более сильно из-за меньшего числа соседей у диффундирующего атома на поверхности, чем в объеме. Таким образом, межкристаллитные границы — это также и каналы для диффузионного перераспределения компонентов поликристаллического материала, в том числе основного (металла) и универсальной примеси (дефектов решетки). Примесные атомы на межкристаллитной границе могут облегчать или затруднять диффузию. Легирование металлов часто направлено на регулиро
вание именно этого свойства внутренней поверхности, а через него на зернистость, прочность, термостойкость, коррозионную стойкость, электрическую проводимость, намагниченность и другие свойства металлов и сплавов. Коррозионная стойкость зависит, например, от скорости диффузии кислорода в металл, а прочность — от скорости диффузии вакансий и других дефектов решетки. Последние диффундируют в область больших механических напряжений, объединяются здесь и образуют зародыш трещины. Уменьшить подвижность и концентрацию дефектов — это значит повысить прочность и термостойкость металла. В сплаве для лопаток газовых турбин реактивных двигателей на основе никеля эту функцию выполняет примесь бора (0,1 % — общее содержание и несколько процентов на межзеренных границах). В дюралюминии (основа А1 + 4,5% Си + 0,5% Mg + 0,5% Мп) — это соединение Си и А1, которое при старении закаленного (т. е. мелкозернистого) сплава образуется на стыках зерен и «пломбирует» каналы диффузии вакансий. Высокий отпуск стали (выдержка при температуре, равной 0,4-0,5 от температуры плавления) позволяет пластинчатым частицам магнетита и карбида железа собраться в округлые частицы, что сильно снижает разупрочняющее действие этих веществ на сталь.
На внешней поверхности адсорбция примесей выражена обычно сильнее, чем на внутренней. На поверхности железа с содержанием фосфора 0,1 % после выдержки при 1450 °C концентрация фосфора достигает 100%. Поверхность никеля, содержащего 99,999% основного вещества, после выдержки при 600-900 °C покрывается монослоем серы, вышедшей на поверхность из объемной фазы металла. В сплаве никель—медь (20 % меди) поверхностный слой на 90 % состоит из меди.
Приведенные примеры показывают, что поверхностная активность и примесей и основных компонентов сплавов весьма велика даже при высоких температурах.
3.4.8. Электрокапиллярность
Влияние взаимодействия между адсорбированными молекулами на поверхностное натяжение наглядно проявляется, если эти молекулы диссоциируют на ионы (ионогенные ПАВ). Взаимное отталкивание адсорбированных одноименно заряженных ионов направлено на увеличение занимаемой ими площади, т. е. действует противоположно силе поверхностного натяжения и, следовательно, уменьшает его. Этот эффект называется электрокапиллярным. Он хорошо изучен. Уравнение, связывающее электрические характеристики поверхности и величину ее натяжения, можно вывести из уравнения Гиббса, применив его к ионам: Г, = -do / dp,.
Адсорбция ионов Г, придает поверхности заряд qs = z,/T„ где F — постоянная Фарадея и z, — формальный заряд (включая знак) адсорбированных ионов. Заряженная поверхность имеет электрический потенциал %, который связан с зарядом qs известной формулой
592
Новый справочник химика и технолога
электростатики: = qs / С, где С — удельная (на единицу поверхности) электрическая емкость поверхности, которую в простейших случаях можно считать постоянной.
Химический потенциал ионов на поверхности включает в себя работу ZiR¥s их переноса из нейтрального раствора на поверхность с потенциалом %, поэтому в случае идеальных растворов изменение химического потенциала при изменении концентрации ионов в растворе будет включать и слагаемое, связанное с сопутствующим изменением величины потенциала поверхности:
d\Xt = ZjFc№s + RT In (de I c).
Тогда, согласно уравнению (3.3.23), можно записать уравнение
do = -Г/ z^d^s + T.RTdc I с. (3.4.23)
Уравнение (3.4.23) можно представить также в следующем виде:
des = -qs c№s + TiRTdc I c.
При постоянстве концентрации ПАВ в растворе уравнение (3.4.23) сводится к уравнению Липпмана — уравнению электрокапиллярности:
des = -qsc№s. (3.4.24)
Если емкость двойного электрического слоя (ДЭС) С не зависит от потенциала Т5, то это уравнение легко интегрируется и дает формулу для электростатического слагаемого <зе поверхностного натяжения:
СТе=-СТ^/2. (3.4.25)
При этом полное натяжение поверхности является алгебраической суммой натяжения о0 незаряженной поверхности и электрокапиллярного эффекта ese:
ез = ез0 + езе. (3.4.26)
Уравнение (3.4.25) как частный случай уравнения (3.4.24) имеет смысл по отношению к мысленному или действительно осуществляемому процессу заряжения поверхности в растворе постоянного состава. Практически этот процесс можно осуществить на границе ртути с раствором электролита путем подачи от внешнего источника разности потенциалов U между ртутью и раствором электролита или ионогенного ПАВ. Полученные этим путем зависимости cs(U) называются элек-трокапиллярными кривыми (рис. 3.33). Они в основном соответствуют уравнениям (3.4.24), (3.4.25) и подтверждают роль отталкивания сорбированных молекул (ионов) в снижении натяжения раствора.
Примечательная особенность электрокапиллярных кривых — наличие на них максимума натяжения. В соответствии с формулой (3.4.26) максимум натяжения дол
жен приходиться на нулевую величину потенциала поверхности. Его смещение на оси потенциалов U означает, что для нейтрализации собственного естественного потенциала поверхности ртути в данном растворе на нее нужно подать определенный внешний потенциал, равный потенциалу нулевого заряда поверхности (рис. 3.33).
Рис. 3.33. Зависимость натяжения границы ртуть—вода от потенциала U, поданного на ртуть:
-----изменение положения максимума при введении в воду анионоактивного ПАВ
Его величина является одной из характеристик электрохимической системы ртуть—раствор и часто называется просто точкой нулевого заряда (ТНЗ). В соответствии с уравнением (3.4.24) на рис. 3.33 слева от ТНЗ поверхность заряжена положительно, а справа — отрицательно. При введении в водную фазу ионогенного поверхностно-активного вещества его поверхностноактивный ион, т. е. органический ион большого размера, будет хорошо адсорбироваться только тогда, когда заряд поверхности ртути противоположен по знаку заряду иона. Соответственно этому натяжение уменьшается только на одной из ветвей кривой (рис. 3.33).
3.4.9. Измерение поверхностного натяжения
В экспериментальных исследованиях дисперсных систем и поверхностных явлений применяется множество различных физических и химических методов. Среди них есть универсальные, такие, например, как измерение концентрации, прозрачности, электрической проводимости растворов, но есть и специфические методы. К ним относится измерение поверхностного натяжения, хотя в большинстве случаев оно сводится к тривиальному измерению силы, действующей на единицу длины некоторого периметра. Тонкости связаны с учетом разнообразных поправок при вычислении натяжения по величине измеренной силы или давления.
Измерение натяжения основано на его макроскопических проявлениях: подъем жидкости в капилляре, избыточное давление в пузырьке газа, втягивание смачиваемой пластинки в жидкость и т. д. В соответствии с этим различают и методы измерения натяжения:
1) метод капиллярного поднятия;
2) метод максимального давления пузырька газа;
Физика, химия и технология дисперсных систем
593
3) метод счета капель жидкости (сталагмометриче-ский метод);
4) метод отрыва кольца;
5) метод капиллярных волн.
Метод капиллярного поднятия. Согласно формуле (3.2.6), при полном смачивании капилляра натяжение о связано с высотой h капиллярного поднятия жидкости формулой
o = Apg/»/tf. (3.4.27)
Здесь К - 21г — кривизна поверхности мениска в полностью смачиваемом капилляре, g — ускорение силы тяжести, Др — разность плотностей контактирующих фаз.
Эта простая формула оказывается неточной вследствие того, что под действием силы тяжести нарушается сферичность мениска — он становится более плоским, т. е. его кривизна на оси капилляра становится меньшей, чем в отсутствие силы тяжести, или фактический радиус мениска г увеличивается по сравнению с радиусом капилляра г0. При полном смачивании соотношение между ними определяется формулой Релея [2]:
г = г0 [1 + (1/3)(г0 / Л) - 0,1288(r0 / h)2 + 0,1312(г0 / Л)3].
(3.4.28)
В методе максимального давления пузырька измеряется давление газа Р, при котором пузырек газа, выдуваемый в жидкость через тонкий капилляр, отрывается от его конца. Это давление должно быть равно капиллярному давлению в газовом пузырьке с радиусом г, равным радиусу капилляра. Поэтому, согласно формуле (3.2.1), между натяжением и давлением газа в пузырьке выполняется соотношение
о = Рг/2. (3.4.29)
По той же причине, что и при капиллярном поднятии, фактический радиус г отрывающегося пузырька оказывается меньше радиуса капилляра г0- Учесть это можно с помощью формулы Шредингера [2]:
г = ГО[1 - (3/2Хг0/Л)-(1/6)(г0/Л)2]. (3.4.30)
Для придания единообразия с формулой Релея здесь давление отрыва пузырька Р выражено через эквивалентную ему высоту столба исследуемой жидкости Л = Р/ pg. Таким образом, если не учитывать искажение формы мениска, то вычисленное натяжение окажется завышенным по сравнению с его фактической величиной. Формула Шредингера (3.4.30), как и формула Релея (3.29), справедлива при достаточно малой величине отношения (г0 / h).
Сталагмометрический метод. Простейшая теория сталагмометрического метода подразумевает, что капля жидкости отрывается от кончика капилляра (трубки), когда ее вес (mg) станет равен силе натяжения 2лг0о,
действующей на линию, длина которой 2лг0 равна длине окружности с радиусом г0, равным радиусу капилляра. Тогда поверхностное натяжение можно вычислить по формуле
<3 = mg/2Ttro. (3.4.31)
Масса капли т вычисляется как средняя величина, полученная из общей массы нескольких капель, вытекших из капилляра. При этом величина натяжения вычисляется довольно грубо по следующим причинам. Фактически перед отрывом капля соединена с капилляром шейкой, радиус которой г меньше радиуса капилляра го- Образование шейки является следствием того, что верхняя часть капли имеет цилиндрическую кривизну, и для создания примерно того же капиллярного давления, что и в нижней части капли, ее радиус должен стать заметно меньше.
Наблюдения показали, что на самом деле отрывается не одна, а две капли. Вторая (меньшего размера) образуется за счет стягивания в каплю сильно растянутой шейки, возникающей при отрыве капли. Поправки на эти эффекты существуют в виде таблиц. Характер этих поправок таков, что вместо величины 1/2л в формуле (3.4.31) появляется переменный коэффициент, величина которого монотонно растет от 0,172 до 0,2559 при уменьшении объема V отрывающихся капель от 5000г03 до О,4ОЗго3 [2].
Метод отрыва кольца (метод дю-Нуи) считается одним из наиболее точных. Он основан на измерении силы F, необходимой для отрыва от поверхности жидкости кольца из тонкой проволоки радиусом R. При этом поверхностное натяжение, сила F и радиус кольца R связаны простым соотношением
o = F/4n/?. (3.4.32)
Точность определения натяжения зависит от смачиваемости кольца и точности его геометрии. В формуле присутствует удвоенная длина окружности кольца 2л/?, так как пленка тянущейся за кольцом жидкости имеет две стороны.
Метод капиллярных волн основан на зависимости скорости и распространения волн по поверхности жидкости от ее натяжения:
и = gA, / 2л + 2ло / рА,. (3.4.33)
Эта формула выведена Томсоном (лордом Кельвином) еще в 1871 г. В эксперименте обычно измеряется длина волны X. Она определяется частотой колебаний f и так как и = А/ то справедливо
a = (р/2 / 2 л) cth (2 л/г / X) А,3 - (pg / 4л2)А,2.(3.4.34)
Формула (3.4.33) предполагает, что глубина h кюветы с жидкостью существенно больше длины волны. В об
594
Новый справочник химика и технолога
щем же случае необходима поправка, равная cth(2?tA / X), которая и внесена в формулу (3.4.34).
Метод капиллярных волн является динамическим, т. е. производимые им результаты должны зависеть от соотношения между частотой возмущений поверхности и временем установления равновесного состава поверхности (см. подраздел 3.4.5). Однако опыт показывает [2], что измеренное натяжение оказывается близким к его статической величине.
Наибольшее различие между статической и динамической величинами натяжения должно иметь место в растворах полимерных поверхностно-активных веществ, каковыми в принципе являются все водорастворимые полимеры. Это обусловлено как медленностью установления равновесия в полимерных системах, так и их особыми реологическими (деформационными) свойствами (см. подразделы 3.9, 3.14, 3.16). В связи с этим следует критически относиться к данным о натяжении полимерных растворов, полученным любым методом.
3.5. Двойной электрический слой
3.5.1. Понятие двойного электрического слоя
Электрические свойства веществ являются функцией их состава и строения, поэтому скачкообразное изменение электрических параметров на межфазной границе является таким же неотъемлемым свойством границы, как и изменение состава. Точнее говоря, электрические величины, как и другие, изменяются постепенно, по мере изменения состава при переходе межфазного слоя конечной толщины (рис. 3.34). Сама возможность видеть границу раздела фаз обусловлена разностью электрических свойств граничащих фаз — диэлектрической проницаемости, проводимости и др.
Исключительно важную роль играет разность электрических потенциалов соприкасающихся фаз. Ее возникновение обусловлено разделением зарядов на границе фаз.
Рис. 334. Диффузный двойной электрический слой:
1 — граница твердой фазы; 2 — раствор электролита; 5 — толщина
Система пространственно разделенных зарядов, в которой заряды одного знака смещены в одну фазу, а другого знака — в другую, называется двойным электрическим слоем (ДЭС). ДЭС существует во всех случаях, когда имеется контакт двух разных фаз: на границе металл—вакуум, металл 1—металл 2, металл—полупроводник, полупроводник 1—полупроводник 2, на границах металлов, полупроводников и диэлектриков с твердыми и жидкими диэлектриками и ионными проводниками (растворами электролитов) и на всех других межфазных границах, если хотя бы в одной из фаз имеются или возникают при взаимодействии фаз носители заряда. Символ «-» (тире) означает наличие общей границы фаз, названных до и после этого символа.
Если одна из фаз является раствором электролита, то двойной слой возникает за счет преимущественной (специфической или избирательной) адсорбции ионов одного знака на границе раздела фаз. На границе твердого вещества с ионным типом связи атомов (золей) ДЭС может возникнуть за счет диссоциации поверхностного слоя вещества твердой фазы. Часто это диссоциация примесей твердого вещества. В дальнейшем под твердой фазой будут подразумеваться в основном малорастворимые неорганические соли или оксиды металлов.
Из раствора электролита специфически адсорбируются на поверхности твердого вещества те ионы, которые образуют с ионами этого вещества малорастворимые в данной жидкой среде соединения. К их числу, в первую очередь, относятся ионы, входящие в состав твердого вещества. Например, на поверхности галогенидов серебра специфически сорбируются галогенид-ионы и ионы серебра, на поверхности силикатных материалов — катионы многовалентных металлов, на поверхности сульфатов и карбонатов щелочноземельных металлов — ионы этих металлов, сульфатные и карбонатные анионы. Большинство оксидов металлов в зависимости от их кислотно-основных свойств сорбируют или, наоборот, десорбируют в результате поверхностной диссоциации гидроксид-ионы или протоны. При этом решающее значение имеет pH раствора как фактор, способствующий или подавляющий проявление кислотно-основных свойств оксидов.
В соответствии со знаком специфически сорбированных ионов поверхность заряжается положительно или отрицательно. Эти ионы называются потенциал-определяющими (ПО). Можно считать, что они расположены в одной плоскости, совпадающей с поверхностью твердого тела (рис. 3.34). Их количество на единицу площади Г( определяет величину удельного (на единицу площади) заряда поверхности qs = ZjFT^ где F— постоянная Фарадея. Нормальная к поверхности координата х будет в дальнейшем отсчитываться именно от этой плоскости. Из раствора к заряженной поверхности притягивается эквивалентное количество противоположно заряженных ионов (противоионов).
Физика, химия и технология дисперсных систем
595
Противоионы, в отличие от локализованных на поверхности ПО ионов, расположены вблизи поверхности более или менее размытым слоем, т. е. диффузно. Эти два слоя противоположно заряженных и пространственно разделенных ионов и являются двойным электрическим слоем. Слой потенциалопределяющих ионов образует внутреннюю часть двойного слоя, а слой противоионов — его внешнюю часть.
Двойной слой, кроме разности потенциалов фаз, поверхностного заряда qs и равного ему по величине, но противоположного по знаку объемного заряда qy внешней части ДЭС, характеризуется толщиной 8, которая фактически является толщиной его внешней части, определяя тем самым объемную плотность р (концентрацию) пространственного заряда этой части двойного слоя.
В растворе присутствуют потенциалопределяющие ионы обоих знаков, так как равновесие жидкой и твердой фаз подразумевает конечную величину растворимости последней и, следовательно, образование насыщенного раствора вещества твердой фазы в жидкой фазе. Вещество твердой фазы имеет очень малую растворимость, и поэтому концентрация насыщенного раствора мала. Следовательно, растворенную часть вещества твердой фазы можно считать полностью диссоциированной, так что в собственном насыщенном растворе концентрации потенциалопределяющих ионов обоих знаков равны между собой и равны растворимости соответствующего вещества. Введение в раствор хорошо растворимой соли, имеющей общий ион с веществом твердой фазы (например, AgNO3, если твердой фазой является AgCl), нарушает указанное равенство, обеспечивает преимущественную адсорбцию введенного ПО иона (в данном случае Ag+) и придает твердой фазе желаемый знак поверхностного заряда. Таким образом, в общем случае заряд поверхности определяется алгебраической суммой зарядов Fz^, по всем видам (/ -индекс вида иона) специфически сорбированных ионов:
qs = F Yz^t = F (z+ Г+- Г_). (3.5.1)
Индексами «+» и «-» здесь и далее помечены абсолютные величины формальных зарядов z катионов и анионов соответственно. В отличие от индексированных величин с конкретным значением индекса («+» или «-») величина zt с неопределенным индексом i включает в себя и знак. Формула (3.5.1) дает пример применения индексов обоих видов. Следует отметить, что адсорбция Г, ионов любого вида — в данном случае величина положительная, так как это адсорбция по Ленгмюру. Знак суммы Е без указания пределов суммирования подразумевает, что оно является полным, т. е. относится ко всему ряду индексированных величин.
При равенстве стехиометрических коэффициентов и пренебрежении гидролизом ионов, степень которого у катионов и анионов может очень сильно отличаться.
Электрический потенциал поверхности Ts (разность потенциалов двух фаз или термодинамический потенциал поверхности) связан с ее удельным (на единицу площади) зарядом qs через электрическую емкость С двойного слоя формулой:
4s = qjC. (3.5.2)
В простейшем случае ДЭС можно представить в виде плоского конденсатора с расстоянием между пластинами, равным толщине 8 ДЭС и удельным зарядом qs одной из пластин. Тогда объемный заряд qv внешней части ДЭС следует приписать второй пластине конденсатора и считать, что он целиком сосредоточен на этой пластине. Удельная (на единицу площади) электрическая емкость плоского конденсатора равна ££0 / 8, где £ — диэлектрическая проницаемость растворителя и £0 — электрическая постоянная (8,85 • 10-12 Ф/м). В таком случае потенциал поверхности связан с ее зарядом и толщиной ДЭС простым соотношением:
% = qs 8 /££0. (3.5.2а)
Двойной слой в целом электронейтрален, что дает еще одно уравнение, связывающее между собой различные его параметры:
qs + qv=Q. (3.5.3)
На самом деле объемный заряд является функцией распределения в пространстве его плотности р и вычисляется интегрированием этой плотности по координате х, направленной по нормали к заряженной поверхности (рис. 3.34):
00
q, = ffxfr. (3.5.4)
О
В свою очередь объемная плотность заряда (его концентрация) р(х) и потенциал Т(х) являются функциями расстояния от поверхности и связаны уравнением Пуассона: АТ = ~р / ££о, где АТ — символическое обозначение оператора Пуассона (суммы вторых производных по всем трем пространственным координатам). В частном случае плоской, бесконечно протяженной заряженной поверхности АТ = 62Т / дх2 (потенциал зависит только от координаты х). Если в уравнении (3.5.4) заменить р с помощью уравнения Пуассона, то получится выражение:
}а2тл
При должной последовательности в описании ДЭС не остается места для сохранения таких понятий, как Гальвани-или Вольта-потенциал.
596
Новый справочник химика и технолога
которое, в свою очередь, можно заменить уравнением
<7g=££0(tM/ / dx)Q. (3.5.5)
Из условия (3.5.3) следует, что удельный поверхностный заряд qs равен той же величине с обратным знаком, т. е. справедливо уравнение
qs = -££о (с№ / dx)0. (3.5.5а)
Первая производная потенциала по координате, взятая с обратным знаком, равна напряженности электрического поля: Е = ~(c№ldx). Кроме того, на бесконечно большом расстоянии от поверхности потенциал, а следовательно, и его производная равны нулю. Таким образом, интегрирование дает отличное от нуля значение только на нижнем пределе, т. е. в формулу (3.5.5) входит напряженность электрического поля на поверхности £0-
£0 = -(б/T / Л)о = -((№ / dx)x=o.
В дальнейшем для краткости первая производная потенциала по расстоянию (без знака «минус») будет называться «напряженностью поля», если это не будет приводить к недоразумениям.
Формула (3.5.5) получена, исходя из общих законов электростатики, и не связана с какими-либо конкретными представлениями о строении ДЭС и о пространственном распределении заряда. Она также справедлива и в отношении любой неполной части внешнего слоя: заряд любой его части, простирающейся от некоторого расстояния х до бесконечности, равен с обратным знаком произведению напряженности поля Е(х) на расстоянии х и абсолютной диэлектрической проницаемости ££о В противоположность этому формула (3.5.5а) не является универсальной — она описывает только те случаи, которые соответствуют частному условию (3.5.3) электронейтральности ДЭС. Более общее условие будет сформулировано позднее, а пока следует выяснить все возможные детали строения двойного слоя в рамках этого простейшего условия. Для этого необходимо решить уравнение Пуассона. Поскольку оно содержит две неизвестные функции — пространственное распределение заряда и потенциала, то для решения задачи требуется еще одно уравнение для тех же функций. Таковым является уравнение Больцмана.
Во внешней части двойного слоя присутствуют ионы обоих знаков: противоионы и коионы — ионы одинакового с потенциалопределяющими ионами знака, которые хотя и отталкиваются от одноименно заряженной поверхности, но в силу теплового движения попадают в двойной слой. Противоионы также участвуют в тепловом движении. Именно по этой причине двойной слой, точнее его внешняя часть, более или менее размыт. Это определяет диффузное строение ДЭС.
Так как противоионы притягиваются к заряженной поверхности, а коионы отталкиваются от нее, то кон
центрация противоионов (с_ — при положительном потенциале поверхности) в двойном слое повышена, а коионов (с+ — при том же потенциале) понижена по сравнению с их концентрациями с0+ и Cq_ в растворе и тем сильнее, чем выше потенциал в данной точке пространства (рис. 3.35).
Рис. 3.35. Распределение противоионов и коионов возле положительно заряженной поверхности:
с- и с+, со. и С(н — концентрации противоионов и коионов в растворе
Несовпадение концентраций катионов и анионов означает наличие пространственного заряда возле поверхности. Его величина (объемная плотность р) равна разности (алгебраической сумме) F(zc. - z с ) зарядов катионов Fzc и анионов Fz с на единицу объема. В случае бинарного симметричного электролита z. = z_ = z, где z — положительное целое число, равное валентности ионов симметричного электролита, и потому р = zF (с+ - с_), а концентрации ионов в растворе с0 ( и с0- одинаковы и равны концентрации с электролита в растворе.
Закон Больцмана связывает концентрацию ионов с, с их потенциальной энергией Epi = z,P¥ в электрическом поле:
ct = cOl exp (-EPi I RT). (3.5.6)
Здесь c( и z, — концентрации и валентности всех ионов, которые присутствуют в растворе.
Кроме потенциалопределяющего электролита раствор может содержать и так называемый индифферентный электролит, который не имеет в своем составе ионов, способных к специфической адсорбции. Таким образом, раствор может состоять более чем из двух видов ионов.
Все дальнейшие вычисления будут относиться только к двум вариантам состава раствора: или это раствор одного бинарного электролита с произвольной комбинацией валентностей катионов и анионов, или это смесь симметричных электролитов, но с одинаковыми валентностями всех ионов. С точки зрения теории строения внешней части ДЭС такая смесь эквивалентна одному симметричному электролиту.
Потенциальная энергия коионов в поле заряженной поверхности положительна, а противоионов — отри
Физика, химия и технология дисперсных систем
597
цательна независимо от знака заряда (потенциала) поверхности, так как первые всегда отталкиваются от поверхности (знак такого взаимодействия всегда будем считать положительным), а вторые — притягиваются (знак отрицательный).
Для сокращения записи удобно ввести безразмерный потенциал W=RV/RT поля и безразмерную энергию ионов и = ZjWв поле.
В симметричном электролите значения последней отличаются для катионов и анионов только знаком, так что z~IV=-zJV= и. При подстановке концентраций из уравнения (3.5.6) в выражение для объемной плотности заряда для произвольного бинарного электролита получится формула:
р = 2/F[exp(-z+ IV) - exp(z_ IV)] I (z+ + z_). (3.5.7)
Здесь i = (z + c0. + z _ c0) /2 — ионная сила раствора. Появление этой величины обусловлено приведением формулы к симметричному, равноправному по отношению к катионам и анионам виду, поскольку исходное выражение:
р = F[z+c0+exp(-z + IV) — z с0 exp(zJf)]
неудобно из-за наличия разных множителей перед экспонентами. При диссоциации бинарного электролита z.c0. = Z-Co-, и z Со можно заменить на z.c0, перед второй экспонентой и затем вынести общий множитель за скобки. В итоге выражение упрощается, но становится несимметричным по отношению к разным ионам, так как содержит только величину zc0:. Его можно привести к симметричному виду, умножив и разделив эту величину на (z+ + z_). Числитель полученного выражения (z 2 с0+ + z_z+Co+) с помощью замены z+c0+ на z. с0 во втором слагаемом приводится к виду (z2 Со+ + z 2_ <?o_). В итоге величина z .c04 после дополнительного умножения и деления на 2 приобретает принятый в формуле (3.5.7) вид 2/ / (z+ + z_), в котором равноправно представлены величины, относящиеся и к катионам, и к анионам. Д ля симметричного электролита z+ = z_ = z, I = z2с и справедлива формула:
р = zFc[exp(-w) - ехр(м)]. (3.5.7 а)
Знак объемного заряда в обеих формулах определяется знаком величин IV или и: при их положительном значении объемный заряд будет отрицательным, в обратном случае он положителен. Формулы (3.5.7) и (3.5.7а) являются по сути дела выражением закона Больцмана (3.5.6) для объемной плотности электрического заряда во внешней части ДЭС. Развернув с помощью той или иной формулы уравнение Пуассона d^/dx2 = -р / ££0, можно получить основное уравнение теории диффузного ДЭС — уравнение Пуассона — Больцмана. В частности, в случае симметричного электролита получается уравнение:
dty/dx2 = -(zFc / ££о) [ехр(-м) - ехр(м)]. (3.5.8)
Потенциал в левой части этого уравнения удобно выразить через безразмерную потенциальную энергию катиона в ДЭС: Т = uRT/ zF, тогда d^/dx2^ (RT/ zF) (du / dx2). Известно, что [exp(-w) - exp(w)] / 2 = sh(w) — гиперболический синус аргумента и, а комбинация постоянных величин в теории электролитов называется параметром Дебая:
x = zF(2c/££o/?7)1/2. (3.5.9)
Тогда уравнение (3.5.8) можно переписать в компактной форме
<?и/ dx2 = -K2sh(u). (3.5.10)
Если представить левую часть уравнения (3.5.10) в виде d(du / dx) / dx и умножить его левую и правую части на du, то оно преобразуется к следующему виду: ydy = -x2sh(w)dw, где у - du / dx. При интегрировании полученного выражения получается следующее уравнение: у2 / 2 = x2ch(w) + Сь где ch(w) — косинус гиперболический аргумента и и С} — постоянная интегрирования. Так как на бесконечном удалении от заряженной поверхности и - 0, а значит у = 0 и ch (и) = 1, то С] = -1. Тогда справедливо уравнение:
(du / dx)2 = 2x2[ch(zz) - 1]. (3.5.10а)
Используя известное преобразование sh2(w / 2) = = [ch (и) - 1] / 2, можно получить уравнение:
du! tfr = -2xsh(z//2). (3.5.11)
Знаки левой и правой частей этого уравнения должны быть различны, так как модуль потенциала и убывает с увеличением расстояния, т. е. d\u\ / dx < 0, a sh(w) является нечетной функцией — ее знак совпадает со знаком аргумента. Выбор знака должен быть сделан при извлечении квадратного корня из обеих частей уравнения (3.5.10а).
Аналогичные преобразования в случае несимметричного бинарного электролита [21] приводят к более общему, чем уравнение (3.5.11), выражению
dW 2
---= х<---- dx I z+ + z.
exp(zJT) -1 exp(zJV) -1
1/2
> .(3.5.12)
Здесь x = F(2I / ££o/?T)1/2. В общем случае это уравнение не интегрируется.
Интегрирование уравнения (3.5.11) осуществляют с помощью табличного интеграла jdu / sh(w) = ln[th(аи / 2)] /а в пределах от х = 0 до произвольного расстояния х и, соответственно, по и от и = us, где us - zR¥, / RT, до произвольной величины и(х). После несложных преобразований получается формула:
598
Новый справочник химика и технолога
th(w / 4) = у(ня)ехр(-хх). (3.5.13)
Функция потенциала поверхности
y(us) = th(wv / 4) = [1 - exp(-wv / 2)] / [1 + exp(-«s / 2)]
изменяется от нуля при us = 0 до 1 при неограниченном увеличении потенциала поверхности. При малой его величине (us «С 1) th(ws / 4) = us 14, при этом и th(w / 4) = = и / 4, так как |«| < |н5|. Такие замены достаточно точны при величине безразмерного потенциала поверхности порядка 1 или потенциале около 25 мВ в случае одновалентных ионов. Поверхность с таким потенциалом считают слабозаряженной. В результате формула (3.5.13) упрощается до
Т = Ч\ехр(-хх). (3.5.13а)
Это выражение для зависимости потенциала от расстояния широко используется в теории двойного слоя, так как позволяет многие задачи теории довести до конечных расчетных формул, что важно для понимания смысла величин и сопоставления их с экспериментальными данными.
Результаты такого анализа часто являются отправной точкой для уточнения теории и представлений о роли тех или иных деталей в достижении соответствия теории и опыта. Зависимость (3.5.13а) графически представлена на рис. 3.36.
Рис. 3.36. Зависимость потенциала от расстояния в двойном слое диффузного строения: 1/х — эффективная толщина ДЭС
Величина 1/х, обратная параметру Дебая х, имеет смысл расстояния, на котором потенциал убывает в е раз по сравнению с потенциалом поверхности Ч\. В теории растворов электролитов аналогичная величина называется радиусом экранирования, или толщиной ионной атмосферы. В случае плоской заряженной поверхности ее уместнее называть эффективной толщиной двойного электрического слоя.
Физическая толщина ДЭС — это расстояние, на котором его внешняя часть полностью нейтрализует поверхностный заряд, т. е. снижает потенциал до нуля. Уравнения (3.5.13) и (3.5.13а) показывают, что потен
циал достигает нулевой величины только при бесконечном удалении от поверхности. ДЭС не имеет определенной физической толщины, как это показано на рис. 3.34, и поэтому в качестве его геометрической характеристики приходится использовать эффективную толщину 5 = 1/х.
При дифференцировании Т по х получается выражение напряженности поля Е = -сЛР / dx слабо заряженного ДЭС:
Е = -х% ехр(-хх). (3.5.14)
При произвольном потенциале или в растворе несимметричного электролита она определяется из уравнений (3.5.11) или (3.5.12) после приведения их левых частей к размерному виду в соответствии с определениями величин и и W.
Напряженность поля, как и потенциал, экспоненциально убывает по модулю с увеличением расстояния от заряженной поверхности, снижаясь до нуля. Практически же электрическое поле заряженной поверхности ощутимо на расстояниях в несколько эффективных толщин. При схематичном изображении строения ДЭС важно показать его соответствие принципу электронейтральности двойного слоя.
Полученные формулы для потенциала и напряженности поля позволяют конкретизировать связь между потенциалом и зарядом поверхности, представленную пока только общими универсальными соотношениями (3.5.2) и (3.5.5). Из формулы (3.5.14), подставив х = О, легко найти величину первой производной потенциала непосредственно на поверхности: (</Р / dx)0 = При подстановке ее в уравнение (3.5.5) получается формула:
^ = ££0х%. (3.5.15)
При сопоставлении формул (3.5.2) и (3.5.15) следует, что электрическая емкость ДЭС определяется формулами:
С ~ ££оХ (3.5.16)
или
С = ££0/8. (3.5.16а)
При сравнении выражения (3.5.16а) с классической формулой емкости плоского конденсатора эффективная толщина 5 ДЭС совпадает с эффективной толщиной плоского конденсатора.
При произвольном потенциале поверхности в растворе симметричного электролита аналогичные преобразования дают формулу:
С = ££Ox[sh(w5 / 2)/(us 12)]. (3.5.17)
В этом случае толщина ДЭС 8 не совпадает с эффективной толщиной плоского конденсатора. Так как значение sh(ws / 2) больше значения его аргумента (us / 2), то емкость сильно заряженного ДЭС больше, чем слабо заряженного, и тогда потенциал поверхности % растет
Физика, химия и технология дисперсных систем
599
с увеличением ее заряда qs слабее, чем при малых потенциалах поверхности.
Связь между потенциалом и зарядом поверхности играет фундаментальную роль в различного рода электрохимических системах — гальванических ваннах, аккумуляторах электрической энергии, электролитических конденсаторах, мембранах нервных клеток и т. д. Поэтому ей уделяется большое внимание в экспериментальных исследованиях прикладного и фундаментального характера. Перечень доступных для таких исследований методов ограничен. Один из них — метод электрокапиллярных кривых (подраздел 3.4.8). Согласно уравнению элекгрокапиллярности, первая производная от поверхностного натяжения по потенциалу равна заряду поверхности (с обратным знаком), а вторая — дифференциальной емкости заряженной поверхности dqs / с№, имеющей слой противоионов и являющейся частью ДЭС. Следовательно, определяемая из электро-капиллярной кривой дифференциальная емкость — это емкость двойного слоя. При малом потенциале поверхности емкость ДЭС не зависит от потенциала и, следовательно, интегральная и дифференциальная емкости совпадают. В общем же случае, представленном формулами (3.5.2) и (3.5.17), не представляет труда вычисление и дифференциальной емкости.
Сопоставление экспериментальных данных с формулами (3.5.16) и (3.5.17) показывает, что формулы дают сильно завышенную величину емкости ДЭС, особенно при больших потенциалах, когда емкость, согласно формуле (3.5.17), неограниченно растет с увеличением потенциала, что заведомо не соответствует действительности. Так как в рассмотренной теории двойного слоя (теория Гун и Чепмена) ионы считаются заряженными математическими точками, то они могут подходить к заряженной поверхности как угодно близко, в том числе располагаться на нулевом расстоянии от нее. Это дает заниженную величину среднего расстояния противоионов от поверхности, т. е. заниженную толщину эффективного конденсатора и, следовательно, завышенную его емкость.
3.5.2. Плотная и диффузная части ДЭС
Получить более точные соотношения можно, если учесть размер ионов. Проще всего это сделать, ограничив сферу применимости уравнения Пуассона — Больцмана (3.5.10) расстояниями х большими, чем диаметр ионов d. На меньших расстояниях присутствие и противоионов, и коионов невозможно, поэтому здесь (при х< d) плотность объемного заряда р равна нулю. Уравнение Пуассона для этой части ДЭС имеет следующий вид: / dx2 = 0, а его первый интеграл —
/ dx = С2, где С2 — константа интегрирования. Постоянство первой производной / dx означает, что здесь потенциал изменяется линейно, или напряженность поля Е = -с№ / dx остается постоянной. Далее эта область (рис. 3.37) называется плотной частью ДЭС (плотным слоем или слоем Гельмгольца), а плоскость, в
которой сосредоточены центры ближайшего слоя противоионов, — плоскостью максимального приближения противоионов. Она расположена на расстоянии х = d от заряженной поверхности. За пределами этой плоскости (х > d) противоионы и коионы распределены диффузно и сохраняют силу основные соотношения теории диффузного слоя Гуи — Чепмена. Для исчерпывающего описания такого ДЭС в дополнение к потенциалу % заряженной поверхности, параметру Дебая х и координате плоскости максимального приближения d нужно ввести еще один параметр, характеризующий ДЭС, — потенциал Т] в плоскости х = d(рис. 3.38).
Для этого систему уравнений теории ДЭС следует дополнить выражением для напряженности поля в плотном слое Ех = (% -4'1)/ которая совпадает с упомянутой выше константой С2 интегрирования уравнения Пуассона для плотного слоя. За пределами плотного слоя распределение ионов и потенциала подчиняется всем закономерностям диффузного слоя, что схематично показано на рис. 3.37 и 3.38.
5--------►
Рис. 3.37. Строение ДЭС при наличии плотного слоятолщиной d‘. 1 — граница твердой фазы; 2 — раствор электролита; 8 — толщина ДЭС
Рис. 3.38. Распределение потенциала в двойном слое при наличии плотной части: 4*1 — потенциал внешней границы плотного слоя
600
Новый справочник химика и технолога
Изложенные представления составляют суть теории Штерна. Деление ДЭС на плотную и диффузную части связано не только с геометрическими различиями в положении ионов разных частей ДЭС.
К плотной относят часть ДЭС, которая расположена в интервале расстояний от нуля до х = d включительно, т. е. в нее входят все потенциалопределяющие ионы и некоторая доля ионов внешней части ДЭС, локализованных в плоскости максимального приближения. Остальная часть двойного слоя — это оставшиеся противоионы и коионы внешней части — относится к диффузному слою. Следует помнить, что в плотном слое потенциал снижается линейно. Одной из проблем теории двойного слоя является отсутствие общего мнения о том, какие ионы внешней части ДЭС следует относить к плотному слою.
В диффузной части ДЭС (х > d) потенциал изменяется в соответствии с полученными ранее экспоненциальным (3.5.13а) или более сложным (3.5.13) законами. Сохраняют силу и некоторые другие из полученных ранее соотношений теории диффузного слоя при замене в них на 'Pi, х на х - d и (d¥ldx) х = 0 на (c№ldx) x = d, например, соотношения:
'Р = 'Pi ехр[-х(х - t/)], (3.5.18)
qv = &&Q(dV / dx)x = d. (3.5.19)
Здесь qv представляет собой заряд всех ионов внешней части ДЭС. Если плоскость максимального приближения пуста, т. е. локализованный в ней заряд равен нулю, то по условию электронейтральности двойного слоя заряд поверхности qs = -qv и поэтому при малом потенциале поверхности справедлива формула:
qs = ЕЕох'Рр (3.5.20)
Если определить емкость С двойного слоя как отношение заряда поверхности к ее потенциалу, то получается формула:
С = ееох'РЛ. (3.5.20а)
При произвольном потенциале справеливы формулы:
qs - ££о к (2RT/zF) sh(ui / 2), (3.5.21)
С — ££0 х sh(W] / 2) / (w4. / 2). (3.5.21а)
В растворе несимметричного электролита в соответствии с уравнением (3.5.12) выполняется уравнение
tflH 2
---- = х<!-------
tic Jx=d I z+ +z.
exp(zH/!) -1 exp(z Щ) -1
z_ z+
1/2
(3.5.22)
Здесь JVi = PPi / RT. Согласно уравнению (3.5.19), qv = = -Е£о(бЛР / dx)x=d, (tiP / dx)x -d = (RT I F)(dW I dx)x = d, a емкость ДЭС С определена как отношение заряда поверхности к ее потенциалу — qsT¥s.
Из формулы (3.5.20а) следует, что при малом потенциале поверхности емкость двойного слоя ионов конечного размера меньше, чем емкость слоя точечных ионов, в (% / 'PJ раз. Это различие полностью зависит от потенциала 'Pi, который пока фигурировал в теории только как дополнительный независимый параметр теории двойного слоя.
Принципиально важна возможность выражения потенциала 'Р] через другие параметры ДЭС. Эта возможность зависит от того, имеются ли еще какие-либо силы, кроме уже учтенных сил электростатического взаимодействия противоионов и коионов с заряженной поверхностью и стерических сил, учтенных введением в уравнения размера ионов. Например, это могут быть силы химического сродства ионов к поверхности. Обычно их называют специфическими. Они проявляют себя в виде адсорбции ионов строго в плоскости максимального приближения ионов в соответствии с теорией адсорбции Ленгмюра, т. е. аналогичны силам адсорбции потенциалопределяющих ионов и отличаются только плоскостью локализации тех и других. Учет специфического взаимодействия ионов внешней части ДЭС с поверхностью и является центральным положением теории Штерна.
Представим уравнение Пуассона в эквивалентном виде: pdx = -££od(d¥ / dx). Здесь pdx — заряд произвольного слоя толщиной dx, -d(d¥ / dx) — приращение напряженности поля на этой толщине — т. е. разность напряженностей поля на «левой» и «правой» сторонах этого слоя. Пусть внутри этого слоя выделена некоторая плоскость d нулевой толщины. Если заряд распределен только по объему слоя dx с плотностью р, то при его уменьшении до толщины d, равной нулю, и при стягивании слоя к плоскости d заряд слоя будет, в соответствии с левой частью уравнения Пуассона, уменьшаться до нуля. Это означает, что плоскость d не несет на себе никакого заряда. Правая часть уравнения также обращается в нуль, а это значит, что скачок напряженности поля d (dV I dx) на плоскости равен нулю, т. е. напряженности поля на «левой» и «правой» сторонах плоскости одинаковы.
Если кроме объемного заряда с плотностью р имеется дополнительный заряд, сосредоточенный в плоскости d с поверхностной плотностью q\, то при уменьшении толщины слоя до нуля его заряд стремится к величине q\, а скачок напряженности поля при переходе через плоскость имеет, согласно уравнению Пуассона, конечную величину,
= <71 / ££0. (3.5.23)
Как уже отмечалось, напряженность поля на «левой» (ближней к поверхности) стороне плоскости максимального приближения ионов (рис. 3.38) в любом случае определяется формулой Е\ = ('Ps-'Pi) / d, а на «правой» — формулой Е2 - хЧ^ (при малом потенциале поверхности). Тогда &Е = Е2- Еь что с учетом формулы (3.5.23) приводит к соотношению:
Физика, химия и технология дисперсных систем
601
Ч'1=(^+Ч\ /(l+xrf). (3.5.24)
кее0 )
В отсутствие специфического взаимодействия противоионов с поверхностью их количество q\ в плоскости максимального приближения равно нулю, и тогда потенциал этой плоскости описывается формулой:
Tj = 4х, / (1 + xd). (3.5.24а)
С учетом формулы (3.5.20а) емкость ДЭС определяется формулой:
С = 8£qx / (1 + xd). (3.5.25)
Напряженность поля на обеих сторонах пустой плоскости максимального приближения ионов при этом одинакова. На зависимости потенциала от расстояния это обстоятельство проявится в отсутствии излома графика на расстоянии х = d(рис. 3.38).
Таким образом, поправка на конечный размер ионов в количественном отношении не дает существенного результата, так как она может сказаться только при xd> 1, что соответствует неприемлемо большим концентрациям электролита. Параметр Дебая х увеличивается пропорционально корню квадратному из концентрации электролита (3.5.9), а емкость двойного слоя асимптотически возрастает, согласно ленгмюровскому закону (3.5.25), до величины Ст = £Ео / d при неограниченном увеличении концентрации электролита. Из структуры этого выражения видно, что Ст представляет собой емкость плоского конденсатора с расстоянием между пластинами, равным размеру ионов. В теории ДЭС эта величина известна как емкость молекулярного конденсатора. При умножении обеих частей формулы Ст = ££о / d на xd легко установить связь емкости Ст молекулярного конденсатора с классической емкостью С/=££ох диффузного ДЭС: Cmxd= 8£ох. С помощью полученного соотношения формула (3.5.25) преобразуется в уравнение:
1 /С= 1/Сг+1 /Ст. (3.5.26)
Изменение обозначения емкости диффузного слоя (Су вместо Q, принятого ранее в формулах (3.5.16) и (3.5.17), продиктовано стремлением сохранить прежнее обозначение С за емкостью всего ДЭС, поскольку выясняется, что в модернизированной теории ДЭС его емкость является комбинацией двух независимых величин: емкости молекулярного конденсатора Ст и емкости диффузного слоя Су. В связи с этим необходимо обратить внимание на важные обстоятельства.
1. В соответствии с законами электротехники формула (3.5.26) означает, что емкость ДЭС равна емкости двух последовательно соединенных конденсаторов, что согласуется с взаимным расположением плотного и диффузного слоев, показанных на рис. 3.37. При таком соединении общая емкость двух конденсаторов не превышает меньшей из двух соединенных емкостей. Это и
позволяет избежать неограниченного роста емкости с увеличением потенциала, что является основным недостатком теории Гуи — Чепмена (рис. 3.39).
2. Формула (3.5.26) является развернутым вариантом формулы (3.5.20а), определяющей смысл понятия «электрическая емкость» для ДЭС, состоящего из ионов конечного размера.
3. В рассмотренном варианте (q^ = 0) молекулярный конденсатор остается пустым — двойной слой не содержит никаких других, кроме диффузно распределенных ионов внешнего слоя и эквивалентного количества потенциалопределяющих ионов на поверхности. Несмотря на это, емкость молекулярного конденсатора приобретает доминирующую роль при больших концентрациях электролита.
Деление ДЭС на плотную и диффузную части и, соответственно, на молекулярный и диффузный конденсаторы не связано со специфической адсорбцией противоионов, как это иногда представляется. Такое деление возможно и при пустом молекулярном конденсаторе, т. е. при полном отсутствии специфического взаимодействия противоионов конечного размера с заряженной поверхностью. Более того, если заряд плоскости qi не равен нулю, то формула для емкости ДЭС, получаемая путем выражения величины потенциала 4х] в формуле (3.5.20а) через потенциал поверхности 4х, с помощью уравнения (3.5.24), не сведется к формуле (3.5.26), означающей последовательное соединение емкостей диффузного и плотного слоев.
Формула (3.5.26) выведена при малом потенциале поверхности. При произвольном потенциале те же рассуждения приводят вместо формулы (3.5.25) к трансцендентному уравнению, которое далее должно использоваться совместно с формулой (3.5.21а) вместо формулы (3.5.20а):
us = U\ + 2xdsh(wi / 2). (3.5.25a)
Наиболее продуктивная сторона теории Штерна заключается, как уже отмечалось, не в разделении ДЭС на две части, а в учете специфического взаимодействия ионов внешней части ДЭС с поверхностью, которое приводит к заполнению плоскости максимального приближения по законам ленгмюровской адсорбции теми же ионами, которые образуют диффузный слой.
Рис. 3.39. Электрическая емкость С диффузного ДЭС как функция потенциала поверхности
602
Новый справочник химика и технолога
Кроме экспериментальных данных о емкости ДЭС, которые указывают на необходимость усовершенствования теории ДЭС, существует еще один массив обширных и достаточно хорошо обобщенных экспериментальных данных. Это данные, полученные с помощью электрокинетических явлений.
Известно, что при движении одной из фаз относительно другой жидкая фаза смещается относительно твердой заряженной поверхности не по границе раздела фаз, а по некоторой плоскости s, которая называется плоскостью скольжения. Очевидно, что при этом перемещаться относительно поверхности будет только заряд qs, расположенный с внешней стороны от плоскости скольжения. Его величина однозначно связана с напряженностью поля Es на плоскости скольжения формулой (3.5.19) при подстановке в нее координаты плоскости скольжения s вместо координаты плоскости максимального приближения противоионов d.
В свою очередь напряженность поля связана с потенциалом на плоскости скольжения Эта связь опи-сывется формулой Es = В электрокинетических экспериментах этот потенциал поверхности s измеряется по скорости относительного движения фаз и поэтому называется электрокинетическим потенциалом. Обычно предполагается, что плоскость s расположена дальше от поверхности, чем плоскость d. В целях упрощения далее можно считать (если это не оговорено специально), что эти плоскости, а следовательно, и потенциалы Т] и Q совпадают.
В рассмотренной выше связи электрокинетического потенциала с другими параметрами ДЭС обычно обращают внимание на то, что с увеличением концентрации индифферентного электролита происходит сжатие двойного слоя. При этом считается, что заряд диффузной части ДЭС уменьшается вследствие перехода части ионов из диффузной в плотную часть двойного слоя. В таком случае электрокинетический потенциал должен монотонно уменьшаться с увеличением концентрации индифферентного электролита.
Эксперимент часто дает зависимость ^-потенциала от концентрации индифферентного электролита, которая не может быть сведена только к эффекту уменьшения дебаевской толщины ДЭС. Так, в ряде случаев наблюдается увеличение электрокинетического потенциала при увеличении концентрации заведомо индифферентного электролита. Усложнение характера зависимости электрокинетического потенциала от состава раствора считается проявлением специфичности (неин-дифферентности) взаимодействия противоионов, т. е. взаимодействия ионов с поверхностью. В связи с этим следует отметить, что в отсутствие специфического взаимодействия противоионов с поверхностью плоскость максимального приближения должна оставаться пустой и, следовательно, по условию электронейтральности ДЭС заряд диффузного слоя (т. е. всего внешнего слоя) не может уменьшаться при сжатии ДЭС. Более того, согласно формулам (3.5.20) и (3.5.24а), при посто
янстве потенциала (при постоянстве концентрации ПО ионов) и умеренной концентрации индифферентного электролита (%d С 1) заряд диффузного слоя должен увеличиваться, равно как и заряд поверхности. Если при этом плоскости 5 и d совпадают, то ^-потенциал будет оставаться равным потенциалу % независимо от концентрации электролита. Иначе говоря, самым очевидным проявлением специфичности противоионов к заряженной поверхности является тривиальное уменьшение электрокинетического потенциала с увеличением концентрации индифферентного электролита.
3.5.3. Специфическая адсорбция и емкость ДЭС
Количественная сторона теории Штерна сводится к двум приведенным ниже уравнениям, первое из которых есть уравнение изотермы адсорбции Ленгмюра, отнесенное к ионам z-го вида, а второе связывает константу к, адсорбционного равновесия этих ионов с потенциалом W- PV / RT в плоскости адсорбции и энергией Ф, их специфического взаимодействия с поверхностью:
/(1+Ла), (3.5.27)
kt= И„ехр (Ф,--ДЭ. (3.5.28)
При их анализе прежде всего нужно иметь в виду, что уравнение (3.5.27) лишь внешне сходно с уравнением Ленгмюра. Фактически это совсем другое уравнение, поскольку величина к, в нем не является константой. Она сложным образом зависит от концентрации ионов с,. Выяснение этой зависимости является основной целью последующего изложения.
Пусть заданы состав раствора, размер d и энергия химического сродства Ф( ионов к поверхности, на которой образуется ДЭС (для краткости будем называть ее электродом), и предельные адсорбции ионов. Уравнения (3.5.27) и (3.5.28) в равной мере могут быть применены как к слою потенциалопределяющих ионов, так и к слою ионов, локализованных в плоскости их максимального приближения (в плоскости d), т. е. к противоионам и коионам. Каждое из выражений (3.5.27) и (3.5.28) в случае бинарного электролита представляет два отдельных уравнения: уравнения адсорбции катионов и анионов по отдельности и уравнения для констант их адсорбционного равновесия. Уравнение адсорбции позволяет вычислить удельный заряд поверхности qs по формуле (3.5.1) qs = FZz^, или заряд <7i плоскости d по той же формуле. Разумеется, что для этого в формулу (3.5.1) должны быть подставлены адсорбции Г; = Гт;(-)( в соответствующих плоскостях локализации ионов — на поверхности или в плоскости максимального приближения, вычисленные по формуле (3.5.27) с использованием подходящих для каждого случая констант (3.5.28) и предельных адсорбций Гт(. Константы адсорбционного равновесия можно найти, только зная потенциалы в соответствующих плоскостях адсорбции. Потенциалы, в свою очередь, определяются искомыми величинами qs или q} через емкость или все
Физика, химия и технология дисперсных систем
603
го ДЭС, или его части соответственно. Емкость ДЭС (или его части) является, в сущности, его геометрической характеристикой, включающей в себя как толщину диффузной и плотной частей, так и распределение заряда между ними, поэтому вычисление заряда поверхности оказывается невозможным без предварительного полного расчета всех параметров внешнего слоя, включая qv и qx. Таким образом, количественные вычисления в рамках уравнений Штерна практически осуществимы только через расчет параметров внешней части ДЭС, что обычно и предполагалось, хотя имели место попытки автономного (независимо от внешней части) расчета параметров поверхности электрода.
Уравнение Штерна (3.5.28) исключает существование индифферентных электролитов, так как при нулевой энергии химического сродства или полной энергии (включая электростатическую) оно дает конечную величину константы адсорбционного равновесия, равную Vn, т. е. предусматривает адсорбцию ионов и в тех случаях, когда ее не должно быть. Чем слабее химическое сродство и меньше потенциал в плоскости адсорбции, тем заметнее ошибка. Это результат хотя и общепринятой, но, тем не менее, не вполне корректной интерпретации самого понятия адсорбции по Ленгмюру. Истоки ошибки находятся в самой логике вывода уравнения изотермы адсорбции: при вычислении константы адсорбционного равновесия скорость адсорбции приравнивается к полному числу столкновений молекул (ионов) сорбата с поверхностью. В действительности из полного числа столкновений нужно исключить упругие столкновения, что приводит к уравнению:
k = Vn exp(-z,JF)[exp(O,) - 1] (3.5.29)
Уравнение (3.5.29) лишено упомянутых недостатков уравнения (3.5.28).
Обоснование общего вида этого уравнения было дано при рассмотрении теории мономолекулярной адсорбции газов (подраздел 3.1.4, формула 3.1.19). Представленный формулой (3.5.29) вариант содержит дополнительные детали, по сравнению с исходной формулой (3.1.19). Они связаны с тем, что полная энергия адсорбции ионов состоит из двух слагаемых: электростатического -ZjW и специфического Ф(, так что экспонента классического уравнения Штерна (3.5.28) может быть представлена в виде двух сомножителей: ехр(-2,1^) и ехр(Ф<), роль которых различна. В теории адсорбции Ленгмюра адсорбционные силы действуют только на молекулы (ионы), которые находятся в плоскости адсорбции и обращаются в нуль при малейшем увеличении расстояния между молекулой (ионом) и поверхностью, что означает ее десорбцию.
Электростатическая энергия иона z:W при бесконечно малом изменении его положения остается практически неизменной, и, следовательно, в соответствии с общими принципами термодинамики электростатическое взаимодействие иона с поверхностью не может быть ни движущей силой, ни причиной мономолеку
лярной адсорбции. Его роль состоит лишь в том, что оно ведет к повышению концентрации противоионов (и понижению концентрации коионов) возле заряженной поверхности в exp(-ZjWz) раз по сравнению с концентрацией в растворе. Понятно, что ленгмюровская адсорбция (число столкновений) определяется концентрацией именно вблизи поверхности, а не в растворе. Поправка же на упругие столкновения (-1) относится только к короткодействующим силам ленгмюровской адсорбции, что и нашло свое отражение в структуре конечного уравнения (3.5.29) для константы равновесия.
Новое уравнение для констант равновесия не снимает упомянутых выше математических трудностей, связанных с зависимостью этих констант от потенциалов, которые можно найти, только зная заряд, рассчитываемый с помощью тех же констант. Этот замкнутый круг разрывается, если в качестве независимой переменной принять потенциал Ч71 плоскости максимального приближения ионов (плоскости d). По этому потенциалу с помощью частных или общих формул (3.5.21а), (3.5.22) теории диффузного слоя однозначно вычисляется его заряд qy и заряд q} специфически сорбированных ионов в плоскости d с помощью формул (3.5.27), (3.5.29), а значит и полный заряд внешней части ДЭС и заряд поверхности. Эти же величины вместе с принятыми значениями d и х позволяют вычислить по формуле типа (3.5.24) потенциал поверхности и далее емкость двойного слоя. Типичные зависимости дифференциальной емкости dq / c№s от потенциала поверхности Ч\, полученные таким путем, представлены графически на рис. 3.39.
Экспериментальные зависимости, полученные методом электрокапиллярности, оказываются, как правило, значительно сложнее. Они отличаются наличием многих экстремумов и ассиметрией расположения катодных и анодных ветвей поляризации электрода. Они отличаются наличием многих экстремумов и асимметрией расположения катодных и анодных ветвей поляризации электрода (рис. 3.40).
Рис. 3.40. Зависимость емкости границы ртуть—водный раствор NaCl от потенциала ртутного электрода при разных концентрациях NaCl
604
Новый справочник химика и технолога
Кроме того, фактическая величина емкости всегда оказывается заметно меньше вычисленных значений. Это означает, что теория ДЭС нуждается в дальнейшем усовершенствовании. Последним достижением в этой области считается [22] модель двойного слоя, предложенная Грэмом [44].
3.5.4. Двухслойная модель плотного слоя
Основная особенность модели Грэма заключается в наличии в двойном слое двух плоскостей локализации ионов в плотной части — ближней (первой) и дальней (второй). Эти плоскости удалены от поверхности электрода на расстояния, равные размерам ионов, причем предполагается, что это ионы одного знака — противоионы, но часть их дегидратирована и поэтому имеет меньший размер d\, а другая часть остается гидратированной и имеет больший размер <4- Локализованным в первой плоскости ионам приписывается способность специфически взаимодействовать с электродом (сорбироваться), тогда как ионы второй плоскости взаимодействуют с электродом только электростатически. Количественная сторона теории Грэма полностью повторяет теорию Штерна и сводится к тем же уравнениям (3.5.27) и (3.5.28) и с теми же параметрами.
В остальном же она носит декларативный характер — задача не доводится до решения уравнений. Теория Грэма заслужила популярность, так как в ней высказано предположение, что при большой концентрации электролита зависимость емкости С двойного слоя от потенциала поверхности является не чем иным, как зависимостью емкости молекулярного конденсатора Ст от потенциала поверхности %. Выше (подраздел 3.5.2) обращалось внимание на то, что при больших концентрациях электролита, согласно формуле (3.5.26), емкость ДЭС равна емкости молекулярного конденсатора Ст. Оригинальность же идеи Грэма в том, что он распространил это очевидное положение на равенство функций Сда=Х%) и С=Х%), описывающих зависимости емкостей от потенциала поверхности.
Считается, что функция Ст молекулярной емкости остается такой же при любых концентрациях, что дает возможность при любых концентрациях электролита рассчитывать функцию полной емкости С=Д%) по формуле (3.5.26) вместе с доступной для расчетов функцией емкости диффузного слоя Cv-fi^Vs) по одной из формул типа (3.5.19)-(3.5.22), (3.5.25а). Таким образом, продуктивная часть теории сводится к обоснованию нового эффективного способа обработки и экстраполяции экспериментальных данных.
Однако формула (3.5.26) последовательного соединения емкостей диффузного и плотного слоев характерна лишь для случая отсутствия специфической адсорбции ионов в плоскости максимального приближения.
Теория Грэма не решает фундаментальных проблем теории ДЭС, а лишь переносит их в область строения и свойств плотной части двойного слоя (молекулярного конденсатора).
Рассмотрим двухслойную модель плотного слоя (рис. 3.41), добавив в нее некоторые изменения.
1. Различия в размерах относятся, прежде всего, к ионам разного знака. Это положение использовалось Фрумкиным [43]. На его основе с помощью эмпирического уравнения он получил практически те же результаты, что и Грэм. Меньший размер может иметь или катион, или анион.
2. Специфическое взаимодействие присуще ионам, локализующимся как в ближней, так и в дальней плоскости.
3. Ионы меньшего размера присутствуют также и в слое, заключенном между первой и второй плоскостями локализации ионов.
Рис. 3.41. Строение ДЭС при наличии разницы в размерах d\ и d2 противоионов и коионов индифферентного электролита
Последнее положение влечет за собой наиболее значительные последствия. Уравнение Пуассона — Больцмана для указанного слоя содержит в правой части только одно слагаемое — объемную плотность заряда, создаваемую в этом слое ионами малого размера:
/ dx2 = -(zi FCl /££0) exp(-Z! ff). (3.5.30)
Здесь ci — концентрация ионов меньшего размера в растворе, Т и W — текущее значение размерного и безразмерного потенциалов на расстоянии х от поверхности электрода.
Уравнение (3.5.30) удобно представить в форме:
(dV /dx)d (с№ /dx) + G ехр(-г1 ff)d (-zt №) (3.5.30a)
Здесь G = C\RT/ ££0. Уравнение (3.5.30a) легко интегрируется и дает уравнение
/ dx)2 / 2 = G exp (-Zl W) + A. (3.5.31)
Физика, химия и технология дисперсных систем
605
Здесь А — константа интегрирования. Они связана с потенциалом первой плоскости % и напряженностью поля в этой плоскости Е\ уравнением (3.5.32а), где Wx = PPj / RT), Ех = (-<F¥ / dx) при х = dx:
А = (1/2) Е? - G exp(-z, Wx). (3.5.32a)
Константу А можно выразить и через аналогичные параметры второй плоскости с помощью формулы
А = (1/2) £22 - G exp(-Z! W2). (3.5.326)
Следует подчеркнуть, что Ех представляет собой напряженность поля на внешней (обращенной к раствору) стороне первой плоскости, а Е2 — на внутренней (обращенной к электроду) стороне второй плоскости. Иначе говоря, это напряженности на внутренних сторонах плоскостей, ограничивающих с обеих сторон слой, доступный только для ионов малого размера. При этом надо иметь в виду, что напряженность поля на плоскости изменяется скачком пропорционально сосредоточенному в плоскости заряду (закон Гаусса).
После извлечения квадратного корня и разделения переменных из уравнения (3.5.31) получается формула
+j2dx = -=----—- —. (3.5.33)
y]G exp(-ZjlF)+ А
Это уравнение имеет два различных решения в зависимости от знака константы А. При А > 0 справедлива формула:
(3.5.34)
При А < 0 следует пользоваться формулой:
(3.5.35)
Здесь /1 = Gexp(-Z]IFi) + A, I2 = Gexp(-z2JF2)+A, В = Fl RT.
Раскрытие интеграла в выражении для заряда слоя
^2
qg = zxcxF Jexp(-Z]lF)<ir
дает формулу:
Я„ = ±V2ss0 (7Л - 7Л). (3.5.36)
Если в данном выражении развернуть величины 1Х и 12, а также величину А с помощью формул (3.5.32а) и (3.5.326), то получится результат столь же очевидный, сколь и бесполезный: qg = ee0(£i - Е2). Он демонстрирует соответствие результатов закону Гаусса, т. е. отсутствие ошибок в проделанных выкладках, но попытка воспользоваться им для вычисления заряда снова воз
вращает нас к уравнению (3.5.36), так как напряженности поля придется выразить через потенциалы на соответствующих плоскостях.
Заряд поверхности qs (заряд внутренней части ДЭС) равен с обратным знаком полному заряду его внешней части qe: qs = -qe, где qe = qx + q2 + qg + qv. Здесь qv = ££o£r— заряд диффузного слоя, выраженный через напряженность поля Еу на внешней стороне второй плоскости локализации ионов, которая вычисляется по известным формулам теории диффузного слоя.
Алгоритм численного решения упомянутой системы уравнений при некоторых выбранных значениях концентрации электролита, размеров dx, d2, уровней специфичности Ф], Ф2 и валентностей катионов и анионов состоит в следующем. Задается произвольная величина потенциала второй плоскости Т2, что позволяет вычислить по уравнениям теории диффузного слоя напряженность поля Еу на внешней стороне этой плоскости (обращенной к раствору) и заряд диффузного слоя qv. По уравнениям (3.5.28) и (3.5.29) вычисляется величина специфической адсорбции ионов большего размера в той же плоскости, соответствующий ей заряд q2 = Qm 02, и далее — напряженность поля на внутренней стороне второй плоскости Е2 = Еу - q2 / ££0. Здесь Qm — заряд плоскости, полностью заполненной сорбирующимися в ней ионами.
По потенциалу и напряженности поля второй плоскости можно вычислить константу интегрирования А и функцию /2. Затем решается относительно функции 1Х одно из уравнений (3.5.34) или (3.5.35) в зависимости от знака константы А. По величинам Ц и А вычисляется безразмерный потенциал Wx плоскости локализации ионов меньшего размера и по величинам потенциалов W\ и W2 — заряд слоя qg, расположенного между плоскостями локализации ионов разного размера. Специфическая адсорбция и заряд ионов qx, локализованных в первой плоскости, вычисляется аналогично заряду второй плоскости q2, что позволяет найти суммарный заряд всей внешней части двойного слоя qe и поверхностный заряд qx = -qe.
Зная значения зарядов различных частей ДЭС, можно вычислить на основе закона Гаусса напряженность поля в промежутке между поверхностью электрода и первой плоскостью локализации ионов: Е, = Е2- (qx + + q2 + <yg) /££0. Это дает возможность найти потенциал поверхности = Т] + Esdx и интегральную емкость двойного электрического слоя С = qs / Т5. Повторение вычислительной процедуры при различных значениях потенциала второй плоскости Т2 дает серию связанных величин и С, а последующее численное дифференцирование заряда по потенциалу поверхности — зависимость дифференциальной емкости электрода от потенциала, которая обычно и приводится при описании экспериментальных данных.
Образцы зависимостей такого рода, полученные с помощью описанной вычислительной процедуры, представлены на рис. 3.42. В области умеренных значений
606
Новый справочник химика и технолога
потенциала поверхности (до 0,5 В по абсолютной величине) они практически идентичны экспериментальным данным (рис. 3.40), относящимся к емкости ртутного электрода в растворах NaCl, и удовлетворительно передают тенденции в изменении величин и положения экстремумов при изменении концентрации электролита на несколько порядков. При сравнении теоретических и экспериментальных результатов следует иметь в виду, что на графиках они выглядят зеркально противоположными, так как в эксперименте по оси абсцисс откладывают отрицательную величину потенциала ртутного электрода.
Рис. 3.42. Зависимость дифференциальной емкости ДЭС dq / dVs от потенциала поверхности при концентрации электролита
При подборе параметров задачи (размер и сродство ионов к поверхности), обеспечивающих наилучшее совпадение теоретических и экспериментальных зависимостей, выявляется необходимость в пониженном значении диэлектрической проницаемости среды в плотной части ДЭС. Для этого есть достаточно оснований: воздействие на растворитель полей очень большой напряженности (порядка 108 В/м), фактическое отсутствие растворителя как сплошной среды между электродом и слоем ионов, расположенных в ближней и дальней плоскостях их локализации.
Последнее обстоятельство позволяет приписать среде, разделяющей поверхность электрода и ближайший слой ионов, относительную диэлектрическую проницаемость, близкую к проницаемости вакуума, тогда как ионы диффузного слоя могут быть окружены средой как с номинальными свойствами, так и с пониженной проницаемостью. При обычной величине диэлектрической проницаемости плотной части двойного слоя его емкость примерно на порядок больше экспериментальных значений. Кроме того, при сильном увеличении концентрации электролита, как правило, необходимо уменьшать размеры ионов (т. е. учитывать их дегидратацию, что вполне оправдано), а также варьировать диэлектрическую проницаемость среды в двойном слое
(что можно считать следствием дегидратации). Увеличение различий в размерах ионов и в диэлектрической проницаемости плотного и диффузного слоев усиливает различие в высоте максимумов при катодной и анодной поляризации электрода.
При равенстве размеров ионов и энергий их специфического взаимодействия катодная и анодная ветви кривых полностью симметричны и дают зависимости, подобные тем, что приведены на рис. 3.39.
Наблюдающееся на практике и показанное на рисунках сильное повышение емкости при анодной поляризации поверхности по сравнению с катодной не вызывает, как можно было думать, уменьшения размера анионов. Более сильное влияние имеет диэлектрическая проницаемость плотного слоя.
В соответствии с формулой (3.5.35) при определенной комбинации параметров (различие размеров катионов и анионов, диэлектрических проницаемостей плотного и диффузного слоев, достаточно большая концентрация электролита) происходит бесконечно большой разрыв зависимости потенциала первой плоскости от потенциала второй плоскости (значение тангенса меняется от +оо до -оо). Так же хаотично меняется и емкость ДЭС вблизи точки разрыва. В некоторых случаях экспериментально установлено чрезвычайно резкое изменение емкости при изменении потенциала поверхности. Разрыв зависимости емкости от потенциала конечной величины имеет место при смене знака постоянной интегрирования. Потенциал и заряд при этом меняются монотонно.
Важно, что соответствие теоретических и экспериментальных зависимостей при сильном изменении концентрации электролита и потенциала достигается при постоянной величине сродства ионов к поверхности. Существенно, что при умеренной концентрации электролита параметры определенного иона можно сохранять неизменными при замене одного электролита на другой, например параметры ионов Na+ при замене NaF на NaCl.
Изложенные представления касаются в основном электрохимической стороны теории двойного слоя. Именно в электрохимии потенциал поверхности электрода выступает в качестве независимой переменной [20]. Ее значение регулируется соединением электрода с внешним источником разности потенциалов, а емкость служит основной равновесной характеристикой электрохимической системы. В дисперсных системах такой способ регулирования электрического состояния поверхности частиц невозможен. В дисперсных системах электрическое состояние частиц задается путем введения в дисперсную систему потенциалопределяю-щего электролита (далее — ПО электролита или ПО иона применительно к ионам).
В рамках рассмотренной модели ДЭС потенциал-определяющий ион — это ион с достаточно большим уровнем специфичности взаимодействия с поверхностью электрода, локализованный при адсорбции непо-
Физика, химия и технология дисперсных систем
607
средственно на границе раздела фаз (d = 0). Одна из задач теории заключается в установлении зависимости потенциала поверхности от состава раствора, в первую очередь от концентрации ПО электролита и в присутствии, как правило, значительного количества индифферентного (фонового) электролита. Таким образом, задача осложняется наличием как минимум двух, а обычно трех разных электролитов: растворенной части самой твердой фазы, фона и потенциалопределяющего электролита. Для ее решения иногда приходится пренебрегать ролью некоторых компонентов раствора при рассмотрении отдельных аспектов проблемы, например, вкладом растворенной части твердой фазы и ПО ионов в ионную силу раствора.
При достаточно сильном сродстве ПО иона к поверхности электрода (Ф, порядка 10ЯТ и более) зависимость потенциала поверхности от концентрации ПО электролита с четко следует логарифмическому закону us = aln(c / со), где us = zFVs I RT — безразмерный потенциал ПО иона с формальным зарядом z на поверхности; со — концентрация, при которой потенциал равен нулю (точка нулевого заряда или ТНЗ поверхности в данном растворе), а коэффициент пропорциональности а близок по величине к RT. Эта зависимость представляет собой не что иное, как закон Нернста, который обычно выводится из законов термодинамики:
us = RT\n(c/со}. (3.5.37)
При снижении сродства Ф, ПО ионов к поверхности линейность зависимости потенциала от логарифма концентрации нарушается (рис. 3.43).
Соответствие закону Нернста результатов, полученных на основе принятой модели строения ДЭС, можно воспринимать как нечто само собой разумеющееся и подтверждающее корректность теории. Однако некоторые «побочные» результаты расчетов вынуждают снова возвращаться к самим основам существующих представлений о природе двойного слоя.
Упомянутые выше расчеты и по модели двухслойной плотной части ДЭС, и по ее упрощенным вариантам неизменно показывают, что при условиях, обеспечивающих соблюдение закона Нернста, адсорбция ПО ионов не зависит от концентрации в растворе потенциалопределяющего электролита. Этот вывод настолько необычен, что его анализу и комментариям посвящен отдельный параграф.
3.5.5. Уравнение Нернста и адсорбция ионов
Уравнение Штерна (3.5.28) для констант адсорбционного равновесия удобно записать в иной форме. В частности для катионов:
k = KKK+, Кк ~ Г„ехр(Фк), K+ = exp(-z+W)
или в виде уравнения:
К+ =[l/cxp(jy)]z' (3.5.38)
Индексы «+» и «к» обозначают принадлежность параметров катионам, причем z, характеризует зарядовое состояние ПО иона на поверхности, которое и фигурирует в уравнении Штерна. Если ввести еще величину z®, аналогичную z , но относящуюся к состоянию того же иона в растворе, то закон Нернста связывает разность потенциалов фаз именно с параметрами ионов в объеме фаз (в растворе).
Выразим с помощью закона Нернста безразмерный потенциал поверхности IV в растворе подходящего электролита через формальный заряд иона z°, концентрацию ПО катионов и ТНЗ поверхности в этом растворе: z°W = In (с+ / с0+) и приведем его к виду ехр(Ж) = (с+ / с0+),/г°.
Если с помощью этого соотношения исключить экспоненциальные функции потенциала из формулы (3.5.38) для константы адсорбционного равновесия, то получается формула:
Рис. 3.43. Зависимость потенциала поверхности от логарифма концентрации ПО электролита при разных уровнях сродства Ф, ПО ионов: 10 (7) и 8 (2)
Произведения £,с(, определяющие, согласно уравнению Ленгмюра (3.5.27), характер зависимости адсорбции, а следовательно, и потенциала от концентрации потенциалопределяющих ионов, для катионов описываются формулой:
k+c+ = Х'ксо с+ . (3.5.40)
Тогда для сопряженных ПО анионов получается формула
к_с_ = Кксо с • (3.5.40а)
608
Новый справочник химика и технолога
Таким образом, при равенстве валентностей ионов до и после адсорбции показатель степени при концентрации равен нулю, и, следовательно, концентрация ионов не будет влиять на величину адсорбции и на заряд поверхности. Это противоречит общепринятым представлениям, но данный вывод строго следует из использованных уравнений и согласуется с результатами расчетов на основе и двухслойной, и классической модели плотного слоя. Формально получающийся парадоксальный результат обусловлен тем, что в рамках использованных уравнений увеличение концентрации ионов точно компенсируется уменьшением значения констант К. и К за счет изменения потенциала поверхности, которое должно произойти в соответствии с уравнением Нернста. При уменьшении концентрации, в том числе неограниченном, происходит то же самое. Поэтому не следует даже при малых концентрациях пренебрегать присутствием в растворе сопутствующего потенциалопределяющего иона. Следует напомнить, что кроме основного ПО иона (например, Ag+ по отношению к AgCl), вводимого в раствор с ПО электролитом (например, AgNO3), в равновесном растворе всегда присутствует сопутствующий ПО ион противоположного знака (СГ), появляющийся из-за растворения вещества твердой фазы (AgCl). Концентрации этих двух сопряженных ионов (основного и сопутствующего) связаны произведением растворимости (ПР) вещества твердой фазы:
ПР=<с?. (3.5.41)
Здесь v+ и V. — стехиометрические коэффициенты соединения Kv+Av_, диссоциирующего в растворе по схеме Kv+Av_ = v+K + v_A. Если задана его растворимость cs, то концентрации ионов связаны с ней формулами: с, = v+cs, С- = v_c4. Величина ПР также может быть выражена через растворимость с помощью уравнения:
nP = <vX+v-. (3.5.41а)
В дополнение к этому по условиям электронейтральности раствора z+ v+ = z_v_.
Результаты, вытекающие из формул (3.5.40) и (3.5.40а), можно привести в соответствие с общепринятыми представлениями, если допустить, что адсорбированные ионы хотя бы частично разряжаются при адсорбции (валентность сорбированных ионов z, должна быть меньше их же валентности в растворе z(°). В этом случае концентрация того и другого войдет в выражения (3.5.40) и (3.5.40а) с дробным положительным показателем степени. Поэтому адсорбция, а с нею и потенциал поверхности должны согласно принятым представлениям увеличиваться (по модулю) по мере увеличения концентрации потенциалопределяющего электролита.
Предположение об уменьшении эффективного заряда (валентности) химически сорбированного иона может восприниматься как тривиальное, поскольку по
определению химическое взаимодействие — это взаимодействие, которое затрагивает электронную конфигурацию иона, т. е. меняет его зарядовое состояние. В этом просматривается некорректность уравнения Штерна, которое является основой количественного описания двойного электрического слоя. Одна из возможных причин некорректности — фактическая неаддитивность химической и электростатической составляющих энергии адсорбции ионов. В уравнении (3.5.28) эти виды взаимодействия являются аддитивными. По крайней мере, в некоторых случаях они должны быть взаимоисключающими: если ион полностью сохранит свое зарядовое состояние, то он не вступит в химическую связь с поверхностью, и наоборот — полный переход в химически связанное состояние означает почти полную потерю заряда. Гипотеза о различных формальных зарядах ионов в растворе и на поверхности простейшим способом учитывает возможность частичного переноса заряда с адсорбированного иона на поверхность (делокализацию заряда).
При концентрации потенциалопределяющего катиона, равной величине ТНЗ, потенциал поверхности по определению равен нулю. Это означает равенство степеней заполнения поверхности катионами и анионами или, в соответствии с законом мономолекулярной адсорбции, равенство произведений Кк К+ с\ и К.ЛК с при с+ = с0+ и с_ = с'о . Если подставить эти значения концентраций в формулы (3.5.40) и (3.5.40а) и исключить затем из них концентрацию анионов с помощью формулы (3.5.41), то можно установить связь величины ТНЗ с константами избирательной адсорбции катионов и анионов с помощью уравнения [13]:
с0+ =ПР2+/а(^+2ЛМ^к) • (3.5.42)
Примечательно, что в формулу (3.5.42) входят только формальные заряды ионов в растворе, что позволяет надеяться на ее применимость вне зависимости от фактического заряда ионов в сорбированном состоянии.
Принцип аддитивности специфического и электростатического взаимодействия ионов с заряженной поверхностью должен сохраняться, например, при адсорбции ионогенных поверхностно-активных веществ. Последние могут адсорбироваться за счет чисто гидрофобных взаимодействий углеводородного радикала с поверхностью электрода, не изменяя, следовательно, электронную конфигурацию ионогенной группы и ее заряд. В таком случае рассмотренная выше возможность согласования системы уравнений Ленгмюра, Нернста и Штерна путем уменьшения заряда сорбированных ионов будет исключена и, следовательно, необходима более глубокая ревизия этой системы.
Парадокс концентрационной зависимости адсорбции ионов, порождаемый использованием уравнения Нернста, в формуле Штерна не снимается заменой последней на ее более корректный вариант (3.5.29). Основная проблема, которая возникает в связи с этим, —
Физика, химия и технология дисперсных систем
609
интерпретация экспериментальных данных по адсорбции ионов, которые как будто свидетельствуют об обратном. Проблема настолько серьезна, что выходит далеко за рамки физики и химии дисперсных систем, и поэтому здесь неуместно в нее углубляться. Важно отметить лишь, что введение понятия адсорбции (и как пространственного избытка, и как временного избытка) в однокомпонентной гетерогенной системе (адсорбции растворенной или газообразной части твердого вещества самого на себя) сталкивается с препятствиями принципиального характера, на что указывал в свое время еще Я.И. Френкель [30]. То же самое относится к экспериментальному определению адсорбции. Суть проблемы в том, что однокомпонентная гетерогенная система имеет пониженное число степеней свободы.
Если химическое состояние молекул некоторого вещества в обеих фазах одинаково (в конденсированной и газообразной фазах или в твердом веществе и в его насыщенном растворе), то при постоянной температуре число степеней свободы равно нулю. Это означает, в частности, отсутствие возможности изменять состав газовой фазы (раствора) и, соответственно, параметры состояния поверхностного слоя (адсорбцию). Если состояние вещества в разных фазах химически различно (например, оно диссоциирует в разреженной фазе), то появляется дополнительная степень свободы и возможность изменения состава разреженной фазы (раствора) введением второго компонента, способного повлиять на химическое равновесие в системе. Однако эта возможность ограничена условием равновесия фаз.
Применительно к двойному слою второй компонент— это потенциалопределяющий электролит, а условие, ограничивающее возможность изменения состава раствора, — это равенство произведения концентраций обоих потенциалопределяющих ионов (катиона и аниона вещества твердой фазы) произведению растворимости.
Экспериментальное исследование адсорбции ПО иона сводится к нахождению разности Ас = ср - са его рецептурной (расчетной) ср и фактической (аналитической) са концентрации в растворе. Предполагается, что последняя может быть определена с помощью тех или иных методов анализа раствора, а разница Ас обусловлена адсорбцией ПО иона на поверхности частиц дисперсной фазы. Действительной причиной уменьшения концентрации может быть уменьшение растворимости вещества дисперсной фазы и выведение ПО ионов из раствора путем образования дополнительного количества вещества твердой фазы.
Уменьшение растворимости при введении электролита, имеющего общий ион с дисперсной фазой, вытекает из условия равновесия твердой фазы и ее насыщенного раствора. Это условие выражается в равенстве произведения фактических концентраций cai и с^ обоих ПО ионов постоянной величине — произведению растворимости вещества ПР. В случае симметричных солей типа Agl, AIPO4 и других. ПР = с*. Расчетные же
концентрации cpi и ср2 вычисляются, если известны растворимость cs вещества дисперсной фазы, первоначальный объем Ко жидкой фазы (насыщенный раствор дисперсного вещества исследуемой системы до добавления к нему ПО электролита), концентрация с и объем V введенного в нее раствора ПО электролита. Тогда концентрацию сР1 вводимого с электролитом ПО иона вычисляют с помощью следующего выражения:
Ср1 = (^о + сКЖо+П,
а концентрацию ср2 сопряженного с ним ПО иона —
cp2 = csK0/(K0+P).
Следует отметить, что эти величины отображают лишь материальный баланс по вводимому и сопряженному ПО ионам, а не фактический состав раствора. Уравнение для фактических концентраций
5^±£К_Дс с^_Дс
-- к0 + и
о
основано на том, что они устанавливаются за счет уменьшения рецептурных величин на одну и ту же величину Ас, так как оба ПО иона выводятся из раствора в равном количестве в виде мало растворимого вещества. Количество этого вещества, образующегося в единице объема раствора, также равно Ас. Выражения в скобках приведенного выше уравнения и есть фактические концентрации ПО ионов, а величина Aci = cpi - са1 может в опыте восприниматься как адсорбция ПО иона, введенного с потенциалопределяющим электролитом.
Решением приведенного выше уравнения относительно Ас является уравнение:
Дс=2сЛ+сК_ If2с/0 +сГY । у (с/0 +сГ)сЛ~ 2(Г0 + К) ^2(K0 + K)J (^о + О2
(3.5.43)
Графически зависимость Ас от V напоминает изотерму мономолекулярной адсорбции, что способствует ошибочной интерпретации выведения иона в осадок как его адсорбции. Фактически это уравнение ложной адсорбции.
В завершение следует напомнить о существовании принципиального запрета на возможность измерения межфазной разности электрических потенциалов. В связи с этим наличие проблем с адсорбцией потенциалопределяющих ионов можно считать его следствием, а их полное устранение означало бы появление возможности обойти указанный запрет. Некоторые соображения общего характера также указывают на то, что адсорбция потенциалопределяющих ионов в принципе не должна зависеть от их концентрации в растворе. Прежде всего, следует принять во внимание, что при любой концентрации ПО ионов система твердая фаза—раст
610
Новый справочник химика и технолога
вор потенциалопределяющего электролита находится в состоянии термодинамического равновесия. Говоря иначе, раствор является насыщенным по отношению к компонентам (ионам), образующим твердую фазу, т. е. любая концентрация с ПО ионов является концентрацией насыщенного раствора cs. С другой стороны, уравнения изотермы адсорбции (уравнение БЭТ, Ленгмюра, адсорбционный потенциал) представляют адсорбцию и адсорбционный потенциал как функцию относительной концентрации (с / cs) или относительного давления компонента (р / ps), которая по указанным выше причинам остается равной единице при любых изменениях концентрации ПО ионов. Следовательно, она не будет изменяться при изменении их концентрации в растворе. В свою очередь адсорбция также не будет изменяться при изменении концентрации ПО ионов.
3.5.6. Электрокинетический и мембранный потенциал
Специфически сорбированные поверхностью по-тенциалопределяющие ионы являются продолжением кристаллической решетки сорбента и поэтому должны быть жестко связаны с ней, тогда как внешняя часть ДЭС (противоионы и коионы), находясь в жидкой фазе, может сохранять свободу движения относительно заряженной поверхности в тангенциальном к ней направлении. В гидродинамическом отношении внешнюю часть ДЭС можно считать слоем заряженной жидкости с объемной плотностью заряда р. В этом случае внешнее электрическое поле, направленное по касательной к межфазной поверхности, вызовет движение этого слоя, которое увлечет за собой и всю жидкую фазу, если ее объем не слишком велик по сравнению с объемом заряженного слоя жидкости. Такие условия создаются, например, в тонких капиллярах, внутренняя поверхность которых несет на себе двойной электрический слой.
Пористые материалы представляют собой совокупность капилляров, поэтому диафрагма из таких материалов, перегораживающая сосуд с жидкостью на две части, действует как насос, перекачивающий жидкость из одной части сосуда в другую при подведении к ним разности электрических потенциалов. Движение жидкости через пористый материал под действием внешнего электрического поля называется электроосмосом.
При обратном соотношении объемов жидкой и твердой фаз, т. е. когда дисперсная система представляет собой сравнительно разбавленную взвесь отдельных частиц в большой массе жидкости, по тем же причинам электрическое поле вызовет движение частиц к противоположно заряженному электроду. Движение частиц дисперсной фазы при действии на дисперсную систему внешнего электрического поля называется электрофорезом.
Электрофорез и электроосмос относятся к прямым электрокинетическим явлениям, состоящим в смещении фаз дисперсной системы в электрическом поле. Существуют и обратные электрокинетические явления —
возникновение разности потенциалов при взаимном смещении фаз дисперсной системы, вызванном механическим воздействием на нее. В частности, если принудительно продавливать жидкость через пористую перегородку, то это приведет к появлению разности потенциалов между входом и выходом потока жидкости в перегородку, т. е. в направлении ее течения. Возникающая при этом разность потенциалов называется потенциалом течения.
Причиной этого является перенос заряда внешней подвижной части ДЭС потоком жидкости. Очевидно, что при положительном заряде поверхности на выходе из перегородки будет скапливаться избыточный отрицательный заряд и, соответственно, появится отрицательный потенциал относительно входа в перегородку. При оседании частиц взвеси встречный поток жидкой среды, омывающий поверхность частиц, «смывает» внешнюю часть двойного слоя, которая, следовательно, отстает от оседающих частиц. Иначе говоря, происходит поляризация частиц и всей дисперсной системы гидродинамическими силами, при которой в направлении оседания частиц (в осадке) накапливается преимущественно заряд того знака, который находится на поверхности частиц, а верхние слои взвеси и освободившаяся от частиц дисперсионная среда обогащаются противоионами. Соответственно такому перераспределению зарядов возникает разность потенциалов в направлении оседания частиц. Она называется потенциалом оседания (потенциалом Дорна). Примечательно, что и после завершения процесса оседания разность потенциалов между осадком и надосадочной средой сохраняется, по крайней мере частично. Эта остаточная разность потенциалов получила название суспензионного, или золь-концентрационного, эффекта. По механизму возникновения он отличается от эффекта Дорна и может быть объяснен «стесненностью» частиц в осадке — выдавливанием противоионов двойного слоя из осадка, поскольку здесь расстояние между частицами меньше, чем толщина двойных слоев на поверхности частиц. Однако оба эффекта обязаны действию на частицы одной и той же силы — силы тяжести, поэтому они связаны не только общим происхождением, но, возможно, и численными значениями эффектов. Иную природу имеет разность потенциалов между растворами двух электролитов, разделенных перегородкой, непроницаемой для одного из ионов, или между набухшим полимером (гелем) и той средой, в которой он набухает (потенциал Доннана). Этот потенциал сохраняется и тогда, когда полимерная сетка сильно разрежена, т. е. исключена стесненность двойных слоев, вызывающая суспензионный эффект. Вместе с тем можно предположить, что суспензионный и доннановский эффекты — это одно и то же, а различие состоит в способах экспериментального осуществления. Для того, чтобы решение этой проблемы приобрело доказательный характер, следует рассмотреть количественную сторону упомянутых выше эффектов.
Физика, химия и технология дисперсных систем
611
Классический вариант количественного описания электрокинетических явлений предполагает наличие в двойном слое плоскости скольжения, расположенной на некотором расстоянии х = d от заряженной поверхности. Эта плоскость якобы отделяет подвижную часть жидкой фазы от ее неподвижной части, связанной с заряженной поверхностью твердого вещества (рис. 3.44). Если радиус капилляра г будет достаточно большим по сравнению с толщиной ДЭС (хг » 1), то поверхность капилляра можно считать плоской, т. е. использовать одномерное уравнение Пуассона cfrV / dx2 =-р / ss0. Согласно уравнениям гидродинамики (Навье — Стокса), при стационарном течении объемная (в данном случае электрическая) сила EpdV, действующая на произвольный элемент среды объемом dV, скомпенсирована силами трения т, действующими на поверхности, ограничивающие этот элемент объема. В случае плоского слоя бесконечно малой толщины dx с единичной площадью больших граней (рис. 3.44) dV= dx, и уравнение Навье — Стокса сводится к равенству dx + pEdx = 0, где dx — разность сил трения на правой (по рисунку) и левой сторонах слоя dx. Имея в виду, что pdx есть заряд dq слоя dx, можно записать соотношение:
dx = -Edq. (3.5.44)
После его интегрирования получается формула:
x(x) = -Eq(x). (3.5.45)
Здесь т(х) — сила трения (вязкое напряжение) в некоторой плоскости, отстоящей на расстояние х от поверхности твердой фазы, q(x) — заряд всей наружной части ДЭС, расположенной правее (рис. 3.44) этой плоскости.
Константа интегрирования равна нулю, так как за пределами ДЭС заряд и вязкие напряжения в среде равны нулю (т. е. градиент скорости течения среды отсутствует).
С помощью закона внутреннего трения Ньютона т = T|tZw / dx, где т| — вязкость жидкости, левую часть уравнения (3.5.45) можно выразить через градиент скорости течения жидкой фазы du / dx. В свою очередь заряд q(x) можно получить, интегрируя величину pdx, где в соответствии с уравнением Пуассона р = -ееос/4? / dx2. Тогда справедливы соотношения:
q (х) = ее0<ЛР / dxnr\du /dx = -еъоЕсМ I dx или
tZ« = -(sso£/n)^. (3.5.46)
Если вязкость т| и диэлектрическая проницаемость е не зависят от потенциала 'Р или скорости течения и (или от координаты х), то последнее уравнение легко интегрируется. В системе координат, связанной с поверхностью твердой фазы, на плоскости скольжения (х = d) скорость движения жидкости относительно твердой фазы будет равна нулю (и = 0), а на бесконечном удалении от нее скорость и равна искомой величине движения жидкой фазы относительно твердой. По
тенциал же на плоскости скольжения равен электро-кинетическому 0? = £), а на бесконечном расстоянии от нее — нулю (4х = 0) независимо от того, с какой фазой — твердой или жидкой — связана система координат. Таким образом, интегрируя выражение (3.5.46) в указанных пределах, получим уравнение:
Рис. 3.44. Распределение потенциала Т и поля скоростей течения и при электроосмосе
В указанной системе координат найденная скорость есть линейная скорость электроосмотического течения среды. Эта же величина, но с обратным знаком, равна скорости электрофоретического движения частиц в поле того же знака. Математическая сторона вывода формулы (3.5.47) при форезе будет отличаться тем, что при этом искомая скорость и — это скорость в плоскости скольжения (х = d), а при бесконечном удалении от поверхности она равна нулю.
При электроосмосе вместо линейной скорости движения жидкости обычно измеряется объемная скорость ее течения v = Su через пористую перегородку (диафрагму), где S — суммарное сечение пор. Вместо напряженности поля Е в этом случае задается (или измеряется) ток I через перегородку. Величина тока I=jS определяется сечением пор S и плотностью тока J в жидкой фазе пористого тела, которая с помощью закона Ома (j = rx.pE) вычисляется через удельную электрическую проводимость жидкости в пбровом пространстве и напряженность поля в нем Е. Если выполнить указанные замены, вместо уравнения (3.5.47) получается следующее выражение:
v=eeo£
ПХР ’
Благодаря наличию на стенках пор двойного слоя концентрация электролита и, соответственно, удельная электрическая проводимость хр порового пространства повышена по сравнению с удельной электрической проводимости) раствора х.
Кратность повышения а = %р / х обычно определяется экспериментально. Она составляет несколько единиц, увеличивается при уменьшении радиуса пор или
612
Новый справочник химика и технолога
увеличении толщины ДЭС и называется коэффициентом эффективности диафрагмы. Заменив кр в формуле для объемной скорости электроосмоса равной ей величиной ах, получим уравнение:
т|ах
Введение в теорию электрокинетических явлений понятия о плоскости скольжения и потенциале на этой плоскости связано с тем, что экспериментально определяемая из скорости электрофореза, электроосмоса или других явлений величина потенциала частиц оказывается меньше (по модулю), чем потенциал поверхности, а иногда отличается от него и по знаку.
В действительности для появления такого отличия совсем не обязательно, чтобы гидродинамически неподвижная часть ДЭС находилась возле заряженной поверхности. Для этого достаточно некоторого понижения подвижности ионов вблизи поверхности, или эквивалентного ему увеличения вязкости среды возле поверхности, или снижения диэлектрической проницаемости вблизи твердой поверхности, например в результате адсорбции неполярных примесей, ориентации молекул воды в сильном поле двойного слоя и т. д. Понижение подвижности противоионов может быть вызвано дискретностью поверхностного заряда.
Так, например, вязкость экспоненциально увеличивается при приближении к заряженной поверхности т|5 = т|[1 + 6ехр(-хх)]. При экспоненциальной же зависимости потенциала от расстояния это выражение будет эквивалентно следующему: т], - т|[ 1 + Ь"¥ / %]. Его подстановка в уравнение (3.5.46) и инте+грирование дают уравнение:
еепТ 1п(1 + М и = Е—2—l—1(3.5.49)
П ь
Это выражение равноценно формуле (3.5.47), если электрокинетическим потенциалом считать потенциал на расстоянии ln[Z> / Ln(l +/?)]/ х от заряженной поверхности, т. е. принять, что С, = %ехр(-хбО = TJn( 1 + b) / Ь, где b — относительное приращение вязкости непосредственно возле поверхности (при х = 0) по сравнению с вязкостью среды. Легко заметить, что с увеличением этого коэффициента от нуля до бесконечности координата эффективной плоскости скольжения изменяется от нуля до бесконечности, а электрокинетический потенциал снижается от потенциала поверхности до нуля.
В концентрированных суспензиях и других дисперсных системах взаимное смещение фаз не всегда можно классифицировать как электрофорез или электроосмос. Наблюдается движение обеих фаз относительно стенок сосуда (точнее относительно электродов, подводящих электрическое напряжение). Вычисляемая по формуле (3.5.47) величина и является в этом случае суммой абсолютных величин линейных скоростей фореза и осмоса.
Переходя к обратным электрокинетическим явлениям, отметим их сходство с пьезоэффектом — появлением разности потенциалов при деформировании некоторых кристаллических веществ — пьезоэлектриков. Отличие состоит в том, что пьезоэффект характеризует изменение равновесного состояния вещества при действии на него механических напряжений, а гидродинамическая поляризация дисперсной системы отражает интенсивность течения необратимого процесса — переноса заряда (электрического тока), который может быть вызван механической силой (градиентом давления) при надлежащих условиях. Величины эффектов, форму их проявления (в виде потенциала или тока гидродинамической поляризации), связь с прямыми элек-трокинетическими эффектами (форез, осмос) можно установить, основываясь на общих положениях термодинамики необратимых процессов.
В отсутствие равновесия в термодинамической системе протекают различные процессы, интенсивность которых характеризуется плотностями потоков экстенсивных величин jik (потоков массы, теплоты, заряда, энтропии и т. д.). Плотность потока — это количество вещества, заряда и т. д., переносимое за единицу времени через единицу площади в нормальном к ней направлении. При небольшом отклонении от равновесия поток любой величины пропорционален всем движущим силам (градиентам интенсивных параметров или обобщенных сил Xk) — градиенту давления, концентрации, температуры, электрического потенциала и т. д.: jtk = Величины a,k этого уравнения называются феноменологическими кинетическими коэффициентами. Суммирование без указания его пределов проводится, согласно принятым в математике обозначениям, по всем величинам с повторяющимся индексом, т. е. по всем обобщенным силам.
В дисперсных системах имеют место потоки массы jm (жидкости, частиц) и заряда jq (тока) под действием механической (Х} = -dP/dx) и электрической (Х2 = -dUtdx) сил:
jm ~ в] 1X1 + 6712^2,
(3.5.50) jq = ^21X1 + а22Х2.
Части этих потоков ju = \Хц j22 = a22X2, пропорциональные сопряженным силам, называются прямыми эффектами, а коэффициенты ац, а22 при этих силах — прямыми коэффициентами. Коэффициент ац при градиенте давления в капиллярах X, = -dP I dx, согласно формуле Пуазейля v = лг4Р / 8т|Р, равен г2 / 8т|, так как линейная скорость течения и = v / яг2, a -dP / dx = Р / /.
Коэффициент при градиенте электрического потенциала, т. е. при напряженности поля Е = -jdU / dx)-U I /, — это электропроводность х внутрипоровой жидкости, а их произведение представляет собой плотность электрического тока в поровом пространстве.
Другие слагаемые потоков: j12 = «12^2 и у21 = «2Л называются перекрестными эффектами, а коэффициен
Физика, химия и технология дисперсных систем
613
ты при силах — перекрестными коэффициентами. Все электрокинетические явления относятся к перекрестным эффектам, причем коэффициент ап = ее0^ I Л, связывающий поток жидкости с градиентом внешнего электрического потенциала, уже известен из формулы (3.5.47) для линейной скорости электроосмоса, поэтому вместо выражений (3.5.50) можно написать уравнения:
• г2 ее0£ / =---------1----£,
8г| dx г|
(3.5.51)
dP
Важное свойство перекрестных коэффициентов (а12 и a2i) заключается в том, что они попарно равны между собой (теорема взаимности Онзагера), т. е. в данном случае а21 = ее0£ / Л, и тогда справедливо следующее выражение:
’ г| dx
Если измерительную ячейку замкнуть накоротко, т. е. соединить цепью с нулевым сопротивлением электроды на входной и выходной сторонах пористой перегородки или в верхней и нижней частях сосуда с оседающей суспензией, то напряженность поля между электродами Е = 0. Тогда приведенное выше соотношение сводится к уравнению, описывающему плотность тока короткого замыкания в цепи:
880£ dP г| dx
(3.5.52)
Разность потенциалов UD = El между образовавшимся на дне сосуда осадком и надосадочной жидкостью будет пропорциональна высоте / столба еще не осевшей части взвеси, согласно уравнению:
(/О=880^ / r|xApg(p I. (3.5.55)
В процессе оседания на дно сосуда высота /, а с ней и величина потенциала Дорна, будет уменьшаться до нуля. Вследствие этого возникает равновесная разность потенциалов осадка и надосадочной среды — суспензионная разность потенциалов.
Электрокинетический потенциал С, не является каким-либо новым независимым параметром, характеризующим ДЭС. В рамках теории Гуи — Чепмена или Штерна он однозначно определяется двумя (х, %) или тремя (х, ^i) другими независимыми характеристи-
ками двойного слоя, если заранее определен смысл этого потенциала. Например, если считать, что С, — это потенциал на каком-то заранее определенном расстоянии d от заряженной поверхности, то его величина легко вычисляется с помощью той или иной зависимости потенциала от расстояния.
Преимущество ^-потенциала перед другими параметрами ДЭС в том, что он доступен измерению с помощью электрокинетических явлений. Это дает возможность воспользоваться обширными экспериментальными данными о зависимости электрокинети-ческого потенциала £ от концентрации электролита с для проверки правильности теории, предсказывающей соответствующую зависимость потенциала от расстояния.
Типичные зависимости электрокинетического потенциала от концентрации электролита представлены на рис. 3.45.
Если эта цепь разомкнута (режим измерения потенциала протекания или оседания), то jq = 0, и то же соотношение преобразуется в уравнение, описывющее напряженность поля между электродами:
£ = ^.—. (3.5.53)
r|x dx
Формулы (3.5.52) и (3.5.53) в большей мере приспособлены для вычисления напряженности поля или потенциала протекания U=El, где I — протяженность пористой перегородки в направлении течения жидкости. В случае оседания частиц ту часть градиента давления, которая вызывает оседание частиц, удобнее выразить через их концентрацию и плотность дисперсной фазы: dP / dx = Apgcp. Здесь Ар = р - р0 — разность плотностей дисперсной фазы р и дисперсионной среды ро, ср — объемная доля диспе рсной фазы в суспензии, g — ускорение свободного падения. Соответственно этому напряженность поля внутри оседающего столба взвеси описывется уравнением:
Рис. 3.45. Электрокинетический потенциал стекла в растворах различных электролитов
Е = 88о<£ / T|xApg(p.
(3.5.54)
614
Новый справочник химика и технолога
В подавляющем числе случаев подобного рода данные не используются для количественного сопоставления с той или иной теорией двойного слоя. Это объясняется невозможностью однозначного толкования физического смысла электрокинетического потенциала.
В то же время его зависимость от концентрации одного из компонентов раствора при постоянстве других условий позволяет получать ценную информацию качественного характера о влиянии исследуемого компонента на параметры ДЭС, о характере взаимодействия этого компонента с поверхностью, в частности о роли специфических адсорбционных сил.
Теория упомянутого выше эффекта Доннана основана, как это обычно подчеркивается при ее изложении [22, 32], на представлении об идеальности растворов электролитов. Поэтому в двухфазной системе фаза 1 представлена гелем — набухшим, слабо диссоциированным, ионогенным полимером (или коллоидным раствором) типа КП, а фаза 2 — разбавленным раствором обычного электролита КА, где К — общий для электролита и геля катион, А — анион и П — полианион (или коллоидная частица с отрицательным зарядом). Достаточно сильное разбавление здесь предусмотрено как средство достижения идеальности раствора.
Содержательная сторона понятия «идеальность» в данном случае означает, что ионные атмосферы поли-электролитных ионов (или заряженных коллоидных частиц) не перекрываются, и тем самым исключается часть суспензионного эффекта, связанная со стесненностью частиц (полиионов). Пусть фазы 7 и 2 имеют одинаковый объем. Фазы геля и раствора электролита приведены в непосредственный контакт, а фазы коллоидного раствора и электролита — через перегородку, непроницаемую только для коллоидных частиц.
В качестве основного параметра, описывающего состояние двухфазной системы, целесообразно выбрать молярную концентрацию эквивалентов ионогенных групп сп в геле и концентрацию cAi коиона А в геле.
Коион — это ион, одноименный по знаку с полимерными ионами П или коллоидными частицами. В исходном состоянии, когда два разных раствора еще только приведены в соприкосновение, его концентрация сА1 = 0, а концентрация сдз в фазе 2 равна начальной концентрации с0 электролита КА в этой фазе. Концентрация полииона сп остается неизменной и отличной от нуля только в фазе 7.
После возникновения контакта двух фаз начинается диффузия коиона А из фазы 2 в фазу 7, в которой он первоначально отсутствовал. При этом появляется разность потенциалов, обусловленная переходом ионов только одного знака из одной фазы в другую. Это влечет за собой и переход противоионов К в том же направлении и в том же количестве, так что обе фазы остаются электронейтральными, а ионные составы фаз связаны уравнениями их электронейтральности
СК2 = СА2, СК1 = сп + Саь (3.5.56)
Здесь cki и Ск2 — концентрации общего иона К в фазах 1 и 2 соответственно. К ним следует добавить и уравнение материального баланса по коиону при равенстве объемов фаз:
с0 = сА1 + са2. (3.5.57)
В состоянии равновесия различие концентраций ионов в двух фазах достигается за счет электростатической составляющей ztR¥f их электрохимического потенциала, где z, — формальный заряд ионов /-го типа и Ту — разность электрических потенциалов фаз. Электрические потенциалы удобно отсчитывать от потенциала фазы 2, который принимается равным нулю. При равенстве электрохимических потенциалов ионов противоположного знака в соседних фазах (при z,= l) справедливы соотношения:
TyF + 7?71п(сК1 / сК2) = 0 — для противоионов,
(3.5.58)
-TyF + 7?Tin (cAi / Сдг) = 0 — для коионов.
После сложения выражений (3.5.58) они преобразуются в уравнения:
1п(сК1сА1 / сК2^А2) — 0 или
СК1СА1= СК2СА2- (3.5.59)
Подстановка соотношений (3.5.56) и (3.5.57) в уравнения (3.5.59) приводит к следующему выражению:
СА1(СП + cAl) = (О) - ^А1)2-
Решением последнего уравнения относительно cAi является выражение:
Cai = Со2 / (2с0 + сп). (3.5.60)
Далее в случае необходимости могут быть вычислены концентрации других ионов в обеих фазах. Следует обратить внимание на то, что сА1 — отнюдь не малая величина. Из фазы 2 в фазу 7 переходит почти половина электролита (а при сп = 0 — равно половина, что само собой разумеется). То же самое относится к противоиону.
Отображенные в уравнениях (3.5.56) и (3.5.57) электронейтральность фаз и баланс по коиону не вполне соблюдаются. На самом деле из фазы 2 в фазу 7 коионов переходит несколько больше, а противоионов несколько меньше, чем это следует из приведенных уравнений. Именно поэтому возникает разность потенциалов фаз 7 и 2. Указанные «излишки» (избытки) коионов в фазе 7 и противоионов в фазе 2 сосредоточены возле разделяющей фазы перегородки, образуя тонкий двойной слой. Формулы (3.5.56)-(3.5.60) относятся только к объемам фаз системы. Они электронейтральны и сбалансированы по количеству ионов. В отличие от тради
Физика, химия и технология дисперсных систем
615
ционного ДЭС этот двойной слой размыт в обеих фазах. Толщина ДЭС в каждой из фаз определяется обычным образом, т. е. концентрацией электролита в соответствующей фазе. Как уже отмечалось, эти концентрации, а следовательно, и толщина ДЭС в каждой фазе близки по величине, так что полная разность потенциалов фаз 1 и 2 складывается из двух примерно равных частей, принадлежащих каждой фазе. Что касается величины потенциала Доннана то при z = 1 она стандартно связана с концентрациями общих ионов в фазах 1 и 2 соотношениями (3.5.58). При замене в них концентраций ионов концентрацией электролита в фазе раствора и концентрацией ионогенных групп в фазе геля с помощью формул (3.5.57) и (3.5.60) получается следующее выражение: Чу = -(RT / F) 1п(1 + сп / с0). Здесь Со — начальная концентрация коиона в растворе. Его равновесная концентрация с при равенстве объемов фаз в 2 раза меньше. Поэтому последнее выражение преобразуется в уравнение:
Чу= (RTIF) 1п( 1 + сп / 2с). (3.5.61)
При малых значениях сп / 2с уравнение (3.5.61) можно заменить приближенным соотношением
cnRT !2cF. (3.5.62)
Экспериментальные данные часто расходятся с результатами, следующими из теории Доннана. По данным Кларенбека [22], концентрация коиона в фазе 1 примерно в три раза превышает рассчитанную по формуле типа (3.5.60), если эквивалентная концентрация сп полимера становится больше, чем концентрация электролита в фазе 2. Кларенбек разработал альтернативную теорию, суть которой сводится к описанному выше «стеснительному» механизму суспензионного эффекта. Результаты расчетов на основе этой теории практически совпадают с экспериментальными данными.
При произвольном соотношении объемов фаз геля И и раствора К2 в системе уравнений (3.5.56)-(3.5.59) уравнение материального баланса (3.5.57) преобразуется в уравнение:
с0И2 = Cai И + скгУг. (3.5.63)
Выражения концентраций ионов в фазе раствора приобретают следующий вид: с^ = с0 - гсм и сК2 = с0 - гсА1, где г = Ki / К2. Их подстановка в уравнение равновесия (3.5.59) приводит к уравнению:
СА1 0 ~ + (2гс<>+ сп) Cai - Cq = 0. (3.5.64)
Его решением относительно cAi при г Ф 1 является соотношение:
ск\
_ Г7 та I
2гс0 + сп I 2гс0 + сп с0 2(1-г2) у[2(1 -г2)] 1-г2
Если гелеобразная фаза присутствует в виде тонких пленок на поверхности частиц дисперсной фазы, находящихся в растворе электролита, то г —> 0, а уравнение (3.5.65) сводится к уравнению:
сА1 = - Сп / 2 + (Сп/ 4 + Cq )1/2. (3.5.66)
При этом существенное значение имеет соотношение между толщиной пленки g и толщиной двойного электрического слоя 5 в ней. Соотношение (3.5.66) имеет смысл только для толстых пленок (g » 5). Полезно установить закон, по которому изменяется потенциал в окрестности границы соприкосновения геля и раствора электролита. Его нахождение, как и в предыдущих случаях, требует решения уравнения Пуассона — Больцмана для фазы геля, в которой концентрация ионогенных групп сП в слое геля не зависит от величины потенциала:
(Л / dx2) = [cnF + 2cFsh(F4' / RT)] I ee0. (3.5.67)
Здесь c — равновесная концентрация 1-1-валентного электролита в растворе. Координата х направлена из фазы геля в фазу раствора по нормали к границе фаз, от этой границы ведется отсчет расстояния х, т. е. в фазе геля х < 0 (рис. 3.46).
Рис. 3.46. Потенциалы тонкого слоя геля
В уравнении (3.5.67) cnF + 2cFsh(F4' / RT) — объемная плотность заряда в геле с обратным знаком. В толстом слое геля вдали от границы фаз она равна нулю. Тогда разность потенциалов Ту фаз геля и раствора описывается уравнением:
^f=(RT/F) Arsh(cn / 2с). (3.5.68)
При сп / 2с < 1 уравнения (3.5.62) и (3.5.68) совпадают.
Тонкая пленка геля (g < 5), ограниченная с одной стороны диэлектрическим субстратом (темная полоса на рис. 3.46), а с другой — раствором электролита, не является электронейтральной. При отмеченных выше условиях она имеет распределенный отрицательный заряд и по отношению к прилегающему раствору элек
616
Новый справочник химика и технолога
тролита играет роль внутренней части двойного электрического слоя. Заряд пленки компенсируется противоположным зарядом внешней части ДЭС — диффузным слоем в фазе раствора, занимающей на рис. 3.46 все пространство при х > 0. Отсюда, согласно теореме Остроградского — Гаусса, следует, что напряженность поля на границе субстрата и пленки равна нулю, т. е. dY / dx = 0 при х = -g.
Потенциал при этом имеет некоторое наибольшее по абсолютной величине значение При х = 0 потенциал равен некоторой величине 'Ро, а значение производной (dY/dx)0 со стороны геля и со стороны раствора одинаково и при малом потенциале равно х'Ро.
При малой величине потенциала sh(FT / RT) ~ RY I RT, что упрощает уравнение (3.5.67) и приводит его к виду
(/Т / dx1) = х2(Т - ХР/). (3.5.69)
Здесь учтено, что IcF1 / ££qRT = к2 и, согласно уравнению (3.5.62), cnRT / 2cF = -Ту. Интегрирование уравнения (3.5.69) приводит к следующему выражению: (dY / dx)2 = х2(*Р - Ту)2 + Ср Упомянутые выше условия на границе с субстратом позволяют найти постоянную интегрирования С\ = -x2(Tg - Ту)2 и законченную форму дифференциального уравнения:
(dY/dx) = ±х[(Т - Ту)2 - (Т^ - Ту)2]1/2. (3.5.70)
Из-за проницаемости границы гель—раствор для ионов на ней не может возникнуть плотная часть двойного слоя, и поэтому в фазе раствора распределение потенциала полностью описывается классической теорией Гуи — Чепмена. Уравнение распределения потенциала в фазе геля получается после интегрирования уравнения (3.5.70) и некоторых преобразований:
U + (С/2 - С/2)1/2 = Н ехр(хх). (3.5.71)
Здесь U = Т - Ту, Ц, = Т* - Т/5 С/о = То - Ту— потенциалы, отсчитываемые от потенциала фазы геля Ту (рис. 3.46), Н = Uo + (С/о2 - U\ )1/2.
Уравнение (3.5.71) передает характер зависимости потенциала Т от расстояния х в слое геля произвольной толщины g при любом значении характерных потенциалов, которые должны соответствовать условию |Ту| > |Tg| > |Т0|. На самом деле только одна пара согласованных значений Tg и То дает распределение, соответствующее электронейтральности двойного слоя. Из условия равенства напряженности поля на левой и правой сторонах границы гель—раствор и уравнения (3.5.70) получается уравнение, описывающее потенциал Ч'о на этой границе:
То = ф i-2Z I (3.5.72)
I 2^f)
Из условия, что при х = -g, где g — толщина слоя геля, U= Ug и уравнения (3.5.71) находят Uo по формуле:
Uo = Ug ch(xg).
(3.5.73)
Решая систему уравнений (3.5.72) и (3.5.73), получают выражение для
4<g = 4'/{[ch2(xg)- l]l/2-ch(xg) + 1}. (3.5.74)
Согласно этому уравнению разность потенциалов двух протяженных фаз — субстрата и раствора, — разделенных тонким слоем геля, увеличивается с увеличением относительной толщины xg этого слоя, асимптотически приближаясь к разности потенциалов Ту массивных фаз геля и раствора (рис. 3.47).
Потенциал То границы гель—раствор составляет при этом некоторую долю от потенциала субстрата, которая уменьшается до 0,5 Tg при большой толщине пленки, но в итоге растет от нуля до 0,5 Ту.
В элекгрокинетических экспериментах величина То является, очевидно, аналогом элекгрокинетического потенциала. Экспериментальные данные (рис. 3.45) в большей мере согласуются с концепцией гель-слоя, чем с классической теорией ДЭС. В частности, первые порции электролита могут вызывать увеличение электро-кинетического потенциала (То), что невозможно объяснить вне этой концепции. Возможность последующего прогрессивного уменьшения этого потенциала с увеличением концентрации электролита с заложена в формуле (3.5.68).
Рис. 3.47. Зависимость относительных потенциалов субстрата и границы гель—раствор То
от относительной толщины xg слоя геля
В завершение этого подраздела укажем на теоретическое и практическое значение исследований описанных выше явлений и закономерностей. Электрокинетический потенциал является одним из немногих параметров двойного электрического слоя, доступных прямому измерению. В связи с этим отметим, что некоторые из этих данных не согласуются с теоретическими представлениями. Так, согласно теории ДЭС и электро-кинетических явлений, электрокинетический потенциал должен монотонно уменьшаться при увеличении концентрации индифферентного электролита, однако в некоторых случаях он увеличивается. По данным Кройта [22] потенциал частиц иодида серебра в растворе
Физика, химия и технология дисперсных систем
617
нитрата бария с концентрацией ПГ’-КГ4 моль/л увеличивается примерно в три раза при добавлении типичного индифферентного электролита — нитрата калия — до концентрации около 102 моль/л. Автор объясняет это устранением эффекта релаксации двойного слоя, величина которого зависит от параметра ха — соотношения размера частиц а и толщины ДЭС 1/х. Максимально эта поправка может дать увеличение скорости электрофореза в 1,5 раза, тогда как опыт обнаруживает ее трехкратное изменение. Очевидно, что оно не объясняется полностью указанной поправкой и, следовательно, дает повод для поиска более адекватных моделей строения ДЭС и электрокинетических явлений.
Электрокинетические явления имеют разнообразные практические применения: электрофорез — для создания тонких слоев произвольного состава на проводящих субстратах, для разделения смеси белков на отдельные компоненты, для введения лекарственных препаратов через кожу пациента; электроосмос — для прецизионного дозирования жидкостей и создания микроманипуляторов; потенциалы оседания (движения) и протекания — как сигналы разнообразных датчиков.
Мембранные потенциалы и соответствующие им концентрационные перепады являются физико-химической основой функционирования разнообразных специализированных клеток живых организмов, в частности нервных. Они же управляют работой и гигантских мембранных опреснителей воды, и миниатюрных ионоселективных электродов, избирательно реагирующих только на ионы заданного типа. Применение мембранных технологий и материалов — характерная черта современной промышленности.
3.6. Расклинивающее давление в тонких пленках
3.6.1. Молекулярное притяжение
Плоскопараллельный зазор толщиной h между двумя телами (твердыми, жидкими, газообразными) может быть заполнен любым флюидным веществом (жидкость, пар) или остаться пустым. Заполняющее зазор вещество, особенно жидкое, может представлять собой электролит, раствор ПАВ или полимера, может состоять из молекул простой (сферы) или сложной (стержни, например) формы, может обладать спонтанной или наведенной анизотропией электрических свойств. Этим определяется большое разнообразие физических состояний слоя вещества в зазоре, в том числе возможная его неоднородность по составу (адсорбция), структуре, электрическому состоянию.
В соответствии с формулами (3.2.28), (3.3.44), (3.3.43) наличие слоя вещества в зазоре между двумя телами влияет на взаимодействие тел в той мере, в какой вещество и его состояние, а также сами тела влияют на натяжение ограниченной этими телами пленки.
Взаимодействие тел через зазор является фундаментальным свойством материального мира. Рассмотренные ранее свойства и явления: капиллярность, смачива
ние, адгезионные свойства веществ, адсорбция — могут считаться различными формами проявления этого взаимодействия через зазор нулевой толщины. Столь же разнообразны и важны проявления этого взаимодействия через зазор конечной величины. В коллоидной химии это, прежде всего, взаимодействие между коллоидными частицами.
Взаимодействие плоских тел, разделенных зазором, является простейшим в математическом отношении и служит необходимой ступенью к рассмотрению взаимодействия коллоидных частиц. Формулы (3.2.28), (3.3.44), (3.3.43) могут быть основой описания подобного взаимодействия, если известны зависимости о(/г) или Г(/г). Если эти зависимости неизвестны, то иногда может быть решена задача непосредственного вычисления энергии взаимодействия тел. Такой подход характерен, например, для вычисления взаимодействия тел, разделенных вакуумом, на основе представлений о силах Ван-дер-Ваальса (молекулярных силах притяжения).
По Гамакеру энергию взаимодействия тел (7) и (2) (рис. 3.48) можно найти, суммируя парные взаимодействия молекул, принадлежащих разным телам.
Этот принцип (принцип аддитивности молекулярных сил) в какой-то мере оправдан для сил дисперсионного типа, которые зависят от случайных отклонений электронной конфигурации молекул от стационарной, т. е. от взаимодействия мгновенных диполей. Однако он применяется и при индукционном, и ориентационном взаимодействии молекул. В любом случае энергия притяжения двух молекул типа i и j описывается следующим законом: t/0 = -Ло/г6, где Ао — константа взаимодействия молекул иг — расстояние между ними.
Рис. 3.48. Пояснение к выводу формулы Гамакера:
1,2 — тела
Если расстояние h между телами заметно превышает размер молекулы, то суммирование парных взаимодействий может быть заменено интегрированием. Технически удобно вначале вычислить энергию взаимодействия какой-либо молекулы одного тела, например (7), со всеми молекулами тела (2). Для этих целей в качестве элементарного объема внутри тела (2) следует взять тонкий сферический слой радиусом г и толщиной dr, центром которого является выбранная в теле (7) молекула. В этом случае все молекулы слоя находятся на
618
Новый справочник химика и технолога
одинаковом расстоянии от выбранной молекулы тела (7) и, следовательно, имеют одинаковую энергию взаимодействия с ней, поэтому энергия взаимодействия dUa выбранной молекулы со сферическим слоем соседнего тела будет просто в dN раз больше энергии парного взаимодействия Uo.
Здесь dN = (NA I — число молекул в слое, И2 — молярный объем вещества, из которого состоит тело (2), dV= 2nr(r - x)dr — объем сферического слоя, х — расстояние от выбранной молекулы до поверхности тела (2), т. е. dUa = 2n(NA I И2) Ur (г - x)dr. При подстановке сюда и0 и интегрировании по г в пределах от г = х до г = оо получается выражение для энергии взаимодействия произвольной молекулы с полубесконечным по протяженности телом, находящимся на расстоянии х от молекулы:
Щх) = —(л/бХТУд / У2) Ао / х3. (3.6.1)
В сущности, это выражение для энергии адсорбционного взаимодействия в духе теории полимолекулярной адсорбции Поляни.
Второй этап решения задачи Гамакера — интегрирование функции U^x) по всему объему тела (2). Физический смысл этой процедуры — сложение взаимодействий каждой молекулы тела (7) с телом (2). Технически это удобно сделать, выделив в окрестности ранее выбранной молекулы тела (7) тонкий плоский слой толщиной dx, большая грань которого параллельна границе тела и имеет площадь, равную единице. Соответственно энергия взаимодействия плоского слоя в теле (7) со всем телом (2) dUm = (NAI V\) Ua(x) dx.
Интегрирование этого выражения по х в пределах от х = h до х = оо дает уравнение, описывающее энергию взаимодействия тел (7) и (2), разделенных плоскопараллельным зазором шириной А:
Um = -Л„ 11г. (3.6.2)
Здесь
Ah = (it/\2)(N2 / V}V2)Al2. (3.6.2а)
Ah — константа взаимодействия тел, или константа Ван-дер-Ваальса — Гамакера. При одинаковой химической природе тел V\ = V2 = Vn, и уравнение (3.6.2а) можно заменить выражением:
Лл = (я/12Х^/(3.6.3)
При сферической форме взаимодействующих тел энергия их взаимодействия Um описывается уравнением:
/
Um (г) = 2лД,
0,5
+ ln(l - q)
(3.6.4)
я
Здесь q = [2r / (h + 2r)]2, h — кратчайшее расстояние между поверхностями сферических частиц, г—их радиус.
Как уже отмечалось, молекулярные силы далеко не всегда аддитивны. Строгая теория взаимодействия тел
через разделяющий их зазор разработана Лифшицем и исходит из того, что взаимодействие тел — это взаимодействие их электромагнитных полей.
Последние всегда существуют, в том числе при абсолютном нуле, и проявляют себя в виде вполне доступных регистрации излучений в ИК, видимом и УФ диапазонах. В силу независимости (некогерентности) излучения разными молекулами и излучения в разных частотных диапазонах полный спектр излучения тел весьма хаотичен и поэтому называется флуктуационным полем. Для пондеромоторного (способного вызвать Механическое перемещение тел) действия флуктуирующих, т. е. случайных по фазе и амплитуде полей необходима синхронизация фаз электромагнитного излучения партнеров по взаимодействию.
Способность тел к взаимному согласованию фаз характеризуется частотной функцией е(Х) — комплексной диэлектрической проницаемостью веществ. Она определяет все особенности поглощения и пропускания веществами электромагнитного излучения в зависимости от длины волны излучения X. Сложный, индивидуальный для каждого вещества вид этой функции делает практически невозможными вычисления по теории Лифшица для реальных веществ. Только с появлением мощных ЭВМ такие вычисления сделаны для немногих простых веществ с хорошо изученными спектральными свойствами (вода, кварц). Теория приводит к полезным выводам общего характера, которые можно сделать на основе расчетов взаимодействия некоторого гипотетического вещества с предельно простой спектральной характеристикой — с одной резонансной длиной волны X в его спектре.
Если расстояние h между поверхностями тел меньше, чем длина волны X, то фазы электромагнитного излучения обоих тел максимально согласуются. В этом случае теория Лифшица дает тот же закон взаимодействия (3.6.2), что и теория Гамакера. Константы взаимодействия также совпадают по порядку величины (около 1О-2оДж). При больших расстояниях между поверхностями (А > X) возникает эффект электромагнитного запаздывания, связанный с конечной скоростью распространения электромагнитных волн: синхронизация фаз излучения запаздывает на время, необходимое для пробега волной зазора А. Это ослабляет взаимодействие тел и изменяет закон этого взаимодействия с квадратичного на кубический. Наличие запаздывающего и незапаздывающего взаимодействий тел подтверждено прямыми измерениями сил притяжения и их зависимости от расстояния.
Ввиду слабости молекулярных сил на дальних расстояниях практический интерес представляют короткодействующие силы, и поэтому в дальнейшем используется формула 3.6.2.
Константа Ау дисперсионного взаимодействия молекул i и j между собой пропорциональна произведению их поляризуемостей а, и а7. В случае взаимодействия молекул одинаковой химической природы Д, = 3hf^j.2 /4 или
Физика, химия и технология дисперсных систем
619
Д, = 3£исс2 /4 , где h — постоянная Планка,/) — частота нулевых колебаний (при О К), ЕИ — энергия ионизации молекул. Последняя по порядку величины составляет я 1 эВ (1,6022 • 10-19 Дж). Поляризуемость, в свою очередь, по порядку величины близка к объему одной молекулы V, / NA, поэтому, согласно уравнению (3.6.3), константа взаимодействия тел Ац = (я/12)3£и / 4 близка к экспериментально полученным величинам (~ Ю-20 Дж).
В случае тел разной химической природы предполагается, что константа их взаимодействия является средней геометрической величиной констант каждого из веществ:
4/ = ГМ,)1/2. (3.6.5)
Если допустить, что формула (3.6.2) остается справедливой при минимально возможном расстоянии между телами, равном размеру молекул d, то при h- d эта формула должна дать энергию когезии WK и адгезии W& для тел одинаковой и разной природы соответственно. Так как они известны из опыта, то это позволяет вычислить константу взаимодействия тел одинаковой и различной природы. Следует, однако, учитывать, что формула (3.6.1) обоснована для дисперсионной составляющей сил Ван-дер-Ваальса, поэтому в расчет следует принимать только часть энергии адгезии (когезии), пропорциональную доле Ь дисперсионных сил в общем их балансе. Для воды b « 0,2, WK = 0,145 Дж/м2 при 293 К и точное значение константы Ван-дер-Ваальса — Гама-кера А = 5,13 • Ю-20 Дж. Из соотношения А / cf'5 = bWK эффективное расстояние d между двумя частями адгезионно разъединяемого столба воды будет примерно 2,3-1010м. Эта величина несколько меньше, чем средняя величина расстояния (И„ / NA)i/3 = 3- 1О1ом, вычисленная из молярного объема воды Vn. Учитывая приближенный характер вычислений, для расчетов оценочного характера будет использована формула d=(Vn / Ул)13- При взаимодействии веществ разной природы это расстояние является средним арифметическим межмолекулярных расстояний каждого вещества, так что d = (Vn\/3 + V„,23)/2N1j4/3 , где VnX и Ии2 — молярные объемы веществ. Таким образом, из уравнений (3.6.2а) и (3.2.5) следует формула для константы взаимодействия твердого тела с жидкостью:
Л12 = ^’^0(1 + cos0). (3.6.6)
Эта формула является альтернативой формуле (3.6.5) и не сводится к ней.
Погружение тел в однородную жидкость не меняет закона их взаимодействия, а изменяет только константу взаимодействия. Сближение двух одинаковых (твердых) тел в жидкости из бесконечности до некоторого конечного расстояния h можно рассматривать как результат замещения равного по объему и конфигурации воображаемого «жидкого тела» (21) «настоящим» твердым телом (7) (рис. 3.49).
Рис. 3.49. Сближение тел (7) в среде (2) до расстояния h между ними
Энергия всей системы в исходном состоянии складывается из энергии взаимодействия U\ -2А\21 й2двух пар разнородных тел (7) и (21), а в конечном состоянии из энергии пары тел (7)-(7) и тел
ип=Ац/ И2 + Ап/ h2.
Изменение энергии системы V\ i - V\ и есть энергия взаимодействия пары тел (7)-(/) в среде (2) Ui2 = А / h2. Это соотношение приводит к уравнению:
= т4ц + А22 — 2^4 )2, (3.6.7)
которое, если учесть уравнение (3.6.5), сводится к выражению:
А = (Ла‘12-А22ш)г. (3.6.8)
Отсюда следует, что константа А всегда положительна, т. е. погружение одинаковых по природе тел в любую жидкость не приведет к их взаимному отталкиванию. Это справедливо, если твердое тело не влияет на структуру и состав жидкости в зазоре, т. е. при его погружении в жидкость не происходит адсорбции, ориентации молекул, появления ДЭС и т. д. Эти возмущения учитываются с помощью введения независимой от молекулярного взаимодействия дополнительной силы отталкивания двойных электрических, адсорбционных и других слоев, возникающих в жидкой фазе вблизи поверхности тела.
Теория взаимодействия тел через тонкую пленку среды, предложенная Дерягиным и Ландау, предполагает, что эти взаимодействия являются суммой независимых слагаемых: молекулярного притяжения и электростатического отталкивания двойных и/или иных слоев. Позже она была систематически изложена Фер-веем и Овербеком [46] и известна как теория ДЛФО.
3.6.2. Электростатическое отталкивание ДЭС
В двойном электрическом слое поверхностный заряд экранирован противоионами на расстоянии порядка толщины 5 ДЭС, поэтому очевидно, что при больших расстояниях между поверхностями (И » 5 или хЛ » 1) заряженные поверхности электростатически не взаимо
620
Новый справочник химика и технолога
действуют, т. е. не отталкиваются при одноименном их заряде. Необходимым условием возникновения сил отталкивания двойных слоев является их перекрывание. Оно влечет за собой изменение распределения потенциала и заряда в ДЭС, а следовательно, и электростатической составляющей поверхностного натяжения каждой из сторон пленки в зазоре между телами, что, согласно общему выражению (3.3.44), и определяет силу отталкивания. В смысле различия натяжений пленка считается тонкой (см. разделы 3.2.2., 3.2.3, 3.3.4), хотя при большой толщине ДЭС она может на многие порядки превышать по толщине пленку мономолеку-лярного адсорбционного слоя ПАВ. Чтобы воспользоваться формулой (3.3.44), необходимо выразить электрическую часть натяжения одной из сторон тонкой пленки через ее толщину, а также через параметры ДЭС — и х. Ограничимся при этом рассмотрением случая слабо заряженного слоя.
Уравнение Пуассона — Больцмана (3.5.10), а также формула (3.5.5а) для заряда поверхности и уравнение (3.4.24) для натяжения справедливы в любом случае, как при перекрывании ДЭС, так и без перекрывания. Задача, следовательно, сводится к нахождению напряженности поля (бАР/б£х)0 на поверхности тел при перекрывании имеющихся на них двойных слоев. Потенциал поверхности не зависит от расстояния h между поверхностями. В противном случае он как характеристика гетерогенной системы лишен смысла, завися от размера и формы поверхности частиц.
Из соображений симметрии распределения потенциала и заряда в зазоре между заряженными поверхностями можно ожидать, что в середине зазора потенциал имеет минимум (рис. 3.50).
Рис. 3.50. Распределение потенциала в слое толщиной h с заряженными стенками: начало координат совмещено с левой границей слоя; й/2 — плоскость симметрии слоя; % — потенциал этой плоскости
Решение уравнения Пуассона — Больцмана для перекрывающихся ДЭС отличается от рассмотренного ранее в подразделе 3.5.1 значением постоянной интег
рирования Ci. Из условия равенства нулю производной / dx в точке х = h/2 (середина зазора) следует, что
Ci = x2chue,
где ие = zP¥e / RT — безразмерный и % — размерный потенциал ДЭС в плоскости симметрии при х = h/2. Тогда справедливо соотношение:
du! dx = x[2(chu - ch ме)]1/2. (3.6.9)
При малых значениях аргумента chy ~ 1 + у2/2, поэтому при малом потенциале поверхности выполняется приближенная формула:
dV/dx-x^2-^2)1^. (3.6.10)
После разделения переменных и интегрирования в пределах от произвольной величины потенциала 4х до Т = % получается следующее выражение:
-х(Л/2 - х) = Archl - Arch('P / %).
Так как Archl = 0, то при переходе от обратной гиперболической функции к прямой можно найти потенциал 4х:
Т = 4/ech[x(A/2 -х)].
В частности, при х = 0 потенциал Т = %, т. е. % = %сЬ(х/?/2). Поэтому потенциал Т и напряженность поля Е = -di/ / dx описываются формулами соответственно:
Т = {ch[x(A/2 - х)] / ch(xA/2)}, (3.6.11)
Е = х% {sh[x(A/2 -х)] / ch(xA/2)}. (3.6.12)
Таким образом, в формуле (3.5.5) Ео = x4/sth(xh/2). При интегрировании уравнения электрокапиллярности (3.4.24) с учетом того, что qs = ееох th(xA/2)T's, получается выражение:
<зе(И) = -О^ееохТ 2 th(xA/2). (3.6.13)
В частности, при h = <х> (толстая пленка) th(xZ?/2) = 1, и уравнение (3.6.13) сводится к формуле:
ое(оо)= -О,5ееохЧ/ ?. (3.6.13а)
При подстановке выражений (3.6.13) и (3.6.13а) в уравнение (3.3.44) удельная (на единицу площади) энергия отталкивания заряженных поверхностей в растворе электролита будет описываться уравнением:
Ue(h) = ееохТ 2 [ 1 - th(xA/2)]. (3.6.14)
Индекс е в формулах (3.6.13), (3.6.13а) и (3.6.14) означает, что они описывают электростатические составляющие межфазного натяжения и энергии отталкивания поверхностей.
При произвольном потенциале поверхности натяжение и отталкивание ДЭС нельзя выразить с помощью простых функций переменных %, х и h.
Физика, химия и технология дисперсных систем
621
3.6.3. Отталкивание адсорбционных слоев
Формулы (3.3.43) и (3.3.44), являющиеся основой для нахождения функции U(h) перекрывания адсорбционных слоев, эквивалентны. Это действительно так, поскольку при h = const в уравнении (3.3.39) Г = -Jo / Jp, а замена в уравнении (3.3.43) Г на -Jo / dp приводит его к следующему виду:
U (Л) = 2
Здесь Jo(/) — изменение натяжения границы толстой пленки, da(h) — изменение натяжения границы тонкой пленки при изменении состава раствора и фиксированной толщине пленок (i и h соответственно).
Если натяжения толстой и тонкой пленок в чистом растворителе равны, т. е. о0(0 = о0(Л), то данное выражение сводится к формуле (3.3.44).
Очевидно, что совпадение о0(/) и о0(Л) возможно для пленок, которые можно считать толстыми в чистом растворителе, например для пленок толщиной 10 9 м в низкомолекулярном растворителе (вода, ацетон, гексан и т. д.). В присутствии молекул ПАВ большой длины (например, 2 • 10~9 м) при неизменной толщине h пленка попадает в разряд тонких. Таким образом, формула (3.3.44) пригодна для пленок достаточно толстых по сравнению с размером молекул растворителя. В более общем случае справедливо уравнение:
ОД = 2{[о(0 - о0(0] - [ОД - <ОД]}. (3.6.15)
Здесь о0(0 и о0(/г) — натяжение границ соответственно толстой и тонкой пленок в чистом растворителе. В дальнейшем будет подразумеваться (если не оговорено обратное), что выполняются условия, позволяющие в чистом растворителе приравнять натяжения толстых и тонких пленок в уравнении (3.6.15), т. е. свести эту формулу к уравнению (3.3.44).
Легко видеть, что подстановка в уравнение (3.3.43) уравнения изотермы адсорбции Ленгмюра, а также соотношения dp = RTdc/c, приводит к выражению:
ОД = 2Я71Гт(/) - ГОД] 1п( 1 + кс). (3.6.16)
Таким образом, получается тот же результат, что и при описании натяжений в уравнении (3.3.44) с помощью формулы Шишковского. В уравнениях (3.6.15) и (3.6.16) предполагается применимость изотерм адсорбции Г(Л) и натяжения о(/?) к тонкому слою раствора, находящемуся в равновесии с его объемной частью, и независимость константы равновесия к от h. При этом зависимость U(h) будет полностью определяться влиянием толщины h пленки на предельную адсорбцию Гт в зазоре. Если предположить, что выполняется простое соотношение: Vm(h) = Гт(/)Л / 25, где h < 25, то энергия отталкивания адсорбционных слоев линейно зависит от ширины зазора, согласно уравнению:
С/(6) = Г.Я71п(1+к)^Л (3.6.17)
О
Если зависимость заполнения зазора от его толщины экспоненциальная, то получается рекомендуемое рядом авторов уравнение:
ОД - ГтЛЛп(1 + кс) ехр(-Л / 25). (3.6.18)
При описании расклинивающего действия ПАВ на качественном уровне обычно выделяют его энтропийную и структурно-механическеую составляющие. Энтропийное отталкивание адсорбционных слоев возникает за счет ограничения числа возможных состояний молекул ПАВ при перекрывании адсорбционных слоев. Этот эффект является основным при малой степени заполнения адсорбционного слоя, т. е. при Г < Гт, так как в насыщенном адсорбционном слое (Г = Гт) число степеней свободы молекул ПАВ и, соответственно, конфигурационная энтропия адсорбционного слоя близки к нулю. Однако именно в этом случае действует структурно-механический фактор отталкивания адсорбционных слоев. Он означает, что адсорбционные слои механически прочные, непроницаемые и поэтому препятствуют сближению покрытых ими поверхностей. Разумеется, что при этом должна быть исключена склонность к притяжению и слипанию самих адсорбционных слоев. Это условие автоматически выполняется, если обращенные к среде концы адсорбированных молекул имеют хорошее сродство к растворителю.
3.6.4. Структурный и стерический эффекты
Взаимодействие поверхности твердого вещества с жидкой фазой кроме адсорбции, т. е. изменения концентрации компонентов в поверхностном слое, может заключаться также в ориентировании молекул растворенного вещества и растворителя в поверхностном слое. Хорошо известный пример такого действия — ориентация стержневидных молекул ПАВ в насыщенном адсорбционном слое. В принципе ориентация может иметь место и без изменения концентрации компонентов раствора в поверхностном слое, а также в однокомпонентной жидкости (чистом растворителе). В этих случаях ориентация вызвана непосредственным воздействием поверхности на молекулы, тогда как при насыщении поверхностного слоя она обусловлена стериче-ским взаимодействием адсорбированных молекул между собой.
Под стерическим подразумевается взаимодействие, связанное с наличием у молекул размера и формы, что накладывает жесткие ограничения на способы их размещения в пространстве. Например, плотнейшая упаковка стержневидных молекул возможна только при их параллельной ориентации перпендикулярно поверхности адсорбента. На принудительное изменение такой ориентации слой отвечает бесконечно сильным сопротивлением при постоянстве адсорбции или конечной величиной сопротивления при десорбции части молекул.
622
Новый справочник химика и технолога
Известный эффект однородного наклона адсорбированных полярных молекул (эффект Толстого [28]) может быть достигнут ценой десорбции небольшого числа молекул. Выигрыш энергии при наклоне возникает за счет уменьшения толщины 5 молекулярного конденсатора, образованного разноименно заряженными концами ориентированных молекул. Убыль энергии конденсатора -SEoT j / 25 компенсирует ее увеличение за счет десорбции части ПАВ с поверхности.
Абсолютно гладкая поверхность твердого тела упорядочивает и молекулы сферической формы — ближайший к поверхности слой этих молекул расположен в одной плоскости, как и противоионы ДЭС в плоскости их максимального приближения (рис. 3.37). Это приводит к упорядоченному расположению следующих слоев вещества. Вдоль нормали к поверхности возникает кристаллоподобный порядок. Очевидно, что только при кратных диаметру молекул толщинах этот слой может иметь нормальную (объемную) плотность и, следовательно, минимум энергии. Отклонения в ту и другую сторону от кратной толщины вызывают понижение плотности и повышение энергии слоя. Это может создавать как отталкивание, так и притяжение поверхностей, ограничивающих толщину слоя [21].
Возникновение большего, чем в газе или жидкости, порядка в расположении молекул принято называть структурированием, а соответствующие слои жидкости и силы, обусловленные соприкосновением или перекрыванием этих слоев, — структурными.
Силы отталкивания поверхностей, вызываемые тем, что поверхности ограничивают свободу движения и ориентации молекул, принято называть стерическими. Очевидно, что и структурные силы могут быть обусловлены стерическими эффектами, поэтому не всегда их можно различить. Если все-таки в этом возникает необходимость, то критерием может служить знак производной dl / dh, где I — параметр порядка, например полнота ориентации молекул. Сближение поверхностей может способствовать ориентации остающиеся в зазоре стержневидных молекул, если их длина больше толщины зазора, т. е. упорядоченность будет нарастать при уменьшении толщины зазора h. Сила отталкивания (стерического) при этом также будет увеличиваться. Структурное отталкивание связано с самопроизвольным упорядочиванием молекул у свободной поверхности (в пленке неограниченной толщины), что является прямым или косвенным результатом взаимодействия молекул с поверхностью (адсорбции). Выдавливание вещества из зазора в объем жидкой фазы, где порядка нет, ведет в этом случае к обратному эффекту — к нарастанию беспорядка в расположении молекул среды или ПАВ.
Стерическое отталкивание не обязательно связано с изменением ориентации молекул в зазоре при изменении ширины зазора. Оно может возникать просто в силу присутствия некоторых веществ в толстом зазоре при той же концентрации, что и в объемной жидкой фазе.
Теория стерического отталкивания удлиненных молекул
Будем считать, что молекулы имеют форму гантелей длиной /, их концентрация с в зазоре та же, что в объеме, и не зависит от ширины зазора h. В толстом слое (h>I) поведение молекул подчиняется законам идеального газа, в частности их давление Р на стенки щели равно cRT.
В тонкой щели (Л < I) молекулы вынуждены располагаться более или менее параллельно стенкам щели. Ее ширина определяет величину угла встречи £2 конца молекулы со стенкой при ее вращательном тепловом движении (рис. 3.51).
Поступательное движение вдоль нормали к поверхности в этих условиях невозможно. Угол между осью молекулы и поверхностью щели связан с ее шириной очевидным соотношением: sin£2 ~ЫI.
В тонкой щели за счет вращательного теплового движения стенка бомбардируется концами гантелеобразных молекул. В пределе h = 0 и sin£2 = 0 концы налетают на поверхность под прямым углом, а так как концентрация концов вдвое больше, чем самих молекул, то и давление в щели P(h) удваивается и достигает величины 2cRT. При конечной ширине слоя вращающийся конец молекулы наносит удар по поверхности под углом £2 к нормали, поэтому нормальная компонента импульса в cos£2 раз меньше, чем полный импульс. Соответственно этому уменьшается избыточное давление P(h) -Р в тонком слое (т. е. его расклинивающее давление П) до величины П(й) = c7?7cos£2. Последнему соотношению эквивалентна формула:
П(й) = cRT [1 - (h I Z)2]1/2 (3.6.19)
Рис. 3.51. Схема взаимодействия срежневидной молекулы со стенками узкой щели:
I — длина стержневидной молекулы; h — ширина щели;
Q — угол столкновения молекулы со стенкой щели
Более последовательный анализ этого эффекта должен учесть неизбежную убыль концентрации молекул в слое, так как повышение давления влечет за собой увеличение химического потенциала, а следовательно, и появление диффузии, направленной на выравнивание химических потенциалов соответствующего компонента в слое и в объеме. По этой причине формула (3.6.19) является иллюстрацией физического механизма стерического отталкивания. Она дает представление о порядке величины этого отталкивания. В связи с этим избы
Физика, химия и технология дисперсных систем
623
точное осмотическое давление при перекрывании диффузных двойных электрических слоев имеет тот же порядок величины:
Пе = 2cRT (chue - 1).
Актуальность проблемы стерической стабилизации связана с широким техническим применением полимеров, их растворов и полимерно-минеральных составов с дисперсным минеральным компонентом. Адсорбция, а в ряде случаев и просто присутствие полимера оказывают очень сильное влияние на устойчивость к коагуляции дисперсного компонента. Представления о сте-рических эффектах занимают центральное место в объяснении свойств полимерных растворов, их расплавов, стабилизирующего и коагулирующего действия на дисперсные системы.
Сложность теплового движения полимерных молекул затрудняет описание стерических эффектов, поэтому целесообразно рассмотреть альтернативный путь, предложенный Фишером. В его основе лежит теория растворов полимеров Флори и Хаггинса. Важнейшим понятием этой теории является параметр взаимодействия полимера с растворителем % = zA(7i2 / RT, где z — координационное число молекул растворителя вокруг звена полимерной молекулы в растворе, ДС/12 = U\2 -- (С/ц + и22) / 2, иу — энергия контакта молекул сорта i и j (растворителя и звеньев полимерной цепи). Взаимодействие полимер—растворитель сказывается, в частности, на величине осмотического давления раствора, которое отражено в уравнении:
Рс = cRT [1 + (0,5 - х) с]. (3.6.20)
Здесь с — концентрация полимера (кмоль/м3). Произведение (0,5 - х) с представляет собой поправку на неидеальность раствора. При хорошем качестве растворителя (х < 0,5) осмотическое давление больше, чем в идеальном растворе, а в плохом растворителе — меньше.
По Фишеру [25] расклинивающее давление при перекрывании адсорбционных полимерных слоев определяется неидеальной частью осмотического давления и описывается уравнением:
П = 2 са2 ЯГ (0,5 - X)- (3-6.21)
Здесь са — концентрация полимера в адсорбционных слоях до их перекрывания.
Таким образом, П не зависит от степени перекрытия адсорбционных слоев, и тогда энергия отталкивания оказывается пропорциональной объему зоны перекрывания адсорбционных слоев. При фиксированном расстоянии между частицами степень перекрытия адсорбционных слоев зависит от их толщины, которая, в свою очередь, зависит от размера адсорбированных молекул.
В отличие от обычных органических веществ, например поверхностно-активных, молекулы полимеров (макромолекулы) не имеют определенного размера.
В силу гибкости длинных полимерных молекул они свернуты в рыхлые клубки, размер и форма которых непрерывно изменяется под действием более или менее независимого теплового движения отдельных звеньев молекулярной цепи. В связи с этим состояние макромолекул характеризуется некоторой среднестатистической величиной их размера, чаще всего — расстояния R между концами полимерной цепи. В растворе этот размер зависит от качества растворителя (0,5 - х), который может быть оценен экспериментально по коэффициенту набухания — относительному увеличению объема образца полимера при его помещении в растворитель.
В статистической теории состояние макромолекул принято характеризовать с помощью линейного коэффициента набухания со = R/ Ro — отношения реального размера R полимерного клубка к размеру Ro в некотором стандартном состоянии. Теория Флори [29] устанавливает связь этого коэффициента с качеством растворителя, которая описывается уравнением:
со5 - со3 - (0,5 - х) К2/И. (3.6.22)
Здесь К2 и И — молярные объемы полимера и растворителя, а /?о — линейный размер клубка в ©-растворителе (тета-растворителе), в котором х = 0,5, т. е. со5 - со3 = 0, что возможно только при со = 1. В хорошем растворителе (х<0,5)со3> 1, а в плохом растворителе со3 < 1, причем коэффициент объемного набухания (относительное приращение объема V образца) А И/ V = со3 - 1.
Характеристика качества растворителя (0,5 - х), полученная экспериментально по набуханию полимера, может быть далее использована при расчетах расклинивающего давления по формулам типа (3.6.21).
Размер полимерных клубков соизмерим с радиусом действия поверхностных сил различной природы, и поэтому присутствие полимерных молекул в зазоре между коллоидными частицами способно повлиять на взаимодействие частиц даже в том случае, когда полимерные молекулы не связаны адсорбционными силами с поверхностью частиц. Такого рода эффекты получили название вытеснительных.
3.6.5. Вытеснительное отталкивание
и притяжение
В тонкой щели между двумя твердыми поверхностями полимерный клубок будет расплющиваться. Это ведет к увеличению свободной энергии Гиббса за счет уменьшения энтропии «расплющенных» молекул.
В силу большого числа звеньев в полимерной цепи одна полимерная молекула может рассматриваться как термодинамическая система, и ее состояние можно характеризовать энтропией молекулы. В соответствии со статистическим определением энтропия пропорциональна логарифму числа возможных микросостояний W системы: S' = Л1пИС Под микросостоянием полимерной молекулы подразумевается то или иное конкретное расположение ее звеньев, т. е. W есть число возможных
624
Новый справочник химика и технолога
конфигураций полимерной цепи. У гибкой молекулы, имеющей N звеньев, таких конфигураций порядка 3\ Соответственно, энтропия, определяемая числом w возможных конфигураций, называется конфигурационной:
SK = *lnw. (3.6.23)
Узкая щель ограничивает величину w, следовательно, и конфигурационную энтропию, что и ведет к увеличению свободной энергии Гиббса, в результате чего молекулы полимера переходят в раствор. При достаточно малом зазоре между непроницаемыми для полимерных звеньев поверхностями в зазоре останется только растворитель. В этой ситуации самопроизвольному уменьшению энергии системы будет способствовать дальнейшее уменьшение ширины зазора h, так как при этом чистый растворитель из зазора смешивается с раствором полимера, который находится вне зазора. При хорошем качестве растворителя смешивание полимера с растворителем (растворение) идет самопроизвольно, т. е. с убылью энергии Гиббса. Аналогичный эффект имеет механизм отталкивания гантелевидных молекул. Различие заключается лишь в способе их описания — механическом или термодинамическом.
Таким образом, присутствие полимерных и других молекул в растворе и в тонком слое между поверхностями само по себе влияет на взаимодействие поверхностей даже в отсутствие адсорбции. Сначала сближение поверхностей сопровождается их отталкиванием за счет деформирования и вытеснения полимерных клубков, а затем оно сменяется притяжением поверхностей из-за выгодности смешивания хорошего растворителя с полимером (рис. 3.51). Эти эффекты, обусловленные вытеснением свободного (неадсорбированного) полимера из тонкого слоя раствора, получили название вытеснительных. В дисперсных системах они приводят как к вытеснительной коагуляции (в разбавленных растворах полимеров), так и к вытеснительной стабилизации (в концентрированных растворах).
Термин «вытеснительная коагуляция» применяют в тех случаях, когда причиной коагуляции является кристаллизация дисперсионной среды или иные фазовые переходы. Так как вхождение инородной частицы в кристаллическую решетку вызывает ее искажение и, следовательно, увеличение энергии решетки, то любые неизоморфные элементы решетки тела (коллоидные частицы, молекулы растворенного вещества, неизоморфные ионы и т. д.) вытесняются растущими кристаллами и оказываются в итоге на межкристаллитных границах, где и слипаются. Это явление лежит в основе очистки кристаллических веществ путем их перекристаллизации, в том числе зонной. Эффект вытеснительной коагуляции фазовым переходом растворителя может быть в принципе выражен через тепловой эффект фазового перехода. Вместе с тем коагуляция происходит и при фазовых переходах, которые не сопровождаются затвердеванием среды. Легко, например, наблю
дать обратимую коагуляцию коллоидного раствора магнетита в классическом жидкокристаллическом веществе — МББА при переходе этого вещества из нормального в жидкокристаллическое состояние.
Отметим, что исчерпывающее количественное описание взаимодействия макроскопических поверхностей и коллоидных частиц во всех рассмотренных в этой главе случаях может базироваться на формулах (3.2.28), (3.3.43), (3.3.44). Проблема в том, что далеко не всегда эти общие формулы можно свести к конкретной зависимости энергии расклинивающего давления от толщины зазора.
3.7. Устойчивость дисперсных систем
3.7.1. Вводные понятия
Та или иная дисперсная система предназначена для выполнения определенных функций: служить исходным материалом для формования строительной конструкции, если это цементная смесь; исполнить роль защитной или декоративной краски, если это суспензия пигмента; подчинить движение жидкости воздействиям магнитного поля, если это коллоидный раствор ферромагнетика, и т. д. Возможность дисперсной системы выполнить предназначенную ей функцию зависит от ее рецептуры — наличия в составе системы частиц вяжущих, окрашенных или магнитных материалов. Однако качество продукта и технологичность его применения и получения определяются общим свойством любых дисперсных систем вне зависимости от их рецептуры — их устойчивостью. Устойчивость — это способность системы сохранять постоянство своих свойств во времени или при достаточно сильном изменении условий. Среди разнообразных свойств всеобъемлющим является равномерность распределения дисперсного материала по всему объему системы. Она определяется многими факторами, к числу которых относится устойчивость к некоторым частным конкретным изменениям состояния системы, среди которых наиболее важна устойчивость против коагуляции и оседания частиц. Терминология, касающаяся устойчивости, сложилась до того, как были выявлены многие детали и варианты изменения состояния взвесей. По этой причине толкование ряда понятий приобрело неоднозначность. Так, «коагуляция» — это слипание частиц и, кроме того, разрушение дисперсной системы, при которой происходит ее разделение на фазы: осадок, дисперсионную среду. Слипание частиц, сопровождающееся не разрушением, а лишь изменением состояния системы, иногда желательным и полезным. Агрегативная устойчивость — способность дисперсной системы противостоять слипанию частиц в том или ином понимании сути этого явления. Слипание может быть разным как по характеру, так и по силе сцепления частиц. Понятие кинетической устойчивости обычно характеризует способность взвеси противостоять расслаиванию (оседанию частиц) за некоторый конечный интервал времени. Термодинамическая устойчи
Физика, химия и технология дисперсных систем
625
вость — это характеристика избыточности энергии дисперсного состояния материала по сравнению с его энергией в монолитном состоянии. В этом смысле типичные дисперсные системы всегда термодинамически неустойчивы и характеризуются временем жизни или кинетикой разрушения. Однако переход в термодинамически выгодное состояние (образование монолита из отдельных частиц) может быть замедлен или даже исключен по ряду причин. Следует иметь в виду, что в литературе понятие устойчивости часто используется в узком смысле — как способность дисперсной системы противостоять коагуляции при введении в нее электролита. Это объясняется тем, что длительное время введение электролитов оставалось единственным способом дозированного воздействия на состояние системы, результаты которого можно было увидеть, зарегистрировать и принять в качестве характеристики устойчивости. В настоящее время благодаря развитию теории и инструментальных средств диагностики состояния дисперсных систем такое узкое толкование понятия «устойчивость» является архаичным.
Требования к устойчивости дисперсных систем могут быть различны. Часто необходима предельно высокая устойчивость дисперсных систем (например, феррожидкостей, многие из которых эксплуатируются без замены десятки лет), в других случаях устойчивость нежелательна (загрязнения воды и воздуха дисперсными частицами), иногда необходим оптимальный уровень устойчивости (при формовании изделий из дисперсных составов). Иначе говоря, устойчивость дисперсных систем должна быть управляемой и поддающейся регулированию доступными средствами в соответствии с функциональной ролью системы.
Устойчивость определяется двумя основными факторами — характером взаимодействия частиц дисперсной системы между собой (агрегативной устойчивостью) и действием на них гравитации. Природа сил, действующих между частицами, та же, что и природа рассмотренных ранее сил взаимодействия различных частей конденсированных фаз через плоские прослойки разделяющей их среды.
Современные представления о природе агрегативной устойчивости дисперсных систем созданы Б. В. Дерягиным [21]. Одна из основополагающих теоретических работ была им выполнена вместе с Л. Д. Ландау. Большие заслуги в систематизации и популяризации этой теории принадлежат Фервею и Овербеку [22, 46]. Теория агрегативной устойчивости дисперсных систем Дерягина, Ландау, Фервея, Овербека получила общее признание под названием «теория ДЛФО».
Основная идея теории ДЛФО состоит в том, что существование взвеси частиц определяется балансом действующих между ними сил, представляющих собой сумму сил молекулярного притяжения частиц и электростатического отталкивания их двойных электрических слоев. Впоследствии теория была дополнена включением в нее и сил отталкивания иной природы.
Принципиально важно, что теория ДЛФО предполагает возможность разложения межчастичных сил на аддитивные слагаемые разной природы.
Одна из характерных особенностей коллоидных растворов заключается в их высокой чувствительности к ионному составу дисперсионной среды. В некоторых случаях даже неконтролируемые (в силу своей малости) изменения ионного состава среды заметно влияют на свойства коллоида. По этой причине почти вся история развития представлений об устойчивости коллоидных растворов — это история экспериментального и теоретического изучения устойчивости этих растворов к действию на них электролитов. Теория ДЛФО, по крайней мере в первичном замысле, была предназначена для решения исключительно этой проблемы, поэтому ее с полным основанием следует определить как теорию устойчивости коллоидов к действию на них электролитов, которая может быть построена на анализе взаимодействия двух частиц. В настоящее время на первый план выдвигаются проблемы изучения тех свойств, которые являются результатом одновременного взаимодействия множества частиц или даже всех частиц дисперсной системы (коллективного взаимодействия). Очевидно, что оно тесно связано со структурой дисперсной системы — характером их взаимного расположения в пространстве и времени.
Проблемы структуры и структурирования дисперсных систем как результата взаимодействия многих частиц постоянно были в поле зрения и экспериментаторов, и теоретиков, однако количественное описание свойств и явлений, связанных со структурой и структурными превращениями, стало возможным только в последнее время благодаря появлению необходимой для этого математической базы — фрактальной геометрии.
3.7.2. Взаимодействие сферических частиц
С известной степенью приближения можно считать, что частицы дисперсных систем имеют сферическую форму. В таком случае в теории устойчивости можно ограничиться анализом сил, действующих между частицами сферической формы. Как уже было отмечено выше, в теории ДЛФО это силы молекулярного притяжения и силы электростатического отталкивания двойных электрических слоев. Формула (3.6.4) для энергии молекулярного притяжения сферических частиц получена тем же методом, что и формула (3.6.2) для взаимодействия плоских поверхностей. Однако для электростатического взаимодействия сфер задача подобным образом не решена (если не считать некоторых весьма частных условий), что порождает необходимость поиска альтернативных путей вычисления энергии или силы взаимодействия двух сферических тел. Такой путь предложен Б. В. Дерягиным и известен под названием «переход Дерягина». Он в настоящее время является единственным универсальным средством преобразования формул расклинивающего давления (или его энер
626
Новый справочник химика и технолога
гии) в плоских межфазных зазорах в формулы для сил и энергии взаимодействия сфер.
Для вывода формулы перехода от энергии взаимодействия Up(h) тел через плоский зазор h к энергии взаимодействия U(a, h) тел сферической формы с радиусом а нужно выделить на обращенных друг к другу сторонах сфер два пояска, расположенные симметрично относительно линии, соединяющей центры частиц (рис. 3.52).
Эти пояски «видны» из центров сфер под углом а к линии центров. Идея Дерягина состоит в том, что при условии а » h взаимодействие таких сферических поясков можно рассматривать как взаимодействие плоских колец некоторой площади dA. Тогда энергия взаимодействия поясков будет: dU(a, h) = U^h') dA, а взаимодействие сфер следует искать как сумму взаимодействий всей совокупности сферических поясков, образующих поверхности сфер, где h' — расстояние между поясками. В пределе бесконечно узких поясков суммирование заменяется интегрированием по поверхности согласно уравнению:
U(a,h) = $Up(h')dA. (3.7.1)
А
Площадь сферического пояска dA = 2nrds, где г = asinct — радиус пояска и ds = ada — его ширина. Необходимо обратить внимание на то, что используется именно площадь сферического пояска:
dA = 27rasina ada,
а не площадь плоского кольца, ширина которого равна dr = acosaJa. Это обосновано тем, что реально взаимодействуют сферические пояски, а не плоскопараллельные кольца, упоминаемые при объяснении сути перехода Дерягина. Расстояние h' между поясками представляет собой сумму кратчайшего расстояния между поверхностями сфер h и двух отрезков величиной а - acosa, т. е.
h’ = h + 2а (1 - cosa).
Приращение расстояния при перемещении по поверхности сфер к очередному пояску будет dh' = 2asinaJa. Эта величина входит и в формулу для площади пояска, так что последняя может быть выражена через приращение расстояния между поясками: dA = itadh*. В итоге подынтегральное выражение формулы (3.7.1) преобразуется к виду
Up(h')dA = naUp(h')dh',
что позволяет получить искомую зависимость энергии взаимодействия сфер простым интегрированием энергии взаимодействия плоских поверхностей по расстоянию согласно уравнению:
2а
и (a, h) = naf Up (h')dh'. (3.7.2)
h
I
Рис. 3.52. Пояснение к выводу формулы (3.7.2) «переход Дерягина»
В этой формуле верхний предел интегрирования можно заменить бесконечностью, так как, в силу условия применимости перехода (3.7.2), диаметр частиц 2а практически равен бесконечности по сравнению с зазором h между ними.
Обращает на себя внимание, что энергия взаимодействия сфер пропорциональна первой степени их размера, а не квадрату размера, как можно было думать, исходя из пропорциональности взаимодействия dU(a, h) поясков их площади.
Формула (3.7.2) универсальна в том отношении, что она применима к любому взаимодействию, если оно пропорционально площади плоских фигур. Применяя ее к молекулярному притяжению плоскостей (3.6.2), без труда получим формулу для притяжения частиц радиусом а:
Um(a, h) = -naA/Jh. (3.7.3)
К такому же результату приводит формула (3.6.4), выведенная непосредственно для притяжения сферических частиц, если ее привести к условию применимости перехода Дерягина а » h, т. е. положить q = 1 - х, где х — малая по сравнению с единицей величина, равная, согласно определению (3.6.4) этого параметра, отношению hl а.
Ненамного сложнее интегрирование выражения (3.6.14) для отталкивания двойных слоев, содержащего функцию [1 - th(x/?/2)], которую можно заменить функцией 2ехр (-Kh) / [1 + exp (-хЛ)]. Ее интегрирование дает выражение для энергии электростатического отталкивания заряженных сферических частиц:
Ue(a, Л) = 2xa£EovE j In [1 + exp (-хЛ)]. (3.7.4)
При большом относительном расстоянии хЛ между частицами (хЛ » 1) это выражение превращается в уравнение:
Ue(a, h) = 2яа££о'¥? exp (-хА). (3.7.4а)
В данном случае относительное расстояние хЛ — это расстояние, выраженное в толщинах двойного слоя 1/х. При большой величине хЛ двойные слои перекрываются слабо, так что распределение потенциала в зазоре может быть с достаточной точностью представлено в виде суперпозиции потенциалов двух противостоящих
Физика, химия и технология дисперсных систем
627
слоев. Непосредственное решение задачи отталкивания двойных слоев в этом приближении также приводит к формуле (3.7.4а).
Исходное выражение для расклинивающего давления в плоском зазоре (3.6.4), следовательно, и формулы (3.7.4) и (3.7.4а) справедливы только при малом потенциале поверхности (около 25 мВ и менее). При произвольном потенциале поверхности и слабом перекрывании двойных слоев сравнительно легко получается следующее выражение:
Ue(a, h) = 32ла££о(/?77 z/OVexp^xA). (3.7.5)
Здесь
у=titzF¥s/4RT) = [1 - exp(-zFP5 /2/?7)] / [1 + exp(-zM\ /2R7)].
С увеличением потенциала поверхности % параметр возрастает от 0 до 1. При малом потенциале формула (3.7.5) переходит в формулу (3.7.4а), так что обе они могут быть представлены одним общим выражением:
Ue = Яехр(-хЛ), (3.7.6)
где В - паВ' нВ' = 32zzq(RT / zF)V- Здесь и в дальнейшем перечень аргументов (a, h) в левой части выражений для энергии того или иного вида взаимодействия сферических частиц опускается.
Максимально возможная величина константы электростатического отталкивания В' равна, согласно уравнению (3.7.5), величине 32££О(7?7’ / zF)2 при у= 1, т. е., при неограниченном увеличении потенциала поверхности. Фактически же уже при потенциале около 250 мВ она достигает предельно большого значения. Это, в частности, означает, что нет никакой необходимости чрезмерно повышать потенциал поверхности, так как это не ведет к увеличению сил отталкивания частиц. Полезно обратить внимание на однотипность вхождения размера частиц (в виде множителя тш) как в формулу (3.7.3) для молекулярного притяжения, так и в формулы (3.7.5), (3.7.6) для отталкивания двойных слоев, а следовательно, и в выражение для полной потенциальной энергии взаимодействия двух частиц U:
U = каВ'ехр (~хЛ) - naAhlh. (3.7.7)
Однотипность и простота влияния размера частиц на оба слагаемых позволяет исключить его из формулы и в дальнейшем рассматривать приведенную энергию U' — энергию взаимодействие частиц с размером а = 1/тс, что упрощает формулу (3.7.7) и приводит к выражению:
U' = Z?'exp(-x/i) -Ahl h. (3.7.8)
Энергия взаимодействия частиц нужного размера получается из приведенной энергии с помощью простой формулы:
Зависимость потенциальной энергии взаимодействия двух частиц от расстояния между ними удобно представлять в виде графика, который принято называть потенциальной кривой (рис. 3.53).
Рис. 3.53. Зависимость энергии взаимодействия двух частиц U, их электростатической Ue и молекулярной Um составляющих от расстояния между частицами h
Характерной особенностью потенциальных кривых является наличие двух экстремумов — достаточно острого максимума, который обычно называют потенциальным барьером, и пологого минимума, который называют потенциальной ямой. Барьер разделяет области ближних и дальних расстояний. В рамках молекулярноэлектростатического взаимодействия, описываемого формулой (3.7.7) или (3.7.8), на ближних расстояниях всегда преобладает притяжение (потенциальная энергия имеет отрицательные значения), причем оно бесконечно велико при расстоянии между поверхностями частиц, равном нулю. Взаимодействие частиц, когда расстояние между ними равно 0, называют контактным взаимодействием. Иногда термин «расстояние между частицами» означает расстояние между их центрами (в формулах оно будет обозначено символом г). Потенциальные же кривые представляют зависимость потенциальной энергии двух частиц от кратчайшего расстояния h между их поверхностями.
На дальних расстояниях (область потенциальной ямы) реально всегда преобладает притяжение, но при некоторых достаточно малых значениях параметра Дебая (больших толщинах ДЭС) потенциальная яма может исчезнуть, т. е. на дальних расстояниях могут преобладать силы отталкивания (потенциальная энергия взаимодействия частиц будет иметь положительное значение).
Одни задачи удобно решать, описывая взаимодействие частиц с помощью потенциальных кривых их взаимодействия (потенциала взаимодействия), решая другие — удобнее использовать силу взаимодействия частиц F(a, h) или F’(a, h). Сила есть производная потенциала U по координате h с обратным знаком, так что уравнение (3.7.8) преобразуется в уравнение приведенной силы
U=naU'.
(3.7.9)
F' = кВ' ехр (-хЛ) -Ah / А2.
(3.7.10)
628
Новый справочник химика и технолога
Силовая функция F взаимодействия частиц произвольного размера а связана с приведенной функцией F’ таким же соотношением, что и энергетические функции 1Ш:
F(a,h) = itaF'(a,h). (3.7.11)
График силовой функции внешне мало чем отличается от графика потенциала парного взаимодействия частиц. Для него характерно наличие силового барьера и силовой ямы, а также бесконечно большая сила сцепления частиц при их контактном взаимодействии.
3.7.3. Устойчивость против коагуляции
Потенциальные и силовые кривые взаимодействия частиц являются важнейшим инструментом теоретического анализа свойств дисперсных систем. При этом, как правило, предполагается, что в исходном состоянии системы частицы находятся на бесконечно большом удалении одна от другой или они равномерно распределены по всему доступному для них пространству, если оно ограниченно. При сближении частиц до их прямого контакта потенциальная энергия взаимодействия достигает абсолютного минимума.
Следовательно, такое состояние частиц дисперсной системы термодинамически наиболее выгодно . Это обстоятельство можно понимать как отражение в теории ДЛФО факта термодинамической неустойчивости дисперсных систем, т. е. невыгодности дисперсного состояния некоторого фиксированного количества вещества по сравнению с его монолитным состоянием. Однако переходу в это состояние, который осуществим только путем сближения частиц, препятствует потенциальный барьер. При достаточно большой величине барьера термодинамически наиболее устойчивое состояние (переход к контактному взаимодействию) оказывается недостижимым, и дисперсная система оказывается в аг-регативно устойчивом состоянии — приобретает способность сохранять неизменным первоначальный размер частиц. Мерой устойчивости при этом считается величина («высота») потенциального барьера АСА
Наличие потенциальной ямы приводит к неконтактному слипанию частиц: между ними сохраняется прослойка среды, толщина которой равна координате минимума на потенциальной кривой. Глубина минимума (потенциальной ямы) является при этом энергетической мерой прочности сцепления частиц. Это наиболее важный для технологии дисперсных систем вид коагуляции, и некоторые его детали будут рассмотрены позже.
Наиболее универсальным и доступным средством регулирования параметров потенциальных кривых (высоты барьера, глубины и положения ямы на оси расстояний) является концентрация электролита в дисперсионной среде.
Считается, что роль энтропии мала.
При этом изменяется параметр Дебая, который по существу определяет радиус действия электростатических сил отталкивания двойных слоев. Увеличение концентрации электролита приводит к сжатию двойного слоя, т. е. к уменьшению радиуса действия сил отталкивания и, следовательно, к уменьшению величины барьера, углублению потенциальной ямы и смещению ее положения в сторону меньших расстояний между частицами (рис. 3.54).
Рис. 3.54. Потенциальные кривые взаимодействия двух частиц при различных концентрациях индифферентного электролита: концентрация увеличивается с увеличением номера кривой
Индифферентные электролиты не влияют на потенциал поверхности и, значит, на константу отталкивания В' или В. Кроме того, молекулярное притяжение частиц не зависит от ионного состава среды.
Таким образом, единственной причиной изменения баланса действующих между частицами сил притяжения и отталкивания в пользу первых при увеличении концентрации индифферентного электролита является уменьшение радиуса действия сил отталкивания двойных слоев. В противоположность этому введение по-тенциалопределяющего электролита изменяет потенциал поверхности и константу отталкивания. Отталкивание усиливается при увеличении потенциала поверхности и ослабляется вплоть до нуля при нулевом значении потенциала поверхности. Толщина двойного слоя и радиус действия электростатических сил могут при этом практически не измениться, если варьирование концентрации потенциалопределяющего электролита происходит в присутствии достаточно высокой концентрации индифферентного электролита, поскольку именно он и будет определять ионную силу раствора и толщину ДЭС. Это дает возможность независимо изменять радиус действия электрических сил либо их амплитуду В' при постоянстве радиуса действия.
3.7.4. Критическая концентрация электролита
Теория ДЛФО объясняет коагуляцию коллоидных растворов при действии на них электролитов, причем коагуляцию тепловую. Тепловая коагуляция происхо-
Физика, химия и технология дисперсных систем
629
дат в результате столкновений частиц между собой в процессе их теплового движения. Интенсивность столкновения характеризуется, как известно, его кинетической энергией. Ее среднее значение равно по порядку величины энергии теплового движения частиц кТ, где к — постоянная Больцмана и Т — температура. Результат столкновения зависит от соотношения между энергией столкновения и максимумом энергии отталкивания частиц, т. е. при постоянстве температуры — от величины потенциального барьера. При большой (по сравнению с кТ) величине барьера столкновения окажутся упругими — частицы будут отскакивать при столкновениях. При недостаточной высоте барьера появляется возможность его преодоления. Формула (3.7.12) отражает вероятность преодоления барьера в зависимости от его высоты:
w « ехр(-Д17 / кТ). (3.7.12)
При отсутствии барьера (Д{/=0, кривая 3 на рис. 3.54) столкновение неизбежно (с вероятностью w = 1) заканчивается слипанием частиц: их сближением до непосредственного контакта (h = 0), который, согласно формулам (3.7.7) и (3.7.8), является бесконечно прочным и поэтому ведет к необратимой коагуляции. Коагуляция, при которой каждое столкновение частиц проводит к их слипанию, называется быстрой. В противоположность этому медленная коагуляция — это коагуляция, при которой только некоторая доля w столкновений приводит к слипанию частиц. Она возникает при умеренной величине барьера (Д17 / кТ < 10). В зависимости от влияния ионного состава на параметры потенциальных кривых следует, что коагуляция может быть вызвана увеличением концентрации любого индифферентного электролита. При некоторой концентрации, которую принято называть критической, потенциальный барьер может быть уменьшен до нуля, и тогда наступит быстрая коагуляция.
Условие перехода к быстрой коагуляции AU = 0 в развернутом виде сводится к системе уравнений:
t/' = 0,F' = 0 (3.7.13)
или
5'ехр(-хй) - Ah / h = 0,
(3.7.13а) хВ'ехр(-хй) - Ah / h2 = 0.
Она является отражением того факта, что при равенстве нулю потенциального барьера (см. рис. 3.54; кривая 3) должно быть равно нулю и значение потенциальной энергии взаимодействия частиц (первое равенство) в точке максимума функции U(h), т. е. при одновременном равенстве нулю ее первой производной, которая равна с обратным знаком силе взаимодействия частиц F(h) (второе равенство).
Иначе говоря, одновременно должно выполняться условие (3.7.13а) равенства нулю правых частей уравнений и (3.7.8), и (3.7.10). Легко заметить, что это воз
можно (система уравнений (3.7.13а) совместна) только при условии:
х = 1 / h или Kh = 1. (3.7.14)
Подставив это значение хй в первое из уравнений (3.7.13а), можно найти по формуле (3.7.15) значение параметра Дебая (его критическую величину хс), при котором потенциальный барьер равен нулю:
Kc = B4eAh. (3.7.15)
Отсюда легко перейти к критической концентрации электролита сс. Следует отметить, что приведенные уравнения и формулы в полной мере справедливы для растворов симметричных электролитов, когда связь параметров раствора выражается простейшей формулой (3.5.9) с = 72/bz\ где b - F (2/s£qRT)112. Тогда критическая концентрация электролита сс вычисляется по формуле:
сс = (В' /zeAhf/b. (3.7.16)
При большом потенциале поверхности константа отталкивания зависит от формального заряда ионов z. Подставив ее в формулу (3.7.16), можно видеть, что критическая концентрация электролита пропорциональна 1/z6. Такого рода закономерность имеет экспериментальное подтверждение: коагулирующая способность электролитов увеличивается в десятки и сотни раз при замене одновалентных ионов на двухвалентные и трехвалентные соответственно (правило Шульце — Гарди). При малом потенциале константа отталкивания пропорциональна его квадрату, и тогда, согласно формуле (3.7.16), критическая концентрация электролита увеличивается пропорционально четвертой степени потенциала поверхности: сс ~ Т*. Эта закономерность наблюдается и на практике. Обычно она формулируется в виде критерия Эйлерса — Корфа: / хс = const, который эквивалентен соотношению сс ~ при условии, что элек-трокинетический потенциал С, и потенциал поверхности % изменяются пропорционально при варьировании устойчивости раствора индифферентным или потен-циалопределяющим электролитом. Это вполне правдоподобно при малых величинах потенциала поверхности. В ряде случаев обнаруживается потеря устойчивости растворов при снижении электрокинетического потенциала до критической величины, равной примерно 25 мВ, что также косвенно подтверждает сильную зависимость критической концентрации электролита от потенциала поверхности. Характерное значение критической концентрации 1-1-валентного электролита равно примерно 0,1 моль/л.
Упомянутые выше эмпирические правила коагуляции являются результатом обобщения и усреднения данных, полученных разными авторами на различных коллоидных растворах. При этом и признаки начала коагуляции при увеличении концентрации электролита
630
Новый справочник химика и технолога
были разными у разных авторов. Коагуляция обычно проявляет себя в помутнении растворов, иногда в изменении окраски, а иногда только в появлении осадка. Учитывая это, можно считать, что теория ДЛФО в целом достаточно хорошо объясняет существующие закономерности коагуляции коллоидных растворов электролитами. Вместе с тем имеется множество фактов аномального (с точки зрения теории) поведения коллоидов при действии электролитов, как то: отсутствие корреляции устойчивости и элекгрокинетического потенциала, сохранение устойчивости при довольно высоких концентрациях электролита (до нескольких моль на литр), сильная задержка начала коагуляции после создания заведомо закритической концентрации коагулятора и другие. Обычно объяснение этих аномалий связывают с наличием сил отталкивания, не учтенных классической теорией устойчивости. К их числу относится легко прогнозируемая и управляемая сила отталкивания адсорбционных слоев ПАВ и полимеров. Часто ссылаются на появление аномальных свойств у поверхностных слоев воды или изменение ее структуры у поверхности частиц.
Далее будет показано, что некоторая модификация классической теории способна устранить многие несоответствия, не прибегая к помощи неэлектростатических сил отталкивания. А сейчас следует обратить внимание на то, что во многих случаях устойчивость определяется не величиной потенциального барьера, а величиной сил, действующих между частицами.
3.7.5. Силовая коагуляция и экстремумы функций взаимодействия частиц
Выше было дано обоснование того, почему тепловую коагуляцию следует анализировать с помощью потенциальных кривых. Оно базировалось на представлении о вероятностном механизме преодоления барьера, заимствованном из молекулярной физики.
В химии эквивалентом потенциального барьера является энергия активации того или иного процесса. Между тем перенос некоторых понятий молекулярнокинетической теории на коллоидные объекты не всегда корректен. Критерием корректности является соотношение между длиной элементарного теплового скачка молекулы (или частицы) и расстоянием, на которое необходимо переместиться молекуле (или частице) для преодоления барьера, т. е. шириной барьера. Для молекул средняя длина элементарного теплового скачка — это длина свободного пробега, а ширина барьера может быть принята равной нулю. Иначе говоря, молекуле всегда достаточно одного скачка для преодоления очень узкого барьера. С коллоидными частицами дело обстоит иначе: длина скачков частицы не больше длины свободного пробега молекул среды, а ширина барьера соизмерима с толщиной ДЭС, т. е. на порядки больше. Поэтому для прохождения барьера необходимы сотни и тысячи броуновских шагов, причем в нуж
ном направлении. Это означает, что встреча коллоидных частиц по характеру взаимодействия не является столкновением. Скорее она похожа на их медленное сближение под действием сил притяжения.
Количественно эти идеи воплощены в теории коагуляции аэрозолей, созданной Н. А. Фуксом и позднее перенесенной на коллоидные растворы. В ней сближение частиц описывается как результат их участия в двух видах движения: диффузии и движения в поле сил взаимного отталкивания (или притяжения). В первом приближении это уменьшает вероятность преодоления потенциального барьера в ка раз:
w = 2хаехр(-ДС/ / кТ).
В соответствии с этой формулой и приведенными выше качественными суждениями вероятность преодоления барьера растет при уменьшении толщины ДЭС (1/х). Влияние размера частиц а менее однозначно, поскольку он входит также и в величину барьера Д//. Очевидно, однако, что при Д U = 0 и типичном для суспензий значении ка » 1 формула дает значение w = 2ка, которое ни с каких точек зрения не может считаться разумным. Искать же причину этого несоответствия в рамках теории тепловой коагуляции не имеет смысла, поскольку это явление не характерно для грубодисперсных систем. В этом можно убедиться, анализируя поведение частиц при критической концентрации электролита.
При равенстве нулю потенциального барьера на силовой кривой сохраняется область преобладания сил отталкивания. Частицы при этом фиксируются в положении минимума потенциальной энергии, и по причине своей броуновской неподвижности вероятностный механизм преодоления барьера для них будет исключен. Коагуляция может быть вызвана действием внешних сил: давлением одного слоя частиц на другой в осадке и тому подобное. Условие коагуляции в этом случае вполне очевидно: сила сдавливания частиц должна превысить силовой барьер (максимум силовой кривой взаимодействия).
При любом механизме коагуляции (тепловом или силовом) ее возможность определяется величиной максимума на соответствующих кривых, а прочность сцепления частиц при безбарьерной коагуляции определяется глубиной минимума на тех же кривых. Эти параметры взаимодействия можно вычислить непосредственно, не прибегая к расчету и построению графиков потенциальной и силовой кривых. В точках экстремума функций производная равна нулю, поэтому, продифференцировав уравнения (3.7.8) и (3.7.10) и приравняв результаты к нулю, можем записать уравнения (3.7.13а):
хВ'ехр(-хА) = Ah / А2 или Ahl h = кйВ'ехр(-хА),
%2В'ехр(-хА) = 2 Ah / h3 или Ahl А2 = 0,5 хАх5'ехр(-хА).
Заменив с помощью этих равенств величины А^ hn Ah I h2 в формулах (3.7.8) и (3.7.10), получим выражения
Физика, химия и технология дисперсных систем
631
(3.7.8а) и (3.7.10а) экстремальных величин энергии Ue и силы Fc соответственно:
Ue = 5'ехр(-хЛе)(1 - xhe), (3.7.8а)
F* = 0,5 5'exp(~x/ze)(2 - %he). (3.7.10а)
Расстояние he в этих формула является координатой максимума или минимума, которые можно найти решением тех же равенств (3.7.13а) относительно величины хЛ. Для этого их удобно представить в виде уравнений:
х2ехр(-х) = (1 / еХх / х<) (3.7.17а)
и
х3ехр(-х) = (2/еХх / хс). (3.7.176)
Здесь х — положительное число, е — основание натуральных логарифмов и хс — критическое значение параметра Дебая. Оно возникает здесь при объединении всех констант, фигурирующих в равенствах (3.7.13а), в единую величину. Число х заменяет собой произведение хЛ. Точнее говоря, замена х = xhe приобретает смысл только после решения уравнения (3.7.17а) или (3.7.176) относительно х. Решение состоит в подборе величины х, при которой экспоненциально-степенная функция будет равна правой части того или иного уравнения. Обе функции имеют максимум (рис. 3.55), поэтому решения существуют при ограниченном сверху значении х = %h — меньшем 0,54 для энергии и меньшем 1,34 для силы.
2 -т Ч -г х е , х е
Рис. 3.55. Универсальные показательно-степенные функции у?ех для нахождения корней уравнения экстремумов энергии (сплошная кривая, п - 2) и силы (штриховая кривая, п = 3) взаимодействия частиц
Этих решений два: первое меньшее 2 и второе большее 2 для экстремумов потенциала, первое меньшее 3 и второе больше 3 для экстремумов силы. Первые при подстановке в формулы (3.7.8) и (3.7.10) дают значение максимума, а вторые — минимума потенциальной и силовой функций соответственно. Первые решения, лежащие в интервале от 1 до 2 для потенциала и в интервале от 2 до 3 для силы, дают отрицательное значе
ние энергии и силы в максимуме. Это значит, что кривые целиком лежат ниже оси абсцисс, хотя и имеют максимум.
Единственные решения xhe = 2 и xhe = 3 дают значения функций U’ и F' в точке их перегиба, a xhe- 1 и xhe = 2 для максимумов соответствуют нулевому значению энергетического и силового барьеров, т. е. условию быстрой тепловой коагуляции и самопроизвольной силовой коагуляции, для которой не требуется воздействие внешней силы для сближения частиц. Напомним, что формулы (3.7.8) и (3.7.10) дают экстремумы приведенных значений энергии Ue и силы Fe. Экстремумы функций Ue и Fe частиц определенного размера а получаются умножением величин Ue и Fe на да.
5.7.6. Эффекты смещения плоскости адсорбции ионов
В расчетах молекулярно-электростатического взаимодействия частиц (или плоских поверхностей) принято, что расстояние между противолежащими слоями потенциалопределяющих ионов, которое и определяет силу отталкивания двойных слоев, совпадает с расстоянием между фазовыми границами частиц. Между тем достаточно беглого взгляда на детальную (на молекулярном уровне) картину строения заряженной поверхности, чтобы убедиться в несовпадении плоскости адсорбции ионов с межфазной границей. Адсорбция потенциалопределяющих (ПО) ионов является, в сущности, процессом достройки кристаллической решетки адсорбента (ее анионной или катионной подрешетки при адсорбции анионов или катионов соответственно), поэтому плоскость локализации сорбированных ионов смещена относительно межфазной границы в глубь раствора на расстояние d/2 порядка половины периода решетки d, а возможно, и на большее. Незавершенность решетки и присутствие в ней преимущественно ионов одного знака (их электростатическое отталкивание) может быть причиной существенного ослабления связи ионов с поверхностью и их дополнительного смещения в сторону раствора. По причине рыхлости этот слой не может дать полноценного вклада в молекулярное притяжение частиц. В итоге молекулярное притяжение частиц (поверхностей) определяется расстоянием h между их фазовыми границами, а электростатическое отталкивание — расстоянием h-d между плоскостями адсорбции ионов, поскольку потенциал именно этих плоскостей входит в константу электростатического отталкивания. Разница этих расстояний d, как уже отмечалось, должна быть порядка периода решетки вещества, служащего дисперсной фазой. Таким образом, в дополнение к энергии специфической адсорбции ионов появляется еще один специфический для каждого вещества параметр — период решетки (или среднее межмолекулярное расстояние в веществе), определяющий структуру ДЭС и его эффективность как фактора устойчивости дисперсной системы. Период решетки прене
632
Новый справочник химика и технолога
брежимо мал в сравнении с толщиной двойного слоя, поэтому сделанное уточнение может показаться малозначительным. На самом деле оно радикально меняет всю картину воздействия электролитов на устойчивость дисперсной системы [12], что и будет продемонстрировано далее.
С учетом разницы в расстояниях между фазовыми границами и плоскостями адсорбции ПО ионов уравнение (3.7.18) описывает молекулярно-электростатическое взаимодействие частиц:
V = В'ехр[-х(й-б/)]-Л/ h. (3.7.18)
Введя новую константу отталкивания В" = В' exp (nd), ту же формулу можно представить в виде уравнения:
U' =B"exp(-xh)-Ah/h. (3.7.19)
Аналогичным образом или дифференцированием выражения (3.7.19) по h получается формула для приведенной силы взаимодействия частиц:
F' = хВ"ехр(-хЛ) - Ah / /г2. (3.7.20)
Одинаковая структура новых и прежних формул (3.7.8), (3.7.10) означает, что условие тепловой коагуляции и уравнения для нахождения экстремумов потенциальной и силовой функций также сохранят прежний вид. Однако условию коагуляции (3.7.15) полезно придать более общий вид:
*>B"leAh. (3.7.21)
Здесь х не является критическим значением параметра Дебая. Условие (3.7.21) означает, что коагуляция будет происходить при концентрации большей, чем ее критическая величина, связанная обычным соотношением с критическим значением параметра Дебая х*. Его можно найти, заменив равенством знак неравенства. В любом случае условие коагуляции является не формулой, а уравнением, которое следует решить относительно параметра Дебая, поскольку последний входит и в константу отталкивания В” = В'ехр(х</)- Выразив ее через классическую константу отталкивания, можно получить соотношение:
х > ехр(хб/)Я' / eAh. (3.7.21а)
После введения величины х = х<7 и имея в виду, что B4eAh = хс — эго значение критического параметра Дебая при d = 0, соотношение (3.7.21а) преобразуется к универсальному и удобному для решения виду
х ехр(-х) = хсd. (3.7.22)
Это уравнение похоже на те, которые использовались ранее при исследовании функций взаимодействия на экстремумы, и решается тем же способом. Правая часть уравнения содержит только известные величины, и задача сводится к подбору величины х, при которой будет удовлетворяться условие неравенства или равен
ства при нахождении критического значения величины х. Это значение является решением уравнения (3.7.22) и будет далее обозначаться символом х*. Тогда х* = х*/ d.
Легко убедиться, что левая часть уравнения (3.7.22) имеет максимум, равный 1/е при х = 1. Это означает, что решения существуют только в том случае, когда значение правой части не превышает величину 1/е, т. е. при условии 1/е > %cd.
Иначе говоря, если смещение плоскости адсорбции ПО ионов превысит некоторую величину, то условие коагуляции не может быть выполнено ни при каких значениях параметра Дебая, т. е. необратимая коагуляция не может быть вызвана увеличением концентрации электролита. Некоторые дисперсные системы ведут себя именно таким образом [34]. Иногда это трактуется как несоответствие теории ДЛФО, хотя на самом деле является ее следствием, а также определением понятия «лиофильная дисперсная система» в рамках теории ДЛФО. Разумеется, что при d = 0 условие необратимой коагуляции всегда может быть выполнено, однако уравнение (3.7.22) в силу чисто математических причин становится непригодным для нахождения критического значения параметра Дебая в этом случае. Оно же в виде выражения (3.7.21а) продолжает выполняться и сводится к тривиальной формуле (3.7.15).
Итак, условие невозможности вызвать коагуляцию дисперсной системы введением электролита формулируется в виде соотношения:
d>Ahlff. (3.7.23)
Если немного изменить форму его записи:
B'>Ahld, (3.7.24)
то становится ясным физический смысл этого условия: коагуляция становится невозможной из-за того, что на расстоянии равном величине d энергия отталкивания частиц превышает энергию их притяжения при любой толщине двойного слоя. Действительно, из формулы (3.7.18) непосредственно следует, что независимо от х при h = dU = В' -Ahl d,a неравенство (3.7.24) означает, что U при этом положительно, т. е. преобладает отталкивание. При большой концентрации электролита на потенциальной кривой в этом месте имеется острый максимум, и он тем острее, чем выше концентрация электролита, а его высота равна В' при любой концентрации электролита (рис. 3.56).
Экстремальные точки потенциальной или силовой кривой взаимодействия частиц при некотором заданном значении параметра Дебая (концентрации электролита) можно искать, не прибегая к предварительному вычислению его критического значения. Независимой переменной при этом будет только межчастичное расстояние. Как уже отмечалось, структура формул (3.7.25)-(3.7.28) для нахождения экстремумов энергии Ue и силы Fe при смещении плоскости адсорбции ПО останется прежней:
Физика, химия и технология дисперсных систем
633
Ue = В" ехр (-*Ле)(1 - %he), (3.7.25)
х2 ехр (-х) = кАИ / В”, (3.7.26)
Fe = 0,5 кВ” ехр (-%М(2 - хЛД (3.7.27) х3 ехр (-х) = 2кАИ / В”. (3.7.28)
Здесь, в отличие от уравнения (3.7.22), х = %h, а корнями уравнений (3.7.26) и (3.7.28) являются величины х* = кИе. Если предварительно вычислено критическое значение параметра к*, то в правых частях этих уравнений может быть произведена эквивалентная замена:
кАи / В” = е~{(к / к*), 2кАн / В” = 2е~1(к / к*).
После нее они примут форму уравнений (3.7.17а) и (3.7.176) для экстремумов классических кривых ДЛФО. Разумеется, что, как и прежде, приведенные значения экстремальных точек Ue и Fe следует при необходимости пересчитать к значениям энергии Ue = па Ue и силы Fe = naFe для частиц конкретного размера а.
В завершение следует рассмотреть условия силовой коагуляции частиц, о которой было упомянуто выше. При положительном значении максимума силы взаимодействия F^ коагуляция произойдет, если внешняя сила, вынуждающая частицы сближаться, превысит максимум отталкивания. При F^ = 0 коагуляция происходит при любом соприкосновении частиц. Это могут быть их броуновские столкновения, а чаще всего это происходит в осадках на дне сосуда или на поверхности электрода в процессе электрофоретического осаждения частиц на электроде.
Согласно уравнению (3.7.27), максимум силы отталкивания равен нулю при кИе = 2 или при he = 2/к. При подстановке этих значений в формулу (3.7.27) можно сформулировать условие силовой коагуляции:
кВ” / е2 = K2Ah / 4
или
Рис. 3.56. Потенциальные кривые парного взаимодействия частей при постоянстве потенциала и концентрации электролита при различной величине сдвига (нм) в жидкую фазу плоскости локализации заряда поверхности: а — 0,Ь — 0,2; с — 0,4
Если выразить В” через классическую константу отталкивания В’, как и в случае тепловой коагуляции, получается уравнение:
х ехр (-х) = 4B'd /e2Ah. (3.7.30)
В нем, как и ранее, х = Kd, а критическое значение параметра Дебая находится из первого корня этого уравнения х*: х* = x*/d. Однако, и не прибегая к этой процедуре, можно исследовать полученный результат. Максимум левой части уравнения (3.7.30) равен, как уже отмечалось, 1/е. Тогда самопроизвольная силовая коагуляция (т. е. без воздействия внешней силы, превышающей силовой барьер) становится невозможной при условии:
d>eAh/4B' (3.7.31)
или
В'> (е/4) Ah / d. (3.7.32)
Из сравнения формул (3.7.23) и (3.7.31) следует, что для предотвращения возможности самопроизвольной необратимой силовой коагуляции плоскость адсорбции ПО ионов должна быть отодвинута от межфазной границы на расстояние, меньшее в е/4 раз, чем при тепловой коагуляции. Под самопроизвольной силовой коагуляцией имеется в виду коагуляция, не требующая воздействия на частицы внешней силы, заставляющей их сближаться. На самом деле, если есть сближение частиц, то есть и ответственная за него сила. В частности, образование осадков подразумевает, что это сила их тяжести Fg = mg, где m = ApV — масса частицы и g — ускорение силы тяжести, причем масса учитывает поправку на закон Архимеда путем умножения объема частицы V на разность плотностей Др дисперсной фазы и среды. Точно так же не бывает тепловых столкновений с нулевой кинетической энергией, что предполагает классическое условие быстрой необратимой тепловой коагуляции. Следует отметить, что величины Д(/ и Un^ эквивалентны.
Описание коагуляции как одного из важнейших процессов, протекающих в дисперсных системах, не сводится к выяснению условий, при которых частицы будут или не будут слипаться, и с какой силой они сцепляются при слипании. Теория ДЛФО и приведенные выше дополнения к этой теории отвечают только на вопрос о результате столкновения (о сближении) двух частиц, однако в реальности явление коагуляции не заканчивается попарным слипанием частиц.
Коагуляция ведет к образованию флокул — объектов из большого числа слипшихся воедино частиц. Флокулы, так же как и частицы, участвуют в тепловом движении и оседании в поле силы тяжести, они также сталкиваются между собой и участвуют в коагуляции, образуя все более сложные и крупные флокулы.
Процесс этот в принципе не ограничен во времени. Очевидно, что сколь угодно полное и точное описание элементарного акта коагуляции — слипания двух час
634
Новый справочник химика и технолога
тиц — не способно выразить те изменения состояния системы, которые могут произойти в ней за достаточно большое время. Для этого необходимо знание структуры флокул и всей дисперсной системы (подраздел 3.13).
3.7.7. Устойчивость многокомпонентных суспензий
В технологической практике часто приходится иметь дело с суспензиями, дисперсная фаза которых состоит из частиц различной химической природы, например из смеси частиц песка и глины. Подобные дисперсные системы далее будут характеризоваться как системы с многокомпонентной дисперсной фазой. С точки зрения устойчивости наиболее существенным их отличием от однокомпонентных суспензий является то, что частицы разной химической природы в общей дисперсионной среде обычно имеют различный по величине или даже по знаку электрический потенциал поверхности. Достаточно очевидно, что наличие у частиц противоположного по знаку заряда ведет к тому, что электрические силы их взаимодействия уже не будут препятствовать коагуляции суспензии, наоборот — они будут способствовать ей. Неизбежность коагуляции не означает, что вмешательство в ход этого процесса бесполезно и регулирование его в желательном направлении невозможно. Детально вопросы эволюции коагулирующих взвесей и средства ее регулирования рассматриваются в подразделах 3.13-3.15. Здесь же обсуждаются важные особенности многокомпонентных систем, в том числе способные повлиять на ход процесса коагуляции и конечное состояние взвеси. Коагуляция, при которой происходит слипание частиц различной химической природы, получила название «гетерокоагуляция», а взаимодействие частиц разной природы далее будет обозначаться как «гетеровзаимодействие».
Прежде всего, следует иметь в виду, что электростатическое взаимодействие частиц гарантированно сводится к отталкиванию только при равном потенциале поверхности двух взаимодействующих частиц. В случае одноименного по знаку, но разного по величине потенциала электростатическое взаимодействие неоднозначно — на больших (по сравнению с толщиной двойного слоя) расстояниях оно положительно (частицы отталкиваются), а на малых расстояниях знак сил меняется, т. е. одноименно заряженные частицы притягиваются. Физическая природа обращения знака электростатического взаимодействия одноименно заряженных частиц проста: она та же, что и природа притяжения легких предметов «наэлектризованным» телом — это индукционное взаимодействие сильно заряженного и незаряженного тел. Достаточно очевидно, что если «незаряженное» тело на самом деле окажется слабо заряженным одноименно с заряженным телом, то это не устранит индукционное взаимодействие. Нечто подобное происходит и при взаимодействии двойных электрических слоев. Полное количественное описание этого взаимодействия приводится в книге Б. В. Дерягина с соавторами [21]. По ряду причин здесь не приводится
полное описание электростатического гетеровзаимодействия частиц, а даны лишь отдельные выдержки из упомянутой книги, иллюстрирующие принципиальные особенности этого взаимодействия. Для технологии интерес представляют поздние стадии коагуляции, когда из первичных частиц образуются крупные составные частицы — флокулы коагулята. Однако на этих стадиях электрическое состояние составных гетерогенных частиц является неопределенным и непредсказуемым, поэтому теория гетеровзаимодействий не может быть использована. Фактически только этап образования двойников из первичных частиц разной химической природы контролируется теми значениями заряда и потенциала, которые приписываются первичным частицам. Но он, как отмечалось, не представляет значительного технологического интереса.
Принципиальные вопросы взаимодействия неодинаково заряженных частиц целесообразно рассматривать в варианте взаимодействия плоских поверхностей. В качестве меры интенсивности взаимодействия принята величина расклинивающего давления П, т. е. удельной (на единицу площади) силы взаимодействия. Ранее уже отмечалось, что во многих случаях силовая функция взаимодействия частиц предпочтительней, чем потенциальная функция. В связи с этим рассмотрение особенностей взаимодействия разнозарядных поверхностей целесообразно начать с формулы (3.7.33) для расклинивающего давления одинаково заряженных поверхностей, которая получается дифференцированием выведенной ранее формулы (3.6.14) для энергии взаимодействия слабо и одинаково заряженных поверхностей:
П = ££0(хЧ/5)2 / [сЬ(хй) + 1]. (3.7.33)
Формула (3.7.34) описывает расклинивающее давление поверхностей с разными величинами потенциалов поверхности Ч*, и ЧЛ:
П = ЕЕо^рТ^гС^хй) - (Ч^2 + Т22)] / sh2(xA). (3.7.34)
При разноименных зарядах поверхностей эта формула всегда соответствует их притяжению. При одноименных, но разных по величине зарядах формула (3.7.34) описывает притяжение на расстояниях меньших величины /?о, определяемой из условия:
сЬ(хй0) = (Ч/12 + Ч/22)/2Ч/1Ч/2
или
хй0 = 1п(Ч/2/Ч#1) при Ч/2 / 4х! > 1. (3.7.35)
На больших расстояниях наблюдается отталкивание частиц, которое максимально при условии:
П^-МхЧ'О2. (3.7.36)
При этом оно определяется только величиной меньшего из двух потенциалов (Ч^), а расстояние hm&x,
Физика, химия и технология дисперсных систем
635
на котором достигается максимальная величина отталкивания, — уравнением:
сЬ(хЛ1Пах) = 'Р2/%. (3.7.37)
Легко также убедиться, что при одинаковых по величине потенциалах эта формула сводится к формуле (3.7.33).
При произвольном потенциале поверхности (в том числе большом) в растворе симметричного электролита с концентрацией с и формальным зарядом ионов z справедливо уравнение:
Птах = bcRT&XzFVi / 2RT). (3.7.38)
Наиболее важные особенности поведения многокомпонентных суспензий обусловлены тем, что они сильно зависят от соотношения концентраций компонентов дисперсной фазы и от соотношения размеров частиц разной химической природы. Влияние электролитов на состояние гетерогенных взвесей также сильно отличается от их влияния на обычные — однофазные дисперсные системы.
Увеличение концентрации электролита, как и в случае одинаково заряженных частиц, ослабляет их электростатическое взаимодействие, но в случае разноименно заряженных частиц это ведет к повышению устойчивости дисперсной системы. В принципе этим путем может быть достигнуто состояние полной устойчивости смеси против коагуляции благодаря тому, что в случае частиц различной химической природы возможно обращение знака их молекулярного взаимодействия. Такая возможность представляется, если энергия взаимодействия частиц со средой больше, чем энергия молекулярного взаимодействия разных по природе веществ многокомпонентной системы. Разумеется, что при этом концентрация электролита должна быть не настолько высокой, чтобы вызвать обычную (гомогенную) коагуляцию одного из компонентов системы. Одновременное выполнение всех этих требований, скорее всего, достижимо только в исключительных случаях и поэтому указанный путь регулирования свойств многокомпонентных смесей не представляет большого интереса. Более обещающим является путь, связанный с влиянием соотношения компонентов, имеющих разный по знаку потенциал, на коагуляцию смеси [11].
С точки зрения устойчивости смеси определяющим является не столько соотношение концентраций компонентов, сколько соотношение их поверхностных зарядов. Заряд компонента Qt пропорционален его поверхности т^:
Qi = miAj Ci'Pi,
где m, — массовая концентрация компонента, Я, — его удельная поверхность и С, — электрическая емкость двойного слоя на поверхности частиц z-ro вида. Наименее благоприятна для устойчивости взвеси ситуация,
когда концентрации, дисперсности и емкости компонентов близки по величине. В этом случае на каждую частицу одного знака приходится одна частица противоположного и равного по величине знака. Попарное слипание частиц противоположного знака приведет к образованию в целом электронейтральных двойников и к дальнейшему развитию процесса коагуляции двойников в лучшем случае по сценарию коагуляции нейтральных частиц. Этот «лучший случай» предполагает, что в двойнике происходит взаимная нейтрализация зарядов частиц, что весьма сомнительно. Более реалистично предполагать, что слипшиеся частицы полностью сохранят свое исходное зарядовое состояние. Следовательно, образуют электрический диполь, момент которого ц = 4тш3СЧ/ определяется параметрами частицы с меньшей величиной заряда Q = ^па^СЧ'. Предположение основано на том, что потенциал поверхности компонента в некоторой среде не зависит от наличия в ней других компонентов и в известной мере — от их взаимного расположения (наличия контактов между частицами). Некоторые изменения электрического состояния частиц, безусловно, происходят в зоне их контакта, но влиянием этого фактора в первом приближении можно пренебрегать.
Образование гигантских (по молекулярным меркам) диполей существенно усилит коагуляцию за счет ди-поль-дипольного притяжения двойников.
Ситуация наиболее благоприятна для получения устойчивой к коагуляции смеси, когда дисперсность и заряд одного из компонентов существенно больше, чем другого. Иначе говоря, один из компонентов представлен крупными частицами, а другой — частицами малых размеров, и их концентрация такова, что суммарный заряд мелких частиц больше заряда грубодисперсного компонента. Такое соотношение может быть легко создано даже при малой плотности поверхностного заряда у мелких частиц. В этом случае мелкие частицы налипают на поверхность крупных сплошным слоем, создавая толстый защитный слой, препятствующий дальнейшему прилипанию мелких частиц и слипанию крупных. Таким образом, процесс коагуляции прекращается (эффект гетеростабилизации). Механизм гетеростабилизации многокомпонентных систем достаточно универсален и не обязательно связан с различием электрических свойств частиц. Стабилизация взвеси крупных частиц мелкими характерна для эмульсий (стабилизация эмульсий порошками), смесей магнитных дисперсных материалов. Общеизвестно стабилизирующее действие мицеллярных растворов ПАВ (защитных коллоидов), а также полимеров, которое тоже может быть интерпретировано описанным выше способом.
Коагуляция, если она не прерывается за счет гетеростабилизации, не ограничивается образованием двойников, тройников и т. д. Процесс укрупнения частиц в определенном смысле ничем не ограничен и ведет к образованию очень крупных, видимых невооруженным глазом хлопьев (флокул). Практический интерес пред
636
Новый справочник химика и технолога
ставляет эта поздняя стадия коагуляции, поскольку именно она определяет конечную структуру взвеси и ее технологические свойства. Рассмотренные выше примеры показали, что в зависимости от рецептурных и геометрических характеристик смеси после начального этапа коагуляции может наступить и ускорение этого процесса (при образовании двойников-диполей), и его полное торможение (гетеростабилизация), и, очевидно, все промежуточные варианты развития процесса. По этой причине расчет взаимодействия составных частиц сохраняет актуальность. Однако, как уже отмечалось, этот расчет невозможен, поскольку неизвестно, какой потенциал «поверхности» следует приписать флокуле даже в том случае, когда точно известно число и характер взаимного расположения первичных частиц разной химической природы в такой флокуле. Вместе с тем можно рассчитывать, что если процесс гетерокоагуляции запущен, то он, за исключением случаев гетеростабилизации, не прервется и на поздних этапах коагуляции. Ход этого процесса, в сущности, не зависит от количественных характеристик взаимодействия фло-кул. Важно лишь преобладание сил притяжения фло-кул, и поэтому он мало чем будет отличаться от коагуляции однокомпонентных суспензий (см. подразделы 3.13 — 3.15).
3.8. Молекулярно-кинетические явления
3.8.1. Регулярное и хаотичное движение частиц
Существуют два разных типа движения частиц и молекул — регулярное и хаотичное. Первое возникает при действии на частицы некоторой внешней силы. Чаще всего это сила тяжести. Под влиянием этой силы возникает оседание частиц. Регулярное движение может быть также вызвано действием центробежной силы, действием электрического поля на заряженные частицы, действием переменного ускорения при вибрировании сосуда с дисперсной системой и силами другой природы. Важно, что величина и направление перемещения частиц в каждый последующий момент времени предсказуемы с любой желаемой степенью детализации. Основной характеристикой регулярного движения частиц является их скорость.
Если интересоваться результатом достаточно длительного воздействия силы F на частицу, то можно пренебречь ее инерционностью и ограничиться уравнением стационарного движения, которое сводится к равенству действующей силы силе трения. При малых скоростях и последняя пропорциональна скорости движения частицы и ее можно описать уравнением:
F=bu. (3.8.1)
Здесь Ь — коэффициент сопротивления движению частицы в вязкой среде. В случае частиц сферической формы и умеренной величины отношения сил инерции вовлекаемой в движение среды к силам трения (числа Рейнольдса) этот коэффициент вычисляется по форму
ле Стокса, в которой ц — вязкость среды и а — радиус частицы:
6 = 6тст]а. (3.8.2)
Наиболее типично движение под действием силы тяжести Fg = FiApg. Здесь V\ = 4тса3/3 — объем частицы, Др = pv - р/— разность плотностей дисперсной фазы и среды соответственно и g — ускорение свободного падения. Подстановка этих соотношений в уравнение (3.8.1) и его решение относительно скорости движения приводит к формуле:
и = 2Apga2 / 9г\. (3.8.3)
По формуле (3.8.4) можно оценить время оседания взвеси ts как время, необходимое для прохождения частицами пути, равного высоте сосуда Н:
ts-H/u. (3.8.4)
Формула (3.8.5) позволяет вычислить размер частиц по времени их осаждения, что используется в седиментационном анализе гранулометрического состава поли-дисперсных взвесей:
a = (9r|/2ApgX/7//,)12. (3.8.5)
Хаотичное движение частицы, в отличие от регулярного, непредсказуемо ни по направлению, ни по величине смещения частицы в любой конкретный момент времени. Это движение имеет тепловую природу и называется броуновским. Оно универсально, неуничтожимо и не зависит от химической природы частиц. Броуновское движение коллоидных частиц можно видеть в микроскоп. Оно выглядит как беспорядочное прерывистое скачкообразное перемещение частиц на небольшие расстояния или как их дрожание. Именно так оно и было открыто.
Непредсказуемость элементарных актов теплового смещения частицы (ее броуновских скачков) приводит на больших интервалах времени или для большого числа частиц к вполне предсказуемым результатам, благодаря усреднению последствий множества элементарных актов движения. Интенсивность теплового движения частиц, молекул, атомов, в сущности, определяет скорость всех физико-химических процессов и саму форму существования материального мира. Тепловое движение — это то, благодаря чему всегда что-то происходит в этом мире, в том числе и то, что нам нужно. Было бы большим самодовольством считать, что это вы, накачивая насосом автомобильное колесо, заталкиваете воздух в камеру. На самом деле воздух туда перетекает сам в результате теплового движения молекул, а вы, давя на насос, всего лишь создаете предпочтительное направление для теплового движения молекул. Причем ваше влияние на движение молекул ничтожно мало — из миллионов тепловых скачков благодаря вашим усилиям только на один-два скачка увеличивается их число в нужном направлении.
Физика, химия и технология дисперсных систем
637
Интенсивность теплового движения характеризуется его средней кинетической энергией <ти2/2>. Угловые скобки означают взятие средней величины (усреднение) от множества значений выражения, заключенного в скобки. Замечательной особенностью этой меры интенсивности хаотичного движения является то, что она не зависит от природы и размера частиц и равна ее температуре, выраженной в энергетических единицах кТ, где к — постоянная Больцмана и Г — температура, К. Эта же величина, но отнесенная не к одной частице (молекуле, иону), а к одному молю вещества, т. е. величина RT=NAkT, уже фигурировала в теории двойного слоя и в теории устойчивости коллоидов. Здесь NA — постоянная Авогадро, которая, кстати, впервые была вычислена по результатам наблюдения за тепловым движением коллоидных частиц.
Строго говоря, средняя кинетическая энергия трехмерного теплового движения частицы равна ЗкТ / 2 или кТ!2 на каждую из трех составляющих этого движения (вдоль трех осей координат).
Другая мера интенсивности теплового движения — это среднеквадратичное смещение г = (<52>)1/2 частиц (молекул, ионов) за некоторое произвольное время t. Его можно представить как векторную сумму последовательности разных по величине и направлению смещений 5, за равные интервалы времени dt, т. е. как расстояние между началом и концом пути, проделанного за п шагов произвольной длины s, в произвольном направлении. При этом каждый шаг делается из точки, в которой частица оказалась на предыдущем шаге (рис. 3.57).
Для выяснения закона броуновских блужданий частицы сначала необходимо рассмотреть результаты случайных перемещений за один шаг. Пусть за один шаг частица проходит путь хг = s. Если бы ее можно было вернуть в исходное положение и снова зарегистрировать длину и направление одного шага, то получилась бы какая-то другая величина. Неоднократное повторение этой воображаемой попытки ведет к тому, что среднее смещение частицы (Es,) / m по всем m воображаемым попыткам окажется равным нулю в силу случайности направлений и длин отдельных смещений. В отличие от этого среднее значение квадрата смещения будет отлично от нуля, так как квадраты смещений имеют положительный знак и, следовательно, взаимно не компенсируются при вычислении среднего.
В статистической теории фундаментальное значение имеет тот факт, что частицы лишены памяти о прошлом. Иначе говоря, поведение частицы (направление и длина ее теплового скачка) на очередном отрезке времени не зависит от того, что предшествовало этому моменту времени. Отсюда следует, что вместо воображаемых опытов с возвратом частицы в исходное положение можно с тем же результатом рассматривать реальные перемещения одной частицы каждый раз из любого нового положения или одновременное перемещение m разных идентичных частиц за один и тот же
промежуток времени. Это положение строго не доказуемо и поэтому в теории фигурирует как эргодическая гипотеза. Таким образом, мерой длины элементарных тепловых скачков может служить среднее за m реальных наблюдений значение <s2> = (S s2)lm квадрата этих скачков или среднеквадратичная величина смещения \Г = (<у,2>)1/2 за один шаг.
Второй шаг длиной s„ сделанный из новой позиции в произвольном направлении, переместит частицу в положение, задаваемое векторной суммой первого шага длиной ]Г и второго шага длиной st. Квадрат этого конкретного смещения относительно первоначального положения будет:
2r2 = ir2 + 52 - 2(xrSj) cosa,
где a — угол между направлениями первого и второго шага. Так как направление скачка (угол а) с равной вероятностью может иметь два любых противоположных направления, то средняя величина 2(ir.s;)cosa равна нулю, и тогда:
<2Г> = / + <s2>. (3.8.6)
В сущности, в формуле (3.8.6) величина ir является произвольной и может представлять собой некоторое реальное перемещение частицы за произвольное число скачков. Иначе говоря, она выражает смещение частицы „г за произвольное число п скачков через величину ее смещения п-\г на предыдущем, п-1 шаге. В свою очередь, смещение „_]Г аналогичным образом можно выразить через величину „_2г и так далее — до первого скачка включительно. В итоге получается уравнение:
У= s2 +52+532+...+52. (3.8.6а)
Реально под числом шагов п подразумевается число отрезков времени фиксированной длины dt, так что У — это средний квадрат смещения частицы за время t = ndt. Далее в обозначении этой величины предваряющий индекс п будет опускаться.
Выражение (3.8.6а) модернизируется в уравнение (3.8.7) путем введения средней из п опытов величины 2 квадрата смещения частицы 5 за интервал времени, равный единице {dt = 1). Тогда г2 = ns2 и п = /.
г2 = s2t. (3.8.7)
Рис. 3.57. Броуновское смещение „г частицы как результат последовательности п более коротких хаотичных смещений s,
638
Новый справочник химика и технолога
Подразумевается, что при достаточно большом числе опытов п величина s2 уже не будет зависеть от их числа (времени наблюдения t) и, следовательно, получит статус новой объективной характеристики интенсивности теплового движения частиц.
Формула (3.8.7) определяет статистический смысл величины s2 (выражает ее через две другие, доступные прямому измерению величины). Задача заключается в нахождении связи вновь введенной характеристики теплового движения со свойствами частиц и среды. Следует предварительно отметить, что аналогичная формула (3.8.8) связывает между собой х, у и z — компоненты трехмерного движения, в частности смещение вдоль оси х:
x2 = sx2t. (3.8.8)
Это следует из того, что г2 = х2 + у2 + Z а в однородном изотропном пространстве x2 = y2 = z2, так что г2 = 3х2. Аналогичные соотношения действуют между 2 смещением 5 за единицу времени и его составляющими вдоль координатных осей: s2 = s2+s2+s2, так что s2 = 3s2x, что и ведет к формуле (3.8.8).
Уравнение (3.8.9) описывет движение частицы под влиянием полученного импульса силы (толчка) в направлении оси х:
m(d2x/ dt2) + b (dx / dt) = 0. (3.8.9)
Оно означает, что движение постепенно замедляется под действием силы трения вязкой среды. Величина полученного импульса скрыта в начальной скорости частицы {dx / dt)t = 0.
Первое слагаемое уравнения (3.8.9) — это сила инерции, действующей на частицу массой т, второе — сила трения о среду (Ь — коэффициент трения частицы). Движение только по инерции происходит при равенстве этих сил, т. е. при равенстве нулю правой части уравнения.
Из формулы (3.8.8) следует, что s2 = dx2 I dt. Для введения величины dx2 I dt в уравнение движения (3.8.9) умножим его на х:
тх (d2x / dt2) + bx (dx I dt) = O
и преобразуем в уравнение (3.8.10), введя х под знак дифференциала второго слагаемого:
тх (d2x / dt2) + (b/2) (dx2 /dt) = O (3.8.10)
Фигурирующая здесь величина dx2 I dt, является средней с точки зрения молекулярно-кинетических представлений, т. е. dx2 / dt= <dx2\dt> = s2.
Запишем тождество d(xdx / dt)/ dt=(dx/ dt)2 + x (cfx I dt2), полученное по правилам дифференцирования произведения, и заменим его левую часть эквивалентным выражением 0,5d(dx2 / dt) dt. Здесь, согласно данному выше определению, d(dx2 / dt)di = ds21 din, следовательно,
d(dx2 I dt)dt = 0, так как величина s2 является константой, характеризующей интенсивность теплового движения частиц, не зависящей от времени. Таким образом, из того же тождества следует, что x(d2x / dt2) = -(dx I dtf, где (dx I dt)2 является средней с молекулярно-кинетической точки зрения величиной, т. е. x(d2x / dt2) = - <(dx I dt)2>. При подстановке этого и полученного выше выражения dx2 / dt= s2 в уравнение (3.8.10) получается соотношение:
m<(dx / dt)2> = (b/2) s2x . (3.8.11)
Величина m<(dx / dt)2> — это удвоенная средняя кинетическая энергия теплового движения вдоль одной из координатных осей, равная кТ. Тогда уравнение (3.8.11) можно заменить уравнением, в котором кТ выражено через средний квадрат смещения за единицу времени:
kT=(b/2)s2. (3.8.12)
Отсюда s2 = 2кТ/ Ьи, согласно уравнению (3.8.8),
x2 = 2Dt. (3.8.13)
Величина
D = kT/b (3.8.14)
называется коэффициентом диффузии частиц, выражение (3.8.14) — формулой Эйнштейна, а выражение (3.8.13) — формулой Смолуховского.
Статистический смысл величины D определен выражением (3.8.13), а более наглядный, макроскопический смысл — уравнением диффузии, которое описывает макроскопические последствия теплового движения частиц (молекул, ионов).
3.8.2. Диффузия
Диффузия компонента — это его перенос в направлении выравнивания концентрации в результате теплового движения частиц (молекул) данного компонента системы. В общем случае диффузия направлена на выравнивание химических потенциалов компонентов системы, которые кроме концентрационного слагаемого включают потенциальную энергию во внешнем поле, в частности в поле гравитационных или центробежных сил. Их роль рассматривается ниже в подразделе 3.8.3. Кроме того, концентрационное слагаемое химического потенциала известным образом зависит от температуры. Здесь и далее рассматривается только изотермическое распределение компонентов дисперсных систем.
Диффузия является макроскопическим проявлением хаотичного движения частиц, и поэтому ее законы можно получить из законов теплового движения. Пусть концентрация с некоторого компонента возрастает в направлении оси х. Рассмотрим два соседних слоя раствора равной толщины dx, расположенных по обе стороны от некоторой плоскости, нормальной к оси х.
Физика, химия и технология дисперсных систем
639
В первом слое концентрация равна с, а во втором с + de. Количество вещества в каждом из них (на единицу площади нормального к оси х сечения) равно cdx и (с + dc)dx соответственно. Согласно уравнению (3.8.13), по истечении времени dt = (dx)2 / 2D, необходимого для перемещения частиц на расстояние dx, все частицы каждого слоя полностью покинут свой слой, поскольку тепловое смещение частиц (молекул) достигнет за это время границ слоя. В силу равной вероятности теплового смещения частиц компонента в прямом и встречном направлениях через плоскость, разделяющую соседние слои, пройдет половина содержимого каждого слоя во встречных направлениях. В итоге результирующий перенос dqx компонента через плоскость вдоль оси х за время dt будет равен разнице половин содержимого соседних слоев: 0,5сб& - 0,5(с + dc)dx или dqx - -0,5dcdx.
Поток компонента qx = dqx I dt — количество вещества, проходящего за единицу времени через единичное сечение — описывается выражением Q,5dcdx / dt, где dt = (dx)2 / 2D, или уравнением:
qx = -D(dc/dx). (3.8.15)
Это соотношение известно как первый закон Фика. Он и определяет макроскопический смысл коэффициента диффузии как потока компонента при единичной величине перепада концентраций на единице длины. Знак «-» указывает на то, что поток компонента направлен в обратном направлении по отношению к направлению увеличения концентрации, т. е. направлен на ее выравнивание.
Если концентрация меняется по всем трем направлениям пространства х, у и z, то ее локальная неоднородность характеризуется градиентом концентрации:
grade = de I dx + de I dy + de I dz.
Градиент — это вектор, и поток компонента будет направлен антипараллельно направлению градиента, т. е. навстречу направлению нарастания концентрации в соответствии с формулой:
dq - -D grade. (3.8.16)
Однако систему координат можно ориентировать так, что ось х совпадет с направлением градиента, и тогда диффузия снова может быть описана одномерным уравнением (3.8.15). Перенос компонента dQ через поверхность с некоторой площадью dA за время dt в dAdt раз отличается от потока через поверхность с единичной площадью и описывается уравнением:
dQ = -Dgy&3cdtdA. (3.8.17)
3.8.3. Равновесное распределение дисперсной фазы
Уникальность коллоидных растворов заключается, в частности, в том, что коллоидные частицы в равной мере подвержены как тепловому, так и регулярному движению, например оседанию в поле силы тяжести
(седиментации). В системах молекулярного уровня дисперсности (газы, истинные растворы) можно пренебрегать действием гравитации. В грубодисперсных взвесях можно полностью пренебречь диффузией частиц. И только в коллоидных растворах нельзя пренебрегать ни тем, ни другим.
Рассмотрим потоки частиц через единичную горизонтальную площадку в вертикально установленном сосуде высотой Н. Свойства находящейся в нем взвеси характеризуются средней по всему сосуду (рецептурной) объемной долей ф дисперсной фазы и массой т одной частицы. Ее стоксова скорость и = mg / bn коэффициент диффузии D = kT / b выражаются при этом через массу частицы, вязкость среды и плотности фаз. Поток частиц dq (3.8.18) через единичное сечение на любой высоте h представляет собой сумму седиментационного (ри и диффузионного Ddy / dh потоков:
dq = (?и + Dd<? I dh. (3.8.18)
Если первоначально взвесь была однородна по составу (подверглась интенсивному перемешиванию), то в ней б/ф / dh = 0. В такой взвеси происходит только седиментация (оседание частиц на дно сосуда). Благодаря оседанию возникает и усиливается перепад концентраций по высоте t/ф / dh и, соответственно этому, увеличивается встречный поток диффундирующих частиц. Поскольку при оседании верхние слои взвеси обедняются частицами, то со временем седиментационный поток ф« ослабляется. По той же причине растет диффузионный поток, так что через некоторое время эти встречные потоки обязательно сравняются, причем на любом расстоянии h от дна сосуда. Иначе говоря, наступит седиментационно-диффузионное равновесие и установится равновесное распределение дисперсной фазы по высоте. При этом, согласно условию равновесия, dq - 0 и фм + Ddy / dh = 0. После разделения переменных и интегрирования этого уравнения в пределах от h = 0 до произвольной высоты h получается соотношение ф = фоехр(-и / D). При подстановке в него и - mg / bn D = kT I b получается уравнение:
ф = Фо exp(-mgh / кТ). (3.8.19)
Здесь фо — концентрация дисперсной фазы на дне сосуда, где она имеет наибольшее при данных условиях значение и экспоненциально уменьшается с увеличением расстояния h от дна.
Произведение mgh есть не что иное, как потенциальная энергия частиц Еп в поле силы тяжести, поэтому закону распределения (3.8.19) можно придать более общий вид ф = фоехр(-£п / кТ). Очевидно, что он является частным проявлением закона распределения частиц по потенциальной энергии, т. е. закона Больцмана, который уже неоднократно использовался выше. В коллоидной химии он известен под названием закона Лапласа — Перрена. Перрен экспериментально установил, что распределение частиц подчиняется тому же закону,
640
Новый справочник химика и технолога
что и распределение давления в атмосфере (закону Лапласа). Последнее свидетельствует о том, что на очень больших расстояниях (сотни метров) становится заметным нарушение однородности газов под действием силы тяжести. В коллоидах же из-за того, что масса частиц намного превышает массу молекул, параметр больцмановского распределения mgh/kT достигает значимой величины уже на расстояниях в доли сантиметра, и становится возможным наблюдение неоднородности концентрации по высоте в сосудах лабораторных размеров. В грубых взвесях масса частиц настолько велика, что резкое падение концентрации должно происходить на расстоянии порядка размера частицы, и тогда уравнение распределения теряет свой смысл.
В законе (3.8.19) предэкспоненциальный множитель фо является неизвестной константой распределения. Ее значение можно установить на основе равенства содержания дисперсного материала ф Н во всем сосуде при его равномерном распределении по высоте ( ф — средняя объемная доля дисперсной фазы в системе, Н — высота сосуда) и после перераспределения по закону Больцмана. Последнее равно yQkT / mg[l -- exp(mgH / кТ)} и получается при интегрировании фс/А в пределах от h = 0 до h = Н. Это условие приводит к уравнению:
Фо = ф mgH / кТ [ 1 - ехр (mgH / кТ)}-1. (3.8.20)
Подобные процедуры называются нормированием распределения. При достаточно большом значении mgHIkT нормирование может дать физически неприемлемое соотношение:
Фо>1. (3.8.21)
Оно означает, что в нижней части сосуда образуется слой осадка некоторой высоты Hs. Таким образом, неравенство (3.8.21) является условием образования осадка в агрегативно устойчивом коллоидном растворе. Над осадком может находиться взвесь с больцмановским распределением частиц по высоте (рис. 3.58).
0 0,5 19
Рис. 3.58. Схема и график равновесного распределения вещества по высоте сосуда h при наличии осадка высотой Hs
Фактически в этих условиях предэкспоненциальный множитель распределения (3.8.19), т. е. величина ф0, должен быть равен концентрации дисперсной фазы в осадке ф„ который для простоты можно считать одно
родным по высоте. В отсутствие коагуляции, что, собственно, и подразумевается при обсуждении равновесия во взвеси, частицы в осадке укладываются плотнейшим образом. В осадке монодисперсных сферических частиц их объемная доля равна 75 %. Остальная часть пространства (25 %) занята полостями между плотно уложенными сферическими частицами.
В полидисперсных взвесях эти полости могут быть заполнены частицами меньших размеров, поэтому концентрация дисперсной фазы в осадках может быть и большей — теоретически до ф5 - 1.
Однако в устойчивых взвесях частицы в осадке разделены прослойками среды, толщина которых определяется толщиной защитных оболочек S на поверхности частиц и приближенно равна удвоенной толщине 2S оболочки на поверхности частиц. Это ведет к снижению доли дисперсной фазы в осадке до следующей величины:
Ф, = [a/(a + S)]3. (3.8.22)
Точнее говоря, расстояние между частицами в осадке определяется из условия равенства силы отталкивания частиц F(2S) силе их сжатия в осадке. Последняя может быть оценена как величина гидростатического давления вышележащего слоя осадка на одну частицу: rca^ApgA, т. е. Tia^vApgA = F(25). Объединение этого выражения с формулой (3.8.22) ведет к уравнению, которое позволяет найти величину S:
7w2ApgA* = (1+6/ a)3F(2S). (3.8.23)
Здесь h* = (Hs -И) — высота слоя осадка над частицами, находящимися на высоте h от дна сосуда (при h < а — радиус частиц, Ар — разность плотностей дисперсной фазы и дисперсионной среды. В дальнейшем в ориентировочных расчетах будет предполагаться, что S не зависит от h и примерно равно толщине двойного (или адсорбционного) слоя. В случае суспензий, где S / а 1, наличием оболочки вообще можно пренебречь и принять фу = 0,75, а для нежестких частиц (флокул) фл = 1.
В уравнении распределения частиц над осадком предэкспоненциальный множитель, как уже отмечалось, фо - ф$, а координата h должна отсчитываться не от дна сосуда, а от поверхности осадка. Таким образом, уравнение (3.8.19) преобразуется в уравнение
Ф = ф4 exp[-mg (А - Hs) / кТ\. (3.8.24)
В нем высота слоя осадка Hs — это новая, неизвестная константа уравнения распределения. Ее можно найти из условия (3.8.25) неизменности количества дисперсной фазы во взвеси при наличии осадка и при равномерном распределении вещества по высоте. Оно отличается от использованного ранее при выводе формулы (3.8.20) тем, что включает слагаемое, учитывающее наличие осадка:
Ф ф.у{Hs + кТ/ mg[l-exp(-mg[T/- Hs] / AT)]}. (3.8.25)
Физика, химия и технология дисперсных систем
641
Это трансцендентное уравнение, которое требует применения специальных методов решения. Наиболее доступен (не требует вычислительной техники) метод последовательных приближений. Начать итерационную процедуру можно, полагая, в качестве нулевого приближения, что все вещество находится в осадке. Тогда Hs = ф Н / (ps. Используя это значение, можно оценить количество вещества во взвеси (величину второго слагаемого в фигурных скобках уравнения (3.8.25)) и затем вычислить более точное значение Hs по формуле:
Я, = (фЯ-Г5)/ф5. (3.8.25а)
Далее итерационный цикл можно повторять до достижения желаемой точности. Следует иметь в виду, что величины <рН, Vs и ф5Я5 дают общее количество вещества, количество вещества во взвеси и в осадке соответственно, приходящееся на единицу площади дна сосуда.
3.8.4. Оседание устойчивых концентрированных суспензий
Формула Стокса (3.8.3) определяет скорость движения частицы относительно той среды, в которой взвешена частица. В реальных же условиях представляет интерес скорость движения (оседания) частиц относительно стенок или дна сосуда, в котором находится взвесь. Например, это может быть емкость для очистки промышленных стоков от взвешенных частиц. Различие между этими двумя скоростями можно не принимать во внимание (что обычно и делается) только в разбавленных суспензиях. В промышленных же условиях всегда актуален вопрос о рациональном использовании оборудования, в том числе емкостей для отделения дисперсной фазы от дисперсионной среды. Самым простым способом увеличения производительности емкостного оборудования является увеличение концентрации тех смесей, которые в них перерабатываются. Но именно при высоких концентрациях частиц различие скоростей их оседания относительно среды us и относительно стенок сосуда ис становится существенным вплоть до несовпадений направлений движения. Причина в том, что при оседании достаточно концентрированной взвеси на этот процесс влияет встречный поток среды, вытесняемой оседающими частицами из нижней части сосуда в верхнюю и, следовательно, замедляющий оседание частиц [12]. В монодисперсной суспензии единственным следствием этого эффекта является замедление процесса оседания, но в полидисперсных взвесях по этой причине происходит качественное изменение характера распределения частиц разного размера как в осадке, так и над ним. Действительно, скорость встречного потока среды может оказаться больше, чем скорость оседания частиц достаточно малого размера, и тогда они потоком среды будут переноситься навстречу основной массе более крупных оседающих частиц. Такая инверсия направления движения сохраняется толь
ко внутри столба оседающей суспензии и исчезает по выходе из него, так как вне столба взвеси среда не движется. Скорость оседания мелких частиц в этой покоящейся среде может оказаться больше, чем скорость опускания верхнего фронта столба основной массы частиц, и тогда мелкие частицы не смогут выйти за пределы верхней границы столба взвеси — они будут накапливаться в узкой зоне вблизи границы. Толщина этой зоны будет расти за счет подвода к ней частиц и снизу, и сверху. Зонное распределение фракций сохранится вплоть до полного оседания взвеси. Очень мелкие частицы, которые в покоящейся среде оседают медленнее, чем основная масса взвеси, создадут над ней слой разреженной взвеси с достаточно четкой границей с соседними слоями того же происхождения. Это явление возможно только при очень хорошей устойчивости полидисперсной суспензии против коагуляции, т. е. при нулевой величине сил сцепления частиц, и экспериментально наблюдается в виде четко разграниченных зон в осадке и образования над плотным осадком нескольких (3-х и более) разреженных слоев взвеси с четкими границами между ними.
Количественное описание упомянутых эффектов удобно строить, представляя полидисперсную взвесь в виде дискретного набора из р отдельных фракций с разным размером частиц. Из условия постоянства объема произвольной части взвеси, простирающейся от дна сосуда до некоторой высоты h, отсчитываемой от дна сосуда, следует уравнение (3.8.26), т. е. поток Vжидкой фазы через верхнюю границу выделенной части взвеси равен встречному потоку дисперсной фазы через ту же границу:
К=Еад>,. (3.8.26)
Здесь иа — скорость оседания частиц z-й фракции на высоте h от дна сосуда относительно стенок сосуда, а символ S без указания пределов суммирования означает суммирование по всем индексированным величинам (в данном случае по величинам, помеченным индексом z).
Линейная скорость и/ встречного течения жидкой фазы определяется долей площади S, приходящейся на эту фазу в любом сечении дисперсной системы: Uf= VIS. Эта доля равна, очевидно, (1 - ЛД где As —доля площади, приходящейся в произвольном сечении на дисперсную фазу. Вклад частиц z-й фракции в величину As можно получить из следующих соображений. Произвольная плоскость будет рассекать все частицы, центры которых удалены от плоскости на расстояние г меньшее радиуса частицы а. Площадь сечения одной частицы указанной плоскостью равна n:(zz2 - г2). Число частиц в тонком слое, расположенном на расстоянии г параллельно секущей плоскости и имеющем толщину dr, равна n,dr, где п, — число частиц z-й фракции в единице объема. Произведение п(а2 - r2}ntdr есть вклад в площадь сечения тех частиц, которые отстоят на расстояние г от секущей плоскости. Общая площадь сечения всех частиц получается при интегрировании этой вели
642
Новый справочник химика и технолога
чины по всем расстояниям от г = -а до г = +а. Она равна объемной доле ф, частиц данной фракции во взвеси, поскольку величина - P)dr есть толщина сферического слоя, вырезанного в теле частицы. Сумма (интеграл по г) толщин этих слоев дает объем Ki одной частицы, который связан с объемной долей этих частиц следующим уравнением: ф, = У1П/..
Аналогично определяется площадь сечения частиц других фракций. Общая площадь сечения частиц произвольной плоскостью равна их суммарной объемной доле Еф, во взвеси. Тогда сечение, через которое проходит поток жидкости, описывается уравнением:
5=1-ЕфЛ (3.8.27)
Линейная скорость встречного течения жидкой фазы Uf= V / S. На эту величину скорость движения ис частиц относительно стенок сосуда будет меньше их стоксовой скорости us (скорости движения относительно жидкой фазы). В монодисперсной взвеси с концентрацией дисперсной фазы ф и скоростью оседания частиц относительно сосуда ис доля свободного сечения равна 1 - Ф, а встречный поток среды равен мсф. Тогда Uf= мсф / (1 - ф), и на эту величину скорость движения частиц относительно стенок сосуда будет меньше их стоксовой скорости ws. Отсюда следуют соотношения:
wc = ws(l - ф), Uf = usq>. (3.8.28)
Скорости UfH us имеют разные знаки. Тем не менее в формулах знак «-» отсутствует, так как он содержится в самой величине и/, которая выше была определена как «скорость встречного течения», что и означает ее обратный знак по отношению к знаку скорости движения частиц.
Стоящая в правой части формулы (3.8.27) сумма — это доля площади поперечного сечения, занятого во взвеси частицами всех размеров. Она совпадает с объемной долей этих частиц. Однако это утверждение справедливо только в геометрическом смысле (если рассматривать мгновенный снимок взвеси). В гидродинамическом смысле это не так, поскольку мелкие частицы могут двигаться, как было показано выше при качественном анализе ситуации, вместе с жидкостью и фактически составлять часть жидкой (точнее сказать флюидной, т. е. текучей) фазы взвеси. Различие в гидродинамической роли крупных и мелких частиц становится очевидным, если обратиться к частному случаю полидисперсной взвеси — бидисперсной системе, где один компонент взвеси — это крупные частицы, а второй — молекулы (или мицеллы) какого-либо красителя, которые следует считать частью флюида. В системах с широким спектром размера частиц столь однозначное разделение невозможно, поэтому практические расчеты расслоения взвесей строятся на основе следующего соглашения: при вычислении скорости оседания частиц k-й фракции она и более крупные частицы относятся к оседающей фазе взвеси, а все частицы меньших разме
ров — к флюидной. Таким образом, каждая фракция взвеси имеет свое значение встречного потока флюида Vk, сечения Sk и линейной скорости флюида Мд. В соответствии с этим при вычислении скорости оседания частиц k-й фракции суммирование в формулах (3.8.26) и (3.8.27) распространяется на фракции от к-й до р-й (индекс i меняется от i = к до i = р). Таким образом, уравнение для скорости движения иск частиц к-й фракции относительно стенок сосуда имеет вид
uck=usk-ufk. (3.8.29)
В нем, в соответствии с формулами (3.8.26) и (3.8.27), Мд определяется суммой
«л=Е«л/(|-2ф.)- (3-8.30)
i=k i=k
Уравнения (3.8.29) и (3.8.30) позволяют получить выражение для скорости движения частиц к-й фракции относительно стенок сосуда (иск) [14]:
Мд(1-1Х)“ Z мс((Р-
«а =--------—Р-----—-------• (3-8.31)
+<Р* i=k
Формула (3.8.31) содержит неизвестные скорости иС1 всех более крупных частиц (/ > к + 1), чем частицы к-й фракции, поэтому вычисление скоростей всех фракций необходимо выполнять последовательно, начиная с самых крупных частиц (к = р). Для этой фракции формула (3.8.31) сводится к формуле (3.8.32), которая идентична формуле (3.8.28) для монодисперсной системы:
ucp = usp(\ -фр). (3.8.32)
Уравнение (3.8.32) получается, если принять во внимание следующие обстоятельства. Для р-й фракции в формуле (3.8.31) последнее слагаемое в числителе равно нулю в силу равенства нулю концентрации частиц больших, чем частицы р-й фракции, а сумма в знаменателе этой же формулы совпадает с величиной ф*.
Присутствие во взвеси мелких частиц влияет на скорость оседания частиц любой более крупной фракции (usk) благодаря загущающему действию мелких частиц на флюидную фазу. Оно может быть учтено с помощью формулы Эйнштейна для вязкости взвеси или более сложных формул этого типа (см. подраздел 3.13).
Таким образом, гидродинамические роли частиц разных фракций различны. Крупные частицы, начиная с некоторой выделенной к-й фракции, оседая под действием силы тяжести, играют роль поршня, который выдавливает флюид в верхнюю часть сосуда. Одновременно крупные частицы являются перегородкой, уменьшающей сечение каналов, по которым флюид перетекает в надосадочное пространство. Более тонкие фракции взвеси являются частью флюидной фазы, по
Физика, химия и технология дисперсных систем
643
вышая ее вязкость по сравнению с вязкостью собственно жидкой фазы взвеси.
Необходимость применения итерационной процедуры для расчета скорости оседания всех фракций по формуле (3.8.31) является не самой трудной проблемой при решении задачи расслоения взвеси. Значительно большие трудности обусловлены тем, что в любой следующий момент времени гранулометрический состав любого слоя будет отличаться от его состава в текущий момент времени. Эти изменения можно предсказать только зная распределение фракций и их скоростей по высоте столба взвеси в данный момент. Таким образом, в каждый момент времени нужно заново решать систему уравнений (3.8.31) и связанных с ней уравнений переноса частиц из слоя в слой. Решение такого рода задач возможно численными методами. Для этого созданы необходимые программы. Здесь же достаточно проанализировать несколько простых случаев.
В качестве иллюстрации полезно рассмотреть оседание бидисперсной системы (р = 2), полагая, что доля мелких частиц (фракция 1) во взвеси незначительна и они являются частью флюидной фазы, а эффект встречного течения среды создается только за счет оседания крупных частиц (фракции 2). Тогда линейная скорость встречного течения среды будет, согласно уравнению (3.8.28), равна одрг. Мелкая фракция, имеющая стоксову скорость ms1, внутри столба оседающей суспензии двигается относительно стенок сосуда со скоростью uci = us\ - Нлгфг» & вне его — со скоростью usl (если ее концентрация настолько мала, что она сама себя практически не тормозит). Если внутри столба основной (тяжелой) фракции легкие частицы остаются неподвижными относительно стенок сосуда, т. е. имеют скорость wc] = 0, то в свободном от частиц пространстве скорость их оседания «я = В частности, она может быть равна скорости оседания столба тяжелой фракции. Тогда wi2(p2 = ««г(1 - фг), фг = 0,5 и usi = us2 / 2. Примечательно, что двукратная разница в стоксовых скоростях оседания достигается при различии размеров частиц всего лишь в у/2 раза.
При таком специфичном соотношении размеров и концентраций обе фракции должны оседать с одинаковой скоростью. В этом случае указанное соотношение скоростей устанавливается сразу с начала оседания и, следовательно, отдельный столб разреженной взвеси мелких частиц над оседающим столбом крупных частиц наблюдаться не будет. С другой стороны, внутри столба крупной фракции мелкие частицы покоятся относительно стенок сосуда. Это означает, что на его дно они не попадают, а концентрируются в узком слое на границе столба оседающей взвеси и столба свободной от частиц жидкости. Этот пример на количественном уровне подтверждает механизм зонного осаждения суспензий, описанный выше.
Направленный вниз седиментационный поток исср дисперсной фазы лишь отчасти определяет скорость накопления осадка на дне сосуда. Дело в том, что верх
няя граница осадка движется навстречу потоку частиц с некоторой скоростью ир и тем самым сокращает время, необходимое частицам для достижения верхней границы осадка, т. е. для перехода в осадок. Его высота Hs, а следовательно, и скорость накопления осадка ир = dHs / di зависит от плотности осадка, характеристикой которой служит объемная доля ф5 дисперсной фазы в осадке. Чем более рыхлый осадок, тем больше его высота и скорость продвижения навстречу седиментационному потоку частиц.
Встречное движение частиц и границы осадка приводит к тому, что высота слоя взвеси над осадком сокращается с суммарной скоростью wc + ир. Количество взвешенного в нем вещества уменьшается соответственно со скоростью ф(мс + ир) и с той же скоростью оно переходит в осадок. Следовательно, высота осадка в произвольный момент времени равна ф(мс + up)t / фу, а скорость его наращивания ир = ф(мс + ир) / фЛ. Отсюда следует формула:
ир = мсф / (ф.у - ф). (3.8.33)
Может показаться, что, согласно этой формуле, скорость накопления осадка становится бесконечно большой при приближении концентрации суспензии ф к ее предельной величине ф,у. На самом деле при этом будет сильно снижаться скорость движения частиц ис относительно стенок сосуда. Если выразить эту скорость через стоксову скорость оседания частиц us, то уравнение (3.8.33) преобразуетя в формулу:
ир = и/р(1 - ф) / (фЛ - ф). (3.8.34)
В частном случае образования предельно плотных осадков (ф5 = 1) уравнение (3.8.34) сводится к формуле:
ир = (pus. (3.8.35)
В этом случае движение фронта осадка навстречу седиментационному потоку точно компенсируется эффектом замедления этого потока встречным течением вытесняемой вверх жидкости. Поэтому скорость накопления осадка описывается уравнением (3.8.35), как будто эффект встречного течения среды отсутствует. Частное проявление этой закономерности состоит в том, что полное оседание частиц из очень разбавленной и очень концентрированной суспензии потребует равного времени. Последнюю ошибочно можно даже принять за готовый осадок, т. е. приписать ей способность мгновенно переходить в состояние осадка. Фактически же в осадок она превратится через то же время, что и разбавленная суспензия, хотя при этом заметного уплотнения взвеси не произойдет. Наиболее существенное различие двух внешне одинаковых состояний взвеси состоит в том, что, будучи суспензией, она обладает заметной текучестью, а после превращения в осадок приобретает свойства твердого тела. В этом легко убедиться, наклоняя прозрачный сосуд с осадком на раз-
644
Новый справочник химика и технолога
них этапах его формирования. Следует иметь в виду, что отмеченные особенности оседания характерны для хорошо стабилизированных против коагуляции суспензий. Их можно распространить и на коагулирующие дисперсные системы, если представить себе, что коагуляция произошла мгновенно и принять, что после этого величина <р в формулах (3.8.33)-(3.8.35) равна концентрации рыхлых флокул коагулята, которая может на порядки превышать концентрацию (объемную долю) дисперсной фазы. Разумеется, что при этом и скорости оседания частиц необходимо заменить скоростями оседания флокул. Коагуляция может протекать достаточно медленно, так что именно скорость коагуляции, а не скорость оседания, будет лимитировать процесс расслоения и другие процессы в дисперсной системе. Для выяснения роли этих двух основополагающих явлений — оседания и коагуляции — необходимо обратится к кинетике коагуляции дисперсных систем (подраздел 3.13).
3.8.5. Седиментационный анализ
В большинстве случаев частицы реальных суспензий, порошков и других коллоидных объектов поли-дисперсны. Данные о размере частиц и их распределении по размерам являются необходимыми сведениями, без которых невозможно делать какие-либо прогнозы свойств и поведения дисперсных систем.
Наиболее доступным способом определения спектра размеров частиц и доли частиц каждого размера в образце дисперсного материала является его седиментационный анализ. Как уже отмечалось, он основан на зависимости (3.8.5) скорости оседания частиц в их взвеси (суспензии) от размера частиц. Он включает в себя собственно измерение, в котором с помощью торсионных весов (рис. 3.59) регистрируется зависимость массы осадка, образующегося в сосуде с суспензией, от времени, и математическую обработку этой зависимости. Обработка результатов измерения проводится обычно графическим методом. Для этого строится седиментационная кривая — график зависимости массы осадка от времени (рис. 3.60). Желательно, чтобы он имел четко выраженный горизонтальный участок в конце графика. Его наличие свидетельствует о том, что все частицы анализируемого образца осели. Тогда ордината горизонтального участка представляет суммарную массу частиц всех размеров. Другое важное требование к виду седиментационной кривой — ее выпуклость на всем протяжении. Иначе говоря, наклон графика максимален в его начале и далее может только уменьшаться или оставаться неизменным на некотором интервале времени. Наличие вогнутости на такой зависимости означает, что в суспензии идет процесс коагуляции (см. подраздел 3.13.4) и результаты измерений непригодны для целей седиментационного анализа.
Обработка седиментационной кривой предполагает, что каждая фракция полидисперсной взвеси вносит свой независимый вклад в общую, регистрируемую в опыте массу осадка Р. Обработка седиментационной
кривой предполагает, что каждая фракция полидисперсной взвеси вносит свой независимый вклад в общую, регистрируемую в опыте массу осадка Р. В любой произвольно выбранный момент времени завершается оседание определенной фракции частиц, а именно такой, которая оседает со скоростью и = Н /t, и, следовательно, имеет размер частиц а, = (Н / Z,S)1/2.
Рис. 3.59. Схема седиментометра:
1 — сосуд с анализируемым препаратом; 2 — весы
Рис. 3.60. Седиментационная кривая и ее деление на части, соответствующие оседанию разных фракций
Здесь Н — глубина погружения чашки весов, принимающей осадок, и S' = 2Apg / 9т| — коэффициент формулы Стокса. В последующие моменты времени данная фракция уже не участвует в накоплении осадка на чашке весов. Поэтому наклон графика dP / dt (скорость накопления осадка) уменьшается на величину, равную вкладу Pfli, / Н данной фракции в общую скорость накопления осадка. Тогда Рдг, / Н= d2P / dt2, а масса фракции определяется уравнением:
Pt = (d2P / dr} tt. (3.8.36)
На практике на седиментационной кривой выбирают несколько (п) произвольно расположенных точек и к каждой из них проводят касательную, наклон которой и дает величины производных dP / dtt в соответствующие моменты времени t,. Фактически это означает деление исследуемого препарата на п фракций с диапазоном размеров частиц от Оо до а\, от а\ до а2,... от ai.l до а,... от ап_\ до ап. При этом Оо > ai > ... а,>а<+1> ... a„_i>a„,Oo—размер
Физика, химия и технология дисперсных систем
645
самых крупных частиц в анализируемом образце дисперсного материала, а ап — самых мелких. Следует принять, что касательная к некоторой точке кривой характеризует наклон той части графика, которая находится правее этой точки, а значение dP / dti Xy относящееся к первой точке седиментационной кривой (z = 1), представляет собой наклон участка кривой, исходящего из начала координат. К условностям такого рода всегда приходится прибегать при замене непрерывной функции конечным числом п ее значений. В этом случае масса Р, фракции с размером частиц от а, _ i до а, определяется уравнением:
Pi = (dPIdti^-dPIdt,) th (3.8.37)
Графически она представляется отрезком на оси ординат между интерцептами касательных к z-й и (z-1 )-й точкам седиментационной кривой (рис. 3.60). Сумма всех отрезков равна общей массе Р% осадка на чашке весов, а гранулометрический состав исследуемого материала характеризуется массовой долей w,-Pt I Р% каждой фракции в данном материале.
Альтернативный способ обработки результатов измерения основан на априорно принятом распределении частиц по фракциям, например на основе функциии распределения Гаусса:
ф(а) = ехр [-(а -а)2/ 2о2] / о (2л)1/2. (3.8.38)
В этом распределении не определены значения параметров распределения: а — средней размер частиц и о — дисперсия (ширина распределения). В принятых выше обозначениях ф(а) = w, / (а, - а; _ ]). Задачей математической обработки является нахождение этих параметров, после чего функция распределения (3.8.38) позволяет исчерпывающим образом описать гранулометрический состав препарата. Ограничение способа состоит в том, что гранулометрический состав реального материала далеко не всегда описывается распределением Гаусса. Простейший и очевидный пример системы, к которой указанный метод неприменим, — это система, состоящая из смеси двух монодисперсных фракций с различным размером частиц этих фракций.
Никаких ограничений на характер распределения частиц по размерам не накладывает метод конечных разностей. В этом методе масса каждой фракции в осадке вычисляется непосредственно по формуле (3.8.36) или (3.8.37), но вместо произвольно выбранных точек седиментационной кривой в ней используются непосредственно результаты измерений, т. е. пары значений t, и P(/j). Фактически это означает замену плавной кривой набором отрезков прямых, а наклона касательных — наклоном этих отрезков
dP / dt, = [Р(4 + 0 - ЛО] / (h +1 -
Каждая фракция при этом считается монодисперсной, и частицам фракции приписывается один из размеров,
соответствующих значениям времен ь /, z, + ь использованных для вычисления первой и второй производных. Недостатком метода является сильное влияние погрешности измерений на плавность распределения. Графические методы обработки результатов измерений всегда включают в себя сглаживание экспериментальной зависимости. Сглаживание происходит при построении плавной седиментационной кривой по ограниченному числу экспериментальных точек, которые содержат в себе и ошибку измерения. Снижение влияния погрешности измерений, однако, трудно реализуемо при машинных способах обработки данных. Наличие сглаженных зависимостей, в сущности, эквивалентно наличию неограниченного числа точных значений tt и P(t^). В этом случае можно перейти от метода конечных разностей к непрерывному дифференцированию и сразу вычислить величину ф(а) = dw, / da, где dwt — массовая доля частиц с размером от а до (а + da). Согласно формуле Стокса, da - ~(HIS){l2dtv2t312. Тогда с помощью формулы (3.8.36) получается уравнение [37]:
<р(а) = 2(S/ Н)т{^Р / dt2) t5'2. (3.8.39)
При конечном числе точек на седиментационной кривой из формулы (3.8.37) следует уравнение:
ф(а,) = 2(S / H)xl2(dP I dt, - dP I dt, + J/,5721 (t, + /,).(3.8.40)
3.9. Электромагнетизм коллоидов
3.9.1. Основы электростатики
В основе представлений об электромагнитных явлениях и свойствах материи лежит несколько фундаментальных законов, являющихся обобщением экспериментальных фактов. Это закон Кулона, сила Лоренца, законы электромагнитной индукции.
По закону Кулона (3.9.1) или (3.9.2) между двумя объектами с зарядами Q\ и Q2 и расстоянием г между ними действует сила F:
F=Q,Q2/4me^, (3.9.1)
Г=е,£, (3.9.2)
E = Q2l4n^,r2. (3.9.3)
Здесь Е — напряженность электрического поля, создаваемого зарядом одного из объектов (в данном примере второго) в точке нахождения другого (первого) объекта (формула (3.9.3)), т. е. сила, с которой источник поля воздействует на единичный положительный заряд, г — расстояние между зарядами. Другие величины, фигурирующие в приведенных формулах, можно рассматривать как вспомогательные и предназначенные исключительно для согласования размерностей силы, расстояния и заряда, но в действительности каждая из них отражает фундаментальные свойства пространства и материи. Так, е0 — это электрическая постоянная, а коэффициент 4л есть максимальная величина телесного
646
Новый справочник химика и технолога
угла в трехмерном пространстве. Величина е — диэлектрическая проницаемость среды — основная электростатическая характеристика той среды, в которой находятся носители заряда. Следует обратить внимание на смысл термина «среда». В контексте описания электромагнитных свойств веществ простого или сложного состава все эти вещества выступают в качестве «среды», в которой существует и действует поле. В контексте описания состава и структуры дисперсного материала «среда» — это один из его компонентов. В необходимых случаях последняя будет заменяться полным термином «дисперсионная среда».
В качестве носителей заряда обычно выступают материальные тела, и поэтому, следует говорить об электрическом взаимодействии тел. Однако для краткости можно говорить о взаимодействии зарядов, в том числе точечных, т. е. не имеющих размера. Здесь не будут рассматриваться электродинамические задачи, связанные с распространением электромагнитных волн, когда поля и электрические токи могут существовать и без каких-либо материальных носителей (кроме самих волн).
Напряженность поля, создаваемого точечным зарядом, убывает обратно пропорционально квадрату расстояния от места локализации этого заряда. Знак заряда определяется по знаку силы в уравнении (3.9.1), исходя из того, что одноименные по знаку заряды отталкиваются, а разноименные — притягиваются. Один из наиболее доступных носителей заряда для разных опытов — это электрон. Ему приписан заряд отрицательного знака величиной 1,6 • 10~19Кл. Напряженность поля Е также может быть положительной и отрицательной. Согласно формуле (3.9.2), напряженность поля положительна, если его источником является положительный заряд. Напряженность поля — векторная величина, т. е. характеризуется не только силой воздействия на единичный положительный заряд, но и направлением этой силы. Направление силы — это направление движения объекта, на который она воздействует. Направление всегда, связано с выбором системы координат. Поскольку такой выбор может быть произвольным, то направление (знак) напряженности поля не определяется только знаком источников поля.
Потенциал V электрического поля в данной точке пространства г — это работа переноса единичного положительного заряда в данную точку поля из бесконечности. При этом подразумевается, что никакие другие силы, кроме кулоновской (3.9.1), на переносимый заряд не действуют. Из определения следует, что потенциал поля равен интегралу от напряженности с обратным знаком (поскольку при таком направлении переноса </г<0). В частности, формула (3.9.4) описывает потенциал точечного заряда Q.
V=Q!4wew. (3.9.4)
Соответственно, напряженность поля — это пространственная производная потенциала с обратным знаком:
E = -dV/dr. (3.9.5)
При фиксированной величине заряда напряженность создаваемого им поля (уравнение (3.9.3)) и его потенциал (уравнение (3.9.4)) зависят от диэлектрической проницаемости среды е. В некоторых случаях необходимо иметь характеристику интенсивности поля, которая не зависит от свойств среды. Таковой является индукция электрического поля D (ее еще называют электрическим смещением). Смысл этой величины формально определен соотношением:
D = £8о£. (3.9.6)
Согласно уравнениям (3.9.3) и (3.9.6), индукция D точечного источника Q равна Q / 4лг2, т. е. это напряженность поля, создаваемого теми же источниками в вакууме.
Удобство введения индукции как характеристики интенсивности электрического поля обусловлено тем, что именно через нее наиболее просто формулируется связь между интенсивностью поля в любой точке пространства и распределением заряда в пространстве. Эта связь выражается теоремой (законом) Остроградского — Гаусса (далее — законом Гаусса):
Т = Q. (3.9.7)
Здесь Т — поток индукции через любую замкнутую поверхность А, охватывающую находящийся внутри ее заряд Q. Поток индукции через элемент поверхности площадью dA — это произведение его площади dA на нормальную к нему компоненту Dn индукции на поверхности. Поток через всю поверхность Т = \DndA получается суммированием (интегрированием) элементарных составляющих потока DndA по всей поверхности А. Из этого закона следует, в частности, что электрическое поле, создаваемое любым центральносимметричным распределением зарядов, например равномерно заряженным шаром, совпадает вне заряженного тела с полем, которое создает тот же заряд, но размещенный в центре симметрии распределения. В дифференциальной форме закон Гаусса входит в систему уравнений электромагнитного поля Максвелла:
VD=p. (3.9.8)
Здесь V = д / дх + д / ду + д / дг — оператор Лапласа, р — объемная плотность пространственного заряда (заряд единицы объема).
Поле, созданное бесконечной заряженной плоскостью с равномерно распределенным по ней зарядом, зависит только от одной координаты х, нормальной к заряженной поверхности. Левая часть уравнения (3.9.8) при этом представлена производной только по координате х, т. е. V£> = dD / dx. В этом смысле поле можно называть одномерным, хотя фактически одномерным (включающим зависимость от одной координаты) явля
Физика, химия и технология дисперсных систем
647
ется уравнение, а не само поле. Одномерным остается и поле, созданное объемно распределенным зарядом, если его объемная плотность р изменяется только вдоль одной оси. Электрическое (или магнитное) поле, напряженность которого не зависит от пространственных координат, называется однородным.
Закон Гаусса применительно к системе зарядов, распределение которых зависит только от одной координаты х, удобно представить в форме:
dE = dQ/EEQ. (3.9.9)
Здесь dQ = pdx — заряд тонкого слоя толщиной dx и площадью равной единице, a dE — разность напряженностей поля на границах этого слоя. Примечательно, что величина dE не изменится, если, не меняя величины заряда слоя, сосредоточить его в одной плоскости или уменьшить толщину слоя dx до нуля — сжать его до математической заряженной плоскости. В этом случае заряд Q будет численно равен поверхностной плотности заряда о. При переходе с одной стороны такой плоскости на другую, т. е. на расстоянии равном нулю, напряженность поля изменится скачком на ту же величину dE = d<5 I EEq. В силу симметрии картины (равноправности обеих сторон плоскости) напряженность поля на каждой из сторон плоскости и вблизи ее будет одинаковой по величине, но противоположной по направлению, т. е. разной по знаку. Отсюда следует формула для напряженности электрического поля возле бесконечной, равномерно заряженной плоской поверхности с поверхностной плотностью заряда о (зарядом единицы площади):
£=о/2е£0. (3.9.10)
С помощью формулы (3.9.5) соотношение (3.9.9) сводится к одномерному уравнению Пуассона, которое, в частности, описывает распределение потенциала возле заряженной плоскости в растворе электролита и используется в теории двойного электрического слоя:
d2Vldx2 = -plEEQ. (3.9.11)
Для изучения электрических свойств различных веществ полезно уметь создавать однородное поле хотя бы в некотором ограниченном пространстве. Это легко делается с помощью двух плоских противоположно заряженных пластин. В пространстве между двумя такими параллельными пластинами поле однородно, а его напряженность (формула (3.9.12)) в 2 раза больше, чем напряженность возле одной пластины (уравнение (3.9.10)):
Е = о/££0. (3.9.12)
Конструкция из двух параллельных проводящих пластин, разделенных зазором 8, называется плоским конденсатором. Его основной характеристикой является емкость — отношение заряда одной из пластин к
разности их потенциалов: С = Q / U. Иначе говоря, емкость — это заряд, который необходимо перенести с одного тела (пластины) на другое, чтобы создать разность потенциалов между ними, равную единице. Вообще, потенциал всегда определяется с точностью до произвольной постоянной величины, поэтому потенциал одной из пластин может быть принят равным нулю, и тогда вместо разности потенциалов можно говорить просто о потенциале другой пластины конденсатора или заряженного тела. Важно, что вне конденсатора электрическое поле отсутствует (не считая его краев). Другое очень важное свойство конденсатора состоит в том, что заряд по его пластинам распределяется равномерно и никаких принудительных мер для обеспечения равномерности принимать не нужно — она соответствует равновесию всей системы зарядов конденсатора, обусловленному притяжением зарядов противоположно заряженных пластин. Электрическое состояние конденсатора удобно характеризовать поверхностной плотностью заряда одной из пластин о = Q / А, где А — площадь пластины (одной ее стороны), и, соответственно, удельной емкостью С = о / U. Следует отметить, что на отдельной проводящей пластине заряды вследствие их взаимного отталкивания будут скапливаться на ее краях. По той же причине заряды электропроводного тела будут целиком сосредоточены на его поверхности, а в случае тел сферической формы заряд распределен по поверхности так же равномерно, как и по пластинам плоского конденсатора. Это свойство подразумевалось при изучении электрических характеристик коллоидных частиц и двойного электрического поля.
Внутри конденсатора поле однородно, а вне конденсатора поле отсутствует, так как пластины дают равные по величине и противоположные по знаку напряженности. Поэтому потенциал в зазоре изменяется в соответствии с уравнением (3.9.5) по линейному закону от потенциала одной пластины до потенциала другой, а напряженность поля везде одинакова и равна (7/8. Все сказанное справедливо, если расстояние 8 между пластинами намного меньше их размера. Типичный пример самопроизвольного упорядочения зарядов по типу плоского конденсатора — это двойной электрический слой. Потенциал заряженной поверхности отсчитывается в этом случае от потенциала раствора электролита, который приравнивается нулю.
Формула (3.9.6) определяет смысл диэлектрической проницаемости £ формально. Чтобы выяснить закономерности, в соответствии с которыми формируются диэлектрические свойства дисперсных систем, необходимо расшифровать физический смысл величины е, т. е. выразить ее через свойства молекул или частиц, составляющих среду, в которой существует и действует поле.
Электрическое состояние отдельной молекулы характеризуется ее дипольным моментом р, который определяется уравнением (3.9.13), т. е. дипольный момент — это произведение заряда Q на расстояние г ме
648
Новый справочник химика и технолога
жду центрами зарядов в паре противоположных по знаку, но равных и связанных зарядов какой-либо частицы (молекулы):
p = rQ. (3.9.13)
Вектор, соединяющий центры зарядов диполя, называется осью диполя. По порядку величины заряд Q в молекулах равен заряду электрона, а расстояние г — размеру атома, так что дипольный момент составляет около 1029 Кл • м.
Дипольный момент может быть постоянным, т. е. его величина не зависит от силы и направления действующего на молекулу (частицу) поля. Вещества, молекулы которых имеют постоянный дипольный момент, называются полярными. Характерной особенностью их химического строения является наличие полярной группы в составе молекулы (-ОН, -NO2, -СООН, -NH2 и т. д.). Сами по себе величины Q и г не имеют значения, важна лишь величина их произведения р. Действие внешнего однородного электрического поля на такие молекулы сводится к ориентации осей диполей вдоль направления поля (путем механического вращения молекул). Ориентации препятствует вращательное тепловое движение молекул. Одновременное ориентирующее действие поля и дезориентирующее влияние вращательной диффузии приводит к зависимости степени ориентации осей диполей L от напряженности поля:
£ = cthz-l/z. (3.9.14)
Здесь z = рЕ / кТ — безразмерная (выраженная в единицах кТ) энергия диполя во внешнем однородном поле. Выражение (3.9.14) называется функцией Ланжевена. Степень ориентации L соответствует среднему значению косинуса угла между направлением поля и направлением оси диполя, так что величина ~р является средней величиной проекции дипольного момента на направление поля:
р=р£. (3.9.15)
Средняя величина проекции дипольного момента на поперечное полю направление равна нулю.
В неполярных молекулах дипольный момент индуцируется действующим на молекулы электрическим полем Е\ оно вызывает поляризацию молекулы — смещение центров положительных и отрицательных зарядов в противоположные стороны, и в итоге молекула приобретает наведенный дипольный момент. Величина момента определяется той же формулой (3.9.13) с той разницей, что в ней расстояние г и, соответственно, дипольный момент р увеличиваются пропорционально напряженности действующего на молекулу поля. Способность молекулы поляризоваться характеризуется ее поляризуемостью а(, а величина наведенного момента вычисляется по формуле:
р^а^оЕ. (3.9.16)
По порядку величины поляризуемость близка к объему молекулы и будет далее называться деформационной поляризуемостью, поскольку она обусловлена деформацией первоначального распределения электронной плотности в молекуле. Направление оси наведенного диполя обычно совпадает с направлением действующего на молекулу поля.
Вещество как совокупность молекул также поляризуется. Поляризованность вещества характеризуется величиной дипольного момента единицы объема вещества Р и поэтому равна сумме дипольных моментов п молекул, содержащихся в единице объема:
Р = пр. (3.9.17)
Следует отметить, что поле с напряженностью Е„ действующее на молекулы внутри поляризуемого вещества, заметно отличается от макроскопического поля Е. Поэтому, согласно уравнению (3.9.16), формула (3.9.17) преобразуется в уравнение:
P = n<xtZoEl. (3.9.18)
Вещество, состоящее из полярных молекул, также поляризуется во внешнем поле, и, как и в предыдущем случае, его состояние характеризуется величиной Р, которая вычисляется по тем же формулам (3.9.16)— (3.9.18). Различие в том, что вместо наведенного дипольного момента в них нужно подставлять среднюю величину проекции р постоянного диполя молекулы на направление поля. Кроме того, при поляризации полярных веществ труднее найти связь между напряженностью Е заданного внешнего поля, в которое помещается исследуемый образец вещества, и напряженностью Е, внутреннего поля, действующего на молекулы этого вещества. В слабых внешних полях аргумент функции Ланжевена z<?cl, и тогда сама функция L=pEt / ЗкТ. Следовательно, в слабом поле величина проекции диполя на направление поля растет пропорционально напряженности поля и, согласно уравнению (3.9.15), ~р =p2Ei / ЗкТ. Тогда ориентационная поляризуемость молекулы (коэффициент поляризуемости) полярного вещества aj = р / г0Е, определяется формулой:
ad = p2/3&okT. (3.9.19)
В итоге, независимо от механизма поляризации вещества — путем деформации молекул или ориентации их постоянных дипольных моментов, — его поляризованность увеличивается пропорционально напряженности поля.
Поляризованность Р вещества как его реакцию на воздействие макроскопического поля можно выразить через электрическую восприимчивость вещества % с помощью уравнения:
Р = Х£о£- (3.9.20)
Действующее на молекулы поле Eh которое подразумевается в формулах (3.9.17)-(3.9.19), в общем слу
Физика, химия и технология дисперсных систем
649
чае не совпадает по величине, а иногда и по направлению, с заданным по условиям опыта макроскопическим полем Е. Причина — дополнительное воздействие на каждую молекулу полей всех других молекул поляризуемого вещества. Только в случае газообразных сред можно считать, что Е, = Ен электрическая восприимчивость вещества равна суммарной восприимчивости всех молекул в единице объема:
Х = «а. (3.9.21)
Поляризуемость молекулы а складывается из ее деформационной а, и ориентационной ad составляющих, т. е. а = а, + ad. В случае конденсированного состояния вещества (жидкого или твердого) связь между практически применяемыми характеристиками веществ % и £ и теоретически предсказуемым свойством молекул — их поляризуемостью а — усложняется. Задача заключается в нахождении этой связи.
Анализ влияния поляризации вещества на напряженность поля внутри вещества удобно проводить с помощью представления о связанных зарядах. В сущности, это альтернативный способ количественного описания поляризации.
Внутри поляризованного вещества, находящегося между пластинами плоского конденсатора, объемный заряд отсутствует, поскольку заряды положительных и отрицательных концов диполей здесь взаимно компенсируются. В прилегающих к пластине конденсатора мономолекулярных слоях вещества такой компенсации нет: к отрицательной пластине конденсатора примыкает слой вещества, на поверхности которого сосредоточены положительные концы молекулярных диполей, а возле положительной пластины на поверхности вещества сосредоточены отрицательные концы молекул. Таким образом, на приэлектродных поверхностях вещества находятся противоположные заряды. Они называются связанными. Поверхностная плотность этих зарядов ст численно равна поляризованности вещества Р. Это следует из того, что дипольный момент PAd слоя вещества толщиной d площадью А одной его стороны равен также <sAd — произведению заряда поверхности стЛ на толщину слоя d. Отсюда вытекает и равенство ст = Р.
Отличие связанных зарядов от свободных в том, что связанные заряды не могут быть отделены от вещества, например их нельзя передать по проводам в виде электрического тока, и в то же время они неизбежно возникают на поверхности вещества при действии на него поля, создаваемого «настоящими» (свободными) зарядами. В остальном их влияние на напряженность и потенциал поля такое же, как и свободных зарядов, поэтому действующее в веществе (в том числе на само вещество) поле в равной мере обязано как свободным, так и связанным зарядам. Наглядно роль тех и других продемонстрирует воображаемый опыт по заполнению воздушного конденсатора (е = 1) жидким диэлектриком
с диэлектрической проницаемостью е > 1. Перед заполнением конденсатор присоединяется к источнику фиксированной разности потенциалов U (батарее) и тем самым между его пластинами создается однородное поле с напряженностью Е - U / d, а на пластинах накапливается удельный (на единицу площади) заряд ст = г^Е. Далее возможны два варианта продолжения опыта: заряженный конденсатор можно опускать в жидкость, предварительно отсоединив его от батареи или не отсоединяя от батареи. В первом случае заполнение идет при постоянстве удельной величины свободного заряда ст на пластинах, а во втором — при постоянстве разности потенциалов пластин, а следовательно, и напряженности поля Е- U / d. Заполнение конденсатора в любом случае ведет к накоплению связанного заряда Р = у$оЕ на границе диэлектрика и пластины конденсатора, который частично компенсирует свободный заряд. В режиме постоянства свободного заряда это влечет за собой уменьшение напряженности поля за счет уменьшения полного заряда (свободный + связанный противоположного знака) в соответствии с формулой (3.9.12). При ее применении нужно проявить осмотрительность: по условиям опыта меняется и полный заряд, и проницаемость среды, однако учитывать при вычислениях напряженности поля следует только изменения одной из этих двух величин. Дело в том, что проницаемость вещества £ сама по себе учитывает непостоянство полного заряда при замене одного диэлектрика на другой, поэтому в первом варианте опыта напряженность изменится от первоначальной величины ст/ео до ст/ее0. Однако новую напряженность поля можно выразить и через полный заряд поверхности Е = (ст - P)Izq, но при этом следует игнорировать изменение проницаемости среды между пластинами, поскольку эффект от этого изменения полностью учтен с помощью полного заряда. Если значения напряженности поля, вычисленные разными путями, равны, то ст - Р = ст / е. Здесь ст = ее0£, а Р - еох^- Из полученных соотношений следует формула:
£=1+Х- (3.9.22)
Замена е на 1 + х в определении индукции (3.9.6) позволяет представить ее в виде двух слагаемых. Первое соответствует вкладу свободных зарядов, а второе — вкладу связанных зарядов в суммарную величину напряженности поля в веществе. Приведенное вслед за формулой (3.9.6) пояснение индукции как напряженности поля в вакууме подразумевает, что в вакууме в качестве источников поля будут действовать и связанные заряды, хотя фактически их нельзя отделить от вещества.
Второй вариант виртуального опыта по заполнению зазора конденсатора жидким диэлектриком предусматривает погружение соединенного с батареей конденсатора в жидкость. При этом напряженность поля в нем останется неизменной за счет перетекания свободного
650
Новый справочник химика и технолога
дополнительного заряда из батареи на пластины в количестве, равном величине связанного заряда. Полный заряд пластины с единичной поверхностью останется равным свободному исходному заряду сг = е^Е, а свободный заряд возрастет до величины сг + Р. Если значения напряженности поля, вычисленные через полный и свободный заряды, равны, то сг / е0 = (сг + Р) / ее0, что снова приводит к формуле (3.9.22). Следует отметить, что здесь, в отличие от предыдущего случая, сг = —
полный удельный заряд пластины конденсатора. Во всех случаях можно руководствоваться простым правилом: в любом выражении для напряженности (или потенциала) поля не могут одновременно использоваться и проницаемость среды, и полный заряд источника поля.
Формула (3.9.22) связывает две альтернативные макроскопические характеристики диэлектрика, а их связь с микроскопической характеристикой — поляризуемостью молекулы (частицы) — остается невыясненной. Для решения этой задачи необходимо иметь описание поля, создаваемого одной поляризованной молекулой, т. е. диполем.
Поле диполя — это сумма полей двух равных зарядов противоположного знака, расположенных на малом расстоянии один от другого. Сложение напряженности полей, создаваемых соседними разноименными зарядами, дает напряженность поля диполя:
Ed=t?(3cos2@ + 1 )1/2 / 4л££оА (3.9.23)
Она зависит от расстояния г между данной точкой поля и центром диполя и от угла О между осью диполя и радиус-вектором точки г. Во многих случаях представляет интерес только проекция Ер этого поля на направление внешнего поля (направление поляризации), определяемая формулой:
Ер = t?(3cos20 - 1) / 4л££ог3. (3.9.24)
Как уже отмечалось, поля, создаваемые объемными зарядами за пределами объемного носителя заряда, идентичны полям точечных зарядов той же величины. При этом расстояния должны отсчитываться от центра «тяжести» зарядов. Сказанное относится и к поляризованным телам конечных размеров, в том числе молекулам и их дипольным моментам. Рассмотренный выше вклад поляризованного вещества в поле конденсатора с этой точки зрения является векторной суммой полей всех диполей, находящихся между пластинами конденсатора. Математический аппарат описания таких полей предполагает, что диполи являются точечными, т. е. расстояние между ними в веществе можно считать большими по сравнению с геометрическими размерами самих диполей. Поскольку поле диполя резко неоднородно (см. приведенные выше формулы), то и поле диполя в веществе сильно неоднородно на расстояниях порядка межмолекулярных. Фигурировавшее выше дополнительное поле в веществе, порождаемое его по
ляризацией, является средним как для точек, занятых молекулами, так и для свободного межмолекулярного пространства. Поляризация молекул определяется, конечно, напряженностью поля в точках, занятых молекулами. Поэтому важной задачей является определение напряженности такого поля, для решения которой используется метод, предложенным Лоренцем.
Этот метод предполагает выделение в однородном поляризованном веществе сферы произвольного радиуса г. При мысленном удалении этого сферического тела из вещества электрическое состояние и вещества, и тела должно сохраниться. Для этого взамен отсутствующих частей тела необходимо разместить на поверхности сферической полости внутри вещества и на поверхности извлеченного тела эквивалентное число связанных зарядов. Их эквивалентность определяется тем, что они создают такое же поле, как и недостающая часть поляризованного тела. Связанный заряд сг(0) распределен по поверхности однородно поляризованной сферы по закону:
q(0) = Pcos0. (3.9.25)
Согласно закону Кулона (3.9.1), каждый элемент заряженной поверхности dA создает в центре сферы поле с напряженностью <j(0)<Z4/47C£or2. Здесь относительная проницаемость среды £ = 1 в соответствии с правилом использования в формулах связанного заряда. Поле, создаваемое узким сферическим поясом, все элементы которого имеют одинаковые величины полярного угла 0, будет состоять только из компонентов, направленных вдоль оси поляризации. Поперечные компоненты поля каждого элемента пояса компенсируются полем элемента, симметрично расположенного на поясе. Тогда напряженность поля, создаваемого сферическим поясом, равна Pcos027cr2sin0cos0J0 / 4ЯЕ0Г2, где 27cr2sin0cos0d0 — площадь пояса. При интегрировании этого выражения по 0 в пределах от 0 = 0 до 0 = л/2 получается формула, описывающая напряженность поля, создаваемого в центре сферы распределенным по ее поверхности связанным зарядом:
£л-/’/Зе0. (3.9.26)
Это поле называют полем Лоренца, что и отмечено индексом £. Оно в рамках использованного метода исчерпывающе отражает действие всей массы вещества на вырезанный из него сферический фрагмент. Так как сфера поляризована однородно, то в любом месте внутри сферы напряженность поля одинакова. С учетом формулы (3.9.20) формула (3.9.26) преобразуется в выражение:
£л-х£/3. (3.9.27)
Кроме поля, создаваемого поляризованным веществом, на сферический фрагмент этого вещества действует еще и поле свободных зарядов £, причем их направле
Физика, химия и технология дисперсных систем
651
ния совпадают, поэтому формула (3.9.28) описывает суммарную напряженность поля внутри сферы:
£, = (1+х/3)£. (3.9.28)
Она не зависит от размера сферы, поэтому в качестве таковой можно взять и отдельную молекулу. Таким образом, формула (3.9.28) дает искомую величину действующего на молекулы поля. При использовании его в формуле (3.9.18) и сравнении полученного результата с выражением (3.9.20) получается выражение:
X = па / (1 - ла/3). (3.9.29)
Соотношение между микроскопической (а) и макроскопическими (х и е) характеристиками поляризуемости вещества часто записывают в виде уравнения Клаузиуса — Мосотти:
па= 3(е - 1) / (е + 2). (3.9.30)
Поскольку символом а была обозначена сумма деформационной и ориентационной поляризуемостей, то уместнее эту формулу называть формулой Дебая [31]. В формуле Клаузиуса — Мосотти должна фигурировать только деформационная поляризуемость молекул.
Формула (3.9.29) показывает, что при иа > 3 восприимчивость становится бесконечно большой, т. е. вещество поляризуется самопроизвольно (отсутствие внешнего поля). Такие вещества (сегнетоэлектрики) действительно существуют, хотя приведенное условие перехода в такое состояние не является достаточно корректным. Причина в том, что самопроизвольная поляризация возможна в веществе, молекулы которого обладают постоянным дипольным моментом, но в этом случае на любую молекулу кроме поля Лоренца действует более сильное поле ближайших соседей (локальное поле), наличие которого формула Клаузиуса — Мосотти не учитывает. Корректный расчет локальных полей требует учета структуры вещества (или дисперсной системы, если речь идет о ее поляризуемости) и дипольного взаимодействия соседних молекул (частиц). Сложность проблемы в том, что структура в свою очередь определяется взаимодействием молекул, так что возникает замкнутый круг двух взаимосвязанных задач, каждая из которых не может решаться отдельно от другой. Существует ряд теорий полярных диэлектриков, в которых постулируется наличие структуры того или иного вида. Разные теории отличаются способами описания структурно зависимой части поля, действующего на каждую молекулу [31]. Это теория локального поля Дебая, теория реактивного поля Онзагера, теория локального поля Кирквуда.
Согласно теории Дебая, в первом приближении (без учета эффекта насыщения ориентации диполей в локальном поле) восприимчивость X/. полярного вещества определяется формулой:
Ь = 1[1-12Ы1 (3.9.31)
Здесь х — диэлектрическая восприимчивость (уравнение (3.9.29)), zL = pEL I kT и EL = ZEd — векторная сумма напряженностей полей, создаваемых ближайшими соседями в центре рассматриваемой молекулы, функция L(zl) определяется формулой (3.9.14). Корректность этого результата определяется лишь точностью вычисления среднего значения локального поля El.
Согласно теории Онзагера, выполняется уравнение:
(е - и2)(2е + п2) / е(и2 + 2)2 = пр2 / Зг()кТ. (3.9.32)
Здесь п — показатель преломления среды. Его появление в этой формуле обусловлено тем, что квадрат показателя преломления равен деформационной поляризуемости молекул данного вещества, точнее говоря — той ее части, которая обусловлена деформацией только электронных оболочек и не включает деформаций типа изменения расстояния между катионом и анионом в молекуле.
Теория Кирквуда приводит к уравнению:
(е- 1)(2е+ \)/9E = n(a,+p2g/3kT)/3e0.(3.9.33)
Здесь g = Z <cosy> + 1, Z— координационное число (число ближайших соседей), <cosy> — среднее значение косинуса угла между направлением осей диполей молекулы и ее ближайших соседей.
3.9.2. Поляризуемость частиц
Выше поляризуемость частиц и молекул фигурирует как некоторое заданное свойство. В молекулярной и коллоидной физике оно подлежит определению исходя из геометрических, электронных и других свойств молекул и частиц. В случае коллоидных частиц считаются известными электрические характеристики дисперсного материала и дисперсионной среды — их диэлектрическая проницаемость, электрическая проводимость, параметры двойного слоя на частицах, подвижности ионов и др. В общем случае нахождение поляризуемости частиц представляет собой сложную задачу. Достаточно отметить, что формула для поляризуемости частиц с двойным электрическим слоем была получена примерно через сто лет после вывода формулы для поляризуемости диэлектрической частицы. Наиболее важные уравнения для определения поляризуемости частиц приводятся ниже без вывода.
Поляризуемость диэлектрической сферы радиуса а с проницаемостью е' в среде с проницаемостью в:
а = еа*(е' - в) / (2в + в')- (3.9.34)
Поляризуемость эллипсоида вращения вдоль длинной оси:
а = еЕ(е' - е)у4тг[е + (в' - е) 7V]. (3.9.35)
Здесь V — объем эллипсоида, N — фактор деполяризации вдоль длинной оси (он равен 1/3 для сферы и снижается почти до 0 при отношении осей, равном 20).
652
Новый справочник химика и технолога
Сумма факторов деполяризации по трем осям эллипсоида равна 1, поэтому поперек оси фактор деполяризации Nt = (1 - N)/2 и поляризуемость а, поперек оси:
а, = £ V (s' - е) / 4л[е + (е ' - е)(1 - Л)/2]. (3.9.36)
Поляризуемость проводящих частиц сферической и эллипсоидной формы в диэлектрике (е / е' = 0):
- сфера
а = ва3, (3.9.37)
- эллипсоид вдоль оси
а = £И/4т< (3.9.38)
- эллипсоид поперек оси
а, = е И/2л(1 -N). (3.9.39)
Поляризуемость диэлектрического эллипсоида в проводящей среде:
- вдоль оси
а = Е V/4it(N- 1), (3.9.40)
- поперек оси
а, = -Е Г/2л(1 + N). (3.9.41)
Поляризуемости проводящей сферической частицы в проводящей среде отвечает формула:
а = (Е V/ 4л)(/7/' -!)/([! +(f//'-l)]. (3.9.42)
Здесь ff — проводимость среды и дисперсной фазы соответственно.
Поляризуемость сферической частицы с двойным электрическим слоем [40]:
а = —(1/2) е а3 [1 - 3Rel / (1 + Rel)]. (3.9.43)
Здесь Rel — приведенное отношение поверхностной (f) и объемной (/) проводимости: Rel =f I af. При больших величинах безразмерных потенциалов плотного слоя (4х! >1) и плоскости скольжения (С > 1), равенстве подвижностей ионов в диффузном слое и в растворе для Rel справедливо уравнение:
Rel = [exp (Й7! /2) + 3 X exp (Q/2)]/xa. (3.9.44)
Здесь
Х = кТеа/4ле2, (3.9.45)
х — параметр Дебая раствора электролита, к — постоянная Больцмана, е — элементарный заряд, ?i и — безразмерные формы потенциалов, обозначаемых теми же символами, но без черты.
Формула (3.9.43) не учитывает концентрационной поляризации, связанной с накоплением ионов возле полюсов поляризации.
При малых величинах указанных потенциалов вкладом ДЭС в поляризацию можно пренебречь: при ка » 1 Rel —* 0, и формула (3.9.44) переходит в формулу для диэлектрической сферической частицы в проводящей среде.
Из приведенных выше формул видно, что легче всего поляризуются частицы электропроводного вещества (металла в частности) в диэлектрической непроводящей среде и, следовательно, суспензии металлов должны иметь наибольшую склонность к самопроизвольной поляризации, т. е. к появлению у них сегнетоэлектрических свойств. Как уже отмечалось в комментарии к формуле (3.9.29), для этого должно выполняться условие па > 3. Так как концентрация частиц п есть величина порядка <р / а3, то в суспензиях металлов, согласно формуле (3.9.37), указанное условие спонтанной поляризации сводится к неравенству <р > 1/8. Тогда суспензия металла должна превратиться в сегнетоэлектрик при объемной доле металлических частиц во взвеси ~<р > 1/е. Однако это предсказание теории не оправдывается. Более того, даже предельно концентрированные суспензии металлов в твердой среде (например парафин, канифоль и др.) ведут себя как обычные диэлектрики с умеренной величиной диэлектрической проницаемости. Разумеется, что при этом должен быть исключен гальванический контакт между частицами, поскольку при этом суспензия станет электропроводной. Следует отметить, что получить суспензию с высокой электропроводностью не менее трудно, чем обеспечить ее полное отсутствие. Для этого нужно совместить наличие хороших контактов между соседними частицами с их высокой концентрацией и равномерным распределением в диэлектрической среде. На самом деле эти требования являются взаимоисключающими, так как наличие контактов означает коагуляцию частиц (их комкование), что не позволяет достичь высокой концентрации и равномерности распределения в среде . Возможно, что сегнетоэлектрическое состояние металлических суспензий не реализуется именно потому, что не удается полностью исключить их электрическую проводимость. Ведь наличие сегнетоэлектрических свойств предполагает, что выделившиеся на некоторых поверхностях заряды не стекают за счет проводимости суспензии. В связи с этим следует обратить внимание на два обстоятельства. Первое связано с тем, что сегнетоэлектрики, как и ферромагнетики, должны иметь доменную структуру, т. е. состоять из областей микроскопических размеров, в пределах которых суспензия поляризована (намагничена) однородно. Поляризация соседних областей при этом различна по направлению. В ферромагнетиках по обе стороны междоменной границы могут сосуществовать как одноименные, так и разноименные «магнитные заряды» — полюса доменов. Очевидно, что в электрических аналогах ферромагне
* См. подразделы, посвященные структурированию и расслоению суспензий.
Физика, химия и технология дисперсных систем
653
тиков — сегнетоэлектриках — сосуществование на одной границе свободных разноименных зарядов невозможно и, следовательно, ставится под сомнение возможность реализации доменной структуры сегнетоэлектрика. В случае связанных зарядов такая возможность сохраняется, но это означает, что дисперсная фаза должна быть диэлектрической, т. е. на основе металлической суспензии создать сегнетоэлектрик невозможно. Второе же обстоятельство связано с тем, что стекание свободных зарядов с полюсов доменов требует времени (время релаксации), определяемого электрической проводимостью суспензии. Это значит, что некоторое время после поляризации суспензии она остается сегнетоэлектриком, и тогда ее фактические электродинамические свойства будут зависеть от частоты электрического поля, а именно — с увеличением частоты диэлектрическая проницаемость должна увеличиваться. Экспериментальные данные свидетельствуют о существовании гетерогенных диэлектриков и с таким, и с иным характером частотной зависимости (дисперсии) диэлектрической проницаемости, в том числе с максимумом при определенной частоте, который, может иметь и совершенно другую природу. Уменьшение проницаемости, обусловленное электрической проводимостью материалов, характеризуют величиной их диэлектрических потерь или мнимой частью диэлектрической проницаемости, являющейся в таких случаях комплексным числом.
Установление связи между взаимодействием молекул, строением и свойствами вещества (в том числе электрическими) является фундаментальной задачей молекулярной физики. Она относительно успешно решается методами статистической механики при небольших концентрациях молекул (частиц) и центральном, характере их взаимодействия. Центральные силы— это силы, не зависящие от ориентации молекул. Диполь-дипольное взаимодействие к их числу не относится, и поэтому такие эффекты, как превращение полярного вещества в сегнетоэлектрик и другие явления в полярных веществах, пока еще не нашли исчерпывающего объяснения современной физикой. Родственная проблема — объяснение ферромагнетизма веществ — существовала в физике магнитных явлений. На атомно-электронном уровне она нашла решение [17] благодаря открытию специфического обменного взаимодействия спинов непарных электронов незаполненных внутренних электронных оболочек некоторых атомов (Fe, Со, Ni и др.). Это взаимодействие выстраивает спины непарных электронов параллельно, что и исчерпывает проблему. В мире электрических явлений такого аналога нет, и поэтому при решении задачи описания электрических свойств полярных веществ можно использовать только классические кулоновские силы (включая дипольные). Разумеется, что они не сводятся к сегнетоэлектричеству.
Можно также добавить, что и экспериментальные методы изучения роли дипольных взаимодействий не
отличаются надежностью, что не способствует развитию теории. В то же время имеются уникальные по своим свойствам объекты, которые как будто специально созданы для экспериментальных исследований влияния дипольных взаимодействий на свойства веществ, — это коллоидные растворы ферритов (известные как «магнитные жидкости»). Каждая частичка такого раствора — это микроскопический магнитный диполь. Взаимодействие таких диполей полностью эквивалентно взаимодействию электрических диполей, поскольку обменное взаимодействие между магнитными коллоидными частицами отсутствует. Кстати, их электрические аналоги — сегнетоэлектрические жидкости — до сих пор не созданы и, по-видимому, не могут быть созданы. Таким образом, магнитные жидкости являются пока единственной удобной моделью для изучения общих свойств полярных веществ. Они также представляют собой уникальные по свойствам материалы для решения ряда проблем машино- и приборостроения, медицины, экологии и т. д. Суспензии и порошки магнитных материалов являются основой производства магнитных носителей информации, магнитных сердечников, постоянных магнитов. Свойства магнитных частиц широко используются в процессах обогащения железной руды. Они в значительной мере определяют географию магнитных полей Земли, на них часто ссылаются при «научных» объяснениях эффектов «омагничивания» воды или лечебного действия магнитных полей и т. д. В связи с этим изучение магнитных свойств веществ, малых частиц и магнитных коллоидов является важной составной частью современной коллоидной химии.
3.9.3. Магнитное поле и магнетизм веществ
Законы электро- и магнитостатики во многом сходны, однако есть и принципиальное различие. Оно обусловлено тем, что не существует магнитных зарядов, и поэтому определение понятия «магнитное поле» не может базироваться на соотношениях типа закона Кулона, который через силу взаимодействия зарядов позволяет определить суть понятия «напряженность электрического поля».
В качестве соотношения, определяющего смысл и количественную меру интенсивности магнитного поля— магнитной индукции В, — можно использовать выражение для силы Лоренца F. Эта сила, согласно формуле (3.9.46), действует только на движущийся заряд Q и зависит от скорости его движения v:
F = 0[vB]. (3.9.46)
Здесь и далее полужирным шрифтом обозначены векторные величины и операторы над ними. В прямые скобки заключены векторные произведения. Численное значение векторной величины, не отражающее ее векторных свойств, будет обозначаться тем же символом, но набранным курсивом.
654
Новый справочник химика и технолога
Для наглядного изображения магнитного поля, как и электрического, используются его силовые линии. Это линии, направление которых совпадает с направлением силы, действующей на положительный заряд. Густота линий качественно характеризует интенсивность поля. Магнитная индукция В, как и скорость, является векторной величиной. Сила также является вектором, а взаимная ориентация этих трех векторов определяется правилом левой руки (точнее — ладони). Если ладонь ориентирована так, что силовые линии магнитного поля входят в ладонь, а вытянутые пальцы указывают направление движения положительного заряда, то отставленный большой палец левой руки укажет направление действующей на заряд силы. Более практично и эквивалентно индукция определяется как сила, действующая на единицу длины проводника, по которому течет ток с силой 1 А, при взаимно перпендикулярной ориентации всех трех векторов: силы, тока и индукции (рис. 3.61) по тому же правилу левой руки.
Измеряется индукция в теслах (Тл). Индукция поля равна 1 Тл, если при движении в нем единичного заряда (1 Кл) со скоростью 1 м/с на него будет действовать сила, равная 1 Н. Эта сила называется амперовой. Такое определение требует предварительного введения понятия «электрический ток», меры его интенсивности и его направления. Электрический ток — это направленное перемещение зарядов.
За направление тока принимается направление движения положительных зарядов, хотя в металлических проводниках фактически это перемещение электронов в противоположном направлении.
Мерой его интенсивности является величина тока — количество электричества (величина заряда), перемещаемого за единицу времени (размерность Кл/с).
Единицей измерения силы тока служит ампер (1 А = 1 Кл/с). Ток в сплошной среде удобнее характеризовать его плотностью J — количеством электричества, перемещаемого за единицу времени через единицу площади, ориентированной перпендикулярно к направлению тока в проводящей среде (размерность — А/м2).
Величина силы Ампера определяется формулой:
F=[JB]. (3.9.47)
Так как плотность тока J = qv, то выражения (3.9.46) и (3.9.47) эквивалентны.
Рис. 3.61. Сила F, действующая на проводник с током положительных зарядов v+ в магнитном поле с индукцией В
Проводники с током являются не только средством обнаружения и измерения магнитного поля, но и средством его создания, т. е. всякий ток порождает магнитное поле. Бесконечно длинный прямой проводник с током создает вокруг себя неоднородное магнитное поле, индукция которого зависит как от силы тока, так и от свойств среды, в которой находится проводник, — от ее абсолютной магнитной проницаемости ца, а также от расстояния г до проводника: В = ца(Но / 4тг)2У / г. Здесь Цо = 4тс • 10-7 Гн/м — магнитная постоянная. При описании магнитных полей, создаваемых с помощью проводников с током (катушками, торами и т. д.), удобнее использовать другую меру интенсивности поля — его напряженность Н. Она связана с индукцией соотношением:
Я=цроЯ. (3.9.48)
Оно по форме аналогично соотношению (3.9.6) между напряженностью и индукцией электрического поля. Однако аналогом напряженности электрического поля является индукция, а не напряженность магнитного поля.
Напряженность магнитного поля не зависит от свойств вещества, в котором расположены источники магнитного поля — проводники с током, так что возле бесконечного проводника с током она описывается формулой:
H=H2nr. (3.9.49)
Силовые линии магнитного поля в этом случае представляют собой концентрические окружности, охватывающие проводник.
Среди других конфигураций проводников с током представляет особый интерес единичный виток (круговой ток, рис. 3.62) с радиусом R. Любой контур с током, в том числе круговой виток, представляет собой магнитный диполь. Его характеристикой является магнитный момент т, величина которого определяется площадью А, огороженной контуром, и силой тока / в контуре:
т = 1А. (3.9.50)
При этом ось диполя совпадает с осью витка с током. Если контур содержит N витков провода и плотность тока в проводнике i\, то z = Мц. Поскольку число витков является безразмерной величиной, то размерности величин i и Nix одинаковы, но последняя называется числом «ампервитков» в контуре (катушке).
Напряженность магнитного поля на оси витка с током на расстоянии X от плоскости витка описывается формулой:
H = 2m/4n(R2+X2f2. (3.9.51)
Здесь R — радиус витка.
Физика, химия и технология дисперсных систем
655
В плоскости витка (Х= 0) поле направлено перпендикулярно плоскости витка. Так как площадь витка А = и т = /А, то в центре витка (А"= 0) напряженности магнитного поля отвечает уравнение:
ZZ = Z/2R. (3.9.52)
На большом расстоянии г от витка (/? X) для напряженности поля на его оси (г = X) справедлива формула:
Н=2т/4пг3. (3.9.53)
Виток с током в последнем случае можно рассматривать как диполь точечных размеров. Ось диполя совпадает с осью контура с током.
В произвольной точке пространства, расположенной на расстоянии г от центра витка, напряженность поля описывается формулой:
Н= т (3cos20 + 1)1/2 / 4лЛ (3.9.54)
Здесь 0 — угол между осью диполя и направлением луча, проведенного из центра витка к заданной точке (рис. 3.62). В этом случае индукции магнитного поля соответствует формула:
В - ЦаРо т (3cos20 + 1)1/2 / 4 л г3. (3.9.55)
Следует обратить внимание на то, как учитывается магнитная проницаемость в этой формуле и диэлектрическая проницаемость в аналогичной формуле (3.9.23) для напряженности поля электрического диполя.
Направление поля можно определить, располагая формулами для напряженности (или индукции) его продольной составляющей Нр (3.9.56) и поперечной составляющей Н, (3.9.57):
Нр = т (3cos2O - 1) / 4 яг3, (3.9.56)
Ht=3 т sin© cos©/4 л г3. (3.9.57)
Первая направлена вдоль оси диполя, а вторая перпендикулярна ей. Тогда направление поля в данной точке с полярными координатами г и 0 можно характеризовать тангенсом угла а между направлением поля и направлением оси диполя или косинусом того же угла (формулы (3.9.58) и (3.9.59)) или другой удобной величиной:
tga = (3cos20 - 1) / 3sin0cos0, (3.9.58)
cosa = (3cos20 - 1) / (3cos20 + 1)’/2. (3.9.59)
В соленоиде — длинной катушке с током — создается однородное магнитное поле. Его напряженность одинакова во всех точках внутри соленоида, удаленных от его концов, и описывается формулой:
Н=п1. (3.9.60)
Рис. 3.62. Магнитный момент m витка с радиусом R, по которому течет ток с силой /, и его магнитное поле с напряженностью Н
Здесь п — число витков проводника, приходящихся на единицу длины соленоида. На концах соленоида напряженность вдвое меньше.
В однородном магнитном поле на магнитный диполь действует механический момент сил М, стремящийся совместить ось диполя с направлением поля. Его величина определяется формулой:
М = [тВ]. (3.9.61)
Механический момент также является вектором. Его направление совпадает с направлением оси вращения магнитного момента.
В неоднородном поле на диполь действует сила, величина и направление которой зависят как от ориентации диполя, так и от направления поля и его градиента. В таких случаях полную силу F, действующую на диполь, удобно представлять ее компонентами Fx, Fy, Fz, параллельными соответствующим осям координат (например Декартовых: х, у, z). Величина каждого компонента, например Fx, определяется градиентом
gradBx = (dBx Idx + dBx /dy + dBx /dz)
одноименной составляющей Bx индукции поля:
Fx = m • gradBx. (3.9.62)
Жирная точка означает скалярное произведение вектора магнитного момента m на каждый компонент градиента поля gradBx.
В однородной по магнитным свойствам среде, в которой проницаемость Ца не зависит от пространственных координат, справедлива формула:
Fx = ЦаЦо ш • grad/Zj. (3.9.63)
Аналогично выглядят формулы для остальных компонентов силы.
Под действием суммарной силы (формула (3.9.64)) диполь втягивается в область максимальной напряжен
656
Новый справочник химика и технолога
ности поля, если его ось совпадает (хотя бы частично) с направлением поля и выталкивается при обратной ориентации диполя:
F = FX+F, + FZ. (3.9.64)
Если диполь имеет возможность ориентироваться параллельно направлению действующего на него поля, то он будет втягиваться в область максимальной напряженности поля и с максимальной силой.
Полная магнитостатическая энергия магнитного момента т во внешнем магнитном поле (формула (3.9.65)) включает в себя как работу перемещения, так и работу ориентации диполя, и зависит, кроме всего прочего, от угла 0 между направлениями осей диполя и магнитного поля (рис. 3.63):
Um = mBcos&. (3.9.65)
Магнитный диполь ориентируется вдоль магнитного поля, так что 0 = 0 и cos© = 1. Выражение mBcos© обозначает скалярное произведение векторов ш и В.
Понятие о магнитно-неоднородной сплошной среде приобрело практический смысл только после создания магнитных жидкостей, поскольку это коллоидные растворы ферритов и они могут претерпевать некоторое расслоение, особенно в сильно неоднородных магнитных полях. В дальнейшем предполагается, что среда (магнетик), в которой создается и действует магнитное поле, является однородной. Она, как уже отмечалось выше, характеризуется магнитной проницаемостью ц, которую можно считать магнитным аналогом диэлектрической проницаемости е.
Соответственно, аналогом электрической поляризуемости веществ является их магнитная восприимчивость х, которая связана с магнитной проницаемостью формулой:
р=1+х. (3.9.66)
Аналогом электрической поляризации вещества является его намагниченность М.
М = кН.
(3.9.67)
Связь магнитной индукции и намагниченности подобна связи между электрической индукцией и поляри-зованностью. Она отражена в формуле (3.9.68), которая получается при объединении уравнений (3.9.66) и (3.9.67):
В = ^(Н+М).
(3.9.68)
Рис. 3.63. Магнитный момент m в магнитном поле с индукцией В
Эти и другие соотношения можно вывести аналогично соответствующим формулам электростатики. По мере необходимости они будут использованы применительно к магнетикам без дополнительных доказательств. При этом следует помнить, что свободных магнитных зарядов не существует, и первичным свойством любого вещества является его намагниченность М, а на атомно-молекулярном уровне — величина магнитного момента т атома или молекулы или коллоидной частицы.
Аналогично электрическим диполям, магнитные диполи могут быть индуцированы воздействием внешнего магнитного поля, а могут быть постоянными, т. е. существующими и в отсутствие внешнего поля. Возникновение тех и других обусловлено молекулярным или атомарным круговым электрическим током (движением электронов по орбитам) или ориентацией электронных магнитных моментов (электронных спинов). Сильное проявление магнетизма веществ связоно с наличием магнитного момента (спина) у электронов. При нечетном числе электронов в электронной оболочке атома он становится постоянным магнитным диполем, а соответствующее вещество — парамагнитным. В дальнейшем будут в основном рассмотрены растворы (дисперсные системы), в которых носителями магнитных свойств являются дискретные элементы (атомы, ионы, молекулы, коллоидные частицы), обладающие постоянным магнитным моментом т. Вне поля они ориентированы хаотично, и вещество не намагничено. Во внешнем поле возникает преимущественная ориентация магнитных моментов вдоль приложенного поля, и вещество в целом намагничивается (рис. 3.64).
Рис. 3.64. Намагничивание ансамбля постоянных магнитных диполей в поле с индукцией В
В сильном поле может наступать насыщение намагниченности. В таком состоянии магнитные моменты всех частиц (молекул, ионов) ориентированы параллельно приложенному полю. Намагниченность насыщения вещества Мх, т. е. магнитный момент единицы объема вещества, равна сумме магнитных моментов т всех п элементарных носителей магнетизма, присутствующих в единице объема:
Ms = nm. (3.9.69)
Намагниченность Л/парамагнитных веществ во внешнем поле составляет некоторую малую долю ЦрупН / кТ) от намагниченности насыщения Ms (формула (3.9.70)). Насыщению препятствует дезориентирующее воздей
Физика, химия и технология дисперсных систем
657
ствие вращательного теплового движения (вращательной диффузии) магнитных диполей:
М = Ms Ц^тН I kT). (3.9.70)
Величина L(pomH I kT) представляет собой упоминавшуюся ранее функцию Ланжевена (рис. 3.65), а выражение в скобках — это аргумент функции Ланжевена. Обычные парамагнетики в любых технически достижимых полях далеки от состояния насыщения. Для них значение аргумента (ротН / kT) 1, поэтому справедливо соотношение:
Ц^тН/кТ) ~ \^тН! ЗкТ. (3.9.70а)
Тогда формулу (3.9.70) для намагниченности можно заменить формулой:
М= М^тН / ЗкТ. (3.9.71)
Для магнитной восприимчивости х, определяемой как отношение намагниченности М к напряженности Н действующего на вещество внешнего поля, можно записать формулу:
к = М^т13кТ. (3.9.72)
Специфическое обменное взаимодействие непарных электронных спинов соседних атомов твердых веществ (как правило, кристаллических) способно установить преимущественно параллельную ориентацию всех атомных моментов вещества и превратить его в ферромагнетик — вещество, самопроизвольно намагниченное до насыщения. Основной магнитной характеристикой таких веществ является их намагниченность насыщения Ms — магнитный момент единицы объема вещества.
Направление самопроизвольного намагничивания вещества более или менее жестко связано с кристаллографическими осями его кристаллической решетки. Жесткость этой связи характеризуется константой магнитной анизотропии вещества К. Она численно равна удельной (на единицу объема) энергии, необходимой для изменения направления намагниченности на противоположное (при наличии одной оси самопроизвольного намагничивания) при действии поля, направленного противоположно намагниченности вещества. Напряженность поля, способного изменить намагниченность на обратную, называется коэрцитивной силой вещества Нс. По порядку величины К = MSHC.
В силу поликристалличности большинства веществ направления самопроизвольной намагниченности отдельных кристаллических областей не совпадают, и поэтому образец вещества в целом не намагничен. В монокристаллических образцах размер областей однородной самопроизвольной намагниченности также ограничен. Эти области называются доменами. Они имеют микроскопические размеры (порядка 10 5 м). Доменная структура ферромагнетика такова, что она
обеспечивает минимум энергии взаимодействия каждой из намагниченных областей вещества с магнитным полем, создаваемым всеми другими областями. Своеобразная доменная структура возникает в тонких магнитных пленках. В частности, это могут быть регулярные полосы макроскопических размеров с противоположным направлением намагниченности соседних полос. Ширина полос составляет около 1 мкм. Под влиянием различных факторов (например формы образца, размеров и формы частиц дисперсного ферромагнетика, механических напряжений и др.) направление самопроизвольного намагничивания доменов, пленок, нитей и дисперсных частиц может не совпадать с направлением кристаллографических осей в домене (частице). В частности, в веществах с малой величиной энергии кристаллографической анизотропии определяющую роль играет форма частиц — вектор самопроизвольного намагничивания располагается вдоль длинной оси частицы. Направление самопроизвольной намагниченности частицы (пленки, домена) называется «легкой осью»
Намагничивание ферромагнетика сводится к вращению направления самопроизвольной намагниченности доменов относительно легкой оси и к движению междоменных стенок. В последнем случае размер доменов, намагниченность которых совпадает с направлением действующего поля, увеличивается за счет уменьшения размеров соседних доменов с неблагоприятным направлением намагниченности. При достаточно большой коэрцитивной силе материала однородная намагниченность всего образца может сохраниться и при выключении внешнего намагничивающего поля, т. е. образец становится постоянным магнитом. При малой коэрцитивной силе вещества (магнитно-мягкий материал) намагниченность уменьшается вплоть до нуля вместе с уменьшением интенсивности намагничивающего поля.
При измельчении ферромагнитного вещества достаточно легко достигается такой размер частиц, при котором каждая из них становится единичным доменом, т. е. превращается в миниатюрный постоянный магнит со всеми вытекающими отсюда последствиями. Эти последствия легко себе представить, если взять в руки два постоянных магнита (лучше в виде пластин, шайб или полос, но не подков) и попытаться манипулировать ими. Достаточно сильное взаимодействие постоянных магнитов ощущается при расстояниях между ними по
658
Новый справочник химика и технолога
рядка размера самих магнитов. Это правило сохраняет силу и для магнитных коллоидных частиц. В магнитных коллоидах это радикально меняет соотношение действующих между частицами сил по сравнению с обычными коллоидами, у которых радиус действия сил на один-два порядка меньше размера частиц. Максимальный размер однодоменных частиц магнитножестких (высококоэрцитивных) материалов может превышать 1 мкм, тогда как в случае магнитно-мягких материалов он существенно меньше.
3.9.4. Дисперсные ферромагнетики
Особенности магнитных дисперсных систем в полной мере проявляются тогда, когда каждая из частиц дисперсной системы является единичным доменом. Однодоменные частицы всегда однородно намагничены до насыщения. Для магнитного момента однодоменной частицы справедлива формула:
m=VMs. (3.9.73)
Здесь V — объем частицы. Следует отметить, что ферромагнетизм является результатом взаимодействия большого числа электронных спинов и потому он исчезает, если частицы становятся слишком малыми. Для ферромагнитных металлов (Fe, Ni, Со) минимальный размер ферромагнитных частиц составляет около 1 нм (т. е. около одной тысячи атомов металла в одной частице). Для ферритов он в несколько раз больше. Например, для магнетита это около 4 нм. Таким образом, размер однодоменных частиц ограничен как сверху, так и снизу.
Основной характеристикой магнитной дисперсной системы является ее намагниченность насыщения М*. Она может быть выражена, как и намагниченность парамагнитного вещества (формула (3.9.69)), через магнитный момент т одной частицы и численную концентрацию частиц п или равноправным образом через объемную долю дисперсного ферромагнетика ср в системе и его намагниченность насыщения Ms:
М* = пт- фЛ^. (3.9.69а)
Если пренебречь магнитным взаимодействием частиц коллоидного ферромагнетика, то взвесь однодоменных частиц можно рассматривать как парамагнитное вещество, т. е. намагниченность взвеси будет описываться уравнением (3.9.70). Существенно, что вследствие большой величины магнитного момента коллоидных частиц аргумент \х^тН / кТ функции Ланжевена даже в слабых полях может стать много больше единицы, и поэтому взвесь легко намагничивается до насыщения. Подчеркивая эту особенность парамагнетизма магнитных коллоидов, их называют суперпарамагнетиками. Один из самых доступных ферромагнитных материалов — магнетит (ЕезОД Его намагниченность насыщения 0,47 • 10б А/м. При размере частицы Ю^м (объеме около 4 • 10“18 м3) ее магнитный момент равен
2 • 1012 А • м2. Тогда в магнитном поле с напряженностью 1 А/м аргумент функции Ланжевена уц}тН/кТ -= 1,2 • Ю-6 • 2 • 10-12/4 • 10 21 = 1000. Намагниченность насыщения взвеси частиц в атмосфере (или выше) с концентрацией 0,1 об. % равна 470 А/м, а начальная магнитная восприимчивость х (восприимчивость в очень слабом поле) достигает, согласно формуле (3.9.72), величины 1,6- 105. Это значение сравнимо с восприимчивостью лучших магнитных материалов. Такая взвесь будет интенсивно поглощать электромагнитные волны, так что несколько тонн распыленного в воздухе ферритового порошка на кубический километр делают зону запыления непроницаемой для радиоволн, а объекты в ней — невидимыми при радиолокации.
Реализуемость такого эксперимента зависит от возможности преодолеть сильное магнитное дипольное притяжение частиц на этапе распыления порошка. Грубая оценка прочности магнитного сцепления частиц размером около 1 мкм дает величину энергии связи порядка ц0Л/2К = 10“12Дж . Это значит, что для разъединения частиц порошок нужно встряхивать с частотой 106 Гц и амплитудой около 1 см или нагревать до температуры 109К. Очевидно, что ни то, ни другое не реально. Этот пример приведен для того, чтобы дать представление о порядке величин, характеризующих силы магнитного взаимодействия частиц, и об уровне технических и технологических проблем, которые нужно решать при работе с дисперсными магнетиками, например при производстве носителей для магнитной записи информации (магнитные ленты, диски и др.). Для того, чтобы наметить рациональные пути решения указанных проблем, необходимо детальнее рассмотреть закономерности дипольного взаимодействия частиц.
В общем случае энергия дипольного взаимодействия двух частиц может быть, согласно формуле (3.9.65), представлена как скалярное произведение дипольного момента т, одной частицы и индукции поля В}, создаваемого другой частицей в месте нахождения первой:
U = -/n^cos0(mB). (3.9.74)
Это в равной мере относится как к магнитным, так и к электрическим диполям, однако для конкретности далее речь будет идти о магнитных диполях и магнитных полях. Символ типа 0(тВ) здесь и далее означает угол между соответствующими векторами. Относительная магнитная проницаемость среды, в которой взвешены частицы, принимается равной единице, так что Bj = роНу, где Hj вычисляется по формуле (3.9.54). Знак «-» указывает, что предоставленные сами себе частицы будут сближаться при их магнитном взаимодействии.
При произвольном взаимном положении двух частиц (рис. 3.66) направления их магнитных осей и полей партнеров по взаимодействию (соседних частиц) не совпадают.
На рис. 3.66 показана ситуация, в которой ось одного диполя (wy) совпадает с направлением поля В, парт
Физика, химия и технология дисперсных систем
659
нера, а ось второго диполя (м,) не совпадает с направлением поля партнера Bj. Согласно формуле (3.9.61), в этой ситуации его ось будет поворачиваться до совпадения ее направления с направлением поля, т. е. до состояния, когда 0(т/Ву) = 0. Однако такая взаимная ориентация диполя и локального поля оказывается неустойчивой. Ведь обе частицы равноправны, и каждая из них должна рассматриваться и как объект, на который воздействует поле партнера, и как источник этого поля. При совпадении направлений векторов т,- и В; такое совпадение, согласно формулам (3.9.58) и (3.9.59), будет отсутствовать у векторов ту и Bf. Это вызовет сближение направлений последних, что нарушит параллельность первой пары векторов и снова повлечет за собой их последующую подстройку. Этот процесс взаимного согласования направлений двух диполей продолжается до тех пор, пока не будет найдено их устойчивое состояние. Ему соответствует минимум потенциальной энергии взаимодействия U двух диполей (как функции углов ©j и ©2 на рис. 3.66). Оказывается, что существует два устойчивых состояния системы из двух диполей (рис. 3.67). Одно соответствует значениям ©! = 0 и О2 = 0 (первое Гауссово положение), а второе — 0, = л/2 и ©2 = -л/2 (второе Гауссово положение). В первом состоянии оси обоих диполей лежат на линии, соединяющей центры диполей (линии центров) и направлены в одну сторону, а во втором — перпендикулярно линии центров и ориентированы антипараллельно один другому (параллельно, но в противоположных направлениях).
Соответственно, в первом состоянии потенциальная энергия взаимодействия двух дипольных частиц описывается формулой:
£/=-Цо2т2/4тгг3, (3.9.75)
а во втором — формулой:
и=-^т2/4лг3. (3.9.76)
Очевидно, в первом состоянии эта энергия вдвое больше (по модулю), чем во втором. Если при этом ничто не мешает частицам сближаться, то такой процесс будет продолжаться до объединения пары частиц в одну составную частицу (двойник). Тогда расстояние г между частицами станет равным диаметру частиц 2а. Потенциальная энергия дипольного взаимодействия частиц в двойнике достигнет абсолютного минимума (при параллельной взаимной ориентации диполей):
м1, /3. (3.9.77)
При антипараллельной ориентации она по абсолютной величине вдвое меньше. Однако это не означает, что параллельная ориентация во всех случаях предпочтительнее антипараллельной. Дело в том, что в реальных условиях двойники не изолированы от взаимодействия с другими частицами или с внешним магнитным
полем. Энергия этого взаимодействия во многих случаях определяется величиной дипольного момента двойника. При параллельной ориентации диполей в их паре дипольный момент этой пары равен сумме магнитных моментов этих диполей, т. е. величине 2т при одинаковом размере частиц. При антипараллельной ориентации диполей результирующий магнитный момент пары равен нулю. Соответственно этому во внешнем однородном поле или в локальном поле соседней частицы с индукцией В потенциальная энергия двойника первой конфигурации может изменяться в пределах от -2тВ до +2тВ в зависимости от взаимной ориентации поля и магнитного момента двойника, а во втором она всегда равна нулю.
В?
Рис. 3.66. Ориентация взаимодействующих магнитных диполей неустойчива при любых значениях углов, кроме ©1 = 02 = 0, ©! = 02 = л/2 0©
О© >
Рис. 3.67. Две устойчивые конфигурации дипольного двойника (квадруполя)
Образование двойников является простейшим примером структурирования магнитной дисперсной системы. Геометрически двойники первого и второго вида неразличимы, но по свойствам они отличаются радикально. Это различие легко обнаруживается по магнитной восприимчивости суспензии однодоменных частиц. Выше уже приводилась численная оценка магнитной восприимчивости взвеси однодоменных частиц. Она настолько высока, что взвесь должна будет намагнититься до насыщения даже в слабом магнитном поле Земли. При образовании двойников с параллельными магнитными моментами, в соответствии с формулой (3.9.72), магнитная восприимчивость взвеси должна стать в два раза больше по сравнению с восприимчивостью взвеси индивидуальных магнитных диполей. В действительности магнитная восприимчивость взвеси в слабом однородном поле на много порядков ниже расчетной величины [5]. Это означает, что преобладающий тип структуры двойников — антипараллельная ориентация магнитных моментов пары частиц. Равенство нулю величины магнитного момента двойников и более крупных блоков частиц объясняет почти нулевую величину фактической магнитной восприимчивости взвеси.
Процессы сближения и взаимной ориентации однодоменных частиц под влиянием их магнитно-диполь
660
Новый справочник химика и технолога
ного взаимодействия не завершаются образованием двойников. Двойники слипаются в агрегаты из четырех частиц (в тетрады, рис. 3.68), тетрады слипаются в октеты и т. д. Образуются и агрегаты промежуточных размеров. Важно, что вторая ступень коагуляции приводит к одному и тому же результату — образованию тетрад с конфигурацией квадрата (или тетраэдра) и нулевой величиной магнитного момента, как при коагуляции двойников первого, так и при коагуляции двойников второго вида. Структурно такая тетрада в обоих случаях может рассматриваться как результат антипа-раллельного соединения двух двойников первого вида, хотя фактически она могла возникнуть и при слипании двух двойников второго вида. Есть еще одна относительно устойчивая конфигурация тетрады — линейная цепь из четырех частиц с ориентацией моментов всех частиц параллельно оси цепи (рис. 3.68), т. е. с магнитным моментом, равным 4w. Она менее устойчива, т. е. ее магнитостатическая энергия выше, чем у тетрады с конфигурацией квадрата и нулевой величиной магнитного момента. В этом легко убедиться путем расчетов потенциальной энергии разных тетрад в дипольном приближении. На последующих этапах коагуляции (образования октетов и далее) взвесь «не помнит» о своем состоянии на первом этапе коагуляции и потому не имеет значения, каким оно было на самом деле. Важно лишь то, что структура коагулята, наследуя в себе основную характеристику устойчивых тетрад — нулевую величину их магнитного момента, — будет иметь примерно ту же величину магнитной восприимчивости взвеси, что и взвесь двойников с антипараллельной ориентацией магнитных моментов пары частиц.
©О 000© О©
Рис. 3.68. Относительно устойчивые конфигурации тетрад
Приведенные выше соображения о структуре флокул коагулята относятся к случаю достаточно сильного магнитно-дипольного взаимодействия частиц ((/а » kT), а оценка магнитной восприимчивости — к слабым магнитным полям. Последнее означает, что напряженность Не внешнего поля намного меньше напряженности локального поля HL, создаваемого на частице всеми ее соседями Z. В случае двойников r = 2a, Z- 1, и напряженность локального магнитного поля достигает, согласно формулам (3.9.54) и (3.9.74), величины порядка М- В общем случае при сильном взаимодействии Z 4- 1 частиц справедливо соотношение:
HL ~ ZM- (3.9.78)
Локальное поле как фактор, определяющий поляризуемость вещества, введено в теоретическую физику Дебаем в его теории поляризации жидких диэлектри
ков. Ее полное и систематичное изложение было дано Френкелем [31]. Далее основные идеи этой теории излагаются в применении к магнитным суспензиям и их магнитной восприимчивости.
Локальные поля имеют случайное направление, а направление магнитного момента каждой частицы совпадает с направлением этого поля. Упрощенно можно считать, что в отсутствие внешнего поля, т. е. в нена-магниченной суспензии, направления магнитных моментов всех частиц распределены поровну между тремя осями координат (г, у, z). Из трети частиц, ориентированных своими моментами вдоль какой-либо оси, половина (шестая часть всех частиц) направлена вдоль этой оси, а другая половина имеет встречное направление и каждая из них равна М* / 6. Намагниченность суспензии М' является вектором, который может быть разложен на параллельные осям координат компоненты М*х, М*, М*. Каждая из трех компонент намагниченности и их векторная сумма М' в ненамагниченной суспензии равны нулю. Систему координат можно выбрать так, чтобы одна из координатных осей, например ось z, совпадала с направлением внешнего поля. Тогда процесс намагничивания в этом поле в его начальной стадии (в слабом поле) сведется к изменению ориентации тех двух третей всех частиц, которые были ориентированы своими магнитными осями вдоль координат х и у. Направление осей магнитных диполей должно совпадать с направлением действующего на них суммарного поля с напряженностью Н = Не + Нг Нетрудно установить, что при наличии слабого внешнего поля синус угла между направлением суммарного поля и первоначальной ориентацией осей частиц приближенно равен отношению Не / HL. Этой же величине равен косинус угла между направлением внешнего поля и новым направлением ориентации упомянутых двух третей всех частиц, поэтому намагниченность суспензии М" (проекция магнитных моментов всех частиц на направление внешнего поля) равна (2/3) М* (Не / HL). Магнитная восприимчивость, экспериментально определяемая как величина М*/ Не, равна в таком случае (2/3)Л/*/ HL. Это один из очевидных результатов, вытекающих из теории локального поля Дебая. Если учесть, что HL = ZMS и М* = <pMs, то получается формула для магнитной восприимчивости флокулированной взвеси однодоменных частиц:
х = 2<p/3Z. (3.9.79)
Здесь (р — объемная доля ферромагнитного материала во взвеси.
Соотношение (3.9.79) дает возможность экспериментально исследовать одну из важнейших структурных характеристик дисперсной системы Z = 2(р / Зх — среднее число ближайших соседей во флокулах коагулированной суспензии. Величина х / (р, обратнопропорциональная Z, является удельной (на единицу' объема
Физика, химия и технология дисперсных систем
661
магнитной фазы) восприимчивостью взвеси. Измерения восприимчивости суспензий гексаферрита бария и у-ок-сида железа дали вполне ожидаемые результаты — с увеличением концентрации удельная восприимчивость монотонно уменьшается, следовательно, обратная ей величина <р / х, равная приближенно среднему числу соседей, увеличивается (рис. 3.69).
Действительно, при стремящейся к 0 концентрации частиц дисперсной фазы суспензии их коагуляция отсутствует. Поэтому среднее число соседей должно стремиться к нулю, а восприимчивость — к бесконечности. Следует отметить, что при измерении магнитной восприимчивости разбавленных коагулирующих суспензий необходимо непрерывное их перемешивание для предотвращения оседания флокул. Само по себе перемешивание способно разрушать флокулы, и поэтому его режим должен оставаться неизменным при изменении рецептуры суспензии. При усилении перемешивания магнитная восприимчивость взвеси однодоменных частиц проходит через максимум. Рост восприимчивости обусловлен разрушением флокул, что ведет к уменьшению среднего числа соседей и соответствующему росту восприимчивости суспензии. Дальнейшее увеличение интенсивности перемешивания после разрушения флокул до единичных частиц препятствует ориентации частиц внешним магнитным полем, т. е. препятствует намагничиванию взвеси. При размере частиц около 1 мкм этот эффект легко наблюдается в суспензиях с достаточно вязкой дисперсионной средой (глицерин, масла).
В сильном магнитном поле (Не » Н{) локальные поля вносят малый вклад в суммарное поле Н, действующее на частицы, так что структура взвеси фактически полностью определяется внешним полем Не. Магнитные моменты всех частиц ориентируются при этом параллельно вектору внешнего поля, поэтому энергия взаимодействия частиц в сильном поле определяется только той компонентой Нр (уравнение (3.9.56)) локального поля частиц, которая параллельна этому полю:
U= -цот2 [3cos20(rHe) - 1] / 4 яг3. (3.9.80)
Здесь 0(гЯс) — угол между направлением линии центров двух частиц и направлением внешнего поля. Минимуму энергии взаимодействия, как функции этого угла, соответствует расположение частиц вдоль силовых линий внешнего поля. В этом случае (гН^ = 0, и формула (3.9.80) сводится к выражению:
U = -(pj^yim2 / г3. (3.9.80а)
Множитель Цо/4л необходим для обеспечения нужной размерности и порядка величин.
Частицы, расположенные вдоль направления поля, сближаются, а расположенные перпендикулярно направлению поля отталкиваются, что ведет к образованию линейных цепочек частиц, вытянутых вдоль силовых линий поля (рис. 3.70).
Рис. 3.69. Зависимость обратной величины начальной удельной магнитной восприимчивости суспензий гексаферрита бария (/) и оксида железа (2) от их концентрации
Н
Рис. 3.70. Образование цепочек из магнитных частиц в магнитном поле
При размере частиц ~1 мкм и более такие цепочки видны невооруженным глазом и поэтому визуализируют картину пространственного распределения поля. Простой опыт с железными опилками, рассыпанными по листу бумаги, и постоянным магнитом является хорошей демонстрацией этого эффекта. В однородном поле взвесь магнитных частиц образует систему параллельных цепей, которые могут иметь неограниченную длину. При размере частиц около 1 мкм невозможно приготовить устойчивый коллоидный раствор ферромагнетика из-за очень сильного магнитного дипольного взаимодействия частиц и быстрой коагуляции взвеси. Между тем представлялось очень заманчивым получить раствор, который обладал бы сильными магнитными свойствами и в то же время вел себя как однородная жидкость. Эта задача была решена в 1962 году в Технологическом институте (Санкт-Петербург). Здесь же к 1964 году были изучены и описаны основные свойства таких жидкостей — концентрированных коллоидных растворов магнетита. Позднее они получили название феррожидкостей и стали материальной основой, по крайней мере, двух новых направлений в науке и технике: физики магнитных жидкостей и феррогидродинамики.
662
Новый справочник химика и технолога
Размер частиц в феррожидкостях, а с ним и уровень их магнитного взаимодействия, на много порядков меньше, чем в суспензиях. Это и обеспечивает возможность существования устойчивой однородной взвеси магнитных частиц, но сильно осложняет количественное описание всех эффектов, связанных с взаимодействием частиц. Качественно же сохраняются все упомянутые выше структурные особенности и их зависимость от соотношения напряженностей внешнего и локального полей.
Однако определяющее значение приобретает при этом отношение Ua I кТ (энергии магнитного взаимодействия Ua к энергии теплового движения частиц кТ). В устойчивом коллоидном растворе этот параметр должен быть порядка единицы или меньше:
Х<1. (3.9.81)
Размер частиц, при котором это условие выполняется, сильно зависит, согласно формуле (3.9.77), от намагниченности насыщения Ms ферромагнитной фазы. Для магнетита и многих ферритов Ms ~ 4,7 • 105 А/м. Оценка радиуса частиц а по формуле (3.9.77) дает при этих условиях а < 5 нм. Действительный размер частиц магнетита в феррожидкостях близок к указанной величине. Что касается другого безразмерного параметра — аргумента функции Ланжевена = \щпНе / кТ во внешнем поле, то он может расти неограниченно с увеличением напряженности поля Не, приближая намагниченность феррожидкости к намагниченности насыщения. Этот же параметр, а следовательно, напряженность поля, определяет характер и силу взаимодействия частиц между собой.
Механизм воздействия внешнего поля на взаимодействие магнитных диполей связан с тем, что вращательное тепловое движение частиц вынуждает флуктуировать магнитные оси диполей вблизи направления суммарного поля Я, действующего на диполь. Средняя величина косинуса угла 0(Нш) между направлением поля Н и диполя тп и есть функция Ланжевена £(£,). В отсутствие внешнего поля при Н = HL, следовательно, Х = ^,. Поэтому при условии (3.9.81) и, согласно формуле (3.9.70а) Ц&) = \wiiHj / ЪкТ. Здесь использованы принятые выше индексированные обозначения магнитного момента т, и локального поля Н} партнеров по взаимодействию. Вращательная тепловая диффузия оси диполя /и, не слишком сильно влияет на напряженность локального поля Hj. Согласно формулам (3.9.75) и (3.9.76), она может меняться всего в 2 раза, так что в среднем можно принять, что Н} = Ьт}/4пг3, где b — средняя (1 < b < 2). Величина числового коэффициента в упомянутых формулах. Можно принять b равным его среднему геометрическому значению, т. е. b = 21/2. Если предположить, что т, = пу = /и, то £(^) = Цо Ьт / 12лг3А7'. Произведение тЦ££) является средней величиной проекции магнитного момента /и, на направление локального поля со средней напряженностью Н} = Ьт / 4лг3, а произведение этих величин — энергией диполь-диполь-
ного взаимодействия двух частиц, вовлеченных в интенсивное вращательное тепловое движение:
U = -(ро/4л) 2/и4 / ЗгвкТ. (3.9.82)
Формула (3.9.82) хорошо известна для энергии взаимодействия молекулярных диполей. Иная по сравнению с формулой (3.9.80) зависимость энергии от расстояния (1/г6 вместо 1/г3) обусловлена нарушением взаимной ориентации диполей под действием их вращательной диффузии.
В другом предельном случае, когда напряженность внешнего поля Не существенно больше напряженности локального поля, можно считать, что И - Не, и тогда значение функции Ланжевена L(^e) определяется ее аргументом = \10тНе / кТ во внешнем поле в соответствии с общим выражением (3.9.70).
Ориентация осей диполей флуктуирует возле направления внешнего поля, так что их взаимодействие между собой определяется формулой (3.9.80) с заменой в ней величины магнитного момента частиц т на среднее значение проекции магнитных моментов на направление внешнего поля, т. е. на тЬ(^е), что приводит к формуле:
U = -(цо/Дл) m27?(^)[3cos20(r/7) - 1] / г3. (3.9.83)
В пределе сильного поля Z(^) = 1 формула (3.9.83) сводится к формуле (3.9.80).
Из сравнения формул (3.9.80) и (3.9.83) следует, что увеличение напряженности внешнего поля увеличивает энергию дипольного взаимодействия частиц на несколько порядков и изменяет структуру агрегатов, которые могут возникать при магнитном слипании частиц. В отсутствие внешнего поля это произвольно ориентированные флокулы, внутри которых несколько преобладает антипараллельная ориентация соседних диполей, а в сильном внешнем поле — это линейные цепочки, намагниченные вдоль их оси и вытянутые параллельно внешнему полю. Эти изменения сказываются на магнитной восприимчивости феррожидкости качественно, так же как и при коагуляции крупных однодоменных частиц в магнитных суспензиях, но в гораздо меньшей степени.
При малой величине магнитного момента частиц уже нельзя считать, что направление их магнитных осей совпадает с направлением локального поля. Локальное поле задает направление, вокруг которого оси частиц сильно флуктуируют. Этим обусловлена отличная от нуля среднестатистическая величина проекции магнитного момента частицы на направление локального поля (локальную намагниченность), равную тЦ&). Действие слабого внешнего поля с напряженностью Не сводится к тому, что оси частиц будут вынуждены флуктуировать вокруг суммарного поля Н. Оно мало отличается по величине и направлению от локального поля, но именно малое изменение направления и средней величины проекции тЦ§ магнитных моментов
Физика, химия и технология дисперсных систем
663
частиц и представляет интерес, определяя намагниченность магнитной дисперсной системы. В рассматриваемой ситуации намагниченность дисперсной системы равна сумме проекций локальных намагниченностей частиц на направление внешнего поля:
ЛГ = М' <L($) cos ®(ННеу>. (3.9.84)
Угловые скобки < > означают операцию усреднения заключенного в них выражения, в данном случае по всем возможным ориентациям суммарного поля относительно внешнего, т. е. по углу ®(ННе). Однако на самом деле независимым параметром, который определяет величины ®(ННе), т, cos®(HHe) и Л(£), является угол а (рис. 3.71) между направлениями внешнего Не и локального HL полей, поэтому через него необходимо выразить все упомянутые величины.
Рис. 3.71. Векторы напряженности магнитных полей, действующих на однодоменную частицу: локального поля HL соседних частиц, слабого внешнего поля Не
и суммарного поля Н
Равновесная ориентация магнитного момента частицы, т. е. ориентация вдоль суммарного поля, означает равенство механических моментов сил \x^mHLs\n®(HHI) и Цо/лЯе81п0(ЯЯе), действующих на нее со стороны локального и внешнего полей соответственно. Отсюда следует формула:
#£sin = //esin 0(Ж)- (3.9.85)
Очевидно, что угол а равен сумме углов, образованных векторами напряженности суммарного и локального полей (0(НЯ£)) и суммарного и внешнего полей (0(ЯЯ<;)), т. е. а = 0(ЯЯ£) + ®(ННе). Тогда, согласно формуле косинуса суммы (разности) углов, можно получить уравнение:
cos0(7///e) = cosa cos0(#H£) + sina
(3.9.86)
Кроме того, согласно правилу сложения векторов, получается формула:
Н=(Н2 + Н2е + 2H£H(;cosa)1/2. (3.9.87)
При слабом внешнем поле, которое обычно применяется при измерении магнитной восприимчивости, HJHl 1, и формула (3.9.87) преобразуется в формулу:
Н = HL [1 + (Не/HL) cosa]. (3.9.88)
Одновременно с этим угол стремится к нулю. Тогда справедливы следующие приближения: cos®(HHL) ~ 1 и sin0(/7/7e) ~ sina, а из условия (3.9.85) получается равенство sin®(HHL) = (Не / H£)sina. В этом случае в формуле (3.9.84) cos®(HHe) можно выразить через тригонометрические функции угла а:
cos ®(ННе) = cosa + (Не / Н{) sin2a. (3.9.89)
Как уже отмечалось, значения функции Ланжевена L в локальном и суммарном поле отличаются незначительно. Это отличие можно выразить через приращение [dLldQfc - ££) локального значения Ll = f(^£) с помощью формулы:
L = LL + (dL/^-^). (3.9.90)
Приращение функции Ланжевена происходит при небольшом увеличении аргумента £ функции Ланжевена от величины S,£ = \wnHL / kT до т = \^тН / kT, т. е. на величину (цо/и / Согласно формуле (3.9.88),
Н-Н]= Hecosa, и тогда
4 - = (Цо"» / kT) 7/(;cosa = ^£ (Не / Я£) cosa.
При подстановке этого выражения в формулу (3.9.90) получается уравнение:
L = Ll + (dL I dty (He IHL) cosa. (3.9.91)
Таким образом, имеется все необходимое для записи в развернутом виде функции Ланжевена в формуле (3.9.84), подлежащей усреднению по углу а:
L(£) cos0(H#e) = [Ll + (dL I d® (He IHL) cosa] x x [cosa + (He IH/) sin2a].
Следует иметь в виду, что значение производной dL'dt относится к локальному полю, т. е. не зависит от угла а. Она характеризует ненамагниченную дисперсную систему и далее будет обозначаться символом (dLld^)L. При усреднении выражения, которое получится после выполнения операции перемножения в правой части приведенной формулы, необходимо учесть, что средние значения cosa и sina равны нулю, а средние значения cos2a= 1/3 и sin2a = 2/3. Кроме того, слагаемое, содержащее множитель пренебрежимо
мало по сравнению с другими слагаемыми. В итоге после исключения пренебрежимо малых слагаемых получается формула:
<L(Q cos ®(ННе)> =
= [(2/3) Ll + (1/3X<& / dt)L ^](Не / HL) (3.9.92)
664
Новый справочник химика и технолога
При дифференцировании функции Ланжевена
A© = cth^-(l/y= 1 -(2LIQ-L2 получается следующее выражение:
(dL/d^L=\-(2LL/^)-L2L.
При его подстановке в формулу (3.9.92) и последующем ее упрощении получается формула:
<Щ) cos®(HHe)> = (£/3) (1 - 4). (3.9.93)
Тогда намагниченность взвеси в слабом внешнем поле, согласно формуле (3.9.84), описывется формулой
М' = М* (pow / 3kT) (1 - 4) Не. (3.9.94)
Начальная магнитная восприимчивость взвеси определяется формулой:
х = хо(1-4)- (3.9.95)
Здесь х0 — классическая начальная магнитная восприимчивость системы невзаимодействующих частиц, определяемая формулой (3.9.72). При сильном взаимодействии частиц А/ = 1 - (1/^l), и формула (3.9.95) сводится к формуле (3.9.96), которая является модификацией формулы (3.9.79):
х = (2/3)Л/;/ HL. (3.9.96)
В магнитном поле умеренной напряженности начинает сказываться эффект приближения намагниченности дисперсной системы к насыщению. Влияние локальных полей при этом слабее, чем следует из формулы (3.9.94). Более точное выражение для намагниченности получается из тех же уравнений, что и формула (3.9.94), но при вычислении величин cos©(///7(,) и dL / d^ с большей точностью — до членов второго порядка малости включительно. Оно имеет следующий вид:
И* = М' (щпНе / ЗкТ) R* - (\ыпНе / ЗА7)37?**/45.
Здесь
«=1-4,
я" = 3[(1 - 4 4 + 3 4) + 4(4 / 4)(2 4 -1) + (4 / 4)2] — поправки на эффект локального поля и на его насыщение.
Рассмотренные эффекты не могут приводить к увеличению намагниченности или магнитной восприимчивости взвеси однодоменных частиц по сравнению с идеальной суперпарамагнитной намагниченностью такой взвеси. Вместе с тем в действительности такие (положительные) отклонения от идеальности наблюдаются, и их природа связана с изменением структуры взвеси, в том числе под действием внешнего магнитно
го поля. Примеры соответствующих структурных изменений уже приводились — это образование линейных цепочек частиц, вытянутых вдоль поля, или стабильных двойников с ориентацией магнитных моментов частиц в одном направлении — параллельно оси двойника. В последнем случае магнитная восприимчивость взвеси удваивается. В феррожидкостях подобное структурирование имеет статистический характер, т. е. цепочки, двойники и другие формы ассоциации частиц имеют ограниченную продолжительность существования. Поэтому положительное отклонение от идеальности выражено слабее, чем в суспензиях, но, тем не менее, оно вполне ощутимо, а главное — имеет принципиальное значение для понимания взаимосвязи магнитных (и диэлектрических) свойств дисперсных систем с их структурой.
В предельном случае — при воздействии на дисперсную систему сильных внешних полей — частицы объединяются в длинные, параллельные полю цепи. Цепи в феррожидкостях отличаются от цепей в суспензиях тем, что в феррожидкостях расстояние между соседними по цепи частицами может быть существенно больше диаметра частиц. Обусловлено это несопоставимо разным вкладом толщины защитной оболочки в равновесные расстояния между соседними частицами цепи в золях (феррожидкостях) и в суспензиях. Защитные оболочки имеют примерно одну и ту же толщину и на частицах феррожидкости, и на частицах суспензии, но в золях она сравнима с размером частиц, а иногда и больше его, а в суспензиях пренебрежимо мала по сравнению с размером частиц. Это и приводит к указанному различию в расположении частиц в цепях феррожидкостей и суспензий. Оно влечет за собой соответствующее различие в напряженности локальных полей, создаваемых на каждой частице ее соседями по цепи, а следовательно, к различию в прочности цепей и их способности неограниченно расти с увеличением напряженности поля, концентрации частиц и времени.
В цепи действующее на частицы поле практически совпадает по направлению с внешним полем, а его напряженность слегка увеличена на величину напряженности локального поля, которое в среднем направлено параллельно внешнему полю, так что Н = Нг + Нг. Намагниченность М' дисперсной системы в этом случае определяется произведением Л/5*£(£), а восприимчивость — формулой (3.9.97), в которой Е, = [цо/и(Яе + + HL)lkT\.
к = M*L(^)/He. (3.9.97)
При этом /W* и х по мере увеличения вклада напряженности локального поля Hl в напряженность суммарного поля Н: Н = Не + HL становятся все больше по сравнению с намагниченностью и восприимчивостью взвеси равномерно распределенных частиц. С увеличением напряженности внешнего поля Не этот эффект стремится к нулю вследствие насыщения намагниченности.
Физика, химия и технология дисперсных систем
665
Таким образом, в зависимости от первоначальной структуры коллоидного раствора и ее чувствительности к действию магнитного поля могут наблюдаться качественно различные отклонения намагниченности раствора от идеальной суперпарамагнитной намагниченности коллоида.
Величина этого эффекта в феррожидкостях мала, и поэтому его можно обнаружить только при дифференциальном способе измерения намагниченности. Измерения проводятся одновременно на двух образцах раствора в двух идентичных измерительных ячейках. Ячейки электрически соединены так, что дают сигнал, пропорциональный разности намагниченности двух образцов раствора. Один из них принимается за эталон, а второй модифицируется тем или иным способом, например введением коагулятора. Таким образом, удается с высокой точностью зарегистрировать изменения намагниченности под влиянием коагулятора или любого иного модификатора свойств жидкости (рис. 3.72). При намагничивании устойчивого коллоидного раствора магнитного материала единственно возможный вид структурирования — это образование цепей, что ведет к увеличению его намагниченности по сравнению с намагниченностью неструктурированного раствора. Этот эффект тем больше, чем меньше расстояние между соседними частицами цепи. Поэтому при уменьшении толщины защитной оболочки на частицах он будет усиливаться, что и наблюдается при небольшом увеличении концентрации электролита в растворе (рис. 3.72). Значительное увеличение концентрации электролита уменьшает толщину защитных оболочек настолько, что коллоидные частицы слипаются еще до воздействия магнитного поля. Внутри образующихся флокул магнитные моменты частиц ориентируются так, чтобы магнитный поток замыкался внутри флокулы, и тогда локальные поля соседних частиц имеют случайное направление по отношению к направлению намагничивающего поля и, следовательно, препятствуют намагничиванию флокул и раствора в целом. На дифференциальных кривых намагничивания это проявляется в виде ухода кривой под ось абсцисс (рис. 3.72). С увеличением концентрации электролита толщина оболочек становится меньше, локальные поля во флокулах усиливаются, и поэтому эффект снижения намагниченности становится больше.
Влияние состава среды на магнитные свойства особенно велико в суспензиях однодоменных частиц. На первый взгляд это кажется неожиданным, потому что введение в среду стабилизаторов или коагуляторов не дает видимого эффекта, поскольку поведение частиц определяется их очень сильным магнитным взаимодействием, которое не изменяется при изменении состава среды. Тем не менее, магнитные свойства суспензии меняются радикально при небольших изменениях состава среды (рис. 3.73). Даже затвердевание (полимеризация) дисперсионной среды (в данном случае стирола) влияет в меньшей мере, чем введение нескольких капель стабилизатора (олеиновой кислоты).
Рис. 3.72. Изменение АЛ/* намагниченности Л/ коллоидного раствора магнетита с намагниченностью насыщения 0,45 кА/м, стабилизированного олеатом натрия, (а) и коллоидного раствора магнетита с намагниченностью насыщения 2,5 кА/м, стабилизированного соляной кислотой, (б) при введении в них NaCl до концентрации 0,05 (/), 0,1 (2) и 0,15 (3) моль/л
Рис. 3.73. Петли гистерезиса суспензии гексаферрита бария при его объемной доле 15 об. %:
1 — в стироле; 2 — в стироле, содержащем 2 % олеиновой кислоты; 3 — после полимеризации стирола
Характер наблюдаемых изменений позволяет понять их причину. Прежде всего, необходимо обратить внимание на принципиально иной, по сравнению с феррожидкостями, характер зависимости намагниченности суспензий от напряженности поля — наличие гистерезиса. Гистерезис — это несовпадение зависимостей свойства (намагниченности) от параметра состояния (напряженности поля), получаемых при увеличении и при уменьшении значения параметра состояния. Гистерезис намагниченности наглядно представляется в виде петли гистерезиса (рис. 3.73). Намагничивание суспензии однодоменных частиц магнитно-жесткого материала при напряженности магнитного поля меньшей, чем коэрцитивная сила частиц, возможно только путем механического поворота частиц в магнитном поле достаточно большой напряженности Н. Она должна быть такой, чтобы крутящий момент [mH], действующий на частицу со стороны магнитного поля, превысил момент
666
Новый справочник химика и технолога
сил ее сцепления с соседними частицами. В отсутствие стабилизатора (олеиновой кислоты в данном случае) контакты между частицами оказываются достаточно прочными для того, чтобы исключить возможность вращения частиц под действием поля, и поэтому суспензия почти не намагничивается. В итоге кривая намагничивания (рис. 3.73; петля 7) суспензии без стабилизатора оказывается качественно почти такой же, как кривая намагничивания (рис. 3.73; петля 3) той же суспензии с твердой средой, где возможность вращения заведомо исключена. Слабое намагничивание в этих случаях обусловлено обратимым отклонением вектора намагниченности частиц от кристаллографической оси легкого намагничивания. Введение стабилизатора уменьшает прочность контактов настолько, что становится возможным необратимое изменение ориентации частиц при сравнительно малой напряженности поля Н, поэтому после уменьшения напряженности поля до нуля суспензия сохраняет почти такую же намагниченность, как и в поле (рис. 3.73, точка пересечения петли 2 и оси намагниченности Л/*). При смене направления намагничивающего поля и увеличении его напряженности до некоторой характерной для суспензии величины Нм происходит практически одновременное изменение ориентации магнитных моментов всех частиц на противоположное и соответствующая смена направления намагниченности. При этом петля гистерезиса намагничивания становится высокой и узкой. Ширина этой петли равна 2НМ. Кривая первоначального намагничивания суспензии (выходящая из начала координат) расположена в основном вне петли гистерезиса. Напряженность поля Hmq, при которой наблюдается самое сильное необратимое увеличение намагниченности, здесь приближенно в 2-3 раза больше, чем Нм. Это означает, что первоначальная структура суспензии (до ее намагничивания) существенно отличается от структуры, формирующейся под воздействием поля. При хорошей стабилизации суспензии, т. е. при полном устранении сил молекулярного сцепления частиц, напряженность поля, при которой становится возможным необратимое изменение ориентации частиц, равно напряженности локального поля, создаваемого всеми соседними частицами.
Различие величин Нм и HMQ показывает, что до намагничивания напряженность локальных полей была приближенно в 2-3 раза больше, чем после намагничивания. Именно таким это различие и должно быть, если первоначальная структура представляет собой систему антиферромагнитно упорядоченных магнитных моментов (рис. 3.74, а), а после намагничивания — ферромагнитно упорядоченных магнитных моментов (рис. 3.74, б). То и другое состояние устойчиво по отношению к слабым возмущениям ориентации магнитных моментов частиц. Ферромагнитное упорядочение при локализации частиц в узлах кубической решетки эквивалентно системе параллельных цепочек, намагниченных в одном направлении вдоль одной из осей решетки. Анти
ферромагнитное упорядочение отличается тем, что соседние цепи имеют противоположное направление намагниченности.
Рис. 3.74. Антиферромагнитное (а) и ферромагнитное (б) упорядочение ориентации магнитных моментов частиц в кубической решетке
При ферромагнитном упорядочении шести ближайших соседей каждой частицы (2 вдоль цепи и 4 в соседних цепях) суммарная напряженность локального поля равна нулю. Тем не менее, напряженность локального поля Hf кубической ферромагнитной решетки отличается от нуля и совпадает с направлением намагниченности благодаря вкладу 8 соседних частиц, расположенных на диагоналях кубической решетки. Этот вклад, согласно формуле (3.9.56), описывется следующим выражением:
Hf= (8/33/2) т [3cos2(k/4) - 1]1/2 / 4пг3.
Здесь г — период решетки. Знаменатель числового множителя (8/33/2) отражает то обстоятельство, что расстояние между частицми на диагонали куба в 31/2 больше периода решетки г. Численно величина /^составляет 4/33/2 от напряженности локального поля HG = 2т/4лг3 на частицах двойника в первом Гауссовом положении (рис. 3.67, конфигурация 1), т. е. около 0,8HG.
При антиферромагнитном упорядочении вклад всех шести ближайших соседей по решетке в напряженность локального поля положителен и равен 4//<;, а вклад диагональных соседей имеет обратный знак и численно равен Hf. Так что напряженность локального поля HL на магнитных моментах частиц при антиферромагнитной ориентации магнитных диполей составляет 3,2HG.
Фактическая напряженность локального поля HL после намагничивания может быть больше 0,8HG, потому что параллельно намагниченные цепи отталкиваются. При этом расстояние между соседними цепями тем больше, чем меньше концентрация суспензии. Это уменьшает, вплоть до нуля, вклад соседних цепей в напряженность локального поля HL внутри отдельной цепочки частиц, и она приближается к величине 2HG.
При цепочечной структуре дисперсной системы с малым размером частиц напряженность локального поля уменьшается в сравнении с величиной 2HG за счет тепловых флуктуаций направления магнитных осей частиц. Этот эффект учитывается так же, как и при описании магнитной восприимчивости — заменой в формуле (3.9.56) постоянного магнитного момента час
Физика, химия и технология дисперсных систем
667
тицы величиной его среднестатистической проекциии на ось цепи, приводящей к формуле:
HL = mL(g)htr\ (3.9.98)
Здесь тЦ£) = <т> — среднестатистическая величина проекции магнитного момента частицы т на продольную ось цепи (см. вывод формулы 3.9.83). Эта формула применима и к суспензиям, с той лишь разницей, что в них г = 2а, а значение функции Ланжевена = 1 практически при любой напряженности поля. В суспензиях магнитно-мягких материалов (карбонильное железо, никель-цинковый феррит и др.) величина <т> однозначно может быть определена при любой напряженности поля из изотермы намагничивания М* =f (Н) по формуле:
<т>=М*/п. (3.9.99)
Здесь п — концентрация частиц суспензии. В любом случае при насыщении намагниченности напряженность локального поля в суспензии приближается к напряженности поля в тонком зазоре между намагниченными до насыщения телами, т. е. к максимально возможной величине, а восприимчивость — к нулю. Поэтому максимум эффекта структурного увеличения намагниченности находится вдали от насыщения, когда магнитная восприимчивость дисперсной системы х остается еще достаточно большой, но цепочечная структура уже сформирована.
Цепочечная структура в большей степени характерна для равновесного состояния суспензий и феррожидкостей в достаточно сильном магнитном поле. В то же время представляет интерес начальная стадия развития процесса структурирования, когда частицы успевают лишь слегка отклониться от своего в среднем равномерного распределения в среде. Это происходит при действии на дисперсную систему кратковременных импульсов поля или высокочастотного переменного поля.
С известной степенью приближения равномерное распределение частиц в среде можно представить в виде их размещения по узлам правильной кубической решетки с периодом г. Если принять также, что внешнее поле направлено вдоль одной из осей кубической решетки, то каждая частица решетки имеет две соседние частицы на линии действия поля и 4 частицы — в плоскости, перпендикулярной направлению внешнего поля. Первые создают на центральной частице поле, напряженность которого определяется формулой (3.9.98), а четверка соседей по плоскости — точно такое же по величине поле, но обратного направления. В этом легко убедиться с помощью формулы (3.9.56). Таким образом, напряженность суммарного поля шести ближайших соседей, которая описывается формулой (3.9.100), равна нулю, так как гцц и — расстояния между центральной частицей и ее соседями, расположенными вдоль поля и поперек поля соответственно, одинаковы и равны постоянной решетки г:
HL = (<т> / тг)[( 1/гцц)3 - (1/гх)3]. (3.9.100)
Начальную стадию перехода к цепочечной структуре можно представить как сближение частиц, расположенных вдоль поля, на величину Дгцц и удаление соседей, расположенных в перпендикулярном направлении, на величину Дгх. Тогда гцц = г - Дгцц, = г + Дг± или Г|||| = Г (1 - £||||), Г± = Г (1 + £j_), где = Ал-ЦП / Г, £± = Дг± / г. Если в уравнении (3.9.100) гцц и выразить через £цц и £_ц соответственно, то получается формула
HL = (<т> /л r3)[ 1/( 1 - £цц)3 - 1/(1 + £±)3]. (3.9.101)
При £цц С 1 и е « 1 справедлива приближенная формула:
HL ~ 3(<т> / л г3)(£|ц| + £±). (3.9.102)
В частности, если расстояние между частицами изменять путем механической деформации упругого материала (резины) с магнитным наполнителем вдоль поля на величину £цц, то при постоянстве объема образца £± = ец||/2, и напряженность локального магнитного поля описывается формулой:
HL = (9/2)£цк (<т> / ti г3). (3.9.103)
Это достаточно сильный эффект, поскольку в отсутствие деформации HL = 0, и он может быть зарегистрирован по изменению намагниченности образца. Имеет место и обратный (магнитострикционный) эффект — изменение размеров образца при его намагничивании.
Таким образом, восприимчивость феррожидкостей, суспензий и магнитнонаполненных материалов в общем случае сложным образом изменяется с увеличением напряженности магнитного поля. Их начальная восприимчивость (при напряженности внешнего поля близкой к нулю) может оказаться в зависимости от ориентации локальных полей и больше, и меньше классической величины. В сильном магнитном поле вклад локальных полей мал, и поэтому магнитная восприимчивость феррожидкости, которой отвечает формула (3.9.104), мало отличается от ее классической величины:
х = М* ЩыпНе / kT)IHe. (3.9.104)
3.9.5. Суперпарамагнетизм Броуна и Нееля
При обсуждении природы суперпарамагнетизма механизм вращательного теплового движения магнитных осей частиц (их вращательной диффузии) не уточнялся. В принципе имеются два разных способа изменения ориентации магнитного момента частицы, как самопроизвольного (при тепловых колебаниях ориентации), так и вынужденного (при действии внешнего магнитного поля на ориентацию магнитного момента). Один способ —. это вращение магнитного момента вместе с самой частицей, а второй — его вращение относительно тела частицы, которое может оставаться неподвижным. Реализация того или иного способа определяется
668
Новый справочник химика и технолога
жесткостью связи направления самопроизвольного намагничивания частицы с ее кристаллической решеткой. Как уже отмечалось, это свойство количественно характеризуется величиной К — константой магнитной анизотропии магнитного вещества или его коэрцитивной силой Нс. Чем больше константа анизотропии, тем жестче магнитный момент частицы привязан к направлениям осей кристаллической решетки и тем предпочтительней становится первый способ изменения ориентации магнитного момента частицы — путем ее механического поворота вместе с «вмороженным» в тело частицы магнитным моментом. Количественной мерой жесткости связи магнитного момента частицы с направлениями осей ее кристаллической решетки является величина KV — энергия магнитной анизотропии частицы объемом V. Это энергия, которую необходимо затратить на изменение направления намагниченности частицы на противоположное. Ее можно также назвать потенциальным барьером вращения магнитного момента относительно тела частицы. Согласно принципу равного распределения энергии теплового движения по всем степеням свободы, направление оси намагниченности частицы колеблется возле своего наиболее выгодного положения с той же средней энергией kT, что и тело частицы относительно пространственных координат. Соответственно этому имеется некоторая вероятность (формула (3.9.105)) преодоления потенциального барьера вращения при тепловом движении частицы и самопроизвольного изменения ориентации магнитного момента частицы относительно ее тела на противоположное:
w = exp (-KV / кТ). (3.9.105)
При малой энергии анизотропии, т. е. при достаточно малом размере частиц, эта вероятность весьма велика, так что магнитные моменты частиц почти свободно вращаются (диффундируют по направлениям) относительно неподвижных частиц. Если среда, в которой взвешены частицы, является твердой, то такой способ вращательной диффузии магнитных моментов становится единственно возможным. Свободная (или почти свободная) вращательная диффузия магнитных моментов частиц в магнитных системах с твердой средой обеспечивает выполнение тех же законов равновесного распределения магнитных моментов частиц по направлениям, что и в жидкой среде. Это значит, что намагничивание взвеси достаточно мелких частиц в твердой среде описывается той же функцией Ланжевена, которая была использована при описании свойств жидких магнитных коллоидов с характерной для них особенностью — гигантской (по сравнению с молекулярными и атомными смесями) парамагнитной восприимчивостью, т. е. суперпарамагнетизмом. Механизм намагничивания, обусловленный вращательной диффузией магнитных моментов относительно тела частиц, открыт Неелем и получил название суперпарамагнетизма Нееля.
Броуновским суперпарамагнетизмом называют явление намагничивания магнитных коллоидов путем ориентации самих частиц вместе с «вмороженным» в их тело магнитным моментом. При подходящих условиях зависимость намагниченности от напряженности поля одинакова как при неелевском, так и при броуновском парамагнетизме. Вместе с тем имеются и существенные качественные различия в поведении систем с твердой и жидкой средой. Неоднозначно влияние температуры на магнитную восприимчивость твердых магнитных коллоидов. С одной стороны, согласно формуле (3.9.105), повышение температуры облегчает вращательную диффузию и тем самым увеличивает магнитную восприимчивость коллоидной системы. Но с другой стороны, это ведет к уменьшению значения аргумента функции Ланжевена в формуле (3.9.104) и к уменьшению восприимчивости. Температурная зависимость восприимчивости (намагниченности) твердых магнитных коллоидов является одним из способов нахождения константы анизотропии или размера магнитных частиц. При достаточно низкой температуре вращательная диффузия магнитных моментов практически отсутствует (магнитные моменты «вмораживаются» в кристаллическую решетку частицы). Это ведет к потере суперпарамагнетизма и к появлению магнитно-жестких свойств — способности вещества сохранять приобретенную в магнитном поле намагниченность и после выключения поля. Благодаря такой особенности некоторые вещества (например, глина с примесью оксидов железа, красный кирпич) сохраняют в себе «отпечаток» геомагнитного поля, действовавшего на них в моменты повышенной температуры (при остывании вулканической породы, при последнем протапливании печи или при пожаре и т. д.). На магнитной памяти веществ основан палеомагнетизм — наука о магнитном поле Земли в геологически отдаленные времена. В структуре дисперсных материалов «зашифрованы» также сведения о физико-химических условиях их возникновения, и это относится не только к магнитным дисперсным системам. Наличие магнитных свойств дает не только дополнительную информацию об условиях возникновения материала, но и дополнительные средства расшифровки его структурного состояния. Осадочные горные породы в свое время сформировались при свободной коагуляции и оседании частиц в сильно разбавленных взвесях морей и океанов. Они представляют собой своеобразную летопись геологических эпох, которая пока еще полностью не расшифрована.
Как уже отмечалось, в случае магнитных дисперсных систем их структура и структура флокул, образующихся при слипании частиц, в сильной мере подвержены влиянию магнитного поля. Другим, мощным и универсальным фактором, определяющим структурное состояние взвесей, в том числе магнитных, являются механические напряжения, возникающие внутри флокул при течении взвесей. Эти вопросы рассматриваются в подразделах 3.11-3.14, посвященных структуре и рео
Физика, химия и технология дисперсных систем
669
логическим свойствам дисперсных систем. Следует отметить, что система параллельных цепочек, образующихся в магнитном поле, является простейшей в геометрическом смысле структурой коагуляционного типа. Именно на ее примере впервые были осознаны закономерности течения структурированных систем [4, 9], введены количественные параметры, характеризующие структурное состояние дисперсной системы в потоке, и выведены уравнения реологии.
3.10. Основы механики деформируемых сред
3.10.1, Предмет реологии
Реология — наука о деформационных свойствах материалов. Она тесно связана с другой областью естествознания — механикой сплошной среды (МСС) и заимствует из нее некоторые основные понятия. МСС устанавливает на основе универсальных принципов механики, термодинамики, геометрии наиболее общие и поэтому справедливые для любых материалов законы их поведения под влиянием деформирующих усилий. Материалы как реологические объекты характеризуются упругостью, вязкостью, прочностью и другими реологическими константами. Наличие у материала тех или иных свойств в МСС постулируется и, исходя из этих свойств, предсказывается его поведение под нагрузкой. В отличие от этого реология является наукой материаловедческой. Ее задача — установить, чем на самом деле окажется материал, изготовленный по определенной рецептуре и технологии: упругим твердым веществом, текучей жидкостью, эластичным (каучукоподобным) телом, пластичным составом или чем-то иным и как рецептура и технология влияют на реологическое состояние и величины констант. Принято считать, что основной путь решения этой задачи — эмпирический, т. е. необходимо опытным путем устанавливать, как поведет себя материал под нагрузкой. Этот путь познания законов реологии ведет к классификации изучаемых объектов и явлений, в данном случае — реологических. Уже повседневный опыт обращения с различными материалами позволяет разделить их на твердые, жидкие и газообразные.
Первые обладают определенной формой и объемом, вторые — только объемом, третьи не имеют ни того, ни другого в отсутствие внешних сил. Твердое тело при действии ограниченной по величине силы деформируется упруго, т. е. при снятии напряжения деформация исчезает, величина деформации пропорциональна деформирующему усилию. Коэффициент пропорциональности (податливость) или обратная ему величина (упругость) и является реологической характеристикой твердого материала. Типичная жидкость при действии любого по величине усилия деформируется неограниченно до тех пор, пока действует это усилие, т. е. течет. В таком случае величина деформации не определяется одной только силой, а зависит также и от времени ее действия. Поведение материала под нагрузкой в этом случае характеризуется деформацией за единицу вре
мени, т. е. скоростью деформации. Эта величина в жидкости пропорциональна деформирующему усилию. Коэффициент пропорциональности (текучесть) или обратная ему величина (вязкость) и является реологической константой, характеризующей жидкость, а также газ. Особенность газа как реологического объекта в том, что он может существовать только в напряженном состоянии — при действии внешней силы, ограничивающей его объем. Реологическое состояние газа в большей мере отражается не деформацией (изменением формы или объема), а интегральной по отношению к ней величиной — самим объемом. При этом уравнение состояния газа pV = RTявляется одновременно и уравнением реологического состояния. Величину RT следует рассматривать как реологическую константу (при обратимых деформациях).
Упругость и вязкость отражают наиболее характерные, но не все свойства твердого и жидкого материалов соответственно. Упругие материалы разогреваются при длительном многократном деформировании. Это значит, что часть работы, совершаемой при деформировании, затрачена на преодоление сил вязкого сопротивления. Жидкость (вода например) проявляет упругость и хрупкость при весьма кратковременных воздействиях силы. В таком материале как битум свойства твердого и жидкого веществ выражены примерно в равной мере. Так, шарик битума, положенный на стол, постепенно расплывается под действием слабой длительно действующей силы собственного веса, т. е. он течет, проявляя свойства жидкости. Тот же шарик раскалывается как хрупкое твердое тело при ударе молотком и проявляет упругость при кратковременном действии умеренной по величине импульсной силы.
Все материалы в большей или меньшей мере подобны битуму, а дисперсные системы обнаруживают и более сложные реологические свойства. Таким образом, понятия «твердого» или «жидкого» материала не являются абсолютными. Проявление тех или иных свойств зависит от режима (скорости) деформирования.
3.10.2. Основные понятия и законы реологии
В технике различают деформации растяжения (сжатия), сдвига, кручения и т. д. В МСС доказывается, что в случае несжимаемых материалов, каковыми являются многие дисперсные системы, основной можно считать деформацию сдвига, а остальные представляют собой различные комбинации этого основного вида деформации.
Количественную меру сдвига можно установить на примере деформации образца в виде прямоугольного параллелепипеда (рис. 3.75). Пусть деформирующее усилие F приложено по касательной к верхней грани площадью А. Оно создает напряжение сдвига во всем образце. Нижняя грань закреплена неподвижно. Деформация материала должна быть выражена величиной, не зависящей от его формы и размера. В данном случае это отношение у = АХ / Y, которое имеет ту же величину (у = dx / dy) и для любого элемента объема внутри об
670
Новый справочник химика и технолога
разца. Соответственно, напряжение r = F / А, приложенное к верхней грани, такое же, как и напряжение в любом элементе объема внутри образца, действующее на его грань, параллельную плоскости xz. Скорость деформации Y = dfldt в данном случае — это скорость сдвига. Так как время t и координата z — независимые переменные, то, изменив порядок дифференцирования по этим переменным, можно получить следующие соотношения: Y - d (dxldt) I dy = du/dy, т. e. скорость деформации при простом сдвиге равна градиенту duldy скорости течения и = dxldt.
Реологические законы должны устанавливать связь между т, у, у' через константы, характеризующие материал. Следовательно, в упругом материале, для которого характерна пропорциональность деформации у напряжению т, справедливо уравнение:
т = Gy.
(3.10.1)
Такого рода связь величин у и т является выражением закона Гука. Величина G — модуль сдвиговой упругости — полностью характеризует реологические свойства идеального упругого материала. Закон Гука следует также рассматривать как определение понятия «упругость» и определение количественной меры G этого свойства.
Необходимо отличать упругость материала от упругости тела (изделия). Так, упругость параллелепипеда (рис. 3.75) равна GA, а упругость нити длиной L на скручивание Gw = TtcfG I 32L, где d — диаметр нити. Эта формула применяется, в частности, при расчете упругого элемента ротационного вискозиметра.
Рис. 3.75. Схема деформации сдвига. Пояснения в тексте
В чисто вязком материале (жидкости) напряжение пропорционально скорости деформации:
Ft = т]уг. (3.10.2)
Реологические свойства жидкости при сдвиге полностью характеризуются ее вязкостью т| (в ламинарном режиме течения). Формально ее можно определить как силу сопротивления относительному перемещению слоев жидкости, приходящуюся на единицу площади слоя и отнесенную к скорости сдвига. Иногда не вполне корректно вязкость определяется как сила сопротивления при скорости сдвига, равной единице. Ошибка здесь в том, что вязкость характеризует свойства жидкости и может быть измерена при любой, а не только
при единичной скорости сдвига. Между этими двумя определениями нет разницы только в том случае, если заранее предполагать, что вязкость не зависит от условий опыта (скорости и напряжения сдвига). В связи с этим постоянство величины ц при изменении т или у' часто воспринимается как основное содержание закона внутреннего трения Ньютона (3.10.2). На самом деле он имеет более глубокий смысл.
Не менее важную роль, чем силы упругости и вязкого трения, играют в технике и силы внешнего (сухого) трения, действующие, например, в подшипнике скольжения. Особенность этих сил в том, что при напряжении, меньшем величины сухого трения т„ деформация (сдвиг) отсутствует, а при напряжении, превышающем силу сухого трения на бесконечно малую величину, сдвиг и скорость сдвига могут быть сколь угодно большими, т. е. справедливы соотношения:
у = 0, у' = 0 при т < т„
у 0, у' 0 при т = т5. (3.10.3)
Иначе говоря, сила сухого трения не зависит от величины и скорости деформации (скольжения) или, что то же самое, к элементу сухого трения (рис. 3.76) невозможно приложить усилие, превышающее величину т.т.
Как только напряжение превысит силу трения, начнется скольжение, скорость которого этим законом не лимитируется, и элемент уже не может оказать дополнительного сопротивления деформированию. По этой причине во второй строке закона (3.10.3) нельзя написать т > тЛ. вместо т = т5. Материалом, свойства которого описываются законом (3.10.3) можно, по-видимому, считать сухой порошок.
Приведенные выше законы исчерпывают перечень фундаментальных эмпирических законов реологии и соответствующих им типов сил, возникающих при деформировании тел разной природы. Это не означает, что с их помощью можно с необходимой полнотой описать поведение любого материала. Реальные материалы ведут себя сложнее, а законы (3.10.1)-(3.10.3) относятся скорее к некоторым идеальным (реологически простым) материалам. Реальные материалы сочетают в себе в разных комбинациях свойства идеального упругого тела, вязкого тела и элемента сухого трения. Ихтак же называют реологически сложными. Описание свойств таких материалов часто строится на основе их механических моделей (эквивалентов). Последние составлены из механических моделей (эквивалентов) простых реологических тел (рис. 3.77 и 3.78).
Р
Рис. 3.76. Механический эквивалент силы сухого трения
Физика, химия и технология дисперсных систем
671
Эквивалентом материала с упругими свойствами является пружина, материала с вязким сопротивлением — поршень, помещенный в жидкость, и сухого трения — груз, лежащий на плоскости.
Класс вязкоупругих материалов в качестве простейших представителей этого класса включает вязко-упругую жидкость (тело Максвелла) и вязкоупругое твердое тело (тело Кельвина). Механическая модель вязкоупругой жидкости представляет собой последовательно соединенные элементы упругого и вязкого сопротивлений, а модель вязкоупругого твердого тела — те же элементы, соединенные параллельно. Примером вязкоупругой жидкости является полиизобутилен, а примером вязкоупругого твердого вещества — набухшая в масле резина.
Если мгновенно создать деформацию тела Максвелла величиной уо (например, переместив цилиндр до упора на рис. 3.78, а) и далее удерживать ее постоянной, то в первый момент времени эта деформация будет целиком обусловлена растяжением пружины, поскольку упругая часть деформации уг не требует для своего развития какого-либо времени. Растянутая пружина будет вызывать движение поршня со скоростью cty/dt = —т / т]. Отрицательный знак означает, что деформация пружины со временем уменьшается, а деформация вязкого элемента увеличивается на ту же величину. Этот процесс является внутренним — невидимым для наблюдателя по условиям опыта (общая деформация, равная сумме упругой и вязкой составляющих, остается неизменной). Если выразить в приведенном выше уравнении напряжение через деформацию с помощью закона Гука т = Gy и решить уравнение при начальном условии уг = уо при т = 0, то получатся уравнения:
у г = y0exp(-G7 / т]), т = тоехр(-б/ / т]). (3.10.4)
Так как т = Gyr и т0 = Gy0, то одновременно с упругой деформацией уменьшается во времени и величина напряжения, необходимого для поддержания первоначальной деформации у0. Этот процесс имеет внешнее проявление в виде уменьшения усилия, создаваемого экспериментатором для поддержания первоначальной деформации. Таким образом, в этом опыте наблюдаемым параметром реологического состояния тела является действующее на него внешнее напряжение, а ненаблюдаемым — внутренние, скрытые от экспериментатора деформации.
По истечении достаточно большого времени по сравнению с величиной т] / G (теоретически бесконечно большого) т становится практически равным нулю и для дальнейшего сохранения деформации у0 уже не требуется приложения силы, т. е. изменение состояния тела становится необратимым. Полная необратимость деформации является признаком жидкости, поэтому тела Максвелла следует относить к жидкостям. Величина /* = т] /G представляет собой время релаксации внешних напряжений или время релаксации упругих внутренних
деформаций. В вязкоупругих жидкостях действуют равные напряжения на ее упругие и вязкие элементы. Деформации этих элементов аддитивны, т. е. складываясь, дают наблюдаемую суммарную величину деформации.
Рис. 3.77. Механические эквиваленты сил упругого (Fr) и вязкого (Fd) сопротивления
Рис. 3.78. Механические эквиваленты вязкоупругих тел Максвелла (а), Кельвина (б) и пластичного тела (в)
Аналогично можно показать, что если в момент времени 0 начать деформацию тела с постоянной скоростью у', то необходимое для этого напряжение будет расти во времени по закону
т = туу' [1 -ехр(-///*)]. (3.10.5)
Напряжение, приложенное к телу Кельвина, распределяется между упругим и вязким сопротивлениями деформированию:
т = (7у+т]у'. (3.10.6)
Иначе говоря, напряжения в нем аддитивны. Деформации обоих элементов сопротивления — упругого и вязкого — в любой момент времени одинаковы и совпадают с наблюдаемой величиной деформации.
Характерные свойства вязкоупругих твердых тел можно обнаружить с помощью следующего воображаемого или реального эксперимента: произвольным усилием т в произвольном режиме вызовем деформацию тела величиной у0 и затем устраним деформирующее усилие, предоставив материалу возможность самопроизвольно изменять свое состояние. Внутренние напряжения (растянутая пружина) будут возвращать тело Кельвина в недеформированное состояние, а вязкие силы будут тормозить этот процесс релаксации деформации в соответствии с уравнением Gy + т]у' = 0 или cty/y = -Gdt/ т|. Его интегрирование при условии у = уо при t = 0 дает закон убывания первоначальной деформации у после разгрузки материала, т. е. при т = 0:
у = yoexp(-G//ц). (3.10.7)
672
Новый справочник химика и технолога
При умножении левой и правой частей формулы (3.10.7) на G получается закон убывания внутренних напряжений тг во времени:
тг = Toexp(-G/ / т|). (3.10.8)
По форме уравнения релаксации (3.10.7), (3.10.8) совпадают с уравнениями (3.10.4), но по содержанию они различны: уравнение (3.10.7) описывает полную, наблюдаемую величину деформации, а уравнения (3.10.4) — невидимый внутренний процесс. Уравнение (3.10.8) описывает ненаблюдаемый процесс релаксации внутренних напряжений, а аналогичная формула в наборе (3.10.4) — внешнего действующего напряжения.
При мгновенном нагружении тела Кельвина постоянной силой т = Cryo деформация растет во времени в соответствии с формулой: у = у0 [1 - ехр( ///*)], а скорость деформации уменьшается от начальной величины у 0 = у<у7* до нуля за бесконечно большое время по закону:
у = y0exp(G7 / ц). (3.10.9)
Здесь по-прежнему величина Z* = ту/ G представляет собой время релаксации напряжений и деформаций, но в вязкоупругом твердом теле Кельвина оно относится к релаксации наблюдаемой деформации и невидимых внутренних напряжений при постоянстве приложенного извне деформирующего усилия. По истечении времени, достаточно большого по сравнению со временем релаксации, деформация достигает постоянной равновесной величины уо, предопределенной величиной действующего усилия т = Gy0. После снятия нагрузки эта деформация исчезнет в соответствии с законами, отражаемыми формулами (3.10.7) и (3.10.8), что и является признаком твердого тела. Время релаксации вязкоупругого твердого тела соответствует времени запаздывания в установлении равновесной деформации.
При кратковременном действии сил реологические свойства тел Максвелла и Кельвина обращаются; первое ведет себя как упругий материал, а второе — как вязкая жидкость. Это обусловлено тем, что за малое время в первом не успевают развиться остаточные деформации, пропорциональные времени, а во втором из-за малости деформации вклад упругих сил в общее сопротивление является несущественным. Таким образом, проявление свойств жидкости или упругого тела зависит от режима деформирования. Особенно наглядно относительность понятий твердого тела и жидкости видна при периодическом деформировании материала. Это один из самых распространенных режимов деформирования, встречающихся на практике. Он важен и как метод определения реологического типа материала и для измерения его реологических констант.
Пусть испытуемый материал подвергается периодическому деформированию с заданной угловой частотой <0 и амплитудой уа по синусоидальному закону: y = yasin<oZ. Тогда сопротивление т такому деформированию также будет меняться по синусоидальному зако
ну с некоторой амплитудой ха, но со сдвигом по фазе Q, обусловленным запаздыванием в установлении величины напряжений, соответствующих текущему значению деформации:
т = Tasin(coz + Q). (3.10.10)
В соответствии с формулой (3.10.6) и с учетом того, что у' = <oyacos<o/, для вязкоупругого твердого тела получается формула:
т = Gyasina)r + <oiqyacos<oZ. (3.10.11)
Если воспользоваться формулой cos<oZ = sin((o/ + л/2) и правилами сложения гармонических колебаний, из уравнения (3.10.11) получается формула (3.10.10), в которой
= Gya [1 + (<о/*)2]1/2, tgQ, = <Щ*. (3.10.12)
Если окажется, что испытуемый материал является вязкоупругой жидкостью, то вместо уравнения (3.10.6) следует воспользоваться уравнением:
(/у = Jt/G + tJz/ц. (3.10.13)
Оно выражает свойство аддитивности упругих dxlG и вязких xdt/x\ деформаций за некоторый промежуток времени dt. Приведя его к виду
Gidy/dt) = (dxldt) + х G / ц
и выразив здесь производные и напряжение через те же тригонометрические функции, снова придем к формуле (3.10.10), в которой, однако, амплитуда напряжений и тангенс угла сдвига фаз зависят от частоты колебаний иначе:
та = coiqyasinQ, tgQ = 1/<oZ*. (3.10.14)
При высокой частоте колебаний <о сдвиг фаз между деформацией и напряжением в этом случае стремится к нулю, а амплитуда напряжений перестает зависеть от частоты, что характерно для идеального твердого материала. В случае же вязкоупругого твердого тела сдвиг фаз при высокой частоте приближается, согласно формуле (3.10.12), к л/2, т. е. напряжения максимальны при максимальной скорости деформации, а не при максимальной амплитуде колебаний. Это означает, что материал ведет себя при данных условиях как жидкость.
Материалы, способные к большим обратимым (т. е. упругим) деформациям, называются эластичными. Мерой эластичности является наибольшая величина обратимой деформации. Превышение этой величины вызывает или развитие необратимых деформаций (течение), или разрушение образца.
Наличие сил сопротивления, подобных сухому трению, придает материалам пластичность — способность деформироваться при умеренных усилиях и сохранять форму (т. е. остаточную деформацию) при малых усилиях, например создаваемых силой собственного веса.
Физика, химия и технология дисперсных систем
673
Это одно из наиболее важных для технологии свойств материалов. Сочетание сил вязкого и сухого трения приводит к появлению вязкопластических свойств. В соответствии с моделью (рис. 3.78, в) скорость деформации (скорость скольжения груза по плоскости на рис. 3.78, в) будет определяться величиной силы, приложенной только к элементу вязкого сопротивления, т. е. у' = (т - т5) /т|*, откуда:
т = т, + п*У' при т >
у' = 0прит<т5. (3.10.15)
Эти соотношения представляют собой уравнения реологии вязкопластических материалов. Первое из них известно как уравнение Шведова — Бингама. Следует, конечно, иметь в виду, что оно имеет смысл только при т>Ь.
Возможны и другие, в том числе более сложные сочетания основных реологических элементов, адекватные реальным материалам. В частности, последовательное соединение тел Максвелла и Кельвина, сочетание вязкоупругости и пластичности в одном материале и т. д.
3.10.3. Вязкость и классификация текучих материалов
Для технологии наибольший интерес представляют материалы с такими реологическими свойствами, которые позволяют придавать им требуемую форму, т. е. превращать материалы в изделия. Эти свойства связаны, прежде всего, с необратимыми (остаточными) деформациями материала. Течение — это процесс увеличения необратимой деформации во времени, иначе говоря — для технологии наиболее приемлемо рассматривать любые материалы как жидкости. Из фундаментальных законов реологии только закон Ньютона (3.10.2) описывает процесс течения, поэтому только он может быть принят за основу при рассмотрении реологических свойств текучих материалов. В связи с этим следует более обстоятельно проанализировать понятие «вязкости» как реологической характеристики материала и понятие «силы трения».
Если умножить левую часть формулы (3.10.2) на (dx/dtdz), а правую — на равную ей величину у', то получится следующее соотношение: xdxldtdz = riy2. Величина idx есть работа силы т на пути dx, которая совершается за время dt при деформировании слоя материала толщиной dz и площадью основания, равного единице (рис. 3.75). Следовательно, левая часть формулы — это потери энергии <?' в единице объема деформируемого материала за единицу времени на преодоление сил трения:
</ = Л(у')2. (3.10.16)
Эта формула представляет собой энергетическую форму записи закона Ньютона. Диссипация энергии, т. е. превращение ее в теплоту, и является причиной возникновения сил вязкого трения при деформировании материала. Зависимость диссипации от скорости
деформирования называется диссипативной функцией деформируемой среды. Сила трения определяется через диссипативную функцию как удельная (на единицу объема) величина потерь энергии, отнесенных к скорости сдвига:
т = ?'//. (3.10.17)
В частности, если q' - т|(у')2, то т = г|у'. В общем случае вид диссипативной функции q' неизвестен
и должен быть установлен неэмпирическим путем, например методами термодинамики необратимых процессов или из молекулярно-кинетической картины процесса деформации. По найденной таким путем функции q' можно с помощью формулы (3.10.16) априори установить реологический закон исследуемого материала.
Закон Ньютона (3.10.2) следует рассматривать и как определение понятия «вязкость». Подобно тому, как второй закон механики F~m (d2xldr) определяет понятие «массы» т, на основании закона (3.10.2) вязкость по Ньютону, или ньютоновская вязкость, есть величина:
т] = т/у'. (3.10.18)
Понятие «массы» количественно характеризуюет материалы по их способности участвовать в основных явлениях переноса подобно коэффициентам диффузии, теплопроводности и электропроводности в уравнениях Фика, Фурье и Ома соответственно и вязкости в законе внутреннего трения (3.10.2).
По признаку зависимости или независимости вязкости от напряжения сдвига все текучие материалы принято делить на ньютоновские и неньютоновские жидкости. Ньютоновскими являются материалы, вязкость которых не зависит от напряжения сдвига, т. е. является постоянным коэффициентом в законе внутреннего трения (3.10.2). К неньютоновским относятся материалы, вязкость которых зависит от напряжения сдвига, т. е. является функцией скорости деформации (или напряжения) в законе (3.10.2). В литературе даются и иные определения понятий ньютоновской и неньютоновской жидкости. Чаще всего говорят, что первая подчиняется, а вторая не подчиняется закону Ньютона. Последнее утверждение ошибочно в принципе. Во-первых, необратимая часть деформации любого материала, а точнее скорость этого процесса, может быть описана уравнением Ньютона (3.10.2), в том числе при переменной вязкости. Более того, не существует других фундаментальных законов и понятий, описывающих взаимосвязь напряжения и скорости деформации и, стало быть, способных описать процесс необратимого деформирования. Во вторых, само сравнение свойств разных жидкостей правомерно только в том случае, если сравниваются одинаковые свойства, например их вязкости по Ньютону. Только сравнив их вязкости по Ньютону (применив этот закон к разным жидкостям) можно получить основания для заключения об их принадлежности к тому или иному типу жидкостей. За неимением
674
Новый справочник химика и технолога
иных возможностей именно так и поступают, приводя в качестве «доказательства» неприменимость закона Ньютона зависимость вязкости неньютоновской жидкости от напряжения сдвига, полученную именно путем применения формулы (3.10.18) или, что то же самое, закона (3.10.2) к результатам измерения величин напряжения и скорости деформации. При такой логике можно утверждать, что и законы ньютоновской механики неприменимы к телам с переменной массой, например к ракетам, поскольку их масса на активном участке траектории полета не остается постоянной.
Деление текучих материалов на ньютоновские и неньютоновские отражает их поведение только в установившемся процессе непрерывного деформирования с некоторой скоростью у'. При рассмотрении зависимости вязкости от скорости сдвига (или от напряжения сдвига) всегда подразумевается, что во времени состояние и свойства системы не меняются. Так называемые реологические кривые — графики зависимости у' от т или q от т (рис. 3.79) — отражают поведение материала в установившемся (стационарном) режиме деформирования.
а) б)
Рис. 3.79. Реологические кривые скорости сдвига (а) и вязкости (б) менее (7) и более (2) вязкой жидкости
Эти графики для ньютоновских материалов с большей (г)]) или меньшей (q2) вязкостью представляют собой прямые, исходящие из начала координат (у', т), или прямые, параллельные оси абсцисс в координатах (т|, т). Графическим образом вязкости в координатах (у', т) является тангенс угла наклона графика к оси у'.
Материалы с сильно выраженными неньютоновскими свойствами имеют разнообразные зависимости у' от т. Простейшая из них — это прямая течения идеального пластика (тела Шведова — Бингама, рис. 3.80). Аналитически она описывается уравнениями (3.10.15). Реологическое поведение идеального пластичного материала исчерпывающе характеризуется двумя константами т5 и т|*. Величина т|* — «пластическая вязкость» — по смыслу отличается от ньютоновской вязкости т|. На основании уравнения (3.10.15) вязкость по Бингаму:
ц* = (т-т5)/у' (3.10.19)
есть тангенс угла наклона графика у'(т) (рис. 3.80) к оси у' и имеет смысл только при т > ту, поскольку при т < материал не течет. Из уравнения (3.10.19) видно, что т|* (точнее у'т|*) учитывает только ту часть полного сопро
тивления деформации т, которая пропорциональна скорости сдвига. Другая часть сопротивления, не зависящая от /, учитывается величиной сдвиговой прочности ту. В отличие от этого ньютоновская вязкость q = т /у' пластичного материала учитывает все сопротивление деформации:
q = q* + x4./y' (3.10.20)
или
г| = г|*т/(т-т4). (3.10.20а)
Она складывается из постоянной величины q *, которая называется пластической вязкостью, и переменного слагаемого / у' (структурной составляющей вязкости), которое в форме вязкости учитывает сопротивление, обусловленное наличием прочности на сдвиг тЛ.
Таким образом, поведение пластичного материала при деформировании может быть представлено в форме уравнения Шведова — Бингама (3.10.15). Тогда оно характеризуется двумя константами xs и q* или в форме уравнения Ньютона (3.10.2). В последнем случае характеристикой реологических свойств материала является переменная ньютоновская вязкость в формуле (3.10.20). Оба способа описания равноправны, но первый имеет преимущество, например, при инженерных расчетах, когда удобнее оперировать двумя константами вместо одной переменной величины. Однако это преимущество полностью теряется для реальных пластиков с нелинейной зависимостью скорости сдвига от напряжения (рис. 3.81); для материалов, обладающих ползучестью (рис. 3.82), и других систем, у которых пластическая вязкость q*, определяемая формулой (3.10.19), становится переменной величиной.
То же самое можно сказать о дифференциальной вязкости t/q/dfy', которая в случае ньютоновских материалов совпадает с «интегральной» ньютоновской вязкостью (3.10.18), а в случае пластичных сред — с пластической вязкостью.
Принципиально важное отличие материалов, обладающих ползучестью (рис. 3.82), заключается в том, что их прочность (предельное напряжение сдвига) является кажущейся. На самом деле материал способен течь и при сколь угодно малых напряжениях, но при его увеличении до некоторой критической величины тс скорость течения начинает резко увеличиваться. Это напряжение называется динамическим предельным напряжением сдвига. В эксперименте не всегда легко определить характер предельного напряжения, т. е. является оно статическим (истинной прочностью) или динамическим (кажущейся прочностью). Дело здесь не в том, что точность (длительность) измерения деформации имеет предел, а в том, существуют ли объективные критерии, позволяющие отличить ползучесть от обычного течения.
Удобство того или иного уравнения является не единственным и не самым важным критерием выбора способа описания реологического поведения. Более
Физика, химия и технология дисперсных систем
675
важна возможность сравнения различных материалов по их реологическим свойствам. Очевидно, что сравнивать можно величины, имеющие один и тот же смысл: ньютоновскую вязкость — с ньютоновской, пластическую — с пластической и т. д. Естественным эталоном сравнения различных текучих материалов служат ньютоновские жидкости, поэтому в качестве сравниваемых величин следует однозначно предпочесть ньютоновские вязкости даже для «неньютоновских» материалов, т. е. отношение напряжения к скорости деформирования. Иногда это отношение называют эффективной вязкостью, что существа дела не меняет, но вносит неоднозначность в терминологию. Не всегда однозначен и смысл предельного напряжения сдвига.
Показанные на рис. 3.80-3.82 реологические кривые представляют собой частный случай более общей зависимости у' от т (рис. 3.83), известной под названием «полная реологическая кривая» (ПРК).
При малых скоростях сдвига ход ПРК совпадает с прямой, отражающей течение ньютоновской жидкости С ВЫСОКОЙ (т]гаах) вязкостью, при высоких скоростях — с прямой низковязкой (т|пип) ньютоновской жидкостью, а в интервале промежуточных скоростей — с ходом зависимости у' от т пластичного материала с некоторым значением динамического предельного напряжения хс.
Экспериментально наименее изучен участок высоких скоростей течения (напряжения порядка тт и более). Он трудно достижим из-за перехода системы к турбулентному режиму течения, при котором теряется смысл понятия вязкости, введенного для описания ламинарного режима течения. Без этого участка ПРК переходит в кривую течения ползучего материала (рис. 3.82). Если, кроме того, вязкость т]тах настолько высока, что начальный участок кривой течения практически сливается с осью абсцисс (материал не течет при т < тД то ПРК переходит в кривую течения пластичного материала (рис. 3.80). Таким образом, реологическое поведение материалов рассмотренного типа относительно полно характеризуется четырьмя реологическими параметрами тс, rm, pmax, rjmin. Они задают две точки (т(., Лтах) И (тто, т]тт), которые вместе с точкой в начале координат позволяют аппроксимировать показанную на рис. 3.83 полную реологическую кривую тремя прямолинейными отрезками. Число реологических констант может быть и меньше (например, для идеального пластика), и больше, когда три упомянутых прямых отрезка переходят в плавную S-образную кривую ПРК. Плавные переходы от одного режима деформирования к другому могут быть результатом «смешивания» однотипных материалов с различными значениями реологических параметров, но могут быть присущи и самому механизму течения материалов определенного типа.
Известен еще один принципиально иной тип реологических кривых, отличающийся тем, что с увеличением напряжения скорость деформирования увеличивается медленнее, чем по линейному закону. Материалы с такими свойствами называют дилатантными.
а)
Рис. 3.80. Реологические кривые скорости деформации (а) и вязкости (б) пластичного материала: т| — вязкость; т|* — пластическая вязкость материала
Рис. 3.81. Реологическая кривая течения нелинейно-пластичного материала
Рис. 3.82. Реологические кривые течения (а) и вязкости (б) псевдопластичного (ползучего) материала
Рис. 3.83. Реологические кривые течения (полная реологическая кривая) (а) и вязкости (б) тиксотропной дисперсной системы
б)
Обобщением формальной стороны закона Ньютона на случаи нелинейной зависимости у' от т является эмпирическая формула (степенной закон):
т = р(уТ+1-
(3.10.21)
676
Новый справочник химика и технолога
Ньютоновская вязкость «степенной» жидкости:
П = Р(/)" (3.10.22)
у дилатантных жидкостей (п > 1) растет с увеличением скорости сдвига и падает у ползучих материалов (п < 1). Здесь Рил — эмпирические константы, отражающие свойства материала. На основании уравнений (3.10.21), (3.10.22) смысл этих констант может быть установлен формально, например Р численно равно вязкости при скорости деформирования / = 1, но отличается от нее по размерности, причем размерность вязкости зависит от показателя п. Величина этого показателя используется как формальный признак принадлежности исследуемых материалов к дилатантному (и > 1) или тиксотропному (п < 1) типу. Константы упомянутой выше линейно аппроксимированной ПРК, которая описывается тремя уравнениями, имеют более определенный смысл:
Т = Птах/ При Т < Тс,
х = тс + т|*/ при хт > х > тс, (3.10.23)
t = Timin/ ПрИТ>Т„,.
Он может быть истолкован с помощью механической модели материала, которая должна быть несколько сложнее рассмотренных ранее (рис. 3.78). В частности, сухое трение должно быть заменено трением через тонкий слой очень вязкой жидкости. С целью физико-химического толкования этих и др. реологических параметров необходимо: установить причины появления пластических и прочих свойств, установить зависимость величины констант от состава и структуры деформируемой среды, выявить пределы применимости тех или иных законов течения и т. д. Для этого необходимо определить физико-химическую сущность самого процесса деформирования дисперсных систем, которая связана, прежде всего, с понятием «структура дисперсной системы» и явлением структурирования. Следует иметь в виду, что не все упомянутые выше параметры, в том числе максимальная вязкость r|max, на самом деле характеризуют исследуемый материал, несмотря на их достаточно широкое применение в научной и технической литературе, а также в программных продуктах ЭВМ для моделирования течения различных жидкостей. Выяснение причин того или иного поведения дисперсных систем на основе их теоретических моделей, а также смысла и области применения различных параметров реологических законов составляет содержание последующих четырех подразделов. В частности, будет показано, что величина максимальной вязкости зависит от конструктивных параметров приборов, на которых она измеряется.
Особого внимания заслуживают тиксотропные системы. Суть явления «тиксотропия» заключается в изменении свойств материала при прикосновении к нему, т. е. под влиянием слабых механических воздействий.
Из опыта давно известно, что при таких воздействиях некоторые из них ощутимо разжижаются (уменьшается их вязкость), что и проявляется в виде нелинейной зависимости скорости сдвига от напряжения (рис. 3.82, 3.83). Поскольку уменьшение вязкости обусловлено изменением (разрушением) структуры дисперсной системы, то в коллоидной химии тиксотропия определяется как способность системы к обратимым изотермическим разрушениям и восстановлениям структуры. Здесь следует обратить внимание на изотермичность превращений, так как термическое разрушение структуры — это плавление материала. Другим видимым проявлением тиксотропности является зависимость вязкости от времени — после механического воздействия (например, встряхивания раствора) материал некоторое время сохраняет высокую текучесть (малую вязкость), что заметно и без применения приборов. По этой причине тиксотропия иногда трактуется как зависимость вязкости от времени. Зависимость свойств от времени в той или иной мере присуща любым материалам и поэтому не может считаться исключительной особенностью материалов одного типа. В связи с этим снова напомним, что здесь и далее обсуждаются закономерности установившегося течения, при котором никаких изменений во времени не происходит. Изучение и описание временной зависимости вязкости и других реологических констант могло бы составить содержание отдельной книги.
3.11. Структура и реология дисперсных систем
3.11.1. Структурная классификация дисперсных систем
Термин «структура» применительно к дисперсным системам и, по-видимому, не только к ним используется для характеристики системы с точки зрения наличия в ней той или иной упорядоченности образующих ее элементов в пространстве. Структурообразующие элементы дисперсной системы — это ее частицы, а иногда и определенные группы частиц, например флокулы. Часто и справедливо утверждается, что именно структура вещества или материала определяет его свойства. Тем не менее, только в редких случаях такие утверждения сопровождаются ссылкой на какие-либо количественные характеристики структуры. В лучшем случае дается чисто качественная визуальная характеристика (фотоснимок, рисунок, схема). Сказанное не относится к структуре кристаллов. Для того, чтобы появилась возможность количественного описания структуры и зависящих от нее свойств дисперсных систем, необходимо ввести параметры, количественно характеризующие структурное состояние дисперсной системы [9, 10].
Структура — это понятие, описывающее состояния дисперсной системы с точки зрения характера взаимного расположения частиц этой системы. Оно может изменяться под влиянием тех или иных воздействий на систему, например нагревания, перемешивания и т. д., поэтому для количественного описания структурного
Физика, химия и технология дисперсных систем
677
состояния необходимо ввести не просто величины, имеющие некоторое численное значение, а параметры состояния, значение которых изменяется и адекватно отражает изменение структурного состояния системы. Здесь и далее «параметр структурного состояния» — это общее наименование величин, с помощью которых можно количественно характеризовать особенности взаимного расположения частиц дисперсной системы или ее отдельных элементов, например флокул, образующихся при коагуляции коллоидного раствора.
Описание структуры дисперсных систем на качественном уровне предполагает три варианта, причем один из них выделяет неструктурированные системы, а два других относятся к состоянию, в котором взаимное положение частиц зафиксировано. В соответствии с такой классификацией структурных состояний вводится понятие «структурирования» — фиксации взаимного положения частиц. Соответственно, неструктурированные системы — это системы, в которых взаимное положение частиц не фиксировано.
1. Неструктурированные системы (это системы, в которых координаты и импульсы частиц независимы, т. е. положение, направление и скорость движения каждой частицы не зависят от положения и скорости других частиц). Такое состояние характерно для разбавленных, устойчивых к коагуляции систем. В физике используются понятия и величины, предназначенные для описания слабо выраженного структурирования молекулярных систем. Это корреляция, радиус корреляции, время корреляции. В обычных жидкостях радиус корреляции имеет величину порядка размера молекул. Это означает, что расстояние между соседними молекулами оказывается равным одной и той же величине чаще, чем это должно быть при их чисто случайном расположении. Время корреляции указывает на продолжительность пребывания пары частиц на упомянутом расстоянии. В идеальных газах корреляция отсутствует по определению, а в общем случае любое столкновение частиц или молекул является элементарным актом их корреляционного взаимодействия. В кристаллах радиус корреляции совпадает с размером кристалла, а время корреляции равно бесконечности.
2. Кристаллоподобные (периодические или квази-кристаллические) структуры (ПКС). В этом состоянии частицы зафиксированы в узлах правильной пространственной решетки. Такое состояние обычно создается путем увеличения концентрации частиц. При достаточно большой концентрации частицы сближаются в силу отсутствия свободного места в растворе на такие расстояния, что в их взаимодействии преобладают силы отталкивания, которые и обеспечивают равноудаленность частиц от ближайших соседей, т. е. происходит их выталкивание в узлы правильной решетки. При строго одинаковом размере частиц решетка настолько регулярна, что она действует на луч света как трехмерная дифракционная решетка, разлагая его на все цвета радуги, что и позволяет обнаружить ПКС.
3. Коагуляционные структуры. Как следует из названия, фиксация взаимного положения частиц в этих системах наступает в результате коагуляции (слипания частиц). При достаточной концентрации дисперсной фазы коагуляция ведет к образованию сплошной рыхлой сетки из взаимосвязанных частиц. Наличие определенной прочности такой сетки ведет к превращению жидкой текучей взвеси в желеобразное или пластичное состояние. Отсюда и название структурированного коллоида — гель (gel) — структурированный коллоидный раствор. Коагуляция — наиболее распространенная причина структурирования. Важным частным случаем коагуляционного структурирования является образование параллельных линейных цепочек из связанных между собой частиц при действии на дисперсную систему магнитного или электрического поля. С их изучения и началось становление современной теоретической реологии дисперсных систем.
В рамках теории устойчивости коллоидов (ДЛФО) коагуляция может происходить с преодолением потенциального барьера отталкивания частиц, а может происходить и без его преодоления при наличии достаточно глубокой потенциальной ямы на дальних расстояниях между частицами. В первом случае возникает непосредственный (фазовый) контакт частиц. Частицы могут при этом спекаться за счет перекристаллизации дисперсной фазы в зоне контакта. Структуры с таким видом связи называются кристаллизационными. Процесс структурирования, как и коагуляция, имеет в этом случае необратимый характер. Дисперсные системы с кристаллизационной структурой обладают свойствами хрупкого твердого тела. Во втором случае (безбарьер-ной коагуляции) связь частиц значительно слабее и она вполне обратима, т. е. легко разрушается и снова восстанавливается. Соответственно этому и состояние системы способно обратимо изменяться. Разрушение связей между частицами, а следовательно, и разрушение структуры, может быть вызвано слабыми механическими воздействиями, например перемешиванием раствора, переливанием его в другой сосуд и т. д. В состоянии покоя разрушенные связи, а с ними и структурное состояние системы полностью восстанавливаются. Количество циклов разрушения и восстановления структуры ничем не ограничено. Способность структурированных систем к обратимым изотермическим разрушениям и восстановлениям структурного состояния называется тиксотропией. Внешним признаком разрушения структуры может быть заметное разжижение взвеси. Восстановление структуры при этом сопровождается ее загустеванием. Этот процесс может занимать достаточно большое время (минуты, часы), а может происходить и практически мгновенно. Частным проявлением тиксотропии служит зависимость вязкости взвеси от времени, если восстановление структуры происходит достаточно медленно. «Мгновенное» тиксотропное восстановление структурного состояния и, соответственно, механических свойств дисперсных
678
Новый справочник химика и технолога
систем проявляет себя в виде другого хорошо известного и технологически чрезвычайно важного свойства материалов — их пластичности.
В определении тиксотропии следует обратить внимание на изотермичность структурных превращений. Дело в том, что связи частиц могут быть разрушены и при нагревании системы. При этом также будет наблюдаться ее разжижение. Термическое разрушение структуры — это процесс плавления. Способность плавиться является универсальной для любых структурированных (твердых) веществ. Изотермические структурные превращения (т. е. тиксотропия) возникают при механических воздействиях. Их роль в технологии исключительно велика.
Приведенная выше классификация не охватывает все возможные варианты структурирования. В соответствии с данным выше общим определением понятия «структура» необходимо уточнить и определение понятия «структурирование». Структурирование — это появление сильных корреляций (согласованности, взаимозависимости) координат и (или) импульсов частиц. Слабые корреляции, как уже отмечалось, могут возникать даже благодаря наличия у частиц размера. Полное же отсутствие корреляций присуще только газу математических точек. С другой стороны, структура — это не обязательно некоторое застывшее, зафиксированное положение частиц. Структурирование может заключаться в появлении согласованности динамических параметров частиц, например их колебания или вращения и т. д. Для пояснения этого обстоятельства полезно обратиться к тем видам структурирования, которые не упоминаются в ряду классических и поэтому требуют более обстоятельного описания.
Ориентация частиц, например параллельная ориентация длинных осей вытянутых частиц, дает самый очевидный пример структурирования, не требующего ни наличия связей между частицами, ни их неподвижности. Он подпадает под данное выше общее определение структурирования и под частное определение, предусматривающее фиксацию взаимного положения частиц. В данном случае это фиксация только угловых координат частиц. В общем случае состояние частиц задается не только их поступательными, но и вращательными степенями свободы.
Ориентация легко наблюдается по различным визуальным эффектам, если частицы отличаются по форме от сферических. Однако это вовсе не означает, что нельзя говорить об ориентационном структурировании в случае строго сферических частиц. Самый очевидный случай — это частицы с постоянным электрическим или магнитным дипольным моментом. Ориентация осей диполей в одном направлении под действием внешнего поля (а в некоторых случаях и без него) — типичный случай ориентационного структурирования, причем такого, которое сильно меняет свойства дисперсной системы. Самое известное свойство, приобретаемое взвесью при однородной ориентации всех ди
польных частиц, — поляризация или намагничивание всей взвеси в целом. Менее очевидно, что при этом изменятся также и механические свойства взвеси. У нее могут появиться те же признаки затвердевания, что и при коагуляционном структурировании, но выраженные слабее. По внешнему же виду ориентационно структурированная взвесь идеально сферических дипольных частиц неотличима от неструктурированной системы. К числу визуально фиксируемых признаков ориентации частиц относится различие оптических свойств коллоидов в разных направлениях (оптическая анизотропия). Оптическая анизотропия указывает на различие оптических свойств частиц (показатель преломления, форма) вдоль разных осей частиц.
Ориентационное структурирование распространено гораздо шире, чем можно было бы думать. Причина этого в том, что существует достаточно широкий круг свойств, явлений и процессов, придающих частицам, в том числе сферическим, анизотропность, т. е. различие свойств (или состояний) вдоль разных направлений относительно тела частицы или относительно пространственных осей координат. Некоторые из них относятся к статическим свойствам, а другие — к динамическим, возникающим в процессе движения (вращения) частиц. Ограничимся примерами разного рода поляризации. Термин «поляризация» в широком смысле слова означает придание частицам анизотропности любого типа. Будем считать, что свойства частицы меняются вдоль одной из ее осей. Те точки на поверхности сферической частицы, которые находятся на противоположных концах оси, назовем северным и южным полюсами частицы (по аналогии с намагниченными телами).
Северное и южное полушария частицы могут иметь различную плотность, и тогда в поле силы тяжести частица будет ориентироваться легким концом вверх. При этом она может иметь сферическую или произвольную форму. Причины появления различий по плотности разнообразны: спекание, слияние, склеивание, соединение в процессе коагуляции частиц разной химической природы. Причиной гравитационной поляризации капель эмульсии и пузырьков пены может быть сползание вниз под действием силы тяжести мелких частиц твердого вещества, налипших на поверхность крупных капель или пузырьков пены. Такое сползание может быть обусловлено разностью температур в окрестностях северного и южного полюсов и оседанием частицы, благодаря которому обтекающие ее потоки среды смывают к «корме» частицы налипшие на нее молекулы и ионы (последнее проявляется в виде потенциала оседания дисперсной системы). При этом большее значение приобретает не градиент плотности частицы, а градиент поверхностного натяжения или электрического потенциала поверхности частицы.
Существуют механизмы, способные вызвать поляризацию частиц при их вращении. Так, вращение сосуда с эмульсией может вызвать деформирование капель центробежными силами. Синхронное вращение частиц
Физика, химия и технология дисперсных систем
679
относительно среды вызывается воздействием вращающегося электрического или магнитного поля на дисперсную систему. Однородное и синхронное вращение возникает также при обычном течении дисперсной системы. Дело в том, что течение всегда связано с наличием градиента скорости течения. Разные слои среды движутся с разными скоростями, и, соответственно, разные стороны частицы, находящейся в неоднородном по скоростям потоке, оказываются под воздействием слоев среды, имеющих противоположное направление течения, что и приводит к закручиванию частиц. Ось такого вращения лежит в плоскости равных скоростей течения среды и ориентирована в этой плоскости перпендикулярно направлению движения среды. Основной эффект, обусловленный свободным вращением частиц в потоке, состоит в уменьшении их гидродинамического сопротивления по сравнению с невра-щающейся частицей. Если каким-либо образом затормозить свободное вращение частиц, например фиксируя их ориентацию внешним полем (при наличии у частиц дипольного момента), то это вызовет увеличение гидродинамического сопротивления взвеси.
Своеобразные эффекты вызывает действие знакопеременного поля на частицы, обладающие постоянным дипольным моментом. При низких и умеренных частотах оси диполей успевают менять ориентацию на противоположную синхронно с изменением направления переменного поля. При жесткой связи оси диполя с телом частицы изменение ориентации возможно только путем механического поворота частиц, причем их поворот происходит синхронно. Однородное вращение частиц будет затруднено из-за трения между соприкасающимися частицами. Сила трения между соседними частицами будет сведена к нулю, если они будут поворачиваться во встречных направлениях — одна налево, а другая — соседняя — направо, не мешая, а даже помогая одна другой синхронно менять свою ориентацию. Одна частица при этом как бы катится по поверхности другой, и проскальзывание в точке контакта отсутствует. Наглядным примером, передающим особенности встречно-синхронного вращения частиц в ПКС, является блок из п зацепляющихся зубчатых колес (шестерней) радиусом г. Если шестерни образуют плоскую квадратную решетку, т. е. каждая из них сцеплена с четырьмя соседними, то помех их одновременному встречному вращению не возникнет. Половина колес будет вращаться в одну сторону, а половина — в другую. Полный момент вращения системы будет равен нулю. Однако если затормозить вращение только одного из зубчатых колес, то весь блок шестерен придет во вращение вокруг заторможенного колеса. Очевидно также, что шестерни, расположенные на диагонали квадратной решетки, вращаются в одну сторону, поэтому, если блок из нескольких колес (частиц) опирается на неподвижную стенку сосуда только диагональными частицами, то весь блок придет в движение относительно опоры (плоскости ВВ на рис. 3.84). Кру
тящий момент, вызывающий движение блока, равен при этом сумме п крутящих моментов т, приложенных к каждой частице (колесу) блока, т. е. это может быть очень мощное воздействие. Суммарная сила, вынуждающая блок двигаться, равна пт / г. На стенку, ориентированную параллельно базовой плоскости АА кристаллоподобной структуры, действуют знакопеременные силы с нулевым суммарным результатом. Такие стенки тормозят направленное движение блока вращающихся частиц (колес).
Концентрированная суспензия в вязкой среде вполне аналогична системе зацепляющихся зубчатых колес. Различие в том, что синхронизация вращательных движений будет здесь обусловлена не жестким зацеплением зубцов, а гидродинамическим взаимодействием частиц — их трением через тонкие прослойки вязкой жидкости между соприкасающимися частицами. Отсутствие абсолютной жесткости такого зацепления допускает полную синхронизацию встречных вращений частиц только в пределах отдельных областей суспензии (вращательных доменов). Вращательное состояние соседних доменов может несколько различаться, а их взаимодействие между собой способно вызвать медленное вращение доменов с частотой прядка cor/R, где со — частота переменного поля и R — радиус доменов.
Взаимодействие доменов со стенками и между собой вызывает видимое организованное движение всей суспензии [3]. Судя по скорости движений, наблюдаемых в поле частотой 50 Гц, размер доменов примерно на два-три порядка превышает размер частиц. В суспензии магнитных частиц в переменном магнитном поле характер движения определяется условиями на границах суспензии. В сосуде со свободной верхней границей суспензии и горизонтальной ориентацией магнитного поля возникает встречное течение суспензии от полюсов электромагнита к середине сосуда. Если между полюсами электромагнита свободно подвесить каплю магнитной суспензии, то в поле она вытягивается в жгут, и на его поверхности возникают движущиеся в произвольных направлениях утолщения (узлы). При встречном движении эти узлы свободно проходят сквозь друг друга, никак не взаимодействуя. Эти и другие динамические структуры являются типичными представителями так называемых диссипативных структур.
Рис. 3.84. Схема согласованного вращения зацепляющихся частиц при синхронном изменении их ориентации
680
Новый справочник химика и технолога
Диссипативная структура — это особое состояние сильно неравновесной системы. В таких системах происходит интенсивный перенос энергии, сопровождающийся ее потерями. Это может быть перенос теплоты от нагретого тела к холодному через слой жидкости или передача механической энергии одного движущегося тела другому через слой жидкости или самой жидкой среде. Это может быть также химическая реакция или передача энергии переменного поля частицам феррита и т. д. Течение этих процессов может принимать своеобразный, регулярный характер. Предпочтительность регулярного течения процесса обусловлена тем, что при прочих равных условиях (например разности температур) скорость переноса энергии увеличивается за счет включения дополнительных механизмов переноса. Классический пример диссипативной структуры — регулярные ячейки конвективных потоков среды при теплопередаче, если нагретое тело расположено внизу, а холодное — вверху. В этом случае теплопередача интенсифицируется за счет конвективного переноса теплоты в дополнение к нормальной теплопередаче неподвижной теплопроводной средой. Обычные волны на поверхности воды служат другим примером диссипативной структуры. Здесь, наряду с пространственной регулярностью возмущений поверхности, возникает и регулярность изменения состояния поверхности во времени. Пример чисто временной регулярности дают некоторые колебательные химические реакции. Внешне периодичность реакции может проявлять себя в том, что цвет раствора периодически с частотой несколько раз в минуту изменяется, например, с красного на синий и обратно. Такие колебания продолжаются до окончания реакции, длящейся десятки минут.
Структурирование отнюдь не ограничено «пожизненной» фиксацией взаимного расположения частиц, т. е. статическими структурами покоя. С другой стороны, и диссипативные структуры более разнообразны, чем принято считать. Самопроизвольная перестройка структуры, направленная на интенсификацию диссипативных процессов, происходит при течении дисперсных систем. В частности, коагуляционная структура нерегулярна по своей природе. В состоянии покоя ее можно представлять как сплошную хаотичную сетку из слипшихся частиц. При течении она разрушается на фрагменты, которые лишь в среднем характеризуются неким общим параметром, например размером. Достаточно очевидно, что чем большее деформирующее усилие приложено, тем сильнее (на более мелкие фрагменты) будет разрушена структура, и тем меньшее сопротивление течению дисперсной системы она будет создавать.
Это интенсифицирует течение, что как раз и характерно для диссипативных структур. Таким образом, динамические структуры при течении являются также и диссипативными, хотя и не обязательно регулярными. Следует также отметить, что возникновение диссипативной структуры в некоторых системах не обязательно
связано с потерями энергии именно в этих системах. Примером могут служить упоминавшиеся выше ячейки конвекции в слое флюидной (текучей) среды, через которые осуществляется перенос теплоты от нижней (теплой) к верхней (холодной) границе слоя. Потери энергии в слое переносчика теплоты составляют малую величину от потерь энергии нагревателя, отдающего ее холодильнику. Гипотетически потери в переносчике энергии могут быть сведены и к нулю. Например, потери энергии в вакууме при прохождении электромагнитных волн равны нулю. В других случаях среда полностью поглощает подводимую к ней энергию. Так обстоит дело при течении вязких жидкостей.
Основная задача теоретической реологии заключается в нахождении законов течения (уравнений реологии) дисперсных систем с известной рецептурой. Из сказанного выше следует, что универсальный алгоритм решения задачи включает в себя получение уравнения структурного состояния дисперсной системы — соотношения, описывающего зависимость параметров структурного состояния от интенсивности процесса деформации. В качестве параметра состояния выступают те величины, от которых при данном типе структуры зависит сопротивление деформированию, а мерой интенсивности процесса деформирования может быть величина действующего в системе напряжения или скорость ее деформирования. Таким образом, структура и структурное состояние при деформировании являются центральным звеном в решении основной задачи реологии.
Завершая обзор различных видов самоорганизации дисперсных систем, можно отметить, что пока эта область физико-химической механики дисперсных систем даже не определилась в части своего содержания. В основном изучается только тот круг проблем, который связан со статическим состоянием структурированных систем и трансляционными степенями свободы.
3.11.2. Неструктурированные системы
Несколько утрируя роль различных признаков того или иного структурного состояния дисперсных систем, можно сказать, что неструктурированная система — это система, которая исчерпывающе характеризуется единственным параметром — концентрацией дисперсной фазы. Отсюда следует, что ее структурное состояние не зависит от интенсивности деформирования и, следовательно, она является ньютоновской жидкостью. Это следует из постулата Ребиндера: изменение вязкости всегда является результатом изменения структуры дисперсной системы. Такой вывод правомерен, если концентрация является параметром структурного состояния системы. Параметр состояния — это величина, от которой зависят реологические свойства дисперсной системы. Полагая концентрацию единственным параметром структурного состояния, мы игнорируем такие факторы, как анизодиаметрия и возможность ориентации частиц в потоке, деформируемость капель эмуль
Физика, химия и технология дисперсных систем
681
сий в потоке и др. Поэтому здесь и далее речь будет идти о взвесях твердых сферических частиц. На это нужно обратить особое внимание, поскольку несфе-ричностью частиц часто спекулируют при объяснении структурирования, неньютоновости и других структурно зависимых свойств систем. Кроме того, с целью решения чисто гидродинамических задач примем, что частицы лишены каких бы то ни было защитных оболочек, которые тоже могут деформироваться, смываться с поверхности частиц потоками среды и т. д.
Присутствие твердых недеформируемых частиц в жидкости по ряду причин ведет к увеличению сил вязкого сопротивления деформированию смеси. Для их понимания важно уяснить различие между скоростью деформации всей дисперсной системы и скоростью деформации той ее части, которая только и может деформироваться — жидкой фазы взвеси. Ее можно представить не в виде множества частиц, а в виде сплошного плоского слоя того же объема, что и все частицы. Этот слой можно мысленно приклеить к одной из пластин прибора, взаимное движение которых и вызывает сдвиговое деформирование заключенного между ними состава. Независимо от того, как мы представляем себе дисперсную фазу, скорость сдвиговой деформации суспензии — это отношение скорости и движения одной пластины относительно другой к расстоянию х между пластинами: у' = и/х. Скорость же деформации жидкой фазы y'f— это отношение той же относительной скорости движения пластин и к толщине f слоя жидкости между ними, поскольку сдвиг происходит только в слое жидкости. Его толщина f=x-d меньше, чем расстояние между пластинами прибора, на толщину d воображаемого слоя твердой фазы. Поэтому y'f= u/(x-d), или с учетом того, что и = ху', можно записать у'у = у'х /(x-d). После деления числителя и знаменателя нах находим:
У7=у7(1-Ф), (3.11.1)
где <р = dlx — объемная доля дисперсной фазы в дисперсной системе. Тот же результат получится, если воображаемый плоский слой твердого вещества не приклеивать к одной из пластин прибора, а поместить между ними и параллельно им. Также ничего не изменится, если один слой разделить на множество тонких параллельных слоев или даже на чешуйки. Отсюда следует, что полученный результат сохраняет силу и в случае взвеси сферических частиц.
Формула (3.11.1) дает возможность альтернативного описания одного и того же эффекта — удельного (на единицу площади пластин прибора) сопротивления т деформированию слоя вещества, заключенного между пластинами прибора. Оно может быть выражено как произведение неизвестной нам вязкости состава т] на скорость его деформации у' или как произведение известной вязкости т|о жидкой фазы на фактическую скорость у у ее деформации в том же составе, часть которого ф приходится на твердые недеформируемые части
цы. Разумеется, что оба дают одну и ту же величину, поэтому т|у' = Т|оу7(1 - ф), откуда:
Т]=Т]о/(1-Ф). (З.П.2)
При ф = 1 формула дает ожидаемый результат — бесконечно большую вязкость. В указанном специфическом случае она абсолютно точна во всем диапазоне концентраций дисперсной фазы, но не учитывает всех факторов, определяющих вязкость взвеси обычных частиц.
Дополнительное сопротивление сдвиговому движению соседних слоев суспензии возникает вследствие того, что при обтекании частиц средой траектории движения струй искривляются, т. е. удлиняется путь, и увеличиваются потери энергии на поддержание заданной скорости деформации дисперсной системы. Решение соответствующей гидродинамической задачи дано Эйнштейном и приводит к уравнению
т| = т|0(1 +аф). (3.11.3)
Здесь а — коэффициент формы частиц, равный для сфер 2,5.
Формула Эйнштейна дает точные результаты только при малых концентрациях дисперсной фазы. Особенно показателен в этом отношении предельный случай Ф = 1, когда вязкость оказывается всего лишь в 3,5 раза больше, чем вязкость среды, тогда как она должна быть равна бесконечности. Из двух формул можно сконструировать их общий прототип:
Т| = т]о/(1 -Ф)а- (3.11.4)
Этот результат получен Бринкменом из формулы Эйнштейна путем вычисления вязкости суспензии объемом И после добавления к ней еще одной частицы известного объема v. Роль т|0 в формуле (3.11.3) в этих расчетах играет вязкость суспензии до введения в нее дополнительной частицы, и, соответственно, роль концентрации ф — величина v/(K+ v). При малой концентрации формула (3.11.4) переходит в формулу (3.11.3). При большой концентрации она дает качественно те же результаты, что и формула (3.11.2). Последняя, в отличие от формулы Эйнштейна (3.11.3), учитывает лишь факт присутствия недеформируемых частиц во взвеси и игнорирует дополнительное увеличение локальных скоростей сдвига за счет обтекания частиц потоком среды.
Механизм повышения вязкости, обусловленный обтеканием частиц средой, становится нагляднее, если рассмотреть задачу о потерях энергии, обусловленных вращением частицы в вязкой среде (рис. 3.85).
Рис. 3.85. Вовлечение частицы во вращательное движение сдвиговым течением среды
682
Новый справочник химика и технолога
Момент силы трения М, действующий на частицу, вращающуюся с угловой скоростью со, равен/йцсо, где v — объем частицы и f — фактор формы. Соответственно, потери энергии в единицу времени на поддержание заданной скорости вращения частицы равны <оЛ/. Потеря энергии q в единице объема (диссипация энергии) будет тогда fnvx\ <о2 (где п — концентрация частиц) или ^=У<рг|«)2, поскольку произведение nv есть объемная доля <р дисперсной фазы. При наличии градиента скорости течения частицы вращаются с частотой со = у' / 2.
Фактор формы для сфер /=6. Подстановка этих значений в выражение для диссипативной функции дает уравнение
? = (3/2)q>iiY')2- (З.И.5)
Потери энергии, обусловленные протеканием различных необратимых процессов, аддитивны. Соответствующие им силы вязкого сопротивления и вязкости, которые получаются по правилам x-q/y' и т| = т/у', также будут подчиняться правилу аддитивности. Поэтому из выражения (3.11.5) можно получить составляющую вязкости г]и = (3/2)фц0, которая обусловлена вращением частиц в среде с вязкостью т|0. Если добавить ее к эффекту присутствия частиц (3.11.2), можно получить полную вязкость дисперсной системы. Нагляднее использовать вариант формулы (3.11.2) при малой концентрации дисперсной фазы:
ц = т]о(1+ф). (3.11.6)
Добавление к этому выражению вращательной части вязкости т|ю = 1,5фт|о приводит к формуле Эйнштейна. Этот результат подтверждает правомочность метода, который использовался для его получения, — разложения сложной гидродинамической картины течения на аддитивные по диссипации энергии составляющие. Он и далее будет применяться, в частности при рассмотрении эффекта вращательной вязкости, создаваемого ограничением вращательной подвижности частиц (см. ниже).
3.11.3. Ориентационное структурирование
В простейшем случае описание ориентационной структуры сводится к констатации самого факта наличия ориентации частиц. Количественная характеристика структурного состояния состоит, как минимум, в указании направления ориентации. Тогда сразу возникает потребность в приписывании дисперсной системе некоторой особой оси (директора), относительно которой можно было бы вести отсчет направлений ориентации частиц. В качестве таковой может выступать направление внешнего поля, действующего на дисперсную систему, направление течения взвеси, направление движения частиц (например в поле силы тяжести), направление температурного градиента и т. д. Направление ориентации отнюдь не всегда совпадает с направлением директора даже в случае электрического поля, в
том числе по причине одновременного воздействия на частицу нескольких ориентирующих факторов, например электрического поля и гидродинамических сил (течения). В ряде случаев кроме направления ориентации для описания структурного состояния необходима и некоторая количественная мера, характеризующая степень ориентированности частиц. В частности, ориентации может препятствовать вращательное броуновское движение частиц (вращательная диффузия). Характеристикой степени ориентации в этом и в других подобных случаях является среднее значение косинуса угла между направлением ориентации частиц и осью анизотропии взвеси. Ось анизотропии — это направление ориентации частиц при отсутствии возмущающих ее факторов (вращательной диффузии, турбулентности потока и др.). Предполагается, что в отсутствие ориентирующего фактора (поля) оси частиц равномерно распределены по азимутальным углам полярной системы координат. Иначе говоря, доля частиц, оси которых лежат в пределах некоторого узкого интервала угловых координат, определяется законом распределения Больцмана по потенциальным энергиям частиц. Их потенциальная энергия зависит только от полярного угла 0. В дальнейшем будет рассматриваться в основном ориентационное структурирование и сопутствующие ему эффекты на примере частиц, имеющих постоянный дипольный момент р. В электрическом поле с напряженностью Е потенциальная энергия частиц равна pEcos®, а энергия частиц с постоянным магнитным моментом т в магнитном поле с напряженностью Н равна Pow//cos0, где Цо — магнитная проницаемость среды, которая в случае обычных (немагнитных) сред практически совпадает с магнитной постоянной вакуума 4л • 10”7 Гн/м. Косинус угла 0 между направлением поля (директора углового распределения) и осью диполя определяет его вклад pcos® в поляризацию (в намагниченность /wcos0) системы, а сумма этих величин по всем частицам дает величину поляризации Р или намагниченности М* взвеси, так что сами эти величины являются мерой ориентационного структурирования. Соответственно средняя величина cos0 имеет смысл относительной величины поляризации P/Ps (намагниченности М * / М*), т. е. отношения поляризации (намагниченности) при данной напряженности поля к поляризации Ps (намагниченности Л/‘) в состоянии насыщения, соответствующем полной ориентации осей частиц параллельно полю. В этом состоянии cos0= 1, и поэтому Ps = пр и М* = пт, где п — концентрация частиц (1/м3).
Зависимость относительной поляризации или намагниченности взвеси жесткодипольных частиц от напряженности поля описывается функцией Ланжевена
L(z) = cth(z) - 1 tz, (3.11.7)
где z=pE/kT при электрической и z=^mHlkT при магнитной поляризации системы. По смыслу эта функция совпадает со средней величиной косинуса угла ме
Физика, химия и технология дисперсных систем
683
жду направлением поля и ориентацией осей диполей, соответственно pL(z) есть средняя величина проекции дипольного момента частицы на направление поля при данном значении z аргумента функции Ланжевена (см. подраздел 3.9).
В сильном поле, т. е. при z » 1, наступает насыщение поляризации (ориентационного структурирования). В этом случае L(z) = 1 и структурное состояние системы характеризуется единственным параметром — направлением ориентации частиц, которое совпадает с направлением действующего на дисперсную систему поля, если оно является единственным фактором ориентационного структурирования. Такая предопределенность состояния дисперсной системы делает его малоинтересным и неинформативным. Оно не зависит ни от напряженности поля, ни от размера частиц, ни от температуры в очень широком диапазоне их варьирования. Например, в суспензии частиц феррита размером 0,1 мкм при нормальной температуре насыщение наступает в поле напряженностью 1-2 А/м, что примерно в 100 раз слабее напряженности геомагнитного поля. Иначе говоря, естественное поле Земли доводит суспензию до такого состояния, что никакое воздействие внешнего поля его уже не изменяет, если не считать возможного изменения ориентации магнитных осей частиц. Разнообразие состояний возникает, если не ограничиваться созерцанием покоящейся в поле взвеси и рассмотреть более сложные условия структурирования, в частности одновременное воздействие магнитного поля и течения. Как уже отмечалось, при течении взвеси на частицы действует момент сил трения Mh, заставляющий частицы непрерывно вращаться в потоке. Величина этого момента:
MA=/vr|(o, (3.11.8)
где со = у72 — частота свободного вращения в потоке, численно равная половине градиента скорости у' сдвигового течения взвеси, f — фактор формы (равен 6 для сферических частиц), v — объем частицы и т] — вязкость среды. Момент силы, действующей на частицу в сильном (в смысле насыщения ориентационного структурирования) электрическом поле:
A^pEsin© (3.11.9)
или в сильном магнитном поле:
Mf= pomHsin©, (3.11.10)
удерживает частицы от вращения. Здесь 0 — текущее (неравновесное) значение угла между направлением поля и оси диполя. Последние две формулы получаются из приведенного выше выражения (3.9.74) для энергии диполей дифференцированием энергии по углу.
Воздействие гидродинамического момента сил на ориентированные полем частицы приводит к тому, что оси диполей будут повернуты относительно направле
ния поля на угол 0S, величина которого в состоянии равновесия определяется из условия равенства действующих на частицу моментов сил:
sin©5 = Уп со / poMsH, 0,v = arcsin(/r| со / \ХоМ3Н). (3.11.11)
Эти формулы относятся только к магнитным частицам. Дискриминация электрического аналога в этих и других формулах будет проводиться и в дальнейшем. Для этого есть ряд веских причин. Первая состоит в том, что имеющаяся во многих случаях идентичность магнитных и электрических эффектов делает излишним дублирование формул. Различие заключается в вычислении энергии и момента сил, которое иллюстрировано приведенными выше формулами, в частности формулами (3.11.9) и (3.11.10). Вторая причина — различие в доступности для экспериментирования ориентационного структурирования в электрическом и магнитном полях. Структурирование электрическим полем достигается только в специальных случаях, а возможность измерения электрической поляризации также сопряжено с рядом трудностей. Измерение статической электрической поляризации и вовсе неосуществимо. Магнитное поле в этих отношениях является предпочтительным. Единственное, о чем необходимо позаботиться, — это подбор дисперсной фазы. Она должна быть магнитной. Никаких других ограничений, в том числе относительно природы среды, не существует. Это может быть диэлектрическая жидкость или раствор электролита высокой концентрации, это может быть даже расплавленный металл, что, кстати, позволяет достичь температуры Кюри магнитного материала и поставить сравнительный эксперимент с одной и той же системой при магнитном и немагнитном состояниях дисперсной фазы. Все эффекты магнитной поляризации и структурирования могут быть реализованы и исследованы экспериментально, тогда как с электрической поляризацией это вряд ли возможно. Наконец, третья причина, по которой далее будет отдаваться предпочтение ферромагнитным системам, — отсутствие трудностей с вычислением и с измерением величины магнитного дипольного момента частиц: в случае однодоменных частиц или в состоянии насыщения многодоменных частиц их магнитный момент легко вычисляется по формуле
m = vMs. (3.11.12)
Именно это соотношение использовано в предыдущей формуле, что позволило вообще исключить из нее величину дипольного момента, оставив вместо него основную магнитную характеристику дисперсной фазы Ms — ее намагниченность насыщения. Примечательно, что, согласно гипотезе Н.А. Толстого, величина постоянного электрического дипольного момента частицы в полярной среде пропорциональна ее поверхности, а не объему, так что влияние размера частиц на эффекты электрической и магнитной поляризаций существенно различается. Вычисление индуцированного электриче
684
Новый справочник химика и технолога
ского дипольного момента частиц требует, как правило, дополнительных исследований свойств среды и дисперсной фазы.
Согласно формуле (3.11.11), в потоке или при вращении сосуда с суспензией в неподвижном магнитном поле, а также во вращающемся магнитном поле направление осей магнитных диполей, т. е. направление намагниченности дисперсной системы, не совпадает с направлением действующего на нее поля. Угол между ними растет с увеличением скорости вращения (течения) и в пределе может достигать 90°. Это максимально возможное значение угла отставания оси диполя от направления магнитного поля. Дальнейшее увеличение скорости вращения до величины со > со*, где
<о, = ЦоИЯ//г| (3.11.13)
— критическое значение частоты вращения, приводит к срыву ориентации, так как при этом гидродинамический крутящий момент превышает магнитный момент сил и поэтому равновесная ориентация частиц становится невозможной. Существенно, что величина ориентирующего магнитного момента сил не испытывает насыщения с увеличением напряженности поля, как это происходит с намагниченностью. Поэтому с увеличением напряженности поля можно выйти из за-критического диапазона скоростей. Следует, однако, иметь в виду, что увеличивать напряженность поля свыше коэрцитивной силы частиц Нс не имеет смысла. Иначе говоря, существует абсолютное значение критической частоты:
<ое = цоИНе//г|, (3.11.14)
выше которой уже невозможно предотвратить срыв ориентационного структурирования увеличением напряженности поля.
Отсутствие равновесия при со > со* не исключает возможности ориентационного структурирования. В слабом поле частицы будут почти свободно вращаться в потоке, как будто поле вовсе отсутствует. Тем не менее, магнитное поле хотя и ненамного, но притормозит вращение частиц в момент прохождения ими угла, близкого к 90°, при котором величина Mf максимальна, и ускорит это вращение при противоположной ориентации оси частицы относительно направления поля. Это означает, что вблизи 90-градусной ориентации ось каждой частицы находится больше времени, чем при противоположной (270°) ориентации. Избыточное время нахождения осей частиц в указанном секторе углов означает, что взвесь будет иметь слабую намагниченность под углом 90° к направлению поля. Усиление поля, замедление вращения (течения), уменьшение вязкости приведет, очевидно, к усилению этого эффекта. В пределе он плавно достигнет намагниченности насыщения, направленной под углом 90° к направлению намагничивающего поля. Таким образом, упомянутый выше срыв ориентации с увеличением скорости враще
ния (переход за критическую частоту вращения) не будет сопровождаться срывом намагничивания. Намагниченность более или менее плавно будет уменьшаться с увеличением частоты вращения поля или скорости сдвига в постоянном поле.
Наличие намагниченности при сверхкритической частоте расширяет определение понятия «структурирование». Дело в том, что задержки осей вращающихся частиц в секторе вблизи 90° происходят несинхронно, т. е. моменты времени, в которые оси разных частиц проходят отметку 90°, не совпадают. Иначе говоря, взаимное положение осей частиц в любой момент времени не зафиксировано, однако статистически предпочтительная ориентация осей частиц существует, поэтому можно говорить о структурировании. Подчеркивая особенность такого структурирования, будем называть его асинхронным. На мгновенном снимке оно отобразится обычным образом: количество частиц, ориентированных своими осями в определенном направлении, будет больше, чем в других направлениях. Аналогично выглядит обычное слабо выраженное статическое ориентационное структурирование в покое, возникающее при значении аргумента функции Ланжевена меньшем единицы. Следовательно, такое структурирование является тоже асинхронным. Напомним, что естественной мерой структурированности в таких случаях является значение функции Ланжевена.
Количественное описание динамического асинхронного структурирования можно сделать на основе уравнения вращательного движения частицы. Удобно считать, что вращается сосуд с магнитной суспензией, а магнитное поле неподвижно и направлено перпендикулярно оси вращения. Уравнение движения частицы получается в данном случае приравниванием ориентирующего момента магнитного поля po^Hsin© и опрокидывающего частицы гидродинамического момента /vr|(co - dQ/dt), который пропорционален скорости вращения частицы относительно среды (со - d&ldt). Последняя равна разности скоростей вращения сосуда и частицы d®/dt относительно неподвижной системы координат. Единственной имеющей значение координатой является в данном случае угол 0 между направлением поля и направлением оси диполя. Таким образом, уравнение движения частицы имеет следующий вид:
wBsin© =/vr|(co- d& / dt). (3.11.15)
Здесь для сокращения введена величина В = —
индукция магнитного поля. Уравнение удобно представить в виде формулы для скорости вращения частиц:
d©/^ = <o-Z>sin@. (3.11.16)
Здесь b = mB/jvx\. В состоянии статического равновесия частицы не вращаются, т. е. dQ/dt = 0. Отсюда и следует выражение (3.11.11) для равновесного угла ориентации осей частиц в потоке (во вращающемся сосуде или вращающемся поле). Равновесие возможно
Физика, химия и технология дисперсных систем
685
только при со = Ь, так как sin© = со/6 должен быть по модулю меньше единицы или, в крайнем случае, равен ей. Если первоначально сосуд с суспензией покоился, а затем начал вращаться, то, согласно формуле (3.11.16), скорость вращения частиц в первый момент времени совпадает со скоростью вращения сосуда, затем замедляясь с увеличением угла 0, достигает при равновесном значении угла нулевой величины, и поэтому дальнейшее вращение прекращается. При закритической частоте (со > b или со > со*) даже при максимальном значении 6sin0, когда sin© = 1, вращение не прекращается, и поэтому статическое равновесие частицы при одновременном действии магнитного и гидродинамического полей невозможно.
Для нахождения количественной меры структурированности (намагниченности) при закритической скорости вращения суспензии найдем время dt прохождения вращающейся частицей некоторого малого углового расстояния <70:
dt= J0 / (со - 6sin0). (3.11.17)
В секторе углов от 0 до л sin© > 0, и поэтому время больше, чем в отсутствии поля (6 = 0). В противоположном секторе, т. е. при значении угла равном (0 + л), sin(0 + л) = -sin© и, согласно уравнению (3.11.17), время прохождения такого же по длине углового отрезка пути <70 в этом секторе сокращается и равно d/ = d©/(co+6sin0). Разность А/ времен прохождения двух противоположных секторов 26sin0d0/(co2 -62sin20) есть избыточное время пребывания оси диполя в окрестности угла 0, избыточная доля которого Az / То и определяет вклад одной частицы rnkt/To в намагниченность с7Л/*(0) = nm&tl То взвеси в этом направлении. Здесь То — период вращения частиц и п — их концентрация. Легко заметить, что скорость вращения частицы минимальна при 0 = л/2, и распределение скоростей симметрично относительно л/2. Следовательно, максимум намагниченности лежит при 0 = л/2, а его величина Л/* есть интеграл от проекции A/*(0)sin0 углового распределения намагниченности Л/*(0) на направление л/2 в пределах сектора от 0 до л:
Л/* = 2(bnml To)bm20J0/(co2 - 62sin20). (3.11.18)
Для его вычисления требуется численный метод интегрирования. Период обращения То частицы вычисляется интегрированием выражения (3.11.17). Соответствующий табличный интеграл существует:
при со2 < Ь2
t = (l/p)ln{[cotg(0/2) + b - Р]I [cotg(0/2) + b + P]} ; ’ , 3 = 1/(co2 - 62)1/2, (3.11.19)
при co2 > b2
t = (2/a) arctg{[cotg(0/2) + 6] / a} ] \,
a = 1 / (62 - co2)1/2. (3.11.20)
Символ 12 означает, что для вычисления времени t поворота оси частицы из положения ©i в положение 02 необходимо найти разность значений выражения при второй (02) и первой (©]) величинах угла. Выражение (3.11.19) имеет смысл применять для вычисления времени установления равновесной ориентации частиц (©1 = 0, 02 = 0S). Выражение (3.11.20) неудобно для вычисления периода вращения частицы (©] = 0, 02 = 2л), так как включает в себя функцию tg(0/2), которая равна нулю на границах этого интервала интегрирования и содержит два бесконечных разрыва внутри него. Значения периода вращения частицы можно легко получить численным интегрированием уравнения (3.11.17), которое удобно выполнять одновременно с нахождением интеграла (3.11.18).
Результаты расчетов показывают, что вблизи со* угловое распределение намагниченности очень острое и намагниченность близка по величине к статической намагниченности. С увеличением частоты распределение размывается, а намагниченность асимптотически приближается к нулю. Время установления стационарной величины намагниченности сравнимо с периодом обращения частицы.
Поведение системы жестких диполей в знакопеременном, как и во вращающемся, поле зависит от соотношения частоты со поля и времени переориентации частицы. В произвольный момент времени на магнитный диполь, ось которого направлена под углом 0 к оси поляризации переменного поля, действует момент сил Mf= wBt,sin©sin(coZ). Здесь Ва — амплитуда индукции переменного поля и Basin(coZ) — ее мгновенное значение в данный момент времени t. Магнитному моменту сил противодействует равный по величине момент сил сопротивления среды Mh=fvx\d®ldt. Время перемещения оси диполя на некоторую угловую величину А0 получается при решении уравнения баланса моментов сил, где sa = тВа!fw\&.
t/0/sin© = -sa sin(cot) <7(cot). (3.11.21)
После интегрирования оно дает уравнение
ln[tg(02/2)/tg(©i/2)] = sa [cos(coZ2) - со8(со^)]. (3.11.22)
Здесь coZ) и wZ2 представляют собой соответствующие моментам времени и Z2 начальную и конечную фазы изменения переменного поля. В частности, изменение угловой координаты оси диполя за время перемены знака поля (coZi = 0, coz2 = л) будет:
tg(02/2)/tg(01/2) = exp(-2sa). (3.11.23)
Наибольшая частота, при которой частица успевает изменить свою ориентацию на противоположную, должна в принципе определяться из условия А0 = л. Однако отмеченные ранее особенности функции tg(0/2) не позволяют этого сделать. Физическая же причина в том, что вблизи 0 = 0 и 0 = л момент ориентирующей
686
Новый справочник химика и технолога
силы Mf стремится к нулю, и поэтому для достижения этих угловых координат требуется бесконечно большое время. По этой причине в качестве двух характерных положений оси диполя, переход между которыми будет характеризовать ориентационную подвижность частиц в поле, целесообразно принять 0 = 30° (л/6) и 0 = 150° (л-л/6). При такой ориентации оси диполя проекция дипольного момента на экваториальную плоскость полярной системы координат составляет половину от его величины. Тогда tg(?r/2 - л/12) / tg(n/l2) = 12, Ln 12 = 2,5, и характеристическая частота поля = тВа / 2,5/vr|. По порядку она близка к критической частоте вращающегося поля mBJfvx\, но несколько отличается от нее в меньшую сторону, так как средняя за период вращения величина действующего на частицу переменного поля меньше, чем постоянного (вращающегося) поля. При численной оценке условий движения частиц в обоих случаях можно пользоваться значением критической частоты, которое дают формулы (3.11.13) и (3.11.14), так что введенный выше параметр sa-mBaljvx\(£> можно представить более удобной формулой: sa= 0^1 ю-
Как уже отмечалось выше, при со < со^ и достаточно сильном гидродинамическом взаимодействии частиц имеет место встречное синхронное вращение частиц, преобразующееся в макроскопические движения разного вида. Практическим критерием уровня гидродинамического взаимодействия является повышенная вязкость суспензии по сравнению с вязкостью дисперсионной среды. Фактически для этого требуется повышение концентрации дисперсной фазы до 20-30 об. % и выше.
Пространственные масштабы возможных макроскопических движений суспензии охватывают весь мыслимый спектр длин — от размера частиц до размера сосуда с суспензией, а наиболее интенсивное (заметное) движение соответствует длинноволновой моде спектра. Кроме упомянутых ранее эффектов встречного синхронного вращения следует отметить разжижение суспензий в переменном поле, отрицательное значение предельного напряжения сдвига (структурной вязкости), которые представляют собой реологическое проявление самодвижения суспензии в переменном поле. На водных суспензиях гексаферрита бария наличие указанных эффектов подтверждено экспериментально [3] в полях с частотой от 50 до 200 Гц.
В высокочастотном поле (со > со^) оси частиц лишь слегка раскачиваются возле своей первоначальной координаты. Амплитуды раскачивания немного отличаются в прямом (в направлении вращения поля) и обратном направлениях. Это видно из формулы (3.11.23), поскольку при со > со^ параметр « L и поэтому отношение тангенсов начальной и конечной угловых координат частиц ненамного отличается от единицы. Если разложить tg(02/2) в ряд Тейлора в окрестности угла ©1/2 и ограничиться его первыми членами, то получится следующее соотношение: tg(02/2) = tg(©i/2) + + 5(0/2) /sin2(©i/2), которое после упрощений дает выражение: tg(02/2)/tg(0]/2) = 1 + 80/sin©!.
Если разложить экспоненту в ряд Тейлора, то можно получить формулу для высокочастотной области:
80 = л’Хсобсо/ - cosco/i)sin©i. (3.11.24)
Такой же результат получится при интегрировании уравнения (3.11.14), если пренебречь изменением sin©i при небольшом изменении угла ©ь
Отклонение оси частицы от первоначального положения ©1 за первый полупериод изменения поля получается при подстановке в формулу (3.11.24) значения со/ = л и со/] = 0:
80a = -25esin01. (3.11.25)
За второй полупериод (ю/ = 2л и а>/] = л) оно имеет ту же величину, но обратный знак. Отклонение оси частицы 8© от произвольного начального положения 0] за время t, отсчитываемое от нулевой фазы колебаний (co/i = 0), определяется формулой
80 = sa (cosarf - l)sin©i. (3.11.26)
Следует подчеркнуть, что отсчет угла 0 в приведенных выше формулах начинается от первоначального направления вектора магнитной индукции синусоидального переменного поля (в момент, соответствующий его фазе a>/i = 0).
Намагниченность М* суспензии в произвольный момент времени есть сумма проекций магнитных моментов т всех п частиц, находящихся в единице объема, на направление магнитного поля в текущий момент времени. Во втором полупериоде направление поля меняется на обратное, и при вычислении проекции следует учитывать, что в этой фазе колебаний угол между направлениями оси частицы и поля равен п + 0. Вклад одной частицы в намагниченность /(/) = mcos©(/) непрерывно меняется во времени из-за изменения угла 0. Если при со » ок изменения 0 малы, то при разложении cos© в ряд Тейлора в окрестности первоначальной угловой координаты 0] оси частицы получается следующее соотношение: cos© = cos©i - sin©]80. При подстановке в него выражения (3.11.26) получается формула
i(t) = т [cos©] - s^sin2©] (cosco/ - 1)]. (3.11.27)
Представляют интерес два параметра, характеризующие магнитное состояние суспензии: среднее значение вклада < i > одной частицы в намагниченность за период колебаний Т и мгновенная величина намагниченности суспензии M*(t) в ее первоначальном структурном состоянии (когда ориентации частиц случайны). Первый из них равен \i(t)dt!T, а второй — 2wn(/)Jsin01cZ01. Функция 2n/7sin0]cZ0) дает число частиц в угловом секторе шириной d&\. Соответствующие вычисления приводят к уравнениям
< i > = m^sin2© (3.11.28)
Физика, химия и технология дисперсных систем
687
и
Л/*(?) = )(1 - coso)?), (3.11.29)
где Л/* = пт — намагниченность насыщения суспензии, соответствующая ориентации всех частиц магнитными осями параллельно полю.
Формула (3.11.28) показывает, что наиболее сильно реагируют на высокочастотное поле те частицы, которые ориентированы перпендикулярно намагничивающему полю (0 = л/2), а частицы с ориентацией параллельной полю (0 = 0) совсем не участвуют в намагничивании суспензии. Это и понятно, поскольку вращательная подвижность последних имеет, согласно уравнению (3.11.21), нулевую величину.
В данном случае намагниченность суспензии Л/*(?)— это проекция магнитных моментов частиц на направление поля, а не на ось пространственных координат. По этой причине она всегда положительна (или равна нулю) и является мерой согласованности ориентаций частиц и поля. Ее основная особенность в том, что максимум намагниченности достигается в той фазе изменения поля {(at = л), когда его напряженность равна нулю. Сближение ориентаций оси частицы с направлением поля продолжается в течение всего полупериода переменного поля. Именно к моменту смены его знака угол между осью частицы и направлением поля минимален, а намагниченность максимальна. Намагничивание такого типа является неупругим, т. е. оно не исчезает при выключении поля. Это справедливо, если поведение частиц исчерпывающе описывается приведенными выше уравнениями. В действительности оно подвержено воздействию и ряда других факторов, находящих свое отражение в реальном поведении суспензий.
Практически важным результатом воздействия высокочастотного переменного поля на магнитную суспензию является, в частности, то, что в таком поле все частицы суспензии через некоторое число циклов перемагничивания (изменения направления намагниченности) ориентируются своими магнитными осями перпендикулярно направлению магнитного поля. Этот вывод не следует из рассмотренных уравнений, в частности из уравнения (3.11.26), которое как раз и дает величину углового смещения оси частицы за полупериод вращения поля, или из более общего уравнения (3.11.23). Действительно, если за первую половину периода (cofi = 0, ®?2 = 7С) ось сместится из произвольного положения 01 в положение 02, то во второй полупериод {(at 1 = л, (at2 = 2л) ее движение в обратном направлении начнется с угла 02 и закончится, согласно формуле (3.11.23), на том же угле Оь с которого начиналось движение в первый полупериод.
Таким образом, за полный период поля положение оси частицы не изменится. Тем не менее, перпендикулярная ориентация магнитных осей частиц к направлению переменного поля является экспериментальным фактом. Она обнаруживается, в частности, благодаря
тому, что обработка концентрированной пастообразной суспензии переменным полем приводит к появлению характерного остаточного эффекта — в ней возникает текстура типа «легкая плоскость». Эта плоскость ориентирована нормально к направлению текстурирующего (перемагничивающего) поля. Термин «текстура», применяемый технологами, эквивалентен термину «структура». Термин «легкая плоскость» означает, что суспензия легко намагничивается в любом направлении, если она лежит в определенной плоскости, а направление, перпендикулярное этой плоскости, является осью наиболее трудного намагничивания. Обычно требуемая текстура изделия из магнитного порошка фиксируется путем отверждения изделия полимеризацией дисперсионной среды, сушкой изделия с последующим обжигом, прессованием и т. д. Отверждение обеспечивает неизменность ориентации частиц при эксплуатации изделия в полях любой ориентации, напряженности и частоты. Направление плоскости легкого намагничивания определяется, по величине остаточной намагниченности изделия после его намагничивания в разных направлениях кратковременным импульсом постоянного магнитного поля с напряженностью, превышающей коэрцитивную силу магнитного материала. Намагничивание в «легкой плоскости» дает большую величину остаточной намагниченности (т. е. после отключения намагничивающего поля), чем в других направлениях. Текстурирование обусловлено медленным дрейфом осей частиц к экваториальной плоскости сферической системы координат. Для его возникновения смещение оси частицы за первый полупериод, когда она сближается с направлением поля, должно быть немного меньше, чем смещение в обратном направлении во второй полупериод. Разность этих смещений Д0, которая зависит от текущей ориентации оси 0, и определит время (число циклов переменного поля), необходимое для достижения требуемой степени текстурирования. Однако, как уже отмечалось, использованные выше уравнения движения осей частиц дают равные величины их смещений за первый и за второй полупериоды колебаний поля, т. е. никакого дрейфа не предусматривают. Существование дрейфа, можно объяснить тем, что максимум намагниченности и, следовательно, минимум средней за период поля магнитной энергии имеют частицы, ориентированные перпендикулярно направлению поля, и поэтому к такой ориентации они и должны стремиться, если для этого имеются предпосылки. Дело в том, что реализация состояния, соответствующего минимуму энергии, возможна только тогда, когда частица реально может оказаться в этом состоянии. Обычно такую возможность частицам предоставляет их тепловое движение (в данном случае вращательная диффузия). Оно и является основным механизмом реализации термодинамически выгодных состояний. И можно быть уверенным в их достижимости, даже если не известны конкретные механизмы перехода из одного состояния в другое. В случае грубодисперсных систем, когда есть
688
Новый справочник химика и технолога
основания пренебрегать эффектами теплового движения частиц, в качестве его «заменителя» действуют различные случайные, не учитываемые факторы: различие размеров и формы частиц, их столкновения между собой, наличие дополнительных (невязкостных) сил сопротивления движению частиц и др.
3.11.4. Вращательная вязкость
Ориентация частиц сказывается на вязкости дисперсной системы благодаря тому, что при этом прекращается свободное вращение частиц в потоке [45]. Схематично механизм возникновения вязкостного эффекта вращения выглядит следующим образом: частица, как шарик, зажатый между двумя параллельными и движущимися в разные стороны плоскостями, почти не оказывает сопротивления их движению, поскольку линейные скорости плоскостей и поверхности частиц в точках их соприкосновения совпадают. В таких условиях отсутствует проскальзывание движущихся с разными скоростями тел (плоскости и шарика) в точках контакта, и поэтому отсутствует трение скольжения. В шарикоподшипниках используется именно этот принцип. Если любым способом предотвратить свободное вращение шарика, то относительное движение плоскостей будет возможно только за счет их проскальзывания относительно поверхности шарика и соответствующего увеличения силы трения. Применительно к суспензии в этой модели плоскости нужно заменить слоями жидкости, прилегающими к поверхности частиц, проскальзывание — локальной величиной градиента скорости течения жидкости и трение скольжения — внутренним трением жидкости. При этом проскальзывание (градиент скорости течения) имеется как при свободном вращении частицы, так и при ее полном торможении, но величина его во втором случае несколько больше, и, соответственно, повышается вязкое сопротивление обтеканию частицы потоками среды. Количественно это различие выражается в том, что при полном торможении вращения частиц вращательная составляющая вязкости возрастает до величины:
П<й = ЗфПо, (3.11.30)
а вязкость в поле — до величины:
Л = Ло(1+4ф). (3.11.31)
Поскольку здесь фигурирует единственная характеристика дисперсной системы — ее концентрация, которая не зависит от режима течения, то может сложиться впечатление, что ориентационное структурирование меняет только величину вязкости, но не закон течения. На самом деле это не так. От режима течения зависит величина числового коэффициента, предшествующего концентрации ф. В формуле Эйнштейна (3.11.3) он был обозначен символом а и равен 2,5 при свободном вращении частиц и 4 при ориентационном структурировании системы. Смена режима, а с ним значения коэффициента и величины вязкости, происходит при увеличе
нии скорости сдвига и момента сил трения, действующих в потоке на частицу, до критической величины.
Уравнение структурного состояния, которое является необходимым звеном вывода реологического закона, в данном случае сводится к соотношениям, которые и были использованы выше:
а = 4 при у' < у'с,
(3.11.32) а = 2,5 при у' » ус.
На рассмотренном ранее примере поведения частиц во вращающемся магнитом поле было показано, что при сверхкритических частотах поля вращение частиц хотя и происходит, но оно замедленно по сравнению с частотой поля и прекращается при очень высоких частотах. При сдвиговом течении в постоянном поле вращается среда вместе с частицами, а поле неподвижно. В остальном картина та же, что и при вращении поля. Это означает, что уменьшение вязкости от величины, соответствующей значению а = 4, до величины, соответствующей значению коэффициента а = 2,5, занимает достаточно широкий интервал сверхкритических скоростей сдвига, т. е. вязкость плавно уменьшается с увеличением скорости сдвига в указанном диапазоне значений [36]. Таким образом, ориентационно структурированная система ведет себя классически — в соответствии с постулатом Ребиндера при разрушении структуры (нарушении ориентации частиц) вязкость снижается.
Воздействием внешнего поля можно не только устранить вращение частиц в потоке, но и ускорить его (по сравнению со свободным вращением) наложением вращающегося поля подходящей частоты и направления вращения. Если вращение поля направлено в ту же сторону, что и вращение частиц под действием сдвигового течения среды, и имеет большую скорость, чем скорость свободного вращения частиц в потоке, то поле уменьшит гидродинамическое сопротивление частиц потоку и приведет к снижению вязкости по сравнению с ее значением вне поля. В предельном случае сильного высокочастотного поля эффект может выразиться в том, что вращательная часть коэффициента а станет отрицательной и равной 3. Формально это значит, что вязкость станет отрицательной — суспензия может течь в отсутствие внешнего деформирующего усилия, и даже в направлении, обратном направлению нормального течения при действии такого усилия. Этот эффект очень просто демонстрируется с помощью стакана с магнитной суспензией или феррожидкостью, помещенного во вращающееся магнитное поле, например в статор трехфазного электродвигателя вместо его ротора.
Аналогично действует и переменное магнитное поле. Оно по понятным причинам менее эффективно, но зато в этом случае нет необходимости заботиться о придании полю и частицам нужного направления вращения. В переменном поле частицы сами выберут его соответственно направлению свободного вращения в потоке
Физика, химия и технология дисперсных систем
689
или соответственно граничным условиям при доминирующей роли согласованности вращений соседних частиц в стесненных условиях. Опыты с переменным полем были описаны во вводном параграфе этой главы.
3.12. Периодическая коллоидная структура
3.12.1. Концентрационное структурирование
Основной параметр, характеризующий периодическую коллоидную структуру (ПКС), — это период 5 кристаллоподобной решетки, т. е. расстояние между центрами ближайших соседних частиц (рис. 3.86). В общем, оно определяется размером частиц и их концентрацией п:
s = n1/3. (3.12.1)
Эта формула отражает равномерность распределения частиц по всему доступному для них объему, т. е. тот факт, что в среднем на одну частицу приходится объем, равный l/п. Тогда 5 является линейным размером пространственной ячейки, занимаемой одной частицей.
Очевидно, что 5 не может быть меньше диаметра частиц 2а, что определяет максимально возможную (предельную) концентрацию частиц пт.
В монодисперсной системе пт есть величина порядка 1/8а3. Обычно концентрация частиц меньше ее предельной величины, поэтому размер ячейки 5 оказывается, согласно формуле (3.12.1), больше диаметра частиц на толщину h прослойки дисперсионной среды, разделяющей соседние частицы дисперсной системы. Это обстоятельство часто используется для оценки толщины упомянутой прослойки по формуле
А = (1/и)1/3-2а (3.12.2)
Поскольку она определяется только рецептурой суспензии — ее концентрацией п, то в дальнейшем этот параметр будет называться рецептурным зазором между частицами. Точность такой оценки величины зазора h зависит от уровня монодисперсности и сферичности частиц, что может быть проконтролировано еще до приготовления дисперсной системы. Однако существует и другие факторы, которые приводят к сильному отличию фактической величины зазора от его рецептурного варианта. Некоторые из них привносятся в процессе приготовления взвеси и поэтому заранее не всегда могут быть предусмотрены. К ним относится и характер взаимодействия частиц, т. е. вид потенциальной или силовой функции их взаимодействия. В рамках теории ДЛФО (см. подраздел 3.7) эта функция определяется природой дисперсной фазы и дисперсионной среды, включая ее ионный состав. Эта теория и будет использоваться далее при количественном описании структурного состояния дисперсных систем. Вместе с тем существуют факторы, не учитываемые классической теорией ДЛФО, но сильно влияющие на взаимодействие частиц. В частности, рассмотренный ранее
эффект смещения плоскости локализации поверхностного заряда частиц. Величина смещения пока может фигурировать в расчетах взаимодействия частиц и структурных параметров системы как свободный подгоночный параметр и, следовательно, априори он не может быть предугадан на основе рецептуры дисперсной системы. В любом случае формула (3.12.2) предполагает, что между частицами действует отталкивание, и радиус действия этих сил ничем не ограничен. На самом деле это не так. Более того, ПКС наиболее характерны для сравнительно грубодисперсных систем, в которых радиус действия сил отталкивания является ничтожно малой величиной по сравнению с размером частиц а. Это значит, что фактически период ПКС в грубодисперсных системах определяется по формуле
5 = 2а. (3.12.2а)
Эта формула совместима с формулой (3.12.1) только при предельно большой концентрации частиц. Следовательно, можно полагать, что в грубодисперсных системах ПКС реализуется только при п = пт. То, что в действительности ПКС возникают и при несколько меньших концентрациях частиц, означает, что структура на самом деле не является строго периодической. Наиболее вероятная причина отклонения от периодичности состоит в том, что ряд узлов кристаллоподобной решетки частицами не занят. Подобные нарушения структуры ПКС, как и обычных кристаллов, принято называть вакансиями (вакантными узлами решетки). Вакансии и другие дефекты решетки кристаллов оказывают, как известно, решающее влияние на свойства реальных твердых тел. Их роль в коллоидных структурах не менее ответственна.
Рис. 3.86. Кристаллоподобная (периодическая) структура: s — период структурной решетки
Важнейшим параметром ПКС является прочность фиксации частиц в узлах решетки. Энергетической мерой прочности фиксации является потенциальный барьер миграции частиц — это работа, которую требуется совершить для перемещения частицы в соседний узел решетки. В простейшем случае она рассчитывается на основе теории ДЛФО как потенциальный барьер Д1/ взаимодействия одной частицы с двумя своими ближайшими соседями, расположенными на общей прямой, т. е. как разность энергий максимума и центрального минимума симметричной функции взаимодействия (рис. 3.87):
U = B {ехр[-А (А - х)] - ехр[ - А (А + х)]} -
— Z> [1 /(А — х) — 1 /(А-ь х)]. (3.12.3)
690
Новый справочник химика и технолога
В формуле (3.12.3) х — виртуальное смещение частицы относительно точки симметрии кривой (узла решетки), В — константа отталкивания и D — константа притя-
жения пары частиц.
взаимодействия частиц в ПКС:
АС/ — мера прочности фикации частиц в углах решетки (а) и изменение вида кривой при увеличении концентрации частиц (б); h — толщина зазора между соседними частицами решетки
Приведенные на рис. 3.87 графики обычно называют потенциальными кривыми взаимодействия трех частиц. Почему следует рассматривать взаимодействие именно трех, а не двух или четырех или большего числа частиц? В разбавленных суспензиях частицы взаимодействуют только путем их столкновений, и обычно редко в столкновении участвует больше двух частиц. Поэтому для выяснения последствий взаимодействия достаточно рассмотреть взаимодействие только двух частиц. В концентрированной дисперсной системе имеет место взаимодействие другого типа — постоянное давление на каждую частицу со стороны ее ближайших соседей. Обычно представляет интерес возможность движения частицы вдоль некоторого направления, поэтому необходимо найти силы, действующие на нее именно вдоль выбранного направления. Эти силы определяются взаимодействием с частицами, лежащими на направлении движения, а их на линии движения как минимум две. Таким образом, в простейшем случае необходимо рассмотреть взаимодействие одной частицы с двумя ее ближайшими соседями.
Следующий очень значимый параметр, характеризующий состояние ПКС, — тип кристаллической решетки, в узлах которой располагаются частицы. Не вдаваясь в детали кристаллографии, можно выделить два типа решеток, присущих периодическим структурам. Это простая кубическая решетка и гексагональная решетка. Первая соответствует сравнительно рыхлой регулярной упаковке частиц — максимальная объемная доля дисперсной фазы <рда при такой укладке составляет 0,52. Она обладает самой простой геометрией и поэтому часто подразумевается при тех или иных численных оценках состояния системы, в частности в приведенных выше формулах (3.12.1) и (3.12.2). Гексагональная упаковка частиц является самой плотной. Доля дисперсной фазы фт при такой укладке достигает 0,75.
Влияние типа упаковки, которая далее характеризуется максимально возможной долей <рда дисперсной фазы,
на величину зазора h между соседними частицами и, соответственно, на прочность их фиксации в узлах решетки можно иллюстрировать следующими расчетами.
При некоторых заданных значениях концентрации п и размера частиц а их объем ф во взвеси равен Дила3 / 3.
Если частицы расположены в узлах периодической решетки, обеспечивающей предельную величину концентрации частиц фда и расстояние между их центрами г, то ф = фт(2а/г)3 и, соответственно, г = 2а(фт/ ф)1/3. При изменении типа решетки, например с гексагональной на кубическую, расстояние г между частицами, а с ним и величина зазора Л, изменится на величину АЛ = Дг. Это изменение можно вычислить по формуле
АЛ = (2а/ф|/3)Д(фм|/3). (3.12.4)
При переходе от гексагональной упаковки к кубической А(фт1/3) ~ —0,1, т. е. зазор между частицами уменьшается не менее чем на 10 %. Такой переход может вызываться сдвиговой деформацией решетки. Если же при плотнейшей (гексагональной) упаковке h = 0, т. е. толщина защитных оболочек пренебрежимо мала по сравнению с размером частиц, то при постоянстве объема системы отсутствует возможность изменения расстояния между частицами, что создает сильное сопротивление деформированию решетки. Если же такая возможность имеется, то деформирование сопровождается увеличением объема дисперсной системы. Это явление называется дилатансией, а дисперсные системы, деформирование которых сопровождается непропорционально сильным увеличением сопротивления при увеличении скорости деформирования, называются дилатантными.
Сопротивление сдвиговому деформирование дилатантных систем обусловлено наличием сил, препятствующих изменению объема системы, т. е. сил, действующих по нормали к плоскостям сдвига и к внешним свободным границам системы. Возникновение нормальных напряжений под действием сдвиговых напряжений является характерной особенностью дилатантных систем. Сопротивление сдвигу обусловлено тем, что увеличение объема, вызванное сдвиговой деформацией, сопровождается затеканием вязкой среды в полости между частицами, так как при кубической упаковке объеме этих полостей (1 - фт) больше, чем при гексагональной. Быстрая деформация и, соответственно, повышенная величина нормальных напряжений сильнее прижимает частицы друг к другу, препятствуя перемещению частиц и сужая каналы, по которым идет перетекание жидкости в межчастичные полости. Это и ведет к непропорционально сильному увеличению напряжения сдвига с увеличением скорости деформации дилатантной системы. Такой механизм дилатантного поведения можно назвать геометрическим, поскольку он обусловлен переходом между геометрически разным расположением частиц. Ниже (см. формулы (3.12.13) и далее) будет показано, что имеются и иные механизмы.
Физика, химия и технология дисперсных систем
691
Приведенные выше соотношения между концентрацией частиц и величиной зазора между ними справедливы, если на всех расстояниях между частицами преобладают силы отталкивания, что довольно проблематично. Более реалистична ситуация, при которой парное взаимодействие частиц характеризуется наличием потенциальной ямы (дальнего минимума). Важную роль при этом играет координата минимума hc на потенциальной кривой парного взаимодействия частиц. Параметры ПКС в этом случае зависят от соотношения между hc и рецептурной величиной межчастичного зазора h, определяемого по формулам типа (3.12.2) или (3.12.4). При концентрации дисперсной фазы Фс= фт/[1 + (hcl2af\ эти два параметра совпадают (Л = he). Потенциальная кривая взаимодействия трех частиц имеет единственный центральный минимум в области отрицательных значений энергии, а его глубина равна удвоенной глубине дальнего минимума парного взаимодействия частиц. При повышении концентрации частиц рецептурный зазор между ними снижается, глубина потенциальной ямы уменьшается, а соответствующая этой яме энергия приобретает положительные значения. При понижении концентрации частиц центральный минимум разделяется на два, а в центре симметричной потенциальной кривой трех частиц возникает небольшой максимум с отрицательным значением энергии частицы. Очевидно, что частицы при этом не могут располагаться в узлах правильной решетки с периодом г = 2а(<рт/ ф)1/3, вычисленным по величине концентрации частиц. Частицы сместятся из узловых точек этой гипотетической решетки к одной из соседних частиц в положение, отвечающее одному из пары минимумов потенциальной энергии. Таким образом, периодичность решетки будет нарушена. Можно предполагать, что возникнут разрывы гипотетической решетки. Тогда вместо сплошной решетки появятся отдельные области периодического расположения частиц, и период решетки в этих областях (доменах) будет определяться координатой минимума hc, а не концентрацией частиц. При незначительной разнице величин tp и фс возможно сохранение сплошной ПКС, а свободное от частиц пространство, возникающее за счет сближения частиц на величину (Л - hc), будет накапливаться в узлах той же самой решетки в виде незанятых частицами узлов — вакансий (рис. 3.88).
Рис. 3.88. Обмен местами вакансий (светлые кружки) и частиц (темные кружки) — элементарные акты процесса течения ПКС в направлении И действия сдвигового усилия
Таким образом, реальные ПКС помимо названных ранее параметров (периода решетки и прочности фиксации в ней частиц) характеризуются еще и концентрацией с вакантных узлов в ней. В рамках приведенных рассуждений ее можно считать предсказуемой величиной. Общее число узлов решетки (1/s3) есть сумма числа п занятых частицами и вакантных (с) узлов:
l/? = w + c. (3.12.5)
Здесь 5 - фактический период решетки, который следует отождествлять с расстоянием между центрами двух частиц, устанавливающимся в процессе их коагуляции в дальний минимум:
s = 2a + hc. (3.12.6)
Эти формулы должны использоваться взамен формулы (3.12.1). Формула (3.12.2а) является предельным случаем формулы (3.12.6) при исчезающе малой величине hc.
3.12.2. Уравнение структурного состояния ПКС
Формулы (3.12.5) и (3.12.6) описывают состояние ПКС в покое. При деформировании дисперсной системы неизменны только размер и концентрация частиц, тогда как два других параметра этих уравнений — период решетки и концентрация вакансий — зависят от интенсивности деформирования. Интенсивность деформирования можно характеризовать скоростью сдвиговой деформации. Применительно к ПКС ее можно представить как отношение разности скоростей движения соседних слоев кристаллической решетки к расстоянию между этими слоями. В первом приближении можно считать, что оно равно периоду решетки. Уточнение требуется, если направление движения слоев не совпадает с направлением кристаллографических осей.
Механизм воздействия деформирования на параметры решетки ПКС, в сущности, очень прост. Частным случаем деформирования является обычное смешивание компонентов дисперсной системы, например порошка и жидкости. Цель перемешивания — достижение равномерности распределения всех компонентов системы по ее объему. Упомянутые выше вакансии кристаллической решетки — равноправный компонент системы. В сущности, они являются сгустками межчастичных пустот и поэтому могут дробиться и распределяться равномерно по всему пространству, как и любой другой компонент дисперсной системы. При постоянстве объема системы (что будет всегда предполагаться, если не делается специальных оговорок) «размазывание» вакансий по межчастичному пространству означает уменьшение их концентрации и соответствующее формуле (3.12.5) увеличение периода решетки.
Количественная связь между интенсивностью деформирования и концентрацией вакансий может быть выражена уравнение структурного состояния. Как уже отмечалось выше, уравнения структурного состояния предназначены для описания не только ПКС, но и всех
692
Новый справочник химика и технолога
других типов структурированных систем без исключения. Впервые они были введены [4] в теорию дисперсных систем при описании состояния магнитных жидкостей при одновременном воздействии на них магнитного поля и течения, а затем при выводе закона течения (реологического уравнения) для того же случая [6].
Количественное описание структурного состояния ПКС как функции скорости деформации можно получить, исходя из того, что при деформировании одновременно протекают два разнонаправленных процесса— исчезновение вакансий и рождение (релаксация) вакансий. Исчезновение вакансий обусловлено их «размазыванием» по межчастичному пространству при деформировании решетки, а рождение — стремлением системы к состоянию с минимальной потенциальной энергией, которому соответствует сближение частиц до равновесного состояния, т. е. к образованию решетки с периодом, присущим состоянию покоя. Динамическому равновесию системы при ее непрерывном деформировании (течении) соответствует равенство скоростей исчезновения и рождения вакансий.
По Френкелю деформирование любого материала происходит путем поочередного скачкообразного перемещения структурных элементов материала (атомов, молекул, частиц, звеньев длинных полимерных молекул) в ближайший вакантный узел регулярной или хаотичной решетки, расположенный вдоль направления силы, действующей на элемент (рис. 3.88). Каждый такой скачок представляет собой временное исчезновение вакантного узла в одном узле решетки и последующее его возрождение в другом. Таким образом, число вакансий, исчезающих в единице объема за единицу времени (dc/dt), равно произведению частоты скачков f совершаемых одной вакансией, и их концентрации с, согласно формуле
(dc/dt) =fc. (3.12.7)
Релаксация вакансий требует перемещения частиц на расстояние, сравнимое с периодом решетки, которое обычно намного превышает радиус действия сил отталкивания. На этом основании можно предполагать, что время релаксации вакансии tr — это время диффузии частицы на расстояние, примерно равное периоду решетки: tr = s2/2D, где D = кТ/6тщ0а — коэффициент диффузии частицы и т|0 — вязкость среды. Если учесть, что период решетки 5 примерно равен 2а, то можно получить формулу
4 = 9лг|оу/ЧГ (3.12.8)
Новые вакансии рождаются из числа вакансий, «размазанных» по межчастичным зазорам. Поэтому скорость рождения вакансий пропорциональна убыли их концентрации. Тогда справедливо соотношение
(dc/dt) = (c0-c)/tr. (3.12.9)
Здесь со — концентрация вакансий в состоянии покоя дисперсной системы.
Приравнивание правых частей уравнений (3.12.7) и (3.12.9) друг другу приводит к формуле
с = с0/(1+ЛД (3.12.10)
В принципе это и есть уравнение структурного состояния ПКС при ее деформировании. Однако интенсивность процесса деформирования здесь присутствует неявно — в виде частоты f перескоков частиц в соседние свободные вакантные узлы. Для получения явной зависимости концентрации вакансий от скорости деформации у' необходимо детально рассмотреть, как из отдельных скачков частиц складывается их непрерывное движение. В связи с этим полезно обратиться к предыстории вопроса. Как уже упоминалось, идея скачкообразного механизма деформирования материалов предложена Френкелем. Позже она была распространена Эйрингом на дисперсные системы и затем неоднократно модернизировалась многими авторами. На этом этапе развития идеи принималось, что скорость движения du слоя частиц относительно ближайшего соседнего слоя равна произведению числа скачков f частицы в единицу времени в направлении действия деформирующего усилия на длину s одного скачка. В действительности это не так. В структурной решетке существует определенное количество вакантных узлов, и перескок частиц может происходить только поочередно в освобождающийся вакантный узел. В решетке можно выделить виртуальную цепочку из v частиц, расположенную вдоль направления их движения, которая начинается от любого вакантного узла и продолжается до ближайшего следующего вакантного узла на линии движения частиц. Вся решетка с вакантными узлами представляет собой в этой модели совокупность параллельных цепей с одним вакантным узлом в каждой. Их средняя длина vs определяется концентрацией вакансий. Она тем короче, чем больше вакантных узлов в решетке. Для того чтобы вся цепь переместилась на расстояние, равное длине одного скачка (периоду решетки 5), каждая из частиц цепи должна совершить один скачок в нужном направлении, т. е. всего потребуется v скачков. Это означает, что действительная скорость движения цепей и, следовательно, всего слоя вещества будет медленнее, чем в теории Френкеля — Эйринга, в v раз [9]. Таким образом, разность скоростей соседних слоев составляет du=fs/v, а скорость деформации у', совпадающая при простом сдвиговом течении с градиентом скорости течения du/dz, где dz = s — расстояние между соседними слоями, описывется формулой
у' =f/v. (3.12.11)
Связь между характеристическим числом частиц в виртуальной цепи v и концентрацией вакансий получается на основе отмеченной выше возможности представить всю ПКС как совокупность таких цепей с одной вакансией на каждую цепочку. В таком случае соотно
Физика, химия и технология дисперсных систем
693
шение между числом занятых и свободных узлов во всей системе будет таким же, как в отдельной цепи, т. е. у = п/с. Тогда справедливо уравнение
f — cf! п. (3.12.12)
Это позволяет преобразовать уравнение структурного состояния (3.12.10) к явной зависимости концентрации вакансий от скорости сдвига. Однако более полезно получить из этих двух выражений зависимость скорости сдвига от частоты скачков, которая учитывает как влияние концентрации вакансий на скорость сдвиговой деформации, так и зависимость этой концентрации от частоты скачков. При подстановке зависимости (3.12.10) в формулу (3.12.12) получается уравнение
у' = с<//«(1+Л). (3.12.13)
Следует иметь в виду, что формальное применение соотношений (3.12.12) и (3.12.13) к системам с большой концентрацией вакансий (с0 > п) дает физически неразумное соотношение между частотой скачков и скоростью деформации — скорость сдвига оказывается больше частоты скачков. В рамках описываемой модели это означает, что при этом длина одного скачка оказывается в с0/п раз больше периода решетки, что маловероятно. Отсюда следует, что применимость рассматриваемой модели ограничена умеренно малой долей (с0/п) вакансий в решетке дефектной ПКС. В системах с высокой концентрацией вакансий регулярность расположения частиц сильно нарушается, и более адекватной моделью такого состояния системы становится ее представление в виде рыхлой коагуляционной сетки.
3.12.3. Уравнение напряжений и уравнение реологии ПКС
Уравнение (3.12.13) дает зависимость скорости деформации от частоты f тепловых скачков одной частицы. Для нахождения закона, выражающего зависимость скорости деформирования от величины деформирующего усилия, необходимо получить зависимость напряжения т от той же величины f. Тогда ее можно будет исключить из этих уравнений и получить искомую закономерность.
Связь напряжения с частотой скачков вытекает из активационного механизма течения, открытого Френкелем. Его суть в том, что частицы (молекулы) считаются фиксированными в узлах регулярной или хаотичной решетки, но благодаря тепловым колебаниям они могут с некоторой частотой f перескакивать в соседний вакантный узел решетки. Эта частота определяется частотой тепловых колебаний частицы и величиной потенциального барьера (энергии активации вязкого течения) Ua, отделяющего частицу от соседнего вакантного узла. Каждый цикл колебательного движения является попыткой преодолеть барьер и перейти в соседний узел.
Вероятность преодоления барьера w = ехр(-С/а/А7) определяется его величиной, так что частота переско
ков ft в соседний узел будет равна wf0. Это выражение дает частоту перескоков в любом направлении. В сущности, оно представляет собой хорошо известный закон Аррениуса, который используется в основном при изучении температурной зависимости различных свойств веществ, в том числе и вязкости. Его преобразование в уравнение напряжений основано на том, что при действии на частицу силы F потенциальный барьер изменяется на величину sF/2, равную работе перемещения частицы этой силой на расстояние s/2, равное половине периода решетки. В направлении действия силы (в прямом направлении) барьер уменьшается, а во встречном направлении увеличивается на указанную величину. В итоге число скачков частицы в прямом направлении будет больше, чем число скачков в обратном направлении, а результирующее число f скачков частицы в нужном направлении за единицу времени будет равно разности этих двух величин:
/=/о {ехр[-(£/Л - sF/2)/kT] - ехр[-((/Л + sF/2)/kT]}
(3.12.14) или
/= 2fish(sF/2kT).
Здесь sh(sF/2kT)=[exp(sF/2kT) - exp(-sF/2kT)]/2 — синус гиперболический аргумента sFI2kT и / = =/>ехр(-(/а/Л7) — частота перескоков частицы в произвольном направлении под действием теплового движения, являющаяся одной из констант для данной дисперсной системы и температуры.
Фигурирующая в формуле (3.12.14) сила F— это та часть напряжения деформации т, которая воспринимается одной частицей. Если разделить напряжение на число частиц 1/s2, приходящихся на единицу площади сечения дисперсной системы плоскостью сдвиговой деформации, то получится следующее выражение: F = xs2. Здесь содержится некоторая неточность, поскольку выражение 1/л2 дает число узлов решетки на единицу площади сечения, часть которых может быть вакантна. Вакансии не воспринимают действующего на решетку напряжения, поэтому оно должно быть поделено на число узлов, занятых частицами. Это несколько увеличит действующую на каждую частицу силу, однако при умеренно малой концентрации вакансий такое уточнение не оказывает существенного влияния на численное значение частоты перемещения частиц. После замены силы напряжением получается формула
f = 2fish(x / 2пкТ). (3.12.15)
В оригинальной теории Френкеля — Эйринга у' =/ поэтому отсюда сразу следует классическое реологическое уравнение при активационном механизме течения [15]:
у'= 2fsh(x / 2пкТ). (3.12.16)
694
Новый справочник химика и технолога
При малых напряжениях s!i(t/2«A:7) = х!2пкТ. Тогда:
у' =fxlnkT, (3.12.17)
а вязкость т] = т/у' описывется формулой
x\ = nkT/f,. (3.12.18)
В общем же случае с увеличением напряжения скорость сдвига растет, согласно формуле (3.12.16), по закону, близкому к экспоненциальному. Таким образом, классическая теория Френкеля — Эйринга предсказывает зависимость, близкую к законам пластического течения. Такой результат не согласуется с опытными данными. Для концентрированных устойчивых к коагуляции суспензий более характерен дилатантный тип зависимости скорости сдвига от напряжения. Учет зависимости скорости сдвига от концентрации вакансий и связи последней с напряжением сдвига с помощью уравнения состояния (3.12.13) и формулы (3.12.15) приводит именно к такому результату [9]:
у' = (с0 / п) 2/sh(x / 2пкТ) / [ 1 + 2/^sh(T / 2л£7)], (3.12.19)
где
/=/оехр(-(/а/£7).
В этом уравнении решающее значение имеет отношение времени релаксации вакансий tr к интервалу времени 1/4 между двумя последовательными тепловыми переходами частицы в вакантный узел решетки. Отношение времен фигурирует в уравнении (3.12.19) в виде произведения t/t. При 1 полученное уравнение совпадает с классическим уравнением (3.12.16) для активационного механизма течения. При //X 1 и малых напряжениях получается зависимость классического типа, т. е. вязкость уменьшается с ростом напряжения, но при достаточно большой величине напряжения произведение ttfeh(x/2пкТ) в уравнении (3.12.19) увеличивается настолько, что единицей в знаменателе можно пренебречь. Тогда скорость деформации перестает зависеть от напряжения. Это означает, что вязкость начинает увеличиваться с увеличением напряжения. Такой смешанный пластично-дилатантный тип зависимости скорости сдвига от напряжения весьма характерен для концентрированных суспензий. При » 1 суспензия практически во всем диапазоне напряжений проявляет дилатантные свойства (рис. 3.89).
Рис. 3.89. Кривые течения ПКС при различных значениях параметраftr'.
1 —fttr «с 1; 2 —fa ~ 1; 3 —ftr» 1
Отметим принципиальную особенность вывода уравнений реологии (3.12.16) и (3.12.19). Он не содержит прямых указаний на то, что сопротивление деформированию ПКС является вязким. Более того, по форме выражение (3.12.17) напоминает уравнение состояния идеального газа. Фигурирующая в нем величина пкТ равна, как известно, давлению газа, а величина F рассматривалась как сила упругого сопротивления, поскольку ее действие вызывало изменение потенциальной энергии частицы в узле решетки. Для сравнения отметим, что вывод формулы Эйнштейна и ее модификаций с самого начала предполагал вязкий тип напряжений. Это выразилось в том, что сопротивление деформированию суспензии определялось как сопротивление вязкой среды, усиленное благодаря особенностям ее течения в присутствии недеформируемой фазы. Примем во внимание, что силы вязкого сопротивления— это силы, обусловленные потерями энергии, подводимой к системе при ее деформировании. Для доказательства того, что сопротивление деформированию является вязким, необходимо выяснить, где и как при деформировании происходит диссипация энергии — ее превращение в теплоту. Ответ содержится в выражении для работы sF/2 упомянутой силы. Согласно этому выражению, деформирующая сила совершает работу, идущую на увеличение потенциальной энергии частицы, только на первой половине (s/2) полного пути 5 частицы из одного равновесного положения в другое. В силу симметричного вида зависимости потенциальной энергии частицы от ее смещения из положения равновесия на второй половине пути сила сопротивления меняет знак на обратный. Следовательно, на второй стадии движения частица не может оказывать сопротивления деформированию. По этой причине в выражении для работы и фигурирует только половина полного пути. Движение частицы на втором отрезке пути идет под действием внутренних сил деформированной решетки, которые не совершают никакой полезной работы, т. е. полученная на первой половине пути энергия теряется. Механизм превращения этой энергии в теплоту не имеет принципиального значения. Можно, например, считать, что она превращается в энергию упругих колебаний частицы возле положения равновесия, которые постепенно передаются всем частицам, превращаясь, таким образом, в их тепловое движение. В таком варианте диссипации не требуется наличия вязкой дисперсионной среды, и поэтому теория применима к описанию вязкостных свойств обычных жидкостей, в которых «дисперсионной средой» является ничто — межмолекулярные пустоты. Для суспензий более подходит схема передачи энергии вязкой дисперсионной среде при самопроизвольном движении в ней частицы на второй части пути. Это важно при вычислении времени релаксации вакансий и величины потенциального барьера движения частиц в решетке, величина которого определяет частоту переходов частиц в соседний узел.
Физика, химия и технология дисперсных систем
695
3.12,4, Время релаксации и потенциальный барьер
Упомянутый выше механизм диссипации энергии упругих напряжений в решетке позволяет определить время релаксации как время перемещения частицы на расстояние, равное половине периода решетки, под влиянием силы внутреннего напряжения F^ возникшей при деформировании решетки. Перемещению противодействует сила вязкого сопротивления среды, так что, в соответствии с формулой Стокса, вместо формулы (3.12.8) получается уравнение
tr = 6KT\0^/Fr. (3.12.20)
Здесь учтено, что приближенно s = 2а, а г|0 — это вязкость среды. Что касается величины силы Fr, то сказать о ней что-либо определенное можно лишь на основании детальных представлений о характере и размерах деформаций решетки, возникающих в окрестности вакантного узла при действии на дисперсную систему деформирующего усилия. Вероятнее всего деформация не сводится к смещению одной, ближайшей к вакансии частицы в сторону этой вакансии. Геометрически это возможно только при простой кубической упаковке частиц и при совпадении направления деформирующего усилия с направлением соответствующей кристаллографической оси. В остальных случаях сама геометрия расположения соседних частиц не допускает этого. Соседние частицы также будут вовлечены в движение (раздвинуты), и такие возмущения решетки могут распространиться на несколько ее периодов. Радиус возмущений Rd мог бы служить в этом случае основной характеристикой локального состояния решетки. Если предположить, что объем, равный объему одной частицы и который первоначально был сосредоточен в вакантном узле, равномерно «размазывается» по зоне деформации решетки, то легко вычислить обусловленное этим увеличение ее периода. Новое значение периода описывается формулой
sd = s[N/(N-1)]1/3. (3.12.21)
Здесь N — число узлов решетки в зоне размером Rd. Оно включает узел, занятый перемещаемой частицей, и вакантный узел, так что минимальное значение N = 2. Соответственно, максимальное значение периода деформированной решетки может в 21/3 раза превышать период равновесной решетки. Разность (sd-s) — это средняя длина пути, который должны проделать (N- 1) частиц для сжатия деформированного фрагмента решетки до нормальной величины периода s и образования вакансии. Энергия активации течения Ua = MJ(sd) -- &U(s) равна в таком случае разности потенциальных барьеров трехчастичного потенциала взаимодействия при увеличенном At/(srf) и нормальном &U(s) значении расстояний между частицами. Очевидно, что эта величина много меньше самих барьеров. Отношение UJ (sd-s) можно принять за величину средней силы Fr в формуле (3.12.20). Тогда и длину пути нужно заме
нить величиной (sd - s), что приводит к формуле для времени релаксации вакансий:
tr = 6ти]0а (sd - s)21 Ua. (3.12.22)
Можно принять, что энергия Ua, рассеиваемая в каждом акте перемещения частицы, не зависит от режима течения, т. е. Ua и tr являются константами реологического уравнения (3.12.19). В нем, однако, остается еще одна нераскрытая константа — частота Уо тепловых колебаний частицы в узле решетки. Если предположить, что колебания являются гармоническими и их энергия т(ХУо)2/2 равна средней кинетической энергии частиц кТ, то можно получить формулу
Уо = (ЛГ/р)1/2 / v5/6. (3.12.23)
Здесь р — плотность дисперсной фазы и v — объем частицы. Эта формула предполагает, что амплитуда колебаний равна размеру частиц.
Независимость потерь энергии в одном акте исчезновения—рождения вакансии от режима течения ведет к тому, что выполнение закона сохранения энергии обеспечивается соответствующим изменением частоты указанных событий при изменении режима течения, так что ftUa = у'т.
Распределение вакансий по межчастичному пространству может привести к тому, что стесненность их состояния исчезнет, т. е. расстояние между ними станет больше радиуса взаимодействия частиц. Такому состоянию благоприятствует достаточно большая концентрация вакансий или малый радиус действия сил отталкивания по сравнению с размером частиц, что типично для технических суспензий. В этом состоянии между частицами никакие силы не действуют, и тогда время формирования ПКС и релаксации вакансий будет определяться временем броуновского смещения частиц на расстояние (sd ~ s). В соответствии с формулой Смо-луховского tr= (sd~s)2/2D, где D = kT/6itr\0a — коэффициент диффузии частиц в дисперсионной среде с ВЯЗКОСТЬЮ Т|о.
3.13. Кинетика коагуляции и структурирования
3.13.1. Введение
Принципиальное отличие неустойчивой к коагуляции системы от устойчивой состоит в том, что состояние первой непрерывно изменяется. Описание свойств такой системы требует введения величин и параметров, которые являются функциями времени. Важнейшая из них — это гранулометрический состав (грансостав) дисперсной системы. Он непрерывно изменяется в сторону увеличения количества крупных частиц за счет уменьшения числа мелких частиц вследствие слипания последних. Типичная постановка задачи нахождения грансостава в произвольный момент времени предполагает, что в исходном состоянии (до начала коагуляции) система была монодисперсной. Допустимость такого упрощения обусловлена тем, что диапазон размеров
696
Новый справочник химика и технолога
частиц коагулированной суспензии в принципе не ограничен, и исходную систему с удовлетворительной точностью можно считать монодисперсной.
Гранулометрический состав коагулирующей дисперсной системы в любой момент времени в принципе может быть вычислен с помощью фундаментальных уравнений кинетики коагуляции Смолуховского. Однако для реализации этой возможности необходимо с помощью других уравнений учесть изменение в процессе коагуляции ряда параметров, влияющих на скорость коагуляции, в некоторых случаях даже на направление этого процесса. Прежде всего, это изменение структуры частиц, поскольку вместо монолитных исходных частиц при коагуляции образуются более или менее рыхлые флокулы. Их размер, гидродинамические свойства, плотность, концентрация, скорость оседания с одной стороны влияют на процесс коагуляции, а с другой стороны они сами определяются ходом коагуляции. Таким образом, коагуляция, структурирование, оседание частиц оказываются разными сторонами единого сложного процесса структурных превращений дисперсной системы. Задача заключается в нахождении параметров и способов, с помощью которых можно описать этот процесс всесторонне. С этой целью первоначально необходимо рассмотреть по отдельности каждый из основополагающих процессов — коагуляцию дисперсной системы, ее структурирование и расслоение под действием силы тяжести — и затем найти связь этих процессов.
3.13.2. Элементы кинетики коагуляции
Скорость коагуляции выражается числом частиц, исчезающих за единицу времени в единице объема, т. е. это производная dnldt от концентрации частиц п по времени t. При быстрой коагуляции, условием которой является равенство нулю потенциального барьера, любое столкновение частиц приводит к их слипанию. В этом случае скорость коагуляции описывается формулой
dnldt = -q. (3.13.1)
Здесь q — число столкновений между частицами в единице объема за единицу времени. Кинетическая теория дает, как известно, q = StiDRh1, где D — коэффициент диффузии частиц и R — радиус взаимного захвата, т. е. расстояние, начиная с которого дальнейшее поведение частиц определяется не их случайными блужданиями, а взаимодействием.
В рамках теории устойчивости коллоидов (теории ДЛФО) радиус захвата — это расстояние между центрами частиц R, которому отвечает максимум на потенциальной кривой их взаимодействия. При этом чаще всего величина зазора между поверхностями частиц много меньше радиуса частиц а, поэтому с хорошей точностью можно считать, что R = 2a. Коэффициент диффузии D = kT/бтща также определяется радиусом частиц, поэтому частота столкновений q оказывается не зависящей от их размера:
<7 = 8и2ШЗт]. (3.13.2)
Именно благодаря этому обстоятельству уравнение (3.13.1) легко интегрируется. В общем же случае частота столкновений зависит от размера частиц.
Если потенциальный барьер взаимодействия частиц не равен нулю, то не каждое столкновение частиц приводит к их слипанию (явление медленной коагуляции). Если ввести долю w эффективных (приводящих к слипанию) столкновений, то получится уравнение кинетики коагуляции
dnldt = -wsn2. (3.13.3)
В этом уравнении s = 8кТ/ 3т| — кинетический коэффициент уравнения кинетики коагуляции. Интегрирование уравнения (3.13.3) по времени в пределах от t = 0, когда концентрация частиц имеет некоторое начальное значение «о, ДО произвольного момента времени t и соответствующей ему концентрации п дает (1/л-1/«о)= w/или:
n = nQl(<\+tlt\ (3.13.4)
где t = wsno — некоторое характерное время процесса коагуляции. Согласно полученному уравнению, при t = t получаем п = п^2, т. е. время, за которое концентрация уменьшается вдвое, и называется оно временем половинной коагуляции. Если выразить коэффициент диффузии с помощью формулы Эйнштейна: D = кТ/бтща, где т] —- вязкость среды, а радиус захвата через радиус частиц a, T.e.R = 2а, то время половинной коагуляции можно описать формулой
/* = Зт| ISkTnow. (3.13.5)
Следует иметь в виду, что левая часть формулы (3.13.4) представляет собой общую концентрацию флокул всех размеров. В их число входят и первичные частицы и флокулы, которые могут состоять из десятков или сотен слипшихся первичных частиц. Каждая из флокул, независимо от размера, выступает как отдельная кинетическая единица, способная сталкиваться с любой другой флокулой или первичной частицей, внося вклад в общую скорость коагуляции.
Состояние коагулирующей взвеси в любой момент времени удобно характеризовать средним числом первичных частиц в одной флокуле m = noln. Согласно формуле (3.13.4),
т=1+(///*). (3.13.6)
Одновременно это число имеет смысл безразмерного времени от начала процесса коагуляции, выраженного в периодах полураспада раствора t. Параметр т является также относительной средней массой флокул и характеристикой их размера.
В рамках простейшего варианта теории Смолуховского, в котором кинетический коэффициент не зависит
Физика, химия и технология дисперсных систем
697
от размера флокул, можно также вычислить концентрацию флокул любого размера в произвольный момент времени. Однако более актуальным является решение другой задачи — нахождение геометрических характеристик флокул, в первую очередь их размера и зависимости кинетического коэффициента от размера флокул.
3.13.3. Фрактальная размерность и структура флокул
В основе разнообразных коагуляционных превращений дисперсных систем лежит фиксация взаимного положения частиц за счет их слипания. Она может охватывать только небольшие группы частиц (флокулы), а может распространяться на все частицы дисперсной системы. В последнем случае образуется сплошная сетка из взаимосвязанных частиц. Флокулы можно в этом смысле считать фрагментами будущей сплошной сетки. В молекулярной физике аналогичная совокупность связанных атомов называется кластером.
Связь между числом частиц в одной флокуле т, размером частиц г и размером флокулы / (рис. 3.90) выражается формулой [27]:
гс = (//г)ф. (3.13.7)
Показатель степени ф этой формулы называется фрактальной размерностью флокул, а саму формулу следует рассматривать как определение понятия «фрактальная размерность». Размер частиц здесь представлен длиной связи г соседних частиц, которая часто совпадает с диаметром частиц 2а. В качестве геометрического размера флокулы выступает диаметр / описанной вокруг флокулы сферы. Для наглядности можно считать, что флокула помещена в сферический контейнер указанного размера с жесткими непроницаемыми стенками.
Фрактальная размерность характеризует плотность заполнения контейнера связанными частицами: чем больше фрактальная размерность, тем более плотную структуру имеет флокула. Физический (точнее, геометрический) смысл, диапазон значений этого параметра и некоторые закономерности можно уяснить из приводимых ниже примеров флокул с регулярной структурой.
При описании структуры флокул целесообразно использовать понятие ее «элементарной ячейки» — блока из минимального числа связанных частиц, который отражает в себе все геометрические особенности более сложных структур, строящихся по типу элементарной ячейки. Простейший вид структуры — это линейная цепочка частиц (рис. 3.91, а). В цепочке любой длины т = 1/г и, следовательно, ф= 1. При заполнении всего объема произвольной трехмерной фигуры по любому периодическому закону — с гексагональной (рис. 3.91, б), кубической (рис. 3.91, в) или иной структурой элементарной ячейки — число частиц т внутри фигуры равно отношению ее объема /3 к объему ячейки г3, занятой одной частицей, т. е. т = (//г)3. В этом случае, согласно формуле (3.13.7), фрактальная размерность ф = 3.
’-----I-------►
Рис. 3.90. Геометрические характеристики флокулы: / — размер и г —длина элементарного звена
Рис. 3.91. Варианты заполнения полости частицами: а — самое рыхлое (ф = 1); б, в — самое плотное (ф - 3)
При заполнении пространства связанными частицами с некоторой промежуточной плотностью упаковки фрактальная размерность флокулы (фрактальность) будет иметь некоторую промежуточную (между 1 и 3) величину. Таким образом, значение фрактальности трехмерных структур должно находиться в диапазоне от 1 до 3.
Понятие «фрактальность» является обобщением понятия размерности пространства, причем она может иметь и дробную величину. Последнее обстоятельство объясняет происхождение терминов «фрактальная размерность», «фракталы» (объекты с фрактальными свойствами) и др. Они происходят от слова fraction — часть (имеется в виду часть целого числа).
По способу образования фракталы — это иерархические объекты, возникающие при связывании в единое целое объектов предыдущего поколения по тем же геометрическим правилам, по которым устроен объект предыдущего поколения. Самым младшим из них является элементарная ячейка, состоящая из монолитных первичных частиц. Старшие (/-е) члены иерархии имеют ту же структуру, что и элементарная ячейка, но ее элементами являются не первичные частицы, а флокулы предыдущего (z-l)-ro поколения. В этом заключается свойство геометрического подобия структур разного порядка.
Примеры регулярных плоских (двухмерных) фрактальных флокул показаны ниже на серии рис. 3.92-3.95. Самый мелкий элемент фрактальной структуры, который выглядит на приведенных рисунках как монолитная частица, может в действительности оказаться фло-кулой и иметь ту же структуру, что и более крупный фрагмент, показанный на рисунках. Конечно, это может оказаться и монолитной элементарной частицей.
698
Новый справочник химика и технолога
а) б)
Рис. 3.92. Построение регулярных фракталов на основе элементарных ячеек со структурой трехлучевой звезды (а) и креста (б). Первая не может, а вторая может образовывать флокулы следующего поколения
Рис. 3.93. Флокула третьего поколения на основе элементарной ячейки гексагональной структуры
Рис. 3.94. Двухмерная схема объемной флокулы третьего поколения с кубической формой полости на основе элементарной 27-частичной кубической ячейки
Рис. 3.95. Двухмерная схема объемной флокулы третьего поколения с кубической формой полости на основе элементарной 8-частичной кубической ячейки
Если структура имеет регулярный характер, то это дает возможность предсказать связь между числом частиц во флокуле и ее размером и, следовательно, вычислить фрактальную размерность флокулы.
Однако не для всяких типов элементарных ячеек это полезно и просто. Чтобы формула (3.13.7) сохраняла силу вплоть до флокулы размером в одну частицу, т. е. и при т = 1, геометрический центр иерархии структур должен быть всегда занят одной частицей. Структура,
показанная на рис. 3.95, этому условию не удовлетворяет — ее геометрический центр частицей не занят (занят полостью между четырьмя частицами). На рис. 3.94 и 3.95 некоторые блоки только намечены, их внутренняя структура идентична структуре полностью прорисованных блоков. Еще более важное требование к структуре флокул состоит в том, что она должна включать только связанные частицы, а это значит, что связь флокул младшего поколения внутри флокулы старшего поколения осуществляется только через непосредственный контакт первичных частиц.
В случае ячейки типа трехлучевой звезды (рис. 3.92, а) это требование оказывается несовместимым со свойством геометрического подобия для флокул третьего поколения и старше. То же самое относится к ее трехмерному аналогу — объемно центрированному тетраэдру (структура молекулы метана и решетки алмаза). Четырехлучевая плоская звезда (рис. 3.92, б) и ее трехмерный вариант (объемно центрированный куб — ОЦК), гексагональная плоская (рис. 3.93) и трехмерная ячейки удовлетворяют всем перечисленным требованиям. Центральная частица ОЦК (рис. 3.92, б) имеет 8 ближайших соседей (координационное число z = 8), а количество частиц в элементарной ячейке q = z + 1 равно 9. Диаметр / сферы, в которую вписывается эта ячейка, равен трем диаметрам частиц (/ = Зг).
Отношение к = П г диаметра / элементарной ячейки к диаметру г частицы равно 3 независимо от структуры элементарной ячейки при условии, что ее центр также занят частицей. Требование связанности всех частиц в элементарной ячейке и равенства числа частиц, связанных с центральной, координационному числу z означает, что центральная частица всегда окружена слоем соседей толщиной в один диаметр частицы и поэтому диаметр ячейки всегда будет в три раза больше диаметра ее ядра — структурной единицы (частицы или контейнера), занимающей центр ячейки.
Структура второго поколения (рис. 3.92, б) сохранит те же пропорции между параметрами этой структуры и структуры предыдущего поколения. В итоге в структуре ОЦК второго поколения (z = 2) число т первичных частиц будет равно 81 (т. е. 92). В общем случае в иерархической структуре z-ro порядка:
m = q'. (3.13.8)
Размер контейнера /„ вмещающего эту структуру из 81 частицы, увеличится еще в к раз по сравнению с размером 4 -1 контейнера предыдущего поколения (/,• = к!,^), поэтому:
Нг = К. (3.13.9)
При логарифмировании формул (3.13.8) и (3.13.9) и исключении из них номера поколения z получается выражение Inm = 1п(/ / г) (Inq / InA) или формула
т = (//г)и/1пА. (3.13.10)
Физика, химия и технология дисперсных систем
699
Отсюда, в соответствии с определением фрактальной размерности (3.13.7), видно, что:
ф = 1п7/Ы. (3.13.11)
Следовательно, для регулярных фрактальных флокул с элементарной структурной ОЦК ф = (1п9 / 1пЗ) = 2. Фракталы с элементарной ячейкой гексагонального типа (к = 3, <7=13) имеют размерность ф = 2,335 = = (1п13 /1пЗ).
Фрактальная структура, которую можно построить из элементарных ячеек не сферической, а кубической формы из 27 частиц (рис. 3.94), имеет размерность ф = (1п27/1пЗ) = 3, что и следовало ожидать, поскольку она соответствует полному заполнению трехмерного пространства частицами. Если ту же кубическую ячейку заключить в описанную вокруг нее сферу и, как и во всех других случаях, создавать структуру следующего поколения из таких элементарных ячеек, заключенных в сферический контейнер, то фрактальность будет заметно меньше: ф = 2,25 [1п27 / 1п(30,5)]. Здесь учтено, что диаметр описанной вокруг куба сферы равен диагонали куба, которая в З0,5 раза больше его ребра. В соответствии с общими правилами структура строится так, чтобы сферы, очерчивающие соприкасающиеся ячейки предыдущего поколения, не пересекались и расстояние между частицами, обеспечивающими связь соседних ячеек, было таким же, как внутри ячейки. В данном случае это означает, что блоки кубической формы связаны между собой вершинами кубов.
Почти все, что сказано о параметрах фракталов, образованных из 27-частичной элементарной кубической ячейки (рис. 3.94), справедливо и для структур, образованных из 8-частичной (рис. 3.95) ячейки. В частности, фрактальность структур из ячеек кубической формы будет равна 3(1п8/1п2). Различие лишь в том, что основное соотношение фрактальной геометрии флокул (3.13.7) будет справедливым только при большом числе частиц во флокуле, поскольку центр кубической элементарной ячейки из 8 частиц не занят частицей. Поэтому случай т = 1 явно не может быть реализован. Впрочем, и во всех других случаях результаты расчетов абсолютно точны, если флокула имеет завершенную структуру, т. е. содержит столько же флокул младшего поколения, сколько первичных частиц элементарная ячейка. Практически это означает, что число частиц во флокуле должно строго соответствовать формуле (3.13.8).
В остальных случаях возникают отклонения в большую или меньшую стороны с примерно равной вероятностью, поэтому в среднем эта ошибка нивелируется. Это обстоятельство (статистически правильные результаты при наличии заметных отклонений в описании свойств отдельных флокул) позволяет перенести выявленные на регулярных структурах закономерности на структуры случайной формы, образующиеся при коагуляции. Статистически принцип подобия структур
младшего и старшего поколений при коагуляционном укрупнении флокул выполняется, так как закон коагуляции остается неизменным по мере укрупнения флокул. Величина фрактальной размерности при этом становится эмпирическим параметром, хотя возможны и ее теоретические расчеты на базе тех или иных предположений о значении параметров q и к в формуле (3.13.11). Эта область физики и химии коллоидов интенсивно развивается, главным образом в направлении компьютерного моделирования процесса коагуляции.
3.13.4. Параметры состояния коагулирующей взвеси и условие структурирования
По мере укрупнения флокулы становятся более рыхлыми. Характеристикой флокул в этом отношении может служить внутрифлокулярная концентрация — объемная доля дисперсной фазы внутри флокулы Ф,„ = тг3 / /3. Размер флокулы /, в соответствии с формулой (3.13.7), выражается через число частиц в ней:
1=гтщ. (3.13.12)
Если раскрыть с помощью этой формулы величину / в выражении для ф(„, то получится уравнение
Ф,„ = /и(ф-3)/ф. (3.13.13)
Рыхлые флокулы занимают во взвеси значительно большую долю объема фт, чем сами частицы дисперсной фазы: фт = л/3, где п — концентрация флокул (1/м3). Выполнив ту же замену, что и в предыдущем случае, а также имея в виду, что произведение п^г3 концентрации первичных частиц л0 на куб их размера г3 — это объемная доля дисперсной фазы во взвеси ф, можно получить формулу
фи = ф/«(3-ф)/ф. (3.13.14)
Как и следовало ожидать ф,„фт = ф.
Согласно формуле (3.13.14), доля объема, занимаемого во взвеси флокулами, растет по мере укрупнения флокул неограниченно. В действительности эта величина не может быть больше единицы.
Условие:
фи=1 (3.13.15)
— это условие заполнения флокулами всего объема дисперсной системы, т. е. условие их слияния в единую, сплошную структурную сетку из взаимосвязанных частиц. Иначе говоря, это условие структурирования взвеси частиц. При этом всякие коагуляционные процессы прекращаются.
Из соотношений (3.13.14) и (3.13.15) можно найти критическое число частиц тс в одной флокуле, при котором происходит структурирование:
тс = (1/ф)ф/(3“ф). (3.13.16)
700
Новый справочник химика и технолога
Формула (3.13.17) позволяет найти соответствующий ему размер флокул 1С, т. е. размер фрактальных доменов, из которых состоит структурная сетка. Этот параметр играет важную роль при рассмотрении реологических свойств дисперсных систем:
7с = г(1/ф)1/(3 ф). (3.13.17)
Время t достижения того или иного состояния можно легко найти с помощью формулы (3.13.6): t = (m- 1)/, подставляя сюда соответствующие величины т и времени коагуляции t. В частности, время tc перехода коагулирующей взвеси в структурированное состояние описывается формулой
tc = (mc~ 1/. (3.13.18)
Структурирование — одно из видимых и важных проявлений процесса коагуляции. Оно возможно, когда этот процесс заходит достаточно глубоко. Другое универсальное проявление коагуляции, которое заметно уже на начальном этапе коагуляции, — это ускорение процесса расслоения взвеси на две или большее число фаз: осадок, взвесь и свободную от частиц среду. Принципиальное отличие оседания коагулирующей взвеси от оседания агрегативно устойчивой взвеси заключается в том, что как скорость этого процесса, так и концентрация оседающей субстанции при коагуляции изменяется во времени. Под субстанцией подразумеваются и первичные частицы взвеси, и флокулы, и сплошная структурная сетка, которая также подвержена оседанию (уплотнению) под действием силы тяжести.
Описание этих двух важнейших для технологии процессов — коагуляции и оседания дисперсных систем — не может быть полным и всесторонним, если их рассматривать по отдельности. Коагуляция через размер флокул и долю занимаемого ими пространства влияет на скорость оседания дисперсной фазы. Скорость коагуляции в свою очередь зависит от концентрации флокул (частиц) и их размера, которые непрерывно меняются как в пространстве, так и во времени, в том числе за счет оседания. Система уравнений, описывающих эти взаимосвязанные процессы, поддается только численному решению на вычислительных машинах. Соответствующие программы существуют, способны функционировать на персональном компьютере под управлением стандартного программного обеспечения и представлять получающиеся зависимости в наглядном виде. Однако это не устраняет необходимости в аналитического описания тех же зависимостей. Только простые и обозримые формулы позволяют проследить влияние различных условий и физических процессов на эволюцию дисперсной системы и на ее состояние в произвольный момент времени и по окончании всех процессов. Однако получить такие зависимости можно при достаточно сильном упрощении задачи.
Очевидно, что многие свойства коагулированных дисперсных систем зависят от гидродинамических характеристик флокул. Наглядная и практически полезная демонстрация такой зависимости приведена ниже на примере оседания флокул в поле силы тяжести.
3.13.5. Скорость оседания флокул
Для описания процесса оседания коагулирующей взвеси необходимо располагать выражением для скорости ит движения флокулы под действием силы тяжести. Естественно при этом предположить, что к флокулам любого порядка т можно применить формулу Стокса
«„ = 2 Ap.gft2/^. (3.13.19)
В ней радиус частиц а заменен на радиус флокул R, а разность плотностей Ар частиц р и среды р0 — на разность плотностей Apw флокул pw и среды. Такая замена предполагает, что рыхлую флокулу можно в гидродинамическом отношении считать сплошным телом. Справедливость этого предположения требует обоснования. В связи с этим необходимо условиться относительно критерия гидродинамической сплошности движущегося в некоторой среде объекта. Для этого введем понятие «облако» как область повышенной концентрации частиц, граничащей с той же средой, которая является частью облака. Очевидное свойство облака — его способность двигаться относительно среды как единое целое, не смешиваясь с ней. В то же время можно наблюдать и такие области повышенной концентрации частиц, которые довольно быстро рассеиваются, смешиваясь со средой (например, выбросы дыма из трубы). На языке количественных понятий это различие означает, что облако способно двигаться относительно окружающей его среды быстрее, чем отдельные частицы, образующие облако. Его можно характеризовать размером R и объемной долей ф/и дисперсного вещества в облаке.
Плотность облака pw, как и любой дисперсной системы, определяется плотностью дисперсной фазы р и ее долей ф,„ в облаке:
Рт = Ро + фшАр, (3.13.20)
где Ар = р - ро — разность плотностей дисперсной фазы и среды р0. С помощью формулы Стокса легко найти отношение скоростей оседания облака, как сплошного объекта, и одной частицы в одинаковой среде. Оно очевидно равно фш/?2 / а2, где а — размер частиц, образующих облако. Отсюда следует, что это отношение больше единицы, т. е. облако будет двигаться быстрее отдельной частицы при условии
<р,„>(а/Я)2. (3.13.21)
Полученное неравенство имеет более общий смысл. Оно выражает условие перехода от миграционного к гидродинамическому режиму движения (смешивания)
Физика, химия и технология дисперсных систем
701
сред разного состава. Например, сточных вод, сбрасываемых в водоем, или выбросов дыма и пыли в атмосферу. В силу своей универсальности условие (3.13.21) может по-разному толковаться в разных ситуациях. В связи с задачей коагуляции оно означает, что флокула при движении обтекается средой как сплошное тело. Ее гидродинамический размер R можно в таком случае приравнять размеру / сферы, вмещающей флокулу. При фрактальной структуре флокулы ее размер R и число первичных частиц т во флокуле связаны, согласно определению понятия «фрактальная размерность» флокулы, соотношением m = (Rla)\ а средняя внутрифло-кулярная концентрация дисперсной фазы в ней
= 7и(ф~3)/ф. Если выразить эту концентрацию через размер флокул <р/„ = (а//?)3~ф и подставить ее в условие (3.13.21), то легко видеть, что оно сводится к неравенству (а/7?)зф> (a/R)2. Так как a<R, то оно будет выполняться при ф > 1, т. е. всегда, за исключением случая линейных цепочечных агрегатов, когда ф = 1 и скорости цепочки и частицы совпадают. На этом основании фрактальная флокула может считаться сплошным телом (только в гидродинамическом смысле), и тогда к ней можно применить формулу Стокса (3.8.3). При выполнении в этой формуле ряда подстановок: R = ат^, R2 = ат2'*, Арт = Арт(ф’зу* и, наконец, /?2Арто = Apt/W*” получаются выражения
ит = (2bpga2/ 9п) т^’,)/ф (3.13.22)
или
ит = ит(^т. (3.13.23)
Здесь и = 2Apgo2/9r| — скорость оседания единичной частицы. Как видно из полученных формул, скорость оседания фрактальных флокул всегда больше в т^~1)/ф раз, чем скорость оседания индивидуальных частиц (кроме случая ф = 1 — простой цепочки), т. е. коагуляция будет ускорять оседание взвеси. Заметим также, что фрактальные флокулы являются, вероятно, наиболее рыхлыми структурами взаимосвязанных частиц. Плотность иных (не фрактальных) флокул, если таковые существуют, будет, по всей видимости, не меньше плотности фрактальных флокул, и поэтому можно полагать, что они при любой структуре оседают быстрее индивидуальных частиц. По этой причине в дальнейшем не будет подчеркиваться, что речь идет о фрактальных флокулах. Их преимущество в том, что имеется однозначная связь между размером и массой флокул.
Итак, относительная масса флокул т (число первичных частиц в одной флокуле) является определяющим параметром по отношению к важнейшим свойствам дисперсных систем и протекающим в них процессам. Особенность этого параметра в том, что он никогда не остается постоянным. В неустойчивых к коагуляции системах он непрерывно и неограниченно растет. Так, по крайней мере, обстоят дела при классическом описании процесса коагуляции, игнорирующем структурные свойства флокул.
3.13.6. Альтернатива: расслоение или структурирование
В процессе оседания частиц коагулирующей взвеси в общем случае формируются минимум три разных слоя: осадок, взвесь и свободная от частиц среда. В дальнейшем будет предполагаться, что плотность дисперсной фазы выше, чем плотность среды. Поэтому слой осадка высотой Нх расположен в нижней части сосуда, слой свободной от частиц среды высотой HL (в дальнейшем «жидкость») остается в верхней части сосуда и собственно взвесь занимает остальную, среднюю часть сосуда. Высота слоя взвеси равна, очевидно, Н - (Hs + HL), где Н — высота сосуда, точнее столба исходной взвеси, с равномерно распределенной дисперсной фазой с концентрацией ср. По умолчанию принимается также, что среда является жидкой, а дисперсная фаза — твердым веществом, т. е. рассматривается расслоение суспензий или золей. Предполагается наличие четкой границы, разделяющей соседние слои суспензии (рис. 3.96). На практике они не всегда видимы, но теоретически существуют, хотя деление на взвесь и осадок может осуществляться по разным признакам. В этом подразделе осадком считается та часть суспензии в нижней части сосуда, где флокулы плотно уложены под действием силы тяжести и образуют сплошную структурную сетку. Иначе говоря, осадок — это часть дисперсной системы, где (рто = 1.
Рис. 3.96. Формирование осадка (Hs) в коагулирующей суспензии, слоя взвешенных над ним флокул, где коагуляция продолжается, и слоя жидкости (Н/), свободной от частиц суспензии: Н — первоначальная высота столба суспензии
Сетка обладает необходимой прочностью и не уплотняется под действием собственного веса. Концентрация дисперсной фазы в осадке и в прилегающем слое взвеси не изменяется скачком, и поэтому визуально эта граница может быть и незаметна. Наоборот, граница взвеси и жидкости должна быть, согласно принятой модели процесса, четко видна. Это согласуется с экспериментально установленными закономерностями оседания коагулирующих взвесей.
Согласно формуле (3.8.35), скорость накопления осадка dHJdt = итут. Раскрыв входящие сюда величины с помощью формул (3.13.14) и (3.13.23), можно получить уравнение dHsldt = uq>m2/\ которое легко интегрируется после замены dt = t dm, вытекающей из равенства т = 1 + tit*. Считая, что начало оседания совпадает с
702
Новый справочник химика и технолога
началом коагуляции, интегрировать следует в пределах от т = 1 (/= 0) до некоторого текущего числа частиц т во флокуле (текущего момента времени). В сущности, величина т здесь выступает как безразмерное время, длительность которого выражается в периодах коагуляции t. В итоге получается выражение для толщины слоя осадка [12]:
Н, = Иф/ [ф/ (ф + 2)] 1];
Ф, = т(ф-3)/ф. (3.13.24)
Здесь же приведена формула для концентрации дисперсной фазы в осадке <ps на высоте Hs.
Она совпадает с концентрацией дисперсной фазы ф|Л (формула (3.13.13)) внутри флокул в момент их соприкосновения с осадком. Таким образом, эта пара формул дает в параметрическом виде зависимость концентрации дисперсной фазы от высоты внутри осадка. Важно, что в отличие от осадков в устойчивых к коагуляции взвесях (см. подраздел 3.8.3) здесь концентрация переменна по высоте, причем на дне <pjw = 1, а на границе со взвесью совпадает с концентрацией дисперсной фазы во взвеси <р. Несмотря на оседание флокул, их концентрация внутри столба взвеси постоянна по высоте, так как все они оседают с одинаковой скоростью.
Скорость продвижения dHJdt границы между взвесью и слоем свободной от частиц жидкости равна, согласно формуле (3.8.28), иш(1-фш). Целесообразно, однако, вычислить сумму скоростей движения этой границы и границы осадок—взвесь: dHJdt = dHs/dt + + dHJdt, которая является скоростью их сближения и одновременно скоростью сокращения высоты слоя взвеси. Она оказывается равной величине ит. Интегрирование выражения (3.13.23) по времени с помощью той же замены dt = t dm, что и в предыдущем случае, дает суммарную толщину Hq слоя осадка и жидкости:
Hq = ut [ф/ (2ф - 1)] [т(2ф"1)/ф - 1], (3.13.25) что позволяет вычислить высоту слоя жидкости над слоем взвеси:
HL = Hq-H3. (3.13.26)
На рис. 3.97. показана зависимость от времени высоты осадка и положения нижней границы слоя чистой жидкости над столбом взвеси, построенная по этим формулам.
Рис. 3.97. Кинетика оседания коагулирующей суспензии:
1 — высота слоя осадка (Я5); 2 — координата нижней границы слоя свободной от частиц жидкости (H-HL)
Очевидно, что процесс оседания завершится, когда взвесь полностью перейдет в осадок, и тогда суммарная толщина Нд слоя осадка и слоя жидкости станет равной высоте Н всего столба суспензии. При подстановке Нд = Н в уравнение (3.13.25) и решении его относительно размера флокул т получается значение числа частиц во флокуле 7ир, при котором завершится процесс оседания:
'/эд -|*/(2Ф-1)
т = ( )----1 (3.13.27)
[_ фм/ J
Соответственно, время завершения процесса расслоения /р будет:
/р = (шр- 1)А (3.13.28)
Полное расслоение или структурирование представляют собой альтернативные варианты завершения эволюции коагулирующей взвеси. Фактически эволюция завершится тем исходом, который наступит раньше. Если /р < tc, то расслоение произойдет до перехода взвеси в структурированное состояние. В обратном случае полного расслоения не произойдет — взвесь перейдет в структурированное состояние и на этом все процессы в дисперсной системе прекратятся. Эквивалентным образом лимитирующий процесс эволюции выбирается по значениям критических значений тр и /ис флокул средних размеров. Таким образом, соотношение (3.13.29) представляет собой условие доминирования структурирования над расслоением:
тй<ту (3.13.29)
К моменту структурирования на дне сосуда образуется слой осадка, толщина которого определяется формулой (3.13.24) при подстановке в нее т = тс. Аналогично с помощью формул (3.13.25) и (3.13.26) вычисляется толщина слоя жидкости над структурированным столбом суспензии и далее высота столба структурированной суспензии. Легко убедиться, что при mz << /Ир толщины осадка и слоя жидкости составляют пренебрежимо малую величину по сравнению с высотой HZ = H-Hq столба структурированной взвеси. В общем случае соотношение между высотами каждого из трех слоев (осадок, структура и жидкость) может быть произвольным (рис. 3.98). Распределение дисперсной фазы в осадке определяется, как уже отмечалось, парой уравнений (3.13.24).
Полученные выше критерии эволюции взвеси основывались на предположении, что флокулы, как и коагуляционная структурная сетка, имеют достаточно высокую прочность. Детально механические свойства флокул и осадков [14] рассматриваются ниже (подраздел 3.14) в разделе реологии тиксотропных систем. Здесь же следует учесть, что флокулы имеют ограниченную прочность, которая определяется прочностью fc контактов соседних частиц во внутрифлокулярных цепочках связанных частиц.
Физика, химия и технология дисперсных систем
703
Рис. 3.98. Распределение дисперсной фазы по высоте по окончании процесса коагуляции в условиях его лимитирования структурированием: Н, — высота слоя осадка;
Яс — высота столба структурированной суспензии; Н— первоначальная высота столба суспензии;
Ф — ее коцентрация
Как было показано выше, плотность флокул, а следовательно, и среднее число контактов на единицу объема флокулы уменьшается с увеличением их размера. Пропорционально этому снижается и прочность флокул по мере увеличения их размера. В то же время на фло-кулу действует сила ее собственного веса, которая при оседании в вязкой среде преобразуется в гидродинамическую силу — силу трения о поверхность набегающих потоков жидкости флокулы. Этот поток смывает с поверхности флокулы ее внешние наиболее рыхлые слои до тех пор, пока сила сцепления частиц не сравняется с силой трения, приходящейся на одну частицу. Последняя оказывается равной силе веса fg одной частицы, так что указанное равенство сил определяет гравитационно-равновесный размер флокул:
т^Ш&. (3.13.30)
В итоге неравенство, выражающее (3.13.29) доминирование структурирования или расслоения в эволюции коагулирующей дисперсной системы, сохраняет свою силу при выполнении двух дополнительных условий:
т^> тс и тъ> тр. (3.13.31)
Если окажется, что ms < тс и mg < тр, то структурирования не произойдет, а будет идти оседание флокул, но уже с неизменной во времени скоростью:
Ug = ит[*~'}/* (3.13.32)
и с постоянной плотностью образующегося на дне осадка. Возможны и другие комбинации трех основных параметров mg, тс и тр, лимитирующих процесс эволюции. В каждом случае его результат будет чем-то отличаться от результатов эволюции при других комбинациях этих параметров.
Несмотря на упрощения в описании кинетики коагуляции, в этом подразделе удалось выявить наличие ряда факторов, лимитирующих процессы коагуляции и оседания. Коагуляция, а с ней и размер флокул могут
быть ограничены или структурированием, или расслоением взвеси, или достижением гидродинамически равновесного размера флокул. Лимитирующий фактор эволюции определяется рецептурно-геометрическими параметрами исходного состояния взвеси по соответствующим формулам. Полный вариант теории включает учет полидисперсности флокул при описании процесса коагуляции, структурирования и оседания с последующим уплотнением осадка и структурной сетки под действием силы тяжести. Описание уплотнения возможно только на основе законов деформирования структурированных дисперсных систем.
3.13.7. Уравнения кинетики коагуляции
Даже если в исходной дисперсной системе, склонной к коагуляции, все частицы первоначально имеют одинаковый размер, через некоторое время она превращается в полидисперсную систему, в которой спектр размеров частиц (точнее флокул) непрерывно изменяется, сдвигаясь в сторону укрупнения флокул.
Коагулирующая дисперсная система может быть представлена как совокупность флокул различного типа. В данном случае тип флокул — это число т первичных частиц во флокуле /и-го типа. Оно может принимать значения от 1 до бесконечности.
Символами вида nt далее обозначаются численные (1/м3) концентрации частиц z-го типа, а символами вида sy — вероятность парного столкновения флокул типа / и j (при их концентрациях равных единице).
Фундаментальное уравнение кинетики коагуляции Смолуховского представляет скорость dnm/dt изменения концентрации пт флокул /и-й фракции как сумму двух слагаемых:
dn 00
=47Г(У5. ml -nSs п). (3.13.33)
ц/и-1) I m-i т im 1 х s
Первое — приращение их концентрации при парных столкновениях флокул /-го и (т - /)-го типов, которые выбраны так, что общее число частиц / + (/»- /) в одной паре столкнувшихся флокул равно числу частиц т во вновь образующейся флокуле /и-го типа. Второе слагаемое — это убыль концентрации флокул /и-го типа за счет их столкновения с флокулами любого другого типа. Вероятность слипания столкнувшихся флокул принимается равной единице (условие быстрой коагуляции).
Основная проблема кинетики коагуляции полидис-персных систем состоит в том, что для описания процесса коагуляции необходимо составить и совместно решить столько уравнений типа (3.13.33), сколько фракций содержит полидисперсная система. Обычные способы математического описания гранулометрического состава предполагают наличие непрерывного спектра размеров, т. е. бесконечное число фракций, а следовательно, и уравнений. Решение системы уравнений в такой постановке невозможно. Эта трудность обходится упомянутым выше способом, т. е. непрерыв
704
Новый справочник химика и технолога
ное распределение представляется как совокупность конечного числа дискретных фракций [14].
Вероятность столкновения частиц непосредственно зависит не от коэффициента их диффузии, а от коэффициента взаимной диффузии частиц, который равен сумме коэффициентов диффузии частиц относительно среды и, следовательно, сумме обратных радиусов частиц (1/7?, + 1/7?у). Радиус захвата сталкивающихся частиц или флокул равен сумме их радиусов (R, + 7?у). Вероятность столкновения пропорциональна произведению этих двух величин, которое может быть выражено через отношение размеров частиц. Это приводит к известному выражению (формуле Мюллера) для вероятности Sy парного столкновения частиц размером 7?, и Rj в процессе их теплового движения:
(3.13.34)
Применение этой формулы к флокулам предполагает, что в гидродинамическом отношении их можно считать сплошными телами. Допустимость такого предположения рассмотрена в подразделе 3.13.5. На основании этого уравнение (3.13.34) можно использовать и в случае рыхлых флокул. Отношение размеров Ri/Rj флокул можно, согласно формуле (3.13.12), заменить отношением числа частиц в них: (z f) в степени 1/ф, а уравнение (3.13.34) — уравнением
кТ L I i I
s.j=T~ 4 + Н
бтггЦ \j)
(3.13.35)
Оно, как и уравнение (3.13.34), при равенстве размеров (z = у) сталкивающихся частиц (флокул) сводится к формуле 5 = 8Л773лг| для кинетического коэффициента уравнения коагуляции монодисперсной взвеси (см. подраздел 3.13.2).
Еще одна проблема чрезвычайно усложняет решение и без того сложной системы множества уравнений кинетики коагуляции с переменными коэффициентами 5,у. Это оседание частиц (флокул). Его роль становится тем сильнее, чем дальше заходит процесс коагуляции и чем шире становится спектр размеров флокул за счет увеличения количества крупных флокул. Их скорость оседания и заметно больше, чем скорость оседания мелких флокул. Различие в скоростях оседания разных флокул приводит к тому, что гранулометрический состав взвеси меняется не только во времени, но и по высоте h столба коагулирующей взвеси (см. подраздел 3.8). Следует отметить, что уравнение оседания полидисперсной взвеси даже без коагуляции не может быть решено аналитическими методами. Тем более это относится ко всей системе уравнений эволюции взвеси, включающей в себя уравнения коагуляции, уравнения оседания и уравнения материального баланса (сохранения) для всех фракций. Уравнения сохранения выражают тот
факт, что суммарное изменение концентрации каждой фракции dnm!dt в некотором слое взвеси складывается из изменения дпщ/dt, обусловленного участием данной фракции в коагуляции, и из переноса (подвода и отвода) данной фракции частиц (флокул) d(nmUn^ldh, обусловленного различием ее потоков на границах слоя:
dnm!dt = dn„Jdt + д(птипУ dh. (3.13.36)
Существуют также концентрационные и временные ограничения применимости уравнений кинетики коагуляции. Они обусловлены возможностью перехода взвеси в процессе коагуляции в структурированное состояние или достижением флокулами гравитационно-равновесного размера (уравнение (3.13.30)). Заранее сформулировать конкретное условие структурирования невозможно, так как выполнимость общего условия структурирования: <ре = 1 (заполнение всего пространства флокулами) не может быть проверена, прежде чем будут выполнены необходимые вычисления. В данном случае величина <ре представляет собой сумму объемных долей <ре„ занимаемых во взвеси флокулами каждого типа, а они могут быть получены только в результате совместного решения уравнений кинетики коагуляции и оседания флокул. Это возможно только численными методами, реализованными в соответствующей компьютерной программе. Алгоритм решения уравнений эволюции взвеси включает в себя корректирование хода вычислений при возникновении тех или иных особенностей состояния взвеси, в том числе й при ее переходе в структурированное состояние. В этом состоянии коагуляционные процессы прекращаются, и уравнения эволюции взвеси сводятся к уравнениям деформации неоднородного по свойствам структурного столба, которые рассматриваются ниже.
3.13.8. Динамика и кинетика уплотнения осадка
Рыхлая пространственная сетка из слипшихся частиц, возникающая в процессе коагуляции частиц и оседания флокул, может подвергаться самоуплотнению под действием собственного веса. Основная проблема, с решением которой связана возможность количественного описания этого процесса, — это проблема реоди-намики структурированных дисперсных систем. Геодинамика представляет собой усложненный вариант гидродинамики. В отличие от гидродинамики реодина-мика имеет дело с неньютоновской деформируемой средой.
Динамика уплотнения осадка [12] в общем случае определяется следующими силами, отнесенными к тонкому слою взвеси толщиной dH в сосуде радиусом R:
1. Его весом TiRldH&pm'¥g, где Т — объемная доля оседающей субстанции, Дрш — разность плотностей оседающей субстанции и среды, g — ускорение свободного падения. Под субстанцией подразумеваются разные формы дисперсной фазы в слое взвеси — в виде отдельных частиц, в виде флокул или в виде фрагмента сплошной структурной сетки.
Физика, химия и технология дисперсных систем
705
2. Разностью сил давления Р верхней и нижней частей взвеси на верхнюю и нижнюю границы слоя nR^dP.
3. Силой трения слоя толщиной dH о стенки сосуда 2nRdHx, где т — удельная (на единицу площади) сила трения о стенки. По своему характеру это статическая сила, подобная сухому трению при скольжении твердого тела по поверхности другого тела. Она не зависит от скорости скольжения.
4. Силой вязкого сопротивления всех частиц (флокул) данного слоя я/?2dHQl1 / vm)bmum, где b„, — коэффициент сопротивления движению частиц (флокул) в вязкой среде, vm — средний объем одной частицы (флокулы), Um — СКОРОСТЬ ДВИЖеНИЯ СЛОЯ И 'Р/Гда — концентрация флокул (их число в единице объема). Такой способ описания сил вязкого сопротивления означает, что взвесь монодисперсна.
Баланс этих сил после сокращения общих величин во всех слагаемых дает уравнение
dP 2т Т
&Pmg4-“--------~ = —Ьтит. (3.13.37)
dH R vm
Для решения задачи необходимо принять во внимание, что столб взвеси, независимо от его массы, не может воздействовать на нижележащие слои взвеси с давлением Р, большим, чем прочность на сжатие Ps этого давящего столба. Более того, при статическом равновесии сжимаемой субстанции в поле силы тяжести давление Р на любой высоте равно прочности Ps субстанции на продольное сжатие. При динамическом же равнове-
_ dP 2т
сии избыточная часть Ар^ч7-----------полного дав-
m dH R
ления столба компенсируется вязким сопро-
тивлением (Ч7 / Уп№тит среды.
Если оседающая субстанция — это флокулы, то величина *Р равна концентрации флокул срто, и тогда, согласно формуле (3.13.14), 4/ = q>we, где т — среднее число частиц в одной флокуле, а е = (3 - ф)/ф. Разность плотностей флокул и среды Арот связана с разностью плотностей Др дисперсной фазы и среды соотношением Дрш = Дртк, поэтому первое слагаемое в уравнении (3.13.37), равно Apgcp.
Основной характеристикой структурированной дисперсной системы является ее прочность на сдвиг т5. Предполагается, что прочность коагуляционной сетки на сжатие Ps пропорциональна прочности той же сетки на сдвиг, т. е. Ps = Kxs. Здесь К — некоторая константа, которая далее будет называться коэффициентом Пуассона, хотя она напоминает его только тем, что отражает соотношение между способностями материала к продольным и поперечным деформация. Указанная связь величин Р, Ps и ts дает возможность заменить градиент давления градиентом прочности структуры.
Если использовать соотношение ту = ттфк (3.14.5) между прочностью ту рыхлой сетки и удельной (на единицу площади) силой сцепления хт соседних частиц, то из уравнения (3.13.37) следует уравнение
е <7фЕ'* 2ттфЕ 1 ЬипГ
--------------= . (3.13.38) е-1 dH R vm
Здесь и далее величина ф обозначает концентрацию дисперсной фазы во взвеси, е = 2/(3 - ф), ф — фрактальная размерность флокул. Второе и третье слагаемые уравнения (3.13.38) отражают влияние прочности структурной сетки на скорость оседания ит слоя взвеси. Причем второе слагаемое — это разность сил давления структурированного столба взвеси на верхнюю и нижнюю границы слоя толщиной dH, а третье — сила трения слоя о стенки сосуда. Роль последнего растет с уменьшением поперечного размера сосуда R.
Если выразить коэффициент трения флокул bm - Ьтх>* через коэффициент трения b одной первичной частицы, объем флокулы vm = через объем v первичной частицы, а также ввести полученную ранее (уравнение (3.8.28)) поправку (1 - фюе) на встречное движение среды, то из уравнения (3.13.38) следует основное уравнение динамики слоя взвешенных коагулирующих частиц:
u(H,t\ = ц(1-фюЕ)юх| 1—фЕ 1 —
v 7 v 7 I ApgflIJ 2(e-l) dH
(3.13.39)
Здесь и = 2Apga2 /9ц — скорость оседания первичных частиц относительно среды (по Стоксу), ц — вязкость среды, % = ——-, а ф = ф(/7, t) — искомое распределение Ф
твердой фазы ф по высоте Н в различные моменты времени t от начала процесса коагуляции и оседания. Координата Н указывает расстояние слоя взвеси от дна
сосуда. Производная ——— < 0 , поэтому в уравнении dH
(3.13.39) обе величины в квадратных скобках положительны.
Фактически выражение (3.13.39) является символической записью двух разных уравнений. В представленной выше форме, но при хт = 0, оно сводится к рассмотренному ранее уравнению оседания монодисперс-ной неструктурированной суспензии. Это состояние реализуется, если объемная доля флокул Ч7 = <рте во взвеси еще не достигла некоторой критической величины Р, при которой происходит структурирование взвеси, т. е. объединение флокул в сплошную сетку. Величина 0 представляет собой объемную долю частиц или флокул при их плотной упаковке. Соответственно (1-0) — это доля свободного пространства, наличие которого необходимо, чтобы взаимное перемещение частиц (флокул) стало хотя бы геометрически возможным.
Условие перехода в структурированное состояние Ч7 = 0 выполняется при увеличении среднего числа частиц т во флокуле до критического значения тс = (р/ф)1/е. Подстановка этой величины вместо т в
706
Новый справочник химика и технолога
уравнение (3.13.39) преобразует первые два сомножителя (1 -ф/яе)/ях этого уравнения в сомножители (1 -Р)(р/ф)е-1. Одновременно взвесь приобретает прочность, т. е. величина хт принимает некоторое конечное по величине значение. В этом случае уравнение (3.13.39) описывает динамику уплотнения структурированной суспензии, которая как раз и характеризуется определенной прочностью т структурной сетки. В нем уже не фигурируют параметры отдельных флокул, и поэтому уравнение динамики уплотнения структурированной суспензии (3.13.40) [12] применимо вне зависимости от выбора модели процесса оседания и структурирования— простой модели монодисперсных флокул или более реалистичной модели полидисперсных флокул:
KRe 2(е-1) dH J
(3.13.40)
Из уравнения динамики (3.13.40) и условия прекращения оседания (м = 0) получается уравнение равновесного распределения вещества в структурированной взвеси с ограниченной прочностью структуры:
2хт ф2 I 2Ч Ф1 J Р|чА7?£/2(е-1)
(3.13.41)
Оно связывает концентрации ф дисперсной фазы и координаты Н двух произвольных слоев, концентрации и координаты которых отмечены нижними индексами 1 и 2. В частности, если приписать этим слоям значения концентраций, равные 0 и р, можно получить уравнение, описывающее максимально возможную высоту Hv слоя переменной концентрации, который может образоваться над предельно уплотненным слоем с концентрацией, равной р:
Tr KR& , f. 2т_р
Hv =—--------In 1----—
2(е-1) ApgT?
(3.13.42)
При R стремящемся к бесконечности, когда исчезает влияние стенок сосуда на распределение вещества в осадке, это выражение сводится к формуле
Apg(e-l)’
(3.13.43)
Такие условия создаются при оседании взвесей в водоемах.
При разумных предположениях относительно величины сил сцепления частиц и их размере порядка 10 6 м толщина переходного слоя между плотным осадком и очищенной от частиц средой колеблется от нескольких миллиметров до десятков сантиметров.
В уравнении динамики (3.13.39) величины ф и т являются функциями координат и времени. При коагуляции они связаны уравнениями кинетики коагуляции, а после перехода в структурированное состояние такая связь теряется. В случае монодисперсных флокул изменение dnldt концентрации частиц п в данном слое складывается из изменения их концентрации за счет коагуляции и за счет переноса частиц из соседних слоев, согласно формуле
= -sn2 + qn . (3.13.44)
Здесь первое слагаемое упрощенно — через средние значения параметров 5 и п коагулирующей взвеси — описывает вклад коагуляции, а второе — вклад переноса вещества в общий эффект изменения числовой концентрации частиц (флокул). Это уравнение является упрощенным вариантом уравнения (3.13.36) из предыдущего подраздела. Коэффициент 5 представляет константу скорости коагуляции: 5 = KkTI Зт), a q — коэффициент переноса, в частности, q = du/dH, если имеется продольный градиент скорости оседания частиц, который и означает сжатие слоя взвеси.
Решение уравнения (3.13.44) дает концентрацию «2 через некоторый интервал времени t, начиная от произвольного момента времени, при котором взвесь имела концентрацию ир
du ( du \ л. —ехр —t ' dH \dH ) п7 =-7---7:
( (du du
4 *AdH ) dH
(3.13.45)
Здесь величина l/s«i является мгновенным значением времени половинной коагуляции t, выраженным через концентрацию флокул «1 в текущий момент времени. В частном случае du/dH —► 0 этот результат сводится к простейшему варианту уравнений кинетики коагуляции, а при 5 —* 0 (отсутствию коагуляции) — к уравнению переноса частиц, являющемуся решением его дифференциальной формы:
, du , dn = n-dt
dH
( du «2=^ expl— t
(3.13.46)
Выражение (3.13.46) можно также рассматривать как уравнение деформации (продольного сжатия столба взвеси), результатом чего и является повышение концентрации в различных слоях столба. В связи с этим следует указать на существование альтернативных способов описания больших деформаций. Принятый здесь способ соответствует деформации по Генки [15], отвечающей требованию аддитивности последовательных ступенчатых деформаций, которое является совершен
Физика, химия и технология дисперсных систем
707
но обязательным при описании процесса уплотнения структурированной суспензии как последовательности локальных сжатий каждого слоя суспензии.
После перехода суспензии в структурированное состояние прекращаются коагуляционные процессы, скорость которых отражается в уравнении (3.13.44) константой s. Указанный переход контролируется соотношением величин т и тс для каждого слоя взвеси при условии, что прочность флокул достаточна для того, чтобы они не разрушались под действием собственного веса. В обратном случае возможное наибольшее число частиц в одной флокуле будет ограничено ее гидродинамически равновесным размером и соответствующим этому размеру числом частиц во флокуле mg. Таким образом, сила сцепления частиц влияет на режим оседание не только в структурированном (через величину т), но и в доструктурном состоянии.
Процесс сжатия описывается системой уравнений (3.13.40) и (3.13.46). Ее решение возможно только численными методами, в том числе в рамках алгоритма, который описан в предыдущем подразделе применительно к решению системы уравнений (3.13.33), (3.13.36) и дополнен расчетом сжатия структурированного столба взвеси. Путем варьирования размера, концентрации и силы сцепления частиц, фрактальной размерности флокул и константы скорости коагуляции (замедления коагуляции) с помощью указанного алгоритма решения системы уравнений (3.13.33), (3.13.40), (3.13.46) можно воспроизвести любой известный из опыта тип эволюции взвеси.
Кинетика, динамика и равновесное состояние взвесей имеют отношение не только к проблемам промышленной переработки дисперсных систем и очистки стоков, но и к глобальным процессам формирования осадочных горных пород, к структуре и концентрационным профилям уже имеющихся отложений на дне морей и океанов, что непосредственно связано с акустическими свойствами донных отложений, т. е. с вопросами навигации, разведки ископаемых и т. д.
3.14. Реология тиксотропных систем
3.14.1. Прочность структурной сетки и флокул
Флокулы и образованная из них структурная сетка имеют ограниченную прочность, которая далее характеризуется величиной xs напряжения сдвига, при котором происходит разрушение структурной сетки по некоторой плоскости сдвига. В литературе эта характеристика часто называется предельным напряжением сдвига. Она определяется прочностью Fc единичных контактов между соседними частицами или флокулами и числом таких контактов ns на единицу площади сечения флокулы или сетки в целом. Контакт двух соседних флокул осуществляется через контакт двух частиц, принадлежащих разным флокулам, и поэтому прочность контакта флокул равна прочности контакта частиц. Последняя равна глубине минимума силовой функции взаимодействия частиц (см. подраздел 3.7.5).
В соответствии с фрактальной моделью [10] коагуляционная структура представляет собой систему плотно упакованных флокул (фрактальных доменов, рис. 3.99) и в этом смысле подобна периодической структуре (ПКС). Каждая флокула находится в контакте с некоторым числом z ближайших соседей. Разъединение такой сетки на две части по произвольной плоскости разрыва разрушает в среднем половину контактов, имеющихся у каждой из ns флокул, попадающих в эту плоскость, так что:
t5 = zhsFc/2. (3.14.1)
Число флокул и5, рассекаемых воображаемой плоскостью с площадью равной единице, равно очевидно 1 / /с2, где Zc — размер фрактального домена, определяемый критическим в смысле перехода в структурированное состояние числом частиц тс во флокуле, поэтому:
ts = zFc/2Zc2. (3.14.2)
Так как размер флокул Z и число частиц в ней т связаны основным соотношением фрактальной геометрии: Z = а тс = (1 /ср) ф/(3 ” ф), то:
=г2/,р2<3-’>,
откуда:
т, = (;Лс/2Г)<г” (3.14.3)
где величина
Tm = zF.!2p, (3.14.4)
по аналогии с формулой (3.14.2), представляет собой прочность системы плотно упакованных частиц с той же силой сцепления и тем же координационным числом z. Таким образом, прочность структурной сетки на сдвиг:
Рис. 3.99. Представление коагуляционной структуры как совокупности плотно упакованных флокул (фрактальных доменов): /с — размер домена
(3.14.5)
Пока оставим в стороне важный вопрос о том, является данная характеристика статической или динамической прочностью структурированной системы. Разли
708
Новый справочник химика и технолога
чие этих понятий в том, что при наличии статической прочности материал сопротивляется необратимому деформированию и при бесконечно медленном (равновесном) увеличении его деформации. Материал, имеющий только динамическую прочность, в некотором диапазоне малых скоростей деформации (диапазоне ползучести) оказывает только вязкое сопротивление, которое растет с увеличением скорости деформирования и стремится к нулю при ее уменьшении. За пределами этого диапазона зависимость скорости деформации от напряжения выглядит так, как будто течение начинается при напряжении, превышающем некоторое предельное значение, которое и получило название предельного напряжения сдвига. Таким образом, сама терминология вынуждает ставить вопрос о статическом или динамическом характере сопротивления деформированию при предельно малых скоростях деформирования. Однако его постановка преждевременна, поскольку ответ может быть найден только после выяснения механизма возникновения кажущейся (динамической) прочности. Это будет сделано позднее на примере цепочечной модели.
Полученная формула для прочности сплошной структурной сетки взаимосвязанных частиц пригодна и для вычисления прочности отдельных флокул, если в формулу (3.14.5) вместо концентрации дисперсной фазы всей дисперсной системы подставить среднюю концентрацию qy дисперсной фазы внутри флокулы. Последняя равна /л(ф’3)/ф, поэтому указанная подстановка дает формулу
т/=т.т-2'*. (3.14.6)
Замена т = (l/r)v* снова возвращает нас к формуле (3.14.2). Однако в ней вместо критического размера флокулы может фигурировать ее произвольный размер:
Tf=zFc/2l2. (3.14.7)
При 7 = 7С, т. е. когда флокулы занимают все пространство, прочности флокул и структурной сетки совпадают. Это означает, что разрушение сетки может с равной вероятностью происходить как по контактам между флокулами, так и по самим флокулам. В таком случае можно было бы думать, что доменная структура коагуляционной сетки не имеет никакого значения. На самом деле это не так. Размер доменов /с, т. е. флокул, из которых сформировалась сплошная структурная сетка, играет важную роль. В частности, он входит в формулы, описывающие состояние и свойства структуры в некоторых нетипичных (см. подраздел 3.14.4) условиях.
Еще одно полезное выражение для прочности флокул получается при замене чу= <р/срт в формуле (3.14.5):
г/=гт(<р/<р„)мз-« (3.14.8)
Связь прочности флокул с их размером и другими параметрами дисперсных систем служит основой для построения уравнения их структурного состояния.
3.14.2. Уравнение структурного состояния и тиксотропия
Формула (3.14.7) показывает, что чем крупнее флокула, тем меньше ее прочность. Флокулы практически всегда находятся в напряженном состоянии. Напряжения возникают под действием силы тяжести, которая при оседании флокул трансформируется в равную по величине силу трения среды. Сила трения действует на флокулы и при течении дисперсной системы. В том и другом случае отношение действующей на флокулы силы к площади ее сечения равно величине создаваемых во флокуле напряжений. Очевидно, что если эти напряжения превысят прочность флокулы, то она будет разрушаться. Диспергирование флокул в потоке и при оседании продолжается до тех пор, пока ее прочность не уравняется с действующим при данном режиме движения напряжением. Если же окажется, что напряжение меньше прочности флокул, то размер последних будет увеличиваться за счет постоянно идущего процесса коагуляции. В любом случае флокулы достигают равновесного размера, при котором напряжение во флокулах равно их прочности. Эта концепция является основой количественного описания всех структурнореологических свойств дисперсных систем и процессов, связанных с их структурными превращениями. Детали определяются тем, каким образом возникающие во флокулах напряжения будут выражаться через действующие на дисперсную систему силы. Здесь возможны варианты, и их соответствие действительности проверяется опытом. Следует лишь заметить, что под «опытом» не обязательно понимать результаты конкретного эксперимента с какой-либо суспензией и прибором. Существует обобщенный опыт в виде известных законов и правил, в том числе эмпирических. Разумеется, что исходные предпосылки и результаты теории не должны им противоречить.
Напряжение xg = mmig/(nP/4), создаваемое во флокуле силой ее собственного веса m}mg, равно, очевидно, отношению этой силы к площади л72 / 4 экваториального сечения флокулы, где т{ — масса одной частицы с поправкой на архимедову силу. Равновесный размер флокулы mg, оседающей в вязкой среде, получается из условия равенства гравитационного напряжения 8wgWig/n во флокуле равновесного размера и ее прочности zFc из формулы (3.14.7). Не претендуя на абсолютную точность результата, можно принять, что:
mg = m\g/Fc. (3.14.9)
Эта формула является уравнением структурного состояния коагулирующей суспензии в поле силы тяжести, в поле центробежных сил и при воздействии низкочастотных вибраций (встряхивания суспензии). Ускорение силы тяжести g следует при этом заменить ускорением центробежных сил или величиной A If2 при встряхивании. Здесь А — амплитуда и/ — частота вибраций. Формула (3.14.9) использовалась при рассмот
Физика, химия и технология дисперсных систем
709
рении факторов, лимитирующих процесс коагуляции (см. подраздел 3.13.6).
В потоке напряжения во флокулах совпадают с величиной напряжений, действующих на дисперсную систему в целом:
Ту = т. (3.14.10)
Замена с помощью этого равенства величины Ху в формуле (3.14.8) сразу ведет к уравнению х - тт(ф/фт)2(3-Ч которое является уравнением структурного состояния флокулированной взвеси в потоке. Оно дает гидродинамически (тиксотропно) равновесную концентрацию (объемную долю) флокул при течении коагулированной дисперсной системы, которая является в данном случае параметром, характеризующим структурное состояние дисперсной системы. Учитывая, что xs = тт ф2/(3 ~ф), находим:
<P„ = (T.,/T)(3-W2. (3.14.11)
Если возникнет потребность вместо объемной доли флокул фт использовать иные параметры состояния, например размер / или число флокул п/т в единице объема, то это легко сделать с помощью основного соотношения фрактальной геометрии флокул т =
Следует иметь в виду, что уравнение (3.14.11) является только частью полной системы уравнений структурного состояния дисперсной системы в потоке. Недостающие части этой системы охватывают случаи малых и больших напряжений. Малыми считаются напряжения, величина которых не превышает прочности структуры, т. е. охватывает диапазон от т = 0 до т = х5. Большие напряжения — это напряжения величиной х > тт. При низких напряжениях структурная сетка остается неразрушенной, и это состояние соответствует величине фт= 1. Оно получается из уравнения (3.14.11) подстановкой минимального значения, при котором величина фт имеет смысл, а именно, при х = х5. При меньших напряжениях уравнение дает физически неприемлемую величину фот > 1. Аналогичным образом, подставляя максимально возможную величину х = тт в уравнение состояния (3.14.11), получим фт = ф, поскольку, согласно формуле (3.14.5), (тх/тт)(3 ф)/2 = ф. Это означает, что при таком напряжении структурная сетка будет полностью разрушена, и взвесь будет состоять из индивидуальных частиц, доля которых во взвеси как раз и равна величине ф. Очевидно, что увеличение напряжения сдвига до величин больших, чем тт, ничего не изменит в состоянии системы и поэтому значение параметра состояния фт = ф останется неизменным. Таким образом, система уравнений структурного состояния состоит из трех уравнений:
фт = 1 при т < т,,
фт = (т5/т)(3’ф)/2прит5<т< тт, (3.14.12)
фот = Ф при X > Хт.
Эти уравнения являются одновременно количественным описанием одного из важнейших свойств структурированных систем — их тиксотропности. Напомним, что тиксотропность — это способность системы к обратимым изотермическим разрушениям и восстановлениям структуры. Обратимость разрушения обусловлена коагуляционным укрупнением фрагментов структуры (флокул) при уменьшении действующего на них напряжения.
3.14.3. Реологическое уравнение тиксотропных систем
При анализе условий оседания флокул было установлено, что в гидродинамическом отношении их можно считать сплошными телами, непроницаемыми для потоков жидкости. Отсюда следует, что реологические свойства взвесей флокул можно описывать с помощью тех же формул, которые применяются для взвесей монолитных частиц, т. е. формулы Эйнштейна и любых ее модификаций, относящихся к концентрированным дисперсным системам. Для этого необходимо только вместо объемной доли ф твердой дисперсной фазы подставлять в них объемную долю фот флокул во взвеси. Пригодность формул для концентрированных систем имеет в данном случае решающее значение, поскольку объемная доля флокул в структурированной системе достигает единицы. Таким образом, вязкость флокулированной или структурированной дисперсной системы можно вычислять по формуле
П = По/(1 -<Ы“. (3.14.13)
Здесь т|0 — вязкость среды и а = 2,5 — коэффициент формулы Эйнштейна. Такое численное значение коэффициента обусловлено тем, что флокулы имеют возможность свободно вращаться в сдвиговом потоке. Принципиальное отличие этой формулы от аналогичной формулы для неструктурированной суспензии в том, что здесь фт есть функция напряжения сдвига, задаваемая системой уравнений (3.14.12). Собственно закон течения (реологическое уравнение) (3.14.14) в данном случае выглядит как закон внутреннего трения Ньютона, в котором, однако, т| есть функция напряжения (уравнение (3.14.13)):
х = т|у'. (3.14.14)
Он вместе с уравнениями (3.14.12) и (3.14.13) образует полную систему уравнений реологии тиксотропных систем [10]. В практических расчетах в качестве независимой переменной удобно принимать напряжение сдвига х, а в качестве функции — скорость сдвига у'. Фрактальная размерность флокул ф, концентрация дисперсной фазы ф, прочность структуры xs и вязкость среды т|о являются параметрами задачи. Непосредственными расчетами легко убедиться, что эта система уравнений дает типичную (рис. 3.100) для ряда тиксотропных систем зависимость скорости сдвига от на
710
Новый справочник химика и технолога
пряжения с тремя четко различимыми режимами течения: отсутствием течения при малых напряжениях, нелинейной пластической деформацией при основном (тиксотропно равновесном) режиме течения и течением с полностью разрушенной структурой при высоких напряжениях сдвига. Если частицы имеют слабую адгезию к стенкам канала, в котором происходит течение, то суспензия способна течь и при малых напряжениях с очень высокой постоянной вязкостью. Эта особенность поведения дисперсной системы называется ползучестью. В теоретической реологии она отождествляется со способностью системы течь без разрушения структуры. Механизм такого течения и количественная характеристика реологических свойств в этом состоянии требует отдельного изучения.
Рис. 3.100. Реологическая кривая течения суспензии с фрактальной структурой коагуляционной сетки: т, — сдвиговая прочность; tm — напряжение полного разрушения структуры
3.14.4. Течение в тонких каналах
С точки зрения традиционных взглядов на роль различных факторов, определяющих структуру и реологические свойства дисперсных систем, приведенные уравнения следовало бы считать полными и исчерпывающими. В действительности это не так. Дело в том, что общепринятый подход к описанию состояния и свойств коллоидов полностью игнорирует роль геометрических характеристик сосудов и каналов, в которых находится или двигается коллоидный раствор. Между тем роль геометрии каналов и сосудов столь же важна, как и роль рецептуры дисперсной системы и других факторов. Примеры влияния высоты сосуда на конечное состояние дисперсной системы, ее коагуляцию и оседание уже были приведены ранее. Здесь же рассмотрено влияние поперечного размера канала, в котором движется или покоится коагулирующая взвесь. При этом достаточно рассмотреть случай плоского канала (щели), размер которого (толщина А) ограничен лишь в одном направлении, перпендикулярном направлению течения. По длине и ширине размер канала считается неограниченным.
Прежде всего, возникает вопрос о критерии, которым следует руководствоваться при классификации каналов, как ограниченных, так и неограниченных в каком-либо измерении. Этот критерий — соотношение соответствующих размеров канала и размера доменов
сплошной структурной сетки. В частности, канал считается узким, если его толщина меньше размера домена, т. е. при условии
Л</с. (3.14.15)
Основное следствие ограниченности размера канала хотя бы в одном измерении заключается в том, что в таких каналах становится невозможным образование сплошной структурной сетки. Это вытекает из того, что размер флокулы не может быть больше, чем меньший из размеров канала (в данном случае его толщина А) и, следовательно, он не сможет достичь критического размера, при котором только и происходит заполнение флокулами всего пространства. Иначе говоря, в узких каналах структура будет фрагментарной. Она представляет собой совокупность не связанных между собой флокул, которые защемлены между стенками узкого канала (рис. 3.101).
Ограничение размера флокул влечет за собой ограничение числа частиц в них величиной:
юа = (А/г)ф (3.14.16)
и соответствующей ему доли объема фЛ, занятого флокулами внутри щели:
<р» = Ф(/1/г)3-*, (3.14.17)
а также ограничение объемной доли твердой фазы ф^ внутри защемленных флокул:
ФЛ = (г/А)’'*. (3.14.18)
Прочность таких флокул = ттф;;,2,п"зависит, как это следует из формул (3.14.18) и (3.14.4), только от толщины щели и силы сцепления двух частиц:
x^zFJlh2. (3.14.19)
В сущности, это та же формула (3.14.7).
Прочность всей фрагментарной структуры tsA получается при умножении прочности защемленных флокул Т/й на площадь их суммарного сечения в плоскости, параллельной широким стенкам щели. Эта площадь совпадает с долей объема фА, занятого флокулами в щели, поэтому tsA = Т/йфл, что после ряда подстановок дает формулу
^ = т™ф(г/А)н. (3.14.20)
Рис. 3.101. Флокула в узком канале
Физика, химия и технология дисперсных систем
711
Подстановка сюда h = /с, которая означает переход от фрагментарной к сплошной структуре, и последующая замена критического размера флокул /с соответствующей функцией концентрации дисперсной фазы преобразует эту формулу в формулу (3.14.5) для прочности сплошной структуры.
Анализируя полученный результат, следует отметить, что прочность фрагментарной структуры растет с уменьшением толщины зазора h между стенками щели и увеличивается пропорционально концентрации дисперсной фазы в первой степени. В неограниченном же пространстве прочность структуры зависит от концентрации в дробной степени. Иначе говоря, при h<lc меняется закон, описывающий концентрационную зависимость прочности. Такого рода изменения в молекулярной физике всегда связывают с фазовыми переходами, например с изменением агрегатного состояния вещества. Необычность структурно-фазового перехода в суспензии в том, что он вызывается изменением геометрических характеристик сосуда, чего никогда не отмечалось в классических фазовых превращениях. Еще ряд закономерностей в поведении коагуляционных структур в щелях и вне их указывает на правомерность отнесения происходящих в щелях изменений к фазовым переходам. Во-первых, как и классические двухфазные молекулярные системы, фрагментарная структура может равновесным образом сосуществовать с другой фазой — обычной коагуляционной структурой или просто с флокулированной суспензией вне щели — и менять параметры своего состояния при изменении условий (толщины щели). Для этого достаточно поместить подходящий щелевидный капилляр в суспензию. Второе важное обстоятельство заключается в том, что структурные характеристики защемленных флокул (размер, плотность, прочность) не зависят от концентрации дисперсной фазы в суспензии. С увеличением концентрации будет пропорционально расти число флокул в щели. Это можно трактовать как возможность изоструктурного изменения концентрации вещества. Аналогом такого поведения молекулярных систем является, например, независимость давления пара от валовой концентрации вещества при наличии в сосуде второй (жидкой) фазы того же вещества. Таким образом, концентрационная изоструктурность суспензии в щелевидных частях сосуда с суспензией говорит о наличии двух разных фаз, одна из которых порождена ограниченностью размера некоторых частей сосуда.
В реальных условиях описанная выше ситуация встречается очень часто: при фильтровании взвесей, в зазорах подшипников, в процессах шлифования и полирования поверхностей в жидкой среде и т. д. Это оправдывает то внимание, которое уделяется состоянию и свойствам дисперсных систем в каналах ограниченных размеров. В последние годы разного рода объекты, имеющие малый размер хотя бы в одном измерении, привлекают особое внимание. Они получили общее название неполномерных объектов. А в общем, если
говорить об обычных материалах, то это известные всем пленки (имеют одно малое измерение), нити (имеется ограничение в двух измерениях) и частицы — основной объект изучения коллоидной химии, у которого малы все три измерения. Некоторые из неполномерных объектов действительно меняют свое физическое состояние с уменьшением размера. Так, очень мелкие частицы феррита или железа при нормальной температуре теряют свои ферромагнитные свойства.
Обращаясь к специфике структурного состояния коагулированной суспензии в узкой щели, важно выяснить, как соотносятся между собой прочности фрагментарной и сплошной структуры одной и той же суспензии. Для этого суспению удобно характеризовать отношением которое легко найти, поделив правые части формул (3.14.20) и (3.14.5):
Wt, = [(г/А)/ф|,(3-«]ф-(3.14.21)
Отсюда следует, что прочность фрагментарной структуры становится больше прочности сплошной структурной сетки при условии, которое с легкостью выполняется при надлежащем уменьшении концентрации дисперсной фазы ср или при увеличении фрактальной размерности флокул ф, т. е. при их уплотнении:
ф < (г/Л)(3-ф). (3.14.22)
В достаточно концентрированных суспензиях с рыхлой структурой флокул прочность фрагментарной структуры может быть меньше, чем сплошной.
Соотношение прочностей фрагментарной и сплошной структур оказывает решающее влияние на возможность осуществления равновесных состояний фрагментарной структуры при ее сдвиговой деформации, т. е. при течении в узких каналах. Она имеет важное техническое и технологическое значение, поскольку определяет возможность устойчивого течения в щелях, например устойчивого скольжения поверхностей подшипника в смазке и т. п. В связи с этим необходимо найти минимальную величину сдвигового напряжения, при котором гидродинамически равновесный размер флокул становится равным (или чуть меньшим), чем ширина щели. Для этого левая часть уравнения состояния (3.14.11) должна содержать величину фЛ = ф(Л/г)3”ф, а правая должна быть равна ей или чуть меньше, т. е. должно быть выполнено условие ф(Л/г)3-ф > (т5/ т)(3-фу2, которое после ряда подстановок из числа приведенных выше формул сводится к условию
(3.14.23)
При меньшем напряжении сдвига, в частности при т = т5л, которое является минимально необходимым для начала сдвигового течения суспензии в тонком зазоре, равновесный размер флокул оказывается большим, чем ширина зазора. Следовательно, в интервале напряжений от т = Tsh до т = т/й гидродинамически равновесное
712
Новый справочник химика и технолога
состояние флокул и соответствующий ему режим течения суспензии неосуществимы.
В силу отмеченных особенностей состояния суспензии зависимость скорости сдвига от напряжения в узком канале будет выглядеть качественно иначе, чем в широком канале. При увеличении напряжения течение будет отсутствовать вплоть до достижения напряжения, равного прочности т5/, фрагментарной структуры. В диапазоне напряжений от прочности структуры до прочности флокул Tft, скорость течения медленно нарастает при неизменности размера и концентрации флокул. При достижении напряжения, превышающего прочность флокул, скорость увеличится скачком за счет перехода зависимости на ветвь тиксотропно равновесного течения при данном напряжении.
Дальнейшее увеличение напряжения вызывает обычное для тиксотропной системы изменение скорости деформации. Последующее уменьшение напряжения сохраняет эту обычную зависимость вплоть до напряжения, равного прочности флокул или несколько меньшего из-за задержки во времени восстановления равновесного размера флокул, после чего скорость должна скачкообразно уменьшиться. Вероятно появление гистерезиса зависимости скорости сдвига от напряжения в области перехода от одного режима течения к другому (рис. 3.102).
Течение в режиме с заданной скоростью деформации по указанной причине может быть неустойчивым и сопровождаться колебаниями показаний прибора и шумом.
Рис. 3.102. Течение с отрицательным значением вязкости на участке 2 реологической кривой
3.14.5. Цепочечная структура
Как уже отмечалось, случай с ф = 1 соответствует наиболее рыхлой структуре. Кроме того, такое значение фрактальной размерности влечет за собой появление и качественно новых свойств дисперсной системы. В частности, в соответствии с формулой (3.14.20) прочность структуры в узкой щели перестает зависеть от ее ширины, а прочность структуры в неограниченном пространстве, согласно формуле (3.14.5), становится линейной функцией концентрации. Последнее может означать, что параметры структурного состояния остаются неизменными с увеличением концентрации. Общее уравнение тиксотропно равновесного состояния (3.14.11) и его щелевой вариант (3.14.17) на первый
взгляд не подтверждают этого, так как параметры срт или <рА, принятые за основную характеристику структурного состояния, в обоих случаях линейно возрастают с увеличением концентрации дисперсной фазы. Однако линейный рост доли объема, занятого в дисперсной системе ее твердыми компонентами, присущ и устойчивым неструктурированным суспензиям. Поэтому указанный характер зависимости не является критерием отсутствия концентрационной изоструктурности. В рассматриваемых особых случаях параметры структурного состояния просто становятся величиной, равной или подобной концентрации дисперсной фазы.
Более важными являются те особенности систем с минимально возможным значением фрактальности, которые могут быть основанием для ревизии самой целесообразности применения фрактального метода в описании состояния дисперсной системы. Следует учесть, что объем, занимаемый фрактальной флокулой, приравнивается к объему описанной вокруг нее сферы. Применительно к простым линейным цепочкам такой подход может быть оправдан, если их ориентация случайна и непостоянна. Тогда действительно они в своем движении, например при вращательной диффузии, очерчивают вокруг себя сферическую полость, которую они якобы всю и всегда занимают. В то же время реально существуют дисперсные системы, в которых ориентация линейных цепей параллельна и неизменна. Это, в частности, линейная цепочечная структура, возникающая при действии магнитного или электрического поля на соответствующие дисперсные системы. В концентрированном коллоидном растворе ферромагнетика расстояния между соседними параллельными цепями могут быть намного меньше их длины. Поэтому нельзя считать, что каждая цепь занимает такой же объем, как сфера с диаметром, равным длине цепи. Главное же обстоятельство состоит в том, что геометрия линейных цепочек настолько проста и предсказуема, что отпадает всякая необходимость рассматривать их как фрактальные объекты. В историческом плане это также оправдано, поскольку основополагающие идеи теоретической реологии, связанные с введением в практику уравнений структурного состояния в потоке, были выдвинуты и развиты [6] на примере цепочечной модели коагуляционных структур задолго до того, как были осознаны и стали применяться возможности фрактальной геометрии в описании коллоидов. В силу геометрической наглядности цепочечная модель позволяет со всей необходимой полнотой понять механизм важнейших реологических эффектов структурирования, поэтому ниже она будет рассмотрена отдельно и детально. Примечательно, что, оставаясь альтернативой фрактальной модели, цепочечная модель дает практически те же результаты, что и фрактальная. Поэтому она может одновременно считаться и частным случаем фрактальной модели. Примечательно, что, оставаясь альтернативой фрактальной модели, цепочечная модель дает результаты, которые в некоторых аспектах сходны с
Физика, химия и технология дисперсных систем
713
результатами, следующими из фрактальной модели. Это важно, поскольку, уяснив на простой модели физическую природу реологических эффектов структурирования, можно перенести эту ясность и на более формальную и менее наглядную фрактальную модель. В то же время цепочечная модель имеет важное принципиальное отличие от фрактальной. Она не предполагает заранее, что структурированная система обладает предельным напряжением сдвига. В цепочечной модели это свойство возникает как следствие модели.
3.14.6. Уравнение состояния и реологический закон цепочечной структуры
При заданном размере и концентрации частиц состояние цепочечной структуры полностью характеризуется длиной / и прочностью цепей Fc. Прочность цепей равна силе сцепления двух частиц. Расстояние г между соседними по цепи частицами может превышать диаметр частиц на величину зазора 8 между ними, который определяется координатой минимума потенциала парного взаимодействия частиц. Число частиц т в цепи, как и во фрактальной флокуле с единичным значением размерности, определяется соотношением т = Пг. Считается, что цепи ориентированы перпендикулярно направлению течения и удерживаются в этом положении достаточно сильным магнитным (или электрическим) полем.
В отсутствие сдвиговых напряжений длина цепей, как и размер флокул при коагуляции вне поля, ничем не ограничена и растет в процессе коагуляции до тех пор, пока концы цепи не упрутся в стенки сосуда или канала. Последний удобно представить в виде узкого канала (щели) шириной h. Остальные его размеры не ограничены. Цепи ориентированы перпендикулярно стенкам канала. Сдвиговое течение в нем создается тем, что одна из его стенок движется относительно другой со скоростью и, задавая тем самым величину градиента скорости течения у = и/h. В потоке цепи разрушаются до гидродинамически равновесной длины, которая меньше ширины канала.
Удобно считать, что цепь состоит из нечетного числа частиц т, так что в середине цепи находится одна из ее частиц (центральная). Это не ограничит общности основных результатов. Частицы нумеруются от середины цепи. В потоке цепь движется как единое целое со скоростью, равной скорости движения дисперсионной среды в той плоскости, в которой расположена центральная частица цепи. Тогда при наличии градиента скорости течения у' все остальные частицы обтекаются средой со скоростью Uj = y'rj, пропорциональной расстоянию rj j-й частицы от середины цепи (рис. 3.103).
На каждую из частиц цепи, кроме центральной, действует сила трения fj = by'rj, пропорциональная скорости ее обтекания. Полная сила FT, действующая на одну (л»-1)/2
половину цепи, равна сумме f указанных сил по
всем (т- 1)/2 частицам половины цепи. Так как под знаком суммы постоянны все величины кроме номера частицы, то суммирование производится только по но-(m-l)/2 (т-1)/2
меру, так что Fm = brfr j ’ причем ;=(m2-iy8.
7=1 7=)
Таким образом, можно записать формулу
FT = Zxy>(w2- 1)/8.
(3.14.24)
Здесь b = 6лт|оа — коэффициент сопротивления Стокса частицы радиусом а, т|0 — вязкость среды.
Рис. 3.103. Цепочка частиц в поле (а), в поле и сдвиговом потоке (б):
Е — напряженность поля; / — длина цепи; г — расстояние между соседними частицами в цепи; и — направление течения;
и, — скорость обтекания потоком среды j-й частицы, находящейся на расстоянии rj от середины цепи
На вторую половину цепи действует такая же сила, но в противоположном направлении, что создает момент сил, стремящийся опрокинуть цепь. Опрокидывания не происходит благодаря ориентирующему действию поля, но в цепи возникают сдвиговые (срезающие) усилия, которые стремятся ее разрушить. Сила сдвига максимальна в середине цепи и равна найденной суммарной силе трения. Если она превысит прочность сцепления частиц Fc, то произойдет разрушение цепи на два почти одинаковых фрагмента («почти» потому, что в цепи, по условию, содержится нечетное число частиц). Если Fy меньше прочности цепи, то последняя будет удлиняться за счет непрерывно идущего процесса коагуляции и вызванного ею присоединения к цепи отдельных частиц или коротких фрагментов. Наличие тех и других подразумевается, а фигурирующая в расчетах величина т является средней для цепей всех размеров.
Описанные выше явления разрушения и удлинения цепей составляют суть тиксотропии цепочечной структуры. В итоге установится тиксотропно (гидродинамически) равновесная длина цепи или равновесное число частиц в ней, которое определяется условием равенства сдвигового усилия в середине цепи и ее прочности на сдвиг (FT = Fc). Последнюю можно отождествить с максимумом силы сцепления Fc, которая, вообще говоря, характеризует прочность цепи на растяжение. Прочность на сдвиг и на растяжение — это разные величи
714
Новый справочник химика и технолога
ны, но применительно к поведению цепей в потоке, некоторая количественная разница этих величин не имеет существенного значения. Указанное равенство в развернутом виде описывается формулой, которая является уравнением структурного состояния простой цепочечной структуры или уравнением тиксотропного равновесия коагулирующей дисперсной системы:
Zry'r(w2- 1)/8 = FC. (3.14.25)
Для полноты картины оно, как и при рассмотрении флокулярной структуры коагулята, должно быть дополнено указанием диапазона его применимости и соотношениями, определяющими размер цепей за его пределами.
Как следует из уравнения (3.14.25), начиная с некоторой достаточно малой скорости сдвига у,, равновесная длина цепи / оказывается больше ширины щели h. Фактически она будет оставаться равной ширине щели при малых скоростях сдвига, так что / = h при у' < у'. Такое значения параметра состояния Z означает, что структура остается неразрушенной, и течение в указанном интервале скоростей сдвига соответствует течению без нарушения сплошности структуры. Возможность течения без разрушения структуры считается характерной особенностью тиксотропных систем. Учет этого условия в уравнении (3.14.25) приводит к формуле
у'( = 8rFc/Ау'А2. (3.14.26)
Уравнение (3.14.26) получается из уравнения (3.14.25), если в последнем также учесть, что т = Ыг и обычно hl г >> 1, т. е. и w2 » 1.
Произведение rFc по порядку величины равно энергии связи частиц, т. е. глубине дальнего минимума Uc потенциала парного взаимодействия частиц. Поэтому формулу (3.14.26) можно заменить формулой
у; = %UJtyW. (3.14.27)
По поводу правомочности такой замены необходимо отметить следующее. Дипольное взаимодействие частиц определяется расстоянием между их центрами, так что F = dU / dr или приближенно U = rF, т. е. такая замена вполне корректна. В случае сил поверхностной природы потенциал и сила взаимодействия определяются величиной зазора между частицами 5, так что F= dlJIdb. Тогда по порядку величины U = 8F, а не rF. Последнее произведение сильно, в г/5 раз, превышает значение Uc в формуле (3.14.25). Однако при сдвиговом смещении одной частицы относительно другой для увеличения расстояния между поверхностями на величину порядка 5 потребуется сдвинуть их в тангенциальном направлении на расстояние порядка размера частиц. Это означает, что сопротивление силе сдвига в г/8 раз слабее, чем сопротивление растягивающему усилию. Можно полагать, что ошибки, внесенные двумя допущениями: использованием в формуле (3.14.26)
прочности на разрыв Fc вместо прочности на сдвиг и последующая замена величины rFc энергией связи частиц Uc компенсируются, так что в итоге формула (3.14.27) оказывается достаточно корректной.
В другом предельном случае, а именно при некоторой достаточно большой скорости сдвига у2, цепь имеет минимальную длину (т = 2). Превышение указанной скорости обеспечит переход системы в состояние полного разрушения структуры, которое означает, что равновесное число частиц т в структурных фрагментах равно единице. Это состояние будет сохраняться при всех скоростях сдвига, превышающих величину у2, являющуюся верхним пределом тиксотропного равновесия в системе с цепочечной структурой. Его значение получается так же, как и значение у' (уравнение (3.14.27)) для нижнего предела тиксотропного равновесия при подстановке в уравнение состояния (3.14.25) величины т = 2:
у' = WJ3br*. (3.14.28)
При сравнении формул (3.14.27) и (3.14.28) видно, что диапазон скоростей деформации, в пределах которого состояние системы остается тиксотропно равновесным, может охватывать несколько порядков. Отношение У2/У1 примерно равно (А/г)2, так что в достаточно типичных случаях (А=10~3м, г=106м) оно составляет около 6 десятичных порядков. Эта величина настолько велика, что ни один из существующих приборов не сможет обеспечить измерение скоростей и напряжений деформации во всем диапазоне значений этих параметров в режиме тиксотропно-равновесного деформирования дисперсной системы.
Таким образом, структурное состояние дисперсной системы с цепочечной структурой с длиной цепей / описывается уравнениями:
/=А, у' < 8С/с/Ау'А2;
/ = (8t/c/Ay')12, 8t/c/Ау'А2 < у'< 8t/c/3Ar2; (3.14.29) l = r, />8Z/c/3AA
Размер цепей и фрактальных флокул может быть также ограничен их тепловым распадом. Вычисление термически равновесных размеров цепей и флокул относится к компетенции статистической физики. Этой проблеме посвящено значительное число работ, однако приемлемый результат пока не получен. Впрочем, при энергии связи частиц, в несколько раз превышающей кинетическую энергию теплового движения, термически равновесный размер цепей и флокул можно считать бесконечно большим и фактически он лимитируется рассмотренными ранее факторами.
При обтекании частиц вязкой средой на каждой из них ежесекундно рассеивается энергия fjUj, равная произведению силы трения на величину перемещения среды относительно частицы, которое равно локальной
Физика, химия и технология дисперсных систем
715
относительной скорости и, течения в окрестности частицы. Рассеяние энергии на всей цепочке вычисляется суммированием по всем частицам цепи так же, как при вычислении суммарной силы трения. Эта операция сводится к суммированию квадратов номеров частиц. Воспользовавшись для этого арифметической формулой (3.14.30) при k = (m-l)/2 и удвоив результат для вычисления потерь энергии qm на обеих половинах одной цепи, можо получить уравнение (3.14.31):
У > = (3.14.30)
1-2-3
26(у>)2 т(т2 -1)
%= \ J-------L- (3-14.31)
3 • о
При умножении этой величины на число цепей в единице объема получается рассеяние энергии (диссипативная функция qc) всей цепочечной структуры:
q.=~b(y'r)2n(m2-l)/i. (3.14.32)
Разделение числового коэффициента (3 • 8) в знаменателе формулы (3.14.31) на два сомножителя 3 и 8 показывает идентичность форм (т2-1)/8, входящих в диссипативную функцию (3.14.25) и в уравнение (3.14.31).
При вычислении диссипативной функции не учитывался вихревой характер обтекания частиц средой, который, как было показано ранее, влечет за собой потери энергии, обусловленные вращением частиц. В соответствии с формулой Эйнштейна для вязкости взвеси эти потери равны аг|оф(у')2- Кроме того, имеются потери в самой среде, примерно равные г|0(/)2- Приближение связано с тем, что среда занимает только часть всего объема дисперсной системы, и скорость деформации среды отличается от скорости деформации дисперсной системы у'. Полные потери энергии q в единице объема за единицу времени составят, следовательно:
q = |(У>)2« (w2- 1)/8 + (1 + аф)цо(у')2. (3.14.33)
Величина вязких сил сопротивления деформированию, в соответствии с энергетической формой записи q = Л С?')2 закона внутреннего трения, равна отношению потерь энергии к скорости деформации, так что:
т = ~b^nq'^m2 - 1)/8 + (1 + аф)г|оу'. (3.14.34)
В таком виде полученное уравнение реологии систем с цепочечной структурой описывает связь напряжения и скорости деформации в любом режиме течения. Первое слагаемое уравнения представляет структурную часть сопротивления, а второе — его эйнштейнову часть, обусловленную лишь присутствием определен
ного количества частиц во взвеси. Исключая структурные параметры из первого слагаемого с помощью уравнения структурного состояния (3.14.25), можно получить частные формы этого уравнения для разных режимов деформирования:
т = [(bnrflXl + г|/г]у' или (тг)2 = А2 при у' < у’;
т = тс + г|£у' или (т2 - 1 )/8 =FJbry’ при у2 > у' > у,;
т = г|£у' или т = 1 при у' > у2. (3.14.35)
Здесь г|а=(1 +афг|о) — эйнштейнова часть вязкости, тс — константа, величина которой определяется прочностью сцепления частиц Fz в цепи и связана с ней формулой
tc = 2wFc/3. (3.14.36)
С точностью до числового коэффициента порядка единицы эта константа равна удельной (на единицу объема) энергии взаимодействия частиц дисперсной системы nUz, так что можно записать формулу
тс = и£7с. (3.14.36а)
Полная реологическая кривая течения простой цепочечной структуры, построенная по уравнениям (3.14.35), представлена на рис. 3.104.
Рис. 3.104. Теоретическая кривая течения (а) и вязкости (б), рассчитанная на основе цепочечной модели тиксотропных систем
Наиболее поучительный результат, вытекающий из цепочечной модели, заключается в том, что в состоянии тиксотропного равновесия структурная часть сопротивления дисперсной системы тс не зависит от скорости деформации. Примечательно, что при этом длина цепочек / может быть намного меньше расстояния h между стенками канала. Тем не менее, они создают сопротивление сдвигу, подобное трению скольжения груза, лежащего на плоскости. Именно такая механическая модель лежит в основе эмпирического уравнения Шведова — Бингама, которое представлено во второй строке системы уравнений (3.14.35). Параметр те уравнений— это предельное напряжение сдвига, причем в общем случае оно является динамическим. Последнее соотношение в каждой строке системы уравнений (3.14.35) — это условие применимости соответствую
716
Новый справочник химика и технолога
щего реологического закона, в том числе закона Шведова — Бингама.
Сопротивление цепочек не зависит от скорости сдвига потому, что чем больше скорость сдвига, тем сильнее разрушены цепи (меньше их длина) и тем меньше их гидродинамическое сопротивление. Обобщая этот результат, можно утверждать, что структурная часть сопротивления дисперсной системы ни в каком случае не может превышать прочности структурных фрагментов или структуры в целом.
Следующий важный результат состоит в том, что зависимость предельного напряжения сдвига цепочечной структуры от концентрации дисперсной фазы оказывается такой же, как и фрактальной структуры с размерностью, равной единице. Более того, совпадают и зависимости гидродинамического объема <рт флокул и цепей от напряжения сдвига, а следовательно, и зависимости скорости сдвига от напряжения, если во фрактальной модели ограничиться концентрациями флокул, допускающими применение формулы Эйнштейна. Таким образом, можно констатировать идентичность законов течения цепочечных структур, полученных из двух разных в математическом отношении моделей строения таких систем. Численные значения основного параметра уравнений — предельного напряжения сдвига — также практически совпадают.
Принципиальное различие цепочечной и фрактальной моделей заключается в том, что цепочечная модель не предполагала наличия предельного напряжения сдвига. Оно возникло как один из результатов теории. В противоположность этому, закон течения фрактальной структуры выводился на основе предположения о наличии у структуры прочности т4 и, соответственно, отсутствия течения при напряжениях меньших, чем прочность структуры.
Это различие нашло свое отражение и в обозначениях однотипных параметров: индекс «$» при величине т5 подчеркивает ее статический характер, индекс «с» при тс напоминает о ее связи с прочностью сцепления частиц Fc. Выявленный выше механизм появления структурного сопротивления цепочечных структур означает, что прочность системы не связанных между собой цепей является кажущейся. Фактически ее предельное напряжение сдвига является динамическим, т. е. проявляет себя только при течении и отсутствует при статической (бесконечно медленной) деформации. Первая строка системы уравнений (3.14.35) как раз и описывает поведение системы при малых скоростях течения, т. е. напряжение растет пропорционально скорости с очень большой величиной коэффициента пропорциональности — вязкости Цщах неразрушенной структуры:
nmax = (W/12 + TU). (3.14.37)
Этот режим течения (ползучесть) реализуется, если концы цепочек не сцепляются со стенками канала (по причине недостаточной длины цепей в покое или нуле
вой величины адгезии частиц к стенкам). Если сила сцепления концевых частиц цепей со стенками не меньше, чем сила сцепления частиц между собой, то может возникнуть статическое сопротивление сдвигу (t.s = тс) и, соответственно, при т < тЛ скорость деформации будет равна нулю. В состоянии тиксотропного равновесия, когда длина цепей меньше размера канала, величина адгезии концевых частиц к стенкам канала не оказывает никакого влияния на законы течения и на значение параметров уравнений реологии и уравнений состояния взвеси. В состоянии покоя длина цепей может быть ограничена их термическим распадом, и тогда во всех формулах вместо h должна фигурировать термически равновесная длина цепей lt. Только в этом случае максимальная вязкость характеризует дисперсную систему и определяется термически равновесным размером цепей (или флокул), который, в свою очередь, зависит от силы сцепления частиц и их концентрации. В иных случаях, т. е. когда Z, > h, максимальная вязкость определяется конструктивным параметром прибора — расстоянием между стенками канала.
Обсуждая роль адгезии частиц к стенкам канала, необходимо принять во внимание различие между сопротивлением сдвиговому и нормальному перемещениям прилипшей частицы относительно стенки. Оно еще сильнее, чем рассмотренное ранее различие этих двух видов сил для двух частиц. Действительно, если стенка абсолютно гладкая, то тангенциальное смещение частиц вообще не будет встречать никакого сопротивления. Иначе говоря, при любых значениях адгезии в идеальном случае (без учета структуры поверхности стенок канала) стенки канала не препятствуют движению цепей. Тогда статическое сопротивление сдвиговой деформации отсутствует.
Эти соображения можно распространить и на медленное течение систем с фрактальной структурой. При деформации со скоростью, превышающей величину у2, структура полностью разрушена, вязкость постоянна и близка по величине к эйнштейновой вязкости которая примерно в (hlrf раз меньше максимальной вязкости неразрушенной структуры.
Таким образом, зависимость скорости сдвига от напряжения графически представляется тремя прямыми (рис. 3.104), описывающими течение в режиме ползучести, в режиме тиксотропно-равновесной деформации (участок пластического течения) и в режиме полного разрушения структуры.
Завершая сопоставление фрактальной и цепочечной моделей, следует отметить, что с точки зрения гидродинамики движения флокул и цепей в этих моделях очень сильно отличаются. Флокула с фрактальностью ф = 1 представляет цепочку частиц как непроницаемую для потоков среды сферу с диаметром, равным длине цепи. В цепочечной же модели среда свободно протекает сквозь цепь частиц, встречая сопротивление только от самих частиц. Действительная картина движения среды возле цепочек будет чем-то средним между при
Физика, химия и технология дисперсных систем
717
нятыми в каждой из моделей вариантами. Несмотря на это, различие конечных результатов той и другой теории сводится к незначительному различию в численных значениях констант реологических уравнений и уравнений структурного состояния. Это означает, что каждая из них корректно и полно учла все факторы, имеющие существенное значение для реологии тиксотропных систем.
3.14.7. Послойное скольжение
Коагуляционная сетка и ее фрагменты в действительности имеют конечную проницаемость для потоков среды, которая далее будет характеризоваться глубиной проникновения 8 потоков в глубь сетки. Это обстоятельство следовало бы, так или иначе, учесть и во фрактальной и в цепочечной моделях тиксотропных систем. Однако более наглядно роль конечной проницаемости проявляется при рассмотрении еще одного механизма течения коагуляционной структуры — ее послойного скольжения. Для этого рассмотрим тонкий, плоский, прочный и пористый слой структурированной суспензии, заключенный между двумя пластинами (стенками прибора), одна из которых неподвижна, а другая движется параллельно первой с некоторой скоростью U (рис. 3.105). Толщина слоя суспензии h равна расстоянию L между пластинами, если он не разрушается при взаимном движении пластин. В общем же случае пространство между пластинами заполнено плоскими, движущимися параллельно друг другу слоями меньшей толщины, чем h. Их суммарная толщина по-прежнему равна L.
Рис. 3.105. Профиль скоростей движения жидкости относительно пористого тела, заключенного между неподвижной а и движущейся со скоростью U пластиной b
Сила адгезии частиц суспензии к пластинам принимается равной нулю, так что сопротивление т взаимному перемещению пластин имеет чисто вязкую природу и определяется вязкостью среды т|0 и градиентом скорости ее течения y'h в некотором тонком слое жидкости между стенкой и слоем суспензии:
ПоТ = т|оУЛ •
(3.14.38)
Величина толщины этого слоя жидкости имеет порядок, близкий к порядку проницаемости слоя 8. Последняя, следовательно, определяется как эффективная глубина проникновения внутрь пористого слоя структурированной суспензии потоков жидкости, увлекаемой движущейся стенкой. Сам слой суспензии, точнее
только ее дисперсная фаза, образующая структурный каркас, движется как единое, жесткое, прочное тело со скоростью, равной половине скорости движения пластины, если h- L. Ось декартовых координат х направлена по нормали к пластинам.
Экспериментатор может не учитывать характер деформации образца в приборе. Для него величина U! L есть не что иное, как экспериментально определяемая скорость деформации у'е (градиент скорости течения) дисперсной системы. Скорость движения слоя суспензии и/, относительно стенки прибора (пластины) связан со скоростью движения пластины уравнением
uh=y'ehl2-Uhl2L. (3.14.39)
В дальнейшем, однако, отсчет скоростей и расстояний ведется от середины движущегося каркаса. Скорость движения среды в его середине (х = 0) относительно самого каркаса равна нулю. Тогда щ в формуле (3.14.39) — это скорость движения жидкости на внешней границе слоя суспензии относительно его середины. На расстоянии х от середины скорость движения жидкости и относительно каркаса меньше, чем на периферии слоя.
Необходимо отметить, что в однородной вязкой среде при ее однородном 1 сдвиговом деформировании полная сила, действующая на слой среды произвольной толщины dx, расположенный параллельно плоскостям сдвигового течения* *2, равна нулю. Это следствие того, что при однородной деформации скорости сдвига одинаковы на обеих сторонах слоя, и поэтому разность сил вязкого трения rjy', действующих на обе его стороны, равна нулю. При наличии в слое толщиной dx некоторого числа ndx частиц, скорость которых на величину и меньше скорости движения жидкости, течение жидкости замедляется в результате ее трения о частицы. Суммарная сила трения частиц f = bundx уравновешена разностью сил трения на границах слоя т|ое/у'. Поскольку у' = duldx, то е/у' = (d~u!dx2)dx, и тогда:
bundx = г|о (d~u!dx2}dx. (3.14.40)
Не следует думать, что формула (3.14.40) представляет частный случай фундаментальных уравнений гидродинамики Навье — Стокса. Последние хотя и относятся к произвольному распределению сил, скоростей и плотностей в среде, но охватывают только те случаи, в которых все частицы (молекулы) данного элемента деформируемой среды имеют равные по величине и направлению скорости, т. е. это уравнения механики сплошной среды. В плане рассматриваемой задачи (и реологии дисперсных систем вообще) сфера компетенции уравнений Навье — Стокса — нахождение распре
*’ Деформирование, при котором градиент скорости одинаков во всех точках среды.
*2 Плоскости равных скоростей, параллельные направлению течения.
718
Новый справочник химика и технолога
деления скоростей среды в одной или в интервале конечного числа частиц при определенном их положении. Положение и размер частиц учитываются при решении уравнений Навье — Стокса в качестве параметров, задающих граничные условия. Здесь эта задача считается решенной. Результатом ее решения является формула Стокса для силы сопротивления движению жидкости Ьи, создаваемой частицей, где b = 6лт]о«.
Раскрыв в уравнении (3.14.40) коэффициент b и выразив концентрацию частиц п через объемную долю ср дисперсной фазы и объем частицы v, т. е. п = cpv, где v = 4тш3/3, находим:
d2u/dx2 = к2и, (3.14.41)
где х2 = Оф/За2.
Интегрирование уравнения (3.14.41) приводит к выражению :
du/dx = (к2и2 + О,/2. (3.14.42)
Знак (+) и константу интегрирования С = (у'о )2 находят, исходя из того, что градиент скорости течения среды должен увеличиваться от середины слоя к его периферии одновременно с увеличением самой скорости. В середине слоя скорость равна нулю, а градиент имеет некоторое, пока неопределенное значение у0. С учетом этого предыдущее уравнение преобразуется к виду
du/dx = к(и2 + м02)1/2. (3.14.43)
Здесь м0 = у0 /х = у08 и 8 = 1 /х — эффективная глубина проникновения потоков среды внутрь пористого структурного каркаса.
После разделения переменных и интегрирования в пределах от х = 0 и w = 0 до произвольного значения величин хим получаются выражения:
Arsh(w/wo) - хх
или (3.14.44)
и = Uq sh(xx).
Здесь и0 = у'о8, х = 1/8, 8 = (а/3)(2/ср)1/2, у0 — неопределенная пока величина градиента скорости течения среды в середине структурного слоя. Ее значение получается из уравнения (3.14.44), если скорость течения щ на границе слоя (х = h/2) выразить с помощью формулы (3.14.39) через его толщину h и макроскопическую скорость сдвиговой деформации у'е так, что:
Yeh/2 = Yo8sh(xA/2)
или (3.14.45)
у о = Ye [(хА/2)/ sh(xA/2)] .
* См. аналогичное по форме уравнение Пуассона — Больцмана в теории двойного слоя.
Эта формула связывает скорость сдвига у'о в середине пористого слоя с его толщиной h.
Выражение для градиента скорости течения du/dx = у' на произвольном расстоянии х от середины пористого слоя получается при подстановке формулы (3.14.44) в уравнение (3.14.43):
у' = xw0[sh2(xx) + 1],/2.
С помощью формул гиперболической тригонометрии оно преобразуется к более удобному виду:
у' = хмосЬ(хх). (3.14.46)
В частном случае при х = h/2 последнее уравнение дает величину скорости сдвиговой деформации уЛ среды на внешней стороне пористого слоя:
уА = хм0сЬ(хЛ/2). (3.14.47)
Так как м0= у'о 8 = у0/х, то, в совокупности с формулой (3.14.45), получается уравнение
уА = Ye [(хЛ/2)/ Л(хЛ/2)]. (3.14.48)
Тогда, согласно формуле (3.14.38), сопротивление сдвиговому деформированию дисперсной системы будет:
т = По Ye [(xA/2)/th(xA/2)]. (3.14.49)
Если, как это отмечалось выше, пористый слой достаточно прочен, то его толщина h остается при движении пластин прибора неизменной и равной расстоянию между пластинами L. В этом случае формула (3.14.45) описывает величину скорости сдвигового течения среды у0 в середине слоя, а формула (3.14.50) — экспериментально определяемую величину вязкости дисперсной системы:
П = По [(хА/2) / th(xA/2)]. (3.14.50)
Следует напомнить, что экспериментатор может ничего не знать о состоянии слоя вещества в зазоре между движущимися пластинами и о характере его деформирования. Для него величина у'е = U! L является обычной скоростью сдвиговой деформации у', а отношение напряжения т к скорости у' — вязкостью дисперсной системы г], находящейся между пластинами прибора.
Все приведенные выше формулы сохраняют свою силу и тогда, когда слой структурированной суспензии разделен на ряд более тонких параллельных слоев, движущихся с разными скоростями, сохраняя свою сплошность. Отличие лишь в том, что толщина слоя h в этом случае непостоянна, зависит от действующего напряжения и в приведенных уравнениях остается пока неопределенной. Число таких слоев равно, L/ h.
Условие равновесия любой части сплошного слоя заключается в том, что действующая на него гидроди
Физика, химия и технология дисперсных систем
719
намическая сила уравновешена силой упругого сопротивления структурного каркаса. Иначе говоря, движение жидкости внутри каркаса создает в нем напряжения. В симметричных относительно середины слоя подслоях толщиной dx эти напряжения равны по величине и противоположны по направлению. Упругие напряжения в каркасе достигают максимума в середине каждого из сплошных слоев толщиной h. Величина этих напряжений может быть найдена интегрированием вклада f каждого подслоя толщиной dx по одной половине сплошного слоя, т. е. в пределах от х = 0 до х = h/2. Величина этого вклада дается формулой (3.14.40). Однако можно поступить и иначе. Формула (3.14.49) дает полную величину т сдвигового усилия, действующего на внешнюю сторону7 сплошного слоя толщиной h. По условию равновесия с тем же усилием одна половина слоя действует на другую. Однако в середине часть этого усилия создается вязким сопротивлением, величина которого равна т|0у0, так что на долю упругих напряжений хг в середине слоя остается величина (т-ЛоУо)-Тогда в соответствии с формулами (3.14.49) и (3.14.45):
тг = По У'е {[(x/i/2) /th(xA/2)] - [(xA/2)/sh(xA/2)]}
или (3.14.51)
т, = По[ch(xA/2) - 1 ][(хЛ/2)/sh(xA/2)].
Слой толщиной h должен расщепляться на два равных более тонких слоя, как только величина упругих напряжений тг в его середине превысит прочность структурного каркаса тЛ. Таким образом, условие хг = или:
т$ = По у; [ch(xhe/2) - 1][W2) / sh^c^)] (3.14.52)
определяет гидродинамически (тиксотропно) равновесную толщину слоев he, на которые распадается сплошной структурный каркас при сдвиговой деформации дисперсной системы с заданной макроскопической скоростью у'е. Иначе говоря, выражение (3.14.52) является уравнением структурного состояния дисперсной системы. Оно вместе с уравнением напряжений (3.14.49) определяет зависимость скорости сдвига от напряжения. При этом в уравнение (3.14.49) следует подставлять равновесную толщину движущихся слоев, полученную решением уравнения (3.14.52) при заданной скорости сдвига. Полная система уравнений реологии в рамках модели послойного скольжения включает условие применимости этих уравнений:
- при xr < xs (течение без разрушения структуры) h = L, - при xr > ts (течение в режиме тиксотропного равновесия) h = he, причем he определяется уравнением (3.14.52).
В любом из этих случаев напряжение сдвига при послойном скольжении:
т = ПоУ'[(^/2) / th(x/i/2)]. (3.14.53)
Здесь опущен индекс «е» при величине /, поскольку в эксперименте не могут фигурировать никакие другие
скорости сдвиговой деформации, кроме макроскопической скорости сдвига, равной в данном случае отношению U/L. Реальный профиль скоростей течения среды осциллирует возле скорости движения слоя структурированной суспензии по мере продвижения от одной пластины прибора к другой (рис. 3.106).
Рис. 3.106. Макроскопически определяемый (пунктир) и реальный профили скоростей течения жидкой фазы при послойном скольжении элементов структуры
Анализ приведенных уравнений реологии возможен только численным методом, поскольку уравнение состояния (3.14.52) аналитически не решается. Численное исследование показывает, что для кривой течения характерно очень резкое увеличение макроскопической скорости деформации U/L при переходе к режиму тиксотропно равновесной деформации при напряжении, немного превышающем статическую прочность т5 структурного каркаса дисперсной системы.
Система уравнений не охватывает течения в режиме полного разрушения структуры. В принятых в этой модели понятиях полное разрушение могло бы означать равенство толщины h движущихся слоев размеру частиц а. Согласно указанию к формуле (3.14.41), произведение хЛ/2 при этом равно 3(<р/2)1/2 или, приближенно, 2(р1/2. При таком малом значении аргумента хЛ/2 гиперболический тангенс практически равен самому аргументу, и тогда, согласно формуле (3.14.53), напряжение сдвига оказывается равным напряжению в чистой дисперсионной среде, что, конечно, не соответствует действительности. На этом основании следует полагать, что механизм послойного скольжения может реализоваться только в режиме, достаточно удаленном от сильного разрушения структурной сетки. Физическая причина такого ограничения понятна — при малой толщине слоев их послойное движение становится неустойчивым, поскольку на каждый элемент слоя действует также момент сил, стремящийся повернуть слой перпендикулярно направлению движения. Более того, среди общих принципов механики сплошной среды есть и принцип попарного равенства касательных напряжений. В данном случае он означает, что в сплошном движущемся слое действуют сдвиговые напряжения и в направлении, перпендикулярном направлению движения слоев, причем они численно равны сдвиговым напряжениям, действующим вдоль направления движения, которые только и были рассмотрены в моде
720
Новый справочник химика и технолога
ли. При большой толщине слоев перпендикулярные к направлению движения усилия скомпенсированы реакцией стенок прибора и соседних слоев, что и оправдывает сделанные в теории упрощения. Это ограничивает область ее компетентности течением при слабых разрушениях структуры. Разрушения можно считать слабыми, если равновесная толщина слоя не слишком мала по сравнению с толщиной всего слоя дисперсной системы или если параметр хЛ/2 » 1.
В заключение заметим, что глубина проникновения 8 движения жидкости в глубь пористых тел ненамного превышает размер частиц, из которых состоят такие тела. Это и оправдывает то, что во фрактальной модели структурированных систем флокулы считаются непроницаемыми для потоков среды.
3.15. Основы вискозиметрии
Реологический эксперимент является важным источником сведений о структуре, взаимодействии частиц и состоянии их поверхности. Вычисление характеристик дисперсной системы из данных реологического эксперимента, как и решение обратной задачи — расчета параметров течения системы на основе данных о поверхностных свойствах частиц, — требует знания наиболее распространенных методов проведения реологического опыта, расчетных соотношений и их возможностей. Приборы, на которых проводятся реологические измерения, называются вискозиметрами. Они могут иметь разные конструкции и принципы действия, но во всех случаях задается или скорость деформации исследуемого материала у' и измеряется соответствующая ей удельная сила сопротивления материала (напряжение) т, или задается деформирующее усилие т, а измеряется соответствующая ему скорость деформации. Тот и другой режим можно реализовать на приборе, который состоит из пары пластин — неподвижной и подвижной, между которыми имеется плоскопараллельный зазор определенной ширины h. Исследуемый препарат помещается в этот зазор и подвергается деформированию путем тангенциального перемещения одной пластины относительно другой при постоянстве h (рис. 3.107). Скорость деформации исследуемого препарата у' = U/ h, где U — скорость перемещения подвижной пластины.
Рис. 3.107. Схема измерения сопротивления сдвиговому деформированию
Напряжение в исследуемом материале t = F/A, где F— сдвиговая сила, приложенная к подвижной пластине (или измеренная, если задана скорость ее перемещения U), и А — ее площадь. Главное достоинство
такого прибора в том, что он обеспечивает однородность напряжений и деформаций в испытуемом материале и в этом смысле является идеальным.
Недостаток прибора в том, что он не может быть реализован конструктивно. В наибольшей мере к идеальному прибору приближаются вискозиметры ротационного типа. В них испытуемый материал находится в зазоре между двумя коаксиальными цилиндрами (рис. 3.108). Один из них (ротор) вращается, а другой (статор) неподвижен. При этом силоизмерительным элементом может быть или ротор, или статор. Измеряется крутящий момент N, действующий на этот элемент, и угловая скорость вращения ротора о).
При условии, что зазор h между ротором и статором много меньше радиуса R ротора (или статора), скорость сдвига вычисляется по формуле (3.15.1), а напряжение —по формуле (3.15.2):
у' = со/?/А, (3.15.1)
t = NI2tzR2H. (3.15.2)
Здесь Н — глубина погружения силоизмерительного элемента прибора в исследуемый раствор.
Конструктивно проще приборы, у которых вращается внутренний цилиндр (вискозиметр с внутренним ротором). Он же обычно служит и датчиком величины крутящего момента. Его величина измеряется по деформации (углу закручивания) упругого элемента (спиральной пружины или торсиона), через который на ротор передается вращательное движение. Эти приборы имеют два недостатка.
Первый состоит в том, что по условиям прилипания испытуемого материала к стенкам ротора и статора внутренние — прилегающие к ротору — слои материала вращаются со скоростью, равной скорости вращения ротора, а внешние слои, прилегающие к статору, неподвижны. Под действием центробежной силы внутренние слои материала отбрасываются к внешней (статорной) стороне рабочего зазора, выдавливая отсюда пристаторные слои вещества по направлению к ротору. Таким образом, возникает движение материала и в радиальном направлении, причем оно имеет ячеистую структуру (зоны движения от ротора к статору и в обратном направлении чередуются). Это нарушает однородность сдвиговой деформации и искажает результаты измерений. Приборы с вращающимся внешним цилиндром (внешним ротором) лишены этого недостатка. При этом упрощается и измерение величины крутящего момента, действующего на внутренний цилиндр, поскольку он неподвижен, но сложнее организовать тер-мостатирование вращающегося стакана (внешнего цилиндра) с исследуемым препаратом.
Второй недостаток присущ всем ротационным вискозиметрам с массивным внутренним цилиндром, выполняющим функцию или ротора, или статора. Он заключается в том, что исследуемый материал заполняет также и донную часть прибора — промежуток между
Физика, химия и технология дисперсных систем
721
дном сосуда (внешнего цилиндра) и нижним торцом внутреннего цилиндра. Эта часть материала вносит свой вклад в общую силу сопротивления деформированию, причем в ряде случаев неизвестную, поскольку скорость деформации здесь непостоянна. Ротационные приборы разных конструкций и фирм, в сущности, отличаются именно способом решения этой проблемы. В частности, этой проблемы фактически не существует в приборах с тонкостенным ротором, у которого площадь нижнего торца ротора, а следовательно, и его вклад в силу трения практически равны нулю.
В приборе с тонкостенным ротором (рис. 3.109), размещенным в зазоре между двумя массивными коаксиальными цилиндрами, исследуемая дисперсная система находится в двух кольцевых зазорах одинаковой толщины h. Скорость сдвига в обоих зазорах одинакова и вычисляется по той же формуле (3.15.1), в которой R — радиус тонкостенного цилиндра. Напряжение сдвига в этом случае:
т = ^/4лЛ2Я (3.15.3)
Прибор такой конструкции, как и прибор с массивным внутренним ротором, пригоден для исследования при низких и умеренных скоростях вращения ротора, так как при высоких скоростях в одном из зазоров (во внешнем, если ротором является тонкостенный цилиндр, и во внутреннем в обратном случае) возникает ячеистое радиальное течение суспензии под действием центробежных сил.
Нижнему торцу внутреннего массивного цилиндра можно придать коническую форму. Тогда зазор hx- сХ между торцом и дном стакана и линейная скорость вращения соАГ увеличиваются с увеличением расстояния X от оси вращения. Поэтому скорость сдвига у' = <йА7 hx не зависит от А" и равна (о / с, где с — тангенс угла Р между дном ротора и образующей конуса (рис. 3.110). Значение этого коэффициента следует выбрать так, чтобы скорости сдвига в коаксиальном рабочем зазоре вискозиметра и в его придонной части были одинаковы, т. е. со/с = d)RIh, где h — зазор между нижней кромкой цилиндра и дном стакана. Отсюда:
c = h!R. (3.15.4)
Сдвиговое напряжение т будет в этом случае одинаково в коаксиальном и придонном слоях материала. Через измеренную величину суммарного крутящего момента, создаваемого обеими частями материала, оно выражается формулой
т = Л'/[8л/?2 (//+2/?/3)]. (3.15.5)
Применяются и приборы типа конус-пластина, в которых вместо внутреннего цилиндра остается тонкая шайба с конической формой ее нижней стороны. Формула для напряжений отличается от предыдущей только тем, что Н = 0. Значение коэффициента с может быть любым, а скорость сдвига у' = со / с.
В ряде случаев можно ограничиться измерением только величины предельного напряжения сдвига тЛ. Для этого имеются простые приборы Ребиндера — Вейлера (рис. 3.111), которые могут быть легко собраны из подручных средств.
А
Рис. 3.108. Схема ротационного вискозиметра:
R — радиус внутреннего цилиндра; h — зазор между внутренним и внешним цилиндром; Н— глубина погружения цилиндра в исследуемый состав
Рис. 3.109. Схема ротационного вискозиметра с тонкостенным ротором:
R — радиус ротора; Н — глубина его погружения в исследуемый состав; h — рабочие зазоры вискозиметра
Рис. 3.110. Схема вискозиметра типа конус-плоскость:
R — радиус; Р — угол конуса;
h — зазор между нижней кромкой цилиндра и дном стакана
Рис. 3.111. Прибор Ребиндера — Вейлера (пластометр):
1 — сосуд; 2 — суспензия; 3 — пластина; 4 — подвижный стол;
5 — шкала; 6 — индикатор; 7 — пружина
722
Новый справочник химика и технолога
Прибор Ребиндера — Вейдера состоит из сосуда 1 произвольной формы и размера, в который помещаются исследуемая суспензия 2 и рифленая пластинка 3, прикрепленная к пружине 7. По удлинению пружины АЛ при медленном вытягивании пластины из суспензии (или опускании столика с сосудом) вычисляется величина действующей на пластину силы F = GAZ и затем определяется прочность исследуемого препарата на сдвиг по формуле
ts = F/2S.
(3.15.6)
Здесь G — константа жесткости пружины и S — площадь одной стороны пластины. Для измерения АЛ прибор снабжен шкалой 5 и стрелкой 6. Деформирование суспензии удобнее производить, опуская столик 4, на котором укреплен сосуд с суспензией.
Картина распределения деформаций в суспензии в этом случае сложна, и поэтому численное значение скорости сдвига остается неопределенным. В связи с этим обычно стремятся к возможно малой скорости деформации, так чтобы она была близка к нулю и в формуле сопротивления сдвигу т = + т|*у' вторым слагаемым можно было пренебречь. При этом т = т5. Такой метод измерения ts неявно предполагает, что измеряемая величина т соответствует участку пластичного течения на реологической кривой, например точкам 1 или 2 (рис. 3.112). Прибор этой конструкции, как и прибор с массивным внутренним ротором, пригоден для исследования при низких и умеренных скоростях вращения ротора, так как при высоких скоростях в одном из зазоров (во внешнем, если ротором является тонкостенный цилиндр, и во внутреннем в обратном случае) возникает ячеистое радиальное течение суспензии под действием центробежных сил. Чтобы убедиться в справедливости такого предположения, следует провести измерения, по крайней мере, при двух различных скоростях деформации (опускания столика), например, отличающихся в два раза. Если при этом разность величин Ti и т2 окажется незначительной, то действительно т5 - Ti = т2. Может, однако, оказаться, что измеренные величины т также будут отличаться примерно вдвое. Это будет означать, что течение отвечает участку ползучести, например точкам 3 и 4, которые далеко отстоят от т5. В этом случае следует увеличивать скорость деформации у' до такой величины, когда т почти не зависит от у'.
Рис. 3.112. К определению предельного напряжения сдвига
В любом случае необходимо убедиться в стационарности процесса деформирования, т. е. в том, что показания силоизмерительного прибора (растяжение пружины) не меняются во времени.
Резко неоднородная деформация создается в шариковых вискозиметрах (рис. 3.113). Шарик 1, укрепленный на штоке 2, вдавливается с заданной силой F в суспензию 3, помещенную в цилиндрический сосуд 4 со строго постоянным по высоте внутренним диаметром. Скорость деформации (вдавливания) вычисляется по времени t прохождения определенного числа делений шкалы 5. Прибор позволяет проводить сравнительные исследования различных материалов, т. е. получать зависимость \/t от F, которая может иметь сходство с зависимостью у' от т, но отличаться от нее численными значениями абсциссы и ординаты. Абсолютные значения т, ts, т|* и у' могут быть определены при калибровке прибора по жидкостям с известными реологическими свойствами.
Разновидностью шарикового вискозиметра является прибор со свободно падающим тяжелым (например, стальным) шариком. Измеряемой величиной является время прохождения шариком определенного пути — между метками на поверхности цилиндрического сосуда, который в этом случае должен быть изготовлен из прозрачного материала. Прибор можно использовать только для изучения прозрачных растворов вследствие визуального способа регистрации времени прохождения отметок на сосуде. Шариковые вискозиметры не пригодны для систем, в которых шарик может вызвать спрессовывание взвеси в нижней части пробирки.
Непостоянство скорости деформации во времени и объеме образца характерно для так называемых пенетрометров. В этом приборе в пластичный материал вдавливается с определенной силой F вершина конуса. По глубине вдавливания Н вычисляется предельное напряжение сдвига по формуле
t^CFIH2. (3.15.7)
Здесь С — константа прибора. Пенетрометр является, по существу, разновидностью стандартного прибора для определения твердости материала по вдавливанию в него призмы или шарика.
Рис. 3.113. Шариковый вискозиметр:
1 — шарик; 2 — шток; 3 — исследуемый раствор;
4 — пробирка; 5 — шкала; F— действующая на шарик сила
Физика, химия и технология дисперсных систем
723
Капиллярный вискозиметр (рис. 3.114), так же как пенетрометр, шариковый прибор и прибор Ребиндера — Вейлера, отличается существенной неоднородностью деформации исследуемого препарата. Однако эта неоднородность вполне определенная, т. е. распределение скоростей в капилляре известно. Прибор позволяет изменять скорость деформации в широких пределах, отличается простотой изготовления и самого процесса измерения, поэтому он является одним из самых распространенных.
Обычно вискозиметр представляет собой два резервуара: 1 — мерный и 2 — приемный. Они соединенны цилиндрическим капилляром 3 с известным радиусом R порядка 1 мм и длиной L около 10 см. Объем верхнего (мерного) резервуара около 1 см3. Процедура измерения сводится к регистрации времени t истечения исследуемой жидкости из мерного резервуара емкостью V от метки А до метки В над и под резервуаром /. Затем вычисляют объемную скорость течения v = VI t. Напряжение деформации т регулируется путем изменения давления Рт газа (воздуха), продавливающего жидкость через капилляр. Для этого мерный резервуар соединен с моностатом, в котором создается нужное давление газа Рт. В простейшем случае (при исследовании заведомо ньютоновской жидкости) можно ограничиться измерением скорости течения v только при одном гидростатическом давления Ph = pgH, где р — плотность исследуемого раствора, g — ускорение свободного падения; Н — средняя разность уровней раствора в резервуарах 1 и 2 за время истечения. В общем же случае напряжение т определяется суммарным давлением Р-Рт + Ph.
Для работы с непрозрачными растворами при визуальном способе регистрации времени их истечения вискозиметр снабжен двумя верхними резервуарами (рис. 3.115).
Один из них / — мерный, известного объема V, заполнен прозрачной вспомогательной жидкостью, которая должна иметь меньшую плотность, чем исследуемая жидкость, и не смешиваться с ней. Исследуемый непрозрачный раствор заполняет второй верхний резервуар, капилляр и приемный резервуар вискозиметра. Регистрируется время истечения I прозрачной жидкости из мерного объема V. Объемная скорость течения v исследуемой жидкости равна при этом измеренной объемной скорости течения вспомогательной жидкости.
Результаты измерений часто представляются в виде графика зависимости объемной скорости течения v от давления Р. Вид этой зависимости в случае неньютоновской жидкости даже качественно отличается от зависимости скорости сдвига у' от напряжения т. Принципиально важно, что параметры кривой течения v = v(P) зависят от геометрии капилляра и поэтому не являются инвариантной характеристикой исследуемой жидкости. В отличие от этого параметры кривой скорости сдвига у' = у'(т) не зависят от конструктивных характеристик вискозиметра, т. е. являются инвариантными по отношению к прибору и поэтому могут применяться
для сравнения между собой свойств различных жидкостей. Для нахождения корректных реологических характеристик исследуемых составов необходимы уравнения преобразования измеряемых величин в их инвариантные значения.
Движение жидкости в капилляре можно представить как движение коаксиальных цилиндрических слоев разного радиуса г с различными скоростями и(г). Диаграмма зависимости скорости движения слоя от его радиуса представляет так называемый профиль скоростей течения (рис. 3.116). Вычисление профиля скоростей и (г) является одним из этапов нахождения уравнений преобразования результатов измерения к инвариантному виду.
Рис. 3.114. Капиллярный вискозиметр:
1 — мерный и 2 — приемный резервуары;
3 — капилляр; А, В — метки
Рис. 3.115. Капиллярный вискозиметр для непрозрачных жидкостей:
] — мерный и 2 — верхний резервуары; А, В — метки
Рис. 3.116. Пуазейлево течение: и(г) — профиль скоростей
Сплошной столб жидкости радиусом г движется под действием силы давления яг2/5 на его торец, пропорциональной площади торца яг2. В установившемся режиме течения эта сила равна силе трения 2 яг Ат цилиндрической поверхности столба 2ягА об остальную часть жидкости. Отсюда находим:
x = rP!2L.
(3.15.8)
724
Новый справочник химика и технолога
Так как напряжение х = ту/ и скорость сдвига у'(г) = -duldr, то:
-duldr = rPI2Lx\. (3.15.9)
Отрицательный знак производной обусловлен тем, что скорость уменьшается с увеличением расстояния г от оси капилляра. При интегрировании этого уравнения при постоянстве вязкости т) в пределах от г = R и и = О до произвольной величины г и и получается уравнение профиля скоростей течения:
w = P/4ZT](l?2-r2). (3.15.10)
Профиль имеет, следовательно, вид параболы (рис. 3.116).
Каждый цилиндрический слой толщиной dr переносит при течении за единицу времени количество жидкости dv = 2nrudr. При интегрировании этого уравнения по радиусу получается выражение объемной скорости течения ньютоновской жидкости по капилляру радиусом R и длиной L под действием давления Р:
v = 7t/?4P/8ni. (3.15.11)
Это выражение известно как формула Пуазейля. Она позволяет непосредственно вычислять инвариантную характеристику исследуемого раствора — его вязкость т|, а при необходимости — напряжение х и скорость сдвига у' по формулам (3.15.8), (3.15.9). Значения т и у' различны на разных расстояниях от оси капилляра, но они укладываются в общую зависимость х = щ'. Это относится и к величине напряжения и скорости сдвига на стенке капилляра, которые часто фигурируют в различных уравнениях (см. ниже).
Простейший с математической точки зрения вариант неньютоновского течения описывается уравнением Шведова — Бингама:
х = х5 + т]*у'. (3.15.12)
Фигурирующая в уравнениях (3.15.9), (3.15.10) вязкость т) описывается с помощью формулы (3.15.12) через напряжение т| = г|*т/(т - xs), что позволяет интегрировать указанные уравнения после перехода в них к новой независимой переменной х вместо г с помощью формулы (3.15.8). При этом следует принять во внимание, что при х < xs деформация невозможна. Поэтому центральная часть столба пластичного материала, где, согласно формуле (3.15.8), напряжения х могут оказаться меньше прочности материала х5, движется без деформации, т. е. как твердый стержень. Его радиус определяется в соответствии с формулой (3.15.8) прочностью материала и давлением:
rs = 2Lxs/P. (3.15.13)
Тогда профиль скорости течения имеет вид усеченной параболы (рис. 3.117), а объемная скорость течения пластичного материала вычисляется как сумма потоков в стержневой и периферийной частях течения.
Рис. 3.117. Течение пластичного материала в трубе радиусом R под действием давления Р: rR — радиус твердообразного ядра течения
Результаты вычислений в этом случае дают формулу
у = лЯ4/78т]7 (1-80/3 + ^/З). (3.15.14)
Здесь Q = xsLIPR, причем v>0 при 0<О,5. Радиус стержневидного ядра течения rs монотонно уменьшается с ростом давления Р. Поэтому зависимость (3.15.14) v = v(P) оказывается нелинейной, тогда как зависимость (3.15.12) у' = у'(х) строго линейна.‘При большом давлении Q << 0,5, радиус стержневидного течения приближается к нулю, а зависимость v = v(P) — к линейной. Если в уравнении (3.15.14) пренебречь величиной 1604/ 3, то получается формула
v = nR*/8x\ L(P-8xsL/3R). (3.15.15)
Из формулы следует, что течение прекращается при уменьшении давления до некоторой критической величины Р = Рс, определяемой формулой
Pc = 8xsL/3R. (3.15.16)
В действительности это происходит при уменьшении давления до величины Ps, определяемой условием 0 = 0,5:
Ps = 2LxJR. (3.15.17)
Давление Рс является кажущимся (динамическим) пределом текучести, который вычисляется экстраполяцией линейного участка зависимости объемной скорости течения от давления к нулевой величине скорости, а давление Ps — истинным пределом текучести по давлению.
В капиллярной вискозиметрии удобно пользоваться значениями напряжения сдвига xR и скорости деформации y'R на стенке капилляра, которые связаны между собой обычным законом внутреннего трения xR = т|у'л. Как следует из формулы (3.15.8), xR = RPI2L. Тогда формула (3.15.14) приобретает вид
г = яЯ3т„/411» [1 -(4/3)(гг/тй) + (1/3)(г,/гя)4].
(3.15.18)
В общем случае неизвестно, каким реологическим уравнением описывается поведение материала при течении, т. е. неизвестна зависимость т) = т](х). Поэтому уравнение (3.15.9) нельзя интегрировать. Тем не менее, имеется возможность получения инвариантных реоло
Физика, химия и технология дисперсных систем
725
гических характеристик и в этом случае, если отказаться от поиска общего решения задачи и ограничиться вычислениями напряжения и скорости сдвига только на стенках капилляра. В любом случае объемную скорость течения можно представить в виде интегрального уравнения
v = 2л jrudr = л judr2. (3.15.19)
При этом интегрирование проводится в пределах от г = 0 до г = R.
По правилам интегрирования по частям получается, что judr2 =-jr2du, поскольку в указанных пределах
значение uR2 = 0. Если провести замену du =— dr = -у'dr, dr
представить dr на основании формулы (3.15.8)
7? . R ,
(г = —т) в виде следующего соотношения: dr = — dx, ~R ~R
то получится уравнение
у = (лЯ7т’) |у'(т)т2^т. (3.15.20)
Величина v/лТ?2 представляет собой среднюю линейную скорость течения в капилляре и, следовательно, v/лТ?3 — среднее значение градиента скорости течения Уо, которое является экспериментально определяемой характеристикой течения при заданном давлении Р. С учетом этого формула (3.15.20) приобретает вид
у0 =(1/4) Jy'CO^tZx. (3.15.21)
Как и формула (3.15.20), она предполагает интегрирование в пределах от т = 0 до т = тЛ. При ее дифференцировании как произведения функций 1/ 4 и |у'(т)т2(/т по верхнему пределу xR дает следующее соотношение:
dl'JdxR = - (3/7?) [(1/) |у'(т)х2й?т] + (1/х^ ) у (т)т2.
Здесь выражение в квадратных скобках есть, согласно формуле (3.15.21), величина у'о. Поэтому после упрощения полученного соотношения получается уравнение [24]:
уЛ (Tr) = 3 у0 + тЛ (d у0 /б7тЛ). (3.15.22)
Этот результат известен как формула Рабиновича — Вайссенберга. При подстановке в нее следующих выражений:
у'о =v/tt7?3 nxR = RP/2L
(в том числе в производной d у^ /<7тЛ) скорость сдвига на стенке сосуда y'R можно выразить через непосредственно контролируемые в эксперименте величины v и Р с помощью формулы
уд (тЛ) = (3 v + Pdv МР)1л&. (3.15.22а)
Аналогичным образом выводится формула для плоского капилляра толщиной h и шириной Ь:
Ул(^) = 4у0 +2xA(tZy0/ dxh). (3.15.23)
Здесь y'h — скорость сдвига, xR — напряжение на стенке, а у'о = v / bh2. Скорость сдвига у0 является экспериментально определяемой величиной, а тЛ (или т/,) — произвольно устанавливаемым с помощью давления Р значением напряжения и у'д (или y'h) — соответствующей ему скоростью сдвига. Таким образом, с помощью уравнения Рабиновича — Вайссенберга удается получить инвариантную характеристику течения жидкости с произвольной и неизвестной заранее зависимостью вязкости от напряжения. Излишне напоминать, что все это возможно благодаря применению закона внутреннего трения Ньютона к «неньютоновским» жидкостям.
Плоские капилляры (каналы) предпочтительны при изучении течения поляризующихся дисперсных систем в электрическом или магнитном поле. Связанные с этим проблемы рассмотрены в подразделе 3.19.3 в связи с описанием воздействия магнитного поля на структуру феррожидкостей.
Электровискозиметры и магнитовискозиметры — приборы для измерения реологических параметров дисперсных систем в электрическом и магнитном поле соответственно — должны удовлетворять следующим требованиям: однородность внешнего поля в рабочем зазоре прибора, однородная поляризация (электрическая или магнитная) по всему объему исследуемой системы, определенность взаимной ориентации внешнего поля, направления течения и градиента скорости течения. Отсюда следует, что это должны быть приборы непроточного типа — ротационные или Ребиндера — Бейлера.
Электровискозиметр типа Ребиндера — Бейлера отличается от обычного прибора (рис. 3.111) прямоугольной формой сосуда 7, материалом, из которого изготовлен сосуд (это должен быть диэлектрик). Измерительная пластина 3 должна быть изготовлена из металла и размещена параллельно паре плоских электродов, погруженных в исследуемую суспензию. Напряженность поля в суспензии E=UIL, где U— разность потенциалов электродов, а А — расстояние между электродами.
Конструирование электровискозиметров по классическим схемам (рис. 3.108, 3.109, 3.110) затруднено в связи с необходимостью вводить скользящий контакт для подвода напряжения. Более удобен прибор с тонкостенным статором и вращающимся сосудом (рис. 3.118).
Статор 7 крепится и центрируется относительно ротора 2 на растяжках 3. При этом определяют крутящий момента по углу закручивания статора. К ротору, являющемуся одновременно сосудом для исследуемого состава, подводится напряжение через скользящий контакт.
726
Новый справочник химика и технолога
Рис. 3.118. Схема ротационного электровискозиметра: Измерительный цилиндр (/) закреплен на растяжках (3) и опущен в щель вращающегося сосуда (2) с суспензией. Разность потенциалов U подана между цилиндром (/) и сосудом (2)
Магнитовискозиметр типа Ребиндера — Вейдера отличается от электровискозиметра тем, что вместо электродов к сосуду с внешней стороны подводятся полюса электромагнита. Все детали прибора должны быть изготовлены из немагнитных материалов.
Схема магнитовискозиметра ротационного типа показана на рис. 3.119. Прибор представляет собой вращающийся электромагнит I с кольцевым зазором между полюсами, в который помещаются исследуемая суспензия и измерительный тонкостенный статор 2, подвешенный на растяжках 3. Магнитное поле создается пропусканием тока по обмотке 4 через токопроводящие щетки 5. Статор и растяжки изготовлены из немагнитных цветных металлов, ротор — из мягкого железа.
Среди методов реологических исследований особое место занимает микрореологический метод [50]. Особенность его в том, что он не требует каких-либо реологических приборов и сводится к измерению статической намагниченности исследуемой системы. Для этого в нее вводится магнитно-жесткий порошок в количестве 0,5-5 % от массы системы (в зависимости от чувствительности магнитометра). Частицы магнитно-жесткого порошка представляют собой постоянные магнитные диполи (единичные домены) с размером частиц около 1 мкм. Они склонны к слипанию, поэтому обеспечить равномерное распределение таких частиц можно только в пластичной среде.
Рис. 3.119. Схема магнитовискозиметра:
1 — вращающийся электромагнит; 2 — измерительный цилиндр;
3 — растяжки для его крепления; 4 — обмотка электромагнита;
5 — скользящие контакты для подачи тока в обмотку
Прочность структуры на сдвиг = ц0 М* Ес / р определяется по напряженности поля Ес, при которой наблюдается наибольшее приращение намагниченности М, т. е. максимум dM/ dE, где М* — намагниченность насыщения используемого магнитного материала и р — фактор формы магнитных частиц (для сфер р = 6).
3.16. Полимеры и растворы полимеров
3.16.1. Введение
Полимеры — это вещества, молекулы которых имеют очень большой размер (молярную массу порядка нескольких тысяч кг/кмоль и более) и состоят из многократно повторяющихся участков — звеньев. За такими молекулами закрепилось название «макромолекулы». Каждое звено макромолекулы — это фрагмент молекулы обычных (молекулярных) размеров. Например, молекула простейшего по составу и строению полимера — полиэтилена — представляет собой линейную цепь связанных метиленовых групп -СН2~. Название «полиэтилен» указывает в данном случае на технологию получения этого полимера — путем полимеризации этилена.
При полимеризации не образуется побочных продуктов химической реакции. Полимеры синтезируются также с помощью реакций поликонденсации. При этом образуется и некоторый простой побочный продукт (вода, соляная кислота и др.). Исходное вещество, участвующее в поликонденсации, должно иметь в своем составе две реакционноспособные группы, например карбоксильную (-СООН) и спиртовую (^СОН). При их взаимодействии образуется сложноэфирный мостик и выделяется вода.
В реакциях полимеризации или поликонденсации могут участвовать исходные вещества разной химической природы. Этими методами синтезируется важнейший класс полимеров — сополимеры. Именно сополимеры, благодаря большому числу возможных комбинаций исходных реагентов, дают неисчислимое множество вариантов химического состава полимеров, включая молекулы ДНК, несущие в себе наследственный код каждого представителя животного и растительного мира. Длина цепи молекул ДНК оценивается величиной порядка сантиметров. Уже из этой оценки видна проблема, которая в первую очередь требует решения при изучении полимеров, — это проблема совместимости гигантских размеров (длин) полимерных цепей и тех реальных размеров пространства, например клетки растения или живого организма, в пределах которого макромолекулы должны размещаться. Иначе говоря, фундаментальной характеристикой любой полимерной молекулы является соотношение между длиной ее цепи и фактическим, чаще всего несоизмеримо меньшим, линейным размером макромолекулы [19]. Очевидно, что при таком соотношении размеров длинные молекулы должны быть свернуты в более или менее плотные клубки. Размер клубка или более удобная
Физика, химия и технология дисперсных систем
727
для расчетов величина — расстояние между концами молекулы — является основной характеристикой ее структурного состояния. Понятие структуры полимерной молекулы не совпадает с понятием химической структуры молекул органических веществ. Последнее описывает последовательность расположения одного-двух или более одинаковых химических фрагментов молекулы вдоль ее цепи. Оно жестко зафиксировано и далее будет обозначаться термином «химическое строение молекулы». Структура же полимерной молекулы — взаимное пространственное расположение ее звеньев — непостоянна. Именно это непостоянство и придает полимерам те уникальные свойства, ради которых эти материалы создаются и изучаются. Среди выдающихся свойств полимеров как конструкционных материалов сразу следует отметить эластичность (рези-ноподобность), а среди физико-химических — способность набухать (сильно увеличивать объем) в подходящем растворителе и превращать жидкие среды в твердообразные (в гели). Полимеры обладают также уникальной по эффективности способностью стабилизировать дисперсные системы. С термином «полимер» логически связан термин «мономер», близкий по смыслу к использованному выше понятию звена полимерной цепи. В химии мономер — это низкомолекулярное вещество, молекулы которого, соединяясь в цепь, образуют макромолекулу полимера. Применительно к полиэтилену мономером является молекула этилена С2Н4, а звеном образовавшейся цепи (далее — «элементарным звеном») — группа -СНг-. В физике полимеров термины «мономер» и «звено» обычно применяются как синонимы, причем, в соответствии с той или иной теоретической моделью полимерной цепи, звеном может считаться участок цепи, состоящий из нескольких элементарных звеньев. Здесь, как и в ряде других случаев, необходимо остановиться на вопросах терминологии, а зачастую вводить и модифицировать термины, потому что современная коллоидная химия — это и химия, и физика, и механика, и технология дисперсных систем. Каждая из этих отраслей выстраивалась в значительной мере независимо одна от другой, и включение принятых в них понятий в одну общую научную дисциплину невозможно без определенной перестройки терминологии.
3.16.2. Гибкость и форма макромолекул
Молекулы полимеров, в отличие от молекул обычных органических веществ, в том числе типичных ПАВ, не имеют определенного геометрического размера. Это обусловлено гибкостью длинных молекул. Различные участки гибкой полимерной молекулы сильно переплетены между собой, так что в целом она имеет форму клубка. Вследствие теплового движения участков цепи размер и конфигурация клубка непрерывно изменяются, поэтому геометрические характеристики таких молекул (размер, объем и т. д.) являются среднестатистическими величинами. В частности, размер по
лимерной молекулы характеризуется величиной среднеквадратичного расстояния R между ее концами.
Простейшим с точки зрения математического описания является представление макромолекулы как свободно-сочлененной цепи, состоящей из М одинаковых звеньев длиной 1\ каждое. Звенья такой цепи имеют неограниченную подвижность и лишены телесности, т. е. не создают препятствий движению других звеньев и не накладывают ограничений на их расположение. Единственным ограничением является связанность всех звеньев в единую, неизменную последовательность. Среднестатистическое расстояние Rq между концами свободно-сочлененной цепи совпадает со среднеквадратичным смещением частицы (молекулы) при ее случайных (броуновских) блужданиях за А] одинаковых по длине /1 шагов:
/?0=/]А]0’5- (3.16.1)
Формула (3.16.1) является точной, если на взаимное расположение звеньев макромолекулы не накладываются упомянутые выше ограничения. Клубок, образованный свободно-сочлененной цепью, называется идеальным, или гауссовым, а представление полимерной молекулы в виде такого клубка — стандартной гауссовой моделью макромолекулы. Название модели связано с именем Гаусса, поскольку его закон нормального распределения случайных величин относится и к распределению молекул по всем возможным случайным размерам клубков со средней величиной, определяемой формулой (3.16.1).
Реальная полимерная цепь не является свободно-сочлененной, так как ее звенья так или иначе ограничены в движении. Ограничения порождаются разными причинами, в том числе непроницаемостью (телесностью) звеньев, неизменностью химического строения при любых изменениях конфигурации молекулярной цепи. Существует ряд моделей строения полимерных молекул, учитывающих наличие ограничений в подвижности их звеньев. Одни модели базируются на незыблемости некоторых свойств молекул, определенных их химическим строением. Другие игнорируют химическую природу молекул, а точнее, «прячут» ее в численное значение некоторого физического свойства макромолекулы. Таковой является персистентная модель — одна из наиболее продуктивных.
Суть ограничений, связанных с химическим строением, можно понять, изучая простейший по химическому составу полимер — полиэтилен. Его можно считать высшим членом гомологического ряда предельных углеводородов нормального строения. Химическое строение молекул полиэтилена сохраняет определенные черты строения первого члена этого ряда — метана. Как известно, все четыре атома водорода в этой молекуле равноценны, а это означает, что их пространственное положение относительно атома углерода одинаково, т. е. они равноудалены один от другого и от атома
728
Новый справочник химика и технолога
углерода. Отсюда, по правилам геометрии, следует, что атомы водорода расположены в вершинах тетраэдра, центр которого занят атомом углерода. Наглядно образованную ими фигуру можно представить себе как тетраэдрическую звезду (рис. 3.120). Угол между лучами этой звезды (между любой парой химических связей С-Н), он же — валентный угол Н-С-Н, равен приближенно 109°.
Рис. 3.120. Строение молекул метана (а), этана (б) и пропана (в). Валентный угол у (« 109°) передается без изменений по наследству следующим членам гомологичекого ряда предельных углеводородов
Существенно, что величина валентного угла почти не изменяется при замещении атома водорода метильной группой -СН3, т. е. при переходе к следующему члену гомологического ряда — этану Н3С-СН3. Иначе говоря, валентный угол С-С-Н остается равным 109°. То же самое относится к валентному углу между соседними связями С-С углеродного скелета С-С-С третьего члена ряда — пропана (рис. 3.120) и всех последующих членов этого ряда углеводородов, в том числе полиэтиленов с разной длиной цепи. Таким образом, непременной деталью химического строения полиэтилена является то, что атомы углерода в его цепи (в углеродном скелете) расположены зигзагообразно. Угол между соседними химическими связями скелета остается равным « 109°, и ни при каких условиях и воздействиях на молекулу не может быть заметно изменен без ее разрушения. Следует иметь в виду, что принятый в органической химии способ написания структурных формул подобных соединений в виде линейной цепи элементарных звеньев не отражает этой важной детали строения молекул. Приведенная выше формула С-С-С углеродного скелета пропана является примером такого упрощенного описания.
Постоянство валентных углов не означает, что углеродный скелет молекулы является жестким и что его конфигурация не может быть в дальнейшем изменена. На самом деле более правильно считать, что полимерная цепь вообще не имеет какой-либо одной определенной формы — она непрерывно изменяется под воздействием случайных факторов, каковым является, в первую очередь, тепловое движение звеньев. На любой фазе этого движения валентный угол не может изменяться, поэтому звеньям доступен только один вид движения — вращение вокруг оси О-O', совпадающей по направлению с соседней связью С-С (рис. 3.121). Вращение всех звеньев приводит к тому, что цепь изгибается и сильно запутывается, как и свободно-сочлененная цепь. Наличие определенного валентного угла
у — угла между соседними звеньями цепи — делает ее более жесткой и, следовательно, увеличивает среднее расстояние между концами цепи. Тем не менее, мгновенный снимок молекулы по-прежнему похож на изломанную в пространстве линию или на траекторию броуновского блуждания частицы (рис. 3.122), что позволяет использовать ту же формулу (3.16.1) и в этом случае, но с некоторыми дополнительными условиями.
При свободном вращении звеньев и произвольной величине валентного угла справедлива формула
R2 = (1 - cosy)/(l + cosy). (3.16.2)
При свободном вращении звеньев и тетраэдрическом угле cosy = -1/3, так что R2 =2 R^.
В действительности вращение не является свободным. Существует потенциальный барьер вращения АС/, обусловленный взаимодействием ближайших групп или атомов (заместителей) соседних звеньев полимерной цепи между собой, а также и с удаленными звеньями. При повороте одного звена цепи относительно другого на угол а расстояние между атомами и их потенциальная энергия 0(a) периодически изменяются (рис. 3.123).
Рис. 3.121. Изменение конфигурации (изгибание) молекулярной цепи путем пространственного вращения вокруг оси О-О' одной части цепи (пунктир) относительно другой. Валентный угол у на всех промежуточных стадиях вращения остается неизменным
Рис. 3.122. Макросостояние молекулярной цепи характеризуется расстоянием R между ее концами
О 120 240 а б)
Рис. 3.123. Вращение звеньев цепи вокруг связи С-С (а) требует затрат энергии. Потенциальная энергия (б) звена Ца) является периодичекой функцией угла а между равновесным положением звена (0) и его смещенным положением (1)
Физика, химия и технология дисперсных систем
729
Разность MJ максимальной и минимальной величин потенциальной энергии взаимодействия атомов и молекул, присоединенных к углеродному скелету цепи, называется потенциальным барьером вращения и является энергетической характеристикой жесткости полимерной цепи. Чем больше потенциальный барьер, тем меньше вероятность w его преодоления, т. е. поворота звена в новое энергетически выгодное положение.
Вероятность преодоления потенциального барьера в результате тепловых колебаний звена экспоненциально зависит от его величины:
w = exp (-AU/kT). (3.16.3)
При Л (7 = 0 вращение происходит свободно. Увеличение барьера или снижение температуры экспоненциально уменьшает вероятность вращательных переходов звеньев между соседними положениями с минимумом энергии, делая молекулу более жесткой и увеличивая ее размер:
^ = ^[b^Y.lt<eos«>.Y (3)64) ^1 + cosy Д 1-< cosa> )
Угловые скобки означают усреднение заключенной в них величины. В соответствии с общими принципами статистики:
2г.
J cosaexp(-l/ / kT)da.
< cosa >= ---------------. (3.16.5)
j ехр(-(/1 kT)da. о
При U = 0 эта формула дает <cosa> = 0. Это вполне ожидаемый результат, так как средняя величина косинуса по всем случайным значениям угла равна нулю. Умеренно большое значение барьера (Д1// кТ=20) при периодической зависимости потенциальной энергии от угла поворота т. е. при U= (Д1//2)(1 - cos3a) дает среднюю величину косинуса угла, равную 0,164. Это увеличивает размер молекулы всего лишь в 1,65 раза по сравнению с размером свободно-сочлененной молекулы. Однако объем клубка больше уже в 4,52 раза по сравнению с объемом свободно-сочлененной молекулы. В коллоидной химии и химической кинетике обычно не представляют интереса значения потенциальных барьеров (энергий активации процесса), большие нескольких десятков кТ. В этом смысле полимерные молекулы обладают уникальными свойствами: их размер очень сильно изменяется при увеличении безразмерной величины потенциального барьера до величин, исчисляемых сотнями и тысячами. Например, при МЛ кТ = 200 <cosa> = 0,664 и только при МЛ кТ порядка тысяч достигает предельной, близкой к единице, величины. Это означает наличие высокой чувствительности полимеров к изменению потенциального барьера или, что то же самое, к изменению температуры в такой области тем
ператур, при которой тепловое движение практически «заморожено» и никакие физико-химические процессы практически не идут. Полимерные молекулы сильно реагируют на изменение температуры и в этом диапазоне, так как незначительные угловые колебания соседних звеньев статистически суммируются по сотням и тысячам таких звеньев вдоль цепи и дают макроскопически значимые изменения состояния макромолекул.
Сильное влияние на вращательную подвижность звеньев полимерной цепи оказывает взаимодействие полимера с растворителем. В хорошем растворителе звенья цепи сольватируются, т. е. увеличиваются в размере, что создает дополнительные помехи вращению звеньев. В плохом растворителе усиливается притяжение звеньев, что также затрудняет их вращение. Универсальный метод учета подобных эффектов и основанное на нем количественное описание свойств полимеров и их растворов разработаны П. Флори [29]. Дальнейшее изложение основано на его идеях.
Размер клубка можно вычислить и по формуле того же типа, что и формула (3.16.1) для свободно-сочлененной цепи, если полимерную цепь из У] звеньев и длиной звена /1 с ограниченной подвижностью эквивалентным образом представить как цепь из меньшего числа N более длинных, но жестких сегментов с неограниченной подвижностью:
Я = со/?0 = гУ1/2. (3.16.6)
Здесь г — длина одного жесткого сегмента цепи. Каждый такой сегмент состоит из п прежних элементарных химических звеньев (мономеров) длиной Д каждое, так что r=nl\, N=Nx/n, и контурная длина молекулы L = 1Л\ = rN остается неизменной. Такая характеристика макромолекул введена Куном, Гутом и Марком и называется куновским эффективным сегментом макромолекулы. Величина со = R/ Ro характеризует кратность увеличения размера R молекулярного клубка по сравнению с размером Ro свободио-сочлененной цепи с одинаковой контурной длиной, т. е. с одинаковой молярной массой. Эта же величина используется как мера набухания полимерного клубка, например, при растворении полимера. Следует отметить, что контурная длина L является инвариантной характеристикой макромолекулы по отношению к разным моделям ее строения. Химическим эквивалентом контурной длины является молярная масса полимера.
Физической основой для введения эквивалентных параметров цепи является то, что через некоторое число п реальных звеньев ориентация звена номер (т + п) теряет какую-либо зависимость от ориентации т-го звена. Взаимная ориентация звеньев характеризуется величиной угла 0 между ними или, что более удобно, косинусом этого угла, поскольку его средняя величина <cos0> при свободном вращении звеньев обладает свойством мультипликативности:
CCOS013> = CCOS01;2> <COS02,3>, (3.16.7)
730
Новый справочник химика и технолога
где ©1,з — угол между первым и третьим звеньями, ®i,2 — угол между первым и вторым и 02,з — между вторым и третьим звеньями. Отсчет ведется от звена с произвольным номером т. Соответственно, для участка цепи, состоящего из звеньев,
<cos0(«)> = (cos0)", (3.16.8)
где Р = л - у, причем у может принимать значения от тс до тс/2. Поскольку cosp < 1, то (cosP)" стремится к 0 при увеличении п. Нулевое значение средней величины означает, что сама эта величина с равной вероятностью принимает любое значение в пределах допустимого диапазона, в данном случае от 0 до 1, а угол 0 — от 0 до л.
Одно из уникальных свойств полимеров — эластичность — можно объяснить в рамках простой гауссовой модели. Эластичность — это способность к большим обратимым деформациям. Механические свойства полимеров, как и других упругих материалов, описываются законом Гука. Однако наибольшая величина деформации, которую материал способен выдержать без разрушения, у полимеров на несколько порядков больше, чем у обычных твердых тел. Предел упругих деформаций стали или стекла составляет несколько процентов, тогда как у эластичного полимера, например каучука, он выражается сотнями процентов. В обычных материалах упругая деформация возникает в результате небольшого (на проценты) изменения межатомных расстояний и углов кристаллической решетки. Очевидно, что эластичность невозможно объяснить таким механизмом деформации. Гигантские величины обратимых деформаций полимерных веществ обусловлены тем, что при действии деформирующего усилия (например, растяжения образца) происходит распрямление молекулярных цепей, а при снятии деформирующего усилия цепи вновь сворачиваются в клубки. Сворачивание в клубки происходит не потому, что в распрямленной цепи возникли какие-либо напряжения (типа тех, что появляются в растянутой стальной пружине). Таковые просто отсутствуют. Состояние и распрямленной, и свернутой в клубок цепи механически одинаково устойчиво. Не существует сил, которые делали бы предпочтительным одно из таких состояний. Причина сворачивания цепи в клубок иная — вероятностная. Существует один способ так расположить звенья цепи, чтобы макромолекула приобрела максимально возможный размер, равный ее контурной длине rN. В то же время имеется множество вариантов (порядка 3N) такого расположения звеньев, при котором расстояние между концами макромолекулы станет равно ее среднестатистической величине R = liN1/2. Каждый из вариантов изогнутого состояния реализуется при тепловом движении звеньев с той же вероятностью (частотой), что и единственное состояние предельно вытянутой молекулы, поэтому растянутый клубок непременно перейдет в одно из многочисленных свернутых состояний под влиянием только лишь теплового движения звеньев.
В рамках гауссовой модели обратимое относительное удлинение полимерного образца может достигать величины (1{N-liNl/2)/lxNU2 х NV2, превышающей тысячи процентов. В реальности оно меньше из-за перепутанности клубков соседних молекул и звеньев одной молекулы внутри своего клубка. Это так называемые стерические эффекты или проявления объемного взаимодействия, т. е. ограничения числа возможных конфигураций цепи из-за того, что звенья занимают определенный объем, и он становится недоступным для размещения других звеньев. Корректный количественный учет стерических и других эффектов взаимодействия звеньев полимерной цепи является одной из центральных задач физики полимеров.
Вероятностный механизм эластичности может быть истолкован термодинамически. Такая возможность основана на связи между вероятностью того или иного состояния системы и ее энтропией S. Эта связь устанавливается фундаментальным законом Больцмана, раскрывающим статистический смысл энтропии:
(3.16.9)
Здесь к — постоянная Больцмана и w — термодинамическая вероятность состояния системы (макромолекулы), т. е. число микросостояний, с помощью которых реализуется данное макросостояние системы. Применительно к полимерной молекуле макросостояние — это состояние с некоторым определенным размером клубка (расстоянием между концами молекулы), а микросостояние молекулы — некоторое одно конкретное взаимное расположение ее звеньев. Как уже отмечалось, предельно вытянутому макросостоянию присуще только одно микросостояние и, следовательно, равная нулю энтропия, а статистически устойчивому макросостоянию клубка — около 3N микросостояний и энтропия порядка kN. Соответственно этому свернутые в клубки макромолекулы имеют минимальную энергию Гельмгольца A = U-TS, а растянутые — максимальную, что и определяет термодинамически выгодное состояние молекул полимера в отсутствие механических напряжений в нем. Здесь U — потенциальная энергия взаимодействия полимерных звеньев, которая, кстати, равна нулю для модели свободно-сочлененной цепи. Следует иметь в виду, что энтропия, определяемая формулой (3.16.9), представляет только ее конфигурационную часть (на одну макромолекулу). Энтропия полимерного вещества включает в себя еще и ее классическую составляющую, связанную с различными комбинациями взаимного расположения молекулярных
Часто употребляется термин «конформация» вместо «конфигурация» и, соответственно, «конформационная» энтропия. В статистической физике термином «конфигурационный» принято обозначать тот параметр состояния системы, который зависит от расположения ее элементов. Это относится и к статистике макромолекул [16].
Физика, химия и технология дисперсных систем
731
клубков. Последнее не меняется при упругих деформациях и может не учитываться. Подчеркивая доминирующую роль энтропии, говорят, что упругость полимерных материалов имеет энтропийную природу, а каждая полимерная молекула представляет собой энтропийную пружину.
Величина потенциального барьера вращения определяется химической природой атомов и молекул, связанных с углеродным скелетом полимерной цепи. Она минимальна, если это атомы водорода, и растет при их замещении более тяжелыми элементами или остатками органических молекул. В поливинилхлориде часть атомов водорода замещена на хлор, в поливиниловом спирте — на гидроксильные группы. Чем полярнее или массивнее заместители водорода, тем больше потенциальный барьер вращения и жестче полимерная цепь. Так, поливиниловый спирт — это твердое хрупкое вещество, тогда как полиэтилен — мягкий, умеренно эластичный материал.
Особого внимания заслуживают полиэлектролиты — полимеры, в которых часть заместителей водорода представляет собой остатки кислот, оснований или солей, сохраняющих способность диссоциировать. Типичным представителем полиэлектролитов является желатин. В его составе содержатся карбоксильные группы -СООН и аммонийное основание -NH3OH. В кислой среде преимущественно диссоциирует аммонийное основание, и звенья полимерной цепи приобретают положительный заряд. В щелочной среде преобладает кислотная диссоциация желатина, и полимерные цепи приобретают отрицательный заряд. В нейтральной среде (точнее, в изоэлектрической точке полиэлектролита) в равной мере диссоциируют кислотные и основные группы, макромолекула в целом электронейтральна, но вдоль цепи чередуются положительно и отрицательно заряженные звенья. Очевидно, что электрическое состояние полимерной цепи сильно влияет на ее конфигурацию: при одноименном заряде звеньев клубок разбухает вследствие электростатического отталкивания звеньев и цепь распрямляется (рис. 3.124, а). В изоэлектрическом состоянии противоположно заряженные звенья притягиваются и цепь сворачивается в плотный клубок (рис. 3.124, б). Таким образом, состояние молекул полиэлектролита в растворе регулируется концентрацией водородных ионов в среде, т. е. величиной pH раствора.
Один из наиболее распространенных типов полиэлектролитов — это полимерные кислоты. Они, как и обычные высшие органические кислоты, практически не растворимы в воде. Однако соли щелочных металлов этих кислот хорошо растворимы, поэтому возникает возможность, регулируя pH раствора (подщелачивая его) изменять свойства растворов и растворимость полимера (от нулевой величины до неограниченной растворимости).
Большое разнообразие химических факторов, влияющих на состояние макромолекул, делает актуаль
ным создание такой модели полимерной молекулы, которая могла бы всех их учесть с помощью некоторого универсального свойства молекулярной цепи.
Рис. 3.124. Разбухание молекулярного клубка (а) при диссоциации одноименных полярных групп и сжатие (б) при диссоциации разноименных групп
Этому требованию отвечает персистентная модель. Ее суть в том, что полимерная молекула представляется в виде однородной истинно упругой тонкой нити. Под «истинной» упругостью подразумевается способность сопротивляться любым деформациям изгиба, т. е. в отсутствие деформирующих усилий истинно упругая нить имеет форму прямой линии. Применимость персистентной модели к реальным макромолекулам обусловлена тем, что в большом по сравнению с размером элементарного звена масштабе не видны детали химической структуры цепи, и длинная молекула полимера действительно выглядит как однородная нить. Природа упругости нити не имеет при этом значения. На молекулярном уровне она может объясняться потерей вращательной подвижности звеньев из-за большой величины потенциального барьера вращения, что иллюстрируется приведенной выше численной величиной размера клубка в комментарии к формулам (3.16.4), (3.16.5). На упругую нить похожа и двойная спираль молекулы ДНК. При больших масштабах ее спиральная структура незаметна, и она представляется в виде сплошной упругой нити. Сопротивление изгибу таких молекул имеет ту же природу, что и у обычных твердых веществ. Небольшой изгиб, обусловленный, например, малым изменением валентных углов между соседними звеньями цепи, складывается при большом числе звеньев и превращает молекулу в упругую гибкую нить. Свойства такой макромолекулы вполне аналогичны свойствам длинного запутанного мотка стальной проволоки. Так, если закрепить один конец мотка, то второй можно перемещать в любом направлении почти без усилий на расстояния порядка диаметра клубка. Иначе говоря, концы достаточно длинного участка упругой нити могут ориентироваться один относительно другого произвольным образом и изменять свою ориентацию практически без приложения внешних усилий, т. е. в результате своего теплового движения.
Минимальная длина 1р участка цепи, на котором ориентация одного конца участка перестает зависеть от ориентации другого, называется персистентной длиной цепи. Грубо говоря, персистентная длина — это минимальная длина участка цепи, который можно согнуть на 180°, почти не прилагая усилия (рис. 3.125), т. е. только за счет тепловых флуктуаций формы цепи. С математической точки зрения более удобно иное количественное
732
Новый справочник химика и технолога
определение этой величины. Оно основано на соотношении (3.16.7), которое применимо и к изотропной упругой нити. Изотропность означает, что сопротивление изгибу не зависит от направления изгиба. Цепь со свободным вращением обладает изотропной упругостью энтропийной природы при достаточно большом расстоянии между концами изгибаемого участка цепи.
Рис. 3.125. Персистентная длина (1р) и длина (г) сегмента Куна гибко-упругой нити
Мультипликативными свойствами обладает экспоненциальная функция, т. е. ехр(ц + Ь) = ехраехр/?. Из сопоставления этого выражения с формулой (3.16.7) следует, что за счет тепловых флуктуаций цепи косинус угла между концевыми участками цепи нарастает экспоненциально с увеличением длины / участка:
<cos0(/)> = ехр(-/ / 1р).
(3.16.10)
Персистентной длиной («экспоненциальной») считается расстояние 1Р вдоль цепи, на котором средняя величина косинуса угла между направлениями начала и конца участка убывает в е раз. Иначе говоря, это расстояние, на котором среднее значение угла достигает величины, равной arccos(l/e) ~ 80°. Тогда участки цепи, расположенные на расстояниях порядка 2-3 персистентных длин, оказываются практически независимыми в отношении их взаимной ориентации. Но это означает, что поведение упругой нити становится таким же, как поведение свободно-сочлененной цепи, и что размер и другие свойства клубка из длинных упругих нитей описываются теми же формулами (3.16.1), (3.16.6). Различие лишь в том, что вместо реальных значений длины /1 и числа элементарных звеньев W| или эффективных звеньев Куна макромолекулу следует представить в виде последовательности прямолинейных жестких участков (сегментов) длиной порядка г, соединенных в свободно-сочлененную цепь (рис. 3.125). Достоинство куновского сегмента как характеристики макромолекулы в том, что он является экспериментально определяемой величиной. Действительно, контурная длина L молекулы (молярная масса полимера) считается известной, а размер клубка R можно найти экспериментально, например по интенсивности рассеяния света или по вязкости раствора полимера. Тогда г вычисляется по формуле (3.16.6). Однако длина куновского сегмента не имеет четкого физического смысла. Например, контурная длина цепи L не равна произведению длины куновского сегмента на их число (rN). Она равна произведе
нию длины дуги плавно изогнутой макромолекулы на число сегментов Куна, если куновские сегменты являются хордами, соединяющими начало и конец соседних изогнутых участков цепи. Можно настаивать и на том, что контурная длина молекулы просто равна сумме длин ее куновских сегментов. Исходя именно из этого, можно определять число эффективных сегментов в упругой гибкой цепи. Однако такое произвольное разбиение плавно изгибающейся молекулярной цепи на прямолинейные жесткие сегменты выглядит сомнительным с позиций физики и химии макромолекул.
Понятно, что прямая, соединяющая концы изогнутого участка цепи, короче его контурной длины, и поэтому длина эффективного жесткого сегмента отличается от персистентной длины (рис. 3.125). Соотношение между ними зависит от степени изгиба участка цепи и не описывается какой-либо простой формулой. В сущности, персистентная длина и длина куновского сегмента применяются параллельно и независимо как две разные величины, характеризующие гибкость макромолекулы. Следует напомнить, что персистентная длина имеет четкий физический смысл, что важно для построения теории полимерных веществ, а длина куновского сегмента доступна экспериментальному определению. Количественная же разница этих альтернативных характеристик гибкости полимерных цепей невелика и практически не зависит от химической природы полимера. Размер куновского сегмента приблизительно в 2 раза больше «экспоненциальной» персистентной длины и меньше показанной на рис. 3.125 180-градусной длины.
Используя гибкость как физико-химическую характеристику полимерных молекул, следует иметь в виду, что это понятие заимствовано из механики и обязывает нас описывать поведение макромолекул в терминах и понятиях механики упругих деформаций (изгиба). Основой такого описания является обобщенный закон Гука: напряжение пропорционально деформации, а коэффициент пропорциональности — модуль упругости (в данном случае на изгиб) — и является характеристикой деформируемого материала (или тела). Следует отметить, что пропорциональность напряжения и деформации имеет место только при небольших деформациях материала. Они могут суммироваться и приводить к большим деформациям тела. Различие в понятиях «материал» и «тело» можно пояснить на примере стальной упругой нити (стержня). Сталь — это материал, и он не может выдерживать больших упругих деформаций. Стержень — это тело, и его деформация может быть большой благодаря суммированию по длине стержня малых деформаций его коротких отрезков, рассматриваемых как небольшие образцы материала. Такой же подход применим и к полимерным молекулам, с той разницей, что имеет смысл говорить только о небольших участках молекулярной цепи вместо «небольших образцов материала» и о модуле упругости цепи, а не о модуле упругости материала. Разумеется,
Физика, химия и технология дисперсных систем
733
что деформации этих малых участков полимерной цепи остаются небольшими даже при больших деформациях клубков, что и обеспечивает применимость закона Гука.
В качестве меры деформированное™ должна выбираться такая величина, которая не изменяется при изменении размеров образца, в данном случае — контурной длины молекулы или контурной длины достаточно большого участка цепи. Другое требование к выбору меры деформированности продиктовано тем, что она должна быть безразмерной величиной. При деформациях сдвига, растяжения (сжатия), кручения такие требования совместимы и даже одно вытекает из другого, а в случае изгиба эти требования несовместимы. Если отдавать предпочтение независимости деформации от размера тела, то таковой становится кривизна % изогнутого участка тела (нити, стержня). Закон Гука для деформации изгиба длинной упругой нити (стержня) может быть тогда записан в виде уравнения
F=GrX. (3.16.11)
Здесь Gr — модуль упругости нити с размерностью энергии (нити, а не материала, из которого она изготовлена!) и F — сила, приложенная к концу деформируемого участка. Согласно этому закону, сила, обеспечивающая заданную величину деформации, не зависит от длины нити. Может показаться, что такая закономерность противоречит интуитивно угадываемой зависимости: чем длиннее упругий стержень, тем легче он поддается деформации. В действительности противоречия здесь нет. Интуитивные представления возникают на основе восприятия деформации как величины отклонения конца стержня от начального положения. Отклонение действительно растет с увеличением длины стержня, но кривизна при этом остается такой же, как и при деформации короткого стержня той же силой. Здесь и далее предполагается, что кривизна постоянна вдоль нити, т. е. при изгибе в одной плоскости нить или ее участок имеет форму дуги окружности. Следует напомнить, что кривизна % линии при ее изгибе в плоскости есть величина, обратная радиусу кривизны линии, в данном случае — радиусу R окружности, частью которой является дуга: % = 1/R. Длина 7 отрезка дуги, величина угла у между направлениями его начала и конца и радиус кривизны R связаны формулой геометрии
l = yR. (3.16.12)
При решении различных задач, связанных с гибкостью полимерных цепей, могут быть полезны некоторые геометрические соотношения для упруго изогнутой нити, приводимые ниже. Предполагается, что один конец нити (стержня) жестко закреплен в начале координат, причем координата X направлена параллельно направлению закрепленного конца, а координата Y — перпендикулярно ему в плоскости изгиба нити (рис. 3.126). Положение свободного конца нити задается его координатами х, у и углом у между направлением
свободного конца нити и осью X. Этот угол совпадает с углом между двумя радиус-векторами R, проведенными из центра кривизны (центра окружности, частью которой является линия изогнутой нити) в начало и конец нити, что непосредственно ведет к формуле (3.16.12). При этих условиях:
x = /?siny, у = /?(1 - cosy). (3.16.13)
Длина пути ds, который проходит свободный конец нити при увеличении угловой меры деформации у на величину dy, описывается очевидным образом через соответствующие приращения dxn dy координат конца, т. е. ds = (tZr2 + dy2)172, что после нахождения и подстановки производных dxldy и dyldy, дает уравнение
Л = (7/у)[1 +(1 -2cosy)/y2 - (2siny)/у]172 dp (3.16.14)
Длина пути здесь выражена через длину дуги 7, так как она остается постоянной при увеличении изгиба дуги. Расстояние 5 по прямой между концами дуги равно 2R sin(y/2) или 5 = 2(7 / y)sin(y/2).
Рис. 3.126. Деформация изгиба.
(Пояснения в тексте)
Следует иметь в виду, что в действительности форма упруго изогнутой нити отличается от формы дуги окружности [23], причем степень отличия зависит от условий на концах нити (стержня). Приведенные соотношения могут использоваться для количественных оценок свойств молекул, но их нельзя применять для аналитического (т. е. математического) исследования проблемы. Кроме того, ими, как и точными формулами, не следует пользоваться для получения соотношения между длиной куновского сегмента и персистентной длиной участка цепи, поскольку последняя связана со среднестатистической величиной угла у между концами цепи.
В общем случае гибкая нить может быть изогнута одновременно в двух взаимно перпендикулярных направлениях. Важный и простой пример такого изгиба демонстрирует винтовая линия. Ее удобнее представлять как нить, намотанную с шагом В на поверхность цилиндра радиусом А. Если ось Z декартовых координат XYZ совместить с осью цилиндра и начало нити закрепить на оси х в точке х = А, у = 0, z = 0, то уравнение этой линии будет: х = 4cos<p, у = 4sinq>, z = В(р/2я.
734
Новый справочник химика и технолога
Угол ф цилиндрической системы координат отсчитывается от оси х. Радиус кривизны винтовой линии R и ее кривизна % описываются формулами
R = (A2 + B2)/A, %=1/R. (3.16.15)
Не представляет также труда вычисление расстояния 5 между двумя точками на винтовой линии. Если одна из них совпадает с началом линии, то 8 = A [4sin2(<p / 2) + (Bip / 2пА )2]1/2. Винтовые линии представляют особый интерес, поскольку именно такова форма молекул ДНК в двойной спирали.
3.16.3. Модуль упругости и персистентная длина макромолекулы
Как было показано выше, упругость полимерных клубков имеет тепловую (энтропийную) природу. Это убеждение сложилось при изучении поведения свободно-сочлененной цепи, но, как оказалось, тот же механизм действует и в случае достаточно жестких цепей, наглядно представляемых в виде тонких упругих нитей. Понятием, которое дает возможность единообразно подходить к изучению макромолекул того и другого типа, является персистентная длина участка молекулярной цепи. Важно также, что оно позволяет абстрагироваться от химической природы полимера, упаковывая ее в численное значение персистентной длины, и связать эту структурную универсальную характеристику полимерной цепи с ее механической характеристикой — модулем упругости.
Для установления связи между силой F, приложенной к свободному концу нити, и ее деформацией вернемся к обсуждавшейся выше задаче об упругой нити (стержне) длиной Z, один конец которой жестко закреплен на опоре. Если сила направлена перпендикулярно оси, то при слабом изгибе стержня [23]:
y = Fl2/3EI. (3.16.16)
Здесь Е — модуль упругости материала, из которого состоит стержень, / — момент инерции поперечного сечения стержня. Для круглого стержня радиусом а I=itaAl^. Как уже отмечалось, существует проблема выбора меры деформированное™, удовлетворяющей требованиям безразмерности и инвариантности по отношению к размерам деформируемого тела. Видно, что формула (3.16.16), взятая из весьма авторитетного источника, просто игнорирует указанные требования в силу их несовместимости и характеризует деформацию нити (стержня) ее абсолютной величиной — координатой у свободного конца стержня. Следует подчеркнуть, что это именно характеристика деформации стержня, а не материала. Между тем, инвариантная мера деформации необходима, так как без нее невозможно перейти к характеристикам вещества (материала), образующего упругую нить (стержень).
На основании того, что при малой величине деформации cosy ~ 1 -//2, вместо формулы (3.16.13) можно
написать: y = Rfl2 или, после подстановки у = //Л, у = /2/2/?или:
у=(/2/2)%, (3.16.17)
где % - 1/Z?. При подстановке этого значения у в уравнение (3.16.16) получается уравнение
F=(3£//2Z)x. (3.16.18)
Сравнивая этот результат с формулой (3.16.11), легко видеть, что модуль упругости нити (стержня) определенной длины Z равен:
G>= 3E//2Z. (3.16.19)
Величина Е является характеристикой (модулем упругости) материала, из которого изготовлен стержень. Характеристикой упругости стержня (нити), не связанной с его длиной, является величина ЗЕ//2. Термины «нить» и «стержень» используются здесь параллельно, поскольку первый в большей мере эквивалентен терминам «макромолекула» или «цепь из молекул», тогда как второй более уместен при построении механического аналога полимерной цепи, основанном на понятиях механики сплошных деформируемых сред, типа модуля упругости Е. Здесь необходимо провести некоторые численные оценки упругости нити, взяв за основу типичные свойства металла. Его модуль упругости Е равен по порядку величины 1012Н/м2. Представим отрезок длиной Z молекулярной цепи как сплошной металлический стержень с вполне реалистичным радиусом а = 1О“10 м.
Тогда / - (р/4) • 10-40 м4 и упругость цепи на единицу ес длины ЗЕ//2 будет равна приближенно 10”28Н/м2. Для оценки правдоподобности этого значения целесообразно перейти от силовой к энергетической характеристике деформации. Потенциальная энергия деформации w получается интегрированием элементарной работы dw перемещения, равной Fdy, по всему пути перемещения конца стержня у от 0 до конечного значения у. Поскольку сила выражена формулой (3.16.11) как функция кривизны отрезка цепи, то и перемещение нужно выразить через ту же величину. Согласно уравнению (3.16.17), dy = (Z2/2)Jx и dw = (ЗЕ 1 U4)xdx. После интегрирования последнего выражения получается: w = (3£/Z/8)x2. Выделение здесь модуля упругости стержня Gr=3EI 121 приводит к формуле
w = Gr (Z2/4)x2. (3.16.20)
Средняя величина потенциальной энергии, запасаемой в изогнутых фрагментах молекулярной цепи, равна средней энергии их теплового движения kT. Для отрезка цепи персистентной длины 1р среднее значение угла у между направлениями начала и конца отрезка равно, как отмечалось ранее, примерно 80° или (я/2,1) ~ 3/2. Обозначим эту величину как ур. Выразив через нее радиус кривизны Rp, длину персистентного отрезка цепи
Физика, химия и технология дисперсных систем
735
lp = ypRp и сделав в формуле (3.16.20) замены %= MRp, w = kT, можно получить выражение для расчета радиуса кривизны Rp персистентного участка цепи:
Я^ЗЕ1ур!Ш. (3.16.21)
При указанных выше численных значениях ЗЕ//2 и YpRp ~ Ю 8 м это вполне правдоподобно. Если принять для «материала», из которого «изготовлена» макромолекула, значение модуля упругости Е на порядок меньшее, чем у металла, то радиус изгиба Rp персистентного участка цепи составляет ~ 10 9 м, что, вероятно, близко к нижнему физически приемлемому значению этого параметра. Через него легко выразить длину персистентного участка:
lP = 4pRP, (3.16.22)
которая примерно в 1,5 раза больше радиуса кривизны.
В рассмотренной механической модели гибкой макромолекулы ее химическое строение и структура зашифрованы в значении условного модуля упругости «материала», из которого «состоит» молекула. Очевидно, что значение этого модуля далеко не равно значению модуля упругости реального полимера, например полиэтилена. В то же время можно ожидать совпадение условного и реального модулей упругости, если в полимерном материале все молекулярные цепи ориентировать параллельно и измерить его упругость на изгиб в направлении, перпендикулярном ориентации цепей.
Известно, что именно таким путем получают полимерные материалы, превосходящие по прочности сталь. Однако, даже при полном успехе на этом направлении, не теряет актуальности задача построения физической модели упругости цепи. Наиболее простая и наглядная физическая модель упругой цепи получается, если по ней распределены электрические заряды, что характерно для макромолекул полиэлектролита. При этом достаточно учесть только влияние заряда на свойства цепи, а ее основа — незаряженная цепь — может считаться идеальной гауссовой цепью, обладающей только упругостью энтропийной природы.
3.16.4. Гибкость молекул полиэлектролита
Пусть на отрезке цепи макромолекулы длиной / равномерно, с шагом b размещены заряды величиной q (рис. 3.127). Заряды экранированы ионной атмосферой, и радиус экранирования равен rD, причем выполняется соотношение /«//)«/. Потенциальная энергия взаимодействия любой пары зарядов = (72схр(-г/г/))/££ог определяется расстоянием г между ними по прямой. При изгибе цепи уменьшается расстояние между зарядами и увеличивается энергия их отталкивания.
Полная энергия некоторого выделенного заряда получается умножением его заряда на суммарный электрический потенциал частично экранированного поля всех зарядов цепи. Для нахождения электростатической энергии we всего отрезка цепи длиной I нужно эту вели
чину умножить на число зарядов на этом отрезке. Кроме того, из потенциальной энергии взаимодействия зарядов изогнутого отрезка цепи нужно вычесть потенциальную энергию тех же зарядов в распрямленной цепи, так как только эта разность Aw(, дает энергию изгиба. Выполнив соответствующие математические операции, находим [19]:
Ли, = (1/8) [(^„)2/ (seoi2)] (у2/Г). (3.16.23)
Поскольку электрическая энергия изгиба может быть также выражена через электрическую составляющую Ge модуля упругости цепи с помощью формулы (3.16.20), то, приравняв правые части формул (3.16.20) и (3.16.23), получим уравнение для нахождения электрической составляющей модуля упругости нити:
Ge U2/4)x2 = (1/8) [W/ W)] (У2//)-
Предварительно необходимо перейти к одинаковой мере деформации в левой и правой частях формулы, выполнив замену % = y/Z, что дает Ge/2/4 = = (1/8) [(.qrf))2 / (ss062)] I и, следовательно,
0, = (<7г„)2/(2ес„б7). (3.16.24)
В силу отмеченных ранее особенностей деформации изгиба модуль упругости зависит от длины I деформируемого участка цепи.
Персистентная длина 1р заряженной цепи получается из формулы (3.16.23), если в ней потенциальную энергию изгиба txwe приравнять энергии теплового движения kT участка изогнутой цепи, т. е. принять, что кТ = = [(^)2/(8eso^2)] ( У2 / 1р) при 1р = I и ур = у.
Рис. 3.127. Цепь заряженных звеньев длиной b в растворе электролита: rD — дебаевский радиус экранирования
Полагая, как и ранее, что ур = 3/2 и у2р ~ 2, получим:
lp = (qrrtf /(4££0Z>2 kT). (3.16.25)
Дебаевский радиус экранирования известным образом связан с концентраций с (моль/л) электролита в растворе: г2 = гг^кТ'! 2cNAq2.
Здесь NA — постоянная Авогадро. Заряд q ионов в растворе равен (по модулю) заряду звена молекулярной цепи. Подставив эти значения в формулу (3.16.25), находим персистентную длину заряженной цепи:
1Р= \/8b2cNA. (3.16.26)
736
Новый справочник химика и технолога
Попутно, из формул (3.16.24) и (3.16.25), получается модуль ее упругости:
Ge = 2kT(lp/l). (3.16.27)
При / = 1(Г8 м, с = 10“5 моль/л (это довольно чистый по ионным примесям раствор) 1Р = 10 7м, что вполне разумно. Кстати, радиус экранирования rD имеет при этих условиях тот же порядок величины. С уменьшением концентрации электролита с персистентная длина растет быстрее (как 1/с), чем радиус экранирования (он растет как 1/с0,5). Соотношение между этими величинами становится ближе к соотношению г ту» «С /, принятому при выводе формул (3.16.23)- (3.16.27). Напомним, что здесь / — произвольный участок цепи, достаточно протяженный по сравнению с персистентной длиной, в том числе во всех формулах этой главы можно принять I = L.
3.16.5. Модуль упругости полимерных материалов
Выше были рассмотрены упругие свойства молекулярных цепей, запутанных в клубки под действием хаотичных по величине и направлению импульсов теплового движения. Для описания макроскопического проявления упругости полимерного материала необходимо выяснить реакцию клубка на вполне определенную по величине и направлению силу, например растяжение клубка. Деформирующее усилие, приложенное к образцу, равномерно распределяется между всеми клубками, находящимися в сечении образца плоскостью, ориентированной перпендикулярно к направлению действия силы. В свою очередь, растягивающая сила / действующая на один клубок, передается без изменений от одного звена цепи к другому. В состоянии равновесия на концы любого жесткого звена длиной г действуют равные и противоположно направленные силы. Направление оси звена в общем случае не совпадает с направлением действующей силы и составляет с ним угол р. Поэтому на каждое звено действует момент силы ц = #sinp, стремящийся совместить ось звена с направлением действия силы (рис. 3.128). В состоянии теплового равновесия этому препятствует только тепловое движение звеньев.
Рис. 3.128. Растяжение цепи силой f эквивалентно действию момента силы fr на каждое звено цепи
Описанная выше ситуация полностью совпадает с ситуацией, создаваемой действием магнитного поля на взвесь частиц, обладающих постоянными магнитными моментами. В том и другом случае результат воздействия хорошо известен: среднее значение косинуса угла а между направлением поля (силы) и оси магнитного момента (звена) (<cosa>) равно функции Ланжевена от
аргумента frlkl\ представляющего собой отношение потенциальной энергии звена fr в поле ориентирующей силы f к энергии теплового движения звена кТ\
<cosa> = cth(fr/kT)-kT/fr. (3.16.28)
Здесь cth(x) — гиперболический котангенс аргумента x=fr/kT. Стандартное обозначение правой части этого выражения — функции Ланжевена: L(x) = cth(x) - 1/х. При малых значениях аргумента L(x) = х/3, а при больших L(x) - 1.
Применительно к звеньям цепи последнее означает, что при достаточно большом растягивающем усилии (fr / кТ 1) все звенья ориентированы параллельно направлению усилия и, следовательно, клубок растягивается в прямую цепь длиной Nr. При промежуточных значениях силы удлинение R клубка:
R = NrL(fr/kT).
(3.16.29)
При малой величине усилия (fr/kT 1) L(frlkT) =fr!3kT. Тогда:
R = (Nr2!3kT)f.
(3.16.30)
Эта формула позволяет определить модуль упругости Gq клубка и полимерного материала в целом при деформации растяжения материала в пределах выполнения закона Гука, т. е. пока деформация R растет пропорционально напряжению:
GG = 3kTiNr\
(3.16.31)
При большой величине усилия зависимость деформации от напряжения становится нелинейной — темп удлинения замедляется, оно достигает (в пределе fr/kT= оо) максимально возможной величины Nr- L и далее практически не меняется при увеличении деформирующего усилия (рис. 3.129).
Одна из особенностей полимерных материалов заключается в том, что нелинейная (негуковская) часть деформации может на порядок превышать величину линейной части деформации. В обычных твердых материалах эти виды деформации близки по величине, что обусловлено различным механизмом деформации полимеров и кристаллических материалов.
Рис. 3.129. Зависимость удлинения цепи R от величины приложенной к ее концам растягивающего усилия frlkT: N — число звеньев в цепи; г — длина звена;
кТ — энергия теплового движения звена
Физика, химия и технология дисперсных систем
737
В приведенных выше формулах величина г — это длина куновского сегмента, R означает не размер, а удлинение клубка. Гауссов клубок очень рыхлый и легко деформируется даже слабыми силами, так что его размер становится много больше размера /?0 невозмущенного клубка. Поэтому разница между удлинением клубка и его размером становится несущественной.
Сильная деформация клубка не означает, что происходит сильная деформация его персистентных участков. Последняя может оставаться очень малой. Здесь следует иметь в виду аналогию между молекулярным клубком и клубком жесткой стальной проволоки. В данном случае нет смысла искать инвариантную по отношению к его размеру меру деформации, так как именно он и является основным параметром, определяющим податливость полимерного материала обратимым деформациям.
3.16.6. Особенности растворения полимеров
Как уже отмечалось, одним из свойств, присущих исключительно полимерам, является их способность набухать в подходящем растворителе. Набухание — это увеличение объема V или массы т образца полимера за счет всасывания в образец растворителя. Это достаточно длительный процесс (часы, сутки). Если ограничить объем образца, поместив его в пористый сосуд, проницаемый только для растворителя, то возникает давление набухания. Его величина может достигать нескольких атмосфер. Мерой набухания является степень набухания X) = (V-Vq)/Vq или (т-т^)/т0. Объем и масса набухшего образца могут в десятки и сотни раз превышать их первоначальные значения. Растворение полимера становится возможным только после его достаточно сильного набухания. Что касается выбора подходящего растворителя, то он производится исключительно эмпирическим путем. Принцип «подобное растворяется в подобном» остается пока единственным ориентиром в выборе растворителей для полимеров. Понятно, что законы, определяющие связь растворимости и химического строения веществ, для полимеров известны не в большей степени, чем для обычных низкомолекулярных веществ, где ориентируются на тот же принцип подобия. Однако если известно, что полимер растворим в некотором растворителе (лучше, если в воде), то различные частности, относящиеся к растворимости в этом конкретном растворителе, могут быть представлены достаточно полно и даже исчерпывающе. В частности, применительно к водорастворимым полимерам важным является вопрос о влиянии концентрации водородных ионов (pH раствора) на растворимость и другие свойства растворов полиэлектролитов.
Кроме правила «подобное растворяется в подобном» существуют и некоторые общие принципы, которым подчиняется процесс растворения полимеров, вытекающие из законов термодинамики. Как всегда, термодинамический анализ процесса или равновесного состояния системы ничего не объясняет, а лишь кон
статирует и количественно фиксирует взаимосвязь разных параметров состояния.
Как известно, возможность и направление протекания того или иного процесса определяются изменением свободной энергии системы в этом процессе, а именно: самопроизвольно идут только те процессы, которые ведут к уменьшению свободной энергии системы. Это относится и к процессам набухания и растворения. Обычно они осуществляются в условиях постоянного давления, и поэтому ход этих процессов контролируется изменениями свободной энергии Гиббса (изобарным потенциалом):
G = H-TS. (3.16.32)
Условие растворения (набухания) сводится к тому, что приращение энергии Гиббса должно быть при этом отрицательным: Д(7 < 0 или:
ДЯ-ГДЗЧО. (3.16.33)
Формально оно в любом случае может быть достигнуто повышением температуры до величины:
Т>ЬН/№. (3.16.33а)
При отрицательном приращении энтальпии Н (экзо-термичности процесса) условие выполняется автоматически, если энтропия 5 при этом не уменьшается, поскольку температура Т в шкале Кельвина имеет только положительное значение и она в любом случае оказывается больше отрицательного значения отношения AH/&S. Однако и отрицательное изменение энтропии не исключено при растворении полярных полимеров в полярных растворителях (воде). Как было показано выше, диссоциация ионогенных групп молекулярной цепи делает ее более жесткой — увеличивает персистентную длину 1р участков цепи. Соответственно этому уменьшается число эффективных свободно-сочлененных звеньев N =Ы 1Р в цепи с контурной длиной L, ас ним — и конфигурационная энтропия полимера Sk ® kN в растворе по сравнению с энтропией в монолитном состоянии. В таком случае отношение АН/NS положительно и представляет собой некоторую критическую температуру. Растворение становится, согласно уравнению (3.16.33а), возможным только при увеличении температуры до величины большей, чем критическая. В ряде случаев такая температура недостижима из-за ограниченной термостойкости полимера или растворителя, и поэтому полимер оказывается нерастворимым.
При растворении неполярных полимеров соответственно в неполярных растворителях изменение энтальпии обычно невелико в силу того, что энергия взаимодействия звеньев цепи между собой и с растворителем одинаково мала. Для неполярных полимеров характерна высокая гибкость макромолекул. В монолите она сильно ограничена стесненностью макромолекул, а при
738
Новый справочник химика и технолога
переходе в раствор становятся доступными все возможные конфигурации макромолекул. Поэтому энтропия растворения велика и играет доминирующую роль в контроле над процессом растворения. Иначе говоря, отношение ЛИ/&S — это малая положительная величина, так что практически при любой технологически приемлемой температуре полимер будет набухать, а возможно, и растворяться.
С точки зрения термодинамики растворения плохой растворитель — это такой растворитель, который не обеспечивает сильного увеличения энтропии при растворении неполярных полимеров или заметной убыли энтальпии при растворении или набухании полярных полимеров. То и другое имеет место, если растворитель не обладает достаточно сильным сродством к полимеру, что как раз и случается, если полимер и растворитель сильно отличаются полярностью.
Набухание образца полимера не всегда переходит в его растворение. Обычно слабо набухающие полимеры не растворяются совсем. Примером может служить маслостойкая резина, из которой изготавливаются различные герметизирующие прокладки автомобильных двигателей, древесина, семена растений (гороха, фасоли). Причиной ограниченного набухания часто является наличие поперечных химических связей между соседними макромолекулами. В резинах такие связи создаются вулканизацией каучука.
Хорошо набухающий полимер растворяется хотя бы частично. Внешне растворение выглядит как уменьшение размера набухшего образца после достижения максимальной степени набухания. При частичном (ограниченном) растворении образуются два несмешиваю-щихся раствора полимера — концентрированный и разбавленный. В сущности, это хорошо известный случай ограниченной взаимной растворимости веществ, характерный для многих бинарных смесей жидкостей. Концентрированная полимерная фаза может представлять собой упругий гель, а может находиться и в состоянии более или менее вязкой жидкости. Вследствие малой величины поверхностного натяжения между двумя полимерными фазами одна из них (чаще более вязкая) легко дробится на мелкие капли. Диспергированная фаза двухфазного раствора полимера называется коацерватом.
При полном растворении полимера объем образца также начинает уменьшаться после достижения максимума, и на некоторой стадии насыщения растворителя полимером происходит смешение двух фаз с образованием однофазного раствора. Процесс растворения вместе с набуханием занимает значительное время. Это объективное свойство процессов с участием полимеров, обусловленное малой диффузионной подвижностью крупных молекул. В производственных условиях стремятся максимально сократить длительность технологического цикла, применяя интенсивное перемешивание, нагревание смеси и т. п. При этом создается видимость растворения полимера, тогда как на самом деле равно
весное состояние системы за короткое время не может быть достигнуто и она остается гетерогенной. Внешние признаки гетерогенности при этом незаметны, так как по плотности, показателям преломления и другим свойствам частицы диспергированного и разбухшего полимера мало отличаются от свойств среды. Со временем, по мере перехода такой смеси в состояние однофазного раствора, ее свойства, в частности вязкость, существенно изменяются. Непостоянство свойств во времени влечет за собой нестабильность технологического процесса и невоспроизводимость свойств получаемого продукта.
3.16.7. Качество растворителя
Статистическая теория растворов полимеров придает столь важному для физико-химии растворов полимеров понятию «качество растворителя» количественный и достаточно прозрачный физический смысл. Количественное описание качества растворителя введено в теорию П. Флори [29]. Несмотря на ряд упрощающих положений, теория Флори качественно правильно описывает свойства растворов полимеров и является базой для усовершенствования теории. Приводимое ниже изложение основ этой теории чередуется с разъяснениями некоторых необходимых для понимания теории соотношений термодинамики и статистической физики растворов [16, 19].
Основным параметром состояния разбавленного раствора полимера является размер молекулярного клубка. Приводившиеся выше данные об этом параметре в основном относятся к состоянию идеального гауссова клубка. Для выяснения влияния растворителя на состояние полимера, необходимо ввести некоторые параметры, характеризующие отклонение клубка от идеальности. В первую очередь таковым является взаимодействие звеньев цепи между собой, которое фактически предопределено выбором растворителя. Взаимодействие звеньев ведет к тому, что размер клубка становится большим или меньшим, чем размер гауссова клубка. Очевидно, что если звенья отталкиваются, то это ведет к разбуханию клубка, а если притягиваются, — то к сжатию. В теории количественно характеризуются величиной отношения со = R / RQ реального размера R клубка к гауссовому Rq и называется параметром набухания.
Это отношение линейных размеров клубков, а не их объемов, которое измеряется экспериментально и выше было определено как степень набухания в. При желании любой из этих параметров набухания можно выразить через другой, но следует иметь в виду, что экспериментально определяемая степень набухания включает в себя не только внутриклубковое набухание, которое характеризуется величиной со, но и межклуб-ковое набухание, сводящееся к увеличению ширины зазоров между соседними клубками. Даже в отсутствие межклубкового набухания 1) не равно относительному приращению объема клубка (со3 - 1), так как последнее отсчитывается от объема идеального гауссова клубка
Физика, химия и технология дисперсных систем
739
/?03 . В экспериментах по набуханию в растворителе отсчет приращения объема образца полимера ведется от объема сухого образца. В этом состоянии эффективный размер Re молекулярных клубков на порядки меньше размера гауссова клубка, так что без межклуб-кового набухания в = (R/Re)3 - 1. Эффективный размер просто равен (Иот/А^)1/3, где Vm — молярный объем полимера и Na — постоянная Авогадро. В сухом образце истинный размер молекулярных клубков близок к размеру Rq гауссова клубка. Но эти клубки сильно переплетены, так что плотность полимера оказывается близкой к плотности мономера (жидкого), т. е. составляет ~103 кг/м3, тогда как плотность гауссовых клубков ближе к плотности мономера в газообразном состоянии (~1 кг/м3).
Равновесная величина параметра набухания (о определяется из условия минимума свободной энергии F клубка, т. е. равенства нулю ее производной по размеру клубка R. Это важно, так как в уравнении, получающемся после приравнивания нулю производной, сократятся постоянные и общие коэффициенты. Поэтому энергию системы можно вычислять с точностью до постоянных и коэффициентов.
Свободная энергия клубка представляется в виде суммы ее идеальной части Fq, вычисляемой из свойств гауссова клубка, и энергии F, взаимодействия звеньев:
F=F(, + F,. (3.16.34)
В контексте рассматриваемой задачи о равновесном размере клубка Fc, представляет собой работу его растяжения или сжатия. Сила/ необходимая для растяжения клубка до заданного размера, определяется формулой (3.16.30). Тогда при интегрировании элементарной работы деформации fdR получается формула
wx = 3kTR?l2Nr2. (3.16.35)
Для нахождения свободной энергии сжатия клубка из А жестких куновских сегментов предварительно необходимо вычислить величину давления Р, которое будет создавать этот клубок, заключенный в сферическую полость диаметром D (рис. 3.130). Размер полости велик по сравнению с длиной сегмента г, но меньше размера Rq невозмущенного гауссова клубка.
Давление создают те звенья цепи, которые соприкасаются со стенками полости, и, следовательно, отыскание давления Р = nkTI D3 сводится к нахождению числа п таких звеньев. Здесь п! DJ — концентрация «идеального газа звеньев» в полости объемом V= D3.
Изогнутую цепь из N звеньев можно разбить на п изогнутых субцепей, расстояние между концами которых как раз и равно D. Число звеньев 5 в субцепи связано с расстоянием между ее концами тем же законом 5 = (£>/г)2, что и для всей гауссовой цепи. Поэтому n = N(r/D)2, а давление, которое оказывает цепь на стенки сферы, описывается формулой
P=kTNr2/D5. (3.16.36)
Рис. 3.130. Гибкая полимерная цепь в полости D, ограничивающей размер молекулярного клубка
Тогда свободная энергия w = PV определяется уравнением
w2 = kT(r/D)2. (3.16.37)
Для нахождения свободной энергии взаимодействия звеньев можно воспользоваться аналогией между газом и подвижными звеньями цепи. Однако в данном случае следует использовать законы реального газа. Свободная энергия реального газа из N молекул (в случае полимера — звеньев) может быть представлена в виде так называемого вириального ряда, представляющего собой разложение функции F = F(N, с, Т...) в ряд по степеням концентрации молекул газа с:
F, = NkT + BcNkT + Cc2NkT + ... (3.16.38)
Первый член ряда — это энергия идеального газа, второй дает энергию парных взаимодействий молекул (звеньев) пропорциональную квадрату их числа cN = N2 / V при произвольном объеме V газа (макромолекулы). Третий член ряда учитывает вклад тройных столкновений частиц. Он становится существенным при больших концентрациях и должен учитываться при сжатии клубка. Постоянные В и С формулы (3.16.38) называются вириальными коэффициентами разложения (второй и третий коэффициенты соответственно, а первый равен 1) и могут быть найдены экспериментально (например, по зависимости давления газа от температуры или от концентрации газа) или на основе статистической теории реального газа. Последняя определяет величину второго вириального коэффициента с помощью уравнения
В(Г) = (1/2)/{| -ехр[-М(г)/ЛГ]}б/3г. (3.16.39)
Здесь и(г) — энергия взаимодействия пары молекул (звеньев) как функция расстояния г между ними, d3r означает, что уравнение (3.16.39) следует интегрировать по всему объему системы (клубка) или трижды интегрировать по расстоянию г. При и(г) = 0 из формулы (3.16.39) следует В = 0. Если и(г) = оо при г < а, где а — радиус молекул, т. е. последние представляются как непроницаемые шары радиусом а, то:
В ~ v, Т*>1, / = (Г-0)/0, (3.16.40)
740
Новый справочник химика и технолога
где v — объем молекулы (шара). В теории он называется исключенным объемом, так как на эту величину уменьшается пространство, доступное для размещения других молекул. Дополнительное условие Т* > 1 означает, что другими эффектами взаимодействия (кроме исключения части объема) можно пренебрегать только при достаточно высокой относительной температуре 7*. Величина © называется ©-температурой (тета-температурой), которая и является основной термодинамической характеристикой качества растворителя. Чем она больше, тем хуже растворитель, т. е. тем сильнее следует нагревать раствор для придания ему свойств, подобных взвеси частиц, взаимодействие которых сводится только к упругим столкновениям. При понижении температуры до Т* < 1 в большей или меньшей степени сказывается влияние сил притяжения, что характерно для плохого растворителя. В этом интервале температур:
В-vT*, 741. (3.16.41)
При Т-© эффекты притяжения и отталкивания звеньев компенсируются, так что второй вириальный коэффициент обращается в нуль. ©-Температура и определяется как температура, при которой притяжение и отталкивание звеньев молекулярной цепи компенсируются. Следует иметь в виду, что в хорошем растворителе второй вириальный коэффициент В равен (в расчете на одну молекулу) объему молекулы, только если ее действительно можно считать жесткой сферой. Для жестких стержнеобразных куновских сегментов это заведомо не так. Однако важно, что физический смысл величины сохраняется — это по-прежнему исключенный объем. Для стержнеобразных молекул он значительно больше их истинного объема, но, очевидно, не может превышать объема сферы с диаметром, равным длине молекулы. Таким образом, всегда имеется возможность оценить физически допустимый диапазон значений второго вириального коэффициента.
При ©, согласно формулам (3.16.35) и (3.16.38), свободная энергия описывается уравнением
F = ЗЛ77?2 / 2Nr2 + kTBN2 /Я3. (3.16.42)
Тогда dFIdR = 3kTR I Nr1 + 3kTBN21 Я4 и
R5 = BN3r2 (3.16.43)
или
/?5 = 5£3/r, (3.16.43a)
где L = Nr — контурная длина цепи.
Если ввести параметр набухания со = R / rNl/2 вместо размера клубка R, то получается формула
со5 = B7V1/2 г3. (3.16.44)
При дифференцировании выражения (3.16.42) величины В, N и г считались постоянными, поскольку рассматривалась вариация энергии клубка при его деформировании. Однако формула (3.16.44), полученная
таким путем, может быть далее использована для анализа влияния упомянутых величин на параметр набухания. Прежде всего, представляет интерес влияние химической природы полимера (гибкости макромолекул) на набухание. Гибкость (а точнее, обратная ей характеристика — жесткость молекулярной цепи) характеризуется длиной куновского сегмента г. При фиксированной контурной длине цепи L размер сегмента определяет число звеньев в цепи N = Ы г и величину второго вириального коэффициента В.
Пусть жесткий сегмент цепи представляет собой п бусинок диаметром а, плотно насаженных на стержень длиной г. Это частный случай одной из стандартных теоретических моделей макромолекулы, известной под названием «бусинки на нити». Бусинку можно считать геометрическим образом элементарного химического звена полимерной цепи (мономера). Если считать, что исключенный объем v сегмента равен среднему геометрическому истинного объема стержня ла3 и объема г3 сферы, очерчиваемой его концами, то v = (го2г3)1'2 = = аг2. Здесь учтено, что п = ria. Так как В = v, то справедлива формула
В=аг\ (3.16.45)
При проведении замен, аналогичных выполненным в уравнении (3.16.43), уравнение (3.16.45) преобразуется в формулу
со5 = а£1/2/г3/2. (3.16.46)
Значение г может меняться от г = а для самой гибкой цепи (неполярного полимера в неполярном растворителе) до г - L в самом жестком полимере, молекула которого подобна длинному жесткому стержню. Соответственно этому параметр набухания изменяется от (£/а)1/2 до (alL). То, что (£/а)1/2 » (а/£), не означает, что в первом случае (предельно гибких молекул) полимерные клубки разбухают сильнее, чем во втором случае (предельно жестких молекул). Все дело в непригодности параметра набухания со для сравнения свойств полимеров разной химической природы и для анализа влияния температуры на набухание. То и другое изменяет размер г жесткого сегмента, а следовательно, и размер Rq = (£г)1/2 стандартного гауссова клубка, от которого ведется отсчет параметра набухания со = 7?//?<>• Больший интерес представляет абсолютная величина набухания, т. е. просто размер R = со7?о набухшего клубка, который доступен прямым измерениям. Его величина определяется непосредственно формулой (3.16.43а), которая после замены В по формуле (3.16.45) дает:
Л5 = аг£3. (3.16.47)
При г = а
7? = £(а/£)2/5, (3.16.48)
а при г = £
7? = £ (а/£)1/5. (3.16.49)
Физика, химия и технология дисперсных систем
741
Из сравнения рассмотренных предельных случаев следует, что размер клубка растет с увеличением жесткости молекулярной цепи, так как (a/Z),/5 > (a/Z)275.
Формула (3.16.49) дает повод для постановки вопроса: почему линейный размер R жесткой стержневидной молекулы оказался существенно меньше ее истинного размера L. Это несоответствие результата нашим геометрическим представлениям связоно с тем, что формула (3.16.45) вводит во все уравнения среднестатистический объем звена v = аг2 вместо его геометрического объема v = га2. Полученное значение размера R стержня с фактической длиной L является эффективным в том смысле, что оно соответствует среднестатистическому (исключенному) объему длинной молекулы, который равен Л3, и является средним между 1? и Zo2.
Согласно формуле (3.16.46), при г -> Z значение параметра набухания со5 —► а/L, т. е. стремится практически к нулю и выходит за пределы изначально предполагавшегося (со > 1) диапазона. При со < 1 следовало бы, согласно уравнению (3.16.41), раскрыть значение второго вириального коэффициента по формуле В = аг^Т* вместо формулы (3.16.45). Это просто добавит во все последующие выражения дополнительный множитель, который частично учтет влияние температуры на коэффициент набухания. В остальном приведенные зависимости размера разбухшего клубка от гибкости макромолекул сохранятся. Необходимо подчеркнуть, что введение в формулы относительной температуры Т* не может полностью раскрыть ее влияние на размеры клубка, так как она более сложно, чем это предписано формулой (3.16.41), влияет на состояние клубка. С повышением температуры, прежде всего, меняется длина ~ 2
куновского сегмента г, а за ней и величины v = га и ^0 = (Zr)1/2.
3.16.8. Вязкость растворов полимеров
Роль полимера во многих случаях сводится к загущению различного рода составов на основе жидкостей. Большая вязкость является одной из самых приметных особенностей полимерных растворов. Объясняется она рассмотренным выше набуханием полимерных клубков. При типичном числе элементарных звеньев в макромолекуле 104 и размере звена 3 • 1(Г10м объем клубка в стандартной гауссовой модели составит 2,7 • 10~23 м, и тогда 1 г полимера (1018-1019 макромолекул) займет в растворе 27-270 см3. Набухание, обусловленное только тем, что каждое звено занимает некоторый (исключенный) объем, увеличит объем клубков еще в 2-3 раза, и тогда на размещение 1 г полимера в растворе потребуется не менее 100 см3 растворителя. При этом свободного растворителя почти не остается — он впитается при набухании внутрь клубков. Иначе говоря, при концентрации полимера в растворе порядка 1 масс. % доля объема, которую занимают в растворе его клубки, близка к единице, что ведет к очень сильному увеличению вязкости и даже к полной потере текучести — превращению раствора в гель.
Реологические свойства полимерных растворов во многом сходны с соответствующими свойствами коллоидных растворов. Они изложены в подразделах 3.12-3.14, посвященных структуре и реологии дисперсных систем. Здесь рассмотрены наиболее важные специфические вопросы реологии растворов полимеров и использованы при этом понятия и соотношения, введенные в упомянутых подразделах.
Вискозиметрия является универсальным и доступным методом изучения состояния макромолекул в растворе. Если ставится задача такого типа, например с целью подбора наилучшего растворителя или определения молярной массы полимера, то самым надежным способом исключить всевозможные вторичные эффекты (типа структурирования) является исследование разбавленных растворов. Критерии разбавленности полимерных растворов могут сильно отличаться от критериев для растворов низкомолекулярных веществ. Приведенная выше количественная оценка показала, что концентрация 1 масс. % может оказаться очень большой с точки зрения ее влияния на вязкость растворов. Заранее критерии разбавленности обычно неизвестны. С другой стороны, работа с сильно разбавленными растворами не обеспечивает требуемой точности измерения вклада полимера в вязкость раствора. По этим причинам практически во всех случаях необходимо исследовать концентрационную зависимость вязкости растворов (находить изотермы вязкости) и затем определять значение констант уравнения изотермы при минимальной концентрации путем экстраполяции изотермы к нулевой концентрации полимера. Отсюда следует, что, во-первых, необходимо располагать уравнением изотермы вязкости и, во-вторых, коэффициенты этого уравнения должны иметь определенный физический смысл, делающий их значения пригодными для суждения о состоянии полимера в растворе. Таковым является уравнение Эйнштейна:
Л = Ло (1 + <*<Р)- (3.16.50)
Здесь <р — объемная доля, занятая в растворе клубками полимерных молекул, а — коэффициент формы, равный 2,5 при сферической форме клубков (частиц). По поводу применения к полимерным растворам формулы Эйнштейна, относящейся к вязкости коллоидных растворов, следует заметить, что основанием для этого могла бы служить непроницаемость молекулярных клубков для потоков жидкой среды. Иначе говоря, должны быть основания считать, что в гидродинамическом отношении полимерные клубки ведут себя как сплошные тела, несмотря на то, что, как было показано выше, они могут на 99 % состоять из жидкой среды. Похожая проблема рассматривалась применительно к рыхлым флокулам коагулята, когда было показано, что она решается положительно в смысле наличия упомянутых оснований. Определяющее значение при этом имели фрактальные свойства флокул. Классическим
742
Новый справочник химика и технолога
примером фрактальных объектов является траектория броуновского движения частиц. Гауссова модель клубка построена на аналогии клубков и броуновских траекторий, т. е. конфигурация макромолекулы и вид траектории броуновского движения частицы похожи. Поэтому макромолекулы также являются фрактальными объектами. Формула (3.16.1) является частным случаем основного соотношения фрактальной геометрии флокул:
7У=(Я/г)ф. (3.16.51)
Оно связывает размер R флокул и число N частиц в ней. Если положить фрактальную размерность ф = 2, то из него следует формула (3.16.1), определяющая размер R свободно-сочлененной цепи. Таким образом, свободно-сочлененной полимерной цепи приписывается фрактальная размерность ф = 2. При ограничении подвижности звеньев размер клубков увеличивается. В рамках гауссовой модели это учитывается путем увеличения размера звена (куновского сегмента), а в рамках фрактальной модели — уменьшением фрактальной размерности при неизменной длине звена. В любом случае фрактальные, или гауссовы, полимерные клубки являются гидродинамическими аналогами фрактальных флокул и на них распространяются вытекающие отсюда результаты, включая доказательство их непроницаемости для потоков жидкой среды. Подчеркивая это свойство молекулярных клубков, уместно величину ф в уравнении (3.16.50) называть гидродинамической объемной долей полимера в растворе.
Второе важное обстоятельство, связанное с применением формулы (3.16.50) к полимерным растворам, заключается в том, что, в отличие от коллоидного раствора, знания рецептуры полимерного раствора, т. е. его массовой концентрации у, недостаточно для вычисления его гидродинамической объемной доли ф в растворе. В связи с этим в вискозиметрии полимеров в формуле Эйнштейна вынужденно используется массовая концентрация вместо объемной доли полимера:
П = По(1 + *Т), (3.16.50а)
где К = од! и тЗ — удельный (на единицу массы) объем полимерных клубков в растворе. В начале этой главы и была проведена оценка величины тЗ (на 1 г полимера). В гипотетическом случае, когда набухание является исключительно межклубковым, растворение не сопровождается увеличением размера клубков, и тЗ = 1/р, т. е. обратной плотности р полимера.
Вискозиметрическое определение константы К сводится к экстраполяции зависимости удельной вязкости 01 ~ ЛоУЛоУ от концентрации полимера у к нулевой величине концентрации:
К = Пт(т|-т|0)/П0У • (3.16.52)
у^О
Найденную таким путем величину К традиционно обозначают символом [ц] и называют характеристиче
ской вязкостью полимера. Такая терминология и символика являются наследием тех времен, когда еще не была в полной мере осознана связь измеряемых величин с формулой Эйнштейна. Величина характеристической вязкости связана с молярной массой полимера М формулой Куна:
[ц] = (ЖЕ. (3.16.53)
Здесь Q и е — эмпирические константы, определяемые по вязкости растворов полимеров с известными молярными массами. Показатель степени обычно составляет десятые доли единицы, а константа Q — порядка 10-4. Их физический смысл и связь со строением полимера можно установить исходя из отмеченного выше тождества [ц] = К и стандартной гауссовой (или более общей — фрактальной) модели молекулярного клубка.
Согласно уравнению (3.16.51), размер клубка R = г.'¥1/ф. Тогда объем одного клубка /?3 = г3\¥3/ф, а объем всех п клубков, присутствующих в единице объема раствора, т. е. гидродинамическая объемная доля ф полимера в растворе, составляет nr3N3\ причем п = (уЛ^ / М). В качестве основных характеристик молекулярной цепи введем молярную массу т элементарного химического звена цепи и молярную массу Мк ее куновского сегмента. Для простоты элементарное звено считается сферической частицей размером а. Тогда:
r = (MK/m)a, N = (M/MK) и
Ф = (yNA / М)(МК / m)3a3(MI MKf*.
Выполнив приведение подобных величин и приняв во внимание, что величина (т/ a3NA) есть не что иное, как плотность полимерного вещества р, получим:
ф = (у / р)(Мк / т)2(М/ Мк). (3.16.54)
Здесь величины сгруппированы так, что они образуют безразмерные комплексы. Подставив полученное выражение в формулу Эйнштейна (3.16.50) и приняв во внимание формулы (3.16.50а) и (3.16.52), находим:
[Л] = (а/р)(А/к/т)2 . (3.16.55)
Приведя этот результат к виду, аналогичному формуле Куна (3.16.53):
[ц] = (аЛ/3(ф-1)/ф / pw2) М(3-ф)/ф, (3.16.55а)
находим, что в формуле (3.16.53):
Q = аЛ/3(ф"1)/ф /рт2, £ = (3-ф)/ф. (3.16.56)
Полученные соотношения содержат две неопределенные величины и ф. В связи с этим распорядиться полученным результатом можно по-разному. 1) Если
Физика, химия и технология дисперсных систем
743
исходить из модели клубка как свернутой свободно-сочлененной гауссовой цепи, то можно принять ф = 2, т. е. е = 0,5, и величина Мк останется единственным подгоночным (эмпирическим) параметром, который может быть найден при сопоставлении расчетной и экспериментальной величин [т|] при известной молярной массе полимера. 2) Если считать, что цепь состоит из элементарных звеньев, т. е. принять, что Мк = да, то в этом случае остается неопределенной величина фрактальной размерности ф полимерного клубка, которую также можно определить экспериментально при известной молярной массе полимера. Для относительно простых полимеров да ~ 20 кг/кмоль, р ~ 1000 кг/м3. При среднем значении ф = 1,5 между ф = 1 (абсолютно жесткая цепь) и ф = 2 (абсолютно гибкая цепь) получим Q—10-4 и е=1. Экспериментальные данные обычно дают значение показателя степени е, промежуточное между 0,5 и 1. Таким образом, с помощью полученных выражений можно правильно оценить интервал возможных значений констант формулы Куна.
Значения параметров Мк/т=\ и ф = 2 соответствуют предельно гибкому состоянию макромолекул. Вязкость при этом минимальна, так как любое увеличение жесткости (уменьшение ф) означает увеличение размера клубков и вязкости соответственно. Однако типичны ситуации, при которых фактическая вязкость раствора оказывается существенно меньше, чем ее минимально возможное теоретическое значение, вычисленное при указанном выше значении параметров. Это, очевидно, означает, что макромолекулы свернуты в клубки, размер которых меньше размера неограниченно (а тем более ограниченно) гибких молекул. Физическая природа уплотнения клубков — аттракционное взаимодействие звеньев цепи. На практике это взаимодействие описывается разными способами, например с помощью понятий о хорошем и плохом растворителе, с помощью представлений об электростатическом взаимодействии разноименно заряженных звеньев цепи и т. п. В рамках фрактальной модели количественным выражением аттракционного взаимодействия любой природы является увеличение фрактальной размерности полимерных цепей до значений ф > 2. В предельном случае ф = 3 плотность клубков совпадает с плотностью монолитного полимера. В физике полимеров молекулярные клубки с увеличенной плотностью принято называть глобулами, а растворы с таким состоянием полимерных молекул — глобулярными. С позиций коллоидной химии глобулярные растворы следует относить к гетерогенным системам, т. е. к коллоидным растворам.
Весьма специфические проблемы возникают при построении картины течения концентрированных растворов полимеров. Под концентрированным подразумевается раствор, в котором соседние клубки совмещаются и переплетаются. Любой участок молекулярной цепи при этом проходит сквозь тело клубка другой молекулы. Иначе говоря, произвольно изогнутая молекулярная цепь оказывается заключенной в туннель, по
вторяющий изгибы молекулярной цепи, и поэтому может двигаться только вдоль этого туннеля, как бы сильно он ни был изогнут. Картина движения молекулы напоминает движение очень длинного железнодорожного состава в сильно изогнутом туннеле. В системе прямоугольных координат на сильно изогнутых участках туннеля направление движения отдельных участков цепи оказывается противоположным (рис. 3.131) [28].
Рис. 3.131. Рептации — механизм движения полимерных цепей при течении концентрированных растворов полимеров
Аналогия с поездом не вполне отражает особенности движения макромолекул по туннелю. Вагоны поезда сцеплены жестко и могут двигаться только все сразу и с одинаковой скоростью. Участки же молекулярной цепи могут удлиняться и сокращаться благодаря гибкости цепи и поэтому могут передвигаться по очереди. Сначала перемещается один участок, растягивая следующий за ним отрезок цепи. Это перемещение продолжается до тех пор, пока сила упругого натяжения соседнего отрезка не уравновесит силу, вызывающую движение первого участка. За этим следует сокращение упруго растянутого отрезка цепи за счет подтягивания его хвостового конца к головному. Хвостовой конец тянет за собой следующий участок цепи, и таким образом по ней передается поочередное перемещение ее звеньев. Такое движение напоминает движение рептилии или червя и поэтому называется рептацией. Диаметр туннеля близок по порядку величины к длине ку-новского сегмента или к персистентной длине цепи.
Туннель, по которому происходит рептация, существует до тех пор, пока в нем находится полимерная молекула. После того как молекула выдернута из туннеля, он распадается и начинается формирование новой системы туннелей вокруг каждой макромолекулы. Очевидно, что направления и конфигурации туннелей случайны и поэтому не каждый из них имеет благоприятную для рептации форму и направление. В движении под действием некоторого деформирующего усилия в каждый момент времени участвует только часть молекул — та, которая случайным образом приобрела благоприятную конфигурацию, т. е. оказалась в туннеле подходящей формы и направления. Таким образом, поочередность движения распространяется не только на отдельные участки цепи, но и на перепутанные полимерные клубки в целом. В соответствии с этим, в рамках модели вязкоупругих жидкостей, полимер или
744
Новый справочник химика и технолога
его концентрированный раствор или гель может характеризоваться двумя и более временами релаксации. Одно из них — время проползания макромолекулы через туннель и второе — время формирования нового благоприятного туннеля. Количественное описание вязко-упругой деформации приведено в разделе «Основные понятия и законы реологии».
Молекулы полиэлектролита могут иметь заряд, который в пределах туннеля не скомпенсирован зарядом противоионов, если радиус туннеля меньше дебаевского радиуса экранирования rD. В таком случае при действии электрического поля макромолекулы полиэлектролита приобретут направленное движение к противоположно заряженному электроду. Это движение, как и движение коллоидных частиц, называется электрофорезом. Электрофорез макромолекул полиэлектролита внутри каналов неподвижной полимерной сетки (геля) называется гель-электрофорезом и является важнейшим методом разделения полиэлектролитов на фракции по их молекулярным массам. Он используется для выделения нужных фракций белков, при медицинской диагностике, идентификации личности по составу ДНК и т. д. Возможности гель-электрофореза основаны на том, что электрофоретическая подвижность макромолекул зависит от их размера, т. е. от молярной массы, контурной длины или числа элементарных звеньев в цепи. Это принципиально отличает гель-электрофорез от обычного электрофореза, поскольку скорость последнего не зависит существенным образом от размера частиц или размера R заряженных клубков (при R » rD) в разбавленном растворе. Причины различия в том, что макромолекулы двигаются по извилистым туннелям в матрице геля и поэтому результирующая движущая сила электрофореза действует только на концы молекулярной цепи, тогда как сила сопротивления — на всю цепь. Поэтому чем длиннее цепь, тем меньше ее подвижность.
Электрическая сила df, - aEdl, действующая на элемент цепи длиной dl, совпадает по направлению с электрическим полем. Здесь о — заряд единицы длины цепи и £ — напряженность электрического поля. Так как движение элемента в туннеле возможно только вдоль его оси, то его скорость будет определяться проекцией силы dfe на локальное направление оси туннеля, т. е. величиной косинуса угла между направлениями поля и локального участка оси молекулы. В изогнутой цепи с равной вероятностью чередуются участки, на которых направление этой силы совпадает с направлением возможного движения цепи и на которых оно противоположно направлению движения. В итоге электрические силы, действующие на разные участки цепи, взаимно компенсируются почти полностью. Некомпенсированной останется только сила:
Fe = a££cos(££), (3.16.57)
пропорциональная расстоянию между концами цепи и косинусу угла между направлениями £ поля и радиус-
вектора R, соединяющего концы цепи. Этот результат является следствием того, что сумма (интеграл) проекций всех отрезков ломаной линии (или плавной кривой) на любое направление равен величине проекции на то же направление радиус-вектора, соединяющего концы линии. Для получения среднестатистической величины электрофоретической силы, действующей на макромолекулу в изогнутом туннеле, нужно усреднить выражение (3.16.57) по углам (ER). Упрощая задачу, заменим среднее значение произведения Rcos(ER) произведением средних величин R и cos(£/?). Первая, согласно формуле (3.16.1), пропорциональна корню квадратному из числа звеньев в цепи и равна rNm, а вторая равна Jsin х cos xdx = 0,5, так что:
<Fe> = (ог/2) £У1/2. (3.16.58)
Сила сопротивления пропорциональна с некоторым коэффициентом ц контурной длине цепи rN и линейной скорости (и) ее движения внутри туннеля:
Fh = prNu. (3.16.59)
Приравняв силы, представленные уравнениями (3.16.58) и (3.16.59), находим макроскопическую скорость движения молекул полиэлектролита:
и = aE/2pNl/2, (3.16.60)
которая равна скорости движения в туннелях, т. е. скорости выползания молекул из них. Что касается коэффициента ц, то его величину можно оценить, если рассматривать туннель как трубу с радиусом Rp, а движение содержимого этой трубы — полимерной молекулы и растворителя — как течение жидкости по трубе с той же линейной скоростью (и). Течение по трубе описывается формулой Пуазейля. Объемная скорость течения V = TtR2pu описывается формулой
К = л^Р/8пА. (3.16.61)
Здесь давление Р, так же как и объемная скорость И, выражено через площадь поперечного сечения tiR2 пуазейлевой трубы и величину силы: Р = Fh/tiR2 . Произведя указанные замены в формуле (3.16.61), получим Fh = 8ttt|£w. При сравнении этого результата с выражением (3.16.59) видно, что:
ц=8лг|. (3.16.62)
Примечательно, что коэффициент сопротивления движению молекул в туннеле не зависит от его радиуса. Окончательно вместо формулы (3.16.60) можно записать:
м = о£/16т1Г|№/2. (3.16.63)
Благодаря такой зависимости скорости гель-электрофореза макромолекул от их молярной массы через некоторое время после начала электрофореза
Физика, химия и технология дисперсных систем
745
происходит четкое разделение полимера по фракциям вдоль направления движения. Присутствие полимера в пленке геля можно визуализировать, и тогда картина распределения фракций по скоростям движения становится видимой и поддающейся визуальному и количественному сравнению с эталонными образцами.
Градация растворов полимеров по их концентрациям особо выделяет случай полуразбавленных растворов. Это растворы, в которых объемная гидродинамическая доля полимера (т. е. доля объема раствора, занятая разбухшими клубками) приближается к единице. Название «полуразбавленный раствор» подчеркивает, что концентрация собственно полимерного вещества в таком растворе может быть малой (порядка 1 масс. %), а концентрация клубков близка к 100 об. %. Раствор в таком состоянии не является структурированным в обычном смысле этого понятия, в том числе не обнаруживает свойств неньютоновских жидкостей. Специфика полуразбавленных неструктурированных растворов полимеров проявляется в виде эффекта Вайс-сенберга. Сущность эффекта обычно излагается как появление свободной поверхности жидкости необычной формы во вращающемся стакане, если в жидкость погрузить симметричный предмет на покоящейся оси, например стержень. При вращении стакана жидкость натекает на стержень, поднимается по нему и тем выше, чем больше скорость вращения. Аналогичное явление наблюдается и при вращении стержня в покоящемся стакане с жидкостью. Опыты с предметами различной формы (трубки, диски и пр.) в общих чертах дают один и тот же результат: жидкость ведет себя так, как будто она притягивается к оси вращения стакана, и тем сильнее, чем больше скорость вращения. Если удалить из жидкости погруженный в нее предмет, то ее поверхность примет обычную форму воронки, обусловленную действием центробежных сил. Таким образом, суть эффекта Вайссенберга заключается в появлении сил, действующих перпендикулярно направлению течения в сторону оси вращения, т. е. радиальных сил.
Эффект Вайссенберга не получил исчерпывающего объяснения, и поэтому о нем, как впрочем и о других пока не понятых явлениях, в учебной литературе не упоминается. В классических трудах по реологии [38] он даже не связывается явным образом с полимерными растворами, но указывается на возможную связь этого эффекта с эластичностью частиц дисперсной фазы. Теоретические вопросы реологии полимеров, в свою очередь, не затрагивают данный эффект.
В объяснении эффекта Вайссенберга необходимо обратить внимание на то, что он возникает в тех случаях, когда линии тока жидкости замкнуты (движение идет по замкнутым кольцевым траекториям). В таком случае появление радиальных сил объясняется тем, что в замкнутых линиях тока возникает натяжение, стремящееся уменьшить длину этих линий. Величина радиального давления Р выражается в таком случае через удельную (Н/м2) силу натяжения Т линий тока пример
но так же, как капиллярное давление Лапласа через поверхностное натяжение при цилиндрической кривизне поверхности:
P=^dR. (3.16.64)
Полная же аналогия имеется между давлением Р в цилиндрическом сосуде с толстыми стенками из эластичного материала и распределением напряжений Т (растяжения) в толще стенки как функции расстояния R от оси сосуда. Можно ожидать, что натяжение Т пропорционально градиенту скорости течения на данном расстоянии R от оси. Тогда задача сводится к отысканию распределения скоростей течения и коэффициента пропорциональности. Причина же возникновения натяжения линий тока представляется достаточно очевидной — это растяжение, а в пределе и распрямление молекулярных клубков под действием сдвиговых напряжений. Известно, что вытянутые частицы (клубки) преимущественно ориентируются своей длинной осью под углом к направлению течения. Далее не сложно убедиться, что растягивающая сила пропорциональна разности скоростей движения жидкой среды у концов растянутой молекулы, т. е. углу наклона ее оси к линии тока. Известно также, что ориентация частиц непостоянна, т. е. частицы вращаются в потоке. Следовательно, в той фазе вращения, когда ось растяжения молекулы совмещена с направлением линии тока, растягивающие силы не действуют и молекула получает возможность свернуться в клубок, сокращая, таким образом, тот отрезок линии тока, частью которого она является. Суммирование этих эффектов и создает макроскопическое проявление натяжения линий тока в виде эффектов Вайсенберга.
3.17. Оптические свойства дисперсных систем
Оптические, в том числе визуальные методы наблюдения являются самым доступным средством изучения, идентификации и диагностики самых разных веществ и явлений. Перечень оптических свойств и методов контроля состава веществ и происходящих в них процессов достаточно обширен. Все они в той или иной мере применимы и к дисперсным системам. Простое перечисление существующих оптических свойств и явлений принесет мало пользы, а приемлемое по полноте описание слишком далеко уведет нас от основного предмета изучения — коллоидов. Поэтому сосредоточим внимание на одном уникальном оптическом свойстве коллоидов — способности рассеивать свет [35]. Если некоторая оптическая среда рассеивает свет, то это однозначно указывает на ее коллоидную природу. Если некоторый состав является коллоидным, то он непременно должен рассеивать свет. Никакие однородные среды не обладают способностью рассеивать свет. Другие оптические явления будут упоминаться только в той мере, в какой это необходимо для понимания тех или иных аспектов светорассеяния.
746
Новый справочник химика и технолога
Прохождение света через любую среду сопровождается в принципе изменением его интенсивности I в соответствии с законом Ламберта — Бугера — Беера:
/=/оехр(-ВД. (3.17.1)
Здесь /0 — интенсивность падающего на образец вещества света, I — интенсивность прошедшего сквозь образец света, L — длина пути луча света в образце (его толщина) и Ко — коэффициент ослабления света данным веществом. Ослабление света складывается из его поглощения и рассеяния, так что коэффициент ослабления является суммой коэффициентов поглощения КА и рассеяния Кл:
K0 = KA + KR. (3.17.2)
Поглощение света заключается в преобразовании энергии электромагнитного излучения в тепловую энергию, так что результатом поглощения является нагревание освещаемого образца. Это явление универсально. Способность поглощать электромагнитные колебания присуща в принципе всем веществам, однако оно может носить ярко выраженный избирательный характер, т. е. поглощаться могут только электромагнитные колебания определенной частоты (длины волны). Спектры поглощения веществ являются, как правило, уникальными, и по ним можно распознавать вещества. Одни вещества практически не поглощают видимую часть спектра электромагнитных колебаний и поэтому представляются прозрачными. Другие поглощают излучение с любой длиной волны и выглядят как черные непрозрачные материалы. Третьи пропускают и отражают часть спектра видимого света и поэтому окрашены.
Рассеяние, как уже отмечалось, является специфическим свойством коллоидных систем. Суть этого явления заключается в том, что световая волна, попадая на коллоидную частицу, изменяет направление своего распространения, причем так, что свет от частицы начинает распространяться во все стороны, т. е. рассеивается. Причина такого поведения световой волны в том, что она, как источник переменного электрического поля, вызывает поляризацию частиц — индуцирует в них переменный (осциллирующий) дипольный момент. Ориентация наведенного диполя совпадает с ориентацией электрической компоненты световой волны, а величина и знак меняются синхронно с напряженностью и знаком электрического поля волны. Поэтому частота осцилляции наведенного диполя равна частоте падающей световой волны. По законам электродинамики, суть которых выражается уравнениями Максвелла, всякий электрический (или магнитный) осциллятор излучает в пространство электромагнитные волны. В данном случае эту функцию выполняет коллоидная частица. Частота излучаемых волн равна частоте падающего на нее света. Пространственное распределение излучения неравномерно (рис. 3.132). Его интен
сивность максимальна в плоскости, перпендикулярной оси диполя, не зависит от азимутального угла и снижается до нуля в направлении оси диполя, т. е. меняется как sin20, где 0 — угол между направлением луча и нормалью к оси диполя. Такое же распределение интенсивности излучения присуще короткой антенне радиопередатчика (длина антенны меньше длины волны). Аналогичное соотношение должно выполняться и для коллоидных частиц, т. е. их размер должен быть заметно меньше длины волны. В противном случае пространственное распределение интенсивности (диаграмма направленности) усложняется.
Описанное выше пространственное распределение излучения относится к случаю освещения частиц поляризованным светом. Электрическое поле естественного (неполяризованного) луча света может быть представлено как сумма полей электромагнитных колебаний двух волн, поляризованных во взаимно перпендикулярных направлениях.
Соответственно этому колебания вектора электрической поляризации освещаемых частиц (их дипольных моментов) также происходят одновременно в двух взаимно перпендикулярных направлениях. Обе эти компоненты осциллирующего диполя излучают электромагнитные волны одновременно. В таком случае пространственное распределение интенсивности излучения получается суммированием излучений каждой компоненты. В итоге диаграмма направленности светорассеяния становится симметричной относительно направления падения луча света (рис. 3.133). Вперед и назад излучение максимально и не поляризовано. В перпендикулярном направлении оно в два раза слабее, но полностью поляризовано. В произвольном направлении рассеивается частично поляризованный свет.
Рис. 3.132. Диаграмма рассеивания поляризованного света частицей (сфера в центре):
1 — направление падающего луча света, поляризованного в вертикальной плоскости. Передняя часть диаграммы отсечена
Рис. 3.133. Диаграмма рассеяния естественного света частицей (сфера в центре координат), свет падает на нее вдоль оси X. В плоскости YZ рассеянный свет полностью поляризован, вдоль оси X— неполяризован. Верхняя часть диаграммы (вдоль оси Z) отсечена
Физика, химия и технология дисперсных систем
747
При падении на частицу луча белого света, представляющего собой смесь световых волн с разной длиной волны, частица также будет излучать на всех длинах волн, но соотношение интенсивностей излучения на разных длинах волн в рассеянном и падающем свете изменяется. Объясняется это тем, что сильнее рассеиваются коротковолновые составляющие белого света. Эта и другие закономерности в компактном виде выражаются формулой Релея для коэффициента рассеяния света коллоидным раствором:
КЙ = (24n3nv2/V)[(и?-п2)/(п? +2л2)} (3.17.3)
Здесь п — концентрация частиц, v — объем частицы, А, — длина волны света, п\ и п0 — показатели преломления дисперсной фазы и дисперсионной среды соответственно, частицы считаются сферическими.
Напомним, что квадрат показателя преломления вещества равен высокочастотной (в области частот световых волн) диэлектрической проницаемости вещества.
Как видно из формулы Релея, рассеяние сильно, пропорционально А,4, увеличивается с уменьшением длины волны света. Это означает, что в спектре рассеяния белого света будут преобладать голубые оттенки, а проходящий без рассеяния луч света будет обогащен красно-оранжевыми компонентами. Иначе говоря, коллоидный раствор бесцветного вещества (например, оксида алюминия, стекла, полистирола) при боковом освещении будет иметь голубоватую окраску, а на просвет — красноватую, т. е. цвет коллоидного раствора зависит от направления падающего света и угла наблюдения. Голубой цвет безоблачного неба днем и оранжевая окраска солнечного диска и луны на их восходе или закате объясняется усиленным рассеянием синеголубой части солнечного света в атмосфере Земли. Если бы земная атмосфера не рассеивала свет, то небо над землей было бы абсолютно черным, а привычные картины земного пейзажа изменились бы до неузнаваемости из-за отсутствия освещения рассеянным светом. Рассеяние света атмосферой происходит на флуктуациях ее плотности. Оно очень слабое по сравнению с рассеянием на коллоидных частицах и становится заметным только при гигантском размере освещаемых объектов (всей земной атмосферы). В обычных же масштабах (сантиметры, метры) однородные среды практически не рассеивают свет и, таким образом, остается справедливым высказанное ранее положение о трм, что светорассеяние — это исключительное свойство коллоидных систем.
Голубоватое свечение (лунный свет) некоторых растворов и веществ при их боковом освещении называется опалесценцией. Опалесценция указывает на коллоидную природу таких веществ, в частности полудрагоценного камня опала, давшего название указанному оптическому явлению. Более четкое макроскопическое проявление светорассеяния можно наблюдать в виде конуса Тиидаля — светящегося конуса, возникающего
при прохождении через кювету с коллоидным раствором узкого расходящегося пучка света (рис. 3.134). Важно, чтобы на кювету при этом не падал свет от каких-либо иных источников, так как блики от стенок кюветы и хаотичное рассеяние света от других источников мешают наблюдению конуса. Если подобные условия создать в микроскопической кювете, то в микроскоп можно будет наблюдать отдельные светящиеся точки — коллоидные частицы, излучающие свет в сторону микроскопа. Их можно сосчитать и таким образом определить концентрацию частиц. Обычный оптический микроскоп, приспособленный для таких наблюдений, называется ультрамикроскопом. Раствор, предназначенный для наблюдения в ультрамикроскоп, должен быть сильно разбавлен, иначе излучение отдельных частиц сольется в сплошной светящийся фон. Примечательно, что свечение отдельных частиц хорошо заметно и в том случае, когда сами они вследствие малости размеров невидимы в микроскоп при обычном освещении.
Рис. 3.134. Конус Тиндаля
При коагуляции число частиц уменьшается, но при этом, как минимум во столько же раз, увеличивается их объем. В итоге, согласно формуле Релея (3.17.3), интенсивность светорассеяния увеличится пропорционально объему частиц (т. е. среднему числу первичных частиц в одной флокуле) который растет, как известно из законов кинетики коагуляции, пропорционально времени. В целом этот вывод подтверждается измерениями зависимости интенсивности светорассеяния от времени, прошедшего от начала коагуляции (введения электролита), а также независимыми прямыми измерениями числа частиц (флокул) в ультрамикроскоп. Имеются, однако, принципиально важные отклонения от прямой пропорциональной зависимости. Отклонения наблюдаются уже на первых этапах коагуляции, и они тем сильнее, чем дальше заходит процесс коагуляции. Интенсивность рассеяния света сильно коагулированным раствором во много раз меньше, чем это следует из формулы Релея. Тому есть ряд причин, и самая очевидная — выход размера флокул за пределы действия закона релеевского рассеяния. Крупные флокулы с размером больше длины волны рассеивают свет совсем по другим законам. В случае очень крупных частиц (флокул) действуют законы геометрической оптики, согласно которым распространение луча света регламентируется явлениями отражения и преломления света на частицах, а не его рассеянием. Однако наиболее важна другая причина нелинейной зависимости светорассеяния от размера (массы) флокул. Она заключается в том, что флокулы коагулята — это рыхлые объекты. В рамках теории Релея это обстоятельство отразится на вели
748
Новый справочник химика и технолога
чине показателя преломления Очевидно, что показатель преломления флокулы как светорассеивающего объекта существенно меньше показателя преломления монолитных частиц, поэтому они рассеивают слабее, чем компактные частицы того же размера. Независимое измерение числа частиц во флокуле и интенсивности светорассеяния позволяет вычислить показатель преломления флокул, а через него и концентрацию дисперсной фазы во флокулах, т. е. их структурную характеристику, образом с фрактальной размерностью флокул. О значимости этого параметра можно судить по тем возможностям (см. подраздел 3.13), который он открывает в построении теории структурирования, коагуляции и реологии дисперсных систем, а также и в оптике коллоидов.
Отклонение формы частиц от сферической дает ряд новых эффектов [28]. В первую очередь это дихроизм — различие в интенсивности рассеяния света при падении на частицы луча света, параллельного и перпендикулярного длинной оси частицы. Практически дихроизм можно наблюдать, если все частицы коллоидного раствора ориентировать параллельно воздействием электрического (или магнитного) поля. При достаточно большой концентрации частиц эффекты их ориентации во внешнем поле могут многократно перекрываться эффектами коагуляции под действием внешнего поля. Примечательно, что коагуляция может быть обратимой по отношению к полю, т. е. при его выключении происходит распад флокул коагулята на исходые частицы и возврат к первоначальной величине коэффициента рассеяния света (см. подраздел 3.19).
Оптические методы контроля и диагностики различных сред часто оказываются единственно доступными и непревзойденными по оперативности и быстродействию. С их помощью ведутся систематические наблюдения за состоянием атмосферы. Они незаменимы для получения информации о быстро протекающих процессах, подобных взрыву. Они незаменимы, когда нет возможности или времени для отбора проб на химический анализ среды и т. д. При этом может быть применен весь спектр оптических явлений, но рассеяние света и прозрачность среды остаются важнейшим показателем ее состояния.
3.18. Получение дисперсных систем
3.18.1. Дробление
Дисперсные системы весьма разнообразны, и столь же разнообразны способы их изготовления, однако все они используют одни и те же принципы в разных комбинациях. В первую очередь это относится к термодинамическому аспекту приготовления коллоидной системы. Исходным продуктом получения суспензий, коллоидных растворов, эмульсий, аэрозолей и других типов дисперсных систем являются, как правило, отдельные компоненты будущей системы в монолитном состоянии. Поэтому первая технологическая задача, подлежащая решению, заключается в превращении од
ного из компонентов в дисперсное состояние. Этот этап технологии всегда связан со значительными энергозатратами, которые лишь отчасти идут на увеличение поверхностной (удельной) энергии дисперсного материала 4^= о/1, где о — натяжение границы дисперсной фазы в той среде, в которой происходит диспергирование, А — удельная поверхность дисперсной фазы. Если диспергируемый материал является твердым веществом, то основная доля энергозатрат идет на его упругие деформации. Действительно, для разрушения материала нужно создать по всему его объему такое напряжение т, при котором упругая деформация материала превысит предельную для него величину ус. В свою очередь напряжение пропорционально деформации т = Су, так что удельная (на единицу объема) работа wr против упругих сил будет пропорциональна модулю упругости G и квадрату предельной величины деформации wr = Gy2 /2 или произведению предела прочности тс на предел деформации wr = тсус/2. В самом благоприятном случае разрушение частицы радиусом а произойдет по ее диаметральному сечению, т. е. площадь вновь возникшей поверхности равна ла2 и, соответственно, соотношение полезной работы (разрушения частицы) и бесполезной работы упругого деформирования будет ла2о/(2ла3Су2/3) или:
wJ/wr=3o/2aGyc2. (3.18.1)
Предел упругих деформаций твердых материалов близок по порядку величины к 0,01, модуль упругости— к 1012 Дж/м3, а натяжение — к 1 Дж/м2, так что указанное соотношение равно примерно 1 / 109а. Нет необходимости доказывать, что энергия упругой деформации после разрушения частицы теряется безвозвратно. Отсюда видно, что доля полезной работы дробления пренебрежимо мала при любом практически достижимом размере частиц. При диспергировании жидкостей вместо работы упругих деформаций появляется работа против вязких сил. Соотношение (3.18.1) сохранит при этом свою структуру, но в нем модуль упругости нужно заменить на вязкость диспергируемой жидкости (или среды), а величину предельной деформации — на некоторую критическую скорость деформации. Существование критической скорости деформации обусловлено тем, что при слишком малой скорости локального деформирования (например, вспучивания гладкой поверхности жидкости) силы поверхностного натяжения успеют сгладить возникшую неровность поверхности, и наращивания деформации до отделения капли жидкости от поверхности сплошной фазы не произойдет.
Скорость деформации границы двух флюидных (текучих) фаз есть отношение относительного приращения радиуса кривизны границы к интервалу времени, т. е. у' = daladt, и, соответственно, величина вязких напряжений равна x\daladt. Им противодействует капиллярное давление 2о / а, так что наименьшую скорость де
Физика, химия и технология дисперсных систем
749
формации межфазной границы можно оценить из условия v\da!adt > 2о / а, или:
daldt>2<slT\ или т > 2о/а или у' > 2&/х\а. (3.18.2)
Это равноценные условия, определяющие минимально приемлемый режим деформирования границы флюидных фаз. Выбор того или иного условия определяется конкретным аппаратурным оформлением процесса, т. е. тем, какой параметр (радиус кривизны, величина создаваемых напряжений или относительная скорость деформации) контролируется. С точки зрения сопоставления условий деформации твердых и жидких веществ следует выбрать последнюю форму условия и подставить в формулу (3.18.1) величину y'c=2v/v\a вместо ус. Это дает:
Зт)а/8о. (3.18.3)
Получается довольно парадоксальное соотношение, по которому работа против сил поверхностного натяжения обратно пропорциональна величине натяжения, а работа против сил вязкого трения обратна вязкости. Это неизбежное следствие того, что последняя, согласно формуле (3.18.2), пропорциональна квадрату скорости деформации, а его критическая величина пропорциональна натяжению границы фаз и обратно пропорциональна вязкости. Это сопоставление правомочно при условии равенства геометрического параметра а процесса диспергирования твердого и жидкого веществ, смысл которого несколько различен в том и другом случае.
Формулу (3.18.3) и приведенный выше комментарий к ней не следует понимать в том смысле, что процесс диспергирования облегчается с уменьшением межфазного натяжения. Работа диспергирования включает в себя как полезную (увеличение поверхности раздела фаз), так и бесполезную работу. Относительная доля последней представлена знаменателем формулы (3.18.3), так что полная работа пропорциональна сумме числителя и знаменателя этой формулы и поэтому увеличивается с увеличением межфазного натяжения и вязкости диспергируемой жидкости. Снижение натяжения способствует, как известно, диспергированию, и это относится также к дроблению твердых веществ.
Уменьшение работы диспергирования под действием поверхностно-активных веществ называется эффектом Ребиндера, или эффектом адсорбционного понижения прочности. Обычно имеется в виду понижение прочности твердых материалов. Можно было бы думать, что данное выше расширенное определение эффекта, т. е. включение в него и работы диспергирования жидкости, тривиально, поскольку из определения когезии (как меры «прочности» жидкостей) следует, что так и должно быть. На самом деле это далеко не так. Напомним, что когезия представляет собой только работу обратимого разделения образца жидкого вещества на две части с соответствующим увеличением поверхности образца. Выше было показано, что в реальном процессе
диспергирования работа совершается в основном против сил вязкого трения и что именно ее доля растет с увеличением межфазного натяжения. Соответственно и действие ПАВ отнюдь не сводится к уменьшению когезии, что и нашло свое отражение в приведенной формулировке сути эффекта адсорбционного понижения прочности. Она может быть более короткой и всеобъемлющей: прочность материала зависит от состава среды, в которой он деформируется.
Энергетический выигрыш от эффекта Ребиндера при дроблении твердых материалов значительно больше того, что можно было бы ожидать от снижения поверхностного натяжения даже до нуля. Как и при распылении жидкостей, он обусловлен снижением затрат энергии на бесполезную часть работы — упругое деформирование материала. В рамках формулы (3.18.1) это объяснить невозможно, так как ни модуль упругости, ни предел обратимых упругих деформаций не зависит от поверхностного натяжения, снижающегося в присутствии ПАВ. Наличие ПАВ в среде влияет на сопутствующие и предшествующие разрушению процессы.
Природа эффекта Ребиндера связана с тем, что при измельчении материал подвергается многократным механическим воздействиям — ударным нагрузкам. В принципе любое воздействие такого типа порождает в материале микротрещины. Их размер может быть очень малым и заключаться в разрыве всего лишь нескольких атомных связей на поверхности кристаллической решетки. После прекращения механического воздействия такие микротрещины бесследно исчезают за счет восстановления разорванных межатомных связей. Для этого есть все необходимые предпосылки — точное восстановление прежнего взаимного положения разъединенных атомов в кристаллической решетке после снятия ее упругой деформации и запас кинетической энергии (возбуждение) атомов, необходимый для преодоления потенциального барьера (энергии активации) реакции восстановления связи. Эта энергия запасается в виде энергии упругих колебаний атомов, возникающих при мгновенном разрушении их связей.
Для залечивания микротрещин важно, чтобы сближение разъединенных атомов прошло прежде, чем затухнут их колебания. Повышение интенсивности колебаний атома или прилегающей к трещине области кристаллической решетки есть не что иное, как локальное повышение температуры. Поэтому условие восстановления связи можно сформулировать в терминах температурной зависимости скорости химической реакции: сближение разъединенных атомов микротрещины должно произойти прежде, чем «остынет» участок разрушенной решетки. Основной механизм охлаждения — распространение энергии колебаний атома по кристаллической решетке. Оно идет со скоростью звука. Энергия колебаний может передаваться и в среду тем же способом. Газообразная среда имеет в несколько раз меньшую скорость распространения механических колебаний (звука) и поэтому не может заметно ускорить
750
Новый справочник химика и технолога
отвод энергии колебаний кристаллической решетки и погасить тем самым реакцию восстановления разорванной связи. Жидкая среда в этом отношении действует эффективнее, так как скорости звука в жидких и твердых материалах близки по величине. Снятие деформирующего усилия после охлаждения разрушенного участка решетки может и не привести к залечиванию трещины. Это обстоятельство оказывается решающим при многократно повторяющихся ударных нагрузках. Если от предыдущего импульса силы останется трещина, то при действии нового импульса она будет расширяться, что, в конце концов, и приводит к разрушению деформируемой частицы. Энергозатраты на дробление могут при этом снижаться многократно.
Охлаждение — не единственный и, видимо, не самый эффективный механизм сохранения микротрещин. Трещина, безусловно, не может исчезнуть, если внутрь нее проникнет хотя бы одна посторонняя молекула из газовой или жидкой среды, так как она не позволит сойтись краям трещины. Не следует думать, что при этом основную роль будет играть размер посторонней молекулы. Он, конечно, определяет угол раскрытия стенок трещины, но если посторонняя молекула не связана достаточно прочно со стенкой трещины, то она покинет трещину с той же вероятностью, с какой попала в нее, после чего края трещины могут сойтись. Таким образом, решающее значение имеет энергия связи (адсорбции) компонентов среды с поверхностью диспергируемого вещества. Надо полагать, что адсорбционная активность всех компонентов среды на стенках свежеобразованной трещины повышена.
Особая эффективность ПАВ как средства уменьшения прочности материалов при их многократном деформировании обусловлена не только тем, что они адсорбируются на стенках микротрещин и просто препятствуют их залечиванию. Основной эффект достигается благодаря тому, что адсорбционные слои ПАВ создают в трещине расклинивающее давление. Оно активно расширяет трещину, действуя как загоняемый в нее клин. Этому подобию и обязан своим рождением термин «расклинивающее давление».
Введение ПАВ в жидкую среду не всегда дает заметный эффект облегчения дробления в этой среде. Закономерности, определяющие правила подбора состава среды для достижения максимального эффекта понижения прочности, те же самые, что и правила подбора среды или адсорбента для достижения наилучшей адсорбции ПАВ: введение ПАВ дает заметный эффект в той среде, которая плохо смачивает измельчаемый материал, например при измельчении угля в воде. Если применяемая жидкость и без поверхностно-активных веществ хорошо смачивает данный материал, то она сама по себе облегчает дробление, и добавление ПАВ оказывается, как правило, бесполезным. Очень эффектно эта закономерность демонстрировалась Ребиндером: упруго изогнутая пластинка цинка с предварительно нанесенной свежей царапиной мгновенно разрушалась
при соприкосновении капли ртути с царапиной. Практически важный аналог этого опыта — хрупкое разрушение латунных деталей (стоек монтажной платы и т. д.), смоченных расплавленным припоем, при любой попытке согнуть деталь в процессе пайки. Приведенные примеры дают повод снова обратить внимание на ограниченность существующих представлений о поверхностно-активных веществах как вод о- или маслорастворимых веществах с дифильным строением молекул. Очевидно, что к средам типа расплавов металлов, солей, оксидов и других это определение не имеет никакого отношения, тогда как смачиваемость, адсорбция, пептизация, диспергирование (в том числе самопроизвольное) в таких средах оказывают решающее влияние на структуру и качество сплавов (см. подраздел 3.4.7), композитных материалов, на основные параметры надежности изделий электроники.
Впечатляющий прогресс на грани тысячелетий в области миниатюризации и повышения быстродействия электронных микросхем не в последнюю очередь обязан грамотному использованию капиллярных свойств материалов при изготовлении микросхем, основным конструктивным элементом которых являются тонкие пленки. В ряду проблем, решение которых определяет возможности миниатюризации изделий микроэлекгро-ники, находится и проблема термодинамической устойчивости тонких пленок. Щукин и Ребиндер [37] нашли условие, при котором возможно самопроизвольное диспергирование вещества (жидкости) в результате тепловых флуктуаций формы межфазной границы. В обобщенном виде оно имеет вид аА < рАТ, где о — межфазное натяжение, А — приращение площади межфазной границы при ее деформировании, р — числовой коэффициент порядка 10, к — константа Больцмана и Т — температура. Флуктуации поверхности можно представить как образование на ней лунок или выступов, имеющих форму шарового сегмента радиусом R и глубиной (высотой) h. Такую форму имеет, например, капля жидкости на твердой поверхности (см. рис. 3.14). При прогибе поверхности раздела фаз на глубину h приращение А площади поверхности равно л/г2 независимо от радиуса прогиба R, в том числе и при образовании капли радиусом R = h. При нормальной температуре и натяжении 0,1 Дж/м2 вполне вероятно возникновение флуктуационных лунок (или выступов) глубиной Ю”10 м (это размер одной молекулы), а при изменении натяжения или температуры глубина флуктуационных лунок и выступов растет пропорционально отношению 77а. Одно из следствий этой закономерности — самопроизвольное диспергирование монолитных веществ (жидкостей) при достаточно низкой величине межфазного натяжения и образование термодинамически устойчивых коллоидных растворов. Термодинамическую устойчивость можно считать следствием того, что приращение поверхностной энергии при диспергировании вещества компенсируется уменьшением свободной энергии системы за счет увеличения энтропии при уве
Физика, химия и технология дисперсных систем
751
личении концентрации частиц [37]. Устойчивость самопроизвольно образующихся коллоидных растворов можно также объяснить тем, что их коагуляция компенсируется постоянным флуктуационным диспергированием вещества, так что в сумме концентрация и размер частиц остаются неизменными.
В случае тонких пленок их целостность будет нарушаться, если глубина флуктуаций становится соизмеримой с толщиной пленки. Понятно, что пленки твердых веществ с большим поверхностным натяжением более устойчивы к их флуктуационным прорывам, однако с уменьшением толщины пленок вероятность прорыва увеличивается и, соответственно, уменьшается надежность электронных чипов.
3.18.2. Физическая и химическая конденсация
Способ получения частиц коллоидного размера альтернативный дроблению основан на конденсации вещества, находящегося первоначально в парообразном или растворенном состоянии. Конденсация, т. е. образование частиц твердого или жидкого вещества из его газообразной фазы или раствора, наступает при перенасыщении пара или раствора. Перенасыщение означает увеличение концентрации сверх той величины, которая присуща веществу при данных условиях (температура, природа растворителя). Перенасыщение может быть создано изменением физических условий (температура, давление газа, диэлектрическая проницаемость растворителя и др.), в которых находится исходная гомогенная фаза (пар, раствор), или проведением химической реакции между компонентами гомогенной фазы, при которой образуется новое вещество, являющееся нелетучим или нерастворимым при условиях проведения реакции. Если гомогенная система находится в мета-стабильном состоянии (перенасыщена, перегрета, переохлаждена), то конденсация вызывается введением зародышей новой фазы или иных центров конденсации. Примеры физической конденсации: образование тумана (взвеси капель воды в воздухе) при охлаждении влажного воздуха, образование коллоидного раствора канифоли в воде при разбавлении водой спиртового раствора канифоли, образование полуколлоидного раствора, сопровождающееся помутнением круто заваренного чая при его охлаждении, проявление треков элементарных частиц в камере Вильсона или в пузырьковой камере. Примеры химической конденсации: образование дыма (взвеси частиц сажи в воздухе) при сгорании топлива, сигнальных, маскировочных и других дымов при срабатывании пиротехнических изделий, красивые реакции образования ярко-синего раствора берлинской лазури (коллоидного раствора гексацианоферрата желе-за(Ш)) и ярко-красного раствора (коллоидного) тиоцианата железа(Ш). Во многих реакциях качественного анализа на присутствие в растворах тех или иных ионов образуются коллоидные растворы.
Образование частиц новой фазы из перенасыщенной гомогенной системы может проходить только в том
случае, когда этот процесс сопровождается уменьшением свободной энергии всей системы в целом, которая по завершении процесса становится гетерогенной. Термодинамика этого процесса рассмотрена в подразделе 3.3. Практически важные закономерности получения мелких частиц конденсацией состоят в следующем.
Конденсацией, в том числе химической, нельзя получить сколь угодно малые частицы. Минимальный размер частиц равен критическому размеру зародыша, величина которого определяется степенью перенасыщения гомогенной системы и формулой Томсона (Кельвина).
Для получения мелких частиц перенасыщение необходимо наращивать резко. В противном случае в начале процесса произойдет образование небольшого числа частиц, и далее они будут играть роль зародышей, на которых будут выкристаллизовываться дополнительные порции продукта реакции, появляющегося по мере увеличения перенасыщения. В итоге появятся крупные частицы, и спектр их размеров окажется широким. Смешивание предварительно растворенных реагентов обеспечивает достаточно быстрое достижение сильного перенасыщения. Вместе с тем следует иметь в виду, что реакция начинается до окончания смешения. Поэтому в различных местах реактора создаются разные условия протекания реакции. Особенно важную роль играет наличие избытка одного или другого реагента в разных местах реактора. Приемы, позволяющие задержать начало реакции до завершения полного смешивания реагентов, исключительно важны для управления процессом получения частиц с заданными свойствами.
Чем меньше растворимость синтезируемого вещества, тем меньше размер его частиц, поскольку степень перенасыщения раствора по отношению к этому веществу равна отношению концентрации исходных реагентов к его растворимости. Концентрация исходных реагентов имеет второстепенное значение, поскольку по порядку величины она одинакова при более или менее стандартных условиях проведения реакций.
Образовавшаяся гетерогенная система неустойчива по отношению к возможному изменению размера частиц. В полидисперсной системе мелкие частицы создают, в соответствии с формулой Томсона, перенасыщение раствора по отношению к крупным частицам. Поэтому размер последних будет постепенно увеличиваться, а мелкие частицы будут растворяться и исчезать за счет прогрессирующего роста их растворимости (явление изотермической перегонки вещества с поверхности мелких частиц на крупные). Монодисперсность лишь замедляет этот процесс, поскольку достаточно небольших флуктуаций размера частиц, чтобы этот процесс начался. Скорость изотермической перегонки может существенно измениться после отделения дисперсной фазы от маточного раствора. Это может быть просто промывка осадка от побочных растворимых продуктов реакции и избытка реагента, это может быть перенос дисперсного материала в другой растворитель
752
Новый справочник химика и технолога
(например, из водного раствора в углеводородную среду) или промывка и высушивание дисперсного материала.
Процесс синтеза частиц может быть совмещен с процессом создания на поверхности частиц защитного слоя, предотвращающего их слипание. В таком случае говорят о получении коллоидного раствора методом конденсации. Для того, чтобы при химической конденсации сразу получился устойчивый коллоидный раствор, необходимо выполнить два условия:
- брать один из реагентов в небольшом избытке по отношению к стехиометрическому соотношению. Это необходимо для того, чтобы по окончании реакции в растворе присутствовал потенциалопределяющий ион, обеспечивающий создание двойного электрического слоя на поверхности частиц (см. подраздел 3.5);
- реакцию следует проводить в разбавленных растворах. При большой концентрации исходных реагентов по окончании реакции в растворе будет большая концентрация хорошо растворимого электролита. При большой концентрации электролита двойной электрический слой имеет малую толщину и не может предотвращать слипание частиц.
Такой метод приготовления коллоидных растворов используется в учебных и некоторых исследовательских целях. Промышленного значения он не имеет, так как производственные мощности используются при этом крайне нерационально.
3.18.3. Пептизация
Пептизация — разрыхление осадка и перевод его в состояние устойчивого коллоидного раствора. Предполагается, что над осадком имеется достаточное количество жидкости, в которую и должны перейти частицы после разрыхления осадка. До пептизации система является двухфазной в коллоидно-химическом смысле. Она состоит из двух фаз: осадка и надосадочной жидкости. После пептизации дисперсная система становится в том же смысле однофазной — образует однородную взвесь частиц в жидкости (коллоидный раствор).
Пептизация как процедура подразумевает более или менее сильное механическое воздействие на двухфазную систему — встряхивание, перемешивание, дробление в мельнице. Эти воздействия приведут к желаемому результату — пептизации — только тогда, когда двухфазная система подготовлена к образованию устойчивой взвеси частиц. Подготовка заключается в оптимизации состава жидкой фазы. Состав должен соответствовать критериям, определяющим устойчивость дисперсной системы против коагуляции (см. подразделы 3.6 и 3.7). Существуют разные способы пептизации, названия которых отражают именно способ оптимизации состава жидкой фазы: адсорбционная пептизация, пептизация поверхностным растворением частиц (диссолюционная пептизация) и пептизация промывкой осадка. Выбор способа зависит от причин, по которым дисперсная фаза оказывается в осадке, а не во взвеси. Таких причин две: отсутствие в растворе компонентов, способных
создать на поверхности частиц защитные оболочки, или присутствие компонентов, способствующих коагуляции.
Адсорбционная пептизация применяется в тех случаях, когда в жидкой фазе отсутствует пептизатор — растворимый в жидкой фазе компонент, способный адсорбироваться на поверхности частиц и тем самым создавать защитную оболочку, препятствующую коагуляции частиц. В таком случае подготовка к пептизации заключается во введении в двухфазную систему пепти-затора. По отношению к системам с водной средой пеп-тизаторами являются электролиты, содержащие в своем составе потенциалопределяющие ионы. Благодаря их адсорбции на частицах образуется двойной электрический слой (ДЭС), обеспечивающий взаимное отталкивание частиц и разрыхление осадка вплоть до перехода частиц во взвешенное состояние. Этому способствует перемешивание двухфазной системы, а часто и дробление в мельницах. Обработка в мельницах практически всегда применяется при промышленном приготовлении дисперсных систем, в том числе суспензий. Для пептизации свежеосажденных осадков в лабораторных условиях бывает достаточно простого перемешивания или встряхивания взвеси.
Иногда пептизация определяется как разрыхление свежеосажденных осадков. Смысл термина «свежеосаж-денный» остается при этом неопределенным. Вероятно, имеются в виду лабораторные условия пептизации, упомянутые выше. В сущности же пептизация является основным промышленным способом приготовления дисперсных систем из порошков. В промышленности процессы производства высокодисперсных материалов и процессы приготовления различного рода дисперсных систем на основе дисперсных материалов обычно разделены. Оксид железа(Ш), оксид кремния (аэросил), карбонильное железо (высокодисперсный порошок железа, получаемый разложением карбонила железа), порошки синтетического алмаза, оксид титана и многие другие дисперсные вещества являются конечным продуктом одних фирм, а фирмы, занятые производством носителей магнитной записи, магнитных компонентов радиоэлектроники, абразивного инструмента, красок и т. д., используют готовые дисперсные материалы как исходное сырье. Самым ответственным этапом изготовления конечного продукта (например, краски) или промежуточного продукта (например, формовочной массы) на основе дисперсных материалов является пептизация — получение однородной, стабильной и хорошо воспроизводимой по свойствам взвеси дисперсного вещества в той или иной дисперсионной среде. Как правило, эта операция не обходится без более или менее длительного измельчения взвеси в растворе пепти-затора. Необходимость помола обусловлена тем, что в исходном сырье — дисперсном веществе — частицы, как правило, образуют достаточно прочные сростки, иногда очень крупные и прочные, так что без их разрушения качественная суспензия не может быть приготовлена. Образование сростков — одно из проявлений
Физика, химия и технология дисперсных систем
753
старения осадков. Поэтому и отдается предпочтение свежеосажденным осадкам при пептизации. Однако основная масса сростков формируется в процессе высушивания осадков (явление агломерации). Разрыхление агломерированных порошков облегчается за счет эффекта понижения прочности, который достигается введением ПАВ в дисперсионную среду. ПАВ выполняют и роль пептизатора. Таким образом, фактически совмещаются два основных промышленных метода получения дисперсных систем — пептизация и диспергирование. Термин «диспергирование» имеет более широкий смысл, чем термин «дробление». По смыслу более близкие к диспергированию понятия «разделение», «разрыхление», «распыление».
Пептизация методом поверхностного растворения осуществляется путем введения в систему кислоты или щелочи. Этим методом пептизируются осадки оксидных материалов (Fe2O3, А12О3, SiO2 и т. п.). Название способа основано на представлении о том, что кислоты и щелочи частично растворяют дисперсный материал, обеспечивая тем самым появление в растворе потен-циалопределяющих ионов (Fe3+, А13+ в кислой среде, А1О 2, SiO з в щелочной). Фактически же роль кислоты и щелочи может сводиться к регулированию pH жидкой фазы и, тем самым, к созданию условий для преимущественной диссоциации гидратированных поверхностных слоев оксидов по типу оснований или кислот. Конечный результат при этом тот же, что и при адсорбционной пептизации — образование двойного электрического слоя.
Промывка осадка применяется в тех случаях, когда причиной образования осадка (вместо устойчивой взвеси) является высокая концентрация коагулятора. Обычно это хорошо растворимый электролит — побочный продукт реакции получения дисперсного вещества методом химической конденсации. В промышленных условиях синтез высокодисперсных веществ этим методом проводится из концентрированных растворов, поэтому и концентрация электролита после завершения реакции оказывается большой. Промывка осадка делается с целью удаления из системы электролита. Толщина двойного слоя и его эффективность как защитной оболочки увеличивается по мере уменьшения концентрации электролита, что и приводит к пептизации. Техника промывки может быть различной: промывка на фильтре, декантация (отстаивание осадка, слив надосадочной жидкости, ее замена чистым растворителем с последующим взмучиванием осадка), отделение осадка от жидкой фазы центрифугированием, которое является разновидностью декантации. Операция замены промывочной жидкости чистым растворителем повторяется многократно до появления признаков пептизации — образования устойчивой взвеси частиц. После промывки может потребоваться введение пептизатора.
Вне зависимости от масштабов и способа пептизации следует руководствоваться правилами пептизации, которые известны как правила осадков Оствальда.
1. Концентрация пептизатора в дисперсионной среде должна быть оптимальной. Эффективность пептизации, выражаемая долей пептизированного осадка, уменьшается как при уменьшении, так и при увеличении концентрации пептизатора.
2. Осадок, постепенно вводимый в раствор пептизатора с оптимальной концентрацией пептизатора, сначала полностью пептизируется, а при его избытке перестает пептизироваться.
С точки зрения современных представлений о природе коллоидов эти правила отражают тот факт, что потенциалопределяющий электролит выступает как пептизатор при малых концентрациях и как коагулятор при высоких. Что касается второго пункта правил, то он является следствием адсорбции потенциалопределяю-щего электролита и убылью его концентрации в растворе при внесении в него большого количества осадка, т. е. сорбента. Есть и другая формулировка правила осадков. Она менее похожа на практические рекомендации и в большей мере акцентирует внимание на различии в закономерностях истинного и коллоидного растворения веществ: коллоидная растворимость (способность пептизироваться), в отличие от истинной растворимости, зависит от соотношения между количеством растворяемого вещества и растворителя и от присутствия в растворителе других растворимых веществ (пептизатора). Все упомянутые правила пептизации являются следствием универсальных закономерностей: увеличение потенциала при введении потенциалопределяюще-го электролита, сжатие ДЭС с увеличением концентрации электролита и соответствующее этому изменение баланса межчастичных сил (см. подраздел 3.7).
Далеко не всегда электролиты являются эффективными пептизаторами. В большинстве случаев, в том числе в неводных средах, удается подобрать эффективный пептизатор из ряда поверхностно-активных веществ или полимеров. Руководствоваться при этом следует также достаточно универсальными закономерностями адсорбции ПАВ на поверхности твердых веществ (см. подраздел 3.4) и зависимости свойств полимеров от их химической природы и природы растворителя (см. подразделы 3.6 и 3.16).
3.19. Феррожидкости. Получение и свойства
3.19.1. Введение
Феррожидкости (магнитные жидкости) — это техническое название концентрированных устойчивых коллоидных растворов ферромагнитных материалов. Сам факт появления такого названия говорит о том, что коллоидные ферромагнетики представляют практический интерес. Причина такого интереса понятна — феррожидкости соединяют в себе свойства двух важнейших для техники типов материалов — жидкостей и ферромагнетиков. Это позволяет создавать новые по принципам действия и назначению устройства, по-новому решать многие традиционные задачи машино- и приборостроения, технологии, медицины и т. д.
754
Новый справочник химика и технолога
С точки зрения коллоидно-химической науки уникальность феррожидкостей в том, что они дают принципиально новые возможности исследования устойчивости коллоидов, связи между силами взаимодействия частиц и свойствами дисперсных систем, закономерностей зарождения и укрупнения коллоидных частиц [8].
Феррожидкости являются на сегодняшний день самой яркой иллюстрацией возможностей коллоидной химии в области создания материалов с уникальными свойствами. Напомним, что эти возможности основаны на способности коллоидно-растворенного материала сохранять физические свойства, присущие ему в монолитном состоянии.
Приведенные выше соображения оправдывают намерение детально рассмотреть некоторые вопросы получения коллоидных растворов ферромагнитных веществ и их свойства. Некоторые параграфы этой главы можно также воспринимать просто как примеры приготовления конкретных коллоидных растворов, иллюстрирующие ранее изложенные общие положения, относящиеся к получению и стабилизации дисперсных систем.
3.19.2. Приготовление магнитных жидкостей
3.19.2.1. Выбор метода и магнитного материала
Совокупность рассмотренных ранее закономерностей (относящихся к доменной структуре дисперсных магнетиков, влиянию размера частиц на их устойчивость к оседанию и коагуляции под воздействием магнитно-дипольных сил и к технике получения частиц малых размеров) с определенностью указывает на то, что магнитные частицы феррожидкости должны иметь размер нанометрового диапазона и, следовательно, технология их получения должна основываться на конденсационных методах . Что касается выбора подходящего магнитного материала для производства магнитных жидкостей, то для этого имеется только один критерий— удобство получения частиц малого размера. И дело здесь не в том, что отдается предпочтение технологичности и доступности продукта, а в наличии ограничений принципиального характера.
Казалось бы, чем сильнее магнетик, используемый для приготовления коллоидного раствора, тем сильнее будут и магнитные свойства этого раствора при прочих равных условиях. Вся подоплека заключается именно в том, что следует подразумевать под «прочими равными условиями». Для наглядности будем иметь в виду типичные магнитные вещества: железо (намагниченность насыщения 1700 кА/м) и магнетит (намагниченность насыщения 470 кА/м). По силе (намагниченности насыщения) они отличаются почти в четыре раза. Сразу же заметим, что рекордная по силе феррожидкость —
В США первые жидкости получены Розенцвейгом (1969 г.) путем длительного (в течение 1000 ч) измельчения в мельнице магнетита в керосине с олеиновой кислотой.
коллоидный раствор магнетита в керосине с олеиновой кислотой в качестве стабилизатора — была получена в СПбГТИ(ТУ) и имела намагниченность насыщения около 90 кА/м. Это соответствует концентрации почти 20 об. % магнетита в керосине. Естественно возникает соблазн получить коллоидный раствор железа с такой же или хотя бы близкой концентрацией в надежде достичь намагниченности феррожидкости около 350 кА/м.
В феррожидкости, как и в любой другой текучей среде, энергия взаимодействия частиц должна быть порядка kT или ниже. Если частицы взаимодействуют сильнее, то это ведет к сильному структурированию взвеси и ее превращению в пасту. Без особых проблем получаются магнитные пастообразные (тиксотропные) суспензии железа с концентрацией последнего около 50 об. % и соответствующими магнитными свойствами. Однако такие системы (суспензии карбонильного железа) в магнитном поле полностью теряют текучесть — они затвердевают, оставаясь подвижными вне магнитного поля. Таким образом, если требовать, чтобы феррожидкость оставалась жидкостью в любом случае, даже в сильном магнитном поле, то энергию взаимодействия магнитных диполей т частиц в ней следует ограничить величиной порядка кТ. Это и есть то самое равное для всех жидкостей условие, которое упоминалось выше.
В сильном магнитном поле максимум (7гаах энергии дипольного притяжения частиц (см. подраздел 3.9.4) с дипольным моментом т определяется толщиной защитной оболочки 5 на их поверхности:
t/raax = 2poW2/[2(a + 5)]3. (3.19.1)
Она же ограничивает и максимально возможную концентрацию частиц птя* ~ 1/(а + J)3, а через нее и намагниченность насыщения коллоида Ms ~ тпт.Лх. Принимая, что Цпах составляет некоторую малую долю X от энергии теплового движения частиц кТ, из формулы (3.19.1) находим минимально приемлемое при таком уровне взаимодействия значение величины (а + б)3:
(а + б)3 = цош2/4ХХ7’, (3.19.2)
где магнитный момент частицы также зависит от ее радиуса: т = (4л/З)а3Л/*). Вводя эту величину в формулу (3.19.2) и решая ее относительно толщины защитного слоя, получим:
6 = («2-va)/v, (3.19.3)
Г 2~|1/3
где v = 9л.кТ/ лц0 (Л/*) , X = t/max/кТ.
Существует некоторый размер однодоменных магнитных частиц, меньше которого заданный уровень
В публикациях и каталогах Ferrofluidics Corporation качество жидкостей характеризуется индукцией насыщения. Для сопоставления с нашими данными их величины следует поделить на 4л.
Физика, химия и технология дисперсных систем
755
магнитного взаимодействия обеспечивается при их непосредственном соприкосновении, т. е. даже без защитной оболочки на поверхности частиц. При нормальной температуре и А, = 0,5 эта величина равна 1,2 нм для железа и 2,7 нм для магнетита. Более крупные частицы должны иметь защитную оболочку для предотвращения их магнитной коагуляции. Данные табл. 3.1 иллюстрируют влияние намагниченности насыщения Л/* дисперсной фазы и размера частиц а на минимально приемлемую толщину 8 оболочки, определяемую ею долю дисперсной фазы <р = а3 / (а + 8)3 и намагниченность насыщения феррожидкости Ms = q>Ms .
Таблица 3.1
Влияние размера частиц на предельно возможную концентрацию феррожидкости при % = 0,5
л/;,а/м а • 109, м 5 • 109, м (а+ 5) 109, м Ф Л/5, А/м
470000 4,15 1,94 6,09 0,32 148000
470000 4,4 2,43 6,83 0,27 125000
470000 4,66 3,02 7,68 0,22 105000
1700000 2,24 1,94 4,18 0,15 261000
1700000 2,35 2,25 4,6 0,13 226000
1700000 2,59 3 5,59 0,10 169000
Защитная оболочка предназначена и для предотвращения обычной коагуляции частиц, обусловленной действием поверхностных сил, и многое определяется ее эффективностью именно в этом аспекте. Типичным представителем эффективных стабилизаторов является олеиновая кислота в неполярных средах и олеат натрия— в водных феррожидкостях. Олеиновая кислота образует в углеводородной среде защитную оболочку толщиной около 2 • 10~9 м. Она может обеспечить намагниченность феррожидкости около 150 кА/м на магнетите и около 260 кА/м на железе (см. табл. 3.1). В действительности, как отмечалось выше, достигнута в полтора раза меньшая намагниченность на магнетите, а коллоидные растворы железа пока несопоставимо слабее по магнитным свойствам. Последнее связано с отсутствием достаточно управляемых методов получения частиц железа требуемого размера непосредственно в жидкой среде. Что касается причины различия теоретического и фактического пределов намагниченности магнетитовых жидкостей, то ее можно считать установленной.
Еще до создания феррожидкостей при изучении тонких магнитных пленок было установлено, что их поверхностный слой является немагнитным. Оценки толщины этого слоя дают разные величины, что связано с влиянием технологии получения пленок на толщину немагнитного слоя. Можно предполагать, что она равна толщине мономолекулярного слоя соответствующего магнитного вещества. Тогда для магнетита она будет равна 0,4 и для железа 0,2 нм. В сумме этот
слой и адсорбционный слой дают несколько большую толщину немагнитной оболочки и, как следствие, уменьшение намагниченности феррожидкости. Вторая и пятая строки табл. 3.1 относятся к этому случаю. Наличие немагнитного слоя вещества той же химической природы, что и у магнитного вещества, требует уточнения некоторых понятий, которые будут использоваться далее. Так, в табл. 3.1 размер а, фигурирующий во втором столбце, представляет собой размер ферромагнитного ядра частицы. Куб этой величины вместе с концентрацией частиц определяет концентрацию магнитной фазы, которая меньше концентрации дисперсной фазы. Последняя аналогичным образом определяется размером частиц дисперсной фазы, включающих в себя и немагнитный поверхностный слой дисперсной фазы. Если предположить, что достигнутая к настоящему времени намагниченность феррожидкости около 90-100 кА/м обусловлена в том числе и наличием немагнитного слоя дисперсной фазы, то поверхностный немагнитный слой магнетита должен иметь толщину около 1 нм (третий столбец табл. 3.1). Вонсовским [17] теоретически показано, что частицы ферромагнитных металлов становятся немагнитными при уменьшении их размера примерно до той же величины (1 нм). Если принять, что на частицах железа имеется немагнитный слой такой толщины, то в равных с магнетитом условиях (по уровню магнитного взаимодействия и толщине немагнитной оболочки) намагниченность железной феррожидкости может достигать примерно 170 кА/м (последний столбец табл. 3.1). Проблема, как отмечалось, в отсутствии приемлемой технологии получения мелких частиц железа или других сильно магнитных металлов. Следует также иметь в виду, что частицы металлов коллоидных размеров чрезвычайно активны химически, поэтому единственным сильно намагниченным веществом, которое легко получить в виде частиц коллоидных размеров, является магнетит.
3.19.2.2. Приготовление магнетитовых феррожидкостей
Получение коллоидного магнетита методом химической конденсации описано Элмором [42]. В его основе лежит химическая реакция соосаждения оксидов двух- и трехвалентного железа из их солей концентрированным раствором щелочи:
2FeCl3 + FeSO4 + 8NaOH =
(3.19.4)
= Fe3O4 + 6NaCl + NaSO4 + 4H2O
Для этого готовятся следующие растворы:
1) 5,4 г FeCl3 • 6Н2О в 150 см3 воды;
2) 2,8 г FeSO4 • 7Н2О в 150 см3 воды;
3) 50 г NaOH в 50 см3 воды.
756
Новый справочник химика и технолога
Количество щелочи, содержащееся в растворе 3, в десятки раз превышает то, что необходимо по уравнению (3.19.4). Избыток щелочи необходим как средство дегидратации первоначально образующихся гидроксидов и их превращения в магнетит и воду. Первые два раствора следует фильтровать через бумажный фильтр и смешать в колбе емкостью 0,5 л, третий — фильтровать через стеклянный фильтр и при перемешивании добавить к смеси первых двух. В колбе образуется обильный осадок магнетита Fe3O4 черного цвета. Расчетный выход магнетита равен 2,3 г. Фактический выход не отличается от расчетного. Осадок следует отделить от маточного раствора (декантацией, на фильтре или в центрифуге) и промыть водой до pH не более 8. Дальнейшая промывка обычно затруднена начинающейся самопроизвольной пептизацией осадка и увеличением потерь магнетита с промывными водами.
Для полной пептизации осадок необходимо перенести в чашку, смочить его 0,1 н раствором НС1 и затем растереть при постепенном добавлении 0,1 н раствора НС1. Обычно удается полностью «растворить» осадок, израсходовав на пептизацию от 50 до 100 см3 раствора соляной кислоты. При этом получается коллоидный раствор с концентрацией 4,5-2,2 масс. %. Этого достаточно, чтобы жидкость видимым образом реагировала на магнитное поле любого постоянного магнита. Частицы магнетита в таком растворе заряжены положительно. Раствор имеет интенсивный красно-коричневый цвет (цвет крепкого кофе или чая).
При синтезе магнетита сульфат железа может быть частично, а иногда и полностью, заменен эквивалентным количеством солей кобальта или других двухвалентных металлов, причем не обязательно ферромагнитных. При полном замещении могут быть получены ферриты соответствующих металлов, однако для завершения процесса ферритизации необходима, как правило, высокотемпературная обработка продуктов соосаж-дения. Без такой обработки эти продукты немагнитны. При частичном замещении ферритизация происходит в нормальных условиях, хотя и может потребовать для своего завершения некоторого времени. Эти процессы легко контролировать по магнитной восприимчивости продуктов реакции.
Впрочем, и магнетит в первые секунды своего рождения не обладает магнитными свойствами. Основной смысл частичной замены железа на другие двухвалентные металлы заключается в стабилизации химической и кристаллохимической структуры магнетита. Это необходимо в силу склонности магнетита к окислению кислородом воздуха.
Основное достоинство описанного способа получения феррожидкости в его экспрессности, доступности и отсутствии специфических требований к условиям проведения процесса. Следует лишь иметь в виду, что концентрированный раствор щелочи опасен, и необходимо исключить его попадание на тело и особенно в глаза. То же самое, хотя и в меньшей мере, относится к смеси
всех трех реагентов, где содержание щелочи достаточно велико.
Недостаток феррожидкости, получаемой указанным способом, в том, что она химически не стабильна. В ней постепенно (дни, недели) происходит окисление магнетита до оксида железа(Ш), цвет раствора изменяется до желтого, магнитные свойства могут и не ослабевать, но частицы, видимо, укрупняются. По этой причине приготовление феррожидкости следует проводить в максимально сжатые сроки, а жидкость хранить в герметичной таре. Поскольку жидкость содержит соляную кислоту, то она химически не пассивна. Тем не менее, именно на феррожидкостях этого состава впервые были установлены их фундаментальные свойства.
Химическая устойчивость и пассивность феррожидкостей на основе неполярных дисперсионных сред несравненно выше. Жидкости на основе керосина хранятся десятилетиями без видимых изменений свойств. В то же время в некоторых индивидуальных углеводородах происходят нежелательные изменения. Особенно отчетливо они наблюдаются в растворах олеиновой кислоты в октане: при его хранении на свету происходит полимеризация олеиновой кислоты на стенках колбы [48].
Возможно, что основную роль в этом процессе играют примеси линолевой и линоленовой кислот, имеющих соответственно две и три двойных связи в своей углеводородной цепи. В растворах ароматических углеводородов признаков ухудшения качества феррожидкостей не наблюдалось. Кроме химической инертности феррожидкости на основе углеводородов могут иметь существенно большую намагниченность, чем жидкости на водной основе. Объясняется это тем, что только в неполярной среде на поверхности частиц можно гарантированно создавать защитные слои, толщина которых не превышает размера молекул поверхностно-активного вещества. В полярных же средах всегда сохраняется опасность возникновения неконтролируемого заряда частиц и появления излишне дальнодейст-вующих сил их отталкивания, которые ограничивают максимально возможную концентрацию частиц. В истории коллоидной химии еще не было случая, когда излишне большая величина сил отталкивания частиц оказывалась нежелательной.
Первоначально технология [7] получения феррожидкостей на основе углеводородов использовала метод замены растворителя. После промывки осадка магнетита от маточного раствора водой следовала промывка от воды каким-либо полярным растворителем (например, ацетоном или этиловым спиртом), который хорошо смешивается и с водой, и с неполярными средами. После удаления воды следовала промывка от полярного растворителя с помощью летучего неполярного растворителя (бензол, гептан и др.) и пептизация осадка в растворе олеиновой кислоты в подходящей дисперсионной среде, например в керосине, при перемешивании и нагревании смеси магнетита и раствора
Физика, химия и технология дисперсных систем
757
пептизатора. При этом удалялись (испарялись) остатки летучих компонентов смеси.
Прочная необратимая адсорбция олеиновой кислоты на частицах магнетита происходит за счет химического взаимодействия между кислотой и оксидом с выделением воды. В молярном соотношении количество выделяющейся воды должно быть равно количеству адсорбированной кислоты, и большая часть воды удаляется в процессе пептизации. Вместе с тем присутствие некоторого количества воды необходимо для нормального течения процесса химической сорбции олеиновой кислоты на магнетите.
Поскольку взаимодействие типа кислота—основание (роль последнего играет гидратированная поверхность магнетита) легче всего осуществляется по ионообменному механизму, то необходимы условия для диссоциации реагентов, что и обеспечивается присутствием воды на поверхности частиц. Отсюда следует, что полное обезвоживание при промывке и пептизации нежелательно, однако критерии, определяющие оптимальную влажность феррожидкости, отсутствуют. Ряд свойств и явлений указывает на то, что влажность влияет на свойства феррожидкостей.
Соосаждение оксидов или гидроксидов двух- и трехвалентного железа подразумевает образование смешанного химического соединения FeO ♦ Fe2O3 (т. е. магнетита Fe3O4), а не их механической смеси. Магнетит можно также рассматривать как феррит железа(И), являющийся продуктом взаимодействия слабого основания гидроксида железа(П) Fe(OH)2 и слабой «железной» кислоты Fe(OH)3. Для того чтобы эти гидроксиды могли проявить свои основные и кислотные свойства соответственно и прореагировать между собой указанным образом, среда не должна быть излишне щелочной или излишне кислой. В сильно щелочной среде оба гидроксида ведут себя как слабые кислоты, т. е. диссоциируют с отщеплением иона водорода, что не способствует образованию магнетита.
Оптимальной является такая концентрация гидрок-сид-ионов (pH раствора), при которой, с одной стороны, надежно идет гидролиз солей железа(П) и желе-за(Ш) до образования их гидроксидов, но с другой стороны не подавляются основные свойства гидроксида железа(П). Кроме того, важно наличие буферных свойств у раствора щелочи. Если раствор обладает буферными свойствами, то водородный показатель не претерпевает сильных изменений в ходе смешивания раствора солей со щелочью и в ходе реакции гидролиза. Всем этим условиям наилучшим образом соответствует водный раствор аммиака.
Промышленное производство магнитных жидкостей использует соосаждение солей железа аммиаком (аммиачной водой). В отличие от натриевой щелочи аммиачной водой можно пользоваться только в специально оборудованной химической лаборатории, поскольку аммиак имеет высокую летучесть и его пары вызывают химическую травму легких, дыхательных путей, глаз.
Промышленный способ приготовления углеводородных феррожидкостей не может быть основан на замене растворителей, так как он связан со значительным их расходом и неприемлем экологически.
Современная технология производства феррожидкостей использует экстракцию частиц магнетита углеводородной средой после их гидрофобизации адсорбционным слоем олеиновой кислоты. Для этого после одно- или двукратной промывки осадка от маточного раствора к водной суспензии магнетита добавляют углеводородный раствор пептизатора (олеиновой кислоты) и все это перемешивают при нагревании. В смеси последовательно протекают следующие процессы: 1) адсорбция олеиновой кислоты на границе двух растворов; 2) взаимодействие адсорбированных молекул кислоты со щелочным водным раствором и образование водорастворимых солей (мыл) олеиновой кислоты; 3) десорбция мыл в водную фазу, их хемосорбция на частицах магнетита и гидрофобизация частиц; 4) взаимодействие адсорбционного слоя олеиновой кислоты непосредственно с частицами магнетита и их гидрофобизация; 5) переход гидрофобизованных частиц магнетита в углеводородную среду с образованием устойчивого коллоидного раствора. Магнетит полностью переходит из водной среды в углеводородную. Процесс экстракции может сопровождаться взаимным эмульгированием двух растворов, что делает необходимой последующую осушку феррожидкости (выпаривание воды).
3.19.3. Свойства феррожидкостей
3.19.3.1. Намагниченность
В общих чертах намагничивание феррожидкостей описывается уравнением Ланжевена (см. подразделы 3.9.3-3.9.5) с типичной для суперпарамагнетиков высокой магнитной восприимчивостью. Имеются, однако, важные детали различной степени сложности и значимости, которые и будут обсуждаться далее.
Прежде всего, оказывается, что намагниченность насыщения магнетитовой феррожидкости примерно вдвое меньше ожидаемой величины при данной концентрации магнетита в феррожидкости. Последняя легко контролируется выпариванием феррожидкости и взвешиванием прокаленного осадка. Наблюдаемый дефицит намагниченности вполне объясняется тем, что поверхностный слой магнетита теряет магнитные свойства. Для уменьшения доли магнитной фазы магнетита до 50 % достаточно, чтобы толщина немагнитного слоя магнетита составила 1/5 от радиуса частицы. Именно такой порядок величин радиуса частиц и толщины немагнитного слоя характерен для частиц магнетитовых феррожидкостей, расчетные характеристики которых приведены в табл. 3.1. Толщина немагнитного слоя дисперсной фазы увеличивается при адсорбции на магнетите олеиновой кислоты из углеводородной среды. Это доказывает химический характер ее адсорбционного взаимодействия с магнетитом [48].
758
Новый справочник химика и технолога
Вторая важная особенность суперпарамагнитных феррожидкостей по сравнению с классическими парамагнитными растворами обусловлена полидисперсностью носителей магнетизма — частиц магнетита. Выше уже отмечалось, что изменение размера частиц всего лишь на 1/5 ведет к изменению магнитного момента частиц почти в два раза и, соответственно, намагниченности насыщения при фиксированной концентрации частиц. Однако если фиксировать концентрацию магнитной фазы, то намагниченность насыщения феррожидкости не будет зависеть от размера частиц. При постоянстве намагниченности насыщения, т. е. концентрации магнитной фазы, начальная магнитная восприимчивость увеличивается пропорционально величине магнитного момента частиц (см. подраздел 3.9.4), поэтому она сильно зависит от гранулометрического состава дисперсной фазы, и последний может исследоваться магнитометрически. Достаточно просто можно оценить диапазон размеров частиц реальной феррожидкости. Такая возможность основана на различии вклада в намагниченность крупных и мелких частиц в слабых и сильных магнитных полях.
В слабых полях намагниченность (см. подраздел 3.9.4) монодисперсной феррожидкости:
M=poMsmHI3kT, (3.19.5)
а намагниченность полидисперсной феррожидкости:
Л/=(цоЧН/ЗВД<р(7и, (3.19.5а)
представляют собой сумму вкладов разных фракций, пропорциональных доле <р( магнитной фазы, приходящейся на частицы с магнитным моментом пц, и самому магнитному моменту частиц. В формуле (3.19.5а) и далее величина <pz представляет долю определенной фракции от общего количества (объемной доли) магнитной фазы в феррожидкости и может быть выражена отношением ф/ = MSII Ms, где Msi — намагниченность насыщения фракции, Ms — общая намагниченность насыщения феррожидкости, которая аддитивно складывается из намагниченностей фракций Ms = и определяется экспериментально по намагниченности в сильном поле.
В сильном магнитном поле намагниченность монодисперсной феррожидкости:
М = Ms( 1 - кТ/ мпН), (3.19.6)
а намагниченность полидисперсной феррожидкости получается суммированием подобных выражений по всем фракциям, что дает:
M=Ms-(kT! \jlqH)^Ms, I т, (3.19.6а)
С помощью этой формулы намагниченность насыщения феррожидкости М5 находят путем экстраполяции зависимости намагниченности от напряженности поля к бесконечно большой напряженности.
В простейшем случае уровень полидисперсности феррожидкости оценивается по значениям магнитных моментов частиц, найденных по начальному (уравнение (3.19.5)) и по конечному (уравнение (3.19.6)) участкам изотермы намагничивания.
Магнитный момент, найденный по начальному участку, обычно в несколько раз больше момента, найденного по конечному участку. Примечательно, что магнитометрическое исследование кинетики феррити-зации осадков магнетита [47] не обнаружило в них присутствия частиц с размером магнитного ядра менее 4 нм даже на этапе их зарождения [8].
3.19.3.2. Эффекты коагуляции и структурирования феррожидкостей
Намагничивание феррожидкости является суммарным результатом намагничивания всех ее частиц. Это тривиальное положение приводится здесь только потому, что существуют и при решении ряда технических задач успешно используются представления о феррожидкости как о сплошной бесструктурной жидкой среде (модель Розенцвейга — Нойрингера). В противовес этому ниже рассматриваются явления и свойства, обусловленные гетерогенностью магнитных жидкостей.
В феррожидкостях, как и в любых других дисперсных системах, могут иметь место различного рода коагуляционные явления и структурные превращения. В данном случае под ними понимаются явления, заключающиеся в нарушении статистически равномерного расположения частиц в пространстве. Классификация и систематическое описание подобных явлений приведено в подразделах 3.11-3.13, а описание их влияния на магнитные свойства — в подразделе 3.9. Здесь рассматриваются экспериментальные данные о влиянии структуры магнитных коллоидов на их оптические и реологические свойства.
Оптические свойства феррожидкостей уникальны. Их прозрачность вдоль магнитного поля многократно уменьшается с увеличением напряженности поля (магнитооптический эффект агрегации). В рамках теории светорассеяния Релея этот эффект объясняется укрупнением центров светорассеяния, в данном случае — образованием и увеличением размеров цепочечных агрегатов. Законы релеевского светорассеяния действуют, пока размер рассеивающих центров (цепочек) не превышает длину световой волны. При сильном увеличении размера флокул вступают в силу законы геометрической оптики, согласно которым агрегация частиц ведет к увеличению светопропускания. Последнее характерно для феррожидкостей, склонных к коагуляции. Обычно же в первый момент при включении поля прозрачность быстро (порядка 0,1 с) и сильно уменьшается до некоторого постоянного значения. При пониженной устойчивости феррожидкости вслед за быстрым падением прозрачности следует период ее медленного (минуты) увеличения вплоть до величины, превышающей исходную прозрачность раствора вне магнитного поля.
Физика, химия и технология дисперсных систем
759
Именно в этом состоянии в микроскоп легко наблюдать присутствие в жидкости крупных эллипсоидальных агрегатов. При выключении поля описанные изменения происходят в обратном порядке, но в короткие сроки (секунды) и с меньшей амплитудой изменения прозрачности. В устойчивых феррожидкостях в поле происходит только резкое падение прозрачности и столь же резкое увеличение при выключении поля (рис. 3.135). Исходная прозрачность при этом полностью восстанавливается. Величина эффекта сильно зависит от наличия коагулятора и растет с увеличением его концентрации в феррожидкости. При оптимальной концентрации коагулятора эффект максимален, а жидкость выдерживает неограниченное число циклов агрегации—дезагрегации магнитным полем и соответствующих изменений прозрачности без накопления каких-либо остаточных явлений. Соответственно этому световой поток стабильно модулируется жидкостью в низкочастотном (десятки герц) магнитном поле. При высокоскоростной развертке на экране осциллографа можно наблюдать небольшой (несколько процентов) всплеск прозрачности в момент включения поля. Он объясняется ориентацией частиц в магнитном поле. В последующие моменты времени этот слабый эффект маскируется сильным эффектом агрегации частиц. В специально подготовленных образцах коллоидного магнетита, в которых размер частиц имеет минимально возможную величину, совместимую с сохранением у частиц магнитных свойств, устойчиво наблюдался только ориентационный эффект.
Наличие цепочечных агрегатов и их ответственность за магнитооптические эффекты в феррожидкостях наглядно демонстрируется с помощью динамо-магнитооптического эффекта. Его суть в том, что цепочечные агрегаты легко разрушаются при механическом воздействии (течении), поэтому происходит ослабление оптического эффекта агрегации магнитным полем. Такие воздействия контролируемой величины легко создаются при сдвиговом течении феррожидкости, например при возвратно-поступательном движении тонкой стеклянной пластинки, помещенной в оптическую кювету с феррожидкостью. Результаты подобных опытов подтвердили эту гипотезу: с увеличением скорости движения пластинки магнитооптический эффект уменьшался ожидаемым образом (рис. 3.136). Теория динамо-магнитооптического эффекта [4] положила начало принципиально новому подходу к проблемам реологии дисперсных систем. Она продемонстрировала возможность количественного описания структурного состояния дисперсных систем как функции скорости или напряжения ее сдвиговой деформации и тем самым ввела в теоретическую реологию понятие об уравнении структурного состояния вместо преобладавших тогда представлений о структуре как о некой качественной, статичной характеристике дисперсной системы. В работах П.А. Ребиндера указывалось, что изменение вязкости неньютоновских жидкостей объясняется измене
нием их структуры. Однако эта закономерность не имела и не могла иметь количественного выражения до тех пор, пока не была найдена количественная характеристика состояния структуры. Цепочечная структура феррожидкостей в магнитном поле предоставила уникальную по простоте и однозначности возможность для введения такой характеристики — равновесной длины цепей и ее зависимости от скорости или напряжения сдвиговой деформации феррожидкости.
О А>
Рис. 3.135. Изменение интенсивности / света, проходящего через слой магнитного коллоида при включении и выключении магнитного поля:
поле направлено параллельно лучу света;
tn — момент включения; /«— момент выключения поля
Рис. 3.136. Приращение относительной прозрачности А///0 коллоидного раствора магнетита при увеличении скорости сдвигового течения у при разных напряженностях поля
Напряженность поля, концентрация коагулятора и прочие факторы также влияют на равновесный размер цепей, но только в качестве параметров (коэффициентов) уравнения структурного состояния. На основе этих уравнений позднее были выведены все известные уравнения реологии, в том числе и с более сложной структурой дисперсной системы. На феррожидкостях и магнитных суспензиях они получили первую количественную экспериментальную проверку.
Реологический эксперимент с феррожидкостями осложнен рядом принципиальных проблем, без надлежащего решения которых теряется ценность результатов измерений.
Первая из них — это размер и положение зоны действия магнитного поля на исследуемый образец. Суть этой проблемы рассмотрим на примере двух самых распространенных методов измерения вязкости жидкостей — капиллярного и ротационного.
В капиллярном методе вязкость вычисляется с помощью формулы Пуазейля по скорости протекания жидкости через капилляр калиброванного диаметра и длины. Практически измеряется время истечения фик
760
Новый справочник химика и технолога
сированного объема жидкости из небольшого (около 1 см3) резервуара, расположенного на несколько сантиметров выше мерного капилляра. Существенно, что отсчет времени ведется по моментам прохождения границей жидкости меток над и под резервуаром.
При подготовке опытов необходимо решить, какую часть вискозиметра помещать в магнитное поле (например, в соленоид). Есть два крайних варианта решения проблемы: 1) поместить в магнитное поле только мерный капилляр вискозиметра; 2) поместить весь вискозиметр внутрь достаточно крупного соленоида.
Первый вариант имеет следующие недостатки: а) жидкость находится в неоднородном поле, и появляется магнитостатическая сила ее втягивания в капилляр, причем это магнитостатическое давление на входном и выходном концах мерного капилляра зависит от напряженности поля на границах жидкости и, следовательно, оно может изменяться в ходе опыта по мере перетекания жидкости из одного резервуара в другой;
б) неоднородность поля ведет к втягиванию магнитных частиц в область максимальной напряженности поля, т. е. в капилляр, из прилегающих областей жидкости и к неконтролируемому увеличению концентрации магнитной фазы в капилляре;
в) вследствие ограниченности времени пребывания жидкости в зоне действия поля параметры ее состояния (например, длина цепей) могут не достигать равновесного значения, т. е. полученные результаты будут относиться к неизвестному состоянию системы.
К недостаткам второго варинта (размещение вискозиметра в магнитном поле) относятся:
а) свободные границы жидкости в магнитном поле неустойчивы — они самопроизвольно искривляются (см. подраздел 3.19.4.2). Может даже произойти дробление жидкости на отдельные иглообразные капли. По этой причине измерение вязкости достаточно сильных магнитных жидкостей в сильном поле становится невозможным; капиллярные эффекты искривленных границ могут повлиять на величину давления, определяющего скорость течения;
б) сильно усложняется регистрация моментов прохождения границей жидкости меток над и под мерным резервуаром из-за того, что весь прибор скрыт внутри соленоида.
Ротационная вискозиметрия требует создания специальных приборов, обеспечивающих наличие однородного магнитного поля в рабочем пространстве вискозиметра — коаксиальном зазоре между ротором и статором. С этим связан ряд проблем конструктивного характера, которые, однако, вполне разрешимы. Принципиально важно, как и в случае капиллярной вискозиметрии, решить вопрос о зоне действия магнитного поля. Если ее сделать широкой, так что весь образец внутри рабочего зазора будет находиться в сильном магнитном поле, то это может вызвать дробление жидкости и ее выталкивание из зазора. Даже в отсутствие явных признаков дробления нет гарантий сохранения
сплошности жидкости в глубине рабочей зоны прибора, поскольку механические возмущения способствуют нарушению устойчивости свободных границ феррожидкостей. Если зону действия поля сделать узкой, так что свободная граница жидкости будет находиться вне поля, то магнитостатическая сила втягивания жидкости в рабочий зазор подавит ее дробление, но одновременно она вызовет неконтролируемое увеличение концентрации магнитной фазы в зоне действия магнитного поля. Это также осложняет интерпретацию результатов, так как разные части жидкости будут находиться в разных условиях.
Сложности магнитной вискозиметрии объясняет то, что опубликовано очень мало достоверных данных о вязкости магнитных жидкостей. Некоторые из них оказали, тем не менее, сильное влияние на развитие физики магнитных жидкостей. Так, Мактейг [45] с помощью капиллярной вискозиметрии открыл эффект вращательной вязкости, с которого в физике магнитных коллоидов начался период осознания того, что это гетерогенные системы.
Исчерпывающая теория вращательной вязкости разработана М.И. Шлиомисом [36]. В ней и в последующих теоретических работах на тему вязкости магнитных коллоидов эффекты агрегирования частиц игнорируются.
Неразрешимость некоторых проблем магнитной вискозиметрии привела к поиску альтернативных, но, тем не менее, эквивалентных вискозиметрии методов исследования свойств феррожидкостей. Таковым стал метод вращающегося магнитного поля [6]. В этом методе небольшой осесимметричный сосуд с жидкостью подвешивается на упругой нити в однородном вращающемся магнитном поле. Нить подвеса является осью вращения поля. Измеряется момент сил, передаваемый полем через жидкость на стенки сосуда. В опытах можно варьировать скорость вращения и напряженность поля, а также состав феррожидкости, в том числе концентрацию коагуляторов.
При течении жидкости цепочечные агрегаты можно в подходящей системе координат считать неподвижными. Они тормозят обтекающие их потоки среды, что и регистрируется как увеличение вязкости. Во вращающемся поле среда неподвижна, а цепочки вращаются, следуя за полем. При этом они передают среде момент сил, действующих на цепи со стороны поля. В среде моменты всех цепей суммируются и передаются стенкам сосуда. Их суммарная величина и регистрируется по углу закручивания упругого подвеса. Во всех случаях разными методами регистрируется один и тот же эффект гидродинамического взаимодействия цепочек или индивидуальных частиц с вязкой средой, поэтому удельная сила трения (на единицу площади) и удельная величина момента сил (на единицу объема) равны по величине и по размерности. Метод вращающегося поля лишен большинства недостатков магнитной вискозиметрии, поскольку исследуемый образец
Физика, химия и технология дисперсных систем
761
полностью находится в однородном поле и при надлежащей конструкции сосуда не имеет свободных границ. Кроме того, этот метод более чувствителен, так как в нем среда неподвижна и отсутствуют те составляющие силы трения, которые связаны со сдвиговой деформацией самой среды. В разбавленных же жидкостях эта составляющая сил трения подавляюще велика по сравнению с эффектами трения частиц и цепей о среду, что делает практически непригодными вискозиметрические методы для изучения разбавленных растворов. Метод вращающего поля практически не накладывает ограничений такого рода.
Зависимости крутящего момента М от частоты вращения со, напряженности поля и концентрации коагулятора количественно и качественно совпадают с аналогичными зависимостями напряжения сдвига от скорости сдвига в цепочечной структуре (рис. 3.104) в режиме тиксотропно равновесного течения:
М— Мс + aqxo. (3.19.7)
В соответствии с цепочечной моделью тиксотропных систем [9] действие поля в том и другом случае сводится к появлению предельного напряжения сдвига или равного ему начального момента сил трения Mc = 2nrFJ3 и к их увеличению предсказуемым образом с увеличением напряженности поля за счет увеличения силы сцепления частиц Fc. Если предполагать, что защитная оболочка на поверхности частиц является жесткой, то расстояние г между центрами соседних частиц в цепи можно считать не зависящим от напряженности поля. Начальный момент сил, как и предельное напряжение сдвига, является в феррожидкостях динамической характеристикой прочности цепей и экспериментально определяется экстраполяцией силы трения (момента сил) к нулевой скорости сдвига или вращения соответственно. Статическая прочность на сдвиг в феррожидкостях отсутствует. Примечательно, что для появления у дисперсной системы динамической прочности не требуется наличия сплошной структуры. Различия в поведении феррожидкости в потоке и во вращающемся поле возникают только в сильно коагулированных системах, в которых вместо линейных цепочек образуются более или менее округлые флокулы. Поэтому величина крутящего момента уменьшается, а не увеличивается при увеличении концентрации коагулятора.
К оптическим методам изучения и диагностики феррожидкостей можно отнести прямые наблюдения за их однородностью, в том числе в магнитном поле. Среди физиков, занимающихся проблемами феррожидкостей, большой популярностью пользуется гипотеза о двухфазности феррожидкостей. Жидкости плохого качества таковыми и являются. Их неоднородность, т. е. наличие крупных, не поддающихся пептизации, иногда видимых невооруженным взглядом хлопьев коагулята, становится особенно заметной в магнитном поле. Сгустки коагулята в поле укрупняются, а в тонком слое
жидкости, ориентированном перпендикулярно направлению поля, они могут, наоборот, дробиться на более мелкие фрагменты. Магнитостатическое отталкивание и дробление хлопьев коагулята в тонком слое приводит к тому, что размеры хлопьев выравниваются, а сами они располагаются в узлах правильной, обычно гексагональной, двухмерной решетки. Это создает довольно привлекательную и видимую невооруженным глазом картину, побуждающую физика к творчеству. Реакция же осведомленного человека, озабоченного созданием качественных жидкостей, на это явление иная — он просто выбрасывает такие образцы жидкости как брак.
3.19.4. Феррогидродинамика
3.19.4.1. Магнитостатическое давление
Описание течения магнитных жидкостей в магнитном поле требует учета множества связанных с этим эффектов. Например, при высоких скоростях течения необходимо принимать во внимание конечную величину скорости намагничивания. В принципе струя вязкой феррожидкости может пересечь межполюсное пространство, не успевая намагнититься, т. е. не реагируя на магнитное поле, а может и остановиться в межполюсном пространстве, если намагниченность и скорость намагничивания достаточно велики. Скорость намагничивания существенным образом зависит от механизма намагничивания частиц. При броуновском механизме скорость определяется вязкостью среды, при неелевском — константой магнитной анизотропии частиц. Магнетитовые феррожидкости намагничиваются практически мгновенно (примерно за 10 8 с) независимо от вязкости среды, а время намагничивания суспензии магнитно-жесткого феррита, например гексаферрита бария, в глицерине (вязкость 1,5 Па-с при 20 °C) в поле с напряженностью порядка 1 000 А/м составляет около 0,001 с, что сопоставимо со временем пребывания суспензии в межполюсном пространстве при ее быстром течении.
Другое универсальное явление, сопровождающее намагничивание феррожидкости, — это магнетокало-рический эффект — нагревание при намагничивании и охлаждение при размагничивании вещества. Температура влияет на многие свойства жидкостей и сама зависит от ряда свойств и условий намагничивания, поэтому уравнения движения жидкости в поле должны учитывать ее теплоемкость, теплопроводность, коэффициент термического расширения, температурную зависимость вязкости и намагниченности, граничные и начальные условия.
Наконец, магнитное поле и скорость течения влияют на структурное состояние и, следовательно, на сами законы течения. В принципе не слишком сложно выписать уравнения, которые формально охватывают все многообразие свойств и явлений при течении феррожидкости в магнитном поле, что иногда и делалось. Одно уравнение может при этом занимать несколько страниц. Такие уравнения приобретают какую-либо
762
Новый справочник химика и технолога
ценность после их конкретизации. В частности, практически важные результаты удается получить, используя простейшее уравнение феррогидродинамики — уравнение Бернулли для феррожидкости:
Р + pgh + рг? /2 - w£71n(sh£ / £) = const. (3.19.8)
Здесь Р — действующее извне давление, р — плотность жидкости, g — ускорение свободного падения, h— высота, и — скорость течения, H&71n(sh£/£) — магнитостатическое давление и ^=\х^тН/кТ— аргумент функции намагничивания Ланжевена £(<;) = cth(Q - 1/£. Последнее слагаемое в левой части формулы (3.19.8) в форме давления учитывает силу втягивания жидкости в магнитное поле. Оно получается интегрированием выражения poMsL(^)dH в пределах от Н = 0 до напряженности поля Н в той области жидкости, к которой относятся остальные параметры состояния жидкости: высота h уровня жидкости и скорость ее течения и, определяющие гидростатический pgh и скоростной рг?/2 напоры соответственно. Выражение poMsL(^)dH — это сила, действующая на тонкий слой жидкости. В поле, которое изменяется только вдоль одной оси х, на единицу объема жидкости действует сила p^MJAQ^dHIdx'), направленная вдоль градиента напряженности поля dH/dx. Сила, действующая на слой толщиной dx и с площадью сечения, равной единице, отличается в dx раз, т. е. пропорциональна величине (dHldx)dx = dx, что и дает упомянутое подынтегральное выражение. Заметим, что жидкость Бернулли лишена вязкости, и факт ее течения учитывается только наличием силы инерции рг?/2. Фактически уравнение пригодно для решения некоторых задач гидростатики феррожидкостей — нахождения равновесной конфигурации границ жидкости и распределения в ней давления в состоянии покоя (и = 0). В частности, в отсутствие внешнего давления (Р = 0) и течения (и = 0) оно дает уравнение зависимости положения (высоты Л) границ жидкости от напряженности поля. Если же пренебречь гидростатической силой, то можно получить величину давления Ртах, необходимого для вытеснения жидкости из магнитного поля. При условии, что одна из границ жидкости находится вне поля, а вторая — в поле, где жидкость намагничена до насыщения, получим:
Ртах=ЦоМН. (3.19.9)
При намагниченности насыщения 105 А/м и напряженности поля 106А/м, которая легко достигается в узком зазоре между полюсами электромагнита, находим, что «пробка» из магнитной жидкости способна выдержать давление порядка 105 Па, т. е. около одной атмосферы. На этом основано большинство идей технического применения феррожидкостей. Приведенная формула дает оптимистическую оценку возможностей феррожидкостных устройств. Реальные возможности зависят, в частности, от эффективности мер, принимаемых для подавления неустойчивости свободной поверхности намагниченной феррожидкости.
3.19.4.2. Неустойчивость свободной границы феррожидкости
О неустойчивости свободной границы феррожидкости уже упоминалось в связи с проблемами вискозиметрии. Под свободной подразумевается поверхность раздела феррожидкости с другой флюидной фазой — немагнитной жидкостью или газом. Суть свойства, обозначаемого понятием «неустойчивость плоской поверхности», с предельной наглядностью демонстрируется видом образца жидкости в поле постоянного магнита (рис. 3.137). В магнитном поле, направленном перпендикулярно свободной поверхности, она сильно искривляется, превращаясь в серию острых, расходящихся шипов [41]. Такое состояние феррожидкости получило поэтичное название «цветок Розенцвейга». Образно говоря, этот «цветок» живой — его шипы шевелятся при малейших механических воздействиях или вариации магнитного поля. В сильном однородном поле, протяженность которого много больше, чем диаметр сосуда с жидкостью, при малой толщине слоя вместо цветка образуется серия изолированных жидких иголок, вытянутых вдоль поля. Они расположены регулярно в узлах гексагональной решетки. Размер игл и период их расположения тем меньше, чем тоньше слой жидкости и выше напряженность поля. В пределе поверхность тонкого слоя жидкости приобретает вид бархатной ткани, т. е. она состоит из множества отдельных очень мелких иголок, неразличимых невооруженным глазом.
Картина радикально меняется, если тонкий слой жидкости ограничить с обеих сторон, например каплю жидкости поместить в плоскую щель между двумя стеклами.
Свободной при этом остается боковая поверхность капли. В магнитном поле, перпендикулярном плоскости слоя, с увеличением напряженности капля вытягивается в произвольном направлении, приобретая форму гантели, которая затем еще больше вытягивается и сгибается посредине примерно под углом 30°. При дальнейшем увеличении напряженности поля цикл удлинения и сгибания повторяется и приводит к образованию тонкой, плотной лабиринтной структуры, в которой, однако, стенки лабиринта нигде не соприкасаются и не пересекаются [33].
Рис. 3.137. Устойчивая форма поверхности феррожидкости на полюсе магнита (а) и в однородном поле (б)
Физика, химия и технология дисперсных систем
763
Описанные выше эффекты являются следствием взаимодействия между собой соседних частей равномерно намагниченного слоя жидкости. Это взаимодействие выражается в том, что любой участок плоского слоя создает в остальных частях слоя магнитное поле, направленное противоположно внешнему намагничивающему полю. В итоге на каждый участок слоя действует суммарное размагничивающее поле всех остальных частей слоя. Его напряженность равна Л//2. В электротехнике при расчетах магнитных цепей размагничивающее действие намагничиваемого тела самого на себя учитывается с помощью фактора размагничивания N. Его величина определяется формой тела, а формальный смысл — уравнением
(3.19.10)
Здесь % — магнитная восприимчивость магнитного вещества при А = 0. Размагничивающий фактор тел сильно вытянутой (вдоль поля) формы равен нулю. Размагничивающий фактор тонких плоских тел максимален при их намагничивании в перпендикулярном к плоскости направлении и равен 1/2. Для частиц сферической формы А = 1/3. Восприимчивость тела ^ь, в отличие от восприимчивости вещества, позволяет выразить его намагниченность М= jj,H непосредственно через напряженность внешнего поля Н. С помощью уравнения (3.19.10) одну из них можно выразить через другую. Так как Х/Л = х(Н- то:
Хб = Х/(1 (3.19.11)
Вспучивание некоторого участка свободной поверхности феррожидкости является результатом перемещения сюда некоторого количества жидкости с соседнего участка, на котором образуется впадина. Напряженность поля в зоне выступа увеличивается, а в зоне впадины уменьшается. Под действием разности напряженностей жидкость перетекает из области с меньшей напряженностью поля в область большей напряженности, увеличивая, таким образом, случайно возникшую неравномерность толщины слоя, что и означает неустойчивость плоской границы намагниченной жидкости. Этот процесс развивается до тех пор, пока магнитостатическое давление, создаваемое перепадом напряженностей поля под выступом и под впадиной, не будет уравновешено нарастающей разностью гидростатических давлений на выступе и впадине в слое жидкости. В том же направлении, т. е. на подавление неустойчивости, действует и сила поверхностного натяжения а, стремящаяся сгладить всякие неровности поверхности. Количественно она выражается величиной капиллярного давления Рк = <зК, где К — кривизна поверхности. Она нарастает при увеличении высоты выступа и глубины впадины. Разность напряженностей также увеличивается с увеличением кривизны поверхности, однако увеличение напряженности имеет предел, равный Л//2. Он определяется разностью размагничивающих факто
ров плоского и сильно вытянутого тел. Таким образом, рано или поздно установится та или иная равновесная конфигурация поверхности намагниченной жидкости.
Неустойчивость возникает при намагничивании жидкости до величины, превышающей некоторое критическое значение Мк [41]:
н^гфгаГр+ац*,)-''2], (31912)
5 = (a/pg)''2
Здесь ц = 1 + х — интегральная магнитная проницаемость, = 1 + х</ — дифференциальная магнитная проницаемость при критической намагниченности феррожидкости, где Xd = (dM/dH) — ее дифференциальная магнитная восприимчивость, s — максимальная величина периода расположения пиков и впадин. Оценки по формулам (3.19.12) дают величину критической намагниченности углеводородной жидкости Мк около 10 кА/м и период s порядка одного сантиметра.
3.19.4.3. Применение феррожидкостей
В большинстве случаев применение феррожидкостей основано на их способности втягиваться в магнитное поле и сопротивляться вытеснению из области наибольшей напряженности поля. Эта способность количественно характеризуется величиной магнитостатического давления, рассмотренного в подразделе 3.19.4.1. Самым известным устройством такого типа являются герметичные вводы вращательного и поступательного движений внутрь разного рода аппаратов (уплотнения). Конструкция уплотнения схематично показана на рис. 3.138. Сильное магнитное поле создается в рабочем зазоре между валом и толстыми полюсными наконечниками в виде шайб, являющихся частью замкнутой магнитной цепи. Эти элементы должны быть изготовлены из мягкого железа. Цепь замыкается постоянным магнитом, также имеющим форму шайбы, намагниченной вдоль своей оси. Зазор заполняется магнитной жидкостью, которая в отсутствие перепада давления распределяется в нем так, что обе границы феррожидкости находятся в областях с равной напряженностью поля, т. е. симметрично относительно плоскости симметрии шайбы, если ее внутренняя кромка имеет симметричную форму (что предпочтительно).
Рис. 3.138. Схема феррожидкостного уплотнения. Положение феррожидкости (обозначено черным) соответствует избыточному давлению Р, близкому к максимально возможному
764
Новый справочник химика и технолога
При наличии перепада давления Р одна из границ феррожидкости вытесняется в область более сильного поля, а другая — в область более слабого поля. Это и создает магнитостатическое давление, определяемое разностью Д(Л/Я) произведений МН намагниченности жидкости на напряженность поля на границах жидкости, так что P = \\q&(MH). Формула (3.19.9) является частным случаем этого выражения, когда на границе, сильно удаленной от середины рабочего зазора, напряженность поля пренебрежимо мала, а жидкость в узкой части зазора намагничена до насыщения. При конструировании уплотняющих элементов такого расположения границ следует избегать, поскольку вытеснение части жидкости в область, где магнитное поле практически не действует, приведет к ее вытеканию из уплотнения и потере. Если определенные потери жидкости допустимы, то таким способом можно достичь автоматического установления нужной степени заполнения рабочего зазора феррожидкостью.
Величину рабочего перепада давлений можно увеличить, придав внутренней кромке полюсных наконечников вид гребенки (рис. 3.139). Каждый выступ гребенки действует как самостоятельный уплотняющий элемент. Феррожидкость должна при этом заполнять только узкие участки профилированного зазора. Широкие участки зазора должны быть обязательно заполнены немагнитной жидкостью, не смешивающейся с магнитной жидкостью. Только в этом случае рабочий перепад давления будет равен сумме перепадов давлений на каждом элементе уплотнения. Проблема подбора несмешивающихся жидкостей достаточно легко решается, если уплотнение разделяет газовые среды с различным давлением. Она усложняется, если одна из сред, в контакте с которой должно работать уплотнение, является жидкой и особенно при высокой скорости вращения вала. Трудности связаны с возможностью эмульгирования феррожидкости и ее уноса. Они сильно усугубляются неустойчивостью гладких границ феррожидкости в сильном магнитном поле. Неустойчивость можно подавить, если граница жидкости находится в области сильно неоднородного магнитного поля. Характер этой неоднородности должен быть таким, чтобы сила втягивания феррожидкости в магнитное поле имела тот же знак, что и гравитационная сила в формуле (3.19.12).
Рис. 3.139. Фрагмент конструкции многоэлементного уплотнения. Наличие немагнитной разделительной жидкости между элементами феррожидкостного уплотнения обязательно
Принцип действия магнитного уплотнения автоматически обеспечивает соблюдение нужного направле
ния градиента напряженности поля. Практически важный вывод заключается в том, что рабочий перепад давления на каждом элементе уплотнения должен быть заметно меньше, чем максимальный перепад давления Ртах = PoMsH, который он может выдержать. Только в этом случае граница феррожидкости находится еще вне зоны максимальной напряженности поля, т. е. под действием градиента напряженности, необходимого для подавления неустойчивости гладкой границы.
По масштабам применения феррожидкостей первое место могут занимать феррогидростатические сепараторы — устройства для разделения измельченных материалов по их плотности р. Принцип действия сепаратора основан на том, что всякое немагнитное тело (а в общем случае тело с меньшей магнитной восприимчивостью, чем у феррожидкости) в неоднородном магнитном поле выталкивается из феррожидкости в направлении наиболее быстрого убывания напряженности поля, а точнее — магнитостатического давления РоМН в феррожидкости (рис. 3.140). Величина выталкивающей силы, действующей на частицу объемом v, вычисляется так же, как и архимедова сила pogv с заменой градиента гидростатического давления pog градиентом магнитостатического давления pograd(A/77). Регулируя напряженность поля, легко заставить всплывать частицы минерала с плотностью pi меньшей, чем эффективная плотность ре феррожидкости в магнитном поле:
pe = po + (Po/g)grad(A//7). (3.19.13)
Феррогидросепарация не пригодна для обработки высокодисперсных продуктов из-за сильной агрегации немагнитных частиц в намагниченной феррожидкости. Немагнитные включения ведут себя в ферромагнитной среде как сильно диамагнитные материалы: намагничиваются в направлении, противоположном направлению намагниченности феррожидкости. Намагничивание ведет к слипанию частиц, в том числе различных по составу, исключая тем самым возможность их разделения. Сила магнитного сцепления увеличивается пропорционально квадрату размера частиц: fd ® роМ2с^/4п. Сила тяжести нарастает с увеличением размера частиц пропорционально их объему v (кубу размера). В той же пропорции нарастает разность сил тяжести (р2 - pi)v, действующих на частицы с разными плотностями р2 и Рь Если эта разность превысит силу диамагнитного сцепления и одновременно эффективная плотность феррожидкости ре превысит плотность менее плотного материала рь то произойдет разделение сцепившихся частиц и всплытие более легкого материала в феррогидросепараторе. Таким образом, для разделения дисперсных материалов, в дополнение к условию ре > р1; должно выполняться условие (р2 - pi)gv > ц0Л/2а2/4л или:
a>MoM2/3(p2-pl)g. (3.19.14)
Физика, химия и технология дисперсных систем
765
Оно показывает, что для разделения предпочтительнее использовать слабомагнитные жидкости, компенсируя их малую намагниченность созданием в сепараторе поля большой напряженности и его градиента. При этих условиях можно осуществить разделение частиц с размером порядка 1 см и меньше. Одновременное создание большой напряженности и градиента напряженности поля возможно лишь в аппаратах с ограниченным размером вдоль градиента напряженности.
Наиболее многочисленная по назначению группа устройств использует возможность удерживать тела в заданном положении с помощью феррожидкости без применения твердых опор. Эти устройства действуют на том же принципе, что магнитостатические сепараторы, но в них регулируется всплывание одной «частицы» — того тела, положение которого нужно зафиксировать. К числу таких устройств относятся подшипники, акселерометры, платформы на газожидкостной подушке, линейные электродвигатели и многие другие приборы и технологические аппараты. В качестве примера на рис. 3.141 схематично показано устройство для перемещения грузов в горизонтальной плоскости. Оно представляет собой платформу, по периметру которой имеется ряд униполярно ориентированных постоянных магнитов. Магниты удерживают сплошной вал феррожидкости по всему периметру платформы. На гладкой поверхности (на полу) этот вал герметично отделяет полость под платформой от окружающей среды, поэтому в полости можно создать избыточное давление, которое и приведет платформу в подвешенное состояние. Грузоподъемность платформы равна произведению ее площади на избыточное давления под платформой, так что удельная грузоподъемность около 1 т/м2 достигается при избыточном давлении 0,1 атм. Вал феррожидкости по периметру платформы одновременно выполняет функцию уплотнения. При указанной грузоподъемности оно должно быть рассчитано на рабочее давление 0,1 атм, что, как было показано выше, вполне реализуется практически. Магниты по периметру платформы способны одновременно выполнять функцию «ротора» линейного электродвигателя, если в полу поместить обмотку статора такого электродвигателя.
Аналогично решается задача перемещения деталей на миниатюрных платформах по гладким направляющим поверхностям постоянной кривизны (вместо пола) в произвольном направлении дискретными шагами.
В завершение обратим внимание на температурную зависимость намагниченности феррожидкостей (уравнения (3.19.5а) и (3.19.6а)). При нагревании жидкости ее намагниченность уменьшается и соответственно этому уменьшается величина магнитостатического давления (сила втягивания жидкости в магнитное поле). Это приводит к тому, что горячие слои неравномерно нагретой жидкости будут выдавливаться из неоднородного поля холодными порциями жидкости. Они, в свою очередь, при наличии источника теплоты после прогрева будут вытеснены новой порцией холодной жидкости
и т. д. Возникнет непрерывное конвективное движение феррожидкости в неоднородном магнитном поле, если в ней с помощью нагревателя и холодильника поддерживать перепад температур. Организовав надлежащим образом движение жидкости по замкнутому контуру (рис. 3.142), можно заставить ее вращать турбину генератора или интенсифицировать теплообмен в каком-либо аппарате.
Отметим, что в невесомости термомагнитная конвекция может заменить обычную гравитационную конвекцию. Последняя обеспечивает на земле привычное течение множества процессов без дополнительных затрат энергии — от кипения воды в чайнике до глобальных атмосферных процессов.
В космосе для поддержания подобных процессов отсутствующая гравитация должна быть в локальных масштабах заменена некоторой искусственно созданной массовой силой: центробежной или магнитостатической.
Рис. 3.140. Схема магнитогидростатического сепаратора.
На тело, погруженное в феррожидкость, в неоднородном поле действует магнитостатическая сила выталкивания (Fm) и сила тяжести (Fg)
111111111111111111
Рис. 3.141. Платформа на воздушной подушке. Избыточное давление воздуха под платформой удерживается феррожидкостным уплотнением по ее периметру: М — магнитное кольцо; Ф — феррожидкость
Рис. 3.142. Схема феррожидкостной тепловой машины: NS — источник магнитного поля; X — холодильник; Т — нагреватель; U — направление потока жидкости; W— турбина
766
Новый справочник химика и технолога
3.19.4.4. Применение эффектов взаимодействия частиц в магнитных суспензиях
Наиболее сильные эффекты, сопровождающие намагничивание дисперсных ферромагнетиков, имеют место в суспензиях магнитно-мягких материалов, в частности в суспензиях карбонильного железа или карбонильного никеля. Карбонильные металлы производятся в промышленных масштабах, и с их помощью легко реализуются все возможности, связанные с эффектами взаимодействия частиц в магнитном поле. Доступны и другие типы магнитно-мягких материалов, в числе которых имеются и ферриты.
Название «карбонильный металл» связано с технологией его получения: из паров карбонилов соответствующих металлов, которые легко разлагаются при нагревании. При этом образуются сферические частицы размером в несколько микрометров. Частицы железа имеют луковичную структуру: состоят из сферических слоев железа, разделенных тонкими прослойками углерода. По магнитной структуре это, безусловно, многодоменные частицы. Сферическая симметрия частиц магнитного материала плохо согласуется с такой доменной структурой, при которой частицы в целом оказываются ненамагниченными. Тем не менее, она существует и отличается исключительной устойчивостью, поскольку после сколь угодно сильного намагничивания частицы карбонильного железа полностью размагничиваются при выключении внешнего магнитного поля. Это, в частности, предопределило применение карбонильного железа как материала для изготовления магнитных сердечников радиотехнических изделий.
Магнитная мягкость карбонильного железа и никеля приводит к тому, что суспензии этих металлов легко намагничиваются и самопроизвольно размагничиваются и, следовательно, обратимо меняют свои свойства, связанные с изменением структуры и сил сцепления частиц при намагничивании—размагничивании. Наиболее сильным проявлением этих изменений служит магнитореологический эффект — затвердевание суспензии в поле и размягчение до первоначального состояния при выключении поля. Точнее говоря, суспензии приобретают в магнитном поле ярко выраженную пластичность с большим предельным напряжением сдвига т4 (сдвиговой прочностью). При цепочечной структуре и магнитной природе сил сцепления частиц сдвиговая прочность равна удельной (на единицу объема) энергии магнитного взаимодействия частиц:
= цоЛ//4лф. (3.19.15)
При намагничивании до насыщения:
т5 = (мо/4лМ М* = (мо/4л)ф(А/; )2. (3.19.16)
Оценка по этой формуле дает при (р = 0,5 прочность порядка 105 Па (1 кг/см2). При малых скоростях сдвига
сопротивление сдвигу практически не зависит от скорости деформации, что позволяет передавать крутящий момент, практически не зависящий от скорости проскальзывания ведущего и ведомого фрикционов. Такая передаточная характеристика может быть полезной в ограничительных и магнитоуправляемых муфтах сцепления, тормозах и других устройствах. Условия применения таких устройств могут быть весьма жесткими при использовании суспензий на основе металлических легкоплавких сред.
Концентрированные суспензии магнитно-жестких ферритов имеют большую величину предельного напряжения сдвига и в отсутствие внешнего магнитного поля, поскольку частицы таких суспензий самопроизвольно намагничены до насыщения. Практический интерес представляет возможность появления в таких суспензиях направленного синхронного вращения частиц в переменном поле (см. подраздел 3.11). При сдвиговой деформации суспензии направление вращения совпадает с направлением сдвига, что проявляется в существенном снижении вязкости суспензии и даже в эффекте, который формально может быть описан как появление отрицательной вязкости. Фактически это означает, что суспензия под действием поля начинает течь и в отсутствие какой-либо внешней, понуждающей к течению силы. Поскольку в отсутствие такой силы суспензия «не знает», в каком направлении следует течь, то течение может принимать различные, иногда причудливые формы [3]: движение от стенок сосуда к его середине, течение одновременно в двух противоположных направлениях. Наличие течения проявляется в том, что на поверхности суспензии регулярно возникают гребни движущихся волн. Суспензия интенсивно нагревается из-за непрерывного вращения частиц в вязкой среде, и поэтому она может использоваться как распределенный тепловыделяющий агент.
В устойчивых феррожидкостях упомянутые выше эффекты практически отсутствуют, что, собственно, и относит их к числу действительно магнитных жидкостей — веществ, вязкость которых в практическом отношении можно считать не зависящей от напряженности магнитного поля. Тем не менее, в них в большей или меньшей степени происходит обратимая агрегация частиц в магнитном поле. Наиболее заметно это эффект сказывается на прозрачности жидкости, что можно использовать для глубокой низкочастотной модуляции света. Выше также отмечалась возможность уменьшения магнитооптического эффекта при воздействии на жидкость сдвиговых напряжений. На этом принципе могут действовать динамо-магнитооптические датчики скорости сдвиговой деформации. Датчики другого типа реагируют на величину, а не на скорость деформации. Чувствительность динамо-магнитооптических датчиков достаточна для создания микрофонов и сенсорных датчиков типа дисплеев карманных компьютеров с сенсорным вводом команд и данных.
ПРИЛОЖЕНИЕ 1
Поверхностное натяжение и поверхностно-активные вещества
1.1. Поверхностное натяжение веществ
1.1.1. Поверхностное натяжение веществ на границе с газовой фазой [1]
Обозначения: а — поверхностное натяжение, Т — температура, Гпл — температура плавления, TKtm — температура кипения. Обозначения состава газовой фазы: взд — воздух, насыщенный паром; пар — насыщенный пар вещества жидкой фазы.
Таблица 1П1.1 Простые вещества
Формула Название Газ Г, °C а, мДж/м2
взд 970 800
н2 995 923
Ag Серебро Н2 1050 916
н2 1100 909
н2 1163 902
Al Алюминий взд 700 840
взд 700 1207
взд 800 1177
Au Золото Н2 1120 1128
н2 1200 1120
н2 1300 1110
н2 300 388
н2 583 354
Bi Висмут Н2 779 343,9
со2 700- 346
800
Br2 Бром взд взд -21 13 62,1 44,1
н2 330 570
Cd Кадмий н2 400 597
н2 600 585
н2 1140 1120
Cu Медь н2 1200 1160
н2 1300 1226
Fe Железо н2 Н2 1267 1310 936 917
Ga Галлий со2 30 358
Ge Германий 959 (Т’пл) 600
Hg Ртуть взд, пар пар, взд 0 10 479,5 477
Продолжение таблицы 1П1.1
Формула Название Газ Г, °C а, мДж/м2
пар, взд 20 475
пар, взд 25 473,5
пар, взд 30 472,5
пар, взд 40 470
пар, взд 50 467,5
Hg Ртуть пар, взд 100 456
пар, взд 150 444
пар, взд 200 433
пар, взд 250 416
пар, взд 300 400
пар, взд 350 381
К Калий пар ПО 95
пар 100 222
Na Натрий пар 250 211
СО2 90 294
взд 50 69,7
Р Фосфор взд взд 54 60,2 68,53 66,95
взд 68,7 64,95
Н2 350 442
Н2 400 438
РЬ Свинец н2 н2 500 600 431 424
н2 800 410
н2 1000 401
Pt Платина взд 2000 1819
Rb Рубидий 39,5 77
взд 119 42
S Сера пар 445 33
(Т’кип)
Sb Сурьма н2 н2 750 1000 368 355
Se Селен взд 217 92,5
Si Кремний Не 1450 725
пар 250 575
пар 500 525
Sn Олово пар н2 600 253 505 526
н2 800 520
н2 1000 497
TI Таллий пар 300-320 375-496
768
Новый справочник химика и технолога
Продолжение таблицы Ш1.1
Формула Название Газ Г, °C ст, мДж/м2
Zn Цинк пар пар Н2 Н2 470 635 510 640 772,2 728,1 785 761
Таблица 2П1.1
Неорганические соединения
Формула Название Газ Г, °C ст, мДж/м2
AgBr Серебра бромид взд 434 (Гпл) 121,4
AgCI Серебра хлорид взд взд взд взд 450 494 501 550 125 121,6 119 113
AsBr3 МышьякаЦП) бромид n2 n2 n2 49,6 121 179,7 49,6 41 36,1
AsCl3 Мышьяка(Ш) хлорид n2 n2 n2 n2 n2 -21 0 20,8 50,2 ПО 43,8 41,4 39,4 36,6 31
BC13 Бора трихлорид nap 20 16,7
ВаС12 Бария хлорид взд 962 (Гпл) 150,5
(CN)2 Дициан nap взд -20 -10 0 10 20 21,9 20,2 18,5 16,8 15,1
CS2 Сероуглерод взд взд взд -30 -20 0 40 38,5 35,5
cs2 Сероуглерод ВЗД ВЗД ВЗД ВЗД 10 20 30 50 33,9 32,4 30,9 27,8
H2S2 Дисульфан ВЗД ВЗД ВЗД ВЗД -20,5 -0,3 14,6 20,5 53,7 50 47,4 46,2
H2O Вода См. отдельную табл.
Продолжение таблицы 2Ш.1
Формула Название Газ r,°c ст, мДж/м2
D2O Тяжелая вода (дейтерия оксид) взд взд взд взд взд взд 15 20 25 30 35 99 110,8 216 73,35 72,6 71,8 71,1 70,3 58,5 56 32,8
КВО3 Калия метаборат N2 992 123,5
КВг Калия бромид n2 n2 n2 взд 775 886,5 920 730 (Т’пл) 85,7 77,8 75,4 91
КС1 Калия хлорид n2 n2 n2 n2 n2 ВЗД 800 885 986 1088 1167 776 (Гпл) 95,8 89,7 82,2 75,2 69,6 98,4
KCN Калия цианид ВЗД 634 (Т’пл) 96,1
KF Калия фторид M2 n2 n2 913 1185 1310 138,4 116,1 104,9
KI Калия иодид n2 n2 n2 737 812 873 75,2 69,2 66,5
KNO3 Калия нитрат n2 n2 n2 n2 380 578 771,6 414 110,4 95,2 80,2 100,7
K2SO4 Калия сульфат n2 n2 n2 n2 1070 1306 1400 1656 143,7 128,8 122,4 106,8
П2Н4 Гидразин 25 40 66,7 60,1
no2 Азота диоксид взд взд 1,6 19,8 29,5 26,6
NaBO2 Натрия метаборат N2 n2 n2 1016 1192 1441 193,7 166,1 126,2
Приложение 1
769
Продолжение таблицы 2П1.1
Формула Название Газ T,°C о, мДж/м2
Na2B4O7 Натрия тетраборат n2 1000 211,9
n2 761 105,8
NaBr Натрия бромид n2 n2 941 1073,5 92,9 84
n2 1166 78
Na2CO3 Натрия взд 851 179
карбонат (Щ
n2 803 113,8
NaCl Натрия n2 960 102,7
хлорид n2 1080 94
n2 1172 88
Nal Натрия иодид n2 n2 n2 705,5 815,5 861 85,6 80,5 77,6
взд 661 93,9
(Т’пл)
NaNO3 Натрия нитрат n2 n2 321,5 513 119,7 108,9
n2 602 103,4
n2 738 93,7
Na2SO4 Натрия сульфат n2 n2 n2 900 990 1077 194,8 188,2 184,7
Никеля 0 17,4
Ni(CO)4 тетракарбо- 20 15,1
НИЛ 50 11,6
n2 -20 45,8
PBr3 Фосфора(Ш) бромид n2 n2 n2 0 20,8 99,8 44,7 43,2 36
n2 170 26,3
n2 -70 37,4
PC13 Фосфора(Ш) хлорид n2 n2 0 35,2 29,3 25,8
n2 75,1 21,9
Ph Фосфора(Ш) иодид n2 n2 75,3 150 56,5 51,4
Фосфора ВЗД 15 32,77
POC13 оксид- взд 49 28,36
трихлорид взд 65 26,57
S2C12 Дисеры дихлорид n2 n2 n2 0 75 121 45,4 34,6 29,4
Продолжение таблицы 2П1.1
Формула Название Газ T,°C ст, мДж/м2
SbCl3 Сурьмы(Ш) хлорид n2 n2 n2 74,5 137 178 49,6 42,6 38,3
SeF4 Селена(1У) фторид -7,6 17,8 89,2 39,06 36,33 27,51
SiCl4 Кремния тетрахлорид n2 20 19,71
SnCl2 Олова(П) хлорид n2 n2 n2 307 405 480 97 89 81,6
T1NO3 Таллия нитрат n2 n2 n2 210 312 430 117,3 109,8 99,5
UF6 Урана(У1) фторид взд взд взд взд 70 80 90 100 16,8 15,6 14,3 13,1
Таблица ЗП1.1
Вода на границе с воздухом
T, °C CT, мДж/м2 T, °C CT, мДж/м2 Д °C ст, мДж/м2
-10 77,1 16 73,34 30 71,15
-5 76,4 17 73,2 35 70,35
0 75,62 18 73,05 40 69,55
5 74,9 19 72,89 45 68,73
6 74,76 20 72,75 50 67,9
7 74,62 21 72,6 60 66,17
8 74,48 22 72,44 70 64,41
9 74,34 23 72,28 80 62,6
10 74,2 24 72,12 90 60,74
11 74,07 25 71,96 100 58,84
12 73,92 26 71,8 110 56,89
13 73,78 27 71,64 120 54,89
14 73,64 28 71,47 130 52,84
15 73,48 29 71,31
Таблица 4П1.1
Смеси Н2О и D2O при 20 °C
w(H2O), масс. % w(D2O), масс. % ст, мДж/м2 w(H2O), масс. % m'(D2O), масс. % ст, мДж/м2
100 0 72,75 8 92 68,1
69 31 71,5 0 100 67,8
34,5 65,5 69,8
no
Новый справочник химика и технолога
Таблица 5Ш. 1
Органические вещества
Таблица 5аП1.1
Алифатические углеводороды
Формула Название Газ Г, °C ст, мДж/м2
С4Н10 Бутан взд взд -20 0 17,34 14,84
СдНю Изобутан взд 10 14,07
с5н8 Изопрен взд 22 22,05
С5н]2 Пентан взд взд взд 20 30 40 16,09 15 13,8
С5н12 Изопентан взд взд 20 30 15 13,93
С6н14 Гексан взд взд взд взд 20 30 40 60 18,41 17,37 16,32 14,33
С6Н14 2-Метилпентан взд взд 20 40 17,38 15,36
СбН14 3-Метил пентан взд взд 20 40 18,12 16,03
С6н14 2,2-Диметилбутан взд взд 20 40 16,3 14,33
С6н14 2,3-Диметилбутан взд взд 20 40 17,37 15,37
С7н16 Гептан взд взд взд 20 30 40 20,29 19,27 18,26
С7Н16 Гептан взд 90 13,6
С7н16 2-Метилгексан взд 20 19,21
С7н16 З-Метилгексан взд взд 20 20 19,82 20,46
С7н16 2,2-Диметилпентан взд 20 18,03
С7н16 2,3-Диметилпентан взд 20 19,82
С7н16 2,4-Диметилпентан взд 20 18,12
С7н16 3,3-Диметилпентан взд 20 19,61
С7н16 2,2,3 -Триметил-бутан взд 20 18,76
с8н18 Октан взд взд взд взд 20 30 40 120 21,78 20,8 19,82 12,6
Нонан взд взд взд 20 30 150 22,96 22,01 11,6
Продолжение таблицы 5аП1.1
Формула Название Газ Т,°С ст, мДж/м2
взд 20 23,89
CioH22 Декан взд 30 22,97
взд 150 12,8
взд 20 24,78
спн24 Ундекан взд 30 23,89
взд 150 13,7
взд 20 25,48
С1гН2б Додекан взд 30 24,6
взд 150 14,7
С1зН28 Тридекан взд взд 20 150 26,13 15,6
С14Н30 Тетрадекан взд взд 20 150 26,69 16,2
С15н32 Пентадекан взд взд 20 150 27,17 16,9
С]бНз4 Гексадекан взд взд 20 150 27,64 17,4
С17Н36 Гептадекан взд взд 20 100 28,06 21,35
С18Нз8 Октадекан взд взд 20 100 28,42 21,75
СгоНдг Эйкозан взд взд 40 100 27,2 23
Таблица 56П1.1
Циклические углеводороды
Формула Название Газ т,°с ст, мДж/м2
взд 15 23,28
с5н10 Циклопентан взд 20 22,65
взд 30 21,32
взд 10 30,24
взд 20 28,87
ед Бензол взд 30 27,5
взд 40 26,13
взд 80 21,2
С6н8 Циклогексадиен взд 20 60 27,32
взд 22,22
взд 20 24,95
С6н12 Циклогексан взд 30 23,75
взд 60 21,35
взд 0 30,93
взд 20 28,53
ед Толуол взд 30 27,33
взд 40 26,13
взд 100 18,93
Приложение 1
771
Продолжение таблицы 56П1.1
Формула Название Газ Т, °C СГ, мДж/м2
СвНю о-Ксилол взд взд взд взд 20 30 40 100 30,03 28,93 27,84 21,27
CsHio .w-Ксилол взд взд взд взд 20 30 40 100 28,63 27,53 26,44 19,87
С8Н10 л-Ксилол взд взд взд взд 20 30 40 100 28,31 27,22 26,13 19,59
С8Н10 Этилбензол взд взд взд взд 20 30 40 100 29,04 27,91 26,78 20
С9Н12 Пропилбензол взд взд взд взд 20 30 40 100 29 27,91 26,83 20,32
С9Н12 Кумол (изопропилбензол) взд 20 28,2
С9Н12 1,2,3 -Трим етил-бензол (гемимеллитол) взд 20 31,27
CjoHg Нафталин взд 80 32,26
СюН]2 Тетралин (1,2,3,4-тетра-гидронафталин) взд 19,3 36,28
СюН18 Декалин (декагидронафталин) взд 19,8 29,89
С12Н10 Бифенил пар пар 129,2 179,7 28,64 24,04
с14н14 1,2-Дифенилэтан пар 108,3 27,86
Ci9H]6 Трифенилметан n2 156 32,7
Таблица 5вП1.1
Галогензамещенные углеводороды
Формула Название Газ Г, °C о, мДж/м2
ССЦ Четыреххлористый углерод пар пар 20 100 25,68 16,48
СНС13 Хлороформ взд пар пар 20 10,2 45,5 27,14 27,62 23,03
СНВгз Трибромметан 20 51
Продолжение таблицы 5вП1.1
Формула Название Газ Г, °C о, мДж/м2
СН2С12 Дихлорметан взд взд взд 15 20 30 28,83 28,12 26,54
СН3С1 Хлорметан взд 20 16,26
С2С14 Т етрахлорэтил ен пар 20 31,74
С2НС13 Трихлорэтилен взд взд взд 15 20 30 29,73 29,1 27,76
С2Н2С14 1,1,2,2-Тетрахлор-этан n2 взд 0 22,5 36,7 36,03
С2Н2Вг4 1,1,2,2-Тетрабром-этан взд 20 49,6
С2Н3С1з 1,1,1 -Трихлорэтан взд взд взд 15 20 30 26,17 25,56 24,25
С2Н3С1з 1,1,2-Трихлорэтан взд взд взд 15 20 30 34,37 33,83 32,78
С2Н4С12 1,1-Дихлорэтан n2 n2 0 30 25,7 22,4
С2Н4С12 1,2-Дихлорэтан n2 n2 0 30 34,1 30,1
С2Н4Вг2 1,2-Дибромэтан пар пар 12,2 45 38,91 34,5
С2Н5Вг Бромэтан пар 20 24,15
С2Н51 Иодэтан n2 20 28,1
c2f3ci3 1,1,2-Трифтортри-хлорэтан (фреон 113) взд 20 17,75
C5F10 Декафторциклопентан 20 11,09
c5F12 Додекафторпентан 20 9,87
c5f12 Додекафторизопентан 20 10,48
С6Н5С1 Хлорбензол взд взд n2 n2 20 30 25 50 33,19 31,98 32,9 29,6
C6H5Br Бромбензол взд n2 n2 25 35,6 71,5 35,75 35,2 31
с6н51 Иодбензол n2 n2 0 25,1 39,1 37,1
C7Fi6 Гексадека-фторгептан 20 12,6
C8Fj8 Октадекафтороктан 20 13,6
772
Новый справочник химика и технолога
Таблица 5гП1.1
Спирты, тиоспирты, фенолы
Таблица 5дП1.1
Простые эфиры, альдегиды, кетоны
Формула Название Газ Г, °C о, мДж/м2
взд 15 22,99
СН4О Метанол взд 20 22,55
взд 30 21,69
n2 -79 30,6
n2 -24 25,2
пар 20 22,03
с2н6о Этанол пар пар 40 60 20,2 18,43
пар 80 16,61
пар 100 14,67
пар 200 3,99
с2н602 Этан-1,2-диол пар 20 46,1
c2H6s Этантиол (этилмеркаптан) взд 20 22,4
взд 15 26,15
С3НбО Проп-2-ен-1-ол взд 20 25,68
взд 30 24,92
с3н8о Пропан-1-ол n2 25 22,9
с3н8о Пропан-2-ол пар 18 21,2
с3н8о3 Глицерин (99,19%) взд взд взд 18 30 90 62,47 62,08 57,65
пар, 0 26,2
взд
С4Н10О Бутан-1-ол пар, взд 20 24,6
пар, 50 22,1
взд
С4Н10О 2-Метилпропан-1 -ол пар 18 22,7
с4н10о 2-Метилпропан-2-ол пар, взд 20 20,7
взд 15 26,03
с5н12о Пентан-1-ол взд 20 25,6
взд 30 24,72
с5н120 З-Метилбутан-1 -ол взд 18 24
взд 50 37,66
С6Н6О Фенол взд 60 36,57
взд 70 35,51
взд 20 26,54
С6Н12О Циклогексанол взд 30 25,22
взд 60 21,54
с7н80 Фенилметанол взд 20 42,76
с8н18о Октан-1 -ол взд 20 27,53
Формула Название Газ Т, °C а, мДж/м2
С2НС13О Трихлоруксусный альдегид пар 20 24,4
С2Н4О Уксусный альдегид пар 20 21,2
С3Н6О Ацетон пар, взд 20 23,7
пар, 20 24,6
с4н8о Бутанон взд
пар 75 17,8
20 35,42
с4н8о2 1,4-Диоксан 30 33,97
90 25,97
пар 20 16,49
С4Н10О Диэтиловый эфир пар 30 15,27
пар 40 14,05
с5н4о2 2-Фур альдегид n2 n2 0 58,3 43,5 37
с5н8о2 Пентан-2,4-дион n2 0 25 31,6 29,2
взд 15 25,87
с5н10о Пентан-3-он взд 20 25,26
взд 30 24,37
С6Н12О3 Паральдегид пар взд 15 20 26,77 25,9
С6Н14О Дипропиловый эфир взд 20 20,55
С7Н6О Бензальдегид пар 15 39,2
C8Fi8O Перфтордибутиловый эфир 20 12,2
С8Н8О Ацетофенон взд 15 40,09
(метилфенилкетон) взд 30 39,15
С8Н18О Дибутиловый эфир взд 20 22,93
С]3НюО Бензофенон (дифенилкетон) пар 33,5 42,38
Таблица 5еП1.1
Кислоты и их производные
Формула Название Газ Г, °C мДж/м2
взд 15 38,13
СН2О2 Муравьиная кислота взд 20 37,58
взд 30 36,48
пар 20 58,2
CH3NO Формамид n2 0 59,6
n2 105 50,1
Приложение 1
773
Продолжение таблицы 5еП 1.1
Формула Название Газ Т,°С ст, мДж/м2
С2НС13О2 Трихлоруксусная кислота n2 80 27,8
С2Н3С1О Ацетил хлорид пар 14,8 25,8
С2Н3С1О2 Хлоруксусная кислота n2 n2 80 150 33,3 26,6
c2h3n Ацетонитрил (нитрил уксусной кислоты) взд взд взд 15 20 30 29,76 29,1 27,8
С2Н4О2 Уксусная кислота взд взд взд 15 20 30 28,26 27,79 26,87
С3Н6О2 Пропионовая кислота взд взд взд 15 20 30 27,21 26,7 25,71
С4Н6О3 Уксусный ангидрид взд взд взд 15 20 30 33,37 32,65 31,22
с4н802 Масляная кислота взд взд взд 15 20 30 27,32 26,74 25,57
С4Н8О2 Изомасляная кислота пар 16 24,99
С5НюО2 Пентановая кислота взд взд взд 15 20 30 27,83 27,36 26,35
СдНюОг 3 -Мети лбутановая кислота взд взд взд 15 20 30 25,78 25,3 24,45
С6Н12О2 Гексановая кислота пар пар 25,7 100 27 21,3
с8н16о2 Октановая кислота пар пар 20 100 28,3 21,6
СюН2о02 Декановая кислота пар пар 31,9 90 27,7 23,4
Ci2H24O2 Лауриновая кислота пар пар 45 103,3 28,5 23,9
С]4Н28О2 Тетрадекановая кислота пар пар 56,8 150 28,6 21,3
С16Н32О2 Пальмитиновая кислота пар пар 65 100 28,6 26,9
Ci8H34O2 Олеиновая кислота взд пар пар 20 65 100 33,3 28,6 26,9
С18Н36О2 Стеариновая кислота 70 150 28,9 22,9
Таблица 5жП 1.1
Сложные эфиры
Формула Название Газ т,°с ст, мДж/м2
С2Н4О2 Метилформиат пар 20 24,62
С2Н4О2 Метилформиат пар пар пар 50 100 200 20,05 12,9 0,87
С3НбО2 Этилформиат взд взд 20 30 23,84 22,38
с3н6о2 Метилацетат взд взд взд 15 20 30 25,2 24,49 23,44
С4Н8О2 Этилацетат взд взд взд 15 20 30 24,36 23,75 22,25
CsHi0O2 Этил пропионат взд взд взд 15 20 30 24,83 24,27 23,16
CsHi0O2 Метилбутират пар пар 10 210 25,56 5,09
CsHi()O2 Метилизобутират пар 10 24,09
СзНюОэ Диэтилкарбонат взд взд 20 30 26,44 25,47
С5НюО3 Этиллактат взд 18 28,8
C6Hi0O3 Ацетоуксусный эфир n2 n2 25 125 32 21,7
C$Hio04 Диэтилоксалат взд взд взд 15 20 30 32,83 32,22 31,03
C6Hi2O2 Бутилацетат взд взд 20 25 24,81 24,3
c6H12o2 Этилбутират взд взд взд 15 20 30 25,09 24,58 23,26
c6H12o2 Этилизобутират n2 0 24,6
C?Hi2O4 Малоновый эфир взд 30 30,56
C7H14O2 Пентилацетат взд взд взд 15 20 30 26,36 25,85 24,8
c8H8o2 Метилбензоат n2 25 37,3
C9Hi0O2 Этилбензоат n2 25 34,6
C9H]o03 Этил-2 -гидроксибензоат n2 0 39,1
C9Hi4O6 Триацетин взд взд 20 100 35,15 27,03
774
Новый справочник химика и технолога
Продолжение таблицы 5жП1.1
Формула Название Газ Г,°C сг, мДж/м2
С15Н2бО6 Трибутирин взд взд 20 100 27,98 21,57
Ci8H36O2 Этилпальмитат 22 100 31,54 24,3
С19Н38О4 Пальмитоил-глицерин взд 100 20,78
CjiHsgOe Тригексаноил-глицерин (трикапроин) взд взд 20 100 26,32 21,68
С21Н42О4 Стеароилглицерин взд 100 18,64
С27Н50О6 Триоктаноил-глицерин (трикаприл ин) взд взд 20 100 25,68 20,68
С51Н98О6 Трипальмитин взд 100 22,57
С57Н104О6 Триолеин взд взд 20 100 28,71 23,17
СзтНцоОб Тристеарин взд 100 24,18
Таблица 5зП1.1
Амины
Формула Название Газ r,°c <5, мДж/м2
n2 -33 24,6
c2h7n Этиламин n2 0 21,4
n2 10 20,4
c2h7n Диметиламин n2 0 18,1
n2 0 26
c3h7n Аллиламин n2 20 23,6
n2 40 21,5
C3H9N Пропил амин n2 20 22,4
n2 0 23,8
C4HhN Бутиламин n2 25 21,2
n2 70 17,4
Изобутиламин n2 0 22,4
C4HhN 55,8
n2 17,7
n2 0 20,8
C4HuN Диэтиламин n2 25 18,2
n2 45 16,6
n2 25 21,9
c5hI3n Пентиламин n2 55 19,2
n2 99,8 4,67
c6h7n Анилин ВЗД 17,5 44,1
взд 20 43,66
C6H15n Дипропиламин n2 0 23,5
n2 50 18,2
Продолжение таблицы 5зП1.1
Формула Название Газ Г, °C сг, мДж/м2
C6H15N Триэтиламин взд взд взд 15 20 30 21,25 20,66 19,62
c7h9on 4-Метоксианилин (и-анизидин) взд 20 44,1
c7h9n 3 -Метиланилин взд 20 39,89
c7h9n о-Толуидин взд 20 40,8
c7h9n .w-Толуидин взд 20 38,3
c7h9n и-Толуидин взд 50 34,9
C8HhN А',А'-Диметиланилин пар, взд пар 20 99 36,6 26,8
c9h13n А'-Пропиланилин взд 20 34,17
с9н13ы А'-Метил-А'-этиланилин взд 20 35,48
C10H15N А-Бутиланилин взд 20 33,76
c„h17n 2-Метил-А',А/-диэтиланилин взд 20 30,19
Ci2HhN Дифениламин пар 77,2 36,66
C14H15N Дибензиламин n2 n2 25 100 38,5 31,7
Q4H23N А-Октиланилин взд 20 32,9
C22H39N АДУ-Диоктиланилин взд 20 31,7
Таблица 5иП1.1
Другие азотсодержащие соединения
Формула Название Газ Г, °C сг, мДж/м2
взд 15 30,9
cn4o8 Тетранитрометан взд 20 30,41
взд 30 29,21
взд 15 37,74
взд 20 36,98
CH3NO2 Нитрометан взд 30 35,51
n2 0 38,1
n2 100 25,4
c2h5no2 Нитроэтан пар 16,6 31,96
взд 15 38,06
c4h5n Пиррол взд 20 37,52
взд 30 36,23
c5h5n Пиридин n2 25 34,9
C5HhN Пиперидин пар 16,5 29,89
C6F15n Перфтортриэтил амин 20 13,6
Приложение 1
775
Продолжение таблицы 5иП1.1
Формула Название Газ Г, °C о, мДж/м2
c6h5no2 Нитробензол взд n2 n2 18 35 100 42,58 41,7 34,4
c6h7n 2-Метилпиридин (а-пиколин) взд взд взд 15 20 30 33,87 33,25 31,96
c7h7no2 1-Метил-2-нитробензол N2 25 40,9
c7h7no2 1-Метил-4-нитробензол n2 60 35,5
c9f21n Перфтортри-пропиламин 20 15
c9h7n Хинолин n2 n2 25 230 44,7 23
c10h14n2 Никотин взд взд 20,5 93,5 38,61 30,99
c12f27n Перфтортрибутил амин 20 16,8
c12h10n2o Дифенилдиазен-оксид N2 55,8 39,3
Таблица 5кП1.1
Прочие органические вещества и масла
Формула Название Газ Г, °C о, мДж/м2
С12Н220ц Тростниковый сахар (расплавленный) взд 160 66,9
С18Н15Р Трифенилфосфин (7™ =80 °C) взд 45,7 40,69
Масло вазелиновое (чистое) взд 20 31,8
Масло касторовое (р = 961,2 кг/м3) взд 18 36,4
Масло оливковое (р = 915,1 кг/м3) взд 18 33,06
Масло сезамовое DAB (р = 921,2 кг/м3) взд 18 31,78
Скипидар (масло терпентинное) (р = 853,3 кг/м3) взд 18 26,79
Керосин (р = 846,7 кг/м3) взд взд взд 0 25 50 28,9 26,4 24,2
Таблица 6П1.1
Сводные данные для ряда распространенных органических растворителей [1]
Вещество м, г/моль р • 10 3, кг/м3 (20 °C) о, мДж/м2, при температуре (°C)
20 30 40 50
Этанол 46,07 0,789 22,4 21,6 20,7 19,9
Пропан-1-ол 60,1 0,804 23,7 22,9 22,2 21,4
Пропан-2-ол 60,1 0,785 21,3 20,5 19,7 19
Бутан-1-ол 74,12 0,81 25,4 24,5 23,6 22,7
Пентан-1-ол 88,15 0,814 25,8 24,9 24 23,2
Пентан-2-ол 88,15 0,81 24 23 22 21
Гексан-1-ол 102,18 0,819 26,2 25,4 24,6 23,8
Гексан 86,18 0,659 18,4 17,4 16,3 15,3
Толуол 92,14 0,867 28,5 27,3 26,1 24,9
Хлороформ 119,38 1,489 27,3 26 24,7 23,4
Четырех- 153,82 1,595 27 25,8 24,6 23,4
хлористый
углерод
Уксусная 60,05 1,049 27,6 26,6 25,6 24,6
кислота
Хлорбензол 112,56 1,107 33,3 32,3 31,1 29,9
Нитробензол 123,11 1,203 43,9 42,7 41,5 40,2
Анилин 93,14 1,022 43,3 42,2 41,2 40,1
Ацетон 58,08 0,791 24 22,9 21,8 20,7
Таблица 7П1.1
Межфазное натяжение воды на границе с другими веществами [1]
Формула Вещество Т,°С о, мДж/м2
c6h7n Анилин 26 30 4,8 5,69
С8Н8О Ацетофенон 30 12,08
С7Н7С1 (Хлорметил)бензол 30 27,1
Бензин 20 48
с6н6 Бензол 25 30 34,1 33,1
с10н14 Бутилбензол 25,13 38,26
СбН14 Гексан 10 51,25
с6н14 Гексан 20 30 40 51,1 50,66 50,48
с7н16 Гептан 25 50,85
С]5Н2бОб Трибутирин 25 11,9
776
Новый справочник химика и технолога
Продолжение таблицы 7П1.1
Формула Вещество Г, °C о, мДж/м2
С57Н104О6 Триолеин 16 20,4
С9Н14О6 Триацетин 25 3,23
С10Н18 транс-Декалин 20 40 51,3 50,44
C10H18 г/ис-Декалин 20 40 51,74 51,12
С10Н22 Декан 20 30 51,24 50,71
c2H4Br2 1,2-Дибромэтан 20 40 37,2 35,03
C8HnN Л^У-Диметиланилин 25 25,57
с8н18 Изооктан 25 49,3
с9н12 Изопропилбензол 25 38,7
Керосин 20 30 48,3 46,3
С5Н10О2 3 -Мети лбутановая кислота 20 2,7
С8Н1бОг Октановая кислота 18 60 8,2 8
С6Н12О2 Гексановая кислота 20 5,2
С12Н24О2 Додекановая кислота 60 10,5
С]8Нз4О2 Олеиновая кислота 20 60 15,7 15
С18Н340з (9Z, 127?)-12-Г идроксиоктадец-9-еновая кислота (рицинолевая кислота) 16 14,2
С11Н20О2 Ундец-10-еновая кислота 25 10,1
С7н14о2 Гептановая кислота 20 6,6
Масло оливковое 20 18,2
Масло парафиновое 25 52,5
Масло хлопковое 30 20,8
Масло хлопковое* 70 28
Масло эвкалиптовое 30 16,1
С21Н42О4 Стеароилглицерин 70 1
c6h5no2 Нитробензол 15,13 25,13 26,65 25,51
c6h5no2 Нитробензол* 30 23,9
c8H18 Октан 10 20 51,01 50,98
С8Н18 Октан* 25 40 50,2 49,58
СбНрОз Паральдегид 30 9,6
с9н12 Пропилбензол 25,13 39,98
Продолжение таблицы 7П1.1
Формула Вещество Г, °C ст, мДж/м2
с5н12о Пентан-1-ол 30 40,11
С4Н10О Бутан-1-ол 25 1,8
с5н12о З-Метилбутан-1 -ол 18 4,42
с5н12о 3 -Мети лбутан-1 -ол * 20 5,4
с4н10о 2-Метилпропан-1 -ол 17 27 37 1,78 1,86 1,8
с8н18о Октан-1-ол 0 16 30 40 7,75 8,7 8,97 9,32
СнН24О Ундекан-1-ол 25 8,61
СюН]2 Тетралин 25 38,6
С7н8 Толуол 25 35,7
с7н8 Толуол* 30 34,4
СС14 Углерод четыреххлористый 25 43,4
ссц Углерод четыреххлористый 30 42,75
С4Н60з Уксусный ангидрид 30 3,6
с5н4о2 2-Фуральдегид 30 5,13
c9h7n Хинолин 30 2,87
С6Н5С1 Хлорбензол 25,13 37,93
СНС13 Хлороформ 30 31,39
С6н12 Циклогексан 20 40 51,01 50,15
с7н14о Гептаналь 0 20 40 60 10,78 13,74 14,82 12,13
С8Ню Этилбензол 25,13 38,26
Ci4Hi2O2 Бензилбензоат 30 23,82
С8Н8О2 Метилбензоат 30 16,91
СюН20О2 Пентилпентаноат 30 21,1
С4Н10О Диэтиловый эфир 10 18 10,19 10,6
С4Н10О Диэтиловый эфир* 20 30 9,7 11,13
С12Н24О2 Этилдеканоат 60 22
с8н16о2 Этилгексаноат 0 20 30 40 21,03 21,29 21,15 21,02
С,4Н28О2 Этилдодеканоат 60 25
Приложение 1
111
Продолжение таблицы 7П1.1
Формула Вещество Т,°С а, мДж/м2
С8Н16О2 Пентилбутират 30 21,92
С6Н12О2 Этилбутират 30 13,31
С1вНзбО2 Этилпальмитат 60 26,5
С12Н1бО3 Пентил-2-гидроксибензоат 30 29,83
С8Н8О3 Метил-2-гидроксибензоат 30 22,3
С7Н14О2 Пентилацетат 30 12,04
С7Н14О2 Пентилацетат* 70 10
СдНюОг Бензилацетат 30 15,15
С6Н12О2 Бутилацетат 25 14,5
С4Н8О2 Этилацетат 25 6,8
С4Н8О2 Этилацетат* 30 6,8
С11Н24О 1 -Этоксинонан 20 23,9
* Данные другого ряда измерений.
Таблица 8П1.1
Межфазное натяжение ртути на границе с водными растворами [1]
Растворенное вещество W, масс. % р-Ю’3, г/см3 Г. °C <7, мДж/м2
формула название
С2Н4О2 Уксусная кислота 5,3 1,006 20 344
С2Н6О Этанол 20 44,5 87,8 98,3 0,969 0,927 0,825 0,795 19-20 19-20 0 19-20 363,2 361,1 366,6 364
CuSO4 Меди сульфат 1,3 6,5 9,6 1,012 1,067 1,103 19-20 19-20 19-20 343,2 334,9 331,7
HCI Соляная кислота 1,15 6,85 24,7 37,8 1,004 1,032 1,122 1,19 19-20 19-20 19-20 19-20 362,8 356,1 342,4 335,7
н2о Вода 20 375
H2SO4 Серная кислота 2,15 10,6 1,015 1,071 19-20 19-20 337,5 319,7
КВг Калия бромид 1* 20 318,5
К2СО3 Калия карбонат 1* 20 381,6
К2С2О4 Калия оксалат Г 1,145 19-20 353,6
КС1 Калия хлорид Г 20 357,5
Продолжение таблицы 8П1.1
Растворенное вещество W, масс. % Р-10’3, г/см3 Т, °C с, мДж/м2
формула название
KI Калия иодид 1* 20 318,5
kno2 Калия нитрит 1* 20 379
KNO3 Калия нитрат 1* 20 338,5
NaC2H3O2 Натрия ацетат 3,1 1,014 19-20 379
NaOH Натрия гидроксид 0,7 7,3 27 1,006 1,079 1,296 19-20 19-20 19-20 407,1 423 429,4
Na2SO4 Натрия сульфат 1,3 6,4 10,7 1,01 1,057 1,098 19-20 19-20 19-20 371,8 371 377,3
ZnCl2 Цинка хлорид 10,4 40,6 56,3 1,094 1,426 1,683 19-20 19-20 19-20 359 328,7 304,7
* Приведена молярная концентрация, моль/л.
Таблица 9П1.1
Межфазное натяжение ртути на границе с органическими жидкостями [ 1 ]
Формула Вещество Т, °C а, мДж/м2
c6h7n Анилин 20 341
С3Н6О Ацетон 20 390,1
с6н6 Бензол 20 25 357 364,3
СюН14 Бутилбензол 25 362,5
С6Н14 Гексан 20 25 378 379,9
С7н16 Гептан 25 378,7
C10H23N Дипентиламин 20 371
С2Н4ВГ2 1,2-Дибромэтан 20 326
С2Н4С12 1,1-Дихлорэтан (хлористый этил идеи) 20 337
С]8Нз4О2 Олеиновая кислота 20 322
С11Н22О2 Ундекановая кислота 20 353
С8Н10 о-Ксилол 20 359
С8Ню м-Ксилол 20 357
С8Ню ц-Ксилол 20 361
СН2С12 Дихлорметан 20 441
c6h5no2 Нитробензол 20 25 350 349,5
778
Новый справочник химика и технолога
Продолжение таблицы 9П1.1
Формула Вещество Т,°С о, мДж/м2
C2H5NO2 Нитроэтан 20 378
Оливковое масло 20 301,8
с8н18 Октан 20 25 375 376
с9н12 Пропилбензол 25 363,1
CS2 Сероуглерод 20 339
С4Н10О Бутан-1-ол 25 372,8
С5н12о 3-Метил бутан-1 -ол 25 373,7
С4Н10О 2-Метилпропан-1 -ол 20 343
С8Н18О Октан-1-ол 20 352
С3Н8О Пропан-1-ол 20 25 368 376,5
СгНбО Этанол 20 25 364 376,9
С2Н2ВГ4 1,1,2,2-Тетраброметан 20 307
С7Н8 Толуол 20 25 359 363,6
ссц Углерод четыреххлористый 20 362
СНС13 Хлороформ 20 357
C2H5I Иодэтан 20 306
С4Н10О Диэтиловый эфир 20 379
Литература
1. Справочник химика. Т. 1. Л.;М.: Госхимиздат, 1962. 1070 с.
1.2. Поверхностное натяжение растворов
Таблица 1П1.2
Изотермы поверхностного натяжения (мН/м) водных растворов полярных веществ [1]
Пояснение'. Метод МД — метод максимального давления пузырька газа, метод ДН — метод Дю-Нуи (отрыв кольца).
Метанол СН4О (метод МД)
с, моль/л Т, °C
10 20 30 40 50 60
0,0244 62,5 60,9 59,3 57,7 56,1 54,5
0,0476 57 55,7 54,4 53 51,9 50,6
0,0909 48,8 47,8 46,8 45,8 44,8 43,8
0,1667 41,3 40,6 39,8 39,1 38,4 37,7
0,25 36,6 36 35,5 35 34,3 33,8
0,3333 33,6 33,1 32,6 32,1 31,6 31,2
0,5 30,1 29,5 28,9 28,3 27,6 27,1
1 23,5 22,6 21,8 20,9 20,1 19,3
Продолжение таблицы 1П1.2
Этанол С2Н6О (метод МД)
с, моль/л т,°с
10 20 30 40 50 60
0,0244 57,6 56 54,5 53 51,4 50,8
0,0476 48,2 46,8 45,5 44,2 42,8 41,7
0,0909 39 38 37 36 35,1 34,2
0,1667 32,1 31,5 31 30,3 29,7 29
0,2 30,8 30,2 29,6 29,1 28,6 28
0,25 29,2 28,3 28,2 27,7 27,2 26,7
0,3333 28 27,3 26,8 26,2 25,6 25
0,5 26,2 25,5 24,8 24 23,3 22,6
1 23,6 22,8 21,9 21 20,1 19,2
Ацетон С3Н6О (метод МД)
с, моль/л Т,°С
0 5 10 15 20 25 30
0 75,7 74,96 74,27 73,51 72,75 71,98 71,21
0,005 64,54 63,84 63,15 62,53 61,99 61,37 60,75
0,011 61,22 60,52 59,7 58,9 58,13 57,42 56,65
0,023 55,12 54,27 53,5 52,6 51,72 51,02 50,26
0,034 51,35 50,5 49,57 48,73 47,8 47,11 46,26
0,054 47,34 46,5 45,72 44,87 44,23 43,41 42,63
0,141 37,7 37,08 36,31 35,77 35,16 34,39 33,85
0,365 30,45 29,91 29,37 28,91 28,14 27,68 27,06
1 26,24 25,48 24,78 24,09 23,62 23 22,39
Диметилформамид C3H7NO (метод МД)
w, масс. % Т,°С
25 40 60 80
0 71,96 69,8 66,2 62,78
10,03 58,647 57,1 54,33 51,703
20,1 55,697 53,538 50,551 47,853
30,23 52,89 50,623 47,56 44,31
39,72 51,349 48,573 45,875 42,81
49,99 48,825 46,306 43,428 39,938
60,24 46,954 44,723 42,024 39,038
69,82 44,19 41,9 39,39 36,52
79,81 41,19 38,89 36,78 34
89,83 38,391 36,52 34,001 31,493
100 37,31 35,55 33,28 31,12
Приложение 1
779
Продолжение таблицы 1П1.2
Глицерин СзН8О3 (метод МД)
Продолжение таблицы 2П1.2
Калия ундеканоат КСцН21О2 (Т= 20 °C, метод ДН)
w, масс. % Т,°С
20 40 60 80 100
0 72,8 69,6 66,2 62,6 58,9
10 72,5 69,4 66,1 62,6 58,9
20 72 69,1 66 62,6 58,8
30 71,4 68,7 65,8 62,5 58,7
40 70,7 68,3 65,5 62,3 58,6
50 69,8 67,7 65,1 62 58,5
60 68,8 67 64,5 61,6 58,3
70 67,6 66 63,8 61,1 58
80 66,3 64,9 62,9 60,5 57,6
90 65 63,4 61,7 59,6 57,1
100 63 61,6 60,2 58,4 56,4
Диоксан С4Н8О2 (метод МД)
w, масс. % Г, °C
20 40 60 80
10 57,77 53,83 49,01 45,92
40 39,67 37,41 35,03 32,97
50 38,36 36,64 33,95 32,46
60 35,15 33,13 31,07 29,56
70 32,52 30,44 28,33
90 33,99 30,42 28,7 25,57
Таблица 2П1.2
Изотермы поверхностного натяжения водных растворов солей жирных кислот [1]
Калия деканоат КСюН19О2 (Т = 20 °C, метод ДН)
w, масс. % о, мН/м w, масс. % о, мН/м
0,00098 70,0 0,125 30,0
0,00195 68,0 0,25 26,0
0,0039 63,0 0,5 25,0
0,0078 56,0 1 24,0
0,0156 48,0 2 26,0
0,0312 37,5 4 26,5
0,0625 32,0
Калия деканоат KC)0Hi9O2 (Г = 50 °C, метод ДН)
w, масс. % ст, мН/м w, масс. % ст, мН/м
4,87 • 10 4 64 6,25 • 10’2 48
9,75 • Ю 4 64 0,125 43
1,95 • 10 3 64 0,25 38
3,90- 10’3 54 0,5 35
7,80- 10’3 54 1 28
1,56- 10’2 54 2 31
3,12* 10’2 54 4 36
w, масс. % ст, мН/м w, масс. % ст, мН/м
2,40 • 104 69 6,25 • 10’2 34
4,90 • Ю4 64 0,125 32
9,80 • 10 4 61 0,25 29
1,85 • 10~3 56 0,5 26
3,90- 10“3 56 1 28
7,80 • 10’3 51,5 2 36
1,56- 102 48,5 4 38
3,12 • 10 2 44
Калия ундеканоат КСцН21О2 (Т = 50 °C, метод ДН)
w, масс. % ст, мН/м w, масс. % ст, мН/м
2,43 • Ю4 63,5 6,25 • 10 2 38
4,87 • 10 4 63,5 0,125 29
9,75 • 10 4 63 0,25 25
1,95 • 10’3 58 0,5 23
3,90- 10’3 56 1 27
7,80 • 10 3 50 2 33
1,56- Ю 2 45 4 34
3,12- 10 2 41
Калия додеканоат КС12Н23О2 (Г= 20 °C, метод ДН)
w, масс. % ст, мН/м w, масс. % ст, мН/м
6,10 • 10 5 67,5 3,12 • 10 2 31
1,22 • 10 4 66 6,25 • Ю 2 34
2,43 • Ю4 64 0,125 27
4,87 • 10 4 62 0,25 29
9,75 • 10 4 57,5 0,5 30
1,95 • 10’3 55 1 31
3,90 • 10’3 55 2 37
7,80 • Ю 3 44 4 35,5
1,56- Ю 2 38
Калия додеканоат KCi2H23O2 (Г = 50 °C, метод ДН)
w, масс. % ст, мН/м w, масс. % ст, мН/м
6,10- 10~5 56 3,12- 10’2 46
1,22 • 10 4 56 6,25 • 10’2 38
2,43 • 10^ 56 0,125 30
4,87 • 104 55 0,25 23
9,75 • 10 4 55 0,5 23
1,95 • 10 3 54 1 32
3,90 • 10’3 53 2 34
7,80 • Ю 3 52 4 34
1,56 • 10 2 48
780
Новый справочник химика и технолога
Продолжение таблицы 2П1.2
Калия тридеканоат КС13Н25О2 (Т = 20 °C, метод ДН)
Продолжение таблицы 2П1.2
Калия пентадеканоат КС15Н29О2 (Т = 20 °C, метод ДН)
xv, масс. % о, мН/м w, масс. % о, мН/м
6,10- 10’5 65 3,12- 10’2 29
1,22 • Ю4 65 6,25 • 10’2 22,5
2,43 • Ю4 60 0,125 22,5
4,87 • 10-4 52 0,25 22
9,75 • Ю 4 50 0,5 26
1,95 • 10’3 41 1 31
3,90 • 10’3 38,5 2 33
7,80 • 10~3 36,5 4 33
1,56- Ю 2 34
w, масс. % о, мН/м xv, масс. % о, мН/м
4,90 • 10-4 58 6,25 • 10’2 47
9,80 • Ю4 57 0,125 44,5
1,95 • 10 3 53 0,25 41,5
3,90 • 10 3 51,5 0,5 40
7,80 • 10 3 51,5 1 37,5
1,56 • 10 2 51,5 2 37,5
3,12 • 10 2 48 4 35,5
Калия тридеканоат КС13Н25О2 (Т = 50 °C, метод ДН)
xv, масс. % о, мН/м w, масс. % о, мН/м
6,10- 10’5 57,5 3,12 • 10'2 34
1,22 • Ю 4 57 6,25 • 10’2 30
2,43 • Ю4 57 0,125 31
4,87 • Ю4 44 0,25 33
9,75 • Ю4 43,5 0,5 34
1,95 • 10’3 41 1 34,5
3,90 • 10’3 37 2 33,5
7,80- Ю 3 36,5 4 32,5
1,56 • 10’2 36
Калия пентадеканоат КС15Н29О2 (Т = 50 °C, метод ДН)
xv, масс. % о, мН/м xv, масс. % о, мН/м
6,10 • ю 5 57 3,12- 10’2 29
1,22 • 10 ’ 57 6,25 • 10’2 25
2,34- 1 О’ 47 0,125 22,5
4,87 • 10 ’ 42 0,25 23,5
9,75 • Ю 4 41 0,5 26
1,95 • Ю 3 40 1 26
3,90 • Ю 3 39 2 30
7,80 • 10 3 38 4 31
1,56 • 10 2 34,5
Калия тетрадеканоат КС14Н27О2 (Т = 20 °C, метод ДН)
xv, масс. % о, мН/м xv, масс. % (7, мН/м
6,10- ю 5 54 3,12 • 10’2 43,5
1,22 • Ю 4 53 6,25 • Ю2 43,5
2,40 • 10 4 52 0,125 37
4,90- 10 4 51 0,25 36,5
9,80- Ю 4 46 0,5 35
1,85 • Ю 3 46 1 35
3,90- Ю 3 45 2 35
7,80 • 10’3 45 4 33
1,56 • 10~2 43,5
Калия тетрадеканоат КС14Н27О2 (Т = 50 °C, метод ДН)
xv, масс. % о, мН/м и', масс. % о, мН/м
2,43 • Ю4 64 6,25 • 10~2 22,5
4,87 • Ю 4 64 0,125 22,5
9,75 • Ю 4 56 0,25 24
1,95 • 10 3 55 0,5 28
3,90 • 10 3 54 1 30
7,80 • 103 42 2 32
1,56 • 10’2 31 4 32
3,12- 10’2 24,5
Калия гексадеканоат КС]6Н31О2 (Т = 20 °C, метод ДН)
xv, масс. % о. мН/м xv, масс. % о, мН/м
7,80 • Ю 3 50,7 0,25 44,5
1,56- 10’2 41 0,5 38,5
3,12 • 10’2 41 1 37,2
6,25 • Ю 2 41,5 2 35
0,125 48 4 35,5
Калия гексадеканоат KCi6H3)O2 (Т = 50 °C, метод ДН)
xv, масс. % (7, мН/м xv, масс. % (7, мН/м
6,10- 10 5 48 3,12 • 10’2 29
1,22- 104 45 6,25 • Ю 2 28
2,34 • 10 4 44 0,125 27
4,87 - 104 43 0,25 25,5
9,75 • 10 4 42 0,5 26
1,95 • Ю 3 39 1 26
3,90 • 10 3 37 2 28
7,80 • 10 3 32 4 30
1,56- 102 30
Приложение 1
781
Продолжение таблицы 2П1.2
Калия гептадеканоат КС17Н33О2 (Т = 20 °C, метод ДН)
w, масс. % о, мН/м w, масс. % ст, мН/м
9,80 • 10"4 63 0,125 53
1,95 • 10’3 62 0,25 51
3,90- 103 59 0,5 51
7,80 • 10’3 59 1 49
1,56 • 10’2 58 2 47
3,12 • 10”2 58 4 44
6,25 • 10“2 57
Калия гептадеканоат КС17Н33О2 (Т= 50 °C, метод ДН)
W, масс. % ст, мН/м w. масс. % ст, мН/м
6,10- 10"5 46 3,12 • 10’2 29
1,22 • 10"4 44 6,25 • 10’2 33
2,34 • 104 43 0,125 32
4,87- 104 39 0,25 30,5
9,75 • 10"4 38,5 0,5 29,5
1,95 • 10“3 34,5 1 28,5
3,90- 10~3 33,5 2 28,5
7,80 • 10 3 32 4 29
1,56- 10"2 30
Калия октадеканоат КС18Н35О2 (Т= 20 °C, метод ДН)
w, масс. % ст, мН/м w, масс. % ст, мН/м
7,80 • 10 3 66,1 0,125 57
1,56- 102 66 0,25 53
3,12 • 10’2 66 0,5 49
6,25 • 10'2 62 1 48
Калия октадеканоат КС18Н35О2 (Т= 50 °C, метод ДН)
w, масс. % ст, мН/м w. масс. % ст, мН/м
6,10- 105 46 1,56- 10~2 36,5
1,22 • 10-4 45 3,12 • 10'2 40
2,34- 10" 43 6,25 • Ю 2 39,5
4,87 • 104 40 0,125 36,5
9,75 • 10"4 43,5 0,25 36
1,95 • 10’3 41,5 0,5 33
3,90- 10“3 40,5 1 34
7,80 • 10’3 38,5 2 34
Натрия децилсульфат NaCioH2i04S (Т= 19 °C, метод ДН)
с 103, моль/л с, мН/м с 103, моль/л ст, мН/м с • 103, моль/л ст, мН/м
0,1 72,8 4 63,1 20 42,2
0,5 72,6 6 53,6 30 37,6
1 72,2 8 54,6 50 39,6
2 68 10 53,6
Продолжение таблицы 2П1.2
Натрия додецилсульфат NaCi2H25O4S (Т = 19 °C, метод ДН)
с • 103, моль/л ст, мН/м с • 103, моль/л СТ, мН/м с 103, моль/л ст, мН/м
0,1 72,1 1 57,8 5 40
0,25 70,9 2 48,4 6 38,8
0,5 69,9 3 48,4 7 38,4
0,75 65 4 40,7 8 38,4
Таблица ЗП 1.2
Изотермы поверхностного натяжения водных растворов катионоактивных ПАВ [1]
На границе с воздухом или органической жидкостью, указанной после знака «—».
Приведена молярная концентрация, моль/л.
Диметил-2-[(2-этилгексил)окси]этиламмоний бромид
C4H9CH(C2H5)CH2OCH2CH2N+H(CH3)2Br
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
4,5 72,4 2,5 62,9 1,5 44,2
3,8 72,5 2,2 59,2 1,2 36,9
3 69,7 2 54,8 1 34,5
2,8 69,6 1,8 51,3 0,9 33,6
Диметил-2-[(2-этилгексил)окси]этиламмоний бромид—гептан
C4H9CH(C2H5)CH2OCH2CH2N+H(CH3)2Br” — С7Н16
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
4,8 48,5 3,5 44,8 2,2 32,1
4,5 48,4 3 43,9 2 28,8
4 47,9 2,8 40,9 1,8 24
3,8 47,1 2,5 37,1 1,5 16,4
Додецилтриметиламмоний бромид [С12Н25ЬГ (СН3)3]Вг
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
4,8 72,6 3,5 68,6 2,2 43,3
4,5 70,7 2,8 62,7 1,6 39
4 71,7 2,5 55,9 1,5 37,6
3,8 69,9
Додецилтриметиламмоний бромид—гептан [C12H25N+(CH3)3]Br~ — С7Н16
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
5,8 50,4 4,5 45 3 36,9
5,5 49,5 4 41,9 2,8 31,9
5 47,5 3,8 42,3 2,5 25
4,8 46,7 3,5 39,5 2,2 14,6
782
Новый справочник химика и технолога
Продолжение таблицы ЗП1.2
Гексадецилтриметиламмоний бромид (ЦТАБ) [C16H33N+(CH3)3]Br- (метод ДН)
с, моль/л мН/м с, моль/л ст, мН/м с, моль/л ст, мН/м
0 72,8 1 66,1 25 47,7
0,1 72,3 5 60,2 100 38,5
0,25 71,6 10 52,6 500 38,1
0,5 66,7 15 52,3
Продолжение таблицы ЗП1.2
Гексадецилтриметиламмоний бромид (ЦТАБ)—гептан-1-о л
[Cl6H33N+(CH3)3] ВГ — С7Н15ОН (метод ДН)
с, моль/л ст, мН/м с, моль/л СТ, мН/м с, моль/л СТ, мН/м
0 9,7 0,5 9 10 3
0,1 9,7 1 9 15 0,5
0,25 9,4 5 9
Гексадецилтриметиламмоний бромид (ЦТАБ)—четыреххлористый углерод
[C]6H33N+(CH3)3]Br — СС14 (метод ДН)
с, моль/л ст, мН/м с, моль/л ст, мН/м с, моль/л ст, мН/м
0 46,3 0,5 42 10 16,2
0,05 45,9 1 32,7 15 14,4
0,1 45,6 5 17,3 25 6,5
0,25 43,6
Тридецилтриэтил аммоний бромид [C13H27N+(C2H5)3]Br-
Гексадецилтриметиламмоний бромид (ЦТАБ)— гептан
[C16H33N+(CH3)3]Br“ — С7Н16 (метод ДН)
с, моль/л ст, мН/м с, моль/л ст, мН/м с, моль/л ст, мН/м
0 50,2 0,5 37,6 10 18,8
0,1 44,1 1 35,6 15 11,5
0,25 42,5 5 19,2 25 7,2
Гексадецилтриметиламмоний бромид (ЦТАБ)— ксилол
[CJ6H33N+(CH3)3]Br — С6Н4(СН3)2
с, моль/л а, мН/м с. моль/л ст, мН/м с, моль/л СТ, мН/м
0 36,1 0,5 29,8 10 10
0,1 33,3 1 28,8 15 1
0,25 30,8 5 20,4
Гексадецилтриметиламмоний бромид (ЦТАБ)—нона-1-ол
[C16H33N+(CH3)3]Br- — С9Н19ОН (метод ДН)
с, моль/л СТ, мН/м с, моль/л ст, мН/м с, моль/л СТ, мН/м
0 10,7 0,25 10,5 1 9
0,1 10,7 0,5 10,4 5 1
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
4,9 71,7 3,9 55 2,3 33,2
4,6 68,2 3,6 39,2 1,9 34,5
4,1 58 2,6 39,3
Тридецилтриэтиламмоний бромид—гептан [C13H27N+(C2H5)3]Br“ — С7Н16
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
5,6 5,1 47,5 46,9 4,9 4,6 45,6 41,5 4,1 3,9 29,5 22,6
Пентадецилтриэтил аммоний бромид
[C15H31N+(C2H5)3]Br-
-Igc ст, мН/м -Igc СТ, мН/м -Igc ст, мН/м
5,1 71,1 4,1 66,5 3,1 50,1
4,9 71,1 3,9 63,6 2,6 39,6
4,6 69,3 3,6 59,1
Гексадецилтриметиламмоний бромид (ЦТАБ)— этилацетат
[C16H33N+(CH3)3]Br“ — С2Н5ОСОСН3 (метод ДН)
Пентадецилтриэти л аммоний бромид—гептан [C15H31N+(C2H5)3]Br — С7Н16
С, моль/л Ст, мН/м с, моль/л ст, мН/м с, моль/л СТ, мН/м
0 7,4 0,5 7,7 10 4
0,1 7,7 1 7,7 15 3
0,25 7,7 5 4-4 25 1
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
5,1 4,9 4,6 71,1 71,1 69,3 4,1 3,9 3,6 66,5 63,6 59,1 3,1 2,6 50,1 39,6
Приложение 1
783
Продолжение таблицы ЗП1.2
Пентадецилтриэтиламмоний бромид в 0,1 М КВг [C15H3IN+(C2H5)3]Br- (Т= 15 °C, метод ДН)
-Igc <7, мН/м -Igc ст, мН/м -Igc о, мН/м
5,9 71,8 4,9 56,4 3,9 38,5
5,6 70,1 4,6 49,2 3,6 38
5,1 60,2 4,1 39
Пентадецилтриэтиламмоний бромид в 0,4 М КВг [Cl5H31N+(C2H5)3]Br- (Т= 14 °C, метод ДН)
-Igc о, мН/м -Igc о, мН/м -Igc о, мН/м
6,6 73,7 5,1 54,9 4,1 36,1
6,1 70,1 4,9 48,8 3,9 34,5
5,9 68,8 4,6 41,5 3,6 34
5,6 64,9
Пентадецилтриэтиламмоний бромид в 0,6 М КВг [Cl5H31N+(C2H5)3]Br- (Г= 14 °C, метод ДН)
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
6,1 73,5 5,1 52,2 4,1 37,2
5,9 68,5 4,9 47,4 3,9 37,3
5,6 62,2 4,6 38,7
Пентадецилтриэтиламмоний бромид в 1,0 М КВг [C15H31N+(C2H5)3]Br- (Т= 12 °C, метод ДН)
-Igc ст, мН/м -Igc ст, мН/м -Igc ст, мН/м
6,1 73,9 5,1 48,8 4,1 37,4
5,9 63,3 4,9 45,3 3,9 37.4
5,6 57,5 4,6 40,7 3,6 35,9
(4-Алкилфенил)триметиламмоний галогениды [C„H2m + 1C6H4N+(CH3)3]X- (Т= 20 °C, метод ДН)
п X
г СГ
с • 105, моль/л ст, мН/м с • 105, моль/л ст, мН/м
530 35,6 2900 37,2
О 220 39,2 1200 40,6
о 72 48,2 390 50,2
13 61,2 15 64,2
3,1 55,2 870 39
1Л 1,3 58,2 250 42,4
1U 0,42 68,4 100 58,8
0,17 72,4 22 68,4
Продолжение таблицы ЗП1.2
п Х‘
г СГ
с • 105, моль/л ст, мН/м с • 105, моль/л ст, мН/м
2,6 50,2 760 38
10 1,1 52,6 62 40,2
1Z 0,35 62,2 25 49,2
0,12 70,8 13 53,8
2,3 46,6 62 37,8
1 А 0,45 46,8 10 41,2
14 0,31 57,4 2,7 50,4
0,1 67,2 1,4 54,2
Таблица 4П1.2
Изотермы поверхностного натяжения водных растворов неионогенных ПАВ, мН/м [1]
Оксиэтилированные жирные спирты
С8Н17О(С2Н4О)„Н
с • 103, моль/л Г, К
283 288 293 298
п = 3
0 71,8
0,002 70,4
0,004 70,9 68,7
0,007 70 66,4
0,01 68,7 65,1
0,02 70,9 66,8 63,2
0,04 72 68,3 64 60,1
0,076 69,9 65,5 60,6 56,9
0,114 68,4 62,9 58,1 55,4
0,152 66,5 62,3 57,2 53,9
0,19 65,2 60,4 56,3 52,5
0,267 63,7 58,4 54,2 50,6
0,38 61,9 57,5 52,8 49,1
0,76 58,4 53,8 49,5 46
1,14 56,2 51,7 47,6 44,1
1,62 54,5 50,4 46,2 42,4
1,9 53,1 49,1 44,5 41,7
2,67 51 47,2 42,9 39,7
3,81 48,9 45,8 40,7 38,3
4,96 47,6 44,3 40,1 37,2
5,34 46,8 43,5 39,8 37
5,72 46,4 42,4 39,3 36,6
6,1 46 42 38,4 36,6
6,87 45,3 41,9 38,1 36,6
7,63 44,4 41 38 36,6
11,45 42,1 40,9 38 36,6
15,26 42,1 40,9 38 36,6
784
Новый справочник химика и технолога
Продолжение таблицы 4П1.2
с 103, моль/л Т, К
283 288 293 298
п = 6
0 70,5
0,012 71,9 66,3
0,025 70,2 68,1 63,5
0,05 69,1 65,8 61,6
0,076 67,7 64,5 59,9
0,101 66,4 63,3 58,8
0,126 70,3 65,2 62 57,9
0,177 68,7 63,8 60,4 56,5
0,253 66,2 62,7 59,1 54,7
0,507 63,5 58,9 55,4 51,8
0,76 61,8 57,4 53,6 50,1
1,01 59,9 56,1 51,7 48,9
1,26 59,2 54,5 51,3 47,9
1,77 57,4 52,9 49,5 46,7
2,53 55,3 51,3 47,6 44,2
3,29 53,6 50,1 46,5 44
3,55 53,2 49,4 46,3 43,5
3,8 52,8 49,2 46,2 43,2
4,06 52,5 48,6 45,7 43,1
4,56 52,1 48 44,6 42,3
5,07 51,4 47,5 43,9 41,8
7,61 50 45,7 42,5 40,5
10,15 47,3 43,9 41,6 40,5
12,69 45,9 43 41,6 40,5
15,22 45,3 43 41,6 40,5
20,3 45,3 43 41,6 40,5
25,38 45,3 43 41,6 40,5
38,07 45,3 43 41,6 40,5
п = 8
0,002 72
0,006 71,3
0,01 71,9 69,8
0,02 71,8 70,7 68,7
0,041 71,8 70,9 69,2 66,3
0,103 69,3 69,1 67,3 64,4
0,207 67,2 66 64,4 61,4
0,415 63,9 63,8 62 58,9
1,037 60,4 60,2 58,7 56,5
2,07 57,5 56,7 54,9 52,6
6,22 51,3 53,5 50,1 49,7
12,44 48,1 48,3 47,5 45,3
13,07 48 45,8 44,1 42,8
13,27 47,8 45,7 43,8 42,3
Продолжение таблицы 4П1.2
с - 103, моль/л Г. К
283 288 293 298
и = 8
13,48 47,6 45,5 43,6 42,1
14,52 47,5 45,3 43,5 42,1
16,59 46,2 45,2 43,4 42,1
20,74 45,9 44,4 43,4 42,1
31,12 45,9 44,4 43,4 42,1
41,49 45,9 44,4 43,4 42,1
п= 10
0,005 72
0,008 71,9
0,017 71,5 71,9 69,8
0,035 69,9 70,5 68,7
0,052 71,5 68,7 68,9 66,8
0,087 69,9 67,1 67,1 64,7
0,122 68 65,9 66 63,6
0,175 66,7 64 64,2 61,9
0,35 64,4 62,1 62,8 60,3
1,75 57,2 55,8 61,7 59,4
5,26 52,8 51,6 59,6 57,5
8,7 50,3 49,3 53,4 49,7
12,28 49,1 48 49,5 47,1
13,15 47,7 47,2 45,7
13,5 48,7 47,6 46 44,5
13,68 47,6 45,7 44,2
14,03 47,2 45,4 44
15,78 46,9 45,4 44
17,54 46,9 46,7 45,3 44
35,08 46,6 46,4 45,2 44
43,85 46,6 46,4 44,7 44
52,63 46,6 46,4 44,7 44
70,7 46,6 46,4 44,7 44
и = 12
0,001 72
0,006 71,9 71,9
0,007 70,5 70,2
0,015 71,8 68,6 69,6
0,03 71,9 70,4 67,1
0,06 66,1 65,6
0,076 69,8 68,1 63,7 63,9
0,151 67,5 65,8 60,8 63,8
0,303 64,7 62,7 58 61,6
0,759 61,2 59,6 55,3 58,9
1,5 58,4 56,7 51,3 55,2
4,55 54,3 52,3 49,9 53,5
Приложение 1
785
Продолжение таблицы 4П1.2
с • 103, моль/л Г, К
283 288 293 298
п= 12
7,59 52,4 51 47,5 49,1
15,1 49,7 48,7 47,2
16,7 45,9
18,2 45,6
19,7 45
21,2 46 45
22,7 47,5 48,3 45,8 45
45,59 47,2 46,7 45,8 45
83,58 47,2 46,7 45,8 45
30,39 47,2 46,7 45,8 45
п = 14
0,001 72
0,006 71,9 69,9
0,013 72 70,4 68,5
0,026 71,7 71,3 69 67,2
0,067 70,6 68,2 66,1 63,9
0,134 68,2 66,1 63,8 61,7
0,402 64,7 62,6 60,4 58,4
0,67 62,8 60,7 58,9 56,9
1,34 60 58,4 56,5 55,1
4,02 56,1 54,5 52,7 51,2
6,7 54,2 52,7 51,8 50
13,4 51,9 50,3 49,2 47,3
26,8 46
28,1 46,7 45,9
30,8 46,5 45,6
33,5 47,7 46,4 45,6
40,2 48,3 46,9 46,3 45,6
53,61 47,7 46,9 46,3 45,6
73,7 47,7 46,9 46,3 45,6
93,83 47,7 46,9 46,3 45,6
Оксиэтилированные жирные спирты
CioH210(C2H40)„H
с- Ю4, моль/л Г, К
283 288 293 298
п = 3
0,0017 71,9
0,0034 71
0,0103 71,8 67,4
0,0172 70,7 65,1
0,0344 71,8 68 62,5
0,069 70,1 65,3 59,2
Продолжение таблицы 4П1.2
с 104, моль/л Г, К
283 288 293 298
п - 3
0,103 71,7 68,2 63,6 56,1
0,172 70 65,9 60,4 53,7
0,344 67,6 62,6 56,5 49,3
0,689 63,7 57,8 51,9 44,5
1,034 61,3 55,1 49,2 41,1
1,72 56,2 51,5 45,2 38
2,41 54,5 48,9 43,2 34,7
3,44 51,3 45,4 40,1 32,8
5,22 47,8 41,7 36,3 30,8
6,89 45,9 37,8 34,8 30,8
10,3 43,1 37,2 34,6 30,8
13,7 39,5 37,2 34,6 30,8
17,2 39,5 37,2 34,6 30,8
24,1 39,5 37,2 34,6 30,8
34,4 39,5 37,2 34,6 30,8
41,3 39,5 37,2 34,6 30,8
с - 103, А
моль/л И -
0,001 70,1
0,002 67,5
0,005 71,5 64,3
0,007 71,8 70,1 63,5
0,011 70,3 68,3 59,4
0,024 71,3 67,7 66,2 56
0,047 68,4 63,9 62,7 53,1
0,071 66,7 62,3 58,8 50,3
0,118 63,6 59,5 56,5 47,8
0,165 61,7 57,1 53,7 45,9
0,236 58,5 54,2 51,9 43,4
0,355 55,9 51,7 49,6 41
0,473 53,8 49,6 46,7 39,5
0,71 52,9 47 44,9 37,7
0,829 50,1 46,1 43 37,4
0,947 48,5 44,5 41,8 37,4
1,18 46,7 43,1 40,5 37,4
1,65 44,6 42,6 40,4 37,4
2,36 44,6 42,6 40,4 37,4
2,84 44,6 42,6 40,4 37,4
4,26 44,6 42,6 40,4 37,4
с • 104, in
моль/л
0,0016 72,2
0,0083 70,5
786
Новый справочник химика и технолога
Продолжение таблицы 4П1.2
с 104, моль/л Г, К
283 288 293 298
л= 10
0,0167 71,8 69
0,034 70,5 66,3
0,05 71,6 69,9 65,2
0,08 70,3 67,8 63,2
0,167 71,4 68,2 65,5 60,3
0,334 68,7 65,9 61,9 57,1
0,501 67,6 64,1 60,1 54,6
0,836 65,5 61,6 57,6 52,5
1,17 63,7 59,3 55,9 50,7
1,67 62 57,7 53,7 49,4
3,34 57,9 53,5 50,2 46
5,01 55,7 51,6 48,1 44,5
6,68 54,3 49,9 46,4 42,9
8,3 53,1 48,3 45,3 42,1
10,03 51,8 47,5 44,3 42,1
П,7 51,2 47,1 43,9 42,1
13,3 50,4 46,2 43,9 42,1
15 49,1 45,7 43,9 42,1
16,7 48,8 45,7 43,9 42,1
20 48,3 45,7 43,9 42,1
30,1 48,3 45,7 43,9 42,1
41,8 48,3 45,7 43,9 42,1
п= 12
0,0014 72,1
0,0072 71,2
0,0145 71,8 69,5
0,0437 71,8 69,7 64,5
0,0728 71,2 68 63,9
0,145 71,4 68,9 67,5 62,1
0,291 69,9 66,8 63 58,5
0,728 66,1 63,4 59,1 54,1
1,45 63,7 60,7 55,9 50,9
2,91 59,8 56,6 52 47,3
4,37 56,8 54,1 50,1 46
7,28 53,9 51,4 47,9 43,4
8,74 53,2 50,5 47,2 43,2
10,2 52,5 50,1 46,4 43,2
11,6 52,1 49,1 45,7 43,2
13,1 51,4 48,3 45,5 43,2
14,5 50,7 47,5 45,5 43,2
17,4 49,6 46,7 45,5 43,2
21,8 48,7 46,7 45,5 43,2
29,1 48,7 46,7 45,5 43,2
Продолжение таблицы 4П1.2
с - 104, моль/л Г, К
283 288 293 298
п= 12
36,4 48,7 46,7 45,5 43,2
40,8 48,7 46,7 45,5 43,2
п= 15
0,0024 58,4 62,5
0,0061 61,9
0,0122 71,5 57
0,0366 70,2 53,6
0,061 71,9 71,9 68,1 51,3
0,122 71,6 70,3 65,4 49,1
0,244 68,4 68,2 61,8 46,3
0,611 65 65,1 57,9 45,8
1,22 61,8 61,5 54,7 45,5
3,66 59,5 55,9 52,5 45,5
6,11 56,3 53,6 50,2 45,5
7,33 55,2 52,5 59,3 45,5
8,55 54,7 51,4 48,2 45,5
9,7 54,1 50,8 47,8 45,5
11 53,9 50,6 47,6 45,5
12,2 52,8 49,9 46,9 45,5
13,4 52,1 49,5 46,6 45,5
14,6 51,4 49,1 46,6 45,5
15,8 51,2 48,8 46,6 45,5
18,3 51,1 47,9 46,6 45,5
20,7 50,9 47,9 46,6 45,5
24,4 49,7 47,9 46,6 45,5
30,5 49,7 47,9 46,6 45,5
34,2 49,7 47,9 46,6 45,5
40,3 49,7 47,9 46,6 45,5
Оксиэтилированные жирные спирты
С12Н25О(С2Н4О)„Н
с - 106, моль/л Г, К
283 288 293 298
п = 3
0,05 70,8
0,1 68,7
0,3 70,1 62,8
0,5 67,8 59,7
1 69,5 63,3 55
2 70,8 64,9 58,8 48,9
3,14 68,6 61,6 54,6 45,5
6,28 63,4 54,7 47,3 38
9,43 60,1 50,8 43,5 35,1
Приложение 1
787
Продолжение таблицы 4П1.2
с • 106, моль/л ГК
283 288 293 298
п = 3
12,5 57,7 47,6 40,4 32,3
15,5 54,5 44,5 37,2 30,5
31 46,9 37,3 33,4 30,5
62 41,1 36,1 33,4 30,5
125 41,1 36,1 33,4 30,5
188 41,1 36,1 33,4 30,5
251 41,1 36,1 33,4 30,5
314 41,1 36,1 33,4 30,5
471 41,1 36,1 33,4 30,5
528 41,1 36,1 33,4 30,5
п = 6
0,1 69,5
0,3 70,3 64,8
0,5 68,4 63,1
1 69,6 65,8 59,5
2 70,3 66 61,4 53,7
4 67,9 62,8 57,7 49,6
8 62,5 58,7 51,5 43,5
11 63,1 56,5 50,2 41,2
22 57,8 50,7 44,6 35,3
33 53,4 47,1 41,2 34,8
44 49,6 44,4 38,7 34,8
88 43,7 41,2 38,2 34,8
133 43,7 41,2 38,2 34,8
177 43,7 41,2 38,2 34,8
222 43,7 41,2 38,2 34,8
333 43,7 41,2 38,2 34,8
444 43,7 41,2 38,2 34,8
п= 8
0,1 68,6
0,3 69,7 63,9
0,5 66,9 61,6
1 69,8 64,1 58
1,8 70,6 67,1 61,7 54,7
3,7 68,5 63,7 57,5 51,2
7 65,8 59,9 53,2 47,5
9 64,1 58,5 51,4 46,1
18 59,8 54,3 47,8 41,9
37 54,7 49,6 43,3 38
55 51,7 46,7 41,1 36,9
74 49,4 44,9 39,3 36,9
111 46,2 42,3 39,3 36,9
148 44,8 42,3 39,3 36,9
Продолжение таблицы 4П1.2
с • 106, моль/л Г, К
283 288 293 298
п = 8
186 44,8 42,3 39,3 36,9
288 44,8 42,3 39,3 36,9
371 44,8 42,3 39,3 36,9
557 44,8 42,3 39,3 36,9
929 44,8 42,3 39,3 36,9
1300 44,8 42,3 39,3 36,9
п= 10
0,01 71,9
0,05 70,5
0,1 69,2 68,3
1 68,7 64,4 59,2
1,5 70,7 66,4 62,7 56,1
3 68,1 63,9 59,2 53
6 64,8 60,6 55,6 50
8 63,9 59,7 54,5 49,7
16 60,4 56,1 50,4 45,9
32 56,8 52,3 47,8 42,3
48 53,9 49,8 45,4 40,4
64 52,7 48,2 43,9 38,9
79 51,4 46,5 42,7 38,9
96 49,3 44,7 41,6 38,9
127 47,6 43,8 41 38,9
159 46,6 43,5 41 38,9
239 45,8 43,5 41 38,9
319 45,8 43,5 41
479 45,8 43,5 41
790 45,8 43,5 41
1110 45,8 43,5 41
«= 12
0,01 69,9
0,1 67,2
0,3 70,2 64,3
0,5 68,1 62,3
1 70,3 66,9 61,1
1,8 68,8 64,7 59,7
3,7 69,7 65,2 61,2 55,5
7 66,3 62,3 58,2 52
9 65,4 61,1 57,5 51,3
18 61,2 56,7 53,6 47,9
37 57,6 53,6 49,9 44,2
55 52,2 48,5 45,4 40,4
74 51,7 47,7 44,5 39,5
111 50,8 46,9 43,7 39,5
788
Новый справочник химика и технолога
Продолжение таблицы 4П1.2
с - 106, моль/л Г, К
283 288 293 298
и= 12
148 49,9 46,4 43,2 39,5
186 48,9 44,9 41,9 39,5
288 46,7 43,8 41,9 39,5
371 46 43,8 41,9 39,5
557 46 43,8 41,9 39,5
929 46 43,8 41,9 39,5
1300 46 43,8 41,9 39,5
п -- 14
0,01 71,9
0,1 70,7
0,3 70,4 67,1
0,5 68,3 64,9
0,7 66,8 63,8
1 70,2 65,6 61,9
1,25 68,9 64,7 61,5
2,5 70,1 66,4 61,9 58
4,9 67,5 63,6 60 54,6
6,2 66 62,3 57,7 53,5
12,4 62,9 58,7 54,5 50,4
25 59,2 54,9 51,6 47,2
49 55,9 52 48,1 44,5
75 53,7 50,1 46,3 42,6
99 52,1 49,2 45,2 41,5
112 51,6 48,5 44,3 41
124 50,8 47,3 43,8 41
187 48,3 45,1 42,3 41
249 46,7 44,3 42,3 41
374 46,2 44,3 42,3 41
623 46,2 44,3 42,3 41
872 46,2 44,3 42,3 41
Оксиэтилированные жирные спирты
С14Н29О(С2Н4О)„Н
с - 106, моль/л Г, К
283 288 293 298
п = 6
0,0104 70,3
0,0209 64,5
0,0627 68,2 58,8
0,104 70,4 64,8 54,3
0,209 66,8 60,4 50,1
0,418 68,2 62,2 55,1 43,6
0,627 65,6 58,7 51,5 40,7
Продолжение таблицы 4П1.2
с- 106, моль/л Г, К
283 288 293 298
п = 6
1,04 61,7 54,1 47,5 35,9
1,25 60,1 52,6 45,6 34,1
1,46 58,3 50,7 43,7 32,4
1,88 56 48,9 41,3
2,09 54,5 47,3 39,7
3,13 49,7 42,7
4,18 45,8 38,4
6,27 41,9 38,4 36,3
10,4 41,9 38,4 36,3
20,9 41,9 38,4 36,3
104 41,9 38,4 36,3
л = 8
0,0017 71,9
0,0088 70,8
0,0176 68,2
0,053 69,7 62,5
0,088 67,4 60,1
0,176 68,9 63,2 54,8
0,353 70,3 65,7 58,6 49,7
0,53 68,1 62,5 54,8 45
0,883 64,9 58,3 50,1 41,7
1,23 62,4 56 46,9 40,1
1,76 58,4 52,2 43,7 36
2,65 54,3 47,8 39,7 34,3
3,53 49,8 44,7 37,6 34,3
5,3 45,2 41,1 37,4 34,3
8,83 43 40,5 37,4 34,3
17,6 43 40,5 37,4 34,3
35,3 43 40,5 37,4 34,3
88,3 43 40,5 37,4 34,3
п= 10
0,0015 71,9
0,0076 68,3
0,0152 70,3 65,2
0,0458 70,6 66,6 59,8
0,0764 69 64,5 56,9
0,152 70,1 68,1 60,9 52,7
0,305 67,8 62,5 57,8 48,8
0,458 65,7 60,2 54,7 46,4
0,764 63,2 57,3 52,4 44,1
1,07 61,1 54,7 49,9 41,9
1,52 58,5 53,1 43,4 39,8
3,05 53,6 47,5 42,2 35,4
Приложение 1
789
Продолжение таблицы 4П1.2
с • 106, моль/л Т,К
283 288 293 298
п= 10
4,58 49,8 43,9 39,1 35,4
6,11 47 41,6 38,4 35,4
12,2 43,6 41 38,4 35,4
15,2 43,6 41 38,4 25,4
30,5 43,6 41 38,4 35,4
76,4 43,6 41 38,4 35,4
п = 12
0,0134 71,8
0,0404 69,7 64,6
0,0673 71,9 68,8 62,4
0,134 70,4 65,4 58,2
0,269 70,6 66,9 60,4 54,7
0,404 68,8 64,8 57,9 51,7
0,673 65,9 61,4 54,3 48
0,943 64,1 59 51,8 46,2
1,34 61,7 56,3 49,2 44,3
2,69 56,5 51,6 44,6 39,9
4,04 53,1 48,2 41,5 37,6
5,39 50,3 45,7 39,8 36,6
6,73 48,3 44,5 39,1 26,6
10,7 44,3 41,8 39,1 36,6
13,4 44,1 41,8 39,1 36,6
16,1 44,1 41,8 39,1 36,6
20,2 44,1 41,8 39,1 36,6
26,9 44,1 41,8 39,1 36,6
67,3 44,1 41,8 39,1 36,6
п = 14
0,006 71,9
0,012 69,3
0,06 71,8 68,3 64,1
0,12 69,6 65,6 60,6
0,24 71,2 65,8 61,4 57,1
0,361 69,6 63,7 58,8 53,8
0,602 66,1 60,3 56,2 51,5
1,2 60,7 55,9 51,6 47,5
2,47 56,3 51,9 48 43,4
3,61 53,3 49,2 45,7 41
6,02 50,2 46,2 42,8 38,2
7,22 48,5 44,7 41,7 37,7
9,63 46,4 43,1 40,5 37,7
12 44,9 42,6 40,5 37,7
14,4 44,5 42,6 40,5 37,7
24 44,5 42,6 40,5 37,7
60,2 44,5 42,6 40,5 37,7
Таблица 5П1.2
Поверхностное натяжение растворов неорганических веществ на границе с влажным воздухом (мДж/м2) [2]
Вещество T,°C Концентрация, масс. %
1 5 10 20 50 75
AgNO3 18 73,5 73,9 74,4 75,25 78,9
A12(SO4)3 18 73,6 74,5
BaCl2 30 71,2 74,35 74,5
СаС12 18 72,72 73,7
CdCl2 15 73,35 73,55 73,82 74
CuSO4 30 71,25 71,8 72,37 73,5
FeSO4 18 73,6 73,62 73,9
HC1 20 72,92 72,46 72,25 71,44
H2SO4 18 74,1 75,2 77,3 72,86
HNO3 20 72,65 71,1 65,43
KBr 18 72,85 73,25 73,9 75,25
KC1 18 72,4 73,6 74,75 77,25
K2SO4 18 72,95 73,6
kno2 20 72,95 73,6 74,35 76 81,7 94,6
KNO3 18 72,7 73 73,6 75
K2CO3 10 74,6 75,8 77 79,2 106,4
LiBr 18 72,95 73,6 74,45
LiCl 18 73,2 74,75
LiNO3 18 73 74,05
Li2SO4 18 73,1 75,27
NH4C1 18 72,6 73,3 74,5
NHiNO-j 100 58,75 59,2 60,1 61,6 67,5 65,2
NH4OH 18 71,65 66,5 63,5 59,3
NaBr 18 72,95 73,6 74,4
NaCI 18 72,7 73,95 75,51
NaClO3 18 72,31 72,6 72,95
NaNO3 30 71,4 72,1 72,8 74,4 79,8
NaOH 20 73,17 74,6 77,3 85,8
Na2SO4 18 72,75 73,82 75,15
MgCl2 18 72,75 73,8
MgSO4 18 73,1 73,75 74,25 77,6
MnSO4 15 73,31 73,65 74,85
SrCl2 30 72,2 72,6 73,27 75,5
ZnSO4 18 72,67 73,25 73,9 75,75
Литература
1. Поверхностно-активные вещества и моющие средства: Справочник / Под ред. А.А. Абрамзона. М.: Изд-во ТОО НТР Гиперокс, 1993.270 с.
2. Справочник химика. Т. 3. М.;Л.: Госхимиздат, 1952.
1192 с.
790
Новый справочник химика и технолога
1.3. ГЛБ и ККМ
1.3.1. Гидрофильно-липофильный баланс веществ
Область применения ПАВ определяется их относительным сродством к фазам, имеющим общую поверхность раздела, на которой и проявляется действие ПАВ. Наряду с такими мерами сродства как растворимость, поверхностная активность, положение молекул в адсорбционном слое, на практике используется величина гидрофильно-липофильного баланса (ГЛБ) — эмпирической характеристики относительного сродства, отражающей непосредственно состояние ПАВ в водной среде и его пригодность для решения той или иной задачи (табл. 1П1.3).
Экспериментально ГЛБ определяется по разным признакам: времени жизни прямых и обратных эмульсий, растворимостям (табл. 1П1.3) и др. Эти методы не стандартизованы, и поэтому отдается предпочтение расчетным методам нахождения ГЛБ. Они тоже различны. Наиболее распространен метод, основанный на вкладе различных групп химических соединений в общий гидрофильно-липофильный баланс молекулы — метод групповых чисел N (табл. 2П1.3). Он имеет определенную теоретическую подоплеку [1].
Расчет ГЛБ молекулы проводится по формуле [1]
ГЛБ = 2А+7, (1П1)
где N — групповые числа из табл. 2П1.1.
Суммирование (S/V) вкладов всех групп ведется с учетом знака группового числа. Он положителен для лиофильных групп и отрицателен для липофильных и некоторых сложных групп (табл. 2П1.3). Например, для олеата натрия C17H33COONa, имеющего лиофильную группу -COONa с групповым числом 19,1 и 17 липофильных групп с групповым числом -0,475, получается ГЛБ = 19,1 - 0,475 17 + 7 = 18,025. По данным Девиса [1], расчетные и экспериментальные значения ГЛБ удовлетворительно совпадают (табл. ЗП1.3).
Таблица 1П1.3
Зависимость состояния ПАВ в воде от величины ГЛБ [ 1 ]
Состояние ГЛБ Применение*
Не смешивается 1-4
Плохо диспергируется 3-6 Эмульгатор в/м
Хорошо диспергируется 6-8 Смачиватель
Устойчивая эмульсия 8-10 Эмульгатор м/в
Высокодисперсная эмульсия 10-13 Эмульгатор м/в
Прозрачный раствор 13 Эмульгатор м/в, солюбилизатор
* в — вода; м — масло.
Таблица 2П1.3
Групповые числа N различных фрагментов молекул ПАВ [1]
Группа N
Лиофильные
-SO4’Na+ 38,7
соок‘ 21,1
COONa 19,1
N (третичный амин) 9,4
Эфир (сорбитановое кольцо) 6,8
Эфир (свободный) 2,4
-СООН 2,1
ОН (свободный) 1,9
—О- 1,3
ОН (сорбитановое кольцо) 0,5
Липофильные
—СН 1 -0,475
-сн2-
сн3
=сн-
Сложные
-СН2СН2О- 0,33
-СН2СН2СН2О- -0,15
Таблица ЗП1.3
ГЛБ поверхностно-активных веществ [1]
ПАВ Экспериментальное Расчетное
Додецилсульфат натрия 40 40
Олеат калия 20 20
Олеат натрия 18 18
Твин-80 (моноолеат сорбитана с 20 группами -СН2СН2О-) 15 15,8
Моноолеат сорбитана с 10 группами -СН2СН2О- 13,5 12,5
Твин-81 (моноолеат сорбитана с 5 группами -СН2СН2О-) 10 10,9
C18H37N(CH2CH2OH)(CH2CH2O)2H 10 10
Монододеканоат сорбитана 8,6 8,5
Метанол 8,4
Этанол 7,9
Пропан-1-ол 7,4
Моноолеат сорбитана с 2 группами -СН2СН2О- 7 7
Бутан-1-ол 7 7
Моноолеат сорбитана 7 7,2
Монопальмитат сорбитана 6,7 6,6
Моностеарат сорбитана 5,9 5,7
Приложение 1
791
Продолжение таблицы ЗП1.3
ПАВ Экспериментальное Расчетное
Спен-80 (моноолеат сорбитана) 4,3 5
Монододеканоат пропан-1,2-диола 4,5 4,6
Дистеарат сорбитана 3,5 3,9
Моностеарат глицерина 3,8 3,7
Моностеарат пропан-1,2-диола* 3,4 1,8
Тристеарат сорбитана 2,1 2,1
Гексадекан-1-ол 1 1,3
Олеиновая кислота 1 1
Тетрастеарат сорбитана 0,5 0,3
* Может содержать примесь мыла.
1.3.2. Критические концентрации мицеллообразования {ККМ)
Подавляющее большинство поверхностно-активных веществ, применяемых в технике и технологии, имеют ограниченную растворимость, причем переход к новому состоянию раствора при повышении его концентрации отличается уникальным своеобразием. Своеобразие заключается в том, что при достижении некоторой характерной для каждого ПАВ концентрации оно не выделяется в виде новой фазы, а образует коллоидный раствор. Эта концентрация называется критической концентрацией мицеллообразования (ККМ). Она является основной термодинамической характеристикой раствора ПАВ. Поверхностно-активные вещества, способные переходить в состояние коллоидного раствора, называются мицеллообразующими или коллоидными поверхностно-активными веществами. Особенность образующихся при этом коллоидных растворов поверхностноактивного вещества в том, что они термодинамически устойчивы, обратимы по отношению к изменению состава раствора и температуры. При валовой концентрации ПАВ большей, чем ККМ, его концентрация в молекулярной форме остается равной ККМ, а все остальное вещество находится в мицеллярной форме. Только мицеллообразующие вещества являются эффективными стабилизаторами суспензий и эмульсий, солюбилизаторами и основным компонентом моющих составов. Критическая концентрация мицеллообразования является и важнейшей технической характеристикой технических поверхностно-активных веществ (табл. 4П1.3).
Таблица 4П1.3
Критические концентрации мицеллообразования (ККМ) типичных представителей поверхностно-активных веществ разного типа [2]
ПАВ Формула ККМ, моль/л
Анионные ПАВ
Деканоат натрия C9H19COONa 9,4 • 10"2
Продолжение таблицы 4П1.3
ПАВ Формула KKM, моль/л
Анионные ПАВ
Додеканоат натрия CnH23COONa 2,5 • 10 2
Миристат натрия C]3H27COONa 6,9 • 10“3
Олеат натрия C17H33COONa 1,1 • 10’3
Олеат калия С17Н33СО(Ж 1,2- 1 O’3
Стеарат калия CI7H35COOK 5 • 104
Додецилсульфат натрия C12H25OSO3Na 8,1 • 103
Додецилсульфат калия C12H25OSO3Na 8 • 10~3
Тетрадецилсульфат натрия C14H29OSO3Na 2,1 • 10“3
Додекан-1 -сульфонат натрия Ci2H25SO3Na 9,8 • 10’3
4-Додецил бензол-сульфонат C12H25C6H4SO3Na 1,2 • IO’3
Катионные ПАВ
Додециламмоний хлорид C12H25NH3C1 1,5 • 1 (Г2
Тетрадецил-аммоний хлорид c14h29nh3ci 2,8- 10"3
Додецилметил-аммоний хлорид C12H25NH2(CH3)C1 1,5 • 10’2
Додецилдиметил-аммоний хлорид CI2H25NH(CH3)2C1 1,6 • 10'2
Додецилтриметил-аммоний хлорид C12H25N(CH3)3C1 2,0 • 10 2
1-Додецил-пиридиний хлорид c12h25nc5h5ci 1,5 • 10’2
Неионогенные ПАВ
Додециловый эфир тетраэтиленгликоля C12H25O(C2H4O)4H 4- 105
Додециловый эфир гексаэтиленгликоля C12H25O(C2H4O)6H 8,7- 10’5
Додециловый эфир октаэтиленгликоля C12H25O(C2H4O)8H 8,3 • 10‘5
Додециловый эфир додекаэтиленгликоля C12H25O(C2H4O)12H 1,4 • 104
4-ги/?еги-Октил-фениловый эфир триэтиленгликоля C8H17C6H4O(C2H4O)3H 1,1 • 10 4
4-трет-Октилфе-ниловый эфир пентаэтиленгликоля C8H17C6H4O(C2H4O)5H 1,3 • 104
4-трет-Октилфе-ниловый эфир гептаэтиленгликоля C8H17C6H4O(C2H4O)7H 1,8 • 10^
792
Новый справочник химика и технолога
1.3.3. Расчет количества ПАВ в дисперсной системе
Простейший расчет количества поверхностноактивного вещества, необходимого для достижения требуемого эффекта, основан на величине так называемой посадочной площадки s молекулы ПАВ. Это площадь, которую занимает одна молекула ПАВ в насыщенном адсорбционном слое этого вещества. Она определяется геометрическими размерами молекулы ПАВ и ее ориентацией в адсорбционном слое.
Для расчета количества ПАВ необходимо знать (или задать) площадь А поверхности дисперсной фазы — частиц суспензии или капель эмульсии. Как правило, максимальный эффект, в частности эффект стабилизации суспензий, эмульсий, пен, достигается за счет образования на поверхности всех частиц (капель, ячеек пены) насыщенного мономолекулярного адсорбционного слоя поверхностно-активного вещества. Для этого в системе должно содержаться, как минимум, такое количество вещества (2тш, которого достаточно для создания такого слоя на всей площади межфазной поверхности А. Оно равно:
л
2min= —, (2П1)
где Na = 6,023 • 1023 1/моль — постоянная Авогадро. Фактическое количество ПАВ Q должно быть увеличено на величину порядка (К • ККМ), где V — объем дисперсионной среды, а ККМ — критическая концентрация мицеллообразования вводимого ПАВ в этой среде. При концентрации ПАВ в среде, равной его критической концентрации, гарантировано образование насыщенного адсорбционного слоя. Не рекомендуется вводить поверхностно-активное вещество в количестве большем, чем ((?mm + V ККМ), так как это иногда ведет к понижению устойчивости дисперсной системы, возможно, за счет образования второго (лиофобизующего) адсорбционного слоя. Необходимые для указанных расчетов свойства поверхностно-активных веществ приведены в табл. 4П1.3 и 5П1.3.
Таблица 5П1.3
Площадь s, занимаемая одной молекулой в насыщенном адсорбционном слое [2]
ПАВ Формула 5 • IO20, m2
Додеканоат натрия CH3(CH2)i0COONa 41
Тетрадеканоат натрия CH3(CH2)12COONa 34
Пальмитат натрия CH3(CH2)I4COONa 25
Стеарат натрия CH3(CH2)16COONa 23
Олеат натрия C8H17CH=CHC7H14COONa 28
4-Додецилбензол-сульфонат натрия C12H25C6H4SO3Na 33
Продолжение таблицы 5П1.3
ПАВ Формула 5 • 1О20, м2
Додецилсульфат натрия C12H25OSO3Na 33
Октилэтиламмо-ний хлорид C8HI7(C2H5)NH2C1 34
1-Гекса-децилпиридиний хлорид c16h33nc5h5ci 46
Октан-1-ол C8H17OH 30
2-Октилокси-этанол C8H17OCH2CH2OH 32
4-Октилфенило-вый эфир октаэтиленгликоля С8Н17СбН4О(С2Н4О)8Н 53
А-трет-Октилфенило-вый эфир октаэтиленгликоля С8Н17С6Н4О(С2Н4О)8Н 58
4-/ире/и-Октил-фениловый эфир декаэтиленгликоля С8Н17С6Н4О(С2Н4О)10Н 74
4-/ире/и-Окгил-фениловый эфир гексадекаэтиленгликоля С8Н17СбН4О(С2Н4О)16Н 80
Литература
1. Эмульсии / Под ред. Ф. Шермана. Л.: Химия, 1972. 448 с.
2. Лабораторные работы и задачи по коллоидной химии / Под ред. Ю.Г. Фролова, А.С. Гродского. М.: Химия, 1986. 216 с.
1.4. ПАВ в неводных средах
1.4.1. Припои и флюсы
Пайка — важнейшая и универсальная технология соединения в одно изделие разнородных элементов. Простейшие электронные приборы, материнские платы компьютеров, электронная часть спутниковой аппаратуры, радиаторы автомобилей и многое другое изготовляются методом пайки. Этим методом можно соединять не только металлические детали, но и детали из керамики, стекла, графита между собой и с металлами.
Процесс пайки заключается в заполнении металлическим расплавом (припоем) зазора между соединяемыми деталями и последующим отверждением расплава при его охлаждении. Решающее значение имеет при этом смачиваемость поверхностей соединяемых деталей припоем. Смачиваемость и поверхностное натяжения расплава известным образом связаны с прочностью соединения (адгезией припоя к поверхности детали).
Приложение 1
793
Кроме того, только при хорошей смачиваемости можно гарантировать заполнение без пропусков стыка деталей, в том числе заполнение участков с нулевой толщиной зазора (в точках соприкосновения деталей). Наличие пропусков не только ослабляет прочность, но и делает невозможным выполнение ряда основных функций спая — обеспечение надежности электрического контакта, обеспечение герметичности соединения. Инородные включения в местах нарушения сплошности, например остатки флюса, способны вызывать коррозию деталей.
Хорошая смачиваемость припоями обеспечивается применением флюсов при пайке. Флюс — это вещество, которое при пайке плавится и растекается по поверхности расплавленного припоя и соединяемых деталей и удаляет пленку оксидов с их поверхности или за счет ее диспергирования, растворения, или путем травления — образования растворимых во флюсе химических соединений оксидов металлов с компонентами флюса. В последнем случае флюс проявляет достаточно сильную агрессивность по отношению к металлу и поэтому должен тщательно удаляться после пайки. Таковыми являются большинство кислотных флюсов, например орто-фосфорная кислота, являющаяся эффективным флюсом при пайке железа. Пример неагрессивного флюса — канифоль, применяемая при пайке изделий из меди.
Для соединения деталей пайкой применяются специальные металлические сплавы — припои. Состав припоя определяется как функциями, которые он должен выполнять, так и природой соединяемых с его помощью материалов. С долей условности припои можно разделить на легкоплавкие и тугоплавкие. Легкоплавкими являются припои общего назначения, и приводимые ниже данные относятся к этой группе припоев и флюсов.
1.4.2. Легкоплавкие припои [1]
Легкоплавкие припои в основном применяются для крепления и создания надежного электрического контакта легких электронных компонентов. Их основой является олово (табл. ИИ.4 и 2П1.4). Большой прочности паяного соединения не требуется, но существуют жесткие ограничения температуры пайки, так как электронные компоненты обычно не выдерживают сильного нагрева.
Кроме того, припой должен иметь высокую электропроводность и устойчивость к коррозии. Припой ПОС 61М может применяться для пайки ювелирных изделий, а ПОСК 50-18 — для пайки металлизированной керамики.
Сплавы на основе олова и цинка (табл. 2П1.4) пригодны для низкотемпературной пайки алюминиевых деталей с подходящим флюсом. Добавление кадмия позволяет резко понизить температуру пайки. Составление тройных и более сложных по составу припоев во многих случаях преследует именно эту цель — регулирование температуры плавления. Понижение темпера
туры технологически выгодно, но обеспечение термостойкости паяного соединения иногда требует повышения температуры плавления (температуры размягчения) припоев.
Требования к прочности, коррозионной стойкости, ограничения по температурным коэффициентам расширения, по санитарно-гигиеническим параметрам ведет к созданию большого числа разнообразных узкоспециализированных припоев.
Отдавая должное коллоидно-химическим аспектам взаимодействия металлических расплавов с поверхностью твердых веществ, следует обратить внимание на легкоплавкие металлы и сплавы как на дисперсионные среды для получения коллоидных растворов и суспензий на их основе. Необходимо сразу исключить из рассмотрения ртуть и сплавы на ее основе по причине их токсичности.
Наиболее известным сплавом с низкой температурой плавления является сплав Вуда. Его состав (масс. %): Bi 50, Pb 25, Sn 12,5, Cd 12,5. Температура плавления 68 °C. На основе этого сплава получены суспензии железа [2]. Концентрация дисперсного (карбонильного) железа в них достигала 50 об. %, так что по намагниченности эти суспензии только в два раза уступают железу. В магнитном поле они обладают сильно выраженным магнитореологическим эффектом.
Таблица 1П1.4
Состав и свойства оловянно-свинцовых припоев (ПОС)
Указано содержание олова и других металлов, масс. %, остальное — свинец.
Марка припоя Sn Другие элементы ТШ1, °с
ПОС 90 89-91 183-220
ПОС 60 60-62 183-190
ПОС 40 39-41 183-238
ПОС 10 9-10 268-299
ПОС 61М 60-62 1,5-2,0 Си 183-192
ПОСК 50-18 49-51 17-19 Cd 142-145
Таблица 2П1.4
Припои на основе олова, цинка и кадмия
Содержание Sn, Zn, Cd, масс. %
Марка припоя Sn Zn Cd Тил., °C
П200А 90 10 200
П250А 80 20 280
Sn70Zn 70 30 315
LSn60Zn 60 40 345
66,5 2,5 31,0 165
57,0 18,0 25,0 190
55,0 25,0 20,0 250
794
Новый справочник химика и технолога
Более низкой температурой плавления обладают сплавы ИНГАС (индия, галлия, олова) с добавками цинка (табл. ЗП1.4).
Сплавы ИНГ АС, как и сам галлий (температура плавления 26 °C), в специальных случаях используются как теплоносители.
Таблица ЗП1.4
Состав и температура плавления сплавов ИНГАС
Ga In Sn Zn Гил, °C
92 8 20
82 12 6 17
76 24 16
67 29 4 13
62 25 13 5
61 25 13 1 3
1.4.3. Флюсы для пайки легкоплавкими припоями [1]
Флюсы классифицируются по их корродирующему действию на металлы. Самым распространенным некорродирующим флюсом является канифоль и растворы канифоли в этиловом спирте (около 30 масс. % канифоли). Растворы применяются при автоматизированной пайке, так как нанесение жидкого флюса на место пайки поддается механизации и автоматизации. Канифольные флюсы пригодны для пайки меди и ее сплавов при температуре пайки до 310 °C. При более высокой температуре канифоль обугливается и может оставлять внутри паяного шва органические включения.
Более универсальными являются флюсы на основе канифоли с добавками слабых кислот (табл. 4П1.4). Их присутствие придает флюсам слабую коррозионную активность. Флюсами с гидразином можно паять медь, никель, кадмий, драгоценные металлы. При температуре пайки свыше 200 °C солянокислый гидразин разлагается, поэтому остаются только нейтральные компоненты флюса и его можно не удалять после пайки.
Таблица 4П1.4
Типовые составы флюсов на основе канифоли
Компоненты флюса w, масс. %
Канифоль 30 97 38 24 20 25
Этанол 60 50 75 79 70
Глицерин 1 35
Уксусная кислота 10
Гидразин гидрохлорид 5 5 5
Анилин гидрохлорид 2 1
Ортофосфорная кислота 12
Цинка хлорид 1
Вода 95 60
Слабой коррозионной активностью обладают флюсы на основе триэтаноламина за счет присутствия в них неорганических солей (табл. 5П1.4). Эти флюсы предназначены для пайки алюминия и его сплавов. Флюсы, состоящие только из твердых компонентов, применяются в виде паст на вазелине.
Самыми универсальным и простым по составу флюсом является водный раствор хлорида цинка (40 масс. %). Многочисленные вариации этого состава сводятся к частичной замене хлорида цинка хлоридами аммония, натрия, калия, меди или соляной кислотой (от долей процента до 80 % хлорида цинка) для снижения температуры плавления и повышения активности флюса. Безводные составы применяются в виде паст на основе вазелина, канифоли, парафина, глицерина и др. Основное назначение этих флюсов — пайка и лужение железа. Остатки флюсов после пайки должны тщательно удаляться в силу их высокой коррозионной активности. Для пайки нержавеющей стали применяется концентрированная ортофосфорная кислота, насыщенный раствор хлорида цинка и его смесь с соляной кислотой (25 масс. % кислоты).
Таблица 5П1.4
Флюсы для пайки алюминия
Компоненты флюса w, масс. %
Триэтаноламин 82,5 82 82 82 2 1,5 1,5
Кадмия тетрафтороборат 10 10
Цинка тетрафторо-борат 2,5 10
Аммония тетрафтороборат 5 8 8
Аммония хлорид 10 18
Олова(П) хлорид 8
Этанол 30 94,5
Глицерин 45
Вода 93,5
Анилин гидрохлорид 5
Салициловая кислота 4
Бензойная кислота 4
Неионогенное ПАВ (ОП-7) 1
Приложение 1
795
Приведенные выше рецептуры припоев и флюсов не исчерпывают всего многообразия возможных их вариантов и являются иллюстрацией принципов составления припоев и флюсов. Отметим также, что существуют приемы безфлюсовой пайки и сварки, например в вакууме или в атмосфере инертных газов (сварка в аргоне), приемы пайки без припоев. Это означает, что некоторые приемы пайки совпадают с приемами сварки. Поэтому здесь описаны составы, относящиеся только к типичной технологии пайки изделий.
Литература
1. Хряпин В.Е., Лакедемонский А.В.: Справочник паяльщика. М.: Машиностроение, 1974. 328 с.
2. Бибик Е.Е., Мазуренко О.М. Способ получения ферромагнитной жидкости на металлическом носителе. А. с. 1015447. Б. И. № 16, 1983.
1.5. Поверхностно-активные вещества, растворимые в углеводородах
1.5.1. Поверхностная активность в неполярной среде
Классическое определение того, что такое поверхностно-активное вещество (ПАВ), основано на его воздействии на водные растворы. Это вещество, способное уменьшать поверхностное натяжение раствора (натяжение границы раствор—воздух или раствор—газ) при увеличении его концентрации в растворе. В то же время техническое применение ПАВ практически никогда не направлено на достижение именно этого эффекта. Кроме того, в подавляющем большинстве случаев применение ПАВ связано с воздействием не на границу раствор—газ, а на границу раздела двух жидкостей типа воды и масла, или на границу раздела твердого вещества и жидкости (раствора ПАВ). При этом ПАВ может быть растворено не в водной, а в масляной среде. Здесь и далее, в соответствии с классификацией жидкостей, принятой при описании эмульсий, под маслом подразумевается любая неполярная жидкость, в том числе летучие растворители.
Изменение поверхностного натяжения водных растворов при наличии в них ПАВ обусловлено, как известно, адсорбцией ПАВ на границе между раствором и газом (воздухом). Именно на явлении адсорбции, а не на изменении натяжения основано практическое применение ПАВ, хотя эти два явления тесно связаны, в частности, уравнением изотермы адсорбции Гиббса. В связи с этим далее будет подразумеваться, что поверхностно-активное вещество — это вещество, которое адсорбируется на границе раздела фаз. Акцент на адсорбции, а не на изменении натяжения, необходим потому, что адсорбция в принципе всегда может быть измерена, тогда как измерение натяжения возможно только на границе двух флюидных фаз (жидкость—газ и жидкость 1—жидкость 2). Натяжение же на границе твердого вещества с любой флюидной фазой не может быть измерено, следовательно, эффективность ПАВ не
может быть проконтролирована и охарактеризована количественно на основании классического определения поверхностной активности как способности изменять натяжение. Отметим, что основная масса производимых поверхностно-активных веществ расходуется именно на регулирование состояния границы твердое тело—жидкость. Это нефтедобыча, основные нефтепродукты (горючее и масла), моющие средства. Во всех этих случаях ПАВ воздействует на поверхность твердого вещества. Таким образом, возникла явная дискриминация методов контроля эффективности ПАВ именно там, где они в основном применяются. Она еще более усиливается в отношении тех сфер применения, где жидкой фазой является не вода, а масло, поскольку нет оснований ожидать, что ПАВ будет понижать натяжение границы масло—газ. Нет и методически единых экспериментальных результатов, которые можно было бы обобщить и на их основе сформулировать эмпирические закономерности воздействия ПАВ на натяжение их масляных растворов. Как неизбежное следствие этого имеющийся огромный массив практических результатов по разработке ПАВ для масляных сред и по созданию разнообразных продуктов не может быть в настоящее время систематизирован на основе фундаментальных представлений о поверхностных явлениях. Даже номенклатура маслорастворимых поверхностноактивных веществ базируется не на их физико-химических свойствах, а на тех функциях, которые они способны выполнять. Соответственно этому вещества, вводимые в смазочные масла и жидкое углеводородное топливо, называют присадками (ингибиторами коррозии, антифрикционными, моющими, депрессорными и т. д.). Производители этих продуктов предпочитают называть их или по технологии получения, или по химическому составу, или даже по происхождению сырья. Ни та, ни другая классификация не отражает в должной мере основные свойства ПАВ, определяющие механизм их воздействия. Очень часто одно и то же вещество выполняет несколько функций, и всегда одну и ту же функцию способны выполнять вещества различного химического строения.
Существует ряд физико-химических характеристик маслорастворимых ПАВ, которые, во-первых, являются универсальными и в равной мере могут быть отнесены как к маслорастворимым, так и к водорастворимым веществам, а во-вторых, объясняют их полезное действие в большинстве случаев применения ПАВ. Это их адсорбционная способность, критическая концентрация мицеллообразования (ККМ), стабилизирующее действие на взвеси, солюбилизирующее действие, величина ГЛБ, влияние на смачиваемость поверхности твердых веществ водой. Разумеется, что названные характеристики взаимосвязаны, причем связующей величиной является адсорбция. Поскольку она различна при разных концентрациях ПАВ, то в качестве более универсальной характеристики следует давать константы уравнения изотермы адсорбции, например константы
796
Новый справочник химика и технолога
уравнения Ленгмюра — величину предельной адсорбции и константу адсорбционного равновесия. Не существует каких-либо принципиальных препятствий к экспериментальному определению этих констант для любой триады веществ: ПАВ, среда, материал. Здесь материал — это то вещество, поверхностные свойства которого модифицируются с помощью ПАВ в данной среде. Например, это бронза, если речь идет о вкладышах подшипников скольжения. Для этого достаточно измельчить материал и далее исследовать на нем адсорбцию стандартными методами.
Некоторые функции ПАВ, например противоизнос-ные, обусловлены их способностью к прочной необратимой адсорбции (хемосорбции). В этих случаях основной характеристикой ПАВ становятся показатели необратимости адсорбции, например процент удаления ПАВ с поверхности под действием хороших растворителей и повышенной температуры.
Из других фундаментальных характеристик ПАВ вполне доступны прямым измерениям ККМ и влияние ПАВ на смачиваемость. К сожалению, фундаментальные свойства маслорастворимых ПАВ указываются в редких случаях. В основном для их характеристики применяются специфические параметры, непосредст
венно связанные с функциями, которые выполняет ПАВ (табл. 1П1.5).
Перечень показателей, которые доступны для прямых измерений и поэтому чаще всего применяются для характеристики эффективности ПАВ как присадок к смазочным маслам и жидким топливам, указаны в табл. 2П1.5. Во многих случаях свойства ПАВ зависят от технологии их получения (например, способа сульфирования и последующей нейтрализации щелочами) и от вида сырья (нефти), используемого для производства ПАВ.
Далее приводятся некоторые свойства поверхностно-активных веществ основных химических классов. Они являются выборочными, т. е. отброшены сведения, которые не связаны с поверхностными или объемными (мицеллообразование) коллоидными явлениями. По указанным причинам они не могут претендовать ни на полноту, ни на систематичность, хотя некоторые из специфических характеристик связаны с упомянутыми фундаментальными свойствами ПАВ. Например, водо-удержание связано с солюбилизирующей способностью и гидрофильностью полярных групп (ГЛБ), водовытес-нение — со смачиваемостью, ингибирование коррозии и противоизносные свойства — с адсорбцией.
Таблица 1П1.5
Основные функции присадок к маслам и топливам [1]
Функция ПАВ Механизм, связь с поверхностной активностью
Регулирование вязкости масел Введение полимеров, в том числе не обладающих поверхностной активностью
Придание смазкам пластичности Структурирование мицеллярных растворов поверхностно-активных веществ
Уменьшение износа и трения Адсорбция ПАВ на поверхности трущихся деталей, в том числе хемосорбция
Уменьшение температурной зависимости вязкости масел Подбор компонентов, имеющих слабую зависимость вязкости от температуры вне связи с поверхностной активностью
Улучшение фильтруемости и морозостойкости топлив Уменьшение размера кристаллов твердой фазы (парафинов) под влиянием ПАВ
Повышение октанового числа топлив Подбор компонентов-антидетонаторов вне связи с поверхностной активностью
Придание маслам моющего действия Солюбилизирующее и стабилизирующее (эмульгирующее) действия ПАВ
Защита металлов от коррозии Основана на создании адсорбционного слоя ПАВ
Снижение окисляемости масел Поверхностная активность не имеет значения, но полярные группы ПАВ могут в этом участвовать
Таблица 2П1.5
Перечень показателей, характеризующих свойства и эффективность ПАВ в смазочных маслах и жидких топливах [1]
Наименование свойства Коллоидно-химические факторы, влияющие на указанные свойства
Критическая концентрация мицеллообразования (ККМ) ККМ
Показатели износа и трения в парах трения Константа адсорбционного равновесия, доля необратимой адсорбции, расклинивающее давление
Потеря массы образцов, площадь поверхности, пораженной коррозией, время до ее появления Избирательность адсорбции на активных центрах определенной химической природы
Приложение 1
797
Продолжение таблицы 2П1.5
Наименование свойства Коллоидно-химические факторы, влияющие на указанные свойства
Моющая способность Солюбилизация, диспергирующее и смачивающее действие ПАВ
Температуры помутнения, потери фильтруемости и застывания топлива
Диэлектрическая проницаемость (е) раствора ПАВ Концентрация полярных веществ (групп), переход в мицеллярное состояние
Приращение проницаемости е как мера водопогло-щения Солюбилизация и гидратация полярных групп
Щелочное число
Зольность
Вязкость
Водовытеснение с поверхности металла Избирательность смачивания
Температура разложения
Электрическое сопротивление масляной пленки на металле Концентрация ионогенных веществ и степень их диссоциации, солюбилизация воды
1.6. Маслорастворимые ПАВ основных химических классов и области их применения [1]
1.6.1. Сульфонаты
Сульфонаты — самый распространенный вид маслорастворимых ПАВ. Это соли сульфокислот, в основном продуктов сульфирования алкилбензолов, т. е. кислот типа RC6H4SO3H, где R — алкильный радикал. Кроме того, производятся алкилфенолсульфокислоты, сульфоалкилянтарные кислоты, разнообразные продукты суль-
фирования нефтепродуктов. В качестве присадок к маслам чаще всего используются их кальциевые соли. Так называемые беззольные присадки представляют собой продукты нейтрализации сульфокислот аминами. Маслорастворимые сульфонаты используются как моющие и противокоррозионные добавки к смазкам, маслам и топливам. Водорастворимые ПАВ (соли щелочных металлов сульфокислот с коротким алкильным радикалом) добавляются в воду, закачиваемую в нефтеносные пласты для увеличения их нефтеотдачи (табл. 1П1.6).
Таблица 1П1.6
Характеристика нефтяных и синтетических сульфонатов [ 11
Продукт Марка продукта Кинематическая вязкость при 100 °C, мм2/с Содержание активного вещества, масс. % Основное назначение
Сульфонат кальция на основе масла ДС-11у ПМСо-а до 45 >18 Моющая присадка к моторным маслам
Сульфонат кальция на основе масла ДС-11, нейтральный СК-3 14-20 15-20
То же, щелочной ВСК-3 15-20
Сульфонат бария на основе масла ДС-11, нейтральный СБ-3 14-20 15-20
То же, щелочной ВСБ-3 15-20
Сульфонат кальция на основе масла АС-6 КСК 20-30 20-25 Ингибитор коррозии
Алкиларилсульфоамид мочевины на основе масла АС-6 БМП 80 20-25
Сульфонат кальция на основе очищенного остаточного масла НГ-104 25-30 28-35 Моющая присадка к моторным маслам
798
Новый справочник химика и технолога
Продолжение таблицы 1П1.6
Продукт Марка продукта Кинематическая вязкость при 100 °C, мм2/с Содержание активного вещества, масс. % Основное назначение
Сульфонаты щелочных и щелочноземельных металлов на основе масла АСВ-5
Лития ИСЛ 15-25 30 Ингибитор коррозии к маслам
Кальция ИСК 20-30 30
Бария ИСБ 30-50 30
Магния ИСМ 20-30 30
Сульфонаты натрия на основе масла АС-6 15 25-30 Основа эмульсолов
Сульфонаты на основе экстрактов из масляных дистиллятов
Кальциевая или бариевая соль ПМСЯ 18-40 25-35 Моющая присадка к моторным маслам
Синтетические сульфонаты на основе алкилбензолов
Кальциевая соль САБ (кальция) до 100 40-45 Ингибитор коррозии и компонент пленочных покрытий
Соль мочевины БМПо-а 25-30 >50 Ингибитор коррозии к топливам и маслам
Карбамидо-кальциевая соль Карбин-3 60-70 >40 Ингибитор коррозии и компонент пленочных покрытий
Кальциево-натриевая соль Консулит-73 70-80 40-50
Кальциевая соль сульфодиалкилнаф-талинов ССК-1 Моющая присадка к моторным маслам
Бариевая соль сульфодиалкилнафта-линов ССБ-1
Литиевые соли и соли щелочноземельных металлов сульфоалкенилянтарной кислоты Ингибиторы коррозии и моющие присадки к маслам
1.6.2. Амины, амиды, имиды
Поверхностно-активные вещества этого класса отличаются большим разнообразием химического строения: анилин, алкилфениламины, алкилфеноламины, алифатические амины, в частности полиэтиленполиамин. Очень разнообразны кислородсодержащие соединения азота. Важнейшим из них являются ПАВ с
общим названием сукцинимиды, которые получаются взаимодействием соответствующих алкенилянтарных ангидоидов с аммиаком и аминами, в том числе полиэтиленполиамином. Как и многие другие ПАВ, эти соединения являются полифункциональными присадками к маслам, смазкам, топливам и другим продуктам. Их характеристики представлены в табл. 2П1.6 и ЗП1.6.
Таблица 2П1.6
Свойства и функциональное назначение присадок к маслам
Показатели Диэлектрическая проницаемость*1 Водоудержа-ние, % Моющее действие, %*2 Водовытес-нение, мм Коррозия пораженной поверхности, %
Олеат трис(2-гидроксиэтил)-аммония 8,7 100 144 2
СольСЖК (Сп-С]2)и дициклогексиламина 2,33 59 20 36 6
Имидозолин (Hol, ЭДА) 2,38 10 20 80 3
Основания Манниха (АФол, ФрмАлд, ДЭТА, Но!) 2,32 4 75 70 4
Приложение 1
799
Продолжение таблицы 2П1.6
Показатели Диэлектрическая проницаемость 1 Водоудержа-ние, % Моющее действие, %*2 Водовытес-нение, мм Коррозия пораженной поверхности, %
Алкенилсукцинимид полиэтиленполиамина 239 15 95 105 55
Алкенилсукцинимид мочевины 2,42 1 90 35 0
Нитроалкенилсукцинимид мочевины 2,6 3 80 52 0
Сульфоксиалкенилсукцинимид мочевины 2,8 2 95 90 4
*' 0,1%-й раствор в бензоле.
*2 5%-й раствор в масле АС-6.
Таблица ЗП1.6
Обобщенная характеристика действия маслорастворимых азотсодержащих ПАВ
Обозначения: э — эффективно, п — положительные изменения, о — отрицательные изменения, х — выраженный негативный эффект.
Типы соединений и их действие в присадках Моющая присадка Антиокислитель Термостабилизатор Антифрикционная присадка Ингибиртор Компонент защитных пленок Водоэмульгатор Термостабилизатор вязкости Присадка дизельного топлива Присадка турбинных масел Присадка авиационных масел Присадка моторных и трансмиссионных масел Компонент пластичных смазок Компонент пленочных покрытий Компонент эмульсолов
Амины алифатичские X X X о X п п о о о X X о о о
Амины ароматические о п о о X п п о о п о о п о п
Соли жирных кислот и аминов X X X о X п э о о о X X о о п
Имидазолины о о X о X п п о о о X о о п п
Основания Манниха п п о II о п о о о о X п п п о
Соли сульфокислот и аминов п п о п о э п о э п X о п п э
Алкенилсукцинимиды полиэтиленполиамина э п о п о о о п п п X э о п о
То же, модифицированные э п п п п п о п э п о э п п о
Алкенилсукцинимид мочевины и модификации п п п п п э п п п э п э э э п
Производные пиромеллитового диангидрида э п э п п э п п э э э э э э п
1.6.3. Кислородсодержащие ПАВ
ПАВ этого класса представлены жирными спиртами, кислотами, эфирами и более сложными по строению веществами, получающимися при окислении нефтепродуктов, или природными соединениями, входящими в состав нефтей. Они часто применяются для защиты металлов от коррозии. Их свойства представлены
в табл. 4П1.6. К этому классу маслорастворимых ПАВ относятся жирные кислоты, в том числе кислоты ряда С17Нз5_„СООН разной степени непредельности: стеариновая (п = 0), олеиновая (п = 2), линолевая (л = 4) и линоленовая (п = 6). В этом ряду только олеиновая кислота является хорошим стабилизатором коллоидных растворов в углеводородных средах. В частности, именно она используется при получении феррожидкостей.
800
Новый справочник химика и технолога
Таблица 4П1.6
Свойства некоторых кислородсодержащих маслорастворимых ПАВ
(5%-й раствор в масле АС-6)
Соединение, относительная молярная масса Диэлектрическая проницаемость 50 % раствора в бензоле Потеря массы (%) при температуре (°C) Коррозия пластин (окисление влажным воздухом), г/м2
200 250 сталь медь свинец
СЖК(С1Г-С20) Разлага- 0,7 24,3 198
Окисленный петролатум 2,26 ется 0,1 12,3 68,1
Алкенилянтарные ангидриды с разной относительной молярной массой на основе полиизобутилена
280 2,97 5,2 24,6 0 4,8 36,3
420 2,83 0,1 5,3 44,4
500 2,75 7,3 38,9 0,2 6,9 93,1
880 2,51 0 3,8 75,6
1300 2,4 8,9 33,4 0,1 4,2 84,2
1470 2,34 о,1 6,7 92,2
Алкенилянтарные ангидриды с разной относительной молярной массой на основе полибутилена (октола)
370 2,68 6,1 21,2 0,1 8,6 93
1000 2,42 12,1 34 0 3,9 68,5
1280 2,32 0,1 4,8 95,2
1680 2,28 0 6,7 42,5
Алкенилянтарный ангидрид на основе полипропилена
300 2,73 4,9 22,3 0 4,3 21,2
Соли алкенилянтарного ангидрида с относительной молярной массой 1000
Лития 2,51 0,1 3,8 36,7
Магния 2,56 0 3,5 22,3
Кальция 2,58 0,2 2,9 37,8
Цинка 2,49
Алюминия 2,41
Эфиры
Бензилянтарной кислоты 3,11 7,8 26,5 0 5,2 45,5
Двухосновной кислоты (моноэфир) 2,86 5,4 20,8 0,1 6,3 70,1
Пентадеценилянтарной кислоты (моноэфир) 2,37 8,3 41 0,3 8,8 88,3
Чистое масло АС-6 0,1 3 36,6
1.6.4. Хлор и серосодержащие соединения
ПАВ этого типа обладают выраженными противо-износными свойствами. Это хлорированные парафины, смесь тетра- и пентахлорбифенилов (савол), сульфиды алкилфенолятов бария, стронция и др.
1.6.5. Сополимеры, содержащие полярные и неполярные звенья [2]
Сополимеры полярных и неполярных мономеров с молярной массой порядка 104 используются в качестве депрессорных присадок к маслам и топливам. Назначе
ние этих присадок — расширить диапазон применения топлив и масел в сторону низких температур. Так как последние представляют собой более или менее широкий набор фракций углеводородов различной молярной массы, то при понижении температуры начинается кристаллизация тяжелых фракций этих смесей, что ведет к их загустеванию. Для топлив первостепенное значение имеет сам факт появления кристалллической фазы, так как это ведет к забиванию топливных фильтров и прекращению подачи топлива в двигатель. Именно в связи с этим важнейшим показателем эффективности депрессор
Приложение 1
801
ных присадок является нижний температурный диапазон фильтруемости топлива. Для экспресс-анализа удобной характеристикой является температура помутнения топлива или масла, которая может быть приблизительно определена визуально.
Простейшей депрессорной присадкой являются сополимеры этилена и винилацетата, содержащие 20-40 % винилацетата. Часть ацетатных звеньев может быть при этом гидролизована или заменена винилхлоридом до полимеризации. Применяются также алкил-метакрилаты вместо винилацетата, используется частичная замена этилена стиролом и многие другие комбинации тройных сополимеров. В ряде случаев эффективны смеси сополимеров разного химического строения, в том числе отличающихся только длиной алкильного радикала. Это разнообразие обусловлено существенным различием состава нефтепродуктов разного происхождения и механизмом действия депрессоров.
Действие депрессоров сводится к сильному уменьшению размера частиц, образующихся при кристаллизации тяжелых компонентов топлива или масла. Этот эффект достигается путем встраивания молекул при
садки в кристаллическую решетку вымерзающего из смеси компонента топлива. Для этого строение неполярной части присадки должно быть подобно строению выпадающего компонента смеси. Разнообразие химического строения тяжелых фракций нефтепродукта требует соответствующего разнообразия в строении присадки.
Встраивание в целом чужеродных молекул присадки в кристаллическую решетку вымерзающего компонента ведет к накоплению дефектов решетки в частицах твердого вещества, которых тем больше, чем крупнее частицы, и, как следствие, к прекращению их роста. Благодаря этому на начальном этапе кристаллизации сохраняется высокая первоначальная степень перенасыщения раствора, что способствует образованию большого количества мелких кристаллов.
Литература
1. Шехтер Ю.Н., Крейн С.Э., Тетерина Л. Н. Маслорастворимые поверхностно-активные вещества. М.: Химия, 1978. 304 с.
2. Тертерян Р.А. Депрессорные присадки к нефтям, топливам и маслам. М.: Химия, 1990. 238 с.
ПРИЛОЖЕНИЕ 2
Дисперсные системы
2.1. Точки нулевого заряда
Точка нулевого заряда (ТНЗ) — электрохимическая характеристика поверхности вещества, например Agl, в определенной среде, например в растворе электролита. Она указывает условие, при котором поверхность вещества в данной среде незаряжена. Величина удельного (на единицу площади поверхности вещества) заряда поверхности, которая также называется поверхностной плотностью заряда, зависит от многих факторов. Среди них важнейшими являются концентрация потенциалопределяющих (ПО) ионов и величина внешней разности потенциалов, подведенной к веществу и той среде, в которой оно находится. В последнем случае вещество исполняет роль одного из электродов электрохимической ячейки. Соответственно этому нулевую величину заряда поверхности можно обеспечить, изменяя концентрацию подходящего электролита, и тогда ТНЗ — это концентрация ПО иона, при которой поверхность не заряжена. Нулевую величину заряда можно также обеспечить, подавая на электрод, изготовленный из исследуемого вещества, электрический потенциал, про-
'Т’ТУта/ЧГГГЧ ГТГЧМТТТТ TTJ ГГП ОТГОТЛЧГ rr/4'T'OtITTTZQ ГГХ7
Л. fAXJVrXlVrJXVrSXVllXJAiri 11V uUvV 1UU1111V1U j XiV X VlXl^llUJX j uv
щества в данной среде. В этом случае ТНЗ — это величина внешней разности потенциалов, подведенной к
веществу и среде, которую более однозначно можно назвать потенциалом нулевого заряда (ПНЗ). Одно и то же вещество может иметь бесчисленное множество потенциалов нулевого заряда, поскольку ПНЗ является также и функцией состава среды. По этой причине ПНЗ не представляет интереса как электрохимическая характеристика вещества. В большей мере он характеризует адсорбционную способность (потенциал) ПО ионов к поверхности данного вещества. Перечень веществ, на которых можно измерять ПНЗ, практически ограничен одной ртутью, и поэтому этим методом можно изучать адсорбционную способность тех или иных ионов только на поверхности ртути. Такие данные представляют ограниченный интерес и далее не рассматриваются. В отличие от этого концентрационная ТНЗ является характеристикой вещества по отношению к любым ионам. Однако фундаментальный интерес представляет значение точки нулевого заряда по отношению к ионам, которые образуют изучаемое вещество (табл. 1П2.1), например к ионам Ag+ и Г, если речь идет о ТНЗ иодида серебра Agl. Однако такого рода данных очень мало. В тех случаях, когда они есть (а это — галогениды се-
даже для классического объекта исследований такого рода — золя Agl.
Таблица 1П2.1
Точки нулевого заряда (моль/л) [1]
Вещество Растворимость, г/100 мл воды ТНЗ по иону серебра ТНЗ по галогенид-иону Произведение ТНЗ
AgCl AgBr Agl Ag 9-10’5 1,2-10’5 3-10~7 0,0001 3-10'7ч-3,98-10^ юМ, 16-1 о6 10"13-* 1,5-10-5 2-106 1,26-10~7+ 1,5-10 6 2,51-10“п-М0“8 2-1O10 4,5-10~13-е-5,01-10"13 7,94-Ю'МО 16
2.2. Константы молекулярного взаимодействия
Несмотря на фундаментальную роль молекулярного притяжения, действующего между двумя телами через плоский узкий зазор толщиной h, и наличие ряда формул для расчета энергии этого взаимодействия (например, A/h2 в случае отсутствия электромагнитного запаздывания), константа А этого взаимодействия не приобрела статуса общепризнанной физической характеристики веществ. В некоторых монографиях, пользующихся общим признанием [1], не приводится ни одного численного значения этой константы, хотя обсуждению общих закономерностей молекулярного притяжения уделяется большое внимание. В работах [2, 3] содержатся как численные значения констант взаимодействия различных веществ, так и обсуждение общих законо
мерностей молекулярного взаимодействия тел. Это указывает на существование различных подходов к проблеме молекулярного притяжения тел.
С точки зрения практической технологии в большинстве случаев желательно иметь не точное значение параметров процесса, например скорости коагуляции, а располагать знанием общих закономерностей, позволяющих предусмотреть в проекте нужные средства его регулирования. Для выбора пределов регулирования зачастую достаточно иметь представление о порядке величины, в данном случае константы притяжения. Вполне реалистичным представляется значение А = 10-21 Дж. Такое значение константы приписывается, в частности, воде. Максимальное значение константы притяжения присуще металлам. Оно примерно на порядок больше указанной выше величины.
804
Новый справочник химика и технолога
Пренебрежение к точному значению константы притяжения А характерно для многих авторов. Оно угадывается в том, как обобщаются данные по порогам коагуляции различных коллоидных растворов электролитами (табл. 1.П2.2, 2.П2.2). Например, один из авторов теории устойчивости — Овербек оперирует их средним значением [1], сопоставляя с теоретическим критерием 1/z6 (z— заряд иона-коагулятора), а не с его более точным вариантом 1Л42?6, который, кстати, может использоваться и для нахождения значения константы притяжения [2].
Следует напомнить, что при отрицательном заряде коллоидных частиц коагулирующая способность электролита определяется валентностью его катиона, а при положительном — валентностью аниона, что и подтверждается приведенными в табл. 1П2.2 и 2П2.2 значениями порогов коагуляции положительно и отрицательно заряженных золей.
Таблица 1П2.2
Пороги коагуляции отрицательно заряженных золей, ммоль/л [1]
Электролит Золь As2S3 Среднее значение Золь Au Среднее значение Золь Agl Среднее значение
А1С13 0,093
A1(NO3)3 0,095 0,067
[А12(8О4)з]/2 0,096 0,091 0,009 0,006 0,068
ВаС12 0,69 0,35
Ba(NO3)2 СаС12 0,65 0,41 2,26
Ca(NO3)2 2,4
Ce(NO3)3 0,08 0,003 0,069
КС1 49,5
KNO3 50 55 25 24 136 142
(K2SO4)/2 La(NO3)3 65,5 23 0,069
LiCl 58
LiNO3 165
MgCl2 Mg(NO3)2 0,72 2,6
MgSO4 0,81
NaCI NaNO3 51 24 140
Pb(NO3)2 2,43
RbNO3 126
SrCl2 Sr(NO3)2 0,635 0,69 0,38 2,38 2,43
Th(NO3)4 0,09 0,09 0,0009 0,0009 0,013 0,013
UO2(NO3)2 0,64
ZnCl2 Zn(NO3)2 0,635 2,5
Таблица 2П2.2
Пороги коагуляции положительно заряженных золей, ммоль/л [1]
Электролит Золь Fe2O3 Среднее значение Золь A12O3 Среднее значение
(ВаС12)/2 9,65
[Ba(NO3)2]/2 14
КС1 9 46
NH4C1 43,5
NaCI 9,25 43,5
KBr 12,5 11,8 52
KSCN 67
KI 16
KNO3 12 60
K2C2O4 0,69
K2Cr2O7 0,195 0,63
K2SO4 0,205 0,3
K3[Fe(CN)6] 0,08 0,08
K4[Fe(CN)6] 0,053 0,053
MgCr2O7 MgSO4 0,22 0,21 0,95 0,63
T12SO4 0,22
Значения констант А молекулярного притяжения частиц дисперсной фазы, найденные из подобного рода экспериментальных данных, приведены в табл. ЗП2.2. Это так называемые сложные константы, которые включают эффект отталкивания частиц, обусловленный молекулярным притяжением дисперсионной среды к частицам, создающим указанный эффект.
Появление множителя 12л в обозначении величин второго столбца табл. ЗП2.2 связано с тем, что компактный способ записи выражения для удельной (на единицу площади) энергии молекулярного притяжения тел Um = A/h2, принятый здесь, отличается от более пространного способа записи того же выражения в большинстве других источников: Um = A /12лй2. Соответственно различаются и численные значения констант притяжения при расчетах по компактной и пространной формулам. В табл. ЗП2.2 приведено увеличенное в 1О2ораз значение константы А*, численно равное величине 12лЛ. Таким образом, при расчетах по компактной формуле Um = A/h2 в нее следует подставлять указанное в табл. ЗП2.2 значение константы после ее деления на величину 12л и еще на Ю20, т. е. А = т4*/(12л-1О20).
Источником наиболее надежных экспериментальных данных о значении констант притяжения веществ являются прямые измерения силы притяжения тел, изготовленных из этих веществ. Такого рода исследования выполнены Дерягиным [3]. Перечень веществ, из которых можно изготовить образцы, пригодные для прецизионных измерений, ограничен. Это в основном металлы, стекло, кварц. В связи с этим результаты пря
Приложение 2
805
мых измерений ценны не как источник сведений о константах взаимодействия, а как прямое подтверждение законов молекулярного взаимодействия частиц. По методическим причинам прямые измерения относятся к расстояниям между поверхностями тел порядка 0,1 мкм и более. Закон притяжения на таких расстояниях отличается вследствие электромагнитного запаздывания от закона взаимодействия на расстояниях порядка 0,01 мкм и менее. Именно эта, ближняя область расстояний важна при коагуляционных взаимодействиях коллоидных частиц. На дальних расстояниях взаимодействие слишком слабо, чтобы проявить себя в реальных процессах и свойствах коллоидов, и поэтому оно далее не рассматривается.
В ряде задач, например связанных с выбором материала дисперсной фазы или дисперсионной среды, важно иметь зависимость константы притяжения от химической природы материала. Самое доступное свойство веществ, через которое может быть выражена константа молекулярного притяжения, — это их показатели преломления п (табл. 4П2.2).
Таблица ЗП2.2
Константы молекулярного притяжения частиц в водной среде
Дисперсная фаза [2] А 12л1О20, Дж Дисперсная фаза [6] А 12я1О20, Дж
Арахиновая кислота 0,03 Графит 3,7
Диоксид кремния 0,5-0,2 Диоксид титана 2,5-10,0
Золото 6 Оксид алюминия 4,2
Иодид серебра 0,5-9 Оксид железа 3,4
Парафин 1,5 Гидроксид алюминия 12,6
Полистирол 0,1-1,1 Гидроксид железа 17,7-20
Селен 0,5-5 Циклогексан 1,5
Платина * 10 Декан 0,56
Золото * 40 Бензол 0,04
Плавленый кварц * 6,5-11,6 Стеариновая кислота 0,08
Кристаллический кварц * 6-6,8 Политетрафторэтилен 0,0024
Боросиликатное стекло * 9 Полиэтилен 0,4
Галогениды таллия * 28,3-40 Полистирол 0,1-1
Слюда * 10 Поливинилацетат 0,54
* Результат прямых измерений силы взаимодействия тел в водной среде.
Таблица 4П2.2
Показатели преломления п при длине волны % = 589,3 нм (желтая линия D натрия) и плотности р твердых веществ при 20 °C [4]
Вещество Формула n p • io3, кг/м3
Алюминия борат А1ВО3 1,64
Алюминия сульфат октадекагидрат A12(SO4)3 • • 18Н2О 1,476 1,69
Алюминия хлорид гексагидрат А1С13 • 6Н2О 1,56 2,4
Алюминия оксид (прокаленный гель) А12О3 1,65 3,9
Алюминия оксид (ромбоэдрический) А12О3 1,768 4
Алюминия оксид тригидрат А12О3 • ЗН2О 1,55 2,44
Аммония нитрат NH4NO3 1,611 1,69
Аммония бромид NH4Br 1,7108 2,429
Аммония иодид nh4i 1,7031 2,511
Аммония сульфат (NH4)2SO4 1,523 1,769
Аммония хлорид nh4ci 1,639 1,53
Аммония перхлорат nh4cio4 1,4833 1,95
Ацетамид ch3conh2 1,54
Бария нитрат Ba(NO3)2 1,572 3,23
Бария бромид дигидрат BaBr2 • 2H2O 1,7266 3,58
Бария силикат BaSiO3 1,82 2,59
Бария карбонат BaCO3 1,676 4,43
Бария фторид BaF2 1,475 4,96
Бария хлорид гидрат BaCl2 • 2H2O 1,646 3,07
Бария хлорид BaCl2 1,7302 3,856
Бария перхлорат гептагидрат Ba(ClO4)2 • • 7H2O 1,533 4,498
Бария гидроксид окгагидрат Ba(OH)2 • 8H2O 1,5017 2,18
Бария оксид BaO 1,98 5,72
1,2-Дифенилэтан-днон C6H5COCOC6H5 1,6588
Бериллия сульфат тетрагидрат BeSO4 • 4H2O 1,472 1,713
Бериллия оксид BeO 1,719 3,02
Борная кислота (орто) H3BO3 1,456 1,435
Бора оксид (стекловидный) B2O3 1,459 1,85
Вольфрамовая кислота H2WO4 2,24 5,5
806
Новый справочник химика и технолога
Продолжение таблицы 4П2.2
Вещество Формула n P • 10’3, кг/м3
Германия(1У) оксид GeO2 1,65 4,703
Гидрохинон СбН^ОНЬ 1,633 1,332
Железа(П) сульфат тетрагидрат FeSO4 • 4Н2О 1,536 2,2
Железа(П) сульфат гептагидрат FeSO4 • 7Н2О 1,4782 1,899
Железа(Ш) сульфат Fe2(SO4)3 1,814 2,1
Железа(П) карбонат FeCO3 1,875 3,8
Железа(П) хлорид FeCl2 1,567 2,98
Железа(Ш) оксид Fe2O3 3,22 5,24
Иттрия сульфат октагидрат Y2(SO4)3 • 8H2O 1,549 2,558
Кадмия сульфид CdS 2,506 4,58
Кадмия сульфат 2,6-гидрат CdSO4 • 2,6H2O 1,564 3,09
Кадмия фторид CdF2 1,56 6,64
Кадмия хлорид 2 5-гм™ат CdCl2 • 2,5H2O 1,6513 3,327
Кадмия оксид CdO 2,49 6,95
Калия нитрат KNO3 1,5038 2,11
Калия бромид KBr 1,5594 2,75
Калия дихромат K2Cr2O7 1,738 2,69
Калия иодид KI 1,667 3,13
Калия фторид KF 1,352 2,48
Калия хлорид KC1 1,4904 1,988
Калия перхлорат KC1O4 1,4737 2,524
Калия дихромат K2Cr2O7 1,7261 2,732
Кальция силикат (а) CaSiO3 1,611 2,905
Кальция силикат (Р) CaSiO3 1,629 2,915
Кальция сульфат (высотемператур-ный) CaSO4 1,576 2,96
Кальция сульфат дигидрат (гипс) CaSO4 • 2H2O 1,5226 2,32
Кальция нитрат Ca (NO3)2 1,498 2,36
Кальция вольфрамат CaWO4 1,92 6,06
Кальция сульфид CaS 2,137 2,18
Кальция карбонат (кальцит) CaCO3 1,55 2,71
Кальция карбонат (арагонит) CaCO3 1,6809 2,93
Продолжение таблицы 4П2.2
Вещество Формула n P • 1 O’3, кг/м3
Кальция ацетиленид (карбид) СаС2 1,75 2,22
Кальция фторид CaF2 1,4339 3,18
Кальция хлорид гексагидрат СаС12 • 6Н2О 1,417 1,68
Кальция гидроксид Са(ОН)2 1,574 2,343
Кальция оксид СаО 1,837 1,837
Кобальта нитрат гексагидрат Co(NO3)2 • 6Н2О 1,4 1,883
Кобальта сульфат гептагидрат CoSO4 ♦ 7Н2О 1,483 1,948
Кобальта карбонат СоСО3 1,855 4,13
Кобальта ацетат тетрагидрат Со(С2Н3О2)2 • • 4Н2О 1,542 1,705
Кремния оксид «-гидрат(опал) SiO2 • «Н2О 1,41 2,2
Кремний Si 3,736 2,4
Кремния карбид SiC 2,654 3,17
Кремния оксид (кварц) S1O2 1 5442 2,65
Кремния оксид (прокаленный гель) SiO2 1,48 2,26
Лития нитрат LiNO3 1,735 2,38
Лития бромид LiBr 1,784 3,464
Лития иодид Lil 1,955 4,061
Лития сульфат Li2SO4 1,465 2,22
Лития фторид LiF 1,3915 2,295
Лития хлорид LiCl 1,662 2,068
Магния сульфид MgS 2,268 2,8
Магния сульфат гептагидрат MgSO4 • 7H2O 1,4554 1,68
Магния ацетат тетрагидрат Mg(C2H3O2)2 • •4H2O 1,491 1,454
Магния фосфат октагидрат Mg3(PO4)2 • • 8H2O 1,52 2,1
Магния фторид MgF2 1,378 3
Магния хлорид MgCl2 1,675 2,325
Магния хлорид гексагидрат MgCl2 • 6H2O 1,507 1,56
Магния гидроксид Mg(OH)2 1,559 2,4
Магния оксид MgO 1,7364 3,65
Марганца(П) сульфат пентагидрат MnSO4 • 5H2O 1,508 2,1
Марганца(П) карбонат MnCO3 1,817 3,125
Приложение 2
807
Продолжение таблицы 4П2.2
Вещество Формула n P • Ю’3, кг/м3
Марганца(П) хлорид тетрагидрат МпС12 • 4Н2О 1,575 2,01
Меди(1) бромид Cu2Br2 2,116 4,72
Меди(1) иодид Cu2I2 2,345 5,63
Меди(П) сульфид CuS 1,45 4,6
Меди(П) сульфат пентагидрат CuSO4 • 5Н2О 1,5368 2,286
Меди(П) сульфат CuSO4 1,733 3,606
Меди(1) хлорид Cu2Cl2 1,973 3,53
Меди(П) хлорид дигидрат CuCl2 • 2H2O 1,684 2,39
Меди(П) оксид CuO 2,84 6,45
Мышьяка(Ш) иодид Asl3 2,59 4,39
Мышьяка(Ш) оксид As2O3 1,755 3,865
Натрия нитрат NaNO3 1,5874 2,257
Натрия бромид NaBr 1,6412 3,205
Натрия иодид Nal 1,7745 3,667
Натрия сульфит Na2SO3 1,565 2,633
Натрия сульфат Na2SO4 1,477 2,698
Натрия карбонат Na2CO3 1,535 2,533
Натрия карбонат декагидрат Na2CO3 • • 10H2O 1,425 1,46
Натрия ацетат NaC2H3O2 1,464 1,528
Натрия фторид NaF 1,3258 2,79
Натрия хлорид NaCI 1,5443 2,163
Никеля сульфат гептагидрат NiSO4 • 7H2O 1,4893 1,948
Никеля хлорид гексагидрат NiCl2 • 6H2O 1,57 2,07
Никеля(П) оксид NiO 2,37 7,45
Олова(1У) оксид SnO2 1,9968 7
Олова(1 V) иодид Snl4 2,106 4,7
Ртути(П) иодид (красный) Hgl2 2,748 6,283
Ртути(П) сульфид (киноварь) HgS 2,854 8,1
Ртути(П) хлорид (сулема) HgCl2 1,859 5,44
Ртути(1) хлорид (каломель) Hg2Cl2 1,9733 7,15
Ртути(И) оксид HgO 2,5 11,14
Рубидия бромид RbBr 1,5528 3,35
Рубидия иодид Rbl 1,6474 3,55
Продолжение таблицы 4П2.2
Вещество Формула n PIO’3, кг/м3
Рубидия фторид RbF 1,396 2,88
Рубидия хлорид RbCl 1,4936 2,76
Самария сульфат октагидрат Sm2(SO4)3 • • 8Н2О 1,5519 2,93
Свинца(П) нитрат Pb(NO3)2 1,7815 4,53
Свинца(П) сульфид (галенит) PbS 3,912 7,5
Свинца(П) сульфат PbSO4 1,8823 6,2
Свинца(П)хлорид PbCl2 2,2172 5,85
Свинца(1У) оксид PbO2 2,229 9,375
Свинца(П) оксид PbO 2,61 8
Селен Se 3 4,8
Сера (аморфная) S 2 1,92
Серебра нитрат AgNO3 1,744 4,352
Серебра бромид AgBr 2,252 6,473
Серебра иодид Agl 2,218 5,67
Серебра хлорид AgCl 2,071 5,56
Стронция нитрат Sr(NO3)2 1,5878 2,986
Стронция гидроксид октагидрат Sr(OH)2 • 8H2O 1,499 1,9
Сурьмы(Ш) иодид Sbl3 2,78 4,768
Сурьмы диоксид Sb2O4 2 4,07
Сурьмы(Ш) оксид Sb2O3 2,087 5,2
Таллия(1) бромид TIBr 2,4 7,557
Таллия(1) иодид TH 2,78 7,09
Таллия(1) хлорид T1C1 2,247 7
Титана(1У) оксид TiO2 2,554 3,84
Тория оксид ThO2 2,2 9,69
Углерод (алмаз) C 2,4173 3,51
Фосфор (желтый) P 2,1168 1,745
Хрома(Ш) сульфат октадекагидрат Cr2(SO4)3 • • 18H2O 1,564 1,7
Хрома(Ш) оксид Cr2O3 2,5 5,21
Цезия нитрат CsNO3 1,55 3,687
Цезия бромид CsBr 1,6984 4,44
Цезия иодид CsI 1,7876 4,51
Цезия фторид CsF 1,578 3,586
Цезия хлорид CsCl 1,6418 3,97
Цинка сульфид (а) ZnS 2,356 4,087
Цинка сульфид (0) ZnS 2,2 4,102
Цинка ацетат Zn(C2H3O2)2 1,494 1,735
Цинка хлорид ZnCl2 1,687 2,91
Цинка оксид ZnO 2,004 5,606
Циркония оксид ZrO2 2,19 5,49
808
Новый справочник химика и технолога
Константа притяжения частиц твердой фазы в жидкой дисперсионной среде, т. е. сложная константа притяжения А, выражается через простые константы притяжения твердой (Л]) и жидкой (А2) фаз в вакууме:
л=(д,,2-42)2.
(1П2)
Для воды значение константы притяжения (А2 = = 3,5 • Ю 20Дж) получено экспериментально на основе данных о равновесных толщинах тонких водных пленок. Для других веществ она рассчитывается по формуле (6П2). Для этого по данным табл. 4.П2.2 находят молярную рефракцию Rm и молекулярную поляризуемость ао дисперсной фазы:
(2П2)
ао ~ э > (ЗП2)
затем характеристическую частоту электронных осцилляторов молекул данного вещества:
постоянную Лондона:
ьо" 2л
(4П2)
Р = (ЗяШа»2 (5П2)
и константу притяжения Ван-дер-Ваальса— Гамакера:
Здесь
(6П2)
(7П2)
— молярный объем вещества, М — его молярная масса и р — плотность, NA — число Авогадро, е — заряд и те— масса электрона, h — постоянная Планка, £0 — диэлектрическая постоянная.
Аналогично вычисляется константа притяжения жидкости (Л2), используемой в качестве дисперсионной среды, по свойствам жидкостей, приведенным в табл. 5П2.2 и 6П2.2. В табл. 5П2.2 приведены также
значения температурных коэффициентов — показате-dT
ля преломления. С помощью температурного коэффициента вычисляется значение показателя преломления пт при температуре Т, отличающейся от 20 °C, к которой относятся приведенные в табл. 5П2.2 значения по
казателей л20. Температурная поправка вводится с помощью формулы:
«г=«?-(Г-20)—. (8П2)
al
Температурный коэффициент показателя преломле-dn
ния — отрицателен, так что с увеличением темпера-
туры его значение уменьшается.
Для расчета молярной рефракции жидкостей необходимо также знание их плотности (табл. 6П2.2).
При использовании жидкостей в качестве среды для дисперсионного анализа, а также для расчетов реологических свойств дисперсных систем требуются значения их вязкости (табл. 7П2.2).
Таблица 5П2.2
Показатели преломления жидкостей при 20 °C и длине волны л = 589,3 нм [5]
Пояснение. --- в интервале температур 15-20 °C
dT
Вещество Формула dn ~~dT
Анилин c6h7n 1,5863 0,00048
Д TTATVMJ г.н.п 1 35Q11 п пппло V, VV V • _Z
Ацетонитрил c2h3n 1,34604 0,00045
Ацетофенон c8H8o 1,53423 0,00041
Бензол c6H6 1,5011 0,00066
Бромбензол С6Н5Вг 1,5601 0,00048
Бутан-1-ол С4Н10О 1,3993
Вода Н2О 1,333 0,00008
Гексан С6Н14 1,37506 0,00055
Гептан С7Н16 1,38764
Глицерин С3Н8О3 1,4744 0,00022
1,4-Диоксан с4н8о2 1,4223
Диэтиловый эфир С4Н10О 1,32575 0,00056
о-Ксилол С8Ню 1,50545
л<~Ксилол С8Ню 1,49722
н-Ксилол С8Ню 1,49582
Метанол СН4О 1,3286 0,0004
Метилацетат С3НбО2 1,3593
2-Метилпропан-1-ол С4Н10О 1,3958
Метил формиат С2Н4О2 1,34201 0,00043
Муравьиная СН2О2 1,3716
кислота
Нитробензол СбН5МО2 1,5524 0,00046
Нитрометан CH3NO2 1,382
Октан ♦’ с8н18 1,3977
Пентан ♦1 С5Н12 1,35769
Пиридин c5h5n 1,51 0,00048
Приложение 2
809
Продолжение таблицы 5П2.2
Вещество Формула п20 nD dn ~~dT
Пропан-1-ол С3Н8О 1,3854
Пропан-2-ол С3Н8О 1,3773
Проп-2-ен-1-ол С3Н7О 1,40911 0,00041
Пропионовая *1 кислота * С3НбО2 1,3869
Сероуглерод CS2 1,628 0,00078
Стирол CrHr 1,5468
(винилбензол)
Тиофен C4H4S 1,5286 0,00044
Толуол С7н8 1,49693 0,00057
Уксусная кислота С2Н4О2
Уксусный альдегид *2 С2Н4О 1,3392
Уксусный ангидрид С4НбО3 1,3877 0,0004
Фснилгидразин c6h8n2 1,6105 0,00024
Фенилметанол с7н8о 1,5404 0,0004
Продолжение таблицы 5П2.2
Вещество Формула п20 nD dn dT
Фенол *3 С6Н6О 1,54
Формамид CH3NO 1,4472
Фтортрихлорметан (фреон-11) *2 CFC13 1,3865
Хлорбензол С6Н5С1 1,5246 0,00058
Хлороформ СНС13 1,4456 0,00059
Циклогексан *‘ с6н12 1,4263
Четыреххлористый углерод ССЦ 1,4603 0,00055
Этан-1,2-диол С2Н6О2 1,4318
Этанол с2н6о 1,3613 0,0004
Этилацетат с4н8о2 1,3726
Этилформиат*1 с3н6о2 1,3603
*’ Линия D гелия.
*2При 18 °C.
*3 При 45 °C.
Таблица 6П2.2
Плотность (р • 10"3, кг/м3) жидкостей при различных температурах [5]
Вещество Т, =С
0 10 20 30 40 50 60
Анилин 1,039 1,0303 1,0218 1,0131 1,0045 0,9958 0,9872
Ацетон 0,8125 0,8014 0,7905 0,7793 0,7682 0,756 0,7496
Ацетонитрил 0,8035 0,7926 0,7822 0,7713
Ацетофенон 1,0364 1,0278 1,0194 1,0106 1,0021 0,9757
Бензол 0,9001 0,8895 0,879 0,8685 0,8576 0,8466 0,8357
Бромбензол 1,5218 1,5083 1,4948 1,4815 1,4682 1,4546 1,4411
Бутан-1-ол 0,8246 0,8171 0,8086 0,802 0,9922 0,988 0,9832
Вода 0,9998 0,9997 0,9982 0,9956 0,9922 0,988 0,9832
Гексан 0,6769 0,6684 0,6595 0,6505 0,6412 0,6318 0,6221
Гептан 0,7005 0,692 0,6836 0,6751 0,6665 0,6579 0,6491
Глицерин 1,2674 1,2642 1,2594 1,2547 1,25 1,2438 1,2376
1,4-Диоксан 1,0338
Диэтиловый эфир 0,7362 0,7248 0,7135 0,7019 0,6894 0,6764 0,6658
о-Ксилол 0,8969 0,8886 0,8802 0,8719 0,8634 0,8549 0,8464
jw-Ксилол 0,8811 0,8726 0,8642 0,8556 0,847 0,8384 0,8297
и-Ксилол 0,861 0,8525 0,8437 0,835 0,8262
Метанол 0,81 0,8008 0,7915 0,7825 0,774 0,765 0,7555
Метилацетат 0,9593 (0,946) 0,9338 (0,920) 0,9075 0,8939 0,88
2-Метилпропан-1 -ол 0,8027
Метилформиат 1,0032 0,9886 0,9742 0,9598 (0,945) 0,9294 (0,913)
Муравьиная кислота 1,2196
Нитробензол 1,2231 1,2131 1,2033 1,1936 1,1837 1,174 1,1638
Нитрометан 1,1382
Октан 0,7185 0,7102 0,7022 0,6942 0,686 0,6778 0,6694
810
Новый справочник химика и технолога
Продолжение таблицы 6П2.2
Вещество Т,°С
0 10 20 30 40 50 60
Пентан 0,6455 0,636 0,6262 0,6163 0,6062 0,5957 0,585
Пиридин 1,003 0,9935 0,9825 0,9729 0,9629 0,9526 0,9424
Пропан-1-ол 0,8193 (0,811) 0,8035 (0,797) 0,7875 (0,780) 0,77
Пропан-2-ол 0,7851
Проп-2-ен-1-ол 0,8681
Пропионовая кислота 0,992
Сероуглерод 1,2927 1,2778 1,2632 1,2482
Стирол (винилбензол) 0,906
Тиофен 1,0647 1,0524
Толуол 0,8855 0,8782 0,867 0,858 0,8483 0,8388 0,8293
Уксусная кислота 1,0697 1,0593 1,0491 1,0392 1,0282 1,0175 1,006
Уксусный альдегид 0,783
Уксусный ангидрид 1,1053 1,093 1,081 1,069 1,0567 1,0443 —
Фенилгидразин 1,0981 1,0899 1,0817 1,0737 1,0653
Фенилметанол 1,0608 1,0532 1,0454 1,0376 1,0297 1,0219
Фенол * 1,0576
Формамид 1,1334
Фтортрихлорметан (фреон-11) 1,5342 1,487 1,462
Хлорбензол 1,1279 1,1171 1,1062 1,0954 1,0846 1,0742 1,0636
Хлороформ 1,5264 1,5077 1,489 1,4706 1,4509 1,4334 1,4114
Циклогексан 0,7879 0,7786 0,7691 0,7596 0,7499 0,7401
Четыреххлористый углерод 1,6326 1,6135 1,5939 1,5748 1,5557 1,5361 1,5165
Этан-1,2-диол 1,113
Этанол 0,8062 0,7979 0,7895 0,781 0,7722 0,7632 0,7541
Этилацетат 0,9244 (0,912) 0,9005 (0,891) 0,8762 (0,867) 0,8508
Этилформиат 0,9168
‘При 41 °C.
Таблица 7П2.2
Вязкость (т| • 103, Па • с) жидкостей при различных температурах [5]
Вещество Т, °C
0 10 20 30 40 50 60
Анилин 10,2 6,46 4,4 3,2 2,35 1,821 1,52
Ацетон 0,397 0,361 0,325 0,296 0,271 0,249 0,228
Ацетонитрил 0,442 0,396 0,357 0,325
Ацетофенон 2,3 1,84 1,51 1,38 1,24
Бензол 0,91 0,755 0,652 0,559 0,503 0,436 0,389
Бромбензол 1,52 1,31 1,13 0,99 0,89 0,79 0,72
Бутан-1-ол 5,19 3,87 2,95 2,28 1,78 1,41 1,133
Вода 1,792 1,308 1,005 0,801 0,656 0,549 0,469
Гексан 0,381 0,343 0,307 0,29 0,253 0,248 0,222
Гептан 0,414 0,373 0,338 0,308 0,281
Глицерин 12100 3950 1500 630 330 180 102
1,4-Диоксан 1,255 1,063 0,917 0,778 0,685
Приложение 2
811
Продолжение таблицы 7П2.2
Вещество Т, °C
0 10 20 30 40 50 60
Диэтиловый эфир 0,284 0,258 0,233 0,213 0,197 0,18 0,166
о-Ксилол 1,108 0,939 0,809 0,708 0,625 0,557 0,501
и-Ксилол 0,8 0,7 0,61 0,55 0,49 0,433 0,403
«-Ксилол 0,74 0,64 0,57 0,51 0,456 0,414
Метанол 0,817 0,69 0,597 0,51 0,45 0,396 0,35
Метилацетат 0,479 0,425 0,381 0,344 0,312 0,284 0,258
2-Метилпропан-1 -ол 8,3 5,65 3,95 2,88 2,12 1,61 1,24
Метилформиат 0,429 0,385 0,348 0,318
Муравьиная кислота 2,262 1,804 1,46 1,29 1,025 0,89
Нитробензол 3,09 2,483 2,034 1,682 1,438 1,251 1,094
Нитрометан 0,85 0,74 0,66 0,595 0,53 0,478 0,433
Октан 0,714 0,622 0,546 0,486 0,435 0,392 0,356
Пентан 0,283 0,259 0,24 0,22
Пиридин 1,33 1,12 0,974 0,83 0,735 0,651 0,58
Пропан-1-ол 3,883 2,897 2,234 2,4 1,129 0,921
Пропан-2-ол 4,6 3,26 2,39 1,77 1,33 1,03 0,8
Проп-2-ен-1-ол 2,145 1,703 1,363 1,07 0,914 0,767 0,657
Пропионовая кислота 1,52 1,29 1,1 0,958 0,84 0,746 0,662
Сероуглерод 0,433 0,396 0,365 0,341 0,319 0,297
Стирол (винилбензол) 1,047 0,879 0,749 0,648 0,565 0,502 0,453
Тиофен 0,871 0,753 0,658 0,582 0,52 0,468 0,424
Толуол 0,77 0,667 0,584 0,517 0,469 (0,425) 0,381
Уксусная кислота 1,45 1,21 1,04 0,9 0,79 0,7
Уксусный альдегид 0,276 0,253 0,225
Уксусный ангидрид 1,245 1,058 0,907 0,787 0,699 0,623 0,55
Фенилгидразин 0,456 0,443 0,404
Фенилметанол 5,8 4,32 3,288 2,574
Фенол 11,6 7 4,77 3,42 2,6
Формамид 7,5 5 3,75 2,94 2,43 2,04 1,71
Фтортрихлорметан (фреон-11) 0,54 0,48 0,44 0,405 0,375 0,346
Хлорбензол 1,056 0,915 0,802 0,708 0,635 0,573 0,52
Хлороформ 0,7 0,63 0,57 0,514 0,466 0,426 0,39
Циклогексан 0,97 0,822 0,706 0,61 0,538
Четыреххлористый углерод 1,33 1,132 0,969 0,843 0,739 0,651 0,585
Этан-1,2-диол 19,9 13,2 9,13 6,65 4,95
Этанол 1,773 1,466 1,2 1,003 0,834 0,702 0,592
Этилацетат 0,582 0,512 0,458 0,403 0,36 0,324 0,294
Этилформиат 0,51 0,45 0,402 0,358 0,329 0,308
Данные об электрической проводимости растворов электролитов необходимы при измерении электрокине-тического потенциала дисперсной фазы (см. подраздел 3.5), а также при экспресс-контроле концентрации электролита в дисперсных системах. В табл. 8П2.2 при
ведена предельная эквивалентная электрическая проводимость ионов в воде при разных температурах.
Предельная эквивалентная электрическая проводимость иона Хоо, — это вклад одного моля эквивалентов данного иона в общую эквивалентную электрическую
812
Новый справочник химика и технолога
проводимость раствора при его бесконечном разбавлении. Предельная эквивалентная электрическая проводимость раствора электролита Хоо вычисляется как сумма предельных эквивалентных электрических проводимостей всех ионов данного электролита по формуле
Хоо= Хоо + + Хоо -• (9П2)
Здесь Хоо+ и Хоо- — предельные эквивалентные электрические проводимости катиона и аниона соответственно.
УДельная электрическая проводимость Хоо сильно разбавленного раствора электролита заданной концентрации с (моль/л) связана с его предельной эквивалентной электрической проводимостью формулой
Хоо = 1 000 Хоо ZC. (10П2)
Здесь z — модуль формального заряда иона (катиона или аниона) с наибольшей валентностью в данном
химическом соединении; zc = сэк — молярная концентрация эквивалентов данного электролита. Формула (10П2) применима для растворов с сэк < 0,01 моль/л, так как с повышением концентрации эквивалентная электропроводность заметно уменьшается. Числовой множитель 1000 необходим для перехода от концентрации электролита, выраженной в моль/л, к концентрации, выраженной в моль/м3.
Пример. Раствору СаС12 при 18 °C и с концентрацией с = 0,004 моль/л соответствует сэк = 0,008 моль/л, Хоо = (50,7 + 66) • Ю 4 = 1,165 • Ю 2 См • моль’1 • м2 и Хоо - 9,336 • 10”2 См • м4.
Удельная электропроводность воды, перегнанной при доступе воздуха, составляет вблизи комнатной температуры (1 - 2) • 10-4 См • м-1, а перегнанной под вакуумом — примерно в 100 раз меньше.
Таблица 8П2.2
Предельная эквивалентная электрическая проводимость (Хоо / * 10"4 См * моль-1 * м2) ионов в водных растворах [5]
Ион T, °C
0 5 15 18 25 45 55 100
Ag+ 33,1 53,5 61,9 175
Ва2+ 34 54,6 63,6 195
Вг- 42,6 49,2 63,1 68 78,1 110,6 127,8
Са2+ 31,2 46,9 50,7 59,5 88,2 180
СГ 41 47,5 61,4 66 76,35 108,9 126,4 212
сю4- 36,9 58,8 67,3 185
Cs+ 44 50 63,1 67 77,2 107,5 123,6
F- 47,3 55,4
Н+ 225 250 300,6 315 349,8 441,4 483,1 630
Г 41,4 48,5 62,1 66,5 76,8 108,6 125,4
к/ 40,7 46,7 59,6 63,9 73,5 103,4 119,2 195
Li+ 19,4 22,7 30,2 32,8 38,6 58 68,7 115
Mg2+ 28,9 44,9 53 165
Na4 26,5 30,3 39,7 42,8 50,1 73,7 86,8 145
nh4+ 40,2 63,9 73,5 180
N(CH3)4+ 24,1 40 44,9
n(c2hs): 16,4 28,2 32,6
N(C3H7)4+ 11,5 20,9 23,4
N(C4H9)4+ 9,6 19,4
N(C5Hn): 8,8 17,4
no; 40 62,3 71,46 195
OH- 105 171 198,3 450
Rb+ 43,9 50,1 63,4 66,5 77,8 108,5 124,2
SO42- 41 68,4 80 260
Приложение 2
813
Продолжение таблицы 8П2.2
Ион Г, °C
0 5 15 18 25 45 55 100
Sr2* СНзСОСГ 31 20,1 50,6 35 59,4 40,9
2.3. Приготовление и свойства некоторых коллоидных систем
2.3.1. Гель кремневой кислоты H2SiO3 [7]
Гель кремневой кислоты получается при взаимодействии силиката натрия Na2SiO3 с соляной кислотой НС1:
Na2SiO3 + 2НС1 = H2SiO3 + 2NaCl.
Для этого 3.4 л раствора Na2SiO3 с плотностью 1400 кг/м3 разбавляют 1 л воды и медленно, при перемешивании вводят 10 н. НС1 до кислой реакции смеси (по тимоловому кислому). Кремневая кислота выделяется из реакционной смеси в виде рыхлого осадка. Его промывают водой до нейтральной реакции промывных вод и сушат 12 ч при 200 °C. При необходимости высушенные комки геля измельчают и рассеивают на фракции с нужным размером гранул геля. В таком виде гель используют как адсорбент.
Монолитные, стекловидные образцы геля SiO2 различной степени гидратированности можно получить гидролизом алкоголятов кремния.
2.3.2. Феррожидкости [8]
Приготовление феррожидкостей описано в подразделе 3.19. Ниже приведены свойства некоторых из них (табл. 1П2.3 и 2П2.3). Все жидкости изготовлены на основе магнетита Fe3O4. Его намагниченность насыщения составляет 480 кА/м, плотность — 5300 кг/м3.
Таблица 1П2.3
Характеристики ферромагнитных жидкостей фирмы «Ferrofluidics Corp.»
Дисперсионная среда Индукция насыщения, Гс Плотность, кг/м3 Вязкость, Па • с о о
Диалкилфталаты 200 1185 0,75 -37
Углеводороды 200 1050 0,03 4
400 1250 0,06 7
Перфторугле-водороды 100 2050 25 -34
Сложные эфиры 200 1150 0,14 -57
одноосновных 400 1300 0,3 -57
кислот 600 1400 0,35 -62
Вода 200 1180 0,07 0
400 1380 1 1 0
Полифенил диэтилового эфира 100 2050 75 10
Большая вязкость некоторых образцов феррожидкостей при сравнительно малой величине намагниченности (индукции насыщения) свидетельствует о структурированности (пастообразности) этих образцов.
Таблица 2П2.3
Характеристика феррожидкостей СПбГТИ(ТУ)
Дисперсионная среда Намагниченность насыщения, кА/м Индукция насыщения, Гс* Начальная магнитная восприимчивость Магнитный момент т • 1019, А • м2 Радиус частиц, нм Объемная доля магнетита, %
Керосин 94 1180 8,1 5 6,2 27
Веретенное масло 70 850 4,9 4 5,9 20
Конденсаторное масло 42 520 2,2 3,5 5,6 12
Трансформаторное 31 390 1,5 3,7 5,7 9
масло
Вакуумное масло ВМ4 41 510 1,8 3,2 5,4 12
Вода 25 310 2,8 6,8 7 7,5
* Индукция насыщения в системе единиц СГСМ, равная 4яА/ Гаусс, где М — намагниченность в той же системе, численно равная намагниченности в системе СИ, выраженной в кА/м. Дана для достижения сопоставимости со свойствами феррожидкостей «Ferrofluidics Corporation» (табл. 1П2.3).
814
Новый справочник химика и технолога
Начальная магнитная восприимчивость найдена экспериментально из изотермы намагничивания фер-
рожидкости м = М(Н) как производная --- от намаг-
dH
ниченности М по напряженности магнитного поля Н на начальном участке изотермы намагниченности. Она в полтора-два раза больше, чем теоретическая начальная
М,т
восприимчивость , где Ms — намагниченность
насыщения и т — магнитный момент частиц, указанные в табл. 2П2.3, к — постоянная Больцмана, Т— температура. Причина в том, что в табл. 2П2.3 дана средняя величина магнитного момента, найденная как среднее арифметическое от значений магнитных моментов, вычисленных по начальной магнитной восприимчивости и по магнитной восприимчивости на ветви насыщения намагниченности. Последняя значительно меньше начальной восприимчивости. Эта разница характеризует уровень полидисперсности различных образцов феррожидкости.
Литература
1. Кройт Г.Р. Наука о коллоидах. М.: ИЛ, 1955. 540 с.
2. Зонтаг Г., Штренге К. Коагуляция и устойчивость дисперсных систем. Л.: Химия, 1973. 152 с.
3. Дерягин Б.В, Чураев Н.В, Муллер В.М. Поверхностные силы. М.: Наука, 1985. 398 с.
4. Справочник химика. Т. 2. М.;Л.: Госхимиздат, 1951. 1148 с.
5. Краткий справочник физико-химических величин / Под ред. К.П. Мищенко, А.А. Равделя. Л.: Химия, 1972.200 с.
6. Лабораторные работы и задачи по коллоидной химии / Под ред. Ю.Г. Фролова, А.С. Гродского. М.: Химия, 1986. 216 с.
7. Брауер Г. Руководство по препаративной неорганической химии. М.: ИЛ, 1956. 896 с.
8. Бибик Е.Е., Бузунов О.В. Достижения в области получения и применения ферромагнитных жидкостей. М.: ЦНИИ Электроника, 1979. 60 с.
ПРИЛОЖЕНИЕ 3
Полимеры
3.1. Гибкость макромолекул
Гибкость — качество, присущее исключительно по
лимерным молекулам, придающее полимерным мате
риалам уникальное свойство — эластичность. Тем не менее, не существует физической величины, непосредственно характеризующей гибкость макромолекул. Есть ряд физико-химических свойств полимерных молекул, влияющих на их гибкость, и ряд физических свойств полимерных материалов, обусловленных гиб
костью. К числу первых относятся величина потенциального барьера вращения, длина эффективного сегмента Куна и температура стеклования. Физическим
свойством полимерного материала, которое можно счи
тать прямым макроскопическим выражением гибкости
мой деформации. У полимеров она необычайно велика
и примерно равна разности контурной длины полимерной цепи и среднестатистического расстояния между ее концами (см. подраздел 3.16). Однако на практике величина предельной деформации не применяется ни как физическая, ни как техническая характеристика полимеров. Обусловлено это тем, что она, как физическая
характеристика полимерною вещества, эквивалентно заменяется двумя названными выше фундаментальными свойствами макромолекул — их контурной длиной и средним расстоянием между концами изогнутой полимерной цепи. Предельная величина деформации, как техническая характеристика, на первый взгляд вполне доступная экспериментальному определению, на самом деле не всегда может быть измерена. Причина в том, что упругая деформация может перейти в необратимую деформацию (течение) еще до полного распрямления полимерных клубков, т. е. до достижения предельной величины обратимой деформации. Кроме того, технический и теоретический интерес обычно ограничивается областью линейной зависимости деформации от напряжения, т. е. областью применимости закона Гука, которая не охватывает область вблизи предельных деформаций. В итоге для характеристики способности полимеров к обратимым деформациям используется модуль их упругости.
Полимерные материалы отличаются от других веществ необычайно сильной зависимостью деформационных свойств от температуры. Упомянутая выше температура стеклования является одной из характеристик этой особенности полимеров. Ниже этой температуры полимер теряет способность к большим обратимым деформациям. Более полной характеристикой является термомеханическая кривая — графическое представление зависимости деформации от температуры при постоянном напряжении. Величина деформации при температуре стеклования является одной из точек этой кривой. Другая характерная температура термомехани
ческой кривой — температура плавления, при которой начинается необратимая деформация полимерного материала (течение). Температура стеклования в равной мере характеризует свойства и макромолекул, и полимерного материала в целом. Температура плавления в большей мере является характеристикой материала, так как она зависит от энергии взаимодействия полимерных молекул между собой, тогда как температура стеклования определяется вращательной подвижностью звеньев в макромолекуле.
Потенциальный барьер вращения звеньев полимерной цепи относительно соседнего звена доступен для определения на молекулах гидрированных мономеров, содержащих ту же связь и в том же окружении. Для полиэтилена это этан, для полипропилена — пропан, и т. д. Он рассчитывается на основании характеристик спектральных линий газообразных мономеров, обусловленных вращательными колебаниями двух частей молекулы мономера относительно связи.
Табл. 1П3.1 дает представление о влиянии химического строения молекул на величину потенциального барьера вращения мономеров. Она характеризует и полимеры соответствующего химического строения.
Непосредственно на полимерных веществах доступны определению реальные размеры R молекулярных клубков, например по их гидродинамическому объему, вычисленному по вязкости разбавленных растворов. Так как размер клубка зависит от качества растворителя, то инвариантной по отношению к свойствам растворителя характеристикой полимера является размер клубков в ©-растворителе. С другой стороны, можно вычислить размер цепи Ro при свободном вращении жестких сегментов Куна (при известной величине контурной длины полимерной цепи). Это дает возможность найти длину г сегментов Куна или параметр набухания <о = —, которые служат мерой гибкости цепи
(табл. 2П3.1). Если известна длина химического звена а, то размер жесткого сегмента можно характеризовать г
числом п = — химических звеньев в одном жестком а
сегменте.
Таблица 1ПЗ. 1
Потенциальные барьеры вращения Ur мономеров [1]
Формула вещества иг, кДж/моль Формула вещества У. кДж/моль
Н3С-СН3 Н,7 Н3С-СН=СН2 8,2
Н3С-СН2СН3 14,3 H3C-CF3 15,5
НзС-СН(СН3)2 16,4 F3C-CF3 18,3
Н3С-С(СН3)з 18,5 Н3С-СНО 4,5
816
Новый справочник химика и технолога
Таблица 2ПЗ. 1
Параметры гибкости полимеров [1]
Полимер со г, нм п
Полидиметилсилоксан 1,4-1,6 1,4 4,9
Полибутадиен 1,7
Натуральный каучук 1,7
Полиизобутилен 2,2 1,83 7,3
Полиэтилен 2,3-2,4 2,08 8,3
Полистирол 2,2-2,4 2,0 7,9
Поливинилхлорид 2,8 2,96 11,7
Полиметилметакрилат 2,2 15,1 6,0
Полигекс илметакрилат 2,4 21,7 8,6
Полиметилакрилат 2,0
Полицетилакрилат 5,0
Полиоктадецилакрилат 6,0
Производные целлюлозы 4,0-4,5 10-25
Полиалкилизоцианаты 100
Поли-и-бензамид 210 320
Биополимеры 240
Таблица ЗПЗ. 1
Температура стеклования полимеров Тс [1]
Полимер Тс, °C
Полидиметилсилоксан Полиэтилен высокого давления Полиэтилен низкого давления Полиоксиэтилен Полиизобутилен Полиизопрен (каучук СКИ) Натуральный каучук Бутилкаучук Полихлоропрен Полибутадиен (каучук СКБ) Поливинилиденхлорид Полиметилакрилат Поливинилацетат Поливинилхлорид Полистирол Полиметилметакрилат Политетрафторэтилен Целлюлоза Нитрат целлюлозы Ацетат целлюлозы Полиакрилонитрил -123 -115 -80 -85 -74 -70 -70 -69 -40 -40 -17 7 28 80 100 100 127 выше температуры разложения
жесткой является
Минимальная величина барьера вращения и, соответственно, максимальная гибкость присуща полиэтиленовым полимерам. Наибольшую жесткость (большую длину сегментов Куна) придает полимерам наличие в цепи атомов азота (полиамины, полиизоцианаты, биополимеры). Если азотсодержащие звенья разделены достаточно длинными звеньями из метиленовых групп, то это возвращает цепям гибкость.
Независимой и доступной прямым измерениям характеристикой подвижности звеньев цепи является температура стеклования полимеров (табл. ЗПЗ.1). Это температура, при которой происходит «замораживание» вращательной подвижности звеньев и, следовательно, потеря гибкости полимерных цепей. Чем выше температура стеклования, тем полимерная цепь.
Температуру стеклования вычислить по формуле [1]:
1 _ тГ"?
Здесь и', и w2 — массовые доли мономерных звеньев разной химической природы в гетерогенной молекулярной цепи и Т] и Т2 — температуры стеклования соответствующих гомополимеров.
С увеличением молярной массы М температура стеклования увеличивается, асимптотически приближаясь к предельной величине Тпт:
Тс сополимеров можно
w2
(1ПЗ)
где к — некоторая постоянная. У полимеров с гибкими цепями (полиизобутилен) температура стеклования приближается к предельному значению уже при М= ЮООг/моль, у полимеров с малой гибкостью температура стеклования становится постоянной, начиная с молекулярных масс М = 120004-40000 и более.
Монотонная зависимость большинства свойств полимеров и их растворов от величины молярной массы нарушается при определенной для каждого типа полимеров величине молярной массы порядка 1000-10000, которая называется критической и далее обозначается символом Мс. Упомянутые выше значения М при описании характера зависимости температуры стеклования от молярной массы М являются критическими значениями Л/с: ход зависимости температуры стеклования от М меняется при достижении величины Мс.
Температура плавления Тт (текучести) монотонно растет с увеличением молекулярной массы полимера, также начиная с критической величины Л/с по уравнению Каргина — Слонимского:
в^~^. (ЗПЗ)
* Vt С'
Приложение 3
817
где В и С — эмпирические константы. Для полиизобутилена В = 374,и С = 268 [1]. С точки зрения этого уравнения критическая величина молярной массы — это величина, при которой на термомеханической кривой отсутствует участок высокоэластичного состояния, т. е. Ту = Тс и полимер сразу плавится, переходя из стеклообразного к текучему состоянию.
По своим деформационным свойствам полимерные материалы отличаются большим разнообразием. В зависимости от химической природы и температуры они могут быть весьма прочными и жесткими (эпоксидные олигомеры), высокоэластичными (каучуки) и вязкотекучими (большинство полимеров при температуре плавления и выше). Для технологии наиболее интересны проявления эластичности и вязкотекучести.
Зависимость упругой деформации от напряжения идеальных каучукоподобных полимеров характеризуется наличием трех участков: участка быстрой обратимой деформации, участка высокоэластической обратимой деформации и участка насыщения упругой деформации. Первый соответствует малым деформациям, не связанным со значительными взаимными перемещениями звеньев молекулярных цепей и, следовательно, с проявлением трения между ними, а поэтому развивается практически мгновенно, характеризуется модулем быстрой деформации — отношением напряжения к величине мгновенной деформации. Второй (основной) связан с перемещениями звеньев гибкой цепи на расстояния порядка размера клубка. Он вносит основной вклад в величину упругой деформации полимера и является участком высокоэластической деформации. Взаимодействие между звеньями цепи на этом участке процесса деформирования препятствует их быстрому взаимному перемещению и проявляет себя как вязкое сопротивление движению звеньев. Это приводит к тому, что достижение равновесной величины упругой деформации требует заметного времени. Часть приложенного к материалу напряжения идет при этом на преодоление вязких сил сопротивления, а часть — на преодоление упругости молекулярных клубков. В итоге модуль эластической деформации GT — отношение приложенного напряжения к величине вызванной им упругой деформации — возрастает по сравнению с модулем быстрой деформации и тем сильнее, чем больше скорость деформации. Иначе говоря, на участке высокоэластической деформации одновременно действуют силы и упругого, и вязкого сопротивления. Количественное описание эластической деформации основано на модели вязкоупругого твердого тела Кельвина.
Модуль эластической упругости в силу его зависимости от скорости деформации не является инвариантной характеристикой полимера. Истинный модуль равновесной высокоэластической упругости G вычисляется путем экстраполяции зависимости модуля GT от скорости деформации к нулевой величине скорости деформации, используя ту или иную теоретическую модель вязкоупругого материала (см. подраздел 3.10).
Проще всего истинный модуль упругости вычисляется как отношение заданной величины напряжения к равновесной величине деформации материала. Деформация изменяется во времени по экспоненциальному закону, и ее равновесное значение определяется как предельная величина деформации, достигаемая по истечении достаточно большого времени по сравнению со временем релаксации. Практически это величина деформации, которая перестает увеличиваться с течением времени. При этом нужно убедиться, что материал не подвержен ползучести — медленной деформации — даже при малых или умеренных напряжениях. Она проявляет себя как медленное, пропорциональное времени увеличение деформации, в отличие от экспоненциального закона приближения деформации к ее предельной величине.
Третий участок зависимости упругой деформации от напряжения обусловлен распрямлением полимерных клубков, которое ведет к нарушению пропорциональности между напряжением и деформацией. Он представляет практический интерес как участок, предшествующий переходу к необратимой деформации (течению), которая, собственно, и используется для изготовления изделий из полимеров. Именно на этом участке упругого деформирования в изделие закладываются упругие напряжения, которые ведут впоследствии к изменению его формы и размеров по сравнению с размерами матрицы для его формования.
Для каучукоподобных полимеров истинный модуль упругости составляет 0,2-10 Па. Участок высокоэластической деформации не является строго линейным, поэтому модуль эластической упругости непостоянен. Иначе говоря, закон Гука на этом участке деформации выполняется приближенно. Более точно зависимость нормального напряжения о от деформации растяжения X описывается эмпирической формулой Муни — Рив-лина [1]:
с = (£j + Е2 X-1)(Х - X2), (4ПЗ)
л I
где Х = — — относительное удлинение деформируемо
« г RTP к
мого образца, Ех =—- — газоподобная упругость Мп
участка молекулярной цепи между узлами структурной сетки, Мп — молярная масса этих участков, р — плотность полимера и £2 — эмпирическая константа для данного полимера, R — газовая постоянная и Г — температура. Тем не менее, расчеты упругой деформации (сдвига) оценочного уровня обычно базируются на законе Гука:
т = Gy. (5ПЗ)
Вследствие некоторого непостоянства модуля эластической упругости его значение не включается в
818
Новый справочник химика и технолога
справочные данные. Для оценки его величины можно пользоваться формулой [1]:
G = ^. (6ПЗ)
ЗЛ/И
Здесь учтено, что для несжимаемых веществ модуль сдвиговой упругости G равен 1/3 модуля упругости Е на растяжение (сжатие). Оценку числа п звеньев полимерной цепи между узловыми точками трехмерной структурной сетки можно сделать на основе формулы Щукина [2]:
<р=-*(3”;— (7пз>
би
где ф — объемная доля полимера в исследуемом материале.
Структурная сетка по Щукину [2] представляет собой совокупность линейных цепочек из сферических частиц, вытянутых вдоль трех координатных осей и имеющих общие точки в узлах кубической решетки. Для полимеров в блоке (ф 1) проблема заключается в выборе элемента полимерной цепи, адекватного частицам Щукина. В полуразбавленных растворах в качестве такового можно принять сегмент Куна. Через число и частиц (звеньев цепи) между узловыми точками выражается молярная масса Мп соответствующего участка цепи:
М„ = пМх, (8ПЗ)
где Мх — молярная масса одного звена. При Мп = 1 кг/моль (1000 г/моль), р = 1000 кг/м3 и Т = 300 К формула (6ПЗ) дает G около 0,8 МПа.
3.2. Вязкость
Вязкость характеризует деформационные свойства полимера не только в жидкотекучем, но и в высокоэластическом состоянии. Как было отмечено выше, процесс высокоэластической упругой деформации сопровождается действием сил вязкого сопротивления. С другой стороны, течение жидкого полимера, даже если оно начинается при сколь угодно малой величине напряжения, сопровождается накоплением в материале внутренних упругих напряжений, вызванных деформацией клубков под действием сил вязкого трения. В том и другом случае величина вязких напряжений в деформируемом материале, в соответствии с законом внутреннего трения Ньютона, пропорциональна скорости деформации. Соотношение между упругими и вязкими напряжениями в простейшем случае описывается в высокоэластичном состоянии уравнением деформации вязкоупругого твердого тела (тела Кельвина), а в состоянии вязкой жидкости — уравнением деформации вязкоупругой жидкости (тела Максвелла).
Для характеристики деформационных свойств вязкоупругих твердых тел, в том числе высокоэластичных
полимеров, не принято определять модуль упругости G и вязкость т| как самостоятельные реологические константы, поскольку в уравнениях кинетики деформирования этих тел они всегда фигурируют в виде отношения rj/G, имеющего размерность и смысл некоторого характерного времени изменения напряжений или деформаций в вязкоупругом материале. В такой же комбинации вязкость и упругость входят и в уравнение кинетики изменения напряжения в вязкоупругой жидкости при фиксированной величине деформации. Для жидкости, однако, характерно непрерывное увеличение деформации во времени при постоянстве напряжения, т. е. течение, и поэтому для нее актуально определение именно вязкости как основной реологической характеристики. Для вязкоупругих твердых тел более приемлемой реологической характеристикой является величина t* = rj/G, которая называется временем релаксации (деформаций). В литературе ее принято также называть временем запаздывания [3]. Имеется в виду, что при задании постоянного деформирующего усилия (напряжения) равновесная величина деформации достигается по прошествии некоторого времени.
Релаксация — это процесс перехода системы из неравновесного состояния в равновесное. При действии напряжения равновесным становится деформированное состояние и, следовательно, время, необходимое для перехода в это состояние, по определению является временем релаксации. Необходимо, однако, уточнить, что в данном случае релаксирует деформация, а не напряжение. Для вязкоупругой жидкости та же величина /* имеет смысл времени релаксации напряжений, по истечении которого уже не требуется воздействие напряжения для сохранения неизменной, первоначально заданной деформации.
Время релаксации деформаций достаточно резко уменьшается с повышением температуры в основном благодаря снижению вязкости. Это означает, что при повышении температуры одна и та же величина деформации при неизменном напряжении будет достигнута за меньшее время, причем эквивалентность влияния температуры и времени на величину деформации распространяется на любое время от начала процесса деформирования и на любые напряжения (в пределах сохранения эластичности). Это важно для технологии. Количественно принцип эквивалентности температуры Т и времени t выражается уравнением Александрова — Лазуркина [1]:
1^0 Т)
(9ПЗ)
где b — постоянная.
Развитие этого принципа выражается уравнением Вильямса — Лендела — Ферри (ВЛФ):
-17,44(7-7;) nJ 51,6 + (7-7с)’
(10ПЗ)
Приложение 3
819
где г| и т]с — вязкости при температурах Т и Тс, Тс — температура стеклования по шкале Кельвина. Это уравнение отражает наличие универсальной зависимости вязкости от температуры: отношение вязкости при произвольной температуре Т к вязкости при некоторой характерной для данного вещества температуре Тс (температуре приведения) является универсальной функцией разности этих температур. Иначе говоря, относительная вязкость 'п/'Пс некоторого полимера при произвольной температуре известна априори, если известна температура приведения данного полимера. Для вычисления абсолютной величины вязкости необходимо измерить вязкость исследуемого полимера при температуре его приведения. В уравнении ВЛФ (10ПЗ) в качестве температуры приведения принята температура стеклования полимера (табл. ЗПЗ.1). На практике применяется температура Ts, которая выше температуры приведения примерно на 50 °C. Это связано с трудностью измерения вязкости т|с при Т = Тс. Разумеется, что уравнение ВЛФ применимо при температуре большей, чем температура приведения. При переходе к другой, отличной от Тс, температуре приведения изменяется значение числовых констант уравнения ВЛФ. Новое значение констант может быть найдено при наличии серии значений температур и вязкостей с помощью обычных методов обработки экспериментальных данных.
Время релаксации высокоэластической деформации определяется и без каких-либо реологических измерений. Оно выражается через энергию активации Ua процесса деформирования:
t* = f 1 exp
(11ПЗ)
Если полагать, что элементарными актами движения участков цепи при ее деформировании являются вращательные переходы звеньев цепи между соседними устойчивыми положениями, то энергия активации Ua равна величине потенциального барьера вращения Ur (табл. 1П3.1), и тогда эти данные могут быть использованы для оценки времени релаксации. Здесь f~ 1012 Гц— частота тепловых колебаний молекул. На практике, однако, температурная зависимость вязкости используется для решения обратной задачи — нахождения энергии активации процесса деформации (или стационарного течения жидкостей). Энергия активации процесса, происходящего в веществе, в том числе его высокоэластической деформации, является общепринятой инвариантной по отношению к температуре характеристикой вещества. При этом обычно обсуждаются отклонения энергии активации от постоянной величины при изменении температуры и причины этого отклонения. Чаще всего причины связаны с изменением структуры вещества при изменении температуры.
Расплавы и растворы полимеров обладают ярко выраженными свойствами неньютоновских жидкостей тиксотропного типа (см. подраздел 3.10). Течение тиксотропных систем обычно имеет три разных режима,
которым соответствуют три участка полной реологической кривой (ПРК). Течение жидкости на первом участке этой кривой, соответствующем малым напряжениям, характеризуется максимальной вязкостью т]тах, остающейся приближенно постоянной на всем протяжении этого участка. Постоянство вязкости тиксотропных коллоидов на первом участке ПРК объясняется тем, что в растворе существует сплошная структурная сетка, которая перекрывает собой все поперечное сечение канала вискозиметра. Она остается сплошной при увеличении скорости течения благодаря тиксотропному восстановлению возникающих в ней разрушений. Сопротивление течению, создаваемое структурной сеткой или ее фрагментами, пропорционально квадрату размера фрагментов или квадрату поперечного размера канала при сохранении сплошности сетки в процессе течения. Это значит, что величина максимальной вязкости должна расти пропорционально квадрату поперечного размера канала, т. е. она не является инвариантной характеристикой жидкости по отношению к измерительному прибору. Несмотря на это, именно максимальная вязкость т]тах используется как основная реологическая характеристика расплавов и концентрированных растворов полимеров. Через нее выражается вязкость при течении в других режимах, влияние температуры и молярной массы на реологические свойства полимеров.
Сравнительно хорошая воспроизводимость величины максимальной вязкости полимеров при ее измерении на различных приборах означает, что свойства сплошной структурной сетки в растворах полимеров принципиально отличаются от свойств структурной сетки типичных гелей гетерогенных коллоидов. Это отличие состоит в том, что положение узлов полимерной сетки на полимерной цепи не фиксировано — они могут сравнительно свободно скользить вдоль цепи под действием приложенного напряжения. Это узлы чисто топологические. В точках контакта цепей силы сцепления между ними не действуют, и поэтому наличие узлов не мешает макромолекулам двигаться в направлении действующей на них силы. В сущности, структурная сетка полимерных расплавов и концентрированных растворов по реологическим свойствам является высоковязкой жидкостью. Механизм ее течения известен — это рептации (см. подраздел 3.16). Ни одна из известных количественных теорий вязкости полимеров не опирается на этот факт, поэтому к приводимым ниже формулам и константам уравнений следует относиться как к эмпирическим, более или менее удачно отражающим экспериментальные данные.
Зависимость вязкости от скорости деформации по теории Ф. Бики [3]:
=l-4Sp^^[2-Av,g)]- (12ПЗ) * Р
Здесь и — вязкость при данной скорости сдвига, По — вязкость растворителя (для расплава По = 0),
820
Новый справочник химика и технолога
fWQ) = ~ СК°Р°СТЬ сдвига, Q =
Р +(У0
Т| — Г|
-1,216-^—Я — максимальное время релаксации, N— число макромолекул в единице объема, — знак суммирования стоящего справа от него выражения по всем целочисленным значениям индекса р вплоть до р = п, где п — число звеньев в полимерной цепи.
Произведение y'Q является безразмерной скоростью деформации — скоростью, в которой время выражено в единицах, равных времени релаксации Q.
Зависимость максимальной вязкости от молярной массы полимера выражается следующими эмпирическими формулами [3]:
Птах = аМа при М < Мс,
(13ПЗ) Птах = ЬМ₽ ПрИ М > MQ.
Здесь а и b — индивидуальные константы для каждого полимергомологического ряда. Константы а=1, р = 3,4-^-3,5. Величина Мс, уже упоминавшаяся ранее, — критическая молярная масса полимера.
В табл. 1П3.2 приведены значения Мс для ряда полимеров.
Формулы (13ПЗ) передают главную особенность зависимости вязкости от молярной массы — она существенно различна для докритических и послекритических величин молярной массы. Более точные формулы предложены Фоксом и Берри:
[\ а
V- ?Р, (14ПЗ)
Хс)
<?><фА <5Г2><СрЛ
где X = п----- — , X = п„------- — , а = 1 при
М \vj Мс \у)
X < Хс и а = 3,4 при X > Хс, — поправочный множи-
тель, который вводится только при X < Хс и тем самым скругляет переход от докритических к закритическим величинам молярной массы. Он является некоторой функцией плотности полимера р. Остальные констан-2
ты: <s > — среднеквадратичным размер молекулярного клубка в ©-растворителе (см. ниже), ф — объемная доля полимера в системе, v — удельный объем системы (раствора, расплава), п и пс — число звеньев в молекулярной цепи произвольного и критического размеров. Значения констант, входящих в формулу Фокса, для ряда полимеров указаны в табл. 2П3.2.
Таблица 1П3.2
Критические величины Мс молярных масс (г/моль) [3]
Полимер л/с
Полиэтилен 4000
Полибутадиен 5600
Полиизобутилен 17000
Полистирол 35000
Полидиметилсилоксан 29000
Поливинилацетат 22500
Полиметилметакрилат 27500
Таблица 2П3.2
Значения констант формулы Фокса [3]
Полимер Г, °C м п V —w м Пс Хс- ю15
Поливинилацетат 155 43 0,880 5,7 570 3,7
Полиметилметакрилат (промышленный) 217 50 0,880 6,2 550 3,9
Полиметилметакрилат (атактический) 217 50 0,880 6,8 630 4,9
Полистирол 217 52 1,038 7,6 600 4,35
Полиизобутилен 217 28 1,123 8,7 540 4,20
Полиэтиленгликоль 80 14,7 0,925 9,8 300 3,20
Полидиметилсилоксан 25 37 1,04 7,2 660 4,60
Политетраметилен-и-фениленсилоксан 218 69 1,0 7,3 582 4,5
Полибутадиен (90 % цис-1,4) 50 18 1,10 1670 8,26
Полибутадиен (80 % цис-1,4) 50 18 1,П 1200 7,20
Полибутадиен (50 % цис-1,4) 27 18 1,11 12,6 330 3,6
Полиэтилен 150 14 1,307 17,0 270 3,5
Поли-а-капроамид 253 16,5 1,0 10,0 310 3,1
Полидиэтиленадипинаг ПО 16,7 0,89 10,5 310 3,6
Полидекаметиленадипинат но 15,8 1,027 11,5 320 3,6
Полидекаметиленсукцинат по 16 0,99 12,5 230 2,9
Полидекаметиленсебацинат по 15,4 1,067 14,7 210 2,9
Полинеопентилсукцинат 100 23,3 1,0 16,0 710 11,4
Приложение 3
821
3.3. Вязкость разбавленных растворов
Вискозиметрия разбавленных растворов полимеров представляет интерес как одно из универсальных средств исследования свойств молекулярных клубков, в том числе как способ определения молярной массы полимера. Вискозиметрическое определение молярной массы основано на формуле Куна: [т|] = KMZ (см. подраздел 3.16). Значения констант К и е этой формулы приведены в табл. ШЗ.З.
Следует иметь в виду, что константа К имеет размерность, обратную размерности концентрации раствора, и, следовательно, ее значение зависит от единиц, в которых выражена концентрация полимера в растворе.
В табл. ШЗ.З приведены значения К • 104, соответствующие традиционному для вискозиметрии полимеров выражению концентрации в граммах на 100 см3 раствора (г/100 см3). Если концентрацию выразить в граммах на литр (г/л), то эти значения следует умножить на 10"5 (л/г), если в граммах на см3 — то на 102 (см3/г).
Таблица ШЗ.З
Константы формулы Куна К (100 см3/г) и а [4]
Полимер Растворитель Т°С К-104 £
Полиэтилен: высокого давления низкого давления «-Ксилол Раствор 2-этилгексан-1-ола (85 %) и декалина (15%) 80 180 10,5 3,26 0,63 0,77
Тетралин 130 4,3- 5,1 0,72-0,76
Полипропилен: изотактический атактический Декалин Толуол Декалин 135 85 135 1,0 16,0 1,58 0,8 0,61 0,77
Полиизобутилен Циклогексанон 20 2,6 0,69
Поливиниловый спирт Вода 25 6,66 0,65
Поливинилацетат Ацетон 20 1,76— 1,02 0,68-0,72
Метанол 20 3,14 0,6
Бутан-2-он 20 1,07 0,71
Полиакриловая кислота 0,5 н. NaCI в воде 20 2,9 0,5
Полиметакрило-нитрил Диметилфор-мамид 20 1,82 0,71
Ацетон 20 8,7 0,545
Поливинилсульфокислота 0,5 н. NaCI в воде 20 2,1 0,65
Продолжение таблицы ШЗ.З
Полимер Растворитель Г, °C Х104 £
Поливинилхлорид Циклогексанон 20 0,14— 0,2 0,56
Поликарбонат Тетрагидрофуран 20 3,99 0,7
Дихлорметан 20 1,11 0,82
Полиакрилонитрил Диметилформ-амид 20 1,66 0,81
Полиметилметакрилат Бензол 20 0,468 0,75-0,77
Полистирол: аморфный изотактический Бензол Бензол 20 30 1,23 1,06 0,72 0,735
Полиацетальдегид Этилацетат 20 5,36 0,65
Полиэтиленгли-кольтерефталат Трифторуксусная кислота 25 4,68 0,68
о-Хлорбензол 25 3-4,2 0,77
3.4. Состояние системы полимер—растворитель
Равновесное состояние смеси полимера и растворителя подчиняется правилу фаз Гиббса П = К + 2 - Ф, где П — число независимых параметров состояния, К — число компонентов системы, Ф — число фаз. Два других параметра состояния — температура и давление. В конденсированных системах (где отсутствует фаза пара) давление не играет существенной роли и обычно постоянно. Поэтому из этих двух параметров остается один — температура. Следовательно, в двухфазных смесях П = 3 - Ф. Состояние смесей полимер— растворитель имеет сходство с состоянием смеси двух ограниченно растворимых жидкостей — в определенном интервале температур и соотношений компонентов смесь является однофазной системой, т. е. образует один истинный раствор, а за пределами этого температурного интервала — двухфазной, причем обе фазы являются истинными, несмешивающимися растворами полимера в растворителе. Первая фаза — это разбавленный раствор полимера, а вторая — раствор с повышенной концентрацией полимера. По аналогии с ограниченно смешивающимися жидкостями говорят, что одна из фаз — насыщенный раствор полимера в растворителе, а вторая — насыщенный раствор растворителя в полимере. Однако следует иметь в виду, что в случае полимеров вторая фаза также может быть разбавленным раствором полимера — содержание растворителя в ней составляет около 90 %. В связи с этим ее не вполне уместно называть насыщенным раствором растворителя в полимере.
Как и смеси жидкостей, полимерные растворы чаще всего распадаются на две фазы при понижении температуры. Минимальная температура, при которой систе
822
Новый справочник химика и технолога
ма остается однофазной при любом составе смеси, называется верхней критической температурой растворения (ВКТР) данной системы. Существуют и смеси, которые распадаются на две фазы при повышении температуры и, соответственно, характеризуются нижней критической температурой растворения (НКТР). Многие системы имеют две критические температуры— верхнюю и нижнюю (табл. 1П3.4). Обычно нижняя температура больше верхней и больше температуры кипения растворителя. С повышением молярной массы полимера его совместимость с растворителями ухудшается, температурный интервал существования однофазной смеси сужается как за счет увеличения ВКТР, так и за счет одновременного повышения НКТР.
Таблица 1П3.4
Критические температуры некоторых систем полимер—растворитель [1]
Система полимер—растворитель ВКТР, °C НКТР, °C
Полистирол—циклогексан 30 180
Полистирол—циклопентан 6 150
Полистирол—этилбензол -5,8 295
Полиизобутилен—бензол 23 160
Поливинилацетат—метанол -3 206
Поливиниловый спирт—вода 231
В некоторых случаях при понижении температуры один их компонентов смеси выделяется в чистом виде (кристаллизуется). Обычно это растворитель с достаточно высокой температурой замерзания, например вода.
Большое практическое значение имеют студни (структурированные, обладающие упругостью растворы полимеров), особенно термически и реологически обратимые, в которых полимерные цепи связаны в структурную сетку физическими силами. Обычно они возникают в растворителях низкого термодинамического качества .
Качество (растворяющая способность) ухудшается при понижении температуры в системах с ВКТР или при добавлении нерастворяющей низкомолекулярной жидкости или электролита. Образование студня (геля) представляет собой незавершенный процесс разделения раствора на две фазы при изменении условий, в котором вторая фаза по кинетическим причинам (большая вязкость) не может достичь равновесного состояния и застывает в виде сетки полимера. Обратимость означает, что студень можно расплавить при повышении температуры или изотермически разрушить при его деформировании, но он восстановится при возврате к начальным условиям (температура, отсутствие механических напряжений). Однако студни не являются термодинамически равновесными системами, как гели — коагуляционные структуры гетерогенных коллоидов.
См. ниже.
Неравновесность состояния студней проявляется, в частности, в том, что они подвержены синерезису — уплотнению полимерной сетки с течением времени и выдавливанию из нее жидкой фазы (первой фазы двухкомпонентной системы).
На практике в качестве количественной характеристики совместимости полимера с различными растворителями (растворимости) широко используется параметр растворимости 5. Он вводится в теории регулярных растворов Гильдебранда, которая относится к смесям низкомолекулярных жидкостей:
3,=^. (15ПЗ)
ДЕ
Величина представляет собой плотность энергии когезии конденсированного вещества (полимера или растворителя). Здесь АЕ, = &HV - TASV — энергия испарения одного моля /-го компонента раствора и И, — его парциальный объем, A//v — теплота (энтальпия) и Д\ — энтропия испарения. Для растворителей энергия испарения доступна прямым измерениям по теплотам испарения, а для полимеров она вычисляется различными методами. В табл. 2П3.4 приведены некоторые из них. Преимущество этого параметра в том, что он характеризует компоненты системы полимер—растворитель, и, следовательно, с помощью конечного числа значений этого параметра, равного числу существующих (или представляющих интерес) растворителей и полимеров, можно составить характеристику системы с любым сочетанием двух компонентов. Такая возможность основана на том, что свойства системы полимер—растворитель зависят от разницы значений параметров растворимости полимера и растворителя. Этот принцип не всегда выполняется, но благодаря своей простоте широко используется для подбора растворителей нужного качества для тех или иных полимеров.
Применение параметра растворимости основано на правиле, согласно которому совместимость полимера и растворителя лучше, если их параметры растворимости близки по величине. Однако гарантировать можно только то, что полимер не будет растворяться в жидкости при слишком большой разнице их параметров растворимости (порядка 10 Дж/л и более). Совместимость при небольшой разнице параметров растворимости возможна, но не гарантируется. Полимер и жидкость могут оказаться взаимно нерастворимыми даже при совпадении параметров растворимости. Дополнительные критерии совместимости связаны с ролью водородной связи и донорно-акцепторных свойств растворителя и химических звеньев полимера, т. е. с химической природой тех и других.
Водородная связь возникает между функциональными группами, одна из которых может выступать донором, а другая — акцептором протонов. В качестве донора (соединения типа А) выступают функциональ
Приложение 3
823
ные группы кислотного типа, а в качестве акцепторов (соединения типа В) — основного. Некоторые могут быть одновременно донором и акцептором водорода (соединения типа АВ). Таковой является вода. Соединения, которые не содержат донорных или акцепторных групп, относят к отдельному классу N. Они хорошо смешиваются друг с другом. Основным видом молекулярного взаимодействия в таких системах являются дисперсионные силы.
Соединения смешиваются, если одно из них является донором, а другое — акцептором протонов.
Донорно-акцепторная связь — это связь между молекулами или группами, способными отдавать или принимать пару электронов. Донорные свойства растворителей характеризуются донорным числом. Они зависят от акцепторных свойств партнера по взаимодействию, в том числе химических звеньев полимера. В табл. ЗПЗ.4 приведены донорные числа ряда растворителей, определенные по отношению к SbCl5.
Корреляция растворимости и химического строения полимеров и растворителей отличается нечеткостью, так что приведенные выше правила и параметры являются ориентировочными. Само значение параметров, полученных из разных источников, может различаться, что видно из сравнения табл. 2П3.4 и ЗПЗ.4.
Таблица 2П3.4
Параметры растворимости растворителей и полимеров [1]
Растворитель 8 • 10"3, (Дж/м3)172 Полимер 8 • 10’3, (Дж/м3)"2
Гексан 14,6 Силиконовый каучук 14,6
Диэтиловый эфир 14,8 Полиизобутилен 15,8
Октан 15,1 Полиэтилен 15,9
Четыреххлористый углерод 17,2 Полипропилен 16,2
Пропилбензол 17,3 Полибутил-метакрилат 17,6
Этилацетат 18,2 Полистирол 18,2
Бензол 18,3 Полиметилметакрилат 18,6
Хлороформ 18,6 Поливинилхлорид 19,1
Бутан-2-он 18,6 Полиэтилен-терефталат 20,2
1,2-Дихлорэтан 19,6 Полиарилат фенолфталеина и терефталевой кислоты 21,4
Тетрагидрофуран 19,8 Полиимид ани-линфталеина и пиромеллитовой кислоты 23,4
Хорошим растворителем поливинилхлорида и других полярных полимеров является тетрагидрофуран.
Ароматические полимеры растворяются в ароматических растворителях и некоторых хлорпроизводных (хлороформ, тетрахлорэтан).
Слабая корреляция между фактической совместимостью полимера с растворителем и совместимостью, предсказанной по величине 5, объясняется тем, что теория Гильдебранда не учитывает ряд существенных обстоятельств, влияющих на взаимную растворимость веществ. Более адекватно термодинамическое качество растворителя по отношению к данному полимеру характеризуется ©-температурой (температурой Флори) системы полимер—растворитель и параметром сродства %1 растворителя к полимеру. Последний введен в теорию Хаггинсом как эмпирический параметр, отражающий влияние взаимодействия компонентов раствора на величину конфигурационной энтропии раствора. Он связан с параметрами растворимости растворителя и полимера соотношением: Xi = (§i - 5г)2 И I RT, а со вторым вириальным коэффициентом А2 осмотического давления раствора соотношением: А2 = pi[(l/2) - XilPiAfj. Из него следует, что хорошему качеству растворителя соответствует Xi < 0,5 (А2 > 0), т. е. относительная близость параметров 5. Значение Xi = 0,5 соответствует образованию идеального раствора (А2 = 0).
Таблица 2П3.4а
Параметры растворимости растворителей
Растворитель 8 • 10~3, (Дж/м3)172
Ацетон 20,0
Гексан-1-ол 20,0
Тетрахлорэтан 20,8
Диметилацетамид 22,2
Диметилформамид 24,2
Этанол 25,4
Метанол 29,0
Вода 46,4
Таблица ЗПЗ.4
Донорные числа D к SbCI5, относительная диэлектрическая проницаемость 8, параметры растворимости 6 растворителей [1]
Растворитель D £ 8, (Дж/м3)172
Нитробензол 4,4 34,8 20,0
Бензонитрил 11,9 25,2 16,8
Ацетон 17,0 20,7 20,0
Этилацетат 17,1 6,0 18,2
Вода 18,0 81,0 47,0
Тетрагидрофуран 20,0 7,6 19,8
Диметилформамид 26,6 36,1 24,2
Диметилацетамид 27,8 38,9 21,6
Диметилсульфоксид 29,8 45,0 25,8
Диэтилацетамид 32,2 19,8
824
Новый справочник химика и технолога
Так как значение параметра сродства зависит от температуры, то состояние идеальности раствора может быть достигнуто не только изменением химической природы растворителя, но и путем изменения температуры раствора. Температура Флори — это температура, при которой разбавленный раствор полимера ведет себя как идеальный. Экспериментально она может быть найдена как критическая температура системы Тт (температура, при которой полимер в данном растворителе может образовывать однофазный раствор) при бесконечно большой величине молярной массы полимера. Она определяется экстраполяцией зависимости критической температуры от величины Л/-1/2 к бесконечно большой величине молярной массы М в соответствии с уравнением:
_L = 1 а
Тт~в + у1м’
(16ПЗ)
где Тт — это или ВКТР, или НКТР, а — некоторый коэффициент, причем для систем с ВКТР а < 0, а для систем с НКТР а > 0. ©-Температура систем с нижней критической температурой называется также температурой Роулинсона.
Параметр сродства и температура Флори О остаются, в основном, средством теоретического описания растворов. В большинстве случаев технологического применения полимерных растворов имеются экспериментально подобранные пары полимер—растворитель требуемого качества. Показательной в этом отношении является технология капсуляции тех или иных продуктов в полимерные оболочки [4]. Методы капсулирова-ния могут быть различными.
В связи с проблемой подбора растворителя требуемого качества интерес представляют накопленные в этой сфере применения полимеров сведения о системах, позволяющие управлять растворимостью полимеров. В частности, при капсуляции (например капель водного раствора в прочную полимерную оболочку) в раствор полимера, в котором взвешен капсулируемый препарат, вводится второй — плохой для данного полимера растворитель (осадитель). Ухудшение качества двухкомпонентного растворителя ведет к выделению полимера из раствора на поверхность капсулируемого препарата (например, на поверхность капель эмульсии) и образованию оболочки. Подбор и дозирование осадителя должны обеспечить плавное изменение качества двухкомпонентного растворителя. Это необходимо для того, чтобы выделение полимера из раствора происходило в виде более концентрированного раствора полимера (второй фазы), способного образовать сплошную оболочку на поверхности капсулируемого препарата и затем переходить в достаточно прочный студень. Учет способности растворителей образовывать водородную связь облегчает решение этой задачи, поэтому ниже, в табл. 4П3.4 и 5П3.4, дана характеристика растворителей (осадителей) также и по силе образуемой ими водородной связи.
На основании параметров растворимости, силы водородной связи и практики микрокапсулирования подобраны растворители и осадители для различных полимеров. Каждый из полимеров может иметь несколько вариантов эффективных пар растворитель—осадитель (табл. 6П3.4). Выбор той или иной пары, а также полимера диктуется совместимостью капсулируемого препарата с растворителем, осадителем и полимером. Например, препарат не должен растворяться ни в одном из компонентов системы и сам не должен растворять полимер. Возможна, однако, и такая технология, при которой капсулируемый препарат первоначально растворен в растворителе или осадителе и выделяется (опережая выделение полимера) при смешивании растворителя и осадителя. Это создает дополнительные требования к химической природе используемых компонентов.
Таблица 4П3.4
Параметр растворимости 5, температуры кипения Тк и плавления Тпл растворителей с различной силой водородных связей [5]
Растворители 5, МПа1/2 Т QC 1 кип» Т °C 1 пл»
Растворители со слабой Н-связью
Дифтордихлорметан 11,3 -28 -160
Бутан 13,9 -0,5 -138
1,1,2-Трифтортрихлорэтан 14,9 47,6 -36,5
Гексан 14,9 68,7 -154
Циклогексан 16,8 80,8 6,6
Дифторхлорметан 17,0 -40,8 -146
Бензонитрил 17,2
Четыреххлористый углерод 17,6 76,8 -23
Декалин 18,0 191,7 -124
Ксилол 18,0
Толуол 18,2 110,6 -95
Бензол 18,8 80,1 5,5
Трихлорэтилен 18,8 87,2 -86
Стирол 19,0 145,2 -30,6
Хлороформ 19,0 61,1 -64
Тетрахлорэтилен 19,0 121,2 -22,4
Хлорбензол 19,4 131,7 -46
Дихлорметан 19,8 40 -97
Нитробензол 20,5 210,8 5,76
Акрилонитрил 21,5
Нитроэтан 22,7 114 -90
Ацетонитрил 24,3 81,6 -46
Нитрометан 26,0 101,3 -28,5
Растворители с Н-связью средней силы
Диэтиловый эфир 15,1 34,5 -116
2,6-Диметилгептан-4-он 16,0 166
Диоктилфталат 16,0
Приложение 3
825
Продолжение таблицы 4П3.4
Растворители 5, МПа1'2 Т °C 1 КИЛ 9 Т °C 1 пл?
Растворители с Н-связью средней силы
1-Хлорпропан 17,4 46,6 -123
Бутилацетат 17,4 126,1 -74
Тетрагидрофуран 18,6 65 -65
Этилацетат 18,6 77,1 -84
4-Гидрокси-4-метилпентан-2-он 18,8 168 -47
Бутан-2-он 19,0 79,5 -87
Дибутил фталат 19,0 280
Бромэтан 19,6 38,4 -119
Метилацетат 19,6 56,3 -98
Хлорметан 19,8 -24 -98
Циклогексанон 20,3 155,7 -16,4
Ацетон 20,3 56,2 -95
Диоксан 20,5 101 11,8
2-Этоксиэтанол 21,5 134
Диметилфталат 21,9
2-Метоксиэтанол 23,3 124,4
Диметилсульфоксид 24,5
Диметилформамид 24,8 153 -61
Диметилсульфон 29,7
1,3-Диоксолан-2-он 30,1
Растворители с сильной Н-связью
Бутиламин 17,8 76,2 -50,5
Уксусная кислота 20,7 117,7 16,6
Анилин 21,1 184,4 -6
Октан-1-ол 21,1 195,3 -5
Уксусный ангидрид 21,1 140 -73
Пиридин 21,9 115,6 -42
Триэтиленгликоль 21,9 278,3 -5
Бутан-2-ол 22,1 99,5 -115
2-(Хлорметил)оксиран 22,5 116,1 -57
Бутан-1-ол 23,3 117,7 -90
Циклогексанол 23,3 161,1 -25,2
Пропан-2-ол 23,5 82,4 -90
Проп-2-ен-1-ол 24,1 97,1 -129
Пропан-1-ол 24,3 97,2 -126
Диэтиленгликоль 24,8 244,3 -10,5
Этан-1,2-диамин 25,2 117 11
Пропан-1,2-диол 25,8 188,2
Этанол 26,0 78,3 -115
Метанол 29,7 64,5 -98
Этан-1,2-диол 29,9 198 -12,6
Глицерин 33,8 290 18,2
Формамид 39,3 210,5 2,55
Вода 47,9 100 0
Таблица 5П3.4
Интервалы параметров растворимости 5 полимеров в растворителях с разной силой водородной связи [5]
Полимер 5, МПа1'2
сл.*1 ср.*2 с.*3
Ацетилцеллюлоза 22,7-25,6 20,3-29,7
Ацетобутират 22,7-26,0 17,3-30,0 26,0-29,7
целлюлозы Этилцеллюлоза 17,3-22,1 19,5-23,4
Цианоэтил- 22,7-26,0 25,0-30,0
целлюлоза Нитрат целлюлозы 22,7-26,0 15,9-30,0 25,5-29,6
Целлюлоза Сополимер акрило- 21,6-2,7 18,9 29,7-33,8
нитрила со стиролом Сополимер акрилонитрила с бутадиеном (Буна N) Алкидный олигомер 17,8-19,0 14,2-24,2 15,1-22 19,4-24,2
на основе фталата глицерина и соевого масла Полиакрилонитрил Поливинилацетат Поливинилхлорид 17,4—19,4 17,4-22,5 24,5-28,6 16,0-21,5
Полигексаметилен-адипамид Поликарбонат 19,4-21,7 19,4-20,5 24,3-29,7
Полиметилмет- 18,2-26,0 17,4-27,2
акрилат Полистирол 17,4-21,7 18,6-19,2
Политетрафторэтилен Полиуретан Полиэтилен Полиэтиленоксид 11,9-13,1 20,0-21,1 15,8-16,8 18,2-26,0 17,4-29,7 19,4-29,7
Полиэтиленфталат 19,4-22,1 19,0-20,3
Каучук натуральный Каучук 16,6-17,4 17,4-21,7 16,0-22,1
хлорированный Сополимер 21,7-22,7 15,9-24,8
винилхлорида и винилацетата Соевое масло 14,3-22,6 15,2-22,1 19,4-24,3
Полифенилсилоксан 17,4-22,7 16,2-25,0 22,3-23,3
*’ сл. — растворители со слабыми Н-связями.
*2 ср. — растворители с Н-связями средней силы.
*3 с. — растворители с сильными Н-связями.
826
Новый справочник химика и технолога
Таблица 6П3.4
Системы полимер—растворитель—осадитель*1 [5]
Полимер Растворитель Осадитель
Полиэтилен Ксилол (сл.) Пропан-1-ол*2 (с.)
Полиэтилен Ксилол (сл.) Полиэтиленгликоль*3 (сл.)
Ксилол Этанол
Толуол (сл.) Пропан-1-ол
Полиизобугилен Бензол (сл.) Ацетон (ср.)
Поливинилацетат Ацетон (ср.) Вода (с.)
Ацетон Гексан (сл.)
Диоксан (ср.) Пропан-2-ол (с.)
Бензол Пропан-2-ол
Бутан-2-он Гексан
Хлороформ Пропан-2-ол
Трихлорэтилен Гептан
Поливиниловый спирт Вода Ацетон
Вода Метанол (с.)
Поливинилхлорид Циклогексанон (ср-) Метанол
Циклогексанон Бутан-1-ол
Тетрагидрофуран (ср.) Вода*4
Поливинилхлорид Циклогексан Этан-1,2-диол
Циклогексанон Гептан (ср.)
Полистирол Бензол (сл.) Метанол
Этилацетат (ср.) Этанол (с.)
Бутан-2-он (ср.) Этанол
Поливинилпирро-лидон Хлороформ (ср.) Метанол
Вода Ацетон
Вода Раствор Na2SO4
Полихлоропрен Бензол Ацетон (ср.)
Бензол Метанол
Полиакриламид Вода Метанол
Полиакрилонитрил Диметилформамид Гептан*5
Полиметилмет-акрилат Ацетон Ацетон, вода
Бензол, тетрахлорэтилен Гексан
Ацетон Гексан (сл.)
Бензол Циклогексан (сл.)
Продолжение таблицы 6П3.4
Полимер Растворитель Осадитель
Полиэтиленоксид Хлороформ Циклогексан (сл.)
Мочевино-формальдегидный олигомер Этанол, вода Метанол
Полиэтиленади-пинат Бутан-1-ол (с.) Гексан (сл.)
Ацетилцеллюлоза Ацетон (ср.) Этанол (с.)
Ацетон Вода
Ацетон Гептан и ацетон
Ацетобутират целлюлозы Ацетон Диизопропиловый эфир
Нитрат целлюлозы Ацетон Гексан
Этилацетат Гептан
Ацетон (ср.) Вода*6
Поливинилстеарат Бензол Ацетон
Хлороформ Бутан-2-он
Керосин Минеральное масло
Поливинилиден-хлорид Тетрагидрофуран Вода
Полистирол Ксилол Петролейный эфир
Ксилол Циклогексан
Тетрагидрофуран Вода
Четыреххлористый углерод Метанол, вода
Сополимер стирола и малеиновой кислоты Этанол, метанол Этилацетат
Этанол, метанол Бутан-2-он
Этанол, метанол Диизопропиловый эфир
Этилцеллюлоза Ксилол Гексан
Этанол Вода
Толуол Петролейный эфир
Ксилол—четыреххлористый углерод Гексан
Толуол—трихлорэтилен Гексан
Диоксан Вода
Бензол Минеральное масло
Бензилцеллюлоза Трихлорэтилен Пропан-1-ол
Приложение 3
827
Продолжение таблицы 6П3.4
Полимер Растворитель Осадитель
Ацетобутират целлюлозы Бутан-2-он Диизопропиловый эфир
Ацетилфтал ил-целлюлоза Диметилсульфоксид Вода, ацетон
Натуральный каучук Бензол Метанол
♦1 Водородные связи: сл. — слабые, ср. — средние, с. —
сильные.
*2 При 90 °C.
*3 При 80 °C, 5 = 18,2-5-26 МПа1/2.
*4 При 50 °C.
*5 При 60 °C.
*6 При 25 °C.
Литература
1. ТагерА.А. Физико-химия полимеров. М.: Химия, 1978. 544 с.
2. Щукин Е.Д., Перцов А.В., Амелина Е.А. Коллоидная химия. М.: Изд-во МГУ, 1982. 348 с.
3. Виноградов Г.В., Малкин А.Я. Реология полимеров. М.: Химия, 1977. 440 с.
4. Лосев И.П., Тростянская Е.Б. Химия синтетических полимеров. М.: Химия, 1964. 640 с.
5. Солодовников В.Д. Микрокапсулирование. М.: Химия, 1980. 216 с.
6. Garden J.L. Encyclopedia of Polymer Science and Technology. V. 3. NY - London - Sidney, Intersience Publ, 1965. P. 853-862.
ПРИЛОЖЕНИЕ 4
Основные физико-химические константы
В качестве химической единицы количества вещества принят кмоль
Наименование Обозначение Значение Единица измерения
Скорость света в вакууме с 2,9979- 10'8 м/с
Заряд электрона е 1,60 • IO-19 Кл
Постоянная Больцмана к 1,38 • IO"23 Дж/К
Постоянная Планка h 6,62 • 10 ’34 Дж • с
Постоянная Планка h = А/2л 1,0546 • 10’34 Дж • с
Ускорение свободного падения g 9,80665 м/с2
Число Авогадро Na 6,02 • 1026 1/кмоль
Универсальная газовая постоянная R 8314 Дж/(кмоль • К)
Постоянная Фарадея F 9,65 • 107 Кл/моль
Объем 1 моля идеального газа при нормальных условиях vm 22,414 м3/кмоль
Диэлектрическая постоянная So 8,85 • IO’12 Ф/м
Магнитная постоянная Цо 1,25664- 106 Гн/м
Тцд льдя 273,16 к
Один электрон-вольт 1,60- IO19 Дж
Одна калория 4,185 Дж/кал
Масса атома водорода 1,67 • IO27 кг
Масса атома водорода 1,00814 а. е. м.
Одна а. е. м. (атомная единица массы) 1,66035 • IO’27 кг
Литература
1. Абрамзон А.А. Поверхностно-активные вещества. Л.: Химия, 1981. 304 с.
2. Адамсон А. Физическая химия поверхностей. М.: Мир, 1979. 568 с.
3. Берданосов Ю.Д., Бибик Е.Е. В кн.: Неравновесные процессы в магнитных суспензиях. Свердловск: УНЦ АН СССР, 1986. С. 32-41.
4. Бибик Е.Е., Лавров И.С. // Коллоид, журн., 1970. Т. 32, № 4. С. 483-488.
5. Бибик Е.Е. // Журн. прикл. химии, 1970. Т. 43, № 3. С. 587-592.
6. Бибик Е.Е., Симонов А.А. // Инж.-физ. журн., 1972. Т. 22, № 2. С. 43-346; Бибик Е.Е., Скобочкин В.Е. И Инж.-физ. журн., 1972. Т. 22, № 4. С. 687-692.
7. Бибик Е.Е. И Коллоид, журн., 1973. Т. 35, № 6. С. 1141-1142.
8. Бибик Е.Е., Бузунов О.В., Грибанов Н.М., Лавров И.С. И Журн. прикл. химии, 1979. Т. 52, № 7. С. 1631-1232.
9. Бибик Е.Е. Реология дисперсных систем. Л.: Изд-во ЛГУ, 1981. 172 с.
10. Бибик Е.Е., Гамов Н.С., Грибанов Н.М. Тез. докл. V Всесоюз. совещания по физике магнитных жид
костей. Пермь: Изд-во ИМСС УрО АН СССР, 1990. С. 28-30.
11. Бибик Е.Е., Грибанов Н.М., Наумов В.Н. // Журн. прикл. химии, 1992. Т. 65, № 6. С. 1261-1263.
12. Бибик Е.Е. // Журн. прикл. химии, 1997. Т. 70, № 8. С.1258-1262.
13. Бибик Е.Е. И Журн. прикл. химии, 1999. Т. 72. № 6. С. 916-924.
14. Бибик Е.Е., Попова Е.А. И Журн. прикл химии, 2000. Т. 73, № 1.С. 19-23.
15. Виноградов Г.В., Малкин А.Я. Реология полимеров. М.: Химия, 1977. 440 с.
16. Волькенштейн М.В. Конфигурационная статистика полимерных цепей. М.; Л.: АН СССР, 1959.
17. Вонсовский С.В. Магнетизм. М.: Наука, 1971.
1032 с.
18. Глазман Ю.М. И Коллоид, журн., 1953. Т. 15. С. 225-227.
19. Гроссберг А.Ю., Хохлов А.Р. Статистическая физика макромолекул. М.: Наука, 1989. 344 с.
20. Дамаскин Б.П., Петрий О.А. Введение в электрохимическую кинетику. М.: Высш, шк., 1975. 416 с.
21. Дерягин Б.В., Чураев Н.В., Муллер В.М. Поверхностные силы. М.: Наука, 1985. 398 с.
830
Новый справочник химика и технолога
22. Кройт Г.Р. Наука о коллоидах. М.: ИЛ, 1955. 540 с.
23. Ландау Л.Д., Лифшиц Е.М.. Теоретическая физика. Т. 7. Теория упругости. М.: Наука, 1987. 248 с.
24. Малкин А.Я., Чалых А.Е. Диффузия и вязкость полимеров. Методы измерения. М.: Химия, 1979. 304 с.
25. Неппер Д. Стабилизация коллоидных дисперсий полимерами. М.: Мир, 1986. 487 с.
26. Русанов А.И. Фазовые равновесия и поверхностные явления. Л.: Химия, 1967. 388 с.
27. Смирнов Б.М. //УФЖ. 1986. Т. 146, № 2. С. 177-219.
28. Толстой Н.А., Спартаков А.А. Электрооптика и магнитооптика дисперсных систем. СПб.: Изд-во СПбГУ, 1996. 244 с.
29. Флори П. Статистическая механика цепных молекул. М.: Мир, 1971. 440 с.
30. Френкель Я.И. // Журн. Рос. физ.-хим. о-ва. Физика. 1924. Т. 56, № 2-3. С. 148-166.
31. Френкель Я.И., Губанов А.И. // Успехи физ. наук, 1940. Т. 24, № 1.С. 68-121.
32. Фридрихсберг Д.А. Курс коллоидной химии. Л.: Химия, 1984. 368 с.
33. Цеберс А.О., Майоров М.М. // Магнитная гидродинамика, 1980. № 1. С. 27-35.
34. Чернобережский Ю.М., Голикова Е.В. // Коллоид, журн., 1972. Т. 34, № 5. С. 793-797.
35. Шифрин К.С. Рассеяние света в мутной среде. М.; Л.: ГИТЛ, 1951.
36. Шлиомис М.И. // Журн. экспер. и теор. физики, 1971. Т. 61, №6. С. 2411-2418.
37. Щукин Е.Д., Перцов А.В., Амелина Е.А. Коллоидная химия. М.: Изд-во, МГУ, 1982. 348 с.
38. Реология / Под ред. Ф. Эйриха. М.: ИЛ. 1962. 824 с.
39. Расчеты и задачи по коллоидной химии / Под ред. В.И Барановой М.: Высш, шк., 1989. 288 с.
40. Электрооптика коллоидов / Под ред. С.С Духина. Киев: Наукова думка, 1977.200 с.
41. Cowley M.D., Rosensweig R.E. // J. Fluid Meeh., 1967. V. 30, N 4. P. 671-688.
42. Elmore W.C. // Phys. Rev., 1938. V. 54. P. 309.
43. Frumkin A. // Trans. Faraday Soc., 1940. V. 36. P. 117-127.
44. Grahame D.C. // Chem. Rev., 1947. V.41, N3. P. 441-501.
45. McTague J.P. //J. Chem. Phys., 1969. V. 51. P. 133-136.
46. Verwey E.J.W., Overbeek J.Th.G. Theory of the stability of lyophobic colloids. Amsterdam, 1948.
47. Бузунов O.B. Физико-химические аспекты применения углеводородных феррожидкостей в магнитножидкостных уплотнениях: Дис. ... канд. хим. наук / ЛТИ им. Ленсовета. Л., 1981.
48. Гермашев В.Г. Стабилизация углеводородных жидкостей поверхностно-активными веществами: Дис. ... канд. хим. наук / ЛТИ им. Ленсовета. Л., 1976.
49. Мазуренко О.М. Влияние переменного магнитного поля и магнитной дисперсной фазы на свойства тиксотропных дисперсных систем: Дис. ... канд. хим. наук / ЛТИ им. Ленсовета. Л., 1986.
50. Скобочкин В.Е. Реологические и магнитные свойства структурированных дисперсных систем с ферромагнитным наполнителем: Дис. ... канд. хим. наук / ЛТИ им. Ленсовета. Л., 1971.
51. Соколова Е.А. Самогрануляция магнитно-твердых материалов в жидких средах: Дис. ... канд. хим. наук / ЛТИ им. Ленсовета. Л., 1973.
СОДЕРЖАНИЕ
Предисловие..................................3
Раздел 1. Электродные процессы...............5
RT Значения коэффициента 2,3026................ 5
F
1.1. Электродные процессы в растворах......6
Таблица 1.1. Стандартные электродные потенциалы в водной среде................................6
1.2. Дополнительные сведения..............34
1.2.1. Стандартные электродные потенциалы в неводных средах............................34
Таблица 1.2.1. Стандартный электродный потенциал....................................34
1.2.2. Вспомогательные электроды и электроды для измерения pH.............................34
Таблица 1.2.2. Поправка для разбавленных водных растворов при различных давлениях............35
Таблица 1.2.3. Стандартный потенциал хингидронного электрода......................35
Таблица 1.2.4. Потенциал каломельного электрода при различных температурах...................36
Таблица 1.2.5. Стандартный потенциал электрода ртуть—сульфат ртути(1).......................36
Таблица 1.2.6. Стандартный потенциал хлорсеребряного электрода....................37
Таблица 1.2.7. Стандартный потенциал бромсеребряного электрода в водной среде........................38
Таблица 1.2.8. Стандартный потенциал иодсеребряного электрода..............38
Таблица 1.2.9. Стандартный потенциал электрода с диоксидом свинца...........................38
1.2.3. Нормальные элементы..................38
1.2.4. Перенапряжение выделения водорода....38
Таблица 1.2.10. Константа для уравнения Тафеля (20 °C)......................................38
1.2.5. Потенциалы нулевого заряда металлов
и амальгам...........................39
Таблица 1.2.11. Потенциалы нулевого заряда металлов.....................................39
Таблица 1.2.12. Потенциалы нулевого заряда металлов восьмой группы периодической системы элементов....................................39
Таблица 1.2.13. Потенциалы нулевого заряда амальгам металлов.....................................40
1.2.6. Стандартные токи обмена металлов.....40
Таблица 1.2.14. Стандартные токи обмена металлов ...40
1.2.7. Дифференциальная емкость двойного электрического слоя на ртути.................40
1.3. Электродные процессы в расплавах........41
Таблица 1.3.1. Равновесные напряжения электрохимических систем с твердыми или расплавленными хлоридами металлов.... 41
Таблица 1.3.2. Электродные потенциалы металлов в расплаве КС1—NaCl...................47
Таблица 1.3.3. Электродные потенциалы металлов в эвтектическом расплаве LiCl—КС1.....47
Таблица 1.3.4. Электродные потенциалы металлов в расплавах индивидуальных галогенидов металлов.....................................47
1.4. Коррозия. Виды коррозии, методы испытаний и способы предотвращения коррозионных повреждений.....................48
1.4.1. Понятие коррозии, основные виды коррозионных повреждений металлов и сплавов.....................................48
1.4.1.1. Способы классификации коррозии........48
1.4.1.2. Химическая коррозия.................50
1.4.1.3. Электрохимическая коррозия..........52
1.4.1.4. Основные виды общей электрохимической коррозии металлов и сплавов..................55
Атмосферная коррозия металлов и сплавов......55
Подземная коррозия металлов и сплавов........58
Морская коррозия металлов и сплавов..........60
1.4.2. Питтинговая коррозия..................61
1.4.3. Коррозионное растрескивание металлов и сплавов......................................64
1.4.3.1. Механизм коррозионного растрескивания.64
1.4.3.2. Основные теории, описывающие природу возникновения склонности материалов к коррозионному растрескиванию...............65
Адсорбционная теория.........................65
Водородная теория............................65
Дислокационная теория........................65
Механохимическая теория......................66
Пленочная теория.............................66
Электрохимическая теория.....................66
1.4.3.3. Коррозионное растрескивание углеродистых и низколегированных сталей....................67
Влияние химического состава стали на склонность к коррозионному растрескиванию........67
Влияние термической обработки на склонность сталей к коррозионному растрескиванию.........69
832
Новый справочник химика и технолога
1.4.3.4. Коррозионное растрескивание в аустенитных сталях...........................70
Влияние растягивающих напряжений на стойкость аустенитных сталей.....................70
Влияние способа задания пластической деформации на скорость анодного процесса в аустенитных сталях..........................71
Влияние скорости нагружения на стойкость к коррозионному растрескиванию хромоникелевых аустенитных сталей......74
1.4.3.5. Коррозионное растрескивание в высокопрочных сталях........................77
1.4.3.6. Коррозионное растрескивание цветных сплавов.......................................78
Коррозионное растрескивание титановых сплавов.78
Коррозионное растрескивание магниевых сплавов.79
Коррозионное растрескивание алюминиевых сплавов................................79
1.4.4. Межкристаллитная коррозия в металлах и сплавах......................................80
1.4.4.1. Влияние основных легирующих и примесных элементов на стойкость к межкристаллитной коррозии аустенитных хромоникелевых сталей.........................80
1.4.4.2. Влияние пластической деформации на стойкость стали к МКК..........................84
1.4.4.3. Пути повышения стойкости хромоникелевых сталей к межкристаллитной коррозии.............86
1.4.4.4. Контроль качества оборудования из аустенитных коррозионностойких сталей после расчетного срока службы с целью обнаружения поврежденных или потенциально склонных к межкристаллитной коррозии зон.................89
1.4.4.5. Межкристаллитная коррозия хромистых нержавеющих сталей............................94
1.4.4.6. Межкристаллитная коррозия алюминиевых сплавов.......................................94
1.4.5. Коррозионностойкие стали и сплавы....94
1.4.5.1. Стали ферритного, мартенситно-ферритного и мартенситного классов.......................95
1.4.5.2. Высокопрочные хромоникелевые стали аустенитно-мартенситного класса...............96
1.4.5.3. Стали аустенитного и аустенитно-ферритного классов.......................................97
1.4.5.4. Кислотоупорные сплавы................99
1.4.6. Коррозия и коррозионная стойкость неметаллических материалов...................102
1.4.6.1. Коррозионная стойкость горных пород.102
1.4.6.2. Коррозионная стойкость керамических и других неметаллических материалов..........103
1.4.6.3. Коррозионная стойкость бетонов......104
1.4.6.4. Коррозионная стойкость древесных материалов...................................106
1.4.6.5. Коррозионная стойкость синтетических изоляционных покрытий........................106
1.4.6.6. Коррозионная стойкость полимерных материалов...................................107
Термическая деструкция полимеров............108
Термоокислительная деструкция полимеров......109
Озонное разрушение каучуков, резин и пластиков.... 109 Фотодеструкция, радиационная деструкция
и светостабилизация полимеров........109
Гидролитическая деструкция, механодеструкция и биологическая деструкция полимеров.........109
1.4.7. Промышленная безопасность оборудования. Способы контроля качества деталей с целью выявления коррозионных дефектов различных типов..............................110
1.4.7.1. Проблемы промышленной безопасности оборудования на современном этапе............110
Эксплуатационная надежность оборудования, отработавшего расчетный срок службы в условиях агрессивного воздействия внешних и рабочих сред.......................110
Особенности разрушения крупногабаритных конструкций..................................112
1.4.7.2. Способы испытаний коррозионной стойкости сталей и сплавов.............................114
1.4.7.3. Испытания на общую коррозию........114
1.4.7.4. Определение склонности сталей и сплавов к питтинговой коррозии.......................115
Химические методы исследования..............116
Способы измерения глубины при анализе питтинговой коррозии.........................116
Электрохимические методы исследования.......117
1.4.7.5. Испытания на межкристаллитную коррозию.....................................117
1.4.7.6. Испытания на стойкость сталей и сплавов к коррозионному растрескиванию...............118
1.4.7.7. Методы неразрушающего контроля крупногабаритного оборудования, отработавшего расчетный срок службы, с целью обнаружения зон, поврежденных различными видами коррозии...................121
1.4.8. Способы защиты металлических и неметаллических материалов от коррозии..................................126
1.4.8.1. Электрохимическая защита...........127
Катодная и протекторная защиты..............127
Некоторые виды катодной защиты..............128
Анодная защита..............................132
1.4.8.2. Ингибиторы коррозии................134
1.4.8.3. Антикоррозионные защитные покрытия..135
Металлические покрытия......................135
Неметаллические покрытия....................136
1.4.8.4. Защита неметаллических конструкций.139
1.4.9. Коррозионная стойкость материалов....141
Содержание
833
Таблица 1.4.62. Коррозионная стойкость материалов..................................142
Таблица 1.4.63. Растворимость водорода в металлах..................................323
Таблица 1.4.64. Коррозия при контактах между металлами и сплавами........................325
Раздел 2. Химическая кинетика и диффузия....329
2.1. Кинетика.............................329
2.1.1. Основные понятия в химической кинетике гомогенных химических реакций...............329
Классификация кинетических кривых...........329
Кинетическая классификация химических реакций ..330
Кинетические и термодинамические параметры химических реакций...................331
Расчет константы скорости по частоте соударений и энергии активации..................334
2.1.1.1. Формальная кинетика химических реакций, протекающих в реакторах периодического и непрерывного действия......................336
Кинетика гомогенных химических реакций в статических условиях...............336
Кинетика гомогенных химических реакций в реакторах идеального перемешивания.........336
Кинетика гомогенных химических реакций в непрерывных процессах.....................341
Кинетика гомогенных химических реакций в режиме идеального вытеснения..............342
Кинетика гомогенных химических реакций в потоке в режиме идеального перемешивания...........344
Мономолекулярные реакции в газовой фазе.....345
Пояснения к таблицам 2.1.15 и 2.1.16........347
Таблица 2.1.15. Константы равновесия химических реакций, протекающих в жидкой фазе....349
Таблица 2.1.16. Кинетические параметры химических реакций, протекающих в жидкой фазе....356
Таблица 2.1.17. Константы скорости гомолитических химических реакций, протекающих в газовой фазе..............................402
2.1.2. Г етерогенные реакции................430
2.1.2.1. Топохимические реакции.............430
Формально-кинетический анализ топохимических реакций.....................................430
Влияние температуры и давления на кинетику топохимических реакций......................437
Кинетика окисления металлов.................446
2.1.2.2. Диффузионная кинетика..............449
2.1.3. Реакции изотопного обмена............453
Таблица 2.1.36. Кинетика реакций изотопного обмена в гомогенной среде..........................453
Таълица 2.1.37. Кинетика реакций изотопного обмена в гетерогенной среде........................468
2.2. Диффузия.............................475
2.2.1. Молекулярная диффузия................475
2.2.1.1. Диффузия в газах......................475
Таблица 2.2.1. Коэффициенты самодиффузии газов при различных температурах и давлении /7=0,1 МПа..................................477
Таблица 2.2.2. Коэффициенты взаимной диффузии в газах и парах £>, полученные различными исследователями при давлении /7=0,1 МПа..................................481
Таблица 2.2.3. Атомные и молекулярные диффузионные объемы для определения АВ..........................496
Таблица 2.2.4. Сводная таблица значений коэффициентов (при 293 К)...................497
Таблица 2.2.5. Зависимость коэффициентов диффузии газов от давления..................497
2.2.1.2. Термическая диффузия в газах....498
Таблица 2.2.7. Данные по термодиффузии в газовых смесях......................................498
2.2.1.3. Диффузия в жидкостях..................513
Таблица 2.2.8. Значения коэффициентов для некоторых веществ, растворенных в воде......................................514
Таблица 2.2.9. Коэффициенты диффузии веществ в водных растворах...................514
Таблица 2.2.10. Коэффициенты диффузии в неводных растворителях.............516
Таблица 2.2.11. Коэффициенты взаимной диффузии в жидких двухкомпонентных органических системах....................................517
Таблица 2.2.12. Диффузия в расплавленных металлах....................................517
Таблица 2.2.13. Диффузия в амальгамах.517
Таблица 2.2.14. Диффузия в расплавленных солях.... 517
Таблица 2.2.15. Экспериментальные значения коэффициентов диффузии различных газов и неэлектролитов в сильноразбавленных водных растворах............................518
Таблица 2.2.16. Коэффициенты взаимной диффузии для Н2, СН4 и СО2 в водных растворах солей.......................................518
Таблица 2.2.17. Экспериментальные значения коэффициентов диффузии в неводных растворителях при бесконечном разбавлении.................................519
Таблица 2.2.18. Аддитивные приращения объемов для расчета молярных объемов растворенных веществ при нормальной температуре кипения......................................519
2.2.1.4. Диффузия в растворах электролитов..520
Таблица 2.2.19. Предельные ионные проводимости в воде при 25 С.............................520
Таблица 2.2.20. Коэффициенты взаимной диффузии неорганических соединений в водных растворах...................................521
834
Новый справочник химика и технолога
Таблица 2.2.21. Коэффициенты взаимной диффузии для водорода и метана в водных растворах солей......................................521
2.2.2. Диффузия в твердой фазе......... 522
2.2.2.1. Общие понятия.....................522
2.2.22. Диффузия в непористых материалах..522
Таблица 2.2.22. Параметры для коэффициента взаимной диффузии металлов и сплавов........525
Таблица 2.2.23. Параметры для коэффициента самодиффузии металлов......................527
Таблица 2.2.24. Концентрационная зависимость констант £)0 и Еа для диффузии металлов в меди при 800 °C..........................527
Таблица 2.2.25. Константы для диффузии металлов в солях....................................528
Таблица 2.2.26. Коэффициент диффузии ионов в кристаллах при различных температурах...............................528
Таблица 2.2.27. Параметры для коэффициента диффузии иона Ag+ в кристаллах......528
Таблица 2.2.28. Коэффициент взаимной диффузии твердых солей.......................528
Таблица 2.2.29. Самодиффузия в твердом водороде ..529
Таблица 2.2.30. Параметры для коэффициента диффузии газов в твердых телах.............529
Таблица 2.2.31. Параметры для коэффициента диффузии малой примеси в серебре....529
Таблица 2.2.32. Параметры для коэффициента диффузии малой примеси в меди..............529
Таблица 2.2.33. Параметры для коэффициента диффузии малой примеси в золоте.....529
Таблица 2.2.34. Параметры для коэффициента диффузии малой примеси в никеле.....529
Таблица 2.2.35. Параметры для коэффициента диффузии различных примесей в металлах III и IV групп..................530
Таблица 2.2.36. Параметры для коэффициента диффузии атомов примесей в щелочных металлах...................................530
Таблица 2.2.37. Параметры для коэффициента диффузии атомов примеси в лантанидах и актинидах................................530
Таблица 2.2.38. Параметры температурной зависимости коэффициента диффузии малой примеси в 0-Ti................531
Таблица 2.2.39. Параметры для коэффициента диффузии ионов в кристаллах солей...531
Таблица 2.2.40. Параметры для коэффициента диффузии примесей в кристаллах солей........531
Таблица 2.2.41. Параметры для коэффициента диффузии атомов водорода и его изотопов в металлах.................................532
Таблица 2.2.42. Параметры для коэффициента диффузии атомов инертных газов в кристаллах солей.........................532
Таблица 2.2.43. Параметры для коэффициента диффузии атомов примеси в полупроводниках..........................532
Таблица 2.2.44. Параметры для коэффициента самодиффузии в некоторых кристаллах.........534
Таблица 2.2.45. Коэффициенты диффузии атомов металлов в амальгамах......................534
Таблица 2.2.46. Коэффициент диффузии в расплавленных металлах...................534
2.2.2.3. Диффузия в капиллярно-пористых материалах.................................534
2.2.2.4. Диффузия в коллоидных
капиллярно-пористых материалах......537
2.2.3. Диффузионная проницаемость через
мембраны............................538
2.2.4. Диффузия в ионообменных смолах......538
Таблица 2.2.47. Значение коэффициентов самодиффузии ионитов.......................539
Раздел 3. Физика, химия и технология дисперсных систем (Коллоидная химия)...................547
3.1. Поверхность. Фундаментальные свойства... 547
3.1.1. Геометрия дисперсной системы.........547
3.1.2. Поверхностный слой...................549
3.1.3. Поверхностное натяжение..............551
3.1.4. Адсорбция газов на поверхности
твердых тел..........................554
3.2. Капиллярные явления..................559
3.2.1. Основные понятия.....................559
3.2.2. Равновесие поверхности твердого вещества
с летучей жидкостью..................563
3.2.3. Равновесие поверхности твердой фазы
с двумя жидкими......................565
3.3. Термодинамика поверхности............569
3.3.1. Фундаментальное уравнение
и термодинамические потенциалы.......569
3.3.2. Уравнения термодинамики гетерогенных
систем...............................571
3.3.3. Работа образования поверхности.......573
3.3.4. Термодинамика тонких пленок..........575
3.4. Поверхностное натяжение и адсорбция..577
3.4.1. Механизм адсорбции из растворов......577
3.4.2. Поверхностно-активные вещества (ПАВ).... 578
3.4.3. Пленки нерастворимых ПАВ.............582
3.4.4. Мицеллярные растворы ПАВ.............584
3.4.5. Градиент поверхностного натяжения....586
3.4.6. Адсорбция из раствора на поверхности
твердого вещества....................588
3.4.7. Адсорбция на внутренней поверхности
поликристаллических веществ..........590
3.4.8. Электрокапиллярность.................591
3.4.9. Измерение поверхностного натяжения...592
3.5. Двойной электрический слой...........594
3.5.1. Понятие двойного электрического слоя.594
Содержание
835
3.5.2. Плотная и диффузная части ДЭС........599
3.5.3. Специфическая адсорбция и емкость ДЭС...602
3.5.4. Двухслойная модель плотного слоя.....604
3.5.5. Уравнение Нернста и адсорбция ионов..607
3.5.6. Электрокинетический и мембранный
потенциал.............................610
3.6. Расклинивающее давление в тонких
пленках...............................617
3.6.1. Молекулярное притяжение..............617
3.6.2. Электростатическое отталкивание ДЭС..619
3.6.3. Отталкивание адсорбционных слоев.....621
3.6.4. Структурный и стерический эффекты....621
3.6.5. Вытеснительное отталкивание
и притяжение..........................623
3.7. Устойчивость дисперсных систем.......624
3.7.1. Вводные понятия......................624
3.7.2. Взаимодействие сферических частиц....625
3.7.3. Устойчивость против коагуляции.......628
3.7.4. Критическая концентрация электролита.628
3.7.5. Силовая коагуляция и экстремумы
функций взаимодействия частиц.........630
3.7.6. Эффекты смещения плоскости адсорбции
ионов.................................631
3.7.7. Устойчивость многокомпонентных
суспензий.............................634
3.8. Молекулярно-кинетические явления.....636
3.8.1. Регулярное и хаотичное движение частиц ...636
3.8.2. Диффузия.............................638
3.8.3. Равновесное распределение дисперсной
фазы..................................639
3.8.4. Оседание устойчивых концентрированных
суспензий.............................641
3.8.5. Седиментационный анализ..............644
3.9. Электромагнетизм коллоидов...........645
3.9.1. Основы электростатики................645
3.9.2. Поляризуемость частиц................651
3.9.3. Магнитное поле и магнетизм веществ...653
3.9.4. Дисперсные ферромагнетики............658
3.9.5. Суперпарамагнетизм Броуна и Нееля....667
3.10. Основы механики деформируемых сред...669
3.10.1. Предмет реологии.....................669
3.10.2. Основные понятия и законы реологии...669
3.10.3. Вязкость и классификация текучих
материалов............................673
3.11. Структура и реология дисперсных систем... 676
3.11.1. Структурная классификация дисперсных
систем................................676
3.11.2. Неструктурированные системы..........680
3.11.3. Ориентационное структурирование......682
3.11.4. Вращательная вязкость................688
3.12. Периодическая коллоидная структура...689
3.12.1. Концентрационное структурирование....689
3.12.2. Уравнение структурного состояния ПКС.691
3.12.3. У равнение напряжений и уравнение
реологии ПКС...........................693
3.12.4. Время релаксации и потенциальный
барьер.................................695
3.13. Кинетика коагуляции
и структурирования.....................695
3.13.1. Введение..............................695
3.13.2. Элементы кинетики коагуляции..........696
3.13.3. Фрактальная размерность и структура
флокул.................................697
3.13.4. Параметры состояния коагулирующей
взвеси и условие структурирования......699
3.13.5. Скорость оседания флокул..............700
3.13.6. Альтернатива: расслоение или
структурирование.......................701
3.13.7. У равнения кинетики коагуляции........703
3.13.8. Динамика и кинетика уплотнения осадка.... 704
3.14. Реология тиксотропных систем..........707
3.14.1. Прочность структурной сетки и флокул..707
3.14.2. Уравнение структурного состояния и
тиксотропия............................708
3.14.3. Реологическое уравнение тиксотропных
систем.................................709
3.14.4. Течение в тонких каналах..............710
3.14.5. Цепочечная структура..................712
3.14.6. Уравнение состояния и реологический закон
цепочечной структуры...................713
3.14.7. Послойное скольжение..................717
3.15. Основы вискозиметрии..................720
3.16. Полимеры и растворы полимеров.........726
3.16.1. Введение..............................726
3.16.2. Гибкость и форма макромолекул.........727
3.16.3. Модуль упругости и персистентная длина
макромолекулы..........................734
3.16.4. Гибкость молекул полиэлектролита......735
3.16.5. Модуль упругости полимерных
материалов.............................736
3.16.6. Особенности растворения полимеров.....737
3.16.7. Качество растворителя.................738
3.16.8. Вязкость растворов полимеров..........741
3.17. Оптические свойства дисперсных систем.... 745
3.18. Получение дисперсных систем...........748
3.18.1. Дробление.............................748
3.18.2. Физическая и химическая конденсация...751
3.18.3. Пептизация............................752
3.19. Феррожидкости. Получение и свойства...753
3.19.1. Введение..............................753
3.19.2. Приготовление магнитных жидкостей.....754
3.19.2.1. Выбор метода и магнитного материала.754
3.19.2.2. Приготовление магнетитовых феррожидкостей...............................755
3.19.3. Свойства феррожидкостей...............757
3.19.3.1. Намагниченность.....................757
836
Новый справочник химика и технолога
3.19.3.2. Эффекты коагуляции и структурирования феррожидкостей...............................758
3.19.4. Феррогидродинамика..................761
3.19.4.1. Магнитостатическое давление.......761
3.19.4.2. Неустойчивость свободной границы феррожидкости................................762
3.19.4.3. Применение феррожидкостей.........763
3.19.4.4. Применение эффектов взаимодействия частиц в магнитных суспензиях................766
Приложение 1. Поверхностное натяжение и поверхностно-активные вещества.............767
1.1. Поверхностное натяжение веществ.....767
1.1.1. Поверхностное натяжение веществ на границе с газовой фазой...................767
Таблица 1П1.1. Простые вещества.............767
Таблица 2П1.1. Неорганические соединения....768
Таблица ЗП1.1. Вода на границе с воздухом...769
Таблица 4П1.1. Смеси Н2О и D2O при 20 °C....769
Таблица 5П1.1. Органические вещества........770
Таблица 5аП1.1. Алифатические углеводороды..770
Таблица 56П1.1. Циклические углеводороды....770
Таблица 5вП 1.1. Галогензамещенные углеводороды.................................771
Таблица 5гП1.1. Спирты, тиоспирты, фенолы...772
Таблица 5дП1.1. Простые эфиры, альдегиды, кетоны.......................................772
Таблица 5еП1.1. Кислоты и их производные....772
Таблица 5жП1.1. Сложные эфиры...............773
Таблица 5зП1.1. Амины.......................774
Таблица 5иП1.1. Другие азотсодержащие соединения...................................774
Таблица 5кП1.1. Прочие органические вещества и масла......................................775
Таблица 6П1.1. Сводные данные для ряда распространенных органических растворителей................................775
Таблица 7П. 1.1. Межфазное натяжение воды на границе с другими веществами..............775
Таблица 8П1.1. Межфазное натяжение ртути на границе с водными растворами..............777
Таблица 9П1.1. Межфазное натяжение ртути на границе с органическими жидкостями........777
1.2. Поверхностное натяжение растворов....778
Таблица 1П1.2. Изотермы поверхностного натяжения водных растворов полярных веществ......................................778
Таблица 2П1.2. Изотермы поверхностного натяжения водных растворов солей жирных кислот.......................................779
Таблица ЗП1.2. Изотермы поверхностного натяжения водных растворов катионоактивных ПАВ..........................781
Таблица 4П1.2. Изотермы поверхностного натяжения водных растворов неионогенных ПАВ.............................783
Таблица 5П1.2. Поверхностное натяжение растворов неорганических веществ на границе с влажным воздухом...........................789
1.3. ГЛБ и ККМ.............................790
1ПЗ. 1. Гидрофильно-липофильный баланс веществ......................................790
Таблица 1П1.3. Зависимость состояния ПАВ в воде от величины ГЛБ.......................790
Таблица 2П1.3. Групповые числа N различных фрагментов молекул ПАВ................790
Таблица ЗП1.3. ГЛБ поверхностно-активных веществ...............................790
1.3.2. Критические концентрации мицеллообразования (ККМ)......................791
Таблица 4П1.3. Критические концентрации мицеллообразования типичных представителей поверхностно-активных веществ разного типа..........................791
1.3.3. Расчет количества ПАВ в дисперсной системе.......................................792
Таблица 5П1.3. Площадь, занимаемая одной молекулой в насыщенном адсорбционном слое.........................................792
1.4. ПАВ в неводных средах.................792
1.4.1. Припои и флюсы........................792
1.4.2. Легкоплавкие припои...................793
Таблица 1П1.4. Состав и свойства оловянно-свинцовых припоев......................................793
Таблица 2П1.4. Припои на основе олова, цинка и кадмия.....................................793
Таблица ЗП1.4. Состав и температура плавления сплавов ИНГ АС...............................794
1.4.3. Флюсы для пайки легкоплавкими припоями.....................................794
Таблица 4П1.4. Типовые составы флюсов на основе канифоли.....................................794
Таблица 5П1.4. Флюсы для пайки алюминия......794
1.5. Поверхностно-активные вещества,
растворимые в углеводородах...........795
1.5.1. Поверхностная активность в неполярной
среде.................................795
Таблица 1П1.5. Основные функции присадок к маслам и топливам..........................796
Таблица 2П1.5. Перечень показателей, характеризующих свойства и эффективность ПАВ в смазочных маслах и жидких топливах.....................................796
1.6. Маслорастворимые ПАВ основных химических классов и области их применения.................................797
1.6.1. Сульфонаты............................797
Содержание
837
Таблица 1П1.6. Характеристика нефтяных и синтетических сульфонатов.................797
1.6.2. Амины, амиды, имиды..................798
Таблица 2П1.6. Свойства и функциональное назначение присадок к маслам................798
Таблица ЗП1.6. Обобщенная характеристика действия маслорастворимых азотсодержащих ПАВ..........................799
1.6.3. Кислородсодержащие ПАВ...............799
Таблица 4П 1.6. Свойства некоторых кислородсодержащих маслорастворимых ПАВ........................800
1.6.4. Хлор и серосодержащие соединения.....800
1.6.5. Сополимеры, содержащие полярные и неполярные звенья.........................800
Приложение 2. Дисперсные системы............803
2.1. Точки нулевого заряда.................803
Таблица 1Ш.2. Точки нулевого заряда.........803
2.2. Константы молекулярного
взаимодействия.......................803
Таблица 1П1.2. Пороги коагуляции отрицательно заряженных золей............................804
Таблица 2П1.2. Пороги коагуляции положительно заряженных золей............................804
Таблица ЗП1.2. Константы молекулярного притяжения частиц в водной среде............805
Таблица 4П1.2. Показатели преломления при длине волны X = 589,3 нм и плотности твердых веществ при 20 °C...........................805
Таблица 5П2.2. Показатели преломления жидкостей при 20 °C и длине волны 589,3 нм............808
Таблица 6П2.2. Плотность жидкостей при различных температурах................809
Таблица 7П2.2. Вязкость жидкостей при различных температурах..........................810
Таблица 8П2.2. Предельная эквивалентная электрическая проводимость ионов в водных растворах..........................812
2.3. Приготовление и свойства некоторых
коллоидных систем....................813
2.3.1. Г ель кремневой кислоты H2SiO3.......813
2.3.2. Феррожидкости........................813
Таблица 1П2.3. Характеристики ферромагнитных жидкостей фирмы «Ferrofluidics Corp.».......813
Таблица 2П2.3. Характеристика феррожидкостей СПбГТИ(ТУ)..................................813
Приложение 3. Полимеры......................815
3.1. Гибкость макромолекул.................815
Таблица 1П3.1. Потенциальные барьеры вращения мономеров...................................815
Таблица 2ПЗ. 1. Параметры гибкости полимеров.816
Таблица ЗПЗ.1. Температура стеклования полимеров...................................816
3.2. Вязкость.............................818
Таблица 1П3.2. Критические величины молярных масс........................................820
Таблица 2П3.2. Значения констант формулы Фокса.......................................820
3.3. Вязкость разбавленных растворов......821
Таблица 1ПЗ.З. Константы формулы Куна.......821
3.4. Состояние системы
полимер—растворитель.................821
Таблица 1П3.4. Критические температуры некоторых систем полимер—растворитель.................822
Таблица 2П3.4. Параметры растворимости растворителей и полимеров............823
Таблица 2П3.4а. Параметры растворимости растворителей........................823
Таблица ЗПЗ. 4. Донорные числа к SbCl5, относительная диэлектрическая проницаемость, параметры растворимости растворителей...............................823
Таблица 4П3.4. Параметр растворимости, температуры кипения и плавления растворителей с различной силой водородных связей...........................824
Таблица 5П3.4. Интервалы параметров растворимости полимеров в растворителях с разной силой водородной связи.............825
Таблица 6П3.4. Системы полимер—растворитель—осадитель.......826
Приложение 4. Основные физико-химические константы............................829
|Мир и (. емья, Профессионал, <116
УШЬГЙ СПРАВОЧНИК ХИМИКА И ТЕХНОЛОГА
Электродные процессы Химическая кинетика и диффузия Коллоидная химия
Научное издание
Издание подготовлено АНО НПО «Профессионал» 191023, Санкт-Петербург, ул. Садовая, 28-30, корп. 35. Телефакс: 321-67-38; 110-59-91; 110-57-93; 115-14-35.
mis@npomis.com http://www.npomis.com http://www.naukaspb.ru
Ответственный за издание: А.А. Полуда Ответственный за выпуск: Н.В. Емельянова Ответственный за подготовку: Л. В. Белканова Научный редактор: В.А. Столярова Редактор-корректор: С.Е. Парфенова, Э.И. Чудновская Компьютерная верстка: Т.А. Бойченко, А.А. Николаева, Т.А. Филина Техническое сопровождение: Т.Н. Жадобина Оператор цифровой печати: А.В. Кеда
Отпечатано в центре цифровой печати АНО НПО «Профессионал» 191023, Санкт-Петербург, ул. Садовая, 28-30, корп. 35. Телефакс: 321-67-38; 110-59-91; 110-57-93; 115-14-35.
Сдано в набор 16.04.2002. Подписано к печати 07.07.2004. Формат 60x90/8. Бумага офсетная, плотность 80 г/м2 Объем 105,5 печ. л. Тираж 1000 экз.
УЪос&ящается 300-летию Санкгп-бТетербурга — Крлыбели отегест&енной киМигескрй науки и промышленности
мовьгй СПРАВОЧНИК ХИМИКА И ТЕХНОЛОГА
Электродные процессы Химическая кинетика и диффузия Коллоидная химия
НПО “Профессионал” Санкт-Петербург 2004
2
2
Г Li 6,941(2)
He 2s’
ЛИТИЙ
lithii
3
3
| Na
22.989770(2)
NeSs’ ’
НАТРИЙ 2
4
5
6
4
5
6
9
57 La ,1
1389055(2) 4
Xe5d'6s2 ’J
ЛАНТАН 8
II
4 Be
9.012182(3)
He2s2
I БЕРИЛЛИЙ
I12 Mg
24.3050(6)
Ne3s* JI
МАГНИЙ | . maanes
5
III
6
IV
VI
VII
VI]
>002602(2)
ГЕЛИЙ
110.811(7) He2s22p* БОР
borum
12.0107(8)
He2sz2p2 4[ [УГЛЕРОД г I_______carboneum
7 | | 4 | 10
|14,OO674(7) »Ml5.9994(3) I i 18,9984032г 5) ».|2O.1797(6)
He2s?2p3 .1 ' He2s22p4 .1 He2s22p5 He 2s’
АЗОТ ?| КИСЛОРОД 2|: ФТОР гИнЕОН
-weeiiej
13
26.981538(2)
He3s23p' ?
АЛЮМИНИЙ 2
I aluminium
14 if15 ! 16 Q | 17
28.0855(3) He3s23p2 4 КРЕМНИЙ ’ silicium №> 30.973761(2) 2 Ne 3s23p5 8 f ФОСФОР г « ohosphorum [ 32.066(6) He3s23p4 8 СЕРА 1 ! 35.4527(9) Ne 3s23p5 7 I ХЛОР г I chlorum
39.948(1)
Ne3s‘
АРГОН
18
,”K
39.0983(1) i
| Ar 4s1 8
КАЛИЙ 2
__________kalium I
20 Ca
40.078(4)
Ar 4s?
[кальций
___________
I21 Sc
44,955910(8)
Ar3d’4s2
[СКАНДИЙ
2
9
29
Cu
63.546(3) Ar3di04s’
МЕДЬ
IB
37 Rb I
J 85.4678(3) 8
i Kr5$' ^8
£ РУБИДИЙ г|
i rubidium!
47 Ag
107.8682(2) i
Kr4d,05s’ 1
СЕРЕБРО
________aroentur
18
18
“ Cs ;|
Xe6s’ ’JI , ЦЕЗИЙ
79
87 Fr «I
A 1 18
(223.0197) 32
Rn 7s’
21
iff.'
ФРАНЦИЙ
58 Ce
140.116(1) 20
Xe4tW ’8
ЦЕРИЙ
90 Th I
232.0381(1) 321
Rn6d:'7s2 ’J
ТОРИЙ
I30 Zn 65,39(2) г] Ar3d’°4s2 ’J: ! ЦИНК 31 69,723(1) 3 Ar 3d’°4s=4p' ’J ГАЛИЙ
i38Sr
87.62(1)
Kr5s2 ’|i
[СТРОНЦИЙ г]
I48 Cd, 112.411(8) is! Kr4d”5s? ’J] [КАДМИЙ ° | I_________cadmium]
56 Ba
137.327(7)
.. 18
Xe 5s2 ’81
БАРИЙ
Au 196.96655(2) 32 Xe4f145d’a6s' 'JI ЗОЛОТО 2| амт.
80 Hg-| [200,59(2) 32 Xe 4fu5d,06s? ’J | РТУТЬ ? hydrargyrum |
88 Ra
[226.0254]
2
8 ]
I8
„........... 321
Rn 7s2 ’«
[РАДИЙ
i radium |
39
188,90585(2) s
Kr4d'5s2 ’J
ИТТРИЙ ’[
yttrium
|22 T. к 23 у | 24 СГ j 25 МП I
И47,867(1) .1 50 9415(1) ?l 51 9961(6) ti 54.938049(9) 2l
Ar3d24s2’°| Ar3d34s2 Ar3d14s’’8| Ar3d54s2 ’||
Г ТИТАН 2| ВАНАДИЙ ?| ХРОМ г| МАРГАНЕЦ ?|
H^^^^btamunJ vanadium^ сОгопиитЦ _^^manaanunil
33 h |34 Ь 35 к
174,92160(2) 5К !78,96(3) б| 79,904(1) ?1
Ar 3d,s4s?4p3 «Ж ArSdWdp4Ar 3d!°4s?4p'‘
МЫШЬЯК г I СЕЛЕН ?| БРОМ
arsenicun*!В f
'*лнтмя1М* твияиявиив*
26
55.845(2)
Ar 3d1
ЖЕЛЕЗО
32
36
72.61(2) 4
Ar 3d”4s24p2 ’J
ГЕРМАНИЙ г
I40 Zr
|91.224(2)
Kr4d25s2
[ЦИРКОНИЙ
10
18
2] irconiunr
42 Mo , 95.94(1) 13
Kr 4ds5s2 ’J
МОЛИБДЕН 2 molvbdaenum
[97.9072] 13 I
Kr4d’5s2 ’J I
ТЕХНЕЦИЙ 2 I
I41 Nb , 192.90638(2) 12
Kr4d45s' ’’
НИОБИЙ 2
43
83.80(1)
Ar 3d’°4s:
КРИПТОН
44
101.07(2)
Kr4d'
РУТЕНИЙ
_________rutt
49 114.818(3) 18 Kr 4dw5s25p’ ’J ИНДИЙ 2 indium 50 118.710(7) is Kr 4d’°Ss25p? ’J ОЛОВО stannum! 51 01 121.760(1) 18 Kr4d'°5s25p3 ’J СУРЬМА г 52 127,60(3) 18 Kr4d’°5s25p4 ’J ТЕЛЛУР 2 tellurium! 53 I 1 126.90447(3) 181 Kr 4d’°5s25pb ’J ИОД 2 iodum ] 54 131.29(2) Kr4d’°5s2 КСЕНОН
57-71 * La-Lu 1 1 72 Hf r 1 10 178,49(2) 32 Xe 4f'45d26s2 ’« ГАФНИЙ ’ hafnium 73 Ta,(| 180,4979(1) 321 Xe4f,‘5d36s! ’J ТАНТАЛ г 74 W 3 ’ ’ 12 183,84(1) 32 Xe 4t,45(f’6s2 ’J ВОЛЬФРАМ “ wolframium 75 Re J 186.207(1) 321 Xe4f’45ds6s2 18 РЕНИЙ rhenium] 76 c 190.23(3) Xe 4f 145d< ОСМИЙ 0
81
18
204.3833(2)
Xe4fl45d't6s26pI ’J I [ТАЛИЙ i___________thallium
89-103 **
Ac-Lr
I59 Pr 140,90765(2) 2?| Xe4(26s2 'Ji ПРАЗЕОДИМ г praseodymium j !eoNd ,1 [144,24(3) 22 Xe4fi6s2 ’8 НЕОДИМ г neodymium!
91 Pa i|
** Vli
231.03586(2) 32 [ Rn iWJs1 « I ПРОТАКТИНИЙ г
92 U J
238,0289(1) 32
Rn5ftd:7s2 181
УРАН
82
83
84
18
18
32
[208.9824]
Xe 4f'45d’°6s26pJ ’«
ПОЛОНИЙ
; 207.2(1)
Xe 4f’45d:06s26p2 ’«
СВИНЕЦ ’
lumbum
208.98038(2)
Xe 4f-45d,06s26p! ’>
ВИСМУТ 2
bismuthum
с I is I 32 I 18 I
a I
85
104
106
18 I [209.9871] 32 I
Xe4f'45d’“6s26p5’8 | АСТАТ astatium
1(263.1125]
RnSf’46d27s2 ’J| РЕЗЕРФОРДИЙ? | rutherfordium I
[262.1144] 32]
Rn5f"6d7s2 ’J|
ДУБНИЙ I
| dubnium!
]266.1219]
Rn 5f'46d47s2 'Jj
СИБОРГИЙ 2
seaborqium;
107Bh d
1264.1247] 32
Rn 5fW ’J БОРИЙ ________baton!
86
[222 0176] Xe 41’45dic6s
РАДОН
108
| [269.1341]
Rn Sf’W ХАССИЙ
6,Pm J
[144,9127] 23
Xetfts2 1«
ПРОМЕТИЙ 2
prcmethium!
93 Np |
[237.0482) 32
RnSfed^s2’8
НЕПТУНИЙ '
62Sm J
150..36(3) 24
Xe4l*6s2 ’a
[САМАРИЙ 8
'Pit il
24
[244.0642] 321
Rn 5f®7s2 ’J|
ПЛУТОНИЙ 51
63 Eu J 151.964(1) 25
| Xe4f'6s? 'J ЕВРОПИЙ 8 __________europium I
[S4Gd
157.25(3) 25
[ Xe It'Sd’es2 ’J
ГАДОЛИНИЙ г qadolinippi i
65 Tb
158,92534(2) 2”
Xe4f36s2 'J
ТЕРБИЙ 2
______
95Am Ji [243.0614] 32
RnSf’/s2 ’»
[АМЕРИЦИЙ г
96 Cm jl
[247,07031 32
I Rn Sfed^s2 ’J
I КЮРИЙ 2
97 Bk
I [247.0703] 32
Rn 5P7S2 ’’
БЕРКЛИЙ г
argon
rtaoon
110
100
103
Rn 51’ Ш'
Н-УННИЛИЙ
{268.1388]
Rn МЕЙТНЕРИЙ
(262,110] 32
Rn Sf’Ws2 «
ЛОУРЕНСИЙ °
174.967(1) 3z
Xe 4f 45d’6s2 1«
ЛЮТЕЦИЙ 2
[258.0984 ] 32
RnStW ’®|
МЕНДЕЛЕВИЙ г
[251.0796] 32
Rn5f1fl7s2 15
КАЛИФОРНИЙ ?
[252.06301 32
RnSf’W 1«
ЭЙНШТЕЙНИЙ I
173.04(3) 32
Xe4f”6s2 'J
ИТТЕРБИЙ г
[259,1011] 32
Rn 5tW ’«
НОБЕЛИЙ г
162,50(3) 28
Xe4f,06s! ’8
ДИСПРОЗИЙ ’
167.26(3) 30
Xe4f’'6s2 ’«
ЭРБИЙ
[257.09511 32
Rn5t,f7s- ’»
1ФЕРМИЙ 2
168.93421(2) 3°
Xe4l,36s2 4 I ТУЛИЙ
164.93032(2 ) 29
Xe4t”6s2 ’«
ГОЛЬМИЙ 2
ронйая Крн:]>(<]
Русское название Латинское название
106,42(1) 18|
Kr4d”
ПАЛЛАДИЙ palladium
2’;й.025П1) 32
Rn StfedW
УРАН
uranium
102,90550(2) 16
Kr4d’5s1 '8
РОДИЙ
rhodium i
Атомный номер
Символ элемента
Распределение электронов по уровням
Атомная масса
серия книг для специалистов
ПРОФЕССИОНАЛ^
www.naukaspb.ru
^0‘ВЫ‘И
СПРАВОЧНИК ХИМИКА и ТЕХНОЛОГА
Электродные процессы Химическая кинетика и диффузия Коллоидная химия Руководители проекта
ХИл<^4.
<<< О <3
А.А. Полуда, Н.В. Емельянова
Под общей редакцией
академика РАЕН, профессора, доктора химических наук
С.А. Симановой
Авторы: проф., д.т.н. Р.Ш. Абиев проф., д.х.н. Е.Е. Бибик проф., д.т.н. Е.А. Власов проф., д.т.н. Б.С. Ермаков проф., Д.т.н. В.С. Зотиков доц., к.т.н. В.А. Иванов акад. РАЕН, проф., д.х.н. С.А. Симанова доц., к.т.н. К.А. Суворов доц., к.х.н. К.А. Хохряков проф., д.т.н. М.А. Яблокова
MMIV Санкт-Петербург 2004