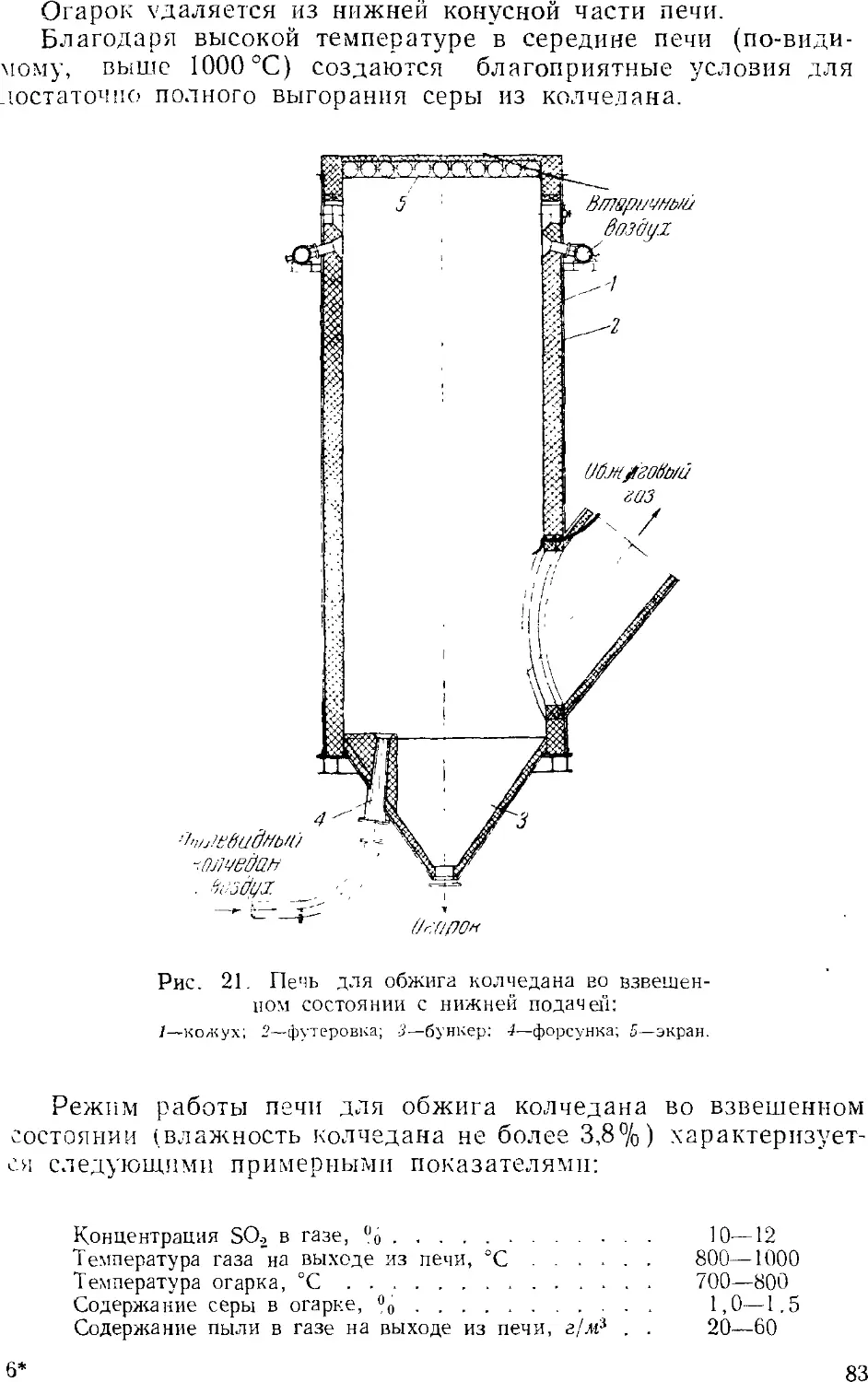
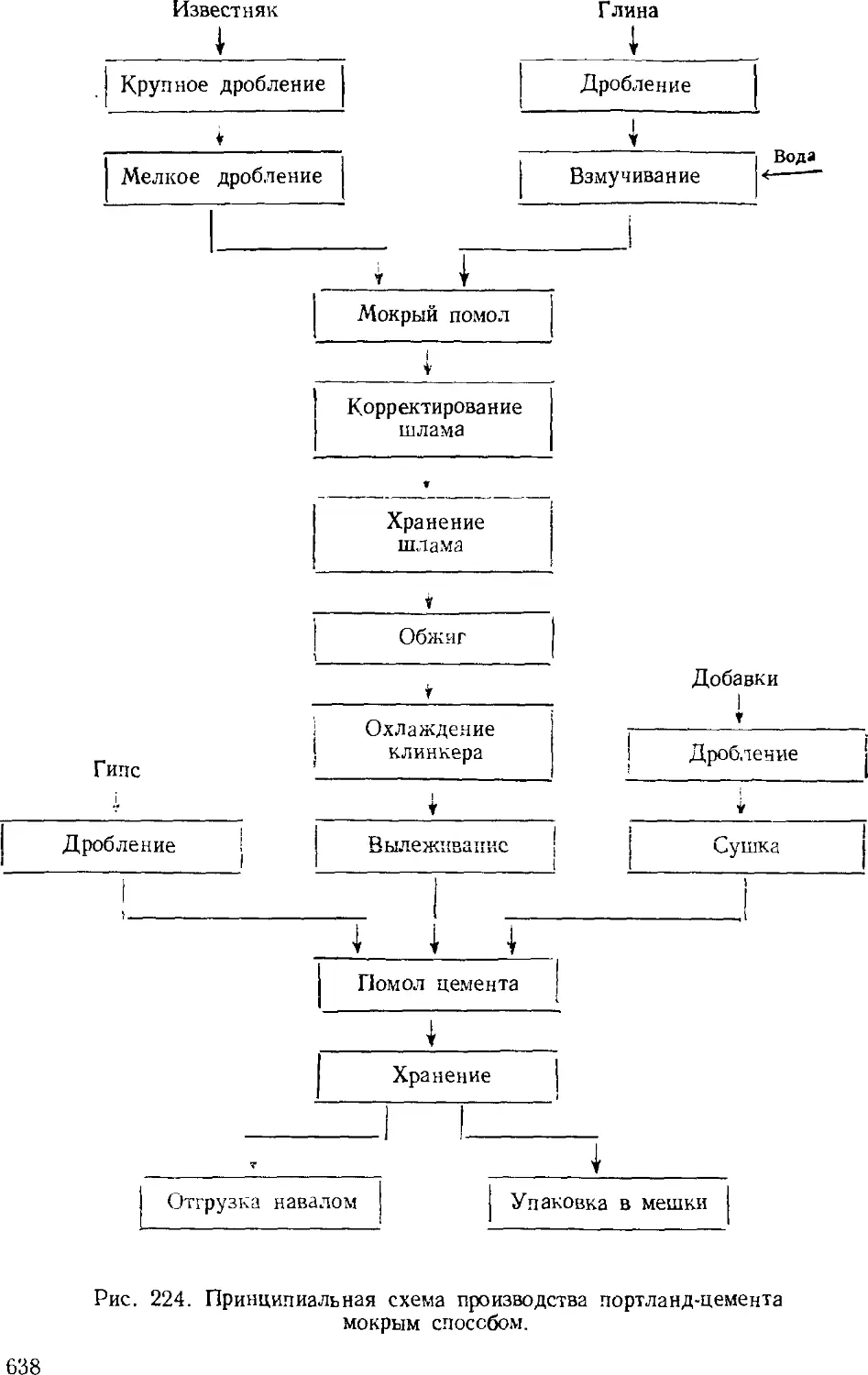
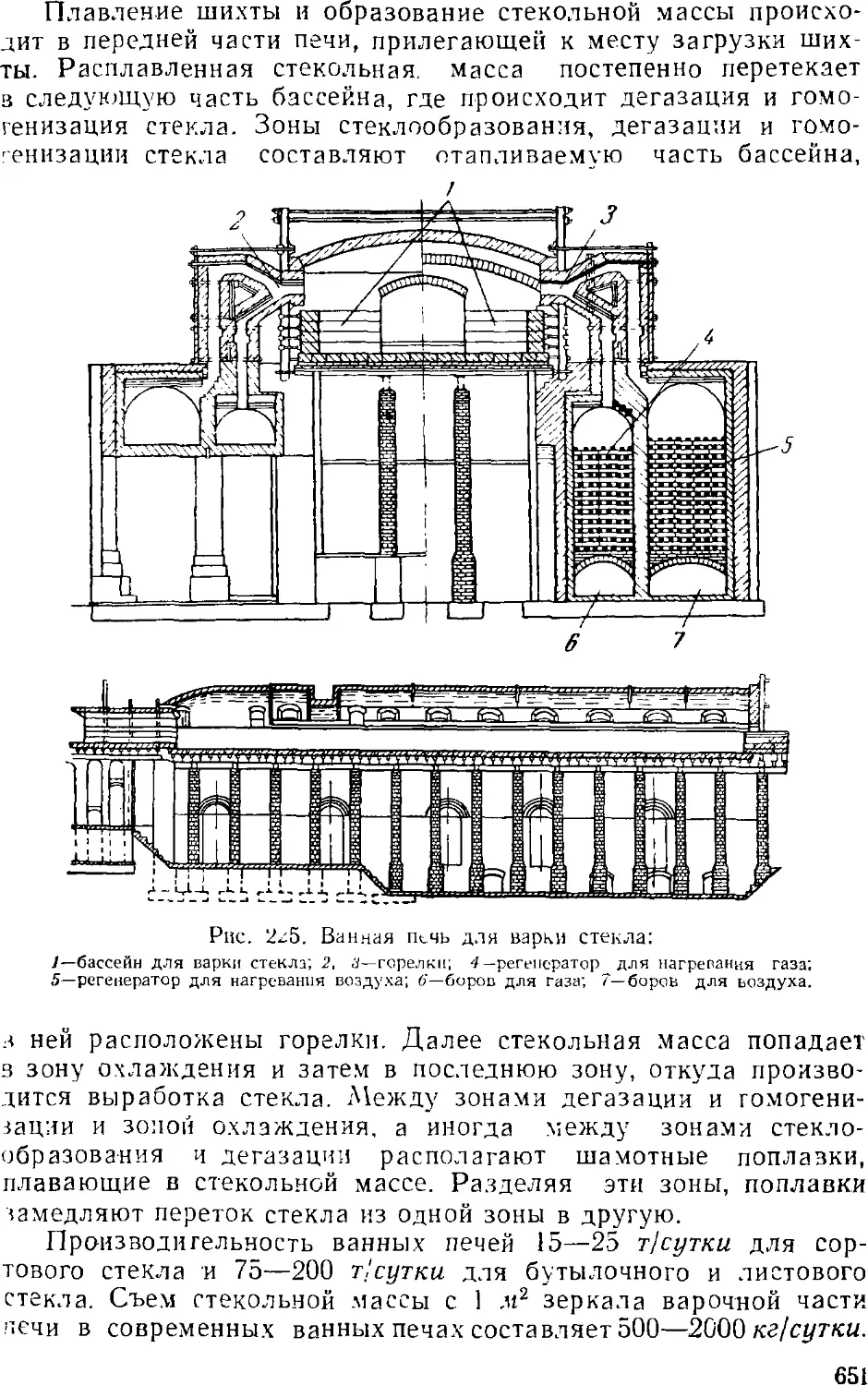
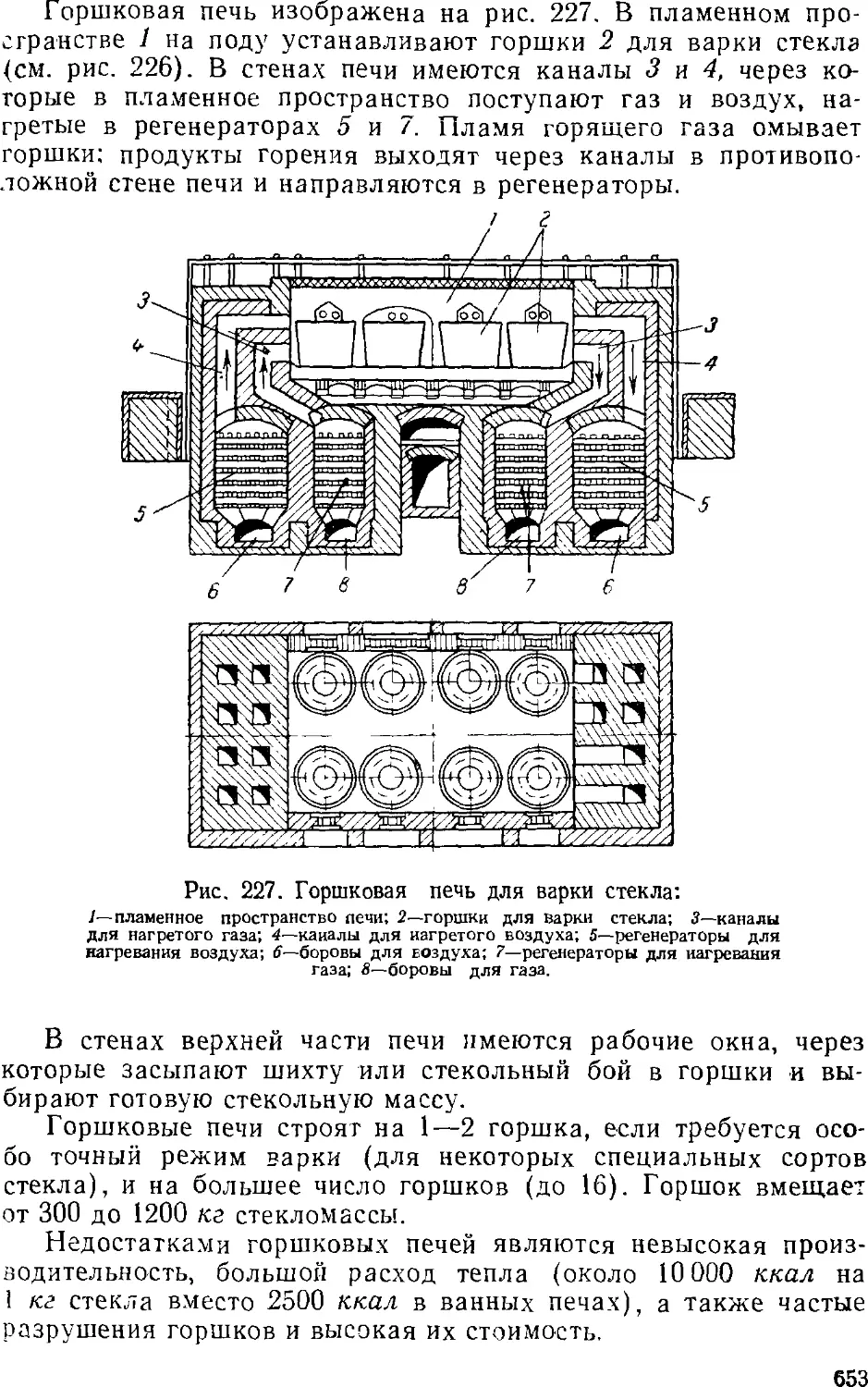
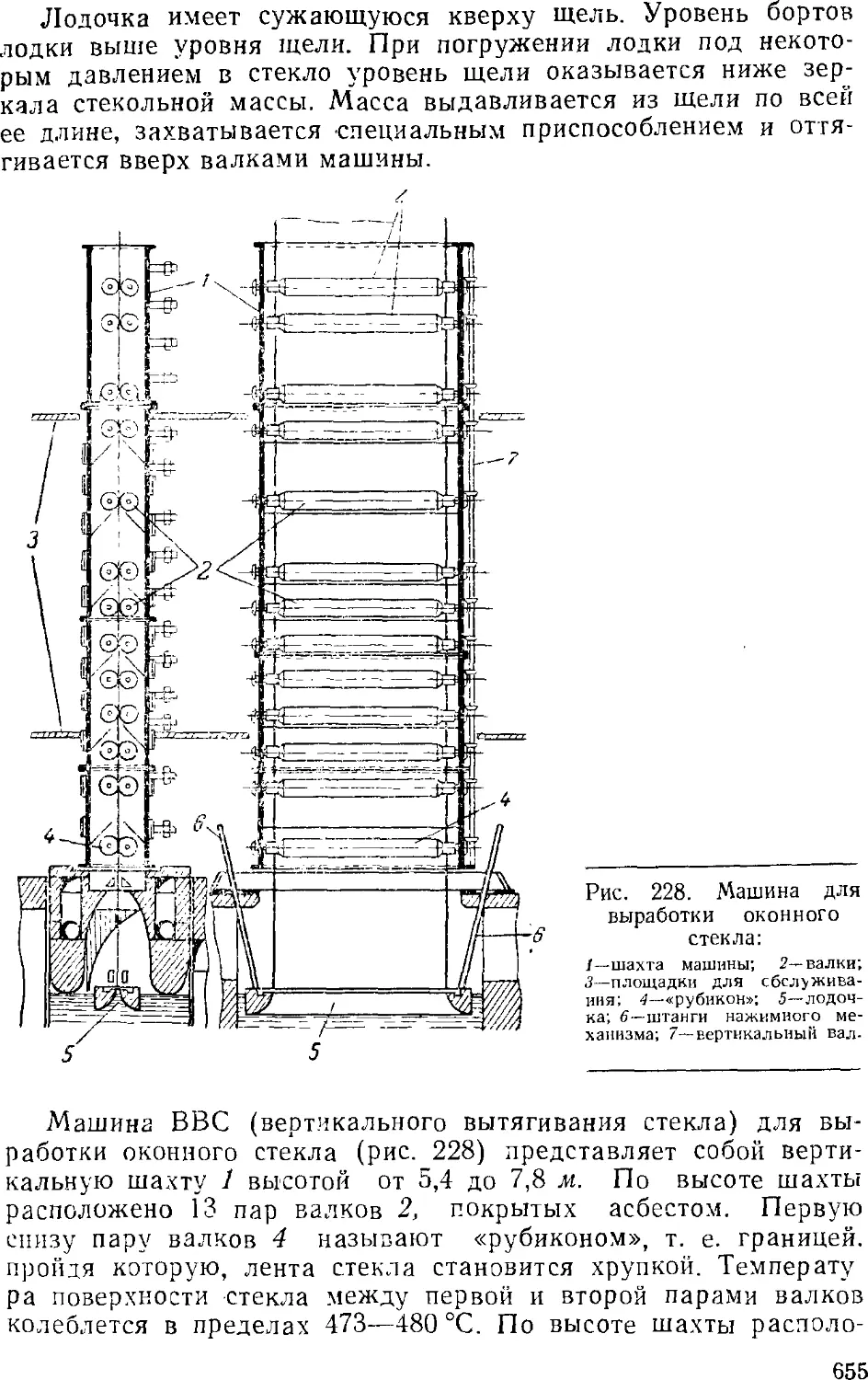
/





















































































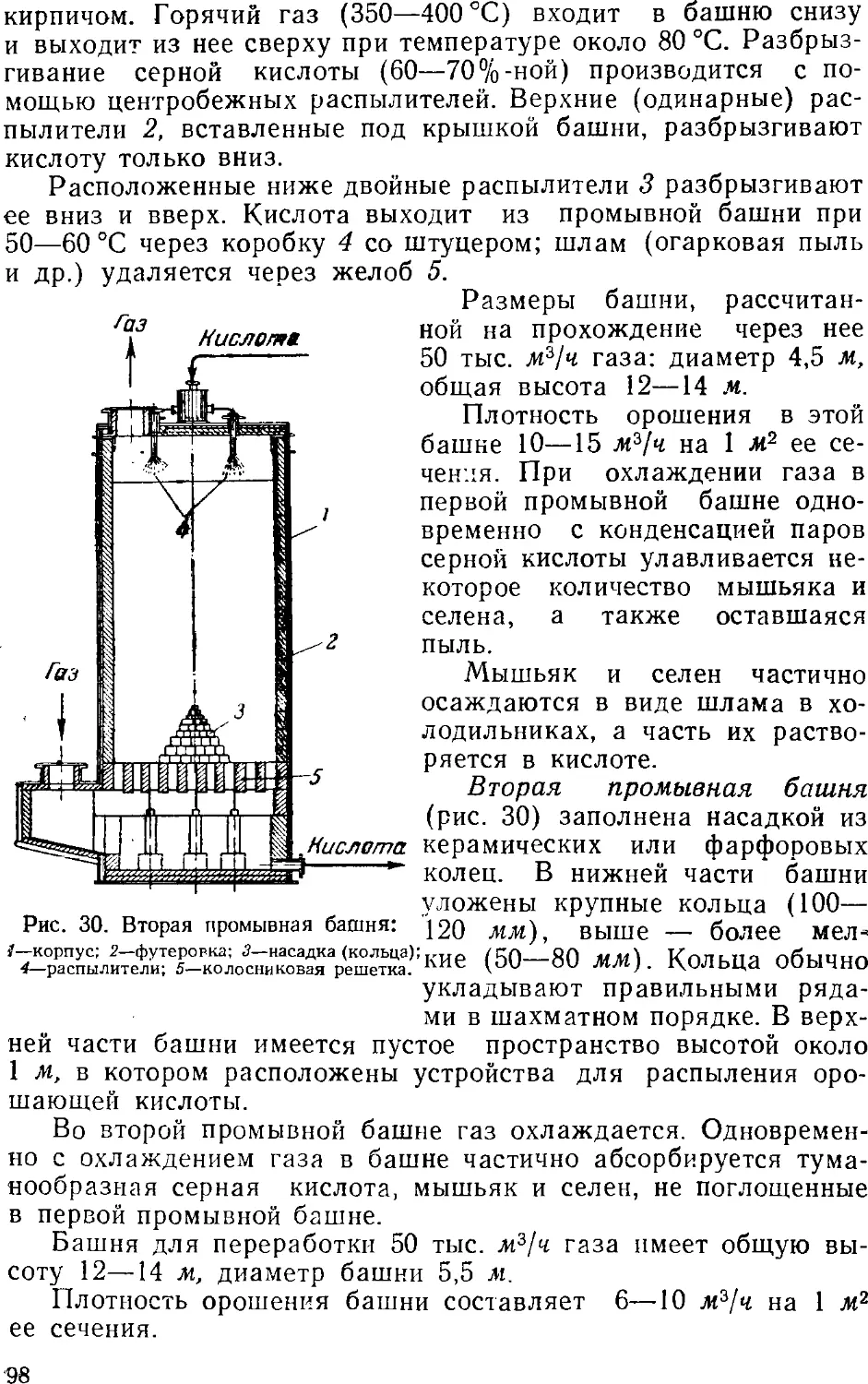

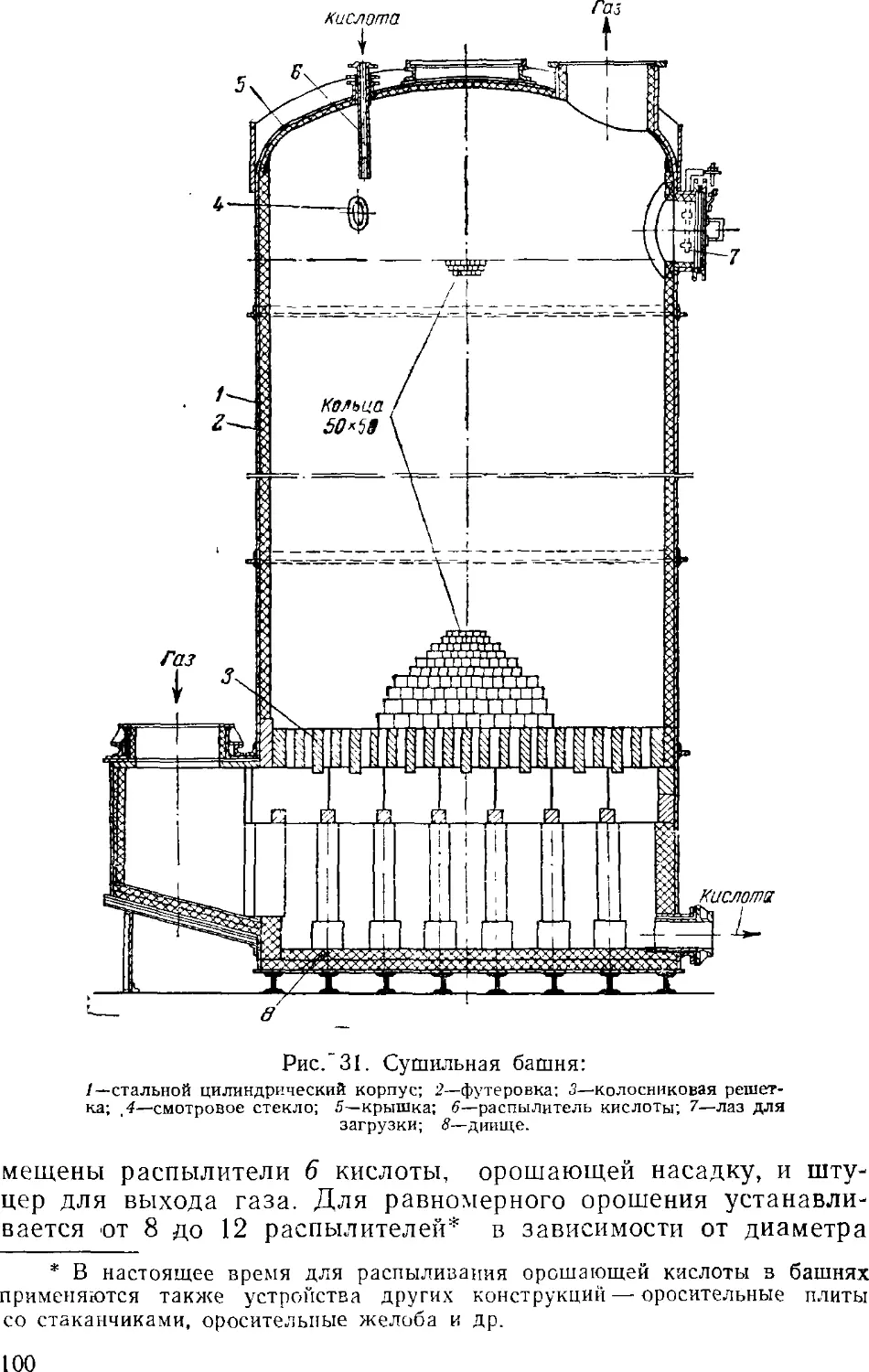



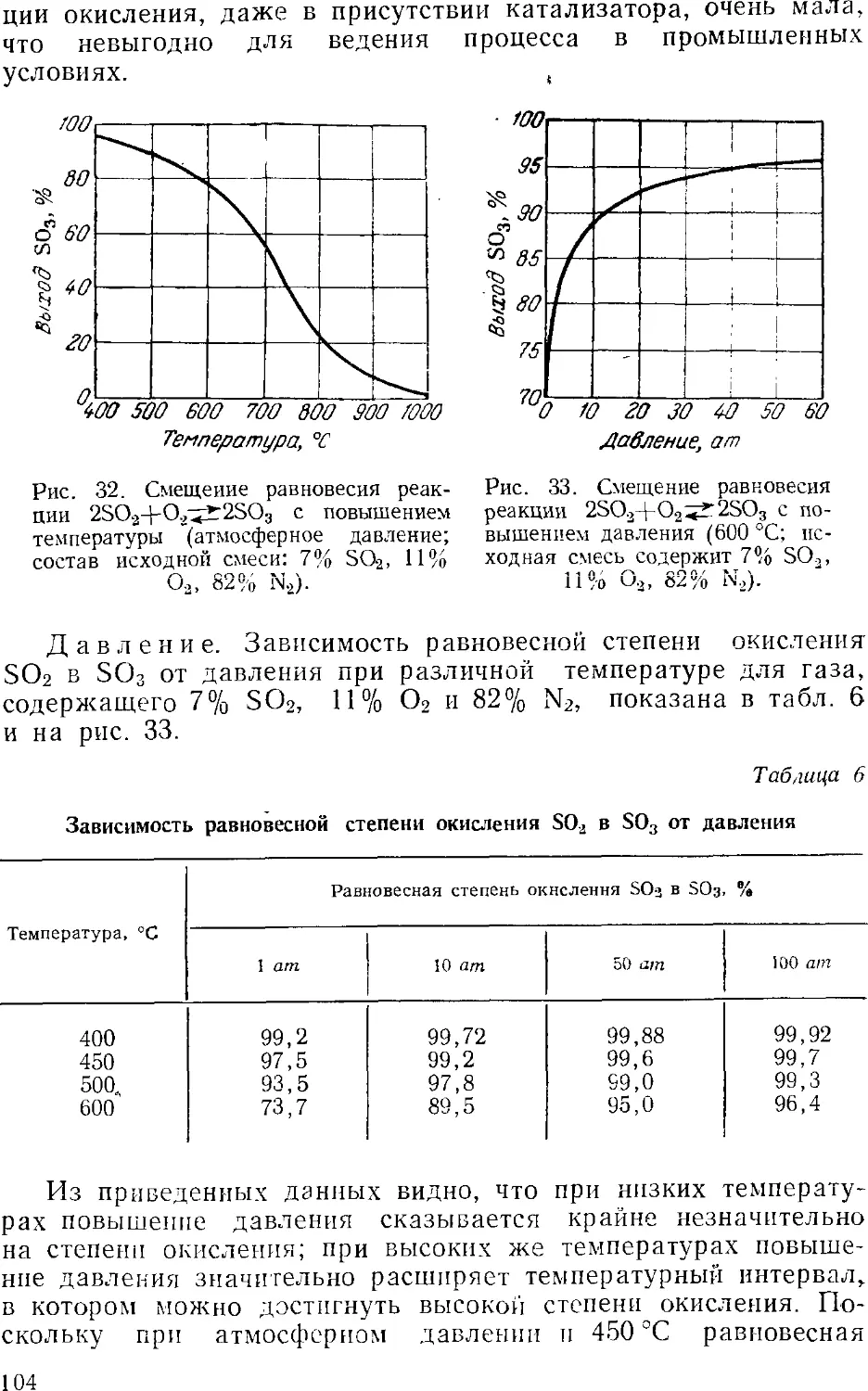




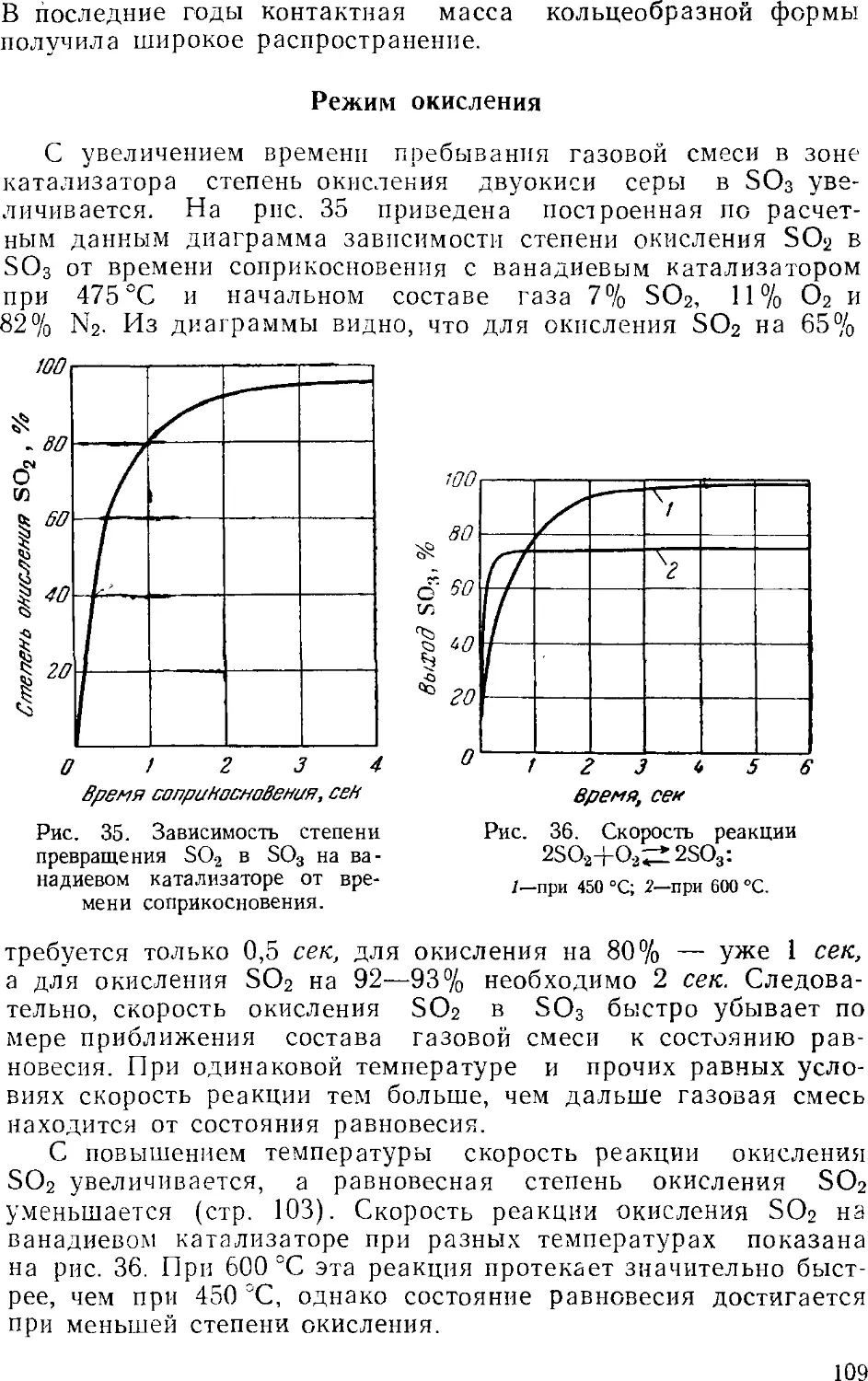




















































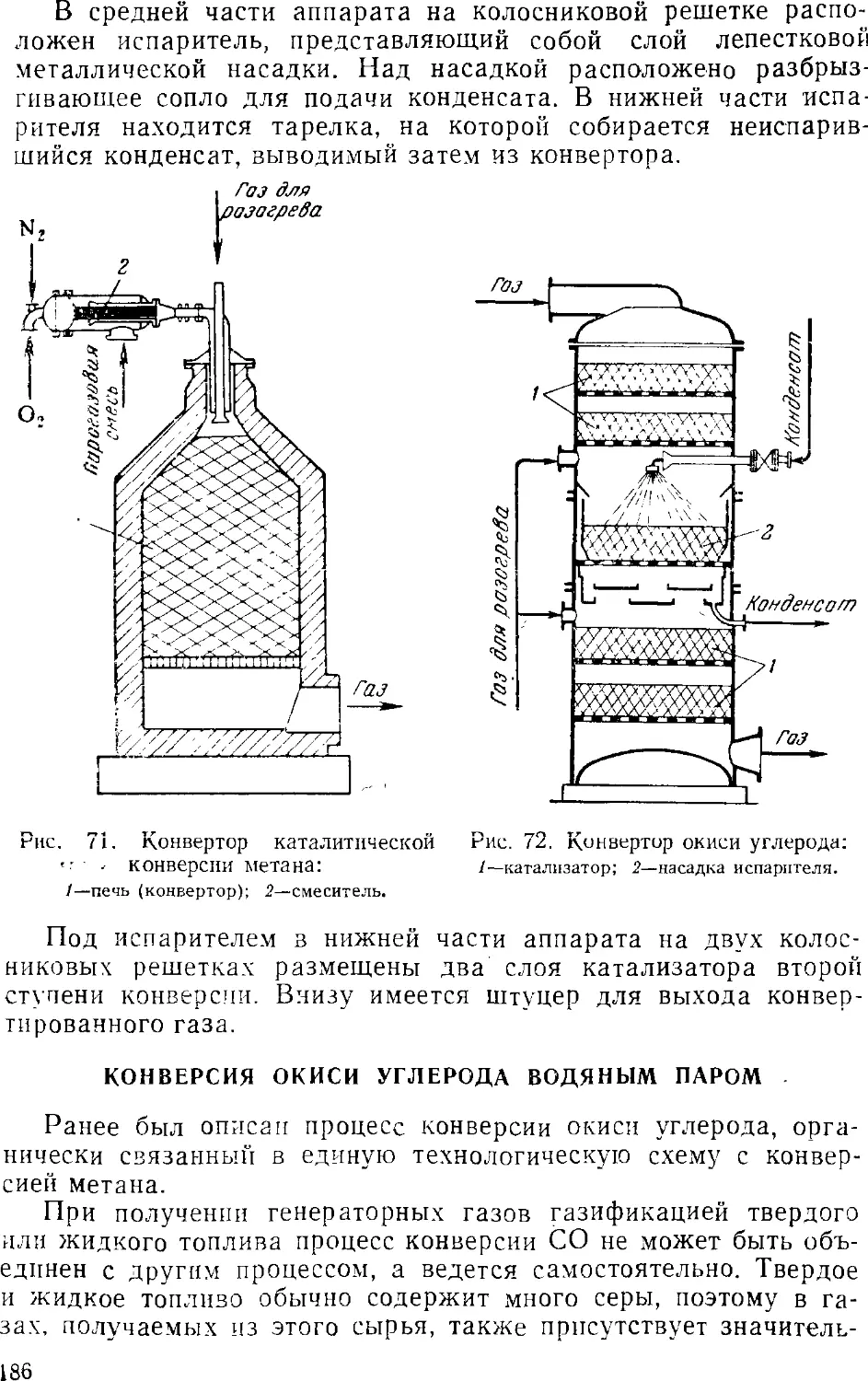


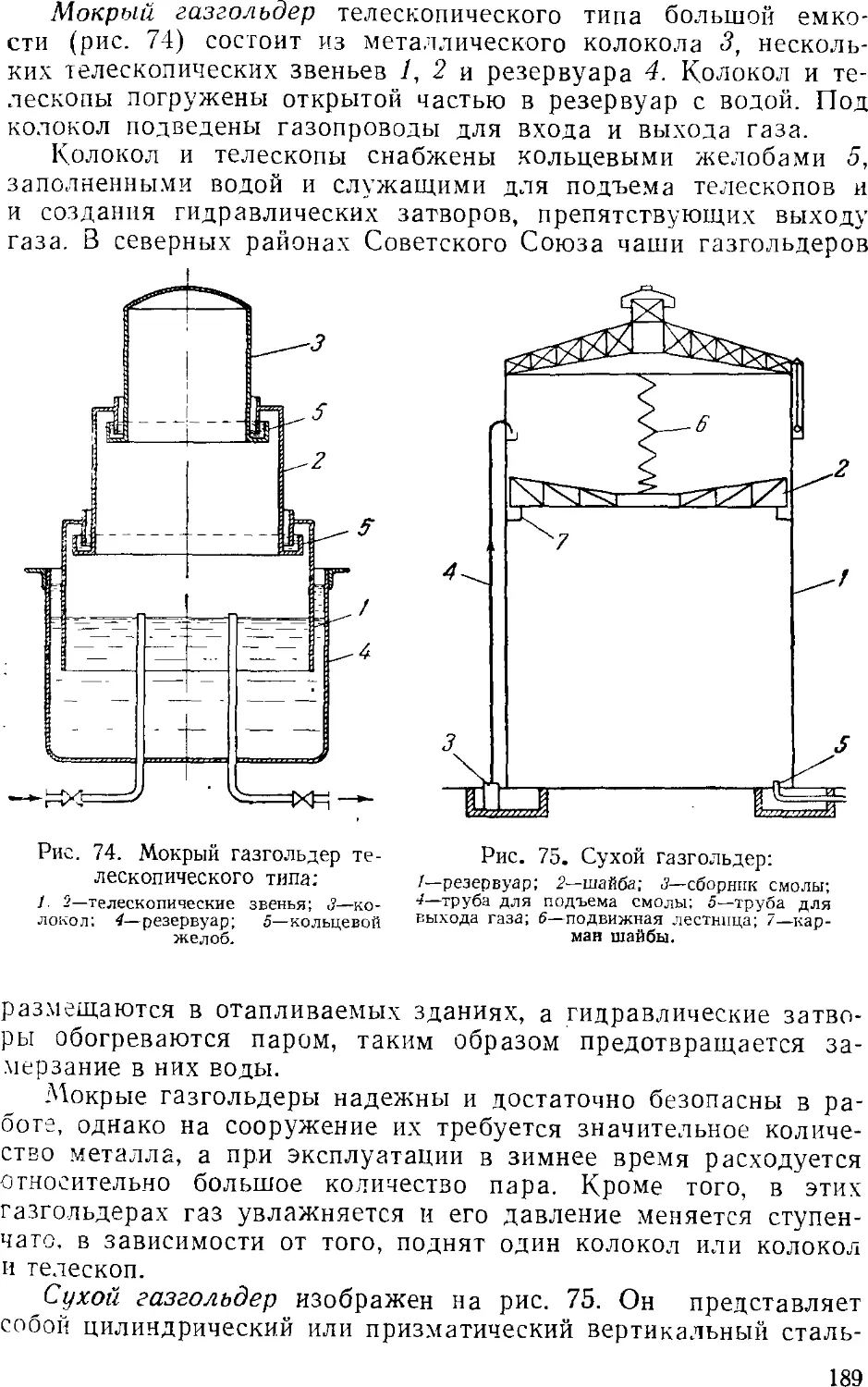



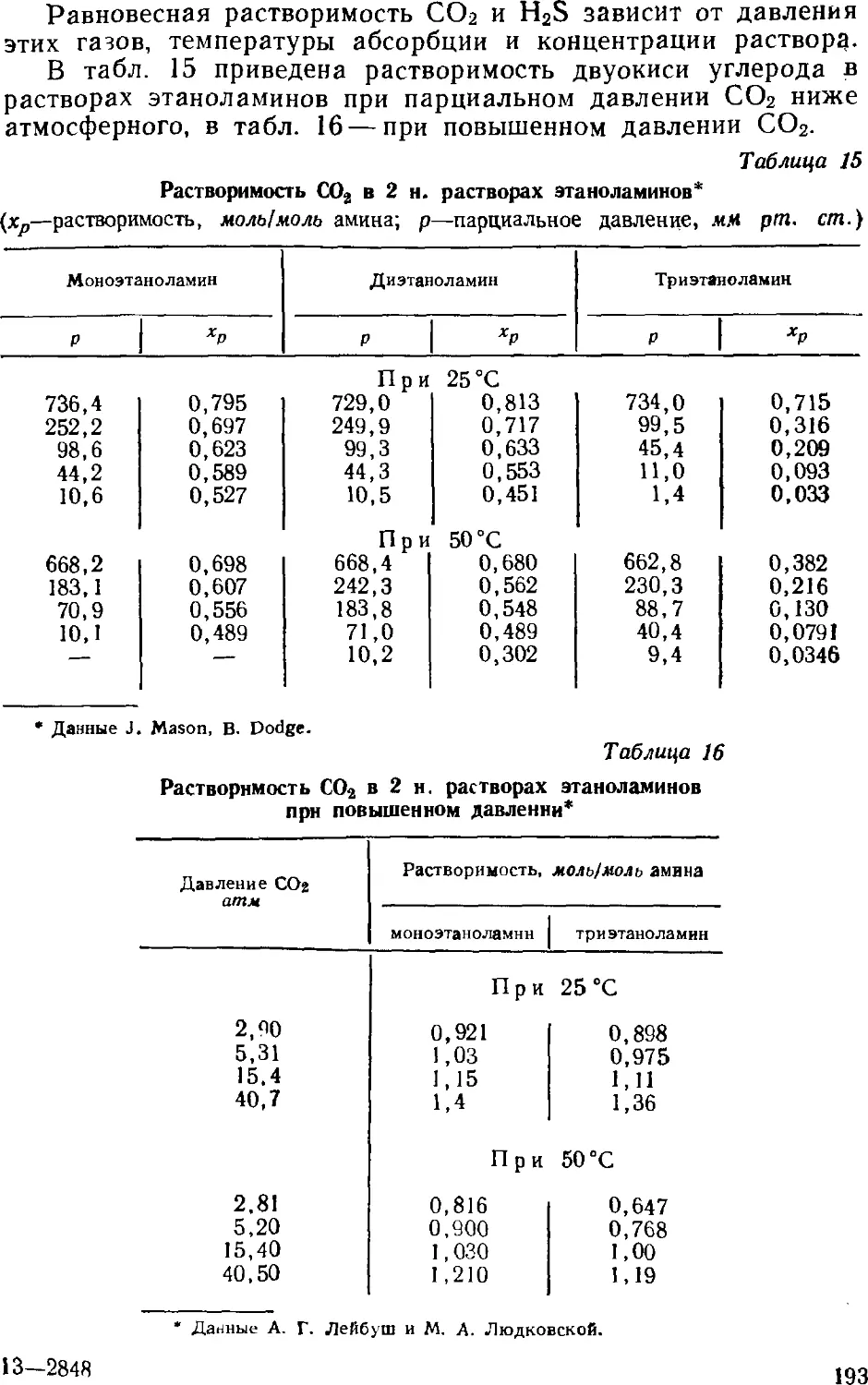


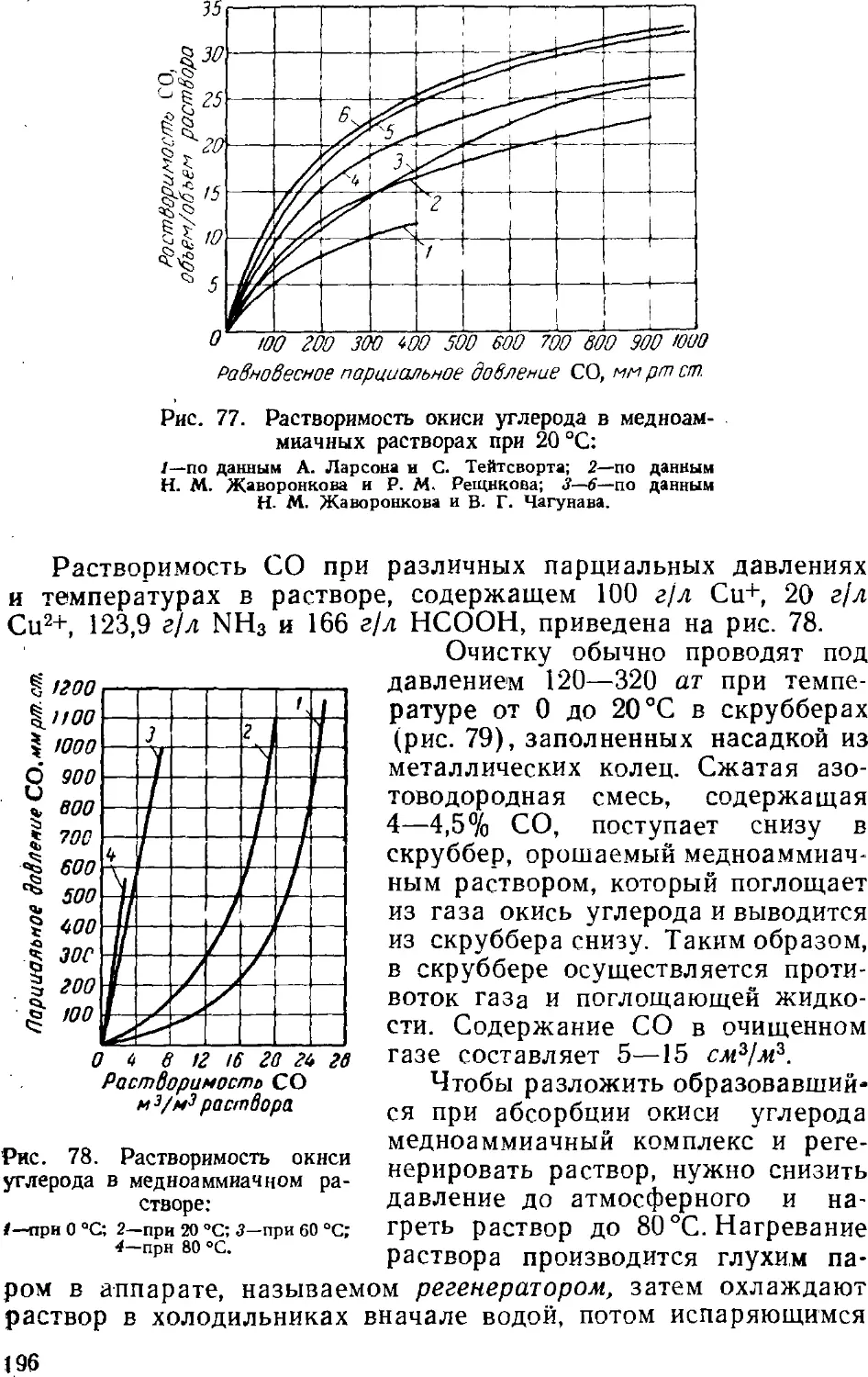








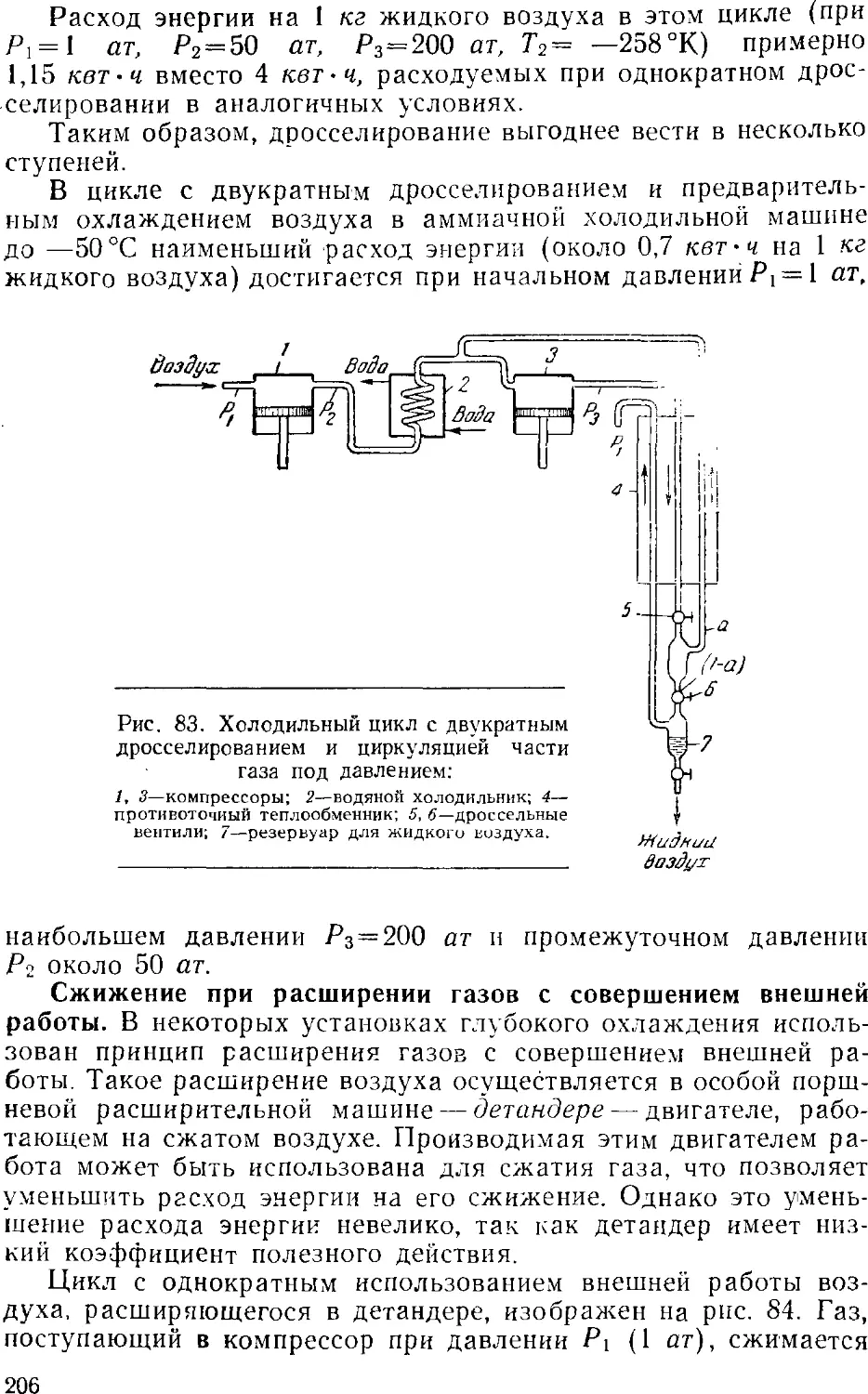






























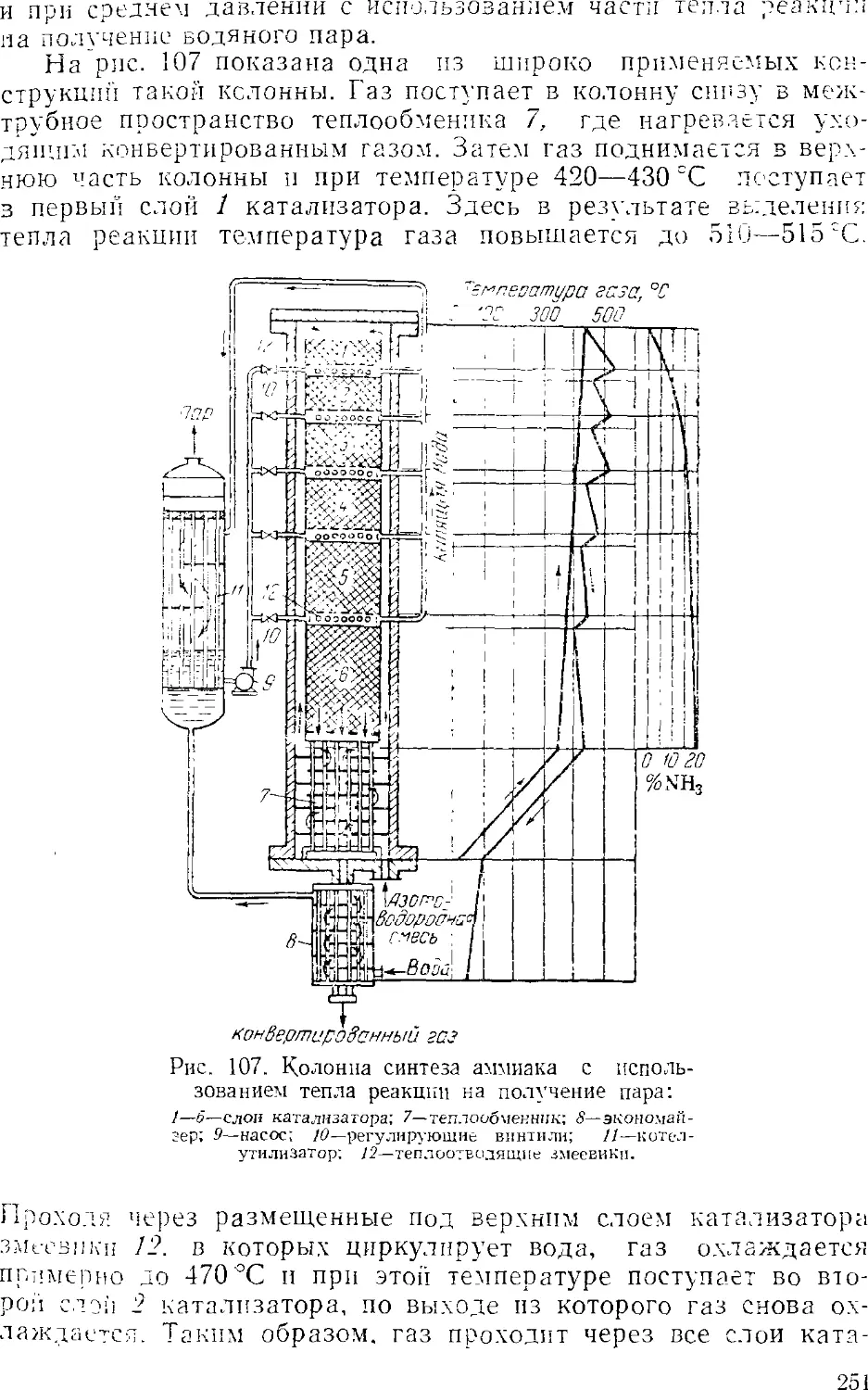


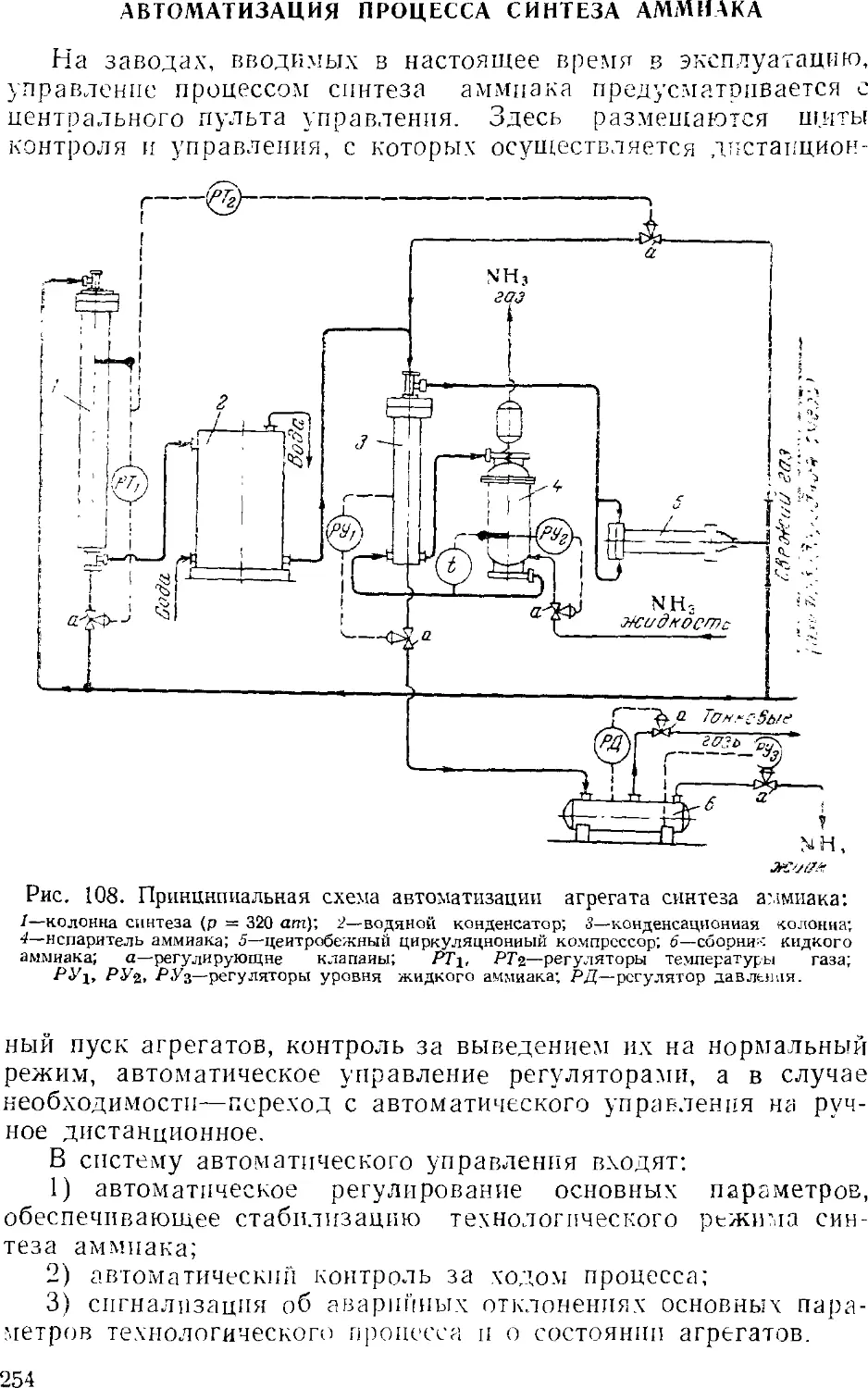





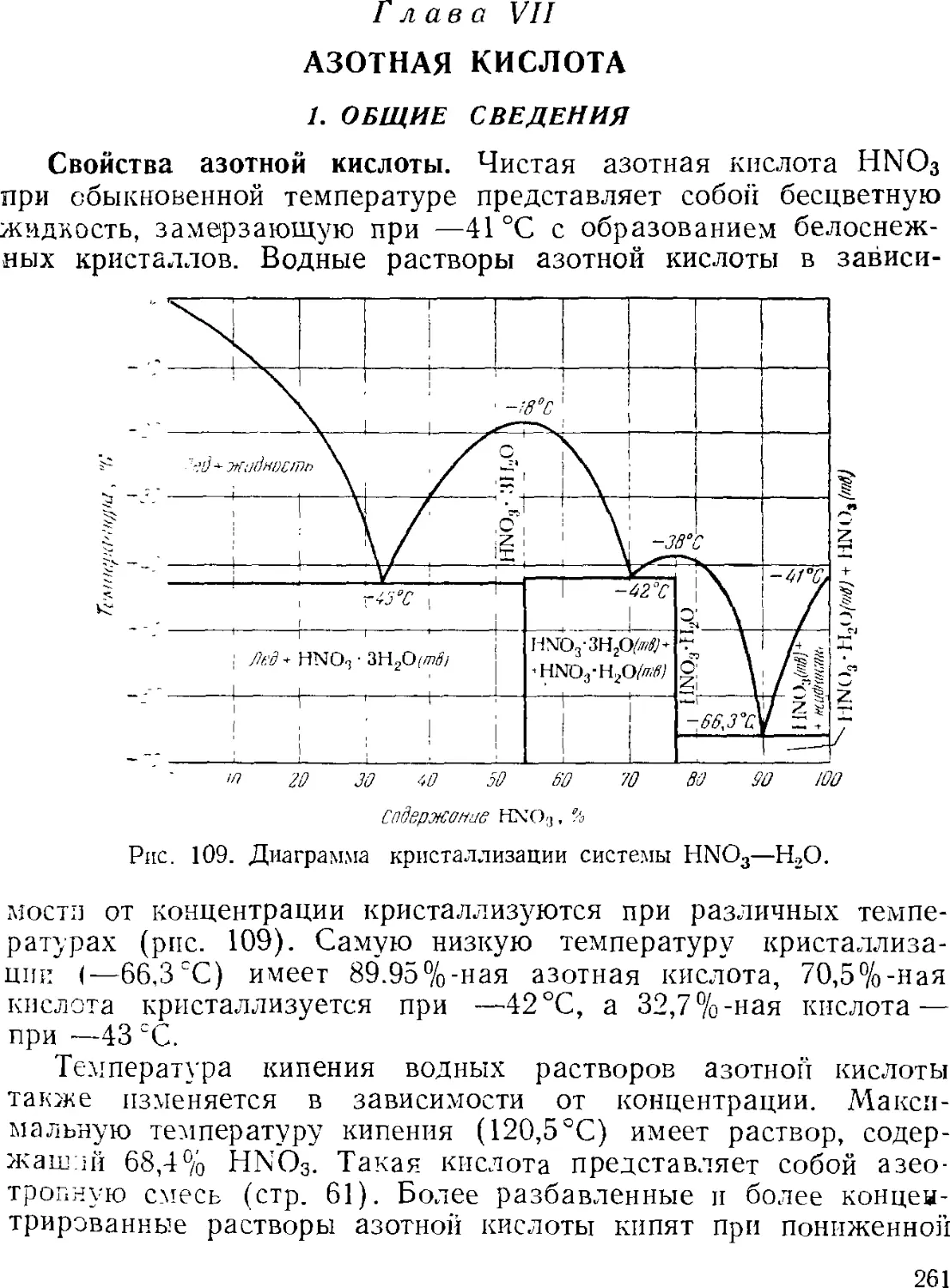













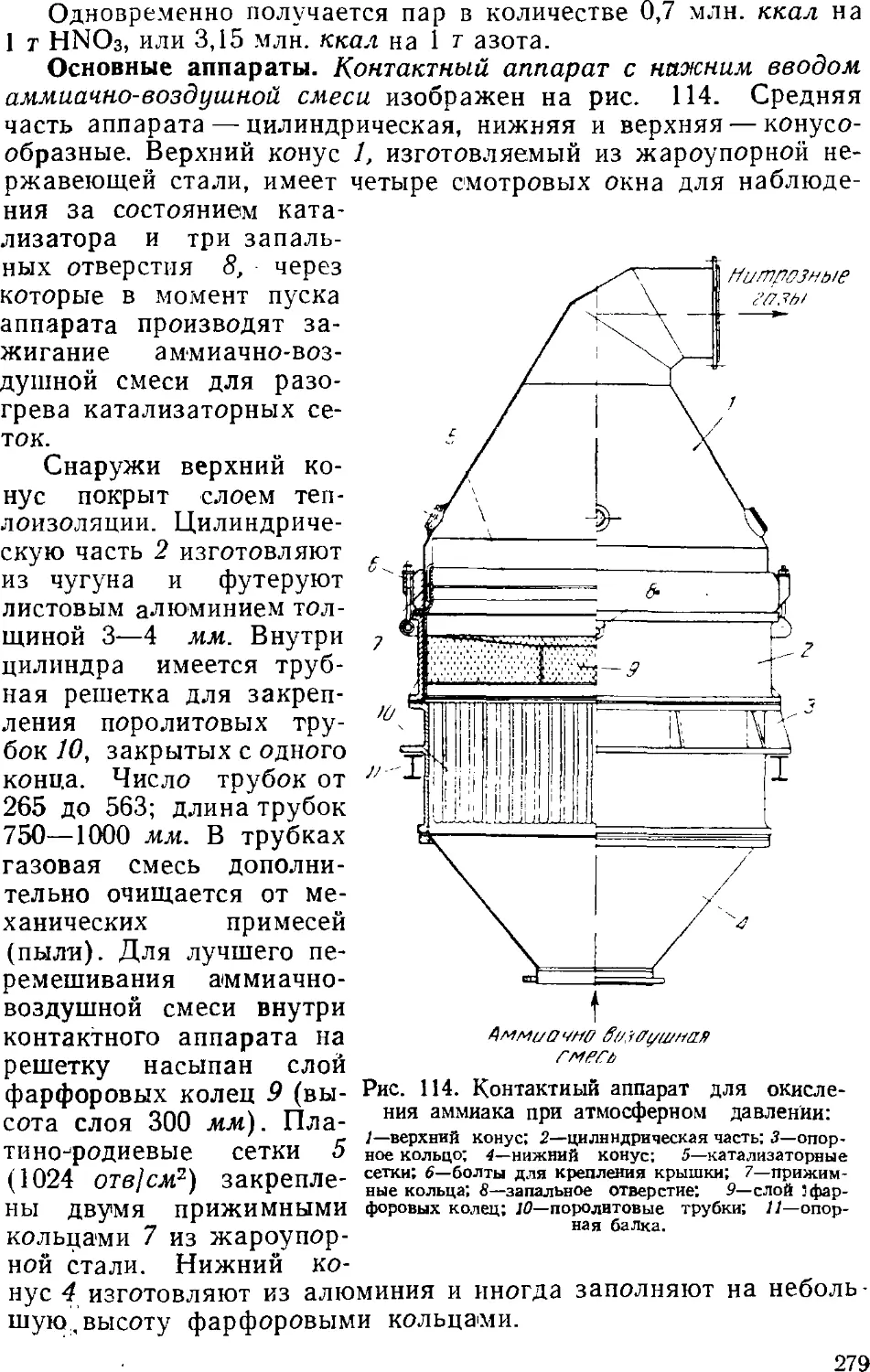

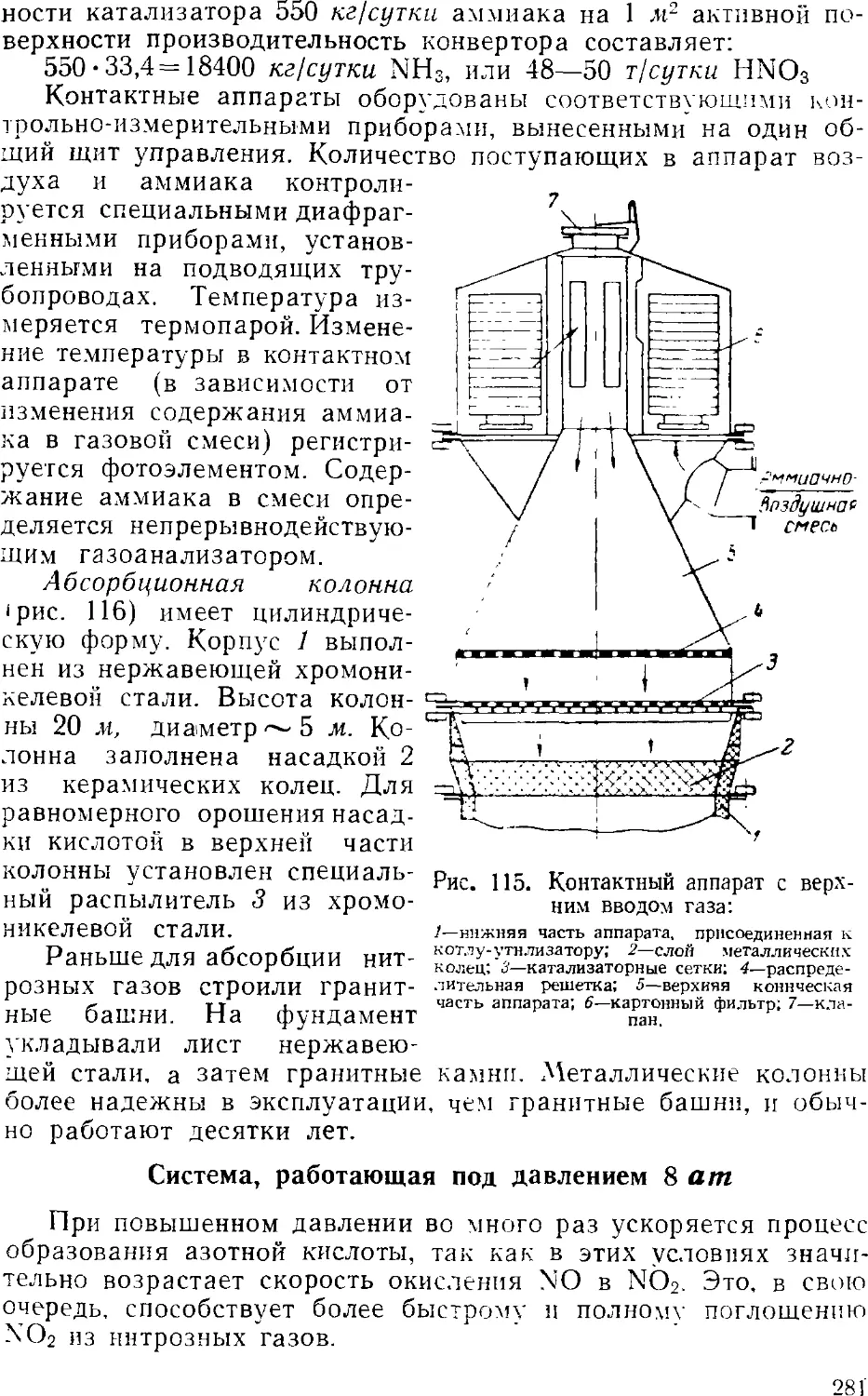
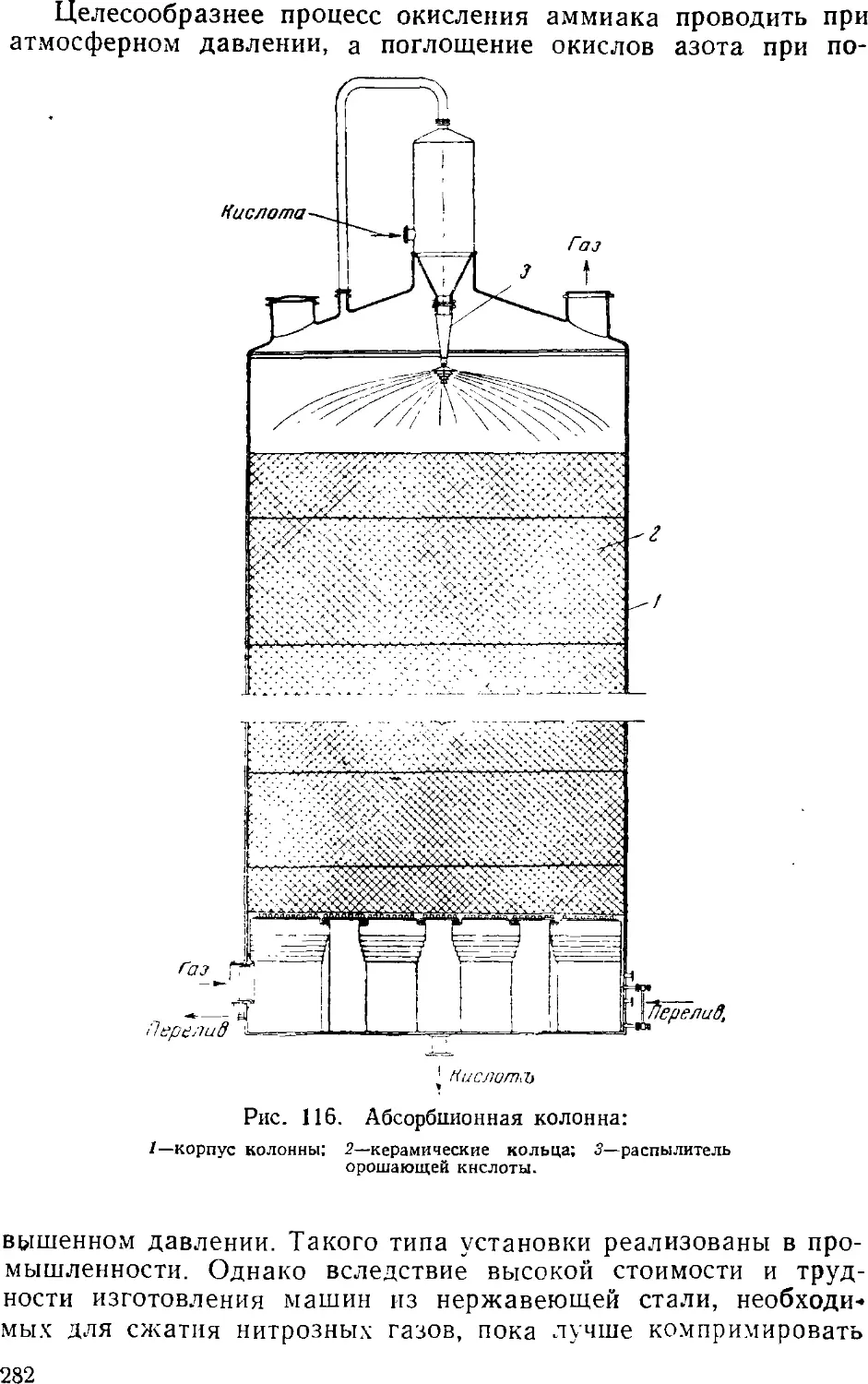

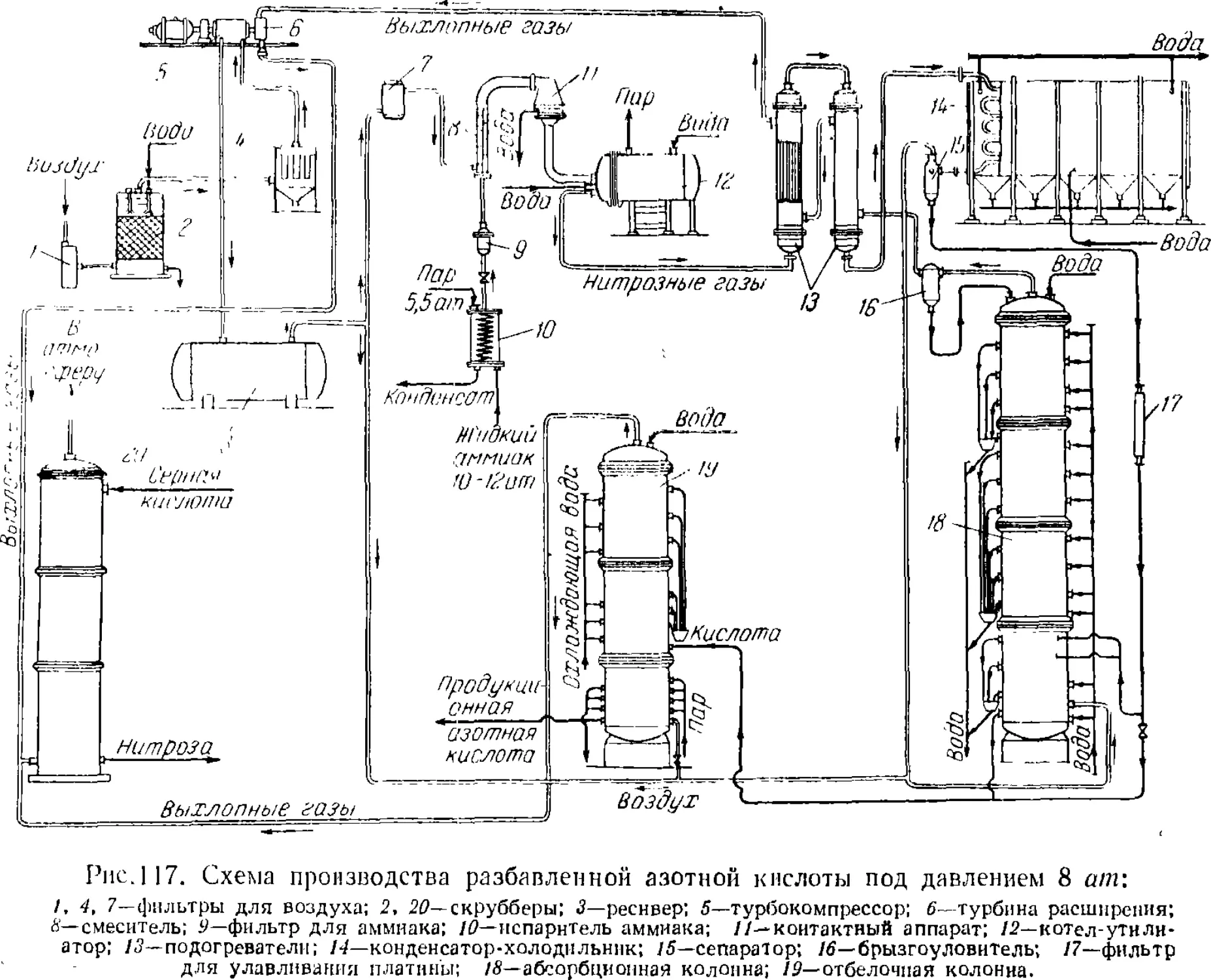































































































































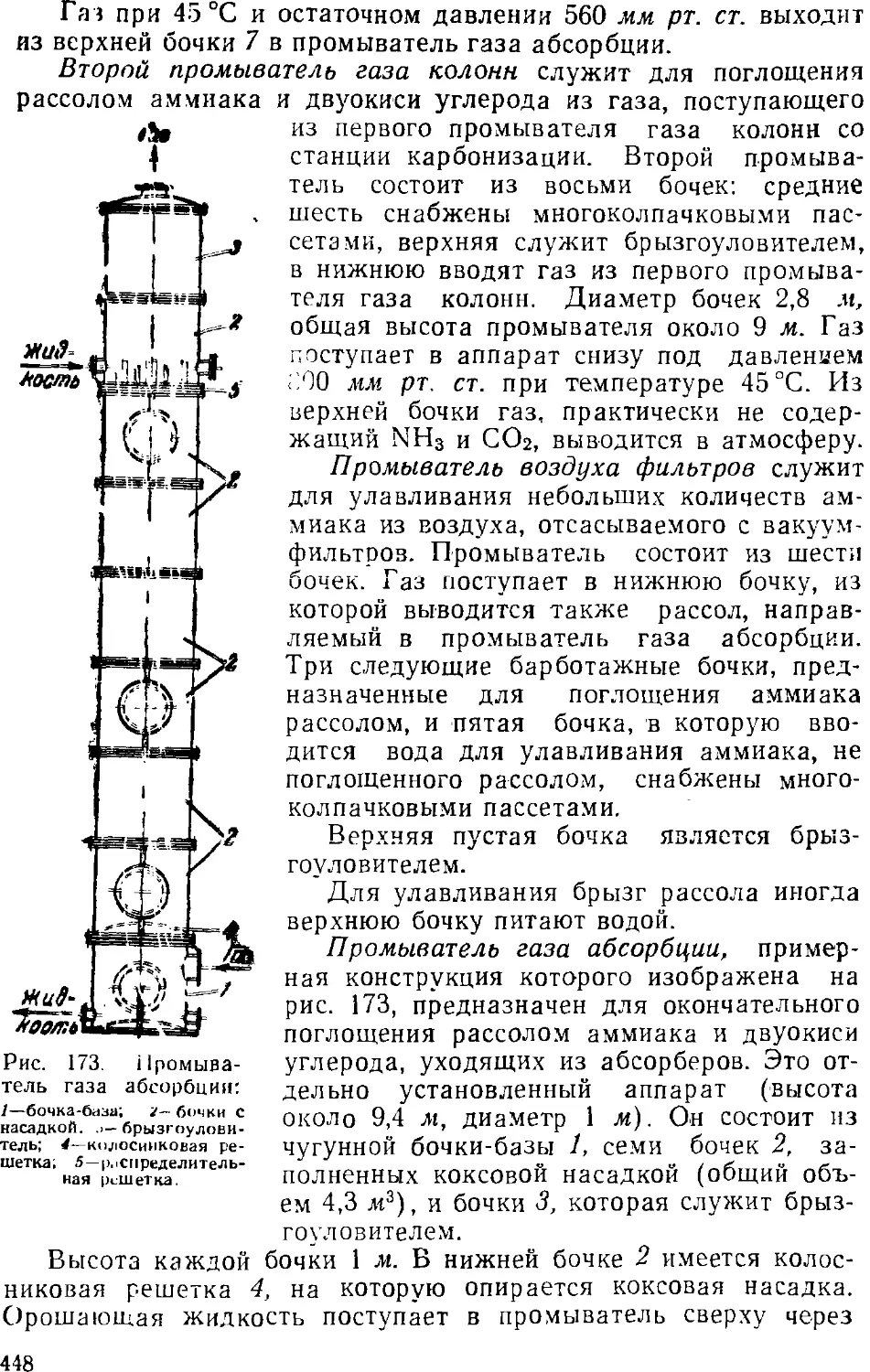

















































































































































































































Text
А. П. ЕГОРОВ, А. И. ШЕРЕШЕВСКИЙ
И. В. ШМАНЕНКОВ
ОБЩАЯ
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
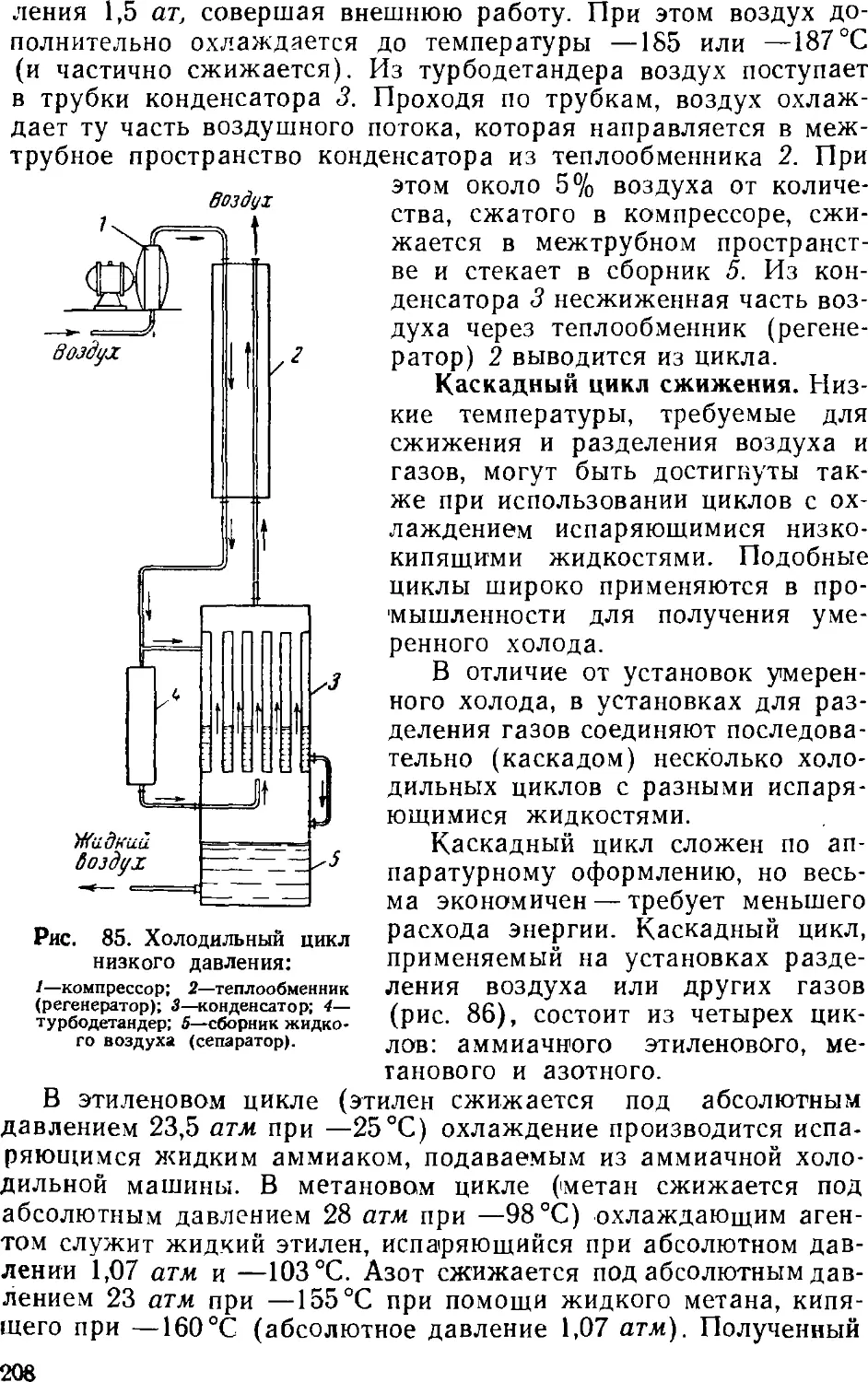
НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
ИЗДАНИЕ ЧЕТВЕРТОЕ, ПЕРЕРАБОТАННОЕ
Допущено Министерством
высшего и среднего специального образования СССР
в качестве учебного пособия
для учащихся техникумов
ИЗДАТЕЛЬСТВО «ХИМИЯ»
Москва 1965 Ленинград
В книге описано производство важнейших неорганических
продуктов (кислот, щелочей, соды, аммиака, минеральных
удобрений, карбида и цианамида кальция) и основных исходных
веществ, применяемых в химической технологии (серы и
сернистого газа, газовых смесей для синтеза аммиака,
электролитического хлора, фосфора). Отдельная глава посвящена
технологии силикатов.
Книга предназначается в качестве учебного пособия для
учащихся химических техникумов. Она может служить также
доступным руководством для широкого круга ииженерно-техни-
ческих работников различных специальностей, желающих
получить краткие сведения о технологии основных неорганических
продуктов.
В четвертом издании материал переработан в соответствии
с современным уровнем химической промышленности и
перспективами ее развития.
Афанасий Петрович Егоров, Абрам Исаакович Шерешевский.
Иван Васильевич Шманенков
Общая химическая технология неорганических веществ (издание четвертое,
переработанное). М., Издательство «Химия», 1965 г.
688 с. УДК 660@75.3)
Редактор Н. С. Абрамова. Техн. редактор В. В. Ксган
Т12268. Подписано к печати 29/ХИ 1964 г. Бумага 60x901/ie=2I,5 бум. л.—43 печ. л.
Уч.-изд. л. 44,67. Заказ 2848 Тираж 10000 экз.- (допечатка) Цена I р. 71 к.
Московская типография № 21 «Главполнграфпрома» Государственного комитета Совета
Министров СССР по печати. Угрешская улица, 12.
СОДЕРЖАНИЕ
Предисловие '
Введение У.
Г лава I. Минеральное сырье 17
Запасы минерального сырья У
Обогащение руд 1!
Испытание минерального сырья 2'
Литература 21
Глава II. Вода 26
1. Общие сведения 2(
2. Водоподготовка 31
Осветление и обесцвечивание воды
Обеззараживание воды
Умягчение и обессоливание воды
Удаление газов из воды
Предотвращение накипи
3. Обезвреживание сточных вод
Литература
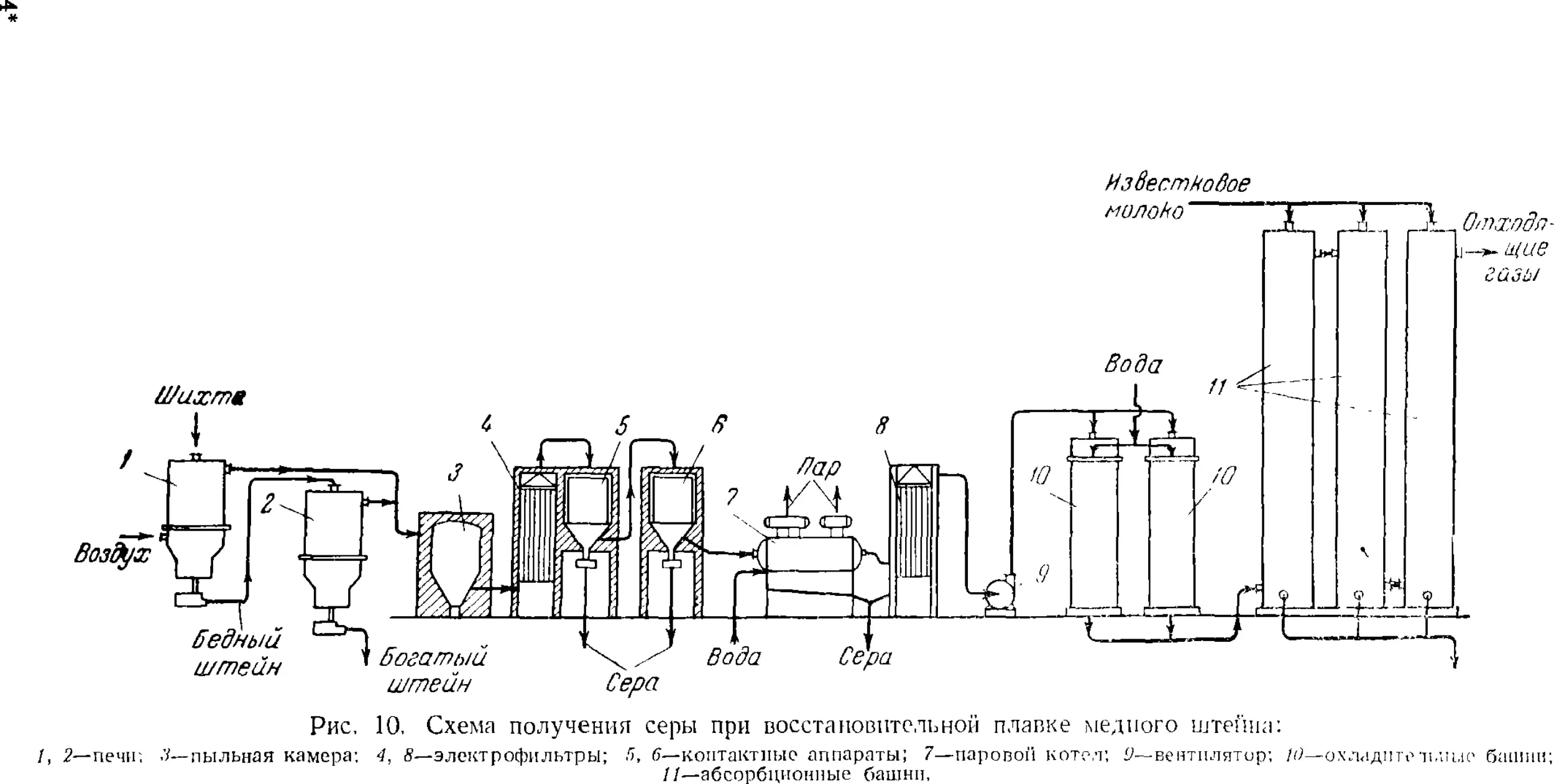
Глава III. Сера ¦
1. Общие сведения
2. Получение серы
Получение серы из самородных руд
Получение серы из сульфидных руд
Извлечение серы из газов
3. Очистка серы
Глава IV. Серная кислота
1. Общие сведения .
Методы производства серной кислоты
Сырье для производства серной кислоты
Технологический процесс производства серной кислоты .
2. Получение сернистого газа
Получение сернистого газа из колчедана
Колчеданные печи
Получение сернистого газа сжиганием серы
Методы очистки сернистого газа от пыли
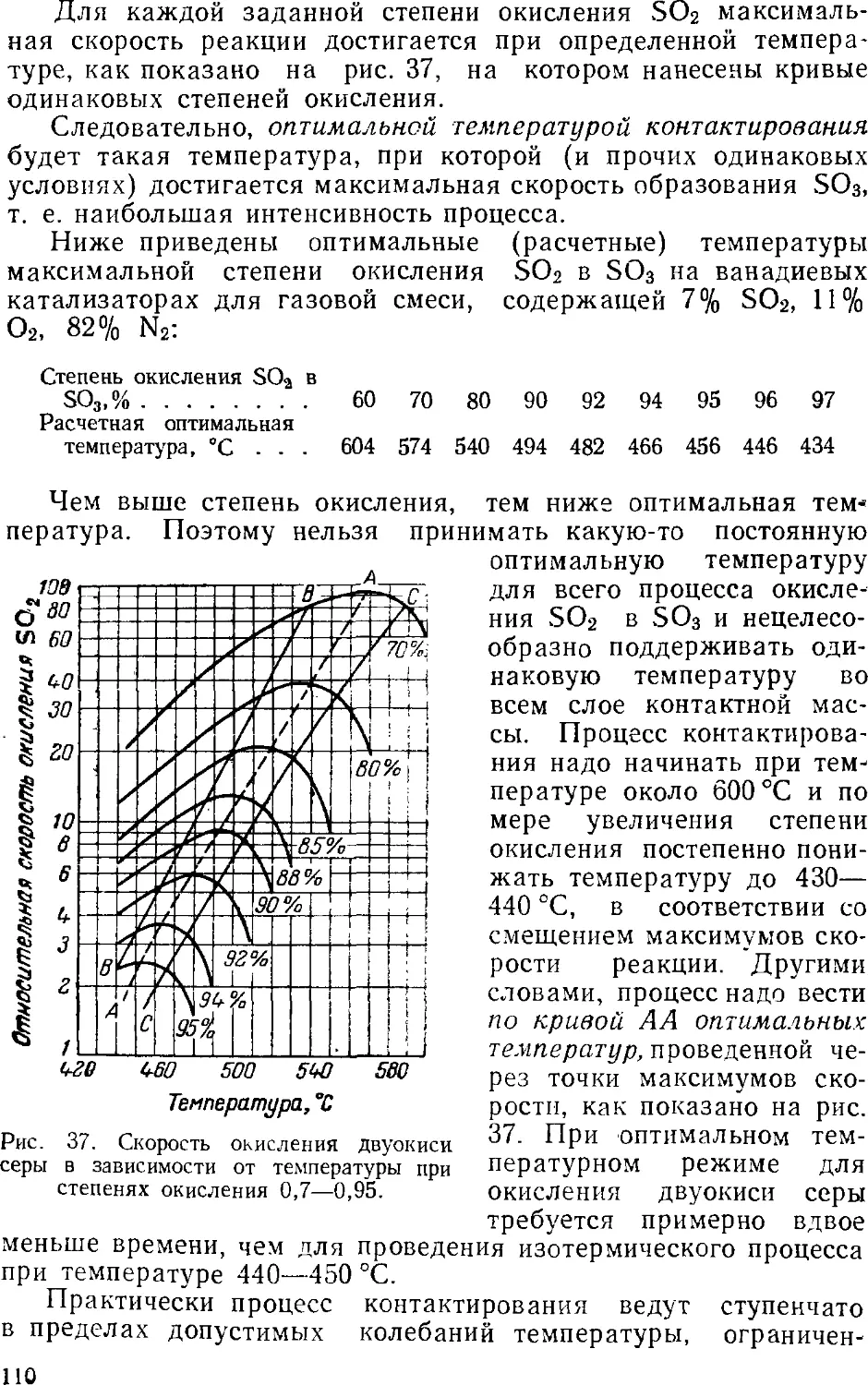
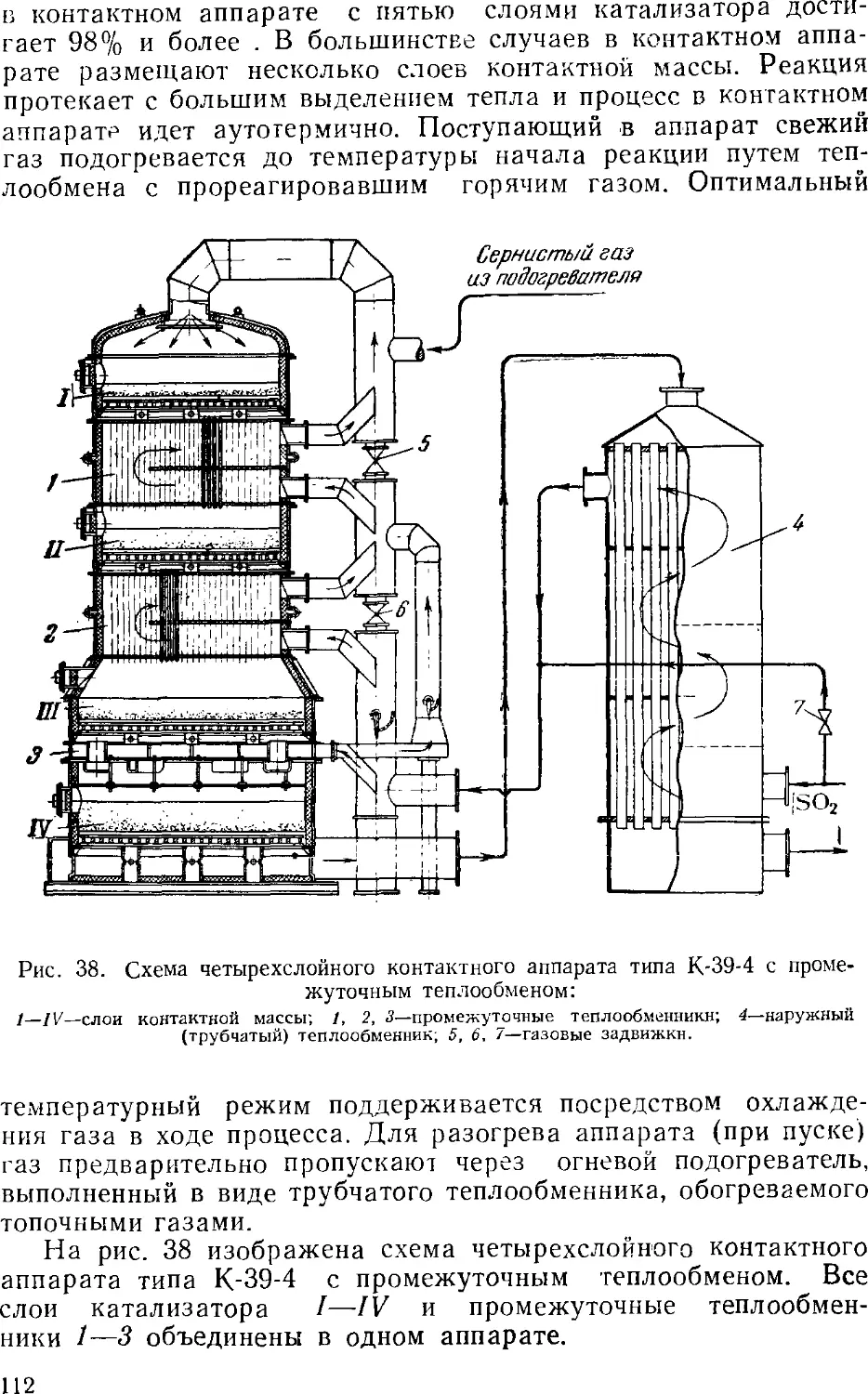
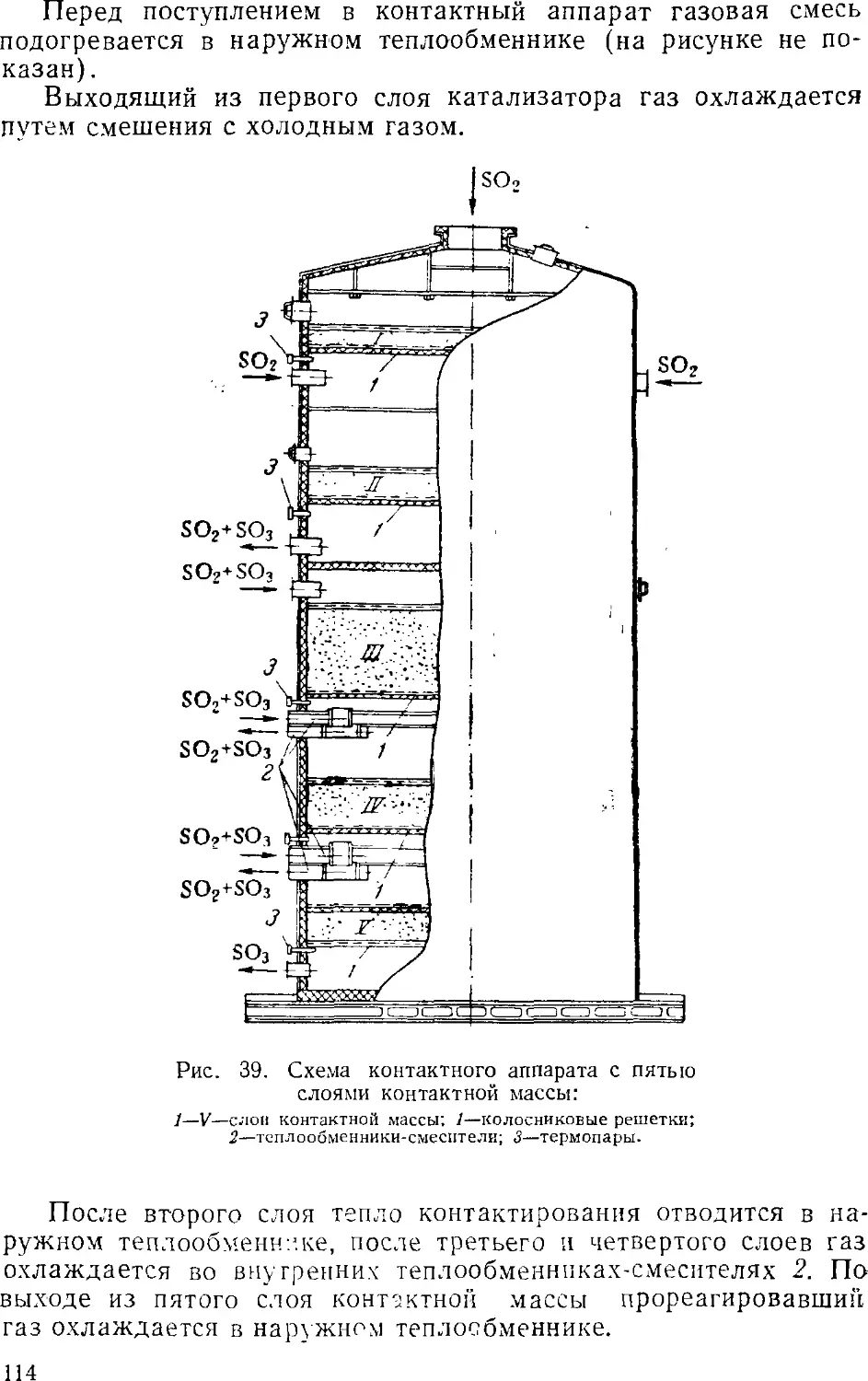
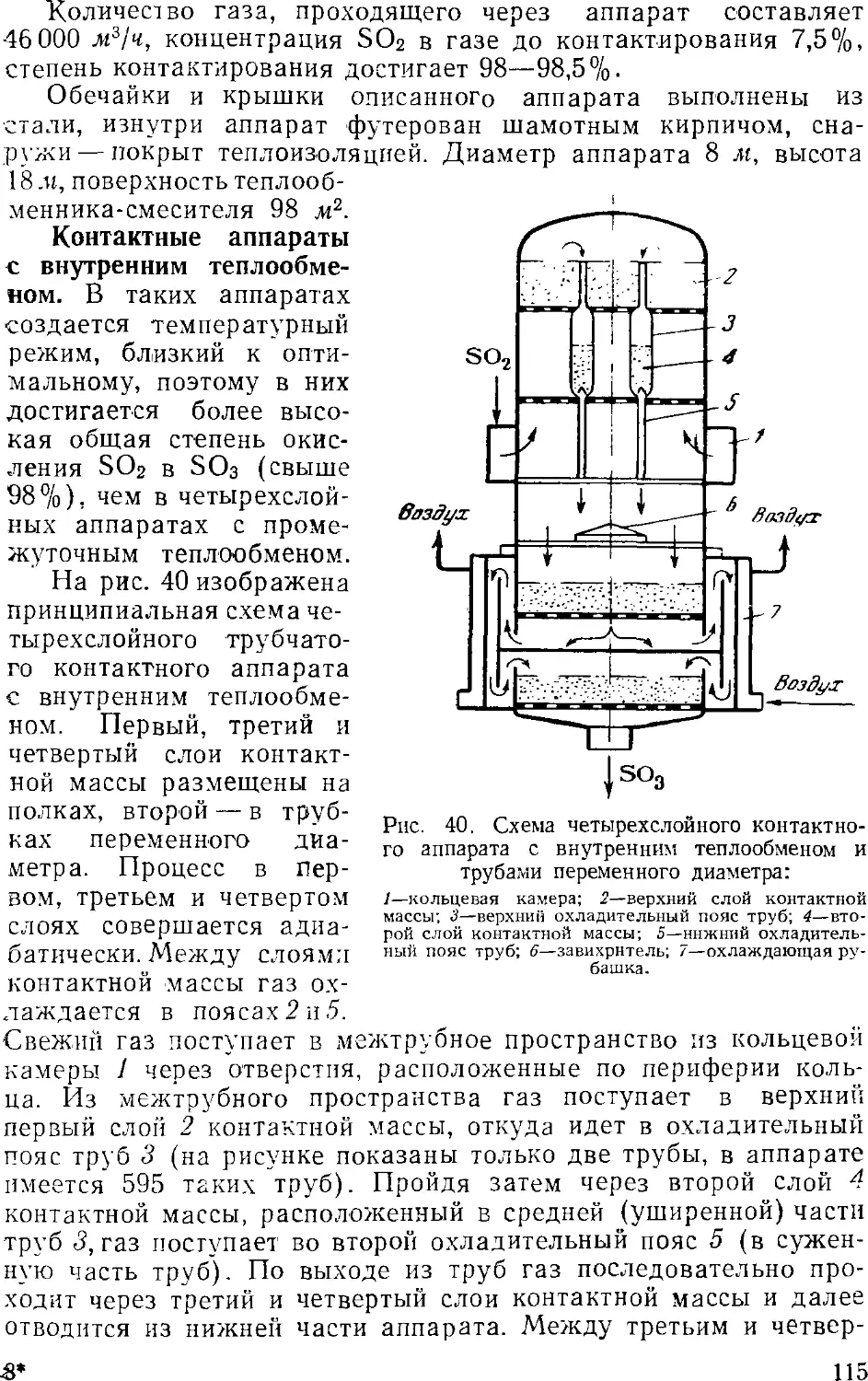
s 3. Контактный метод производства серной кислоты
Принципиальная схема производства
Очистка сернистого газа
Осушка сернистого газа
Контактное окисление SO2 в SO3
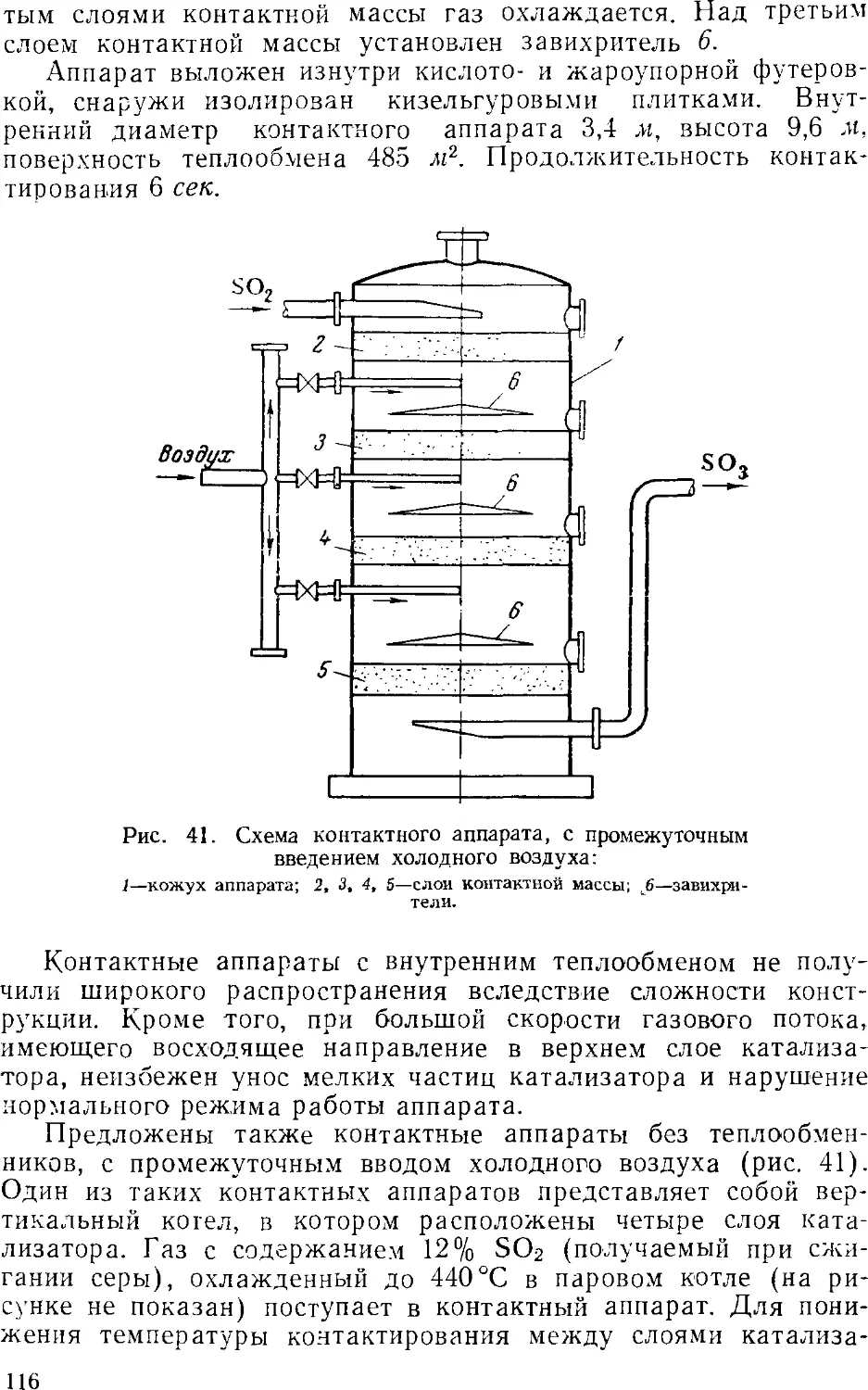
Контактные аппараты
Абсорбция серного ангидрида
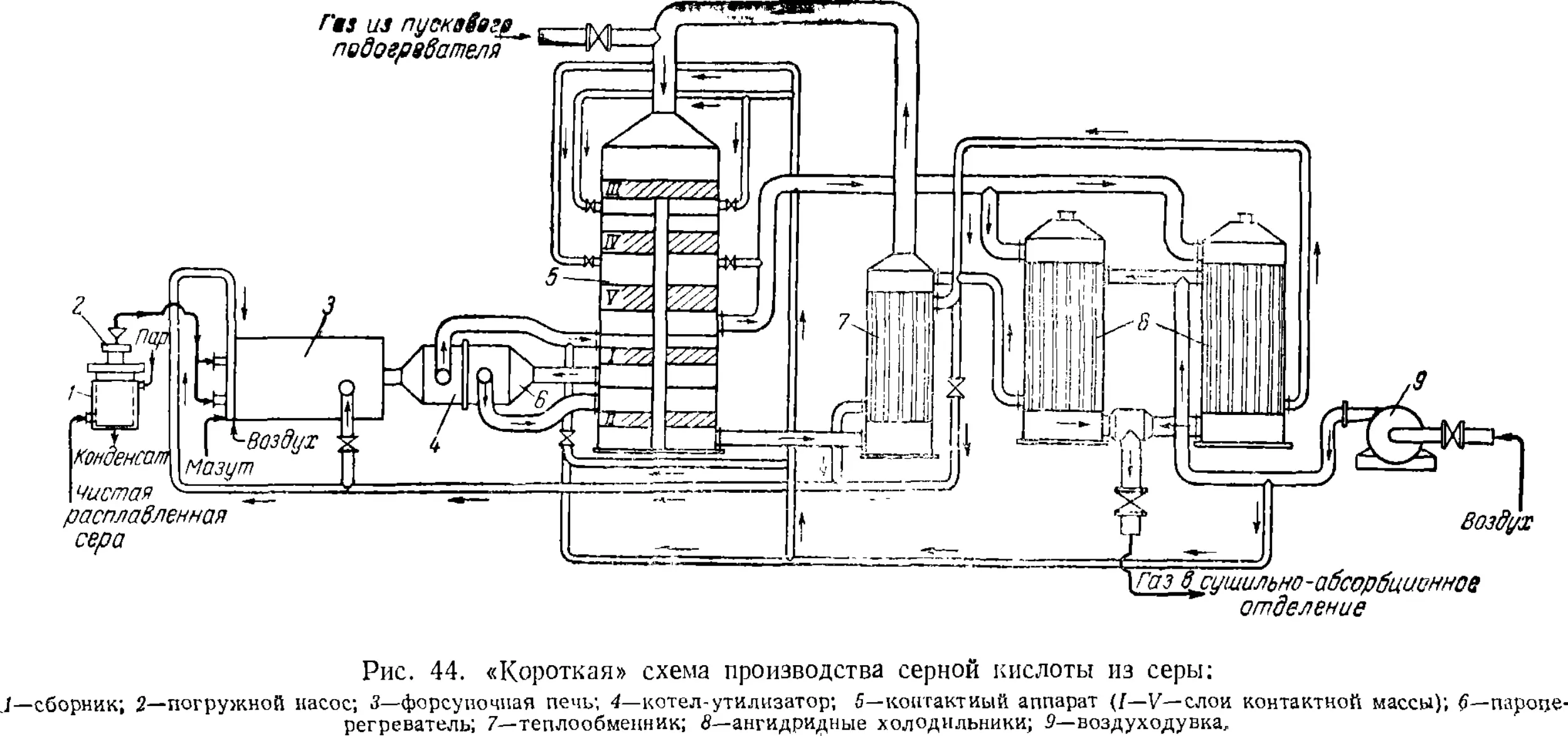
Производство контактной серной кислоты из серы . . .
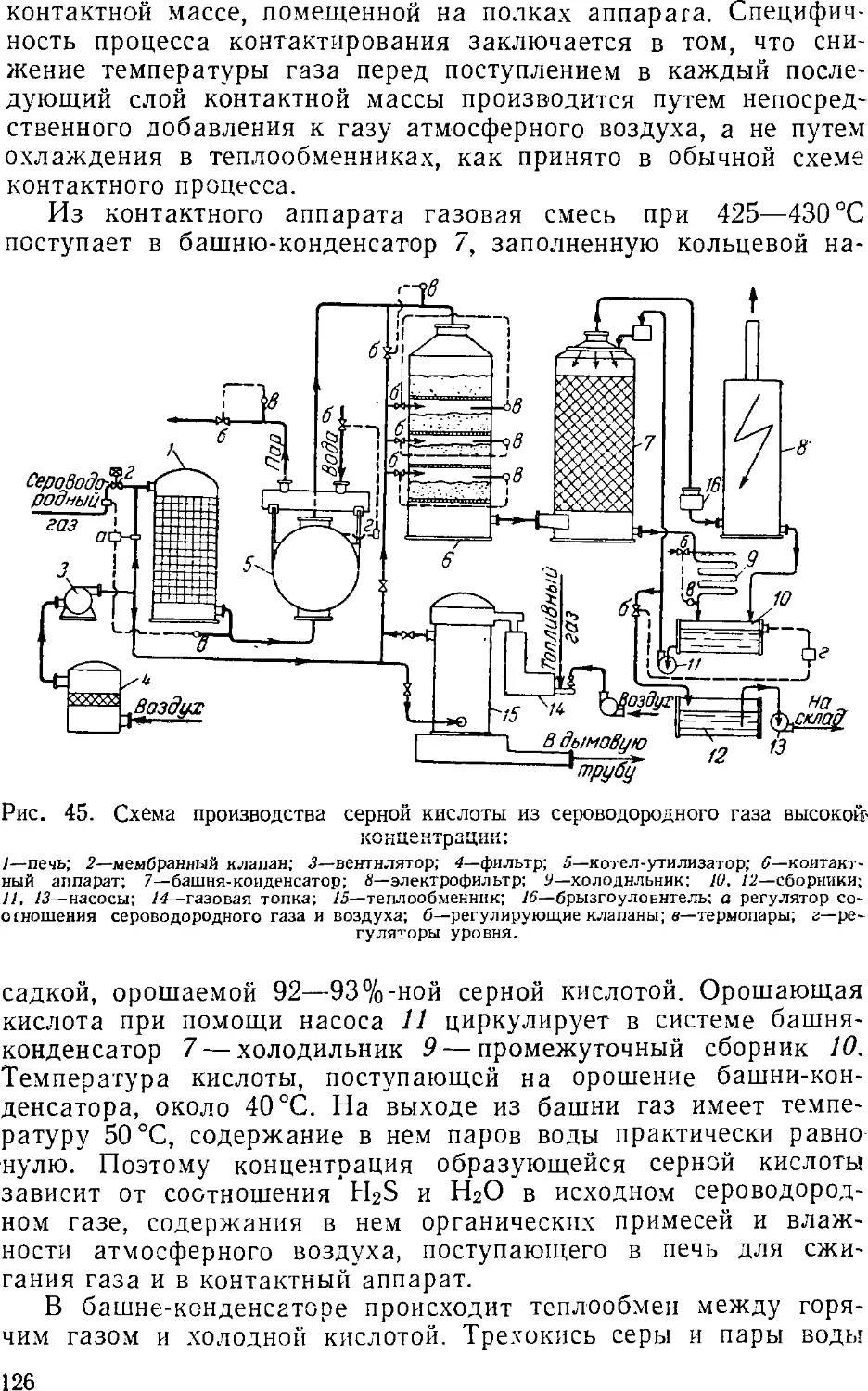
Производство контактной серной кислоты из
сероводорода 12
Основная аппаратура 12
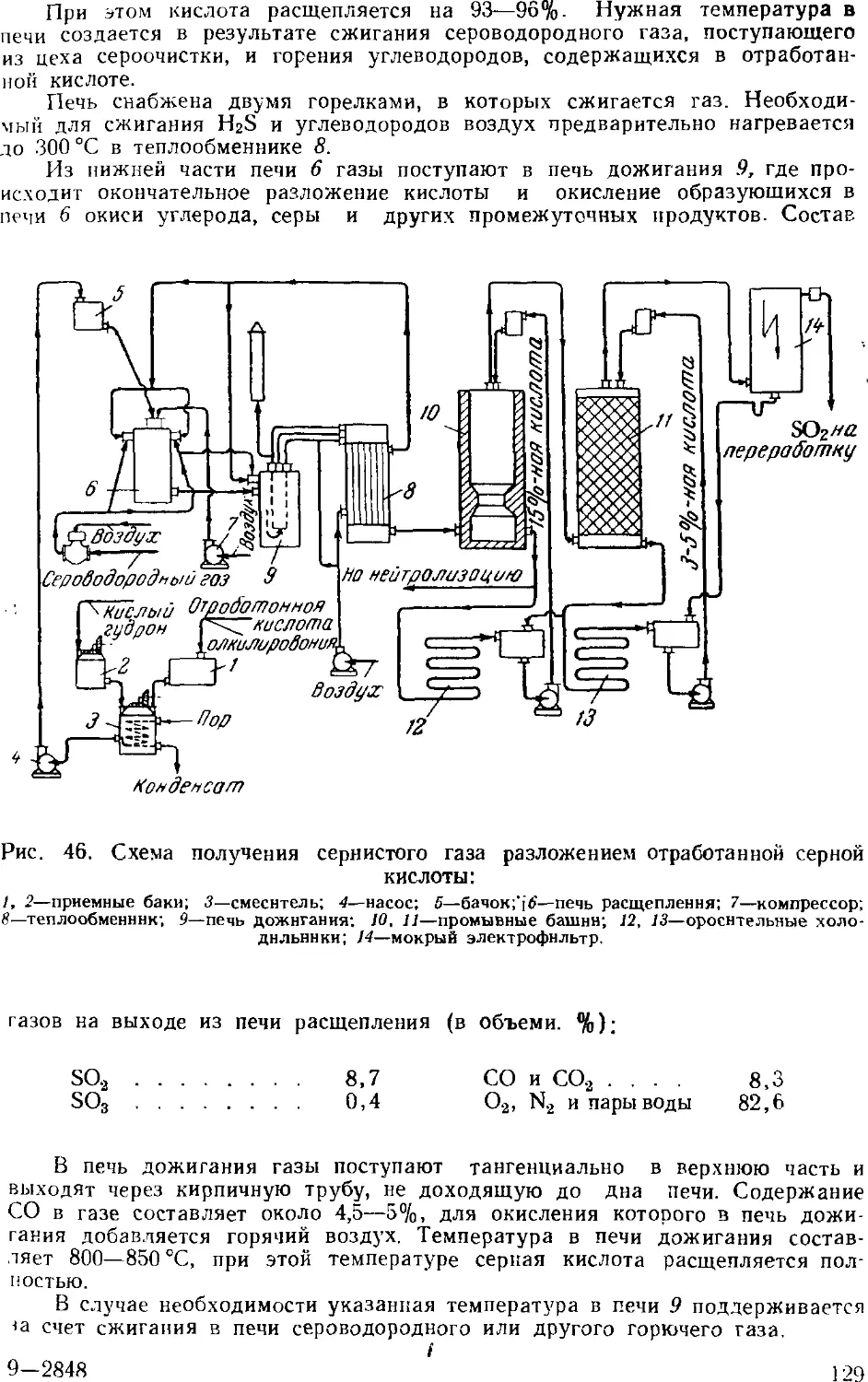
Производство олеума из отработанной серной кислоты . 12:
4. Нитрозный метод производства серной кислоты 13!
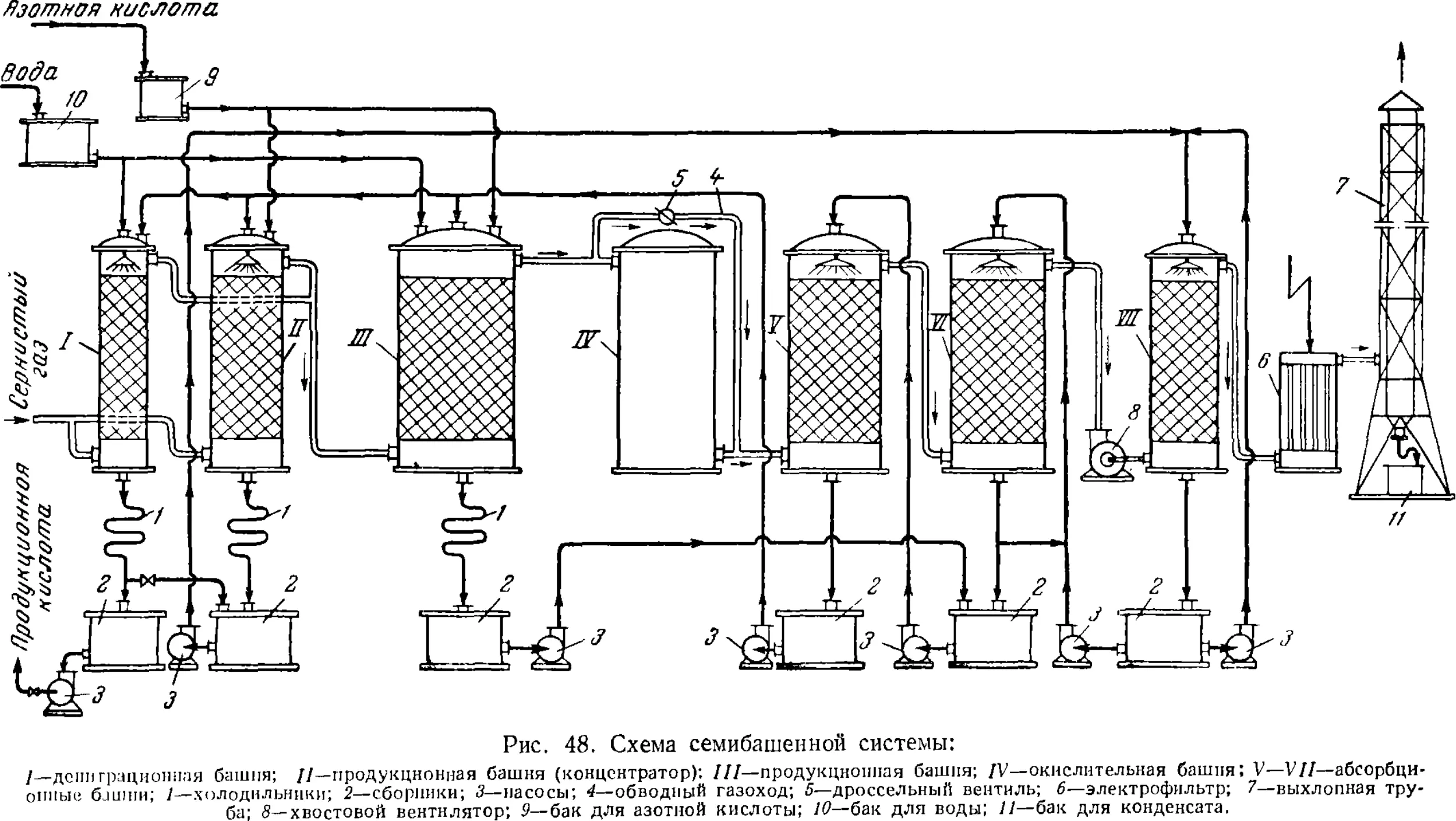
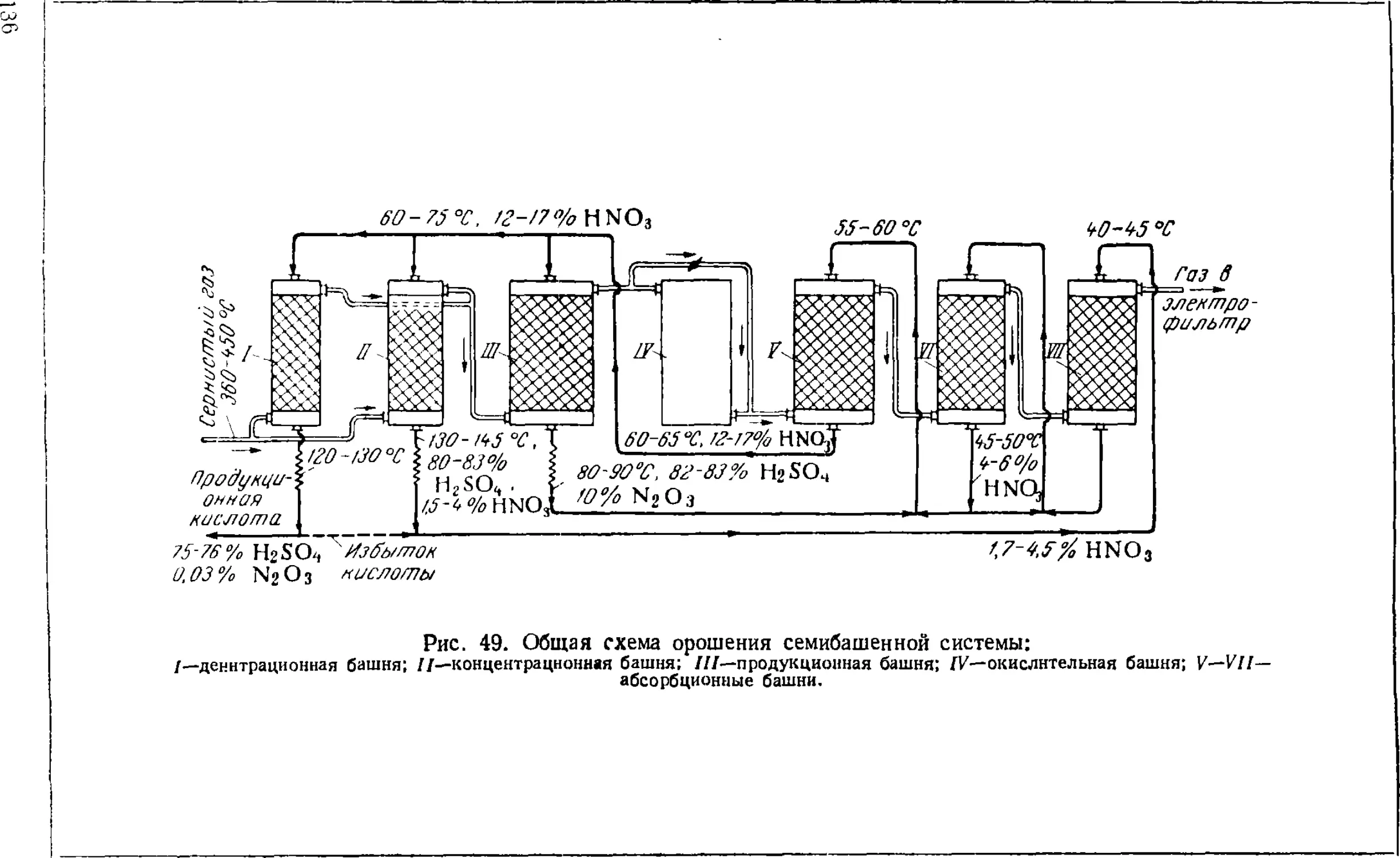
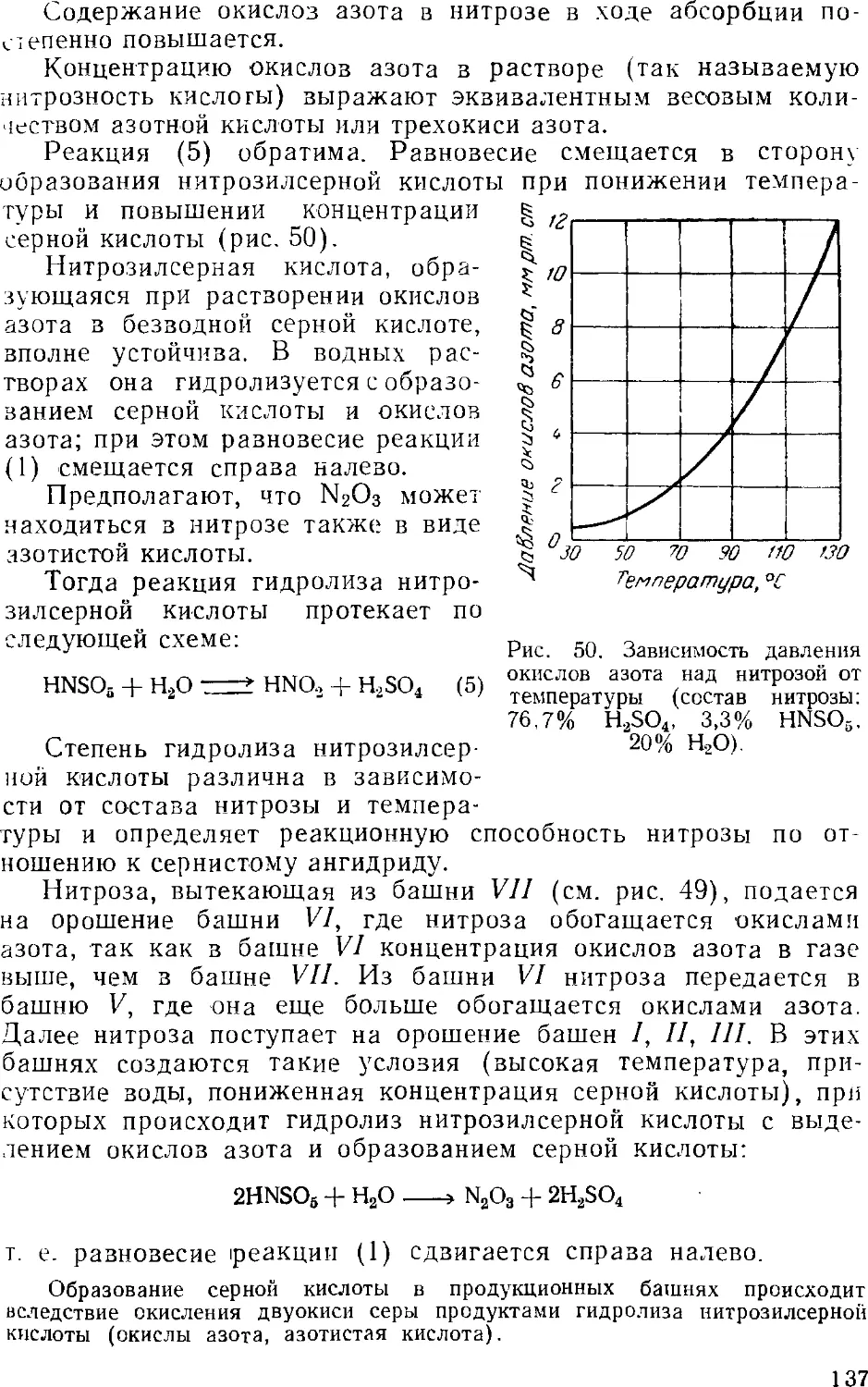
Принципиальная схема производства 13!
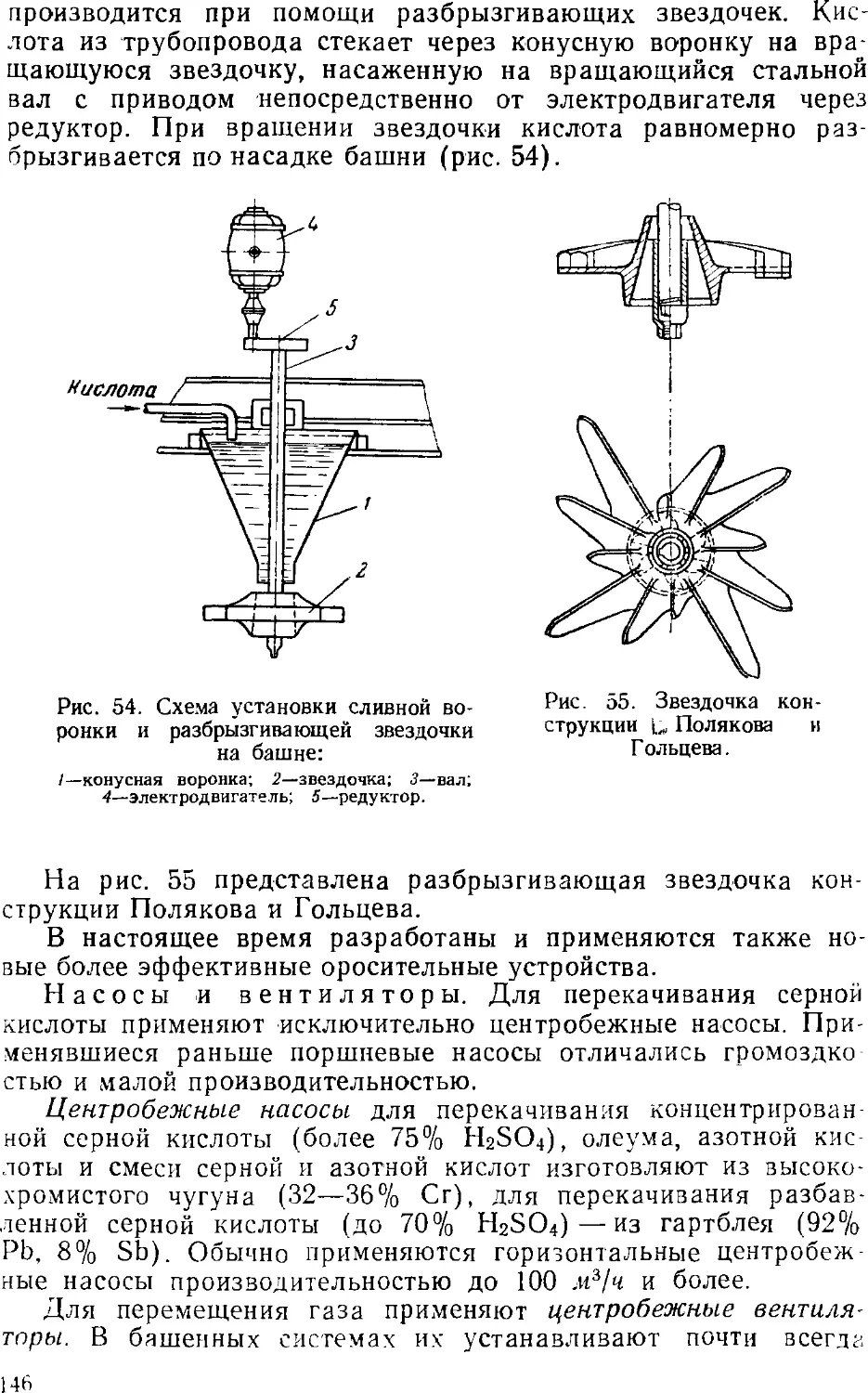
Технологический режим башенной системы 13i
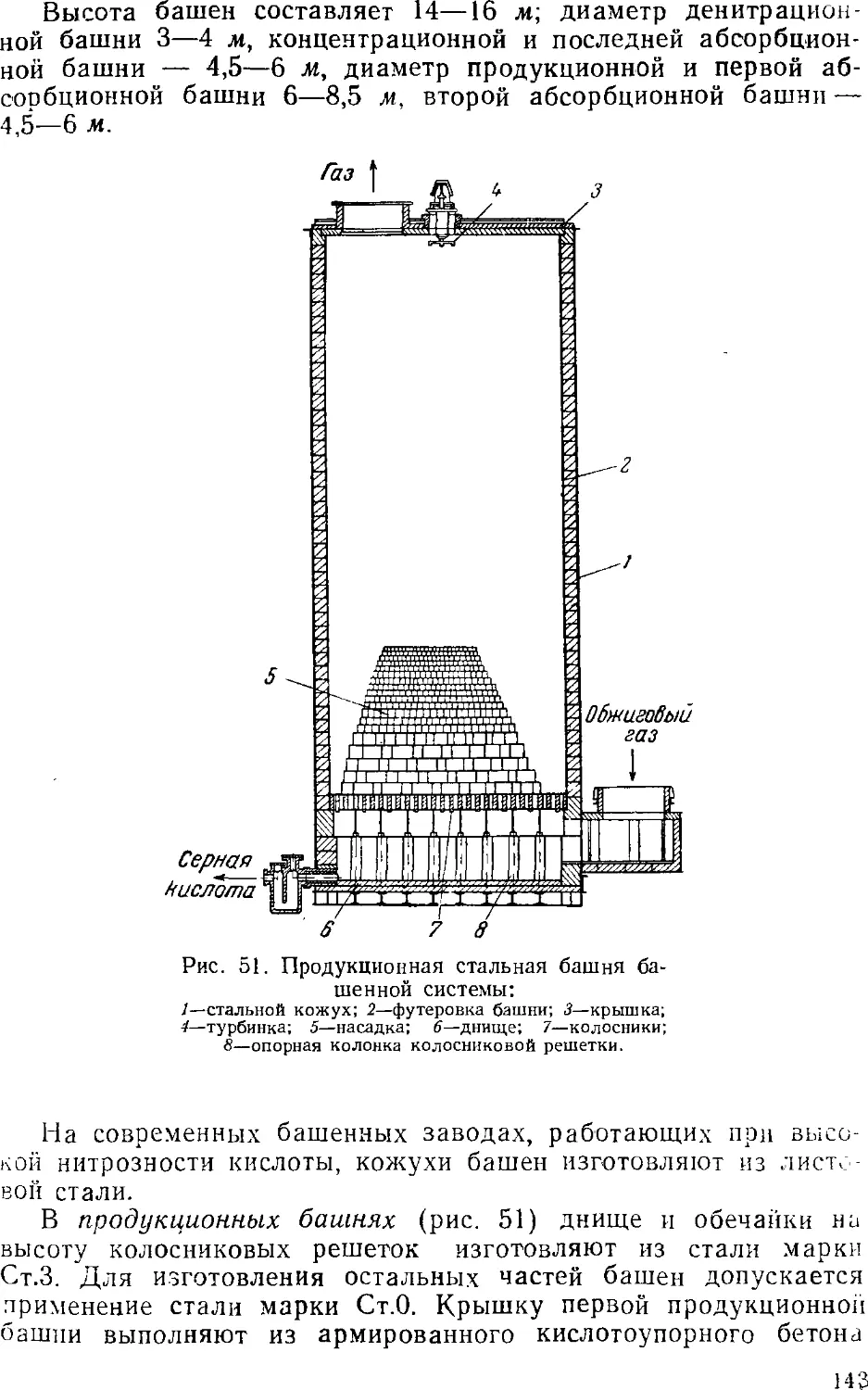
Аппаратура башенных систем 141
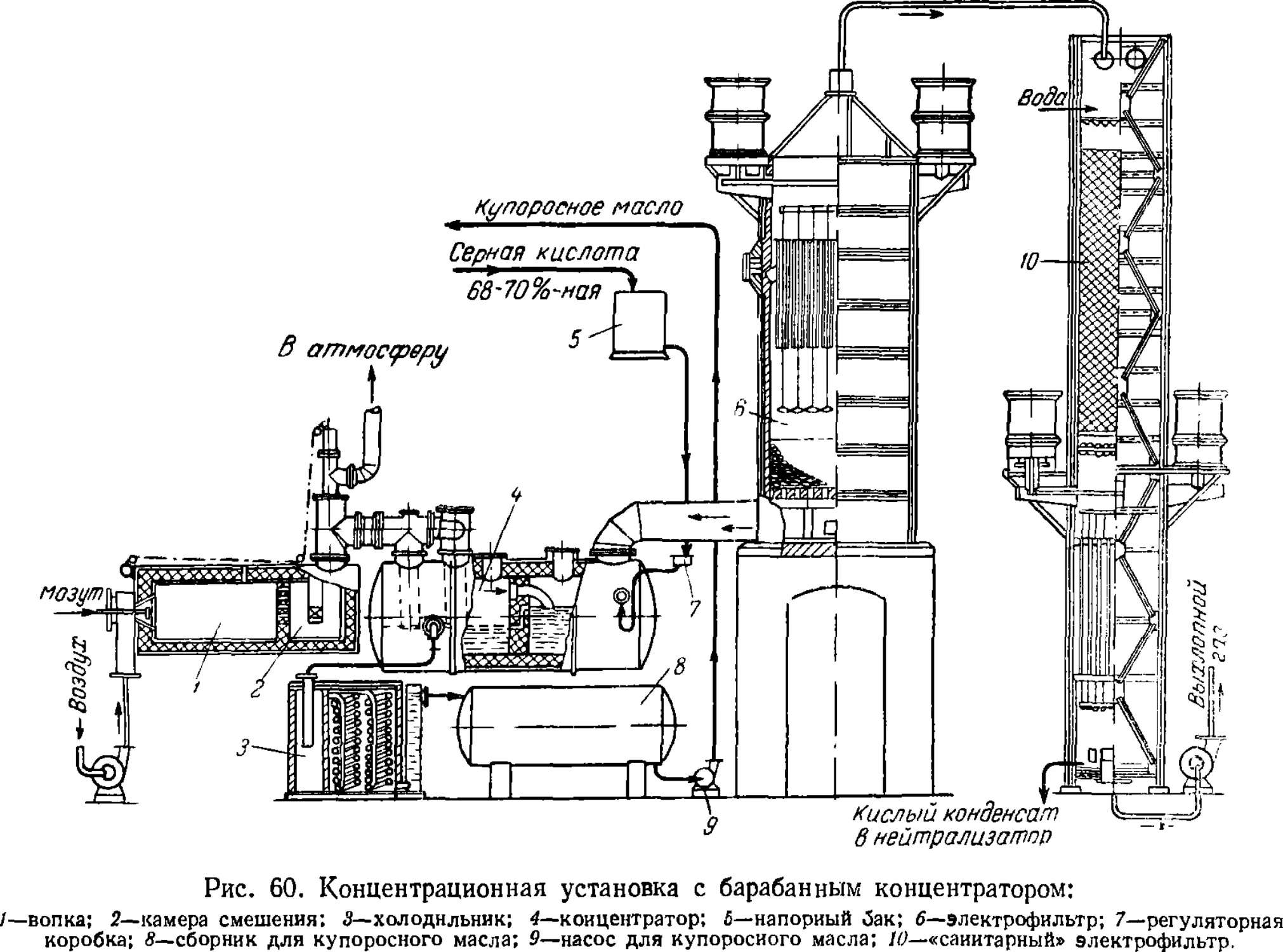
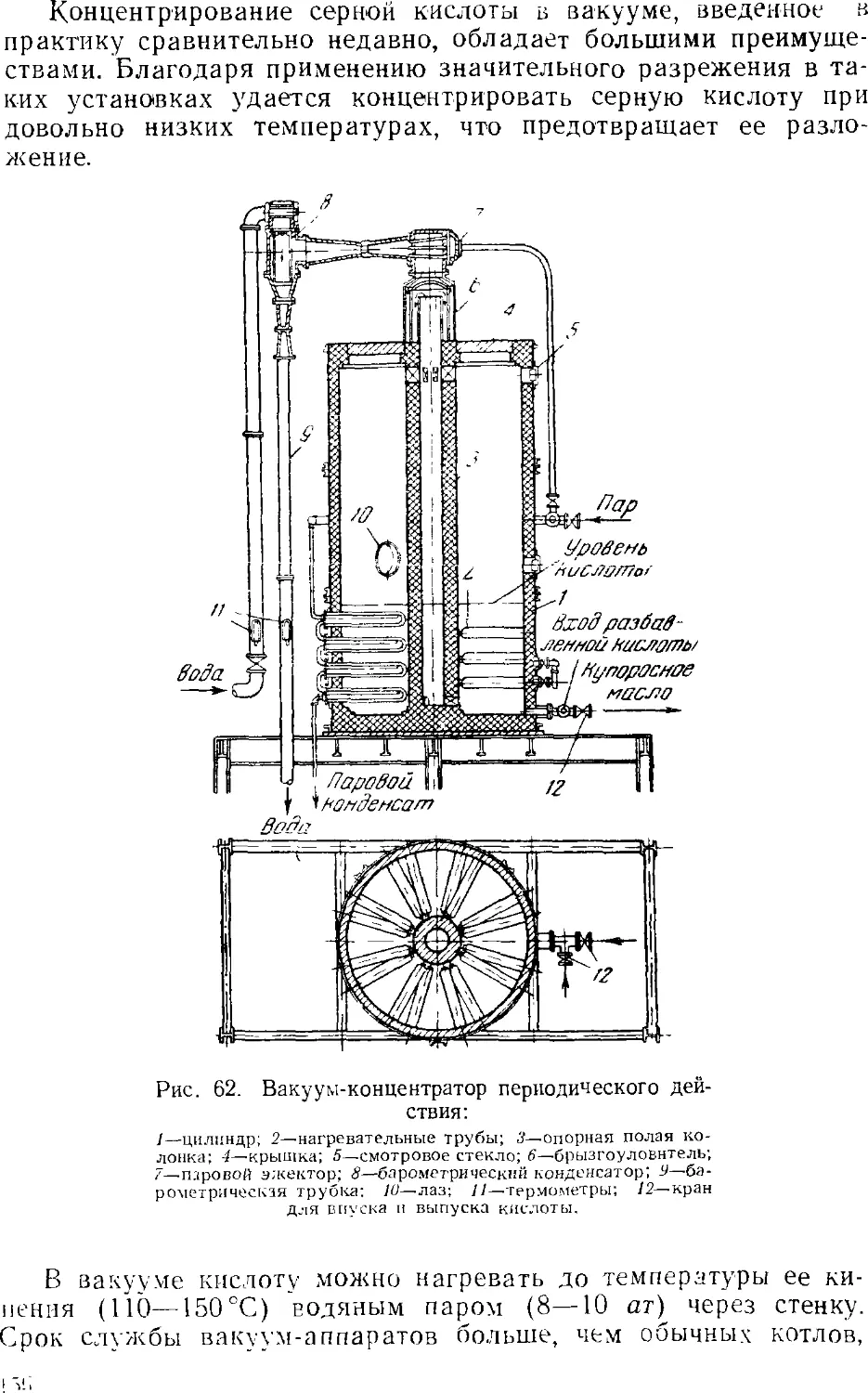
5. Концентрирование серной кислоты 14?
Установки для концентрирования 151
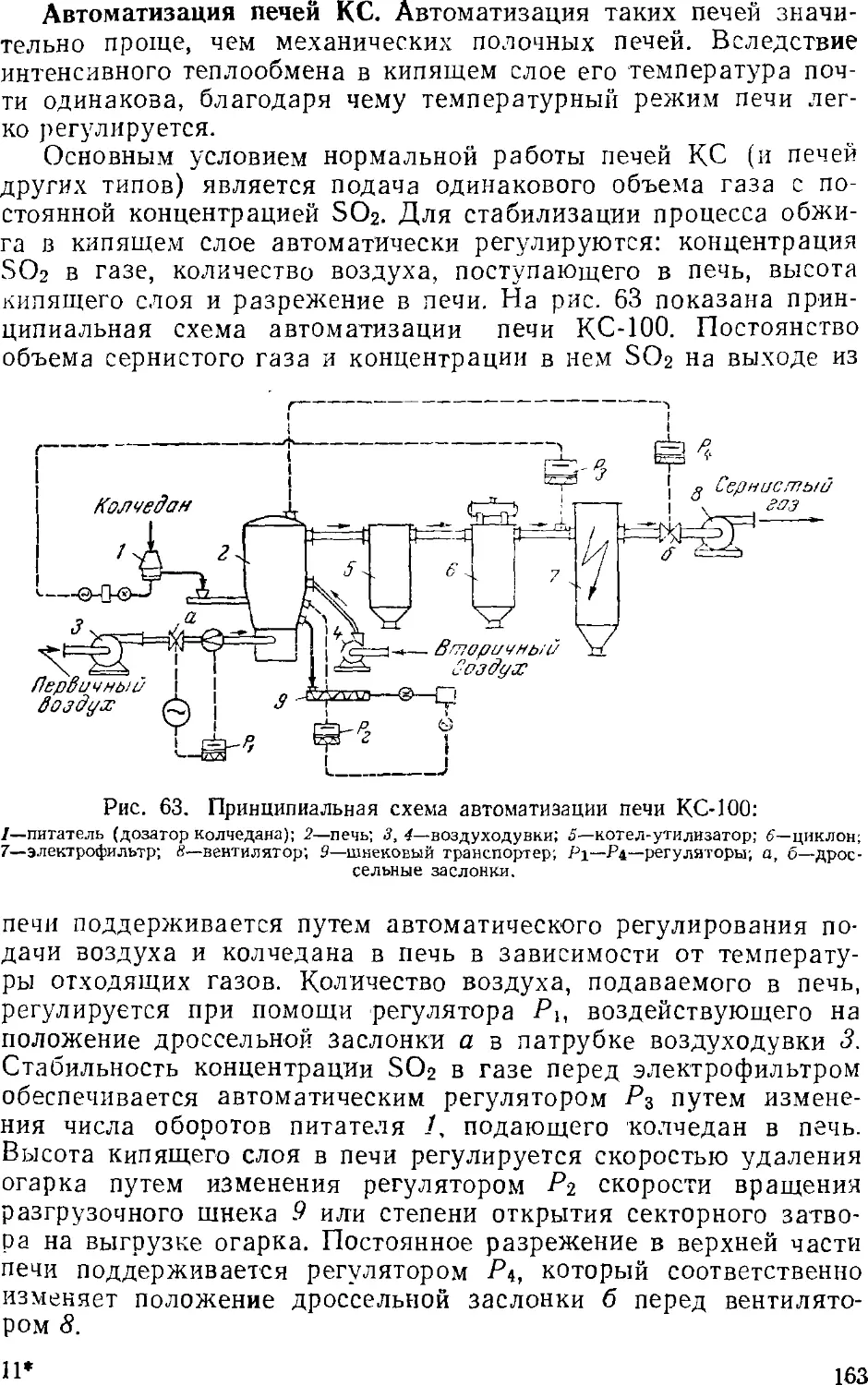
6. Контроль и автоматизация в производстве серной кислоты 160
Автоматизация контактного процесса 16<
Автоматизация производства серной кислоты из
сероводорода lGf
Автоматизация ба'иенного процесса 167
Литература (к главам III и IV) 164
Глава V. Производство водорода, азота и кислорода 170
1. Общие сведения 170
2. Производство водорода конверсией углеводородных газов . 175
Паровая каталитическая двухступенчатая конверсия
метана и окиси углерода 177
Каталитическая парокислородная и парокислородовоз-
душная конверсия метана 179
Высокотемпературная (некаталитическая) конверсия
метана кислородом или обогащенным воздухом 182
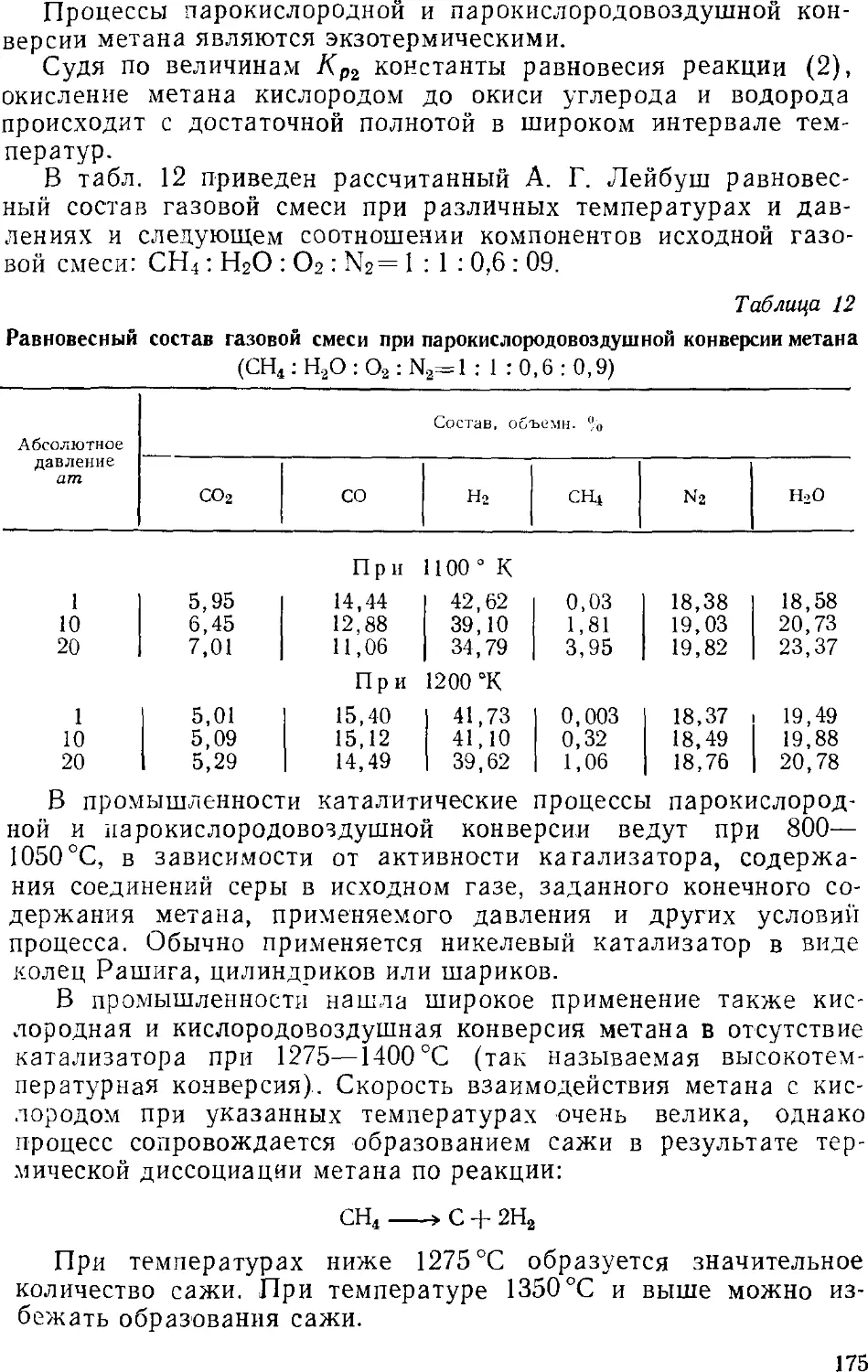
Основная аппаратура 1S4
Конверсия окиси углерода водяным паром 186
Газгольдеры 188
3. Очистка азотоводородной смеси 19!
Удаление двуокиси углерода и сероводорода 191
Удаление окиси углерода 19о
4. Производство гзота, кислорода и водорода разделением
газовых смесей глубоким охлажденном 202
Сжижение воздуха 204
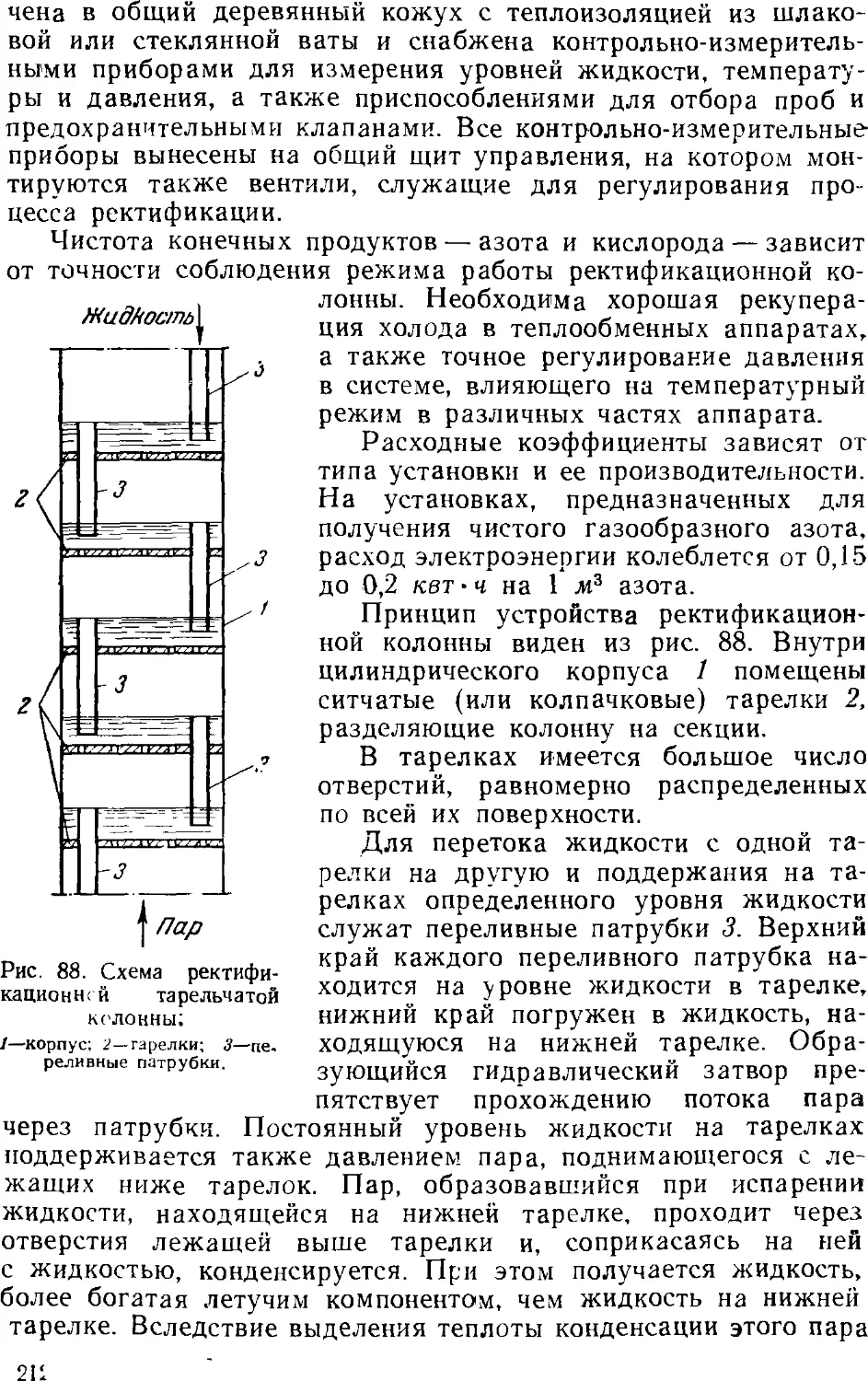
Ректификация жидкого воздуха 209
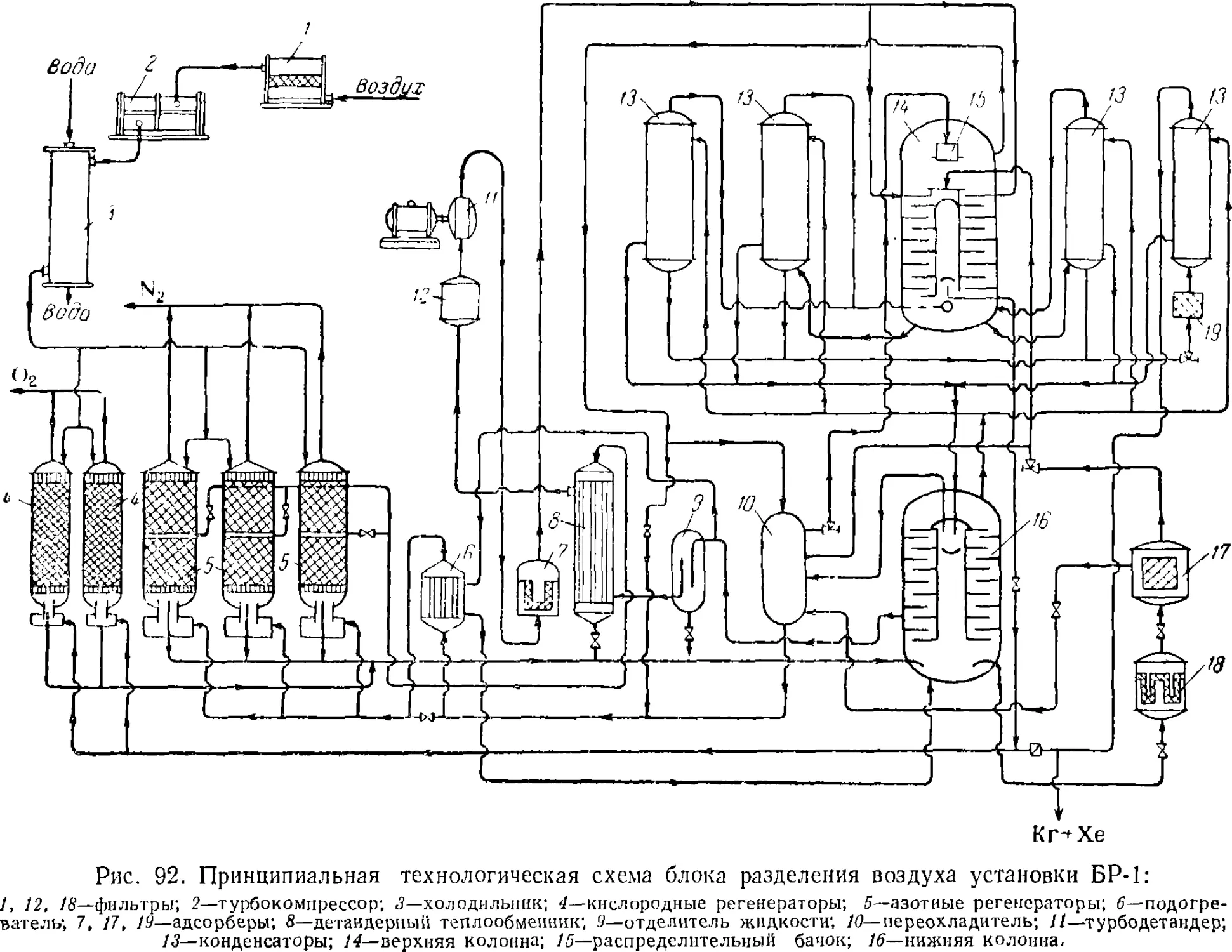
Установки для разделения воздуха 213
0. Производства водорода (азотоводородной смеси)
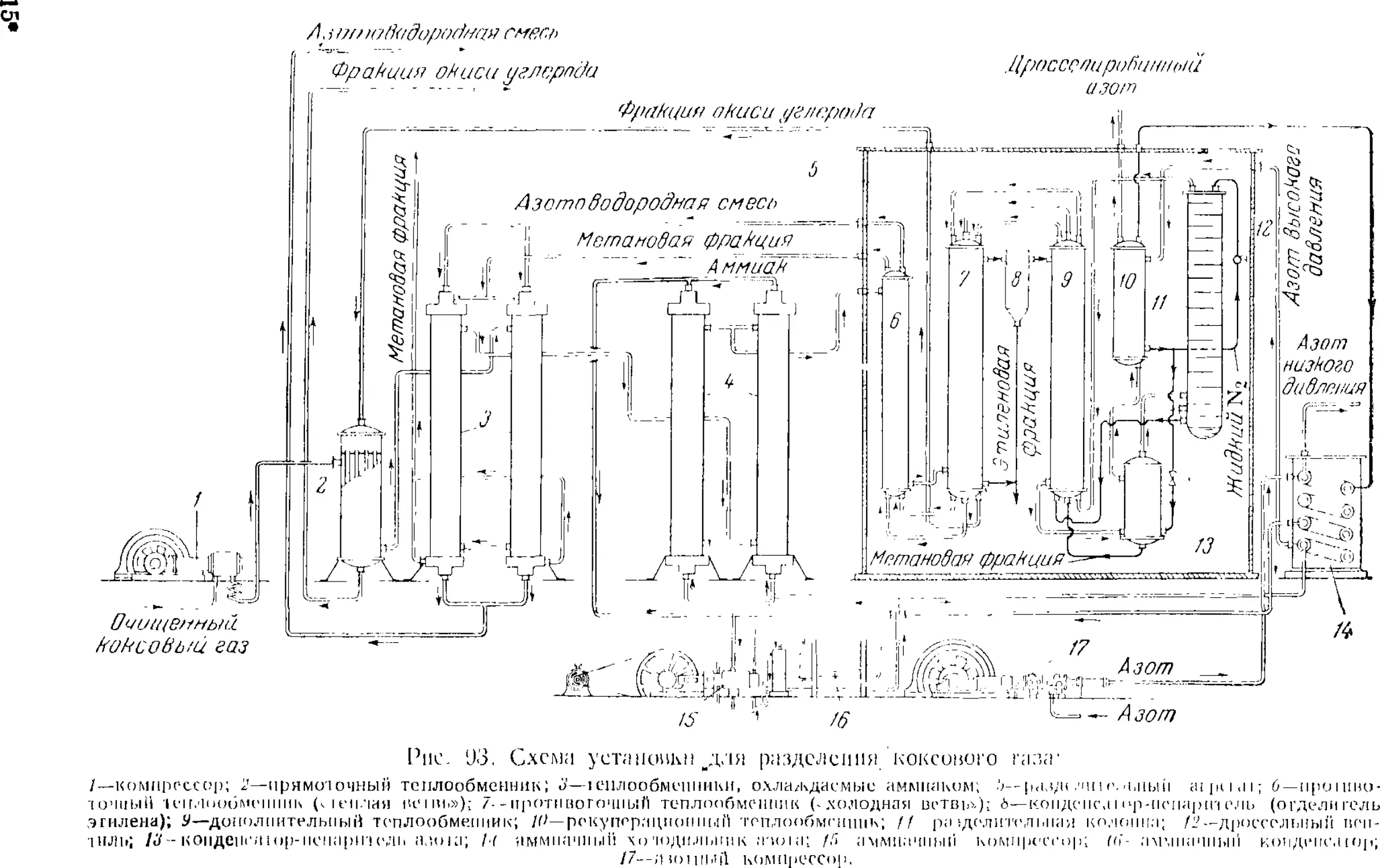
разделением коксового газа 223
Низкотемпературное разделение коксового газа 225
Автоматизация процесса разделения коксового газа . . . 230
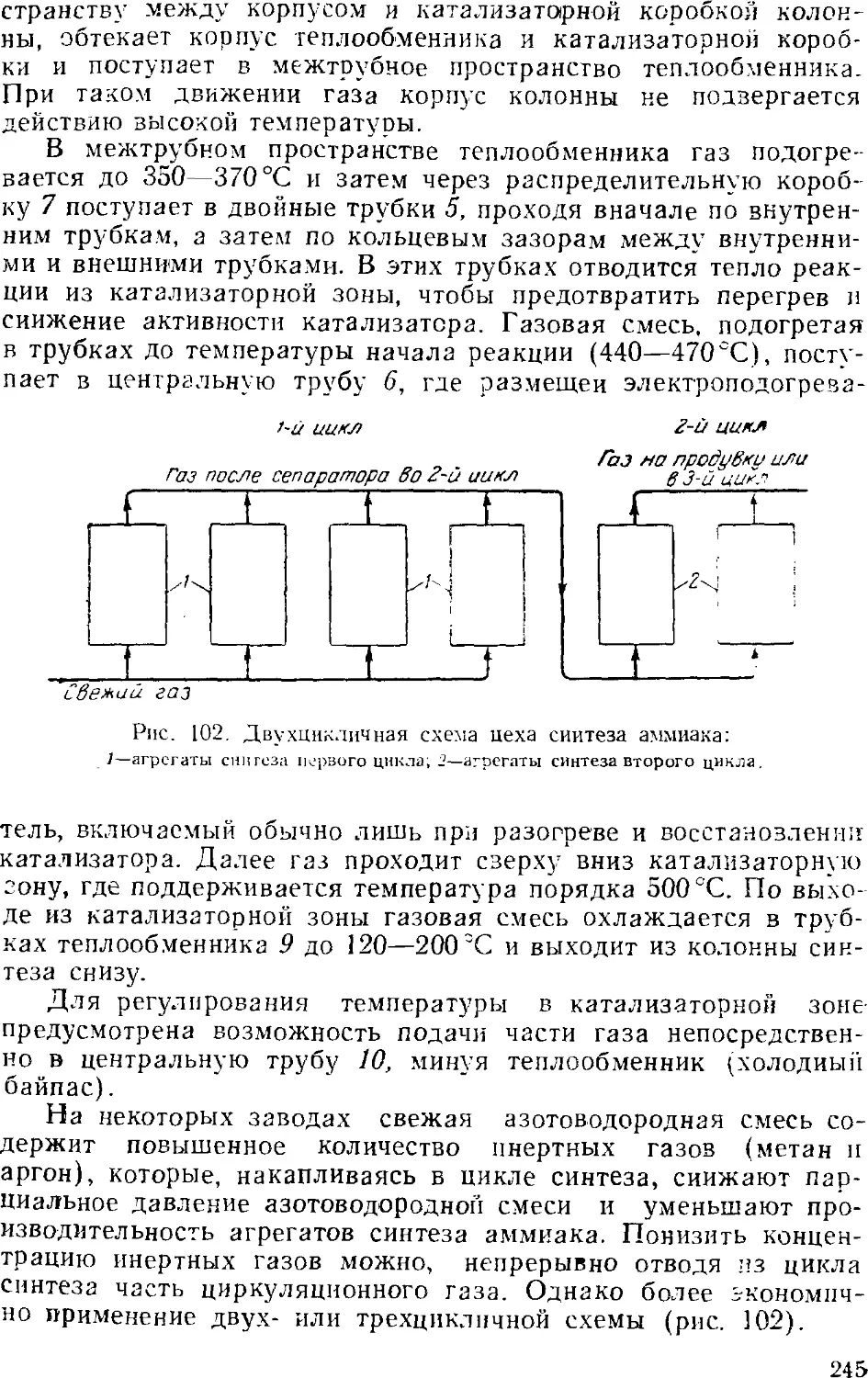
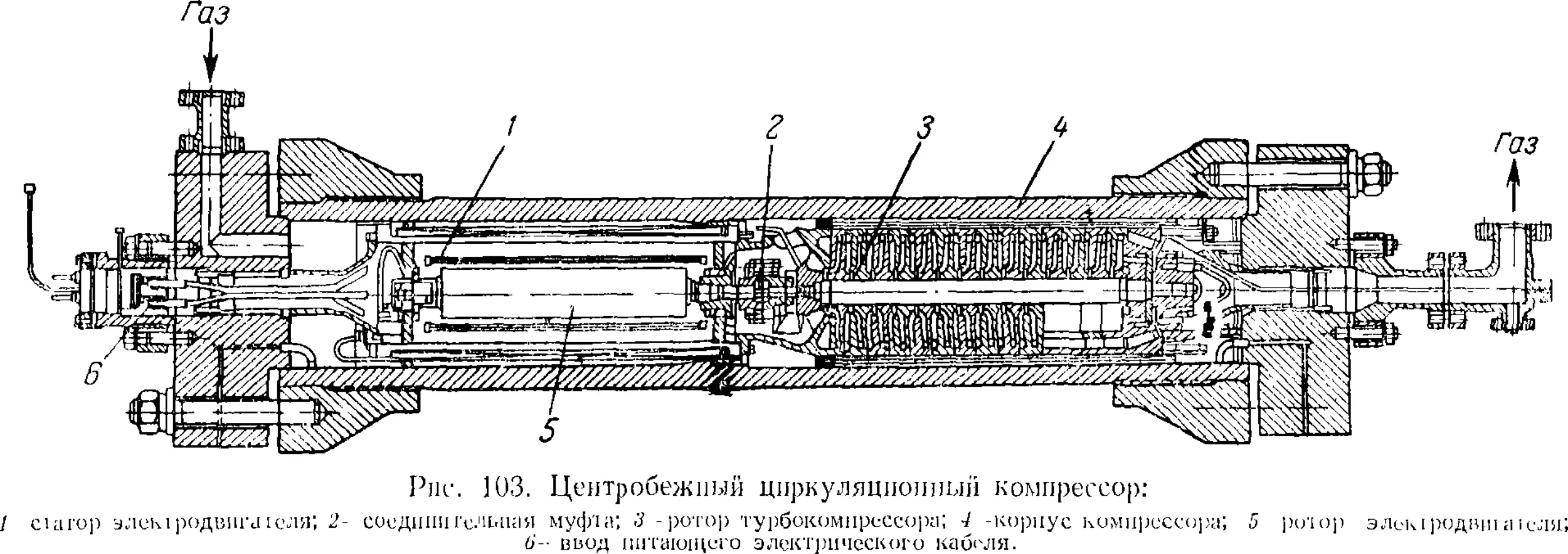
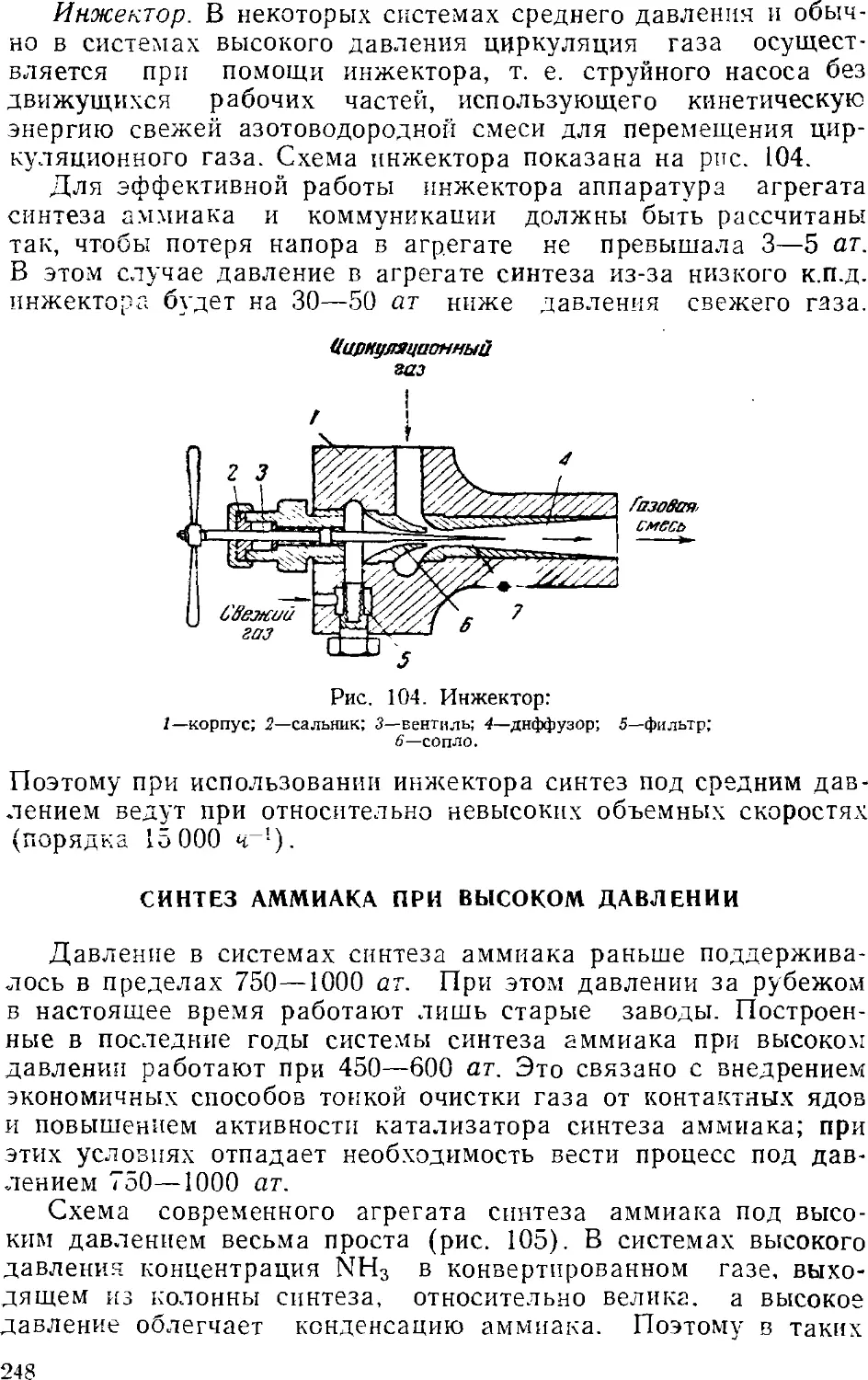
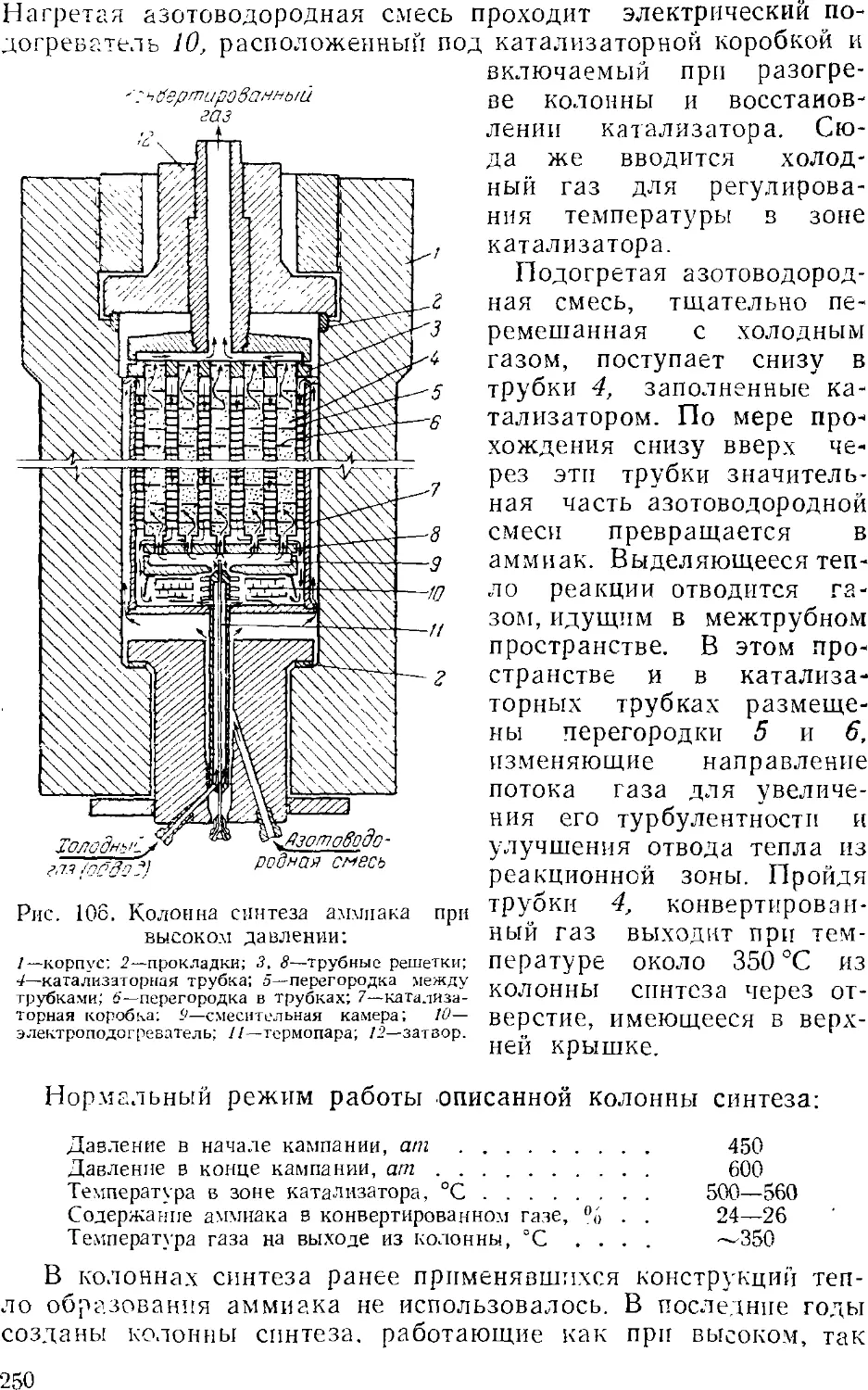
Глава VI. Аммиак 231
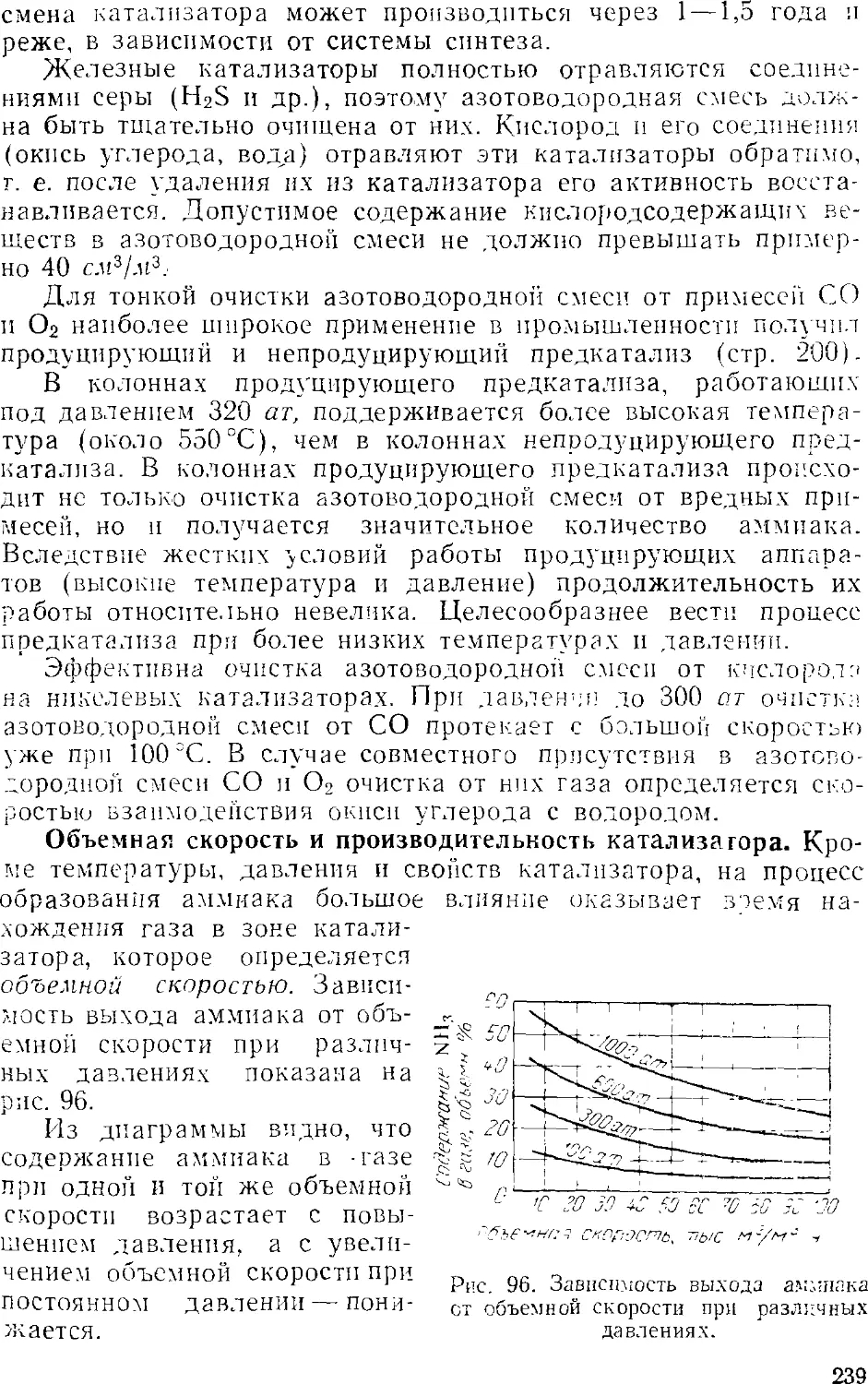
1. Общие сведения 231
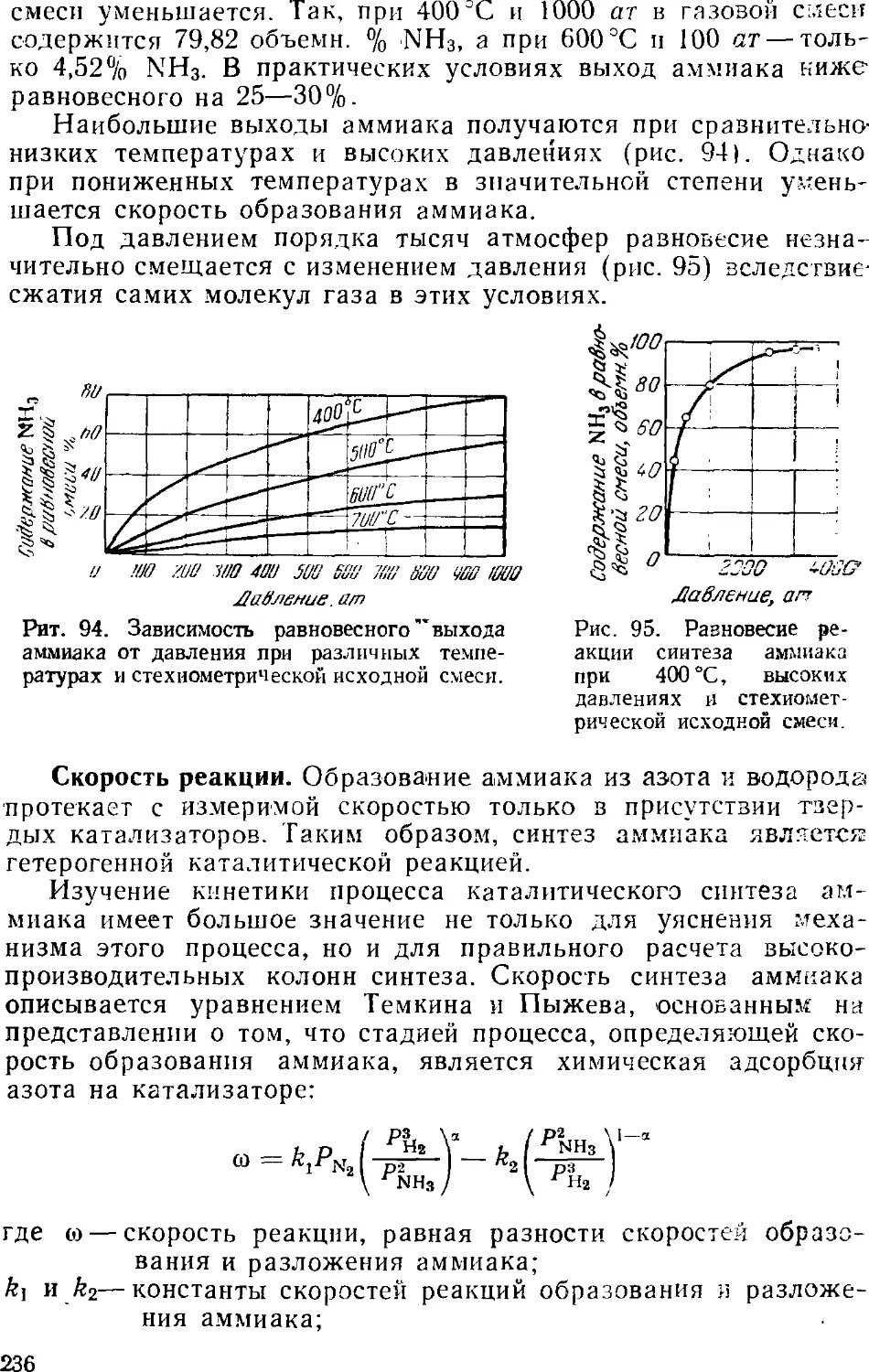
2. Физико-химические основы процесса синтеза аммиака . . . 235
4
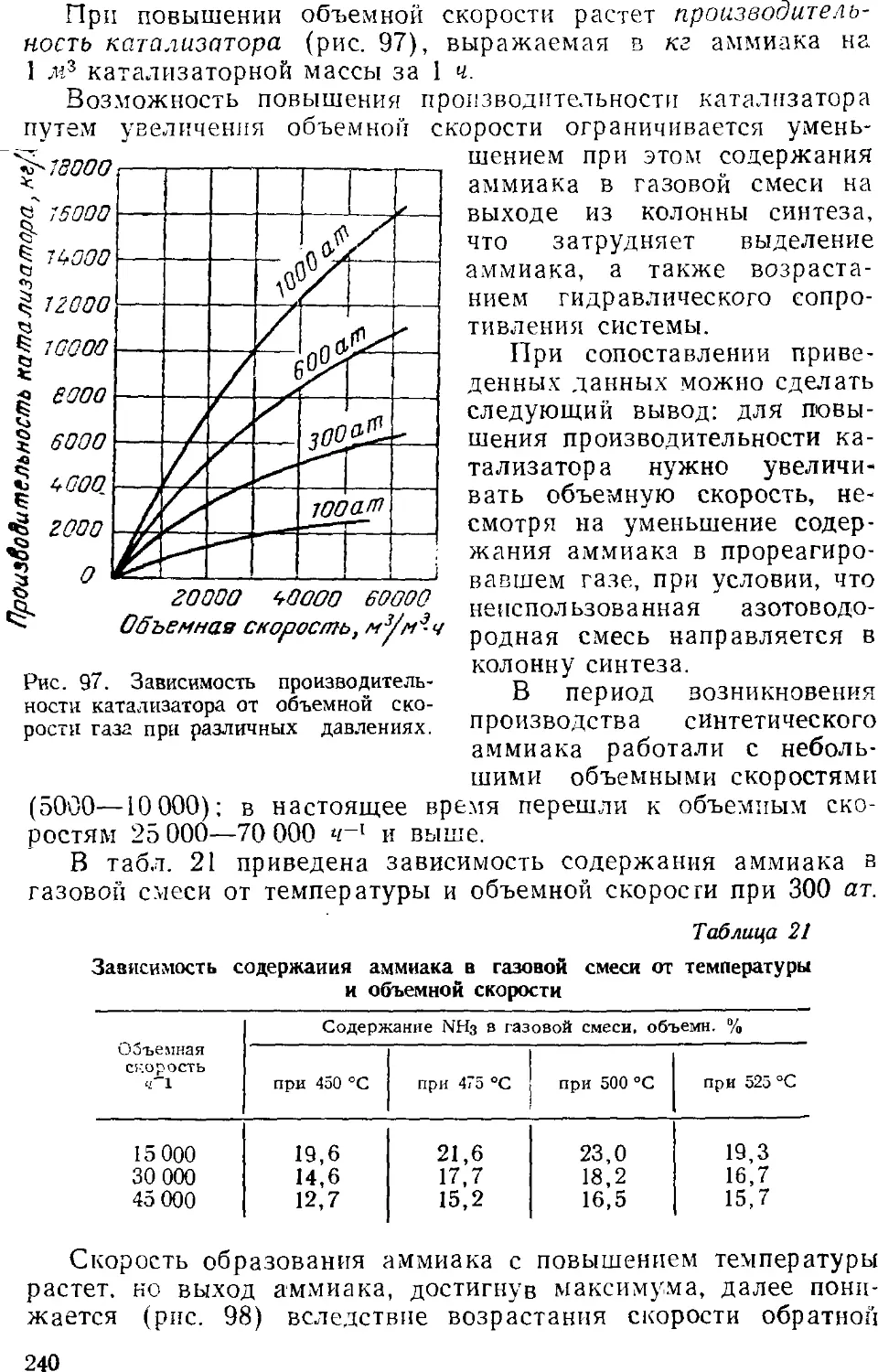
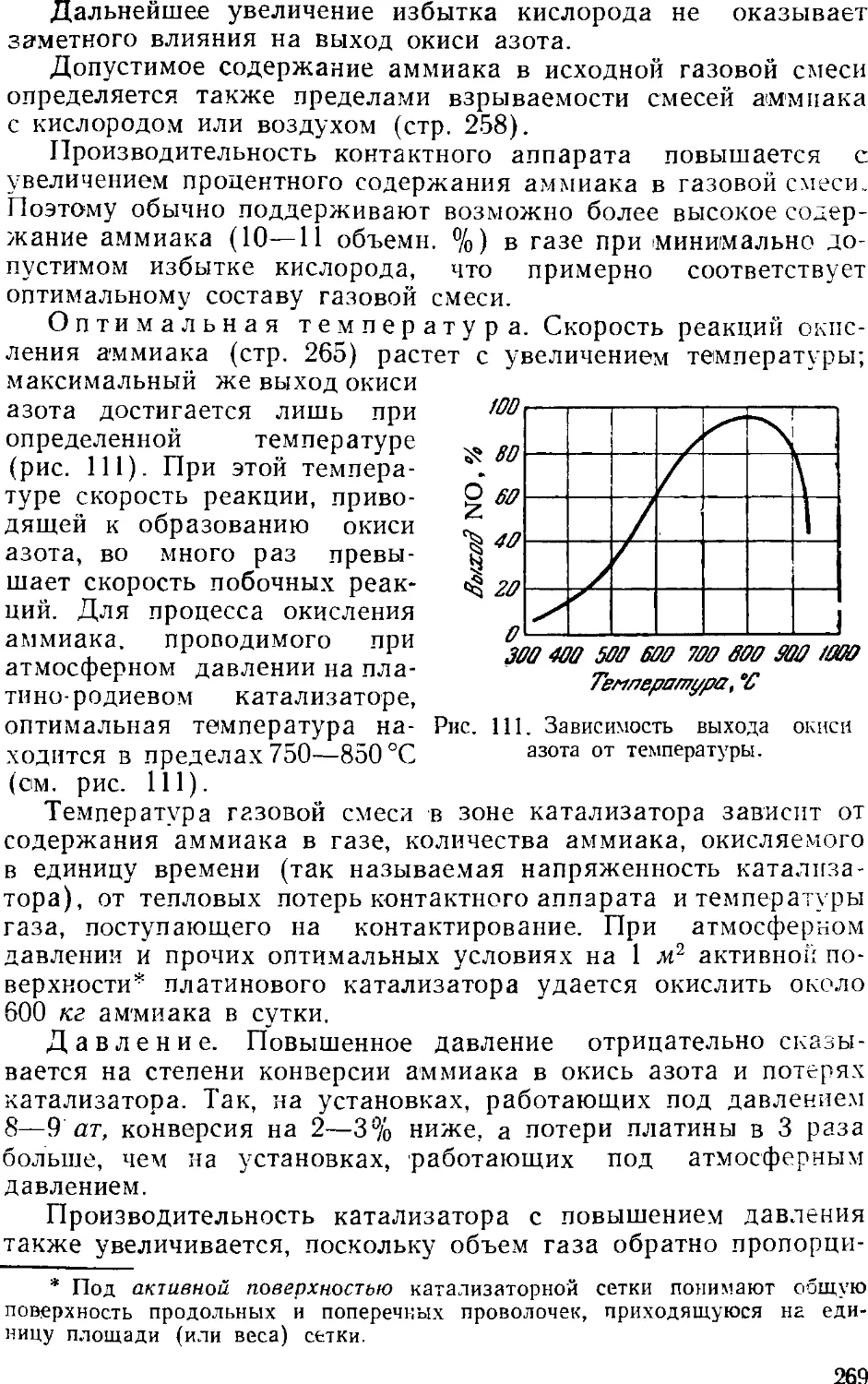
3. Промышленный синтез аммиака 24'i
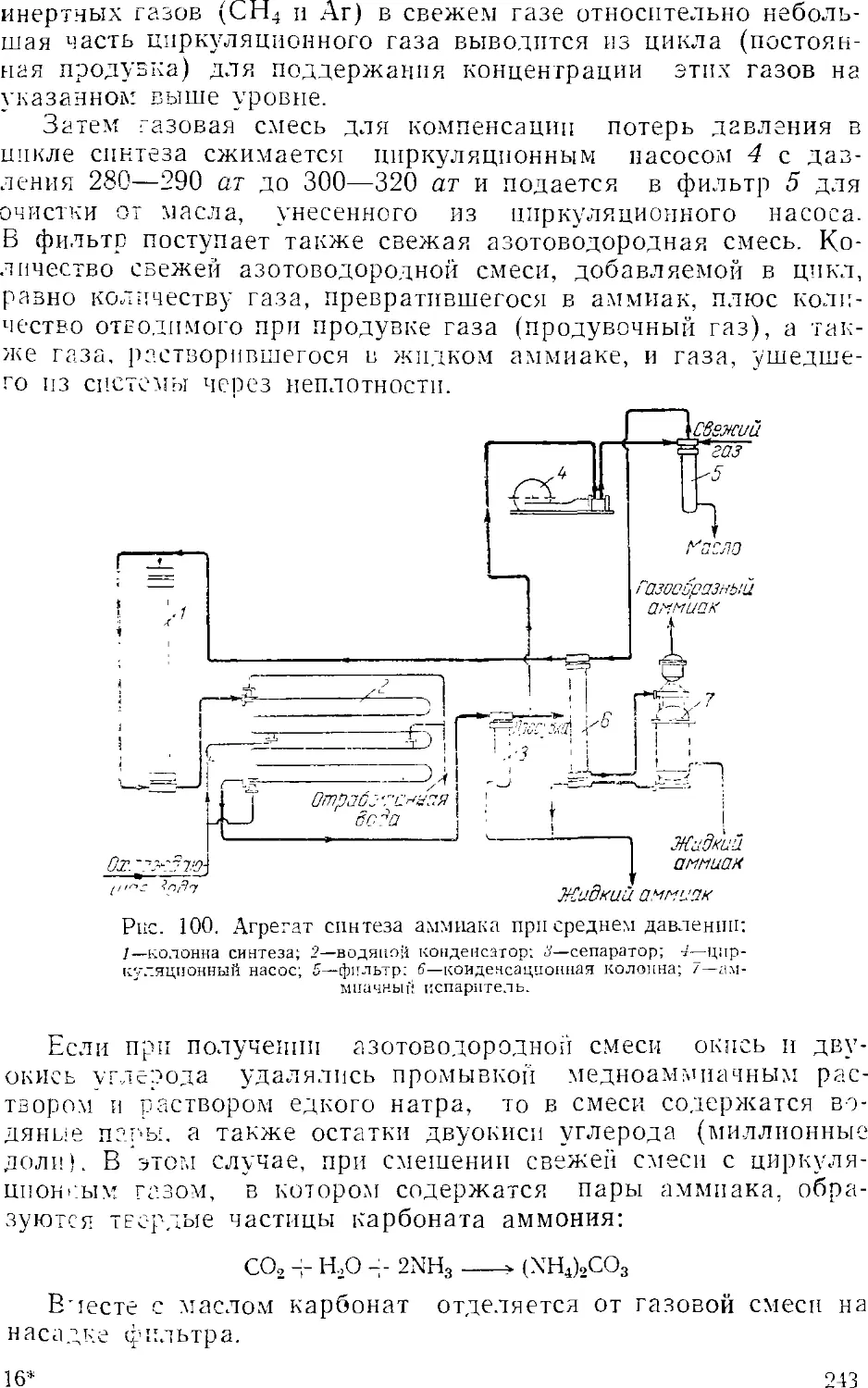
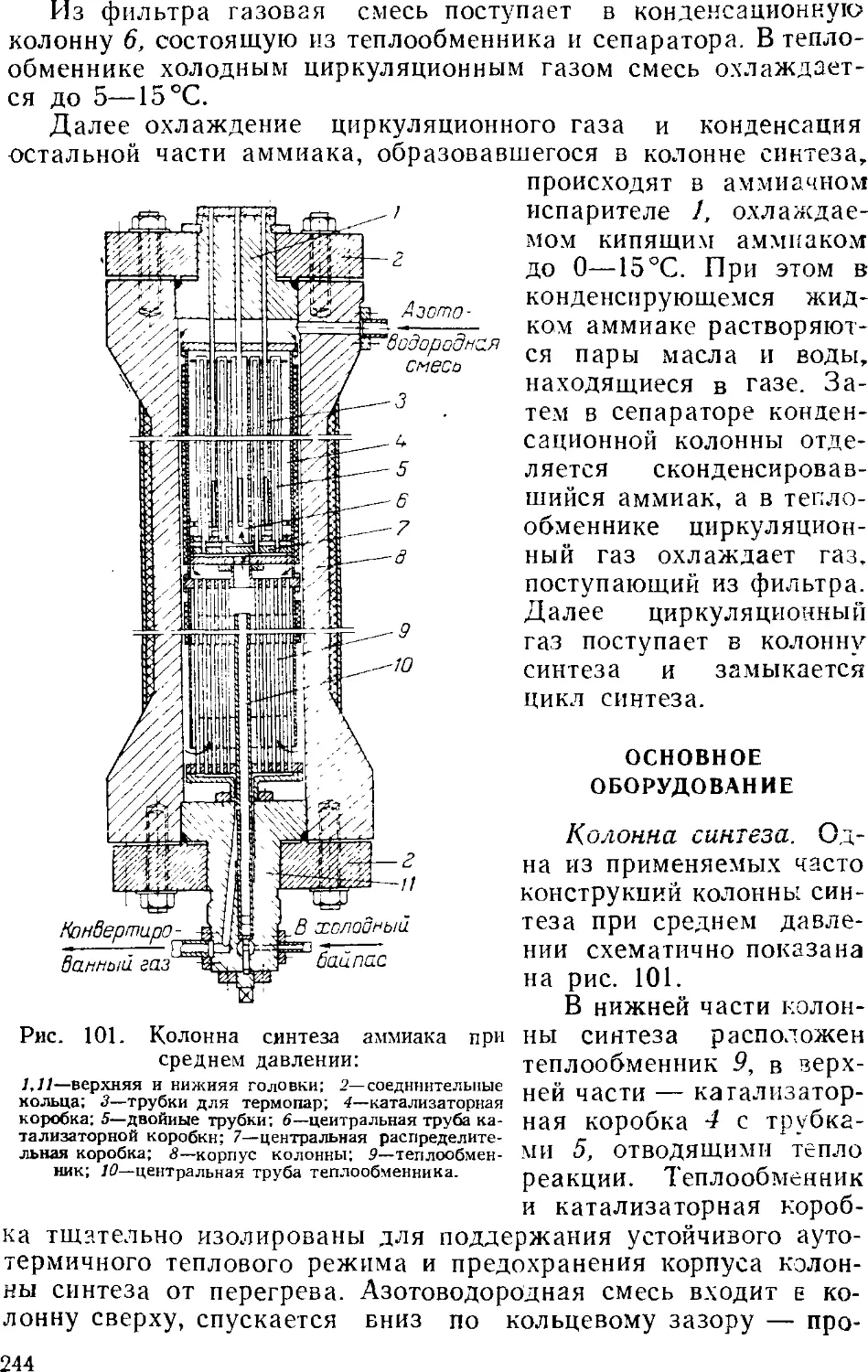
Синтсз аммиака при среднем давлении 242
Основное оборудование 241
Синтез аммиака при высоком давлении 248
Автоматизация процесса синтеза аммиака 25i
Приготовление аммиачной воды 257
Глава VII. Азотная кислота 261
1. Общие сведения 261
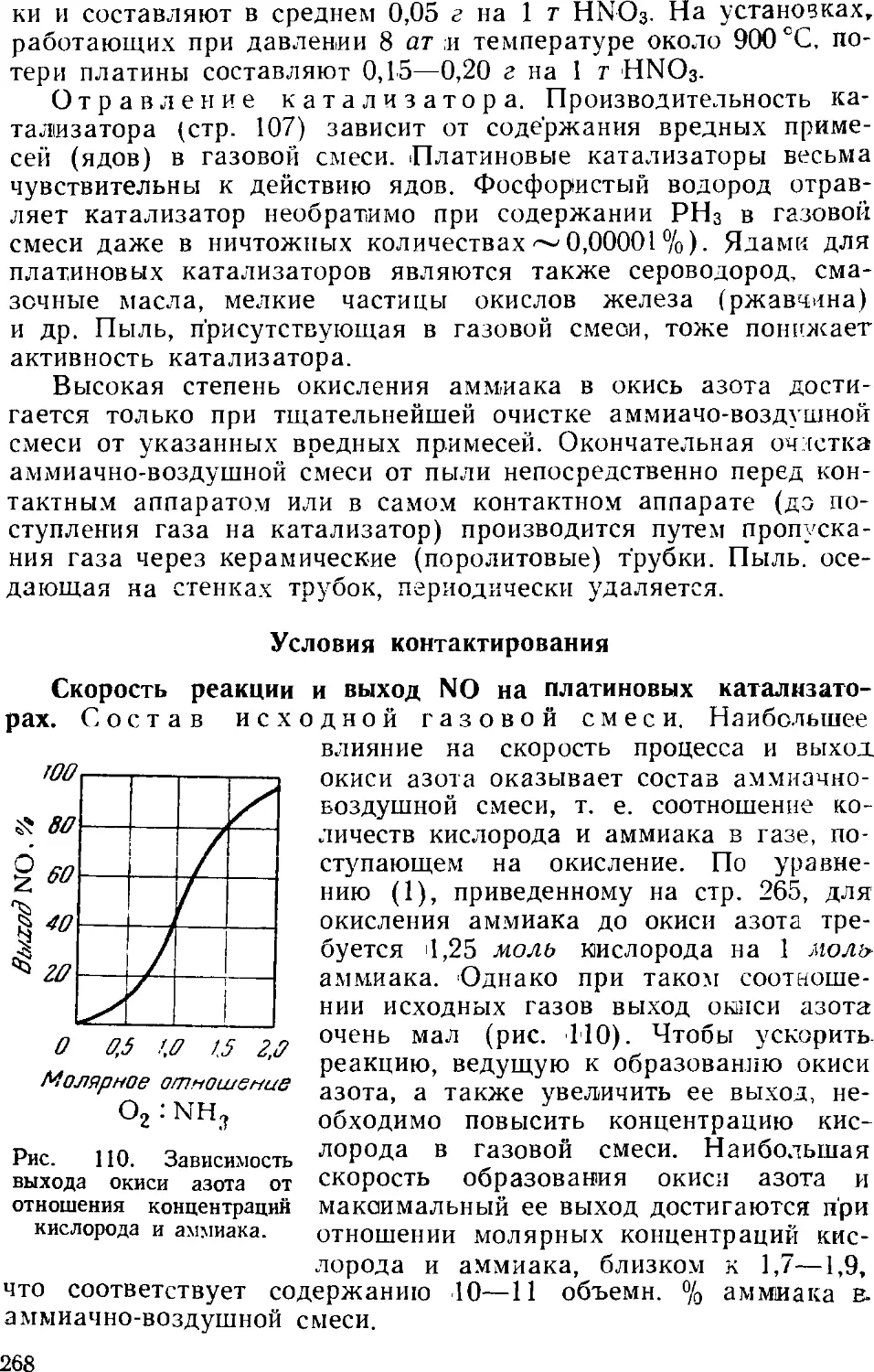
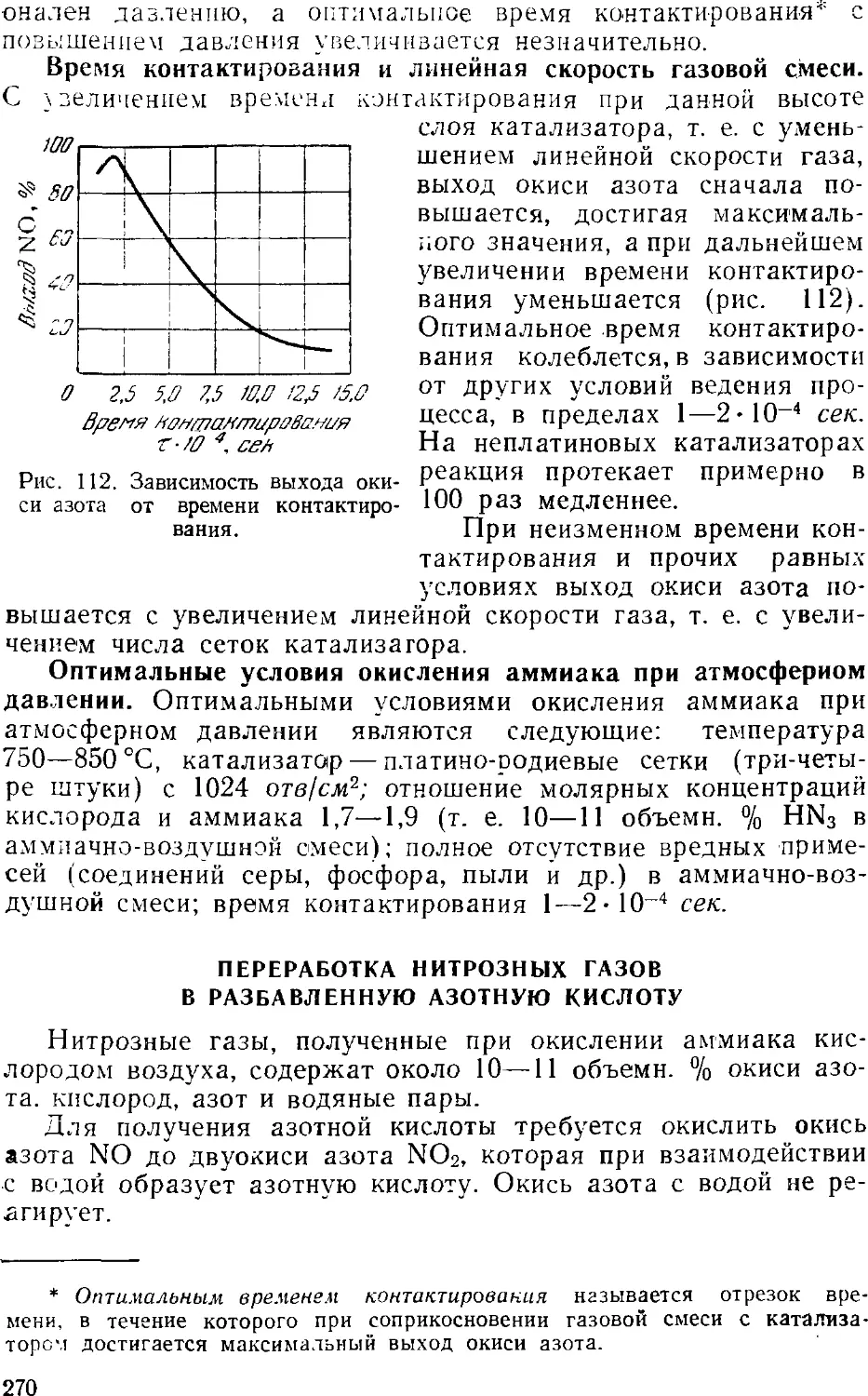
2. Производство азотной кислоты окислением аммиака .... 2Ь4
Контактное окисление аммиака 265
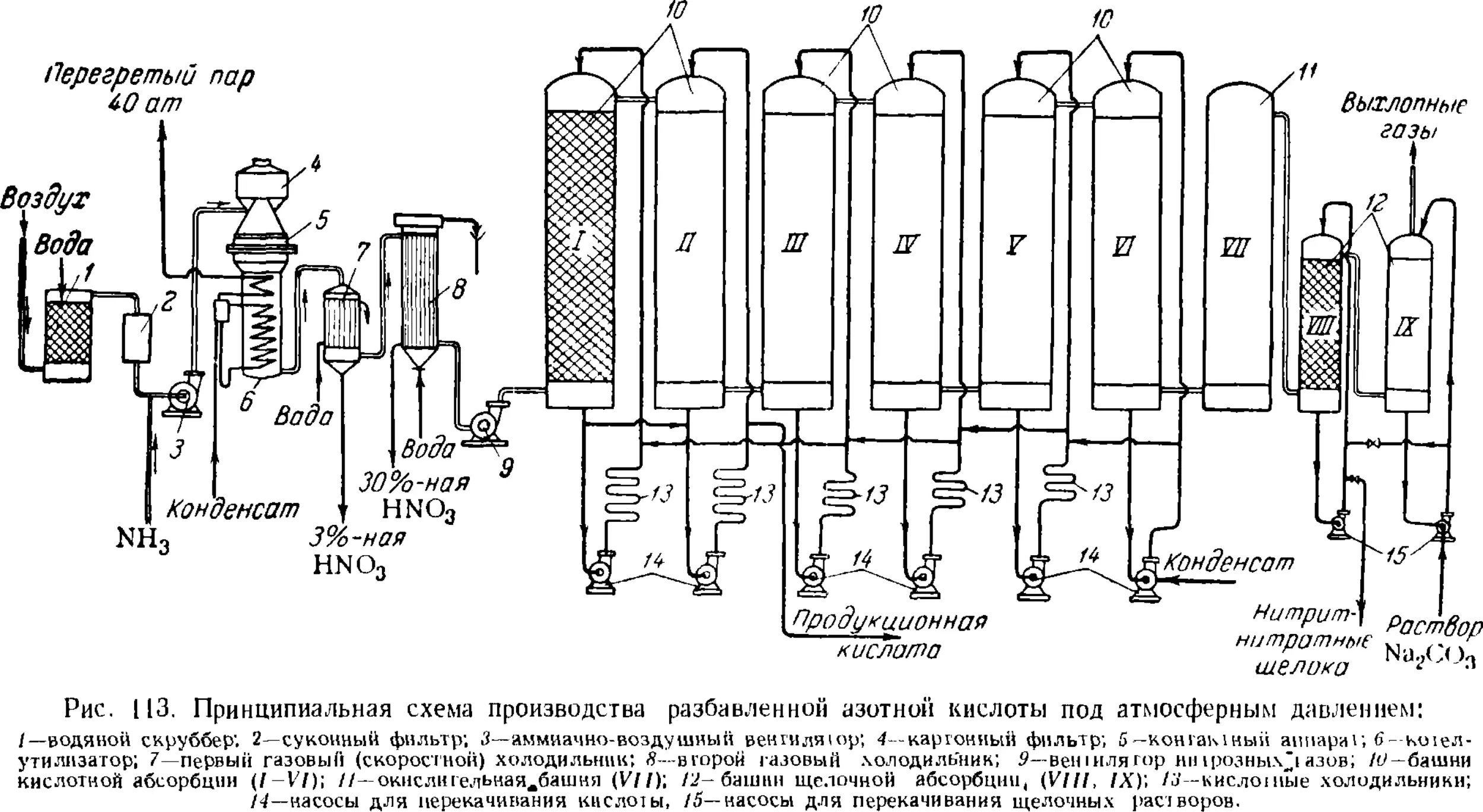
Переработка нитрозных газов в разбавленную азотную
кислоту 270
Технологические схемы производства разбавленной
азотной кислоты 273
3. Концентрирование азотной кислоты 20]
Схемы установок для концентрирования 295
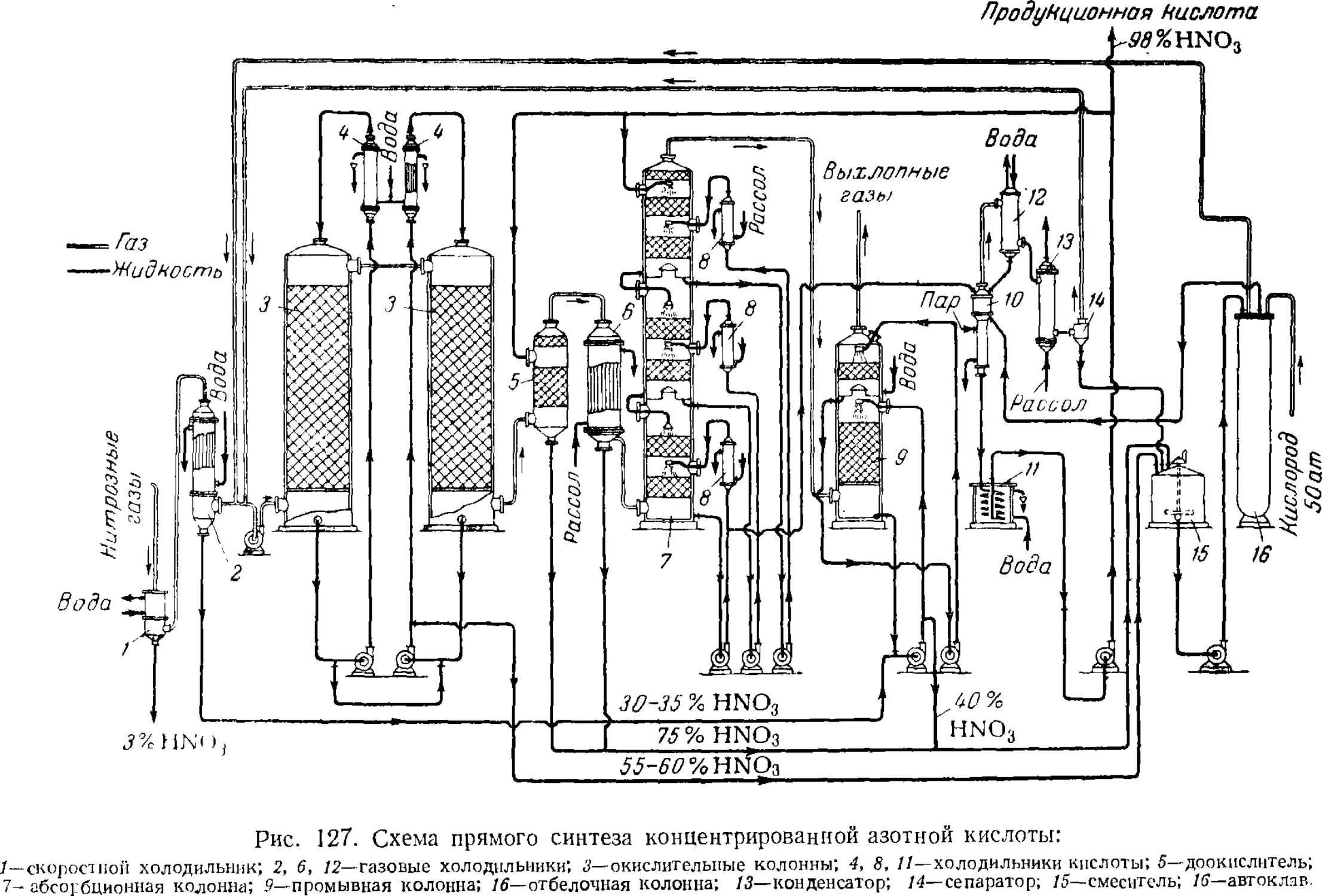
4. Прямой синтез концентрированной азотной кислоты 299
5. Совместное получение серной и азотной кислот 304
Литература (к главам V, VI, VII) 306
Глава VIII. Электрохимические производства 307
Основные сведения по электрохимии 308
!. Электрохимическое производство водорода и кислорода . . 314
Теоретические основы процесса электролиза воды .... 315
Электролизеры 317
2. Электрохимическое производство хлора и щелочи 324
Методы производства хлора 324
Свойства и применение хлора и щелочи 32'i
Сырье для производства хлора н щелочи 331
Приготовление и очистка рассола 333
Теоретические основы процесса электролиза с твердым
(стальным) катодом 33G
Диафрагмы 340
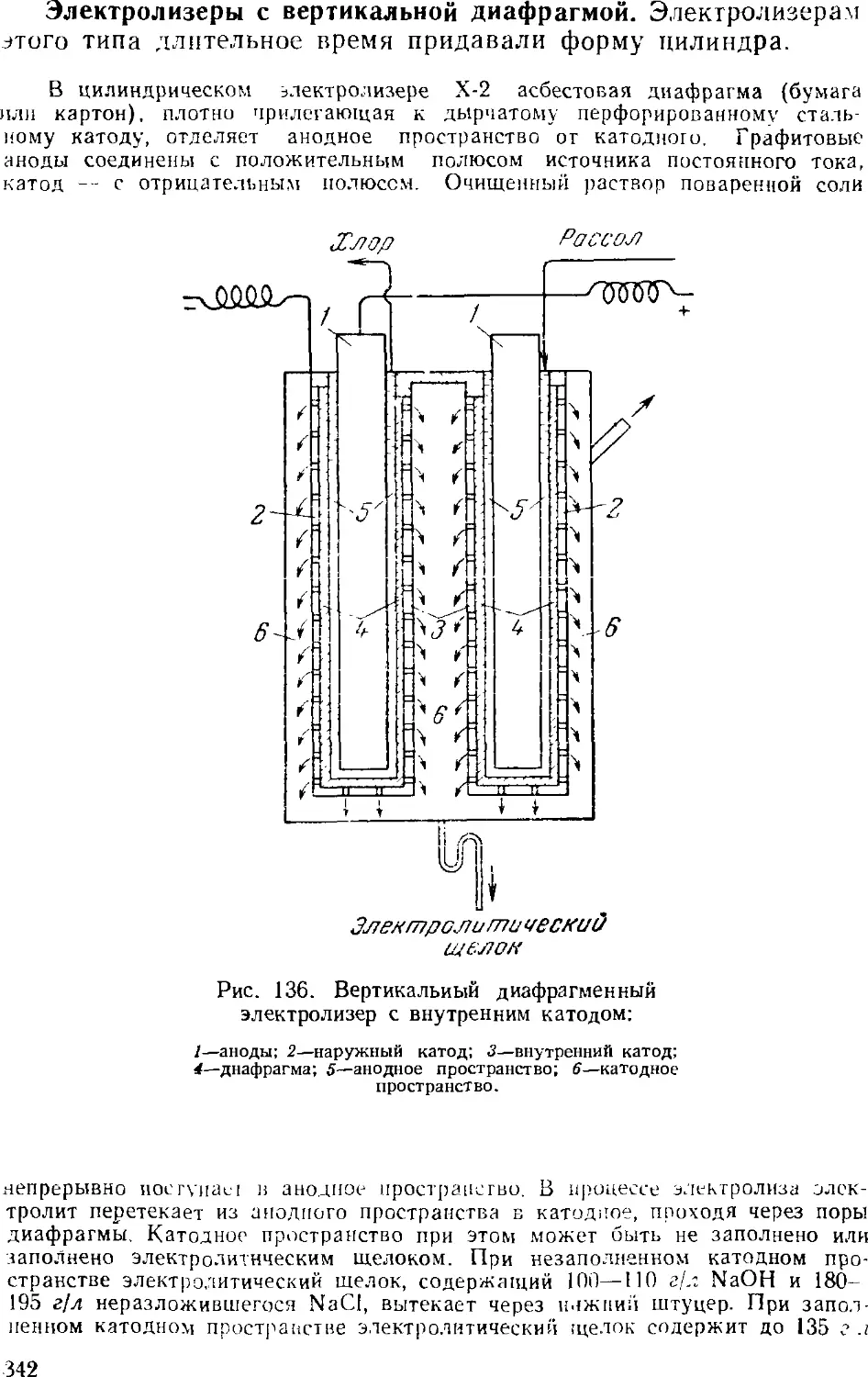
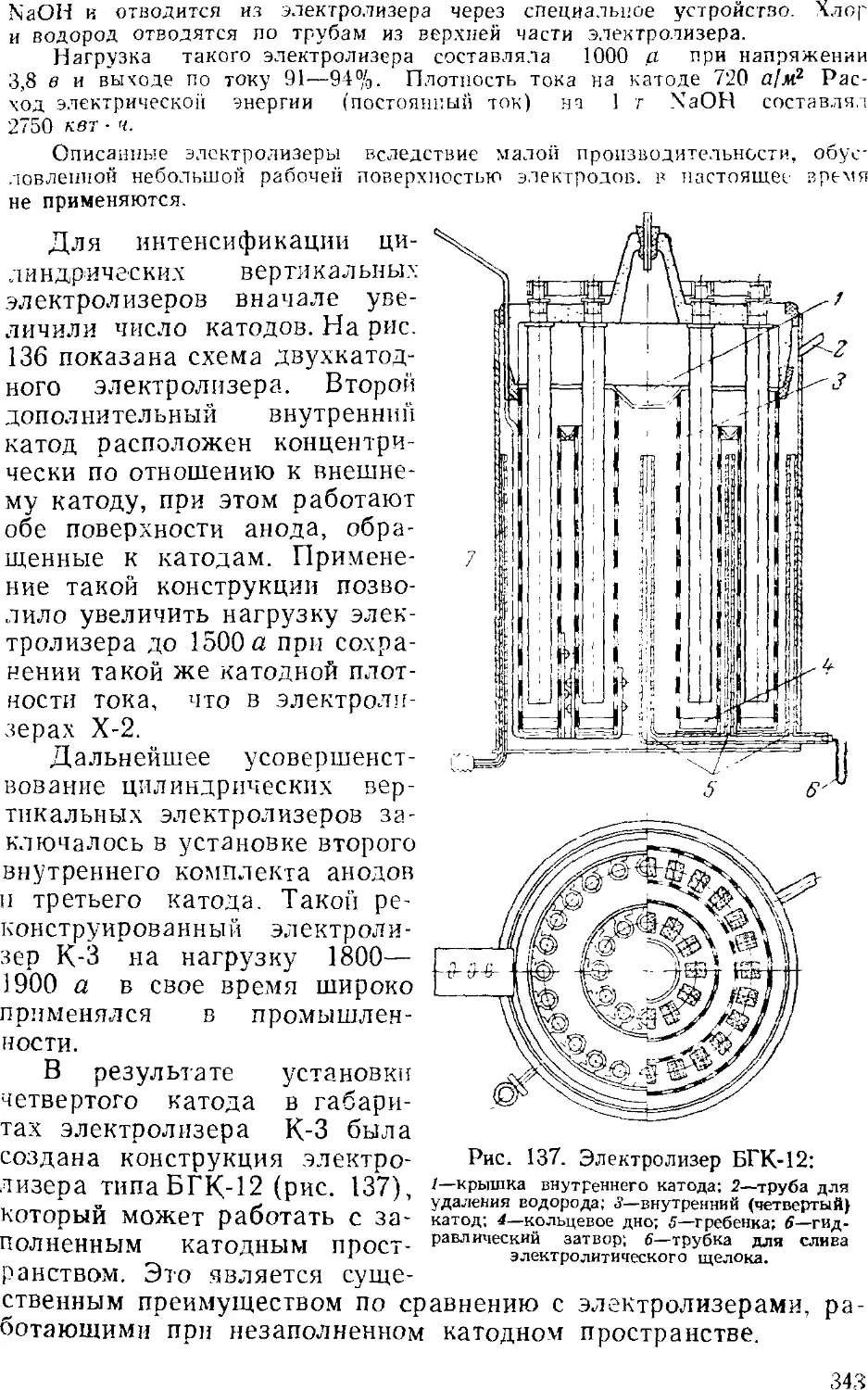
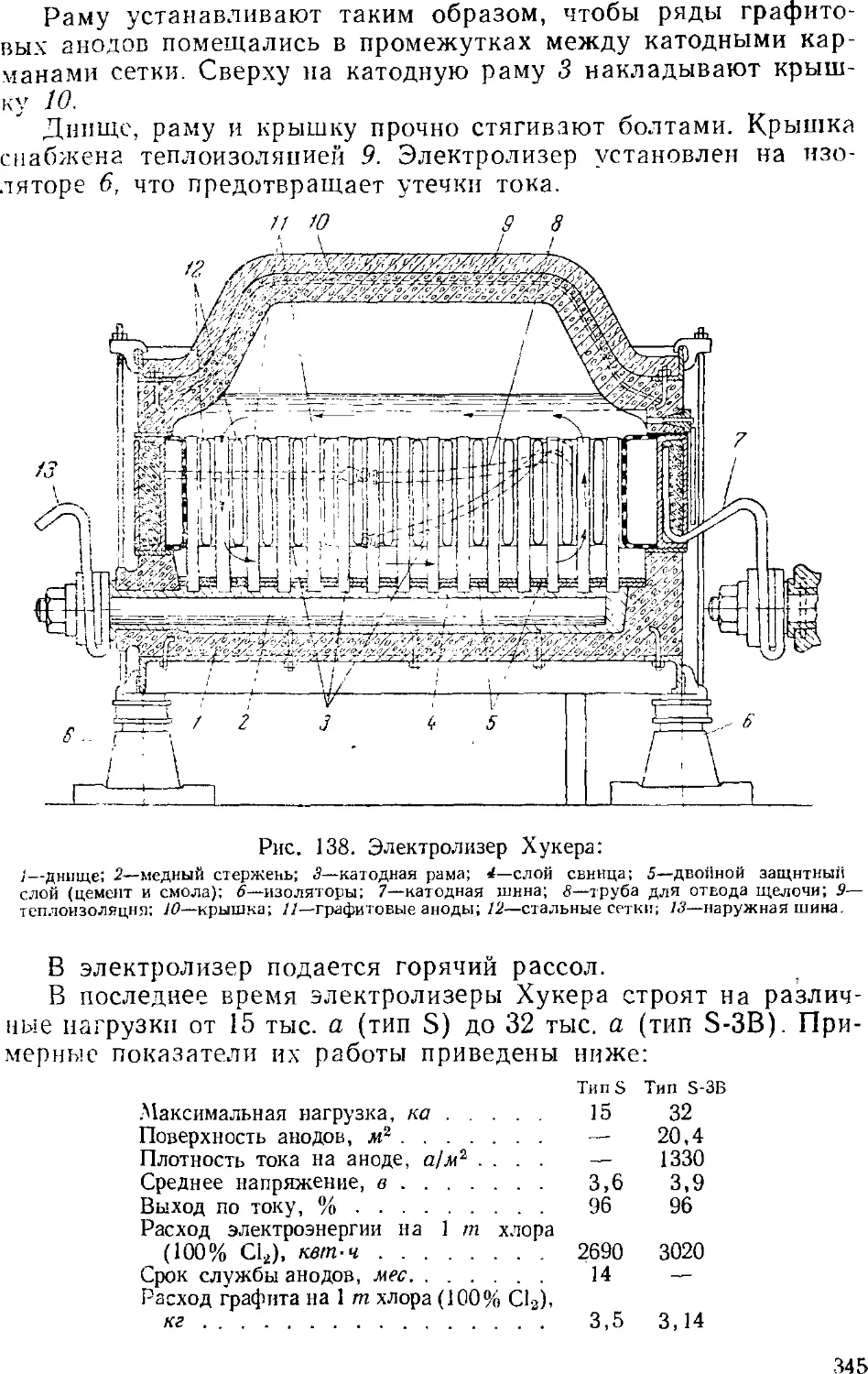
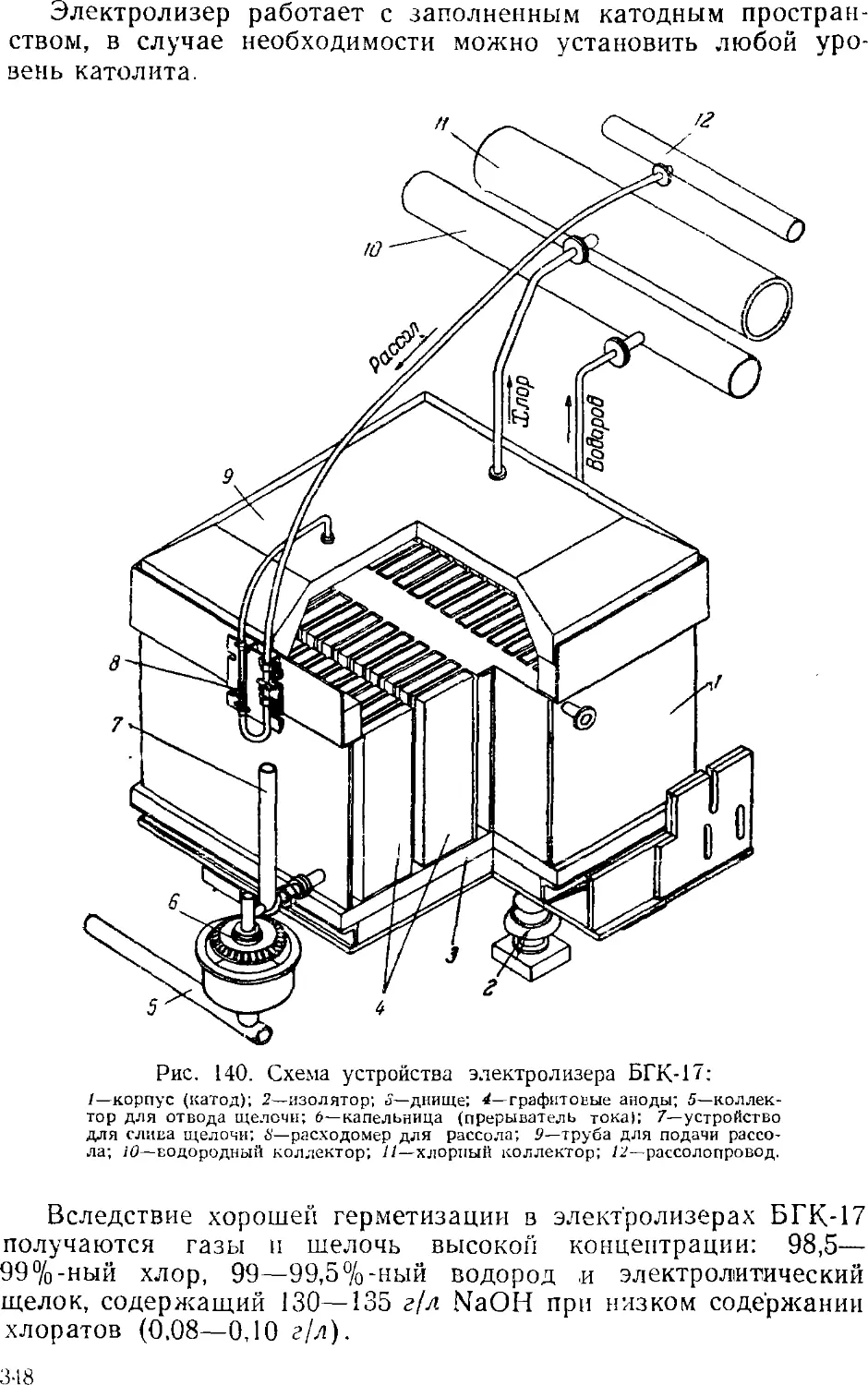
Диафрагменные электролизеры 341
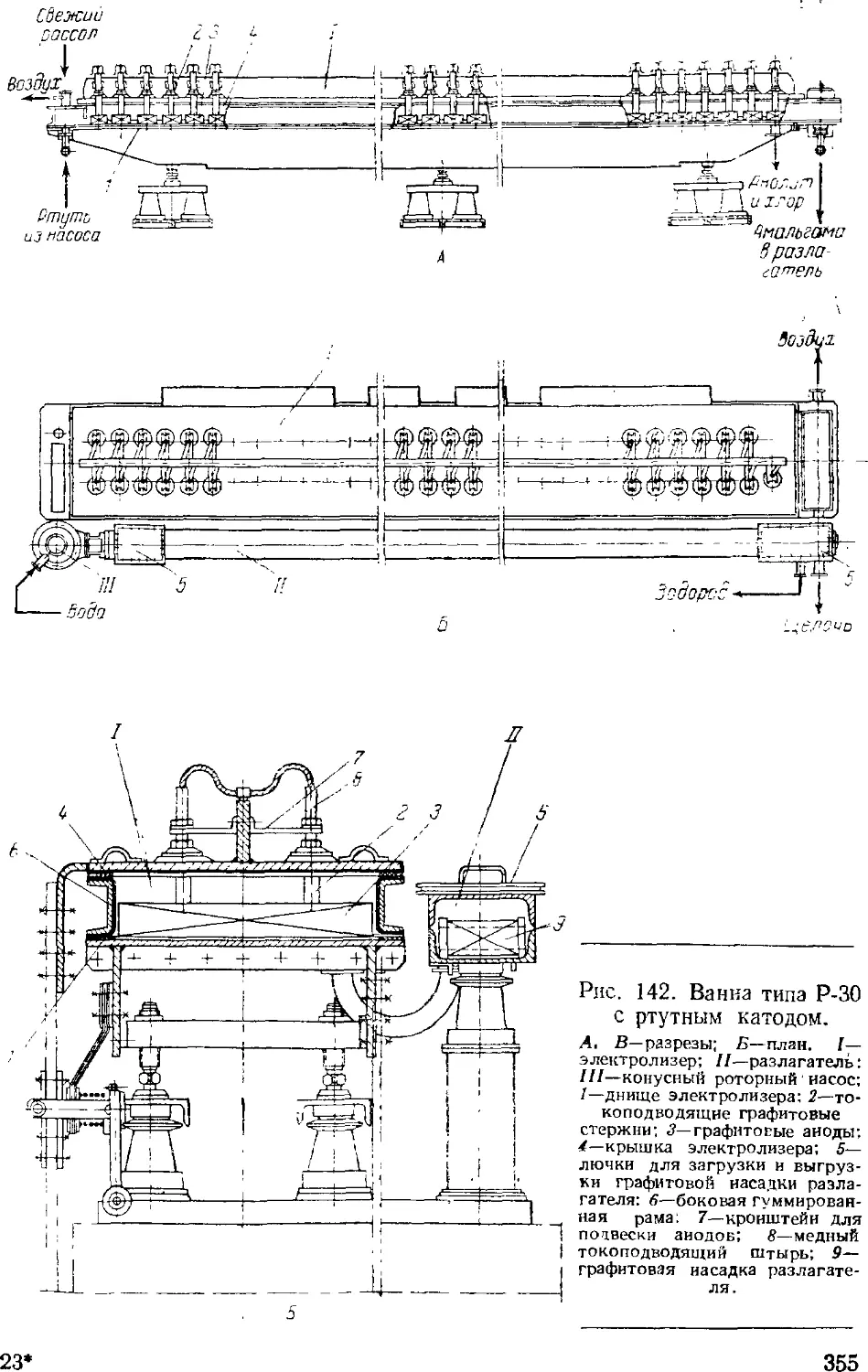
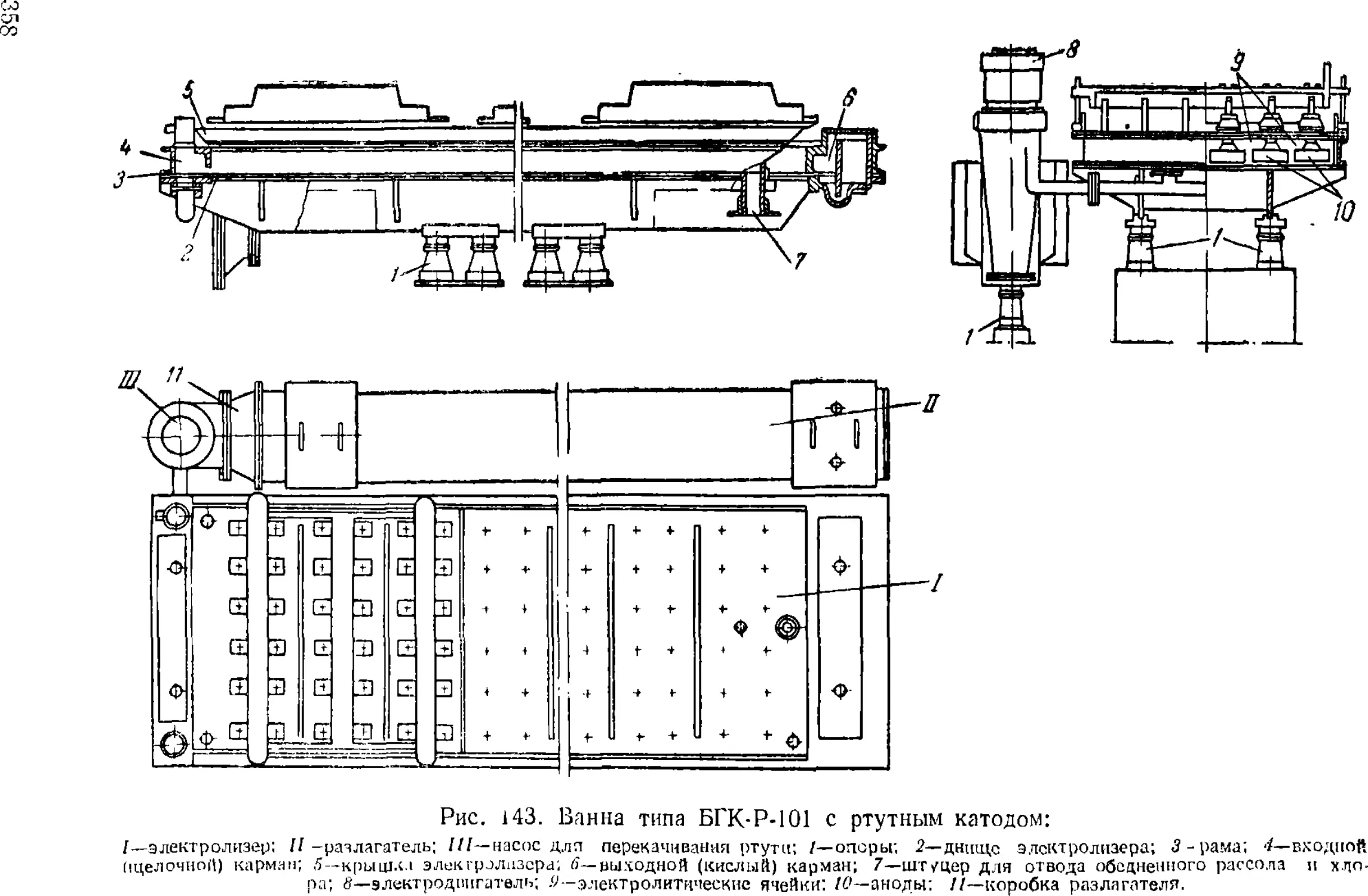
Электролиз с ртутным катодом 351
Ванны с ртутным катодом 354
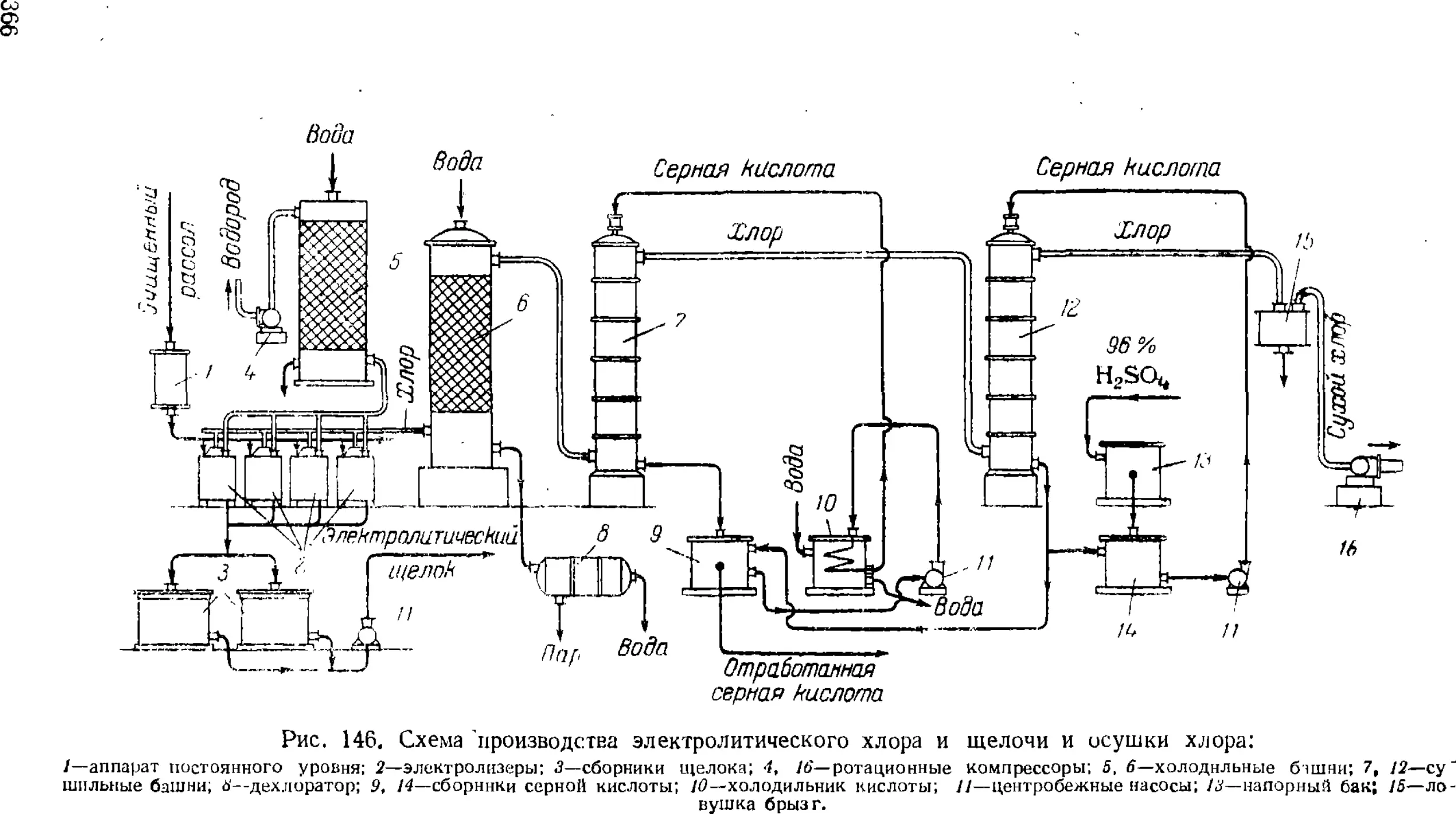
Технологическая схема производства хлора и щелочи . . 365
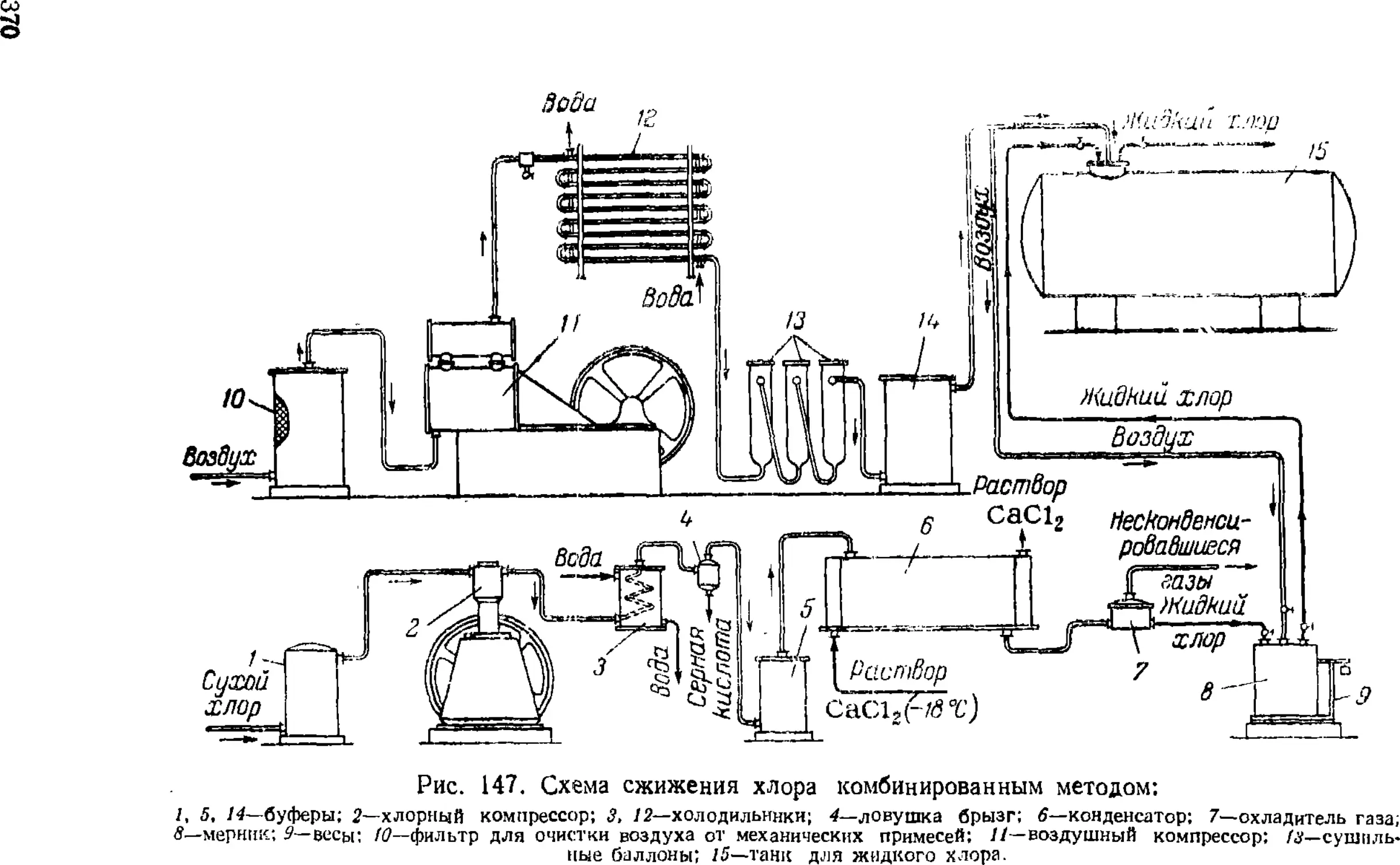
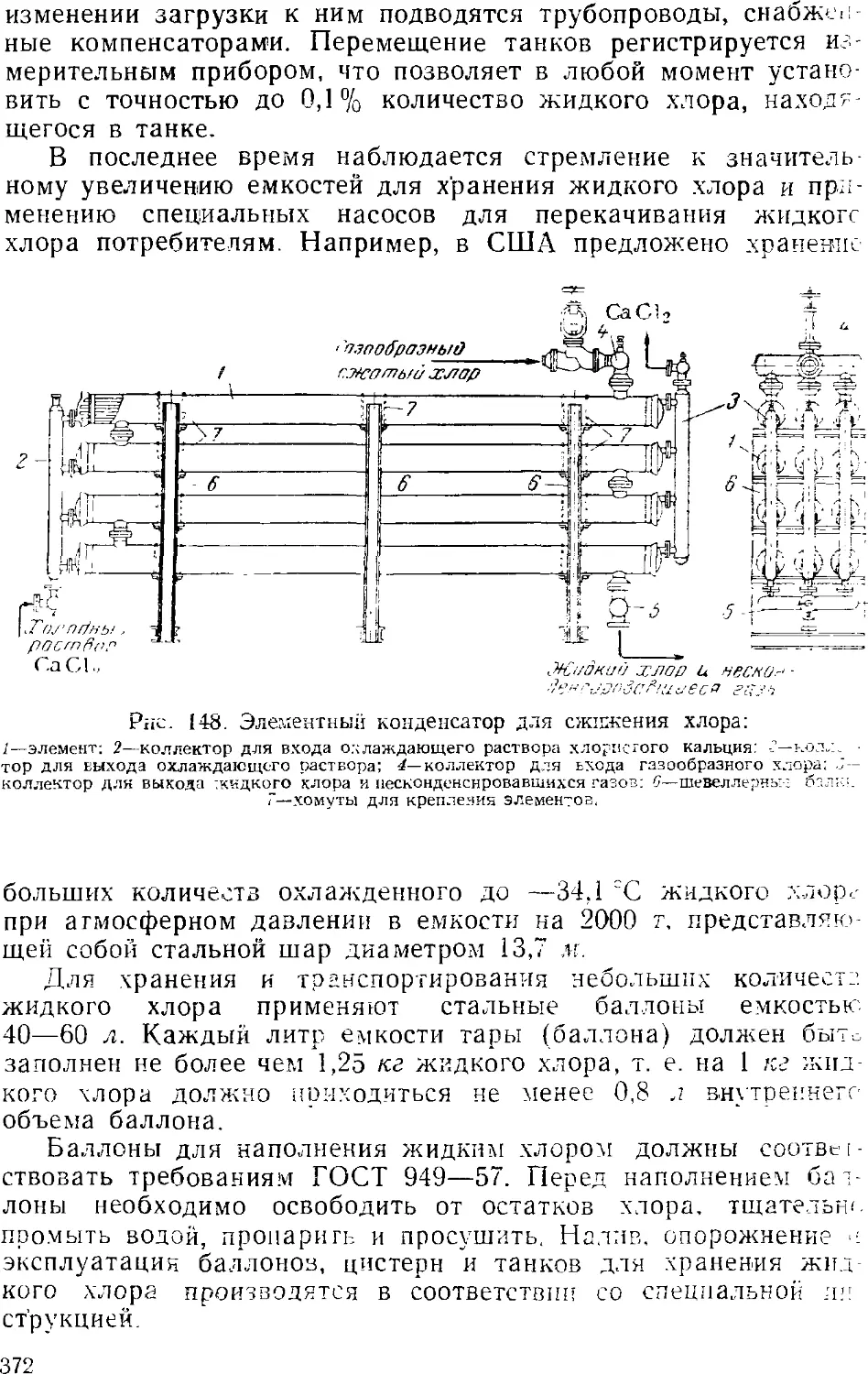
Сжижение .хлора 3GS
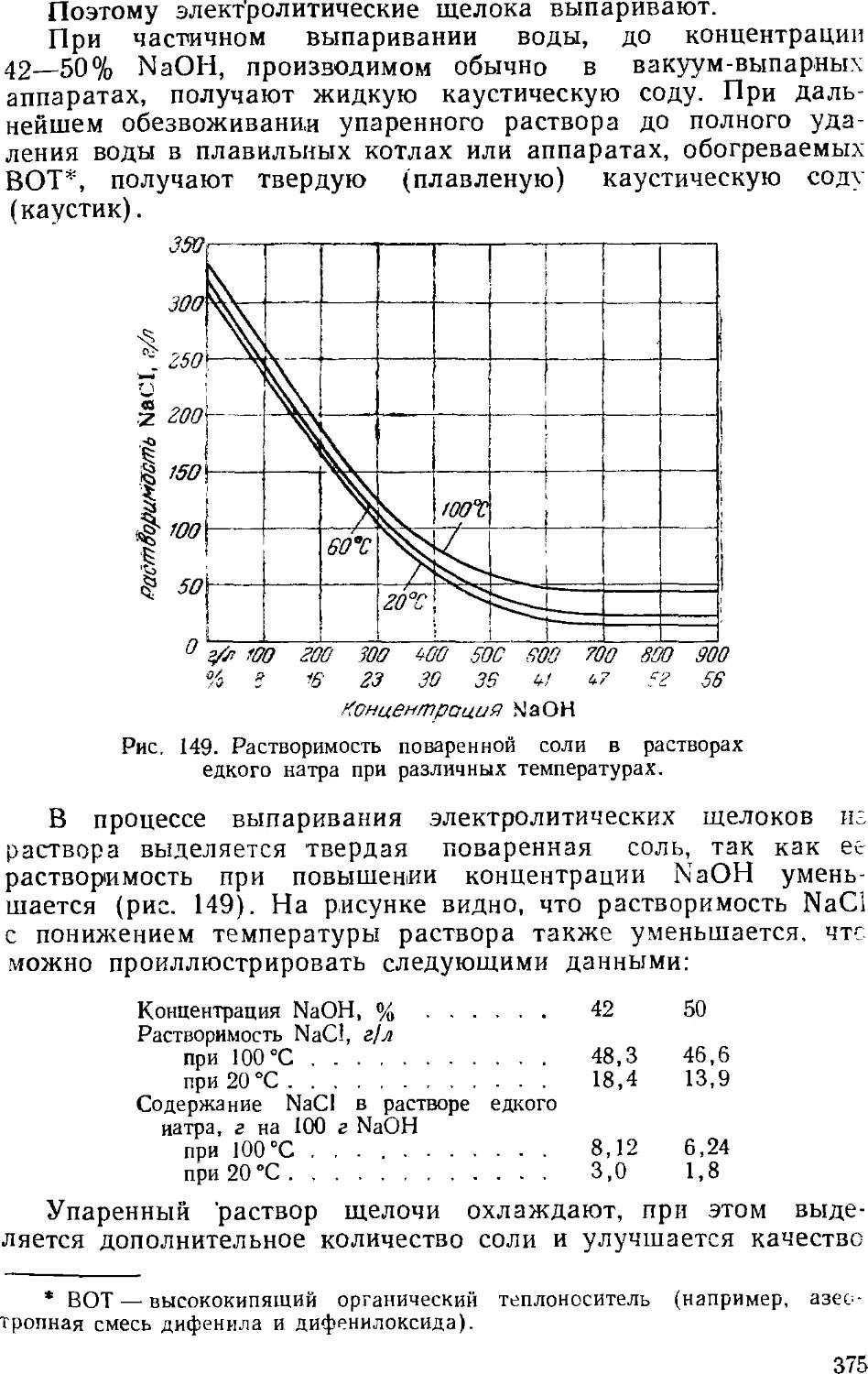
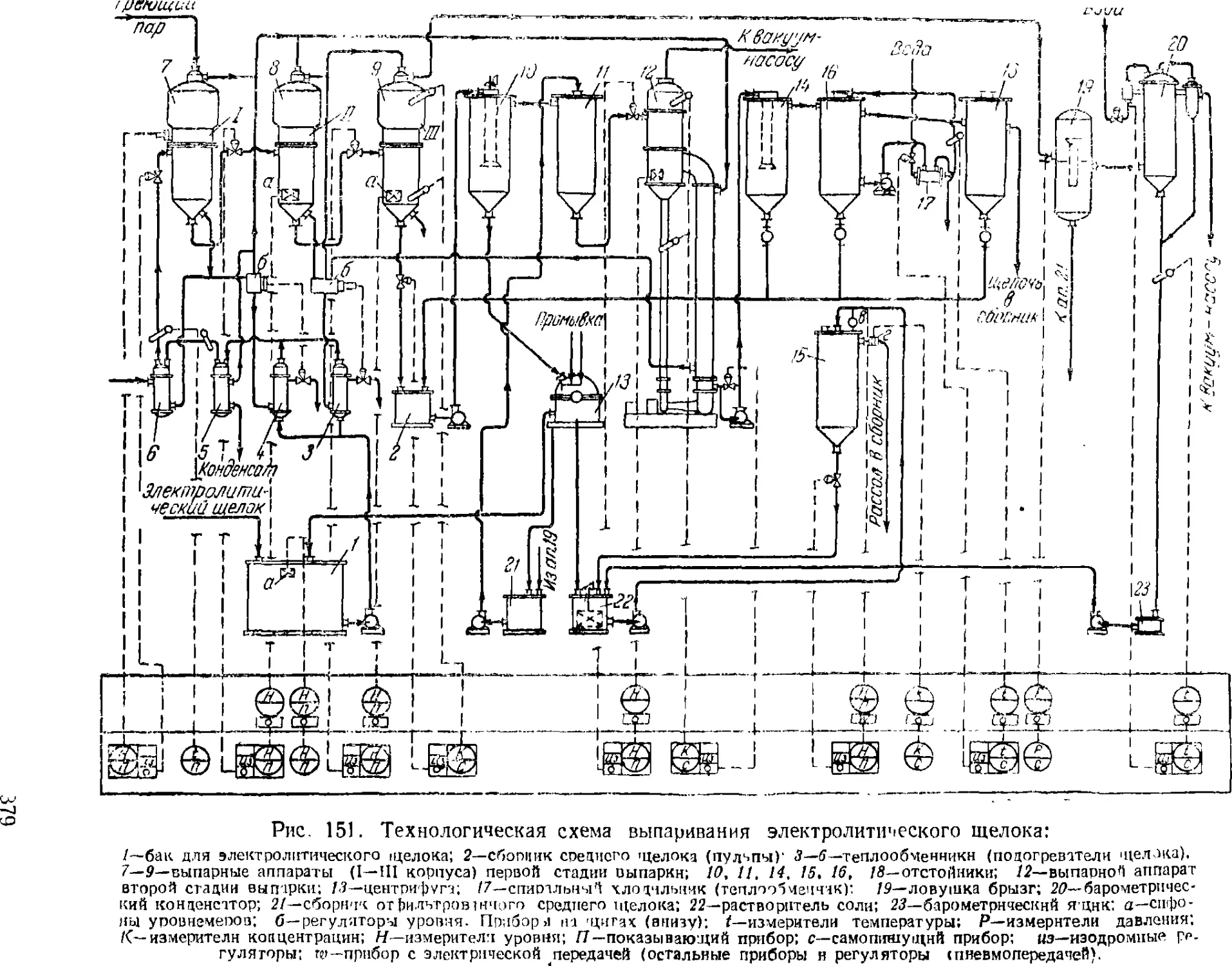
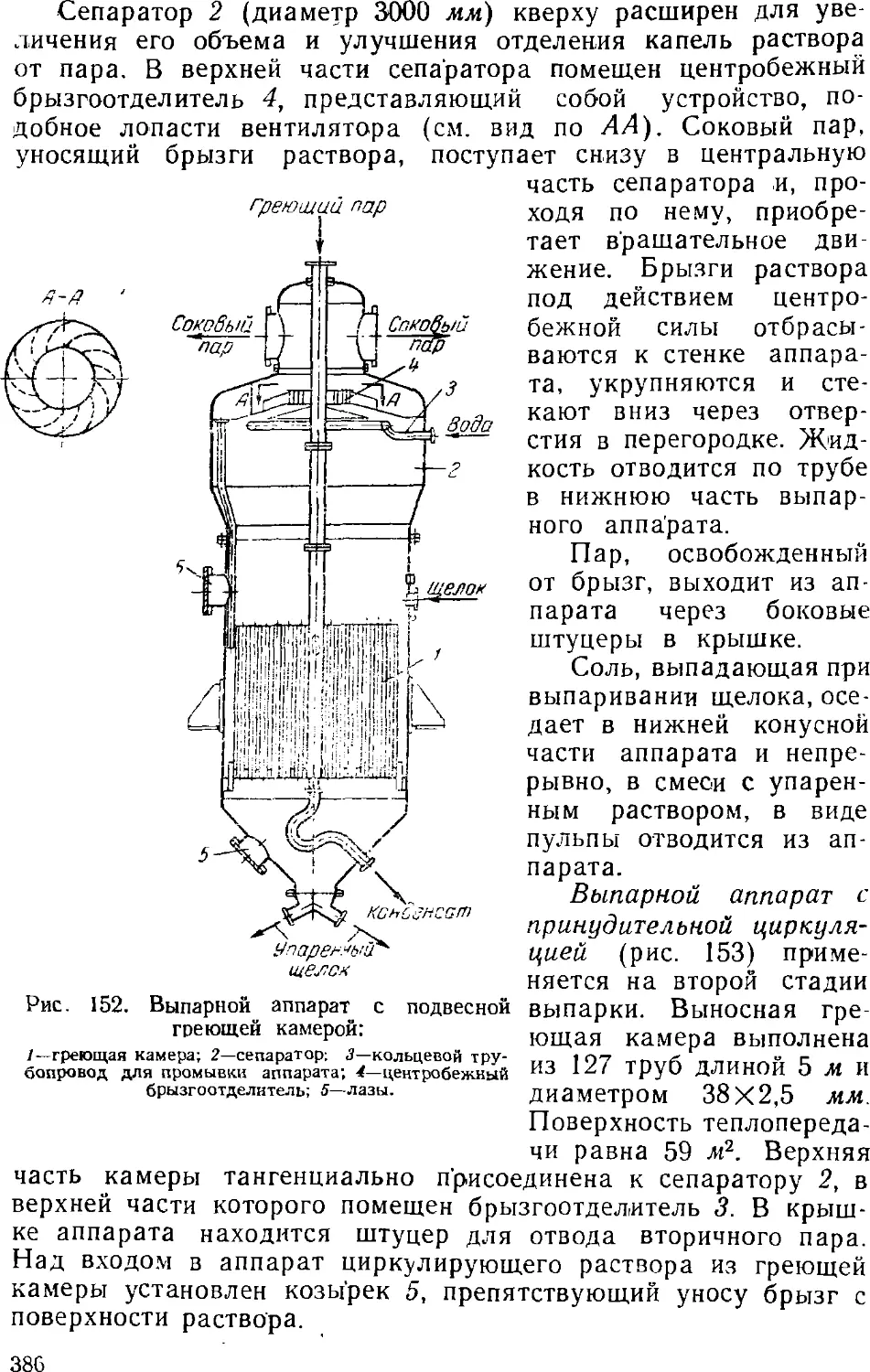
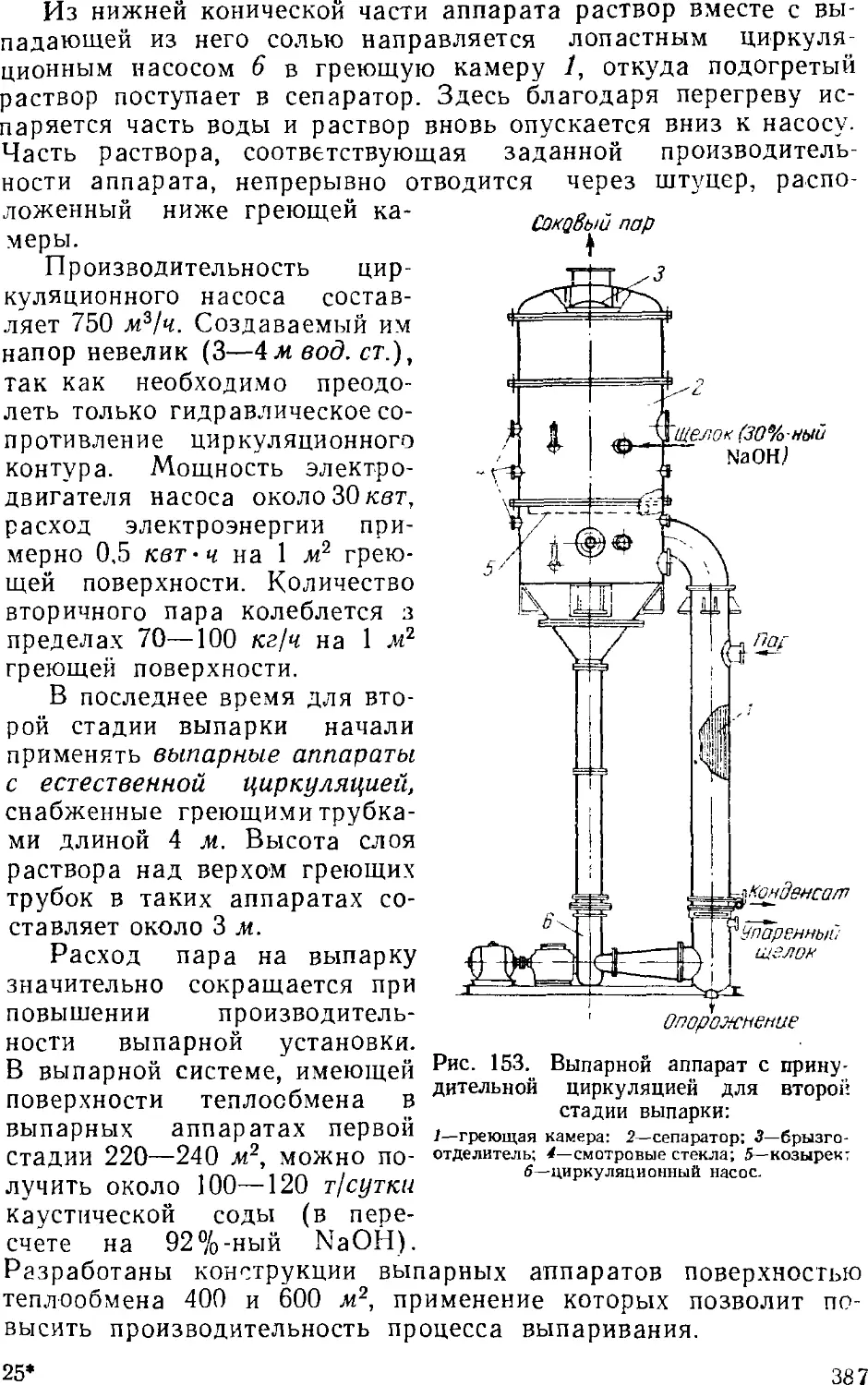
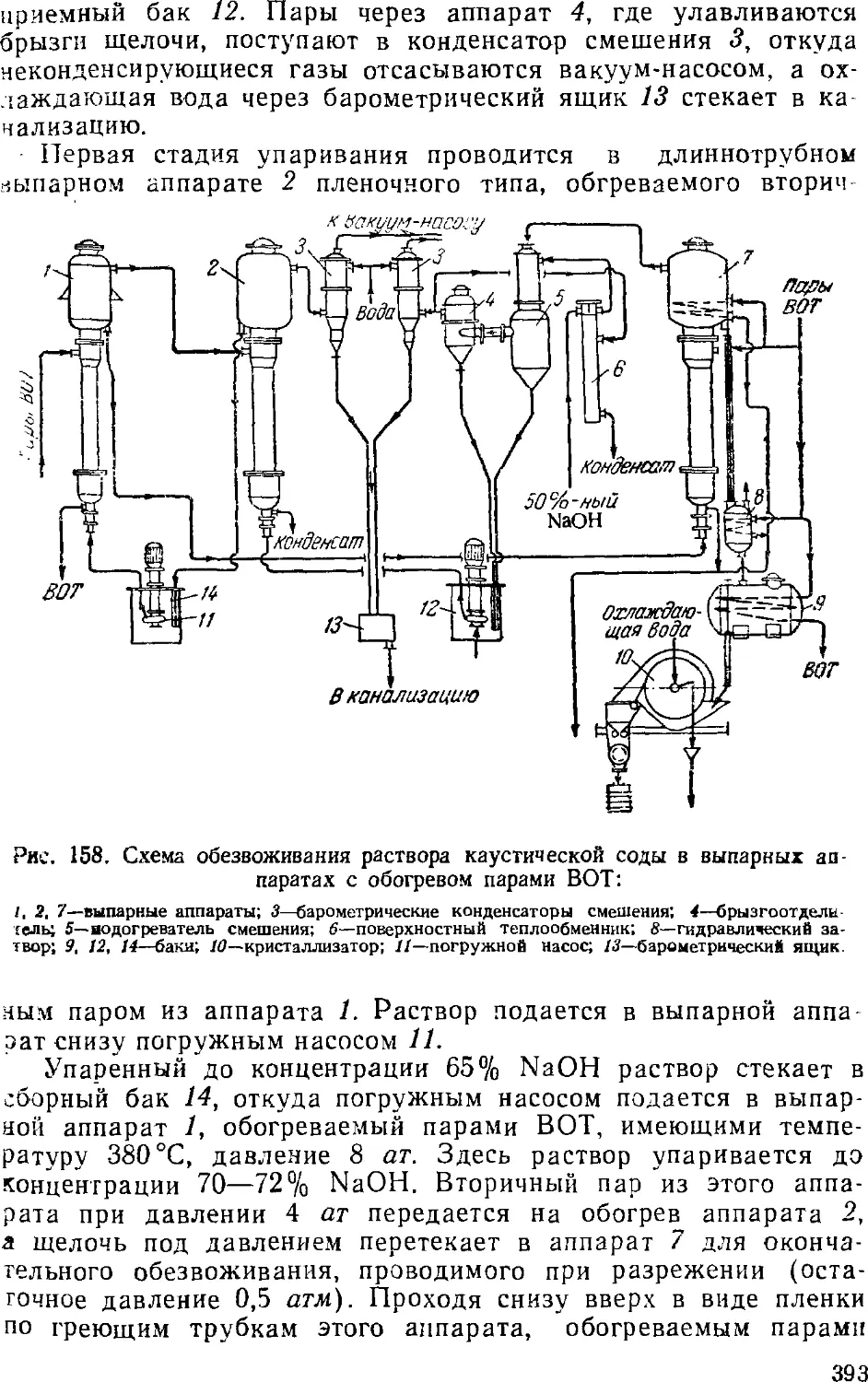
3. Выпаривание электролитического щелока 374
Двухстадинная выпарка 377
Автоматизация процесса ьыпарки 382
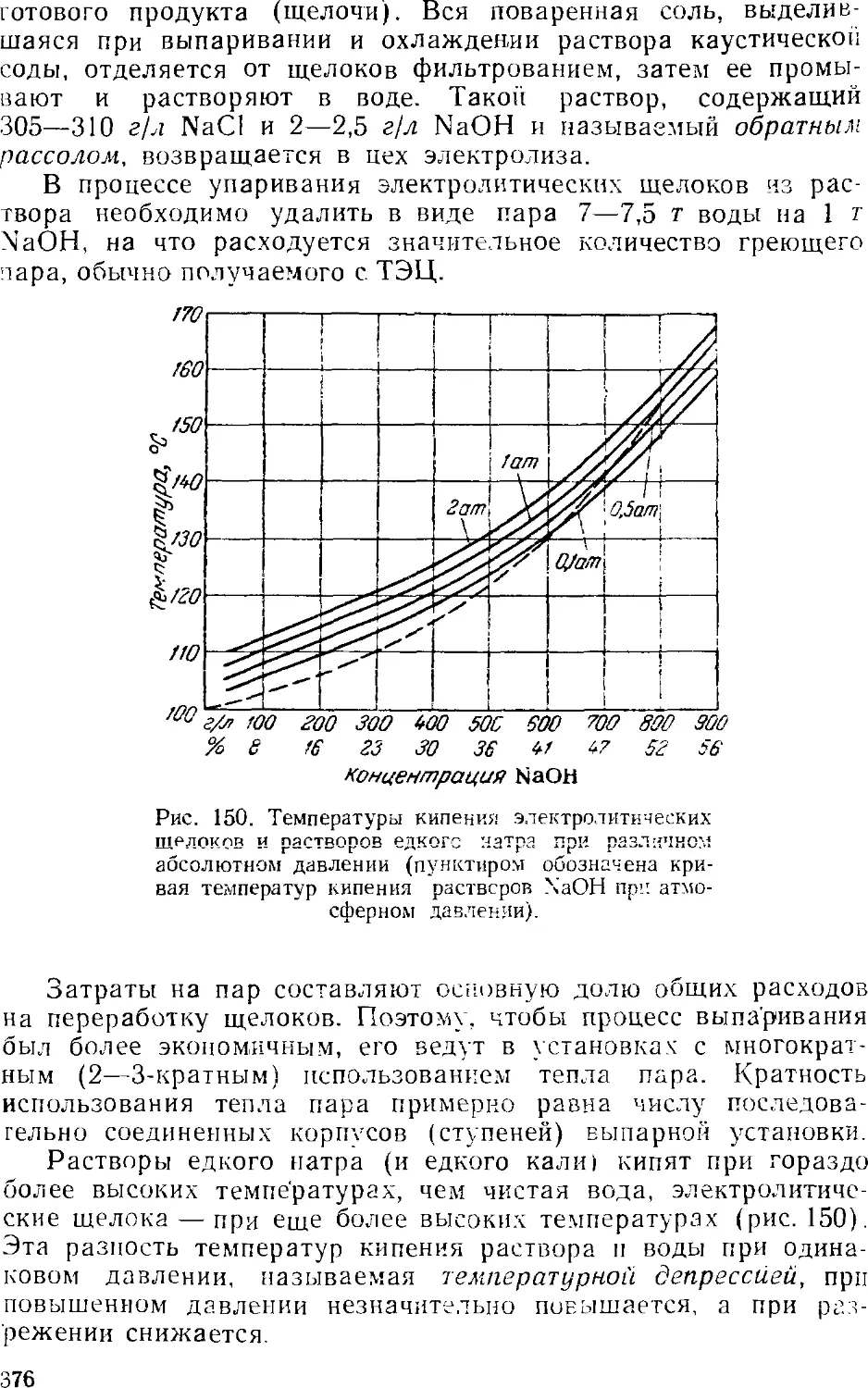
Основная аппаратура выпарных установок 384
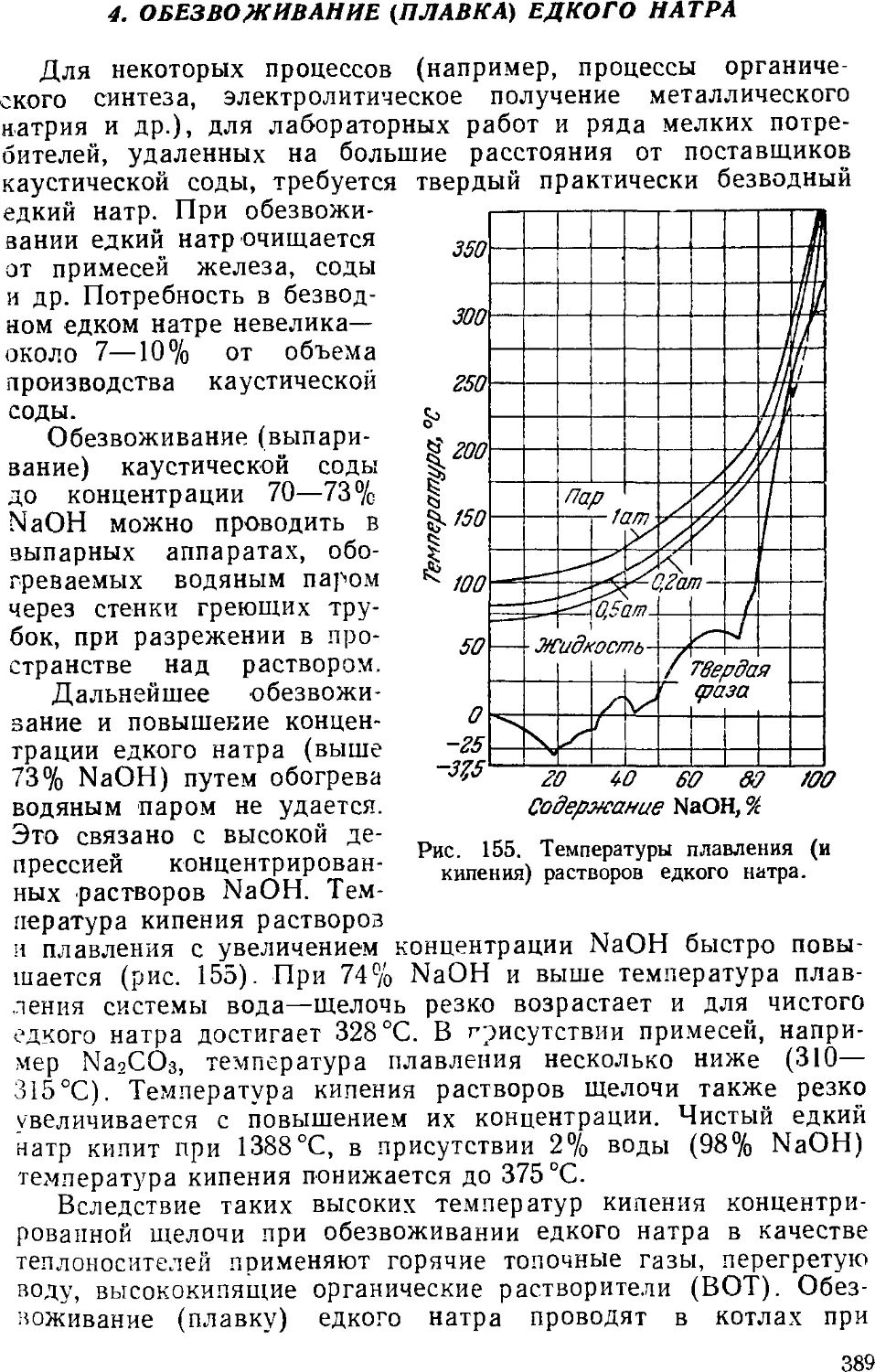
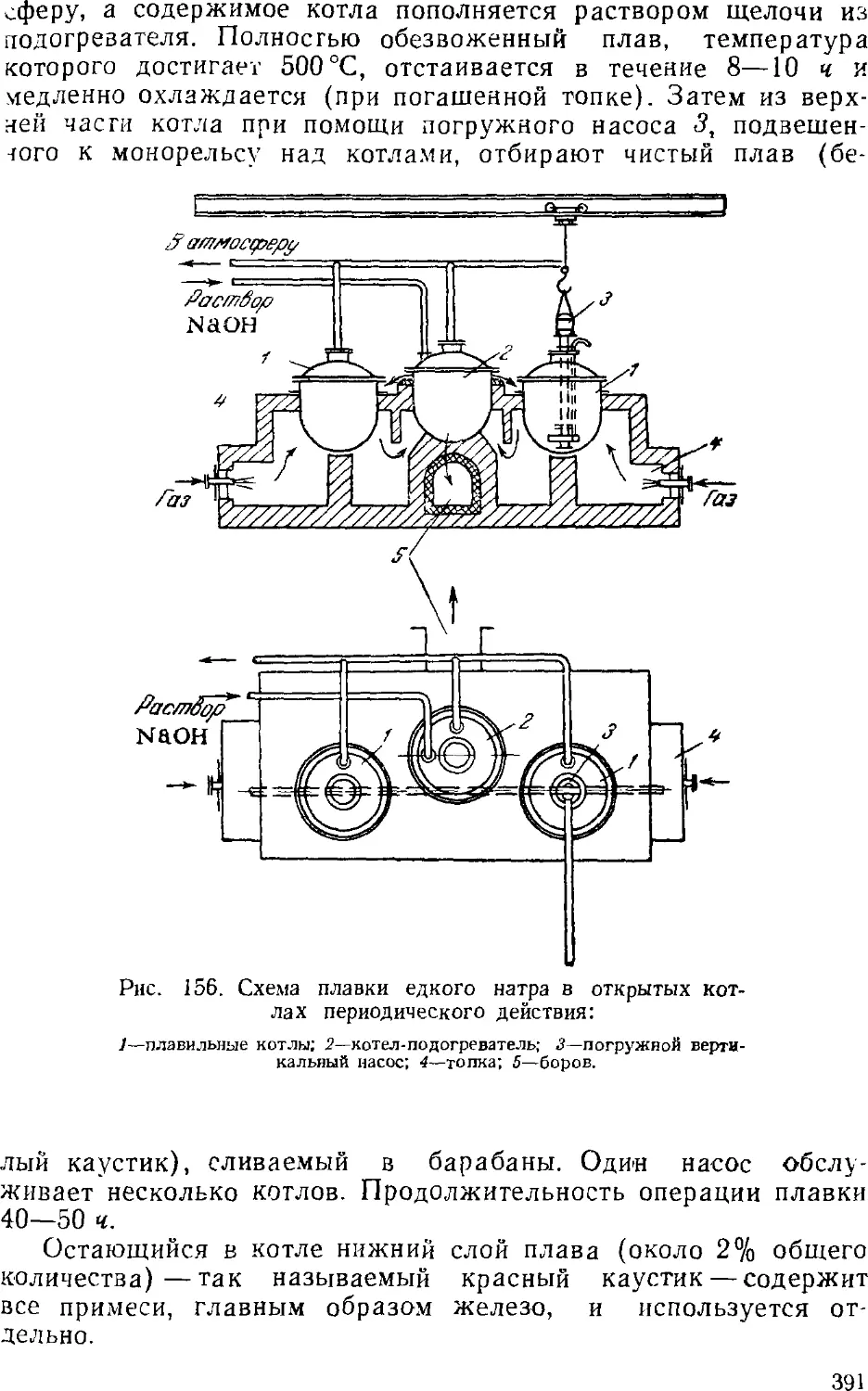
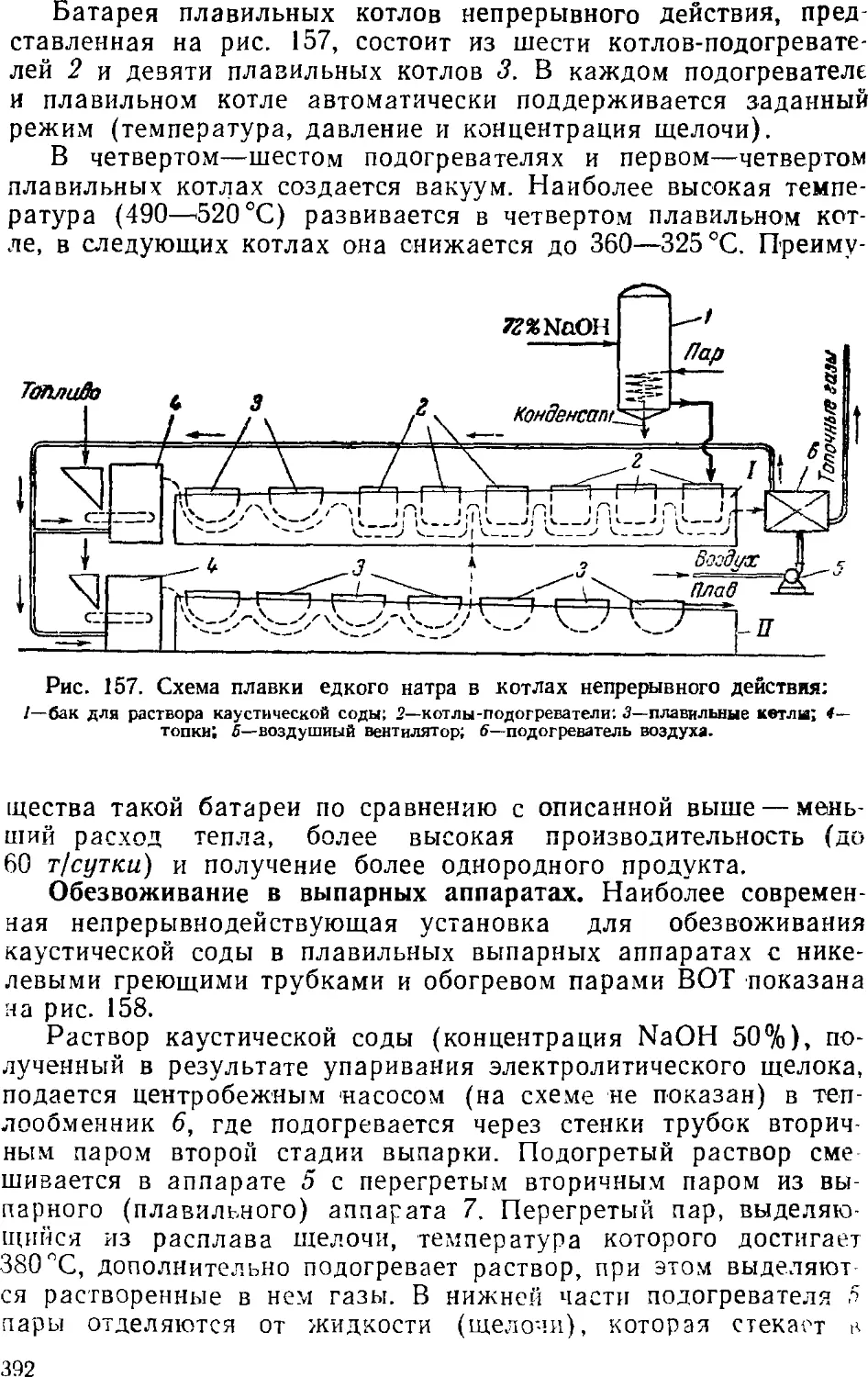
4. Обезвоживание (плавка) едкого натра 389
Глава IX. Переработка электролитического хлора 390
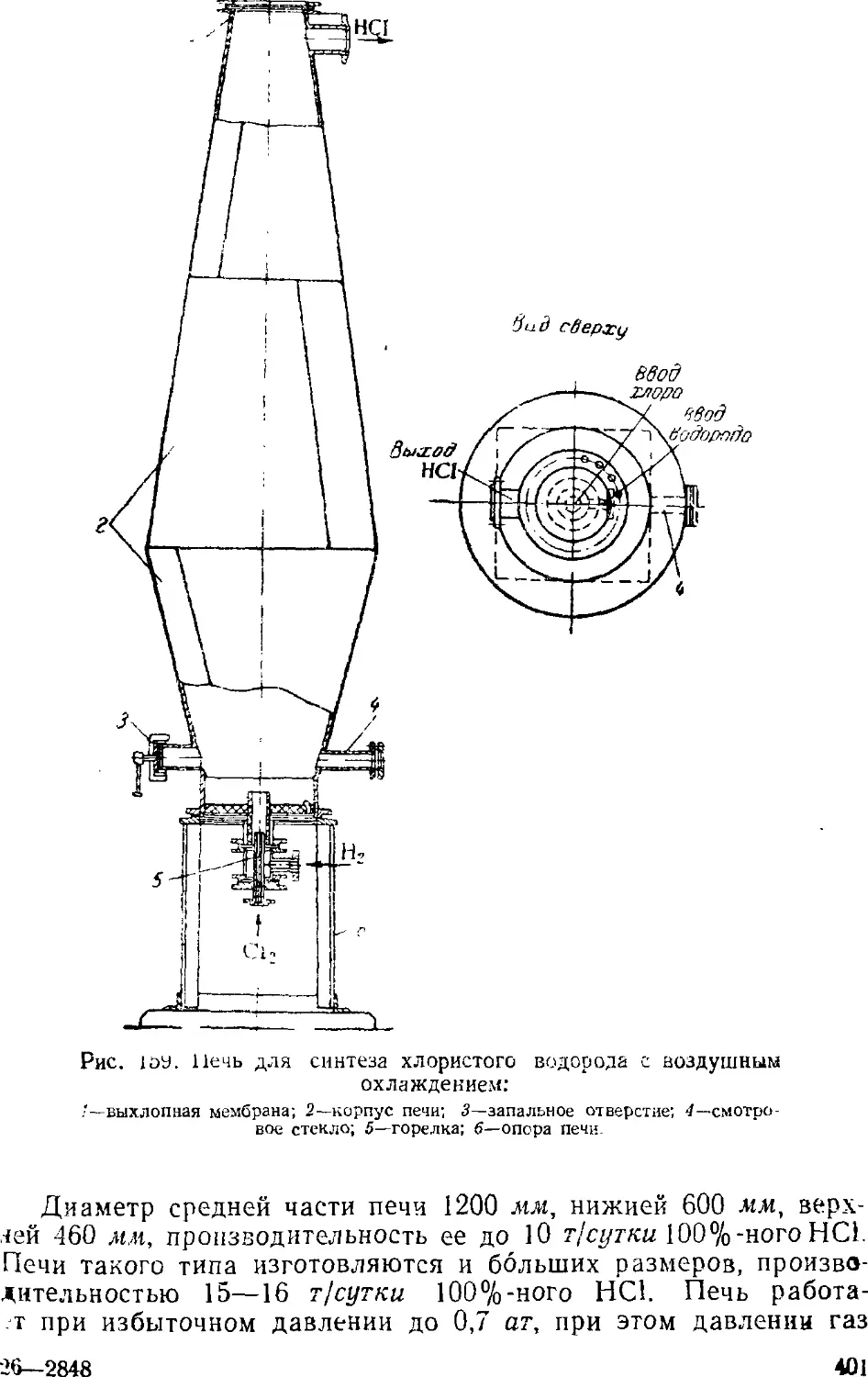
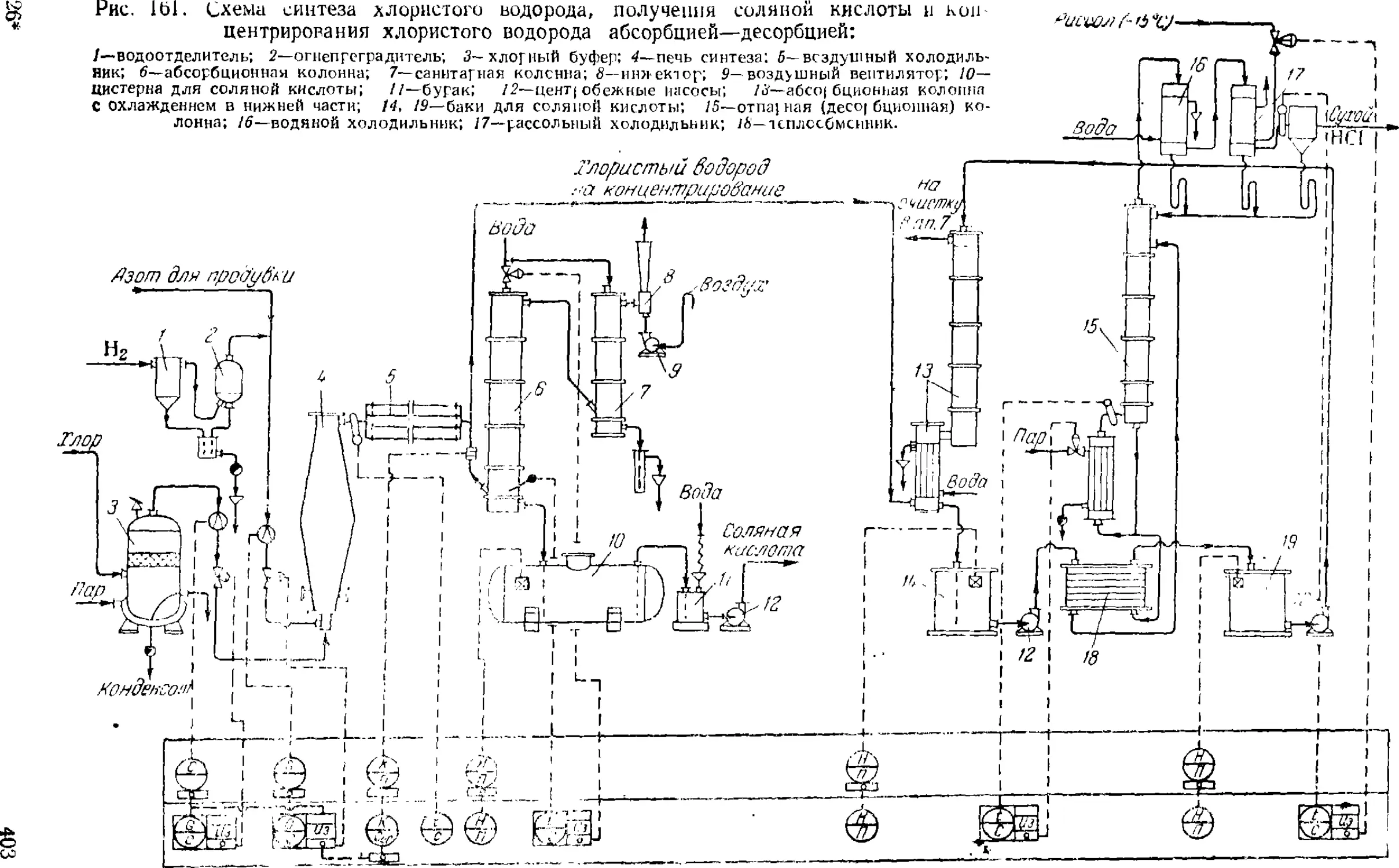
1. Производство хлористого водорода и соляной кислоты . . . 39G
Способы производства хлористого водорода и соляной
кислоты 397
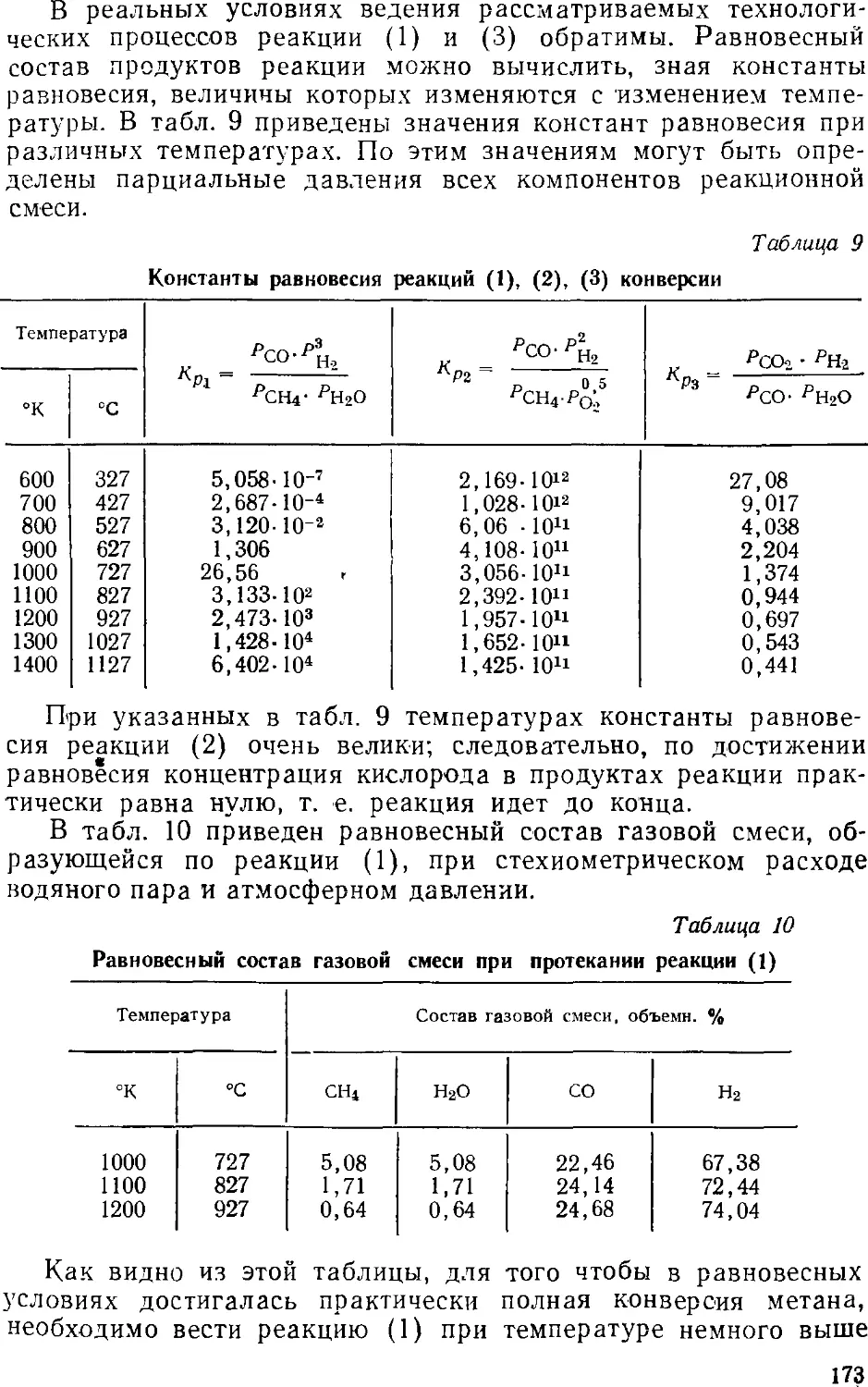
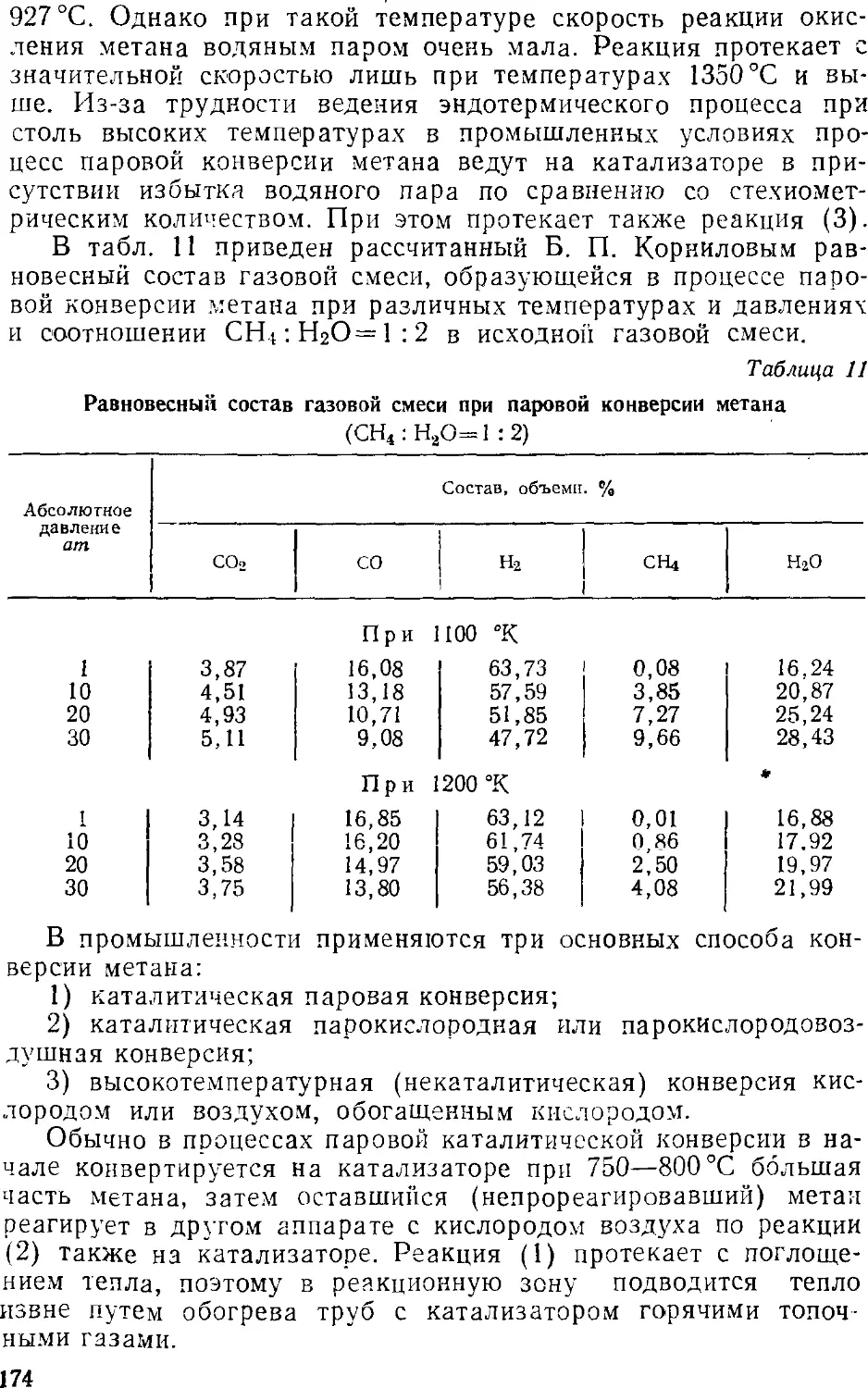
Синтез хлористого водорода 31)9
Получение соляной кислоты 404
Концентрирование хлористого водорода 405
5
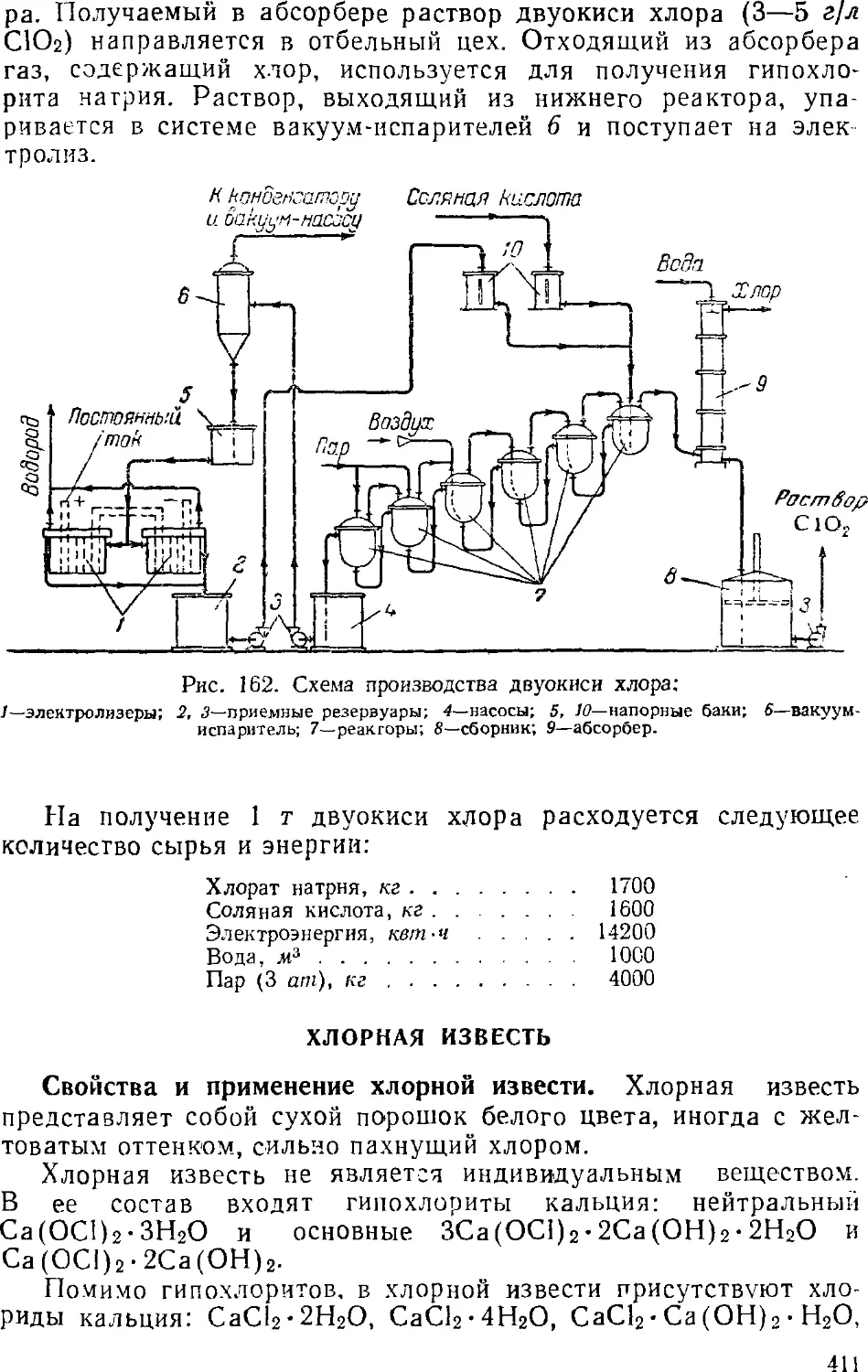
2. Производство кислородсодержащих хлебопродуктов 40S
Двуокись хлора 408
Хлорная известь 4] 1
Гипохлорит кальция 417
Белильные растворы 41Э-
Литература (к главам VIII и IX) 420
Глава X. Содовые продукты 421
1. Кальцинированная сода. Общие сведения 421
Синтетические способы получения кальцинированной содь: 423-
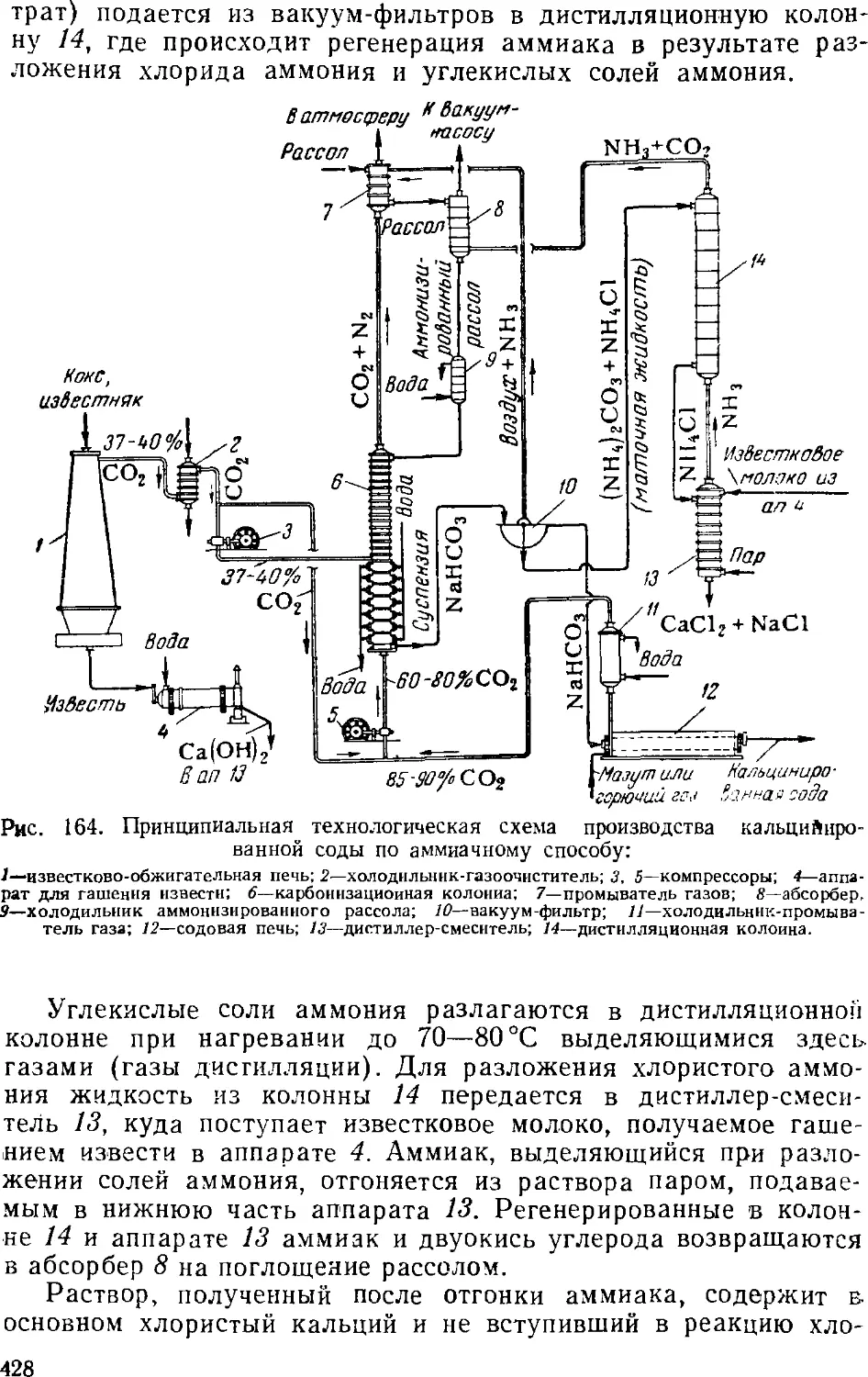
2. Производство кальцинированной соды аммиачным
способом 425
Принципиальная схема производства кальцинированной
соды 427
3. Важнейшие операции и аппаратура производства
кальцинированной соды 430
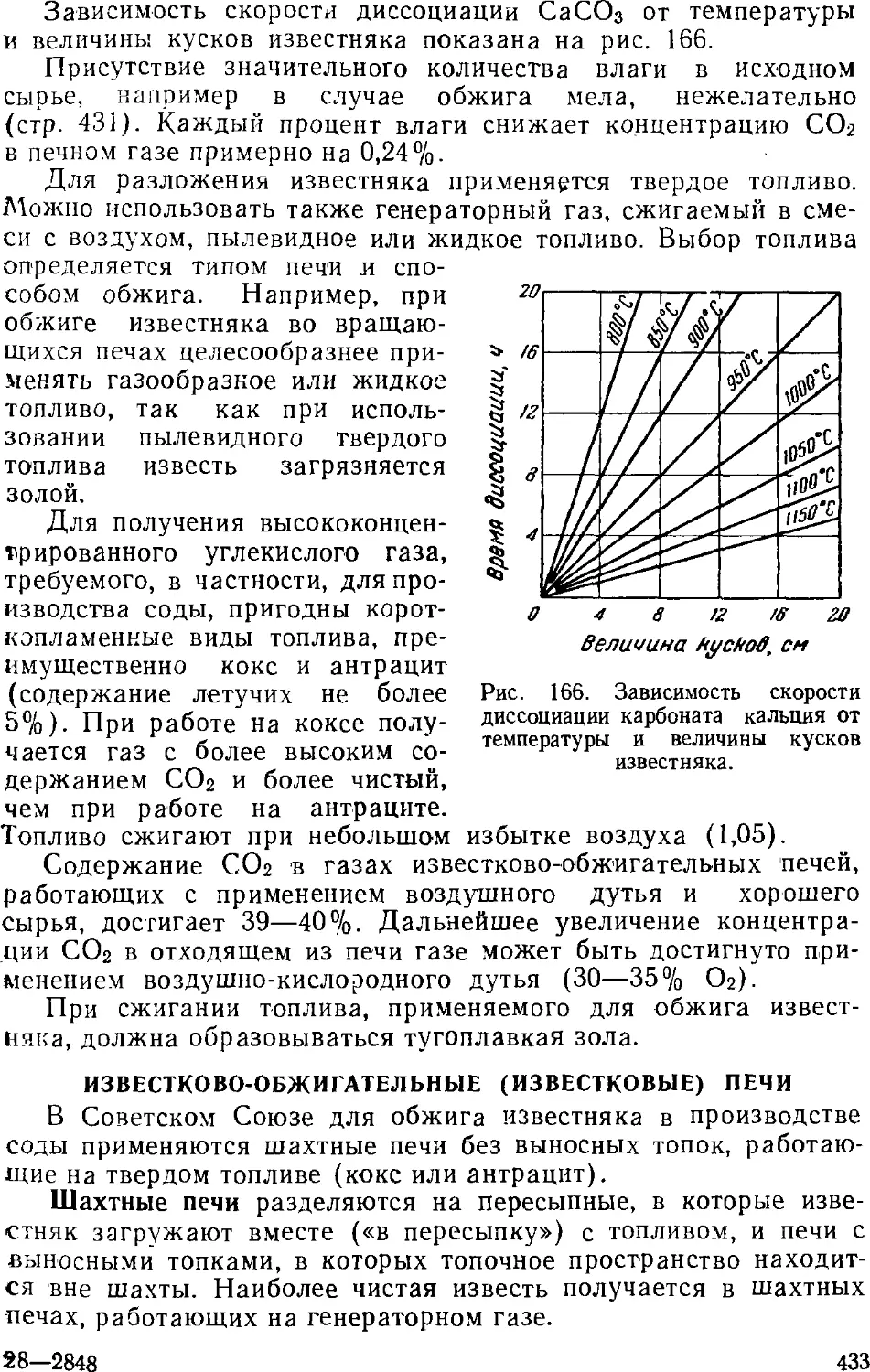
Получение извести и двуокиси углерода 430
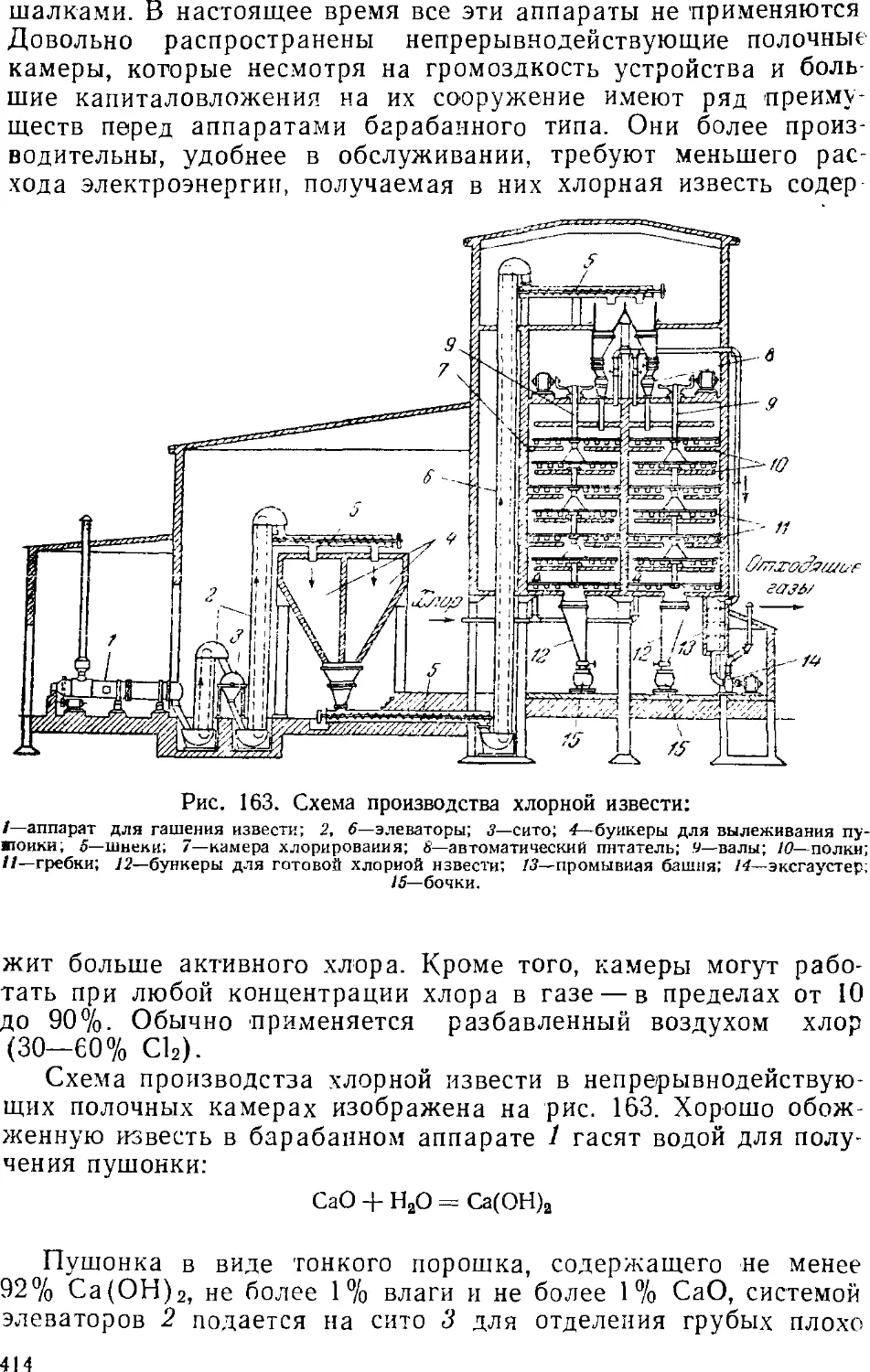
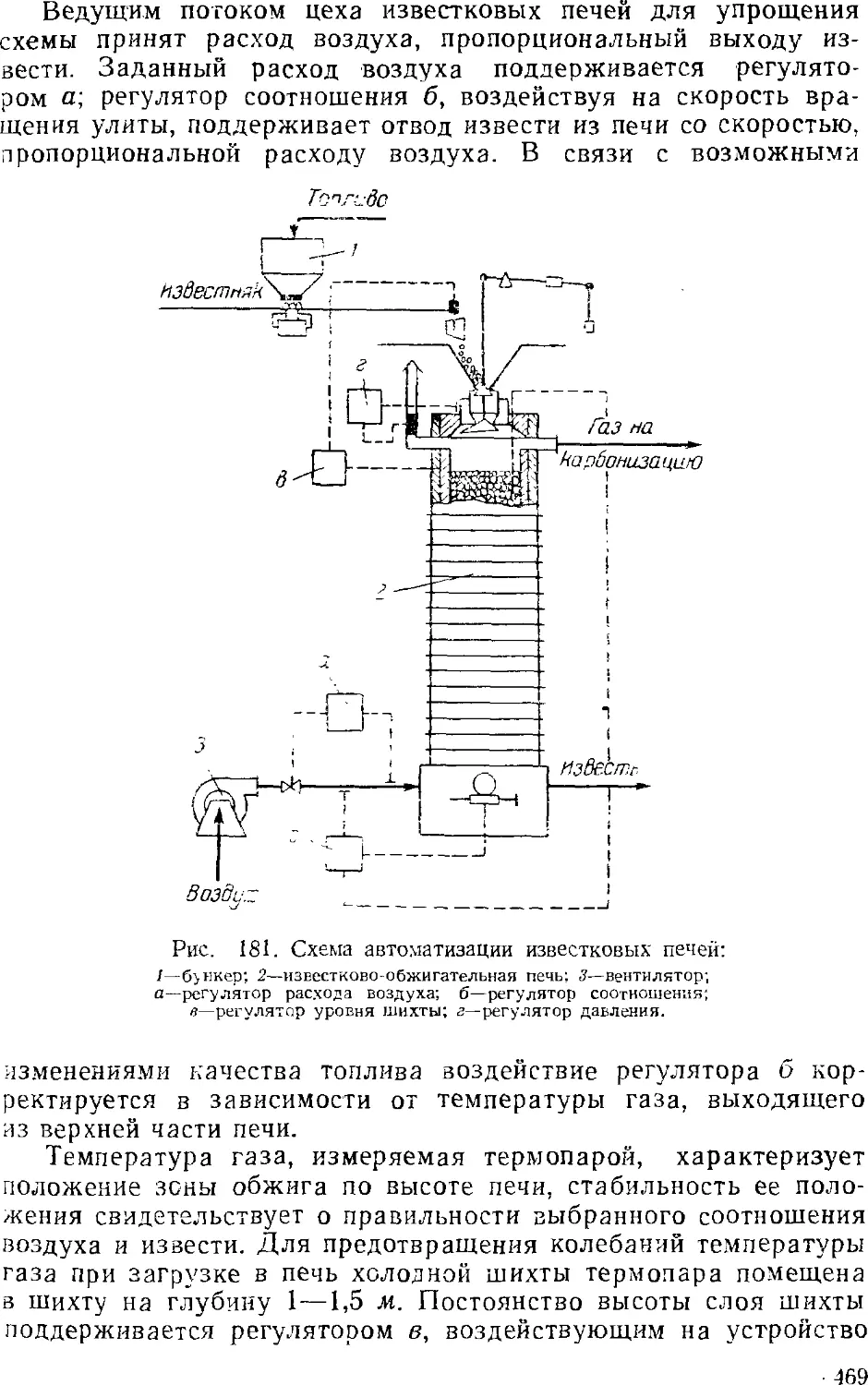
Известково-сбжигательные (известковые) печи 433-
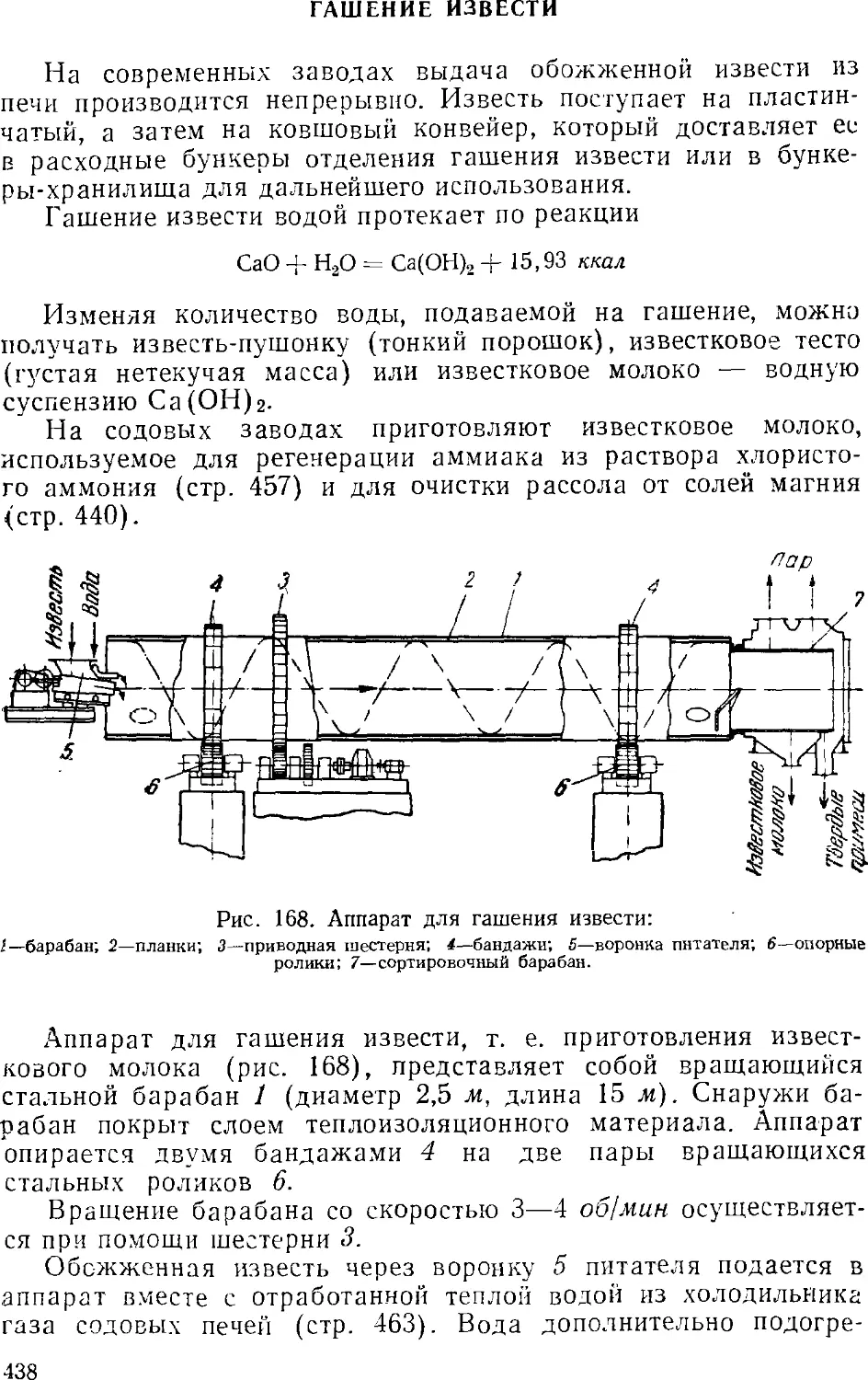
Гашение извести 438
Приготовление и предварительная очистка рассола . . . 439
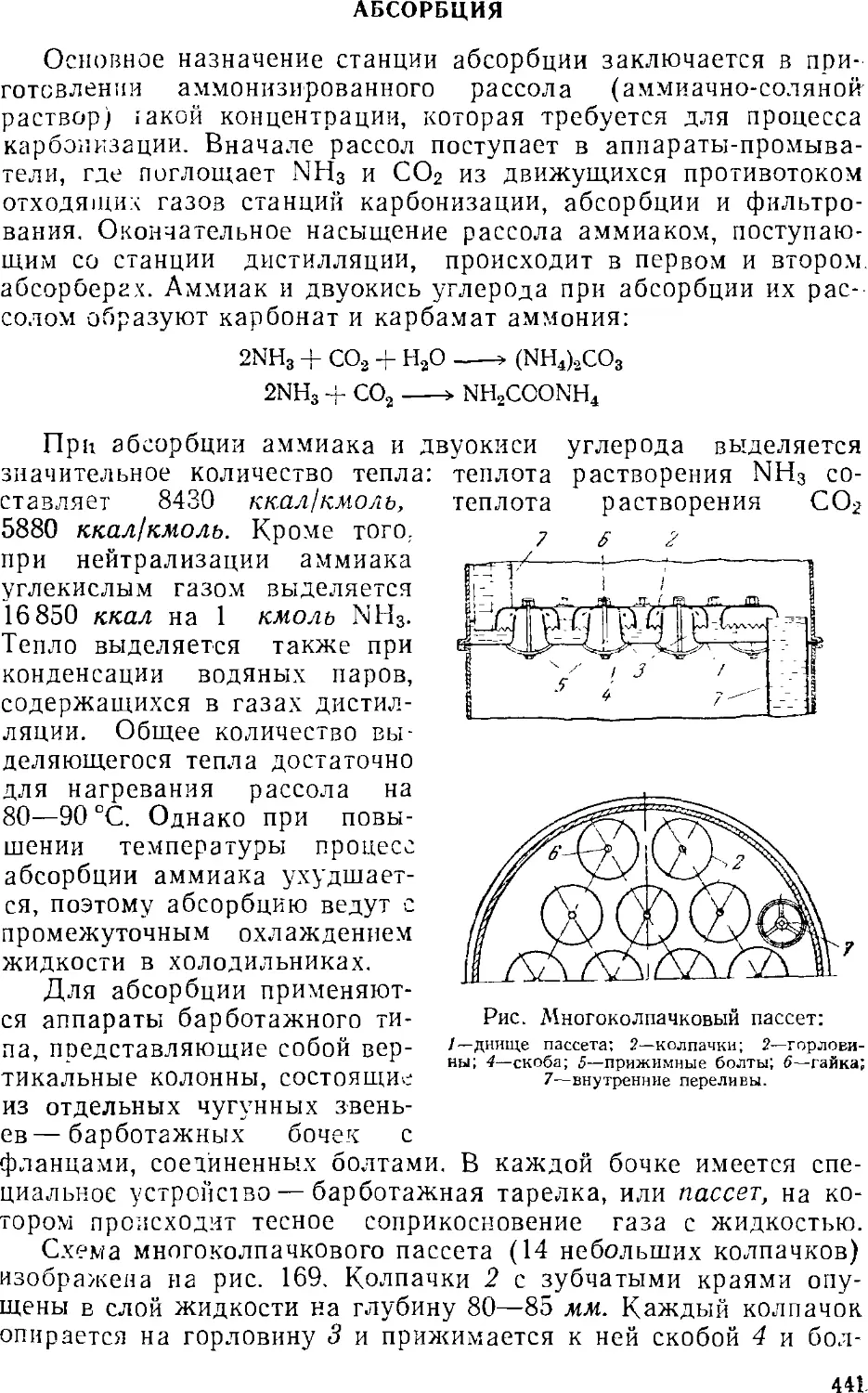
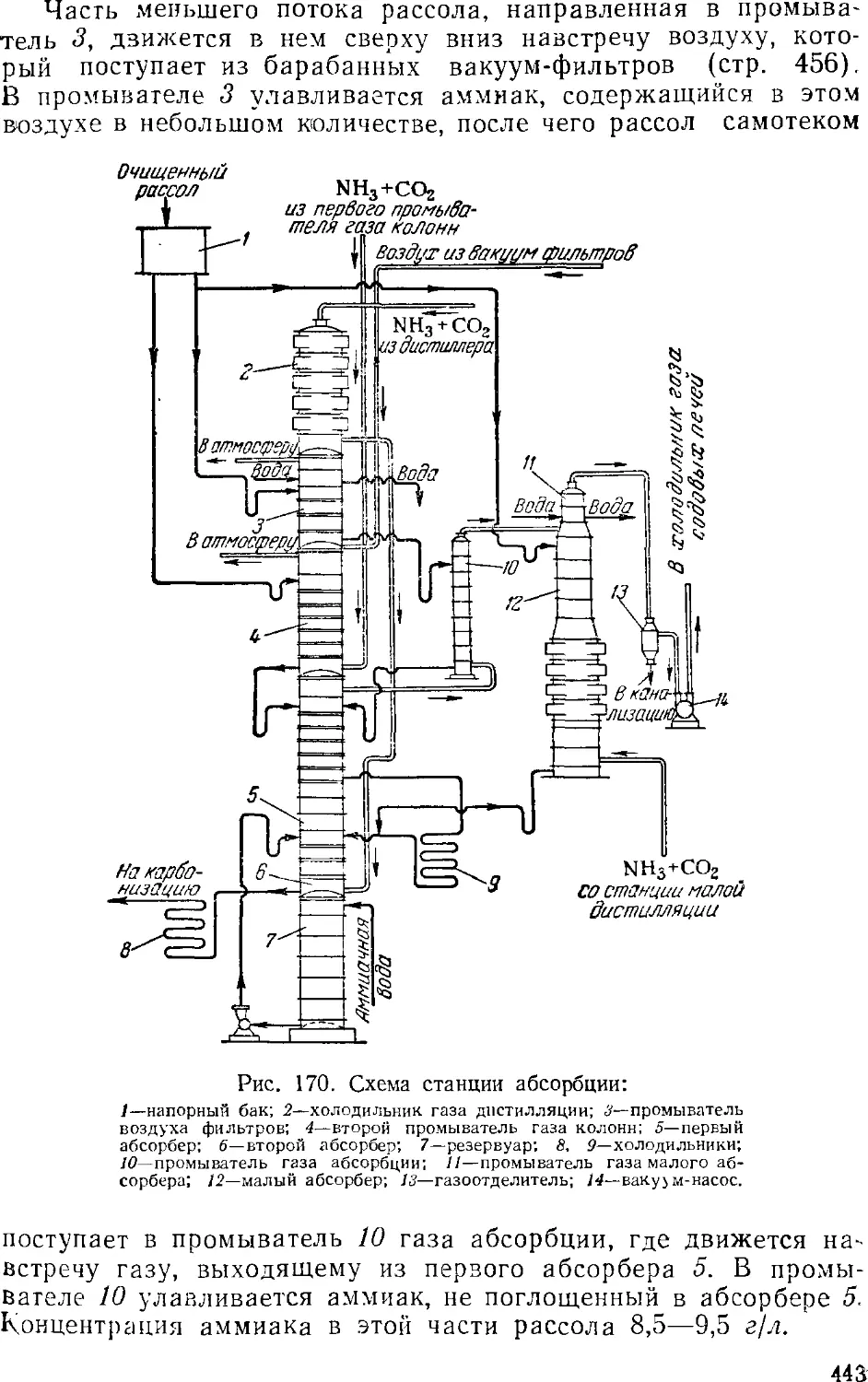
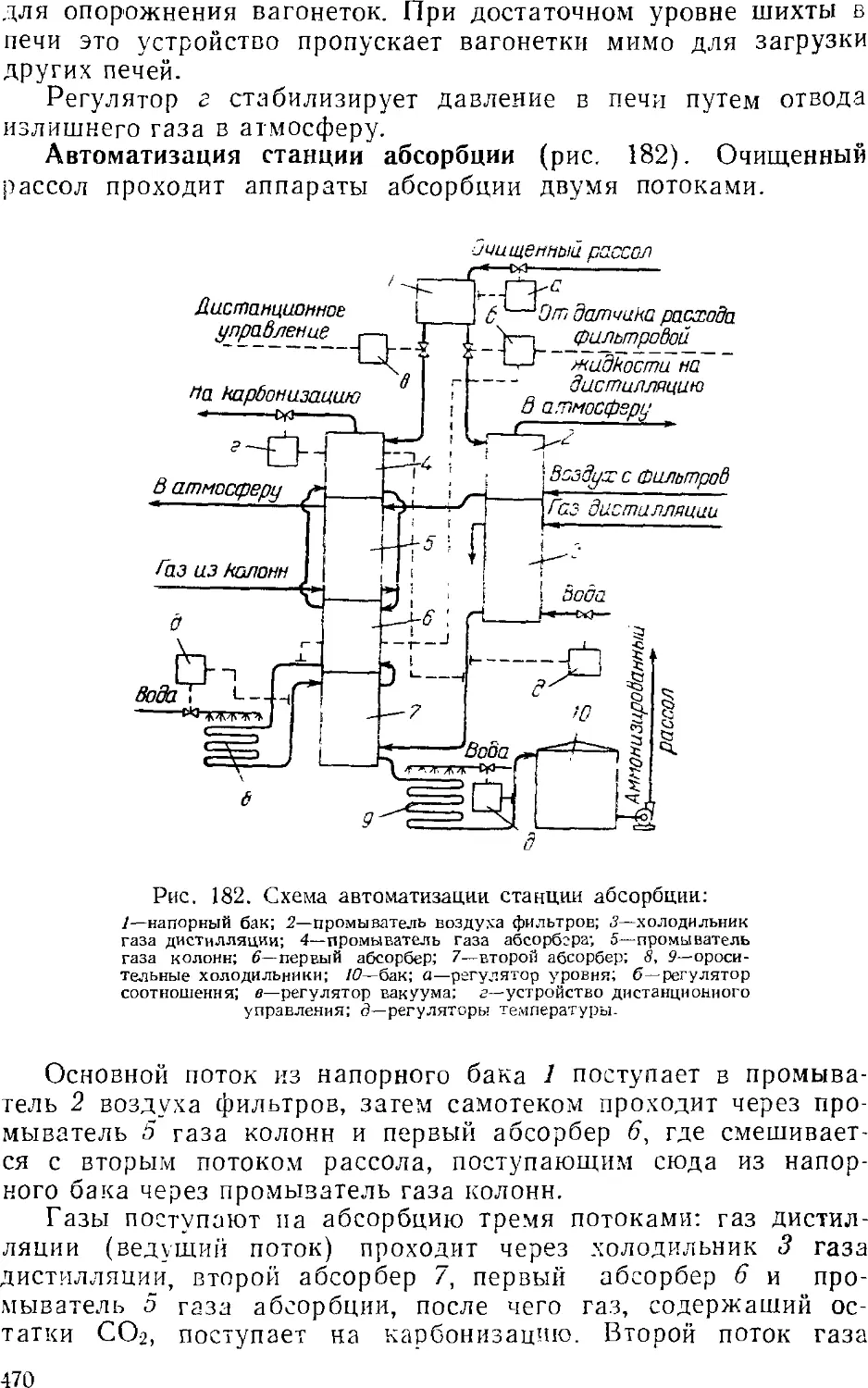
Абсорбция 441
Основная аппаратура станций абсорбции 445
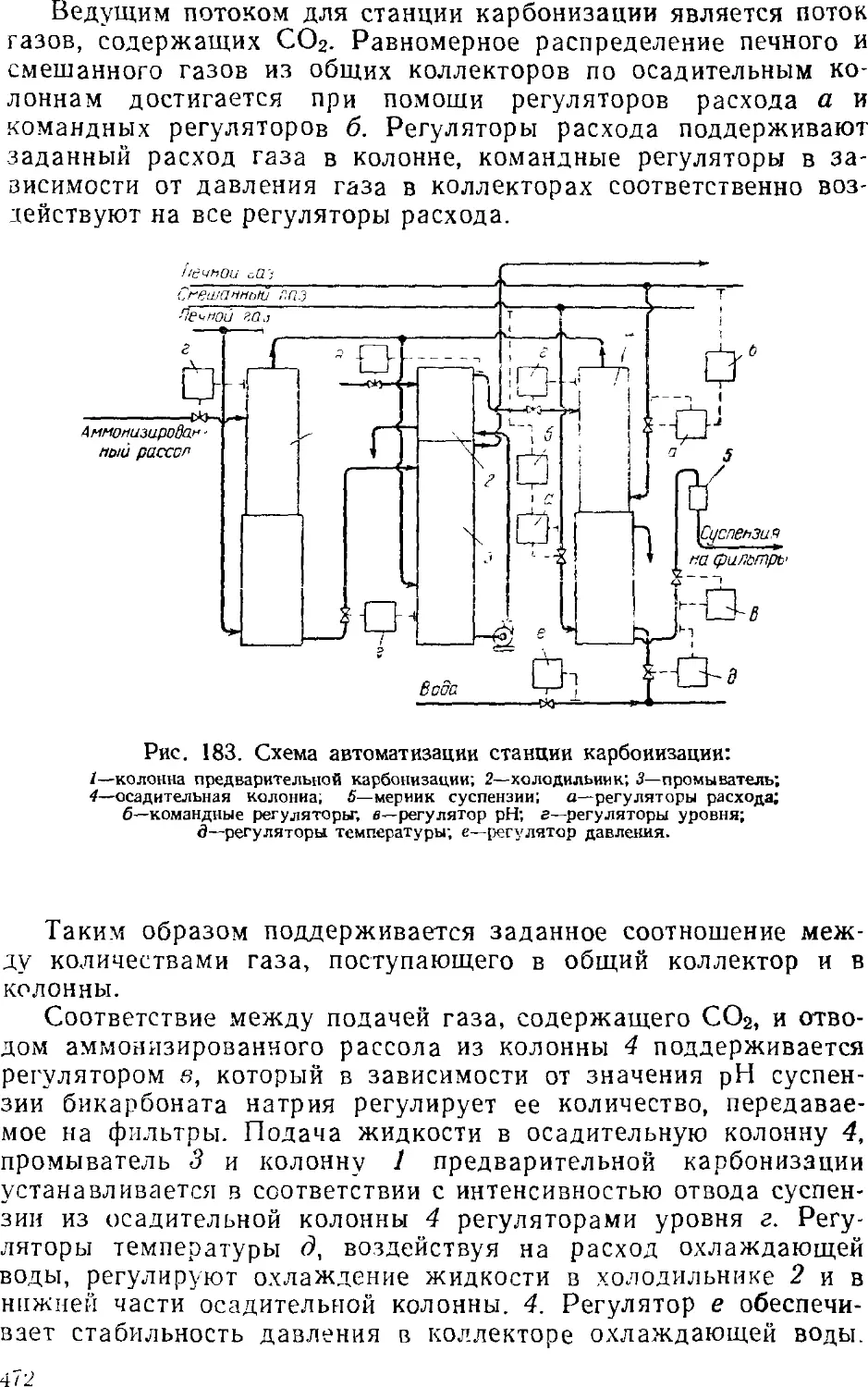
Карбонизация 449
Карбонизационные колонны 452
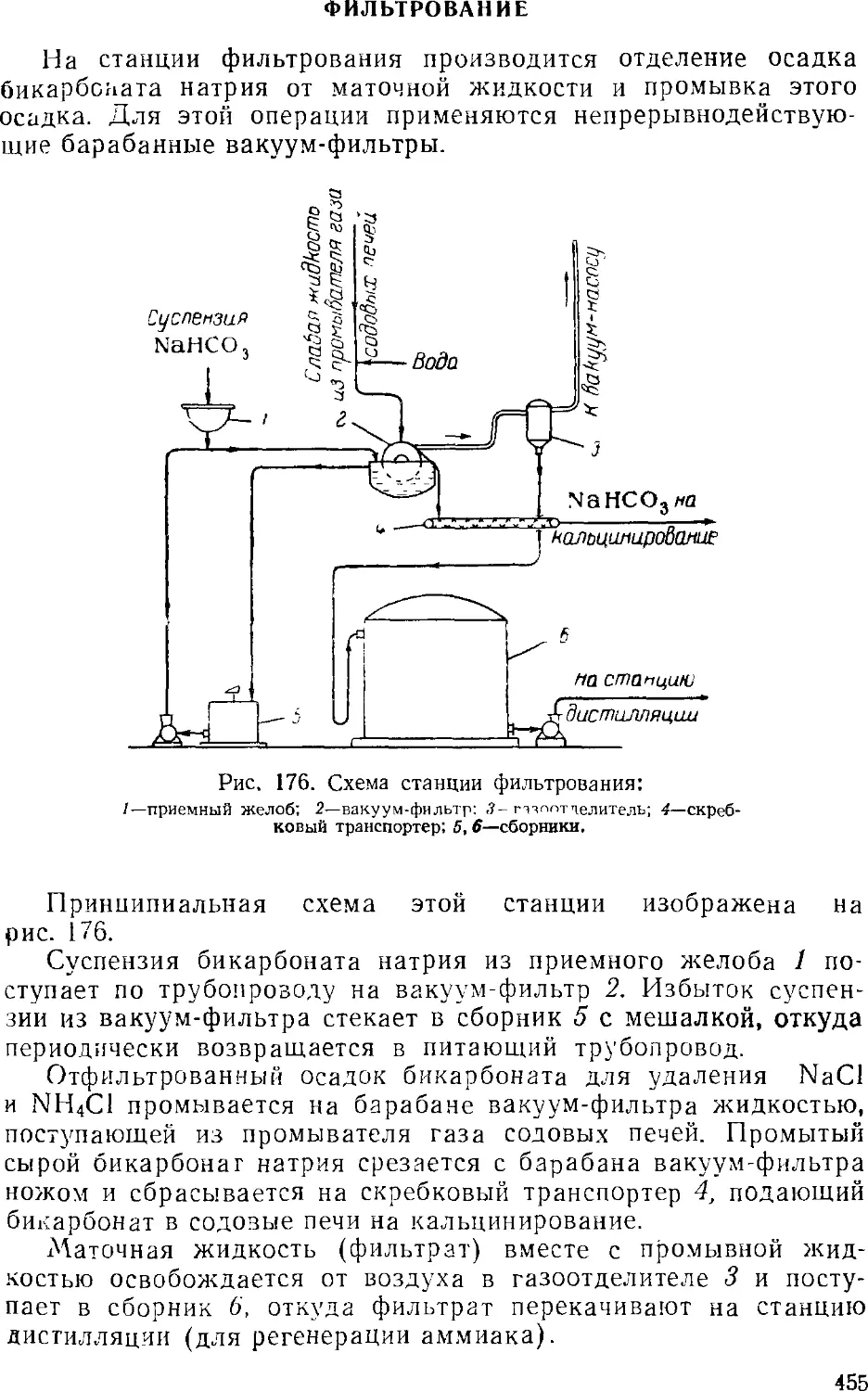
Фильтрование 455
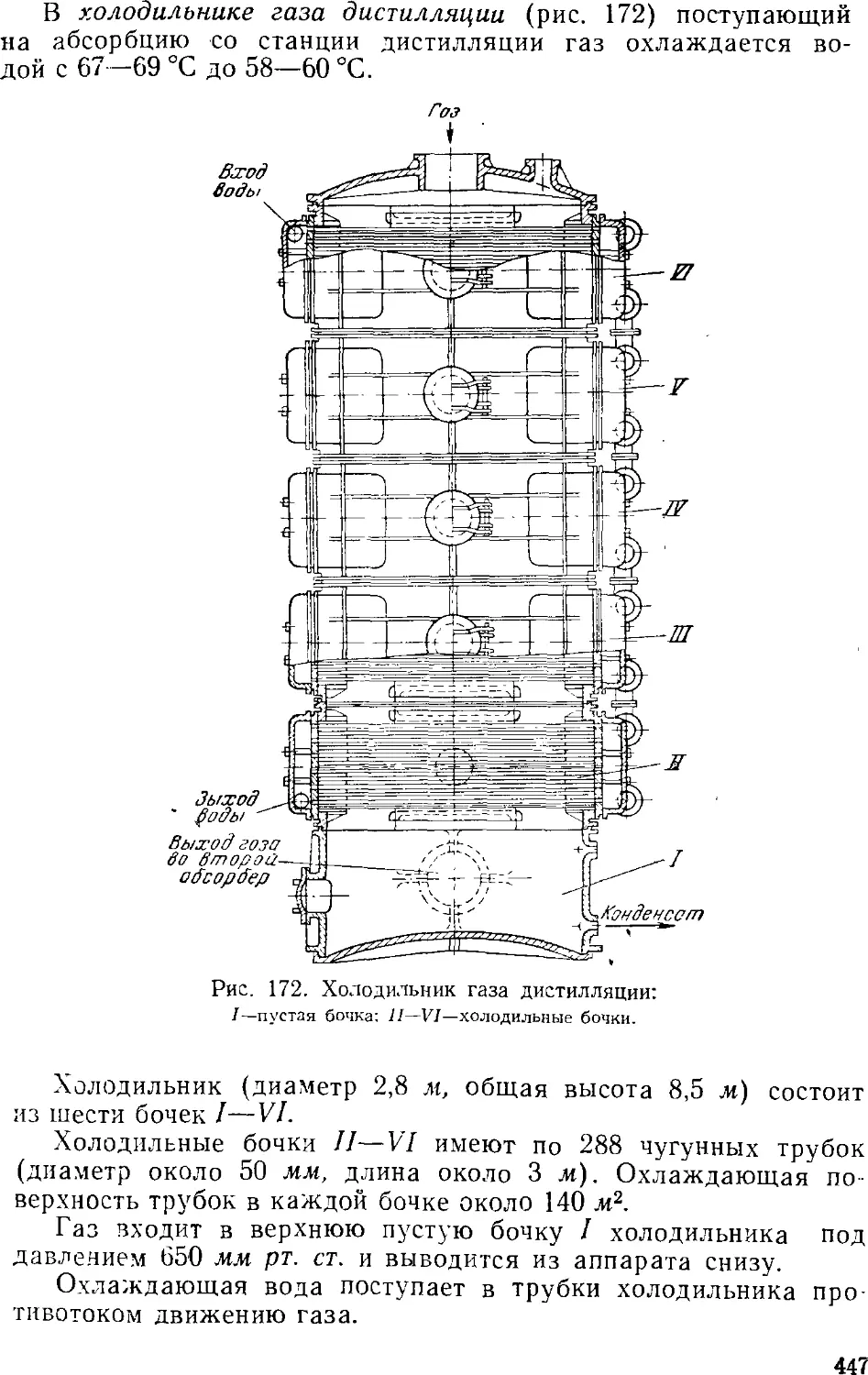
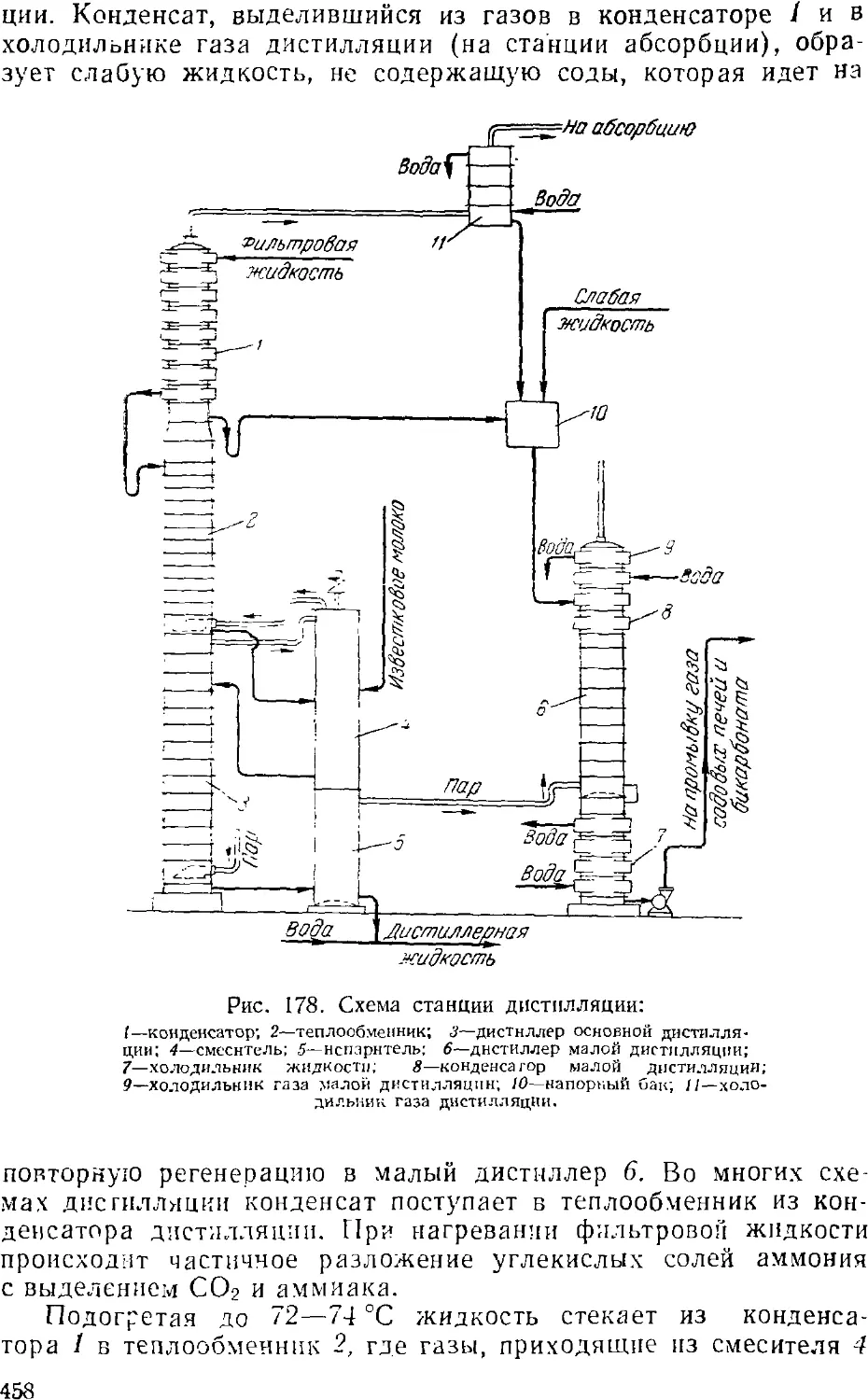
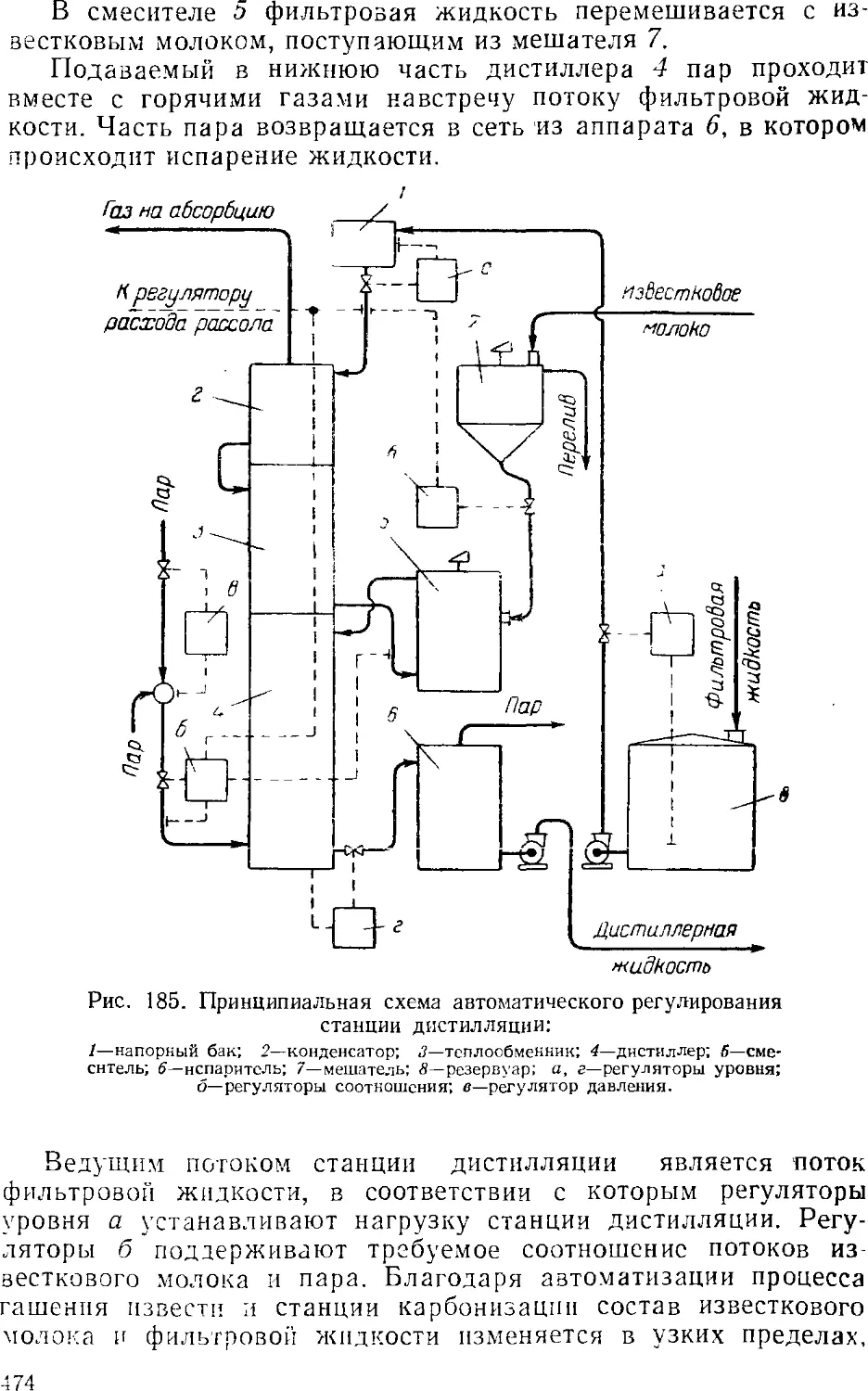
Дистилляция (регенерация аммиака) 457
Дистилляционные колонны 460
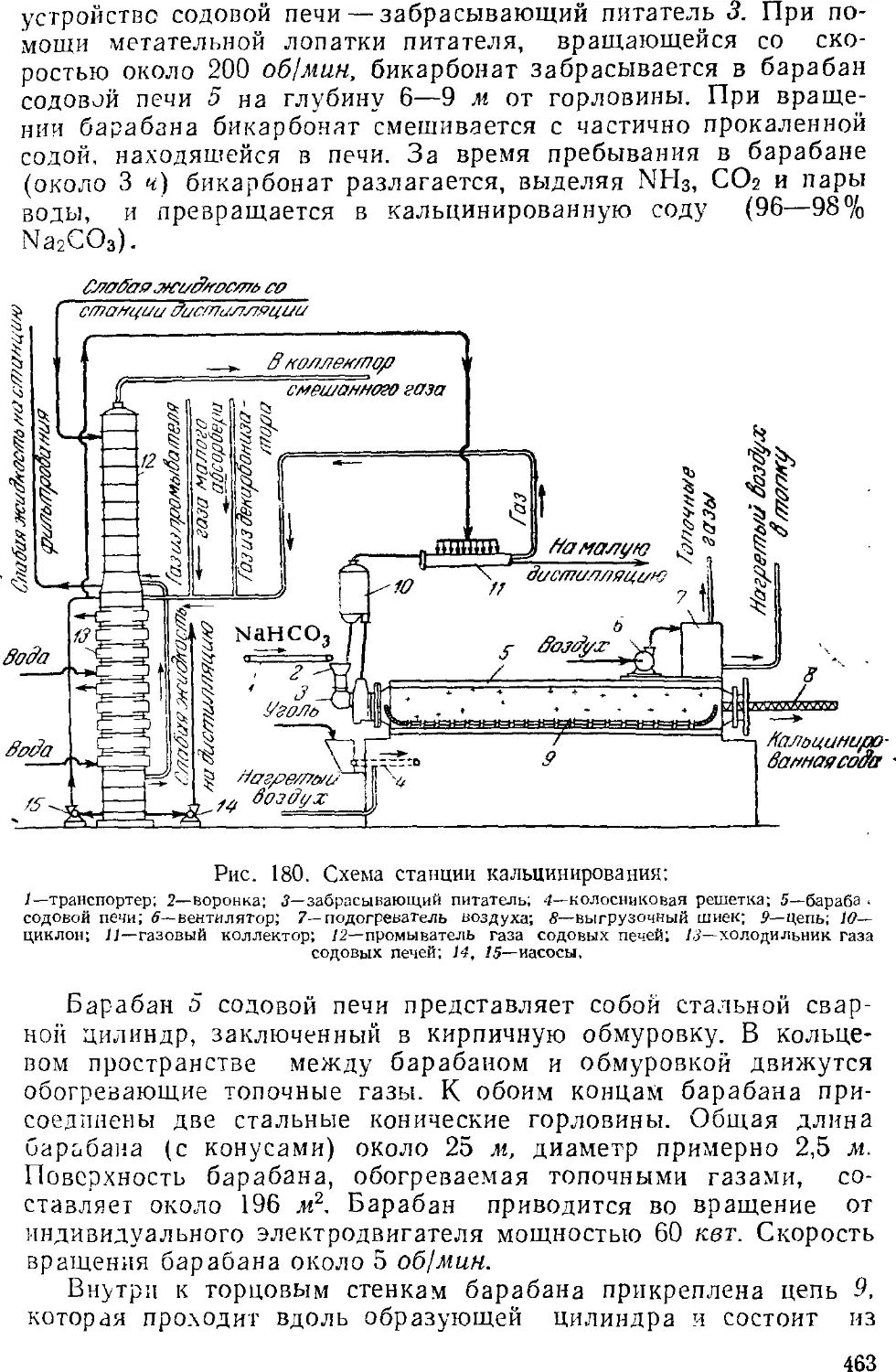
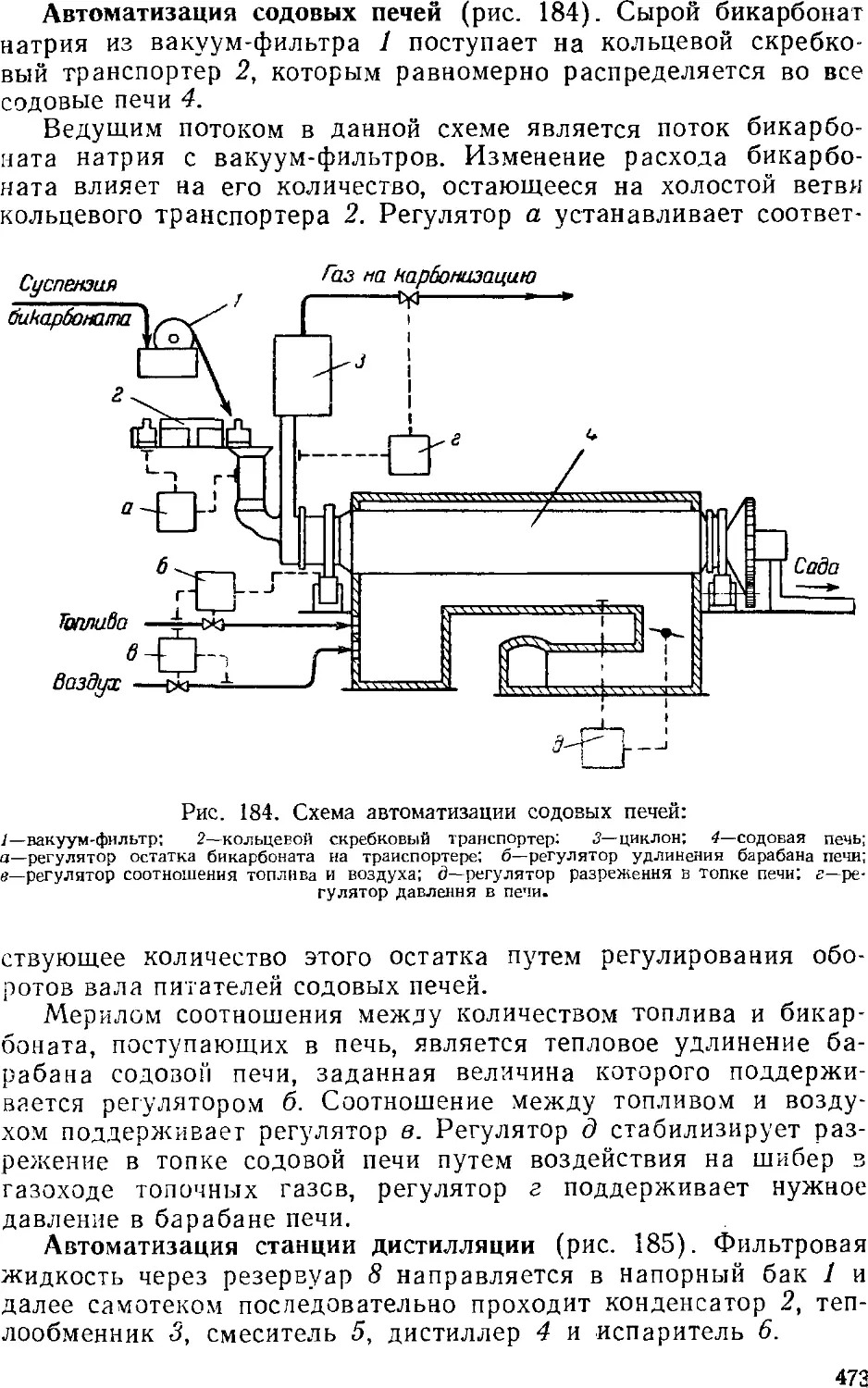
Кальцинирование 462
4. Автоматизация производства кальцинированной соды .... 468
5. Бикарбонат натрия 475
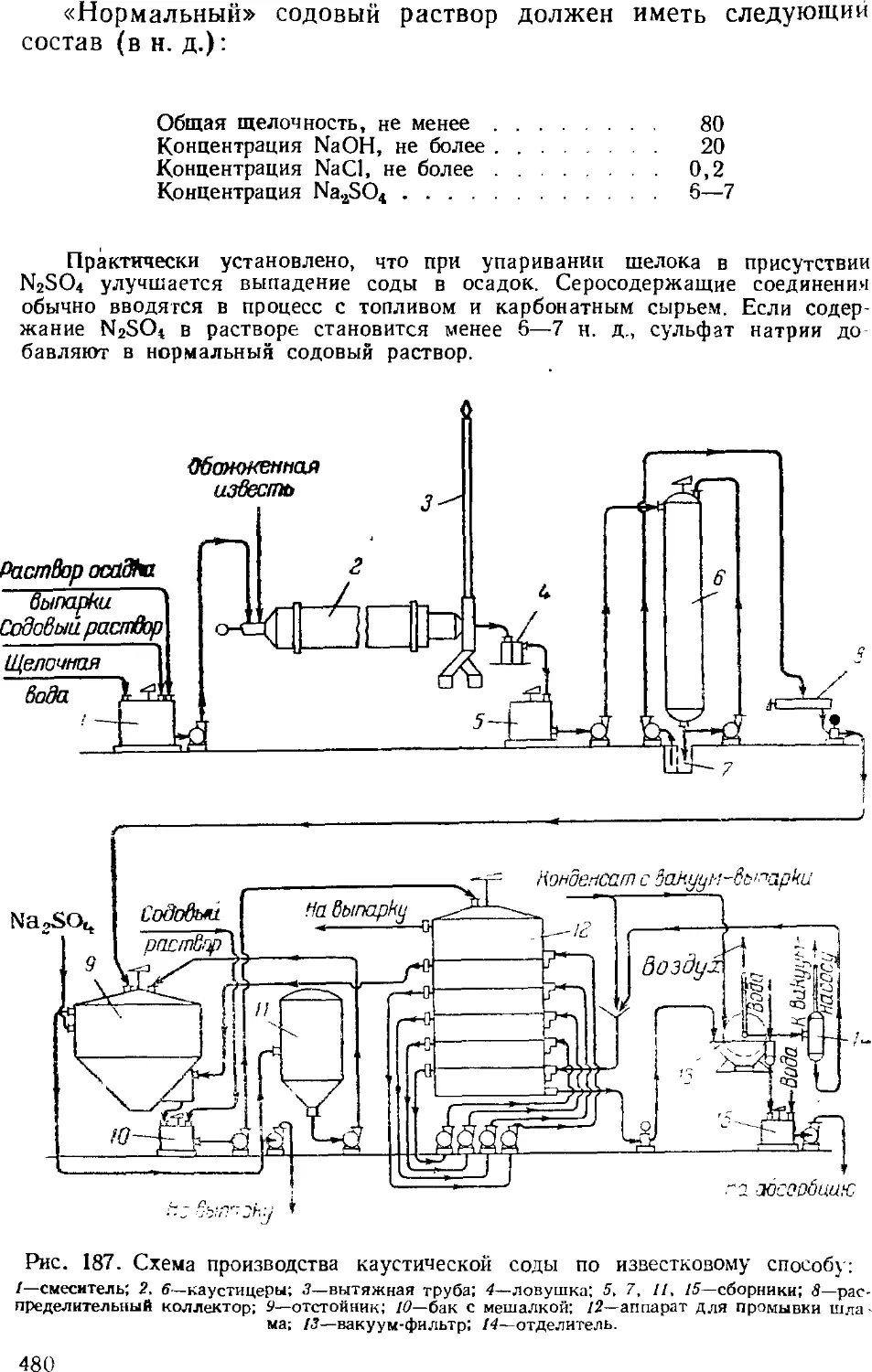
6. Производство каустической соды химическими способами . . 476
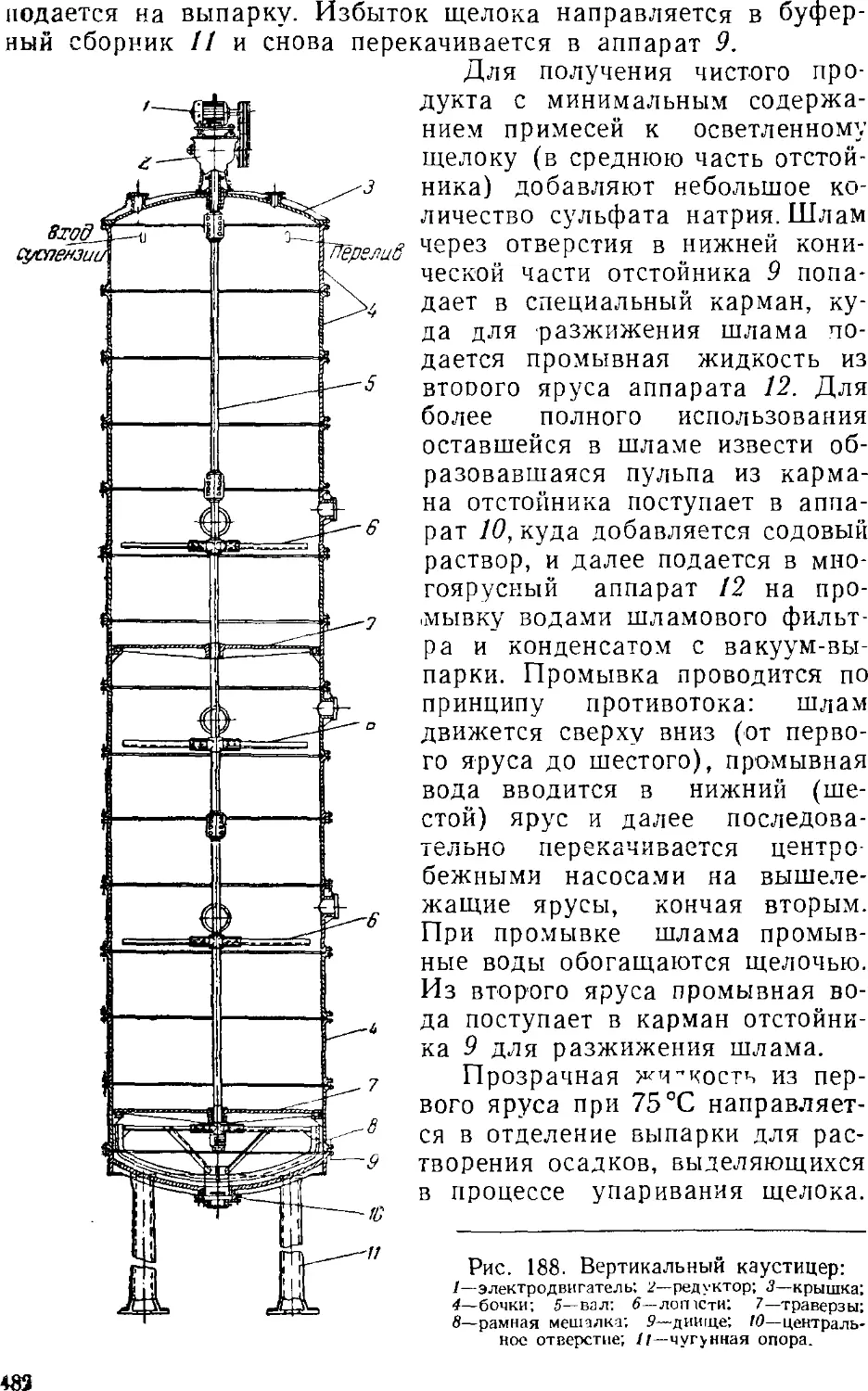
Известковый способ производства каустической соды . . 477
Ферритный способ производства каустической соды. . . 484
Литература 489
Глава XI. Фосфор и фосфорная кислота 490
1. Фосфор. Общие сведения 490
2. Желтый фосфор 491
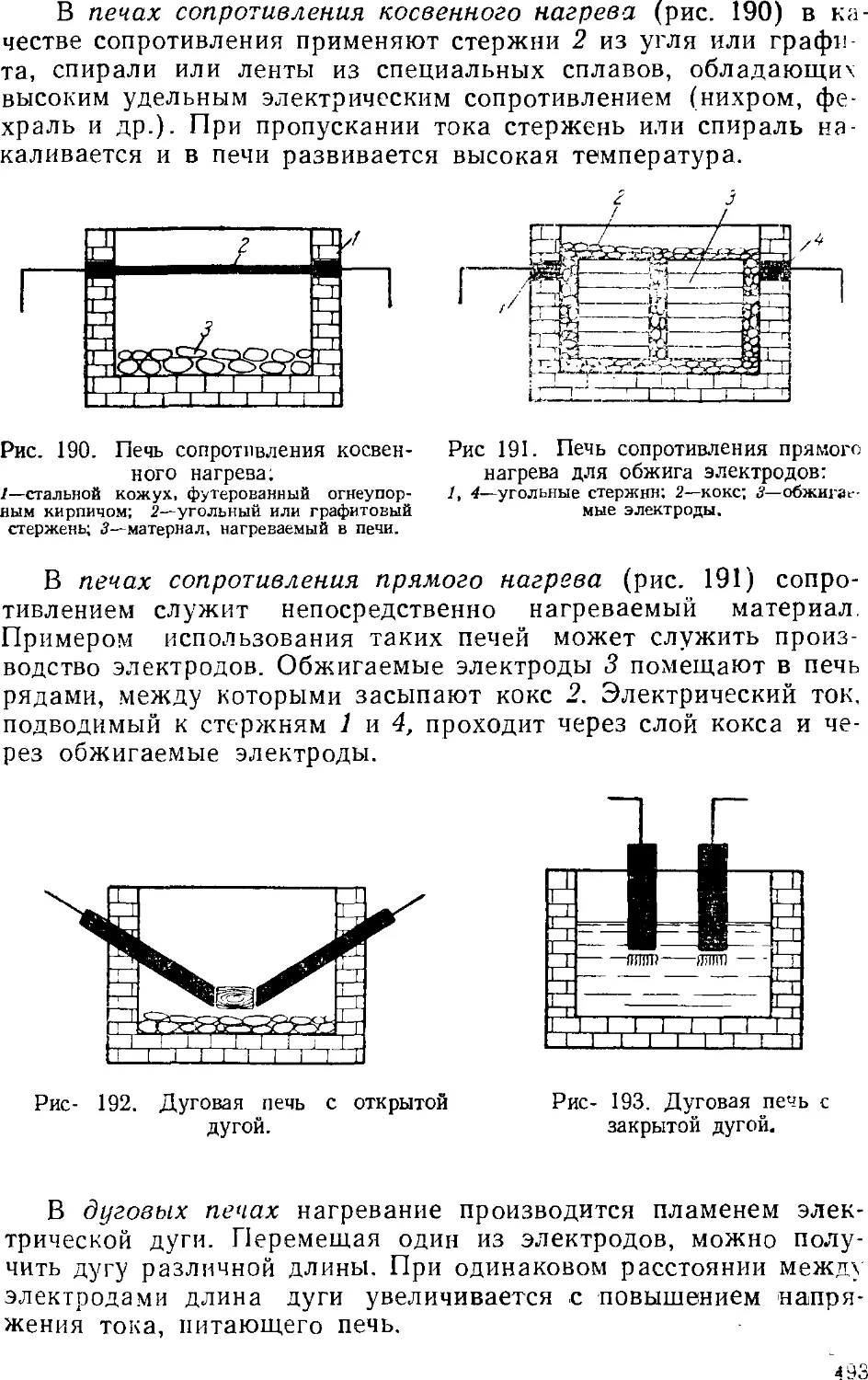
Некоторые сведения из электротермии 491
Электрические печи 492
Возгонка фосфора в электрических печах 497
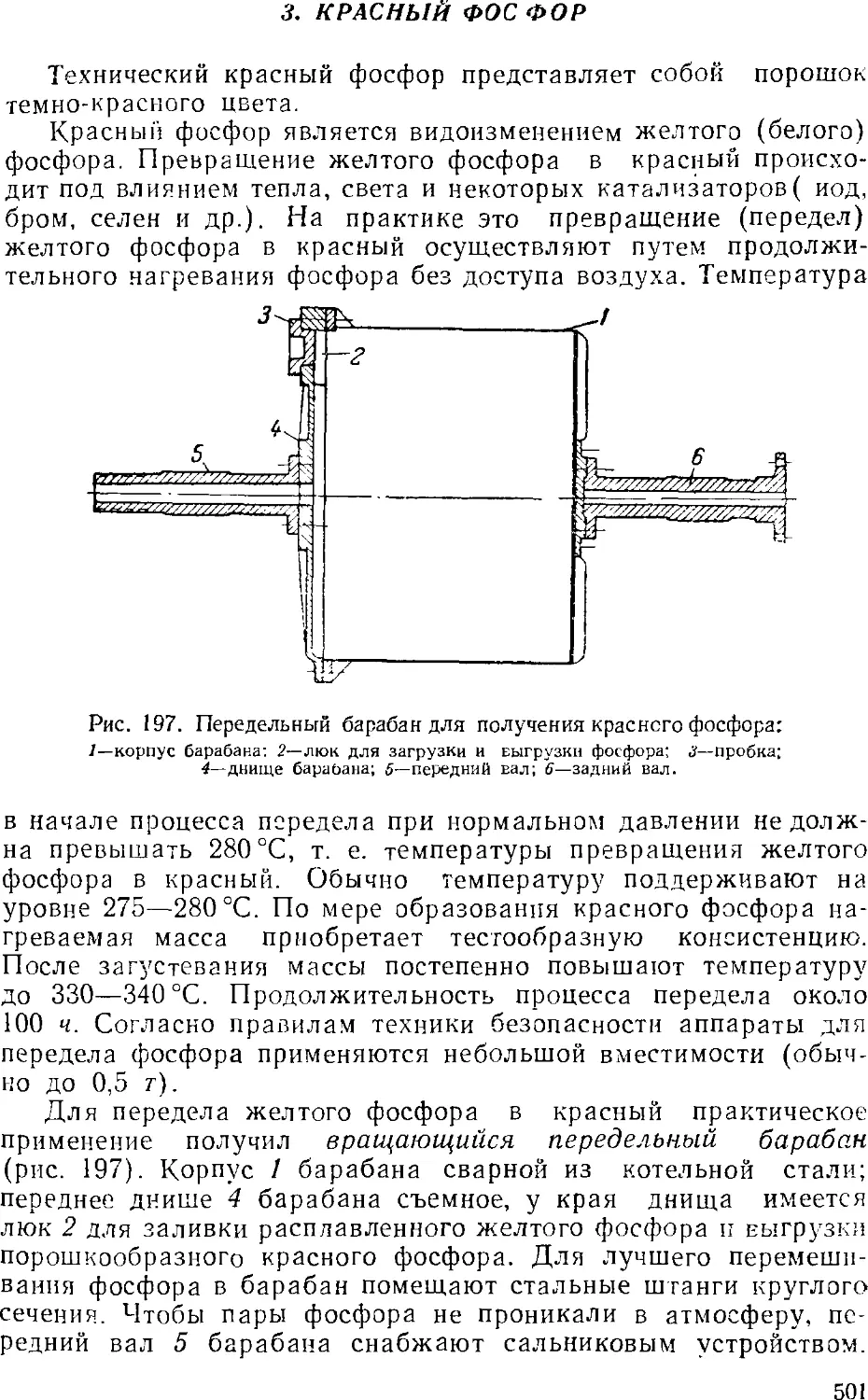
3. Красный фосфор 501
4. Фосфорная кислота 502
Экстракционная фосфорная кислота 503
Упаривание фосфорной кислоты 507
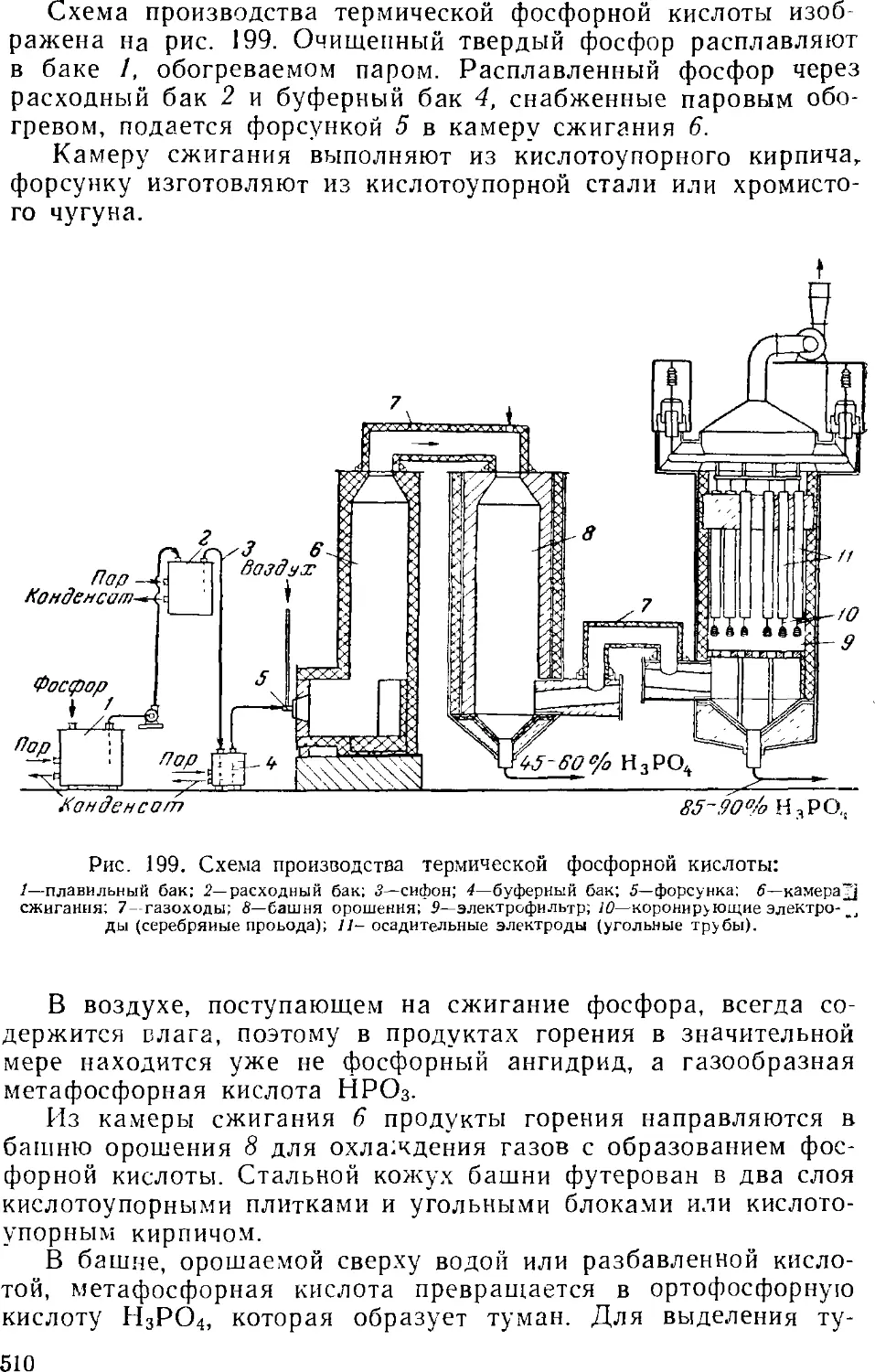
Термическая фосфорная кислота 509
Литература 512
Г ла за XII. Минеральные удобрения 514
1. Обшие сведения 514
2. Фосфорные удобрения 520
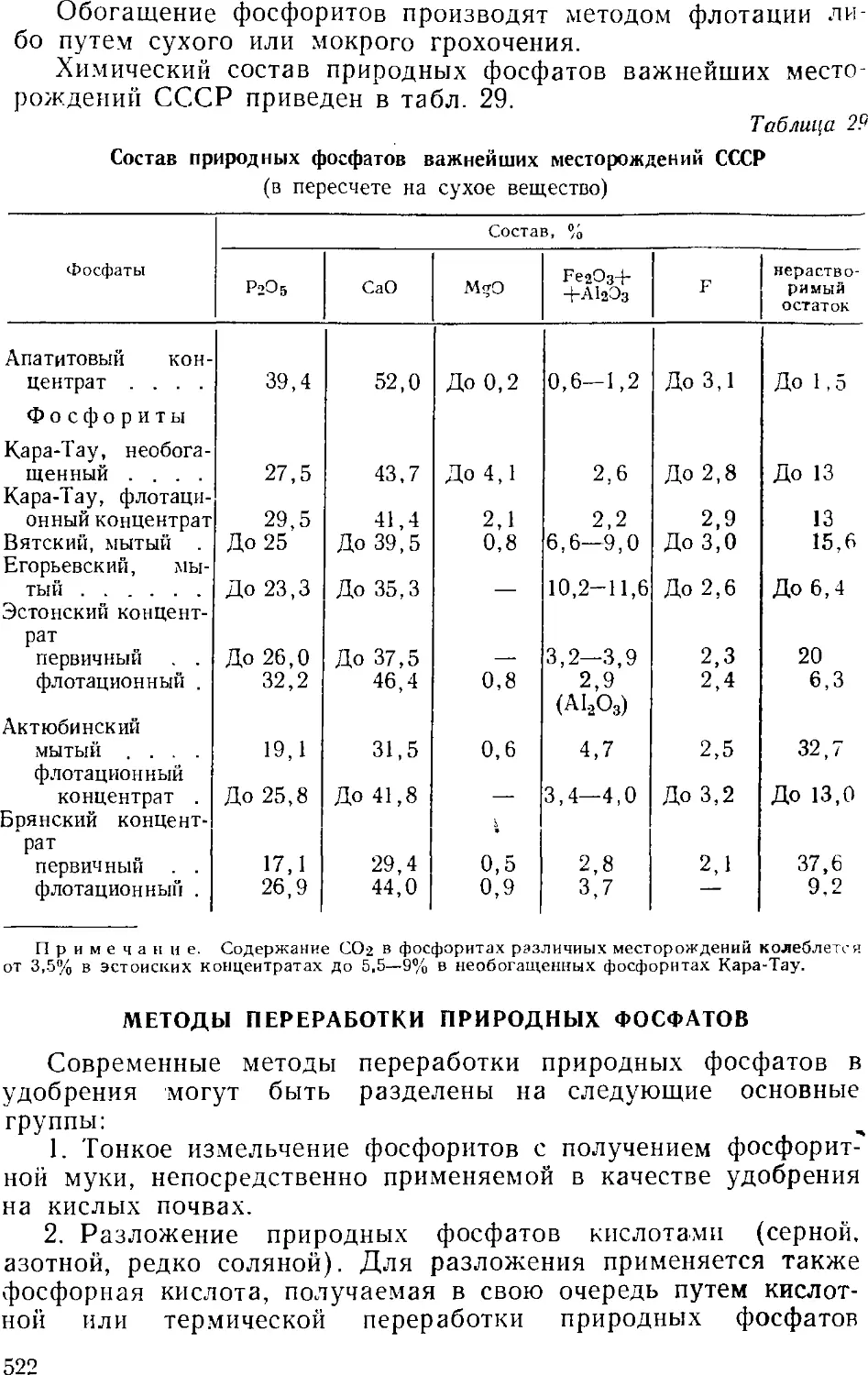
Сырье для производства фосфорных удобрении 520
Методы переработки природных фосфатов 522
Фосфоритная мука 52.J
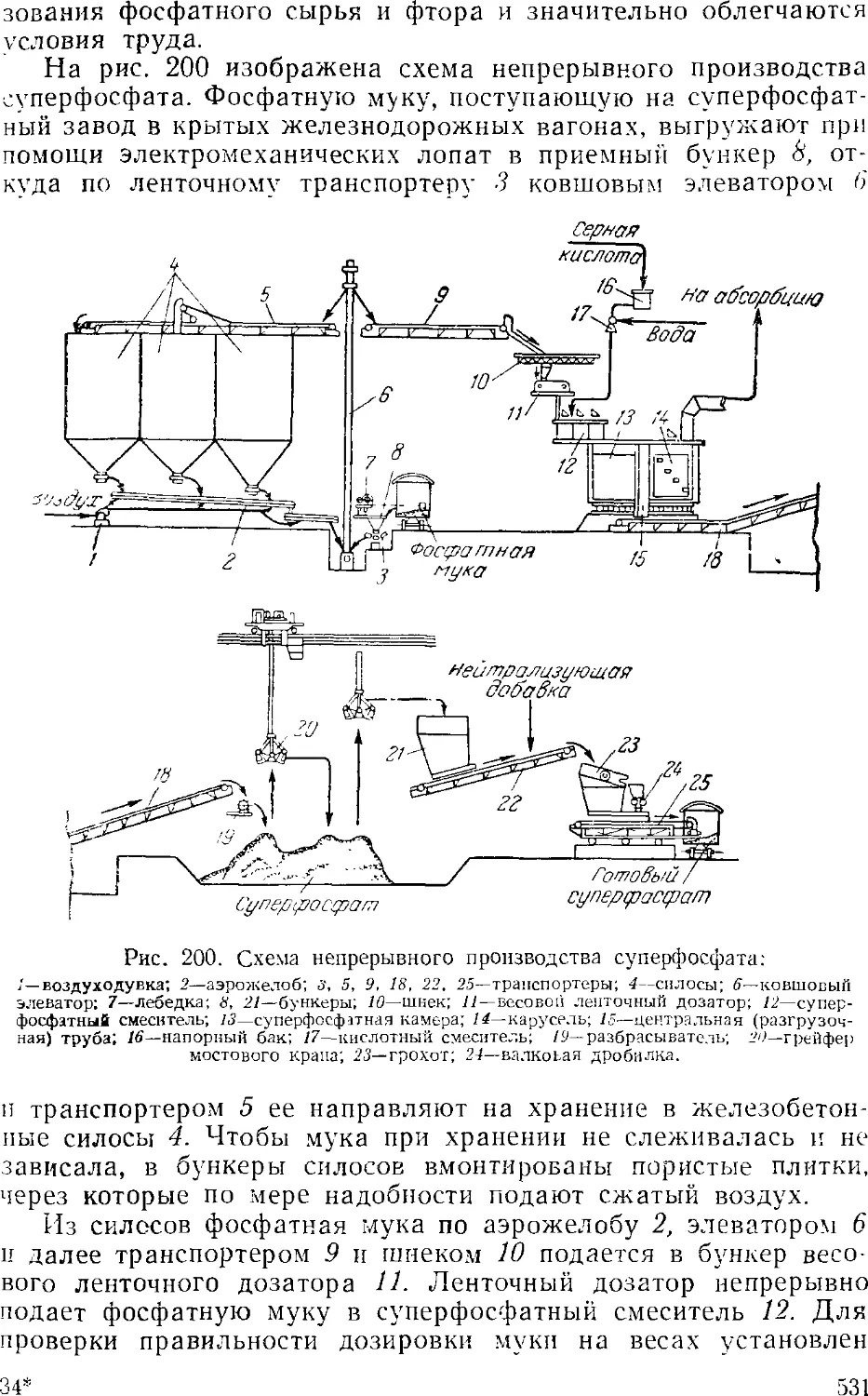
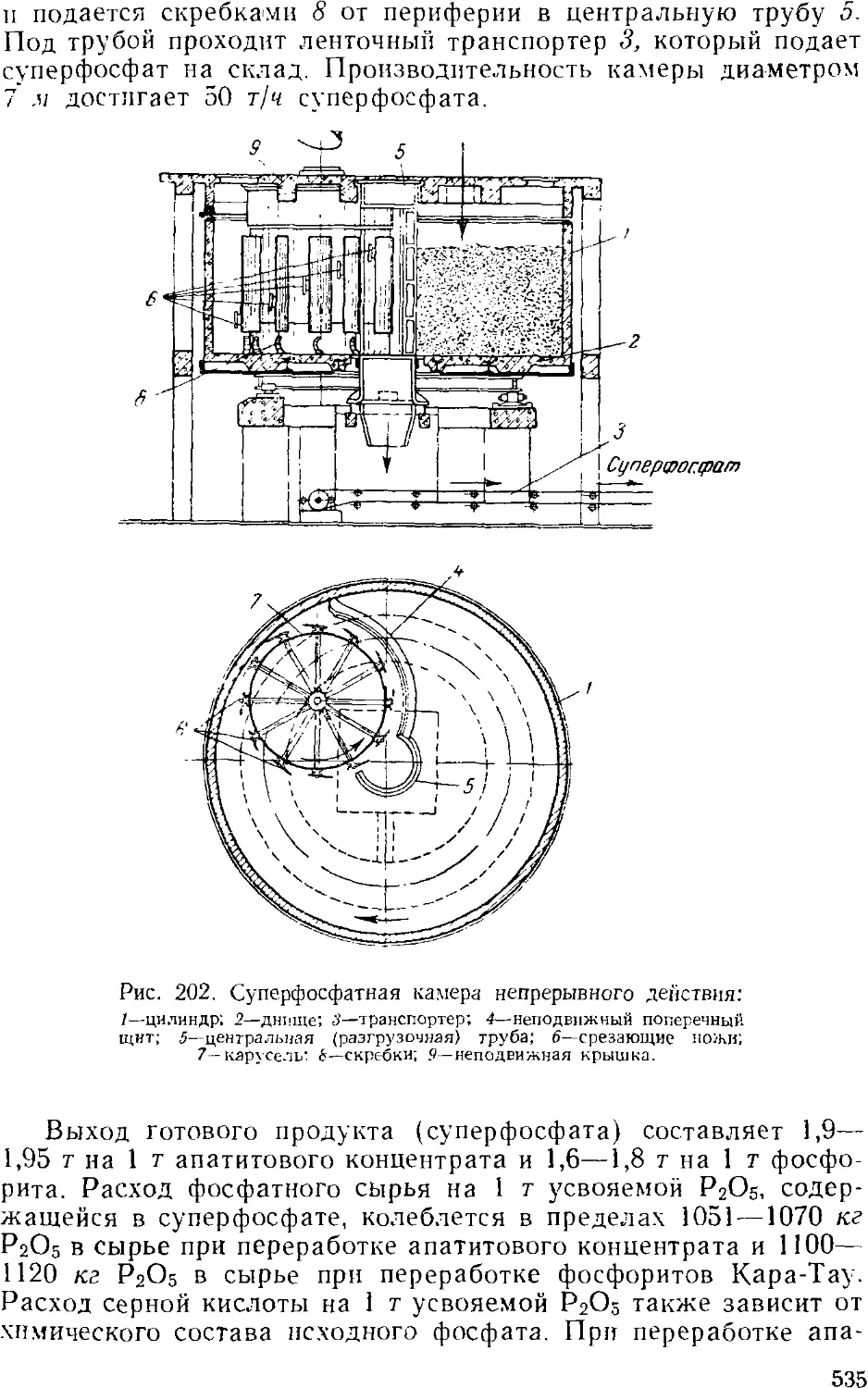
Суперфосфат 524
Гранулированный суперфосфат . 537
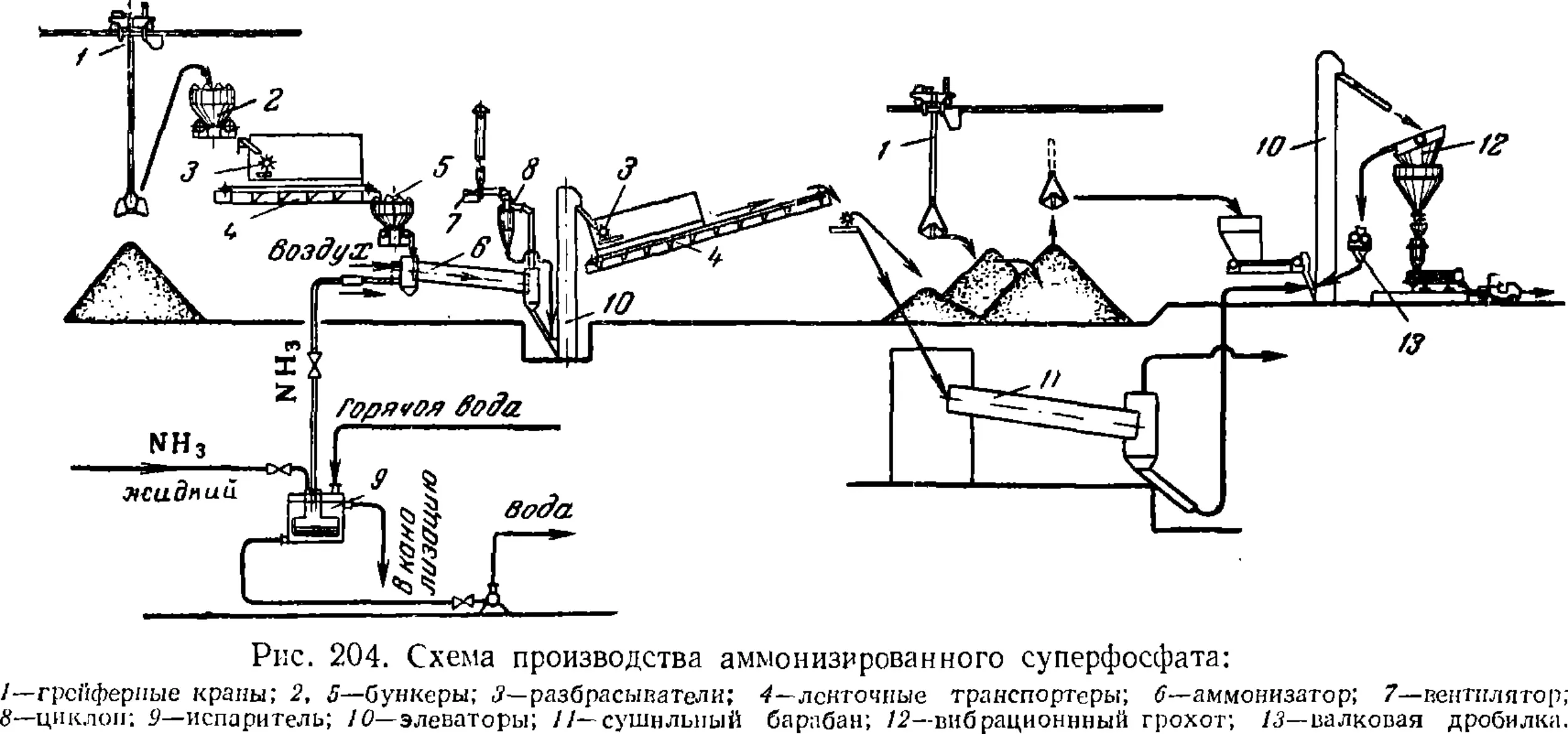
Аммонизированный суперфосфат 539
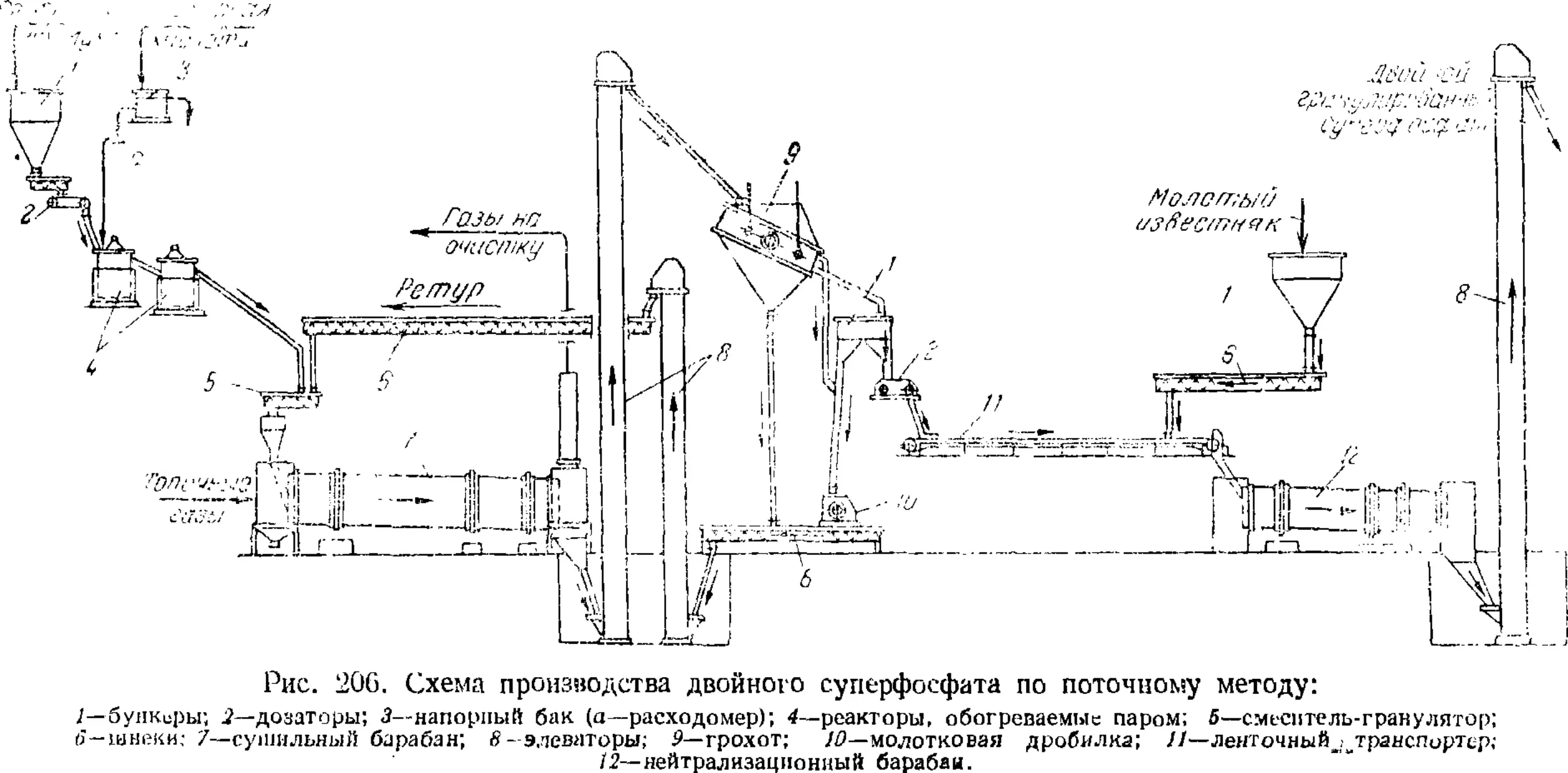
Двойной суперфосфат 545
Преципитат 549
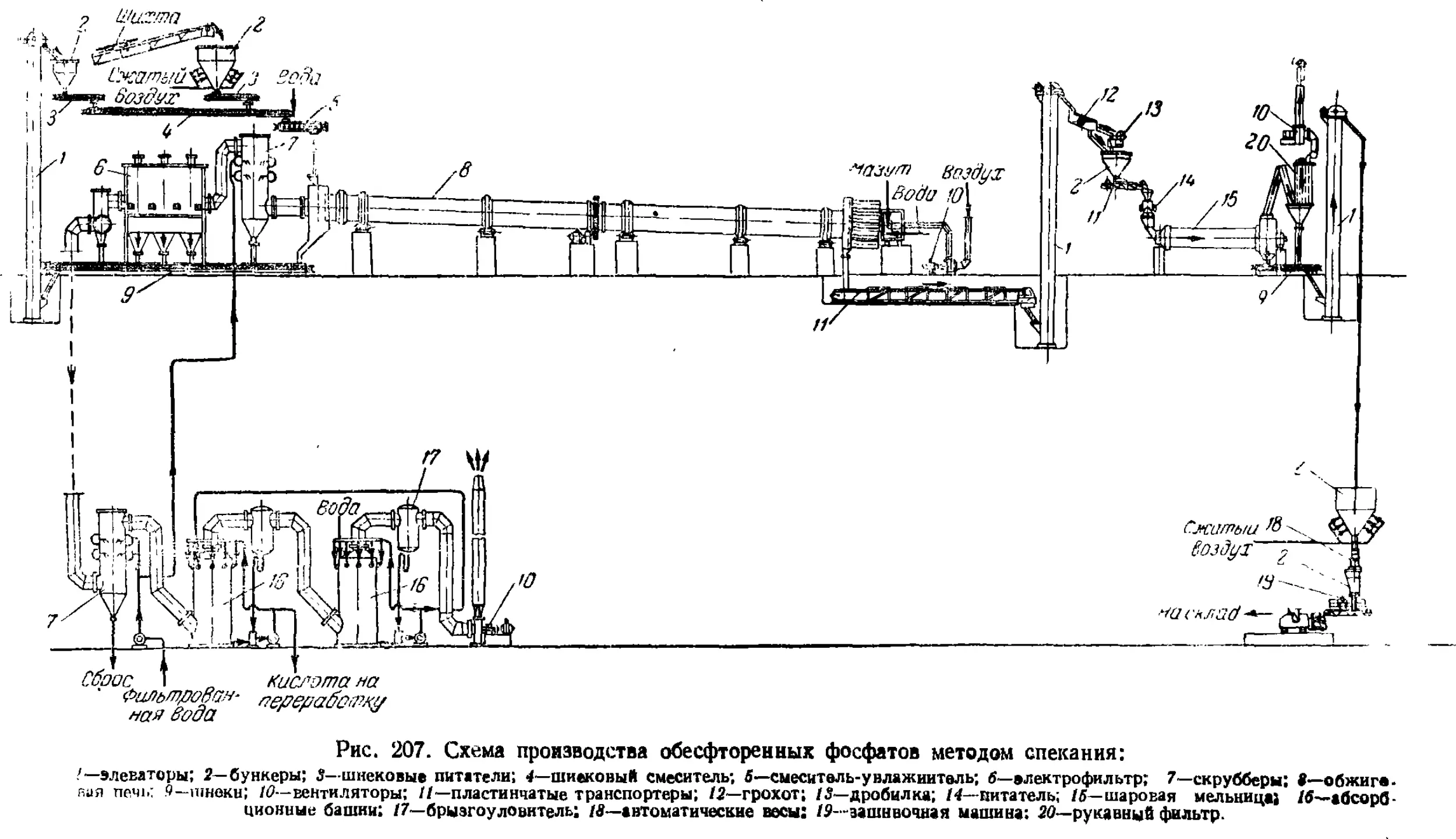
Обесфторенные фосфаты 550
3. Азотные удобрения 555
1 Аммиачная селитра 555
Натриевая и калиевая селитра 562
Кальциевая селитра 561
Сульфат аммония 560
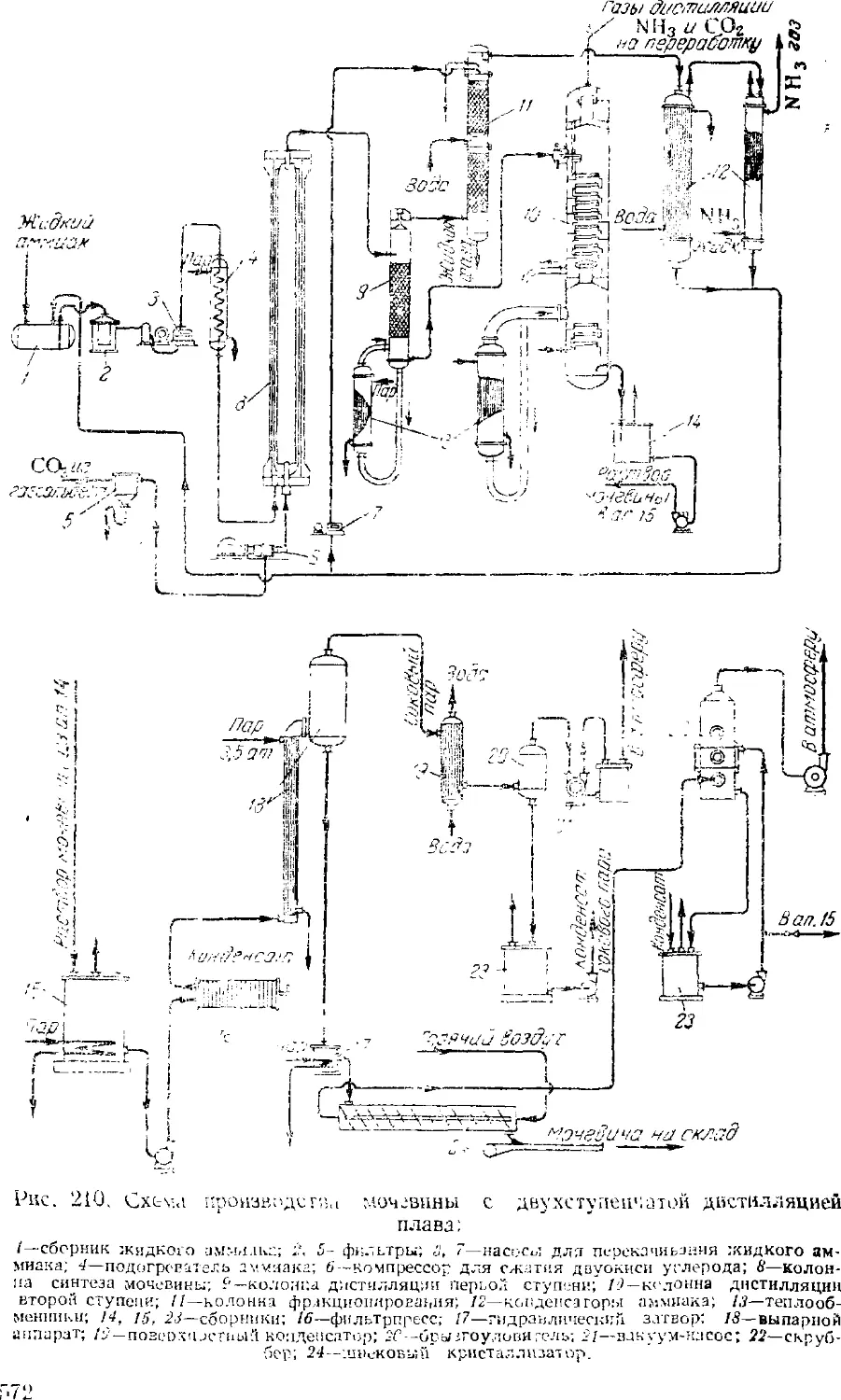
Мочевииа 568
Жидкие азотные удобрения 576
4. Калийные удобрения 57"
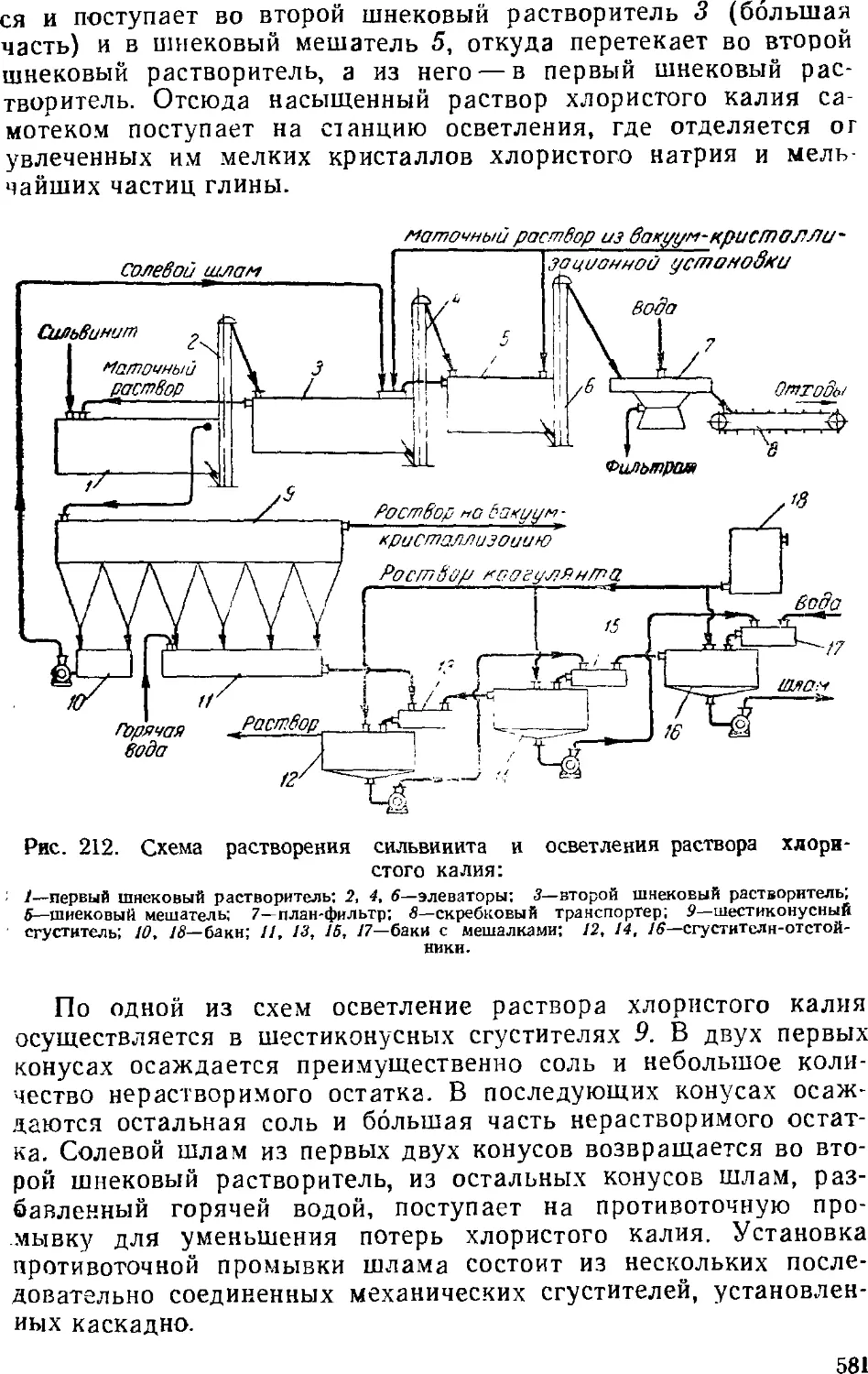
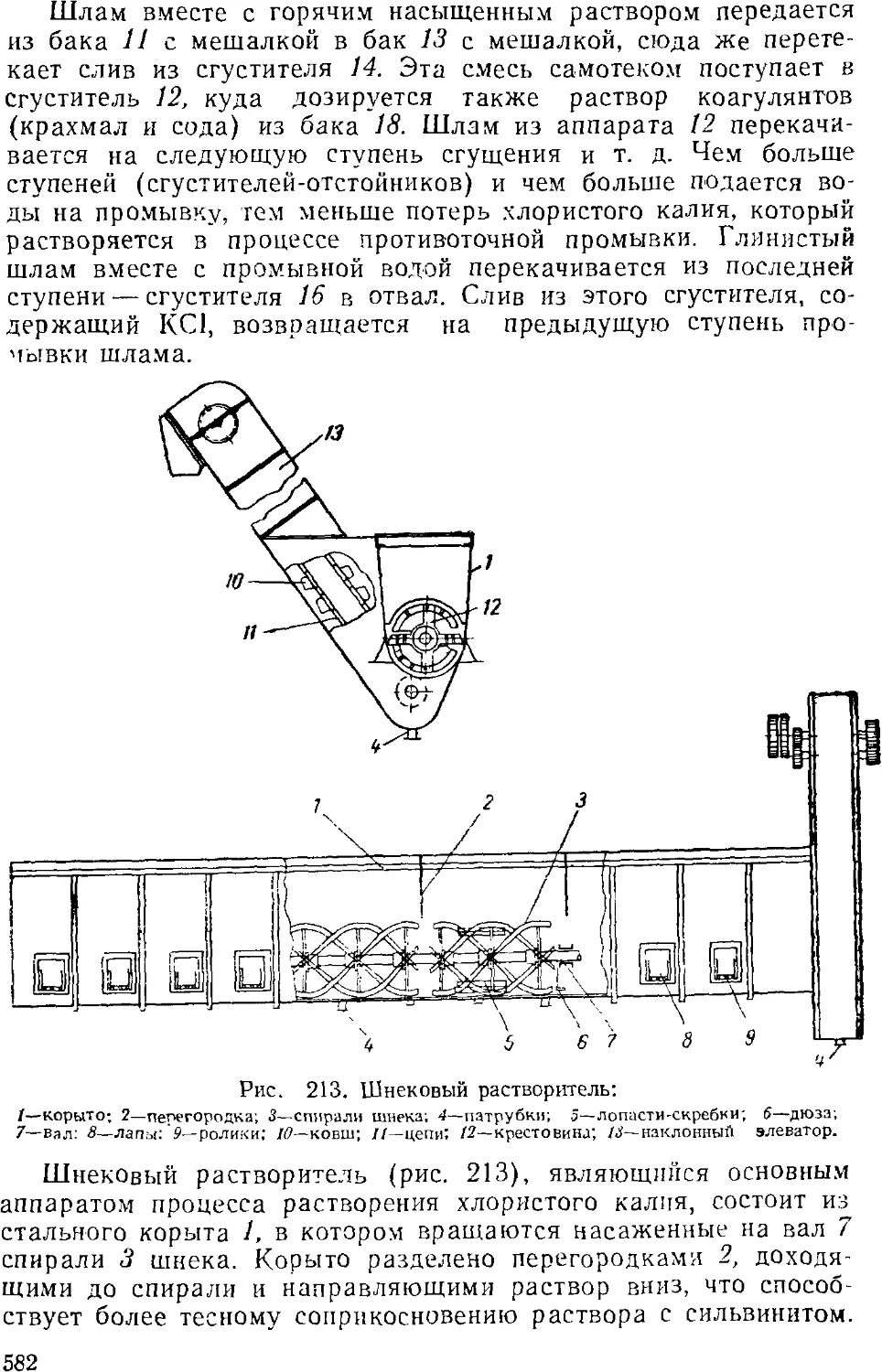
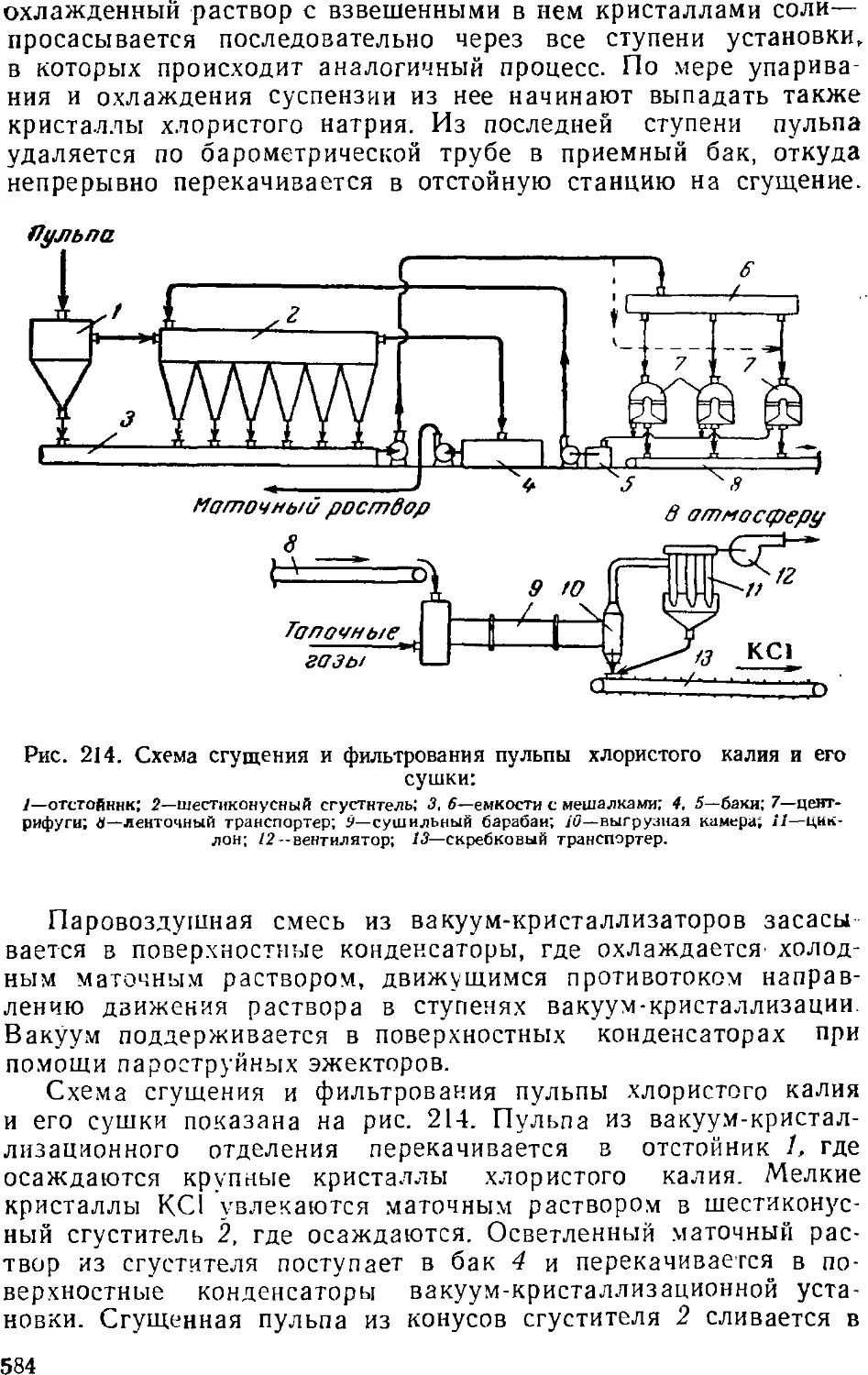
Хлористый калий 579
Сульфат калия 586
Каинит 587
5. Комплексные удобрения 587
Сложные удобрения 588
Смешанные удобрения 596
6. Микроудобрения 598
Литература 600
Глава XIII. Карбид и цианамид кальция 001
1. Карбид кальция 601
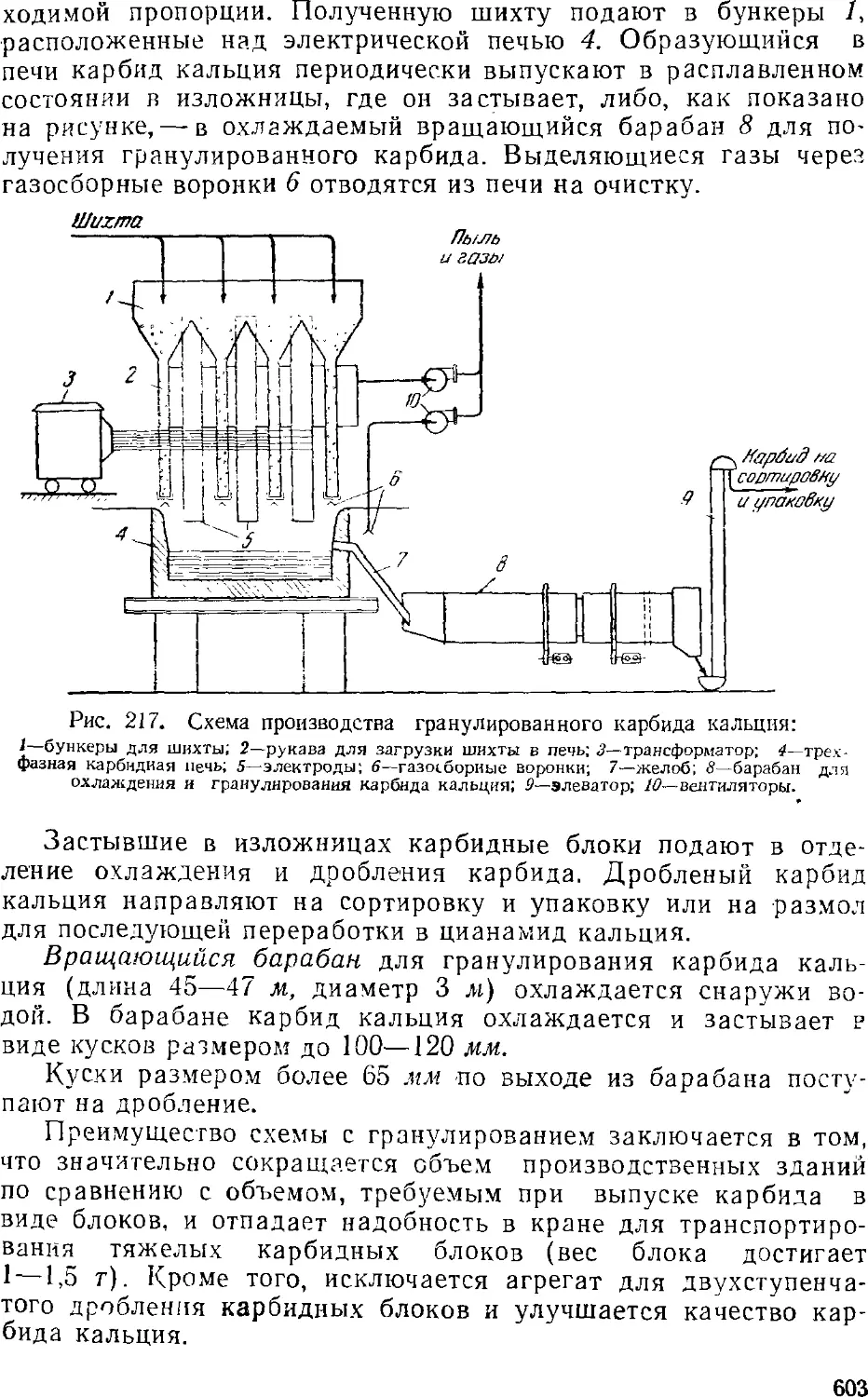
Схема производства G02
Карбидные печи вО4
Брикетированный карбид калышя 605
2. Цианамид кгльция 606
Циапамидные печи 608
.Литература . . . 610
Глава XIV. Технология силикатов 011
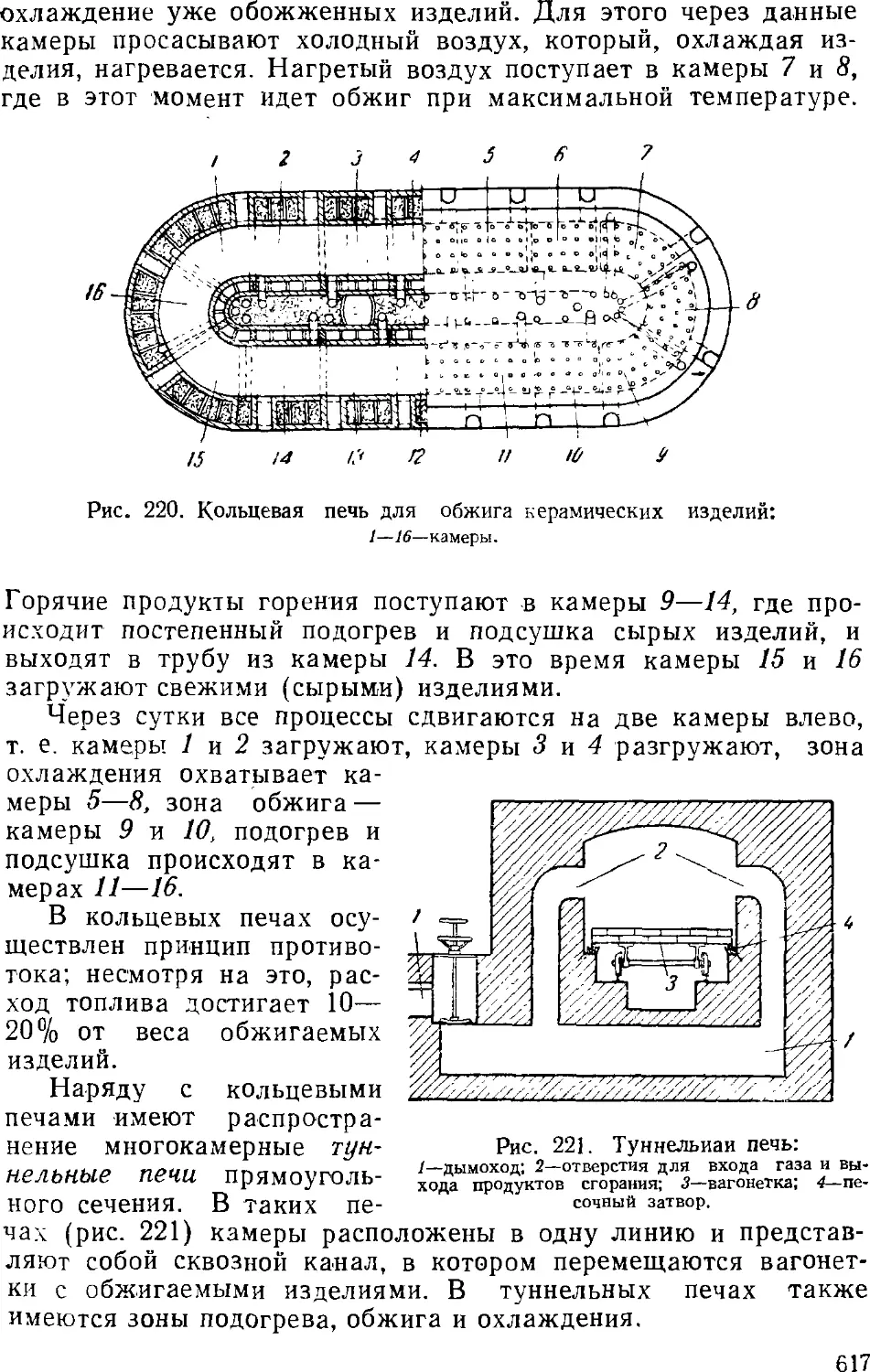
1. Керамика 611
Сырье для керамических изделий 012
Производство керамических изделий 614
Фарфор и фаянс 619
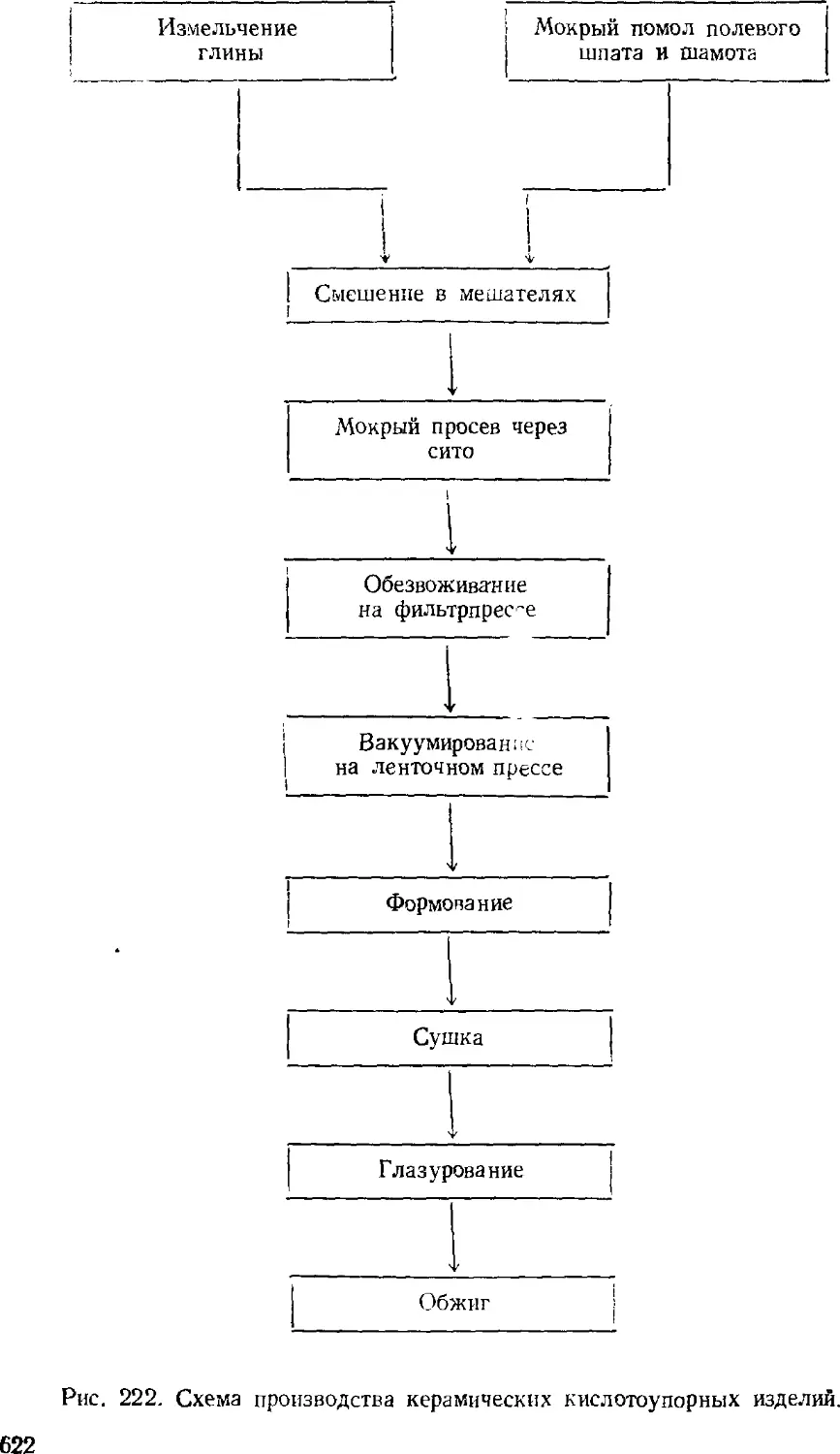
Кислотоупорные изделия 620
Керамические строительные материалы 023
Огнеупоры 624
2. Минеральные вяжущие вещества 62'Э
Воздушные вяжущие вещества ЬЗО
Гидраплпческне вяжущие вещества 034
Материалы и изделия на основе вяжушнх вешеств .... 043
3. Стекло
Сырье для производства стекла
Производство стекольной массы
Стекловаренные печи
Изготовление стеклянных изделий .
4. Растворимое стекло и силикагель ,
Растворимое стекло
Силикагель
5. Плавленые горные породы и природные кислотоупоры .
6. Ситаллы
Литература
П d е д м е т н ы ii указатель
ПРЕДИСЛОВИЕ
В процессе подготовки к четвертому изданию книга «Общая
химическая технология неорганических¦ веществ» подверглась
значительной переработке. Это было вызвано необходимостью
отразить в ней достижения технического прогресса бурно
развевающейся химической промышленности в области
технологии важнейших неорганических продуктов.
При переработке книги авторы стремились не только
осветить вопросы внедрения новой техники, методы интенсификации
существующих и создания новых производств, но и, по
возможности, показать перспективы дальнейшего развития основной
химической промышленности на ближайшие годы.
Наиболее существенно переработаны главы, посвященные
технологии серы и серной кислоты, производству газов (азота,
водорода, кислорода), технологии связанного азота, производству
электролитического хлора и щелочей, переработке хлора,
производству минеральных удобрений. Некоторые из этих
разделов книги заново написаны или переработаны специалистами,
дополнительно привлеченными в состав авторского
коллектива.
В книге учтены также замечания и пожелания
преподавателей техникумов и отзывы читателей.
Глава «Металлы» исключена из четвертого издания в связи
с тем, что содержащиеся в ней сведения не входят в программу
техникумов по курсу общей химической технологии и
изучаются з курсе металловедения. В этом издании опущена также
глаза «Минеральные соли», поскольку технология
разнообразных и специфических солевых производств относится к
специальным дисциплинам, которые изучаются по программам,
соответствующим определенным профилям средних
специальных учебных заведений.
Введение, глава II и разделы 1 и 6 главы XIV написаны
И. В. Шманенковым, главы I, XI, XII, XITI и разделы 2—5
главы XIV— А. И. Шерешевским. Главы III, IV, разделы 1, 4 и 5
главы V, разделы 1, 2 главы VI, раздел «Основные сведения по
электрохимии» и раздел 1 (за исключением сведении о
свойствах и применении едкого натра), а также раздел 2
главы VIII, раздел 2 главы IX (за исключением подраздела
«Двуокись хлора») и главу X написал А. П. Егоров.
Разделы 2 и 3 главы V и раздел 3 главы VI написал
Е. Я- Мельников. Глава VII существенно переработана и
дополнена С. И. Каргиным. Подраздел «Едкий натр (каустическая
сода)», а также разделы 3 и 4 главы VIII, раздел 1 и
подраздел «Двуокись хлора» главы IX написал С. М. Круглый.
Авторы считают своим приятным долгом выразить глубокую
благодарность за большую помощь в работе над книгой
Г. С. Дурассу, проф. Б. А. Сасс-Тисовскому, В. Н. Антонозу,.
И. Е. Авцину, А. Г. Сокальскому, П. Т. Остапенко, А. А.
Егорову, М. Н. Степанову, А. Е. Калинчуку, Ю. П. Бернацкому,
А. Н. Костюковскому, М. П. Пятницкому.
Критические замечания и пожелания читателей будут с
признательностью приняты авторами.
Авторы
ВВЕДЕНИЕ
Химическая технология изучает процессы переработки
природных материалов в средства производства (продукты и
полупродукты, применяемые в промышленности и сельском
хозяйстве) и б предметы народного потребления. В химической
технологии рассматриваются процессы, связанные с изменением
химического состава перерабатываемых материалов, в отличие
ют механической технологии, изучающей процессы, при
которых изменяются лишь форма или внешний вид
обрабатываемого материала.
Примерами процессов, относящихся к химической
технологии, являются получение синтетического аммиака из азота и
водорода, получение кальцинированной соды из поваренной
соли и известняка и др.; примерами процессов, относящихся к
механической технологии, — прокат металла, прядение хлопка
и т. д.
В химической технологии неорганических веществ
изучаются процессы переработки материалов минерального
происхождения: производство серной кислоты из колчедана, серы,
серосодержащих газов; синтез аммиака из азота воздуха и
водорода, получаемого, например, электролизом воды; производство
силикатных материалов и т. д.
В химической технологии органических веществ
рассматриваются процессы синтеза углеродсодержащих веществ из
продуктов переработки угля, нефти и природных газов, а также
процессы переработки материалов растительного или
животного происхождения (производство сахара, бумаги, дубление
КОЖ)! И ДР.).
В связи с все большим расширением комплексного
использования сырья и комбинированием отдельных производств з
настоящее время деление на органические и неорганические
производства становится в значительной мере условным.
Поясним некоторые понятия, которые будут встречаться при
описании различных технологических процессов.
Под сырьем понимают исходные материалы, которые служат
для получения того или иного продукта. В технологических
процессах часто используют несколько видов сырья, смешан-
иых з определенном отношении. Если такая смесь состоит из
твердых Материалов, ее называют шихтой.
11
Сырье во многих случаях проходит несколько стадий
переработки. Продукт, полученный в последней стадии
технологического цикла, носит название готового продукта, полученный
в какой-либо промежуточной стадии — полупродукта.
При производстве того или иного вещества часто
получаются различного рода побочные продукты и отходы. Если эти
отходы не используются, то они называются отбросами. Иногда
отходы подвергают дополнительной обработке для извлечения
неиспользованного сырья и возвращения его в технологический
процесс; такая операция носит название регенерации.
Одной из важнейших задач современной техники является
комплексная переработка сырья с целью наиболее полного
использования отходов. Так, например, отходами производства
серной кислоты из колчедана являются огарок, пыль и шлам.
Из этих отходов в настоящее время получают ценные
продукты. Так, из огарка можно выделить железо, из пыли —
германий, таллий, кадмий и др., из шлама — селен и тел-
¦уур-
При комплексном использовании сырья необходима высокая
техника пылеулавливания и очистки газов и сточных вод.
Одновременно при этом уменьшаются или ликвидируются вредные
выделения в атмосферу и сброс вредных стоков в водоемы, что
приьодиг к значительному оздоровлению условий жизни в
промышленных районах.
Р> истории развития химической промышленности известны
факты, когда отбросы становились главными продуктами, а
главные продукты — побочными. Например, при производстве
соды по способу Леблана сырьем служил сульфат натрия,
который получали разложением хлористого натрия серной кислотой,.
причем выделявшийся хлористый водород долгое время являлся
обременительным отбросом. После того как нашли применение
водному раствору хлористого водорода — соляной кислоте, она
стала главным продуктом, а сульфат натрия — побочным.
Долгое время изыскивались области применения хлора,
образующегося при электрохимическом производстве едкого натра. Теперь
хлор широко используется в синтезе разнообразных химических
продуктов, имеющих огромное народнохозяйственное значение,
в производстве титана, ниобия и др.
Расход сырья и количество получаемых в технологическом
процессе продуктов, полупродуктов и отходов определяют на
основе технических расчетов. Результаты этих расчетов обычно
сводят в материальный баланс, который составляют в расчете-
на единицу готового продукта или исходного сырья (на I кг
или 1 г) или на количество продукта, получаемого з единицу
времени (час, сутки).
В качестве примера в табл. 1 приведен материальный
баланс обжига колчедана, графически изображенный на рис. 1_
12
Таблица 1
Материальный баланс обжига колчедана
(на 1 т двуокиси серы)
Приход
Колчедан сухой
Влага колчедана ....
Кислород воздуха ....
Азот воздуха ....
Влага воздуха .....
Всего . .
Количество
кг
1210
42,5
1126
3790
41,5
6210
%
19,4
0,6
18,1
61,3
0,6
100
Расход
Двуокись серы
Кислород
Азот . .
Водяной пар
Всего . .
Коли! естпо
КС
1000
440
3790
84
896
6210
16,1
7,1
61,3
1,2
14,3
100
По данным материального баланса вычисляют коэффициент
использования сырья, представляющий собой отношение
количества полезного вещества в готовом продукте к количеству
полезного вещества в затраченном сырье. Так, например, если
при выплавке серы из 100 кг руды, содержащей 40% S,
получено 38 кг сырой серы,
содержащей 90% S, то коэффициент
использования сырья составит:
38-90-100
40-100
= 85,5%
На основе расчетов
прихода и расхода тепла при
осуществлении технологических
процессов составляют
тепловые балансы. Приход
определяют, суммируя тепло,
выделяющееся при протекании
экзотермических химических
реакций, тепло, приносимое реа-
кислород и25 кг
азот 37?0кс
Влага 4/l5hs
¦6210 нг A00%)-
газ
двуокись серь/ ,'ШОнг
кислород
азот
водяной пар 84 нг
гируюшими веществами, и теп- о„„ , п ,
" J-, Рис. 1. Диаграмма материального ба-
ЛО, ПОДВОДИМое извне. В СО- Ланса получения 1000 кг двуокиси се-
ответствии с законом сохране- ры сжиганием колчедана.
ния энергии приход тепла
должен быть равен его расходу. Расход тепла складывается из
телла, требуемого для протекания эндотермических реакций,
тепла, выводимого с продуктами реакции, и потерь тепла в
окружающую среду.
Пример теплового баланса представлен в табл. 2 и
графически изображен на рис. 2.
13
Таблица 2
Тепловой баланс продукционной башни-концентратора в производстве
серной кислоты
(на 1 т H2SO4, содержащейся в башенной кислоте)
Приход тепла
С сернистым газом
D00 °С)
С циркуляционной
кислотой D5 °О
Теплота кислотообразова-
ния
Всего. . .
Количество
ккал
165000
30000
55000
250000
%
66
12
22
100
Расход тепла
С отходящим газом
A20°С)
На концентрирование
кислоты
С концентрированной
кислотой A40°С)
Потери
Всего. . .
Количестве
ккал
500ОО
95000
72500
32500
250000
%
20
38
29
13
100
Коэффициентом использования электроэнергии называют
отношение количества энергии, которое теоретически требуется
затратить при получении весовой единицы продукта, к количе-
На нот/ептрированив
95000 чнал
С концентрированной
кислотой 72500'кн
Рис. 2. Диаграмма теплового баланса продукционной
башни (на 1 т H2SO4, содержащейся в кислоте).
ству практически затраченной энергии. Так, например,
теоретически при электролизе хлористого натрия на получение 1 г
хлора должно быть затрачено 1656 квт-ч электроэнергии.
Практически же расход электроэнергии гораздо больше. Если
практический расход электроэнергии составляет 2800 квт-ч,
коэффициент использования энергии будет равен:
1656
2800
• 100 = 59,1%
Производительностью аппаратов называют количество
продукции, полученной з них за единицу времени (например, за
сутки). В зависимости от характера аппарата производитель-
14
ность его можно отнести к единице объема или рабочей
поверхности аппарата или к единице площади поперечного сечения.
Эту относительную величину часто называют съемом.
При проведении технологических процессов стремятся
достигнуть максимальной производительности аппаратов.
Проектную производительность аппаратов устанавливают с учетом
оптимальных условий ведения технологического процесса и
называют мощностью.
Технологические процессы разделяются на периодические я
непрерывные.
При периодическом процессе в аппарат загружают нужную
порцию сырья и в течение определенного времени создают и
поддерживают заданные условия ведения процесса или
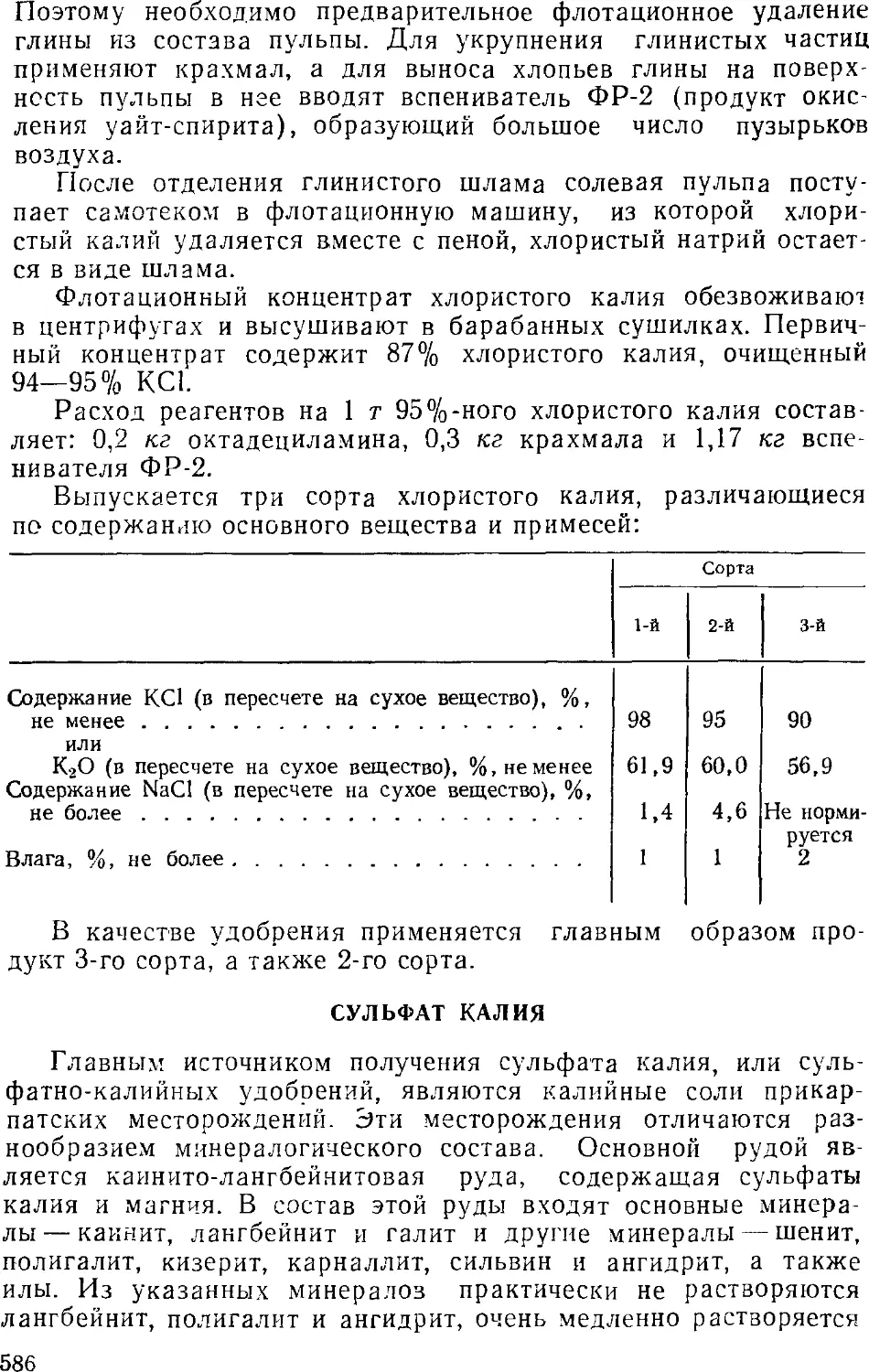
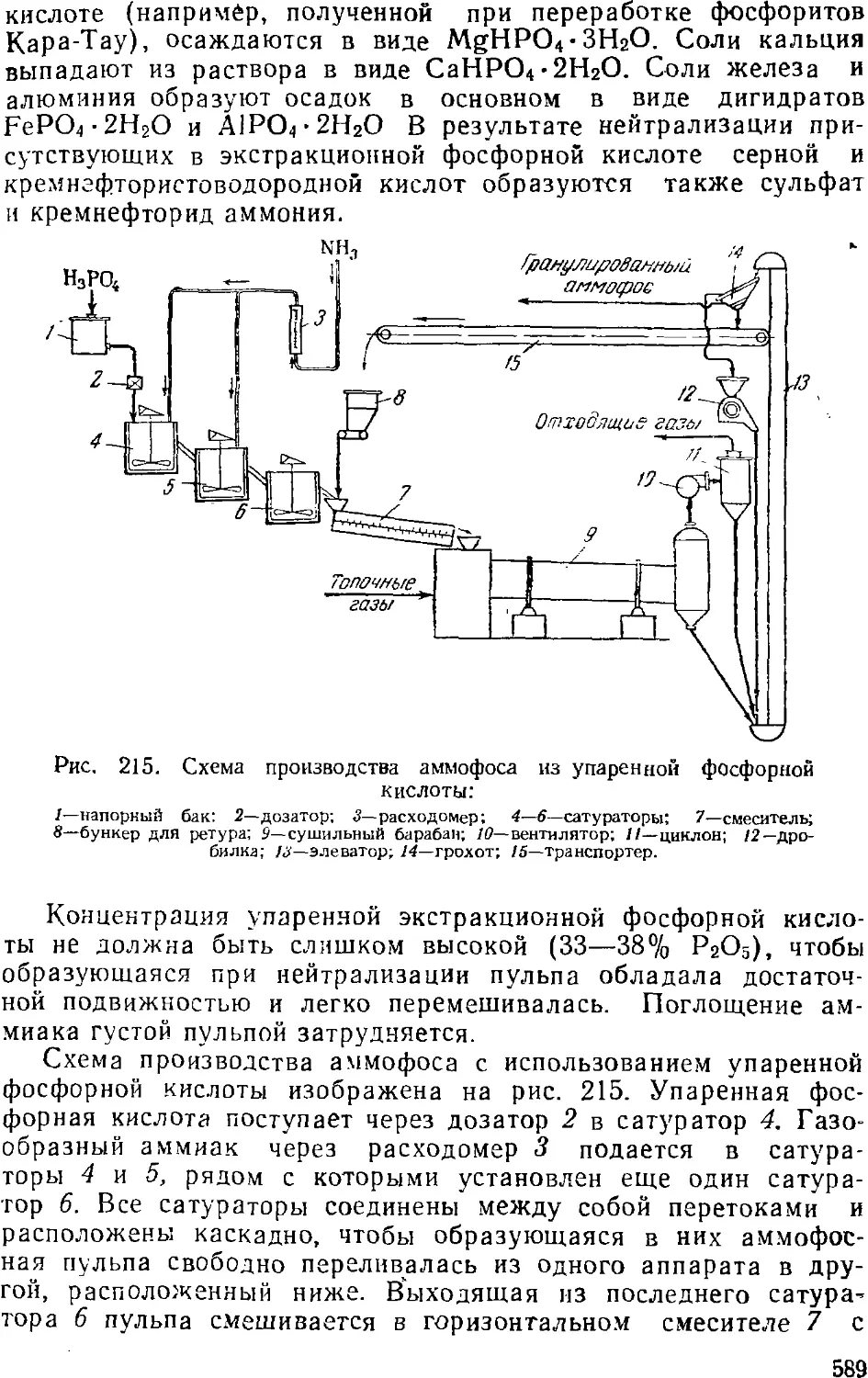
изменяют их соответственно программе его ведения. При этом
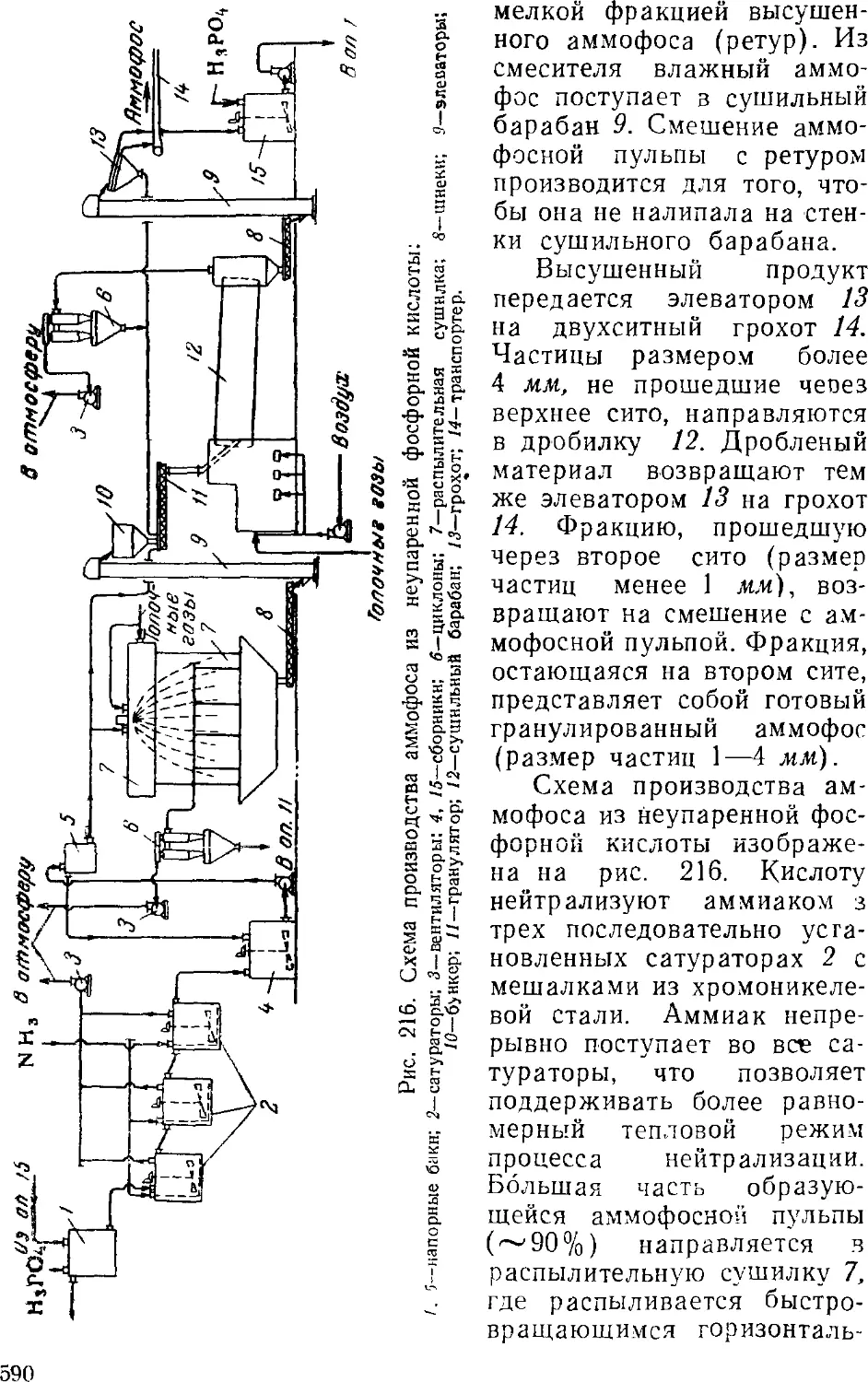
соотношение и концентрация реагентов и получаемых продуктов,
температура реакционной массы, давление и другие условия
изменяются во времени, а все стадии данного процесса
последовательно протекают во всем объеме реакционной массы,
находящейся в аппарате. После завершения процесса из аппарата
выгружают продукт реакции и снова повторяют все операции.
В непрерывных процессах загрузка сырья производится
непрерывно или периодически, определенными порциями по мере
освобождения некоторого объема аппарата. Таким образом, ъ
непрерывных процессах часть сырья только загружается в
аппарат, часть находится в стадии переработки, а часть уже
прореагировала и выводится из процесса. В отличие от
периодических процессов все стадии непрерывного процесса
одновременно протекают в различных зонах реакционной массы.
Причем в каждой зоне в любой момент времени сохраняются
неизменные условия данной стадии процесса, и для осуществления
всего процесса в целом необходимо непрерывное перемещение
реакционной массы по зонам.
Примерами периодических процессов могут служить
азотирование карбида кальция в цианамидных печах, обжиг
керамических изделий в кольцевых печах; в качестве примеров
непрерывных процессов можно назвать обжиг колчедана или
известняка, электролиз растворов хлоридов и др.
Процессы, в которых непрореагировавший исходный
материал вместе с новой порцией свежего материала возвращается
в начальную стадию процесса, называются круговыми
процессами. Таким образом, при круговом процессе часть исходного
сырья непрерывно циркулирует в замкнутом цикле. Примером
кругового процесса может служить синтез аммиака из водорода
и азота.
В современной технике при производстве крупнотоннажных
продуктов стремятся заменять периодические процессы
непрерывными процессами, отличающимися значительными преиму-
15
щестзами. Эти преимущества заключаются в лучшем
использовании выделяющегося тепла, возможности полной (или
комплексной) механизации и автоматизации управления процессом
и, следовательно, в большей устойчивости и постоянстве
технологического режима, в связи с чем получается продукция
лучшего качества. Кроме того, при непрерывных процессах
улучшаются условия труда и уменьшаются капиталовложения
(в результате уменьшения размеров аппаратуры, а
следовательно, и размеров производственных зданий).
В химической технологии широко применяется так
называемый принцип противотока. Например, для улавливания
аммиака из коксового газа устанавливают четыре скруббера
(башни), в которых вода, поглощающая аммиак, движется навстречу
коксовому газу. Свежий коксовый газ поступает в первый
скруббер, чистая вода — в четвертый скруббер, где извлекает
из газа незначительные количества аммиака, не поглощенные
в предыдущих скрубберах. В первый скруббер попадает вода,
уже содержащая аммиак; встречая богатые аммиаком газы,
ока поглощает еще некоторое количество аммиака. Таким
образом достигается наиболее полное извлечение аммиака и
получается более концентрированный раствор. По принципу
противотока работает также большинство теплообменников
(холодильники, подогреватели, конденсаторы, регенераторы и др.).
В последние годы в химической промышленности широко
внедряются комплексные механизация и автоматизация
производственных процессов с применением различных средств
автоматического управления. Системой комплексной автоматизации
осуществляются автоматические контроль, регулирование,
сигнализация, а также защита и блокировка. При этом
автоматически поддерживается заданный режим, т. е. достигается
стабилизация процессов. Особое внимание в химических
производствах уделяется разработке и применению средств
дистанционного управления процессами, предотвращающего и даже
исключающего различные вредные воздействия (высокая
температура, агрессивные среды, выделяющаяся пыль и др.) на
обслуживающий персонал.
В ряде химических производств при необходимости
регулирования большого количества параметров требуется
математическая обработка получаемых данных. В этих случаях
целесообразно применение управляющих
счетно-вычислительных машин. При помощи таких машин возможно создание
систем автоматической оптимизации процессов, т. е. определение
и поддержание такого технологического режима, который
обеспечивает максимальную технико-экономическую
эффективность данного производства.
Глава I
МИНЕРАЛЬНОЕ СЫРЬЕ
ЗАПАСЫ МИНЕРАЛЬНОГО СЫРЬЯ
В земной коре* степень распространения отдельных
химических элементов весьма различна. Как видно из приведенных
в табл. 3 данных, 99,61% земной коры состоит из соединений
только 15 химических элементов: кислорода, кремния,
алюминия, железа, кальция, натрия, магния, калия, водорода, титана,
углерода, хлора, фосфора, серы и марганца. Содержание
остальных химических элементов в земной коре составляет
0,39%. Некоторые элементы, содержащиеся в земной коре в
незначительных или очень малых количествах, имеют большое
значение в современной технике. К таким элементам относятся.
например, ртуть, бром, иод, бор, германий, индий, литий, цезий
и др.
Таблица 3
Средний химический состав земной коры
(по данным акад. А. Е. Ферсмана)
Элемент ь[
Содержание j;
вес. %\)
Элементы
Содер-|
жание
вес. %1|
Элементы
Содержание
вес. %
Кислород .
Кре>ннй .
Алюминий
Железо . .
Кальций .
Натрий . .
Магний . .
Калий . .
Водород .
49,13
26,00
3,25
1,00
Титан . .
Углерод .
Хлор . .
Фосфор .
Сера . .
Марганец
Фтор . .
Барий . .
Азот . .
0,61 | Стронций
0,35 j Хром . .
0,20 ! Цирконий
0,12 J Ванадий .
0,10 I Никель .
0,10 Цинк
0,08 I Медь
0,05 I Прочие элементь;
0,04 '
0,035
0,03
0,025
0,02
0,02
0,02
0,0!
0,06
Участки земной коры, где имеются скопления тех или иных
элементов или их соединений (минералов), называются
месторождениями минерального сырья. При оценке месторождения
* Земной корой условно называют слой земли толщиной около 16 км.
2—2848 1 7
определяют запасы минерального сырья, его свойства,
химический состав, условия залегания и доступность для разработки.
Кроме определения величины запасов сырья и качественной
их характеристики, для полной оценки месторождения имеют
также значение н экономические факторы — близость к водным
или железнодорожным путям сообщения, наличие источников
воды, топлива или других видов энергии, климатические
условия, отдаленность от районов потребления сырья нли готовой
продукции из данного сырья.
Наличие месторождений минерального сырья имеет
исключительно важное значение для экономического развития сгра-
ны. СССР обладает многочисленными месторождениями
минерального сырья; многие их них имеют мировое значение.
В зависимости от вида добываемого полезного ископаемого
и характера горных работ участки месторождений, на которых
ведется добыча минерального сырья, называют рудниками
(железные, медные, фосфоритные, калийные рудники), промыслами
(промыслы самосадочной соли, нефтяные промыслы),
приисками (золотые, платиновые прииски), карьерами (добыча
известняка, гипса, песчаника, глины и др.).
ОБОГАЩЕНИЕ РУД
Горные породы, из которых можно добывать
промышленными способами минеральное сырье, называются рудами.
Руды обычно содержат, кроме полезных элементоз,
некоторое количество вредных или бесполезных примесей, так
называемую пустую породу. В большинстве случаев содержание
примесей относительно велико, поэтому руду приходится
обогащать.
Обогащение заключается в отделении от полезных
минералов большей части примесей. Если в руде содержится несколько
полезных компонентов, применяют такие способы обогащения,
которые позволяют не только отделить пустую породу, но и
произвести разделение полезных компонентов.
Поступающее на обогащение минеральное сырье называется
исходной рудой, полученный после обогащения продукт —
концентратом. Концентрат содержит данного полезного зещества
больше, чем исходная руда. Если в минеральном сырье
содержится несколько полезных компонентов, при его обогащении
часто получают несколько видов концентратов. Например, из
уральского медного колчедана получают медный, цинковый и
пиритный концентраты. Остатки после обогащения руды
называются хвостами.
Обычно в хвостах остается некоторое количество полезных
веществ. Отношение количества концентрата к количеству
исходной руды, выраженное в весовых процентах, называется
18
выходом. Отношение количества полезного компонента в
полученном концентрате к количеству полезного компонента,
содержавшегося в исходной руде, принято называть степенью
извлечения. Отношение же процентного содержания полезного
компонента з концентрате к процентному содержанию полезного
компонента в исходной руде называют степенью обогащения.
Наиболее полное комплексное использование руд — не
только концентратов, но и хвостов — задача большого
народнохозяйственного значения.
Существует много способов обогащения руд. Все они
основаны на различии физических (реже химических) свойств
полезных минералов и пустой породы.
Ручная рудоразборка заключается в ручном отборе породы
по внешним признакам — цвету, блеску, размеру и форме
кусков или зерен. Это очень простой метод, которым, однако,
нельзя достигнуть достаточно полного обогащения.
Для ускорения ручной рудоразборки применяют ленточные
транспортеры или вращающиеся круглые столы, на которых
материал движется с такой скоростью, чтобы рабочие успевала
отобрать нужную руду.
Грохочение (обогащение по крупности) заключается в
пропускании руды через грохоты — сига различных систем. Так,
например, при обогащении фосфоритных руд фосфорит, как
более крупный материал, задерживается грохотом, а пустая
порода проходит через отверстия сиг. Если руду необходимо
разделить по крупности более чем на два сорта, ее пропускают
через несколько последовательно установленных грохотов с все
уменьшающимися размерами отверстий в ситах. Различают
грохочение сухое и мокрое; в последнем случае грохот
орошается водой, увлекающей пустую породу, благодаря чему
одновременно происходит ее отделение от руды.
Разновидностью грохочения является обогащение,
основанное на различии формы частиц полезных минералов и пустой
породы. Некоторые минералы (сернистые соединения свинца,
цинка, железа) образуют включения в форме куба или шара
и поэтому могут быть легко отделены от пустой породы,
состоящей из пластинчатых кусков, на ситах с отверстиями
определенной формы.
Обогащение, основанное на различии характера
поверхности (по трению), заключается в том, что зерна разной
гладкости поя действием внешних сил перемещаются по поверхности
с различной скоростью. Этим способом можно обогащать руды,
разделяя их на легко скользящие гладкие зерна и зерна с
неоднородной шероховатой поверхностью, скользящие более мед
ленно. Косзенное влияние оказывает в данном случае не только
характер поверхности, но и форма зерна. Так, при движении
раздробленной асбестовой руды, состоящей из волокон асбеста
2* 19
и твердой горной породы — змеевика, шероховатые волокна
асбеста перемещаются медленнее, чем зерна змеевика с гладкой
поверхностью.
Для обогащения по трению применяют спиральные или
винтовые сепараторы.
Избирательное дробление основано на различной твердости
минералов, входящих в состав руды. Так, фосфорит имеет
меньшую твердость, чем кварц, и при дроблении руды
измельчается более тонко.
Комбинируя размол н рассев, можно получить фракции,
более богатые фосфоритом.
Гидравлическое, или мокрое, обогащение основано на
различной скорости осаждения в жидкости (обычно в воде) зерен
разной величины или зерен одинаковой величины, но различной
плотности. Наиболее простым способом гидравлического
обогащения является промывка руды водой. Так, при мокром
обогащении фосфоритной руды более легкие частицы глины и песка
уносятся струей воды. Промывку руды ведут в гравиемойках и
других аппаратах.
Гравиемойка представляет собой наклонный желоб с зра-
щающимся шнеком. Внутрь этого желоба подают воду,
исходную руду загружают в нижнюю часть желоба (корыто), а
отмытую руду выгружают из верхней его части.
В аппарате другого типа, представляющем собой корыто,
дно которого имеет небольшой уклон, навстречу друг другу
вращаются два вала с лопатками, насаженными на вал по
винтовой линии. При вращении валов материалу, находящемуся
з корыте, сообщается поступательное движение вверх к
разгрузочному концу корыта. Вода, подаваемая в верхнюю часть
корыта, течет к нижнему концу, навстречу руде. Мелкие частицы
глины и песка уносятся водой, отмытая руда выгружается
возле верхнего конца корыта.
Промывка руды обычно является предварительной
операцией, предшествующей другим обогатительным процессам.
Для разделения руды на различные по крупности частицы
одинаковой плотности применяют различные гидравличиеские
классификаторы. Один из таких аппаратов представляет собой
большой ящик, нижняя часть которого разделена
вертикальными перегородками на отдельные ячейки. В ящик в
горизонтальном направлении поступает взвесь руды в воде. Вследствие
разной скорости осаждения в первых по ходу жидкости ячейках
осаждаются наиболее крупные частицы, а в последних —
наиболее мелкие. В зависимости от скорости движения воды,
глубины ящика, расстояния между перегородками, их высоты и.
количества можно получить разное число фракций,
отличающихся по размерам частиц. Для облегчения выгрузки
осажденных частиц руды ящику придают сужающуюся книзу форму.
20
Применяют также оолее сложные гидравлические
классификаторы, в которых поток воды движется зигзагообразно по
серии каналов, образуя восходящие и нисходящие струи, что
дает возможность регулировать скорость осаждения частиц.
Гидравлическое обогащение рыхлых мелкозернистых пород
по крупности зерен осуществляют большей частью на
наклонных столах-концентраторах, где твердый материал под
действием горизонтально направленной струи воды, поступающей с
одной стороны стола, расслаивается соответственно крупности
зерен. Для гидравлического обогащения, основанного на
различии плотности минералов, входящих в состав руды, применяют
так называемые отсадочные машины.
Отсадочная машина представляет собой ящик с решетчатым
дном, в котором материал подвергается многократному
воздействию восходящих и нисходящих струй воды, причем зерна
материала различной плотности падают с разной скоростью,
образуя слои частиц, обладающих соответствующей плотностью.
Таким образом обогащают, например, руды, содержащие
сернистые металлы (сернистый свинец, сернистое железо,
сернистый цинк), отделяя их от кварца.
Воздушное обогащение основано на различных скоростях
падения твердых частиц в воздушном потоке в зависимости ог
их размеров и плотности. Этот способ применяют для
обогащения сырья, состоящего из частиц размером не более 1,5 лш_
Аппараты, в которых производится воздушное обогащение,
называются сепараторами. Различают воздушные и центробежные
сепараторы.
Воздушный сепаратор представляет собой аппарат, в
котором отделение легких частиц от более тяжелых производится
при помощи струи воздуха. Процесс разделения частиц
регулируют изменением скорости воздушной струи, что достигается
различным комбинированием количества и размеров лопастей
вентилятора и скорости их вращения.
В центробежных сепараторах частицы сырья попадают на
диск, вращающийся в горизонтальной плоскости с большой
скоростью. Наиболее крупные и тяжелые частицы под действием
центробежной силы отбрасываются на более далекое
расстояние, чем легкие и мелкие частицы.
Магнитное или электромагнитное обогащение основано на
различии магнитной проницаемости минералов, входящих в
состав сырья. Размолотую руду пропускают через магнитное
поле, создаваемое магнитами или, чаще, электромагнитами.
Частицы более намагничивающегося минерала, например,
магнитного железняка, отделяемого при обогащении песков,
проходя через магнитное поле, отклоняются в большей или
меньшей степени от своего первоначального пути. Частицы слабо
намагничивающихся или совсем ненамагничивающихся мине-
2 К
ралов не отклоняются. Аппараты, применяемые для такого
обогащения, называют соответственно магнитными или
электромагнитными сепараторами. Наиболее распространено
электромагнитное обогащение сухих материалов. Для сильно магнитных
минералов применяют иногда мокрое электромагнитное
обогащение, при котором через магнитное поле пропускают взвесь
руды в воде.
Обогащение в тяжелых жидкостях и суспензиях. В
лабораторных исследованиях широко применяется метод обогащения,
основанный на разделении минералов, имеющих различную
плотность, в жидкостях, обладающих промежуточной
плотностью. Минералы с меньшей плотностью, чем жидкость,
всплывают, более тяжелые минералы оседают на дно. Применяемые
для разделения тяжелые жидкости не должны вступать в
химическое взаимодействие с минералами и обладать высокой
вязкостью. В промышленности этот метод обогащения не
применяется вследствие высокой стоимости тяжелых жидкостей к
трудности их регенерации.
В промышленной практике получил распространение метод
обогащения полезных ископаемых в тяжелых суспензиях.
Средой для разделения минералов с различной плотностью в этом
случае служит взвесь твердых минералов (утяжелителей) в
воде, обладающая повышенной плотностью, определенной
вязкостью, текучестью и упругими свойствами. В качестве
утяжелителей применяют кварцевый песок и магнетит (для
обогащения легких материалов) или ферросилиций (для обогащения
тяжелых материалов). Ферросилиций является наиболее
распространенным утяжелителем. Он обладает высокой твердостью
E—6), частицы его медленнее истираются при обогащении и
потому образуют более устойчизые суспензии.
Расход ферросилиция на 1 г обрабатываемой руды
составляет 100—500 г.
Флотационное обогащение основано на различной
смачиваемости частиц разных минералов водой и способности несмачи-
ваемых или плохо смачиваемых частиц всплывать в виде пены
вместе с пузырьками специально подаваемого воздуха*.
Обычно при обогащении руд методом флотации в смесь
воды и измельченных минералов вводят небольшие количества
так называемых флотационных реагентов (пенообразователи,
коллекторы и др.). Пенообразователи уменьшают поверхностное
натяжение воды и тем самым способствуют образованию пены.
Коллекторы образуют на поверхности частиц минералов
тончайшую гидрофобную (несмачиваемую водой) пленку, что
способствует лучшему прилипанию частиц к пузырькам воздуха.
* Механизм действия воздушных пузырьков можно образно
представить, если сравнить пузырек, удерживающий прилипшую к нему частицу
минерала, с аэростатом, поднимающим корзину.
Расход флотационных реагентов при флотации незначителен.
Схема процесса флотационного обогащения состоит в
следующем. Исходную руду подвергают последовательно
дроблению и тонкому измельчению. Измельченную руду смешивают
с водой и флотационными реагентами, полученную пульпу
обрабатывают воздухом в специальных флотационных машинах.
В результате такой обработки в пульпе образуется большое
количество воздушных пузырьков, которые увлекают
флотируемые минералы вверх и вместе с ними образуют пену,
выходящую из машины. Нефлотируемые минералы оседают на дно и
удаляются из машины в виде шлама.
Чаще всего полезные минералы находятся в пене, в этих
случаях пену направляют в специальные отстойники, где из нее
оседают извлеченные минералы. Отфильтрованный и
высушенный осадок представляет собой готовый концентрат. Если
полезные минералы находятся в шламе, его подвергают
отстаиванию, фильтрации и сушке.
В настоящее время флотационный метод применяют для
обогащения многих руд: колчеданов, полиметаллических руд,
самородной серы, апатита, плавикового шпата, баритов и др.
В зависимости от особенностей перерабатываемых руд и
целей обогащения на практике применяются разнообразные
схемы этого процесса, обычно сочетающие несколько способов
обогащения в различных комбинациях.
ИСПЫТАНИЕ МИНЕРАЛЬНОГО СЫРЬЯ
Обычно определяют химический и гранулометрический
состав минерального сырья. В ряде случаев исследуются
некоторые специфические свойства сырья — плотность, твердость,
магнитная и электрическая проницаемость и др. Наряду с
химическими и физико-механическим методами исследования:
сырья применяются микроскопические, термические,
электрометрические, рентгеноскопические и другие методы его
исследования.
Отбор средней пробы. При переработке минерального сырья
очень важно правильно отобрать среднюю пробу; состав
средней пробы должен отвечать составу всего материала, от
которого взята проба.
Методика взятия средней пробы сводится к отбору
некоторой определенной части от всего количества испытуемого-
сырья. Отбор проб на месте хранения сырья производят в
нескольких точках, находящихся на определенных расстояниях
друг от друга. Отбор проб из потока, например с движущегося
транспортера, производится через определенные промежутки
времени. Число точек для отбора проб и глубина слоя, из
которого берут пробы, зависят от характера сырья. Чем менее
23
однородно сырье по минералогическому составу и по крупности
кусков, тем больше должно быть отобрано проб.
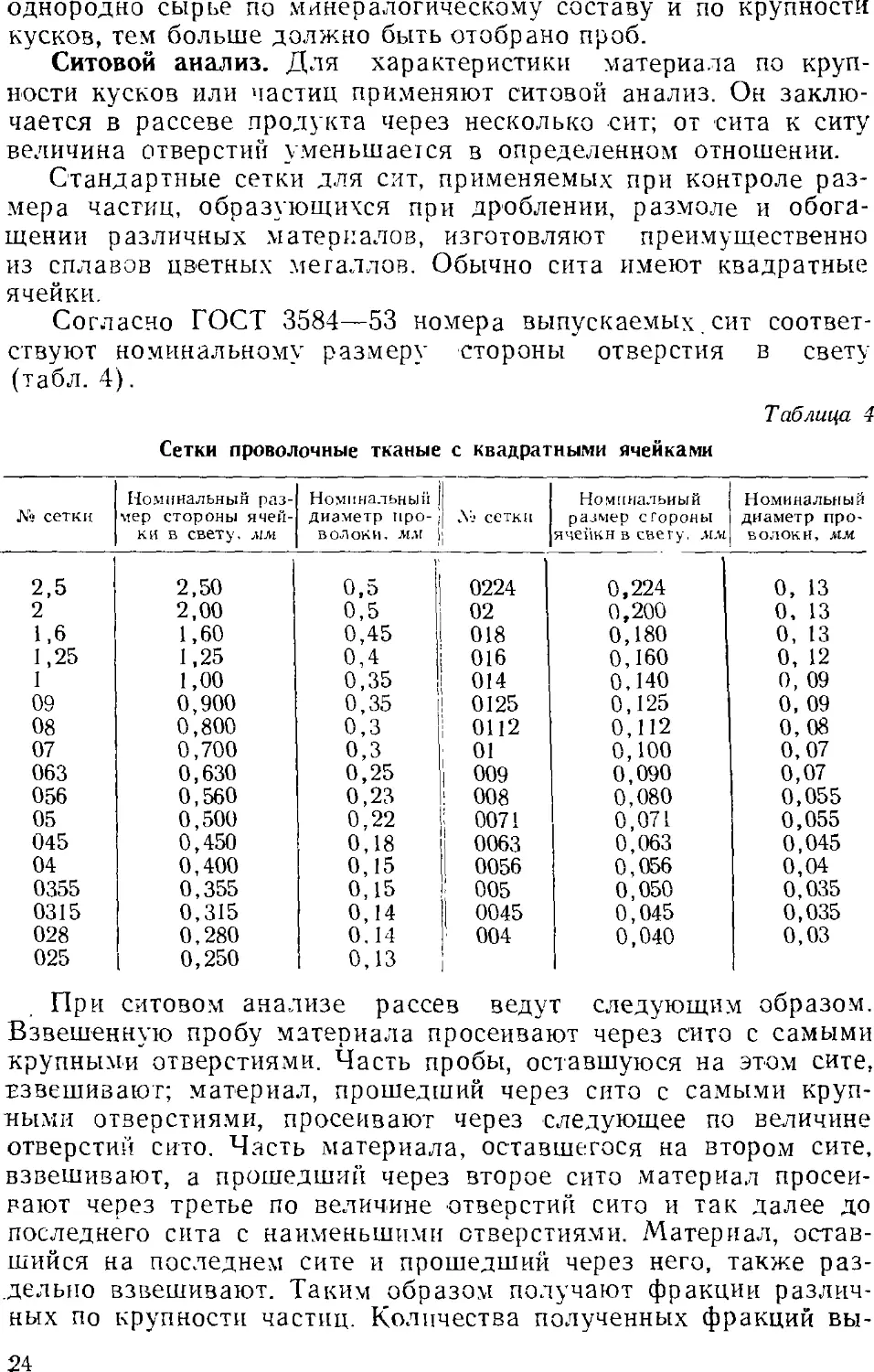
Ситовой анализ. Для характеристики материала по
крупности кусков или частиц применяют ситовой анализ. Он
заключается в рассеве продукта через несколько сит; от сита к ситу
величина отверстий уменьшается в определенном отношении.
Стандартные сетки для сит, применяемых при контроле
размера частиц, образующихся при дроблении, размоле и
обогащении различных материалов, изготовляют преимущественно
из сплавов цветных металлов. Обычно сита имеют квадратные
ячейки.
Согласно ГОСТ 3584—53 номера выпускаемых, сит
соответствуют номинальному размеру стороны отверстия в свету
(табл. 4).
Таблица 4
Сетки проволочные тканые с квадратными ячейками
№ сетки
2,5
2
1,6
1,25
1
09
08
07
063
056
05
045
04
0355
0315
028
025
Номинальный
размер стороны
ячейки в свету, мм
2,50
2,00
1,60
1,25
1,00
0,900
0,800
0,700
0,630
0,560
0,500
0,450
0,400
0,355
0,315
0,280
0,250
Номинальный
диаметр
проволоки, Л1ДГ
0,5
0,5
0,45
0,4
0,35
0,35
0,3
0,3
0,25
0,23
0,22
0,18
0,15
0,15
0,14
0.14
0,13
ЛЬ сетки
0224
02
018
016
014
0125
0112
01
009
008
; 007!
0063
0056
005
0045
004
Номинальный
размер стороны
ячейки в снегу, мм
0,224
0,200
0,180
0,160
О.НО
0,125
0,112
0,100
0,090
0,080
0,071
0,063
0,056
0,050
0,045
0,040
1
Номинальный
диаметр
проволоки, мм
0, 13
0, 13
0, 13
0, 12
0, 09
0,09
0,08
0,07
0,07
0,055
0,055
0,045
0,04
0,035
0,035
0,03
При сотовом анализе рассев ведут следующим образом.
Взвешенную пробу материала просеивают через сито с самыми
крупными отверстиями. Часть пробы, оставшуюся на этом сите,
взвешивают; материал, прошедший через сито с самыми
крупными отверстиями, просеивают через следующее по величине
отверстий сито. Часть материала, оставшегося на втором сите,
взвешивают, а прошедший через второе сито материал
просеивают через третье по величине отверстий сито и так далее до
последнего сита с наименьшими отверстиями. Материал,
оставшийся на последнем сите и прошедший через него, также
раздельно взвешивают. Таким образом получают фракции
различных по крупности частиц. Количества полученных фракций вы-
24
ражают в процентах от общего количества пробы. Классы
частиц, оставшихся на сите, обозначают знаком плюс, а классы
частиц, прошедших через сито, — знаком минус.
Обычно для проведения ситового анализа пользуются
набором лабораторных стандартных сит, вставляемых одно в
другое. Сита встряхивают вручную или механическим путем. Прл
ручном встряхивании рассев ведут в некоторых случаях до
получения постоянного веса остатка на каждом сите. Это очень
трудоемкая операция, особенно при массовых анализах. В
других случаях рассев считают удовлетворительным, если при
повторном встряхивании в течение 1 мин через сито проходит не
больше 1% от ранее оставшегося на нем материала. При
применении механических встряхивателеи устанавливают
требуемую продолжительность встряхивания для каждого вида сырья.
ЛИТЕРАТУРА
С. И. В о л ь ф к о в и ч, А. П. Егоров, Д. А. Э п ш т е й н, Общая
химическая технология, т. I, Госхимиздат, 1953.
М. А. Э й г е л е с, Обогащение неметаллических полезных ископаемых, Пром-
стройиздат, 1952.
Г. А. X а н, Опробование, контроль и автоматизация процесса обогащения,
Металлургиздат, 1958.
Глава И • ?
ВОДА
ОБЩИЕ СВЕДЕНИЯ
Вода играет очень важную роль в жизни человека.
Кроме расходования воды на бытовые нужды, огромное
количество ее потребляется промышленностью, транспортом и
сельским хозяйством. В химической промышленности воду
применяют для самых разнообразных целей. Значительные
количества водяного пара используют для обогрева аппаратов, а
также для получения синтез-газа, применяемого в производстве
аммиака, метанола и ряда других химических продуктов.
В районах с дешевой электроэнергией вода может служить
сырьем для получения водорода и кислорода методом
электролиза. В технологических процессах воду применяют для
охлаждения и нагревания реагентов, в качестве растворителя, для
промывки продуктов с целью отделения растворимых в воде
примесей и т. д.
Природные воды обычно содержат примеси различных
минеральных и органических веществ и растворенные газы.
Различают механические примеси, взвешенные в воде, и
химические примеси — растворенные в воде соли и газы. Кроме
того, в воде часто содержатся коллоидные частицы глины,
гидроокиси железа и др., органические примеси, например гумино-
вые вещества, а также присутствуют простейшие живые
организмы: бактерии, грибки и т. п.
Химические примеси состоят главным образом из
бикарбонатов кальция и магния, хлоридов и сульфатов натрия, калия,
кальция, магния, железа и алюминия, некоторых силикатов
(CaSiO3, MgSiO3 и др.).
Из газообразных веществ обычно растворены в воде азот,
кислород и двуокись углерода.
Рудничные шахтные или промысловые воды, откачиваемые
при добыче полезных ископаемых, часто содержат соли
цветных и редких металлов, иод, бром, сероводород,
азотсодержащие вещества, комплексные органоминеральные соединения
и др.
26
При исследовании качества воды обращают внимание
на ее цвет, залах, вкус, прозрачность воды (или ее мутность),
изменения при хранении, определяют температуру, содержание
взвешенных и растворенных примесей, в том числе ССЬ,
жесткость, окисляемость органических и неорганических веществ,
активную реакцию воды (кислотность или щелочность),
электропроводность, а также бактериальную и радиоактизную
загрязненность.
О количестве растворенных в воде примесей можно судить
по так называемому сухому, или плотному, остатку,
образующемуся в результате испарения 1 л воды и высушивания
остатка при 110°С до постоянного веса. Если этот остаток прокалить
при температуре темно-красного каления, то по потере веса
после прокаливания можно судить о количестве органических
примесей.
При оценке качества воды чаще всего предъявляют
требования в отношении ее жесткости. Под жесткостью понимают
содержание растворенных в воде солей кальция и магния,
выраженное в миллиграмм-эквивалентах ионов кальция или
магния. Единица жесткости отвечает содержанию 20,04 мг/л Са2+
или 12,16 мг/л Mg2+.
Раньше жесткость воды выражали в градусах жесткости,
причем за единицу (немецкий градус) принимали содержание
в 1 л воды 10 мг окиси кальция или 7,19 мг окиси магния. При
пересчете величины жесткости в мг-экв/л Са2+ или Mg2+ число
немецких градусов жесткости следует разделить на 2,8 или
умножить на 0,357.
Различают временную, постоянную и общую жесткость.
Временная (карбонатная) или устранимая, жесткость
характеризуется содержанием бикарбонатов кальция Са(НСОз)г
и магния Mg(HCO3J, которые при кипячении воды
превращаются в карбонаты и выпадают в виде накипи. Постоянная
(некарбонатная) жесткость воды зависит от содержания в ней
сульфатов, хлоридов и других солей кальция и магния, при
кипячении остающихся в растворе. Общей жесткостью называют
суммарную величину временной и постоянной жесткости.
Принята следующая классификация технической воды по
жесткости:
мг-экв/л мг-экв/л
Са (или Mg) Ca (или Mg)
Очень мягкая. . . . 0—1,5 Жесткая 6—10
Мягкая 1,5—3 Очень жесткая ... 10 и более
Средняя - 3—6
Кислотность или щелочность воды характеризуется
концентрацией водородных ионов, которую принято выражать через
водородный показатель рН. Величина рН природных вод
обычно колеблется в пределах 6,8—7,3, т. е. реакция их близка к
нейтральной.
27
Для контроля качества воды удобны походные
гидрохимические лаборатории. С помощью такой лаборатории,
укомплектованной набором аппаратуры, посуды и реагентов, определяют
прозрачность, цвет, запах и вкус воды, концентрацию
водородных ионов, общую и карбонатную жесткость, содержание
сероводорода, свободной углекислоты, закисного и окисного
железа, ионов аммония, кальция, магния, натрия, хлора, карбонат-
ионов и бикарбонат-ионов.
Классификация природных вод. В природе совершается по
¦стоянный круговорот воды. Под действием солнечного тепла она
испаряется с земной поверхности, образуя туман и облака, а
затем снова зоззращаегся на землю в виде атмосферных
осадков.
Различают следующие виды природных вод: атмосферную,
грунтовую, поверхностную и морскую.
Атмосферная вода (дождевая, снег, град) — наиболее
чистая. В сельских местностях она почти не содержит примесей,
в промышленных районах в ней содержатся примеси,
извлеченные из воздуха: частицы пыли и угля, различные газы. При
исследовании дождевой воды в районе некоторых химических
заводов было найдено до 70 мг серной кислоты в 1 л воды.
Сухой остаток после выпаривания 1 л дождевой воды в
среднем составляет 40 мг.
Грунтовая вода (из колодцев, подземных ключей,
артезианских скважин) содержит различные органические примеси
(растворимые вещества растительного и животного
происхождения) и неорганические примеси (бикарбонаты, хлориды и
сульфаты натрия, калия, магния, кальция и др.) Примеси
вымываются из слоев почвы и горных пород, через которые
проходят грунтовые воды. От состава этих горных пород зависит
химический состав грунтовых вод, который вследствие этого
может быть весьма различным.
Поверхностная вода (ручьи, реки, озера) также отличается
большим разнообразием химического состава; обычно она
содержит растворенных солей меньше, чем подземная, зато
загрязнена механическими примесями, особенно во время
весенних и осенних паводков.
Морская вода содержит много солей, особенно хлоридов.
Количество сухого остатка, получаемого при испарении 1 л
морской воды, колеблется от 30 до 40 г.
В зависимости от назначения потребляемой воды
различают:
1) питьевую воду;
2) техническую' воду, потребляемую в промышленности,
сельском хозяйстве, транспорте, коммунальном хозяйстве
(бани, прачечные, поливка территории, плавательные бассейны
и др.). для противопожарных нужд и т. д.;
28
3) минеральные воды, содержащие целебные примеси (соли,
газы) и используемые для лечебных целей.
Требования к качеству питьевой воды регламентируются
ГОСТ 2875—54. Особо жесткие нормы установлены на
содержание вредных примесей в питьевой воде, например, мышьяка,
свинца, фтора, меди, цинка и др.
Количество колоний бактерий в 1 мл воды после 24-часового
выращивания при 37 °С должно быть не более 100, количество
кишечных палочек в 1 л воды—не более 3. Содержание в
питьевой воде взвешенных веществ (мутность) не должно превышать
2 лг/л.
Требования к качеству технической воды, потребляемой в
различных отраслях народного хозяйства, весьма многообразны
и устанавливаются в зависимости от условий и характера
применения воды — для обогащения руд, в гидрометаллургических,
химических и других технологических процессах, для
получения пара, охлаждения, использования в гидравлических
устройствах, теплообменниках и т. д.
Некоторые требования к качеству технической воды
установлены более высокими, чем для питьевой воды, например, по
содержанию ряда примесей.
Наличие примесей в воде для питания паровых котлов
вызывает образование на стенках котла накипи из плотной
массы солей.
Теплопроводность стенок котла при этом значительно
ухудшается, а вынужденные перегревы приводят к его
преждевременному износу.
Накипь может образоваться в результате следующих
реакций:
Са(НСОз)., -г MgSO4 = CaSO4 -f- Mg(OH)., -f- 2CO2
Ca(HCO3J = CaCO3 -f- CO2 + H2O
CaCO3 -r MgSO4 ~c * CaSO4 + MgCO3
Ca(HCO3), — Na,SO4 -—* CaSO4 -f Na2CO3 -f- CO., + H3O
Первые две реакции практически необратимы, остальные
обратимы. Образующиеся в результате этих реакций гипс п
карбонаты кальция и магния вместе с присутствующими в воде
силикатами и механическими примесями образуют накипь.
Для удаления накипи котлы приходится периодически
чистить и промывать.
Содержащиеся в воде примеси вызывают коррозию
металлических частей котла и его арматуры, а также «бросание»
воды з котлах, вспенивание и, как следствие этого, загрязнение
паропроводов, цилиндров паровых машин и лопаток турбин,
что может приводить к авариям. Вспенивание воды в паровых
29
котлах вызывается примесями, находящимися в коллоидном
состоянии, илистыми органическими веществами, щелочами
и др.
Наиболее сильную коррозию вызывает вода, содержащая
свободные минеральные кислоты и растворенные газы
(кислород, хлор, двуокись углерода). В настоящее время борьбу с
коррозией ведут путем очистки воды; кроме того, для
изготовления котолов применяют специальные марки стали.
Жесткость воды для питания котлов высокого давления не
должна превышать 0,01 мг-экв/л, содержание в ней кислорода
должно быть не более 0,03 мг/л, кремневой кислоты не более
0,3 мг/л (в пересчете на SiCb), общее содержание солей
1—2 мг/л. Еще более чистая вода (содержание солей не более
0,2 мг/л) требуется в производстве полупроводниковых
материалов, люминофоров и ряда других химических
продуктов.
После использования воды в производственных процессах
и для хозяйственно-бытовых нужд образуется большое
количество сточных вод (хозяйственно-фекальные и промышленные
стоки), отводимых в канализацию. Часто промышленные
сточные воды после охлаждения, отстаивания или специальной
очистки снова возвращаются в производственный цикл. В этом
случае они называются оборотной водой.
Обычно коммунальные и промышленные предприятия, а
также другие крупные потребители воды — в сельском
хозяйстве, на транспорте и др. — имеют комплекс сооружений,
необходимых для водоснабжения (насосные станции, сети
трубопроводов, резервуары и водонапорные башни, очистные
сооружения и т. д.) и канализации стоков (прием сточных вод и
транспортировка их к очистным сооружениям, сброс очищенных
вод в водоем или возврат в производство).
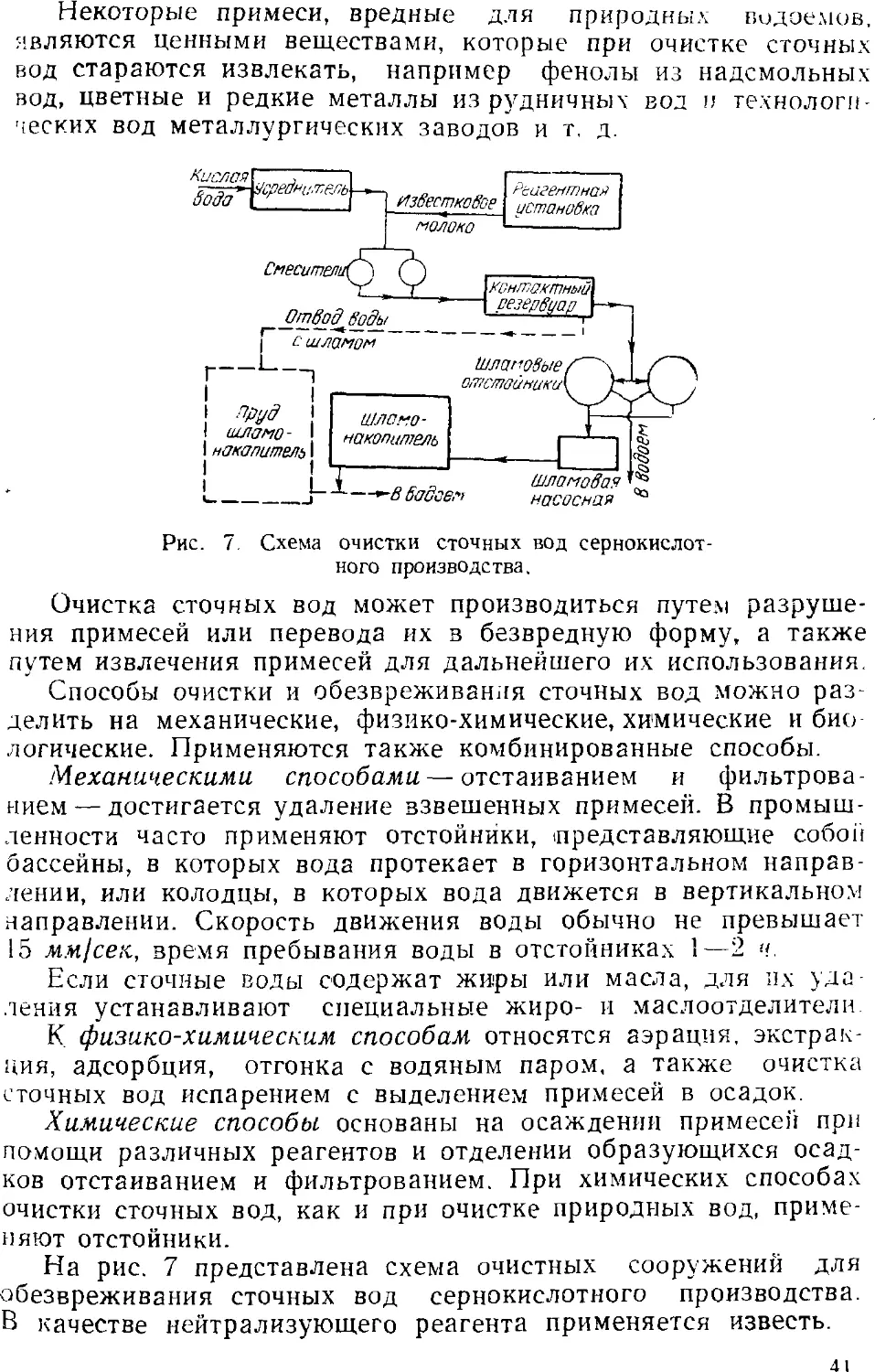
2. ВОДОПОДГОТОВКА
Под технологией воды в узком смысле понимают водоподго-
товку, т. е. очистку природной воды от примесей с целью
использования ее для питания котлов, в технологических
процессах, для питья, а также обезвреживание сточных вод. Водо-
подготовка чаще всего заключается в удалении механических
и нежелательных химических примесей, а также в
освобождении воды от микроорганизмов.
Подготовка воды включает следующие основные процессы:
1) осветление и обесцвечивание путем отстаивания и
фильтрования;
2) обеззараживание — уничтожение микроорганизмов
действием химических реагентов и физическими методами;
30
3) умягчение и обессоливание воды химическими методами,
с применением ионообменных фильтров и физическими
методами;
4) дегазация — удаление газов (двуокиси углерода,
сероводорода и др.).
ОСВЕТЛЕНИЕ И ОБЕСЦВЕЧИВАНИЕ ВОДЫ
Удаление механических примесей путем простого
отстаивания и фильтрования воды в настоящее время применяется
редко, гак как этот процесс протекает очень медленно и применим
только к слабоокрашенной жидкости, содержащей крупные
взвешенные частицы. Для осветления и обесцвечивания воды
чаше всего употребляют различные коагулянты: сульфат
Рис. 3. Cxe:va водопроводной станции с очистными сооружениями:
/—водоем; i—колодец; У, 12—насосы; 4—смеситель; 5—дозатор; 5—распределитель;
7—реакционная камера; 8—отстойник; S—напорный бак: /0—фильтр; //—сборник.
алюминия, сульфат и хлорид железа и др. Добавляемые
к воде коагулянты гидролизуются, образуя соответствующие
гидроокиси (алюминия, железа) в виде хлопьевидных частиц,
обладающих развитой активной поверхностью. Эти частицы
адсорбируют содержащиеся в воде коллоидные примеси и
увлекают их в осадок. После отстаивания небольшое количество
неосевших частиц удаляется фильтрованием.
Гидроокись алюминия образуется также при взаимодействии
¦сульфата алюминия с присутствующими в воде бикарбонатами
кальция и магния:
A12(SO4K + ЗСа(НСО3J = 3CaSO4-г 2А1(ОНK т 6СО,
На рис. 3 представлена одна из схем осветления и
обесцвечивания воды. Вода из водоема 1 поступает в приемный
колодец 2 и насосом 3 подается в смеситель 4, где обрабатывается
коагулянтом, который поступает из дозатора 5. Вода,
содержащая коагулянт, через распределитель 6 направляется далее
в реакционную камеру 7. В результате гидролиза в камере
образуются хлопьевидные частицы гидроокиси алюминия и же-
31
леза,. которые вместе с примесями оседают в отстойнике о. Из
отстойника вода через напорный бак 9 поступает на песчано-
гравийный фильтр 10 для окончательного осветления.
Осветленная вода яз сборника // насосом 12 подается
потребителем.
Для ускорения образования крупных хлопьев и разрушения
коллоидных структур применяются флокулянты— коллоидная
кремнекислота, высокомолекулярные органические вещества
(крахмал, полиакриламид, поливиниловый спирт и др.). Расход
высокомолекулярных флокулянтов при осзетлении воды не
превышает 1,5 мг/л.
ОБЕЗЗАРАЖИВАНИЕ ВОДЫ
Для полной дезинфекции профильтрованную воду чаще
всего хлорируют или озонируют. Иногда обеззараживание воды
производится действием ионов серебра, ультрафиолетовыми
лучами, ультразвуковыми волнами. Небольшие количества воды
обеззараживают кипячением (термический способ).
Хлорирование. Раньше для хлорирования воды применяли
почти исключительно хлорную (белильную) известь, теперь
широко применяют гипохлорит, хлорамин и жидкий хлор. При
введении в воду гипохлорита кальция протекает следующая
реакция: ^._
Са(ОС1J — СОа + Н.2О = СаСО3 -j- 2НОС1
Образующийся карбонат кальция выпадает в осадок, а
хлорноватистая кислота отщепляет кислород, окисляющий
органические примеси и убивающий микроорганизмы.
Расход хлорной извести на очистку питьевой воды
составляет около 4 мг/л. Избыток хлора удаляют прибавлением к
воде «антихлора» — тиосульфата или же сульфита натрия. При
применении жидкого хлора в качестве хлорирующего агента
потери его меньше, чем в случае применения белильной
извести, так как жидкий хлор удается точнее дозировать.
Озонирование. Обработка воды озоном применяется для
обеззараживания, обесцвечивания и устранения прлзкусов и
запахов.
Выделяющийся при разложении озона атомарный кислород
О3 'с—* О2 + О
являясь сильным окислителем, убивает микроорганизмы. Так,
например, бациллы тифа и холеры при озонировании гибнут
через 2 мин. Для обработки воды применяют воздух,
насыщенный озоном. Озон получают в специальных аппаратах —
озонаторах.
32
Для устранения привкусов и запахов воды применяют
также аэрирование — продувку воздухом. При этом из воды
удаляются железо и марганец, присутствующие в ней в виде
двухвалентных ионов. Они окисляются кислородом воздуха и
образуют гидроокиси Fe(OHK и Мп(ОНL, выпадающие в осадок.
УМЯГЧЕНИЕ И ОБЕССОЛИВАНИЕ ВОДЫ
Полное или частичное удаление солей кальция и магния
называют водоумягчением, а удаление или понижение
концентрации всех солей — обессоливанием или опреснением воды.
Химические способы умягчения воды основаны на том, что
под действием различных реактивов содержащиеся в воде
бикарбонаты и сульфаты превращаются в труднорастворимые
соли, выпадающие в осадок (шлам). В качестве реактивов
применяют известь, кальцинированную соду, едкий натр, тринат-
рийфосфат и др.
Вода, применяемая для питания котлов высокого давления, не должна
содержать даже незначительных количеств кремневой кислоты, которая
может образовывать плотную силикатную накипь. Для удаления
кремневой кислоты воду подкисляют серной или соляной кислотой до рН = 7,2—7.0
и подщелачивают алюминатом натрия. При этих условиях кремневая
кислота переходит в коллоидное состояние, затем коагулирует и выпадает з
осадок, который отделяют фильтрованием.
К физическим способам относятся термический способ
(кипячение), дистилляция и вымораживание. Термический способ
основан на уменьшении растворимости карбонатов
щелочноземельных металлов при повышении температуры, вследствие
чего эти соли выпадают в осадок. Кроме того, находящиеся з
зоде бикарбонаты кальция и магния при нагревании
разлагаются, образуя нерастворимые карбонаты. Для более полного
удаления солей надо длительно кипятить воду под давлением
Дистиллированную воду, не содержащую солей, получают
в испарительных установках. В районах с жарким климатом
для дистилляции воды может быть использована солнечная
энергия (гелиоопреснение). Для получения опресненной воды
методом вымораживания используют холодильные установки
Существуют также электрохимические способы, основанные
на использовании электродиализа и электроосмоса.
Применяются и комбинированные способы, например термический способ
комбинируют с известково-содовым.
В последнее время все более широкое распространение
приобретают ионообменные (ионитовые) способы умягчения и обес-
еоливания воды. Они основаны на способности некоторых
твердых малорастворимых веществ к обменным реакциям с
катионами или анионами, содержащимися в воде.
Наиболее распространенными способами умягчения воды
являются известково-содовый и катионитовый.
3-2848 33
Известково-содовый способ умягчения воды заключается в
обработке ее известью и содой. При применении извести и соды
содержащиеся в воде кальциевые соли превращаются в
нерастворимый карбонат кальция, магниевые соли — в гидроокись
магния. Известь удаляет из воды бикарбонаты и свободную
двуокись углерода, сода — остальные соли.
Реакции, протекающие при обработке воды известью, могут
быть выражены следующими уравнениями:
СО3 + Са(ОНJ = СаСО3 + Н2О
Са(НСО3J + Са(ОНJ = 2СаСО3 + 2Н2О
MgCO3 + Са(ОНJ = Mg(OHJ + CaCO3
MgSO4 + Са(ОНJ = Mg(OHJ + CaSO4
При удалении примесей из воды с помощью соды протекают
реакции:
CaSO4 -|- Na2CO3 = CaCO3 + Na2SO4
MgSO4 + Na2CO3 = MgCO3 + Na2SO4
MgCl2 + Na2CO3 = MgCO3 -)- 2NaCl
Частичное удаление солей магния достигается при
обработке воды известью, более полное — при применении соды.
Обычно для полноты очистки реактивы вводят с некоторым
избытком A—2%) по сравнению с теоретически необходимым
количеством. При нагревании воды до 60—70 °С расход извести
уменьшается, так как при этом происходит разложение части
"бикарбонатов. Очень важное значение имеет правильная
дозировка извести и соды, так как необходимо обеспечить возможно
бс~^ полное умягчение воды, не добавляя большой избыток
соды. В случае избытка соды при кипячении воды образуется
едкий натр, что вызывает коррозию стенок и труб котла и
арматуры. В последнее время в котлостроении получили применение
специальные щелочеупорные стали, стойкие и при избытке соды
более 2%.
Водоумягчительная установка, работающая по известково-
содовому способу, состоит из следующих основных аппаратов:
¦распределителей сырой воды, аппарата для приготовления
известкового молока (так называемый сатуратор), прибора для
дозировки реактивов, смесителя и отстойного бака.
На рис. 4 изображена одна из схем водоумягчительной
установки. Подаваемая на умягчение вода через систему
водораспределителей 1 и 2 поступает через воронку в сатуратор 10 и
на гидразличсское колесо 3 раздатчика реактивов. Часть воды
поступает непосредственно через смеситель 4 в отстойник 7.
Bhvtoii сатуратора 10 имеется коническая перегородка 12,
разделяющая его на верхнюю и нижнюю камеры. В верхней
части сатуратора помещен фильтр 9. Струя воды из воронки
34
рнсктивы
g
>o
поступает по полому валу, на который насажены мешалки //,
в нижнюю часть сатуратора. Здесь вода смешивается с
известковым молоком, поступающим в нижнюю часть верхней
камеры. Отработанное известковое молоко периодически выводится
из сатуратора через нижний
патрубок. При перемешивании
известкового молока с
очищаемой водой образуется
насыщенный раствор. Он
заполняет всю нижнюю камеру
сатуратора и нижнюю часть
верхней камеры, где
встречается со свежей порцией
известкового молока. Затем
насыщенный известковый раствор
поднимается вверх, проходит й
через фильтр 9 и по трубе
поступает в смеситель 4. 7
Хорошо растворимые в
воде реактивы (сода или едкий
натр и др.) поступают в
раздатчик, представляющий собой
небольшой сосуд, разделенный
перегородками на ряд
отделений. В одном отделении
вращается под действием
произвольно изменяемой струи воды
маленькое гидравлическое
колесо 3, в других отделениях
находятся реактивы. С колесом
связаны черпачки, которые
передают реактивы в смеситель.
Смеситель имеет ряд
наклонных перегородок,
расположение которых позволяет не- Рис
сколько раз резко изменять
направление
\
и ::/</-
'}Г;П1вП0Ш;ЫХ
Схема водоумягчительной
установки:
ЖИДКОСТИ И '¦ г—В0Д°РасгФеДелители; Л—гидравлическое
жидиис1и и колесо раздатчика реактивов; 4-смеснтель;
УДЛИНИТЬ ее ПУТЬ. Благодаря 5—желоб для умягченной воды; 6, 9—фильт-
,- ры; 7—отстойник; 8—коническая труба; 10—
ЭТОМУ ДОСТИГаеТСЯ ООЛее ПОЛ- сатуратор; //-мешалки; 12—коническая пе-
НОР ГМРШИВЯНИР ПеаКТИВОВ С Регородка; И—вентиль для периодического
ное смешивание реак1ивив v_ спуска отработанного известкового молока,
водой.
Поступающая из смесителя
вода проходит по конической трубе 8 и затем медленно
поднимается вверх в отстойник 7. Нерастворимые осадки удаляются
из нижней части отстойника. Через фильхр 6 умягченная вода
попадает в кольцевой желоб 5, из которого отводится в сборные
резервуары и далее подается потребителям.
35
Фосфатное водоумягчение ввиду сравнительно высокой
стоимости тринатрийфосфата применяется главным образом в
комбинированных схемах. В этих схемах основная масса солей из
воды удаляется известью, содой или едким натром, а доумяг-
чение осуществляется при помощи фосфатов. Это позволяет
получать хорошо очищенную воду, пригодную даже для питания
чотлов высокого давления.
Ионообменный способ умягчения и обессоливания воды.
С помощью ионообменивающих веществ может быть достигнуто
умягчение воды — удаление из нее катионов кальция и магния
(катионятовый способ очистки воды)—или полное ее обессо-
ливание (деионизация). Ионообменные способы приобретают
все большее распространение как при подготовке воды,
предназначенной для питания паровых котлов, так и при удалении
нежелательных ионов из воды, используемой в химической,
пищевой промышленности и др.
Ионообменивающие вещества — иониты делятся на катиони-
ты и аниониты. К катионитам относятся, например,
алюмосиликаты типа Na2O • А!2О3 • 2SiO2-nH2O (обменивающие ионы
натрия на ионы магния и кальция растворенных в воде солей)
и органические понпгы, содержащие кислотные группы.
Органические катпониты обменивают на катионы растворенных в
воде солей водород и потому называются Н-ионитами.
В качестве анионообменивающих веществ применяются
органические, преимущественно синтетические полимеры,
обменивающие ионы гидроксила на анионы солей, растворенных в
воде. Такие вещества носят название ОН-ионитов.
Процессы ионного обмена обратимы. Поглотивший катионы
1или анионы) ионит можно регенерировать, пропуская через
него раствор щелочи (или кислоты).
Процессы, протекающие при ионном обмене, можно
схематически представить следующим образом:
катионный обмен
[Кат.] Н + MaCi 7 » [Кат.] Na + HCI
2[Кат.] Н + Са(НСОз),. 7=2 [Кат.],Са + 2СО, + Н,0
или
2[Кат.] Na + М§(НСО3)-Л 7—* [KaT.].2Mg -f 2NaHCOg
2[Кат.] Na + CaSO4 - » [Кат.].Са + Na.SO,
анионный обмен
[Ал.] O1-I -|- HCI » [Дн.] С1 г Н.,0
2[Ан.]0Н-|- H,SO4 zmiAH.joSOt , 2H..O
Последовательным пропусканием воды через хатионптозып
и аннонитозый фильтры достигается ее полное обессолявание.
36
Аппарат для катионитовой очистки (умягчения) воды изо-
')ражен на рис. 5.
Из неорганических катионитов применяют природные
алюмосиликаты (глауконит, волконскоит и др.) или искусственно
лркготовленные (пермутит и др.). В качестве органических ка-
гнонитов применяют некоторые сорта угля, содержащего
кислотные остатки, сульфированный уголь и главным образом син-
гетические полимеры (сополимеры стирола и дивинилбензола,
Рис. 5. Катионитовый фильтр.
феноло-формальдегидные полимеры и др.), в которых имеются
еульфогруппы или карбоксильные группы. Для регенерации ка-
тионит промывают обычно раствором поваренной соли или
кислоты.
При выборе катионита учитывают его полную и рабочую
обменную способность.
Под полной обменной способностью при умягчении воды
понимают количество ионов кальция и магния (в г-экв),
которое может быть поглотано единицей объема катлонита (в см5
1-лп as3) до прекращения процесса ионообмена, в данном случае
ло исчерпания возможности дальнейшего умягчения воды.
Рабочая обменная способность выражается количеством ионов
хальпия I! магния (в г-экв), поглощаемом единицей объема ка-
тиок;:та (в см3 пли мг) до момента появления эг,!х понов в
жиг.мостм, прошедшей через катионитовый фильтр. Обычно
количество поглощенных ионов колеблется от 0,3 ;ю 3% от песа
катионита.
37
Основными факторами, влияющими на обменную
способность, являются скорость фильтрования воды, степень ее
жесткости, величина зерен кагионита и температура воды. Обменная
способность уменьшается при увеличении размеров зерен ка-
тионита, понижении температуры и жесткости воды.
Преимущества катионитового способа по сравнению с из-
вестково-содовым: 1) компактность и простота аппаратуры;
2) отсутствие необходимости подогрева воды; 3) более
глубокое умягчение воды (до 0,035—0,07 мг-экв/л); 4) простота
обслуживания и контроля; 5) дешевизна.
-со,
2 , . 3
вода
S
Обессоленная
Рис. 6. Схема установки для обессоливания воды иони-
тами:
/—бак с раствором кислоты; 2—Н-катионитовый фильтр; Л—аннонн-
товый фильтр; 4—бак с раствором щелочи; 5—дегазатор; 6—насос;
/"—сборник обессоленной воды.
Преимущества кагионитового метода перед другими
методами очистки настолько значительны, что он приобретает все
более широкое распространение.
На рис. 6 представлена схема устанозки для обессоливания
воды ионообменным способом. Очищаемая вода поступает на
Н-катионитовый фильтр 2, где происходит удаление из воды
катионов — ионов кальция, магния и натрия. Затем вода
поступает в аниомитозый фильтр 3, в котором из нее удаляются
анионы кисло г. Далее вода освобождается от двуокиси углерода в
дегазаторе 5 и поступает в сборник 7, откуда подается
потребителю. Для регенерации катионита применяется разбавленный
раствор серной или соляной кислоты, поступающий в фильтр 2
из бака /, для регенерации анионита — раствор щелочи,
подаваемый в фильтр 3 из бака 4.
Иониты с высокой обменной способностью нашли
применение в электрохимических процессах обессоливания воды в
качестве материала для изготовления диафрагм электролизеров.
Подбирая соответствующие иониты, можно изготовить
диафрагмы, пропускающие только анионы или только катионы. Кроме
того, размеры пор ионитовых диафрагм соизмеримы с
размерами ионов, что позволяет устранить диффузионные процессы в
члектролизерах и получить воду высокой чистоты.
УДАЛЕНИЕ ГАЗОВ ИЗ ВОДЫ
Большое значение имеет удаление двуокиси углерода и
кислорода из воды, применяемой для питания паровых котлов, так
как эти газы разрушающе действуют на котельную сталь.
Газы удаляют из воды химическими и физическими
способами. В первом случае применяют гашеную известь и железо,
которые образуют с СО2 и Ог нерастворимые соединения:
Са(ОНJ + СО2 = СаСО3 + Н2О
4Fe + ЗО2 = 2Fe2O3
Обычно для удаления двуокиси углерода к воде добавляют
известковое молоко, а для удаления кислорода химическими
способами применяют фильтры, заполненные железными
опилками, стружками или порошкообразным железом.
Физические способы удаления газов основаны на выделении
из воды значительной части растворенных в ней газов при
аэрации или нагревании воды в вакууме.
ПРЕДОТВРАЩЕНИЕ НАКИПИ
Чтобы предотвратить образование плотных осадков на
стенках котла, используют так называемые антинакшшны,
добавление которых к воде способствует образованию осадков в виде
рыхлого шлама, сравнительно легко удаляемого из котла
промывкой. Антинакипины можно разделить на три группы: 1)
минеральные, 2) органические и 3) смешанные. К минеральным
антинакипинам относятся различные смеси соды, едкого натра,
хлористого бария, фосфатов натрия и др., к органическим
антинакипинам— графит, смесь глицерина и щелочи, гуминовые
вещества и др. Смешанными антинакипинами являются смеси
соды или других минеральных солей с органическими веществами.
К антинакипинам относят также различные обмазки,
состоящие из масла, дегтя, асфальта, канифоли и графитовой муки,
которые не предотвращают образования накипи, но облегчают
удаление ее со стенок котла.
Применение антинакипинов является менее эффективным
способом борьбы с накипью, чем умягчение воды, вследствие
коррозионного действия составных частей антинакипинов на
котельную сталь и сильного вспенивания воды.
Известен электролитический способ предотвращения
образования накипи. Через воду пропускают постоянный ток
низкого напряжения, при этом образуются пузырьки водорода,
которые покрывают стенки котла и предотвращают образование
накипи.
39
В настоящее время разрабатываются новые методы
подготовки воды для питания котлов, основанные на применении
аппаратов и приборов, в которых образуются магнитные,
ультразвуковые и электростатические поля.
3. ОБЕЗВРЕЖИВАНИЕ СТОЧНЫХ ВОД
Сточные воды, образующиеся в ходе производственных
процессов, а также бытовые сточные воды загрязнены различными
зешествами растительного, животного и минерального
происхождения. В санитарном отношении наиболее вредны
органические загрязнения, так как они являются питательной средой
для различных бактерий и грибков. Вследствие гниения и
брожения органических примесей вода приобретает зловонный
запах и становится совершенно непригодной к спуску в водоемы.
Особенно богаты органическими примесями бытовые сточные
воды, воды пищевых предприятий, текстильных фабрик,
кожевенных заводов и др.
Вредные неорганические примеси содержатся в сточных
водах промышленных предприятий. Эти воды часто содержат
значительное количество растворенных солей, свободных кислот,
сероводорода, соединений мышьяка, фенолов и др. Без
предварительной очистки такие воды нельзя спускать в реки и другие
водоемы, так как ядовитые примеси сточных вод могут
уничтожить животный мир водоемов и прибрежную растительность.
Действующее в СССР законодательство предусматривает
строгую санитарную охрану естественных водоемов. Спуск
промышленных сточных вод в водоемы должен производиться в
соответствии с санитарными правилами.
Ниже приводятся следующие предельно допустимые
концентрации некоторых вредных веществ в воде водоемов (в мг/л)
после смешения ее со сточными водами:
Барий
Бензин .....
Бензол
Дихлорэтан . . .
Железо
Кадмий
Капролактзу. . . .
Керосин
Кобальт
Медь
Мышьяк . . .
Нефть
. . . 4,0
. . . 0,1
0 5
. . . 2,0
. . . 0,5
. . . 0,01
. . 1,0
. . . 0,3
. . . 1,0
. . . 0,1
. . . 0,05
. . . 0,3
Никель . . . .
Ртуть
Свинец ....
Селен
Сульфиды . . .
Фенолы ....
Формальдегид .
Фтор
Хлор
Хром
Цианиды . . .
Цинк
0,1
0,005
0,1
0,01
Отсутствие
0,001
0,5
1,5
Отсутствие
0,1—0,5
0,1
1,0
В сточных водах могут содержаться газообразные, жидкие
11 твердые примеси во взвешенном, коллоидном или
растворенном состояния.
Некоторые примеси, вредные для природных водоемов,
являются ценными веществами, которые при очистке сточных
вод стараются извлекать, например фенолы из надсмольных
вод, цветные и редкие металлы из рудничных вод и
технологических вод металлургических заводов и т, д.
известковое
пололо
установка
Смеет
?твоеводы
I с шламом
резервуар
Г ' 1
I I
I Пруд I
I 1Ш1ОМ0- I
I накопитель I
I I
! г" х—-Вбадоеп
Шлаковые
о/ястойнцки
Шлапо-
накопитель
Шламовая f*5
насосная
Рис. 7. Схема очистки сточных вод
сернокислотного производства.
Очистка сточных вод может производиться путем
разрушения примесей или перевода их з безвредную форму, а также
путем извлечения примесей для дальнейшего их использования.
Способы очистки и обезвреживания сточных вод можно
разделить на механические, физико-химические, химические и
биологические. Применяются также комбинированные способы.
Механическими способами — отстаиванием и
фильтрованием— достигается удаление взвешенных примесей. В
промышленности часто применяют отстойники, представляющие собой
бассейны, в которых вода протекает в горизонтальном
направлении, или колодцы, в которых вода движется в вертикальном
направлении. Скорость движения воды обычно не превышает
15 мм/сек., время пребывания воды в отстойниках 1—2 ч.
Если сточные воды содержат жиры или масла, для их уда
ления устанавливают специальные жиро- и маслоотделители
К физико-химическим способам относятся аэрация,
экстракция, адсорбция, отгонка с водяным паром, а также очистка
сточных вод испарением с выделением примесей в осадок.
Химические способы основаны на осаждении примесей при
помощи различных реагентов и отделении образующихся
осадков отстаиванием и фильтрованием. При химических способах
очистки сточных вод, как и при очистке природных вод,
применяют отстойники.
На рис. 7 представлена схема очистных сооружений для
обезвреживания сточных вод сернокислотного производства.
В качестве нейтрализующего реагента применяется известь.
41
В связи с развитием атомной энергетики и расширением
областей применения радиоактивных изотопов в народном
хозяйстве возникает необходимость обезвреживания
радиоактивных сточных вод. Для этого требуется сооружение сложных
очистных устройств и применение специфических методов
очистки.
Биологические способы применяют для очистки главным
образом фекальных вод, т. е. сточных вод населенных пунктов.
Биологические способы основаны на разрушении
органических загрязнений в результате жизнедеятельности
микроорганизмов. Так, например, некоторые бактерии перерабатывают
выделяющийся при разложении органических веществ
сероводород в серную кислоту, а образующиеся аммиак и
органические соединения азота — в азотистую и далее в азотную
кислоту.
Кислоты образуют безвредные минеральные соли, и
сточная вода может быть сброшена в водоемы. Таким образом,
целью биологических методов очистки сточных вод является
создание благоприятных условий для размножения полезных
в данном случае бактерий. Эти условия могут быть созданы
при доступе или без доступа кислорода воздуха. В первом
случае будут развиваться так называемые аэробные бактерии и
в процессе окисления органические вещества будут
превращаться в минеральные. Во втором случае развиваются анаэробные
бактерии, которые в процессе гниения будут разрушать
органические вещества с образованием аммиака п газообразных
углеводородов.
В настоящее время биологические способы начинают
применять для очистки от фенолов сточных вод коксохимических
заводов и других предприятий.
Биологическая очистка сточных вод осуществляется на
полях орошения и фильтрации или на биологических станциях.
Поля орошения представляют собой удаленные от
населенных пунктов площади земли с водопроницаемой почвой,
которые заливают сточными водами. Примеси, содержащиеся в
сточных водах, задерживаются почвой и служат для нее
удобрением. На этих полях обычно разводят овощные культуры.
Поля фильтрации представляют собой открытые водоемы
с водопроницаемым грунтом. Прошедшая через грунт вода
освобождается от загрязнений и удаляется через дренажные трубы
в отводные каналы.
Поля орошения и фильтрации должны быть открыты для
доступа воздуха и света. Они должны быть расположены
вблизи естественных водоемов для спуска очищенных сточных вод
и ограждены от проникания в них весенних талых вод.
Биологическая станция представляет собой довольно
сложное сооружение, состоящее из устройства (решетки) для улав-
42
ливания крупных взвешенных частиц, отстойника,
приспособлений для накапливания и подсушки осадков, окислителя или
фильтра, вторичного отстойника или крупнопесочного фильтра
н устройства для дезинфекции очищенных вод.
После очистки сточное воды дезинфицируют путем
хлорирования, озонирования или ультрафиолетового облучения.
Последние два способа применяются редко.
Для удаления двуокиси углерода и других газов, а также
для частичного окисления органических веществ через воду
пропускают воздух или же разбрызгивают воду при помощи
сопла.
ЛИТЕРАТУРА
А. М. К о и ю ш к о в, С, В, Яковлев, Водоснабжение п к.чналпзлшя. Г".'-
стройиздат, 1960.
Журн. ВХО им. Д. И. Менделеева, № 6 A960); № 2 A961).
А. А. Кастальский, Д. М. Минц, Подготовка воды для питьевого ч
промышленного водоснабжения, Изд. «Высшая школам, 19G2.
М. С. Шкроб, Водоподготовка, Госэиергоиздат, 1950.
11, Л. Гордон, Водоподготовка в тепловом хозяйстве и промышленности.
Энергоиздат, 1940.
А. И. Жуков и др., Проектирование сооружений для очистки промыи.
ленных сточных вод, Стройиздат, 1949.
Л. Л. К у л ь с к и й, Химия и технология обработки йоды, Пзд,
Министерства коммунального хозяйства РСФСР, 1954,
ч. А. Резников, А. Е. М у л и к о в с к а я, И. Ю. Соколов. Методы aiij-
лиза природных вод, Госгеолиздат, 1963.
А. Ф. Ш а б а л и н, Очистка сточных вод предприятий черной металлурги!!.
Металлургиздат, 1953.
Ю Ю, Лурье, А. И. Рыбникова, Химический анализ производственны.
сточных вод, Госхимиздат, 1963,
Глава [II
СЕРА
/. ОБЩИЕ СВЕДЕНИЯ
Применение серы. Сера приобрела особое значение в XII в.,
когда ее начали применять в качестве составной части черного
пороха. С конца XVIII в. серу начали использовать для
производства серной кислоты.
В настоящее время основным потребителем элементарной
серы является производство серной кислоты. Значительные
количества серы используют в резиновой и целлюлозно-бумажной
промышленности, в промышленности органического синтеза
(получение сернистых красителей, сероуглерода и др.), в
химико-фармацевтической промышленности, в производстве спичек
и др. В сельском хозяйстве сера применяется в качестве
средства для борьбы с вредителями растений (виноградника,
хлопчатника и др.).
Общее потребление серосодержащего сырья в
капиталистических странах в 1959 г. достигло 17,9 адлн. г S, из них
5,95 млн. т было израсходовано в США.
Широкое использование элементарной серы в химической
и других отраслях промышленности вызвало исключительно
быстрый рост ее мирового производства в последние
десятилетия. В 1962 т. з капиталистических странах было
произведено более 10 млн. г элементарной серы, в том числе свыше
6 млн. т в США.
Свойства серы. Средняя атомная масса серы 32,064.
Элементарная сера состоит из смеси стойких изотопов: S32 (95,018%).
S33 @,750%), S34 D,215%), S36 @,017%). При обычной
температуре сера представляет собой твердое вещество желтого
цвета. С понижением температуры цвет серы постепенно светлеет
и при температуре ниже —50 °С она обесцвечивается.
Сера кристаллизуется в двух модификациях: ромбической
и моноклинной. Ромбическая сера (а-модификация) устойчива
при температурах ниже 95,6°С. Моноклинная сера (р-модифи-
кация) устойчива в интервале 95,6—119,3°С и кристаллизуется
в форме длинных игл. По цвету и прозрачности моноклинная
сера сходна с ромбической. Температура плавления ромбиче-
44
ской серы (при быстром нагревании) 112,8 С, моноклинной
серы 119,3°С. Температура кипения серы 444,6СС (под
атмосферным давлением).
При повышении температуры (выше точки плавления)
вязкость жидкой серы сначала понижается, но при 160 °С
начинается загустевание, а при 250 °С жидкая сера вновь
приобретает текучесть. В связи с этим серу выплавляют из руд
при температуре около 150°С.
Практически чистая сера обладает весьма малой
электропроводностью и является превосходным изолятором.
2. ПОЛУЧЕНИЕ СЕРЫ
Элементарную серу получают несколькими способами: из
самородных серных и сульфидных руд, а также из природных
и промышленных газов, содержащих сероводород и другие
соединения серы (отходящие газы цветной металлургии, газы
нефтепереработки, коксовый газ, генераторные газы).
ПОЛУЧЕНИЕ СЕРЫ ИЗ САМОРОДНЫХ РУД
Самородные серные руды. Месторождения серных руд
образуют продукты осадочного или вулканического происхождения,
имеются также залежи серной руды в шляпах соляных
куполов. Наиболее распространены относительно небогатые серой
руды осадочного типа, содержащие 15—30% серы. К
осадочному типу относятся, например, месторождения серы в Италии
(о. Сицилия и др.). Крупные месторождения серы
вулканического типа находятся в Южной Америке, Японии,
месторождения меньшего значения — в Турции, Объединенной Арабской
Республике, Эфиопии, Греции. Залежи серных руд в шляпах
соляных куполов на глубине 150—500 м имеются в США
(штаты Луизиана и Техас) и в Мексике; содержание серы в рудах
этих месторождений составляет от 27 до 70%.
В зависимости от условий залегания серных руд добыча их
производится открытым способом (в карьерах) или в
неглубоких шахтах. Особенности условий залегания серных руд в США
позволяют производить подземную выплавку серы.
Запасы самородной серы в дореволюционной России
определялись в несколько тысяч тонн, 80% из них приходилось на
долю каракумских месторождений. Однако даже эти запасы
не использовались, и вся потребность страны в сере
удовлетворялась лишь за счет импорта.
За годы Советской власти в СССР открыты месторождения
серы в Средней Азии, Поволжье, в Крыму, на Урале, на Алтае.
В середине 30-х годов были построены и введены в
эксплуатацию серные рудники, обогатительные фабрики, сероплавильные
45
заводы. В последнее десятилетие в разных районах Советского
Союза выявлены новые крупные запасы самородной серы.
Значительные месторождения серы сосредоточены на Западной
Украине (Предкарпатский сероносный район), в Средней Азии,
Поволжье.
Крупные месторождения самородной серы на Западной
Украине (Язовское, Роздольское и др.) относятся к осадочному
типу и представляют собой пластовые залежи известняковых
или глинистых серных руд.
Пласт залегает неглубоко, что позволяет осуществить его
разработку открытым способом.
Содержание элементарной серы в серных рудах колеблется
в широких пределах примерно от 16 до 30%.
По запасам самородной серы, в несколько раз
превышающим запасы сульфидной серы, Советский Союз занимает одно
из первых мест в мире.
Извлечение серы из руд. Выбор метода переработки
самородных серных руд определяется прежде всего содержанием в
них серы, а также составом и свойствами пустой породы. Руды
с относительно высоким содержанием серы (свыше 25%) можно
непосредственно перерабатывать без предварительной
подготовки. Более бедные руды предварительно обогащают путем
флотации. По методу флотации, разработанному ГИГХС*, при
обогащении получается тонкодисперсный флотационный
концентрат, содержащий до 75% серы.
Сущность этого метода, применяемого для получения серы
из руды Роздольского месторождения., заключается в
следующем.
Серная руда подается из карьеров в приемные бункеры,
откуда поступает на грубое дробление в щековые дробилки.
Затем пластинчатые питатели подают руду на колосниковые
грохоты, и далее она поступает на предварительное
измельчение в молотковые дробилки. Отсюда ленточные конвейеры
направляют руду в стержневые мельницы, затем она подается: на
окончательное измельчение в шаровые мельницы, работающие
в замкнутом цикле с классификаторами.
По достижении требуемой тонины помола серная руда
поступает на флотацию. В качестве флотационных реагентов
применяются жидкое стекло, керосин и спирты С7—Сд.
Флотационный серный концентрат, содержащий 75% серы, сгущается до
соотношения твердой и жидкой фаз (Т:Ж), равного 1:1. Из
сгущенного серного концентрата в автоклавах выплавляют серу.
Для выплавки серы применяются автоклавы емкостью 50 тыс. л.
В автоклавы загружают концентрат, добавляют небольшие ко-
* Государственный научно-исследовательский ингтитут горнохнмиче-
ского сырья.
46
личества реагентов (керосин, сода, триполифосфат натрия
NasPsOlo) и подают острый пар.
Из автоклавов расплавленная сера передается по трубам
на бетонированную площадку с бортами, где разливается в
формы (штабели). Застывшие охлажденные блоки серы грузят
в железнодорожные вагоны и отправляют потребителям.
Готовый продукт содержит примерно 99,5% серы.
В процессе плавки в автоклавах сера неполностью
извлекается из руды. После слива расплавленной серы в автоклане
остаются так называемые хвосты, в которых содержится
35—40% серы. Эти остатки выгружают из автоклавов, сгущают
(см. выше) и подвергают дополнительной флотации,
проводимой в присутствии флотореагентов (сода, жидкое стекло,
сосновое масло).
В результате флотации хвостов получается концентрат,
содержащий 70—75% серы. Его смешивают с концентратом,
полученным при флотации исходной
серной руды, и подают смесь в
автоклавы для выплавки серы.
Описанным способом удается
извлечь 85% серы от общего
содержания ее в руде.
Для извлечения серы из руды,
кроме флотационного метода, применяет-
ся нагревание руды водяным паром
под давлением или перегретой водой;
можно также экстрагировать серу
растворителями (сероуглерод,
дихлорэтан и т. д.).
Выплавка серы из измельченной
суды при помощи пара и перегретой
воды проводится в автоклавах.
Автоклав периодического действия (рис. 8)
представляет собой вертикальный
стальной цилиндр, рассчитанный на ^~
работу под избыточным давлением до Рис & Автоклав для зы^
5 ат. В автоклав загружают измель- ' плавки серы,
ченную серную руду, затем подают
воду в количестве примерно 15% от веса руды, после чего снизу
вводят острый пар, нагревающий содержимое автоклава до
150°С.
Процесс плавления в автоклаве длится несколько часов.
Выплавленная жидкая сера скапливается в нижней части
аппарата, оттуда ее выгружают под давлением через нижний
патрубок и по желобам разливают в ящики-вагонетки, а затем в
формы. Пар выводят из автоклава через верхний патрубок, снижая
давление в аппарате до атмосферного. Затем из аппарата сли-
47
вают воду н выгружают отработанную породу. При выплавке
извлекается до 55% серы, содержащейся в руде.
Применяемый в США метод подземной выплазкп- серы из
богатых руд, залегающих на значительной глубине (метод Фра
ша), заключается в расплавлении серы перегретой водой и
выдавливании ее на поверхность земли сжатым воздухом
(давление примерно 25 ат). Недостатком этого метода является
невысокая степень извлечения серы из руды B5—30%. в отдельных
случаях до 50%)-
Комплексная переработка серных руд. В настоящее время
разрабатывается комплексный метод переработки
известняковых серных руд.
Технологический процесс переработки состоит из следующих
операций:
1) дробление руды на зерна размерами до 5 мм;
2) сушка руды до остаточной влажности 0,2% (средняя
влажность исходной руды около 3%);
3) возгонка серы из руды в трубчатых ретортах
непрерывного действия с наружным обогревом;
4) очистка паров серы от механических примесей (пыль) и
конденсация паров серы;
5) разливка жидкой серы в металлические формы;
6) переработка клинкера ка цемент (стр. 637).
Схема установки изображена на рис 9. Серная руда,
измельченная в дробилках 2 и 3, элеватором 4 подается в
бункер 5, откуда поступает в шнеки 6, обогреваемые паром.
Отсюда расплавленная руда шнеком-питателем 7 подается в
непрерывно действующую вращающуюся трубчатую реторту 8,
помещенную в обмуровку и обогреваемую горячими топочнымч
газами.
Внутри реторты поддерживается температура около 500°С.
при которой сера испаряется,
На участке между ретортой и конденсационно-охладитель-
ной аппаратурой должна сохраняться постоянная температура
(—'400°С), поэтому вся система для очистки паров серы от
механических примесей помещена в общий с ретортой печной
блок*. Выходящие из реторты пары серы последовательно
поступают в пылеуловители 9, электрофильтр 10 и
котел-конденсатор 11. При конденсации паров выделяется значительное
количество тепла, затраченного ранее на нагревание и возгонку
серы.
Образовавшаяся жидкая сера подается на
барабан-кристаллизатор 12, где превращается в чешуйчатую серу, или ми-
* Обычные сталь и чугун непригодны для изготогления аппаратуры,
работающей в среде серы. Наиболее стойкой в этих условиях является
специальная сталь марки Х25Ю5.
48
нуя барабан 12, поступает на разливку в металлические формы.
Отходящие газы, пройдя холодильники 13, вентилятором 14
отводятся в атмосферу.
Нагретый остаток (пустая порода) ссыпается из
пылеуловителя 9, образуя затвор, препятствующий прониканию паров
серы в атмосферу и одновременно наружного воздуха в
реторту.
Серная руда
п
6
Рис. 9. Схема установки для комплексной переработки серных руд:
1, 5, 16, 17, 18, 23—буккеры; 2,'3—дробилки; 4, 15, 24—элеваторы; 6—шнекн; 7—
шнек-питатель; 8— реторта; 9—пылеуловители; 10—электрофильтр;
//—котел-конденсатор; 12—барабанный кристаллизатор; 13, 27—холодильники; 14—вентилятор:
19—глиномялка; 20—весы; 21—шаровая мельница; 22— силос;
25—циклоны-подогреватели; 26—печь; 28—транспортер.
Элеватором 15 сыпучий остаток подается в бункер 16,
откуда через промежуточный бункер 17 направляется на
переработку в цемент по сухому или мокрому способу. Необходимая
для этого процесса глина подается в бункер 18, откуда через
глиномялку 19 и дозировочные весы 20 поступает вместе с
сыпучим остатком в шаровую мельницу 21. Измельченная смесь
4—2848
49
обоих компонентов через корректирующий силос 22 подается
в бункер 23, откуда элеватором 24 через серию
циклонов-подогревателей 25 вводится во вращающуюся цементную печь 26,
в которой поддерживается температура около 1450 °С.
Выходящий из печи клинкер охлаждается в холодильнике 27 и
транспортером 28 подается на дальнейшую обработку по
обычной схеме (стр. 637 ел).
ПОЛУЧЕНИЕ СЕРЫ ИЗ СУЛЬФИДНЫХ РУД
Источником получения серы могут служить сульфидные
руды, содержащие пирит.
Из способов, предложенных для получения элементарной
серы непосредственно из пирита, промышленное применение
получил только способ восстановительной плавки медного
колчедана.
Процесс возгонки серы из пирита не нашел распространения
в промышленности вследствие несовершенства и
нерентабельности.
При восстановительной плавке медного колчедана в ватер-
жакетных печах одновременно получают медный штейн и серу.
Схема получения серы из колчедана по этому методу
изображена на рис. 10. В печь / загружают шихту, состоящую из
медного колчедана, кокса, кварца и известняка. Воздух подают
в печь вентилятором через расположенные внизу фурмы с та-
ким расчетом, чтобы в отходящих газах отсутствовал кислород.
Газ, выходящий из печи при температуре 420—450 °С, содержит
пары элементарной серы, двуокись серы, сероуглерод, сероокись
углерода и сероводород. Образующийся в печи / бедный штейн
перерабатывают в печи 2 на богатый штейн, содержащий
45—50% меди.
Газы, образующиеся при плавке бедного штейна,
присоединяют к газам первой плавки и направляют на очистку от пыли
сначала в камеру 3, а затем в электрофильтр 4.
Освобожденные от пыли газы поступают в первый контактный аппарат 5,
заполненный катализатором, (боксит или особым образом
приготовленная гидроокись алюминия). В этом аппарате при 450СС
в присутствии избытка SO2 происходит разрушение побочных
продуктов (COS и CS2) и выделение элементарной серы по
реакциям
4COS + 2SOa с 6S -f 4CO.;
2CS3 + 2SO, ¦?=! 6S + 2СО,
В указанных условиях контактирования равновесие в обеих
системах смещено вправо, и реакции протекают с высокими
выходами.
50
Известковое
молоко'
Bosdf/x
U/ахтш
I
Бедный ^^~\
штейн ' Богатый
штейн
Сера
Рис, 10. Схема получения серы при восстановительной плавке медного штейна:
/, 2—печи; Я— пыльная камера: 4, 8—электрофильтры; Л, 6—контактные аппараты; 7—паровом1 коте.т, й—вентилятор; М—охллдмто-п.'.шс башни;
//—абсорбционные башни,
Сероводород, содержащийся в газе, при этих условиях не
реагирует с двуокисью серы.
Из первого контактного аппарата 5 газы, содержащие серу,
незначительные количества двуокиси серы и сероводород,
поступают во второй контактный аппарат 6. Здесь при более ннз-
кой температуре (около 250°С) протекает с высоким выходом
реакция:
2H3S + SOjj = 2Н2О + 3S
Элементарная сера выделяется из газов и отводится
отдельно из каждого контактного аппарата. Из второго контактного
аппарата 6 газы, содержащие небольшое количество серы и
незначительные примеси SO2 и H2S, направляют в паровой
котел 7. При охлаждении газов с 250 до 130°С в котле происходит
конденсация оставшейся в газах серы, и за счет тепла газов
образуется пар. Не сконденсировавшаяся в котле сера
осаждается в электрофильтре 8. Из электрофильтра газ поступает в
охладительные башни 10, а затем в башни //, орошаемые
известковым молоком для связывания остатков SO2 и H2S, после
чего отводится в атмосферу.
Просасывание газов через систему производится при помощи
вентилятора 9.
По этому способу наряду с богатым медным штейном
удается выделить из 1 т сульфидной руды около 340 кг серы. Потерн
серы (в виде различных соединений) с выхлопными газами
составляют около 5%.
ИЗВЛЕЧЕНИЕ СЕРЫ ИЗ ГАЗОВ
Промышленные газы (генераторный, коксовый, газы
нефтепереработки, попутные нефтяные газы и др.), а также
природный газ, добываемый в настоящее время в огромных
количествах, содержат серу главным образом в виде сероводорода,
который является нежелательной примесью этих газов. Поэтому
все газы перед их использованием в промышленности
подвергаются обязательной специальной очистке от серы.
Коксовый газ, образующийся наряду с коксом и смолой при
коксовании каменных углей, содержит в среднем 19—20 г/м'1
сероводорода. Кроме сероводорода, в коксовом газе содержатся
также органические соединения серы—сероокись углерода,
меркаптаны, тиофены и др. В зависимости от способа очистки
коксового газа можно получить элементарную серу или
сероводород.
Другим важным источником получения сероводорода
являются газы нефтепереработки. Они используются главным
образом для синтеза органических продуктов и должны быть
полностью очищены от H2S.
52
Значительные количества серы в виде сероводорода
содержатся также в газогенераторных газах, получаемых
газификацией угля при 750—1000 °С в присутствии воздуха, кислорода
или смеси их с водяным паром. Сероводород содержится также
в попутных нефтяных и природных газах.
Коксохимические и нефтеперерабатывающие заводы
являются не только поставщиками серосодержащего сырья в виде
сероводородного газа, но и потребителями серной кислоты,
необходимой, например, для извлечения аммиака из коксового газа.
Поэтому использование извлекаемого из газов сероводорода
для переработки его на месте в серную кислоту позволит
освободить транспорт от встречных перевозок серной кислоты
и обеспечить указанные заводы собственной серной
кислотой.
Из отходящего сероводородного газа может быть получена
элементарная сера или серная кислота методом мокрого ката
лиза (стр. 124).
Многочисленные методы очистки горючих газов от
сероводорода можно разделить на две группы: сухие методы,
основанные на применении различных твердых поглотителей, и мокрые
методы, в которых используются жидкие поглотители
(растворы). Применяемые в промышленности различные варианты
обоих методов различаются по составу поглотителя, способам
регенерации, конструкциям используемой аппаратуры.
Сухая очистка газов производится болотной рудой,
активированным углем, гашеной известью.
Очистка газов болотной рудой. На некоторых заводах еще
до сих пор применяется очистка газов от сероводорода
болотной рудой*, смешанной для разрыхления с древесными
опилками. При поглощении сероводорода протекают следующие
реакции:
3H2S + Fe2(OHN = Fe2S3 — 6Н,0 ~', 14,Щккал
3H2S + Fe2(OHN = 2FeS -J- S -К,6Н2О
Одновременно протекают побочные реакции (при иных
соотношениях реагирующих компонентов) и реакции связывания
других вредных примесей, присутствующих в горючих газах.
В процессе очистки газа от сероводорода газоочистительная
масса постепенно обогащается серой и поглотительная
способность массы уменьшается. Для регенерации ее поливают водой
и разрыхляют. При этом сернистое железо окисляется
кислородом воздуха и выделяется элементарная сера:
2Fe.,S3 + ЗО2 + 6Н2О = 2Fe2(OHN -f 6S 4- 289,98 икал
Осадочная порода, содержащая гидроокись железа Fe2(OHN.
53
Очистка газов активированным углем является более
совершенным способом, который заключается в следующем. Газ,
содержащий, например, 5—25 г/м3 сероводорода, смешивают с
3—4% воздуха и пропускают через фильтр, заполненный
активированным углем. Уголь загружают в фильтр слоями: нижний
слой — крупные зерна размером около 10 мм, средний — зерна
размером 2—4 мм, верхний — зерна размером 1—2 мм.
Сероводород окисляется на фильтре по реакции
2H2S + О2 = 2S + 2Н2О -fill ккал
Реакция ускоряется в присутствии аммиака, который
вводят в газ з количестве до 0,3 г/м3. Оптимальная температура
процесса около 40°С.
Роль активированного угля двояка: с одной стороны, он
является катализатором процесса окисления H2S, с другой —
адсорбентом серы, образующейся в процессе окисления.
Когда активированный уголь в фильтре насытится серой, гал
направляют во второй фильтр. В это время уголь в первом
фильтре обрабатывают раствором сернистого аммония.
Сернистый аммоний растворяет серу с образованием
многосернистого аммония:
(NH4JS + nS =
Насыщенный раствор многосернистого аммония поступает
в кипятильник, где разлагается на аммиак, сероводород и серу
(NH4JSn+1 = 2NH3 + H2S + «S
поступающие далее в конденсатор.
Выделившаяся сера является товарным продуктом
хорошего качества (около 99% S).
Активированный уголь сушат и снова возвращают в процесс.
Длительность рабочего периода очистки, в зависимости от
содержания сероводорода в газе, колеблется от 1 до 3 недель.
Активированный уголь может служить адсорбентом без замены
около двух лет,
Достоинством сухих методов является очень высокая
степень очистки газов от сероводорода, недостатком — громоздкая
аппаратура периодического действия. В промышленности эти
методы используются для окончательной очистки от
сероводорода после предварительной очистки их более дешевыми
мокрыми методами.
Мокрая очистка газов. Основное достоинство мокрых
методов очистки газов от сернистых соединений заключается в
относительной простоте и непрерывности процессов очистки,
одновременно обычно достигается достаточно высокая степень
очистки газа от сероводорода. Вследствие этого мокрые методы
очистки в настоящее время получили наибольшее
распространение.
54
Применяемые в промышленности методы мокрой очистки
газов можно разделить на две группы. Для первой группы
характерны химические превращения сероводорода, в результате
которых образуется элементарная сера, сульфат аммония и
другие продукты, к второй группе относятся такие процессы
очистки, при которых извлекается газообразный сероводород в смеси
с другими газами. Все методы мокрой очистки газов от
сероводорода проводятся по
следующей схеме. Очищаемый газ
поступает в башню-скруббер,
которая орошается раствором,
поглощающим сернистые
соединения. Из скруббера
поглотительный раствор поступает
в башню-регенератор, где из
раствора выделяется сера,
регенерированный раствор
снова возвращается на орошение
скруббера.
В качестве поглотителей
сероводорода применяют
растворы соды (мышьяково-содо-
вый и вакуум-карбонатный
способы), этаноламины и др.
По мышьяково-содовому
Рис. 11. Схема мышьяково-содовоп
очистки от сероводорода:
СПОСОбу СерОВОДОрОД ПОГЛО- /-абсорбер; 2-бак для отработанного рас-
ЩаеТСЯ ИЗ ГаЗОВ разбаВЛеННЫЫ тв0Ра: S-регенератор; 4—пеноотделитель;
содовым раствором,
содержащим мышьковистый ангидрид.
Кислород окситиомышьяковокислого натрия, образовавшегося
в мышьяково-содовом растворе, при взаимодействии с
сероводородом замещается серой:
Na3AsS3O -'г H3S = Na3AsS4 -г Н,0
В процессе регенерации сера снова вытесняется кислородом:
2Na3AsS4 -г О3 = 2Na3AsS3O + 2S
Абсорбция и регенерация проводятся в установке башенного
типа (рис. 11).
Поступающий на очистку газ проходит через абсорбер /,
орошаемый мышьяково-содовым раствором. Отработанная
жидкость стекает в бак 2, откуда ее перекачивают насосом в
нижнюю часть регенератора 3. Одновременно в регенератор снизу
подают сжатый воздух. Образующаяся серная пена
отделяется от регенерированного раствора в пеноотделителе 4.
Регенерированный раствор поступает в абсорбер /.
55
После фильтрования пены получается очень чистая сера и
весьма мелкодисперсная (значительное количество частиц имеет
диаметр от 1 до 5 мк, максимальный диаметр частиц 15 мк).
Такая сера успешно применяется для борьбы с вредителями и
болезнями растений.
Для растворения белого мышьяка вместо раствора соды
можно применять аммиачную воду.
Сущность вакуум-карбонатного способа, обычно
применяемого для очистки коксового газа от сероводорода, состоит в
обработке газа (в башне с насадкой) раствором соды или
поташа, обладающим большей поглотительной способностью.
Далее поглотительный раствор регенерируют нагреванием в
вакууме.
При поглощении сероводорода раствором поташа протекает
следующая реакция;
H,S -ь К,СО3 = KHS 4- КНСО3
В процессе регенерации поглотительного раствора из него
выделяется сероводород:
KHS -г КНСО3 ^ H2S -f К2СО3
Для отгонки сероводорода из раствора применяют пар при
температуре 70—71 СС и остаточном давлении 120 мм рт. ст.
Вакуум-карбонатным способом удается извлечь из
коксового газа примерно 90% содержащегося в нем сероводорода.
Такая степень очистки достаточна лишь в том случае, если газ
используется как энергетическое топливо в металлургической
промышленности. Если же необходима более тщательная очи
стка от сероводорода, например при использовании водорода
коксового газа для синтеза аммиака, вакуум-карбонатный
способ непригоден.
Для очистки некоторых газов от серы и СО2 широко
применяют этаноламиновый способ (стр. 192). От ранее описанных
способов он отличается отсутствием окислительных реакций ь
процессе очистки газа. Сероводород и двуокись углерода обра
зуют с этаноламинами соли, которые при нагревании
разлагаются с выделением концентрированных сероводорода,
двуокиси углерода и исходного этаноламина.
3. ОЧИСТКА СЕРЫ
Серу, полученную обычными способами и содержащую
96,5—99,8% S, очищают в рафинировочных печах.
Рафинировочная печь (рис. 12) состоит из чугунного котла 1
для расплавления серы, перегонной реторты 2 и кон'ден-
56
сационной камеры 4. Реторта обогревается горячими газами,
полученными при сжигании топлива в топке 3. Загружаемая
в котел сера плавится и по боковой трубе стекает в реторту 2,
где возгоняется. Часть возогпанной серы осаждается в виде
серного цвета на стенах конденсационной камеры 4, остальная
сера в жидком виде стекает через отверстие 5 в приемник 6.
Потери серы при рафинировании составляют от 2 до
2.5%. |
Рафинированную серу выпускают в продажу в виде серного
цвета, а также в виде черенковой серы, получаемой при
застывании расплавленной
серы в формах.
Чешуйчатая сера.
Сера, выпускаемая в виде
блоков или черенков,
непригодна для ряда
потребителей. Ее
приходится измельчать, что
связано с дополнительным!:
расходами и потерей
серы. Для обеспечения
таких потребителей на
некоторых заводах
вырабатывают товарную серу в
виде лепестков-чешуек.
Для этого жидкую серу
Рис. 12, Печь для рафинирования серы:
У—котел; 2— перегонная реторта; 3—топка; 4—кон-
денсационная камера; 5—отверстие для выпуска
серы; 6—приемник серы.
при 130 °С заливают в
стальное корыто, в
которое опущен медлешю
вращающийся барабан
(~50 об/ч), изнутри
охлаждаемый водой A3°С). На наружной поверхности
вращающегося барабана, имеющей температуру около 25—30 °С,
образуется тонкая пленка кристаллической серы, снимаемая ножом
и далее ссыпающаяся в бункер. Средняя толщина
образующихся чешуек 1.3 мм. насыпной вес 0,96 г/.и3.
Сорта серы
Элементарную серу — природную и газовую, получаемую
описанными выше способами, выпускают в виде молотой и
комовой серы. В зависимости от содержания основного вещества
и примесей элементарная техническая сера разделяется на
сорта.
Элементарная сера должна соответствовать следующим
показателям (в пересчете лл е\\ос вещество):
Г,7
Содержание
Содержание
битумы
мышьяк
зола .
Влажность,
Кислотность
серы, %, не менее . . .
примесей, %, не более
%, не более
, %, не более
Природная
1-й
сорт
99,9
0,2
0,002
0,3
0,2
- 0,005
2-ii
сорт
98
0
0
0
0
0
5
6
003
7
3
005
сера
3-й
сорт
97,5
1,0
0,003
2,0
0,5
0,01
Газовая сера
1-й
сорт
99,6
0,01
0,2
0,2
0,02
2-й
сорт
98,6
0,05
0,7
0,5
0,03
3-й
сорт
96,5
0,5
2,0
1,0
0,03
Упаковка, перевозка и хранение серы
Комовую серу перевозят навалом в крытых
железнодорожных вагонах, предварительно тщательно очищенных и
выметенных. Молотую серу упаковывают в многослойные битумиро-
ванные бумажные мешки. Предназначенная для резиновой и
медицинской промышленности молотая сера упаковывается б
два вложенных друг в друга многослойных бумажных мешка
(верхний слой должен быть покрыт битумом).
Поставляемую сельскому хозяйству молотую серу
упаковывают в пятислойные бумажные мешки (вес нетто по 5 и 1 кг),
помещенные в бумажные пятислойные мешки (вес нетто по
20 кг). Мешки зашивают на машине.
При отправке серы мелкими партиями или смешанным
(железнодорожным и водным) транспортом бумажные мешки
дополнительно упаковывают в фанерные барабаны, деревянные
ящики, бочки или джутовые мешки. При перевозке молотой
серы автомобильным и гужевым транспортом мешки должны
быть накрыты брезентом.
Молотую серу хранят в сухих крытых помещениях,
защищенных от проникания влаги. Упакованный продукт размещают
на деревянных подмостках. Расстояние между мешками и
стенами складского помещения должно быть не менее 0,5 м.
Не допускается хранение мешков с серой вблизи
водопроводных и канализационных труб и отопительных приборов.
Глава IV
СЕРНАЯ КИСЛОТА
/. ОБЩИЕ СВЕДЕНИЯ
Свойства серной кислоты. Серную кислоту H2SO4 следует
рассматривать как соединение одной молекулы трехокиси серы
SO3 с одной молекулой воды Н2О. Безводная серная кислота
содержит 81,63% SO3 и 18,37% Н2О. Молекулярная масса
серной кислоты 98,076.
Безводная серная кислота представляет собой бесцветную
маслянистую жидкость плотностью 1,8305 г/см3 (при 20 °С),
кипящую при 296,2 °С под давлением 760 мм рт. ст. и
кристаллизующуюся при 10,45°С. При нагревании выше 150—200°С
безводная серная кислота заметно разлагается по реакции:
H.,SO41—' SO3 -f Н,0
При температуре выше 440 °С и атмосферном давлении
серная кислота практически полностью разлагается,
выделяющаяся трехокись серы также частично разлагается.
2SO3 ^=± 2SO3 + О,
В технике серной кислотой называют ее водные растворы
различной концентрации (H2SO4 + nH2O). Серная кислота
смешивается с водой в любых отношениях и образует гидраты
следующего состава: H2SO4 • Н2О (84,48% H2SO4) H2SO4-2H2O
G3,13% H2SO4) и H2SO4-4H2O E7.67% H2SO4).
С трехокисью серы серная кислота образует два соединения
H2SO4.SO3 и H2SO4 2SO3.
Раствор трехокиси серы в серной кислоте (H2SO4 + SO3)
называют олеумом. Состав олеума характеризуется содержанием
в нем свободной трехокиси серы (сверх 100% H2SO4),
выраженным в весовых процентах. Общее содержание трехокиси
серы в олеуме складывается из содержания свободной SO3 и
связанной с водой в виде H2SCV
Температура кристаллизации. Гидраты серной кислоты
имеют определенные температуры кристаллизации (рис. 13).
Из диаграммы видно, что по мере повышения содержания
серной кислоты (от 0 до 38% H2SO4) в растворе температура
59
кристаллизации постепенно понижается до точки минимума,
которая называется эвтектической точкой. Сооответствующии ей
состав системы H2SO4—Н2О называется эвтектикой. Из рас
творов, содержащих менее 38% H2SO4. выпадает лед. В эвтек-
40
40 ffO во /00 20 40 SO вО /00
ЪОг(свод\вес%
Рис. 13. Диаграмма кристаллизации системы Н^О—SO3-
гической точке кристалл:;.!) с тс я смесь, содержащая 38% H2SO4
и состоящая из кристаллов льда и тетрагндрата НаЬСЛ •-гН:О.
И; растворов с более высоким содержанием серном кислоты
(до 73% H-SO...I крлсталлилетс? се тетр:;п.'драт, а темпера-
гура крнстлллп:)ПЦ1Н! по^клппется дг то;п<|! максимума,
соответствующей температуре кргстллтацпи этого гидрата.
Сорта серной кислоты, выпускаемые промышленностью, по
составу близки соответствующим эвтектическим смесям, т. с.
характеризуются наиболее низкими температурами
кристаллизации. Так, 75%-ная H2SO4 кристаллизуется при —41 °С;
'•!-),3%-нзя H2SO4 при —37,85 °С; олеум с содержанием SO,
! своб.) 18,07% при —17,05°С. Некоторые растворы серной кис-
юты кристаллизуются при довольно высокой температуре (на-
тример, 98%-ная серная кислота при +0,1 °С), что необходимо
учитывать при производстве и транспортировании кислоты.
Температура кипения. При нагревании растворов серной
кислоты, содержащих менее 70% H2SO4, вплоть до температуры
кипения практически выделяются только пары воды. Над
растворами серной кислоты более высокой концентрации (до 93%)
наряду с парами воды присутствуют также пары серной
кислоты, причем и при температурах кипения и при более низки-;
температурах отношение Н2О : H2SO4 в парах выше, чем в
жидкости. Над еще более концентрированными растворами кислоты
(свыше 98%) отношение Н2О : H2SO4 в парах, наоборот, ниже,
чем в жидкости.
Растворы серной кислоты под атмосферным давлением
кипят при температуре выше 100°С, температура кипения
повышается с увеличением содержания H2SO4 до 98,3%- При
дальнейшем увеличении содержания H2SO4 в растворе температура
его кипения понижается. При содержании в растворе 98,3%
серной кислоты и 1,7% зоды образуется азеотропная смесь'* >¦
постоянной температурой кипения 338,6 °С. Поэтому при кипя
чении водных растворов серной кислоты (с отводом паров:
содержание H2SO4 в жидкости в конечном итоге становится
равным 98,3%.
Температура кипения олеума с увеличением содержания п
нем SO3 понижается, а давление паров над олеумом
возрастает. Над олеумом, содержащим больше 20% SO3 (своб.),
практически находятся только пары SO3; содержание H2SO4 в
парах крайне незначительно.
Товарные сорта серной кислоты имеют при давлении
760 мм рт. ст. следующие температуры кипоп-я (в °С):
Башенная G5 % H,SO4) . . . 187,8
Регенерированная (91% HnSO4) . . .... 275,9
Техническая (92,5% H,S0'4) 286,9
Техническая улучшенная (92,5—S4,0% H.,SO,> . . . 286,9—2°8,4
Олеум A8,5% SO3 спсб.) " _ 173.5
Теплоты образования и разбавления. Реакция образования
серной кислоты протекает с большим выделением тепла. Тет:-
* Азсотропгые смеси жидкостей кипят при постоянной температуре без
изменения состлеа. От чистых жидкостей азеотропные смеси отличаются
тем. что с изменением давления мопяются не только температ\!ры их кнпе-
-ия, но и состаз.
ловой эффект образования H2SO4 из SO2, О2 и Н2О при 25 °С,
по расчетным данным, составляет 52,12 ккал/моль H2SO4, или
532 ккси/кг H2SO4.
При получении водных растворов серной кислоты, кроме
теплоты образования, выделяется также теплота разбавления
серной кислоты водой. Теплота разбавления H2SO4 до
концентрации башенной кислоты G5% H2SO4) составляет 94 000 ккал
на 1 г H2SO4. Выделение тепла происходит и при смешении
кислот различных концентраций. Этот тепловой эффект,
называемый теплотой смешения (QCm.), можно определить по
формуле:
QcM. = Qs ("i + n2)
где Qi и Q2 — теплоты разбавления исходных кислот;
Q3 — теплота разбавления полученной кислоты;
П) и п-2 — количество молей H2SO4 в исходных кислотах,
взятых для смешения.
Теплоемкость. С повышением концентрации кислоты
теплоемкость водных растворов H2SO4 уменьшается, с повышением
температуры — увеличивается. Безводная серная кислота имеет
минимальную теплоемкость @,338 кал/г ¦ град): теплоемкость
олеума с повышением содержания SO3 (своб.) увеличивается.
В технологических расчетах обычно пользуются значениями
средней теплоемкости в определенном интервале температур.
Теплопроводность серной кислоты уменьшается с
повышением концентрации и понижением температуры.
Применение серной кислоты. Серная кислота является одним
из важнейших продуктов химической промышленности и имеет
огромное народнохозяйственное значение. Крупнейшим
потребителем серной кислоты является производство минеральных
удобрений, ежегодно потребляющее миллионы тонн H2SO4 для
получения суперфосфата, комплексных удобрений, сульфата
аммония и др. Значительные количества серной кислоты
расходуют в производстве соляной, плавиковой, фосфорной, уксусной
и других кислот из солей, а также для концентрирования
азотной кислоты. В металлообрабатывающей промышленности
серная кислота применяется, например, для очистки
(травления) поверхности стали от окислов перед нанесением на нее
металлических покрытий (цикка, олова, хрома, никеля).
Образующиеся при этом травильные растворы содержат до 25%
FeSO4.
Большое количество серной кислоты расходуется на очисткч
органических веществ, в частности нефтепродуктов (бен-
чина, смазочных масел и др.), от сернистых и непредельных
соединений. В настоящее время внедряются новые методк
очистки нефтепродуктов, без применения серной кислоты,
однако сернокислотный метод пока наиболее распространен. В ка-
честве отхода сернокислотной очистки нефтепродуктов
получается так называемый кислый гудрон, представляющий собой
очень сложную смесь различных сульфосоединений,
окисленных углеводородов и других веществ; в этой смеси содержится
от 20 до 50% серной кислоты.
Концентрированная серная кислота и олеум применяются
для сульфирования органических соединений, используемых в
качестве полупродуктов при синтезе красителей, лекарственных
веществ, ионообменных смол. Концентрированная серная
кислота (особенно олеум) необходима для производства
взрывчатых веществ (нитроглицерина, пироксилина, тротила и др.), а
также дымообразующих средств. Серная кислота широко
используется также в промышленности основного
органического синтеза.
Из этого далеко не полного перечня областей применения
серной кислоты видно, как огромно ее значение для нужд
народного хозяйства. С развитием промышленности и
совершенствованием техники некоторые области применения серной
кислоты теряют значение и возникают новые, но в целом
потребность в серной кислоте, особенно в концентрированной, из года
в год растет, а технология ее производства совершенствуется.
В 1900 г. во всем мире было произведено 4,2 млн. т серной
кислоты (в пересчете на 100%-ную), в 1937 г. (без СССР) оно
возросло до 18,8 млн. т, в 1954 г. увеличилось до 34,3 млн. т,
в 1960 г. мировое производство серной кислоты составило
45,8 млн. т. Производство серной кислоты за 1959—1962 гг., в
отдельных странах характеризуется следующими данными
(в млн. т 100%-ной H2SO4):
Страны
США ....
СССР ....
Япония . . .
ФРГ ....
Англия . . .
Франция . . .
Италия . . .
Испания . . .
Канада . . .
1959 г.
15,97
5,08
4,19
2,94
2,47
1,82
—
1,13
—
1960 г.
16,22
5,4
4,45
3,17
2,74
1,98
3,7
1,08
1,5
1962 г.
17,3
6,9A963г.)
4,9
3,1
2,7
2,15
2,6
—
—
МЕТОДЫ ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
Серная кислота производится двумя методами: нитрозным
и контактным. Производство серной кислоты по нитрозному
методу (стр. 130 ел.) быстро развивалась с начала XIX в. в свя-
!и с ростом производства соды. Первый завод для получения
серной кислоты нитрозным методом с применением камерной
системы был построен в России в 1805 г. Вначале серную
кислоту получали в свинцовых камерах периодического действия,
полнее перешли к непрерывному процессу.
63
Все возраставшая потребность в серной кислоте вызвала
необходимость интенсификации камерных систем. Это было
достигнуто заменой громоздких полых камер, в которых съем
кислоты с единицы реакционного объема был невелик, более
производительными башнями с насадкой, орошаемой
циркулирующей кислотой. Таким образом возник башенный способ
производства серной кислоты, давший возможность не только
резко повысить производительность сернокислотных установок,
но и получать более концентрированную серную кислоту
G5%-ную вместо 65%-ной в камерных системах).
С ростом потребления серной кислоты промышленностью
органического синтеза и другими производствами появилась
необходимость в выпуске больших количеств чистой серной
кислоты высокой концентрации и олеума. Это стало возможным
только при осуществлении контактного метода получения
серной кислоты (стр. 93 ел.), возникшего в конце XIX в.
При контактном методе достигаются лучшие
технико-экономические показатели, чем при нитрозном, более надежна
аппаратура, проще управление процессом и лучше условия
труда. Кроме того, серная кислота контактных систем может быть
получена более высокой концентрации н достаточной чистоты.
Несмотря на существенные преимущества контактного
метода, башенная кислота сохраняет еще свое значение для
производств, не требующих концентрированной и чистой серной кис
лоты (производство минеральных удобрений и др.). В случае
необходимости башенную кислоту концентрируют путем
упаривания.
СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
В качестве сырья для производства серной кислоты
применяются колчедан (флотационный и серный) элементарная сера,
сернистый газ из отходящих газов металлургических печей,
сероводород, извлекаемый из газов нефтепереработки,
коксового и других газов. Кроме того, для получения серной кислоты
можно использовать железный купорос из травильных растзо-
ров. а также отработанную серную кислоту и кислый гудрон.
Важнейшие виды серосодержащего сырья, применяемого в
отечественной сернокислотной промышленности, приведены нп-
/ке (в %):
Сырье 1963 г. 1965 г.
Колчедан 51 45
Сера 26 24
Газы, содержащие SO., !0 23
Сероводород 4 8
Из этих данных следует, что доля колчедана, применяемого
в Советском Союзе для производства сеппон кислоты, будет
постепенно уменьшаться. Во все больших масштабах в
ближайшие годы будут использоваться в производстве серной кислоты
сернистый ангидрид и сероводород, содержащиеся в отходящих
промышленных газах.
Серный колчедан. Во многих странах сырьем для
производства серной кислоты служит пирит, или серный колчедан, сЪ-
стоящий в основном из двусернистого железа FeS2. Серный
колчедан представляет собой минерал желтоватого или
желтовато-серого цвета с металлическим блеском; плотность его
колеблется в пределах от 4,95 до 5,0 г/см3.
В химически чистом двусернистом железе FeS2 содержится
53,46% S и 46,54 Fe. Однако в природе двусернистое железо
в совершенно чистом виде не встречается. Среднее содержание
серы з природном серном колчедане составляет от 40 до 50%,
содержание железа 35—44%; остальное — примеси, из которых
основными являются сернистые соединения меди, цинка,
свинца, мышьяка, селена, теллура, углекислые и сернокислые соли
кальция и магния, тальк, кварц и часто в незначительных
количествах золото и серебро.
Мышьяк присутствует главным образом в виде FeAsS
(мышьяковый колчедан), медь в виде CuFeS2 (медный
колчедан), Cu2S (медный блеск) и CuS( коввелин). Если меди
содержится не менее 0,7—1,0%. ее экономически выгодно
выплавлять из колчедана.
Мышьяк и селен являются вредными примесями при
производстве серной кислоты контактным методом, поэтому
присутствие их в сырье нежелательно.
Советский Союз богат месторождениями серного колчедана.
Крупнейшие месторождения его расположены на Урале.
Сырьем для получения сернистого газа может также
служить магнитный колчедан, или пирротин, залегающий в
значительных количествах в Карелии (Кольский полуостров). Состав
пирротинов чаще всего отвечает формуле Fe7Se; содержание
серы в них около 30%¦
В большинстве месторождений Урала колчедан добывают
шахтным способом. Применяется также метод открытых
разработок. С рудников колчедан поступает в виде кусков
размером 50—400 мм. Серный колчедан, содержащий примеси
сернистых соединений цветных металлов, размалывают и
обогащают путем флотации.
Флотационный колчедан. Наиболее совершенным способом
комплексного использования сернистых руд с содержанием
0,7—1% меди является их флотационное обогащение. При
флотационном обогащении медная руда разделяется на две части—
медный концентрат с содержанием 15—20% Си и отходы,
называемые флотационными хвостами, или флотационным
колчеданом.
5—2848 65
Флотационный колчедан представляет собой
мелкодисперсный порошок (около 46% частиц размером менее 0,05 мм). Он
содержит те же примеси, что и серный колчедан. Из 100 т
колчедана, подвергаемого флотации, получается 80—85 т
флотационного колчедана и 15—20 т концентрата.
Содержание серы зо флотационном концентрате составляет
35—42%. Для повышения содержания серы флотационный
концентрат можно подвергнуть вторичной флотации и, отделив
пустую породу, получить пиритный концентрат D8% S и более).
Полученный флотационный концентрат содержит 12—15%
влаги. Сырой флотационный концентрат подвергают сушке в
барабанных сушилках, в результате чего содержание влаги в
нем снижается до 3—3,8%. В таком виде флотационный
колчедан отгружается сернокислотным заводам. В летнее время
допускается отгрузка потребителям колчедана с влажностью до
8%. В осенне-зимний период содержание влаги в отгружаемом
флотационном колчедане во избежание его смерзания в пути п
на сернокислотных складах не должно превышать 3,8%.
Примерный состав колчеданов для производства серной
кислоты приведен в табл. 5.
Таблица 5
Примерный состав колчеданов, применяемых для производства серной кислоты
Колчедан
Серный
Флотационный
Пиритный концентрат . .
Состав, %
S
40—50
35—42
48—50
Fe
44,0
35,0
44,0
А12о3
1,0
¦ 4,5
0,9
СаО
0,02
1,2
MgO
0,05
0,6
0,03
Си
0,73
0,8
0,2
Zn
0,7
1,0
SiO-з
1,2
18,7
1,7
Отходящие газы металлургических печей. При выплавке
цветных металлов (меди, цинка и др.) из сульфидных руд в
механических обжиговых печах, в ватержакетных и отражательных
печах, а также в конверторах получаются большие количества
газов, содержащих SO2.
Например, при получении 1 г черновой меди одновременно
образуется 7,3 г SO2. Из этого количества двуокиси серы
можно получить примерно 11 т серной кислоты.
Использование отходящих газов цветной металлургии
имеет большое народнохозяйственное значение, так как позволяет
избежать потерь ценных продуктов и исключить операцию
дробления и обжига серного сырья в производстве серной
кислоты. Кроме того, благодаря улавливанию двуокиси серы,
которая губит растения и вредна для здоровья людей, не
загрязняется атмосферный воздух.
66
Топочные газы. При сжигании угля в топках котельных
установок содержащаяся в угле сера превращается в двуокись серы,'
котооля до сего времени вместе с отходящими топочными
газами выбрасывается в атмосферу. Хотя содержание SO2 в
топочных газах невелико @,2—0,5%), улавливание его (особенно в
населенных районах) необходимо прежде всего из санитарно-
гигиенических соображений. При этом представляет большой
интерес использование улавливаемой двуокиси серы.
Однако практическое использование топочных газов для
получения серной кислоты связано с большими трудностями
вследствие низкого содержания SO2 в таких газах. В
настоящее время проводятся исследования с целью разработки
экономичного способа использования SO2 топочных газов для
переработки в серную кислоту.
Сера. Лучшим сырьем для производства серной кислоты
является элементарная сера (самородная и газовая). Особые
преимущества создаются при использовании для производства
серной килоты контактным способом самородной серы, не
содержащей примесей мышьяка и селена. Сжиганием самородной
серы получают концентрированный сернистый газ, не
требующий громоздких и дорогостоящих операций специальной
очистки (стр. 97 ел.).
Сероводород. При очистке горючих газов получаются
большие количества сероводородного высококонцентрированного
газа, который является весьма ценным серосодержащим
сырьем.
При сжигании сероводорода образуются сернистый газ или
сера, используемые для производства серной кислоты.
Другие виды сырья. При травлении металлов в качестве
отхода получаются травильные растворы, содержащие 2—4%
серной кислоты и до 25% сульфата железа. Термическим
разложением железного купороса, выделяемого из травильных
растворов, можно получить сернистый газ н регенерировать таким
образом серную кислоту.
При сульфировании органических соединений и очистке
нефтепродуктов в качестве отходов получается загрязненная
органическими примесями и разбавленная серная кислота —
отработанная кислота. Методы использования этих отходов
зависят от степени их загрязнения и разбавления. Отработанные
кислоты либо концентрируют и очищают от примесей, либо
подвергают термическому разложению с получением
сернистого газа для последующей переработки его в серную кислоту.
Для получения сернистого газа целесообразно использовать
также кислый гудрон. При термическом разложении кислого
гудрона, проводимого во вращающихся печах, выделяется вода
н образуются SO2, кокс и углеводороды. Полученный сернистый
газ можно перерабатывать в серную кислоту (стр. 128).
5* 67
В некоторых зарубежных странах в качестве сырья для
производства серной кислоты применяется гипс CaSO,). При
термическом разложении гипса выделяется двуокись серы, которую
обычным способом перерабатывают в сернокислотных системах.
ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС ПРОИЗВОДСТВА
СЕРНОЙ КИСЛОТЫ
В промышленности исходным зеществом для производства
серной кислоты служит сернистый газ. В технике сернистым
газом называется газовая смесь, содержащая обычно только
7—15% двуокиси серы. Количество остальных компонентов
сернистого газа (кислород, азот и др.) различно в зависимости от
состава и качества исходного сырья и методов его обжига.
Процесс переработки сернистого газа в серную кислоту
состоит чз двух основных стадий:
1. Окисление двуокиси серы SO2 в трехокись серы SO3:
so2 + ~y о2 = so3 + Q
2. Абсорбция (поглощение) трехокиси серы водой с
образованием серной кислоты:
SO3 + Н2О = HaSO4 4- Q
Этот процесс может быть выражен следующим суммарным
стехиометрическим уравнением:
SO2 + -g- Oa + гсНаО = H,SO4 -г (п — 1) Н3О
В промышленных масштабах процесс получения серной
кислоты осуществляется двумя различными методами —
контактным и нитрозным.
По контактному методу двуокись серы окисляется
кислородом воздуха на поверхности твердого катализатора. По нитроз-
ному методу окисление двуокиси серы производится
кислородом воздуха при помощи окислов азота, являющихся его
передатчиком. При этом высшие окислы азота (N2O3, NO2), окисляя
SO2, восстанавливаются до окиси азота N0, которая вновь
окисляется кислородом с образованием высших окислов азота.
2. ПОЛУЧЕНИЕ СЕРНИСТОГО ГАЗА
Независимо от метода производства серной кислоты первой
стадией процесса всегда является получение двуокиси серы.
В большинстве случаев для этого специально сжигают
серосодержащее сырье — серный колчедан, элементарную серу,
сероводород и др., или используют двуокись серы, получаемую в
качестве отхода при обжиге руд на заводах цветной
металлургии.
68
Свойства двуокиси серы. Двуокись серы, или сернистый
ангидрид, SO2 представляет собой бесцветный газ с характерным
резким запахом, сильно раздражающий слизистые оболочки
глаз и дыхательных путей.
В одном объеме воды при 20°С растворяется около 40
объемов SO2; при этом выделяется 8,2 ккал/моль тепла. При
взаимодействии SO2 с водой образуется малоустойчивая сернистая
кислота
SO2 -г Н2О = H2SO3
существующая только в виде растворов.
С повышением температуры растворимость SO2 в воде
уменьшается. Растворимость SO2 в серной кислоте 'меньше, чем
в воде. С повышением концентрации серной кислоты
растворимость SO2 вначале уменьшается, достигая минимума при 85%
H2SO4, затем вновь увеличивается.
Двуокись серы легко превращается в жидкость при
атмосферном давлении с одновременным охлаждением газа до
—10,1 °С. Критическая температура SO2 157°С, критическое
абсолютное давление 78 атм.
Жидкая двуокись серы замерзает при —73 °С. Давление
паров SO2 над жидкой фазой составляет 3,25 атм при 20°С п
8,4 атм при 50 °С.
Жидкая двуокись серы применяется в холодильной
технике, в целлюлозной промышленности, в производстве некоторых
органических продуктов, а также используется как
консервирующее вещество (например, при хранении и перевозке
фруктов и ягод). Транспортируется жидкая двуокись серы в
стальных баллонах или цистернах.
Двуокись серы вредна для человеческого организма, так как
вызывает раздражение кожи, верхних дыхательных путей,
слизистых оболочек носоглотки, глаз. По существующим нормам*,
допустимое содержание SO; в воздухе рабочих помещений не
должно превышать 0,01 мг/л. Поэтому в сернокислотных цехах
необходимы тщательная герметизация аппаратуры и хорошая
вентиляция помещений.
ПОЛУЧЕНИЕ СЕРНИСТОГО ГАЗА ИЗ КОЛЧЕДАНА
Колчедан (флотационный п рядовой) сохраняет свое
значение в качестве одного ил основных видов сырья для
производства серной кислоты, хотя доля колчедана в балансе
серосодержащего сырья постепенно уменьшается (стр. 64).
* Правила и нормы техники безопасности и промышленной санитарии
для проектирования, строительства и эксплуатации производства серной
кислоты контактным и башенным способами, Госхимиздат. 1961.
69
Рядовой (кусковой) колчедан перед обжигом подвергают
дроблению на щековых дробилках и просеивают на
барабанных грохотах. При переработке флотационного колчедана
(флотационные хвосты) операция дробления является излишней и
он подвергается только сушке.
Сушка флотационного колчедана. Флотационный колчедан,
взятый непосредственно из отвалов медеплавильных заводов:
колчедан
В г,еччое отделение
Рис. 14. Сушильная установка для флотационного колчедана:
1—топка; 2—камера смешения; 3—бункер; 4—питатель; 5—течка; 6—бандажи; 7—сушильный
барабан; 8—зубчатое колесо; 9— разгрузочная камера; 10—циклон; У/—электрофильтр; 12—Еен-
тилятор; 13—моло1кОЕая дробилка; 14—ленточный транспортер.
содержит около 15% влаги. Перевозить колчедан с таким
содержанием влгги нецелесообразно; кроме того, влажный колчедан
зимой смерзается, что весьма затрудняет его перевозку и
использование. Поэтому флотационный колчедан предварительно
подсушивают до влажности ~ 3,8%. Сушку флотационного
колчедана, применяемого для сжигания в механических печах и в
печах пылевидного обжига, производят непосредственно
топочными газами в непрерывно действующих сушилках
барабанного типа.
Схема сушильной установки для флотационного колчедана
изображена на рис. 14. Топочные газы из топки 1 поступают в
камеру 2, где смешиваются с воздухом. Смесь воздуха и газа
поступает в наклонно расположенный (под углом 3—4°)
стальной сушильный барабан 7. Для увеличения поверхности
соприкосновения материала с топочными газами и, следовательно,
повышения производительности сушильной установки внутри
барабана имеется специальная насадка.
Влажный колчедан поступает в сушильный барабан из
бункера 3 через питатель 4 и течку 5. При вращении барабана
колчедан постепенно перемещается к разгрузочной камере 9,
70
расположенной на другом конце барабана. Соприкасаясь с
горячими газами, влажный колчедан в барабане высушивается.
При сушке колчедан комкуется, поэтому на выходе его из
сушилки в разгрузочной камере 9 установлена молотковая
дробилка 13. Готовый продукт направляется ленточным
транспортером 14 в печное отделение. Для освобождения от пыли
газ нз рагрузочной камеры проходит последовательно
циклон 10 и электрофильтр //, после чего отводится через
выхлопную трубу в атмосферу. Отсасывание газа производится
вентилятором 12.
Барабан приводится во вращение электродвигателем через
систему передач и зубчатое колесо 8, укрепленное на
барабане. Стальными бандажами 6 барабан опирается на ролики.
Скорость вращения барабана 3,5—5 об/мин; примерные
размеры: длина 11 —14 м, диаметр 1,1—1,6 м. Производительность
сушильного барабана 3—6 т/ч (в зависимости от влажности
колчедана).
Температура газов на входе в барабан 600—650 °С, на
выходе 100—110°С, температура выгружаемого колчедана 80—
90 ЭС. Вследствие довольно высокой влажности колчедана,
поступающего на сушку, он не загорается при соприкосновении
с горячими топочными газами на входе в барабан.
Расход энергии на сушку 1 т колчедана зависит от
содержания в нем влаги и составляет примерно 6—10 квт-ч.
Устройство и работа аппаратов для очистки газа от пыли
(циклона и электрофильтра) описаны на стр. 88 ел.
Обжиг серного колчедана
Процесс горения серного колчедана протекает с
образованием окиси железа:
4FeS, — 1102 = 8SO2 + 2Fe2O3 + 815,2 ккал A)
либо закись-окиси железа:
3FeS, -f 8O2 = 6SOa -{- Fe3O4 - 581,9 ккал B,
Обе реакции идут с большим выделением тепла.
Выделяющегося тепла вполне достаточно для сжигания колчедана без
подвода тепла извне, поэтому топливо здесь требуется только
для разогрева печи в период ее пуска.
Благоприятными условиями для образования закись-окиси
железа являются: повышенное содержание двуокиси серы в
газе, высокая температура и относительно большое содержание
несгоревшего колчедана.
Стемюметрические уравнения A) и B) лишь суммарно
выражают процесс обжига серного колчедана. В
действительности этот процесс состоит из ряда более простых промежуточ-
71
ных реакции, механизм которых еще не вполне точно
установлен.
Упрощенно ход процесса можно представить приводимыми
ниже реакциями. При высокой температуре, достигающей
800—900 °С, двусернистое железо диссоциирует:
FeS.3 = S + FeS — Q C)
а выделившаяся парообразная сера сгорает в газовой фазе:
S+O2 = SO2 + Q D)
Сернистое железо реагирует с кислородом с образованием
закиси железа
2FeS + ЗО3 = 2FeO + 2SO2 + Q E)
которая далее окисляется через закись-окись железа в окись
железа:
4FeO + О3 = 2Fe2O3 + Q F)
Сложиз уравнения C) и F), получим приведенное выше
суммарное уравнение A).
Некоторое количество SO2, уже в печи, в присутствии
окислов железа (F3O4), действующих как катализатор, переходит
в SO3:
2SO3 + О3 = 2SO3
Присутствующие в колчедане сернистые соединения других
металлов сгорают с образованием соответствующих окислов и
двуокиси серы. Окислы этих металлов, реагируют с трехокисью
серы, образуют сульфаты.
Если в колчедане имеются карбонаты магния и кальция, они
разлагаются с выделением двуокиси углерода и образованием
окислов, которые затем превращаются в сульфаты:
СаО + SO3 = CaSO4
MgO + SO., = MgSCX,
Содержащиеся в колчедане примеси мышьяка и селена
также вступают в химические реакции. Соединения мышьяка
образуют парообразную трехокись мышьяка As2O3, селен —
двуокись селена SeO2.
Твердый остаток после обжига — так называемый огарок —
состоит в основном из Fe2O3. Кроме того, в огарке могут
содержаться FeS, РезС>4, FeSj, негорючие примеси, например SiCb,
CaSO4, MgSO4 и др.
Содержание SO2 в печном газе в зависимости от состава
колчедана, коэффициента избытка воздуха, конструкции печи
и т. д. составляет 9—15%.
Запыленность печного газа колеблется в очень широких
пределах A0—300 г/м3) и зависит от способа и
интенсивности обжига, состава колчедана и др.
79
Скорость обжига колчедана. Реакция горения
колчедана является гетерогенным процессом, так как протекает
между твердым и газообразным веществами. Этот сложный
700 750 800
Температура, °С
Рис. 15. Зависимость скорости
диссоциации пирита от
температуры.
Диаметр кусков колчедана, ////
Рис. 16 Зависимость скорости
обжига колчедана от размеров
кусков.
процесс включает несколько последовательно и параллельно
протекающих реакций, скорость которых различна. Скорость
суммарного процесса горения определяется скоростью наиболее
медленной реакции.
Диссоциация пирита начинается при температуре около
500'С и быстро возрастает с повышением температуры
(рис. 15). Скорость реакция
окисления одно- и двусерня-
стого железа значительно
меньше скорости диссоциации
пирита. Начальная скорость
этих реакций обратно
пропорциональна размерам кусков
колчедана (рис. 16) и увели- _
чивается с повышением кон- 600 700 ^Т
ЦеНТрЭЦИИ КИСЛОрОДа В гаЗОВОЙ Температура,
фазе. При добавлении К ВОЗ- Рис. 17. Зависимость скорости горения
духу кислорода (обжиг С При- сернистого железа от температуры,
менением обогащенного
кислородом воздуха) реакция
горения колчедана ускоряется. С повышением температуры
скорость горения сернистого железа возрастает (рис. 17).
При температуре выше 900 °С скорость обжига серного
колчедана уменьшается, что объясняется его спеканием,
затрудняющим выделение газообразных веществ и доступ кислорода.
Температура, при которой начинается спекание, зависит от качест-
73
ва колчедана и размера его частиц. С уменьшением размера
частиц спекание начинается при более низкой температуре.
Условия обжига колчедана в механических печах способствуют
его спеканию; при обжиге во взвешенном состоянии и в кипящем
слое (стр. 75 и 82) наблюдается меньшее спекание отдельных
частиц.
Обжиг колчедана с размерами частиц 6—8 мм проводят при
температуре около 850°С; обжиг флотационного колчедана во
взвешенном состоянии можно проводить при более высокой
температуре.
Оптимальные условия обжига. Оптимальными
условиями процесса обжига колчедана считаются такие, при
которых достигается наибольшая скорость горения
колчедана. При обжиге тонко измельченного колчедана во
взвешенном состоянии достигается очень высокая скорость
горения.
Температура процесса горения не должна превышать
температуру спекания твердого материала (для колчедана эта
гемпература приближается к 900 °С) и должна равномерно
поддерживаться во всех стадиях процесса горения. При
сжигании колчедана во взвешенном состоянии температуру
поддерживают на 100—150 °С выше, чем при обжиге кускового
материала в полочных, печах, т. е. проводят процесс при
температуре, несколько превышающей температуру плавления
материала.
Для достижения достаточной скорости процесса и
поддержания оптимальной температуры горения обжиг серного сырья
ведут при избытке воздуха. При обжиге колчедана в кипящем
слое требуется избыток воздуха в количестве до 125% от сте-
хиометрического. При этом содержание SO2 в газах, уходящих
из печей, составляет около 15%. В процессе обжига колчедана
в полочных механических печах необходим избыток воздуха
до 160% от стехиометрического количества, содержание SO2 в
уходящих печных газах составляет примерно 10%.
Применение воздуха, обогащенного кислородом,
интенсифицирует процесс обжига и дальнейшую переработку газа, но
пока этот метод еще не получил широкого распространения.
КОЛЧЕДАННЫЕ ПЕЧИ
В настоящее время для обжига колчедана в
промышленности применяют печи трех типов:
1) печи для обжига колчедана в кипящем слое (КС);
2) полочные механические печи, в которых колчедан
сгорает, передвигаясь по поверхности обжига;
3) печи для обжига пылевидного колчедана во взвешенном
состоянии.
74
Обжиговый
гоз
Печи для обжига колчедана в кипящем слое
Промышленное использование высокоинтенсивного метода
обжига серного сырья в кипящем слое является крупным
усовершенствованием этого процесса. Печь (рис. 18) для обжига
колчедана в кипящем слое представляет собой стальную
цилиндрическую (или прямоугольную) камеру /, футерованную
огнеупорным материалом 2. В нижней части печи расположена
решетка 3, через которую снизу вверх подается воздух.
Пылевидный или тонко измельченный
колчедан, помещенный на решетку, при
равномерном продувании через него
воздуха (с определенной скоростью)
образует над решеткой подвижный
полувзвешенный (кипящий) слой 4,
иначе называемый псевдоожиженным
слоем.
Отличительная особенность такого
метода обжига состоит в том, что
вследствие очень тесного
соприкосновения колчедана с воздухом
выгорание серы в кипящем слое протекает
очень быстро, в результате чего
достигается высокая интенсивность обжига.
Турбулентный кипящий слой
практически состоит из обожженного
материала (огарка). В кипящий слой
непрерывно поступает воздух,
загружается колчедан и отбирается
твердый продукт реакции — огарок. Из
верхней части печи отводится
обжиговый газ, причем часть огарка
уносится газом в виде пыли. Количество
пыли, уносимой из печей для обжига в
кипящем слое, достигает 90—95% от
веса огарка. Запыленный газ
пропускают через 1—2 циклона, а затем через электрофильтр.
Степень очистки газа при этом достигает 99,5% и более; таким
образом, содержание пыли в газе снижается до 0,2 г/.и3 и менее.
Частицы горящего колчедана в кипящем слое тесно
соприкасаются друг с другом. Чтобы избежать их слипания,
необходимо поддерживать в кипящем слое температуру 620—640 °С,
а в верху печи до 850—900 "С. Требуемая температура в
кипящем слое поддерживается за счет тепла реакции и почти
одинакова во всем объеме слоя.
Разность температур в различных точках слоя не превышает
5—10°С.
\ f \Огарок
Рис. 18. Принципиальн ан
схема печи для обжига
колчедана в кипящем слое:
1—камера; 2—огнеупорная
футеровка; 3—колосниковая решетка:
4—КИПЯ1ДИН СЛОЙ.
75
Высота кипящего слоя может быть различной. Время
соприкосновения воздуха с горящим колчеданом зависит от высоты
кипящего слоя. Чем выше слой, тем полнее выгорает сера.
Однако для поддержания над решеткой более высокого кипящего
слоя требуется увеличить давление подаваемого в печь воздуха,
что связано с дополнительной затратой энергии. Потери
тепла в окружающую среду в печах с кипящим слоем
относительно невелики (около 3%). Поэтому для поддержания
оптимальной температуры обжига необходимо отводить из реакционного
пространства большое количество тепла. Для этого в кипящем
1 слое располагают водяные теплообменники или секции труб
парового котла (на рис. 18 не показаны). Кипящий слой
обжигаемого колчедана характеризуется высоким коэффициентом
теплопередачи B00—300 ккал/м2 • ч • град).
При указанном способе отбора тепла из печи кипящего слоя
процесс сжигания колчедана совмещается с получением пара.
Печи с кипящим слоем применяются для обжига не только
измельченного серного колчедана (рядового или
флотационного), но и других сульфидных руд цветных металлов, для
сжигания элементарной серы, газоочистительной массы и
различных материалов, содержащих серу.
Обжигаемые материалы предварительно измельчают на
частицы размером до 6 мм.
В кипящем слое можно проводить также другие процессы,
требующие подвода тепла, например сушку.
На рис. 19 схематично изображена промышленная печь с
кипящим слоем для обжига флотационного концентрата,
производительность печи 100 т/'сутки. Печь представляет собой
шахту внутренним диаметром около 4 м и высотой около 9 м
(без опор и выхлопной трубы). Общая высота печи составляет
19 м, объем печи около 90 м3, вес 14 т. Кожух печи стальной,
изнутри футерованный огнеупорным материалом. В нижней
части печи расположена подовая плита 6 с большим
количеством отверстий, снабженных колпачками для подачи воздуха в
зону кипящего слоя.
Количество подаваемого воздуха около 6000 м3[ч.
Выше зоны кипящего слоя через коллектор 10 может быть
подан вторичный воздух для дожигания мелких частиц
колчедана, уносимых газом. Вторичный воздух поступает в печь
через щели, расположенные ниже ее конической части. В
кипящем слое размещены водяные охлаждающие элементы 7,
выполненные из жаростойких металлических труб. Пучки труб
присоединены к системе принудительной циркуляции парового
котла-утилизатора.
В загрузочной камере 9 также находятся охлаждающие
элементы 5.
76
Обжиговый,
газ
Уровень спокойного
слоя
Рис. 19. Схема промышленной печи для обжига
флотационного концентрата в кипящем слое:
/—опорная рама; 2—обечайка; 3—конус; 4—бункер под провальной
решеткой загрузочной камеры; 5, 7—охлаждающие элементы;
6—подовая плита; 8—форсунки; 9—загрузочная камера; 10—коллектор;
И—выхлопная труба.
77
Высушенный или влажный (до 8—10% влаги)
флотационный колчедан из дозатора непрерывно поступает в загрузочную
камеру 9, воздух подается в печь снизу через стояк.
Для розжига печи установлены форсунки, которые могут
работать на газе или жидком топливе (керосин, мазут и др.).
Температура обжигового газа на выходе из печи
поддерживается в пределах 850—900°С, температура огарка,
уходящего из печи, около 800°С.
Огарок удаляется в бункер 4, расположенный под
провальной решеткой загрузочной камеры 9. При подаче воздуха в
бункер для огарка его температура снижается до 50—60 °С.
Такой охлажденный огарок можно подавать в
железнодорожные бункеры скребковыми транспортерами.
Концентрация SO2 в печном газе составляет 14—15%. При
добавлении кислорода к воздушному дутью возможно
дальнейшее повышение концентрации SO2 в газе. Содержание пыли в
газе составляет до 300 г/ж3, содержание серы в огарке
0,5—1,0%.
Установка снабжена автоматическими регуляторами
температуры, давления, количества и концентрации газов.
Автоматическое регулирование обеспечивает устойчивый режим
обжига, постоянные концентрацию SO2 и количество газа.
В последнее время для обжига флотационного колчедана
в кипящем слое начали применять печи К.С-200 суточной
производительностью 200 г в сутки (считая на условный колчедан
с 45% серы).
Характеристика печи:
Общий объем, ж3 222
Площадь, жа
решетки (дутьевого пода) 20,1
верхнего сечения 30,0
нижнего сечения 17,7
Поверхность охлаждающих змеевиков, м2 • 34,0
Показатели процесса обжига колчедана в печи КС-200:
Интенсивность* обжига, кг/лр-сутки До 1ОЭ0
Температура, °С
в кипящем слое 750
в верхней части печи 900
газа, выходящего из печи 900
Концентрация SO3 в печном газе, °„ 14—14.5
Содержание серы в огарке, % 1,0
* Под интенсивностью печи понимают количество котюдан i (в п-рсгчегс
на колчедан, содержащий 45% серы), сжигаемого п^сутки па 1 .:;- и i pajo-
чей поверхности (или! м^ объема) печи.
Достоинством печей с кипящим слоем является
относительная простота их устройства (отсутствие движущихся деталей
и др.), возможность полной автоматизации управления про-
78
цессом обжига (стр. 163), что позволяет получать газ с
устойчивой концентрацией SO2 (до 15%) и низким содержанием SO3
и одновременно обеспечивает также малое содержание серы
в огарке. Кроме того, использование тепла отходящих газов
дает возможность получать при сжигании каждой тонны
колчедана 1,2—1,3 т и более пара давлением до 44 ат. При
эксплуатации печей КС уменьшается теплоотдача в окружающую
среду, улучшаются и облегчаются условия труда (устраняется
загазованность печного отделения и др.).
Недостатки печей КС: относительно высокий расход
электроэнергии на воздушное дутье A2—14 кет • ч на 1 т
сжигаемого колчедана), высокое содержание пыли в печном газе (до
300 г/ж3) и др. Однако экономический эффект внедрения печей
КС в сернокислотную промышленность значительно
превосходит дополнительные затраты, связанные с отмеченными выше
недостатками.
Широкое внедрение в промышленность печей КС для
обжига колчедана является одним из важных путей увеличения
производства серной кислоты, снижения ее себестоимости и
улучшения других технико-экономических показателей
сернокислотных систем.
Получение концентрированного сернистого газа при взаимодействии пирита
с окисью железа
В настоящее время исследуется возможность получения
высококонцентрированного сернистого газа путем взаимодействия пнрнта и окиси железа
FeS2 + 16Fe.2O3 = 1 lFe3O4 + 2SO2 — 107,6 ккал A)
Эта реакция протекает при 675 °С с поглощением тепла.
Образующаяся закись-окись железа окисляется кислородом зоздуха при
790 СС до Fe2O3:
HFe3O4 + 2,750, == 16,5Fe2O3 + 305,25 ккал B)
Суммарный положительный тепловой эффект реакций A) и B),
равный 197,65 ккал!моль FeS2, достаточен для поддержания аутотермичпоп»
процесса.
Высушенный флотационный колчедан (влажность до 1%) пробовали
обжигать в кипящем слое с циркулирующим в аппарате огарком (Ре2Оз).
Образующийся при этом высококонцептрированный сернистый газ (почти 100%
SO2) после очистки его от пыли в циклонах (стр. 88) охлаждается в
котле-утилизаторе и затем очищается от остатков пыли в электрофильтре.
Отсюда часть газа направляется на переработку в серную кислоту, остальное
количество газа под небольшим даэлением подается под решетку
реакционного аппарата для создания кипящего слоя. Аппарат состоит
из двух частей — реактора и регенератора, совмещенных в одном
корпусе. Кратность циркуляции огарка составляет 28,5 по отношению к
количеству поступающего в аппарат флотационного колчедана. Избыток
тепла процесса окисления FeS2 (за счет РегОз) до РезО4 и последующего
окисления Fe3O4 снова до Fe2O3 отводится холодильными элементами,
вмонтированными в зону кипящего слоя регенератора.
79
Получение серной кислоты из концентрированного газа (почти 100%
SO2) вместо обычного разбавленного газа G—10% SO2) представляет
большой интерес, так как позволяет во много раз увеличить интенсивность
сернокислотных систем.
Полочные механические печи
Основным типом полочных механических печей в СССР
является печь ВХЗ (Воскресенского химического завода). На
отдельных заводах установлены печи, отличающиеся от печей
ВХЗ некоторыми конструктивными особенностями и
производительностью.
Ускорение процесса горения колчедана в печах полочного
типа достигается путем постоянного перемешивания твердого
материала.
Печь ВХЗ (рис. 20) представляет собой вертикальный
стальной цилиндр диаметром около 6 м, общей высотой около 8 м,
футерованный изнутри огнеупорным фасонным кирпичом. По
высоте печи расположены семь рабочих сводов (подов) /—VII
для обжига и один для подсушки колчедана. Общая
поверхность рабочих сводов 140 м2. В центре печи установлен
вертикальный полый вал 3 (диаметром около 0,9 м), в гнезда
которого на каждом своде вставлено по два гребка-4, снабженных
съемными зубьями.
С помощью приводного механизма 7 вал, а вместе с ним и
гребки с зубьями приводятся в медленное вращение (около
1 об/мин).
Для обеспечения надежной и длительной работы при
высоких температурах зубья и гребки изготовляют из специальных
жароупорных материалов (высокохромистый чугун).
При вращении вала и гребков с зубьями колчедан
перемещается на нечетных сводах печи от центра к периферии, а на
четных сводах и на сушильном (нулевом) своде — от
периферии к центру. Такое перемещение колчедана достигается
установкой зубьев под соответствующим углом к оси гребка.
Измельченный колчедан из верхнего бункера поступает на
сушильный свод, где подсушивается, перемещаясь от
периферии к центру, и через питательную тарелку 2 в центре печи
пересыпается на первый рабочий свод. На первом рабочем
своде начинается горение колчедана. В процессе работы колчедан
передвигается гребками от центра к периферии и через
отверстия 5 в сводах, расположенные по периферии печи, ссыпается
на второй рабочий свод; здесь колчедан перемещается от
периферии к центру и через отверстия 5 около вала пересыпается
на третий рабочий свод и т. д. С последнего свода VII
обожженный материал (огарок) ссыпается через течку 6 на
транспортер.
80
Воздух подается в печь противотоком движению колчедана,
что позволяет поддерживать возможно большую концентрацию
кислорода в конце процесса, когда его доступ к поверхности
сернистого железа наиболее затруднен.
Использование противотока позволяет также снизить
содержание серы в огарке.
Колчедан
воздух для охлаждения
вала и гребков
Рис. 20. Печь ВХЗ:
/_1///_рабочие своды; /—питатель; 2—питательная тарелка; S—вал печи,
4—гребки; 5—отверстия в сводах; 6—течка; 7—приводной механизм.
6—2848
81
Воздух, необходимый для горения колчедана, засасывается
через отверстия («воздушники»), расположенные на уровне
нижнего свода. «Воздушники» имеют задвижки, с помощью
которых можно регулировать количество подаваемого в печь
воздуха.
Полый вал и гребки охлаждаются воздухом, для подачи
которого установлен вентилятор.
Нормальный режим работы печи ВХЗ характеризуется
следующими показателями:
Концентрация SO2 в газе на выходе из печи, % 10*
Температура газа на выходе из печи, °С 550—650
Наибольшая допустимая температура газа на третьем-четвер-
том сводах. °С 850—900
Температура воздуха, выходящего из вала печи, °С .... —200
Температура огарка, °С 450—550
Содержание серы в огарке, % 1,5—2,0
Содержание пыли в газе на выходе из печи, г/м3 10—15
* Концентрация SO-2 в обжиговом газе на выходе из печи должна быть
постоянной; на передовых заводах отклонения концентрации составляют ±0,1% SO-2.
Интенсивность печей ВХЗ в настоящее время доведена до
240 кг/м2 в сутки. При такой интенсивности в одной печи
может быть обожжено 32—35 т колчедана (с содержанием 45% S)
в сутки.
На ряде сернокислотных заводов полочные механические
печи реконструированы в печи с кипящим слоем.
Печи для обжига пылевидного колчедана
Такие печи применяются для сжигания флотационного
колчедана (величина зерен не более 0,05—0,07 мм). Для обжига
пылевидного колчедана во взвешенном состоянии применяются
два типа печей отечественной конструкции: цилиндрические и
прямоугольные печи с нижней или верхней подачей
колчедана.
Цилиндрическая печь с нижней подачей колчедана (рис. 21)
представляет собой высокую шахту диаметром 4,2 м, общей
высотой 10,8 м. Печь выложена из огнеупорного кирпича
(футеровка 2) и заключена в стальной кожух /. Взвесь колчедана
в воздухе подается в печь снизу форсункой 4; горение
колчедана происходит во всем объеме печи. Для более полного
сжигания колчедана в верхнюю часть печи дополнительно
подается воздух (вторичный воздух). Чтобы устранить налипание
частиц колчедана на верхний свод печи (образование
«козлов»), этот свод защищают экраном 5 из труб, охлаждаемых
водой.
82
Огарок удаляется из нижней конусной части печи.
Благодаря высокой температуре в середине печи
(по-видимому, выше 1000 °С) создаются благоприятные условия для
достаточно полного выгорания серы из колчедана.
¦ вшричньш
вазду.1
Шм/говый
газ
Рис. 21. Печь для обжига колчедана во
взвешенном состоянии с нижней подачей:
/—кожух; 2—футеровка; 3—бункер: 4—форсунка; 5—экран.
Режим работы печи для обжига колчедана во взвешенном
состоянии (влажность колчедана не более 3,8%)
характеризуется следующими примерными показателями:
Концентрация SO., в газе, "о 10—12
Температура газа на выходе из печи, °С 800—1000
Температура огарка, °С 700—800
Содержание серы в огарке, % 1,0—1.5
Содержание пыли в газе на выходе из печи, г/ж3 . . 20—60
6* 83
Важнейшим условием нормальной работы печи для
пылевидного обжига является равномерная подача колчедана и
воздуха и правильное соотношение их, что достигается при
автоматизации питания печей колчеданом.
Колебания концентрации SO2 в газе и температуры в печи
вызываются различным содержанием влаги в колчедане,
поступающем на сжигание.
В настоящее время печи для обжига пылевидного
колчедана снабжены автоматическими устройствами для
стабилизации содержания SO2 в печном газе.
Газоанализатор, контролирующий содержание SO2 в газе,
выходящем из печи, при отклонении концентрации от нормы
подает питателю импульс, вызывающий увеличение или
уменьшение подачи колчедана в печь.
Достоинствами печей пылевидного обжига являются:
1) большая производительность;
2) возможность получения газа с содержанием 10—12% SO^;
3) простота устройства;
4) быстрый пуск C—4 ч вместо 3—4 суток для механических
печей) и др.
Существенными недостатками таких печей являются:
1) высокая температура огарка;
2) большое содержание пыли в газе.
В установках, оборудованных печами пылевидного обжига,
тепло образующихся газов используется для получения пара.
Для этого устанавливают котлы-утилизаторы.
В результате исследований, проводившихся в течение
последнего времени, установлены преимущества печей с верхней
подачей пылевидного колчедана. Предложена конструкция печи
с центральным выводом обжигового газа, что позволяет
уменьшить его запыленность, повысить степень использования серы
и охлаждать огарок технологическим воздухом, подаваемым в
печь.
При этом упрощается механизация удаления огарка и
используется его физическое тепло, благодаря чему снижаются
теплопотери печи.
Интенсивность процесса обжига в подобных печах достигает
1200—1400 кг/м3 в сутки.
Удаление огарка
В настоящее время при сжигании колчедана на
сернокислотных заводах получаются огромные количества огарка (до
72—75% от веса колчедана). На современных заводах
применяются механизированные и автоматические способы
удаления огарка, что способствует улучшению санитарно-гигиени-
84
ческих условий труда. Наиболее разработанными в
конструктивном отношении являются способы охлаждения и удаления
огарка при помощи холодильных барабанов, холодильно-транс-
портных труб и холодильных шнеков.
По первому из названных способов огарок из печи
ссыпается во вращающийся барабан, орошаемый сверху водой и
закрытый кожухом. В барабане огарок охлаждается до 50—60 СС
и ссыпается на ленточный транспортер, который поднимает его
в бункер. Транспортер также закрыт специальным кожухом.
Из бункера огарок выгружают в железнодорожные вагоны и
удаляют с территории завода.
Такой способ охлаждения и удаления огарка целесообразно
применять при сжигании колчедана в печах ВХЗ и печах
пылевидного обжига.
Преимущества холодильных шнеков заключаются в
возможности одновременного увлажнения н транспортирования
огарка, а также в компактности и удобстве обслуживания. Ввод в
эксплуатацию шнековых устройств для охлаждения и
удаления огарка также значительно облегчает и улучшает условия
труда в печных отделениях.
На новых и реконструированных сернокислотных заводах
огарок из-под печи, парового котла и циклонов удаляется при
помощи конвейеров с погружными скребками.
Огарок должен быть увлажнен перед поступлением его на
ленточный транспортер, подающий огарок в бункеры.
Уменьшение загазованности и запыленности в печных отделениях
достигается путем тщательной герметизации шнеков, отвода пара
и др.
Использование огарка
Колчеданные огарки являются ценным материалом,
содержащим до 47—48% железа, а также небольшие количества
меди, серебра, золота, цинка, кобальта, таллия и других
элементов. Наличие меди в колчеданных огарках позволяет
использовать их в доменных печах для выплавки
высококачественного медистого чугуна. Для получения такого чугуна
огарки подвергают предварительному спеканию (агломерация),
нередко в смеси с железной рудой. В процессе агломерации из
колчеданных огарков удаляется сера. Агломерированный
огарок, представляющий собой кусковый материал с высоким
содержанием железа (до 60%), вполне пригоден для доменной
плавки.
Для извлечения из колчеданных огарков наряду с железом
остальных ценных элементов огарок подвергают комплексной
переработке путем хлорирующего обжига и другими методами.
Сущность метода хлорирующего обжига заключается в
нагревании при 850—900 °С смеси колчеданных огарков с 5—10%
85
поваренной соли. Полученную после обжига массу
выщелачивают, при этом водорастворимые хлориды меди переходят в
раствор, направляемый на дальнейшую переработку (путем
электролиза или др.) для выделения металлов. Наряду с
хлоридами металлов образуется также сульфат натрия.
Выщелоченный огарок после флотации содержит до 63% железа и
используется в доменном процессе.
На сернокислотных заводах к настоящему времени
скопилось большое количество колчеданного огарка. Для
переработки его методом хлорирующего обжига на одном из крупных
металлургических заводов строится мощная установка.
Кроме металлургии, огарки используются в цементной
промышленности, а также в сельском хозяйстве в качестве
удобрений, содержащих медь, особенно на низменных участках
торфянистых почв, богатых известью. Небольшие количества
колчеданного огарка могут быть использованы для получения
минеральных красок — сурика и мумии.
В настоящее время ведутся изыскания новых методов
комплексной переработки колчеданного огарка, которые
позволили бы более широко использовать его в народном хозяйстве.
ПОЛУЧЕНИЕ СЕРНИСТОГО ГАЗА СЖИГАНИЕМ СЕРЫ
Сжиганием серы получают сернистый газ, используемый в
производстве серной кислоты и в целлюлозно-бумажной
промышленности. Реакция
S+ O2 = SO2J- 17 ккал
протекает быстро и до конца. При сжигании чистой серы, не
содержащей ядовитых для катализатора примесей (мышьяк,
битумы и др.), получается газ, который может быть
использован непосредственно (без очистки) для производства серной
кислоты контактным методом.
Тепло, выделяющееся при сгорании серы, используется для
получения водяного пара.
Для сжигания серы применяются печи различных
конструкций. В настоящее время широкое распространение получили
печи, в которых сжигается распыленная сера.
Схема установки для сжигания серы в распыленном
состоянии изображена на рис. 22. Серу загружают в плавильник 1,
обогреваемый паровыми змеевиками (на рисунке не показано).
Расплавленная сера подается погружным насосом 3 в фильтр 4,
где она очищается от примесей, затем поступает во второе
отделение плавильника. Из этого отделения погружной насос 2
подает расплавленную серу через разбрызгивающие форсунки 5
в печь 6 на сжигание. Печь представляет собой стальной,
изнутри футерованный горизонтальный цилиндр. Для более пол-
86
ного соприкосновения серы с воздухом в печи имеются две
перегородки. Сера, сжигаемая в распыленном состоянии, должна
быть чистой.
Разбрызгивающие форсунки (распылители) для серы
изготовляют из жаростойкой стали.
Рис. 22. Схема установки для сжигания серы в распыленном
состоянии:
/—плавильник; 2, 3—погружные насосы; 4, 8—фильтры; 5—форсунка; б—печь; 7—
осушительная башня; 4—котел-утнлизатор.
Воздух, поступающий в печь, должен быть освобожден от
механических примесей и влаги.
Для этого его предварительно пропускают через фильтр 8
и осушительную башню 7, орошаемую концентрированной
серной кислотой.
Сернистый газ, получаемый в результате сжигания серы,
поступает в паровой котел-утилизатор 9 для использования
избыточного тепла и далее направляется непосредственно на
производство контактной серной кислоты по «короткой"
технологической схеме: печь — контактный аппарат — абсорбер
(стр. 122).
Распыленная сера, поступающая на сжигание, не должна
содержать органических примесей (углеводородов). В
противном случае при сжигании серы образуются пары воды,
которые, соединяясь с SO3, образуют туманообразную серную
кислоту, вызывающую нарушение нормального технологического
режима и коррозию аппаратуры.
Загрязненную бигумом серу можно также использовать в
производстве башенной серной кислоты (стр. 130 ел.) путем
сжигания в печах кипящего слоя. При этом исключается
необходимость предварительного плавления серы, ее
транспортирования и распыления в жидком виде. В качестве носителя
кипящего слоя могут оыть использованы песок или пиритные
огарки. Сера, попадая в кипящий слой, частично сгорает, а в
основном испаряется. Пары серы сгорают в объеме печи, окисляясь
87
воздухом, поступающим под решетку. Необходимая
температура в кипящем слое (850—900 °С) поддерживается путем
отвода тепла паровыми змеевиками, расположенными в зоне
кипящего слоя. Примеси, присутствующие в сере (зола, битумы),
а также песок (или пиритные огарки) уносятся из печи газом.
Перед поступлением в башенную систему газ, содержащий
13—14% SO2 и имеющий температуру 850—900 °С,
подвергается очистке от пыли в циклонах и разбавляется до концентрации
9—10% SO2.
МЕТОДЫ ОЧИСТКИ СЕРНИСТОГО ГАЗА ОТ ПЫЛИ
При сжигании колчедана с сернистым (печным) газом
уносится некоторая часть колчедана и огарка в виде пыли.
Содержание пыли в газе зависит от способа обжига колчедана и от
степени его измельчения (стр. 72).
Отложение пыли на поверхности катализатора в
контактных аппаратах ведет к понижению активности катализатора и,
следовательно, к понижению производительности аппаратов.
Тщательная очистка печного газа от пыли необходима также
во избежание загрязнения насадок промывных, денитрацион-
ных и абсорбционных башен, ухудшения тяги и загрязнения
готовой кислоты.
В сернокислотной промышленности применяют
механический и электрический методы очистки газа от пыли.
Механический метод очистки основан на способности
пылинок отделяться от газа под действием силы тяжести или
центробежной силы, а также при фильтрации газа через пористый
материал. При электрическом способе очистки пылинки
отделяются от газа под действием электростатического поля
высокого напряжения.
В обоих случаях газ очищают от пыли в горячем состоянии,
без предварительного охлаждения, что определяется условиями
дальнейшей переработки сернистого газа в серную кислоту.
Механическая очистка. В' качестве пылеочистительных
аппаратов в производстве серной кислоты применяют
центробежные пылеотделители — циклоны. Очистка в таких аппаратах
основана на выделении пылинок из газа под действием на них
центробежной силы, развивающейся при движении газа по
окружности.
Циклон (рис. 23) состоит из стального цилиндра 1,
оканчивающегося в нижней части коническим бункером 3. Внутрь
цилиндра / вставлен цилиндр 2. Газ, содержащий пыль,
поступает в циклон через трубу и проходит пространство между
цилиндрами. Совершая в кольцевом пространстве движение по
окружности, газ под действием центробежной силы
освобождается от пыли и уходит из аппарата через внутренний ци-
Очищенный
газ
т
Запылеи-^-i
ный газ \f—
/ /
г -"
1—
1
\
\
линдр -¦ Выделившаяся пыль собирается в коническом
бункере 3, откуда ее периодически выгружают.
Нормальная скорость газа в циклоне 7—20 м\сгк.
Циклоны применяются для предварительной очистки
печных газов от крупных частиц пыли; их используют также для
пылеулавливания в дробнлыю-раз-
мольном отделении и в отделении
сушки колчедана.
Электрическая очистка. Впервые
электрическая очистка газов от пыли
была применена в 1908 г. в цементной
промышленности. В настоящее время
этот метод нашел широкое
применение в сернокислотной,
металлургической, целлюлозной и других отраслях
промышленности.
Принцип работы электрофильтров
заключается в следующем.
Если отрицательный полюс
источника постоянного электрического тока
высокого напряжения присоединить к
тонкому металлическому проводу,
расположенному в центре трубы, по
которой проходит газ, то вокруг
провода создается электрическое поле.
В слое газа, окружающем провод,
происходит ионизация и возникает
характерное голубоватое свечение. Это
явление называется явлением
коронного разряда, а провод, вокруг
которого возникает коронный разряд,—
коронирующим электродом.
Образующиеся возле электрода ионы,
встречаясь с пылинками очищаемого газа,
заряжают их отрицательными
зарядами, одноименными с зарядом
провода. Поэтому заряженные пылинки
с большой скоростью отталкиваются
от центрального провода и попадают
на стенку заземленной трубы, потенциал которой равен нулю.
Отдавая свой заряд, пылинки оседают на стенки трубы и затем
под действием силы тяжести падают вниз.
Принципиальная схема действия электрофильтра
изображена на рис. 24. В центре трубы / расположен провод 2,
соединенный с отрицательным полюсом выпрямителя тока (на
рисунке не показан). Положительный полюс выпрямителя и
труба / заземлены. Газ подается в электрофильтр сбоку и, прохо-
Запылен- г
наш газ !
Рис. 23. Центробежный пы-
леотделитель (циклон):
/—внешний цилиндр;
2—внутренний цилиндр; 3—конический
бункер.
Постоянный ток
напряжения
Очищенный
'03
дя трубу снизу вверх, выходит очищенным из ее верхней части.
Пылинки, содержащиеся в газе, оседают на внутренней
поверхности трубы; при ее периодическом встряхивании пыль
ссыпается в бункер 3.
Для питания аппарата применяют выпрямленный ток
высокого напряжения F0 000 в и выше).
В производстве серной кислоты применяют сухие и мокрые
электрофильтры. Первые служат для удаления из газа пыли;
вторые — для осаждения примесей,
находящихся в газе в туманообраз*
ном состоянии.
Огарковый сетчатый
электрофильтр марки ХК-45 (рис. 25),
относящийся к сухим
электрофильтрам, представляет собой кирпичную
камеру 1 длиной 5,52 м, шириной
4,49 м и высотой 10,7 м. В нижней
части камеры имеется бункер 6.
Внутри камеры находится 18
сетчатых (осадительных) электродов 2,
заряженных положительно. Между
сетками подвешены рядами тонкие
коронирующие электроды 3, заря*
женные отрицательно.
Удаление пыли с сеток
производится встряхиванием их при
помощи механизма 4. Пыль ссыпается
в бункер 6 и периодически
выгружается из него.
При нормальной работе
электрофильтра содержание пыли в газе в результате очистки
уменьшается до 0.25 г\смъ (при скорости газа в активном сечении
0,5 м/сек).
Температура газов в электрофильтре 475—275°С.
Электрофильтр марки ХК-45 может обеспечить очистку газа,
полученного при сжигании 50—55 т 45%-ного колчедана в сутки (при
содержанки 7% SO2 в газе).
Мышьякозистый ангитряд п селен не отделяются в таком
электрофильтре, так как печные газы поступают на фильтр
горячими, а трехокись мышьяка и дзуокись селена при высокой
температуре находятся в парообразном состоянии. При
охлаждении газа до 30—35 °С содержащиеся в нем соединения
мышьяка и селена прекращаются в туман и полностью
удаляются из газа при пропускании его через так называемый мокрый
электрофильтр. Такие электрофильтры, применяемые в
контактном способе производства серной кислоты, названы мокрыми
потому, что охлажденные газы, очищаемые от
мышьяковистоРис. 24. Принципиальная схема
действия электрофильтра:
/—труба; 2—провод; 3— бункер.
90
го ангидрида и двуокиси селена, предварительно увлажняют в
промывных башнях.
В последние годы на сернокислотных заводах большое
применение получили многопольные электрофильтры, обеспечиваю-
Очищепнш
газ
Рис. 25. Огарковый электрофильтр:
/—камера- 2—сетчатые осадигельные электроды; .?—проволочные коронн-
рующие электроды; 4—встряхивающий механизм; 5—газораспределительная
решетка; 5—бункер.
щие более полную очистку газа от пыли. В этих электофильт-
рах очищаемый газ последовательно проходит в
горизонтальном направлении через три или четыре секции с
самостоятельным электрическим полем в каждой секции. В огличне от
описанного электрофильтра (ХК-45), многопольные
электрофильтры оборудованы устройством для механизированного удаления
пыли с электродов.
91
Рис. 26. Схема очистки газа, получаемого при сжигании колчедана в печах
кипящего слоя:
У—печь; .2—бункер для колчедана; 3—котел-утилизатор; 4—циклоны; 5—электрофильтры; 6—
воздуходувка; 7—скребковые конвейеры.
Очистка газа от пыли на установках, оборудованных печами
КС, осуществляется по схеме, показанной на рис. 26. Для
очистки газа применены
горизонтальные электрофильтры с
тремя или четырьмя полями.
Пластинчатые осадительные
электроды изготовлены из
стали и снабжены
приспособлениями для механического
встряхивания; коронирующие
электроды, выполненные из
нихромовых проволочек
диаметром 2 лш, снабжены
вибрационным встряхивателем.
Удаление пыли с
газораспределительных решеток
механизировано. Корпуса электродов и
бункеры — стальные, без
внутренней футеровки, но
с наружной теплоизоляцией.
Для грубой очистки газа от
пыля перед электрофильтрами
устанавливается группа
циклонов типа ЦН-15 диаметром
Рис. 27. Секционный газотрубный
котел-утилизатор к печи КС-100;
.'—секции труб; 2—барабаи-сепаратор;
J—перегородка (ложная секция).
92
от 600 до 800 мм. Содержание пыли в газе после очистки в
циклонах снижается с 300 до 20 г\мъ.
При скорости газа в активном сечении электрофильтра до
0,6 м/сек газ очищается в трехпольных электрофильтрах до
содержания пыли ОД—0,2 г/м3, в четырехпольных
электрофильтрах — до 0,05—0,1 г,'м3.
Для использования тепла очищаемого печного газа
применяются котлы-утилизаторы различных конструкций.
На рис. 27 изображен секционный газотрубный
котел-утилизатор к печи КС-100. Для получения пара используется вода
(около 100 °С), поступающая в котел из охлаждающих трубок,
расположенных в кипящем слое печи. Дополнительное
количество воды поступает в котел из сети. Трубки котла двойные
(труба в трубе). По наружным трубкам движется вода, по
внутренним — горячий газ (~850°С) из печи КС. Газ огибает
ложную секцию и по выходе из трубок поступает на очистку
от пыли.
Часовая производительность котла — 7,3 т насыщенного пара
B55°С) или 4,8—5,3 т перегретого пара (до 450 °С).
Поверхность нагрева котла 258 м2, диаметр 3 м, общая
высота 14,5 м, диаметр барабана 1,58 м.
3. КОНТАКТНЫЙ МЕТОД ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
Контактный метод призводства серной кислоты был
предложен в 1831 г. и впервые реализован в промышленности в
1875 г. Применение контактного метода в его современном
виде стало возможно лишь после того, как были установлены
оптимальные условия окисления SO2 в SO3 и найдены причины
отравления катализатора. Широкое распространение
контактный метод получил только в конце прошлого столетия, когда
в связи с развитием промышленности синтетических красителей
и взрывчатых веществ, возросшим потреблением серной
кислоты для очистки нефти и для других нужд возникла потребность
в больших количествах концентрированной серной кислоты.
В дореволюционной России производство серной кислоты
контактным методом впервые было осуществлено в 1903 г. на
Тентелевском заводе*, где был самостоятельно освоен
контактный метод, получивший широкую известность под названием
тентелевского. Этот метод затем был реализован в США,
Японии и многих других странах.
Важным этапом в развитии контактного метода была
замена в 20—30-х годах XX в. первоначально применявшихся
платиновых катализаторов значительно более дешевыми
ванадиевыми контактными массами.
* Ныне завод «Красный химик» в Ленинграде.
93
Быстрое развитие контактного метода производства
объясняется возможностью получения чистой концентрированной
серной кислоты и олеума—продуктов, имеющих большое
промышленное значение. По контактному методу получается более
чистая серная кислота, чем башенная, так как в этом случае
сернистый газ подвергается очень тщательной очистке от вредных
примесей мышьяка и селена, отравляющих катализатор.
Очистка сернистого газа требует большой затраты
электроэнергии и воды, а также громоздкой аппаратуры. Степень
использования серы в исходном сырье при контактном методе
несколько меньше, чем при нитрозном. Расход колчедана (в
пересчете на 45% S) на 1 т башенной кислоты составляет
790—815 кг, на 1 т контактной кислоты достигает 830—840 кг.
Однако общие затраты на выработку 1 т контактной кислоты
всего на несколько процентов выше, чем на 1 т башенной
кислоты.
Дальнейшее улучшение технологических показателей
контактного процесса и снижение себестоимости контактной
серной кислоты может быть достигнуто путем упрощения методов
очистки газа и абсорбции трехокиси серы и дальнейшего
укрупнения контактных агрегатов, так как это позволяет снизить
затраты на строительство и эксплуатацию сернокислотных систем.
Технологические схемы производства контактной серной
кислоты несколько различаются по аппаратурному
оформлению, однако сущность процесса всегда одинакова и сводится
к следующему. Сначала двуокись серы SO2 окисляется в трех-
окись серы $Оз в присутствии твердого катализатора:
2SO2 + О2 = 2SO3
Затем трехокись серы поглощается серной кислотой.
ПРИНЦИПИАЛЬНАЯ СХЕМА ПРОИЗВОДСТВА
Производство серной кислоты контактным способом состоит
из следующих основных стадий: 1) очистка газа от примесей,
вредных для процесса контактного окисления SO2 в SO3;
2) окисление SO^ в SO3 на поверхности твердого катализатора;
3) поглощение трехокиси серы серной кислотой с получением
концентрированной серной кислоты и олеума.
Отличия между отдельными технологическими схемами
заключаются в способах очистки газов от вредных примесей
(мышьяк, селен и др.), в применяемых катализаторах и их
носителях, а также в конструктивных особенностях отдельных
аппаратов и в технологическом режиме.
На рис. 28 изображена принципиальная схема контактного
цеха, работающего на колчедане. Обжиговый газ C50—400 °С)
через огарковый электрофильтр / поступает в промывную баш-
94
ню 2, где охлаждается примерно до 80 °С орошающей башню
60—70% -ной серной кислотой. Кислота, вытекающая из баш»
ни, охлаждается в водяном оросительном холодильнике 8 и
снова перекачивается на орошение башни 2. Из первой про-
мывпой бчшни газ поступает во вторую промывную башню 3,
Нислота
на склад
10
II К
Отходящие
топочные
газы
-С
8
Е
j
-
V
л
\i
I .Г
Таз в
5 f—I Хатмо-
P\U| ctpepy
/в
Олеум
на склад
Рис. 28. Принципиальная схема производства серной кислоты контактным
методом:
¦/—огарковый электрофильтр; 2, 3—промывные башни; 4, 6—мокрые электрофильтры; 5—
увлажнительная башня; 7—сушильная башня; 8—оросительные холодильники; 9—сборники;
10—турбокомпрессор; 11—фильтр; 12—пусковой подогреватель; 13—теплообменник;
14—контактный аппарат; 15—трубчатый холодильник; 16—олеумная башня; 17—кислотная башня;
18—башня для получения солей аммония; 19—брызгоотделитель.
орошаемую 30%-ной серной кислотой, где дополнительно
охлаждается примерно до 30 °С. Вытекающая из башки 3
кислота, пройдя холодильник 8, вновь подается на орошение этой
б?шч:'
В промывных башнях газ освобождается от остатков пыли;
здесь из трехокпсн серы, всегда присутствующей в обжиговом
газе, образуются также пары серной кислоты. При
конденсации паров в обьеме кислота переходит в туманообразное
состояние. Трехокись мышьяка при охлаждении газа переходит
непосредственно в твердое состояние. Туманообразная серная
кислота и трехокись мышьяка только частично улавливаются
в промывных башнях.
95
Для окончательной очистки газа от туманообразной серной
кислоты, соединений мышьяка и селена газ последовательно
пропускают через мокрые электрофильтры 4 и 6. Перед
вторым электрофильтром б газ увлажняется в башне 5, орошаемой
5%-ной серной кислотой. Благодаря увлажнению мелкие
частицы тумана, не уловленные в электрофильтре 4, укрупняются и
полностью оседают,в электрофильтре 6.
Кислота, вытекающая из увлажнительной башни,
охлаждается в холодильнике 8 и снова подается на орошение башни. Из
электрофильтра 6 очищенный газ при температуре около 30 °С
направляется на осушку от влаги в башню 7, орошаемую
концентрированной серной кислотой (93—-95%-ной)*. После
осушки газ должен содержать не более 0,15 г/м3 водяного пара.
Кислота из башни 7, пройдя холодильник 8, снова направляется на
орошение башни. Часть кислоты отводится на склад.
Очищенный сухой газ турбокомпрессором 10 через фильтр 11
подается в теплообменник 13, где подогревается горячими
газами, выходящими из контактного аппарата 14. "Подогретый
газ из теплообменника 13 поступает в контактный аппарат, где
двуокись серы окисляется до трехокиси серы на 97—98,5% и
более**.
В период пуска системы газ предварительно нагревается в
подогревателе 12 топочными газами, поступающими из
расположенной рядом топки (на схеме не показана).
Горячий газ, выходящий из контактного аппарата, в
теплообменнике 13 отдает тепло свежему газу, подаваемому
турбокомпрессором, далее поступает в водяной трубчатый
холодильник 15, где охлаждается до 60 °С, а затем направляется на
абсорбцию.
Абсорбция SO3 происходит в двух последовательно
расположенных башнях 16 и 17. Первая по ходу газа башня 16
орошается олеумом, содержащим 18,5—20% SO3 (своб.). Вторая
башня 17 орошается серной кислотой (98,3% H2SO4). Серная
кислота и олеум, вытекающие из башен 16 и 17, охлаждаются в
водяных оросительных холодильниках 8 и вновь подаются на
орошение башен. Часть олеума отводится на склад. В башнях
16 и 17 улавливается до 99,9% SO3.
Отработанные газы, содержащие остатки двуокиси серы,
перед удалением в атмосферу целесообразно пропускать через
специальную башню 18, снабженную насадкой и орошаемую
аммиачной водой.
* Над растворами серной кислоты такой концентрации давление паров
воды очень мало.
** В пятислойных контактных аппаратах увеличивается степень
окисления SO2 в SO3, что позволяет снизить содержание сернистого газа в
отходящих газах
96
В этой башне образуются растворы солей аммония:
сульфита аммония (NH4):>SO3, бисульфита аммония NH4HSO3 и
сульфата аммония (NH4JSO4.
Полученные в башне 18 растворы солей аммония могут быть
использованы для получения 100%-ной двуокиси серы (путем
отгонки SO2 из раствора паром) или переданы на переработку
в другие химические производства.
ОЧИСТКА СЕРНИСТОГО ГАЗА
Степень очистки обжигового газа, достигаемая в огарковом
электрофильтре, недостаточна для контактного способа
производства серной кислоты.
После обычной очистки от
пыли здесь необходима
специальная тщательная
очистка от остатков пыли
и примесей, которые
отравляют катализаторы,
применяемые в
контактном процессе. Такими
вредными примесями
являются соединения
мышьяка и селена, тумано-
образная серная кислота,
влага, брызги масла (из
турбокомпрессора).
При контактном
способе производства серной
кислоты специальная
очистка газа проводится в
промывных башнях и
мокрых электрофильтрах.
Для полноты очистки
перед вторым по ходу газа
электрофильтром
устанавливают
увлажнительную башню.
Осушку газа
производят в сушильной башне,
орошаемой 93—95%-ной
серной кислотой.
Первая промывная
башня (рис. 29)
представляет собой полый
стальной цилиндр 1,
футерованный кислотоупорным
7—2848
Рис. 29. Первая промывная башня:
/—стальной корпус; 2—распылители одинарные; 3—
распылители двойные; 4—коробка со штуцером для
выхода кислоты; 5—желоб для спуска шлама;
6—коробка для ввода кислоты; 7—футеровка.
97
Нислотя
кирпичом. Горячий газ C50—400 °С) входит в башню снизу
и выходит из нее сверху при температуре около 80 °С.
Разбрызгивание серной кислоты F0—70%-ной) производится с
помощью центробежных распылителей. Верхние (одинарные)
распылители 2, вставленные под крышкой башни, разбрызгивают
кислоту только вниз.
Расположенные ниже двойные распылители 3 разбрызгивают
ее вниз и вверх. Кислота выходит из промывной башни при
50—60 °С через коробку 4 со штуцером; шлам (огарковая пыль
и др.) удаляется через желоб 5.
Размеры башни,
рассчитанной на прохождение через нее
50 тыс. М3/ч газа: диаметр 4,5 м,
общая высота 12—14 м.
Плотность орошения в этой
башне 10—15 м3/ч на 1 м2 ее се-
ченля. При охлаждении газа в
первой промывной башне
одновременно с конденсацией паров
серной кислоты улавливается
некоторое количество мышьяка и
селена, а также оставшаяся
пыль.
Мышьяк и селен частично
осаждаются в виде шлама в
холодильниках, а часть их
растворяется в кислоте.
Вторая промывная башня
(рис. 30) заполнена насадкой из
шин
Кислота керамических или фарфоровых
колец. В нижней части башни
уложены крупные кольца A00—
Рис. 30. Вторая промывная башня: j20 мм), выше — более мел-
?—корпус; 2—футеровка; 3—насадка (кольца); „ир (СП on м м\ I/nnf,Tin nfiuuun
4—распылители; 5—колосниковая решетка. КИе Vou ои ММ). 1\ОЛЬЦЭ ООЫЧНО
укладывают правильными
рядами в шахматном порядке. В
верхней части башни имеется пустое пространство высотой около
1 м, в котором расположены устройства для распыления
орошающей кислоты.
Во второй промывной башне газ охлаждается.
Одновременно с охлаждением газа в башне частично абсорбируется тума-
нообразная серная кислота, мышьяк и селен, не поглощенные
в первой промывной башне.
Башня для переработки 50 тыс. м3/ч, газа имеет общую
высоту 12—14 м, диаметр башни 5,5 м.
Плотность орошения башни составляет 6—10 м3/ч на 1 м2
ее сечения.
98
Мокрые электрофильтры служат для очистки газа от брызг
и тумана серной кислоты, трехокиси мышьяка, двуокиси селена
и тонкой огарковой пыли. Количество примесей зависит от
состава сырья, способа его сжигания, температурного режима
промывной башни и концентрации орошающих кислот.
Высокая степень очистки достигается при проведении ее в две
ступени. Очищенный газ должен содержать тумана серной
кислоты не более 0,005 г/м3, мышьяк и селен должны отсутствовать
в газе.
Очистка газа в мокрых электрофильтрах основана на том
же принципе, что и очистка его от пыли в огарковых (сухих)
электрофильтрах. В отличие от сухих электрофильтров корони-
рующие электроды мокрых электрофильтров изготовляются из
стальных освинцованных проводов, осадительные электроды
обычно представляют собой свинцовые шестигранные трубы.
Установка для очистки газа состоит из увлажнительной
башни* и двух электрофильтров (двухсекционного типа). В первой
ступени очистки температуру газа обычно поддерживают в
пределах 30—35 °С, во второй — не выше 25—30 °С. Скорость
газа 1 м/сек.
Двухсекционные свинцовые аппараты типа М
устанавливают при разрежении в системе до 300 мм вод. ст. На открытых
площадках при разрежении в системе до 500 мм вод. ст.
устанавливают односекционные электрофильтры типа ШМК с
шестигранными свинцовыми электродами. Стальные
цилиндрические корпуса электрофильтров изнутри защищены от коррозии
двумя слоями полиизобутилена и кислотостойкой футеровкой.
При установке электрофильтров на открытых площадках
наружную поверхность корпуса покрывают слоем теплоизоляции,
а стоки кислоты снабжают электроподогревом. Разработаны
конструкции фильтров типа ШМК с активной площадью
сечения от 2,2 до 6,6 м2.
Основным показателем нормальной работы мокрых
электрофильтров является отсутствие сернокислотного тумана в
очищенном газе. Для своевременного обнаружения тумана серной
кислоты в газе устанавливают туманомеры (стр. 161).
ОСУШКА СЕРНИСТОГО ГАЗА
Осушка газа производится в башне, орошаемой 93—95%-ной
серной кислотой.
Сушильная башня (рис. 31) представляет собой стальной
цилиндр 1, футерованный кислотоупорным кирпичом 2, с
днищем 8 и сферической (или плоской) крышкой 5. В нижней части
башни расположена колосниковая решетка 3. В крышке раз-
* Применение увлажнительной башни необязательно.
7* 99
Рис/ 31. Сушильная башня:
/—стальной цилиндрический корпус; 2—футеровка: 3—колосниковая
решетка; ч4—смотровое стекло; 5—крышка; 6—распылитель кислоты; 7—лаз для
загрузки; S—днище.
мещены распылители 6 кислоты, орошающей насадку, и
штуцер для выхода газа. Для равномерного орошения
устанавливается от 8 до 12 распылителей* в зависимости от диаметра
* В настоящее время для распиливания орошающей кислоты з башнях
применяются также устройства других конструкций — оросительные плиты
со стаканчиками, оросительные желоба и др.
100
башни. В верхней части башни имеется лаз 7 для загрузки и
выгрузки н&садки и два стекла 4 для освещения и осмотра.
Башня заполнена керамическими (или фарфоровыми)
кольцами.
Плотность орошения насадки составляет от 6 до 10 м5/ч на
1 м2 сечения башни. После осушки газ должен содержать не
более 0,15 г/м3 влаги, т. е. не выше 0,02 объемн. % Н2О. Для
достижения такой степени осушки газа необходимо
непрерывное и равномерное орошение насадки 93—95%-ной кислотой.
Концентрация кислоты, вытекающей из башни, должна быть
на 0,3—0,5% H2SO4 ниже концентрации орошающей кислоты.
Для постоянного поддержания требуемой концентрации
орошающей кислоты в сборник сушильной кислоты непрерывно
добавляется концентрированная серная кислота, избыток
сушильной кислоты одновременно перекачивается насосом в
абсорбционное отделение. Температура кислоты на входе в
башню 30—40 °С, после башни 45—50 °С.
Чаще устанавливают последовательно две сушильные
башни, реже одну.
При двух последовательно включенных башнях первую из
них орошают 90—93%-ной, вторую 94—95%-ной серной
кислотой.
Сопротивление аппаратуры очистного отделения колеблется
в широких пределах (от 100 до 500 мм вод. ст.) в зависимости
от типа аппаратов, состояния насадки башен и количества
проходящего газа.
Регулирование концентрации газа. Оптимальная
концентрация двуокиси серы в газе, поступающем на
контактирование, составляет 7,3±0,2%- Газы, получаемые после
сжигания серного сырья, содержат от 9 до 15% SO2. Чтобы не
увеличивалось гидравлическое сопротивление аппаратуры
очистного отделения, сохраняют указанную концентрацию SO2 в
газе, а перед сушильными башнями разбавляют газ
атмосферным воздухом, открывая задвижку на газоходе.
Извлечение селена из сернистого газа
Среднее содержание селена в рядовом серном колчедане составляет
80—100 г/г, во флотационном 45—50 г/т. Практически установлено, что при
обжиге колчедана в механических печах содержание селена в огарке
составляет 20 г/7, при обжиге флотационного колчедана в таких же печах 10 г/т.
Остальное количество селена поступает в виде SeO2 вместе с печными
газами в очистное отделение (в контактных системах) и улавливается в
промывных башнях и электрофильтрах в виде шлама. Шлам периодически
отбирается раздельно из отстойников первых промывных башен и
электрофильтров. Процесс извлечения селена из шлама промывной кислоты
заключается в следующем. Промывную кислоту, содержащую взвесь селенового
шлама и растворенные в ней соединения селена (SeCb и др.), нагревают
острым паром до 90—100 °С с одновременной обработкой сернистым газом
101
для восстановления селена из его соединении. Этот процесс протекает полнее
в присутствии добавок-восстановителей (например, сульфата двухвалентного
железа).
Образующуюся при этом пульпу отфильтровывают. Фильтрат
(промывная кислота) передается на склад для использования, остаток на
фильтре — шлам, содержащий селен; промывают водой, нейтрализуют 5%-ным
раствором соды и высушивают в сушильных шкафах на противнях.
Высушенный селеновый шлам упаковывают и отправляют на централизованную
переработку для извлечения элементарного селена.
Примерно также извлекается селеновый шлам из конденсата мокрых
электрофильтров. При очередной чистке и пропарке их шлам смывается с
электродов и стенок электрофильтра циркулирующим при помощи насоса
конденсатом. Затем конденсат отфильтровывают. Фильтрат (слабая
промывная кислота) передается в сборник второй промывной башни, осадок
промывают, нейтрализуют, высушивают, упаковывают и отправляют на"
централизованную переработку.
КОНТАКТНОЕ ОКИСЛЕНИЕ SO2 в SO3
Реакция окисления двуокиси серы в трехокись
2SOa + О2 = 2SO3 + Q
протекает с уменьшением объема и с выделением тепла.
Равновесный выход трехокиси серы зависит от состава исходной
газовой смеси, температуры и давления. С понижением
температуры и повышением давления равновесие реакции
смещается в правую сторону.
Для достижения наибольшей степени окисления двуокиси
серы в трехокись серы применяют газ с содержанием 7—7,5%
SO2.
Количество выделяющегося тепла зависит от температуры
реакции:
Температура, °С 25 450 600
Теплота реакции, кал/моль .... 22989 22657 22335
В интервале 400—700 °С тепловой эффект реакции
окисления SO2 может быть подсчитан с достаточной точностью по
уравнению
Q = 24205 — 2.21Г
где Q —¦ тепловой эффект реакции, кал;
Т—абсолютная температура.
Равновесный выход SO3. Для газовой смеси, имеющей
состав 7% SO2, 11% О2 и 82% N2, расчетная равновесная
степень окисления двуокиси серы при 475 °С равна 95,8%. Более
высокие выходы трехокиси серы достигаются при разбавлении
печных газов, так как степень окисления SO2 в SO3 (при поо-
чих равных условиях) возрастает с увеличением содержания
кислорода и уменьшением содержания SO2 в исходной
газовой смеси.
102
Состав исходной газовой смеси влияет на равновесную
степень окисления SO2 в SO3 (при атмосферном давлении и
475 °С) следующим образом:
Содержание SO2, объемн. % 5 6 7 8 9 10
Содержание О2, объемн. % 13,86 12,43 11,0 9,58 8,15 6,72
Равновесная степень
окисления SOa в SO3, % . . . . 96,5 96,2 95,8 95,2 94,3 92,3
Таким образом, оптимальный состав газовой смеси должен
соответствовать 7—7,5% SO2, 11% О2, 82% N2. При более
высоком содержании SO2 значительно уменьшается
равновесный выход SO3 (вследствие уменьшения содержания О2 в
газе); при меньшем содержании SO2 понижается
производительность контактной системы. Поступающий в контактный
аппарат газ, содержащий 7—7,5% SO2 при установившемся
процессе предварительно подогревается до требуемой
температуры. Для нагревания используется тепло
прореагировавших газов, выходящих из контактного аппарата. В этом случае
процесс окисления SCh в SO3 протекает аутотермично, т. е.
без подвода тепла извне, за счет тепла реакции.
Температура. С понижением температуры равновесие
реакции окисления SO2 в SO3 смещается в сторону
образования трехокиси серы. Если известен состав исходной
газовой смеси, поступающей на контактирование, т. е. содержание
в ней SO2 и О2, можно вычислить состав газа для
равновесного состояния при различных температурах реакции.
Влияние температуры на состояние равновесия для
исходной газовой смеси состава 7% SO2, 11% О2 и 82% N2 при
атмосферном давлении сказывается следующим образом
(расчетные данные):
Температура, "С . . . 400 450 475 500 530 550 600 1000
Степень окисления SO3
в SO3,% 99,2 97,5 95,8 93,5 89,2 85,6 73,7 5,6
На рис. 32 показано, как смещается равновесие реакции
окисления SO2 с повышением температуры и при прочих
равных условиях.
Из приведенных данных следует, что для достижения
высокой степени окисления SO2 в SO3 необходимо вести процесс
при возможно более низкой температуре.
На практике для достижения наиболее высокой степени
окисления (до 98%) стремятся проводить реакцию, по
крайней мере к моменту ее завершения, при температуре ниже
440 °С.
При температурах ниже 400°С равновесная степень
окисления SO2 в SO3 близка к 100%, но при этом скорость реак-
103
ции окисления, даже в присутствии катализатора, очень мала,
что невыгодно для ведения процесса в промышленных
условиях. ,
/00
80
60
\
\
ч
(Л
40ff S00 600 700 300 900 /000
Температура, °С
Рис. 32. Смещение равновесия
реакции 2SOa+O.,^t:2SO3 с повышением
температуры (атмосферное давление;
состав исходной смеси: 7% SO2, 11%
О2, 82% N2).
• /00
95
w 85
&s Я О
* 75
7П
/
/
/
у'
1
1
1
i
i
=
/7 .
О /О 20 30 40 SO 60
Давление, am
Рис. 33. Смещение равновесия
реакции 2SO2+O2^?2SO3 с
повышением давления F00 °С;
исходная смесь содержит 7% SO2,
11% О3, 82% N,).
Давление. Зависимость равновесной степени окисления
SO2 в SO3 от давления при различной температуре для газа,
содержащего 7% SO2, 11% О2 и 82% N2, показана в табл. 6
и на рис. 33.
Таблица 6
Зависимость равновесной степени окисления SO2 в SO3 от давления
Температура, °С
400
450
500,
600
Равновесная степень окнслення SO-з в 5Оз- %
1 am
99,2
97,5
93,5
73,7
10 am
99,72
99,2
97,8
89,5
50 am
99,88
99,6
99,0
95,0
100 am
99,92
99,7
99,3
96,4
Из приведенных данных видно, что при низких
температурах повышение давления сказывается крайне незначительно
на степени окисления; при высоких же температурах
повышение давления значительно расширяет температурный интервал,
в котором можно достигнуть высокой степени окисления.
Поскольку при атмосферном давлении и 450 °С равновесная
104
степень окисления SO2 в SO3 теоретически может достичь
97,5%. то необходимость в применении давления отпадает.
Небольшое увеличение степени окисления с повышением
давления не компенсирует затрат, связанных с удорожанием
аппаратуры и коммуникаций и увеличением расхода энергии
на сжатие газа.
Скорость реакции окисления SO2 в SO3. Скорость этой
реакции зависит от применяемого катализатора (в отсутствие
катализатора реакция практически не идет), начального
состава газа, температуры контактирования. Кроме того,
скорость этой реакции зависит от степени окисления SO2 в SO3—
чем выше становится концентрация SO3 в газовой смеси, тем
медленнее протекает реакция.
Катализаторы
Окисление SO2 в SO3 является гетерогенно-каталитической
реакцией. Независимо от свойств катализатора можно считать,
что эта реакция протекает в несколько последовательных
стадий:
1) адсорбция кислорода из газа поверхностью
катализатора; при этом связь между атомами адсорбированного
кислорода нарушается (активированная адсорбция);
2) связывание молекул двуокиси серы с атомами
кислорода на поверхности катализатора;
3) перегруппировка, приводящая к образованию на
поверхности катализатора групп SO3:
SO2 • О • катализатор * SO3-катализатор
4) удаление (десорбция) образовавшихся молекул SO3 с
поверхности катализатора в газовый объем.
Раньше в сернокислотной промышленности в качестве
катализатора повсеместно применялась очень тонко
измельченная платина, нанесенная на пористые инертные материалы —
«носители» (волокнистый асбест, сернокислый магний, гель
кремнекислоты). На некоторых заводах в качестве
катализатора применяли окись железа. В настоящее время все заводы
работают на ванадиевых катализаторах*.
Платина является наиболее активным катализатором, на
котором реакция идет уже при 400 °С. В присутствии окиси
железа реакция протекает достаточно быстро только при
температурах выше 600 °С. Ванадиевые катализаторы по
активности несколько уступают платиновым, но более дешевы и
* Ванадиевые катализаторы введены в СССР в практику на основе
работ Г. К. Борескова и других советских ученых.
105
менее чувствительны к действию примесей в газовой смеси
(ядов), снижающих активность катализаторов.
Чистая пятиокись ванадия V2O5 обладает невысокой
каталитической активностью е отношении реакции окисления SO»
в SO3.
Рис. 34. Формы частиц ванадиевой контактной массы:
а—таблетки; б, в—гранулы; г—кольца; д—гранулы для кипящего слоя.
Активные ванадиевые катализаторы Есегда содержат
наряду с пятиокисью ванадия и другие соединения.
Каталитически активными соединениями в промышленных
ванадиевых катализаторах являются сульфованадаты щелочных
металлов (например, КгО • V2O5 • SO3), нанесенные на
поверхность двуокиси кремния. На отечественных заводах применяют
бариево-алюмованадиевые (БАВ) контактные массы в Еиде
белых или розоватых таблеток, гранул, колец (рис. 34).
Контактную массу предварительно обрабатывают 0,3—0,5%-ным
SO2, так как действие на нее обжигового газа, содержащего
7—7,5% SO2, сопровождается значительным выделением тепла,
что может вызвать спекание и потерю активности катализатора.
После такой обработки контактная масса приобретает желтый
цвет, насыпкой вес ее возрастает до 0,60—0.65 г/см3, истинная
плотность — до 2,72 г/см3.
Катализатор СВД (сульфо-ванадиевый на доломите) не
требует предварительного насыщения сернистым газом, обладает
достаточной механической прочностью, высокой активностью,
термической устойчивостью и относительно низкой
температурой зажигания (стр. 108) на входе газа — от 410 до 415 СС.
106
Процесс изготовления катализатора СВД несложен.
Высушенную инфузорную землю смешивают с гипсом и
размалывают в шаровой мельнице. Предварительно измельченную
пятиокнсь ванадия смешивают с раствором бисульфата
калия. Исходные компоненты — смесь инфузорной земли с
гипсом и пульпа пятиокиси ванадия (в растворе бисульфата
калия) тщательно перемешивают и гранулируют, после чего
прокаливают при температуре 500 °С. Охлажденные после
прокаливания гранулы подвергают грохочению и размолу.
Отсев возвращается на операцию перемешивания и
гранулирования.
Наиболее рационально приготовление катализаторов,
несколько различающихся по составу и оптимальных для
различных температур и степеней окисления SO2 в SO3. Для
первой половины верхнего слоя катализатора его оптимальный
состав 4% V2O5.
В нижнюю часть первого слоя целесообразно загружать
катализатор, содержащий 6% V2O5.
Для остальных слоев контактной массы оптимальный состав
катализатора должен соответствовать также примерно 6%
V2O5.
Степень ускорения реакции, вызываемого данным
катализатором, характеризует его активность. Кроме того, в технике
учитывают производительность катализатора, под которой
подразумевают весовое количество продукта, полученное в час
при прохождении газа через катализатор с данной объемной
скоростью.
Объемной скоростью называется объем газовой смеси (в
л3)*, прошедший через 1 Л13 контактной массы за 1 ч (мг/мг • ч,
или <Н). Так, при объемной скорости 1000 ч-1 через 1 мг
катализатора в течение 1 ч должно быть пропущено 1000 м3 газа.
Чем активнее катализатор и чем выше его
производительность, тем большее количество SO2 можно окислить в SO3 на
одном и том же количестве катализатора (при прочих равных
условиях).
Активность ванадиевых катализаторов проявляется в
довольно узком интервале температур. Нижняя температурная
граница активности ванадиевых катализаторов БАВ D30—
440 °С) обусловлена превращением при этой температуре
активного ванадиевого соединения, содержащего
пятивалентный ванадий, в неактивное — сульфат ванадила:
V2O5 + SO2 + SOS = 2VOSO4
* Имеется в виду объем газовой смеси, приведенный к нормальным
условиям (О "С и 760 мм рт. ст.).
107
При высокой концентрации SO3 (т. е. когда уже большая
часть SO2 окислилась) сульфат ванадила может
образоваться при температуре ниже 380 °С.
Верхняя граница активности ванадиевого катализатора
БАВ 600—620 "С. Активность катализатора БАВ, работающего
при этой температуре, постепенно снижается из-за нарушения
его структуры и уменьшения пористости (см. ниже). Снижение
активности ванадиевых катализаторов после длительной
эксплуатации при высокой температуре может быть связано
также со спеканием контактной массы вследствие загрязнения
ее сульфатом железа, образующимся в результате коррозии
аппаратуры.
Помимо активности катализатора, пригодность его в
производственных условиях определяется также минимальной
температурой газа, при которой возможен быстрый разогрев
и дальнейшее аутотермическое течение реакции. Эту
минимальную температуру входящего газа принято называть
температурой зажигания. Для газа, содержащего 7% SO2 и
11% Ог, температуру зажигания на входе в слой катализатора
СВД 410—415 °С; для ванадиевого катализатора БАВ
температура зажигания выше D30—440 °С). При одновременном
повышении концентрации двуокиси серы и кислорода температура
зажигания снижается.
При уменьшении же концентрации кислорода в газе
температура зажигания возрастает.
Качество катализатора определяет важнейшие показатели
технологического процесса: его интенсивность, выход продукта,
длительность непрерывной работы установки, расход энергии
на преодоление гидравлического сопротивления и др. Для
уменьшения гидравлического сопротивления при
конструировании аппаратов стремятся создать слой контактной массы
наименьшей высоты, газ должен проходить через этот слой с
минимальной скоростью. Сопротивление слоя катализатора
газовому потоку в значительной степени зависит от размера его
зерен. В слое из более крупных зерен создается меньшее
сопротивление.
Каталитическая активность зависит также от величины
внутренней поверхности катализатора и ее доступности для
реагирующих веществ (в данном случае SO? и Ог), что
определяется пористостью катализатора. Современные
промышленные катализаторы, применяемые в производстве серной
кислоты и других продуктов, отличаются весьма развитой
поверхностью.
Для снижения гидравлического сопротивления слоя
катализатора и увеличения его активной поверхности
целесообразно применять контактную массу в виде колец (см. рис. 34)
вместо мелкозернистого и гранулированного катализатора.
108
В последние годы контактная масса кольцеобразной формы
получила широкое распространение.
Режим окисления
С увеличением времени пребывания газовой смеси в зоне
катализатора степень окисления двуокиси серы в SO3
увеличивается. На рис. 35 приведена построенная по
расчетным данным диаграмма зависимости степени окисления SO2 в
SO3 от времени соприкосновения с ванадиевым катализатором
при 475 °С и начальном составе газа 7% SO2, 11% О2 и
82% N2. Из диаграммы видно, что для окисления SO2 на 65%
0 12 3 4
Время соприкосновения, сеИ
Рис. 35. Зависимость степени
превращения SO2 в SO3 на
ванадиевом катализаторе от
времени соприкосновения.
2 3 « 5 6
время, сек
Рис. 36. Скорость реакции
2SO2+O^2SO:
2+3^3
/—при 450 °С; 2—при 600 °С.
требуется только 0,5 сек, для окисления на 80% — уже 1 сек,
а для окисления SO2 на 92—93% необходимо 2 сек.
Следовательно, скорость окисления SO2 в SO3 быстро убывает по
мере приближения состава газовой смеси к состоянию
равновесия. При одинаковой температуре и прочих равных
условиях скорость реакции тем больше, чем дальше газовая смесь
находится от состояния равновесия.
С повышением температуры скорость реакции окисления
SO2 увеличивается, а равновесная степень окисления SO2
уменьшается (стр. 103). Скорость реакции окисления БОг на
ванадиевом катализаторе при разных температурах показана
на рис. 36. При 600 °С эта реакция протекает значительно
быстрее, чем при 450 °С, однако состояние равновесия достигается
при меньшей степени окисления.
109
Для каждой заданной степени окисления SO2
максимальная скорость реакции достигается при определенной
температуре, как показано на рис. 37, на котором нанесены кривые
одинаковых степеней окисления.
Следовательно, оптимальной температурой контактирования
будет такая температура, при которой (и прочих одинаковых
условиях) достигается максимальная скорость образования SO3,
т. е. наибольшая интенсивность процесса.
Ниже приведены оптимальные (расчетные) температуры
максимальной степени окисления SO2 в SO3 на ванадиевых
катализаторах для газовой смеси, содержащей 7% SO2, 11%
О2, 82% N2:
Степень окисления SO2 в
SO3,% 60 70 80 90 92 94 95 96 97
Расчетная оптимальная
температура, °С . . . 604 574 540 494 482 466 456 446 434
Чем выше степень окисления, тем ниже оптимальная
температура. Поэтому нельзя принимать какую-то постоянную
оптимальную температуру
109
О1 80
60
ЬО
30
го
I
1
В
А
<
у
>
f.
С
t
i
У
;
\
/
7
\
1
{
&
/
ф
\
г
9Ь%
1
-у
f
?<
V
7
/
с
А
\Ь
f
s
>
\
¦8У.
вОУо
90%
Уа
А
/
'/о
r-
-[TV
/ '
7QC/Pi
80%
j !
\—
'
j
i
для всего процесса
окисления SO2 в SO3 и
нецелесообразно поддерживать
одинаковую температуру во
всем слое контактной
массы. Процесс
контактирования надо начинать при тем-
пературе около 600°С и по
мере увеличения степени
окисления постепенно
понижать температуру до 430—
440 °С, в соответствии со
смещением максимумов
скорости реакции. Другими
словами, процесс надо вести
по кривой АА оптимальных
температур, проведенной
через точки максимумов
скорости, как показано на рис.
37. При оптимальном
температурном режиме для
окисления двуокиси серы
требуется примерно вдвое
меньше времени, чем для проведения изотермического процесса
при температуре 440—450°С.
Практически процесс контактирования ведут ступенчато
в пределах допустимых колебаний температуры,
ограниченно
580
игв (+60 500 5Ь0
Температура, °С
Рис. 37. Скорость окисления двуокиси
серы в зависимости от температуры при
степенях окисления 0,7—0,95.
ных кривыми ВВ и СС. В зоне этих температур скорость
окисления SO2 близка к оптимальной.
Температура газа на входе в контактный аппарат,
загруженный контактной массой БАВ, обычно составляет около
440°С. Вследствие выделения тепла реакции температура газа
быстро повышается до 600—620 °С. Далее тепло отводится
для понижения температуры к концу процесса до 440 °С и
ниже.
Достаточно высокая степень окисления SCb в SO3 (до
97—98%) достигается при следующем составе газовой смеси:
7—7,5% SO2, 11% О2 и 82% N2, ведении процесса по кривой
оптимальных температур и отсутствии в газе катализаторных
ядов (As, Se, туман серной кислоты, влага).
Наиболее вредным ядом для ванадиевых катализаторов
является мышьяк. В присутствии As2O3 или AsH3 в контакта-
руемом газе при температуре выше 550 °С образуется летучее
соединение V2Os • As2Os, что приводит к снижению
каталитической активности. Содержащаяся в газе влага не влияет на
активность БАВ при температурах, превышающих температуру
конденсации серной кислоты. При более низких температурах
образуется серная кислота, вызывающая разрушение
контактной массы.
КОНТАКТНЫЕ АППАРАТЫ
Известны два способа контактирования:
1) в несколько стадий с охлаждением газовой смеси после
каждой стадии (контактные аппараты типа К-39-4 с
промежуточным теплообменом);
2) при непрерывном отводе тепла через теплообменные
трубки (контактные аппараты типа ВТ-3 и П-3 с внутренним
теплообменом), помещенные в контактном пространстве; при
этом тепло реакции используется для подогрева свежего газа,
поступающего на контактирование.
Контактные аппараты с промежуточным теплообменом.
Если процесс окисления SO2 и SO3 проводить, например, в
две стадии в контактном аппарате с промежуточным
теплообменом, то создаются условия, недостаточно близкие к
оптимальному режиму, т. е. процесс протекает не по кривой
оптимальных температур. В начале процесса температура газа
слишком низка, в конце — слишком высока. Поэтому
конечная степень окисления SO2 в SO3, достигаемая в таком двух-
стадийном контактном аппарате, относительно невелика
(94-95%).
Режим, более близкий к оптимальному, создается, если
окисление SO2 в SO3 ведут в несколько стадий (четыре, пять
и более). В этом случае степень контактирования, например,
111
в контактном аппарате с пятью слоями катализатора
достигает 98% и более . В большинстве случаев в контактном
аппарате размещают несколько слоев контактной массы. Реакция
протекает с большим выделением тепла и процесс в контактном
аппарата идет аутогермично. Поступающий в аппарат свежий
газ подогревается до температуры начала реакции путем
теплообмена с прореагировавшим горячим газом. Оптимальный
Сернистый газ
из подогревателя
Рис. 38. Схема четырехслоиного контактного аппарата типа К-39-4 с
промежуточным теплообменом:
/—IV—слои контактной массы; /, 2, 3—промежуточные теплообменники; 4—наружный
(трубчатый) теплообменник; 5, 6, /—газовые задвижки.
температурный режим поддерживается посредством
охлаждения газа в ходе процесса. Для разогрева аппарата (при пуске)
газ предварительно пропускают через огневой подогреватель,
выполненный в виде трубчатого теплообменника, обогреваемого
топочными газами.
На рис. 38 изображена схема четырехслоиного контактного
аппарата типа К-39-4 с промежуточным теплообменом. Все
слои катализатора /—IV и промежуточные
теплообменники /—3 объединены в одном аппарате.
112
Слои катализаторной массы размещены на четырех
колосниковых решетках. Между слоями находятся трубчатые
теплообменники.
Сернистый газ, поступающий на контактирование при 45—
50 =С, нагревается в межтрубном пространстве наружного
теплообменника 4 до 230—240 °С горячими контактными
газами, движущимися противотоком по внутренним трубам
теплообменника. Далее газ последовательно проходит в
межтрубном пространстве теплообменников 3, 2, 1 (начиная с
нижнего), где нагревается до температуры начала реакции
(около 440°С), и далее поступает сверху в первый слой
катализатора. Здесь окисляется большая часть сернистого
ангидрида (около 70%), так как концентрации SO2 и О2 в газе
велики и соответственно высока скорость реакции.
Температура газа в первом слое повышается до 590—600 °С, далее он
охлаждается до 450—460 °С в трубах промежуточного
теплообмена 1 и поступает во второй слой катализатора, где
окисляется еще около 20% SO2. Затем газ охлаждается в
теплообменнике 2 до 440°С и проходит через третий слой
катализатора, где окисляется около 6% SO2. После охлаждения в
теплообменнике 3 до 415—418 °С газ проходит четвертый слой
катализатора, здесь окисляется еще 2% SO2. Таким образом,
суммарная степень окисления SO2 в SO3 составляет около 98%.
В третьем и четвертом слоях катализатора реакция протекает
значительно медленнее, так как концентрации реагирующих
веществ малы и степень окисления приближается к
равновесной. Поэтому в третьем и четвертом слоях помещают в два
раза больше катализатора, чем в верхних слоях. Для снижения
гидравлического сопротивления в третьем и четвертом слоях
катализаторной массы нижняя часть контактного аппарата
расширена.
Чтобы повысить скорость реакции окисления SO2 в SO3 в
многослойном контактном аппарате, целесообразно загружать
ка каждую полку катализатор различного состава (стр. 107),
наиболее соответствующий условиям процесса на данном слое.
В настоящее время на крупных сернокислотных заводах
установлены контактные аппараты производительностью 120,
180 и 360—400 т/сутки* (считая на 100%-ную H2SO4).
На рис. 39 изображена схема контактного аппарата
производительностью 360 т/сутки с 5 слоями контактной массы
(четыре верхних слоя — кольцеобразная контактная масса, пятый
нижний слой — гранулированная). Под каждым слоем
катализатора находится слой кварца.
* При использовании газа, полученного сжитанием серного колчедана,
производительность контактного аппарата 360 т/сутки; если газ получается
сжиганием серы, производительность контактного аппарата возрастает до
400 TJcyrKU.
&—2848 113
Перед поступлением в контактный аппарат газовая смесь
подогревается в наружном теплообменнике (на рисунке не
показан).
Выходящий из первого слоя катализатора газ охлаждается
путем смешения с холодным газом.
SO2+so3
SO2+SO3
Рис. 39. Схема контактного аппарата с пятью
слоями контактной массы:
У—V—слои контактной массы; /—колосниковые решетки;
2—теплообменники-смесители; 3—термопары.
После второго слоя тепло контактирования отводится в
наружном теплообменнике, после третьего и четвертого слоев газ
охлаждается во внутренних теплообменниках-смесителях 2. По
выходе из пятого слоя контактной массы прореагировавший
газ охлаждается в наружном теплообменнике.
114
Количество газа, проходящего через аппарат составляет
46 000 м3/ч, концентрация SO2 в газе до контактирования 7,5%,
степень контактирования достигает 98—98,5%.
Обечайки и крышки описанного аппарата выполнены из
стали, изнутри аппарат футерован шамотным кирпичом,
снаружи— покрыт теплоизоляцией. Диаметр аппарата 8 м, высота
18 м, поверхность
теплообменника-смесителя 98 ж2.
Контактные аппараты
с внутренним
теплообменом. В таких аппаратах
создается температурный
режим, близкий к
оптимальному, поэтому в них
достигается более
высокая общая степень
окисления SO2 в SO3 (свыше
98%), чем в четырехслой-
ных аппаратах с
промежуточным теплообменом.
На рис. 40 изображена
принципиальная схема
четырехслоиного
трубчатого контактного аппарата
с внутренним
теплообменом. Первый, третий и
четвертый слои
контактной массы размещены на
полках, второй — в
трубках переменного
диаметра. Процесс в
первом, третьем и четвертом
слоях совершается
адиабатически. Между слоями
контактной массы газ
охлаждается в поясах 2 и 5.
Свежий газ поступает в межтрубное пространство из кольцевой
камеры У через отверстия, расположенные по периферии
кольца. Из межтрубного пространства газ поступает в верхний
первый слой 2 контактной массы, откуда идет в охладительный
пояс труб 3 (на рисунке показаны только две трубы, в аппарате
имеется 595 таких труб). Пройдя затем через второй слой 4
контактной массы, расположенный в средней (уширенной) части
труб 3, газ поступает во второй охладительный пояс 5 (в
суженную часть труб). По выходе из труб газ последовательно
проходит через третий и четвертый слои контактной массы и далее
отводится из нижней части аппарата. Между третьим и четвер-
Рпс. 40. Схема четырехслоиного
контактного аппарата с внутренним теплообменом и
трубами переменного диаметра:
1—кольцевая камера; 2—верхний слой контактной
массы. 3—верхний охладительный пояс труб;
4—второй слой контактной массы; 5—нижний
охладительный пояс труб; 6—завихрнтель; 7—охлаждающая
рубашка.
115
тым слоями контактной массы газ охлаждается. Над третьим
слоем контактной массы установлен завихритель 6.
Аппарат выложен изнутри кислою- и жароупорной
футеровкой, снаружи изолирован кизельгуровыми плитками.
Внутренний диаметр контактного аппарата 3,4 м, высота 9,6 м,
поверхность теплообмена 485 м2. Продолжительность
контактирования 6 сек.
Воздух
Рис. 41. Схема контактного аппарата, с промежуточным
введением холодного воздуха:
/—кожух аппарата; 2, 3, 4, 5—слои контактной массы; ,6—завихри-
тели.
Контактные аппараты с внутренним теплообменом не
получили широкого распространения вследствие сложности
конструкции. Кроме того, при большой скорости газового потока,
имеющего восходящее направление в верхнем слое
катализатора, неизбежен унос мелких частиц катализатора и нарушение
нормального режима работы аппарата.
Предложены также контактные аппараты без
теплообменников, с промежуточным вводом холодного воздуха (рис. 41).
Один из таких контактных аппаратов представляет собой
вертикальный когел, в котором расположены четыре слоя
катализатора. Газ с содержанием 12% SO2 (получаемый при
сжигании серы), охлажденный до 440°С в паровом котле (на
рисунке не показан) поступает в контактный аппарат. Для
понижения температуры контактирования между слоями катализа-
116
тора вводится холодный воздух, предварительно очищенный от
механических примесей и осушенный. С помощью особых за-
ЕПЛ'рителей добавляемый воздух перемешивается со всем
газовым потоком. По расчетным данным конечная степень
окисления SO2 в SO3 превышает 99%.
Новые контактные аппараты. В настоящее время
разработана конструкция пятислойного контактного аппарата с
встроенными в него теплообменниками из горизонтальных трубок, а
также контактного аппарата с вертикальными
теплообменниками, расположенными в центральной части аппарата. Такие
аппараты удобны при эксплуатации и ремонте.
Для новых контактных сернокислотных установок,
работающих на колчедане (мощность установки до 1000 т/сутки, или
~320 тыс. т!год), намечена установка аппаратов суточной
производительностью по 500 т серной кислоты.
Передача газа в контактной систиме
Сопротивление контактных систем достигает 1500—
2000 мм вод. ст. Для транспортирования газа в контактных
системах раньше устанавливали турбокомпрессоры
производительностью 40—65 тыс. м3/ч (скорость вращения 3000 об/мин)
при напоре 2000—2600 мм вод. ст., мощность электродвигателя
350—600 кет.
На вновь проектируемых крупных сернокислотных заводах
мощностью 500—1000 т/сутки A00%-ной H2SO4) предусмотрена
установка мощных турбокомпрессоров производительностью до
100 тыс. м3/ч при напоре 2000—2800 мм вод. ст., мощность
электродвигателя до 1100 кет.
Расход электроэнергии, потребляемой турбокомпрессором
контактной системы, составляет примерно 40 кет • ч на 1 т
100%-ной H2SO4. При уменьшении сопротивления системы,
прежде всего сопротивления контактной массы, возможно
снижение расхода электроэнергии.
АБСОРБЦИЯ СЕРНОГО АНГИДРИДА
Последней стадией производства контактной серной
кислоты является абсорбция (поглощение) серного ангидрида из
газовой фазы с образованием серной кислоты. Абсорбция SOs
серной кислотой производится в башнях. Серный ангидрид
растворяется в серной кислоте и взаимодействует с содержащейся
в ней водой по реакции:
raSO3 + НаО = H2SO4 -f (га — 1)SO3 -f Q
В зависимости от соотношения трехокиси серы и воды
получается серная кислота различной концентрации. При п>\
1 7
wo
т
О
4
"
85
80.
1
1
/
1
70
образуется олеум, при п=\ получается 100%-ная серная
кислота, при п<1—серная кислота меньшей концентрации. На
контактных сернокислотных заводах выпускают олеум и
купоросное масло.
Для абсорбции серного ангидрида в контактных системах
обычно устанавливают две башни. В первой абсорбционной
башне, называемой олеумным абсорбером, SO3 поглощается
18,5—20%-ным олеумом, во второй башне (моногидратный
абсорбер) —98,3%-ной серной кислотой.
Давление паров SO3 над 98,3°/о-ной кислотой практически
равно нулю, а потому она поглощает
SO3 лучше, чем более
концентрированная кислота и олеум. Над
разбавленными растворами серной кислоты имеются
водяные пары, при соприкосновении
таких растворов с SO3 образуется трудно
улавливаемая туманообразная серная
кислота.
Поэтому для абсорбции
трехокисисеры нецелесообразно применять серную
кислоту с содержанием менее 98,3%
H2SO4."
На рис. 42 показана зависимость
степени абсорбции серного ангидрида от
концентрации серной кислоты (по
опытным данным) при температуре 30°С.
Абсорбцию серного ангидрида в
настоящее время проводят в скрубберах с
насадкой, орошаемой серной кислотой.
Кроме скрубберов с насадкой, для
абсорбции серного ангидрида могут применяться также аппараты
барботажного типа, в которых газ барботирует через слой
серной кислоты*. В обоих случаях серная кислота абсорбирует
только трехокись серы, остальная часть газа, пройдя
абсорберы, удаляется в атмосферу. Поглощение SO3 сопровождается
выделением значительного количества тепла, вследствие чего
орошающая кислота нагревается, что ведет к ухудшению
абсорбции.
Для создания по всей высоте абсорбера более
благоприятных условий поглощения SO3 следует орошать насадку
возможно большим количеством холодного олеума или холодной
98,3%-ной H2SO4.
В олеумных абсорберах с насадкой поглощается 40—70%
поступающего з систему серного ангидрида.
8D 90
H2SO4.%
Рис. 42. Зависимость
степени абсорбции серного
ангидрида от
концентрации серной кислоты по
опытным данным прн
температуре 30 °С.
* Абсорберы барботажного типа пока не получили широкого
распространения.
118
Олеумньш абсорбер (рис. 43) представляет собой
вертикальный стальной цилиндр / с крышкой и днищем, в нижней
части которого на стальных балках помещена колосниковая
решетка 2. На решетке расположена насадка 3 (керамические
кольца), служащая для увеличения поверхности
соприкосновения газа с жидкостью. Для орошения насадки олеумом в
верхней части абсорбера установлены стальные распылители 4.
Сверху насадка орошается
олеумом, содержащим 18,5 —
20% SO3 (своб.).
При прохождении олеума
через абсорбер содержание в
нем SO3 (своб.)
увеличивается до 22%. В сборнике олеум
непрерывно разбавляется
небольшим количеством
кислоты, поступающей из
моногидратного абсорбера, до
первоначальной концентрации, т. е.
до 18,5—20% SO3 (своб.).
После охлаждения олеум снова
подается насосом на
орошение абсорбера. Часть олеума
в виде готового продукта
выводится из цикла и
направляется на склад.
Моногидратный абсорбер по
устройству не отличается от
сушильной башни (см. рис. 31).
Он орошается в среднем
98,3%-ной кислотой. Проходя
абсорбер, кислота поглощает
SO3, при этом ее
концентрация повышается до 98,7—99%
H2SO4. В моногидратном
абсорбере кислота разбавляется
водой или сушильной кислотой
до концентрации 98,3—98,4%
H2SO4 и через холодильник
вновь подается на орошение
моногидратного абсорбера.
Размеры моногидратного
абсорбера и количество моногидрата, подаваемого на
орошение обычно такое же, как в олеумном абсорбере (или несколько
больше).
Размеры абсорбционных башен устанавливают на
основании технологических расчетов в зависимости от заданной про-
Крепкии
олеум
Рис. 43. Олеумнын абсорбер:
/—стальной цилиндр; 2—колосниковая
решетка; 3—насадка (керамические кольца);
4—распылитель олеума; 5—лаз для загрузки и раз-
грузки абсорбера; б—смотровое стекло.
119
изводительности системы. Абсорбционная башня
производительностью около 300 т кислоты в сутки (считая на 100%
H2SO4) имеет внутренний диаметр 5,5 м и общую высоту
12,8 м.
Описанный метод абсорбции SO3 олеумом (серной
кислотой) имеет ряд недостатков: громоздкость аппаратурного
оформления, значительный расход электроэнергии на
перекачивание орошающих кислот, низкий коэффициент
теплопередачи A50—250 ккал/м2-ч-град) оросительных холодильников
и др.
Известный интерес представляет применение барботажных
абсорбционных аппаратов. Для абсорбции серного ангидрида
предложено два типа барботажных аппаратов с ситчатыми
тарелками: с перекрестным током газа и жидкости и с
провальными, тарелками. В аппаратах барботажного типа удается
несколько повысить интенсивность процесса массопередачи и
уменьшить расход электроэнергии. Совмещение процесса массо-
и теплопередачи в одном абсорбционном аппарате позволяет
также сократить некоторую часть выносных оросительных
холодильников. Однако тепло реакции абсорбции приходится
отводить путем установки охлаждающих змеевиков на ситчатых
тарелках, что приводит к усложнению конструкции и
обслуживания аппаратуры абсорбционного отделения.
Технологические схемы абсорбции и аппаратурное
оформление их на различных сернокислотных заводах мало
отличаются друг от друга. Примерный технологический режим
абсорбционного отделения контактной системы, включающей
ангидридные холодильники:
Температура газа, °С
на входе в олеумный абсорбер 80—90
на выходе из олеумного абсобрбера 65—80
на выходе нз моногидратного асорбера . . . Не выше 60
Концентрация орошающей кислоты
в олеумном абсорбере, % SO3 (своб.) .... 18,5—20,0
в моногидратном абсорбере, % H,SO4 .... 98,3±0,3%
Степень абсорбции, % ' 99,97—99,99
При степени абсорбции 99,99% содержание тумана серной
кислоты в выхлопных газах не превышает 0,02 г/м3
{0,0005 объемн. % H2SO4).
Расходные коэффициенты на 1 т контактной серной
кислоты (считая на 100% H2SO4):
Колчедан D5% серы), кг 815—820
или сера, кг 350—375
Электроэнергия, кет • ч 60—70
Вода оборотная (при 15 °С), ж3 . ¦ ... 35—70
i20
Серный ангидрид
При обычных условиях серный ангидрид—бесцветный газ.
на воздухе мгновенно вступающий в реакцию с парами воды;
при этом образуется туманообразная серная кислота. При
44,8 °С SO3 сжижается в бесцветную жидкость плотностью
1,9224 г/см3.
Серный ангидрид является сильным водоотнимающим
средством и сильным окислителем.
Твердый SO3 может существовать в четырех модификациях,
плавящихся соответственно при температурах 16,8; 31,5; 62,5
и95°С.
Жидкий серный ангидрид склонен к полимеризации, при
этом он переходит в твердое состояние, что сильно затрудняет
его перевозку и употребление. Полимеризация жидкого SCb
предотвращается при хранении его при температурах выше
30СС или при введении стабилизатора (ингибитора).
Наилучшим ингибитором процесса полимеризации БОз является
высушенный при 110 СС борный ангидрид. Минимальное
количество вводимого ВгОз 0,3—0,5%.
Стабилизованный SO3 не подвергается полимеризации в
течение многих месяцев, даже при температуре ниже 0 °С, если
он не абсорбировал влагу.
Серный ангидрид обладает высокой химической активностью
и широко применяется в качестве сульфирующего агента в
производстве многих органических продуктов.
Стабилизированный серный ангидрид может использоваться для получения
имидосульфоновой, сульфаминовой, хлор- и фторсульфонозои
кислот, сульфурплхлорида и других неорганических ве-
щестз.
Кроме того, серный ангидрид применяется для
приготовления олеума любой концентрации, в качестве дегидратирующего
агента при получении безводной пятиокиси азота,
высококонцентрированной азотной кислоты и др.
Серный ангидрид можно получить тремя способами:
1) отгонкой трехокиси серы из обычного 20%-ного олеума
в аппаратах, обогреваемых топочными газами;
2) окислением двуокиси серы чистым кислородом на
катализаторе;
3) отгонкой трехокиси серы из олеума, циркулирующего в
абсорбционной системе контактной установки, с
использованием физического тепла газов, поступающих из контактного
отделения.
Аппаратуру установок для получения SO3 предпочтительнее
изготовлять из коррозионностойких материалов. В качестве
защитных покрытий лучше всего применять фторопласт-4
(политетрафторэтилен).
J21
Стабилизированный SO3 хранят и перевозят в тщательно
закупоренных (неполных) стальных бочонках емкостью 20 и
32 л. Большие количества серного ангидрида перевозят в
специальных железнодорожных цистернах, изготовленных из
малоуглеродистой стали.
В некоторых случаях вместо 100%-ного серного ангидрида
пользуются высокопроцентным олеумом, содержащим 60—65%
SO3 (своб.). Олеум такой концентрации кристаллизуется при
температуре несколько ниже 0°С.
Высокопроцентный олеум получается насыщением
технического 20%-ного олеума газообразным 100%-ным SO3,
полученным описанными ранее способами.
ПРОИЗВОДСТВО КОНТАКТНОЙ СЕРНОЙ КИСЛОТЫ ИЗ СЕРЫ
Природная сера, не содержащая мышьяка и селена,
должна быть предварительно очищена от минеральных и
органических примесей (битумы и др.). При сжигании серы
содержащиеся в ней битумы и другие органические примеси образуют
ряд продуктов и воду, которая при взаимодействии с серным
ангидридом дает туманообразную серную кислоту. Туманооб-
разная серная кислота плохо улавливается в отделении
абсорбции и ухудшает санитарные условия работы; кроме того,
она оказывает сильное коррозионное действие на аппаратуру
и коммуникации.
Природная сера, не содержащая As и Se и очищенная от
органических и минеральных примесей и золы,
перерабатывается в серную кислоту по «короткой» технологической схеме
(рис. 44). Природная сера 1-го сорта должна содержать не
более 0,05% минеральных примесей (зола). При сжигании серы
часть золы будет в виде пыли уноситься с газом из печи.
Поскольку в «короткой» схеме не предусмотрена очистка
газов, сера должна быть очищена до сжигания, чтобы
предотвратить засорение форсунок печи и контактной массы. Для этого
природную серу предварительно расплавляют, после чего
расплав отстаивается в течение 60 ч при 140 СС и затем
отфильтровывается (на рис. не показано). Чистая расплавленная сера
самотеком поступает в обогреваемый паром сборник /, откуда
погружным центробежным насосом 2 подается в форсуночную
печь 3 для сжигания. Печь (диаметр 4,5 м, длина 15 м)
выполнена из стали, изнутри футерована огнеупорным кирпичом,
снаружи покрыта теплоизоляцией. В печи имеются две
перегородки, в передней стенке печи установлены три серных
форсунки и одна нефтяная или мазутная (для розжига).
Образующиеся в печи горячие газы A1 —12% SO2, ~11% О2) при
1000—1100°С поступают в котел-утилизатор 4, установленный
непосредственно за нечью. Отсюда газ, содержащий 7,5% SO2
122
{чистая ч
расплавленная
сера
Воздух
Л Газ д^сшппльно -абсорбционное
отделение
Рис. 44. «Короткая» схема производства серной кислоты из серы:
/—сборник; 2—погружной насос; 3—форсуночная печь1, 4—котел-утилизатор; 5—контактный аппарат (/—V— слои контактной массы);
$—пароперегреватель; 7—теплообменник; 8—ангидридные холодильники; 9— воздуходувка.
после разбавления осушенным воздухом, при температуре
около 440°С входит в первый слой контактной массы пятислойного
контактного аппарата 5 (диаметр 8 м, высота 17,5 м). Далее
газ при 600 °С поступает в пароперегреватель 6, охлаждается
здесь до 500 СС, входит в зыносной воздушный теплообменник
и при 450°С направляется во второй слой контактной массы,
где нагревается до 520 °С. Затем в наружном теплообменнике 7
газ охлаждается до 440 °С воздухом, проходящим в
межтрубном пространстве теплообменика.
Дальнейшее охлаждение газа до 430 °С после третьего и
четвертого слоек контактной массы происходит при добавлении
холодного осушенного воздуха.
Степень окисления SO2 в SO3 в контактном аппарате
достигает 97,5—98,5%. Из контактного аппарата газ поступает
в ангидридные холодильники 8 и далее в сушильно-абсорб-
ционное отделение, где по обычной технологической схеме
(стр. 95) перерабатывается в серную кислоту или олеум
Для первоначального пуска системы в работу установка
имеет пусковой подогреватель с топкой и дымососом. Воздух
подается в систему воздуходувкой 9.
Преимущества «короткой» системы: простота и
компактность, зозможносгь автоматизации процесса, небольшие
капиталовложения, низкая стоимость переработки сырья и высокий
коэффициент использования серы (96—97%), высокая степень
использования тепла. Тепло сжигания серы и окисления SO2
в SO3 используется в этом процессе для получения
перегретого пара высокого давления, который можно направлять з
гурбины.
ПРОИЗВОДСТВО КОНТАКТНОЙ СЕРНОЙ КИСЛОТЫ
ИЗ СЕРОВОДОРОДА
В результате сероочистки промышленных газов (коксовый
газ, газы нефтепереработки и др.) получаются значительные
количества сероводородного газа высокой концентрации
(85—98% H2S). Сероводород целесообразно использовать для
получения элементарной серы в качестве готового продукта
или перерабатывать ее по «короткой» схеме в серную кислоту.
Кроме этого, сероводород можно непосредственно
перерабатывать в серную кислоту по методу мокрого катализа. Сущность
этого метода заключается в том, что окисление SO2,
полученного при сжигании сгроводорода проводится на ванадиевой
контактной массе в присутствии паров воды.
Процесс производства серной кислоты этим методом
состоит из следующих стадий:
124
1. Получение двуокиси серы сжиганием сероводорода
H,S -f -у О2 = SO2 4- Н2О + 123,94 икал/моль
2. Охлаждение сернистого газа в котлах-утилизаторах.
3. Окисление двуокиси серы в трехокись серы на
ванадиевом катализаторе
SO2 + ~y Oj = SO3 + 22,97 ккал/моль
4. Конденсация серного ангидрида и паров воды с
образованием серной кислоты
SO3 (газ) + Н2О (пар) = H2SO4 (пар) + 29,83 ккал/моль
H2SO4 (пар) = H»SO4 (жидк.) + 11,98 ккал/моль
5. Охлаждение серной кислоты.
6. Очистка отходящих газов от тумана серной кислоты.
Сжигание сероводорода проводится при температуре около
100 °С при минимальном избытке воздуха (или даже при его
недостатке). При сжигании H2S выделяется большое
количество тепла, поэтому перед поступлением на контактирование
газовая смесь предварительно охлаждается в котле-утилизаторе
до 470—488 °С. Значительная часть тепла используется для
получения водяного пара. На 1 т кислоты A00% H2SO4)
получается 0,8 т пара давлением 40 ат. Сероводородный газ,
поступающий на контактирование, не содержит вредных
примесей, отравляющих ванадиевый катализатор, и потому не
подвергается специальной очистке, что значительно упрощает
технологическую схему производства серной кислоты. При
охлаждении газовой смеси, выходящей из контактного
аппарата, образуются пары серной кислоты, которые при
дальнейшем охлаждении газа конденсируются; образовавшийся
конденсат отделяется от газовой смеси.
Схема производства серной кислоты из сероводородного
газа высокой концентрации показана на рис. 45.
Сероводородный газ (85—98% H2S) поступает на сжигание в печь / через
мембранный клапан 2, который автоматически отключает
подачу сероводородного газа в печь в случае внезапного
прекращения подачи воздуха вентилятором 3. Воздух, поступающий
в систему, очищается в фильтре 4.
В печи / сероводород сгорает с образованием SO2.
Выходящий из печи газ имеет температуру 950—975 °С. Перед
поступлением в контактный аппарат газ охлаждается до 470—
480 °С в паровом котле-утилизаторе 5. На входе в контактный
аппарат температура газа составляет 440 °С.
В четырехслойном контактном аппарате типа МК-40
влажная двуокись серы окисляется в трехокись серы на ванадиевой
125
контактной массе, помещенной на полках аппарата.
Специфичность процесса контактирования заключается в том, что
снижение температуры газа перед поступлением в каждый
последующий слой контактной массы производится путем
непосредственного добавления к газу атмосферного воздуха, а не путем
охлаждения в теплообменниках, как принято в обычной схеме
контактного процесса.
Из контактного аппарата газовая смесь при 425—430°С
поступает в башню-конденсатор 7, заполненную кольцевой на-
t
13
Рис. 45. Схема производства серной кислоты из сероводородного газа высоко»
концентрации:
/—печь; 2—мембранный клапан; 3—вентилятор; 4—фильтр; 5—котел-утилизатор;
6—контактный аппарат; 7—башня-коиденсатор; 8—электрофильтр; 9—холодильник; 10, 12—сборники;
//, /3—насосы; 14—газовая топка; 15—теплообменник; 16—брызгоуловнтель; а регулятор
соотношения сероводородного газа и воздуха; б—регулирующие клапаны; в—термопары;
г—регуляторы уровня.
садкой, орошаемой 92—93%-ной серной кислотой. Орошающая
кислота при помощи насоса // циркулирует в системе башня-
конденсатор 7— холодильник 9 — промежуточный сборник 10.
Температура кислоты, поступающей на орошение
башни-конденсатора, около 40 СС. На выходе из башни газ имеет
температуру 50 СС, содержание в нем паров воды практически равно
нулю. Поэтому концентрация образующейся серной кислоты
зависит от соотношения H2S и Н2О в исходном
сероводородном газе, содержания в нем органических примесей и
влажности атмосферного воздуха, поступающего в печь для
сжигания газа и в контактный аппарат.
В башне-конденсаторе происходит теплообмен между
горячим газом и холодной кислотой. Трехокись серы и пары воды
126
образуют пары серной кислоты, которые затем конденсируются.
При быстром охлаждении газа часть паров в
башне-конденсаторе конденсируется в объеме с образованием тумана
серной кислоты. Очистка газа от тумана производится в хвостовом
электрофильтре 8, откуда сконденсировавшаяся кислота
стекает в сборник 10. Продукционная кислота поступает в
сборник 12 и насосом 13 перекачивается на склад.
Для разогрева системы перед пуском установлен пусковой
подогреватель, состоящий из газовой топки 14 и
теплообменника 15.
По описанной схеме получается 92,5—93%-ная серная
кислота. Одновременно побочно получается водяной пар
давлением 40 ат.
ОСНОВНАЯ АППАРАТУРА
Печь для сжигания сероводорода представляет собой
стальной цилиндрический котел диаметром 5 м и высотой 11,2 м
(толщина стенки 10 мм), футерованный огнеупорным кирпичом.
В печи имеется колосниковая шамотная насадка.
Сероводородный газ поступает в верхнюю часть печи, продукты
сгорания отводятся из печи снизу. Печь снабжена
предохранительным взрывным клапаном, закрытым заглушкой из тонкой
листовой стали @,8 мм) и листового асбеста. В случае
воспламенения взрывоопасной газовоздушной смеси заглушка
вырывается силой взрыва из фланца, что предотвращает разрушение
печи.
Контактный аппарат. На описанной установке мокрого
катализа применяется четырехслойный контактный аппарат
производительностью 180 т/сутки. Внутренний диаметр верхней
части аппарата 3,6 м, нижней 4,5 м, высота 10,2 м,
гидравлическое сопротивление 400 мм вод. ст.
Расчетная степень контактирования в таком аппарате
превышает 0,98, что достигается благодаря добавлению в
контактный аппарат большого количества воздуха для охлаждения
газа. Это создает благоприятные условия контактирования —
снижается концентрация SO2 в газе и соответственно
увеличивается концентрация кислорода. На входе в контактный
аппарат концентрация SO2 составляет 6—6,5%; температура
газа на входе в первый слой контактной массы около 440°С;
на выходе из четвертого слоя 425—428 °С.
Башня-конденсатор, в которой охлаждается газ после
контактного аппарата и происходит конденсация серной кислоты,
по устройству сходна с башнями, используемыми для осушки
сернистого газа и абсорбции SO3 (см. рис. 31, стр. 100) в
обычной схеме производства контактной серной кислоты.
Электрофильтры устанавливают для улавливания
сернокислотного тумака из газа. Они имеют осадительные электроды
127
из чугунных труб и коронирующие электроды из стальной
проволоки. Электрофильтры работают весьма эффективно: при
скорости газа в электрическом поле около 0,65 м/сек в
хвостовых газах отсутствуют даже следы SO3, при скорости
0,8—0,9 м/сек степень улавливания тумана остается в
пределах нормы.
Примерные расходные коэффициенты на 1 г серной кислоты
A00% H2SO4), получаемой по описанной схеме:
Сероводородный газ (92,5% H2S), .и3 . . . 256*
Электроэнергия, квт-ч 46—50
Вода, ж3 ... 70
Контактная масса, г 160
* Объем газа, приведенный к нормальным условиям
Схема производства серной кислоты по методу мокрого
катализа из сероводородного газа высокой концентрации
довольно проста и компактна. Поэтому возможна полная автоултиза-
пкя процесса.
Для получения серной кислоты по методу мокрого катализа
сероводородного газа высокой концентрации строятся
установки производительностью до 300 т,/сутки H2SO4 A00 тыс. т,'год).
Такая установка состоит из двух систем, каждая
производительностью по 150 т/сутки серной кислоты.
ПРОИЗВОДСТВО ОЛЕУМА ИЗ ОТРАБОТАННОЙ СЕРНОЙ КИСЛОТЫ
В последние годы введены в действие контактные сернокислотные
системы, в которых получается 18,5—20%-ный олеум путем разложения
отработанной серной кислоты (из цехов алкилирования) и кислого гудрона.
Примерный состав отработанной серной кислоты (после алкилирования)
и кислого гудрона (в %):
Отработан- Кислый
ная кислота гудрон
H2SO4 83 50
Углеводороды . . 12 46
Вода 5 4
Кислый гудроц можно использовать, если в смеси гудрона и кислоты
отсутствует твердый осадок.
Схема технологического процесса получения сернистого газа
представлена на рис. 46. Отработанная кислота из цехов алкилирования
перекачивается в приемный бак 1, жидкий гудрон — в приемный бак 2. Далее они
самотеком поступают в смеситель 3, снабженный паровым змеевиком и
мешалкой, откуда смесь насосом 4 перекачивается в напорный бачок 5. Затем
кислота самотеком поступает через распылитель (на рис. не показано) в печь
6. Агрегат для разложения кислоты состоит из печи расщепления 6, печи
дожигания 9 и теплообменника 8.
Кислота распыляется воздухом, подаваемым компрессором 7 под
абсолютным давлением 5 (it.
В печи 6' поддерживается температура 700—750 СС, необходимая для
разложения кислоты по реакции:
HaSO4 => SO, + -у- О., + Н.,0
128
При этом кислота расщепляется на 93—96%. Нужная температура в
печи создается в результате сжигания сероводородного газа, поступающего
из цеха сероочистки, и горения углеводородов, содержащихся в
отработанной кислоте.
Печь снабжена двумя горелками, в которых сжигается газ.
Необходимый для сжигания H2S и углеводородов воздух предварительно нагревается
до 300 °С в теплообменнике 8.
Из нижней части печи 6 газы поступают в печь дожигания 9, где
происходит окончательное разложение кислоты и окисление образующихся в
печи 6 окиси углерода, серы и других промежуточных продуктов. Состав
Конденсат
Рис. 46. Схема получения сернистого газа разложением отработанной серной
кислоты:
/, 2—приемные баки; 3—смеситель; 4—насос; 5—бачок;'[?—печь расщепления; 7—компрессор;
8—теплообменник; 9—печь дожигания: 10, 11—промывные башнн; 12, 13—оросительные
холодильники; 14—мокрый электрофильтр.
газов на выходе из печи расщепления (в объеми. %);
SO2
SO3
8,7 СО и СО2 .... 8,3
0,4 О2, N2 и пары воды 82,6
В печь дожигания газы поступают тангенциально в верхнюю часть и
выходят через кирпичную трубу, не доходящую до дна печи. Содержание
СО в газе составляет около 4,5—5%, для окисления которого в печь
дожигания добавляется горячий воздух. Температура в печи дожигания
составляет 800—850 °С, при этой температуре серная кислота расщепляется
полностью.
В случае необходимости указанная температура в печи 9 поддерживается
*а счет сжигания в печи сероводородного или другого горючего газа.
9-2848
190
Из печи дожигания газ при 800—850 °С поступает в обдуваемый
воздухом газоход из стали марки 1Х18Н9Т. Здесь газ охлаждается до^ОО С
и далее в теплообменнике 8 дополнительно охлаждается до 300 °С
воздухом, имеющим температуру 120 С
Нагретый в теплообменнике воздух
подается в печь расщепления и печь
дожигания.
Охлажденный до 300 °С печной
газ поступает на промывку в две
последовательно установленные
башни 10 к 11.
Кислота, орошающая промывные
башни, охлаждается в оросительных
холодильниках 12 и 13.
В полученном сернистом газе от
сутствуют селен, мышьяк и другие
вредные примеси, газ очищается
только от туманообразной серпом
кислоты в мокром
электрофильтре 14.
Дальнейшая переработка серии
стого газа с целью получения из
него стандартного 18.5—20%-ного
олеума производится по обычной
технологической схеме, описанной на
стр. 94 ел.
Печь для разложения отработан
ной кислоты (рис. 47) представляв!
собой полый стальной цилиндр
диаметром 3,6 м, футерованный
шамотным и теплоизоляционным
(диатомовым) кирпичом. Общая высота пе-
Рис. 47. Печь для разложения
отработанной серной кислоты:
/—обечайка; 2—футеровка; 3—днище;
4—защитный экран; 5—предохранительный
мембранный клапан; б—крышка; 7—выхлопной
люк.
6,4 м.
На 1 т серной кислоты A00%
H2SO4), получаемой описанным
способом, расходуется примерно 1,23 т
отработанной кислоты, свыше 0,3 т жидкого кислого гудрона и около 30 л3
концентрированного сероводородного газа.
4. НИТРОЗНЫЙ [МЕТОД ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
Сущность нитрозного метода заключается в том, что
двуокись серы, находящаяся в очищенном от пыли обжиговом
(сернистом) газе, окисляется в присутствии окислов азота,
являющихся в данном процессе передатчиком кислорода.
Двуокись серы сначала поглощается нитрозой — серной
кислотой, содержащей окислы азота, а затем окисляется окис-
тами азота до серной кислоты.
Нитрозилсерная кислота, содержащаяся в нитрозе,
образуется по реакции:
N,O3 + 2H.,SO4 ~ » 2HNSO5 + Н.,0 A)
Эта реакция обратима, т. е. окислы азота содержатся в
нитрозе одновременно в двух формах, находящихся между
собой в равновесии: часть окислов азота химически связана с
130
серной кислотой в виде нитрозилсерной кислоты HNSO5,
другая часть остается в свободном растворенном состоянии.
Серная кислота поглощает окислы азота в виде
эквимолекулярной смеси N0 и N02, условно обозначаемой N2O3.
Таким образом, процесс образования серной кислоты в нитроз-
ном процессе схематически можно представить следующим
уравнением:
SOa + NaO3 + Н20 = H2SO4 + 2N0f B)
При окислении SO2 двуокись азота N02 восстанавливается
до окиси азота N0, которая выделяется из нитрозы, так как
плохо растворима в ней. Часть окиси азота снова окисляется
до NO2 кислородом, присутствующим в реакционной зоне:
2N0 + 08 = 2NO2 C)
Двуокись азота образует с непрореагировавшей окисью
азота азотистый ангидрид
NO + N02 = N2O3 D)
который вновь поглощается серной кислотой с образованием
нитрозы. Далее по реакции A) образуются новые количеств;)
серной кислоты и т. д.
Таким образом, в процессе окисления SO2 окислы азота
играют роль передатчиков кислорода и теоретически не
расходуются, а возвращаются в цикл. Однако вследствие
неполного поглощения окислов азота серной кислотой они частично
уносятся отходящими из системы газами. Кроме этого,
небольшое количество окислов азота, вследствие неполной
денитрации (см. ниже), остается в продукционной кислоте и
также теряется.
Процесс производства серной кислоты нитрозным
способом состоит из следующих стадий:
1. Абсорбция двуокиси серы нитрозой.
2. Окисление двуокиси серы нитрозой до серной кислоты.
3. Выделение окислов азота из нитрозы (денитрация).
4. Окисление выделившейся из нитрозы окиси азота до
двуокиси азота с образованием эквимолекулярной смеси N0 ч
N02.
5. Абсорбция окислов азота с образованием нитрозы.
Нитрозный метод производства серной кислоты, возникший
более 200 лет назад, на практике может осуществляться двумя
способами — камерным и башенным. В настоящее время
применяется только башенный способ производства серной кислоты
по нитрозному методу. По этому способу все процессы
переработки двуокиси серы в серную кислоту проводятся в башнях,
заполненных насадкой и орошаемых серной кислотой.
В большинство систем теперь включены также полые башни
(без насадки).
9* 131
Башенный способ получения серной кислоты всесторонне
изучался в СССР целый ряд лет. Были тщательно разработаны
физико-химические основы процесса образования серной
кислоты, что дало возможность достигнуть высокой интенсивности
работы башенных систем, найдены соответствующие материалы
для аппаратурного оформления процесса, созданы конструкции
мощных кислотоупорных насосов, налажено их изготовление,
а также введен ряд других усовершенствований башенного
процесса, позволивших увеличить интенсивность
сернокислотных башенных установок во много раз.
Производство серной кислоты нитрозным способом является
сложным процессом, включающим ряд параллельно и
последовательно протекающих реакций.
ПРИНЦИПИАЛЬНАЯ СХЕМА ПРОИЗВОДСТВА
На рис. 48 показана схема семибашенной сернокислотной
системы.
Для выделения тумана серной кислоты из отходящих
газов в конце системы установлен электрофильтр. В отличие
от контактного метода в производстве серной кислоты по нит-
розному методу сернистый газ предварительно освобождается
только от механических примесей (пыли). Очистки газа ог
мышьяка, селена и других примесей не требуется, так как они
не влияют на течение процесса. Очищенный от пыли сернистый
газ (концентрация SO2~ 9°/o) поступает в башенную систему
при 360—450 °С непосредственно из огарковых (сухих)
электрофильтров и проходит через все башни сернокислотной системы.
Вначале сернистый газ поступает в башни / и //. Около
30% от общего количества обжигового газа поступает в баш-
ню-денитратор /, остальные 70%—в башню-концентратор //.
По выходе из этих башен оба потока газа объединяются, и газ
поступает последовательно в продукционную башню ///,
окислительную башню IV и абсорбционные башни V—VII.
В некоторых башенных системах, работающих при высокой
температуре сернистого газа D00—450 °С), башню-денитратор
включают в шунт с башней-концентратором. В этом случае
30% газа сначала поступают в денитрационную башню. Пройдя
денитратор, газ присоединяется к потоку газа, поступающему
в концентрационную башню.
В башне / происходит денитрация — выделение окислов
азота из кислоты, орошающей эту башню. Денитрованная
серная кислота, имеющая температуру 120—130°С, через
холодильник У поступает в сборник 2, откуда перекачивается на
склад готовой продукции. При избытке продукционной
кислоты и в период пуска башенной системы кислота из башни /
может быть передана в сборник 2 при башне II.
132
Язотноя кислота
3
Рис. 48. Схема семибашенной системы:
/—дсппграциошшя башня; //—продукционная башня (концентратор): ///—продукционная башня; /V—окислительная башня;
V—V//—абсорбционные б-шши; /—холодильники; 2— сборники; 3— насосы; 4— обводный газоход; 5—дроссельный вентиль; 6—электрофильтр; 7—выхлопная
труба1 Я—хвостовой вентилятор; 9—баи для азотной кислоты; 10—бак для воды; II—бак. для конденсата,
В денитрационной башне / при высокой температуре газз
происходит интенсивное выделение окислов азота из кислоты,
содержание в ней окислов азота понижается до 0,03% (считая
на N2O3).
Одновременно с денитрацией часть SO2 поглощается серной
кислотой и окисляется окислами азота и азотистой кислотой
в серную кислоту. Кроме этого, в денитрационной башне
происходит конденсация паров серной кислоты с образованием
тумана и частичное поглощение его орошающей кислотой.
В башенном процессе готовую продукционную серную
кислоту G5—76%-ную H2SO4) выводят из денитрационной башни,
поэтому кислота загрязнена примесями (пыль, мышьяк, селен
и др.), оставшимися в сернистом газе.
В концентрационной башне // происходит абсорбция SO2
из газа и окисление его нитрозой до серной кислоты.
Вытекающая из этой башни наиболее концентрированная серная
кислота (80—83%-ная H2SO4), содержащая 1,5—4% HNO3,
подается на орошение последней абсорбционной башни VII.
Благодаря повышенной концентрации серной кислоты,
орошающей башню VI/, улучшается абсорбция окислов азота в
этой башне.
Если количество кислоты из башни // недостаточно для
полного смачивания насадки башни VII, то часть вытекающей из
этой башни кислоты вновь поступает на ее орошение, в этом
случае последняя абсорбционная башня частично орошается
«на себя».
Температура кислоты на выходе из башни // составляет
130—145°С, после охлаждения в холодильнике / она
понижается до 40—45 °С.
В продукционной башне ///, орошаемой
концентрированной нитрозой A0—17% HNO3), окисление SO2 в серную
кислоту протекает с большой скоростью. В этой башне почти
заканчивается процесс образования H2SO4. Температура
кислоты на выходе из башни /// поддерживается в пределах
85—100°С, а после охлаждения она понижается до 55—60 °С.
Из сборника 2 кислота подается на орошение абсорбционной
башни V.
Окислы азота, выделяющиеся из нитрозы в процессе
окисления SO2 в H2SO4, частично поглощаются в верхней части
продукционной башни /// орошающей ее нитрозой. Большая
часть окислов азота поступает вместе с газом в полую
окислительную башню* IV. Здесь происходит окисление окиси
азота до NO2.
Степень окисления должна быть такой, чтобы соот-
* Иногда окислительную башню заполняют насадкой и орошают «на
себя» 60%-ной серной кислотой.
134
ношение N0 и NO2 приближалось к эквимолекулярному:
NOa + NO = NgO3
Для поддержания этого соотношения окислов азота в
башне IV часть газа, минуя окислительную башню, пропускают по
обводному газоходу 4 (байпас).
Количество байпасного газа регулируется дроссельным
вентилем 5.
Из окислительной башни газ последовательно проходит
башни V, VI и VII, где происходит абсорбция окислов азота
серной кислотой, орошающей эти башни.
Из последней абсорбционной башни VII газ направляется з
электрофильтр 6 (или в циклон-брызгоуловитель) для
отделения брызг и тумана серной кислоты, после чего отходящие газы
удаляются через выхлопную трубу 7 в атмосферу.
Передача газа в башенной системе осуществляется
вентилятором 5, устанавливаемым между абсорбционными
башнями VI и VII.
Таким образом, в первых шести башнях создается некоторое
разрежение, а в абсорбционной башне VII и электрофильтре 6
(или в циклоне-брызгоуловителе)—небольшое давление.
Неизбежные потери окислов азота (с отходящими газами,
продукционной кислотой и др.) компенсируются подачей в де-
гштрационную и продукционную башни свежей азотной
кислоты, меланжа* или окислов азота, получаемых при
контактном окислении аммиака (стр. 265 ел.).
Вода, необходимая для образования серной кислоты,
вводится из бака 10 в денитрационную башню /ив
продукционную башню ///.
Схема орошения
Схема орошения, т. е. передачи кислоты в башнях, зависит
от технологического режима башенной системы и количества
башен. На рис. 49 показана схема орошения семибашенной
системы, включающей полую окислительную башню.
Вытекающая из денитрационной башни / продукционная 75—
76%-ная серная кислота, содержащая 0,03% N2O3, отводится
на склад.
Наиболее концентрированная и денитрованная серная
кислота из башни // подается на орошение последней
абсорбционной башни VII, в которой окислы азота поглощаются
80—83%-ной серной кислотой. По мере протекания абсорбции
-"¦решающая кислота превращается в нитрозу по реакции:
N2O3 + 2H.,SO4 " * 2HNSOS + Н2О + 20,6 ккал A)
* Меланжем называется смесь концентрированной серной и азотной
¦ нслот, содержащая, 87—89% HNO3 и не менее 7,5% H2SO4.
135
60- 75 °c. 12-/7% Н NO3
55-60 "С
75-76% WiSO^Убыток
0.03% N2 О з кислоты
Газ в
электрофильтр
Рис. 49. Общая схема орошения семибашенной системы:
/—деннтрационная башня; //—концентрационная башня; ///—продукционная башня; IV— окислительная башня; V—VII-
абсорбциониые башни.
Содержание окислоз азота в нитрозе в ходе абсорбции
постепенно повышается.
Концентрацию окислоз азота в растворе (так называемую
нитрозность кислоты) выражают эквивалентным весовым
количеством азотной кислоты или трехокиси азота.
Реакция E) обратима. Равновесие смещается в сторону
образования нитрозилсернои кислоты при понижении
температуры и повышении концентрации | ,г
серной кислоты (рис. 50). ^
Нитрозилсерная кислота, обра- ^ ,0
зующаяся при растворении окислов *_
азота в безводной серной кислоте, | в
вполне устойчива. В водных рас- ^
творах она гидролизуется с образо- ^ 6
занием серной кислоты и окислов ^
азота; при этом равновесие реакции ^ 4
A) смещается справа налево. Q
Предполагают, что N2O3 может § i
находиться в нитрозе также в виде J п
азотистой кислоты.
Тогда реакция гидролиза
нитрозилсернои кислоты протекает по
следующей схеме:
/
/
1
/
30
50 70 90 ttO >30
Температура, °С
HNSO5
HNO., + H2SO4 E)
Рис. 50. Зависимость давления
окислов азота над нитрозой от
температуры (состав нитрозы:
76,7% H3SO4, 3,3% HNSO5.
20% Н2О).
Степень гидролиза нитрозилсер-
ной кислоты различна в
зависимости от состава нитрозы и
температуры и определяет реакционную способность нитрозы по
отношению к сернистому ангидриду.
Нитроза, вытекающая из башни VII (см. рис. 49), подается
на орошение башни VI, где нитроза обогащается окислами
азота, так как в башне VI концентрация окислоз азота в газе
выше, чем в башне VII. Из башни VI нитроза передается в
башню V, где она еще больше обогащается окислами азота.
Далее нитроза поступает на орошение башен /, //, ///. В этих
башнях создаются такие услозия (высокая температура,
присутствие воды, пониженная концентрация серной кислоты), при
которых происходит гидролиз нитрозилсернои кислоты с
выделением окислоз азота и образованием серной кислоты:
2HNSO6 + HaO
NaO3-
¦ 2H2SO4
т. е. равновесие реакции A) сдвигается справа налево.
Образование серной кислоты в продукционных башнях происходит
вследствие окисления двуокиси серы продуктами гидролиза нитрозилсернои
кислоты (окислы азота, азотистая кислота).
137
Наиболее вероятную схему образования серной кислоты в нитрозном
процессе можно представить в виде следующих реакций:
1) поглощение сернистого газа жидкостью:
SO3 + Ц?> = H2SO3
2) реакции в жидкой фазе:
2NO (раств.) + О2 = 2NO2 (раств.)
HNSO5 + НаО = HaSO4 + HNOa
NO + NOa + HaO = 2HNOa
H2SO3 + 2HNOa = 2HaSO4 + 2NO + HaO
3) выделение окислов азота из жидкой фазы:
2HNSO5 + Н2О > 2H2SO4 + NOS + NO; NO (раств.) > NO>a3)
Возможно, что очень небольшая часть двуокиси серы окисляется
непосредственно в газовой фазе:
SOa + NOa = SO3 + NO
Трехокись серы, реагируя с водой, превращается в серную кислоту:
SO3 -f HaO = H2SO4
Выделяющаяся окись азота частично окисляется кислородом в
продукционных башнях с образованием высших окислов азота, которые вновь
принимают участие в образовании серной кислоты.
Скорость окисления двуокиси серы в жидкой фазе
возрастает с увеличением содержания SC*2 в газе, с повышением
температуры и нитрозности кислоты. Максимальная скорость
окисления достигается при поглощении SCb нитрозой,
содержащей 57,6% H2SO4. При таком содержании H2SO4 в нитрозе
нитрозилсерная кислота полностью гидролизована.
Денитрация серной кислоты является важной стадией нит-
розного процесса, так как позволяет уменьшить потери окислов
азота и выделить их из продукционной серной кислоты, т. е.
улучшить ее качество. Скорость денитрации серной кислоты
уменьшается с повышением ее концентрации и возрастает с
повышением температуры. Практически в денитрационнои
башне не удается полностью освободить серную кислоту от
окислов азота. Для некоторых же потребителей серной
кислоты присутствие в ней окислов азота недопустимо. Уменьшить
содержание окислов азота в продукционной кислоте можно
путем добавления к ней некоторых реагентов (сульфат аммония
и др.), которые, взаимодействуя с окислами азота,
находящимися в кислоте, разлагают их с выделением элементарного азота-
Газ, уходящий из продукционной башни /// (см. рис. 49).
содержит выделившиеся из нитрозы окислы азота
преимущественно в виде N0. Чтобы полностью связать окислы азота
серной кислотой в абсорбционных башнях V, VI и VII, газ, по-
138
ступающий в башню V, должен содержать окислы азота,
степень окисления которых соответствует эквимолекулярному
соотношению N0 и NO2, т. е. N2O3. Для достижения этого
необходима окислительная башня IV, где окись азота окисляется
кислородом по реакции
2N0 + 02 » 2N0, -f Q
В условиях процесса производства серной кислоты эта
реакция практически необратима. Скорость ее увеличивается с
понижением температуры, что является одним из немногих
исключений из общего правила.
Образовавшаяся двуокись азота образует с непрореагиро-
вавшей окисью азота азотистый ангидрид:
NO2=N2O3+Q
Эта реакция протекает практически мгновенно, но ее
равновесие при атмосферном давлении почти полностью смещено
влево.
При поглощении окислов азота серной кислотой азотистый
ангидрид быстро реагирует с ней, а окись и двуокись азотг
практически не реагируют. Взамен прореагировавших молекул
азотистого ангидрида мгновенно образуются новые молекулы
N2O3.
«Грязный» цикл орошения. При недостаточно тщательной
очистке газа содержащаяся в нем пыль загрязняет насадку
башен, что ведет к ухудшению условий процесса, увеличению
сопротивления системы, засорению холодильников.
Чтобы предотвратить проникание в средние башни системы
пыли, содержащейся в газе, на некоторых заводах применяется
так называемый грязный цикл орошения. Газ отмывается от
пыли в первой башне, которую орошают малонитрозной
кислотой. Грязная кислота циркулирует только между первой и
последней башнями системы. Вследствие малой нитрозности
кислоты, орошающей первую башню, в ней связывается
меньшее количество двуокиси серы и процесс кислотообразования
сдвигается к концу системы. При таком ходе процесса
повышается расход азотной кислоты.
При очистке газа до содержания пыли 0,1 г/м3 нет
необходимости применять «грязный» цикл орошения.
ТЕХНОЛОГИЧЕСКИЙ РЕЖИМ БАШЕННОЙ СИСТЕМЫ
Одним из обязательных условий нормальной работы
башенной системы является постоянство объема поступающего газа
и концентрации SO2 в нем.
Установлено, что интенсивность работы продукционных
башен растет с увеличением концентрации двуокиси серы в газе.
139
Содержание SCb в газе на входе в систему должно составлять
9—9,5%, а на выходе из последней продукционной башни
должно быть не более 0,15%.
Основные процессы, протекающие в продукционных
башнях в жидкой фазе, ускоряются при повышении температуры
(до известных пределов) и увеличении нитрозности
орошающей кислоты. Для последующего же процесса абсорбции
окислов азота благоприятны возможно более низкая температура
и малая нитрозность орошающей кислоты. Поэтому для
нормальной работы башенной системы должны быть выбраны
оптимальные температура и нитрозность орошения.
При достаточно высокой температуре и нитрозности
кислоты переработка двуокиси серы идет интенсивнее в случае
применения 80—83%-ной серной кислоты, которая хорошо
связывает также окислы азота в абсорбционных башнях.
Увеличение нитрозности кислоты сыграло наибольшую роль
в интенсификации башенных систем. Чем выше интенсивность
системы, тем больше должно быть содержание НРТОз в
нитрозе. При интенсивности системы 200—260 кг/м3 в сутки (при
прочих равных услозиях) содержание HNO3 в нитрозе,
орошающей вторую башню, должно составлять 12—17%.
Процессы денитрации и взаимодействия SO2 с нитрозой
протекают тем интенсивнее, чем выше температура газов,
поступающих в систему. Обычно температура газов на входе з
систему колеблется в пределах 360—450 СС.
Для бесперебойной работы башенной системы требуется
количество орошения*, необходимое для хорошего смачивания
насадки, и равномерное распределение газа и кислоты по
всему сечению башен.
Количество окислов азота, выделяющихся б газовую фазу
(азотооборот) в башенных «системах интенсивностью
220—Й60 кг/м3 в сутки, колеблется от 700 до 1000 кг (в
пересчете на НРТОз) на 1 т продукционной серной кислоты.
Содержание окислов азота (NO и РТОг) в газе, выходящем из
последней продукционной башни, в современных башенных
системах составляет 12—17% (в пересчете на НГТО3).
В абсорбционных башнях окислы азота должны
улавливаться возможно полнее, что важно для повышения экономиче
ской эффективности процесса, так как стоимость азотной
кислоты в несколько раз превышает стоимость серной кислоты.
* Количеством орошения называется объем кислоты, поступающей в
башню в единицу времени; выражается в м3/ч. Кратность орошения —
отношение количества орошения, подаваемого в единицу времени во все башни,
включая абсорбционные, к количеству готовой кислоты, выводимой за это
время из системы. Количество орошения, приходящееся на 1 м2
горизонтального сечения башни, заполненной насадкой, называют плотностью
орошения.
140
Окислы азота (и азотная кислота) теряются главным образом
в результате неполноты абсорбции (потери с хвостовыми газа-
мл), а также вследствие неполной денитрации кислоты и
недостаточной герметичности аппаратуры и трубопроводов.
Удельный расход азотной кислоты составляет на передовых заводах
10—14 кг на 1 т серной кислоты.
Скорость и полнота поглощения окислов азота
увеличиваются также с повышением концентрации орошающей серной
кислоты и с понижением ее нитрозности. При этом, однако,
приходится, учитывать, что кислота из первой (по ходу газа)
абсорбционной башни поступает в продукционные башни, где
для интенсивного протекания процессов требуется орошение
менее концентрированной, но более высоконитрозной серной
кислотой. Нитрозность кислоты, орошающей последнюю
абсорбционную башню, при содержании в кислоте 80—83% H2SO1
должна быть 0,5—1,5% (считая на HNO3).
Газ, уходящий из последней абсорбционной башни,
содержит, кроме окислов азота (до 0,15%), значительное количество
брызг серной кислоты и некоторое количество сернокислотного
тумана (около 5 г H2SO4 на 1 ж3 газа). Удаление в атмосферу
газа с таким высоким содержанием брызг и тумана серной
кислоты недопустимо по санитарным условиям. Для очистки
отходящих газов от брызг и тумана серной кислоты в конце
башенных систем устанавливают санитарные
электрофильтры.
Процессы образования серной кислоты, окисления окиси
азота и абсорбции окислов азота сопровождаются выделением
значительного количества тепла. Кроме того, большое
количество тепла вводится в систему с сернистым газом. Для
поддержания оптимального температурного режима процесса
избыточное тепло отводят путем охлаждения кислоты,
вытекающей из башен, з водяных холодильниках.
В современных башенных системах установлены семь
башен, это позволяет достигать высокой интенсивности процесса
с одновременным снижением удельного расхода азотной
кислоты. Однако башенный процесс получения серной кислоты
может проводиться в большем или меньшем числе башен. Чем
меньше число башен, тем труднее поддерживать нормальный
технологический режим процесса; с увеличением числа башен
технологический режим устойчивее.
При нормальной работе башенных систем степень
использования серы составляет не менее 95%.
В последние годы на ряде отечественных сернокислотных
заводов в башенные системы включены полые (безнасадочные)
абсорбционные башни. Это позволило повысить интенсивность
сернокислотных систем и уменьшить удельный расход азотной
кислоты.
141
Примерные расходные коэффициенты на 1 г башенной
серной кислоты (считая на 100% H2SO4) при интексивности
системы 220—260 кг/м3 в сутки:
Серный колчедан D5% S), кг 790—810
Азотная кислота A00% HNO3), кг . . . 10—14
Вода (для охлаждения), л3 40—50
Извлечение селена из башенной кислоты
До недавнего времени весь селен из башенных систем полностью
терялся, так как двуокись селена, содержащаяся в печном газе, растворяется в
орошающей кислоте с образованием селенистой кислоты НгБеОз, которая
вместе с продукционной кислотой отводилась на склад. В настоящее время
разработан способ извлечения селена из продукционной башенной кислоты.
Сущность этого способа заключается в том, что через кислоту,
находящуюся в сборнике, пропускают сернистый газ и добавляют 25%-ный раствор
поваренной соли. При этом растворимые в кислоте соединения селена
восстанавливаются и выпадают в осадок. Селеновый шлам отфильтровывают,
промывают водой, нейтрализуют содовым раствором и сушат.
Высушенный шлам отправляется на централизованную переработку для извлечения
элементарного селена.
АППАРАТУРА БАШЕННЫХ СИСТЕМ
Основными аппаратами башенных систем являются башни,
снабженные приспособлениями для разбрызгивания
орошающей кислоты. Кроме того, в башенных системах устанавливают
холодильники, сборники для кислоты, насосы для
перекачивания кислот, вентиляторы для протягивания газа через систему,
аппараты для получения газообразных окислов азота
окислением аммиака, брызгоуловители (циклоны) для улавливания
брызг кислоты из отходящих газов.
Основная аппаратура башенных систем до их
интенсификации выполнялась из свинца. В условиях работы интенсивных
башенных систем (повышенная температура кислот,
орошающих продукционные башни, а также более высокая нитрозность
газов и кислот) свинец быстро разрушается, но достаточно
стойки черные металлы.
Коррозия чугуна и стали в 80—83% -ной серной кислоте,
обычно циркулирующей в башенных системах, замедляется с
повышением температуры и нитрозности кислоты, что
объясняется образованием в этих условиях пассивирующей пленки на
поверхности металла.
В настоящее время почти вся аппаратура башенных систем
выполняется из углеродистой стали, чугуна и неметаллических
кислотоупорных материалов.
Башни. При определении общего объема башен исходят
из интенсивности системы, которую принимают в настоящее
время не менее 200 кг/м3 в сутки (в расчете на 100% H2SO4).
142
Высота башен составляет 14—16 м; диаметр денитрацион-
ной башни 3—4 м, концентрационной и последней
абсорбционной башни — 4,5—6 м, диаметр продукционной и первой
абсорбционной башни 6—8,5 м, второй абсорбционной башнн —
4,5—6 м.
Серная
кислота1
Обжиговый
газ
в
Рис. 51. Продукционная стальная башня
башенной системы:
1—стальной кожух; 2—футеровка башни; 3—крышка;
4—турбинка; 5—насадка; 6—днище; 7—колосники;
8—опорная колонка колосниковой решетки.
На современных башенных заводах, работающих при
высокой нитрозности кислоты, кожухи башен изготовляют из
листовой стали.
В продукционных башнях (рис. 51) днище и обечайки на
высоту колосниковых решеток изготовляют из стали марки
Ст.З. Для изготовления остальных частей башен допускается
применение стали марки Ст.О. Крышку первой продукционной
башни выполняют из армированного кислотоупорного бетона
143
толщиной 100—120 мм, крышки других башен — из листовой
стали марки Ст.О.
Стальной кожух продукционных башен защищен от
действия горячей нитрозы кислотоупорной футеровкой (толщина
A50—450 мм), служащей также термоизоляцией, которая
необходима для поддержания в башнях требуемой температуры.
Сохранение тепла в продукционных башнях желательно, так
как с повышением температуры увеличивается скорость
окисления SO2 в SO3 и, кроме того, снижается растворимость
окислов азота, вследствие чего улучшается денитрация нитрозных
кислот.
Рис. 52. Керамические кольца для насадки башен:
а—навалом (засыпка); б—правильными рядами (укладка).
В качестве футеровочного материала используют
кислотоупорный кирпич или камни, вытесанные из природных кислого-
упоров (андезит, бештаунит, см. стр. 666). Связующим
материалом служит кислотоупорная, быстросхватывающаяся
замазка (молотый андезит или бештаунит на жидком стекле с
добавкой кремнеф гористого натрия).
Для увеличения поверхности соприкосновения жидкости с
газом продукционные башни заполняют насадкой из
керамических колец (рис. 52) размерами 50X50 мм, 80X80 мм,
100X100 мм и 120X120 мм. Иногда применяются фасонные
керамические насадки (рис. 53). Насадку укладывают на
колосниковую решетку из прямоугольных брусьев, вытесанных
из природного андезита или бештаунита. В стенах башен
имеются люки для осмотра и очистки подколосникового пространства.
Башни устанавливают на кирпичном или бетонном фундаменте.
На зыходе кислоты из башни имеется гидравлический
затвор.
Окислительная башня изготовляется из стали толщиной
6 мм. Башня имеет цилиндрическую форму и не снабжается ни
футеровкой, ни насадкой. Газы могут поступать в
окислительную башню сверху или снизу. Степень окисления газа
регулируют при помощи обводного газохода (см. рис. 48), по
которому газы можно полностью или частично направлять
непосредственно из второй продукционной башни в первую абсорб-
144
цнонную, минуя окислительную оашню. В обводном газоходе
имеется дроссельный вентиль, регулирующий количество га-
;ов, направляемых мимо окислительной башни.
Газопроводы между башнями изготовляют сварными из ста-
. in марки Ст.О толщиной 6—8 мм. Скорость газа в
газопроводе 6—10 м/сек. Газопровод между первой и второй
продукционными башнями футеруют изнутри кислотоупорными плитками
для защиты от коррозии.
6 в г
Рис. 53. Фасонные керамические насадки:
и—седлообразная; б—кольца с перегородками; в—спиральные кольца.
г—пропеллерная.
Для защиты от коррозии абсорбционные башни изнутри
футеруют кислотостойкими материалами. В качестве насадки
башен применяют керамические кольца таких же размеров.
как в продукционных башнях.
Электрофильтры. Для очистки отходящих из
башенных установок газов, содержащих брызги и туман серной
кислоты в количестве 2—5 г/ж3, примерно 0,15% окислов азота и
до 0,1% SCb устанавливают на открытых площадках
электрофильтры типа МТ-9,5. Осадительные электроды выполняются
из стальных труб, размещенных в стальном цилиндрическом
корпусе, футерованном кислотоупорным кирпичом и плиткой,
коронирующие электроды — из ферросилида. В электрофильтре
обеспечивается очистка газа от брызг и тумана серной кислоты
до содержания 40 мг/м3. На некоторых заводах после
электрофильтра дополнительно устанавливают трубы высотой до 120 м,
через которые выхлопные газы отводятся в верхние слон
атмосферы.
Вспомогательная аппаратура. Разбрызгиватели.
Обязательным условием интенсивной работы башен является
равномерное, достаточно интенсивное и бесперебойное орошение. На
некоторых башенных сернокислотных системах орошение башен
10—2848
145
производится при помощи разбрызгивающих звездочек.
Кислота из трубопровода стекает через конусную воронку на
вращающуюся звездочку, насаженную на вращающийся стальной
вал с приводом непосредственно от электродвигателя через
редуктор. При вращении звездочки кислота равномерно
разбрызгивается по насадке башни (рис. 54).
Кислота
Рис. 54. Схема установки сливной
воронки и разбрызгивающей звездочки
на башне:
/—конусная воронка; 2—звездочка; 3—вал;
4—электродвигатель; 5—редуктор.
Рис. 55. Звездочка
конструкции [„ Полякова и
Гольцева.
На рис. 55 представлена разбрызгивающая звездочка
конструкции Полякова и Гольцева.
В настоящее время разработаны и применяются также
новые более эффективные оросительные устройства.
Насосы и вентиляторы. Для перекачивания серной
кислоты применяют исключительно центробежные насосы.
Применявшиеся раньше поршневые насосы отличались громоздко
стью и малой производительностью.
Центробежные насосы для перекачивания
концентрированной серной кислоты (более 75% H2SO4), олеума, азотной
кислоты и смеси серной и азотной кислот изготовляют из
высокохромистого чугуна C2—36% Сг), для перекачивания
разбавленной серной кислоты (до 70% H2SO4)—из гартблея (92%
РЬ, 8% Sb). Обычно применяются горизонтальные
центробежные насосы производительностью до 100 м3/ч и более.
Для перемещения газа применяют центробежные
вентиляторы. В башенных системах их устанавливают почти всегда
146
Горячая
кислота
Охлажденное
нисло/па
между абсорбционными башнями (хвостовые вентиляторы).
Роторы вентиляторов целесообразно изготовлять из
кислотоупорной стали.
Производительность вентиляторов для перемещения сернк
сгого газа составляет 65 000—100 000 м6\ч, статический напор
до 1225 мм вод. ст. Для вращения вентилятора
производительностью 65 000 м3/ч требуется электродвигатель мощностью
500 кет, совершающий 950 об/мин.
Холодильники. Для охлаждения кислоты, вытекающей
из первой продукционной башни (денитратора), применяют
логружные холодильники. Кислота, вытекающая из остальных
башен, охлаждается в
погружных или оросительных
холодильниках.
Схема простейшего
погружного змеевикового
холодильника с одним
внутренним стаканом
изображена на рис. 56. Внутри
стальной обечайки 1
холодильника установлен
цилиндрический стакан 2, высота
которого равна высоте
холодильника. Диаметр стакана
выбирают таким, чтобы з Рис 56- ПогружнслГзмеевиковый холо-
ЛрОМежуТКе Между СТеНКа- /^обечашсаЛ-^така^ 5-змеевик.
ми холодильника и
стенками стакана помещался
змеевик 3 и оставался некоторый зазор для протекания
кислоты. Поступающая на охлаждение кислота входит в аппарат по
трубе снизу; охлажденная кислота отводится из верхней
трубы. Охлаждающая вода входит в змеевик сверху и вытекает
¦лз нижней части аппарата.
Таким образом, горячая кислота и охлаждающая вода
движутся противотоком. Внутренняя поверхность стальной
обечайки 1 футерована кислотоупорными плитками.
Обечайки погружных холодильников изготовляют из стали
марки Ст.З. Змеевики для холодильников первой
продукционной башни выполняются из свинцовых труб (свинец марки С2)
или из хромоникелевой стали; в остальных холодильниках
устанавливают стальные змеевики из цельнотянутых труб.
Недостатком погружных змеевиковых холодильников
является низкий коэффициент теплопередачи (от 50 до
150 ккал/м2-ч- град), чтоб обусловлено малой скоростью
движения кислоты в аппарате и быстрым загрязнением поверхности
змеевика. В результате этого увеличивается расход воды на
охлаждение.
10*
47
Вместо малопроизводительных погружных змеевиковых
холодильников устанавливают оросительные холодильники, в
которых кислота течет по трубам, а вода орошает эти трубы
снаружи.
Коэффициент теплопередачи в холодильниках этого типа
выше, чем в змеевиковых, что объясняется большей скоростью
протекания кислоты в трубах @,7—0,8 м/сек) и меньшей
возможностью их загрязнения.
Схема устройства оросительного холодильника показана на
рис. 57. Холодильник состоит из ряда расположенных друг
под другом, с незначительным наклоном, чугунных или сталь-
вода
Рис. 57. Оросительный^холодильник:
'—трубы; 2—калачи; 3—желоб; 4—козырек; 5—поддон.
ных (пассивированных) труб /, соединенных между собой на
фланцах полукруглыми патрубками 2 (калачами). Над
трубами установлен желоб 3 (с зубчатыми краями) для орошения
труб охлаждающей водой, а внизу — корытообразный
поддон 5 для сбора воды. На каждую трубу надевают козырек 4
с зубчатыми краями для равномерного слива охлаждающей
воды на расположенную ниже трубу. Поступающая на
охлаждение горячая кислота протекает внутри труб / и выводится
сверху в сборник (на рисунке не показан). Оросительные
холодильники отличаются простотой устройства и легко доступны
для осмотра и очистки труб.
При охлаждении кислоты в чугунных оросительных
холодильниках целесообразно к продукционной кислоте,
вытекающей из башни, предварительно добавлять уже охлажденную
кислоту и подавать такую смесь в холодильник при 70—80°С,
так как при этой температуре чугун вполне устойчив.
148
В последние годы получили распространение более
компактные спиральные холодильники. Такой холодильник
(рис. 58) изготовляется из двух длинных стальных листов,
свернутых в спираль. При этом образуются два узких спиральных
канала, по одному из них движется кислота, по другому —
охлаждающая вода. Благодаря большой скорости
движения кислоты A,2—1,5 м/сек) коэффициент теплопередачи
достигает 1800 ккал/м2 ¦ ч ¦ град.
Достоинства спиральных холодильников — компактность,
полная герметичность и меньшая стоимость по сравнению с
оросительными холодильниками. .. ^
Недостатком спиральных холо- охлаждающей вос/ы
дильников является необходи- горячая ^Шк^ногретая
мость применения очищенной 1<ис-лота^Ж^^^Ж^л??^?
воды для охлаждения, трудность ~ ^^^ШШг^ в /ход
чистки и ремонта, большее гид- J^H1^S^>охлажденной
равлическое сопротивление, чем I кислоты
в оросительных холодильниках. '
Для охлаждения безнитрозной Рис. 58. Схема спиргльногсГхоло-
кислоты нашли применение хо- Дильника (пространство, занятое
лодильники из специальных ста- кислотой, заштриховано),
лей и из антегмита*,
обладающего хорошей теплопроводностью и высокой химической
стойкостью к действию серной кислоты (до 76% H2SO4). Антегмиг
марки АТМ-1 применяется и при изготовлении холодильников
промывных кислот в производстве контактной серной кислоты.
5. КОНЦЕНТРИРОВАНИЕ СЕРНОЙ КИСЛОТЫ
Полученную в башенных системах 75%-ную серную кислоту
применяют глазным образом в производстве минеральных
удобрений и других продуктов, а частично подвергают
концентрированию. Концентрируют также разбавленную серную
кислоту, получаемую в качестве отхода при концентрировании
слабой азотной кислоты, в ряде процессов органического син-
геза и др. При концентрировании получают кислоту,
содержащую 92—94% H2SO4.
Серную кислоту концентрируют путем упаривания. При
нагревании разбавленной серной кислоты до кипения вначале
испаряется только вода, и концентрация кислоты непрерывно
повышается, пока содержание H2SO4 в растворе не достигнет
70%. При дальнейшем нагревании вместе с парами воды
начинают выделяться пары серной кислоты. По мере повышения
концентрации упариваемого раствора увеличивается и
количество испаряющейся серной кислоты. При этом температура
* Антегмит — пластическая масса \\г основе феноло-формальдегидных
чолимеров и графита.
149
кипения раствора непрерывно повышается до тех пор, пока
концентрация кислоты не достигнет 98,3% H2SO4.
На рис. 59 приведена диаграмма кипения серной кислоты
при атмосферном давлении и разрежении. Верхняя кривая
показывает зависимость содержания H2SO4 в парах от
температуры кипения кислоты. Из диаграммы видно, что при
концентрации кислоты менее 98,3% H2SO4 относительное количество
воды в парах больше, чем в жидкости. Из этого следует, что
350
О !0 20 30 40 50 50 V SO ffP '00
Содержание H_,SO.,. esc %
Рис. 59. Диаграмма кипения серной кислоты
при атмосферном давлении и разрежении.
при нагревании водных растворов, содержащих меньше 98,3%
H2SO4, концентрация кислоты будет повышаться, если удалять
выделяющиеся пары воды. Сконцентрировать раствор серной
кислоты обычным упариванием до содержания H2SO4 более
98,3% невозможно, так как 98,3%-ная кислота представляет
собой азеотропную смесь, которая при кипении перегоняется
без изменения состава. Под атмосферным давлением азеотроп-
ная смесь, содержащая 98,3 вес. % H2SO4, кипит при 338,8°С;
безводная серная кислота кипит при более низкой температуре,
выделяя трехокись серы и превращаясь при этом так же в
98,3%-ный водный раствор HiSO4. При нагревании до 440°С
серная кислота полностью разлагается. Под давлением меньше
атмосферного, т. е. при разрежении, концентрирование серной
150
кислоты может быть проведено при более низких температурах.
Так, температура кипения 75%-ной серной кислоты при 1 атм
равна 187,8 С, а при вакууме* 7,5 мм рт. ст. лишь 75 °С.
УСТАНОВКИ ДЛЯ КОНЦЕНТРИРОВАНИЯ
Серную кислоту концентрируют в аппаратах Двух типов:
1) при непосредственном соприкосновении горячих топочных
газов с концентрируемой кислотой; 2) нагреванием кислоты
горячими газами через стенку.
К аппаратам первого типа относятся концентраторы, в
которых осуществляется поверхностное соприкосновение горячих
газов с жидкостью или барботирование горячих газов через
слой выпариваемой кислоты. Одним из таких аппаратов
является продукционная башня сернокислотной системы, в
которой для концентрирования башенной кислоты используется
тепло печных газов (например, при получении купоросного масла
з башенных системах).
Концентраторы, работающие на основе поверхностного
соприкосновения горячих газов с концентрируемым раствором
серной кислоты, вследствие небольшой производительности,
тяжелых условий труда, громоздкости, высоких расходных
коэффициентов и других недостатков почти полностью вышли из
употребления.
Установка с барабанным концентратором. В настоящее
время для концентрирования серной кислоты в основном
применяются барабанные концентраторы, в которых горячие
топочные газы барботируют через кислоту, благодаря чему
создаются хорошие условия теплопередачи.
Установка барабанного типа (рис. 60) состоит из топки,
концентратора, электрофильтра для улавливания тумана серной
кислоты, сборников кислоты и холодильника. Серная кислота
из напорного бака 5 через регуляторную коробку 7 поступает
в концентратор 4. Концентратор представляет собой стальной
футерованный барабан, разделенный перегородкой из андезита
на две камеры, каждая диаметром 2,8 м и длиной 7,8 м. Первая
камера служит для окончательного ее упаривания. Уровень
кислоты в первой камере на 350 мм ниже, чем во второй. Штуцеры
для входа и выхода кислоты в каждой из камер расположены
на одном уровне. Обычно на концентрирование поступает
68—70%-ная серная кислота. После предварительного
упаривания во второй камере кислота через внутренний или внешний
переток (на рисунке не показан) поступает в первую камеру
для окончательного концентрирования.
* Здесь и далее величина вакуума означает остаточное давление, т. е.
разность между давлением 760 мм рт. ст. и разрежением в системе. В
данном случае остаточное давление равно 760—752,5=7,5 мм рт. ст.
151
Серная кислота
68-70%-ная
Кислый конденсат _
в нейтрализатор
Рис. 60. Концентрационная установка с барабанным концентратором:
/—вопка; 2—камера смешения: 3— холодильник; 4—концентратор; fi—напориый бак; 6—электрофильтр; 7—регуляторная
коробка; *—сборник для купоросного масла; 9—насос для купоросного масла; 10—«санитарный» электрофильтр.
Топливо (газообразное или жидкое) в смеси с воздухом,
подаваемым вентилятором, сжигается в топке /, где
поддерживают температуру около 1000 °С. В камере смешения 2 газы
разбавляют воздухом, вследствие чего температура их понижается
до 750—800 "С.
Горячие топочные газы под избыточным давлением
1000—1750 мм вод. ст. сначала поступают в первую камеру
концентратора, барботируя через кислоту, концентрируют ее до
купоросного масла и одновременно обогащаются парами воды.
Затем газы, имеющие температуру около 230 °С, барботируют
через кислоту во второй камере. Здесь концентрация кислоты
повышается до содержания 80—83% H2SO4, а газы,
дополнительно поглотившие пары воды и охладившиеся до 135—150 °С,
по газопроводу поступают з электрофильтр 6 для улавливания
тумана серной кислоты, увлеченного из концентратора.
Конденсат собирается на дне электрофильтра в виде 68—
70%-ной серной кислоты, которая возвращается во вторую
камеру концентратора (на схеме не показано).
Таким образом, в барабанном концентраторе между
обогревающими газами и кислотой осуществлен противоток.
Наиболее горячие газы в камере окончательного упаривания
соприкасаются с более концентрированной кислотой, а несколько
охладившиеся газы подогревают разбавленную кислоту в
камере предварительного концентрирования.
Концентрированная кислота (92—94% H2SO4), имеющая
температуру 220°С, поступает из первой камеры в
холодильник 3, где охлаждается до 35—40 °С, и затем самотеком
направляется в сборник 8.
Отсюда кислота насосом 9 перекачивается на склад или в
потребляющие цехи.
В электрофильтре 6 не удается полностью уловить из газа
туманообразную серную кислоту, поэтому на установках
барабанного типа ставят еще добавочный «санитарный»
электрофильтр 10. Поступающие в него газы предварительно
промываются водой. Кислый конденсат, образующийся в
электрофильтре 10, передают на нейтрализацию, а газы
вентилятором отводятся в атмосферу. Из отходящих газов
улавливается 6—13% всей кислоты, поступающей на
концентрирование.
Потери концентрируемой кислоты составляют 1—2%.
Уменьшить образование туманообразной серной кислоты (и,
следовательно, унос ее в атмосферу) с 35—40 до 11 г/м3
влажного газа можно при дополнительном параллельном вводе
горячего топочного газа во вторую камеру. При замене мазута,
содержащего примеси серы, природным или коксовым газом
также значительно снижается содержание серной кислоты и
ЬО2 в выхлопных газах.
153
Производительность концентратора барабанного типа
составляет 150—200 т/сутки при начальной концентрации кислоты
68—70% H2SO4 и конечной концентрации 93%. При
концентрировании 68%-ной серной кислоты, поступающей на установку
при 25 °С, расходуется примерно (в пересчете на 1 г
H2SO4):
Мазут, кг
Пар (Рабс- = ^ иш), кг
Электроэнергия, квпг-ч
Вода, м3
50—55
20—25
20—25
5-6
Недостатками описанной установки для концентрирования
серной кислоты являются некоторая громоздкость
аппаратурного оформления, значительные капитальные затраты и др.
Вода
смаВая
Нислота
Рис. 61. Концентрационная установка с дефлегматором:
/—котел; 2—мешалка; 3—топка; 4—холодильник; 5—конденсатор: 6—лоЕушка;
7—дефлегматор; 6—напорный, бак для слабой кислоты.
Концентрирование в аппаратах с внешним обогревом. На
рис. 61 изображена схема концентрационной установки, в
которой нагревание кислоты осуществляется через стенку аппарата.
Установка работает при атмосферном давлении. Она состоит
154
из котла 1, изготовленного из кислотоупорного чугуна и
снабженного мешалкой 2, и тарельчатого дефлегматора 7. Котел
обогревается газами, поступающими из топки 3. В котле при
установившемся процессе кипит концентрированная серная
кислота, которая непрерывно стекает в холодильник 4, а оттуда в
сборник (на рисунке не показан). В дефлегматор 7 из
напорного бака 8 непрерывно поступает сверху 68—70%-ная
кислота; содержание H2SO4 в парах над такой кислотой
практически равно нулю (см. рис. 59). Из котла пары кислоты,
имеющие такой же состав, как и кипящая кислота, поступают на
первую снизу тарелку дефлегматора 7, где кипит более
разбавленная кислота, чем в котле. На этой тарелке содержание
серной кислоты в парах, находящихся в равновесии с кипящей
жидкостью, будет меньше, чем в парах, поступающих из
выпарного котла, а потому часть серной кислоты из паров
перейдет в жидкость, и в выпарной котел будет стекать более
концентрированная кислота, чем поступает на первую тарелку с
второй. Наоборот, в парах, переходящих с первой тарелки на
вторую, содержание серной кислоты будет меньше, чем в парах,
поступающих на первую тарелку из котла. Таким же образом
протекает процесс и на следующих тарелках дефлегматора.
Если на какой-нибудь тарелке кислота содержит
менее 70% H0SO4, то в парах, находящихся в равновесии с
жидкостью, даже при температуре кипения будет содержаться
только вода.
Теоретически в парах, отходящих из дефлегматора, не
должно быть серной кислоты. Практически в этих парах лишь в
незначительном количестве присутствуют увлеченные ими брызги
кислоты, которые задерживаются в ловушке 6. Пары воды из
ловушки поступают в конденсатор 5, где конденсируются,
смешиваясь с водой; конденсат удаляется в канализацию.
Благодаря такому способу конденсации в аппаратуре поддерживается
давление несколько ниже атмосферного.
Преимуществом установок этого типа является возможность
получения более концентрированной кислоты (95—98% H2SO.i)i
к недостаткам их относятся большой расход топлива и
сравнительно малый срок службы котлов.
Концентрирование в вакууме. Более совершенным и
экономичным способом концентрирования серной кислоты, по
сравнению с описанными, является концентрирование в вакууме с
передачей тепла через стенку. Для этого процесса разработаны
конструкции аппаратов с литыми нагревательными элементами
(трубчатка), выполненными из материала, стойкого к действию
кипящей серной кислоты при температурах 300—320°С
(температуры греющего агента). В качестве такого материала
может быть применен никелькремнистый сплав A1,2—12% Si,
4—4,5% Си и до 0,8% других примесей).
\ 5=>
Концентрирование серной кислоты б вакууме, введенное в
практику сравнительно недавно, обладает большими
преимуществами. Благодаря применению значительного разрежения в
таких установках удается концентрировать серную кислоту при
довольно низких температурах, что предотвращает ее
разложение.
вода
Рис. 62. Вакуум-концентратор периодического
действия:
1—цнлпндр; 2—нагревательные трубы; 3—опорная полая
колонка; 4—крышка; 5—смотровое стекло; 6—брызгоуловнтель;
7—плровой эжектор; 8—барометрический конденсатор;
У—барометрическая трубка: 10—лаз; 11—термометры; 12— кран
для впуска и выпуска кислоты.
В вакууме кислоту можно нагревать до температуры ее
кипения (Гш—150 °С) водяным паром (8—10 ат) через стенку.
Срок службы вакуум-аппаратов больше, чем обычных котлов,
благодаря значительно менее жестким условиям (в смысле
коррозии). Кроме того, в вакуумных концентраторах меньше
теряется серной кислоты с отходящими парами.
Вакуум-концентратор периодического действия (рис. 62)
чредставляет собой стальной цилиндр 1 с крышкой 4,
футерованный изнутри листовым свинцом и кислотоупорной керамикой
щи андезитом. В центре аппарата для жесткости установлена
полая керамическая колонка 3, служащая опорой для брызго-
уловителя 6. Брызгоуловитель состоит из двух керамических
фильтров, в которых создается лабиринтный ход для паров,
выходящих из концентратора. Улавливаемые брызги кислоты
стекают в виде капель обратно в концентратор. По
окружности корпуса аппарата радиально расположены 16 секций
нагревательных труб 2 (по восемь труб в секции) из
кислотоупорного чугуна A1,2—12% Si). Греющий глухой водяной пар
F—8 ат) подается параллельно во все секции сверху вниз.
Серная кислота F8—70% H2SO4) поступает в концентратор
снизу по трубе с краном 12. По окончании концентрирования
через этот же кран из аппарата отводится купоросное масло.
Пары из концентратора отсасываются паровым эжектором 7
в барометрический конденсатор 8, где конденсируются при
смешении их с холодной водой. Кислый конденсат стекает в
приемник через барометрическую трубу 9. Воздух удаляется из
конденсатора при помощи двухступенчатого парового. эжектора
(на рисунке не показан) и удаляется в атмосферу.
Концентрирование 68%-ной кислоты в аппарате начинается
при 110°С и остаточном давлении около 100 мм рт. ст.,
поддерживаемом при помощи барометрического конденсатора 8.
По мере повышения концентрации кислоты остаточное
давление в аппарате уменьшается.
Вакуум-концентратор производительностью 100 г серной
кислоты в сутки (при начальной концентрации кислоты 68%
H2SO4) имеет диаметр около 5 ж и высоту 6 м. Поверхность
греющих труб 100 м2.
Примерный режим работы такого вакуум-концентратора:
Температура исходной кислоты F8% H9SO4), °С 75
Температура упаренной кислоты (93% H2SO4), °C 149
Содержание H2SO4 в конденсате, % 0,032
Потери кислоты, % 0,5—2
Примерные расходные коэффициенты в расчете на 1 /
H2SO4 (при начальной концентрации кислоты 68%):
Пар (8 am), m 1,2
Вода B0°С), м3 56,5
Электроэнергия, квпг-ч .... До 0,2
При наличии двух вакуум-аппаратов процесс
концентрирования можно осуществлять непрерывно. В этом случае п пер-
вом (по ходу кислоты) концентраторе кислоту упаривают с
68 до 81% H2SO4, а во втором —с 81 до 93% H2SO4. Горячая
кислота, непрерывно вытекающая из второго концентратора,
охлаждается в холодильнике, откуда поступает в сборник и
далее на склад или непосредственно на использование.
Сорта серной кислоты
В настоящее время в Советском Союзе выпускается
контактная, башенная и регенерированная серная кислота. В
соответствии с ГОСТ основные сорта серной кислоты должны
удовлетворять следующим техническим требованиям:
Содержание. %. не менее
HaSO4 ..'.....
свободный SO3 . . .
Примеси, %, не более
окислы азота (в
пересчете на N2O3) . .
прокаленный" остаток
железо
мышьяк
НС!
техническая
92,5
—
—
—
Контактная
улучшенная
92,5—94
0,0001
0.02—0,03
0,01—0,015
0,0001
0,0005—
—0,001
олеум
технический
—
18,5
0,0005
0,02
0,0075
0,0001
Башенная
75
0,03
—
—
Регенерированная
91
0,01
—
—
В производстве искусственного волокна потребляется
только контактная улучшенная серная кислота. В контактной
технической серной кислоте, поставляемой пищевой
промышленности, содержание мышьяка не должно превышать 0,0001%.
содержание окислов азота (в пересчете на N2O3)—не более
0,0001%.
В зимний период (с 1 ноября по 15 апреля)
заводы-изготовители обязаны отгружать башенную кислоту, содержащую в
среднем 74—75% H2SO4, контактную и регенерированную
кислоту с содержанием не более 94% H2SO4, олеум с содержанием
свободного SO3 не более 22%. Это обусловлено температурами
кристаллизации серной кислоты (стр. 60).
Кроме перечисленных сортов серной кислоты, выпускается
аккумуляторная серная кислота (92—94% H2SO4), применяемая
для заливки свинцовых аккумуляторов. В зависимости от
содержания примесей аккумуляторная кислота подразделяется на
два сорта А и Б.
158
0,03
0,00005
0,006
0,00005
0,00005
0.00005
0,
0,
0,
0,
0,
0,
05
0001
012
0001
0001
0005
Допустимое количество примесей в аккумуляторной кислоте
в %, не более):
Нелетучий от < го;* .
Марганец ...
Железо
Мышьяк
Окислы азог;!
НС1
Анализ серной кислоты. Готовую (продукционную) серную
кислоту подвергают анализу для определения всех показате-
тей, предусмотренных ГОСТ. Концентрацию серной кислоты
определяют титрованием 0,5 н. раствором щелочи и выражают
в весовых процентах H2SO4. Концентрацию кислоты можно
также найти по таблицам, определив плотность кислоты (в г/см3).
В олеуме определяют содержание SO3 (своб.) и примесей.
Хранение и перевозка серной кислоты
Для хранения готовой кислоты на каждом сернокислотном
¦;азоде предусматривается склад. На складе серную кислоту
хранят в закрытых цилиндрических (горизонтальных или
вертикальных) стальных резервуарах емкостью до 1000 мъ,
размещаемых в здании, под навесом или на открытой площадке.
Башенную и контактную кислоту, а также олеум хранят в
стальных нефутерованных баках, кислоту, содержащую менее 75%
H2SO4, — в стальных баках, футерованных кислотоупорным кир-
шчом. В северных районах Советского Союза, где температура
а зимнее время может быть ниже температуры
кристаллизации серной кислоты, хранилища размещают в отапливаемых
помещениях. В средней полосе и южных районах страны в
отепленных складах хранят только олеум.
Техническую серную кислоту перевозят в железнодорожных
стальных цистернах грузоподъемностью до 50 т. Отгрузка
мелких партий серной кислоты допускается в контейнерах, бочках
емкостью до 1 т и в стеклянных бутылях емкостью 30—45 л.
Перевозка олеума производится в специальных цистернах,
снабженных теплоизоляцией. Улучшенную серную кислоту,
предназначенную для производства искусственного волокна, следуе-
перевозить в цистернах из нержавеющей стали, приписанных
к заводам-изготовителям кислоты. Аккумуляторную кислоту
перевозят в специальных контейнерах или стеклянных бутылях.
Перед заливкой серной кислоты в цистерны, контейнеры,
бочки и бутыли вся тара должна быть осмотрена и проверена
но отсутствие загрязнений и посторонних веществ. Заливку кис-
юты и выгрузку ее из цистерн производят при помощи сифона.
159
Цистерны снабжаются воздушным куполом на случай
теплового расширения кислоты при повышении температуры. Для
внутреннего осмотра цистерны в ней имеется лаз.
Бутыли с кислотой помещают в ивовые плетеные корзины
или в деревянные обрешетки, по высоте достигающие уровня
горла бутыли. Снизу и с боков бутыли тщательно обкладывают
соломой или мелкой древесной стружкой. Бутыли
закупоривают стеклянными или обожженными глиняными пробками,
которые заливают алебастром или замазкой.
6. КОНТРОЛЬ И АВТОМАТИЗАЦИЯ В ПРОИЗВОДСТВЕ
СЕРНОЙ КИСЛОТЫ
Автоматизация производства серной кислоты позволяет
значительно улучшить основные технико-экономические показатели
технологического процесса; сократить расход сырья и
катализатора, удлинить сроки службы основного оборудования
(печи, контактные аппараты, насосы, холодильники и др.),
увеличить производительность систем вследствие достижения
более высокой степени контактирования и улучшения переработки
газа в продукционных башнях, а также дает возможность
уменьшить количество обслуживающего персонала, улучшить
и облегчить условия труда.
Затраты на автоматизацию сернокислотного производства
окупаются в течение короткого времени (за 1—1,5 года).
Контрольно-измерительные приборы
Производство серной кислоты является непрерывным
процессом, поэтому для его контроля применяют в основном
автоматические постояннодействующие приборы, показывающие и
записывающие, а также подающие соответствующие сигналы
при отклонении параметров процесса от заданных значений.
Автоматические приборы могут быть установлены на
значительном расстоянии от точки замера параметров, показания
приборов выносятся на щит управления.
В производстве серной кислоты контролируются следующие
параметры процесса: концентрация SO2 в печном газе;
концентрация серной кислоты, олеума, окислов азота и тумана серной
кислоты в газе; давление и разрежение в системе; температуры
газов и кислоты, уровни кислоты в сборниках и др. В качестве
контрольно-измерительных приборов применяются
газоанализаторы, концентратомеры, туманомеры, термометры,
уровнемеры и т. д.
Определение концентрации сернистого ангидрида в печном
газе и в газе на входе в контактный аппарат производится
автоматическими газоанализаторами со шкалой 0—20% SO2.
160
определение концентрации сернистого ангидрида в газе,
выходящем из контактного аппарата, —газоанализаторами со
шкалой 0—1 и 0—1,5% SO2.
Принцип действия этих газоанализаторов основан на
изменении теплопроводности анализируемой газовой смеси в
зависимости от ее химического состава.
Перед поступлением в автоматический газоанализатор
сернистый газ предварительно очищается от пыли в керамическом-
фильтре и от тумана серной кислоты в специальном
электрофильтре.
Концентрация тумана серной кислоты в газах после Мокрых
электрофильтров автоматически определяется
фотоэлектрическим туманомером со шкалой 0—0,05% H2SO4. При помощи
этого прибора измеряется прозрачность анализируемого газа,
изменяющаяся при различном содержании в нем туманообраз-
ной серной кислоты.
Для автоматического измерения концентрации окислов
азота в выхлопных газах башенных систем применяются
фотоэлектрические колориметры. Относительная погрешность
фотоэлектрического колориметра составляет ±7%.
Уровни серной кислоты измеряются в сборниках
промывного и сушильно-абсорбционного отделений и в хранилищах
кислоты на складах готовой продукции. Принцип действия
уровнемеров основан на измерении гидростатического давления
столба жидкости в сосудах. Конструкции приборов отличаются друг
от друг,а лишь способом передачи показаний на расстояние.
Практически применяются приборы с электрической и
пневматической передачей показаний.
Для измерения расхода серной кислоты в сушильно-абсорб-
ционных отделениях контактных систем и количества
продукционной кислоты, поступающей на склад, используются
щелевые расходомеры типа РМ. Принцип действия такого
расходомера основан на измерении напора кислоты над сливной щелью,
через которую кислота поступает в приемник, находящийся под
атмосферным давлением. Расходомеры РМ выпускаются на
следующие количества кислоты: 10, 20, 30 и 50 жг\я.
Погрешность приборов по техническим условиям не должна превышать
±3,5% максимального значения шкалы.
Концентрация серной кислоты определяется по ее плотности.
Для этих измерений используются пьезометрические
плотномеры со шкалой 66—71 % H2SO4 (относительная погрешность
±4%). Прибор устанавливается на технологическом кислото-
проводе при расходе кислоты через датчик до 50 мг/ч.
Контроль кислотности сточных вод из оросительных
холодильников осуществляется при помощи рН-метров. Основная
погрешность прибора при рН = 2—7 и температуре воды до 30 °С
составляет ±0,15 измеряемой величины рН.
П-2848 161
Сигнализаторы кислотности сточных вод работают по
принципу изменения электропроводности водных растворов при
попадании в них кислот.
При наливе кислот и олеума в железнодорожные цистерны
применяются сигнализаторы уровня.
Автоматические регуляторы
Для автоматического регулирования температуры газа к
кислот на сернокислотных заводах применяются схемы
регулирования, оснащенные серийными датчиками и регуляторами.
Автоматическое регулирование концентрации серной
кислоты осуществляется электрическими регулирующими приборами,
работающими с концентратомерами. Кроме электрических
регуляторов, в сернокислотной промышленности применяются
также пневматические регуляторы с электронными
потенциометрами и мостами. В комплекте с термопарами и термометрами
сопротивления эти приборы используются в схемах
регулирования температуры.
Для автоматического регулирования уровня кислоты
применяются электронные регулирующие приборы, работающие з
комплекте с электрическим уровнемером.
В последнее время все большее применение получают также
автоматические регуляторы агрегатной унифицированной
системы (АУС).
АВТОМАТИЗАЦИЯ КОНТАКТНОГО ПРОЦЕССА
Схема комплексной автоматизации контактной
сернокислотной системы, работающей на серном колчедане, включает
автоматизацию печного, сушильно-абсорбционного и контактного
отделений.
Схема включает также автоматизацию процесса сушки
колчедана.
Автоматизация сушки колчедана. Основным назначением
автоматизации в отделении сушки колчедана является
обеспечение постоянной влажности колчедана после сушильных
барабанов (в пределах 1,5—3,0%). Эго достигается путем
установки специальных питателей, равномерно подающих в барабан
строго определенные количества колчедана, а также
автоматическим поддержанием постоянной температуры в топке
сушильного барабана. Для поддержания постоянства температуры
топочных газов автоматически регулируется количество
сжигаемого топлива и воздуха, подаваемого в топку.
Термопары, установленные в барабане, подают через
соответствующие устройства импульсы регуляторам подачи топлива
и воздуха.
162
Автоматизация печей КС Автоматизация таких печей
значительно проще, чем механических полочных печей. Вследствие
интенсивного теплообмена в кипящем слое его температура
почти одинакова, благодаря чему температурный режим печи
легко регулируется.
Основным условием нормальной работы печей КС (и печей
других типов) является подача одинакового объема газа с
постоянной концентрацией SO2. Для стабилизации процесса
обжига в кипящем слое автоматически регулируются: концентрация
SO2 в газе, количество воздуха, поступающего в печь, высота
кипящего слоя и разрежение в печи. На рис. 63 показана
принципиальная схема автоматизации печи КС-100. Постоянство
объема сернистого газа и концентрации в нем SCb на выходе из
Г
Первичный
воздух
Рис. 63. Принципиальная схема автоматизации печи КС-100:
/—питатель (дозатор колчедана); 2—печь; 3, 4—воздуходувки; 5—котел-утилизатор; б—циклон;
7—электрофильтр; 8—вентилятор; 9—шнековый транспортер; Pi—Pi—регуляторы; а,
б—дроссельные заслонки.
печи поддерживается путем автоматического регулирования
подачи воздуха и колчедана в печь в зависимости от
температуры отходящих газов. Количество воздуха, подаваемого в печь,
регулируется при помощи регулятора Р\, воздействующего на
положение дроссельной заслонки а в патрубке воздуходувки 3.
Стабильность концентрации SO2 в газе перед электрофильтром
обеспечивается автоматическим регулятором Р% путем
изменения числа оборотов питателя /, подающего колчедан в печь.
Высота кипящего слоя в печи регулируется скоростью удаления
огарка путем изменения регулятором Pi скорости вращения
разгрузочного шнека 9 или степени открытия секторного
затвора на выгрузке огарка. Постоянное разрежение в верхней части
печи поддерживается регулятором Рц, который соответственно
изменяет положение дроссельной заслонки б перед
вентилятором 8.
11*
163
Автоматизация контактного отделения. Для достижения
наибольшей производительности контактного аппарата при
заданном составе и количестве газа необходимо поддерживать такой
режим контактирования, при котором обеспечивается
наибольшая степень окисления SO2 в SO3. Это возможно при ведении
процесса в условиях, близких к оптимальному температурному
режиму, когда реакция окисления протекает с наибольшей
скоростью. Отклонение от оптимальной температуры на 10°С
приводит к изменению скорости реакции почти на 10%. Эти
изменения температуры обусловлены колебаниями концентрации
SO2 в газе. При изменении концентрации SO2 на 1%
температура газа повышается почти на 30 °С.
Рис. 64. Принципиальная схема автоматизации четырех-
слойного контактного аппарата:
1—-сушильная башня; 2—брызгоуловитель; 3—турбокомпрессор; 4—
наружный теплообменник; 5—контактный аппарат; а—термопары;
6—регуляторы температуры; в—исполнительные механизмы; г—кла.
Для более эффективной работы контактного аппарата
необходимо одновременно регулировать концентрацию SO2 в
поступающем газе с точностью до ±0,3% и температуру газа с
точностью ±2°С. Такая точность достигается при
автоматическом регулировании концентрации. Принципиальная схема
автоматизации контактного аппарата, применяемая на
сернокислотных заводах, изображена на рис. 64.
Постоянная температура газа на входе в контактный
аппарат поддерживается следующим образом. Термопара,
измеряющая температуру входящего газа, воздействует при помощи
соответствующих устройств на клапан г, изменяющий
количество холодного газа, поступающего по байпасной (обводной)
линии, помимо теплообменника 4. При постоянной температуре
газа на входе в первый слой контактной массы температура
газа по выходе из этого слоя (при постоянном объеме газа)
164
зависит только от концентрации SO2 в поступающем газе. Эта
зависимость используется для поддержания постоянной
концентрации SO2.
Система регулирования температуры газа на входе в
аппарат состоит из термопары а, регулятора б и исполнительного
механизма в, перестанавливающего клапан г на линии
холодного газа. Система регулирования температуры газа на выходе,
из первого слоя состоит из термопары а, регулятора б и
исполнительного механизма в, управляющего подсосом воздуха
из атмосферы через клапан г (заслонку) перед сушильной
башней 1.
При постоянной температуре газа после первого слоя
контактной массы и постоянной концентрации SO2 в газе
температурный режим устойчив и регулирование его требуется в
редких случаях.
Рис. 65. Схема автоматизации абсорбционносушильного отделения:
1> 2—рушильные башни; 3—моногидратный абсорбер; 4—олеумный абсорбер; 5—сборники;
6—насосы; 7—напорные бачки; Pi—P4—регуляторы концентрации кислоты1.
Р5—Pg—регуляторы уровней кислоты; К\—Кд—регулирующие клапаны.
Автоматизация абсорбционно-сушильного отделения. Осушка
сернистого газа в сушильных башнях и абсорбция серного
ангидрида в абсорберах являются отдельными стадиями
технологического процесса, однако вследствие тесной связи между
этими процессами* и однотипности аппаратуры схемы
автоматизации этих отделений объединены.
На рис. 65 изображена принципиальная схема
автоматизации абсорбционно-сушильного отделения системы, работающей
при переменной концентрации SOs в газе. Важнейшим парамет-
* Между сушильным и абсорбционным отделениями существует
постоянный переток кислот; сушильная кислота поступает в сборник моногидрат-
ного абсорбера для разбавления кислоты, которая поступает в сборник
сушильной башни для укрепления сушильной кислоты.
165
ром, определяющим ход технологического процесса в этом
отделении, является концентрация орошающих кислот. На входе
в первую сушильную башню оптимальная концентрация
кислоты составляет 93,3% H2SO4.
Постоянная концентрация олеума поддерживается
регулятором Р\, воздействующим на клапан К[, установленный на линии
подачи моногидрата в циркуляционный сборник 5 олеумного
абсорбера. Постоянство концентрации кислоты, орошающей
моногидратный абсорбер 3, обеспечивается регулятором Р-г
путем изменения подачи сушильной кислоты через клапан Кг ч
воды в циркуляционный сборник 5 через клапан Кэ- Вода
подается в этот сборник в том случае, когда поток сушильной
кислоты не разбавляет моногидрат до нужной (оптимальной)
концентрации. При снижении концентрации кислоты,
орошающей абсорбер 3, сначала полностью закрывается клапан Кэ,
затем клапан Кг-
Концентрация сушильной кислоты в башне 2
поддерживается регулятором Рз, воздействующим на клапан Л'з, через
который кислота подается в сборник 5 этой башни.
Концентрация сушильной кислоты в цикле башни / поддерживается
регулятором Pt путем взаимосвязанного регулирования подачн
олеума и сушильной кислоты в соответствующие сборники 5.
Одновременно с изменением подачи олеума в сборник 5 через
клапан Ка регулятор Р\ подает импульс на изменение подачи
кислот в сборники 5 через клапан Кг и через клапан Ке-
Постоянный уровень в олеумном сборнике поддерживается
регулятором Рв путем передачи олеума на склад (через клапан
Ks)- Постоянный уровень в моногидратном сборнике
обеспечивается регулятором Ре путем передачи кислоты в сборник
первой сушильной башни (через клапан Д'б). Регулятор уровня
Pi в сборнике сушильной башни 2 управляет передачей
кислоты из цикла этой башни в сборник кислоты первой сушильной
башни (через клапан Кт). Регулятор уровня Р8 в сборнике
сушильной башни 1 управляет подачей кислоты на склад (через
клапан Kg).
Описанная схема автоматизации сушильно-абсорбционного
отделения осуществлена на практике и позволяет поддерживать
концентрацию кислот с точностью до ±0,2% и уровни их в
сборниках с точностью ±5 см.
АВТОМАТИЗАЦИЯ ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
ИЗ СЕРОВОДОРОДА
На принципиальной технологической схеме (см. рис. 45,
стр. 126) процесса получения серной кислоты из серозодород-
ного газа высокой концентрации показаны регулирующие
166
устройства. Непременным условием нормальной работы
установки является постоянство количества поступающего в
систему газа и концентрации в нем H2S. Определенное соотношение
между количеством поступающего в печь сероводородного газа
и воздуха поддерживается регулятором а. Изменение
концентрации H2S в сероводородном газе вызывает изменение
температуры газа на выходе из печи. Термопара в, измеряющая
температуру в газоходе после печи, через особые устройства
воздействует на регулятор а, который увеличивает или уменьшает
количество поступающего воздуха на единицу объема
сероводородного газа. Таким образом поддерживается заданная
концентрация SOo в газе на выходе из печи. Постоянство
температуры газа на входе в контактный аппарат поддерживается
путем воздействия термопары в на клапан б, регулирующий
количество воздуха, добавляемого к газу. Поддержание
заданных (оптимальных) температур газа на входе во второй, третий
и четвертый слои контактной массы достигается путем
блокировки термопар, измеряющих температуры газа в этих слоях,
с клапанами б, регулирующими количество воздуха,
добавляемого к газу.
Поддержание постоянного температурного режима процесса
образования серной кислоты в башне-конденсаторе 7
обеспечивается воздействием термопары, измеряющей температуру
кислоты, на ктапан, регулирующий количество воды, которое
подается на орошение холодильника 9. Регулирование
количества продукционной серной кислоты, перекачиваемой на склад,
обеспечивается воздействием регулятора г уровня кислоты в
сборнике 10 на клапан б, изменяющий количество выводимой
из системы кислоты. Поддержание постоянного уровня воды в
паровом котле-утилизаторе 5 достигается воздействием
регулятора уровня г на клапан б, изменяющий количество свежей
воды, поступающей в котел-утилизатор.
АВТОМАТИЗАЦИЯ БАШЕННОГО ПРОЦЕССА
Для нормальной работы башенных систем необходимо
постоянство количества и концентрации SO2 в печном газе,
постоянное содержание серной кислоты и окислов азота в нитрозе
н постоянство температуры и плотности орошения башен. В
интенсивных башенных системах изменение концентрации SO2 в
газе на 1% (с 8 до 9%) вызывает увеличение содержания окиси
азота в выхлопных газах на 0,6—0,8%.
Схемой автоматизации башенной системы
предусматриваются автоматический контроль следующих параметров:
концентрации SO2 перед первой башней н расхода газа через всю
систему башен, температуры газа и разрежения на входе в
башни и выходе из них, плотности (концентрации) орошающих
167
кислот и расхода нитрозы, количества продукционной кислоты,
поступающей на склад, и уровней кислоты в сборниках. Кроме
того, автоматизированы подготовка окислов азота к абсорбции
и регулирование содержания их в выхлопных газах.
При обжиге колчедана в механических печах концентрация
SO2 в газе стабилизируется путем регулирования степени
разбавления печного газа воздухом по показаниям электрического
газоанализатора.
Концентрация H2SO4 в продукционной кислоте, содержащей
около 0,03% окислов азота, определяется концентратомерами
со шкалой 72—78 % H2SO4.
Количество нитрозы, подаваемой на орошение башен,
автоматически измеряется щелевыми расходомерами, плотность
нитрозы определяется плотномерами, количество воды и
меланжа— ротаметрами. Для автоматического измерения
концентрации окислов азота применяются фотоколориметры.-
Техника безопасности в производстве
серной кислоты
В производстве серной кислоты должны быть созданы такие
условия труда, при которых исключается возможность
отравления работающих вредными газами (SO2, SO3, окислы азота),
парами серной кислоты и вдыхания пыли (колчеданной и огар-
ковой), а также возможность ожогов кислотами.
По существующим правилам и нормам техники безопасности
и промышленной санитарии в производстве контактной и
башенной серной кислоты, предельно допустимые концентрация
вредных газов в воздухе рабочей зоны производственных
помещений не должны превышать следующих величин (в мг/м3):
Сернистый ангидрид .... 10 Сероводород 10
Серный ангидрид 1 Окислы азота (в пересчете
Двуокись селена 0,1 на NaO5) 5
Мышьяковистый ангидрид . 0,3
Предельно допустимое содержание пыли колчедана и
огарка в воздухе рабочей зоны производственных помещений 2мг/м3.
В производстве серной кислоты контактным и башенны\г
способами необходимы установки для очистки газов от пыли,
паров, брызг и тумана кислоты.
Во всех помещениях сернокислотного производства должна
предусматриваться исправно действующая естественная,
искусственная и смешанная вентиляция.
Для уменьшения вредных выделений в воздух рабочих
помещений аппаратура и трубопроводы должны быть тщательно
168
герметизированы и в случае необходимости снабжены
теплоизоляцией. Оборудование для перемещения, дробления и
просеивания сыпучих материалов необходимо снабжать уплотнениями
и приспособлениями для отсоса пыли.
Обслуживающий персонал в производстве серной кислоты
обеспечивается спецодеждой, спецобувью и средствами
индивидуальной защиты (противогазы, респираторы, шлемы,
защитные очки, маски и др.) в соответствии с действующими
правилами и нормами и условиями работы.
При повышении концентрации газов сверх допустимых
санитарных норм (например, при внезапной остановке системы,
авариях и пр.) все работающие в цехе должны надеть
противогазы. Операции, при которых возможно соприкосновение с
кислотой (вследствие проливов, брызг и пр.), необходимо
выполнять в защитных очках, в шерстяной или суконной спецодежде
и резиновых перчатках.
На работах, связанных с возможностью поражения
электрическим током, применяются специальные защитные
(изолирующие) средства и приспособления.
ЛИТЕРАТУРА
(к главам III и IV).
С. И. В о л ь ф к о в и ч, А. П. Егоров, Д. А. Эпштейн, Общая
химическая технология, т. I, Госхимиздат, 1952.
Самородная сера, Труды Государственного научно-исследовательского
института горнохимического сырья (ГИГХС), вып. 6 Госгортехиздат,
1960.
К. М. Малин и др., Технология серной кислоты, Госхимиздат, 1950.
И. Н. Кузьминых, Технология серной кислоты, Госхимиздат, 1955.
А. Г. Амелин, Производство серной кислоты, Госхимиздат, 1956.
А. Г. А м е л и н, Производство серной кислоты из сероводорода по методу
мокрого катализа, Госхимиздат, 1960.
Г. К- Б о р е с к о в, Катализ в производстве серной кислоты, Госхимиздат,
1954.
А. Г. Амелин, Л. Г. П л и с к и н, Н. Н. Ш у м и л о в с к и й, Основы
автоматизации производства серной кислоты контактным методом,
Госхимиздат, 1961.
Г, М. Ф и а л к о, Автоматизация производства серной кислоты,
Машиностроение, 1964.
Правила и нормы техники безопасности и промышленной санитарии для
проектирования, строительства и эксплуатации производства серной
кислоты контактным и башенным способами, Госхимиздат, 1961.
Глава V
ПРОИЗВОДСТВО ВОДОРОДА, АЗОТА И КИСЛОРОДА
/. ОБЩИЕ СВЕДЕНИЯ
Водород, азот и кислород являются исходными веществами
для синтеза важнейших химических продуктов — аммиака,
азотной кислоты и др. Водород получают переработкой природного
и попутных нефтяных газов, коксового газа, мазута, кокса,
угля, газов нефтепереработки, электролизом воды, при которое
получается также кислород. Разделением воздуха получают
азот и кислород.
Водород применяют в огромных количествах для
производства синтетического аммиака, гидрирования жидких топ л ив
(мазут н др.), гидрирования жидких жиров для получения
твердых жиров, используют во многих процессах органического
синтеза, при сварке и резании металлов и т. д. Большие
количества водорода в виде смеси с окисью углерода (так
называемый синтез-газ) применяются для получения метилового и
высших спиртов.
Азот применяется для получения азотоводородной смеси,
необходимой в синтезе аммиака, используется в производстве
цианамида кальция (стр. 606) и в других процессах
азотирования, употребляется как инертный газ при тушении пожаров
(стр. 260) и для создания безопасной среды в аппаратуре и
коммуникациях ряда производств.
Кислород используется в разнообразных процессах
окисления, газификации топлива, конверсии углеводородных газов
(стр. 179 ел.), применяется в качестве дутья в металлургических
процессах для интенсификации выплавки металлов, а также
для многих других целей.
В данной главе рассматриваются главным образом методы
производства водорода (и азотоводородной смеси) для синтеза
аммиака. Применяются следующие промышленные методы
производства водорода:
1. Каталитическая конверсия природного газа под
давлением 1,9, 16 и 30 ат и высокотемпературная конверсия его под
давлением до 30 ат.
2. Газификация твердого и жидкого топлива с получением
генераторных газов (водяной и полуводяной газ).
170
3. Глубокое охлаждение коксового газа (стр. 223 ел.).
4. Электролиз воды (стр. 314 ел.).
В отдельных случаях для небольших установок может быть
использован Еодород, получаемый в качестве побочного
продукта при электролизе раствороз поваренной соли (стр. 336).
До недавнего времени на некоторых старых заводах синтетического
аммиака водород получали железо-паровым способом. В настоящее время
этот способ вследствие дороговизны, малой производительности и других
недостатков потерял промышленное значение и потому здесь не описывается.
До 1940 г. основное количество водорода для синтеза
аммиака получали из водяного и коксового газов и электролизом
воды. В настоящее время все возрастающее значение в
качестве источника сырья для азотной промышленности
приобретают природный газ, мазут и газы нефтепереработки (табл. 7).
Таблица 7
Источники сырья азотной промышленности в капиталистических странах
Источники сырья
Доля, %
1929 г.
1939 г.
1956 г.
1959 г.
Газы, получаемые газификацией твердого топлива .
Коксовый газ
Электролиз воды
Природный газ
Мазут и газы нефтепереработки
Прочие источники
65
16
18
54
27
18
1
26
23
10
37
4
21
20
8
36
15
В азотной промышленности Советского Союза
используются различные источники исходного технологического газа
(табл. 8).
Таблица 8
Источники сырья азотной промышленности в СССР
Источники сырья
3,0ля, %
1955 г.
45
34
21
1958
44,
34,
0,
20.
-¦
3
7
6
4
I960
23
40
25
9
г.
9
7
6
8
1965 г.
14,6
17,5
62,6
J
5,3
Газы, получаемые газификацией твердого топлива .
Коксовый газ
Природный и попутный газы
Остаточные газы производства ацетилена из
природного газа
Прочие источники
В связи с бурным ростом добычи природного газа и
кооперированием азотной промышленности с другими химическими
производствами синтетический аммиак получается в СССР з
171
основном из газового сырья (природный газ, попутные
нефтяные газы, коксовый газ, а также остаточные газы производства
ацетилена окислительным пиролизом метана).
Использование для получения водорода природного и
попутных нефтяных газов значительно удешевляет производство
синтетического аммиака, так как исключается сложный процесс
газификации твердых топлив, а вследствие малого содержания
соединений серы в природных газах значительно упрощается
очистка азотоводородной смеси для синтеза аммиака.
Расчетные технико-экономические показатели производства
синтетического аммиака при использовании различных видоз
сырья приведены ниже (в %):
Удельные Заводская
капитало- себестои-
вложения мость 1 т
(на I m аммиака
аммиака)
Газификация кокса 100 100
Газификация бурого угля 115 95
Переработка коксового газа 85 60
Переработка природного газа 80 50
Электролиз воды , . , 160 125
Таким образом, наиболее низкие себестоимость и удельные
капиталовложения достижимы при синтезе аммиака из
продуктов переработки природного газа.
2. ПРОИЗВОДСТВО ВОДОРОДА КОНВЕРСИЕЙ УГЛЕВОДОРОДНЫХ
ГАЗОВ
В настоящее время производство водорода методом
конверсии углеводородных газов приобретает важное значение, так
как ресурсы природного и попутного газов весьма велики, а
переработка их в синтетический аммиак дешевле переработки
других видоз сырья в синтез-газ.
Конверсию углеводородных газов, в частности метана, для
получения водорода, необходимого при синтезе аммиака,
ведут, применяя в качестве окислителя водяной пар или
кислород. Основные реакция конверсии:
СН4 + Н2О = СО + ЗН4 — 49,27 ккал A)
СН4 + 0,5О2 = СО + 2Н4+8,3 ккал B)
СО + Н2О = СО8 + Н2 + 9,8 ккал C)
СН4 + СО2 = 2СО + 2Н2 — 59,3 ккал D)
Реакция D) здесь не рассматривается, так как синтез-газ,
образующийся при ее протекании, применяется для получения
метанола и других кислородсодержащих органических
соединений, но менее пригоден по составу для синтеза аммиака.
Как видно из приведенных выше реакций, вначале получают
смесь СО и Н2, затем окись углерода дополнительно конверт
тируется водяным паром с образованием водорода и CO
172
В реальных условиях ведения рассматриваемых
технологических процессов реакции A) и C) обратимы. Равновесный
состав продуктов реакции можно вычислить, зная константы
равновесия, величины которых изменяются с изменением
температуры. В табл. 9 приведены значения констант равновесия при
различных температурах. По этим значениям могут быть
определены парциальные давления всех компонентов реакционной
смеси.
Таблица 9
Константы равновесия реакций A), B), C) конверсии
Температура
"К
600
700
800
900
1000
1100
1200
1300
1400
°С
327
427
527
627
727
827
927
1027
1127
¦рсо-рн,
KPl = —
5,058-Ю-7
2,687-Ю-4
3,120-Ю
1,306
26,56
3,133-Ю2
2,473-Ю3
1,428- Ю4
6,402-Ю4
2
у СО' Н2
АР2 0 5
2,169-1012
1,028-1012
6,06 -ЮН
4,108-ЮИ
3,056-ЮН
2,392-10»
1,957-104
1,652-10»
1,425- ЮН
рСОо • РНг
Р3 РС°- РН2°
27,08
9,017
4,038
2,204
1,374
0,944
0,697
0,543
0,441
При указанных в табл. 9 температурах константы
равновесия реакции B) очень велики; следовательно, по достижении
равновесия концентрация кислорода в продуктах реакции
практически равна нулю, т. е. реакция идет до конца.
В табл. 10 приведен равновесный состав газовой смеси,
образующейся по реакции A), при стехиометрическом расходе
водяного пара и атмосферном давлении.
Таблица 10
Равновесный состав газовой смеси при протекании реакции A)
Температура
°К
1000
1100
1200
°с
727
827
927
5,08
1,71
0,64
Состав
Н2О
5,08
1,71
0,64
газовой смеси,
СО
22,46
24,14
24,68
объемн. %
н2
67,38
72,44
74,04
Как видно из этой таблицы, для того чтобы в равновесных
условиях достигалась практически полная конверсия метана,
необходимо вести реакцию A) при температуре немного выше
173
927°С. Однако при такой температуре скорость реакции
окисления метана водяным паром очень мала. Реакция протекает с
значительной скоростью лишь при температурах 1350 °С и
выше. Из-за трудности ведения эндотермического процесса при
столь высоких температурах в промышленных условиях
процесс паровой конверсии метана ведут на катализаторе в
присутствии избытка водяного пара по сравнению со стехиомет-
рическим количеством. При этом протекает также реакция C).
В табл. 11 приведен рассчитанный Б. П. Корниловым
равновесный состав газовой смеси, образующейся в процессе
паровой конверсии метана при различных температурах и давлениях
и соотношении CHt: НгО= 1 : 2 в исходной газовой смеси.
Таблица 11
Равновесный состав газовой смеси при паровой конверсии метана
(СН4:Н8О=1:2)
Абсолютное
давление
1
10
20
30
1
10
20
30
Состав, объемп. %
СО2
3,87
4,51
4,93
5,11
3,14
3,28
3,58
3,75
СО
При 1
16,08
13,18
10,71
9,08
При 1
16,85
16,20
14,97
13,80
Н2
аи
100 "К
63,73
57,59
51,85
47,72
0,08
3,85
7,27
9,66
200 °К
63,12
61,74
59,03
56,38
0,01
0,86
2,50
4,08
Н2О
16,24
20,87
25,24
28,43
*
16,88
17.92
19,97
21,99
В промышленности применяются три основных способа
конверсии метана:
1) каталитическая паровая конверсия;
2) каталитическая парокислородная или парокислородовоз-
душная конверсия;
3) высокотемпературная (некаталитическая) конверсия
кислородом или воздухом, обогащенным кислородом.
Обычно в процессах паровой каталитической конверсии в
начале конвертируется на катализаторе при 750—800°С большая
часть метана, затем оставшийся (непрореагировавший) метан
реагирует в другом аппарате с кислородом воздуха по реакции
B) также на катализаторе. Реакция A) протекает с
поглощением тепла, поэтому в реакционную зону подводится тепло
извне путем обогрева труб с катализатором горячими
топочными газами.
174
Процессы парокислороднои и парокислородовоздушной
конверсии метана являются экзотермическими.
Судя по величинам КР2 константы равновесия реакции B),
окисление метана кислородом до окиси углерода и водорода
происходит с достаточной полнотой в широком интервале
температур.
В табл. 12 приведен рассчитанный А. Г. Лейбуш
равновесный состав газовой смеси при различных температурах и
давлениях и следующем соотношении компонентов исходной
газовой смеси: СН4: Н2О : О2: N2= 1 : 1 : 0,6 : 09.
Таблица 12
Равновесный состав газовой смеси при парокислородовоздушной конверсии метана
(СН4 : Н2О : О2: N3= 1:1:0,6:0,9)
Абсолютное
давление
am
1
10
20
1
10
20
со2
5,95
6,45
7,01
5,01
5,09
5,29
СО
При
14,44
12,88
11,06
При
15,40
15,12
14,49
Состав, объемн. %
н2
100° К
42,62
39,10
34,79
1200 "К
41,73
41,10
39,62
0,03
1,81
3,95
0,003
0,32
1,06
N2
18,38
19,03
19,82
18,37
18,49
18,76
НоО
18,58
20,73
23,37
19,49
19,88
20,78
В промышленности каталитические процессы
парокислороднои и парокислородовоздушной конверсии ведут при 800—
1050°С, в зависимости от активности катализатора,
содержания соединений серы в исходном газе, заданного конечного
содержания метана, применяемого давления и других условий
процесса. Обычно применяется никелевый катализатор в виде
колец Рашига, цилиндриков или шариков.
В промышленности нашла широкое применение также
кислородная и кислородовоздушная конверсия метана в отсутствие
катализатора при 1275—1400 °С (так называемая
высокотемпературная конверсия).. Скорость взаимодействия метана с
кислородом при указанных температурах очень велика, однако
процесс сопровождается образованием сажи в результате
термической диссоциации метана по реакции:
СН,
. С + 2Н2
При температурах ниже 1275°С образуется значительное
количество сажи. При температуре 1350 °С и выше можно
избежать образования сажи.
175
Реакции A) и B) протекают с увеличением объема газа.
Далее переработка конвертированного газа в азотоводородную
смесь, а затем в аммиак проводится под давлением, поэтому
ведение описанных выше процессов конверсии метана также
под давлением позволяет снизить расход электроэнергии на
сжатие газа в компрессорах, сократить их количество,
уменьшить габариты теплообменных и контактных аппаратов и
улучшить использование тепла. Следует отметить, что при
проведении процесса каталитической конверсии метана под
давлением требуется в соответствии с принципом Ле-Шателье
повысить температуру процесса, чтобы сдвинуть в желательном
направлении состояние равновесия реакции A).
Высокотемпературную некаталитическую конверсию метана
под давлением ведут при той же температуре, что и под
атмосферным давлением, так как при столь высоких температурах
равновесные концентрации метана ничтожно малы.
Сопутствующая процессам конверсии метана реакция C)
конверсии окиси углерода водяным паром протекает частично.
Эта реакция является желательной, поскольку дальнейшая
переработка конвертированного газа в азотоводородную смесь
осуществляется также путем конверсии окиси углерода.
Как видно из табл. 9, при температурах процесса
каталитической конверсии метана лишь часть окиси углерода окисляется
на катализаторе водяными парами до двуокиси углерода с
образованием водорода. Реакция конверсии СО протекает с
выделением тепла и без изменения объема. Поэтому, согласно
принципу Ле-Шателье, понижение температуры
благоприятствует белее полной конверсии, а изменение давления не
сказывается на состоянии равновесия.
Рабочая температура для большинства современных
катализаторов конзерсии СО составляет 400—500 °С. Поэтому по
завершении процесса конверсии метана конвертируют окись
углерода при 400—500 °С на специальном катализаторе в
отдельном аппарате. Обычно стремятся к возможно более
полному превращению СО в СОг как для лучшего использования
газа, так и потому, что остающаяся в газе окись углерода
отравляет катализатор процесса синтеза аммиака, а удаление
окисн углерода из газовой смеси является сложной и дорогой
операцией. Разрабатывают также катализатор, на котором
конверсию окиси углерода можно вести при 200—250 °С. В этом
случае содержание СО снижается до 0,3%, что позволяет
значительно упростить очистку газа, применяя для этого лишь
предкатализ (стр. 200).
Судя по величинам константы равновесия реакции
конверсии окиси углерода при 400—500°С, при этих температурах
достаточно полная конверсия СО достигается лишь при большом
избытке водяного пара по сравнению со стехиометрическим его
176
количеством. Обычно применяют 1,1—2,5-кратныи избыток
водяного пара, в зависимости от начальной концентрации окиси
углерода. При этом содержание окиси углерода в газе после
конверсии не превышает 3—3,5%.
Ниже описываются наиболее распространенные схемы
процессов конверсии метана.
ПАРОВАЯ КАТАЛИТИЧЕСКАЯ ДВУХСТУПЕНЧАТАЯ КОНВЕРСИЯ
МЕТАНА И ОКИСИ УГЛЕРОДА
Схема процесса показана на рис. 66. Поступающий на
конверсию природный газ нагревается в трубчатом
теплообменнике 2 до 380—400 °С. Катализатор конверсии метана,
применяемый в этом процессе, сильно отравляется соединениями серы,
поэтому обычно природный газ предварительно тщательно очи-
Природнып газ
к горелкам лечи
{Конвертированный газ
Рис. 66. Схема паровой каталитической двухступенчатой конверсии метана
и окиси углерода при давлении, близком к атмосферному:
/—поглотитель соединений серы; 2—теплообменник; 3—пароперегреватель; 4—конвертор
окиси углерода; 5—смеситель; 5—котел-утилизатор; 7—увлажнитель; 8—конвертор метана
второй ступени; 9—трубчатая печь (конвертор метана первой ступени).
щают от сероводорода и органических соединений серы.
Содержание соединений серы в природном газе, направляемом в
конвертор, не должно превышать 2—3 мг/мъ. Очистку газа от серы
ведут в аппарате /при 380—400 °С на поглотителе,
приготовленном на основе окиси цинка.
Водяной пар, необходимый для процесса конверсии, также
нагревается до 380—400 °С в пароперегревателе 3 и затем
смешивается с очищенным природным газом в объемном
соотношении газ : пар = 1 : 2,5. Это соотношение поддерживается с
помощью автоматического регулятора. Парогазовая смесь при
380 °С поступает в трубчатую печь 9, где конвертируется
большая часть метана, содержащегося в природном газе. В печи
парогазовая смесь проходит по трубам, заполненным никелевым
12-2848 177
катализатором. Вследствие эндотермичности реакции
конверсии СН4 трубы снаружи обогреваются продуктами сгорания
природного газа, движущимися в межтрубном пространстве
печи. Греющие газы, имеющие начальную температуру
примерно 1000 °С, покидают трубчатую печь при 800—850 °С. Тепло
этих газов используется далее в котле-утилизаторе (на рисунке
не показан) для получения пара давлением до 40 ат.
В зоне катализатора парогазовая смесь нагревается до 700—
750°С, при этом примерно 75% исходного метана
конвертируется. Объемная скорость процесса (стр. 107) 500—600 час1 в
пересчете на природный газ.
По выходе из трубчатой печи газовая смесь поступает в
конвертор метана второй ступени 8, куда добавляется воздух.
Количество вводимого воздуха определяется необходимостью
последующего получения азотоводородной смеси для синтеза ам-
мнака, в которой соотношение Н2: N2 должно составлять 3:1.
Это соотношение точно регулируется при помощи регулятора
соотношения газа и воздуха.
В верхней части конвертора 8, заполненного никелевым
катализатором, кислород воздуха окисляет часть СН4 по
реакции B). Вследствие экзотермичности этой реакции температуре
газовой смеси повышается до 950—1000 °С. В остальной части
аппарата протекает эндотермическая реакция A) между
метаном, не прореагировавшим в трубчатой печи 9, и водяным
паром и частично идет реакция C).
Конвертированный газ охлаждается в котле-утилизаторе 6
до 400 °С — температуры конверсии окиси углерода. Для более
точного регулирования процесса часть газа может поступать
в увлажнитель 7, минуя котел. В увлажнитель впрыскивается
паровой конденсат, при этом снижается температура газа и он
насыщается водяными парами. Количество впрыскиваемого
конденсата регулируется автоматически, что позволяет
поддерживать на заданном уровне температуру газа на входе в
конвертор 4 окиси углерода.
Перед поступлением в конвертор 4 к газовой смеси
добавляется в смесителе 5 водяной пар для поддержания
определенного соотношения между паром и газом @,35:1). При этом
избыток пара (сверх стехиометрического количества)
составляет примерно 1,5, что вполне достаточно для проведения
конверсии СО, содержание которой в газе до ее конверсии
составляет 13,7% (стр. 179).
В конверторе 4 на железохромовом катализаторе протекает
реакция C) окисления окиси углерода водяным паром до
двуокиси углерода с одновременным образованием водорода в
результате восстановления водяного пара. Обычно конверсию
окиси углерода ведут в две ступени. В первой ступени
конвертируется большая часть СО при более высокой температуре,
178
так как процесс еще далек от состояния равновесия, а с
повышением температуры увеличивается скорость реакции.
Температура газов иа входе в первую ступень 400°С, на выходе —
близка к 500 СС. Затем газовая смесь охлаждается до 400—
420 °С, для чего впрыскивается конденсат водяного пара; при
испарении конденсата на насадке испарителя, размещенного в
аппарате 4 между слоями катализатора первой и второй
ступеней, газ охлаждается и одновременно дополнительно
насыщается водяными парами.
Во второй ступени конверсия СО проводится при 420—440 °С,
благодаря этому газовая смесь удаляется от состояния
равновесия, что способствует дополнительному конвертированию
окиси углерода.
Выходящий из конвертора окиси углерода газ разделяется
на два параллельных потока, направляемых в теплообменник
2 и пароперегреватель 3 для подогрева исходного природного
газа и водяного пара. Далее конвертированный газ поступает
на переработку в азотоводородную смесь (очистка от СО2 и
СО). Ниже приведен примерный состав газовой смеси (считая
на сухой газ) на различных стадиях описанного процесса:
Состав, объемн. %
Газ CHj СО Н-2 СО2 N2
Природный 98 — — 0,4 1,6
После трубчатой печи 10 10,8 68,9 9,8 0,5
После конвертора метана второй ступени 0,5 13,7 56,1 7,2 22,5
После конвертора окиси углерода . . . 0,4 4,0 59,8 15,2 20,6
В промышленности применяется также процесс
каталитической конверсии под давлением до 30 ат.
КАТАЛИТИЧЕСКАЯ ПАРОКИСЛОРОДНАЯ
И ПАРОКИСЛОРОДОВОЗДУШНАЯ КОНВЕРСИЯ МЕТАНА
В зависимости от выбранного способа тонкой очистки газа
от окиси углерода применяется парокислородная или парокис-
лородовоздушная конверсия метана. Парокислородную
конверсию применяют в случае удаления СО из конвертированного
газа промывкой его жидким азотом (стр. 197). При этом из
газовой смеси удаляется также остаточный метан, а водород
насыщается испаряющимся азотом. Поэтому содержание
метана в конвертированном газе после парокислородной конверсии
может достигать 2%. Если применяется парокислородовоздуш-
ная конверсия метана, промывка азотом не может быть
использована, так как газовая смесь после конверсии содержит N2
и Н2 в стехиометрическом соотношении C:1). В этом
случае очистка газа от СО производится медноаммиачным
раствором под высоким давлением (стр. 195). Он хорошо растворяет
12* 179
лишь окись и двуокись углерода, в связи с чем необходима
более глубокая конверсия метана—до остаточного
содержания СН4 в конвертированном газе в пределах 0,3—0,5%.
В процессе парокислородовоздушной конверсии метана
удельный расход (на 1 т NH3) дорогостоящего кислорода,
получаемого разделением воздуха, на 40% ниже, чем при
парокислородной конверсии, так как в первом случае используется
также кислород воздуха. Однако объем аппаратуры для паро-
кислородовоздушной конверсии метана, конверсии окиси
углерода и для очистки газа от СОг должен рассчитываться на
проход больших количеств газовой смеси, содержащей также азот.
Вода
Кислород или
кислородовоз-
душная смесь
Природ-1 & 1!==
Отработанная
вода
Рис. 67. Схема каталитической парокислородной конверсии метана:
/—сатурационная башня; 2—теплообменник; 3—смеситель; 4—конвертор метана;
5—увлажнитель; 6—конвертор окиси углерода; 7—котел-утилизатор; 8—водонагревательный
теплообменник; 9—конденсационная башня; 10—насос.
что удорожает перечисленные стадии процесса. Экономичность
того или иного процесса во многом определяется конкретными
условиями строительства и эксплуатации азотного завода.
На рис. 67 показана схема процесса каталитической
парокислородной конверсии природного газа и связанной с ней
конверсии окиси углерода под давлением, близким к
атмосферному. Для этого процесса допустимо обычное содержание
соединений серы в природном газе B0 мг/м3). Природный газ под
абсолютным давлением 1,9 ат поступает в сатурационную
башню /, орошаемую горячей водой (82 °С), нагревается до 78—
80 °С и насыщается водяным паром до объемного соотношения
пар : газ = 0,35 : 1.
Далее в парогазовую смесь добавляется водяной пар до
соотношения пар:газ=1:1. Затем парогазовая смесь,
подогретая в теплообменнике 2 до 500—600 °С, тщательно
смешивается с кислородом или с воздухом, обогащенным кислородом,
в смесителе 3 и поступает в конвертор метана 4, заполненный
никелевым катализатором.
180
Кислород вводится в таком количестве, чтобы процесс
протекал аутотермично и поддерживалась требуемая температура
реакции. Тепло, выделяющееся при экзотермической реакции
B), поглощается в ходе эндотермической реакции A). Обычно
на 1 м3 природного газа добавляется 0,55 м3 кислорода.
Чтобы избежать воспламенения газовой смеси в свободном
объеме перед катализатором (это приводит к образованию
сажи), скорость газового потока в смесителе и во входном
канале конвертора метана должна быть значительно больше
скорости распространения пламени. В этих условиях реакция
взаимодействия метана с кислородом происходит только на
катализаторе.
Водяной пар зводится в процесс для предотвращения саже-
образования и для увеличения выхода водорода по реакции A).
Конверсия метана зедется на ннкелевом катализаторе при
объемной скорости 300 ч~1 (считая на природный газ). Температура
газов на выходе из конвертора равна 850 °С при содержании
в газе соединений серы не более 10 мг/м3. При более высоком
содержании серы температура выходящих газов соответственно
повышается.
Конвертированный газ далее проходит через увлажнитель
5, где охлаждается до 750 °С благодаря испарению
впрыскиваемого конденсата, и насыщается водяными парами. Далее
парогазовая смесь в теплообменнике 2 охлаждается до 400—
420 °С и поступает в двухступенчатый конвертор окиси
углерода 6. Перед входом в аппарат к газовой смеси добавляется
водяной пар (при парокислородовоздушной конверсии до
соотношения пар : газ = 0,45 : 1, при парокислородной конверсии
это соотношение составляет 0,65 : 1).
Конструкция конвертора окиси углерода такая же, как для
паровой каталитической двухступенчатой конверсии (стр. 185).
Выходящий из конвертора окиси углерода газ охлаждается
до 180 °С в котле-утилизаторе 7, где получается пар низкого
давления D—Ъат).
Затем в теплообменнике 8 газ подогревает воду, подаваемую
в сатурационную башню /, и окончательно охлаждается в
конденсационной башне 9. Далее газ сжимается в
компрессорах до давления, требуемого з последующих процессах очистки
от двуокиси углерода, тонкой очистки от остатков окиси
углерода и в процессе синтеза аммиака.
В табл. 13 приведен состав газовой смеси (в пересчете на
сухой газ) на различных стадиях процесса.
В промышленности процесс конверсии метана ведут также
под давлением 16 и 30 ат до конечного содержания метана
з конвертированном газе, равного 2%. Температуры газов на
выходе из конвертора метана в этих процессах поддерживаются
соответственно 1050 и 1150°С.
181
Таблица 13
Состав газовой смеси в процессах парокислородной и парокислородовоздушной
конверсии метана
Процесс
Парокислородная конверсия
после конвертора метана ....
после конвертора окиси углерода .
Парокислородовоздушная конверсия
после конвертора метана ....
после конвертора окиси углерода .
Состав, объемн. %
н2
66,1
71,6
51,8
57,5
СО
23,4
3,7
17,7
3,7
СОг
8,5
23
7,3
18,3
СН4
0,5
0,4
0,5
0,4
Аг
0,9
0,8
0,4
0,3
N2
0,6
0,5
22,3
19,8
ВЫСОКОТЕМПЕРАТУРНАЯ (НЕКАТАЛИТИЧЕСКАЯ) КОНВЕРСИЯ
МЕТАНА КИСЛОРОДОМ ИЛИ ОБОГАЩЕННЫМ ВОЗДУХОМ
В Советском Союзе этот процесс намечено применить для
производства газа, используемого в синтезе аммиака.
Конверсия осуществляется под давлением до 30 ат. На рис. 68
показана схема процесса, проводимого под давлением 18 ат.
Природный газ нагревается до 450 °С в подогревателе 2
топочными газами., кислород — в подогревателе / до 350 СС.
Оба газозых потока направляются в горелку 3 конвертора ме-
Газ но подогрев
воды и конденсата
f!
о2
Рис. 68. Схема высокотемпературной конверсии природного газа под
давлением:
/—подогреватель кислорода; 2—подогреватель природного газа; 3—горелка; 4—конвертор
метана; 5—иасос; if—башня сажеочисткн; 7—теплообменник; 8—конвертор окиси углерода;
9 — котел-утнли затор.
182
тана 4, тщательно смешиваются и при температуре примерно
400 °С поступают в реакционную зону конвертора. По реакции
B), приведенной на стр. 172. объемное отношение кислорода к
метану должно быть 0,5. В реальных условиях оно составляет
0,67, что обусловливается теплопотерями и недостаточно
высокой температурой газов до реакции. Указанное соотношение
кислорода и газа поддерживается автоматически.
В горелке газы весьма тщательно смешиваются, так как
иначе могут возникнуть зоны с недостатком кислорода,
вследствие чего образуется сажа. Лишь при температуре в реакцион
ной зоне 1350 °С и выше в горелках лучших конструкций
удается вести процесс без образования сажи. При использовании
некоторых типов горелок содержание сажи при указанных
температурах составляет 10—15 мг\мъ. При понижении температуры
процесса, например, до 1275 °С оно увеличивается до
нескольких граммоз на 1 м3 полученной газовой смеси. Описываемая
схема рассчитана на переработку газа, который может
содержать сажу.
В реакционной зоне конвертора происходит весьма
интенсивное частичное окисление метана и его гомологов.
Конвертированный газ при 1400 °С поступает в увлажнитель,
конструктивно совмещенный с конвертором метана. В увлажнитель
впрыскивается конденсат, при этом газ охлаждается до 300 °С
и далее поступает в башню сажеочистки 6. Конвертор снабжен
рубашкой, в нее подается конденсат и получается пар.
Здесь газ соприкасается с большим количеством
циркулирующей воды и насыщается паром, соответственно охлаждаясь
до точки росы, в данном случае до 172 °С. Соотношение пар : газ
при этом составляет 1,15 : 1.
Иногда для очистки от сажи применяют трубу Вентури. з
которую впрыскивается вода. Мельчайшие частицы сажи
сталкиваются с каплями воды и отделяются от газа в мокром
циклоне.
Далее парогазовая смесь нагревается в теплообменнике 7
до 390°С газом, выходящим из конвертора окиси углерода.
Нагретая до температуры процесса конверсии парогазовая
смесь поступает в конвертор 8 окиси углерода, где при
начальной температуре 390 °С и объемной скорости 800—850 ч~х
протекает реакция окисления окиси углерода кислородом водяного
пара до двуокиси углерода. Остаточное содержание СО в
конвертированном газе 3,2%.
Конвертор окиси углерода имеет двухступенчатую
конструкцию с промежуточным испарением (стр. 178).
По выходе из конвертора окиси углерода
конвертированный газ охлаждается до 235 °С в теплообменнике 7. Для
дальнейшего использования тепла конвертированный газ
направляется в котел-утилизатор 9, в котором получается пар
183
давлением 2,4 ат. Затем тепло газа используется для подогрева
воды, питающей котел-утилизатор, и для нагревания
конденсата.
Обычно высокотемпературную конверсию проводят с
применением дутья, содержащего 98—99% кислорода, и
последующей очисткой газа от СО промывкой жидким азотом. В
качестве дутья может быть также использована
кислородовоздушная смесь, в этом случае тонкая очистка газа от СО
производится медноаммиачным раствором.
Состав газовой смеси (в пересчете на сухой газ) на
отдельных стадиях технологического процесса приводится в табл. 14.
Таблица 14
Состав газовой смеси в процессах высокотемпературной конверсии метана
(без катализатора)
Процесс
Кислородная конверсия
после конвертора метана
после конвертора окиси углерода . . .
Кислородовоздушная конверсия
после конвертора метана
после конвертора окиси углерода . . .
Состав, объемн. %
Н2
62,1
70,9
47.5
57,0
со
34,8
3,7
26.4
3,7
СО2
1,8
24,4
2,4
19,9
N2
1.0
0.8
23,2
19,0
сн4
0,3
0,2
0,5
0,4
ОСНОВНАЯ АППАРАТУРА
Трубчатая печь. В прямоугольной печи, выложенной из
огнеупорных материалов и разделенной на три секции, вертикально
подвешены 66 труб диаметром 273 мм из жаропрочной стали.
Внутри каждой такой трубы размещается труба диаметром
108 мм (рис. 69). Кольцевое пространство между внешней и
внутренней трубами заполнено катализатором. Печь снабжена
горелками, в которых сжигается природный газ.
Тепло сгорания передается путем радиации реакционным
трубам. Парогазовая смесь входит в верхнюю часть
реакционных труб и движется вниз через слой катализатора. По мере
движения сверху вниз смесь нагревается до температуры
процесса конверсии за счет обогрева наружных труб и
теплообмена с конвертированным газом, движущимся по внутренним
трубкам. Затем газовая смесь поступает в сборный коллектор
и направляется далее.
Отработанные греющие газы через газоход выводятся из
печи и направляются в котел-утилизатор.
Конвертор метана второй ступени. Цилиндрический
вертикальный стальной аппарат (рис. 70) имеет внутреннюю огне-
184
упорную футеровку /. В нижней части конвертора
расположена колосниковая решетка 3 из огнеупорного кирпича, на
которой находится слой катализатора 2. В верхней части аппарата
имеются штуцеры для ввода воздуха и газовой смеси.
Конвертированный газ выходит из аппарата
через нижний боковой штуцер.
газ
Газ
Рис. 70. Конвертор
метана второй ступени:
1—футеровка; 2—катализатор;
3—колосниковая решетка; 4—
штуцеры для термопар.
Конвертор парокислородной и па-
рокислородовоздушной конверсии
метана. Аппарат для конверсии,
проводимой без давления (рис. 71), по
конструкции близок к конвертору метана
второй ступени. Конверторы с верхним
подводом исходной парогазокислороднои смеси вверху имеют
смесительный кннал, в который поступает кислород и газовая
смесь из выносного смесителя. К каналу подведен также азот
для продувки. Из смесительного канала парогазокислородная
смесь поступает на катализатор.
Конвертор окиси углерода (конверсия без давления).
Цилиндрический вертикальный стальной аппарат (рис. 72) в
верхней части снабжен штуцером для входа парогазовой смеси.
Вверху на колосниковых решетках размещаются два слоя
катализатора первой ступени конверсии.
Рис. 69. Двухходовая
реакционная труба:
I—внешняя труба;
2—внутренняя труба; 3—катализатор;
4—решетка; 5—щели для газа.
185
В средней части аппарата на колосниковой решетке
расположен испаритель, представляющий собой слой лепестковой
металлической насадки. Над насадкой расположено
разбрызгивающее сопло для подачи конденсата. В нижней части
испарителя находится тарелка, на которой собирается неиспарив-
шийся конденсат, выводимый затем из конвертора.
U Газ для
ajoepe6a
roj
Конденсат
Рис. 71. Конвертор каталитической Рис. 72. Конвертор окиси углерода:
¦¦¦¦ . конверсии метана: /—катализатор; 2—насадка испарителя.
/—печь (конвертор); 2—смеситель.
Под испарителем з нижней части аппарата на двух
колосниковых решетках размещены два слоя катализатора второй
ступени конверсии. Внизу имеется штуцер для выхода
конвертированного газа.
КОНВЕРСИЯ ОКИСИ УГЛЕРОДА ВОДЯНЫМ ПАРОМ
Ранее был описан процесс конверсии окиси углерода,
органически связанный в единую технологическую схему с
конверсией метана.
При получении генераторных газов газификацией твердого
или жидкого топлива процесс конверсии СО не может быть
объединен с другим процессом, а ведется самостоятельно. Твердое
и жидкое топливо обычно содержит много серы, поэтому в
газах, получаемых из этого сырья, также присутствует значитель-
186
ное количество сероводорода и органических соединении серы
(главным образом COS). Для улучшения условий работы
катализатора конверсии СО и уменьшения коррозии газы
предварительно очищают от H2S (стр. 52 ел., 191 ел.). Очистка от
органических соединений серы, ввиду сложности этого
процесса, обычно не проводится. При конверсии окиси углерода
большая часть COS реагирует с водородом при 400—500 °С на
катализаторе, образуя сероводород:
COS + Н2 = H2S + СО
После конверсии окиси углерода количество H2S в газах
составляет 0,5—1 г/м3.
На рис. 73 показана схема двухступенчатой конверсии СО.
Газодувка 1 подает в нижнюю часть сатурационной башни 2
Воде
Газ на
конверсию СО
Вода
Горячая водо
Рис. 73. Схема двухступенчатой конверсии окиси углерода:
'—газодувка; 2—сатурациониая башия; 3—теплообменник; 4—конвертор окиси углерода;
5—водонагревательная башня; 6—конденсационная башня; 7—иасос.
генераторный газ при температуре 20—30 °С. Башня
заполнена деревянной хордовой насадкой и орошается горячей водой
G8—82 °С). Проходя через башню, газ нагревается до 75—
78 ЬС и насыщается водяным паром. Охлажденная до 65 °С
вода из сатурационной башни циркуляционным насосом 7
подается в водонагревательную башню 5. К парогазовой смеси
добавляется пар в количестве, необходимом для процесса
конверсии (объемное соотношение пар:газ=1—1,2:1); для этого
установлен автоматический регулятор соотношения. Затем
парогазовая смесь нагревается в теплообменнике 3 до
температуры начала конверсии C90—400°С) и поступает в
конвертор 4 на первую ступень конверсии СО. На выходе из зоны
катализа первой ступени температура газовой смеси составляет
187
500°С. Конструкция конвертора окиси углерода описана выше
(стр. 185), Пройдя испаритель и охладившись здесь до 420—
440 °С, конвертированный газ поступает во вторую ступень
конверсии СО и при температуре около 440°С возвращается в
теплообменник 3. Объемная скорость в процессе конверсии
330—350 ч-\ считая на исходный сухой газ. В теплообменнике
газ охлаждается до 160—180°С, нагревая парогазовую смесь,
идущую на конверсию. Затем конвертированный газ
направляется в башню 5, нагревает здесь воду до 78—82 °С и
поступает в конденсационную башню 6, орошаемую холодной водой.
Конструкция водонагревательнои и конденсационной башен
аналогична конструкции сатурационной башни.
Из конденсационной башни газ, охлажденный до 20—30 СС,
поступает на компрессию. К газопроводу подключен
газгольдер.
Состав сухого газа до и после конверсии СО приведен ниже
(в объема. %):
До После
конверсия конверсии
СО2 6—7 28,0
СО 33—34 3,5
Н2 38,2 51,5
N2 21.5 16,8
СН4 0,3 0,2
В процессе конверсии СО обычно применяется среднетем-
пературный железохромовый катализатор, активный при 400—
500 °С. Срок службы катализатора составляет два года.
Как указывалось ранее, давление не смещает равновесие
реакции конверсии СО в сторону образования водорода.
Однако в ряде случаев целесообразно проводить конверсию под
повышенным давлением, так как при этом повышается
объемная скорость, уменьшаются габариты аппаратуры, расход
металла на аппаратуру и электроэнергии на сжатие газа в
компрессорах, улучшается использование тепла конвертированного
газа и появляется возможность создания более мощных
высокопроизводительных агрегатов. В связи с этим в процессе
конверсии СО часто применяется давление 12, 18 и 30 ат.
ГАЗГОЛЬДЕРЫ
Для хранения ггза и равномерной и бесперебойной подачи
его потребителям строят газохранилища, называемые
газгольдерами. В газгольдере газ всегда находится под некоторым
избыточным давлением, во избежание подсосов воздуха и
образования взрывоопасных газовоздушных смесей.
В промышленной практике применяются мокрые и сухие
газгольдеры, а также газгольдеры постоянного объема.
188
Мокрый газгольдер телескопического типа большой
емкости (рис. 74) состоит из металлического колокола 5,
нескольких телескопических звеньев 1, 2 и резервуара 4. Колокол и
телескопы погружены открытой частью в резервуар с водой. Под
колокол подведены газопроводы для входа и выхода газа.
Колокол и телескопы снабжены кольцевыми желобами 5,
заполненными водой и служащими для подъема телескопов и
и создания гидравлических затворов, препятствующих выходу
газа. В северных районах Советского Союза чаши газгольдеров
.5
-2
-в- 4
Рис. 74. Мокрый газгольдер те- Рис. 75. Сухой газгольдер:
лескопического типа: /—резервуар; 2—шайба; J—сборник смолы;
/. 2—телескопические звенья; 3—ко- •'—труба для подъема смолы; 5—труба для
локол; 4— резервуар- 5—кольцевой выхода газа; 6—подвижная лестница; 7—кар-
желоб' йб
д
ман шайбы.
размещаются в отапливаемых зданиях, а гидравлические
затворы обогреваются паром, таким образом предотвращается
замерзание в них воды.
Мокрые газгольдеры надежны и достаточно безопасны в
работе, однако на сооружение их требуется значительное
количество металла, а при эксплуатации в зимнее время расходуется
относительно большое количество пара. Кроме того, в этих
газгольдерах газ увлажняется и его давление меняется
ступенчато, в зависимости от того, поднят один колокол или колокол
и телескоп.
Сухой газгольдер изображен на рис. 75. Он представляет
собой цилиндрический или призматический вертикальный сталь-
189
ной резервуар 1 с плоским дном. Внутри газгольдера имеется
подвижной диск 2 (шайба), плотно прилегающий к стенкам
газгольдера и являющийся его верхней крышкой. Уплотнение
между шайбой и стенкой газгольдера обеспечивается
прокладками, орошаемыми густой смазкой и прижимаемыми к стенкам
специальным устройством, либо при помощи масляного
(смоляного) затвора.
Смола (газгольдерное масло) подается насосами снизу на
верхнюю плоскость шайбы, Некоторая часть смолы протекает
через неплотности, смазывает стенки и собирается на дне
газгольдера, откуда стекает в сборник 3, из которого
автоматически насосом по трубе 4 подается наверх. Шайба с помощью
специальных роликов устанавливается строго горизонтально и
не должна иметь перекосов.
Газ подводится снизу под шайбу и, заполняя газгольдер,
поднимает шайбу вверх. При выпуске газа из газгольдера
шайба опускается вниз.
Для сооружения сухих газгольдеров требуется меньшее
количество металла, они могут изготовляться весьма большой
емкости (несколько сот тысяч м3) и не нуждаются в обогреве
паром. Газ не увлажняется в сухих газгольдерах, и давление
его почти не зависит от высоты подъема шайбы. Однако при
их эксплуатации возможно образование взрывоопасных
газовоздушных смесей вследствие проникания газа в пространство
над шайбой.
Поэтому необходимо точное изготовление шайбы и
соответствующая обработка внутренних стенок газгольдера. В
азотной промышленности обычно используются мокрые
газгольдеры.
Газгольдер постоянного объема представляет собой
герметически закрытый шаровой или цилиндрический металлический
резервуар, в котором газ хранится под избыточным давлением
от 4 до 18 ат. Вследствие относительно небольшой емкости
такие газгольдеры в некоторых случаях устанавливают группами.
Из-за большого расхода металла на сооружение газгольдеров
постоянного объема они не получили заметного
распространения в азотной промышленности.
При эксплуатации сухих и мокрых газгольдеров необходимо
следить за их герметичностью, отсутствием перекосов шайбы
или колокола н за тем, чтобы газгольдеры не переполнялись
сверх определенных пределов и излишне не опорожнялись. При
переполнении газгольдера возможен выброс лишнего газа в
атмосферу, при опорожнении в газгольдере может образоваться
вакуум, что приведет к смятию колокола и телескопов под
давлением атмосферного воздуха.
В отечественной промышленности разработаны и находят
применение также схемы работы без газгольдеров.
190
3. ОЧИСТКА АЗОТОВОДОРОДНОЙ СМЕСИ
Азотоводородная смесь для синтеза аммиака должна быть
тщательно очищена от контактных ядов (СО2, H2S, CO, О2).
УДАЛЕНИЕ ДВУОКИСИ УГЛЕРОДА И СЕРОВОДОРОДА
Газ, полученный из природного газа конверсией метана и
окиси, углерода, как правило, практически освобожден от
сероводорода и органических соединений серы. Газы, полученные
газификацией тзердого или жидкого топлива, очищают от
сероводорода до конверсии окиси углерода. Однако после
конверсии СО в них также содержится сероводород в количестве 0,5—
I г/.м3 (стр. 187). Двуокись углерода относительно хорошо
растворяется в воде, растворимость возрастает с повышением
давления и понижением температуры воды. Поэтому СО2 уда-
/О 20 30 W SO" SO 70 SO 90 700
Абсолютное давление, on
Рис. 76. Растворимость СОо в воде:
1—при О -С; 2—при 12,5 СС; 3—ппн 21 :С; 4—при 5и СС:
5—при 75 °С; 6—при 100 =С.
ляют из газовой смеси путем отмывки ее водой под давлением
16 и 28 ат. Зависимость растворимости СО2 от давления и
температуры показана на рис. 76 (объем СО2 в см3 приведен к
0°С !t 760 мм рт. ст.). При очистке газов от СО2 удаляется
также сероводород, растворимость которого тоже возрастает с
повышением давления и понижением температуры.
Отмывка СО2 водой под давлением. Этот процесс ведут
следующим способом. Газ поступает в нижнюю часть водяного
скруббера, представляющего собой вертикальный цнлидриче-
ский стальной аппарат диаметром 2,5—3 м и высотой 20—22 м,
заполненный насадкой из керамических или стальных колец.
Вода подается насосом в верхнюю часть скруббера под
давлением, несколько превышающим давление газов в аппарате.
191
Количество воды, поступающей на орошение скруббера,
обычно колеблется от 5 до 10 м3 на 100 м3 исходного газа в
зависимости от применяемого давления и температуры воды. Стекая
по насадке скруббера сверху вниз, вода поглощает из газовой
смеси СО2 и H2S Затем вода для использования ее давления
подается в гидравлическую турбину, скомпонованную на одном
валу с насосом и электродвигателем. Рекуперированная в
турбине энергия (примерно 40% энергии, затраченной в насосе на
подачу воды) используется для вращения вала насоса.
Вследствие снижения давления в турбине большая часть
растворенных в воде СО2 и H2S выделяется из воды. Вода
отделяется от газа в экспанзерах (газоотделители),
установленных после турбины. В экспанзерах получается газ, содержащий
80—95% СО2 и более. Он пригоден для синтеза мочевины,
получения соды, углекислых солей аммония, твердой и жидкой
углекислоты и т. д.
Примерный состав азотоводородной смеси после водной
очистки (в объемн. %):
СО2 . . 1,5—2 СО . . 4,5—5
Н2 . . 70 СН4 . . 0,3—0,5
N2 . . 23—23,5
Очистка этаноламинами. Для очистки от сероводорода и
двуокиси углерода широко применяются также водные растворы
этаноламинов, в первую очередь растворы моноэтанол-
амина CH2CH2(OH)NH2. Реже применяется диэтаноламин
(CH2CH2OHJNH и еще более редко триэтаноламин
(CH2CH2OHKN. Моноэтаноламин (МЭА) обладает
максимальной абсорбционной способностью по отношению к СО2 и H2S,
что позволяет очень тщательно очистить газы от этих примесей.
При 25—35 °С амины образуют с кислыми газами (СО2 и
H2S) соли — карбонаты, бикарбонаты, сульфиды, бисульфиды.
Например, моноэтаноламин образует следующие соединения:
2RNH2 + Н2О + СО.2 < » (RNH3JCO3
RNHa + Н2О + СОа j=n RNH3HCO3
2RNH2 + H2S ^r=f (RNH3)aS
RNH2 + H2S <—* RNH3HS
где R —группа —CH2CH2OH.
Соответственно реагируют ди- и триэтаноламин.
При нагревании до 105—125 °С растворов аминов,
поглотивших СО2 и H2S, происходит распад образовавшихся солей с
выделением поглощенных ранее газов. После охлаждения такие
растворы снова могут поглощать кислые газы. Таким образом,
очистка газов от СО2 н H2S растворами аминов является
циклическим процессом, в котором осуществляется кругооборот
поглотительного раствора.
192
Равновесная растворимость ССЬ и H2S зависит от давления
этих газов, температуры абсорбции и концентрации раствора.
В табл. 15 приведена растворимость двуокиси углерода в
растворах этаноламинов при парциальном давлении СОг ниже
атмосферного, в табл. 16 — при повышенном давлении СО2.
Таблица 15
Растворимость СОа в 2 н. растворах этаноламинов*
(хр—растворимость, моль/моль амина; р—парциальное давление, ми рпг. ст.)
Моноэтаноламин
р ХР
Диэтаноламин
Р ХР
Триэтаноламин
Р ХР
736,4
252,2
98,6
44,2
10,6
668,2
183,1
70,9
10,1
0,795
0,697
0,623
0,589
0,527
0,698
0,607
0,556
0,489
При
729,0
249,9
99,3
44,3
10,5
При
668,4
242,3
183,8
71,0
10,2
25 °С
0,813
0,717
0,633
0,553
0,451
50 °С
0,680
0,562
0,548
0,489
0,302
• Данные J. Mason, В. Dodge.
734,0
99,5
45,4
11,0
1,4
662,8
230,3
88,7
40,4
9,4
Таблица 16
Растворимость СО2 в 2 н. растворах этаноламинов
при повышенном давлении*
Давление COj
атм
2,00
5,31
15.4
40,7
2.81
5,20
15,40
40,50
Растворимость,
моноэтаноламнн
При
0,921
1,03
1,15
1,4
При
0,816
0,900
1,030
1,210
моль/моль амина
триэтаноламин
25 °С
0,898
0,975
1,11
1,36
50 "С
0,647
0,768
1,00
1,19
0,715
0,316
0,209
0,093
0,033
0,382
0,216
0,130
0,0791
0,0346
Данные А. Г. Лейбуш и М. А. Людковской.
13—2848
Растворимость сероводорода в растворах этаноламинов при
давлении FbS ниже атмосферного давления характеризуется
данными табл. 17.
Таблица 17
Растворимость H2S в растворах этаноламинов*
Растворимость H2S
моль/моль амина
2,5 н. раствор
0,061
0,019
0,25'
0,35
0,688
0,846
2н. раствор
0,035
0,175
0,342
0,465
0,624
0,78Ь
Парциальное давление H2S
мм ртп.
при 25 °С
~тп.
при 50 °С
моноэтаноламина
0,160
0,45
1,71
7,15
31,4
104
0,64
1,84
7,25
30,6
121
348
диэтаноламина
0,167
12Д
23,7
68,5
219
0,565
11.3
40,8
86,5
.
—
* Данные А. Г. Лейбуш и А. Л. Шнеперсон.
Принципиальная схема очистки газа раствором
моноэтаноламина заключается в следующем.
Конвертированный газ поступает в нижнюю часть
абсорбера, насадка которого орошается 15—20% раствором
моноэтаноламина, поглощающим двуокись углерода и сероводород.
Сверху из абсорбера выходит очищенный газ, который, во
избежание уноса брызг и паров раствора, промывается на 2—3
тарелках холодным конденсатом.
Раствор, поглотивший СО2 и H2S, выходит из абсорбера
снизу и нагревается в теплообменнике до 95—100 °С. Далее
раствор поступает в верхнюю часть регенератора (отгонная
колонна), в нижней части которого расположен трубчатый
кипятильник, обогреваемый водяным паром. Раствор, стекая вниз по
насадке регенератора, нагревается парами, поднимающимися
снизу, при этом из раствора выделяются СО2 и H2S.
Окончательная регенерация растворов происходит в кипятильнике при
температуре примерно 120 °С.
Сверху из регенератора выходит ССЬ, H2S и пары воды.
Часть водяных паров конденсируется, конденсат направляется
на орошение регенератора для отмывки газа от паров и брызг
раствора амина. Регенерированный раствор поступает из реге-
194
нератора в теплообменник, где нагревает раствор,
направляемый на регенерацию. Затем регенерированный раствор
охлаждается в холодильнике и насосом подается на орошение
абсорбера. Получаемая при этаноламиновой очистке
высококонцентрированная двуокись углерода (99% СО2) может быть
использована для производства мочевины (стр. 568), соды (стр. 425),
сухого льда.
УДАЛЕНИЕ ОКИСИ УГЛЕРОДА
Содержание окиси углерода в азотоводородной смеси,
применяемой для синтеза аммиака, не должно превышать 0,001 —
0,002%.
Для тонкой очистки газа от окиси углерода применяют
следующие способы:
1) промывка газа медноаммначным раствором под
давлением 120 или 320 ат;
2) промывка газа жидким азотом под давлением 28 ат;
3) каталитическая очистка газа.
Медноаммиачная очистка газа широко применяется в
отечественной промышленности. Для этой цели применяют
растворы муравьинокислой, уксуснокислой или углекислой солей
одновалентной меди в аммиачной воде.
Примерный состав применяемого для очистки от СО медно-
аммиачного раствора (в г/л):
Си4 . . .
Си2+ . . .
115—130
16—20
NH3
НСООН . . .
или СНзСООН
145—160
60
140
Применяют также растворы другого состава.
Процесс абсорбции СО может быть описан следующим
уравнением:
Cu(NH3)^OOCH + СО > [СиAЧН3)„СО]ООСН
Растворимость СО увеличивается при повышении его
парциального давления и понижении температуры раствора.
Растворимость СО в медноаммиачных растворах при 20°С
и различном парциальном давлении окиси углерода
представлена на рис. 77. Кривые растворимости /—6 на этом рисунке
соответствуют следующим составам поглотительных растворов:
Кривые Состав поглотительного рветвора, г/л ¦ • ' I
на рис. 77
/ . . . .
2
з\
4 ....
5
6 . ...
Си
53
119
94
91
103
123
-р
,3
,8
,1
,1
Си2'
2,5
13
12
26
16
NH3
ПО
117,1
136,8
148,9
141,3
145,2
НСООН
142
166
—.
71,0
87,5
162,5
соа
85,4
52,0
79,5
1
г
А
—|
Гг
к-"
i
§30
; ^ /О
^ 5
О 100 200 300 iOO 500 600 700 600 900 Ю1H
Равновесное парциальное довление СО, мм рт ст.
Рис. 77. Растворимость окиси углерода в медноам-
миачных растворах при 20 °С:
I—no данным Л. Ларсона и С. Тейтсворта; 2—по данным
Н. М. Жаворонкова и Р. М. Рещнкова; 3—6—по данным
Н- М. Жаворонков» и В. Г. Чагунава.
Растворимость СО при различных парциальных давлениях
и температурах в растворе, содержащем 100 г/л Си+, 20 г/л
Си2+, 123,9 г/л NH3 и 166 г/л НСООН, приведена на рис. 78.
Очистку обычно проводят под
давлением 120—320 ат при
температуре от 0 до 20°С в скрубберах
(рис. 79), заполненных насадкой из
металлических колец. Сжатая азо-
товодородная смесь, содержащая
4—4,5% СО, поступает снизу в
скруббер, орошаемый медноаммиач-
ным раствором, который поглощает
из газа окись углерода и выводится
из скруббера снизу. Таким образом,
в скруббере осуществляется
противоток газа и поглощающей
жидкости. Содержание СО в очищенном
газе составляет 5—15 см3/м3.
Чтобы разложить
образовавшийся при абсорбции окиси углерода
медноаммиачный комплекс и
регенерировать раствор, нужно снизить
давление до атмосферного и
нагреть раствор до 80°С. Нагревание
раствора производится глухим
паром в аппарате, называемом регенератором, затем охлаждают
раствор в холодильниках вначале водой, потом испаряющимся
196
§' 1200
%Н00
| Ю00
0 900
<ъ В00
1 70C
•^ 600
t 500
I Ш
ЭОС
гоо
юо
!
!
I
3
1
/
/
* /
и
1
I
/
и
У
/
/
/
I Mi
г j |т
\\ \
/ /
/ /
/ 1 /
/
/
/
/
о 4 в 12 /б гв гь гв
Растборимоств СО
м 3/м3 раствора
Рис. 78. Растворимость окнси
углерода в медноаммиачном
растворе:
#—при 0 "С; 2—при 20 "С; 3—при 60 °С;
4—прн 80 °С.
Раствор
аммиаком. После этого регенерированный раствор снова
подают под давлением 120—320 ат на орошение скрубберов.
Применяется также регенерация в вакууме (абсолютное
давление 0,1 атм) при нагревании до 45 °С. Регенерированный
раствор также направляется в холодильник.
Для использования энергии отработанного раствора
применяют поршневые вертикальные
расширительные (экспансионные) машины.
Газ, получаемый при регенерации
раствора, содержит 60% СО, 27—28%
СО2, 12—13% (H2+iN2). Он
возвращается на конверсию окиси углерода
для получения водорода или
используется в органических синтезах,
например добавляется в газ,
направляемый на синтез метилового спирта.
После медноаммиачной очистки в
азотоводородной смеси еще остается
0,01—0,05% СОг, которая является
ядом для катализатора синтеза
аммиака. Поэтому применяется тонкая
очистка газа от СОг путем щелочной
промывки азотоводородной смеси под тем
же давлением A20—320 ат) в
скруббере. Для промывки используется
7%-ный раствор едкого натра или
20%-ная аммиачная вода. После
тонкой очистки газа остаточное
содержание СО2 в нем составляет 0,0005—
0,001%.
Промывка жидким азотом. В
последние годы начинает приобретать
широкое распространение очистка
газа от окиси углерода промывкой жид- Раствор^
ким азотом. Вместе с окисью
углерода при этом удаляются также метан
и аргон. В результате получается азо-
товодородная смесь, очень хорошо Рис- 79- Скруббер для очист-
очищенная от контактных ядов и ки газа от со-
инертных газов (СН4 и Аг). Газ, поступающий на промывку
жидким азотом, должен быть предварительно тщательно
очищен от СОг во избежание забивки аппаратуры глубокого
холода твердой двуокисью углерода. Обычно газ очищают от СОг
промывкой раствором этаноламина, а затем раствором едкого
натра до содержания в газе не более 0.0005—0,001% СО2.
Кроме того, газ должен содержать в среднем не более
0,02 см3/м3 окислов азота (в пересчете на NO). При низких тем-
197
Фракция СО
¦-ГГ""'Г1
/о
Рис. 80. Очистка газа промывкой жидким азотом:
1—предсарительиые теплообменники; 2. 13—влагоотделители; 3, 12—аммиачные холодильники; 4, 14—осушители; 5. в, 7, 15.
16—теплообменники; 4-нгнаритель; 'J— промывная колонна; 10—теплообменники азота высокого давления; 11, 17, 18. 20—фильтры; 1У—адсорберы.
пературах окислы азота образуют с конденсирующимися
непредельными углеводородами (примеси которых присутствуют
в конвертированном газе) нитросоединения, способные
самопроизвольно взрываться.
Жидкий азот, растворяющий окись углерода, должен быть
высокой чистоты, чтобы получалась чистая азотоводородная
смесь. Обычно азот содержит не более 0,004% СЬ и не свыше
0,01% Аг.
Схема процесса промывки показана на рис. 80.
Конвертированный газ, сжатый до абсолютного давления 28 ат,
охлаждается в одном из предварительных теплообменников / до
—20 °С обратным потоком азотоводородной смеси и, пройдя
влагоотделитель 2, поступает в аммиачный холодильник 3, где
охлаждается до —45 °С испаряющимся аммиаком. При этой
температуре газ проходит осушитель 4 и фильтры 17 и далее
в теплообменниках 5, 6, 7, охлаждаемых азотоводородной
смесью, и в испарителе 8 фракции окиси углерода охлаждается до
—190 °С. По выходе из теплообменников 5 и 6 газ
освобождается от NO в адсорберах 19 и от механических примесей в
фильтрах 20. Затем при температуре —190°С газ поступает в
промывную тарельчатую колонну 9, орошаемую жидким азотом.
Жидкий азот для промывки газа получают следующим
путем. Газообразный азот сжимается в компрессорах до
давления 200 ат и охлаждается до —20 °С в одном из
теплообменников 10, охлаждаемых выходящей фракцией окиси углерода
(см. ниже), затем до —45°С в одном из аммиачных
холодильников 12. При этой температуре азот проходит
влагоотделитель 13, очищается от масла в фильтре // и осушается в
аппарате 14.
Дальнейшее охлаждение азота в теплообменнике 15
производится фракцией окиси углерода (см. ниже), затем азот
дросселируется с 200 ат до абсолютного давления 28 ат,
конденсируется в испарителе 8 и направляется в колонну 9 для
промывки газа. При недостатке холода часть азота высокого
давления охлаждается в теплообменнике 16 азотоводородной
смесью, дросселируется и дозируется в азотоводородную смесь.
Перетекая в промывной колонне 9 с тарелки на тарелку,
жидкий азот поглотает окись углерода, а также метан и аргон.
Часть жидкого азота испаряется н переходит в газовую смесь.
Из верхней части промывной колонны выходит чистая азото-
подородная смесь, которая затем поступает в теплообменники
для охлаждения подаваемого на промывку газа.
Из нижней части промывной колонны вытекает жидкий азот,
содержащий растворенные окись углерода, метан, аргон и
водород. Холод этой фракции, называемой фракцией окиси
углерода, используется для охлаждения газовых потоков, как
указывалось выше.
199
Примерный состав газообразной фракции окиси углерода
(в объемн. %):
СО 22 СН4 . . . . 13
На 7 Аг 3
Na 55
Предкатализ. После очистки газа от окиси углерода
карбонатным медноаммиачным раствором в азотоводородной смеси
остается до 0,05% СО. Эта концентрация слишком высока для
работы на активном катализаторе синтеза аммиака, поэтому
азотоводородную смесь дополнительно очищают в агрегате
предкатализа.
В связи с появлением низкотемпературного катализатора
конверсии СО (стр. 176) предкатализ начинают широко применять
вместо медноаммиачной очистки и промывки газа жидким
азотом.
Схема предкатализа состоит в следующем: азотоводородная
смесь в колонне предкатализа проходит последовательно
теплообменник (предварительный подогрев), электроподогреватель
(нагрев до температуры процесса) и поступает в зону
предкатализа. Проходя через катализатор в этой зоне,
азотоводородная смесь освобождается от контактных ядов.
На железном или никельхромовом катализаторе при
давлении 320 ат протекает следующая реакция:
СО + ЗНа = СН4 + Н2О
Если в газе содержится кислород, он реагирует с водородом:
О2 + 2Н2 = 2Н3О
Температура реакции на железном катализаторе 300—350°С,
на никельхромовом катализаторе 200°С.
Очищенная азотоводородная смесь отдает свое тепло газу,
идущему на предкатализ, в теплообменнике и выходит из
колонны предкатализа. После предкатализа смесь охлаждается в
водяном холодильнике, затем проходит сепаратор для
отделения образовавшейся воды.
Далее газовая смесь, содержащая в случае предкатализа на
железном катализаторе не более 0,002% СО и 0,002% О2,
направляется в агрегаты синтеза. На никельхромовом
катализаторе достигается значительно более тонкая очистка газа.
Компрессия газов
Сжатие газов до требуемого давления обычно производится
в поршневых компрессорах. На больших заводах в некоторых
случаях сжатие газа до 12—30 ат производится в
турбокомпрессорах большой производительности, а с 12—30 ат до
конечного давления—в поршневых компрессорах. Применение
турбокомпрессоров позволяет сократить количество агрегатоз
для сжатия и уменьшить расходы на ремонтные работы.
200
В настоящее время созданы первые образцы
высокопроизводительных турбокомпрессоров G0—150 тыс. м3/ч)
сжимающих газ до давления 160—250 ат.
Для сжатия газа с 1 до 320 ат обычно устанавливаются ше-
стиступенчатые компрессоры, для сжатия до 700—1000 ат —
семиступенчатые. Обычно степень сжатия газа в одной ступени
поршневого компрессора принимается 2,5—3,5 т е давление
Рис. 81. Компрессор на оппозитной базе.
газа после сжатия в этой ступени должно быть в 2,5—3 5 раза
выше давления перед сжатием. При таких относительно
невысоких степенях сжатия уменьшается расход энергии на сжатие
газа^ а температура сжатого газа не превышает 150—165 °С
После каждой ступени сжатия газ охлаждается в водяном
холодильнике, где одновременно конденсируются влага и пары
масла. Для отделения влаги и брызг масла от газа
устанавливают влагомаслоотделители, из которых жидкость периодически
выводится. В водяные холодильники ступеней низкого давления
часто встроены сепарационные устройства для выделения из
газа брызг жидкости.
201
Охлажденный газ поступает в следующую ступень сжатия
непосредственно либо после прохождения технологической
аппаратуры (например, для очистки от СО2, СО и т. д.).
Для сжатия азотоводородной смеси широко применяются
компрессоры типа 1Г-266/320. Производительность компрессора
16 000 м3/ч при начальном давлении J ,02 ат и конечном 320 ат,
мощность электродвигателя 4000 кет, количество оборотов
125 в мин.
В настоящее время намечен выпуск более совершенных
быстроходных компрессоров со встречно движущимися поршнями
(так называемые компрессоры на оппозитной базе). Такие
компрессоры (рис. 81) значительно компактнее, легче и дешевле
горизонтальных тихоходных- компрессоров. Применение
компрессоров этого типа позволяет повысить количество оборотов
и довольно полно уравновесить инерционные усилия и,
следовательно, облегчить фундаменты.
4. ПРОИЗВОДСТВО АЗОТА, КИСЛОРОДА И ВОДОРОДА
РАЗДЕЛЕНИЕМ ГАЗОВЫХ СМЕСЕЙ ЛУБОКИМ ОХЛАЖДЕНИЕМ
В современной технике широко пользуются методом
разделения газовых смесей путем их сжижения с последующей
ректификацией полученных жидкостей и методом
фракционированной конденсации.
Для сжижения таких газовых смесей, как воздух, коксовый
газ, газы крекинга углеводородов и др., необходимо
охлаждение до очень низких температур (табл. 18).
Таблица IS
Температура кипения сжиженных газов
(при атмосферном давлении)
Газ
Формула
Температура кипения
°С
Газ
Формула
Температура кипения
°С
н-Бутан
Аммиак
Пропан
Пропилеи . . . .
Двуокись углерода
Ацетилен
Этан .
Этилен
С4Н10
NH3
с3н8
C3HG
с,н,
C,HG
с,н4
—0,5
—33,35
—42,1
—47,7
—78,5
(возг.)
—83,6
(возг.)
—88,6
—103,7
Метан ....
Кислород . . .
Аргон ....
Окись углерода
Воздух ....
Азот
Водород
Гелий
СН4
O-j
Аг
СО
Н,
Не
— 161,6
—183
—185,9
— 191,6
— 192,0
—195,8
—252,8
—268,9
Область низких температур принято делить на две части:
1) умеренный холод (jo минус 70 — минус 100°С) и 2) глубо-
202
кий .холод (минус 100°С и ниже). Для охлаждения в области
умеренного холода обычно пользуются в качестве холодильных
агентов сжиженными газами — аммиаком, двуокисью серы,
пропаном, бутаном (сжиженные углеводородные газы) и др.,
т. е. веществами с низкими температурами кипения. При
использовании таких холодильных агентов охлаждение
происходит вследствие поглощения тепла испаряющейся жидкостью.
Газы, образующиеся при испарении хладоагентов, вновь
сжижают путем сжатия с последующим охлаждением в водяных-
холодильниках.
Умеренное охлаждение применяется в различных отраслях
химической н пищевой промышленности, при горных работах
и др.
Глубоким холодом пользуются при сжижении воздуха для
выделения из него азота, кислорода, аргона. Глубокое
охлаждение применяют также для выделения водорода из коксового
газа, этилена из газов крекинга углеводородов и др.
Для достижения очень низких температур (глубокого
холода) применяют холодильные машины, работа которых основана
на свойстве реальных газов (например, воздуха) охлаждаться
при расширении в определенных условиях.
Расширение идеальных газов. Реальные газы не
подчиняются в точности законам Бойля—Мариотга, Гей-Люссака, Авогад-
ро, Джоуля, причем наблюдаемые отклонения тем больше, чем
ниже температура и выше давление, под которым находится
газ.
При расширении реального газа без совершения внешней
работы и без теплообмена с окружающей средой совершается
работа по преодолению сил притяжения между молекулами,
вследствие чего температура газа понижается. Это явление
Таблица 19
Понижение температуры At (в °С) при дросселировании воздуха
(рк—конечное избыточное давление, am)
Начальная
температура
СС
+30
— 10
—50
—90
— 130
Рк
0
0
0
0
0
50
ю,
14,
21,
34,
9,
3
8
9
2
6
Начальное давление
100
Рк
0
0
0
0
34
М
20,0
28,4
43,8
69,6
13,6
Рк
0
0
0
18,3
35,2
, am
150
27,
40,
61,
60,
13
3
5
2
6
8
р.
0
0
0
26,4
35,9
200
Л
34
50
72
54
12
(
,0
,3
1_
,3
,6
203
(эффект Джоуля —Томсона) и используется при получении
низких температур в большинстве аппаратов для разделения
воздуха и коксового газа.
Понижение температуры при дросселировании* воздуха для
разных перепадов давления показано в табл. 19 (стр. 203).
СЖИЖЕНИЕ ВОЗДУХА
Чистый газообразный азот для синтеза аммиака,
производства цианамида кальция и т. д. получают исключительно из-
воздуха путем глубокого охлаждения. Для получения чистого
азота воздух необходимо сна-
ВозЗих чала превратить в жидкость, а
—- затем ректификацией
разделить на азот и кислород.
Превратить воздух, как и всякий
газ, в жидкость возможно,
только понизив его
температуру ниже критической**.
Критическая температура воздуха
равна —140,7°С, критическое
абсолютное давление 37,2 атм.
При атмосферном давлении
для превращения воздуха в
жидкость необходимо охладить
его до — 192Х.
Сжижение воздуха при
расширении без совершения
внешней работы. Данные табл. 19
показывают, что понижение
температуры при
Жидкий воздух
Рис. 82. Простейший холодильный
цикл:
U ^??? рУР Р рр
ный вентиль; 5—резервуар для жидкого воз- ВаНИИ НевелИКО ДЭЖе При бОЛЬ-
духа' шой разности давлений.
Поэтому однократным
дросселированием, несмотря на весьма высокие начальные давления,
нельзя понизить температуру газа настолько, чтобы его можно^
было превратить в жидкость. Однако путем многократного
дросселирования удается достигнуть весьма низких температур,
применяя так называемый регенеративный принцип. Сущность
этого принципа состоит в непрерывном использовании холода,
получаемого при дросселировании, для охлаждения новых
порций воздуха. Сжатый воздух, идущий к дроссельному
вентилю, охлаждают в противоточном теплообменнике за счет хо-
* Дросселированием, называют расширение газа (переход от высокого
давления к низкому (без совершения внешней работы.
** Критической температурой называют такую температуру, выше
которой вещество не может находиться в жидком состоянии; давление пара
над жидкостью при этой температуре называется критическим давлением..
204
лода уже дросселированного газа. Таким образом понижается
начальная температура воздуха до его расширения. На рис. 82
изображена принципиальная схема простейшей установки для
сжижения воздуха с однократным дросселированием. Воздух
сжимается компрессором / от давления Pi до давления Р2
(при этом температура воздуха повышается от Т\ до ТА) и
охлаждается в водяном холодильнике 2 до начальной
температуры Т\. Далее сжатый воздух поступает в противоточный
теплообменник 3, где охлаждается до температуры Г2 более
холодным дросселированным воздухом, идущим в
противоположном направлении. Пройдя теплообменник 3, охлажденный
сжатый воздух дросселируется в вентиле 4 до начального дав
ления Р\\ при этом температура его понижается до Т3.
Расширившийся воздух, проходя через противоточный
теплообменник 3, охлаждает воздух высокого давления до температуры Г2.
а сам нагревается до температуры Т\ и затем выходит из
установки. При установившемся режиме в резервуаре 5 собирается
жидкий воздух.
Холодопроизводительность* установок глубокого
охлаждения без совершения сжатым воздухом внешней работы
приблизительно пропорциональна разности давления, а затрата работы
на сжатие газа пропорциональна логарифму отношения
давления сжатого газа к давлению газа до сжатия. Поэтому часть
газа целесообразно не дросселировать до абсолюного
давления 1 ат, а возвращать в компрессор при более высоком
давлении, чтобы уменьшить расход работы на сжатие этой части
газа.
Схема цикла с двукратным дросселированием и
циркуляцией части газа под давлением изображена на рис. 83. Воздух
при давлении Pi поступает в компрессор /, где сжимается до
среднего давления Р2. К сжатому и охлажденному в водяном
холодильнике 2 воздуху присоединяется из цикла некоторое
количество воздуха, охлажденного дросселированием от давления
Р3 до того же давления Р2. В компрессоре 3 воздух сжимается
до давления Рз и поступает в противоточный теплообменник 4.
Здесь воздух охлаждается идущими навстречу холодными
газами, дросселированными до давления Р2 и Р\. По выходе из
теплообменника воздух дросселируется в вентиле 5 до
давления Р2.
Часть воздуха (а) поступает непосредственно в
противоточный теплообменник; другая часть A—а) дросселируется в
вентиле 6 до давления Рг. Сжиженную часть воздуха
выводят из цикла, несжиженная направляется через противоточный
теплообменник.
* Холодопроизводительность характеризуется количеством тепла (в
ккал), которое отводится холодильным агентом от охлаждаемого вещества
или тела. <
205
Расход энергии на 1 кг жидкого воздуха в этом цикле (при
Р, = 1 ат, Р2 = 50 ат, Р3=200 ат, Т2= — 258°К) примерно
1,15 квт-ч вместо 4 кет • ч, расходуемых при однократном
дросселировании в аналогичных условиях.
Таким образом, дросселирование выгоднее вести в несколько
ступеней.
В цикле с двукратным дросселированием и
предварительным охлаждением воздуха в аммиачной холодильной машине
до —50°С наименьший расход энергии (около 0,7 квт-ч на 1 кг
жидкого воздуха) достигается при начальном давлении Pi = 1 ат.
даэдух
Рис. 83. Холодильный цикл с двукратным
дросселированием и циркуляцией части
газа под давлением:
1, 3—компрессоры; 2—водяной холодильник; 4—
противоточиый теплообменник; 5, 6—дроссельные
вентили; 7—резервуар для жидкого виздуха.
{
ваэдут
наибольшем давлении Р3 = 200 ат и промежуточном давлении
Р2 около 50 ат.
Сжижение при расширении газов с совершением внешней
работы. В некоторых установках глубокого охлаждения
использован принцип расширения газов с совершением внешней
работы. Такое расширение воздуха осуществляется в особой
поршневой расширительной машине — детандере — двигателе,
работающем на сжатом воздухе. Производимая этим двигателем
работа может быть использована для сжатия газа, что позволяет
уменьшить расход энергии на его сжижение. Однако это
уменьшение расхода энергии невелико, так как детандер имеет
низкий коэффициент полезного действия.
Цикл с однократным использованием внешней работы
воздуха, расширяющегося в детандере, изображен на рис. 84. Газ,
поступающий в компрессор при давлении Pi A ат), сжимается
206
до давления Р% D0—50 ат). После сжатия газ охлаждается
сначала в водяном холодильнике 2, а затем газами низкого
давления в противоточном теплообменнике 3. Пройдя
теплообменник 3. сжатый газ разделяется на два лотока: один из них
поступает в детандер 4, другой через противоточные
теплообменники 5 и б направляется к дроссельному вентилю 7.
Сжиженная часть воздуха выводится из цикла, холодный несжи-
женный воздух используется в
теплообменниках 6, 5 и 3 для
охлаждения сжатого воздуха. К,
воздуху, выходящему из
теплообменника 6, присоединяется
воздух, расширившийся в детандере,
и также проходит
последовательно теплообменники 5 и 3.
Обычно в детандер
направляют 75—80% воздуха, а на
дросселирование 20—25%. В этих
условиях на сжижение
затрачивается минимальное количество
энергии (около 0,9 кет• ч на 1 кг
воздуха).
Недостатками цикла с
однократным расширением и
совершением внешней работы
являются.- плохое использование работы
поршневой расширительной
машины, трудность ее смазки, боль- .
шое вредное пространство и др. воздух
Холодильный цикл низкого Рис 84 холодильный цикл с рас-
давления. В описанных выше хо- ширением газа и совершением
лодильных циклах газ сжимает- внешней работы:
СЯ ДО ВЫСОКОГО Давления (ОТ 40 ^-компрессор; 2-холодильник; 3, 5 6-
„ . _ 1по^7 ,, теплообменники; 4—расширительна*!ма-
ДО 200 ат). В 19^7 Г. В Институте шина (детандер); 7—дроссельный вентиль;
фиЗИЧеСКИХ Проблем Академии «-резервуар для жидкого газа.
наук СССР П. Л. Капица
разработал холодильный цикл низкого давления.
В холодильном цикле низкого давления (рис. 85) воздух,
сжатый в компрессоре / до избыточного давления 6 ат,
проходит через теплообменник 2 особого устройства (регенератор^
см, стр. 216), где охлаждается до —155 или —160°С. Для охт
лаждения используется несжиженный воздух, выходящий из
установки. По выходе из теплообменника воздух разделяется
на два потока. Меньшая часть воздуха направляется в
межтрубное пространство конденсатора 3, где сжижается, и
стекает в сборник (сепаратор) 5. Большая часть воздуха
направляется в турбодетандер 4, где расширяется до избыточного давт
207
воздух
воздух
ления 1,5 ат, совершая внешнюю работу. При этом воздух
дополнительно охлаждается до температуры —185 или —187 °С
(и частично сжижается). Из турбодетандера воздух поступает
в трубки конденсатора 3. Проходя по трубкам, воздух
охлаждает ту часть воздушного потока, которая направляется в
межтрубное пространство конденсатора из теплообменника 2. При
этом около 5% воздуха от
количества, сжатого в компрессоре,
сжижается в межтрубном
пространстве и стекает в сборник 5. Из
конденсатора 3 несжиженная часть
воздуха через теплообменник
(регенератор) 2 выводится из цикла.
Каскадный цикл сжижения.
Низкие температуры, требуемые для
сжижения и разделения воздуха и
газов, могут быть достигнуты
также при использовании циклов с
охлаждением испаряющимися низко-
кипящими жидкостями. Подобные
циклы широко применяются в
промышленности для получения
умеренного холода.
В отличие от установок
умеренного холода, в установках для
разделения газов соединяют
последовательно (каскадом) несколько
холодильных циклов с разными
испаряющимися жидкостями.
Каскадный цикл сложен по
аппаратурному оформлению, но
весьма экономичен — требует меньшего
Рис. 85. Холодильный цикл Расхода энергии. Каскадный цикл,
применяемый на установках
разделения воздуха или других газов
низкого давления:
/—компрессор; 2—теплообменник
(регенератор); 3—конденсатор; 4— СПИР QR\ рпгтпит Ич ЧРТЫПРХ пнк-
турбодетандер; 5—сборник жидко- 1РИС- 00)> СОСТОИТ ИЗ че1ЬфСЛ ЦИК
го воздуха (сепаратор). ЛОВ! ЭММИаЧНОГО ЭТИЛеНОВОГО, Ме-
ганового и азотного.
В этиленовом цикле (этилен сжижается под абсолютным
давлением 23,5 атм при —25°С) охлаждение производится
испаряющимся жидким аммиаком, подаваемым из аммиачной
холодильной машины. В метановом цикле (метан сжижается под
абсолютным давлением 28 атм при —98 °С) охлаждающим
агентом служит жидкий этилен, испаряющийся при абсолютном
давлении 1,07 атм и —103 °С. Азот сжижается под абсолютным
давлением 23 атм при —155°С при помощи жидкого метана,
кипящего при —160°С (абсолютное давление 1,07 атм). Полученный
208
жидкий азот дросселируют и направляют в ректификационную
колонну для разделения жидкого воздуха.
В каскадной установке с большим числом циклов можно
превратить в жидкость даже наиболее трудно сжижаемый газ —
гелий (темп. кип. —268,9 °С при абсолютном давлении I атм).
Азот
Вода , II ,
Аммиак Этилен Метан Жидшй азот
в рентшринациоту/о
налоту
Рис. 86. Каскадный холодильный цикл:
/—компрессоры; 2—водяной холодильник; J—теплообменники-конденсаторы; 4—
дроссельные вентили.
РЕКТИФИКАЦИЯ ЖИДКОГО ВОЗДУХА
Процесс разделения жидкого воздуха на азот и кислород
основан на различии температур кипения азота и кислорода;
под атмосферным давлением азот кипит при —195,8 °С,
кислород при —183 °С. Разница в 12,8 °С вполне достаточна для
того, чтобы жидкий воздух, кипящий при —192°С, разделить
ректификацией на чистые азот и кислород.
Жидкие кислород и азот выделяют в чистом виде из
жидкого воздуха путем многократного испарения и конденсации. Пары
более высококипящей жидкости — кислорода конденсируются
при соприкосновении с орошающим колонну жидким азотом,
имеющим более низкую температуру кипения. За счет теплоты
конденсации паров кислорода происходит испарение жидкого
азота. Если многократно и последовательно повторять процесс
конденсации и испарения, то пары обогащаются азотом, а
жидкость— кислородом; при этом происходит относительно полное
разделение смеси на составные части.
Над поверхностью земли сухой воздух имеет следующий
состав (в объемн. %):
N» 78,03 COS 0,03
О2 20,99 Н4 0,01
Ar 0,933
Кроме того, воздух содержит водяные пары @,02—2 вес. %),
а также некоторое количество пыли (до нескольких
миллиграммов в 1 .и3 воздуха) и незначительные количества неона, гелия,
14—2848 209
Чистый
газообразный
Обогащенная
кислородом
Чистый
газообразный
кислород
криптона и ксенона. При разделении воздуха методом
ректификации его всегда предварительно очищают от пыли и двуокиси
углерода и удаляют водяные пары. Очистка необходима во
избежание закупорки аппаратуры льдом, образующимся
вследствие замерзания воды и жидкой
углекислоты.
Ацетилен, обычно
содержащийся в воздухе на территории
химических заводов,
представляет особую опасность, так как
образует в аппаратуре глубокого
охлаждения взрывчатые
соединения.
Разделением воздуха в
современных установках (например,
установки типа БР-1, стр. 218)
получают азот, содержащий не
более 0,02% О2 и применяемый
главным образом для синтеза
аммиака, и кислород (с примесью
не более 1,5% N2), используемый
в химических и металлургических
производствах, а также для
сварки и резания металлов.
Для разделения воздуха
применяют двухколонные
ректификационные аппараты.
Двухколонный
ректификационный аппарат (рис. 87) состоит
из нижней разделительной
колонны 4, работающей под
избыточным давлением 6 ат, и верхней
разделительной колонны /,
работающей при атмосферном
давлении. Между этими колоннами
помещен конденсатор 2,
являющийся одновременно испарителем
для верхней колонны. Воздух,
сжатый до давления 50—100 ат
и охлажденный б нротивоточном теплообменнике (на рисунке
не показан), проходит через змеевик 5 кипятильника 6 и
вызывает испарение жидкого обогащенного кислородом воздуха,
стекающего с тарелок колонны 4. По выходе из змеевика
воздух высокого давления проходит через дроссельный вентиль 7.
При этом давление его понижается до 6 ат, после чего жидкий
воздух поступает в нижнюю колонну. В нижней колонне при
перетекании по тарелкам 10 из жидкости испаряется более нпз-
Рис. 87. Двухколонный
ректификационный аппарат для
разделения воздуха:
/—верхняя колонна;
2—конденсатор-испаритель; 3—карманы для
жидкого азота; 4—нижняя
колонна; 5—змеевик; 6—кипятильник;
7, 8, 9—дроссельные вентили;
10—тарелки.
210
кокипящнй азот. Чистый азот собирается в карманах 3 верхней
части колонны 4, а на дно ее стекает жидкий воздух,
обогащенный кислородом C0—40% О2).
Температура кипения кислорода при атмосферном давлении
—183СС. При этой температуре жидкий азот кипит только под
абсолютным давлением 3,6 атм. Поэтогму, если в нижней
колонне поддерживать давление несколько выше 3,6 атм, богатые
азотом пары конденсируются в конденсаторе-испарителе 2 при
охлаждении испаряющимся жидким кислородом, который
стекает из верхней колонны / в межтрубное пространство
конденсатора 2.
Конденсатор состоит из ряда закрытых сверху трубок; в его
межтрубном пространстве кипит при атмосферном давлении
жидкий кислород, в трубках конденсируется азот, стекающий
вниз. Часть жидкого азота собирается в карман 3, другая часть,
стекая с тарелки на тарелку в нижней колонне 4, постепенно
обогащается кислородом. Эту обогащенную кислородом
жидкость из нижней части кипятильника 6 через дроссельный
вентиль 8 подают на средние тарелки верхней колонны 1, а жидкий
азот из карманов 3 через дроссельный вентиль 9 поступает в
верхнюю часть колонны /, откуда он стекает вниз навстречу
поднимающимся парам кислорода.
Испарение кислорода в межтрубном пространстве
конденсатора 2 происходит за счет тепла конденсации паров азота,
имеющих более высокую температуру (примерно на 3°С).
Пары чистого азота отводят из верхней части колонны 1 и
направляют в теплообменник (на рисунке не показан), чистый
газообразный кислород отбирают в верхней части
конденсатора 2.
Для безопасной работы ректификационной установки очень
важно пе допускать накопления в системе ацетилена, всегда
содержащегося в небольшом количестве в исходном
атмосферном воздухе. Для удаления ацетилена обогащенную
кислородом жидкость из нижней колонны, до подачи ее в верхнюю
колонну, пропускают через ацетиленовый адсорбер (на
рисунке не показан), заполненный твердым поглотителем —
активированными силикагелем или алюмогелам (стр. 215). Далее эту
жидкость пропускают через керамические фильтры для очистки
(п частиц твердой углекислоты.
Ректификационная колонна выполнена из красной
электролитической меди. Колонны производительностью 500—800 м3/ч
азога имеют одинарные ситчатые тарелки (с отверстиями
диаметром 0,8 мм), которые расположены на расстоянии 60 мм
друг от друга; в колоннах производительностью от 2000
до 5000 мъ)ч устанавливают двойные ситчатые тарелки. Внешний
диаметр высокопроизводительных колонн равен 1,2—1,5 м (в
верхней части), общая высота колонн 7—10 м. Колонна заклю-
14* 211
Жидкость
:тА
чена в общий деревянный кожух с теплоизоляцией из
шлаковой или стеклянной ваты и снабжена
контрольно-измерительными приборами для измерения уровней жидкости,
температуры и давления, а также приспособлениями для отбора проб и
предохранительными клапанами. Все контрольно-измерительные-
приборы вынесены на общий щит управления, на котором
монтируются также вентили, служащие для регулирования
процесса ректификации.
Чистота конечных продуктов — азота и кислорода — зависит
от точности соблюдения режима работы ректификационной
колонны. Необходима хорошая
рекуперация холода в теплообменных аппаратах,
а также точное регулирование давления
в системе, влияющего на температурный
режим в различных частях аппарата.
Расходные коэффициенты зависят от
типа установки и ее производительности.
На установках, предназначенных для
получения чистого газообразного азота,
расход электроэнергии колеблется от 0,15
до 0,2 кет ¦ ч на 1 м3 азота.
Принцип устройства
ректификационной колонны виден из рис. 88. Внутри
цилиндрического корпуса / помещены
ситчатые (или колпачковые) тарелки 2,
разделяющие колонну на секции.
В тарелках имеется большое число
отверстий, равномерно распределенных
по всей их поверхности.
Для перетока жидкости с одной
тарелки на другую и поддержания на
тарелках определенного уровня жидкости
служат переливные патрубки 3. Верхний
край каждого переливного патрубка
находится на уровне жидкости в тарелке,
нижний край погружен в жидкость,
находящуюся на нижней тарелке.
Образующийся гидравлический затвор
препятствует прохождению потока пара
через патрубки. Постоянный уровень жидкости на тарелках
поддерживается также давлением пара, поднимающегося с
лежащих ниже тарелок. Пар, образовавшийся при испарении
жидкости, находящейся на нижней тарелке, проходит через
отверстия лежащей выше тарелки и, соприкасаясь на ней
с жидкостью, конденсируется. При этом получается жидкость,
более богатая летучим компонентом, чем жидкость на нижней
тарелке. Вследствие выделения теплоты конденсации этого пара
-3
-J
\Пар
Рис. 88. Схема ректифи-
кационш й тарельчатой
колонны;
/—корпус; 2—гарелки;
3—переливные патрубки.
211
жидкость па лежащей выше тарелке кипит и образует пар, еще
более богатый легколетучим компонентом по сравнению с
жидкостью. Этот пар поднимается на следующую тарелку, где
также конденсируется, образуя жидкость, обогащенную летучим
компонентом, и т. д. Избыток жидкости, образовавшейся при
конденсации пара, стекает с каждой тарелки через переливные
патрубки S на тарелку, расположенную ниже.
Вследствие многократного испарения и конденсации на
тарелках в верхней части колонны получается пар чистого легко-
легучего компонента, в нижней части колонны собирается
жидкость— чистый, менее летучий компонент.
УСТАНОВКИ ДЛЯ РАЗДЕЛЕНИЯ ВОЗДУХА
Для получения азота и кислорода разделением воздуха а
промышленности применяют главным образом установки с
дросселированием сжатого воздуха (в один или два цикла) и с
предварительным аммиачным охлаждением, а также установки
высокого и низкого давления с регенераторами и турбодетанде-
рами. Различные установки для производства азота и
кислорода отличаются друг от друга главным образом способами
сжижения воздуха, схемой ректификации, способом очистки
воздуха от двуокиси углерода и паров воды, а также
конструктивным оформлением.
Установки с дросселированием воздуха и предварительным
аммиачным охлаждением. Такие установки оснащены
дополнительным оборудованием (аммиачные холодильные машины).
Несмотря на это, они более экономичны, чем описанные ранее,
и входят в состав агрегатов средней и большой
производительности.
Схема установки для разделения воздуха на азот и
кислород с дросселированием и предварительным аммиачным
охлаждением показана на рис. 89. Воздух, очищенный от
механических примесей в фильтре /, поступает в воздушный
четырехступенчатый компрессор 2. В первых двух ступенях компрессора
воздух сжимается приблизительно до 8—10 ат и направляется-
для отделения двуокиси углерода в аппарат 3, орошаемый
10%-ным раствором NaOH. Затем воздух снова возвращается
в компрессор, где в третьем и четвертом цилиндрах сжимается;
до рабочего давления 35 или 60 ат.
Удаление влаги из сжатого воздуха на небольших
установках производят в осушительных батареях, заполненных твердым
едким натром. В последнее время для осушки воздуха
применяют также силикагель или алюмогель. На более крупных
установках влагу чаще всего удаляют путем вымораживания
испаряющимся жидким аммиаком.
213
Сжатый воздух проходит сначала через теплообменники 4,
куда поступает холодный азот из колонны 9, а затем через
аммиачный холодильник 5, где вымораживается содержащаяся
в нем влага.
Аммиачный холодильник 5 состоит из трех отдельных
змеевиков с двойными трубками (на рисунке показан один змеевик).
Внутри трубок движется воздух, в межтрубном пространстве —
эммиак (или наоборот). В первом (пс ходу воздуха) змеевике
воздух охлаждается до температуры примерно 2°С (при этом
Раствор
NaOH
Рис. 89. Схема разделения воздуха на азот и кислород с дросселированием
и предварительным аммиачным охлаждением:
/—фильтр; 2—компрессор; 3—аппарат для отделения углекислоты; 4—теплообменники;
5—аммиачный холодильник с тремя змеевиками; 6—аммиачный компрессор; 7—конденсатор; 8—
змеевиковый теплообменник; 9—двойная ректификационная ко.-шнна; 10—испаритель.
часть водяных паров конденсируется), а во втором до
температуры минус 25 — минус 40 °С. При этой температуре водяные
пары превращаются в лед, постепенно забивающий воздушные
проходы, что вызывает уменьшение коэффициента
теплопередачи. Чтобы процесс протекал непрерывно, в холодильнике
параллельно второму змеевику устанавливают третий змеевик.
Когда замороженный змеевик разогревается, в работу
включают другой змеевик. Переключение змеевиков производят один-
два раза в сутки. Для оттаивания льда используют
газообразный аммиак при давлении 7,5—12 ат и температуре 50—80 °С.
Освобожденный от водяных паров сжатый воздух поступает
в противоточный змеевиковый якорный теплообменник 8 с
длиной змеевика около 20 м, смонтированный вокруг
ректификационной колонны 9. Здесь воздух охлаждается чистым азотом
и кислородом, поступающими из колонны.
Из теплообменника 8 воздух поступает в испаритель 10,
расположенный внутри ректификационной колонны, где
дополнительно охлаждается испаряющимся жидким воздухом,
обогащенным кислородом. Эта жидкость стекает с тарелок нижней
части ректификационной колонны. Далее сжатый воздух
проходит дроссельный винтпль и поступает приблизительно в
середину нижней колонны. При этом абсолютное давление воздуха
214
снижается до 6 ат (давление в нижней колонне). Затем жидки»
воздух подвергается обычной ректификации.
Азот из верхней разделительной части колонны
направляется в теплообменник 4, где охлаждает поступающий
противотоком сжатый воздух. Далее азот и кислород поступают в
соответствующие газгольдеры.
При нормальной работе ректификационной колонны в
зависимости от условий получаются 99,95—99,97%-ный азот и 90—
92%-ный кислород или чистый (99,5—99,7%-ный) кислород и
азот с повышенным содержанием кислорода.
Можно одновременно получать и чистый азот и чистый
кислород. Для этого из нижней части верхней колонны выводят
часть газа, богатого аргоном. Такой газ, называемый сырым
кислородом, может быть использован для получения аргона.
При работе с осушительными батареями установка для
разделения воздуха работает под давлением примерно 60 ат;
разница между температурами сжатого воздуха и продуктов
разделения на теплом конце теплообменника (на входе газа)
составляет 4—5°С. При работе с аммиачным холодильником
рабочее давление в крупных агрегатах равно 100—120 ат.
Для сжатия воздуха применяют четырех- и пятиступенчатые
компрессоры производительностью от 180 до нескольких тысяч
мг1ч. Воздух после каждой ступени проходит холодильник и
маслоотделитель (на рис. 89 не показаны).
Осушка воздуха. Несмотря на тщательную осушку воздуха
от влаги, часть ее проникает в разделительный аппарат, где
выделяется в виде льда на поверхности трубок теплообменника и
тарелок колонны, что ухудшает теплообмен и процесс
ректификации. При этом нормальный режим работы аппарата
нарушается и его приходится останавливать на размораживание.
В последние годы для глубокой осушки воздуха и других
газовых'смесей (коксового, природного, попутного газов) в
качестве адсорбента применяется активированный алюмогель
(активная окись алюминия). Количество влаги, остающейся в
газе после адсорбции ее активированным алюмогелем,
уменьшается в 13—14 раз по сравнению с влажностью газа после
осушки вымораживанием.
Кроме активированного алюмогеля, начали применять
активированные силикагель и бокситы, в качестве сорбентов
предложены также искусственные цеолиты. Осушку газов твердыми
адсорбентами можно проводить как в адиабатических, так и в
изотермических условиях.
Адсорбционная установка непрерывного действия состоит из
двух стальных баллонов, заполненных активированным
алюмогелем. Между этими адсорберами расположен
электронагреватель для подогрева азота, поступающего на регенерацию
адсорбента. Адсорберы н подогреватель заключены в общий ко-
215
жух, снабженный теплоизоляцией. Сжатый и охлажденный
воздух попеременно пропускают через баллоны с адсорбентом.
В" то время как в одном баллоне происходит осушка воздуха,
в другом производится регенерация адсорбента (удаление из
него влаги) путем нагревания и продувки сухим азото.-.i После
охлаждения регенерированного адсорбента до 25--30°С баллон
снова переключается на осушку воздуха. Рабочий цикл одного
адсорбера продолжается 8—12 ч.
Аппараты различной производительности непрерывно
работают от 20 до 100 суток. После остановки аппарата намерзшие
примеси удаляют продуванием теплого воздуха, подогретого
паром до 50°С. Продолжительность отогрева малых агрегатов
4—5 ч, высокопроизводительных — до 48 ч. Охлаждение
аппарата после оттаивания занимает от 6—8 до 20 ч.
В описанной выше разделительной установке теплообмен
осуществляется через стенку, разделяющую газовые потоки.
Это требует больших поверхностей теплообмена и служит
причиной значительных потерь давления в системе. В ряде
случаев для получения дешевого обогащенного кислородом
воздуха (например, для 'металлургических процессов) или при полу
чении только чистого кислорода вместо трубчатых
теплообменников устанавливают регенераторы.
Регенераторы. В кислородных установках регенераторы
применяются для охлаждения основного потока воздуха, идущего
на разделение в ректификационную колонну, а также для
очистки его от двуокиси углерода и от влаги. Для непрерывной
работы требуются два аппарата. Регенераторы представляют
собой цилиндрические сдвоенные аппараты (рис. 90),
заполненные насадкой. В одном из этих аппаратов уходящие из системы
газы отдают свое тепло (или холод) насадке, в это же время
в другом регенераторе газы, поступающие в систему,
нагреваются (или охлаждаются), соприкасаясь с горячей (или
холодной) насадкой. Периодический ввод холодного и теплого
газа в регенераторы (через 0,5—3 мин) осуществляется
автоматически при помощи переключающих механизмов.
Насадка (рис. 91) выполняется из гофрированных
алюминиевых лент (высота ленты 35 мм, толщина 0,2—0,4 мм),
свернутых в виде дисков. Такие диски, уложенные в корпусе
регенератора, образуют теплоемкую насадку с весьма развитой
поверхностью. Поверхность теплообмена на 1 м3 такой насадки
¦составляет 20О0—4000 м2 в зависимости от типа ленты.
Преимуществами регенераторов по сравнению с
теплообменниками являются значительно большая поверхность
теплообмена и незначительное гидравлическое сопротивление, что
обусловливает весьма малые потери давления при пропускании
через аппарат больших объемов газов. Кроме того, при
использовании регенераторов отпадает необходимость предвари-
216
тельнои очистки воздуха от двуокиси углерода и его осушки..
Остающиеся на насадке регенератора при охлаждении воздуха
твердая двуокись углерода и лед удаляются обратными
потоками газов. К недостаткам регенераторов следует отнести
загрязнение продуктов разделения воздухом, который
пропускают через регенератор до поступления в него газов,
уходящих из системы. Загрязнение тем больше, чем выше давление
газа, пропускавшегося до переключения регенераторов. Для
уменьшения загрязнений и потерь воздуха на регенераторах
устанавливают перепускные
клапаны, выравнивающие давление
перед переключением.
Воздух
воздух
Рис. SO. Схема
регенераторов.
Рис. 91. Насадка регенераторов
из гофрированной ленты
Регенераторы применяются в крупных установках для
получения 96—98%-кого газообразного кислорода.
Азотная установка Г-6800. В этой установке применены два
цикла давлений воздуха и аммиачное охлаждение.
Производительность установки 6800 м3/ч воздуха, при этом получается
5400 м3/ч азота и около 1200 м3/ч 99%-ного кислорода.
Основное количество воздуха (80%) сжимается до избыточного
давления 5—5,5 ат, остальное — до 100—120 ат. Во время
пускового периода поддерживается максимальное давление B00 ат).
Эта установка более экономична, чем описанная выше
установка с о^"" циклом давления и аммиачным охлаждением.
Характеристика установки с двумя циклами давления и
аммиа' ,i охлаждением при различной производительности
приведена ниже:
Производительность, л'/ч кислорода .
Расход энергии, квт-чЫ3 кислорода
Расхоц охлаждающей воды, М3/ч . .
200 5С0 1000
0,82 0,72 0,7
13 30 35
217
В последние годы в аппаратуру установки внесен ряд
конструктивных усовершенствований. Так, вместо кожухотрубных
теплообменников низкого давления начали применять змееви-
ковые теплообменники, вместо якорных теплообменников —
змеевиковые теплообменники высокого давления. Изменены
также конструкции ректификационных тарелок,
конденсатора и т. д.
Кислородная установка БР-1 низкого давления. В настоящее
время для получения больших количеств так называемого
технологического (96—98%-ного) кислорода, используемого в
металлургических процессах, для газификации твердого топлива
и др., широко применяются установки низкого давления
F—6.5 ат) с регенераторами и турбодетандером. В таких
установках применяются турбодетандеры реактивного действия,
впервые разработанные акад. П. Л. Капица в 1937 г.
Реактивный турбодетандер имеет высокий адиабатический к. п. д.
@,82—0,83). Крупная установка БР-1 для получения
технологического кислорода работает по циклу низкого давления и
рассчитана на выработку 12 500 м3/ч 96—98%-ного кислорода.
На рис. 92 показана принципиальная технологическая схема
блока разделения воздуха установки БР-].
Воздух, очищенный в фильтре / от механических примесей
и сжатым п турбокомпрессоре 2 до абсолютного давления 6—
6,5 ат, поступает параллельно через холодильник 3 в два
кислородных регенератора 4 и в три азотных регенератора 5, где
охлаждается и освобождается от двуокиси углерода и влаги.
Выпадающие в регенераторах твердая двуокись углерода и лед
удаляются током азота, который одновременно охлаждает
регенераторы. Регенераторы переключаются через каждые
3 мин.
Холодный воздух, выходящий из регенераторов, делится на
два неравных потока. Больший поток направляется в куб
нижней колонны 16, меньший, предназначенный для
дополнительного охлаждения насадок регенераторов, через специальный
клапан сначала поступает в холодные концы регенераторов для
создания здесь разности температур в 5—6°С между прямым и
обратным потоками. Из середины регенераторов воздух
отводится для охлаждения в детандерный теплообменник 8 и далее
присоединяется к большему потоку воздуха, идущему в
нижнюю колонну.
Разделительный аппарат состоит из двух отдельных
колонн— нижней колонны 16, верхней колонны 14 и четырех
конденсаторов 13. Около 25% воздуха из нижней колонны
направляется через отделитель жидкости 9 в детандерный
теплообменник 8, где подогревается до минус 156—157°С A16—
117 °К), и через детандерный фильтр 12 поступает в
турбодетандер.
218
вода
Рис. 92. Принципиальная технологическая схема блока разделения воздуха установки БР-1:
1, 12, 18—фильтры; 2—турбокомпрессор; 3—холодильник; 4—кислородные регенераторы; 5—азотные регенераторы;
6—подогреватель; 7, 17, /9—адсорберы; 8—детамдерньш теплообменник; 9—отделитель жидкости; /0—переохладитель; //—турбодетаидер;
/J—конденсаторы; 14— верхняя колонна; /5—распределительный бачок; 16—нижняя колонна.
После расширения в турбодетандере и очистки от ацетилена
б адсорбере 7 воздух направляется в верхнюю колонну 14.
Из нижней колонны кубовая жидкость, представляющая
собой жидкий воздух, обогащенный кислородом, последовательно
проходит через керамический фильтр 18, где сначала
освобождается от твердых частиц двуокиси углерода, затем в
адсорбере 17 очищается от ацетилена.
Отсюда жидкий азот проходит переохладитель 10 и
дросселируется в распределительный бачок 15, откуда подается на
тарелки колонны 14.
Пары азота, уходящие из нижней колонны 16, поступают
в трубы конденсаторов 13, где сжижаются. За счет теплоты
конденсации азота в трубах аппаратов 13 происходит кипение
кислорода в межтрубном пространстве.
Пары азота, уходящие из верхней колонны, сначала
проходят переохладитель 10, затем подогреватель 6, где
температура азота повышается до —178° (95 °К) вследствие конденсации
части воздуха, поступающего из нижней колонны. Благодаря
подогреву азота улучшается удаление двуокиси углерода из
насадки регенераторов. Далее азот проходит регенераторы 5 и
освобождает их насадку от твердой двуокиси углерода и льда,
а затем удаляется в атмосферу.
Кислород, очищенный от ацетилена в адсорбере 19,
отводится из верхней части конденсатора 13 в кислородные
регенераторы 4 и далее поступает в газгольдер.
Поскольку на установке БР-1 перерабатываются большие
количества воздуха, из продуктов его разделения
целесообразно извлекать криптон и ксенон.
Удельный расход энергии на описанной установке составляет
0.43—0,47 квт-ч на 1 мг газообразного кислорода.
Кроме установки БР-1, для получения технологического
кислорода применяются также установки низкого давления
производительностью 3500 и 5000 мг/ч кислорода.
Установки с двумя циклами давления, регенераторами и
турбодетандером применяются для получения до 3600 м3/ч
97%-ного кислорода. Охлаждение производится при помощи
турбодетандера и холодильного цикла высокого давления
(Рабс.= 150—200 ат).
Расход энергии на 1 м3 газообразного кислорода составляет
0,53—0,56 квт-ч.
На установках с регенераторами и турбодетандером можно
также получать 99—99,5%-ный кислород или 99,8-ный азот.
Для снижения себестоимости кислорода на агрегатах с
регенераторами и турбодетандером целесообразно устанавливать
дополнительную колонну для извлечения криптоно-ксеоновой
фракции.
220
Хранение и перевозка кислорода
Значительная часть кислорода не используется на месте его
производства, а транспортируется потребителям. Газообразный
кислород хранят и перевозят в стальных бесшовных баллонах
средней или малой емкости под давлением 150 или 200 ат (до-
пустимое отклонение ±5 ат при 20°С).
Предварительно осушенный кислород нагнетается в баллоны
кислородным компрессорам через наполнительные рамы.
Осушка кислорода производится в специальных баллонах,
заполненных хлористым кальцием, или в адсорберах с насадкой из
активированного глинозема. Вес стальных баллонов в 8,5—9 раз
превышает вес содержащегося в них кислорода A,36 кг),
вследствие чего стоимость перевозки кислорода в баллонах довольно
высока.
Поскольку кислород находится в баллонах под высоким
давлением, необходимо обращаться с ними весьма осторожно и
соблюдать необходимые правила техники безопасности. При
сборке вентиля баллона и при эксплуатации баллонов не
допускается применение органических веществ для уплотнения
и смазки, так как они смогут воспламеняться при
соприкосновении со сжатым кислородом. Баллоны, находящиеся в
эксплуатации, через каждые пять лет подвергают периодическому
осмотру и пневматическим и гидравлическим испытаниям.
Хранение и перевозка жидкого кислорода. Значительно
экономичнее хранение и перевозка кислорода в жидком состоянии.
Так, для хранения 2400 м3 C430 кг) газообразного кислорода
требуется 400 баллонов емкостью по 6 и3 и общим весом 27 т.
Для хранения такого же количества жидкого кислорода
требуется один небольшой танк, собственный вес которого 1,8 т.
Для перевозки указанного количества газообразного кислорода
необходимы шесть 5-тонных автомашин, а для перевозки его
е жидком виде — одна такая автомашина. Однако следует
учитывать, что хранение жидкого кислорода, наполнение им
тары и слив в газификаторы сопровождаются значительными
потерями газа, в отдельных случаях достигающими 30—40%
кислорода, что снижает преимущества транспортирования
кислорода в жидком состоянии. Поэтому для потребителей
небольших количеств кислорода целесообразнее получать его с
кислородных установок в газообразном виде.
Для хранения жидкого кислорода, отбираемого
непосредственно из разделительного аппарата, применяют
стационарные танки емкостью от 1000 до 12000 л, которые
устанавливают рядом с разделительным аппаратом (обычно за
кирпичной стеной). Танк состоит из внутреннего шарового сосуда,
наружного сосуда и цилиндрического кожуха, служащего
опорой. Внутренний шар, изготовляемый из тонкой листовой ла-
221
туни или меди, при помощи болтов и цепей крепится к
наружному стальному сосуду. Пространство между обоими сосудами
заполняется теплоизоляционным материалом. Жидкий
кислород поступает в танк по трубе, соединенной с конденсатором
разделительного аппарата. Испаряющийся в танке кислород
направляется в газгольдер. Избыточное давление е танке не
должно превышать 0,3—0,4 ат. Танк снабжен
предохранительным клапаном, установленным на предельное избыточное
давление 0,6 ат.
При отборе жидкого кислорода из разделительного
аппарата увеличивается расход холода на установке. Следовательно,
в этом случае больше энергии расходуется на предварительное
сжатие воздуха до более высокого давления.
Жидкий кислород перевозят потребителю в транспортных
танках на автомашинах. По устройству транспортный танк во
многом сходен со стационарным. Емкость транспортных танков
от 660 до 3000 л.
Для хранения и перевозки больших количеств жидкого
кислорода применяются цистерны с теплоизоляцией,
вместимость цистерн 10; 13.5 и 32 т. Цистерны на 32 т кислорода
изготовляют из алюминиевого сплава и снабжают наружным
стальным кожухом. Пространство между кожухом и цистерной
заполнено теплоизоляционным материалом (толщина 350—
400 мм). Потери кислорода при хранении его в цистернах
обычно составляют 3—5% в сутки. Выпускаются цистерны двух
типов на избыточное давление 0,7 и 0,5 ат, монтируемые на
железнодорожных платформах соответствующей грузоподъемности.
Стационарные цистерны аналогичной конструкции
устанавливаются на крупных заводах и используются для приема и
хранения жидкого кислорода.
Для хранения небольших количеств кислорода (от 2 до 50 л)
применяются сосуды Дьюара с вакуумной изоляцией.
Сорта кислорода и азота
Выпускаемый промышленностью газообразный (технический
и медицинский) кислород, согласно ГОСТ 5583—58, должен
отвечать следующим требованиям:
Высший 1-й 2-й
сорт сорт сорт
Содержание кислорода, объемн. %, не
менее 99,5 99,2 98,5
Содержание влаги, г;лг, не более 0,07 0,07 0,07
Технический газообразный и жидкий азот, получаемый из
атмосферного воздуха путем его сжижения с последующей
222
ректификацией, согласно ГОСТ 9293—59 должен удовлетворять
таким показателям:
Содержание
менее . .
Содержание
не более
азота, o6beN4H. %,
кислорода, объемн.
не
%,
Газообразный
вакуумный
99,9
0,1
1-го
сорта
99,5
0,5
азот
2-го
сорта
99
1,0
Жидкий
азот
96
4
Примечания. 1. Содержание влаги может соответствовать полному
насыщению водяным паром при температуре газа в баллонах.
2. Настоящий ГОСТ не распространяется иа газообразный и жидкий азот,
применяемый в производстве синтетического аммиака.
Газообразный азот отпускают потребителям в стальных
бесшовных баллонах средней или малой емкости под давлением
150 кгс/см2 (отклонения а:5 кгс/см2 при температуре 20 °С).
Жидкий азот отпускают в металлических сосудах Дьюара или
передают в транспортные емкости (плотность жидкого азота
при температуре кипенич под давлением 1 атм составляет
0,81 кг\л).
Контроль работы и автоматизация установок
Для нормальной эксплуатации каждая установка глубокого
холода снабжается контрольно-измерительными приборами
(манометры, дроссельные вентили, указатели уровня,
переключатели, предохранительные клапаны и др.), которые монтируются
на щите управления разделительного агрегата. В настоящее
время разрабатываются схемы дистанционного управления и
автоматизации установок глубокого холода, в частности
кислородной установки низкого давления. Блок разделения этой
установки оснащен восемью автоматическими регуляторами
(четыре регулируют тепловой режим регенератора,
остальные—режим разделительной колонны). В качестве датчиков служат
термометры сопротивления с различной градуировкой, диафрагмы
и дифференциальные манометры, газоанализаторы.
Регулирующими органами являются дроссельные заслонки, жидкостные
вентили, двухседельные клапаны различного диаметра и др.
Автоматизация агрегатов глубокого холода позволяет не
только увеличить их производительность по азоту п кислороду,
но и уменьшить расход энергии на компримирование газов.
5. ПРОИЗВОДСТВО ВОДОРОДА (А30Г0В0 ДОРОДНОЙ С МЕСИ)
РАЗДЕЛЕНИЕМ КОКСОВОГО ГАЗА
При коксовании каменных углей получаются кокс, коксовый
газ и каменноугольная смола. В зависимости от состава и
свойств каменного угля, подвергающегося коксованию, выход
223
коксового газа (считая на сухой уголь) составляет 15—20%,
выход каменноугольной смолы колеблется в пределах 3—4,5%
от количества полученного металлургического кокса.
В зависимости от качества угольной шихты и условий
коксования коксовый газ после выделения из .чего аммиака, сырого
бензола и смолы (так называемый обратный газ) имеет
примерно следующий состав (в объемн. %):
Водород 55—61
Метан (и этан) 24—28
Олефины (этилен, пропилен) 1,5—3,0
Окись углерода 6—9
Двуокись углерода 2—4
Азот 2—7
Кислород . 0,3—0,5
Сероводород 0,2—1,2
Кроме того, обратный коксовый газ содержит органические
соединения серы D00—1000 мг/м3), небольшие количества
бензола, нафталина, синильной кислоты, ацетилена, окиси азота.
В химической промышленности коксовый газ используется
как источник получения водорода для синтеза аммиака
и ряда органических продуктов. Выделяемая из коксового газа
этиленовая фракция используется в синтезах полиэтилена,
окиси этилена, этанола, дивинила, этилбензола, изопропилбен-
зола, этиленгликоля, дихлорэтана и других важных химических
продуктов, потребление которых непрерывно возрастает.
Сероводород, получаемый в результате очистки коксового
газа, перерабатывается в серную кислоту или в элементарную
серу (стр. 52"сл., 124 ел.).
Водород (азотоводородную смесь для синтеза аммиака) из
коксового газа можно получать двумя принципиально
различными способами: низкотемпературным разделении коксового
газа и конверсией содержащегося в газе метана (стр. 172 ел.).
При наличии достаточно больших ресурсов коксового газа
целесообразна схема низкотемпературного разделения газа,
предварительно очищенного от сероводорода, двуокиси углерода
и окиси азота. По этой схеме в азотоводородную смесь
переходит до 95% водорода, содержащегося в коксовом газе,
одновременно выделяется этиленовая фракция (до 50—60% С2Н4).
Остаточный, так называемый богатый газ, содержащий метан
и окись углерода, используется как топливо для
металлургических печей.
Конверсионный метод переработки коксового газа позволяет
значительно увеличить выход водорода, так как при этом в
азотоводородную смесь переходит не только водород,
содержащийся в газе, но и водород, образующийся в результате
конверсии углеводородов коксового газа. Однако для
каталитической конверсии требуется более тонкая очистка газа от серы.
224
Кроме того, при конверсии не используются олефины коксового
газа.
При включении азотного завода, использующего водород
коксового газа, в состав металлургического и коксохимического
комбината целесообразно предусматривать общую установку
разделения воздуха. Получаемый на ней азот высокой чистоты
будет использоваться в синтезе аммиака, кислород — в
металлургических процессах (для интенсификации доменного
процесса выплавки чугуна, мартеновского процесса производства
стали).
При кооперировании азотных и металлургических
предприятий себестоимость получаемого аммиака снижается. S)ro
позволяет отнести коксовый газ к одним из дешевых видсз сырья
для производства связанного азота.
НИЗКОТЕМПЕРАТУРНОЕ РАЗДЕЛЕНИЕ КОКСОВОГО ГАЗА
Предварительно очищенный от бензола, нафталина, окислов
азота, аммиака, 'цианистых и сернистых соединений, а также
двуокиси углерода и водяных паров коксовый газ направляется
на разделительную установку. Разделение коксового газа
методом глубокого (низкотемпературного) охлаждения основано
на большой разнице температур кипения компонентов газа.
Глубоким охлаждением коксового газа (примерно до
—196 °С) с последующей промывкой его жидким азотом зь;де-
ляют газообразную азотоводородную смесь (при абсолютном
давлении 1 атм температура кипения водорода —252,8 °С,
азота— 195,8°С), используемую для синтеза аммиака, и жидкие
фракции — метановую, окиси углерода, этиленовую, пропиле-
новую. Эти жидкие фракции испаряют, теплоту их испарения
используют для охлаждения и конденсации коксового газа.
Практически для разделения коксового газа путем
глубокого охлаждения используют метод ступенчатой
(фракционированной) конденсации. Обычно из коксового газа выделяют
3—4 фракции; каждая фракция конденсируется при
охлаждении газа до определенной температуры.
Фракционированную конденсацию целесообразно проводить
при повышенном давлении, испарение фракции — при
атмосферном давлении. Например, чистый метан кипит под
атмосферным давлением при —161,6 °С, а конденсируется при
абсолютном давлении коксового газа 10 атм уже при —150"С.
При охлаждении коксового газа, кроме конденсации его
компонентов, происходит растворение еще не сжиженных газов в
образовавшемся конденсате. Чем ниже температура и выше
дазление коксового газа, тем больше растворимость газов и,
следовательно, больше потери водорода с другими фракциями.
Так, при охлаждении коксового газа до —185°С при атмосфер-
15—2848 225
ном давлении конденсируется около 25% начального объема
газа, причем в конденсате растворяется около 2% Н2. При
давлении 10 атм и той же температуре конденсируется около 42%
газа, а в конденсате растворяется 4% Н2.
В агрегатах разделения коксового газа для компенсации
потерь холода предусматривается специальная холодильная
установка, в которой рабочей средой является азот. Для
промежуточного охлаждения коксового газа и сжатого азота служит
аммиачная холодильная установка.
Технологический процесс состоит из следующих операций:
1) очистка коксового газа от нафталина, сероводорода, окислов
азота и удаление водяных паров; 2) сжатие газа (обычно до
избыточного давления 12—13 ат); 3) освобождение от бензолг
(охлаждением); 4) удаление углекислоты; 4)
промежуточное охлаждение до —45 °С; 6) глубокое охлаждение и
фракционированная конденсация.
Схема разделения коксового газа изображена на рис. 93.
Коксовый газ, предварительно полностью очищенный (см.
выше), сжимается в компрессоре 1 до давления 12—13 ат, про
ходит теплообменник 2, затем теплообменники 3. В
теплообменнике 2 коксовый газ охлаждается фракцией окиси углерода, в
теплообменниках 3— метановой фракцией и азотоводороднои
смесью, выходящими из разделительного агрегата 5.
Температура коксового газа при этом понижается примерно до —25 °С.
Далее газ охлаждается в теплообменниках 4 до —45 °С
кипящим жидким аммиаком, полученным в аммиачной холодильной
установке. Охлажденный до —45 °С коксовый газ поступает в
разделительный агрегат 5 на фракционированную конденсацию
н промывку жидким азотом.
Разделительный агрегат глубокого охлаждения состоит из
ряда теплообменников (б, 7, 9, 10).
конденсаторов-испарителей 8 и 13 и тарельчатой разделительной колонны //,
устроенной по типу ректификационной колонны.
По мере охлаждения из коксового газа последовательно
конденсируются компоненты, обычно отводимые в виде четырех
фракций: пропиленовой, этиленовой, метановой и фракции окиси
углерода. Смешивая все эти фракции, получают богатый газ
с теплотворной способностью около 6000 ккал/м3.
Углеводородные фракции применяют также в качестве исходного сырья для
синтеза органических продуктов.
В разделительном агрегате 5 в газообразном состоянии
остаются только водород, азот и небольшое количество окиси
углерода н метана. Окончательное освобождение
азотоводороднои смеси от остатков СО и СН4 достигается промывкой газа
в разделительной колонне 11 жидким азотом.
Разделение коксового газа в агрегате 5 осуществляется
следующим образом. Вначале газ охлаждается до —453С,
226
A, 1
Очищенный
коксовый газ
15 *' /6
Рис, lJ3, Схема уетлиоикп мдля разделения'kokcoisoi'o \'ivmv
/—компрессор; 2—прямо'! очным теплообменник; о—i еплообмепиики, охлаждаемые аммиаком; '¦>—- р<и/и /и! i г. -uibiii ai pu л i; 0 — про i и но-
точны!! 'K4r.'iotjuiMt4iiini\ (ut'ii/iHH in'iiib»); 7-- проти ног пчп bifi тс^лообмсчнш к (-холодная dc-tbij>.); Ь—koi I дсч ica 11 >р -исч lap ii'i ель (отдели гель
этилена); У—дополнительный теплообменник-; /^— рркупрр.'щиоипип теплообмен пи к; // ра ^с-литсльпая ко.чгмша; f'2— дроссельный неп-
'iiuiii; lii - КО||ДСЦе;11 up-1 кч i;i pi i'i e;iii a,ioia; !¦{ иммначны!'! \о юднлышк a-ioi а; /Л аммилчиьи! компресс op l (й- амммачп]>|Г| кочдепеа irjp;
17--л (oi nui'l компрессор,
Затем газ поступает сверху в межтрубное пространство
теплообменника 6 («теплая ветвь»), внутри которого по
трубкам проходят снизу вверх холодная азотоводородная смесь и
метановая фракция. Температура газа понижается примерно
до—105 °С, при этом из него конденсируются пропилен и
другие углеводороды, кипящие при более высокой температуре
(пропиленовая фракция).
Далее газовая смесь проходит снизу вверх теплообменник 7
(«холодная ветвь»), где охлаждается выходящими из
разделительного агрегата азотоводородной смесью, метановой
фракцией и фракцией окиси углерода до —145 °С. При этой
температуре из газа конденсируется этилен. Основное
количество сконденсировавшегося этилена стекает вниз, растворяет
выделившиеся в твердом виде компоненты газ (высшие
углеводороды, водяные пары, СО2) и собирается внизу «холодной
ветви». Часть этилена, увлеченная газом, задерживается в
отделителе 8. Смесь конденсатов из теплообменника 7 и
отделителя 8 образует этиленовую фракцию.
В следующем, дополнительном теплообменнике 9 газ
движется в межтрубном пространстве сверху вниз; навстречу ему
движутся по трубам азотоводородная смесь, метановая
"фракция и фракция окиси углерода. Здесь газ охлаждается до
—180°С, при этом конденсируется метановая фракция, которая
вместе с газовой фазой поступает в азотный испаритель 13.
В испарителе 13 коксовый газ движется по трубам снизу вверх.
Здесь при температуре —190 °С, создаваемой за счет кипения
азота в межтрубном пространстве (под абсолютным давлением
около 1,5 ат), происходит окончательная конденсация
метановой фракции и частичная конденсация окиси углерода.
Метановая фракция и часть окиси углерода стекают по трубкам вниз
и вместе с конденсатом, идущим из дополнительного
теплообменника, собираются в нижнем резервуаре, расположенном под
азотным испарителем 13 (на рисунке не показано). Метановая
фракция из нижнего резервуара проходит последовательно
теплообменники 9, 7 и 3, охлаждая поступающий навстречу
коксовый газ.
Газовая смесь, выходящая из испарителя азота 13, содержит
в основном водород и следующие примеси (в объемн. %).
N2 . . . . 5—10 СН4 . . 0,2—1,0
СО. ... 3—4 О2 . . . 0,3
Газ такого состава непригоден для синтеза аммиака. Для
удаления окиси углерода, метана и кислорода газ направляют
в разделительную тарельчатую колонну 11, где он промывается
поступающим сверху жидким азотом. Из верхней части
промывной колонны выходит сырая азотоводородная смесь под
избыточным давлением около 10 ат, содержащая ~15% N2 и
228
-35% Н2. Содержание водорода в смеси далее снижают до 75%
л добавления в нее чистого азота.
Лзотосодородная смесь направляется в теплообменники 9,
7 и 6 для охлаждения газа и выходит из разделительного
агрегата 5. В нижней части разделительной колонны // собирается
фракция окиси углерода (состоящая из СО2, N2 и небольшого
количества метана), которую направляют из разделительной
колонны в теплообменник 9 и далее через теплообменники 7
и 2 выводят из цикла.
Для разбавления газовой смеси применяют азот,
полученный ректификацией жидкого воздуха (стр. 209). Его сжимают
з пятиступенчатом компрессоре 17 до 200 ат и затем охлаждают
до —45 ЭС в змеевиковом аммиачном холодильнике 14. После
этого азот высокого давления дополнительно охлаждается в
рекуперацпонном теплообменнике 10 парами азота,
поступающие из испарителя 13. Из рекуперациокного теплообменника
10 часть жидкого азота через дроссельный вентиль 12
направляется на орошение в разделительную колонну //, другая часть
используется для охлаждения испарителя 13.
В описанной установке азотоводородная смесь выходит из
разделительного агрегата под избыточным давлением 10 ат.
Содержание окиси углерода в азотоводородной смеси
колеблется ? пределах 0,001—0,005%.
Разделительный аппарат периодически выключают и
разогревают для удаления накопившихся в системе твердых
отложений (льда, двуокиси углерода, бензола, смол). Кроме того,
в установках для разделения коксового газа постепенно
накапливаются окислы азота, которые, вступая во взаимодействие с
непредельными углеводородами, образуют неустойчивые
взрывчатые нитросоединения, которые необходимо удалять из
разделительного аппарата (стр. 259).
Для получения 1 м3 азотоводородной смеси, состоящей из
25% N2 и 75% Н2, расходуется 1,5—1,55 мг коксового газа и
примерно 0.25 м3 азота.
Этилен, содержащийся в коксовом газе, выделяют из
этиленовой фракции, получаемой при фракционированном
разделении коксового газа или же непосредственно извлекают из кок-
-созого газа. Этиленовая фракция, содержащая примерно 25—
35% С2Н4, 3—6% С3Н6, до 1% С2Н2, 35—45% СН4, до 1% О2, до
3% СО, 5—6% N2 и переменное количество водорода, после
испарения в разделительном аппарате вновь конденсируется
при —160СС и небольшом давлении. Конденсат разделяется на
три фракции: 98—70%-ный этилен (выход до 90%), 90%-ный
метан (выход 50%) и 60%-ный пропилен (выход до
85%).
Расход энергии в этом процессе составляет около 3700 кет • ч
ча 1 г 97%-ного этилена.
229
Метод фракционированной низкотемпературной
конденсации используется для получения олефпнов из газов
нефтепереработки, а также для очистки сырой азотоводородной смеси,
получаемой из газов конверсии метана.
Для непосредственного извлечения этилена из коксового
газа используются процессы избирательной адсорбции,—
например, движущимся слоем поглотителя (гиперсорбция) и др.
АВТОМАТИЗАЦИЯ ПРОЦЕССА РАЗДЕЛЕНИЯ КОКСОВОГО ГАЗА
Автоматизация установок для низкотемпературного
разделения коксового газа на фракции осложняется необходимостью
регулирования параметров процесса при низких температурах
(до •—200 °С) и высоких давлениях (до 200 ат). При этом во
многих случаях серийные автоматические приборы непригодны,
и приходится применять специальные приборы и аппаратуру.
Схемой автоматизации агрегата разделения коксового газа
на одном из отечественных заводов предусматриваются
следующие системы автоматического регулирования и управления:
1) регулирование температуры коксового газа ка выходе
из испарителя путем изменения подачи жидкого азога в
испаритель; заданная температура коксового газа автоматически
корректируется по концентрации СО на одной из тарелок
колонны;
2) контроль очистки азотоводородной смеси от СО путем
изменения расхода азота на промывку в зависимости от
концентрации окиси углерода на одной из тарелок колонны;
3) регулирование температуры коксового газа перед
дополнительным теплообменником путем изменения подачи азота в
сырую азотоводородкую смесь;
4) регулирование уровня жидкости в испарителе изменением
подачи в него азота;
5) регулирование уровня в «холодной ветви» и в этиленовой
ловушке путем изменения количества сливаемого этилена;
6) автоматическое переключение и продувка фракционных
и аммиачных теплообменников, змеевиков и др.
Глав о Г/
АММИАК
/. ОБЩИЕ СВЕДЕНИЯ
Свойства и применение аммиака. При комнатной
температуре аммиак ХН3 представляет собой бесцветный газ с
удушливым резким запахом и едким вкусом, раздражающе деиствую-
ашй на слизистые оболочки.
Основные физико-химические константы аммиака приведены
.чиже:
Плотность при нормальных условиях, г'л 0,77
Температура кипения, °С —33,35
Температура плавления, °С —77,75
Критическая температура, °С 132,4
Критическое давление, атм 111,5
Удельная теплоемкость ср при 25°С. кал'(моль-град) ..... 8,523
Теплота испарения NFL, жидк,, кал]г
при — 50 СС ..." 337,97
при 0=С 300,52
при 50 =С 252,29
Аммиак очень хорошо растворим в воде, при комнатной
температуре и атмосферном давлении в 1 л воды растворяется
около 750 л газообразного аммиака. Несколько хуже он
растворим в органических растворителях (спирт, ацетон, хлороформ,
бензол). Для многих соединений, содержащих азот, аммиак
является хорошим растворителем.
Водные растворы аммиака (аммиачная вода),
водно-аммиачные растворы солей и жидкий аммиак широко применяются &
сельском хозяйстве для непосредственного внесения в почву а
качестве жидких азотных удобрений.
При обычной температуре аммиак устойчив. Диссоциация
аммиака на N2 и Н2 в газозой фазе становится заметной при
1200—1300СС и выше, в присутствии катализатора аммиак
диссоциирует при 300—400 °С. Аммиак весьма реакционноспособ-
ное вещество, вступающее в реакции присоединения,
замещения и окисления.
При непосредственном соединении аммиака с кислотами
получаются соли, например нитрат аммония (стр. 556), сульфат
¦аммония (стр. 566). При взаимодействии жидкого аммиака с
231
двуокисью углерода получается мочевина (стр. 569). При
замещении атомов водорода в МНз металлом образуются
нитриды, имиды и амиды металлов.
В обычных условиях аммиак довольно устойчив х действию
окислителей, но при поджигании горит с образованием азота и
воды.
На поверхности твердого катализатора, например
сплава платины с родием или сплава платины с палладием п родием,
при 750—800 "С аммиак окисляется до окиси азота. На этой
реакции основан промышленный способ производства азотной
кислоты (стр. 265, 270 ел.).
Окислением аммиака и метана воздухом получается
синильная кислота HCN.
Сухие смеси аммиака с воздухом при определенных
условиях способны взрываться. Пределы взрываемости* аммиачно-
воздушных смесей (при 18°С): верхний предел 27 объемн. %
NH3, нижний предел 15,5 объемн. % NH3. Таким образом,
смеси, содержащие меньше 15,5 или больше 27 объемн. % NH3,
при зажигании их искрой не взрываются. При повышении
температуры пределы взрываемости аммиачно-воздушных смесей
расширяются.
В настоящее время аммиак в огромных количествах
применяется для производства азотной кислоты, твердых и жидких
азотных удобрений (стр. 555 ел., 576), используется в
производстве соды по аммиачному способу (стр, 425 ел.) и ряда других
зажных химических продуктов. Азотсодержащие соединения
являются исходными веществами во многих процессах
органического синтеза, в том числе в синтезах весьма ценных
полимеров (полиамиды, полиуретаны, полиакрилонптркл н др.).
Отсюда ясно, что производство связанного азота имеет
исключительно важное народнохозяйственное значение.
Производство связанного азота. В природе азот встречается
в свободном состоянии как составная часть воздуха, а также
в виде нитратов (селитры), главным образом в виде клтрата
натрия, и в виде органических соединений в торфе и каменном
угле. Получение соединений азота из воздуха, несмотря на
неограниченные количества его, было сопряжено с такими
большими трудностями, что впервые это производство /далось
создать только в начале XX в. Запасы же связанного азота
в органических ископаемых (углях) и в форме селитры
довольно малы.
* Существуют нижний и верхний пределы взрываемости смесей горючих
газов (паров) с воздухом. Нижним предеяо.ч начыпие-'ся наименьшее
содержание горючего газа или пара в смеси с воздухом, при котором может
произойти взрыв, верхним пределом — наибольшее содержание горючего
газа или пара з смеси з воздухом, при котором мо;к> т произойти чрыв.
232
До 1903 г. важнейшим источником получения соединении
азота были природные залежи натриевой селитры, запасы
которой сравнительно невелики и сосредоточены главным образом
б Чили (Южная Америка). Другим источником соединении
азота является аммиак, содержащийся в коксовом газе. В
настоящее время все коксовые печи снабжены установками для
улавливания аммиака из отходящих газов. Однако количество
получаемых из коксохимического аммиака азотных соединений,
главным образом в виде сульфата аммония (стр.
567)—минерального азотного удобрения, относительно невелико.
В связи с ограниченностью запасов природной селитры и
отдаленностью их от основных потребителей решение задачи
использования свободного азота приобрело в целом ряде стран
первостепенное значение. Однако вследствие химической
инертности азота попытки фиксации его из атмосферного воздуха
длительное время оставались безуспешными. Лишь в начале
XX в. удалось использовать азот воздуха путем его
окисления кислородом в электрической дуге при 3500 — 4000 °С
(дуговой метод) по реакции
N2 + О, = 2NO — 42,8 /скал
В результате этого процесса получались газы, содержащие
2—3% окиси азота, которая в дальнейшем перерабатывалась
в азотную кислоту. Получение связанного азота таким методом
требовало затраты очень большого количества электроэнергии
F0 тыс. квт-ч на 1 т связанного азота).
Дуговой метод связывания атмосферного азота не
получил значительного распространения и был полностью оставлен
в конце 20-х годов.
В последнее время стали разрабатывать способы получения
окиси азота из воздуха при 2200—2300 °С с использованием
тепла горения газов или вольтовой дуги. При этом получаются
нитрозные газы, содержащие не более 2,5% окислов азота,
поэтому для дальнейшей переработки их необходимо
концентрировать. С этой целью можно применять адсорбцию на
силикагеле с последующей десорбцией более
концентрированного газа.
Описанный вариант дугового метода может оказаться
достаточно экономичным для использования его в промышленных
условиях.
Одновременно с дуговым методом был разработан и освоен
цианамидный метод связывания азота, основанный на
способности карбида кальция СаС2 присоединять азот при температуре
около 1000 °С. Расход электроэнергии на получение 1 г
связанного азота в форме цианамида кальция CaCN2 значительно
меньше, чем при дуговом методе, и составляет 9—10 тыс. квт-ч.
233
Этим методом и в настоящее время получают цианамид
кальция (стр. 607), однако доля цианамидного метода з мировом
производстве связанного азота невелика и постепенно
уменьшается.
В 1913 г. в Германии на основе работ Габера и Боша
впервые удалось осуществить в промышленном масштабе синтез
аммиака из азота и водорода.
Этот метод оказался экономичнее дугового и цпанампдного
методов и потому быстро получил широкое распространение.
На 1 т связанного азота в виде синтетического аммиака
расходуется 4—5 тыс. кзт¦ ч электроэнергии (включая расход на
получение водорода).
Производство связанного азота продолжает интенсивно
развиваться и в настоящее время. Так, в 1940 г. объем мирового
производства связанного азота (без СССР) составлял
примерно 5 млн. т, в 1958 г. он возрос до 8,5 млн. i, а в 1960 г.
превысил 11 млн. г.
Производство аммиака в некоторых зарубежных
капиталистических странах в I960—1962 гг. характеризуется примерно
следующими данными (в тыс. г. 100%-пого МНз):
Ц>60 г. 1901 г.
США 4543 5300
ФРГ 1647 1600
Япония 1288 1500
Франция SSO 1125
Италия 879 1010
Англия 569 800
В СССР производство связанного азота развилось з
крупную отрасль химической промышленности за годы Советской
власти.
В 30-х гг. были построены и введены в эксплуатацию
мощные азотные предприятия, и к 1940 г. Советский Союз занял
одно из первых мест в мире по объему производства
связанного азота.
Развитие отечественной азотной промышленности з
последние годы характеризуется не только все возрастающим
увеличением мощностей, но и существенными качественными
изменениями. Значительно усовершенствовано технологическое г
аппаратурное оформление процессов, все больше внедряется их
автоматизация, непрерывно увеличивается использование в
качестве исходного сырья углеводородных газов, комплексная
переработка которых привела к значительному расширению
ассортимента продукции азотных предприятии на базе их
кооперирования с производствами основного органического синтеза.
Все это позволяет значительно снизить себестоимость
получаемой азотной продукции п уменьшить удельные капитальные
затраты.
234
2. ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА СИНТЕЗА
АММИАКА
Равновесие реакции. Синтез аммиака из азота и водорола
возможен только при больших давлениях и высокой
температуре в присутствии катализатора. Аммиак образуется по реакции
. , ЗН, + N, т * 2NH3 — 26,64 ккал
При протекании этой реакции на 1 моль образующегося
аммиака выделяется 13,32 ккал (при 500°С и 300 ат).
Реакция образования аммиака обратима, идет с выделением
тепла и значительным уменьшением объема (из трех объемов
водорода и одного объема азота получаются два объема
аммиака). Поэтому низкие температуры и высокое давление
благоприятствуют смещению равновесия реакции в правую
сторону, т. е. в сторону образования аммиака. При повышении тем,-
пературы и понижении давления равновесие реакции
сдвигается в левую сторону, происходит диссоциация аммиака на азот
и водород, вследствие чего содержание аммиака в равновесной
смеси уменьшается. С повышением давления при неизменной
температуре содержание аммиака, находящегося в равновесии
с азотом и водородом, увеличивается.
Количественное определение равновесия реакции синтеза
аммиака было проведено Габером и Нернстом.
Содержанне аммиака в равновесной газовой смеси в
зависимости от температуры и давления по расчетным данным для
чистой стехпометрниескон смеси (три объема водорода на один
объем азота) приведено в табл. 20.
Таблица 20
Содержание аммиака в равновесной газовой смеси
(при стехиэчетрическом отношении азота п подорода)
Температура, °С
400
450
500
550
600
Содержание NH3. объемн. % при различном давлении
100 am
25,12
16.43
10,61
6,82
4,52
ЗСО am
47,00
35,82
26,44
19,13
13,77
1000 am
79,82
69,69
57,47
41,16
31,43
1500 am
88,54
84.07
2U01' 1ЛШ
93,07
89, S3
3500 сил
97,73
97,18
Данные таблицы показывают, чго по мере повышения
температурь: г понижения давления содержание аммиака в газовой
смеси уменьшается. Так, при 400"С и 1000 ат в газовой смеси
содержится 79,82 объемы. % NH3, а при 600 °С и 100 ат —
только 4,52% NH3. В практических условиях выход аммиака ниже
равновесного на 25—30%.
Наибольшие выходы аммиака получаются при сравнительно-
низких температурах и высоких давлениях (рис. 94). Однако
при пониженных температурах в значительной степени
уменьшается скорость образования аммиака.
Под давлением порядка тысяч атмосфер равновесие
незначительно смещается с изменением давления (рис. 95) вследствие-
сжатия самих молекул газа в этих условиях.
к
1
——'
d
—
.—-—
—•
=-.—
zoo.
\
г—
5Jl?,
с |
mrc___
7ШГС —
-
,—
—J
41 -
s.%
го
и № Ш> W 40U 50В ШУ 7вв Ш Ш IBUL
Давление, а/л
Рит. 94. Зависимость равновесного "выхода
аммиака от давления при различных
температурах и стехиометрической исходной смеси.
Давление, an
Рис. 95. Равновесие
реакции синтеза aiVMnaica
при 400 °С, высоких
давлениях и
стехиометрической исходной смеси.
Скорость реакции. Образование аммиака из азота к водорода
¦протекает с измеримой скоростью только в присутствии тзер-
дых катализаторов. Таким образом, синтез аммиака является
гетерогенной каталитической реакцией.
Изучение кинетики процесса каталитического синтеза
аммиака имеет большое значение не только для уяснения
механизма этого процесса, но и для правильного расчета
высокопроизводительных колонн синтеза. Скорость синтеза аммиака
описывается уравнением Темкина и Пыжева, основанным на
представлении о том, что стадией процесса, определяющей
скорость образования аммиака, является химическая адсорбция
азота на катализаторе:
со =
!k_Y
PNH3/
I—i
где со — скорость реакции, равная разности скоростей
образования и разложения аммиака;
k\ и k2—константы скоростей реакций образования и
разложения аммиака;
236
PN , Рн„ и -Pnh3—парциальные давления газов;
а—коэффициент, изменяющийся от 0 до 1, в
зависимости от степени покрытия поверхности
катализатора адсорбированным азотом.
Для обычных промышленных катализаторов а=0,5. В
состоянии равновесия ы = 0. при этом отношение k\llk2 = K
является константой равновесия реакции синтеза аммиака.
Величины k\ и кг при высоких давлениях несколько
изменяются вследствие отклонения реальных газов в этих условиях
от законов идеальных газов и других причин.
Катализаторы
Основные требования к катализаторам, применяемым в
промышленности, состоят в следующем. Катализаторы должны
быть достаточно активными, т. е. обеспечивать необходимую
скорость реакции при возможно более низких температурах, и
обладать избирательным действием — ускорять основную
реакцию, не ускоряя нежелательных побочных реакций. Кроме того,
промышленные катализаторы должны быть дешевыми,
механически прочными и достаточно стойкими к действию ядов,
содержащихся з реакционной смеси (см. ниже).
Из большого количества предложенных и испытанных
катализаторов для синтеза аммиака наиболее пригодным
оказалось железо с добавкой небольшого количества активирующих
веществ (промотированное железо). В промышленности в
качестве катализатора применяют губчатое железо, промогиро-
ванное окисью калия и окисью алюминия. Такой катализатор
устойчив и обладает высокой активностью, сохраняющейся в
течение длительного времени. При введении в состав
катализатора третьего промотора (СаО) повышается термическая
стойкость катализатора. К преимуществам такого
промотированного катализатора относятся его дешевизна н доступность
исходных веществ.
Катализатор изготовляют из железной магнетитовой руды
или железа, предварительно окисленного в токе кислорода.
Окислы железа плавят в электрической печи вместе с
промоторами, полученную массу дробят и просевают. Зерненый
материал загружают в контактный аппарат — колонну синтеза,
где восстанавливают его азотоводородной смесью,
пропускаемой через аппарат в течение нескольких суток. Рентгеновские
исследования показывают, что частицы катализатора состоят
из кристаллитов, внутренняя часть которых представляет собой
чистое железо, промоторы содержатся в поверхностных слоях.
Поверхность промотированного катализатора примерно в
десять раз превышает поверхность чистого железного
катализатора и составляет 10—15 м2/г.
237
Испытан в промышленном масштабе метод восстановления
катализатора вне колонны синтеза. Это позволяет сократить
период пуска колонны и получить более активный катализатор.
Примерный состав одного из промышленных катализаторов
(до его восстановления): 31,4% FeO, 62,1 %Fe2O3, 1,67% А12О3,
1,1% К2О. 0,0006% Р, 0,001% S, 0,03% SiO2. Оптимальное
отношение закисиого и окнсиого железа в катализаторе близко к
0.5. максимум активности соответствует соединению РезО4.
Обычно размер зерен катализатора составляет 4—6 мм.
Процесс синтеза аммиака на катализаторе ведут при 450—
525СС, при более высокой температуре активность
катализатора снижается.
Наиболее медленной стадией синтеза аммиака на
железном и некоторых других катализаторах является химическая
адсорбция азота на поверхности катализатора. Некоторое
увеличение скорости реакции достигается при избытке азота в
азотоводородной смеси. Адсорбированный азот образует на
поверхности железного катализатора соединения с атомами
железа.
Повышение активности катализатора является важнейшим
способом увеличения производительности контактных
аппаратов. Дальнейшее усовершенствование методов получения
активных катализаторов позволит значительно увеличить
производительность колонн синтеза аммиака.
Катализаторные яды Катализатор при протекании реакции
синтеза аммиака химически почти не изменяется. Однако
практически в процессе работы активность катализатора
понижается вследствие его «старения», что вызывает необходимость
замены отработанного катализатора свежим. Явление «старения»
.можно объяснить главным образом действием ядов.
Скорость реакции образования аммиака из азота и водорода
с повышением температуры растет, в то время как содержание
аммиака в равновесной газовой смеси с повышением
температурь? уменьшается. Температура, при которой скорость
образования аммиака является оптимальной в условиях
промышленного процесса синтеза, зависит от свойств применяемого
катализатора и чистоты азотоводородной смеси. Чем,выше
активность катализатора, тем ниже температура, при которой
процесс может протекать с достаточной скоростью. Однако
понижение температуры процесса вызывает необходимость более
полной очистки азотоводородной смеси, так как чем больше
активность катализатора и ниже температура реакции, тем
сильнее действуют на катализатор вредные примеси (яды).
Вредными примесями являются: окись углерода, сероводород,
сероокись углерода, пары воды, кислород, пары масла и др.
При хорошей предварительной очистке азотоводородной
смеси и соблюдении оптимального технологического режима
238
смена катализатора может производиться через 1 — 1,о года л
реже, в зависимости от системы синтеза.
Железные катализаторы полностью отравляются
соединениями серы (H2S и др.), поэтому азотоводородная смесь
должна быть тщательно очищена от них. Кислород и его соединения
(окись углерода, вода) отравляют эти катализаторы обратимо,
г. е. после удаления их из катализатора его активность
восстанавливается. Допустимое содержание кислородсодержащих
веществ в азотоводородной смеси не должно превышать
примерно 40 с.1'3/.н3.
Для тонкой очистки азотоводородной смеси от примесей СО
н О2 наиболее широкое применение в промышленности получил
продуцирующий и непродуцирующий предкаталнз (стр. 200).
В колоннах продуцирующего предкаталнза, работающих
под давлением 320 ат, поддерживается более высокая
температура (около 550°С), чем в колоннах непродуцирующего пред-
катализа. В колоннах продуцирующего предкатализа
происходит не только очистка азотоводородной смеем от вредных
примесей, но и получается значительное количество аммиака.
Вследствие жестких условий работы продуцирующих
аппаратов (высокие температура и давление) продолжительность их
Работы относительно невелика. Целесообразнее вести процесс
предкатализа при более низких температурах и давлении.
Эффективна очистка азотоводородноп смеси от кпелорол:»
на никелевых катализаторах. При давлен1:» до 300 ат очистки
азотоводородной смеси от СО протекает с большой скоростью
уже при 100 °С. В случае совместного присутствия в азотсво-
дородной смеси СО и О2 очистка от них газа определяется
скоростью взаимодействия окиси углерода с водородом.
Объемная скорость и производительность катализатора.
Кроме температуры, давления и свойств катализатора, на процесс
образования аммиака большое влияние оказывает зтемя
нахождения газа в зоне
катализатора, которое определяется
объемной скоростью. Зависи-
мость выхода аммиака от
объемной скорости при
различных давлениях показана на
рис. 96.
Из диаграммы видно, что
содержание аммиака в -газе
при одной и тон же объемной
скорости возрастает с
повышением давления, а с
увеличением объемной скорости при
постоянном давлении —
понижается.
'Г 30
~бъв -
50 гс 7о ~с я"
¦Jcrvt, ~ыс м -/м -"
Рис. 96. Зависимость выхода ам;шпка
от объемной скорости при различных
давлениях.
239
^73000
При повышении объемной скорости растет
производительность катализатора (рис. 97), выражаемая в кг аммиака на
1 м3 катализаторной массы за 1 ч.
Возможность повышения производительности катализатора
путем увеличения объемной скорости ограничивается
уменьшением при этом содержания
аммиака в газовой смеси на
выходе из колонны синтеза,
что затрудняет выделение
аммиака, а также
возрастанием гидравлического
сопротивления системы.
При сопоставлении
приведенных данных можно сделать
следующий вывод: для
повышения производительности
катализатора нужно
увеличивать объемную скорость,
несмотря на уменьшение
содержания аммиака в
прореагировавшем газе, при условии, что
20000 ЬОООО 60О00 неиспользованная азотоводо-
Объемнавскорость, м3/м-v родная смесь направляется в
колонну синтеза.
В период возникновения
производства синтетического
аммиака работали с
небольшими объемными скоростями
E000—10 000); в настоящее время перешли к объемным
скоростям 25 000—70 000 ч~1 и выше.
В табл. 21 приведена зависимость содержания аммиака в
газовой смеси от температуры и объемной скорости при 300 ат.
Таблица 21
Зависимость содержания аммиака в газовой смеси от температуры
и объемной скорости
Рис. 97. Зависимость
производительности катализатора от объемной
скорости газа при различных давлениях.
Ооъемная
скорость
15 000
30 000
45 000
Содержание NH3
при 450 °С
19,6
14,6
12,7
при 475
21,6
17,7
15,2
в газовой смеси
°С
при 500
23,0
18,2
16,5
, объемн. %
°С
при 525 °С
19,3
16,7
15,7
Скорость образования аммиака с повышением температуры
растет, ко выход аммиака, достигнув максимума, далее
понижается (рис. 98) вследствие возрастания скорости обратной
240
реакции — разложения аммиака. Чем выше содержание
аммиака в газовой смеси, тем ниже температура, при которой
скорость синтеза аммиака максимальна. Поэтому в колоннах
синтеза стремятся снижать температуру в направлении потока
газа путем отвода тепла реакции. Наибольшая
производительность аппарата достигается в том случае, если по всей высоте
слоя катализатора температуру поддерживают в соответствии
с линией оптимальных температур, показанной на рис. 98.
Таким образом, процесс синтеза аммиака целесообразно начинать
при высокой температуре и по мере увеличения содержания
аммиака в газовой смеси понижать температуру.
Выбор объемной скорости в каждом отдельном случае
может быть сделан только на основании разностороннего анализа
процесса. Повышение производительности катализатора путем
увеличения объемной
скорости ограничивается
стремлением вести процесс ауто-
термично (без
дополнительного подвода тепла извне).
Кроме того, с увеличением
объемной скорости
возрастает гидравлическое
сопротивление системы и
уменьшается выход аммиака (см.
рис. 98 и табл. 21).
50
/A
у
f
л
€'
Y
у
30
Ь75 500 525
Температура, "С
Рис. 98. Зависимость выхода аммиака
от температуры ведения процесса
синтеза при различных объемных
скоростях и давлении 300 am.
1 го
гоо <too ooo soo /ооо
Давление, am
Рис. 99. Зависимость выхода
аммиака от давления при
прочих равных условиях.
С повышением давления выход аммиака растет (рис. 99).
Следовательно, для увеличения производительности контактного
аппарата синтез аммиака нужно проводить при высоких
давлениях и пониженных температурах. Оптимальную для определен-
16—284 R
241
ного катализатора температуру устанавливают по данным,
полученным при изучении скорости реакции. Необходимо
также работать с большими объемными скоростями и с возможно
более чистым газом.
3. ПРОМЫШЛЕННЫЙ СИНТЕЗ АММИАКА
Эксплуатируемые в промышленности системы синтеза
аммиака можно разделить в зависимости от применяемого
давления на две группы:
1. Системы, работающие при высоких давлениях порядка
450—1000 ат.
2. Системы, работающие при средних давлениях порядка
250—360 ат.
Установки, работавшие ранее при давлении до 100 ат,
впоследствии стали работать при давлении 160 ат.
В настоящее время в мировой азотной промышленности
большая часть синтетического аммиака производится в
системах среднего давления. В СССР наибольшее распространение
получили системы, работающие под давлением 300—320 ат.
Системы синтеза аммиака отличаются также схемами
циркуляции непрореагировавшей азотоводороднон смеси,
способами выделения из азотоводороднон смеси полученного аммиака,
параметрами технологического режима (температура,
объемные скорости, концентрации инертных газов в цикле),
конструкциями аппаратов, типами машин для циркуляции газов и др.
Ниже рассмотрены наиболее распространенные схемы
агрегатов синтеза аммиака.
СИНТЕЗ АММИАКА ПРИ СРЕДНЕМ ДАВЛЕНИИ
Схема агрегата синтеза аммиака под давлением 550—360 ат
приведена на рис. 100.
Азотоводородная смесь F4—66% Н2, 21—22% Х2, до 6%
СН4, до 5% Аг и 2—3% -\ТН3) поступает в колонну синтеза /, :s
которой частично превращается на катализаторе в аммиак.
Конвертированный газ на выходе из колонны синтеза
имеет следующий состав: 52—57% Н2, 17,5—19% №, 12—18% \Н3,
6,6% СН4. 5,5% Аг, Концентрация аммиака после колонны
синтеза зависит в основном от чистоты исходного газа,
применяемой объемной скорости, активности н длительности работы-
катализатора и конструкции катализаторной коробки з колонне
синтеза.
Из колонны синтеза газ при температуре 120—200 СС
поступает г. водяной конденсатор 2, где при охлаждении до 25—
35СС сжижается большая часть аммиака, полученного в
колонне синтеза. Сконденсировавшийся аммиак отделяется от
циркуляционного газа в сепараторе 3. При высоком содержание
242
инертных газов (СН4 и Аг) в свежем газе относительно
небольшая часть циркуляционного газа выводится из цикла
(постоянная продувка) для поддержания концентрации этих газов на
указанном выше уровне.
Затем газовая смесь для компенсации потерь давления в
цикле синтеза сжимается циркуляционным насосом 4 с даз-
ления 280—290 ат до 300—320 ат и подается в фильтр 5 для
очистки от масла, унесенного из циркуляционного насоса.
В фильтр поступает также свежая азотоводородная смесь.
Количество свежей азотоводородной смеси, добавляемой в цикл,
равно количеству газа, превратившегося в аммиак, плюс
количество отводимого при продувке газа (продувочный газ), а
также газа, растворившегося ь жидком аммиаке, и газа,
ушедшего из системы через неплотности.
Свежий
Жидкий
аммиак
Жидкий аммиак
Рис. 100. Агрегат синтеза аммиака при среднем давлении:
;—колонна синтеза; 2—водяной конденсатор: Л—сепаратор;
J—циркуляционный насос; 5—фильтр: 6—конденсационная колонна; /—
аммиачный испаритель.
Если при получении азотоводородной смеси окись и
двуокись углерода удалялись промывкой медноаммначным
раствором и раствором едкого натра, то в смеси содержатся
водяные пг'Т'Ы. а также остатки двуокиси углерода (миллионные
доли). В этом случае, при смешении свежей смеси с
циркуляционным газом, в котором содержатся пары аммиака,
образуются твердые частицы карбоната аммония:
СО, -;- Н.,0 -- 2NH3 > (ХН4JСО3
Вместе с маслом карбонат отделяется от газовой смеси на
насадке фильтра.
16*
243
'водородная
смесь
Из фильтра газовая смесь поступает в конденсационную
колонну 6, состоящую из теплообменника и сепаратора. В
теплообменнике холодным циркуляционным газом смесь
охлаждается до 5—15 °С.
Далее охлаждение циркуляционного газа и конденсация
остальной части аммиака, образовавшегося в колонне синтеза,
происходят в аммиачном
испарителе /,
охлаждаемом кипящим аммиаком
до 0—15°С. При этом в
конденсирующемся
жидком аммиаке
растворяются пары масла и воды,
находящиеся в газе.
Затем в сепараторе
конденсационной колонны
отделяется
сконденсировавшийся аммиак, а в
теплообменнике
циркуляционный газ охлаждает газг
поступающий из фильтра.
Далее циркуляционный
газ поступает в колонну
синтеза и замыкается
цикл синтеза.
ОСНОВНОЕ
ОБОРУДОВАНИЕ
Колонна синтеза.
Одна из применяемых часто
конструкций колонны
синтеза при среднем
давлении схематично показана
на рис. 101.
В нижней части колон-
Рис. 101. Колонна синтеза аммиака при НЫ синтеза расположен
среднем давлении: теплообменник 9, в зерх-
1,11—верхняя и нижняя головки; 2— соединительные НРн иягтм 1-ягяч1И"тпп
кольца; 5-трубки для термопар; 4-катализаторная НеИ Чс1С1И hd I d.IHJaIop-
коробга: S—двойные трубки; б—центральная труба ка- ная коробка 4 с трубка-
тализаторной коробки; 7—центральная распределите- г
льная коробка; 8— корпус колонны; 9—теплообмен- МИ О, ОТВОДЯЩИМИ ТсПЛО
ник; /0-центральная труба теплообменника. реЭКЦИИ. ТеПЛООбмеННИК
и катализаторная
коробка тщательно изолированы для поддержания устойчивого ауто-
термичного теплового режима и предохранения корпуса
колонны синтеза от перегрева. Азотоводородная смесь входит е
колонну сверху, спускается вниз по кольцевому зазору — про-
244
странству между корпусом и катализаторной короокол
колонны, обтекает корпус теплообменника и катализаторной
коробки и поступает в межтрубное пространство теплообменника.
При таком движении газа корпус колонны ке подвергается
действию высокой температуры.
В межтрубном пространстве теплообменника газ
подогревается до 350—370°С и затем через распределительную
коробку 7 поступает в двойные трубки 5, проходя вначале по
внутренним трубкам, а затем по кольцевым зазорам между
внутренними и внешними трубками. В этих трубках отводится тепло
реакции из катализаторной зоны, чтобы предотвратить перегрев и
снижение активности катализатора. Газовая смесь, подогретая
в трубках до температуры начала реакции D40—470сС),
поступает в центральную трубу 6, где размещен электроподогрева-
1-й UUKJI
Газ после сепаратора во 2-й иикм
2-й цикл
Газ на продувки usu
в 3-й uuf.f
свехшп газ
Рис. 102. Двухцикличная схема цеха синтеза аммиака:
/—агрегаты синтеза первого цикла; 2—агрегаты синтеза второго цикла.
тель, включаемый обычно лишь при разогреве и восстанозлении
катализатора. Далее газ проходит езерху вниз катализаторную
зону, где поддерживается температура порядка 500°С. По
выходе из катализаторной зоны газовая смесь охлаждается в
трубках теплообменника 9 до 120—200 ~С и выходит из колонны
синтеза снизу.
Для регулирования температуры в катализаторной зоне-
предусмотрена возможность подачи части газа
непосредственно в центральную трубу 10, минуя теплообменник (холодный
байпас).
На некоторых заводах свежая азотоводородная смесь
содержит повышенное количество инертных газов (метан н
аргон), которые, накапливаясь в цикле синтеза, снижают
парциальное давление азотоводороднон смеси и уменьшают
производительность агрегатов синтеза аммиака. Понизить
концентрацию инертных газов можно, непрерывно отводя из цикла
синтеза часть циркуляционного газа. Однако более
экономично применение двух- или трехциклнчной схемы (рис. 102).
245
Сущность двухцикличнон схемы заключается в следующем.
Большая часть агрегатов синтеза работает в первом цикле, а
остальные агрегаты — во втором цикле. Свежий газ подается
только в агрегаты синтеза первого цикла. Часть
циркуляционного газа из первого цикла непрерывно отводится в агрегаты
второго цикла. Поэтому в циркуляционном газе первого цикла
поддерживается относительно невысокая концентрация
инертных газоз (приблизительно 5%). В циркуляционном газе
второго цикла содержание инертных газов выше A5—20%).
Вследствие этого колонны синтеза первого цикла работают
с значительно более высокой производительностью, чем
колонны второго цикла. Количество отдуваемого циркуляционного
газа при двухцпкличной схеме относительно невелико, что
делает ее экономичной в связи с малыми потерями азотоводород-
ноп смеси высокого давления, имеющей высокую стоимость.
При трехцикличной схеме отдуваемый циркуляционный газ
из второго цикла поступает в агрегаты третьего цикла.
Циркуляционные компрессоры. На действующих азотных
заводах обычно применяются поршневые циркуляционные
компрессоры, при помощи которых давление циркуляционного
газа повышается на 20—30 ат, т. е. компенсируется потеря
давления в аппаратуре и трубопроводах цикла синтеза.
Недостатками поршневых компрессоров являются загрязнение газа
смазочным маслом, отравляющим катализатор, а также
значительные затраты труда па ремонт и большие потери азотоводо-
родной смеси из-за недостаточной плотности сальников
высокого давления.
В настоящее время разработана конструкция
центробежного циркуляционного компрессора высокого давления (ЦЦЮ.
Компрессор и электродвигатель этой машины (рис. 103)
размещены внутри сосуда высокого давления, поэтому
отсутствуют сальники высокого давления, которые обычно
недостаточно надежны. Собственно циркуляционный компрессор
является обычным многоколесным турбокомпрессором, но его
электродвигатель имеет специальную конструкцию,
рассчитанную на работу в среде азотоводородной смеси высокого
давления.
При применении центробежного циркуляционного
компрессора схема агрегата синтеза существенно упрощается
благодаря тому, что отпадает необходимость установки фильтра и
сепаратора после водяного холодильника, поскольку газ не
загрязняется маслом в таком компрессоре. Исключение из схемы
указанны:; аппаратов позволяет снизить па 25% расход металла
на изготовление аппаратуры агрегатов синтеза аммиака. ¦
Центробежный циркуляционный компрессор компенсирует
потерн давления в цикле синтеза, повышая давление
циркуляционного газа на 10—20 ат.
246
^ ___|^__Л1_^:
^-y^t^'^'^^yi^
Рис. 103. Центробежный циркуляционный компрессор:
/ ciarop злим родвш-j юля; 2- соидшшгслыкш муфт; 3 -ротор TyiNoKuMiipcccop;i; 4 -корпус компрессора; 5 ро'юр элиыроднш.пи.-ш;
б1-- 13150Д тггующиго электрического кабеля.
Инжектор. В некоторых системах среднего давления и
обычно в системах высокого давления циркуляция газа
осуществляется при помощи инжектора, т. е. струйного насоса без
движущихся рабочих частей, использующего кинетическую
энергию свежей азотоводородной смеси для перемещения
циркуляционного газа. Схема инжектора показана на рис. 104.
Для эффективной работы инжектора аппаратура агрегата
синтеза аммиака и коммуникации должны быть рассчитаны
так, чтобы потеря напора в агрегате не превышала 3—5 ат.
В этом случае давление в агрегате синтеза из-за низкого к.п.д.
инжектора будет на 30—50 ат ниже давления свежего газа.
Циркуляционный
газ
Рис. 104. Инжектор:
/—корпус; 2—сальник; 3—вентиль; 4—диффузор; 5—фильтр;
в—сопло.
Поэтому при использовании инжектора синтез под средним
давлением ведут при относительно невысоких объемных скоростях
(порядка 15 000 чгх).
СИНТЕЗ АММИАКА ПРИ ВЫСОКОМ ДАВЛЕНИИ
Давление в системах синтеза аммиака раньше
поддерживалось в пределах 750—1000 ат. При этом давлении за рубежом
в настоящее время работают лишь старые заводы.
Построенные в последние годы системы синтеза аммиака при высоком
давлении работают при 450—600 ат. Это связано с внедрением
экономичных способов тонкой очистки газа от контактных ядов
и повышением активности катализатора синтеза аммиака; при
этих условиях отпадает необходимость вести процесс под
давлением 750—1000 ат.
Схема современного агрегата синтеза аммиака под
высоким давлением весьма проста (рис. 105). В системах высокого
давления концентрация NH3 в конвертированном газе,
выходящем из колонны синтеза, относительно велика, а высокое
давление облегчает конденсацию аммиака. Поэтому в таких
248
системах, в отличие от систем среднего давления, конденсация
аммиака из газовой смеси производится лишь при охлаждении
водой. Свежая азотоводородная смесь поступает в инжектор /,
куда подается и циркуляционный газ из сепаратора 4.
Затем газовая смесь направляется з колонну синтеза 2, где при
температуре 500—530 ЭС и объемной скорости газа 30 000—
45 000 ч-'1 на катализаторе образуется аммиак.
Концентрация алшнака в газе после колонны синтеза составляет
20—25%. Конвертированный газ, выходящий из колонны, при
температуре 240—300 °С охлаждается до 30—35 СС в водяном
f—
I
]
>
Отработанноя беса
Рис. 105. Агрегат синтеза аммиака при высоком давлении:
1—инжектор; 2—колонна синтеза; ?—водяной конденсатор; 4—сепгргтор.
конденсаторе 5. Сконденсировавшийся аммиак отделяется от
газа в сепараторе 4 и поступает в сборник, а затем передается
на склад. Циркуляционный газ подается в инжектор /, в
котором за счет кинетической энергии свежего газа давление
циркуляционного газа повышается на величину, равную потере
давления газа в цикле синтеза.
Колонна синтеза высокого давления часто применяемой
системы Клода изображена на рис. 106.
Газовая смесь при 30—35 °С под давлением 500—600 ат
поступает в колонну синтеза через отверстие в нижней крышке
лорпуса / и поднимается вверх по кольцевому пространству
между корпусом колонны и катализаторной коробкой 7. Таким
образом корпус колонны предохраняется от перегрева.
Далее азотоводородная смесь проходит межтрубное
пространство в катализаторной коробке, нагреваясь путем отвода
тепла реакции из зоны катализатора, размещенного в трубках 4.
249
Нагретая азотоводородная смесь проходит электрический
подогреватель 10, расположенный под катализаторной коробкой и
включаемый при
разогреве колонны и
восстановлении катализатора.
Сюда же вводится
холодный газ для
регулирования температуры в зоне
., катализатора.
Подогретая азотоводород-
2 ная смесь, тщательно пе-
'3 ремешанная с холодным
¦4
5
в
газом, поступает снизу в
трубки 4, заполненные
катализатором. По мере
прохождения снизу вверх
через эти трубки
значительная часть азотоводородной
смеси превращается в
аммиак. Выделяющееся
тепло реакции отводится
газом, идущим в межтрубном
пространстве. В этом
пространстве и в катализа-
торных трубках
размещены перегородки 5 и 6,
изменяющие направление
потока газа для
увеличения его турбулентности и
улучшения отвода тепла из
реакционной зоны. Пройдя
трубки 4,
конвертированный газ выходит при
температуре около 350 °С из
колонны синтеза через
отверстие, имеющееся в
верхней крышке.
Рис. 106. Колонна синтеза аммиака при
высоком давлении:
/—корпус: 2—прокладки; 3. 8—трубные решетки;
4—кагализаторная трубка; 5—перегородка между
трубками; 6—перегородка в трубках; 7—катализа-
торная коробка; 9—смесительная камера; 10—
электроподогреватель; //—термопара; 12—затвор.
Нормальный режим работы описанной колонны синтеза:
Давление в начале кампании, am
Давление в конце кампании, am
Температура в зоне катализатора, °С
Содержание аммиака в конвертированном газе, °'а
Температура газа на выходе из колонны, °С . .
450
600
500—560
24—26
—350
В колоннах синтеза ранее применявшихся конструкции
тепло образования аммиака не использовалось. В последние годы
созданы колонны синтеза, работающие как при высоком, так
250
и при среднем давлении с использованием части тепла реа
на получение водяного пара.
На "рис. 107 показана одна из широко применяемых
конструкций такой кслонны. Газ поступает в колонну снизу в
межтрубное пространство теплообменнка 7, где нагревается
уходящим конвертированным газом. Затем газ поднимается в
верхнюю часть колонны и при температуре 420—430 "С поступает
з первый слой / катализатора. Здесь в результате выделения
тепла реакции температура газа повышается до 510—515 X.
тгг>г.евптцра газа, °С
100 500
конвертированный газ
Рис. 107. Колонна синтеза аммиака с
использованием тепла реакции на получение пара:
1—6—слон катализатора; 7— теплообменник;
5—экономайзер; 9—насос; 10—регулирующие винтили; //—котел-
утилизатор; 12—теплоотЕодящне змеевики.
Проходя через размещенные под верхним слоем катализатора
змеовикн 12. в которых циркулирует вода, газ охлаждается
примерно до 470 °С и при этой температуре поступает во
втором слои 2 катализатора, по выходе из которого газ снова ох-
лаждаетсгт. Таким образом, газ проходит через все слои ката-
251
.лизатора, поступает в трубки теплообменника 7, затем в трубки
экономайзера 8 и идет из колонны в водяной конденсатор.
Циркуляция воды б системе осуществляется прк помощи
насоса 9. Давление воды в змеевиках 12 равно давлению газа в
колонне синтеза аммиака. Вследствие выделения тепла
реакции в колонне вода нагревается в змеевиках до 370 °С. По
выходе из змеевиков вода поступает в межтрубное пространство
котла-утилизатора И, ..где охлаждается до 220 °С. При этом в
трубах котла образуется пар давлением 20 ат, поступающий
далее в заводскую сеть на использование. Из межтрубного
пространства котла-утилизатора вода подается насосом 9 в
змеевики 12 колонны синтеза и затем снова возвращается в
котел. Вода, циркулирующая в системе, должна быть очень
чистой, поэтому в данном случае применяется бидистиллят
(дважды дистиллированная вода).
Как видно на рис. 107, высота слоев катализатора и,
следовательно, его объем увеличиваются по направлению хода газа
(сверху вниз), так как скорость реакции снижается по мере
приближения газовой смеси к состоянию равновесия. Такая
конструкция катализаторной коробки позволяет поддерживать
оптимальные температуры в зоне катализа и соответственно
повышать производительность катализатора. При подобной
конструкции насадки колонны синтеза на 1 т вырабатываемого
аммиака получают примерно 0,8 т пара давлением 20 ат.
На рис. 107 (справа) показаны также изменения
температуры газовой смеси и концентрации NH3 в ней по мере
прохождения газа через различные части колонны синтеза.
Применяются также другие способы использования тепла
реакции для получения водяного пара. Например, газ из зоны
катализа поступает в котел-утилизатор, а затем в
теплообменник, или же часть газа, пройдя катализатор, направляется в
котел, а остальной газ отдает тепло в теплообменнике.
В некоторых системах синтеза при высоком давлении котел-
утилизатор устанавливают на линии конвертированного газа
после теплообменника, т. е. перед водяным конденсатором.
Корпуса колонн синтеза и других аппаратов высокого
давления раньше изготовляли только коваными. Центральная
часть металлического слитка после термической обработки в
горячем состоянии обсаживалась (выбивалась) или
высверливалась, а слиток подвергался ковке также в горячем
состоянии. В настоящее время, наряду с коваными корпусами
высокого давления, применяют сварные корпуса из тонкостенных
листов, образующих многослойные конструкции.
В некоторых странах корпуса сосудов высокого давления
изготовляют путем навивки на тонкостенную обечайку ряда
слоев профилированной ленты. Разрабатывается также
технология изготовления корпусов большого диаметра (внутренний
252
диаметр более 1000 мм) и длиной до 14—18 м путем сварки
отдельных кованых толстостенных царг.
Колонны синтеза аммиака при среднем давлении
изготовляются из низколегированных хромоникельмолибденовых
сталей C—4% Сг, 0,25—0,6% Мо, 0,15—0,35% С, до 0,5% №).
Насадки колонн синтеза выполняют из хромоникелевой
стали лзарки 1Х18Н9Т. Все остальные аппараты агрегатов синтеза
аммиака изготовляют из углеродистой стали.
Промышленные способы синтеза аммиака к настоящему
врекени достигли высокого технического уровня. Дальнейшее
усовершенствование процесса заключается в его
интенсификации, укрупнении оборудования и автоматизации.
Интенсификация агрегатов синтеза аммиака достигается
путем улучшения предварительной очистки газа от катализа-
торных ядов, улучшения теплового режима в зоне катализа,
увеличения объемной скорости, применения более активных
катализаторов. Укрупнение оборудования позволяет снизить
капмталовложения в строительство азотных заводов и
повысить производительность труда.
В настоящее время строятся агрегаты мощностью 100—
120 тыс. т аммиака в год, удельный расход металла для
агрегата синтеза годовой мощностью 100 тыс. г аммиака на 20%
ниже, чем для агрегата мощностью 50 тыс. г.
Ведутся работы в области повышения давлений в процессе
синтеза аммиака до 450—500 ат. Это облегчает создание агре-
гатоз повышенной мощности и позволит получать весь аммиак
в жидком виде, что особенно важно для производства
мочевины (стр. 571).
Расходные коэффициенты
На производство 1 г NH3 расходуют 2750—2900 мъ азото-
водородной смеси (считая на объем, приведенный к
нормальным условиям) в зависимости от содержания в смеси
инертных газов.
Расходные коэффициенты зависят от вида сырья и
технологии его переработки. Ниже приведены основные расходные
коэффициенты на 1 т аммиака при получении водорода из
природного газа методами каталитической парокислородной
конверсии (расход электроэнергии и воды дан с учетом
производства кислорода):
Конверсия Конверсия под
без давления давлением
Природный газ, м3 840 860
Электроэнергия, квпг-ч . . . 1500 1250
Вода, м3 370 300
Водяной пар, яг 1,2—1,5 0,6—1,0
Катализаторы, кг
конверсии метана ... 0,18 0,07
конверсии окиси углерода 0,61 0,40
синтеза аммиака .... 0,08 0,08
253
АВТОМАТИЗАЦИЯ ПРОЦЕССА СИНТЕЗА АММИАКА
На заводах, вводимых в настоящее время в эксплуатацию,
управление процессом синтеза аммиака предусматривается с
центрального пульта управления. Здесь размешаются ш.иты
контроля и управления, с которых осуществляется дпетапцион-
Рис. 108. Принципиальная схема автоматизации агрегата синтеза аммиака:
/—колонна синтеза (р =; 320 am)', 2—водяной конденсатор; 3—конденсационная колонна;
4—испаритель аммиака; 5—центробежный циркуляционный компрессор; 6—сборник кидкого
аммиака; а—регулирующие клапаны; РТ\, РТ&—регуляторы температуры газа;
РУ\, РУ%, РУъ—регуляторы уровня жидкого аммиака; РД—регулятор давленая.
ныи пуск агрегатов, контроль за выведением их на нормальный
режим, автоматическое управление регуляторами, а в случае
необходимости—переход с автоматического управления на
ручное дистанционное.
В систему автоматического управления входят:
1) автоматическое регулирование основных параметров,
обеспечивающее стабилизацию технологического режима
синтеза аммиака;
2) автоматический контроль за ходом процесса;
3) сигнализация об аварийных отклонениях основных
параметров технологического процесса и о состоянии агрегатов.
254
При аварийном отклонении от заданного режима
процесса синтеза зажигаются сигнальные лампы на световых
табло.
Поскольку в цех синтеза аммиака поступает азотоводород-
ная смесь, имеющая постоянные давление и состав, схемой ав-
томптизацпн агрегата синтеза (рис. 108) предусмотрена
стабилизация температурного режима в каталпзаторной зоне
колонны синтеза, концентрации аммиака в газе перед колонной
синтеза, уровня жидкого аммиака в конденсационной колонне,
давления п уровня жидкого аммиака в сборнике.
В каталпзаторной коробке в зоне наиболее высокой
температуры (так называемая горячая точка) пометена термопара.
Температурный режим в катализаторной зоне стабилизируется
путем поддержания постоянной оптимальной температуры в
«горячен точке». Это достигается воздействием регулятора РТи
получающего импульс от термопары, на клапан, регулирующий
количество газа, поступающего в катализаторную зону
колонны синтеза /, минуя теплообменник по холодному байпасу
(стр. 245). При этом температура газовой смеси на входе в ка-
талчзатор соответственно изменяется в результате
смешения газа, прошедшего через теплообменник, с холодным
газом. Объемная скорость процесса в данном случае не
меняется.
В начале кампании агрегата синтеза катализатор обладает
максимальной активностью, и по холодному байпасу проходит
значительная часть газа (до 25—30% от общего количества).
По мере понижения активности катализатора доля газа,
направляемого через холодный байпас, автоматически
уменьшается. При полном закрытии регулирующего клапана на
холодном байпасе автоматически включается регулятор
температуры РТ2, который воздействует на регулирующий клапан, по-
меЩсннып на байпасной линии центробежного
циркуляционного компрессора 5. В этом случае температура «горячей
точки» катализатора автоматически регулируется в результате
изменения объемной скорости процесса.
Заданная концентрация аммиака в циркуляционном газе
поддерживается путем автоматического регулирования уровня
аммиака регулятором РУ2 в испарителе 4 с соответствующей
коррекцией по температуре газа, выходящего из
конденсационной колонны 3. Постоянство концентрации аммиака в
циркуляционном газе, т. е. до колонны синтеза, облегчает стабилизацию
температурного режима процесса синтеза аммиака.
Автоматическое регулирование уровня жидкого аммиака ь
конденсационной колонне 3 (регулятор РУ\) и в сборнике
жидкого аммиака 6 (регулятор РУ3) и поддержание в нем
постоянного давления регулятором РД также обеспечивают
стабилизацию режима работы агрегата синтеза аммиака.
255
В настоящее время ведутся исследования и проектно-кснст-
рукторские работы в области оптимизации всего
технологического процесса производства аммиака при помощи
счетно-решающих машин.
Сорта, перевозка и хранение аммиака
Аммиак жидкий синтетический выпускают двух сортов,
которые должны соответствовать следующим требованиям:
1-й сорт 2-й сорт
Содержание NH3, %, не \;енее . . 99,9 99,6
Примеси, не более
влага, % 0,1 0,4
масло, мг/л 10 35
Железо, Мг/л 2 Не нормируется
Продукт 1-го сорта предназначен для холодильных машин,
аммиак 2-го сорта — для получения газообразного аммиака,
азотной кислоты, азотных удобрений и др. Для некоторых
потребителей выпускается аммиак особой чистоты.
Небольшие количества жидкого аммиака перевозят в
стальных баллонах, испытанных под давлением 60 ат. Рабочее
давление аммиака в баллоне 30 ат. Баллоны окрашены в желтый
цвет и снабжены черной надписью «Аммиак». Норма
наполнения — не более 1 кг аммиака на 1,76 л емкости. Емкость
баллонов различна, обычно применяют баллоны емкостью 30 и
35 л.
Станция наполнения баллонов жидким аммиаком и склад
наполненных баллонов размещаются, как правило, в
отдельном здании, располагаемом на окраине заводской площадки.
Для перевозки больших количеств жидкого аммиака служат
специальные железнодорожные цистерны низкого давления
(Рабс. = 4,5 ат) или высокого давления (не более 20 ат)
емкостью до 50 тыс. л. Цистерны снабжены предохранительными
клапанами и манометрами. Цистерны низкого давления
покрыты слоем теплоизоляции. Жидкий аммиак хранят в стальных
резервуарах вместимостью 30—50 т под избыточным
давлением, обычно не превышающим 16 ат. Хранилища снабжены
предохранительными клапанами, указателями уровня и
-обогревающими устройствами для испарения аммиака.
Хранилища, не рассчитанные на повышенное давление
(обычно вертикальные резервуары емкостью до 100 т),
должны быть снабжены теплоизоляцией, чтобы не происходило
излишнего испарения аммиака. Такие хранилища снабжают
также гидравлическими затворами, чтобы предотвратить
повышение давления сверх допустимых пределов.
256
Склады для хранения аммиака должны быть удалены от
производственных зданий на расстояние не менее 30—45 м.
Количество аммиака на складе не должно превышать 1000 т.
Перекачивание жидкого аммиака производится
специальными центробежными насосами. Для подачи газообразного
аммиака применяются аммиачные компрессоры.
ПРИГОТОВЛЕНИЕ АММИАЧНОЙ ВОДЫ
Аммиачную воду получают растворением аммиака в иоде:
NH3 -Ь Н2О -—» NH4OH
Эта реакция протекает с выделением тепла.
Обычно аммиачную воду приготовляют путем абсорбции
газообразного аммиака водой в вертикальных абсорберах,
снабженных тарелками или насадкой из колец Рашига. Можно
получать аммиачную воду также растворением жидкого аммиака
з воде или поглощением аммиака из танковых и продувочных
газов аммиачного производства.
Растворимость аммиака в воде тем больше, чем выше
давление и ниже температура. В связи с этим приготовление
аммиачной воды обычно ведут под давлением до 16 ат с
отводом тепла реакции в холодильниках, охлаждаемых водой. Б
абсорберах с тарельчатой насадкой холодильники размещаются
на тарелках и имеют форму змеевиков. В абсорберах с
насадкой из колец Рашига процесс абсорбции обычно идет в
несколько ступеней. Такие абсорберы снабжены одним или
несколькими выносными холодильниками для охлаждения
циркулирующей аммиачной воды.
Аммиачная вода имеет большое применение в различных
отраслях промышленности, особенно для получения жидких
удобрений. Стандартная аммиачная вода высшего и первого
сорта содержит не менее 25 вес. % NH3, аммиачная вода
второго сорта — 20 вес. % NH3.
Аммиачная вода хранится в горизонтальных и вертикальных
цилиндрических резервуарах. В больших складах
применяются вертикальные хранилища емкостью по 2000 .ад3 и более.
Аммиачную воду перевозят в железнодоржных цистернах.
Мелким потребителям аммиачную воду отгружают в
стеклянных бутылях, помещенных в корзинах или в деревянных
обрешетках.
Техника безопасности в производстве
а м м и а к а
Производство синтетического аммиака является огне- ir
взрывоопасным. Оборудование системы синтеза работает под
давлением порядка сотен атмосфер, при высоких и весьма
низких температурах.
П-2848 257
Аппараты, работающие под давлением, должны быть
рассчитаны, изготовлены, установлены и освидетельствованы
согласно нормам Госгортехнадзора, а аппаратура и
коммуникации высокого давления должны также удовлетворять
требованиям действующих норм и технических условий.
В производстве аммиака получают и перерабатывают
газовые смеси, содержащие водород, окись углерода, аммиак,
углеводороды и другие газы, образующие с воздухом
взрывоопасные смеси, а также являющиеся токсическими
веществами (табл. 22).
Таблица 22
Взрывчатые и токсические свойства некоторых газов
Газы
Водород . .
Азот ....
Окись
углерода . . .
Двуокись
углерода . .
Метан . . .
Аммиак . .
Сероводород
Температура
воспламенения, °С
510
.610
645
780
290
Пределы взры-
ваемостн смеси
с воздухом
объемн. %
нижний
4,1
12,5
4,9
15,5
4,3
верхний
74,2
75,0
15,4
27,0
45,5
Действие на организм
Физиологически инертный газ
При большой концентрации уменьшает
парциальное давление кислорода в
легких, вызывая удушье
Отравляющее действие вследствие
образования (при вдыхании) соединений с
гемоглобином крови
Наркотическое и удушающее дейстие,
вытесняет кислород из зоны дыхания
В больших концентрациях наркотическое
действие
Острое раздражение слизистых оболочек,
слезотечение, удушье
Жидкий аммиак производит сильные
ожоги кожи
Сильный яд, при высоких концентрациях
вызывает потерю сознания и смерть
Газовая смесь может взорваться при концентрациях
горючего газа пли пара, находящихся между нижним и верхним
пределами взрываемости (стр. 232), в случае соприкосновения с
открытым огнем, при возникновении электрической искры,
искры от удара или трения, а также при нагревании газовой
смеси (или ее части) до определенной температуры,
называемой температурой самовоспламенения.
258
При нарушениях технологического режима в производстве
аммиака или при авариях возможно значительное выделение
газов, в результате этого могут возникнуть пожары, взрывы,
произойти химические и термические ожоги, отравления и
обмораживания обслуживающего персонала. Поэтому при
эксплуатации оборудования необходимо тщательно выполнять все ра-
оочие инструкции, а также инструкции по технике
безопасности, промышленной санитарии и противопожарные
инструкции и обеспечизагь полную герметичность оборудования и
коммуникаций.
При получении азота и кислорода разделением воздуха
(стр. 209) могут создаваться взрывоопасные условия в случае
попадания ацетилена или других непредельных углеводородов в
жидкий и сжатый кислород. Соприкосновение кислорода или
ьэздуха, обогащенного кислородом, с необезжиренной
аппаратурой и коммуникациями, с маслом и некоторыми
органическими веществами также может привести к пожару и взрыву.
При получении водорода разделением коксового газа
(стр. 223 ел.) взрывоопасные условия могут создаться, если
окислы азота, обычно присутствующие в коксовом газе
(миллионные доли N0), проникают в аппаратуру глубокого холода, где
образуют со смолами и углеводородами ззрывчатые нитросое-
динения. Поэтому количество окислов азота в коксовом газе за
время пробега агрегата разделения этого газа (время от пуска
до остановки агрегата на размораживание) строго
регламентировано. Кроме того, регламентируются также количество 'л
максимальная концентрация окислов азота в газе,
поступающем на промывку жидким азотом.
Вредные и горючие газы (содержащие NH3, CO, H2S. H2
и др.), выделяющиеся в атмосферу из предохранительных
устройств и при периодических продувках аппаратуры, должны
отводится на высоту не менее 3 м над коньком крыши по
специальным продувочным трубопроводам, снабженным огнепре-
градителями.
Все движущиеся и вращающиеся части машин и аппаратов
должны быть оборудованы надежными ограждениями.
Помещения, в которых могут выделиться газы, образующие
взрывчатые смеси с воздухом, являются взрывоопасными.
В таких помещениях запрещается разведение открытого огня
и производство сварочных работ. Сварка может быть
разрешена лишь при соблюдении определенных правил.
Во взрывоопасных помещениях электрооборудование
должно устанавливаться во взрывобезопасном или взрывозащишен-
ном исполнении.
Все здания в производстве синтетического аммиака должны
быть снабжены механической, естественной или смешанной
вентиляцией, обеспечивающей требуемые по кормам техники
17* 259
безопасности и промышленной санитарии кратности
воздухообмена.
Концентрация токсических веществ в воздухе рабочей
зоны производственных помещений не должна превышать
следующих пределов (в мг/м3):
Аммиак ...... 20 Сероуглерод .... 10
Сероводород .... 10 Бензол 20
Окись углерода ... 20
Для устранения вредных воздействий токсических веществ
на эксплуатационный персонал рекомендуется при
строительстве новых цехов синтеза аммиака всю аппаратуру и основные
коммуникации размещать вне зданий, а в помещениях
располагать лишь машины и пульты управления.
Эксплуатационный персонал должен быть снабжен
исправными противогазами и обучен методам пользования
индивидуальными средствами защиты от действия токсических
веществ, веществ, вызывающих ожоги и другие поражения.
Подтягивать болты фланцевых соединений трубопроводов, а
также проводить ремонтные работы на оборудовании,
находящемся под давлением, не допускается. На всех
взрывоопасных участках, а также при ремонте и очистке аппаратов и
емкостей внутри них следует работать только инструментом
из металлов или сплавов, которые при ударах не дают искры
(медь, бронза и др-)-
Во избежание несчастных случаев при ремонте
трубопроводов, по которым транспортируется жидкий аммиак, необходимо
опорожнить их от аммиака, продуть и вести работы, применяя
противогазы и резиновые перчатки.
Если в результате истечения через неплотности газ
воспламенится, то, кроме спуска давления в системе, необходимо ту-
шпть пламя азотом из азотного трубопровода пли из баллонов.
Глава VII
АЗОТНАЯ КИСЛОТА
/. ОБЩИЕ СВЕДЕНИЯ
Свойства азотной кислоты. Чистая азотная кислота HNO3
при обыкновенной температуре представляет собой бесцветную
жидкость, замерзающую при —41 °С с образованием
белоснежных кристаллов. Водные растворы азотной кислоты в зависи-
70
во
МО
« 20 30 40 50 SO
Содержание НХО;], %
Рис. 109. Диаграмма кристаллизации системы HNO3—Н2О.
мостл от концентрации кристаллизуются при различных
температурах (рис. 109). Самую низкую температуру
кристаллизации I—66,3 СС) имеет 89.95%-ная азотная кислота, 70,5%-ная
кислота кристаллизуется при —42°С, а 32,7%-ная кислота —
при —43 СС.
Температура кипения водных растворов азотной кислоты
также изменяется в зависимости от концентрации.
Максимальную температуру кипения A20,5 °С) имеет раствор,
содержаний 68,4% HNO3. Такая кислота представляет собой азео-
тропную смесь (стр. 61). Более разбавленные и более
концентрированные растворы азотной кислоты кипят при пониженной
261
температуре. Так, 50%-ная азотная кислота кипит при 117 С,
90%-ная при 102сС и т. д. (стр. 294).
Теплота образования HNO3 (в жидкой фазе) из простых
веществ ('/г^-Н/гОгЧ- 7гН2) равна 41,7 ккал/моль. Теплота
растворения HNO3 в воде при бесконечном разбавлении
7,5 ккал/моль, теплота плавления 9,5 кал/г, теплота испарения
115 кал/г.
Применение азотной кислоты. Азотную кислоту применяют
в огромных количествах для производства минеральных
удобрений, красителей, взрывчатых веществ, для получения адипи-
новой кислоты (исходный мономер в синтезе смолы найлон)
и других продуктов. Много азотной кислоты расходуется в
производстве серной кислоты по нитрозному методу (стр. 130 ел.).
В качестве удобрений, получаемых на основе азотной
кислоты, широкое применение имеет аммиачная селитра, в
незначительном количестве потребляются также натриевая и
кальциевая селитра.
Азотная кислота применяется, как и серная кислота, для
переработки фосфоритов и апатита в удобрения (стр. 524 ел.,
591 ел.). Азотная киелста широко используется, кроме того, в
производстве нитролаков, кинопленки и др.
Из минеральных кислот азотная кислота по масштабу
производства и потребления уступает только серной кислоте.
В связи с быстрым ростом производства минеральных
удобрений в СССР увеличиваются и мощности по производству
азотной кислоты, необходимой для получения азотных и
комплексных удобрений.
Сорта азотной кислоты
Промышленность выпускает разбавленную (слабую)
азотную кислоту трех сортов и концентрированную (крепкую)
азотную кислоту двух сортов.
Разбавленная азотная кислота должна удовлетворять
следующим требованиям:
Сорта
] -й 2-й 3-й
Содержание HNO3, вес. %, не менее 55 47 45
Твердый (прокаленный) остаток, %, не более. ... 0,05 0,1 0,1
Разбавленная азотная кислота используется
преимущественно в производстве азотнокислых солей, применяемых
главным образом в качестве удобрений (стр. 555 ел.).
Концентрированная азотная кислота должна иметь
следующие показатели:
Сорта
1-й 2-й
Содержание HNO3, вес. %, не менее 1- 98 97
Содержание окислов азота (в пересчете на N2O4), %, не более ' 0,3 0,4
Содержание HaSO4, %, не более '. 0,08 0,12
Твердый остаток, %, не более 0,03 0,05
262
Концентрированную азотную кислоту применяют главным
образом в виде смесей с серной кислотой (так называемые
нитрационные смеси) для нитрования органических веществ.
Путем нитрования их получают разнообразные нитросоедине-
ния. применяемые в качестве полупродуктов при синтезе
красителей и многих других ценных веществ. Некоторые нитро-
соединения применяются в качестве взрывчатых веществ.
Продукты нитрования целлюлозы используются для получения
кинопленки, порохов, целлулоида и др.
Способы производства азотной кислоты. До 20-х годов
настоящего столетия азотную кислоту получали главным образом
из нитрата натрия и серной кислоты по реакции
NaNO3 -f H3SO4 = HNO3 + NaHSO4
Этот способ давно не применяется.
Перечисленные выше цианамидный и дуговой способы
получения аммиака (стр. 233) не могли обеспечить широкого
развития производства синтетической азотной кислоты. К тому же
стоимость аммиака, полученного этими способами, значительно
превышала стоимость синтетического аммиака.
Развитие промышленных способов синтеза аммиака,
позволившее значительно снизить его стоимость, дало возможность
создать мощную азотную промышленность.
В настоящее время практически вся вырабатываемая
азотная кислота получается окислением газообразного
синтетического аммиака.
Процесс окисления аммиака в азотную кислоту изучался
еще до осуществления промышленного способа синтеза
аммиака. Первоначально в качестве сырья использовался аммиак,
извлекаемый из коксового газа.
В России еще в 1915 г. инженер И. И. Андреев впервые
поставил вопрос «о получении азотной кислоты из аммиачных вод
коксовых печей». Андреев подробно изучил реакцию окисления
аммиака на изготовленных под его руководством
катализаторах (сплавы платины с иридием), предложил конструкцию
контактных аппаратов для окисления аммиака и построил
опытную промышленную установку для получения азотной кислоты.
Положительные результаты, полученные на этой установке,
были положены им в основу проекта первого отечественного
завода для производства азотной кислоты окислением аммиака.
Строительство контактного азотнокислотного завода в Юзовкс
(ныне Донецк) было начато в 1916 г., а в июле 1917 г. на
заводе уже работали 14 контактных аппаратов.
В настоящее время в производство азотной кислоты внесен
ряд существенных усовершенствований: улучшены методы
очистки аммиачно-воздушной смеси, внедрены новые катализа-
263
торы для окисления аммиака, усовершенствованы методы
концентрирования слабой азотной кислоты, разработаны и
реализованы в промышленности методы получения двуокиси азота
и концентрированной азотной кислоты прямым синтезом
и др.
2. ПРОИЗВОДСТВО АЗОТНОЙ КИСЛОТЫ ОКИСЛЕНИЕМ
АММИАКА
Азотная кислота получается из аммиака в две стадии.
Первой стадией является окисление аммиака кислородом в
присутствии катализатора. При этом протекают две реакции:
основная, приводящая к образованию окиси азота
4NH3 + 5О2 = 4NO + 6Н2О -i- 216,7 ккал
и нежелательная реакция окисления NH3 до элементарного
азота:
4NH3 -г ЗО2 = 2N2 -f 6НаО ^ 303,1 ккал
В отсутствие катализатора окись азота из амлшака
практически не образуется; продуктами реакции являются
элементарный азот и пары воды. При окислении аммиака кислородом
воздуха в заводских установках 96—'98% NH3 превращается в
окись азота, остальные 4—2% теряются, образуя молекулярный
азот.
Вторая стадия процесса заключается в превращении
полученной окиси азота (так называемые нитрозные газы) в
азотную кислоту. Окись азота окисляется в высшие окислы азота,
которые при взаимодействии с водой образуют азотную кислоту
и окись азота. Упрощенно эти процессы можно представить
следующими уравнениями:
2NO -f O2 = 2NOa + 26,92 ккал
3NO2(газ)-ЬН2О(жидк.) = 2HNO3(жидк.) + Ж)(газ) + 32,53 ккал
Как видно из последнего уравнения, при абсорбции
высших окислов азота водой вновь выделяется окись азота,
которую далее опять окисляют в двуокись азота для превращения
в азотную кислоту. Таким образом, в абсорбционной
аппаратуре чередуются реакция окисления окиси азота и реакция
взаимодействия полученной двуокиси азота с водой до тех пор,
пока концентрация окиси азота в нитрозных газах не
понизится настолько, что дальнейшее ее использование становится
экономически нецелесообразным. При этом получается
разбавленная 50%-ная и 60%-ная азотная кислота.
Первоначально процесс получения азотной кислоты вели при
атмосферном давлении. Такие установки широко распространет
ны и сейчас.
264
Установки, в которых применяется повышенное давление,
можно разделить на два типа. В установках первого типа обе
стадии процесса — окисление аммиака и окисление окиси азота
с последующей абсорбцией высших окислов азота — ведут под
давление',; 6—8ат. Ко второму типу относятся комбинированные
установки, в которых окисление аммиака проводят при
атмосферном давлении, а вторую стадию процесса — окисление N0
и поглощение высших окислов азота — при повышенном
давлении.
Концентрированную азотную кислоту получают
несколькими способами. Более старый способ заключается в перегонке
разбавленной азотной кислоты в смеси с серной кислотой
(купоросным маслом). В настоящее время построены и работают
промышленные установки для получения концентрированной
азотной кислоты взаимодействием жидкой двуокиси азота с
водой (практически пользуются слабой азотной кислотой) в
присутствии кислорода. Процесс протекает при давлении 50 ат и
температуре около 70 °С по суммарному уравнению
2N2O4 + 2Н2О + О2 = 4HNO3 4-14,2 ккал
По этому способу, называемому прямым синтезом, сразу
получается 98%-ная азотная кислота.
КОНТАКТНОЕ ОКИСЛЕНИЕ АММИАКА
Реакции окисления аммиака. В настоящее время азотная
кислота получается в промышленности почти исключительно
окислением синтетического аммиака воздухом, к которому
иногда добавляют чистый кислород. Вместо воздуха применяют
также смеси кислорода с водяным паром.
В отсутствие катализаторов аммиак практически
окисляется всегда до элементарного азота. При окслении аммиака
кислородом в присутствии катализаторов в зависимости от условий
могут получаться окись азота, закись азота или элементарный
азот:
4NH3 + 5О2 = 4NO + 6Н2О + 216,7 ккал A)
4NH3 + 4О2 = 2N2O + 6Н2О + 263,9 ккал B)
4NH3 + ЗО2 = 2N2 + 6Н2О + 303,1 ккал C)
Таким образом, окисление аммиака является сложным
процессом.
Степень превращения аммиака в окись азота зависит от
состава катализатора, концентрации кислорода в
воздушно-аммиачной смеси, продолжительности соприкосновения газа с
катализатором, температуры контактирования и содержания в
газе вредных примесей (H2S, PH3 и др.), являющихся ядами
для катализатора.
265
Реакция окисления аммиака кислородом относится к
классу гетерогенно-газовых каталитических реакций. Применяя
различные катализаторы и регулируя температуру и другие
условия процесса, можно получить в качестве основного продукта
реакции окись азота или закись азота. При 250—300 °С в
присутствии марганцевого катализатора 70% аммиака окисляется
до N2O. На действующих азотнокислотных заводах выход
окиси азота на платиновом катализаторе (с добавками Rh, Pd)r
достигает 96—98%, остальной аммиак окисляется до
элементарного азота и, следовательно, безвозвратно теряется.
Равновесие реакции. Окисление аммиака при атмосферном
давлении на платиновом катализаторе протекает при высоких
температурах G50—850°С). В этих условиях образование
закиси азота по реакции B) не происходит. Реакции же A) и-C)г
как показывают термодинамические расчеты изменения
свободной энергии реакций, протекают практически полностью слева
направо, т. е. необратимы. Вычисленные для этих реакций
константы равновесия при 750 °С имеют ничтожно малые
значения:
Кг = S^ e 1(Г*° I*™ Pea™»1 (!I
= 10~7* [для реакции C)]
Следовательно, превращение аммиака в конечные продукты
при 750 °С достигает почти 100%.
Суммарный результат реакций окисления аммиака, как "ir
всякого сложного, практически необратимого процесса,
зависит в основном от соотношения скоростей образования
каждого из продуктов при данных условиях. Поэтому состав
конечных продуктов будет определяться в первую очередь
избирательностью действия катализатора.
Для успешного осуществления процесса надо подобрать
такой катализатор, который при оптимальных условиях
окисления аммиака (состав газовой смеси, температура, время
контактирования) в большей степени ускоряет основную
реакцию A) образования окиси азота, чем побочную реакцию C):.
в результате которой образуется элементарный азот.
Катализаторы
Для производственных условий требуется дешевый
катализатор, стойкий к ядовитым примесям, находящимся в контакт-и-
руемых газах. Катализатор должен обеспечивать высокий
выход NO, сохранять длительное время активность и легко 'ее
восстанавливать. Таким требованиям в большей или меньшей
266
степени отвечают платина и ее сплавы с металлами
платиновой группы (платиновые катализаторы), а также окислы
кобальта,' железа, марганца и некоторых других металлов
(неплатиновые катализаторы). В промышленности применяют
катализаторы обоих видов.
Неплатиновые катализаторы дешевле, но выход окиси
азота на них ниже, чем на платиновых. Существенным
недостатком неплатиновых катализаторов является также их
сравнительная недолговечность и относительно быстрое понижение
актизности, особенно при наличии вредных примесей в
газовой- смеси, поступающей на контактирование. Поэтому в
промышленности до настоящего времени в основном
применяются платиновые катализаторы» несмотря на их высокую
стоимость.
Платиновые катализаторы, преимущественно сплавы
платины с родием E—10% Rh), применяются обычно в виде сеток
с числом отверстий 1024 C2X32) на 1 см2. Сетки
изготовляются из нитей диаметром от 0,06 до 0,09 мм. Применение такого
сетчатого катализатора позволяет достигать более высоких вы-
ходоз окиси азота, чем на мелкодисперсном платиновом
катализаторе. С увеличением числа сеток выход окиси азота
повышается. При окислении аммиака под атмосферным давлением
применяют обычно пакеты из трех-четырех платино-родиевых
сеток, свободно наложенных одна на другую. На некоторых
установках диаметр сеток составляет 3 м.
Максимальный выход, получаемый на таком катализаторе
при наиболее благоприятных условиях ведения процесса,
достигает 98%. Остальной аммиак окисляется до азота.
На установках для контактного окисления аммиака при
давлении 6—8 ат применяют пакеты из 16—18
платино-родиевых сеток; диаметр сеток может достигать 1 м.
: Скорость окисления аммиака на платиновых катализаторах
чрезвычайно велика: для протекания ее требуется всего
1—2 • 10~4 сек. Это одна из самых быстрых гетерогенно-газовых
каталитических реакций.
Разрушение катализатора. В процессе работы
катализатор подвергается механическим разрушениям; мельчайшие
частицы сплава уносятся током газовой смеси. Ллатино-родие-
вые катализаторы обладают большей механической прочностью,
чем чистая платина. С повышением температуры и
увеличением давления потери платины возрастают. Для уменьшения
потерь катализатора платиновые сетки периодически (после
12—16 месяцев работы) отправляют на переплавку, кроме
того, часть платиновой пыли, уносимой током газа, улавливают в
специальных фильтрах.
Потерн платины на установках, работающих при атмосфер-
гном давлении и температуре 750—850 СС. относительно невели-
267
ки и составляют в среднем 0,05 г на 1 т HNO3. На установках,
работающих при давлении 8 ат и температуре около 900 °С,
потери платины составляют 0,15—0,20 г на 1 г НЫОз-
Отравление катализатора. Производительность
катализатора (стр. 107) зависит от содержания вредных
примесей (ядов) в газовой смеси. Платиновые катализаторы весьма
чувствительны к действию ядов. Фосфористый водород
отравляет катализатор необратимо при содержании РНз в газовой
смеси даже в ничтожных количествах ~0,00001 %). Ядамк для
платиновых катализаторов являются также сероводород,
смазочные масла, мелкие частицы окислов железа (ржавчина)
и др. Пыль, присутствующая в газовой смеси, тоже понижает
активность катализатора.
Высокая степень окисления аммиака в окись азота
достигается только при тщательнейшей очистке аммиачо-воздушной
смеси от указанных вредных примесей. Окончательная очхстка
аммиачно-воздушнои смеси от пыли непосредственно перед
контактным аппаратом или в самом контактном аппарате (до
поступления газа на катализатор) производится путем
пропускания газа через керамические (поролитовые) трубки. Пыль,
оседающая на стенках трубок, периодически удаляется.
Условия контактирования
Скорость реакции и выход NO на платиновых
катализаторах. Состав исходной газовой смеси. Наибольшее
влияние на скорость процесса и выход
окиси азота оказывает состав аммиачно-
воздушной смеси, т. е. соотношение
количеств кислорода и аммиака в газе,
поступающем на окисление. По
уравнению A), приведенному на стр. 265, для
окисления аммиака до окиси азота
требуется 1,25 моль кислорода на 1 молы
аммиака. Однако при таком
соотношении исходных газов выход оыпси азота
очень мал (рис. 110). Чтобы ускорить
реакцию, ведущую к образованию окиси
ГОО
80
I
А
О 0,5 !,0 15 2,0
Молярное отношение
О
азота, а также увеличить ее выход, не-
'г-1Ч1Пз обходимо повысить концентрацию кис-
Рис. ПО. Зависимость города в газовой смеси. Наибольшая
выхода окиси азота от скорость образования окиси азота и
отношения концентраций максимальный ее выход достигаются при
кислорода и аммиака. отношении молярных концентраций
кислорода и аммиака, близком к 1,7—1,9,
что соответствует содержанию 10—11 объемн. % аммиака в.
аммиачно-воздушнои смеси.
268
Дальнейшее увеличение избытка кислорода не оказывает
заметного влияния на выход окиси азота.
Допустимое содержание аммиака в исходной газовой смеси
определяется также пределами взрываемости смесей аммиака
с кислородом или воздухом (стр. 258).
Производительность контактного аппарата повышается с
увеличением процентного содержания аммиака в газовой смеси.
Поэтому обычно поддерживают возможно более высокое
содержание аммиака A0—11 объемн. %) в газе при минимально
допустимом избытке кислорода, что примерно соответствует
оптимальному составу газовой смеси.
Оптимальная температура. Скорость реакций
окисления аммиака (стр. 265) растет с увеличением температуры;
максимальный же выход окиси
азота достигается лишь при
определенной температуре
(рис. 111). При этой
температуре скорость реакции,
приводящей к образованию окиси
азота, во много раз
превышает скорость побочных
реакций. Для процесса окисления
аммиака. проводимого при
W0
S? 80
Vе
<§. 20
О
/
/
/
1
1
300 400 500 600 700 SOO
Температура, °С
/ООО
атмосферном давлении на пла-
тино-родиевом катализаторе,
оптимальная температура на- Рис. 111. Зависимость выхода окиси
ходнтся в пределах 750—850 °С азота от температуры.
(ом. рис. 111).
Температура газовой смеси в зоне катализатора зависит от
содержания аммиака в газе, количества аммиака, окисляемого
в единицу времени (так называемая напряженность
катализатора), от тепловых потерь контактного аппарата и температуры
газа, поступающего на контактирование. При атмосферном
давлении и прочих оптимальных условиях на 1 м2 активной
поверхности* платинового катализатора удается окислить около
600 кг аммиака в сутки.
Давление. Повышенное давление отрицательно
сказывается на степени конверсии аммиака в окись азота и потерях
катализатора. Так, на установках, работающих под давлением
8—9 ат, конверсия на 2—3% ниже, а потери платины в 3 раза
больше, чем на установках, работающих под атмосферным
давлением.
Производительность катализатора с повышением давления
также увеличивается, поскольку объем газа обратно пропорци-
* Под активной поверхностью катализаторной сетки понимают общую
поверхность продольных и поперечных проволочек, приходящуюся не
единицу площади (или веса) сетки.
269
С
too
80
<§
\
\
1
\
О 2,5 5,0 7,5 W.0 /2J 15,0
нан/ран/пир
т-/04, се*
онален давлению, а оптимальное время контактирования* с
повышением давления увеличивается незначительно.
Время контактирования и линейная скорость газовой смеси.
увеличением времени контактирования при данной высоте
слоя катализатора, т. е. с
уменьшением линейной скорости газа,
выход окиси азота сначала
повышается, достигая
максимального значения, а при дальнейшем
увеличении времени
контактирования уменьшается (рис. 112).
Оптимальное время
контактирования колеблется, в зависимости
от других условий ведения
процесса, в пределах 1—2-10~4 сек.
На неплатиновых катализаторах
Рис. 112. Зависимость выхода оки- реакция протекает примерно в
си азота от времени контактиро- ЮО Раз медленнее.
вания. При неизменном времени
контактирования и прочих равных
условиях выход окиси азота
повышается с увеличением линейной скорости газа, т. е. с
увеличением числа сеток катализатора.
Оптимальные условия окисления аммиака при атмосферном
давлении. Оптимальными условиями окисления аммиака при
атмосферном давлении являются следующие: температура
750—850 °С, катализатор — платино-родиевые сетки
(три-четыре штуки) с 1024 отв/см2; отношение молярных концентраций
кислорода и аммиака 1,7—1,9 (т. е. 10—11 объемн. % HN3 в
аммначно-воздушной смеси); полное отсутствие вредных
примесей (соединений серы, фосфора, пыли и др.) в аммиачно-воз-
душной смеси; время контактирования 1—2-10~4 сек.
ПЕРЕРАБОТКА НИТРОЗНЫХ ГАЗОВ
В РАЗБАВЛЕННУЮ АЗОТНУЮ КИСЛОТУ
Нитрозные газы, полученные при окислении аммиака
кислородом воздуха, содержат около 10—11 объемн. % окиси
азота, кислород, азот и водяные пары.
Для получения азотной кислоты требуется окислить окись
азота NO до двуокиси азота NO2, которая при взаимодействии
с водой образует азотную кислоту. Окись азота с водой не
реагирует.
* Оптимальным временем контактирования называется отрезок
времени, в течение которого при соприкосновении газовой смеси с
катализатором достигается максимальный выход окиси азота.
270
Равновесие реакции окисления N0 в N02. Реакция окисления
окиси азота
2N0 -f 02 ~ * 2NO2 + 26,92 ккал D)
обратима, протекает с уменьшением объема и выделением
тепла. Следовательно, понижение температуры нитрозных газов и
повышение давления будут сдвигать равновесие реакции в
правую сторону, т. е. в сторону образования двуокиси азота.
Константа равновесия реакции Kv— N02 ¦P^- очень силь-
/'NOa
но зависит от температуры:
Температура, °С . . . 20 100 200 500 800 900
^(вычисленная). . 8,07-10"i5 5,5-Ю-9 1,35-10-» 1,18 177 662
Из приведенных данных видно, что, например, при 800—
900°С (Кр велика) равновесие реакции сдвинуто в сторону
образования окиси азота, а при температурах до 100°С (Кр мала)
скись азота может практически полностью окислиться до
двуокиси азота, которая в этих условиях не диссоциирует. Из
сказанного ясно, что в горячих нитрозных газах, выходящих из
контактного аппарата при 750—850 °С. двуокись азота
отсутствует. Чтобы получить двуокись азота, необходимую для
образования азотной кислоты, нитрозные газы приходится
охлаждать ниже 100 °С.
Скорость реакции окисления NO в NO2. Скорость этой
реакции зависит от концентрации NO; при низких концентрациях
NO в газе она очень мала. Так как при производстве азотной
кислоты приходится перерабатывать нитрозные газы с
относительно небольшим содержанием NO, то для окисления NO в
Ж>2 нужны огромные реакционные объемы.
Реакция окисления NO в NO2 является одной из немногих
известных тримолекулярных реакций, т. е. таких реакций, для
осуществления которых необходимо одновременное
столкновение трех молекул — в данном случае двух молекул NO и одной
молекулы О2. Другой особенностью этой реакции является то,
что скорость ее с повышением температуры уменьшается. При
0°С реакция протекает в 12 раз медленнее, а при 300 °С в 30
раз медленнее, чем при —130 °С. Следовательно, для увеличения
скорости окисления NO в NO2, иными словами, для
уменьшения реакционного объема аппаратуры, необходимо вести
реакцию при возможно более низких температурах. Поэтому
реакционные аппараты усиленно охлаждают водой.
Увеличение давления также повышает скорость окисления
окиси азота. Как показывает опыт, скорость окисления NO
(если ее выразить через концентрацию NO2 в объемных
процентах в данный момент реакции) пропорциональна кубу общего
давления газовой смеси, а время, необходимое для дости-
271
жения данной степени окисления N0 в N02, обратно
пропорционально квадрату давления. При применении давления
реакционный объем, необходимый для окисления данного
количества N0, уменьшится по двум причинам: во-первых,
вследствие уменьшения объема газа и, во-вторых, вследствие
уменьшения времени, требуемого для окисления. Так как время
окисления обратно пропорционально квадрату давления,
реакционный объем будет обратно пропорционален кубу давления
(если применяемые давления относительно невелики).
Этими соотношениями и определяется выгодность окисления
N0 з N02 под давлением.
Полимеризация N02 и образование N2O3. Кроме основной
реакции окисления NO в NO2, в газовой фазе протекают еще
две реакции. Первая реакция — полимеризация
2NO2 ' * N2O4 +13,6 ккал E)
протекает с выделением тепла и уменьшением объема. Поэтому
понижение температуры и повышение давления сдвигают
равновесие реакции в сторону образования димера (N2O4). При 0°С
равновесие значительно сдвинуто в сторону образования
димера, а при 100°С, наоборот, — в сторону образования
мономера (NO2). Скорость этой реакции велика, и равновесие
устанавливается практически мгновенно.
Можно считать, что в интервале температур от 0 до 100°С
всегда имеется равновесная смесь NO2 и N2O4.
Вторая реакция—образование азотистого ангидрида
NO + NO2 = N.,O3 + 9,6 ккал
протекает также практически 'мгновенно.
Азотистый ангидрид — очень неустойчивое соединение и
может присутствовать в нитрозных газах при обычных условиях
лишь в ничтожных количествах. Поэтому в производстве
азотной кислоты эта реакция не имеет существенного значения.
Азотный ангидрид N2O5 в условиях процесса производства
азотной кислоты не образуется; он может быть получен путем
окисления четырехокиси азота озоном.
Взаимодействие нитрозных газов с водой. При взаимодей-
ствтгл двуокиси азота с водой образуются азотная и азотистая
кислота:
2NO2 + Н2О = HNO3 + HNO., + 27,73 ккал F).
N2O4 + Н2О = HNO3 + HNO2 +14,13 ккал G)
Азотистая кислота неустойчива и распадается по реакции
3HNO2 = HNO3 + 2NO + Н2О —18,13 ккал (8)
Из уравнений F) и (8) получаем:
3NO2 (газ) + Н2О (жидк.) > 2HNO3 (жидк.) + N0 (газ) + 32,53 ккая (9)
272
Реакция (9) обратима; состояние равновесия зависит от
концентрации образующейся азотной кислоты, давления и
температуры. Если образуется очень разбавленная азотная кислота,
то она полностью поглощает МОг с одновременным
выделением окиси азота, в то время как при образовании 60%-нон
азотной кислоты связывается только небольшая часть двуокиси
азота. 3 производственных условиях обычно не стремятся
достигнуть состояния равновесия реакции (9), так как скорость ее
по мере приближения системы к равновесному состоянию
быстро понижается. Скорость образования азотной кислоты
увеличивается с повышением температуры, но при этом равновесие
реакции (9) сдвигается в левую сторону. Поэтому повышение
температуры для ускорения реакции допустимо только при
одновременном применении давления, способствующего
смещению разновесия ракции (9) слева направо. При повышенном
давлении F—8 ат) и температуре 50—80°С получается 58—
60%-лая азотная кислота (в таких условиях равновесие
полностью смещено слева направо), тогда как при атмосферном
давлении и обычной температуре можно получить только
50%-н\ю азотную кислоту.
В промышленных установках степень превращения окислов
азота з азотную кислоту составляет при атмосферном давлении
примерно 92% от исходного количества окиси азота, а при
6—8 ат достигает 98%. Это объясняется тем, что с повышением
давления скорость реакции окисления N0 в NO2 возрастает в
десятки раз. Понижение температуры до 0—5°С также
приводит к значительному увеличению интенсивности процесса.
Скорость реакции (9) можно также повысить путем увеличения
поверхности соприкосновения газовой и жидкой фаз, что
достигается заполнением абсорбционных колонн насадками.
Окислительный объем в поглотительной системе
рассчитывают на окисление не только количества NO, которое
содержится в газах, поступающих из контактного аппарата, но и
того количества NO, которое образуется по реакции (9). Из
1 моль исходной окиси азота после ее окисления до двуокиси
азота и последующего поглощения образуется еще Уз моль
окиси азота, из которой после повторного окисления и
поглощения получается, в свою очередь, еще '/з-1/з моль окиси азота
и т. д.
Таким образом, всего потребуется окислить:
1 -г -g- + gTg + g^g -\ ^1,5 моль NO
Следовательно, абсорбционная зона должна быть
рассчитана на окисление полуторного количества поступающей окиси
азота.
18—284в 273
При получении азотной кислоты без применения
избыточного давления оставшиеся в газе окислы азота улавливают
раствором соды (щелочная абсорбция):
Na2CO3 + NO, -f NO = 2NaNO, -f CO2 A0)
Na2CO3 -f 2NO, = NaNO, -f NaNO3 + COa A1)
В щелочных башнях улавливается б—7% окислов азота от
общего их содержания в нитрозных газах, поступающих на
переработку, вследствие чего степень использования окиси азота
повышается приблизительно до 98—99%. Газы, уходящие из
щелочных башен, должны содержать около 0,1% окиси азота.
Для более полной очистки отходящих (хвостовых) газов от
окислов азота в последнее время стали применять
каталитическое расщепление окислов при температуре 600—700 °С на
катализаторе. Отходящие газы смешивают с природным газом и
сжигают. При сжигании метан окисляется кислородом,
присутствующим в газах и связанным с азотом в виде NO и NO2.
В результате сжигания метана окислы азота расщепляются с
выделением свободного азота, а кислород образует с
водородом и углеродом соотвественно воду и двуокись углерода.
Возможно также образование небольшого количества окиси
углерода.
На некоторых заводах для более полной очистки отходящих
газов применяют промывку их купоросным маслом, которое
значительно лучше поглощает NO и NO2, чем растворы
щелочей. Получаемая при этом нитрозилсерная кислота
используется в производстве серной кислоты башенным способом.
Выхлопные газы, состоящие в основном из азота, после
соответствующей очистки от окислов азота и кислорода можно
использовать, например, для производства цианамида
кальция (стр. 606) и др.
В результате щелочной абсорбции получаются растворы
нитрата и нитрита натрия, содержащие около 250 г/л NaNO2 n
около 50 г/л NaNO3. Эти растворы (так называемые нитрит-
нитратные щелока) используют для получения нитрата натрия,
(стр. 278, 563). Для этого щелока обрабатывают 50%-ной
азотной кислотой, в результате чего нитрит инвертируется в нитрат:
3NaNOg + 2HNO3 = 3NaNO3 + 2NO + Н,0 — 7,39 кка.г A2>
Полученные растворы, содержащие небольшое количества
HNO3. нейтрализуют щелочью и упаривают.
Газ, образующийся при окислении NaNO2, является
источником получения концентрированной окиси азота. При
добавлении к этому газу кислорода с одновременным охлаждением
можно получить жидкую двуокись азота.
274
Зместо раствора соды для улавливания окислов азота из
газов применяют также более дешевое известковое молоко; в
этом случае получаются растворы Ca(NO3J и Ca(NO2J-
Образующийся нитрит кальция превращают в нитрат:
3Ca(NOaJ + 4HNO3 = 3Ca(NO3J + 4NO -f 2H,0 A3)
Получаемая при этом кальциевая селитра используется в
качестве азотного удобрения (стр. 564).
ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ ПРОИЗВОДСТВА
РАЗБАВЛЕННОЙ АЗОТНОЙ КИСЛОТЫ
Существуют два типа промышленных установок для
получения разбавленной азотной кислоты. К первому типу
относятся установки, в которых окисление аммиака и поглощение
окислов азота водой проводят под атмосферным давлением; при
этом получается 50%-ная азотная кислота. В установках второго
типа окисление аммиака и поглощение окислов азота водой
проводятся под повышенным давлением (от 2,5 до 8 ат). На
таких установках получается 60%-ная и более
концентрированная азотная кислота.
Окисление аммиака можно также проводить при
атмосферном давлении, а поглощение окислов азота — под повышенным
давлением 6—8 ат. Преимущества этого процесса очевидны:
проводя окисление аммиака при атмосферном давлении,
удается уменьшить потери платины, а процесс поглощения окислов
азота под давлением значительно интенсифицируется.
Система, работающая под атмосферным давлением
На рис. 113 представлена схема получения разбавленной
¦азотной кислоты под атмосферным давлением.
Очищенный газообразный аммиак подается во
всасывающий патрубок вентилятора 3, где смешивается с воздухом.
Очистку воздуха от посторонних газов кислого характера обычно
производят посредством промывки в скруббере 1 водой или
разбавленным раствором соды. Для отделения механических
примесей воздух фильтруется через матерчатый фильтр 2.
Аммиак и очищенный воздух, смешанные в соотношении
10—12 объемн. ч. аммиака на 90—88 объемн. ч. воздуха,
вентилятором 3 подаются в систему.
При наличии общей вентиляторной станции в цехе во все
контактные аппараты подается смесь, имеющая постоянный и
•одинаковый состав, что обеспечивает стабильный режим
окисления аммиака и позволяет упростить контроль и
автоматическое управление процессом. Если используется отбросный
кислород, его подмешивают к воздуху до очистки, при этом
содержание амлшака в смеси можно повысить до 12.5%.
U* 275
Перегретый пар
i-0 am
Воздух
Вода
Выхлопные
газы
Вода'
30%-на я
HNO3
3%-ная
HNO3
Продукционная
кислота
телика
Рис. 113. Принципиальная схема производства разбавленной азотной кислоты под атмосферным давлением:
/—водяной скруббер'. 2—суконный фильтр; 3—аммиачно-воздушный венгилятр; 4—картонный фильтр; 5—конгамньш аппарат, 6--кшел-
утилизатор; 7—первый газовый (скоростной) холодильник; 8—второй газовый холодильник; 5—вешнлятор им ip°3Hbi.\j азов; 10—"
кислотной абсорбции (/-V/); //—окнслигельная.башня (V//): /2-башни щелочной абсорбции, (VIII, IX); 13—кислошые холодил!
14—насосы для перекачивания кислоты, /5—насосы для перекачивания щелочных растворов.
—башни
;ьники;
Газовую смесь до поступления на катализатор еще раз
фильтруют через поролитовый фильтр (выносной или
расположенный в нижней части конвертора под платиновыми сетками)
или через картонный фильтр 4, помещенный в верхней части
контактного аппарата.
Аммиак окисляют кислородом воздуха на катализаторе з
контактном аппарате 5 при температуре в пределах 750—850 °С.
Нитрозные газы при этой температуре поступают в
котел-утилизатор 6 на охлаждение. Конвертор монтируется
непосредственно на котле-утилизаторе, благодаря чему потери тепла в
окружающую среду сокращаются до минимума.
В котле 6 прямоточного типа смонтирован змеевик, по
которому проходит вода, при нагревании превращающаяся в
пароводяную эмульсию, разделяемую далее в сепараторе на пар и
жидкость. Пар из сепаратора поступает в змеевиковып
перегреватель, расположенный в верхней части котла, и направляется
в паровую сеть. Температура пара 450 °С, давление 40 ат.
Дальнейшее охлаждение нитрозных газов, имеющих
температуру 160°С, производится в двух последовательно
включенных водяных холодильниках 7 и 8. Первый по ходу газов —
холодильник скоростного типа, в котором газы охлаждаются и
получается з виде конденсата возможно более разбавленная
кислота. При температуре китрозных газов на выходе из
холодильника 40 °С образуется конденсат, содержащий 2—3% HNO3.
При охлаждении нитрозных газов содержащаяся в них окись
азота начинает быстро окисляться кислородом воздуха, и
температура газов снова повышается. Далее газы проходят второй
холодильник 8, в котором выделяется конденсат, содержащий
25—30% HNO3. Второй газовый холодильник устанавливают
вблизи вентилятора 9, подающего нитрозные газы в
абсорбционную систему.
Охлажденные и частично обезвоженные нитрозные газы
поступают в абсорбционную систему из 6—8 башен 10, в
последнюю башню подается вода противотоком движению газов.
Каждая башня орошается кислотой соответствующей концентрации.
Циркуляция кислоты осуществляется при помощи
центробежных насосов 14, подающих ее через холодильники 13,
охлаждаемые водой для отвода выделяющегося в башнях тепла. По
мере повышения концентрации HNO3 кислота передается из
башни в башню навстречу ходу газа и по достижении заданной
концентрации HNO3 отводится из системы. Обычно
продукционную кислоту выводят из второй по ходу газа башни //,
первая башня / служит для подготовки газов к абсорбции, т. е.
для окисления NO в ЫОг-
Нитрозный вентилятор 9 должен подсасывать в систему
такое количество воздуха, чтобы содержание кислорода в
выхлопных газах поддерживалось в пределах 5—5,5%.
277
Концентрация окислов азота в нитрозных газах,
поступающих в поглотительную систему, составляет ~ 10% и перед по
следней башней снижается до 0,6—0,8%, что соответствуег
поглощению 92—94%) окислов азота в системе кислотной
абсорбции. Остатки окислов азота улавливаются в башнях 12,
орошаемых щелочными растворами. На выходе из системы
кислотной абсорбции степень окисленности нитрозных газов
колеблется в пределах 25—35%, в зависимости от температурного
режима и числа ступеней абсорбции.
Для процесса щелочкой абсорбции наиболее благоприятно
соотношение NO2:NO=1:1, т. е. степень окисления нитрозных
газов, равная 50%. Для достижения такого соотношения N0 и
NO2 в нитрозных газах между системами кислотной и
щелочной абсорбции устанавливают пустотелую неорошаемую
башню //, в которой NO окисляется в NO2.
В системе щелочной абсорбции также осуществлен принцип
противотока: во вторую по ходу газов башню подается свежий
раствор соды B00—250 г/л Na2CO3) или известковое молоко
[80—100 г/л Са(ОНJ]. В течение некоторого времени раствор
циркулирует в этой башне, а затем передается на орошение
первой по ходу газа башни VIII. где окончательно
перерабатывается в нитрит-нитратные щелока (стр. 274).
Отводимые из башни нитрит-нитратные щелока далее
обрабатывают азотной кислотой для превращения азотистокислых
солей в азотнокислые (инверсия). После упаривания
инвертированных щелоков получается сухая соль, содержащая 98—99%
NaNO3. Выделяющаяся при инверсии окись азота в смеси с-
воздухом возвращается в абсорбционную систему.
Готовая азотная кислота, получаемая описанным методом,
содержит 47—50% HNO3.
Удельный объем системы кислотной абсорбции при
получении 50%)-ной азотной кислоты и степени абсорбции окислов
азота 92% составляет 28—32 м3 на 1 т HNO3, получаемой з
сутки. Реакционный объем аппаратуры щелочной абсорбции
(башни с насадкой из колец Рашига) составляет 15—30% от
объема системы кислотной абсорбции.
Ниже приведены расходные коэффициенты в производстве
азотной кислоты по описанной схеме:
На 1 m HNO3 На 1 m азота
Аммиак, m 0,287-0,295 1,29—1,33
Платина, г 0,049 0,221
Электроэнергия, кв/пч 120 540
Пар, млн. ккал 0,09 0,405
Охлаждающая вода, м3 150 ¦ 675
Вода очищенная, м3 1,9 8,55
278
Одновременно получается пар в количестве 0,7 млн. ккал на
1 г HNO3, или 3,15 млн. ккал на 1 т азота.
Основные аппараты. Контактный аппарат с нижним вводом
аммиачно-воздушной смеси изображен на рис. 114. Средняя
часть аппарата — цилиндрическая, нижняя и верхняя —
конусообразные. Верхний конус /, изготовляемый из жароупорной
нержавеющей стали, имеет четыре смотровых окна для
наблюдения за состоянием
катализатора и три
запальных отверстия 8, через
которые в момент пуска
аппарата производят
зажигание
аммиачно-воздушной смеси для
разогрева катализаторных
сеток.
Снаружи верхний
конус покрыт слоем
теплоизоляции.
Цилиндрическую часть 2 изготовляют ,
из чугуна и футеруют
листовым алюминием
толщиной 3—4 мм. Внутри
цилиндра имеется
трубная решетка для
закрепления поролитовых
трубок 10, закрытых с одного
конца. Число трубок от
265 до 563; длина трубок
750—1000 мм. В трубках
газовая смесь
дополнительно очищается от
механических примесей
(пыля). Для лучшего
перемешивания аммиачно-
воздушной смеси внутри
контактного аппарата на
решетку насыпан слой
фарфоровых колец 9 (вы- Рис- I'4- Контактный аппарат для окисле-
сота слоя 300 лик). Пла- ния аммиака ПРИ атмосферном давлении:
' г /—верхний конус; 2—цилиндрическая часть: 3—опор-
ТИНО-фОДИеВЫе сеТКИ О ное кольцо; 4—нижний конус; 5—катализаторные
П094 птв1гм2\ ЧЯКПРП71Р- сетки; 6—болты для крепления крышки; 7—прижим-
[lUZt 0T8JLM ) закреиле Ные кольца; «-запальное отверстие: Р-слой !фар-
НЫ ДВуМЯ прИЖИМНЫМИ форовых колец; 10—поролитовые трубки; //—опор-
кольцами 7 из жароупор- ная баЛка-
ной стали. Нижний
конус 4 изготовляют из алюминия и иногда заполняют на
небольшую, высоту фарфоровыми кольцами.
279
нал
Общая высота контактного аппарата достигает 4,1 м;
высота нижнего конуса составляет 0,9 м, верхнего—1,45 м.
Диаметр контактного аппарата 2 м, диаметр входного патрубка
0,75 м, выходного ~ 0,6 м.
В одном аппарате, имеющем три сетки, окисляется около
10 т аммиака в сугки. При тщательной очистке и подогреве
газовой смеси степень контактирования достигает 97—98%.
В последние годы стали чаще применять контактные
аппараты с верхним вводом аммиачно-воздушной смеси, в которых
создаются более благоприятные условия для контактирования.
Катализаторные сетки в таких аппаратах лежат на опорах и
не прогибаются под напором газового потока, вследствие чего
процесс контактирования протекает более равномерно по всей
поверхности сетки.
На рис. 115 представлен контактный аппарат с верхней
подачей аммпачно-воздушной смеси, работающий под
атмосферным давлением. Аппарат является частью комбинированного
агрегата, состоящего из картонного фильтра, собственно
конвертора и котла-утилизатора. ,
Картонный фильтр, расположенный над верхней конической
частью аппарата (диаметры конуса 3 и 0,85 м, высота 1,5 м),
состоит из ряда касет, через которые проходит газовая смесь.
Через окна она поступает во внутренний цилиндр и далее —
во входной конус конвертора.
В месте перехода конуса 5 в цилиндрическую часть
конвертора расположена решетка 4 диаметром 2,9 м, служащая для
равномерного распределения потока газовой смеси,
поступающей на катализаторные сетки 3. Это способствует
равномерной нагрузке катализатора и выравниванию температуры
по всей поверхности сетки и, в конечном счете, повышению
степени конверсии аммиака.
По периферии цилиндрической части аппарата между
решеткой 4 и катализаторными сетками 3 расположены четыре
смотровых стекла, штуцеры для розжига, для взятия проб газа
на содержание аммиака и для присоединения манометра.
Рабочий диаметр катализаторных сеток 2,8 м; они
опираются на колосники, ниже которых находится слой 2
металлических колец Рашигз (толщина слоя 250 мм). При наличии
такого слоя, выполняющего роль обратного теплового
аккумулятора, облегчается розжиг аппарата после кратковременной
остановки. Кроме того, в слое колец -могут задерживаться
наиболее крупные частицы платины, которую затем собирают.
Нижняя часть аппарата футерована жаростойким кирпичом
и присоединена непосредственно к верхней части
котла-утилизатора.
Поверхность трех катализаторных сеток из нитей толщиной
0,09 мм с 1024 отв/см2 равна 33,4 м2. При средней напряжен-
280
ности катализатора 550 кг/сутки аммиака на 1 м2 активной
поверхности производительность конвертора составляет:
550-33,4=18400 кг/сутки NH3, или 48—50 т/сутки HNO3
Контактные аппараты оборудованы соответствующими
контрольно-измерительными приборами, вынесенными" на один
общий щит управления. Количество поступающих в аппарат
воздуха и аммиака
контролируется специальными диафраг-
менными приборами,
установленными на подводящих
трубопроводах. Температура
измеряется термопарой.
Изменение температуры в контактном
аппарате (в зависимости от
изменения содержания
аммиака в газовой смеси)
регистрируется фотоэлементом.
Содержание аммиака в смеси
определяется непрерывнодействую-
щим газоанализатором.
Абсорбционная колонна
'рис. 116) имеет
цилиндрическую форму. Корпус 1
выполнен из нержавеющей хромони-
келевой стали. Высота
колонны 20 м, диаметр'—'5 м.
Колонна заполнена насадкой 2
из керамических колец. Для
равномерного орошения
насадки кислотой в верхней части
колонны установлен
специальный распылитель 3 из хромо-
никелевой стали.
Рис. 115. Контактный аппарат
ним вводом газа:
верх-
1—нижняя часть аппарата, присоединенная к
РаНЬШе ДЛЯ абсОпбцИИ НИТ- ко™У->т™-™затору; 2-слой металлических
опиш^ длл оишрицпи пи! колец; 3—катализаторные сетки:
4—распределительная решетка; 5—верхняя коническая
часть аппарата; 6—картонный фильтр;
7—клапан.
розных газов строили
гранитные башни. На фундамент
укладывали лист
нержавеющей стали, а затем гранитные камни. Металлические колонны
более надежны в эксплуатации, чем гранитные башни, и
обычно работают десятки лет.
Система, работающая под давлением 8 am
При повышенном давлении во много раз ускоряется процесс
образования азотной кислоты, так как в этих условиях
значительно возрастает скорость окисления NO в NO2. Это, в свою
очередь, способствует более быстрому и полному поглощению
NO из ннтрозных газов.
281
Целесообразнее процесс окисления аммиака проводить при
атмосферном давлении, а поглощение окислов азота при
поГаз
щШ1ШШШ1
Y: -¦—-.v.^
^ Кислота>
Рис. 116. Абсорбционная колонна:
/—корпус колонны; 2—керамические кольца; 3—распылитель
орошающей кислоты.
вишенном давлении. Такого типа установки реализованы в
промышленности. Однако вследствие высокой стоимости и
трудности изготовления машин из нержавеющей стали,
необходимых для сжатия нитрозных газов, пока лучше компримировать
282
аммиачно-воздушную смесь до контактирования. В этом
случае можно применять турбокомпрессоры, выполненные из
углеродистой стали.
Схема производства разбавленной азотной кислоты при
абсолютном давлении 8 ат изображена на рис. 117. Жидкий
аммиак, поступающий из весового танка под давлением 10—12 ат,
подогревается до 70 °С в испарителе 10.
Далее газообразный аммиак через фильтр 9 поступает в
смеситель 8.
Воздух, необходимый для окисления аммиака, должен быть
тщательно очищен. Грубая очистка от пыли производится в
висциновом фильтре 1, который состоит из отдельных кассет,
заполненных керамическими кольцами, смазанными висцино-
вым маслом*.
Далее воздух промывается водой в насадочном скруббере 2,
после чего фильтруется в матерчатом фильтре 4, где
задерживаются более мелкие частицы пыли. Очищенный воздух
сжимается в турбокомпрессоре 5 до абсолютного давления 9 ат,
нагреваясь при этом до 135 °С, и поступает в ресивер 3,
служащий для смягчения толчков воздуха и для дополнительной
очистки от смазочных масел и водяных паров. Большая часть
сжатого воздуха через дополнительный фильтр 7, заполненный
волокнистым асбестом**, поступает в смеситель 8; остальной
воздух направляется в колонны 18 и 19.
Все трубопроводы от газовых фильтров 7 и 9 до контактного
аппарата 11 изготовлены из хромоникелевой стали во
избежание загрязнений очищенных воздуха и аммиака продуктами
коррозии.
В аппарате 8 образуется аммиачно-воздушная смесь
A1 — 12% NH3, 19—20% О2 и 69—70% N2), имеющая
температуру около 115°С (так как воздух нагрелся в
турбокомпрессоре до, 135 °С). Смесь поступает сверху в контактный
аппарат //, где на платино-родиевом катализаторе A6—18 сеток)
аммиак окисляется до окиси азота. При окислении в
контактном аппарате развивается температура до 900 °С. Во
избежание размягчения металла под действием высокой температуры
нижнюю часть контактного аппарата и соединительную трубу
охлаждают водой. Образовавшиеся в контактном аппарате нит-
розные газы A0,5—11,0% NO, 6—8% О2, 14—15% Н2О и
68—70% N2) охлаждаются в котле-утилизаторе 12 примерно до
390 °С и направляются в подогреватели 13, где нагревают от
25 до 150°С выхлопные газы, поступающие из абсорбционной
колонны 18.
* Висциновое масло представляет собой смесь машинного масла,
технического глицерина и 10%-ного раствора NaOH.
** На некоторых установках применяются фильтры из поролитовых
трубок.
283
Вода
llUJl/l/J
Рис.117. Схема производства разбавленной азотной кислоты под давлением 8 am:
I, 4, 7—фильтры для воздуха; 2, 20— скрубберы; 3—ресивер; 5—турбокомпрессор; 6— турбина расширения;
8— смеситель; К—фильтр для аммиака; 10— испаритель аммиака; //—контактный аппарат; 12—котел-утили-
атор; 13—подогреватели; 14—конденсатор-холодильник; 15—сепаратор; 16—брызгоуловнтель; 17—фильтр
для улавливании платины; 18—абсорбционная колонна; 19— отбелочная колонна.
Выхлопные газы направляются в турбину расширения 6,
установленную на одном валу с турбокомпрессором 5 и
электродвигателем. Таким образом достигается рекуперация 15—
20% энергии, затраченной на сжатие воздуха. Из турбины
расширения газы поступают в скруббер 20, орошаемый серной
кислотой, для связывания окислов азота в нитрозилсерную
кислоту, используемую в башенном процессе получения серной
кислоты (стр. 130). Пройдя скруббер, выхлопные газы
удаляются з атмосферу.
Из подогревателя 13 нитрозные газы при температуре
около 150°С поступают в холодильник-конденсатор 14, где
охлаждаются до 35—40°С циркулирующей водой. При этом
образуется 53—56%-ная азотная кислота в количестве до 50% от
общей выработки агрегата. Азотная кислота отделяется от
газов в сепараторе 15, проходит фильтр 17 для улавливания
платины и направляется в абсорбционную колонну IS для
укрепления или в колонну 19, где освобождается от
растворенных окислов азота путем продувания сжатым воздухом.
Далее азотная кислота направляется на склад готовой
продукции.
Нптрозные газы из сепаратора 15 входят в нижнюю часть
абсорбционной тарельчатой колонны 18, орошаемой водой
(паровом конденсат). Добавочный воздух B0% от основного
количества) вводится в конденсатор-холодильник 14 либо, как
показано на схеме, в нижнюю часть абсорбционной
колонны 1С. В колонне 18 нитрозные газы барботируют через слой
кислоты на тарелках (высота слоя на тарелке около 50 мм).
Кислота стекает по тарелкам навстречу газам, поглощая
окислы азота, и укрепляется до концентрации 58—60% HNO3. Для
отвода тепла, выделяющегося при поглощении окислов азота
водо!1. на тарелках колонны уложены змеевики, в которых
циркулирует охлаждающая вода.
Из абсорбционной колонны 18 газы, содержащие 0,1—0,2%
окислов азота, через брызгоуловить 16 поступают в
подогреватели 13 и далее в турбину расширения 6.
При указанном содержании в отходящих газах окислов
азота степень превращения их в азотную кислоту составляет
97-98%.
Примерные расходные коэффициенты для описанной
системы при выпуске 60%-ной азотной кислоты:
На 1 т На 1 т
НМОз азота
А:.:.\:иак. т 0,285—0,295 1.29—1,33
Платина, г ..... 0,15—0,20 0,675—0,90
Электроэнергия, квт-ч 350—500 1580—2250
Пар, т 0,3—0,5 1,35—2,25
Вода, мя 110—160 495—720
Нитрознсе
Вода
Рис. 118. Контактный аппарат для окисления аммиака под
давлением:
/—колпак; 2—усеченный конус (чаша); J—катализаторные сетки:
4—зажимные кольца; 5—гильза термопары; 6—водяная рубашка; 7—труба для отвода
нитрозных газов; 8—люк.
Основные аппараты. Контактный аппарат для окисления
аммиака под давлением схематично изображен на рис. 118.
Контактный аппарат состоит нз двух соединенных основаниями
усеченных конусов. Верхняя часть аппарата — колпак / — изго-
286
товлена из никеля, нижняя часть — усеченный конус 2 — из
хромистой стали.
Катализатором служат платино-родиевые сетки 3 (до 18
сеток), зажатые в кольца 4. Отводная труба охлаждается водой,
подаваемой в водяную рубашку 6.
Контактные аппараты, работающие под давлением, имеют
диаметр до 1 м.
Поступающая на контактирование аммиачно-воздушная
смесь содержит 11 —12% NH3. Температура контактирования
870—900 °С.
Степень контактирования около 95%. В зависимости от
условий процесса концентрация кислоты, получаемой после
абсорбции, колеблется от 58 до 60% НЫОз-
Абсорбционная колонна (рис. 119) представляет собой
сварной цилиндрический вертикальный аппарат, выполненный из
стали марки 1Х18Н9Т.
Диаметр колонны 3 м, высота ~45 м. Насадка колонны
тарельчатая. Ситчатые тарелки в количестве 40 шт. расположены
в строго горизонтальном положении и на различных
расстояниях друг от друга по высоте; в нижней части интервал между
тарелками 1,2 м, в верхней—-1 м.
При прохождении через отверстия тарелки газов со
скоростью 0,3—0,4 м/сек (в свободном сечении) на тарелках
образуется пена, имеющая весьма развитую поверхность раздела
фаз, что способствует интенсивному массообмену в
абсорбционной колонне.
Для перетока жидкости с вышележащей тарелки на
нижележащую служат переточные трубки диаметром 57/3,5 мм.
Нижний конец переточной трубки опущен в приемный карман
нижележащей тарелки, из которого жидкость растекается по
тарелке, направляясь к переточным трубкам, расположенным в
противоположной стороне от входа жидкости.
Для отвода тепла, выделяющегося в процессе абсорбции
нитрозных газов, на тарелках размещены охлаждающие
змеевики из труб диаметром 38/2,5 мм, в которых циркулирует
вода. Суммарная поверхность охлаждения в колонне 500 м2.
Для ремонта внутренних деталей колонны в ее стенке на
каждой тарелке, снабженной охлаждающими змеевиками,
имеются лазы. По высоте колонны расположены штуцеры для
ввода кислоты, образующейся в системе и подаваемой в
колонну ка укрепление, и штуцеры для взятия проб кислоты из
колонны.
Через нижний штуцер в колонну вводятся нитрозные газы,
через штуцер в крышке удаляются выхлопные газы, через
второй штуцер на крышке подается водяной конденсат.
Вес колонны 58 т, объем 316 м3, что соответствует
удельному объему 2,34 м3 на 1 г кислоты, получаемой в сутки.
287
Охлаждающая
вода
Диаметр отверстий
2 мм, шаг 9 пм
Рис. 119. Ситчатая абсорбционная колонна:
/—охлаждающие змеевики; //—переточные трубки; I—40—ситчатье
тарелки.
Система, работающая при повышенном давлении
и без подвода энергии
Системы, работающие при повышенном давлении,
отличаются большей интенсивностью по сравнению с системами,
работающими под атмосферным давлением, и потому более
перспективны. Однако при повышенном давлении требуются
значительные затраты энергии на сжатие газов. В то же время при
окислении аммиака выделяется такое количество тепловой энергии,
которой при правильном использовании хватило бы для сжатия
газов до требуемого давления.
В описываемой схеме* получения разбавленной азотной
кислоты под давлением 6—8 ат (рис. 120) вся энергия,
необходимая для сжатия воздуха, потребляемого в производстве
азотной кислоты, получается в системе за счет тепла реакции
окисления аммиака.
Образующийся при этогл перегретый пар (давление 27,5 ат,
температура 286 °С) используется в турбине, приводящей в
движение турбокомпрессор, который сжимает воздух до нужного
давления.
Атмосферный воздух, очищенный в фильтре /, сжимается
в турбокомпрессоре первой ступени до давления 2 ат,
разогреваясь при этом до 95 °С. Тепло сжатого воздуха после первой
ступени компрессии используется для подогрева хвостовых
газов до 75 °С в теплообменнике 6. Воздух, выходящий ич этого
теплообменника при температуре 54 °С, далее охлаждается
водой в аппарате 8 до 32°С и затем сжимается
турбокомпрессором 4 второй ступени до 8 ат, нагреваясь при этом примерно до
168 °С.
Турбокомпрессоры обеих ступеней работают по
адиабатическому циклу, т. е. с минимальными потерями тепла в
окружающую среду.
Воздух, сжатый во второй ступени компрессии, подогревает
в аппарате 7 хвостовые газы до 95 °С, охлаждаясь до 86°С.
Через теплообменник 7 проходит лишь часть воздуха,
направляемая в абсорбционную систему. Основной поток воздуха,
имеющего температуру 168°С, поступает на приготовление ам-
миачно-воздушной смеси.
Горячие нитрозные газы, образующиеся при окислении
аммиака в конверторе 12, проходят котел-утилизатор 13, где
получается перегретый пар, поступающий в турбину 2,
насаженную на вал воздушных турбокомпрессоров 3 и 4. Нитрозные
газы, имеющие по выходе из котла-утилизатора достаточно
высокую температуру, подогревают в теплообменнике 14
хвостовые газы (с 95 до 170°С), охлаждаясь при этом до 115°С.
* Пат. США 2955917 КЛ 23—162 от 11 октября 1960 г.
19—2848 289
Далее нитрозные газы используются для подогрева
конденсата в экономайзере 15 и при температуре, близкой к точке
росы, поступают в холодильник 16, где охлаждаются водой с
одновременным образованием азотной кислоты. Затем
нитрозные газы проходят окислитель-холодильник П. Здесь при
охлаждении водой происходит окисление N0 в N02, в
результате чего степень окисленности газов, поступающих в
абсорбционную колонну 18, максимально повышается.
Хвостовые газы из колонны 18 последовательно поступают
в подогреватели 6, 7 и 14 и при температуре 170°С
направляются в газовую турбину расширения 5, отдавая часть
энергии на вал турбокомпрессоров 3 и 4. Таким образом,
турбокомпрессоры приводятся в движение паровой турбиной 2 и газовой
рекуиерационной турбиной 5. Получаемой при этом энергии
достаточно для сжатия до 6—8 ат всего количества воздуха,
потребляемого в системе.
пар на хонде»- т„а выглоп
сааию
Рис. 120. Принципиальная схема производства разбавленной азотной
кислоты под давлением 6—8 am без подвода энергии извне:
1, 10—фильтры для воздуха; 2—паровая турбина расширения; 3—вогдушный
турбокомпрессор первой ступени; 4—втодутлный турбокомпрессор второй ступени; 5—газовая
турбина расширения; 6, 7, 14—теплообменники (подогревлтелн хвостовых газов); 8—водяной
холодильник; 9—фильтр д-яя аммиака; 11—смеситель аммиака и воздуха; 12—конвертор;
13—котел -утилизатор; 15—экономайзер; 16—конденсатор-холодильник; 17—окнслнтель-
холодильннк; 18—абсорбционная колонна.
Наличие паровой трубины позволяет легко осуществить
пуск системы, получив необходимый для этого пар из
общезаводской магистрали.
На описанной установке можно получать 57—60%-ную
азотную кислоту.
290
Система, работающая под давлением 2,5 и 5,5 ат
По данным фирмы «Монтекатини», разработавшей эту
схему, ее сущность сводится к следующему (рис. 121).
Жидкий аммиак подается в испаритель 1, далее
газообразный NH3 подогревается паром в аппарате 2 и через фильтр 3
поступает на смешение с воздухом в смеситель 6. Атмосферный
воздух, пройдя фильтр 4, сжимается в турбокомпрессоре 16 до
избыточного давления 2,5 ат, еще раз фильтруется в аппарате 5
и смешивается с аммиаком в аппарате 6.
Аммиачно-воздушная смесь сжигается на платино-родиевом
катализаторе под избыточным давлением 2,5 ат в конверторе 7,
соединенном с котлом-утилизатором 8. Получаемый здесь при
теплообмене с реакционными газами пар поступает в
сепаратор 9, перегревается в котле-утилизаторе и используется в
турбине 14, приводящей в движение турбокомпрессор.
| шслова
Рис. 121. Схема производства разбавленной азотной кислоты под давлением
2,5 и 5,5 ат:
1—испаритель аммиака; 2—подогреватель аммика: 3—фильтр для аммиака; 4, 5—фильтры
для воздуха; б—смеситель аммиака с воздухом; 7—контактный аппарат;
S—котел-утилизатор; 9—сепаратор пара; 10. 11—окислители N0; 12—абсорбционная колонна; 13—отбелочная
колонна; 14—паровая турбина расширения; /5—газовая турбина расширения;
/б—воздушный двухступенчатый турбокомпрессор; 17—газовый турбокомпрессор.
Нитрозные газы по выходе из котла-утилизатора отдают
остатки тепла выхлопным газам, используемым в турбине
расширения 15, затем окисляются в двух полых аппаратах 10.
Окисленные газы сжимаются турбокомпрессором 17 до 5,5 ат,
отдают тепло сжатия выхлопным газам и поступают во вторые
окислительные аппараты 11.
Подготовленные таким образом нитрозные газы
поглощаются водой в абсорбционной колонне 12. Полученная кислота
проходит отбелку в колонне 13, а выхлопные газы, подогретые в
двух теплообменниках, поступают в турбину 15. При этом
рекуперируется часть энергии, затраченной на сжатие газов.
19*
291
Турбокомпрессоры 16 и 17 для сжатия воздуха и нитрозных
газов насажены на один общий вал с газовой турбиной 15 и
паровой турбиной 14.
В описанной системе конверсия аммиака проводится на
четырех катализаторных сетках под избыточным давлением
2,5 ат, абсорбция окислов азота ведется под вдвое большим
давлением 5,5 ат. Такие условия благоприятны для стадии
переработки нитрозных газов и позволяют вести процесс с
умеренными потерями платинового катализатора.
В данной системе получается количество пара, достаточное
для работы паровой турбины, которая вместе с газовой
турбиной расширения полностью обеспечивают потребность в
энергии для сжатия воздуха и нитрозных газов до указанных выше
давлений. Степень кислотной абсорбции достигает 99% при
концентрации кислоты 65% HNO3 и удельном объеме
абсорбционной колонны 2 м3 на 1 т кислоты в сутки.
Система, работающая под давлением 1 и 4 am
В 1957 г. за рубежом в промышленность была внедрена
новая схема получения азотной кислоты, разработанная
французской фирмой «Кюльман» (фр. пат. 1090510 от 10 октября 1954 г.).
Вытлолнь/е газы
Дммиачно-
воздушная
смесь
Рис/122. Схема производства азотной кислоты под
давлением 1 и 4 am по способу фирмы «Кюльман»:
I—контактный аппарат; 2—котел-утилизатор; 3—газовый
холодильник; 4—газовый теплообменник; 5—турбокомпрессор; 6—турбина
расширения; 7—конденсатор-окислитель; 8—абсорбционные
тарельчатые колонны.
Сущность этой схемы, представленной на рис. 122, заключается
в следующем.
Конверсия аммиака проводится под атмосферным
давлением в контактном аппарате /. Тепло, выделяющееся при
окислении аммиака, используется для получения пара в
котле-утилизаторе 2. Дальнейшее охлаждение нитрозных газов произво-
292
дится в водяном холодильнике 3, охлажденные газы
сжимаются турбокомпрессором 5 до абсолютного давления 4 ат и
затем проходят трубчатый теплообменник 4, где отдают тепло
выхлопным газам. Последние проходят турбину 6, насаженную
на один вал с турбокомпрессором 5, при этом рекуперируется
часть энергии, затраченной на сжатие нитрозных газов.
Далее нитрозные газы охлаждаются водой в аппарате 7, где
происходит конденсация реакционной воды и окисление окиси
азота в двуокись азота. Охлажденные и достаточно
окисленные нитрозные газы последовательно поступают в
абсорбционные колонны 8, которые снабжены тарелками особой
конструкции (фр. пат. 1092289 от 10 ноября 1954 г.).
В колоннах 8 на этих тарелках реакция 2NO + O2 = 2NO2
протекает главным образом в жидкой фазе с окислением N0
растворенным в кислоте кислородом воздуха. Скорость этой
реакции в жидкой фазе во много раз больше, чем в газовой
фазе. Благодаря этому сильно интенсифицируется весь процесс
переработки окислов азота в азотную кислоту.
Для лучшего растворения кислорода в кислоте в нижнюю
часть ее слоя на каждой тарелке дополнительно подается
воздух. Этот процесс растворения происходит в специальном
секторе тарелки, отделенном перегородками от остальной ее части.
Кроме того, для улучшения абсорбции производится частичное
охлаждение колонны аммиаком.
На таких тарелках абсорбционной колонны оба процесса
(окисление N0 и абсорбция NO2) идут одновременно, при этом
исключается вредное влияние окиси азота, присутствующей в
нитрозных газах, на скорость абсорбции NO2 и достигается
возможность получения азотной кислоты повышенной
концентрации. На описанной установке можно получать азотную кислоту,
имеющую концентрацию до 70% HNO3, и повысить степень
кислотной абсорбции до 99—99,5%, что в абсорбционных
колоннах обычной конструкции не представляется возможным.
3. КОНЦЕНТРИРОВАНИЕ АЗОТНОЙ КИСЛОТЫ
Путем упаривания разбавленной азотной кислоты ее
концентрацию можно повысить только до 68,4°/о HNO3. При такой
концентрации раствор азотной кислоты имеет наивысшую
температуру кипения, равную 120,5 °С.
На рис. 123 представлена диаграмма кипеняя водных
растворов азотной кислоты при атмосферном давлении.
Верхняя кривая соответствует составу пара, нижняя —
составу жидкости при кипении азотной кислоты различной
концентрации.
Из диаграммы видно, что при перегонке водных растворов
кислоты, содержащих менее 68,4% HNO3, в дистиллят пере-
293
'го
115
ходит смесь с меньшим содержанием HNO3, чем в кипящей
жидкости. При ведении процесса концентрирования такой
кислоты в ректификационной колонне с дефлегматором можно
разделить раствор на воду и азеотропную (постояннокипящую)
смесь, содержащую 68,4% HNO3, т. е. можно довести
концентрацию HNO3 до 68,4% (практически до 65%) с получением
3%-ной кислоты в дистилляте. Если же перегонять азотную
кислоту концентрацией вы-
125,—,—,—,—,—,—¦—,—,—,—, ше 68,4% HNO3, то в
дистиллят будет переходить
азотная кислота большей
концентрации, чем кипящая
жидкость, до тех пор, пока
концентрация кипящей
жидкости не снизится до 68,4%
HNO3.
Если упаривание азотной
кислоты проводить в
вакууме, температура кипения
раствора понизится: оста-
.• точному давлению порядка.
о W го зо но so so 7о во до шо 50—60 мм рт. ст. соответст-
Содержание HNO3, вес.% вует темп. кип. 68,4%-ной
§.100
I
90
85
80
/
f
ПаР
•"'
у
\
^Жадность ^
\
\
\
\ 11
\
\
\
ч
Рис. 123. Диаграмма кипения системы
HN03—Н3О.
HNO3 около 70 °С. Это
выгодно также потому, что
при более низких
температурах материалы, из
которых выполняются аппараты для концентрирования, устойчивее
к действию кипящей азотной кислоты.
Для получения кислоты, содержащей более 68,4% HNO3.
перегонку следует вести с водоотнимающими веществами.
Таким веществом обычно служит серная кислота (купоросное
масло). При добавлении концентрированной серной кислоты к
раствору азотной кислоты понижается давление водяных паров,
в то время как давление паров азотной кислоты над смесью
почти не изменяется. Поэтому при нагревании будет отгоняться
азотная кислота.
Концентрирование азотной кислоты при помощи
купоросного масла проводят в тарельчатых дистклляционных
колоннах из ферросилида, тарелки колонн снабжены колпачками и
переливными трубами. Применяются также колонны с
насадкой из колец; производительность таких колонн выше, чем
тарельчатых, вследствие меньшего сопротивления аппарата
газовому потоку. Смешение разбавленной азотной кислоты и
купоросного масла проводится чаще всего непосредственно в
колонне (в некоторых установках это делают предварительно). Смесь
нагревают острым паром.
294
СХЕМЫ УСТАНОВОК ДЛЯ КОНЦЕНТРИРОВАНИЯ
Принципиальная схема установки для концентрирования
азотной кислоты изображена на рис. 124. Купоросное масло
подают на шестнадцатую снизу тарелку колонны 2. Часть
разбавленной азотной кислоты E0—60% от общего количества)
по алюминиевому трубопроводу поступает в обогреваемый
паром D—6 ат) испаритель 1. Предварительное испарение части
азотной кислоты позволяет уменьшить расход серной кислоты
вода
Разбавленная
Рис. 124. Схема концентрирования разбавленной азотной кислоты:
/—испаритель; 2—концентрационная колонна; Л—конденсатор-холодильник:
4—холодильник азотной кислоты; 5—абсорбционная колонна; 6—вентилятор.
на концентрирование и одновременно увеличить
производительность колонны. Из испарителя пары азотной кислоты
поступают на десятую тарелку колонны 2. Остальная часть
разбавленной азотной кислоты поступает непосредственно на
тринадцатую тарелку колонны 2.
Купоросное масло, перетекая с тарелки на тарелку,
смешивается с разбавленной азотной кислотой, в результате чего
образуется тройная смесь серной кислоты, азотной кислоты и
воды.
Для денитрации отработанной серной кислоты в нижнюю
часть колонны (под первую тарелку) подается острый
перегретый водяной пар, имеющий температуру около 250СС. Пар
нагревает тройную смесь и из нее начинает испаряться наиболее
летучий компонент — азотная кислота. Выходящие in колонны
пары азотной кислоты, имеющие температуру 70—85°С, кон-
295
денсируются в ферросилидовом холодильнике 3. При этом
образуется 98—99%-ная азотная кислота (температура кислоты,
около 30°С), которая содержит значительное количество
растворенных окислов азота.
Присутствие окислов азота в парах азотной кислоты,
поступающих на конденсацию, объясняется частичным разложением
HNO3 в колонне при высокой температуре:
4HNO3 = 4NOa + О2 + 2Н2О
Кроме того, в разбавленной азотной кислоте, поступающей
на концентрирование, обычно растворено 0,2—0,6% окислов
азота.
Концентрированная азотная кислота возвращается из
конденсатора-холодильника 3 в верхнее звено колонны 2 и,
проходя последовательно сверху вниз две или три тарелки,
продувается парами азотной кислоты для освобождения от
растворенных окислов азота. Затем концентрированная азотная
кислота выводится из колонны, охлаждается в холодильнике 4
и далее поступает на склад готовой продукции.
Несконденсировавшиеся пары азотной кислоты и непогло-
щснттые окислы азота из конденсатора-холодильника 3
поступают в абсорбционную колонну 5, орошаемую водой или
разбавленной азотной кислотой. В колонне 5 постепенно
образуется разбавленная азотная кислота E0% НМОз), также
поступающая на концентрирование з колонну 2, инертные газы
отводятся в атмосферу вентилятором 6, который поддерживает
необходимое разрежение е системе.
Отработанная серная кислота (около 65% H2SO4) на выходе
из колонны 2 имеет температуру 160°С и, вследствие
продувки острым перегретым водяным паром, содержит весьма
незначительное количество окислов азота (не более 0,03%).
Обычно горячую отработанную серную кислоту из колонны
передают непосредственно на концентрирование до купоросного
масла.
Основным аппаратом цеха концентрирования является фер-
росилидовая колонна для концентрирования кислоты (рис. 125),
представляющая собой цилиндр (высота около 9 м, внутренний
диаметр 1 м), обычно состоящий из 20 отдельных звеньев,
скрепленных анкерными болтами. Для герметичности между
звеньями колонны имеются асбестовые прокладки. Внутри
колонны расположены барботажные тарелки 2, покрытые
колпаками 4 с зубчатыми краями. Тарелки снабжены переливными
трубками 1. Устройство всех звеньев колонны одинаково.
Для лучшего соприкосновения паров и газов со смесью
кислот зубчатые края каждого колпака погружены в слой кислоты.
Газы и пары в каждом звене проходят через отверстие 3,
расположенное в центре тарелки. В звене, пз которого выходит
296
. Пары
HNO3
Концентрированная
HNO3
Концентрированная
HNO3
Купоросное
масло
Разбавленная
¦*— HNO,
Разбавленная
_HNO3
Отработанная
"* H2SO4
Рис. 125. Колонна для концентрирования
разбавленной азотной кислоты:
/—переливные трубки; 2—барботажные тарелки; 3—отверстие
для прохода газа и паров; 4—колпак.
концентрированная HNO3 после отдувки из нее окислов азота
(второе или третье звено сверху), отверстие в переливной
трубке закрыто заглушкой. Купоросное масло входит через штуцер
на шестнадцатую тарелку, заполняет звено до уровня трубки 1
и, переливаясь через ее края, попадает в расположенное под
ней звено и т. д.
Пары азотной кислоты уходят через штуцер в крышке
колонны.
297
Нормально концентрационная колонна работает при 110—
130 °С и разрежении 20—60 мм рт. ст. Получаемая азотная
кислота содержит 96—98% НМО3, не более 0,4% окислов азота
(а пересчете на N2O4) и не содержит серной кислоты. Для
контроля процесса систематически отбирают пробы для
определения концентрации азотной кислоты и отработанной серной
кислоты. Кроме того, делают качественный (иногда и
количественный) анализ отработанной серной кислоты на содержание
в ней окислов азота, так как горячая серная кислота,
содержащая окислы азота, разрушает свинцовую аппаратуру.
Для перекачивания кислот применяют центробежные
насосы, изготовленные из ферросилида или гартблея (сплав свинца
с сурьмой).
Примерные расходные коэффициенты при концентрировании
50%-ной азотной кислоты:
На I m На 1 m
HNO3 азота
Разбавленная азотная кислота (в
пересчете на 100% HNO3), m 1,015—1,02 4,57—4,58
Серная кислота (в пересчете на 100%
H2SO4), m Около 3 13,5
в том числе безвозвратные потери, кг 10—12 45—54
Вода (на охлаждение), м3 30—50 135—225
Перегретый пар (давление 6 am), m . . . 0,5—0,6 2,25—2,7
Электроэнергия, квш-ч 10—12 45—54
Если принять за единицу расход серной кислоты (считая на
100%) H2SO4) на 1 г азотной кислоты, имеющей начальную
концентрацию 65% HNO3, то относительное увеличение расхода
H2SO4 в зависимости от концентрации азотной кислоты
выразится следующим образом:
HNOS1 % 65 60 55 50 45 40
Расход H2SO4 1,0 1,4 1,9 2,5 3,3 4,2
Концентрирование азотной кислоты путем ее дистилляции
в смеси с серной кислотой громоздко и дорого, кроме того, при
упаривании отработанной серной кислоты значительная часть
ее в виде тумана уносится с газами, отравляя окружающую
атмосферу. В связи с этими недостатками описанного способа
изыскиваются возможности применения других водоотннмаю-
щих средств при концентрировании азотной кислоты.
В этом отношении представляет интерес способ, по которому
в качестве водоотнимающего средства используется
азотнокислый магний (англ. пат. 724229 от 16 февраля 1955 г.).
Технологическая схема работы такой установки приведена
на рис. 126 и заключается в следующем:
Разбавленная E5%-ная) азотная кислота подается в
верхнюю часть отпарной тарельчатой колонны /. Несколько выше
ввода кислоты в колонну поступает раствор нитрата магния
G0—72%-ный). Нижняя часть отпарной колонны соединена
2 8
Вода
-пая
с трубчатым кипятильником 2, который обогревается паром.
Через кипятильник циркулирует часть раствора нитрата
магния, благодаря чему обеспечивается подвод необходимого
количества тепла в отпарную колонну. В нижней части колонны
температура поддерживается в пределах 140—190 °С. Пары,
выходящие из отпарной колонны и содержащие около 87%
HNO3 и 13% Н2О, поступают в тарельчатую дпстилляционную
колонну 3. Жидкая
фракция из колонны 3 G5%
HNO3 и 25% Н2О)
возвращается в отпарную
колонну 1, пары,
выходящие из этой колонны,
имеют температуру 86 °С
и содержат 98—99%
HNO3 и 1—2% Н2О.
Пары поступают в
водяной
конденсатор-холодильник 4. Конденсат,
представляющий собой
концентрированную
азотную кислоту (до 99%
HNO3), отводится на
склад, а часть кислоты в
виде флегмы
возвращается на верхнюю тарелку
дистплляционной
колонны 3.
Жидкая фракция,
вытекающая из отпарной Рис- 126- Принципиальная схема концентри-
, -_,_ рования азотной кислоты при помощи нитрата
колонны /, содержит 5о— v магния- F p
70 /о НПТрата МаГНИЯ И Не ;_отпарная колонна; 2—кипятильник; 3—дистнлля-
()ОЛее 0 1 % СВобоДНОЙ ционная колонна; 4—холодильник-конденсатор; 5—ба-
„ ' / рометрическнй конденсатор; 6—вакуум-нспаритель;
ЭЗОТНОИ КИСЛОТЫ (ОСТЭЛЬ- 7—подогреватель раствора нитрата магния; 8—насос.
ное вода) и имеет
температуру 140—190°С, в зависимости от давления и концентрации
Mg(NO3)?. Часть этой жидкости возвращается в кипятильник 2,
остальное подвергается закуум-концентрированию в системе
аппаратов 5, 6 и 7. Упаренный раствор нитрата магния
насосом 8 снова подается в отпарную колонну 1.
4. ПРЯМОЙ СИНТЕЗ КОНЦЕНТРИРОВАННОЙ АЗОТНОЙ
КИСЛОТЫ
Прямой синтез концентрированной азотной кислоты
заключается во взаимодействии жидкой четырехокиси азота с водой
в присутствии газообразного кислорода.
299
Реакция образования азотной кислоты протекает
следующим образом:
2N2O4 (жидк.) + 2Н2О (жидк.) + О2 (газ) = 4HNO3 (жидк.) + 18,84 ккал
При атмосферном давлении или под давлением не более
8 ат реакция образования азотной кислоты
3NO2 + Н2О = 2HNO3 + NO
заканчивается, когда концентрация азотной кислоты достигает
65—70% HNO3. В присутствии же кислорода и под давлением
50 ат равновесие реакции сдвигается вправо вплоть до
образования 98—99% -ной азотной кислоты.
В этом процессе кислород играет роль окислителя окиси
азота, выделяющейся при абсорбции двуокиси азота водой.
Скорость реакции окисления N0 кислородом при давлении
50 ат очень велика, время, необходимое для окисления окиси
азота, обратно пропорционально квадрату давления. Поэтому
при окислении окиси азота под давлением 50 ат требуется в
2500 раз меньше времени, чем при окислении под атмосферным
давлением. Для ускорения реакции взаимодействия двуокиси
(четырехокиси) азота с водой температуру процесса следует
поддерживать в пределах 70—80 °С. При температуре выше
80 °С процесс образования кислоты ускоряется, но
одновременно увеличивается и коррозия алюминия, из которого
выполняется аппаратура.
На рис. 127 изображена схема прямого синтеза
концентрированной азотной кислоты из жидких окислов азота
абсорбционным методом. Нитрозные газы, получаемые так же, как при
производстве разбавленной азотной кислоты под атмосферным
давлением (стр. 277), пройдя через котел-утилизатор, посту
пают в скоростной холодильник 1, где охлаждаются до
40—45 °С. При этом конденсируется около 75%) водяных паров,
содержащихся в газе, и происходит частичное окисление NO
в NO2. Образующийся водный конденсат, содержащий 2—3%
HNO3, используется в производстве разбавленной HNO3 для
орошения абсорбционных башен. Из скоростного холодильника
нитрозные газы поступают в обычный газовый холодильник 2,
где происходит дальнейшая конденсация паров воды,
оставшихся в газе, а также частичное окисление NO в NO2.
Образующаяся при этом 30—35%-ная азотная кислота направляется в
колонну 9, где используется для промывки выхлопных газов.
Из газового холодильника нитрозные газы вентилятором
подаются последовательно в две окислительные колонны 3, где
при 30—35 °С окись азота примерно на 93°/о окисляется в NO2
кислородом, поступившим в систему вместе с аммиаком. В
окислительных колоннах образуется 55—60%-ная азотная кислота.
Эти колонны частично орошаются «на себя», избыток кислоты
300
Продукционная кислота
Вода
Рис. 127. Схема прямого синтеза концентрированной азотной кислоты:
7—скоростной холодильник; 2, 6, 12— газовые холодильники; 3— окислительные колонны; 4, 8, 11—холодильники кислоты; 5—доокпслитель;
7- абсорбционная колонна; 9—промывная колонна; 16— отбелочная колонна; 13—конденсатор; 14—сепаратор; /5—смеситель; /5—автоклав
передается в смеситель 15. Для отвода тепла,
выделяющегося в колоннах, циркулирующая кислота охлаждается в
холодильниках 4. Из колонн 3 нитрозные газы поступают в доокис-
литель 5, орошаемый 98%-ной азотной кислотой. Здесь при
30—35 °С происходит окисление оставшегося в газе N0:
NO -f 2HNO3 = 3NO2 -f H20
Выходящая из доокислителя 75%-ная HNO3 также
передается в смеситель 15. Нитрозные газы по выходе из
доокислителя охлаждаются в рассольном холодильнике 6 до —10 °С и
далее поступают в абсорбционную колонну 7, где двуокись
азота поглощается 98% -ной азотной кислотой. Тепло,
выделяющееся при абсорбции, отводится путем интенсивного охлаждения
кислоты (до —10 °С) в рассольных холодильниках 8.
Выхлопные газы из колонны 7, содержащие около 0,15% (NO2+N2O4),
проходят промывную колонну 9, орошаемую 30—35%-ной
азотной кислотой, после чего удаляются в атмосферу.
Раствор окислов азота в 98%-ной азотной кислоте из
абсорбционной колонны 7 передают в отбелочную колонну 10,
состоящую из двух секций — нижней, обогреваемой паром, и верхней,
с водяным охлаждением. Кислота подается в верхнюю секцию
и нагревается в нижней секции глухим паром до 85 °С, при
этом из раствора отгоняется газообразная двуокись азота.
В отбелочную колонну, кроме кислоты из абсорбционной
колонны 7, поступает на отбелку также продукционная кислота
из автоклава 16, в которой содержится до 20% растворенной
двуокиси азота. Освобожденная от окислов азота (отбеленная)
чистая продукционная азотная кислота охлаждается в змееви-
ковом холодильнике 11 до 30°С. Часть продукционной
(98%-ной) кислоты перекачивается на склад, остальное
возвращается на орошение абсорбционной колонны 7 и в доокис-
литель 5.
Газообразная двуокись азота из колонны 10 через водяной
холодильник 12 поступает в два последовательно установленных
конденсатора 13 (на рисунке показан один аппарат). В первом
по ходу газа аппарате двуокись азота охлаждается водой
(—20 °С); во втором — рассолом (—10 °С).
Жидкая двуокись азота передается в смеситель 15, нескон-
деисировавшиеся при охлаждении газы возвращаются в
окислительную колонну 3.
Оптимальный состав «сырой» смеси (в аппарате 15), при
котором достигается максимальное превращение ее
компонентов в концентрированную азотную кислоту, примерно
следующий (в вес. %): 70% N2O4, 18% HNO3, 12% Н2О. Такая смесь
из аппарата 15 поступает в непрерывнодействующий
автоклав 16, куда под давлением 50 ат, создаваемым кислородным
302
Ог Кисло/по
компрессором, подается газообразный кислород. Выходящая из
автоклава азотная кислота, содержащая до 25% растворенной
двуокиси азота, поступает в
отбелочную колонну 10, где разделяется
на окислы азота и
концентрированную 98%-ную азотную кислоту.
Автоклав (рис. 128)
представляет собой толстостенный
вертикальный стальной цилиндр
(корпус /) емкостью 8—12 Л13. Изнутри
он футерован листовым алюминием
(стакан 2) для защиты корпуса
автоклава от разрушающего действия
азотной кислоты, которое особенно
сказывается при повышенных
температурах. Крышка автоклава
изнутри также покрыта листовым
алюминием. Внутрь корпуса
автоклава вставлен алюминиевый
реакционный сосуд 3, закрепленный
во фланце корпуса. В сосуде
происходит реакция образования
азотной кислоты. Давление внутри
алюминиевого стакана компенсируется
давлением кислорода, подаваемого
в кольцевое пространство между
стенками автоклава и
алюминиевым сосудом.
В реакционный сосуд свободно
вставлена насадка, обеспечивающая
непрерывный режим работы
автоклава. Насадка представляет собой
алюминиевые ситчатые тарелки 4,
собранные в виде этажерки и
скрепленные между собой
алюминиевыми штангами 5.
В тарелках имеются отверстия
диаметром по 2 ми (шаг между Рис 128 Автоклав для прямого
отверстиями 30 мм, количество синтеза азотной кислоты:
ИХ 650 ШТ.) . 1—корпус автоклава; 2—защитный ста-
Кппмр ттттянг ЦРПРЧ тяПРЛКИ ппп кан; 5—реакционный сосуд; 4—ситча-
J\pOMe ШТаш.Черес! тарелки Про- тые тарелки; 5—распорные штанги: 5—
ХОДЯТ Две ТРубы 6, Служащие Ка- чехольные трубы (каналы); 7—перелив-
* ные трубы.
налами для трубопроводов,
подводящих кислород и отводящих
кислоту. Труба, по которой подается кислород под давлением
50—52 ат, опущена под нижнюю тарелку; труба, отводящая
готовую кислоту из автоклава, опущена еще ниже, почти до
303
дна реакционного сосуда. Конец трубы, через которую в
автоклав поступает сырая смесь, находится в пространстве между
крышкой автоклава и верхней тарелкой насадки.
Через крышку автоклава проходят еще две трубы длиной
1500 и 800 мм, служащие для перелива излишка жидкости в
приемную емкость (на ссас насоса).
Кислая смесь, состоящая из N2O4, HNO3 и воды, поступает
на верхнюю тарелку автоклава и переливается на
нижележащие тарелки противотоком движению кислорода, который бар-
ботирует снизу вверх через слой кислоты. Продувочные газы
отводятся из пространства над верхней тарелкой по
специальной трубе (на рисунке не показана).
Давление в аппарате поддерживается около 50 ат,
температура 80 °С.
Примерные расходные коэффициенты на описанной
установке прямого синтеза азотной кислоты:
Аммиак, m
Кислород, м3
Платина, г
Электроэнергия, квт-ч
Пар, т
Вода, м3
Холод, тыс. /скал
б. СОВМЕСТНОЕ ПОЛУЧЕНИЕ СЕРНОЙ И АЗОТНОЙ КИСЛОТ
Наиболее медленной стадией процесса получения азотной кислоты
является окисление окиси азота:
2NO + О2 = 2NOa
Скорость этой реакции особенно быстро уменьшается с понижением
содержания NO в газе (пропорционально квадрату ее концентрации).
Следовательно, для увеличения скорости реакции необходимо, чтобы содержание
NO в газе было возможно более высоким. Если в газовую смесь с высоким
содержанием окислов азота ввести газ, содержащий SO2 (печной газ после
сжигания колчедана), то двуокись серы будет быстро окисляться до SO3 с
образованием в дальнейшем серной кислоты. Серная кислота при 35—40 °С
достаточно полно связывает окислы азота, образуя нитрозилсерную кислоту.
N4O3 + 2H2SO4 = 2HSNO5 + Н.2О
При высокой температуре, создаваемой за счет тепла печных газов,
нитрозилсерная кислота освобождается от окислов азота с образованием
серной кислоты и концентрированных нитрозных газов. Денитрованную
78%-ную серную кислоту (готовый продукт) выводят из процесса.
Концентрированные окислы азота направляют на дальнейшую пегсеработку
обычным способом для получения из них азотной кислоты нужной концентрации
F0% и 98% НШ)
304
На 1 от
HNO3
0,29
150
0,05
270
0,6
200
400
На 1 т
азота
1,31
675
0,225
1210
2,7
900
1800
Хранение и перевозка азотной кислоты
Разбавленную азотную кислоту D7—60% HNO3) хранят
в резервуарах из нержавеющей стали. Склады азотной кислоты
обычно имеют небольшую емкость, так как разбавленную
кислоту большей частью перерабатывают на этом же предприятии
в соли азотной кислоты или концентрируют и отправляют
потребителям.
При отправке разбавленной азотной кислоты потребителям
в зависимости от количества и назначения кислоты ее
перевозят в стеклянных бутылях емкостью по 30—50 л или в
железнодорожных цистернах из нержавеющей стали. Для
предотвращения пожара при случайной проливке кислоты стеклянные
бутыли упаковывают в корзины или в деревянную обрешетку
с обкладкой соломой или стружками, пропитанными хлористым
кальцием или другими огнестойкими веществами.
Концентрированную азотную кислоту хранят и перевозят в
алюминиевых цистернах. Чаще всего ее смешивают с
небольшим количеством купоросного масла G,5% H2SO4). Такую
смесь, называемую меланжем, отправляют потребителям в
цистернах из углеродистой стали.
Перевозка концентрированной азотной кислоты в
стеклянных бутылях с деревянной обрешеткой не допускается.
Техника безопасности в производстве
азотной кислоты
Для обеспечения безопасной работы в производстве азотной
кислоты должны соблюдаться следующие основные правила.
Концентрация окислов азота в воздухе производственных
помещений не должна превышать 5 мг/м3 (в пересчете на
N2O5), концентрация аммиака — 20 мг/м3. Поэтому в
производственных помещениях обязательно устанавливается мощная,
хорошо работающая вентиляция. Если содержание окислов
азота или аммиака в помещении превысит допускаемые нормы,
работающие должны надеть противогазы и не снимать их до
создания нормальных условий.
Весь обслуживающий персонал должен быть снабжен
индивидуальными средствами защиты (противогазы, очки,
резиновые перчатки и др.). Помимо индивидуальных противогазов,
в цехе должны иметься запасные противогазы марок К, КД, В
н специальные шланговые противогазы.
При попадании на кожу концентрированной азотной
кислоты необходимо немедленно смыть ее сильной струей воды и
смазать пораженное место мазью от ожогов.
При производстве азотной кислоты прямым синтезом в
автоклав не должны попадать горючие и воспламеняющиеся
вещества, так как это может привести к "взрыву. Поэтому нуж-
20—2848 305
но очень осторожно производить смазку подшипников насосов
для свежей смеси, редукторов на мешалках, а также не
применять кислород, содержащий примеси водорода, ацетилена или
других легковоспламеняющихся газов.
Противопожарные средства в производственных
помещениях должны находиться в постоянной готовности и регулярно
проверяться.
ЛИТЕРАТУРА
(к главам V, VI, VII)
С. И. В о л ь ф к о в и ч, А. П. Егоров, Д. А. Э п ш т е й н, Общая
химическая технология, т. I, Госхимиздат, 1952.
Д. А. Э п ш т е й н, Основы химической технологии, Изд. Академии
педагогических наук РСФСР, 1957.
С. Я. Г е р ш, Глубокое охлаждение, ч. I, Госэнергоиздат, 1957, ч. II, Гос-
энергоиздат, 1960.
Д. Л. Глизманенко, Кислород и его получение, Госхимиздат, 1956.
В. Б. И о ф ф е, Основы производства водорода, Гостоптехиздат, 1960.
И. И. Г е л ь п е р и н, Г. М. 3 е л и к с о н, Л. Л. Рапопорт, Справочник
по разделению газовых смесей методом глубокого охлаждения,
Госхимиздат, 1963.
Е. Б л а с я к и др., Технология связанного азота. Синтетический аммиак,
перев. с польск., Госхимиздат, 1961.
Е. Я. Мельников, Современные схемы производства технологического
газа для синтеза аммиака, Труды ЛИЭИ, вып. 37, Изд.
Ленинградского университета, 1961, стр. 76.
В. М. Ордынцев, Система автоматического управления технологическими
процессами крупного аммиачного производства, Журн. ВХО
им. Д. И. Менделеева, № 5, 515 A961).
Б. П. Самарин, Модернизация технологического процесса и основного
оборудования производства аммиака, Журн. ВХО им. Д. И.
Менделеева, № 1, 39 A961).
В. И. Атрощенко, С. И. К а р г и н, Технология азотной кислоты,
Госхимиздат, 1962 г.
В. И. Атрощенко, И. И. Литвиненко, Кинетика растворения окислов
азота в водных растворах азотной кислоты, сб. «Химия и химическая
технология», Изв. вузов, вып. 4, Изд, Ивановского хим.-технол.
института, 1958, стр. 71.
И. Н. Кузьминых, А. И. Родионов, Ю. С. Мищенко, Абсорбция
окислов азота в барботажной колонне, сб. «Химия и химическая
технология^, Изв. вузов, вып. 2, Изд. Ивановского хим.-технол
института, 1959, стр. 287.
В. И. Атрощенко, В. М. К а у т, Кинетика поглощения окислов азота
концентрированной азотной кислотой, ЖПХ, 31, 3, 352 A958).
Правила и нормы техники безопасности и промышленной санитарии для
проектирования, строительства и эксплуатации производства
синтетического аммиака и сырца-метанола, Госхимиздат, 1960.
Првила и нормы техники безопасности и промышленной санитарии для
проектирования, строительства и эксплуатации производства
разбавленной азотной кислоты, концентрированной азотной кислоты и
купоросного масла. Госхимиздат, I960.
Глава VIII
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОИЗВОДСТВА
Электрохимическими процессами называются химические
процессы, протекающие под действием постоянного
электрического тока. До 90-х годов XIX в. электрическая энергия
находила ограниченное применение в химической промышленности,
развитие промышленной электрохимии началось лишь с 1870 г.
после изобретения динамомашины.
Применение сравнительно простых и относительно дешевых
электрохимических методов позволяет получать прямым путем
многие весьма важные продукты и материалы.
В химической технологии электролиз применяется для
производства хлора, едких щелочей, чистых водорода и кислорода,
фтора, перманганата калия, перекисных соединений, например
надсерной кислоты, используемой для получения перекиси
водорода, и др. Электрохимические методы используются и в
некоторых процессах органического синтеза
(электровосстановление, электроокисление, электрогалоидирование и др.).
Огромное значение электролиз имеет в металлургии. Так,
цветные и легкие металлы, большинство редких металлов
получают электролизом водных растворов или расплавов солей.
Гидроэлектрометаллургические процессы применяются для
получения и рафинирования меди, никеля, свинца, цинка,
получения рения, индия, таллия, галлия, кадмия и др. Электролизом
из расплавов производятся алюминий, магний, натрий.
Электрохимическими методами удалось осуществить производство
таких ценных металлов, как бериллий, церий, литий, сурьма,
получить безуглеродистый марганец и хром.
Защита металлов и их сплавов от коррозии путем
электроосаждения на их поверхности тонкого слоя другого металла
(хромирование, никелирование, оцинкование и др.) позволяет
ежегодно экономить многие тысячи тонн металла и
одновременно удлинять срок службы металлических изделий.
Из электрохимических процессов наибольшее
распространение в химической промышленности получили электролиз
водных растворов хлористого натрия (поваренной соли) с
одновременным получением хлора, каустической соды и водорода,
а также электролиз воды для получения чистых водорода и кис-
20* 307
лорода и электрохимические процессы получения некоторых
окислителей (хлораты, перхлораты, пербораты и др.).
Электрохимические производства относятся к числу весьма
энергоемких процессов. Например, на получение 1 т хлората
натрия (стр. 410) расходуется примерно 7000 кет • ч
электроэнергии. Отсюда следует, что для широкого развития
технической электрохимии необходимы достаточные ресурсы
дешевой электрической энергии.
ОСНОВНЫЕ СВЕДЕНИЯ ПО ЭЛЕКТРОХИМИИ
Проводники. Проводники электрического тока делятся на
проводники первого и второго рода.
В проводниках первого рода, к которым относятся металлы,
сплавы, уголь, графит и некоторые другие материалы, передача
электричества обусловлена движением свободных электронов,
причем прохождение электрического тока не сопровождается
химическими изменениями проводника. Электронной
проводимостью обладают также полупроводники.
Проводниками второго рода, называемыми также
электролитами, являются некоторые растворы кислот, солей и
оснований, а также расплавы ряда солей.
При прохождении постоянного электрического тока через
проводники второго рода происходят химические превращения
их — разложение и образование новых веществ.
Электропроводность проводников второго рода обусловлена движением
заряженных частиц — ионов.
Сопротивление проводников первого рода, за исключением
графита и аморфного угля, с повышением температуры
увеличивается; сопротивление же проводников второго рода при
повышении температуры понижается.
Явление электролиза. Разложение электролитов под
действием постоянного электрического тока с образованием новых
химических веществ на электродах, подводящих ток, называется
электролизом. Аппараты, в которых проводится электролиз,,
называют электролизерами (электролитические ванны или
просто ванны).
При прохождении постоянного электрического тока череа
электролиты положительно заряженные ионы (катионы)
движутся к катоду, отрицательно заряженные ионы (анионы) —
к аноду. На электродах эти ионы могут разряжаться. Ионы
щелочных, щелочноземельных и некоторых других металлов
в водных растворах не разряжаются на твердом кагоде, так
как потенциал их разряда значительно превышает потенциал
разряда ионов водорода Н+. Например, при электролизе
водного раствора хлористого натрия на аноде выделяется хлор, на
308
катоде — водород, а в растворе (около катода) образуется
едкий натр. Поскольку в водном растворе присутствуют также
ионы ОН", они будут разряжаться на аноде с выделением
кислорода. Доля электрического тока, расходуемого на разряд ио~
нов ОН~, будет тем меньше, чем ниже относительная
концентрация их в растворе.
Законы электролиза. При электролизе зависимость между
количеством выделившихся на электродах веществ и
количеством электричества, протекающего через электролит,
определяется двумя законами Фарадея.
Согласно первому закону Фарадея, количества веществ,
выделяющихся на электродах, прямо пропорциональны
количеству электричества, протекающему через электролит.
Согласно второму закону Фарадея, при прохождении одного
и того же количества электричества через различные
электролиты количества веществ, выделяющихся на электродах,
пропорциональны химическим эквивалентам этих веществ, т.е. для
выделения 1 г-экв любого вещества требуется затратить одно
и то же количество электричества. Установлено, что это
количество электричества составляет примерно 96 500 кулонов (чис-
. , , „. 96500
ло Фарадея, или 1 фарадеи), или ¦ 36Q0 = /Ь,Ь ампер-часа (а-ч).
Количество вещества, выделяющееся при прохождении через
электролит 1 а-ч электричества, называется электрохимическим
эквивалентом вещества.
При прохождении 1 а-ч электричества через водный
раствор поваренной соли, согласно второму закону Фарадея, на
аноде должно выделиться 1,323 г СЬ, а на катоде 1,4925 г NaOH
и 0,0376 г Н2.
Указанные количества веществ подсчитаны следующим
образом;
35,45
Для хлора =1,323
26,8
40,0
Для едкого натра = 1,4925
26,8
1,008
Для водорода = 0,0376
26,8
Здесь 35,45; 40,0 и L008 — грамм-эквиваленты хлора, едкого
натра и водорода.
Однако практически при электролизе водного раствора NaCl
1 а • ч электричества выделяет на аноде и катоде несколько
меньшие количества веществ, чем подсчитано, исходя из второго
закона Фарадея.
Это объясняется тем, что часть электрического тока
расходуется на побочные электрохимические процессы.
309
Выход по току. Зная количество полученного продукта
электролиза, можно подсчитать, какая часть электрического тока
израсходована полезно. Отношение фактического количества
выделенного вещества к количеству, которое должно было бы
выделиться согласно законам Фарадея, называется
коэффициентом использования тока. Это отношение, выраженное в
процентах, называется выходом по току.
Количество вещества G, которое теоретически должно
выделиться при электролизе согласно законам Фарадея, равно:
G — Е I-
zb,8
где Е — химический эквивалент данного вещества г;
I — сила тока, а;
х — время прохождения тока, ч.
Тогда выход по току (в %) составит:
Оф-26,8-100
—
тде Сф — количество фактически полученного вещества, г.
Например, при электролизе раствора поваренной соли в электролизере,
работавшем в течение 24 ч при силе тока 30 000 а, было получено 8500 л
электролитической щелочи, содержащей 120 г/л NaOH. Подсчитаем выход по
току (для щелочи).
Теоретически по законам Фарадея должно быть получено следующее
количество едкого натра (в кг):
40-30 000-24
G= 26,8-1000 — 1075 ^
Фактически получено:
120-8500
= Ш2° кг
Выход по току составит:
¦<] = -^--100 = -jjj^p ЮО =* 94,9%
Напряжение разложения. При протекании процесса
электролиза разность потенциалов между электродами должна
достигать определенной для данного электролита величины,
называемой напряжением разложения. Если напряжение тока,
питающего электролизер, меньше напряжения разложения данного
электролита, электролиз не происходит.
Напряжение разложения всякого электролита складывается
из разности потенциалов между анодом и раствором,
называемой анодным потенциалом, и разности потенциалов между
раствором и катодом, называемой катодным потенциалом. Анодный
и катодный потенциалы называются также потенциалами
разряда аниона и катиона.
310
Потенциал электрода. Минимальный потенциал
электрода, необходимый для разряда данного иона при
концентрации ионов 1 г-экв (точнее, при активности ионов, равной
единице), называется нормальным (или стандартным) электродным
потенциалом. Расположив нормальные потенциалы (?о),
измеренные при 25 °С по отношению к нормальному водородному
электроду*, в порядке возрастания их алгебраических величин,
получим ряд напряжений (табл. 23).
Таблица 23
Ряд напряжений, в воде при 25 °С
Электродная реакция
Na+ + е ^± Na
Mg2+ + 2е z^ Mg
Als+ + 3e i^ Al
Zn2+ + 2e zgi Zn
Fe2+ + 2e ^ Fe
№ + e ^ V2H3
Нормальный
электродный
потенциал ?о
в
-2,92
—2,71
—2,37
— 1,66
—0,763
—0,44
0,00
Электродная реакция
Cu2+ + 2е м
Hg2++2e-
2ОН" — 2е
2Вг~—2е z?
2СГ—2е z?
2F~—2e z^i
А Си
^Hg
*+~нг2о3
rBf.,j (жидк.)
: С13 (газ)
F3 (газ)
Нормальный
электродный
потенциал Eq
в
+0,337
+0,798
+0,401
+ 1,07
+ 1,36
+2,87
Из таблицы видно, что на электродах сначала будут
разряжаться те ионы, потенциал разряда которых имеет
наименьшую абсолютную величину. Это справедливо в условиях
обратимого процесса. С изменением концентрации данного иона в
растворе минимальный теоретически необходимый потенциал
(обратимый потенциал электрода) существенно изменяется.
Обратимые потенциалы разряда катиона и аниона, в
зависимости от концентрации ионов при 18 °С, вычисляются по
следующим уравнениям:
для катиона
с- с- , 0,058,
'¦]ёС
для аниона
где ?т — обратимый потенциал электрода при концентрации
ионов, равной С, в;
Ео — нормальный электродный потенциал, в;
п — число зарядов, которое получает или отдает данный
ион;
С—концентрация ионов, г-экв/л раствора.
* Потенциал нормального водородного электрода условно принят за
нуль, активность ионов — за единицу.
311,
Если концентрация ионов С=1, то потенциал электрода
становится равным нормальному электродному потенциалу, т. е.
Например, теоретическое напряжение разложения 1 н.
раствора хлористого цинка составит:
?т = 1,36 — (—0,763) = 2,123 в
где 1,36 — нормальный электродный потенциал для хлора, в;
—0,763 — нормальный электродный потенциал для цинка, в.
Перенапряжение. Потенциал электрода, необходимый для
разряда иона, в действительности больше теоретического
обратимого потенциала, определяемого по приведенным выше
уравнениям. Разность между действительным потенциалом
выделения ионов при электролизе и значением теоретического
(обратимого) электродного потенциала в состоянии равновесия
называется перенапряжением на данном электроде.
Практически под перенапряжением понимается разность
между фактической и теоретической величинами напряжения.
В случае разряда на электродах ионов металлов
перенапряжение относительно невелико, при выделении же на электроде
газообразных веществ перенапряжение более значительно и
является переменной величиной, зависящей от ряда условий:
материала электрода и характера его поверхности, плотности
тока*, температуры электролита, продолжительности процесса
электролиза. Так, перенапряжение выделения водорода на
гладкой платине 0,0063 в, на никеле 0,087 в, на ртути 0,860 в.
Перенапряжение можно уменьшить путем понижения
плотности тока, а также повышением температуры электролита
(при этом повышается проводимость электролита) и другими
приемами. Так, величину перенапряжения, зависящую главным
образом от материала электродов и состояния их поверхдости,
можно существенно снизить, например, путем предварительной
пескоструйной или наждачной обработки поверхности
электрода. При электролизе воды F0—80 °С, плотность тока 1000 а/м2,
концентрация NaOH 16%) со стальным катодом и никелевым
анодом удается снизить потенциал выделения водорода на 0,2 в
в результате пескоструйной обработки катода. При электролизе
растворов поваренной соли G5°С, плотность тока 1000 а/м2,
концентрация NaCl 310 г/л) указанная обработка стального
катода дает снижение катодного потенциала на 0,15—0,20 в.
Целесообразно также покрытие поверхности электрода губчатыми
металлами (железо, никель или его сплавы).
* Плотность тока определяется как сила гока (в а), отнесенная к
единице площади электродов. Обычно плотность тока выражают в а/м*.
Анодная плотность тока может отличаться от катодной.
312
Снижение потенциалов выделения катодных и анодных
продуктов электролиза на 0,2—0,4 s приводит к значительной
экономии электроэнергии (до 10% при электролизе растворов
хлорида натрия).
Исходя из величин перенапряжения, выбирают материал
электродов для проведения электрохимических процессов в
промышленных условиях.
Баланс напряжения. Напряжение на клеммах электролизера
определяется не только величинами напряжения разложения и
перенапряжения, но и дополнительным омическим
сопротивлением, возникающим при прохождении тока через электролит,
а также сопротивлением электродов, контактов, шин
(проводники первого рода).
Таким образом, общее напряжение Е на элекролизере в
реальных условиях электролиза можно выразить следующим
уравнением:
где ?а и Ек — фактические потенциалы анода и катода с
учетом перенапряжения, в;
I — сила тока, а;
Яэл. — сопротивление электролита, ом;
ИЕП — падение напряжения в проводниках первого
рода, в.
Для понижения общего напряжения на электролизере
стремятся повысить электропроводность электролитов (путем
повышения его концентрации и температуры), уменьшить по
возможности расстояние между электродами и др.
Расход электроэнергии при электролизе
Важнейшим показателем, определяющим экономику
электрохимических производств, является удельный расход
электроэнергии на весовую единицу получаемых продуктов. С расходом
электроэнергии непосредственно связан выход по току,
который, однако, еще не характеризует фактического
расхода электроэнергии на электролиз. Фактический расход
электроэнергии, обычно выражаемый в кет ¦ ч на 1 кг (или на
1 т) полученного вещества, равен произведению количества
затраченного электричества (в а ¦ ч) на напряжение на
электролизере (в в). Минимальный расход электрического тока
определяется законом Фарадея, минимальное
напряжение—напряжением разложения электролита. Отношение количества
электроэнергии, теоретически необходимого для ведения данного
процесса электролиза, к фактически затраченному в этом процессе
называется выходом по энергии.
313
Для определения расхода электроэнергии на 1 т продуктов
электролиза (например, хлора, едкого натра, водорода при
электролизе раствора NaCl) надо знать выход по току и
среднее рабочее напряжение на электролизере.
Расход электроэнергии при электролизе определяется по
формуле:
UP =-^-—-100.1000
л -ц
где W — удельный расход электроэнергии, кат ¦ ч\т,
К — электрохимический эквивалент, данного вещества,
г/а • ч;
V — среднее рабочее напряжение на электролизере, в;
т) — выход по току, %.
Пользуясь этой формулой, определим фактический расход
электроэнергии №ф на получение 1 т хлора, если среднее напряжение на
электролизере 3,35 в, а выход по току 96%:
1 3 35
1ГФ= j—223-—gg— -100-1000 = 2637 квт-ч
Если принять выход по току за 100%, то при теоретическом
напряжении разложения NaCl, равном 2,17 в, теоретический расход энергии WT на
1 т хлора составит:
П7'"= Г323'2'17'1000= 1640 квт'4
В этом случае выход по энергии составит:
1640-100
2637
¦^62,2%
Выход по энергии может быть вычислен более просто:
2,17-0,96-100
3,35
— 62,2%
При получении хлора электролизом раствора NaCl выход по энергии
колеблется от 60 до 66%
/. ЭЛЕКТРОХИМИЧЕСКОЕ ПРОИЗВОДСТВО ВОДОРОДА
И КИСЛОРОДА
Электролиз воды является самым простым способом
производства водорода и кислорода и используется при
необходимости высокой чистоты этих газов. Однако вследствие
значительной энергоемкости этого процесса в настоящее время для
получения больших количеств водорода (например, для
синтеза аммиака) электролиз воды почти не применяется. Большие
количества водорода получаются другими способами — конвер-
314
сией углеводородных газов (метана) и окиси углерода,
глубоким охлаждением коксового газа и др. (стр. 172 ел.).
Электролиз воды может быть использован также для
получения тяжелой воды, потребность в которой увеличивается с
развитием атомной техники. Однако в последние годы
электрохимические способы получения тяжелой воды вытесняются
другими более дешевыми методами.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА ЭЛЕКТРОЛИЗА ВОДЫ
Чистая перегнанная вода имеет ничтожную
электропроводность @,04-Ю ом~] • емг1 при 18°С), обусловленную низкой
степенью диссоциации Н2О на ионы. Поэтому при электролизе
воды используются растворы щелочей (NaOH, КОН) или
кислот (например, H2SO4), обладающие гораздо большей
электропроводностью, чем вода. При пропускании постоянного
электрического тока через такие растворы, в которые помещены
нерастворимые электроды (например, мягкое железо), происходит
разложение воды:
2Н2О -^ZL 2Н+ + 2ОЬГ
При этом на катоде выделяется водород:
2Н+ + 2е => Н2
на аноде кислород:
2ОН" — 2е =-^-О2-Ь НгО
Согласно закону Фарадея, при прохождении через
электролит 26,8 а-ч должно выделиться 11,2 л водорода и 5,6л
кислорода (объемы сухих газов при 0 °С и 760 мм рт. ст.).
Следовательно, для получения 1 ж3 водорода и 0,5 м3 кислорода
теоретически требуется:
2Ё^? = 2383,8 в-ч
Напряжение разложения. Теоретическое напряжение
разложения воды равно 1,23 в при 18 °С, с повышением
температуры оно уменьшается и при 80 °С составляет 1,18 в. Фактическое
напряжение на электролизере превышает теоретическое и
составляет 1,9—2,5 в. Расход электроэнергии можно уменьшить
путем снижения напряжения способами, изложенными на
стр. 312.
Практически температуру электролиза воды поддерживают
в пределах 70—80°С, так как при более высокой температуре
с уходящими из электролизера газами уносится большое
количество электролита, одновременно значительно увеличивается
коррозия аппаратуры под действием горячих щелочных или
кислых электролитов.
315
Для уменьшения внутреннего сопротивления электролита
пользуются растворами, имеющими большую
электропроводность. В настоящее время применяют почти исключительно
растворы щелочей в очищенной воде A6—18% NaOH или
25—29% КОН), обладающие достаточно высокой
проводимостью и менее агрессивные, чем кислые растворы.
Снижение внутреннего сопротивления достигается также в
результате уменьшения расстояния между электродами (до
5 мм в современных электролизерах).
Расход энергии. Расход электрической энергии W
(в кет • ч) пропорционален напряжению ? (в в) и количеству
электричества Q (в а • ч):
EQ
W =
1000
Количество электричества, необходимое для выделения 1 мъ
водорода и 0,5 м? кислорода, составляет 2383,8 а • ч.
Отсюда теоретический расход электрической энергии на 1 мъ
водорода и 0,5 м? кислорода (объемы сухих газов при
нормальных условиях) равен:
w — ' квпг ¦ч
где 1,23 — теоретическое напряжение разложения воды, в.
В современных ваннах для электролиза воды выход по
току близок к 100%, поэтому можно считать, что расход
энергии практически зависит только от рабочего напряжения на
электролизере и возрастает пропорционально этому напряжэ-
нию Среднее рабочее напряжение превышает теоретическое в
1,5—2 раза; следовательно, фактический расход электрической
энергии на 1 м3 водорода и 0,5 м3 кислорода (сухие газы при
нормальных условиях) составляет 4,5—6 кет ¦ ч.
Расход воды. Теоретически на образование 1 м3 водорода
и 0,5 м5 кислорода при нормальных условиях должно
расходоваться 804 г воды. Фактический расход воды в электролизере
больше вследствие уноса ее в виде паров с уходящими газами.
Количество уносимых водяных паров возрастает с повышением
температуры электролиза. С учетом испарения воды и других
потерь практически ее расход на получение 1 м3 водорода
составляет 0,85—0,9 л.
Качество воды. Поскольку в процессе электролиза воды
происходят ее разложение и унос в виде пара с газами, в
электролизер непрерывно или периодически добавляются новые
порции воды. Подаваемая на электролиз вода должна быть
предварительно очищена от механических примесей и растворимых
в ней минеральных солей. Прежде всего требуется удаление
316
хлоридов и карбонатов, так как даже при небольшом
содержании хлоридов в воде возрастает износ электродов, а наличие
карбонатов ведет к снижению электропроводности электролита.
Максимально допустимое содержание хлоридов составляет
25 мг/л, карбонатов не более 1% от содержания в электролите
NaOH (или КОН). Удаление солей производится перегонкой,
электроосмотической или ионообменной очисткой. Удельное
электрическое сопротивление электролита должно быть не
менее 60 ТЫС. ОМ • CM.
ЭЛЕКТРОЛИЗЕРЫ
При получении водорода и кислорода в электролизерах
необходимо отделять катодные продукты электролиза от анодных.
Для этого в электролизерах устанавливают асбестовую
диафрагму.
В качестве материалов применяют для катода главным
образом мягкое железо с шероховатой поверхностью, для анода—
мягкое железо с гальваниче-
ским покрытием слоем никеля.
Существуют электролизеры
различных типов:
монополярные с двойными плоскими
электродами, биполярные,
конструируемые по типу фильтр-
прессов, и др.
В монополярных
электролизерах электроды расположены
параллельно. Половина всех
Рис. 129. Принципиальная схема
биполярного электролизера:
электродов параллельно сое- , „ „ ,
r r „ I—анод; 2—катод; 3—биполярные электроды;
ДИНеНЭ С ПОЛОЖИТеЛЬНОИ ШИ- <—электролитическая ячейка.
ной электрической цепи
(аноды), другая половина соединена аналогичным образом с
отрицательной шиной (катоды). Напряжение на электролизере
определяется разностью потенциалов на одной паре электродов
(анод и катод) и не зависит от числа электродов; величина
тока пропорциональна поверхности всех электродов одноименной
полярности.
В биполярных электролизерах (рис. 129) ряд параллельно
расположенных электродов 3 последовательно включен в
электрическую цепь. Электрический ток подводится только к
крайним электродам — аноду / и катоду 2. С анода электричество
переносится катионами к первому промежуточному электроду,
где катионы разряжаются, принимая от этого электрода
электроны. Промежуточный электрод приобретает при этом
положительный заряд, т. е. становится анодом по отношению к
раствору, находящемуся по другую сторону промежуточного
электрода, и т. д. Таким образом, все промежуточные электроды
317
являются биполярными электродами, последовательно
включенными в электрическую цепь.
Крайние электроды ванны монополярны, т. е. один из них
является только анодом, другой — только катодом. Одна
сторона каждого биполярного электрода служит анодом, другая—
катодом.
Ток, протекающий в биполярных электролизерах, одинаков
на всех электродах и не зависит от их количества. Напряжение
на биполярном электролизере определяется разностью
потенциалов между соседними биполярными электродами и прямо-
пропорционально их количеству. При разности потециалов
между анодом и катодом, например 2 в, напряжение на
электролизере, изображенном на рис. 129, составляет 2-4 = 8 в.
Монополярные электролизеры. Для промышленного
электролиза воды получил применение монополярный электролизер
с двойными плоскими электродами, схематически
изображенный на рис. 130.
В стальном кожухе 1 (размеры 0,62x1,0x1,3 м),
установленном на опорах-изоляторах, подвешены двойные плоские
электроды 2, погруженные в электролит — водный раствор-
едкого натра.
Каждый электрод состоит из двух плоских железных листов
размером 1000X 1000 мм, толщиной 2 мм, расположенных
параллельно друг другу на расстоянии 6 мм. Для сохранения
плоской формы электродов при относительно небольшой их
толщине на листах имеется несколько штампованных ребер,,
сообщающих электродам необходимую жесткость. Расстояние
между такими двойными электродами составляет 50 мм.
При помощи стержней 7 электроды подвешены к газовым
колоколам 4, которые в свою очередь подвешены к стальному
кожуху /.
Колоколы 4 предстазляют собой узкие коробки, имеющие
небольшие куполы 5 с отверстиями 10 для выхода газа. К
нижнему краю каждого колокола 4 прикреплена диафрагма 3 из-
асбестовой ткани, окружающая электрод. Диафрагма отделяет
анодные продукты электролиза от катодных.
Постоянство уровня жидкости поддерживается
автоматически путем добавления электролита через питатель,
снабженный поплавком, который погружен в жидкость.
Водород, собирающийся в куполах 5, выходит через
отверстия 10 и собирается в одном общем сборном колоколе 9,
которым накрыты все куполы водородных колоколов. Из сборного
колокола водород направляется в сборный трубопровод.
Подобным же образом с помощью сборного колокола 8 собирают
и выводят кислород. Газосборные колоколы 8 и 9 нижними
краями погружены в электролит и образуют таким образом
затворы для газов.
318
В электролизере поддерживают температуру 60—70 С,
охлаждая аппарат снаружи холодной водой.
пислорвд
Рис. 130. Электролизер с двойными плоскими электродами:
/—стальной кожух ванны; 2—электроды; 3—асбестовый мешок (диафрагма); 4—газовые ко-
локолы; 5—куполы колоколов: 6—медные шины; 7—стальной стержень; в—сборный
колокол для кислорода; 9—сборный колокол для водорода; 10—отверстия в куполе колокола
для выхода газа.
Ток к электродам подводится никелированными медными
шинами 6. Шина прикреплена к листам электродов и проходит
через колокол 4; от колокола и электролита она изолирована
эбонитовой втулкой и цементной футеровкой, обеспечивающей
герметичность.
Электролизеры с двойными плоскими электродами строят
на нагрузку от 6 до 14 ка. Плотность тока на электродах
от 400 до 600 а/м7, напряжение 2,1—2,3 в.
Концентрация получаемого водорода 99,6—99,9%,
концентрация кислорода 99,2—99,6%.
Монополярный электролизер другого типа оборудован
электродами, составленными из полос, образующих решетку,
катоды заключены в диафрагменные мешки. Такие электролизеры
строят на нагрузку до 18 ка. При нагрузке более 10 ка
предусматривается охлаждение электролита в холодильниках.
Среднее рабочее напряжение при такой нагрузке составляет
2,19 в; расход электроэнергии на 1 м3 водорода примерно
5 кет • ч.
319
Концентрация получаемых газов: 99,88% Нг, 99,65% О2.
В США, Канаде, Австралии и некоторых других странах
получили распространение электролизеры, отличающиеся особой
конструкцией крышки, которая выполняется из монолитного
армированного бетона. В крышке имеются камеры для
раздельного сбора газов и крепления асбестовых диафрагм.
Катоды выполняются из стали, аноды — из никелированной стали,
форма катодов и анодов одинакова. Токоподводящие шины
проходят через специальные герметизированные отверстия в
крышке. Нормальная нагрузка составляет 10 ка. Обычно в
электролизерах имеется 15, 17 или 21 электродная пара.
Длина аппарата 1,3 м, высота 1,4 м, ширина в зависимости
от количества электродов 0,75—1 м. В случае необходимости
электролизер можно охлаждать путем орошения водой одной
из его боковых стенок.
Расход воды на 1 м3 водорода составляет 0,88 л.
Биполярные электролизеры часто конструируют по типу
фильтрпрессов.
Фильтрпрессный биполярный электролизер (рис. 131)
состоит из стальных рам / прямоугольного или круглого сечения,
между которыми помещены биполярные электроды 2,
отделенные от рам изолирующими и уплотняющими прокладками 3.
Рамы и электроды плотно прижаты к прокладкам при помощи
стягивающих болтов.
Для получения больших количеств водорода применяются
биполярные фильтрпрессные электролизеры с выносными
электродами.
Благодаря удачной конструкции электролитических ячеек
такие электролизеры более совершенны.
На рнс. 132 показано устройство электролитической ячейки
с выносными электродами. Каждая ячейка представляет собой
стальную раму /, сваренную из балок специального профиля.
К полке 2 рамы прикреплена диафрагма 3 из прочной
асбестовой ткани.
Каждый электрод состоит из основного электрода 4 —
сплошного стального листа и двух выносных электродов — дырчатых
(перфорированных) листов 5 и 6, прикрепленных к основному
электроду болтами. Один из выносных электродов работает
как анод, другой — как катод.
Выносной катод 5 укреплен на большем расстоянии от
основного электрода, чем выносной анод 6. Различие расстояний от
выносных электродов до основного электрода соответствует
различным объемам выделяющихся на электродах водорода и
кислорода. Газы выходят из ячейки по стальным трубам 7. Один
конец такой трубы вварен в отверстие рамы, другой —
приварен к стальным кольцам 8, образующим каналы для выхода
газов.
320
Между рамами и основными электродами, а также между
стальными кольцами 8 помещены изоляционные уплотняющие
прокладки.
Фильтрпресснып электролизер с выносными электродами в
собранном виде показан на рис. 133. В середине электролизера
расположена так называемая средняя камера, предназначенная
для охлаждения электролита, сбора газа и
отделения его от электролита. С обеих
сторон камеры расположены
электролитические ячейки /, зажатые стяжными
болтами 2 между концевыми нажимными
плитами 3.
Охлажденный электролит поступает из
средней камеры в ячейки через питающий
канал 17, соединенный с каждой
ячейкой.
кислород
водород '
i
1 .
( !
тз
Т i
i
н2|ог
1
1
1
Г
+
+
+
+
+
_
-
-
-
1
L
1
f
II
!
1
\
\
_
1
t
1
1
12
1
1
1
1, ,
\
> -
* -
й 2
1
1
1
1
1
1
1
,
+
+
+
+¦
+
1
вода
Рис. 131. Схема биполярного фильтрпрессно-
го электролизера:
1—стальные рамы; 2—биполярные электроды;
3—изолирующие и уплотняющие прокладки.
Рис. 132.
Электролитическая ячейка с выносными
электродами:
/—рама; 2—полка;
3—диафрагма; 4—сплошной электрод;
5, 6—выносные дырчатые
электроды; 7—трубы для выхода
газов; 8—кольца.
Образующиеся газы собираются в газосборных цилиндрах 5
и 6, соединенных трубой 7 с газосборными цилиндрами средней
камеры.
Газосборные цилиндры снабжены штуцерами 8 с
трехходовыми кранами 9, позволяющими направлять газы в
сборные коллекторы через трубу 10 или в атмосферу через трубу //.
Для предотвращения утечек электричества выходные
трубопроводы снабжены стеклянными изоляторами 12.
21-2848
321
Рис. 133. Фильтрпрессныи электролизер с выносными электродами:
/—электролитические ячейки; 2—стяжные болты; 3—нажимные плиты; 4—стальная тарельчатая пружина; 5. 6—горизонтальные газосборные
цилиндры; /—труба, соединяющая цилиндры с газовым куполом: 8—штуцеры, 9—трехходовые краны; 10—труба, ведущая в сборные коллекторы;
;/—труба для выпуска газа в атмосферу, /2—стеклянные изоляторы; lit—резервуар для поддержания постоянного уровня электролита; 14,
15—фарфоровые изоляторы; 16—фильтр для очистки электролита; 17— питающий канал; 18—трубопровод с вентилем для подачи воды; 19—шины для
подводки тока к крайним электродам.
Под газосборными цилиндрами имеется резервуар 13 для'
поддержания постоянного уровня электролита при вытеснении
его газом во время пуска электролизера.
Под средней камерой находится фильтр 16 для очистки
циркулирующего электролита от механических загрязнений
(асбестовые волокна диафрагмы и др.) .
Электролизер установлен на фарфоровых изоляторах 14, на
которые опираются нижние стяжные болты 2. На этих болтах
установлены диафрагменные рамы, каждая на двух
фарфоровых изоляторах 15. Ток подводится шинами 19 только к
крайним электродам. Ячейки электролизера включены
последовательно. Напряжение на каждой ячейке 2,18—2,2 в.
В качестве электролита обычно применяют раствор КОН,
температура электролизера поддерживается в пределах 80—
85 °С. Такой электролизер, рассчитанный на нагрузку 7500 а,
дает возможность получать около 500 м3/ч водорода.
Концентрация получаемого водорода 99,95%, концентрация
кислорода 99,8%; выход по току составляет 96%.
Обслуживание электролизера весьма просто и состоит В
периодическом добавлении воды и регулировании водяного
охлаждения для поддержания заданной температуры процесса.
Электролиз воды под давлением
Представляет интерес электролиз воды под давлением
8—20 ат. Электролизеры, работающие под давлением,
занимают меньше места и удобны для обслуживания. Энергия,
затрачиваемая на сжатие газов, не теряется, так как газы в
дальнейшем используются в процессах, проводимых под давлением
(например, заполнение баллонов сжатым газом и др.).
Для получения небольших количеств водорода и кислорода
(например, для сварки и резания металлов) применяют
установки, работающие под давлением 8—10 ат.
Кроме стационарных установок для электролиза воды под
давлением, разработаны и эксплуатируются передвижные
установки, смонтированные на платформах автомашин.
Электролизер питается постоянным электрическим током от динамо-
машины, приводимой в действие двигателем автомобиля.
Такого рода установки предназначаются для обслуживания
монтажных работ при прокладке труб, например газопроводов,
нефтепроводов и т. п.
Контроль производства
Производство водорода и кислорода путем электролиза воды
является взрывоопасным. Пределы взрываемости смесей
водорода с кислородом 4,65—93,9 объемн. % Н2, пределы
взрываемости смесей водорода с воздухом 4,1—74,2 объемн. % Н2.
21* 323
Поэтому необходим систематический контроль состава
получаемых газов, осуществляемый обычно автоматическими
газоанализаторами с сигнализирующими устройствами, действие
которых основано, например, на изменении теплового эффекта
при каталитическом сжигании водорода, на изменении
теплопроводности или плотности газов.
Основными примесями в газах являются пары воды и туман
электролита. От тумана газы освобождаются путем промывки
водой в особых аппаратах, промывная вода используется далее
для питания электролизеров.
2. ЭЛЕКТРОХИМИЧЕСКОЕ ПРОИЗВОДСТВО ХЛОРА И ЩЕЛОЧИ
МЕТОДЫ ПРОИЗВОДСТВА ХЛОРА
В природе хлор в свободном виде не встречается, зато
широко распространены его соединения с другими элементами,
главным образом хлориды NaCl, KC1, MgCU и др.
Хлор можно получать из его соединений химическими и
электрохимическими методами.
Химические методы основаны на окислении хлористого водорода
различными окислителями, например двуокисью марганца, или на разложении
хлористого натрия (калия) азотной кислотой.
До 90-х годов XIX в. хлор получали исключительно окислением
хлористого водорода двуокисью марганца (с последующей ее регенерацией) или
кислородом воздуха в присутствии катализаторов. Исторически и
экономически эти способы были связаны с производством соды по способу Леблана
(стр. 423).
Взаимодействие хлористого водорода с двуокисью марганца протекает
по реакции
4HCI + Мп62 = Ci, 4- МпС13 + 2Н2О
Для регенерации двуокиси марганца щелок, содержащий хлористый
маргагад, обрабатывали известковым молоком и продували воздух через
образовавшуюся массу:
MnCi, + Са(ОНK = Мп(ОН)а + СаС1.4
2Мп(.ОНK + О4 = 2МпО2 + 2Н2О
Регенерированную двуокись марганца возвращали в процесс для
окисления новых порций хлористого водорода.
Окисление хлористого водорода кислородом воздуха а присутствии
катализатора (солен меди) проводилось при температуре около 400СС:
4HCI + Оа = 2С12 + 2Н,0
В настоящее время химическими методами получают оч".нь небольшое
количество хлора.
При разложении хлористого натрия или калия азотной кислотой (так
называемый нитрозил-хлоридный способ) одновременно с хлором образуется
соответствующий нитрат » хлористый нитрозил:
3NaCi + 4HNO3 - Cia + 3NaNO3 + NOCI + 2H.,0
324
Ввиду возможности получения одновременно с хлором азотного удоо-
рения — натриевой или калиевой селитры без расхода соды или щелочи,
jtot способ представляет некоторый интерес. Однако он не получил
распространения в промышленности, так как производство нитратов
осуществляется другими более дешевыми способами (стр. 562), а побочно
образующийся хлористый нитрозил длительное время не находил применения. В
последнее время его предложено использовать в синтезе капролактама —
исходного продукта для производства полиамидов.
Электрохимические методы получения хлора заключаются
в электролизе водных растворов хлоридов щелочных металлов
или в электролизе расплавленных хлоридов с одновременным
получением в первом случае хлора, водорода и щелочи, во
втором — легких металлов и хлора.
Электролиз водных растворов хлоридов щелочных металлов
осуществляется в промышленном масштабе с 1890 г., причем
уже тогда обнаружились большие преимущества этого метода
перед химическими, а к концу первого десятилетия XX в. он
почти полностью вытеснил старые химические методы. В
России хлор начали получать в промышленном масштабе
электрохимическим путем с 1900 г.
Электрохимическим методом — путем электролиза водных
растворов хлоридов натрия пли калия — в настоящее время
производится более 99% хлора, вырабатываемого во всем мире.
Соли щелочных металлов легко растворимы в воде, растворы их
хорошо проводят электрический ток, что дает возможность
пропускать через небольшие объемы растворов большие количества
электричества при невысоком напряжении.
Электрохимический метод производства хлора достиг в
настоящее время высокого технического уровня. Этим методом
получают хлор высокой концентрации и чистоты и
одновременно весьма ценные продукты — щелочь (едкий натр или едкое
кали) и водород. При электролизе поваренной соли на 1 г
хлора образуется 1,139 г едкого натра и до 300 м3 водорода (объем
при 0°С и 760 мм рт. ст.).
Электрохимическое производство хлора и щелочи
осуществляется в промышленности двумя способами:
1) с применением твердого стального катода;
2) с использованием в качестве катода жидкой ртути.
В обоих случаях применяются графитовые аноды. Эти
способы принципиально отличаются процессами, протекающими на
катодах. На твердом катоде (стали) происходит разряд
ионов водорода, а в растворе образуемся щелочь, на ртутном
катоде разряжаются ионы натрия, образующего с ртутью
амальгаму.
Регенерация хлора. Возможно также получение хлора
путем регенерации хлористого водорода. Большие количества его
побочно образуются прь синтезе разнообразных органических
хлебопродуктов, производство и ассортимент которых непре-
325
рывно расширяются. В связи с чтим использование
отбросного хлористого водорода является очень важной проблемой, для
решения которой предложены различные способы:
I. Окислительное хлорирование углеводородов, например
бензола, на катализаторе:
1 катализатор
с6н6 -:- — о2 - нс1 —^—*¦ 0,н8с1 -;- н,о
2. Каталитическое окисление хлористого водорода:
катализатор
4НС1 4- О., *~ 2С12 — 2Н2О
3. Электролиз водных растворов хлористого водорода
(стр. 350).
Производство хлора и щелочи электролизом водных
растворов хлоридов обычно состоит из следующих основных
процессов:
1) приготовление н очистка раствора хлорида (рассола);
2) электроллз и осушка хлора, перекачивание хлора;
3) сжижение хлора;
4) выпаривание электролитической щелочи.
Кроме электрохимических методов получения щелочи
(одновременно с хлором), существуют также менее распространенные
и менее экономичные химические методы производства едких
щелочен — известковый и ферритный (стр. 476 ел.).
СВОЙСТВА И ПРИМЕНЕНИЕ ХЛОРА И ЩЕЛОЧИ
Хлор. В обычных условиях хло-р представляет собой газ
желто-зеленого цвета с резким удушливым запахом. При
нормальных условиях @°С и 760 мм рт. ст.) плотность
газообразного хлора составляет 3.214 г/л, плотность хлора по отношению
к воздуху равна 2,486.
При атмосферном давлении и —34,1 °С хлор превращается
в жидкость плотностью 1,5575 г/см3. При охлаждении до
—101,5 GC жидкий хлор переходит в твердое состояние.
Хлор хорошо растворим в воде, при 10 °С и 760 мм рт. ст.
в 1 л воды растворяется 9.97 г хлора. При более низкой
температуре из насыщенного раствора выпадают кристаллогидраты
С12-8Н2О. С повышением температуры его растворимость
значительно уменьшается. При растворении в воде хлор частично
гпдролизуется
С1., — Н„О -(—* НС1 -I- HOC1
с образованием так называемой хлорной воды. Равновесие этой
реакции устанавливается медленно (при 25°С через 48 ч).
326
Растворимость хлора в водном растворе поваренной соли
невелика и значительно понижается с увеличением
концентрации NaCl в растворе и повышением температуры (табл. 24).
Таблица 24
Растворимость хлора в воде и растворах NaCl
Концентрация
NaCl
г/л
0 (вода)
229
297
20
7,
2,
1,
°с
3
90
96
Растворимость, г/л
40
4,
1,
1,
°С
6
8
0
60 °С
2,6
0,95
0,54
8С
1,
0,
0,
°с
2
42
25
Хлор относится к наиболее активным химическим элементам
и может вступать в многочисленные химические реакции:
непосредственно соединяется с металлами, особенно с
щелочными, с металлоидами (сера, фосфор и др.); с окислами и
гидроокисями металлов хлор образует соли хлорокислородных кислот.
Влажный хлор, хлорная вода и соли хлорноватистой
кислоты (гипохлориты) являются сильными окислителями.
Взаимодействие хлора с водородом по реакции
Н2 + С12
2НС1
и темноте при обычной температуре практически не происходит.
На прямом солнечном свете и при высокой температуре смесч
хлора и водорода при содержании от 4 до 96% Н2 в хлоре
взрываются. С наибольшей силой взрывается смесь равных
объемов Н2 и С12. Однако при равномерном поступлении обоих
газов в замкнутое пространство они могут гореть друг в друге.
Этот процесс используется в промышленности для получения
хлористого водорода (стр. 399).
С органическими соединениями, в частности с
углеводородами, хлор легко вступает в разнообразные реакции
присоединения и замещения, образуя многочисленные хлорпроизводные.
Хлор обладает токсическими свойствами. Получасовое
вдыхание воздуха, содержащего 0,003—0,005 мг/л хлора, вызывает
сильное раздражение верхних дыхательных путей, а при
вдыхании воздуха, в котором содержится 0,1—0,2 мг/л хлора, может
наступить смерть. В очень небольших концентрациях хлор
оказывает эффективное бактерицидное действие (стр. 32).
Наряду с серной кислотой, аммиаком и содой хлор
является одним из важнейших продуктов химической промышленности
и производится в больших количествах. Мировое производство
хлора непрерывно возрастает: в 1960 г. оно составило 8,2 млн. т,
а в 1961 г. достигло 9 млн. т (данные без СССР).
327
Производство хлора в некоторых зарубежных странах
характеризуется следующими данными (в тыс. г).
1950 г. 1961г.
США 4206,6 4176,0
ФРГ 657,8 724,0
Япония 685,0 700,0
Англия 560,0 600,0
Франция 331,5 355,7
Канада 292,1 322,1
Италия 270,2 —
Хлор, обладающий токсическими и стерилизующими
свойствами, применяется для обеззараживания технической и
питьевой воды и сточных вод, для дезинфекции и т. п. На
окислительных свойствах хлора основано его применение в
текстильной и целлюлозно-бумажной промышленности в качестве
отбеливающего средства. Широкое применение как активны-г
окислители имеют хлорокислородные соединения — гипохлори-
ты, хлораты, перхлораты, двуокись хлора, белильная известь.
Соляная кислота (водный раствор хлористого водорода)
является одной из наиболее распространенных минеральных кислот
и находит разнообразное применение в различных отраслях
народного хозяйства. Хлор используется также для получения
трех- и пятихлористого фосфора, хлорокиси фосфора,
хлористого алюминия, хлорного железа и др.
Однако основным направлением использования хлора в
настоящее время является химическая переработка его в
различные хлороргаиические продукты, имеющие важнейшее значение
в современной технике.
Хлорированием метана получают хлористые метил н
метилен, хлороформ, четыреххлористый углерод, являющиеся
хорошими растворителями и исходными веществами для синтеза
ряда ценных продуктов, в том числе полимерных материалов.
Так, хлористый метил используется для синтеза кремнийорга-
нических полимеров, обладающих высокой термической
стойкостью и другими ценными свойствами.
Четыреххлористый углерод является полупродуктом для
производства синтетического волокна энант и ряда других
синтезов.
Путем хлорирования непредельных углеводородов получают
такие ценные растворители и органические полупродукты, как
тетрахлорэтан, трихлорэтилен, пентахлорэтан, тетрахлорэтилен,
дихлорэтан.
В результате гндрохлорирования ацетилена или омыления
дихлорэтана получается хлористый винил. Полимеры
хлористого винила (поливинилхлорид) и его сополимеры широко
применяются в качестве ценных электроизоляционных,
конструкционных и других материалов. Хлорированием полнвинил-
328
хлорида получают перхлорвиниловую смолу, которая
используется в производстве антикоррозионных лаков и
синтетического волокна хлорин, обладающего высокой химической
стойкостью и другими ценными свойствами.
Хлор используется также в производстве окиси этилена и
хлоропренового каучука (наирит), отличающегося высокой
стойкостью к действию масел, тепла, окислителей и других
химических реагентов.
Значительные количества хлора употребляются в
производстве ядохимикатов — ДДТ, гексахлорана и других препаратов,
широко применяемых в сельском хозяйстве как химические
средства защиты растений. Непрерывно возрастает
использование хлора в производстве синтетических моющих средств,
получаемых из продуктов сульфохлорирозания (действие СЬ п
SO2) или хлорирования парафиновых углеводородов С]2—C1S
(керосин и другие нефтяные фракции).
Большое количество хлора расходуется на производство
хлорбензола — одного из важнейших полупродуктов для
синтезов фенола, анилина, нитробензола и др. Существенным
потребителем хлора становится производство синтетического
глицерина через хлористый аллил, получаемый хлорированием
пропилена.
Кроме хлорпроиззодных, большое значение приобретают
различные фторхлорзамещенные метана и этана — так
называемые фреоны, применяемые в холодильных установках в
качестве малотоксичных, взрывобезопасных и не вызывающих
коррозию хладоагентов.
Перечисленные примеры далеко не исчерпывают применения
хлора в производстве разнообразных хлоропродуктов,
ассортимент которых непрерывно расширяется, а объем производства
возрастает.
Сравнительно новой областью использования хлора
является так называемая хлорная металлургия. Высокотемпературным
хлорированием руд или их концентратов (например, в
расплаве) получают такие ценные металлы, как титан, ниобий и др.
Хлорированием кремннйсодержащего сырья производится че-
гыреххлорнстый кремний, из которого получают один из
важнейших полупроводников — металлический кремний.
Едкий натр (каустическая сода). Чистый твердый едкий натр
NaOH представляет собой белое или слегка окрашенное
непрозрачное вещество (плотность 2,13 г/см3), сильно гигроскопичное
и потому расплывающееся на воздухе. Температура плавления
безводного едкого натра при атмосферном давлении 328 °С,
плотность расплава при 340—500 °С составляет 1,8—1,9 г/см6
(теплота плавления NaOH 40 ккал/кг). Под давлением ниже
атмосферного и в присутствии примесей (в том числе воды)
температура плавления едкого натра снижается. Так, при со-
329
держании 1,6% Na2CO3 и 0,9% влаги температура плавления
едкого натра понижается до 310 °С. Температура кипения
чистого едкого натра 1388 °С (при 760 мм рт. ст.).
Едкий натр полностью растворим в воде; с повышением
температуры его растворимость возрастает (см. также рис. 149.
стр. 375):
Температура, °С 10 20 30 50 100
Концентрация NaOH, % .... 48,6 52,0 54,4 60,7 77,3
Плотность водных растворов NaOH при 30 °С составляет:
Концентрация NaOH,% ... 20 30 40 50
Плотность раствора, г/см3 . . 1,21 1,32 1,42 1,52
При растворении едкого натра в воде, а также при
разбавлении водных растворов выделяется большое количество тепла,
что в некоторых случаях приводит даже к закипанию раствора.
Например, при растворении 40 г NaOH в 54 г воды с
образованием 42,5%-ного раствора NaOH выделяется около 13,4 кка.?
тепла (теплота растворения). Соответствующее количество
тепла должно быть подведено при выпаривании (обезвоживании)
растворов едкого натра.
Температура кипения растворов NaOH сильно возрастает
с повышением концентрации:
Концентрация NaOH, % 10 20 30 40 50 60 70
Температура кипения
раствора при 1 шпм,
"С 103 107,7 116,2 128,0 144,0 162,0 181,5
В органических растворителях (метиловый, этиловый спирт,
глицерин) едкий натр также растворяется.
Едкий натр являе1ся одной из самых сильных щелочей и
широко используется в разнообразных процессах
нейтрализации и гидролиза. При нейтрализации кислот едким натром
образуются натриевые соли, как правило, хорошо растворимые
в воде.
Растительные и животные жиры сравнительно легко омы-
ляются едким натром с образованием солей жирных кислот
(мыла). На этом свойстве едкого натра основано его
применение в мыловаренной промышленности, которая является
крупным потребителем каустической соды. Однако с развитием
производства синтетических моющих средств потребление
каустической соды (и пищевых жиров) в мыловарении будет
сокращаться, что имеет важное значение, так как каустическая сода
весьма широко применяется во многих отраслях
промышленности, а потребление ее непрерывно возрастает.
330
Наибольшее количество каустической соды (до 30% ее
мировой выработки) расходуется в производстве искусственных
волокон (вискозный шелк). Качество волокна и получаемой из
него ткани существенно зависит от чистоты едкого натра.
Полому для этих целей применяется преимущественно более
чистая каустическая сода, получаемая ртутным электролизом.
Значительные количества каустической соды используются
в текстильной промышленности—в процессах отбеливания и
отделки (мерсеризации) тканей, в целлюлозно-бумажной
промышленности каустическая сода применяется для обработки
целлюлозы и бумажной массы.
В химической промышленности, потребляющей большие
количества каустической соды, она используется в производстве
органических красителей и полупродуктов, пластических масс,
инсектицидов, лекарственных веществ, синтетического фенола,
глицерина, хлористого винила и во многих других процессах
основного органического синтеза, газоочистке и др. Для
очистки нефти, нефтепродуктов, минеральных масел также
расходуются большие количества каустической соды.
Она является распространенным реагентом во многих
гидрометаллургических процессах — при получении окиси алюминия
(глинозема) из бокситов, при извлечении из минерального
сырья (выщелачивании) ряда ценных металлов (Zr, Tlh, V
и др.) и т. д. Едкий натр может быть использован для обессе-
ривания стали в расплавленном состоянии.
Из этого далеко не полного перечня областей применения
каустической соды следует, что она, наряду с хлором,
относится к важнейшим видам химической продукции.
В 1960 г. мировое производство каустической соды
электрохимическими и химическими способами составило примерно
11 млн. т (без СССР). Объем производства каустической соды
¦:, некоторых странах приведен ниже (в тыс. т):
США .
Англия
Япония
ФРГ
Франция
Италия
Канада .
I960 г.
4500
840
847
776
500
... 426
338
1961 г.
4300
922
810
—
446
378
СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА ХЛОРА И ЩЕЛОЧИ
Основным сырьем для получения электролитического хлора
11 щелочи является поваренная соль. Природная поваренная
соль, кроме NaCl, содержит примеси хлоридов и сульфатов
кальция, магния и др. Помимо поваренной соли, сырьем для
производства хлора могут служить хлориды калия и магния.
331
Хлористый натрий залегает в земной коре в виде пластов
каменной соли (минерал галит), состоящей почти целиком из
NaCl. Большие количества хлористого натрия содержатся в воде
океанов, морей и некоторых озер, а также в соляных
источниках, образуемых подземными водами, растворяющими соли
горных пород.
Поваренная соль, выделяющаяся в твердом виде на дне
соляных озер и неглубоких морских заливов, называется
самосадочной солью. Естественные рассолы в озерах и заливах
называют рапой.
Методы добычи поваренной соли зависят от условий ее за-
легания. Добычу твердой каменной соли ведут горными
выработками или получают из нее подземный рассол, нагнетая воду
в пласт каменной соли через буровые скважины.
Крупнейшие месторождения каменной соли находятся в
Донбассе (Славянское и Артемовское месторождения), богатые
месторождения каменной соли имеются также на Урале (Бе-
резниковско-Соликамское месторождение), в районе Усолья.
Иркутска, Илецка и др. Илецкая соль содержит свыше 99%
NaCi и по чистоте не имеет себе равных.
Источниками самосадочной соли в Советском Союзе
являются многочисленные озера: Баскунчак, Эльтон, Павлодарские
озера и др. По запасам самосадочной соли Советский Союз
занимает первое место в мире.
Во многих районах Советского Союза имеются подземные
рассолы с большим содержанием хлористого натрия. Они
используются непосредственно на месте их добычи или
перекачиваются потребителям на значительные расстояния. В
подземных рассолах Березниковского и Славянского месторождений
содержится (в г/л):
NaCl 300—310 Mg2+ . . . 0,14—1,4
Са2+ около 2 БОГ ¦ ¦ • 3,9—4,4
Подземные рассолы на глубине 1000—1500 м имеются в
районе Москвы и г. Горького. В Московском месторождении
(Боенская скважина) содержание NaCl составляет 225 г/л.
В настоящее время на заводах-потребителях,
расположенных вблизи месторождений каменной соли, рассолы
приготовляются путем подземного растворения (выщелачивания)
пласта каменной соли методом гидровруба. Этот метод
экономичен и дает возможность получать рассолы, содержащие
до 310 г/л NaCl.
В воде морей и океанов содержится до 35 г/л NaCl, а также
значительные количества сульфатов магния, кальция и других
солей. Добыча соли из морской воды производится в районах
с сухим, жарким климатом, в мелких заливах, где вследствие
332
естественного испарения воды образуется концентрированный
раствор, из которого далее выделяется поваренная соль.
В странах, не имеющих собственных месторождений
поваренной соли и вынужденных импортировать ее, для нужд
содовой и хлорной промышленности разработаны способы
концентрирования морской воды с использованием ионообменных смел
п постоянного электрического тока.
ПРИГОТОВЛЕНИЕ И ОЧИСТКА РАССОЛА
Подземные рассолы — естественные или искусственные —
по трубопроводам направляют в сборники к месту потребления.
На заводах, использующих твердую поваренную соль для
приготовления рассола, растворение ее производят обычно в
заглубленных (подземных) баках. В них сверху загружается соль,
а снизу подается вода, проходящая через слой соли и
растворяющая ее. Циркуляция рассола, осуществляемая
центробежным насосом, ускоряет растворение загруженной в бак соли.
Концентрированный рассол, содержащий 310—315 г/л NaCl,
подается из баков в сборники сырого рассола, поступающего
затем на очистку.
Сырой рассол содержит механические примеси (песок и др.),
а также растворимые в воде сульфаты кальция, магния и
другие соли. Поэтому для нормального протекания
процесса электролиза и достижения высоких выходов по току
рассол должен быть тщательно очищен от примесей.
Очистку рассола проводят обычно содово-каустическим
способом. Для осаждения ионов Са2+ рассол обрабатывают
раствором кальцинированной соды в рассоле:
Са2+ + Na2CO3 > СаСО3 + 2Na+
Для удаления ионов магния сырой рассол смешивают с
обратным рассолом (стр. 376), содержащим 2—2,5 г/л NaOH:
Mg2+ + 2NaOH » Mg(OH)., + 2Na+
Выпадающие в осадок карбонат кальция и гидроокись
магния удаляются из раствора путем отстаивания и последующего
отфильтровывания.
Для частичного использования щелочи, содержащейся в
обратном рассоле, и сокращения расхода соляной кислоты на ее
нейтрализацию целесообразно для осаждения ионов Са2+
вместо кальцинированной соды применять карбонизацию
обратного рассола, используя отбросный углекислый газ, например,
известково-обжигательных печей (стр. 433).
Если хлорный завод расположен близко от содового, очистка
рассола для обоих заводов производится
содово-известковым способом, по которому для удаления ионов магния рассол
обрабатывают известковым молоком:
Mg2+ + Са(ОНJ > Mg(OHJ + Саг+
333
Кальций осаждают кальцинированной содой. В этом случае
иа хлорном заводе производится только подогрев и
фильтрование (доочистка) рассола.
Для полноты очистки рассол подогревают до 40—50 °С, а
реактивы вводят в некотором избытке. Нейтрализацию избытка
NaOH производят соляной кислотой, добавляемой в рассол в
таком количестве, чтобы после нейтрализации он содержал не
менее 0,05 и не более 0,1 г/л NaOH, а избыток Na2CO3
составлял 0,2—0,3 г/л.
Ранее очистка рассола проводилась преимущественно
периодическим способом в нескольких баках. Этот способ громоздок,
для осуществления его требуются большие емкости,
автоматизация процесса невозможна, что затрудняет поддержание
постоянства состава рассола и др. На современных хлорных и
содовых заводах принята непрерывная очистка рассола.
Схема непрерывной очистки рассола с его карбонизацией
изображена на рис. 134. Сырой рассол C10—315 г/л NaCl),
подогретый в теплообменнике 4 до 40—50 °С, поступает в
смеситель 6. Обратный рассол из сборника / подается насосом
в верхнюю часть скруббера 3, заполненного насадкой.
Снизу в скруббер противотоком подается углекислый газ.
Выходящий из скруббера обратный рассол, содержащий
некоторое количество соды, поступает в сборник 2, откуда идет в
теплообменник 5. Отсюда подогретый до 40—50 °С рассол посту-
ОЬратныи
рассол
Пар
(Неё
Но
¦щ.
9
6
? ,
нденсат
Шлам на
промывку'
Рассол ha
электролаз
Рис. 134. Схема непрерывной очистки рассола с карбонизацией:
/, 2, л"—сборники; 4. 5, 9—теплообменники; 6—смеситель; 7—отстойник: /'—иасадочные
вертикальные фильтры.
334
пает в смеситель 6 и далее в отстойник 7, куда для
нейтрализации избытка щелочи подается соляная кислота. Осветленный
раствор из отстойника 7 непрерывно поступает в сборник 8 и
после подогрева в теплообменнике 9 до 70—80 °С направляется
в насадочные вертикальные фильтры 10. Отсюда очищенный
осветленный рассол подается на электролиз.
Отстаивание рассола от образовавшихся осадков
производится в непрерывно действующих отстойниках-резервуарах
диаметром 18 м, высотой 6,5 м с днищем, имеющим уклон к центру.
Отстойник снабжен медленно вращающейся гребковой
мешалкой, перемещающей осадок к расположенному в центре днища
выходному отверстию. Содержание взвешенных частиц в
рассоле, выходящем из отстойника, составляет около 20 мл!л.
Окончательная очистка рассола от примесей осуществляется в
вертикальных насадочных фильтрах диаметром 3 м, заполненных
зерненым антрацитом или мраморной крошкой, высота слоя
насадки 0,9—1,2 м.
Для непрерывной очистки рассола может найти применение
аппарат типа ЦНИИ МПС, совмещающий реактор и
осветлитель. Аппарат работает по принципу укрупнения
мелкодисперсных частиц осадка, образующихся при прохождении через слой
рассола со взвешенным в нем ранее образовавшимся осадком.
Восходящий поток очищенного рассола поддерживает во
взвешенном состоянии частицы осадка, играющие роль «затравки»,
на которой нарастают осаждающиеся частицы примесей.
Аппарат не имеет мешалки и весьма прост по своей конструкции и
в обслуживании.
Интенсификация процесса непрерывной очистки рассола
достигается в аппарате вертикального типа (ОВР), в котором
происходит фильтрование через взвешенный слой осадка. Этот
аппарат имеет большую производительность, чем описанный
выше, прост и легко монтируется.
Для электролиза в ваннах с ртутным катодом применяется
рассол, содержащий 305—310 г/л NaCl. Его приготовляют
путем донасыщения твердой солью анолита из ртутных ванн.
Чтобы очистку можно было вести в среде, не оказывающей
коррозионного действия на аппаратуру, и наиболее полно осадить
примеси, анолит подкисляют соляной кислотой и удаляют из
него при разрежении основное количество растворенного хлора
(обесхлориванне). Остальной хлор удаляется из анолита при
последующей продувке его сжатым воздухом и обработке
сульфидом натрия. При этом рассол освобождается и от
растворенной в нем ртути и примесей тяжелых металлов, которые
оказывают вредное влияние на процесс электролиза с ртутным
катодом (наиболее вредно присутствие в рассоле хрома,
молибдена, ванадия, германия). Обесхлоренный анолит
(концентрация NaCl 260—275 г/л), имеющий температуру 60—70 °С, до-
335
насыщают твердой поваренной солью, очищают обычными
методами от примесей и дважды фильтруют.
Примеси ионов кальция (до 1—1,5 г/л), магния (до 0,1 г(л)
и сульфатов (до 4—5 г/л) не мешают процессу электролиза
с ртутным катодом. Поэтому во многих случаях удаляют лишь
часть этих примесей из рассола или очищают только часть
рассола, следя за тем, чтобы количество примесей в нем не
превышало указанных выше пределов.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА ЭЛЕКТРОЛИЗА
С ТВЕРДЫМ (СТАЛЬНЫМ) КАТОДОМ
В водном растворе поваренной соли содержатся ионы
натрия, хлора, водорода и гидроксильные ионы:
NaCl = Na+ + СГ
При прохождении через водный раствор постоянного
электрического тока к аноду движутся отрицательно заряженные
ионы С1~ и ОН", к катоду — положительно заряженные ионы
с наименьшим потенциалом разряда. Потенциалы разряда ионов
зависят также от условий электролиза.
Катодный процесс. Нормальный потенциал разряда иона
Na+ на твердом катоде в нейтральном растворе NaCi равен
—2,71 в. Обратимый потенциал разряда водорода в тех же
условиях составляет только —0,415 в. Вследствие такого большого
различия потенциалов на твердом катоде, даже при
значительном перенапряжении, будут разряжаться только ионы
водорода*
2Н+-|-2е = 2Н
с последующим выделением водорода в виде газа:
2Н *Н2
Ионы Na+ разряжаться не будут.
Выделение водорода нарушает равновесие
=1 Н+ + ОН-
вследствне чего_ все новые количества воды диссоциируют на
ионы Н и ОН . Около катода ионы ОН~ и Na+ образуют
раствор едкого натра.
* По некоторым современным представлениям, в щелочном растворе на
катоде происходит разложение молекул воды, которое в суммарном виде
может быть представлено следующим уравнением:
Н
336
Анодный процесс. На аноде возможен разряд ионов О или
ОН". В нейтральной среде при 18 °С обратимый потенциал
разряда ионов ОН" равен +0,84 в. Обратимый потенциал разряда
нонов С1~ в насыщенном растворе NaCl при той же
температуре равен +1,33 в. Можно было бы ожидать, что на аноде
будет происходить разряд ионов ОН" с выделением кислорода:
4ОН- — Ае = 2Н2О + 0.2
Однако вследствие высокого перенапряжения кислорода
(даже на платинированной платине, на которой хлор разря-
жае_тся почти без перенапряжения) потенциал разряда ионов
ОН" становится более положительным, чем потенциал разряда
ионов С1~. Поэтому из концентрированных растворов хлористого
натрия на платинированной платине разряжаются только ионы
.хлора с последующим выделением молекулярного хлора:
2СГ — 2е = 2С1
2С1 > С\,
Таким образом, при электролизе водных растворов
хлористого натрия на твердом катоде в качестве первичных
продуктов образуются едкий натр и водород, а на аноде — хлор.
Водород и хлор выделяются в виде газов, едкий натр остается
в растворе.
Побочные процессы. В промышленности при электролизе в
качестве анода применяют графит. Ввиду значительного
перенапряжения хлора на графите величины потенциалов разряда
ионов СГ и ОН~ взаимно приближаются, а потому
одновременно с хлором на аноде может выделиться кислород:
2ОН- — 2е = Н2О + -^- О.,
Выделяющийся на аноде хлор частично гидролизуется в
растворе:
С12 + Н2О -<—* НОС1 + НС1
Образующаяся при этом хлорноватистая кислота слабо
диссоциирована на ионы. Однако она может взаимодействовать
с ионами ОН :
НОС1 + ОН" ^=2; ОС1- + Н.,0
Потенциал разряда иона ОС1 па аноде значительно ниже,
чем иона хлора, поэтому ионы ОС1 , даже при незначительном
содержании их в растворе, будут разряжаться на аноде вместе
с ионами СГ. При разряде на аноде ионы ОСГ в присутствии
нонов ОН" окисляются:
6ОСГ + 6ОН~ — бе = 2СЮ:Г + 4СГ + 30 + ЗН2О
22-2848 337
Выделяющийся при этом кислород может реагировать с
углеродом графитового анода:
С + О = СО
2СО -f О2 == 2СО2
Присутствующая в растворе хлорноватистая кислота
взаимодействует также с образующейся около катода щелочью:
HOCl -f NaOH = NaOCl + Н2О
Кроме того, в растворе могут образовываться другие соли
хлорокисдородных кислот:
NaOCl + 2HOC1 > NaClO3 + 2HC1
2NaOCl > NaC10a + NaCl
NaC102 + NaOCl > NaC103 + NaCl
Если не отделять катодные продукты электролиза от
анодных, то едкий натр (катодный продукт) будет реагировать с
хлорноватой и соляной кислотами (анодными продуктами):
2НСЮ3 4- 4НС1 + 6NaOH > 2NaC103 + 4NaCl -f 6Н2О
Таким образом, вторичные реакции приводят к образованию
нежелательных побочных веществ —¦ гипохлоритов и хлоратоЕ
и к непроизводительному расходу электроэнергии. Образование
гипохлорнтов н хлоратов вызывает разрушение графитовых
анодов н загрязнение получаемого хлора значительными
количествами окиси и двуокиси углерода.
Чтобы предотвратить протекание побочных реакций и
смешение хлора с водородом, образующих взрывоопасные смеси,
необходимо отделять катодные продукты электролиза (щелочь
и водород) от анодных продуктов (хлора). Это достигается при
установке в электролизере пористой перегородки —
фильтрующей диафрагмы (стр. 340)
Если вести электролиз хлорида натрия (или калия) без
диафрагмы при температуре не выше 20 °С, создаются условия,
благоприятные для образования гипохлоритов. При
температуре около 70—75 °С без диафрагмы в слабокислой среде и при
большом содержании в электролите исходных хлоридов
образуются преимущественно хлораты.
Напряжение разложения. Теоретический потенциал
разложения насыщенного водного раствора хлористого натрия с
образованием хлора, едкого натра и водорода определяется как
алгебраическая разность обратимых анодного и катодного
потенциалов:
Е= 1,33 —(—0,84) = 2,17 в
где 1,33 — обратимый потенциал анода в насыщенном
растворе NaCl, в;
—0,84 — обратимый потенциал катода в электролизере с
твердым катодом, в.
338
Фактическое напряжение разложения водных растворов
NaCl всегда больше теоретического (стр. 312).
В электролизерах с диафрагмой и твердым (стальным) ка
тодом оно составляет от 3,35 до 3,8 в, в зависимости от усло-
р,ий процесса электролиза.
Степень разложения NaCl. При электролизе водных
растворов хлористого натрия только часть его превращается в едкий
натр.
Неразложившаяся часть NaCl удаляется из электролизера
вместе с раствором каустической соды (электролитическим
щелоком).
Отношение количества NaCl, превратившегося при
электролизе в едкий натр, к количеству хлорида натрия, поступившему
в виде раствора на электролиз, называется степенью
разложения хлорида натрия.
При нормальном режиме работы электролизера с
диафрагмой степень разложения хлористого натрия колеблется в
пределах 45—55%.
Выбор оптимальных условий электролиза. Для уменьшения
удельного расхода электроэнергии в процессе электролиза (на
весовую единицу получаемого продукта) стремятся к созданию
таких условий процесса, при которых достигается
максимальный выход по току и возможно более низкое рабочее
напряжение на электролизере.
Важнейшими условиями достижения высоких выходов по
току являются следующие:
1. Возможно более полное разделение анодных и катодных
продуктов электролиза; при этом уменьшается возможность
образования нежелательных побочных продутов (пшохлори-
тов, хлоратов и др.).
2. Концентрация хлористого натрия в растворе, близкая к
насыщению C10—315 г/л). Чем концентрированнее раствор
\таС1, тем меньше растворимость хлора и, следовательно, ниже
равновесная концентрация НОС1 и затруднен разряд ионов
ОС1 . Кроме того, в концентрированных растворах электролита
затрудняется перенос щелочи в анодное пространство и
образуется меньше побочных продуктов электролиза.
3. Проведение электролиза при высокой температуре
электролита (85—97 °С), что позволяет снизить растворимость хлора
а электролите, уменьшить проникание ионов ОН~ в анолит и
тем самым увеличить выход хлора и едкого натра. С
повышением температуры и концентрации электролита уменьшается
также перенапряжение при разряде ионоз на электродах и
сопротивление электролита.
4. Материал анода должен обладатьнаименьшим
перенапряжением для разряда ионов С1 . материал катода — для разряда
ионов Н+.
22* 339
ДИАФРАГМЫ
В электролизерах с твердым (стальным) катодом анодное
и катодное пространства разделены фильтрующей диафрагмой.
Диафрагма препятствует механическому смешению анодных
и катодных продуктов электролиза (анолита и католнта), но
не оказывает большого сопротивления прохождению
электрического тока.
Раствор хлористого натрия непрерывно поступает в
анодное пространство и проходит через диафрагму в направлении
от анода к катоду, что препятствует обратному движению ионов
ОН" от катода к аноду. Однако даже при таком разделении
продуктов электролиза не удается полностью предотвратить
проникание ионов ОН" в анодное пространство электролизера.
Диафрагма, обычно наносимая на катод, должна быть
стойкой к действию щелочи (католита) и к действию слабокислого
анолита, содержащего растворенный хлор. Кроме того,
диафрагма должна обладать определенной протекаемостью и быть
достаточно дешевой. В настоящее время диафрагмы изготовляют
из асбеста, удовлетворяющего перечисленным требованиям.
В электролизерах старых конструкций диафрагма
выполнялась из двух-трех слоев асбестовой бумаги толщиной 0,7 мм,
накладываемой на катодную сетку (электролизеры с бумажной
диафрагмой).
В диафрагменных электролизерах современных конструкций
катод имеет развитую поверхность и сложную конфигурацию.
Обкладка таких катодов асбестовой бумагой практически
невозможна, поэтому диафрагму осаждают непосредственно на
катоде путем насасывания на его поверхность слоя асбеста.
Такие электролизеры получили название электролизеров с
осажденной диафрагмой.
Покрытие катода сложной конфигурации осажденной
асбестовой диафрагмой производится путем насасывания на его
поверхность тонкого слоя асбестового волокна. Для осаждения
диафрагмы на катоде в анодное пространство электролизера
заливают заранее приготовленную пульпу, содержащую 6—8 г/л
сухого асбестового волокна, 60—80 г/л NaOH и 200—240 г/л
NaCI, затем создают вакуум в катодном пространстве, и пульпа
просасывается через поверхность сетчатого катода. При этом
асбестовое волокно осаждается тонким слоем на поверхности
катода, который выдерживают в пульпе до достижения
требуемого вакуума, что указывает на безупречно плотное покрытие
асбестом всей поверхности катодного комплекта и достаточную
толщину покрытия. После этого пульпу удаляют из
электролизера, а диафрагму подсушивают сначала на воздухе в
течение 25—30 мин, затем в течение 6—7 ч непрерывно
просасывают через нее горячий воздух (80—110°С).
340
Протекаемость диафрагмы и ее сопротивление
прохождению электрического тока изменяются в процессе электролиза.
При длительной работе электролизера протекаемость
диафрагмы уменьшается, что ведет к снижению выхода по току.
Для нормальной работы диафрагмы и достижения высоких
выходов по току необходима тщательная очистка рассола,
подаваемого на электролиз (стр. 333), иначе поры диафрагмы
быстро засорятся частицами примесей и уменьшится ее про-
текясмость.
ДИАФРАГМЕННЫЕ ЭЛЕКТРОЛИЗЕРЫ
Современные промышленные электролизеры с фильтрующей
диафрагмой работают непрерывно, с проточным электролитом.
В зависимости от устройства и расположения диафрагмы в
электролизере различают электролизеры с вертикальной или
горизонтальной фильтрующей диафрагмой.
Электролизеры с горизонтальной диафрагмой. На рис. 135 показам
схема такого электролизера. Раствор поваренной соли (рассол) поступает в
электролизер по трубке, опущенной в электролит. Электролит движется из
Рассол
\тийг=
Электролитический
щелок
Рис. 135. Электролизер с горизонтальной
диафрагмой:
/—анод'. 2—диафрагма; 3—катод; 4—анодное
пространство; 4—катодное пространство.
анодного пространства 4 в катодное пространство 5 через диафрагму 2.
Образующийся около катода 5 раствор щелочи (электролитический щелок)
непрерывно отводится по трубе снизу, водород — сбоку. Выделяющийся на
графитовом аноде / хлор отбирается сверху.
Работа электролизера с горизонтальным расположением диафрагмы
основана па принципе естественного расслоения электролита по плотности.
Чем ближе слой электролита к катоду, тем выше концентрации щелочи.
Расположение диафрагмы между горизонтальными анодом и катодом
предотвращает проникание водорода из катодного пространства в аиодиое
и задерживает движение шелочи от катода к аноду.
Вследствие громоздкости и неудовлетворительных показателен по
напряжению и расходу электроэнергии электролизеры с горизонтальной
диафрагмой в настоящее время почти не применяются.
341
Электролизеры с вертикальной диафрагмой. Электролизерам
лого типа длительное время придавали форму цилиндра.
В цилиндрическом электролизере Х-2 асбестовая диафрагма (бумага
или картон), плотно чрилегающая к дырчатому перфорированному
стальному катоду, отделяет анодное пространство от катодного. Графитовые
аноды соединены с положительным полюсом источника постоянного тока,
катод -- с отрицательным иолюссм. Очищенный раствор поваренной соли
Zjtop
7CC0J?
2~-
r
t
A
/'
j:.
t
t
' -
< (
I
i
\
i
1
-
if-
\ ;
/
F4 if'
~\ <'
у *
~-\ A
г >"
ь ?
3^ Ai
\
i
1
I
i i
¦
'\
v>
¦\
:>
: 4
:>
/?
-2
-6
Электролитический
щелон
Рис. 136. Вертикальный диафрагменный
электролизер с внутренним катодом:
—аноды; 2—наружный катод; 3—внутренний катод;
—диафрагма; 5—анодное пространство; 6—катодное
пространство.
непрерывно nocrvnaui и анодное пространство. В процессе электролиза
электролит перетекает из анодного пространства в катодное, проходя через поры
диафрагмы. Катодное пространство при этом может быть не заполнено или
заполнено электролитическим щелоком. При незаполненном катодном
пространстве электролитический шелок, содержащий 100—ПО г/.: NaOH и 180-
195 г/л неразложившегося NaCI, вытекает через нижним штуцер. При
заполненном катодном пространстве электролитический щелок содержит до 135 г .1
342
NaOH и отводится из электролизера через специальное устройство. Хлор
и водород отводятся по трубам из верхней части электролизера.
Нагрузка такого электролизера составляла 1000 р. при напряжении
3,8 в и выходе по току 91—94%. Плотность тока на катоде 720 а/м?
Расход электрической энергии (постоянный ток) нч 1 т NaOH составлял
2750 кет ¦ ч.
Описанные электролизеры вследствие малой производительности,
обусловленной небольшой рабочей поверхностью электродов. i< настоящее время
не применяются.
Для интенсификации
цилиндрических вертикальных
электролизеров вначале
увеличили число катодов. На рис.
136 показана схема двухкатод-
ного электролизера. Второй
дополнительный внутренний
катод расположен
концентрически по отношению к
внешнему катоду, при этом работают
обе поверхности анода,
обращенные к катодам.
Применение такой конструкции
позволило увеличить нагрузку
электролизера до 1500 а при
сохранении такой же катодной
плотности тока, что в
электролизерах Х-2.
Дальнейшее
усовершенствование цилиндрических
вертикальных электролизеров
заключалось в установке второго
внутреннего комплекта анодов
п третьего катода. Такой
реконструированный
электролизер К-3 на нагрузку 1800—
1900 а в свое время широко
применялся в
промышленности.
В результате установки
четвертого катода в
габаритах электролизера К-3 была
создана конструкция электро- Рис. 137. Электролизер БГК-12:
ЛИЗера ТИПа БГК-12 (РИС. 137), '-крышка внутреннего катода; 2-труба для
который может работать с
заполненным катодным прост-
ранством. Это является
существенным преимуществом по сравнению с электролизерами,
работающими при незаполненном катодном пространстве.
343
Средние показатели электролизера БГК-12:
Нагрузка, а 2200
Среднее напряжение, в 3.35
Катодная плотность тока, а/м2 625
Расход электроэнергии (постоянный ток) на 1 п NaOH, квт-ч 2410
Средний выход по току, % 93,0
Концентрация NaOH в электролитической щелочи, г/л .... 115
Концентрация С13 в хлоргазе, % 95—97
Срок службы диафрагмы и анодов, мес. 6—8
Электролизеры этого типа в настоящее время применяются
очень редко.
Электролизеры с вертикальной осажденной диафрагмой.
Вследствие трудности эксплуатации цилиндрических
электролизеров и небольшой производительности их в дальнейшем
перешли к разработке конструкции укрупненных прямоугольных
электролизеров на гораздо большие нагрузки, чем в
цилиндрических ваннах. В прямоугольных электролизерах удалось
значительно увеличить рабочие поверхности электродов (путем
изменения конфигурации катода) с одновременным
уменьшением площади, занимаемой электролизером на единицу
продукции. При одинаковой мощности хлорного завода
прямоугольных электролизеров требуется во много раз меньше, чем
цилиндрических, число которых в крупном цехе электролиза
может достигать сотен и даже тысяч.
В течение последних десятилетий мощные прямоугольные
ванны с вертикально расположенными электродами и
осажденной асбестовой диафрагмой получили широкое применение.
Путем снижения плотности тока благодаря максимальному
развитию рабочей поверхности электродов и повышению
температуры электролита до 94—97°С использование энергии в таких
электролизерах удалось повысить до 66%.
Схема электролизера Хукера (тип S) с вертикальной
осажденной диафрагмой показана на рис. 138. Днище / и
крышка 10 электролизера выполнены из бетона. Через днище
пропущен медный стержень 2 (анодная шина, соединенная с
наружной шиной 13), служащий для питания электролизера током.
На днище электролизера параллельно установлены
графитовые аноды 11, представляющие собой плиты размером
30x160x680 мм. Стержень 2 и концы анодов // залиты слоем
свинца 4, благодаря чему между ними достигается хороший
электрический контакт. Для предохранения свинца от действия
кислого анолита поверхность его покрыта двойным защитным
слоем 5 из цемента и смолы. Катод 3 состоит из прямоугольной
рамы (швеллерная балка) и толстой стальной сетки 12. Сетка
натянута в виде параллельных плоских карманов и приварена
к раме 3. К катодной раме приварена также катодная
шина 7.
344
Раму устанавливают таким образом, чтобы ряды
графитовых анодов помещались в промежутках между катодными
карманами сетки. Сверху на катодную раму 3 накладывают
крышку 10.
Днище, раму и крышку прочно стягивают болтами. Крышка
снабжена теплоизоляцией 9. Электролизер установлен на
изоляторе 6, что предотвращает утечки тока.
/J
Рис. 138. Электролизер Хукера:
;—днище; 2—медный стержеьь; 3—катодная рама; 4—слой евннца; 5—двойной защитный
слой (цемент и смола); 6—изоляторы; 7—катодная шнна; 8—труба для отвода щелочи; 9—
теплоизоляция; 10—крышка; //—графитовые аноды; 12—стальные сетки; 13—наружная шина.
В электролизер подается горячий рассол.
В последнее время электролизеры Хукера строят на
различные нагрузки от 15 тыс. а (тип S) до 32 тыс. а (тип S-3B).
Примерные показатели их работы приведены ниже:
Тип S Тип S-3B
.Максимальная нагрузка, ка 15 32
Поверхность анодов, м2 — 20,4
Плотность тока на аноде, а/м2 .... — 1330
Среднее напряжение, в 3,6 3,9
Выход по току, % 96 96
Расход электроэнергии на 1 т хлора
A00% CIJ, квт-ч 2690 3020
Срок службы анодов, мес 14 —
Расход графита на 1 т хлора A00% С13).
кг 3,5 3,14
345
Относящиеся к этому же типу электролизеров диафрагмен-
ные хлорные ванны фирмы «Даймонд», по опубликованным
данным, имеют нагрузку до 36 ка и выход по току до 96,5%.
Рис. 139. Электролизер БГК-13:
/—анодные шинки; 2—асбоцементная крышка; г—графитовые
аноды; 4—бетонная футеровка; 5—кожух; 6—сетчатый
гребенчатый катод; 7—трубка для слива электролитического
щелока; 8—капельница; 9—катодная шина.
При плотности тока 920 а/м2 и нагрузке 33 ка среднее
напряжение 3,94 в. На 1 т хлора расходуется 2800 кет ¦ ч
электроэнергии, срок службы анодов 7,4 мес, диафрагмы 3.7 мес.
346
Электролизер БГК-13 с осажденной диафрагмой и развитой
катодной поверхностью показан на рис. 139. Электролизер
рассчитан на нагрузку 5000 а и обычно работает с заполненным
катодным пространством.
Корпус электролизера БГК-13 выполнен и виде
прямоугольного кожуха 5 с приваренным изнутри каркасом, на
который натянут сетчатый гребенчатый катод 6. Гребенку образуют
плоские пустотелые выступы — катодные карманы, между
которыми находятся графитовые аноды 3, размещенные в два
ряда. Крышка электролизера плоская (или в виде купола).
Более мощным п совершенным отечественным
электролизером с осажденной диафрагмой является электролизер БГК-17.
рассчитанный на нагрузку 25 ка или ка 50 ка, при плотности
тока до 1200 а/м2. Электролизер отличается простотой
устройства, компактностью и экономичностью и может применяться
для электролиза растворов хлоридов натрия или калия.
Электролизер БГК-17, схематически изображенный на
рис. 140, состоит из ряда вертикальных параллельно
расположенных электролитических секций, в которых помещены
плоскопараллельные электроды в виде пропитанных графитовых
плит. Катодом является стальная сетка с развитой
поверхностью, на которую насасывается асбестовая диафрагма. В
собранном виде катод представляет собой многоячейковую
систему из ряда параллельных электролитических секций,
образуемых стенками корпуса катода и внутренними полыми
перегородками, на которых закреплены полые перфорированные
карманы. Все электролитические секции сообщаются между со-
ooii по анодному и катодному пространствам.
Снаружи электролизер покрыт теплоизоляцией, что
позволяет не только снизить расход тепла на предварительный
подогрев рассола, по и вести электролиз при оптимальной
температуре (94—98°С).
Специальные устройства позволяют работать с. заполненным
катодным пространством и в случае надобности устанавливать
любой уровень катоЛ'Мта в электролизере. Давление хлора и
водорода в электролизере п трубопроводах автоматически
регулируется. Оптимальная концентрация NaOH в католите A30—
135 г/л) поддерживается автоматически — саморегулированием
гидростатического напора па диафрагму. При этом достигаются
высокие выходы по току и длительная работа анодов.
Показатели работы электролизеров БГК-17:
Нагрузка, ка . . . 25 50
Плотность тока на электродах, а/м- 750 1000
Выход по току, % 96 96
Напряжение на электролизере, в 3,45 3,65
Расход электроэнергии на 1 m NaOH, квш-ч 2410 2550
347
Электролизер работает с заполненным катодным
пространством, в случае необходимости можно установить любой
уровень католита.
Рис. 140. Схема устройства электролизера БГК-17:
/—корпус (катод); 2—изолятор; S—днище; 4—графитовые аноды;
5—коллектор для отвода щелочи; 6—капельница (прерыватель тока): 7—устройство
для слива щелочи; 6—расходомер для рассола; 9—труба для подачи
рассола; 10—Еодородный коллектор; II—хлорный коллектор; 12—рассолопровод.
Вследствие хорошей герметизации в электролизерах БГК-17
получаются газы и щелочь высокой концентрации: 98,5—
99%-ный хлор, 99—99,5%-ный водород и электролитический
щелок, содержащий 130—135 г/л NaOH при низком содержании
хлоратов @.08—0,10 г/л).
3-18
Эксплуатация диафрагменных электролизеров
Современные диафрагменные электролизеры работают с
проточным электролитом, перетекающим от анода к катоду.
Температура электролита в процессе электролиза
поддерживается в пределах 80—90°С, в мощных электролизерах новых
конструкций она достигает 98 °С. При повышении температуры
рассола снижается напряжение на электролизере и,
следовательно, уменьшается удельный расход энергии, кроме того,
возрастает выход по току.
Для уменьшения утечки тока горячий рассол поступает в
электролизеры через специальные прерыватели струй, через
подобные же прерыватели отводится электролитический щелок.
Корпуса электролизеров выполняются из стали, часто в
комбинации с бетонными, керамическими, стеклянными деталями.
Каждый электролизер снабжен трубопроводами для подачи
рассола и отвода хлора, водорода и щелока. Хлоропроводы,
как правило, керамические, остальные трубопроводы —
стальные. Иногда рассолопроводы монтируются из фарфоровых или
стальных гуммированных изнутри труб, так как раствор
поваренной соли вызывает заметную коррозию стали.
В большинстве случаев в анодном и катодном пространствах
электролизеров поддерживают небольшое разрежение
(несколько мм вод. ст.). Чтобы предотвратить проникание воздуха
в ванны, которое может привести к образованию взрывоопасных
газовых смесей, разбавлению получаемых газов и к другим
осложнениям, электролизеры тщательно герметизируют в
местах стыков деталей и по ребрам крышек. Для этого
применяется специальная замазка, стойкая к действию хлора и
повышенной температуры (90 °С и выше).
Электролизеры располагают сериями и включают их в
серии последовательно. Серии же включаются в электрическую
цепь параллельно. Число электролизеров в серии зависит от
напряжения на клеммах источника постоянного тока и
среднего напряжения на электролизере, которое обычно составляет
3,3—3,7 в. Нагрузка диафрагменных электролизеров различных
конструкций — от 2 до 50 ка и более.
Подводка тока к электролизерам осуществляется медными
шинами или гибкими медными проводами. В последнее время
медь стали заменять алюминием и омедненным алюминием.
Преобразование тока. Для нормальной работы цеха
электролиза необходимо равномерное и бесперебойное снабжение
его постоянным током и поддержание требуемой нагрузки
электролизеров. Переменный ток, подаваемый электрическими
сетями, преобразуется в постоянный на специальных
преобразовательных подстанциях, включаемых обычно в состав хлорного
завода. Для преобразования тока могут применяться мотор-
349
генераторы при рабочем напряжении постоянного тока 250—
270 в, имеющие сравнительно низкий к. п. д. — 82—88%, и
ртутные выпрямители при рабочем напряжении 800—825 в, к. п. л.
которых несколько выше (92—94%).
Для питания постоянным током крупных цехов электролиза,.
оборудованных мощными электролизерами на нагрузку от 2о
до 200 ка (ванны с 'ртутным катодом, стр. 354), целесообразно
устанавливать контактные преобразователи — механические
выпрямители, отличающиеся высоким к. п. д. (до 98%), а также
полупроводниковые выпрямители, достоинствами которых,
кроме высокого к. п. д., являются отсутствие вращающихся частей,
компактность, надежность и простота эксплуатации.
Следует отметить, что с повышением рабочего напряжения
постоянного тока улучшается использование
электрооборудования преобразовательных подстанций, а в некоторых случаях
и повышаются технико-экономические показатели цехов
электролиза.
Электролиз соляной кислоты
Одним из рациональных способов использования отбросного
хлористого водорода, образующегося при хлорировании
органических соединений, является регенерация из него хлора путем
электролиза водных растворов соляной кислоты. Получаемый
при этом хлор снева используется для хлорирования и других
синтезов.
Электропроводность растворов соляной кислоты в несколько
раз больше электропроводности применяемых рассолов, а
напряжение разложения НС1 примерно на 0,8 в ниже, чем
для хлористого натрия, что благоприятно для процесса
электролиза.
Электролиз соляной кислоты можно проводить в
монополярных или биполярных электролизерах. Рамы электролизеров
фильтрп'рессного типа (стр. 320) изготовляются из фаолита,
поливинилхлорида или графита, диафрагмы — из поливинил-
хлоридпой ткани, весьма устойчивой к действию соляной
кислоты, катоды и аноды выполняются из графита. В качестве
электролита применяют соляную кислоту, имеющую концентрацию
180—200 г/л НС1, пр,я которой раствор обладает наилучшей
электропроводностью.
Процесс электролиза ведут при температуре до 80—90°С
и плотности тока 600 — 1000 а/м2. Напряжение на электролизере
поддерживается в пределах 2,15—2,4 в. Выход по току
составляет 90—94%. Расход электроэнергии на 1 т хлора достигает
1800—2000 квт-ч.
Получаемый хлор отличается высокой чистотой (до 99,7%
С12), содержание примесей СО2 в нем не превышает 0,2—0,4%.
350
ЭЛЕКТРОЛИЗ С РТУТНЫМ КАТОДОМ
В отличие от электролиза в диафрагменных ваннах,
электролиз растворов хлорида натрия (или калия) с ртутным
жидким катодом проводится в двух аппаратах — в электролизере
л связанном с ним разлагателе амальгамы натрия. В
электролизере происходит оазложение хлорида с выделением хлора и
образованием амальгамы натрия (или соответственно калия)
2NaCl + 2nHg > Cl3 + 2NaHgn
которая перетекает в разлагатель. В нем проводится
разложение амальгамы водой (на графитовой насадке) с выделением
водорода и образованием щелочи:
2NaHg,n + 2Н2О » 2NaOH + Н, + 2rcHg
Регенерированная ртуть возвращается в электролизер. Та-
кшм образом, процесс электролиза осуществляется с
движущимся ртутным катодом при непрерывной циркуляции ртути
и амальгамы; при этом отсутствует необходимость отделять
катодный продукт (хлор) от анодных (щелочи и водорода).
Хлор
Отработанный
рассол
, —
(анопигп
Наустическая
сода
Рис. 141. Принципиальная схема электролиза
с ртутным катодом:
/—электролизер; //—разлагатель. 1— графитовый анод: 2—
ртутный (жидкий) катод; 3—насос для перекачивания ртути.
Принципиальная схема электролиза с ртутным катодом
изображена на рис. 141. Ванна состоит из двух отдельных
аппаратов: электролизера /, в котором образуются хлор и
амальгама натрия, и разлагателя //, где происходит разложение
амальгамы горячей водой с образованием концентрированного
раствора каустической соды, водорода и 'ртути. По наклонному
дну ртуть тонким слоем непрерывно течет в направлении,
указанном стрелкой. В электролизере над ртутью расположены
графитовые аноды. Предварительно очищенный рассол, содер-
351
жащий 308—310 г/л NaCl, непрерывно поступает в электро
лизер. В процессе электролиза часть поваренной соли
разлагается, отработанный рассол вытекает из электролизера,
образующийся хлор отводится в хлорный коллектор. По мере
движения ртути по наклонному дну электролизера она
постепенно обогащается натрием, ионы которого разряжаются на
катоде. На выходе из электролизера содержание натрия в
амальгаме составляет 0,1—0,25%.
В разлагателе, куда подается чистая горячая вода,
взаимодействующая с амальгамой, последняя течет по наклонному дну
в направлении, обратном ее направлению в электролизере.
В разлагателе уложены графитовые плиты
(индифферентный катод), что способствует снижению перенапряжения
выделения водорода на ртути и, следовательно, ускоряет разложение
амальгамы.
Содержание натрия в амальгаме снижается в процессе
разложения до тысячных долей процента.
Регенерированная ртуть подается насосом в электролизер.
Из разлагателя отводится чистый концентрированный раствор
щелочи — 50%-ная каустическия сода, представляющая собой
товарный продукт. Водород удаляется в цеховой коллектор.
Катодный процесс. В электролизерах с жидким ртутным
катодом катодный процесс, заключающийся в разряде ионов
натрия на ртути, приводит к образованию амальгамы:
Na+ + «Hg + e = Nagn
Разряд иона Na+, а не иона водорода, возможен вследствие
большего перенапряжения разряда иона Н+ на ртути
(например, 1,066 в при плотности тока 770 а/м2). Это перенапряжение
растет с увеличением плотности тока.
Сколько-нибудь значительное выделение водорода на ртути
из нейтрального 'раствора, в котором обратимый потенциал
водорода равен —0,415 в, теоретически возможно лишь пр.и
потенциале катода около —1,20 в. На практике в процессе
электролиза при больших плотностях тока и незначительной
концентрации натрия в амальгаме выделение водорода на катоде
в заметных количествах может происходить лишь при
потенциале около —2 в.
Ионы натрия разряжаются на ртутном катоде при
потенциале ниже обратимого вследствие так называемого явления
деполяризации. Потенциал разряда ионов Na+ в растворе
понижается тем сильнее, чем меньше концентрация натрия в ртутной
амальгаме. При содержании натрия в амальгаме в пределах
0,1—0,05% нон Na+, для которого нормальный потенциал в
'растворе его соли равен —2,71 в, разряжается в заметных
количествах уже при потенциале ртутного катода —1,2 в.
352
Анодный процесс. На графитовом аноде электролизера с
ртутным катодом происходит разряд ионов хлора
2СГ — 2е = 2С1
с выделением газообразного хлора:
2С1 > С12
Кроме того, протекает побочный процесс разряда гидро-
ксильных ионов с образованием газообразного кислорода:
4ОН" — 4е > О3 + 2НаО
Часть выделившегося кислорода вступает в реакцию с
углеродом анода, образуя двуокись углерода:
С + О2 »соа
Для подавления побочной реакции образования СО2 и,
следовательно, увеличения выхода по току следует подавать в
электролизер концентрированный рассол C08—310 г/л NaCl),
концентрация хлорида натрия в уходящем из электролизера
обедненном рассоле должна быть не менее 260—270 г/л. При
таком составе свежего и отработанного рассола интенсивность
разряда гидроксильных ионов, зависящая от соотношения ионов
С1~ и ОН~ в растворе, еще незначительна. Отметим, что
повышение плотности тока также благоприятствует уменьшению
этого побочного процесса и. следовательно, увеличению выхода
по току на аноде.
Разложение амальгамы. Образовавшаяся амальгама натрия
разлагается водой, при этом образуются едкий натр, водород
и ртуть:
NaHgra + Н.О = NaOH + -у- Н3 + nHg
Этот процесс слагается из следующих реакций:
NaHgn — е «z=: Na+
Н2О + е » -у На + ОН-
Na+ + ОН" TTZ? NaOH
Для ускорения процесса разложения амальгамы натрия ее заставляют
работать в качестве растворимого анода гальванического элемента,
построенного по схеме: амальгамный электрод — щелочной раствор —
индифферентный электрод. Электродвижущая сила такого элемента около 1 в.
В качестве индифферентного электрода в промышленности применяют
графит. При замыкании элемента протекают следующие реакции:
на амальгамном аноде
2Na » 2Na+ + 2e
на индифферентном катоде
2Н+ + 2е » На
1Ъ —2848 353
Напряжение разложения раствора хлористого натрия при
электролизе с ртутным катодом определяется как
алгебраическая разность нормальных электродных потенциалов анода и
амальгамного катода. При 25 °С нормальный потенциал анода
составляет 1,33 в, а теоретический катодный потенциал
выделения Na на амальгамном катоде —1,83 в. Таким образом,
теоретический потенциал разложения Nad равен.-
?"т = 1,33 —(—1,83) = 3,16 в
т. е. почти на 1 в больше, чем при электролизе с твердым
стальным катодом.
Фактическое напряжение разложения хлористого натрия при
электролизе с ртутным катодом значительно выше
теоретического и колеблется в пределах 4,4—4,8 в, в зависимости от
конструкций ртутных электролизеров :и условий их эксплуатации.
Следовательно, в процессе электролиза с ртутным катодом
расходуется большее количество электрической энергии, чем в
диафрагменных электролизерах. Достоинством электролиза
ртутным катодом является возможность получения чистой кон
центрированной щелочи (до 650—760 г/л NaOH).
ВАННЫ С РТУТНЫМ КАТОДОМ
В производстве хлора и щелочи электролизом с ртутным
катодом применяют различные электролизеры и разлагатели:
Электролизеры — с горизонтальным ртутным катодом, с
вертикальным вращающимся стальным катодом, смоченным
ртутью, и с вертикальным катодом, орошаемым ртутью.
Разлагатели — горизонтальные, скрубберные с графитовой
насадкой, орошаемой амальгамой или погруженной в нее.
Независимо от типа разлагателя вода и амальгама
движутся в нем противотоком при получении 50%-ной щелочи и
прямотоком при получении 73%-ной щелочи.
В настоящее время наиболее распространены ванны с
горизонтальным ртутным катодом, различающиеся по конструкции.
нагрузке, размерам и расположению основных частей.
Ванны с горизонтальными катодом н разлагателем. До
недавнего времени применялись электролизеры этого типа, рас
считанные на нагрузку 15—30 ка. В последние годы введены
в эксплуатацию мощные ванны на нагрузку 100—200 ка,
которые более компактны и проще в обслуживании.
¦ Ванна Р-30. На рис. 142 схематично показана ванна Р-30,
рассчитанная на нагрузку 30 ка при катодной плотности тока
около 5000 а/л12. Основные аппараты—электролизер I, разла-
гатель II и насос III для перекачивания ртути. Корпус
электролизера длиной 8,2 м состоит из плоского стального днища /,
укрепленного на несущей стальной конструкции, и стальных
354
Свежий
рассол
воздух
j
из насоса
вразла-
сОтвПЬ
доз
Рис. 142. Банка типа Р-30
с ртутным катодом.
А. В—разрезы; В—план. I—
электролизер; //—разлагатель:
///—конусный роторный ¦ насос;
/—днище электролизера: 2—то-
коподводящне графитовые
стержни; 3— графитосые аноды;
t—крышка электролизера; 5—
лючки для загрузки и
выгрузки графитовой насадки разла-
гателя: в—боковая
гуммированная рама; 7—кронштейн для
поавески анодов; в—медный
токоподводящий штырь; 9—
графитовая насадка разлагате-
ля.
23*
355
гуммированных боковых 'рам 6, образующих стенки ванны.
Крышка 4, через которую ток подводится к анодам 3
электролизера, также гуммированная. К каждому графитовому аноду
ток подводится при помощи двух графитовых стержней 2, в
которые ввернуты медные штыри 8. Уплотнение графитовых
стержней в крышке достигается при помощи мембран из
пластмассы.
Аноды подвешены на кронштейнах 7. По мере износа
анодов их можно опускать вращением гаек, благодаря чему между
анодами и жидким катодом, протекающим по дну разлагателя,
всегда сохраняется одинаковое расстояние C—5 мм).
Стальной разлагатель // выполнен в форме стальной
коробки, длина которой равна длине электролизера, а ширина
несколько меньше. В разлагателе помещена насадка 9 в виде
пакетов тонких графитовых плиток, которые загружают и
разгружают через лючки 5.
Для перекачивания ртути из разлагателя в электролизер
служит конусный роторный насос ///.
Свежий очищенный рассол (до 310 г/л NaCl) подается в
верхний конец электролизера через жидкостной ротаметр
(измеряющий количество подаваемого на электролиз рассола) и
трубку, присоединенную к крышке электролизера. Обедненный
электролит B60—275 г/л NaCl) удаляется вместе с хлором
через гуммированную трубку, проходящую сквозь дно
электролизера в нижнем его конце. Для поддержания минимального
уровня электролита в электролизере конец трубки для вывода
рассола расположен выше дна.
По выходе из электролизера хлор отделяется от электролита
и поступает в хлорный коллектор, отработанный электролит
направляется б сборник, откуда поступает на обесхлоривание.
донасышение и очистку (стр. 335) и затем возвращается на
электролиз.
Очищенная вода подается для разложения амальгамы
через жидкостной ротаметр в нижний конец разлагателя.
Образующийся в разлагателе концентрированный раствор щелочи
F50—700 г/л NaOH и более) выводится через гуммированную
трубку в дне разлагателя, конец которой расположен также
выше дна.
Водород, собирающийся под крышкой, отводится из
разлагателя через трубу.
Примерные показатели работы ванны Р-30:
Нагрузка, ка 30
Среднее напряжение, в 4,6
Катодная плотность тока, а/м2 5350
Выход по току (на NaOH), % 95—97
Расход электроэнергии (постоянный ток) на
1 т NaOH, квт-ч 3240
Количество ртути на J000 а нагрузки, кг . 23
356
Ванна БГК-Р-Ю1. Более мощная ванна с ртутным
катодом типа БГК-Р-Ю1 на нагрузку 100 ка при плотности тока
5000 а/м2 представлена на рис. 143.
Она также состоит из электролизера /, разлагателя // и
насоса /// и питается подогретым до 60 °С рассолом,
содержащим 310 г/л NaCl. Днище 2, рама 3 и крышка 5 электролизера
выполнены из стали. Поверхность рамы, соприкасающейся с
рассолом, газом я 'ртутью, гуммирована. По дну, имеющему
некоторый уклон A0 мм/м), течет ртуть—жидкий катод. Ток
подводится к катоду через днище. В среднюю часть
электролизера, являющуюся электрохимической ячейкой, через
крышку 5 опущены графитовые аноды 10, под которыми протекает
ртуть.
По мере износа аноды можно опускать, не отключая тока.
Свежий рассол и ртуть поступают в электролизер через
входной (щелочной) карман 4 и протекают по уклону к
нижнему концу.
Обедненный рассол вместе с хлором выводится из
электролизера через штуцер 7. После отделения от хлора рассол
поступает на донасыщение поваренной солью и очистку и
возвращается в электролизер.
Разлагатель амальгамы, 'расположенный с уклоном 20 мм/м,
состоит из стального корпуса прямоугольного сечения и
коробки //. В верхней части разлагателя имеются три
герметически закрывающихся люка, через которые производится
загрузка <ц выгрузка графитовой насадки и очистка
¦разлагателя.
Вода, необходимая для разложения амальгамы, подается
через насос /// и стекает из него в коробку 11,
предназначенную для создания гидравлического затвора (что
предотвращает проникание водорода в полость насоса) и для подачи
ртути, вытекающей из разлагателя, во всасывающий патрубок
насоса.
Отбор водорода и щелочи производится через штуцер,
введенный снизу в верхний (по уклону) конец разлагателя. Сюда
же самотеком поступает амальгама из электролизера.
В разлагателе амальгама и раствор каустической соды
движутся противотоком.
Освобожденная от натрия ртуть перекачивается в
электролизер, таким образом осуществляется ее непрерывная
циркуляция между электролизером и разлагателем.
Показатели работы ванны БГК-Р-Ю1:
Нагрузка, ка 100
Среднее напряжение, в 4,3
Катодная плотность тока, а/м2 5000
Выход по току (на NaOH), % 96
Количество ртути на 1000 а нагрузки, кг . 17—20
357
Рис. 143. Ванна типа БГК-Р-101 с ртутным катодом:
/—электролизер; //-рачлагатель; ///—насос длл перекачивании ртути; /—опоры; 2—дкшцс электролизера; 3 - рама; 'i—входной
(щелочит!) кармам; 5—Kpuui.ii элекгрллизсра; 6— выходной (кислый) карман; 7—шт/цер для отвода обедненного рассола и хпл-
ра; 8—электроднмгатель; .'--электролитические ячейки: 10—аноды: //—коробка разлаглтеля.
Ванны Сольве. К наиболее мощным горизонтальным
ваннам с ртутным катодом относятся также ванны Сольве типа
V-200 на нагрузку до 200 ка и аналогичные им ванны типа
V-100 на нагрузку 100 ка. Кроме того, за рубежом
применяются ванны Матисона Е-8 на нагрузку 30 ка, горизонтальные
ванны Уде E0 и 100 ка), горизонтальные ванны Де-Нооа
D0, 80 и 120 ка) и др.
Ванна Сольве типа V-200 состоит из горизонтальных
электролизера и разлагателя, который для уменьшения площадь-
пола, занимаемой ванной, расположен под электролизером.
Ванны размещаются рядом друг с другом, без проходов между
ними, что приводит не только к экономии производственных
площадей, но и позволяет значительно сократить "расход меди
на ошиновку и уменьшить потери тока в проводах.
Электролизер ванны Сольве типа V-200 (рис. 144)
представляет собой прямоугольный ящик длиной 25,6 м и шириной
Кб м. Днище 1 электролизера — незащищенный стальной лист.
Рис. 144. Схема ванны Сольве с ртутным катодом:
Боковые стенки 3 выполнены из привернутых к днищу
гуммированных уголков, образующих раму.
Стальная крышка 5, гуммированная изнутри, состоит из
нескольких частей, опирающихся роликами 9 на борт ванны. На
роликах можно передвигать части крышки к любому концу
359
электролизера. Края крышки уплотняются при помощи
резиновых трубок 8, надуваемых воздухом. Ток подводится к анодам
через стойка 4 медными шинами 6, присоединенными к
графитовым втулкам 7. К анодным стойкам ток подводится медными
шинами, соединенными с шинами 10, идущими от катода
предыдущей ванны. Сверху над электролизером уложены съемные
мостики 11 для обслуживания ванны. Графитовые 'решетки
разлагателя можно с помощью рычага повернуть в ту или
другую сторону для удаления твердых отложений.
Общий вид зала электролиза, обгрудоваиного горизонтальным;] ваннами с
ртутным катодом.
Показатели работы ванны Сольве типа V-200:
Максимальная нагрузка, ка 200
Среднее напряжение, в 4,56
Катодная плотность тока, а/м2 6250
Выход по току, % 96
Расход электроэнергии (постоянный ток) на
1 т NaOH, квт-ч 3180
Количество ртути на 1000 а нагрузки, кг . 19,5
Количество ртути в электролизере на ! г мощности на '/з
меньше, чем в горизонтальных электролизерах других типов
с ртутным катодом, применяемых ча рубежом. Благодаря
зео
централизованному контролю работы описанных ванн
количество обслуживающего персонала сведено к минимуму.
Ванны с вертикальными катодом и разлагателем.
Горизонтальные ванны большой мощности с ртутным катодом все же
представляют собой громоздкие тяжелые агрегаты, занимающие
большую площадь, что связано с большими капитальными
затратами на сооружение цехов электролиза.
В связи с этим, наряду с усовершенствованием ртутных ванн
горизонтального типа, были предприняты попытки создания
занн с вертикальным расположением ртутного катода. В
настоящее время в ряде зарубежных стран эксплуатируются
некоторые типы вертикальных ртутных ванн, из которых следует
отметить Еанну Де-Нора со стекающей пленкой ртути (рис. 145).
Электролизер / выполнен в виде стального корпуса с
гуммированными стенками и незащищенным стальным днищем.
В него помещены 17 анодных плит толщиной 50 мм и
поверхностью каждая около 1 м2, расстояние между плитами 12 мм.
Каждый анод заключен в диафрагму из пластмассы,
предотвращающую сильное завихрение аколита около катода и
соприкосновение газообразного хлора с амальгамой. При плотности
тока 2500 а/м2 потеря напряжения в диафрагме составляет
около 0,07 в. Выход по току в ваннах с диафрагмой 97%, прп
отсутствии диафрагмы только 80%.
Между каждой парой анодных плит строго вертикально
установлена стальная проволочная катодная сетка,
закрепленная с боков и снизу в стальной рамке. Верхняя часть катодной
сетки пропущена в щель, вырезанную в стенке трубки, через
которую поступает ртуть (амальгама, содержащая 0,06% Na),
стекающая по сетке на дно электролизера тонким равномерным
слоем толщиной до 1 мм. Амальгама, находящаяся на дне ванны,
через затвор поступает в приемный карман, расположенный под
разлагателем, откуда забирается насосом 3. Большая часть
амальгамы подается на разложение в вертикальный скруббер-
ный разлагатель 2, меньшая часть смешивается с ртутью,
выходящей из разлагателя, и поступает в электролизер на
орошение катодной сетки.
По опубликованным в литературе данным, показатели
работы ванны Де-Нора таковы:
Нагрузка, ка 40
Среднее напряжение, в 4.3
Катодная плотность тока, а/м?- 1900
Выход по току, % До 97
Расход электроэнергии (постоянный ток) .па
1 т NaOH, квт-ч 3040
Количество ртути на 10С0 а нагрузки, кг . 25,9
361
Рис. 145. Схема ванны Де-Нора со стекающей пленкой
ртути:
/—электролизер: 2—скрубберный разлагатель; 3—центробежный
насос для перекачивания ртути; *—сборник амальгамы.
Направления технического прогресса в области электролиза
с ртутным катодом заключаются в увеличении мощности
электролизеров и их интенсификации путем повышения плотности
тока на электродах и понижения рабочего напряжения, а также
362
в развитии поверхности ртутного катода, разработке
конструкций вертикальных и многоэтажных электролизеров, замене
графитовых анодов другими малоизнашивающимися анодами.
Весьма важна также разработка эффективых методов
очистки рассола от вредных примесей (Cr, Mo, V, Ge и др.),
присутствие которых даже в ничтожном количестве вызывает
усиленное разложение амальгамы в электролизере. Это
приводит к значительному выделению в нем водорода и,
следовательно, к повышению содержания Н2 в хлоргазе, что может,
в конечном счете, вызвать взрыв газовой смеси.
Для уменьшения возможности образования в электролизере
гипохлоритов и хлоратов и уменьшения износа анодов
целесообразно подавать на электролиз горячий слабокислый рассол,
содержащий около 310 г/л хлористого натрия.
Сокращение удельного расхода ртути (на единицу готовой
продукции) может быть достигнуто в результате разработки и
применения эффективных способов ее улавливания и
регенерации из шлама после фильтрации рассола.
Сравнение методов электролиза
Главное преимущество метода электролиза с ртутным
катодом перед диафрагменным методом заключается в том, что
по ртутному методу сразу получают концентрированную
щелочь— едкий натр или едкое кали высокой чистоты.
Концентрация щелочи, получаемой по ртутному методу, в 5—6 раз
больше, чем по диафрагменному методу. Такая щелочь может
быть непосредственно использована, например, в производстве
искусственного волокна, без предварительного выпаривания и
очистки.
В связи с этим на получение щелочи по ртутному методу
расходуется гораздо меньше пара.
Основные недостатки ртутного метода: более высокий
удельный расход электроэнергии постоянного тока (примерно на
800 кет- ч на 1 т 92%-ного NaOH) и гораздо большие
капитальные затраты, чем при диафрагменном методе. Они на 40%
выше, чем при строительстве цехов, оборудованных диафраг-
менными электролизерами, без очистки получаемой в них
каустической соды, и на 15% больше в случае очистки ее
аммиачным способом (стр. 382).
Кроме того, ртутный метод электролиза отличается большей
вредностью для обслуживающего персонала в связи с
возможным прониканием паров ртути в атмосферу производственных
помещений.
В зависимости от метода электролиза, режима процесса,
конструкции электролизеров и других условий расходные
коэффициенты могут значительно колебаться.
363
Ниже приведены примерные расходные коэффициенты на
1 т щелочи (считая на 92%-ный NaOH), полученной по диа-
фрагменному и ртутному методу:
Поваренная соль, m . . . .
Кальцинированная сода, кг .
Соляная кислота, кг . . . .
Серная кислота, кг ....
Графитовые ьлектроды, кг .
Асбестовое волокно, кг . .
Едкнй натр, кг
Сернистый натрий, кг . . .
Ртуть, кг
Пар, млн. ккал
Вода, м3
Электроэнергия, квпг-ч
постоянный ток ....
переменный ток ....
Дифраг-
менный
метод
1,65
20,0
2,0
20,0
8,0
0,7
4,25
120
2500
170
Ртутный
метод
1,6
—
6,0
20,0
4,0
14,0
2,0
0,38
0,75
40
3350
165
В табл. 25 приведены для сравнения основные показатели
.'лектролтеров с диафрагмой и с ртутным катодом.
Таблица 25
Показатели работы электролизеров с диафрагмой и с ртутным катодом
„ . „ |С ртутным ка-
С диафрагмой | к годом
Максимальная нагрузка на электролизер, ка
Катодная плотность тока, а/м2
Среднее напряжение, в
Выход по току, %
Срок службы диафрагмы, мес
Срок службы графитовых анодов, мес. . . .
Количество ртути на 1000 а нагрузки, кг .
Концентрация NaOH в щелоке, г/л . . . .
25—50
700—1200
3,3—4,2
94—96
до 8
8—12
110—140
30—200
2400—6200
4,2—4,6
94—96
—
8—12
17—25
650—760
Выбор метода электролиза определяется соотношением
стоимости получаемых- с ТЭЦ электроэнергии и пара. При вы
сокой стоимости водяного пара и низкой стоимости
электроэнергии целесообразнее ртутный метод производства
технической каустической соды. В тех случаях, когда не требуется
чистая каустическая сода, экономичнее диафрагменный метод
электролиза.
В связи с ростом потребности в каустической соде высокой
чистоты для производства искусственного волокна и других
продуктов, а также благодаря усовершенствованию ртутного
метода электролиза во многих странах увеличивается доля
каустической соды, вырабатываемой по этому методу.
364
В настоящее время доля мирового производства хлора,
вырабатываемого различными методами, характеризуется
примерно следующими цифрами (в %):
Диафрагменный электролиз .... 49,4
Ртутный электролиз 47
Электролиз расплавов 3
Химические методы 0,6
ТЕХНОЛОГИЧЕСКАЯ СХЕМА ПРОИЗВОДСТВА ХЛОРА И ЩЕЛОЧИ
Примерная технологическая схема производства хлора и
щелочи методом диафрагменного электролиза раствора
поваренной соли показана на 'рис. 146.
Очищенный рассол, подогретый до температуры около 80 °С.
поступает в аппарат 1 постоянного уровня и распределяется по
электролизерам 2.
Из электролизеров по стальным трубам стекает
электролитический щелок в сборники 3, откуда перекачивается на
упаривание. Выделяющийся в электролизерах влажный хлор,
имеющий температуру 90—80 °С, поступает в общий керамический
коллектор и направляется в орошаемую холодной водой
башню 6 с насадкой, где охлаждается до 15—20 °С. При этом
из хлора выделяется около 90% влаги. Охлаждение хлора
можно производить также в трубчатых оросительных
холодильниках из пропитанного графита (стр. 149).
Потери хлора при растворении в воде сравнительно
невелики. Однако вода, стекающая из башни, содержит небольшое
количество хлора, а также НС1 и НОС1 (образуются в
результате взаимодействия хлора с водой). Поэтому перед удалением
в канализацию воду пропускают через дехлоратор 8.
Удаление хлора из воды, отводимой в канализацию,
проводится в две стадии: сначала воду подогревают и продувают
острым паром, затем пропускают через дробленый графит;
при взаимодействии графита с НОС1 образуется двуокись
углерода:
2НОС1 + С > СОа + 2НС1
После такой обработки вода содержит лишь 0,1—0,2 г/л
хлора.
Для окончательного освобождения хлора от влаги его
пропускают через две последовательно включенные сушильные
башни 7 и 12, заполненные насадкой из керамических колец
и орошаемые серной кислотой. Башни работают по принципу
противотока: наиболее концентрированная серная кислота
(96% H2SO4) орошает последнюю по ходу газа башню 12 и
соприкасается с хлором, содержащим меньшее количество
влаги. Хлор из холодильной башни 6 поступает снизу в сушиль-
365
Серная кислота
Серная кислота
L_L_1__JZ1
и
Отработанная
серная кислота
Рис. 146. Схема производства электролитического хлора и щелочи и исушки хлора:
/—аппарат постоянного уровня; 2—электролизеры; Л—сборники щелока; 4, 16— ротационные компрессоры; 5, в—холодильные б'шши; 7, 12—су'
шильные башни; а--дехлоратор; 9, 14—сборники серной кислоты; 10—холодильник кислоты; //—центробежные насосы; IS—напорный бак;
/5—ловушка брызг.
ную башню 7 и поднимается вверх противотоком движению
серной кислоты, стекающей по насадке. Из башни 7 хлор
поступает в нижнюю часть башни 12 и соприкасается здесь с
концентрированной серной кислотой, почти полностью
освобождаясь от влаги. Содержание влаги в осушенном хлоре не
должно превышать 0,05%, или 1,5 г/м3. По выходе из
башни 12 осушенный хлор освобождается в ловушке 15 от брызг
серной кислоты и поступает в ротационный компрессор, откуда
под абсолютным давлением до 1,2 ат подается в стальной
коллектор и направляется на сжижение или передается на
переработку в другие цехи.
Серная кислота, стекающая по насадке в башнях 7 и 12
навстречу хлору, непрерывно циркулирует в системе при по-
мсщи насосов //. Из башни 7 кислота поступает в сборник 9,
затем охлаждается в холодильнике 10 и снова подается в
башню 7 на орошение насадки (орошение «на себя»). Во
вторую сушильную башню 12 концентрированная (96%-ная)
серная кислота из сборника 13 через бак 14 подается на орошение
насадки башни без промежуточного охлаждения. Количество
кислоты, орошающей насадку башен, должно быть не менее
5 м3/ч на 1 м2 сечения башни. Свежая 96% -ная серная кислота
подается со склада в напорный бак 13, откуда самотеком
поступает в сборник 14. Избыток кислоты, циркулирующей в
системе башни 7, самотеком перетекает в сборник 9, откуда
избыток отработанной серной кислоты G4—76%-ная H2SO4)
удаляется на склад. В отработанной серной кислоте содержится
около 25 г/л растворенного хлора. Удаление хлора из кислоты
осуществляется продувкой через нее сжатого воздуха;
отдуваемый хлор улавливается водой или щелочью.
Водород по выходе из электролизеров поступает в
холодильную башню 5 с насадкой из керамических колец,
орошаемой водой, и затем ротационным компрессором 4 под
избыточным давлением до 1 ат направляется в стальной коллектор,
откуда передается потребителям.
Водород, полученный в электролизерах с ртутным катодом,
всегда содержит небольшое количество паров ртути. Поэтому
перед использованием газ очищают от них. Надежная очистка
водорода от паров ртути достигается пропусканием
охлажденного газа через слой активного угля, предварительно
обработанного разбавленной азотной кислотой.
Усовершенствование процессов охлаждения и осушки хлора
может быть достигнуто осуществлением замкнутой системы
циркуляции жидкостей — хлорной воды « серной кислоты,
применением холодильников из пропитанного графита или
из титана, увеличением плотности орошения башен и скорости
газовых потоков в них, применением более эффективных насэ-
док (например, седловидной вместо кольцевой) и др.
367
Практический интерес представляет замена существующих
абсорбционных насадочных башен для охлаждения и осушки
хлора аппаратами пенного типа, в которых коэффициент
теплопередачи в 10 раз выше, чем в холодильниках смешения, и
достигает 40 000 ккал/м2 • ч • град.
Использование пенных аппаратов для осушки хлора
позволяет значительно повысить коэффициент абсорбции водяных
паров серной кислотой.
Для перекачивания хлора вместо жидкостных ротационных
компрессоров целесообразно применять турбокомпрессоры
большой производительности, а для перекачивания водорода —
винтовые компрессоры.
СЖИЖЕНИЕ ХЛОРА
Производство жидкого хлора на хлорном заводе относится
к крупным потребителям газообразного хлора и является
промежуточным буферным цехом. Наличие такого цеха-буфера на
хлорном заводе позволяет превращать газообразный хлир п
жидкий продукт, хранить его в течение любого времени \\
обеспечивать внутризаводских потребителей
высококонцентрированным испаренным хлором или жидким хлором,
транспортируемым по трубопроводам. Кроме того, это дает возможность
регулировать производство и потребление хлора и
поддерживать постоянное давление в сети хлоропроводов. Использование
жидкого хлора, который можно перевозить в баллонах и
цистернах, облегчает снабжение потребителей хлора, удаленных
от места его производства.
Жидкий хлор. Впервые хлор был сжижен Фарадеем в 1823 г.
Жидкий хлор представляет собой маслянистую жидкость
темно-зеленого цвета, кипящую под атмосферным давлением при
—34,1 °С
Хлор относится к числу легко сжижаемых газов. Его
сжижение можно проводить при обычной или повышенной
температуре. Критическая температура для хлора 144°С,
критическое давление 76,1 атм, критическая плотность 0,573 кг/л.
С повышением температуры соответственно должно быть
увеличено и давление для сжижения газообразного хлора.
Зависимость абсолютного давления паров над жидким
хлором от температуры приведена ниже:
Температура Давленио Температура Дтленпе
С'С атм DC атм
—50 0,46 -40 4,95
—34,1 1,00 4-20 6,62
—10 2,63 4-30 8,75
О 3,66
368
Жидкий хлор, не содержащий влаги, как и сухой
газообразный хлор, не оказывают коррозионного действия на сталь.
В присутствии влаги хлор сильно разрушает металлы. Поэтому
леред сжижением весьма важна тщательная осушка
газообразного хлора.
Поступающий на сжижение газообразный хлор должен
содержать не менее 96% С12, не более 1% Н2 и не выше 0,1%
злаги. Эффективность процесса сжижения характеризуется
коэффициентом сжижения, т. е. отношением количества хлора,
перешедшего в жидкое состояние, к количеству хлора,
поступившего на сжижение. Этот коэффициент, зависящий от
концентрации хлора в сжижаемом газе, температуры и давления,
тем больше, чем выше концентрация С1о и давление и ниже
температура.
Методы сжижения хлора. Известны три промышленных
метода сжижения газообразного хлора:
1) метод высокого давления — сжатие до избыточного
давления 12 ат без применения искусственного охлаждения;
2) метод так называемого глубокого охлаждения — сжатие
ди избыточного давления 0,8 ат с искусственным охлаждением
до минус 35 — минус 45 °С;
3) комбинированный метод — сжатие до избыточного дав-
тсния примерно 5 ат при умеренном искусственном охлажде-
:пш (не ниже —18 °С).
Метод высокого давления характеризуется простотой техно-
югического оборудования (отсутствие специальной
холодильной установки), низкими удельными затратами энергии (на 1 т
жидкого хлора расходуется 80—100 квтч электроэнергии).
Кроме того, при сжижении хлора под высоким давлением
отпадает необходимость теплоизоляции коммуникаций. Опнако
главное достоинство этого метода заключается в возможности
комбинировать сжатие газа высокопроизводительными
компрессионными агрегатами с процессами его сжижения и пере-
..ачивания и обеспечивать широкие пределы давлений хлора,
необходимые его разнообразным потребителям.
Метод глубокого охлаждения также довольно прост по
аппаратурному оформлению и отличается высоким
коэффициентом сжижения хло'ра, но требует тщательной
теплоизоляции всех коммуникаций. При использовании этого метода обес-
ачиваются вполне безопасные и гигиеничные условия работы.
Комбинированный метод, по которому достигается высокий
коэффициент сжижения, имеет значение главным образом для
концентрирования разбавленного хлоргаза, получаемого,
например, при электролизе расплавленных хлористых солей.
Умеренное охлаждение газа осуществляется при помощи аммиачных
холодильных установок. Недостатком метода чвляется
некоторая сложность применяемого оборудования.
---2548 369
Рис. 147. Схема сжижения хлора комбинированным методом:
1, 5, 14—буферы; 2—хлорный компрессор; 3, 12—холодильники; 4—ловушка брызг; 6—конденсатор; 7—охладитель газа
8—мерник; 9— весы; /<7—фильтр для очистки воздуха от механических примесей; //—воздушный компрессор:
«—сушильные баллоны; /5—танк для жидкого хлора.
На одном из хлорных заводов в США сжижение хлора
производится по так называемой короткой схеме. Сухой хлор
сжимается компрессором до давления 2,8—3,2 ат я поступает
сначала в скруббер, где промывается жидким хлором от брызг
серной кислоты (смазка поршней компрессора) и органических
примесей, затем идет в конденсатор, где превращается в
жидкость. Из конденсаторов жидкий хлор передается в танки-
хранилища.
Технологическая схема сжижения хлора комбинированным
методом изображена на рис. 147. Осушенный хлор поступает
через буфер / в двухступенчатый компрессор 2, где сжимается
до избыточного давления 5 ат. После охлаждения в водяном
холодильнике 3 сжатый хлор освобождается в ловушке 4 от
брызг концентрированной серной кислоты, которой
смазываются поршни компрессора. Далее через буфер 5 хлор поступает
в межтрубное пространство горизонтального стального
элементного конденсатора 6, где, охлаждаясь до температуры
—18 °С, сжижается. Хладоагентом, подаваемым в трубки кон-
дер.сатора, служит холодный рассол (раствор СаСЬ),
поступающий с аммиачной холодильной установки.
Для сжижения хлора могут применяться также
вертикальные холодильники, охлаждаемые фреонами (стр. 329).
Конденсатор устанавливают на таком уровне, чтобы
жидкий хлор через отделитель 7 несжиженных газов поступал
самотеком в мерники 8, установленные на весах 9. Из мерников
жидкий хлор направляют в танки-хранилища 15, Не
сжиженная в конденсаторе часть газообразного хлора с примесью
воздуха, СО2 и Н2 направляется для использования на месте,
например для получения белильных растворов (стр. 419).
Из хранилищ жидкий хлор под давлением очищенного
воздуха, предварительно сжатого з компрессоре // до давления
10—12 ат и затем охлажденного и осушенного, передают в
железнодорожные цистерны или (в отдельных случаях) в
стальные баллоны.
Элементный конденсатор для сжижения хлора схематично
показан на рис. 148.
Отпускаемый потребителям жидкий хлор должен содержать
не менее 99,5% С12 и не более 0,05% влаги.
Хранение и перевозка жидкого хлора
Ж'идкий хлор хранится в специально оборудованных сталь
пых танках емкостью 50 т и более и транспортируется в же-
1езнодорожных цистернах емкостью до 50 т, предварительно
испытанных на давление 22,5 ат. На современных заводах
крупные танки для хранения жидкого хлора устанавливаются
ча весах. Для компенсации перемещения весовых танков при
-Ч 371
изменении загрузки к ним подводятся трубопроводы, снаояат;
ные компенсаторами. Перемещение танков регистрируется из-
мерительнбгм прибором, что позволяет в любой момент
установить с точностью до 0,1% количество жидкого хлора,
находящегося в танке.
В последнее время наблюдается стре!мление к
значительному увеличению емкостей для хранения жидкого хлора и
применению специальных насосов для перекачивания жидкого
хлора потребителям. Например, в США предложено .хранение
\Т(и'П/1нЫ ,
poem fit.
Г.аС1.,
Ж,/о к и О хлор и и ее но.- ¦
Рис. 148. Элементный конденсатор для сжижения хлора:
1—элемент: 2—коллектор для входа охлаждающего раствора хлористого кальция: ^—кол*:.
тор для выхода охлаждающего раствора; 4—коллектор для ьхода газообразного хлора: .; —
коллектор длк выхода жидкого клора и песконденснровавшихся газов: 0—шевеллернь:: блл;:ч
Г—хомуты для креплег-шя элементов,
больших количеств охлажденного до —34,1 "С жидкого хлор./
при атмосферном давлении в емкости на 2000 г,
представляющей собой стальной шар диаметром 13,7 м.
Для хранения и транспортирования небольших количеств
жидкого хлора применяют стальные баллоны емкостью
40—60 л. Каждый литр емкости тары (баллона) должен быто
заполнен не более чем 1,25 кг жидкого хлора, т. е. на 1 кг жил
кого хлора должно приходиться не менее 0,8 л внутреннего
объема баллона.
Баллоны для наполнения жидким хлором должны соотве \ -
ствовать требованиям ГОСТ 949—57. Перед наполнением
баллоны необходимо освободить от остатков хлора, тщательн'
промыть водой, пропарить и просушить, Налив, опорожнение ¦•
эксплуатация баллонов, цистерн и танков для хранения жид
кого хлора производятся в соответствии со специальной nv.
ст'рукцией.
372
Техника безопасности в производстве
хлора и щелочей
Для создания безопасных условий работы на хлорных
заводах должны прежде всего учитываться свойства продуктов
электролиза — взрывоопасность смесей водорода с хлором и
воздухом, а также токсичность хлора (стр. 327),
раздражающее и обжигающее действие щелочи на кожу и слизистые
оболочки. Неосторожное обращение с реактивами,
применяемыми для очистки рассола и осушки хлора (соляная и серная
кислота, кальцинированная сода и др.). тоже может служить
источником травматизма и заболеваний.
Следует также отметить возможность поражений
обслуживающего персонала электрическим током, особенно при
применении повышенного напряжения в токопроводах E00 в и
более) , и значительные тепловыделения с поверхности
электролизеров.
Согласно санитарным нормам, предельно допустимая
концентрация хлора в рабочей зоне производственных помещений
не должна превышать 1 мг\м6. Необходимо также исключить
возможность заражения хлором атмосферы в районе
расположения хлорного завода.
В качестве противоядий при отравлении хлором
рекомендуется вдыхание паров этилового спирта, эфира и аммиака,
полезно также вдыхать кислород (при абсолютном покое
пострадавшего) .
Особо тщательные меры предосторожности необходимы в
цехах электролиза с ртутным катодом вследствие токсического
действия паров ртути на живые организмы. Ядовитое действие
паров ртути на организм человека является кумулятивным,
т. е. суммируется и постепенно возрастает. Поэтому при
получении хлора по ртутному методу электролиза должна
быть исключена возможность проникания в атмосферу паров
металлической ртути. По существующим санитарным
нормам, содержание ртути в воздухе рабочей зоны
помещений не должно превышать 0,01 мг/мг воздуха.
Нормальные и безопасные условия труда на хлорных
заводах обеспечиваются в результате тщательной герметизации
аппаратуры и технологических (газовых и жидкостных)
трубопроводов, автоматического поддержания в них
соответствующего вакуума, исключающего возможность проникания
вредных и опасных паров и газов в атмосферу, а также путем
тепловой изоляции поверхности электролизеров (особенно
крупных ванн). Для создания таких условий необходима,
кроме того, мощная приточно-вытяжная и егт^тв^нная
вентиляция производственных помещений с многократным часовым
373
обменом воздуха. Усиленная вентиляция предусмотрена в цехах
электролиза A5—30-кратный часовой воздухообмен) и
отделениях осушки хлора E—6-кратный часовой обмен воздуха),
а также на установке сжижения хлора.
Средством, предотвращающим возможность поражения
электрическим током, является надежная электрическая
изоляция от земли электролизеров, токоведущей ошиновки, трубо~
проводов, обслуживающих площадок, щитов, уложенных в
проходах между аппаратами, и других деталей а оборудования,
которые находятся или могут оказаться под напряжением.
Состояние и исправность электрической изоляции в цехах
электролиза систематически контролируются. При ремонте и других
работах с токоведущими элементами необходима
дополнительная злектроизоляция — специальные диэлектрические коврики,
диэлектрические резиновые обувь и перчатки и защитные очки.
Весь персонал, обслуживающий хлорное производство,
должен быть снабжен личными противогазами. В помещениях, где
возможно проникание хлора в воздух, необходимо иметь
неприкосновенный запас противогазов .и защитных масок
(респираторов). Кроме того, помещения производственных цехов
должны быть снабжены противопожарным оборудованием и
средствами пожаротушения (пожарные гидранты и краны с
рукавами и брандспойтами, сухие огнетушители, ящики с песком
и лопатами и пр.).
Поскольку водород образует с хлором и воздухом взрыве
опасные смеси, производство хлора по степени пожарной
опасности согласно действующим нормам относится к
категории А. В производстве электролитического хлора допускается
установка электрооборудования и осветительной арматуры
только во взрывобезопасном исполнении.
3. ВЫПАРИВАНИЕ ЭЛЕКТРОЛИТИЧЕСКОГО ЩЕЛОКА
Электролитические щелока, получаемые в результате
электролиза растворов поваренной соли в ваннах с диафрагмой,
содержат 100—140 г/л NaOH, 160—200 г/л NaCl и около
900 г/л воды. Вследствие низкой концентрации едкого натра и
большого содержания поваренной соли в растворе эти щелока
не могут быть использованы в других производствах. При
транспортировании электролитического щелока на 1 т щелочи
пришлось бы перевозить около 9 т балласта (вода и соль), что
связано с непроизводительными затратами и излишней
загрузкой транспорта.
Поваренную соль, содержащуюся в электролитических
щелоках в количестве около 1,5 г на 1 т едкого натра, необходимо
выделить и возвратить в производственный цикл, чтобы
избежать безвозвратных потерь соли.
374
Поэтому электролитические щелока выпаривают.
При частичном выпаривании воды, до концентрации
42—50% NaOH, производимом обычно в вакуум-выпарных
аппаратах, получают жидкую каустическую соду. При
дальнейшем обезвоживании упаренного раствора до полного
удаления воды в плавильных котлах или аппаратах, обогреваемых
ВОТ*, получают твердую (плавленую) каустическую соду
(каустик).
^
со
Z
f
OJ<J
300
4
6OU\ ^
150
ЮО
50
-
1
4 —
\
\
ffO'C
[
IOO°C
го°с\ ^
т гоо то ш soc soo
23 зо зб w
Концентраиия NaOH
soo эоо
?г ss
Рис.
149. Растворимость поваренной соли в растворах
едкого натра при различных температурах.
В процессе выпаривания электролитических щелоков иг
раствора выделяется твердая поваренная соль, так как ее
растворимость при повышении концентрации NaOH
уменьшается (рис. 149). На рисунке видно, что растворимость NaCi
с понижением температуры раствора также уменьшается, чтс
можно проиллюстрировать следующими данными:
Концентрация NaOH, % 42 50
Растворимость NaC!, г/л
при 100°С 48,3 46,6
при20°С 18,4 13,9
Содержание NaCI в растворе едкого
иатра, г на 100 г NaOH
при 100 °С 8,12 6,24
при20°С 3,0 1,8
Упаренный 'раствор щелочи охлаждают, при этом
выделяется дополнительное количество соли и улучшается качество
* ВОТ — высококипящий органический теплоноситель (например, азес-
тропная смесь дифенила и дифенилоксида).
375
готового продукта (щелочи). Вся поваренная соль,
выделившаяся при выпаривании и охлаждении раствора каустической
соды, отделяется от щелоков фильтрованием, затем ее
промывают и растворяют в воде. Такой раствор, содержащий
305—310 г/л NaCl и 2—2,5 г/л NaOH и называемый обратным
рассолом, возвращается в цех электролиза.
В процессе упаривания электролитических щелоков из
раствора необходимо удалить в виде пара 7—7,5 г воды на 1 т
NaOH, на что расходуется значительное количество греющего
пара, обычно получаемого с. ТЭЦ.
170
/60
/50
Sam
\ f
lam
У
ж
Ж,
УУЛЛ ':a5am\
f
I
i
1
I
1
\зо
Ik к
/ю,
!0° г/л ЮО 200 300 Ш 5ОС 600 700 800 900
% 8 96 23 30 36 V 47 52 56
концентраций NaOH
Рис. 150. Температуры кипения электролитических
щелоков и растворов едкого натра при различном
абсолютном давлении (пунктиром обозначена
кривая температур кипения растворов NaOH при
атмосферном давлении).
Затраты на пар составляют основную долю общих расходов
на переработку щелоков. Поэтому, чтобы процесс выпаривания
был более экономичным, его ведут в установках с
многократным B—3-кратным) использованием тепла пара. Кратность
использования тепла пара примерно равна числу
последовательно соединенных корпусов (ступеней) выпарной установки.
Растворы едкого натра (и едкого кали) кипят при гораздо
более высоких температурах, чем чистая вода,
электролитические щелока — при еще более высоких температурах (рис. 150).
Эта разность температур кипения раствора п воды при
одинаковом давлении, называемая температурной депрессией, при
повышенном давлении незначительно повышается, а при
разрежении снижается.
376
Вследствие большой температурной депрессии растворов
едкого натра D0—50СС и более при высокой концентрации
NaOH) сильно снижается общая полезная разность
температур* в выпарном аппарате, что делает неэкономичным
выпарные установки с числом корпусов более трех. Для
увеличения общей полезной разности температур в последнем
выпарном корпусе поддерживают разрежение порядка 0,8—0,9 ат
(остаточное давление 0,2—0,1 ел). Но даже при разрежении,
используя греющий пар с абсолютным давлением 5—5,5 ат,
на установке с трехкратным использованием тепла пара можно
упаривать щелока только до концентрации 25—30% NaOH.
При дальнейшем повышении концентрации щелоков
уменьшается полезная разность температур из-за увеличения
депрессии и соответственно снижается производительность выпарки.
Поэтому при указанном давлении пара применяют двухста-
дийную выпарку: на первой стадии с трехкратным
использованием тепла пара (система триплекс) щелока упаривают до
концентрации 25—26% NaOH, на второй стадии для обогрева
выпарного аппарата используется вторичный (соковый) пар
давлением около 3 ат из первого корпуса первой стадии и
раствор упаривается до концентрации 42—50% NaOH.
При использовании греющего пара низкого давления
B—4 ат) первая стадия выпарки работает с двухкратным
использованием тепла пара (система дуплекс), а выпарной
аппарат второй стадии обогревается свежим паром. При наличии
пара давлением 8—10 ат A75—183 °С) можно выпаривать
электролитические щелока до концентрации 50% NaOH в одну
стадию и таким образом снизить расход греющего пара. В
случае необходимости упаривания раствора до концентрации
70—72% NaOH устанавливают еще один выпарной аппарат,
обогреваемый свежим паром, т. е. ведут выпарку в три стадии.
ДВУХСТАДИИНАЯ ВЫПАРКА
На рис. 151 показана схема двухстадийного процесса
выпарки электролитических щелоков с трехкратным
использованием тепла пара на первой стадии. Следует отметить, что
схемы концентрирования электролитических щелоков и
растворов едкого натра, полученных химическими способами из
кальцинированной соды (стр. 476 ел.), по существу одинаковы.
Электролитические щелока из цеха электролиза при
температуре 60—70 °С поступают в сборники, рассчитанные на
хранение примерно суточного запаса и установленные вне зданий.
* Разность температур греющего пара, поступающего в первый корпус
выпарной установки, и вторичного пара, выходящего из пос-ледне-о ко-пуса,
за вычетом температурных потерь (температурная, гидравлическая,
гидростатическая депрессия).
377
Из этих баков электролитические щелока передаются в
расходные баки, установленные в цехе выпарки.
Подогрев электролитического щелока производится в
теплообменниках 3—6. Щелока подаются параллельно в
теплообменники 3 и 4, где подогреваются до 90—100 °С. В
теплообменнике 3 щелок нагревается конденсатом из II корпуса 8 первой
стадии выпарки и из выпарного аппарата 12 второй стадии вы-
яарки, в теплообменнике 4 — конденсатом из I корпуса
выпарки 7. Далее в теплообменнике 5 щелок подогревается до
ПО—115°С вторичным паром из I корпуса выпарки 7 и в
теплообменнике 6 греющим (свежим) паром до 130—135 °С
Такая система подогрева позволяет нагреть щелок до
температуры, близкой к температуре его кипения в I корпусе, теплом
конденсата и вторичного пара, вследствие чего снижается
расход свежего пара. Конденсат собирается в баки (на схеме не
показаны) и возвращается на ТЭЦ.
Первая стадия выпарки осуществляется в трехкорпусной
выпарной установке, состоящей из последовательно соединенных
(по пару и раствору) выпарных аппаратов 7—9 с подвесной
греющей камерой. Подогретый щелок поступает в выпарной
аппарат 7 A корпус). Тепло, необходимое для выпаривания
зоды из раствора, подводится свежим паром (рабс.= 5—5,5 ат),
поступающим в греющую камеру.
Частично упаренный щелок вместе с выпавшей из раствора
поваренной солью перетекает из конического днища I корпуса
вследствие разности давлений во II корпус, обогреваемый
вторичным паром из I корпуса (давление пара 2,7—3 ат).
Вторичный пар из I корпуса передается также на обогрев
аппарата 12 второй стадии выпарки и подогревателя щелоков 5.
Общее количество вторичного пара, отбираемого .из I
корпуса, в 2—2,5 раза больше, чем из II и III корпусов, и потому
поверхность теплообмена в I корпусе должна быть также
соответственно больше, как показано на рис. 151.
Если желательна установка однотипных аппаратов первой
ступени выпарки, то вместо одного I корпуса устанавливают
2—3 одинаковых параллельно работающих выпарных аппарата.
Поверхность теплообмена в каждом таком аппарате должна
быть разна поверхности теплообмена II -и III корпуса первой
стадии выпарки.
Из конического днища II корпуса щелок вместе с выпавшей
при упаривании солью перетекает в III корпус, обогреваемый
вторичным паром II корпуса (давление пара 1,0—1,4 ат).
Очистка вторичного пара от брызг щелочи и конденсация.
Последний III корпус выпарной установки работает при
разрежении (остаточное давление 0,1—0,2 ат). Вторичный пар
III корпуса содержит 2—3% брызг раствора щелочи, поэтому
перед конденсацией он проходит ловушку 19, в которой капли
378
IfJBKJUCUit
1Пар
го
]конденсо/П
Электролитический щелок
Рис. 15). Технологическая схема выпарывания электролитического щелока:
/-—бак для электролитического щелока; 2—сбопиик срецчсго щелока (пульпы)' 3—6—теплообменники (подогреватели 'пел.жа).
7— 9—выпарные аппараты (I—III корпуса) первой стадии оыпаркн; 10, 11. 14, 15, 16, /8—отстойники; 12— выпарноИ аппарат
второй стадии выпфкп; /.!—центрифуп; /7—спио1Льич1 хлодчльник (тзплто1чемччк): 19—ловушка брызг; 20—
барометрический конценстгор; 2/—сборнтч отЬил^тровтчого среднего щелока; 22—растворитель соли; 23—барометрнчесинй яцнк;
а—сифоны угкшнгмепоп; б—регуляторы уровня. ПппЗор j па 'цнгзх (внизу); t—измерители температуры; Р—измерители давления;
К— измерители концентрации; Н—измерителе уровня; Я—показывающий прибор; с—самопишущий прибор; из—иэодромные
регуляторы; w—прибор с электрической передачей (остальные приборы и регуляторы (пвевмопередачей).
раствора отделяются олагодаря изменениям направления
движения пара. Отделившийся раствор, содержащий 300—350 г/л
NaOH, стекает в сборник 21, откуда возвращается на
упаривание. Очищенный вторичный пар поступает в конденсатор
смешения 20, орошаемый водой. Неконденсирующиеся газы
отводятся водокольцевым вакуум-насосом в атмосферу. Теплая
вода C5—40 °С) из конденсатора смешения 20, располагаемого
на 12—15 м выше уровня земли, через гидравлический затвор
(барометрический ящик 23) сливается в лотковую
канализацию.
Открытая лотковая канализация применяется для удобства
очистки ее стенок от солей жесткости (стр. 27), выпадающих
из воды при нагревании и взаимодействии с щелочью, частично
проникающей в конденсатор вместе с паром, несмотря на его
предварительную очистку от брызг щелочи в аппарате 19.
Часть теплой воды из конденсатора (так называемая
барометрическая вода) подается для растворения соли в
аппарат 22 или -используется, например, для приготовления
рассола.
Фильтрование щелока и растворение соли. После первой
стадии выпарки получается так называемый средний щелок,
содержащий примерно 26% NaOH C40 г/л). Вместе с
выпавшей из раствора и взвешенной б нем солью щелок в виде
пульпы (Т:Ж=1:3) из III корпуса 9 стекает в сборщик 2.
Сюда же собирается пульпа из отстойников 14, 16, 18 щелока
второй стадии выпарки. Содержимое сборника 2 перекачивается
в отстойник 10, из верхней части которого осветленный
средний щелок перетекает в отстойник 11.
Отсюда щелок подается на дальнейшее упаривание (вторая
стадия выпарки).
Сгущенная пульпа (Т:Ж=1:1) из нижней конической
части отстойника 10 самотеком поступает на автоматическую
полунепрерывную горизонтальную центрифугу 13 для
отделения соли. Маточный раствор (средний щелок) из центрифуги
стекает в бак 21 и затем перекачивается в отстойник 11. На
центрифуге 13 соль промывается последовательно
электролитическим щелоком и водой.
Промывная жидкость .из центрифуги стекает в расходный
бак /.
Отфильтрованная и промытая соль автоматически
выгружается из центрифуги (срезается ножом) в растворитель
соли 22, представляюший собой аппарат с мешалкой, в который
подается теплая барометрическая вода. Соль не успевает здесь
полностью раствориться, смесь полученного рассола и твердой
соли (пульпа) перекачивается в отстойник 15. Из нижней его
части пульпа по мере накопления снова возвращается в
аппарат 22 для окончательного растворения.
380
Осветленный, так называемый обратный рассол, содержащий
305—310 г/л NaCl и 2—2,5 г/л NaOH, направляется для
повторного использования в цех электролиза.
Вторая стадия выпарки. Средние щелока из сборника //
B6—30% NaOH и 70—100 г/л NaCl) упариваются до
содержания 42—50% NaOH з аппарате 12 второй стадии выпарки,
называемом также аппаратом окончательного упаривания.
На рис. 151 был показан аппарат с принудительной
циркуляцией (АПЦ) для окончательного упаривания щелока.
Аппарат этого типа работает более интенсивно, чем выпарные
аппараты с естественной циркуляцией (АЕЦ). Для обогрева
аппарата окончательного упаривания щелока используется
вторичный пар из I корпуса 7 первой стадии выпарки, что позволяет
снизить расход свежего греющего пара.
Вторичный пар из аппарата АПЦ, работающего при
разрежении (остаточное давление 0,1—0,2 ат), поступает в систему
очистки и конденсации, аналогичную такой же системе III
корпуса первой стадии выпарки (аппараты 19, 20, 23). На схеме
эта вторая система не показана.
Раздельные системы конденсации первой и второй стадий
выпарки удобны для поддержания высокого и постоянного
вакуума в аппаратуре.
Охлаждение упаренного щелока. Упаренный щелок из
аппарата 12 вместе с выпавшей из раствора поваренной солью
непрерывно перекачивается в отстойник 14. Из верхней его части
осветленный горячий щелок самотеком передается в
отстойник 16. Для выделения из упаренного раствора возможно
большего количества соли он охлаждается водой в
спиральном теплообменнике 17 и при температуре 15—20°С
возвращается в отстойник 16.
Из верхней части этого отстойника раствор при 25—30 ЭС
перетекает в аппарат 18, а отсюда подается в складские
резервуары и затем отгружается потребителям каустической
соды.
Во многих случаях в цехах выпарки из раствора удаляют
сульфат натрия, содержащийся в исходном рассоле. Для этого
соль из аппаратов второй стадии выпарки и из холодильников,
¦з которых выпадает в осадок наибольшее количество сульфата
натрия, подвергают фракционной промывке щелоками и
циркулирующим рассолом при температуре 20—25°С. В
результате получают рассол, содержащий 270—280 г/л NaCl и
70—80 г/л N2SO4, из которого при охлаждении до 0°С или
нагревании до 100 °С выделяют твердый сульфат натрия.
Для повышения качества каустической соды раствор можно
упаривать до содержания в нем 50% NaOH, при этом
содержание NaCl снижается до 2—2,2%, в 42%-ном 'растворе NaOH
содержится примерно 4% NaCl.
38\
Глубокая очистка каустической соды от поваренной соли,
хлоратов и окислов железа достигается экстракцией этих
примесей из раствора жидким аммиаком при 60 ат и 60—80 °С.
Расходные коэффициенты. В описанной выпарной установке
при получении 42%-ного раствора NaOH (каустическая сода)
расходуются примерно следующие количества сырья и энергии
на 1 т продукта в (пересчете на 92%-ный NaOH):
Электролитический щелок
A05—110 г/л NaOH) в
пересчете на 100% NaOH, кг 945—950
Пар E am), m 4,5—5,0
B,75—3,0 млн. ккал)
Электроэнергия, кет-я . . . 70—80
Вода B5—26 "С летом), м3 . 80—110
Потери хлористого натрия с вторичным паром составляют
15—20 кг. Потери NaOH с вторичным паром и обратным рас
солом 25—30 кг на 1 т продукта (в пересчете на 92% NaOH)
На ТЭЦ стремятся вернуть возможно большее количество
конденсата, не загрязненного солью и щелочью. Возврат конден
сата достигает 140—150% от количества израсходованного
(свежего) пара. Качество конденсата, передаваемого на ТЭЦ.
непрерывно контролируется.
Расходные коэффициенты могут быть значительно
уменьшены при повышении концентрации электролитического щелока
получаемого в процессе электролиза. Например, при повышении
концентрации электролитического щелока на 10 г/л NaOH
расход пара на 1 т каустической соды снижается на 0,4 т (около
0,25 млн. ккал). Соответственно сокращается расход воды и
электроэнергии и повышается производительность установки. По
этому в цехах электролиза стремятся получать щелок с
высокой концентрацией NaOH — 130—140 г/л.
Для дальнейшего снижения расхода пара, стоимость кото
рого составляет основную долю расходов на переработку
щелоков, целесообразно применение одностадийной схемы
выпарки с полным трехкратным использованием тепла греющего
пара. Более глубокий вакуум в III корпусе (остаточное давление
до 0,1 ат) также способствует увеличению полезной разности
температур и повышению производительности выпарной
установки. Расход греющего пара при одностадийной схеме выпар-
'ки составляет около 2,3 млн. ккал на 1 т 92%-ного NaOH (при
мерно 3,7—4 т).
АВТОМАТИЗАЦИЯ ПРОЦЕССА ВЫПАРКИ
П'роцесс выпаривания электролитического щелока является
непрерывным ,и полностью автоматизирован. Автоматизация
его связана с трудностями, обусловленными присутствием в
растворах взвеси кристаллов хлорида натрия, которые осаж
382
даются в коммуникациях и аппаратуре и забивают
трубопроводы и вентили. При достаточной скорости раствора в
трубопроводах, регулярной промывке, а также в результате других
мероприятий обеспечивается нормальная работа контрольно-
измерительных приборов.
На описанной выше технологической схеме (см. рис. 151)
показаны основные точки автоматического контроля н
регулирования процесса.
Подогрев электролитических щелоков контролируется
термометрами сопротивления, установленными после
подогревателей 5 л 6, и регулируется вручную по их показаниям. Выход
конденсата из греющих камер выпарных аппаратов
осуществляется автоматически при помощи регуляторов уровня б,
сблокированных с вентилями, регулирующими количество
отводимого конденсата.
В каждом корпусе первой стадии выпарки автоматически
контролируются давление в греющей камере и в паровом
пространстве и температура раствора, а также 'регулируется
уровень раствора в выпарных аппаратах путем воздействия изо-
дромного регулятора, получающего импульс от сифона
уровнемера, на пневматический клапан, установленный на входе
раствора в данный корпус.
Кроме того, в III корпусе автоматически «оддержяваетск
концентрация щелока около 26% NaOH. При более высокой
концентрации открывается клапан, через который щелок
отводится из III корпуса в сборник 2, при этом уровень
раствора в аппарате 9 понижается и открывается клапан для
входа раствора из аппарата 8 (II корпус). Вследствие
перетока раствора в III корпус уровень раствора во II корпусе
снижается, тогда регулятор уровня подает импульс изодром-
ному регулятору с пневмопередачеи, который открывает клапан
на линии входа раствора из I во II корпус. Уровень раствора
=s I корпусе при этом восстанавливается путем воздействия
регулятора уровня на клапан входа щелока в I корпус.
Описанная система автоматического регулирования,
находясь в состоянии подвижного равновесия, поддерживает
заданный режим процесса выпарки.
На второй стадии выпарки по достижений нужной
концентрации щелочи в растворе D2—50% NaOH) из аппарата 12
автоматически отбирается упаренный раствор и в соответствии
с понижением у'ровня в этом аппарате туда вводится средний
щелок из отстойника //.
Измерение и регулирование концентрации растворов
производится по величине депрессии, определяемой как разность
температур раствора в аппарате и вторичного пара,
увлажненного конденсатом (или конденсата при давлении вторичного
пара)
383
Для поддержания нужного вакуума и уменьшения расход .
воды в конденсаторах автоматически регулируется ее подача
в зависимости от температуры сточной (барометрической»
воды.
Измеряется также расход пара, воды, щелоков, давление
в трубопроводах, аппаратах и напор, создаваемый
насосами. Кроме того, регулируются уровни растворов в баках,
причем указатели уровня сблокированы с насосами, которые
включаются или выключаются по достижении максимального
или минимального уровня в данном аппарате.
Центрифуга работает автоматически по заданной
программе: загрузка—фильтрование—промывка—осушка
осадка—выгрузка.
Основные показания приборов контроля и регулирования
вынесены на центральный щит управления, с которого можно
также дистанционно управлять процессом.
При использовании описанной системы регулирования
снижаются затраты труда, облегчаются условия работы
обслуживающего персонала, уменьшаются отклонения от нормального
хода процесса, что позволяет уменьшить 'расход пара, воды и
энергии и повысить производительность выпарной установка.
ОСНОВНАЯ АППАРАТУРА ВЫПАРНЫХ УСТАНОВОК
Выпарные аппараты. Процесс выпаривания
электролитических щелоков затрудняется из-за отложения на стенках
греющих трубок твердой поваренной соли, выпадающей из раствора
по мере его упаривания, а также вследствие значительного
увеличения вязкости раствора при повышении концентрации
NaOH. Так, вязкость раствора, содержащего 26% NaOH, при
60°С в шесть раз, а при концентрации NaOH 50% в 15 ра
больше вязкости воды. Вследствие отложения соли и повыше
ния вязкости раствора снижается коэффициент теплопередачи
и уменьшается производительность выпарных аппаратов и всеп
установки.
Для устранения этих отложений, которые обычно выпадают
в зоне кипения раствора, применяют выпарные аппараты с так
называемой вынесенной зоной кипения и затопленной
поверхностью теплопередачи. В греющих трубках таких аппаратов
раствор циркулирует с высокой скоростью, а уровень раствора
превышает верхние концы греющих трубок.
В аппаратах с принудительной циркуляцией зона кипения
вынесена из греющих трубок, так как раствор при помощи
циркуляционного насоса непрерывно и многократно
циркулирует через трубки греющих камер со скоростью 2—2.5 м/сек,
нагреваясь при этом лишь на 1—1,5 °С. При таком подогреве
раствор может закипеть только в верхной части трубок нг,
384
уровне сепаратора. Таким образом удается почти полностью
предотвратить отложение солей (инкрустацию) на греющих
поверхностях и одновременно достигнуть высокого
коэффициента теплопередачи A800—2200 ккал/м2-ч•град).
Следует, однако, отметить, что конструкции выпарных
аппаратов с принудительной циркуляцией очень сложны; кроме
того, требуется дополнительный расход электроэнергии на
циркуляцию раствора @,4—0,5 кет на 1 м2 поверхности
теплообмена). Поэтому по возможности избегают применения этих
аппаратов и используют аппараты с естественной
циркуляцией (АЕЦ): например, на первой стадии выпарки, когда
вязкость щелоков еще относительно невелика, а в некоторых
случаях АЕЦ применяют и на второй стадии выпарки.
В аппаратах с естественной циркуляцией скорость движения
раствора составляет 1—2 м/сек, а коэффициент теплопередачи
на 15—20% меньше, чем в аппаратах с принудительной
циркуляцией (при одинаковых условиях). Высокая скорость
естественной циркуляции достигается за счет напора, создаваемого
вскипающим слоем жидкости (парожидкостная эмульсия), при
низком гидравлическом сопротивлении циркуляционного
контура.
При этом кипение жидкости должно происходить выше
греющих трубок (затопленная поверхность теплопередачи).
Это достигается, если сечение циркуляционного контура
примерно в 3—4 раза больше сечения греющих трубок, а над ними
находится достаточно большой слой жидкости, гидравлическое
давление которого предотвращает кипение раствора в трубках.
Ниже описаны аппараты, наиболее широко применяемые
при выпаривании электролитических щелоков.
Выпарной аппарат с подвесной греющей камерой (рис. 152)
является аппаратом с естественной циркуляцией и
применяется преимущественно на первой стадии выпарки.
Греющая камера 1, размещенная в нижней части корпуса,,
выполнена мз легированной нержавеющей стали марки
1Х18Н9Т. Камера представляет собой цилиндр диаметром
2000 мм с двумя плоскими трубными решетками, в которых
развальцованы 614 трубок диаметром 57X3,5 мм и длиной
2300 мм. При этом поверхность теплопередачи составляет
244 м2. Камера свободно лежит на опорах, находящихся в
нижней части корпуса, диаметр которого составляет 2400 мм.
При такой конструкции легко достигается компенсация
теплового расширения греющих трубок.
Греющий пар поступает сверху по центральной трубе
диаметром 250 мм, выходящей наружу через сальник в крышке
аппарата.
Конденсат отводится по трубе с компенсатором,
проходящей через нижнюю трубную решетку.
25-2848 385
Сепаратор 2 (диаметр 3000 мм) кверху расширен для
увеличения его объема и улучшения отделения капель раствора
от пара. В верхней части сепаратора помещен центробежный
брызгоотделитель 4, представляющий собой устройство,
подобное лопасти вентилятора (см. вид по АА). Соковый пар,
уносящий брызги раствора, поступает снизу в центральную
часть сепаратора и, про-
греюшийпар ходя п0 Нему,
приобретает вращательное
движение. Брызги раствора
под действием
центробежной силы
отбрасываются к стенке
аппарата, укрупняются и
стекают вниз через
отверстия в перегородке.
Жидкость отводится по трубе
в нижнюю часть
выпарного аппарата.
Пар, освобожденный
от брызг, выходит из
аппарата через боковые
штуцеры в крышке.
Соль, выпадающая при
выпаривании щелока,
оседает в нижней конусной
части аппарата и
непрерывно, в смеси с
упаренным раствором, в виде
пульпы отводится из
аппарата.
Выпарной аппарат с
принудительной
циркуляцией (рис. 153)
применяется на второй стадии
Рис. 152. Выпарной аппарат с подвесной выпарки. Выносная гре-
гоеющей камерой: ющая камера выполнена
/-греющая камера; 2-сепаратор; J-кольцевой тру- 197 -rnvfi лпнипй ^ „ „
бопровод для промывки аппарата; 4—центробежный ил 1Л1 'РУи Длиичи О М И
брызгоотделнтель; 5-лазы. ДИЭМетрОМ 38X2,5 ММ
Поверхность
теплопередачи равна 59 м2. Верхняя
часть камеры тангенциально присоединена к сепаратору 2, в
верхней части которого помещен брызгоотделитель 3. В
крышке аппарата находится штуцер для отвода вторичного пара.
Над входом в аппарат циркулирующего раствора из греющей
камеры установлен козырек 5, препятствующий уносу брызг с
поверхности раствора.
386
Упаре/-уыи
щелок
Соковый пар
'Щелок C0%-ный
NaOH)
Из нижней конической части аппарата раствор вместе с
выпадающей из него солью направляется лопастным
циркуляционным насосом 6 в греющую камеру 1, откуда подогретый
раствор поступает в сепаратор. Здесь благодаря перегреву
испаряется часть воды и раствор вновь опускается вниз к насосу.
Часть раствора, соответствующая заданной
производительности аппарата, непрерывно отводится через штуцер,
расположенный ниже греющей
камеры.
Производительность
циркуляционного насоса
составляет 750 м3/ч. Создаваемый им
напор невелик C—4 м вод. ст.),
так как необходимо
преодолеть только гидравлическое
сопротивление циркуляционного
контура. Мощность
электродвигателя насоса около 30 кет,
расход электроэнергии
примерно 0,5 кет • ч на 1 м2
греющей поверхности. Количество
вторичного пара колеблется з
пределах 70—100 кг/ч на 1 м2
греющей поверхности.
В последнее время для
второй стадии выпарки начали
применять выпарные аппараты
с естественной циркуляцией,
снабженные греющими
трубками длиной 4 м. Высота слоя
раствора над верхом греющих
трубок в таких аппаратах
составляет около 3 м.
Расход пара на выпарку
значительно сокращается при
повышении
производительности выпарной установки
Опорожнение
В выпарной системе, имеющей Рис- 153- Выпарной аппарат с прину-
поверхности теплообмена в д™ Ж^Е^-Г ^
ВЫпарнЫХ аппаратах ПерВОИ ,_греЮ1цая камера: 2-сепаратор: 3-брызго-
СТЭДИИ 220—240 М2, МОЖНО ПО- отделитель; 4—смотровые стекла; 5—козырек:
1 пп 1 пл / 5—циркуляционный насос.
лучить около 100—120 т/сутки
каустической соды (в
пересчете на 92%-ный NaOH).
Разработаны конструкции выпарных аппаратов поверхностью
теплообмена 400 и 600 м2, применение которых позволит
повысить производительность процесса выпаривания.
25*
387
Центрифуга (рис. 154). Для отделения соли от средних
щелоков поименяются горизонтальные фильтрующие
автоматические центрифуги полунепрерывного действия типа АГ-1800
(цифра обозначает диаметр корзины в миллиметрах).
Корзина 2 центрифуги делает 500—700 об/мин. Из напорного бака
через вентиль 8 с гидравлическим приводом в центрифугу при
вращении ротора подается солевая пульпа, по окончании
загрузки центрифуги вентиль закрывается и в течение 1 мин
производится фильтрование. Центробежной силой пульпа
отбрасывается к стенкам корзины, при этом соль задерживается на
Пульпа
Сола на
растбореиие Маточный
раствор
Рис. 154. Горизонтальная автоматическая центрифуга' типа АГ-1800
полунепрерывного действия:
/—корпус; 2—корзина (ротор); 3—фильтрующая сетка: 4—иож для срезания соли;
5—гидравлический привод иожа; 6—приемный бункер для соли; 7—коробка для
отвода маточных и промывных растворов; 8—загрузочный вентиль с гидроприводом.
никелевой фильтрующей сетке 3, а раствор проходит через нее
я через клапанную коробку 7 стекает в приемный бак.
После этого в течение 10—15 сек производится промывка
осадка соли электролитическим щелоком. Затем осадок
подсушивается и снова промывается водой. Отфильтрованная про
мытая соль срезается ножом 4, приводимым в движение
гидроприводом 5. и сбрасывается в бункер 6, откуда поступает на
растворение. Одновременно в бункер автоматически подается
воча, что способствует более быстрому удалению соли.
4. ОБЕЗВОЖИВАНИЕ (ПЛАВКА) ЕДКОГО НАТРА
Для некоторых процессов (например, процессы
органического синтеза, электролитическое получение металлического
натрия и др.), для лабораторных работ и ряда мелких
потребителей, удаленных на большие расстояния от поставщиков
каустической соды, требуется твердый практически безводный
едкий натр. При
обезвоживании едкий натр очищается
от примесей железа, соды
и др. Потребность в
безводном едком натре невелика—
около 7—10% от объема
производства каустической
соды.
Обезвоживание
(выпаривание) каустической соды
до концентрации 70—73%
NaOH можно проводить в
выпарных аппаратах,
обогреваемых водяным паром
через стенки греющих
трубок, при разрежении в
пространстве над раствором.
Дальнейшее обезвожи-
350
300
250
!
I
too
50
о
-25
-37,5
„——'
Пар
—
7/77
"^\ *•
nSnml
1
ч
/"
/
\s
у
А
/
///
у
у-
/
/
\
'/А
]
\
}
J
/Твердая
С фаза
!
1
h
//
1ш
if
(j
20 ЬО 60 80
Содержание NaOH, %
/00
Рис. 155. Температуры плавления (и
кипения) растворов едкого натра.
вание и повышение
концентрации едкого натра (выше
73% NaOH) путем обогрева
водяным паром не удается.
Это связано с высокой
депрессией
концентрированных растворов NaOH.
Температура кипения растворов
и плавления с увеличением концентрации NaOH быстро
повышается (рис. 155). При 74% NaOH и выше температура
плавления системы вода—щелочь резко возрастает и для чистого
едкого натра достигает 328 °С. В г;>исутствии примесей,
например Ыа2СОз, температура плавления несколько ниже C10—
315°С). Температура кипения растворов щелочи также резко
увеличивается с повышением их концентрации. Чистый едкий
натр кипит при 1388°С, в присутствии 2% воды (98% NaOH)
температура кипения понижается до 375 °С.
Вследствие таких высоких температур кипения
концентрированной щелочи при обезвоживании едкого натра в качестве
теплоносителей применяют горячие топочные газы, перегретую
воду, высококипящие органические растворители (ВОТ).
Обезвоживание (плавку) едкого натра проводят в котлах при
389
атмосферном давлении или разрежении периодическим или
непрерывным способом.
Для снижения температуры кипения растворов едкого натра
применяют аппараты, работающие при разрежении. Однако
понижение давления более чем на 0,5 ат над
концентрированными растворами (82% NaOH и более) недопустимо, так как
раствор в этих условиях затвердевает. Кривая температур
кипения раствора на рис. 155 под давлением, например 0,2 атм
в области высоких концентраций NaOH пересекает кривую
температур плавления и проходит ниже ее (см. пунктирную
линию), т. е. в области твердой фазы.
Поэтому периодический процесс плавки при
концентрации выше 82% NaOH ведут под атмосферным давлением.
Непрерывное обезвоживание проводят при остаточном давлении
не ниже 0,5 атм.
В последнее время для обезвоживания применяются более
совершенные и экономичные вакуум-выпарные аппараты
непрерывного действия из никеля. В качестве теплоносителя в
таких аппаратах применяются ВОТ (стр. 375).
Обезвоживание в котлах. Котлы для обезвоживания
технического едкого натра выполняют из щелочестойкого чугуна с
добавкой до 1 % никеля. Котлы имеют объем 9—18 м3 и
замуровываются в кирпичную кладку печи группами по
нескольку котлов (батареи). Их обогревают горячими топочными
газами, полученными сжиганием угля, мазута или горючего
газа.
Для защиты внутренних стенок котлов от коррозии и
осветления каустической соды к обезвоживаемому раствору
добавляют селитру и серу.
При обезвоживании чистой каустической соды, полученной
из электролизеров с ртутным катодом (стр. 351), применяют
котлы из чистого никеля меньшего (чем чугунные котлы)
объема и обогреваемые электрическим током. Таким образом
предотвращается загрязнение продукта. Применение топочных
газов в этом случае недопустимо, так как содержащаяся в газах
сера разрушает стенки никелевого котла.
Одна из наиболее типичных плавильных батарей,
работающих периодически, схематически показана на рис. 156. Батарея
состоит из трех котлов — среднего подогревателя 2,
расположенного выше остальных, и двух крайних плавильных котлов /.
Раствор каустической соды, упаренный до концентрации
50—70% NaOH, поступает из напорного бака в
котел-подогреватель. Здесь щелочь нагревается отходящими топочными
газами и непрерывно перетекает по сифону или штуцеру из
верхней части этого котла в плавильные котлы. В плавильном
котле, обогреваемом топочными газами, происходит
обезвоживание раствора каустической соды. Влага удаляется в атмо
390
сферу, а содержимое котла пополняется раствором щелочи из
подогревателя. Полностью обезвоженный плав, температура
которого достигает 500°С, отстаивается в течение 8—10 ч и
медленно охлаждается (при погашенной топке). Затем из
верхней части котла при помощи погружного насоса 3,
подвешенного к монорельсу над котлами, отбирают чистый плав (бе-
Рис. 156. Схема плавки едкого натра в открытых
котлах периодического действия:
1—плавильные котлы; 2—котел-подогреватель; 3—погружной
вертикальный насос; 4—топка; 5—боров.
лый каустик), сливаемый в барабаны. Один насос
обслуживает несколько котлов. Продолжительность операции плавки
40—50 ч.
Остающийся в котле нижний слой плава (около 2% общего
количества)—так называемый красный каустик — содержит
все примеси, главным образом железо, и используется
отдельно.
391
Батарея плавильных котлов непрерывного действия,
представленная на рис. 157, состоит из шести
котлов-подогревателей 2 и дезяти плавильных котлов 3. В каждом подогревателе
и плавильном котле автоматически поддерживается заданный
режим (температура, давление и концентрация щелочи).
В четвертом—шестом подогревателях и первом—четвертом
плавильных котлах создается вакуум. Наиболее высокая
температура D90—520 °С) развивается в четвертом плавильном
котле, в следующих котлах она снижается до 360—325 °С. Преиму-
Твплц/Ь
Рис. 157. Схема плавки едкого натра в котлах непрерывного действия:
/—бак для раствора каустической соды; 2—котлы-подогреватели: 3—плавильные кетли; 4—
топки; 5—воздушный вентилятор; 6—подогреватель воздуха.
щества такой батареи по сравнению с описанной выше —
меньший расход тепла, более высокая производительность (до
60 т/сутки) и получение более однородного продукта.
Обезвоживание в выпарных аппаратах. Наиболее
современная непрерывнодействующая установка для обезвоживания
каустической соды в плавильных выпарных аппаратах с
никелевыми греющими трубками и обогревом парами ВОТ показана
на рис. 158.
Раствор каустической соды (концентрация NaOH 50%),
полученный в результате упаривания электролитического щелока,
подается центробежным насосом (на схеме не показан) в
теплообменник 6, где подогревается через стенки трубок
вторичным паром второй стадии выпарки. Подогретый раствор сме
шивается в аппарате 5 с перегретым вторичным паром из
выпарного (плавильного) аппарата 7. Перегретый пар,
выделяющийся из расплава щелочи, температура которого достигает
380 °С, дополнительно подогревает раствор, при этом выделяют
ся растворенные в нем газы. В нижней части подогревателя 5
пары отделяются от жидкости (щелочи), которая стекает и
392
приемный бак 12. Пары через аппарат 4, где улавливаются
брызги щелочи, поступают в конденсатор смешения <?, откуда
неконденсирующиеся газы отсасываются вакуум-насосом, а
охлаждающая вода через барометрический ящик 13 стекает в
канализацию.
Первая стадия упаривания проводится в длиннотрубном
йыпарном аппарате 2 пленочного типа, обгреваемого вторим-
ВОТ
Рис. 158. Схема обезвоживания раствора каустической соды в выпарных
аппаратах с обогревом парами ВОТ:
/, 2, 7—выпарные аппараты; 3—барометрические конденсаторы смешения; 4—брызгоотдели
сель; 5—аодогреватель смешения; 6—поверхностный теплообменник; 8—гидравлический
затвор; 9, 12, 14—баки; 10—кристаллизатор; 11—погружной насос; 13—барометрический ящик.
ным паром из аппарата /. Раствор подается в выпарной
аппарат снизу погружным насосом //.
Упаренный до концентрации 65% NaOH. раствор стекает в
сборный бак 14, откуда погружным насосом подается в
выпарной аппарат /, обогреваемый парами ВОТ, имеющими
температуру 380°С, давление 8 ат. Здесь раствор упаривается до
концентрации 70—72% NaOH. Вторичный пар из этого
аппарата при давлении 4 ат передается на обогрев аппарата 2,
а щелочь под давлением перетекает в аппарат 7 для
окончательного обезвоживания, проводимого при разрежении (оста-
гочное давление 0,5 атм). Проходя снизу вверх в виде пленки
по греющим трубкам этого аппарата, обогреваемым парами
393
ВОТ, раствор полностью обезвоживается (содержание
твердого вещества 99,9%). Плав стекает через гидравлический
затвор 8 в обогреваемый сборный бак 9, откуда непрерывно
подается в барабанный кристаллизатор 10.
Каустическая сода кристаллизуется из плава на наружных
стенках охлаждаемого барабана, снимается с него ножом и
ссыпается в приемный барабан через течку, в которую встроена
дробилка. При этом получается продукт в виде мелких чешуек
(чешуированная каустическая сода).
Можно передавать обезвоженный плав непосредственно в
барабаны, минуя кристаллизатор. В этом случае в барабане
помещается почти в два раза больше продукта, чем чешуиро-
ванной каустической соды. Однако применение такого
продукта, представляющего собой после застывания монолитную
массу, гораздо менее удобно для потребителей.
Вследствие сильного коррозионного действия, которое
оказывает плавленая каустическая сода на стальную и чугунную
аппаратуру, описанная установка выполняется из никеля, что
обусловливает гораздо меньшие расходы на ремонт аппаратов.
Для защиты никеля от разрушения хлоратами, содержащимися
в электролитическом щелоке, в раствор, подаваемый на
обезвоживание, добавляют сахарозу, которая окисляется хлоратами
с выделением СО2, образующего в щелочной среде карбонат
натрия.
Производительность описанной установки составляет
100 т/сутки твердой каустической соды. Расход топлива при
начальной концентрации раствора 50% NaOH в 1,5—2 раза
меньше, чем в батарее, состоящей из плавильных котлов, и
составляет около 220—240 кг условного топлива G000 ккал/кг)
на 1 т получаемой безводной щелочи.
Сорта каустической соды
Промышленность вырабатывает каустическую соду — техни
ческий едкий натр — в твердом и жидком виде.
Твердый едкий натр технический выпускается двух марок —
А и Б. Продукт марки А — химический A-й и 2-й сорт)
получается из кальцинированной соды химическими способами,
продукт марки Б — диафрагменный — из жидкого едкого натра,
образующегося в результате электролиза растворов хлористого
натрия в ваннах с диафрагмой.
Жидкий едкий натр технический выпускается пяти марок.
Продукт марок А, Б и В получают электрохимическим
методом, продукт марок Г и Д — химическими.
Согласно ГОСТ 2263—59, качество технического едкого
натра по содержанию основного вещества (NaOH) и
примесей должно соответствовать следующим показателям:
394
Содержание NaOH,
%, не менее . .
Содержание
примесей, %, не более
Na2CO3 ....
NaCl
окислы Fe, Al,
Мп (суммарно)
железо (в
пересчете на Fe2O3)
Твердый едкий
марка А
(химический)
i сорт
-
96
2,0
1,0
0,3
Не
нормируется
1
Copied
95
3,0
1,5
0,2
иатр
марка
Б
афраг-
нный
92
2,5
3,75
Не i
0,2
марка
А
~3
н
о.
42
0,6
0,05
Жидкий едки
марка
Б
111
¦t§?=
з JJ 4 2
50
1,0
2,2
ормируетс
0,0015
0,04
марка
В
афраг-
нный
Д1
42
2,0
4,0
я
0,04
й иатр'
марка
Г
марка Д
химический
43
2,0
1,0
0,05
0,02
42
2,5
2,0
Не
нормируется
0,2
* Содержание примесей отнесено к 100% NaOH.
Жидкий едкий натр марки А (ртутный) должен
удовлетворять следующим дополнительным требованиям по содержанию
примесей (в % не более):
Сульфаты 0,02
Кальций 0,006
Тяжелые металлы (Pb, Sn и др.) 0,003
Кремнекислота 0,008
Упаковка и перевозка каустической сод
ы
Жидкий едкий натр транспортируют в железнодорожны.ч
цистернах, металлических контейнерах для перевозки
жидкостей и в стальных бочках. Твердый едкий натр перевозят в
барабанах из кровельной стали емкостью по 50 и 100 л. Твердый
едкий натр, выпускаемый в виде пластин-чешуек,
транспортируют в барабанах из кровельной стали с гофрированными
корпусами емкостью 25—100 л.
Чешуированный едкий натр загружают также в
железнодорожные цистерны и в них же растворяют на месте потребления.
При этом уменьшаются расходы на тару и перевозку продукта.
Глава IX
ПЕРЕРАБОТКА ЭЛЕКТРОЛИТИЧЕСКОГО ХЛОРА
Электролитический хлор, получаемый в цехе электролиза,
используется на этом же заводе для производства жидкого
хлора (сгр. 368), отбеливающих средств (хлорная известь, ги-
похлорит кальция, гипохлорит натрия), хлоратов, различных
хлоридов (хлорное железо, четыреххлористый кремний, четы
реххлористый титан, хлористый алюминий, хлористый аммо
ннй и др.), а также направляется на синтезы различных хлор
органических соединений (стр. 328), которые в данной книге
не рассматриваются. Цехи, в которых перерабатывается хлор,
называются хлоропотребляющими. В составе хлорного завода
почти всегда организуется также производство синтетического
хлористого водорода и соляной кислоты.
/. ПРОИЗВОДСТВО ХЛОРИСТОГО ВОДОРОДА И СОЛЯНОЙ
кислоты
Свойства и применение HCI. Хлористый водород — бесцвет
ный газ с резким запахом, плотность его при нормальных
условиях 1,639 кг/м3, температура кипения —85°С, температура
плавления —1!4,2°С. Хлористый водород хорошо растворяется
в воде с выделением значительного количества тепла, на
воздухе он дымит, образуя с влагой воздуха туман. Водный
раствор хлористого водорода называется соляной кислотой.
Растворимость хлористого водорода в воде зависит от тем
пературы. Минимальная растворимость при температуре 110°С
составляет около 20% НС1, при этом образуется азеотропная
смесь. Максимальная растворимость при температуре 20 °С и
давлении НС1 709 мм рт. ст. достигает 42% НС1. В
промышленности применяют соляную кислоту, содержащую от 27,5 до
37% НС1.
Соляная кислота представляет собой бесцветную или жел-
говатого цвета прозрачную жидкость. Желтый цвет вызывается
присутствием примесей, главным образом солей железа.
Соляная кислота относится к числу наиболее сильных одноосновных
кислот (степень диссоциации 95—96%) и энергично растворяет
многие металлы, реагирует с их окислами.
В промышленности широко применяются как хлорисгьш
водород, так и соляная кислота. Хлористый водород
используется главным образом для производства хлорорганических
продуктов путем гидрохлорирования непредельных
углеводородов. Взаимодействием хлористого водорода с ацетиленом
получают хлористый винил, с этиленом — хлористый этил. Может
быть осуществлен также процесс окислительного хлорирования
органических соединений хлористым водородом (стр. 326).
Соляная кислота применяется для получения хлоридов
цинка, аммония, бария, магния, кальция, железа, для травления
при пайке, при лужении, в гальванопластике, для очистки
паровых котлов от накипи, используется в гидрометаллургии
платины, золота, серебра, в нефтяной промышленности — при
бурении нефтяных скважин. Кроме того, соляная кислота
применяется при гидролизе древесины, при дублении и окраске
кожи, при крашении тканей, в производстве красителей,
уксусной кислоты, пластических масс и т. д.
Общий масштаб потребления соляной кислоты весьма
значителен и непрерывно возрастает.
СПОСОБЫ ПРОИЗВОДСТВА ХЛОРИСТОГО ВОДОРОДА
И СОЛЯНОЙ КИСЛОТЫ
В промышленности хлористый водород можно получать
несколькими способами.
Сульфатный способ заключается в действии 92—93%-ной
серной кислоты на поваренную соль при 500—550°С:
2NaCl + HaSO4 = Na.jSO4 + 2НС1
Процесс протекает в печах, обогреваемых топочными газами,
имеющими температуру 950—1000°С. Вследствие негерметич
ности печей концентрация получаемого хлористого водорода
составляет 30—50% НО. Газ загрязнен брызгами серной
кислоты и другими примесями. Поэтому абсорбцией хлористого
водорода водой можно получить только техническую соляную
кислоту, концентрация которой не превышает 28—31 % НС1.
Этот способ применялся раньше главным образом для
получения сульфата натрия. В связи с открытием природных
месторождений сульфата натрия этот способ почти всюду полностью
утратил свое значение, новые производства по этому способу
в настоящее время не организуют.
Синтез хлористого водорода из хлора и водорода нашел
широкое применение во всех странах мира и в настоящее
время является основным. По этому способу получается чистый
кониентрированный хлористый водород, содержащий 95—96%
НО. Абсорбцией синтетического хлористого водорода получают
397
техническую соляную кислоту высокой чистоты, а в некоторых
случаях — химически чистую кислоту.
Хлористый водород получается также в качестве отхода при
хлорировании насыщенных углеводородов:
RH -f С12 > RC1 + НС1
Концентрация побочно образующегося (отбросного)
хлористого водорода весьма различна и колеблется в широких
пределах (от 25—35 до 95—98% НС1). Содержание примесей
зависит от свойств хлорируемых веществ и получаемых при
хлорировании продуктов и условий процесса.
Производство хлорорганических продуктов непрерывно
развивается, в связи с этим увеличивается и количество
отбросного хлористого водорода. В настоящее время его получается
в 1,5—2,5 раза больше, чем синтетического хлористого
водорода. Для правильного использования отбросного хлористого
водорода разработаны способы его очистки и регенерации в
элементарный хлор (стр. 326, 350).
Абсорбцией неочищенного отбросного хлористого водорода
водой получают техническую соляную кислоту, загрязненную
органическими примесями. Она находит ограниченное
применение в некоторых отраслях промышленности, не
предъявляющих высоких требований к качеству соляной кислоты.
Отбросный хлористый водород до абсорбции водой може(
быть очищен путем поглощения примесей органических
соединений и хлора некоторыми высококипящими органическими
растворителями (например, зеленое масло*, хлорированный
керосин и др.). Очистку можно осуществить также
сорбцией примесей, например на активированном угле. Однако эти
методы очистки хлористого водорода сложны и не всегда
дают удовлетворительные результаты, а потому пока редко
используются.
Широкое применение получает метод очистки и
концентрирования хлористого водорода, заключающийся в абсорбции
его 20%-ной соляной кислотой (азеотропная смесь). При этом
получается 32—35%-ная соляная кислота, из которой затем
отпаривается чистый концентрированный хлористый водород.
Кубовый остаток (азеотропная смесь) возвращается на
абсорбцию НС1.
При абсорбции азеотропной смесью хлористый водород
разбавляют воздухом, поэтому легколетучие органические примеси
удаляются с отходящими газами. Высококипящие,
труднолетучие органические примеси при абсорбции поглощаются и
могут загрязнить соляную кислоту. Однако при отгонке
хлористого водорода из кислоты в ректификационной колонне
* Зеленое масло — отход переработки нефтепродуктов.
398
эти примеси остаются в кубе колонны, а отгоняется чистый
хлористый водород (99,5—99,9% НС1) без примесей. Этим
способом концентрируют также синтетический хлористый
водород.
Регенерированный НС1 можно использовать для
окислительного хлорирования или гидрохлорирования. Разработаны
методы регенерации отбросного хлористого водорода
каталитическим окислением или электролизом с получением хлор-
газа.
СИНТЕЗ ХЛОРИСТОГО ВОДОРОДА
Производство синтетического хлористого водорода возникло
лишь в 20-х гг. текущего века. В последнее десятилетие резко
возросла его выработка, в десятки раз увеличена мощность
печей для синтеза, усовершенствован технологический процесс.
Для производства синтетического хлористого водорода
использую г хлор и водород, получаемые при электролизе водных
растворов хлоридов щелочных металлов.
Реакция между хлором и водородом протекает с выделением
тепла и без изменения объема:
Н2 + С12 » 2HCI + 44 ккал
При обычных температурах в темноте хлор и водород не
взаимодействуют. На свету или при нагревании реакция в смес<1
этих газов сопровождается взрывом. Спокойное протекание
реакции между хлором и водородом (без взрыва) возможно пои
непрерывном поступлении газов в реакционную зону и высокой
температуре (обычно 2000—2400°С), которая поддерживается
за счет выделения реакционного тепла. Реагирующие газы
образуют светящийся факел бледно-лунного цвета.
Точная дозировка газов невозможна, поэтому один из
реагентов подают в избытке. При избытке хлора режим работы
называют хлорным, при избытке водорода — водородным.
Применяется преимущественно водородный режим (избыток
водорода 3—10%), при этом получаются хлористый водород и
соляная кислота, не содержащие хлора, и уменьшается
коррозия аппаратуры.
Процесс проводят з аппаратах, называемых печами синтеза,
конструкции которых в зависимости от способа отвода тепла
реакции можно разделить на две большие группы: печи с
воздушным охлаждением и печи с водяным охлаждением.
Охлаждение стенок печи синтеза водой, необходимое для
отвода весьма значительного тепла реакции (около 600 тыс. ккал
на 1 т НС1), позволяет значительно интенсифицировать
процесс теплопередачи, благодаря чему возрастает
производительность печи без увеличения ее размеров.
399
Наиболее широкое применение нашли печи из углеродистых
или легированных жароупорных сталей. Материалами для
печей синтеза могут служить также кварц, углеродистые стали,
футерованные керамикой, диабазом, шамотом или платирован-
ные свинцом, а также графит. Однако из-за низкой
теплопроводности неметаллические материалы для этих целей в
настоящее время применяются редко, только в случае необходимости
получения чистой соляной кислоты. Печи, изготовленные из этих
материалов, имеют низкую производительность — 2—3 т/сутки
100%-ногоНС1.
Хлористый водород при температуре выше 120—150°С
(т. е. выше точки росы содержащейся в нем влаги) не
оказывает коррозионного действия на сталь. При водородном режиме
(восстановительная среда) на внутренней стороне стенки печи
синтеза образуется защитная пленка хлористого железа. Она
разрушается при температуре ниже 80—100°С, когда на стен
ках конденсируется влага, или при температуре выше 600 °С,
когда хлористое железо начинает возгоняться.
Поэтому производительность печей из углеродистой стали с
воздушным охлаждением должна быть такой, чтобы
температура стенок печи не превышала 450—500 °С, а при водяном
охлаждении температура стенки печи не должна быть ниже
100—120 °С (соответственно температура охлаждающей воды
может быть только на 10—15°С ниже температуры стенки). На
рис. 159 показана двухконусная печь для синтеза хлористого
водорода с воздушным (естественным) охлаждением.
Форма корпуса 2 печи близка к форме факела пламени ре
агирующих хлора и водорода, благодаря чему достигается
равномерная температура всей поверхности стенок. Печь
устанавливается на раму днищем или подвешивается на лапах,
приваренных сверху (во избежание местных перегревов стенок
приваривание к ним лап не допускается).
В днище печи установлена горелка 5, представляющая собой
две концентрически расположенные трубки. Внутренняя трубка,
по которой подается хлор, снабжена кварцевым наконечником,
наружная выполнена из стали. По кольцевому пространству
между трубками в горелку поступает водород. На выходе ич
горелки газы смешиваются и спокойно реагируют, образуя
факел, температура которого достигает 2000—2400 °С. Наблюдение
за пламенем осуществляется через смотровое стекло 4. На
противоположной стороне печи имеется штуцер с заглушкой,—
запальное отверстие 5 для зажигания печи. В верхней части
установлена выхлопная мембрана 1 из тонкой жести,
защищенная изнутри листом асбеста.
При установке такой мембраны предотвращается
разрушение стенок печи в случае неравномерного горения пламени
(хлопки).
400
Рис. ЮУ. Печь для синтеза хлористого водорода с воздушным
охлаждением:
.'—выхлопная мембрана; 2—корпус печи; 3—запальное отверстие; 4—
смотровое стекло; 5—горелка; 6—опора печи.
Диаметр средней части печи 1200 мм, нижней 600 аш,
верхней 460 мм, производительность ее до 10 т/сутки 100%-ногоНС1.
Печи такого типа изготовляются и больших размеров,
производительностью 15—16 т/сутки 100%-ного НС1. Печь
работает при избыточном давлении до 0,7 ат, при этом давлении газ
¦26—2848
401
Вода
поступает потребителям. Если хлористый водород
направляется на абсорбцию, давление в печи снижается почти до
атмосферного.
На рис. 160 показана печь с водяным охлаждением.
Основные ее детали такие же, как печи с воздушным охлаждением.
В отличие от последней, корпус печи заключен в резервуар с
проточной водой, в котором расположен также змеевик 2 для
охлаждения газа,
выходящего из печи.
Производительность такой печи, в
зависимости от размеров, достигает
25—35 т/сутки 100%-ного
хлористого водорода.
На рис. 161 приведена
технологическая схема синтеза
хлористого водорода с
последующей адиабатической
абсорбцией его для получений
технической соляной кислоты
и концентрированием
хлористого водорода путем отпарю;
соляной кислоты.
Водород из цеха электро
лиза (98% Н2) проходят
водоотделитель / и гравийный ог-
непреградитель 2,
предотвращающий проникание пламени
в трубопровод, и
направляется в горелку печи синтеза 4
Вода из аппаратов 1 и 2
через регулятор уровня
сливается в канализацию.
Хлоргаз, поступающий
также из цеха электролиза (94—
96% С!г), проходит буфер S
для смягчения толчков газа
и отделения жидкого хлора,
который может образоваться
в трубопроводе в зимнее
время, и брызг серной кислоты,
увлекаемых газом из колонн
осушки хлора, и подводится к
горелке печи синтеза.
Собирающийся в буфере 3 жидкий хлор испаряется при
обогреве паром (днище буфера снабжено паровой рубашкой) и
возвращается в хлоропровод.
Серная кислота отводится через продувочную трубку.
402
воаа
Рис. 160. Печь для синтеза
хлористого водорода с водяным охлаждением:
/—корпус печи; 2—змеевик для охлаждения
хлористого водорода; 3— охлаждающий ре-
вервуар; 4—выхлопная мембрана; 5—горелка;
6—смотровое стекло.
Рис. 101. Схема синтеза хлористого водорода, получения соляной кислоты и кои
центрирорания хлористого водорода абсорбцией—десорбцией:
/—водоотделитель; 2—огнепгеграднтель; 3- хлорный буфер; 4—печь синтеза; 5— воздушный
холодильник; б—абсорбционная колонна; 7—санитарная колснна; в—ии»ектор; 9—воздушный вентилятор; 10—
цистерна для соляной кислоты; //—бурак; /2—цент| обежные насосы; 18—абсо| бционьая колошш
С охлаждением в нижней части; 14, /9—баки для соляной кислоты; /5—отпа] пая (десо| бциопная)
колонна; /5—водяной холодильник; 17—рассольный холодильник; 18— тсплссбмснник.
Тлор
Полученный в печи синтеза хлористый водород, на выходе
из печи имеющий температуру около 250—350 °С, охлаждается
в воздушном холодильнике 5 до 120—150 °С и далее може~
быть использован для производства соляной кислоты путем
абсорбции его водой или направлен на концентрирование до
содержания 99,5—99.9% НС1 и осушку.
ПОЛУЧЕНИЕ СОЛЯНОЙ КИСЛОТЫ
В процессе абсорбции хлористого водорода водой с
образованием раствора соляной кислоты выделяется значительное
количество тепла — около 17,5 ккал/моль, или около 140 тыс.
ккал на 1 т 31%-ной соляной кислоты. Этого количества тепла
достаточно, чтобы нагреть образующуюся кислоту до кипения.
Поэтому в процессе абсорбции необходим отвод тепла, в
противном случае не удается получить кислоту нужной
концентрации, так как растворимость хлористого водорода с повышением
температуры уменьшается.
Процесс абсорбции НС1 ведут в абсорберах с отводом теплг
через стенку (изотермическая абсорбция) или в абсорберах ¦
отводом тепла путем испарения части воды (адиабатическая
абсорбция). Вследствие того, что соляная кислота имеет
сильное коррозионное действие, подбор конструкционных мате
риалов для аппаратуры изотермической абсорбции очень ело
жен. Неметаллические материалы (керамика, стекло, фарфор,
кварц, диабаз, фаолит) имеют низкую теплопроводность и не
достаточно высокие механические свойства (хрупкость и др.).
Устойчивы к действию соляной кислоты графит и тантал,
однако дороговизна этих материалов и некоторые другие
недостатки ограничивают их применение.
Для проведения процесса адиабатической абсорбции, раз
работанного впервые в Советском Союзе, не требуется
применения теплопроводных материалов. Абсорбция осуществляется
в простых аппаратах колонного типа (см. рис. 161),
изготовляемых из фаолита и заполненных цилиндрическими кольцами
Хлористый водород поступает в колонну 6 снизу, вода
движется противотоком газу сверху. В нижней части колонны об
разующаяся соляная кислота охлаждается и донасыщаетск
хлористым водородом. По мере движения газа вверх концен
трация стекающей кислоты уменьшается, поэтому количестве
выделяющегося гепла непрерывно возрастает. В средней части
колонны кислота кипит при температуре около 110°С, концен
трация ее здесь составляет 20% НС1, что соответствует составу
азеотропной смеси. Вследствие испарения азеотропной смеси
происходят охлаждение кислоты в верхней части колонны и
абсорбция хлористого водорода водой, поступающей в верхнюю
часть колонны. Отхолящие гачы, содержащие 1 —1,5% HCl
направляются в колонну 7, орошаемую водой или известковым
молоком. Жидкость, выходящая нз колонны 7, содержит 0,2—
0,5% НС1 н направляется в канализацию кислых стоков.
Отходящие из колонны 7 газы, освобожденные от хлористого
водорода, отводятся в атмосферу воздушным инжектором 8.
Воздух подается в инжектор вентилятором 9.
Температура соляной кислоты, вытекающей из колонны 6,
составляет примерно 70°С, концентрация 31—31,5% НС1.
Путем адиабатической абсорбции не удается получить более
концентрированную кислоту даже при пониженной температуре.
что является недостатком данного метода. Если необходимо
повысить концентрацию кислоты, устанавливают дополнительный
абсорбер, выполненный, например, из графита (см. аппарат 13
на рис. 161). Тепло отводится в нем через стенку, а кислота
одновременно донасыщается НС1.
Полученная соляная кислота направляется в стальные
цистерны 10, гуммированные и футерованные поверх гуммировки.
Отсюда соляная кислота через сифон отбирается при помощи
вспомогательного сосуда // (так называемый бурак) и
перекачивается на склад центробежным насосом, изготовленным из
пластмассы, или передается потребителям.
При пуске насоса сосуд // заполняют водой, таким образом
заливают сифон и насос. Цистерны для соляной кислоты не
имеют нижних штуцеров, так как при разрушении штуцера
вследствие коррозии произойдет утечка большого количества,
кислоты.
КОНЦЕНТРИРОВАНИЕ ХЛОРИСТОГО ВОДОРОДА
Для получения концентрированного сухого хлористого во
дорода газ из печей синтеза или отбросный хлористый водород
(отход производства хлорорганических продуктов) направляют
з колонну 13 (см. рис. 161) на абсорбцию азеотропной смесью.
Из нижней части колонны 13 вытекает 31—35%-ная соляная
кислота, нз верхней части выходят отходящие газы,
направляемые в санитарную колонну (на схеме не показана),
аналогичную колонне 7. Кислота собирается в бак 14, через сифон
подается в теплообменник 18, где подогревается до 60—70 °С, и
поступает в отпарную графитовую колонну 15. В кипятильнике
этой колонны, также выполненном из графита, кислота
нагревается паром для удаления НС1. Из нижней части колонны 15
вытекает 20%-ная соляная кислота (азеотропная смесь). В те-
алообменнике 18 она охлаждается до 35—40°С, нагревая
кислоту, поступающую на отпаривание, и направляется в бак 19.
откуда перекачивается на абсорбцию в колонну 13.
Хлористый водород, выходящий из верхней части колон-
чьт 15, охлаждается водой в холодильнике 16 и холодным рас
солом (—15 С) в холодильнике 17. После охлаждения газ
содержит не более 0,1—0,5 г/ж3 влаги и практически является
сухим. Осушка газа происходит вследствие того, что давление
паров воды над соляной кислотой значительно меньше, чем над
водой.
Производство хлористого водорода и соляной кислоты
является взрывоопасным и сопряжено с сильной коррозией
аппаратуры и трубопроводов. Для предотвращения аварий и
несчастных случаев в этом производстве необходимо
соблюдение специальных мер предосторожности и правил техники
безопасности.
Аппаратура и оборудование монтируются вне зданий, на
открытом воздухе и обслуживаются со щита управления,
располагаемого в помещении. На водородопроводе устанавливают
гравийные огнепреградители, печи синтеза снабжают
выхлопными мембранами, к водородной линии подводится азот для
продувки системы перед пуском.
Автоматические непрерывные контроль и регулирование
процесса также обеспечивают надежную и безаварийную
работу производства.
Для производства 1 г синтетического хлористого водорода
расходуется около 1 т газообразного хлора и около 330 мэ
водорода (в пересчете на 100%-ные газы), 12—15 мъ воды и
15—25 кет ¦ ч электроэнергии.
Для регенерации отбросного хлористого водорода (отхода
хлорорганических производств) и для концентрирования
синтетического хлористого водорода (на 1 г НС1) расходуется 1,5—
3,0 т пара, примерно 20—25 тыс. ккал на получение холодного
рассола и небольшое количество электроэнергии и воды.
Расходные коэффициенты в производстве соляной кислоты
зависят от заданного содержания в ней хлористого водорода.
Сорта соляной кислоты
Выпускаются следующие виды технической соляной кислоты:
1) кислота соляная техническая A-й и 2-й сорт),
получаемая из хлористого водорода, образующегося при
взаимодействии серной кислоты и хлористого натрия;
2) кислота соляная синтетическая (техническая),
получаемая из хлористого водорода, образующегося при взаимоденст
вии электролитических хлора и водорода.
На стр. 407 приведены требования к качеству технической
соляной кислоты, регламентируемые соответствующими ГОСТ.
Соляная кислота, получаемая из отбросного хлористого
водорода (отход производства хлорорганических продуктов),
должна содержать не менее 27,5% НС1, не более 0,03%
примесей железа и не свыше 0,1% свободного хлора.
406
Содержание HCI, %,
Содержание примесей,
серная кислота (в
SO,) . . . .
железо . . .
мышьяк
Нелетучий остаток
не менее . .
%, не более
пересчете на
Кислота соляная техническая
ГОСТ 1382—42
1-й сорт
27,5
0,4
0,03
0,01
2-й сорт
27,5
0,6
) Не норми-
J руется
Кислота соляная
синтетическая
ГОСТ 857—57
31
0,005
0,02
0,0002
0,2
Выпускается также ингибированная соляная кислота, в
которую для уменьшения ее коррозионного действия вводят
около 1% ингибитора ПБ-5 (продукт конденсации анилина с
уротропином) и 0,01—0,05% соли мышьяка, усиливающего
защитное действие ингибитора коррозии. Ингибитор образует на
поверхности резервуара с соляной кислотой защитную пленку,
вследствие чего скорость коррозии в ингибированной соляной
кислоте в 150—200 раз меньше, чем в отсутствие ингибитора.
Содержание хлористого водорода в ингибированной соляной
кислоте должно быть в пределах 19—25%, железа не более
0,01 %, свободного хлора до 0,1 %.
К соляной кислоте, предназначенной для пищевой
промышленности, предъявляются очень жесткие требования в
отношении содержания примесей.
Хранение и перевозка соляной кислоты
Для хранения больших количеств соляной кислоты приме
няют стальные цистерны емкостью до 50 м3 и баки емкостью
100 м3 и более. Хранилища изнутри гуммируют и футеруют
поверх гуммировки диабазовой или другой кислотоупорной
плиткой. Штуцеры для отбора кислоты из нижней части хранилищ
отсутствуют.
Кроме футерованных, как указано выше, емкостей для
хранения небольших количеств соляной кислоты применяют также
баки из фаолита, фарфора и керамики. Керамические емкости
используют главным образом для хранения чистой соляной
кислоты.
Техническую соляную кислоту, имеющую концентрацию НС1
31%, перевозят в стальных, гуммированных изнутри цистернах
емкостью до 50 т, в стальных гуммированных контейнерах или
бочках, а также в фаолитовых или керамических контейнерах
емкостью до 2000 л.
407
Ингибированную соляную кислоту можно перевозить в
стальных (нефутерованных) цистернах.
Небольшие количества соляной кислоты перевозят в
стеклянных бутылях емкостью до 40 л, закрытых
пришлифованными или обожженными глиняными пробками на специальной
герметизирующей мастике, не содержащей железа. Горловины
закрытых пробкой бутылей обертывают плотной тканью или
крафтцеллюлозной бумагой и обвязывают шпагатом.
Бутыли, предназначенные для перевозки соляной кислоты,
обкладывают стружкой или соломой и помещают в открытые
деревянные ящики (обрешетки) или в ивовые корзины с
прочными ручками.
Пищевую соляную кислоту разливают в стеклянные бутыли
емкостью до 30 л, закрываемые чистыми деревянными
пробками.
Жидкий хлористый водород
В некоторых производствах, например в промышленности органического
синтеза, для получения ряда продуктов требуется чистый газообразный или
жидкий хлористый водород.
При охлаждении и сжатии газообразный хлористый водород может
быть сжижен. Критическая температура хлористого водорода +51,3 "С,
критическое давление 81,6 атм, теплота испарения 3,850 каг/моль, плотность
жидкого НС1 при температуре кипения 1,194 кг/л.
Сжижение проводится следующим образом. Хлористый водород,
полученный синтезом из Н2 и С12, после тщательной осушки сжимают до
давления 60 ат.
При охлаждении водой компримироваиный хлористый водород
превращается в жидкость, котор>ю разливают в стальные баллоны. Жидкий
хлористый водород содержит 99,9% НС1.
2. ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ
ХЛОРОПРОДУКТОВ
ДВУОКИСЬ ХЛОРА
Двуокись хлора применяется как отбеливающее средство
в виде водного раствора, содержащего 5—10 г\л СЮг.
Белящее действие двуокиси хлора обусловлено выделением кисло
рода по реакции
2С1Оа + НгО > 2НС1 + 2yOj
Преимущество двуокиси хлора перед другими
отбеливающими средствами заключается в способности СЮг окислять лигнин
и вещества, окрашивающие волокно без его разрушения.
Поэтому при использовании для этих целей двуокиси хлора
достигается высокая белизна волокна без ухудшения его
механических свойств.
408
Причем белизна с течением времени сохраняется, тогда
как в случае отбелки, например, гипохлоритом кальция
наблюдается постепенное уменьшение степени белизны волокна.
Двуокись хлора представляет собой желто-оранжевый газ
с резким запахом. При +11 °С и давлении 760 мм рт. ст.
двуокись хлора сжижается, а при —59 °С затвердевает. Плотность
жидкой СЮг при 5 °С составляет 1,635 г/см3, плотность
твердого вещества при —60 °С достигает 1,979 г/см3.
Растворимость двуокиси хлора в воде зависит от темпера
туры и парциального давления (р) С1О2:
Температура Растворимость СЮг. г/л
"С р = 50 мм pm. cm р= 100 мм рт. ст.
20 7,5 16,5
25 5 10
35 3,5 7,5
Двуокись хлора весьма токсична и оказывает такое же
вредное действие на человека, как хлор.
Двуокись хлора — неустойчивое соединение. В газообразном
состоянии при температуре выше 65 °С и повышенном
давлении, при соприкосновении с горючими веществами или под
действием солнечного света двуокись хлора разлагается со взрывом:
2С1О3 = С1а 4- 2О2
В кислых водных растворах и в темноте двуокись хлора бо
лее устойчива.
Вследствие невозможности транспортирования неустойчивой
двуокиси хлора ее получают только на месте потребления пу
тем восстановления хлоратов натрия, калия или кальция.
Наиболее распространены способы, основанные на восстановлении
хлората натрия в сильнокислой среде. В качестве
восстановителей применяют двуокись серы, метанол или сульфат хрома
з среде серной кислоты или же соляную кислоту.
Уравнения реакций восстановления хлоратов различными
реагентами:
2NaC103 + SO3 = 2СЮа + NaaSO4
4NaC103 -f CH3OH + 2HaSO4 = 4С1Оа + HCOOH + 2NajSO4 + 3H2O
NaClO3 -f 2HC1 = ClOa -f -gT Cle -f NaCl + HaO
Преимущества способа восстановления хлората натрия
соляной кислотой заключаются в доступности исходных веществ
и возможности вести замкнутый циклический процесс почти бе?
подвода сырья.
4OQ
Принципиальная схема процесса:
t Электролиз
NaC103+3H2
Упаривание
I г
f Восстановление 1 | |
NaC103 + 2HC1 *¦ C10a + -y Cla+ Nad + H2O
1
1, + Н.2—2НС1 2NaOH + С13 » NaOCl -+- NaCI -+- Н.3О
.3
I1
I Электролиз На отбелку
CIa -f H, + 2NaOH ¦< 2NaCl + 2H,0
Исходный хлорат натрия получают путем электролиза хло-
рид-хлоратного раствора, выходящего из реакторов для синтеза
двуокиси хлора. Этот раствор содержит всего 20—30 г/л .МаСЮз
и сильно разбавлен водой, выделяющейся при реакции
восстановления и вводимой с соляной кислотой. Поэтому до
электролиза раствор предварительно упаривают. Получаемая в
процессе восстановления хлората натрия двуокись хлора
направляется на абсорбцию водой, а побочно образующийся хлор
используется для получения гипохлорита натрия. В цехе электролиза
имеются также ванны, в которых получаются хлор и водород,
направляемые на синтез НС1, необходимой для восстановления
хлората натрия. Получаемый в этих же ваннах едкий натр
используется в производстве гипохлорита натрия,
направляемого на отбелку тканей.
Технологическая схема производства СЮ2 показана на
рис. 162.
Раствор хлората натрия из бака 2 центробежным насосом
подается в напорный бак 10. В такой же напорный бак
подается соляная кислота. Из напорных баков жидкости непрерывно
дозируются в верхний реактор установки, состоящей из шести
каскадно расположенных аппаратов 7 для получения С!О2.
Перетекая последовательно из одного реактора в другой, раствор
нагревается от 40 до 100—102 °С. Для подогрева раствора в
два нижних реактора подается острый пар. В остальных
реакторах раствор подогревается вторичным паром, который
движется противотоком раствору и барботирует через него.
Образующаяся в растворе цзуокись хлора выдувается и
разбавляется воздухом во избежание возможности образования
взрывоопасной концентрации СЮ2. Воздух поступает в третий
снизу реактор. Парогазовая смесь, состоящая из С1О2, хлора,
воздуха и паров воды, поступает из верхнего реактора в
абсорбер 9, орошаемый холодной водой, поглощающей двуокись хло-
410
pa. Получаемый в абсорбере раствор двуокиси хлора C—5 г/л
СЮ2) направляется в отбельный цех. Отходящий из абсорбера
газ, содержащий хлор, используется для получения гипохло-
рнта натрия. Раствор, выходящий из нижнего реактора,
упаривается в системе вакуум-испарителей 6 и поступает на
электролиз.
К конденсатору
и бакуум-насйсу
Соляная кислота
Раствор
СЮ2
Рис. 162. Схема производства двуокиси хлора:
/—электролизеры; 2, а—приемные резервуары; 4—насосы; 5, 10— напорные баки; 5—вакуум-
испаритель; 7—реакторы; 8—сборник; 9—абсорбер.
На получение 1 г двуокиси хлора расходуется следующее
количество сырья и энергии:
Хлорат натрия, кг . . .
Соляная кислота, кг . .
Электроэнергия, квш-ч
Вода, ж3
Пар C am), кг , . . .
1700
1600
14200
1000
4000
ХЛОРНАЯ ИЗВЕСТЬ
Свойства и применение хлорной извести. Хлорная известь
представляет собой сухой порошок белого цвета, иногда с
желтоватым оттенком, сильно пахнущий хлором.
Хлорная известь не является индивидуальным веществом.
В ее состав входят гипохлориты кальция: нейтральный
Са(ОС1J-ЗН2О и основные ЗСа(ОС1J-2Са(ОНJ-2Н2О и
Са(ОС!J-2Са(ОНJ.
Помимо гипохлоритов, в хлорной извести присутствуют
хлориды кальция: СаС12-2Н2О, СаС12-4Н2О, СаС12-Са(ОНJ ¦ Н2О,
411
СаС12-ЗСа(ОНJ* 12Н2О, а также свободная гидроокись
кальция.
В водной среде входящий в состав хлорной извести
неустойчивый гилохлорит кальция разлагается:
Са(ОС1)а + Н8О = Са(ОН)ОС1 + НОС1
Са(ОН)ОС1 + Н4О = Са(ОНL + НОС1
Образующаяся при этом хлорноватистая кислота оказывает
окислительное действие. Под влиянием прямого солнечного
света, в присутствии веществ, способных легко окисляться, а
также солей некоторых металлов (например, солей Со, Ni, Mn.
Fe и др.) хлорноватистая кислота практически нацело
разлагается по реакции
НОС1 = НС1 + О
Белящие и дезинфицирующие свойства хлорной извести
оцениваются по содержанию в ней активного хлора.
Количественно активный хлор определяют путем обработки испытуемого
вещества соляной кислотой, при взаимодействии которой с
гипохлоритом кальция выделяется свободный хлор:
Ca(OCl), + 4HC1 = 2С1а + СаС1а -f 21^0
Из этого уравнения следует, что одна молекула гипохлорита
кальция эквивалентна двум молекулам активного хлора, т. е.
окислительный эквивалент Са@С1)г равен 4. Следовательно,
содержание активного хлора в чистом безводном гипохлорите
кальция теоретически должно составлять
4-35,45
143
-.100 = 99,2 %
где 35,45 и 143 — соответственно атомный вес хлора и
молекулярный вес гипохлорита кальция.
Кристаллизующийся из воды нейтральный гипохлорит
кальция Са(ОС1J-ЗН2О содержит 72,6% хлора. Таким образом,
содержание активного хлора является лишь условной оценкой
окислительной способности продукта и не соответствует
фактическому содержанию в нем хлора, которое, например, для
Са(ОС1J составляет 49,6%.
Поскольку хлорная известь, кроме гипохлорита кальция, со-
1ержит много других соединений, содержание в ней активного
хлора невелико. Товарная хлорная известь обычно содержит
32—35% активного хлора.
Хлорная известь — неустойчивый продукт, сравнительно
легко теряющий свой активный хлор. Стойкость ее зависит от
содержания хлористого кальция, воды, от температуры, наличия
примесей, входящих в состав исходных продуктов (п\шонки
412
и ллора) и других факторов. При хранения на открытом
воздухе хлорная известь постепенно разлагается с выделением
элементарного хлора. В присутствии влаги и при повышенной тем-
лературе разложение хлорной извести ускоряется. При храж-
чип высококачественной хлорной извести в надлежащих уело
;чях потери активного хлора обычно незначительны.
До недавнего времени хлорная известь находила широкое
применение в различных областях народного хозяйства. С
расширением производства жидкого хлора и гипохлоритов кальция
i натрия доля хлорной извести в общем балансе выпускаемых
фомышленноегью кислородсодержащих хлоропродуктов сни
жается.
Основным потребителем хлорной извести является сейчас
целлюлозно-бумажная промышленность (отбеливание
целлюлозы и бумажной массы). В коммунальном хозяйстве хлорную
язвесть наряду с жидким хлором применяют для дезинфекции
сточных и питьевых вод, мест общего пользования и т. п.
Хлорная известь употребляется также для уничтожения
возбудителей некоторых заболеваний растений (в овощехранилищах и а
лочве) и для обеззараживания почвы около зернохранилищ.
Получение хлорной извести
Исходным сырьем для получения хлорной извести служат
газообразный хлор и известь-пушонка Са(ОНJ — продукт
гашения водой обожженной извести СаО, так называемой ки-
яелки.
Реакция образования хлорной извести может быть весьма
приближенно выражена следующим уравнением:
2Ca(OH)g + 2С18 > Са(ОС1)8 + СаС12
При этом на каждый килограмм тюглощенного хлора
выделяется около 250 ккал тепла.
Абсолютно сухие гидроокись кальция и хлор не вступают
з реакцию; хлор только адсорбируется известью. В
производственных условиях хлорируют пушонку, содержащую до 1 %
влаги; применяемый газообразный хлор также должен
содержать небольшое количество влаги.
Процесс образования хлорной извести может быть интенси
фицирозан путем увеличения поверхности соприкосновения
реагентов, что достигается непрерывным перемешиванием
пушонки.
Вначале хлорную известь получали в периодически дейст-
зующих свинцовых камерах, позднее — в негарерывнодействую-
Щих, но малопроизводительных барабанах с механическими ме-
413
шалками. В настоящее время все эти аппараты не применяются
Довольно распространены непрерывнодеиствующие полочные
камеры, которые несмотря на громоздкость устройства и
большие капиталовложения на их сооружение имеют ряд
преимуществ перед аппаратами барабанного типа. Они более
производительны, удобнее в обслуживании, требуют меньшего
расхода электроэнергии, получаемая в них хлорная известь содер
Рис. 163. Схема производства хлорной извести:
/—аппарат для гашения извести; 2, б—элеваторы; 3—сито; 4—бункеры для вылеживания пу-
шоики, о—шнеки; 7—камера хлорирования; 8—автоматический питатель; У—валы; 10—полки;
//—гребки; 12—бункеры для готовой хлорной извести; 13—промывная башня; 14—эксгаустер;
15— бочки.
жит больше активного хлора. Кроме того, камеры могут
работать при любой концентрации хлора в газе — в пределах от 10
до 90%. Обычно применяется разбавленный воздухом хлор
C0—60% СЬ).
Схема производстза хлорной извести в непрерывнодействую-
щих полочных камерах изображена на рис. 163. Хорошо
обожженную известь в барабанном аппарате / гасят водой для
получения пушонки:
СаО + НаО = Са(ОН)а
Пушонка в виде тонкого порошка, содержащего не менее
92% Са(ОНJ, не более 1% влаги и не более 1% СаО, системой
элеваторов 2 подается на сито 3 для отделения грубых плохо
414
обожженных частиц, а затем в промежуточные бункеры 4, где
пушонку выдерживают 4—5 суток, в течение которых состав
продукта становится более или менее однородным. Из бункеров
пушонка шнеками 5 и элеватором 6 подается сверху в камеру
хлорирования 7. Снизу в камеру поступает газообразный хлор.
Камера хлорирования по устройству сходна с механической
печью для сжигания колчедана (стр. 81). Она представляет
собой восьмигранную железобетонную башню (внутри
цилиндрическую), рабочая высота ее 9,6 м, внутренний диаметр 5,5 м.
По высоте каждая камера разделена на восемь этажей семью
полками 10 с отверстиями в центре или по периферии.
Через центры полок камеры проходят металлические,
медленно вращающиеся вертикальные валы 9 с гребками* 11.
Вращающиеся валы с гребками перемешивают пушонку на полках
и перемещают ее с одной полки на другую. На одних полках
гребки передзигают пушонку от центра к периферии, на
других— от периферии к центру. Во избежание коррозии все ме-
галличэские части внутри камеры покрыты химически стойким
лаком. Для удобстза обслуживания и экономии площади
камеры соединяют в группы (блоки) по 4, 6, 8, 10, 12 и даже по
16 камер.
Температуру в камерах 7 поддерживают не выше 35 °С.
Для регулирования температуры служат вмонтированные в
межэтажные полки камеры змеевики, в которые подают
холодную воду или рассол, охлаждаемый в холодильной установке
(па рисунке не показана). Пушонка подается на верхнюю полку
камеры автоматическими питателями 8, хлор поступает по трубе
на вторую полку снизу и проходит вверх противотоком
движению пушонки.
Отходящие газы, содержащие небольшое количество не-
логлощенного хлора @,1—0,3%), перед удалением в атмосферу
пропускают через промызную башню 13, орошаемую
известковым молоком. На некоторых заводах вместо известкового
молока применяют раствор едкого натра B2% NaOH), при этом
получается гипохлорит натрия, содержащий около 15%
активного хлора, который используется, например, для отбелки.
Через камеру хлорирования и промывную башню газ
просасывается эксгаустером 14, создающим во всей системе
небольшое разрежение.
Готозая хлорная известь поступает в бункеры 12 и оттуда
а бочки 15.
Хлорирование пушонки в камере происходит как при
перемещении ее по полкам, так и при пересыпании с одной полкч
на другую. В последнем случае условия протекания гетероген-
ной реакции между хлором и пушонкой гораздо более благо-
* В последнее время применяют гребки из пластмассы на основе фено-
ло-формальдегидных полимеров.
413
приятны, так как пушонка в это время находится в распылен
ном состоянии, следовательно, поверхность соприкосновения
фаз и скорость реакции хлорирования возрастают. Для интен
сификации этого процесса следует сокращать время
пребывания пушонки на полках, что достигается увеличением скорости
вращения мешалок, например с 0,3 до 0,49 об/мин. В
результате значительно увеличивается поверхность соприкосновения
реагентов и возрастает производительность камер хлорирования
в среднем на 34—35%. Сокращение времени пребывания шихты
в камере приводит к уменьшению разности между содержанием
общего и активного хлора в хлорной извести при условии, чтс
продолжительность этого времени будет достаточна для про
цесса хлорирования.
При неизменной производительности камер увеличение
скорости вращения мешалок приводит к уменьшению высоты слоя
шихты на полках, что улучшает условия хлорирования и отвод
тепла реакции. Снижение высоты слоя шихты позволяет также
уменьшить высоту гребков с 300 до 200 мм и таким образом
сэкономить значительное количество конструкционных
материалов.
Расходные коэффициенты на 1 г хлорной извести,
содержащей 35% активного хлора:
Обожженная известь (в пересчете на 100% СаО), пг. 0,5
Хлор A00% С]а), пг 0,36—0,38
Вода, м3 17,5—20
Электроэнергия, квт-ч 11—20
Промышленность выпускает хлорную известь трех марок
Согласно ГОСТ 1692—58, качество' ее должно соответствовать
следующим требованиям:
I
Марки
Внешний вид и цвет
Содержание активного хлора, %, не более . . .
Разница между содержанием общего и активного
хлора, %, не более
Содержание влаги, %, не более
яри длительном хранении
при текущем потреблении
Порошкообразный продукт
белого цвета
35*
2
2
2
35
2
10
32
4
Не
нормируется
Не
нормируется
• Завод-поставщик обязан безвозмездно заменить хлорную известь марки А, если в течс-
ияе трех лет ее хранения со дня отгрузки потребителю потери активного хлора превысят 4%
Кроме описанных полочных камер, на некоторых
зарубежных заводах для получения хлорной извести нашли применение
аппараты других конструкций. Например, наклонно располс
416
женные вращающиеся барабаны длиной до 20 м, охлаждаемые
снаружи водой (в этих барабанах газообразный хлор и пушон-1
ка движутся противотоком), шнековые аппараты особой
конструкции, аппараты для хлорирования в вакууме и т. д.
Упаковка и хранение хлорной извести ;
Хлорную известь марки А, а также хлорную известь
марки Б, предназначенную для длительного хранения, упаковы
вают в стальные барабаны емкостью 100 л. В остальных случаях
хлорную известь упаковывают в деревянные бочки емкостью
50 и 275 л или в фанерные бочки и барабаны емкостью 50 я
100 л. По договоренности с потребителем хлорную известь, не
предназначенную для длительного хранения, разрешается
упаковывать в исправные, бывшие в употреблении сухие
деревянные бочки емкостью от 50 до 275 л.
Упакованную хлорную известь до отправки потребителям
хранят в крытых складах или под навесом.
При недостаточной герметичности тары хлорная известь
иногда полностью теряет активный хлор в течение 1 года (и
даже быстрее), а при повышенной температуре D0—45 °С) —
в течение 1—2 мес. В странах с жарким климатом применяют
более стабильную хлорную известь, содержащую 34—35%
активного хлора и 1—2% влаги. Хлорную известь можно под^
сушивать во взвешенном состоянии воздухом, нагретым до
150—200 °С, при очень кратковременном соприкосновении
продукта с горячим воздухом во избежание разложения хлорной'
извести. Можно также вести подсушку при разрежении.
ГИПОХЛОРИТ КАЛЬЦИЯ
В последние десятилетия начали получать твердый
устойчивый гипохлорит кальция, содержащий значительно больше
активного хлора, чем хлорная известь.
Химически чистый безводный гипохлорит кальция Са(ОС1J
теоретически должен содержать 99,2% активного хлора
(стр. 412). Из водных растворов он кристаллизуется в виде
Са(ОС1J • ЗН2О- Чистый гипохлорит кальция сравнительно
хорошо растворим в воде.
Технический товарный продукт не является чистой
кальциевой солью хлорноватистой кислоты. Он примерно соответствует
формуле ЗСа(СЮJ-2Са(ОНJ-2Н2О и содержит 47—52%-
активного хлора и 18—20% Са(ОНJ. Кроме того, в техническом
гипохлорите кальция всегда присутствуют примеси (СаС12,'
СаСО3, SiOo, хлорат кальция и др.), количество и состав
которых зависят от способа производства. Вследствие присутствия
примесей технический гипохлорит кальция полностью в воде
не растворяется, а образует суспензии.
27-2848 417
Процесс получения гипохлорита кальция основан на
взаимодействии хлора с известковым молоком, содержащим 400—
410 г/л Са(ОНJ.
Гипохлорит кальция представляет собой белый
кристаллический порошок, слегка пахнущий хлором. По окислительному
и хлорирующему действию он аналогичен хлорной извести;
применяется для отбеливания бумажной массы, хлопчатобумажных
тканей, шерсти, для дезинфекции питьевых и сточных вод и т. п.
Гипохлорит кальция обладает некоторыми преимуществами
перед хлорной известью. Содержание в нем активного хлора
и, следовательно, окислительная способность почти вдвое
больше, чем для свежеприготовленной хлорной извести.
Вследствие низкой гигроскопичности (влажность
гипохлорита кальция 2% и ниже) он более устойчив и лучше сохраняется
в кристаллическом (порошкообразном) состоянии, а потому
стабильность его в несколько раз выше стабильности хлорной
извести. Вследствие лучшей растворимости гипохлорита кальция
в воде растворы его содержат больше активного хлора.
В отношении перевозки гипохлорит кальция также
представляет большие преимущества. Балласт (включая тару)
составляет всего 60—90% в расчете на активный хлор, в то время как
в случае перевозки жидкого хлора в баллонах балласт
составляет 100—200% (учитывая возврат баллонов), а при перевозке
хлорной извести достигает 250—300%.
Производство твердых гипохлоритов кальция довольно
сложно, в особенности по аппаратурному оформлению. Поэтому
их целесообразно получать, когда потребителю необходимы
твердые гипохлориты или когда они предназначены для
длительного хранения. Если же гипохлориты используются в виде
водных растворов, лучше насыщать щелочные растворы хлором.
Гипохлорит кальция выпускается в виде дветретиосновной
соли ЗСа(С1ОJ-2Са(ОНJ-2Н2О, обозначаемой ДТС ГК, и
двуосновного гипохлорита кальция Са(С1ОJ-2Са(ОНJ,
обозначаемого ДС ГК. Качество гипохлорита кальция должно
удовлетворять следующим требованиям:
Содержание активного хлора,
%, не менее
Общее содержание хлора,
%, не более
Содержание гигроскопической
влаги, %, не более . . .
1-й сорт
52
акт. хлор
2
2
ДТС ГК
2-й сорт
47
акт. хлор , й
2
2
ДС ГК
39
акт. хлор . _
2
2
Продукт 1-го сорта упаковывают в оцинкованные стальные
барабаны емкостью 25, 33 и 50 л, окрашенные изнутри
натуральной олифой, продукт 2-го сорта и ДС П< — в стальные
барабаны, окрашенные изнутри химически стойким лаком, или же в
тару для 1-го сорта. Упакованный гипохлорит кальция следует
хранить в сухих неотапливаемых помещениях.
БЕЛИЛЬНЫЕ РАСТВОРЫ
Белильные растворы — растворы гипохлоритов кальция или
натрия — применяются в текстильной промышленности для
отбелки тканей, для очистки сточных вод и питьевой воды. Для
этих целей применяется также двуокись хлора (стр. 408).
Отбелка представляет собой процесс окисления
хлорноватистой кислотой (ион СЮ') различных загрязняющих вещестз,
обычно язляющихся восстановителями, по схеме
С1О~ + Восстановитель > С1~ + Окисленный восстановитель
Белильные растворы получают пропусканием газообразного
хлора через растворы Са(ОНJ или NaOH. Реакцию между
известковым молоком и хлором
2Са(ОНJ + 2С13 = Са(ОС1)а + СаС12 + 2Н2О
проводят при температуре не выше 35—45 °С, охлаждая
раствор. Получаемый в этих условиях белильный раствор
содержит 2—10% активного хлора. Если пропускать хлор,
разбавленный воздухом, через раствор, циркулирующий
противотоком газу, можно не охлаждать раствор.
Хлорирование ведут до содержания в растворе 5—15 г/л
щелочи.
По одному из способов растворы гипохлорита натрия
приготовляют, пропуская хлор через раствор соды:
2Na,CO3 + С12 + Н2О = NaOCJ + NaCl + 2NaHCO3
В процессе дальнейшего хлорирования бикарбонат натрия
разлагается с выделением двуокиси углерода и образованием
хлорнозатистой кислоты:
NaHCO3 + С12 = NaCl + СО2 + НОС1
Выделяющаяся двуокись углерода разлагает
хлорноватистую кислоту с образованием соляной кислоты и кислорода.
Поэтому пропускание хлора прекращают прежде, чем начнется
выделение СОг. При хлорировании растворов едкого натра.
2NaOH + С1а = NaOCl
получаются устойчивые растворы гипохлорита натрия,
-содержащие 15—17% -NaOCl при избытке до 4 г/л NaOH,
27* 419
Растворы гипохлорита натрия, содержащие избыточную
щелочь, можно сохранять в соответствующей таре до 10 суток.
Стойкость растворов может быть повышена при добавлении
специальных стабилизирующих органических веществ.
В промышленности растворы гипохлорита натрия с большим
содержанием активного хлора получают в резервуарах,
снабженных мешалками и охлаждающими змеевиками.
Перемешивание необходимо, чтобы избежать местных перегревов,
ведущих к быстрому разложению продукта. Хлорируют обычно
10—30%-ный растворы NaOH при температуре не выше 35°С,
особенно в конце процесса. Хлорирование можно вести также
в башнях при непрерывной циркуляции растворов.
Белильные растворы можно получать также обменным
разложением гипохлорита кальция содой или сульфатом натрия:
Са(ОС1J + Na2CO3(Na2SO4) = 2NaOCl + CaCO3(CaSO4)
Гипохлорит натрия выпускают в виде водных растворов
двух марок:
Содержание, г/л Марка 1 Марка II
Активный хлор 100—140 Не менее i70
Щелочь 100—140 Не более 20
Железо, не более 0,1 0,075
Продукт марки I применяется как окислитель в цветной
металлургии, продукт марки II — в анилинокрасочной
промышленности. Растворы гипохлорита натрия отпускают
потребителям в стальной таре, изнутри покрытой винипластом или
гуммированной, при температуре раствора не выше 25 °С.
ЛИТЕРАТУРА
. '-¦ (к главам VIII и IX)
С. И. В о л ь ф к о в и ч, А. П. Егоров, Д. А. Э п ш т е й н, Общая химиче'
екая технология, т. I, Госхимнздат, 1952.
Л. С. Гения, Электролиз растворов поваренной солн, Госхимиздат, 1960.
Ж- Биллнтер, Промышленный электролиз водных растворов, перев. с ием.
под ред. Л. М. Якименко, Госхнмиздат, 1959.
Н. П. Федотьев н др., Прикладная электрохимия, Госхимиздат, 1962.
B. В. Стендер и др., Прикладная электрохимия, Изд. Харьковского
университета, 1961.
В Г. Хомяков, В. П. Машовец, Л. Л. Кузьмин, Технология
электрохимических производств, Госхнмнздат, 1949.
А. Г. Симон, П. Г. X а и н, Л. М. Якименко, Пути интенсификации
технологических процессов в производстве хлора, Жури. ВХО
нм. Д. И. Менделеева, № 1, 16 A961).
Хим. наука и пром., № 4 A958).
П. Г. X а и и, А. Г. Симон, С. М. Круглый. Основные технические
решения в проектах хлорного производства; Вести, техн. и экоиом. ииф..
№ 7, 3 A958).
C. Я. Файнштейн и др., Основные направления техники производства
хлора за рубежом, Хим. пром., № 4, 51 A957).
Глава X
СОДОВЫЕ ПРОДУКТЫ
/. КАЛЬЦИНИРОВАННАЯ СОДА. ОБЩИЕ СВЕДЕНИЯ
Применение соды. Под общим названием «сода»
промышленность производит ряд продуктов: кальцинированную, или
углекислую, соду Na2CC>3; очищенный бикарбонат натрия, или
двууглекислый натрий NaHCO3, называемый также питьевой
содой; кристаллическую соду ЫагСОз-ЮНгО и ЫагСОз-ЫгО;
едкий натр, или каустическую соду NaOH. Выпускается также
специальный сорт соды — так называемая тяжелая сода
(насыпной вес 0,9—1,0 т/м3).
Важнейшими содовыми продуктами являются
кальцинированная и каустическая сода. Кальцинированная сода служит
исходным веществом в производстве каустической соды,
получаемой химическим способом, бикарбоната натрия, тяжелой
соды. Кальцинированная сода применяется в мыловаренной,
стекольной, нефтяной, текстильной, целлюлозно-бумажной,
пищевой, лакокрасочной промышленности, в черной и цветной
металлургии, в кожевенном производстве и т. д.
Очищенный бикарбонат натрия применяют в пищевой,
кондитерской и химико-фармацевтической промышленности, в
производстве искусственных минеральных вод, в медицине, в
качестве зарядной массы для огнетушителей и т. п. Десятиводная
и одноводная кристаллическая сода используются
преимущественно в домашнем хозяйстве.
Тяжелая сода применяется наряду с обычной содой в
различных отраслях промышленности (металлургической,
стекольной и др.).
Разнообразное и все возрастающее потребление
кальцинированной соды во многих отраслях народного хозяйства
обусловливает непрерывное увеличение ее производства. Возрастает
также производство каустической соды электрохимическим
методом (стр. 324 ел.). Доля каустической соды, получаемой
химическими способами (стр. 476), в дальнейшем будет
сокращаться.
Мировое производство кальцинированной соды (без СССР)
в 1960 г. составило примерно 15,2 млн. т, в 1961 г. — 16 млн. т.
421
Объем ее производства в ряде стран мира за эти годы составил
(в тыс. г):
19С0 г. 1961 г.
США 4888 —
СССР 1900 2330*
Англия (оценка) 1570 —
ФРГ 1117 1063
Франция 848 850
Япония 791 800
Италия 565 569
Индия 145 177
* 1962 г.
Свойства кальцинированной соды. Чистый углекислый натрий
'представляет собой белый кристаллический порошок
плотностью 2,53 г/см3, плавящийся при 854 °С, объемный
(насыпной) вес его 0,5—0,55 т/м3. На воздухе кальцинированная
сода поглощает СО2 и влагу, превращаясь при этом частично в
бикарбонат натрия. С водой углекислый натрий образует ряд
кристаллогидратов. Так. при температуре ниже 32,5 С из
насыщенного раствор Na2CO3 выделяется декагидрат
Na2CO3-10Н2О, в интерзале температур 32,5—36 °С — гепта-
гидрат Na2CO3-7H2O, при более высоких температурах
образуется Na2CO3-H2O. Кальцинированная сода хорошо
растворяется в воде, причем с повышением температуры
растворимость ее увеличивается:
Температура Растворимость КагСОз
"С г/100 г воды
0 6,8
25 28,52
100 44
При растворении соды в воде выделяется тепло. Так,
растворение ! моль Мз2СО3 в 400 моль воды сопровождается
выделением 5,64 икал тепла. Водные растворы кальцинированной
соды обладают сильно щелочной реакцией.
Природная сода. Сода была известна и применялась за
несколько тысячелетий до нашей эры. Ее добывали из природных
залежей или из золы, образующейся при сжигании некоторых
морских водорослей. Сода встречается в природе в виде троны.
Na2CO3- NaHCO3-2H2O, называемой также сесквикарбонатом,
или полуторной содой, а также в виде патрона —
кристаллического декагидрата Na2CO3-10H2O и термонатрита —
порошкообразного гидрата Na2CO3-H2O.
В растворенном состоянии Na2CO3 содержится в различных
щелочных минеральных водах. Значительные количества
природной соды находятся в рапе некоторых содовых озер в СССР,
США, ОАР и в других странах. В СССР содовые озера
промышленного значения расположены з Западной Сибири
(группа озер Тантар) и в Кулундинской степи (Павлодарская
область).
422
В начале XIX в. высокая стоимость соды и ограниченный
масштаб ее добычи из природных источников начали тормозить
развитие таких важных отраслей промышленности, как
стекольная, мыловаренная, кожевенная и другие потребители соды.
В связи с этим возникла необходимость получения соды
синтетическим путем.
СИНТЕТИЧЕСКИЕ СПОСОБЫ ПОЛУЧЕНИЯ
КАЛЬЦИНИРОВАННОЙ СОДЫ
Способ Леблана. В 1791 г. французский врач и химик Ле-
блан осуществил синтетический способ производства соды из
широко распространенного в природе минерального сырья —
поваренной соли, известняка и угля. В России еще до Леблана
предпринимались попытки получения соды восстановлением
сульфата натрия древесным углем, не реализованные, однако,
в промышленных масштабах.
Сущность способа Леблана заключалась в сплавлении при
950—1000 °С смеси сульфата натрия (полученного
взаимодействием серной кислоты с поваренной солью), известняка и угля
во вращающихся печах. Образовавшийся в печах содовый плаз
(сырая сода) дробили и выщелачивали водой. При этом сода
переходила в раствор, а в осадке оставались сульфид кальция,
непрореагировазший карбонат кальция и другие примеси.
Полученный содовый раствор (щелок) обрабатывали двуокисью
углерода Для превращения частично образовавшегося едкого
натра в соду и для разложения примеси сульфида кальция.
После отстаивания щелок выпаривали. Полученный продукт
прокаливали (кальцинирование) и измельчали.
Реакции, протекающие при получении соды по способу
Леблана, можно представить следующими уравнениями:
2NaCl + H2SO4 = Na2SO4 + 2НС1
NaaSO4 + 2C + CaCO3 = Na.2CO3 + CaS -f 2CO2
Кроме этих реакций, при температуре около 1000°С
происходит диссоциация карбоната кальция:
СаСО3 -с—* СаО + СО3
При обработке двуокисью углерода твердых остатков,
полученных после выщелачивания содового плава, выделяется
сероводород
CaS + СО2 + НгО = H2S + CaCO3
который в присутствии раскаленной окиси железа или боксита
окисляется при избытке воздуха до элементарной серы:
2H2S + О2 =- 2S + 2Н2О
423
Образовавшуюся серу далее перерабатывали в серную
кислоту.
Таким образом, процесс получения соды по способу Лебла-
на можно рассматривать как круговой процесс, в котором в
качестве исходных веществ применяются хлористый натрий
(поваренная соль), известняк, уголь и вода, а конечными
продуктами являются сода и хлористый водород.
Одна из трудностей осуществления этого процесса в то время
заключалась в невозможности использовать большие
количества хлористого водорода, который по санитарным условиям
нельзя выпускать в атмосферу. После разработки
промышленных способов получения хлора из хлористого водорода
рентабельность способа Леблана несколько повысились. Однако
вскоре хлор стали получать электролизом водных растворов
хлористого натрия, что привело к резкому сокращению
производства соды по способу Леблана.
Существенными недостатками описанного способа являются
также большой расход топлива, связанный с необходимостью
проведения основной реакции (спекания) при высокой
температуре, громоздкость аппаратурного оформления, трудоемкость
процесса, невысокое качество получаемого продукта и др.
Предложенный Сольве в 60-х годах XIX в. более
совершенный 'И экономичный аммиачный способ производства соды уже
в конце прошлого века полностью вытеснил из практики способ
Леблана.
В СССР был разработан способ получения соды на основе
природного сульфата натрия, близкий по характеру реакций к
способу Леблана, отличающийся от него по схеме процесса и
аппаратурному оформлению.
Аммиачный способ. Химические реакции, протекающие при
получении соды по аммиачному способу, могут быть выражены
следующими суммарными уравнениями:
NaCl + NH3 + COa + Н2О 7= NaHCO3 + NH4CI A)
2NaHCO3 < * Na2CO3 + CO2 + H2O B)
Обратимая реакция A) при невысоких температурах (около
30 °С) протекает в желательном направлении, т. е. слева
направо; при температуре около 70°С и выше реакция идет в
обратном направлении. Реакция B) также обратима: слева
направо идет при высоких температурах, но на холоду
возможно поглощение кальцинированной содой двуокиси углерода и
воды с образованием бикарбоната натрия.
Требуемая для процесса двуокись углерода получается
обжигом известняка или мела; используется также двуокись
углерода, образующаяся при прокаливании ЫаНСОз по реакции B).
Обжиг известняка или мела:
СаСО3 = СаО + СО2
424
производится в непрерывнодеиствующих известково-обжига-
тельных (известковых) печах. Известь, полученная наряду с
двуокисью углерода, при обработке избытком воды
превращается в известковое молоко
СаО + Н2О = Са(ОНJ
которое используют для регенерации аммиака из раствора
хлористого аммония:
2NH4C1 + Са(ОНJ = 2NH3 + СаС12 + 2Н.2О
Регенерированный аммиак возвращается в процесс.
Таким образом, исходным сырьем для производства соды
по аммиачному способу служат естественные или
искусственные концентрированные растворы поваренной соли и известняк
(или мел). Аммиак, находящийся все время в круговороте,
теоретически не расходуется. Практически неизбежные небольшие
потери аммиака компенсируются путем ввода в систему
соответствующих количеств 18—20%-ной аммиачной воды.
Взаимную связь между отдельными стадиями процесса
получения соды по аммиачному способу можно представить
следующей схемой:
СаСО3
СаО + Н2О
CaO + CO2; NaCl + NH3
> Ca(OHJ; Ca(OHJ +
2NaAcO3 >
+ C0-
2NH4C1
f
Na2CO3 -
hHaO
—>
f co2
4-
t * NH4C) -
1
2NH3 + aClg
+ H20
1- NaHCO3
1
+ 2H2O
Некоторое количество содовых продуктов получается из
отходов производства глинозема путем переработки нефелина
.n(Na,KbO • А12О3- mSiC>2. Отходами этого производства
являются щелока, содержащие до 10% Na2CO3 и 3—4% К2СО3. Путем
упаривания щелоков при различной температуре раздельно
получают соду и поташ. С увеличением объема переработки
нефелинового сырья этот способ может стать перспективным,
так как требует значительно меньших затрат для производства
соды, чем аммиачный способ.
2. ПРОИЗВОДСТВО КАЛЬЦИНИРОВАННОЙ СОДЫ
АММИАЧНЫМ СПОСОБОМ
При разработке теоретических основ процесса получения
кальцинированной соды по аммиачному способу одним из
основных вопросов являлось установление предельного выхода
бикарбоната натрия при насыщении двуокисью углерода аммиач-
425
но-соляного раствора и зависимость выхода NaHCO3 от
различных переменных условий: концентрации и температуры
растворов поваренной соли, степени насыщения двуокисью
углерода аммиачно-соляного раствора и др.
Насыщенный двуокисью углерода а.ммиачно-соляной
раствор представляет собой сложную двухфазную систему: жидкая
фаза является раствором солей NaCl, NH4C1, NH4HCO3.
NaHCO3, (МН^гСОз; твердая фаза (осадок) состоит из
NaHCO3. Причем количество бикарбоната натрия, выпавшего
в осадок и находящегося в растворе, эквивалентно количеству
NH4CI в растворе. По выходе раствора из аппаратов для
насыщения двуокисью углерода эта система находится в
состоянии, близком к равновесному.
Степень осаждения бикарбоната натрия, в основном
определяющая выход кальцинированной соды, зависит при данной
температуре от следующих условий:
1) соотношения количеств NaCl и NH3 в исходном растворе;
2) количества воды в исходном растворе;
3) степени насыщения раствора двуокисью углерода.
С уменьшением отношения количества хлористого натрия к
количеству аммиака степень осаждения бикарбоната натрия,
в соответствии с законом действия масс, возрастает. С
увеличением количества воды в исходной жидкости больше
растворяется бикарбоната натрия и, следовательно, уменьшается его
выход.
Степень насыщения раствора двуокисью углерода должна
быть как можно ближе к отношению СО2 : NH3 в молекуле
бикарбоната аммония. Чем ближе степень насыщения к этому
стехиометрическому отношению, тем большее количество
NaHCO3 осаждается из раствора. Для правильного выбора
таких соотношений исходных компонентов (NaCl, NH3 и НгО),
при которых достигается наибольший выход NaHCO3, т. е.
выпадение в осадок наибольшего количества бикарбоната натрия,
надо знать растворимость солей в сложной многокомпонентной
системе:
NaCl—NH4HCO3—(NH4JCO3—H3O
Эта система была изучена рядом авторов (П. П. Федотьев,.
Е. И. Орлов, А. П. Белопольский и др.).
По данным Федотьева, наибольший достижимый выход
NaHCO3 при 15 °С не превышает 78,8%, при этом коэффициент
использования NaCl составляет 78,8%, аммиака — 85,1%.
Более высокий выход NaHCO3, но не выше 84%, по Федотье-
ву, может быть получен при 30—32 °С.
Для аммиачного способа получения соды такой выход
NaHCO3 язляется теоретически возможным пределом, к
которому стремится практика.
426
Длительное время надеялись путем различных
усовершенствований достигнуть почти полного превращения поваренной
соли в бикарбонат натрия. Исследования равновесия
указанной солевой системы, проведенные Федотьевым, показали, что
при сохранении существа данного процесса такого рода
попытки не могут увенчаться успехом.
ПРИНЦИПИАЛЬНАЯ СХЕМА ПРОИЗВОДСТВА
КАЛЬЦИНИРОВАННОЙ СОДЫ
Принципиальная технологическая схема производства
кальцинированной соды по аммиачному способу показана на
рис. 164.
Водный раствор поваренной соли, содержащий 307—310 г\л
NaCl (рассол), предварительно очищенный от солей кальция
и магния, самотеком поступает в промыватель 7, где
поглотает двуокись углерода из газов, покидающих
карбонизационные колонны 6, и аммиак из газов, поступающих с вакуум-
фильтров 10. Отработанный газ (в основном азот) удаляется
в атмосферу.
Из промывателя газов 7 рассол поступает в абсорбер 8, где
поглощает аммиак и СО2, содержащиеся в газах дистилляцион-
ной колонны 14. Уходящие из абсорбера 8 газы направляются
вакуум-насосом в промыватель 7. Аммонизированный рассол,
предварительно охлажденный в холодильнике 9, непрерывно
поступает в верхнюю часть карбонизационной колонны 6, почти
доверху заполненной жидкостью. Здесь рассол встречается с
промытым и очищенным в аппарате 2 газом
известково-обжигательных печей / C7—40% СОг) и смешанным газом (смесь
газа известково-обжигательных печей и газа содовых печей,
содержащего 85—90% СОг). Концентрация смешанного газа
60—80% СО2 при двух вводах газа в карбонизационную
колонну и около 50% СО2 при одном вводе. В большинстве случаев,
как показано на рис. 164, имеется два ввода газа в
карбонизационные колонны: для смешанного газа F0—80% СОг)
и для газа иззестково-обжигательных печей C7—40% СО2).
Газы подаются в колонну 6 компрессорами 3 и 5.
В карбонизационной колонне 6 протекает основная реакция
превращения хлористого натрия и двуокиси углерода в
бикарбонат натрия. Из верхней части этой колонны газ, содержащий
некоторое количество СОг, поступает в промыватель 7, где
двуокись углерода поглощается свежим рассолом. Образующаяся
з карбонизационных колоннах суспензия кристаллического
бикарбоната натрия в растворе хлористого аммония и непроре-
агировавшего хлористого натрия подается в вакуум-фильтр 10
для отделения бикарбоната натрия. Маточная жидкость (филь-
427
трат) подается из вакуум-фильтров в дистилляционную
колонну 14, где происходит регенерация аммиака в результате
разложения хлорида аммония и углекислых солей аммония.
В атмосферу ^ Вакуу»-
I насосу
Рассол
Кокс,
известняк
Известь
Са(ОНJ
в ал 13
СО2
Нальцинира-
г я оо да
-Мазут или
горючий г?,<
Рис 164. Принципиальная технологическая схема производства кальцийнро-
ванной соды по аммиачному способу:
i—известково-обжигательная печь; 2—холодильник-газоочиститель; 3, 5—компрессоры;
4—аппарат для гашения извести; 6—карбонизационная колонна; 7— промыватель газов; в—абсорбер.
9—холодильник аммонизированного рассола; 10— вакуум-фильтр; 11—холодильник-промыва-
тель газа; 12—содовая печь; W—-дистиллер-смеситель; /4—-дистилляционная колонна.
Углекислые соли аммония разлагаются в дистилляционноп
колонне при нагревании до 70—80 °С выделяющимися здесь
газами (газы дистилляции). Для разложения хлористого
аммония жидкость из колонны 14 передается в
дистиллер-смеситель 13, куда поступает известковое молоко, получаемое
гашением извести в аппарате 4. Аммиак, выделяющийся при
разложении солей аммония, отгоняется из раствора паром,
подаваемым в нижнюю часть аппарата 13. Регенерированные в
колонне 14 и аппарате 13 аммиак и двуокись углерода возвращаются
в абсорбер 8 на поглощение рассолом.
Раствор, полученный после отгонки аммиака, содержит в-
основном хлористый кальций и не вступивший в реакцию хло-
428
ристый натрий. Этот раствор, называемый дистиллврной
жидкостью, является отходом производства, рациональное
использование которого имеет очень важное значение (стр. 465 ел.).
Промытый на вакуум-фильтре 10 осадок NaHCO3 (сырой
бикарбонат натрия) подается во вращающуюся содовую печь/I'
на кальцинирование — разложение с образованием соды, воды
и двуокиси углерода. Кальцинированная сода из печи 12
поступает на склад и далее на упаковку.
В результате разложения бикарбоната натрия в содовых
печах образуется газ, содержащий 85—90% СОг. Пройдя холо-
дильник-промыватель 11, очищенный газ содовых печей
поступает в компрессор 5 и смешивается с промытым и
охлажденным газом известково-обжигательных печей 1. Смешанный газ
F0—80% СОг) компрессор 5 под избыточным давлением около
3 ат подает в нижнюю часть карбонизационной колонны 6.
Производство кальцинированной соды по аммиачному
способу осуществляется в промышленности почти по одинаковой
в принципе технологической схеме. Различны лишь
конструкции, внутреннее устройство, размеры и производительность
отдельных аппаратов. Весьма сложный процесс производства
соды можно разделить на несколько стадий (так называемые
станции или отделения), каждая из которых проводится в
одном или нескольких соединенных между собой аппаратах. На
заводах, вырабатывающих кальцинированную соду,
предусматриваются следующие станции:
1) предварительная очистка ^рассола от солей кальция И!
магния;
2) абсорбция — насыщение рассола аммиаком и частично
двуокисью углерода путем поглощения газов дистилляции;
3) карбонизация—насыщение аммиачно-соляного раствора
двуокисью углерода с образованием бикарбоната натрия в виде
кристаллической взвеси — суспензии (к станции карбонизации
относится также отделение газовых компрессоров); •
4) фильтрование — разделение суспензии бикарбоната
натрия после карбонизации;
5) дистилляция — регенерация аммиака и двуокиси
углерода из жидкостей, образующихся в процессе производства соды;
6) кальцинирование (кальцинация) — разложение бикарбо-.
ната натрия на углекислый натрий (кальцинированную соду),
воду и двуокись углерода.
Кроме этих основных операций получения
кальцинированной соды, проводятся также следующие вспомогательные
процессы:
1) обжиг известняка для получения двуокиси углерода и
извести;
2) гашение извести для получения известкового молока;
3) упаковка соды и складские операции.
429
Схема материальных потоков сложного процесса
производства кальцинированной соды аммиачным способом
изображена на рис. 165.
ТоплиВй
-Дутье
— в- ?jwpL'c!7?trtiJ изльций —*-
Рис. 165. Схема материальных потоков процесса производства
кальцинированной соды аммиачным способом.
3. ВАЖНЕЙШИЕ ОПЕРАЦИИ И АППАРАТУРА ПРОИЗВОДСТВА
КАЛЬЦИНИРОВАННОЙ СОДЫ
Операции и аппаратура производства соды будут
описываться последовательно по ходу технологического процесса. Вначале
рассмотрим подготовительную операцию получения извести и
двуокиси углерода, используемых в производстве содовых
продуктов.
ПОЛУЧЕНИЕ ИЗВЕСТИ И ДВУОКИСИ УГЛЕРОДА
Известь используется в производстве кальцинированной
соды и при получении едкого натра из соды (каустификация),
двуокись углерода применяется в производстве
кальцинированной соды и бикарбоната натрия.
Кроме содовой промышленности, известь широко применяется в
производстве карбида кальция, хлорной извести и гипохлоритов кальция,
кальциевой селитры, многих удобрений, инсектицидов, а также в
многочисленных органических синтезах (в процессах гидролиза, дегидратации,
нейтрализации и т. д.). В сельском хозяйстве иззесть применяют в больших
количествах для известкования почв. Огромные количества извести
потребляются промышленностью строительных материалов (для изготовления силикат-
430
ного и динасового кирпича, смешанных цементов, керамики и др.) и
непосредственно в строительстве в качестве вяжущего (стр. 630).
Двуокись углерода, ломимо производства кальцинированной соды,
применяется для получения различных углекислых солей карбамида (стр. 568),
в сахарном производстве, для изготовления минеральных вод, сухого льда
и др.
Сырье. Сырьем для промышленного получения извести и
двуокиси углерода являются известняки и мел — горные породы,
содержащие карбонат кальция. Карбонатные горные породы в
виде известняка и мела часто образуют целые горные хребты.
Месторождения известняка и мела широко распространены з
различных районах СССР. В горных породах карбонату кальция
сопутствуют примеси SiO2, CaSO4, A12O3, Fe2O3, MgCO3 и др.
Для производства соды в качестве карбонатного сырья
предпочтительнее применять известняк, а не мел. Вследствие
большего содержания влаги в меле по сравнению с известняком
увеличивается расход топлива на обжиг мела и, следовательно,
расход воздуха на сжигание. Увеличение расхода воздуха,
который содержит балластный азот, приводит к получению
обжигового газа с меньшим содержанием СО2. Кроме того,
механическая прочность мела меньше, чем известняка, а при обжиге
мел более склонен к растрескиванию.
Примерный состав извесгняка, применяемого в производстве
соды (в %):
СаСО3 92—94
MgCO3 1,5—2,5
SiO, Не более 3,0
Fe2O3+ А1,О3 0,2—0,6
СаСО4 . . " 1,0
« Влага 0,5
Механизированная добыча известняка (или мела) ведется
главным образом методом открытых разработок в карьерах.
Добытый известняк транспортируется на дробление и
сортировку.
Дробление твердого известняка производится щековыми
дробилками, работающими по принципу раздавливания. Для
дробления мела применяют Еалковые дробилки. Раздробленный
материал путем грохочения разделяют на фракции. Мелкие
фракции мела D0—50 мм) идут в отвал и пока находят
ограниченное применение. Мелкие фракции известняка (менее
40 мм) после вторичного дробления используются в
производстве цемента, отсев B0—50 мм) применяется в строительстве.
Крупная фракция — куски размером 50—120 мм в зависимости
от расстояния, отделяющего карьер от содового завода,
подается к известково-обжигательным печам по подвесной дороге а
вагонетках, либо на открытых железнодорожных платформах,
или в саморазгружающихся гондолах. Известняк,
транспортируемый в вагонетках, разгружается непосредственно в,
обжиговую печь. С железнодорожных платформ известняк сначала по-
431
ступает на заводской прирельсовый склад, откуда вагонетками
подвесной дороги подается в обжиговые печи.
Насыпной вес кускового известняка 1,3—1,4 т/м3. Вследствие
высокой плотности он впитывает лишь до 1% влаги. Насыпной
вес кускового мела около 1 т/л3.
При обжиге карбонатных пород образуются газы,
содержащие СО2. Потеря веса при прокаливании известняка и мела
составляет 41—43%.
Обжиг известняка. При высоких температурах происходит
разложение карбоната кальция
СаСО3 1 > СаО + СОа — 42,52 ккал/моль
Равновесие этой эндотермической реакции смещается в
правую сторону при повышении температуры и понижении
парциального давления СО2-
Зависимость парциального давления СО2 над карбонатом
кальция от температуры характеризуется следующими
данными:
Температура, °С . . . 587 701 749 800 852 898 990,6
Парциальное давление,
мм pm. cm 1,0 23,9 72 183 381 760 2053
Из этих данных видно, что при давлении СО2, равном 1 атм,
разложение СаСОз происходит при 898 °С.
Разложение известняка начинается при температуре около
600 °С. Достаточная скорость диссоциации достигается лишь
при температуре выше 900—950 °С. Поэтому в зоне обжига в
известково-обжигательных печах поддерживают температуру
а пределах 1000—1300°С в зависимости от состава исходного
материала. Примерная температура обжига карбонатного
сырья: при небольшом количестве примесей 1300°С, при более
значительном количестве примесей 1050—1150°С. Повышать
температуру обжига можно только до определенного предела.
Повышение температуры сверх этого предела, вследствие
наличия в обжигаемом материале примесей (кремнезема, окислов
железа, глинозема и др.), взаимодействующих с СаО, приводит
к спеканию массы. Образуются так называемые «козлы»,
прилипающие к поверхности футеровки печи, нарушается
нормальный ход обжига и ухудшается качество извести. Чем больше
таких примесей, тем медленее протекает обжиг и меньше
выход извести.
Кроме температуры, ход обжига известняка зависит также
от физической структуры (плотности) н размероз обжигаемых
кусков. По истечении определенного времени при прочих
одинаковых условиях разложение СаСО3 в более мелких кусках
известняка может закончиться, тогда как внутри более крупных
кусков останется неразложившийся карбонат кальция.
432
Зависимость скорости диссоциации СаСО3 от температуры
и величины кусков известняка показана на рис. 166.
Присутствие значительного количества влаги в исходном
сырье, например в случае обжига мела, нежелательно
(стр. 431). Каждый процент влаги снижает концентрацию СО2
в печном газе примерно на 0,24%.
Для разложения известняка применяется твердое топливо.
Можно использовать также генераторный газ, сжигаемый в
смеси с воздухом, пылевидное или жидкое топливо. Выбор топлива
определяется типом печи и
способом обжига. Например, при
обжиге известняка во
вращающихся печах целесообразнее
применять газообразное или жидкое
топливо, так как при
использовании пылевидного твердого
топлива известь загрязняется
золой.
Для получения высококонцен-
Г'рированного углекислого газа,
требуемого, в частности, для
производства соды, пригодны корот-
кэпламенкые виды топлива,
преимущественно кокс и антрацит
(содержание летучих не более Рис. 166. Зависимость скорости
5°/о). При работе на коксе полу- диссоциации карбоната кальция от
чается газ с более высоким со- темпеРатУРы JL*™" КУСКОВ
держанием СО2 и более чистый,
чем при работе на антраците.
Топливо сжигают при небольшом избытке воздуха A,05).
Содержание СОг в газах известково-обжигательных печей,
работающих с применением воздушного дутья и хорошего
сырья, достигает 39—40%. Дальнейшее увеличение
концентрации СОг в отходящем из печи газе может быть достигнуто
применением воздушно-кислородного дутья C0—35% СЬ).
При сжигании топлива, применяемого для обжига
известняка, должна образовываться тугоплавкая зола.
ИЗВЕСТКОВО-ОБЖИГАТЕЛЬНЫЕ (ИЗВЕСТКОВЫЕ) ПЕЧИ
В Советском Союзе для обжига известняка в производстве
соды применяются шахтные печи без выносных топок,
работающие на твердом топливе (кокс или антрацит).
Шахтные печи разделяются на пересыпные, в которые
известняк загружают вместе («в пересыпку») с топливом, и печи с
выносными топками, в которых топочное пространство
находится вне шахты. Наиболее чистая известь получается в шахтных
печах, работающих на генераторном газе.
28—2848 433
4 в 12 1в 2О
величина Мускод, см
Большое распространение получили вертикальные
(цилиндрические) шахтные печи постоянного сечения или слегка
расширяющиеся книзу. Шахтные пересыпные печи большой
производительности часто имеют
форму цилиндра, в нижней
части переходящего в усеченный
конус. В шахтных пересыпных
печах шихта (смесь
известняка и топлива), постепенно
опускаясь вниз, проходит
последовательно три зоны:
подогрева, обжига и охлаждения.
На рис. 167 схематично
изображена шахтная
пересыпная печь с вращающимся
подом. Печь представляет собой
цилиндр (высота около 17 м,
внутренний диаметр около
4,5 м). Шахта 4 печи выложена
из красного глиняного кирпича
(лучше из диатомитового
кирпича, стр. 623) и футерована
изнутри огнеупорным
(шамотным) кирпичом 6. Для
уменьшения потерь тепла путем
лучеиспускания между
футеровкой и наружной кирпичной
кладкой имеется слой
теплоизоляционного материала 5.
Снаружи шахта печи
заключена в стальной кожух 3 или
стянута стальными кольцами.
Кладка печи опирается на
металлическое кольцо,
расположенное на чугунных колоннах,
которые установлены на
железобетонном фундаменте.
На содовых заводах подача
известняка и топлива в печь
производится
преимущественно из вагонеток. Шихта
загружается в печь автоматически
через загрузочное устройство/
с двойным затвором и рассеивающим конусом, который
равномерно распределяет шихту по сечению печи. Клапаны
загрузочного устройства открываются и закрываются автоматически.
Коэффициент заполнения объема печи составляет 0,9.
Рис. 167. Известково-обжигательная
печь с вращающимся подом:
1—загрузочное устройство; 2—труба для
отвода газов; о—стальной кожух; 4—шахта из
глиняного кирпича; 5—теплоизоляционный
слой; 6—футеровка из огнеупорного кирпича;
7—колпак; 8—вращающийся под; S—стол;
10—вращающийся транспортер; 11—привод;
12—выгрузочная течка; 13—лопастной
питатель.
434
Обжигозый газ отводится из верхней части печи по трубе 2.
Выгрузка обожженной извести производится из нижней
части печи при помощи медленно вращающегося пода 8
специальной конструкции. Под представляет собой круглую
спиралеобразную ступенчатую плиту («улиту»). В верхней части пода
имеется колпак 7, под который в печь подается воздух,
необходимый для горения топлиза. Обожженная известь падает с
вращающегося пода на круглый неподвижный стол 9 и
поступает на выгрузочный вращающийся кольцевой транспортер 10.
При вращении транспортера неподвижно закрепленный нож
сбрасывает известь в течку 12. Отсюда известь при помощи
лопастного питателя 13 непрерывно выгружается из печи.
Процесс обжига извести по высоте печи протекает
следующим образом. В верхней части печи — зоне подогрева,
занимающей примерно 25% объема печи, шихта постепенно
подогревается и подсушивается горячими газами, поднимающимися
снизу. При температуре выше 700 СС начинается горение
топлива. В средней части печи — зоне обжига, которая занимает
около 50% объема печи, при 900—1300°С происходит
диссоциация известняка с образованием СаО и ССЬ и сгорание большей
части топлива. При более высокой температуре возможно
образование «козлов» и частичный обжиг известняка «намертво»—
пережог известняка (пережженная известь плохо гасится
водой). К концу зоны обжига температура несколько понижается.
Продукт обжига — известь спускается в нижнюю часть
печи, в зону охлаждения, и охлаждается поступающим в печь
снизу холодным воздухом, который при этом нагревается. Чем
более охлаждена известь к моменту выхода из печи и чем ниже
температура выходящих газов, тем благоприятнее тепловой
баланс печи.
При нормальном протекании процесса обжига из печи
должна выгружаться хорошо обожженная известь при температуре
не выше 40—50 °С.
Условия нормальной работы печи: равномерная подача
воздуха и шихты, постоянство состава шихты, точное соблюдение
температурного режима обжига, определенная величина кусков
известняка E0—120 мм) и топлива C0—60 мм).
Шахтные печи являются одними из наиболее эффективных
непрерывнодействующих тепловых агрегатов. В них весьма
рационально осуществлен принцип противотока между сырье-
ьым материалом (шихтой) и отходящими газами и между
готовой продукцией (обожженной известью) и воздухом,
подаваемым для сжигания топлива.
Пересыпные шахтные печи отличаются простотой
конструкции, сравнительной легкостью обслуживания и большой
производительностью. Расход топлива (кокса) в пересыпных извест-
ковс-обжлгательных печах меньше, чем во вращающихся печах
Ж 435
и в шахтных печах других типов. На 1 т 85%-ной извести,
получаемой из известняка, расходуется 125—135 кг кокса, расход
антрацита при обжиге мела составляет 150—180 кг в
пересчете на условное топливо G000 ккал/кг) с учетом отходов мелочи.
Средняя производительность отечественных шахтных печей
составляет от 150 до 200 г извести в сутки. Установлены также
печи суточной производительностью более 300 г извести. Нз
новых и реконструируемых содовых заводах устанавливаются
мощные известково-обжигательные печи диаметром свыше 6 м,
высотой более 22 м и производительностью 500—600 т/сутки.
Интенсивность процесса обжига известняка в шахтных
пересыпных печах может быть увеличена следующими способами:
1) повышением температуры обжига (более интенсивная
подача д>тья, применение в качестве дутья воздуха, обогащенного
кислородом, сжигание топлива, образующего золу с высокой
температурой плавления);
2) применением сортированного карбонатного сырья в виде
небольших кусков одинакового размера;
3) более равномерным распределением шихты по всему
сечению печи.
В шахтные печи, работающие на генераторном газе, его
вводят через специальные отверстия. Воздух поступает в печь
через слой обожженной извести на уровне 7з—U высоты от
пода печи. В печи газ смешивается с воздухом и сгорает.
Вращающиеся печи. При обжиге известковой мелочи в
шахтных печах создается большое сопротивление току воздуха, что
нарушает процесс нормального горения топлива*. Этого
недостатка лишены вращающиеся известковые печи, в которых
обычно и производится обжиг известковой мелочи или щебня
(куски размером 10—50 мм). По устройству вращающиеся печи
для обжига известковой мелочи сходны с вращающимися
печами, применяемыми в производстве портланд-цемента
(стр. 640).
Вращающаяся печь одного из применяемых типов
представляет собой стальной барабан, изнутри футерованный
огнеупорным кирпичом. Барабан, снабженный стальными бандажам»
с роликовыми опорами, медленно вращается вокруг
горизонтальной оси. Для перемещения кусков обжигаемого известняка
вдоль печи барабан устанавливают с уклоном 5°. Известняк
загружают в приподнятую часть печи; при вращении печи он
постепенно перемещается, обожженная известь выходит из
барабана с противоположной стороны. При скорости вращения
барабана 1,2 об/мин материал проходит через печь примерно
за 2 ч.
* Известковую мелочь можно использовать для обжига в шахтных
печах в виде брикетов (так называемые черные брикеты).
436
Тепло, необходимое для проведения процесса обжига,
подводят в печь путем сжигания пылевидного топлива (уголь,,
нефтяной кокс и др.), вдуваемого сильной струей подогретого
воздуха в нижнюю часть барабана. Между обжигаемым
материалом и горячими топочными газами осуществляется
противоток. Температура в печи достигает 1000—1100°С.
Размеры вращающихся печей: длина от 20 до 100 м,
внутренний диаметр от 1,8 до 2,5 м. Производительность 60—120 т
извести в сутки. Вращающиеся печи конструкции Уралмаш-
завода имеют объем 275 мъ и производительность около 100 т
извести в сутки. Лучшее использование тепла достигается, если
отходящие газы применяются для обогрева паровых котлов
или для нагревания воды.
Во вращающихся печах другого типа охлаждение извести
и подогрев воздуха производятся в дополнительно
установленных вращающихся барабанах (от одного до трех) значительно
меньших размеров, чем барабан для обжига известняка.
Кольцевые печи находят все меньшее применение для
обжига известняка. Принцип устройства таких печей и режим их
работы описаны б главе XIV (стр. 617). В настоящее время
новых кольцевых печей для обжига известняка не строят.
Охлаждение и очистка газа
известков о-о бжигательных печей
Выходящие из печей газы, имеющие температуру 120—
160 °С, в случае использования СОг для получения соды и
других химических процессов должны быть охлаждены и очищены
от пыли. Охлаждение я очистка газа производятся в мокрых
электрофильтрах, коксовых фильтрах или в турбулентных про-
мывателях (грубы Вентури). В нижней скрубберной части
мокрого электрофильтра, заполненной деревянной хордовой
насадкой, газ проходит снизу вверх навстречу орошающей воде.
При этом он охлаждается до 30 °С и затем поступает
собственно в электрофильтр.
Пыль с осадительных и коронирующих электродов
электрофильтра периодически смывается водой и удаляется
через скрубберную часть электрофильтра. Во время промывки
электродов очистка газа производится в запасном
электрофильтре.
Газ, поступающий на очистку, содержит около 700 мг/мг
пыля, после очистки в скрубберной части электрофильтра
содержание пыли понижается до 250—300 жг/ж3, а после
электрической очистки составляет 3—10 мг/м3. При прохождении
газа через скруббер концентрация СОг в газе несколько
уменьшается (на 0,3—0,4%) вследствие растворения СОг в воде, а
также подсоса воздуха через неплотности аппаратуры.
437
ГАШЕНИЕ ИЗВЕСТИ
На современных заводах выдача обожженной извести из
печи производится непрерывно. Известь поступает на
пластинчатый, а затем на ковшовый конвейер, который доставляет еи
в расходные бункеры отделения гашения извести или в
бункеры-хранилища для дальнейшего использования.
Гашение извести водой протекает по реакции
СаО + Н2О = Са(ОН), + 15,93 ккал
Изменяя количество воды, подаваемой на гашение, можно
получать известь-пушонку (тонкий порошок), известковое тесто
(густая нетекучая масса) или известковое молоко — водную
суспензию Са(ОН)г.
На содовых заводах приготовляют известковое молоко,
используемое для регенерации аммиака из раствора
хлористого аммония (стр. 457) и для очистки рассола от солей магния
{стр. 440).
Рис. 168. Аппарат для гашения извести:
I—барабан; 2—планки; 3—приводная шестерня; 4—бандажи; 5—воронка питателя; 6—опорные
ролики; 7—сортировочный барабан.
Аппарат для гашения извести, т. е. приготовления
известкового молока (рис. 168), представляет собой вращающийся
стальной барабан / (диаметр 2,5 м, длина 15 м). Снаружи
барабан покрыт слоем теплоизоляционного материала. Аппарат
опирается двумя бандажами 4 на две пары вращающихся
стальных роликов 6.
Вращение барабана со скоростью 3—4 об/мин
осуществляется при помощи шестерни 3.
Обожженная известь через воронку 5 питателя подается в
аппарат вместе с отработанной теплой водой из холодильника
газа содовых печей (стр. 463). Вода дополнительно подогре-
438
вается до 60—80 С водяным паром, выходящим из аппарата
для гашения. Перемещаясь внутри барабана, известь
постепенно превращается в известковое молоко, которое вытекает из
противоположного конца аппарата. Температура реакционной
массы в барабане 95—98 °С. Для более быстрого передвижения
и лучшего перемешивания материала внутри барабана имеются
планки 2, расположенные винтообразно. Водяные пары
отводятся через две вытяжные трубы, установленные на кожухе;
на одной из труб смонтирован подогреватель воды (на рисунке
не показан).
Концентрация Са(ОНJ з известковом молоке составляет
280—294 г/л. Известковое молоко отделяется от крупных кусков
недожога во вращающемся сортировочном барабане (сито),
смонтированном в разгрузочном конце аппарата. Для
окончательной очистки от песка, мелких частиц пережженной извести
и других примесей известковое молоко на выходе из барабана
поступает на мелкое вибрационное сито, откуда направляется
в отделение дистилляции. Крупные куски недожога,
отделенные от известкового молока в сортировочном барабане 7, снова
возвращают в печь на обжиг, мелкие примеси с
вибрационного сига направляют в отброс.
Количество мелких отбросов, удаляемых с вибрационного
сита барабанного гасителя, составляет 60—100 кг на 1 т
извести.
Суточная производительность описанного гасителя
достаточна для обеспечения известковым молоком завода,
вырабатывающего 1000—1200 т соды в сутки.
Примерные расходные коэффициенты на 1 т 85%-ной
извести в виде известкового молока:
Известняк (94% СаСО3), m . . . 1,73—1,75
Кокс, пг 0,13
Вода, мл 4,5
Электроэнергия, квт-ч 10,2
ПРИГОТОВЛЕНИЕ И ПРЕДВАРИТЕЛЬНАЯ
ОЧИСТКА РАССОЛА
В производстве кальцинированной соды по аммиачному
способу перерабатывают водный раствор поваренной соли
(рассол), получаемый из естественных источников или растворением
твердой каменной соли в воде (стр. 332 ел.)
Для приготовления рассола используется также отбросная
поваренная соль — отход производства хлористого калия из
сильвинита (стр. 579 ел.).
439
На 1 г кальцинированной соды расходуется около 5 мг
сырого рассола, содержащего 305—310 г/л NaCl*, поэтому
содовые заводы строят обычно вблизи месторождений поваренной
соли. При выборе места для строительства содовых заводов
учитывают также наличие сырья для получения двуокиси
углерода и извести (известняк, мел), топливные и водные ресурсы и др.
Содержащиеся в рассоле небольшие примеси солей кальция
и магния F—7 г/л) при введении в раствор аммиака и
двуокиси углерода выпадают в осадок в виде карбоната кальция
и гидроокиси магния. Чтобы избежать засорения аппаратуры
и загрязнения готового продукта (соды) и не усложнять
конструкцию основных аппаратов, эти осадки возможно полнее
удаляют из раствора. Для этой цели на содовых заводах
введена предварительная очистка рассола от ионов кальция и
магния, проводимая преимущественно содово-известковым
методом (стр. 333). Рассол обрабатывают известковым молоком
я раствором соды, при этом нежелательные примеси выпадают
з осадок.
После отстаивания образовавшихся осадков рассол, почти
полностью очищенный от кальциевых и магниевых солей,
направляется на абсорбцию аммиака.
При кооперировании содового и электрохимического
производств, использующих отходы поваренной соли с калийных
комбинатов, рассолоснабжение может быть организовано по
следующей схеме. Подаваемый по трубопроводам с калийного
комбината сырой рассол, приготовленный из отбросного NaCl,
подвергается предварительной очистке от Са и Mg и поступает
на дополнительную очистку путем отстаивания, подогрева и
фильтрования. Очищенный рассол подается на электролиз.
Отработанный электролит (обратный рассол) из ртутных ванн
обесхлоривают, очищают от ртути и донасыщаюг обратной
•солью, выпавшей из упаренного электролитического щелока
цехоз диафрагменного электролиза. Далее очищенный рассол,
имеющий нужную концентрацию NaCl, поступает на
производство кальцинированной соды.
По такой схеме может быть получен дешевый рассол
хорошего качества.
Предварительная очистка рассола вне аппаратуры содового
производства способствует повышению культуры аммиачно-
содового процесса, позволяет улучшить работу основных
станций содового производства (абсорбции, карбонизации,
фильтрования).
* На содовых заводах концентрацию жидкостей принято выражать не
в г/л, а в так называемых нормальных делениях (сокращенно н, д.). Одно
нормальное деление соответствует содержанию '/го г-экв вещества в 1 л
раствора. Концентрация применяемого рассола C05—310 г/л NaCl)
соответствует 104,3—106 н. д.
440
АБСОРБЦИЯ
Основное назначение станции абсорбции заключается в
приготовлении аммонизированного рассола (аммиачно-соляной-
раствор) такой концентрации, которая требуется для процесса
карбонизации. Вначале рассол поступает в аппараты-промыва-
тели, где поглощает NH3 и СО2 из движущихся противотоком
отходящих газов станций карбонизации, абсорбции и
фильтрования. Окончательное насыщение рассола аммиаком,
поступающим со станции дистилляции, происходит в первом и втором,
абсорбергх. Аммиак и двуокись углерода при абсорбции их
рассолом образуют карбонат и карбамат аммония:
2NH3 + СО2 + Н2О > (NH4JCO3
2NH3 + СО2 ¦> NH2COONH4
растворения СО2
При абсорбции аммиака и двуокиси углерода выделяется
значительное количество тепла: теплота растворения NH3
составляет 8430 кклл/кмоль,
5880 ккал/кмоль. Кроме того,,
при нейтрализации аммиака
углекислым газом выделяется
16 850 ккал на 1 кмоль NH3.
Тепло выделяется также при
конденсации водяных паров,
содержащихся в газах
дистилляции. Общее количество
выделяющегося тепла достаточно
для нагревания рассола на
80—90 °С. Однако при
повышении температуры процесс
абсорбции аммиака
ухудшается, поэтому абсорбцию ведут с
промежуточным охлаждением
жидкости в холодильниках.
Для абсорбции
применяются аппараты барботажного
типа, представляющие собой
вертикальные колонны, состоящие
из отдельных чугунных
звеньев— барботажных бочек с
фланцами, соединенных болтами. В каждой бочке имеется
специальное устройство —барботажная тарелка, или пассет, на
котором происходит тесное соприкосновение газа с жидкостью.
Схема многоколпачкового пассета A4 небольших колпачков)
изображена на рис. 169. Колпачки 2 с зубчатыми краями
опущены в слой жидкости ка глубину 80—85 мм. Каждый колпачок
опирается на горловину 3 и прижимается к ней скобой 4 и бол-
Рис. Многоколпачковый пассет:
/—днище пассета; 2—колпачки;
2—горловины; 4—скоба; 5—прижимные болты; 6—гайка;
7—внутренние переливы.
441
том 5 (в центре колпачка) с гайкой 6. Газ входит под
колпачки 2 через горловины 3, барботирует через слой жидкости
и уходит в верхнюю бочку.
Для перетекания жидкости из одной бочки в другую
каждая из них снабжена двумя внутренними переливными
трубами 7, выступающими на 175 мм над тарелкой (над днищем
пассета).
На содовых заводах применяются многоколпачковые пас-
сеты и реже одноколпачковые.
При введении многоколпачковых пассетов с внутренними
переливными трубами вместо ранее применявшихся однокол-
пачковых пассетов в три раза увеличилась поверхность
соприкосновения газа с жидкостью, уменьшилась глубина барботажа
и, таким образом, понизилось гидравлическое сопротивление
прохождению газа.
Разработан и применяется новый аппарат—абсорбер с
внутренним охлаждением жидкости. Процесс абсорбции
аммиака в этом аппарате протекает одновременно с охлаждением
жидкости при помощи холодильных трубок, расположенных в
нижней части аппарата и являющихся насадкой.
По выходе из таких аппаратов аммонизированный рассол
дополнительно охлаждается в пластинчатых водяных
холодильниках.
Одна из схем станции абсорбции изображена на рис. 170.
Очищенный рассол самотеком поступает на абсорбцию из
напорного бака /, расположенного на высоте около 50 м.
Постоянный уровень жидкости в напорном баке поддерживается при
помощи поплавка и троса, связанного с вентилем на
нагнетательной линии рассола. Пройдя через регистрирующие
расходомеры (на рисунке не показаны), рассол разделяется на два
потока. Большая часть рассола направляется во второй про-
мыватель 4 газа карбонизационных колонн, в который газ
поступает из первого промывателя (см. рис. 174, стр. 450).
Меньший поток рассола, в свою очередь, разделяется на две части:
одна направляется в промыватель 3 воздуха, поступающего из
вакуум-фильтра, другая — в малый абсорбер 12.
Во втором промывателе 4 рассол подается в верхнюю бар-
ботажную бочку и перетекает вниз навстречу газовому потоку.
Здесь из газа улавливаются содержащиеся в нем аммиак и
частично двуокись углерода.
Газ, выходящий из второго промывателя 4, после
освобождения от брызг рассола и промывки водой (на рисунке не
показано) отводится в атмосферу.
Рассол, частично насыщенный аммиаком (около 21,5 г/л) и
СО2 (около 8,2 г/л), из второго промывателя 4 газа колонн
поступает в первый абсорбер 5 на дальнейшее насыщение
аммиаком.
442
Часть меньшего потока рассола, направленная в промыва-
тель <?, дзижется в нем сверху вниз навстречу воздуху,
который поступает из барабанных вакуум-фильтров (стр. 456).
В промывателе 3 улавливается аммиак, содержащийся в этом
воздухе в небольшом количестве, после чего рассол самотеком
Очищенный
рассол NH3+CO2
1 из первого проныва-
—• теля газа колонн
Воздух из вакуум фильтров
NH3+CO2
со станции мамой
дистилляции
Рис. 170. Схема станции абсорбции:
/—напорный бак; 2—холодильник газа дистилляции; 3— промыватель
воздуха фильтров; 4—второй промыватель газа колонн; 5—первый
абсорбер; 6—второй абсорбер; 7—резервуар; 8, 9— холодильники;
10—промыватель газа абсорбции; //—промыватель газа малого
абсорбера; 12—малый абсорбер; 13—газоотделитель; /4—ваку} м-насос.
поступает в промыватель 10 газа абсорбции, где движется
навстречу газу, выходящему из первого абсорбера 5. В
промывателе 10 улавливается аммиак, не поглощенный в абсорбере 5.
Концентрация аммиака в этой части рассола 8,5—9,5 г/л.
443
По выходе из промывателя 10 рассол также поступает в
перзый абсорбер 5, в верхней части которого оба потока
рассола соединяются вместе.
В первом абсорбере 5 рассол, стекая навстречу газу,
поступающему из второго абсорбера 6, поглощает аммиак и двуокись
углерода. Вследствие экзотермичности процесса абсорбции,
сопровождающегося образованием (NH4JCO3 и NH4OH, в
абсорбере 5 жидкость нагревается до 70 °С. Для достижения
требуемой концентрации аммиака в рассоле во втором абсорбере 6
рассол, содержащий уже 61—63 г/л аммиака и 27,5 г/л
двуокиси углерода, по выходе из первого абсорбера 5 охлаждается
до 30 °С в холодильнике 9 и самотеком поступает во второй
абсорбер 6.
Часть рассола, поступившая в малый абсорбер 12, движется
здесь навстречу газу, который подается из аппаратов малой
дистилляции (стр. 458). Охлаждение рассола до 30 °С
производится по данной схеме в трубчатых холодильниках,
расположенных в малом абсорбере. Выходящий из абсорбера 12 рассол
присоединяется к охлажденному аммонизированному рассолу,
поступающему в абсорбер 6 из холодильника 9.
Во втором абсорбере 6 рассол проходит последовательно
все барботажные бочки сзсрху вниз навстречу газу,
поступающему со станции дистилляции и предварительно охлажденному
в холодильнике 2. Во втором абсорбере происходит основной
процесс абсорбции аммиака и частичное поглощение двуокиси
углерода из газа дистилляции. Температура аммонизированного
рассола повышается при этом до 70°С. Рассол, содержащий
около 87 г/л NH3 и около 40 г/л СОг, охлаждается в
холодильнике 8 до 30 °С, самотеком направляется в резервуар-хранилище
и далее на карбонизацию.
Абсорбция газов рассолом проводится при разрежении,
создаваемом вакуум-насосом 14. Всасывающая линия вакуум-
нгсоса присоединена через газоотделитель 13 к верхней части
промывателя // газов малого абсорбера 12. Отсасываемые
вакуум-насосом газы идут в холодильник газа содовых печей.
Потери аммиака в производстве соды восполняются
введением свежей аммиачной воды (обычно 18—20%-ной)* во
второй абсорбер 6. Аммиачная вода из хранилища перекачивается
в цеховый резерзуар 7, являющийся постаментом одной из
колонн абсорбции, и центробежным насосом подается в верхнюю
барботажную бочку второго абсорбера 6. Для уменьшения
коррозии аппаратуры и улучшения качества продукции к
очищенному рассолу, поступающему в первый абсорбер 5, иногда
добавляют небольшое количество раствора сернистого натрия.
* При сравнительных расчетах количество аммиачной воды пересчиты-
вается в условные единицы — на содержание 25,5% аммиака.
444
Примерный состав газов в основной аппаратуре станции
абсорбции приведен в табл. 26.
Таблица 26
Примерный состав газов
Газы
На входе во второй абсорбер 6
На выходе нз первого абсорбера 5
На входе в промыватель 4 газа колонн . . .
Состав газа, объемн. %
NH3
65
11
18
СО2
12
60
6
воздух
1
21
70—71
пары
воды
22
8
5-6
Применявшиеся ранее аппараты барботажного типа,
например, промыватель газа абсорбции и др., на новых заводах
заменены скрубберами с насадкой, благодаря чему снизились
затраты энергии на просасывание газов через абсорбционную
систему.
ОСНОВНАЯ АППАРАТУРА СТАНЦИЙ АБСОРБЦИИ
Ока смонтирована в виде большой чугунной колонны
(общая высота около 41 м, диаметр 2,8 м). Колонна состоит из
следующих аппаратов, установленных друг на друге (снизу
вверх): резервуар для хранения аммиачной воды, конденсата
из холодильника газа дистилляции и других жидкостей,
второй абсорбер, первый абсорбер, второй промыватель газа
колонн, промыватель воздуха вакуум-фильтров и холодильник
газэ дистилляции.
Абсорберы, второй и первый (рис. 171) предназначены для
поглощения рассолом аммиака и двуокиси углерода,
содержащихся в газе, который поступает со станции дистилляции.
Второй абсорбер (общая высота около 7,4 м, диаметр 2,8 м)
состоит из семи бочек /—VII с толщиной стенок 30 мм. Средние
три барботажные бочки //—IV снабжены многоколпачковыми
пассетами. Над барботажными бочками расположены три
пустые бочки V—VII, служащие брызгоуловителями. Нижняя
бочка / используется для входа газов и выхода жидкости. Газ
поступает во второй абсорбер при давлении 630 мм рт. ст. и
температуре 58 °С. Первый абсорбер отделяется от второго
чугунным днищем, по конструкции и размерам он аналогичен
второму абсорберу. Из семи бочек I—7 верхняя бочка 7
служит брызгоуловителем, остальные, снабженные
многоколпачковыми пассетами, являются барботажными. Рассол поступает
в верхнюю барботажкую бочку 6 при 28—30 °С, выводится из
нижней бочки 1 при 70 СС.
445
Вход
рассола аз """
про мы да теля
so за колон*
ерЗь/й
odcopdep
Второй
adcopdep
ЯммонизироЗон-
ttbtu россол
8холодильник j/ci/д-
кости Второго
aocopdepa
Газ
в прамывотель
газа ао~сар$ции
Рассол
из промыва теля
воздуха,
фильтров
Э+СиЗхасть
8хо& газа
дистиллер
с л ado й
Им мо низаро$онмый
*. рассол
6 эсоло&ильник
docmu первого
adcopdepa.
гага
диет илляцаи
Рис. 171. Второй и первый абсорберы:
1-—VU-— бочки второго абсорбера; 1—7— бочки первого абсорбера.
В холодильнике газа дистилляции (рис. 172) поступающий
на абсорбцию со станции дистилляции газ охлаждается
водой с 67—69 °С до 58—60 °С.
Вход
водь/
Зыход
Выход го за
во второй-
оо~соро~ер
Рис. 172. Холодильник газа дистилляции:
/—пустая бочка: II—VI—холодильные бочки.
Холодильник (диаметр 2,8 м, общая высота 8,5 м) состоит
из шести бочек /—VI.
Холодильные бочки //—VI имеют по 288 чугунных трубок
(диаметр около 50 мм, длина около 3 м). Охлаждающая
поверхность трубок в каждой бочке около 140 м2.
Газ входит в верхнюю пустую бочку / холодильника под
давлением 650 мм рт. ст. и выводится из аппарата снизу.
Охлаждающая вода поступает в трубки холодильника про
тивотоком движению газа.
447
Гач при 45 °С и остаточном давлении 560 мм рт. ст. выходит
из верхней бочки 7 в промыватель газа абсорбции.
Второй промыватель газа колонн служит для поглощения
рассолом аммиака и двуокиси углерода из газа, поступающего
из первого промывателя газа колонн со
станции карбонизации. Второй
промыватель состоит из восьми бочек: средние
шесть снабжены многоколпачковыми пас-
сетами, верхняя служит брызгоуловителем,
в нижнюю вводят газ из первого
промывателя газа колонн. Диаметр бочек 2,8 м,
общая высота промывателя около 9 м. Газ
поступает в аппарат снизу под давлением
;!00 мм рт. ст. при температуре 45 °С. Из
верхней бочки газ, практически не
содержащий NH3 и СОг, выводится в атмосферу.
Промыватель воздуха фильтров служит
для улавливания небольших количеств
аммиака из воздуха, отсасываемого с вакуум-
фильтров. Промыватель состоит из шести
бочек. Газ поступает в нижнюю бочку, из
которой выводится также рассол,
направляемый в промыватель газа абсорбции.
Три следующие барботажные бочки,
предназначенные для поглощения аммиака
рассолом, и пятая бочка, в которую
вводится вода для улавливания аммиака, не
поглощенного рассолом, снабжены
многоколпачковыми пассетами.
Верхняя пустая бочка язлястся
брызгоуловителем.
Для улавливания брызг рассола иногда
верхнюю бочку питают водой.
Промыватель газа абсорбции,
примерная конструкция которого изображена на
рис. 173, предназначен для окончательного
поглощения рассолом аммиака и двуокиси
углерода, уходящих из абсорберов. Это
отдельно установленный аппарат (высота
около 9,4 м, диаметр 1 м). Он состоит из
чугунной бочки-базы 1, семи бочек 2,
заполненных коксовой насадкой (общий
объем 4,3 м3), и бочки 3, которая служит
брызгоуловителем.
Высота каждой бочки 1 м. В нижней бочке 2 имеется
колосниковая решетка 4, на которую опирается коксовая насадка.
Орошающая жидкость поступает в промыватель сверху через
Рис. 173.
Промыватель газа абсорбции:
1—бочка-база; 2— бочки с
насадкой. ..— брызгоулови-
тель; 4—колосниковая
решетка; 5
—распределительная ришетка.
448
распределительную решетку о и выходит из нижней
бочки-базы 1. Газ движется снизу противотоком к орошающей
жидкости и выходит через штуцер бочки-брызгоуловителя 3 в промы-
ватель газа малого абсорбера.
Контроль процесса аммонизации. Нормальный режим
процесса аммониэации рассола поддерживается при
систематическом наблюдении за работой аппаратуры и точном
регулировании потоков рассола, охлаждающей воды и вакуума в
системе. Систематически контролируется содержание аммиака в
жидкости и газе из абсорберов, промывателя газа
карбонизационных колонн и периодически — из промывателей газа абсорбции
и газа из вакуум-фильтров. Кроме этого, регулярно
измеряется температура рассола и вакуум во всей системе.
В настоящее время контроль и управление процессом ам-
монязацин рассола автоматизированы на подавляющем
большинстве содовых заводов.
КАРБОНИЗАЦИЯ
В процессе карбонизации аммонизированный рассол
насыщается СО2 с образованием суспензии бикарбоната натрия.
Карбонизация проводится последовательно в трех
аппаратах: в колонне предварительной карбонизации, в первом про-
мывателе газа колонн, в осадительной колонне.
Процесс предварительной карбонизации совмещается с
промывкой карбонизационной колонны от осевшего на ее стенках
бикарбоната натрия.
Реакции, протекающие при растворении бикарбоната,
осевшего на стенках колонн, можно представить следующими
уравнениями:
в колонне предварительной карбонизации
2NaHCOs + 2NH4OH = Na2CO3 + (NH4)aCO3 + 2H3O
в осадительной колонне
Na2CO3 + СОа + Н2О = 2NaHCO3
Продолжительность работы каждой колонны в качестве
аппарата предварительной карбонизации около суток, в
качестве осадительной колонны от 3 до 4 суток. Таким образом, 3—
4 осадительные колонны обслуживаются одной колонной
предварительной карбонизации.
Обычно группа карбонизационных колонн состоит из пяти
колонных аппаратов (четыре осадительные колонны и одна
промывная).
По окончании промывки колонна работает как осадитель-
ная. При этом аммонизированный рассол донасыщается
двуокисью углерода, т. е. происходит процесс основной
карбонизации.
29—2848 449
Схема станции карбонизации изображена на рис. 174.
Аммонизированный рассол со станции абсорбции (стр. 443) при тем-
ператург 30 °С подается в верхнюю часть колонны 2, которая
является в данном цикле колонной предварительной
карбонизации. Очищенный и охлажденный газ известково-обжигатель-
ных печей, содержащий 37—40% СО2, подается компрессором /
Рис. 174. Схема станции карбонизации:
/—компрессор; 2—колонна предварительной карбонизации; 3—первый промыватель "газа ко-
лоии; 4—сборник; 5—осадительиая карбонизационная колонна; 6—холодильные бочки;
7—мериик.
в нижнюю бочку колонны. В этих условиях достигается быстрое
растворение осадка и полная абсорбция двуокиси углерода
аммонизированным рассолом.
Жидкость, выходящая из колонны предварительной
карбонизации, содержит 60,5—66,0 г/л СО2 и 83—85 г/л NH3 и имеет
температуру 38—39°С. Колонну предварительной карбонизации
можно охлаждать, но к этому прибегают обычно только в
жаркое время года.
Из колонны предварительной карбонизации под избыточным
давлением 700—800 мм рт. ст. выходит газ, содержащий 5—6%
COi- л 25% NH3 и направляемый в первый промыватель 3 газа
колонн. Промыватель орошается жидкостью, поступающей из
колонны предварительной карбонизации 2. В промывателе
жидкость несколько донасыщается двуокисью углерода,
которая приходит сюда с газами, выходящими из осадительных
карбонизационных колонн 5 (на рисунке показана одна колон-
450
на) и из колонны предварительной карбонизации 2. Благодаря
этому полнее улавливается двуокись углерода и меньше
аммиака уносится газом из жидкости.
Из первого промывателя 3 газ направляется зо второй про-
мыватель газа колонн на станцию абсорбции, жидкость через
сборник 4 центробежным насосом подается в осадитсльную
колонну 5.
Для поддержания в колонне предварительной карбонизации
постоянного уровня жидкости в осадительной колонне и в
сборнике 4 установлены поплавковые регуляторы (на рисунке не
показаны), связанные с дроссельным клапаном на
нагнетательной линии.
В осадительную карбонизационную колонну 5 газ поступает
в двух местах: через ввод над верхней холодильной бочкой
подается газ пзвестково-обжигательных печей, содержащий
37—40% СО2, в нижнюю бочку колонны — смесь газа содовых
и известково-обжигательных печей — смешанный газ,
содержащий 60—80% СОг. Из верхней части карбонизационных
колонн 5 газ отводится по общему трубопроводу в первый про-
мыза^ель 3 газа колонн.
Реакция карбонизации протекает с значительным
выделением тепла, вследствие чего температура жидкости в средней
части карбонизационной колонны повышается до 60—65 °С.
Поэтому нижняя часть колонны (десять холодильных бочек 6)
охлаждается водой. Охлаждающая вода поступает двумя
параллельными потоками в первую и вторую (считая снизу)
холодильные бочки карбонизационной колонны. Таким образом,
один поток охлаждающей воды проходит все нечетные
холодильные бочки колонны, второй поток—все четные. Нагретую
воду удаляют через общий коллектор. Применяются колонны
~л других конструкций и различные схемы ввода газа и воды
в холодильные бочки колонн.
Охлажденная до 26—28 °С жидкость*, содержащая 59,5—
61 г!л связанного аммиака (эквивалентно количеству
осажденного NaHCO3), вместе с взвесью бикарбоната натрия
выводится из нижней части колонны и через мерник 7 самотеком
направляется на вакуум-фильтры для отделения бикарбоната
натрия.
Основная реакция, протекающая в карбонизационной
колонне, схематически может быть выражена уравнением
NaCl + NH3 + СО3 + Н2О ; > NaHCO3 + NH4C1
или
NaCl + NH4HCO3 -^=± NaHCO3 -f NH4C1
* В летнее время температура жидкости выше и показатели работы
колонн хуже.
29* 451
Газ
Выход бикарбоната натрия
в аммиачном процессе
производства соды зависит от
содержания NH3 и NaCl в
аммонизированном рассоле,
концентрации СОг в газе,
поступающем на карбонизацию, и
температуры процесса. Чем
выше содержание NaCl и NH3
в жидкости и СОг в газе и чем
ниже температура в нижней
части колонны, тем больше
выход NaHCCb, т. е. выше
коэффициент использования
натрия.
КАРБОНИЗАЦИОННЫЕ
КОЛОННЫ
Осадительная
карбонизационная колонна (рис. 175),
применяемая на одном из
содовых заводов, представляет
собой цилиндрический
чугунный аппарат барботажного
типа диаметром 2,7 м и общей
высотой около 26 м. Колонна
состоит из бочки-базы 1,
десяти холодильных бочек 4,
двадцати пяти барботажных
бочек 5 (с тарелками), двух
газовых бочек 7 (без тарелок).
Газ движется противотоком,
проходя через всю толщу
аммонизированного рассола,
перетекающего сверху вниз из
одного поглотительного
устройства в другое, и
уходит через верхние штуцеры
колонны.
Рис. 175. Осадительная
карбонизационная колонна (предкарбонизатор):
/_бочка-база; 2—конический отражатель; з—
барботажные тчрелки; 4—холодильные бочки;
Л—барботажные бочки; 6—колокол;
7—газовые бочки; 8— отверстие для прохода газа и
жидкости; 9—отверстие овальной формы; 10—
водомерное стекло- //—предохранительный
клапан.
452
Бочка-база / высотой около 0,9 м имеет штуцеры для входа
смешанного газа (под избыточным давлением 2,8 ат) и выхода
жидкости из колонны. Над бочкой-базой для равномерного
распределения газа по сечению колонны установлена барботажная
тарелка 3, а также конический отражатель 2.
Холодильные бочки 4 (высота каждой бочки 0,95 м)
снабжены трубками, по которым снизу вверх проходит вода
(максимальное избыточное давление воды в трубках 2 ат). Общая
поверхность охлаждения в холодильных бочках колонны около
1000 м2. Карбонизованный раствор движется в межтрубном
пространстве сверху вниз. Между холодильными бочками также
установлены барботажные тарелки 3.
Каждая барботажная бочка снабжена тарелкой 3
линзообразной формы с колоколом 6, имеющим зубчатые края. Для
прохода газа и жидкости в тарелке имеется центральное
отверстие 8 диаметром около 0,3 м, вокруг него восемь овальных
отверстий 9 и на периферии тарелки 72 конусных отверстия (на
рисунке не показаны), расположенных в шахматном порядке
в два ряда. Такое же устройство имеют тарелки, установленные
между холодильными бочками над бочкой-базой.
Газ из известково-обжигательных печей поступает в нижнюю
барботажную бочку, расположенную над холодильными
бочками, а жидкость вводится в верхнюю барботажную бочку*.
Высота столба жидкости в колонне около 24 м. Две верхние
газовые бочки 7 (высота каждой 1,35 м) снабжены водомерными
стеклами 10 для наблюдения за уровнем жидкости в колонне.
В крышке колонны имеются предохранительный клапан И и
два штуцера для выхода газа из колонны.
Продолжительность прохождения жидкости через
карбонизационную колонну 2—2,5 ч.
Содержание СО2 в газе, выходящем из карбонизационной
колонны, не должно превышать 6—7%. На 1 г соды
поглощается около 650 кг СОг, из этого количества около 155 кг СОг
поглощается в процессе предварительной карбонизации.
Образование крупных кристаллов бикарбоната натрия,
которые в дальнейшем хорошо отфильтровываются, достигается
при соблюдении примерно следующего температурного режима
карбонизации:
•С
Газ на выходе из колонны 45—48
Жидкость:
на входе в колонну 36—38
в середине колонны • . . 60—65
на выходе из колонны 25—28
Охлаждающая вода на выходе из холодильных бочек . . 40—45
* Во время работы колонны в качестве предкарбонизатора жидкость
подается во вторую сверху барботажную бочку.
453
Производительность колонны также зависит от
температурного режима карбонизации, содержания СОг в газе и других
условий.
С увеличением концентрации СОг в газе возрастает
скорость реакции образования NaHCO3 в нижней зоне колонны, а
при хорошем охлаждении колонны одновременно увеличивается
и скорость кристаллизации бикарбоната натрия.
При нормальной работе производительность колонн
указанных выше размеров составляет около 190 г
кальцинированной соды в сутки.
Коэффициент использования натрия при этом составляет
около 72%.
Степень использования натрия в процессе карбонизации
определяется разностью между общим содержанием аммиака
в колонной жидкости и содержанием его в жидкости,
покидающей колонны.
Производительность карбонизационной колонны может быть
повышена при использовании отбросного углекислого газа с
более высоким содержанием СО2 (например, 85%), побочно
образующегося при очистке азотоводородяой смеси,
получаемой из газов конверсии окиси углерода водяным паром
(стр. 177 ел.).
Коэффициент использования натрия увеличивается при этом
до 75%, расход рассола снижается на 4—5%, уменьшаются
потери аммиака и т. д.
Новые карбонизационные колонны диаметром 3 м имеют
производительность.250 -т/сутки кальцинированной соды.
Расстояние между верхними барботажными бочками
увеличено, что позволило уменьшить скорость карбонизации в
начале процесса (до кристаллизации бикарбоната) и увеличить
ее в конце кристаллизации.
При этом получаются кристаллы бикарбоната натрия
хорошего качества.
Контроль процесса карбонизации. На станции карбонизации
непрерывно контролируется с помощью самопишущих приборов
содержание СО2 в газе, поступающем в колонны, и в газе,
выходящем из осадительных колонн; по показаниям
автоматических самопишущих приборов регулируется отбор жидкости из
карбонизационных колонн.
Большое значение для нормального режима работы
карбонизационных колонн имеет разность температур входящей в
колонну жидкости и уходящего из колонны газа, а также
температура в средней части колонны, где начинает
кристаллизоваться бикарбонат натрия.
В зависимости от температуры колонной жидкости
регулируют подачу охлаждающей воды в холодильные бочки.
454
ФИЛЬТРОВАНИЕ
На станции фильтрования производится отделение осадка
бикарбоната натрия от маточной жидкости и промывка этого
осадка. Для этой операции применяются непрерывнодействую-
щие барабанные вакуум-фильтры.
Рис. 176. Схема станции фильтрования:
/—приемный желоб; 2—вакуум-фильтр: 3- тпптчелитель;
4—скребковый транспортер; 5,6—сборники.
Принципиальная схема этой станции изображена на
рис. 176.
Суспензия бикарбоната натрия из приемного желоба /
поступает по трубопроводу на вакуум-фильтр 2. Избыток
суспензии из вакуум-фильтра стекает в сборник 5 с мешалкой, откуда
периодически возвращается в питающий трубопровод.
Отфильтрованный осадок бикарбоната для удаления NaCl
и NH4CI промывается на барабане вакуум-фильтра жидкостью,
поступающей из промывателя газа содовых печей. Промытый
сырой бикарбонат натрия срезается с барабана вакуум-фильтра
ножом и сбрасывается на скребковый транспортер 4, подающий
бикарбонат в содозые печи на кальцинирование.
Маточная жидкость (фильтрат) вместе с промывной
жидкостью освобождается от воздуха в газоотделителе 3 и
поступает в сборник 6', откуда фильтрат перекачивают на станцию
дистилляции (для регенерации аммиака).
455
Вакуум на фильтрах создается вакуум-насосами,
установленными на станции абсорбции. Воздух, отделенный от
маточной жидкости в газоотделителе 3, поступает в промыватель
воздуха фильтров на станции абсорбции (стр. 442 ел.).
Вакуум-фильтр состоит из вращающегося барабана, корыта
с мешалкой для фильтровой жидкости, промывного устройства,
вращающихся прижимных роликов, ножа для срезания
бикарбоната натрия и аппарата для отделения жидкости от воздуха.
Вода
для промывки
I Зоэдух 9ля ошдувш
_^ NaHCXK на
кальцинирование
воздух для продувки
фильтрующей
ткани
Рис. 177. Схема работы вакуум-фильтра:
I—вакуумные ячейки; 2—ячейка отдувки: 3—промежуточная
ячейка; 4—слой бикарбоната; 5—нож для срезания осадка;
6—секция промывки: 7—центральный вал; 8—секция
всасывания распределительной головки; 9—ячейка продувки
(очистки фильтра), 10—мешалка.
Принцип работы вращающегося вакуум-фильтра (рис. 177)
заключается в следующем. Бикарбонат натрия осаждается на
ткани, натянутой на дырчатый барабан длиной 1 м и
диаметром 1,8 м (поверхность фильтрации 5,6 м2). Барабан вращается
на горизонтальном полом валу 7 со скоростью около
1,1—3,4 об/мин. Через вал из вакуум-фильтра отсасывают
воздух, газы и жидкость. Одновременно осадок промывается
жидкостью, поступающей из промывателя газа содовых печей.
Чтобы частицы осадка не забивали поры фильтрующей ткани,
фильтр периодически продувают сжатым воздухом,
подаваемым в секции (ячейки) изнутри. Бикарбонат срезается с ба
рабана стальным ножом 5, установленным под углом 45° на
расстоянии 5,8 мм от поверхности фильтрующей ткани.
Отфильтрованный осадок содержит около 14 % влаги.
Производительность одного вакуум-фильтра 160—200 г соды
з сутки. Вакуум-фильтры, обслуживающие производство NaOH
456
и очищенного NaHCO3, имеют меньшую производительность —
до 130—140 г соды в сутки. В этих случаях для лучшей
промывки осадка уменьшают число оборотов барабана.
ДИСТИЛЛЯЦИЯ (РЕГЕНЕРАЦИЯ АММИАКА)
На станции дистилляции производится регенерация аммиака
из жидкости, поступающей с вакуум-фильтров (фильтровая
жидкость), и из слабых жидкостей, в которых аммиак
содержится в виде (ЬЛ-ЦЬСОз. Фильтровая жидкость содержит,
кроме NaCl, главным образом хлорид аммония (связанный
аммиак), а также карбонат и бикарбонат аммония (так
называемый поли связанный аммиак). Фильтровая жидкость при
температуре около 30 °С поступает на дистилляцию в
количестве 5,7—6,2 м3 на 1 т соды.
Полусвязанный аммиак легко выделяется при нагревании
жидкости до 72—74 °С:
(NH4JCO3 * 2NH3 -f CO2 + Н2О
Для выделения же связанного аммиака из хлористого
аммония требуется обработка фильтрата известковым молоком
при нагревании:
2NH4C1 + Са(ОНJ * 2NH3 + СаС12 -'г 2Н2О
Выделившийся аммиак отгоняется из жидкости водяным
паром.
Раствор неиспользованного хлористого натрия и
образовавшегося хлористого кальция (дистиллерную жидкость)
целесообразно перерабатывать для получения хлоридов кальция,
аммония, магния (стр. 465 ел.) или перекачивать в скважины
для законтурного обводнения нефтяных месторождений.
Регенерация аммиака из фильтровой жидкости (основная
дистилляция) и из слабых жидкостей (малая дистилляция)
чаще производятся раздельно.
Схема станции дистилляции показана на рис. 178.
Фильтровая жидкость со станции фильтрования подается в напорный
бак, расположенный на высоте около 50 м, и самотеком
поступает в конденсатор дистилляции /, где подогревается до
72—74 СС газами, выходящими из аппаратов дистилляции (на
рисунке ке показано).
В конденсаторе / фильтровая жидкость движется по
трубкам -и последовательно проходит через все восемь бочек
аппарата, поступая в нижний штуцер каждой бочки и выходя через
ее верхний штуцер. Противотоком жидкости в межтрубтм
пространстве движутся газы, выходящие из аппаратов дистилля-
457
ции. Конденсат, выделившийся из газов в конденсаторе i и в
холодильнике газа дистилляции (на станции абсорбции),
образует слабую жидкость, не содержащую соды, которая идет на
¦на абсорбцию
Вода
Вода \мистилле{7ная
жидкость
Рис. 178. Схема станции дистилляции:
/—конденсатор; 2—теплообменник; 3—дистиллер основной
дистилляции; 4—смеситель; 5— испаритель: 6—дистиллер малой дистилляции;
7—холодильник жидкости; 8—конденсатор малой дистилляции;
9—холодильник газа малой дистилляции; /О—напорный Сак;
/;—холодильник газа дистилляции.
повторную регенерацию в малый дистиллер 6. Во многих
схемах дистилляции конденсат поступает в теплообменник из
конденсатора дистилляции. При нагревании фильтровой жидкости
происходит частичное разложение углекислых солей аммония
с выделением СО2 и аммиака.
Подогретая до 72—74 °С жидкость стекает из
конденсатора / в теплообменник 2, где газы, приходящие из смесителя 4
458
при 85 С, охлаждаются до 70—72 С. Дальнейшее охлаждение
этих газов и паров жидкости происходит в холодильнике газа
дистилляции 11.
В теплообменнике 2 происходит полное разложение всех
углекислых солей аммония, оставшихся в фильтровой жидкости
после нагревания в конденсаторе /. Практически полное
разложение происходит при нагревании жидкости до 93—95°С
парами, поступающими из дистиллера 3.
Из теплообменника 2 жидкость, освобожденная от
углекислых солей аммония и содержащая связанный аммиак в виде
хлорида аммония, поступает в среднюю часть смесителя 4,
куда подается также горячее или холодное известковое молоко
в количестве до 2,35 м3 на 1 г соды. В смесителе при высокой
температуре и перемешивании происходит разложение
хлористого аммония с выделением NH3. Большая часть
выделившегося аммиака растворяется в жидкости, газообразный аммиак
присоединяется к потоку парогазовой смеси, проходящей
через теплообменник 2 и конденсатор / и направляемой
далее на станцию абсорбции. Из нижней части смесителя 4
жидкость, содержащая некоторый избыток известкового
молока, поступает в третью (сверху) бочку дистиллера 3. Здесь
аммиак отгоняется из жидкости острым водяным паром,
подаваемым в нижнюю часть дистиллера.
Для дистилляции аммиака в производстве соды большей
частью применяется отработанный пар газовых компрессоров
с добавлением отборного или мятого пара турбин
электростанций. В зависимости от принятого режима работы пар
поступает в дистиллер под давлением до 2 ат и при температуре
от 93 до 120 °С. Расход пара на дистилляцию составляет 1,5—
1,8 г на 1 т соды.
После отгонки аммиака дистиллерная жидкость, имеющая
температуру 107—111 °С, поступает в испаритель дистилляции 5,
работающий при разрежении. Вследствие снижения давления
над жидкостью в испарителе происходит значительное
выделение водяных паров, которые направляются в дистиллер 6.
Дистиллерная жидкость разбавляется водой и удаляется в
отброс или направляется на использование.
Дистилляция слабых жидкостей. Слабая жидкость и
флегма из конденсатора / (см. рис. 178) и холодильника газа
дистилляции // перерабатываются в колонне малой
дистилляции 6. Из напорного бака 10 смесь этих жидкостей
поступает в теплообменные трубы верхней теплообменной
бочки конденсатора 8, в межтрубном пространстве которого снизу
поднимается парогазовая смесь из дистиллера 6. Газ из
конденсатора малой дистилляции 8 поступает в межтрубное
пространство теплообменных бочек холодильника 9, где охлаждается
до 56—58 °С водой, подаваемой в трубы этих бочек. Охлажден-
459
спесь d холодильник
ныи газ направляется на стан
цию абсорбции, в третью бар-
ботажную бочку первого
абсорбера (на рис. 178 не
показано).
Жидкость, подогретая в
конденсаторе 5 малой
дистилляции, стекает в дистиллер 6.
В нижнюю его часть навстречу
потоку жидкости поступает из
испарителя 5 пар, к которому
добавляют отработанный пар
турбин. В малом
дистиллере 6 происходит полное
разложение углекислых солей
аммония. Жидкость,
освобожденная от аммиака и двуокиси
углерода, охлаждается в
холодильнике 7 и используетсч
затем для промывки газа
содовых печей и бикарбоната
натрия на вакуум-фильтрах.
Дистилляцию слабых
жидкостей обычно проводят под
давлением: в нижней части
дистиллера до 1300 ммрт.ст.,
в верхней части
теплообменника до 870 мм рт. ст.
ДИСТИЛЛЯЦИОННЫЕ КОЛОННЫ
Основным аппаратом
станции регенерации аммиака
является дистилляционная
колонна (рис. 179). Она состоит
из следующих аппаратов,
установленных друг на друге:
дистиллера /,
теплообменника // и конденсата ///.
Рис. 179. Днстилляционная колонна:
/—дистиллер; (/—теплообменник;
///—конденсатор: /—бочка-база; 2—нижняя бочка
дистиллера; J—барботажные бочки
дистиллера (а—зубчатый колокол; б—переливы);
4—газовые бочки дистиллера; 5—нижняя бочка
теплообменника; б—барботажные бочки
теплообменника; 7 —газовая бочка
теплообменника; S—переходная бочка; 9—газовая бочка
конденсатора; 10—теплообменные бочки;
// — крышка.
4OU
Общая высота колонны ~ 46 м, диаметр дистиллера и
теплообменника 3 м, диаметр конденсатора 2,8 м.
Дистиллер, предназначенный для отгонки аммиака из
жидкости водяным паром, представляет собой чугунный аппарат
барботажного типа (общая высота около 17 м). Он состоит из
бочки-базы / со штуцером для более полного опорожнения,
нижней бочки 2, служащей для входа пара и выхода жидкости,
одиннадцати барботажных бочек 3, каждая из которых имеет
барботажную тарелку с зубчатым колоколом а. Глубина бар-
ботажа 50 мм.
Над барботажными бочками расположены две газовые
бочки 4, служащие брызгоуловителями. Для лучшего отделения
парогазовой смеси от брызг жидкости газовые бочки
снабжены специальными отбойными устройствами.
В настоящее время на крупных содовых заводах дистилля-
ционные колонны перерабатывают более 130 м3[ч фильтровой
жидкости каждая. Дистиллер отделяется от теплообменника
промежуточным днищем с заглушённым отверстием; днище
расположено над верхней газовой бочкой.
Теплообменник дистиллера предназначен для разложения
углекислых солей аммония и отгонки СОг из фильтровой
жидкости при помощи водяного пара, поступающего с газом из
дистиллера. Применяются теплообменники двух типов: с
барботажными тарелками и с насадкой (деревянная хордовая или
коксовая). Теплообменник барботажного типа представляет
собой чугунный аппарат общей высотой около 16 м, в котором
имеются нижняя бочка 5, служащая для входа парогазовой
смеси и выхода жидкости; двенадцать барботажных бочек 6,
каждая из которых снабжена барботажной тарелкой с
четырнадцатью колпачками. В верхнюю барботажную бочку
поступает фильтровая жидкость из конденсатора. Над верхней бар-
ботажной бочкой помещена газовая бочка 7 с деревянной хор-
дозой насадкой для отделения от газа брызг жидкости.
Над газовой бочкой помещена переходная бочка 8, которая
отделяется от конденсатора промежуточным чугунным днищем.
Конденсатор предназначен для подогрева фильтровой
жидкости, поступающей на дистилляцию. Это — чугунный аппарат
диаметром 2,8 м и высотой около 12 м. Он состоит из газовой
бочки 9, восьми теплообменных бочек 10 и крышки 11 со
штуцером для выхода парогазовой смеси. Все теплообменные
бочки имеют отводы для газов, выделяющихся из фильтровой
жидкости (СОг и NH3), присоединенные к общему коллектору
в верхней части аппарата. Поверхность теплообмена каждой
бочки 10 около 100 м2. В межтрубном пространстве конденса-
тор-j проходит снизу вверх парогазовая смесь; образующийся
конденсат стекает вниз и отводится из нижней бочки
конденсатора.
461
Фильтровая жидкость проходит по трубам последовательно
через все теплообменные бочки сверху вниз.
Примерный состав парогазовой смеси, выходящей из
дистиллера, теплообменника и конденсатора дистилляционной
колонны (в объемн. %):
NH3 СОг
Дистиллер ....
Теплообменник . .
Конденсатор . . .
28
38
52
—
5,5
11
Температурный режим работы колонны:
°С
Температура фильтровой жидкости:
на входе в колонну 29
в нижней части дистиллера 115
Температура парогазовой смеси на
выходе из конденсатора, °С 68
Контроль процесса дистилляции осуществляется путем заме-
роз и анализов, производимых различными автоматическими
указывающими и регистрирующими приборами, показания
которых дистанционно передаются на щит управления станцией
дистилляции. Температурный режим процесса дистилляции
обычно контролируется при помощи термометров
сопротивления с указывающими и регистрирующими устройствами.
Все точки контроля снабжаются также обычными стеклянными
термометрами.
Контроль режима давления производится при помощи
поплавковых мановакуумметров с дистанционной передачей
показаний. Для этого применяются также ртутные манометры и
ртутные U-образные мановакуумметры, располагаемые
непосредственно в точках замера.
Аналитический контроль осуществляется в основном
объемными методами и выполняется аппаратчиками и цеховой
лабораторией.
КАЛЬЦИНИРОВАНИЕ
На станции кальцинирования сырой бикарбонат натрия
превращается в карбонат натрия, т. е. в кальцинированную соду.
Для этого бикарбонат натрия нагревают до 140—170 °С. При
нагревании протекают следующие реакции:
2ЫаНСО3(тв.) > Ка2СО3(тв.) + СО2+ Н2О (пар) — 29,95 ккал
(NH4JCO3 > 2NH3 + СО, + Н2О
Выделяющийся аммиак и СО2, образующиеся при
разложении аммонийных солей, возвращаются в производство.
Схема станции кальцинирования показана на рис. 180.
Сырой бикарбонат натрия подается транспортером 1 через
воронку 2, снабженную вертикальной мешалкой, в загрузочное
462
устройстве содовой печи — забрасывающий питатель 3. При
помощи метательной лопатки питателя, вращающейся со
скоростью около 200 об/мин, бикарбонат забрасывается в барабан
содовой печи 5 на глубину 6—9 м от горловины. При
вращении барабана бикарбонат смешивается с частично прокаленной
содой, находящейся в печи. За время пребывания в барабане
(около 3 ч) бикарбонат разлагается, выделяя NH3, СОг и пары
воды, и превращается в кальцинированную соду (96—98%
Na2CO3).
Рис. 180. Схема станции кальцинирования:
/—транспортер; 2—воронка; 3—забрасывающий питатель; 4—колосниковая решетка; 5—бараба .
содовой печи; 6—вентилятор; 7—подогреватель воздуха; 8—выгрузочный шиек; 9—Цепь; 10—
циклон; //—газовый коллектор; 12—промыватель газа содовых печей; 13—холодильник газа
содовых печей; 14, /5—иасосы.
Барабан 5 содовой печи представляет собой стальной
сварной цилиндр, заключенный в кирпичную обмуровку. В
кольцевом пространстве между барабаном и обмуровкой движутся
обогревающие топочные газы. К обоим концам барабана
присоединены две стальные конические горловины. Общая длина
барабана (с конусами) около 25 м, диаметр примерно 2,5 м.
Поверхность барабана, обогреваемая топочными газами,
составляет около 196 м2. Барабан приводится во вращение от
индивидуального электродвигателя мощностью 60 кет. Скорость
вращения барабана около 5 об/мин.
Внутри к торцовым стенкам барабана прикреплена цепь 9,
которая про.чодит вдоль образующей цилиндра и состоит из
463
тяжелых и легких звеньев, шарнирно связанных между собой.
При вращении содовой печи цепь совершает волнообразные
движения, разрезая и разбивая корку соли на куски. Таким
образом предотвращается прилипание бикарбоната натрия к
стенкам печи.
Образовавшаяся б печи сода при помощи разгрузочных
ковшей и шнека 8 выводится из печи при температуре около 180°С
в сборный шнековый транспортер, проходящий вдоль
выгрузочных концов всех содовых печей, и после охлаждения
поступает на склад для упаковки.
На 1 т соды в содовых печах образуется около 400 кг СО2.
Углекислый газ, выходящий из печи вместе с парами воды,
небольшим количеством аммиака и содовой пыли,
освобождается от нее в циклоне 10. Выгрузка содовой пыли из циклона
механизирована и производится при помощи рамной мешалки
и гребков, расположенных внутри циклона. Пыль по желобу
возвращается в содовую печь.
Интенсификация описанных содовых печей достигается
увеличением линейных скоростей газа путем сужения газоходов
и усиления тяги.
В некоторые содовые печи, не оборудованные устройствами
для забрасывания сырого бикарбоната, подается смесь готовой
(ретурной) соды и бикарбоната, что предотвращает
комкование и спекание продукта в процессе кальцинирования.
Применяются также мощные содовые печи производительностью до
220 т/сутки, работающие с безретурным питанием. За рубежом
•распространены горизонтальные вращающиеся содовые печи,
обогреваемые паром высокого давления C0—40 ат) и питаемые
смесью ретура и сырого бикарбоната. Разработана
отечественная конструкция паровой содовой печи, в которой процесс
кальцинирования осуществляется в кипящем слое.
Выходящий из циклона 10 газ — 95%-ный СО2 (см. рис. 180)
направляется в коллектор 11 газа содовых печей, орошаемый
изнутри жидкостью, поступающей через специальные брызгалки
из холодильника 13 газа содовых печей. Жидкость далее
перекачивается на малую дистилляцию для отгонки аммиака и
двуокиси углерода.
Газы из коллектора 11, имеющие температуру 125 °С,
охлаждаются до 40—45 °С в холодильнике 13 газа содовых печей.
Газы движутся в межтрубном пространстве, куда подается
некоторое количество слабой жидкости. Охлажденный газ
далее поступает в нижнюю часть промывателя 12 газа содовых
печей, заполненного деревянной хордовой насадкой. Промыва-
тель орошается охлажденной жидкостью, из которой в
аппаратах малой дистилляции отогнан аммиак. Циркуляция
жидкости в холодильнике 13 осуществляется центробежным
насосом /5, который подает также жидкость в коллектор 11. Из
464
промывателя 12 газ направляется в коллектор смешанного
газа, где присоединяется к газу известково-обжигательных печей.
В качестве топлива для наружного обогрева содовых
печей может применяться мелкий уголь (горошек), мазут,
природный газ. Сжигание угля производится в топках,
оборудованных движущейся беспровальной колосниковой решеткой 4.
Воздух, поступающий в топку, на некоторых заводах
предварительно подогревается до 300 °С путем теплообмена с
отходящими из топок содовых печей газами. Вентилятор 6 подает
воздух в подогреватель 7, расположенный непосредственно на
обмуровке содовых печен в месте выхода топочных газов.
Нагретый воздух направляется в топку, топочные газы в
дымовую трубу. Вместо подогревателя воздуха можно
устанавливать котел-утилизатор для получения пара (до 420 кг на 1 г
соды), используемого в паровых компрессорах, при регенерации
аммиака и для других нужд.
Нормальная работа содовых печей зависит главным образом
ог качества сырого бикарбоната натрия и теплового режима
процесса кальцинирования. Подаваемый в печь сырой
бикарбонат должен содержать 82—84% NaHCO3, не более 0,2—0,4%
NaCl и не свыше 13—15% влаги.
Выход кальцинированной соды из такого продукта
составляет 52—54%, при повышенном содержании влаги в
бикарбонате (до 18%), выход снижается до 50,5%-
Примерные расходные коэффициенты на I т
кальцинированной соды приведены ниже:
Очищенный рассол C10 г/л NaCl), м? . 4,84—5,15
Аммиачная вода B5,5% NH3), кг 12—14
Известь (85% СаО), пг 0,7—0,71
Электроэнергия, квт-ч 36—102
Пар, млн. ккал 1,28—1,32
Топливо G000 ккал/кг), кг 123,5—145
Вода, м3 82—140
Контроль процесса кальцинирования заключается в
определении содержания ССЬ в газах при помощи автоматических
газоанализаторов, снабженных самопишущими устройствами,
непрерывно регистрирующими на диаграмме результаты
анализа. Отвод топочных газов регулируется автоматически.
Необходимый режим давления в коллекторе газа содовых печей
автоматически поддерживается струйным регулятором.
Утилизация отходов производства соды
Отвод дистиллерной жидкости, содержащей СаС12 и NaCl,
в специальные обвалованные хранилища (так называемые
белые моря) с последующим сбросом жидкости в водоемы
приводит к их засолонению и целому ряду других осложнений.
30—2848 465
На каждую тонну полученной кальцинированной соды
сбрасывается в «белые моря» 9 м3 раствора хлористого
кальция. На крупных содовых заводах в «белых морях»
накапливаются миллионы кубометров раствора хлористого кальция.
Однако при рациональной переработке дистиллерной жидкости
производство кальцинированной соды может быть осуществлено
без всяких отбросов.
П>тем двухстадийного упаривания дистиллерной жидкости
можно вначале выделить хлорид натрия, после отмывки
которого от примесей получается пищевая поваренная соль.
Проводя вторую стадию выпарки при глубоком вакууме, можно
получить порошкообразный хлористый кальций, содержащий
более 70% СаСЬ.
Хлористый кальций используют в производстве хлорида бария, для
коагуляции латекса, в синтезах некоторых красителей, в
химико-фармацевтической промышленности, в системах кондиционирования воздуха. В
связи с большой гигроскопичностью СаСЬ его часто применяют в качестве
осушителя газов и жидкостей. Он используется также для получения
металлического кальция электролизом расплава кальциевых солей и в
производстве баббитов. Низкие температуры замерзания водных растворов
СаС1г обусловливают применение их в качестве хладоагентов и
антифризов (для двигателей в авиации, автомобильном транспорте). Хлористый
кальций употребляется в строительстве как добавка к бетону при его
кладке в зимних условиях и др. За рубежом значительное количество
хлористого кальция используется для обеспыливания грунтовых и щебеночных
дорог, а также при строительстве дорог.
Хлористый кальций выпускают в виде раствора (не менее 32% СаС12).
плавленого продукта (не менее 67% СаС12) и в виде безводной соли (не
менее 92% СаС1г) Плавленый СаС12 перевозят в стальных барабан:;:-..
Возможно также комбинирование производства соды и
хлористого аммония методом вымораживания последнего из
маточной жидкости после отделения бикарбоната натрия на
фильтрах (стр. 455). Маточную жидкость донасыщают
аммиаком и СОг и направляют на низкотемпературную
кристаллизацию в присутствии NaCl для более полного выделения хлорида
ам,мония.
Путем повторной кристаллизации получают достаточно
чистый продукт.
Хлористый аммоний используется при пайке, лужении, цинковании
металлов, в текстильной и химико-фармацевтической промышленности, для
наполнения гальванических элементов и др. Он находит также известное
применение в качестве удобрения на нейтральных и насыщенных
основаниями почвах под такие культуры, как лен, конопля, овес, озимая рожь.
При использовании для регенерации аммиака суспензии
Mg(OHJ, т. е. доломитового молока вместо известкового
молока, можно получать, наряду с содой, .хлористый магний. Ди-
стиллерная жидкость в этом случае содержит, кроме СаСЬ,
взвесь окиси магния.
466
При карбонизации жидкости
СаС1г + MgO + COa •
MgCl2 + CaCO8
в осадок выпадает карбонат кальция, который можно
использовать в производстве извести, цемента (стр. 634 ел.) и для
других целей. В результате упаривания раствора выделяется
шестиводный хлорид магния (бишофит), применяемый для
производства металлического магния, хлората магния
(эффективный дефолиант), магнезиальных огнеупоров (стр. 627).
Сорта, упаковка и перевозка
кальцинированной соды
В СССР производится кальцинированная (синтетическая)
года трех марок: техническая, фотографическая и оптическая,
различающаяся по степени чистоты. Требования, которым,
согласно ГОСТ 5100—49, должна удовлетворять
кальцинированная (синтетическая) сода, приведены ниже:
Общая щелочность в пересчете на Na2CO3,
%, не менее
Потери веса при прокаливании, %, не более
Содержание примесей, %, не более
нерастворимые в воде вещества ....
NaCl
Na2SO4
Fe . . . ...
Техническая
95
3,5
Допускается
легкая
муть
1
Не
определяется
Не
нормируется
Фотографическая
95
3,5
0,1
1
0,1
' 0,01
Оптическая
96
2,5
0,3
0,5
0,05
0,005
К технической кальцинированной соде, потребляемой
различными отраслями промышленности, предъявляются
некоторые дополнительные требования. Так, сода, предназначенная
для никелевой промышленности, по требованию потребителя
должна содержать не более 0,05% карбоната магния;
содержание сульфата натрия в соде, предназначенной для алюминиевой
промышленности,- должно быть не выше 0,1%; содержание
железа в соде, применяемой в стекольной промышленности и для
эмалирования, — не более 0,02%.
Для производства высококачественного оптического стекла
применяется кальцинированная сода, соответствующая нормам
ГОСТ 83—41.
Кальцинированную соду упаковывают в 4—6-слойные
бумажные мешки, вес нетто не более 50 кг. На таре четко обоз-
30 *
467
начается название завода-изготовителя, название и марка
продукта, номер партии и номер ГОСТ. Перевозится
кальцинированная сода в крытых железнодорожных вагонах.
4. АВТОМАТИЗАЦИЯ ПРОИЗВОДСТВА
КАЛЬЦИНИРОВАННОЙ СОДЫ
Производство кальцинированной соды является одним из
наиболее сложных процессов химической технологии.
Автоматизация этого процесса позволяет повысить культуру произ
водства, увеличить степень использования сырья и энергии,
облегчить условия труда, достигнуть длительной
бесперебойной работы аппаратуры, снизить себестоимость и улучшить
качество готовой продукции и т. д. В настоящее время для
всех технологических станций содово-аммиачного процесса
разработаны и для большинства станций внедрены в производство
системы комплексной автоматизации, исключающие
необходимость непосредственного участия обслуживающего персонала
в регулировании процесса производства соды и являющиеся
составными частями общей схемы его автоматического управ
ления.
Все системы автоматизации отдельных станций разработаны
по общей единой методике, сущность которой заключается в
следующем.
Из всех материальных потоков (см. рис. 165, стр. 430),
поступающих на станцию и выходящих из нее, выбирается один
поток, величина которого определяет нагрузку данной станции
Этот ведущий поток является независимой переменной в
системе ее регулирования. Все остальные потоки подчинены
ведущему потоку, и системой регулирования приводятся в соответствие
с ним в каждый данный момент времени. При этом регуляторы
соотношения потоков воздействуют через вторичные приборы
и исполнительные органы на подчиненные потоки. Заданное
соотношение потоков с течением времени соответственно
изменяется, так как состаз и величина материальных потоков
также изменяются во времени.
Ниже описаны системы автоматического регулирования
важнейших станций производства соды по аммиачному способу.
Автоматизация известковых печей (рис. 181). Известняк или
мел вагонетками подвесной канатной дороги подается в изве-
стково-обжигательную печь 2. Перед печью установлены
автоматические весы, дозирующие в каждую вагонетку из
бункера 1 определенную порцию топлива (кокс или антрацит).
Шихта поступает в печь через автоматически открывающийся
двойной затвор, воздух подается в нижнюю часть печи
вентилятором 3
468
Ведущим потоком цеха известковых печей для упрощения
схемы принят расход воздуха, пропорциональный выходу
извести. Заданный расход воздуха поддерживается
регулятором а; регулятор соотношения б, воздействуя на скорость
вращения улиты, поддерживает отвод извести из печи со скоростью,
пропорциональной расходу воздуха. В связи с возможными
воэди-
Рис. 181. Схема автоматизации известковых печей:
/—б>нкер; 2—известково-обжигательная печь; 3—вентилятор;
а—регулятор расхода воздуха; б—регулятор соотношения;
в—регулятор уровня шихты; г—регулятор давления.
изменениями качества топлива воздействие регулятора б
корректируется в зависимости от температуры газа, выходящего
из верхней части лечи.
Температура газа, измеряемая термопарой, характеризует
положение зоны обжига по высоте лечи, стабильность ее
положения свидетельствует о правильности выбранного соотношения
воздуха и извести. Для предотвращения колебаний температуры
газа при загрузке в печь холодной шихты термопара помещена
в шихту на глубину 1—1,5 м. Постоянство высоты слоя шихты
поддерживается регулятором в, воздействующим на устройство
¦ 469
для опорожнения вагонеток. При достаточном уровне шихты в
печи это устройство пропускает вагонетки мимо для загрузки
других печей.
Регулятор г стабилизирует давление в печи путем отвода
излишнего газа в атмосферу.
Автоматизация станции абсорбции (рис. 182). Очищенный
рассол проходит аппараты абсорбции двумя потоками.
очищенный рассол
Дистанционное
управление
жидкости на
дистилляцию
В атмосферу
Ряс. 182. Схема автоматизации станции абсорбции:
1—напорный бак; 2—промыватель воздуха фильтров; 3—холодильник
газа дистилляции; 4—промьшатель газа абсорбера; 5—промыватель
газа колонн; 6—первый абсорбер; 7—второй абсорбер; 8,
9—оросительные холодильники; 10—бак; а—регулятор уровня; б—регулятор
соотношения; в—регулятор вакуума; г—устройство дистанционного
управления; д—регуляторы темлературы-
Основной поток из напорного бака / поступает в
промыватель 2 воздуха фильтров, затем самотеком проходит через
промыватель 5 газа колонн и первый абсорбер 6, где
смешивается с вторым потоком рассола, поступающим сюда из
напорного бака через промызатель газа колонн.
Газы поступают на абсорбцию тремя потоками: газ
дистилляции (ведущий поток) проходит через холодильник 3 газа
дистилляции, второй абсорбер 7, первый абсорбер 6 и
промыватель 5 газа абсорбции, после чего газ, содержащий
остатки COj, поступает на карбонизацию. Второй поток газа
470
(из карбонизационных колонн) проходит через промыватель с-
и отводится в атмосферу. Третий поток газа (воздух с
фильтров), пройдя промыватель 2, также удаляется в атмосферу.
Путем регулирования расхода очищенного рассола система
автоматизации процесса абсорбции должна обеспечить полное
поглощение всего аммиака, поступающего с газами, и
заданную концентрацию 1МНз в аммонизированном рассоле. В систему
автоматического регулирования станции абсорбции входят
следующие автоматические устройства:
регулятор а уровня рассола в напорном баке 1;
регулятор соотношения б, поддерживающий
соответствующую нагрузку процесса дистилляции по фильтрату и процесса
абсорбции но очищенному рассолу. Состав этих жидкостей
может изменяться, поэтому предусмотрена корректировка
настройки регулятора б по температуре жидкости, выходящей из
абсорбера 7. Температура жидкости зависит только от
концентрации аммиака в рассоле, так как очищенный рассол имеет
постоянную температуру в течение длительного времени;
регулятор вакуума в, регулирующий отсос газа
вакуум-насосами на карбонизацию;
регуляторы температуры д. соответственно изменяющие
расход охлаждающей воды.
Второй поток очищенного рассола поддерживается
постоянным. В исключительных случаях изменение его производится
при помощи устройства дистанционного управления г.
Автоматизация станции карбонизации. Для упрощения на
рис. 183 показаны две колонны: колонна 1 предварительной
карбонизации и осадительная (карбонизационная) колонна 4.
Каждая колонна в течение 3—4 суток работает как
осадительная, а затем на сутки переключается на предкарбонизацию, во
время которой ее промывают. Коммуникации, необходимые для
переключения колонн, на рис. 183 для упрощения схемы не
показаны.
Аммонизированный рассол поступает в колонну 1
предварительной карбонизации и затем последовательно проходит
промыватель 3, холодильник 2, осадительную колонну 4 и мерник
суспензии 5. Газ поступает на карбонизацию Тремя потоками:
газ известково-обжигательных печей (печной газ) под
повышенным давлением подается в нижнюю часть колонны /;
смешанный газ поступает в нижнюю часть осадительной
колонны 4;
печной газ вводится также в среднюю часть колонны 4.
Газы колонн собираются в общем коллекторе и поступают
в низ промывателя 3, откуда направляются на абсорбцию. При
поглощении двуокиси углерода аммонизированный рассол
нагревается. Охлаждение его происходит в трубчатом водяном
холодильнике 2 и в нижней части осадительной колонны 4.
471
Ведущим потоком для станции карбонизации является поток
газов, содержащих СОг. Равномерное распределение печного и
смешанного газов из общих коллекторов по осадительным
колоннам достигается при помогли регуляторов расхода а и
командных регуляторов б. Регуляторы расхода поддерживают
заданный расход газа в колонне, командные регуляторы в
зависимости от давления газа в коллекторах соответственно
воздействуют на все регуляторы расхода.
Рис. 183. Схема автоматизации станции карбоиизации:
/—колонна предварительной карбонизации; 2—холодильник; 3—промыватель;
4—осадительная колонна, 5— мерник суспензии; а—регуляторы расхода;
б—командные регуляторы, в—регулятор рН; в—регуляторы уровня;
в—регуляторы температуры; е—регулятор давления.
Таким образом поддерживается заданное соотношение
между количествами газа, поступающего в общий коллектор и в
колонны.
Соответствие между подачей газа, содержащего СО2, и
отводом аммонизированного рассола из колонны 4 поддерживается
регулятором в, который в зависимости от значения рН
суспензии бикарбоната натрия регулирует ее количество,
передаваемое на фильтры. Подача жидкости в осадительную колонну 4,
промыватель 3 и колонну 1 предварительной карбонизации
устанавливается в соответствии с интенсивностью отвода
суспензии из осадительной колонны 4 регуляторами уровня г.
Регуляторы температуры д, воздействуя на расход охлаждающей
воды, регулируют охлаждение жидкости в холодильнике 2 и в
нижней части осадительной колонны. 4. Регулятор е
обеспечивает стабильность давления в коллекторе охлаждающей воды.
472
Автоматизация содовых печей (рис. 184). Сырой бикарбонат
натрия из вакуум-фильтра / поступает на кольцевой
скребковый транспортер 2, которым равномерно распределяется во все
содовые печи 4.
Ведущим потоком в данной схеме является поток
бикарбоната натрия с вакуум-фильтров. Изменение расхода
бикарбоната влияет на его количество, остающееся на холостой ветви
кольцевого транспортера 2. Регулятор а устанавливает соответ-
Суспенэив
Газ па Нарбоншацию
ТоплиВа
Воздух
Рис. 184. Схема автоматизации содовых печей:
/—вакуум-фильтр; 2—кольцевой скребковый транспортер: г—циклон; 4—содовая печь;
а—регулятор остатка бикарбоната на транспортере; б—регулятор удлинения барабана печи;
в—регулятор соотношения топлива и воздуха; б—регулятор разрежения в топке печи;
г—регулятор давления в печи.
ствующее количество этого остатка путем регулирования
оборотов вала питателей содовых печей.
Мерилом соотношения между количеством топлива и
бикарбоната, поступающих в печь, является тепловое удлинение
барабана содовой печи, заданная величина которого
поддерживается регулятором б. Соотношение между топливом и
воздухом поддерживает регулятор в. Регулятор д стабилизирует
разрежение в топке содовой печи путем воздействия на шибер з
газоходе топочных газсв, регулятор г поддерживает нужное
давление в барабане печи.
Автоматизация станции дистилляции (рис. 185). Фильтровая
жидкость через резервуар 8 направляется в напорный бак / и
далее самотеком последовательно проходит конденсатор 2,
теплообменник 5, смеситель 5, дистиллер 4 и испаритель 6.
473
В смесителе 5 фильтровая жидкость перемешивается с
известковым молоком, поступающим из мешателя 7.
Подаваемый в нижнюю часть дистиллера 4 пар проходит
вместе с горячими газами навстречу потоку фильтровой
жидкости. Часть пара возвращается в сеть из аппарата 6, в котором
происходит испарение жидкости.
Газ на абсорбцию
К регулятору
расхода рассола
жидкость
Рис. 185. Принципиальная схема автоматического регулирования
станции дистилляции:
/—напорный бак; 2—конденсатор; 3—теплообменник; 4—дистиллер;
В—смеситель; 6—испаритель; 7— мешатель; в—резервуар; а, г—регуляторы уровня;
б—регуляторы соотношения; в—регулятор давления.
Ведущим потоком станции дистилляции является поток
фильтровой жидкости, в соответствии с которым регуляторы
уровня а устанавливают нагрузку станции дистилляции.
Регуляторы б поддерживают требуемое соотношение потоков
известкового молока и пара. Благодаря автоматизации процесса
гашения извести и станции карбонизации состав известкового
молока и фильтровой жидкости изменяется в узких пределах.
474
поэтому соотношение их расхода колеблется редко.
Корректировка соотношения потоков фильтровой жидкости и пара
производится по температуре жидкости, выходящей из
теплообменника 3, или по температуре газов, выходящих из
конденсатора 2.
Регулятор в поддерживает соответствующее давление в
паровом коллекторе, что позволяет использовать пар после
компрессоров и восполнить его недостаток паром из турбин.
Регулятор уровня г предотвращает возможность утечки пара через
испаритель 6. Датчик расхода фильтрата на дистилляцию
связан с регулятором подачи рассола на абсорбцию.
Внедрение на содовых заводах частичной автоматизации
позволило улучшить технико-экономические показатели
производства кальцинированной соды.
При внедрении комплексной автоматизации
технико-экономические показатели процесса в целом еще более улучшатся.
5. БИКАРБОНАТ НАТРИЯ
Бикарбонат натрия NaHCO3 представляет собой белый
кристаллический порошок, растворимый в воде. При
взаимодействии с кислотами и при нагревании он разлагается с
выделением двуокиси углерода.
Очищенный бикарбонат натрия находит широкое
применение в пищевой промышленности (хлебопечение, изготовление
кондитерских изделий, безалкогольных шипучих напитков,
искусственных минеральных вод), в резиновой промышленности в
качестве порообразователя при изготовлении микропористых
резин, а также для наполнения жидкостных и пенных
огнетушителей, в медицине, для бытовых нужд, в текстильной
промышленности (для мойки шерсти, шелка) и т. д.
Выпускается очищенный бикарбонат натрия двух сортов
(медицинский и технический), а также сырой бикарбонат.
Бикарбонат натрия должен удовлетворять следующим
требованиям:
Содержание,1;.;,
NaHCO3, не менее . . .
NaXO3, не более . . .
NaCl, » » . . .
Влага » » . . .
Тзхннческнп
. . 98,5
. . 1,2
. . 0,05
. . 1,0
Медицинский
98,5
1,0
0,05
1,0
Сыр oi
72
4,5
0,5
20
В готовом продукте должны полностью отсутствовать также
соли аммония, тяжелые металлы и мышьяк.
Исходным сырьем для производства бикарбоната натрия
является содовый раствор, который готовят двумя способами:
из готовой кальцинированной соды или из сырого бикарбоната
475
натрия, получаемого на фильтрах в качестве промежуточного
продукта при производстве кальцинированной соды (стр. 456).
По первому способу исходную кальцинированную соду
растворяют в воде. При приготовлении раствора по второму
способу бикарбонат натрия должен быть тщательно отделен от
раствора хлористого аммония и хорошо промыт. Сырой
бикарбонат натрия размешивают в воде и в содержащих ЫаНСОз
маточных щелоках, полученных от предыдущей операции. Пульпу
подают в декарбонизатор, где под действием пара происходит
разложение бикарбоната:
2NaHCO3 = Na2CO3 + СО3 + Н2О
Одновременно разлагаются содержащиеся в пульпе
аммонийные соли; аммиак удаляется вместе с двуокисью углерода.
Полученные таким образом содовые растворы после
фильтрования подвергаются карбонизации. При этом образуется
осадок чистого бикарбоната натрия:
Na,CO3 + СО2 + Н2О = 2NaHCO3
Выпавший з осадок бикарбонат натрия отфильтровывают,
центрифугируют, сушат горячим воздухом и измельчают.
Измельчению подвергается только часть продукционного
бикарбоната натрия при получении его специальных сортов.
В некоторых случаях бикарбонат натрия отделяют от
маточного раствора непосредственно на центрифугах и сушат во
вращающейся горизонтальной сушилке барабанного типа,
обогреваемой водяным паром. Фильтрат после выделения из него
бикарбоната натрия используют для растворения новых порций
сырого бикарбоната.
Готовый бикарбонат натрия упаковывают в четырехслойные
мешки (вес не более 50 кг), в деревянные ящики или фанерные
барабаны (вес не более 80 кг), выложенные изнутри
оберточной бумагой, а также в стеклянные банки различной емкости.
Упакованный продукт хранят в сухих помещениях.
б. ПРОИЗВОДСТВО КАУСТИЧЕСКОЙ СОДЫ ХИМИЧЕСКИМИ
СПОСОБАМИ
Кроме эле?строхимических методов получения каустической
соды (одновременно с хлором), имеющих в настоящее время
наибольшее значение, в промышленности для получения
каустической соды применяются также химические способы —
известковый и ферритный. По известковому способу
производится основное количество химического едкого натра.
Химические способы различаются лишь процессами
приготовления разбавленных растворов едкого натра — щелоков.
Дальнейшая переработка щелоков для получения более концен-
476
грлрованных растворов NaOH и твердого едкого натра
одинакова как для химических, так и для электрохимических
методов и описана в главе VIII (стр. 374 ел.).
известковый способ производства
каустической соды
Сущность более старого и распространенного известкового
способа производства каустической соды заключается в
обработке 18—20%-ного раствора кальцинированной соды B10—
220 г/л ЫагСОз) негашеной известью при перемешивании и
нагревании (каустификация). В растворе соды негашеная известь
превращается в гидроокись кальция, которая реагирует с
содой, образуя едкий натр:
Na.2CO3 + Са(ОНJ i—» 2NaOH + CaCOs
Реакция образования едкого натра обратима, и при
определенных условиях процесса (концентрация и температура)
наступает состояние равновесия.
С увеличением содержания Na2CO3 в растворе степень
превращения ее в NaOH понижается вследствие совместной
растворимости СаСОз и Са(ОНJ в щелочном растворе.
Зависимость степени превращения (каустификации)
кальцинированной соды в едкий натр от начального содержания
Na2CO3 в исходном растворе при 100 °С приведена ниже:
Содержание NaaCO3
в растворе, % .... 5,10 9,96 13,77 16,67 19,58 21,48 22,58
Степень каустификации,% 99,2 97,0 93,6 89,2 84,5 80,7 78,1
Практически степень каустификации не превышает 90% в
равновесном состоянии.
Повышение температуры вызывает понижение выхода
едкого натра. Поэтому некоторые заводы проводят процесс
получения едкого натра при 60—70°С. Однако реакцию выгоднее
вести при температуре около 100°С, так как в этих условиях
одновременно увеличивается скорость реакции и скорость
осаждения шлама {СаСОз и другие примеси) вследствие понижения
вязкости раствора. Увеличению скорости каустификации
способствует также интенсивное перемешивание суспензии, так как
при этом возрастает скорость растворения Са(ОН)а.
Обычно щелока, получаемые после отделения осадка
карбоната кальция, содержат 90—135 г/л NaOH, т. е.
концентрация их примерно такая же, как при электрохимическом способе
электролиза с твердым катодом (стр. 374). Дальнейшая
переработка таких щелоков заключается в их концентрировании
'путем упаривания и плавления (обезвоживания).
477
щелока; 8) обезвоживание
Парогазовая смесо
Конденсат
МаНСО,
— ~? У
Технологический процесс производства NaOH по
известковому способу включает следующие основные операции: 1) при-
готоЕление содового раствора разложением технического
бикарбоната натрия паром (декарбонизация); 2) приготовление так
называемого нормального раствора; 3) взаимодействие
«нормального» содового раствора с гидроокисью кальция (первая
каустификация); 4) отделение шлама, содержащего непроре-
агировавшую активную известь, после первой каустификации;
5) обработка полученного шлама жидкостью из декарбонатора
(вторая каустификация); 6) промывка шлама; 7) упаривание
едкого натра.
Приготовление
содового раствора. Для
получения каустической соды
по известковому способу
бикарбонат натрия
обычно разлагают паром в
водном растворе. Этот
процесс называется дв
карбонизацией.
Бикарбонат натрия з
водном растворе уже при
95—98°С быстро разла-
^ гается по реакции
2NaHCO3 = Na3CO3+ CO2+HaO
При нагревании
раствора в течение
нескольких минут до 95—98°С
превращается в соду
около 75% бикарбоната
натрия. Для превращения
остальных 25% NaHCO3
требуется значительно
больше времени.
Разложение ускоряется при
пропускании в раствор
водяного пара, который подогревает жидкость до 105—110°С и
уносит оставшуюся двуокись углерода, понижая таким
образом ее давление над раствором. При декарбонизации
одновременно с двуокисью углерода отгоняется паром и аммиак,
содержащийся в сыром бикарбонате натрия в виде углекислого
аммония.
Декарбонизация проводится по схеме, изображенной на
рис. 186. Со скребкового транспортера /, подающего сырой
бикарбонат на кальцинирование (стр. 463), часть бикарбоната
поступает через питатель в вертикальный смеситель 2. Сюда же
Пар
Рис. 186. Схема декарбонизации:
/—скребковый транспортер; 2—смеситель с
питателем; 3, 5—сборники; 4—декарбонатор.
478
из напорного бака непрерывно подается жидкость (конденсат),
поступающая из цеха выпарки растворов едкого натра.
Образующаяся в смесителе 2 бикарбонатная пульпа перетекает в
сборник 3, снабженный мешалкой, откуда перекачивается в де-
карбонатор 4. В верхней части декарбонатора имеются барбо-
тажные тарелки; нижняя (скрубберная) часть колонны
заполнена насадкой.
В нижнюю часть декарбонатора подается отработанный пар
(давление не ниже 1,5 ат), который движется противотоком к
бикарбонатной суспензии, нагревая ее. При этом происходит
разложение NaHCO3 с образованием раствора, содержащего
около 305 г/л Na2CO3. Раствор соды стекает в нижнюю бочку-
базу декарбонатора и далее через сборник 5 перекачивается на
каустификацию.
Парогазовая смесь, выходящая из верхней части
декарбонатора, направляется в холодильник газа содовых печей
(стр. 463), где присоединяется к потоку двуокиси углерода,
поступающему из печей.
Декарбонатор представляет собой чугунную колонну бар-
ботажного или барботажно-скрубберного типа диаметром 2,8 м
и высотой около 31 м. Верхняя часть колонны состоит из
семи барботажных бочек с одноколпачковыми тарелками. Под
барботажными тарелками расположены восемнадцать скруб-
берных бочек, заполненных коксовой насадкой. Пар подается
в третью бочку снизу; две нижние бочки — переливные.
Бикарбонатная суспензия поступает на разложение в верхнюю часть
колонны, содовый раствор выходит из нижней бочки-базы.
В нижней части декарбонатора поддерживается давление
порядка 1,4—1,5 ат и температура 105—110°С для уменьшения
растворимости СО2 и увеличения скорости разложения NaHCOs
и степени декарбонизации.
Технологический режим процесса декарбонизации:
Общая щелочность суспензии, н. д 105—115
Состав готового раствора, н. д.:
Na.,CO3 90—100
NaHCO3 10—15
NaCI 0,3
Степень декарбонизации, %, не менее 85
Температура газа на выходе из декарбонатора, "С .... 90—92
Расход тепла в процессе декарбонизации около 600 ккал на
1 кг соды в растворе.
Дальнейшие стадии процесса производства каустической
соды известковым способом показаны на рис. 187.
Каустификация. Требуемый для первой каустификации
«нормальный» содовый раствор приготовляют в смесителе /, куда
подаются дозируемые количества декарбонизованного
содового раствора, раствора осадка после выпарки щелока и шелоч-
'ioii воды.
479
«Нормальный» содовый раствор должен иметь следующий
состав (в н. д.):
Общая щелочность, не менее 80
Концентрация NaOH, не более 20
Концентрация NaCl, не более 0,2
Концентрация NaaSO4 6—7
м Д1Рактически установлено, что при упаривании шелока в присутствии
N2SO4 улучшается выпадение соды в осадок. Серосодержащие соединения
обычно вводятся в процесс с топливом и карбонатным сырьем. Если
содержание N2SO, в растворе становится менее 6—7 н. д., сульфат натрии до
бавляют в нормальный содовый раствор.
Оботкенная
известь
Раствор осй&к
выпарки
Содовый раствор
Щелочная
Вова
! ¦—.
Рис. 187. Схема производства каустической соды по известковому способу:
/—смеситель; 2, ff—каустицеры; Л—вытяжная труба; 4—ловушка; 5, 7, //, /5—сборники- Л—пас
пределнтелышй коллектор; 9—отстойник; /0-бак с мешалкой; /2-аппарат для промывки
шлама; /J—вакуум-фильтр; 14—отделитель.
480
Температура «нормального» содового раствора должна быть
не выше 80 °С, при повышении температуры (свыше 80°С};
раствор охлаждают.
Первая стадия каустификации проводится при избытке из
вести (около 10%) во вращающемся аппарате —
горизонтальном каустицере, представляющем собой стальной барабан
диаметром 2,5 м и длиной 15 м. Внутри барабана имеется
стальная рубашка толщиной 10 мм, предохраняющая стенки
аппарата от истирания. Барабан вращается со скоростью 1,6 об/мин.
С торцов барабан закрыт стальными крышками с отверстиями,
через одну из этих крышек загружают известь и «нормальный»-
содовый раствор, с противоположного торца выгружают
полученную суспензию (щелок со шламом) и отбросы, получай
мые при гашении извести. Каустнцер устанавливают с
небольшим уклоном в сторону выгрузки.
Производительность каустицера обеспечивает выработку
150 т каустической соды в сутки. Степень каустификации рас
твора на первой стадии достигает 75%.
По практическим данным на первой стадии каустификации
требуемый объем каустицеров составляет 1,0—1,5 м3 на .1 г
каустической соды в сутки. :
В каустицер 2 (см. рис. 187) поступает обожженная известь
(куски размером 20—40 мм) и дозируемое количество «нор
малького» содового раствора из смесителя 1. Полученная ь
результате каустификации щелочная суспензия отделяется в
ловушке 4 от мелких кусков извести и кокса и поступает з
сборник 5 с мешалкой. На выходе суспензии из каустицера 2
установлена зыгяжная труба 3 для удаления выделяющихся
паров в атмосферу. Суспензия из сборника 5 перекачивается в
систему вертикальных каустицеров 6, состоящую из двух-трех
последовательно работающих аппаратов (на схеме показан
один). В вертикальных каустицерах проводится вторая стадия
каустификации — степень каустификации содового раствора
доводится до 85—90%. Далее жидкость стекает в сборники 7, где
отделяется от примесей (песка), и подается затем в
распределительный коллектор 8.
Постоянный уровень жидкости в каустицерах 6
поддерживается при помощи поплавкового регулятора, связанного с
дроссельным клапаном на линии вывода суспензии из каустицерг
(на рисунке не показано). Перемешивание суспензии в
каустицере производится мешалкой, а в случае ее остановки — путем
циркуляции с помощью центробежного насоса.
Осветление щелоков. Из распределительного
коллектора 8, снабженного шнековой мешалкой, суспензия плунжерным
насосом подается в отстойник 9 для осветления. Из: средней
части отстойника осветленный щелок (концентрация около
135 г/л NaOH) при 80 °С поступает в общий коллектор, откуда
31—2848 481
подается на выпарку. Избыток щелока направляется в
буферный сборник // и снова перекачивается в аппарат 9.
Для получения чистого
продукта с минимальным
содержанием примесей к осветленному
щелоку (в среднюю часть
отстойника) добавляют небольшое
количество сульфата натрия. Шлам
сра?еюии\ Ъгеремв чеРез отверстия в нижней
конической части отстойника 9
попадает в специальный карман,
куда для разжижения шлама
подается промывная жидкость из
втооого яруса аппарата 12. Для
более полного использования
оставшейся в шламе извести
образовавшаяся пульпа из
кармана отстойника поступает в
аппарат 10, куда добавляется содовый
раствор, и далее подается в
многоярусный аппарат 12 на
промывку водами шламового
фильтра и конденсатом с
вакуум-выпарки. Промывка проводится по
принципу противотока: шлам
движется сверху вниз (от
первого яруса до шестого), промывная
вода вводится в нижний
(шестой) ярус и далее
последовательно перекачивается
центробежными насосами на
вышележащие ярусы, кончая вторым.
При промывке шлама
промывные воды обогащаются щелочью.
Из второго яруса промывная
вода поступает в карман
отстойника 9 для разжижения шлама.
Прозрачная жч-кость из
первого яруса при 75 °С
направляется в отделение выпарки для
растворения осадков, выделяющихся
в процессе упаривания щелока.
Рис. 188. Вертикальный каустицер:
/—электродвигатель; 2—редуктор; 3—крышка;
4—бочки; 5—вал; 6—лотсти; 7—траверзы;
в—рамная мешалка; 9—днище; 10—централь-
нос отверстие; /(—чугунная опора.
482
Выходящий из нижнего яруса шлам поступает на
вращающийся вакуум-фильтр 13, где окончательно отмывается от
щелочи конденсатом с вакуум-выпарки.
Фильтровая жидкость и промывные воды из
вакуум-фильтра 13 поступают на промывку шлама в аппарат 12. Промытый
шлам продувается на вакуум-фильтре воздухом, разбавляется
водой в сборнике шлама 15, снабженном мешалкой, к перека
чивается на станции абсорбции и дистилляции (стр. 443, 458)
Отсюда шламовые воды отводятся вместе с дистнллерной жил.
костью.
Основная операция известкового способа — получение
едкого натра из кальцинированной соды — проводится в
горизонтальном и вертикальном аппаратах-каустицерах.
Вертикальный каустицер (рис. 188) представляет собой
чугунный цилиндрический аппарат с мешалкой. Диаметр его 3 м,
высота свыше 13 ж. Объем рабочей части 94,5 ж3. Аппарат уста
новлен на опорах //. Цилиндрическая часть аппарата
смонтирована из четырнадцати бочек 4 высотой по 0,95 м. В
верхней бочке имеются два штуцера: для входа суспензии и для
перелива. Содовый раствор выводится снизу через отверстие 10
в днище 9. По всей высоте аппарата проходит вал 5, к нижней
части которого присоединена рамная мешалка 8; кроме того, к
валу на разной высоте прикреплены три лопасти 6. Вал
вращается со скоростью 4 об/мин. Чугунные траверзы 7
препятствуют изгибу вала и смещению его по осн. На крышке
аппарата установлен привод мешалки с электродвигателем / и
редуктором 2. Снаружи аппарат покрыт слоем теплоизоляции.
Примерные расходные коэффициенты на 1 т каустической
соды (92% NaOH), получаемой известковым способом:
Бикарбонат натрия (95 % Na,CO3), tn . 1,35
Известь (85% CaO), m . . '. 1,0
Пар D — 6 am), млн. ккал 4,0—4,5
Вода техническая, м3 100
Электроэнергия, квт-ч 90—100
К наиболее медленным процессам получения каустической
соды по известковому способу относятся отстаивание и
промывка шламов каустификации, проводимые в громоздкой и
дорогостоящей аппаратуре (отстойники, многоярусные промыватели
и т. д.). Скорость отстаивания шлама возрастает при
добавлении крахмала, солей железа, сульфата натрия. Скорость
промывки шлама можно ускорить при замене громоздких
многоярусных промывателей более компактными вращающимися
вакуум-фильтрами. Использование извести (вместо известкового
молока) для каустификации позволяет повысить концентрацию
содового раствора и, следовательно, снизить расход тепла на
его выпаривание. Количество промывных вод можно уменьшить,
применяя фильтрование шлама первой каустификации.
31* 483
Повторный обжиг во вращающихся печах или печах
кипящего слоя значительного количества карбонатного шлама,
пока являющегося отходом производства, позволит удлинить
сроки эксплуатации карьеров карбонатного сырья и строить
установки для производства химической каустической соды, не
связанные территориально с сырьевой базой.
Применение повторного обжига шлама даст возможность
снизить также себестоимость каустической соды при
одновременном улучшении ее качества (вследствие снижения
содержания примесей). При комплексной автоматизации производства
будут улучшены все технико-экономические показатели
процесса.
ФЕРРИТНЫЙ СПОСОБ ПРОИЗВОДСТВА
КАУСТИЧЕСКОЙ СОДЫ
Основным сырьем при получении каустической соды фер-
ритным способом служит кальцинированная сода.
Применяемая в этом процессе окись железа может быть только условно
отнесена к сырью, так как она не входит в состав готового
продукта и теоретически не должна расходоваться. Для
прокаливания смеси кальцинированной соды и окиси железа в качестве
топлива применяют мазут.
При получении едкого натра ферритным способом сухую
кальцинированную соду смешивают с измельченной окисью
железа. Общая щелочность сухой смеси в пересчете на Кта2СОз
(или «титрсмеси») должна составлять не менее 25%. Остальные
75% приходятся на долю окиси железа и примесей. Влажность
смеси может колебаться от 12 до 15%.
Приготовленную смесь кальцинированной соды и окиси
железа прокаливают в горизонтальных вращающихся печах при
температуре выше 1000 °С. При этом образуются твердые
крупинки феррита натрия ЫагО • Fe2O3, кристаллизующегося в
ромбической системе.
Образование феррита натрия протекает по реакции:
Na2CO3 + Fe2O3 = NaaO • Fe2O3 + CO2 — 38,33 ккал
По нормам технологического режима содержание N2O в
феррите (в пересчете на NaOH) должно составлять 20—23%,
содержание ЫагСО3 не более 3%, степень каустификации
87—90%.
Под действием горячей воды (выщелачивание) феррит
натрия разлагается с образованием раствора едкого натра и окиси
железа по следующему наиболее вероятному уравнению
реакции:
Na2O ¦ Fe2O3 + 2Н2О = 2NaOH + Fe2O3 • Н2О
Окись железа отделяют от раствора и возвращают в
производственный процесс.
Образовавшийся при выщелачивании феррита натрия
раствор, содержащий 360—380 г/л NaOH, поступает на
концентрирование (при получении жидкой каустической соды) или на
последующее обезвоживание (плавление) до образования
твердого продукта.
Технологический процесс производства каустической соды
по ферритному способу включает следующие основные стадии:
1) приготовление шихты (смешение соды и окиси железа);
2) прокаливание шихты и получение феррита натрия; 3)
гашение и выщелачивание феррита натрия; 4) упаривание
раствора NaOH до концентрации не менее 610 г/л и кристаллизация
солей (Na2CO3, Na2SO4); 5) фильтрование упаренного щелока
и отделение солей; 6) окончательное упаривание и плавление
едкого натра; 7) осветление и слив расплавленного едкого
натра.
На рис. 189 изображена принципиальная схема
производства каустической соды по ферритному способу.
Из бункера 5 кальцинированная сода через дозирующее
устройство 6 транспортером 7 направляется в смеситель 10, где
смешивается с промытой окисью железа, подаваемой из
бункера 9 транспортом 7 через другое дозирующее устройство 6.
Кроме кальцинированной соды и окиси железа, в смеситель 10
подается также свежая железная руда (для компенсации
потерь окиси железа в производственном процессе) и содовая
пыль, уносимая топочными газами из ферритных печей и
улавливаемая в электрофильтре /*. Содовая пыль подается
транспортером 2 и элеватором 3 в бункер 4 и через дозирующее
устройство 6 транспортером 7 направляется в смеситель 10.
Для предотвращения пыления в смеситель подают также
небольшое количество воды и осадок солей, выпавших при
упаривании раствора едкого натра. Тщательно перемешанная
шихта транспортером 12 подается в дозирующее устройство 13,
откуда элеватором 14 загружается во вращающуюся феррит-
ную печь 15. Здесь при сжигании жидкого топлива (мазут) в
смеси с воздухом создается высокая температура, необходимая
для разложения соды и образования феррита натрия. Из фер-
ритной печи продукты горения, а также двуокись углерода,
образующаяся в результате разложения кальциннпорянн^й соды,
и пыль (сода и окись железа) поступают в электрофильтр 1.
Осевшая в электрофильтре пыль возвращается
транспортером 2 и элеватором 3 в бункер пыли 4 и снова поступает в
феррптную печь.
* На некоторых установках для улавливания содовой пыли вместо
электрофильтров применяют пыльные камеры.
485
Газ
в атмосферу
В атмосферу
Слабая
жидкость
20
газы
Промытая смесь
Рис. 189. Принципиальная схема производства едкого натра по ферритному способу:
г /—электрофильтр; 2, 7. II, 12—транспортеры; 3, 8, 14, 17— элеваторы; 4, 5. 9—бункеры; 6, IS—дозирующие устройства;
" ю—смеситель; 15— печь; 16—аппарат для гашения; 18— вытяжная трубт; 19—вагонетка; 20—напорный б^к; 21—аппарат
для выщелачивания; 22—отстойник.
L
Из печи 15 распыленный феррит натрия выгружается в
аппарат для гашения 16, куда из напорного бака 20 подается
слабая жидкость (водный раствор NaOH и ИагСОз небольшой
концентрации). Выделяющийся при гашении феррита
водяной пар удаляется в атмосферу. Гашеный феррит элеватором 17
подается в подвижную вагонетку 19, откуда выгружается в
батарею вертикально установленных аппаратов 21 для
выщелачивания (на рисун"ке показан один такой аппарат).
Процесс выщелачивания феррита натрия осуществляется по
принципу противотока. В аппарат, загруженный свежей
массой феррита натрия, поступает концентрированный щелок из
соседнего аппарата, а в аппарат с почти выщелочным
ферритом натрия подают чистую воду. Щелок, выходящий из
последнего аппарата, загруженного свежим гашеным ферритом,
содержит 360—380 г/л NaOH и периодически перекачивается
в отстойник 22 для осветления.
Промытую окись железа перед выгрузкой из аппарата 21
отделяют от слабой (спускной) жидкости и транспортером //
и элеватором 8 подают в бункер 9, откуда она поступает в
смеситель 10 на повторное использование. Спускная жидкость
подвергается сначала декантации, а затем фильтрованию через
слой кварцевого песка (на рисунке не показано). Осветленная
таким образом жидкость перекачивается в напорный бак 20,
откуда распределяется по аппаратам для выщелачивания и
гашения.
Осветленный щелок из отстойника 22 направляют в
выпарную установку (стр. 377 ел.).
Примерные расходные коэффициенты на 1 т каустической
соды (92% NaOH), получаемой по ферритному способу:
Кальцинированная сода (95% NaaCO3). m . . . 1,35—1,4
Окись железа, кг 28—30
Мазут, m 0,56—0,63
Вода техническая, .и3 около 50
Электроэнергия, квтч 95—150
Пар E—6 am), млн. ккал 1,9
Пар A0—12 am), млн. ккал 0,2
Комовая сера и натриевая селитра (для
очистки плава в котлах), кг 0,6—1,0
(поровну)
Интенсификация и рационализация феррнтного способа
производства каустической соды направлены на увеличение
производительности агрегатов, уменьшение расхода сырья и
топлива, механизацию и автоматизацию трудоемких процессов
и улучшение условий труда.
Использование тепла выгружаемого феррита для подогрева
воздуха, подаваемого на сжигание топлива в ферритных печах,
даст возможность снизить расход топлива, а использование
487
тепла газов, отходящих из ферритных печей, в
котлах-утилизаторах позволит получить около 0,95 т пара и на 1 т каустической
соды. Полученный пар может быть использован при
выпаривании слабых щелоков и для других заводских нужд. Замена
мазута в ферритных печах природным газом приведет не только
к снижению себестоимости готовой продукции (так как при*
родный газ примерно в два раза дешевле мазута в пересчете
на условное топливо), но и к упрощению внутризаводского
топливного хозяйства. При этом отпадает необходимость в
резервуарах для хранения мазута, помещениях для его слива,
промывки цистерн и других операций.
На 100 т каустической соды, вырабатываемой в сутки,
внутрицеховым транспортером перевозится более 1950 г сырья и
полупродуктов, поэтому механизация транспортирования,
погрузки и выгрузки сырья и готовой продукции позволит не только
снизить себестоимость соды, но значительно повысить общую
культуру производства и улучшить условия труда.
Техника безопасности в производстве
содовых продуктов
В производстве содовых продуктов вредное действие на
организм человека оказывают, кроме каустической соды,
также кальцинированная сода, известковое молоко, содовый
раствор. При вдыхании содовой пыли раздражаются слизистые
оболочки носа и дыхательных путей. В летнее время (а в
горячих цехах и зимой) содовая пыль может вызывать
заболевания кожных покровов (экзема). Пыль негашеной извести
также раздражает слизистые оболочки и вызывает кашель.
Очень опасны ожог;: глаз известью. Обслуживающий персонал
в производстве содовых продуктов обязан работать в
спецодежде и пользоваться предохранительными приспособлениями
(очки, шлемы, респираторы, перчатки и т. д.).
По степени пожарной опасности отделения ферритных
печей и выщелачивания относятся к категории Г, все остальные
отделения производства каустической соды по известковому и
ферритному способу, склады готовой продукции и другие
вспомогательные помещения отнесены к категории Д. В
соответствии с действующими санитарными нормами ширина
озелененной и благоустроенной санитарно-защитной зоны на территории
производства каустической соды химическими способами
должна быть не менее 300 м.
В холодное время года температура во всех
производственных помещениях должна поддерживаться не ниже 16°С.
Вентиляция должна обеспечивать трехкратный обмен воздуха в
час, в помещении топок часовой обмен воздуха должен быть
не менее пятикратного.
488
На предприятиях, вырабатывающих содовые продукты,
кроме правил техники безопасности и охраны труда,
предусмотренных в законодательном порядке, при проектировании,
строительстве и эксплуатации должны соблюдаться правила и
нормы, учитывающие специфику производства этих продуктов и
отражаемые в рабочих инструкциях и производственных регла-1
ментах.
Строгое соблюдение технологического режима,
своевременный планово-предупредительный ремонт оборудования,
автоматизация и механизация производственных процессов,
точное выполнение мероприятий по технике безопасности и
охране труда способствуют повышению производительности труда
и улучшению его санитарно-гигиенических условий.
ЛИТЕРАТУРА
С. И. В о л ь ф к о в и ч, А. П.. Егоров, Д. А. Э п ш т е й н, Общая
химическая технология, т. I, Госхимиздат, 1952.
В. Ф. Чернов, Производство кальцинированной соды, Госхимиздат, 1956.
М. Б 3 е л и к и н и др., Производство кальцинированной соды,
Госхимиздат, 1959.
Г. И. М и кули н, И. К. Поляков, Дестилляция в производстве соды,
Госхимиздат, 1956.
М. И. Рабинович, Современная содовая промышленность и пути ее
развития в СССР, Журн. ВХО им. Д. И. Менделеева, № 1, 35 A962).
В. И. Максюта, Комплексная автоматизация производства
кальцинированной соды, Журн. ВХО им. Д. И. Менделеева, № 5, 517 A961).
М. Б. 3 е л и к и н, Производство каустической соды .химическими
способами, Госхимиздат, 1961.
Глава XI
ФОСФОР И ФОСФОРНАЯ КИСЛОТА
/. ФОСФОР. ОБЩИЕ СВЕДЕНИЯ
Свойства и применение фосфора. Чистый фосфор при
обычной температуре представляет собой твердое бесцветное
вещество, напоминающее воск. Под влиянием света фосфор
приобретает желтую окраску вследствие образования
незначительных количеств красного фосфора. Такой фосфор называют
желтым фосфором.
Желтый фосфор плавится при 44,2 °С. На воздухе он
быстро окисляется и загорается, образуя густой белый дым
фосфорного ангидрида. Поэтому все операции с фосфором
проводят под водой. При нагревании без доступа воздуха до 270—
300 °С желтый фосфор превращается в более устойчивую
модификацию— красный фосфор, который при обыкновенной
температуре на воздухе ке загорается.
При взаимодействии фосфора или фосфорного ангидрида с
хлором образуются хлорсодержащие соединения, из которых
наиболее распространены треххлористый фосфор РС1з, пяти-
хлористый фосфор РС!5 и хлорокись фосфора РОС13. Из
фосфора получают его сернистые соединения — пятисернистый
фосфор P2S5 и полуторасернистый фосфор P4S3. Хлорсодержащие
и сернистые соединения фосфора используют для производства
различных фосфорорганических соединений, применяемых в
качестве пластификаторов, инсектицидов, флотореагентов,
исходных веществ для синтезов (например, для синтеза полифосфо-
нитрилхлорида) и др.
Из желтого фосфора готовят красный фосфор, фосфорный
ангидрид, фосфорную кислоту. Он может применяться также
в качестве дымообразующего вещества.
Красный фосфор применяют главным образом в
производстве спичек. Смесь фосфора с клеем и толченым стеклом
наносят на боковые поверхности спичечной коробки. Масса на
головках спичек не содержит фосфора и состоит из смеси
сернистой сурьмы и хлората калия (бертолетовой соли) или
перекиси свинца. При трении головки спички о шероховатую
поверхность коробки происходит местное повышение температуры,
490
вызывающее реакцию между фосфором и нанесенным на
головку составом, богатым кислородом, поэтому спичка вспыхивает.
Красный фосфор применяют также в производстве
фосфористых бронз, фосфида цинка и в пиротехнике.
2. ЖЕЛТЫЙ ФОСФОР
В основе производства фосфора лежит процесс
восстановления фосфата кальция углеродом в присутствии окиси
кремния при 1400—1600 °С по реакции
Са3(РО4J + 5С + 2SiO2 = Р2 + 5СО + 3CaO-2SiO2
Окись кремния понижает температуру реакции, а также
ускоряет реакцию.
Для осуществления этого процесса требуется затрата
большого количества тепла. На практике процесс проводят в
электрических печах (электротермический процесс), в качестве
восстановителя применяют кокс или антрацит.
НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ЭЛЕКТРОТЕРМИИ
Значение электротермических процессов в современной
технике чрезвычайно велико, так как нагревание электрическим
током имеет в ряде случаев значительные преимущества перед
другими способами нагрева. Электрический нагрев дает
возможность получать очень высокие температуры,
сосредоточивать большое количество тепла в незначительном объеме, что
ускоряет нагревание и, следовательно, химический процесс.
Благодаря этому упрощается конструкция аппаратов,
облегчается регулирование процесса, возникает возможность
получения более чистого продукта.
Развитие электротермических производств зависит в
значительной степени от наличия ресурсов электроэнергии,
потребляемой при электротермических процессах в очень больших
количествах.
В химической промышленности электротермические
процессы применяются в производстве карбида и цианамида кальция
F01 ел.), фосфора, сероуглерода, карборунда (карбид
кремния), при изготовлении изделий из плавленых горных пород
(кварц, диабаз, базальт, стр. 665 ел.) и др. Широкое
применение, как известно, электротермические процессы получили и в
металлургии.
Готовые продукты электротермических производств
получаются в расплавленном виде (например, карбид кальция) или
в спекшемся состоянии (цианамид кальция), либо возгоняются
и выводятся из печей в газообразном состоянии (фосфор,
сероуглерод).
491
Превращение электрической энергии в тепловую. Между
количеством выделенного электрическим током тепла Q (в
кал), током / (в с), сопротивлением проводника R (в ом),
временем / прохождения тока по проводнику (в сек) имеется
следующая зависимость (закон Джоуля):
Q = 0,239PRt
Применяя закон Ома
/ Е
1
(Е— напряжение в з), можно представить эту зависимость в
более удобном виде, так как практически легче измерять
напряжение, чем сопротивление проводника.
Q = 0.239ЕЛ
Произведение El выражает работу электрического тока за
I сек, т. е. электрическую мощность.
Однако эта формула справедлива лишь для постоянного
тока. Промышленные же электрические печи работают на
переменном токе — однофазном или, чаще, трехфазном.
Количество тепла, выделяемого переменным током, не соответствует
величине El. Для переменного тока различают кажущуюся
мощность El, которая выражается в з- а, и действительную
мощность W, которая выражается в зт. Отношение действительной
мощности к кажущейся мощности называется коэффициентом
мощности и выражается математически через
W
-w =COS(p
Поскольку не вся электрическая мощность превращается в
тепло, cos ф всегда меньше единицы.
Действительная мощность однофазного переменного тока
(в зт) определяется выражением El cos ср. Чем ближе cos cp к
единице, тем лучше работает установка. Практически при
работе электрических печей величина cos ср колеблется от 0,8 до
0,98. Мощность трехфазного тока выражается более сложной
формулой:
1^3EI cos ф = 1,73EI cos ф
ЭЛЕКТРИЧЕСКИЕ ПЕЧИ
В химической промышленности применяют электрические
печи следующих типов:
1) печи сопротивления — прямого и косвенного нагрева;
2) дуговые печи — с открытой и закрытой дугой;
3) комбинированные печи.
492
В печах сопротивления косвенного нагрева (рис. 190) в
качестве сопротивления применяют стержни 2 из угля или
графита, спирали или ленты из специальных сплавов, обладающих
высоким удельным электрическим сопротивлением (нихром,
фехраль и др.)- При пропускании тока стержень или спираль
накаливается и в печи развивается высокая температура.
Рис 191. Печь сопротивления прямого
нагрева для обжига электродов:
/, 4—угольные стержни; 2—кокс;
3—обжигаемые электроды.
Рис. 190. Печь сопротивления
косвенного нагрева.
/—стальной кожух, футерованный огнеупор- . .
ным кирпичом; 2—угольный или графитовый мые электроды,
стержень; 3—материал, нагреваемый в печи.
В печах сопротивления прямого нагрева (рис. 191)
сопротивлением служит непосредственно нагреваемый материал.
Примером использования таких печей может служить
производство электродов. Обжигаемые электроды 3 помещают в печь
рядами, между которыми засыпают кокс 2. Электрический ток,
подводимый к стержням / и 4, проходит через слой кокса и
через обжигаемые электроды.
Рис- 192. Дуговая печь с открытой
дугой.
Рис- 193. Дуговая печь с
закрытой дугой.
В дуговых печах нагревание производится пламенем
электрической дуги. Перемещая один из электродов, можно
получить дугу различной длины. При одинаковом расстоянии между
электродами длина дуги увеличивается с повышением
напряжения тока, питающего печь.
4 93
В печах с открытой дугой (рис. 192) пламя дуги
находится над реагирующей массой и тепло передается нагреваемым
материалам путем излучения.
В печах с закрытой дугой (рис. 193) дуга образуется
между электродом и реакционной массой.
В комбинированных печах (рис. 194) нагревание
производится дугой и сопротивлением. В этом случае сопротивлением
служит расплавленный материал.
Типы электрических печей. Современные электрические
(комбинированные) печи разделяются на однофазные и
трехфазные. В однофазных печах один электрод
соединен с одной фазой источника
трехфазного тока; вторая фаза соединена с
нижним (подовым) электродом.
Трехфазные печи имеют три
электрода, к каждому из которых подводится
ток от одной из фаз.
Электрическая печь состоит обычно
из стального кожуха толщиной 8—15 мм
с огнеупорной футеровкой. Под и
боковые стенки печи на уровне реакционной
зоны в ряде случаев (например, при
производстве фосфора, некоторых ферро-
Рис. 194. Комбинирован- сплавов) выполняют из угольных бло-
ная электрическая печь ков» уложенных на графитовой замазке,
или из утрамбованной электродной
массы. Выпуск расплава производится через
специальные отверстия в стенках печи, расположенные
напротив электродов. Применяются электрические печи открытого,
полузакрытого и закрытого типов.
Величина реакционного пространства в электрической печи
определяется размерами электродов и сферой их действия, т. е.
расстоянием, на которое электродами передается большое
количество тепла. Расстояние от электрода до стенки печи
должно быть таким, чтобы стенки находились по возможности вне
воздействия высокой температуры, развивающейся в печи.
В однофазных печах электрод подвешивают на середине
печи. В трехфазных печах электроды располагают по
треугольнику или в ряд. Расположение электродов определяет форму
электрической печи: однофазные печи и трехфазные с
расположением электродов по треугольнику делают цилиндрическими,
трехфазные печи с расположением электродов в ряд —
прямоугольными или эллиптического сечения.
Электроды. Существенным элементом устройства дуговых и
комбинированных электрических печей являются электроды.
Для изготовления электродов применяют почти исключительно
углерод, как очень стойкий при высоких температурах мате-
494
риал, обладающий к тому же высокой электропроводностью,
которая возрастает при нагревании. Изготовляются угольные
электроды (из так называемого аморфного углерода) и
графитовые электроды (из кристаллического углерода — графита).
В химической промышленности большей частью применяют
угольные электроды, изготовляемые из смеси углей (антрацит,
спекающийся каменный уголь) или из искусственных
углеродных материалов (кокс, ретортный уголь, остатки электродов),
в качестве связующего служит каменноугольная смола или
пек. Смесь прессуют и обжигают без доступа воздуха.
Допустимая электрическая нагрузка на электроды, т. е.
плотность тока в а/см2, зависит от качества электрода,
температурного режима печи и других факторов. На практике
работают с плотностью от 2 до 12 а/см2.
Применяются угольные электроды сменные и непрерывного
действия.
Сменные электроды обычно имеют круглое сечение. При
большой величине тока, когда нужно большое сечение
электрода, применяют так называемые пакетные электроды —
отдельные электроды, собранные в четырехугольный пакет и
склеенные. К пакету прикрепляется электрододержатель, к
которому подводится электрический ток от трансформатора.
Недостатком пакетных электродов является то, что при их замене на
новые приходится выключать на время электрическую печь.
Кроме того, в электрододержателе остаются неиспользованные
огарки B0—30% от веса пакета).
Электроды непрерывного действия можно разделить на три
группы:
1) предварительно обожженные;
2) самоспекающиеся;
3) смешанного типа.
Электроды непрерывного действия лишены недостатков
сменных электродов.
Предварительно обожженные электроды, изготовляемые на
специальных заводах, представляют собой брусья
цилиндрической формы длиной 1—2 м и диаметром до 1 м и более. Они
укреплены в электрододержателе с помощью контактных плит,
охватывающих электроды сбоку. Наращивание электродов
производится с помощью ниппеля — угольного бруска с винтовой
нарезкой. В торцах электродов имеются углубления с винтовой
нарезкой, соответствующей нарезке ниппеля. При наращивании
электрода в печи в углубление на верхнем торце до половины
ввертывают ниппель и на свободную половину ниппеля
навертывают новый электрод. Для обеспечения хорошего контакта в
месте соединения электродов поверхность ниппеля и
углублений в торцах электродов покрывают слоем электродной замаз-
495
чи.По мере сгорания электрода его опускают в печь, ослаоляя
контактные плиты электрододержателя.
Для операций свинчивания электродов и подвозки электрод*
ных блоков к печам применяют краны с устройствами для за-
хвата электродов. Недостатком электродов этого типа являются
сравнительно частые поломки в местах соединений и
обрушивание нижнего электрода в печь.
Непрерывно действующий самоспекающийся электрод
представляет собой стальной кожух, набиваемый электродной
массой. Масса состоит из крупнозернистого прокаленного
антрацита (частицы 15—20 мм) и тонко измельченных кокса и
прокаленного антрацита; связующим является смесь
каменноугольной смолы и пека. Электродную массу изготовляют обычно на
том же заводе, где применяют электроды. Электрод укрепляют
при помощи контактных плит в электрододержателе и по мере
сгорания опускают вниз, ослабляя контактные плиты
электрододержателя. Сверху загружают новые порции электродной
массы, при опускании электрода постепенно происходят
нагревание и обжиг электродной массы.
Недостатком самоспекающихся электродов является
наличие в них около 5% железа (из кожуха), что для некоторых
производств нежелательно.
Электроды смешанного типа состоят из предварительно
обожженных наружных блоков и необожженной электродной
массы, составляющей внутреннюю часть электрода. Сечение
таких электродов достигает значительных размеров (диаметр
До 6 м). Угольные блоки прикрепляют к стальным балкам,
составляющим каркас электрода, электрический ток подводится
к контактным плитам, охватывающим электрод. Вместе с
электродом расходуется и каркас. При применении электродов
смешанного типа расходуется 10—12% железа от веса электродов.
Подводка электрического тока. От электростанций заводы
обычно получают электрический ток высокого напряжения.
С центральной трансформаторной подстанции ток поступает в
обмотку трансформатора, установленного непосредственно у
электрических печей, и трансформируется с высокого
напряжения на низкое. На электрических печах большей частью
применяют напряжение от 80 до 300 в, на более мощных печах
до 500 в. При высоком напряжении повышается коэффициент
мощности электрического агрегата. Трансформатор
устанавливают возможно ближе к печи, чтобы уменьшить длину медных
шин, по которым ток низкого напряжения подводится к печи.
и тем самым снизить потери электроэнергии и расход меди.
Шины гибкими медными кабелями соединены с электрододер-
жателями, от которых ток идет по медным трубам,
охлаждаемым водой, к контактным пластинам, прилегающим к
электродам.
4 36
Напряжение и величину тока контролируют с помощью
измерительных приборов, установленных в специальном помеш.е«
нии возле печей. Работу печи регулируют путем изменения
напряжения на электродах (переключая ступени
трансформатора), а также путем изменения тока (поднимая или опуская
электроды). Электроды подвешивают в печах на стальных
тросах, подъем и опускание электродов производят с помощью
лебедок. Работу лебедки можно регулировать вручную или с при-
менением автоматических устройств.
ВОЗГОНКА ФОСФОРА В ЭЛЕКТРИЧЕСКИХ ПЕЧАХ
Схема электротермического получения фосфора
изображена на рис. 195. Исходные материалы — природный фосфат
(стр. 520) и восстановитель — раздельно дробят, сушат и отсе-
Вода
Шихто
Шлам
Шлан и
meppoipoccpop
Желтый
фоарор
Рис. 195. Схема электротермического получения желтого фосфора:
/—трехфазная электрическая печь для возгонки фосфора; 2— электрод; 3—конденсаторы
фосфора; 4—приемник фосфора-сырца; 5—баки для очистки фосфора-сырца.
вают от пыли. Затем их смешивают и загружают в
электрическую печь / через воронки в крышке печи. Мелкую руду, отходы
от дробления и грохочения руды агломерируют. Можно также
применять мелкую руду в виде брикетов, спрессованных вместе
с восстановителем.
Если и природном фосфате содержится недостаточное
количество кремнезема, в шихту добавляют дробленый кварцит
или крупный песок. Оптимальное соотношение SiO2:CaO в
шихте составляет 0,85—0,9: 1. Восстановление трикальцийфосфата,
содержащегося в природных фосфатах, происходит внизу печи
у концов опушенных в нее электродов 2, где развивается
высокая температура. Полученный фосфор возгоняется и в виде
паров непрерывно удаляется из печи вместе с окисью углерода.
32—2848 497
При возгонке фосфора в нижней части электрической печи
образуется большое количество шлака, состоящего из силиката
кальция и некоторых окислов, а также небольшого количества
неразложившейся РгО5 @,5—1,5%). Вследствие
восстановления окислов железа, содержащихся в шихте, и
взаимодействия части фосфора с железом, в печи образуется
некоторое количество феррофосфора, содержащего ~24% Р. Расплав
феррофосфора, более тяжелый, чем шлак, собирается на дне
электрической печи. Расплавленные шлак и феррофосфор
выпускают из печи через отверстие в стенке у пода. По мере
выхода шлака из печи шихта, загруженная в печь, постепенно
опускается и в верхнюю часть печи загружают новые порции
шихты. Таким образом процесс идет непрерывно.
Степень восстановления фосфора, который отводится с
газами и переходит в феррофосфор, достигает 96—97%, выход
фосфора, уходящего с газами, составляет 88—92%.
Выходящие из печи газы, содержащие фосфор, поступают в
конденсаторы 3— полые стальные цилиндры, орошаемые
снаружи и изнутри водой. При охлаждении фосфор
конденсируется и оседает под водой в приемниках 4. Степень конденсации
фосфора достигает 96—97%. Из приемников фосфор-сырец
поступает в обогреваемые баки 5, где отстаивается. Более
тяжелый фосфор оседает на дно, примеси вместе с водой образуют
взвесь. Фосфор, оставшийся в шламе, извлекают раствором
бихромата калия в серной кислоте. Очищенный фосфор
заливают в тару или используют непосредственно, например для
получения термической фосфорной кислоты (стр. 509).
Для получения чистого фосфора газы, выходящие из печи,
до поступления в конденсаторы очищают от пыли в
электрофильтрах, установленных в обогреваемых камерах
Возгонку фосфора обычно проводят в трехфазных
электрических печах комбинированного типа.
В отличие от печей некоторых других производств печь
должна быть абсолютно герметичной, чтобы фосфор не
окислялся воздухом и пары фосфора не проникали в помещение.
Кроме того, чтобы предотвратить проникание
фосфорсодержащих газов в производственные помещения, в бункеры для
шихты вдувают инертный газ.
Трехфазная печь (рис. 196) состоит из стального кожуха 1,
футерованного в нижней части угольными блоками 3, а в
верхней часгн огнеупорным шамотиьгм кирпичом 5; печь имеет
съемную крышку 7. Шихту из бункера 15 по течкам
равномерно распределяют вокруг всех трех электродов 9.
Газы, содержащие фосфор, отводятся через отверстие в
крышке печи или в верхней части стенки печи по газоходам 13.
Шлак выпускают через летку 2. При выпуске шлака
феррофосфор оседает, его собирают в специальной ловушке.
498
Рис. 196. Трехфазная электрическая печь для возгонки фосфора (продольный и П(.ш.речный разрезы):
/—кожух печи; 2- летка для выпуска шлака; ,?—футеровка (угольные блоки); 4—трансформатор; 5—футеровка (огнеупорный кирпич);
5—шины от трансформатора; 7—съемная крышка печи; S— сальниковые кольца; 9— электроды; 10—электрододержатели; //—гибкие мед-
иые кабели; /2—щит управления и контроля; 13—газоходы; 14—лебедка для подъема электродов; /5—бункеры с течками для шихты.
Электроды 9 укреплены в электрододержателях 10. Между
электродом и электрододержателем должен быть хороший
контакт.
Ток подводится к электрододержателям от понижающего
трансформатора 4 по шинам 6 и гибким кабелям //.
Трансформатор имеет обычно несколько ступеней напряжения.
Работу печи регулируют, изменяя напряжение тока на
электродах переключением ступеней трансформатора, а также
изменяя величину тока подъемом или опусканием электродов
посредством лебедок 14.
Мощность современных трехфазных электрических печей
для возгонки фосфора составляет от нескольких тысяч
киловатт до 50 тыс. кет.
В зависимости от мощности печи и состава исходного сырья
расход электроэнергии на 1 т желтого фосфора составляет
12—18 тыс. кет- ч.
Давление газов в электрических печах можно регулировать
автоматически.
Очищенный готовый продукт содержит не менее 99,7—99,9%
фосфора и не более 0,1—0,3% веществ, нерастворимых в
сероуглероде. Расплавленный желтый фосфор заливают в
стальные бочки емкостью не более 250 л или в специальные
железнодорожные цистерны. Желтый фосфор в виде болванок или
палочек упаковывают в банки из оцинкованной стали,
вмещающие не более 12,5 кг фосфора. Над поверхностью фосфора
должен быть слон незамерзающего раствора хлорида кальция
или натрия (высота слоя не менее 5 см в бочках или банках
и 30 см в цистернах).
Над незамерзающим раствором должно быть свободное
пространство (не менее 5% объема бочки или банки и 10%
объема цистерны).
Использование отходов
электротермической возгонки фосфора
Газы фосфорных печей после конденсации паров фосфора содержат:
% г/мз
H,S 3—6
РН3 .... 1 — 1,5
Р ... . 1—2
COS 0,8—1,0
Эти газы непосредственно используются в качестве топлива для сушки
шихты или выпускаемых заводом удобрений. После специальной очистки
газы могут быть применены в других производствах.
Шлаки фосфорных печей можно использовать в производстве цемента
(стр. 643), в качестве легких заполнителей в строительных материалах
(стр. 644), при строительстве дорог и т. д.
Феррофосфор применяют в металлургической промышленности при
производстве некоторых сортов стали с повышенным содержанием фосфора.
500
СО. . .
со2 . . .
н2 . . . .
N.....
. . 80—90
до 1
до 8
до 11
3. КРАСНЫЙ ФОС ФОР
Технический красный фосфор представляет собой порошок
темно-красного цвета.
Красный фосфор является видоизменением желтого (белого)
фосфора. Превращение желтого фосфора в красный
происходит под влиянием тепла, света и некоторых катализаторов( иод,
бром, селен и др.). На практике это превращение (передел)
желтого фосфора в красный осуществляют путем
продолжительного нагревания фосфора без доступа воздуха. Температура
щ
p^. 5 7^
i!
4
Рис. 197. Передельный барабан для получения красного фосфора:
1—корпус барабана: 2—люк для загрузки и Еыгрузкн фосфора; 3—пробка;
4—днище барабана; 5—передний вал; 6—задний вал.
в начале процесса передела при нормальном давлении не
должна превышать 280 °С, т. е. температуры превращения желтого
фосфора в красный. Обычно температуру поддерживают на
уровне 275—280 °С. По мере образования красного фосфора
нагреваемая масса приобретает тестообразную консистенцию.
После загустевания массы постепенно повышают температур}'
до 330—340 °С. Продолжительность процесса передела около
100 ч. Согласно правилам техники безопасности аппараты для
передела фосфора применяются небольшой вместимости
(обычно до 0,5 т).
Для передела желтого фосфора в красный практическое
применение получил вращающийся передельный барабан
(рис. 197). Корпус / барабана сварной из котельной стали;
переднее днише 4 барабана съемное, у края днища имеется
люк 2 для заливки расплавленного желтого фосфора и выгрузки
порошкообразного красного фосфора. Для лучшего
перемешивания фосфора в барабан помещают стальные штанги круглого
сечения. Чтобы пары фосфора не проникали в атмосферу,
передний вал 5 барабана снабжают сальниковым устройством.
501
Для нагревания фосфора передельные барабаны помещают
в так называемую печь передела. Основным элементом такой
печи является муфель — гнездо, предназначенное для барабана.
Имеются печи, в которых каждый муфель нагревается
самостоятельно, и печи, в которых муфели нагреваются
последовательно или параллельно от общего источника тепла. Муфели
обогреваются топочными газами или электрическим током. По
окончании процесса передела вращающиеся барабаны
охлаждают путем частичного погружения в периодически сменяемую
воду.
При расфасовке красный фосфор просевают через сито с
ячейками размером 1—1,5 мм и упаковывают по 5 и 10 кг в
банки из кровельной стали с двойной крышкой. Для
получения 1 т товарного красного фосфора расходуется 1,1 т желтого
фосфора.
Готовый продукт содержит (в %):
Красный фосфор, не менее • 96,5—98,7
Примеси, не более:
желтый фосфор 0,005—0,03
фосфорная кислота Н3РО4 0,6—1,5
(Н3РО3+Н3РО2) 0,2
нерастворимый в азотной кислоте
остаток 0,4—1,5
сера 0,002—0,03
Температура воспламенения красного фосфора должна быть
не ниже 250—270 "С.
Разрабатываются также способы непрерывного передела
желтого фосфора в красный с применением катализаторов
этого процесса.
4. ФОСФОРНАЯ КИСЛОТА
Фосфорная кислота Н3РО4 является важнейшим
полупродуктом для производства ряда концентрированных удобрений
(двойной суперфосфат, аммофос, преципитат, нитрофоска, ни-
трофос и др.), а также технических фосфатов аммония,
натрия, кальция, марганца, алюминия. Фосфорную кислоту
применяют также в синтезе ряда органических продуктов и в
производстве активированного угля.
Очищенную фосфорную кислоту, так называемую пищевую
фосфорную кислоту, применяют в производстве газированных
вод, фармацевтических препаратов, для получения чистых
солей, используемых при изготовлении печенья, питательных
сухарей, кормового преципитата и др.
Техническая фосфорная кислота выпускается в виде
растворов различной концентрации и содержит примеси.
.502
По способам получения различают экстракционную и
термическую фосфорную кислоту. Экстракционную фосфорную
кислоту получают при разложении природных фосфатов серной
кислотой, термическую фосфорную кислоту — восстановлением
трикальцийфосфата, содержащегося в природных фосфатах, до
элементарного фосфора с последующим окислением его в
фосфорную кислоту.
Термическая фосфорная кислота отличается от
экстракционной фосфорной кислоты более высокой концентрацией н
чистотой. Для получения экстракционной фосфорной кислоты
непригодно фосфатное сырье с повышенным содержанием
полуторных окислов, растворимых в кислотах, особенно окиси
железа, а также соединений магния. Для производства
фосфора и, следовательно, термической фосфорной кислоты можно
применять фосфатное сырье с такими примесями.
ЭКСТРАКЦИОННАЯ ФОСФОРНАЯ КИСЛОТА
Процесс производства экстракционной фосфорной кислоты
заключается в обработке измельченных природных фосфатот:
серной кислотой и в отделении образующейся фосфорной
кислоты от осадка (гипса) на фильтрах.
Действие серной кислоты на важнейший фосфатный
минерал— фторапатит выражается в общем виде уравнением:
Ca5(PO4KF + 5H,SO4 + 10H2O = ЗН3РО4 + 5(CaSO4-2H3O) + HF
В фосфоритах обычно содержится некоторое количество
карбонатных минералов (кальцит, доломит и др.), которые также
реагируют с серной кислотой:
СаСО3 + H2SO4 + Н2О = CaSO4-2H2O + СО2
CaCO3-MgCO3 + 2H2SO4 = CaSO4-2H,2O + MgSO4 + 2CO2
Растворимость гипса CaSO4-2H2O в фосфорной кислоте
сравнительно невелика, поэтому кальций почти полностью
переходит в осадок. Магний же переходит в раствор, так как
сульфат магния растворим в фосфорной кислоте.
При разложении серной кислотой природных фосфатов
содержащиеся в них полуторные окислы железа и алюминия
частично образуют растворимые сульфаты; в раствор переходят
также сульфаты натрия и калия и некоторое количество
растворимой кремнекислоты.
Выделяющийся при разложении природных фосфатов
фтористый водород взаимодействует с кремнеземом, содержащимся
в природных фосфатах:
6HF + SiOa = H3SiF6 + 2Н2О
503
При этом образуется кремнефтористоводородная кислота,
которая переходит в раствор вместе с фосфорной кислотой.
Часть фтора, выделяющегося в газообразном состоянии в
виде S1F4, удаляется вместе с двуокисью углерода.
Раствор фосфорной кислоты, содержащий растворенные
примеси, отделяют от осадка. Осадок (фосфогипс) состоит из гипса
и неразложившихся компонентов природного фосфата.
Для образования легко отфильтровывающегося или хорошо
отстаивающегося осадка важно, чтобы гипс получался по
возможности в виде крупных кристаллов. Кроме того,
крупнозернистые осадки удерживают лишь незначительное количество
жидкости, и их удается промыть небольшим количеством воды.
Это дает возможность получать более концентрированную
фосфорную кислоту, поскольку промывные растворы возвращают
в процесс в виде так называемого раствора разбавления,
циркулирующего в системе.
Раствор разбавления вводят в реактор одновременно с
фосфатной мукой и серной кислотой, поддерживая при этом
определенное отношение между жидкой и твердой фазами
(Ж : Т) в реакционной пульпе. Обычно экстракцию фосфорной
кислоты ведут при отношении Ж:Т в пульпе B—3) : 1, что
обеспечивает хорошее смешение реагентов, облегчает
перекачивание пульпы и способствует образованию хорошо
отфильтровываемых крупных кристаллов гипса.
Поддержание температуры в пределах 70—80 °С
благоприятно влияет на скорость роста кристаллов гипса; при этой тем*
пературе кристаллы гипса вырастают до наибольших размеров.
Кристаллизация гипса производится из пересыщенного
раствора. Степень пересыщения выражается отношением
С-Со
где Со—концентрация насыщенного раствора;
С — концентрация пересыщенного раствора.
Это отношение следует поддерживать в пределах 0,2—0,5.
Для получения крупных однородных кристаллов гипса важно
также, чтобы разложение фосфатов проводилось при
небольшом избытке в растворе ионов SO42~ При избытке ионов Са2+
образуются тонкие, удлиненные, неоднородные кристаллы.
Весовое количество абсолютно сухого фосфогипса,
образующегося из весовой единицы природного фосфата,
называется гипсовым числом. Практически гипсовое число находится в
пределах 1,3—1,6.
Для экстракции фосфорной кислоты чаще всего применяют
93%-ную серную кислоту. Применение более разбавленной
кислоты нецелесообразно, так как ведет к снижению
концентрации получаемой фосфорной кислоты.
50 4
Схема производства экстракционной фосфорной кислоты
изображена на рис. 198. Процесс экстракции проводится
непрерывно в системе реакционных аппаратов ¦— экстракторов 10.
Они представляют собой стальные чаны, футерованные
изнутри кислотоупорными плитками и кирпичом, уложенным на по-
лиизобутилене. Аппараты снабжены двухлопастными
стальными гуммированными мешалками и соединены между собой
перетоками.
Росфзгилс
Рис. 198. Схема производства экстракционной фосфорной кислоты: ¦
/—элеваторы; 2— бункер; if—питатель; 4—шнек; б, 7, 9—дозаторы; 6—напорные баки;
в—сборник фильтрата; 10—экстракторы; //—воздуходувка; 12—сборник продукционной
кислоты; 13—вакуум-приемники; 14—ленточный вакуум-фильтр; 15— бак для взмучивания
шлама.
Фосфатная мука транспортирующей системой /—4 через
дозатор 5 непрерывно подается в первый экстрактор 10, куда
непрерывно вводится заданное количество серной кислоты.
Часть серной кислоты иногда подается и во второй
экстрактор. В первый экстрактор непрерывно поступает также
раствор разбавления, представляющий собой смесь второго
фильтрата, образующегося после промывки фосфогипса во второй
зоне вакуум-фильтра и некоторого количества первого
фильтрата, отводимого из перьой зоны фильтра (так называемая
оборотная фосфорная кислота). Количество оборотной фосфорной
кислоты, подаваемой в экстракционную систему, соответствует
соотношению Ж •" Т в экстракционной пульпе в пределах
B,5—3) : 1.
Для предотвращения перегрева пульпы и образования
осадка полуводного гидрата сульфата кальция CaSO4-0,5H2O в
один или несколько экстракторов воздуходувка // подает воз-
505
дух. барботирующий через пульпу и охлаждающий ее. Можно
охлаждать также пульпу путем подачи ее в вакуум-испаритель
(на схеме не показано), где вследствие испарения части воды
температура пульпы понижается. Чтобы образовались крупные
кристаллы гипса, продолжительность пребывания реагентов в
первом экстракторе должна быть не менее 1—2 ч.
Соответственно этому устанавливают реакционный объем данного аппарата.
Общая продолжительность реакции, т. е. время пребывания
пульпы в экстракторах, составляет 5—6 ч. Из последнего
(четвертого) экстрактора пульпа, представляющая собой
суспензию фосфогипса в растворе фосфорной кислоты, непрерывно
поступает через верхний штуцер в питающий лоток непрерыв-
нодействующего вакуум-фильтра 14.
Для фильтрования экстракционной пульпы и промывки
осадка фосфогипса применяются ленточные, карусельные или кон-
вейерно-лотковые вакуум-фильтры. Пульпа и промывные
растворы наливаются на их фильтрующую поверхность, что
позволяет производить фильтрование и полную промывку осадка
в одном аппарате. На таких фильтрах фильтрующая ткань,
каналы и трубопроводы, отводящие фильтраты, почти не
забиваются осадком, так как в конце каждого цикла промываются
водой. В первой зоне ленточного вакуум-фильтра 14
происходит фильтрование пульпы и отсос первого основного
фильтрата (продукционная фосфорная кислота). Осадок
фосфогипса на ленте по мере ее движения проходит поочередно три зоны
промывки и зону подсушки.
Фильтрование и промывка осадка производятся по четырех-
фильтратной противоточной схеме. В четвертую зону фильтра
(третья зона промывки) подается заданное количество
нагретой воды. Отсасываемые раздельно четвертый и третий
фильтраты, пройдя соответствующие вакуум-приемники 13,
непрерывно подаются насосами глубокого всасывания
непосредственно на промывку фосфогипса в третью и вторую зоны фильтра.
Фильтрат, отсасываемый из второй зоны, полностью
возвращается из второго вакуум-приемника в первый экстрактор.
Первый фильтрат в основном направляется в сборник 12
продукционной фосфорной кислоты, меньшая часть фильтрата
возвращается в экстракционную систему (оборотная кислота).
С приводного барабана фильтра промытый осадок
фосфогипса, имеющий влажность около 40%, свободно сбрасывается в
бак 15, где взмучивается водой, стекающей из корыта фильтра
(после промывки фильтрующей ткани), и удаляется шламовым
насосом в отвал.
Несущая лента вакуум-фильтра изготовляется из резины с
прокладками из синтетической ткани (анид, лавсан).
Фильтрующая перегородка выполняется из хлориновой ткани, вакуум-
камера— из нержавеющей стали.
506
Экстракционная фосфорная кислота, полученная из
апатитового концентрата, содержит 32—33% Р2О5, из фосфоритов
Кара-Тау — 26—27% Р2О5. Примесями в экстракционной
фосфорной кислоте являются ионы SOI , Al3+, Fe3+ и SiFg , а также
Mg2+ (при переработке фосфоритов Кара-Тау). Кроме того,
фосфорная кислота содержит небольшое количество мелких
частиц фосфогипса, прошедших через фильтровальную ткань.
Взвешенные частицы придают экстракционной фосфорной
кислоте мутный желто-зеленый цвет.
Основные технологические показатели процесса
производства экстракционной фосфорной кислоты из апатитового
концентрата и фосфоритов Кара-Тау:
Сырье
апатитовый фосфориты
концентрат Кара-Тау
Количество H2SO4, % от СаО + MgO .... 100 102
Степень разложения сырья, % 98 97
Степень отмывки Р.,О5, % 98 97
Выход Р2О5, % . ." 96 94
Съем сухого осадка с 1 м2 фильтрующей
поверхности, кг/ч 800—850 600—700
В последнее время разрабатывается так называемый ангидритный
процесс получения экстракционной фосфорной кислоты, содержащей около 45%
P2Os. По этой схеме сульфат кальция, образующийся при экстракции
фосфорной кислоты, выпадает в осадок в виде ангидрита CaSO4, а не дигид-
рата CaSO4 • 2НзО. Ангидритный процесс проводится при более высокой
температуре A00 —105 °С).
При производстве экстракционной фосфорной кислоты из некоторых
фосфатов, содержащих 100—300 г/г урана, он переходит в раствор. В этом
случае экстракционная фосфорная кислота содержит до 200 мг/л U3Oe.
Несмотря на незначительное содержание урана в природных фосфатах, иногда
его целесообразно извлекать, так как количество перерабатываемого
фосфатного сырья очень велико. На некоторых зарубежных заводах уран
извлекают из фосфорной кислоты 1—2%-ным раствором алкилфосфатов в
керосине и после разделения фаз осаждают из раствора (в вице фторида)
фтористоводородной кислотой. Регенерированный экстрагент возвращается в
производственный цикл вместе со свежими порциями растворителя. Состав
фосфорной кислоты после выделения урана почти не изменяется.
УПАРИВАНИЕ ФОСФОРНОЙ КИСЛОТЫ
Экстракционную фосфорную кислоту, полученную из
апатитового концентрата, упаривают до содержания 55—56% Р2О5.
Кислогу из фосфоритов Кара-Тау, вследствие большого
содержания в ней растворимых солей и повышенной вязкости, можно
упаривать только до содержания 33—34% Р2О5.
Упаривание фосфорной кислоты сопряжено с техническими
трудностями. Экстракционная фосфорная кислота содержит
значительное количество растворенных солей, которые при
упаривании выпадают в осадок и отлагаются на греющих
поверхностях и в корпусе выпарного аппарата, вследствие чего испа-
507
рительная способность аппарата уменьшается. Иногда осадок
солей настолько забивает аппаратуру, что процесс упаривания
сильно замедляется.
При упаривании фосфорной кислоты значительно возрастает
давление паров содержащейся в ней кремнефтористоводороднои
кислоты. Поэтому, кроме водяных паров, при упаривании
выделяются также фторсодержащие газы, что весьма усложняет
антикоррозионную защиту аппаратуры для упаривания
фосфорной кислоты и для поглощения отходящих газов.
Упаривание фосфорной кислоты производят в аппаратах
барботажного типа или в вакуум-испарителях.
В барботажном аппарате горячие топочные газы
интенсивно барботируют через слой упариваемой кислоты, отдавая ей
свое тепло. Аппарат состоит из одной камеры, выполненной из
стали и футерованной диабазовыми плитками и
кислотоупорным кирпичом. Топочные газы, получаемые сжиганием мазута
или газа, поступают под давлением через вертикальную бар-
ботажную трубу в слой кислоты, находящейся в камере.
Давление создается воздуходувкой, подающей воздух, необходимый
для горения топлива.
Как и при упаривании серной кислоты, непосредственный
контакт газов с упариваемой фосфорной кислотой приводит к
образованию туманообразной кислоты, которую можно
улавливать в электрофильтре. Однако упаривание фосфорной
кислоты протекает легче упаривания серной кислоты, вследствие
значительного давления водяного пара над раствором
фосфорной кислоты.
Автоматическое регулирование процесса упаривания
осуществляется по принципу автотерморегулировки. Подача слабой
кислоты регулируется по температуре в аппарате,
поступление воздуха на разбавление топочных газов — по температуре
газов в барботажной трубе, подача мазута в топку — по
температуре в топке, а количество пара для подогрева мазута — по
его температуре в расходном баке. Автоматическое
регулирование процесса позволяет поддерживать устойчивый
технологический режим, строго соблюдать заданную концентрацию
упаренной фосфорной кислоты. Для охлаждения упаренной
кислоты можно применять колонку с провальными тарелками.
В вакуум-испарителе пар поступает в систему
горизонтальных медных труб, снаружи тщательно освинцованных,
кислота движется в межтрубном пространстве. Разрежение в
аппарате создается водоструйными эжекторами-конденсаторами,
которые служат одновременно и для конденсации сокового пара.
Эжекторы рассчитаны на вакуум около 600 мм рт. ст. Чтобы
выпадающий при упаривании осадок не отлагался на стенках и
трубах аппарата, кислоту с помощью специального насоса
заставляют интенсивно циркулировать. Этот аппарат конструк-
508
тивно сложнее барботажного. Для изготовления
вакуум-испарителя требуется большое количество свинца и кислотостойкой
стали, а работает он с меньшей интенсивностью и менее удобен
в обслуживании, особенно при очистке аппарата от осадков.
Использование фосфогипса
Фосфогипс отличается от природного гипса главным образом тем, что
содержит остаток неразложенного фосфата и небольшую часть неотмытой
фосфорной кислоты.
Из фосфогипса могут быть получены различные воздушные и
гидравлические вяжушие материалы (стр. 629 ел.) — штукатурный фосфогипс, фос-
фоангидритовый цемент и др.
Фосфогипс может быть переработан на портланд-цемент с
одновременным получением сернистого газа. При этом удается частично
регенерировать серную кислоту, затраченную на производство минеральных удобрений.
Для получения портланд-цемента к фосфогипсу добавляют около 15—
20% глины н 5—6% металлургического кокса, служащего восстановителем.
Такую смесь (шихту) обжигают в обычных вращающихся цементных печах
при 1450—1550 °С. Выделяющийся при обжиге сернистый газ можно
использовать для получения серной кислоты (стр. 93 ел.). Материал, выходящий
из печи (клинкер), после некоторого вылеживания измельчают в шаровых
мельницах, добавляя туда 2—3% высушенного фосфогипса. Полученный
портланд-цемент после схватывания обладает высокой механической прочностью.
ТЕРМИЧЕСКАЯ ФОСФОРНАЯ КИСЛОТА
Термическая фосфорная кислота получается путем
окисления фосфора. Этот процесс осуществляется при
непосредственном сжигании сконденсированного из газов фосфора. При
достаточном количестве воздуха фосфор сгорает с образованием
фосфорного ангидрида*
Р4 + 50, = РА,
который, взаимодействуя с парами воды при температуре ниже
1000 °С, образует сначала газообразную метафосфорную
кислоту
Р4О10 + 2Н2О = 4НРО3
превращающуюся при охлаждении и достаточном количестве
воды в ортофосфорную
НРО3 + Н2О = Н3РО4
Окисление фосфора при недостатке кислорода приводит к
образованию фосфористой кислоты Н3РО3 и фосфорноватистой
кислоты Н3РО2.
* Обычно фосфорный ангидрид обозначают формулой Р2О5: в
действительности молекула фосфорного ангидрида соответствует формуле Р4О10, a
молекула фосфора Р4.
509
Схема производства термической фосфорной кислоты
изображена на рис. 199. Очищенный твердый фосфор расплавляют
в баке /, обогреваемом паром. Расплавленный фосфор через
расходный бак 2 и буферный бак 4, снабженные паровым
обогревом, подается форсункой 5 в камеру сжигания 6.
Камеру сжигания выполняют из кислотоупорного кирпича,
форсунку изготовляют из кислотоупорной стали или
хромистого чугуна.
Пор
Конденсат
Ханденсат
85-90% Н,РО,,
Рис. 199. Схема производства термической фосфорной кислоты:
/—плавильный бак: 2—расходный бак; 3—сифон; 4—буферный бак; 5—форсунка; 6—камераТ]
сжигания; 7-газоходы; 8— башня орошения; 9—электрофильтр; J0—коронирующие электро- _,
ды (серебряные проьода); 11- осадительные электроды (угольные трубы).
В воздухе, поступающем на сжигание фосфора, всегда
содержится влага, поэтому в продуктах горения в значительной
мере находится уже не фосфорный ангидрид, а газообразная
метафосфорная кислота НРОз.
Из камеры сжигания 6 продукты горения направляются в
башню орошения 8 для охлаждения газов с образованием
фосфорной кислоты. Стальной кожух башни футерован в два слоя
кислотоупорными плитками и угольными блоками или
кислотоупорным кирпичом.
В башне, орошаемой сверху водой или разбавленной
кислотой, метафосфорная кислота превращается в ортофосфорную
кислоту Н3РО4, которая образует туман. Для выделения ту-
510
манообраьной фосфорной кислоты газы, выходящие из башни
орошения, направляются в трубчатый электрофильтр 9,
сложенный из кислотоупорного кирпича.
В качестве осадительных электродов применяют угольные
трубы, в качестве коронирующих электродов — серебряные
провода. Температура газа в электрофильтре около 150°С. Степень
осаждения фосфорной кислоты в электрофильтре достигает
98—99%.
Чтобы предотвратить проникание фосфорных газов в
атмосферу, в нижней части башни орошения и в электрофильтре
имеются гидравлические затворы (на рисунке не показаны).
Вытекающая из электрофильтра фосфорная кислота содержит
85—90% Н3РО4. Небольшая часть туманообразной фосфорной
кислоты A0—20%) поглощается водой или разбавленной
кислотой в башне орошения, из которой отбирается фосфорная
кислота, содержащая 45—60% Н3РО4.
Обычно кислоту, вытекающую из электрофильтра и из
башни орошения, смешивают вместе и получают продукт со
средним содержанием 70—85% Н3РО4.
На некоторых новых заводах фосфор сжигают в
верхней части башни (факел форсунки направлен сверху вниз).
Стенки башни непрерывно орошаются холодной разбавленной
фосфорной кислотой. Вследствие выделения тепла при
сгорании фосфора кислота упаривается и на выходе из башни
содержит 86—88% Н3РО4. Стекая по стенкам башни, кислота
предохраняет их от разрушающего действия фосфорного пламени и
горячей метафосфорной кислоты. Охлажденный в нижней части
башни газ, содержащий туман фосфорной кислоты,
направляется в электрофильтр на очистку.
Термическая фосфорная кислота содержит мало примесей,
поэтому из нее получают концентрированные удобрения,
кормовой преципитат и ряд технических фосфатов. Часть
термической фосфорной кислоты очищают и выпускают в виде
пищевой и реактивной кислоты.
Полифосфорная кислота. Путем сжигания фосфора и
абсорбции образующегося фосфорного ангидрида концентрированной
фосфоррой кислотой в последнее время начали получать
полифосфорную кислоту — высококонцентрированную фосфорную
кислоту, называемую иногда также суперфосфорной кислотой.
Полифосфорная кислота, наряду с оптофосфорной кислотой
H.^POi, содержит пирофосфорную Н4Р2О7, триполифосфорную
Н5Р3О0 и тетраполифосфорную H6P4Oi3 кислоты. Общее
содержание Р2О5 в полифоссЬорной кислоте составляет около 76%,
что в пересчете на Н3РОд соответствует 105%. В отличие от
обычной термической фосфорной кислоты, полифосфорная
кислота менее агрессивна, имеет низкую температуру замерзания
и поэтому более удобна для перевозки и хранения.
511
Полифосфорную кислоту целесообразно использовать для
получения концентрированных удобрений — полифосфатов
аммония, комплексных жидких удобрений и др.
Техника безопасности в производстве
фосфора и фосфорной кислоты
Желтый фосфор — сильный яд. Смертельная доза его для
человека составляет 0,05—0,1 г. Чистый красный фосфор не
ядовит, но присутствие примесей желтого фосфора делает его
опасным. Продолжительная работа в атмосфере, содержащей
пары желтого фосфора, без соблюдения правил техники
безопасности и гигиены, приводит к хроническому отравлению. При
попадании на кожу горящий желтый фосфор причиняет
болезненные, долго не заживающие ожоги, при проникании фосфора
в кровь нарушаются процессы обмена веществ в организме.
В случае ожогов фосфором необходимо немедленно
обильное промывание пораженных участков кожи.
При отравлении обязательно промывание желудка
(недопустим прием молока и жиров, так как они растворяют
фосфор).
Вредность производства фосфорной кислоты определяется
токсическими свойствами фосфора и фосфорного ангидрида и
огнеопасностью фосфора, возможностью термических ожогов
фосфором и химических ожогов кислотами — фосфорной и
серной, применяемой для разложения фосфатов. Кроме того,
следует учитывать, что производство фосфора и термической
фосфорной кислоты связано с применением электрического тока
высокого напряжения.
Основные меры предотвращения травматизма и отравлений
в указанных производствах: механизация процессов загрузки,
выгрузки и транспортирования фосфора, полная герметизация
оборудования и коммуникаций, устройство общей и местной
вытяжной вентиляции, пользование индивидуальными мерами
защиты (спецодежда, спецобувь, противогазы и др.), строгое
соблюдение правил личной гигиены. Прием пищи и курение в
производственных помещениях запрещается.
ЛИТЕРАТУРА
С. И, Вольфкович. 3. А, Роговин, Ю, П, Руденко. И. В. Ш м а-
н е н к о в. Общая химическая технология, т. II, Госхичичдат. 1959.
В. Ваггаман, Фосфорная кислота, фосфаты и фосфорные удобрения,
перев с англ., Госхимиздат, 1957.
Л. Я. Марковский, Д. Л. Оршанский, В, П. Прянишников,
Химическая электротермия, Госхимиздат, 1952.
512
Н. Н, Постников, Состояние и тенденции фосфорной промышленности
за рубежом, Хим. пром., № 6, 57 A958).
B. А. Кононов, С. Д. Э в е н ч и к, Модернизация оборудования и
интенсификация производства концентрированных фосфорных удобрений,
Жури- ВХО им. Д. И. Менделеева, № 1, 2 A961).
C. К- Воскресенский, Концентрированные фосфорные удобрения, Хим.
наука и пром., № 2, 129 A956).
Н. Н. Постников, Современное состояние и тенденции производства
фосфорной кислоты (термической) за рубежом Хим. пром., № 6, 82
A959).
Н. Н. Постников, М. Г. Френкель, Производство фосфорной кислоты
и концентрированных удобрений на основе электротермической
переработки фосфатов, Журн. ВХО им. Д. И. Менделеева № 5, 500 A962).
Правила и нормы техники безопасности и промышленной санитарии ДЛЯ
проектирования, строительства и эксплуатации производств желтого
фосфора и термической фосфорной кислоты, Госхимиздат, 1962.
33—2848
Глава XII
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
/. ОБЩИЕ СВЕДЕНИЯ
Главнейшими элементами, входящими в состав растений,
являются: углерод, кислород, водород, азот, фосфор, сера, калий,
кальций, магний, железо. Воду и минеральные вещества
растение получает из почвы, углерод — из углекислоты воздуха.
Многие минеральные вещества находятся в почве в
достаточном количестве, некоторых же, в частности азота, фосфора
и калия, часто недостает, особенно в виде соединений,
усвояемых растениями. Часть недостающих растениям химических
элементов, а также гуминовые органические вещества и
полезные бактерии вносят в почву с навозом. Однако количество
питательных веществ в навозе недостаточно для полного питания
растений, поэтому некоторые химические элементы, главным
образом азот, фосфор и калий, а в некоторых случаях бор, магний
и др. вносят в почву с минеральными удобрениями.
Применение минеральных удобрений способствует не
только получению высоких и устойчивых урожаев, но и повышению
качества сельскохозяйственных продуктов: улучшению свойств
волокна хлопчатника, льна и других культур, увеличению
содержания сахара в сахарной свекле и винограде, крахмала в
картофеле, белков в злаках и кормовых травах и т. д.
Минеральные удобрения вносят перед посевом (основное
удобрение), во время посева (припосевное удобрение) и в
период роста растений (подкормка). При основном внесении удо-
брения разбрасывают туковыми сеялками равномерно по
всей площади поля, при последующей обработке почвы
удобрения перемешиваются с нею. При внесении удобрении во время
посева их вводят сосредоточенно в непосредственной близости
к вносимым семенам. Применяемые для этого
комбинированные сеялки вносят в почву удобрения одновременно с
семенами. При подкормке удобрения также вносят в почву в
непосредственной близости к растениям. Иногда применяется
внекорневая подкормка: опрыскивание растений растворами удобрении
или опыливание порошкообразными удобрениями.
Дозы минеральных удобрений выражают в килограммах
азота, Р2О5 и К2О на 1 га посевной площади (табл. 27). Тре-
514
буемые дозы зависят от характера почв, вида удобряемых
культур, времени и техники внесения удобрений.
Таблица 27
Дозы минеральных удобрений
Внесение удобрений
Основное
Припосевное
Подкормка
Дозы удобрений, кг/га
N
30—90*
10—15
15—30
р2о5
30—90*
10—20
20-30
К2О
40—120
10—15
20—40
• Под хлопчатник, члп, з шогрзд н цчтрусов^г культуры вчосят и большие дозы.
Для достижения наибольшей эффективности -минеральных
удобрений их необходимо применять в неразрывном комплексе
с другими агротехническими мероприятиями (хорошая
обработка почвы, сочетание минеральных удобрений с
органическими, улучшение водного режима почвы путем ирригационных
и мелиоративных работ, применение химических средств
защиты растений для борьбы с вредителями и возбудителями
болезней растений, для уничтожения сорняков и др.).
Агрохимическая наука и многолетняя практика, в особенности опыт
передовых колхозоз, совхозов и целых районов, подтверждают
исключительно важную роль удобрений для получения
устойчивых высоких урожаев.
По данным НИУИФ, при правильном применении
минеральных удобрений в комплексе с другими агротехническими
мероприятиями прирост урожая сельскохозяйственных культур
можно характеризовать величинами, приведенными в табл. 28.
Таблица 28
Прирост урожая при внесении минеральных удобрений
(на 1 т N, P2Ofi, K2O)
Культуры
Хлопчатник (хлопок-сырец)
Сахарная свекла:
клуони
сахар
Лен (волокно)
Картофель (клубни)
Озимая пшеница (зерно)*
Прирост, т
N
12
100
16
2,5
120
20
Р2О5
6
70
10
2
80
25
К2О
2
40
6
1,5
60
4-
Азот в подкормку, фосфор в рядки.
33*
515
Применение полных удобрений, содержащих азот, фосфор
и калий и вносимых в нужных количествах, повышает урожай
сельскохозяйственных культур в среднем в 1,5—2 раза.
Промышленное производство фосфорных и калийных удобрений
возникло по второй половине XIX в., а азотных удобрений в XX в. До этого в
качестве азотного удобрения применяли природную чилийскую (натриевую)
селитру. В XX в. промышленность минеральных удобрений получила
большое развитие во многих странах. В настоящее время мировое производство
минеральных удобрений составляет около 120 млн. г.
В дореволюционной России имелись лишь небольшие разработки
фосфоритов, кустарное производство фосфоритной муки и несколько небольших
суперфосфатных заводов; азотные и калийные удобрения не производились.
В годы первой мировой войны производство минеральных удобрений в
России почти прекратилось. Оно было создано заново в нашей стране в 30-х гг.,
когда были построены крупные предприятия, изготовляющие фосфорные,
азотные и калийные удобрения. Быстро растущая потребность сельского
хозяйства в минеральных удобрениях обусловила дальнейшее развитие
производства минеральных удобрений. Ниже приведены данные о
производстве минеральных удобрений в СССР (в тыс. т стандартных удобрений):
Годы
1913. . .
1928 . . .
1932 . . .
89*
135
921
Годы
1937. . . .
1950. . . .
1955. . . .
3238
5497
9669
Годы
1958. .
1960. .
1962. .
1963 . .
12420
13867
17262
19900
* На территории в современных границах.
В Программе КПСС намечено увеличение общего объема
сельскохозяйственной продукции за 10 лет примерно в 2,5 раза,
а за 20 лет — в 3,5 раза. Успешное решение этой важной задачи
неразрывно связано с широкой всесторонней химизацией
сельского хозяйства, в результате которой значительно повысится
урожайность, улучшится качество сельскохозяйственной
продукции, уменьшатся потери урожая от сорняков и вредителей
растений, расширятся ресурсы кормовых средств для
животноводства, повысится питательность кормов и т. д. При
глубоком внедрении разнообразных химических средств в сельское
хозяйство оно станет гораздо менее зависимым от природных
и климатических условий. Для обеспечения устойчивых,
возрастающих урожаев необходим дальнейший значительный рост
производства минеральных удобрений. Так, в 1965 г.
производство минеральных удобрений в СССР предстоит довести до
33,5 млн. т. Развитие отечественной промышленности
минеральных удобрений будет сопровождаться применением новых, более
прогрессивных методов и процессов. Расширится ассортимент
удобрений, особенно концентрированных, преимущественно
сложных и смешанных удобрений, микроудобрений, а также
жидких удобрений. В СССР исследования в области удобрений
приняли исключительно большой размах, особенно широкое
развитие получили исследования по агрохимии, химии и технологии
минеральных удобрений.
516
В результате планомерных геологоразведочных работ
найдены и изучены многочисленные месторождения полезных
ископаемых, служащих сырьем для промышленности минеральных
удобрений.
Классификация удобрений. По происхождению удобрения
подразделяются на минеральные, органические и органо-мине-
ральные.
К минеральным удобрениям относятся продукты
неорганического происхождения, специально получаемые на заводах;
эти продукты называют также искусственными удобрениями.
В органических удобрениях питательные элементы
находятся главным образом в форме органических соединений. К
органическим удобрениям относятся в первую очередь навоз, а
также различные продукты переработки веществ животного и
растительного происхождения.
Органо-минвральные удобрения, как показывает название,
содержат и органические и минеральные вещества. В качестве
органических веществ в состав таких удобрений входят торф,
бурый уголь и различные отбросы органического
происхождения.
По агрономическому назначению различают прямые и
косвенные минеральные удобрения.
Прямые удобрения предназначены для
непосредственного питания растений и содержат необходимые питательные
элементы: азот, фосфор, калий, а также магний, серу,
микроэлементы (бор, медь, марганец, молибден, цинк и др.),
требующиеся в небольших дозах. Прямые удобрения разделяют на
односторонние и комплексные, называемые также
многосторонними удобрениями.
Односторонние удобрения содержат один питательный
элемент. К ним относятся: азотные, фосфорные, калийные,
магниевые удобрения, борные и другие микроудобрения.
Азотные удобрения различают по форме соединений азота:
нитратные, аммиачные, амидные и содержащие азот в
различных формах, например аммиачно-нитратные, аммиачно-амид-
ные, аммиачно-нитратно-амидные. Азотные удобрения
растворимы в воде. В районах избыточного увлажнения и
орошаемого земледелия азотные удобрения частично вымываются из
почвы. В последнее время разработаны новые формы невымы-
ваемых, труднорастворимых азотных удобрений, например мо-
чевино-формальдегидные удобрения.
• Фосфорные удобрения различают по их способности
растворяться в почвенных растворах. Фосфорнокислые соли,
растворимые в воде, легко всасываются корнями растений и хорошо
ими усваиваются. Некоторые фосфорнокислые соли
нерастворимы в воде, но в большей или меньшей степени растворяются
в слабокислых соках, выделяемых корнями растений.
517
Чтобы судить о растворимости фосфорных удобрений в
почвенных растворах, а следовательно, о их способности
усваиваться растениями, было предложено пользоваться
специальными искусственно подобранными растворами. По
растворимости в таких растворах фосфорные удобрения
подразделяются на следующие группы:
1) воднорастворимые удобрения, в которых большая часть
фосфорных соединений растворима в воде и, следовательно,
наиболее легко усваивается растениями (суперфосфат, двойной
суперфосфат, аммофос);
2) иитратнорастворимые удобрения, в которых содержатся
соединения фосфора, растворимые в аммиачном растворе
лимоннокислого аммония (цитрата аммония). Фосфорная
кислота из таких соединений обычно легко усваивается
растениями.
К цитратнорастворимым удобрениям относятся преципитат,
обесфторенный фосфат из некоторых фосфоритов;
3) лимоннорастворимые удобрения, нерастворимые в во
де и аммиачном растворе цитрата аммония, но растворимые в
2%-ном растворе лимонной кислоты. Эти удобрения
(обесфторенный фосфат, томасшлак) усваиваются растениями
предпочтительно на кислых почвах;
4) малорастворимые удобрения, фосфорные соединения
которых в значительной своей части нерастворимы ни в воде,
ни в аммиачном растворе цитрата аммония и лишь частично
растворяются в лимонной кислоте (фосфоритная мука),
Несмотря на малую растворимость, эти вещества являются для
ряда кислых почв хорошими удобрениями. Фосфорные
соединения этих удобрений медленно переходят в почвенный раствор,
и действие их длится ряд лет.
Комплексные удобрения содержат два и более питательных
элементов. Различают двойные удобрения (например, азотно-
фосфорные, азотно-калийные, фосфорно-калийные) и тройные
удобрения (например, азотно-фосфорно-калийные), которые
называют также полными минеральными удобрениями. В
состав комплексных удобрений могут входить также
микроэлемента!, добавки ростовых веществ и др.
По способу производства комплексные удобрения
разделяются ка смешанные и сложные.
Смешанные удобрения представляют собой механические
смеси различных удобрений, состоящие из различных по
составу частиц или агрегатов таких частиц в виде гранул.
Сложные удобрения содержат несколько питательных
элементов в однородных по составу частицах. Их получают в
результате химических процессоз, а также путем совместной
кристаллизации или сплавления основных компонентов
удобрения.
518
Косвенные удобрения вносят для улучшения свойств
почвы или мобилизации ее питательных веществ. К косвенным
удобрениям относятся вещества, применяемые для
нейтрализации кислотности почв, например молотые известняк,
доломит* (известкование), для мелиорации солонцов, например
молотый гипс (гипсование), служащий в данном случае
источником кальция, а также удобрения, предназначенные для кис-
лования почв с целью улучшения растворимости фосфорных
удобрений (бисульфат натрия).
По влиянию на реакцию почвы различают физиологически
кислые, физиологически щелочные и нейтральные минеральные
удобрения. К физиологически кислым относятся удобрения,
катионы которых поглощаются растениями более интенсивно, чем
анионы, а анионы подкисляют почвенный раствор (например,
сульфат аммония, хлористый калий). К физиологически
щелочным принадлежат удобрения, анионы которых быстрее
ассимилируются растениями, а катионы, постепенно накапливаясь
в почве, подщелачивают ее (например, кальциевая селитра,
натриевая селитра, томасшлак). Физиологически нейтральные
удобрения не изменяют реакцию почвы (например, известково-
аммиачная селитра).
Большую часть минеральных удобрений выпускают в
твердом виде (порошкообразные и гранулированные удобрения).
Гранулированные минеральные удобрения, выпускаемые в виде
гранул определенных размеров, слеживаются меньше
порошкообразных продуктов и лучше рассеваются. Гранулированные
воднорастворимые фосфорные удобрения полнее используются
на кислых почвах, чем порошкообразные. В последнее время
получают распространение жидкие минеральные удобрения
i аммиачная вода, жидкий аммиак, аммиакаты).
Качество удобрений. Важнейшим показателем качества
минеральных удобрений является содержание в них питательных
веществ, выражаемое обычно в процентах. Содержание
важнейших питательных элементов — азота, фосфора и калия
выражают в процентах N, P2O5 и КгО. Чем выше концентрация
питательных веществ в удобрениях, тем они ценнее.
Применение концентрированных удобрений позволяет сократить
расходы на перевозку, тару и складирование, а также на внесение
удобрений в почву.
Удобрение должно быть сухим, рассыпчатым, удобным для
рассева механическим:! сеялками и не должно слеживаться в
сплошные глыбы или крупные комки. Частицы удобрения
должны быть не слишком крупными (чтобы растение могло извлечь
из них все питательные вещества) и не слишком мелкими
(чтобы их не уносило ветром). Для оценки физических свойств
* Допомнт з ряде случаев является и прямым магнитным удобрением.
519
удобрений в ряде случаев определяют содержание в них влаги.
Повышенное содержание влаги вызывает комкование или
слеживание удобрений. Кроме того, избыточная влага в удобрении
является балластом, понижающим содержание питательных
веществ в единице веса удобрения. Весьма важным показателем
качества удобрений является их гигроскопичность, которая
определяет возможность применения удобрений, условия их
хранения, характер тары.
Гигроскопичность характеризуется так называемой
гигроскопической точкой, указывающей относительную влажность
воздуха, при которой удобрение не поглощает и не теряет
влагу. При этой влажности давление паров воды над удобрением
равно давлению паров воды в окружающем воздухе.
Удобрения, гигроскопическая точка которых ниже 60%,
считают сильногигроскопичными, от 60 до 65 %
—гигроскопичными, от 70 до 75%—слабогигроскопичными, от 75 до 80% —
почти негигроскопичными, выше 80% —негигроскопичными.
2. ФОСФОРНЫЕ УДОБРЕНИЯ
СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА ФОСФОРНЫХ УДОБРЕНИЙ
Сырьем для производства фосфорных удобрений и других
фосфорсодержащих продуктов служат природные
фосфорсодержащие руды. Промышленное значение имеют два типа
природных фосфатов — апатиты и фосфориты. Фосфор находится в
них главным образом в виде минерала фторапатита
Cas(PO4KF, нерастворимого в воде. Теоретическое содержание
РгО5 во фторапатите составляет 42,24%.
Апатиты обычно представляют собой изверженные породы
кристаллического строения и являются первоисточником
фосфора в земной коре. Однако месторождения апатитов, имеющие
промышленное значение, встречаются редко.
Основные запасы апатитов не сосредоточены в виде
крупных залежей, а распространены в форме мельчайших примесей
к изверженным породам. Эти огромные количества
распыленного апатита служили исходным материалом для образования
осадочных фосфатов. В больших водных бассейнах
происходило постепенное накопление принесенных с суши фосфатов,
которые обычно содержатся в организмах морских животных,
в растениях и тем или иным путем выпадают в осадок. Эти
большие скопления образовали в дальнейшем осадочные
породы, называемые фосфоритами. В зависимости от интенсивности
накопления фосфорных соединений на морском дне образуются
породы с содержанием Р2О5 от долей процента до 34—36%.
Фосфориты залегают в виде сплошных пластов либо в виде
желваков — кусков неправильной формы, различного размера,
с неровной бугристой поверхностью.
520
Фосфатное вещество фосфоритов состоит преимущественно
из высокодисперсного фторапатита. Кроме фосфатного
вещества в состав фосфоритных руд входят кварц, глауконит,
доломит и другие минералы. При добыче фосфориты загрязняются
обломками сопутствующих пород (пески, глины, известняки,
доломиты и др.).
Наиболее крупные в мире месторождения апатита
находятся в Хибинской тундре (Кольский полуостров). Апатито-иефе-
линовая порода этого месторождения состоит в основном из
мелкозернистого апатита, среди которого распределены мелкие
зерна нефелина и примеси других нефосфатных минералов.
Главнейшие месторождения фосфоритов в СССР
находятся в хребте Кара-Тау (Казахская ССР), большое значение
имеют также Вятско-Камское, Воскресенско-Егорьевское
(Московская обл.), Эстонское и Актюбинское фосфоритовые
месторождения, а также группа месторождений в Курской, Брянской
и Калужской областях.
Большинство месторождений природных фосфатов в нашей
стране открыто и изучено за годы Советской власти.
Способы разработки месторождений апатитов и фосфоритов
зависят от строения и условий залегания фосфатных руд.
Разработка хибинских апатитов производится взрывным способом,
открытыми или подземными работами. Открытая разработка
фосфоритных месторождений ведется при помощи
экскаваторов, подземная — с помощью отбойных молотков и взрывным
способом.
Примеси, находящиеся в фосфатных рудах, значительно
понижают содержание в них Р2О5, некоторые примеси оказывают
вредное влияние на процессы кислотной переработки природных
фосфатов и качество получаемых из них удобрений. Наиболее
нежелательными примесями природных фосфатов при их
кислотной переработке являются полуторные окислы (Fe2O3, А1аО3)
и карбонаты, особенно если они содержат много магния.
Чтобы повысить содержание основного полезного компонента
руды— фосфата и максимально возможно удалить вредные
примеси, фосфатные руды подвергают обогащению.
Апатито-нефелиновую руду, содержащую от 12 до 30% Р2О5,
обогащают методом флотации (стр. 22). Получаемый в
результате флотационного обогащения апатитовый концентрат
представляет собой мелкий рассыпчатый светло-серый
порошок, состоящий в основном из фторапатита. Согласно ГОСТ
3277—54, содержание Р2О5 в апатитовом концентрате (в
пересчете на сухое вещество) должно быть не менее 39,4%, влаги
не более 1%, остаток на сите с отверстиями размером 0,15 мм
не более 14%. Апатитовый концентрат является наиболее
богатым фосфатным сырьем в мире и важнейшим отечественным
фосфатным сырьем.
521
Обогащение фосфоритов производят методом флотации
либо путем сухого или мокрого грохочения.
Химический состав природных фосфатов важнейших
месторождений СССР приведен в табл. 29.
Таблица 29
Состав природных фосфатов важнейших месторождений СССР
(в пересчете на сухое вещество)
Фосфаты
Апатитовый
концентрат ....
Фосфориты
Кара-Тау, необога -
щенный ....
Кара-Тау,
флотационный концентрат
Вятский, мытый
Егорьевский,
мытый
Эстонский
концентрат
первичный . .
флотационный .
Актюбинский
мытый ....
флотационный
концентрат .
Брянский
концентрат
первичный . .
флотационный .
Р
До
До
До
До
-°5
39
27
29
25
23
26
32
19
25
17
26
,4
,5
,5
,3
,0
,2
,1
,8
,1
,9
СаО
52,
43,
41,
До 39,
До 35,
До 37,
46,
31,
До 41,
29,
44,
0
7
4
5
3
5
4
5
8
4
0
Со ста
м?о
До 0,2
До 4,1
2,1
0,8
—
0,8
0,6
—
i
0,5
0,9
з. %
Fe2O 4-
+А1аЭз
0,6—1,2
2,6
2,2
6,6—9,0
10,2-11,6
3,2—3,9
2,9
(А1.2О3)
4,7
3,4—4,0
2,8
3,7
До
До
До
До
До
F
3,1
2,8
2,9
3,0
2,6
2,3
2,4
2,5
3,2
2,1
—
нерастворимый
остаток
До 1,5
До 13
13
15,6
До 6,4
20
6,3
32,7
До 13,0
37,6
9,2
Примечание. Содержание СОг в фосфоритах различных месторождений колеблется
от 3,5% в эстонских концентратах до 5,5—9% в необогащенных фосфоритах Кара-Тау.
МЕТОДЫ ПЕРЕРАБОТКИ ПРИРОДНЫХ ФОСФАТОВ
Современные методы переработки природных фосфатов в
удобрения могут быть разделены на следующие основные
группы:
1. Тонкое измельчение фосфоритов с получением
фосфоритной муки, непосредственно применяемой в качестве удобрения
на кислых почвах.
2. Разложение природных фосфатов кислотами (серной,
азотной, редко соляной). Для разложения применяется также
фосфорная кислота, получаемая в свою очередь путем
кислотной или термической переработки природных фосфатов
522
(стр. 502). В результате кислотной переработки природных
фосфатов получаются главным образом воднорастворимые
удобрения (суперфосфат, двойной суперфосфат, аммофос), а также
удобрения, содержащие РгО5 как в воднорастворимой, так и в
цитратнорастворимой форме (нитрофос, нитрофоска), или
только в цитратнорастворимой форме (преципитат).
3. Гидротермическая переработка природных
фосфатов—обработка водяным паром при высокой температуре,
сопровождающаяся удалением фтора и переводом фосфата в цитратно-
растворимую или лимоннорастворимую (усвояемую) форму.
При этом получаются так называемые обесфторенные фосфаты.
4. Разложение природных фосфатов при высокой
температуре путем спекания или сплавления их с солями натрия, калия,
магния и других щелочных и щелочноземельных металлов.
3 результате получают цитратнорастворимые или лимоннорас-
творимые удобрения (термофосфаты, плавленые фосфаты).
К этой группе можно условно отнести и процессы получения
томасшлака и других металлургических фосфатных шлаков.
ФОСФОРИТНАЯ МУКА
Фосфоритная мука представляет собой сухой, пылящий,
гонкий порошок буровато-серого цвета. Ее изготовляют путем
измельчения фосфоритов, фосфатное вещество которых
растворяется в почвенных растворах и усваивается растениями на
кислых подзолистых и торфянистых почвах, на оподзоленных
и выщелоченных черноземных почвах и на красноземах. Чем
тоньше измельчены фосфориты, тем эффективнее их
удобрительное действие. Апатиты и некоторые фосфориты с хорошо
окристаллизованным фосфатным веществом (например,
фосфориты Кара-Тау) даже при очень тонком измельчении почти не
усваиваются растениями и потому не применяются в виде
фосфоритной муки.
Куски фосфоритной руды величиной более 10—15 мм
предварительно дробят в щековых дробилках. Для тонкого
измельчения фосфоритов применяют кольцевые, валковые или
трубчатые мельницы. Тонкую муку отделяют в воздушных
сепараторах, которые комбинируют с мельницей. Мука, не
доведенная до требуемой тонины, поступает обратно в мельницу.
Фосфоритная руда содержит обычно 10—25% влаги, что
затрудняет, а в ряде случаев делает невозможным ее размол нз-
ла налипания влажного фосфорита на внутренние части
мельниц. Поэтому перед измельчением фосфориты сушат во
вращающихся сушильных барабанах до содержания не более 2%
влаги. При производстве фосфоритной муки выделяется много
пыли, поэтому все оборудование должно быть герметически
< а крыто.
523
Чтобы уменьшить выделение пыли, через размольное и
транспортное оборудование на некоторых заводах пропускают
ток воздуха, уносящего пыль в циклоны или рукавные
матерчатые фильтры, где она улавливается. Качество фосфоритной
муки определяют по содержанию РгО5, влажности и тонине
помола.
Фосфоритную муку выпускают трех сортов: высшего
(флотационный концентрат), первого и второго, а также более
бедную по содержанию P2Os муку из щигровского фосфорита.
Ниже приведен состав фосфоритной муки (в %):
Из щи-
гров-
Высший 1-й 2-й ского
сорт сорт
фосфорита
Р2О5, в пересчете на сухое вещество,
не менее 25 22 19 16
Влага, не более 3 3 3 3
Остаток на сите с размерами отверстий
0,18 мм, не более 20 20 20 20
Фосфоритную муку перевозят обычно навалом в
железнодорожных вагонах.
СУПЕРФОСФАТ
Суперфосфат является распространенным универсальным
воднорастворимым фосфорным удобрением. Он получается
путем разложения серной кислотой измельченных природных
фосфатов — апатита и фосфоритов и представляет собой
рассыпчатый продукт серого цвета.
Кислотное разложение фосфатов. Разложение природных
фосфатов серной кислотой можно рассматривать как
разложение отдельных минералов, входящих в состав фосфоритовых
или апатитовых руд. Разложение основного из этих минералов—
фторапатита — может быть представлено следующим
уравнением:
10Ca5(PO4KF + 35H2SO4 + 15H2O = 15Са(Н2РО4J-Н2О + 35CaSO4 + 10HF
Это уравнение отражает лишь конечный результат
разложения. Практически разложение протекает в две стадии. В
первой стадии происходит реакция между фторапатитом и серной
кислотой:
7Ca5(PO4KF + 35H2SO4 = 21H3PO4 + 35CaSO4 + 7HF
При этом из раствора выделяется большое количество
сульфата кальция, так как растворимость его в фосфорной кислоте
очень мала. Эта стадия при разложении апатитового концентра-
524
та заканчивается через 30—40 мин. Сульфат кальция выпадает
сначала в виде полугидрата CaSO4-0,5H2O, который в условиях
последующего вызревания в камерах при температуре
несколько выше 100 °С превращается в течение примерно первых 10 мин
в ангидрит CaSO4.
Образующаяся в первой стадии процесса фосфорная
кислота реагирует с остальной частью фторапатита.
Вторую стадию разложения можно представить в виде
следующей реакции:
3C?5(PO4KF |- 2Ш3РО4 + 15НаО = 15Са(Н,РО4J.НгО + 3HF
Образовавшийся монокальцийфосфат в отличие от сульфата
кальция не выпадает тотчас из раствора. Он постепенно
насыщает раствор фосфорной кислоты и начинает
выкристаллизовываться в виде Са(Н2РО4J"Н2О, когда раствор становится
пересыщенным. Взаимодействие фторапатита с фосфорной
кислотой протекает с постепенно убывающей скоростью, и для
полного завершения процесса разложения фторапатита требуется
более или менее длительное время. Поэтому в
свежеприготовленном суперфосфате содержится повышенное количество
свободной фосфорной кислоты и часть неразложившегося
фторапатита. После выдерживания такого суперфосфата в течение
5—20 суток (в зависимости от свойств фосфатного сырья и
технологического режима производства) разложение фторапа-
тнта считают практически законченным, хотя и в этом случае
в суперфосфате еще остается небольшое количество
неразложившегося фосфата и некоторое количество свободной
фосфорной кислоты.
Чтобы ускорить окончательное разложение (дозревание)
апатита, суперфосфат охлаждают путем распыления и
перелопачивания. Происходящее при этом испарение влаги также
способствует охлаждению суперфосфата. Понижение температуры
и повышение концентрации монокальцийфосфата в фосфорной
кислоте благоприятствует его кристаллизации.
После выпадения монокальцийфосфата из раствора реакция
разложения апатита фосфорной кислотой ускоряется.
Переохлаждение суперфосфата замедляет эту реакцию.
Дозревание суперфосфата лучше всего протекает при
температуре 35—45 °С.
Из нефосфатных примесей, находящихся в фосфатных
рудах, серной кислотой разлагаются карбонаты и некоторые
минералы, содержащие полуторные окислы железа и алюминия,
например глауконит в фосфоритах, нефелин* в апатитовой
руде и др.
* Глауконит — силикат Fe, К, AI, Mg, нефелин — алюмосиликат К и Na.
525
Присутствие этих примесей вызывает увеличение расхода
серной кислоты при производстве суперфосфата. Разложение
примесей протекает по следующим реакциям:
СаСО3 + H2SO4 = CaSO4 -f- CO. + H2O
Fe2O3 -f H2SO4 -f Са(Н2РО„J = 2FePO4 + CaSO4 -j- 3H3O
A12O3 + H2SO4 + Са(Н2РО„J = 2A1PO4 + CaSO4 + 3H,O
Фосфаты алюминия лучше растворимы, чем фосфаты железа,
и выпадают в твердую фазу при меньшей кислотности
суперфосфата. Фосфаты железа и алюминия образуют устойчивые
пересыщенные растворы и кристаллизуются из них очень
медленно в виде FePO4-2H2O и А1РО4*2Н2О. Поэтому при
хранении суперфосфата, полученного из природных фосфатов,
содержащих значительное количество полуторных окислов, особенно
Fe2O3, содержание воднорастворимой P2Os постепенно
снижается. Процесс перехода P2Os из воднорастворимой формы
в цитратнорастворимую или еще менее растворимую форму
называется ретроградацией Р2О5. Если отношение Fe2C>3 : P2Os
в природном фосфате не превышает 0,08—0,1, ретроградации
Р2С>5 в суперфосфате практически не происходит.
Разложение природных фосфатов, содержащих доломит
(Ca,Mg) (СО3J, например фосфоритов Кара-Тау, в которых
присутствует до 4% MgO, может быть представлено в виде
следующих трех стадий:
Ca5(PO4KF -f 5H2SO4 = 5CaSO4 + ЗН3РО4 -J- HF A)
(Ca.Mg) (CO3)a + 2H2SO4 = CaSO4 + MgSO4 + 2H2O + 2CO2
Ca5(PO4KF + 5MgSO4 + 7Н3РО„ + 10H2O = 5CaSO4+5Mg(H2PO,J-2H2O-(-HF B)
Ca5(PO4KF -j- 7Н3РО„ + 10H2O = 5Са(Н2РО„J • 2H2O + HF C)
Растворимые соли магния, накапливаясь в жидкой фазе,
задерживают разложение природного фосфата. Понижение
температуры не приводит к кристаллизации солей магния, а лишь
уменьшает скорость взаимодействия фосфорной кислоты с
фосфатом. Поэтому богатый магнием суперфосфат,
изготовленный из фосфоритной муки Кара-Тау, дозревает лучше при
более высокой температуре. Чем больше магния в суперфосфате,
тем больше в нем жидкой фазы и тем он гигроскопичнее. Такой
суперфосфат мажется, частицы его слипаются при хранении и
перевозке и применение его в сельском хозяйстве
затруднительно.
Содержание магния в фосфоритах оценивают не по
абсолютному его количеству, а как отношение MgO : Р2О5, выраженное
в процентах. Для производства суперфосфата пригодны
фосфориты, для которых отношение MgO : Р2Об не превышает 7—8%.
При более высоком содержании магния фосфориты должны
быть подвергнуты обогащению.
.526
Содержащаяся в суперфосфате РгО5 находится в основном
в воднорзстворимой форме в виде монокальцийфосфата
Са(Н2РО4J-Н2О (иногда также в виде мономагнийфосфата,
входящего главным образом в состав жидкой фазы) и
свободной Н3РО4. Небольшая часть P2Os находится в цитратнораство-
римой форме в виде фосфатов железа и алюминия и в трудно
растворимой форме в виде неразложившегося трикальцийфос-
фата. Значительную часть суперфосфата составляет сульфат
кальция, преимущественно в виде ангидрита.
Воднорастворимые н цитратнорастворимые соединения
фосфора составляют усвояемую растениями часть удобрения.
Содержащуюся в таких соединениях Р2О5 принято называть
усвояемой Р2О5. Содержание всех соединений фосфора в
суперфосфате, включая неразложившийся фторапатит,
пересчитанное на Р2О5, называют общим содержанием Р2О5. Качество
суперфосфата оценивается по содержанию в нем усвояемой Р2О5,
выраженному в процентах.
Фтористый водород, образующийся при разложении
фосфатных минералов, легко вступает в реакцию с двуокисью
кремния, которая всегда присутствует в природных фосфатах. В
результате получается газообразный фтористый кремний:
4HF + SiO2 = SiF4 + 2R.O
Поэтому в газах, выделяющихся при разложении фосфатов,
фтор содержится в виде SiF4. Часть фтора остается в
суперфосфате в виде кремнефтористоводородной кислоты H2SiFc,
образующейся по реакции
SiF4 + 2HF = H2SiF6
Кремнефтористоводородная кислота частично связывается с
основаниями, находящимися в природном фосфате.
Практически из общего количества фтора, содержащегося в исходном
природном фосфате, в газообразное состояние переходит
примерно 35—45%.
Большая часть приведенных выше реакций протекает с
выделением тепла, поэтому образование суперфосфата
сопровождается разогреванием реакционной массы до 115—120°С
и испарением некоторого количества влаги.
В результате образования твердых солей
(кристаллизующихся в виде гидратов), испарения влаги и выделения газов
смесь фосфатной муки с серной кислотой довольно скоро
затвердевает. Выделение из смеси паров и газов придает
затвердевающему суперфосфату пористую структуру.
Количество кислоты. Количество (норма) серной
кислоты, требуемое для разложения природного фосфата,
можно рассчитать по приведенным выше уравнениям реакций
разложения фторапатита и карбонатов. Находящиеся в природ-
527
ных фосфатах минералы, содержащие полуторные окислы,
обладают различной растворимостью в кислотах, вследствие чего
количество кислоты, нужное для их разложения, не
рассчитывают. Необходимую норму серной кислоты для разложения
всей массы природного фосфата уточняют опытным путем и
выражают в процентах от расчетной нормы. Практически норма
серной кислоты обычно составляет 105—115% от расчетного
количества, необходимого для разложения фторапатита и
карбонатов. В производстве суперфосфата из апатитового
концентрата норма серной кислоты составляет 68 вес. ч. (в расчете на
100% H2SO4) на 100 вес. ч. апатитового концентрата. В случае
недостаточной емкости склада для дозревания суперфосфата
или необходимости срочной отгрузки суперфосфата
устанавливают более высокую норму серной кислоты (до 74 вес. ч.
H2SO4) для ускорения процесса разложения фторапатита.
Получаемый при этом суперфосфат имеет высокую свободную
кислотность, которую перед отгрузкой удобрения нейтрализуют
путем добавки к нему костяной муки, легко разлагаемой
фосфоритной муки или других реагентов.
Концентрация и температура кислоты,
вводимой в процесс, оказывают существенное влияние на скорость
разложения природного фосфата и на структуру суперфосфата.
При низкой концентрации серной кислоты скорость разложения
фосфата высока. Однако применение серной кислоты с низкой
концентрацией H2SO4 недопустимо, так как при этом в
реакционную массу вводится слишком большое количество воды и
получается влажный мажущийся суперфосфат с пониженным
содержанием Р2О5, а иногда суперфосфатная пульпа,
образующаяся при разложении фосфата, вовсе не затвердевает нз-за
недостаточного содержания в ней твердой фазы. С повышением
концентрации серной кислоты скорость разложения фосфата
вначале замедляется и достигает минимума, а затем снова
возрастает. В области концентраций серной кислоты,
соответствующих минимальной скорости разложения фосфата (зона
пассивации), на зернах фосфата образуется плотная
непроницаемая пленка полугидрата сульфата кальция CaSO4-О.бНгО,
очень медленно превращающегося в безводный ангидрит CaSO4.
Скорость диффузии кислоты через пленку постепенно
уменьшается, вследствие чего реакция разложения фосфата
замедляется. Это вызывает ухудшение физических свойств
суперфосфата.
При производстве суперфосфата применяют серную кислоту
более высокой концентрации, чем в зоне пассивации. В этих
условиях реакция разложения фосфата протекает с большой
скоростью, полугидрат сульфата кальция быстро превращается
в ангидрит, благодаря чему на зернах фосфата образуется
менее плотная пленка, через которую кислота легко диффунди-
52S
рует, и получается суперфосфат хорошей структуры. С
повышением концентрации применяемой серной кислоты
соответственно понижается влажность суперфосфата и повышается
содержание в нем Р2О5. Уменьшению влажности суперфосфата
на 1 % соответствует увеличение содержания в нем Р2О5
примерно на 0,2%. Однако при слишком высокой концентрации
серной кислоты скорость разложения фосфата замедляется и
ухудшаются физические свойства суперфосфата. Для природных
фосфатов различного состава (см. табл. 29) опытным путем
устанавливают соответствующие оптимальные концентрации
серной кислоты, необходимые для нормального протекания
процесса разложения данного фосфатного сырья.
При непрерывном смешении реагентов можно применять
более концентрированную кислоту, чем при периодическом
смешении, так как серная кислота поступает в прореагировавшую
суперфосфатную пульпу, находящуюся в непрерывнодействую-
щем смесителе большого объема. Содержащаяся в пульпе
свободная фосфорная кислота разбавляет серную кислоту до ее
оптимальной концентрации. В производстве суперфосфата из
апатитового концентрата по непрерывному способу применяют
68—68,5%-ную серную кислоту, по периодическому способу —
63—64%-кую.
Повышение температуры кислоты способствует ускорению
реакции разложения природного фосфата в начале процесса,
более интенсивному выделению фторсодержащих газов и
большему испарению воды, т. е. снижению влажности суперфосфата.
Однако при очень высокой температуре, как и при слишком
высокой концентрации H2SO4, ухудшаются физические свойства
суперфосфата. Обычно вводят серную кислоту при
температуре 60—70 °С. Следует иметь в виду, что более высокой
температуре кислоты должна соответствовать меньшая
концентрация ее (в допустимых пределах).
Тонина помола фосфатов значительно влияет на
скорость их разложения. Мелкие фракции измельченного сырья
разлагаются с большой скоростью, а крупные частицы (80—
100 мк) разлагаются медленно. Быстрому разложению
мелких частиц способствует также то, что фосфатная мука
в первую очередь реагирует с более активной серной кислотой,
во второй стадии процесса оставшиеся крупные частицы
фосфата медленно разлагаются менее реакционноспособной
фосфорной кислотой. Кроме тонины помола фосфатов, на
скорость их разложения влияет также физическая структура
природного сырья, которая характеризуется его общей удельной
поверхностью. При одинаковой степени измельчения многие
фосфориты, обладающие высокоразвитой общей удельной
поверхностью, химически более активны, чем апатиты, имеющие
низкую общую удельную поверхность. Фосфориты разных ме-
34-2848 529
сторождений имеют различную общую удельную поверхность.
С возрастанием тонины помола и общей удельной поверхности
фосфата повышается степень его разложения и быстрее
протекает дозревание суперфосфата. Соответственно снижается
расход серной кислоты и фосфата на 1 г усвояемой Р2О5 в
суперфосфате и повышается качество удобрения.
Однако с повышением тонины помола фосфата уменьшается
производительность и повышается стоимость размольных
агрегатов, а также увеличивается расход электроэнергии на
измельчение. Поэтому для каждого вида природного фосфата
практически устанавливают оптимальную тонину помола, т. е. более
экономически выгодную степень измельчения данного сырья.
Применяемые для производства суперфосфата апатитовый
концентрат и фосфоритную муку из месторождений Кара-Тау
обычно измельчают до тонины помола, соответствующего не
более 14% остатка на сите с отверстиями размером 0,15 мм.
Перемешивание. Важным условием наиболее полного
разложения природных фосфатов является интенсивность и
продолжительность перемешивания их с серной кислотой. При
разложении апатита необходимо более длительное E—6 хин) и
интенсивное перемешивание, чем для фосфоритов A—2 мин).
Интенсивное перемешивание достигается в смесителях
специальной конструкции, снабженных мощными, быстроходными
мешалками.
Схема производства суперфосфата. В СССР все
суперфосфатные заводы получают фосфатное сырье в измельченном виде.
Производство суперфосфата раньше осуществлялось
периодическим способом. Отдельные порции фосфатной муки и
серной кислоты после перемешивания в смесителе загружали в
суперфосфатную камеру, где масса постепенно твердела
(схватывалась). По истечении 0,5—2 ч суперфосфат выгружали из
камеры и передавали в отделение доразложения. Затем снова
заполняли суперфосфатную камеру.
В настоящее время преимущественное распространение
получил более совершенный, непрерывный способ производства
суперфосфата. На некоторых заводах реакционные отделения
реконструированы частично: дозирование и перемешивание
реагентов производится непрерывно, а суперфосфатные камеры
работают периодически. Такая схема производства является
промежуточной между непрерывной и периодической схемами
и иногда называется полунепрерывной.
Непрерывный способ производства суперфосфата имеет
существенные достоинства. Вследствие применения более
концентрированной серной кислоты и несколько более полного
разложения фосфата получается суперфосфат лучшего качества,
причем на его дозревание требуется меньшее время. Кроме того,
снижается расход серной кислоты, повышается степень исполь-
530
зованик фосфатного сырья и фтора и значительно облегчаются
условия труда.
На рис. 200 изображена схема непрерывного производства
суперфосфата. Фосфатную муку, поступающую на
суперфосфатный завод в крытых железнодорожных вагонах, выгружают при
помощи электромеханических лопат в приемный бункер S,
откуда по ленточному транспортеру 3 ковшовым элеватором в
на абсорбци/о
нейтрализующая
JU_ добавка
Сулерроарагл
Готовый /
суперсроссрат
Рис. 200. Схема непрерывного производства суперфосфата:
/—воздуходувка; 2—аэрожелоб; 3, 5, 9, 18, 22. 25—транспортеры; 4—силосы; 5—ковшовый
элеватор: 7—лебедка; а, 21—бункеры; /0—шнек; 11 — весовой ленточный дозатор;
/^—суперфосфатный смеситель; ^—суперфосфатная камера; И—карусель; IS—центральная
(разгрузочная) труба; 16—напорный бак; 17—кислотный смеситель; 19— разбрасыватель; 20—грейфер
мостового крапа; 23— грохот; 24—валкоьая дробилка.
и транспортером 5 ее направляют на хранение в
железобетонные силосы 4. Чтобы мука при хранении не слеживалась и не
зависала, в бункеры силосов вмонтированы пористые плитки,
через которые по мере надобности подают сжатый воздух.
Из силосов фосфатная мука по аэрожелобу 2, элеватором 6
и далее транспортером 9 и шнеком 10 подается в бункер
весового ленточного дозатора И. Ленточный дозатор непрерывно
подает фосфатную муку в суперфосфатный смеситель 12. Для
проверки правильности дозировки муки на весах установлен
34*
531
контрольный бункер, в который периодически направляется
фосфатная мука (на рисунке не показано).
Серная кислота из напорного бака 16, снабженного
змеевиком для охлаждения кислоты водой в случае надобности,
непрерывно поступает в кислотный смеситель 17, где разбавляется
водой до концентрации 63—68,5% H2SO4 (в зависимости от
температуры кислоты и вида перерабатываемого фосфата).
Температура и концентрация серной кислоты после ее
разбавления, а также количество кислоты, поступающей в
суперфосфатный смеситель за единицу времени, регулируются с
помощью трех приборов — регуляторов температуры, концентра-
томера и расходомера. Эти приборы, будучи настроены на
заданный режим, работают автоматически и взаимосвязанно.
Достаточно совершенный прибор для автоматического
регулирования заданного соотношения фосфатной муки и серной
кислоты еще не разработан, поэтому ограничиваются
автоматическим дозированием муки и кислоты по отдельности.
В суперфосфатном смесителе 12 фосфатная мука
перемешивается в течение нескольких минут с серной кислотой.
Образующаяся при смешении густая сметанообразная пульпа
(температура пульпы ПО—115°С) непрерывно поступает в
суперфосфатную камеру 13, где продолжается начавшаяся в
смесителе реакция разложения фосфата серной кислотой. Камера
медленно вращается. За один оборот в течение 1 ч пульпа в
камере успевает настолько затвердеть (схватиться), что
образующуюся суперфосфатную массу можно выгружать из
камеры путем срезания тонких слоев массы ножом карусели 14,
расположенной внутри камеры. Срезанный рассыпающийся
суперфосфат падает в центральную трубу 15 камеры и
ленточным транспортером 18 подается в отделение доразложения
(склад). С транспортера суперфосфат падает на
разбрасыватель 19, разбивающий комки суперфосфата. При этом часть
влаги, содержащейся в суперфосфате, испаряется, а
суперфосфат охлаждается.
С помощью грейфера 20 мостового крана суперфосфат
укладывают в кучи высотой 6—8 м, где выдерживают 6—20 и более
суток. В течение этого времени суперфосфат с помощью того
же грейфера несколько раз перелопачивают для охлаждения
и обновления поверхности, что ускоряет доразложение фосфата.
Выделившиеся из смесителя 12 и суперфосфатной камеры 13
газы, содержащие фтористый кремний, вентилятором
отсасываются в абсорбционную систему, где SiF4 поглощается водой
с образованием раствора кремнефтористоводородной кислоты.
Вызревший суперфосфат с помощью грейфера 20 мостового
крана подают в бункер 21, откуда суперфосфат поступает по
ленточному транспортеру 22 на грохот 23. Для нейтрализации
избыточной свободной кислоты, содержащейся в суперфосфате.
532
на транспотер 22 подают костяную или фосфоритную муку
(нейтрализующая добавка) в количестве 2—4% от веса
суперфосфата. Прошедший через грохот суперфосфат поступает на
передвижной транспортер 25, снабженный разбрасывателем,
который распределяет суперфосфат по железнодорожному
вагону. Комки суперфосфата, не прошедшие через грохот,
измельчаются в валковой дробилке 24 и также направляются на
транспортер 25.
Суперфосфатный смеситель непрерывного действия (рис. 201)
представляет собой горизонтальный прямоугольный аппарат.
Рис. 201. Суперфосфатный смеситель
действия:
непрерывного
/—сообщающиеся цилиндры; 2—мешалки; 3—переливная коробка;
4—шибер; 5—входной штуцер.
Корпус смесителя состоит из нескольких (трех-четырех)
сообщающихся цилиндров / и переливной коробки 3. По оси
каждого цилиндра подвешены лопастные мешалки 2. Окружная
скорость на концах вращающихся лопастей 6—7 м/сек.
Фосфатная мука и серная кислота через входной штуцер 5 в
крышке аппарата поступают в один конец смесителя. Образующаяся
533
пульпа протекает по всей длине смесителя и через переливную
коробку 3 поступает в суперфосфатную камеру. Между
последней по движению пульпы мешалкой и переливной коробкой
установлен чугунный шибер 4, регулирующий уровень пульпы
в смесителе.
Стальной корпус суперфосфатного смесителя футеруют
кислотоупорным кирпичом. Мешалки и шибер выполняют из
чугуна. Для защиты от коррозии мешалки обматывают стальной
проволокой или асбестовым шнуром и покрывают диабазовой
замазкой.
Суперфосфатная камера непрерывного действия. Из
суперфосфатных камер непрерывного действия наиболее
распространена вращающаяся цилиндрическая камера (рис. 202). Она
представляет собой вертикальный железобетонный цилиндр /
со стальным кожухом и бетонным днищем 2. Камера вращается
вокруг внутренней неподвижной чугунной трубы 5,
установленной на фундаменте. Труба прочно соединена с неподвижной
бетонной крышкой 9 камеры и с неподвижным поперечным
чугунным щитом 4, расположенным внутри камеры и
подвешенным к крышке. В местах соприкосновения с днищем и
стенкой камеры к щиту прикреплены гибкие листы специальной
стали, которые при вращении камеры скользят по стенкам и
днищу, отгибаясь и создавая уплотнение.
Внутри камеры, на вертикальном валу подвешен механизм 7
для вырезки суперфосфата (карусель), вращающийся в
направлении, противоположном вращению камеры. Стальным
кольцом, прикрепленным к днищу, камера опирается на
фундамент. При вращении камеры опорное кольцо катится по
роликам. Вращение камеры осуществляется при помощи
электродвигателя через червячную передачу.
Крышка камеры служит рабочей площадкой. На ней
установлены смеситель для фосфатной муки и серной кислоты и
электродвигатель с приводом (на рисунке не показаны).
Уплотнение между крышкой и вращающейся камерой достигается
при помощи резиновой прокладки. Зазор между днищем камеры
и центральной трубой 5 уплотнен специальным сальником
В крышке камеры имеется отверстие для отсоса фторсодержа-
щих газов.
Камера работает следующим образом. Густая
суперфосфатная пульпа, выходящая из смесителя, поступает в кольцевое
пространство через отверстие в крышке камеры возле
неподвижного щита 4 и перемещается вместе с корпусом камеры.
Суперфосфатный пирог при этом скользит вдоль стенки
центральной неподвижной трубы 5. Скорость вращения камеры
должна быть такой, чтобы за время ее полного оборота
суперфосфат достаточно затвердел и был готов для вырезки.
Созревший к концу оборота камеры суперфосфат срезается ножами (>
534
п подается скребками 8 от периферии в центральную трубу о.
Под трубой проходит ленточный транспортер 3, который подает
суперфосфат на склад. Производительность камеры диаметром
1 м достигает 50 т/ч суперфосфата.
| Cyneptpoctpam
Рис. 202. Суперфосфатная камера непрерывного действия:
/—цилиндр; 2—днище; S—транспортер; 4— неподвижный поперечный
щит; 5—центральная (разгрузочная) труба; 6—срезающие ножи;
7—карусель: 6—скребки; .«—неподвижная крышка.
Выход готового продукта (суперфосфата) составляет 1,9—
1,95 т на 1 т апатитового концентрата и 1,6—1,8 т на 1 т
фосфорита. Расход фосфатного сырья на 1 т усвояемой Р_2О5,
содержащейся в суперфосфате, колеблется в пределах 1051 —1070 кг
РгО5 в сырье при переработке апатитового концентрата и 1100—
1120 кг Р2О5 в сырье при переработке фосфоритов Кара-Тау.
Расход серной кислоты на 1 т усвояемой Р2О5 также зависит от
химического состава исходного фосфата. При переработке апа-
535
титового концентрата он составляет 1790—1840 кг, при
переработке фосфоритов Кара-Тау — около 2500 кг H2SO4.
Непрерывный способ производства суперфосфата можно
осуществлять с некоторыми изменениями описанной выше
принципиальной схемы. Так, на ряде заводов фосфатная мука
хранится не в силосах, а в складах, заполняемых путем
сбрасывания муки с верхнего продольного ленточного транспортера
через телескопические трубы. Из таких складов фосфатная мука
подается в реакционное отделение скреперами и затем
элеваторами и ленточными транспортерами. Однако при заполнении
и разгрузке складов такими способами образуется много пыли,
что ухудшает условия труда.
Схемы непрерывного производства суперфосфата
различаются также по оформлению устройств, дозирующих фосфатную
муку и серную кислоту, по конструкции суперфосфатной
камеры и способу дообработки суперфосфата. Для
транспортирования суперфосфата вместо грейферов иногда применяются
подвесные самоходные вагонетки, а для перелопачивания и
погрузки суперфосфата — экскаваторы.
Характерным технико-экономическим показателем
производства суперфосфата является степень разложения фосфата,
отражающая полноту перехода Р2О5 исходного сырья в
усвояемую форму. Степень разложения выражается отношением
процентного содержания усвояемой РгО5 к общему содержанию ее
в суперфосфате и составляет (в %):
Суперфосфат
Сырье из камер товарный
Апатитовый концентрат - 85—87 93—96
Фосфоритная мука Кара-Тау 82—84 84—-89
Качество суперфосфата согласно существующим стандартам
должно соответствовать следующим требованиям:
Из апатитового Из фосфорит-
концентрата ной муки
высший 1-й Кара-Тау
сорт сорт
Содержание, %
Р2О5 усвояемая, не менее . . . 19,5 19,0 14,0
влага, не более 12,0 13,0 15,0
свободная кислота (в пересчете
на Р2О5), не более 5 5 5,5
В суперфосфате, полученном из апатитового концентрата по
непрерывному способу, содержание усвояемой Р2О5 составляет
20% и более."
Суперфосфат должен быть рассыпчатым п не слеживаться
в плотные или мажущиеся комки.
Обычно суперфосфат перевозят навалом в крытых
железнодорожных вагонах, оборудованных плотными щитами,
закрывающими обе двери.
536
ГРАНУЛИРОВАННЫЙ СУПЕРФОСФАТ
Обычный суперфосфат плохо рассевается сеялками
вследствие некоторой пластичности и способности комковаться и
слеживаться. Это отрицательное свойство суперфосфата в
настоящее время устраняется путем переработки его в
гранулированный продукт, состоящий из небольших сухих гранул — частиц
размером примерно 1—4 мм.
Гранулированный суперфосфат имеет и агрохимические
преимущества, особенно при применении его на кислых почвах,
богатых окислами железа и алюминия. Мелкие частицы негра-
нулированного суперфосфата полнее соприкасаются с почвой,
вследствие чего значительная часть содержащейся в них водно-
растворимой Р2О5 легко реагирует с окислами и образует
труднорастворимые фосфаты, которые плохо усваиваются
корнями растений. Воднорастворимая РгО5 в гранулированном
суперфосфате, состоящем из более крупных частиц, не
фиксируется так быстро почвой, и корневая система растений
успевает извлечь из удобрения большую часть воднорастворимой
Р2О5. Кроме того, гранулированный суперфосфат можно вносить
в почву при помощи рядовых сеялок равномерно и на требуемую
глубину, а также в междурядные борозды или одновременно
с семенами.
Таким образом, гранулирование приводит к увеличению
эффективности действия суперфосфата, и в настоящее время все
большие количества суперфосфата выпускаются в виде
гранулированного продукта.
Схема производства гранулированного суперфосфата
изображена на рис. 203.
Готовый суперфосфат (после дозревания) предварительно
смешивают с небольшим количеством молотого известняка или
мела для нейтрализации свободной кислоты до содержания ее
1—2,5% в готовом продукте (в пересчете на Р2О5).
Нейтрализованный суперфосфат дозируется ленточным питателем 3 в
барабанный гранулятор (агломератор) 4. Одновременно в гра-
нулятор поступает также мелкая фракция (отсев)
гранулированного суперфосфата после классификации на грохотах. В гра-
нуляторе смесь увлажняется до содержания около 16% влаги
при помощи специальных форсунок, тонко разбрызгивающих
воду и позволяющих регулировать степень увлажнения. В
барабанном грануляторе в течение 8—9 мин происходит
укатывание суперфосфата. Кроме барабанных, применяют также
тарельчатые грануляторы. Из гранулятора суперфосфат в виде
сырых гранул разного размера поступает в барабан 5,
обогреваемый топочными газами, где продукт подсушивается до
содержания 3% влаги. В результате высушивания содержание
усвояемой Р2О5 в суперфосфате повышается.
537
При нагревании суперфосфата может произойти частичная
ретоградация Р2О5, поэтому сушка ведется в
низкотемпературном режиме прямотока высушиваемых гранул и топочных
газов. Температура гранулированного суперфосфата не должна
превышать 85°С. Из сушильного барабана выходят сухие
твердые гранулы различных размеров.
нейтрализованный i~~l
cj/neptpoapam 7
¦у у iv v iffl
Водо
Рис. 203. Схема производства гранулированного суперфосфата:
/ -транспортер; 2, 15—бункеры; 3—ленточный питатель; 4—гранулятор; 5—сушильный барабан;
6—шнек; 7—циклоп; S—грохот; 9—элеватор; 10—валковая дробилка; //— вентилятор;
12—мокрый пылеуловитель (скруббер); 13—транспортер; 14—абсорбционная башня; 15—брызгоулови-
тель; П—автоматические весы; 18—зашивочная машина.
Газы, выходящие из сушильного барабана, поступают в
сухой циклон 7 для грубой очистки от пыли и далее
вентилятором И, через мокрый скруббер—пылеуловитель 12,
абсорбционную башню 14 и брызгоуловитель 15 отводятся в атмосферу.
Для получения суперфосфата в виде гранул определенного
размера высушенный гранулированный продукт подается
элеватором 9 на двухеитный вибрационный грохот 8, где разделяется
на три фракции. Крупная фракция (частицы крупнее 4 мм), не
прошедшая через верхнее сито, поступает в валковую
дробилку 10 и затем снова возвращается на грохот. Мелкая фракция
(частицы менее 1 мм), прошедшая через второе сито,
возвращается шнеком 6 в бункер гранулятора. Мелкая фракция может
быть использована также как особый сорт сухого
суперфосфата. Средняя фракция, представляющая собой товарный
продукт, передается транспортером 13 в бункеры 16,
продуваемые воздухом для охлаждения гранул. При помощи
автоматических весов 17 и зашивочной машины 18 гранулированный
суперфосфат расфасовывается в многослойные битумирован-
ные мешки или прорезиненные бумажные мешки. Упакованный
суперфосфат развозится автопогрузчиками по складу и
укладывается з штабели или погружается в железнодорожные вагоны.
538
При точном регулировании влажности продукта и
нормальном ведении технологического процесса готовый гранулирован
ный суперфосфат имеет достаточно однородный
гранулометрический состав, при этом достигается незначительный отсев
мелкой фракции.
Содержание усвояемой РгО5 в гранулированном
суперфосфате из апатитового концентрата составляет 19,5—21%, в
суперфосфате из фосфоритной муки Кара-Тау — около 14%.
Описанный метод производства гранулированного
суперфосфата характеризуется высокой производительностью н
позволяет получать прочные гранулы заданного размера при разной
степени нейтрализации продукта. Однако этому методу
присуши некоторые недостатки: значительное выделение пыли
при просеве, дроблении и транспортировании гранулированного
суперфосфата, выделение фторсодержащих газов при сушке
продукта. Эти газы продолжают выделяться и при хранении
гранулированного суперфосфата, что приводит к разрушению
тары, в которую упаковано удобрение. К недостаткам
метода следует отнести также необходимость увлажнения шихты
при гранулировании и затраты топлива на испарение
увлажняющей воды. В связи с перечисленными недостатками
данного метода целесообразно проводить гранулирование
суперфосфата без сушки. При этом на гранулирование подается
камерный суперфосфат, обладающий высокой кислотностью. Кислые
гранулы обрабатывают легкоразлагаемыми фосфатами,
например тонко измельченным обесфторенным фосфатом (стр. 550).
в результате получается нейтральный продукт в виде
достаточно прочных гранул, не требующих сушки.
АММОНИЗИРОВАННЫЙ СУПЕРФОСФАТ
Аммонизированный суперфосфат получают путем обработки
суперфосфата аммиаком.
Аммиак взаимодействует прежде всего со свободной
фосфорной кислотой, содержащейся в суперфосфате, затем с моно-
кальцийфосфатом, образуя моноаммонийфосфат и дикальций-
фосфат:
NH3 -;- H3POj = NH4H2PO4
NH3 -r Ca(H2PO4)., --= NH4H,PO4 -f GjHPOj
В результате этих реакций часть воднорастворимой РгО5 пе-
[К'ходнт в цнтратнорастворимую форму (дикальцийфосфат);
количество усвояемой Р2О5 при этом не уменьшается.
При аммонизации вводят 2—3% аммиака от веса
суперфосфата. Более высокое содержание азота в аммонизированном
суперфосфате (около. 7%) достигается в тех случаях, когда для
яммонизашш применяются аммиакаты (стр. 576).
539
Аммонизированный суперфосфат не мажется, хорошо рас-
севается, не содержит свободной кислоты и не выделяет фтора,
а потому не оказывает разрушающего действия на тару и
сеялки. Аммонизированный суперфосфат представляет собой
высокоэффективное удобрение и благодаря отсутствию в нем
свободной кислоты может являться ценным компонентом смешанных
удобрений, особенно если в смеси присутствует аммиачная
селитра.
Аммонизации следует подвергать лишь вызревший
суперфосфат. При аммонизации недозревшего суперфосфата дораз-
ложение фосфата в нем приостанавливается. При
нейтрализации аммиаком суперфосфат нагревается до 65—70 °С и из него
испаряется влага в количестве, примерно равном количеству
введенного аммиака. Во время постепенного охлаждения
аммонизированного суперфосфата из жидкой фазы медленно
выкристаллизовываются фосфаты кальция, магния и аммония,
вследствие чего продукт несколько схватывается. Чтобы
предотвратить такое схватывание, аммонизированный суперфосфат во
время хранения перелопачивают несколько раз или
подсушивают до содержания в нем 2—4% влаги. При такой влажности
аммонизированный суперфосфат совсем не схватывается.
Схема производства аммонизированного суперфосфата
представлена на рис. 204. Вызревший суперфосфат подается
грейферным краном / в бункер 2, снабженный колосниковой
решеткой, которая задерживает крупные куски продукта. Из бункера
суперфосфат подается ленточным питателем на
разбрасыватель 3 барабанного типа. Измельченный разбрасывателем
суперфосфат подается ленточным транспортером 4 в бункер 5
с ленточным дозатором, непрерывно питающим аммонизатор 6,
который представляет собой горизонтальный вращающийся
барабан, установленный с небольшим уклоном.
Жидкий аммиак из хранилищ поступает в испаритель 9.
Отсюда газообразный аммиак непрерывно подается через диа-
фрагменный дозатор в аммонизатор 6 под слой суперфосфата.
В передний торец аммонизатора непрерывно засасывается
воздух вентилятором 7. При прохождении через аммонизатор
воздух уносит с собой испаряющуюся влагу. Суперфосфатная пыль,
уносимая из аммонизатора, улавливается в циклоне 8.
Выходящий из аммонизатора суперфосфат проходит второй
разбрасыватель 3, измельчающий комки, и охлаждается на
транспортере 4. Далее аммонизированный суперфосфат
поступает на промежуточное хранение, где подвергается
перелопачиванию, либо направляется в сушильный барабан //.
Перелопаченный или подсушенный и охлажденный аммонизированный
суперфосфат (холодильная труба на рисунке не показана)
поступает на вибрационный грохот 12 с отверстиями размером
4 мч в сите. Частицы продукта, не прошедшие через сито,
5*0
Рис. 204. Схема производства аммонизированного суперфосфата:
/—грейферные краны; 2, S—бункеры; 3— разбрасыватели; 4—ленточные транспортеры; б—аммонизатор; 7—вентилятор;
8—циклоп; 9—испаритель; 10—элеваторы; //—сушильный барибан; 12—внбрационнный грохот; 13—валковая дробилка.
размалываются в вадковой дробилке 13 и возвращаются на
грохот. Аммонизированный суперфосфат упаковывают в битуми-
рованные бумажные мешки и автопогрузчиком отправляют на
склад или подают на погрузку в железнодорожные вагоны.
Использование фтора
при производстве суперфосфата
Газы, выделяющиеся в процессе производства
суперфосфата из апатитового концентрата, содержат около 6 кг фтора в
виде S1F4 на 1 т полученного удобрения.
Улавливание фтора из отходящих промышленных газов
необходимо как по санитарным требованиям*, так и по
экономическим соображениям, поскольку фторсодержащие газы могут
служить источником фтора, соединения которого находят
разнообразное все возрастающее применение во многих отраслях
народного хозяйства.
На суперфосфатных заводах фторсодержащие газы
перерабатывают главным образом на кремнефтористый натрий Na2SiFe,
который раньше широко применялся для борьбы с вредителями
сельскохозяйственных культур. К настоящему времени в связи
с появлением других более эффективных препаратов,
преимущественно хлорорганических (стр. 329), кремнефтористый
натрий утратил свое значение в качестве инсектицида и
используется преимущественно в производстве кислотоупорных бетона
и замазок, при фторировании бетона**, в производстве эмали
и стекла и др. На некоторых суперфосфатных заводах из крем-
нефтористого натрия получают фтористый натрий,
употребляемый в основном в качестве антисептика для пропитки
древесины, предохраняющей ее от гниения. Небольшие количества
фторсодержащих газов используются для производства
криолита, кремнефтористого аммония и кремнефтористого калия,
намечено также производство других солей (кремнефториды
магния и цинка, фториды алюминия и кальция).
В настоящее время изыскиваются также рациональные пути
получения фтористоводородной кислоты из отходящих
фтористых газов.
Общее содержание фтора в газах, выделяющихся из
суперфосфатной камеры, составляет 15—35 г/ж3, температура газов
на выходе 65—75°С.
* По санитарным нормам, содержание соединений фтора в
воздушном бассейне вблизи химических предприятии не дочжно превышать
0,03 .мг/л!3 в пересчете на элементарный фтор.
** Фторирование бетона — обработка газообразным фтористым кремнием
бетонных изделии для увеличения их механическом прочности и стойкости
в агрессивных средах. Кремнефтористый натрии применяется в данном
случае для получения сухого концентрированного фтористого кремния.
542
Если в газах присутствуют пары воды, то при охлаждении
газов до температуры ниже 60 °С фтористый кремний
взаимодействует с водой и образуются кремнефтористоводородная и
кремневая кислоты:
3SiF4 + 4Н2О = 2H2SiFG + SiO.r2H2O
Кремнефтористоводородная кислота получается в жидком
виде, кремневая — в виде студенистых хлопьев.
Поглощение фтора. Для поглощения фтористого кремния
фторсодержащие газы и водяные пары, выделяющиеся в
суперфосфатных смесителе и камере, отсасываются в абсорбционную
систему хвостовым вентилятором, создающим в
суперфосфатной камере разрежение порядка 15—20 мм вод. ст.
В
атмосферу
из
суперфосфатной
кам
Рис. 205. Схема поглощения фтористых газов:
/— абсорбционная камера; 2—абсорбционная башня; 3—брызгоулоиитель; 4-
вентилятор.
Процесс абсорбции фтористых газов проводится в две
ступени (рис. 205). Основное количество фтора (около 90%)
поглощается в полой кирпичной камере /, снабженной
разбрызгивающими валками, частично погруженными в жидкость.
Остальное количество фтора улавливается в полон стальной
башне 2, орошаемой при помощи форсунок. Камера и башня
изнутри выложены полиизобутиленом и поверх него
футерованы кислотоупорным кирпичом на андезитовой замазке.
Выходящие из абсорбционной башни газы проходят брызгоулови-
тель 3 и отводятся через выхлопную трубу в атмосферу.
В башню 2 подается свежая вода в количестве,
соответствующем образованию 10%-ной кремнефтористоводородной
кислоты. Для создания надлежащей плотности орошения башня
орошается «на себя». Концентрация
кремнефтористоводородной кислоты, циркулирующей в башне, составляет всего 0,1- —
0,2%. Избыток кислоты, соответствующий количеству вводимой
свежей воды, непрерывно подается в абсорбционную камеру 1.
Вытекающая из нее 10%-ная кремнефтористоводородная кнсло-
га направляется на переработку.
543
Переработка фтора. Из осветленного путем отстаивания
раствора кремнефтористоводородной кислоты осаждают кремнефто-
ристый натрий насыщенным раствором хлористого натрия:
H2SiF6 + 2NaCl = Na2SiF6 + 2НС1
При этом 90—95% образующегося кремнефтористого натрия
выпадает в виде мелкокристаллического осадка.
Отделение кремнефтористого натрия от жидкости
производят декантацией и центрифугированием. Выходящий из
центрифуги кремнефтористый натрий содержит 10—14% влаги и
высушивается до содержания не более 1 % влаги.
Кремнефтористый натрий представляет собой мелкокристаллический
порошок белого цвета.
Выход готового кремнефтористого натрия составляет 8—9 кг
на 1 г суперфосфгта.
Вырабатываемый суперфосфатными заводами
кремнефтористый натрий должен иметь следующий состав (в %):
Na2SiF6, не менее 95,0
Свободная кислота (в пересчете на НС1), не
более 0,15
Влага, не более 1
Остаток на сите с отверстиями 0,06 мм
(тонина помола), не более 15
Технический кремнефтористый натрий упаковывают в
выложенные изнутри многослойной бумагой фанерные барабаны
емкостью 20—50 л или в деревянные бочки емкостью 40—75 л.
Путем взаимодействия кальцинированной соды с кремнефто-
ристым натрием
Na2SiF6 + 2Na2CO3 = 6NaF + SiO3 + 2CO2
на суперфосфатных заводах получают фтористый натрий.
Этот процесс, осуществляемый содово-суспензионным
методом, проводят при 70:—80 °С и перемешивании содовой
суспензии с кремнефтористым натрием. Фтористый натрий отделяют
от маточного раствора на центрифуге. Маточный раствор,
содержащий до 3,5% NaF, используется для растворения соды,
таким образом процесс ведется в замкнутом цикле.
Высушенный фтористый натрий содержит 70—75% NaF и 20—25%
примеси SiO2. При необходимости получения высокопроцентного
фтористого натрия (более 94% NaF) двуокись кремния удаляют
из продукта путем растворения ее в растворе едкого натра.
Образующееся жидкое стекло (стр. 662) отделяется от осадка
фтористого натрия, который промывают водой и высушивают.
544
ДВОЙНОЙ СУПЕРФОСФАТ
Двойной суперфосфат — концентрированное воднораствори-
мое фосфорное удобрение, получаемое разложением молотого
природного фосфата фосфорной кислотой*. Подобно простому
суперфосфату, двойной суперфосфат содержит воднораствори-
мую Р2О5 главным образом в виде монокальцийфосфата, а
также некоторого количества свободной фосфорной кислоты, но,
в отличие от простого суперфосфата, он не содержит примеси
сульфата кальция. Двойной суперфосфат имеет такое название
потому, что он получается в результате двукратного
разложения природного фосфата: одна (большая) часть природного
фосфата разлагается серной кислотой или восстанавливается до
элементарного фосфора для получения экстракционной или
термической фосфорной кислоты, другая (меньшая) часть
природного фосфата разлагается фосфорной кислотой, в результате
чего получается данное удобрение — двойной суперфосфат.
В зависимости от качества природного фосфата и метода
его переработки двойной суперфосфат содержит 42—48%
усвояемой Р2О5 (из них 85—95% в воднорастворимой форме), т. е.
в 2,5—3 раза больше усвояемой Р2О5, чем простой суперфосфат;
поэтому в некоторых зарубежных странах двойной
суперфосфат называют также тройным суперфосфатом.
Вследствие высокого содержания питательных веществ в
двойном суперфосфате его целесообразно применять даже при
необходимости перевозки на дальние расстояния, а также при
внесении в почву путем распыливания с самолетов.
Основные реакции разложения природных фосфатов
фосфорной кислотой можно выразить следующими уравнениями:!
Cas(PO4KF + 7Н3РО4 + 5Н2О = 5[Са(Н2РО4J-Н2О] + HF
СаСОз + 2Н3РО4 = Са(Н2РО4J-Н2О + СО,
Са, Mg) (COSJ + 4Н3РО4 + Н2О = Са(Н2РО4J ¦ Н2О + Mg(H2PO4)a • 2Н,О + 2СО9
F&A + 2Н3РО4 + Н,0 = 2[FePO4-2H,O]
А1аОв + 2Н3РО4 + Н,О = 2[А1РО4-2Н2О]
Содержащаяся в природных фосфатах двуокись креиния
связывается с HF, образуя кремнефтористоводородную
кислоту, и частично выделяется в виде газообразного SiF4.
Количество (норма) фосфорной кислоты, необходимое для
разложения природного фосфата, может быть рассчитано по
приведенным выше уравнениям. Кроме того, должна быть
учтена и степень нейтрализации фосфорной кислоты (особенно
экстракционной) примесями. В этих случаях количество фосфорной
* Продукт, получаемый разложением природного фосфата смесью
серной и фосфорной кислот, называют обогащенным суперфосфатом
35-2848 545
кислоты следует рассчитывать не по содержанию в ней Р2О5, a
по содержанию свободных (первых) водородных ионов кислоты,
способных вступать в реакции*. Степень разложения
природного фосфата возрастает с уменьшением размера его частии
и повышением концентрации фосфорной кислоты. Повышение
температуры несколько ускоряет реакции разложения
природного фосфата. Практически, в зависимости от состава
природного фосфата, степени его измельчения и других факторов,
оптимальные технологические условия процесса (количество,
концентрация, температура фосфорной кислоты, интенсивность
и длительность смешивания реагентов, длительность
схватывания суперфосфата и др.) устанавливают опытным путем.
Двойной суперфосфат выпускают в виде гранулированного
продукта для непосредственного применения в качестве
удобрения и в виде порошкообразного вещества, применяемого для
производства концентрированных смешанных удобрений.
Различают два основных метода производства двойного су
перфосфата. Схема производства двойного суперфосфата по
первому методу аналогична схеме непрерывного производства
суперфосфата (см. рис. 200, стр 531) с дозреванием продукта
на складе. По этому методу применяют концентрированную
фосфорную кислоту; например, для разложения апатитового
концентрата употребляют кислоту, имеющую концентрацию
54—57% Р2О5, т. е. термическую или упаренную
экстракционную фосфорную кислоту. Степень разложения природного
фосфата такой кислотой составляет 87—97%. Гранулирование
двойного суперфосфата осуществляют по такой же схеме, как
и простого суперфосфата (стр. 537 ел.).
Главными недостатками схемы производства двойного
суперфосфата с дозреванием продукта на складе являются:
неполная непрерывность процесса и необходимость упаривания эк
стракционной фосфорной кислоты.
Экстракционная фосфорная кислота, получаемая из необо
тащенных фосфоритов Кара-Тау, бывает настолько загрязнена
примесями (магний и др.), что ее нецелесообразно упаривать
и, следовательно, применять для производства двойного
суперфосфата по данной схеме. Схему с дозреванием продукта на
складе применяют для производства двойного суперфосфата из
трудноразлагаемых природных фосфатов, содержащих
относительно небольшое количество примесей, например из апатито
вого концентрата.
По второму методу процесс производства двойного
суперфосфата полностью осуществляется по непрерывной схеме, от-
* Количество свободного первого водородного иона и фосфорной
кислоте определяют титрованием ее шелочью в присутствии индикатора бром-
крезолзеленого или метилового желтого.
546
падает необходимость длительного дозревания продукта на
складе, продукт получается в гранулированном виде. Этот метод
производства двойного суперфосфата называют поточным
методом. По поточному методу можно применять фосфорную
кислоту низкой концентрации: неупаренную B5—32% Р2О5) или
слегка упаренную (до 38% Р2О5).
Схема производства двойного суперфосфата по поточному
методу изображена на рис. 206. Разложение фосфатной муки
фосфорной кислотой осуществляется в двух последовательно
расположенных реакторах 4, обогреваемых паром. Пульпа,
выходящая из второго реактора, смешивается в горизонтальном
двухвальном смесителе-грануляторе 5 с тонко измельченным
высушенным продуктом (ретур) и в виде сырых гранул
поступает в прямоточный сушильный барабан 7. Высушенный
гранулированный материал рассевается на двухситном
вибрационном грохоте 9 на три фракции, например +4 мм (крупная),
—4-^+0,5 мм (средняя) и —0,5 мм (мелкая). Мелкая фракция
(ретур) возвращается в смеситель-гранулятор. Средняя
фракция делится на два потока: часть, соответствующую заданной
производительности цеха, нейтрализуют молотым известняком
во вращающемся барабане 12 и затем направляют на
расфасовку и упаковку.
Большая часть средней фракции измельчается в молотковой
дробилке 10 н присоединяется к ретуру.
Существенным недостатком поточной схемы является
необходимость дробления, рассева н транспортирования
значительного количества ретура, которое в зависимости от типа
обрабатываемого фосфатного сырья составляет 4,5—10 вес. ч. на
1 вес. ч. готового продукта; при этом необходимо устройство
довольно громоздкой пылеулавливающей системы. Для умень
шения количества ретура разработана несколько
видоизмененная поточная схема: большая часть пульпы поступает из
второго реактора в дисковую распылительную сушилку,
высушенный материал смешивается с остальной частью пульпы в
горизонтальном смесителе, смесь высушивается в сушильном
барабане. Затем высушенный материал подвергается рассеву и
обработке, аналогичной описанной выше.
При производстве двойного суперфосфата по поточному
методу из трудноразлагаемых природных фосфатов, например
из апатитового концентрата, степень разложения апатита
фосфорной кислотой на 20—25% ниже, чем по методу с
дозреванием суперфосфата на складе. По поточной схеме получают
двойной суперфосфат главным образом из относительно легко
разлагаемых природных фосфатов и неупаренной фосфорной
кислоты.
Двойной суперфосфат можно аммонизировать так же, как
простой суперфосфат.
35*
547
Liu
Рис. 20G. Схема произнодства двойного суперфосфата по поточному методу:
бункеры; 7—дозаторы; Л—напорный бак (а—расходомер); 4—реакторы, обогреваемые паром; 5—смеситель-гранулятор;
шьлжи,- 7—сумиильный барабан; 8-элеваторы; 9—грохот; 10—молотковая дробилка; //—ленточный^транспортср;
12— нейтрализационный барабан.
Расходные коэффициенты на 1 т двойного суперфосфата:
Экстракционная фосфорная кислота (из апатитового
концентрата), ш Р2О5 0,34—0,36
Фосфоритная мука (эстонские, егорьевские, вятские
фосфориты), m 0,4—0,48
известняк, m . . . . . 0,02—0,05
ПРЕЦИПИТАТ
Преципитатом* называется фосфорное удобрение, состоящее
в основном из дикальцийфосфата СаНРО4-2Н2О. Преципитат,
полученный из фосфоритов Кара-Тау, содержит также
некоторое количество димагнийфосфата MgHPO4-3H2O. В
преципитате пятиокись фосфора РгО5 находится в цитратнорастворимой
форме и потому его применяют только для основного
предпосевного внесение.
Преципитат представляет собой сухой пылящий белый
порошок, не слеживающийся, не комкующийся н не
гигроскопичный. Преципитат получается при взаимодействии фосфорной
кислоты с молотым известняком и известковым молоком по
реакции:
ЗН3РО4 + 2СаСО3 = СаНРО4-2Н2О + Са(Н3РО4J + 2СО.,
г садок ргствор
Са(Н4РО4).л + Ca(OH)z = 2СаНРО4-2Н2О
осадок
По некоторым схемам для осаждения дикальцийфосфата
применяют лишь молотый известняк.
Осадок дикальцийфосфата отделяют от раствора
фильтрованием и высушивают.
Так как отделение от раствора осуществляется
фильтрованием или декантацией, то важно получить
крупнокристаллический осадок. Для получения крупных кристаллов известковое
молоко следует медленно вводить в реакционную среду и
поддерживать температуру реакции 60°С и рН раствора не выше
4,6—4,8. При осаждении известняком кристаллическая
структура преципитата значительно улучшается, но осаждение
происходит медленнее. Присутствие полуторных окислов ухудшает
кристаллическую структуру преципитата.
Для производства преципитата целесообразно использовать
фосфорную кислоту, полученную разложением фосфатов
дешевой соляной кислотой, являющейся отходом других
производств (стр. 398). Солянокислотная экстракция фосфатов
значительно проще сернокислотной экстракции, так как побочно
образующийся при этом хлористый кальций, в отличие от гипса.
'¦ Слово «приципитат» п переводе означает осадок. Такое название
¦'игноесо данному удоОреишо потом\. что оно получается путем осажде-
- 1-я hj раствора.
переходит в раствор фосфорной кислоты, вследствие чего отпа
дает сложная операция отфильтровывания фосфогипса. После
отделения преципитата фильтрованием пульпы хлористый
кальций остается в растворе.
Производство преципитата может быть осуществлено и на
основе разложения фосфатов азотной кислотой. После отфиль
тровывания преципитата раствор нитрата кальция нейтрализуют
аммиаком и упаривают, при этом из него кристаллизуется
кальциевая селитра. Однако на основе продуктов азотнокислот-
ного разложения природных фосфатов целесообразнее
изготовлять не преципитат и кальциевую селитру, а сложные
удобрения (стр. 591 ел.). Для получения чистого дикальцийфосфата,
применяемого для подкормки животных (кормовой
преципитат), лучше использовать более дорогую, но зато более чистую
термическую фосфорную кислоту.
Качество преципитата определяется содержанием в нем ци-
тратнорастворимой Р2О5. Технический продукт, в зависимости
от состава исходного сырья и метода его переработки, в раз
личной степени загрязнен другими солями и содержит 33—40%
цитратнорастворимой Р2О5. Чистый двуводный дикальцийфос-
фат содержит 41,27% РгО5.
Готовый преципитат должен полностью проходить через
сито с отверстиями размером 2 мм. Преципитат упаковывают в
четырехслойные бумажные мешки.
ОБЕСФТОРЕННЫЕ ФОСФАТЫ
Обесфторенный фосфат — высокоэффективное удобрение
при основном внесении в дерново-подзолистые почвы, оно
эффективно также на черноземных почвах. В обесфторенных
фосфатах РгО5 находится в основном в лимоннорастворимой
форме и частично в цитратнорастворимой форме.
Обесфторенные фосфаты являются продуктами
гидротермической переработки природных фосфатов, т. е. действия на них
паров воды при высокой температуре и в присутствии
небольшого количества кремнезема. Сущность гидротермического
процесса заключается в следующем. Фторапатит
взаимодействует с водяным паром при 1450—1550°С, в результате
происходит замещение иона фтора гидроксилом с образованием
гидроксилапатита;
Са5(РО4)зР + Н2О = Са6(РО4KОН -f HF
Нестабильный гидрокенлапатит при температуре выше
1440 °С распадается на три- и тетракальцийфосфат:
2Са5(РО4KОН = 2Са3(РО4)а + Са4Р3О9 + Н2О
В присутствии кремнезема образуются лимоннорастворимые
силикофосфаты различного состава в зависимости от
соотношения между фосфатом и кремнеземом.
550
Реакцию между гидроксилапатитом и кремнеземом можно
выразить следующей схемой:
2nCa6(PO4KOH + mSiO2 = иН,О +
где п и т — переменные величины, зависящие от состава
исходной шихты.
Большинство природных фосфатов содержит кремнезем в
количестве, достаточном для этого процесса. При недостатке
кремнезема его вводят в шихту в виде измельченного песка.
Так, при получении обесфторенного фосфата из апатитового
концентрата к нему добавляют 2—3% песка.
Обесфторенные фосфаты могут быть получены путем
спекания или плавления. По методу спекания следует применять
тугоплавкую шихту во избежание чрезмерного оплавления
материала в печи. Тугоплавкой является, например, шихта из
апатитового концентрата с небольшой добавкой кремнезема. В
процессе производства обесфторенных фосфатов методом спекания
из фосфоритов Кара-Тау в шихту необходимо вводить около
40% известняка для повышения температуры ее плавления.
Поскольку карбонаты при высоких температурах процесса
разлагаются с выделением двуокиси углерода, вес известняковой
добавки при спекании уменьшается почти вдвое, благодаря
чему не происходит значительного снижения содержания Р2О5
в готовом продукте.
При получении обесфторенных фосфатов по методу
плавления целесообразнее применять легкоплавкую шихту, чтобы
избежать необходимости ведения процесса при слишком высоких
температурах.
При нормальном ведении процесса в обесфторенном фосфате
остается очень небольшое количество фтора. Весь полученный
продукт практически растворим в 0,4%-ной соляной кислоте,
т. е. легко усваивается животными. Поэтому обесфторенный
фосфат применяют не только как удобрение, но и для
подкормки скота и птицы. При содержании фтора более 0,2%
применение обесфторенных фосфатов в качестве кормовых средств
недопустимо.
Схема производства обесфторенных фосфатов методом
спекания апатитового концентрата изображена на рис. 207.
Шихта—смесь апатитового концентрата с небольшим количеством
измельченного песка увлажняется до содержания 12—14% воды
и подается в горизонтальную вращающуюся печь 8,
установленную с небольшим уклоном и отапливаемую мазутом,
поступающим с противоположной стороны печи. Для отопления печи
можно использовать и другое водородсодержащее топливо,
например природный газ. При сжигании такого топлива
образуются пары воды, необходимые для обесфторнвания фосфата.
Температура факела пламени в печи 1520—1560°С, температура
551
Ши~Ш
' ФильтроНач-
пая еода
Рис. 207. Схема производства обесфтореннык фосфатов методом спекания:
.'—элеваторы; 2— бункеры; S— шнековые питатели; 4—шиековый смеситель; S—сиеснтвль-увлажиитвль; в—электрофильтр; 7—скрубберы; в—обжиге-
пия прч1.: 9—шноки; 10— Еентиляторы; //—пластинчатые транспортеры; 12—грохот; IS—дробилка; И—витатель; 16— шарогая мельница! /S—«бсорО
ционные башни; 17—брызгоуловнтель; IS—«втоыатическне весы: /S--зашивочная машина: 20—рукавный фильтр.
материала в зоне спекания несколько ниже, температура газов
на выходе из печи 400—800 °С.
В зоне спекания и охлаждения клинкера печь футерована
чромомагнезитовым кирпичом, а в остальной части — высоко-
глиноземистым кирпичом (стр. 628).
Клинкер обесфторенного фосфата, выходящий из печи,
охлаждается и измельчается в шаровой мельнице 15 до тонины,
соответствующей прохождению частиц через сито с отверстиями
размером 0,!5 мм (допускается остаток на сите в количестве
5%). Готовый продукт упаковывается в бумажные мешки.
Выходящие из печи газы, содержащие фтор в основном в
знде HF и частично в виде SiF4, после охлаждения в полом
скруббере-испарителе 7 очищаются от пыли в электрофильтре 6
и поступают в систему абсорбционных башен 16 с хордовой
насадкой, в которых почти полностью улавливается фтор.
Получаемая в результате абсорбции смесь фтористоводородной и
кремнефтористоводородной кислот может быть использована
для получения синтетического фторида кальция.
При производстве обесфторенных фосфатов из апатитового
концентрата степень обесфторивания составляет 96—99%>
количество РгО5, переходящей в лимоннорастворимую форму,
достигает 90% и более.
Состав готового продукта (в %):
Р2О5 лимоинорастворимая до 36
Р„О5, растворимая в 0,4%-иой НС1 . 38иболее
СаО —52
Фтор . 0,1—0,2
Обесфторенные фосфаты можно также получать методом
плавления в шахтных печах; в последнее время осваивается
применение для этой цели циклонных печей.
Метод плавления особенно эффективен для
обесфторивания фосфоритов Кара-Тау и других месторождений, так как при
этом отпадает необходимость добавления в шихту известняка,
что дает возможность не только удешевить производство, но
и повысить содержание Р2О5 в продукте. При !450—1500 °С
расплав фосфоритов Кара-Тау обладает невысокой вязкостью
и хорошей текучестью. Степень обесфторивания фосфоритов
Кара-Тау составляет 96—98%, степень перехода Р2О5 в
лимоннорастворимую форму 90—95%.
Из фосфоритов Кара-Тау, содержащих 26,5% Р2О5,
получается обесфторенный плавленый фосфат примерно
следующего состава (в %):
Р,О5 лимоинорастворимая 26—28
Р,О5, растворимая в 0,4%-ной НС) ... 27—29
Фтор . . 0,05—0,15
553
Техника безопасности в производстве
фосфорных удобрений
Источниками травматизма и профессиональных заболеваний
в производстве фосфорных удобрений может явиться пылеоб-
разование при разгрузке и транспортировке фосфатного сырья
и различных порошкообразных добавок (молотый известняк,
мел), проникание в воздух тонкодисперсной пыли получаемого
удобрения, выделение фторсодержащих газов. Кроме того, при
неосторожном обращении с кислотами, горячей пульпой,
содержащей серную и фосфорную кислоты, *а также с горячим
суперфосфатом или другим фосфорным удобрением,
выгруженным из аппаратуры, возможны химические и термические
ожоги.
Фосфатная пыль, выделяющаяся при выгрузке фосфоритной
муки и апатитового концентрата из железнодорожных вагонов,
при сбрасывании фосфатного сырья на склад и
транспортировании по цеху, не токсична, но вызывает раздражение
слизистых оболочек и кожи и увеличивает восприимчивость к
заболеваниям дыхательных органов. Запыленность воздуха в
рабочей зоне производственных помещений не должна превышать
2 мг/м3.
Фторсодержащие газы, выделяющиеся при разложении
природных фосфатов, весьма вредны для организма, вызывают
сильное раздражение слизистых оболочек, действуют на
центральную нервную систему, вызывают, удушье и другие
неприятные симптомы. Наибольшее выделение газов возможно при
нарушениях технологического процесса и авариях, а также
при транспортировании горячего удобрения, например
камерного суперфосфата на склад. Предельно допустимая
концентрация фтористого водорода в воздухе рабочего помещения не
должна превышать 0,5 мг/м3.
Важнейшими условиями создания нормальных и безопасных
условий труда в производстве фосфорных удобрений являются
тщательная герметизация аппаратуры, оборудования и
коммуникаций, механизация и автоматизация процессов, связанных
с выделением пыли, вредных газов и паров, устройство мощной
хорошо работающей местной и общей вентиляции и
поддержание ее в полной исправности. Для уменьшения пылевыделений
в некоторых случаях прибегают к увлажнению фосфатного
сырья перед подачей его в производственный корпус. Для
увлажнения в элеватор подается небольшое количество воды
(летом) или острого пара (зимой). Чтобы предотвратить про
никание фторсодержащих газов в воздух рабочих помещений,
в аппаратах для смешения фосфатного сырья с кислотами
необходимо поддерживать небольшое разрежение, следить за
надежностью и исправностью соответствующих уплотнений, си-
554
схематически очищать и промывать газоходы. Площадки для
обслуживания аппаратуры, в которых проводится кислотное
разложение фосфатов, должны быть оборудованы приточно-
бытяжной вентиляцией.
Индивидуальными средствами работающих служат проти-
вопылевые респираторы, фильтрующие и изолирующие
противогазы марки БКФ (с белой полоской), в некоторых случаях
шланговые противогазы, а также предохранительные очки. Для
предотвращения химических и термических ожогов необходимо
пользоваться шерстяной спецодеждой, резиновыми обувью и
перчатками и защитными очками. При попадании кислоты на
кожу следует немедленно смыть кислоту большим количеством
воды и обратиться в медпункт. Мерой первой помощи при
отравлении фтористым водородом является ингаляция содовым
раствором.
В помещениях с химически агрессивной средой
устанавливается электрооборудование в закрытом исполнении с
химически стойкой изоляцией, в помещениях с пыльной средой должны
устанавливаться закрытые пыленепроницаемые
электродвигатели и другое электрооборудование в таком же исполнении.
Электродвигатели, которые обслуживают механизмы и аппараты,
работающие в непрерывном потоке, должны быть
сблокированы для обеспечения пуска оборудования в определенной
последовательности потока и, при необходимости, быстрой
остановки его.
3. АЗОТНЫЕ УДОБРЕНИЯ
До того как был осуществлен в промышленности прямой
синтез аммиака из азота и водорода, в качестве азотных
удобрений применяли лишь сульфат аммония, натриевую и
кальциевую селитру ,и цианамид кальция. В дальнейшем по мере
развития азотной промышленности стали производить и
другие азотные удобрения: аммиачную селитру, сульфат-нитрат
аммония, калиевую селитру, мочевину и т. д.
В данном разделе рассматриваются процессы производства
важнейших азотных удобрений.
АММИАЧНАЯ СЕЛИТРА
Аммиачная селитра, или нитрат аммония, NH4NO3
представляет собой белое кристаллическое вещество, содержащее около
35% азота в аммиачной и нитратной формах. Аммиачная
селитра существует в нескольких кристаллических модификациях,
устойчивых в определенных температурных интервалах.
Превращение одной модификации в другую сопровождается
значительным изменением объема и тепловым эффектом.
Аммиачная селитра хорошо растворяется в воде и весьма
гигроскопична. При содержании до 0,1% влаги негранулиро-
555
ванная аммиачная селитра слеживается незначительно, но с
повышением влажности и изменением температуры слежи-
ваемость ее увеличивается. При изменении температуры
происходит увлажнение или подсыхание продукта. Подсыхание
вызывает кристаллизацию соли из ее насыщенного раствора на
поверхности частиц, что способствует сцеплению кристаллов.
Изменение температуры может вызвать перекристаллизацию
аммиачной селитры, т. е. переход из одной кристаллической
модификации в другую.
Сухая аммиачная селитра может взрываться под действием
сильного детонатора; влажная соль, содержащая более 3% вла
ги, не взрывает. Многолетняя практика применения аммиачной
селитры показала, что при соблюдении определенных условий
хранения, перевозки и обращения возможность взрыва может
быть исключена. Аммиачная селитра не чувствительна к
толчкам, ударам, трению и не самовоспламеняется. Пыль аммиач»
ной селитры с примесями органических веществ взрывоопасна.
Аммиачная селитра — очень распространенное
концентрированное и эффективное азотное удобрение универсального
действия. Она применяется на всех почвах и под все культуры как
для основного внесения, так и для подкормки растений.
Аммиачная селитра используется также для производства
взрывчатых веществ, применяемых при горно-взрывных работах.
Впервые аммиачная селитра была широко применена в
качестве азотного удобрения в СССР и в последующем получила
большое распространение в качестве азотного удобрения и &
других странах.
Аммиачная селитра образуется в результате процесса ней
трализации — взаимодействия аммиака с азотной кислотой:
Обычно для производства аммиачной селитры применяют
газообразный аммиак и 47—49%-ную азотную кислоту. В
случае употребления жидкого аммиака последний
предварительно испаряют на специальной установке.
Реакция образования нитрата аммония протекает с
выделением значительного количества тепла, вследствие чего
возникает опасность разложения азотной кислоты и азотнокислого
аммония с образованием окислов азота. Чтобы предотвратить
потери HNO3 и NH4NO3, необходимо отводить или использовать
выделяющееся тепло. Поэтому процесс получения аммиачной
селитры ведут с использованием тепла реакции для испарения
воды. Реакцию нейтрализации азотной кислоты аммиаком
целесообразно проводить в слабокислой среде (с некоторым
-избытком Н1МОз) для уменьшения потерь азота.
На рис. 208 изображена схема производства аммиачной
селитры с использованием тепла нейтрализации. По этой схеме
556
Pacmffop
NH4NO}
fc4
/?Op($G-T?}-^ .
=•=-.== ./.¦¦ _A , -«.-4
/S
i ///ill И\
/ / I | I i и \
Рис. 208. Схема производства аммиачной селитры;
/—неПтрализаюи (аппарат ИТН); 2—донейтрализатор; 3, 12, 17—напорные баки; *—выпарной аппарат первой ступени; 5, //. Я-сепар»-
торы; б-баромртрнческие конде!!саторы; 7-промежуточный сборник (гидравлический затвор); в-барометрические ящики; 9-
-вакуум-насосы; Ю—расширитель конденсата; 13—выпарной аппарат второй ступени; 15—желоб для плава; 16—вентилятор; /«—разбрызгиватель
плава (гранулятор); 19—грануляционная башня; 20—ленточный транспортер.
централизацию азотной кислоты аммиаком проводят при а,
мосферном давлении; тепло нейтрализации используют для
упаривания образующихся растворов аммиачной селитры.
Газообразный аммиак и азотная кислота поступают в
централизатор / (аппарат ИТН*), изготовленный из специальной
стали; азотную кислоту с добавками, способствующими
уменьшению слежизаемости аммиачной селитры (стр. 560), вводят во
внутренний цилиндр этого аппарата. Образующийся
слабокислый раствор нитрата аммония переходит во внешний
цилиндр аппарата. Тепло реакции используется для
концентрирования раствора. Выделяющийся соковый пар может быть
использован для испарения жидкого аммиака и обогрева
выпарных аппаратов.
Процесс нейтрализации полностью автоматизирован, что
позволяет поддерживать заданную кислотность раствора
(избыток азотной кислоты колеблется в пределах 1—2 г/л).
Автоматическое управление осуществляется регулятором давления
аммиака, регуляторами-стабилизаторами количества аммиака
и клапанами, корректирующими подачу азотной кислоты по
импульсам от ячейки рН-метра.
Раствор аммиачной селитры, поступающий на последующее
упаривание, должен быть нейтральным (во избежание
коррозии выпарных аппаратов и потерь азота). Поэтому из
нейтрализатора раствор поступает в периодически действующие
аппараты 2 (донейтрализаторы), куда для окончательной
нейтрализации вводят аммиак при интенсивном перемешивании.
Раствор аммичной селитры, содержащий менее 0,5 г/л
избыточного аммиака и примерно 64% NH4NO3, перекачивают из до-
нейтрализатора в напорный бак 3. Отсюда раствор самотеком
поступает в непрерывнодействующий выпарной аппарат 4,
изготовленный из нержавеющей стали.
Чтобы уменьшить расход свежего греющего пара,
упаривание растворов амм;иачной селитры ведут в многокорпусных
выпарных аппаратах (стр. 377 ел.) в две, рейсе в три ступени, с
использованием на первой ступени выпарки тепла сокочого пара
из аппаратов ИТН и из расширителя конденсата второй
ступени выпарки. Для возможности использования сокового пара
первую стадию выпарки проводят при разрежени:: 600—
650 мм рт. ст.
Малоконцентрированные растворы аммиачной селитры,
поступающие в первую ступень, обычно упаривают в аппаратах
пленочного типа, в которых тонкий слой раствора движется с
большой скоростью по внутренней поверхности трубок,
обогреваемых снаружи паром.
При входе в трубку раствор закипает и постепенно образует -
* Начальные буквы слов: «использование тепла нейтрализации».
558
ся парожидкостная эмульсия, поступающая из трубок в
сепаратор 5 для отделения сокового пара, который конденсируется
затем в аппарате 6.
Раствор, упаренный в первой ступени до концентрации 82—¦
84% NH4NO3, через сборник 7 перекачивается на вторую
ступень выпарки, проводимую при разрежении 500—550 мм рт. ст.
в горизонтальных аппаратах 13, обогреваемых свежим греющим
паром давлением не свыше 9 ат. Во второй ступени раствор
упаривается до состояния плава (98—98,5% NH4NO3),
соковый пар отделяется от капель жидкости в сепараторах 14 и //.
причем часть жидкости через сборник 7 возвращается на
упаривание. Конденсат г'рею'цсто пара поступает в расширитель 10
и .используется для получения пара, дополнительно
подаваемого на обогрев выпарных аппаратов первой ступени.
При использовании в процессе нейтрализации более
концентрированной азотной кислоты E6—58% HNO3) получается
раствор аммиачной селитры, содержащий 82—84% NH4NO3, кото*
рый можно упаривать до состояния плава в одну ступень.
Концентрированный плав аммиачной селитры из сепаратора
поступает на кристаллизацию, которая в данном процессе яв*
ляется весьма важной операцией, определяющей качество
готового продукта. Для уменьшения слеживаемости аммиачной
селитры кристаллизацию проводят таким образом, чтобы
получался продукт в виде гранул. Плав через желоб 15 поступает в
центробежный гранулятор 18, установленный в верхней части
башни 19 (диаметр 16 м, вь сота цилиндрической части 25 м),
и разбрызгивается по всему ее сечению. Капли плава при
падении вниз охлаждаются, закристаллпзовываются и слегка
подсушиваются встречным потоком воздуха, который засасывается
вентилятором 16 через отверстия в нижней части башни.
Отсюда гранулы поступают на ленточный транспортер 20,
подающий продукт на склад.
На некоторых заводах кристаллизацию высококонцентриро-
панного плава аммиачной селитры проводят на поверхности
охлаждаемых изнутри вращающихся барабанов. Образующаяся
на наружной поверхности барабана корка соли толщиной 1 —
1,2 мм срезается ножом. Срезаемая соль представляет собой
агрегаты кристаллов, плотно связанных друг с другом в виде
чешуек, которые слеживаются гораздо больше, чем гранулы.
Гранулированная аммиачная селитра состоит из гранул
округлой формы размером 1—3 мм: чешуйчатая — из частиц
размером 1—5 мм.
Для уменьшения слеживаемости чешуйчатого продукта его
сушат во вращающихся барабанах воздухом, нагретым в кало
рифере до 105—110°С. Можно проводить сушку охлажденным
воздухом с одновременным охлаждением аммиачной селитры.
Гранулированную аммиачную селитру, содержащую обычно не
559
более 1,5% влаги, не сушат и направляют непосредственно на
упаковку.
На получение 1 г аммиачной селитры расходуется около
0.21 т аммиака и 0,8 т азотной кислоты (считая на 100%
NH3 и НМОз).
Для уменьшения слеживаемости аммиачной селитры при
хранении стремятся к тому, чтобы температура гранул на
выходе из башни была возможно более низкой. Для этой цели
предложено, например, охлаждать гранулы до 30—35 °С в кипя
щем (псевдоожиженном) слое в нижней части
грануляционной башни. Слеживаемость аммиачной селитры уменьшают
также в процессе ее изготовления путем добавления в аппарат
ИТН нитратов кальция и магния. Для этого вместе с азотной
кислотой в аппарат вводят небольшое количество пульпы,
полученной азотнокислотным разложением природных фосфатов,
доломита или известняка. Эти добавки вводят из расчета со
держания в готовом продукте 0,5—1% Р2С>5 или 0,5—1%
СаО + MgO.
На некоторых заводах слеживаемость аммиачной селитры
устраняют путем добавления в плав аммиачной селитры 1,5%
метасиликата кальция или 10%-ного водного ра'стзора
красителя амаранта @,03% от веса сухой аммиачной селитры).
В настоящее время установлено, что процесс нейтрализа
ции азотной кислоты аммиаком можно проводить в таких
условиях, чтобы все реакционное тепло использовалось на испаре
ние воды из раствора с непосредственным получением сухой
аммиачной селитры без выпарки. Этот так называемый без-
упарочный процесс проводится в трубчатом
реакторе-нейтрализаторе при температуре до 230°С и стехиометрическом
соотношении реагентов — аммиака и 59—60%-ной азотной кислоты.
Образующийся в этих условиях 98%-ный плав аммиачной
селитры находится в реакторе очень непродолжительное время и
потому не успевает разложиться под действием высокой
температуры. Далее плав продувается горячим воздухом для почти
полного удаления влаги и поступает на кристаллизацию.
Можно проводить безупарочный процесс, под давлением 3,5—4,5 ат
с использованием тепла сокового пара (из нейтрализатора) для
подогрева реагентов и дополнительным обезвоживанием плава
в вакуум-испарителе до содержания 99% HN4NO3.
Аммиачную селитру выпускают трех сортов: сорт А — мел
кокристаллическая, сорт Б — чешуйчатая и кристаллическая и
сорт В — гранулированная и чешуйчатая. Сорта А и Б
предназначены для применения в промышленности, сорт В — в
качестве азотного удобрения (оно должно быть
рассыпчатым).
Качество аммиачной селитры должно удовлетворять требо
заниям, приведенным на стр. 561.
560
Содержание NH4NO3 (в пересчете на
сухое вещество), %, не менее . .
Влажность, %, не более
Реакция
Сорта
99,5 99,5
0,5 | 0,8
Нейтральная
97,0
1,5
Кислотность
менее 0,02%
в пересчете
на HNO,
Аммиачную селитру упаковывают в бумажные пятислойные
дитумированные мешки весом по 42—48 кг и перевозят любым
видом транспорта.
Из удобрений на основе аммиачной селитры следует
отметить известково-аммиачную селитру — сплав или смесь
аммиачной селитры с молотым известняком или мелом.
Это удобрение особенно эффективно на кислых почвах, так
как содержащаяся в нем известь нейтрализует вредную для
растений избыточную кислоту в почве. При сплавлении
получается более однородный гранулированный неслеживающийся
продукт. В промышленности мзвестково-аммиачную селитру
изготовляют только методом сплавления. Методом смешения
известково-аммиачную селитру можно приготовить на местах
потребления.
Для получения гранулированной швестково-аммиачной
селитры плав, упаренный до концентрации 95% NH4NO3,
смешивают с молотым известняком или мелом и с пылевидной извест-
ково-аммиачной селитрой, частично образующейся в процессе
этого производства. Далее известково-аммиачную селитру,
имеющую консистенцию плава, подают при температуре
125—-130 °С в верхнюю часть грануляционной башни, где плав
разбрызгивается (см. рис. 208).
Для получения однородных гранул требуемого размера B—
5 мм)^ и для уменьшения слеживаемости селитры
гранулированный продукт подвергают дроблению и рассеву, охлаждают
воздухом и припудривают известковой пудрой.
Готовый продукт содержит 18—20,5% азота.
В некоторых зарубежных странах находят известное
применение в качестве удобрений также смеси и сплавы аммиачной
селитры с сульфатом аммония (сульфат-нитрат аммония) или
с хлористым калием (калийно-аммиачная селитра).
Техника безопасности в производстве
аммиачной селитры
В производстве аммиачной селитры при неисправности an
паратуры, отклонении от нормального технологического режима
и авариях возможно выделение вредных газов и паров (аммиак,
36-2848
пары азотной кислоты, окислы азота). Предельно допустимые
концентрации токсичных газов и паров в воздухе производствен
ных помещений в цехах аммиачной селитры составляют не
более 0,02 мг/л NH3 и не свыше 0,005 мг/л окислов азота (в
пересчете на N2O5)- Кроме того, при повышении температуры
или в присутствии некоторых примесей (свободная кислота,
хлориды, органические вещества) может произойти термическое
разложение аммиачной селитры и ее растворов, протекающее
также с выделением вредных газов и паров и даже со
взрывом.
Заметное разложение чистой аммиачной селитры
начинается при 200°С; в присутствии некоторых примесей или кислот
температура разложения аммиачной селитры понижается.
В отделениях выпарки и нейтрализации возможны
проливы и разбрызгивание горячих растворов и плава, что может
привести к ожогам.
Ожоги могут произойти также при пропаривании, разборке
и чистке трубопроводов и аппаратов. Во время
транспортирования, охлаждения и упаковки аммиачной селитры выделяется
пыль.
Основными мерами защиты от вредных воздействий в
производстве аммиачной селитры являются: герметизация
аппаратуры и коммуникаций, автоматизация технологических
процессов, устройство общей и местной приточно-вытяжнон и
естественной вентиляции, теплоизоляция горячих поверхностей ап
паратов и трубопроводов, пользование средствами
индивидуальной защиты (противогазы, респираторы, защитные очки,
спецодежда, спецобувь, перчатки, рукавицы и др.).
Во избежание взрыва не следует допускать нагревания
аммиачной селитры или ее растворов выше установленной
регламентом температуры и загрязнения их посторонними
примесями.
НАТРИЕВАЯ И КАЛИЕВАЯ СЕЛИТРА
Химически чистый нитрат натрия NaNO3 представляет собой
бесцветные прозрачные кристаллы и мало гигроскопичен.
Однако техническая натриевая селитра при хранении в
неблагоприятных условиях иногда слеживается.
Нитрат натрия применяется для производства калиевой се
литры, нитрита натрия, нитрата бария и других солей,
используется в пищевой промышленности как консервирующее
средство, а также в производстве стекла и в процессах закалки
стали.
Значительная часть вырабатываемой натриевой селитры
применяется в качестве азотного удобрения, особенно
эффективного при внесении под сахарную свеклу, на которую натрий
оказывает положительное влияние.
562
Природная натриевая селитра добывается в Чили.
Синтетический нитрат натрия может быть получен нейтрализацией азот
ной кислоты содой. Однако чтобы не расходовать готовую
азотную кислоту, в промышленности натриевую селитру
обычно получают в качестве побочного продукта при производстве
азотной кислоты — путем щелочной абсорбции хвостовых ни-
трозных газов (стр. 274).
Полученные при этом растворы (так называемые нит'рит-ни-
тратные щелока) обрабатывают азотной кислотой для
превращения (инверсии) NaNO2 в NaNO3. Инвертированный раствор
нейтрализуют содой и направляют последовательно на вакуум-
выпарку, кристаллизацию, центрифугирование и сушку
воздухом.
При абсорбции нитрозных газов раствором едкого кали или
известковым молоком могут быть получены соответственно
калиевая селитра или кальциевая селитра (стр. 275).
Возможно также получение нитрата натрия (или калия)
методом катионного обмена, который заключается в
пропускании раствора кальциевой селитры через аппараты,
заполненные катионитом (стр. 36 ел.), поглощающим ,из раствора ионы
кальция:
Ca(NO3J + 2Na [Кат.] - > 2NaNO3 -f Ca [Кат.]2
Техническая натриевая селитра выпускается двух сортов.
Продукт 1-го сорта, применяемый для технических целей,
должен содержать не менее 99% NaNOe и не более 0,02% NaNO;
и других окисляемых веществ и не свыше 1,5% влаги. В
продукте 2-го сорта, используемом как азотное удобрение,
содержание NaNO3 должно быть не менее 98% A6,1% N),
содержание нитрита натрия и других окисляемых веществ должно быть
не более 0,25%, влаги до 2% (содержание NaNO3 дано в
пересчете на сухое вещество). Натриевую селитру упаковывают
в деревянные бочки или в бумажные пятислойные битумиро-
ванные мешки.
Наиболее распространенный метод получения калиевой
селитры, являющейся эффективным безбалластным
негигроскопичным удобрением, — конверсионный метод, т. е. обменное
разложение нитрата натрия и хлорида калия:
NaNO3 + КС1 ^^ KNO3 + NaCl
Выпадающий в условиях процесса конверсии осадок
хлорида натрия отфильтровывают, а раствор направляют на
кристаллизацию из него нитрата калия.
Нитрат калия широко применяется также для технических
целей — в производстве черных (дымных) порохов, стекла, в
пищевой промышленности, в пиротехнин. Выпускаются три
36* 563
сорта калиевой селитры, различающиеся по содержанию
нитрата калия и примесей. Продукт 3-го сорта, применяемый в
сельском хозяйстве, должен содержать не менее 98% KNO3 и не
более 2% влаги. Калиевую селитру упаковывают в деревянные
бочки, фанерные барабаны, деревянные ящики, изнутри
выложенные бумагой, или в многослойные бумажные мешки.
КАЛЬЦИЕВАЯ СЕЛИТРА
Нитрат кальция, или кальциевая селитра, существует в виде
безводной соли Са (N03b или в виде ди-, три- и тетрагидрата
Наиболее устойчива при обычной температуре четырехводная
соль Ca(NO3J-4H2O.
Кальциевую селитру с добавкой около 5% аммиачной
селитры выпускают в виде гранул или чешуек. Содержание азота
в таком продукте составляет 15,5—17%, влаги — около 13,5%.
Кальциевая селитра весьма гигроскопична и способна
слеживаться, аммиачная селитра снижает гигроскопичность
кальциевой селитры. В герметичной таре, особенно в гранулированном
виде, кальциевая селитра не слеживается.
Кальциевая селитра применяется в качестве удобрения на
кислых почвах, так как содержащаяся в ней известь оказывает
полезное нейтрализующее действие на такие почвы.
Кальциевую селитру получают методом абсорбции хвостовых
нитрозных газов известковым молоком (стр. 275) и в качестве
побочного продукта пр,и разложении природных фосфатов
азотной кислотой в производстве сложных удобрений или
преципитата.
Абсорбция нитрозных газов. При абсорбции известковым
молоком окислов азота, содержащихся в хвостовых нитрозных
газах, образуется нитрит-нитратный щелок — раствор нитрита
в имтрата кальция:
Са(ОН)а -f NO + NO2 = Ca(NOa)a + НаО
2Са(ОН)а + 4NO2 = Ca{NO3)a + Ca(NO2J + 2H2O
Этот раствор поступает на инверсию азотной кислотой для
окисления нитрита в нитрат:
3Ca(NOa)a + 4HNO3 = 3Ca(NO3)9 + 2Н2О + 4NO
Выделяющиеся в процессе инверсии окислы азота
направляются в абсорбционную систему производства слабой азотной
кислоты (стр. 277).
В инвертированном растворе нитрата кальция остается
избыток азотной кислоты, которую нейтрализуют аммиаком в до-
нейтрализаторе. При этом образуется некоторое количество
аммиачной селитры, присутствие которой в кальциевой селитре
полезно. Далее раствор поступает на упаривание до содержа-
564
ния в плаве 18—20% воды. Упаривание раствора кальциевой
селитры проводится в однокорпусных или многокорпусных
выпарных установках. В присутствии аммиачной селитры
скорость кристаллизации кальциевой селитры увеличивается в
1,5—2 раза.
Кристаллизация кальциевой селитры проводится в
грануляционных башнях с получением гранулированной кальциевой
селитры или на барабанах, охлаждаемых изнутри водой, с
получением чешуйчатой кальциевой селитры. Чтобы получить
продукт в виде частиц, однородных по величине, его подвергают
рассеву и дроблению, мелкие фракции возвращают в цикл.
Перед загрузкой в тару кальциевую селитру охлаждают во
вращающемся барабане, через который продувается охлажденный
воздух.
Получение кальциевой селитры из продуктов азотнокислот-
ного разложения фосфатов. При разложении природных
фосфатов азотной кислотой образуется раствор фосфорной кислоты
и азотнокислого кальция:
Ca5(PO4KF + 10HNO, = ЗН3РО4 + 5Ca(NO3)a + HF
Применяя для нейтрализации этого кислотного раствора
различные реагенты и разные методы его переработки, можно
получать разнообразные удобрения, в том числе кальциевую
селитру.
По одному из вариантов процесса производства сложных
удобрений (нитрофоса, нитрофоски) для удаления из
кислотного раствора избытка кальция (стр. 593) кальциевую селитру
выделяют путем вымораживания. При этом раствор
охлаждается в системе кристаллизаторов, а выпавшие кристаллы
кальциевой селитры отделяются на центрифуге. Для нейтрализации
азотной кислоты, оставшейся на кристаллах, кальциевую
селитру смешивают с измельченным известняком, в результате
образуется дополнительное количество кальциевой селитры. Для
получения кальциевой селитры более высокого качества
кристаллы из центрифуги растворяют, нейтрализуют свободную
азотную кислоту аммиаком и перерабатывают далее раствор
аналогично переработке раствора, полученного при абсорбции
китрозных газов.
По другому варианту из раствора, образовавшегося при
разложении природных фосфатов азотной кислотой, выделяют
и отфильтровывают преципитат, а фильтрат, представляющий
собой 30—35%-ный раствор кальциевой селитры, нейтрализуют
аммиаком и упаривают.
При этом из раствора выкристаллизовывается обычная каль
циевая селитра.
Кальциевую селитру упаковывают в пятислойные бумажные
битумированные мешки.
565
СУЛЬФАТ АММОНИЯ
Сульфат аммония (NFLibSCu представляет собой
кристаллический тонкий порошок белого или серого цвета. Он хорошо
растворим в воде, слабо гигроскопичен, мало слеживается,
хорошо рассевается. Сульфат аммония — эффективное азотное
удобрение на почвах, насыщенных основаниями (черноземы,
сероземы, каштановые почвы), и на известкованных
подзолистых почвах.
Получение сульфата аммония основано на взаимодействии
аммиака с серной кислотой:
2NH3 + H2SO4 = (NH4JSO4
Сульфат аммония является недостаточно
концентрированным удобрением, а для его производства требуется большое
количество серной кислоты, которую целесообразнее
использовать на получение других более концентрированных удобрений.
В Советском Союзе ограничиваются получением сульфата
аммония главным образом из аммиака коксового газа на
коксохимических заводах. Такой аммиак, без тщательной очистки от
органических примесей, непригоден для производства более
концентрированного и универсального удобрения — аммиачной
селитры. Синтетический аммиак применяется для производства
сульфата аммония лишь при использовании отработанной
серной кислоты отдельных производств.
На некоторых зарубежных заводах для поглощения аммиака
из коксового газа применяют фосфорную кислоту вместо
серной. При этом получается двойное азотно-фосфорное
удобрение— диаммонийфосфат (стр. 588).
Схема производства сульфата аммония из аммиака
коксового газа изображена на рис. 209. Газ по выходе из коксовых
печей охлаждают в первичных газовых холодильниках до 25—
30 °С и эксгаустером подают в электрофильтры для очистки от
смолы и удаления части воды с растворенными в ней
аммиаком и солями аммония. Очищенный газ нагревается в
трубчатом подогревателе до 60—80 °С горячим коксовым газом и
поступает через барботажную трубу 2 в сатуратор /, заполненный
78%-ной серной кислотой.
Сатуратор представляет собой стальной сосуд с
конусообразным дном, выложенным изнутри свинцом или
кислотоупорными плитками. Сатуратор снабжен ловушкой 5 для
улавливания брызг серной кислоты из отходящих газов.
Вследствие нагревания кислоты поступающим газом и
выделения значительного количества тепла при реакции
нейтрализации в сатураторе происходит испарение воды, содержавшейся
в коксовом газе и серной кислоте. В аппарате
образуется насыщенный раствор сульфата аммония и начинается
566
выпадение его в виде кристаллов. Кристаллы собираются в
конической части сатуратора, откуда мх инжектором 3 передают
в приемник 6. Из приемника кристаллы периодически
загружают в центрифугу 7, маточный раствор возвращают через
циркуляционный бак 4 в сатуратор 1. Отфугованный продукт
поступает на склад готовой продукции.
Конденсат, образовавшийся при охлаждении коксового газа,
содержит смолу и аммиачную воду, содержащую также аммо-
Сулырат
аммония
Рис. 209. Схема производства сульфата аммония из
аммиака коксового газа:
/—сатуратор; 2—барботажная труба сатуратора; 3—инжектор;
^-циркуляционный бак; 5—ловушка; 6—приемник кристаллов;
7—центрифуга.
нийные соли. После отделения в отстойниках смолы
аммиачную воду обрабатывают известковым молоком. При этом
выделяется аммиак, который отгоняют с паром и присоединяют
к очищенному коксовому газу, поступающему в сатуратор.
Теоретический расход серной кислоты для получения 1 т
сульфата аммония составляет 0,742 г (считая на 100% H2SO4).
практически расход серной кислоты составляет 0,750—0,752 г
H2SO4, расход аммиака достигает 0,3 т.
Производство сульфата аммония из синтетического аммиака
принципиально не отличается от производства его из аммиака
коксового газа. В этом случае исключаются лишь операции,
связанные с очисткой коксозого газа, и применяются сатураторы
больших размеров.
Сульфат аммония может быть получен из отхода
производства капролактама—растворов, содержащих до 33% (NH4JSO4
и около 1,5% NH4NO3. Путем вакуум-кристаллизации из таких
растворов выделяют сульфат аммония, который затем отфуго-
вывают. сушат и охлаждают.
567
Технический сульфат аммония содержит 20,5—20,8% азота
(в пересчете на сухое вещество), 0,05—0,2% свободной H2SO4
и 1 —1,5% влаги. Готовый продукт упаковывают в бумажные
мешки.
МОЧЕВИНА
Свойства и применение мочевины. Мочевину, или карбамид,
CO(NH2J можно рассматривать как амид угольной кислоты или
карбаминовой кислоты. Чистая мочевина содержит 46,6% азота
и представляет собой бесцветные кристаллы. Технический
продукт имеет белый или желтоватый цвет. Мочевина хорошо
растворяется в воде с частичным образованием карбоната аммония.
При нагревании водных растворов мочевины выше 80 °С
начинается ее гидролиз и превращение в карбамат аммония
CO(NHa)a + HgO = HaN—CO—ONH4
который в разбавленном растворе почти полностью
превращается в карбонат аммония.
При температуре выше 130 °С мочевина в водном растворе
диссоциирует на аммиак и двуокись углерода. При нагревании
водных растворов мочевины одновременно с гидролизом
происходит ее термическое разложение с выделением аммиака и
образованием биурета
2H,N—CO—NH, > HjN—CO—NH—CO—NH, + NH3
биурет
который является нежелательной примесью мочевины, так как
оказывает вредное действие на растения.
В условиях обычной температуры и влажности воздуха
мочевина негигроскопична, а при высокой относительной
влажности воздуха (95%) становится очень гигроскопичной. Слежи-
ваемость мочевины незначительна.
Благодаря высокой концентрации азота и хорошим
физическим свойствам мочевина считается одним из лучших азотных
удобрений.
Она применяется не только для внесения в почву, но и
является оптимальной формой азотного удобрения для внекормо-
вой подкормки растений (внесение на листья в виде 1%-ного
водного раствора).
Мочевину используют и как жидкое удобрение в виде ам
миакатов (стр. 576).
Весьма эффективно применение мочевины в качестве
азотсодержащей добавки к кормам жвачных животных. В рубце
желудка таких животных имеются микроорганизмы,
синтезирующие из азота мочевины и углеводов, находящихся в корме,
белки, которые затем усваиваются организмом животного.
568
В промышленности мочевину используют для получения мо-
чевино-формальдегидных полимеров (карбамидные смолы),
применяемых в производстве ценных пластических масс,
древесностружечных плит, синтетических клеев, составов для пропитки
тканей.
Мочевина употребляется также при изготовлении
фармацевтических препаратов, очистке нефтяных масел, а в
последнее время и в производстве синтетического волокна урилон.
Продукты поликонденсации мочевины с формальдегидом
начали применять также в качестве высококонцентрированного
азотного удобрения (до 40% N).
Мочевино-формальдегидные удобрения (МФУ) отличаются
длительным действием вследствие малой растворимости в
почвенной влаге.
В настоящее время мировое производство мочевины
превышает 2 млн. г в год. Значительно увеличивается производство
мочевины и в СССР.
Гранулированная мочевина предназначена для применения
в сельском хозяйстве, кристаллическая мочевина марок А и
Б — для технических целей.
Качество мочевины должно соответствовать следующим
показателям:
Содержание азота (в пересчете на
сухое вещество), %, не менее . . .
Примеси,- %, не более:
биурет
железо
свободный аммиак
сульфаты .
нерастворимые примеси
Влажность, %, не более
Техническая
марка А
46,3
0,2
Не
определяется
0,005
0,003
0,02
0,2
марка Л
46,3
0,8
0,005
0,015
0,02
0,02
1,0
Для сельского
хозяйства
46,0
1,0
1 Не опре-
1 деляется
0,3
Синтез мочевины. Производство мочевины осуществляется
путем синтеза ее из аммиака и двуокиси углерода. Синтез
протекает в две стадии, в первой стадии из аммиака и двуокиси
углерода образуется карбамат аммония:
2NHS + СОа : * H4N—CO—ONH4
В оптимальных условиях процесса реакция образования
карбамата аммония протекает почти нацело и с большой
скоростью. Во второй стадии происходит отщепление воды от карб
амата аммония и превращение его в мочевину:
HaN—CO—ONH< '—> HjN-CO—NH5 + Н,0
569
Дегидратация карбамата аммония протекает медленно и не
полностью и ускоряется лишь в жидкой фазе. Поэтому для
успешного проведения процесса синтеза мочевины необходимо,
чтобы карбамат аммония находился в расплавленном
состоянии. Оптимальными условиями процесса получения мочевины
являются: температура 185—200 °С, давление около 200 ат,
избыток аммиака не менее 100% сверх стехиометрического
количества, продолжительность процесса 1 ч. Выход мочевины при
этом составляет 60—70%.
Процесс производства мочевины состоит лз следующих
стадий: синтез мочевины из аммиака и двуокиси углерода,
дистилляция продуктов синтеза (плава мочевины) и улавливание
газов дистилляции (аммиака и двуокиси углерода), переработка
растворов мочевины в гранулированный или кристаллический
продукт. В качестве источника двуокиси углерода обычно
применяют экспанзерный газ (стр. 192), содержащий ^-95% СОг.
могут быть также использованы газы .известково-обжигатель-
ных печей (стр. 433).
Технологические схемы производства мочевины отличаются
главным образом способами улавливания и использования
газов дистилляции. Схемы, в которых не превращенные в
мочевину аммиак и двуокись углерода вновь используются для
получения мочевины, т. е. схемы с рециркуляцией непрореагировав-
ших газов, называются замкнутыми. Схемы, по которым непре-
вращенные в мочевину газы используются для получения других
продуктов (аммиачной селитры или иных солей), называют
разомкнутыми. При возвращении части газов
дистилляции в цикл синтеза мочевины производство ее
осуществляется по полузамкнутой схеме (схема с частичным рециклом
газов).
По разомкнутой схеме дистилляция плава мочевины
осуществляется в одну ступень и весь аммиак, содержащийся в газах,
отгоняемых из плава, поглощается азотной кислотой с
образованием аммиачной селитры. Этот наиболее простой и старый
способ производства мочевины отличается низким
использованием реагентов по прямому назначению, т. е. на синтез
мочевины (степень использования аммиака составляет лишь 30—
45%), и большим расходом тепла на упаривание получаемых
60%-ных растворов аммиачной селитры. При одноступенчатой
дистилляции плава на 1 т вырабатываемой мочевины приходит
ся получать 5—7,5 т аммиачной селитры, что приводит к
необходимости сочетать производство мочевины с производством
аммиачной селитры, значительно превышающим его по мощности.
Разомкнутая схема с одноступенчатой дистилляцией плава
целесообразна в том случае, когда сравнительно небольшое
производство мочевины комбинируется с мощным
производством аммонийных солей.
S70
Замкнутые схемы производства мочевины, различающиеся
по способам возвращения газов дистилляции в цикл синтеза
мочевины, более экономичны и потому находят все большее
применение. Методы переработки и использования газов
дистилляции в разных схемах различны. В одних схемах из
отходящих газов только выделяется аммиак, который далее ком-
примируется и используется для получения мочевины. По
другим схемам аммиак и двуокись углерода, содержащиеся в
газах дистилляции, используются без предварительного
разделения. По замкнутым и полузамкнутым схемам дистилляция
плава мочевины проводится в две ступени.
Схема синтеза мочевины с двухступенчатой дистилляцией
плава показана на рис. 210. Жидкий подогретый аммиак
подается в колонну синтеза 8 плунжерным насосом 3 под
давлением 200 ат. Газообразная двуокись углерода, предваритель
но очищенная от соединений серы, освобождается от
механических загрязнений в матерчатом фильтре 5, сжимается
пятиступенчатым компрессором 6 до 200 ат и под этим давлением
подается в колонну 8.
Синтез мочевины в колонне 8 проводится под давлением
180—200 ат при температуре 180—200°С и 125%-ном избытке
аммиака (сверх стехиометрического количества). В результате
взаимодействия аммиака с двуокисью углерода образуется плав
мочевины, содержащий до 35% CO(NH2J, до 35% NH3, около
20% карбамата аммония и примерно 10% воды.
По выходе из колонны синтеза плав дросселируется до
давления 25 ат и поступает в колонну .9 дистилляции первой
ступени, где отгоняется большая часть избыточного аммиака в
смеси с незначительным количеством двуокиси углерода и паров
воды. Газы первой дистилляции направляются в колонну
фракционирования 11, орошаемую жидким аммиаком и водой, где
происходит конденсация паров воды и некоторой части
аммиака и растворение аммонийных солей в аммиачной воде.
Жидкая фаза из колонны фракционирования поступает в
отделение переработки газов дистилляции или возвращается в
колонну 9, а газовая фаза, состоящая примерно на 40% из аммиака
и примесей двуокиси углерода, паров воды>и азота,
направляется в конденсаторы 12. Часть сконденсировавшегося здесь
аммиака направляется насосом 7 на орошение колонны фракцио
нирования 11, большая часть передается в сборник / и
возвращается в колонну синтеза 8.
Плав, выходящий из колонны 9 дистилляции первой
ступени, содержит 46—47% мочевины, до 25% карбамата аммония.
13—14% воды и 15—16% NH3. Этот плав дросселируется до
давления 1.2 ат и поступает в колонну 10 дистилляции второй
ступени, где происходит разложение непрореагировавшего
карбамата и других солей аммония на аммиак ,и двуокись
57!
газы т/слнтяиии
X, »а переработку
Рис. 210, Cxtv.,i
МОЧЛ1ШЫ
плава:
с двухступенчатой дистилляцией
/—сборник жидкого ciMM.i.ik::; ;?, 5- фн.'ьтры; S, "—насосет для пирокачиьапия :кидкого
аммиака; 4—подс^грератс^ь 2\'.Mnaita; ?*—компрессор для сжатия двуокиси углерода;
6'—колонна синтеза мочо-вины; 9— колонка дистилляции перьол ступени;" 10 — кслонна дистилляции
второй ступени; //—колонна фр^кциоидрое^ьня; 12—кс.пдеисагорм пммнакэ;
13—теплообменники; 14, is, 23—сборники; 16—фильтрпресс; 17—гидравлический затвор; 18—выпарной
аппарат; ti — повсвиисгииИ конденсатор; X--брьлгоуловнгсль; v/—яакгум-шсос; 22—
скруббер; 24—:ш1>_-ковай кристаллизатор.
углерода >и отгонка оставшегося избыточного аммиака и пре-
дуктов разложения. Для более полного удаления аммиака в
колонну дистилляции второй ступени подается пар.
Газы второй дистилляции содержат примерно 56—57% Nl-Ц.
32—33% СО2 и 10—11% водяных паров. Эти газы могут быть
направлены на разделение и возвращены в цикл синтеза (замк-
г'.\тая схема) и частью использованы для получения аммиачной
селитры или других продуктов (полузамкнутая схема). При
этом на 1 т мочевины получается 2—2,5 т аммиачной селитры.
т. е. значительно меньше, чем по разомкнутой схеме.
Из дистилляционной колонны 10 второй ступени вытекает
раствор мочевины, содержащий 65—85% CO(NH2h, который
отстаивается в сборнике 14 от примесей масла и затем
передается на переработку. Этот раствор можно непосредственно
использовать в качестве жидкого азотного удобрения. Переработка
раствора мочевины заключается в освобождении его от
механических примесей на фильтрпрессе 16, упаривании и
последующей кристаллизации продукта из упаренного раствора.
При получении кристаллической мочевины упаривание рас-
твсра проводится в одну ступень при разрежении 500 мм рт. ст..
а процесс кристаллизации продукта — в шнековых аппаратах,
как показано на рис. 210. В шнековый кристаллизатор 24
подается раствор мочевины, упаренный до содержания 92—93%
COfNP^b, и нагретый до 60—65 °С воздух, который движется
противотоком перемещающемуся в аппарате продукту. Воздух
затем очищается от пыли мочевины в скруббере 22 и отводится
з атмосферу. Орошающая скруббер жидкость периодически
отводится в сборник 15. Кристаллическая мочевина передается
транспортером на склад.
Переработка раствора мочевины в гранулированный продукт,
применяемый в качестве удобрения, отличается тем, что
упаривание раствора проводится в две ступени, а кристаллизация
мочевины — в грануляционных башнях. Выпарка первой
ступени и очистка раствора от примесей происходят в таких же
аппаратах и таких же условиях, как и при получении
кристаллической мочевины. Вторую ступень выпарки ведут при
разрежении 350 мм рт. ст. и упаривают раствор до состояния плава
\99,5% мочевины).
Плав подается насосом на верх грануляционной башни я
разбрызгивается. В нижнюю часть башни вентилятором
подается воздух, предварительно подогретый до 40—50 °С. Гра-
нулнровгнная мочевина выгружается из нижней части башгея
и транспортером передается на склад.
Мочевину упаковывают в многослойные бумажные мешки
весом по 40—50 кг.
Возвращение в цикл синтеза аммиака и двуокиси углерода,
содержащихся в газах дистилляции, может осуществляться
573
Плад мочевину
различными способами, например по схемам с жидкостным
рециклом. По одному из вариантов такой схемы аммиак,
выделяющийся при дросселировании плава мочевины, возвращается
в колонну синтеза, а газы дистилляции поглощаются водой с
образованием концентрированного
раствора солей аммония, который
также направляется в колонну
синтеза. По другому варианту
жидкостного рецикла газы поглощаются
минеральным маслом с
образованием суспензии карбамата
аммония, подаваемой в колонну синтеза.
Выходящая :из колонны смесь
мочевины, масла, воды и карбамата
аммония дросселируется и
подается в колонну для разложения
карбамата и разделения жидкой и
газовой фаз. Отделенное от раствора
масло вновь используется для
абсорбции газов.
Применяются также способы
раздельного возврата в цикл NH3 и
СО2, основанные на избирательной
абсорбции этих газов. Одним из
таких способов является
селективная абсорбция двуокиси углерода
раствором моноэтанола мина или
поташа, который после выделения
поглощенной СО2 снова поступает
на абсорбцию. Аммиак
конденсируется из газов и возвращается в
цикл, как и двуокись углерода,
выделенная из поглотительного
раствора.
Колонна синтеза
мочевины (рис. 211) состоит из корпуса
/ высокого давления с днищем 6
и крышкой 8. Внутри корпуса
имеются два вертикальных
цилиндра 2 и 3, в нижней части колонны
— устройство для перемешивания
реакционной массы, выполненное в виде дисковых перегоро-
юк 4 с прорезями. Жидкий аммиак поступает через боковые
штуцеры в нижнюю часть колонны » поднимается вверх по
кольцевому пространству между корпусом / и наружным
цилиндром 2, затем опускается вниз по кольцевому пространству
между цилиндрами 2 я 3 и через центральное отверстие днн-
574
NHg чиптый
NH3 возвратный
Рис.
ГО,
211. Колонна
мочевины:
синтеза
/—корпус высокого давления:
2—наружный вставной цилиндр;
3—внутренний вставной цилиндр;
4—перегородки с прорезями; 5— внутреннее
сферическое днище; 6—днище корпуса
колонны; 7—термопара; 8—крышка.
ща 5 поступает в реакционную зону. При такой схеме
движения жидкого аммиака корпус / колонны высокого давления не
соприкасается с агрессивной реакционной средой и не
подвергается действию высокой температуры. В некоторых случаях
часть аммиака вводят через боковой штуцер вверху корпуса
высокого давления. Двуокись углерода поступает в
реакционную зону по штуцеру, проходящему через днища 5 и 6, и сме
шивается с жидким аммиаком.
Реакционная масса постепенно поднимается по колонне
вверх и проходит через прорези перегородок. При этом поток
жидкости многократно меняет свое направление и хорошо
перемешивается, что способствует тесному соприкосновению
реагентов и улучшению условий теплообмена. Плав мочевины
отводится из колонны через штуцер в крышке.
Все части колонны, соприкасающиеся с реакционной мае
сой, изготовляются из хромоникельмолибденовой стали.
Вследствие необходимости ведения процесса при высоком
давлении и соответственно при большом расходе энергии
технологическая схема и аппаратурное оформление процесса
производства мочевины более сложны, чем в производстве других
твердых азотных удобрений. В последние годы
технологические схемы производства мочевины усовершенствованы,
сконструирована новая высокопроизводительная аппаратура,
снижены энергетические затраты, успешно решены вопросы
антикоррозионной защиты. Это позволило снизить капитальные
затраты и эксплуатационные расходы.
Техника безопасности в производстве
мочевины
При нарушениях технологического режима или авариях в
воздух производственных помещений может выделяться
газообразный аммиак, вследствие этого возможны возгорания
газов, взрывы, а также отравления обслуживающего персонала
Возможно также выделение в воздух СОг и окиси углерода,
входящей в состав экспанзерного газа в количестве около
2 объемн. %. Предельно допустимые концентрации этих газов:
NH3> мг/м3 20
СО, мг/м3 20
СО.,, объемн. %
при кратковременном пребывании ... 0,2
при продолжительном пребывании . . . 0,125
При переработке раствора мочевины, а также при
расфасовке и хранении готового продукта возможно выделение пыли
мочевины в случае неисправности оборудования и вентиляции.
При выделении аммиака в помещениях в количествах,
превышающих допустимую его концентрацию или вызывающих
575
образование взрывоопасной концентрации NH3 в воздухе, рабо
ту прекращают до ликвидации аварии (снижают давление в
аппаратах и прекращают все проводимые в это время
ремонтные работы). При проливе жидкого аммиака необходимо
включать аварийную вентиляцию и пользоваться
индивидуальными средствами защиты.
Безопасная работа колонны синтеза мочевины
обеспечивается путем блокировки регулятора давления с линиями подачи
реагентов в колонну. Насосы и компрессоры, подающие сырье
на синтез, также блокируются между собой. Для
предотвращения резких повышений давления в дистилляционной системе
нри аварийных выбросах плава из колонны синтеза должна
быть исключена возможность значительного снижения
давления воздуха, питающего пневморегуляторы выхода плава.
Об авариях агрегатов разделения газов дистилляции должна
извещать автоматическая сигнализация. При этом в отделениях
синтеза и дистилляции мочевины производится аварийный
выброс газов дистилляции в атмосферу. Отделения или агрегаты
синтеза, связанные с вышедшим из строя агрегатом
разделения газов дистилляции, останавливаются.
ЖИДКИЕ АЗОТНЫЕ УДОБРЕНИЯ
Кроме твердых удобрений, все большее применение находят
жидкие удобрения, главным образом азотные. В настоящее
время используют три основных типа жидких азотных
удобрений: жидкий аммиак, аммиачную воду и аммиакаты —
растворы азотсодержащих веществ в аммиаке «или аммиачной
воде.
Жидкий аммиак—самое концентрированное азотное
удобрение; он содержит около 82% азота, Поскольку жидкий
аммиак обладает высоким давлением паров, его хранят и
транспортируют в стальных цистернах или баллонах, рассчитанных
на давление не менее 16 ат.
Аммиачная вода представляет собой водный 20—25%-ный
раствор аммиака и содержит около 20% азота. Вследствие
сравнительно малого давления пара аммиачную воду можно
хранить и транспортировать в обычных цистернах и стальных
емкостях.
Аммиакаты могут быть получены в твердом или жидком
состоянии. Твердые аммиакаты представляют собой комплексные
соединения, образующиеся при взаимодействии некоторых
твердых солей с жидким или газообразным аммиаком. Аммиакаты
в виде растворов изготовляют путем насыщения аммиаком
водных растворов солей аммиачной селитры, кальциевой селитры,
мочевины или их смесей или же путем растворения этих
веществ в аммиачной воде. Такие аммиакаты имеют вид светлых
S76
жидкостей с сильным запахом аммиака. Содержание азота в-
аммиакатах в зависимости от их состава может изменяться от
30 до 45%. Давление паров над аммиакатами значительно ниже
давления паров жидкого аммиака. Аммиакаты хранят и
перевозят в специальных цистернах или баллонах, рассчитанных на
небольшое давление.
Азот аммиака усваивается растениями так же, как азот
обычных твердых удобрений. Во избежание потерь аммиака
жидкие азотные удобрения необходимо вносить в почву на глу->
бину не менее 10—12 см, поскольку почва обладает сорбцион-
ной способностью.
Жидкие азотные удобрения вносят в почву специальными
машинами, подобными культиваторам-растениепитателям,
применяемым для жидкой подкормки растений. Такие машины
обеспечивают заделку жидких удобрений на достаточную
глубину без контакта удобрения с воздухом. Все операции по ист
пользованию жидких азотных удобрений полностью
механизированы. Жидкие удобрения доставляются с завода в
железнодорожных цистернах в пункты назначения, где перегружаются
в соответствующие хранилища. Из хранилищ жидкие
удобрения доставляются непосредственно к месту внесения в почву
в автоцистернах, из которых жидкость перекачивается под
давлением в резервуары указанных выше машин.
При использовании жидких азотных удобрений отпадает
необходимость придания удобрениям улучшенных физических
свойств (исключаются гранулирование, сушка), упаковки в
тару, загрузки в ящики туковых сеялок и др. В целом применение
жидких удобрений в сельском хозяйстве менее трудоёмко по
сравнению с применением твердых удобрений. Жидкие
удобрения более равномерно распределяются в почве, чем твердые.
Применение жидкого аммиака или аммиачной воды для
внесения в почву позволяет получать удобрения по укороченной
схеме, без переработки аммиака и затрат на строительство
цехов азотной кислоты и аммичной селитры или мочевины.
Следует отметить, что для хранения жидких удобрений
требуется сооружение резервуароз большой емкости, так как
удобрения используются лишь в течение относительно короткого
зремени. Жидкие азотные удобрения можно применять также
для аммонизации при производстве гранулированных
смешанных удобрений (стр. 597).
4. КАЛИЙНЫЕ УДОБРЕНИЯ
В качестве калийных удобрений в сельском хозяйстве ещё
давно пользовались золой растений, в состав которой входит
калий. Однако этот источник калия не мог удовлетворить все
37-2848 577
возрастающий спрос на калийные удобрения. Получение мине'
ральных калийных удобрений стало возможным лишь в 40-х
годах XIX в. с открытием месторождений калийных солей.
Впервые товарный хлористый калий появился в 1861 г. С тех
пор мировая калийная промышленность выросла в крупную
отрасль промышленности. В настоящее время свыше 90% ми"
ровой добычи калийных солей применяется в качестве
удобрений, остальное количество используется в производстве едкого
кали, хромата калия, йодистого и бромистого калия,
бертолетовой соли, цианистого калия и других химических продуктов.
СССР обладает крупнейшими в мире разведанными
запасами калийных солей. Самое значительное калийное
месторождение—Верхнекамское представляет собой мощные пласты
калийных солей, залегающие в верхней части соляной толщи.
Большие запасы калийных солей сосредоточены также в Ста-
рОбинском месторождении (Белорусская ССР), в Прикарпатье,
в месторождениях мощной соленосной толщи Прикаспийской
впадины (Казахская ССР), в Гаурдакском месторождении
(Туркменская ССР) и др.
Советская калийная промышленность, возникшая лишь в
1933 г., после открытия Верхнекамского калийного
месторождения, в настоящее время является одной из крупнейших
отраслей химической промышленности.
Природные калийные соли. Важнейшими калиевыми
минералами являются сильвин КС1 и карналлит КС1 • MgCl2 • 6Н2О.
Значительно меньше используется в промышленности минерал
каинит КС1 • MgSO4 • ЗН2О.
По внешнему виду сильвин трудно отличить от поваренной
соли. Чистый сильвин бесцветен, но примеси окрашивают его
в красноватый и желтоватый цвет. Вкус сильвина
горько-соленый. Разрабатываемые залежи представляют собой смесь
сильвина и каменной соли, так называемый сильвинит, следующего
состава (в %):
КС1 . . 20—40
NaCl . - 58-78
Кроме того, сильвинит содержит некоторое количество
примесей MgCl2, CaSO4 и др.
Чистый карналлит бесцветен, примеси соединений железа и
других элементов обычно придают ему окраску от лимонно-
желтой и оранжевой до красной. Вкус карналлита горький.
Вследствие сильной гигроскопичности карналлит на воздухе
расплывается.
Менее распространены в природе сульфаты калия. Из них
наибольшее значение имеют лангбейнит K2SO4 • 2MgSO4, шенит
578
K2SO4 • MgSO4 • 6H2O, глазерит 2K2SO4 • Na2SO4, полигалит
K2SO4-MgSO4-2CaSO4-2H2O. Эти соли или продукты их
переработки применяются преимущественно для питания растений,
чувствительных к действию хлора (табак, виноград, чай,
цитрусовые, плодово-ягодные культуры).
ХЛОРИСТЫЙ КАЛИЙ
В качестве удобрений используются природные минералы,
содержащие хлористый калий, или продукты их переработки.
Исходные (сырые) соли содержат от 15 до 22% калия в
пересчете на КгО-
Из природных солей чаще всего применяют в качестве
удобрения молотый сильвинит. Породу размалывают до величины
зерен 3—4 мм. Молотые соли несколько гигроскопичны и могут
слеживаться при хранении.
Кроме сырых калийных солей, выпускают 30- и 40%-ные
калийные удобрения, содержащие соответственно 30—32% и
40—42% КгО. Они получаются путем механического смешения
размолотых природных калийных пород (преимущественно
сильвинита) с техническим хлористым калием 3-го сорта.
Технический хлористый калий применяется непосредственно
в качестве удобрения и служит полупродуктом для
изготовления 30- и 40%-ных калийных удобрений, калийной селитры,
калийно-аммиачной селитры, нитрофоски и других удобрений,
а также для производства калийных соединений методом
электролиза.
Хлористый калий получают почти исключительно из
сильвинита. Первая стадия переработки сильвинита заключается в
его дроблении и измельчении. Исходный сильвлнит направляют
в дробилки на предварительное (крупное) дробление.
Раздробленный на куски величиной до 25 мм сильвинит поступает на
грохоты, где разделяется на крупную (более 5 мм) и мелкую
фракции. Крупную фракцию измельчают на вальцах и вместе
с прошедшей через грохот мелкой фракцией направляют
ленточными транспортерами на переработку, либо в
железнодорожные вагоны или на склад.
Переработка калийных солей, заключающаяся в отделении
хлористого калия от хлористого натрия, может осуществляться
методами галургии*, флотации или комбинированными
способами.
* Галургия — отрасль химической технологии, занимающаяся
получением и переработкой солей действием на их растворы тепла (нагревание,
охлаждение, испарение).
37* 579
Метод галургии.основан на изменении растворимости солей
в зависимости от температуры. Флотация основана на
различной смачиваемости минералов водой. Комбинированные
способы представляют собой сочетание методов галургии и фло
тации или одного из этих методов с сепарацией в поле
центробежных сил, основанной на различной плотности минералов.
Производство хлористого калия методом галургии.
Выделение КС1 из сильвинита основано на том, что растворимость
хлористого калия с повышением температуры значительно
возрастает, тогда как растворимость хлористого натрня почти не
изменяется.
Процесс разделения этих солей заключается в основном в
следующем. Сильвинит обрабатывают при повышенной
температуре насыщенным на холоду раствором обеих солей. При
этом раствор дополнительно насыщается хлористым калием,
часть хлористого натрия переходит в осадок и отделяется
фильтрованием. Из охлажденного раствора выкристаллизовывается
только хлористый калий. Кристаллы отделяют на центрифугах
от маточного раствора и высушивают в сушильных барабанах.
Подогретый маточный раствор применяют для выщелачивания
хлористого калия из новой порции сильвинита. В случае
присутствия в сильвините значительной примеси хлористого
магния процесс несколько усложняется.
Производство хлористого калия по методу галургии состоит
из следующих стадий: растворение сильвинита, осветление
насыщенного раствора, охлаждение раствора и кристаллизация
хлористого калия, отделение хлористого калия от маточного
раствора (сгущение и фильтрование), сушка хлористого калия.
Схема процесса растворения сильвинита и осветления
насыщенного раствора хлористого калия изображена на рис. 212
Измельченный сильвинит поступает в первый шнековый
растворитель /, где начинает растворяться в горячем маточном
растворе при параллельном токе соли и раствора. Нерастворив-
шийся остаток передается отсюда элеватором 2 в шнековый
растворитель 3, где растворение в горячем маточном растворе
происходит при противотоке. Нерастворившийся остаток
передается далее элеватором 4 в шнековый мешатель 5,
растворение здесь протекает также при противотоке. Твердый остаток из
шнекового глешателя подается элеватором 6 на план-фильтр 7.
откуда промытые горячей водой отходы (хлористый натрий,
загрязненный примесями) сбрасываются на скребковый
транспортер «S. Аппараты для растворения сильвинита не имеют
нагревательных устройств, потери тепла восполняются подачей
горячего маточного раствора и острого пара через дюзы (на
рисунке не показано).
Маточный раствор после отделения хлористого калия на
вакуум-кристаллизационной установке (см. ниже) подогревает-
580
ся и поступает во второй шнековый растворитель 3 (большая
часть) и в шнековый мешатель 5, откуда перетекает во второй
шнековый растворитель, а из него — в первый шнековый
растворитель. Отсюда насыщенный раствор хлористого калия
самотеком поступает на станцию осветления, где отделяется or
увлеченных им мелких кристаллов хлористого натрия и
мельчайших частиц глины.
маточный раствор из вакуум-кристолли-
солевой шлам
Рис. 212. Схема растворения сильвинита и осветления раствора
хлористого калия:
¦ У—первый шнековый растворитель; 2, 4, 5—элеваторы; 3— второй шнековый растворитель;
6—шиековый мешатель; 7—план-фильтр; в—скребковый транспортер; 9—шестиконусный
сгуститель; 10, IS— баки; 11, 13, 15, 17—баки с мешалками; 12, 14, 16—сгустителн-отстой-
ники.
По одной из схем осветление раствора хлористого калия
осуществляется в шестиконусных сгустителях 9. В двух первых
конусах осаждается преимущественно соль и небольшое
количество нерастворимого остатка. В последующих конусах
осаждаются остальная соль и большая часть нерастворимого
остатка. Солевой шлам из первых двух конусов возвращается во
второй шнековый растворитель, из остальных конусов шлам,
разбавленный горячей водой, поступает на противоточную
промывку для уменьшения потерь хлористого калия. Установка
противоточной промывки шлама состоит из нескольких
последовательно соединенных механических сгустителей,
установленных каскадно.
581
Шлам вместе с горячим насыщенным раствором передается
из бака // с мешалкой в бак 13 с мешалкой, сюда же
перетекает слив из сгустителя 14. Эта смесь самотеком поступает в
сгуститель 12, куда дозируется также раствор коагулянтов
(крахмал и сода) из бака 18. Шлам из аппарата 12
перекачивается на следующую ступень сгущения и т. д. Чем больше
ступеней (сгустителей-отстойников) и чем больше подается
воды на промывку, тем меньше потерь хлористого калия, который
растворяется в процессе противоточной промывки. Глинистый
шлам вместе с промывной водой перекачивается из последней
ступени — сгустителя 16 в отвал. Слив из этого сгустителя,
содержащий КС1, возвращается на предыдущую ступень
промывки шлама.
Рис. 213. Шнековый растворитель:
/-корыто; 2-перегородаа; 3-спнрали шнека; 4-патрубки; 5-лопастн-скребки; S—дюза;
/ вал. «-лапы: S-ролики: л?-ковш; //-цепи; /2-крестовинд; /i-наклонный элеватор.
Шнековый растворитель (рис. 213), являющийся основным
аппаратом процесса растворения хлористого калия, состоит из
стального корыта /, в котором вращаются насаженные на вал 7
спирали <3 шнека. Корыто разделено перегородками 2,
доходящими до спирали и направляющими раствор вниз, что
способствует более тесному соприкосновению раствора с сильвинитом.
582
На вал насажены также крестовины 12, к которым
прикреплены спирали. Между отдельными элементами спиралей
поставлены лопасти-скребки 5, которые поднимают руду с дна корыта,
распределяют ее в растворе и замедляют передвижение руды.
В корыте растворителя имеются дюзы 6 для ввода острого
пара, что необходимо для поддержания определенной температуры
раствора. Удаление из растворителя твердого остатка
осуществляется наклонным элеватором 13, смонтированным вместе
с корытом. Ковши 10 элевзтора имеют сетчатые стенки, через
которые раствор стекает в аппарат. Для освобождения корыта
и приямка элеватора от руды и раствора имеются спускные
патрубки 4.
Осветленный раствор хлористого калия подается на вакуум-
кристаллизационную установку для охлаждения раствора и
кристаллизации КС1 из раствора. На некоторых заводах эти
процессы осуществляются последовательно в две стадии: в
вакуум-установке и в охладительной деревянной башне очень
большого объема, в верхней части которой разбрызгивается
раствор хлористого калия. Двухстадийный процесс охлаждения
и кристаллизации имеет существенные недостатки. В
охладительной башне происходит быстрое понижение температуры
раствора при большой разности температур охлаждаемого
раствора и охлаждающей среды, вследствие чего образуется
большое количество мелких загрязненных кристаллов и
безвозвратно теряется тепло раствора. Целесообразнее получать
возможно более крупные кристаллы хлористого калия, что
облегчает проведение последующих процессов отстаивания,
фильтрования и сушки. Кроме того, крупные кристаллы соли меньше
слеживаются, а при перевозке и применении хлористого калия
в сельском хозяйстве это имеет очень важное значение.
Более совершенным является одностадийный процесс
охлаждения и кристаллизации хлористого калия в многоступенчатых
вакуум-кристаллизационных установках при относительно
глубоком вакууме. Длительное пребывание образовавшихся
кристаллов в растворе при небольшой разности температур
охлаждающей среды и охлаждаемого раствора и
перемешивании раствора способствует росту кристаллов. Кроме того, по
этому способу возможно максимальное использование тепла
раствора.
Вакуум-кристаллизационная установка для охлаждения
раствора и кристаллизации соли в одну стадию состоит из 14
ступеней, заключенных в одном вертикальном и шести
горизонтальных корпусах. Горячий насыщенный раствор подается из
расходного бака в первую ступень установки — вертикальный
вакуум-корпус, где начинает кипеть. Вследствие испарения воды
и частичного охлаждения раствора из него выделяются
кристаллы хлористого калия. Образующаяся при этом суспензия —
583
охлажденный раствор с взвешенными в нем кристаллами соли—
просасывается последовательно через все ступени установки,
в которых происходит аналогичный процесс. По мере
упаривания и охлаждения суспензии из нее начинают выпадать также
кристаллы хлористого натрия. Из последней ступени пульпа
удаляется по барометрической трубе в приемный бак, откуда
непрерывно перекачивается в отстойную станцию на сгущение.
Пульпа
маточный роствор
8
газы
Рис. 214. Схема сгущения и фильтрования пульпы хлористого калия и его
сушки:
/—отстойник: 2—шестиконуеный сгуститель; 3. 6—емкости с мешалками; 4. 5—баки;
7—центрифуги; «—ленточный транспортер; 9—сушильный барабан; 10—выгрузная камера;
ii—циклон; 12-вентилятор; 13— скребковый транспортер.
Паровоздушная смесь из вакуум-кристаллизаторов засасы
вается в поверхностные конденсаторы, где охлаждается
холодным маточным раствором, движущимся противотоком
направлению движения раствора в ступенях вакуум-кристаллизации
Вакуум поддерживается в поверхностных конденсаторах при
помощи пароструйных эжекторов.
Схема сгущения и фильтрования пульпы хлористого калия
и его сушки показана на рис. 214. Пульпа из
вакуум-кристаллизационного отделения перекачивается в отстойник /, где
осаждаются крупные кристаллы хлористого калия. Мелкие
кристаллы КС1 увлекаются маточным раствором в шестиконус-
ный сгуститель 2, где осаждаются. Осветленный маточный
раствор из сгустителя поступает в бак 4 и перекачивается в
поверхностные конденсаторы вакуум-кристаллизационной
установки. Сгущенная пульпа из конусов сгустителя 2 сливается в
584
емкость 3 с мешалкой, взмучивается здесь и далее
перекачивается в бак 6 с мешалкой, откуда самотеком передается на
центрифуги 7. Минуя бак 6, пульпа может также
непосредственно поступать в центрифуги. Фильтрат, перетекающий из
центрифуг в бак 5, содержит некоторое количество мелких
кристаллов и потому возвращается з шестиконусный сгуститель 2.
Влажный хлористый калий, содержащий 5—7% влаги,
сбрасывается из центрифуг 7 на ленточный транспортер 8 и
направляется на сушку, необходимую для предотвращения сильного
слеживания продукта. Сушка хлористого калия производится
в прямоточных сушильных барабанах 9, обогреваемых
топочными газами. Газы, выходящие из сушильных барабанов,
просасываются через циклон 11 для улавливания увлеченной ими
солевой пыли и отводятся вентилятором 12 через выхлопную
трубу в атмосферу. Высушенный хлористый калий
пересыпается из выгрузной камеры 10 на скребковый транспортер 13,
передающий готовый продукт на склад или к железнодорожным
вагонам.
Упаковка хлористого калия производится в пятислойные
бумажные мешки.
Сушку хлористого калия можно вести более интенсивным
способом — в кипящем слое, конечная влажность продукта в
этом случае понижается до 0,2%. При сушке в кипящем слое
газом уносится 10—15% соли, которую необходимо улавливать.
Степень извлечения хлористого калия из сильвинитовой руды
методом галургии составляет примерно 90%, содержание
хлористого калия в отходах до 1,65%.
Недостатки производства хлористого калия методом
галургии: энергоемкость процесса, громоздкость оборудования,
необходимость антикоррозионной защиты аппаратуры и
образование недостаточно крупных кристаллов хлористого калия, что
приводит к слеживанию продукта.
Производство хлористого калия методом флотации имеет
ряд преимуществ перед методом галургии: процесс протекает
при обычной, а не при позышенной температуре, требуются
меньшие капитальные затраты на сооружение установок,
получаются несколько более крупные кристаллы хлористого калия,
который поэтому меньше слеживается.
Флотация калийных солей проводится в их насыщенных
растворах, так как в воде эти соли хорошо растворяются. В
качестве флотационного реагента в этом процессе применяют кати-
оиные сорбенты—первичные алифатические амины, содержащие
от 12 до 20 атомов углерода, в частности солянокислый октаде-
циламин. Однако такой флотореагент адсорбируется иа
частицах глины, присутствующей в сильвинитовой руде.
Вследствие этого образуется устойчивая пена, что затрудняет
флотацию крупных частиц сильвинита и повышает расход амина.
585
Поэтому необходимо предварительное флотационное удаление
глины из состава пульпы. Для укрупнения глинистых частиц
применяют крахмал, а для выноса хлопьев глины на
поверхность пульпы в нее вводят вспениватель ФР-2 (продукт
окисления уайт-спирита), образующий большое число пузырьков
воздуха.
После отделения глинистого шлама солевая пульпа
поступает самотеком в флотационную машину, из которой
хлористый калий удаляется вместе с пеной, хлористый натрий
остается в виде шлама.
Флотационный концентрат хлористого калия обезвоживают
в центрифугах и высушивают в барабанных сушилках.
Первичный концентрат содержит 87% хлористого калия, очищенный
94—95% КС1.
Расход реагентов на 1 т 95%-ного хлористого калия
составляет: 0,2 кг октадециламина, 0,3 кг крахмала и 1,17 кг вспе-
нивателя ФР-2.
Выпускается три сорта хлористого калия, различающиеся
по содержанию основного вещества и примесей:
Сорта
1-й
2-й
3-й
Со держание КС1 (в пересчете на сухое вещество), %,
не менее
или
К2О (в пересчете на сухое вещество), %, не менее
Содержание NaCl (в пересчете на сухое вещество), %,
не более
Влага, %, не более
98
61,9
1.4
1
95
60,0
4,6
1
90
56,9
Не
нормируется
2
В качестве удобрения применяется главным образом
продукт 3-го сорта, а также 2-го сорта.
СУЛЬФАТ КАЛИЯ
Главным источником получения сульфата калия, или
сульфатно-калийных удобрений, являются калийные соли
прикарпатских месторождений. Эти месторождения отличаются
разнообразием минералогического состава. Основной рудой
является каинито-лангбейнитовая руда, содержащая сульфаты
калия и магния. В состав этой руды входят основные
минералы— каинит, лангбейнит и галит и другие минералы — шенит,
полигалит, кизерит, карналлит, сильвин и ангидрит, а также
илы. Из указанных минералоз практически не растворяются
лангбейнит, полигалит и ангидрит, очень медленно растворяется
586
кизерит, остальные минералы легко растворяются. В породах
эксплуатируемых месторождений содержание калия в
пересчете на КгО составляет 10—12%.
Для получения сульфата калия требуется сложная
переработка указанного полиминерального сырья. Растворимые
минералы извлекаются горячими оборотными щелоками,
концентрированный горячий щелок осветляется и подвергается
дальнейшей переработке, нерастворившийся остаток поступает на
флотацию. Сульфат калия получается при переработке
осветленного концентрированного щелока методом конверсии, т. е.
путем обменного разложения по реакции:
2КС1 + MgSO4 = K2SO4 + MgCla
Полученный таким путем сульфат калия содержит около
53% КгО. Попутно получают другие калийные удобрения,
например калимагнезию, содержащую в основном сульфаты калия
и магния. В продукте 1-го сорта содержится около 31% КгО,
в продукте 2-го сорта 16—18% КгО.
КАИНИТ
Каинит — бедное калийное удобрение, представляющее
собой измельченную руду западноукраинских месторождений. Он
содержит двойную соль КС1 -MgSC^ • ЗН2О и механические
примеси каменной соли, CaSO4, MgSO4 и др. Каинит легко
растворим в вэде, содержит 10—12% КгО и 7—10% MgO. Продукт
перевозят навалом.
5. КОМПЛЕКСНЫЕ УДОБРЕНИЯ
Комплексные удобрения (сложные и смешанные) имеют ряд
преимуществ перед простыми удобрениями. При рассеве таких
удобрений затрачивается меньше времени и труда, чем при
раздельном внесении в почву простых удобрений. Кроме того,
применение комплексных удобрений позволяет распределить
полезные элeiMeнты в почве более равномерно.
Комплексные удобрения изготовляют с разнообразным
соотношением основных питательных элементов: азота, фосфора
и калия, применительно к различным почвам,
сельскохозяйственным культурам и условиям применения удобрений.
В СССР целесообразно применение комплексных удобрений
примерно с такими соотношениями питательных веществ:
P.O.
1
1,5
2
К2О N
1 1
1,5 1
Р2О5
1
При производстве смешанных удобрений возможно
получение продуктов с более разнообразным соотношением
питательных веществ, чем в сложных удобрениях.
СЛОЖНЫЕ УДОБРЕНИЯ
Ассортимент сложных удобрений весьма разнообразен. Ниже
описаны наиболее известные сложные удобрения.
Аммофос — сокращенное название технических фосфатов
аммония. Это высококонцентрированное удобрение, содержащее
от 57 до 63% питательных веществ (N + P2O5).
Фосфаты аммония применяют также для пропитки
древесины и тканей (придание огнестойкости), в пищевой
промышленности и пр.
Аммофос получают нейтрализацией фосфорной кислоты
аммиаком. При этом могут быть получены три аммониевые соли
фосфорной кислоты:
моноаммонийфосфат
НЭРО4 + 2NH3 = (NH4JHPO4
диаммонийфосфат
Н3РО4 + 3NH3 = (NH4KPO4
триаммонийфосфат
Триаммонийфосфат в качестве удобрения не применяют, так
как он представляет собой неустойчивую соль, из которой при
температуре около 30 СС выделяется аммиак:
(NH4KPO4 = (NH4JHPO4 + NH3
Диэммонийфосфат более устойчив, чем триаммонийфосфат,
однако при температуре около 70 СС он также начинает
выделять аммиак:
(NH4JHPO4 = NH4HaPO4 + NH3
Моноаммонийфосфат является вполне устойчивой солью.
В диаммонийфосфате отношение N : Р2О5 более
благоприятно для растений, чем в моноаммонийфосфате.
Фосфаты аммония, получаемые из термической фосфорной
кислоты, отличаются высокой чистотой, почти не содержат
примесей и используются преимущественно в пищевой и
фармацевтической промышленности и для других технических
нужд. В производстве аммофоса используется упаренная или
неупаренная экстракционная фосфорная кислота.
Нейтрализацию фосфорной кислоты аммиаком ведут с таким расчетом,
чтобы образовалось примерно 80—90% моноаммонийфосфата
и 20—10% диаммонийфосфата. При введении большего
количества аммиака возможны потери его при сушке аммофоса.
Соли магния, присутствующие в экстракционной фосфорной
588
кислоте (например, полученной при переработке фосфоритов
Кара-Тау), осаждаются в виде MgHPO4-ЗН2О. Соли кальция
выпадают из раствора в виде СаНРСч-2Н2О. Соли железа и
алюминия образуют осадок в основном в виде дигидратов
FePO4-2H2O и AlPCv2H2O В результате нейтрализации
присутствующих в экстракционной фосфорной кислоте серной и
кремнгфтористоводородной кислот образуются также сульфат
и кремнефторид аммония.
NH3
Н3РО4
аммосрое
Рис. 215. Схема производства аммофоса из упаренной фосфорной
кислоты:
/—напорный бак: 2—дозатор; 3—расходомер; 4—6—сатураторы; 7—смеситель;
S—бункер для ретура; 9—сушильный барабан; 10— вентилятор; //—циклон; 12
—дробилка; ID—элеватор; 14—грохот; /5—транспортер.
Концентрация упаренной экстракционной фосфорной
кислоты не должна быть слишком высокой C3—38% РгО5), чтобы
образующаяся при нейтрализации пульпа обладала
достаточной подвижностью и легко перемешивалась. Поглощение
аммиака густой пульпой затрудняется.
Схема производства аммофоса с использованием упаренной
фосфорной кислоты изображена на рис. 215. Упаренная
фосфорная кислота поступает через дозатор 2 в сатуратор 4.
Газообразный аммиак через расходомер 3 подается в
сатураторы 4 и 5, рядом с которыми установлен еще один
сатуратор 6. Все сатураторы соединены между собой перетоками и
расположены каскадно, чтобы образующаяся в них аммофос-
ная пульпа свободно переливалась из одного аппарата в
другой, расположенным ниже. Выходящая из последнего сатура'
тора 6 пульпа смешивается в горизонтальном смесителе 7 с
589
мелкой фракцией
высушенного аммофоса (ретур). Из
смесителя влажный
аммофос поступает з сушильный
барабан 9. Смешение аммо-
фосной пульпы с ретуром
производится для того,
чтобы она не налипала на
стенки сушильного барабана.
Высушенный продукт
передается элеватором 13
на двухситный грохот 14.
Частицы размером более
4 мм, не прошедшие чевез
верхнее сито, направляются
в дробилку 12. Дробленый
материал возвращают тем
же элеватором 13 на грохот
14. Фракцию, прошедшую
через второе сито (размер
частиц менее 1 мм),
возвращают на смешение с ам-
мофосной пульпой. Фракция,
остающаяся на втором сите,
представляет собой готовый
гранулированный аммофос
(размер частиц 1—4 мм).
Схема производства
аммофоса из неупаренной
фосфорной кислоты
изображена на рис. 216. Кислоту
нейтрализуют аммиаком з
трех последовательно
установленных сатураторах 2 с
мешалками из хромоникеле-
вой стали. Аммиак
непрерывно поступает во все
сатураторы, что позволяет
поддерживать более
равномерный тепловой режим
процесса нейтрализации.
Большая часть
образующейся аммофосной пульпы
('--'90%) направляется в
распылительную сушилку 7,
где распыливается быстро-
вращающимся горизонталь-
590
ным диском или форсунками. В сушилку поступают также
топочные газы, имеющие температуру примерно 650 °С. Газы,
выходящие из сушилки при температуре около 100 °С, очищаются
от пыли в батарейных циклонах 6. Высушенный материал
подвергается в шнековом аппарате 11 гранулированию, для чего
смешивается с остальной частью аммофосной пульпы (~ 10%)
и ретуром (мелкая фракция). Из гранулятора 11 аммофос
поступает в прямоточный сушильный барабан 12, откуда подается
элеватором 9 на дзухситный вибрационный грохот 13, где рас-
севается на три фракции. Отходящие из сушильного барабана
газы также очищаются в циклонах 6. Крупную фракцию (более
4 мм) растворяют в фосфорной кислоте в сборнике 15 и
возвращают в бак /. Мелкая фракция (менее 1 мм) направляется в
гранулятор 11. Фракция с размером частиц 1—4 мм является
готовым продуктом.
Примерный состав аммофоса приведен в табл. 30.
Таблица 30
Примерный состав аммофоса (в %)
Исходный фосфат
Апатитовый
концентрат
Фосфориты Кара-Тау .
РгО5
общая
53
48
усвояемая
52
47
водно-
растворимая
48
36
N
11,5
10,7
so3
4,2
6—6,5
MgO
5,6—6
Полуторные
окислы
R2O3
3
3
Влага
1,5—2
2—2,5
Нитрофос и нитрофоска. Нитрофос представляет собой
азотно-фосфорное удобрение, нитрофоска — азотно-фосфорно-
калийное удобрение. Эти удобрения выпускаются в
гранулированном виде и сэстоят в основном из дикальцийфосфата,
аммиачной селитры и некоторого количества фосфатов аммония
(нитрофос), а также солей калия (нитрофоска). Таким образом
значительная часть Р2О5 находится в этих сложных удобрениях
в цитратнорастзоримой форме в виде дикальцийфосфата и
некоторая часть в воднорастворимой форме в виде фосфатов
аммония.
Нитрофос и нитрофоску получают на основе продуктов
разложения природных фосфатов азотной кислотой. Азотная
кислота используется в этих процессах не только для
разложения фосфатов, но и как источник нитратного азота,
входящего в состав сложного удобрения.
591
Реакция разложения фторапатита— основного фосфатного
минерала — азотной кислотой выражается уравнением:
Ca6(PO4KF + 10HNO3 = ЗН3РО4 -+¦ 5Ca(NO3)a + HF
При наличии карбонатов в природных фосфатах протекает
реакция
(Са, Mg) (СО.,) + 2HNO3 = (Са, Mg) (NO3J + СО2 + Н2О
Кроме того, в раствор переходят (неполностью) окислы
железа и алюминия
R2O3 + 6HNO3 = 2R(NO3K +
и незначительные примеси солей некоторых редкоземельных
металлов (при переработке апатита). Фтористоводородная
кислота, выделившаяся в результате разложения фторапатита,
реагирует с двуокисью кремния, присутствующей в фосфатах:
6HF + SiO2 = HaSiFe + 2Н2О
Таким образом, в результате разложения природных
фосфатов азотной кислотой получается азотнокислотная
вытяжка— раствор, содержащий в основном фосфорную кислоту и
нитрат кальция, а также примеси кремнефтористоводороднои
кислоты, нитратов железа, алюминия. Кроме того, в растворе
содержится свободная азотная кислота. В азотнокислотной
вытяжке остается также небольшое количество нерастворимого
остатка (шлам), который может быть ртделен от раствора в
отстойной центрифуге. Возможна переработка азотнокислотной
вытяжки и без отделения шлама.
^Для разложения измельченных природных фосфатов обычно
применяют 47—55%-ную азотную кислоту. Количество азотной
кислоты должно быть не менее ее стехиометрического
количества, необходимого для разложения природного фосфата и
его примесей. В зависимости от заданного соотношения
N: Р2О5 в получаемом сложном удобрении азотную кислоту
вводят с определенным избытком сверх стехиометрического
количества B—5%), что предотвращает также замедление
процесса разложения фосфатов. Для перехода всех
компонентов природного фосфата в раствор требуется 2—2,5 ч.
Разложение природных фосфатов азотной кислотой
проводится непрерывным способом в нескольких последовательно
соединенных реакторах, расположенных каскадом.
Азотнокислотная вытяжка самотеком переходит из одного реактора в
другой. В каждом реакторе имеется мешалка, непрерывно
перемешивающая реагенты. Реакторы и мешалки изготовляют из
хромоникелевой стали.
Разработано несколько схем производства нитрофоса и
нитрофоски на основе продуктов разложения природных
фосфатов азотной кислотой. Эти схемы различаются главным
образом способом выделения или связывания избытка кальция,
592
содержащегося в азотнокислотной вытяжке. В природных
фосфатах и, следовательно, в азотнокислотной вытяжке отношение
СаО:РгО5 больше, чем нужно для связывания кальция со
всем количеством Р2О5 в виде дикальцийфосфата. В
апатитовом концентрате отношение СаО: Р2О5= 1,32, в фосфоритах
около 1,6, а в дикальцийфосфате это отношение составляет
0,79. Отношение СаО : Р2О5 в растворе должно быть меньше
0,79 для того, чтобы часть свободной фосфорной кислоты затем
можно было связать с аммиаком для образования воднорас-
творимой формы Р2О5 в виде фосфата аммония. Для условий
земледелия СССР желательно, чтобы не менее 50% Р2О5,
входящей в состав сложного удобрения, находилось в воднорас-
творимой форме. По одним схемам производства сложных
удобрений избыточное количество кальция выводится в виде
Ca(NO3J путем охлаждения вытяжки (схемы с
вымораживанием части кальциевой селитры). По другим схемам избыток
кальция связывается фосфорной или серной кислотой в виде
дикальцинфосфата или сульфата кальция. Можно также
связывать избыток кальция газообразной двуокисью углерода,
используя дешевый (отбросный) углекислый газ.
Ниже рассмотрены некоторые схемы производства нитро-
фоса и нитрофоски.
Разложение фосфатов азотной кислотой с
вымораживанием кальциевой селитры.
Выделение нитрата кальция из раствора основано по этой схеме на
изменении растворимости СаAМО3J в кислотном растворе.
Азотнокислотная вытяжка после отделения нерастворимого
остатка поступает в кристаллизаторы, где раствор постепенно
охлаждается до температуры минус 5 — минус 10 °С. При этом
выделяется осадок четырехводного нитрата кальция
Ca(NO3J*4H2O. Кристаллы кальциевой селитры отлагаются
также на поверхности охлаждающих змеевиков, что
значительно ухудшает условия теплопередачи, поэтому через каждые
2—3 ч в змеевики кристаллизаторов вводят пар. При этом
кристаллы плавятся на поверхности змеевиков в течение
нескольких минут, благодаря чему быстро восстанавливается
нормальный теплообмен.
Из последнего по ходу раствора кристаллизатора маточный
раствор вместе с кристаллами кальциевой селитры поступает
r центрифугу для отделения их от раствора.
Отфугованный кристаллический осадок далее перерабаты
вают обычным способом в кальциевую селитру (стр. 565), а
раствор направляют в сатураторы на нейтрализацию аммиаком,
которая протекает примерно по следующей реакции:
2Н3РО4 + 1,5Ca(NOsJ + 0,5HNO3 + 4,25NHS =
= 3,5NH4NO3 + 1,5CaHPO4 + 0,25NH4H2PO4 + 0,25(NH4)eHPO4
38—2848 593
При нейтрализации происходит разогревание раствора и
испарение воды. Чтобы предотвратить потери аммиака в
процессе нейтрализации, рН маточного раствора вначале доводят
до 3,5—3,8, во второй стадии нейтрализации предварительно
упаренный раствор донасыщают аммиаком до рН = 6—6,5.
После нейтрализации пульпа, содержащая взвесь дикаль-
цийфосфата и не более 14% влаги, передается в
грануляционный шнек-смеситель. При изготовлении нитрофоски в шнек-
смеситель вводят также хлористый калий. При этом
происходит реакция обменного разложения:
КС1 + NH4NO3 = KNO3 + NH4C1
Вместо КС1 в нитрофоску можно вводить сульфат калия.
В шнек-смеситель поступает также мелкая фракция нитро-
фоса или нитрофоски (ретур), полученная после рассева
высушенного материала. Количество добавляемой мелкой фракции
определяется из расчета уменьшения влажности пульпы до
4%. При такой влажности масса не налипает на стенки
сушильного барабана в процессе последующей сушки.
В сушильном барабане масса высушивается до содержания
1 % влаги. Высушенный продукт подается на двухситный
вибрационный грохот, где рассевается на три фракции. Крупная
фракция направляется в дробилку и затем снова на рассев,
мелкая фракция возвращается в шнек-смеситель, средняя
фракция (размер частиц 1—4 мм) представляет собой готовый
продукт. Он охлаждается в барабане до 25—35 °С для
уменьшения слеживаемости удобрения и поступает на упаковку в
бумажные битумированные мешки.
По указанной схеме нитрофос и нитрофоска могут быть
получены с разным соотношением питательных веществ. При
отношенли Ы:РгО5=1:1 нитрофос, полученный из
апатитового концентрата, содержит примерно по 20% N и Р2О5.
Нитрофоска при отношении N : Р2О5: К2О=1 : 1 : 1 содержит
примерно по 16% N, Р2О5 и К2О.
По другому варианту описанной схемы азотнокислотную
вытяжку после частичного вымораживания кальциевой селитры
нейтрализуют аммиаком. Полученная пульпа подвергается
упариванию, кристаллизации и сушке в такой же аппаратуре,
как для производства аммиачной селитры (стр. 557 ел.). Во
избежание забизки выпарных аппаратов осадком соли и
затруднений при кристаллизации образующийся плав должен
содержать избыток аммиачной селитры. При этом получается
сложное удобрение типа нитрофоса с преобладающим
содержанием азота по сравнению с Р2О5.
Разложение фосфатов смесью азотной и ф о с
фор ной кислот. По этой схеме фосфорная кислота взо
дится в процесс разложения в таком количестве, чтобы избыто
594
СаО можно было связать в дикальцийфосфат и получить
нитрофос или нитрофоску с определенным отношением N : РгО5
или N : Р2Об : КгО, причем не менее 50% Р2О5, содержащейся в
удобрении, должно быть в воднорастворимой форме.
Полученную при разложении природного фосфата пульпу
нейтрализуют аммиаком по реакции
1, 5Ca(NO3J -f 3H3PO4 -f 4, 5NH3 = 1, 5СаНРО4 + 1, 5NH4H2PO4 + 3NH4NO3
Как видно из этого уравнения, получаемый продукт
содержит 50% РгО5 в виде моноаммонийфосфата, т. е. в
воднорастворимой форме.
В случае необходимости увеличить содержание азота в
сложном удобрении вводят дополнительное количество азотной
кислоты и соответствующее количество аммиака для ее
нейтрализации либо добавляют аммиачную селитру.
Пульпу, образовавшуются после нейтрализации кислотной
вытяжки, перерабатывают так же, как в схеме с
вымораживанием кальциевой селитры.
При отношении N:P2O5 = 1 : 1 нитрофос, полученный по этой
схеме, содержит примерно по 20% N и Р2О5, а при отношении
N : Р2О5: КгО=1 : 1 : 1 нитрофоска содержит по 14—15% N,
Р2О5 и К2О.
Разложение фосфатов смесью азотной и
серной кислот. Данная схема производства сложных
удобрений аналогична предыдущей схеме. Разница заключается в
том, что в состав полученного удобрения входит дополнительный
компонент — гипс, являющийся балластом и снижающий
содержание питательных веществ в готовом продукте. Полученный
по этой схеме нитрофос содержит примерно по 14% N и Р2О5,
нитрофоска примерно по 12% N, P2O5 и КгО.
Вместо серной кислоты для связывания избытка кальция
можно добавлять к азотнокислотной вытяжке сульфаты
аммония или калия в виде сухой соли или раствора. При этом
готовый продукт также будет содержать гипс.
Схемы разложения природных фосфатов смесями азотной
и фосфорной кислот или азотной и серной кислот и разложения
фосфатов азотной кислотой с последующим добавлением
сульфатов называют упрощенными схемами производства сложных
удобрений, так как в этих схемах не требуются сложные
операции отделения осадка, промывки и вымораживания. По этим
схемам сложные удобрения получаются без побочных
продуктов и все исходные компоненты входят в состав готового
продукта.
Однако применение некоторых из упрощенных схем (азот-
носернокислотной, азотносульфатной) приводит к снижению
концентрации питательных веществ в удобрении, так как в нем
остается избыток кальция и нерастворимые примеси
38* 595
СМЕШАННЫЕ УДОБРЕНИЯ
Для получения смешанного удобрения хорошего качества
необходимо, чтобы компоненты, употребляемые при смешении,
были достаточно сухими, имели близкую по величине плотность
и примерно одинаковый размер частиц. При смешении
компонентов, сильно отличающихся по плотности и величине частиц,
не удается получить достаточно однородное смешанное
удобрение.
Не все удобрения можно смешивать друг с другом, так как
при смешении некоторых удобрений между ними протекают
нежелательные химические реакции или ухудшаются физические
свойства продукта. В результате таких реакций часть
питательных веществ при смешении удобрений может перейти в
неусвояемую или менее усвояемую форму или же теряется
(например, выделяется часть аммиака).
Часто в состав смешанных удобрений вводят добавки (так
называемые наполнители), которые нейтрализуют избыточную
кислоту, содержащуюся в смешанном удобрении, и улучшают
его физические свойства. Некоторые добавки, кроме того,
содержат полезные для растений элементы.
В качестве добавок применяют известняк, доломит, жмых,
костяную муку и др.
Ассортимент смешанных удобрений весьма разнообразен,
смеси можно готовить с различным соотношением питательных
веществ. Каждый питательный элемент может входить в смесь
в виде того или иного компонента, например Р2О5 в виде
суперфосфата, преципитата, томасшлака и др. В состав смешанных
удобрений можно вводить также микроудобрения (стр. 598).
Техника получения смешанных удобрений проста, смешение
компоиентов производят как непрерывным, так и
периодическим способом.
По непрерывной схеме исходные компоненты непрерывно
подают в смеситель через специальные дозаторы. Для
нормальной работы дозаторов необходимо, чтобы смешиваемые
компоненты были предварительно просеяны через сита с
определенными размерами отверстий. Материал, ие прошедший через
сито, поступает в аппарат для измельчения и затем
возвращается иа рассев. Из смесителя непрерывно выгружается
готовый продукт—смешанное удобрение.
При работе по периодической схеме смешиваемые
компоненты также пропускают через сито, а не прошедший через сито
остаток направляют на измельчение. Затем взвешивают на
весах требуемое количество каждого из компоиентов и загружают
навески в смеситель. Загруженный материал смешивают в
течение нескольких мииут и выгружают из смесителя готовый
продукт — смешанное удобрение.
596
В настоящее время производство смешанных удобрений
стремятся совмещать с аммонизацией и гранулированием смеси.
Кроме сыпучих минеральных удобрений и аммиака или
аммиакатов, в смесь можно вводить кислоты, а также измельченные
природные фосфаты и микроэлементы. При введении в смесь
компонентов, богатых питательными веществами (мочевина,
фосфаты аммония, концентрированная фосфорная кислота,
хлористый калий, сульфат калия), получаются концентрированные
гранулированные смешанные удобрения.
Непрерывное смешение компонентов, их взаимодействие и
гранулирование могут проводиться в одном совмещенном
аппарате— аммонизаторе-грануляторе. Одновременно в нем
происходит испарение влаги из смеси вследствие выделения тепла
реакций. Если в смесь вводят жидкие компоненты, требуется
дополнительная операция сушки. Готовый продукт получается
в виде однородных гранул, содержащих несколько питательных
элементов.
При производстве аммонизированных гранулированных
смешанных удобрений суперфосфат, азотные и калийные соли и
мелкая фракция готового продукта (ретур) непрерывно
дозируются в аммонизатор-гранулятор, снабженный устройством для
перемешивания компонентов. Одновременно в аппарат через
соответствующие дозаторы поступают под слой сыпучих
материалов аммиак и вода или аммиакаты, а также кислоты
(фосфорная или серная). Гранулированный продукт поступает
в сушильный барабан, где подсушивается до содержания влаги
менее 3%, затем охлаждается воздухом во вращающемся
барабане и направляется на рассев. Крупная фракция направляется
на дробление, мелкая фракция (ретур) возвращается в
аммонизатор-гранулятор, средняя фракция — товарный продукт
поступает на упаковку в крафт-целлюлозные мешки.
Количество ретура при производстве различных смешанных
удобрений составляет 0,1—0,9 вес. ч. на 1 вес. ч. готового
продукта. Время пребывания материала в
аммонизаторе-грануляторе не превышает 10 мин, температура смеси в процессе аммо-
низации повышается до 70 °С и более.
Температурный режим и соотношение в смеси жидкой и
твердой фаз (Ж: Т) регулируют подачей в
аммонизатор-гранулятор охлаждающего воздуха, ретура и в случае
необходимости пара.
В зависимости от состава компонентов, вводимых в смесь,
можно получать удобрения с разнообразным соотношением и
содержанием питательных веществ. Содержание воднораствори-
мой Р2О5 в продукте составляет 70—90% от количества
усвояемой Р2О5. Гранулы получаются прочные и не слеживающиеся
при хранении.
597
6. МИКРОУДОБРЕНИЯ
Микроудобрения содержат такие элементы, как бор, медь,
марганец, молибден, цинк и др., которые требуются растениям
в очень небольших количествах и потому называются
микроэлементами. Так, борные удобрения вносят на 1 га при посевах
из расчета около 1 кг бора, а при внекорневой подкормке
около 0,2 кг бора в случае опрыскивания растений и около
0,5 кг при оиыливании растений.
Микроэлементы участвуют в важных жизненных процессах
роста и плодоношения растений. Поэтому, несмотря на малые
дозы внесения микроудобрений, в присутствии их значительно
повышаются урожаи, улучшается качество
сельскохозяйственной продукции, а также предотвращаются некоторые
заболевания растений.
Борные удобрения. Наибольшее значение среди
микроудобрений имеют борные удобрения. Они применяются на почвах
с низким содержанием подвижных форм бора, особенно на
дерново-подзолистых известкованных почвах. В боре
нуждаются главным образом семенные посевы клевера и люцерны,
сахарная свекла, лен, плодово-ягодные, овощные и некоторые
другие культуры. Ниже описаны отдельные виды борных и
боро-фосфорных удобрений.
Бормагниевое удобрение получают из маточного раствора —
отхода производства борной кислоты, вырабатываемой из маг-
нийсодержащего сырья. Маточный раствор, содержащий 21—
24% MgSO4, 2—2,5% Н3ВО3, 0,2—0,5% H2SO4, 0,2—1,2%
полуторных окислов R2O3, нейтрализуют молотым известняком или
мелом и обезвоживают в распылительной сушилке, где
жидкость распыливается вращающимся диском. Бормагниевое
удобрение содержит 6—7% Н3ВО3 и 65—75% MgSO4
(содержание MgO около 7з от веса удобрения).
Борат магния осажденный получается в результате
обработки технической окисью магния маточного раствора (отход про-
изводстза борной кислоты), содержащего MgSO4 и Н3ВО3.
Вследствие небольшой растворимости образующегося при этом
бората магния практически весь бор выделяется в осадок,
который отфильтровывают, сушат и измельчают. Выпускаемый
борат магния содержит не менее 9% бора (в пересчете на
Н3ВО3) и 20—35% MgO в виде бората и сульфата магния.
В этом удобрении бор содержится в лимоннорастворимой
форме, усвояемой растениями, магний также находится в
усвояемой растениями форме.
Борнодатолитовое удобрение получается путем
разложения датолитового концентрата* 45—47%-ной серной
* Минерал датолит — боросиликат состава 2СаО • В2О3 • 2SiO2 • Н2О.
содержащий 21,8% В2О3.
598
кислотой в аппаратуре, применяемой для производства
суперфосфата (стр. 531 ел.)- При производстве этого удобрения не
требуется вылеживание продукта для его доразложения, а
также исключается процесс гранулирования. При наличии в дато-
литовом концентрате 15% В2О3 борнодатолитовое удобрение
содержит около 12—13% Н3ВО3 в воднорастворимой форме.
Бородвойной суперфосфат получается обработкой
экстракционной неупаренной фосфорной кислотой измельченной дато-
литовой руды или смеси датолитового концентрата с
фосфоритной мукой. Схема производства бородвойного суперфосфата
аналогична поточной схеме производства двойного
суперфосфата (сгр. 548). Бородвойной суперфосфат, полученный из да-
толитовой руды, содержит около 36% усвояемой Р2О5 в водно-
растворимой форме и б—8% бора (в пересчете на Н3ВО3).
Удобрение, полученное из датолитового концентрата и
фосфоритной муки, содержит 40—42% усвояемой Р2О5 и 6—6,5%
бора в растворимой форме (в пересчете на Н3ВО3).
Боросуперфосфат получают путем смешения суперфосфата
с борнодатолитовым удобрением, взятых в нужных
соотношениях. Удобрение, полученное по одному из вариантов
смешения, содержит 16—17% усвояемой Р2О5 и около 2% бора
(в пересчете на Н3ВО3) в воднорастворимой форме.
Медные удобрения. Медные удобрения оказывают
значительный эффект на многих торфяных, песчаных и
дерново-подзолистых почвах. При освоении болотных почв использование их
зачастую невозможно без применения удобрений, содержащих
медь.
В качестве медьсодержащих удобрений в СССР применяют
пиритные колчеданные огарки (стр. 85, 86), в которых, кроме
меди @,3—0,6%), содержатся цинк, кобальт, молибден и
другие микроэлементы.
Марганцевые удобрения. Потребность растений в марганце
чаще всего проявляется на карбонатных и других нейтральных
или слабощелочных почвах. В качестве марганцевых удобрении
используются преимущественно нерастворимые в воде
марганцевые шламы — отходы обогащения марганцевых руд,
содержащие 15—18% Мп. Кроме того, применяются
легкорастворимый сульфат марганца — отход нефтяной промышленности,
содержащий 21—22% Мп, а также марганизированный
суперфосфат. Его изготовляют путем добавления 10—15% марганцевого
шлама к суперфосфату перед гранулированием последнего.
Марганизированный суперфосфат содержит 17,5—18%
усвояемой РгО5 и 1,5—2,5% марганца.
Молибденовые удобрения. Молибден, необходимый для
жизни всех растений, особенно нужен бобовым культурам. В
молибдене нуждаются кислые, а также железистые почвы,
вследствие низкого содержания в них подвижных форм молибдена.
50Q
В качестве молибденовых удобрений применяют молибдат
аммония, главным образом в виде раствора, для предпосевной
обработки семян и опрыскивания растений (внекорневая
подкормка), а также молибденизированный суперфосфат,
содержащий 18—20% усвояемой Р2О5 и 0,05—0,2% Мо.
При изготовлении молибденизированного гранулированного
суперфосфата сырьевую смесь увлажняют в грануляторе
раствором молибдата аммония, разбрызгиваемого форсунками.
ЛИТЕРАТУРА
М. Е. П о з и н, Технология минеральных солей, Госхимиздзт, 1961.
Справочник по минеральным удобрениям, Сельхозгиз, 1960.
В. Ваггаман, Фосфорная кислота, фосфаты и фосфорные удобрения, пер.
с англ., Госхимиздат, 1957.
М. Л. Ч е п е л е в ец к и н, Е. Б. Бруцкус, Суперфосфат,
физико-химические основы производства, Госхимиздат, 1958.
B. С. Демин, Производство фосфоритной муки, суперфосфата и крем-
нефтористого натрия, Госхимиздат, 1955.
C. В. Кузнецов, Автоматизация производства суперфосфата,
Госхимиздат, 1956.
М. Н. Н а б и е в, А. А. Вишнякова, Аммонизированный суперфосфат нз
фосфоритов Кара-Тау, Изд. АН УзбССР, 1960.
В. А. Кононов, С. Д. Э в е н ч и к. Модернизация оборудования и
интенсификация производства концентрированных фосфорных удобрений.
Журн. ВХО им. Д И. Менделеева, № 1, 2 A961).
B. А. Клевке, Н. Н. Поляков, Л. 3. Арсеньева, Технология
азотных удобрений, Госхимиздат, 1963.
К- А. Бело в, Улавливание химических продуктов коксования, Металлург-
издат, 1948.
А. Н. Андреичев, А. Б. Нудельман, Добыча и переработка калийных
солей, Госхимиздат, 1960.
C. И. Вольфкович, Ф. Г. Марголис, Н. Н. Поляков, Комплексные
удобрения на основе азотнокислотного разложения фосфатов, Хим.
пром., № 1, 34 (I960).
А. М. Дубовицкий, Ф. Г. Марголис, Технология сложных и
смешанных удобрений за рубежом, Научно-технический информационный
бюллетень НИУИФ, № 4 П957).
М. В. К а т а л ы м о в, Т. П. У н а н я н ц, Производство н применение
микроудобрений в СССР и за рубежом, ВНИТИ, 1960.
Журн. ВХО им. Д. И. Менделеева, № 5 A962).
Глава Xlll
КАРБИД И ЦИАНАМИД КАЛЬЦИЯ
/. КАРБИД КАЛЬЦИЯ
Химически чистый карбид кальция СаСг кристаллизуется в
виде бесцпетных кристаллов. Технический карбид кальция дает
кристаллический излом серого цвета с различными оттенками
в зависимости от чистоты продукта.
Карбид кальция широко применяется в технике, главным
образом для получения ацетилена, а также для получения
цианамида кальция. Ацетилен образуется при разложении
карбида кальция водой:
ОС, + 2Н2О = QHa + Ca(OHJ
Процесс разложения карбида кальция водой протекает с
выделением тепла.
Ацетилен получают не только из карбида кальция, но и
другими методами: путем термического крекинга, электрокрекинга
и окислительного пиролиза углеводородных газов (природный
газ, нефтяные газы).
Ацетилен является исходным продуктом для производства
разнообразных органических веществ: ацетальдегида,
перерабатываемого на этиловый спирт, уксусную кислоту, бутадиен,
этилацетат, н-бутанол и другие продукты, а также для синтеза
хлорорганических соединений (хлористый винил, хлоропрен)
и других веществ (винилацетат, виниловые эфиры, акрилонит-
рил и др.). используемых в качестве растворителей, мономеров,
в производстве синтетических полимеров и т. д. Значительное
количество ацетилена применяется для резания и сварки
металлов.
Качество карбида кальция определяется количеством
ацетилена, получаемого при разложении водой 1 кг продукта. Чем
чище карбид кальция, тем больше ацетилена выделяется из
одного и того же количества карбида. Количество литров
ацетилена, выделяемого 1 кг карбида кальция при его полном
разложении водой, называется литражом карбида кальция. Из
1 кг химически чистого карбида кальция при разложении водой
получается 346,8 л ацетилена (при 0°С и 760 мм рт. ст.). Ли-
601
траж технического карбида кальция практически составляет от
320 до 235 л. Чем крупнее куски карбида, тем больше
ацетилена выделяется при его разложении.
Карбид кальция получается сплавлением обожженной
извести с антрацитом и коксом. К исходному сырью предъявляют
жесткие требования в отношении содержания примесей, так как
они ухудшают качество готового продукта, а в некоторых
случаях присутствие примесей нарушает нормальный ход процесса
получения карбида. Особенно вредна примесь фосфора,
образующего фосфористый кальций СазРг, который при
последующем разложении карбида водой дает ядовитый и в смеси с
ацетиленом взрывоопасный газ — фосфористый водород РН3.
Вредной примесью является также сера, которая образует
сернистый кальций CaS, а при разложении карбида водой —
сероводород H2S; последний при сжигании ацетилена сгорает с
образованием сернистого газа SO2, вызывающего коррозию
металлов. Примеси окислов магния и алюминия делают карбид
кальция более тугоплавким.
Известняк, применяемый для обжига, должен содержать не
менее 97—98% СаСО3, не более 0,008% Р и не свыше 0,1% S.
Антрацит и кокс должны содержать не более 0,04% Р и до
1,5% S.
Процесс образования карбида кальция
СаО + ЗС = CaGj + СО — 108 ккал
протекает только при высокой температуре и с поглощением
большого количества тепла. Начальная температура
образования карбида кальция находится в пределах 1700—1800 °С.
В карбидной печи температура достигает 2000 °С, при
повышении температуры до 2200°С начинается испарение карбида
и разложение его на металлический кальций и углерод.
В процессе получения карбида кальция реакция происходит
между двумя твердыми фазами, поэтому размеры кусков
шихты, равномерность их измельчения и качество смешения
оказывают большое влияние на скорость и полноту взаимодействия
реагентов. На реакцию образования карбида кальция
расходуется большое количество электрической энергии.
Практический расход электрической энергии значительно превышает
теоретический расход (на 53—54%) вследствие протекания
побочных реакций — разложения карбида и остатков СаСО3 (в
извести), образования ферросилиция из примесей кремния и
железа в шихте, а также вследствие значительных потерь тепла.
СХЕМА ПРОИЗВОДСТВА
Процесс производства карбида кальция заключается в
следующем (рис. 217). Обожженную известь, антрацит и кокс
дробят на куски определенной величины и смешивают в необ-
602
ходимой пропорции. Полученную шихту подают в бункеры /,
расположенные над электрической печью 4. Образующийся в
печи карбид кальция периодически выпускают в расплавленном
состоянии в изложницы, где он застывает, либо, как показано
на рисунке,— в охлаждаемый вращающийся барабан 8 для
получения гранулированного карбида. Выделяющиеся газы через
газосборные воронки 6 отводятся из печи на очистку.
UJuzma
pd на
' сод/тшрввну
и упаковку
Рис. 217. Схема производства гранулированного карбида кальция:
/—бункеры для шихты; 2—рукава для загрузки шихты в печь; 3— трансформатор;
^трехфазная карбидная печь; 5—электроды; 6—газосборные воронки; 7—желоб; 8—барабан для
охлаждения и гранулирования карбида кальция; 9—элеватор; 10— вентиляторы.
Застывшие в изложницах карбидные блоки подают в
отделение охлаждения и дробления карбида. Дробленый карбид
кальция направляют на сортировку и упаковку или на размол
для последующей переработки в цианамид кальция.
Вращающийся барабан для гранулирования карбида
кальция (длина 45—47 м, диаметр 3 м) охлаждается снаружи
водой. В барабане карбид кальция охлаждается и застывает р
виде кусков размером до 100—J20 мм.
Куски размером более 65 мм по выходе из барабана
поступают на дробление.
Преимущество схемы с гранулированием заключается в том,
что значительно сокращается объем производственных зданий
по сравнению с объемом, требуемым при выпуске карбида в
виде блоков, и отпадает надобность в кране для
транспортирования тяжелых карбидных блоков (вес блока достигает
1 —1,5 г). Кроме того, исключается агрегат для
двухступенчатого дробления карбидных блоков и улучшается качество
карбида кальция.
603
Карбид кальция выпускают двух сортов, каждый сорт
подразделяется на четыре класса в зависимости от крупности
кусков (от 2—8 до 25—80 мм). Содержание мелочи, т. е. частиц
размером меньше 2 мм, в мелком классе не должно превышать
5%, в остальных классах не более 2%. Каждый класс
продукта должен соответствовать определенным требованиям в
отношении литража: при 20 °С и 760 мм рт. ст. 255—235 л для
наиболее мелкого класса и 285—245 л для крупных классов.
В ацетилене, получаемом из карбида кальция, допустимое
содержание РН3 не должно превышать 0,08 объемн. %,
соединений серы (в пересчете на сероводород) не более 0,15 объемн. %.
Чтобы предотвратить разложение карбида под действием
влаги воздуха, продукт следует хранить и транспортировать в
герметичной таре. Обычно карбид кальция упаковывают в
герметичные стальные барабаны вместимостью по 50—130 кг.
КАРБИДНЫЕ ПЕЧИ
Для производства карбида кальция применяют преимущест
венно трехфазные электрические печи. Карбидные печи строя"
на разную мощность — от 2 до 60 тыс. кет.
Существуют карбидные печи открытого, полузакрытого и
закрытого типов. В открытых печах окись углерода,
выделяющаяся при образовании карбида кальция, сгорает на
поверхности шихты. При этом выделяется большое количество тепла
и много пыли, что ухудшает условия обслуживания печи. В
настоящее время мощные механизированные закрытые печи с
автоматическим обслуживанием вытесняют печи открытого
типа. Применение таких закрытых печей позволяет снизить расход
сырья, энергии, электродов, уменьшить трудозатраты на
обслуживание агрегатов, повысить их производительность, а также
значительно улучшить условия труда и предотвратить
загрязнение воздушного бассейна печными газами. Закрытые печи
имеют подвижную ванну, которая медленно вращается или
совершает возвратно-поступательные движения. В печах с
подвижной ванной шихта лучше разрыхляется, что ускоряет ее
расплавление.
В зависимости от конструкции карбидной печи и режима
процесса на 1 т карбида кальция получается 150—250 мъ газа
(считая на объем при нормальных условиях) с теплотворной
способностью 2200—2800 ккал\мъ. Выделяющийся из
карбидных печей газ может быть использован после очистки в
качестве топлива или для синтезов органических продуктов.
Состав газа (в объемн. %):
СО ... 70-85 СО„ . . 3—5
Н2 . . . 6-15 О2 . . . 0,5-1,0
604
Остальное составляют азот и некоторые примеси.
Газ отделяют от пыли мокрым или сухим способом, в
результате чего содержание пыли снижается с 30—40 до 5 мг/м3.
Получаемый газ можно также использовать для обжига до 60%
извести, необходимой в процессе получения карбида кальция.
В современных карбидных печах на 1 т технического кар-
била кальция расходуется (в кг):
Известь 900—960
Антрацит 450
Кокс 140
Расход электроэнергии на 1 т карбида (в пересчете на
250-литровый) на мощных печах составляет 2400—.2800 квт-ч.
на малых печах достигает 3150—3200 квт-ч.
БРИКЕТИРОВАННЫЙ КАРБИД КАЛЬЦИЯ
Брикетированный карбид кальция изготовляется из мелких
фракций карбида, которые обычно под действием влаги воздуха
быстро разлагаются, вследствие чего не может быть обеспечено
равномерное выделение газа в ацетиленовом генераторе.
Производство брикетированного карбида кальция несложно.
Мелкий карбид (размеры частиц от 2 до 7 мм) смешивают в
подогретом состоянии с небольшим количеством F—8%)
хлорированного пека; полученную массу брикетируют. Вес
брикетов 0,5 и 1 кг. Для образования на брикетах поверхностного
слоя, защищающего карбид от воздействия влаги, брикеты
помещают в сетчатые ковши, которые на полчаса погружают в
ванну с разогретым минеральным маслом. Затем брикеты
вынимают и упаковывают в стальные барабаны. Выход ацетилена
из брикетированного карбида кальция составляет 260—270 л.
При хранении брикетированного карбида в течение 3—4 недель
без герметической упаковки не наблюдается заметного
уменьшения выхода ацетилена. Брикетированный карбид кальция
весьма удобен для применения в мелких механических
мастерских, на машинно-тракторных станциях и т. д.
Техника безопасности в производстве
карбида кальция
На карбидных заводах техника безопасности имеет очень
важное значение, так как при соприкосновении влаги с
карбидом кальция получается ацетилен, который образует с воздухом
взрывоопасную смесь. Кроме того, работа у карбидной печи
ведется при высокой температуре и сопровождается
выделением пыли и газа. Для улучшения условий труда все процессы
60
следует но возможности механизировать, аппаратуру
герметизировать и обеспечить рабочие места вентиляцией. Токонесущие
части установки должны быть надежно изолированы.
Необходимо следить за исправностью системы водяного
охлаждения электрододержателей, так как проникание в
карбидную печь значительного количества воды может вызвать
взрыв с выбрасыванием горячей шихты и даже расплавленного
карбида кальция; при этом обслуживающий персонал может
получить сильные ожоги. Чтобы предохранить работающих от
действия тепла, излучаемого электрической печью, в местах
загрузки шихты и выгрузки карбида кальция устанавливают
защитные экраны в виде цепных завес.
Рабочие, креме спецодежды, должны пользоваться
респираторами и защитными очками, а при заправке фаз, опускании
электродов на верхней площадке карбидной печи и прожиге
леток для слива расплавленного карбида кальция — также
сеткой для защиты лица от ожогов.
При возникновении пожара в отделении карбидных печей
и в других помещениях, где находится карбид кальция, для
тушения можно применять только сухой песок и сухие
огнетушители. Ни в коем случае нельзя гасить огонь водой.
2. ЦИАНАМИД КАЛЬЦИЯ
Цианамид кальция СаСИг представляет собой соль
цианамида NH2CN.
Химически чистый цианамид кальция образует бесцветные
кристаллы. Технический продукт представляет собой порошок
или гранулы темно-серого цвета размером по 2—3 мм.
В годы первой мировой войны цианамид кальция служил
источником получения аммиака и, следовательно, азотной
кислоты, необходимой для производства взрывчатых веществ.
В последующие годы, в связи с развитием промышленности
синтетического аммиака, значение цианамида кальция
уменьшилось. В настоящее время цианамид кальция применяется в
качестве полупродукта для получения цианистых соединений
(цианплава), дициандиамида, тиомочевины и др. Цианплаз
используется в золотодобывающей промышленности, в цветной
металлургии, для закалки специальных сталей, а также для
протравливания зерна, для борьбы с грызунами и пр. Дициан-
диамид применяется в производстве пластических масс, а
также для получения меламина, гуанидина, его солей и др.
Цианамид кальция является эффективным дефолиантом,
применяемым для предуборочного удаления листьев
хлопчатника (при механизированной уборке), в незначительных
количествах он употребляется в качестве азотного удобрения. При
606
хранении и внесении в почву цианамид кальция под действием
двуокиси углерода и влаги разлагается, образуя
последовательно цианамид, дицианамид, мочевину и аммиак.
Основным сырьем для производства цианамида кальция
являются карбид кальция и азот. Поэтому обычно на каждом
цианамидном заводе имеются печи для получения карбида
кальция и установка для получения азота разделением воздуха
(стр. 213 ел.).
Получение цианамида кальция основано на взаимодействии
газообразного азота с тонко размолотым карбидом кальция
при высокой температуре:
CaQ + Na = CaCN2 + С + 72 ккал
Для снижения температуры азотирования карбида кальция
и увеличения скорости реакции к карбиду добавляют
некоторое количество плавикового шпата или хлористого кальция.
Добавление хлористого кальция менее рационально, так как
цианамид кальция получается в этом случае более
гигроскопичным.
Процесс получения цианамида кальция ведут при 800—
1150°С в печах периодического или непрерывного действия.
Наибольшее распространение получил периодический метод
получения цианамида кальция.
Доставляемые из карбидного цеха блоки карбида кальция
измельчают в дробилке и размалывают с добавкой плавикового
шпата или хлористого кальция в трубчатой мельнице. Для
безопасности работы через мельницу и аппаратуру для
перемещения и хранения размолотого карбида кальция непрерывно
продувают азот.
Размолотую шихту загружают в цианамидную печь и
подают в нее азот .В результате азотирования карбида в печах
образуется цианамид кальция в виде спекшихся блоков. Блоки
охлаждают до температуры окружающего воздуха в
неотапливаемом здании с хорошей вентиляцией. Охлажденные блоки
цианамида кальция измельчают в дробилке и размалывают в
мельнице.
Цианамид кальция, предназначенный для дефолиации, после
размола обрабатывают во вращающихся стальных барабанах
водой и минеральным маслом. Вода разлагает оставшийся я
цианамиде карбид кальция, масло добавляется для того, чтобы
продукт получался менее пылящим.
Для получения непылящего цианамида кальция его иногда
гранулируют. Гранулированный цианамид кальция получают
замешиванием тонко размолотого продукта с водой во
вращающемся барабане или шнеке с последующей сушкой и
сортировкой образующихся гранул.
607
ЦИАНАМИДНЫЕ ПЕЧИ
Наиболее распространены цианамидные печи
периодического действия с непосредственной загрузкой шихты.
Цианамидная печь с непосредственной загрузкой шихты
(рис. 218) представляет собой
стальной цилиндр 1, футерованный
шамотным кирпичом 2. Кладка
печи книзу несколько сужается на
конус для облегчения выгрузки
образующегося цианамидного блока.
Печь закрывается крышкой 10 с
песочным затвором 9. В нижней
части печи имеется коробка 4, в
которую по трубопроводу 6 подается
азот, поступающий далее в шихту
по нескольким вертикальным кана
лам 6. В центре печи в шихте
оставляют канал для установки
нагревательного электрода 7. Нижний
конец электрода упирается в изолиро
ванную от печи контактную плиту 5,
к которой подводят электрический
ток, верхний конец электрода при
жат к заземленной крышке 10.
Перед загрузкой шихты в иечн
устанавливают трубы, после
удаления которых в шихте остаются
каналы 8 и центральный канал для
электрода 7. Для выгрузки
цианамидного блока на дно печи
устанавливают чугунный конус 3 с
продолговатой прорезью. В прорези
укрепляют конец штанги, с
помощью которой цианамидный блок
извлекается из печи.
Нагревание шихты производится
электрическим током (напряжение
70—90 в, ток 70—100 а).
Раскаленный электрическим током электрод
нагревает окружающую его шихту
до температуры, необходимой для начала реакции
азотирования карбида кальция. Затем ток выключают, и в дальнейшем
требуемая температура шихты поддерживается за счет тепла
реакции. По окончании реакции подачу азота прекращают.
Несколько остывший, но еще горячий блок цианамида кальция
извлекают из печи и направляют на охлаждение.
608
Рис. 218. Цианамидная печь:
/—цилиндрический кожух:
2—шамотная футеровка; 3—чугунный конус;
4—зольная коробка; 5—контактная
плита; б—трубопровод для подачи
азота; 7—электрод; 8—каналы; 9—
песочный затвор: 10—крышка;
II—штуцеры.
Степень превращения карбида в цианамид кальция
составляет 83—89% от теоретической.
Цианамидные печи с непосредственной загрузкой шихты
строят на производительность 1,7; 3 и 5 т цианамида кальция
за один цикл. Продолжительность одного цикла 1,7-тонной печи
состазляет 48—50 ч, из которых непосредственно на
азотирование приходится 36 ч.
На некоторых заводах: применяют канальные цианамидныг
печи непрерывного действия с газовым или электрическим
обогревом. Внутреннее пространство канала в печи разделено на
четыре зоны: зону предварительного подогрева, зону нагрева,
реакционную зону и зону охлаждения. Шихту загружают в
специальные ящики, устанавливаемые на тележки, которые
вкатывают в печь по рельсам. Постепенно тележки с шихтой
проходят все четыре зоны. Азот вводят в печь противотоком
движению материала. Для регулирования температуры в печи азот
можно вводить также в разных точках канала.
Кроме канальных печей, применяются также горизонтальные
вращающиеся цианамидные печи непрерывного действия. Они
менее громоздки, чем канальные, но обладают тем недостатком,
что с течением времени внутри печи образуется кольцо из
спекшегося продукта. Поэтому вращающиеся печи приходится
периодически останавливать для чистки, представляющей собой
трудоемкую операцию.
Примерные расходные коэффициенты на 1 т цианамида
кальция, содержащего 18% связанного азота:
Карбид кальция, m 0,559
Азот, м3 342
Плавиковый шпат, m . . . 0,0144
Электроэнергия, квт-ч ... 63
Цианамид кальция выпускают двух марок. Продукт
марки А используется для химической переработки, продукт
марки Б — для предуборочного удаления листьев хлопчатника.
Состав цианамида кальция и требуемая тонина его помола
приведены ниже:
Марка А Марка Б
1-й сорт 2-й сорт
Азот, %, не менее 20 18,5 19
Карбид кальция, %, не более 1 2 0,2
Минеральное масло, % — — 0,5-—1,5
Остаток на сите, %, не более
0,071 мм — _ 5,0
0,6 мм 0,5 0,5 —
Цианамид кальция упаковывают в стальные барабаны
емкостью 50—100 л или в бумажные пятислойные мешки весом
по 35 кг.
39—2848 609
Техника безопасности в производстве
цианамида кальция
При производстве цианамида кальция наиболее вредное
действие на человеческий организм оказывает цианамидная пыль.
Для защиты от действия цианамидной пыли работающие
должны надевать спецодежду, пользоваться респираторами,
защитными очками, смазывать лицо и руки вазелином.
Обязательными условиями безопасной работы являются: исправное
состояние вентиляции, наличие отсосов в местах выделения пыли,
герметизация оборудования.
Цианамид кальция содержит остаточный карбид кальция,
который при взаимодействии с влагой воздуха образует
ацетилен. Поэтому во избежание взрыва все производственные
операции с цианамидом проводят в токе инертного газа, обычно
азота, и систематически определяют содержание кислорода в
газе, заполняющем мельницы для измельчения карбида и
цианамида кальция. Кроме того, з транспортирующей аппаратуре и
в мельницах устраивают предохранительные взрывные люки.
ЛИТЕРАТУРА
С. И. Вольфкович, 3. А. Роговин, Ю. П. Руденко, И. В. Ш м а-
ненков, Общая химическая технология, т. II, Госхимиздат, 1959.
Л. Я. Марковский, Д. Л. Оршанский, В. П. Прянишников,
Химическая электротермия, Госхимиздат, 1952
Л. А. Кузнецов, Производство карбида кальция, Госхимиздат, 1950.
В. А. Клевке, Н. Н. Поляков, Л. 3. А р с е н ь е в а, Технология
азотных удобрений, Госхимиздат, 1963.
Ю. Я. Туров, Е. Е. Юдович, Современное состояние и перспективы
производства и потребления карбида кальция, Журн. ВХО
им, Д. И. Менделеева, № 1, 81 A962).
П. Е. К а з а р я н, Современное состояние и перспективы производства
цианамида кальция, Жури. ВХО им. Д. И. Менделеева, № 1, 87 A962).
Глава XIV
ТЕХНОЛОГИЯ СИЛИКАТОВ
Технологию силикатов обычно подразделяют на производи
ство керамических изделий, производство вяжущих материалов
и производство стекла.
Новой отраслью силикатной промышленности является
петрургия, производящая химически стойкие строительные и
другие материалы путем литья расплавленных горных пород
(диабаз, базальт) и металлургических шлаков. В последние
годы путем сочетания этого метода со специальной термической
обработкой стали получать стеклокерамику.
В современной технике приобретают все большее значение
материалы на основе кремнийорганических полимеров. Они
сочетают ценные свойства органических полимеров
(эластичность, пластичность) с преимуществами силикатных
материалов (теплостойкость, морозостойкость, малая усадка).
/. КЕРАМИКА
Керамикой обычно называют изделия из различных видов
минерального сырья, главным образом из глины, а также из
магнезита, доломита, кварцита, талька и тому подобных
материалов, изготовленные путем формования или отливки с
последующей сушкой и обжигом до спекания. Керамические изделия
широко применяются в различных отраслях техники, в
строительстве и в быту. Наиболее распространенным видом
керамических изделий является кирпич, широко применяемый в
строительстве для кладки стен, печей, газоходов и др.
В химической промышленности используют кислотоупорные
и щелочеупорные керамические изделия. Кирпичами и
плитками футеруют различные аппараты, например башни и желобы
в сернокислотном производстве. Керамическими кольцами и
другими видами насадок заполняют скрубберы и
абсорбционные аппараты. Большое применение имеют керамические
холодильники, насосы, вентиляторы, реторты, трубопроводы для
передачи кислых жидкостей и газов, пористые керамические
фильтровальные плитки и трубки.
Керамические материалы широко применяются также в
металлургической и электротехнической промышленности
(огнеупорные изделия, фарфоровые изоляторы).
39* 611
Керамические изделия при значительном сопротивлении
истиранию отличаются относительной хрупкостью. В настоящее
время ведутся работы по улучшению механической прочности
керамических изделий. Так, например, при добавлении к
керамической массе плавленого глинозема удалось изготовить
весьма прочные крылья для центробежных насосов. Весьма
перспективны в этом отношении методы ситаллургии (стр. 667).
Для изготовления керамических изделий сырьевые
материалы измельчают и приготовленную шихту смешивают с водой до
образования пластичной массы. Из этой массы формуют или
отливают изделия, которые далее подвергают сушке и обжигу
(получение черепка).
По свойствам черепка керамические изделия делят на два
основных класса:
1) изделия с пористым, непрозрачным черепком,
обладающие землистым изломом и всасывающей способностью (вплоть
до пропускания воды);
2) изделия с плотным, спекшимся черепком, имеющие
глянцевитый излом и не обладающие всасывающей способностью.
К первому классу относят строительную керамику (кирпич,
черепица, гончарный товар и др.) и огнеупорные изделия; ко
второму классу — каменный товар (кислотоупорная
аппаратура, канализационные трубы, метлахские плитки и др.) и
фарфор.
СЫРЬЕ ДЛЯ КЕРАМИЧЕСКИХ ИЗДЕЛИЙ
Основным видом сырья для керамических изделий
являются глина и содержащие глину горные породы. Чистая глина
состоит из минерала каолина, химический состав которого
соответствует формуле А1гО3 • 2S1O2-2H2O.
Каолин содержит (в % вес):
А12О3 39,7
SiO3 46,4
Н2О 13,9
Обычно для изготовления керамических изделий применяют
различные глины, в которых, кроме каолина, имеются примеси
кварца, полевого шпата, карбонатов кальция и магния,
соединений железа и др.
По назначению глин для изготовления тех или иных изделий
различают фарфоровые глины, огнеупорные, терракотовые,
кирпичные, черепичные, клинкерные и т. д.
Важнейшим свойством глины как керамического сырья
является пластичность, т. е. ее способность в смеси с
определенным количеством воды принимать любую форму и сохранять
ее после сушки и обжига. Различают высокопластичные, сред-
непластичные, умереннопластичные, малопластичные и непла-
612
стичные глины. Непластичные глины не образуют пластичного
теста. Высокопластичные (жирные) глины при замешивании с
водой образуют очень пластичное тесто, легко поддающееся
формованию. Однако изделия из таких глин при сушке и
обжиге дают большую усадку, что ведет к сильным деформациям
и образованию трещин. При замешивании с водой
малопластичных и непластичных (тощих, или сухих) глин получается менее
пластичное тесто, но при сушке и обжиге изделий наблюдается
меньшая усадка и деформация.
В зависимости от свойств исходного материала и-рода
изделий на практике приходится повышать или уменьшать
пластичность глин. Для уменьшения пластичности к высокопластичным
глинам добавляют сухие глины или так называемые отощаю-
щие добавки: шамот, квэрц, полевой шпат и др. Увеличение
пластичности глин достигается при выветривании их,
вылеживании, вымораживании, препарировании массы, добавлении
электролитов и органических коллоидов или высокопластичных
глин.
Процесс выветривания заключается в том, что глину
оставляют лежать на воздухе в виде небольших грядок в течение
года и дольше. Вылеживание глины производится в сырых
подвалах з течение нескольких недель при 3—7°С. В обоих
случаях происходит присоединение воды к окиси алюминия,
благодаря чему увеличивается пластичность глины. Электролиты,
увеличивающие пластичность глин, должны содержать ионы
ОН". Наличие ионов Н+ уменьшает пластичность. Из
органических коллоидных веществ для повышения пластичности
применяют гумус, сахарную патоку, декстрин и др.
Пластичность глин повышают также путем дезаэрации, т. е.
вакуумирования, для уменьшения количества находящегося в
массе воздуха и другими методами.
Огнеупорность глин зависит от их химического и
минералогического состава. По огнеупорности (температуре размягчения
изделий) различают огнеупорные глины (показатель
огнеупорности выше 1580СС), тугоплавкие глины (показатель
огнеупорности от 1350 до 1580 °С) и легкоплавкие глины (показатель
огнеупорности ниже 1350 Г'С).
При изготовлении изделий с плотным черепком,
обладающих хорошей механической прочностью, кислотоупорностью и
другими ценными свойствами, к глинам добавляют плавни —
полевой шпат, доломит, магнезит ,и т. п. Наибольшее значение
имеет калиевый полевой шпат (т. пл. около 1200°С). При
обжиге керамических изделий калиевый полевой шпат плавится,
образуя вязкую стеклообразную массу, которая заполняет
промежутки между зернами остальных компонентов керамической
массы, благодаря чему после остывания получается
монолитный материал. При производстве мягкого и твердого фарфора,
613
фаянса, кислотоупорных, изделии, метлахских и облицовочных
плиток, канализационных труб, а также при изготовлении
глазурей и эмалей полевой шпат является необходимым
компонентом шихты.
К пластичным керамическим материалам относятся также
тальковые горные породы, основой которых является минерал
стеатит, или плотный тальк.
Тальк содержит (в % вес):
MgO 31,8
SiO2 63,6
Н2О 4,6
ПРОИЗВОДСТВО КЕРАМИЧЕСКИХ ИЗДЕЛИЙ
Керамические массы бывают природные, получаемые
обработкой одной какой-либо горной породы, и искусственные,
получаемые смешением нескольких видов материалов.
Из природных керамических масс изготовляют так
называемую грубую керамику: гончарные изделия, строительный
кирпич, шамотный товар, дренажные трубы, облицовочные плитки
и др. Сюда относятся также кислотоупорные изделия.
Из искусственных керамических масс изготовляют главным
образом фарфоровые и фаянсовые изделия — тонкую
керамику.
Подготовка сырья. Подготовка сырья производится
различными способами, в зависимости от свойств сырья, условий
производства и характера получаемых керамических изделий, и
заключается в дроблении, смешении и увлажнении. При
производстве фарфора и фаянса каолин часто обогащают, т. е.
освобождают от излишних примесей (песка, железосодержащих
минералов и др.) отмучиванием и воздушной сепарацией. В
большинстве случаев сырье перед дроблением сушат, а при
изготовлении шамотных изделий обжигают.
Для дробления сырья применяют щековые дробилки,
дезинтеграторы, вальцевые и шаровые мельницы, бегуны и др.
После измельчения материалам придают определенный
гранулометрический состав*.
Приготовление керамических масс. Этот процесс может
осуществляться несколькими путями:
1) при пластичном способе формовки изделий — смешением
измельченных материалов с последующим увлажнением их и
разминанием до тестообразного состояния;
2) при отливке изделий — размешиванием тонко
измельченных материалов в воде;
* Под гранулометрическим составом материала понимают
определенное процентное содержание в нем зерен различной крупности.
614
3) подсушкой материалов до 8—12% влажности — при
полусухом способе формовки ,или сушкой до полного удаления
влаги — при сухом способе.
Смешение производят в аппаратах различных типов: в
вертикальных и горизонтальных глиномялках, а при больших
масштабах производства — в мешателях периодического действия.
При существующих способах формовки содержание влаги в
керамической массе должно составлять от 4 до 40%. Если после
смешения керамическая масса содержит избыточную влагу, ее
удаляют на фильтрпрессах. При производстве ответственных
огнеупорных изделий или тонкой керамики полученную
керамическую массу дополнительно обрабатывают на специальных
массомялках для придания ей большей однородности и для
возможно более полного удаления пузырьков воздуха.
Формование изделий. Приготовленная керамическая масса
поступает на формование.
Основными способами формования керамических изделий
являются: пластичный способ — формовка керамических масс
в пластичном (тестообразном) состоянии (фарфоровые и
фаянсовые изделия, керамические трубы, изоляторы и др.); сухой и
полусухой способы — прессование керамических масс (кирпич,
облицовочные плитки и др.); отливка изделий сложной формы
из суспензий тонко измельченных керамических масс.
В настоящее время применяют главным образом отливку и
машинную формовку изделий как из пластичных, так и из сухих
и полусухих керамических масс.
Отливка изделий производится следующим образом.
Керамическую массу размешивают с водой до густоты сливок и
выливают в толстостенную форму из гипса. Вода впитывается
формой, и на внутренней поверхности формы образуется корка
твердой массы. Избыток керамической массы выливают. При
затвердевании в результате последующего испарения влаги
происходит усадка массы, поэтому отформованное изделие
довольно хорошо отстает от формы. Методом отливки изготовляют
фаянсовые, кислотоупорные и другие изделия.
Машинная формовка керамических изделий производится
на ленточных или ящичных прессах. Для пластичного
прессования служат ленточные прессы. Ящичные прессы используются
главным образом для сухого и полусухого прессования.
В ленточных прессах (рис. 219) керамическая масса из
загрузочной коробки / через вальцы 2 поступает в прессовальный
барабан 3 и при помощи шнека 4 выдавливается через
выходное отверстие мундштука 5 в виде ленты 6. На резательной
машине 7 или на столике лента разделывается на изделия.
Вода для смачивания мундштука (смазка) подается из бачка.
При формовке в ящичных прессах керамическая масса
выдавливается в формы при помощи штампа.
615
Обжиг изделии. После формовки керамические изделия
подвергают естественной или искусственной сушке, а затем обжигу
в специальных печах.
Обжиг — самая ответственная операция технологического
процесса изготовления керамических изделий. При обжиге
происходит ряд сложных физических и химических процессов. Для
удаления влаги и превращения керамической массы в твердый
Керамическая
масса
Рис, 219. Ленточный пресс:
/—загрузочная коробка; 2—вальцы для подачи керамической массы в
барабан; Л—прессовальный барабан; 4—шнек; 5— мундштук; 6— пресованная
керамическая масса; 7—резательная машина.
черепок требуется определенный тепловой режим,
устанавливаемый для каждого вида материала в отдельности.
Нарушение этого режима приводит к образованию трещин, рванин,
короблению и сплавлению готовых изделий. Максимальная
температура обжига также различна для разных видов
изделий. Например, кирпич из обыкновенной глины обжигают при
900—1100°С, кирпич из огнеупорной глины при 1300—1450 °С,
кислотоупорные изделия при 1250—1350 °С, магнезитовый
кирпич при 1550—1680 °С.
Обжиг керамических изделий производят обыкновенно в
кольцевых или туннельных печах. Наиболее распространены
непрерывно действующие кольцевые печи.
На рис. 220 схематически изображен разрез шестнадцати-
камерной кольцевой печи. Печь состоит из кольцевого канала,
разделенного на 16 камер. Каждая камера соединена с
центральным дымоходом. В работающей печи имеются три зоны:
подогрева, обжига и охлаждения.
Если в шестнадцатикамерной печи полный цикл обжига
керамических изделий проводят в течение семи суток, то печь
должна работать по следующей схеме. В то время как из
камер 1 и 2 выгружают изделия, в камерах 3—6 происходит
616
охлаждение уже обожженных изделий. Для этого через данные
камеры просасывают холодный воздух, который, охлаждая
изделия, нагревается. Нагретый воздух поступает в камеры 7 и 8,
где в этот момент идет обжиг при максимальной температуре.
Рис. 220. Кольцевая печь для обжига керамических изделий:
/—16—камеры.
Горячие продукты горения поступают в камеры 9—14, где
происходит постепенный подогрев и подсушка сырых изделий, и
выходят в трубу из камеры 14. В это время камеры 15 и 16
загружают свежими (сырыми) изделиями.
Через сутки все процессы сдвигаются на две камеры влево,
т. е. камеры / и 2 загружают, камеры 3 и 4 разгружают, зона
охлаждения охватывает
камеры 5—8, зона обжига —
камеры 9 и 10, подогрев и
подсушка происходят в
камерах //—16.
В кольцевых печах осу- /
ществлен принцип
противотока; несмотря на это,
расход топлива достигает 10—
20% от веса обжигаемых
изделий.
Наряду с кольцевыми
печами имеют
распространение многокамерные тун- Рис. 221. Туннельиаи печь:
ИР/thHhlP ПРЧи nnannvmni. У—дымоход; 2—отверстия для входа газа и вы-
нельные печи прямоуГОЛЬ- хода продуктов сгорания; J-вагонетка; 4-пе-
ного сечения. В таких пе- сочный затвор,
чах (рис. 221) камеры расположены в одну линию и
представляют собой сквозной канал, в котором перемещаются
вагонетки с обжигаемыми изделиями. В туннельных печах также
имеются зоны подогрева, обжига и охлаждения.
617
Обжигаемые изделия подаются вагонетками 3 в зону
подогрева. Платформы вагонеток плотно примыкают друг к другу.
образуя сплошной под печи. Продольные края вагонеток имеют
щитки, которые продвигаются по песочным затворам 4,
расположенным вдоль рельсового пути по обеим сторонам канала.
Благодаря этому оси и колеса вагонеток, а также рельсы
защищены от воздействия высокой температуры; кроме того, они
охлаждаются воздухом, подаваемым вентилятором по
трубопроводу, расположенному между рельсами.
Через определенные промежутки времени (от 30 мин до 3 ч)
цепь вагонеток проталкивается вперед на расстояние одной за-
гонетки. При этом в начале зоны подогрева освобождается
место для загрузки новой вагонетки, а из зоны охлаждения
выходит вагонетка с готовыми изделиями.
Для создания нужных температурных условий в зону
обжига подают горячие топочные газы или генераторный газ.
Воздух, необходимый для сжигания генераторного газа,
поступает через зону охлаждения, где он подогревается, охлаждая
обожженные изделия. Продукты сгорания из зоны обжига
поступают в зону подогрева, а затем через отверстия 2 уходят
в дымоход /.
Туннельные печи имеют ряд преимуществ по сравнению с
кольцевыми печами: более удобная загрузка и разгрузка;
лучшие условия работы (в помещениях с нормальной
температурой и хорошей вентиляцией); меньший расход топлива, чем
в кольцевых печах, благодаря лучшему использованию тепла
продуктов горения и др.
Нанесение рисунков и глазури на керамические изделия.
Рисунки наносят на обожженные изделия различными
способами: ручной живописью, раскраской с помощью пульверизатора
по трафарету, раскраской штампом, декалькоманией и
фотокерамикой. Наиболее распространенный способ раскраски —
декалькомания состоит в том, что рисунок, отпечатанный на
гуммированной бумаге керамическими красками, переводят на
изделия (как детские переводные картинки). После
закрепления рисунка путем специальной обработки изделия сушат и
затем обжигают.
По способу фотокерамики на обожженные,
преимущественно глазурованные изделия наносят светочувствительный слой
и фотографируют рисунок, затем изделия обжигают при низкой
температуре.
В качестве керамических красок употребляют главным
образом окислы различных металлов и их соли: силикаты, бораты
и алюминаты. Окись железа дает много оттенков от светло-
желтых до темно-коричневых: окись хрома — зеленый; смесь
окиси хрома и окиси олова — розово-красный; окись кобальта—
синий, а в присутствии свинца и натрия — фиолетовый; окись
618
калия — ярко-синий; окислы сурьмы — желто-оранжевые
оттенки; золото—пурпурный.
Некоторые керамические изделия покрывают глазурями,
которые придают поверхности изделия плотность (газо- и
водонепроницаемость), твердость, блеск и стойкость к химическим
воздействиям. Глазури наносятся тонким слоем и представляют
собой стекловидные силикаты. Применяют главным
образом щелочноземельные глазури (для покрытия кислото- и
щелочеупорной керамики) и соляные глазури, а также
бессвинцовые глазури.
Температура плавления глазури должна соответствовать
температуре обжига керамических изделий. Поэтому в состав
глазури вводят большое количество плавней, например
полевого шпата.
Глазурь для фарфора имеет примерно следующий состав
(в % вес):
Каолин 9
Кварц 27
Полевой шпат . . 64
Коэффициенты расширения глазури и черепка, на который
ее наносят, должны быть по возможности равны, иначе при
обжиге глазурь трескается.
Глазурь наносят на изделия различными способами:
погружением изделий в сосуд с глазурью, обливанием изделий,
намазыванием кистью и обрызгиванием из пульверизатора
Изделия покрывают глазурью до обжига или после обжига (в этом
случае их вторично обжигают). Последний способ применяют
для глазирования тонкой керамики (фарфора и фаянса).
Для каменного товара применяют соляную глазурь,
которая получается при действии паров хлористого натрия на
обжигаемые изделия. В топку печи перед концом обжига
загружают некоторое количество поваренной соли, соль возгоняется
и ее пары реагируют с соединениями кремния и алюминия,
содержащимися в черепке обжигаемых изделий, и парами воды,
образуя силикаты и алюмосиликаты натрия.
ФАРФОР И ФАЯНС
Фарфор и фаянс отличаются друг от друга физическими
свойствами черепка: фарфор имеет плотный черепок, фаянс—
пористый. Фарфоровые и фаянсовые изделия обычно
покрывают глазурью. В химической промышленности большое
применение имеют изделия из фарфора вследствие их высокой
кислото- и щелочестойкости, механической прочности, термической
устойчивости и достаточной огнеупорности.
В состав шихты для изготовления фарфора и фаянса входят
пластичные материалы (каолины, белые глины), непластичные,
619
или отощающие, материалы (кварц, обожженные глины, бой
фарфора или фаянса) и плавни (полевой шпат, мел и др.)-
Изготовление фарфоровых изделий отличается от
технологии обычных керамических изделий более тщательной
подготовкой сырья, тонким измельчением его, хорошим смешением
пластичных и непластичных материалов. Формовку изделий
производят пластичным способом, отливкой или холодной
штамповкой. Сушка готовых изделий, ранее производившаяся на
стеллажах, в настоящее время проводится в специальных
сушилках.
Различают твердый фарфор, обжигаемый при 1350—1400 СС,
и мягкий фарфор, обжигаемый при 1200—1350 °С и содержащий
поэтому большее количество плавней, чем твердый фарфор.
Примерный состав твердого и мягкого фарфора (в % вес):
Твердый Мягкий
фарфор фарфор
Каолин 50 25—40
Кварц 25 30—45
Полевой шпат 25 30—35
Мягкий фарфор применяют главным образом для
изготовления хозяйственной посуды, художественных изделий,
низковольтных изоляторов. Твердый фарфор широко используется
при производстве химической посуды, высоковольтных
изоляторов и других технических изделий.
Фаянс также бывает мягкий и твердый. Мягкий фаянс
отличается от твердого меньшей плотностью и более мягким
черепком. Обычно мягкий фаянс изготовляют из пластичных глин
(до 80%) н кварца (до 20%). Плавни (полевой шпат)
добавляют редко и в малом количестве. Из мягкого фаянса делают
пористые сосуды и фильтры, а также хозяйственную посуду.
Твердый фаянс изготовляют из глин D5—55%) и кварцевогэ
песка B5—30%) с добавками 10% боя (черепка) и 5—8%
полевого шпата. Из твердого фаянса вырабатывают
глазурованные плитки для облицовки стен и санитарно-технические
изделия (умывальники, ванны и т. п.).
КИСЛОТОУПОРНЫЕ ИЗДЕЛИЯ
В химической промышленности применяют грубые и тонкие
кислотоупорные даделия из керамики. К грубым изделиям
относятся кислотоупорный кирпич, облицовочные плитки, кольца
для насадок в абсорбционных башнях и т. д., к тонким
изделиям— детали реакционных аппаратов, насосов и др.
Сырьем для получения кислотоупорных изделий обычно
служат легкоплавкие (низкоспекающиеся) и среднепластич-
'20
ные глины, при обжиге-которых получается плотный и
однородный черепок. В качестве отощающей добавки применяют
обожженную глину. В шихту вводят также до 10% полевого
шпата.
Глины не должны содержать примесей гипса и соединений
железа, образующих при обжиге легкоплавкие включения, что
приводит к появлению в изделиях раковин и пузырей.
Во избежание резких деформаций черепка при обжиге
содержание окиси кальция в шихте не должно превышать 1—2%.
Для увеличения механической и термической прочности,
теплопроводности, кислотоупорности и щелочеупорное™ в шихту
иногда добавляют карборунд SiC и окись циркония ZrO2.
Сушку и обжиг кислотоупорных изделий необходимо вести
очень осторожно, не допуская их деформации и образования
трещин.
Кислотоупорные изделия покрывают соляной глазурью или
глазурью из легкоплавких глин.
На рис. 222 приведена одна из применяемых схем
производства керамических кислотоупорных изделий.
Для изготовления щелочестойких изделий часто применяют
магнезит, доломит и шамот.
Для футеровки реакторов и других кислотоупорных
аппаратов применяют керамические кислотоупорные плитки (тип К)
и керамические термохислотоупорные плитки (тип ТК),
качество которых согласно ГОСТ 961—57 должно соответствовать
следующим показателям:
Тип К Тип ТК
Кислотостойкость, %, не менее, для плиток толщиной:
до 10 мм 98 —
от 10 до 30 мм 97 97
свыше 30 мм 96 96
Водопоглощение, %, не более, для плиток толщиной:
до 30 мм 6 7
свыше 30 мм 7 9
Предел прочности, кгс/см2, не менее
при сжатии 300 300
при изгибе 150 150
Термическая стойкость, количество теплосмен, не менее 2 8
Примечание. Водопоглощеиие определяют как привес образца после
2-часового кипячения в воде, кислотостойкость—как разность весов испытуемого образца до
и после 1-часового кипячения в концентрированной серной кислоте. Термическая
стойкость характеризуется числом резких изменений температуры (от 1300 "С до
температуры проточной охлаждающей воды), которое выдерживает изделие до потери 20%
первоначального веса.
621
I Измельчение
I глины
Мокрый помол полевого
шпата и шамота
Смешение в мешателях
Мокрый просев через
сито
Обезвожива-ние
на фильтрпрес-е
Вакуумированис
на ленточном прессе
Формование
Сушка
Глазурование
Обжиг
Рис. 222. Схема производства керамических кислотоупорных изделий.
КЕРАМИЧЕСКИЕ СТРОИТЕЛЬНЫЕ МАТЕРИАЛЫ
К керамическим изделиям этой группы относятся
обыкновенный глиняный (красный) кирпич, силикатный кирпич,
пустотелый и пористый кирпич, применяемые для кладки стен,
строительная терракота (орнаментные и фигурные изделия), а
также глиняные плитки, черепица, дренажные трубы. Сырьем для
керамических строительных материалов могут служить
валунные суглинки и тощие и жирные глины с добавками песка,
шамота, а иногда органических веществ для придания
керамическим изделиям пористости. Такое сырье весьма
распространено в природе.
Для изготовления обыкновенного глиняного кирпича
применяют глины, содержащие 53—81% SiO2, 7—23% А12О3, 2—8%
Fe2O3, до 14% СаО и некоторое количество MgO, окислов
щелочных металлов и других примесей.
Кирпич формуют главным образом в глиномялках с
мундштуком, а также в прессах. Перед обжигом кирпич-сырец
подвергают естественной или искусственной сушке. Естественную
сушку на стеллажах часто комбинируют с сушкой в камерах
или туннельных сушилках. Обжиг кирпича производят в печах
различного типа (стр. 617) при 900—1000°С в случае
пластичного способа формования и при 950—1100°С, если формование
ведут полусухим способом.
Размеры готового кирпича 250X120X65 мм, поверхность
его должна быть ровной, без трещин и свищей. Предел
прочности кирпича при растяжении и сжатии в зависимости от
сорта от 150 до 75 кгс/см2, плотность 2,3—2,68 г/см3, водопоглоще-
ние не ниже 8 и не выше 20%.
Кирпич при испытаниях должен выдерживать 15-кратное
попеременное замораживание и оттаивание без последующих
деформаций.
Силикатный кирпич, применяемый в настоящее время в
больших количествах, изготовляют безобжиговым способом из
увлажненной смеси песка и извести. Отформованный на
прессах кирпич обрабатывают в течение 8—14 ч паром под
давлением около 10 ат. Таким же способом изготовляют шлаковый
кирпич из доменных шлаков.
Пористый кирпич, обладающий хорошими
теплоизоляционными свойствами, изготовляют из диатомитов (диатомитовый
кирпич) и трепелов — горных пород пористой структуры, или
из глины с добавкой компонентов, выгорающих при обжиге
(древесные опилки и др.).
Производство черепицы, дренажных труб и терракоты
сходно с производством обыкновенного глиняного кирпича, но в
этом случае требуются лучшие сорта однородных пластичных
глин.
623
ОГНЕУПОРЫ
Огнеупорами называются керамические изделия,
сохраняющие свои механические свойства при высокой температуре и не
разрушающиеся при этой температуре под воздействием [Таз-
личных газов, расплавленных шлаков и металлов и др.
При оценке качества огнеупоров, кроме огнеупорности,
учитывают также следующие показатели: предел прочности при
растяжении, сжатии и изгибе при обычных и высоких
температурах, истираемость, термическую устойчивость, постоянство
объема, газопроницаемость, химическую стойкость, тепло- и
электропроводность.
Огнеупорность обычно характеризуется температурой
размягчения керамических изделий. Эти изделия состоят из
сложных смесей различных компонентов и потому не имеют
определенной температуры плавления, а переходят из твердого в
жидкое состояние в некотором интервале температур,
достигающем иногда нескольких сот градусов.
Огнеупорные изделия должны иметь температуру
размягчения не ниже 1580 °С, высокоогнеупорные изделия — не ниже
1770°С, изделия высшей огнеупорности — не ниже 2000 °С.
Сопротивление действию высокой температуры зависит не только
от температуры рязмягчения огнеупоров, но и от условий их
эксплуатации, особенно от воздействия механических нагрузок
и реакционной среды.
По химическому составу огнеупоры разделяются на кислые,
основные и нейтральные.
Наиболее важными видами огнеупоров являются шамотные
изделия (кислые, основные, нейтральные), динас (кислые
изделия) и магнезиальные (основные) изделия.
Шамотные изделия. Шамотом называется прокаленная и
измельченная огнеупорная глина. Шамотные изделия
изготовляют из шамотных масс, т. е. из смеси шамота и глины в
соотношении от 1 : 1 до 1 :3.
Производство шамотных изделий заключается в следующем.
Сначала глину обжигают, затем размалывают и составляют
смесь требуемого гранулометрического состава.
Гранулометрический состав шамота и глины сильно сказывается на
огнеупорности и механических свойствах готовых шамотных изделий.
Приготовленный шамот смешивают с глиной, увлажняют
смесь до получения пластичной массы и формуют. Во
избежание деформаций отформованные изделия тщательно сушат в
течение 10—20 суток. Обжиг изделий проводят при 1300—
1425°С.
Нейтральный, или нормальный, шамот изготовляют из
обычной огнеупорной глины, основной (глиноземистый)—с
добавкой окиси алюминия, кислый (кварцевый) — из глины с
частичной или полной заменой обожженной глины кварцевым песком.
624
Шамотные изделия довольно легко выдерживают резкие
колебания температуры, но вследствие большой пористости они
газопроницаемы, что ограничивает их применение. Особенно
газопроницаемым является кварцевый шамот, содержащий
крупные зерна кварца.
Химический состав шамотных глин и кирпича приведен в
табл. 31.
Таблица 31
Средний химический состав шамотных глин и кирпича
Шамотные глины н кирпич
Глины
Боровнчская . . .
Глуховецкая . . .
Кыштымская . . .
Кирпич
Основной
Кислый
SiO2
44,2
47,0
50,0
52,5
73,8
Al2o3
39,4
33,1
36,6
43,6
22,8
Состав, вес
Fe2O3
1,6
1,2
1,9
1,6
1,9
СаО
0,2
0,7
1,4
0,6
0,5
. %
MgO
0,10
1,0
0,6
0 5
0,7
Na2O,
К2О
0,8
1,2
0,3
Н2О
13,7
17,0
9,5
Изделия из шамота широко применяют для футеровки печей
и газогенераторов, для изготовления тиглей, реторт, муфелей,
стекловаренных горшков и др.
В зависимости от огнеупорности различают шамотные
изделия следующих четырех классов:
Класс
О .
А .
Б .
В .
Огнеупорность
°с
Выше 1750
Не ниже 1730
з> » 1670
» » 1610
При хранении шамотных изделий нельзя допускать их
увлажнения и последующего замораживания (в зимнее время),
укладывать их следует в штабели высотой 2—2,5 м. Перевозка
шамотных изделий должна производиться в крытых вагонах.
Динасовые изделия. Для сооружения промышленных печей,
работающих при высоких температурах (мартеновские,
электрические сталеплавильные, стекловаренные, коксовые печи,
бессемеровские конверторы и др.), широко применяют
динасовые изделия.
Динас содержит не менее 92% SiO2. Благодаря этому на
него не оказывают действия кислоты, кроме плавиковой, но
0—2848
625
разрушающе действуют окислы металлов и основные силикаты,
образующиеся в печах при высокой температуре.
Главное достоинство динаса заключается в том, что при
достаточной огнеупорности (обычно 1710—1750°С) он
сохраняет форму почти до самого момента плавления и не дает
усадки. Газопроницаемость динасовых изделий меньше, чем
шамотных. При эксплуатации печей из динасовых изделий
необходимо избегать резких колебаний температуры, так как при
этом появляются трещины, что приводит к разрушению кладки.
Основным сырьем для получения динаса являются
кварциты — горные породы, состоящие преимущественно из кварца:
Для получения высококачественного динаса применяют
кварциты, содержащие не менее 96% SiO2, при изготовлении динаса
для электрических сталеплавильных печен кварцит должен
содержать не менее 97,5% SiO2. Кварциты измельчают до
величины зерен не более 3 мм, причем фракции зерен размером
менее 0,5 мм должны составлять 50—60% от веса
измельченного кварцита. Затем из кварцитов различных сортоз
составляют шихту, которую смешивают с известковым молоком и
обрабатывают на смешивающих (мокрых) бегунах до достижения
определенного гранулометрического состава смеси.
Известь играет роль связующего вещества и
минерализатора, при обжиге она ускоряет превращение кварца в
устойчивые его модификации. Кроме извести, минерализаторами могут
служить окислы железа. В качестве органических связующих
веществ при производстве динаса могут применяться патока,
меласса и сульфитцеллюлозные щелока.
Из приготовленной таким образом динасовой массы
формуют изделия, которые после сушки в специальных сушилках
при 120°С обжигают при температуре около 1500 °С.
Обжиг — самая ответственная операция при получении
динаса. Температурный режим обжига устанавливают в
зависимости от качества кварцитов и системы печей. Обычно
нагревание продолжается 7—10 суток, а охлаждение 4—6 суток. На
рис. 223 представлена примерная температурная кривая
обжига динаса.
Динасовый кирпич, предназначенный для кладки
мартеновских печей, должен удовлетворять следующим требованиям:
SiO2, %
А12О3, % ¦
СаО, %
Огнеупорность, °С, не ниже
Температура (в °С) начала деформации при иа-
грузке 2 кгс/см2
Плотность, т/м3
Пористость, %, не более
Предел прочности при сжатии, кгс/смг, не менее
(j26
1-й класс
94,0
1,5
3,0
1710
1650
2,38
32
178
2-й класс
92,0
1,8
3,5
1690
1620
2,42
25
150
1600
BOO
/
/
\
,
ч
—
0
12
4 6 8//?
время, CL/тни
Рис. 223. Температурная кривая
обжига динаса.
Магнезиальные изделия представляют собой
высокоогнеупорные материалы. Чистая окись магния плавится при температуре
около 2800°С. Она применяется при изготовлении специальных
огнеупорных изделий, например тиглей для плавки основных
силикатов. Огнеупорность магнезиальных изделий, содержащих
обычно не менее 90% MgO, составляет 1825—1850 °С. При
2000°С окись магния восстанавливается углеродом до
металлического магния, который
испаряется. Магнезиальные
изделия плохо переносят
колебания температуры.
Для получения
магнезиальных изделий жженую
магнезию смешивают с водой или
органическими связующими ве-
ществами. Если при
изготовлении массы применяют воду,
то надо выдержать
замешанную массу во влажной атмо-
сфере 5—7 суток, чтобы успела
произойти полная гидратация
окиси магния. Полученную массу формуют под давлением
около 4 ат, сушат при 120°С и обжигают.
В промышленности применяют магнезию, получаемую
путем обжига минерала магнезита MgCOa. Обожженный минерал,
часто неправильно называемый просто магнезитом, бывает
двух видов: каустический магнезит, применяемый для
приготовления магнезиального цемента и в качестве изоляционного
материала, и металлургический порошок, применяемый для
изготовления огнеупорных изделий.
Каустический магнезит (объемный вес 1,2—1,3 т/м3)
получают в результате обжига природного магнезита при 800—
900 СС. Содержание посторонних окислов (S1O2, А12О3, ИегОз и
СаО) в каустическом магнезите не должно превышать 10%.
Металлургический порошок — обожженный при 1500—1600 °С
магнезит — имеет объемный вес 1,8 т/м3.
Для изготовления магнезиальной массы металлургический
порошок увлажняют 5—6% воды и оставляют на несколько
суток во влажной атмосфере.
Затем порошок смешивают с густым крахмальным
клейстером до получения хорошо формующегося теста. Форму
изнутри обкладывают бумагой. После формования изделие
подсушивают и обжигают при 1600°С.
Обожженные изделия выдерживают нагревание до 1800 °С.
Если исходным материалом служит чистая окись магния,
полученная осаждением в водной среде, ее следует прокалить,
а при замешивании массы добавить к ней 2—5% глинозема.
40*
627
В качестве заменителя магнезиальных изделий широко
применяют изделия из доломита, представляющего собой двойную
углекислую соль кальция и магния MgCO3 • СаСО3 с примесью
SiO2, Fe2O3, Мп3О4 и др.
Специальные огнеупорные материалы. Специальными
огнеупорными материалами называют материалы, обладающие
очень высокой огнеупорностью и стойкие к действию различных
реагентов. К таким материалам относятся хромитовые,
высокоглиноземистые, углеродсодержащие, форстеритовые,
карборундовые и циркониевые огнеупоры, а также некоторые нитриды
и карбиды.
Хромитовые огнеупорные материалы обладают стойкостью
по отношению к кислым и щелочным реагентам, поэтому из них
изготовляют тигли для выплавки металлов из различных руд.
Исходным сырьем служит хромистый железняк, который
смешивают со связующими веществами — глиной, гипсом,
сульфатом алюминия, формуют и затем обжигают. Температура
размягчения хромитовых огнеупоров около 2000 °С.
Высокоглиноземистые огнеупоры содержат более 45% А12О3-
Они обладают хорошей стойкостью к действию шлаков.
Углеродистые огнеупорные изделия изготовляют обычно из
прессованного древесного угля, кокса или графита. Эти
изделия выдерживают температуру до 3500 °С, не разрушаются
шлаками и другими силикатами. Однако при нагревании в
окислительной среде (например, в воздухе) такие огнеупоры
быстро изнашиваются. Наибольшее распространение из
углеродистых изделий имеют графитовые тигли, а также
графитовые электроды.
Форстеритовые огнеупоры состоят в основном из минерала
форстерита Mg2SiO4. Они изготовляются из природных
силикатов магния, содержащих MgO и БЮг в соотношении примерно
0,94:1,33. Форстеритовые изделия отличаются хорошей
термической устойчивостью и стойки к воздействию основных
шлаков. Температура их размягчения выше 1700°С.
Карборунд, или технический карбид кремния, является
химическим соединением кремния и углерода SiC. Его
температура плавления около 2500 °С, однако уже при 2000 °С он
начинает разлагаться и диссоциировать на углерод и кремний.
Карборунд хорошо выдерживает колебания температуры, обладает
хорошей теплопроводностью и весьма прочен, что позволяет
изготовлять из него тонкостенные изделия.
Выше 1000°С карборунд легко поддается воздействию
расплавленных металлов и основных силикатов; кислые силикаты,
особенно чистый кремнезем, оказывают на него меньшее
действие, поэтому карборунд применяют при плавлении кварца.
Изготовление изделий из карборунда сопряжено с трудностями
из-за плохой формуемости материала.
628
Обычно карборундовые изделия изготовляют путем
формования- смеси тонко измельченного карборунда с различными
органическими связующими веществами (например, с
крахмалом) и последующего обжига, главным образом в
электрических печах. Карборунд служит также для изготовления
специального кирпича и тиглей.
Изделия из двуокиси циркония относятся к
высокоогнеупорным— выдерживают температуру до 2000 °С; они устойчивы
к действию кислот и щелочей и почти не электропроводны. Эти
свойства позволяют применять циркониевые изделия в
лабораторной практике; из двуокиси циркония изготовляют трубки и
тигли для намотки молибденовых и вольфрамовых
сопротивлений, тигли для плавления различных шлаков и т. п. Изделия
изготовляют путем формования смеси двуокиси циркония со
связующими веществами, например с крахмалом.
Отформованные изделия осторожно обжигают при 1500°С.
Корунд представляет собой чистую окись алюминия. Он
плавится при температуре около 2050СС, нечувствителен к
резким изменениям температуры. Огнеупорные изделия из корунда
с примесью глины носят название динамадоновых.
Нитриды и карбиды некоторых элементов применяются как
особо огнеупорные материалы (высшей огнеупорности).
Нитрид бора выдерживает температуру выше 3000 °С, смесь
нитрида титана и карбида титана плавится при 3222°С, смесь
4 моль карбида таллия и 1 моль карбида циркония — при
3932 °С, смесь 4 моль карбида таллия и 1 моль карбида таф-
ния —при 4215 °С.
Кроме того, для специальных целей в настоящее время
употребляются различные виды высокоогнеупорных материалов:
титановые, вольфрамовые, танталовые, урановые, ториевые
и др. Их применяют, например, для изготовления тиглей, в
которых проводится плавка чистых редких металлов, а также для
получения искусственных кристаллов, используемых в
радиотехнике.
2. МИНЕРАЛЬНЫЕ ВЯЖУЩИЕ ВЕЩЕСТВА
Вяжущими веществами называются материалы,
применяемые в качестве связующих и обладающие способностью
переходить из жидкого или тестообразного состояния в твердое.
Вяжущие вещества разделяются на минеральные и
органические. Органические вяжущие вещества — смолы, битумы,
клеи и др.— в настоящей книге не рассматриваются.
Минеральные вяжущие вещества делят на воздушные и
гидравлические. Воздушные вяжущие вещества твердеют лишь
на воздухе. К ним относятся воздушная известь, гипсовые и
магнезиальные вяжущие вещества. Гидравлические вяжущие
вещества могут твердеть и длительно сохранять прочность как
629
на воздухе, так и в воде. К ним относятся гидравлическая
известь, роман-цемент, портланд-цемент, пуццолановые и
шлаковые цементы, цементы с микронаполнителями, глиноземистый
цемент и некоторые новые виды цемента.
В связи с исключительно быстрым развитием строительства
в СССР потребность в минеральных вяжущих веществах
непрерывно возрастает и производство их увеличивается.
В 1960 г. было произведено 45,4 млн. т цемента, в 1963 г.
61 млн. т. В 1965 г. и в последующие годы предусматривается
дальнейший рост производства цемента.
ВОЗДУШНЫЕ ВЯЖУЩИЕ ВЕЩЕСТВА
Воздушная известь является важнейшим воздушным
вяжущим веществом и очень широко применяется в строительстве.
Ее получают обжигом чистых или магнезиальных известняков
по возможности до полного выделения СОг (см. стр. 430 ел.),
но без спекания материала.
Различают следующие виды строительной воздушной
извести:
1. Негашеная комовая известь, или кипелка, — продукт
обжига известняка, состоящий главным образом из СаО и
имеющий вид пористых кусков белого или серого цвета.
2. Негашеная молотая известь — измельченная комовая
известь.
3". Гидратная известь, или пушонка,— тонкий порошок,
получаемый в результате гашения комовой извести определенным
количеством воды и состоящий в основном из Са(ОН)г.
4. Известковое тесто — материал пластичной консистенции,
получаемый при гашении комовой извести избытком воды и
состоящий главным образом из Са(ОНJ и воды.
Процесс гашения извести сопровождается выделением
значительного количества тепла:
СаО + Н.2О = Са(ОНJ +15,5 ккал
Поэтому при хранении и перевозке негашеной извести
следует предохранять ее от воздействия влаги во избежание
сильного разогрева продукта и возможного при этом
воспламенения деревянных частей склада или вагона.
Гашение извести производят в гидраторах различных
конструкций при энергичном перемешивании всей массы.
Простейшим видом гидратора непрерывного действия является
гасильный шнек, представляющий собой закрытый желоб, в
котором вращается лопастной винт.
В качестве гидраторов применяются также вращающиеся
гасильные барабаны.
630
Процесс затвердевания известкового раствора заключается
в испарении избыточной воды и образовании карбоната
кальция в результате поглощения двуокиси углерода из воздуха:
Са(ОНJ -f СО3 = СаСО3 + Н2О
Превращение гидроокиси кальция в карбонат кальция
происходит в течение длительного времени, так как образующаяся
наружная корка СаСОз затрудняет проникание воздуха внутрь
массы и выделение из нее воды.
Гипсовые вяжущие вещества получаются путем обжига гипса
CaSO4-2H2O. В зависимости от температуры обжига гипс
способен терять различное количество кристаллизационной воды.
Гипсовые вяжущие вещества делят на две основные группы:
низкообжиговые и высокообжиговые. Низкообжиговые
вяжущие вещества, получаемые обжигом гипса при низких
температурах, состоят главным образом из полуводного сульфата
кальция CaSO4-0,5^0 и быстро твердеют. Вяжущие вещества,
получаемые обжигом при высоких температурах, твердеют
медленно. Наиболее распространены низкообжиговые материалы
(например, строительный гипс).
Строительный гипс получается путем обжига гипса при
температуре материала 140—190°С до перехода его в полуводный
сульфат кальция. Материал измельчают до или после обжига
или в процессе обжига. Тонко размолотый строительный гипс,
называемый формовочным гипсом, схватывается быстрее.
Гипс можно обжигать в котлах, в аппаратах для
совмещенного помола и обжига, во вращающихся печах, в установках
для обжига во взвешенном состоянии и в других аппаратах.
Тонина помола строительного гипса должна быть такова,
чтобы остаток на сите с отверстиями размером 0,2 мм не
превышал 15—25% (в зависимости от сорта). Начало схватывания
гипса наступает не ранее 4 мин, конец схватывания — не ранее
6 мин и не позднее 30 мин после начала затворения гипсового
теста. Время от начала затворения гипсового теста до конца
кристаллизации гипса составляет не менее 12 мин. Предел
прочности при сжатии образцов строительного гипса не менее
40—55 кгс/см2 через 1,5 ч после схватывания; образцов, высу-
щенных до постоянного веса, — не менее 75—100 кгс/см2.
Гипс применяется в основном для производства
строительных деталей, известково-гипсовые вяжущие вещества — для
штукатурных работ. Строительный гипс вместе с асбестом и
другими материалами входит в состав термоизоляционных
композиций. Гипс применяется для изготовления форм при
отливке керамических изделий, скульптурных и архитектурных
деталей, а также используется в медицине для наложения
гипсовых повязок, зубоврачевания и пр.
631
Вяжущее вещество, получаемое путем обжига гипса при
высокой температуре F00—700 °С) и состоящее главным
образом из безводного сульфата кальция CaSO4, носит название
ангидритового цемента. Ангидритовый цемент получают
обжигом гипсового камня в шахтных или вращающихся печах с
последующим измельчением в тонкодисперсный порошок (остаток
не более 15% на сите с отверстиями размером 0,08 мм) и
добавлением некоторых минеральных веществ (главным образом
1—5% извести), ускоряющих процесс твердения цементного
камня. Ангидритовый цемент можно получать также
путем.измельчения природного ангидрита.
При нагревании выше 400°С гипс полностью теряет воду и
образует нерастворимый ангидрит CaSO4 (так называемый
мертвообожженный), который почти не схватывается и не
твердеет. Поэтому обожженный гипс подвергают тонкому размолу
совместно с различными минеральными добавками —
бисульфатом или сульфатом натрия в сочетании с медным или
железным купоросом, известью или обожженным доломитом,
доменным шлаком и др. Благодаря способности ангидрита вступать
в комплексные соединения на поверхности частиц
ангидритового цемента в присутствии воды образуются неустойчивые
сложные гидраты типа (Соль-тСа5О,г лН2О), которые в
процессе затвердевания раствора на основе ангидритового цемента
распадаются на кристаллогидрат (Соль-лгН2О) и дигидрат
сульфата кальция CaSO4-2H2O. По мере протекания процессов
гидратации и перекристаллизации ангидритовый цемент
приобретает все большую прочность.
Ангидритовый цемент схватывается медленнее, чем
строительный гипс. Начало схватывания ангидритового цемента
должно наступать не ранее чем через 30 мин, а конец — не
позднее чем через 24 ч от начала затворения. При твердении он
практически не увеличивается в объеме. Предел прочности при
сжатии образцов ангидритового цемента, изготовленных при
весовом отношении цемента к заполнителю (песок, шлак) 1:3,
на седьмые сутки составляет 25—100 кгс/см2 (в зависимости
от марки цемента).
Ангидритовый цемент применяется для получения
строительных растворов в смеси с песком или со шлаком,
изготовления различных теплобетонных блоков, обыкновенного бетона
низких марок (стр. 644) и др.
Высокообжиговый гипс, или эстрих-гипс, получают путем
обжига гипса или природного ангидрита в шахтных или
вращающихся печах при 800—1000 °С с последующим тонким
измельчением продукта обжига.
При обжиге часть сульфата кальция разлагается:
2CaSO4 * 2СаО + 2SO2 + О2
632
Твердение высокообжигового гипса происходит вследствие
превращения СаО в гидроокись и затем в карбонат кальция и
гидратации безводного сульфата кальция. Затвердевший
высокообжиговый гипс отличается высоким сопротивлением
истиранию. Он применяется для изготовления бесшовных полов,
кладочных и штукатурных растворов и бетонов для надземных
сооружений, а также для изготовления искусственного
мрамора и различных строительных деталей.
Магнезиальные вяжущие вещества — каустический магнезит
и каустический доломит—изготовляют путем обжига
магнезита MgCO3 или доло!мита CaCO3-MgCO3 в шахтных или
вращающихся печах при 750—850 °С.
Целью обжига является разложение MgCO3 в результате
диссоциации:
MgCO3 * MgO + СО2
Содержащийся в доломите карбонат кальция СаСО3 в этих
условиях почти не изменяется, так как для его диссоциации
требуется более высокая температура (до 1000°С).
Продукт, полученный в результате обжига, размалывают в
шаровых мельницах.
Магнезиальные вяжущие вещества, в отличие от других
вяжущих веществ, затворяют не водой, а главным образом
раствором хлористого магния MgCl2. При затворении водой
магнезиальные вяжущие вещества затвердевают очень медленно, а
в затвердевшем виде обладают небольшой прочностью. При
затворении же раствором хлористого магния магнезиальный
цемент быстро затвердевает и приобретает высокую прочность.
Магнезиальные вяжущие вещества хорошо связывают
значительные количества добавляемых к ним наполнителей и
применяются в производстве ксилолита, фибролита, искусственных
жерновых камней, точильных кругов и др.
Ксилолит — камневидная спрессованная масса, состоящая из
древесных опилок и магнезиального цемента; применяется для
настила полов, изготовления лестничных ступеней,
подоконников и т. п., а также в качестве тепловой изоляции.
Фибролит применяется в качестве строительного тепло- и
звукоизоляционного материала. Фибролитовые плиты получают
пресеованием смеси древесных стружек и магнезиального
цемента с последующей сушкой.
Искусственные жерновые камни для мельничных поставов
изготовляют из различных минеральных наполнителей
(наждак, песок, гравий и др.), сцементированных магнезиальным
цементом.
Аналогично изготовляются и точильные круги; в качестве
наполнителей применяют наждак, корунд, кварцевый песок.
633
ГИДРАВЛИЧЕСКИЕ ВЯЖУЩИЕ ВЕЩЕСТВА
Гидравлическая известь получается путем обжига
мергелистых известняков при 900—1100°С без доведения материала до
спекания. Обжиг известняка ведут в шахтных печах
непрерывного действия. Известняки, применяемые для производства
гидравлической извести, содержат от 6 до 20% глинистых
веществ.
При смачивании водой гидравлическая известь, вследствие
содержания в ней свободной окиси кальция, гасится и
рассыпается, а при обработке достаточным количеством воды
образует тесто, которое начинает твердеть на воздухе и продолжает
твердеть под водой без доступа воздуха.
Гидравлическую известь применяют главным образом в
гидротехнических сооружениях.
Роман-цемент получают обжигом мергелей при 1000—1100°С,
без доведения материала до спекания. Процесс обжига
проводят в печах различного типа, обычно в шахтных или кольцевых.
Для получения роман-цемента применяют мергели, в которых
отношение между известковой и глинистой частями таково, что
в обожженном продукте вся или почти вся известь связывается
в силикаты, алюминаты и ферриты кальция: 2CaO-SiO2,
СаО-А12О3, 5СаО'ЗА12О3 и 2CaO-Fe2O3.
Продукт обжига тонко размалывают. При смачивании
водой он не гасится, а в воде твердеет.
На основе роман-цемента приготовляют растворы для
заполнения швов кладки и некоторых видов бетона.
Портланд-цемент получают путем обжига до спекания
шихты, содержащей 75—80% карбоната кальция (известняк, мел)
и 25—20% глины или мергелистых известняков
соответствующего состава. Спекшийся материал, представляющий собой
камневидные мелкие и крупные куски, называется клинкером.
Клинкер далее тонко размалывают.
Состав портланд-цемента обычно несколько отличается от
состава клинкера. При помоле к клинкеру добавляют до 3%
гипса для замедления схватывания и до 15% гидравлических
добавок (доменные шлаки, зола, различные вулканические или
осадочные породы) для связывания свободной извести,
выделяющейся при затвердевании цемента, что способствует
повышению его качества.
Химический состав обычного портланд-цементного клинкера
колеблется в следующих пределах (в % вес):
СаО . . . . 60—67 SO3 0,3—1,0
SiO3 . . . 17—25 K3O-bNa,O . . 0,5—1,3
А12О3 . . . 3—8 • TiO, ..... 0,2—0,5
Fe2O3 . . . 0,3—6 P2Os 0,1—0,3
MgO . . . 0,1—4,5
634
Для ускорения образования кристаллов химических
соединений, входящих в состав клинкера, в шихту в ряде случаев
добавляют плавиковый шпат в количестве 0,5—1% от веса
получаемого клинкера. Это повышает производительность
цементных печей в среднем на 10% и снижает расход топлива на
5-7%.
На качество портланд-цемента, кроме химического состава
клинкера, оказывают влияние и другие факторы, в частности —
физическая структура и минералогический состав сырья,
влияющие на течение процесса образования клинкера при обжиге;
характер топлива и его зольность, влияющие на состав
клинкера и условия его спекания; температура и длительность
обжига, от которых зависит степень завершения процесса
образования клинкера; скорость охлаждения клинкера; тонина
помола портланд-цемента, определяющая его удельную
поверхность и, соответственно, скорость его гидратации при смешении
с водой.
Состав портланд-цементного клинкера характеризуется как
данными химического анализа, так и содержанием образующих
клинкер соединений — клинкерных минералов. Важной
характеристикой состава клинкера являются также соотношения
между главнейшими окислами, которые выражают в виде
различных коэффициентов (модулей).
Важнейшими минералами, входящими в состав портланд-
цементного клинкера, являются силикаты (трехкальциевый
силикат 3CaO-SiO2 алит и двухкальциевый силикат 2CaO-SiO2
белит) и плавни (трехкальциевый алюминат ЗСаО • АЬОз и че-
тырехкальциевый алюмоферрит 4СаО • А12Оз .Fe2O3, иначе
называемый браунмиллеритом).
Из соотношений между окислами важнейшим является
коэффициент насыщения известью кремнезема
портланд-цементного клинкера. Этот коэффициент выражает отношение
количества окиси кальция, остающейся после полного связывания
окислов железа и алюминия, к тому количеству окиси кальция,
которое необходимо для связывания всего кремнезема до трех-
кальциевого силиката. На заводах обычно изготовляют порт-
ланд-цементный клинкер с коэффициентом насыщения 0,80—0,95.
Для характеристики соотношения между кислотными
окислами введены два модуля: кремнеземный и глиноземный.
Кремнеземный модуль представляет собой отношение
процентного содержания кремнезема в клинкере к процентному
содержанию глинозема и окиси железа:
%SiOa
Величина кремнеземного модуля определяет относительное
содержание в клинкере минералов-силикатов и минералов-
635
плавней, поэтому его часто называют также силикатным
модулем. Для обычного портланд-цемента величина кремнеземного
модуля колеблется в пределах от 1,7 до 3,5.
Глиноземный модуль представляет собой отношение
процентного содержания глинозема в клинкере к процентному
содержанию окиси железа:
%А12О3
Величина глиноземного модуля определяет соотношение в'
клинкере алюмината кальция и железосодержащего браунмил-
лерита. Поэтому глиноземный модуль называют еще алюминат-
ным модулем. Для обычных портланд-цементов величина
глиноземного модуля колеблется в пределах от 1 до 3.
По современной теории акад. А. А. Байкова в процессе превращения
портланд-цемента в твердое камкевидкое вещество — цементный камень —
различают три периода.
В первый период — период растворения при смешении портланд-цемента
с водой основная составная часть цемента — алит гидролитически
диссоциирует с образованием гидроокиси кальция и гидросиликата кальция, менее
основного, чем трехкальциевый силикат (алит):
3CaO-SiO2 + «Н2О = л;Са(ОНJ + г/СаО • SiO2 • niiifi
где х+у=3.
При взаимодействии другой составной части цемента — белита с
ограниченным количеством воды свободная известь почти не образуется:
2СаО • SiO2 -f гсН2О = 2СаО • SiO2- пН2О
Алюминаты и алюмоферриты кальция при гидратации образуют
гидроалюминаты и гидроалюмоферриты кальция.
В течение первого периода твердения портланд-цемента вода
постепенно насыщается растворимыми продуктами (щелочами, известью, гипсом,
растворяющимися клинкерными минералами). После насыщения жидкой фазы
начинается второй период — период коллоидации — выделение мельчайших
частиц твердых продуктов реакции и образование коллоидной системы—
геля (студня). В течение второго периода твердения цементное тесто
утрачивает текучесть и подвижность — схватывается, ко не приобретает еще
прочности, свойственной затвердевшему цементному камню.
Третий период процесса твердения — период кристаллизации
характеризуется тем, что из геля, образовавшегося в пэриод коллоидации,
выделяются укрупненные, а потому менее растворимые кристаллические сростки.
В результате тесного пространственного переплетения этих кристаллов и
образуется затвердевшая масса, отличающаяся высокой прочностью,—
цементный камень.
Процесс твердения цемента скачала идет быстро, а затем все более
замедляется.
Производство портланд-цемента состоит из следующих
главных процессов: приготовление сырьевой смеси, ее обжиг,
размол обожженного клинкера. Существуют два основных способа
производства портланд-цемента — мокрый и сухой,
различающиеся методом приготовления сырьевой смеси.
636
По мокрому способу сырые материалы измельчают и
смешивают в присутствии большого количества воды. При этом
получается сметанообразная пульпа — шлам, содержащий 32—
45% воды. По сухому способу сырьевые материалы
высушивают, затем измельчают и смешивают. В результате получается
тонкий сухой порошок, называемый сырьевой мукой. Для
уменьшения уноса сырьевой муки с продуктами горения при
обжиге муку увлажняют или гранулируют.
При мокром способе производства для обжига сырьевой
смеси применяют вращающиеся печи, при сухом способе обжиг
чаще проводится также во вращающихся печах и реже в печах
шахтного типа. В шахтных печах труднее получить
высококачественный клинкер, так как работу таких печей сложнее
регулировать; кроме того, при обжиге клинкера в шахтных
печах необходимо только твердое топливо определенного
качества.
Выбор способа производства портланд-цемента
определяется главным образом свойствами сырьевых материалов. Мокрый
способ применяют при переработке мягких материалов,
обладающих высокой естественной влажностью, легко
диспергирующихся водой или имеющих очень разнообразный химический
состав; сырьевую смесь из таких материалов, подготовленную
мокрым способом, легче гомогенизировать. Сухой способ
целесообразен при переработке сырьевых материалов с низкой
естественной влажностью и более однородным химическим
составом.
Мокрый способ производства портланд-цемента более
распространен на отечественных и зарубежных заводах.
Схема производства портланд-цемента по мокрому способу
представлена на рис. 224. Добываемый в карьере твердый
известняк подвергается дроблению, в зависимости от крупности
кусков в две или три стадии — в щековой и молотковой
дробилках до получения кусков размером 8—10 мм. Глину,
поступающую из карьера, измельчают в валковой дробилке на куски
размером менее 100 мм и взмучивают с водой >в болтушках.
Глинистый шлам, содержащий 60—70% воды, и дробленый
известняк совместно измельчают в многокамерных шаровых
мельницах.
При использовании в мокром способе мягких известняковых
пород (мел, известняковый туф и др.) их после дробления
взмучивают в болтушках вместе с глиной, затем измельчают в
мельницах. Если в качестве сырья применяют только твердые
материалы (например, известняк, мергель, глинистые сланцы),
а не глину, то из схемы производства исключается стадия
взмучивания в болтушках, все материалы поступают в дробилки и
затем совместно измельчаются в мельницах, куда добавляют
воду
637
Известняк
Крупное дробление
Глина
1
Дробление
Мелкое дробление
Взмучивание
\
Мокрый помол
ьода
1
Корректирование
шлама
*
Хранение
шлама
V
Обжиг
Добавки
i Охлаждение
клинкера
Гипс '
t
Дробление
i I i
Дробление !
1
Вылеживание
i
\
Сушка
Помол цемента |
1
Хранение
Отгрузка навалом
1
Упаковка в мешки
Рис. 224. Принципиальная схема производства портланд-цемента
мокрым способом.
638
Шлам, отбираемый из мельницы, представляет собой
недостаточно однородную сырьевую смесь, непригодную для
осуществления правильного хода технологического процесса и
получения высококачественного портланд-цемента. Поэтому из
мельницы шлам транспортируется насосами в шламовые
бассейны, где тщательно перемешивается сжатым воздухом. После
определения химического состава шлама (главным образом
содержания СаСОз, а в отдельных случаях и содержания
некоторых примесей) его состав корректируют, т. е. исправляют
в нужную сторону. Колебания состава шлама зависят от
качества сырья, добываемого в карьерах. Для исправления состава
шлама приготовляют в специальном коррекционном бассейне
шлам с повышенным (или соответственно пониженным)
содержанием СаСОз и затем основной и корректирующий шлам из
обоих бассейнов соединяют в соответствующих пропорциях в
третьем бассейне. После тщательного перемешивания и
проверки химического состава шлам можно перекачивать в другие
бассейны — хранилища запасов шлама. Из этих бассейнов шлам
по мере необходимости передают во вращающуюся печь для
обжига. Запасы шлама создаются для обеспечения
бесперебойной работы цементных печей, пуск которых после
длительной остановки является продолжительной и трудоемкой
операцией.
При производстве портланд-цемента сухим способом
известняк и глина, поступающие из карьеров, подвергаются
дроблению и затем подаются в сепараторную мельницу для
одновременной сушки и помола. Тонко размолотая сухая смесь
(сырьевая мука) подается пневматическими устройствами в
смесительные силосы, где перемешивается сжатым воздухом. При обжиге
сырьевой смеси в виде мелких прочных гранул их изготовляют
в специальных грануляторах тарельчатого или барабанного
типа. '
Шлам, полученный по мокрому способу, равно как сырьевую
муку или гранулированный материал, полученные по сухому
способу, обжигают во вращающихся печах.
При обжиге протекает последовательно несколько
процессов: при 100—120°С происходит испарение влаги, при
температуре около 500 °С сгорают содержащиеся в шихте
органические вещества, при 650—1000 °С диссоциируют карбонаты
магния и кальция:
MgCO3 = MgO + СОа
СаСО3 = СаО + СО2
Двуокись углерода СО2 удаляется из печи с греющими
газами, окись кальция при дальнейшем повышении температуры до
1000—1300°С реагирует с SiO2, A12O3 и Fe2O3 с образованием
силикатов, алюминатов и ферритов кальция. При 1300°С на-
639
ступает стадия спекания, в этой стадии температура материала
постепенно повышается до 1450 СС, а затем снова понижается
до 1300°С.
У выходного конца печи находится зона охлаждения,
выходящий из нее спекшийся материал (клинкер) имеет
температуру 800—1000 °С.
Цементная вращающаяся печь представляет собой стальной
барабан длиной 118—185 м и внутренним диаметром 3,3—5 м,
футерованный шамотным, хромомагнезитовым и тальковым
кирпичом. Барабан печи снабжен бандажами для установки на
опорные ролики, расположенные на железобетонных
фундаментах. Печь приводится во вращение электродвигателем через
редуктор и венцовую шестерню, скорость вращения 0,5—1,2
об/мин. Печь установлена с наклоном около 4° для того,
чтобы при ее вращении материал продвигался вдоль барабана.
Поднятый конец печи входит в пылеосадительную камеру.
Нижний конец печи снабжен рекуператорами, в которых клинкер,
поступающий через отверстия в барабане, охлаждается
воздухом.
В торце нижней части печи имеются отверстия для
форсунок, по которым подается топливо. В качестве топлива
применяются пылевидный уголь, нефть, газ. Выходящие из печи
газы поступают в электрофильтр для тонкой очистки от пыли
и затем отводятся в атмосферу через дымовую трубу.
Производительность одной цементной печи б зависимости от ее
размеров составляет 400—1800 т/сутки.
На современных заводах обжиг сырьевой смеси
автоматически контролируется, что позволяет управлять процессом
образования клинкера и предотвращать нарушения
технологического режима. В результате автоматизации улучшается
качество клинкера, повышается производительность труда и
увеличивается выпуск цемента.
Полученный после обжига клинкер охлаждается в
барабанном или колосниковом холодильнике до 50—200°С воздухом,
движущимся противотоком материалу. Воздух, нагретый в
рекуператорах и барабанном холодильнике, поступает во
вращающуюся печь, где используется для сжигания топлива.
Охлажденный клинкер направляется на склад для
вылеживания (магазинирования) в течение 10—15 суток. На складах
клинкер перелопачивают при помощи грейферных мостовых
кранов для лучшего контакта материала с воздухом. При вы-
леживгшга клинкер охлаждается, а также происходит
гашение свободной извести, оставшейся в клинкере после его
обжига, влагой воздуха. При этом клинкер становится более
рыхлым, что облегчает его помол, а при твердении бетона
происходит более равномерное изменение объема
портландцемента.
640
Измельчение клинкера после вылеживания обычно
производят вместе с добавками (гипс, гидравлические добавки) в
многокамерных трубных мельницах. Чем теньше помол
клинкера, тем выше активность получаемого портланд-цемента и
тем быстрее он твердеет. Однако с возрастанием тонины
помола увеличивается расход электроэнергии на измельчение
продукта и уменьшается пропускная способность мельниц. Помол
является наиболее энергоемкой операцией в производстве
портланд-цемента.
Более тонкое измельчение достигается в мельницах
вибрационного помола, в которых размалывание происходит под
действием частых ударов вибрирующих мелющих тел (около
1500—3000 вибраций в 1 мин). Однако из-за низкой
производительности вибромельннцы не получили пока широкого
применения в цементной промышленности. В некоторых случаях их
применяют для домалывания цемента. Готовый цемент хранят
в железобетонных или стальных силосах большой емкости.
Общая вместимость склада должна быть такова, чтобы
обеспечить хранение портланд-цемента в количестве его 20-суточного
выпуска для окончательного гашения свободной извести
влагой воздуха и для охлаждения цемента. Чтобы портланд-цемент
не слеживался при хранении, в нижнюю часть силоса через
специальное аэрирующее днище периодически нагнетается воздух,
разрыхляющий цемент. Аэрирование облегчает также разгрузку
цемента из силоса.
В настоящее время испытывается рювый эффективный сухой
метод обжига сырьевой смеси в кипящем слое.
Разработан и внедряется в промышленность способ
получения портланд-цемента при комплексной переработке
нефелина. После выщелачивания глинозема из продукта спекания
нефелина с известняком остается шлам, содержащий белит с
небольшой примегью глинозема и щелочей и до 40% воды.
Совместным помолом шлама с добавками небольших количеств
известняка, боксита и колчеданного огарка (вместо двух
последних компонентов в шихту можно вводить минерализующие
добавки фторида кальция или кремнефторида натрия)
получают сырьевую шихту, которую обжигают в
высокопроизводительных вращающихся печах. Клинкер перерабатывают в
цемент описанным выше способом.
Требования к качеству портлан д-ц е м е н т а
По степени прочности портланд-цемент разделяют на пять
марок: 250, 300, 400, 450 и 500. Эти числа обозначают предел
прочности при сжатич (в кгс/см2) образцов, изготовленных из
раствора с весовым отношением цемента к песку 1 : 3 и
испытанных через 28 суток после их изготовления.
41—2848 641
Начало схватывания портланд-цемента должно наступать не
ранее чем через 45 мин, конец схватывания — не позднее чем
через 12 ч после начала смешения с водой.
При слишком медленном схватывании цемента для
достижения требуемой прочности конструкций, изготовляемых из
цементных смесей, требуется более длительное время, что
привело бы к замедлению темпов строительства. Если же цемент
схватывается слишком быстро, он превращается в камневидное
тело прежде, чем его успевают использовать.
Тонина помола портланд-цемента должна быть такова,
чтобы остаток на сите с отверстиями размером 0,08 мм не
превышал 15%. Цементные заводы в ряде случаев выпускают
портланд-цемент более тонкого помола (остаток на указанном сите
8-12%).
Портланд-цемент упаковывают в 4—6-слойные бумажные
мешки. Для расфасовки цемента применяют упаковочные
машины различного типа. Портланд-цемент перевозят также
навалом в специальных саморазгружающихся цементовозах
железнодорожным и автомобильным транспортом или же в
специально оборудованных железнодорожных вагонах или судах.
Как вяжущий материал, обладающий еысокой прочностью
и сравнительно быстрым ее нарастанием при твердении,
портланд-цемент очень широко применяется в строительной
промышленности. Его используют при строительстве самых
разнообразных наземных, подземных, подводных бетонных и
железобетонных сооружений, возводимых в различных климатических
условиях и для различных условий эксплуатации, применяют
в дорожном строительстве и для других целей.
Пуццолановый цемент представляет собой продукт
совместного измельчения гидравлической (пуццоланической) добавки
с портланд-цементом или известью. Его получают также путем
тщательного смешения этих материалов, предварительно
раздельно измельченных. Гидравлическими, или активными,
минеральными добавками называют такие вещества, которые при
смешении в измельченном виде с воздушной известью придают
ей способность твердеть под еодой. При смешении с
портландцементом гидравлические добавки повышают его еодостойкость.
В качестве гидравлических добавок наиболее часто применяют
активный кремнезем (кремнеземистые отходы), трепелы и
диатомиты, измельченные вулканические породы (трасс, пепел,
пемзы, туфы). Содержание гидравлических добавок в пуццола-
новом цементе составляет 20—45%.
Шлаковые цементы получаются в результате совместного
измельчения вяжущих материалов с доменными шлаками.
В доменных шлаках содержится значительное количество таких
окислов, как CaO, SiOL>, A12O3, и сравнительно небольшое
количество некоторых других окислов. Различают шлакопорт-
642
ланд-цемент, известково-шлаковый цемент и
сульфатно-шлаковые цементы.
Шлакопортланд-цемент является продуктом совместного
измельчения портланд-цементного клинкера и гранулированного
доменного шлака или тщательного смешения этих же
раздельно измельченных сухих материалов. Содержание шлака в шла-
копортланд-цементе колеблется в пределах 30—60%.
Известково-шлаковый цемент получают путем совместного
измельчения высушенного шлака с известью или путем
тщательного смешения этих же раздельно измельченных
материалов.
Сульфатно-шлаковые цементы образуются при совместном
измельчении высушенного шлака и добавок, влияющих на
процесс твердения. Среди таких добавок преобладают сульфаты
(гипс, ангидрит).
Глиноземистый цемент получают путем сплавления смеси
тонкомолотого материала, богатого глиноземом (например,
боксита), с известняком и последующего тонкого измельчения
сплава. В качестве глиноземистого цемента используют также
богатые глиноземом шлаки, получаемые при выплавке чугуна
в доменных печах, а также шлаки из газогенераторов с жидким
шлакоудалением.
Глиноземистый цемент содержит главным образом
алюминаты кальция и частично силикаты кальция. Примерный
состав глиноземистого цемента (в %):
А!,О3 . . . ~40 SiO, ... До 10
СаО . . . ~40 FeO+Fe.2O3 До 10
Бетон, изготовленный на основе глиноземистого цемента,
характеризуется быстрым нарастанием механической прочности.
Уже на вторые сутки после начала схватывания такого бетона
предел его прочности при сжатии достигает 500—600 кгс/см2.
Другой особенностью изделий из глиноземистого цемента
является щелочестойкость и значительная устойчивость к
действию морской воды и грунтовых вод, содержащих сульфаты
и магниевые соли.
Глиноземистый цемент значительно дороже
портландцемента и поэтому применяется в тех случаях, когда указанные
его свойства имеют решающее значение.
МАТЕРИАЛЫ И ИЗДЕЛИЯ НА ОСНОВЕ ВЯЖУЩИХ ВЕЩЕСТВ
На основе вяжущих веществ изготовляют ряд строительных
материалов. Наибольшее распространение имеют бетоны и
железобетон.
41* 643
Бетон представляет собой искусственный камень,
получаемый в результате твердения затворенной водой смеси, в состав
которой входят щебень или гравий, песок и другие заполнители
и вяжущее вещество — цемент. Вследствие химической реакции
между частицами цемента и водой (стр. 636) происходит
образование цементного камня и сцепление его с заполнителями,
образующими жесткий скелет бетона.
В зависимости от свойств заполнителей различают
обыкновенные, легкие и теплоизоляционные бетоны.
В качестве заполнителей в обыкновенных бетонах
применяются песок и гравий или щебень, в легких бетонах —
пористые материалы — керамзит (получается путем
кратковременного обжига глин во вращающихся печах при 1150—
1250 °С), аглопорит (получают спеканием глин, смешанных с
углем и древесными опилками, на агломерационных лентах),
шлаки, пемзу, туф и др. Теплоизоляционные (ячеистые) бетоны
получают путем введения в цементное тесто газообразующих
или пенообразующих добавок, в результате чего получается
бетон крупнопористой структуры. По способу изготовления и
укладки бетон подразделяется на жесткий, пластичный и литой.
Кислотоупорный бетон изготовляют на основе
кислотостойких заполнителей (андезит, бештаунит, стр. 666) с добавкой
10—15% жидкого стекла и 1—2% кремнефторида натрия.
Кислотоупорный бетон обладает хорошей механической прочностью
и высокой кислотостойкостью. Его применяют для сооружения
продукционных башен в производстве серной кислоты и других
аппаратов и резервуаров для химических производств, а также
для изготовления полов, перекрытий и некоторых других
строительных конструкций, подвергающихся при эксплуатации
действию кислых сред.
Жароупорный бетон отличается от кислотоупорного бетона
составом заполнителей, в качестве которых применяются
хромистый железняк, шамот и др. Жароупорный бетон обладает
высокой термической устойчивостью и кислотостойкостью. Он
употребляется для футеровки колчеданных печей, применяемых
в производстве серной кислоты (стр. 75 ел.), и других
химических аппаратов, работающих при высокой температуре. В
ряде случаев печи из жароупорного бетона можно изготовлять без
металлического кожуха.
Железобетон представляет собой залитый бетоном каркас
из стальной арматуры (стальные пруты, трубы и др.). Бетон
обладает большим сопротивлением раздавливанию и малым
пределом прочности при растяжении. Стальная армировка
увеличивает предел прочности конструкции при растяжении, так
как имеет почти одинаковый коэффициент расширения с
бетоном; сила же сцепления стали с бетоном почти равна силе
сцепления частиц самого бетона.
644
Устойчивость железобетонных конструкций к
растягивающим и изгибающим нагрузкам значительно повышается в
случае применения стальной арматуры, предварительно
подвергнутой растяжению. В таком напряженно армированном бетоне
после затвердения бетонной массы арматура освобождается от
натяжения, вследствие чего внутренние силы упругости
сжимают бетон и при изгибе трещины в нем не появляются.
Широкое распространение в строительстве получил сборный
железобетон — изготовленные заводским способом крупные
железобетонные панели и блоки, из которых на строительных
площадках монтируются элементы и узлы зданий и
сооружений. Применение крупных сборных железобетонных и бетонных
деталей позволило в значительной степени механизировать и
ускорить строительство.
3. СТЕКЛО
Свойства стекла*. Стекло представляет собой продукт
совместного плавления нескольких минеральных веществ.
Характерной особенностью стекла является то, что при охлаждении в
определенных условиях расплавленной стекольной массы
обычно образуется прозрачное аморфное твердое тело. При очень
медленном охлаждении расплав стекла затвердевает с
образованием кристаллов.
Наряду с высокой твердостью для стекла характерна
хрупкость; чтобы уменьшить ее, стекло подвергают специальной
термической обработке. Стекло мало теплопроводно и обладает
низкой электропроводностью.
Ценными свойствами стекла являются устойчивость его к
воздействию химических реагентов, а также однородность,
прозрачность, лучепреломление, отражение, рассеивание,
поглощение света и другие ценные оптические свойства.
Состав стекла выражают соотношением трех групп окислов:
щелочных, щелочноземельных и кислотных.
В общем виде формула стекла может быть выражена
следующим образом:
где R2O — окислы щелочных металлов Na2O, K2O, Li2O н др.;
RO — окислы щелочноземельных и тяжелых металлов и
окислы, обусловливающие окраску стекла СаО,
MgO, BaO/SrO, PbO, ZnO, FeO, MnO и др.;
RO2 — кислотный окисел SiO2; сюда же относят А12О3.
В2О3, P2O5 и др.
* Стекло известно человечеству с глубокой древности. В Египте
производили стекло почти за 6000 лет до нашей эры.
R4K
Свойства стекла зависят от характера и количественного
соотношения окислов. Так, кислотные окислы сообщают стеклу
высокую термическую, химическую и механическую стойкость.
Окислы щелочных металлов понижают вязкость и температуру
плавления стекла, а также уменьшают его твердость и
ухудшают термические и химические свойства стекла.
Содержание SiO2 в стекле составляет 60—75%, за
исключением .некоторых специальных стекол и хрусталя, где оно ниже,
и кварцевого стекла (плавленый кварц), в котором содержание
SiO2 достигает 100%.
Содержание окислое щелочных металлов в обычных сортах
стекла составляет 10—13%, в отдельных сортах понижается до
4% и позышается до 18%.
Окислы щелозноземельной группы придают стеклу
требуемую вязкость и повышают его химическую стойкость.
Содержание окислов щелочноземельных металлов в стекле
обычных сортов составляет 10—15%, достигая в некоторых
сортах стекла 25%.
Наиболее распространенным типом стекла является система
из трех весьма доступных и дешевых окислов: Na2O—СаО—SiO2.
Стехиометрический состав такого стекла приближается к
формуле Na2O • СаО • 6SiO2.
СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА СТЕКЛА
Важнейшим сырьем для стекловарения являются кварцевый
песок, известняк или мел и сода или безводный сульфат натрия.
Эти вещества принято называть стеклообразующими
материалами.
В зависимости рт сорта производимого стекла к качеству
сырых материалов предъявляются соответствующие
требования.
Так, песок должен содержать не менее 98% SiO2, на ряде
заводов применяют местный песок с более низким содержанием
двуокиси кремния.
Вредной примесью в сырых материалах являются
окислы железа, так как они придают стеклу зеленый оттенок. В
песке для производства оптического стекла содержание Fe2O3 не
должно превышать 0,01%, для листового стекла — не более
0,05%, при производстве темного бутылочного стекла
допустимо применение песка, содержащего несколько процентов Fe2O3.
В известняке и меле, применяемых при получении стекла для
сортовой посуды, Fe2O3 должно содержаться не более 0,03%,
для полированного стекла — не выше 0,1%, для оконного
стекла — не более 0,2%.
Во многих случаях песок, предназначенный для стекольной
шихты, подвергают обогащению, главным образом для
удаления окислов железа. В зависимости от характера примесей
64G
железа и требовании к качеству песка применяются
соответствующие методы обогащения. Методом магнитной сепарации
обогащают пески, в которых примеси железа содержатся в
виде минералов, обладающих магнитными свойствами.
Промывка водой применяется для обогащения песков, загрязненных
преимущественно ожелезненными глинистыми примесями
(содержащими значительное количество окислов железа).
Если железосодержащие примеси в песках находятся в виде
пленок гидроокиси железа, обволакивающих кварцевые зерна,
обогащение проводят методом оттирки — путем взаимного
трения частиц песка друг о друга.
Наибольшее распространение получил флотооттирочный
метод обогащения песков. Он заключается в последовательном
проведении трех операций: флотации, оттирки пленки и
промывки.
В результате этих процессов удаляются три группы
примесей: тяжелые минералы, пленки гидроокиси железа и
глинистые примеси.
В качестве компонента стекольной шихты сода имеет ряд
технических преимуществ по сразнению с сульфатом натрия.
Сода плавится при 851 °С, сульфат натрия при 885 °С. Кроме
того, сода уже при плавлении начинает реагировать с SiC>2
с выделением двуокиси углерода, а сульфат натрия плавится
без разложения и начинает диссоциировать лишь при
температуре выше 1200 °С. Поэтому при плавке шихты, содержащей
сульфат натрия, необходимо поддерживать более высокую
температуру з печи, что, в свою очередь, вызывает повышенный
расход топлива и быстрый износ огнеупорной футеровки печи.
При применении сульфата натрия з шихту вводят
восстановитель (древесный уголь, антрацит, кокс), ускоряющий процесс
разложения сульфата натрия по реакции:
2Na2SO4 + С = 2Na2SO3 -f СОЯ
Образующийся сульфит натрия диссоциирует значительно
легче. Преимущество соды заключается также в том, что она
содержит 58,5% Na2O, а сульфат натрия лишь 43,6% Na2O.
Таким образом, для введения в стекло одного и того же
количества ЫагО тпебуется больше сульфата натрия, чем соды.
Однако вследствие дефицитности соды, кроме нее, в стекольной
промышленности применяют значительное количество
сульфата натрия.
Окись калия вводится в стекольную шихту в виде поташа
К2СО3. Поташ применяют для изготовления лучших сортов
посудного стекла, хрусталя, оптических стекол. Калийные стекла
обладают блеском и прозрачностью. Окись свинца применяют
в виде сурика Plb3O4 при изготовлении хрусталя, оптических
стекол и др. Свинцовое стекло сильно преломляет лучи света
647
и отличается сильным блеском. Борный ангидрид вводят в виде
борной кислоты или буры при изготовлении борных стекол,
применяемых в оптике и для выделки химической посуды.
В стекольном производстве применяют также
металлургические шлаки и различные изверженные горные породы,
содержащие окислы, входящие в состав стекла, например полевые
шпаты, андезит, пемзу, вулканический пепел, нефелин.
Кроме перечисленных стеклообразующих материалов, в
шихту для ускорения варки вводят стекольный бой в количестве
25—30%.
Весьма эффективными ускорителями процесса
стекловарения являются фтористые соединения, которые добавляют в
количестве до 1 % от веса шихты.
Для лучшего осветления стекломассы, т. е. удаления из нее
газовых пузырей, в шихту вводят 0,5—1% так называемых
осветлителей (трехокись мышьяка, натриевая селитра, иногда
если аммония).
Для получения окрашенного стекла в шихту вводят
соответствующие краски, большей частью окислы различных
металлов, которые растворяются в стекле. Так, в окислительном
пламени окись железа окрашивает стекло в желто-зеленый
цвет, окись марганца — в фиолетовый. В восстановительном
пламени окись железа переходит в закись и окрашивает стекло
в сине-зеленый цвет, закись марганца — в светло-желтый или
розовый цвет. Окислы никеля и кобальта растворяются в
стекле только в виде закиси. Окислы никеля придают стеклу,
содержащему Na2O, цвет от красно-коричневого до серого, стеклу,
содержащему РЪО и К2О, — фиолетовый цвет различных
оттенков. Окислы кобальта окрашивают стекло в синий цвет. Цвет
стекла зависит также от количества введенного окисла,
толщины изделия и состава стекла.
Реже применяется способ коллоидного распределения
красящего вещества в стекле. В этом случае для окраски
применяют не окислы, а металлы. Так, например, золото и медь
окрашивают стекло в красный цвет, селен — в розовый, серебро —
в желтый.
Для получения молочного и опалового стекла в массу
вводят так называемые глушители. Частицы глушителя могут
быть сами по себе прозрачны и бесцветны, но должны
отличаться от стекла по величине коэффициента лучепреломления.
В качестве глушителей применяют фториды и фосфаты, а
также соединения олова и сурьмы.
В тех случаях, когда исходные материалы загрязнены
окислами железа, придающими стеклу желтоватый или
зеленоватый оттенок, з шихту вводят обесцвечивающие вещества. Эти
вещества должны давать окраску, являющуюся дополнительной
к окраске, вызванной железом, в результате чего получается
648
бесцветное стекло. В качестве обесцвечивающих веществ
применяют окислы никеля и кобальта, селен, МпО2 и др.
ПРОИЗВОДСТВО СТЕКОЛЬНОЙ МАССЫ
Сырые материалы, входящие в состав шихты, промывают,
сушат, измельчают и просеивают. При промывке песка вода
отделяет содержащиеся в нем глинистые частицы и
органические примеси и уносит их с собой. С глиной удаляется также
большая часть окислов железа, содержащихся в песке.
Сушку песка, мела и других материалов производят во
вращающихся сушильных барабанах с циклонами для улавливания
пыли. Для предварительного дробления материалов применяют
обычно шековые дробилки, для окончательного измельчения—
бегуны и молотковые дробилки. Все материалы, составляющие
стекольную шихту, обязательно пропускают через сито, так как
крупные частицы не успевают расплавиться в печи, что
приводит к образованию включений («камня») в стекле.
Подготовленные указанным образом материалы смешивают в
определенных весовых соотношениях до получения вполне
однородной смеси, которую загружают в печь для варки стекла.
Для получения стекла требуемого качества рассчитывают
рецептуру шихты на основе химического анализа применяемого
сырья с учетом его примесей. При расчете шихты учитывают
также потери некоторых компонентов шихты, улетучивающихся
в процессе варки стекла.
В процессе стекловарения различают пять стадий:
1) Силикатообразозание. При постепенном разогревании
шихты сначала испаряется гигроскопическая влага, затем его-
рают примеси органических веществ, при более высоких
температурах удаляется кристаллизационная вода. При 600—700 °С
начинается медленное взаимодействие между еще не
расплавившимися компонентами шихты. К концу этой стадии (для
обычных стекол при 800—900 °С) шихта превращается в
спекшуюся массу, состоящую из силикатов и кремнезема.
2) С теплообразование. При дальнейшем нагревании масса
начинает плавиться. Одновременно происходит взаимное
растворение силикатов и кремнезема. К концу этой стадии (для
обычных стекол при 1200°С) стекольная масса становится
прозрачной. Однако при этой температуре она еще пронизана
большим количеством газовых пузырьков и неоднородна по
химическому составу и свойствам.
3) Дегазация. При дальнейшем нагревании (для обычных
стекол до 1400—1500 °С) вязкость стекольной массы снижается
и из нее начинают выделяться газообразные включения;
происходит освобождение стекла от видимых пузырьков, т. е. его
осветление.
649
4) Гомогенизация. В этой стадии стекольную массу для
достижения однородности ее химического состава длительно
выдерживают при высокой температуре. Для обычных стекол
стадия гомогенизации может быть завершена при более низкой
температуре, чем стадия осветления.
5) Охлаждение (студка). Температуру стекольной массы
понижают на 200—300 °С. При этом она приобретает вязкость,
необходимую для формования стекла.
На практике некоторые стадии протекают не в строгой
последовательности, а одновременно, например совмещаются
стадия силикатообразования и стеклообразования или стадии
дегазации и гомогенизации.
СТЕКЛОВАРЕННЫЕ ПЕЧИ
Для варки стекла применяют ванные и горшковые печи,
которые обычно обогреваются генераторным или природным
газом и имеют регенераторы для нагревания воздуха и газа.
В ванных печах стекло варится непосредственно в самой
печи, поедставляющей собой бассейн или ванну, выложенную
из огнеупорных брусьев. Бассейн печи в зоне высоких
температур обычно выкладывают из высокоглиноземистых огнеупоров,
з состав которых вводят до 35% двуокиси циркония.
Стены и свод печи выполняют из динасовых огнеупоров,
етудочные и выработочные части печи, а также дно — из
шамотных брусьев.
В горшковых печах стекло варится в специальных
огнеупорных керамических горшках, установленных в этих печах.
Ванные печи значительно более распространены, чем горшковые.
В настоящее время горшковые печи применяются лишь для
производства специальных сортов стекла (оптического, для
художественных изделий и др.).
Ванные печи являются большей частью печами непрерывного
действия. Одна из конструкций ванной печи изображена на
рис. 225. В верхней части печи находится бассейн 1, в котором
производится варка стекла, и два ряда горелок 2 и 3. Газ и
воздух нагреваются в регенераторах 4 и 5 (камеры с насадкой из
огнеупорного кирпича) и поступают в пламенное пространство
печи через горелки 3. Продукты горения газа проходят над
стекольной массой поперек печи и через каналы горелок 2
направляются в противоположную пару регенераторов. Горячие газы
нагревают насадку регенераторов и уходят в дымовую трубу.
Периодически производится переключение регенераторов: газ
и воздух сжигают в горелках 2, а горячие газы направляют
в регенераторы 4 и 5.
Шихта загружается в печь с одного конца бассейна через
загрузочное окно или загрузочный карман, а из другого конца
печи стекло непрерывно отбирается на выработку.
650
Плавление шихты и образование стекольной массы
происходит в передней части печи, прилегающей к месту загрузки
шихты. Расплавленная стекольная, масса постепенно перетекает
з следующую часть бассейна, где происходит дегазация и
гомогенизация стекла. Зоны стеклообразовання, дегазации и
гомогенизации стекла составляют отапливаемую часть бассейна,
Рис. 2^5. Ванная пьчь для варки стекла:
/—бассейн для варки стекла; 2, а—горелки; 4—регенератор для нагрепання газа;
5—регенератор для нагревания воздуха; 6—боров для газа; 7—боров для ьоздуха.
л ней расположены горелки. Далее стекольная масса попадает
з зону охлаждения и затем в последнюю зону, откуда
производится выработка стекла. Между зонами дегазации и
гомогенизации и зоной охлаждения, а иногда между зонами стекло-
образования ч дегазации располагают шамотные поплазки,
плавающие в стекольной массе. Разделяя эти зоны, поплавки
замедляют переток стекла из одной зоны в другую.
Производительность ванных печей 15—25 т/сутки для
сортового стекла и 75—200 т.'сутки для бутылочного и листового
стекла. Съем стекольной .массы с 1 м2 зеркала варочной части
мечи в современных ванных печах составляет 500—2000 кг/сутки.
651
В ванных печах непрерывного действия трудоемкий процесс
выработки стекла полностью механизирован.
Наилучшая стабилизация режима стекловарения в ванной
печи достигается при автоматическом управлении ею.
Основными объектами автоматического управления являются
давление газовой среды в печи, количество и соотношение топлива
и воздуха для его горения, температуры подогрева газа и
воздуха в регенераторах и уровень стекломассы в печи. На
большинстве отечественных стекольных заводов проведена полная
или частичная автоматизация стекловаренных печей.
Горшковые печи. В горшковых печах процесс варки стекла
происходит периодически, причем последовательно
производится разогрев печи, загрузка шихты, собственно варка стекла,
дегазация массы, охлаждение ее и выборка стекла для
выработки изделий.
Во время разогрева печи в горшки (рис. 226) засыпают
стекольный бой, так как он плавится при более низкой
температуре, чем шихта. После того как бой расплавится, засыпают
шихту. Объем шихты при плавке уменьшается (шихта «садит
ся»), поэтому загрузку производят в два-три приема.
Рис. 226. Горшки для варки стекла.
По окончании варки производят дегазацию стекломассы,
нагревая ее до более высокой температуры. Одновременно
производят «бурление» стекла, которое также способствует
скорейшему выделению пузырьков газа. Для этого в расплавленное
стекло вводят кусок мокрой, хорошо пропитавшейся водой дре
весины. Вода сразу превращается в пар, который, стремясь
выйти из стекломассы, вызывает ее «бурление».
После дегазации стекломассы температуру в печи понижают,
чтобы стекло приобрело вязкость, необходимую для выработки
соответствующих изделий. Для этого прекращают приток газа
и воздуха в печь. Во время выработки из стекла изделий в
горшковой печи поддерживают температуру, при которой
стекломасса сохраняет требуемую вязкость.
Весь цикл продолжается около суток.
652
Горшковая печь изображена на рис. 227. В пламенном
пространстве / на поду устанавливают горшки 2 для варки стекла
(см. рис. 226). В стенах печи имеются каналы 3 я 4, через
которые в пламенное пространство поступают газ и воздух,
нагретые в регенераторах 5 и 7. Пламя горящего газа омывает
горшки; продукты горения выходят через каналы в
противоположной стене печи и направляются в регенераторы.
Рис. 227. Горшковая печь для варки стекла:
У—пламенное пространство печи; 2—горшки для варки стекла; 3—каналы
для нагретого газа; 4—каналы для нагретого воздуха; 5—регенераторы для
нагревания воздуха; 6—боровы для ЕОздуха; 7—регенераторы для нагревания
газа; 8—боровы для газа.
В стенах верхней части печи имеются рабочие окна, через
которые засыпают шихту или стекольный бой в горшки и
выбирают готовую стекольную массу.
Горшковые печи строят на 1—2 горшка, если требуется
особо точный режим варки (для некоторых специальных сортов
стекла), и на большее число горшков (до 16). Горшок вмещает
от 300 до 1200 кг стекломассы.
Недостатками горшковых печей являются невысокая
производительность, большой расход тепла (около 10 000 ккал на
! кг стекла вместо 2500 ккал в ванных печах), а также частые
разрушения горшков и высокая их стоимость.
653
ИЗГОТОВЛЕНИЕ СТЕКЛЯННЫХ ИЗДЕЛИЙ
Формование стекла осуществляют путем прессования,
выдувания, вытягивания, прокатки и центробежного формования.
Стеклянные изделия изготовляются самых разнообразных
форм и размеров. Различают следующие группы стеклянных
изделий: листовое оконное, листовое полированное, безопасное
стекло (триплекс и закаленное), архитектурно-строительное
стекло, пеностекло, стеклянное волокно, стеклопластики,
стеклянные трубы, тарное, посудное и хозяйственное,
электровакуумное, химико-лабораторное, светотехническое, оптическое
стекло, эмали.
Все стеклянные изделия обязательно подвергаются отжигу—
термической обработке, необходимой для снятия или
ослабления внутренних напряжений, возникающих в стекле при более
или менее быстром неравномерном его охлаждении, главным
образом во время выработки. Температура отжига должна быть
такой, чтобы частицы стекла сохраняли подвижность,
достаточную для быстрого выравнивания напряжений, без
деформации изделия. Отжиг штучных изделий производят в туннельных
печах.
Листовое стекло. Листовым оконным стеклом называют
прозрачные неполированные листы стекла толщиной от 2 до
6 мм. Производство листового оконного стекла в настоящее
время механизировано. Наиболее распространен способ
вертикального вытягивания ленты стекла с поверхности
расплавленной стекольной массы специальной машиной через лодочку.
В шахте машины производится и отжиг непрерывной
стеклянной ленты.
Стекло варят в ванной печи, соединенной перешейком с
каналом, расположенным перпендикулярно к продольной оси печи.
В больших современные ванных печах для непрерывного
производства листового стекла способом вертикального
вытягивания ширина бассейна достигает 10 м, длина 40 м и глубина до
1,5 м. В зоне максимального нагрева в печи поддерживают
температуру 1455—1475 °С, в месте перехода к перешейку 1220—
1230 °С. Канал разделен на ряд отдельных подогревательных
камер шамотными мостами, погруженными в расплавленную
стекольную массу. Температура стекломассы в
подогревательных камерах 1020—1120 °С.
Над каналом установлены 10 или 12 машин,
вырабатывающих стекло. Пространство под каждой машиной называется
подмашинной камерой. Между подмашинными камерами
находятся подогревательные камеры, в которых при помощи
специальных горелок поддерживается определенная температура
стекломассы. Подмашинаые камеры являются вырабэточноп
частью канала, и в них помещаются шамотные лодочки.
654
Лодочка имеет сужающуюся кверху щель. Уровень бортов
лодки выше уровня щели. При погружении лодки под
некоторым давлением в стекло уровень щели оказывается ниже
зеркала стекольной массы. Масса выдавливается из Щели по всей
ее длине, захватывается специальным приспособлением и
оттягивается вверх валками машины.
Рис. 228. Машина для
выработки оконного
стекла:
/—шахта машины; 2—валки;
3—площадки для сбслужива-
иия; 4—«Рубикон»;
5—лодочка; б—штанги нажимного
механизма; 7—вертикальный вал-
Машина ВВС (вертикального вытягивания стекла) для
выработки оконного стекла (рис. 228) представляет собой
вертикальную шахту 1 высотой от 5,4 до 7,8 м. По высоте шахты
расположено 13 пар валков 2, покрытых асбестом. Первую
снизу пару валков 4 называют «рубиконом», т. е. границей,
пройдя которую, лента стекла становится хрупкой. Температу
ра поверхности стекла между первой и второй парами валков
колеблется в пределах 473—480°С. По высоте шахты располо-
655
жены площадки 3 для обслуживания машины. На верхней
площацке машины (выше последней пары валков) от
движущейся вверх бесконечной ленты отрезают листы стекла.
Температурный режим в шахте машины определяет качество
отжига ленты стекла. Колебания температуры при оттягивании
ленты стекла из Щели лодочки 5 и при отжиге ее в шахте не
должны превышать 10—25 СС. Нарушение правильного
температурного режима вызывает растрескивание ленты в шахте
машины. Регулирование температуры производится большим
или меньшим открыванием люков, расположенных в шахте
машины. Температура поверхности ленты стекла на выходе из
верхней пары валков составляет 100—130СС, а в месте
отрезания листа 60—70 СС. Полезная ширина вытягиваемой
стеклянной ленты от 1800 до 3000 мм.
Большое значение при механизированном способе
выработки оконного стекла имеет вязкость стекломассы при ее
втягивании в машину и скорость нарастания вязкости при
прохождении ленты стекла в машине. При поддержании требуемой
вязкости стекломассы производительность машины возрастает
и качество стекла улучшается. Чтобы ленту стекла можно было
вытягивать с достаточной скоростью, необходимо в состав
стекла вводить до 2% АЬОз и до 3,5% MgO. При введении этих
добавок уменьшается возможность кристаллизации стекла при
температуре выработки и повышается скорость его твердения.
На отечественных стекольных заводах производительность
машин ВВС значительно повышена. Скорость вытягивания
ленты стекла в настоящее время составляет обычно 80—100 м(ч,
а иногда достигает 145 м/ч.
На некоторых заводах применяется безлодочное
вытягивание стекла, позволяющее сразу получать стекло с ровной и
гладкой поверхностью, без последующей шлифовки и
полировки. Для равномерного вытягивания стекломассы у места ее
отбора устанавливают специальную огнеупорную решетку, не
погруженную в расплав. Состав шихты при безлодочном
способе подбирают так, чтобы стекломасса затвердевала несколько
быстрее, чем при вытягивании с помощью лодки. При
безлодочном способе используются машины ВВС, описанные выше.
Листовое полированное стекло, применяемое для остекления
витрин, окон в вагонах, самолетах, автомашинах и др., для
изготовления зеркал и т. п., выпускают в виде листов толщиной
от 4 до 9 мм. Изготовляются и более толстые листы.
Производство полированного стекла состоит из двух
самостоятельных процессов:
1) выработка листового стекла путем проката;
2) обработка стекла — его шлифовка и полировка.
При современном непрерывном способе проката
стекломасса из ванной печи поступает через специальный желоб в валь-
656
цы Тфокатнои машины и далее в виде непрерывной ленты
движется в туннельную отжигательную печь. Далее стеклянная
лента поступает на валики транспортера, где дополнительно
охлаждается и разрезается на листы. Их направляют в цех
конвейерной шлифовки и полировки стекла.
Для изготовления зеркал стекло серебрят, покрывая одну
его поверхность тончайшим отражающим слоем серебряной
амальгамы. При серебрении аммиачный раствор нитрата
серебра восстанавливают различными органическими
восстановителями (инвертный сахар, сегнетова соль и др.).
Безопасное стекло, не образующее при разбивании острых
осколков или удерживающее их, выпускают в виде стекла
триплекс и закаленного стекла.
Триплекс (трехслойное), или безосколочное стекло, состоит
из двух стеклянных листов, склеенных прозрачным и
эластичным слоем или пленкой органического полимера. Выпускают
плоский и гнутый триплекс. Его изготовляют путем
прессования в автоклаве трехслойного пакета и фацетирования, т. е.
обточки краев триплекса водо-песочной смесью.
Закаленное небьющееся стекло получают путем специальной
термической обработки листового стекла — закалки. В
результате такой обработки стекло приобретает механическую
прочность, во много раз превышающую прочность обычного стекла,
и очень высокую термическую устойчивость. При особенно
сильном ударе такое стекло все же распадается на мелкие осколки,
которые, однако, не имеют острых, режущих краев. Закаленное
стекло также выпускают плоским и гнутым.
Безопасные стекла применяются главным образом длч
остекления кабин автомашин, самолетов. В случае аварий
возможность ранения такими стеклами почти исключена.
Архитектурно-строительное стекло применяют в качестве
строительного и декоративного материала для стен, потолков
и полов, для архитектурного оформления зданий и других
целей.
Из разнообразного ассортимента
архитектурно-строительного стекла можно указать на стеклянные блоки, изготовляемые
путем сварки двух размягченных половинок блока и
последующего отжига. При остывании блока внутри него создается
закуум, что улучшает тепло- и звукоизоляционные свойства
такого элемента.
К этому виду стекла относятся детали стекложелезобетон-
ных перекрытий и армированное стекло, изготовляемое путем
закладки металлической сетки в стеклянные листы.
Пеностекло представляет собой пористое стекло,
изготовляемое в виде блоков, плит и других деталей. Оно применяется я
качестве строительного, теплоизоляционного,
звукоизоляционного и конструкционного материала.
42—2848 657
Пеностекло изготовляют следующим образом. Измельченное
стекло и пенообразователь, например кокс, тщательно
перемешивают. Полученную смесь засыпают в формы и нагревают в
туннельной печи, обогреваемой очищенным генераторным
газом. Печь имеет несколько температурных зон. При
температуре около 600 °С частицы стекольного порошка слипаются и
образуют полости, в которых замкнуты частицы
пенообразователя. При дальнейшем повышении температуры поверхностные
пленки стекла начинают растягиваться газами, выделяемыми
пенообразователем. Процесс вспенивания продолжается строго
определенное время при температуре не выше 830°С. В
следующей зоне печи температура резко снижается. Вязкость
стекла при этом значительно возрастает, и пена становится
устойчивой. Образовавшийся в форме блок или другое изделие из
71еностекла постепенно охлаждается.
Пеностекло — легкий материал (объемный вес от 100 до
400 кг/м3). Механическая прочность его увеличивается с
повышением объемного веса. Предел прочности при сжатии
термоизоляционного пеностекла с объемным весом 250—300 кг/м3,
составляет около 30 кгс/см2. Пеностекло с объемным весом
400 кг/м3 имеет более высокую прочность. В зависимости от
объемного веса коэффициент теплопроводности пеностекла
меняется в пределах 0,04—0,12 ккал[м-ч-град.
Пеностекло легко подвергается механической обработке и
склеивается с другими материалами. Его можно пилить, резать,
сверлить и обтачивать на токарных станках.
Стеклянное волокно отличается большой прочностью при
растяжении, высоким модулем упругости, малой
гигроскопичностью, хорошими диэлектрическими свойствами, химической
устойчивостью, влагостойкостью, негорючестью и
неспособностью к гниению. Лучшие диэлектрические характеристики,
высокую механическую прочность и химическую устойчивость имеет
стеклянное волокно, изготовленное из бесщелочного и
малощелочного алюмоборосиликатного стекла. Различают два
основных вида стеклянного волокна:
1) текстильное, перерабатываемое в пряжу и ткани (длина
непрерызной нити 20 км и более), и штапельное (длина нити
от 5 до 50 см);
2) теплоизоляционное, представляющее собой стеклянный
¦войлок и стеклянную вату (в виде непрерывных нитей к в виде
штапелек).
Диаметр стеклянного волокна колеблется от 3 до 30 мк,
ультратонкое стеклянное волокно имеет диаметр до 1 мк.
Непрерывное волокно по внешнему виду напоминает натуральный
и искусственный шелк, штапельное волокно — хлопок и шерсть.
Стеклянное волокно изготовляют главным образом фильер-
ны\! :i дутьевым способами.
658
По фильерному способу вытягивания расплавленная
стекломасса под давлением собственного веса вытекает из фильер
(отверстия диаметром 1—3 мм) в виде капель, которые, падая
вниз, растягиваются и образуют волокна. Эти волокна
захватываются быстро вращающимся барабаном, вытягивающим их
до заданной толщины. Этим способом получают непрерывное
текстильное стеклянное волокно диаметром от 3 до 10 мк.
Пучок волокон собирается в прядь и склеивается при помощи
замасливающего приспособления. Нити волокон настолько
эластичны, что из них вырабатывают ткани на обычных
текстильных машинах. Предел прочности при растяжении
стекловолокна диаметром 3—б мк составляет 200—400 кгс/'мм2, т. е.
значительно выше, чем для обычного стекла (предел прочности при
растяжении стеклянных палочек 5—б кгс/мм2) ,и даже
высокопрочной стали. Из непрерывного стеклянного волокна
изготовляют различные технические ткани. Так, стеклянную ткань
применяют для оплетки кабелей и в качестве изоляции
электродвигателей. При нагревании такой ткани до 500°С ее
изоляционные свойства не ухудшаются, что позволяет почти на
половину снизить вес электродвигателя.
Теплоизоляционное непрерывное стеклянное волокно
диаметром от 10 до 30 мк, изготовляемое фильерным способом, имеет
меньшую прочность, чем текстильное стеклянное волокно.
С увеличением диаметра стеклянного волокна с 2 до 20 мк
прочность его снижается почти в 10 раз.
Дутьевым способом стеклянное волокно получают в
результате раздувания струи расплавленной стекломассы паром или
горячим воздухом, выходящим с большой скоростью из
дутьевого устройства (сопла). Этим способом получают штапельное
теплоизоляционное и текстильное стеклянное волокно,
применяемое главным образом для выработки теплоизоляционных и
звукоизоляционных изделий и фильтровальных тканей, стойких
к действию различных химических реагентов.
Стеклопластики. Очень большое распространение получили
стеклопластики, представляющие собой композиции из
синтетических полимеров (чаще всего полиэфиров), армированных
стеклянным волокном. Стеклопластики изготовляют путем
горячего прессования (в формах) стекловолокна или стеклянной
ткани, пропитанных раствором полимера, отверждающегося
при нагревании. Вследствие высокой механической прочности
и малой плотности стеклопластики являются прекрасными
конструкционными материалами, в ряде случаев с успехом
заменяющими алюминий и сталь.
Стеклопластики находят самое разнообразное применение.
Их используют в строительстве для устройства кровель и
внутренних перегородок, из стеклопластиков изготовляют кузозы
автомашин, детали вагонов, корпуса лодок и катеров, корпуса
42* 659
аккумуляторных батарей, трубы для транспортирования
химических и нефтяных продуктов и мног.ие другие изделия и
детали. Из стекловолокна, пропитанного битумными смолами,
изготовляют также рулонные гидроизоляционные материалы.
Стеклянные трубы изготовляют различных диаметров — от
тончайших капилляров и тонких трубок до массивных
толстостенных труб диаметром 150 мм. Стеклянные тонкостенные
трубы малого диаметра и стеклянные палочки используют главным
образом при изготовлении электровакуумных изделий,
лабораторного оборудования, ампул и пр. Толстостенные трубы
большого диаметра применяют для транспортирования
агрессивных жидкостей и пищевых продуктов, при скрытой
прокладке электропроводки и пр. Стеклянные трубы изготовляют путем
вытягивания расплавленной стекломассы различными
механизированными способами.
Тарное стекло по назначению и способам изготовления
разделяется на два основных вида: узкогорлую тару с внутренним
диаметром горла до 30 мм (главным образом бутылки) и ши-
рокогорлую тару для хранения полужидких и твердых
продуктов (глазным образом консервные банки). При производстве
тарного стекла определенное количество расплавленной
стекломассы (соответствующее весу вырабатываемого изделия)
подается при помощи питателя или засасывается
непосредственно из печи сначала в черновую, а затем в чистовую форму
машины для формования тары. Из машины конвейер доставляет
отформованную тару к отжигательной печи. В СССР
применяются разнообразные стеклоформующие автоматические
машины для выработки стеклянной тары.
Посудное и хозяйственное стекло. Ассортимент посудного и
хозяйственного стекла весьма разнообразен. Сюда относятся
разнообразные стаканы, рюмки, бокалы, вазы, графины,,
кувшины и т. п. Посудное и хозяйственное стекло чаще
изготовляют путем выдувания, реже — путем прессования. В ряде
случаев поверхность изделий обрабатывают различными
способами: наносят рисунки или узоры шлифовальными колесами или
путем травления плавиковой кислотой, шлифуют с помощью
кварцевого песка на чугунной шайбе (гранение) и др.
Электровакуумное стекло представляет собой полуфабрикат,
предназначенный для изготовления ламп накаливания,
газоразрядных ламп, рентгеновских трубок .и других подобных
изделий. Для электрических ламп изготовляют две детали: колбу
(баллон) и ножку, в которой закрепляется светящаяся нить.
Ввиду особых требований, предъявляемых к
электровакуумному стеклу, для его изготовления применяют
многокомпонентную шихту. Кроме обычных трех главнейших окислов SiO2,
CaO, Na2C), эти стекла содержат также окислы магния, свин-
иа, алюминия, калия, бария, борный ангидрид. Стеклянные
660
колбы электрических ламп изготовляют выдуванием.
Производительность автомата, вырабатывающего колбы среднего
размера, составляет 50—70 тыс. штук в сутки. Детали ножки из-
гоюзляются при помощи машин вертикального или
горизонтального вытягизания.
Химико-лабораторное стекло должно обладать высокой
стойкостью к действию различных химических реагентов, высокой
термической стойкостью и малой способностью к
кристаллизации, что дает возможность обрабатывать стекло на
стеклодувной горелке. Поэтому такое стекло тоже имеет сложный
многокомпонентный состав. Кроме обычных стеклообразующих
окислов, в него почти всегда вводят борный ангидрид, окислы
алюминия, цинка, бария.
Светотехническое стекло можно разделить на три группы:
1) Рассеивающие стекла (молочные или опаловые),
получаемые при введении в состаз шихты глушителей, главным
образом фтористых солей. Такие стекла применяются для
изготовления электроосветительной арматуры.
2) Призматические стекла применяются для изменения
направления светового потока, например при передаче световых
сигналов на дальние расстояния.
3) Стекла с избирательным поглощением (светофильтры)
применяются для полного или частичного поглощения
отдельных участков спектра. Из таких стекол изготовляются,
например, защитные очки, поглощающие ультрафиолетовые,
инфракрасные или другие части видимых лучей спектра.
К этому типу относится также увиолевое стекло, которое
отличается способностью пропускать ультрафиолетовые лучи,
оказывающие благоприятное влияние на организм.
Прохождению ультрафиолетовых лучей через обыкновенное стекло
препятствует повышенное содержание в нем окислов железа, осо-
¦бенно в виде Fe2O3. В увиолевом стекле содержание Fe2O3 не
должно превышать 0,01%, что достигается применением
исходных материалов, не содержащих примесей. Увиолевое стекло
применяют в медицине при ультрафиолетовом облучении, в
некоторых случаях для изготовления оконного стекла,
пропускающего ультрафиолетовые лучи, используемого, например, для
остекления больниц.
Оптическое стекло должно обладать высокой прозрачностью
и отзечать определенным требованиям в отношении
лучепреломления и светорассеяния. Обязательным условием является
высокая однородность оптического стекла по химическому
составу и физическим свойствам. Производство оптического
стекла весьма сложно.
В состав оптического стекла входят различные окислы в
разнообразных сочетаниях. Особенно большое значение имеет
чистота исходных материалов (отсутствие соединений железа,
661
хрома, сульфатов, .хлоридов и др.). Оптическое стекло
выпускают в виде прямоугольных кусков различных размеров и в
виде заготовок, имеющих форму оптических деталей (линзы,
призмы), с припусками для точной обработки.
Эмали представляют собой легкоплавкие глухие или
окрашенные стекла, наплавляемые на металлы для предохранения
их от коррозии и придания металлическим изделиям красивого
вида. Разработаны многочисленные составы эмалей различного
назначения. Наиболее распространены эмали для покрытия
чугунной и стальной аппаратуры, применяемой в химической и
пищевой промышленности, а также холодильной и газовой ап
паратуры, посуды и др.
Для эмалирования стали и чугуна применяют две эмали:
грунтовую и покровную.
Грунтовая эмаль должна прочно сцепляться с металлом
Она содержит 10—20% борного ангидрида и так называемые
сцепляющие окислы кобальта и никеля. Покровная эмаль
более легкоплавка, чем грунтовая, и содержит соединения
фтора (глушитель). В состав цветных эмалей вводят различные
красители — пигменты. В зависимости от назначения
покровная эмаль должна обладать химической стойкостью к действию
определенных реагентов.
Шихту для эмали приготовляют так же, как для стекла.
Изделия, покрытые эмалью, обжигают в туннельных или
муфельных печах.
Эмалируют также изделия из золота, серебра, меди и ее
сплавов. Для этой цели применяют эмали, представляющие
собой особо легкоплавкие, окрашенные, прозрачные или глухие-
стекла, содержащие окись свинца.
4. РАСТВОРИМОЕ СТЕКЛО И СИЛИКАГЕЛЬ
РАСТВОРИМОЕ СТЕКЛО
Растворимое стекло состоит из силикатов щелочных
металлов переменного состава, выражаемого общей формулой
R2O>nSiO2, где ИгО— окись натрия Na2O или калия К2О.
Наибольшее распространение имеет натриевое растворимое стекло.
Отношение числа молей SiO2 к числу молей R2O называется:
модулем растворимого стекла. В приведенной общей формуле
растворимого стекла буква п обозначает его модуль.
Выпускаемые сорта растворимого стекла имеют модуль от 1 до 4,2.
Стекла с модулем ниже 3 называются щелочными, с более
высоким модулем — нейтральными.
По многим свойствам растворимое стекло может быть
отнесено к настоящим стеклам. От последних оно отличается
растворимостью в воде и способностью образовывать коллоидные
растворы. Водные растворы растворимого стекла называются
жидким стеклом.
662
Все сорта растворимого стекла можно разделить на три
труппы.
К первой группе относятся твердые растворимые стекла,
получаемые путем сплавления кремнезема с содой или сульфатом
натрия и выпускаемые обычно в виде кусков, напоминающих
обыкновенное стекло, или в виде порошка. Такое стекло не
содержит воды или содержит ничтожное количество ее в
поверхностном слое и носит название силикат-глыбы или, просто,
силиката.
Вторую группу составляют твердые сорта растворимого
стекла, содержащие большее или меньшее количество воды.
Такое стекло, называемое гидратированным стеклом, хорошо
растворимо в воде. Будучи твердым, как силикат-глыба, гидра-
тированное стекло обладает некоторой эластичностью.
К третьей группе относятся растворы жидкого стекла
различных концентраций и состава. Это обычно густые, вязкие
жидкости.
Растворимые стекла применяют в строительстве в качестве
связующего вещества и для изготовления кислотоупорных
силикатных композиций. Растйсфимое стекло применяется также
в производстве абразивных материалов, для брикетирования
различных порошкообразных материалов, в качестве клеящего
и пропитывающего вещества, флотационного агента при
обогащении руд, наполнителя при мыловарении, компонента
синтетических моющих средств, для изготовления силикатных
красок, силикагеля и др.
Растворимые стекла обычно употребляют в виде растворов
большей или меньшей концентрации.
Наиболее распространенный метод получения растворимого
стекла заключается в сплавлении кремнезема (обычно
кварцевого песка) с содой или сульфатом натрия, в последнем случае
в присутствии угля. Омесь плавят в ванной печи, как при варке
обычного стекла. Температура процесса, в зависимости от
модуля стекла, составляет 1300—1500°С.
Чем больше щелочей содержит стекло, тем ниже
температура плавления. Расплавленную массу выпускают из печи и
охлаждают.
Другой метод получения растворимого стекла основан на
растворении природного или искусственного аморфного
кремнезема в растворе едкой щелочи:
2NaOH + nSiO3 = Na,O-nSiO.2 + ШЭ
Для ускорения процесс проводят в автоклаве под
давлением.
Гидратированное стекло получают обработкой водой
силикат-глыбы пли концентрированием раствора жидкого стекла.
663
СИЛИКАГЕЛЬ
Силикагель представляет собой ангидрид кремневой
кислоты в особом агрегатном состоянии, с сильно развитой
поверхностью и потому является весьма ценным адсорбентом.
Силикагель имеет вид твердых прозрачных или матовых
зерен. Величина зерен в зависимости от сорта составляет
2,8—7,0; 1,5—3,5; 0,25—2,0 и 0,2—0,5 мм.
В зависимости от величины пор различают силикагель
мелкопористый с радиусом пор около 15А* и крупнопористый
с радиусом пор более 50А-
Мелкопористый силикагель применяют для поглощения
паров воды из воздуха при низкой его влажности, а также для
•сорбции некоторых других паров и газов. Крупнопористый
силикагель применяют преимущественно для сорбции паров и
газов при высокой их концентрации, а также для очистки
жидкостей (осветление минеральных масел, керосина, сырого
бензола, очистка нефтепродуктов от серы и др.). При насыщении
сорбента адсорбционная способность его может быть
восстановлена путем продувки горячим воздухом или сушкой
(регенерация). Силикагель применяют также в качестве
катализатора, носителя для катализаторов и др.
При производстве силикагеля сначала получают золь крем-
некислоты путем взаимодействия силиката натрия с серной
кислотой плотностью 1,16—1,18 г/см3 по реакции:
Na2O-nSiOa -j- H2SO4 = nSiO2 + Н2О + Na2SO4
Полученную жидкость сливают в стальной освинцованный
ящик-коагулятор. Коагуляция золя, т, е. переход его в гель
(застудневание), протекает при 18—26°С в течение 5 суток. За
это время сформировывается сеть капилляров силикагеля, от
чего зависит его строение и сорбционная способность.
Для получения однородных кусков готовый гель выгружают
из коагулятора и продавливают через сита с отверстиями
размером 45 мм в деревянные корзины с днищем из мешковины
или другой фильтровальной ткани. Высота слоя геля в корзине
не должна превышать 100 мм.
Далее гель отмывают от электролитов (сульфат натрия,
избыток серной кислоты). Продолжительность промывки 4—7
суток. По окончании промывки продукт высушивают на воздухе,
затем загружают на противни из нержавеющей стали слоем
40—50 мм. Противни устанавливают в камерную сушилку.
Сушка продолжается 16—18 ч, при постепенном (через каждые 2 ч)
повышении температуры от 120 до 400—425°С.
* А— ангстрем, равен 10-8 см.
664
После сушки силикагель дробят и рассевают на фракции.
Мелкопористый силикагель упаковывают в мешки весом
нетто по 30—35 кг, крупнопористый в такие же мешки весом
нетто по 18—22 кг.
5. ПЛАВЛЕНЫЕ ГОРНЫЕ ПОРОДЫ И ПРИРОДНЫЕ
КИСЛОТОУПОРЫ
При плавлении некоторых горных пород получается масса,
пригодная для изготовления различных изделий.
Так, плавленый кварц, или кварцевое стекло, является
прекрасным огнеупорным материалом, выдерживающим резкие
изменения температуры.
Существует два вида кварцевого стекла — прозрачное и
непрозрачное. Прозрачное кварцевое стекло изготовляют из
горного хрусталя плавлением его в индукционных электрических
лечах. Непрозрачное кварцевое стекло изготовляют плавлением
чистого кварцевого песка в печах сопротивления. Из
прозрачного стекла изготовляют главным образом лабораторное
оборудование, из непрозрачного — аппараты для заводских
установок.
Каменным литьем называют изделия из некоторых
плавленых горных пород, главным образом базальта и диабаза
(изверженные горные породы).
Примерный состав изверженных горных пород (в % вес):
SiO2. .
А1гО3 .
FeO3 .'
. 49—50
. 11—16
6—15
5—9
CaO . .
MgO. .
Na,0 .
K2O . .
3—11
2—10
. Около З
. 1—1,5
Каменное литье отличается большой химической стойкостью
и высоким сопротивлением истиранию. Поэтому плитки из
каменного литья применяют для футеровки химической
аппаратуры, работающей в агрессивных средах; из каменного литья
изготовляются различные отливки деталей химической
аппаратуры, шары и футеровочные плиты для шаровых и
трубчатых мельниц, плиты для полов, лотки каналов
гидрозолоудаления и др. Промышленное производство химически стойких
строительных и других материалов путем литья расплавленных
горных пород или металлургических шлаков получило
название петрургии.
Производство каменного литья заключается в основном в
плавлении породы, отливке изделий и их термической
обработке. Сырье поступает в плавильную печь в виде кусков
величиной до 200 мм.
Плавильные печи бывают пламенные с регенераторами,
отапливаемые мазутом или газом, и электрические. Ванна печи
выполняется из магнезита, свод — из динаса. Температура ван-
665
ны 1400—14оО С. Масса, расплавленная в печи, непрерывно
перетекает через отапливаемый сливной канал в так
называемый «копильник». Последний представляет собой футерованный
магнезитом стальной барабан, вращающийся на роликах.
Копильник является как бы промежуточным сборником, в
котором расплавленная масса приобретает большую однородность.
Средняя температура массы, выходящей из копильника, 1180—
1220 °С.
Расплавленную массу заливают в предварительно нагретые
формы, изготовленные из формовочной земли. Нагревание
форм предотвращает быстрое остывание массы и
способствует заполнению всех деталей формы, а также образованию
кристаллической, а не стеклообразной структуры изделия.
Форму с отливкой загружают в круглую печь с вращающимся
подом и шамотными колосниками для укладки форм. В печи
температура в течение 45 мин снижается с 1050—1100 до
850—900 5С.
Эти условия способствуют кристаллизации массы и
отвердеванию изделий. Отвердевшие изделия в формах
поступают на отжиг и охлаждение в туннельные печи. В этих
печах происходит дальнейшее равномерное охлаждение
изделий для устранения в них внутренних напряжений.
По выходе из туннельных печей изделия освобождают от"
форм и затем очищают стальной щеткой от приставшей
формовочной земли.
Кроме изделий из плавленых горных пород, в химической
промышленности применяются также природные
кислотоупорные материалы.
К ним относятся горные породы — андезит и бештауннт,
которые добывают на Кавказе (Боржомское и Казбекское
месторождения), а также гранит.
Казбекский андезит имеет примерно следующий
химический состав (в % вес):
SiO, .... 63,2 СаО .... 5,1
ТЮ„ ... 0,5 MgO ... 2,5
А1,б3 . . . 17,0 КоО .... 2,0
Fef,O3 ... 2,5 Na,O ... 4,0
FeO . . 2,5 Прочие . . 0,6
-МпО ... 0,1
Бештаунит, по химическому составу близкий к андезиту,
отличается более высокой кислотостойкостью.
Андезит и бештаунит, добываемые в виде брусков и блоков,,
применяют для изготовления решеток электрофильтров и
сернокислотных башен, а также в качестве футеровочне.го
материала.
В виде щебня н муки андезит и бештауннт используют дли
приготовления кислотоупорного бетона.
666
Месторождения гранитов имеются на Кольском
полуострове, в Карелии, на Украине, на Кавказе и на Урале.
Средний химический состав гранитов (в % вес):
SiO2 .... 70,2 MgO ... 0,9
TiO2 .... 0,4 CaO .... 2,0
A12O3 . . . 14,5 NaX) ... 3,5
F&jOa ... 1,6 K..0 .... 4,1
FeO .... 1,8 Прочие . 0.9
MnO ... 0,1
Благодаря кислотоупорности гранит также пригоден как
материал для химической аппаратуры, например в
производстве брома и иода. Однако плохая термическая устойчивость
гранита ограничивает его применение.
6. СИТАЛЛЫ
В последнее время начало развиваться производство новых
силикатных материалов, обладающих рядом важных
технических свойств — особенно высокими термической стойкостью,
механической прочностью и устойчивостью к коррозии. Эти
материалы, получившие наименование ситаллов (в США они извест-
зы под названием «пирокерамы»), представляют собой
стеклокерамику, в которой количество стекловидной фазы колеблется
в пределах 40—95%. Ситаллы получаются в результате
плавления керамических материалов, последующего охлаждения
и термической обработки по специальному режиму,
обеспечивающему как необходимое соотношение кристаллической и
стекловидной (аморфной) фаз, так и нужные структурные,
физико-химические и химические свойства материала.
Термическая обработка проводится при температуре 700—
1300°С. В результате этого процесса в материале образуется
кристаллическая фаза, состоящая из частиц размером менее
1 мк. Чем меньше размеры кристаллов, тем выше
механическая прочность, жаростойкость, термическая и химическая
устойчивость и лучше другие важные свойства ситаллов.
По механическим свойствам ситаллы превосходят сталь,
уступая ей лишь в отношении ударной вязкости. В настоящее
время получены стеклокерамические материалы с
коэффициентом теплового расширения, близким к нулю, пределом
прочности при сжатии до 9000 кгс/см2 и при изгибе до 1800 кгс/см2,
устойчивые к действию смеси азотной и_серной кислот при
температурах до 300°С и выдерживающие резкие (до 1000°С)
перепады температур.
Стеклокерамические изделия изготовляются по технологии,
аналогичной процессам производства стеклянных изделий
(отливка, прессование, прокатка, центробежное литье,
вытягивание, выдувание).
Работами И. И. Китайгородского выявлена возможность
изготовления ситаллов из огненножидких шлаков, являющихся
многотоннажными отходами металлургической промышленности.
Из них методом ситаллургии можно изготовлять многие
строительные материалы (панели, плиты для настила полов и
лестниц, блоки и др.,), термоизоляционный пеноситалл,
трубопроводы, сантехнические изделия, износо- и коррозионностойкую
футеровку для дробилок и другого оборудования, различную тару
и др. Ситаллургические методы можно использовать также для
производства большого количества дешевых микроудобрений*
на основе расплавленных металлургических шлаков.
Добавки некоторых редких и редкоземельных металлов в
шихту для производства ситаллов придают им ряд ценных
свойств. Например, в США нашли применение пирокерамы с
добавкой лития, обладающие низким коэффициентом
расширения.
ЛИТЕРАТУРЛ
С. И. В о л ь ф к о в и ч, 3. А. Роговин, Ю. П. Р у д е н к о, И. В. Ш м а-
н е н к о в, Общая химическая технология, т. II, Госхимиздат, 195Э.
Ю. М. Б у т т, Г. Н. Д у д е р о в, М. А, Матвеев, Общая технология
силикатов, Госстройиздат, 1962.
П. П. Б у д н и к о в и др., Технология керамики и огнеупоров,
Госстройиздат, 1962.
Ю. М. Б у т т. Технология цемента и других вяжущих материалов, Пром-
стройиздат, 1956.
В. А. Воробьев, В. С. Колокольников, Производство минеральных.
вяжущих, Госстройиздат, 1960.
Ю. С. Лурье, Портланд-цемент, Госстройиздат, 1959.
Н. А. Т о р о п о в, Химия цементов, Промстройиздат, 1956.
Л. М. Бутт, В. В. По.тля к. Технология стекла, Госстройиздат, 1960.
И. И. Китайгородский и др., Технология стекла, Госстройиздат, 196!.-
Б. С. Темкин, Технология стекла и стеклоизделий, Ростехиздат, 1962.
Журн. ВХО им. Д. И. Менделеева, № 2 A960); N° 2 A963).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорберы 120, 442 ел., см. также
Абсорбционные колонны
олеумные М8, 119
Абсорбционные колонны (башни)
ПО, 118 ел., 132 ел., 281,
282, 287, 288, 293, 444 ел.
Абсорбция
адиабатическая 402, 404
автоматизация 165, 470 ел.
аммиака 16
— и двуокиси углерода 427 ел,
441 ел.
двуокиси углерода 191 ел., 574
окиси углерода 195 ел.
окислов азота 131 ел., 140 ел.,
272 ел., 277, 278, 285, 291,
293, 563, 564
сероводорода 55 ел., 191 ел.
трехокиси серы 96, 117 ел.
фтора 543 ел.
хлористого водорода 397, 404
Автоклавы 47, 303
Автоматизация 16
известково-обжигательных печей
468
контактных аппаратов 164, 255
печей КС 163
производства
аммиака 254 ел.
аммиачной селитры 557
кальцинированной соды 468 ел.
каустической соды 382 ел.
серной кислоты 160 ел.
суперфосфата 532
процессов
абсорбции 165, 470, 471
выпаривания 382 ел., 508
дистилляции 473 ел.
карбонизации 471 ел.
конверсии метана 177
мокрого катализа 166
осушки сернистого газа 165
разделения коксового газа 230
стекловарения 652
сушки колчедана 162
содовых печей 473
холодильных установок 223
Автоматические регуляторы 162, 379,
383, 471 ел., 532, 557
Аутотермичные процессы 103
Агломерация огарка 85
Аглопорит 644
Агрегаты
разделения коксового газа 226 ел.
синтеза аммиака 243, 249, 254
Агротехника 515
Адсорбенты 53, 54, 177, 215, 502, 664
Адсорбция
активированная 105
влаги 213, 215
сероводорода 53 ел.
химическая 238
хлора 413
Азеотропные смеси 61, 150, 375, 398
Азот
двуокись см. Двуокись азота
жидкий 179
получение 209 ел.
применение 197 ел., 225
окисление 233
окислы см. Окислы азота и Ни-
трозные газы
окись см. Окись азота
применение 170, 235 ел., 607 ел.
связанный 232 ел.
сорта 223
четырехокись 272, 300
Азотирование карбида кальция 607,
608
Азотистая кислота 138, 272
Азотистый ангидрид 131, 137, 272
Азотная кислота 232
концентрированная 262 ел., 293 ел.
образование 272
отбелка 302
перевозка и хранение 305
применение 262, 274, 278, 324, 367,
550, 556 ел., 565, 591 ел.
производство см. Азотная
кислота, производство
разбавленная (слабая) 262, 270 ел.
свойства 261
сорта 262
669
Азотчая кислота, производство
263 ел.
дуговым методом 233
концентрированной 265, 293 ел.
под давлением 281 ел., 289 ел.
при атмосферном давлении 275 ел.
прямой синтез 265, 299 ел.
разбавленной (слабой) 264, 270 ел.
расходные коэффициенты 278, 285,
304
сезместно с серной кислотой 304
техника безопасности 305
Азотно-фосфоркые удобрения 539,
565, 588 ел.
Азотные удобрения 231, 257, 517,
555 ел.
амидные 568 ел., 606
аммиачно-нитратно-амидные 576
аммиачно-нитратные 555, 561, 576,
593 ел.
аммиачные 566 ел., 576, 577,
588 ел.
гранулированные 559, 561
жидкие 231, 232, 257, 568, 573,
576, 577
нитратные 562 ел.
Азотоводородная смесь 170
очистка 191 ел., 238, 239
получение 170 ел., 223 ел.
Азотооборот 140
Аккумуляторная серная кислота 158
Активированный уголь 54, 502
Активный хлор 412, 416 ел.
Алит 635, 636
Алюминаткый модуль 636
Алюминаты 618, 643
Алючогель 215
Амальгама
натрия 351, 353
серебра 657
Амидные удобрения 568 ел., 606
Аммиак
абсорбция 16, 427 ел., 441 ел.
выход при синтезе 239 ел.
дистилляция 457 ел., 570 ел.
жидкий 256
окисление см. Контактное
окисление аммиака
перевозка и хранение 256
применение 213, 214, 217, 232, 257,
263 ел., 382, 424 ел. 539
556 ел., 566, 569, 576,' 588,'
593, 597
регенерация 425
свойства 231
синтез см. Синтез аммиака
сорта 256
Аммиакаты 519, 568, 576
Аммиачная вода 195, 231, 444, 519,
576
Аммиачная селитра 231, 561
кристаллизация 555, 559
применение 556, 561, 576, 595
производство 556 ел., 570
свойства 555 ел.
удобрения на ее основе 561
Аммиачно-нитратные удобрения 555,
561, 576, 593 ел.
Аммиачные удобрения 566 ел., 576,
577, 588
Аммоний
карбамат 568, 569
нитрат см. Аммиачная селитра
молибдат 600
сульфат 231, 233, 566 ел., 595 _
углекислые соли 425 ел., 449, 45!,
457, 472
фосфаты 588 ел.
хлорид 424 ел., 466
Аммонизация
рассола 441 ел.
удобрений 539 ел., 547, 577, 588 ел.,
597
Аммофос 588 ел.
Анализ
серной кислоты 159
ситовой 24
Ангидридные холодильники 123
Ангидрит 505, 643
Ангидритовый цемент 632
Андезит 648, 666
Аниониты 36
Анодный потенциал 310
Антегмит 149
Антинакипины 39
Апатиты 520, 521
Архитектурно-строительное стекло 657
Асбестовые диафрагмы 317, 338, 340
Атмосферная вода 28
Ацетилен 211, 328, 601, 604
Аэрирование 33, 39, 43, 641
Базальт 665
Байпас 135, 245
Баланс
напряжения 313
материальный 12, 13
тепловой 13, 14
Баллоны
аммиачные 256
кислородные 221
хлорные 372
Барабанные концентраты 151 ел.
Барабаны
грануляционные 537, 603
передельные 501
670
Барботажные
бочки 441 ел.
концентраторы 508
Башенная серная кислота 61, 63, 64
производство 130 ел., 139 ел.
Башни
абсорбционные 100, 118 ел.,
133 ел., 281, 282, 287, 288,
293, 442 ел.
грануляционные 558, 559, 573
денитрационные 133, 143
окислительные 133, 136, 144
продукционные 133, 143
промывные 97, 98
сушильные 99 ел.
Безопасное стекло 657
Безупарочное производство
аммиачной селитры 560
Белильные растворы 419, 420
Белит 635, 636
«Белые моря» 465
Бетон 644
фторирование 542
Бештаунит 666
Бикарбонат натрия 475, 476
кальцинирование 462 ел.
осаждение 426
получение 451 ел.
разложение 429, 478
Биологическая очистка воды 42
Биполярные электролизеры 317,
320 ел.
Биурет 568, 569
Бишофит 467
Болотная руда 53
Бора нитрид 629
Бораты 598, 618
Борная кислота 598, 648
Борные удобрения 598, 599
Бочки барботажные 441 ел.
Браунмиллерит 635
«Бросание» воды 29
Бура 648
Вакуум-испарители 508
Вакуум-карбонатная газоочистка 56
Вакуум-концентраторы 156
Вакуум-кристаллизация 583, 584
Вакуум-фильтры 456, 505, 506
Ванадиевые катализаторы 105 ел.
Ванны электролитические см.
Электролизеры
Ванные печи 650 ел., 665 ел.
БВС машина 655
Вентиляторы центробежные 146, 147
Взрываемости пределы газовых
смесей 232, 258, 327
Вода 26 ел.
аммиачная 195, 231, 444, 519, 57G
атмосферная 28
жесткость 27, 30
обеззараживание 32, 328
осветление и обесцвечивание 31
очистка 30 ел., 316
предотвращение накипи 39
природная, классификация 28
сточная 40 ел.
удаление газов 39
умягчение 33 ел.
хлорная 326
электролиз 171, 314 ел., 323
Воднорастаоримые удобрения 518
Водоподготовка 30 ел.
Водород
очистка 367
получение
конверсией метана 172 ел.
— окиси углерода 186 ел.
разделением коксового газа
223 ел.
электролизом воды 314 ел.
применение 170, 234 ел.
фосфористый 602
фтористый 527
хлористый см. Хлористый
водород
Водяной пар при конверсии
метана 172, 177, 179
окиси углерода 172, 176, 178 ел.,
1"86 ел.,
Возгонка
серы 48
фосфора 497 ел.
Воздух
дросселирование 203, 213
жидкий, ректификация 209 ел.
осушка 213, 2M
разделение 213 ел.
сжижение 204 ел.
состав 209
Воздушное обогащение 21
Воздушные вяжущие вещества 509,
629
гипсовые 631 ел.
известковые 630
магнезиальные 633
Воздушные сепараторы 21
Волокно стеклянное 658, 659
Вращающиеся печи 80 ел.. 436. 463.
609, 640
Время контактирования 270
Вспениватели 586
Вторичный пар 377
Выветривание и вылеживание глин
613
671
Вылеживание клинкера 640
Вымораживание кальциевой селитрь
565, 593 ел.
Выпаривание 377 ел.
растворов
аммиачной селитры 557 ел.
мочевины 573
фосфорной кислоты 507 ел.
электролитических щелоков 374 ел
Выпарные аппараты 378, 381, 384 ел.
392 ел.
АЕЦ 387
АПЦ 386
горизонтальные 559
пленочные 557, 558
с подвесной греющей камерон 381
Выплавка серы из руд 46 ел.
Высокообжигозый гипс 632
Вытягивание стекла 655
безлодочное 656
фильеркое 659
Выход
по току 310, 356 ел.
— энергии 314
Выщелачивание 487
Вяжущие вещества 509, 629 ел.
воздушные см. Воздушные
вяжущие вещества
гидравлические см. Гидравличе
ские вяжущие вешества
минеральные см. Минеральные
вяжущие вещества
Газгольдеры 188 ел.
Газификация топлива 170 ел.
Газоанализаторы 160, 161
Газоочистительнаи масса 53, 54
Газоочистка 12, 243
азотоводородной смеси 191 ел.,
238, 239
водорода 367
жидким азотом 197 ел., 225
коксового газа 52, 224
медноаммиачная 179, 195 ел.
механическая 88 ел., 132
мокрая 54 ел., 191 ел., 195 ел.
от аргона 197
— двуокиси углерода 191 ел., 197
— метана 197
— мышьяка 90, 98
— окиси углерода 179, 195 ел.
• — пыли 88 ел., 139, 437
— селена 98, 101
— серы 52 ел., 177, 191, 192
: — фтора 527, 543 ел.
предкатализом 200, 239
сернистого газа 88 ел.. 97 ел., 132
сухая 53 ел., 177
672
Газоочистка 12, 243
хлористого водорода 398
электрическая 89 ел., 99, 127, 145,
437
этаноламинами 192 ел., 574
Газ(ы)
взрываемость смесей 232, 258, 327
генераторные 171
глубокое охлаждение 202 ел.,
225 ел., 369
идеальные 203 ел.
известково-обжигательных печен
427, 433, 437
коксовый см. Коксовый газ
компрессия 200 ел.
конверсии метана 173 ел., 179, 182,
U84, 188
металлургических печей 66
нефтепереработки 52, 171
нитрозные см. Нитрозные газы
осушка 97, 99 ел., 213, 215, 367
очистка см. Газоочистка
попутные нефтиные 52, 171
природный 52, 171 ел.
растворимость см. Растворимость
сернистый см. Сернистый газ
сероводородные 67. 124 ел., 166
сжиженные см. Жидкие газы
токсичность 258, 327
топочные 67
транспортирование 117, 221 ел.,
256, 371
углеводородные 171 ел.
удаление из воды 39
хранение 188 ел., 221, 256, 371
экспанзернын 197
Галургия 579 ел.
Гафния карбид 629
Гашение извести 414, 429, 438, 439,
630
Гелиоопреснение воды 33
Гигроскопическая точка 520
Гигроскопичность удобрений 520, 555,
564, 566, 568
Гидравлическая известь 634
Гидравлические вяжущие вещества
509
глиноземистые 643
портланд-цемент 634 ел.
пуццолановые 642
роман-цемент 634
шлаковые 642, 643
Гидравлические добавки 642
Гидравлическое (мокрое)
обогащение 20
Гидратная известь 630
Гидраторы 630
Гидровруба метод добычи соли 332
Гидроизоляционные стеклопластики
660
Гидрокснлапатит 550
Гидролиз
нитрозилсерной кислоты 137
хлора 326
Гидрометаллургия 331
Гидротермические процессы 550
Гидрофобные коллекторы 22
Гидрохлорирование 328, 397
Гидроэлектрометаллургия 307
Гилохлориты 327
кальция 32, 411 ел.
натрия 410, 419
Гипс 631, 643
Гипсовое число 504
Гипсовые вяжущие вещества 631 ел.
Глазерит 578
Глазури 619
Глауконит 525
Глинозем 331, 425, 641, 643
Глиноземистые
вяжущие вещества 643
огнеупоры 624, 628
Глиноземный модуль 636
Глины 612 ел, 620, 621, 623, 625
Глиняный кирпич 623
Глубокое охлаждение газов 202 ел,
225 ел, 369
Глушители стекол 648, 661, 662
Гомогенизация стекольной массы 650
Гончарный товар 612
Горшки стекловаренные 652
Горшковые печи 652 ел.
Гравиемойка 20
Гранит 666, 667
Гранулирование
аммиачной селитры 559
двойного суперфосфата 546, 547
известково-аммиачиой селитры 561
карбида кальция 603
мочевины 573
суперфосфата 537 ел.
цяанамида кальция 607
Гранулометрический состав 614
Грануляционные башии 558, 559, 573
Графитовые
огнеупоры 628
холодильники 149
электроды 495
Грохочение 19
«Грязный» цикл орошения 139
Гидрон кислый 67, 128 ел.
Давление критическое 204
Дате, тит 598
Двойной суперфосфат 545 ел.
Двуокись азота 131, 138
жидкая 274
полимеризация 272
получение 270
Двуокись серы 69, 103, 160
см. также Сернистый газ
окисление 96, 102 ел, 109
применение 66, 67, 86, 409
Двуокись углерода 224
абсорбция 191 ел, 427 ел, 441 ел,
574
получение 429 ел, 433 ел.
применение 423 ел, 431, 569
растворимость 191, 193
удаление из воды 43
Двуокись хлора 408 ел.
Двуокись циркония 621, 629
Двууглекислая сода см. Бикарбонат
натрия
Дегазации
воды 39, 43
стекольной массы 649
Дезаэрация 613
Дезинфекция воды 32, 43
Действительная мощность 492
Декалькомания 618
Декарбонизация 478
Денитрационные башни 133, 143
Денитрация 131, 132, 134, 138, 295
Деполяризация 352
Депрессия температурная 376, 383
Детандер 206
Дефолианты 467, 606
Джоуля закон 492
Джоуля—Томсона эффект 203
Диабаз 665
Диаграмма
кипения серной кислоты 150
— азотной кислоты 294
кристаллизации серной кислоты
60
— азотной кислоты 261
материального баланса 13
теплового баланса 14
Диаммонийфосфат 588
Диатомовый кирпич 434, 623
Диафрагмы электролизеров 317, 338,
340
Дикальцийфосфат 539, 549
Димагнийфосфат 549
Динамидоновые огнеупоры 629
Динас 625, 626
Дистанционное управление 16, 223,
384
Дистиллерная жидкость 429, 457
переработка 465 ел.
Дистиллированная вода 33
43—2848
673
Дистилляционные колонны 458,
460 ел, 571, о72
Дистилляция
автоматизированная 473 ел.
азотной кислоты 294 ел.
в производстве мочевины 570 ел.
кальцинированной соды 428,
429, 457 ел.
Дифенильная смесь (ВОТ) 375, 393
Диэтаноламин 192 ел.
Добавки гидравлические 642
Доломит 596, 628
каустический 633
Дробление 514, 637, 639
избирательное 20
известняка 431
карбида кальция 607
клинкера 641
колчедана 70
фосфоритов 523
хлористого калия 579
Дросселирование 204, 571, 574
Дуговой метод фиксации азота 233
Дуговые печи 493, 494
Дуплекс система выпарки 377
Дутьевой способ получения
стекловолокна 659
Дьюара сосуды 222, 223
Едкий натр
выпаривание растворов 374 ел.
жидкий 394, 395
кристаллизация 394
обезвоживание (плавка) 389 ел.
применение 330, 331, 333, 410, 419
производство см. Едкий натр,
производство
свойства 329, 330, 389
сорта 394, 395
твердый 394
упаковка, перевозка 395
чешуйчатый 394
Едкий натр, производство 331, 410
известковым способом 477 ел.
расходные коэффициенты 343, 344,
347, 356, 357, 359, 361, 382,
483, 487
техника безопасности 373, 374
ферритным способом 484 ел.
химическими методами 476 ел.
электрохимическое 336 ел.
Жароупорный бетон 644
Железные катализаторы 200, 237, 238
Железобетон 644
Желе.чохромовые катализаторы 178,
188
674
Желтый фосфор 490, 491
передел 501
получение 497 ел.
Жерновые камни 633
Жесткость воды 27, 30
Жидкие (сжиженные) газы 202 ел.,
225 ел.
азот 197, 199, 223
аммиак 256, 572, 576
воздух 209 ел.
двуокись серы 69
кислород 221, 222
трехокись серы 121
Жидкие удобрения 231, 232, 257,
568, 573, 576, 577
Жидкое стекло 662
Жирные глины 613
Закаленное стекло 657
Закон(ы)
Джоуля 492
Ома 492
Фарадея 309
Заполнители бетона 644
Звездочки разбрызгивающие Мб
Зеленое масло 398
Зеркальное стекло 657
Змеевиковые холодильники 147
Идеальные газы 203 ел.
Избирательное дробление 20
Известково-аммиачная селитра
Известковое молоко
получение 429, 438
применение 39, 275, 333, 4!
хлорирование 418, 419
Известково-обжигательные
433 ел.
автоматизация 468
Известково-шлаковый цемент
Известняк 431 ел.
обжиг 424, 432, 630, 634
применение 423, 425, 549, 55
596, 602 ел., 637
разложение 432
Известь
воздушная 630
получение 424, 432, 534
гидравлическая 634
негашеная 630
обжиг 414, 424, 425, 429 с~
применение 34, 39, 477 ст., 6
пушонка 413, 630
хлорирование 413 ел.
хлорная см. Хлорная лззе
Извлечение селена из г?зсз и
ты 98, 101, 142
56!
9, 5-19
печи
643
1. 561,
16
ть
КИС.'Ю-
Извлечение
серы из руд 46 ел., 50 ел.
газов 52 ел., 177
Извлечения степень 19
Нзмельченке см. Дробление
Изодромные регуляторы 379, 383
Инверсия китрит-нитратных щелоков
274, 278, 563, 564
Ингибированнзя соляная кислота
406 ел.
Инжекторы 248
Интенсивность печей 78
Интенсификация
сернокислотных систем 140
синтеза аммиака 253
Иониты 33, 36, 37
Ионообменные процессы 36 ел., 563
ИТН аппарат 557, 558
Кажущаяся мощность 492
Каинит 57S, 587
Калиевая селитра 563, 564
Калий
месторождения 578
нитрат 563, 564
сульфаты 578, 586, 587, 594, 595
хлорид см. Хлористый калий
Калийно-аммиачная селитра 561. 579
Калийные удобрения 577 ел., 587
бесхлор кые 563, 564, 586, 587
природные соли 578, 586, 587
производство 580 ел., 585 ел.
Калимагнезия 587
Кальциевая селитра 275, 550, 563 ел.,
576
выморажизание 565, 593 ел.
Кальций
алюминат 643
алюмоферрит 635
гипохлорит 32, 411, 417 ел.
карбид см. Карбид кальция
карбонат см. Известняк
нитрат см. Кальциевая селитра
силикаты 635, 636
сульфаты 524, 578, 586, 587, 634,
643
фосфаты 524, 525, 526, 539 545
549
фо;фид 602
фторид 607
хлорид 370, 466, 607
цианамид 233, 274, 606 ел.
Кальцинирование 429, 462 ел.
Кальцинированная сода 421 ел,
мистификация 477 ел.
применение 34, 55, 274 333,
419 ел., 477, 484, 646, 647
природная 422, 423
Кальцинированная сода
производство см.
Кальцинированная сода, производство
свойства 422
синтетическая 423 ел.
сорта 467
Кальцинированная сода,
производство 422
автоматизация 468 ел.
аммиачным способом 424 ел.
расходные коэффициенты 465
способом Леблана 12, 423
схема 427 ел.
техника безопасности 488
утилизация отходов 465
Каменное литье 665
Каменный товар 612, 619, 620 ел.
Камеры
полочные 414
пыльные 485
суперфосфатные 534, 535
Канальные печи 609
Каолин 612, 619, 620
Капицы, холодильный цикл 207
Капролактам 567
Карбамат аммония 568, 569
Карбид см. Мочевина
Карбамидные смолы 669
Карбид гафния 629
Карбид кальция 233, 601
азотирование 607, 608
брикетированный 605
гранулированный 603
применение 601, 605, 607 ел.
производство 602 ел.
Карбидные печи 604
Карбиды таллия, титана, циркония
629
Карбонизационные колонны 450,
452 ел.
Карбонизация 429, 449
автоматическая 471 ел.
рассола 334, 335
Карборунд 621, 628, 629
Карналлит 578
Каскадный цикл 208
Катализ мокрый 53, 67, 124 ел., 16S
Катализатор(ы)
активная поверхность 269
активность 107, 108, 237. 238, 24S,
266
БАВ 106
ванадиевые 105 ел.,
железные 200, 237, 238
железохромовые 178. 188
конверсии метана 175, 180
— окиси углерода 178, 183
никелевые 175, 239
1шкельхромовып 200
43*
675
Катализатор(ы)
окисления аммиака 232, 263,
266 ел.
— двуокиси серы 105 ел.
окисные 267
отравление 177, 197, 238, 268
платиновые 105, 232, 263, 267
производительность 107, 239 ел.
промотированные 237
разрушение 267
СВД 106 ел.
синтеза аммиака 237 ел.
Каталитические процессы
конверсии метана 174, 177, 179
окисления аммиака 265 ел.
— двуокиси серы 102 ел., 125 ел.
синтеза аммиака 235 ел.
Катиониты 36, 37
Катионообменные процессы
водоочистки 36 ел.
получения нитрата натрия 563
Катодный потенциал 310
Каустификация 477 ел.
Каустицеры 481 ел.
Каустическая сода см. Едкий натр
Каустический доломит 633
Каустический магнезит 627, 633
Кварц 619, 620, 625
плавленый 665
Кварцевое стекло 665
Кварциты 626
Керамзит 644
Керамика 611 ел.
грубая 612, 614, 620, 623 ел.
тонкая 612, 614, 619 ел.
Керамические изделия
глазурование 619, 621
дикасовые 625, 626
кирпичные 623 ел.
кислотоупорные 612, 620 ел.
классификация 612
магнезиальные 627
обжиг 616 ел., 623, 626, 627, 629
огнеупорные 612, 624 ел.
производство 614 ел.
раскраска 618
строительные 612, 623
сырье 612 ел., 614
фарфоровые и фаянсовые 619 ел.
формовка 615
шамотные 624, 625
щелочестойкие 621
Керамические массы 614, 615
Кипелка 630
Кипящий слой 75 ел., 87
Кирпич 612
кислотоупорный 620
огнеупорный 624 ел.
строительный 623
Кислород 222, 224
жидкий 221, 222
применение 170, 172, 177, 179 сл.г
182 ел.
производство 209 ел., 314 ел.
Кисдотоупоры
бетон 542, 644
керамика 612, 620 ел.
природные 665 ел.
Классификаторы гидравлические 20
Классификация
керамических изделий 612
природных вод 28 ел.
процесов 15
удобрений 517
Клинкер цементный 48, 50, ьоЗ. 834-
635
Коагулянты 31, 582
Коагулятор 664
Коксовый газ 52, 171, 566
очистка 52, 224
разделение 225 ел.
Количество орошения НО
Коллекторы гидрофобные 22
Колонны см. также Башни
абсорбционные 100, 118 ел., 132 ел.
281, 282, 287, 288, 293,
444 ел.
дистилляционные 460, 572
карбонизационные 450, 452 ст.
концентрационные 296, 297
осадительные 452
предкатализа 239
ректификационные 210 ел., 227
синтеза аммиака 244, 249 ел.
— мочевины 572, 574
тарельчатые 210
Колориметры 161
Колчедан серный (Пирит) 50, 64, 65
обжиг 13, 71 ел.
сушка 70
флотационный 65 ел.
Колчеданный огарок 75, 85 ел.. 599
Кольцевые печи 437, 616, 617
Комбинирование производств 11. 466,
570
Комбинированные печи 492, 494
Комовая сера 57, 58
Комплексная переработка 11, 12
нефелина 425, 641
огарка 85 ел.
серных руд 47 ел., 65
Комплексные удобрения 518, 5S7 ел.
двойные 588 ел., 591
производство 589 ел., 593 с". 596
сложные 588 ел.
смешанные 596 ел.
тройные 591, 597
С75
Компрессоры
многоступенчатые 200
оппозитные 201, 202
центробежные 246, 247
Конверсия
коксового газа 224
метана 172 ел.
высокотемпературная 182 ел.
каталитическая 179 ел.
нитрата натрия 563
окиси углерода 172 ел., 178,
186 ел.
Конвертированный газ 173 ел., 178,
179
Конверторы 178, 181, 183, 184 ел.,
244, 249 ел.
Конденсаторы 372, 461
Конденсация фракционированная 225
Константы
критические 204
равновесия реакций 173, 266, 271
Контактирование 111 ел.
время 270
температура ПО
Контактное окисление аммиака
аппаратура 279, 281, 285, 286
без давления 275, 281, 292
под давлением 283, 289, 291
реакции 265
условия 268 ел.
Контактные аппараты см. также
Конверторы
автоматизация 164
окисления аммиака 279, 281, 286
— двуокиси серы 11 ел., 125 ел.
Контроль процессов
производства соды 449, 462, 465
— серной кислоты 160 ел.
холодильных установок 223
электролиза воды 323
Концентрат 18
датолитовый 598
флотационный 46, 66
Концентраторы
барабанные 151 ел.
барботажные 508
вакуумные 115 ел.
с внешним обогревом 154
Концентрации предельно допустимые
вредных веществ
в воде 40
в воздухе 168, 260, 305
¦Концентрирование
азотной кислоты 293 ел.
серной кислоты 149 ел.
фосфорной кислоты 507 ел.
хлористого водорода 398, 403,
405 ел.
электролитических щелоков 374 ел.
Копильник 666
Кора земная, состав 17
Кормовые добавки 551 ел., 568
Коронный разряд 89
Корунд 629, 633
Косвенные удобрения 518
Косинус ф 492
Котлы
плавильные 47, 390 ел.
утилизаторы тепла 92, 93
Коэффициент (ы)
избытка воздуха 78
использования
натрия 454
сырья 13
тока 310
энергии 14
мощности 492
насыщения 635
расходные см. Расходные
коэффициенты в производстве
Краски керамические 618
Красный кирпич 623
Красный фосфор 490, 501, 502
Кратность орошения 140
Кремневая кислота 664
удаление из воды 30
Кремнеземный модуль 635
Кременефтористоводородная кислота
503, 527, 592
Кремний фтористый 527, 542, 543
Кремнийорганические полимеры 328,
611
Кристаллизация
аммиачной селитры 556, 559
бикарбоната натрия 453
гипса 504
дикальцийфосфата 549
едкого натра 389, 394
¦мочевины 573
сульфата аммония 567
хлористого калия 583, 584
Критические константы 204
Круги точильные 633
Круговые процессы 15
Ксилолит 633
Купоросное масло 61
получение 118 ел., 127
Лангбейнит 578
Леблана способ производства соды
12, 423
Легки!! бетон 644
Ленточный
пресс 615, 616
фильтр 505, 506
Ле-Шательв принцип 176
Лимоннорастворимые удобрения 518,
523
677
Листовое стекло 654
Литраж карбида кальция 601
Литье каменное 665
Лодочка 655
Магазинированпе 640
Магнезиальные
вяжущие вещества 633
огнеупоры 627
Магнезит 627, 633
Магний
борат 598
карбонат 596, 633
сульфат 578, 587, 598
фосфат 549
Магнитное обогащение 21
Магнитные сепараторы 22
Марганцевые удобрения 599
Материальный баланс 12, 13
Машина отсадочная 21
Машинная выработка стекла 654 ел.
Медноаммиачная очистка газа 179,
195 ел.
Медные удобрения 599
Медный колчедан 50
Мел 431, 538, 561
Меланж 135, 305
Месторождения 17
андезита, бештаунита 666
гранита 667
калийные 578
натриевой селитры 233, 563
серного колчедана 65
серных руд 45, 46
соды 422
фосфатов 521
Металлургический порошок 627
Металлургия хлорная 329
Метан 224
конверсия 172 ел.
разложение 175
свойства 202, 225
удаление из газов 197
Механическая технология II
Микроудобрения 596, 598 ел., 668
Минеральное сырье 17 ел.
Минеральные вяжущие вещества 629
воздушные 630 ел.
гидравлические 634 ел.
гипсовые 631
изделия из них 643 ел.
магнезиальные 633
Минеральные удобрения 514 ел.
азотные см. Азотные удобрения
борные 589, 599
гранулированные 519. 559. 56!,
569, 573, 597
лозы 515
Минеральные удобрения
жидкие 231, 232, 257, 568, 573,
576, 577
калийные см. Калийные
удобрения
кислотность и щелочность 519
комплексные см. Комплексные
удобрения
концентрированные 545, 549, 568,
576, 587 ел.
косвенные 518
марганцевые, медные,
молибденовые 599
объем производства 516
односторонние 517
прямые 517
сложные 519, 539,550,565,588 ел.
смешанные 519, 596, 597
фосфорные см. Фосфорные
удобрения
Модули
алюминатный и глиноземный 636
кременеземный 635
растворимого стекла 662
Мокрая газоочистка 54., 191 ел.,
195 ел.
Мокрое (гидравлическое)
обогащение 19, 20
Мокрые газгольдеры 188
Мокрый катализ 53, 67, 124 ел., 166
Молибденовые удобрения 599, 600
Молотая сера 57, 58
Моноаммонийфосфат 588
Моногидратные абсорберы 118, 119
Монокальцийфосфат 524, 525, 527,
545
Монополярные электролизеры 317 ел.
Моноэтаноламин 192 ел.
Мочевина 232, 576
дистилляция плава 571, 572
применение 568, 569
производство 569 ел.
свойства 568
сорта 569
Мочевино-формальдегидные
удобрения 569
Мощность
аппаратов 15
электрическая 492
Мука фосфоритная 523, 524
Муфель 502
Мышьяк, извлечение из газов 90, 98
Мышьяково-содовая газоочистка 55
Накипь 29
удаление 39
Напряжение разложения
338, 354
310, 315,
678
Напряжения
баланс 313
ряд 311
Напряженно армированный бетон 645
Насадки
керамические 144, 145
колонн синтеза аммиака 244, 250,
251, 253
регенераторов 217
Насосы центробежные 146
Насыщения коэффициент 635
Натриевая селитра 562, 563
получение 274
применение 263, 648
природная 233, 563
Натрий
бикарбонат см. Бикарбонат
натрия
гидроокись см. Едкий натр
гипохлорит 410, 419
карбонат см. Кальцинированная
сода
кремнефторид 542, 544
нитрат см. Натриевая селитра
нитрит 274, 563
окситиоарсенат 55
силикат 664
сульфат 646
тиоарсенит 55
феррит 484
фосфаты 36, 39, 47
хлорат 410
хлорид см. Хлористый натрий и
Поваренная соль
Натрон 422
Небьющееся стекло 657
Негашеная известь 630
Нейтрализаторы 557, 558
Нейтрализация аммиаком
азотной кислоты 556 ел.
азотнокислотной вытяжки
фосфатов 593
серной кислоты 566
фосфорной кислоты 588
Непрерывные процессы 15, 16
Нефелин 525
комплексная переработка 425, 641
Никелевые катализаторы 175, 239
Никельхромовый катализатор 200
Нитратные удобрения 562 ел.
Нитраты
аммония см. Аммиачная селитра
калия 563, 564
кальция см. Кальциевая селитра
магния 299
натрия см. Натриевая селитра
Нитриды бора и титана 629
Нитрит-нитратные щелока 274, 278,
563, 564
Нитроза 130
Нитрозилсерная кислота 130, 137, 274
Нитрозные газы 264
абсорбция 272 ел., 277, 278, 285,
291, 293, 563, 564
переработка в азотную кислоту
270 ел.
Нитрофос 565, 591 ел.
Нитрофоска 565, 591, 594 ел.
«Нормальные деления» 440
Нормальный потенциал 31!
«Нормальный» раствор соды 480
Обезвоживание едкого натра 389 ел.
Обезвреживание сточных вод 40 ел.
Обеззараживание воды 32
Обессоливание воды 33 ел.
Обесфторенные фосфаты 550 ел.
Обесцвечивание
воды 31 ел.
стекла 648
Обжиг
доломита 633
известняка 429, 432, 630, 634
керамических изделий 616 ел., 623,
626, 627, 629
колчедана 13, 71 ел.
магнезита 633
цементного шлама 639
электродов 493, 495
Обменная способность ионитов 37
Обогащение 18 ел.
в тяжелых жидкостях 22
магнитное 647
медных руд 65
степень 19
флотационное 22, 521, 647
электромагнитное 21
Обратимый потенциал 311
Обратный рассол 376, 440
Объемная скорость 107, 178, 239
Огарок колчеданный 75, 599
утилизация 85 ел.
Огнеупорность 613, 624, 625, 626 ел.
Огнеупоры 612
динасовые 625
доломитовые 628
кварцевые 665
магнезиальные 627
специальные 628, 629
шамотные 624
Односторонние удобрения 517
Однофазные печи 494
Озонирование воды 32
Окисление
азота 233
аммиака см. Контактное окисление
аммиака
дзуокиси серы 102 ел., 109 ел.
6 9
Окисление
метана см. Конверсия метана
окиси азота 131, 138, 270 ел., 300
сероводорода 125
серы 122
фосфора 509 ел.
Окислительные башии 133, 136, 144
Окислы азога см. также Нитрозные
газы
в производстве серной кислоты
130 ел.
окисление 131, 138, 270 ел., 300
расщепление 274
предельная концентрация 168, 305
Окись азота
концентрированная 274
получение см. Контактное
окисление аммиака
Окись железа 79
Окись углерода 224
конверсия 172 ел., 178, 186 ел.
предельная концентрация 260
растворимость, 196
удаление из газов 179, 195 ел.
Олеум 59, 61, 94, 158
высококонцентрированный 122
получение 118 ел., 128 ел,
Олеумные абсорберы 118, 119
Опреснение воды 33
Оптимальная температура
контактирования БОг ПО
окисления аммиака 269
синтеза аммиака 241
Оптимизация процессов 16
Оптическое стекло 661
Органические вяжущие вещества 629
Органические удобрения 514, 517
Органо-минеральные удобрения 517
Оросительные холодильники 148
Орошение
башенных систем 135 ел.
«грязный» цикл 139
количество и кратность 140
плотность 98, 140
Осаждение
асбестовых диафрагм 340
бикарбоната натрия 426
дикальцийфосфата 549
примесей сточных вод 41
Осветление
воды 31
щелоков 481 ел., 487
Осушка
воздуха 213, 215
сернистого газа 97, .99 ел.
хлора 365 ел.
Отбелка
азотной кислоты 302
волокна 408, 419
Отбросы 12
Отжиг стекла 654
Отливка керамических изделий 615
Отравление катализаторов 177, 197,
238, 268
Отсадочная машина 21
Отходы 12
переработка 425, 465 ел., 484, 500,
509, 542, 567, 641
Очистка
воды 30 ел., 316
газов см. Газоочистка
жидкостей 664
каустической соды 382
кислоты от селена 142
рассолов 333 ел., 381, 427, 429. 440
серы 56 ел., 122
сокового пара 378. 380
сточных вод 12, 40 ел.
Пакетные электроды 495
Пассеты 441, 442
Пассивации зона 528
Пенообразователи 22
Пеноситалл 667
Пеностекло 657, 658
Передельные барабаны 501
Перенапряжение 312
Периодические процессы 15
Перхлорвиниловая смола 329
Петрургия 611, 667 ел.
Печи
ванные 650 ел., 665 ел.
вращающиеся 80 ел., 436, 463,
609, 640
ВХЗ 80 ел.
горшковые 652 ел.
для разложения кислоты 128 ел.
— рафинирования серы 57
— сжигания сероводорода 126,
127
серы 86
дуговые 493, 494
известково-обжигательные 433 ел.
интенсивность 78
канальные 609
карбидные 604
кипящего слоя 75 ел., 87, 163
колчеданные 74 ел.
кольцевые 437, 616, 617
комбинированные 494
косвенного нагрева 493
однофазные 494
плавильные 665
полочные механические 80 ел.
прямого нагрева 493
пылевидного обжига 82
синтеза хлористого водорода
399 ел.
680
Печи
содовые 463
сопротивления 493
стекловаренные 650 ел.
трехфазные 494, 498, 604
трубчатые 184
туннельные 617, 618, 666
цементные 640
цианамидные 608
¦ шахтные 433
электрические 493 ел., 604, 608
Пирит см. Колчедан серный
Пирокерамы 667, 668
Плавиковый шпат 607
Плавильные
батареи 390 ел.
печи 665
Плавка (плавление)
едкого натра 389 ел.
медного колчедана 50 ел.
фосфатов 551, 553
Плавленые горные породы 665 ел.
Плавни 613, 635
Пластичность глины 612
Платиновые катализаторы 105, 232.
263, 267
Плитки
каменные 665
керамические 621
стеклокерамические 667
Плотномеры 161
Плотность
орошения 98, 140
тока 312, 343 ел., 356 ел., 360 ел.,
364
Побочные продукты 12
Поваренная соль см. также
Хлористый натрий
добыча 332
очистка растворов 333 ел., 381,
427, 429, 440
Подземные рассолы 332
Погружные холодильники 147
Полевой штат 613, 614, 619, 620, 648
Полезная разность температур 377
Поливинилхлорид 328
Полигалит 578
Полимеризация
двуокиси азота 272
серного ангидрида 656
Полированное стекло 656
Полифосфорная кислота 511
Полупроводники 308, 329
Полупродукты 12
Поля орошения и фильтрации 42
Пористые
бетон 644
кирпич 623
стекло 657
Портланд-цемент 49, 509, 634 ел.
качество 635, 641
производство 636 ел.
Посудное стекло 660
Поташ 425, 647
Потенциал
рязряда ионов 310
электрода 311
Пределы взрываемости газовых
смесей 232, 258, 327
Предельные концентрации вредных
веществ
з водоемах 40
— воздухе 168, 260, 373, 542, 554,
575
Предкатализ 200, 239
Преобразование тока 349, 350
Пресс
ленточный 615, 616
ящичный 615
Прессование
керамических изделий 615 ел.
стеклопластиков 659
Преципитат 549, 550
Природный газ 52, 171 ел.
Проводники 308
Производительность
аппаратов 14
катализаторов 107, 240
Промоторы 237
Промыватели газов 442 ел., 447, 463
470
Промывные башни 97, 98
Протекаемость диафрагмы 341
Противоток 16
Прямые удобрения 517
Пуццолановый цемент 642
Пушонка 413, 630
рН-метры 161
Равновесие реакций
окисления NO 271
— NH3 266
— SO2 102 ел.
синтеза аммиака 235
Разбрызгивание кислоты 145, 146
Разделение
воздуха 213 ел.
коксового газа 223 ел.
Разложение
амальгамы 351, 354 ел., 361 ел.
бикарбоната натрия 429, 478
отработанной серной кислоты
128 ел.
солей аммония 459
фосфатов кислотами 503, 524 ел.
азотной 565, 591 ел.
серной 524, 526 ел., 532
фосфорной 525, 545
681
Рапа 332
Раскраска керамических изделии 618
Рассев пробы 24
Рассол (ы)
аммонизация 441 ел.
обратный 376, 440
очистка 333 ел., 381, 427, 429, 440
подземные 332
приготовление 33 ел., 439 ел.
Раствор разбавления 504
Растворимое стекло 662 ел.
Растворимость
аммиака 231
двуокиси серы 69
— углерода 191, 193
— хлора 409
едкого натра 330
карбоната натрия 422
окиси углерода 195, 196
сероводорода 194
фосфатных удобрений 516
хлора 327
хлористого натрия 375
хлористого водорода 396
Растворитель
заглубленный 333
шнековый 582
Расходные коэффициенты в
производстве
азотной кислоты 278, 285, 287, 301
аммиака 233, 234, 253
аммиачной селитры 560
двуокиси хлора 411
извести 436, 439
кальцинированной соды 465
карбида кальция 605
каустической соды 343, 344, 347,
356, 359, 361, 382, 483, 487
серной кислоты 94, 121, 128. 130,
142, 154, 157
сульфата аммония 567
суперфосфата 536, 549
фосфора 502
хлора 345
хлората натрия 308
хлористого водорода 406
хлорной извести 416
цианамида кальция 609
Расходомеры 161
Рафинирование серы 56
Регенеративный принцип 205
Регенераторы 196, 216, 653
Регенерация 12
аммиака 425, 428, 457 ел.
двуокиси марганца 324
ртути 351, 353
сорбентов 55, 56, 194, 196, 664
хлора 325, 350
хлористого водорода 399
Регенерированная серная кислота 61,
158
Регуляторы автоматические 162,
468 ел., 508, 532, 557
изодромные 379, 383
Ректификационные колонны 210 ел.,
227
Ректификация жидкого воздуха
209 ел.
Ретроградация Р2О5 526 «
Ретур 464, 590, 594, 597
Роман-цемент 634
Ртутный катод 351
Рудоразборка 19
Руды 18
калийные 578
обогащение см. Обогащение
серные 45 ел.
сульфидные 50
фосфатные 520 ел.
Ряд напряжении 311
Сажа 175, 183
Самовоспламенения температура 258
Самосадочная соль 332
Сатуратор 566, 567
Сборный железобетон 645
Светотехническое, стекло 661
Светофильтры 661
Селен
извлечение 98, 101, 142
двуокись 168
Селитра
аммиачная см. Аммиачная селитра
нзвестково-аммиачная 561
калиевая 563, 564
кальциевая см. Кальциевая
селитра
натриевая см. Натриевая селитра
Сепараторы 21, 22
Сера 44 ел., 67
двуокись см. Двуокись серы и
Сернистый газ
очистка 56 ел., 122
получение 45 ел., 50 ел., 124
применение 44, 122 ел.
свойства 44, 45
сжигание 86 ел., 122 ел.
сорта 57, 58
трехокись см. Серным ангидрид
удаление из газов 52 ел.; 177,
191, 192 ел.
Серебрение зеркал 657
Серная кислота 59 ел.
анализ 159
башенная 61, 63. 64. 132, 15S
денитрация 295
контактная 64, 94 ел., 158
682
Серная кислота 59 ел.
концентрирование 149 ел.
отработанная 67, 128 ел.
перевозка 159
применение 62, 128 ел., 158,
263, 274, 295 ел., 304, 367,
503, 504, 524 ел., 545, 566,
595, 598, 664
производство см. Серная
кислота, производство
свойства 59 ел.
сорта 61, 158
Серная кислота, производство 63, 64,
68
автоматизация 160 ел.
из отработанной кислоты 128 ел.
— сероводорода 124 ел.
— серы 122 ел.
контактный метод 93 ел.
«короткая» схема 122
мокрым катализом 124
нитрозный метод 130 ел.
расходные коэффициенты 94, 121,
128, 130, 142, 154, 157
совместно с азотной 304
сырье 64 ел.
техника безопасности 168
Сернистый газ 14
контактирование 111 ел.
концентрация SO2 72, 74, 78, 82,
101
концентрированный 79
металлургических печей 6G
очистка 88 ел., 97 ел.
получение 68 ел., 79, 86 ел.,
128 ел.
Серные руды 45 ел.
Серный ангидрид 121, 122
абсорбция 96, 117 ел., 165
получение 102 ел.
Сероводород 52, 67, 224
мокрый катализ 53, 124 ел., 166
применение 52 ел., 423
растворимость 194
сжигание 125
удаление из газов 52 ел., 177,
191, 192
Сесквикарбоиат 422
Сетки проволочные (сита) 24
Сжигание
сероводорода 125
серы 86 ел.
фосфора 509 ел.
Сжижение
воздуха 204 ел.
хлора 368 ел.
хлористого водорода 408
Сигнализаторы 162
Силикагель 664
Силикат-глыба 663
Силикатный кирпич 623
Силикатный модуль 636
Силикатообразование 649
Силикаты 598, 611 ел., 618, 635, 636,
643, 664
Силикофосфаты 550, 551
Сильвин 578
Сильвинит 578, 579
Синтез аммиака 234 ел.
автоматизация 254 ел.
двухцикличная схема 245
при высоком давлении 248 ел.
— среднем давлении 242 ел.
расходные коэффициенты 233, 234,
253
техника безопасности 257 ел.
Синтетические моющие средства 329
Сита, размеры 24
Ситаллы 667, 668
Ситовой анализ 24
Скоростью объемная 107, 178, 239
Скорость реакций
конверсии метана 173 ел.
обжига колчедана 73, 74
окислении двуокиси серы 105, 109
— окиси азота 271
синтеза аммиака 236
Скрубберы 16, 197
Слеживаемость удобрений 556,
560 ел., 564, 566, 568
Сложные удобрения 519, 565, 588 ел.,
593 ел.
Смеситель суперфосфатный 533
Смешанные удобрения 519, 596 ел.
Сода
двууглекислая см. Бикарбонат
натрия
кальцинированная см.
Кальцинированная сода
каустическая см. Едкий иатр
полуторная 422
природная 422
тяжелая 421
Содовые печи 463
автоматизация 473
Соковый пар 377
Соли жесткости 27
Соляная глазурь 619
Соляная кислота
перевозка и хранение 407
применение 395, 409, 410, 412, 549
производство 397, 403
свойства 395
синтетическая 406
сорта 406, 407
электролиз 350
Сольве способ производства соды 424
Сосуды Дьюара 222, 223
683
Спекание фосфатов 551 ел.
Спиральные холодильники 144
Средняя проба 23
Станция биологическая 43
Стеатит 614
Стекло 645 ел.
армированное 657
архитектурно-строительное 657
безопасное 657
выработка 654 ел.
гидратированное 663
жидкое 662
закаленное 657
зеркальное 657
кварцевое 665
листовое 654 ел.
обесцвечивание 648
окрашенное 648
оптическое 661
отжиг 654
полированное 656
порястое 657, 658
посудное 660
производство 649 ел., 654 ел.
растворимое 662 ел.
светотехническое 661
свойства 645, 646
сырье 646 ел.
тарное 660
триплекс 657
увиолевое 661
химико-лабораторное 661
хозяйственное 660
электровакуумное 660
Стекловаренные печи 650 ел.
Стекложелезобетон 657
Стеклокерамика 611, 667, 668
Стеклообразование 649
Стеклообразующие материалы 646 ел.
Стеклопластики 659, 660
Стеклянное волокно 658, 659
Стеклянные трубы 660
Стекольная масса, получение 649 ел.
Степень
извлечения 19
каустификации 477
обесфторивания фосфатов 553
обогащения 19
окисления SO2 103, 104, 109, ПО
пересыщения раствора 504
Сточные воды 30
очистка 12, 40 ел.
Строительная керамика 612, 623
Строительный гипс 631
Студка стекла 650
Сульфат аммония 231, 233, 566 ел.,
595
Сульфат-нитрат аммония 561
Сульфатно-шлаковый цемент 643
Сульфаты
калия 578, 586, 587, 594, 595
кальция 524, 578, 586, 587, 631,
634, 643
магния 578, 587, 598
марганца 599
натрия 647
Суперфосфат
аммонизированный 593 ел.
бородвойной 599
гранулированный 537 ел.
двойной 545 ел.
марганнзированный 599
молибденизированный 600
обогащенный 545
простой 524 ел.
Суперфосфорная кислота 511
Сурик 647
Сушильные башни 99 ел.
Сушка
аммиачной селитры 559
колчедана 70, 162
хлористого калия 585
Сухая очистка газов 53 ел., 177
Сухое грохочение 19
Сухой газгольдер 189
Съем продукции 15
Сырье минеральное 11, 17 ел.
испытания 23 ел.
коэффициент использования 13
Таллия карбид 629
Тальк 614
Танки для сжиженных газов 221, 222,
256, 371, 372
Тарельчатые колонны 211
Тарное стекло 660
Твердение вяжущих веществ 631 ел.,
634, 636, 642
Темкинр и Пыжева уравнение 236
Температура
зажигания 108
критическая 204
оптимальная ПО, 241, 269
самовоспламенения 258
Температура кипения
азотной кислоты 261, 294
олеума 61
растворов едкого натра 376, 389
серной кислоты 61, 150
серы 45
сжиженных газов 69, 121, 202, 225,
231, 326, 396, 409
Температура кристаллизации
азотной кислоты 261
едкого натра 389
сериой кислоты 59, 60
серы 44, 45
684
Тепловой баланс 13, 14
Теплоизоляционный бетон 644
Термическая фосфорная кислота
50 ел., 588
Тер]«онатрит 422
Терракота строительная 623
Тесто известковое 630
Тетракальцийфосфат 550
Техника безопасности в производстве
азотной кислоты 305
аммиака 257 ел.
аммиачной селитры 561, 562
карбида кальция 605, 606
мочевины 575, 576
серной кислоты 168
содопродуктов 488
фосфора и фосфорной кислоты 512
фосфорных удобрений 554
хлора и щелочей 373, 374
цианамида кальция 610
Титана карбид и нитрид 629
Топочные газы 67
Точильные круги 633
Травильные растворы 62, 67
Трепел 623
Трехокись серы см. Серный ангидрид
Трехфазные печи 494, 498, 501, 604
Триаммонийфосфат 588
Трикальцийфосфат 550
Тринзтрийфосфат 37
Триплекс
система выпарки 377
стекло 657
Триполифосфат натрия 46
Триэтаноламин 192, 193
Трона 422
Трубы
керамические 612
стеклянные 660
Туманомер'ы 161
Туннельные печи 617, 618, 666
Турбодетандеры 213, 218
Увиолевое стекло 661
Углеводороды
конверсия 172 ел., 179 ел., 182 ел.,
224
хлорирование 328, 329, 398
Углекислый газ см. Двуокись
углерода
Углеродистые огнеупоры 628
Угольные электроды 495, 511
Удобрения
азотные см. Азотные удобпения
влияние на урожай 514, 515
внесение 514. 515, 549., 550, 568,
577
дозы 515
Удобрения
гигроскопичность 520, 555, 564,
566, 568
гранулированные 519, 559, 561,
" 569, 573, 597
длительного действия 569
жидкие 231, 232, 257, 568, 573,
576, 577
калийные см. Калийные
удобрения
качество 519
классификация 517, 518
комплексные см. Комплексные
удобрения
микроудобрения 596, 598 ел., 668
минеральные см. Минеральные
удобрения
органические 514, 517
органо-1Минеральные 517
растворимость 518
слеживаемость 556, 560 ел., 564,
566, 568
сложные 519, 564 ел,, 568, 588 ел.
смешанные 519, 596 ел.
фосфорные см. Фосфорные
удобрения
Умягчение воды 33 ел.
Упаривание см. Выпаривание
Управление
автоматическое см. Автоматизация
дистанционное 16, 223, 384
Уравнение Темкина и Пыжева 236
Уровнемеры 161
Фарадея законы и число 309
Фарфор 612, 619, 620
Фаянс 619, 620
Феррофосфор 498 ел.
Ферритный способ производства
соды 484 ел.
Фибролит 633
Фиксация азота 233 ел.
Фильерное вытягивание стекла 659
Фильтрование
бикарбоната натрия 455 ел., 476
преципитата 549
сточных вод 41
фосфогипса 506
щелоков 380, 388
Фильтровая жидкость 457
Фильтрпрессные электролизеры
320 ел., 350
Фильтр (ы)
вакуумные 456, 505, 506
катионитовый 37, 38
электрические см. Электрофильтры
Флокулянты 31
Флотационный колчедан 65
685
Флотация 22, 647
апатито-нефелиновых руд 521
калийных солей 585 ел.
колчедана 65, 66
серных руд 46
фосфоритов 522
Флотореагенты 22, 46, 585
Формование керамических изделий
615
Формовочный гипс 631
Форстерит 628
Фосфаты
аммония 588 ел.
кальция 524, 525, 526, 539, 545,
549
магния 549
натрия 36, 39, 47
обесфторенные 550 ел.
плавление 551, 553
природные 520 ел.
разложение кислотами 503, 524 ел.
спекание 551 ел.
тонина помола 552
Фосфогипс 504, 509
Фосфор
возгонка 497 ел.
желтый см. Желтый фосфор
красный 490, 501, 502
окисление 509 ел.
применение 490, 491
пятиокись 514, 515, 519, 526 ел.,
537 ел., 545, 550, 553, 592 ел.
свойства 490, 501, 502
техника безопасности в
производстве 512
Фосфористый водород 602
Фосфоритная мука 523, 524
Фосфориты 520 ел.
Фосфорная кислота
выпаривание 507 ел.
нейтрализация аммиаком 588
применение 502, 545, 549, 550, 588,
594, 559, 599
производство 503 ел., 509 ел.
техника безопасности 512
термическая 509 ел., 588
экстракционная 503 ел., 588 ел.,
599
Фосфорные удобрения 517, 518,
520 ел.
воднорастворимые 518
гранулированные 537 ел.
двойной суперфосфат 545 ел.
концентрированные 545 ел., 550
лимоннорастворимые 518, 523
малорастворимые 518
мука фосфоритная 523, 524
обесфторенные 550 ел.
преципитат 549
Фосфорные удобрения
производство
схемы 530 ел., 537 ел., 540 ел.
546 ел., 551 ел.
сырье 520 ел.
техника безопасности 554
растворимость 517, 518
суперфосфат см. Суперфосфат
цитратнорастворимые 518, 523
Фотокерамика 618
Фотоэлектрические приборы 161
Фракционированная конденсация 225
Фраша метод выплавки серы А'й
Фреоны 329
Фтор
абсорбция 542 ел.
переработка 544 ел.
удаление из фосфатов 527, 530 ел.
Фторапатит 503, 520, 550
Фторирование бетона 542
Фтористый
водород 524 ел., 545, 550, 592
кальций 607
кремний 527, 542, 543
натрий 542
Хвосты флотационные 18, 66
Химико-лабораторное стекло 661
Химическая технология 11
Химические средства защиты
растений 329
Хлор
адсорбция 413
активный 412, 416 ел.
двуокись 408 ел.
жидкий 368
осушка 365 ел.
переработка 396 ел.
применение 12, 32, 328, 329, 413,
418, 419
производство см. Хлор,
производство
регенерация 325, 350
свойства 326, 327, 368
сжижение 368 ел.
хранение и перевозка 371 ел.
Хлор, производство 328
расходные коэффициенты 345
схема 365 ел.
сырье 331 ел.
техника безопасности 373, 374
химический метод 324
электрохимические методы 325,
326 ел., 351 ел.
Хлораты
магния 467
натрия 410
686
Хлориды
аммония 466
кальция см. Хлористый калий
кальция 371, 466, 607
натрия см. Поваренная соль и
Хлористый натрий
магния 466, 467
Хлорирование
воды 32
иззесткового молока 418, 419
окислительное 326, 397
углеводородов 328, 329, 398
Хлористый винил 328
Хлористый водород
жидкий 408
концентрирование 402, 405 ел.
окисление 326
применение 324, 328, 397
производство 397, 399 ел.
регенерация 325, 350
свойства 396
Хлопнстый калий 579 ел.
кристаллизация 583 ел.
месторождения 578
применение 565, 578, 579, 594
производство 580 ел., 585 ел.
сорта 586
Хлористый натрий
месторождения 332
отделение от КС1 579 ел.
щелока 380
пэнменение 14, 333 ел., 397,
423 ел., 439. 440, 466
вгетворимость 375
электролиз растворов 336 ел.,
351 ел.
Хлорная вода 326
Хлорная известь
получение 413 ел.
езойства 411, 412
сорта 416
упаковка и хранение 417
Хлорная металлургия 329
Хозяйственное стекло 660
Холодильники
аммиачные 214
ангидридные 123
газа дистилляции 448
графитовые 149
змеевиковые 147
оросительные 148
погружные 147
спиральные 149
хлорные 372
Холодильные циклы 204 ел.
Холодопроизводительность 205
Хромнтовые огнеупоры 628
Хромомагнезитовые oriievnopbi 553
Цемент 48, 50
ангидритовый 632
глиноземистый 643
портланд-цемент см.
Портландцемент
пуццолановый 642
роман-цемент 634
шлаковый 642, 643
Центрифуги автоматические 388
Центробежные
вентиляторы 146, 147
компрессоры 246, 247
насосы 146
пылеотделители 89
сепараторы 21
Цианамид кальция 233, 274, 606 ел.
Цианамидные печи 607
Цикл (процесс) замкнутый 15
Циклы холодильные 204 ел.
Циркониевые огнеупоры 629
Цитратнораствориыые удобрения 518,
523
Черепица 612, 623
Черепок керамический 612
Четырехокись азота 272, 300
Чешуйчатые
аммиачная селитра 559, 560
едкий натр 394
сера 57
Число Фарадея 309
Шамот 624, 625
Шахтные печи 433 ел.
Шенит 578
Шихта 11
Шлаки 667
Шлаковый
кирпич 623
цемент 642, 643
Шлакопортланд-цемент 643
Шла мы
марганцевый 599
селеновый 98, 101
солевой 581. 582
цементный 639
Шлифование стекла 660
Шнековый растворитель 582
Шпат
плавиковый 607
полевой 613, 614, 619, 620, 648
Щелок
выпаривание 374 ел.
осветление 481 ел, 487
687
Щелочестойкая керамика 621
Щелочная абсорбция 274, 275, 278
Эквивалент электрохимический 309
Экспансионные машины 197
Экстракционная фосфорная кислота
503 ел., 588 ел,. 599
Электрические печи 492 ел., 604,
608 ел.
Электровакуумное стекло 660
Электроды 308
биполярные 317, 321
графитовые 495
коронирующие 89
монополярные 317, 319
обжиг 493, 495
обработка 312
осадительные 90
пакетные 495
ртутные 351 ел.
стальные 336 ел.
угольные 495, 511
Электролиз 308
воды 171, 314 ел., 323
расплавов 307
растворов хлористого натрия
336 ел., 351 ел., 410
расход энергии 313 ел., 316
соляной кислоты 350
Электролизеры 308
БГК-Р-Ю1 357, 358
БГК-12 343
БГК-13 346
БГК-17 347 ел.
биполярные 317, 320 ел.
вертикальные 342 ел., 361 ел.
горизонтальные 341, 354 ел.
ДеНора 361, 362
Электролизеры 308
диафрагменные 340 ел.
монополярные 317 ел.
Р-30 354 ел.
с ртутным катодом 351, 354 ел.
Сольве 359, 360
фильтрпрессные 320 ел., 350
Хукера 345
цилиндрические 342 ел.
эксплуатация 349
Электромагнитное обогащение 2i
Электроосаждение 307
Электропроводность 308, 315, 350
Электротермические процессы 491,
497 ел.
Электрофильтр (ы) 89 ел., 127. 145
мокрые 99, 437
огарковый 90
Электрохимические процессы 33,
307 ел., 314 ел., 336 ел,
350 ел.
Электрохимический эквивалент :09
Элементный конденсатор 372
Элементы химические,
распространенность 17
Эмали 662
Эстрих-гипс 632
Этаноламины 56, 192 сл„ 574
Эффект Джоуля—Томсона 203
Яды катализаторные (контакичые)
177, 197, 238, 268
Ячеистый бетон 644
Ящичный пресс 615