/
















































































































Author: Полуэктов Н.С. Мищенко В.Т. Кононенко Л.И. Бельтюкова С.В.
Tags: аналитическая химия неорганическая химия химия
Year: 1978




Text
АКАДЕМИЯ НАУД СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ»
АНАЛИТИЧЕСКАЯ ХИМИЯ
СТРОНЦИЯ
Н. С. Полуэктов, В. Т. Мищенко, Л. И. Кононенко, С. В. Бельтюкова
В
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1978
УДК 543:546.4
Серия: «Аналитическая химия элементов'»
Главный редактор член-корреспондент АН СССР Ю. А. Золотов
Редакционная коллегия:
И. П. Алимарин, Ю. И. Беляев, А. И. Вусев, М. П. Волынец, А, II. Ермаков, В. М, Иванов, А. В. Карякин, Н. М, Кузьмин, С. В, Саввин, Н. М. Ростоцкая (ученый секретарь)
Редактор тома «Аналитическая химия стронция» доктор химических наук Ф. П. Горбенко
Адрес редколлегии: 117334. Москва, Воробьевское шоссе, 47а Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
П
20506-522
130-78
© Издательство «Наука», 1978 г.
055 (02)-78
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернад ского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов» — составит около 50 томов. Потребность в подобного рода издании назрела давно. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом возникло настоящее издание — серия «Аналитическая химия элементов», которое осуществляется впервые. Издание серии было начато по инициативе академика А. П. Виноградова, который с 1958 по 1975 г. был ее главным редактором.
Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы, как физические, так и физико-химические и химические, излагаются применительно к количественному определению данного химического элемента начиная с анализа сырья, далее типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
S
Монографии содержат обширную библиографию, доведенную до последних лет; они рассчитаны на широкий круг химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйств, а также на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, плутонию, бериллию, никелю, редкоземельным элементам и иттрию, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, нептунию и трансплутониевым элементам, селену и теллуру, алюминию, платиновым металлам, германию, родию, магнию, рению, золоту, кадмию, фосфору, марганцу, ртути, кальцию, рубидию и цезию, серебру, цинку, олову, кремнию, сере, литию, вольфраму, мышьяку, барию, азоту. Готовятся к печати монографии но аналитической химии углерода, хрома, меди, сурьмы.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Стронций и его соединения находят все возрастающее применение в различных областях деятельности человека. Они используются в металлургической, химической, керамической и стекольной промышленности, радиоэлектронике, пиротехнике и др. Радиоактивный изотоп 90Sr представляет опасность для живых организмов. Другой его изотоп — 87Sr используется при датировании образования горных пород [429].
Все это выдвигает задачу систематизации сведений о методах определения стронция и анализа его соединений. В литературе имеются обзорные статьи или разделы монографий, посвященные аналитической химии стронция или какому-либо методу его определения, например спектральному или пламеннофотометрическому [15, 56, 71, 95, 149, 152, 279, 327, 332, 453, 728, 932], однако приводимый в них материал в некоторой части устарел и нуждается в дополнении.
За последние два десятилетия аналитическая химия стронция развивалась во многих направлениях. Для открытия и определения стронция был предложен ряд новых окрашенных органических реагентов, применение которых позволило существенно повысить чувствительность и селективность реакций на стронций. В дополнение к старым методам отделения стронция от других элементов, основанным на осаждении, предложены новые. Экстракция органических соединений стронция, в частности р-дикетонатов и комплексов с азо-азоксисоединениями, позволяет производить быстрое его выделение и отделение от других щелочноземельных и редкоземельных элементов. Ионообменная хроматография с использованием как катионитов, так и анионитов нашла широкое применение в анализе для тех же целей. Распределительная и тонкослойная хроматография пригодны для отделения микрограммо-вых количеств стронция. Для этой же цели находит применение электрофорез.
Из физических и физико-химических методов количественного определения стронция широкое применение при анализе объектов окружающей среды и при геохимических исследованиях находит спектрографический (искровой и дуговой) метод. Эмиссионная фотометрия пламени является простым и надежным методом определения стронция в различных объектах как при малых, так
я
и при больших его содержаниях. В последнее время все более широко используется атомно-абсорбционная спектрофотометрия с использованием как пламени, так и беспламенных испарителей.
Для определения изотопного состава стронция находит применение эмиссионный спектральный и масс-спектрометрический методы. Недеструктивный анализ ряда объектов на содержание стронция может быть быстро выполнен с помощью активационного и рентгенофлуоресцентного методов. Они являются наиболее перспективными при анализе объектов на стронций. Однако не потеряли своего значения гравиметрический и титриметрический (комплексонометрический) методы определения стронция, которые используются при стандартизации исходных растворов солей стронция, а также в случаях определения больших его количеств. Все эти методы описаны в соответствующих разделах монографии.
В главах, посвященных определению стронция в различных объектах и анализу его соединений и препаратов на примеси посторонних элементов, приведены примеры анализа сложных объектов.
Важной областью анализа, имеющей значение для геохимии, атомной энергетики, биохимии, является радиоаналитика стронция. Этому вопросу посвящена специальная глава монографии.
Литература по аналитической химии стронция весьма обширна. В связи с ограниченным объемом монографии авторам пришлось отказаться от цитирования многочисленных источников, которые уже устарели или носят вспомогательный характер.
Авторы выражают искреннюю признательность доктору химических наук Ф. П. Горбенко, кандидатам химических наук Ф. И. Павлоцкой и Н. С. Фруминой за замечания и дружеские советы и глубокую благодарность Н. Н. Александровой, Л. П. Пономаренко и Е. И. Целик — за помощь при оформлении рукописи.
Авторы будут благодарны читателям монографии за все критические замечания.
Авторы
6
Глава I
ФИЗИКО-ХИМИЧЕСКАЯ И ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКИ СТРОНЦИЯ И ЕГО СОЕДИНЕНИЙ
ОБЩИН СВЕДЕНИЯ
Краткие исторические данные
Стронций — элемент второй группы периодической системы элементов Д. И. Менделеева с атомным номером 38. Вместе с кальцием, барием и радием он образует подгруппу щелочноземельных элементов.
Стронций был открыт в 1790 г. английским химиком А. Кроуфордом. Свое название элемент получил от г. Стронциана в Шотландии, вблизи которого был найден минерал стронцианит. В изучение химии стронция большой вклад внес русский ученый Т. Е. Ловиц.
Нахождение стронция в природе
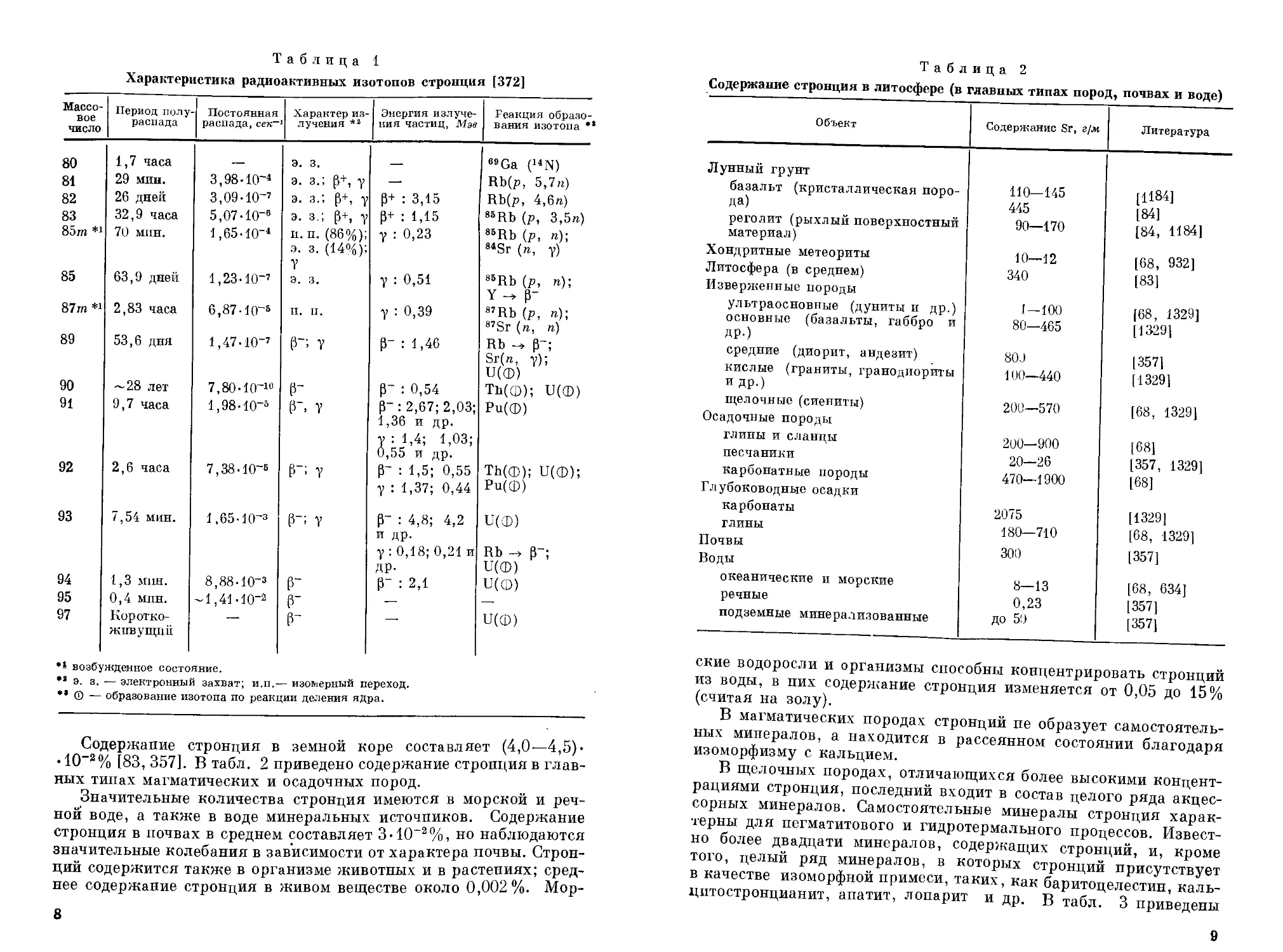
В природе стронций встречается в виде смеси четырех стабильных изотопов: 84Sr, 86Sr, 87Sr и 88Sr, содержание их в природной смеси соответственно составляет 0,56; 9,86; 7,02; 82,56%, причем содержание 87Sr колеблется, так как он образуется при распаде радиоактивного долгоживущего изотопа 87Rb [932]. Последний имеет период полураспада (6,2 + 0,3)-Ю10 лет и составляет 27% от общего содержания рубидия в земной коре.
Кроме стабильных изотопов получены искусственные радиоактивные изотопы стронция с массовыми числами от 80 до 97 (табл. 1) Из них следует отметить долгоживущие изотопы 89Sr и ®°Sr, образующиеся с большим выходом в ядерных реакциях деления урана, в частности при ядерных взрывах. Изотоп 90Sr относится к числу наиболее опасных радиоактивных изотопов. Наряду с большим периодом полураспада (~ 28 лет) он имеет свойство прочно удерживаться в организме, в основном в костях, и медленно выводится из него, являясь источником постоянного облучения костного мозга [210].
7
Таблица 1
Характеристика радиоактивных изотопов стронция [372]
Массовое число Период полураспада Постоянная распада,сект Характер излучения *2 Энергия излучения частиц, Мэв Реакция образования изотопа **
80 1,7 часа — Э. 3. — e9Ga (14N)
81 29 мин. 3,98-10-* э. з.; р+, у — Rb(p, 5,7«)
82 26 дней 3,09-10-’ э. з.’, р+, у р+ : 3,15 Rb(p, 4,6n.)
83 32,9 часа 5,07-10-* э. з.; р+, у Р+ : 1,15 86Rb (p, 3,5n)
85m *’ 70 мин. 1,65-Ю"1 и. п. (86%); э. з. (14%); У у : 0,23 85Rb (p, «); 8*Sr (ra, y)
85 63,9 дней 1,23-10-’ э. з. у : 0,51 85Rb (p, n); Y- P-
87m*1 2,83 часа 6,87 -Иг5 и. п. у : 0,39 8’Rb (p, «); 8’Sr (n, n)
89 53,6 дня 1,47-10-’ Р~; у р- : 1,46 Rb p-; Sr(ra, v); U(®)
90 ~28 лет 7,80-10-10 г р- : 0,54 Ть(ф); U(®)
91 9,7 часа 1,98-10-° Р". у Р" : 2,67; 2,03; 1,36 и др. У : 1,4; 1,03; 0,55 и др. Ри(ф)
92 2,6 часа 7,38-10-« Р~ у р- : 1,5; 0,55 у : 1,37; 0,44 ТЪ(ф); U(®); Ри(ф)
93 7,54 мин. 1,65-IO-3 г; т Р~ : 4,8; 4,2 и др. Y : 0,18; 0,21 и ДР- и(Ф) Rb р-; U((D)
94 1,3 мин. 8,88-10-з Г р- : 2,1 и(ф)
95 0,4 мпн. ~1,41-10-2 г .— —
97 Короткоживущий — г —• и(ф)
возбужденное состояние.
** э. з. — электронный захват; н.п.— изомерный переход.
*• © — образование изотопа по реакции деления ядра.
Содержание стронция в земной коре составляет (4,0—4,5) • • 10-г% [83, 357]. В табл. 2 приведено содержание стронция в главных типах магматических и осадочных пород.
Значительные количества стронция имеются в морской и речной воде, а также в воде минеральных источников. Содержание стронция в почвах в среднем составляет 3-10~2%, но наблюдаются значительные колебания в зависимости от характера почвы. Стронций содержится также в организме животных и в растениях; среднее содержание стронция в живом веществе около 0,002%. Мор-
8
Таблица 2
Содержание стронция в литосфере (в главных типах пород, почвах и воде)
Объект Содержание Sr, г/м Литература
Лунный грунт
базальт (кристаллическая поро- 110—145 [Ц84]
да) 445 [84]
реголит (рыхлый поверхностный материал) 90—170 [84, 1184]
Хондритные метеориты 10—12 [68, 932]
Литосфера (в среднем) Изверженные породы 340 [83]
ультраосновные (дуниты и др.) 1—100 [68, 1329]
основные (базальты, габбро и ДР-) 80—465 [1329]
средние (диорит, андезит) 809 [357]
кислые (граниты, гранодиориты и др.) 100—440 [1329]
щелочные (сиениты) Осадочные породы 200—570 [68, 1329]
глины и сланцы 200—900 [68]
песчаники 20—26 [357, 1329]
карбонатные породы Глубоководные осадки 470—1900 [68]
карбонаты 2075 [1329]
ГЛИНЫ 180—710 [68, 1329]
Почвы Воды 300 [357]
океанические и морские 8—13 [68, 634]
речные 0,23 [357]
подземные минерализованные до 59 [357]
ские водоросли и организмы способны концентрировать стронций из воды, в них содержание стронция изменяется от 0,05 до 15% (считая на золу).
В магматических породах стронций не образует самостоятельных минералов, а находится в рассеянном состоянии благодаря изоморфизму с кальцием.
В щелочных породах, отличающихся более высокими концентрациями стронция, последний входит в состав целого ряда акцессорных минералов. Самостоятельные минералы стронция характерны для пегматитового и гидротермального процессов. Известно более двадцати минералов, содержащих стронций, и, кроме того, целый ряд минералов, в которых стронций присутствует в качестве изоморфной примеси, таких, как баритоцелестин, каль-цитостронцианит, апатит, лопарит и др. В табл. 3 приведены
9
Таблица 3
Минералы стронция
Класс Название минерала Химический состав Содержание SrO, %
О кислы Пандаит (Ba, Sr)2(Nb, Та, Ti)2Oe-H2O 6,4
Фториды Ярлит NaF(SrF2)3(AlF3)3 35,6
Карбонаты Стронцианит SrCO3 70,2
Бербанкит Na2(Ce, Ba, Sr, Ca)4(CO3)6 19,42
Карбоцернаит (Na,Ca,P39,Sr,Ba) CO3 12,43
Анкилит (амбатоаринит) SrCe(CO3)2(OH)-H2O <22
Сульфаты Целестин SrSO4 56,42
Фосфаты Беловит (Sr,Ce,Na)10(PeO24)(OH)2 33,6
Стронций-апатит SreCa4(PeO24)(F, OH)2 46,06
Ферморит (Ca, Sr)5 [(P, As)O4]3(F, OH) 9,93
Беггильдит Na2Sr2Al2(PO4)F9 31,89
Палермоит (Li,Na)4SrAl9(PO4)8(OH)e 9,2
Гояцит SrAl3(PO4)2(OH)6-H2O <22,45
Сванбергит (тих-винит, соколовит) SrAl3(SO4)(PO4)(OH)5 24,43
Люсюнгит (Sr, Pb) Fe3(PO4)2(OH)5-H2O ~ 14
Ванадаты Дельриоит CaSrV2O6(OH)2-2H2O’ <25,0
Сантафеит Na2Mn2(Mn, Sr)6V6O26-8H2O 6,0
Бораты Витчит SrBe01o'2II20 <32,45
Кургантаит Стронциоджино- (Sr,Ca)2(B4O8) • H2O (Sr,Ca)2(B14O23) • 8H2O 37,48
рит (волковит) <20
Стронциогильгар- (Ca, Sr)2[B5O8(OH)2Cl] 20,25
ДИТ С тронциоборит [(Ca,Sr)O]4(MgO)2(B2O3)i2-9H2O 21,6
Силикаты Лампрофиллит Na4Sr2Fe2Ti4Si6O25F2 <14,6
Нордит Na3CeSrMn2Si6O18 7,4
Бревстерит (Sr, Ba, Ca)(AlSi3O8)2-5H2O 8,99
стронциевые минералы [68, 263]. Большинство из них встречается в природе крайне редко и пе представляет промышленного интереса. Промышленными являются только два минерала — целестин (SrSO4) и стронцианит (SrCO3).
Способы переработки минералов
Целестиновые и стронцианитовые руды обогащают, получаемые при этом концентраты содержат соответственно не менее 95% SrSO4 и около 85% SrCO3.
Основными промышленными способами переработки целестиновых концентратов являются карботермический и содовый [385].
10
Карботермический способ основан на переводе сульфата стронция в растворимый в воде сульфид путем нагревания с углем при 1100° С с последующим выщелачиванием его и выделением из раствора солей стронция [385].
Для получения гидроокиси стронция сульфид обрабатывают горячей водой, при этом происходит гидролиз сульфида. При охлаждении из раствора выпадает гидроокись, которую отделяют центрифугированием. Из оставшегося гидросульфидного раствора путем обменной реакции можно выделить различные соли стронция. Для выделения карбоната стронция через гидрс-сульфидный раствор пропускают углекислый газ
Sr (HS)2 + СО3 + Н2О -» SrCO3 + 2H2S.
Извлечение стронция составляет 80—85%.
Содовый способ вскрытия целестина [385] основан на взаимодействии сульфата стронпия и углекислого натрия по реакции
SrSO4 + Ne2CO3 — SrCOj л- Na2SO4.
Образующийся карбонат стронция отфильтровывают, промывают горячей водой до удаления сульфат-ионов и растворяют в HNO3. Из раствора осаждают гидроокиси железа и алюминия действием NH4OH. Раствор упаривают на 80%, выпавший азотнокислый стронций отделяют от маточного раствора. Извлечение стронция составляет около 80%.
Переработка стронцианитовых концентратов [385, 762] основана на переведении карбоната в гидроокись. Используется либо обжиг SrCOs при 1200° С с последующим выщелачиванием окиси стронция водой, либо разложение SrCOs перегретым (500— 600° С) паром. Применяют также обработку SrCOs соляной кислотой с последующим выделением гидроокиси щелочью. Стронцианит вскрывают и азотной кислотой, образующийся при этом азотнокислый стронций выделяют из раствора либо кристаллизацией, либо высаливанием избытком конц. HNO3.
Получение стронция
Существуют три группы методов получения металлического стронция: термическое разложение некоторых соединений стронция, электролиз и восстановление окиси или хлорида стронция.
Способы получения стронция разложением гидрида и нитрида стронция не нашли промышленного применения вследствие образования мелкодисперсного легковоспламеняющегося порошка.
Электролитический способ получения стронция также не нашел широкого применения в промышленности вследствие малого выхода по току и загрязненности стронция его соединениями.
Основным промышленным методом получения металлического стронция является термическое восстановление его окиси алюминием [298] с последующей дистилляцией полученного металла и конденсацией его на охлаждаемой поверхности.
11
Применение стронция
Основными областями применения стронция, его сплавов и соединений является пиротехника, радиоэлектроника, металлургическая, химическая и керамическая промышленности [68, 298].
Металлический стронций пока имеет ограниченное применение в технике. Его используют для раскисления меди и бронзы и как поглотитель газов в электровакуумной технике. Уменьшению хрупкости титана и его сплавов при высоких температурах способствует введение в них 0,1% стронция. Добавка стронция к магниевым сплавам придает им вязкость, достаточную для прокатки. Для закалки литых изделий и предохранения их от раковин применяют сплав Си—Sr. 0,2—0,6% Sr в комбинации с 0,2—2% Sn добавляют к свинцу, применяемому для аккумуляторных пластин, сплав Sr—Cd используют для гальванических элементов. Sr входит в состав некоторых сплавов с сильно пирофорными свойствами (например, Mg—Sr). Сплавы Sr с Mg, Pb, Al отличаются высокой твердостью, антифрикционными и другими ценными свойствами. Стронций применяют как катализатор при синтезе органических веществ.
Радиоактивный изотоп 89Sr используется для обнаружения повреждения телеграфных кабелей, 90Sr является источником P-излучения и применяется в атомных электрических батареях, характеризующихся постоянством напряжения и длительным сроком службы [210].
Более широко в технике используются соединения стронция. Так, стронцианит и карбонат Sr используются для очистки высокосортных сталей от серы и фосфора. В производстве чистой каустической соды целестин служит адсорбентом примесей Fe и Мп. Широко распространено применение соединений стронция в пиротехнике благодаря их свойству окрашивать пламя в интенсивный красный цвет. Нитрат, карбонат и оксалат стронция входят в составы, используемые для изготовления сигнальных и осветительных ракет, а также трассирующих и зажигательных пуль и снарядов.
, Углекислый стронций используют в производстве ферромагнитных и люминесцентных материалов, катализаторов, при очистке едкого натра, каустической соды, цинка, в производстве высококачественной стали. Соединения Sr используют для замены ядовитой окиси свинца в производстве стекол, эмалей, глазурей, стойких к атмосферным воздействиям. Окись стронция входит в состав оксидных покрытий катодов радиоламп, отличающихся высокими эмиссионными свойствами. Гидроокись стронция применяют для выделения сахара из патоки. Хромат Sr используют для грунтовки и изготовления художественных красок. Титанат Sr входит в состав керамических масс с полупроводниковыми свойствами, а также используется в ювелирном деле. Стронциевые соли жирных кислот, так называемые «стронциевые мыла», при
12
меняют при изготовлении специальных видов консистентных смазок. Галогениды стронция используют в холодильной промышленности, медицине и косметике.
Фторид стронция — перспективный материал для оптики и лазерной техники [435], в производстве полупроводниковых пленок, квантовых счетчиков и люминесцентных материалов, при хемосорбции осколочных элементов, производстве смазочных материалов. Из фторида стронция может быть получена оптическая керамика, отличающаяся большой механической прочностью и устойчивостью к резким перепадам температур. Сульфид стронция входит в состав люминофоров, характеризующихся длительным послесвечением.
ФИЗИЧЕСКИS И ХИМИЧЕСКИЕ СВОЙСТВА СТРОНЦИЯ
Стронций—мягкий серебристо-белый металл, ковкий и пластичный; он полиморфен. Установлено существование трех модификаций стронция: ос, |3, у. 'До 215° С устойчива кубическая гранецентрированная модификация (tx-Sr), между 215 и 605° — гексагональная модификация (|3-Sr), выше 605° — кубическая, объемноцентрированная модификация (y-Sr).
Ниже приведены основные физические константы стронция [1110].
Атомный вес 87,63
Атомный объем, см3/г-атом 34
Атомный радиус, А 2,15
Ионный радиус (по Гольдшмидту), А 1,27
Плотность (20 °C), г/см3 2,63
Электрохимический эквивалент, мг/кулон 0,45404
Удельное электросопротивление, ом-см-1 22,76-10-°
Термический коэффициент электросопротивления (0 —100 °C) 0,00240
Т.пл., °C 770
Т.кип., °C 1380
Теплота плавления, кал/г 25
Теплота испарения, кал/г 383
Удельная теплоемкость (20 °C), кал/г-град 0,176
Давление пара, мм рт.ст. 10-3 (462 °C),
Поверхностное натяжение, дин/см 1 (733 °C), 100 (1092 °C) 165
Модуль упругости, кГ/мм? 1600
Модуль сжатия, кГ/мм2 1220
Твердость по Бринеллю, кГ/ммг 13
Сечение захвата тепловых нейтронов, барн/атом 1,16
Энергия ионизации (ов): Sr0—*Sr+ 5,692
Sr+ Sr2+ 11,027
13
По химическим свойствам стронций сходен с кальцием и барием, занимая промежуточное положение между ними. В соединениях стронций двухвалентен. В ряду напряжений стронций находится среди наиболее электроотрицательных металлов, что обусловливает его большую химическую активность. Стронций является сильным восстановителем, его нормальный электродный потенциал равен —2,89 в. В соответствии с величиной нормального потенциала стронций легко вытесняет водород не только из разбавленных кислот, но и из воды, а также все тяжелые металлы из растворов их солей.
Металлический стронций быстро окисляется на воздухе, образуя на поверхности желтоватую пленку, в которой, наряду с нормальной окисью SrO, частично содержится перекись Sr02 и нитрид Sr2N2; при нагревании на воздухе воспламеняется, а порошкообразный стронций самовозгорается при комнатной температуре.
С неметаллами стронций соединяется весьма энергично с выделением значительного количества тепла. При повышенных температурах стронций взаимодействует с водородом (выше 200° С), с азотом (выше 400° С), с фосфором, серой и галогенами. Стронций почти не подвергается действию концентрированных растворов едких щелочей. Концентрированные кислоты (HNOS и H2SO4) на стронций действуют слабо; разбавленные — растворяют стронций с образованием соответствующих солей, последние бесцветны. Соли минеральных кислот с анионами С1~, Вг~, J~ в NO3 легкорастворимы, с анионами F“, SO|“, COg~ и РО4“ — труднорастворимы в воде.
СОЕДИНЕНИЯ СТРОНЦИЯ
Неорганические соединения стронция
Окись стронция представляет собой белое тугоплавкое вещество с температурой плавления 2430° С. Получают SrO в аморфном состоянии путем прокаливания карбоната стронция при 1100° С. Ее можно получить также в кристаллическом состоянии (кубические кристаллы, плотность 4,7 г/см3).
Гидроокись стронция Sr(0H)2 получается при взаимодействии аморфной окиси стронция с водой с выделением большого количества тепла. С избытком воды образует гидраты, наиболее богатым водой является октагидрат Sr(OH)2-8H2O. Моногидрат полностью обезвоживается при нагревании до 100° G, выше 400° С образуется SrO. Октагидрат гидроокиси стронция — бесцветные тетрагональные кристаллы, плотность 1,90 г/см3. Он растворяется в воде с отрицательным тепловым эффектом (—14,6 ккал/молъ), в то время как при растворении в воде безводной гидроокиси выделяется 11,6 ккал/молъ тепла.
14
Растворимость Sr(OH)2 в воде сильно возрастает с повышением температуры и при 100° С составляет 21,83 г в 100 г воды. Водный раствор гидроокиси поглощает СО2, при этом выпадает в осадок SrCOs.
Перекись стронция SrO2 образуется при окислении окиси стронция кислородом при высоком давлении. Гидрат перекиси стронция (SrO2-8H2O) получается при действии Н2О2 или Na2O2 на раствор гидроокиси стронция; при нагревании до 100 -130 °C гидрат обезвоживается. При 700 —800° С перекись стронция разлагается с выделением кислорода; в воде перекись растворяется очень мало (0,008% при 20° С), а разбавленные кислоты ее разлагают.
Галогениды стронция образуются при взаимодействии стронция с галогенами, а также при растворении SrO или SrCO3 в галогеноводородных кислотах. Среди галогенидов фторид занимает особое положение, поскольку он почти нерастворим в воде и в разбавленных кислотах.
Фторид стронция можно получать в виде белого осадка путем обработки гидроокиси или карбоната плавиковой кислотой или при осаждении ионов Sr2+ ионами F-. SrF2 — бесцветные кубические кристаллы, труднорастворимые в воде (0,1173 г!л при 17,4 °C). Растворяется в горячей соляной кислоте.
Хлорид, стронция кристаллизуется из раствора при температуре ниже 60° С в виде гексагидрата SrCl2-6H2O, представляющего собой гексагональные иглы, расплывающиеся на воздухе. При температуре выше 60° С кристаллизуется дигидрат в виде прямоугольных пластинок. Нагревание дигидрата выше 145° С (при давлении выше атмосферного) приводит к образованию моногидрата SrCl2-H2O. Последний при нагревании до 250° С полностью обезвоживается, причем наблюдается частичный гидролиз. В воде SrCl2 растворяется очень легко (43,5 г в 100 з воды при 0° С).
Бромид стронция — бесцветные ромбические кристаллы, хорошо растворяются в воде (50,6% при 20° С). Из водных растворов при температуре ниже 18,62° С кристаллизуется гексагидрат SrBr2*6H2O, а выше этой температуры — дигидрат SrBr2-2H2O. Бромид стронция растворим в этаноле.
Иодид стронция представляет собой бесцветные кристаллы. При температуре ниже 83,9° С из водных растворов кристаллизуется SrJ2-6H2O, при более высокой температуре — дигидрат SrJ2-2H2O. Иодид стронция растворим в этаноле.
Сульфид стронция — бесцветные кубические кристаллы. В воде подвергается гидролизу. Получается при восстановлении SrSO4 углеродом. Особо чистый SrS (для люминофорных составов) получают действием сероводорода на SrCOs при 900° С.
Гидрид стронция SrH2 — бесцветные кристаллы. При нагревании до 800° С разлагается, при нагревании в атмосфере азота превращается в желтый имид SrNH.
15
Нитрид стронция Sr3N2 — черные кристаллы, легко разлагаются водой
Sr3N2 + 6Н2О = 2NH3 + 3Sr (ОН)2.
Растворы металла стронция в жидком аммиаке имеют синий цвет, после отгонки из них аммиака получается аммиакат Sr(NH3)e медно-красного цвета с золотистым блеском. Последний в присутствии катализатора (например, платины) разлагается с образованием белого амида стронция Sr(NH2)2, который при нагревании в вакууме превращается в имид. При нагревании имида стронция в высоком вакууме получается красновато-коричневый кристаллический пернитрид Sr2N4, разлагаемый кислотами с выделением азота. С углеродом стронций образует карбид SrC2, который, как и СаС2, разлагает воду с выделением ацетилена.
Из кислородсодержащих солей стронция наибольшее значение имеют карбонат, нитрат, сульфат.
Карбонат стронция SrCO3 — бесцветные ромбические кристаллы. Получается при обработке сульфида стронция водным раствором соды или при осаждении карбонатом аммония из растворов стронциевых солей. Карбонат стронция очень трудно растворим в воде (1,1 мг в 100 г воды при 20° С). Его раствор вследствие частичного гидролиза имеет щелочную реакцию. Присутствие в воде углекислоты значительно повышает его растворимость вследствие образования бикарбоната. SrC03 растворим в растворах аммонийных солей, легко разлагается кислотами.
При температуре 929° С ромбический SrCO3 переходит в гексагональный, который плавится при 1497° С (при давлении С02 60 атм).
Сульфат стронция — бесцветные ромбические кристаллы. Является практически нерастворимой солью (растворимость 41,4 мг SrSO4 в 100 г воды при 18° С); с сульфатами щелочных металлов образует труднорастворимые двойные соли.
При нагревании выше 1152° С обычная ромбическая модификация сульфата стронция переходит в другую, вероятно, моноклинную. Сульфат стронция плавится при температуре 1580° С, предварительно теряя SO3.
Нитрат стронция — бесцветные кубические кристаллы. Его растворимость составляет 70,8 г в 100 г воды при 20° С. При низкой температуре из водных растворов Sr(NO3)2 кристаллизуется его моноклинный тетрагидрат Sr(NO3)2-4H2O, который при нагревании выше 100° С легко теряет воду. При дальнейшем повышении температуры отщепляется кислород, образуется нитрит и только при сильном прокаливании происходит переход последнего в окисел.
В абсолютном этаноле нитрат стронция растворяется плохо (1 : 8,5-103) и еще труднее в смеси равных объемов этанола и этилового эфира (1 : 6-104). Этим пользуются для отделения стронция от кальция.
16
Таблица 4
Произведение растворимости (ПР) некоторых соединении стронция [1248J
Соединение t, °C —1g ПР Соединение t, °C -IgHP
SrCrO4 25 4,65 Sr g(AsO4)2 20 17,79
SrCO3 25 9,96 SrSO4 25 6,24
SrC2O4 20 6,4 SrSeO4 25 4,8
SCgfPO^ 19—20 27,39 SrF2 25 8,61
Помимо указанных соединений, стронций образует со многими элементами ряд бинарных соединений, таких, как бориды, силициды, арсениды, сульфиды и др.
В табл. 4 приведены данные о произведении растворимости некоторых неорганических соединений стронция.
Органические соединения стронция
Ион стронция образует комплексы с большим числом органических соединений. Подробно изучено взаимодействие стронция с аминополикарбоновыми кислотами [134, 456]: этилендиаминтетра-уксусной [89, 411, 914, 956, 964], иминодиуксусной [773] и ее производными [18, 869], нитрилотриуксусной [503, 914, 1005], гексаметилендиаминтетрауксусной [122] и другими кислотами этого типа [608, 764, 948, 993, 1062, 1091, 1092].
Исследованы комплексные соединения стронция с 8-оксихи-нолином и его производными [389, 738, 876, 1074, 1078], с купфероном [1361] и его флуореновым аналогом [1095], а также с N-фенилбензоилгидроксиламином [640].
Установлено, что стронций образует комплексы с некоторыми |3-дикетонами, такими, как ацетилацетон [394], дибензоилметан [1338], 2-фуроилтрифторацетон [1027], 2-фуроил-2-теноилметан [1338], 2,2,6,6-тетраметил-3,5-гептандион [542, 1297] и их производным бнс-ацетилацетонэтилендиамином [19].
Изучены соединения стронция с органическими кислотами, а именно: уксусной [1002], лимонной [395, 407, 914, 1112], янтарной [66], глутаровой и триоксиглутаровой [66, 273], молочной [1341], адипиновой [66], яблочной и винной [66], пикриновой [559], гидроксамовой [504], малоновой и ее алкилзвмещенными [1102], стеариновой [667], глутаминовой и аспарагиновой [983] и рядом других [121, 422, 606].
С фосфорорганическими кислотами стронций также образует комплексные соединения, из которых подробно изучены комплексы с дигексиловым эфиром фенилсульфониламидофосфорной [468], 2,2'-диаминодиэтиловым эфиром-N, N, N', N'-тетраметилфосфо-новой [420], диэтил ентриамин-N, N, N', N', N''-пентаметилфосфо-
17
Таблица 5
Константы устойчивости комплексов стронция [1248]
Лиганд t, °C 1g кх
он- 25 0,85
N03 18 0,82
pof 20 4,18
р,о*- 25 5,4
sof 25 1,14
Кислоты
уксусная 25 0,44
щавелевая 18 2,54
глутаровая 25 0,6
аскорбиновая 25 -0,35
молочная 25 0,98
винная 25 1,59
лимонная 25 2,85
иминодиуксусная 20 2,23
нитрилотриуксусная 26,3 4,91
этилендиаминтетра уксусная 25,3 8,53
метилендифосфоновая 25 4,48
Тайрон 25 4,55
8-Оксихинолин 25 2,7
5,7-Дибром-8-оксихинолин 18 7,1
Дибензоилметан 30 6,40
новой [419], оксиалкилидендифосфоновой [215], оксиэтилендифосфоновой [183], нитрилотриметиленфосфоновой [123], ди-и-(1,1,3,3-тетраметилбутил)фенилфосфорной [1254], аденозинтри-фосфорной [903] и аденозин-5-фосфорной [904] кислотами, а также окисью метил-бпс-(диоксифосфинилметил)фосфина [182].
Соединения с органическими аминами, иминами, нитросоединениями описаны рядом авторов [355, 1015]; изучено комплексообразование с эфирами [870], в частности, с макроциклическими полиэфирами [958, 1116]. В работе [403] рассматривается взаимодействие стронция с мочевиной.
В литературе описан ряд соединений стронция с различными органическими лигандами, такими, как 2-окси-1-нафтальдегид [530], а-карбокси-р-метилтрополон [793], диацетилоксимсалицил-альгидразон и диацетилоксимсалицилоилгидразон [1343], N-ok-сид пиридина [1183], 1,8-нафтиридин [560], порфин [498].
Сольватация ионов стронция спиртами изучена авторами [132, 133]. Комплексообразование сахаров со стронцием описано в работе [507]. В] табл. 5 приведены константы устойчивости комплексов Sr с некоторыми неорганическими и органическими лигандами.
18
Глава II
КАЧЕСТВЕННОЕ ОБНАРУЖЕНИЕ СТРОНЦИЯ
Описанные в литературе качественные реакции на стронций — реакции осаждения, цветные, флуоресцентные и микрокристалло-скопические, а также наблюдения окрашивания пламени являются малоспецифичными и в большинстве случаев могут быть применены после отделения стронция от других щелочноземельных и многих нещелочноземельных металлов. Наиболее надежно обнаружение стронция в природных и промышленных объектах может быть выполнено с помощью спектроскопических методов.
РЕАКЦИИ ОСАЖДЕНИЯ
В реакциях образования нерастворимых соединений поведение иона Sr2+ сходно с поведением ионов других щелочноземельных элементов — Са2+ и Ва2+. Большинство из них входит в перечень реакций, составляющих основу качественного химического анализа Sr [11, 14, 410, 728, 1201]. Для группового выделения щелочноземельных элементов кроме классической (сероводородной) схемы полного анализа катионов [11] используют ряд вариантов (бессероводородной) кислотно-щелочной схемы, обзор которых приведен в работе [476]; при этом осаждение производят в виде карбонатов [11, 887, 1301], сульфатов [476, 887], фторидов [887, 976], оксалатов [1145]. Эти же соединения и ряд других используют для обнаружения одного стронция (табл. 6).
В ряде случаев обнаружение стронция производят в присутствии этилендиаминтетрауксусной кислоты (ЭДТА) как комплексообразующего агента [512, 712, 1089]. Предложен, например, метод, основанный на вытеснении щелочноземельных элементов из их комплексонатов ионами Со(П) с одновременным осаждением их ЗО^-ионами. Метод позволяет определять Sr (или Ba + Sr) в присутствии избытка Са и разделять Ва и Sr при их соотношении 1 : 200 [1089]. Для быстрого качественного обнаружения Sr в смеси с другими элементами связывают ионы многих тяжелых металлов (при pH 4,5) этилендиаминтетрауксусной кислотой, а ионы Ва2+ избирательно осаждают кроконатом калия.
19
Таблица 6
Обнаружение стронция при использовании реакций осаждения в водных растворах
Осадитель Образующееся соединение Чувствительность, мг/мл Мешающие элементы Литература
H2SO4 (растворимые сульфаты) SrSO4 0,04—0,1 Ba, Са, Pb(II) [11, 980, 1145]
(NH4)2C2O4 SrC2O4-H2O 0,1 Са, Ba, Pb(II), Ag, Hg(I), Bi, Cu(II), Cd [И, 14, 410]
К2СгО4 SrCrO4 1,2* Ba, Pb(II) и др. [410,619, 728, 887, 1314]
(NH4)2CO3 SrCO3 0,02 Ba, Са и др. [11]
Na2HPO4 SrHPO4 0,1 Ba, Са и др. [728]
Пикролоновая кислота Желтый осадок 0,05 Ca, Cu(II), Mn(II), Ba, Pb [1234]
2-Окси-4-нитро-халкон Желто-оранжевый осадок ~50 Mg, Ca, Ba [499]
3,5-Диметилпикриновая кислота Желтый осадок <100 Ba. Pb(II) и др. [1057]
* В присутствии этанола — 0,02 мг(мл.
К 5 каплям анализируемого раствора хлоридов щелочноземельных элементов добавляют 10 капель реагента А (30 г| ЭД ТА 140 .ил. 14 N NH40H + 200 мл лед. СН3С00Н) и 5 капель раствора кроконата калия (30 г/л). Выпадает желтый осадок, который отфильтровывают. К фильтрату добавляют 10 капель реагента А и 3 капли насыщенного раствора (NH4)2SO4, при кипячении выпадает осадок SrS04 [712].
Для растворения сульфата стронция в процессе разделения Pb(II), Са, Sr и Ва используют 5%-ный раствор ЭДТА [512].
Элементы осаждают в виде сульфатов из водно-этанольного раствора, затем CaS04 растворяют в воде, PbSO4 — в насыщенном растворе NaCl. К нерастворившемуся осадку прибавляют 1 мл раствора ЭДТА, 1 каплю раствора индикатора (смесь равных объемов этанольных растворов: 0,2%-ного метилового красного и 0,1%-ного метиленового голубого) и по каплям добавляют 0,05 А раствор NaOH до зеленого окрашивания. Нагревают в водяной бане 5 мин., центрифугируют и обнаруживают в растворе Sr2+ с помощью 2 А раствора H2SO4 или CdS04.
Для обнаружения следов Sr можно использовать индуцированное осаждение PbSO4 [1339].
В одну из двух пробирок, содержащих по 2 мл раствора PbS04 (500 г CH3COONH4 растворяют в минимальном количестве воды, взбалтывают
20
с 60 г PbSO4 в течение 12 час., фильтруют через бумажный фильтр и разбавляют водой до 1 л; перед использованием раствору дают постоять 2 недели), прибавляют 1 мл анализируемого раствора, в другую — 1 мл воды. В обе пробирки добавляют по 2 мл этанола. Появление осадка в анализируемой пробе на 30 сек. раньше, чем в холостой, указывает на присутствие Sr. Чувствительность реакции 0,2 мкг Sr, предельное разбавление 1 : 5-10е. Определению мешают ионы Ва2+, большие количества Са2+ и F".
Ионы стронция взаимодействуют с рядом органических соединений, например: 2-флуореновым аналогом купферона [1095]; 8-ок-сихинолин-5-сульфокислотой, которая образует со стронцием осадки двух типов — Sr(HA)2 (белый) и SrA (желтый) [1074]; р-се-микарбазон изатина (1%-ный раствор реагента в 35%-ном этаноле, 0,2%-ном по NaOH) вызывает образование желтого осадка соединения стронция, подобно Ва, Са и другим элементам [855]. В качестве реагентов на стронций изучались пиридин-2,3,4-трикарбоновая и пиридин-3,4-дикарбоновая кислоты [1070]. Помимо стронция, они осаждают Ag, Pb(II), Hg(I), Cu(II), Cd, Bi(III), Ca, Mn(II). В присутствии тартрата К—Na (в качестве маскирующего реактива) первый реагент осаждает только Са и Sr, а второй— только Са (pH 5). При определенных условиях винная кислота осаждает Sr [71]. Для определения Ва, Са и Sr при их совместном присутствии предложен [743] диоксим щавелевой кислоты (Са и Ва при разных значениях pH образуют осадки характерной кристаллической формы, a Sr при содержании > 0,75 мг/мл осаждается в виде аморфного осадка). Все эти реакции малочувствительны.
ЦВЕТНЫЕ РЕАКЦИИ
Ионы стронция взаимодействуют с рядом окрашенных органических реагентов с изменением окраски последних. Высокую чувствительность реакции на стронций проявляют бпс-азопроиз-водные хромотроповой кислоты, содержащие в о-положении к азогруппе сульфо- или фосфоновую группу, оксиазокрасители — производные фенолфталеина, красители трифенилметанового ряда ,. содержащие аминодикарбоновые группировки и некоторые другие (табл. 7). Специфические цветные реакции на стронций отсутствуют.
2,7-бис-Азопроизводные хромотроповой кислоты способны давать с ионами стронция три типа реакций [318, 320, 321, 362, 364, 365, 366, 368, 589, 591, 1384], в которых комплексообразование протекает с участием оксигруппы хромотроповой кислоты, азогруппы и солеобразующей группы (z) (I тип), двух оксигрупп хромотроповой кислоты (II тип) и оксиазогруппы с переходом второй части молекулы в хинонгидразонную форму (III тип). С одним и тем же реагентом в различных условиях возможно образование комплексов по двум и даже трем типам реакций (табл. 8).
Растворы комплексов, образующиеся по реакциям I типа, имеют два максимума поглощения. При реакциях III типа эти макси-
21
Таблица 7
Цветные реакции иона стронция
Реагент Окраска Чувствитель-ность, мкг/мл Литература
реагента | комплекса
2,7-бис-(2,2'-Дисульфо-фенилазо)хромотропо-вая кислота (ортанило-вый С, сульфоназо III) Фиолетовокрасная Синяя 0,01 (в 60%-ном ацетоне, PH 3,5) [129, 362, 365]
2,7-£мс-(2,2'-Дисульфо-4,4'-динитрофенилазо) хромотроповая кислота (нитроортаниловый С, нитрохромазо) Фиолетовая Сине-зеленая 0,02 (в 60%-ном ацетоне PH 4,3) [129, 212]
2,7-бис-(2,2'-Дисульфо-4,4'-диметил фенилазо) хромотроповая кислота (диметилсульфоназо III) Фиолетовокрасная Синяя 0,4 (в 35-40%-ном ацетоне) [591]
2,7-бкс-(2,2'-Фосфон- 4,4'-дихлорфенилазо) хромотроповая кислота (хлорофосфоназо III) Фиолетоворозовая Фиолетовосиняя пли сине-зеленая 0,1 (pH 2,5- 5,5) [247, 589]
2'-Оксинафталин-(1 '-азо-2)-нафталин-1-суль-фокислота Оранжевая Малиновая (суспензия) 0,5 [222]
3,3'-бис-(Хромотропа-зо) фенолфталеин Синяя Красная 0,05 (pH 11,3) [919]
3,3'-бис-(2-Окси-3-кар-боксинафтилазо)фенол-фталеин Сине-фиолетовая » 0,3 (pH 13,5— 14) [1319]
Металлофталепн (о-кре-золфталеинкомплексон, фталеинкомплексон, метилфталексон, фталеиновый пурпуровый) Бледно-розовая К расно-фиолетовая ОД [1141]
Эриохром черный Т Синяя Красная 0,05—1,0 [177, 896 , 1324]
Мурексид (пурпурат аммония) Сине-фиолетовая » 0,07 [1141, 1231]
Родизонат натрия Коричневая Оранжевая (буро-красная) 0,1 [427, 630, 631]
9,1О-Фенантренхинон-2-сульфокислота Темно-коричневая Зеленая 0,5 [1103]
Пирогаллол, а также пирогаллолкарбоновая и пирогаллолсульфо-кислоты Светло-коричневая Синяя или сине-фиолетовая 0,1 [396, 851, 1097]
22
Таблица 8
Реакции иона Sr2+ с 2,7-бис-азопроизводными хромотроповой кислоты
Тип реакции
Комплексообразующая группировка
Состав комплекса
(М : L)
pH раствора
Чпах, НЛ1
8.10-*
II
III
Z—Sr
О о-н
Sr/2—О О------► Н
6—7
8-10
3-5
600-610;
640—650
600—620
620-640;
690—710
0,6-0,8
0,4
1-1,4
I
мумы расположены при больших значениях длин волн. В случае же реакций II типа спектры имеют лишь один широкий максимум при 600—620 нм (для реагентов с НО2-группами до 660 нм). Добавка органических растворителей (этанола, ацетона, диоксана и др.) к водным растворам способствует увеличению чувствительности реакции. При этом увеличивается оптическая плотность в максимуме поглощения комплекса и уменьшается в максимуме поглощения реагента. Это наиболее характерно для реакций I типа. При реакциях III типа при переходе от водной среды к водноорганической может наблюдаться обратный эффект — оптическая плотность растворов комплекса в более длинноволновом максимуме зачастую уменьшается.
Некоторые из 2,7-бис-азозамещенных хромотроповой кислоты, используемые для фотометрического открытия и определения Sr, представлены в табл. 7. Чувствительность в водных растворах составляет 0,1—0,4 мкг Sr/мл, в 60 %-ном ацетоне — примерно на порядок выше. Наиболее часто из них применяются ортаниловый С, нитроортаниловый С и хлорофосфоназо III.
Прочность комплексов с ортаниловым и нитроор-таниловым С уменьшается в ряду Ba Д- Sr >• Ca Д- Mg, поэтому одно- и двукратные количества Са не мешают определению Sr. Не мешают также 100-кратные количества NH^, Li+, Na+, С2Н5ННз, 30-кратные К+, трехкратные Со2+, пятикратные Mg+2. Мешают Ва и РЬ. Присутствие в исследуемых растворах ионов Zr, Ti, Та и некоторых других металлов приводит к снижению интенсивности окраски комплексных соединений стронция [129].
23
В случае хлорфосфоназо III порядок изменения прочности комплексов другой: Са > Sr Ва [322]. Поэтому при pH 2,5—3,5 можно определять Sr в присутствии бария [223]. Применение комплексона III и сульфата натрия как маскирующих агентов позволяет определять стронций в присутствии многих катионов, в том числе кальция [247] или же бария [212].
Солеобразующую группировку
SO„H ОН
1 J-
N=N—
имеет также 2'-о ксинафтали н(1'-а з о-2)-н а ф т а л и н-1-сульфокислота (см. табл. 7) [222]. Аналогично стронцию с ним реагируют Be, Mg, Y, Со, Fe(II) и другие элементы. Заслуживают внимания и такие реагенты, как 2-(2'-т рихлорме-тилфенилаз о)х ромотроповая кислота, которая в присутствии Sr2+ и ионов других щелочноземельных металлов изменяет окраску от розовой до темно-красной (pH И) [705], а также 2-(2'-о ксиазобензо л)х ромотроповаякис-л о т а [421].
Для обнаружения стронция предложены реагенты, получаемые путем нитрования фенолфталеина и фенолсульфофталеина (или их производных), восстановления нитрогруппы до аминогруппы, диазотирования и последующего сочетания полученного бис-диазосоединения с хромотроповой кислотой, Р-кислотой или 2-окси-З-нафтойной кислотой. Из них наибольшей чувствительностью обладают производные фенолфталеина [919].
Комплексоны — красители трифенилметанового ряда взаимодействуют с ионами стронция с изменением окраски. Показано [525], что о-к резолфталеин ком плексон образует с двухвалентными металлами комплексы трех типов: MF4-, M2F2-и MHF3~ (F8-—анион реактива). При pH 9 Са, Sr и Ва образуют только комплексы MF4~ и M2F2- темно-красного цвета. Окраска комплексов Mg слабее, чем комплексов Са, Sr и Ва, а Be не реагирует совсем.
Обнаружение стронция капельным методом, с помощью о-крезолфталеин~ комплексона. Каплю анализируемого раствора наносят на фильтровальную бумагу, прибавляют 1 каплю 5 N NH4C1 и 1 каплю 0,2%-ного этанольного раствора о-крезолфталеинкомплексона и выдерживают пятно над конц. раствором NH40H. В присутствии Sr появляется красно-фиолетовая окраска. Открываемый минимум 0,1 мкг Sr; Mg, Са и Ва дают такую же реакцию
Кроме о-крезолфталеинкомплексона [453, 525] с ионами стронция взаимодействуют также тимолфталексон [546], т и-м о л ф т а л е к с о н S [216], метилтимоловый синий [120, 439] и ксиленоловый оранжевый (в присутствии четвертичных аммониевых оснований) [1295].
24
Отмечена также способность к цветным реакциям со стронцием таких реагентов трифенилметанового ряда, как хромазурол S [1312] и 2', 3', 6', 7'-тетраоксифлуоран и его производные [268].
Другие органические реагенты. Эриохром черный Т и ряд реагентов, имеющих в своем составе комплексообразующую группировку
ОН ОН ОН
V_Z
Омег а-х ром черно-синий G [487 ], эриохром серый 3BL — супра [921] в основном применяются как метал-лохромные индикаторы при комплексонометрическом определении стронция (pH 10—12,5). Как одноцветный индикатор используют и смесь кислого ализаринового SN с эриохром красным В [741]. Для качественного определения стронция на капельной пластинке используют омега-хром черно-синий G [487].
Мурексид взаимодействует со стронцием при pH 11,3. Мешающими элементами являются Mg, Са, Ва, Ag и ряд других металлов. Используется также производное мурексида — т е т-раметилмурексид [926].
Реакция стронция сродизоновой кислотой является одной из наиболее известных реакций на этот элемент, хотя ряд других металлов также взаимодействует с этой кислотой. Дают чувствительные реакции Ва2+ (оранжевая окраска), Ag+ (сине-черная), РЬа+ (коричнево-пурпурная), Fe3+ (черная окраска, переходящая в синюю), Ni2+ и Со2+ (оранжевая). Менее чувствительны реакции родизоновой кислоты с Zn2+ (оранжевая окраска), А13+, Сг3+, UO*+ и Th4+ (коричневая окраска, переходящая в желтую) и др. [1065].
Спектрофотометрическим методом изучена устойчивость растворов родизоновой кислоты и ее соединений со стронцием и другими металлами. Устойчивость кислоты максимальная при pH 6,6; добавка комплексона III, а также ацетона (до 20% по объему) стабилизирует ее раствор. В органических растворителях прочность соединений стронция возрастает [620].
Обнаружение стронция в присутствии бария. На фильтровальную бумагу, пропитанную 0,5%-ным раствором хромата калия, помещают каплю исследуемого раствора. Спустя минуту смачивают пятно каплей водного раствора родизоната натрия. Появление буро-красного пятна или кружка указывает на присутствие стронция [427].
Реакция стронция с родизонатом натрия используется для его обнаружения в сернокислых и углекислых минералах [279].
25
Аналогично родизоновой кислоте ведет себя по отношению к стронцию реагент сходного строения — тетраоксихинон [1251].
Пирогаллол [396] и его производные. Пирогаллол-карбоновая [1097], пирогаллолсульфо- и п и-р огаллолдисульфокислоты [8511 реагируют со щелочноземельными элементами в щелочной среде, образуя интенсивную синюю или сине-фиолетовую окраску. Разработан [851] метод обнаружения стронция и бария при взаимном присутствии: в щелочной среде пирогаллолдисульфокислота взаимодействует с обоими металлами, а в присутствии ЭО^-ионов — только с» стронцием. Чувствительность реакции 1 : 1 • 104.
Реакции свиолуровой кислотой [685, 706], т е т-рацианохинондиметанидом [6851, 2,6-д и хлор-фенолиндофенолом в присутствии нитрата серебра [527], алюминоном [11661, ализарином S [6111, кв ар цетином [13031 используются для обнаружения зон стронция после разделения методами бумажной или тонкослойной хроматографии (см. стр. 70, 72). В литературе имеются указания на то, что стронций дает чувствительную реакцию с 2,З-д и-оксихинолином в присутствии основного фуксина или метиленовой сини [1267].
Стронций в сикативах, лаках и маслах может быть обнаружен по реакции с э о з и н о м. [571]. Последний используют в виде 0,003 %-ного раствора в ксилоле. В присутствии стронция появляется красная окраска и осаждается эозинат. В качестве растворителей применяют бензол, керосин или четыреххлористый углерод.
Аналогичную реакцию дает большое число других элементов.
Реакции с использованием экстракции органическими растворителями
При экстракции хлороформом раствора соли стронция, содержащего 8-оксихинолин и 1-о ксиантрахинон, наблюдается ярко-оранжевая окраска экстракта при слабой желто-зеленой окраске раствора холостого опыта [130]. Аналогичную реакцию дают ионы кальция и магния.
Реакции с органическими электроотрицательными лигандами и родамином С. Стронций может быть обнаружен по окраске экстрактов ионных ассоциатов с красителем ксантенового ряда — родамином С и реагентами: 5,7-д и б р о м-8-о ксихинолином, фенилантраниловой кислотой или теноил-трифторацетоном. Оптимальные значения pH раствора 9—13 (для различных реагентов). Экстракция производится бензолом, толуолом или ксилолом. Соотношение компонентов в соединениях Me : реагент : родамин С = 1 : 3 : 1. Чувствительность 0,01—0,02 мкг 8т2+/мл. Аналогичную реакцию дают кальций и магний [34, 329].
26
Стронций сдиметилсульфоназо (ДАЛ) образует экстрагируемые бутанолом окрашенные в синий цвет комплексы, которые используются для его спектрофотометрического определения [590] (см. стр. 89).
Для открытия стронция в сложных смесях с различными катионами используют метод кольцевой бани. Стронций в зонах чаще всего идентифицируют с помощью родизоната натрия (630, 631, 756, 757, 1365].
ФЛУОРЕСЦЕНТНЫЕ РЕАКЦИИ
Для определения стронция с использованием флуоресцентной реакции в растворах наиболее часто применяется флуорексон (кальцеин) (табл. 9). Подробное описание условий применения, этого реагента для определения Sr и других щелочноземельных элементов приводится в монографии [431. Сам флуорексон в разбавленных слабокислых и нейтральных растворах проявляет сильную желто-зеленую флуоресценцию. В щелочной среде флуоресценция ослабевает и в 0,025 М растворе едкого кали совершенно исчезает, но возникает вновь в присутствии ионов Са2 + , Sr2+ и Ва2+. Катионы, мешающие определению стронция, могут быть маскированы цианидом калия (например, Cu2 + , Fe3+, Со2+, Ni2+) или триэтаноламином (Fe3+, Мп3+). Другие щелочноземельные элементы должны быть отделены. Флуорексон может содержать примесь флуоресцеина, которая увеличивает интенсивность флуоресценции контрольного опыта.
Примерно одинаковую чувствительность имеют реакции стронция с морином [774] и кур кумином — красящим началом корня куркумы [6581. Предельное разбавление при этом составляет 1:2 -104.
Голубой цвет люминесценции дают 2 реагента: 3,5'-бис-(р и-карбокси метиламинометил) -4,4'-д и о к с и - транс-стильбен [592] и 1,5-бмс-(д икарбоксиметил амино-мет и л)-2,6-д иоксинафталин [5931, образующие со стронцием и другими щелочноземельными элементами комплексы состава Me : L = 2 : 1. Чувствительность реакции во втором случае примерно в 20 раз выше, чем в первом. Из других реагентов, имеющих дикарбоксиметиламинометильную группировку, следует отметить замещенную в положении 1 2-о к с и-3-н афтой-ную кислоту, которая позволяет открыть 3 мкг&с/мл по сине-зеленой флуоресценции (pH 14) [10691.
На бумаге и других носителях обнаружение стронция производится при облучении ультрафиолетовым светом с помощью ряда веществ: морина [7531, виолуровой кислоты [12331, 4-о ксибензти а з ол а [7151, 8-о ксихинолина [43, 406, 604, 683, 742, 823, 1013, 1260] или смеси последнего с к о й-е в о й кислотой [543, 701], а также 8-о ксихинолин-5-с ульфокислоты [1074].
27
Таблица 9
Флуоресцентные реакции, используемые для открытия иона стронция
1 еагент Условия проведения реакции Чувствительность, мкг/мл Мешающие элементы Литература
Флуорексон (каль- цеин) — бнс-(дикарбок-симетиламинометил) флуоресцеин В 0,025 М растворе КОН (желто-зеленая флуоресценция) 9 Са, Ва, Hg(II), Pb(II), Fe(III), частично Mg и др. [938, 1369]
Морин (3, 5, 7, 2', 4'-лентаокси флаван) В пентаноле 50 Be и ряд тяжелых металлов [43, 753, 774]
3,5'-бнс-( Дикарбоксиметиламинометил)- 4,4'-диокси-огрдис-стил ьбен В воде, pH 10,9 (голубая флуоресценция) — РЗЭ, Mg, Са, Ва, Zn, Cd, Be, Al [592]
1,5-бнс-(Дикарбокси-метиламинометил)-2, 6-диоксинафталин В воде, pH 11,7 (голубая флуоресценция) 0,001 Al, Ba, Be, Ca, Mg [593]
Куркумин—бис-(4-окси-3-метоксициннамоил) метан Этанольно-водная щелочная среда (желто-зеленая флуоресценция) 50 [658, 774J
При наблюдении в УФ-свете можно также идентифицировать Sr2+ при использовании в качестве реагентов хлораниловой [528] иродизоновой [10361 кислот.
В ряде случаев флуоресцентные реакции используются для по-луколичественного [753] и количественного [938, 10131 определения стронция.
МИКРОКРИСТАЛЛОСКОПИЧЕСКИЕ РЕАКЦИИ
Для открытия стронция предложен ряд микрокристаллоско-пических реакций (табл. 10). Для обнаружения стронция также используют метаниловый желтый [724], 5-н и т р о-барбитуровую кислоту [724], нафтоловый желтый [397], |3-о ксинафтойный альдегид [124, 224], озазон диск сив инной кислоты [397].
ОКРАШИВАНИЕ ПЛАМЕНИ
Летучие соли стронция окрашивают бесцветное пламя горелки в характерный карминово-красный цвет. Для определения стронция в нелетучих солях (карбонатах, сульфатах и др.) последние замешивают в кашицу с конц. НС1 и в ушке платиновой проволоки вносят в пламя горелки [И].
28
Таблица 10
Микрокристаллоскопические реакции, предложенные для обнаружения стронция
Реагент Образующееся соединение Характеристика соединения ПpeдeJ обнаружения, мкг Мешающие] элементы Литература
HNO3 (6М) Sr(NO3)2 Октаэдры, шестиугольники и треугольники 0,08 Mg, Са, Ва, РЬ(П), Nb(V), Mo(VI), W(VI) [354]
H2SO4 SrSO4 Крестики (прямоугольники, шестиугольники—при перекристаллизации из большого количества горячей НС1) 0,2 Са, Ва, Pb [208]
К2СгО4 SrCrO4 Желтые иглы (пучкообразные или веникообразные кристаллы) 0,8 Са, Ва, Pb(II) [208, 1129]
KJO3 Sr(JO3)2- •6H2O Иглы с расширением посередине, иногда ромбические призмы и короткие бипирамиды 0,1 Са, Ва, Ag(I) [208, 561]
Ni(NO3)2 + -|- kno2 (pH 6,5) K2SrNi- •(NO,). Светло-желтые сильно преломляющие кубики 0,1 Ва, Са — в большом избытке [836]
Cu(NO3)2 4-4- KNO2 K2SrCu- •(NO3). Кубики, интенсивно окрашенные в сине-зеленый цвет 0,1 Са, Ва, РЬ(П), Ан, Pd, Se(iV), Te((lV),V(V), Mo(VI), W(VI) [354]
K3[Fe(CN)e] + -|- уротропин KSr[Fe- •(CN).b •2C.HJ2N4- •12H2O Октаэдры желтого цвета — Mo, Ca, Ba [208]
H2C2O4 или (NH4)2C2O4 SrC2O4 Октаэдры (крупного размера и мелкого — при образовании кристаллогидрата SrC2O4-3H2O, четырехлучевые розетки 0,8 Ca, Ba, Pb(II), Zn, Mn(II) [208J
Тартрат калия-натрия (KNaC4H4Oe) SrC4H4O. Призмы, шестиугольники, треугольники 0,4 K, Rb, Cs, Ca [208]
Виолуровая кислота Розовые иглы, часто в пучках 5 Cs, Rb, Li, T1(I), Ca, Ba, Pb(II), Cd, Zn, Co, UO22+ [970]
29
ДРУГИЕ МЕТОДЫ
Предложен [198] способ определения малых количеств сульфата стронция в сульфате бария, основанный на резком различии в растворимости этих солей. При их взаимодействии с катионитом СБС в Н+-форме (RH) выделившиеся по реакции 2RH + Sr2+ — = R2Sr + 2Н+ ионы водорода характеризуют наличие и количество стронция в смеси сульфатов.
Отмечена [749] возможность быстрого определения поглощенного на ионообменной смоле Sr2+, как и других катионов (Na+, К+, NH*, Са2+ и Ва2+), с помощью метода дифференциального термического анализа (термическая устойчивость смолы зависит от характера иона, поглощенного активными группами смолы).
Г лава III
МЕТОДЫ ОТДЕЛЕНИЯ СТРОНЦИЯ ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
Используются различные методы отделения стронция в зависимости от того, необходимо ли определение его больших количеств, например, в минералах или его соединениях, или малых •— при анализе горных пород, биологических материалов и др. Ряд методов предложен также для его отделения и определения в ходе качественного анализа смеси катионов.
Выделение стронция из различных материалов обычно включает следующие три этапа:
1. Отделение элементов III—V аналитических групп, осаждающихся в виде нерастворимых хлоридов, сульфидов, гидроокисей или других соединений.
2. Выделение группы щелочноземельных элементов.
3. Выделение стронция из смеси щелочноземельных элементов.
МЕТОДЫ ОСАЖДЕНИЯ
Методы, основанные на осаждении стронция
Эти методы используются главным образом для выделения смеси щелочноземельных элементов и для отделения стронция от других элементов второй аналитической группы (Са, Ва и Ra). Сводка важнейших методов представлена в табл. 11.
В ходе анализа часто необходимо, как уже отмечалось, отделение элементов других групп, кроме щелочных металлов.
Отделение стронция в виде нитрата
Отделение стронция азотной кислотой основано на образовании Sr(NO3)2, труднорастворимого в конц. HNO3 или органических растворителях. Метод применяется в основном для выделения стронция из различных объектов анализа и отделения его от кальция.
В табл. 12 приведены данные о растворимости нитратов Са, Sr, Ва, РЬ и Се в HNO3 различной концентрации. Она уменьшается
31
Таблица 11
Отделение стронция путем осаждения
Реагент Среда Состав осадка Соосажда-емые элементы Отделяемые элементы Литература
Азотная кислота (79-81%-ная) — Sr(NO3% Ba, Ra, Pb Ca |95, 210, 1370]
Карбонат аммония pH 8—9 SrCO3 Ca, Ba Щелочные металлы [77, 377]
Сульфат аммония 50%-ный этанол SrS04 Ca, Ba, Pb « [95, 513]
Оксалат аммония pH 3-7 (50%-ный этанол) SrC2O4-H2O Ca, Ba « [45, 46 , 728, 1087, 1216]
Хромат аммония pH 10 (50%-ный этанол) SrCrO4 Ba, Ra, Pb Ca [887]
Фосфат аммония pH 10 Sr3(PO4)2 Ba, Ca Щелочные металлк [233, 759, 862]
Родизонат калия pH 6,0 (водная или вод-но-ацетоно-вая) SrC6O6 Ba, Pb Ca [824, 854, 1357]
Таблица 12
Растворимость (г/л) азотнокислых солей стронция и некоторых других элементов [210]
Соль Концентрация HNOs, % Соль Концентрация HNOs, %
67 75 82 67 75 F2
Ca(NO3)2 Sr(NO3)2 Ba(NO3)2 25,2 0,056 0,007 8,56 0,009 0,001 1,75 0,003 0,0006 Pb(NO3)2 Ce(NO3)3 0,280 63,0 0,036 27,5 0,010 2,92
с увеличением концентрации кислоты, причем для щелочноземельных металлов и РЬ примерно в одинаковой степени.
Отделение Sr осаждением азотной кислотой описано в работе [95].
К 10 мл анализируемого раствора нитратов прибавляют из бюретки по каплям при механическом помешивании 26 мл 100%-ной (дымящей) HNO3 и дают постоять 30 мин. Затем фильтруют через пористый тигель, осадок переносят на фильтр с помощью 80%-ной HNO3 и промывают 10 раз той же кислотой порциями в 1 мл. Осаждение можно проводить при любой температуре в пределах от 20 до 70° С. Если присутствуют <^5лгг Sr, раствор надо перемешивать в течение 45 мин. В присутствии больших количеств Са необ
32
ходимо увеличить общий объем раствора. При содержании Са > 50 мг проводят переосаждение: осадок растворяют в горячей воде, подкисленной HNO3, и вновь его осаждают описанным выше способом [05].
Предложенный метод используют для отделения Sr от Са [24, 87, 258, 417, 461, 601, 617, 798, 1289], Mg, Be и 25 других элементов [1371], в том числе от редкоземельных элементов [416, 1360], Fe и др. Поскольку Ва практически полностью осаждается 76 %-ной, а РЬ — 84%-ной HNO3, то эти элементы, а также Ra соосаждаются вместе со Sr [95, 210, 755, 759, 848, 935, 991,1161, 1246, 1286, 1360, 1370]. Основную массу Sr можно отделить от Ва путем избирательного выщелачивания первого из осадков смеси их нитратов 5N HNO3 (в 3 стадии). Конечный продукт содержит 92% Sr и 8% Ва.
Следует заметить, что при больших соотношениях Са : Sr (100 и более) метод не обеспечивает полноты отделения от Са.
Стронций можно отделить от Са также путем выщелачивания сухих нитратов конц. HNO3 или органическими растворителями— пентанолом [210, 728, 1054], ацетоном [28, 69, 449, 1087], смесью этанола и эфира [95, 377, 602, 728 ] и др. При использовании HNO3 поступают следующим образом:
К 0,1—0,5 г сухих нитратов добавляют 5—10 мл (дымящей) HNOS с уд. массой 1,42—1,46 г!см3, перемешивают, после отстаивания сливают прозрачную жидкость через фильтрующий стеклянный тигель № 4 и промывают 3—4 раза той же кислотой порциями по 3—5 мл.
Из органических растворителей чаще всего используют спирты и ацетон. В табл. 13 приведена растворимость нитратов стронция и кальция в этих растворителях и бутилцеллозольве [95, 175].
Отделение Sr от Са с помощью ацетона. Растворы нитратов, содержащие до 50 мг суммы Sr и Са, выпаривают досуха, высушивают при 130—140° С в течение 60 мин., охлаждают 15—30 мин. с эксикаторе над Р2О5, добавляют 10 мл ацетона, экстрагируют 60 мин. при периодическом перемешивании, фильтруют через небольшой фильтр, смоченный ацетоном; остаток промывают 5—10 мл ацетона, растворяют в теплой воде, после добавления нескольких
Таблица 13
Растворимость нитратов Sr и Са в некоторых органических растворителях
Растворитель Растворимость при 25° С, % Растворитель Растворимость при 25° С, %
Sr(NO3), Ca(NO,)s Sr(NO,)s Ca(NO3)2
Этанол 0,02 52,0 Бутилцеллозольв 0,0009 3,6
Пропанол 0,02 36,5 (бутиловый эфир
Изобутанол 0,01 25,0 этиленгликоля) 37,0
Пентанол 0,003 13,3 Смесь этанола и эфира (1 :1) 0,00069
Ацетон 0,02 58,5
2 Н. С. Полуэктов я др.
33
капель HNO3 полученный раствор выпаривают для повторного экстрагирования. При четырехкратном повторении описанной операции метод позволяет отделить ~15 мг Sr от 500 мг Са с выходом 90% (содержание Са в выделенном Sr <[2%) [28]. Вместо фильтрования может быть применено центрифугирование с отсасыванием раствора нитрата кальция в ацетоне. При трехкратном проведении такой операции около 9,5% Са остается в осадке со Sr [69].
Отделение Sr от Са с помощью этанола и эфира [95]. При малых содержаниях стронция в смеси с кальцием (Sr ~10 мг, Са ~100 мг) к сухим нитратам в колбе приливают 10-кратное по массе количество абсолютного этанола, колбу закрывают, осторожно перемешивают и оставляют на 1—2 часа. Затем прибавляют равный объем абсолютного эфира и оставляют на 14— 16 час. Нерастворившийся нитрат Sr (и Ва) переносят на фильтр из тонкого' асбеста или мелкораздробленной платины и осторожно отсасывают. При использовании бумажного фильтра размеры его должны соответствовать величине осадка и скорости его промывания. Промывной жидкостью служит смесь этих же растворителей (1:1). Если нерастворившийся остаток оказался значительным, его промывают, высушивают и растворяют на фильтре в горячей воде; раствор выпаривают, высушивают остаток в колбе, как в первый раз, и повторяют обработку этанолом и этиловым эфиром.
Методы с применением пентанола [728, 1054] и изобутанола [728], а также ледяной уксусной кислоты [523], метилэтилкетона, содержащего уксусный ангидрид, уксусную и азотную кислоты [565, 980], не находят широкого применения.
При выделении малых количеств стронция (чаще всего его радиоактивных изотопов) в качестве соосадителя используют нитраты бария и неактивного стронция (глава VII).
Выделение стронция путем соосаждения с Ba(NO3)2 производят из 16V HNO3 при 55° С (чтобы избежать загрязнения осадка нитратами алюминия и железа) при перемешивании в течение 1 часа. При кристаллизации Sr отделяется от Са. Например, при соотношении Са : Sr в исходном растворе, равном 1 : 1 и 1 : 4, коэффициент очистки составляет 85 и ИЗ соответственно, перекристаллизацию полученного осадка производят из 16V HNO3 при 35° С. Из осадка после второй кристаллизации нитрат стронция избирательно выщелачивается в три стадии 57V HNO3, поскольку растворимость нитратов стронция и бария в 100 г 57V HNO3 составляет 12,45 и 0,17 г соответственно. Конечный продукт содержит 92% Sr(NO3)2, 8% Ba(NO3)2, примесей < 0,5%. Общий выход нитрата стронция 84% [1085].
Отделение стронция путем соосаждения с нитратом свинца используют при анализе горных пород и минералов. [392].
Навеску образца, содержащую 10—40 мкг Sr, разлагают смесью HF + НС104. После окончания разложения и удаления фтора (выпариванием с НС1О4 до получения влажных солей) остаток растворяют в 8 мл 10%-ной HNO3 и раствор переносят в кварцевый стакан емкостью 100 мл. Затем добавляют 2 мл раствора нитрата свинца (20 л«гРЬ2+/л«л) и при энергичном пере-
34
мешиванпи приливают 30 мл 99 %-ной HNO3. Через 30 мин. выпавший осадок Pb(NO3)2, содержащий Sr, отделяют от раствора центрифугированием в кварцевой пробирке с притертой) пробкой,) осадок растворяют в 5 мл 10%-ной HNO3 и снова осаждают Pb(NO3)2 добавлением 15 мл 99%-ной HNO3 и центрифугируют. Затем осадок растворяют при нагревании в 10 мл 2,5 N НС1, раствор охлаждают, выпавший азотнокислый свинец отделяют. Стронций остается при этом в растворе. Его дополнительная очистка производится на ионообменной колонке.
Отделение стронция в виде карбоната
Карбонатный метод широко применяется для группового выделения всех щелочноземельных элементов в ходе анализа, но не для их разделения [77, 461, 759, 935, 1054, 1202, 1303]. В отсутствие других щелочноземельных элементов карбонатный метод применяется для выделения одного стронция.
Наиболее благоприятной средой для выделения щелочноземельных элементов является раствор с pH 8 —9, так как при pH <) 8 уменьшается полнота их осаждения, а при pH 9 вместе с карбонатами щелочноземельных элементов осаждаются значительные количества магния. Необходимое pH раствора создают добавлением аммиака и аммонийно-аммиачного буферного раствора с требуемым значением pH. Чаще всего осаждение ведут карбонатом аммония. Нагревание раствора до 60—70° С ускоряет созревание осадков карбонатов и способствует превращению гидрокарбонатов в карбонаты [77].
Раствор (10 мл) нагревают до 50—70° С, прибавляют 20—30 капель раствора, содержащего по 150 г (NH4)2CO3 и NH4C1 в 500 2.V NH40H,
дают отстояться и фильтруют. Осадок карбонатов щелочноземельных элементов промывают раствором осадителя, разбавленным в 10 раз [377].
При использовании этилендиаминтетрауксусной кислоты в качестве конкурирующего лиганда возможно отделение Sr (в виде карбоната) от Са, поскольку последний образует с ЭДТА более прочные комплексы, чем Sr [45, 46, 12861. Разделение происходит при двукратном осаждении SrCO3 в присутствии раствора четырехзамещенного этилендиаминтетраацетата натрия [1286].
Рекомендуется [45, 46] предварительно определить количество кальция в пробе путем титрования его раствором комплексона III (динатриевой соли этилендиаминтетрауксусной кислоты) в присутствии мурексида. Затем берут вторую аликвотную часть той же пробы и проводят операции по отделению Sr (Sr + Ва) от Са. Если необходимо, в пробу вводят носитель Ва, упаривают до небольшого объема, добавляют рассчитанное по результатам титрования количество’комплексона III и подщелачивают. Образующиеся осадки гидроокисей; удаляют фильтрованием. Затем осаждают карбонаты стронция и бария 30 мл 1М раствора NasCO3 при’кипячении. После охлаждения осадок отфильтровывают, промывают водой (50 мл), в которую'предварительно добавляют 3—4 капли конц. NH40H.
2*
35
Осаждение карбоната стронция можно производить путем вытеснения его из этилендиаминтетрацетатного комплекса с помощью солей меди(П) в присутствии карбоната натрия.
Карбонат кальция используется для соосаждения малых количеств Sr [260] и его радиоактивных изотопов [297, 1188]. Теоретические аспекты метода рассмотрены в работах [50, 51, 913, 916]. Изучено [916] соосаждение стронция, меченного 89Sr, с осадком СаСОд различных модификаций, получаемых при различных условиях эксперимента (температуры осаждения, времени перемешивания, скорости добавления осадителя). При осаждении из растворов 0,17V СаС12 и 0,00017V SrCl2 раствором карбоната натрия (17И) получают (в зависимости от температуры) осадки СаСО3 в четырех модификациях: геля (< 10° С), фатерита (20—30° С), вальцита (40° С) и арагонита (50° С), причем осаждение протекает к меньшей степени с кальцитом и в большей степени с арагонитом. При превращении последнего в кальцит (после 70 час. перемешивания при 60° С) происходит почти полное выделение из осадка первоначально соосажденного Sr. Количество переходящего в раствор стронция приблизительно пропорционально образующемуся количеству кальцита.
Отделение стронция в виде сульфата
Сульфатный метод является классическим методом выделения суммы щелочноземельных элементов [95, 440, 476, 511, 512, 513, 526, 610, 825, 960, 980].
Анализируемый раствор может быть нейтральным или слабо подкисленным НС1 или HNO3 [95]. Прибавляют разбавленную (1 : 1) H2SO4 в 10-кратном избытке и4 объема этанола,[перемешивают, оставляют на 12 час., фильтруют и промывают 75%-ным этанолом.
Затем можно проводить дальнейшие операции по разделению щелочноземельных элементов.
Для переведения сульфатов в раствор их сплавляют с НагСО3, плав выщелачивают водой, осадок промывают и растворяют в НС1 или HNO3 [95]. В других случаях сульфаты переводят в карбонаты путем их кипячения с раствором Na2CO3 [887]. Применяется также выпаривание с NH4J и конц. НС! [960] или KJ иНС104 [980]. При этом сульфат-ион восстанавливается до суль-фид-иона, который удаляется в виде H2S. .
Вместе с Ва, Sr и Са в виде сульфатов выделяется РЬ [476, 511] и Ra. Эти металлы отделяются от Sr вместе с Ва путем осаждения в виде хромата; свинец может быть отделен от группы щелочноземельных элементов в виде сульфида [960], а также путем растворения его сульфата при нагревании в насыщенных растворах уксуснокислого аммония или хлористого натрия.
Из смеси сульфатов Ва, Sr, Са и Mg последний удаляют раство-рением^его сульфата в смеси этанол — вода.
S6
Осадок обрабатывается небольшим объемом воды при нагревании, прибавляют 5 капель конц. H2SO4 и 150—180 мл этанола, хорошо перемешивают и оставляют на ночь. Нерастворившиеся сульфаты щелочноземельных металлов отфильтровывают и промывают 50%-ным этанолом.
Сульфатный метод может быть применен для отделения Sr [728, 887, 1122, 1246, 1305] от Са или смеси Са и Mg. Используется то обстоятельство, что Са не образует осадка при действии на растворы его солей раствора (NH4)2SO4. Однако разделение смесей металлов, а также осаждение стронция является неполным [728]. Например, при осаждении Ва и Sr в присутствии Са в осадок выделяется 99% Ва, 60% Sr и 10% Са. Поэтому требуется повторное переосаждение осадков. Количественное отделение Sr от Са и частичное разделение Ва и Sr достигается при использовании диметилсульфата для осаждения Sr по методу возникающих реагентов [703, 727, 772].
В последнее время сульфатный метод разделения щелочноземельных элементов существенно улучшен благодаря применению в качестве маскирующего лиганда лактозы [416] и в особенности этилендиаминтетрауксусной кислоты [21—23, 28, 177, 412, 513, 539, 541, 712, 887, 1058, 1246, 1299, 1305, 1318]. Метод основан на конкуренции в образовании сульфатов и этилендиаминтетрацетатов щелочноземельных элементов при различной кислотности раствора.
Разделение Pb2+, Са2+, Sr2+ и Ва2+, заключающееся в их выделении из спиртово-водного раствора в виде сульфатов, растворении CaSO4 в воде, PbSO4 — в насыщенном растворе NaCl и SrSO4 — в 5%-ном растворе этилендиаминтетрацетата натрия, описано в [513].
К 2 мл анализируемого раствора прибавляют 3 мл этанола и по каплям 2 N H2SO4, нагревают 1 мин. в кипящей водяной бане, охлаждают и центрифугируют. Осадок обрабатывают 1 мл воды и центрифугируют. Нерастворившиеся сульфаты свинца, стронция и бария экстрагируют 1 мл насыщенного раствора NaCl, отделяя свинец. К осадку прибавляют 1 мл 5%-ного раствора комплексона III, 1 каплю раствора индикатора (смесь равных объемов спиртовых растворов метилового красного и метиленового голубого 0,2%- и 0,1%-ных соответственно) и по каплям — 0,05A\NaOH до зеленого окрашивания; затем нагревают на водяной бане 5 мин. Нерастворившийся сульфат бария отделяют путем центрифугирования. Разделение сопровождается качественным определением анализируемых элементов.
Разделение Sr и Са может быть достигнуто при pH 4—5 [21—23].
Смесь окислов стронция и кальция растворяют в HNO3 или НС1, прибавляют этилендиаминтетрацетат натрия (из расчета 800 мг на каждые 100 мг окислов), воду до объема 30—35 мл и доводят раствором NH40H до pH 4,5. Приливают 10 мл ацетатного буферного раствора с таким же pH (250 мл 98%-ной СН3СООН и 100 мл конц. NH40H на 1 л), 10 мл 10%-ного раствора (NH4)2SO4 и 50 мл 96%-ного этанола. Через сутки выпавший осадок отфиль
37
тровывают, промывают 5—6 раз 50%-ным этанолом с небольшим количеством (NH4)2SO4.
Соотношение компонентов Sr : Са может изменяться от 1 : 100 до 2 : 1 (абсолютные количества Sr от 20 до 1 мг). Относительная ошибка определения стронция не превышает 10%.
В ряде случаев разделение основано на вытеснении Sr из его комплексов с этилендиаминтетрауксусной кислотой ионами других металлов, например, Mg2+ [5411, Cu2+[1056J или Со2+[]1093, 1318]. Освободившиеся ионы Sr2+ осаждаются посредством S04~-ионов. С использованием Mg успешно отделяют Sr от Са при их весовом соотношении от 1 : 1 до 1 : 9. Сумму Ва и Sr отделяют от Са тем же способом при добавлении Co(NO3)2 в присутствии этанола [13181.
Предложено применять в качестве маскирующего агента диок-сиэтилглицин, комплексы которого со стронцием менее устойчивы, чем с кальцием. Поэтому при добавлении (NH4)2SO4 и этанола осаждается только Sr [1302].
Недостатком всех методик, связанных с применением комплексона при отделении Sr от других щелочноземельных элементов сульфатным методом, является необходимость предварительной оценки соотношения количеств этих элементов.
При выделении малых количеств стронция используется его соосаждение с сульфатами бария или свинца.
Теоретические основы метода отделения стронция соосажде-нием с сульфатом бария изучены в [595, 7721 и других работах. Чтобы вместе со Sr не соосаждался Са, последний связывают комплексоном III [9431.
При анализе образцов горных пород и минералов метод определения стронция с использованием выделения его путем соосаж-дения с сульфатом бария (пламеннофотометрическое окончание) был сопоставлен с масс-спектральным методом анализа. Удовлетворительное совпадение результатов свидетельствует о надежности операций по выделению стронция обсуждаемым методом [139].
Отделение стронция соосаждением с сульфатом с в и н-ц а применяется для выделения микроколичеств Sr и Ва перед их определением спектральным методом [187].
Отделение стронция в виде оксалата
Осаждение Sr в виде оксалата используется для его выделения совместно с Са и Ва в ходе анализа минералов. В присутствии больших количеств кальция стронций выделяется вместе с ним в достаточно полной степени. Однако при малых количествах кальция выделение стронция неполное и необходимо добавление этанола для понижения растворимости оксалата стронция. В присутствии посторонних солей, таких, как NH4C1 [3381, а также КС1, KNОз и CH3COONH4 [27], растворимость оксалата стронция по
38
вышается, что может привести к неполноте его выделения. Отделение Sr от больших количеств Fe, Ni, Со и Bi в присутствии ЭДТА описано в [859].
Выделение стронция из водного раствора в присутствии солей кальция. К 100—400 мл раствора, содержащего щелочные и щелочноземельные элементы, добавляют раствор NH40H дс pH 7, нагревают до кипения и осаждают оксалаты щелочноземельных элементов, добавляя горячий насыщенный раствор (NH4)2C204. Через 12 час. осадок отфильтровывают и промывают несколько раз холодным полунасыщенным раствором осадителя [1216].
Отделение стронция в присутствии этанола. К 250—260 мл нейтрального раствора, содержащего Sr и Са, прибавляют 50 мл 6,5%-ного раствора (NH4)2C2O4, 100 мл 95%-ного этанола и выдерживают 3 часа (Sr-|-Са
10 мг) или 18 час. (Sr + Са < 10 мг), фильтруют и промывают осадок оксалатов стронция; и кальция 0,25%-ным раствором (NH^C^ [1087].
Были предприняты попытки использовать различную растворимость оксалатов и сульфатов щелочноземельных элементов для их разделения. Использовались обработка смеси оксалатов сульфатом аммония (для переведения бария в BaS04) или кипячение со смесью сульфата и оксалата для переведения стронция в SrS04, причем кальций переводится в СаС2О4, растворяющийся в НС1. Эти методы не дают удовлетворительных результатов [522, 728.]
Вопросы теории соосаждения стронция с оксалатом кальция рассмотрены в работах [203, 538, 1016]. Изучена зависимость степени соосаждения стронция от концентрации осадителя, температуры, pH раствора, порядка прибавления реагентов и присутствия комплексона III. Установлено [538], что добавление последнего в раствор увеличивает степень соосаждения стронция, что обусловлено изменением условных значений произведений растворимости оксалатов кальция и стронция и замедлением осаждения СаС204.
Соосаждение производят из слабощелочной или нейтральной среды. Полученные оксалаты промывают и растворяют в НС1 или HNOз (или же сначала переводят оксалаты в окислы прокаливанием, а затем растворяют в НС1). Полученный раствор может быть использован для пламеннофотометрического определения Sr и Са. Сравнение с результатами масс-спектрального определения Sr в тех же образцах показало [139], что соосаждение с оксалатом кальция является надежной операцией по количественному выделению Sr.
[Отдетение стронция в виде хромата
Хроматный метод в основном используется для отделении Ва от Sr путем осаждения первого. Для отделения Sr от Са хроматный метод считается мало пригодным ввиду неполного осаждения Sr, хотя он и находит применение в качественном анализе. При этом для понижения растворимости SrCrO4 прибавляют этанол [887].
39
Отделение стронция от кальция. К центрифугату, полученному после отделения осадка ВаСгО4 из раствора окислов щелочноземельных элементов (10 .иг) в смесп 6М СН3СООН и ЗМ CH3COONH4, прибавляют раствор 6М NH4OH до щелочной реакции, 1 каплю 1М раствора К3СгО4 и равный объем этанола. Осадок SrCrO4 отделяют центрифугированием, ионы кальция остаются в растворе.
ОтделеннеДстронция в виде фосфата
Несмотря на некоторое различие в значениях pH, при которых происходит полное осаждение фосфатов Ва (9,2), Sr (10) и Са (>10) [233], разделить эти элементы при использовании солей фосфорной кислоты не удается. Фосфаты применяются для отделения группы щелочноземельных от щелочных элементов.
Осаждение фосфатов производят при pH 8—11. Осадок отфильтровывают через плотный]складчатый фильтр или отделяют центрифугированием, затем промывают раствором NH4OH (1 : 1) или водой [759, 862].
В зависимости от дальнейших операций по разделению щелочноземельных элементов осадок фосфатов может быть растворен в разб. НС1 или HNO3, в конц. СН3СООН или же действием конц. HN03 эти элементы могут быть переведены в нитраты (с последующим отделением Sr от Са нитратным методом [759, 9991).
Отделение стронция в виде родизоната
Осаждение стронция родизонатом калия или натрия при pH 5,8—6 используется для отделения Sr от Са. В работе [1357] изучены условия отделения Sr (50 мг) от больших количеств Са (11 г).
К 50 мл анализируемого раствора прибавляют 0,2%-ный свежеприготовленный раствор родизоната калия (соотношение молярных концентраций родизоната калия: Sr = 4 : 1). Осадок центрифугируют и промывают водой (45 мл). Выход стронция 80%, полнота отделения кальция 99%.
Метод разделения родизонатом используют для объемного определения Sr и Са в смеси (Sr ~ 5 мг, Са 0,5—5 мг) [824].
Рекомендуется производить разделение Sr и Са в присутствии N-оксиэтилэтилендиаминтриуксусной кислоты, которая связывает Са в растворимую комплексную соль [804].
В нейтральный раствор, содержащий до 80 мг суммы Са и Sr в виде хлоридов или] нитратов, добавляют раствор N-оксиэтилэтилендиаминтри-уксусной кислоты (24 мл 52%-ного раствора ее тринатриевой соли разбавляют водой до 80 мл, вводят НС1 (1 : 1) до pH 5,8 и разбавляют раствор водой до 100 мл) из расчета 1,3—2,5 моля на 1 г-иоч кальция, нагревают до кипения, приливают этанольный раствор родизоновой кислоты (2 мг!мл) до сильного помутнения, прекращают нагревание и снова добавляют раствор осадителя до появления желтой окраски раствора. Затем прибавляют этанол до конечной концентрации его 75%, охлаждают до комнатной температуры, осадок отфильтровывают и промывают этанолом.
40
При соотношении Sr : Са в исходном растворе 1 : 2 в фильтрате остается ~ 0,2% Sr, однако при соотношении Sr : Са = 1 : : 10 в фильтрате остается 4% Sr, а в осадок переходит 2% Са.
При определении стронция в костях окончательное его выделение производят смесью 0,2%-ного водного раствора родизоната натрия с ацетоном (4 : 1) при pH 8 [854].
Отделение стронция соосажденнем j Гс гидроокисями металлов]
Соосаждение стронция с гидроокисью железа для целей отделения стронция описано в довольно значительном числе работ [135, 266, 267, 285-288, 1227].
Изучено [266, 267] поведение малых количеств стронция при соосаждении с гидроокисью железа в зависимости от pH, концентрации NH4NO3, NH4OH и Sr2+, продолжительности соприкосновения раствора с осадком, количества носителя, температуры и т. д. Установлено, что соосаждение стронция при его концентрации 5,4-10-5 г-экв!л начинается при pH ~ 7,2 и достигает 96% при pH 9,5; при количествах соосадившегося Sr до 83% наблюдается прямолинейная зависимость степени соосаждения от pH раствора (рис. 1). При уменьшении концентрации Sr в растворе степень его соосаждения возрастает. Присутствие NH4NO3 в растворе в концентрации 0,05 М понижает степень соосаждения Sr тем больше, чем выше концентрация NH4NO3. Полнота соосаждения зависит также от порядка смешения растворов. Наименьшая величина соосаждения наблюдается при добавлении раствора Sr(NO3)2 к раствору, содержащему уже осажденную гидроокись железа, наибольшая — при добавлении Fe(NO3)3 к раствору, содержащему Sr(NO3)2 и избыток NH4OH. С повышением температуры степень соосаждения уменьшается и возрастает при увеличении продолжительности соприкосновения осадка с раствором, концентрации соосадителя (NH40H) и скорости его добавления.
При соосаждении стронция с заранее приготовленной гидроокисью железа с повышением температуры степень соосаждения увеличивается. Установлено, что в области насыщения осадка стронцием образуется соединение типа ферритов с соотношением Sr2+: Fe(OH)3 = 1 : 2 (pH 12) [135].
При практическом использовании выделения или отделения стронция от других элементов с помощью гидроокиси железа рекомендуется [285—288] для создания необходимого pH раствора (12—13) вместо NH4OH применять КОН. Контроль pH осуществляют рН-метром.
К кислому раствору пробы (0,1М по HNO3 или НС1) добавляют растворы нитрата или хлорида железа (5 мг железа на 20 мл конечного объема раствора) и нитрата или хлорида аммония до концентрации 1М в конечном объеме. Затем при энергичном перемешивании путем добавления NaOH выделяют Sr с гидроокисью железа при pH > 12, осадок после центрифугирования
4t
растворяют в 5 мл 0,iM HNO3 (или НС1); осаждают гидроокись железа при pH 3—4,
Рис. 1. Соосаждеиие стронция с гидроокисями титана, железа, алюминия и бериллия (Sr 100 мкг; NH4OH 0,5 М; объем 20 мл; время 30 мин.; температура 18° С)
1,2 — 10 мг ТЮ, в 0,4 и 1 М
NH4NO3 соответственио;
3 — 10 ли Ре2ОзЗ в 0,05 М NH.NO>;
4,5 — 6,4 мг Л12О> в 0,05 .и 1 М NH4NO3 соответственно;
6— 3,2 мг ВеО в 0,05 М NH4N0s
из полученного раствора вновь а стронций остается в растворе.
Аналогичным образом производится очистка 90Sr от радиоактивного изотопа}рубидия до желаемой степени чистоты [288].
Соосаждеиие «стронция с гидроокисью титана. На рис. 1 показано влияние pH на соосаждеиие Sr с гидроокисью титана при двух концентрациях NH4NO3 (кривые 1 и 2). Из рисунка следует, что’с’гидроокисью титана стронций соосаждается при более низких значениях pH и’более полно, чем с гидроокисью железа [266, 267]. При этом выделяется осадок состава SrTiO3-Гидроокись титана (TiO2-H2O) получают гидролизом TiCl4, эфиров титановой кислоты или K2TiO(C2O4)2 [558].
Соосаждеиие стронция с гидратированной двуокисью марганца применяется для выделения радиоизотопа 90Sr [136, 1240]. Поскольку гидрат двуокиси марганца МнО2>Н2О проявляет свойства слабой кислоты, a Sr(OH)2 — явно выраженное основание, то в щелочной среде они образуют химическое соединение с соотношением Sr : Мп = 1:2 [135] (состава SrMn2O5-nH2O, где п = 3,3ч-3,5) [356]. Выделение микроколичеств e0Sr в присутствии микроколичеств Ва и Са показало, что Ва сильнее Са подавляет сорбцию Sr и что логарифм коэффициента распределения последнего линейно зависит от отношения общего количества его аналога к сорбенту [136].
Применение в качестве соосадителей Sr гидроокиси алюминия и бериллия оказалось’неэффективным (см. рис. 1) [266, 267, 285, 286].
Методы, основанные на осаждении посторонних элементов
Элементы III—V аналитических групп отделяют от стронция и других щелочноземельных элементов обычными методами — осаждением сероводородом, сульфидом аммония или аммиаком.
42
Таблица 14
Методы отделения стронция от других элементов, основанные на осаждении посторонних элементов
Реагент Среда Отделяемые элементы Литература
Соляная кислота Бутанол или этанол Ва (728, 893,
1289]
Сульфат аммония Этиловый эфир Ва [935]
ЭДТА] (pH 6,4-6,8 Ва [21-23,
или — 8) 1299, 1318]
Хромат аммония pH 4-5,5 Ва, Ва, РЬ [728, 1122]
Фосфат аммония ЭДТА (pH 4,5-10,0) Ва [523, 717]
pH 6,9—7,0 Al, Сг, Fe, [944]
Zr, Nb
Ферроцианид калия ЭДТА' (pH 5,0) uo*+ [1315]
Кремнефтористоводо- pH 9,0 Ca ] 99]
родная кислота —- Ba [728]
При этом оказывают мешающее действие сульфат-, фосфат- и арсенат-ионы, с которыми стронций выделяется в осадок, хотя в отдельных случаях эти анионы могут быть использованы для отделения от стронция посторонних элементов (табл. 14).
Отгеление{бария в виге хлорида и сульфата
Барий от стронция может быть отделен осаждением в виде хлорида смесью HG1 и этанола.
Смесь нитратов бария и стронция, содержащую 15 мг Ва и 30 мг Sr, растворяют в 4—5 мл воды, прибавляют 25 мл смеси НС1 и этилового эфира (5:1), перемешивают при охлаждении 2 мин. и центрифугируют. Раствор сливают, растворяют осадок в 4—5 мл воды и повторяют операцию по отделению Sr от В а [935].
Для отделения Ва может быть использовано также осаждение его сульфатом в присутствии комплексона III.
Опыты по выбору оптимальных условий разделения бария и стронция показали, что верхняя граница pH количественного осаждения BaSO4 и SrSO4 в присутствии комплексона III смещается для обоих элементов в зависимости от их концентрации и концентрации осадителя (NH4)2S04 в растворе. Чем больше концентрация осадителя и металлов, тем при большем pH наблюдается, осаждение элементов. При суммарной концентрации металлов ~ 1 мг!мл их разделение возможно путем осаждения бария при
4г
pH 6,4—6,8, если концентрация осадителя в растворе равна 5 мг/мл [21—23].
Раствор, содержащий не более 50 мг Ва и Sr, помещают в стакан емкостью 100 мл и добавляют комплексон III в количестве, достаточном для связывания обоих металлов. Раствор подщелачивают разбавленным аммиа-ком'по феноловому красному до появления розовой окраски, избегая избытка аммиака, и прибавляют 5 мл 10%-ного (NH4)2SO4. Общий объем раствора доводят до 100 мл, после чего нагревают раствор на водяной бане. При этом pH раствора снижается и Ва осаждается в виде сульфата. Нагревание при 80—90° С продолжают до тех пор, пока раствор над осадком не станет прозрачным (цветиндикатора при этом переходит в лимонно-желтый). В зависимости от концентрации Ва в растворе и от условий нагревания равновесие устанавливается за 1,5—3 часа. Во время нагревания в стакан добавляют воду до первоначального объема. Раствор с осадком оставляют на ночь, после чего сульфат бария отфильтровывают через плотный фильтр и промывают 5—6 раз водой, содержащей небольшое количество (NH4)2SO4. При необходимости переосаждения BaSO4 его растворяют в аммиачном растворе комплексона III, затем осаждение производят тем же способом.
Отделение Ва от Sr может быть достигнуто также путем переведения этих элементов в комплексы с этилендиаминтетрауксусной кислотой с последующим применением Со2+ в качестве вытесняющего катиона. Хорошие результаты получаются, когда Ва значительно меньше Sr, например при соотношении Ва : Sr = 1 : 200 [1093].
Отделение бария в виде хромата
Метод осаждения бария хроматом был описан еще в 1871 г. [728]. G тех пор он широко применяется для разделения бария и стронция при проведении как качественного, так и количественного анализов [240, 261, 302, 306, 377, 404, 440, 447, 449, 594, 601, 623, 681, 684, 717, 771, 792, 802, 846, 979, 980, 1054, 1094, 1122, 1150, 1182, 1289, 1370]. При этом вместе с Ва могут выделяться другие элементы, образующие с хромат-ионами труднорастворимые осадки,—Ran Pb [950, 1246], a Sr в растворе может оставаться вместе с Са; для их разделения требуется применение других методов.
При выделении Ва описываемым методом [261] из смеси со Sr последний частично соосаждается при низких значениях pH и может осаждаться полностью при pH ^>10 (рис. 2). Поэтому осаждение Ва рекомендуется проводить при pH 4—4,5, причем, как правило, производится переосаждение хромата бария.
Для осаждения бария берут такое количество хромат-ионов, чтобы их отношение к Ва было 1,2 : 1 [594]. Очень часто нужное значение pH создают с помощью буферной смеси уксусная кислота—ацетат аммония [95, 240, 306, 447, 449, 684, 728, 979, 980, 991, 1122, 1182]. Иногда вместо СН3СООН применяют HNO3 [681].
44
Рис. 2. Осаждение BaCrO4 (1) и соосаждение SrCrO4c ВаСгО4 (2) в зависимости от pH раствора (2,46 .10“4 Л/ Ва2+; 2,51.
•1О-*ЛГ Sr2+)
Для уменьшения соосаждаемости SrCrO4 с ВаСгО4 в рабочий раствор добавляют ряд веществ. Так, в работе [261] было показано, что в присутствии 0,01 % желатины степень соосаждения Sr с Ва снижается примерно наполовину. Благоприятное влияние оказывает и добавление в раствор мочевины [771, 1087], обеспечивающей получение легко фильтруемого крупнокристаллического осадка; за счет этого соосаждение стронция доведено до минимума. Разделение Ва и Sr в этом случае производят следующим образом [771].
К 250 мл раствора, содержащего 0,1 г Ва2+, добавляют 10 г мочевины и 6 г CH3COONH4, разбавляют до 150 мл и, добавляя НС1 или NH4OH, устанавливают pH 1,7—1,8. Затем прибавляют 20 мл 10%-ного раствора K2Cr20i, раствор нагревают при’95—98° С 2—5 час., поддерживая постоянный объем жидкости. Время обработки можно сократить, так как выделение осадка ВаСгО4 обычно заканчивается в течение 1 часа. Раствор с осадко охлаждают; значение pH раствора должно составлять 5,7. Нагретый раствор отфильтровывают, осадок промывают 0,5%-ным раствором К2Сг2О7 (pH 5,7) и водой. Найдено, что ~ 100 мг Ва и ~40 мг Sr разделяются при однократном осаждении ВаСгО4, а при равных количествах этих металлов требуется пере-осаждение осадка [771].
В работе [523] подробно изучено поведение хроматов бария и стронция в присутствии избытка этилендиаминтетрауксусной кислоты и установлено, что количественное разделение достигается при pH 4,5—5,2. Добавление ЭДТА в раствор рекомендуется и в других работах [717, 1316].
Описан [717] метод отделения Ва от относительно больших количеств Sr (и РЬ). Ионы бария постепенно вытесняются из его комплекса с ЭДТА при медленном добавлении раствора MgCl2, вследствие чего образующийся осадок ВаСгО4 состоит из крупных кристаллов, легко фильтруется и не содержит Sr (и других элементов). С помощью радиоизотопов 140Ва и 90Sr показано, что при разделении равных количеств Ва и Sr в осадок переходит 99,7 % Ва и < 0,6 % Sr.
К 200 мл раствора хлоридов, содержащих ~70 мг Ва, прибавляют избыток 0,2 М раствора ЭДТА, раствор аммиака до pH ~ 10, 1 ммолъ К2Сг2О7,
45
разбавляют водой до —400 мл, нагревают до 90—95° С и при постоянном перемешивании медленно прибавляют 0,02 М раствор хлорида магния (1 капля в 4—6 сек.) в количестве, необходимом для связывания свободного комплексона и количественного'вытеснения из комплекса Ва, Выделившийся осадок отфильтровывают через пористый тигель, промывают сначала 0,001 М раствором K^Cr-jOv (слабо подщелоченном раствором NH4OH) и затем 3 раза водой.
Для удержания больших количеств Sr в растворе в качестве комплексообразующих веществ рекомендовано также применять оксиэтилиминодиуксусную или нитрило-триуксусную кислоту [404].
К аммиачному анализируемому раствору, содержащему не более 1 ммоля суммы Ва2+ и Sr2+ и большой избыток маскирующего реактива, прибавляют КгСггО?, нагревают 10 мин. на кипящей водяной бане, охлаждают 1 час льдом и центрифугируют; осадок ВаСгО4 отделяют и промывают. По этой методике можно легко отделить <0,1 ммоля Ва от 0,8 ммоля Sr с небольшим соосаждением.
Нитрилотриуксусная кислота обладает большей маскирующей силой, чем оксиэтилиминодиуксусная, но более чувствительна к изменению pH среды. Ионы щелочных металлов не мешают; мешают анионы-осадители стронция (F~, СО|", С2О4Д РО4~ и др.).
Отделение посторонних элементов в виде фосф поз
Отделение Al, Cr, Fe путем осаждения фосфатов [944]’основано на различии произведений растворимости фосфатов указанных элементов и Sr. Zr и Nb могут быть отделены от Sr путем осаждения фосфатов указанных элементов в кислом растворе [1010]. При выделении урана в виде ураниламмонийфосфата при pH 5,0 Sr и другие щелочноземельные элементы связывают в комплекс с ЭДТА.
Отделение Са и Ва ферроцианидом
Ферроцианидный метод, по данным работы [99], с успехом может конкурировать с нитратным методом отделения Са от Sr. Осаждение производят при значении pH 9,0 из горячего раствора насыщенным раствором ферроцианида калия. Метод, применим для отделения от Sr не только больших количеств Саг но и малых количеств Ва.
Раствор нитратов стронция и кальция (радиоизотоп 90Sr), содержащий Sr 0,4 г, Са — 0,4—40 г, доводят до слабощелочной реакции буферной смесью (NH4OH 4- NH4C1) и нагревают до кипения. К горячему раствору приливают горячий конц. раствор K4Fe(CN)6 (в 50%-ном избытке). Осадок ферроцианида кальция отделяют и промывают разбавленным раствором СН3СООН. Ошибка, при отделении Sr составляет не более 2,5% [99].
46
Другие методы отделения посторонних элементов
Для отделения'Ва от Sr можно использовать также кремне-фтористово д\о родную кислоту [728] и другие осадители.
Отделение Sr от $е производят путем соосаждения последнего с Те.
В исходную смесь добавляют 1 мл 1 %-ного раствора ТеОа в 6 TV НС1 и восстанавливают его раствором хлористого олова. Осадок теллура вместе с соосажденным селеном промывают несколько раз 6 N НС1 [270].
Из органических осадителей, применяющихся для отделения некоторых элементов от стронция, следует отметить: д и п и-криламин (для отделения Cs) [799], бриллиантовый желтый (для отделения Mg при отношении Mg : Sr = 1 : 3) [352], оксифениларсоновую кислоту (для отделения РЬ) [336], диоксимы 1,2 - дикетонов в присутствии комплексообразующих веществ (для отделения Bi) [531], фталаниловую, и-толилфталаниловую и а-н афтилфталаниловую кислоты (для отделения Pd2+ при pH 2—4,5) [660], пиро меллитовую кислоту (для отделения равных количеств Ва) [79], нитрилотриметиленфосфоновую кислоту (для отделения Be) [123], 8-оксихинолин в присутствии мочевины (для отделения Y) [600] и др.
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Методы, основанные на экстракции стронция
В табл. 15 приведены методы, используемые для отделения стронция экстракцией.
Стронций можно проэкстрагировать из растворов перхлоратов, иодидов или роданидов трибутилфосфатом (ТБФ).
Из 2М раствора перхлората натрия при pH 1—4 и равных объемах органической и водной фаз Sr экстрагируется на 92, Са — на 99,5, Ва — на 83%. Одновременно со щелочноземельными элементами экстрагируется ряд тяжелых металлов, однако отделение от них возможно, если проводить экстракцию при pH 1,4 в присутствии этилендиаминтетрауксусной кислоты. Показана возможность такого отделения от Pb, Си, Bi, Zn, Zr. Реэкстрак-ция Sr может быть произведена 3tV НС1 [113].
Экстракция иодида стронция трибутилфосфатом проходит [236] в широком интервале кислотности — от 0,5 N НС1 до pH 8. Степень экстракции стронция из 4 М NaJ равна 74, Са 86, Ва 65 %:
Роданид стронция экстрагируется трибутилфосфатом из кислых растворов при концентрации роданида 2 М. Сте-
4Т
Таблица 15
Экстракционные методы отделения
Реагент Органический растворитель Экстрагируемое соединение / Назначение Литература
Перхлораты (2ЛГ NaClO,, pH 1—4) Трибутилфос-фат (ТБФ) Sr(C104)2-4TE<? Отделение группы щелочноземельных элементов [ИЗ, 235]
Йодиды (0,5 N НС1, pH 8-9) То же 8г12-4ТБФ То же [236]
Полииодиды Нитробензол — Отделение Sr от Са [952]
Роданиды (2,4М NaSCN, 0,1М НС1) Трибутилфосфат 8г(8СХ)2-4ТБФ То же [103, 108, 110, 229, 1198]
Теноилтри-фторацетон (ТТА), pH 8 Метилизобутил-кетон Sr(TTA)2-НТТА Выделение Sr (после отделения Y бензолом при pH 7) [907]
1-Фенил-З-ме-тил-4-каприл-пиразолон-5 То же —‘ Выделение Sr (после отделения Cd, Y, La) [1041,1042, 1043]
1-Фенил-З-ме-тил-4- бензоил-пиразолон-5 Циклогексанон этилацетат — Выделение щелочноземельных элементов [163, 164, 165]
р-Изопропил-трополон (НА) pHJ 9-9,5 Ди-м-бутил-' фосфорная, ди-2-этилгексил-фосфорная кислоты (НА) Хлороформ МеА2-НА То же [694]
pH 4-5 Циклогексанон Ме(НА2)2(НА)п (п от 2 до 4) » [686, 697, 875, 929, 931, 969, 1051]
pH 2-3,8 Керосин, толуол — Отделение Sr от Са [601, 875]
8-Оксихино-лин (НА) Хлороформ МеА2-2НА Выделение щелочноземельных элементов [695]
Купферон (pH 3) Трибутилфосфат — Выделение Sr [181]
Глиоксаль-б uc-2-оксиани л Гексанол, хлороформ — То же [1334]
Азо-тазокси БН (Н2А), 0,8—12V NaOH СС14 (80%) + + ТБФ (20%) МеА-2ТБФ Отделение Sr [104, 112, ИЗ, 116, 276]
Азо-азокси -ФМП (Н2А), 0,2—0,5AfNaOH То же МеА-2ТБФ То же [101, 106, 116, 276]
Таблица 15 (окончание)
Реагент Органический растворитель \ Экстрагируемое соединение Назначение Литература
Хелант БТ (НгА) Циклогексан — ТБФ (1 : 1) 8ГА-2ТБФ Выделение Sr (щелочноземельных элементов) [86, 117а]
пень экстракции Ga, Sr и Ва составляет 98, 80 и 72% соответственно при равных объемах фаз и кислотности раствора около 0,1 N [229]. Необходимо проведение двукратной или трехкратной экстракции, как и в предыдущих методах. Возможно отделение от таких металлов, как Fe, Ni, Со, Pb, Zn, Cr, Ti, Zr, РЗЭ и др. При реэкстракции Sr 0,35 М раствором лимонной кислоты Sr можно отделить от Са, который остается в органической фазе.
Используя экстракцию Sr в виде роданидных комплексов, успешно отделяют его от редкоземельных элементов, Се и Y [101].
Доводят pH анализируемого раствора, содержащего 0,5—5,0 мг Sr и 10—100 мг РЗЭ, до 4 (по индикаторной бумаге), после чего приливают несколько капель раствора металлохромного индикатора ксиленолового оранжевого и добавляют 0,24 М раствор комплексона III до исчезновения розовой окраски и небольшой избыток его. После этого добавляют NaSCN до концентрации 2,0—2,4 М и НС1 до pH 2—4. Общий объем раствора составляет 25 мл. Приливают такой же объем трибутилфосфата и встряхивают 1 мин. После расслаивания водный слой переносят в другую делительную воронку, создают те же условия и повторно экстрагируют. Экстракты объединяют и отмывают захваченный комплексон III встряхиванием с кислым раствором NaSCN (с той же концентрацией NaSCN и НС1). Щелочноземельные элементы реэкстрагируют НС1 и определяют их.
Анализ искусственных смесей показал, что степень экстракции щелочноземельных элементов при двукратном извлечении достигает 99—99,85% [101, 229]. Показана возможность использования этого метода и при определении щелочноземельных элементов в магнии и его соединениях [112].
Экстракция полииодидов щелочных металлов нитробензолом используется для их разделения и в особенности для отделения 137Gs от других продуктов деления. В работах [952, 1258] показано, что при этом экстрагируются также Sr и Са. Sr экстрагируется лучше, чем Са, разделение улучшается в присутствии этилендиаминтетрауксусной или циклогександиамино-тетрауксусной кислоты; в присутствии последней фактор разделения Sr и Са достигает 220. Подходящие условия для экстракции: 0,26 М LiJ, 0,8 М J2, pH 4—5. Коэффициент распределения Sr между органической и водной фазами при этом достигает 60. При экстракции следовых количеств Sr 0,2 М раствором иода в нитробензоле увеличение концентрации иодйдов в водной фазе снижает коэффициент распределения до 1-10~2.
49
Растворы многих р-дикетонов в органических растворителях экстрагируют'Эг при высоких значениях pH. Однако растворы аце-тилацетона в бензоле или хлороформе не экстрагируют Sr.
0,1 М раствором бензоилацетона в бензоле экстрагируется — 50 % Sr при pH 11,5, а раствором дибензоилмета н а в бензоле — 80% Sr при pH 12. Заметное снижение pH экстракции наблюдается при использовании в качестве реагента т е н о’и лтрифторацетона (ТТА). G его помощью в бензол экстрагируется (pH водной фазы 8—10): Са на 99, Sr на 80% [563], Ва на 50—60% [907, 1149]. В качестве растворителя используют также метилизобутилкетон [494, 687, 725, 874, 907, 930]. При этом коэффициент распределения элементов возрастает [494]. <
Более 99% Sr экстрагируется 0,2 М раствором теноилтри-фторацетонав метилизобутилкетоне при pH 10—12 (при pH 6 — 50% Sr). Стронций может быть также проэкстрагирован 0,1 М раствором ТТА в четыреххлористом углероде, содержащем ТБФ в концентрации 0,1 М (pH 7,0).
Изучение экстракции комплексов стронция с ТТА описано в [494, 874, 930].
4-Ацилпроизводные 1-фенил-З-метилпира-золона-5, содержащие функциональную группировку |3-ди-кетонов, являются хорошими экстрагентами для стронция. 1 - Ф е-н'ил’-3-м’етил-4-бе н з о и л п и р а зол он-5 (ФМБП), равновесные формы которого изображаются следующим образом:
[СН3-C=N
СвН5—С—СН—С
II II
СН3— C=N
С6Н5—с=с-с
I II он о
о
экстрагирует стронций, будучи растворенным в изопентаноле, при pH 5 (рис. 3). Максимальная экстракция достигается при pH 6,9— 9,7, логарифм коэффициента распределения 1g Етах = 1,38 [164].
В качестве растворителей могут быть взяты также циклогексанон и этилацетат (1g Ета^ = 1,54 и 1,22 соответственно). Растворами ФМБП<в хлороформе и бензоле стронций не экстрагируется. Кальций экстрагируется при несколько меньших значениях pH.
Растворами’ФМБП экстрагируется также много других металлов (в большинстве случаев в более кислой среде). Например, скандий экстрагируется в области pH от 2 до 6 и, таким образом, при pH 2—4 его^можно отделить от стронция.
Другое соединение этого класса — 1-фенил-3-метил-4-каприлпи р’а’з’о л о н - 5 (ФМКП) в растворе в метилизобутилкетоне также применяется для отделения Sr. Экстракция производится при pH 9,4. С этим реагентом экстрагируется и ряд других элементов,..которые могут быть отделены от Sr экстракцией из
.50
более кислых растворов. Так, Y может быть отделен экстракцией при pH 3,8, Gd—при pH 5,4. При pH 9 Ва экстрагируется в меньшей степени, что может быть использовано для его отделения от Sr [1044]. ФМКП нашел применение для выделения радиоизотопов без носителей и их разделения [725, 1042, 1043, 1044].
По свойствам близко примыкает к Р-дикетонам и [3 - и з о-пропилтрополон, с которым щелочноземельные элементы образуют комплексы типа МеА2-НА [694]. При этом Са экстрагируется на 99 (pH 8,5), Sr — на 97 (pH 9,0—9,5) и Ва — на 50% (pH 10). Растворитель — хлороформ. Реактив не селективен.
Алкилфосфорными кислотами щелочноземельные элементы экстрагируют из слабокислой среды [556, 601, 720, 815, 1120, 1253]. При экстракции эквивалентным количеством реагента обычно образуются соединения типа МеА2, слабо растворимые в органических растворителях; при экстракции избытком диалкилфосфорной кислоты образуются комплексы типа Ме(НА2)2, Ме(НА2)2-НА или Ме(НА2)2’(НА)2 и другие, растворимость которых в органических растворителях на несколько порядков выше [686, 911, 929, 931, 1022, 1023, 1024, 1051, 1119, 1252]. Наиболее изученными реагентами этого типа являются д и-«-б утилфосфорная и ди-2-этилгексилфосфорная кислоты.
Экстракция ди-«-бутилфосфорной кислотой щелочноземельных элементов снижается в ряду растворителей: циклогексан, гексан, керосин, четыреххлористый углерод, бензол, хлороформ [697]. Она не нашла широкого применения, однако используется для отделения РЗЭ от щелочноземельных элементов [693, 720]. Для этой же цели применялись и диоктилфосфорная [815, 1120], д и-н-к резилфосфорная [337] и монооктил-анилинобензилфосфорная [826] кислоты. Экстракция проводится из «С 0,1—0,05 М HG1 или HNO3 0,005— 0,01 М растворами экстрагентов в бензоле, петролейном эфире или хлороформе.
Ди-2-этилгексилфосфорная кислота (ДЭГФК) многократно изучалась как экстрагент для выделения щелочноземельных элементов [686, 873, 912, 929, 931, 1022, 1023,
51
1024, 1051, 1119]. При экстрагировании из водных солянокислых растворовУкоэффициенты распределения двухзарядных катионов уменыпаются’в таком порядке: UO22+, Zn РЬ > Мп > Си Cd, Ni Mg, Ra Ba, Sr > Ca [576]. В присутствии 0,5 M нитрата натрия экстрагируемость металлов бензольными растворами ДЭГФК распределяется следующим образом: Be г>> Са > Mg « Sr Ва, а при возрастании концентрации нитрата натрия до 4 М степень’экстракции Mg становится больше, чем Sr [1022, 1023, 1024]. Состав экстрагирующихся соединений щелочноземельных элементов может изменяться, как и для других алкилфосфорных кислот, от МеА2 до МеА2-4НА [686, 929, 1022, 1023, 1024, 1051]. На состав экстрагирующихся соединений оказывает влияние род растворителя. В работе [686] найдено, что в случае алифатических растворителей (гексан, октан, декан, додекан, тетрадекан, циклогексан) состав соединения МеАа-^НА, в случае ароматических растворителей (бензол, толуол, ксилол), а также СС14 — МеА2-НА и, наконец, в случае полярных растворителей (гексанол, деканол, трибутилфосфат, три-2-этилгексилфосфат) образуются соединения типа МеА2 и МеА2-2НА.
Установлено, что экстрагирующая способность ди-2-этилгек-силфосфорной кислоты по отношению к щелочноземельным и некоторым другим элементам существенно увеличивается в присутствии в растворе Zr, Hf и Ti. Увеличение степени экстракции элементов зависит от концентрации Zr в исходном растворе и от анионного состава последнего. Влияние Zr на улучшение экстракции других элементов объясняется тем, что его комплексы с ДЭГФК гидролизованы и водород ОН~-групп, связанных с Zr, способен легче обмениваться на катион экстрагируемого металла, чем водород исходного экстрагента [1353].
Наилучшая экстракция стронция наблюдается при pH 4—5 [875, 931]. При pH 5 0,64 М раствором ди-2-этилгексилфосфорной кислоты в толуоле Са извлекается практически полностью [912, 1119], Sr — на 99% [601], Ва — на 1 % [912]. Поэтому в процессе экстракции Sr вместе с ним в экстракт попадает Са и в меньшей мере — Ва.
Отделение Sr от Са может быть достигнуто путем экстракции 0,4 М раствором ДЭГФК в керосине, содержащем 0,2 М трибутилфосфата. Из органической фазы Sr избирательно реэкстрагируется 1 М раствором лимонной кислоты с pH 2—2,4, Са и другие примеси остаются в органическом растворителе [875].
В другом методе’[601] сначала экстрагируют Са раствором ди-2-этилгексилфосфорной кислоты в толуоле при pH 3,6—3,8. Затем повторно экстрагируют Са при pH 2,6—2,8и, наконец, экстрагируют Sr при pH 4,8—5. Необходимо дополнительное отделение Sr от Са путем осаждения в виде нитрата конц. HNO3; степень выделения Sr 85 %.
При использовании в качестве экстрагента д и -1,1,3,3- тетра-метилбутилфенилфосфорной кислоты в то-
52
Рис. 4. Зависимость коэффициентов распределения Са (7) п Sr (2) в присутствии этиленди-аминтетрауксусной кислоты от pH водной фазы при экстрагировании ди-1,1,3,3-тетраме-тилбутилфенилфосфорной кислотой в толуоле [0,1 М ЭДТА; ионная сила 0,1 (NaNO3)l
.луоле в присутствии этилендиаминтетрауксусной кислоты при pH 3,5 Sr экстрагируется в большей степени, чем Са. При pH < < 3,5 зависимость обратная (рис. 4).
Коэффициент распределения при pH 5,0 для Sr — 100, для Са 10 [969].
При выделении Sr из раствора, содержащего РЗЭ, Fe, РЬ, Zr, Nb и Ru для маскирования последних используют этилен-диаминтетрауксусную [596, 646, 875], диэтилентриаминпентаук-сусную [646, 1226] или винную [852] кислоты. Для экстракции применяют 0,4 М раствор ДЭГФК с 0,2 М трибутилфосфатом в •очищенном керосине, в разбавителях Шелла [1226] или «Амско 125-82» [852].
Наряду с ди-2-этилгексилфосфорной кислотой для экстрагирования щелочноземельных металлов могут быть успешно применены и такие производные фосфорной кислоты, как д и и з о-октилфенилфосфорная, н-октилфенилфос-ф о р н а я и н-о ктилхлор метилфосфорная кислоты. Степень экстракции элементов изменяется в порядке Мц < Са > Sr > Ва [1119].
Дигексиловый эфир фенилсульфонил-амидофосфорной кислоты количественно экстрагирует щелочноземельные металлы [469—472, 576, 601, 646, 885, 995, 1126, 1226]; pH 50 %-ной экстракции для раствора в бензоле и изопентаноле составляет соответственно для Са 3,77 и 3,27, для Sr 4,31 и 3,45, для Ва 4,37 и 3,38. Реактив не селективен для этих элементов.
Для группового экстрагирования щелочноземельных элементов может быть использован 8-о ксихинолин (оксин) в Хлороформе [619, 695, 981, 982, 1266]. Экстракция происходит при^значительном избытке реагента, в этом случае экстрагируется соединение типа SrOx2-2HOx [1266]. Благоприятно влияют на экстракцию амины, в присутствии которых образуются соединения типа R(NH3)2(MeOx4) [125]. При недостатке 8-оксихинолина в растворе образуется плохо экстрагирующийся гидратированный комплекс SrOx2-2H2O [695]. В присутствии ТБФ растворимость оксинатов в органической фазе увеличивается, а коэффициент
53
распределения повышается по сравнению с экстракцией хлороформом [339]. Это же относится к экстракции стронция в виде соединений с дигалоидопроизводными оксина трибутилфосфатом [341].
Способ группового извлечения щелочноземельных металлов в виде оксинатов имеет ряд недостатков, основными из которых являются малая избирательность, так как, помимо Sr, Са и Ва, оксин взаимодействует по крайней мере еще с 50 элементами [394]. С некоторыми из них (Sc, Nd и Th) Sr соэкстрагируется [454]. Кроме того, экстракция оксихинолинатов щелочноземельных элементов происходит при сравнительно высоких значениях pH (9—12) (рис. 5), вследствие чего многие присутствующие элементы гидролизуются и затрудняют экстракцию щелочноземельных металлов. Несмотря на это, 8-оксихинолин в ряде случаев используется для выделения щелочноземельных элементов. Элементы, которые экстрагируются вместе со щелочноземельными, отделяются от последних путем реэкстракции щелочноземельных элементов из хлороформного раствора при pH 5—8.
Чаще же оксихинолинатный метод используют для отделения элементов других групп, экстрагирующихся при более низких значениях pH, чем Sr [394].
Отделение Са от Sr и Ва может быть достигнуто повторной экстракцией при последовательном изменении концентрации 8-окси-хинолина и бутиламина в растворе. При одной экстракции (pH 11,3 ± 0,1) совместно с кальцием экстрагируется 10 % Sr и 3 % Ва. Реэкстракция в водную фазу 0,2 N НС1 (конечное значение pH — 8,0) и повторная экстракция раствором 8-оксихинолина и бутиламина в хлороформе (pH 11,3) при двукратном повторении этой операции дает отделение Са от Sr и Ва [1333].
Показана [130] возможность экстрагирования щелочноземельных металлов хлороформом в виде смешанных комплексов с 8 -оксихинолином и 1-оксиантрахиноном.
Согласно данным работы [181], стронций может быть проэкстра-гирован из водных растворов с pH 3,0 в виде купфероната с помощью ТБФ. Реэкстракцию производят раствором НС1. При pH 8 Са экстрагируется на 95, Sr на 80 и Ва на 55% [277а]. Однако купферонаты чаще используются для экстракционного отделения других металлов от стронция: Ti (смесью изопентанола и бензола (1 : 1)) [324], Fe (хлороформом из 1 N НС1), Th (этилацетатом из IN НС1), U(IV) (хлороформом при pH 1,8), РЗЭ и некоторых других элементов (хлороформом, pH 4) [905]. При определении стронция в солях железа, меди и висмута эти металлы предварительно отделяют экстракцией с помощью купферона при pH 1 [101].
Sr, подобно Са, экстрагируется из 0,08 N раствора NaOH раствором глиоксаль-бпс-2-оксианила в смеси гексанола с хлороформом. Из 0,8 N NaOH в присутствии 0,02 N Na2CO3 Sr и Ва не экстрагируются и таким путем могут быть отделены от Са [1334].
54
Рис. 5. Зависимость степени экстракции элементов II группы периодической системы Д. И. Менделеева 0,5 М раствором 8-оксихинолина в хлороформе от pH раствора
1 — Ве; 2 — Mg;
з — Са; 4 — Sr;
Л — Ва
Рис. 6. Зависимость степени .экстракции Са (7), Sr (2) и Ва (3) с помощью экстрагента АТ от концентрации NaOH в растворе
Отделение стронция от других щелочноземельных элементов (101, 102, 104—106, 111, 112, 116, 119, 276] может быть достигнуто с помощью реагентов азо-азокси БН и аз(о-а з о к с и ФМП, которые для экстракции щелочноземельных элементов применяются в виде растворов в смеси четыреххлористого углерода с 20 объемн. % трибутилфосфата (экстрагенты АТ и АФТ).
При использовании экстрагента АТ стронций экстрагируется в виде комплекса состава ЗгААБН>2ТБФ [116]:
OR OR OR OR OR OR
\№N N=N/
! О
Как'следует из рис. 6, экстракция щелочноземельных элементов экстрагентом АТ происходит в щелочной среде, причем Са практически полностью извлекается при концентрации NaOH 0,05 N, a Sr в этих условиях экстрагируется всего лишь’на 10—
055
15%. Ва в изученном интервале щелочности (0,005—10,0 7V) экстрагируется весьма незначительно. Примесь Са из соединений Sr можно экстрагировать из 0,05—0,1 7V раствора щелочи, а примесь Sr из соединений Ва — в интервале 0,8—2,0 7V щелочи. Фактор разделения Са и Sr равен 300, Sr и Ва 900 (табл. 16).
^Таблица 16
Коэффициенты распределения Са, Sr и Ва при экстракции их экстрагентом АТ в оптимальных для разделения условиях
Элемент Концентрация NaOH, N Коэффициент распределения (Е)
Кальций 0,01 1,00 99,0 999,0
Стронций 0,05 1,00 0,33 9,0
Барий 0,05 1,00 0,001 0,010
Помещают 25 мл нейтрального раствора, содержащего 50—500 мкг Са, Sr и Ва (бария могут быть и большие количества), в делительную воронку, добавляют NaOH до 0,05 N раствора (0,25 мл 20%-ного NaOH), равный объем экстрагента АТ и экстрагируют кальций 1—2 мин. После расслоения водный слой отделяют и переносят в кварцевый стакан емкостью 250 мл, а органический — переносят в делительную воронку и проводят переэкстрак-цию. Для этого к экстракту приливают 25 мл 0,01 N раствора НС1 и встряхивают 1 мин., затем, не разделяя слои, нейтрализуют кислоту, добавляют NaOH до получения 0,05 N раствора и снова встряхивают. После расслоения водный слой отделяют и присоединяют к раствору, полученному при первой экстракции (помещенному в кварцевый стакан), а органический слой подвергают еще одной переэкстракции.
Далее к органическому слою приливают 25 мл 0,01 N раствора НС1 и реэкстрагируют кальций встряхиванием. Реэкстракт отделяют, переносят в коническую колбу и определяют содержание [Са комплексонометрическим микрометодом с индикатором глиоксаль-бис-2-оксианилом. Водный слой выпаривают в кварцевом стакане до объема 10—15 мл, помещают в делительную воронку, доводят объем до 25 мл водой и добавляют NaOH до 0,8—1,0 7V. Экстрагируют равным объемом экстрагента дважды. Из объединенных экстрактов Sr реэкстрагируют 25 мл 0,01 N раствора НС1 и определяют era содержание комплексонометрическим методом с метилтимоловым синим. Содержание Ва определяют в водном слое (после извлечения Са и Sr) тем же методом.
Метод позволяет разделять и определять Са, Sr и Ва с достаточной точностью. Он позволяет определять микропримесь одного-щелочноземельного элемента в соединениях другого. Особенна
56
это успешно осуществляется при таком соотношении элементов в смеси: Са < Sr < Ва. Здесь возможно определение Sr с чувствительностью 1-10~3—1-10~5%. Погрешность определения 50— 500 мкг Sr в смеси щелочноземельных элементов не превышает 2,4—10 отн. % Время анализа 2 часа.
С применением экстрагента АТ разработаны методики определения Са, Sr и Ва в ферритах, солях магния, железа, висмута, меди, цинка, кадмия и свинца [101, 103, 112]. При определении примеси^Эг (Sr + Са) в углекислом барии Sr отделяют от Ва экстракцией экстрагентом АТ из 1ЛГ раствора NaOH. Экстракт трижды промывают для удаления захваченного бария 1А раствором щелочи (40 мл). Затем уже производят разделение стронция и кальция и дальнейшее их определение [119].
Другим, еще более эффективным экстрагентом для стронция является экстрагент АФТ [101, 106, 116, 276]. Комплексообразующий реактив а з о-a зокси ФМ П (2-(1"-ф е н и л-3"-м е т и л-5"-п и р а з о л о н-(4"-а з о-2'>-ф е н и л-а з о к с и) - 4'-м е т ил-фен о л) образует со стронцием комплекс такого же состава, как и реагент азо-азокси БН, а именно: ЭгААФМП-2 ТБФ.
Преимущества реагента АФТ перед АТ следующие: первым из них Sr экстрагируется практически полностью, а вторым — лишь на 93 %, причем экстракция Sr экстрагентом АФТ протекает в менее щелочной среде (pH 50 %-ной экстракции 11,8 и 13,15 для АФТ и АТ соответственно) и в более широком интервале щелочности. Однако эффективность отделения Са от Sr является более высокой, если для этой цели применяется экстрагент АТ, а не АФТ. Поэтому при анализе щелочноземельных элементов рекомендуется сначала отделять Са с помощью экстрагента АТ (из 0,05 N раствора NaOH), а затем уже при более высокой щелочности (0,2 N NaOH) отделяют Sr от Ва с помощью экстрагента АФТ. При этом фактор разделения этих элементов достигает 4800.
Метод использован для отделения Sr от Ва и Y [276]. При разделении Sr экстрагируется экстрагентом АФТ (pH 12,6—14), Y маскируется сегнетовой солью. При соотношении Sr : Ва = = 1 : 500 и Sr : Y = 1 : 300 ошибка не превышает 10% (отн.). Продолжительность разделения и определения стронций 20— 30 мин.
Показано [117], что экстракционную емкость ААБН (и других аналогичных реагентов) можно увеличить путем экстракции металла в присутствии твердого реагента. Экстракционная емкость ААБН в этих условиях увеличивается от 1 • 10_3 до 1 • 10-1 г-ион/л, т. е. в 100 раз. Несмотря на то, что метод имеет некоторые недостатки (в частности, необходимо отделение избытка твердого реагента после проведения экстракции), в отдельных случаях он может оказаться^очень полезным.
Для выделения и разделения макроколичеств щелочноземельных элементов предложен реагент - хелант БТ [5-а м и н о-2-о к с и ф е н и л-4-(5-х л о р-2- оксифени л-а з o)-2N-6 е н з о-
57
триазол], обладающий высокой избирательностью по отношению-к щелочноземельным элементам и высокой экстракционной емкостью [114]. Благодаря этому он пригоден для извлечения этих элементов обычной экстракцией [117а].
Стронций экстрагируется раствором реагента БТ в смеси растворителей ТБФ и циклогексана в объемном отношении 1 : 1 (экстрагент БТ-1) в виде комплекса ЭгА-2ТБФ (где А — БТ). Строение комплекса аналогично строению комплексов щелочноземельных элементов с ААБН. Объем экстрагента обычно составляет 25 мл и равен объему водной фазы. Нужное pH (11,5) создают аммиаком. Стронций реэкстрагируют 25 мл 0,1 М раствора НС1 и определяют его содержание комплексонометрически. Этим методом можно извлекать и определять 5—15 мг Sr с ошибкой 0,4— 0,2% (отн.). Экстракция стронция с использованием реагента БТ протекает в менее щелочной области, чем с другими реагентами (по сравнению с реагентом ААФМП на 1,5 единицы pH ниже, а по сравнению с ААБН — на 2,5 единицы). Это важно для экстракции больших количеств стронция в присутствии других, особенно тяжелых металлов.
Использование экстрагента БТ-1 позволяет не только экстрагировать макроколичества щелочноземельных элементов, но и разделять их смеси. Так, например, бинарная смесь Са и Sr при соотношении компонентов 1 : 1 разделяется с фактором обогащения 5sr/ca — 0,1, a Sr и Ва — с S^a/sr = 0,033. Экстракции не мешают граммовые количества щелочных элементов, а также элементов, образующих амфотерные окислы. Не мешают миллиграммовые количества элементов, образующих аммиакаты (Си, Ni, Со). Мешает Zn (^> 1 мг), который сам экстрагируется в этих условиях. Мешающее влияние легко гидролизующихся элементов устраняют маскированием, либо их предварительным экстракционным отделением в виде диэтилдитиокарбаминатов или купферонатов.
Из щелочных растворов (1 М NH4OH, 1 М NaNO3) щелочноземельные элементы экстрагируются растворами салицил-альдоксима в ТБФ или другом растворителе — ДАМФК; коэффициенты распределения между органической и водной фазами составляют для Са 10—20, Sr 0,3—0,8, Ва 0,17—0,18. Метод использован для разделения пар Са—Sr и Sr—Ва с помощью противоточной экстракции [340].
Экстракция комплексов Sr и Y растворами пикролоно-вой кислоты в метилциклогексане изучена в работе [1353].
Sr и Y экстрагируются нафтеновыми кислотами (мол. масса —200), причем Y экстрагируется при меньших значениях pH (4,5), чем Sr (pH 6,5). Наибольшие коэффициенты разделения наблюдаются при pH 5,5 [483]. Ва экстрагируется при. меньших значениях pH (5,8), Са — при больших Q>7,2) [4].
Изучена также экстракция Sr и других щелочноземельных элементов растворами алифатической монокарбоновой кислоты («в е р с а т о в а я кислота 1519»^
58
в N-октане [1025], масляной кислотой и ее растворами в хлороформе [91], расплавленной стеариновой кислотой [431]; экстракция нитробензолом дипикрилам и-натов и |тетрафенилборатов щелочноземельных элементов [1135, 1235] и комплекса стронция смышьяковой кислотой и пиро к’а т е х и н о м [947].
Исследована экстракция щелочноземельных элементов из растворов HG1 и NaGl растворами динонилнафталинсуль-фокислоты в гептане [1364]. Показано, что коэффициент распределения уменьшается в ряду Ва Sr Са. При экстракции из растворов хлористого натрия в присутствии ЭДТА различия в коэффициентах распределения увеличиваются. При концентрации Sr и Ва —1 • 10~4 М они могут быть разделены экстракцией из 0,1 *М NaGl, содержащего 0,01 М ЭДТА; при pH 5,5 фактор разделения ^>100. Полнота разделения достигается многократной экстракцией.
Отделение Sr от Са и Ва экстракцией нитробензолом в присутствии полиэтиленгликоля и борсодержащего гидрофобного аниона использовано для его радиометрического определения [479].
Методы, основанные на экстракции посторонних элементов
Как следует из некоторых приведенных выше примеров, посторонние элементы могут быть отделены от Sr путем экстракции их в виде различных соединений.
Показана возможность отделения Са от Sr экстракцией Са из роданидного раствора с pH 4 смесью (1 : 1) трибутилфосфата и четыреххлористого углерода [230].
Установлено [826], что Y может быть количественно отделен от Sr путем экстракции Y раствором монооктиланили-нобензилфосфоната (в петролейном эфире) из 0,01— 0,1 М раствора HG1 (рис. 7). Количество Sr не влияет на экстракцию Y.
Для отделения урана от щелочноземельных металлов и других продуктов деления экстрагируют его нитрат с помощью эфира [81, 830, 1159] или с помощью раствора трибутилфосфата в керосине (1 + 10) при pH 5 [1139]. Рассматривается также возможность отделения урана путем экстрагирования с помощью нитрата трикаприлметиламмония [920] и н-т р и о к т илам и н а с НС1 в четыреххлористом углероде [418].
Th можно отделить от Sr экстракцией диантипирил-метанроданидного комплекса [141]; Gu, Fe, Ni, Со и другие — экстракцией дитизонатов [588, 1375]. Для этой же цели используется раствор 8-м еркаптохино-л и н а в хлороформе [1035]. 95Zr и 96Nb могут быть отделены от щелочноземельных элементов в виде ионных ассоциатов с щ а-велевой кислотой и триизооктиламином — при экстрагировании ксилолом [1292], Ru — с помощью т р и-»-
5»
Рис. 7. Зависимость степени экстракции Sr (7) и Y (2) раствором монооктиланилинобен-зилфосфоната (5 .10 ~6 М) в пет-ролейном эфире от концентрации НС1 в водной фазе (1 • •10-4 М Sr2+; 3,3-10-’ М Y3+)
октила мина иэ азотнокислых растворов [466], а при экстрагировании пиридином из раствора NaOH + NaOCl удается полностью отделить 106Ru от 80Sr и других продуктов деления [906]. Высокоэффективное отделение Sb от щелочноземельных элементов достигается при двукратной экстракции ее из 2N солянокислого раствора равным объемом 1 %-ного раствора д и ф е-нилкарбазид.а в пентаноле или изопентаноле [458]. Раствор изооктиламина вметилизобутилке тоне используется для отделения Cd от продуктов деления, в том числе и Sr [1045].
Следует также отметить возможность отделения Bi3+ и других элементов от Sr путем экстрагирования их в виде иодидных комплексов циклогексаном [439] или растворами вторичных аминов (мол. масса 335—353) в ксилоле [1008]. Экстракция LiCl w-бутанолом используется [751] для отделения Li от Sr.
Вопросы теории и практики экстракционного отделения щелочноземельных элементов или одного Sr от других элементов рассмотрены также в работах [5, 118, 465, 500, 582, 587, 786, 953, 1025, 1059, 1077, 1173, 1204, 1258, 1305].
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Ионообменная хроматография
Ионообменная хроматография нашла широкое применение для концентрирования микроколичеств стронция, отделения стронция от элементов других групп и от других щелочноземельных элементов. Используется как катионообменная, так и анионообменная хроматография.
60
Применение катионитов
Для разделения смесей металлов на катионитах часто применяются растворы их простых солей — хлоридов или нитратов, а также НС1 и HNO3. В табл. 17 приведены значения равновесных коэффициентов распределения (Kv) щелочноземельных и других элементов второй группы Периодической системы элементов — Mg и Be в растворах соляной кислоты разных концентраций, а в табл. 18 сопоставлены Кр четырех металлов в НС1 и HNO3 одинаковой концентрации. Как видно из данных таблиц, с ростом концентрации кислоты в растворе значения коэффициентов распределения снижаются, при этом в растворах НС1 они выше, чем в растворах HNO3.
Показано [691], что фактор разделения Са и Sr (элюент — 0,48— 2,04 N НС1, катиониты типа Дауэкс-5О\У) имеет максимум при использовании катионита со средней степенью поперечной связанности (8% поперечных связей) и монотонно увеличивается' с ростом температуры.
Возможности разделения большой группы элементов, в том числе щелочноземельных, при использовании в качестве элюента
Таблица 17
Коэффициенты распределения катионов Be, Mg, Са, Sr и Ва при различной концентрации НС1 (катионит Дауэкс-50Х8) [1278]
Катион Кр при концентрации HCI, N
0,1 0,2 0,5 1,0 2,0 3,0 4,0
Ве2+ 255 117 42 13 5 3 2
Mg2+ 1720 530 88 21 6 4 4
Са2+ 3200 790 151 42 12 7 5
Sr2+ 4700 1070 217 60 18 10 8
Ва2+ >10* 2930 590 127 36 19 12
Таблица 18
Сравнение коэффициентов распределения щелочноземельных элементов , и магния в 1 М НС1 и HNO3 [1278]
Катион Кр в среде Катион Кр в среде
НС1 HNO,1 НС1 jHNO,
Mg2+ 21,0 24,0 Sr2+ 60,2 50,3
Са2+ 42,3 39,6 Ва2+ 126,9 68,4
61
Таблица 19
Ионообменные методы отделения стронция с помощью катионитов при использовании водных растворов минеральных кислот и их солей в качестве элюентов
Катионит Элюент Отделяемые элементы Литература
Дауэкс-50Х8 0,02—6 N НС1 Th, Mg, Be, 1570, 639,
Fe(III) и др. 894]
0,1—1 М HF Al, Cd, Mo, Nb и др. [731]
12—5 М НВг (60° С) Mg, Be, Ra, Ba [1081]
0,1—6 М HNO3 Al, РЗЭ, UO22+ [378, 730]
0,26—0,46 М (NH4)2SO4 Mg, Ca, Ba [639]
Формиатный буферный Na, K, Mg, Ca, [1324]
раствор Ba
Дауэкс-5Э\¥Х12 1,5—2 М CH3COONH4 Mg, Ca [380, 1094|
Дауэкс-50) Смесь НС1 и Н N О3 РЗЭ, UO22+ [941J
КУ-1 1 М NH4C1 Th [147]
КУ-2 0,5—2 М НС1, HNO3 (рЩ2) Ce, As [299, 374]
Амберлит [CG-120 0,8—4 М НС1, 2,5— Mg, Ca, Ba, [193, 577,
3 М NaCl, 0,1 М NagSgOs Pb(II) 1020, 1021]
Диолит-С62, С63, С65 0,1—6 М HNO3 РЗЭ, e6Zr, [795]
esNb, loeRu, ioeRh
Вофатит KPS-200 3,4% (NH4)2SO4, 2N HNO3 Ca [638]
Вионит CS-2 (Н+-фор-ма) 1,5—2 M CH3COONH4 UO22+, Y, Eu [1004]
Повер хностно-сульфи- 0,02—6 N HC1 eey, 137 Cs 1255]
рованная смола (Н+-форма)
0,1—0,5 М раствора перхлората этилендиаммония исследованы в [732].
Табл. 19 и 20 содержат перечень работ, в которых изучалось отделение Sr от щелочноземельных и некоторых других элементов при использовании растворов кислот или их солей в воде или в органических растворителях. Последовательное элюирование щелочноземельных элементов может быть достигнуто применением одной и той же минеральной кислоты различной концентрации, например НС1 (рис. 8) [393].
Более надежные результаты получаются при использовании комплексообразующих веществ (см. табл. 21), повышающих коэф-
62
Рис. 8. Выходные кривые Mg (I), Са (2), Sr (3) и Ва (4) при хроматографировании на катионите КУ-2Х6
Элюент: 0,055 (Г), 0,75 (г), 0,95 (3)и 1,35 (4) М НС1
фициент разделения металлов. Усиление комплексообразования наблюдается в ряду Ra—Be,.
В качестве комплексообразующих веществ используются оксикарбоновые и аминополикарбоновые кислоты — молочная, лимонная, этилендиаминтетрауксусная, д и а м и н о ц и к л о г е кс а нт ет р а у к с у с н а я и др. (табл. 21). Наиболее часто применяются соли этилендиаминтетра-уксусной кислоты. Хотя, как показали сравнительные опыты по разделению пар Sr—Са и Sr—Ва [1282], наилучшие факторы разделения получены для пары Sr—Са при использовании диами-ноциклогексантетрауксусной кислоты, затем этиленгликоль-бпс-(Р-аминоэтилэфир)нитрилотетрауксусной, а потом уже этилен-диаминтетрауксусной кислот. Для пары Sr—Ва второй и третий комплексоны меняются местами.
Отделение Sr от Са в присутствии этилендиаминтетрауксусной кислоты описано в [65].
Т а б лица 20
Ионообменные методы отделения стронция с помощью катионитов и органических растворителей
Катионит Элюент Органический растворитель Отделяемые элементы Литература
Дауэкс-50\УХ8 0,4—0,5 М CHsC00NH4 Метанол (30— 50%) ©оу [899]
Формиатный буферный раствор, pH 3,5—6,0 Метанол (10%) Fe, Al, Mg, Са, Ва [408]
Дауэкс-50Х8 0,7—1,6 N НС1 Метанол (20%) Mg, Са, Ва [408]
AG-50WX8 3 М НС1 Этанол (20%) Ва [1279]
63
Таблица 21
Ионообменные методы отделения стронция с помощью катионитов при использовании растворов органических комплексообразующих веществ
Комплексообразующее вещество Катионит Отделяемые элементы Литература
«-Оксиизомасляная кислота пли ее аммонийная соль 0,5 М, pH 3—5 КУ-2 Дауэкс-50Х8 РЗЭ, 90Y Са, Ва, La, Се, у so у [1242, 1285J [585, 1121, (304, 1374 J
Молочная кислота 0,2—0,5 М, pH 2,9— 7,5 Дауэкс-SOWXe Be, Mg, Са, Ва, Ra, РЗЭ, 90Y (из продуктов деления) [918, 943, 1038, 1106, 1143, 1176, 1177, 1236, 1317J
Лимонная кислота 1—5%-ная, pH 3,0— 7,0 Дауэкс-50Х12 Дауэкс-50Х8 Пермутит-50 Амберлит IR-120, КУ-2 РЗЭ, Y (9°Y), Sc, Th, Mn(II), Ca, Ba, Cu, Cd, Al, Fe(III), In [157,879, 882,996, 1029, 1263, 1280]
Смесь лактата (1 М) и цитрата (0,05 М) аммония Цеокарб-225 Ca [975[
Гликолевая кислота 0,5 М, pH 3,0—4,6 Дауэкс-50 WX8 90y [1177J
Малоновая кислота 0,2-0,7 N, pH 7,0 Дауэкс-50Х8 Al, Fe, Ti, Be, Mg, Ca [1281, 1283]
Щавелевая кислота 1%-ный раствор, pH 6,5 Дауэкс-50Х8 9°Y [1293]
Этилендиаминтетра-уксусная кислота, pH 3,0—10,0 Дауэкс-50Х8 КУ-2Х15 Цеокарб-225 Вофатит-F 9°Y Be, Mg, Ca, Ba [555, 985, 1284] [369,882, 1039] [185, 690] [65 , 568 , 572 , 665, 1180]
Моксиэтилэтиленди-аминтриуксусная кислота, pH 3,5—5,0 — Pb, Zr, Nb, Fe, Ru, РЗЭ, 90Y [1100, 1366]
Диаминоциклогексан-гетрауксусная кислота, pH 5,0—12,0 Дауэкс-50Х8 (50X12), Амберлит IR-120 Ca, Ba [595, 1088, 1152, 1153, 1214, 1287, 1385]
К 2—3 мл анализируемого раствора, содержащего 0,3—3 мг Sr и Са, добавляют 25 мл 0,02 М раствора аммонийной соли этилендиаминтетрауксус" ной кислоты (I) и необходимое для достижения pH 5,1 количество НС1 или 1ЧН4ОН. Полученную смесь пропускают со скоростью 2 мл!мин через катио-нитовую колонку диаметром 9 мм и длиной 300 мм, содержащую слой Вофа-тита-F (равный 100 мм с размером зерен 0,10—0,25 мм), переведенного
<64
в NH^-форму пропусканием раствора I названной выше концентрации с pH 10 и предварительно промытого 0,02 М раствором I с pH 5,1 ± 0,1 до такого же значения pH в вытекающем растворе. После пропускания анализируемого раствора колонку промывают 150—180 мл раствора I с pH 5,1 для вымывания Са, а затем вымывают сорбированный Sr с помощью 20 мл 0,1 М раствора того же элюента с pH 10. Метод обеспечивает количественное разделение смесей при отношении Са : Sr от 10 : 1 до 100 : 1.
Выделение стронция из морской воды [505]. 1 л анализируемой воды пропускают через колонку с 25 г смолы Дауэкс-50Х12 в Са2+-форме. Колонку промывают 20 мл 0,01 М раствора CH3COONH4 с pH 5 и элюируют Са 500— 600 мл 0,02 М раствора диаминоциклогексантетрауксусной кислоты (I) с pH 5,0. Затем элюируют Sr 60 мл 0,02 М раствора I с pH 6,5; элюат разбавляют до 500 мл, в полученном растворе определяют Sr пламеннофотометрическим методом.
Сорбция 20 ионов металлов, в том числе и Sra+, на сильнокислотном катионите flayaKC-50WX8 из раствора, содержащего три-этилентетраамингексауксусную кислоту, изучена в [1379].
Описано применение катионообменной смолы КУ-2 для отделения Sr и других катионов от Р04 при анализе фосфатных минералов [398].
Применение анионитов
Наиболее часто используемые методы отделения стронция с применением анионообменников представлены в табл. 22.
Сорбция Sr на анионообменниках из НС1 и НВт, либо из нейтральных нитратов с органическими растворителями изучалась в [939, 1053], а из сернокислых растворов на анионитах в ОН“-и ЗО4”-формах — в [1211].
Sr не сорбируется из растворов в соляной кислоте на анионитах в Cl-форме, что позволяет отделить его от ряда элементов, например, Fe, U [585], Rh, Zr, Cd, Mo, Sn и других [1242]. Сорбция Sr также не наблюдается на анионите в ОН~-форме, вследствие чего возможно его отделение от Zr,’ Се и Rh [1271]. Са может быть осажден на анионообменнике в ОНГ-форме, Sr — извлечен раствором Са(ОН)2 перед его полярографическим определением [394].
Стронций способен сорбироваться на анионитах вНО3-форме из растворов, содержащих HNO3 и органический растворитель — ацетон, метилэтилкетон, метанол и др. При этом возможно отделение от ряда элементов, не сорбирующихся на анионите, таких как Cs [148], Th и U [42], Са [735]. Sr можно отделить от Re, Mg и Са после сорбции на анионообменнике в виде нитрата последовательным элюированием растворами HNO3h KNO3, содержащими различные органические растворители [226].
Из карбонатных растворов радиоизотопы 80Y и 140La в индикаторных количествах; сорбируются на анионите, 80Sr и 140Ra практически не, поглощаются, что позволяет разделять эти эле-
3 Н. С. Полуэктов и др.
65
Таблица 22
Ионообменные методы отделения стронция с помощью анионитов
Анионит Форма анионита Элюент Отделяемые элементы Литература
Дауэкс-1,2 AG-1X8 С1- CI- 1 М НС1 Rh, Ag, Zr, Cd Fe, U [1242] [585]]
Амберлит ХН-1002 NO3- 0,25 М HNO3 в 95%-ном метаноле (этаноле) Mg, Ca [735]
Амберлит А-29 NO3- Смесь HNO3 + KNO3 с органическими растворителями Be, Mg, Ca [226, 227]
АВ-17 NO3- NO3- 0,5 М HNOS в 95%-ном ацетоне или 85%-ном метаноле 0,5 М HNO3 в 90%-ном метилэтилкетоне Cs, Ce 234Th, U [148] [42]
Дауэкс-2Х8 ОН- Фильтрование раствора через колонку Zr, Ce, Ru (изотопы) [1271]]
Дауэкс-1Х8 он- СОЛ НСО3- SO42- Насыщ. раствор Са(ОН)2 0,02—0,32М (NH4)2CO3 Вода (вымывают Sr, Y на колонке) 50%-ный метанол Ca "Sr от MY 90у 90Y в элюате [934] [1046] [1096] [1381]
Дауэкс-1Х8 Этилендиа-минтетра-ацетатная 0,01 М, 0,1 М раствор ЭДТА, pH 11,2; 8,0 Са, Y [325]
Дауэкс-1Х8 2,6-пиридиндикар-боксилат- ная 0,1 М раствор 2,6-пи-ридиндикарбоксилата аммония, pH 7,1—7,6 Mg, В а [540]
Дауэкс-1 Х4 Нитрило-триацетатная 0,1 М раствор НТА, pH 6,1 Pb, Y,Bi (радиоизотопы) [1349]
Дауэкс-1 Цитратная 0,05 М цитрат аммония, pH 7,5 Ва, Са [1080]
менты. Анионит в бикарбонатной форме сорбирует как 90Sr, так и 90Y, однако стронций может быть десорбирован промыванием водой [1096]. На колонке, содержащей анионит в SOF-форме, 90Sr сорбируется из раствора, содержащего метанол и HNO3, 80Y при этом не удерживается [1381].
Ряд методов основан на применении растворов комплексов стронция с органическими кислотами. Са и Sr могут быть разделены после сорбции на анионите в этилендиаминтетраацетатной форме; этим же методом можно отделить 90Sr от 90Y. Используют
66
анионит АН-1, десорбируют] сначала 90Sr, а затем 90Y 0,01 М раствором ЭДТА с pH 8 [3251.
Сходным образом из растворов комплексов с нитрилотриуксус-ной кислотой (НТA) Sr можно отделить от Pb, Y и Bi. Смесь элементов сорбируют на колонке с ионообменником, насыщенным НТА, затем элюируют 0,1 М раствором НТА (pH 6,1) последовательно Sr, РЬ, Y, Bi [1349]. От других щелочноземельных металлов стронций отделяется при использовании растворов цитратных комплексов [1080]. По сорбируемости цитратные комплексы располагаются в последовательности: Be >• Mg > Са > Sr > > Ва^> Ва. Смесь хлоридов Ва, Са, Sr и Mg, поглощенная в верхней части колонки, анионит которой находится в цитратной форме, промывается 0,05 М раствором цитрата аммония с pH 7,5; Ва, Sr и Са последовательно переходят в элюат, Mg остается на колонке.
Для отделения стронция от других элементов применяют также и хелатообразующие ионообменные смолы, содержащие различные функциональные группы [434, 545, 557, 827, 963].
При использовании анионита в 2,6-пиридиндикарбоксилатной форме возможно отделение стронция от Mg и Ва [540]. При разделении смеси Ва—Sr барий вымывают 0,002 М раствором 2,6-пи-ридиндикарбоксилата аммония, a Sr — 0,1 М раствором того же реагента.
Применение целлюлозы для отделения Sr от Fe описано в [168, 169]; фосфат целлюлозы предложен для отделения щелочноземельных элементов от щелочных, редкоземельных элементов и алюминия [1219].
Аниониты нашли применение также для отделения стронция и других катионов от анионов кислот — фосфорной [800] и родизо-новой [824].
Отделение стронция от преобладающих количеств кальция рассмотрено в работе [735].
Анализируемую пробу, содержащую 0,0005—0,1 ммоля Sr и 0,5 ммоля Са, вводят в колонку (22 X 1 еле2), заполненную анионитом Амберлит XN-1002 с размером зерен 60—100 меш, предварительно промытую 40—50 мл 0,25 М HNO3 в 95%-ном СН3ОН. Элюирование производят таким же раствором HNO3 в СН3ОН, пропуская его со скоростью 0,5—0,6 мл!мин. Первые 10 мл, не содержащие определяемых катионов, отбрасывают и собирают 80— 100 мл (в зависимости от соотношения Са : Sr) Са-содержащей фракции. Sr, оставшийся на колонке, десорбируют 50 мл 95%-ного СН3ОН. Содержание Sr в растворе определяют комплексонометрическим либо спектрофотометрическим методом.
Применение неорганических ионообменных материалов
Для отделения стронция могут быть также использованы синтетические неорганические ионообменные материалы. Широкое распространение получили труднорастворимые соли фосфорной
3*
67
Таблица 23
Коэффициенты распределения и коэффициенты разделения Sr и Rb на фосфате циркония в зависимости от концентрации элюирующего раствора
Концентрация Kp(Rb) KP(Sr) Sr “Rb Kp(Rb) K'p(Sr) Sr “Rb Kp(Rb) KP(Sr) Sr “Rb
раствора,
М HC1 CH.COONHi NH4C1
0,01 24 2,4 0,1 1,1 49,0 44,6 6,1 2,6 0,43
0,1 7,4 з,з 0,45 1,1 10,1 9,2 0,72 0,19 0,26
0,5 0,96 — — 0,19 1,3 6,8 0,15 0,04 0,27
1,0 0,45 — .— 0,07 0,72 10,3 0,09 0,02 0,22
2,0 0,30 —— — 0,05 0,47 9,4 0,05 0,02 0,40
з,о 0,23 — — 0,04 0,32 8,0 0,04 0,02 0,50
4,0 0,07 — — 0,04 0,19 4,7 0,02 0,01 0,50
5,0 0,14 — — 0,03 0,14 4,7 0,01 0,01 1,0
6,0 0,07 — — 0,03 0,12 4,0 -— — —
кислоты — фосфат олова [584, 866], фосфат циркония [17, 145, 516, 1212], фосфоровольфраматы аммония и калия [946, 1014], фосфоромолибдат аммония [578, 580, 1259].
Исследование условий отделения микроколичеств Rb от макроколичеств Sr на фосфате циркония проведено в [145]. В табл.23 приведены значения коэффициентов распределения (А?Р) и разделения (оснь) Rb и Sr для растворов с различными электролитами. Целесообразным считается применение фосфата циркония для разделения образцов с высокой концентрацией радиоизотопов, поскольку этот катионит проявляет в данных условиях высокую устойчивость.
Сорбция стронция на силикагеле используется для его концентрирования при анализе вод [475].
Показана возможность разделения смеси Sr и Ва на колонках, содержащих арсенат олов a(IV) [1170] или окись алюминия [524], смеси Sr и La — на колонке из в о л ь-фрамата тория [1169]. Ферроцианид титана [966, 967] применяется для отделения Sr от других щелочноземельных элементов и Cs.
Разделение щелочных и щелочноземельных металлов на ионо-обменпиках типа гетерополикислот (молибдатах и вольфраматах циркония и титана), а также двуокиси титана изучалось в [231, 603, 656]. Окись циркония, содержащая молибдатные обменные группы, использовалась в [1307] для разделения Sr и Ва. Cd и Sr разделяют на колонке, заполненной вольфраматом циркония, элюируя первый из них 6 мл раствора (0,2 М NH4J + 0,1 М ацетат-
68
ный буферный раствор с pH 4,5 +20% ацетона), а стронций — 18 мл 1 М раствора NH4C1 [1213].
Разделение магния, кальция и стронция на кристаллах молибдата циркония описано в [603].
Раствор (5 мл), содержащий < 0,05 ммоля щелочноземельных элементов, пропускают со скоростью 0,75 мл!мин через колонку, заполненную свежеприготовленными кристаллами молибдата циркония, промытыми 10 мл 2 М NH4NO3 для повышения обменной емкости. Колонку промывают 10 мл воды (если анализируемый раствор содержит комплексон III, то колонку промывают 20 мл воды). Mg десорбируют с помощью 7 мл 0,14 М (NH4)2SO4 в 60%-ном метаноле, Са — 20 мл смеси (0,2 М по NH4NO3 и 0,005 М по HNO3), a Sr вымывают пропусканием 5 мл 1 М NH4NO3. В полученном элюате соответствующие катионы определяют комплексонометрическим методом.
Окислительно-восстановительный ионит, состоящий из гелеобразного оксигидрата циркония, насыщенного ди-, тионитом натрия, используют для отделения 90Sr от 106Ru [1373].
Активированный древесный уголь марки Б А У применяется для концентрирования микроколичеств стронция [413, 414]; на окисленном угле проводят очистку солей стронция от РЬ, Си и Fe [414].
Находят применение методы отделения стронция от других элементов посредством тонкослойной хроматографии на бумаге, содержащей ионообменник. Разделение Са, Sr и Ва проводят с применением катионообменной бумаги SA-2 [517], а отделение Sr от Cs — с применением анионообменной бумаги, содержащей Амберлит IRA-400 [843]. С помощью тонкослойной хроматографии на бумаге, содержащей гипофосфат циркония, Sr может быть отделен от Со, Ni и других элементов [933]. Бумага, пропитанная солями гетерополикислот — кремне-молибдатом пиридина и оксина [961], к р е м-невольфраматом аммония [1003], фосфороволь-фраматом аммония [1154], находит применение при отделении Sr от Cs и ряда других элементов.
Распределительная хроматография
Отделение стронция от других, в основном щелочноземельных элементов, в этом методе достигается за счет различия в коэффициентах распределения между неподвижной и подвижной фазами. Наиболее часто используется хроматография на бумаге, реже — тонкослойная хроматография и хроматография на колонках.
Хроматография на бумаге
Чаще всего используется одномерная восходящая хроматография, реже — нисходящая и двухмерная. Находит применение также метод радиальной хроматографии [523, 543, 1098, 1348].
69
В качестве подвижной фазы при распределительной хроматографии предлагались разнообразные смеси органических растворителей с водой, с минеральными и органическими кислотами, комплексообразующими веществами, солями щелочных металлов и др. Состав некоторых из используемых смесей приведен в табл. 24. Другие подвижные растворители описаны в [443, 682, 689, 872, 1072, 1291].
Для характеристики достигаемого разделения в методе распределительной хроматографии на бумаге пользуются величиной Rf, равной отношению расстояний, пройденных зоной металла и фронтом растворителя. В табл. 24 приведены значения Rf для Sr, других щелочноземельных элементов, а также Be и Mg, получаемые с наиболее часто используемыми растворителями.
Обычно значения R/ для Sr находятся между значениями Rf для Ва и Са. Значения RT для Mg и Be самые большие.
Влияние различных факторов на величину Rf для стронция — диэлектрической проницаемости, подвижности растворителя, рода и концентрации кислот изучено в работах [1168, 1260].
Для проявления зон стронция на хроматограмме используют ряд реагентов: родизонат натрия или калия [158, 180, 630, 747, 748, 962, 988, 1019, 1048, 1049, 1142, 1291], 8-о к си-хин о л и н (оксин) [650, 1098, 1146, 1260, 1348] или его смесь с койевой кислотой (1г койевой кислоты -Ь 5 г оксина в 1 л 60%-ного этанола) [543, 701] или аналог оксина— 4-о к с и-бензтиазол [701] (зти реактивы вызывают желтую окраску, а под действием ультрафиолетовых лучей пятна флуоресцируют желто-зеленым или светло-голубым цветом). Используют также пирогаллол (0,3 г пирогаллола растворяют при нагревании до кипения в 10 мл этанола с добавлением 1—2 мл конц. NH4OH) [396, 701], кверцетин (в 60%-ном этаноле) [1303], бромкрезоловый зеленый (в 20%-ном этаноле, нейтрализованном NaOH) [ИЗО], виолуровую [706] и хлораниловую [528] кислоты. При хроматографировании хлоридов зоны можно локализовать раствором нитрата серебра [443, 484, 527, 1321] с добавкой 2,6- дихлорфе-нолиндофенола и этанола (бледно-розовые пятна, становящиеся на свету коричневыми) [527]. После отмывания избытка AgNO3 пятна могут быть обработаны также сульфидом аммония [443, 484].
Для количественного определения стронция можно сравнить величину пятен на хроматограмме с величиной стандартных пятен, получаемых аналогичным образом [484, 1048, 1049]. Предложена [206] следующая эмпирическая формула, связывающая концентрацию металла с площадью пятна: S = a\g с Ь.
Интенсивность пятен может быть определена при визуальном сравнении со шкалой [1165] или с помощью фотометра (пятно ро-дизоната стронция фотометрируют при 410 нм) [689]. Техника количественной хроматографии на бумаге описана в [473].
70
Таблица 24
Разделение Be, Mg, (Ja, Sr и Ba методом распределительной хроматографии на бумаге
Подвижный растворитель К^-100 Литература
ве Mg Са 8Г Ва
Метанол — конц. НС1 — Н,0 (80 : 10 : 10) Метанол—конц. НС1 (95 : 5) — роданид аммония (0,5 вес. %) 85 70 47 26 11 [630, 1260]
— — 77 52 26 [1165]
Метанол—СН3С00Н (98 : 2) 88 74 66 52 39 [ИЗО]
Метанол—этанол (50 : 50) — 80 62 49 28 [527]
Метанол—н-бутанол — коллидин—6 N СН3СООН (40 : 20: : 20 : 20) — 71—81 64—71 30—47 11—26 [988]
Метанол—изобутанол— СН3СООН-Н2О (51 : 30 : 2 : : 15)—формиат натрия (2,5 вес. %) — 75 60 45 30 [1142[
Метанол—изопропанол—муравьиная кислота (50 : 30 : 20) 77 59 45 31 21 [650]
Метанол —тетрагидрофуран— 8.V НС1 (20 : 10 : 70) — 8-ок-сихинолин (0,1 вес. %) — 85 72 60 44 [1348]
Этанол—2 N HNO3 (2 : 1) 81 60 45 31 16 [1075]
Этанол—15—25% Н2О—2% LiCl (LiBr, LiJ, LiNO3) — 77 62 36 16 [396]
Этанол (96%)—2.V CH3COOH (80 : 20) 86 76 б8 55 43 [706[
Этанол—вода—пропионовая кислота—конц. NH.OH (84 : 8.4: 4) — 93 82 56 23 [10491
Этанол—пиридин—1,5 N СН3СООН (40 : 40 : 20) — 72—80 46—50 23—25 7—9 [158]
Бутанол—6 А НС1 (50 : 50) — 60 50 20 10 [1291]
Ацетон—21% НС1—Н2О (70 : : 10 : 20) — 55 31 18 0,0 [748]
Тетрагидрофуран—30% 1,2— 3,7 N НС1 или 70% 7,4—12 N НС1 — 70 36 21 2 [811]
Коллидин — 0,4 .V HNO3 (50 : 50) — 65 52 40 26 [701]
Отрезок бумаги, содержащий пятно стронция, можно выщелочить 2N НС1 и в полученном растворе определить количество металла спектрофотометрически с помощью о-крезолфталеинкомп-лексона [1142], а также объемным комплексонометрическим методом [176].
71
Метод бумажной хроматографии использовался при определении стронция в летучих продуктах, образующихся на оксидном катоде [689], при анализе облученного урана [653], природных вод [1165], «золы Бикини», выпавшей 1.III. 1954 г. при испытании водородной бомбы [1072].
Приводим описание одного из методов отделения Sr от Be, Mg, Са и Ва [988] с использованием нисходящей хроматографии.
На полоску фильтровальной бумаги размером 15 X 60 см наносят 0,01 мл исследуемого раствора, высушивают на воздухе и разделяют зоны в течение 16—18 час. смесью метанола, н-бутанола, н-коллидина и уксусной кислоты (см. табл. 24). После перемещения подвижного растворителя на расстояние 40—50 см полоску высушивают и проявляют пятна Sr и Ва раствором родизоната натрия.
Находит применение и хроматография на целлюлозной колонке, например, для отделении радиобария от радиостронция [723], U от Sr [1067] и т. д. Показано, что разделение Mg, Са и Sr на колонках различной длины, заполненных микрокристаллической целлюлозой, может быть успешно проведено с использованием растворителя, содержащего метанол, соляную кислоту и воду в различной комбинации (ошибка опыта не более 4,3%). Элементы вымываются в порядке возрастания их атомных номеров. Метод может быть использован при нейтронно-активационном определении щелочноземельных элементов [733].
Тонкослойная хроматография
Техника работы по этому методу описана в [25, 473]. Для получения тонкого активного слоя на пластинку из стекла или на пленку из полимерного материала наносят силикагель, целлюлозу, крахмал, окись алюминия или другие вещества.
В табл. 25 приведены используемые сорбенты, растворители и значения Rf при разделении щелочноземельных элементов, Be и Mg методом тонкослойной хроматографии.
Как и в хроматографии на бумаге, для качественного и количественного определения стронция используют его цветные и люминесцентные реакции, упомянутые выше.
Разделение Са, Sr и Ва на] слое силикагеля, закрепленном крахмалом, описано в [25].
Наносят по 0,001 мл растворов ацетатов разделяемых металлов, содержащих ледяную уксусную кислоту, и для сравнения такие же количества растворов Ga, Sr и Ва в отдельности. Растворитель — смесь: этанол—пропанол—ледяная уксусная кислота—ацетилацетон—вода (37,5 : 37,5 : 5 : 1 : : 20). Хроматограмму обрызгивают 1,5%-ным раствором виолуровой кислоты и сушат при 100° С в течение 20 мин. Сравнивая с положением зон Са, Sr и Ва в отдельности, находят на хроматограмме смеси элементов зоны разделяемых катионов.
72
Таблица 25
Разделение Be, Mg, Са, Sr и Ва методом тонкослойной хроматографии
Сорбент Растворитель (в скобках объемн. %) Вг100 Литература
Be Mg Са »г в.
Целлюлоза Диоксан—конц. НС1—Н2О (58 : 12 : 30) 95 77 50 30 12 [742]
Метанол—конц. НС1—Н2О (73 : 12 : 15) 86 67 48 32 20 [742]
Метанол—конц. НС1—Н2О (80 : 10 : 10) — 75 50 27 10 [726]
Микрокристаллическая Метанол - 10А НС1-Н2О (80 : 12 : 8) — 80 55 35 15 [579]
пеллюлоза Изопропанол—Н20—1М НС1 (40 : 40 : 20) Коллидин—0,4 М HNO3 (50 : : 50) — — 73 66 55 [1383].
— — 64 56 46 [1383]
Окись алюминия Ацетон-4М НС1 (92 : 8) — 70 — 40 00 [814]
Силикагель 0,01 М НС1 —— — —. 53 55 [960]
Крахмал Ацетон-ЗМ НС1 (50 : 50) — 50 26 15 7 [604]
Хроматография с обращенными фазами
В этом методе органическая фаза закреплена на носителе, а подвижной фазой служат водные растворы солей или кислот. Описание техники работы и особенности метода приведены в [192].
Отделение стронция производят как на гидрофобизированной бумаге (при восходящем и нисходящем вариантах), так и на колонках при использовании в качестве наполнителей различных веществ —гидрофобизированных порошков целлюлозы [616], силикагеля [877], полихлортрифторэтилена [968, 1098], тефлона [922, 1034] и других веществ. В качестве органической фазы используют трибутилфосфат [441, 922], дибутилфосфорную кислоту в хлороформе [655], ди-(2-этилгексилфосфинил)метан [1098], три-октиламин [1034], триоктилфосфиноксид в циклогексане [615], теноилтрифторацетон в метилизобутилкетоне [891] и др.
В ряде работ представлены прописи для отделения стронция от щелочных и других щелочноземельных элементов [441, 612, 922, 968], Zn, Cd, Fe, Al, Tl, U [613, 1034], Y [192, 615, 1098]. В качестве примера на рис. 9 показано положение зон Sr и некоторых других элементов [613] при использовании неподвижного органического растворителя 0,1 М раствора ди-(2-этилгексил)фос-
73
2
0
©
0
5r
8a
Рис. 9. Положение зон Sr и некоторых других элементов при хроматографировании с обращенными фазами на гидрофобизпрованной бумаге
Подвижный растворитель 7,5 (7) и 8,0 (2) М НСГ, неподвижный растворитель 0,1 М ДЭГФК в циклогексаноне
форной кислоты в циклогексаноне и раствора соляной кислоты как подвижного растворителя.
Разделение щелочноземельных металлов на колонке с ди-(2-этилгексил) фосфорной кислотой описано в работе [877].
Колонку диаметром 0,3 см, высотой 13 см, заполненную гидрофобизированным силикагелем (размер зерен 0,032—0,058 мм), содержащим 13% ДЭГФК, предварительно приводят в равновесие с водным раствором, имеющим pH 3,0 или 3,2, и добавляют 0,05 мл раствора анализируемой смеси. Последовательно элюируют Ва 10 мл 0,5 М раствора NaNO3 с pH 3,0, Sr — 5 мл того же раствора с pH 2,0 и Са — 2,5 .« раствора с pH 0,8. Метод позволяет отделить от Са и Ва 90Sr без носителя.
ЭЛЕКТРОФОРЕЗ
Метод основан на различной подвижности ионов стронция и отделяемых
от него ионов других металлов под воздействием электрического поля (табл. 26).
Техника работы и используемая аппаратура подробно представлены в [482]. В качестве носителя применяется чаще всего бумага (реже — порошкообразные наполнители, например, силикагель и т. д.).
На полоску фильтровальной бумаги, смоченной электролитом, со стороны, близкой к аноду, наносят разделяемые соли металлов. Целесообразно поместить разделяемые соли в виде полосы, перпендикулярной к направлению прохождения тока. Для этого
Таблица 26
Подвижность ионов (U) при разных условиях проведения эксперимента [788]
Ион СТ* 10s, см2* в-1 •сек-'1 Ион
растворитель 0,75 М муравьиная кислота; pH 2,0; и == ЮО в/см растворитель 0,1 М карбонат аммония; pH 8,9; v == 80 в/см растворитель 0,75 М муравьиная кислота; pH 2,0; v =, 100 в /с«м растворитель 0,1 М карбонат аммония; pH 8,9; •у = 80 в
Ве2+ 17,2 0,0 Sr2+ 25,94 15,83
Mg2+. Са2+ 24,6 29,06 15,62 13,64 Ва2+ 27,6 16,45
Примечание. Бумага Ватман № 3 ММ, t = 11° С; v — градиент напряжения.
74
пропитывают раствором солей узкую полоску фильтровальной бумаги или хлопчатобумажную нить, которую прижимают к требуемому месту бумаги. Концы бумаги опускают в кюветы, содержащие электролит, или же присоединяют к графитовым электродам и накрывают сверху стеклянной пластинкой (во избежание испарения электролита). Лист бумаги, на котором проводят разделение, находится на холодильнике, выполненном в виде плоской коробки, через которую пропускается вода для охлаждения. К электродам, прикрепленным на бумаге или находящимся в кюветах с электролитом, подводят напряжение постоянного тока и проводят электрофорез. Величина напряжения зависит от расстояния между электродами, рода электролита и других факторов и составляет в различных работах от 200 в до 4,0 кв, падение напряжения 3,5—100 в!см, сила тока 1—16 ма на 1 см ширины полоски бумаги; продолжительность электрофореза зависит от градиента напряжения на 1 см длины полоски.
Местонахождение зоны элемента после электрофореза определяют с помощью реагентов или (в случае разделения радиоизотопов) радиометрическим методом.
В табл. 27 приведены некоторые растворители, используемые для отделения стронция от других металлов методом электрофореза. Ряд систем можно найти в [482].
В качестве примера ниже описано разделение смесей Sr—Ga и Sr—Ва [211].
На середину полоски хроматографической бумаги Ватман (например, № 3) длиной 58 см и шириной 2,5 см, пропитанную 0,05 М раствором лимонной кислоты с pH 8, наносят 0,005—0,01 мл раствора цитратных комплексов элементов с концентрацией 0,5 мг/мл. После проведения разделения (8,5 в!см, 30 мин.) ионы идентифицируют с помощью раствора фенола М; Sr мигрирует к аноду на 1 см, Са и Ва на 2,9 и 2,1 см соответственно.
При непрерывном электрофорезе раствор смеси солей поступает в разделительную камеру, заполненную кварцевым песком, в которой перпендикулярно движению электролита прикладывается электрическое поле и каждый ион смеси образует собственную дорожку. Наклон последней по отношению к линии старта определяется подвижностью ионов, градиентом потенциала и скоростью движения раствора. На выходе из установки отбираются индивидуальные компоненты.
При использовании в качестве электролита 0,05 %-ного раствора комплексона III успешно разделяются смеси Са и Sr, содержащие эти элементы в соотношениях от 100 : 1 до 100 000 : 1 [462]. Наилучший эффект разделения наблюдается при pH 4,8. Для подавления сорбции Sr на кварцевом песке в электролит добавляют носитель Sr—Ва в количестве 1 мкг/л. При соотношении Sr : Са ~ 1 : 5700 выход Са составляет 99, a Sr 73% [462].
Для разделения катионов Li+, К.+, Na+, Са2+, Sr2+ и Ва2+ рекомендован [856] быстрый электрофоретический метод с не-
75
Таблица {27
Отделение стронция от других элементов методом электрофореза
Разделяемые элементы Условия электрофореза Литература
Са, Sr HPO3; pH 1,5 [251]
Sr, Ва 0,1 M HC1O4; 100 в; 8 час. [1167]
Mg, Са, Sr 0,1 М NH4C1 [997]
Са, Sr или Mg, Sr 0,1 М HNO3 [997]
Sr, Ba 0,5 М НС1; 500 в; 8 мин. [482]
UOf, Sr 0,5 М (СН3СООН; pH |3,56—4,05; 900 в [1001]
Ca, Sr, Ba, Cs, Ce, La, Y, Zr Раствор 10 объемн. % пиридина и 1 объемн. % СН3СООН; pH 6,0 [1123]
Sr, Ba Метанол—ацетон—НС1 в различных соотношениях [251]
Li, Na, K, Ca, Sr, Ba 0,8 М а-оксиизомасляная кислота, 0,05%-ная по СН3СООН; 62 в/см (непрерывное разделение) [856]
K, Rb, Cs, Li, Na, Mg, 0,007—0,15 М лимонная кислота; pH 5,5-8,0 или5М NH4OH,8,5 — [211, 405,
Ca, Sr, Ba, Fe, Co, Ni, 519, 788,
Pb, Zn, Y, Cr, Mn, Al, Cd, Bi, Sn, Sb, As, Hg, Ag, Cu 160 в[см', от 9 мин. до 8 час. 1073, 1167]
Sr (Ca, Ba), Li, Na, K, Rb, Cs 0,05 М ди- или триаммонийцитрат (pH 5,2 и 5,9 соответственно); 5 в/см\ 3 часа [708]
Li, Na, K, Rb, Cs, Ca, 0,005 М насыщ. раствор ЭДТА, [462, 519,
Sr, Ba, Y, Sm, Pr, Ce, La, Nd либо в смеси с NH4OH; pH 1,7—10; 5—120 в!см\ 10—72 мин. 677, 708]
Rb, Y, Zr, Nb, Mo, Mg, Ca, Sr, Ba 0,05 М N-2-оксиэтилиминодиуксус-ная кислота; pH 5,5—10; 15 е/см', 1 час [886]
прерывным потоком буферного раствора (0,005 %-пая СН3СООН) со скоростью 1—2 мл/мин по вертикально укрепленной полоске бумаги, к которой подведено напряжение, а проба нанесена у верхнего конца бумажной полосы. Электромиграционные зоны, пройдя бумагу, поочередно элюируются. Метод применен для анализа растворимых солей в строительных материалах.
Разделение может быть проведено с помощью колонки из бумажных дисков, к концам которых прикладываются электроды [155].
В методе фокусирующего ионного обмена используется противоток комплексных анионов и простых катионов разделяемых металлов в электрическом поле (рис. 10). Поскольку устойчивость комплексных соединений различных металлов различна, каждый
76
Рис. 10. Схема установки для проведения разделения методом фокусирующего ионного обмена
1 — бумажная полоска;
2 — анолит;
<3 — католит;
4 — зона разделения
Рпс. 11. Электрофореограмма разделения радиоизотопов Sr, Ва и их дочерних изотопов (Y и La)
о) Sr : Ва = 1 : 1; б) Sr : Ва = 50 : 1
из них будет концентрироваться (фокусироваться) на бумаге в отдельной узкой зоне, положение которой определяется градиентом концентрации комплексообразующего вещества и константой устойчивости комплексного соединения.
В качестве католита обычно используют растворы ЭДТА [460, 1048] (иногда содержащие 0,5 М NaOH [1229] или ацетат натрия с pH 5 [460]) или смесь ЭДТА с лимонной кислотой [463] (концентрация ЭДТА 0,02—0,1 М, лимонной кислоты 0,03—0,05 М). Используется также нитрилотриуксусная кислота (0,03—0,3 М, pH 5—7) [464, 482, 1114]. В качестве анолита обычно применяется соляная кислота с концентрацией 0,03—1 М или буферный раствор, например, гликолевый буфер с pH 2,9—3,5 [460]. Напряжение при проведении электрофореза выбирается 20—50 в/см, время разделения 5—15 мин.
На рис. 11 в качестве примера приведена электрофореграмма, полученная при разделении радиоизотопов 90Sr, 140Ва, и дочерних изотопов 90Y, 140La [460].
Иногда метод электрофореза используется в комбинации с методом распределительной хроматографии [987, 1233]. Электро-хроматофорез используется также в комбинации с разделением на ионитах [430].
77
ЭЛЕКТРОДИАЛИЗ
Выделение стронция электродиализом применяется часто для разделения радиоизотопов 90Sr — 90Y [775, 776, 787, 989], разделения Cs и Sr [832, 1355], выделения 90Sr из почв [783] и т. д.
При электродиализе в приборе с пятью анионитными мембранами с применением 0,001 М раствора ЭДТА (pH 5,5) ионы Y3+ образуют анионный комплекс, а ионы Sr2+ остаются в свободном состоянии. Коэффициент очистки "Y составляет 106 [787].
При электродиализе раствора, содержащего ЭДТА и NaOH (pH 11), Cs остается в виде катиона, Sr образует анионный комплекс.
90Sr можно выделить из почв, помещая 5—10 а пробы в мембранный мешочек, расположенный между двумя электродами, соединенными с источником постоянного тока с разностью напряжения 100 в. Катионы, перешедшие в диализат из почвы, затем подвергают дальнейшему разделению [783].
ЭЛЕКТРОЛИЗ
Электролитический метод отделения больших количеств Ва от малых количеств Sr (Ва : Sr = 500 : 1) путем восстановления Ва на ртутном катоде при контролируемом потенциале (—2,03 в) разработан в [971]. Из смеси Sr и Zn последний выделяют на никелевом катоде (pH 4,0—4,2) при плотности тока 0,01—0,02 а/см1. Время разделения 90 мин. [146].
Для качественного и количественного определения стронция после его отделения от других элементов методами, основанными на использовании электрического тока, применяют радиохимический метод, а также методы полярографии [971], фото- [1233] или рентгене- [1363] флуоресценции, фотометрии пламени [708] и др.
Г лава IV
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СТРОНЦИЯ
ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Гравиметрические методы
Гравиметрические методы применяются для определения стронция в большинстве случаев после его отделения от других щелочноземельных элементов, однако некоторые из них могут быть применены в присутствии последних. Сводка этих методов анализа приведена в табл. 28.
В работе [1117] термогравиметрически изучены весовые формы соединений стронция. Полученные кривые пиролиза представлены на рис. 12.
Таблица 28
Гравиметрические методы определения стронция
Выделяемое соединение Весовая форма t°, C Литература
Сульфат SrSO4 500 [95, 415, 703, 1087, 1299, 1370'
Карбонат SrCO3 600—950 [918, 1201]
Оксалат SrCoOi 200—400 [359]
Хромат SrCrO4 100—400 [1201j
Фторид SrF2 50—95*0 [1110]
Иодат Sr(JOs)2 157—600 [1110]
Перйодат Sr6(JOe)2 748—880 [1110]
Пироарсенат Sr2As2O7 412 [1110]
Тартрат SrCO3 810 [1201]
Тетрафторборат Sr(BF4)2 140—150 [360]
Пикролонат Sr(C10H7N4O6)-8H2O 20—25 [317, 1118, 1234]
Эмбелат Sr C 17H24O4. 110—115 [547]
5-Нитробарбитурат Sr(C4H2O6N3)2 160—180 [765]
Броманилат SrC6O4Br2 110 [801]
79
Рис. 12. Кривые пиролиза соединений стронция
1 — сульфат; 2 — карбонат; з — хромат; 4 — оксалат; 5 — тартрат. Цифры у кривых — темпе-ратура(°С)
/ ' Определение стронция в виде сульфата
Метод применяют после отделения Sr от Са и Ва [1117].
Раствор хлорида стронция должен быть по возможности нейтральным, к нему прибавляют 10-кратный избыток разб. H2SO4 и этанол в объеме, равном объему раствора. Осадок оставляют на 12 час., фильтруют, промывают 50%-ным этанолом, содержащим немного H2SO4, затем — 95%-ным этанолом до удаления кислоты. Взвешивание осадка рекомендуется производить после высушивания при температуре 100—300° С. При более высоких температурах наблюдается разложение сульфата стронция с выделением SO3. Формулы для пересчета: SrSO4-0,477 = Sr; SrS04- 0,563 = SrO.
В [1299] предложено осаждение сульфата стронция в присутствии комплексона III. Метод позволяет производить количественный анализ
смеси Sr, Са и Ва без ее предварительного разделения.
Смесь карбонатов Са, Sr и Ва растворяют в небольшом количестве разб. НС1 и доводят объем водой до 100—250 мл.
Аликвотную часть раствора, содержащую не более 200 мг окисей щелочноземельных элементов, разбавляют водой до 50 мл, прибавляют комплексон III (из расчета 800 мг на каждые 100 мг смеси окисей), конц. NH4OH до сильного запаха, 10 мл 10%-ного раствора (NH4)2SO4 и нагревают на водяной бане до исчезновения запаха аммиака. При pH ~ 8 из раствора выпадает BaSO4.
Через 3 часа осадок отфильтровывают, промывают 0,1%-ным раствором (NH4)2SO4 и прокаливают при ~800° С. Переосаждают BaSO4, для чего к остатку после прокаливания прибавляют комплексон III (5—10 мг на 1 мг остатка), 20 мл 10%-ного раствора NH4OH, нагревают до растворения, разбавляют водой до ~50 мл, прибавляют 10 мл 10%-ного раствора (NH4)2SO4 и снова осаждают BaSO4, как указано выше.
Оба фильтрата объединяют, упаривают до объема 40 мл, прибавляют 1 каплю раствора метилового красного и раствор NH4OH до перехода красной окраски в желтую. Затем прибавляют 1 N H2SO4 до обратного перехода желтой окраски в красную, вводят 10 мл буферного раствора с pH 4,5 (250 мл СН3СООН + 100 мл конц. NH40H в 1 л) а 50 мл 96%-ного этанола. Через 24 часа отфильтровывают выделившийся SrSO4, промывают 5—6 раз 0,1 %-ным раствором (НН4)гЯ04 в 50%-ном этаноле и прокаливают при ~700° С. Переосаждают SrSO4, растворяя остаток при нагревании с комплексоном III (5—10 мг на 1 мг остатка) и 20 мл 10%-ного раствора NH4OH. К раствору
80
прибавляют H2SO4 до кислой реакции по метиловому красному, 5 мл буферного раствора (pH 4,5), 5 мл 10%-ного раствора (NH4)2SO4, разбавляют водой до —25 мл а добавляют равный объем этанола. Выделившийся SrSO^ отфильтровывают, прокаливают и взвешивают. Метод используется при анализе рассолов.
Спектрохимическое изучение чистоты осажденного SrSO4, полученного в ходе анализа после отделения от Ва и Са, проведено в работе [1370]. Найдено, что со SrSO4 соосаждается <0,01—0,02 % Сг (если Ва предварительно выделялся в виде хромата), а также Na и другие катионы в количестве 0,01—0,58%. Потери вследствие растворимости сульфата стронция составляют от 0,1 до 2,35%.
Осаждение стронция при использовании метода возникающих реактивов может быть произведено с помощью диметил с_ у л’ь-ф а т а в присутствии метанола [703] и диоксана [415].
Нейтральный раствор, содержащий от 20 до 80 мг Sr в виде хлорида, раз-, бавляют водой до 25 мл, прибавляют 50 мл диоксана, нагревают до 60—70° С. и при помешивании вводят 5 мл диметилсульфата. Через несколько минут выделяется кристаллический осадок, смесь нагревают на водяной бане 15— 20 мин., после охлаждения осадок отфильтровывают через плотный фильтр и промывают на фильтре 3—4 раза диоксаном (по 10—12 мл). Фильтр с осадком сушат в тигле и прокаливают до постоянного веса; взвешивают в виде SrS04 [415].
Определение стронция в виде карбоната
При определении стронция в виде карбоната осаждение проводят карбонатом аммония в присутствии аммиака. Осадок отфильтровывают, промывают водой, содержащей карбонат аммония и аммиак, прокаливают при темно-красном калении. Взвешивают в виде карбоната.
По данным [1117], карбонат стронция до температуры 400° С не сохраняет постоянного веса, в интервале температур 410— 950° С он устойчив.
Если предварительно перед осаждением карбоната было проведено осаждение Ва в виде хромата, то осадок карбоната будет содержать хромовую кислоту. В этом случае осадок растворяют в НС1 и производят осаждение сульфата [1201].
Взвешивание карбоната и полученной из него окиси может быть использовано для косвенного определения Sr в бинарной смеси с Са или Ва.
Осаждают смесь карбонатов из раствора, отфильтровывают, фильтр высушивают и сжигают, нагревают остаток с (NH4)2CO3, последний удаляют при нагревании. Взвешивают карбонаты. Для получения веса окислов карбонаты вносят во взвешенный тигель с расплавленной бурой и после удаления СОа по привесу определяют вес окислов. Содержание компонентов рассчитывают по специальным формулам [1201].
81
Определение стронция в виде оксалата
Стронций практически полностью осаждается оксалатом аммония из раствора, содержащего 20 % этанола. Полученный осадок прокаливают и взвешивают.
Термогравиметрическое изучение [1117] показало, что оксалат стронция начинает обезвоживаться при 41° С, при 177° С теряет молекулу воды. Безводный оксалат может быть взвешен после прокаливания при температуре 177—400° С. Разложение оксалата происходит при температуре от 400 до 540° С. При дальнейшем увеличении температуры до 950° С вес осадка остается постоянным; Sr находится в форме карбоната.
Расчетный метод определения Sr и Са (без предварительного их разделения), основанный на осаждении оксалатов и переведении их в окислы Sr и Са, описан в [359].
Анализируемую пробу сплавляют с содой, плав растворяют в 10%-ной НС1. К двум аликвотным порциям прибавляют 10 мл 10%-ной СН3СООН и 25—30 мл 0,2 N Н2С2О4, разбавляют водой до 70—80 мл и осаждают аммиаком оксалаты Sr и Са. Для полноты осаждения SrC2O4 к раствору прибавляют 40—50 мл этанола. Через 12 час. осадки отфильтровывают и промывают 50%-ным этанолом; 1-й осадок прокаливают при 1000° С до SrO и СаО; 2-й сушат для удаления этанола при 60—70° С, прибавляют 60—70 мл воды, 15—20 мл 10%-ной H-2SO4, нагревают до 80° С и титруют ионы С,О4“ 0,1 N раствором КМпО4.
Обозначая вес СаО через С, вес SrO через S, вес СаО + SrO, определенный после прокаливания, через Р, количество 0,1 У Н2С2О4, израсходованное на'осаждение СаС2О4 и SrC2O4, через А, и, принимая во внимание мол. массы СаО, SrO и Н2С2О4, вычисляют количество СаО по уравнению С = 0,00611А — 1,1794Р, а количество SrO — по уравнению S — Р — С. При малом содержании одного из элементов к пробе добавляют известное количество этого элемента. Метод применим для определения Sr и Са в известняках, гипсе и др.
Определение стронция в виде тартрата и других соединений
Осаждение стронция в виде тартрата производят из уксуснокислого раствора 10%-ным раствором винной кислоты.
Согласно [1117], при нагревании до 200° С не наблюдается полного удаления воды из осажденного моногидрата, при более высокой температуре наблюдается разложение остатка винной кислоты. Постоянный вес осадка достигается при прокаливании (Г = 810° С) вследствие превращения тартрата в карбонат.
Менее распространенными являются методы определения стронция в виде хромата [1201] итетрафторбората [360].
82
Анализируемые растворы в последнем случае дважды выпаривают с 2 лл 35%-ной HBF4. Чтобы перевести остаток в весовунУ форму, его высушивают до постоянного веса при 140—150° С.
Для гравиметрического определения стронция пригодны также осадки его пикролоната [317, 1118] иэмбелата [547].
Аликвотную часть анализируемого раствора разбавляют до 100 мл, с помощью ацетатного буферного раствора устанавливают pH 6,5—7,0, нагревают до 60° С и приливают избыток 1%-ного раствора эмбелина в горячем этаноле. Смесь выдерживают на паровой бане 30 мин. Выпавший бледнофиолетового цвета осадок отфильтровывают, промывают водой и этанолом, сушат 1,5 часа при 110—115° С и взвешивают в виде соли SrC17H24O4.
Возможность гравиметрического определения Sr, Са и Ва после осаждения их в виде солей 5-н итробарбиту-р о в ой кислоты исследована в [765]. Авторами установлено, что Sr количественно осаждается 0,05 М раствором кислоты в 50 %-ном этаноле. Выделенный осадок высушивают при 160— 180° С и взвешивают.
Титрииетрические методы
Титриметрическое определение стронция может быть произведено после его отделения от всех или большинства мешающих элементов. Наибольшее распространение нашел комплексонометрический метод [35Ц. Менее распространены методы, основанные на титровании аниона, связанного со стронцием, в том случае, если стронций предварительно выделен в виде соединения определенного состава. Сводка методов приведена в табл. 29.
Гидроокись стронция может быть определена путем титрования соляной, азотной, щавелевой или серной кислотами при добавлении фенолфталеина в качестве индикатора. При использовании того же индикатора нейтральный раствор хлорида стронция, нагретый до кипения, может быть оттитрован хроматом калия или аммония.
Прямое титрование Sr в водно-ацетоновом растворе раствором оксалата натрия использовано в [785]. В качестве индикатора применяют несколько капель 0,1 N раствора КМпО4. В точке эквивалентности окраска раствора исчезает вследствие восстановления перманганата.
Используется ряд методов косвенного определения стронция. При определении стронция в карбонате навеску последнего растворяют в избытке соляной кислоты, избыток кислоты оттитровы-вают карбонатом натрия, по результатам титрования находят содержание стронция [1201]. Такой же принцип расчета содержания металла положен в основу ряда других косвенных методов определения стронция. Он используется и в том случае, когда стронций выделяется в виде осадка, например, фосфата [942]. Избыток реагента оттитровывают солью цинка.
83
Т’а блица 29
Титриметрические методы определения стронция
Метод Анализируемое соединение или реагент Индикатор Литература
Алкалиметрический Sr(OH)2, SrCO3 Фенолфталеин, метилоранж [1201]
Перманганатометрический SrC2O4 или SrCrO4 (после восстановления хрома избытком (NH4)2.[Fe(SO4)2] КМпО4 [1201]
Арсенатно-иодометрический Sr3(AsO4)2 J2 [455]
Осаждения Na2C2O4 КМпО4 [785]
SrHPO4 — Г942]
Na2SO4 Вариаминовый синий [833]
Комплексономет- Комплексон Ш Эриохром черный Т, [39, 114, 120,
рический метилтимоловый синий, фталеинкомп лексон и др. 351, 439,824, 850, 1208, 1298]
Спектрофотомет- Комплексон III Эриохром черный Т, [734, 818,
рический арсеназо I 1108]
Na2SO4 Нитхромазо [213]
Потенциометрический Циклогександиамин-тетрауксусная кислота (или комплексон III)+Hg(NO3)2 — [900-902]
Комплексон Ш Индикаторные Ag-или МпО2-электроды [729, 1326]
Гидроокись тетра этиламмония — [214]
Na2SO4 — [813]
Кондуктометри- Лимонная кислота — [567]
ческий НС1 — [437]
Амперометрический Н2С2О4 — [53]
Спектрополяриметрический D-(—}-транс-1,2-циклогександиамин-тетрауксусная кислота [520]
Стронций может быть определен по результатам титрования оксалат-иона, выделившегося в осадок вместе со стронцием [1201]. Титрант — перманганат калия. Титрование проводят в присутствии H2SO4 или НС1, содержащей соль марганца.
Арсенатно-иодометрический метод предусматривает осаждение стронция в виде арсената, растворение осадка в НС1, добавление к раствору бензола, KJ и титрование тиосульфатом нат
84
р и я выделившегося иода. Посторонние элементы (Fe, Al, Ti) маскируют лимонной кислотой.
К анализируемому раствору прибавляют несколько капель конц. HNO3, нагревают несколько минут и после охлаждения до комнатной температуры прибавляют 10 мл 10%-ного раствора лимонной кислоты, 20 мл 0,5 М раствора арсената натрия или аммония, раствор разбавляют до ~70 мл и производят осаждение арсената стронция добавлением конц. NH4OH. Затем прибавляют избыток конц. NH4OH (25—30 мл) и отфильтровывают осадок. Его промывают 3%-ным раствором NH4OH, помещают вместе с фильтром в коническую колбу и растворяют в 25—30 мл НС1 (1 : 1). К раствору прибавляют 25 мл бензола, 3 мл 2 A K.J, взбалтывают 20—30 сек., добавляют 20 мл воды и титруют раствором Na2S2O3 до обесцвечивания слоя бензола. 1 ли 0,1 N Na2S2O3 соответствует 0,004381 г Sr [455].
Наиболее широкое применение нашел метод комплексонометрического титрования с использованием реагента комплексо-н а III. Титрование Sr проводят в щелочной среде, при этом в обычных условиях титрования может выпасть осадок труднорастворимого карбоната, вследствие чего определение почти всегда проводят по методу вытеснения или по методу обратного титрования. Для индикации точки эквивалентности при комплексонометрическом определении Sr используют эриохром черный Т [351], метилтимоловый синий [39, 114, 120, 439], флуорексон, фталеинкомплексон (металлфталеин) [1208], эриохромазурол S, тимолфталексон, кальцеин, метилкальцеин, хром фиолетовый для меха, хром прочный синий RLL, кислотный ализариновый черный SN, 4-(2-пиридилазо)резорцин, 1-(2-пиридилазо)-2-наф-тол, родизоновую кислоту, умбелликомплексон, люминол [1298].
Сравнительное изучение пригодности индикаторов метилтимолового синего, арсеназо М и карбоксиарсеназо показало, что лучшим из них является последний [337а].
При определении стронция по методу вытеснения с применением комплексоната магния в присутствии эриохром черного Т получается хороший переход окраски [351].
Анализируемый раствор соли стронция нейтрализуют едким натром и на каждые 100 мл раствора прибавляют 10 мл 0,1 N раствора комплексоната Mg и 2 ж аммиачного буферного раствора (54 a'NH4Cl и 350 мл конц. NH4OH в 1 л воды). Вытесненный магний" титруют комплексоном III с эриохром черным Т до перехода окраски от красной к чисто синей. 1 мл 0,01 М раствора комплексона III соответствует 0,8763 мг Sr [351].
Смешанный индикатор дает лучший переход окраски. Рекомендуется [351] смесь, состоящая из 0,1 г фталеинкомплексона, 0,005 г метилового красного и 0,05 г диаминового зеленого В. Четкость перехода окраски повышается в 50 %-ном этанольном растворе. Однако в присутствии этанола легко выделяются карбонаты, вследствие чего титрование растягивается. Поэтому рекомендуется обратное титрование.
85
К 100 мл анализируемого раствора (- 0,001 М Sr) прибавляют 0,3 мл раствора смешанно го ^индикатора и 5—10 мл конц. NH40H. Затем титруют раствором комплексона III до резкого исчезновения красной окраски (если раствор в точке эквивалентности слишком интенсивно окрашен, уменьшают pH раствора прибавлением хлорида аммония). Прибавляю^ небольшой избыток комплексона III, вносят в раствор столько этанола, чтобы его окончательная концентрация стала 50%-ной. При этом серая окраска раствора переходит в зеленоватую. Избыток комплексона титруют 0,01 М раствором хлорида бария до четкого перехода окраски в красную [351].
Другая смесь индикаторов (0,1 г о-крезолфталеинкомплексона, 0,05 г метилового красного и 0,05 г бромкрезолового зеленога в 100 мл воды) предложена в [824]; в [1208] применяют смесь фталеинового пурпурного, метилового красного и эрио зеленого.
[Индикация точки эквивалентности физико-химическими методами
При титриметрическом определении стронция используются также физико-химические методы индикации точки эквивалентности.
Спектрофотометрическое титрование Sr раствором комплексона III описано в [818, 1108, 1208]. После хроматографического выделения стронция его определяют титрованием в ультрафиолетовой области спектра (при 225 нм, без индикатора) или в видимой области (при 630 нм, с индикатором эриохромом черным Т). В первом случае получаются менее совпадающие результаты. Мешающее влияние Fe3+ устраняют его восстановлением до Fe2+ аскорбиновой кислотой, Сн2+ и другие мешающие ионы связывают в цианидный комплекс в щелочной среде [818].
К анализируемому раствору добавляют 1 мл 0,001 М MgCl2, 1 мл 0,2%-ного раствора тартрата аммония, 0,05 мл свежеприготовленного 1 %-ного раствора аскорбиновой кислоты, 2 мл NH40H и 1 мл 0,4%-ного раствора KCN, нагревают до 80° С, охлаждают до комнатной температуры, прибавляют 0,1 мл раствора индикатора (50 мг эриохром черного Т и 450 мг NH2OH -НС1 в 10 мл метанола), разбавляют до 100 мл и титруют 0,0025 М раствором комплексона III при 630 нм. Содержание стронция рассчитывают по разности между результатами титрования смеси Sr + Mg и одного Mg. Метод применен при анализе карбонатных пород.
Возможность применения арсеназо I или смеси индикаторов (фталеинового пурпурного, метилового красного и эрио зеленого) при спектрофотометрическом титровании Sr комплексоном III показана в [734, 1208]. С индикатором нитхромазо спектрофотометрическое титрование стронция проводят раствором Na2SO4 в среде 65 %-ного ацетонитрила [213].
Потенциометрическое титрование Sr комплексоном III с применением индикаторного Ag-электрода предложено в [729, 1275, 1276] (в присутствии 1 • 10-6 М Ag+ в растворе), в [1326] ис
86
пользуют МпО2-электрод (при добавлении в раствор комплексоната Мп2+), в [289] — висмутовый электрод.
Разработан метод обратного титрования бинарных смесей стронция (а также Ва и Mg) с РЬ, Си, Со или Ni при использовании раствора комплексона III [900, 902] или циклогександи а-минтетрауксусной кислоты [901], избыток которых оттитровывают раствором Hg(NO3)2. Метод основан на том, что при pH 8—9 Hg(NO3)2 легко вступает в обменную реакцию с комплексонатами Ва, Sr и Mg и не взаимодействует с комплек-сонатами РЬ, Си, Со и Ni. При повышении pH до 11 Hg(NO3)2 не реагирует ни с одним из перечисленных комплексонатов [900].
К 5—10 мл ’раствора анализируемой смеси прибавляют 15 мл 0,01 М раствора комплексона III, 40 мл буферного раствора с pH 8 (смесь 1 М NH4NO3 и 1 М NH4OH) и титруют потенциометрически ~0,05 М раствором Hg(NO3)z. Разность между добавленным количеством комплексона III и оттитрованным обратно соответствует содержанию РЬ, Си, Со или Ni в бинарной] смеси. Затем проводят такое же титрование при pH 11. Количество связанного комплексона, найденное в этом случае, соответствует суммарному содержанию двух элементов в бинарной смеси. По разности результатов 1-го и 2-го титрований определяют содержание 2-го элемента в бинарной смеси. Ошибка определения Sr < 1,3%.
Методы потенциометрического титрования в смеси метанола с ацетоном (1 : 2,7) хлоридов и нитратов Sr, а также смесей Sr с рядом металлов (Li, Си, Ag, Mg, Zn, Cd и др.) разработаны в [214]. В качестве титранта используют гидроокись тет-разтиламмония.
Потенциометрическое титрование Sr раствором Na2SO4 в воднодиоксановых (50—70% диоксана) растворах при pH 5—7 применено в [813]. Метод использован для определения стронция в органических образцах после мокрого сожжения.
Кондуктометрическое титрование Sr л и м о н н о й кисло-т о й, полностью нейтрализованной триэтаноламином, использовано в [567]. В [437] показана возможность хронокондуктометриче-ского титрования Sr(OH)2 раствором 0,3 N НС1. Условия кондуктометрического титрования стронция комплексоном III приведены в [438].
В работе [1347] описан метод определения стронция амперометрическим титрованием с применением двух поляризованных платиновых электродов и этилендиаминтетрауксус-ной кислоты в качестве титранта. Метод биамперометрического титрования Стронция раствором щавелевой кислоты в среде изопропанола с использованием вращающегося Cu-микроэлектро-да А Cu-электрода сравнения описан в [53].
Для титрования берут 2—5 мл 0,1—0,01 М раствора пробы, прибавляют 5—10 мл изопропанола, устанавливают напряжение 1 в и титруют 0,1 М раствором Н2С2О4 в изопропаноле.
87
Использование хелатообразующих агентов при биамперомет рическом титровании Sr изучено в [1346].
Спектрополяриметрический метод определения щелочноземельных элементов, основанный на их титровании раствором оптически активной Т)-(-)-т ран с-1,2-ц и к л о г е к с а н д и а м и н-тетрауксусной кислоты, разработан в [520]. Титрование проводят при pH 8—11 при использовании монохроматического света с длиной волны 365 нм. Образующиеся в процессе титрования комплексы металлов имеют положительный угол оптического вращения. Сам титрант имеет отрицательный угол оптического вращения, что обеспечивает четкое определение точки эквивалентности.
Радиометрическое определение точки эквивалентности при комплексонометрическом титровании стронция основано на применении в качестве радиоактивного индикатора осадка U0AgJO3, растворяющегося после окончания титрования стронция [62].
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Спектрофотометрические методы
Спектрофотометрические методы определения стронция можно разделить на прямые и косвенные. Прямые методы основаны на реакциях образования окрашенных соединений при действии реактивов на ионы стронция. В косвенных методах стронций осаждается в виде труднорастворимого соединения с окрашенным реактивом, присутствующим в избытке, осадок отделяют и по количеству несвязанного реактива определяют концентрацию стронция в пробе.
Для спектрофотометрического определения стронция применяются многие из реактивов, представленные в главе «Качественное определение стронция».
Прямые методы определения стронция
Определение стронция с нитроортаниловым С (нитрохром аз о) или ортаниловым С рассмотрено в работах [129, 212, 362]. Записи спектров поглощения растворов нитроортанилового С и ортанилового С, а также их комплексов со стронцием показаны на рис. 13. Оптимальные условия определения стронция с нитроортаниловым С [129] следующие: 60 %-ный этанольный или ацетоновый раствор с pH 2,8 (устанавливается до введения этанола или ацетона), содержание стронция 2—20 мкг в 25 мл раствора, фотометрируют при 650 нм. В этих условиях определению стронция не мешают: Na, NH+4 — до-100-кратного избытка; К — до 30-кратного; Са — до 1,5-кратного; Mg — до 5-кратного. Мешают Ва и РЬ(П), давая с реагентом цветную реакцию. Zr, Ti, Та и некоторые другие элементы приводят
88
к резкому занижению результатов. Их удаляют путем осаждения в виде гидроокисей при pH ~ 4.
При определении стронция в солях бария последний осаждают в виде сульфата, предварительно добавив в раствор ацетатный буфер.
Для определения малых количеств стронция рекомендуется концентрирование соосаждением его комплекса с нитрохромазо и дифенилгуани-динием с индифферентным соосади-телем (2-нафтолом). Последующее фотометрирование проводят в среде 80%-ной уксусной кислоты [221].
С ортаниловым С фотометрическое определение стронция производится в 50—70%-ном ацетоне или этаноле при pH 2—8 и при 640 нм. Чувствительность <20,05 мкгЗт/мл [362].
Определение стронция с д и м е-тилсульфоназо III и д и -метилсульфоназо описано в [590, 591]. С диметилсульфоназо III [591] производится спектрофото-
Рис. 13. Спектры поглощения водных растворов7 ортаниловэ-го С (1), нитроортанилового С (2) и комплексов стронция с этими реагентами 3 п 4 соответственно (1-10-6 М Sr2+(3); 2,5-10-6 М Sr2+ (4); I = 1 см)
метрическое определение Sr и Ва, при их совместном присутствии как в водной, так и водно-ацетоновой (40%-ной по ацетону) среде. Применение последней дает выигрыш в чувствительности, так как
молярный коэффициент погашения комплекса стронция при этом значительно возрастает (от 600 в воде до 34300 в водно-ацетоновом растворе). Для бария эти величины равны 13 900 и 28 800 соот
ветственно.
Исследуемый раствор доводят до щелочной реакции по метиловому красному. Отфильтровывают осадок, если он образуется, и помещают аликвотные части раствора, содержащие 10—100 мкг Ва и Sr, в две мерные колбы емкостью 25 мл. Добавляют 1 мл буферного раствора (1,4 г уротропина в 100 мл воды) и 5 мл 0,0001 М раствора диметилсульфоназо III в каждую колбу и в одну из них — 10 мл ацетона. Разбавляют до метки водой и перемешивают. Через 5 мин. измеряют оптическую плотность в кювете 1 см при 665 нм против холостых растворов, приготовленных таким же образом без Sr и Ва.
Содержание Ва и Sr вычисляют при помощи уравнений, содержащих данные об измеренной оптической плотности в водном и водно-ацетоновом растворе и значения молярных коэффициентов погашения комплексов обоих металлов в водной и водно-ацетоновой среде. При определении 14—82 мкг Ва и 9—87 мкг Sr ошибка < 2,5 и 7,5% соответственно.
89
Элементы III—V аналитических групп должны быть удалены. Количество аммонийных солей и щелочных металлов должно быть не более 10 мг, Mg < 100 мкг. Сульфаты и фосфаты мешают, если их больше 0,03 ммоля. Избыток реактива по отношению к определяемым катионам должен быть примерно двукратным.
В дианилиде диметилсульфоназо (диметилсульфоназо ДАЛ) [590] две гидрофильные сульфогруппы остатка хромотроповой кислоты блокированы. Данный реактив предложено использовать для экстракционно-фотометрического определения Sr или Ва.
К 10 мл анализируемого раствора, содержащего 5—30 мкг Sr, добавляют 0,1—0,2 мл 0,01 М раствора комплексона III, подкисляют по метиловому красному, добавляют 10 мл бутанольного раствора диметилсульфоназо ДАЛ, перемешивают 3—5 мин. После отстаивания отделяют органическую фазу и фотометрируют экстракт при 670 нм в кювете 1 см, используя экстракт контрольного опыта в качестве раствора сравнения. Для приготовления раствора диметилсульфоназо ДАЛ 87 мг реагента растворяют в 250 мл 0,1 М NaOH, добавляют 1 л и-бутанола и ~ 250 мл 0,5 М НС1, смесь перемешивают 2— Змин., отделяют органическую фазу и промывают ее 3—4 раза водой до pH водной фазы 5—6. Раствор реагента в и-бутаноле устойчив на протяжении нескольких месяцев.
Определению мешают многие металлы, в том числе Са и Mg, если их содержание в пробе 0,3 мкмоля, а Сп(П) 0,25 мкмоля. Более чем 0,1 мкмоля сульфата, сульфида или цианида недопустимо. Большие количества мешающих металлов могут быть удалены экстракцией с помощью ацетилацетона и хлороформа (1 : 1) при различных значениях pH. После экстракции водная фаза упаривается досуха, остаток прокаливается при 300—500° С и используется для определения Sr или Ва.
Определение стронция с хлорфосфоназо III рассмотрено в работах [247, 248]. Хлорфосфоназо III (см. табл. 7) взаимодействует с ионами стронция при pH 2,5—5,5, при этом его окраска в зависимости от концентрации стронция переходит из’фиолетоворозовой в фиолетово-синюю или сине-зеленую (рис. 14); окрашенные растворы фотометрируют при 660 нм. Окраска устойчива не менее суток. Предел обнаружения составляет 0,5 мкг Sr в 5 мл раствора. Закон Бера соблюдается в интервале от 0,5 до 10 мкг Sr.
В условиях определения стронция хлорфосфоназо III образует окрашенные соединения также с Са, Ва, Mg, Cu(II), Zr, Al, Fe(IH), Со, Ni, Be, Pb(II), UOt, Y, Th, Sc, Nd, Ce. Комплексон III и сульфат натрия резко повышают избирательность реакции, хотя при этом чувствительность определения стронция понижается. В этих условиях не мешают 50—100-кратные количества Li, Rb, Cs, Ag, Cd, As(III), Se(IV), Ge, Mo(VI), Mn(II), Sc, Sm, Pb(II), Zn; 10—20-кратные количества Au, Nd, Ni, Co, In, Al, Ba, Mg; 5-кратные количества Cr(III) и (VI), Hg(I), Y, Ce(III), Sb(III), Ga, Fe(III), равные количества Ca, Th, Sn(II). He мешают также
90
Рис. 14. Кривые светопогло-щения раствора хлорфосфоназо III (1) и его комплекса со Sr2+ (2) (50 мкг Sr; pH 4,6; I = = 1 см)
40 мг хлоридов, 25 мг нитратов, 20 мг сульфатов, 10 мг фторидов, 7 мг фосфатов.
Погрешность определения 1,67% стронция в материалах на основе титана, циркония, свинца и висмута не превышает 7,6 отн.%.
Определение стронция с.карбоксинитразо изучено в ряде работ [320, 321, 367]. Реакция стронция с карбоксинитразо является одной из наиболее чувствительных (в 80 %-ном этаноле молярный коэффициент погашения равен 140 000) [321]. С помощью этой реакции определяют 0,08—0,6 мкгБг/мл.
На воспроизводимость результатов определения стронция большое влияние оказывает последовательность добавления органического растворителя к анализируемому раствору. Рекомендуется пользоваться заранее приготовленными водно-спиртовыми смесями.
В мерную колбу емкостью 25 мл помещают анализируемый раствор с содержанием Sr от 2 до 14 мкг, затем добавляют 20 мл этанола, 0,5 мл 0,1 %-ного раствора реагента, разбавляют до метки водой и фотометрируют при 710 нм.
Определению 0,4 мкгЗг/мл не мешают 150-кратные количества Na, 300-кратные — К, NH^, винной и лимонной кислот, а также 4000-кратные количества щавелевой кислоты. В присутствии сульфосалициловой кислоты уменьшаются помехи со стороны некоторых металлов: не мешают 175-кратные количества А1 и 80-кратные — Fe(III) (вместо 9- и 4-кратных Количеств соответственно в отсутствие маскирующего агента) [321, 367].
Рекомендовано [319] определять Sr в присутствии Са при маскировании последнего в виде оксалата.
Определение стронция с о-к резолфталеинкомплек-соном и другими производными фенолфталеина описано в ряде работ [373, 919, 1141].
Раствор соли стронция помещают в мерную колбу емкостью 100 мл, добавляют 25 мл буферного раствора (0,24%-ный раствор NH4C1 в 5%-ном водном растворе NH4OH) и разбавляют водой до — 90 мл, перемешивают
91
и прибавляют 10 мл раствора о-крезолфталеинкомплексона (0,1%-ный раствор реактива в смеси 28 мл упомянутого выше буферного раствора и 72 мл воды). Перемешивают, доводят объем до метки водой и фотометрируют в кювете с I = 1 см при 575 нм. Калибровочный график прямолинеен до концентрации 2,8 mkzSt/мл [1141].
Влияние ацетата и перхлората натрия на точность определения стронция описано в [1194].
Определение стронция смурексидом предложено [1141, 1203]. Стабилизация растворов мурексида и его комплекса со стронцием достигается добавлением этиленгликоля [1203].
Анализируемый раствор выпаривают досуха, остаток растворяют в небольшом количестве воды, прибавляют 15 мл этиленгликоля, 3 мл 0,05 М раствора NaOH, разбавляют водой до ~ 22 мл, перемешивают и охлаждают льдом. Затем добавляют 3 мл 0,05%-ного раствора мурексида в 80%-ном этиленгликоле, разбавляют водой до 25 мл и фотометрируют в кювете с I = = 2 см при 510 им, применяя раствор контрольного опыта в качестве раствора сравнения. Закон Вера соблюдается для растворов с концентрацией 8 мкг8г/мл.
При определении 93,8 мкг Sr присутствие Mg (300), Са (2), Ва (50), Li (75), К (150) или Ag (10 мкг) приводит к положительной ошибке ~5%.
Вместо мурексида для спектрофотометрического определения стронция и других щелочноземельных элементов может быть применено его производное — тетраметилмурексид [926].
Определение стронция с8-оксихинолином рассмотрено в работе [394].
В анализируемом растворе устанавливают pH ~ 9 и экстрагируют мешающие ионы несколькими порциями 0,1 М раствора оксина в хлороформе до тех пор, пока окраска органической фазы не перестанет изменяться. К водной фазе добавляют 0,1 N раствор едкого натра, чтобы установить pH 11,5—12. Экстрагируют стронций равным объемом 0,5 М раствора оксина в хлороформе. Повторяют экстракцию и измеряют светопоглощение объединенных органических экстрактов при 400 или 380 нм.
Многие ионы, мешающие определению, можно удалить предварительно экстракцией в виде оксинатов при более низких значениях pH.
Определение стронция сэриохромом серым 3 В L - с у п-р а описано в работе [921].
В мерную колбу на 25 мл вводят 0,45 мл 0,1 %-ного водного раствора эриохрома серого 3BL-cynpa, раствор соли металла и доводят до метки буферным раствором (pH 11,3). Через 30 мин. измеряют поглощение света с помощью фотометра с оранжевым светофильтром в кювете 2 см. Концентрацию металла находят по калибровочному графику, который прямолинеен в интервале до 2 мкг Sr/мл.
92
Катионы РЬ, Си, Ni, Со, Fe(II и III), Mn(Il), Al, Zn образуют аналогичную окраску при pH 7,7—8,0; Mg, Ca и Cd — при pH 11,3.
Определение стронция с родизоновой кислотой [323, 689, 1048, 1165]. При выполнении реакции на стронций с родизоновой кислотой в разбавленных растворах образуется окрашенная взвесь, по интенсивности окраски которой можно проводить его колориметрическое определение. Для стабилизации взвеси обычно прибавляют желатину или гуммиарабик. Определению стронция этим методом мешают многие металлы (см. стр. 25), которые должны быть предварительно удалены [323].
В большинстве случаев полуколичественное и количественное определение стронция производят на бумаге путем фотометриро-вания розовых пятен родизоната стронция [689, 988, 1165].
При обработке розового пятна 2—3 каплями раствора хлорида диметил-аммония (приготовленного путем разбавления его насыщенного раствора равным объемом 95%-ного этанола) оно становится фиолетово-синим и достигает максимальной интенсивности через 20 мин. (окраска сохраняется 2 часа). Розовое пятно родизоната бария, обработанное таким же образом, становится ярко-красным [1181].
По интенсивности пятен родизонатного комплекса стронция на бумаге определяют его содержание при анализе природных вод [1165] и летучих продуктов, образующихся на оксидном катоде [689].
Количественное определение сульфата и нитрата стронция в смеси с соответствующими солями бария можно производить при использовании инфракрасных спектров поглощения [628, 629]. Используются полосы поглощения SrSO4 при 993 см-1, Sr(NO3)2 при 737 слг1. Соли прессуют в таблетки с КВт. Соотношение BaSO4: SrSO4 может быть от 20 : 1 до 1 : 20 [628].
Косвенные методы определения стронция
Предлагавшиеся ранее косвенные спектрофотометрические методы определения стронция в настоящее время не находят применения (ввиду их малой селективности) и будут здесь лишь упомянуты.
8-Оксихинолиновый метод [323] основывается на осаждении стронция в виде оксихинолината и определения содержания оксихинолина в осадке по цветной реакции с ионами Fe(III). Стронций можно осадить с помощью хлораниловой кислоты и фотометрически определить избыток последней [978]. На аналогичном принципе основан метод с дилитуровой (5-нитробарбитуровой) кислотой [190].
При использовании пикролоновой кислоты стронций также выделяют в виде ее нерастворимой соли, а избыток осадителя в фильтрате определяют путем переведения его в краси
93
тель пиразолоновый синий при действии персульфата калия [6].
Определение стронция с помощью хромата описано в 1377, 1094].
Методы спектрофотометрического титрования описаны в разделе «Титриметрические методы» настоящей главы.
Электрохимические методы
Полярографический метод
Ион Sr2+ дает хорошо выраженный диффузионный ток на фоне '0,17V водного раствора Lid (потенциал полуволны = — 2,1 в) и в этанольно-водных растворах (в 50%-ных растворах по этанолу Е>1г = — 2,0 в) на фоне иодида тетраэтиламмония. При достаточно высокой концентрации последнего между величиной диффузионного тока Sr2+ и его концентрацией наблюдается прямая пропорциональность [218]. Полярографическое определение стронция в водных растворах без применения этанола изучено в [371].
В условиях определения стронция ион Ва2+ дает волну с Е^г на 200 ме более положительную, поэтому стронций может быть определен в присутствии небольших количеств бария. Потенциал восстановления Са2+ близок к потенциалу восстановления Sr24-, вследствие этого последний в присутствии больших количеств кальция определять нельзя.
Полярографическое определение бария и стронция при их совместном присутствии возможно также при замене этанола на изобутанол. В 80%-ном изобутаноле Е1/г равны —1,43 и —1,62 в для Ва2+ и Sr2+ соответственно (фон — 0,1 М раствор тетраметил-аммония) [637].
Найдено, что подходящим фоном для определения малых количеств Sr является 0,02 М (G2H5)4NBr в абсолютном этаноле. для Sr и Ва равны соответственно —1,97 и —1,74 в относительно донной ртути. В присутствии приблизительно равных концентраций Mg и Са определение Sr невозможно [1157].
Если соотношение Ва : Sr в смеси равно 500 : 1, а Са : Sr равно 450 : 1, то В а отделяют путем его восстановления на ртутном катоде при контролируемом потенциале, равном —2,03 в [971], а Са отделяют на сильноосновном анионите в ОН_-форме; Sr элюируют насыщенным раствором Са(ОН)2 [934]. В обоих случаях Sr определяют по полярограмме, имеющей Е^ = —2,17 в (относительно нас. к.э.). Подлежат отделению также К и Na имеющие Е1/г = —2,17 и —2,15 в соответственно [934].
Однако может быть применена техника работы, позволяющая определять Sr в присутствии Na и К [1157]. В абсолютном этаноле сначала записывают суммарный пик Sr + Na(K), затем добавляют несколько микролитров 0,02 М раствора сульфата тетраэтиламмония и снова снимают полярограмму, вызываемую катионами Na+ или К+. Высоту пика Sr определяют по разности.
94
Дифференциальный полярографический метод дает возможность определять малые количества Sr в присутствии больших количеств Na и К. Метод применен для анализа кристаллов NaGl и КС1 (чувствительность — 0,0001 моль Sr/моль соли) [1322].
Инверсионная полярография (амальгамная полярография с накоплением) позволяет определить стронций в очень малых концентрациях (10“5—10“8 М), если его сначала сконцентрировать в капле ртути путем электролиза, а затем подвергнуть ее анодному растворению. Используется осциллографическая техника. Для получения амальгамы при анализе растворов с концентрацией до 10"’ М достаточно 10 мл анализируемого раствора подвергнуть электролизу при силе тока 25 ма в течение 5 мин. При снижении концентрации анализируемого раствора необходимо увеличить его объем и продолжительность электролиза. Определение проводят в 0,05 М растворе иодида тетраметиламмония. Средняя ошибка составляет 3—5% [895].
Описан [797] метод косвенного определения стронция, основанный на реакции вытеснения Cd2+ из его комплексоната с последующим определением Cd2+ с помощью квадратно-волновой полярографии. При анализе смеси Mg и’Sr суммарное содержание металлов определяют в 5 М растворе NH4OH, затем определяют Sr в растворе 5 М по NH4OH и 1 М по КОН (Mg в этих условиях образует гидроокись и не реагирует с комплексонатом кадмия). Определению не мешают Al, Zn, Fe(III), Th, Си, Ti, Bi, Ce; мешают Ba и Pb.
В другом случае [1318] количество стронция находят (с ошибкой <3%) по уменьшению полярографической волны комплексоната серебра, при взаимодействии которого с сульфатом стронция серебро замещается на стронций.
Кондуктометрический метод
Для количественного определения стронция используют также кондуктометрический метод (после отделения Sr методом электрофореза от группы элементов Li, К, Na, Са и Ва, входящих в растворимые соли строительных материалов) [856].
ФИЗИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Спектральные методы
Спектрографический (искровой и дуговой) метод
Спектрографические методы широко используются при определении Sr в водах, породах, почвах, растениях. Условия определения Sr в различных объектах приведены в руководствах [56, 58, 357, 493].
Спектр стронция, наблюдаемый в источнике возбуждения, характеризуется линиями, образующимися при возбуждении атома
95
Sr(I) и иона Sr(II). Для спектра Sr(I) характерны две системы термов: синглеты и триплеты. Упрощенная схема возможных переходов электронов на различные уровни энергии и соответствующие им спектральные линии для атома Sr представлена на рис. 15 [209, 1058]. । |
Характеристика основных линий спектра Sr приведена в табл. 30. Наиболее интенсивные линии стронция лежат в видимой области спектра: 4607,33; 4077,71 и 4215,52 А, причем две последние находятся в области циановых полос. Поэтому при использовании для анализа дуги с угольными электродами эти линии менее пригодны. Указанные линии могут быть использованы для определения Sr в рудах при содержании его в тысячных и десятитысячных долях процента. Линия стронция 4607,33 А отличается сильным самопоглощением, поэтому рекомендуется использовать ее при определении лишь малых концентраций Sr (ниже 0,1%). При высоких его содержаниях используются линии Sr 4811,88 и 4832,08 А, а также 3464,46 А.
В ультрафиолетовой области спектра используются значительно более слабые линии 3464,46 и 3380,71 А, последняя из них расположена в области спектра, обладающего фоном.
При определении стронция в различных объектах необходимо учитывать возможные помехи вследствие наложения линий посторонних элементов на аналитические линии стронция (табл. 31).
Положение стронция в ряду летучести элементов зависит от рода соединений, в которых он находится [357].
Окись стронция является тугоплавкой и довольно медленно испаряется в угольной дуге после сгорания концов электродов и появления окислов на поверхности угля. Образования свободного металла в угольной дуге из окисла стронция не наблюдается.
В ряду летучести окислов в угольной дуге стронций расположен в середине. При испарении окисных и силикатных руд стронций поступает в пламя дуги обычно после испарения хорошо летучих составных частей руд (щелочных металлов, Zn, Cd, As), но одновременно с элементами, расположенными в центре ряда летучести окислов в угольной дуге (Fe, Go, Ni, Si, Al, V).
При нахождении металлов в пробе в виде хлоридов стронций испаряется после щелочных металлов, Mg и Са.
Карбонат стронция при испарении из электрода постепенно теряет двуокись углерода и превращается в окисел.
Сульфат стронция частично восстанавливается до сульфида частично превращается в окисел.
В ряду летучести фосфатов стронций стоит возле А1 и Сг и предшествует расположенным в конце ряда Th, Zr и элементам группы редких земель. Фосфат стронция поступает в пламя дуги вместе со свободным фосфором, который образуется частично при восстановлении фосфата углеродом и может испаряться из расплава фосфата до улетучивания щелочноземельного элемента.
96
3 св S &g «
h—Г ж Е?
М л
4 Н. С. Полуэктов и др.
Т а б л и ц а 30
Аналитические линии для определения стронция [357]
Длина волны, Л Потенциал возбуждения, эв ^отн Чувствительность определения, %
дуга искра
2152,84 II 7,56 15 15 0,1
2165,96 П 7,56 257? 15 0,1
2322,355 II 8,37 8 5 0,1
2428,095 1 5,10 ЮЛ — 0,1
2471,597 П 8,05 8 5 0,1
2569,469 I 4,82 257? 5 0,1
2931,830 I 4,23 30 8 0,1
3301,734 I 5,53 100 10 0,1
3307,534 I 5,54 200 10А 0,1
3329,988 I 5,52 100 10 0,1
3351,246 I 5,54 300 15 0,03
3366,333 1 5,53 100 10 0,1
3380,711 II 6,61 150 200 0,01
3464,457 П 6,64 200 200 0,01
3474,887 II 6,64 80 50 0,1
4077,714 II 3,04 400г 50017 0,001
4215,524 II 2,94 300г 40017 0,001
4305,447 II 5,91 40 — 0,01
4319,12 I 5,13 50 20 0,1
4337,89 I 5,13 150 50 0,1
4607,331 I 2,69 10007? 507? 0,0003
4722,278 I 4,42 30 — 0,1
4741,922 I 4,39 30 — 0,1
4811,881 I 4,42 40 — 0,1
4832,075 I 4,34 200 8 0,1
4872,493 I 4,34 25 — 0,1
4876,325 I 4,39 200 60 0,1
4962,253 I 4,35 40 — 0,1
5256,903 I 4,63 90 25 0,1
5480,84 I 4,53 100Л 30 0,1
6408,473 I 4,21 50 20 0,1
При меча ни е. т — слабое самообращение; R — сильное самообращение; h — линия диффузная; S — линия асимметричная (смещение в сторону коротких длин волн); W—линия уширена или состоит из нескольких компонент; 10тН—относительная интенсивность линии.
Кривые испарения стронция из ряда соединений приведены на рис. 16 [357].
Поскольку стронций и его соединения отличаются средней ле-тучестью, наиболее подходящими элементами внутреннего стан-98
Рис. 16. Кривые испарения стронция из различных соединений [357]
1 — SrS04; 2 — SrCO3; з — Sr3(PO4)2. М — относительное количество вещества, поступающего в дугу
дарта являются Са, Сг, V, Pd, Ti, а также Y, Zr, Li, Cs. Длины волн некоторых линий элементов, наиболее часто используемых в качестве внутреннего стандарта при определении стронция, приведены в табл. 32. Помимо указанных в таблице, в качестве элементов сравнения применяют Pt [56], Ni [1345], Mo [554], Со [252].
В некоторых случаях в качестве внутреннего стандарта используется фон вблизи линий исследуемого элемента [1].
При определении Sr необходимо учитывать влияние валового состава пробы на интенсивность линий стронция.
Если изменения содержания Fe и Si в пробе не оказывают заметного влияния на интенсивность линий Sr, то увеличение содержания Na повышает интенсивность дуговых линий и несколько понижает интенсивность искровых [56]. Такое же влияние Са и Mg [13, 361, 1328]. Влияние солей щелочных металлов на интенсивность спектральных линий щелочноземельных элементов устраняют путем изменения техники работы [38]. При использовании специальных буферных смесей можно устранить влияние Са, Mg и Na [674]. Влияние «третьих» компонентов на интенсивность спектральных линий стронция рассматривается также в [304, 515, 598].
Для стабилизации температуры горения дуги и достижения более высокой точности определения Sr используют буферные смеси. В зависимости от состава анализируемых пород в качестве буферов используют NaCl [56, 242, 1011], Na2SO4 [58], K2SO4 [56, 256], СаС2О4 и Са3(РО4)2 [1354], СаСО3 и К2СО3 [1124, 1290, 1328], ВаСО3 [1037]; Fe2(SO4)3 и (NH4)2SO4 предложены в [501, 553, 648, 1162].
В ряде случаев в качестве основы применяют SiO2 [207], смесь SiO2 с NaCl [205], SiO2 с Na2CO3 [1068]. Соли лития (LiF, Li2CO3) предложены в [88, 1031].
Влияние NaF и СнС12 на интенсивность спектральных линий Sr исследовалось в [12].
4*
99
Таблица 31
Возможные помехи при спектральном определении Sr [493]
Элемент Длина волны, A 7qth Элемент Длина волны, A А)ТН
тъ 4607,81 4 VI 4077,977 2
Fe I 4607,654 50 Dy 4077,974 150r
Мп 4607,625 50 Tb 4077,97 25
Nd 4607,381 25 Er 4077,970 205
Au I 4607,34 30 Hgl 4077,811 150
Sri 4607,331 1000B u 4077,786 15
Co 4607,33 2Л Ta 4077,721 4
La 4607,3 3dh Sn 4077,72 2
Ce 4607,290 3 Cu 4077,716 5
V 4607,226 4 Sr II 4077,714 400r
Ce 4607,087 2 Pr 4077,69 4
Mo 4607,075 8 Mo 4077,682 §
Pr 4606,920 3 Cr 4077,677 30
Ru 4606,833 4 Nd 4077,620 8
Eu 4078,231 3 RhI 4077,57 5
Pr 4078,16 5 Cell 4077,470 18
W 4078,124 7 Col 4077,406 100 Wh
Re 4078,124 10 YI 4077,366 50
Mo 4078,074 4 Dy 4077,35 4
Ho 4078.00 3 Lail 4077,340 600
Pr 4077,99 10 Yb 4077,27 30
Til 4077,153 18
При мечание. Обозначения см. в табл. 30.
Различные параметры дуги постоянного тока, различные зоны плазмы, распределение излучающих атомов в дуговом разряде, способы введения вещества в разряд с целью нахождения оптимальных условий анализа исследованы в [38, 140, 514, 515, 973, 1199, 1332].
При анализе образцов на стронций в качестве источника возбуждения спектра применяется в основном дуга постоянного или переменного тока, а также искра и высокочастотный разряд.
При использовании дуги постоянного тока образцы смешивают с угольным порошком или графитом в отношении образец: угольный порошок 2 : 1 [796], 1 : 1 [56, 292, 1195, 1206], 1 : 2 [52, 56], 1 : 3 [863], 1 : 5 [56]. В некоторых случаях применяют сульфидирование пробы [314]; образцы смешивают с AgCl и CdS и испаряют либо в дуге постоянного тока, либо в разрядных трубках с полым катодом. При анализе растворов их наносят на уголь-100
Таблица 32
Длины волн некоторых линий элементов, используемых в качестве элемента сравнения при определении Sr [56, 58]
Длина волны Sr, А Потенциал возбуждения, эв Длина волны элемента сравнения, А Потенциал возбуждения, 9в
Sr I 4607,33 2,69 Са I 4581,40 Са I 4526,94 CrI 4254,35 CrI 4274,80 Cr I 4600,75 V I 4379,24 V I 4384,72 V I 4389,97 V I 4395,23 VI 4400,58 Y I 4643,69 Li I 4602,86 Cs I 4593,18 Pd I 4473,59 Pd I 3460,77 Pd I 3481,1 5,23 5,44 2,91 2,90 3,69 3,13 3,11 3,10 3,08 3,08 2,67 4,54 2,70 4,40 4,81
Sr II 4215,52 8,63* Ba II 4554,04 7,93*
Sr II 4077,71 8,73* Zr II 4149,20 Ca II 3933,67 Mo 4070,0 10,74* 9,26 *
Sr II 3464,5 12,31 * Pdl 3460,8 4,40
Sr II 3380,71 12,30 Till 3383,76 Ti II 3387,84 10,50* 10,52 *
В потенциал возбуждения линий ионизованного атома включена также величина потенциала ионизации соответствующего атома, т. е. приведенные величины соответствуют общей энергии, необходимой для возбуждения данной линии.
ный порошок [131]. В работе [1380] при определении Sr из растворов предложено применять медные электроды.
При возбуждении спектров в дуге переменного тока образцы также смешивают с угольным порошком [1, 56, 488], просыпка пробы при одновременном воздушном дутье применяется в [56, 253, 680].
Для полуколичественного определения Sr в кернах буровых скважин применяют латунные электроды [56], при этом в спектре наблюдается большое число линий Си и Zn, которые могут быть использованы как линии сравнения. При анализе растворов вещество вводится в разряд с помощью фульгуратора [56, 58] либо в виде сухого остатка на торце электрода [201, 881].
101
Для устранения полос циана определение Sr проводят в аргоне [633, 740, 883, 1197, 1243] либо переводят пробы во фтористые соединения [56].
Чувствительность определения Sr в дуге составляет 5-10“5— 1 • 10-4%, относительная ошибка определения +4 — 15%.
Применение импульсного дугового разряда большой силы тока в аргоне [1296] позволяет значительно повысить чувствительность определения Sr (3-10-12а).
При определении Sr с использованием возбуждения искрой применяется различная техника. В ряде случаев образец переводится в раствор и наносится на торцы графитового [679, 965] или медного [56, 202, 1256] электродов. Применяется введение раствора в искровой разряд с помощью вращающегося дискового электрода [782, 937, 945] либо через графитовый пористый электрод [58], а также ряд других методов [56]. Порошкообразные образцы анализируют путем просыпки через конусообразный медный колпа-чек на нижний плоский медный электрод [242]. Для локального анализа вещество испаряют с помощью лазера, спектры полученных паров возбуждают в искре [1050].
Предварительное испарение пробы в испарителе на угольный стержень с последующим анализом конденсата с использованием дуги переменного тока либо искрового разряда предложено в [127, 128].
Установка для анализа a-активных материалов с помощью искрового разряда описана в [554].
Чувствительность определения Sr в искре составляет (1—5)-•10-4%, в ряде случаев 2,5-10~7% [1221, 1222]. Ошибка определения ±4—6%.
Ряд авторов [56, 58] используют высокочастотную искру при подаче раствора в искровой промежуток с помощью фульгуратора. Однако чувствительность определения Sr в этом случае несколько ниже, чем при использовании дуги.
Возбуждение плазмы высокочастотного безэлектродного разряда в атмосфере аргона при определении Sr изучено в [31, 915]. Достигнута чувствительность 8-10-12 г.
Предварительное ионообменное концентрирование и дальнейшее спектральное определение Sr непосредственно в смоле применено в [779, 1290].
Определение Sr в различных объектах может быть произведено с помощью фотоэлектрической регистрации интенсивности излучения при использовании квантометров [35, 92, 529, 780, 1313].
С целью повышения точности и абсолютной чувствительности анализа, а также устранения влияния мешающих линий посторонних элементов в [282] предложено использовать интерферометр, скрещенный со спектрографом.
Соли стронция в некоторых случаях используются в качестве буфера при определении других элементов спектральным методом 102
[56, 1230]. Как уже отмечалось, соединения щелочноземельных элементов обладают средней летучестью и их буферное действие обусловлено тем, что они испаряются в течение более длительного» времени, чем другие элементы. Они не подавляют испарение других элементов. Буферное действие щелочноземельных элементов ослабляется при наличии в пробе больших количеств щелочных элементов [56].
Эмиссионная фотометрия пламени
Благодаря своей простоте и надежности пламенно фотометрический метод определения стронция находит широкое применение, особенно при анализе горных пород и минералов, природных и сточных вод, биологических и других материалов [56, 64, 70, 327, 328, 831, 1017]. Он пригоден для определения как малых, так и больших содержаний элемента с достаточно высокой точностью (1—2 отн. %) и чувствительностью, причем в большинстве случаев определение стронция может быть выполнено без отделения от других элементов.
Основные линии и молекулярные полосы в спектре пламени, содержащего стронций, представлены в табл. 33.
Для анализа наиболее часто используют резонансную линию атома стронция, равную 460,7 нм.
Используемые пламена и аппаратура. Для определения стронция в основном находят применение высокотемпературные пламена, такие, как пламя смеси ацетилен—воздух, пропан 4- бутан—кислород, ацетилен—кислород и ацетилен—закись азота [296, 327, 349, 502, 928, 1064, 1131].
Наиболее важным параметром пламени является его температура. Она существенным образом влияет на физико-химические процессы, в которых участвуют введенные в него вещества; с ее повышением возрастает интенсивность излучения атомов стронция. Из используемых пламен наилучшим является пламя смеси ацетилена и закиси азота, температуры которого порядка 3000 °C [739, 1064].
Таблица 33
Спектр излучения стронция в пламени [327]
Линии; молекулярные полосы Энергетический переход Длина волны, НЛ1 Энергия возбуждения, эв
Резонансная линия атома Линии иона Sr+ Наиболее интенсивные канты молекулярных полос SrO и Sr ОН _ ©-©«> 1 1 а, а, 1 Й « Ю 1 । ' К о - со со со ,-t ел ел LO LO Ю 460,7 421,6 407,8 610 и 670 2,69 2,94 3,04
103
Анализируемый раствор вводится в пламя в виде аэрозоля обычно с помощью пневматического распылителя, работающего под действием сжатого газа — воздуха, кислорода или закиси азота.
Разрабатывается техника получения аэрозоля с помощью ультразвукового распылителя [675]. Растворитель с капелек аэрозоля может быть удален путем пропускания через трубчатую кварцевую печь, нагретую до 400° G, с последующей его конденсацией. Горючая смесь может состоять из водорода и воздуха [495] или же используется плазма высокочастотного разряда (9000° К) [675].
Изучается техника импульсного испарения содержащих стронций проб — с поверхности угольных [343—346], пирографитовых [348, 349] или вольфрамовых [1270] микрозондов или танталовой лодочки [296]. Раствор пробы с помощью микрошприца наносится на поверхность зонда, испаряется и высушивается, а затем вводится в пламя на несколько секунд.
Техника работы и аппаратура, используемая для пламеннофотометрического эмиссионного метода, подробно описаны в упомянутых выше руководствах.
Фотометры для пламени состоят из горелки с приспособлением для введения вещества в пламя, спектрального прибора и регистрирующего устройства. Их можно разделить на 2 типа: фотометры со светофильтрами и спектрофотометры.
Подавляющее число работ по эмиссионной пламенной фотометрии стронция выполнено с применением спектрофотометров, обеспечивающих получение более высокой чувствительности и специфичности. Применение сканирования спектра и самопишущего потенциометра позволяет вести регистрацию интенсивности излучения как при одной длине волны, так и с разверткой спектра.
Чувствительность метода зависит от ряда факторов, основными из которых являются тип (температура) пламени и разрешающая способность аппаратуры. Наиболее высокая чувствительность достигается при использовании аппаратуры с автоматической записью спектра и высокотемпературных пламен. Так, при использовании кислородно-водородного пламени чувствительность метода найдена равной 0,007 мкгЗт/мл [1139], ацетилено-воздушного 0,005 мкгЗг/мл [75], смеси закись азота—ацетилен 0,0001 — 0,0005 мкгЗг/мл [636, 928, 1164] и ВЧ-плазмы 0,00002 мкгЗг/мл [711].
При импульсном методе испарения абсолютный предел обнаружения Sr составляет 1-10'13—2-1СГ12 г (пламя смеси ацетилен— закись азота) [343, 345, 346] и 8-Ю”12 г (ВЧ-плазма) [915]. При достаточно больших количествах пробы (~10 мг) относительный предел определяемого содержания стронция снижается до 1-10~7%, в то время как при введении раствора пробы в пламя с помощью распылителя он равен всего 3-10“5%.
Влияние концентрации и состава раствора. Прямолинейная зависимость между интенсивностью излучения (7) стронция при 104
Рис. 17. Логарифмическая зависимость интенсивности излучения от концентрации Sr (М) в пламени светильный газ—воздух (7, 3) и ацетилен—воздух (2, 4) при использовании резонансной линии 460,7 нм (7, 2) и молекулярной полосы 670 нм (3, 4)
Рис. 18. Зависимость интенсивности излучения Sr (100 мкг Sr/мл) от концентрации кислот в растворе (пламя ацетилен—воздух)
1 — Н3РО4; 2 — H2SO4 в 1 N HNOa; 3 — НВг; 4 — НС1; 5 — HNO»; 6 — СНаСООН
460,7 нм и его содержанием в растворе наблюдается в области концентрации (с) до 100—200 мкг/мл [334]. При более высоких концентрациях стронция вследствие самопоглощения график зависимости I — с загибается к оси абсцисс.
При построении графиков в логарифмических координатах (рис. 17) тангенс угла наклона равен 1 при с — 0,0001—0,0012 М и ~0,5 — при с = 0,0012—1 М (пламя ацетилен—воздух). Для пламени светильный газ—воздух график имеет почти такой же вид, однако в случае больших концентраций (0,1—1,0 М) тангенс угла наклона <0,5. В случае молекулярной полосы SrOH (X = 670 нм) как при пламени ацетилена, так и пламени светильного газа наблюдается прямая пропорциональность I от с во всех изученных областях концентраций, кроме самых больших (0,5—1,0 71/), где линии несколько изгибаются в оси абсцисс [331].
Органические растворители, такие, как, например, этанол, метанол, ацетон и изопропанол или смесь этанола с бутанолом, увеличивают интенсивность излучения стронция.
Значительное влияние на интенсивность излучения могут оказывать анионы (анионный эффект). Он наблюдался для хлоридов и фторидов, азотной, серной, фосфорной и кремневой кислот и заключается в том, что интенсивность излучения стронция в присутствии названных кислот или их анионов значительно понижается (рис. 18). Однако органические кислоты (например, уксусная) усиливают интенсивность излучения.
105
Меньше всего снижает интенсивность излучения стронция азотная кислота, затем — галоидсодержащие кислоты. Значительно сильнее действует серная и в особенности фосфорная кислоты. Все эти кислоты препятствуют испарению стронция из частиц аэрозоля, а серная и фосфорная кислоты, кроме того, образуют с ним малолетучие соединения определенного состава в момент испарения частиц аэрозоля. Такое заключение подтверждается тем, что кислоты совсем не оказывают влияния на стронций при введении их в пламя через другой распылитель [327].
Состав соединений, образуемых стронцием с фосфорной кислотой в пламени, исследован в [327, 865]. Оказалось, что он соответствует соотношению ЗМе : 2Р (пламя светильный газ — воздух) и 2Ме : Р (пламя ацетилен — воздух). Стронций и серная кислота образуют труднолетучее соединение с соотношением 1 : 1 (пламя водород—кислород).
Соли ряда металлов, таких, как Al, U, Ti, Zr, Sn, V, Та, Mo, W, Mn, Fe, также снижают интенсивность излучения стронция [196, 330, 334, 502, 739, 928, 1064, 1139, 1200].
Опыты с двумя распылителями показывают, что соли постороннего элемента гасят излучение стронция, лишь находясь в одном растворе с последним, и не влияют при введении гасящего элемента в пламя через другой распылитель. Поэтому можно заключить, что вероятной причиной гашения излучения является образование не успевающих улетучиться при прохождении через пламя труднолетучих соединений стронция с металлами. Состав этих соединений был определен с помощью метода изомолярных серий.
Было найдено, что алюминий со стронцием образует в пламени соединения с соотношением Al : Sr = 2 : 1 (SrAi2O4), Zr, Ti, V, Mo и Cr образуют соединения с соотношением компонентов 1 : 1 (SrZrO3, SrTiO3, Sr2V2O7, SrMoO4, SrCrO4), а уран — с соотношением U : Sr = 2 : 3 [330, 334].
В большинстве случаев определение Sr проводят с помощью двух методов: 1) с применением эталонных растворов, имитирующих состав пробы, и 2) методом добавок [327, 634]. В обоих случаях можно получить удовлетворительные результаты с достаточно хорошей точностью определения Sr.
Устранения влияния гасящих катионов и анионов можно достичь двумя способами: 1) путем удаления их из раствора, например, осаждением [7, 400, 910], методом ионного обмена [131, 575, 1115, 1144] и экстракцией [106, 195] или, в случае легколетучих кислот, выпариванием; 2) путем добавления к раствору так называемых освобождающих агентов, которые способствуют разрушению труднолетучих соединений стронция, в результате чего последний переходит в газы пламени.
Очень часто для устранения влияния Al, Fe и других элементов, находящихся вместе со стронцием в горных породах и минералах, добавляют соль кальция [196, 548, 670]; влияние Al, Zr, V, В, Be, Cr, Fe, Mn, W, SiOl~, РО|“, SO4~ можно устранить путем до-106
бавления соли редкоземельного элемента — La [1139,1378], Pr (La, Nd) [678] или же La вместе с 8-оксихинолином. Раствор последнего может применяться в качестве буфера и самостоятельно [76, 1336] — при определении стронция в силикатах, железосодержащих минералах и др. Кроме того, буферирующие свойства могут проявлять также ацетилацетон, салициловая и щавелевая кислоты [1336], а также комплексон III [668].
Другой вид взаимного влияния элементов связан с ионизацией атомов стронция в пламени, так как потенциал ионизации стронция (5,69 эв) таков, что его атомы в значительной степени ионизируются в пламенах. Показано, что при концентрации стронция в растворе 5,5 мкг/мл степень ионизации стронция в газах пламени составляет 13% (в пламени смеси ацетилена с воздухом) и 84% (в пламени смеси ацетилен—закись азота) [274, 502]. В присутствии другого металла, также способного ионизироваться, увеличение концентрации электронов в газах пламени приводит к смещению равновесия ионизации стронция по уравнению:
Sr° Si+ -|- с,
в результате чего концентрация нейтральных атомов возрастает, а ионов — снижается.
Для подавления ионизации можно вводить в растворы соли легкоионизирующихся металлов, таких, как Na, К, Rb [76, 1336]. Однако концентрация добавляемого металла не должна быть велика, так как в противном случае могут возникнуть дополнительно помехи, связанные с увеличением фона пламени и наложением излучения добавляемого элемента на излучение стронция. По этой причине в эмиссионной фотометрии пламени не рекомендуется для подавления ионизации применять один из наиболее легкоионизирующихся металлов — Cs, линия излучения которого (X = 459,3 нм) близка к резонансной линии атома Sr (X = 460,7 нм) [64]. Са также может способствовать уменьшению ионизации Sr, о чем свидетельствует усиление в его присутствии резонансной линии атома Sr и одновременное ослабление линии иона Sr+ при 407,8 нм.
Эмиссионный пламеннофотометрический метод определения стронция находит применение для косвенного определения сульфатов и фосфатов. При определении первых ЗО^-иоиы осаждают хлоридом стронция в присутствии 50% этанола, понижающего растворимость осадка, а затем или определяют концентрацию оставшегося в растворе стронция [599] или же, наоборот, отделяют осадок сульфата стронция, растворяют в растворе, содержащем ЭДТА и NH4OH и определяют стронций [752].
По результатам пламеннофотометрического определения не-осажденного (избыточного) стронция определяют и фосфор (0,1—1 лгР2О3/.юг) [228], а по молекулярной полосе SrF (580 нм) в пламени водород—кислород рекомендуется определять ион F~ при его концентрации от 5 до 5000 мкг в 100 мл раствора [794].
107
Атомно-абсорбционная спектрофотометрия
Атомно-абсорбционный метод в последнее время находит все более широкое применение в анализе различных объектов. Принцип метода и используемая аппаратура описаны в руководствах [250, 327, 387, 704, 1156]. Указывается, что наибольшая точность обеспечивается при использовании двулучевых приборов, в которых влияние нестабильности источников света снижается примерно в 40 раз и воспроизводимость результатов измерения оптической плотности составляет несколько десятых долей процента.
Определение стронция производится путем измерения поглощения света его атомами. Наиболее часто используется линия стронция 460,7 нм, с меньшей чувствительностью стронций может быть определен по линиям 242,8; 256,9; 293,2; 689,3 нм. При использовании высокотемпературных пламен стронций можно определять также по ионной линии 407,8 нм (ионно-абсорбционная спектроскопия) [518].
В качестве источников излучения применяют трубки с полым катодом, содержащие металлический стронций, а также безэлек-тродные высокочастотные лампы [13771. Питание трубок с полым катодом может производиться пульсирующим током, при этом достигается увеличение яркости резонансной линии стронция в 600 раз по сравнению с питанием трубки постоянным током [666]. Находят применение также лампы со сплошным спектром излучения, в частности, ксеноновые [1026], водородные или вольфрамовые [927].
При работе по атомно-абсорбционному методу обычно используют горелки для предварительно смешанных газов, питаемые распылителями с камерами распыления. Наиболее широкое применение нашло пламя смеси ацетилена с воздухом или закисью азота [327]. Используются также некоторые другие пламена, например, пламя смеси ацетилена с воздухом, окруженное кислородом [1269], пламя смеси закиси азота с водородом [1372], плазма высокочастотного разряда [32]. В кюветах для отходящих газов пламени смеси водород—воздух концентрация атомов стронция быстро снижается на расстояниях больше 3 см. Такие кюветы не обеспечивают выигрыша в чувствительности [506].
Распределение свободных атомов стронция в пламени смеси ацетилена с воздухом описано в [1178]. Благодаря неравномерному распределению атомов стронция в пламени использование линзовых диафрагм для просвечивания оптимальных зон ведет к повышению чувствительности его определения [842].
Чувствительность метода зависит от аппаратурных факторов (эффективности распыления, длины горелки, наличия приспособления для расширения шкалы поглощения света и др.), а также типа пламени. Для пламени смеси ацетилена с воздухом чувствительность составляет 0,01—0,2 мкгЗг/мл [663, 1109], для пламени смеси закись азота—апетилен 0,006—0,04 мкг5г/мл.
108
Таблица 34
Величина ошибки при атомно-абсорбционном определении стронция в присутствии ряда посторонних ионов
Посторонние ионы Величина ошибки, % Посторонние ионы Величина ошибки, %
пламя ацетилен— воздух пламя закись азота—ацетилен пламя ацетилен — воздух пламя закись азота — ацетилен
А13+ —87,4 —0,7 (СОО)22- —10,8 —2,6
Ве2+ —24,7 —0,5 F- —5,8 +0,3
Th1+ —24,6 +1,6 HAsO42- —18,7 +0,5
ТВ+ —93,5 +1,3 —11,0 —1,9
ив+ —59,7 +1,2 SiO32- —54,4 —0,3
\74+ —62,6 —3,7 VO3- —75,9 +2,3
Zr4+ —62,3 + 1,7
Калибровочные графики зависимости оптической плотности от концентрации прямолинейны до концентрации 20—50 лкгЗг/лы [409, 485, 550, 867].
При использовании в качестве источника излучения ксеноновой лампы со сплошным спектром [1026] искривление калибровочного графика к оси абсцисс начинается уже при невысоких значениях поглощения.
Как и в эмиссионной фотометрии пламени, различаются два вида помех при атомно-абсорбционном поглощении стронция. Первый вид помех связан с образованием труднолетучих соединений и проявляется в пламени смеси ацетилена с воздухом. Наиболее часто отмечается влияние катионов Al3+, Ti4+, Zr4+ и других, анионов РО|~ и SiOt-
В качестве примера в табл. 34 приведены величины ошибок при атомно-абсорбционном определении стронция в присутствии тринадцати посторонних элементов [990]. Содержание стронция составляет 1 • 10“4 М, в присутствии 6-кратной концентрации посторонних ионов. Остальные 39 исследованных катионов и анионов не мешают. Как видно из табл. 34, эти помехи в значительной степени устраняются при переходе к более горячему пламени смеси закись азота—ацетилен.
Другой вид помех — вследствие ионизации атомов стронция, например, за счет влияния Са и Ва [1083], увеличения атомного поглощения от присутствия Na и К [868, 1164] и др.
Для устранения помех в атомно-абсорбционной фотометрии пламени рекомендуются те же приемы, что и в эмиссионной фотометрии пламени. Образование труднолетучих соединений (химические помехи) устраняется добавлением различных веществ, препятствующих их образованию.
109
Так, например, влияние ионов алюминия и кремневой кислоты может быть устранено добавлением солей кальция в 15- и 25-н'рат-ных количествах [409]. Однако наибольшее применение полечило добавление раствора LaCl3 в концентрации 10 мг!мл [1249]. /Рекомендуется также добавление La в смеси с 8-оксихинолином /и НС1 [1052] или аммонийной соли этилендиаминтетрауксусной кйслоты [1033]. '
Как уже было отмечено, эти помехи меньше или вообще отсутствуют при использовании пламени смеси закись азота—ацетилен [609]. Устранение помех со стороны железа путем подбора состава газов в пламени описано в [739]. Помехи со стороны Ti, Zr и Hf отсутствуют, если они находятся в виде комплексных циклопента-диенильных соединений [1215]. Что же касается помех вследствие ионизации, то они эффективно устраняются добавлением 0,1—1 % солей натрия, калия или цезия (конечная концентрация 1—10 мг/мл).
Снижение оптической плотности пламен, содержащих стронций, в присутствии посторонних веществ использовано для разработки косвенных методов определения Si [781], РО|~ [1250] и Ti [618].
При определении стронция, растворенного в несмешивающих-ся с водой органических растворителях, для приготовления стандартов пользуются бутиратом стронция [1156].
В последнее время разрабатываются беспламенные методы определения стронция при испарении с металлических зондов танталовой [1012], угольной или рениевой [845] нити, а также метод импульсного испарения с петли вольфрамовой проволоки, вносимой в разреженное пламя [1270]. Чувствительность обнаружения стронция 1-10-4—4-10-12 г.
Находит применение графитовая трубчатая печь, которая позволяет определять стронций в незначительных количествах материала с высокой чувствительностью [535, 822, 998].
Спектральное определение изотопного состава стронция
Анализ изотопного состава стронция представляет интерес в связи с распространением рубидиево-стронциевого метода определения возраста горных пород. Для этой цели в образце определяют относительное количество нечетного изотопа 87Sr, так как соотношение концентраций других изотопов остается постоянным.
Спектральный анализ изотопного состава стронция с фотоэлектрической регистрацией спектра разработан в [142, 143]. Аналогичный метод с применением фотографической регистрации предложен в [700, 719]. Описание метода приведено также в руководствах [149, 150, 152].
Изотопические смещения спектральных линий стронция на столько малы, что линии четных изотопов сливаются. Линии нечетного изотопа 87Sr, спин ядра которого составляет 9/2, обнару-110
живают сверхтонкую структуру [837]. Это дает возможность, измерив соотношение интенсивностей двух компонент линии изотопа ®?Sr и линии, принадлежащей четным изотопам, определить относительное содержание нечетного и четных изотопов.
В \качестве аналитической используется в основном линия Sr II л = 4077,8 А. В работе [719] с фотографической регистрацией сдектра применяется линия Sr I X = 6791,1 А. Линия Sr II % = 4077,8 А соответствует переходу 2St/2 — 2Р>/2. Расщепление терма 2Л’з/2 пренебрежимо мало, сверхтонкая структура этой линии представляется дублетом, определяемым расщеплением терма 28ч„ расстояние между компонентами которого равно 0,158 см"1 [837].
Схема термов и расположение компонентов сверхтонкой и изотопной структур этой линии представлены на рис. 19. Компоненты А и В — это составляющие сверхтонкой структуры линии 87Sr, компонента X представляет собой слившиеся линии 84Sr + 8eSr + 4~ 88Sr. Для нахождения относительного содержания 87Sr измеряют относительную интенсивность компонентов А и В к X: {1А + 1В)ИХ.
Возбуждение спектра Sr осуществляется в разрядной трубке с полым катодом, охлажденным водой либо жидким азотом в атмосфере неона. Стронций вводится в полный катод в виде капли азотнокислого или солянокислого раствора, который затем высушивается.
Зависимость относительной интенсивности компонентов от давления газа, силы разрядного тока, навески и способа введения пообы в полый катод, ширины щели монохроматора, времени горения пробы и скорости сканирования интерференционной картины изучалось в работе [142].
Структура аналитической линии Sr разрешается при помощи интерферометра Фабри-Перо и регистрируется фотоэлектрически с помощью установки, описанной в работе [151]. Расстояние между зеркалами интерферометра выбирается так, чтобы составляющие сверхтонкой структуры линии 87Sr из соседних порядков не накладывались друг на друга. Этому соответствует толщина интерферометра 32 мм, что достигается путем использования стандартных распорных колец. В качестве монохроматора применяется спектрограф ИСП-51 при ширине щели 0,4 мм. Интерференционная картина проектируется на плоскость выходной диафрагмы (d = 2,4 мм) объективом с фокусным расстоянием 120 мм. Интерферометр имеет многослойные диэлектрические покрытия с коэффициентом отражения 86 %.
На рис. 20 представлены регистрограммы структуры линии Sr II X = 4077,8 А. Содержание Sr определяют по градуировочному графику, либо по расчетной формуле. Образец градуировочного графика приведен на рис. 21. Полученное в [142] хорошее соответствие между экспериментальным и теоретическим ходом градуировочного графика позволило авторам перейти к
111
z р
Рис. 19. Схема термов (а) и рас- % положение компонентов (б) линии Sr II = 4077,8 А [152]
А — »’St; В — »’Sr, X — ««Sr + "Sr + "Sr (Л4К — милликайзеры — 1 • 10-• см-*)
а
б
В X Л 1/7 |77
1—
1, 158 .
Рис. 21. Градуировочный график для определения стронция по изотопу878г
Рис. 20. Регистрограммы структуры линии Sr II % = 4077,8 А при концентрации 87Sr 7,0 (а) и 40(6) %
безэталонному методу анализа с помощью следующей формулы:
с,,7Чтч = 1,107 (—:--;—j — 1,04'.А .
("&г)выч ’ \ 1 -f- IxlIA + ZB /
Сравнение результатов анализа масс-спектрометрическим и спектральным методами показывает удовлетворительное совпадение данных [142].
Относительная ошибка определения зависит от содержания стронция в пробе и составляет 2—7%. При содержании 8,Sr 15— 50% ошибка определения не превышает 3%, при его содержании 7—15% ошибка определения возрастает до 5—7% [143]. Природа ошибок при спектральном анализе изотопного состава Sr изучена в [9, 144]. Показано, что путем выбора оптимальных условий определения ошибку анализа можно снизить до 0.2—1,5%.
112
Таблица 35
Изотопы, используемые в активационном методе анализа [255, 372]
Радиоизотоп, по которому проводится измерейие Период полураспада Т1Д Энергия у-излу-чения, Мэв Ядро-мишень Содержание изотопа в природной смеси, % Реакция Литература
83Sr 32,9 часа 0,17; 0,04 84Sr 0,56 п, 2п [Д
86Sr 63,9 дня 0,515 84Sr 0,56 п, у [581, 1225]
86Sr 9,86 У» и [61]
85mgr 70 мин. 0,150; 84Sr 0,56 71, У [4,61]
0,225 88Sr 9,86 п, 2п [4]
87mgr 2,83 часа 0,391 8eSr 9,86 п-> У [840]
8,Sr 7,02 у, У' [199]
88Sr 82,56 п, 2п [4, 241]
U9Sr 53,6 дня 1,462* 88Sr 82,56 d, р [858]
. 88Sr 82,56 71, У
* Энергия (3-излучения.
Активационный метод
Активационный метод нашел применение при определении Sr в различных объектах. Сводка использовавшихся ядерных реакций и свойства образовавшихся радиоактивных изотопов, по которым производится измерение активности, приведена в табл. 35.
Наибольшее распространение нашел метод определения по активности 87mSr. В большинстве случаев определение производится по измерению активности после радиохимического выделения Sr, которое проводится с использованием методов осаждения, экстракции или ионного обмена.
При определении стронция в морской воде [858] 1—2 мл образцов в ампулах облучают в реакторе потоком тепловых нейтронов в течение 5—7 мин. Добавляют носитель — соль стронция (40 мг Sr2+) и производят радиохимическое отделение Sr, которое включает осаждение карбонатов, растворение их в НС1, осаждение и удаление гидроокисей тяжелых металлов, двойное осаждение нитратов. В заключение проводят последовательное осаждение в виде хромата, сульфата и подсчитывают активность стронция.
В методе [2] отделение Sr после облучения производится экстракцией из щелочного раствора 1 М раствором 8-оксихинолина в хлороформе. Для определения выхода Sr в облученный водный раствор добавляют радиоизотоп 85Sr (E-f = 0,51 Мэв) с известной активностью и измеряют его активность после выделения основной массы стронция.
ИЗ
До облучения нейтронами Sr может быть выделен вместе с Са в виде оксалата [1179]. После облучения воды и добавления носителя Sr применяется также его осаждение в виде SrCO3 [1262].
Предварительное отделение стронция от основной массы образца методом ионного обмена используется в [759] при анализе кристаллов галогенидов щелочных металлов. >
Образец (0,2—0,5 г) растворяют в 30 мл воды, щелочноземельные элементы сорбируют на катионите Биорад AG 50W-X8, после чего колонку промывают водой, извлекают смолу, сушат ее, облучают в реакторе вместе со стандартом. Из облученной смолы вымывают поглощенные катионы горячим 4 М раствором НС1, содержащим носитель (Sr), и вводят в раствор 86Sr для последующего определения химического выхода. Из полученного раствора последовательно осаждают карбонаты, фосфаты, нитраты стронция и бария и измеряют активность 87mSr (£? =0,391 Мэе). Чувствительность определения '0,01 мкг Sr.
Применение у-спектрометра с высоким разрешением позволяет повысить точность метода и сократить число операций по отделению, так как возможно определение Sr в присутствии ряда посторонних элементов.
Определение стронция в горных породах описано в работах [433, 659, 716, 840, 841].
Образец (0,1—0,5 г) облучают в полиэтиленовой капсуле в течение времени от 15 сек. до 30 мин. в реакторе потоком нейтронов / = 1012—1013 ней-трон/см2 сек вместе с эталонами, в качестве которых используют, например, бумагу, пропитанную 0,001 мл раствора соли стронция с концентрацией 23 лггЗг/лгл. Облученные образец и эталоны выдерживают до 30 мин., переводят в раствор, добавляют носитель, затем следует обычное радиохимическое выделение Sr и измерение активности.
Для определения выхода Sr используется также выдерживание до полного распада 87mSr, повторное облучение в нейтронном реакторе и измерение активности того же изотопа при сравнении -с эталонами, содержащими количества Sr, соизмеримые с количеством добавленного носителя [840].
Определение Sr в биологических материалах по активности 87mSr описано в работах [197, 573, 795, 844, 991, 1210, 1309].
Пробу предварительно озоляют, облучают образец потоком нейтронов с энергией 1012—1,5-1013 нейтрон! см2-сек, проводят химическое выделение Sr одним из общепринятых способов, измеряют активность 87™Sr. В [991] используется предварительное концентрирование Sr путем осаждения оксалатов и нитратов. Полученные нитраты растворяют в воде, добавляют в качестве внутреннего стандарта медь, облучают в полиэтиленовой ампуле в течение 1 часа в потоке 1013 нейтрон!см2-сек. Через 30 мин. после облучения добавляют носитель Sr, очищают Sr осаждением нитратов и хромата, медь отделяют экстракцией дитизоном. Содержание Sr определяют по активности 87mSr и 64Сп (внутренний стандарт).
114
Во многих работах описано определение стронция методом активационного анализа без разрушения (деструкции) исследуемого образца.
Определение стронция в образцах, содержащих его большие количества, например в минералах и солях, описано в [244].
Образец (10—12 мг) облучают 2 мин. в реакторе потоком нейтронов с энергией 1,8-1013 нейтрон!см2-сек, охлаждают 10—20 мин. до полного распада 28А1 и 37S и измеряют у-активность 87mSr по пику с энергией 0,391 Мэв. Чувствительность метода 1-10-2% Sr.
Методика недеструктивного определения примеси Sr в кормовых фосфатах [241] заключается в следующем.
Пробы облучают в течение 10 мин. в нейтронном генераторе с потоком ~ 5-10s нейтрон!см2-сек. После облучения пробу выдерживают 20—25 мин. для распада короткоживущих радиоизотопов и измеряют активность 87mSr по у-пику (0,391 Мэв). Чувствительность определения 2,5-10-3 г Sr.
Описано [40] недеструктивное определение Sr в нефтях путем облучения потоком нейтронов от генератора, в котором присутствуют как быстрые нейтроны с энергией 14 Мэв, так и тепловые нейтроны. Выход генератора 109 нейтрон!сек, время облучения 20 мин. В спектре излучения [Ge(Li) спектрометр] наблюдается линия как 8;mSr, так и 85n1Sr.
Чувствительность определения стронция в шести матричных материалах (кровь, молоко, вода и т. д.) при недеструктивном активационном анализе (облучение в реакторе 30 мин., у-спектро-метрия) приведена в работе [889]. Определяемый минимум Sr снижается с уменьшением содержания в объекте анализа Na и С1 и составляет, например, в водопроводной воде 3 мкг!мл (содержание Na 57, Cl 110 мкг/мл), в дистиллированной воде 0,009 мкг/мл.
В работах [200, 1174] описан недеструктивный метод определения ряда элементов, в том числе и Sr, в алмазах. Время облучения в реакторе 1 час, определение производится по 87mSr, чувствительность метода 1,95-10~5% [200].
Определение Sr путем возбуждения метастабильного состояния по реакции у,у' в водах, рассолах, рудах и продуктах их переработки описано в [199, 770, 1340]. В [199] для облучения используют тормозное у-излучениес энергией 4,2 Мэв, получаемое в линейном ускорителе электронов. Тормозные у-кванты образуются в свинцовой мишени. Регистрацию наведенной у-активности осуществляют при помощи сцинтилляционного у-спектрометра с кристаллом NaJ(Tl). Чувствительность определения 0,3 мг Sr (6-10~5 г/г).
Содержание Sr в рассолах [1340] определяют путем облучения источником с 9700 кюри 60Со. Мощность дозы у-излучения составляла 106 рентген!час. Измерение активности 87mSr производят при помощи сцинтилляционного датчика и одноканального анализатора. Чувствительность метода невелика.
115
Использование изотопа 8BSr и 8BmSr для нейтроно-активационного определения Sr описано в работах [96, 272, 581, 758, 1^25].
В методе [1225] при'определении ряда элементов, в том числе и Sr,/в биологических материалах пробу запаивают в кварцевую ампулу, обличают 24 часа' вместе с эталонами в реакторе потоком тепловых нейтронбв — 5* • 1013 нейтрон! см2 сек, выдерживают 2—3 дня после облучения. Затем’облу-ченную пробу подвергают мокрому сожжению путем нагревания со смесью конц. HNO3 и 30%-ной Н2О2, далее проводят радиохимическое отделение Sr, включающее осаждение благородных металлов SnCl2, экстракцию из водного раствора Fe, Ga, Sb, Sn и Th метилизобутилкетоном, экстракцию других посторонних элементов — ТБФ, осаждение сероводородом, осаждение сульфатов, отделение Сг и La, осаждение карбонатов. Измеряют радиоактивность на сцинтилляционном у-спектрометре с кристаллом NaJ(Tl).
При определении Sr в арсениде галлия [96] для отделения макроосновы от примесных элементов после облучения образца в реакторе производят отгонку мышьяка в виде бромида и экстракцию Ga, Au, Fe и Т1 р,Р'-дихлордиэтиловым эфиром из 10А НС1. Далее проводят последовательные экстракции хлоридом диметил-бензилалкиламмония в 1,2-дихлорэтане, ТБФ, а-«-нонил-пиридин-N-оксидом в бензоле, снова ТБФ. После отделения РЗЭ в водной фазе остаются щелочные и щелочноземельные элементы, Мн, Ni и Gr, которые определяют у-спектрометрически без дальнейшего разделения.
Методика ^-спектрометрического определения стронция в минеральных водах описана в [272]. В этом случае пробы воды облучают в реакторе потоком тепловых нейтронов мощностью 1019 нейтрон!см2 • сек. Измерения проводят через 2 месяца после прекращения облучения на сцинтилляционном 4л-у-спектрометре.
Возможность полуколичественного определения Sr в алюминии по долгоживущему изотопу 85Sr показана в [758].
Недеструктивный метод определения Sr и других элементов в стандартных горных породах описан в [581].
Пробу (100 мг) и стандарты сравнения заворачивают в листки Al-фольги, 5 проб, 12 стандартов и 2 холостые Al-пробы упаковывают в цилиндрический Cd-контейнер, облучают 2 дня в ядер ном реакторе потоком тепловых нейтронов 1,5-1013 нейтрон!см2-сек. Снимают у-спектры облученных проб и стандартов 2 раза через 5—7 и 20—25 дней после конца облучения при помощи у-спектрометра с Ое(Ы)-детектором. Метод позволяет определять содержание в образцах стандартов горных пород 1,6—6,2-10“2%.
Активационный метод используют для определения стронция в дентине и эмали зубов [1187], крови и продуктах жизнедеятельности человека [1191], в сплавах с кремнием [41], а также при определении возраста земных горных [1351, 828] и лунных [1350] пород.
116
Масс-спектрометрический метод
Масс-спектрометрия используется для определения изотопного состава стронция, знание которого необходимо при вычислении геологического возраста образцов по рубидиево-стронциевому методу и при определении микроколичеств стронция в различных объектах методом изотопного разбавления. Определению изотопного состава Sr посвящено значительное число работ [3, 94, 137, 262, 496, 710, 713, 714, 819, 834, 858, 1132, 1189].
В качестве источника ионов применяют окись стронция, нагреваемую на платиновой проволоке, хлорид или сульфат стронция, нагреваемые на вольфрамовой проволоке, покрытой никелем, или проводят ионизацию паров металла, нагретого до 700° G, медленными электронами; ленточные термоионные источники используются для ионизации сульфата [137], нитрата [137, 1189], оксалата [713] или гидрата окиси стронция, впитанного алюмосиликатным ионообменником [3].
В искровом источнике, используемом для масс-спектрометрического определения следовых количеств стронция в кристаллах KG1, применены танталовые электроды.
Анализируемый образец (2x2x5 мм) укрепляют в держателе из танталовой фольги [768]. Обыскривание стержня из анализируемого металла — натрия, помещенного внутри графитового стакана, при]использовании контрэлектрода] из Au-проволоки проводят в [544]. Электроды предварительно охлаждают жидким азотом до температуры —190° С. Другой вариант методики заключается в использовании двух экземпляров из исследуемого образца в виде стержней, охлаждение электродов осуществляется до —50° С.
Искровой источник ионов при определении Sr используется и рядом других авторов [552, 1063]. Предельная абсолютная чувствительность определения Sr масс-спектральным методом вакуумной искры составляет 9 • 10-11 г [450].
Конструкция двухколлекторной системы для масс-спектраль-ного прецизионного измерения отношения 87Sr/86Sr предложена в [817]. Использован нулевой компенсационный метод регистрации отношения изотопов с записью показаний на ленте электронного потенциометра. Проведено сопоставление предложенного метода с одноколлекторным методом измерений относительной распространенности изотопов.
Типичный масс-спектр стронциевого объекта, содержащего Rb, показан на рис. 22 [3]. Пик массы 85 принадлежит 85Rb, пик массы 87 — составной (87Sr + 87Rb).
I Возможные наложения линий других ионов на линии двухзарядного иона Sr, а также требуемое для их разделения значение разрешающей * силы масс-спектрометра (М/АМ) приведены в табл. 36’[450].
Методика определения примеси Sr в солях натрия путем изотопного разбавления пробы образцами, обогащенными 86Sr,
117
Рис. 22. Масс-спектр стронция (цифры над пиками — кратность увеличения записи)
и определение в смеси относительной концентрации изотопов 88Sr/86Sr на масс-спектрометре описаны в [1189].
Проба, предварительно прошедшая специальную обработку и переведенная в раствор азотнокислых солей, наносится на W-ленту. После предварительной тренировки источника ленту нагревают до 1500° С, записывают до 50 регистрограмм, из которых рассчитывают среднее значение отношения интенсивностей. Чувствительность определения составляет 10~9 г Sr, квадратичная ошибка единичного определения 0,05%.
86Sr в качестве изотопного разбавителя использован также в [713]. Для той же цели применен 84Sr [94]. Определение Sr в морской воде [858] проводят с помощью двух индикаторов 84Sr и 88Sr.
При определении стронция в силикатных породах и метеоритах его предварительно отделяют от других элементов, используя различные приемы [1066, 1244].
В платиновом тигле выпаривают растворы стабильных, изотопно обогащенных индикаторов определяемых элементов, затем вносят образец (100 мг), разлагают с помощью HF и НС104, раствор выпаривают досуха, остаток растворяют в 2 мл 2,5 М НС1. Полученный раствор пропускают через катионообменную колонку с ионообменником Цео-Карб-225 в Н+-форме и элюируют щелочные металлы 2,5 М НС1. Затем колонку промывают водой и проводят элюирование 2,07 М раствором HNO3, собирая фракцию от 20 до 140 мл, содержащую стронций. Полученную фракцию подвергают дополнительной очистке на колонке с анионитом Де-Ацедит-FF и анализируют на масс-спектрометре [1066].
Метод изотопного разбавления используется для анализа силикатных стекол [1060], изучения продуктов ядерных реакций [1127, 1128, 1368], определения Sr в тектитах [1132], лепидолитах [497, 834], изверженных породах [710]. Определение стронция в лунном грунте масс-спектральным методом проведено в [84, 1184].
Возможность определения шестидесяти элементов, представляющих определенный геологический интерес, с помощью масс-спектрального метода вакуумной искры найдена в [1063]. В порядке подготовки к анализу лунных образцов проведен анализ базальтовых пород. Определение чувствительности метода выполнено с. помощью стандартного образца диабаза.
118
Таблица 36
Наложение линий масс-спектра, образованных различными ионами
Массовое число Ион Изотопная распространенность элемента,% Точная масса м/лм (для пары различных ионов)
9,9---- 42,95468
2000
86- 2+
43-
44
с3‘н
Са
А1'6О-
с16о
Sr-
С2‘н416о±
•16л
I о
с/н/о^
99,1
42,9764461
43,0183879
43,0547122
1000
1200
430
2,1---- 43,9554950
92,0---- 43.971843
98,9--- 43,9898282
44,0262125
с3'н8
44,0625968
2700
2400
1200
1200
18 000
1300
810
-600
2300-
600
400
410
I
Пробы перед анализом высушивают при 110° С, добавляют 2,5-10~2% In в виде IibOj, (в качестве внутреннего стандарта) и смешивают с угольным порошком (1 : 1) или Ag (1 : 9). Из смеси прессуют диски толщиной 1—2 мм, которые затем разрезают на четыре части, полученные доли устанавливают плоскими торцами друг к другу в Та-держателях источника ионов. Получена хорошая сходимость с данными нейтронно-активационного анализа.
G целью изучения условий формирования катода для электронных ламп проведено исследование продуктов, выделяющихся при прогреве карбонатов щелочноземельных металлов в ионном источнике масс-спектрометра при 1100—1800° G [671].
Рентгенофлуорес дентпый метод
Рентгенофлуоресцентный метод определения стронция в последнее время находит возрастающее применение. Преимуществом его является возможность производить анализ без разрушения
119
образца и быстрота выполнения (анализ продолжается 2—5 мин.). В методе исключается влияние основы, его воспроизводимость + 2—5%. Чувствительность метода (1 • 10-4 — 1-10~3% Sr) достаточна для большинства целей [1356]. Характеристика рентгеновского излучения Sr, а также Y и Rb, часто используемых в качестве элементов внутреннего стандарта, приводится ниже (табл. 37).
Таблица 37
Характеристика рентгеновского излучения Sr, Y и Rb [977]
Элемент Атомный номер Z Критическое напряжение, кв ка,А К₽,А К *, А Углы Брэгга 2 0, град (кристалл-анализатор LiF)
Rb 37 15,2 0,926 0,827 0,814 26,60 23,71
Sr 38 16,1 0,877 0,783 0,770 25,16 22,42
Y 39 17,0 0,831 0,740 0,727 23,82 21,18
* Край поглощения.
Линия стронция La. совпадает с линией железа Ка, поэтому в общем не используется в анализе, однако находит применение при определении больших содержаний стронция в смеси карбонатов отронция и кальция [388]. В большинстве спектрометров применяется излучение рентгеновской трубки с вольфрамовым анодом (40—50 кв, 20—50 ма), однако для определения малых содержаний стронция выгоднее применение излучения трубки с молибденовым анодом [632]. Используется также рентгеновское (52 кэв) и у-из-лучение (84 кэв) изотопа 170Тш [153, 154, 239]. Вакуумирование спектрофотометра для удаления воздуха, поглощающего излучение стронция, приводит к повышению чувствительности метода.
Обычно в спектрофотометрах применяются кристаллы-анализаторы LiF, реже кварц или топаз. Сцинтилляционные счетчики в соединении с усилителем и дискриминатором, входящие в конструкцию современных спектрометров, позволяют повысить чувствительность метода благодаря увеличению эффективности счета и снижению фона. Находят применение полупроводниковые Ge(Li)-[154] и Si (Ы)-детекторы [153, 949].
Техника измерения обычно заключается в измерении интенсивности линии стронция Ка (скорости счета импульсов) и фона у основания линии с обеих ее сторон. Применение внутреннего стандарта снижает ошибки вследствие поглощения рентгеновского флуоресцентного излучения образцом. В качестве такого стандарта кроме Y [736, 977, 1163] и Rb [644, 1274] используют также Мо 120
{Z = 42) [736, 1300, 1356] и As (Z = 33) [1300]. При этом фон измеряют между линиями стронция и элемента внутреннего стандарта.
Исследуемое вещество (например, горные породы) может быть помещено в виде порошка непосредственно в кюветы прибора [803, 977,1356]. Указывается, что на результаты анализа влияет размер зерен образца [641, 642]. Распространена техника подготовки, образца путем его прессования в таблетки диаметром 12—25 мм. В качестве связывающего вещества при прессовании таблеток применяются борная кислота [644], хлорид калия [1125], полистирол [72, 281], терефталевая кислота или метилцеллюлоза [651], сахар [767], коллодий [1192]. Порошок образца может быть помещен в держатель из плексигласа и запрессован под полимерной пленкой [1111] или нанесен на клейкую пленку [1155].
Анализируемые растворы могут быть выпарены на фильтровальной бумаге [647, 1076], однако выпаривание на полимерной пленке (при добавлении поверхностно-активного вещества для лучшего смачивания ее) дает в 10 раз меньший фон [791, 1274].
Большое значение при анализе рентгенофлуоресцентным методом имеет исключение влияния состава образца на результаты анализа вследствие поглощения в нем рентгеновского излучения. Используются различные методы. Поправка на поглощение может быть введена расчетным путем [481] исходя из содержания главных составных частей образца [621, 803, 925], или же поглощение линии стронция матрицей может быть определено экспериментально для образца на том же приборе [767].
Целесообразно применение техники внутреннего стандарта, о чем уже упоминалось выше. Во многих случаях используют синтетические стандарты, по составу имитирующие анализируемые объекты с известным содержанием Sr [281, 651, 977, 1125, 1155] или же для приготовления стандартов берут образцы анализируемого материала с известным содержанием стронция, к которым добавляют определенные его количества [849, 940, 1155]. Может быть применен также метод добавок [1107].
Рентгенофлуоресцентный метод определения стронция нашел применение при анализе разнообразных образцов — воды [384, 761, 1342], в частности, водопроводной [1274] и морской [1330], горных пород [153, 154, 239, 627, 632, 736, 803, 849, 977, 1155, 1300], вулканических выбросов [878], каолинов и глин [940], силикатов [925] и их стандартных образцов [1107], почв [1337], руд и минералов [281], геохимических проб [1356], озерных отложений [651], биологических материалов [621, 767, 1076, 1111, 1125, 1192, 1342], в том числе растительных [622, 1111], налетов в электронных трубках [1171], вольфрамовых материалов [994], бинарных смесей титанатов кальция и бария [583].
Глава V
ОПРЕДЕЛЕНИЕ СТРОНЦИЯ В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
При определении Sr в природных и промышленных объектах наибольшее применение нашли спектральные методы — эмиссионный спектрографический и пламеннофотометрический. В последнее время широко используется атомно-абсорбционный метод. Фотометрический метод, требующий предварительного отделения стронция от других элементов, используется сравнительно редко. По той же причине, а также в связи с длительностью хода анализа в настоящее время почти не используются гравиметрические и титриметрические методы.
АНАЛИЗ ВОД
Перечень различных работ, в которых приведено описание методов определения стронция в водах, представлен в табл. 38.
При фотометрическом определении стронция в водах находит применение о-крезолфталеинкомплексон [379, 424, 565]. Стронций предварительно отделяют вместе с кальцием в виде оксалата, а затем отделяют от кальция ионообменной хроматографией или же с помощью радиальной хроматографии на бумаге. Косвенным фотометрическим методом определяют стронций в рассолах [377].
Рассол (10 мл) нагревают до 50—70° С, прибавляют 20—30 капель раствора, содержащего 150 г (NH4)2CO3 и 150 г NH4C1 в 500 мл 2 N NH4OH, и фильтруют. Осадок карбонатов промывают разбавленным раствором (NH4)2CO3 (раствор осадителя разбавляют в 10 раз) и растворяют в минимальном количестве НС1 (1 : 1). К полученному раствору прибавляют 1—2 мл эфирного раствора НС1 (перемешивают 354 мл 12 N НС1, 46 мл воды и 100 мл эфира) и центрифугируют. Осадок ВаС12 промывают раствором осадителя, а прозрачный раствор и промывную жидкость выпаривают почти досуха, прибавляют 5—10 капель конц. HNO3 и снова выпаривают. Остаток высушивают 15 мин. при 140—160° С, по охлаждении прибавляют 3—4 мл смеси (1 : 1) этанола и эфира (для экстрагирования Са) и центрифугируют. Раствор отбрасывают, а осадок растворяют в 10 мл этанола (1 : 1), прибавляют 5 мл 0,04 М К2СгО4, нагревают 15 мин. на водяной бане, по охлаждении прозрачный 122
раствор сливают в кювету с I = 1 см и спектрофотометрируют при 460 нм, Относительная ошибка определения Sr не превышает ±5%. Продолжительность анализа 3 часа.
Спектрографический анализ вод проводится с применением разнообразной техники. Анализируемая вода может быть введена в разряд дуги переменного тока с помощью фульгуратора [59, 60] или же наносится на поверхность электрода в количестве нескольких капель [37, 881, 1256]. Для устранения влияния различия в составе и количестве растворенных солей к воде может быть добавлен буферный раствор, содержащий хлориды Na, Са и La и комплексон III [1256].
При использовании фотоэлектрической регистрации с одновременным определением других 18 элементов воду предварительно концентрируют выпариванием [937, 945]. Высокую чувствительность дает вдувание пробы морской воды в зону разряда дуги переменного тока [973].
В ряде методов определение стронция в водах производится из сухого остатка. Раствор может быть нанесен на поверхность медного электрода и выпарен [1222]. При получении сухого остатка, чтобы он не поглощал водяные пары из воздуха, электроды с пробами оставляют стоять на плитке до проведения съемки спектра [796]. Применяется также переведение солей (в сухих остатках от выпаривания вод) в сульфаты [36, 201, 264]. Для устранения влияния состава воды при выпаривании возможно добавление 10—20% смеси NaCl и Na2CO3, а также 10—25% SiO2 [633].
При анализе минеральных вод стронций может быть предварительно выделен вместе с кальцием путем осаждения карбонатом аммония в аммиачной среде (после предварительного удаления SiO2). Смесь окислов прокаливают, смешивают с угольным порошком в соотношении 1 : 3 и 22—24 мг этой смеси испаряют в дуге постоянного тока [863].
Предложены методы анализа вод на содержание стронция и других микрокомпонентов, в которых стронций предварительно отделяется при использовании геля кремнекислоты [475], ионооб-менника Церолита-225 [598] или же с помощью органических реагентов [597].
При эииссиоином пламеннофотометриче.ском методе определения стронция предварительная подготовка образца чаще всего заключается в ионообменном отделении стронция от сопутствующих щелочных и щелочноземельных элементов [505, 645, 784, 853, 888, 1138, 1190], а также от мешающих анионов, или же в отделении стронция путем осаждения в виде оксалата [26, 375, 1335]. Однако при наличии спектрофотометра достаточно высокой разрешающей силы с автоматической записью спектра возможно прямое определение при использовании метода добавок или же при добавлении соли кальция в качестве «радиационного» буфера [1172].
Определение стронция в речной воде описано в [376].
123
Таблица 38
Определение стронция в водах
Объект анализа Метод определения Литература
Речные воды Спектрографический Пламеннофотометрический Атомно-абсорбционный [37, 58—60, 264, 633, 674, 796, 937, 1256, 1140] [26, 245, 350, 375, 376, 853, 888, 1335] [718]
Морские воды Спектрофотометрический Спектрографический Пламеннофотометрический Атомно-абсорбционный [379, 424, 565] [973] 1505, 634, 1190] 1551, 607]
Подземные и пластовые воды Спектрографический Пламеннофотометрический [36, 201, 246] [8, 90, 246]
Рассолы и рапа нефтяных промыслов Спектрофотометрический Гравиметрический Пламеннофотометрический Атомно-абсорбционный [377] [1299] 1б45] [550, 744]
Минеральные и лечебные воды Спектрографический Гравиметрический Пламеннофотометрический [475, 597, 598, 863, 881] [87] [1138]
Промышленные воды Спектрографический [204, 945, 1221, 1222]
Пробу (1—2 л) подкисляют 2—3 мл конц. HNO3, выпаривают до объема 35—40 мл и, разбавив до 50 мл, анализируют по методу добавок.
Те же авторы [375] при определении стронция в реках Подмосковья осаждают Sr вместе с Са (для отделения от Na) и после растворения осадка в НС1 фотометрируют. Прямое пламеннофотометрическое определение Sr в природной воде описано в [1335].
Образец (50 мл) смешивают с 10 мл «радиационного» буферного раствора, содержащего 10% НС1 и 5% двухзамещенного цитрата аммония, разбавляют до 100 мл и определяют Sr по резонансной линии 460,7 нм, одновременно определяя фон при 458 нм. Стандартные растворы содержат 1; 3; 5; 7 и 10 мкг Sr/мл и 10 % по объему «радиационного» буфера [853]. В качестве буфера можно применять и растворы солей кальция, которые, как и соли других элементов (Li, Na, К, Rb, Си, Zn и Ni), мало влияют на результат определения Sr.
Для определения Sr в рапе предложена следующая методика [645].
124
Три равные аликвотные части пробы помещают в три мерные колбы емкостью 50 мл, добавляют в первую и вторую из них соответственно 1 и 2 мг Sr (в виде SrCl2), вводят в каждую колбу по 15 мл этанола, разбавляют водой до метки. Вводя полученные растворы в пламя, регистрируют для каждого из них 3 раза фон пламени при 690 нм и интенсивность излучения Sr при 680 нм. Вычитают первую величину из второй и определяют содержание стронция графически, как обычно при работе по методу добавок. Чувствительность определения Sr увеличивается в 3—5 раз при добавлении этанола. При малых содержаниях Sr определяют по линии 460,7 нм, отсчитывая фон при 465 нм. Описанным методом в пробах рапы найдено до 1500 мкг Sr/мл, чувствительность метода 1 мкг Sr/мл.]
В подземных водах с минерализацией 5 г/л точность определения Sr 7—10%. Анализ ведется по методу трех эталонов или по методу трех добавок. В водах нефтяных и газовых месторождений >0,1 мг Sr/мл определяют с относительной ошибкой 10% [8].
В морской воде стронций определяют после его отделения от других металлов с помощью ионообменного метода [505, 1190] или же производят определение, не прибегая к предварительной обработке воды [335, 634].
Готовят ряд проб разбавленной морской воды, причем к части из них добавляют Sr в количестве 2,5; 5,0 и 7,5 мкг/мл. Для этого порции [морской воды по 25 мл, к которым добавлено 0; 125; 250 и 375 мкг Sr, разбавляют дистиллированной водой до 50 мл. Растворы фотометрируют, получая отсчеты при 460,7 нм\ из найденных величин вычитают фон пламени при 454 нм [634].
Концентрацию Sr в воде Черного моря определяют также по методу добавок [335].
Фотометрируют разбавленную вдвое морскую воду без добавки и с добавками 2; 4 и 8 мкг Sr/мл, записывая участок спектра в области 459,0—465 нм,. и по высоте пика линии 460,7 нм рассчитывают концентрацию Sr. В шести параллельных пробах образца воды найдено 6,8; 7,2; 6,84; 6,4; 6,92 и 6,92 мкг Sr/мл или в среднем 6,84 мкг'^г/мл. Максимальное отклонение составляет ± 0,4 мкг Sr/мл, среднее отклонение 0,168 мкг Sr/мл (2,4 отн.%).
Предварительное концентрирование стронция из разбавленных растворов можно производить путем соосаждения его с оксалатом кальция [26, 1335], или же с помощью ионного обмена [853].
В последнем случае 500 мл воды пропускают со скоростью 1—1,5 мл/мин через колонку, содержащую 10 мл катионита Амберлит IR-120, промывают колонку 50 мл воды и элюируют Sr 50 мл смеси 2 М CH3COONH4 и 1 М СН3СООН (pH 5,4). Собирают 50 мл элюата и фотометрируют, сравнивая со стандартными растворами, полученными путем проведения известных количеств стронция (50—500 мкг) через операцию отделения.
Атомно-абсорбционным методом стронций определяют в различных типах вод.
125
К 10 мл пробы речной воды добавляют 1 мл раствора, содержащего в виде хлоридов 10% La и 1% К (для устранения помех, связанных с влиянием химического состава и ионизацией атомов Sr в пламени). Такое же количество La и К вводят в стандартные растворы. Используется пламя смеси ацетилена с воздухом. Чувствительность метода 0,02 мг 8т/л [718].
При анализе морской воды [607] определение стронция производят при непосредственном распылении образца, стандартные растворы содержат, кроме стронция, основные составляющие морской воды приблизительно в той же концентрации. В рассолах нефтяных месторождений Sr определяют после разбавления их приблизительно в 100 раз с тем, чтобы концентрация Sr находилась в интервале 0,1—50 мкг/мл, который соответствует прямолинейному участку калибровочного графика. Коэффициент вариации 2% [550].
В водопроводной воде стронций можно определить рентгено-флуорес рентным методом [1274].
Аликвотную часть образца, содержащего 20—30 мкг Sr, концентрируют до объема 100 мл, добавляют 20 мл 8%-ного раствора щавелевой кислоты, NH4OH до pH 4. Выдерживают 1 час при 60° С, осадок отфильтровывают, фильтр сжигают при 500° С в кварцевой чашке. Карбонаты растворяют в 30%-ной HNO3, выпаривают досуха. Остаток растворяют в 1 мл раствора RbCl (содержащего 200 мкг Rb), добавляют 2 капли (1 : 60 000) раствора анионного смачивающего агента для понижения поверхностного натяжения; 2 капли полученного раствора помещают на пленку, высушивают в вакуум-эксикаторе 20 мин.
Стандарты готовят из раствора Sr(NO3)2 с добавлением 1 мл раствора RbCl (200 мкг Rb), раствор выпаривают до 1 мл; далее проводят подготовку, как описано выше.
Полученный на поверхности пленки остаток помещают в прибор. Измерение скорости счета производят на линиях Ка стронция и рубидия (установка гониометра 25,09° и 26,58°); фонизмеряют при 25,6°. Подсчет ведется в течение 100 сек. Из отсчета для линии вычитают величину фона и определяют отношение интенсивностей Для использованного прибора справед-
ливо соотношение cSr/cRb-0,96 = л'а(иг)'Л'а(11Ь)> гДе с — концентрация. Линейная зависимость сохраняется в интервале содержаний 50 —1000 'мкг Sr.
АНАЛИЗ МИНЕРАЛОВ, ГОРНЫХ ПОРОД, РУД И ПОЧВ
При определении стронция в этих объектах используются в основном спектральные методы — эмиссионный спектрографический, фотометрия пламени, атомно-абсорбционная спектрофотометрия. Сводка работ, посвященных анализу этих объектов, приведена в табл. 39.
При использовании гравиметрического метода [22, 449, 880] стронций определяют в виде сульфата. Предварительно выделяют группу щелочноземельных элементов; Sr отделяют от Ва и Са применением описанных ранее методов, например, путем осажде-126
Таблица 39
Определение стронция в рудах, минералах, горных породах и почвах
Объект анализа Метод определения Литература
РУДЫ Гравиметрический Спектрофотометрич еский Спектрографический Пламеннофотометрический Атомно-абсорбционный [390] [247, 363[ [И [76, 220, 295, 678] [661]
Минералы Г равиметри ческий Спектрографический Пламеннофотометрический Атомно-абсорбционный [22, 283, 449, 880] [56, 58, 202, 446, 501, 679, 740, 779, 812, 1037, 1327, 1380] [283, 428, 448, 789] [745, 1033]
Породы С пектрографический П л ам еннофотометр и ч ески й Атомно-абсорбционный Р ентгенофлуор есцентный [13, 54-56, 92, 205, 252,253, 265, 294, 515, 699, 709, 745, 780, 816,883, 1011, 1068, 1124, 1195, 1243] [44, 137, 139, 196, 219, 400,. 425, 548, 635, 669, 722] [178, 205, 486, 534, 688, 847, 1030, 1052, 1134] [977]
Почвы Спектрофотометрический Пламеннофотометрический [33, 35, 167, 680, 1206] [30, 1216]
ния Ва в виде хромата и последующего выделения Sr в виде нитрата. Sr может быть также отделен от Са осаждением сульфата в присутствии комплексона III, удерживающего Са в растворе.
Определение стронция в апатитово-нефелиновой руде спектрофотометрическим методом с хлорфосфоназо III рассмотрено в работе [247].
Тонко измельченную руду (0,1—0,2 г) разлагают при кипячении с разбавленной HNO3 (1 : 1), раствор выпаривают досуха сначала на водяной бане, а затем на песчаной. Для отделения Sr от большей части Са сухой остаток обрабатывают ацетоном, затем растворяют в воде, раствор переносят в мерную колбу. Аликвотную часть раствора разбавляют водой до объема 2,1 мл, прибавляют 2 мл раствора ацетатной буферной смеси (pH 4,6), 0,5 мл 5%-ного раствора комплексона III и 0,4 мл 0,02 %-ного водного раствора хлорфосфоназо III. Измеряют оптическую плотность при X 660 нм, количество Sr находят по калибровочному графику.
Спектрографический метод анализа применяется наиболее широко [56, 57, 58, 357, 493]. При прямом методе обычно используется дуга постоянного тока 7—17 а [294, 1195].
127
Пробу смешивают с угольным порошком [1223] и помещают в отверстие угольного электрода, например, диаметром 1,6 мм и глубиной 6—7 мм. Нагревают сведенные электроды 10 сек. при силе тока 3 а и фотографируют -спектры при силе тока 7 а с экспозицией 60 сек (спектрограф с кварцевой оптикой) или с экспозицией 120 сек (спектрограф со стеклянной оптикой). Внутренний стандарт — In и Pd [63, 492, 821, 1195].
В других методах применяется дуга переменного тока [13, 252, 265]. Эталоны готовят на основе пород, близких по составу к анализируемым объектам [252, 265]. В качестве внутреннего стандарта используют ряд элементов, например Ст [13, 358], V [1] и Со [252].
Применение метода добавок при определении стронция описано в [294].
Во многих методах применяется буферирование пробы разными веществами, что устраняет влияние различия в составе анализируемых образцов. Некоторые используемые буферные вещества приведены в табл. 40.
Для устранения влияния валового состава проб используется также предварительная химическая обработка образца, например, превращение в сульфаты [1011], нитраты (в случае легкорастворимых кальциевых минералов) [202], фосфаты [515], фториды [491].
Таблица 40
Применение буферных веществ в анализе горных пород спектральным методом
Объект анализа Соотношение образец: буфер: графит (С) Литература
Слюды, полевой шпат Образец: ВаСО3 : С = 1 : 1 : 2 [1037]
Силикатные породы Образец: CaSO4 : С = 1 : 3 : 4 [812]
Силикаты, карбонаты Образец: NaCl : СаСО3 = 1:1:2 [19]
Образец: СаСО3 : буф. смесь* = = 1 : 2 : 18 [357]
Силикатная фаза метеоритов Образец: LiF : С = 1 : 2 : 3 [88]
Горные породы Образец: СаСО3 = 1:1 [1328]
Образец: SiO2 : С = 1 : 1 : 2 [1068]
Образец: SiO2 : NaCL = 1:1:1 [205]
Образец: СаСО3 : С = 2 : 1 : 1 [1124]
Радиоактивные минералы Образец: (NH4)2SO4 =1:9 [501]
Силикатные и карбонатные породы Образец: K2SO4 : С = 1 : 1 : 2 [816]
* Буферная смесь — графит, содержащий 10% Na2CO3 и 1,5% Сг2О3.
128
Применяют также сплавление с карбонатом натрия [782] или со щелочью [679]. В этих случаях сплав растворяют в соляной кислоте и анализируют раствор. После сплавления с безводным LiBOa (или Na2B4O7) сплав измельчают, смешивают с угольным порошком в соотношении 1 : 2 и испаряют в дуге постоянного тока [883].
В некоторых методах при определении микрокомпонентов в горных породах образец переводят в раствор и концентрируют стронций с помощью ионного обмена [699, 779] или осаждают вместе с кальцием в виде оксалата, остаток прокаливают и спект-рографируют [861].
Для определения стронция в породах может быть использована фотометрическая регистрация излучения с помощью квантометра [92, 779, 780, 1313].
Сцинтилляционный метод определения стронция при использовании ацетилено-воздушного пламени и вдувании растертого образца в пламя описан в [305]. В работах [205, 253] стронций определяют в образцах при вдувании порошка в дуговой разряд.
Пламеннофотометрический метод определения стронция при анализе горных пород и минералов находит широкое применение.
Прямое пламеннофотометрическое определение стронция в силикатах (граните и диабазе) производят следующим образом [721, 722].
Образец (1 г) растворяют в смеси 5 мл НС1О4 и 20 мл HF при нагревании на водяной бане в течение 2 час. Раствор выпаривают досуха на песчаной бане. К остатку прибавляют 3 мл НС1О4 и повторно выпаривают. При малом содержании Са в образце к остатку прибавляют Ю’.и.г раствора СаС12, содержащего в 1 мл 10 мг Са (для исключения гасящего действия А1 на интенсивность излучения Sr), и разбавляют водой до 50 мл. К 25 мл полученного раствора прибавляют SrCl2 (250 мкг SrO) и разбавляют водой до 50 мл (раствор образца с добавкой). Оставшиеся 25 мл раствора также разбавляют водой до 50 мл (раствор образца). Оба раствора фотометрируют при 460,7 нм с помощью спектрофотометра Бекман DU (кислородно-водородное пламя). При содержании Sr 0,01—0,14% относительная ошибка определения 2—3%.
Применение солей кальция в качестве буфера описано и в других работах [137, 139, 196, 219, 333, 400, 548], однако оно ограничено наличием в препаратах Са следов Sr и трудностью учета вызываемого ими непрерывного фона. Особенно значительны эти помехи при высоких концентрациях Са. Поэтому при определении Sr в различных природных материалах рекомендуется так производить разложение последних, чтобы большая часть Са была отделена [220]. Хорошие результаты получены при определении Sr в силикатных и фосфатных рудах, а также в минералах с использованием Pr (La, Nd) для устранения мешающего влияния посторонних элементов [333, 425].
После удаления SiO2 с помощью HF остаток переводят в раствор (конечная концентрация 10—20 мкг8т/мл), смешивают с раствором соли празеодима
5 Н. С. Полуэктов и др.
129
(молярное отношение Рг и элемента, снижающего интенсивность излучения Sr, должно составлять ~ 25 : 1), вводят НС1О4 до конечной концентрации 5%, разбавляют водой и фотометрируют по линии 460,7 нм, измеряя фон при 454 и 466 нм. Стандартные растворы Sr должны содержать такие же количества Рг и НС1О4 [333].
Разработаны методы определения стронция в железосодержащих минералах, в которых для повышения чувствительности определения и устранения влияния других элементов предложено добавлять 8-оксихинолин к анализируемым и стандартным растворам [76, 668, 669, 670, 789].
Образец (0,5 г) разлагают нагреванием с HF и H3SO4, остаток после выпаривания растворяют в НС1, раствор разбавляют до 100 мл. Отбирают 50 лл, прибавляют 25 мл раствора 8-оксихинолина (200 г последнего растворяют в 400 мл ледяной СН3СООН и разбавляют водой до 1 л), 20 мл НС1 (1 : 1), разбавляют до 100 лиг и фотометрируют, сравнивая с соответствующими стандартными растворами, также содержащими 8-оксихинолин и НС1.
Очень часто элементы, мешающие определению Sr в горных породах и минералах, удаляют из раствора, например, с помощью ионообменного метода [1115, 1287]. В последнем случае определяют стронций пламеннофотометрическим методом с ошибкой 1 % (при соотношении Sr : Ва = 1 : 300). При переработке фосфатных [398J или боратных минералов [399] ионный обмен используют для отделения металлов от анионов РО4~ и ВО3 соответственно, также мешающих пламеннофотометрическому определению стронция.
Наконец, в части работ предусматривается отделение полуторных окислов от стронция (или от всей группы щелочноземельных металлов) путем их осаждения в виде гидроокисей [7, 44, 75, 9Ч, 139, 426].
При определении стронция в минералах, содержащих кальций и сульфаты [333], пробу разлагают сплавлением со смесью К2СО3 и Na3CO3 (1 ; 1).
Навеску образца 0,1 г сплавляют в платиновом тигле с 3 г смеси карбонатов указанных металлов, сплав растворяют в горячей воде и фильтруют, а осадок промывают 0,5%-ным раствором Na3CO3. Затем осадок помещают в стакан и растворяют в 25 мл 5 N НС1, промывая фильтр, на котором находились карбонаты. Полученный раствор нейтрализуют аммиаком в присутствии индикатора метилового оранжевого до появления желтой окраски. Раствор нагревают до осаждения гидроокисей, охлаждают, разбавляют до 250 мл, фильтруют и фотометрируют по линии 460,7 нм, сравнивая со стандартными растворами, содержащими 25—250 мкг5т1мл (в виде хлоридов).
Метод дает возможность определять Sr при его содержании в пробе от 0,05 до 60%. При малом содержании Sr (менее 5%) рекомендуется для большей точности получать отсчеты для фона по обеим сторонам от линии 460,7 нм. Среднее значение отсчетов для
130
фона вычитают из отсчетов для растворов как пробы, так и стандартных растворов. При большом содержании полуторных окислов в породе, ввиду возможного захватывания ими стронция, необходимо переосаждение гидроокисей перед фотометрированием раствора.
Если содержание Sr <^0,05%, производят его концентрирование путем упаривания фильтрата, полученного после промывания осадка гидроокисей [93].
Определению стронция в горных породах и минералах пламеннофотометрическим методом посвящен также ряд других работ {138, 237, 446, 448, 533, 722, 790, 1264, 1265].
Ход анализа при атомно-абсорбционном определении стронция в горных породах и минералах, как уже отмечалось, зависит от рода используемого пламени и состава образца [295, 534, 537, 661, 847, 1030, 1033, 1052, 1134]. Описано определение стронция (в диапазоне концентраций 0,0025—0,21%) в эталонных образцах гранита (G-1) и диабаза (W-1) [857].
Навеску образца (1 г) растворяют в HF, HNO3 и НС1О4, упаривают до прекращения выделения паров НС1О4, остаток растворяют в НС1, разбавляют до 100 мл раствором НС1 (1 : 20). Перед анализом к аликвотным частям раствора (2,5 или 10 мл) прибавляют LaCl3 (1% La), НС1, разбавляют растворы до 25 мл и фотометрируют в воздушно-ацетиленовом пламени.
При использовании пламени смеси закиси азота с ацетиленом образец силикатной породы переводят в раствор обработкой кислотами и фотометрируют при добавлении хлорида калия до концентрации 1 мг К/мл. Стандартные растворы готовят по составам, близким к анализируемым. С этой целью используют образец гранита с известным низким содержанием Sr (например, 0,0015%), который переводят в раствор. Аликвотные части полученного раствора смешивают с различными добавками Sr. Коэффициент вариации 1,7% [534, 857].
Сплавление с метаборатом лития рекомендуется при анализе силикатов в [1030].
Мелкоизмельченный образец (0,1 г) смешивают с 0,5 г LiBO2 и сплавляют в графитовом тигле 10—15 мин. при 1000° С, сплав растворяют в 40 мл 3%-ной HNO3. 10 Л1Л полученного раствора смешивают с 10 мл 2%-ного раствора соли лантана и фотометрируют.
При определении стронция в баритах (0,5—8,0 96 SrO) поступают следующим образом.
Минерал (0,1 г) сплавляют с 1—1,5 г Na2CO3 в платиновом тигле при 000° С. Сплав обрабатывают водой, осадок отфильтровывают, промывают 0,5%-ным раствором Na2CO3, затем водой и растворяют в 15 мл горячей НС1 (1 : 3). Для устранения влияния Ti и А1 прибавляют 3 мл раствора СаС12 (1 мл содержит 10 мг Са), разбавляют до 100 мл и фотометрируют [1033] (рис. 23).
5*
131
Рис, 23. Влияние катионов на поглощение линии стронция (X = 460,73 нм) в пламени ацетилен—воздух
1 — Си; г — Fe, Са, Ва;
3 — Mg; 4 — Al;
5 — Ti
В фосфатах, например в апатитах, стронций определяют после переведения образца в раствор и отделения от РО4“ с помощью катионита [8471 или анионита — с использованием метода добавок [661].
Образец апатита (1 г), содержащего 2—3% SrO, дважды обрабатывают 5 мл конц. НС1 с выпариванием раствора досуха, остаток растворяют в 1 мл той же кислоты и 50 мл горячей воды, раствор отфильтровывают, фильтр промывают водой. Полученный раствор пропускают со скоростью 3 мл1мин через колонку с катионитом Леватит-S 100 в Н+-форме (10—12 мл), промывают колонку 150 мл 0,01 N НС1, элюируют катионы 250 мл kN НС1, элюат разбавляют водой до 500 мл. В три мерные колбы емкостью 100 мл отбирают по 25 мл полученного раствора, добавляют в одну из колб 10 мл, а в другую 20 мл отандартного раствора SrCl2 (1 мгЗ?1мл), разбавляют водой до метки и фотометрируют.
Рентгенофлуоресцентный метод применяется для определения стронция в осадочных породах нефтяных месторождений [977].
Образец анализируемого ангидрита (CaSO4) измельчают до 200 меш' 100 мг его смешивают со 100 мг матричной смеси (матричная смесь — 0,5% УгОд в CaSO4), помещают образец в кюветы и сканируют спектр в области 20 = 20—27° (кристалл-анализатор L1F).
Стандарты готовят из ангидрита, содержащего 0; 0,1; 0,2; 0,3 и 0,4% Sr, добавляя при их изготовлении стронций в виде 8гСО3. Затем эти образцы смешивают с матричной смесью в соотношении 1:1.
Содержание Sr вычисляют по формуле: cSr (%) = 0,6920-Ка(8г)/Ка(у)— 0,3602, где A’a(Sr/-^a(Y) — отношение интенсивностей.
Определение стронция в почвах производят обычно эмиссионным спектрографическим методом [33, 35, 167, 680, 1206] или методом фотометрии пламени [30, 1133, 1216]. Используется также атомно-абсорбционный метод [661]. Общий ход анализа такой же, как в случае горных пород и минералов. При спектральном определении стронция возможно также применение квантомет-ра [35].
132
АНАЛИЗ БИОЛОГИЧЕСКИХ МАТЕРИАЛОВ
Ряд работ посвящен определению стронция в биологических объектах (в морском планктоне, костях, животных тканях, растениях и др.). Преимущественно используются спектрографический [85, 131, 207, 242, 256, 890, 1354, 1382], пламеннофотометрический [195, 347, 574, 614, 702, 810, 910, 1009, 1218, 1306] и атомно-абсорбционный [274, 387, 432, 536, 661,662,664, 672, 737, 867, 1040, 1055, 1277, 1320, 1352] методы. Определение стронция титриметри-ческим методом в моче описано в [1305].
Общий ход анализа биологических материалов заключается в озолении материала и в дальнейшем непосредственном анализе золы по одному из спектрографических методов, описанных для горных пород; или в переведении золы в раствор с последующим анализом также по одному из приведенных выше методов эмиссионной или атомно-абсорбционной фотометрии пламени.
Определение стронция в золе костей спектральным методом описано в работе [890].
Образец (15 мг) смешивают с 135 мг CuSO4 и 150 мг графита, изготовляют 5 брикетов, помещают на поверхность специального электрода и полностью сжигают в дуге постоянного тока силой 7 а. Аналитическая линия Sr 4607,33 А, линия сравнения Си I 4704,59 А. Используют дифракционный трехметровый спектрограф.
Определение стронция в золе растений, молока и других пищевых продуктов эмиссионной фотометрией пламени [890].
Золу переводят в раствор, выделяют оксалат кальция (вместе со Sr), высушивают. 36,5 мг оксалатов растворяют при нагревании в 0,5 мл конц. НС1 и 1 мл дистиллированной воды, охлаждают, разбавляют водой до 10 мл и фотометрируют. Стандартные растворы содержат Са, Н2С2О4, НС1, Sr.
В других случаях определение стронция производят после предварительного выделения его с помощью ионообменной хроматографии [575, 1284].
Определение стронция в золах пищевых продуктов и мягких тканей методом] атомно-абсорбционной спектрофотометрии [661].
Материал озоляют 4 часа при 500° С в кварцевой чашке, остаток (0,3— 0,5 г золы) выпаривают дважды с 5 мл 6 N НС1, растворяют в 10 мл 0,1 N НС1, раствор фильтруют и разбавляют до 50 мл водой. 10 мл полученного раствора пропускают через колонку с анионитом (50 мл) Де-Асидит FF (d — = 1,8 см) в СН3СОО "-форме, промывают небольшими порциями 0,1 N СН3СООН, отбрасывают первые 9 мл элюата, остальной элюат собирают в мерной колбе емкостью 25 мл, затем измеряют поглощение света в пламени с помощью атомно-абсорбционного фотометра. Если необходимо, анализируемый раствор разбавляют водой до получения отсчета не более 12 делений шкалы.
133
Определение стронция в сыворотке крови рассмотрено в работе 1535].
Сыворотку (20 мкл) помещают в печь, высушивают 2 мин. при 100° С, сжигают при 110° С, атомизируют при 9 в 20 сек. Стандартные кривые строят на количествах от 5 до 40 мкл раствора SrCI2 (концентрация 0,05 мкг/мл). Определяемые содержания 0,01—0,05 мкг Sr/мл.
Определение стронция в костях животных рентгенофлуоресцентным методом [1192].
Золу анализируемых костей (2 г) измельчают до 200 меш, размешивают с 1 мл ацетона с добавлением 2 капель раствора коллодия. Высушенный порошок прессуют в диск в соответствующей форме. Стандартные образцы готовят из золы костей с добавлением Sr в концентрации 0,01—0,08%. Измеряют интенсивность линии стронция Ка и фона. При построении калибровочного графика из найденной интенсивности линий вычитают интенсивность фона и интенсивность счета Sr, входящего в состав золы. Ошибка определения ±0,001%.
АНАЛИЗ МЕТАЛЛОВ И СПЛАВОВ
В табл. 41 приведена сводка опубликованных методов. Приведен обзор атомно-абсорбционных методов [892].
Сплавы А1 — Sr с содержанием стронция 0,8—4% добавляют в качестве модификаторов в некоторые алюминиевые сплавы (например, А1 — Si и Al — FeAl3).
Сплав Al — Sr (2,5 г) растворяют в 200 мл 5 N НС1, добавляют HNOS, раствор кипятят, если нужно фильтруют и разбавляют водой до 1 л. 10 мл полученного раствора помещают в 100 мл колбу, добавляют 5 мл раствора LaCl3 (267 г LaCl3-7 Н2О в 1 л), 10 мл раствора KNO3 (260 г/л), разбавляют до метки водой и распыляют в пламя закись азота — ацетилен. Измерение атомного поглощения ведут по линии Sr 4607,33 А. Калибровочный график строят по растворам, содержащим А1 250, Sr 0—10 мкг/мл; La 5, К 10 мг/мл [820].
Определение примеси стронция в порошке металлического молибдена спектрографическим методом [692].
Порошок молибдена (1 г) смешивают с 0,28 г графитового порошка, содержащего 4%’порошка кобальта. Смесь помещают в кратер электрода и сжигают в дуге постоянного тока (300 в; 7,5 а). Время экспозиции 45 сек. Аналитическая линия Sr 4607,33 х, линия сравнения Со 7085,0 А. Определяемое содержание 0,001—0,01% Sr. Коэффициент вариации равен 10%.
Более высокая чувствительность достигается при использовании предварительного испарения, которое производят при температуре 2000° С.
Пробу (МоО3) смешивают в отношении 2:1с угольным порошком, содержащим 0,03% Li в виде Li2CO3, а также элемент внутреннего стандарта — Au (0,001%). Испарение производят из угольных стаканчиков, прием-
134
Таблица 41
Мет оды определения стронция неметаллах и сплавах
Объект анализа Метод определения Литература
Алюминиевые сплавы Атомно-абсорбционный [820]
Молибден Спектрографический [128, 692]
Свинец Спектрографический Пламеннофотометрический [187] [769]
Стали и сплавы Спектрографический [391]
Уран Спектрографический [16, 649]
Пламеннофотометрический [1139, 1151]
ником служит угольный стержень. Концентрат анализируют с помощью искрового разряда. Линия Si 4077,71 1, линия сравнения Au 2675,95 А. Область определяемых концентраций Sr 3-10"6 — 1-10~3% [128].
При спектральном определении стронция в хромо-никелевой стали [391] образец переводят в раствор, наносят на медный электрод, высушивают и возбуждают спектры в низковольтной искре; аналитическая пара линий Sr составляет 4077,71 А и Си 4062,7 А.
Определение стронция (и других щелочноземельных, а также щелочных металлов) в металлическом уране ядерной степени чистоты производится пламеннофотометрическим методом после предварительного отделения урана экстракцией.
Уран или его соединения (1—3 г) обрабатывают азотной кислотой для переведения в нитрат уранила. Раствор упаривают досуха, остаток растворяют в 20 мл 5 N HNO3, полученный раствор переносят в специальный экстрактор непрерывного действия. Уран экстрагируют, пропуская через экстрактор 2 00 мл раствора трибутилфосфата в керосине (1 : 10). Водную фазу выпаривают досуха, остаток растворяют в 10 мл 1%-ной НС], в полученном растворе определяют стронций пламеннофотометрическим методом. Для устранения влияния Al, Zr и V добавляют раствор LaCl3 с таким расчетом, чтобы конечная концентрация соответствовала 800 мкгка/мл. Чувствительность метода порядка 2,5 -IO-6 — 1.10~5% Sr [1139].
В работе [1151] предварительное отделение урана производится экстракцией уранилнитрата эфиром.
АНАЛИЗ ДРУГИХ ОБЪЕКТОВ
В табл. 42 представлена сводка объектов анализа, не включенных в предыдущие части главы, с указанием использовавшихся методов.
135
Таблица 42
Определение стронция в различных химических соединениях, технических, строительных и других материалах
Объект анализа Метод определения Литература
Окисли металлов
MgO Спектрографический [314, 315]
А12О3 » [1345, 1367]
Sc2O3 [275]
УЬ2О3 [1367]
Мп02 » [310, 1031]
wo3 » [127, 1367]
и308 » [514]
Ba2Zn2_xMnx Fe12022,BaFe120le » [52]
(гекса ферриты) Ва, Са (окисные налеты на де- Спектрографический [965]
талях электронных ламп) Пламеннофотометрический [489]
Окисные ванадийсодержащие Пламеннофотбметрический, [67, 161]
материалы атомно-абсорбционный
ВаО Гравиметрический [1136]
ВаО2 и другие объекты Соли и другие соединения » [1087]
металлов
ВаС12 Спектрофотометрический [212, 249]
NaCl, КС1 Полярографический [1322]
NaCl.KCl, СаСО3, Ba(NO3)2 Пламеннофотометрический [342]
NaCl Атомно-абсорбционный [1257]
ВаСО3 Рентгенофлуоресцентный [436]
Номплексонометрическое титрование 1119, 234]
Ангидрит (CaSO4) Пламеннофотометрический [829]
Соединения Na, Са, Ва, Fe, Комплексонометрическое тит- [1061
Со, Ni, РЬ рование
Соли тяжелых редкоземель- То же [109, ИЗ,
ныл элементов, Bi, Си, РЬ, Zn, Zr 234, 276]
Ферриты » [ЮЗ]
у-Активационный [74]
Соли щелочных металлов Спектрографический [38, 52]
CaF2 » [313]
BaF2 » [1367]
BaSO4 » [488]
MnSO4 » [488 , 311]
Неорганические неметалличв- > [553, 648,
ские материалы 1162]
Таблица 42 (окончание)
Объект анализа Метод определения Литература
Активные материалы 140Ва и др.) Спектрографический [271, 554, 1193]
Электролит для хромирования Пламеннофотометрический [186]
Катализаторы для хромирования сталей > [326]
Коньячный спирт, дубовая древесина Спектрографический [243]
Бумага » [423]
Дизельные масла » [78]
Виски Пламеннофотометрический [1158]
Мазуты и нефтяные коксы » [1241]
Строительные материалы По электропроводности элюата при непрерывном электрофорезе [856]
Цемент Спектрографический [529]
Пламеннофотометрический [274, 673]
Атомно-абсорбционный [608, 857 1196, 1308]
Стекло Титриметрический [306]
Атомно-абсорбционный [370, 490, 839]
При определении стронция в окислах металлов наибольшее распространение нашел спектрографический метод.
Двуокись марганца. Образец смешивают с 1% LiaCO3 и добавляют равный вес графита. 30 мг смеси помещают в графитовый электрод (углубление 4,15 мм), сжигают в дуге постоянного тока 10 а, съемка спектра 10 сек. Аналитическая линия Sr составляет 4077,71 А; линия внутреннего стандарта Li 2741,3 А. Чувствительность метода 0,01% Sr [1031].
Окись алюминия. Смешивают образец с никелем и графитом в соотношении 2:1:3. Используется дуга постоянного тока 13 а. Проба испаряется из графитового электрода (анод), канал глубиной 4 мм, диаметром 3 мм. Аналитическая линия стронция 4607,33 А, линия внутреннего стандарта никеля 4714 А [1345].
Трехокись вольфрама. Используется предварительное концентрирование примеси стронция и других элементов путем их испарения. Стаканчик с пробой зажимают между графитовыми щетками испарителя (рис. 24), нагревают в течение 1,5 мин. до 2300” С и продолжают испарение вше 1,5 мин. При этом температура поднимается до 2400° С. Приемник — угольный электрод, расположенный над стаканчиком (диаметр 6 мм, длина 12 мм). Спектр конденсата возбуждают в дуге переменного тока 4 а, экспо-
137
Рис. 24. Закрепление стаканчика с пробой между графитовыми щетками испарителя
а — приемник конденсата — капсула; б — угольный стаканчик; в — графитовые щетки (размеры в лон)
зиция 50 сек. Верхним электродом также служит капсула с конденсатом. Аналитическая линия стронция 4607,33 Л. Чувствительность метода 5 .10"4% Sr, средняя ошибка 14%. Максимальная определяемая концентрация Sr 5-10~2% [127].
Закис ь—о кись урана (U3O8). Для повышения чувствительности определения стронция к образцу прибавляют 2% UF4. 50 мг смеси помещают в графитовый электрод (углубление диаметром 3,97 мм, глубиной 5,6 мм), служащий'анодом. Съемку спектра производят при сжигании в дуге постоянного тока 13 а в течение 35 сек., при этом фотометрируется прикатод-ная область (0,2 ллщниже катода). Чувствительность метода 5 .10-5% Sr (в отсутствие UF4 — 1 -10—3% Sr) [514].
Окись маг ни я. Используется дуга постоянного тока или трубка с полым катодом. Для устранения влияния основы добавляется носитель. При использовании дуги постоянного тока (/ = 15 а) в углубление электрода диаметром 4 и глубиной 12 мм помещают 106 мг образца в смеси с 7 мг AgCl и 7 мг CdS. Экспозиция равна 1 мин. При использовании трубки с полым катодом в качестве носителя применяется один AgCl (1 мг). Навеска анализируемого образца равна 25 мг. Спектры возбуждают в атмосфере гелия (давление 20 мм рт. ст.), сила тока разряда 0,5 а (30 сек.) и затем 1 а (также 30 сек.). Аналитическая линия стронция 4607,33 А. Чувствительность метода в дуге с носителем 1.10-3% Sr, в полом катоде с носителем 1.10-4% Sr [314].
Окислы алю ми ни я, иттербия и вольфрама. Используется испарение микроколичеств вещества с поверхности порошкообразных проб с помощью рубинового лазера и возбуждение спектра искрой (2 кв, 20 мкф, 100 лкг«). Электроды располагаются горизонтально над пробой на высоте 1,8 мм. Аналитическая линия стронция 4077,71 А. Чувствительность метода 5 .10-4% Sr (в A12OS). Показано, что влияние различных основ при таком методе значительно меньше, чем в дуге постоянного тока [1367].
138
В работах [67, 161] изучено влияние ряда элементов на точность определения стронция эмиссионным и атомно-абсорбционным пламеннофотометрическим методами. Порядок влияния элементов в первом методе Mg ]> Са ]> Ва, во втором Ва > Си > Cd > Sr > Mg.
Для анализа перекиси бария предложен гравиметрический метод [1087], который включает растворение образца в НС1 и осаждение ВаСгО4 при постепенном понижении pH (путем кипячения с мочевиной). После отфильтровывания осадка ВаСгО4 производится осаждение Sr и Са в виде оксалатов из водно-эта-нольной среды, оксалаты прокаливают до окислов, последние обработкой HNO3 переводят в нитраты. С помощью ацетона отделяют Sr(NO3)2 от Ca(NO3)2 и определяют Sr в виде’сульфата. Определяемые содержания стронция 1—2%.
Ниже рассматриваются соли и другие соединения металлов. В работе [212] описан анализ хлорида, нитрата или ацетата бария.
Навеску 0,19—0,22 г соли растворяют в воде в мерной колбе емкостью 250 мл. Переносят 1 мл полученного раствора в пробирку на 20 мл с притертой пробкой, добавляют 0,5 лл 0,055%-ного водного раствора нитроортанило-вого С, 3,5 мл ацетатного буферного раствора (10 мл ХМ раствора CH3COONa и 3 мл ледяной; СН3СООН в 1 л) и 1 мл 0,074%-ного раствора Na2SO4 (для осаждения Ва), перемешивают и оставляют на 20 мин. В течение этого времени окраска меняется от синей к фиолетовой. Затем вводят 4 мл ацетона, вновь перемешивают и после полного отстаивания (или центрифугирования) раствора фотометрируют в кювете с I = 1 см при 650 им. Определению 0,3— 2,4 мкг St/мл не мешает до 60 мкг Ва/л«л, а также содержание в анализируемой соли до 3% Са. Стандартное отклонение составляет 2,7—9,7%. В отдельных случаях ошибка достигает 50% и более (при высоких соотношениях Ва : Sr).
Хлориды натрия и калия. Применен дифференциальный полярографический метод (с использованием двух синхронно капающих ртутных электродов).
По 1 г анализируемой и чистой соли растворяют в 5 мл воды, в полярографические сосуды помещают по 2 мл раствора, 5 капель 0,5%-ного раствора желатина и снимают дифференциальную полярограмму. Определяемый минимум 1 •10"4М Sr в 1 М растворе анализируемой соли [1322].
Нитраты и хлориды Pb, Zn, Си, Bi и ферриты Zr. Используется предварительная экстракция роданидных или перхлоратных комплексов щелочноземельных металлов трибутилфосфатом при связывании мешающих элементов комплексоном III. Затем (в случае анализа ферритов) применяют реэкстракцию соляной кислотой и разделяют щелочноземельные элементы экстракцией с помощью реагента АТ (стр. 55) и каждый металл определяют комплексонометрически [103, ИЗ].
139
В присутствии 2 М раствора перхлората натрия и ЭДТА щелочноземельные элементы экстрагируют трибутилфосфатом из растворов солей РЬ, Си, Bi, Zn и Zr (pH 4). Реэкстракцию проводят 3 М НС1, сумму Са, Sr и Ва определяют комплексонометрически с индикатором метилтимоловым синим. Определяемые содержания— 0,5 мг Sr в присутствии 20 мг нитратов или хлоридов ряда металлов
При определении Са, Sr и Ва в солях Fe, Си и Bi основные компоненты предварительно отделяют с помощью купферона в смеси изоамилового спирта и бензола (pH 1). Затем последовательно экстрагируют при различной кислотности Са и Sr, а Ва остается в водном растворе. При содержании стронция 0,5 мг относительная ошибка его определения не превышает 4% (отн.) 1101, 113].
Углекислый барий. Образец (0,5 г) растворяют в НС1 (1 : 1), выпаривают до начала образования солей, остаток растворяют в воде и разбавляют до 100 мл. К 10 мл полученного раствора приливают 4 мл 20%-ного раствора NaOH, экстрагируют Са и Sr двумя порциями по 20 мл реагента АТ, экстракты объединяют и промывают 3 раза порциями по 40 мл 1 N NaOH. Реэкстрагируют Са и Sr 20 мл 0,1 7VHC1 и отделяют Са от Sr экстракцией из 0,05 N раствора NaOH, для чего реэкстракт нейтрализуют по универсальной индикаторной бумаге, добавляют 0,25 мл 5 N раствора NaOH, равный объем реагента АТ и содержимое воронки встряхивают.
После расслаивания водный слой, содержащий основную массу Sr, переносят в кварцевую чашку, а органический слой, содержащий Са и частично Sr, переносят в другую делительную воронку и проводят операцию реэкстракции. Для этого приливают 20 мл 0,1 N раствора НС1 и встряхивают, после чего, не разделяя фаз, водный слой нейтрализуют до pH ~ 7, приливают 0,25 мл 5 N раствора NaOH и снова встряхивают.
Органический слой переносят в другую делительную воронку и повторяют операцию переэкстракции. Водные растворы после переэкстракции соединяют с основной массой Sr в кварцевой чашке. Раствор в кварцевой чашке упаривают до 20 мл, переносят в коническую колбочку емкостью 100 мл, прибавляют 20 мл раствора NH4OH, индикатор метилтимоловый синий и титруют раствором комплексона III до изменения окраски индикатора от голубой до серой [119, 234].
При анализе галогенидов щелочных металлов в спектрографическом методе анализируемое вещество вводится в электрический разряд в виде тонкого слоя на электроде.
Йодид натрия, калия, рубидия или цезия (1,25 г) переводят в водный раствор объемом 25 мл. На угольные электроды с плоской поверхностью, пропитанной 0,05 Л4Л 3%-ного раствора полистирола в толуоле, [после высушивания наносят по 0,03 мл раствора эталонов или анализируемых образцов. После повторного высушивания,’ фотографируют спектры при силе тока дуги переменного тока 12 а. Эталонные растворы содержат 5% чистой соли и 1. • 10-4 — 3-10-2Bec.%Sr по отношению к соли. Аналитическая линия Sr/I.
.140
4607,33 А. Содержание Sr определяют по калибровочному графику. Чувствительность метода 2-10-4% Sr [166].
Для повышения чувствительности метода может быть использовано кристаллизационное концентрирование [38].
Атомно-абсорбционный метод был применен для изучения распределения стронция (1 ч-2,9)-10-4% во фракциях, получаемых при перекристаллизации хлорида натрия. Концентрация поваренной соли в анализируемом растворе составляла 10% [1257].
При анализе сульфата бария и гексаферритов (Ва2, Zn2-xMnx-Fe12O2 2 и BaFe12O19) образцы смешивают с угольным порошком в соотношении 1:1 — 1:5, помещают в канал угольного электрода и испаряют в дуге переменного или постоянного тока. При анализе концентратов BaSO4 анализ проводят по линиям: Sr II 3380,71, Ва II 2771,35 А. Эталоны готовят на основе BaSO4. Определяемые содержания 1—3% SrSO4. В гекса-ферритах1 примесь стронция определяется при его содержаниях 0,005-1% [52, 488].
Анализ фторида кальция рассмотрен в работе [313]
Образец (60 мг) помещают в канал угольного электрода (анод) и сжигают в дуге постоянного тока (12 а). Аналитическая линия стронция 4607,3 А. Эталоны готовят смешиванием SrO с CaF2. Чувствительность метода 1 -10-2% Sr [313].
Анализ неорганических материалов методом желез но го флюса описан в ряде работ [648, 1162, 605].
Образец (10 мг) смешивают с 10-кратным количеством смеси сульфата аммония и сульфата железа любой валентности (1 : 1). Прессуют таблетки весом 10 мг и диаметром 3 мм. Таблетку помещают на плоский торец медного электрода (контр-электрод также медный). Спектры возбуждают в дуге постоянного тока 7 а (электрод с таблеткой— катод). Чувствительность определения около 0,01% Sr [648, 1162].
Цемент. Образец (1 г) смачивают 10 мл холодной воды, добавляют 10 мл конц. НС1 и нагревают до полного растворения. Раствор выпаривают досуха, добавляют 10 мл НС1 (1 : 1) и дают постоять 10 мин. Разбавляют горячей водой (10 мл) и отфильтровывают выделившуюся кремневую кислоту, осадок промывают на фильтре горячей НС1 (1 : 99). Фильтраты и промывные воды разбавляют водой до 100 мл. Полученный исходный раствор разбавляют в 10 раз водой (при добавлении соли калия до концентрации 1 мг/мл или лантана до концентрации 10 мг/мл) и определяют стронций атомно-абсорбционным методом с использованием пламени закись азота—ацетилен. Определяемые содержания стронция 0,05—0,3% [605].
Аналогичный метод с использованием пламени смеси ацетилена с воздухом при добавлении LaCl3 описан в [857].
В методе [1196] образец растворяют в смеси НС1 и HF и фотометрируют непосредственно полученный раствор после добавления
141
Н3ВО3 (для связывания фтор-иона) и хлорида лантана (как освобождающего агента).
Образец (0,5 г) помещают в полиэтиленовый или фторопластовый стакан емкостью 100 мл. Обмывают стенки стакана 20 мл воды, добавляют 10 конц. НС1 и (после растворения образца) 1 мл 40%-ной HF. Перемешивают смесь до растворения выделившейся SiO2, добавляют 50 мл 4%-ного раствора Н3ВО3, 20 .ил раствора LaCl3 (50 мг Ьаз+/лл), разбавляют до 200 мл водой. Определяют стронций атомно-абсорбционным методом, используя пламя смеси закись азота—ацетилен. Стандартные растворы содержат 0—12,5 мкг SrO/мл и 5 mzL3?+/ma. Для приготовления раствора LaCl3 58,6 г его окиси растворяют при нагревании в 800 .ил 20%-ной НС1 и разбавляют водой до 1 л.
Стекла Мелко измельченный образец (0,1 г) в платиновой чашке смешивают с 2—11> мл воды, 1 мл НС1О4и 5—10 мл HF. Нагревают сначала на водяной бане до разложения образца и затем на плитке до полного удаления хлорной кислоты. Добавляют 1 мл НС1О4 и снова выпаривают почти досуха. Остаток смешивают с 5 каплями НС1О4, растворяют в воде и разбавляют до 100 мл. К определенному объему полученного раствора прибавляют буферный раствор, содержащий тетрааммонийнуюсоль этилендиаминтетрауксусной кислоты и перхлорат лантана, и разбавляют водой так, чтобы концентрация стронция в пересчете на SrO составляла 5 мкг/мл, (ГШ4)4ЭДТА 0,01 М и Ьа2О3 2 мг/мл. Определяют стронций атомно-абсорбционным способом с использованием пламени смеси Н2—О2. Для приготовления исходного буферного раствора 20—37,5 г ЭДТА растворяют в 100 мл воды при добавлении достаточного количества NH4OH для получения аммонийной соли ЭДТА. Отдельно растворяют 12,5 г La2O3 в 15 мл конц. НС1О4 и разбавляют до 100 мл водой. Сливают оба раствора при непрерывном перемешивании, добавляют, если нужно, NH4OH до получения щелочного раствора по метиловому оранжевому и разбавляют до 500 мл водой [490].
Глава VI
АНАЛИЗ СТРОНЦИЯ И ЕГО СОЕДИНЕНИЙ
ОПРЕДЕЛЕНИЕ ПРИМЕСИ ЭЛЕМЕНТОВ В СТРОНЦИИ И ЕГО СОЕДИНЕНИЯХ
Для определения примеси элементов в металлическом стронции и в его соединениях предложено большое число методов, сводка которых приведена в табл. 43.
Определение неметаллов
Обзор методов определения кислорода, углерода, азота и водорода, которые используются при анализе стронция (или других щелочноземельных и щелочных металлов), приведен в [97, 225].
Определение кислорода в стронции методом ртутной экстракции. Метод основан на том, что металлическая ртуть растворяет металлический стронций, окись же стронция со ртутью не взаимодействует и остается на поверхности ее вследствие большой разницы плотности ртути и окиси стронция.
Образец (50—100 мг) очищают в аргоновой камере от поверхностной пленки и после взвешивания на торзионных весах помещают в колонку экстрактора, которую предварительно тщательно промывают. Колонку откачивают на вакуумной установке при нагревании до давления 1.10~4 мм рт. ст., а затем помещают в аргоновую камеру. (Размеры колонки выбраны с таким расчетом, чтобы вместе с воронкой для ртути ее можно было поместить в аргоновую камеру).
Колонку экстрактора (рис. 25) и приемник амальгамы подсоединяют к вакуумной установке с помощью вакуумных шлангов (через отростки на кранах 4 и 10). Сначала производят откачку воздуха форвакуумным насосом из приемника амальгамы 8, затем — аргона из колонки экстрактора. Дальнейшая откачка прибора до давления 1 10~4 мм рт. ст. осуществляется масляным диффузионным насосом типа ЦВЛ-100.
В воронку 1 заливают около 30 мл перегнанной ртути. Примерно 10 мл ртути из этой воронки заливают через центральный канал крана 2 в колонку экстрактора. Пламенем газовой горелки колонку со ртутью и образцом стронция нагревают до 80—100° С при непрерывном покачивании колонки (для
143
Таблица 43
Методы определения примесных элементов в стронции и его соединениях
Объект анализа Определяемые элементы (ионы) Метод определения Чувствительность, % Литература
On ределение п римеси неметаллов
Металлический стронций И z о d О Ртутная экстракция, вакуумная дистилляция, вакуум-плавление с оловянной ванной 6-10-3 [225]
SrF2, SrCls o2 Вакуум-плавление и восстановительное плавление в потоке инертного газа, изотопное разбавление, радиоактивацион-ный (4—5)-IO-* [97, 98[
SrJ2 H20 и 0H--ионы Титриметрический (реактив Фишера) 2-10-2 [254]
SrO Cl- Потенциометрическое титрование (AgNO3) — [1137]
SrCO3 N03- Спектрофотометрический 1 • 10-1; 5-Ю-3 [532,778]
NO3- Полярографический 2-10-1 [778]
S2", SO42- ~М0“5 [402]
Смесь SrSO4 c PbSO4 и SiO2 SiO2 On ределе! Гравиметрический iue примеси металлов — [442[
Металлический стронций Fe, Cr, Zn, Sb, Si, Cu, Sn, Pb, Bi, Cd Спектрографический 5-10~6— 1•10~3 [292,293]
Mg, Ca, Al, Ti, Химико-спектрогра- 2.10-’— [188,189, 257, 290, 291,312, 316]
Cr, Mn, Fe, Co, Ni, Cu, Ag, Zn, Cd, In, Sn, Sb, Bi, Pb фический 1 • ю-3
Zn, Cd, Pb, Cu, Sb, Bi Пол яро графический 2-10-5 [80]
SrO Mn, Zn, Pb, Cu Щелочные металлы » Пламеннофотометрический [1137] [1137]
SrCO3 Ba, Ni, Mn, TI, Cr, Fe, Sb, Sn Полярографический МО-5 [402]
Na Радиоактивацион-ный МО-2 [763]
Fe, Al, Ca, Cu, Mg, Ba Спектрографический МО-4 [179,316]
144
Таблица 43 (окончание)
Объект анализа Определяемые элементы (ионы) Метод определения Чувствительность, % Литература
SrF2 Sr J 2 Sr(NO3)2 Sr(NO3)2 Соли (нитраты, карбонаты, хлориды, бораты и др.) SrMoO4 Смесь Са и Sr Смесь Ва, Sr, Са и др. Fe, Си, Со, Са, Mg и др. La, Sm, Gd, Tb, Dy, Ho, Er, Tu, Lu, Y, Yb Eu, Ce, Pr, Nd Cu, TI Eu Cr, Mg, Ca, Si Fe, Al, Ti, V, Mo и др. Mg Na Ca Ba Cu Cd, Si, Cu, Pb, Cr, Zn Al, Bi, Ga, Fe, In, Mn, Cu, Mo, Ti и др. Микропримеси различных металлов Са Ва Спектрографический Химико-спектрографический Спектрографический » Атомно-абсорбционный Спектрофотометрический То же Атомно-а бсор бцион-ный То же Потенциометрическое титрование щелочью (после обмена Na+ на Н+ на ионообменной колонке) Комплексонометрическое титрование Атомно-абсорбционный Каталитический Спектрографический Химико-спектрогра -фический Масс-спектральный Радиоизотопное разбавление Химико-спектрографический 1-10-6— МО'3 1 -до-5 (2—8)-IO"2 (2—8)-10-1 10-s—10-’ МО"’ 5-10-в-1-10.-4 5-Ю-з l-10-з 1-Ю-4— l-10-з 3-10-2 1-Ю"5 5-10-4— МО"8 1-10-8— 1 - 10-4 l-10-e— 1-10-4 (179,477) [444] [191] ’[191] [451] [381] [316] [170] [171] [897] [104] [1071] [835] [293] [308,309, ё121 [676] [452] [1310]
145
Рис. 25. Ртутный экстрактор
1 — воронка для ртути;
2,4,6, ю — краны;
з — разбрызгиватель ртути;
5 — кварцевая колонка;
7 — пробка;
8 — приемник амальгамы;
9 — ложечка
Рис. 26. Установка для титрования реактивом Фишера
лучшего смачивания ртутью образца стронция).В зависимости от веса образца время амальгамирования колеблется от 1,5 до 2 час. Растворение образца производится в 2—3 порциях ртути. Амальгаму сливают в приемник 8 после того, как она остынет до комнатной температуры. Каждый раз в колонке остается около всей порции ртути.
После растворения всего образца производят отмывку амальгамы. Для этого в колонку заливают несколько порций ртути по 10 мл так, чтобы она омывала стенки колонки; после отстаивания в течение ~2 мин. ртуть сливают в приемник; часть ее (около 2 мл) каждый раз оставляют в колонке. Анализ проводят при непрерывной откачке прибора.
После 5—6 циклов берут пробу ртути на отсутствие стронция. Для этого . в ложечку 9 через кран 6 наливают около 0,5 мл ртути из колонки экстрактора; в приемник амальгамы впускают воздух при перекрытом кране 6, ложечку вынимают из приемника и производят пробу на стронций (с фенолфталеином). В случае отсутствия стронция в ртути отмывку амальгамы прекращают, в колонку впускают воздух. Нерастворимые в ртути примеси окиси и нитрида стронция переводят водой в раствор, объем которого доводят до 25 или 50 мл в мерной колбе. Аликвотную часть раствора титруют для определения количества стронция, соединенного с азотом и кислородом. Зная содержание азота в стронции (определяется заранее), рассчитывают количество кислорода в анализируемом образце. Расход ртути на одно определение 50— 100 мл.
146
Определение примеси азота, углерода и водорода может быть произведено методами, предложенными для анализа металлического лития [225].
Определение кислорода во фториде стронция методом изотопного разбавления [97, 98]. В установку для анализа газов в металлах загружают от 1 до 5 г SrF2 и от 10 до 50 мг дозировочного образца (смесь SrF2 со SrO, обогащенную-кислородом — 18О). Предварительно в течение 1—2 час. производят дегазацию графитового тигля при 2000—2200° С до достижения вакуума в печи 1 -Ю-6 мм рт. ст. Параллельно с дегазацией тигля дегазируют ампулы с цеолитом, предназначенные для сбора газа на изотопный анализ.
Перед проведением процесса обмена температура снижается до 600— 800° С. Печь отключают от вакуумной системы. В тигель помещают анализируемый и дозировочный образцы. Температуру тигля поднимают, пробу и дозировочный образец выдерживают в расплавленном состоянии некоторое время для достижения равномерного распределения изотопов кислорода в расплаве, затем температуру опять повышают для выделения газа на изотопный анализ. Газ собирают в ампулы с цеолитом, охлаждаемые жидким азотом. Изотопный анализ производят с помощью масс-спектрометра. Чувствительность метода 4—5-10-4 вес.%жО2.
Определение воды и гидроксильных ионов в иодиде стронция [254]. Использован метод, основанный на титровании реактивом Фишера раствора SrJ2 в метаноле. Уравнение реакции
J2 + SO2 + Н2О + СНзОН + 3C5H5N CHsOSO3~ + 2J~ + 3C6H6NH+.
Наряду с этим пиридиний-ион, содержащийся в реактиве Фишера, вступает в реакцию нейтрализации с окислами и гидроокисями металлов, в результате чего выделяется вода, взаимодействующая с титрантом по представленному уравнению.
Установка для титрования состоит из следующих деталей (рис. 26): бюретки 1 емкостью 5 мл\ сифона 2 с приспособлением для автоматической установки нуля в бюретке; сосуда 3 для титрования емкостью 150 мл с наружным шлифом (во время титрования сосуд закрывают пробкой, в которую впаян удлиненный конец бюретки, а при растворении проб — отдельной пробкой, не связанной с установкой для титрования); магнита 4, запаянного в стеклянную пробирку; магнитной мешалки 5; темной склянки 6 для реактива Фишера емкостью 1000 мл с пришлифованной пробкой; резиновых колец 7 для плотного закрывания склянок 3 и 6; колонки 8, заполненной безводным хлоридом кальция; резинового баллончика 9 для нагнетания воздуха при заполнении бюретки реактивом Фишера; крана 10, с помощью которого установку соединяют с атмосферой, чтобы снять избыточное давление после заполнения бюретки реактивом Фишера.
Анализируемый образец тщательно перемешивают и растирают в агатовой ступке в боксе с сухим воздухом. Около 2 г препарата SrJ2 взвешивают с точностью до 0,0002 г в бюксе с пришлифованной крышкой и всыпают (через воронку для сыпучих веществ) в колбу для титрования. После этого взве-
147
Рис. 27. Спектр поглощения нитрат-иона
Г — 0,675 мг NO3 и 5 мл НС104 в 100 мл;
2 — 0,675 г NO3 и 5 мл НС1О< в 100 мл; (I = 1 сл)
шивают бюкс с указанной выше точностью и по разности определяют навеску. Для растворения препарата в колбу добавляют 10 мл метанола, присоединяют колбу к бюретке, включают магнитную мешалку, затем титруют полученный раствор реактивом Фишера до появления бурой окраски иода. Одновременно проводят холостой опыт, титруя 10 мл метанола.
Содержание воды (вес.%) рассчитывают по формуле
(7о-7х).Г.Ю0
где 70 — объем реактива Фишера, израсходованного на титрование анализируемой пробы; — объем реактива Фишера, израсходованного на титрование в холостом опыте; Т — титр реактива Фишера (г/мл): G — навеска препарата (г). Чувствительность метода 2-10-2 вес.%Н2О. Относительная ошибка 12—20%.
Определение нитратов в смеси карбонатов Са, Sr и Ва может быть произведено по поглощению нитрат-иона при 210—220 нм (рис. 27).
Образец (1 г)\ помещают в стакан емкостью 100 мл, заливают водой и добавляют по каплям хлорную кислоту до растворения образца. При этой операции стакан накрывают часовым стеклом, затем смывают стенки стакана водой и перемешивают до прекращения выделения СО2. Полученный раствор или его аликвотную часть смешивают с 5 мл HCIO4, разбавляют до 100 мл, затем определяют оптическую плотность при 210 или 220 нм. Определяемое содержание NOj-ионов 0,1—1% [532].
В другом методе щелочноземельные элементы предварительно отделяют на катионите и затем определяют нитрат-ионы по реакции образования нитросалициловой кислоты.
Пробу 100 мг) помещают в специальный сосуд с механической мешалкой, вносят 25 мл катионообменной смолы (предварительно промытой водой, подкисленной до pH ~5), добавляют 10 мл воды и размешивают 10 мин. Затем раствор выпускают из сосуда и к оставшейся смеси пробы с катионитом снова добавляют воду (5 раз) с последующим перемешиванием и фильтрованием. Полученный раствор подщелачивают до pH 9—11 и упаривают до 5—10 мл. К упаренному раствору прибавляют 5 мл 0,1 М раствора салицилата натрия,
148
0,5 мл 1 М раствора карбоната натрия и выпаривают досуха. К сухому •остатку после охлаждения прибавляют 2 мл конц. H2SOi и в течение 30 мин. периодически перемешивают. Затем реакционную смесь разбавляют водой до ~60 мл, добавляют 25 мл 5 М NaOH, разбавляют водой до 100 мл и измеряют •оптическую плотность раствора при 405 нм в кювете с I = 2 или 4 см. Метод позволяет определять 5-10“®% NOg с ошибкой <15% [778].
Определение металлов
Для определения в препаратах стронция примеси щелочных металлов и магния в основном используются пламеннофотометрический и атомно-абсорбционный методы.
Примеси тяжелых металлов преимущественно определяют спектрографическим методом, который применяется как непосредственно к объекту анализа, так и после предварительного химического обогащения. В отдельных случаях успешно используются полярографический, масс-спектральный и другие методы (см. табл. 43).
Анализ металлического стронция прямым спектрографическим методом (292, 293]. Навеску металла обрабатывают водой (50-кратным количеством), полученный раствор гидроокиси стронция насыщают СО2. Раствор упаривают досуха, высушенные при 105—110° С осадки 8гСО3*используют для анализа при добавлении графита (50 мг образца + 30 мг графитового порошка). Применяется дуга переменного тока 8 а, 220 в. Экспозиция 30 сек. (2 раза). Чувствительность определения примесей и аналитические линии приведены в табл. 44. Эталоны готовят из чистого карбоната стронция путем добавления микроколичеств солей соответствующих элементов.
Таблица 44
Чувствительность определения примесей в металлическом стронции спектрографическим методом
Аналитиче- ская линия, А Чувствительность, % (n-10~’) Аналитическая линия, А Чувствительность,% (п-10-3)
в карбонате стронция в пересчете на металлический стронций в карбонате стронция в пересчете на металлический стронций
Fe 3020,64 1 1,7 РЬ 2833,07 0,1 0,17
€d 2288,02 0,1 0,17 Сг 2835,63 1 1,7
Si 2516,12 1 1,7 Zn 3345,02 5 8
€u 3274,54 0,1 1,7
Спектрографические методы с предварительным химическим обогащением. Осаждением стронция в виде сульфата [189, 316] достигается обогащение примесей металлов в 20—30 раз и чувствительность определения 17 из них составляет 1 • 10~4— 5-10~6%.
149
Навеску пробы металла (3—5 г) растворяют в HNO3 (1 : 10), помещенной в кварцевую колбу. Полученный раствор нагревают до кипения. Затем в него прибавляют по каплям при энергичном перемешивании 50 мл H2SO4 (1 : 10). Раствору с осадком дают отстояться в течение нескольких часов. Маточный раствор сливают в предварительно прокаленную и взвешенную кварцевую чашку и упаривают его.
Осадок SrSOa подвергают перекристаллизации путем нагревания на воздушной бане с 25 мл HNO3 (1 : 1) в течение 3 час. Кислоту объединяют с маточным раствором, упаривают досуха, остаток прокаливают 30 мин. при 550° С и определяют вес полученного концентрата. Вычисляют коэффициент обогащения примесей как отношение веса исходной навески к весу полученного концентрата (в пересчете на металлический Sr). При съемке спектра ис-
Таблица 45
Чувствительность спектрографического определения примесей металлов при их концентрировании путем осаждения стронция в виде сульфата
Элемент и линия, А Чувствительность, % (п-10-4) Элемент и линия, X Чувствительность, % (n-10~‘)
Mg 2795,5 0,3 Ni 3050,8 2
Са 3933,7 3 Си 3247,5 0,2
А1 3082,2 1 Ag 3280,7 0,2
Ti 3234,5 0,5 Cd 2288,0 0,2
Сг 4274,8 3 In 3256,1 0,2
Мп 2794,8 0,05 Sn 2840,0 0,5
Fe 2720,9 2 Sb 2598,1 2
Со 3405,1 2 Bi 3067,7 1
Na 5890,0 1
паряют 40 мг пробы в дуге постоянного тока (12 а), экспозиция 2,5 мин. Аналитические линии элементов — примесей и чувствительность их определения представлены в табл. 45.
В других методах предварительного концентрирования тяжелых металлов используется осаждение гидроокисей и основных солей на гидроокиси алюминия, соосаждеиие сульфидов и диэтил-дитиокарбаминатов с использованием в качестве носителя сульфида кадмия [308, 309, 312], экстракция диэтилдитиокарбаминатов [290, 291, 444].
Определение примесей в металлическом стронции с концентрированием путем экстракции. Анализируемый металл (2 г) помещают в кварцевый стакан на 50 мл, покрытый часовым стеклом, и осторожно растворяют в 10— 12 мл воды, дважды перегнанной в кварцевой аппаратуре и прокипяченной для удаления СО2. Воду вливают постепенно. В полученный раствор приливают по каплям дважды перегнанную НС1 до pH 5—6 по универсальному индикатору.
150
Раствор переводят в кварцевую делительную воронку емкостью 25—50 мл, приливают 1 мл 2%-ного водного раствора диэтилдитиокарбамината натрия и 2 мл хлороформа. Воронку энергично встряхивают в течение 1 мин.
После расслаивания органический слой переводят в другую кварцевую делительную воронку, оставляя около Vs части органического слоя вместе с водным. К последнему приливают 1 мл раствора диэтилдитиокарбамината натрия и 2 мл хлороформа. Экстракцию повторяют] 4—6 раз в тех же условиях. Последнюю экстракцию производят только хлороформом.
Объединенный экстракт промывают 2—3 раза водой порциями по 10 мл. Органический слой (после тщательного отделения от водного) переливают в кварцевую чашку, содержащую 50 мг угольного порошка, в котором спектрографическим методом не обнаружено содержание определяемых примесей. Выпаривают на водяной бане. Полученный концентрат примесей поступает на спектрографический анализ.
Предлагаемый метод может быть использован также при анализе хлорида стронция или карбоната (после переведения в хлорид) [290].
Анализ фторида стронция. Материал (1 г) помещают в платиновую чашку, добавляют 60—70%-ную НСЮ4 (5 мл) и нагревают на плитке в течение ~1 часа (при этом кислота почти полностью удаляется). Остаток растворяют в воде (30—40 мл). Раствор нейтрализуют раствором аммиака до pH 5. Добавляют 2%-ный раствор диэтилдитиокарбамината натрия (1 мл). Полученный раствор нагревают в течение 2—5 мин. до 80—90° С, затем],быстро охлаждают, переносят в делительную воронку, вводят хлороформ (5 мл) и энергично смешивают жидкости в течение 1 мин.
После расслаивания органический слой переводят в другую воронку. Экстракцию карбаминатов повторяют еще 2 раза. Экстракты объединяют и 3 раза промывают водой (по 5 мл), затем переносят в кварцевую чашку.
Для окисления элементарной серы добавляют 3—4 капли HNOS и упаривают вначале на водяной бане (до удаления органического растворителя), затем на плитке до прекращения выделения паров серной кислоты. После повторной обработки HNO3 остаток растворяют в воде (10—15 мл). Добавляют угольный порошок (30 мг), раствор упаривают досуха.
Спектры определяемых металлов возбуждают в дуге постоянного тока (10—12 а). При добавлении к концентратам (30—50 мг) 2 мг поваренной соли чувствительность метода повышается (табл. 46).
Определение в плавленном фториде стронция с применением автоматического фотометрирования [477]. Образец, смешанный с графитовым^порошком в соотношении 1:1, испаряют в дуге постоянного тока (10 а). В табл. 47 приведены аналитические линии и чувствительность метода.
Применение спектрографа высокой разрешающей силы (ДФС-8) при фотографической регистрации спектра дает выигрыш в чувствительности по сравнению с прибором средней дисперсри (ИСП-28) за счет] снижения сплошного фона фторидов. Автоматическое фотометрирование слабых спектральных линий (вид получаемой
151
Таблица 46
Чувствительность спектрографического определения примесей в металлическом стронции и во фториде стронция при их концентрировании методом экстракции [290, 444]
Элемент Металлический Sr SrF,
аналитическая линия, A чувствительность,% (n«10~e) аналитическая линия, A чувствительность, % (n«10“*)
Bi I 3067,1 0,7 — —
Fel 2599,39 0,2 2483,27 10
Cdl 2288,02 0,2 — —
MnI, 11 2576,10 0,2 2794,8 0,6
Cui 3247,54 0,2 3247,54 0,6
Pbl 2833,07 0,7 2833,07 2,5
SbI 2598,06 0,2 — —
Zn I 3345,57 7 — —
Nil — — 3002,49 2,5
Col — — 3044,0 2,5
Cr II — — 2843,24 2,5
Таблица 47
Чувствительность определения примесей спектрографическим методом с применением автоматического фотометрирования
Элемент и линия, А Чувствительность, % (п.10-*) Элемент и линия, А Чувствительность, % (n-10->)
Fe I 2483,27 10 Cr II 2835,63 1
Си I 3247,54 0,5 Mg II 2795,53 0,1
Со I 3044,0 10 Al 3082,155 0,5
Ni I 3050,82 10 Ca II 3179,33 1
Мп I 2794,8 0,8 Sil 2506,899 1
регистрограммы приведен на рис. 28) позволяет повысить чувствительность определения Си, Со, Ni, Са, Мп в 2—3 раза.
Определение калия в смешанных титанатах состава Ba1_a,Sra.TiO3; (0,01—2%). Тонко растертую пробу (0,1 г) сплавляют в платиновом тигле с 0,5 г Н3ВО3 и 0,4 г ZnO в течение 1,0—1,5 часа. По охлаждении к плаву прибавляют 5 мл 70%-ной НС1О4 и по каплям вводят 10 мл 30%-ной Н2О2. Раствор разбавляют до 50 мл и фотометрируют в воздушно-ацетиленовом пламени с помощью фотометра, например, с интерференционным светофильтром. Калибровочный график строят для растворов, содержащих от 0 до 15 мг К и полученных с использованием тех же количеств плавней и реагентов. [1090].
152
Линия темнового тона
+
Рис. 28. Вид регистрограммы участка спектра
Гф и Гл+ф — интенсивности фона и линии + фона соответственно
Рис. 29. Полосы поглощения 5,7-дибром-8-оксихинолина (I) п его комплекса с европием (2)
Определение примеси европия в монокристалле иодида стронция [381]. После переведения образца в раствор примесь европия, добавляемого в качестве активатора, определяется экстракционнофотометрическим методом по окраске 5,7-дибром-8-оксихинолино-вого комплекса, раствор которого имеет максимум поглощения около 400 нм (рис. 29).
Измельченный образец (0,25—0,5 г) нагревают в кварцевом тигле с 10 мл HNO3 до отгонки иода, затем раствор выпаривают досуха. Остаток растворяют в воде, раствор разбавляют водой до 50 мл. Аликвотную часть раствора, содержащую 50—100 мкг Ен, разбавляют водой до 25 мл, прибавляют одну каплю фенолового красного, 2 мл этанола и нейтрализуют раствором NH4OH до розовой окраски индикатора. Затем прибавляют 10 мл аммонийно-ацетат-ного буферного раствора (pH 8,7) и экстрагируют Ен двумя порциями по 5 мл 0,5%-ного раствора 5,7-дибром-8-оксихинолина в хлороформе. Экстракты собирают в мерную колбу емкостью 25 мл, разбавляют хлороформом до метки и фильтруют в кювету с I = 2 см. Измеряют оптическую плотность полученного раствора относительно холостого раствора. Градуировочный график строят, проводя известные количества европия через стадию экстракции из аммонийно-ацетатных растворов [381].
АНАЛИЗ ПРЕПАРАТОВ, СОДЕРЖАЩИХ СТРОНЦИЙ
Для определения стронция в содержащих его препаратах преимущественно используются комплексонометрический и спектрографический методы. Спектрофотометрический и другие методы нашли ограниченное применение.
Приводимый (табл. 48) перечень анализируемых объектов содержит в основном сложные неорганические соединения, представляющие практический интерес.
153
Таблица 48
Методы определения стронция в его препаратах
Объект анализа Метод определения Литература
Иодид стронция Комплексонометрический [39]
Стронций-кальциевый титанат » [115]
Стронций-бариевый титанат » [Ю7]
Стронций-кадмиевый титанат » [324]
Стронций-бариевый титанат Рентгенофлуоресцентный [48, 49]
Стронций-висмутовый титанат Комплексонометрический [439]
Стронций-кальций-висмуто- Рентгенофлуоресцентный [47 , 49J
вый титанат Комплексонометрический [277]:
Стронций-кальций-висмуто-вый титанат с добавкой церия Спектрографический [478]
Стронций-бариевый ниобат » [467]
Кальций-стронций-свинцовый ниобат Комплексонометрический [120]
Комплексные соли (ацетил-ацетонаты, аминоэтилмеркап-тиды, нитрилотриацетаты) » [1325]
Определение основного вещества в иодиде стронция [39]. Навеску препарата (2 г SrJ2) растворяют в бидистиллированной воде, раствор переводят в мерную колбу емкостью 100 мл и разбавляют водой до метки. Для определения стронция отбирают 20 мл полученного раствора, прибавляют 80—100 мл воды. Затем прибавляют 25 мл NH^OH (уд. вес 0,91) и около 0,1 г индикаторной смеси (метилтимоловый синий, разбавленный хлоридом натрия «х.ч.», в отношении 1 : 100). Титруют 0,05 М раствором комплексона III до перехода интенсивно синей окраски в серую.
Содержание иодид-иона определяют аргентометрическим методом с индикатором флуоресцеином.
Определение составляющих в сложных титанатах (Sr—Са, Sr—Ва, Sr—Cd, Sr—Bi) [107, 115, 324,439]. Образец растворяют в НС1 или в H2SO4 (в случае стронций-висмутового титаната), титан отделяют экстракцией в виде купфероната (смесью равных объемов изобутанола и бензола), висмут — в видегодида (экстрагент— циклогексанон). Стронций определяют комплексонометрически в ряде случаев совместно с другим присутствующим металлом, последний определяют каким-либо методом и рассчитывают содержание стронция по разности. Титан определяют из отдельной навески спектрофотометрическим методом. Для примера приводим методику анализа стронций-бариевого титаната [107].
Пробу (0,2 г) растворяют при нагревании в 80 мл конц. НС1, раствор упаривают до 40—50 мл, прибавляют 10 мл 6%-ного| раствора купферона в 154
Рис. 30. Зависимость интенсивности излучения Sr (1), Ва (2) и Са (3) от концентрации Nb в растворе
экстрагируют купферонат титана 30 мл смеси (1 : 1) изопентанола с бензолом; экстракцию повторяют. Водный раствор кипятят 10—15 мин. и по охлаждении разбавляют до 200 мл (раствор А).
Для определения бария к 100 мл раствора прибавляют несколько капель метилового оранжевого и нейтрализуют конц. раствором NH4OH до перехода окраски в желтую, затем прибавляют 10 мл 5%-ной СН3СООН и по каплям 10 мл 0,65’ЛГ раствора К2СгО4. Пробу выдерживают 2 часа на водяной бане, осадок отфильтровывают, отмывают его от раствора К2СгО4 (до обесцвечивания промывных вод) и растворяют в 50 мл НС1 (1 : 1), промывая фильтр 5—6 раз водой.
К раствору^прибавляют 3 г KJ и через 15 мин. титруют выделившийся иод 0,01’А раствором Na2S2Os (индикатор — крахмал). Для определения суммы Ва + Sr к 15 мл раствора А прибавляют 300 мл воды, 100 мл конц. раствора NH4OH, 3—5 мл 5%-ного раствора комплексоната магния, 0,1 г метилтимолового синего и титруют 0,01 N раствором комплексона III до перехода голубой окраски раствора в серую. Содержание стронция рассчитывают по разности. Титан определяют в отдельной навеске (0,1 г) дифференци ально-спектрофотометрическим методом по реакции с перекисью водорода. Относительная погрешность определения стронция, бария и титана 0,5— 1,5 отн. %. Продолжительность анализа 5—6 час.
Анализ ниобатов CaieSryBa[i_(iB+y)](NbO3)2 [474]. Для определения щелочноземельных металлов используется пламеннофотометрический метод. Ва предварительно отделяется в виде сульфата. Ввиду влияния Nb на интенсивность излучения щелочноземельных металлов (рис. 30), определение Са и Sr проводят в присутствии избытка соответственно Sr или Са.
Навеску (0,15 г) тонко измельченной пробы сплавляют с 1 г NaOH в серебряном тигле в муфельной печи при 750—800° С. Плав выщелачивают водой с добавлением 8—10 мл конц. НС1 и 0,5 мл Н2О2. Полученный раствор переводят в мерную колбу емкостью 100 мл и доводят водой объем до метки (раствор Б). Из аликвотной части (50 мл) раствора В определяют Ва. Его осаждают серной кислотой, осадок BaSO4 отфильтровывают, промывают разбавленной (1 : 20) H2SO4. Фильтр с осадком переносят в стакан емкостью 200 мл, обрабатывают аммиачным раствором комплексона III (10 мл 0,1 М ЭДТА +
155
10 мл аммиака) при нагревании до кипения. Раствор фильтруют в мерную, колбу емкостью 100 мл, доводят водой объем до метки. Проводят пламенно-фотометрическое определение бария при 553,5 нм.
Фильтрат и промывные воды после отделения Ва объединяют и используют для определения Nb весовым методом. Раствор упаривают под инфракрасной лампой до влажного остатка, приливают 20—30 мл воды, добавляют NH4OH до слабого запаха, нагревают до кипения и осторожно кипятят 2—3 мин., накрыв стакан часовым стеклом. Затем раствор охлаждают и фильтруют через-двойной фильтр (синяя лента), на который предварительно нанесена бумажная пульпа. Осадок тщательно промывают 150—200 мл горячего 2%-ного-раствора NH4NO3, фильтр с осадком высушивают и прокаливают до постоянной массы в платиновом тигле в муфельной печи при 1000° С.
Для определения Sr 1 мл раствора Б переносят в мерную колбу емкостью-25 мл, добавляют раствор СаС12 до концентрации 250 мкгСа/мл, объем доводят водой до метки.
Для определения Са 5 мл раствора Б помещают в мерную колбу емкостью-25 мл, прибавляют раствор SrCl2 до концентрации 250 мкг Sr/мл, объем доводят до метки водой.
Определяют Са, Sr и Ва по методу добавок или по калибровочным графикам в координатах I — с, где I — интенсивность аналитической линииjb условных единицах (излучение фона пламени учитывают графически на диаграммной ленте самописца), с — концентрация элемента в эталонном растворе /мкг/мл). Коэффициент вариации не превышает 3%.
Определение бария в смешанных титанатах Ba^^Sr^TiOg. [846]. Используется титриметрический метод, предусматривающий осаждение хромата бария.
Образец (0,5 г) в платиновом тигле сплавляют с 0,5 г Na2COs и 0,25 а HSBO3. Плав после охлаждения обрабатывают 4 мл НС1 (1 : 1). Содержимое тигля переводят в делительную воронку, разбавляют водой до 20 мл, приливают 17 мл насыщенного водного раствора купферона, 30 мл пентанола и 10 мл бензола и встряхивают. Органическую фазу, содержащую купферонат титана, промывают дважды 20 мл 0,1 N НС1. Промывную жидкость присоединяют к водному раствору, добавляют 21 мл конц. НС1 и выпаривают досуха. К остатку приливают немного воды, 25 мл 0,05 N К2Сг2О7 и NH4OH до полного осаждения хромата бария. Осадок отфильтровывают на стеклянном фильтре, растворяют в разбавленной хлорной кислоте, прибавляют 5 мл конц. Н3РО4 и 20 мл 0,04 N раствора Fe2+ в Н3РО4 (для восстановления хромат-иона). Непрореагировавший избыток Fe2+ титруют 0,02 N раствором КМпО4, используя в качестве индикатора ферроин.
Определение стронция во внутрикомплексных соединениях (ацетилаце-тонате, аминоэтилмеркаптиде, нитрилотриацетате) [1325]. Анализируемую пробу (10—20 мг) помещают в пробирку, добавляют 10 капель смеси 62%-ной HNO3 и 60%-ной НСЮг (3 : 1). Осторожно при непрерывном встряхивании нагревают микрогорелкой и упаривают раствор до небольшого объема^ В полученном растворе (после разбавления водой) содержание Sr определяют комплексоно метрически.
Глава VII
ОПРЕДЕЛЕНИЕ РАДИОАКТИВНОГО СТРОНЦИЯ
ОБЩИЕ СВЕДЕНИЯ
Радиоактивные изотопы стронция с массовыми числами 89, 90 и 91 принадлежат к числу изотопов, на которые приходится один из двух максимумов кривой зависимости выхода продуктов деления тяжелых ядер от их массового числа (рис. 31). При делении урана максимальный выход одного из изотопов стронция составляет 6,5%, при делении плутония — 2,4% (табл. 49).
Таблица 49
Выход изотопов стронция (%) при делении урана и плутония [1229а]
Изотоп стронция Выход изотопов Sr при делении ядер и вид нейтронов (указывается в скобках)
2,,Ц (термин.) >«и (14 Мэе) 2“»и (быстр.) 21»l u (термин.) «»Ри (быстр.>
SSSr 6,5 4,5 2,9 1,9 -
90Sr — 4,5 3,2 — 2,3
91Sr — 4,9 — 2,4 —
Превращения атомов, непосредственно приводящих к образованию изотопов с массовыми числами 89 и 90, приведены на рис. 32 [1205].
Наибольшую опасность для организма людей и животных представляет долгоживущий изотоп "Sr. Изотоп 89Sr сравнительно быстро распадается и еще быстрее распадается изотоп 81Sr. Данные о периоде полураспада других изотопов имеются в табл. 1.
Для определения 89Sr в присутствии 90Sr можно использовать разницу в верхней границе энергии спектра [J-частиц, излучаемых ими (90Sr 0,5 Мэе, 89Sr 1,5 Мэе). Частицы, выделяемые 90Sr, можно задержать с помощью поглотителя — алюминиевой фольги (100 мг/см2).
Как следует из рис. 32, 9°Sr превращается в 90Y, который явля-
157
Рис. 31. Распределение осколков деления 235U по массовым числам (атомным весам) при расщеплении его термическими нейтронами (1) и нейтронами с энергией 14 Мэв (2) [1229а]
Рис. 32. Схемы превращений изотопов 89Вг и 90Вг, приводящих к образованию изотопов 89Sr и 90Sr
Периоды полураспада изотопов представлены в прямоугольниках
+ нейтрм
<ется Р-излучателем с энергией р-частиц до 2,3 Мэв (период полураспада 64 часа).
При определении 80Sr имеются три метода устранения помех or дочерней активности:
1. После отделения стронция дают установиться радиоактивному равновесию между 90Sr и 90Y. Для этого требуется прохождение по меньшей мере десяти периодов полураспада иттрия. Обычно выдерживают 14—20 дней. При этом отношение 90Sr/90Y становится постоянным и может быть измерена их суммарная активность.
2. Из смеси изотопов отделяют 90Y одним из описываемых ниже методов, затем измеряют активность 90Sr (до накопления заметных количеств 90Y).
3. Наиболее часто используемый метод заключается в выделении 90Sr, который затем оставляют до накопления 90Y. Последний отделяют и измеряют его р-активность.
158
Содержание радиоактивного стронция в образце рассчитывают по формуле [300]
Z7 = — —---»10~12 кюри/кг или кюри/л,
2,
где .7 — содержание 90Sr в единицах радиоактивности; А — измеренная на счетчике радиоактивность (имп/мин)', К — коэффициент перехода к абсолютному счету; и — константы распада 90Sr и 90Y (дни-1 и часы-1 соответственно), — время от момента отбора пробы до измерения активности, (дни); t3 — время от момента отделения иттрия от стронция до измерения активности (часы); гх и i2 — химический выход носителей — стронция и иттрия соответственно; М — количество образца, взятого для ана- 1 лиза (кг или л).
При отсутствии равновесия между 90Sr и 90Y равновесное количество последнего рассчитывается по формуле [300, 1288]
| __g—Xsfa
где A1 — активность, измеренная на счетчике через t3 дней от начала накопления 90Y (имп/мин)', 2.3 — константа распада 90Y (дни"1).
В образцах стронция, подвергшихся облучению нейтронами, содержатся изотопы 86mSr, 86Sr, 87mSr, 89Sr; может присутствовать также 89mSr, который мало изучен. Единственным [3-излучателем в этой группе изотопов является 89Sr, другие изотопы излучают только у-кванты и их скорость распада может быть'измерена сцинтилляционным счетчиком. Поэтому дают разложиться короткоживущим изотопам, после чего измеряют активность 89Sr и 86Sr. Последний как у-излучатель может быть легко идентифицирован с помощью сцинтилляционного счетчика.
Продуктами распада могут быть и ряд других изотопов стронция. По названному выше принципу измеряют активность в смеси изотопов 89Sr и 90Sr. Изотопы с более высокой массой — 91Sr и 92Sr, кроме р-частиц, излучают у-кванты и также могут быть определены с помощью сцинтилляционных счетчиков. Короткий период полураспада (см. табл. 1) имеют р-излучатели — 93Sr, 94Sr, 96Sr и 97Sr. Поэтому их активность во внимание не берется.
В операциях выделения стронция из естественных или искусственных объектов используются различные аналитические методы: осаждение, экстракция, ионный обмен и др. Ход анализа состоит в добавлении носителя, неактивного стронция (25—50 мг), а также носителей других радиоактивных элементов, и в отделении стронция от посторонних элементов с применением ряда операций. Затем гравиметрическим, титриметрическим или пламеннофотометрическим методом определяют выход стронция и вводят поправку в измеренную активность на степень выделения.
159'
Если анализируемые образцы содержат заметные количества неактивного стронция, они также должны быть проанализированы на его содержание (чтобы можно было точно рассчитать выход стронция).
Для определения выхода стронция к анализируемому раствору можно также добавить 89Sr или 85Sr, определение активности которых проводят по у-излучению. Выделение радиоактивного стронция из растворов продуктов деления описано в работе [860].
К анализируемому раствору добавляют носители Sr и Ва и раствор Na2CO3. Фильтрат отбрасывают, а осадок SrCO3 + ВаСО3 растворяют в HNO» и добавляют дымящую HNO3. Вновь отделяют фильтрат, а осадок Ba(NO3)2 + Sr(NO3)2 переосаждают, растворяют в воде, добавляют носитель Fe и осаждают Fe(OH)3. Осадок отфильтровывают, а в раствор добавляют ацетатный буфер и Na2CrO4. Выпавший осадок ВаСгОа отбрасывают, в раствор добавляют Na2CO3. Затем отделяют фильтрат, а образовавшийся осадок SrCO3 растворяют b’HNOs, вновь вводят носитель Ва и повторяют осаждение ВаСгОа. Осадок отфильтровывают, а в раствор добавляют Na2CO3. Полученный осадок SrCOs отделяют от фильтрата, растворяют в кислоте, добавляют носитель Fe, кипятят до удаления СО2 и добавляют NH4OH, фиксируя время. Затем осадок Fe(OH)3 отделяют, раствор нагревают с твердым (NH<)2CO3, фильтрат отделяют и отбрасывают, а осадок SrCO3 взвешивают и измеряют активность 8s Sr + 903г’(активность 90Sr измеряют по активности дочернего продукта "Y, который накапливается в течение 10—15 дней и отделяется от материнского изотопа).
Операция отделения Sr от Са осаждением нитратов дымящей HNOS может быть опущена путем подсчета черенковского излучения изотопов 89Sr и 90Sr с использованием современных жидкостных сцинтилляционных счетчиков, так как максимальная энергия 45Са (0,258 Мэв) ниже черенковского порога в воде (0,263 Мэв) [1185].
ОТДЕЛЕНИЕ t0Y ОТ 93Sr
Для отделения дочернего 90Y от "Sr и получения 90Y, свободного от носителя, предложено большое число методов, основная сводка которых представлена в табл. 50. Ряд ионообменных методов разделения этих изотопов можно найти в главе III (табл. 20— .22). Перспективными являются также экстракционные методы. Как следует из табл. 50, сорбируемым на твердой поверхности, выделяемым в виде осадка, экстрагируемым в органическую фазу или удерживаемым на ионите может быть каждый из разделяемых изотопов. Применение метода электроосаждения базируется на выделении иттрия.
Экстракционные методы
При использовании в качестве экстрагента трибутилфосф а т а коэффициент распределения 90Y составляет ~14 (для 11—12 N HNO3) и 6,5 (11—12 N HG1); соответствующие
160
Таблица 50
Методы, используемые для отделения 90Y от "Sr
Метод Реагент, поверхностно-активный материал или выделяемое соединение Выделяемый (удерживаемый) элемент Литература
Сорбция BaS04 (прокаленный осадок, находящийся в смеси растворов Н2О, С2Н6ОН, НС1, K2SO4 и носителя Y) Sr [896]
SrSO4 (активированный!0,5 N H2SO4)J Sr [807]
Стекло (предварительно обработанное растворами кислоты и гидроокиси стронция) Y [194, 480]
Окись алюминия (1,6 М NH4NO3) Y [1082]
Винилхлоридная лента (pH 8,0—8,4) Y [864]
Осаждение Фосфат, оксалат Sr [999]
Нитрат Sr [1209]
Соосаждеиие СаС2О4-Н2О (в присутствии комплексона III) Sr [203]
Fe(OH)3(pH 3—4), осаждение аммиаком или пиридином в присутствии 2% K2S04 Y [278, 288, 923, 924]
Экстракция Трибутилфосфат [(10—12 N HNO3 или НС1)| Y [280, 284]
Диалкил- и диарилфосфорные кислоты (растворители: бензол, толуол, хлороформ и др.) Y [337, 693, 696, 995]
0,005 М монооктиланилино-бензилфосфонат в петролей-ном эфире (0,01— 0,1 М НС1) Y [826]
Теноилтрифторацетон (0,5 г/л в бензоле, pH 6,5—7,0) Y [725, 907]
1-Фенил-З-метил -4-каприлпи-разолон-5 (в метилизобутил-кетоне (pH 3,8) Y [1041|
Ионообменная Дауэкс-50Х8; 1,4 М |HNO3 Sr [1176]
хроматография Дауэкс-50Х8; 0,55 М CH3COONH4+ 0,001 М HN03 в 30%-ном СН3ОН Sr [899]
Дауэкс-50Х8; 0,2—0,5 М молочная кислота (pH 2,0— 3,45) или 0,5 М гликолевая кислота (pH 3,18—3,77) Sr [1176, 1177]
Дауэкс-50Х8; 1%-ный раствор (NH4)2C2O4 (pH 6,5) Sr [1293]
6 н. G. Полуэктов и др.
161
Табл и ц а 50 (продолжение)
Метод Реагент, поверхностно-активный материал или выделяемое соединение Выделяемый (удерживаемый) элемент Литература
Дауэкс-50Х8; 5%-ная лимонная кислота (pH 3,2) Sr [566 J
КУ-2; цитрат аммония (pH 5,0) или 0,77%-ная а-окси-масляная кислота Sr [1285]
Хелатообразующий ионит на основе 8-оксихинолина (ацетатный буфер, pH 3,0) Y [545]
Ионит, содержащий функциональные группы: -CH2-CHfO-PO(OH)2]-, .u-COOHC6H4C(OH)[PO(OH)2]- •С6Н4СНСН2 (0,2 TV НС1) Y [1007|
АН-1 (в этилендиаминтетра-ацетатной форме); 0,01 М ЭДТА (pH 8) Y [325]
Дауэкс-1Х8 (НСОй~-форма); элюэнт — вода Y [1066]
Дауэкс-1Х8 (8О24~-форма); сорбция из 50%-ного СН3ОН+ + 6% HNO3; элюэнт — 50%-ный СН3ОН] Y [1381]
Неорганические ионообмен-ники: пирофосфат Zr, фосфат Sn(IV); элюэнт — разб. HNO3 Y [654, 866 J
Бумажная хроматография Ватман Р-20; 1 М а-оксиизо-масляная кислота (pH 5,0) Sr [755]
Бумага импрегнированная: Амберлитом IR-4B (элюэнт — вода) или Амберлитом IR-120 (элюэнт — 5%-ная лимонная кислота, pH 3,8) [1268|
фосфатом Zr (элюэнт— смесь СН3СООН + + CH3COONa, pH 4,5) —' [1358]
фосфатом Sn(IV) (элюэнт— 1,0 М NH4NO3) — [445]
кремневольфраматом аммония (элюэнт — 0,1 М HNO3) — [10001
фосфоромолибдатом аммония (элюэнт — 0,1 М NH4NO3) [1224]
162
Таблица 50 (продолжение)
Метод Реагент, поверхностно-активный материал или выделяемое соединение Выделяемый (удерживаемый) элемент Литература
Распределительная хроматогра- Растворители: метано л—этанол —2N НС1 [777]
фия на бумаге (3:3:4) этанол—бутанол—10 %- [908]
ный р-р NH4SCN (5:5:2) метанол—этанол—разб. [754]
Метод кольцевой бани НС1—лимонная кислота 0,1-0,2 NH4SCN — [1261]
Тонкослойная Силикагель; подвижная фа- [805]
хроматография за — этанол + 10%-ный р-р NH4SCN (5 : 3) Силикагель и крахмал; подвижная фаза 0,01—l,0Y H2SO4 Фторопласт-4, покрытый [951|
Хроматография Y* [238]
с обращенными фазами пленкой ди-2-этилгексилфос-форной кислоты (элюэнт 0,1 — 0,15 М НС1) Бумага Ватман № 1, пропи- [984]
тайная ди-2-этилгексилфос-форной кислотой (элюэнт— 0,5 М НС1О4) Асбестовая бумага, обрабо- |510]
Электрофорез на бумаге тайная конц. растворами ]НС1 hHNO3 (растворитель — 1 N НС1) Электролиты: 0,001 М НС1 [898]
0,5 М NaNO3 — [1247[
K4[Fe(CN)6] (2%-ный) — [986]
0,044 М СН3СООН — [957]
Соли аммония: 0,1 М аце- [184]
тат (pH 5), 0,01 М малонат (pH 5), 0,1 М формиат (pH 5), 0,05 М цитрат (PH 2,7) 0,1 М CH3COONH4 или [677]
0,1 М комплексон III 0,15 М лимонная кислота [269]
Фокусирующий 0,1 М нитрилотриуксусная — [1113, 12281
электрофорез на бумаге кислота (католит), 0,05— 0,1 М НС1 (анолит)
6*
163
Таблица 50 (окончание)
Метод Реагент, поверхностно-активный материал или выделяемое соединение Выделяемый (удерживаемый) элемент Литература
0,02—0,3 М этилендиамин-тетрауксусная кислота (католит), 0,1— 0,5 М НС1 (анолит) — [1018, 1228)
Электродиализ через ионообменную мембрану Катионит Амберплекс С-1 или анионит Амберплекс А-1; 0,001 М НС1 или NH4C1 Sr [775]
Аниониты; раствор H2SO. (pH 3) — [776]
Электроосажде- Аниониты; 0,001 М ЭДТА (PH 5,5) Электролиты: [787]
NH4NO3, HNO3 (pH 2,5± + 0,3) Y [Э54|
NH4CI, NH4NO3 или (NH4)2SO4 (pH 5-6) Y [806]
Флотация Желатиновая пена в присутствии Fe(III) (pH 6,0) Y [353]
Желатиновая пена в присутствии ВаС12, Na2SO4 или lA’ H2SO4 Sr [353]
* В этом случае разделены изотопы ”mSr и ”Y.
значения для 90Sr равны 0,0003 и 0,01. Процесс включает экстракцию "Y, два промывания экстракта 12 N HNO3, трехкратную реэкстракцию водой (по 200 мл) при 50—60° С и промывание реэкстрактов четыреххлористым углеродом. Степень извлечения 90Y из исходного раствора составляет 97 %, а его радиохимическая чистота достигает 99,999% [280].
Выделение "Sr из смеси продуктов деления [907] экстракцией раствором теноилтрифторацетона (ТТА) проводят следующим образом. Предварительно производят экстракцию 0,05 М раствором ТТА в бензоле при pH 7,0. При этом извлекаются все продукты деления, кроме Sr, Cs и Ru. Затем pH водной фазы доводят до 8,0, экстрагируют стронций 0,05 М раствором ТТА в метилизобутилкетоне. Присутствие комплексообразующих веществ (ЭДТА, лимонная кислота) препятствует выделению стронция.
При выделении 88Y из Sr-мишени, облученной в циклотроне, растворяют образец стронция в соляной кислоте, устанавливают кислотность ~0,05 М, экстрагируют иттрий|2 мин. равным объемом 5• 10~3 М раствора монооктиланилинбензил-фосфоната в петролейном эфире [826].
164
Хроматографические методы
Применение метода ионного обмена основано на различной способности разделяемых элементов к комплексообразованию с рядом лигандов.
Исходный раствор [1176] выпаривают досуха, остаток растворяют в нескольких каплях разб. HNO3, Sr и Y сорбируют на колонке (0,58 X 3,3 см), заполненной катионитом Дауэкс-50АУХ8 (200—400 меги), 9oSr вымывают 20 мл) 1,4 М раствора HN03 со скоростью ~1 см!мин, после чего вымывают ®°Y ЗМ раствором HNO3 (10 мл). При многократном выделении 9°Y из одного и того же препарата 9oSr—9oY проводят избирательное элюирование 9oY 0,5 М раствором лактата аммония с pH 2,9—3,45 (9oSr остается на колонке).
В других случаях при использовании того же катионита (100— 400 меш) для разделения 90Sr и "Y последний вымывают из колонки 0,55 М ацетатом аммония в 30%-ном метаноле [899], 1%-ным раствором (NH4)8C8O4,; при pH 6,5 [1293], 5%-ным раствором лимонной кислоты (pH 3,2) [566], 0,5 М растворами гликолевой (pH 3,18—3,77) или молочной (pH 3,05—3,45) кислот [1177]. Из 0,2 М молочной кислоты на колонке сорбируется "Sr; "Y при этом не задерживается (pH 4,6).
Ионообменное разделение Y и Sr на катионите КУ-2 в ХН4-форме путем элюирования раствором цитрата (pH 5,0) или 0,77%-ным раствором а-оксиизомасляной кислоты предложено в [1285].
Хелатный катионит, синтезированный на основе 8-оксихиноли-на [545], обладает высокой радиационной устойчивостью и пригоден для аналитических разделений при большой интенсивности радиации. С помощью ионита в Н+-форме осуществлено отделение "Y от "Sr. Последний не сорбируется смолой в присутствии ацетатного буфера (pH 3,0).
Селективные сорбционные свойства полимерных катионообмен-ников, содержащих фосфор в своей функциональной группе, использованы для отделения "Y от "Sr в [1007]. "Y избирательно сорбируется катионитом из 0,2 N НС1.
Разделение при помощи ионообменной бумаги проведено в [755, 1268]. Стронций, сорбированный на катионообменной бумаге (Ватман Р-20), может быть элюирован 2 N НС1 или 5 N HNOS [755]. Предварительная обработка бумаги раствором NH4C1 ,поз-воляет четко разделить "Sr, "Y и 137Cs при использовании в качестве элюента 1 М раствора а-оксиизобутирата аммония (pH 5).
Каплю'раствора, содержащего изотопы обоих металлов в IN НС1, наносят на полоску хроматографической бумаги, пропитанной смолой Амберлит IR-120 в Ха+-форме. Элюэнтом служит 5%-ный раствор лимонной кислоты с pH 3,8 (Rf для 9о Sr равен 0; Rf для 9°Y равен 0,75). Хорошие результаты получены также с другой модификацией той же смолы (IR-4B) в ОН_-форме; проявление проводится водой (Rf для Sr равен 0,87; Rf для Y равен 0) [1268],
165
Рис. 33. Разделение радиоизотопов стронция (7) и иттрия (2) на анионите АН-1
В ряде работ для разделения 90Sr — 90Y предложено использовать неорганические ионообменники, нанесенные на полоски бумаги: фосфат циркония [1358] и олова (IV) [445], кремневольфрамат [1000] и фосфоромолибдат [1224] аммония. Например, при использовании бумаги, импрегнированной фосфатом олова, удовлетворительные результаты получаются при элюировании 1 71/ раствором NH4NO3 (7?/ равны 0,80 и 0,02 для 90Sr и "Y соответственно).
Стронций от иттрия может быть отделен с помощью анионитов Дауэкс-1Х8 [1096, 1381], АН-1 [325] и др.
Заполненную анионитом АН-1 колонку (12 X 0,5 см) в этилендиаминте-траацетатной форме промывают водой и вносят 0,1 мл раствора, содержащего 9<1Sr и90У. Десорбцию производят 0,01 М раствором комплексона III при pH 8 (рис. 33).
При разделении изотопов стронция и иттрия методом бумажной хроматографии используют различные проявляющие растворители. Так, в смеси, содержащей 30% метанола, 30% этанола и 40% 2 N НС1, значения Rf равны 0,42 и 0,63 для стронция и иттрия соответственно. Успешное разделение элементов наблюдается при содержании каждого элемента в пробе не выше 20 мкг.
Смесь, состоящая из этанола и бутанола с добавлением раствора NH4SCN, применялась в [908, 909]. Показано, что изменение концентрации NH4SCN от 6 до 18% не влияет на эффективность разделения элементов. Rf для Y без носителя равен нулю, а для Sr 0,7—0,8. Минимальное время, требуемое для разделения, составляет ~30 мин. Влияние лимонной кислоты при разделении некоторых продуктов деления урана исследовано в [754]. Разде
166
ление 90Sr и 90Y методом бумажной хроматографии описано в [1079].
Метод тонкослойной хроматографии на силикагеле предложен в [805]. В качестве подвижной фазы используется смесь этанола и 10%NH4SCN (5:3), величина Rf составляет 0,83 (90Sr) и 0,07 (90Y). Хроматографирование в тонком слое, содержащем SrSO4, силикагель и крахмал, изучено в [951].
0,005 мл 0,01 М раствора по НС1, содержащего смесь 9oSr—90Y, наносят на расстоянии 25 мм от одного из краев пластины и через 20 мин. осуществляют хроматографирование восходящим методом в течение 16—45 мин., используя в качестве подвижного растворителя 0,01—1 N раствор H2SO4-.R/ для9°У равен ~,0,9,9oSr удерживается на месте нанесения пробы. Увеличение концентрации H2SO4 (до 1 N) способствует более четкому отделению 90У от 9°Sr.
Возможность определения примеси иттрия в препаратах радиоактивного стронция показана в [984]. Хроматографическое разделение проводят на бумаге с применением ди-2-этилгексилфос-форной кислоты в качестве неподвижной фазы и 0,5 Л/ НС1О4 в качестве подвижной. Фторопласт-4 (30—40 меш), покрытый пленкой ди-2-этилгексилфосфорной кислоты (0,4 г на 1 г фторопласта), использован для получения 87mSr без носителя [238].
Облученную Sr-мишень растворяют в 0,1—1,5 М НС1, доводят кислотность до 0,1—0,15 М по НС1 и пропускают через колонку с фторопластом. S’Y практически полностью сорбируется на колонке, стронций и другие возможные радиоактивные примеси остаются в растворе. Колонку промывают 100 мл 0,1 М НС1 и оставляют для накопления 87mSr (результат распада *’Y). Затем колонку промывают 5—8 мл 0,1 N НС1, в которую переходит 70—80% 87mSr. Через 9 час. по восстановлении равновесия между 87Y и 87™Sr вновь вымывают 87mSr. Метод позволяет получать 87n,Sr с чистотой 99,99% .
Распределительная хроматография при помощи асбестовой бумаги использована в [510].
Электрофоретические и другие методы
При электрофоретических методах на полосках бумаги или на активном тонком слое может быть достигнуто быстрое разделение "Sr и 90Y. Так, например, при градиенте потенциала 30 в!см в растворе уксусной кислоты разделение достигается за 30— 40 мин, [957].
В [806] иттрий осаждают на катоде из платиновой фольги электролизом раствора, содержащего смесь 9oSr—9oY при pH 5—6. Анодом служит вращающаяся платиновая проволока. Радиохимическая чистота иттрия ~99,9%. Электролитами; служат растворы NH4C1, NH4N03 или (NH4)2SO4. Добавка комплексантов улучшает чистоту иттрия, однако снижает его выход. Оптимальные условия электролиза: pH 2,5 ± 0,3; t ~ 50° С, плотность тока - 20 ма!смг. Путем повторного осаждения (катод предыдущего осаждения
167
используют в качестве анода для переведения иттрия в раствор) можно снизить концентрацию 9oSr в 9oY до 10-4% [954].
В некоторых случаях применяют выделение "Y путем осаждения в виде гидроокиси на носителе Fe(OH)3 с последующей его радиохимической очисткой [288, 278, 923, 924]. Используется также сорбция на неорганических сорбентах или осаждение 90Sr в виде фосфата, оксалата, нитрата [203, 999, 1209] (табл. 51).
ОПРЕДЕЛЕНИЕ СТРОНЦИЯ В РАЗЛИЧНЫХ^ОБЪЕКТАХ
Анализ вод
Количество воды, необходимое для проведения анализа, зависит от содержания радиоактивных веществ. При активности 10-10 кюри!л достаточно не более 10 л воды, при меньшей радиоактивности — 50—100 л или даже (при анализе морской воды) 200—300 л. Добавляют носитель — соль неактивного стронция — обычно до конечной концентрации 5—10 жгйг/л, а также носители для других элементов (Cs, Са, Ва, Y) и производят концентрирование стронция. При малых объемах раствора (3—9 л) концентрирование производят простым упариванием [46, 461, 562] до объема ~200 мл. Если необходимо брать большие объемы, используют методы осаждения, сорбции, ионного обмена и др.
Осаждение карбонатов [10, 46, 172, 174, 307, 386, 457, 562, 643, 681,766,802,808, 885, 935, 936, 1047, 1054, 1061, 1286, 1304]. Воду нейтрализуют аммиаком или едким натром и добавляют избыток карбоната натрия. Для удержания магния в растворе добавляют хлорид аммония [457]. Предложено осаждение карбонатом аммония [1054]. Часто осаждение карбонатов щелочноземельных элементов объединяют с выделением Cs в виде смешанных ферроцианидов цезия и щелочноземельных элементов [10, 174].
Осадок затем обрабатывают соляной кислотой и отфильтровывают от остатка, содержащего цезий. Далее применяют последовательные операции отделения загрязнений путем осаждения с Fe(OH)3 осадка гидроокисей, выделения хромата бария, осаждения нитрата стронция и т. д.
Осаждение оксалатов щелочноземельных металлов описано в [974, 992]. Осаждение производят из слабощелочной среды. Осадок оксалатов прокаливают до окислов, растворяют в НС1, после добавления носителя (соли иттрия) осаждают Y(OH)3, активность которого измеряют после прокаливания осадка.
Осадок оксалатов можно также растворить в HNO3, выделить нитрат стронция осаждением дымящей HNO3, затем отделить примеси соосаждением с Fe(OH)3, осадить ВаСгО4, после чего следует осаждение оксалата Sr и измерение его активности.
Ионообменный метод нашел широкое применение для выделения радиоизотопов стронция [137, 508, 509, 569, 570, 578, 624,
168
626, 808, 918, 943, 1029, 1237, 1238, 1239, 1263, 1271, 1376]. Водут pH которой предварительно доводят до 2—5 или 1—1,5, пропускают через колонку, заполненную катионитом в NH*- или Н+-форме (КУ-2, Даузкс-50\¥Х8 или др.). Для связывания Са к воде добавляют ЭДТА [943]. Производят элюирование катионов соляной кислотой [569] или же последовательно элюируют различные элементы. Например, гликолятом аммония различной концентрации можно последовательно элюировать Cs и Sr [1237], гликолятом или лактатом аммония — Cs и Се после чего Sr элюируют раствором цитрата аммония [1238, 1239].
Катионит можно озолить, после чего золу обрабатывают кислотами, затем следует обычный ход анализа с применением осаждения гидроокисей (Се и других РЗЭ), хромата бария, нитратов (отделение от Са) [508].
Для определения 90Sr в дождевой воде [578] последняя собирается в течение месяца в специальной воронке и последовательно фильтруется через колонку, содержащую слои асбестового волокна, фосфоромолибдата аммония, анионит в ОН~-форме и катионит в Н+-форме. Затем колонку разбирают на составные части и 9°Sr вымывают азотной кислотой, очищают осаждением нитрата и определяют по дочернему 90Y после его накопления.
В ряде других работ для предварительного выделения стронция использовались сорбция на сульфате бария, активированном кальцием [217], фосфоромолибдате аммония [580], фосфатированной целлюлозе [73], геле гидроокиси алюминия [750]; соосаждение с гидроокисью марганца(1У) [1246]; осаждение в виде фосфата [862], родизоната [162]; электродиализ [1104]. Применяются также минеральные сорбенты, лучшим из которых оказался вермикулит [624].
Описаны упрощенные методы определения 90Sr в морской воде, основанные на удалении из образца радиоизотопов посторонних элементов, накоплении дочернего 90Y, отделении последнего и измерении его активности. В методе [1245] к 20 л морской воды добавляют соль железа(Ш), осаждают и отделяют гидроокиси. Оставляют раствор на 2 недели для установления равновесия между 90Sr и "Y. Затем 90Y выделяют соосаждением с Fe(OH)3, неактивными Y(OH)3 и Се(ОН)3. Проводят очистку Y путем осаждения оксалата, экстракции Y из 14 М HNO3 трибутилфосфатом, отделения следов Се и Th соосаждением с йодатом циркония. В заключение выделяют оксалат иттрия, активность которого измеряют.
В [382] также удаляют мешающие радиоизотопы соосаждением с Fe(OH)3, оставляют раствор для накопления "Y, выделяют "Y путем осаждения с носителем иттрия в виде гидроокиси (осадок переосаждают) и оксалата, прокаливают последний до окиси и измеряют активность на p-спектрометре. Если предполагают наличие в морской воде радиоизотопов 14°Ва —140 La, то после первого осаждения Y(OH)3 осаждают Ва и La из насыщенного раство
169
ра сульфата калия в виде BaSO4 и двойного сульфата La и К (см. также [1101]).
Эффективное выделение 90Y при анализе морской воды проводится путем его экстракции с помощью теноилтрифторацетопа [725] и ди-2-этилгексилфосфорной кислоты [995].
Экспрессный метод определения 90Sr по его дочернему изотопу 90Y, основанный на использовании высокочувствительного p-спектрометра, описан в [383].
Ниже приведены две методики определения радиоактивного стронция в поверхностных водах.
КЗ л анализируемой воды добавляют 2 мл 0,1 М раствора SrCl2 и мл 0,01 М раствора ВаС12, выпаривают воду до объема 200 мл, прибавляют необходимое для связывания Са2+ количество комплексона III, рассчитанное на основании результатов титрования в отдельной пробе. После этого прэ >у подщелачивают и отфильтровывают осадки гидроокисей. Добавляют 30 мл 1 М Na2CO3, кипятят, охлаждают и фильтруют. Осадок карбонатов стронция и бария промывают водой (50 мл}, подщелоченной 3—4 каплями конц. раствора NH4OH и растворяют в 5—10 мл 1 М НС1; нейтрализуют раствор в присутствии метиловог о оранжевого конц. раствором NH4OH, прибавляют 10 мл 1 М СН3СООН, 4 г NH4C1, 5 мл 10%-ного раствора К2СгО4. Осадок хромата бария оставляют на ночь, отфильтровывают, промывают водой, этанолом, эфиром. Фильтрат нейтрализуют аммиаком, добавляют 1 мл 1 М СН3С00Н~и 10 мл 10%-ного раствора К2С2О4, кипятят. Образовавшийся осадок SrC2O4-H2O фильтруют, промывают водой (50 мл), затем этанолом. Растворяют в 10 мл ) м НС1 при нагревании, повторно осаждают оксалат стронция. Осадок промывают водой, этанолом, эфиром, высушивают в эксикаторе, измеряют его вес и активность (в помощью сцинтилляционного счетчика). Абсолютную активность стронция находят с помощью эталона 9oSr 4-+ 90Y [45].
Пробе (50—80 л) морской или речной воды дают отстояться в течение 2 —3 суток, декантируют, подкисляют НС1 до pH 2—3, вводят носители (Sr и Cs) до конечной концентрации 10 и 3 мг!л соответственно, приливают раствор, содержащий 150—200 г соли k4Fe(CN)<i-3H2O в минимальном количестве анализируемой воды, и отстаивают 3—4 часа. Затем при перемешивании последовательно прибавляют раствор, содержащий 200 г NH4C1 в небольшом количестве воды] и 400 г Na2CO3. Перемешивают 20—30 мин. и выдерживают 24 часа. Выпавший осадок карбонатов и ферроцианидов отфильтровывают, переносят в стакан, растворяют при кипячении с минимальным количеством 0,5—1,0 М НС1 в течение 1—2 час. и отфильтровывают нерастворпв-шийся остаток.
В фильтрате определяют 9°Sr. Для этого к нему прибавляют 200 м Fe3+ (осаждают примеси с Fe(OH)3), осадок отделяют, из раствора выделяют экса-латы при pH 3—4. Осадок оксалатов прокаливают при 80 С, остаток растворяют в 2 М НС1, к раствору прибавляют 50 мг носителя Y, выдерживают 14—16 дней (для накопления дочернего 9oY), осаждают Y(OH)3 аммиаком (без СО2). При этом фиксируют время отделения Y от Sr. Осадок Y(0H)3 растворяют в 2 М НС1, осаждают Y2(C2O4)3 с помощью щавелевой кислоты, 170
осадок отфильтровывают, высушивают, прокаливают, взвешивают для опре-деления химического выхода и измеряют f-активность на малофоновой счетной установке. Радиохимическую чистоту выделенного 9oY контролируют по периоду полураспада [10].
Анализ воздуха, осадков
Анализируемый воздух в количестве от нескольких десяткой до 40 000 л3 просасывают через специальные фильтры или собирают выпадения («сухие» и «мокрые») в сосуды в течение длительного срока (месяц и более); «сухие» выпадения (5—10 г атмосферной пыли) собирают также в специальные планшеты.
Для разрушения органических веществ и переведения в раствор стронция (и других катионов) собранные осадки подвергают химической обработке: нагревают со смесью HNO3 и Н2О2 [1359], HNO3 + Вг2 [1006], НС1 + HF [625], сплавляют остаток после озоления с Na2CO3 [917, 1360], со смесью Na2B4O7 и Na2CO3 [972] и т. п. После этого применяют обычный ход радиохимического отделения стронция с добавлением носителей и выделения Sr2+ в виде карбоната [917, 1006, 1359], нитрата [625, 1360], оксалата [1207]. Или используют метод ионного обмена [29, 871, 1323].
Выделение стронция методом хроматографии на бумаге [1359]. Фильтр после пропускания через него пробы воздуха] обрабатывают HNO3 (1 : 1), добавляют по 1 мл растворов металлов-носителей, содержащих 5 мг!мл Fe, 40 мкг!мл Sr, Ва, Се, La, Y, Zr и Nb. Для разложения органических веществ приливают конц. HNO3, 30%-ную Н2О2 и упаривают до объема 25 мл. Затем 3 раза выпаривают с 10 мл конц. HF и 10 мл конц. HNO3 (для удаления SiО2)?
Остаток растворяют в 25 мл конц. НС1 при нагревании, раствор нейтрализуют с помощью NH40H до pH 5,0, прибавляют 3 мл насыщенного] раствора Na2CO3 и раствор NH4OH до pH 9. Образовавшийся] осадок отделяют фильтрованием и растворяют в НС1. Разделяют] катионы Ва, Sr, La, Се и Y хроматографированием на бумаге смесью этанола, метанола, 2N НС1 и ацетилацетона (16 : 16 : 8 : 1). Зону Sr идентифицируют люминесцентным методом после обработки хроматограммы 0,5%-ным раствором 8-оксихинолина в 60%-ном этаноле и выдерживания ее в течение 2 мин. в атмосфере NH3. Вырезают зону Sr и обрабатывают смесью конц. HNO3 (50 мл) и НС1О4 (5 мл)', полученный раствор упаривают до 1—2 мл и добавляют 10 мл раствора носителя. 9°Sr выделяют на осадке SrCO3 и определяют по [[-излучению.
Применение кольцевой бани [972]. Воздух (4,5 .и3) пропускают через бумажный фильтр, смачивают его растворами носителей, раствором смеси Na2B4O7 и Na2CO3 (1 : 1) и помещают в платиновую спираль, нагреваемую электрическим током, в которой происходит озоление фильтра и образование перла. Полученный перл растворяют при слабом нагревании в 4 каплях iN НС1, прибавляют 6 капель воды, выпаривают раствор на фильтре, находящемся на кольцевой бане, и разрезают фильтр с образовавшейся кольцевой зоной на равные секторы. Секторы используют для определения отдельных радиоизотопов путем осаждения необходимого элемента в виде нерастворимого соединения и вымывания остальных элементов. Стронций осаждают в виде
171
сульфата. Измерение активности изотопов на фильтрах производят методом авторадиографии.
Метод ионообменного выделения стронция [1323] предусматривает предварительное его выделение из образца (совместно с цезием) на катионите Амберлит CG-120 и дальнейшее разделение Sr и Сана катионите Дианон-ВК. Показана возможность [29] концентрирования 90Sr из растворенной части атмосферных осадков на катионите КУ-2 с последующим вымыванием 6 М НС1. Растворы носителей при этом не добавляют.
Анализ почв
Радиоизотопы стронция, выпадая на поверхность земли, удерживаются в почвенном покрове (главным образом за счет процессов ионного обмена) и извлекаются из почвы разбавленными растворами минеральных кислот (в среднем степень перехода в раствор порядка 90%). Для оценки уровня загрязнения почв наиболее распространена обработка 6 М НС1 [232, 258, 259, 300, 302, 303, 401] и HNO3 [159], хотя в последнем случае в раствор переходит больше органических веществ, что усложняет анализ. 90Sr и 89Sr извлекаются растворами нейтральных солей (главным образом 1 N раствором ацетата аммония). |
Выщелачивание соляной кислотой [302]. Почву (500 г) предварительно высушивают при 105—110° С в течение 6—12 час., растительную часть пробы (корни, стебли) взвешивают и озоляют при 400—500° С. К объединенной пробе (почвы и золы) добавляют 2 мл раствора) Sr(NO3)2 (300 мг Sr) и обрабатывают 500 мл 6 М НС1, перемешивают в течение 30 мин., затем отфильтровывают, промывают. Остаток повторно обрабатывают 500 мл 6 М НС1. Солянокислые экстракты и промывные воды объединяют и производят необходимые операции по выделению стронция.]
В другом варианте [1205] 100 г почвы помещают в стакан, добавляют 250 мл 11 М НС1 (в случае известковых почв) или 500 мл той же кислоты (при анализе почв других типов), нагревают до 50° С и оставляют на 8 час. при перемешивании, отфильтровывают, промывают. Повторно обрабатывают остаток таким же образом. Второй фильтрат присоединяют к первому и концентрируют путем выпаривания. Раствор нейтрализуют 10 М NaOH и осаждают карбонаты. При этом железо связывают в комплекс оксалат-ионами. Фильтрат удаляют, осадок переосаждают 4 раза, растворяя его в HNO3 при кипячении. Затем аммиаком осаждают Fe и А1, Са и Sr осаждают карбонатом аммония или натрия. Осадок растворяют в НС1 и раствор поступает на измерение активности 9oSr.
Обработка соляной кислотой производится также после озоления почвы [160, 1186].
Сухую почву (400 г) озоляют при 500° С 2 часа, полученную золу встряхивают 4 часа с 1200 мл 10%-ного раствора НС1 при добавлении 50 мл раствора соли стронция (40 мг Sr в 1 мл).
т
Далее следует выделение сульфатов Sr и Ва, осаждение с Ре(ОН)з, отделение Ва осаждением хромата и т. д. [1186].
Метод извлечения стронция раствором ацетата аммония (метод ионного обмена) имеет ряд недостатков, к которым следует отнести необходимость применения больших количеств экстрагента и наблюдаемое в ряде случаев различие в степени извлечения Sr и Са, что приводит к сдвигу наблюдаемого соотношения между ними в почве и в экстракте. Последнее обусловлено тем, что Sr обменивается в почве на другие ионы в большей степени, чем Са, особенно в карбонатных почвах [232, 586]. Количество Са, определенное путем извлечения 6 М НС1, в 2—3 раза выше его количества, определенного путем извлечения ацетатом и лактатом аммония [232]. Существенное значение этот факт имеет при выражении содержания 90Sr в стронциевых единицах, т. е. в количестве мкмккюри 90 Sr на 1 г Са. Если извлечение этих элементов из почв проводится по методу ионного обмена, то отношение 90Sr/Ca может получиться более высоким, чем при более полном их извлечении соляной кислотой или сплавлением с содой.
Метод электродиализа также используется для выделения стронция из почв [783]. Преимуществом этого метода является получение почвенных экстрактов, содержащих небольшое количество Fe, Al, Мп и еще меньшие количества щелочных металлов.
Однако полнота извлечения "Sr при этом способе меньше в сравнении с методом кислотной экстракции. В зависимости от типа почв это уменьшение составляет от 5 до 25 % (по сравнению с экстракцией 6 М НС1 [301]).
Почву (100 г) помещают в электродиализную ячейку и покрывают водой. Если почва содержит мало солей, вместо воды применяют 0,05 М раствора Н3ВО3. Электродиализ проводят в течение 12 час. при напряжении 100 в. По окончании диализа водную фракцию удаляют отсасыванием, электроды и электродиализную ячейку промывают небольшим количеством разбавленной кислоты. Почвенный экстракт (объединенный с промывными водами) поступает на дальнейший анализ [783].
Разложение почв путем сплавления со смесью КОН + KNO3 + +КгСО3 (2:1:1) описано в [1032]. Сплав выщелачивают водой, остаток обрабатывают 6 М НС1. Из полученных солянокислых вытяжек производится выделение мешающих элементов (Si,Се и др.), а затем Sr.
В [401] из экстрактов предварительно выделяют Fe в виде Fe(OH)3, вводят носитель (100 мг Sr), затем карбонатом’аммония осаждают[СаСО3 и SrCO:), осадок высушивают, взвешивают и оставляют до накопления 9°Y.
Выделение Sr в виде карбоната описано также в других работах [99, 1032].
Для отделения микроколичеств Sr от больших количеств Са используют [99] избирательное осаждение Са раствором K*Fe(CN)e.
173
Анализируемый раствор слабо подщелачивают аммиачно-хлоридной буферной смесью, осаждают Са при нагревании 1,5-кратным избытком горячего раствора K4Fe(CN)6. Из фильтрата осаждают Sr карбонатом аммония, осадок прокаливают, взвешивают и определяют химический выход Sr. Далее осадок растворяют в 2 N HNO3, вводят носитель (Y), раствор выдерживают 2 недели для накопления 90Y, выделяют иттрий и измеряют его активность.
Выделение Sr из экстрактов в виде оксалата предложено в [258, 259, 301, 303], в виде су льфатав [160].
Почвенную вытяжку выпаривают почти досуха, разбавляют водой и в присутствии этанола осаждают SrSO4 при помощи 5%-hohH2SO4. Осадок переводят в раствор кипячением с Na2CO3. Двукратным осаждением HNO3 и ВаСгО4 очищают раствор Sr от Са, Ва, Ra и РЬ. Затем после отделения дочернего 9oY [путем внесения в раствор соли иттрия и осаждения Y(0H)3 аммиаком] повторно осаждают SrSO4, осадок отфильтровывают, прокаливают и взвешивают. Прокаленный сульфат стронция переводят в раствор, добавляют носитель Y, через 12 дней отделяют 90Y, измеряют его активность и рассчитывают содержание 90Sr.
Выделение сульфатов непосредственно из экстрактов серной кислотой проводят в [1186]. Далее сульфаты переводят в карбонаты, затем осаждают гидроокись Fe, хроматы Ва и Ra. Повторно осаждают карбонаты и растворяют их. После накопления осаждают 90Y на Fe(OH)3 и измеряют активность. В фильтрате определяют химический выход Sr путем осаждения оксалатов.
Предварительное выделение ряда элементов (Fe, Со, Ni, Zn, РЗЭ, Ru, Zr, Cu, As) путем осаждения их сульфидов или гидроокисей из экстрактов предложено в [1205]. При определении стронция в почвенных экстрактах, полученных методом электродиализа [783], используют ионообменное отделение.
Са, Mg и некоторые другие элементы связывают в комплексы путем добавления ЭДТА при pH 10. Затем подкисляют раствор до pH 5,5 и пропускают его через колонку с катионитом Дауэкс-50Х12. Mg, Са и редкоземельные элементы вымывают с колонки смесью 1%-ного раствора лимонной кислоты и 0,75%-ного раствора ЭДТА. Сорбированный на катионите 90Sr остается для накопления "Y.
В [707] также используется ионообменное отделение Sr от РЬ, Са, Ва и Ra.
Анализ биологических материалов
Основные объекты анализа — пищевые продукты, кости, моча, молоко. Проводят озоление образца при 550—600° С, золу обрабатывают кислотами — соляной [156, 302, 459, 652, 854, 955, 1126], азотной [100, 126, 600, 1084, 1331], фтористоводородной и хлорной [258] или хлорной и серной [1175]. Применяется также сплавление с карбонатами [459, 1148]. Далее из полученного раствора выделяют (после добавления носителей) стронций.
174
Первичной стадией является осаждение нитратов [809, 1084, 1288], карбонатов [156, 1288, 1331], фосфатов [126, 258, 259], оксалатов [302, 955] или сульфатов [173, 459, 1175]. В [1344] выделение сульфатов производится в присутствии желатины, препятствующей выпадению сульфата кальция. Используется выделение на катионите [854, 1106], в присутствии больших количеств Са при добавлении ЭДТА (при pH 3,8 Са находится в анионной форме и не сорбируется ионитом, Sr присутствует как катион и поглощается смолой [1148]). В [564] сорбция на анионите из 12 М НС1 применяется для удаления из раствора и отделения от Sr элементов Fe, Zn, Zr, Nb и др.
Заслуживают внимания быстрые методы определения 90Sr по дочернему изотопу 90Y, который непосредственно выделяется из раствора золы образца экстракцией трибутилфосфатом [100], ди-2-этилгексилфосфорной кислотой [1126] или последовательным осаждением в виде оксалата и гидроокиси [549].
Определение стронция в золе костей. Навеску (3 г) золы костей растворяют в 20 мл HNO3 (1 : 1), добавляют 2 мл раствора носителя Ва и 3 мл носителя Sr (по 10 мг Ме2+/л1л). Раствор нагревают до полного улетучивания свободного иода, затем прибавляют 40 мл раствора молибдата аммония (15 г/л), 1—2 мл 10%-ного раствора NH4OH, смесь умеренно нагревают и охлаждают. Центрифугируют и выбрасывают осадок.
К раствору прибавляют 25 мл 20%-ного раствора NaOH, образующийся осадок отделяют центрифугированием и отбрасывают. К фильтрату прибавляют несколько капель метилового оранжевого и титруют разбавленным раствором NaOH до перехода окраски от красной к желтой. Смесь упаривают до объема 30 мл и устанавливают pH 8, центрифугируют, подщелачивают до pH 9 и нагревают. Прибавляют равный объем насыщенного раствора K4Fe(CN)6, образующийся осадок ферроцианида кальция центрифугируют.
Фильтрат подщелачивают 5 мл 20%-ного раствора NaOH п осаждают Ва, добавлением 3 мл насыщенного раствора Na2CrO4, оставляют на 30 мин., центрифугируют. Осадок растворяют в 10 мл буферного раствора (6 М СН3СООН + 6 М CHjCOONH, 1 : 2), нерастворенную часть отделяют центрифугированием и отбрасывают. К раствору прибавляют 10 мл 20%-ного раствора NaOH и 3 мл насыщенного раствора Na2CO3, смесь нагревают, охлаждают 1 час, выпавший осадок SrCOs отделяют; фильтрат отбрасывают. Осадок дважды промывают водой, оставляют на воздухе.
К фильтрату после отделения ВаСгО4 добавляют 3 мл насыщенного раствора Na2CO3, выделенный осадок промывают водой, отделяют центрифугированием.
Оба осадка карбоната стронция соединяют в один, промывают водой количественно переносят в алюминиевые чашечки и высушивают. После высушивания определяют выход Sr в виде SrCO3 весовым методом. 90Sr измеряют радиометрически [1331].’
Определение 90Sr по 90Y. Костную золу (10 г) растворяют в 17/лл НС1 и 85 мл Н2О при нагревании, охлаждают, доводят разб. раствором NH4OH pH до 1,0. Переносят полученный раствор в делительную воронку, добавляют 50 мл 0,45 М раствора ди-2-этилгексилфосфорной кислоты и экстрагируют
17S
"Y в течение 1 мин. Отделяют водную фазу и отбрасывают. Экстракт промы вают 3 раза 0,5 М НС1 и реэкстрагируют иттрий 3 раза 9 М НС1 (порциями по 15 мл). Все три солянокислых реэкстракта объединяют в делительной воронке и производят очистку от изотопов Zr и U путем их экстрагирования 50 мл 30%-ного раствора хлористого метилтрикаприламмония в толуоле (в течение 1 мин.). Водную фазу, содержащую "Y, выпаривают досуха, остаток озоляют при 600° С в муфельной печи (10 мин)., растворяют в 5 мл HNO3, и, упарив раствор до 1 мл, переносят в мишень, высушивают.[Определяют активность 9oY и пересчитывают ее на активность 93Sr [1126].
Определение стронция в зерне и овощах. Золу, полученную при сжигании 1 кг зерна или 5—10 кг картофеля, обрабатывают при кипячении в течение 30 мин. 6 М НС1 в присутствии носителя (250—300 мг Sr2+). Остаток отфильтровывают, промывают 100 мл горячей воды и снова обрабатывают 6 М НС1 (в случае большого содержания сульфат-ионов перед второй обработкой кислотой остаток кипятят в течение 30 мин. с насыщенным раствором соды). Солянокислые фильтраты и промывные воды объединяют, нагревают до 50° С и нейтрализуют NH4OH по метиловому оранжевому. Прибавляют кристаллическую Н2С2О4 до кислой реакции, затем избыток 5—10 г, нагревают до кипения и добавляют 4%-ный раствор (NH4)2C2O4 до полного осаждения ок» салатов. Осадок фильтруют, промывают водой и продолжают анализ ана-логично анализу образцов почв [301, 303, 1186].
Для определения содержания стабильного Sr и Са золу (200 г зерна или 1 кг картофеля) обрабатывают 50 мл 6 М НС1, как и в случае извлечения 90Sr. Раствор’ разбавляют до 0,3 М по НС1 и пропускают через катионит КУ-2 в Н+-форме. Промывают катионит последовательно 400 мл 0,3 М НС1 и ЗОО-илЗМ раствора Н2С2О4(для удаления К, Fe, Al, Р и других элементов), Затем через колонку пропускают 300 мл 3 М НС1 для вымывания Са и S» Из раствора осаждают оксалаты, осадок прокаливают, окиси растворяют в определенном объеме горячей 1 М НС1, содержание Sr определяют пламеннофотометрически.
Быстрый метод определения 89Sr, не требующий его предварительного выделения из биологического материала, предложен в [652].
Золу образца растворяют в НС1, разбавляют водой до 100 мл, полученный раствор наносят на диск из стекловолокна, сушат 3 часа под ИК-лампой. Сухой диск помещают в счетную кювету, затем измеряют активность 89Sr на жидкостно-сцинтилляционном спектрометре.
Предварительное выделение рутения тиоацетамидом из раствора пробы предложено в [564]. Затем раствор выпаривают с азотной и соляной кислотами, пропускают через анионит. Дау-экс-2Х8 в ОН~-форме, на котором сорбируются Fe, Go, Zn, Zr, Np, Nb. Из раствора, прошедшего через колонку, осаждают нитрат стронция.
Определение стронция в молоке. Определение стронция производится в золе остатка после выпаривания молока досуха [521, 1105,1140, 1232] или же в осадке оксалата кальция, полученном из
176
сыворотки молока после осаждения белка. Катионы из молока можно выделить с помощью ионного обмена [760, 12201. Находит применение метод определения 90Sr по "Y, при этом молоко перед анализом должно сохраняться по крайней мере 15 дней (при 0-1° С) для накопления 90Y [521, 1099, 1147].
Определение S0Sr по "Y (выделение' на анионите) [1147]. К 1 л молока прибавляют 1 мл раствора цитрата иттрия с pH 7 (около 5 мг Y) и пропускают последовательно через колонки с катионитом Дауэкс-50АУХ8 в Ха+-форме (50—100 меш, диаметр 5 см, объем смолы 140 мл) и с анионитом Дауэкс-1Х8 в СВ-форме (50—100 меш, диаметр 3 см, объем смолы 30 мл) со скоростью 10 мл!мин. Промывают 300 мл горячей воды и разъединяют колонки. Иттрий с колонки анионита вымывают 200 мл 2 М НС1 со скоростью 2 мл/мин. Его фракцию (35 мл) начинают собирать, после того как pH вытекающего раствора станет равным единице (после протекания 14—24 мл). К раствору прибавляют 5 мл 1 М раствора Н2С2О4 и конц. раствор NH4OH до] pH 2 (2,5 мл). Осадок переосаждают, фильтруют через взвешенный бумажный фильтр, промывают 10 мл воды, 3 раза по 10 мл этанола и эфира и, высушив до постоянной массы, рассчитывают химический выход и измеряют активность 9oY.
В методе [1099] для выделения иттрия используют его экстракцию теноилтрифторацетоном в бензоле.
Определение 90Sr по 90Y (экстракция иттрия трибутилфосфатом [521]). Золу молока (15 г) обрабатывают смесью 70 мл HNO3 (уд. масса 1,33) и 30 г NH4NO3 в присутствии 1,5 мл носителя Y (10 мг/мл), отфильтровывают. Фильтрат встряхивают с 200 мл трибутилфосфата (приведенного в равновесие с HNO3) в течение 5 мин.; через 5 мин. водный слой отбрасывают. Органический слой промывают HNO3 (2 раза по 50 .ил), затем экстрагируют Н2О (3 раза по 100 .ил). Водные фазы объединяют, выпаривают, остаток растворяют в 10 мл 6 М HG1, добавляют 10 мл Н2О и нейтрализуют аммиаком по, метанило-вому желтому. Нагревают до 80° С, добавляют 20%-ный раствор Н2С204. Осадок отфильтровывают, промывают водой, этанолом, сушат под ИК-лам-пой, проводят отсчет P-излучения £0Y.
Определение стронция в моче. Прямое выделение нитратов из растворов озоленных образцов обычно дает низкий выход стронция [258]. Поэтому щелочноземельные металлы предварительно концентрируют осаждением их в виде фосфатов, затем смесь фосфатов растворяют в кислоте и производят выделение и очистку Sr [1160, 1362].
В работе [1362] фосфаты растворяют в HNO3, выделяют Sr(NO3)2, удаляют примеси соосаждением с ВаСгОд, отделяют Y(OH)3, который переводят затем в оксалат и измеряют его активность. В [1160] после осаждения хроматов выделяют SrCO3, отделяют Y(OH)3, затем снова осаждают SrCO3 и измеряют активность "Sr. Для измерения содержания другого изотопа — 90Sr карбонат растворяют, вносят носитель Y, выдерживают^ 10 дней для накопления 90Y, осаждают его в виде гидроокиси, затем переводят в оксалат и измеряют его выход и активность.
177
Используется также предварительное выделение Sr в виде фосфата [258, 1288].
К пробе (300— 500 мл) добавляют 50 мл 16 М HNO3, 10 мл раствора носителя Sr и 1 мл раствора носителя Ва. Раствор выпаривают в (кварцевой чашке досуха, прокаливают в течение 15 мин. при 500° С. Остаток растворяют в минимальном количестве конц. HNO3, добавляют еще 2 мл HNO3 и 20 мл воды, фильтруют, промывают фильтр водой, фильтрат собирают и доводят до объема 250—300 мл. К фильтрату добавляют 5 мл сиропообразной Н3РО4 п избыток NH4OH. Раствор центрифугируют, маточный раствор отбрасывают, осадок промывают 100 мл промывного раствора (в 2 л Н2О 3 мл сиропообразной Н3РО4 и 20 мл 17 М NH4OH). Осадок растворяют в 20 мл дымящей HNO3, затем добавляют еще 40 мл той же кислоты, охлаждают проточной водой в течение 30 мин. Смесь центрифугируют, маточный раствор отбрасывают. Осадок растворяют в 40 мл воды, добавляют 90 мл дымящей HNO3 и охлаждают, помешивая в течение 30 мин. Центрифугируют, раствор отбрасывают, осадок дважды промывают 20—30 мл воды, промывные воды собирают, остаток на фильтре отбрасывают. Полученный азотнокислый раствор стронция поступает на дальнейший анализ [258].
Предложен [698] метод избирательного осаждения SrSO4 в присутствии комплексона III непосредственно из мочи.
К 300 мл мочи прибавляют 1 мл раствора SrCl2 (100 мг носителя Sr), 60 мл насыщенного раствора комплексона III и 20 г (NH4)2SO4, устанавливают pH 4,5 прибавлением 2 М II2SO4 и нагревают до 80° С. После охлаждения отделяют образовавшийся осадок SrSO4 центрифугированием, промывают 0,1 М раствором (NH4)2SO4, затем водой и растворяют в 15 мл 0,2 М раствора комплексона III, 1,0 М по NaOH.
К полученному раствору прибавляют 1 мл раствора ВаС12 (20 .иг носителя Ва), 1 мл 1 М раствора Na2CrO4n 2жл конц. раствора NH4OH, раствор подкисляют 6 М раствором СН3СООН и добавляют 1 мл избытка кислоты. Для коагуляции осадка ВаСгО4 раствор нагревают 15 мин. на водяной бане, затем осадок отделяют центрифугированием, промывают водой (5 мл) и присоединяют промывные воды к основному раствору. Раствор подщелачивают конц. NH4OH, добавляют 1 мл избытка, вводят 5 мл насыщенного раствора Na2CO3 и 3 мл 2 М раствора СиС12, нагревают 10 мин. на водяной бане для коагуляции SrCO3, после охлаждения центрифугируют, промывают осадок 0,1 М раствором NH4OH и растворяют в 5 мл 2 М НС1.
Раствор нагревают па водяной бане для удаления СО2, разбавляют водой до 20 мл, прибавляют 1 мл раствора Y(NO3)3 (5 мг носителя Y), осаждают Y (ОН)3 аммиаком, осадок отделяют центрифугированием, промывают 0,1 М раствором NH4OH и присоединяют промывные воды к основному раствору. Из полученного раствора осаждают SrCO3 прибавлением 5 мл насыщенного раствора Na2CO3, осадок промывают 2 раза водой, затем 95%-ным этанолом, переносят осадок в счетную тарелку, сушат под ИК-лампой, прокаливают 10 мин. при 500° С, измеряют радиоактивность осадка на низкофоновом р-счетчике (не позднее чем через 1 час после осаждения Y (ОН)3), затем взвешивают осадок для определения химического выхода стронция.
178
При очень низком содержании 90Sr используют пробу объемом 1500 мл и производят предварительное концентрирование Sr. Для этого из мочи соосаждают Sr и Са в виде оксалатов, осадок прокаливают до получения карбонатов, растворяют в НС1, осаждают SrSO4 в присутствии комплексона III и анализируют далее по описанной выше методике.
Жидкий катионообменник, содержащий ди-2-этилгексилфос-форную кислоту в микропорах порошка полиэтилена, предложен в [1311] для выделения из мочи смеси 90Sr и 90Y. Дальнейшее разделение этих изотопов проводят на хроматографической колонке путем вымывания "Y 6 М HNO3.
Анализ продуктов деления и облученных препаратов
Общий ход анализа заключается в переведении образца в раствор, добавлении носителей и выделении стронция последовательным применением описанных ранее операций осаждения или экстракции. В табл. 51 приведен обзор используемых методов выделения стронция.
При определении 91Sr и 52Sr в выбросных газах реактора [1182] производят предварительную сорбцию их на активированном угле. Для разделения Y, Ва и Sr применяют обычные химические методы; J2 отгоняют при сжигании угля в токе О2. Золу, оставшуюся после сжигания, сплавляют с NaaCO3, плав растворяют в HNO3 и разделяют Ва и Sr путем осаждения ВаСгО4 в присутствии ацетатного буферного раствора; Sr затем осаждают в виде оксалата.
Для разделения смеси продуктов деления используют также ионообмен-ники, например, катионит Дауэкс-50Х8 (Н+-форма). После вымывания Ru, Sb, Те, Cd, Sn, Zr и Nb промыванием 0,9 M раствором CH3COONH4 в 0,8 М СН3С00Н выделяют редкоземельные элементы и цезий. Затем колонку вымывают водой и 0,01 М раствором комплексона III в 0,1 М CII3COONH4 с постепенно возрастающим pH, выделяя сначала "Y, а затем 90Sr [1039].
В [657] анализируемый раствор с носителями пропускают через колонку, заполненную анионитом Амберлит IRA-400 (в ОН_-форме). Из смеси элюата и промывных вод осаждают стронций добавлением 1—2 г твердой СО2 (или пропускают ток СО2), осадок отделяют, растворяют в НС1, осаждают оксалаты и определяют активность осадка.
Использование анионита Дауэкс-2Х8 (в ОН-- и 32--форме) описано в [1272, 1273].
Определение 89Sr и 90Sr [860]. В центрифужную пробирку объемом 40 мл вносят по 2 мл носителей Sr (20 мг/мл) и Ва (10 мг!мл) и аликвотную часть исследуемого раствора. Раствор разбавляют водой до 20 мл, нагревают и осаждают ВаСО3 и SrCO3 добавлением 2 М раствора Na2CO3. Отделенный центрифугированием осадок растворяют в минимальном количестве 6 М HNO3 и, обрабатывая раствор дымящей HNO3 (по 30 мл), дважды переосаждают нит-
179
Таблица 51
Определение радиоактивного стронция в продуктах деления и облученных препаратах
Объект анализа Метод выделения стронция * Литература
Продукты деле- Многократное переосаждение нитратов [1086]
ния урана Последовательное осаждение сульфатов, карбонатов, нитратов, отделение хромата бария Последовательное выделение нитратов, карбонатов, осаждение Fe(OH)3 и ВаСгО4, выделение SrC2O4 Выделение оксалатов, осаждение аммиаком 90у Поглощение из выбросных газов реактора на активированном угле, осаждение ВаСгО4 и SrC2O4 [82, 1028]
[860, 1159, 1288] [746] [1182]
Облученные Ионообменное отделение То же (с последующим осаждением карбонатов, Fe(OH)3, ВаСгО4) [657, 1039] [1272]
Уран Выделение нитратов, Fe(OH)3, ВаСгО4 [232, 1288,
торий То же [1288]
висмут » [1288]
золото [1288]
хлористый рубидий Осаждение сульфидов Cu, Zn, Fe(OH)3, SrCO3 Осаждение оксалатов [1288] [1288]
графит Ионообменное отделение, осаждение карбонатов, отделение ВаСгО4 [1273]
* Определение ”Sr, #,Sr и 92Sr проводят прямым методом; ’“Sr — как прямым, так и по дочернему изотопу ’“У.
раты Ва и Sr (при охлаждении на ледяной бане в течение 5—10 мин.). После растворения осадка в 5—10мл воды раствор очищают осаждением Fe(OH)3 и последовательно добавляют 1—2 капли раствора индикатора бромфенолового синего, 6 М HNO3 (до изменения окраски индикатора) и 3 мл ацетатного буферного раствора. Затем, добавив к раствору 1 .ил 1,5 М Na2CrO4, осаждают при нагревании ВаСгО4 и отделяют (он может быть использован для определения радиоактивного Ва). Раствор обрабатывают 2М Na2COs (при нагревании), SrC03 растворяют в минимальном объеме 6M(HNO3, разбавляют водой до (Олли после добавления 1 мл носителя Ва повторяют осаждение ВаСгО4.
Из раствора осаждают SrC03 (при нагревании), осадок промывают водой и растворяют вбМ HNO3. После добавления 1—2 капель Н2О2 (для восстановления Сгв+ до Сг3+) и 1 мл Fe3+ (5 мг/мл} раствор очищают (предварительно удалив СОа) осаждением Fe(OH)3. Добавив 0,5 г твердого (NH4)2CO3 (при нагревании), выделяют SrCO3, который последовательно промывают 5 мл воды
180
и СН3ОН. Осадок высушивают, взвешивают и измеряют суммарную активность 89Sr и 90Sr.
Выдержав образец в течение 10 дней, растворяют SrCOs в 2М HN03, добавляют 2 мл носителя Y (20 мг/мл) и, удалив СО2, осаждают Y (ОН)3 свежеприготовленным 6М раствором NHjOH. Отделенный центрифугированием осадок растворяют в 2 мл 2 М HNO3, добавляют 1 мл носителя Sr, разбавляют до объема 10 мл и дважды переосаждают Y(OH)3. Промытый водой осадок растворяют в 1 мл 6 М HNO3, раствор разбавляют водой до объема 10 мл, нагревают до кипения и, добавляя 15 мл насыщенного раствора Н2С2О4, осаждают оксалат иттрия. Отделенный осадок промывают СН3ОН, высушивают, прокаливают при 800° С в течение 10 мин. до Y2O3 и измеряют активность 90 Y.
Облученный уран [1288]*. К аликвотной части азотнокислого раствора пробы добавляют по 10 мг Sr, Ва и доводят объем (путем выпаривания) до 1,5 мл, добавляют 12—13 мл дымящей HNO3, охлаждают 5 мин., растворяют осадок в 1 мл воды и переосаждают ~ 13 мл дымящей HNO3, центрифугируют. Осадок нитратов растворяют в 5 мл воды, добавляют 2 мг Fe2+ и осаждают Fe(OH)3 аммиаком (без СО2). Повторно осаждают Fe(OH)3 (добавив 1 мг Fe3+), раствор нейтрализуют конц. HNO3 по метиловому оранжевому, добавляют 4 мл 6 М СН3СООН, 2 мл 6 М CHsCOONH4, нагревают до кипения, добавляют 1 мл 1,5 М Na2CrO4, выдерживают в течение 1 мин., отделяют ВаСгО4.
К раствору добавляют 2 мл конц. NH4OH, нагревают и осаждают SrCOs насыщенным раствором Na2CO3, выдерживают 2 мин., центрифугируют. Осадок растворяют в 6 М НС1, удаляют СО2 кипячением. Подщелачивают раствор аммиаком, добавляют насыщенный Na2C2O4, нагревают, раствор охлаждают, отфильтровывают осадок. Осадок промывают по Зраза водой (5 .ил), этанолом (5 мл) и эфиром (5 мл), сушат и взвешивают в виде SrC2O4-H2O, определяя выход стронция. Затем измеряют его активность.
Облученный-хлористый рубидий [1288]. Облученную пробу через 5 дней обрабатывают 0,1 М НС1. Раствор фильтруют, доводят кислотность его до 0,3 М по НС1. Осаждают медь сероводородом и отделяют осадок. К фильтрату добавляют 50 мг Zn (ZnCl2), раствор подщелачивают аммиаком и осаждают цинк сероводородом, осадок отделяют. Фильтрат подкисляют и концентрируют до объема ~25 мл. Добавляют 50 мг железа (FeCl3), равные объемы карбонатного реагента (9 М (NH4)2CO3b 6N NH4OH) и этанола, полученный раствор доводят до кипения. Осадок гидроокиси железа (совместно с соосажден-нымикарбонатами) отфильтровывают и промывают 3 М NH4OH. Осадок растворяют в 1 М НС1, добавляют SOjwaRbCl, переосаждают гидроокись железа равным объемом карбонатного реагента, осадок отделяют центрифугированием, повторно переосаждают Fe(OH)s. Осадок растворяют в 6 М НС1 и из раствора несколькими порциями эфира проводят экстракцию железа. Свободный от Fe раствор выпаривают досуха и нагревают до удаления NH4C1. Остаток растворяют в разбавленной НС1.
К небольшим порциям раствора добавляют по 10 мг Zn, Y, Rb и Sr. Zn и Y осаждают сероводородом и аммиаком, Sr осаждают из фильтрата карбонатным реагентом в присутствии этанола; определяют его выход и активность. * Методика применима, когда активного Sr значительно больше, чем активного Ва.
ЛИТЕРАТУРА
1. Абашидзе Н. Ф., Махарадзе Л. В., Тодрадзе Л. Г. Бюл. науч.-техн, информ. ВИМС, № 4 (48), 81 (1963).
2. Абдуллаев А. А., Хакимов С., Хасанов А. С. Радиохимия, 14, 496 (1972),
3. Акишин П. И., Панченков Г. М., Васильев Н. Н., Никитин О. Т. Ж. физ. химии, 30, 1387 (1956).
4. Алекперов Р. А., Гейбатова С. С., Маков Н. Н. Ж. неорган. химии, 14, 542 (1969).
5. Алекперов Р. А., Маков Н. Н. Радиохимия, 10, 241 (1968).
6. Александров А., Васильева-Александрова П. Науч. тр. Высш, ин-т хранит, и вкус, пром-сти (Пловдив), 10, 255 (1963).
7. Александров Г. П., Демкив О. Т., Шевченко Ю. В. Укр. хим. ж., 29,. 623 (1963).
8. Александров Г. П., Шереметьев С. X., Захаренко Э. И. Сб. «Геология и геохимия нефтяных и газовых месторождений», К» 3. Киев, «Науко-ва думка», 1965, стр. 140.
9. Александрук В. М., Жиглинский А. Г., Хлопина Т. Н. Ж. аналит. хи- ' мии, 22, 841 (1967).
10. Алексанъян О. М. Гидрохимические материалы, 53, 163 (1972).
11. А лексеев В. Н. Курс качественного химического полумикроанализа, М., «Химия», 1973, стр. 187.
12. Алексеев М. А., Кычкин И. С. Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, стр. 397.
13. Алексеева В. М., Ильясова Н. В., Русанов А. К. Сб. «Спектральный анализ в геологии» (Материалы IV семинара по спектральному анализу для работников организаций Министерства геологии СССР). М., изд. ВИМС, 1971, стр. 42.
14. Алимарин И. П., Архангельская В. Н. Качественный полумикроанализ. М.—Л., Госхимиздат, 1952, стр. 93.
15. Анализ минерального сырья. Под ред. Ю. Н. Книпович и 10. В. Мора-чевского. Л., Госхимиздат, 1956.
16. АнтонЦевиК В. Г., КлискаМ. С., МаринковиИ М. Гласник хем. друшт-ва (Београд), 38, № 3—4, 321 (1973).
17. Араки С. Бунсэки кагаку, 14, 1163 (1965); РЖХим, 1966, 15Г58.
18. Арсланова С. С., Сорочан А. М., Сенявин М. М., Рахимов X. Р-Узб. хим. ж., № 4, 32 (1969).
19. Астахов К. В., Вуколов И. Е. Ж. неорган. химии, 7, 2082 (1962).
20. Атаманова С. П. Сб «Методы анализа минерального сырья». Апатиты,. Кольский филиал АН СССР, 1971, стр. 122.
21. Афанасьева Л. И. Ж. аналит. химии, 14, 294 (1959).
22. Афанасьева Л. И. Ж. аналит. химии, 15, 564 (1960).
23. Афанасьева Л. И. Ж. аналит. химии, 18, 712 (1963).
24. Афанасьева Л. И., Цикунов В. А. Тр. Гос. океаногр. ин-та, вып. 77,
83 (1964).
25. АхремА. А., Кузнецова А. И. Тонкослойная хроматография. М., «Наука», 1964.
26. Бабина И. М., Гошпаренко Л. Л. Доклады Межвузовской научной
182
конференции по спектроскопии и спектральному анализу. Изд-во Томского ун-та, 1960, стр. 12.
27. Бабкин М. П. Ж. аналит. химии, 11, 337 (1956).
28. Баранов В. И., В иленский В. ! Ж. аналит. химии, 17, 295 (1962).
29. БарановВ. И., В иленский В. Д. Радиохимия, 4, 486 (1962).
30. Баситова С. М., Засорина Е. Ф., Пометун Е. А., Боярова В. И. Изв. АН ТаджССР. Отд. физ.-техн, и хим. наук, № 2 (15), 57 (1964).
31. Бачурина Л. Г., Девяткин И. И., Крестьянинов А. Г., Перминова В. М., Цемко А. И., Чуприна Л. К. XVII Всесоюзный съезд по спектроскопии. Тезисы докладов. Атомный спектральный анализ. Минск, «Наука и техника», 1971, стр. 13.
32. Бачурина Л. Г., Перминова В. М., Савостин С. А. Зав. лаб., 40, 1348 (1974).
33. Белицина Г. Д., Зырии Н. Г. Сб. «Роль микроэлементов в сельском хозяйстве». Изд-во МГУ, 1961, стр. 285.
34. Бельтюкова С. В., Полуэктов Н. С. Ж. аналит. химии, 25, 1714(1970).
35. Беляев 10. И., Костин Б. И., Тамонтъев В. П. Агрохимия, № 5, 130 (1964).
36. Березина М. Д. Тр. Гос. ин-та по проектир. и исслед. работам нефте-доб. пром-сти. Гипровостокнефть, вып. 3, 85 (1961).
37. Берман Е.Л. Сб. «Спектроскопия. Методы и применение». М., «Наука», 1964, стр. 67.
38. Бланк А. Б., Афанасиади Л. И., Золотовицкая Э. С., Яковенко Е. И. XVII Всесоюзный съезд по спектроскопии. Тезисы докладов. Атомный спектральный анализ. Минск, «Наука и техника», 1971, стр. 19.
39. Бланк А. Б., Николенко А. Я., Манжелий Л. С. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты», ч. 2. Харьков, изд. ВНИИ монокристаллов, 1971, стр. 213.
40. Бланков Е. Б., Бланкова Т. И., Русяев В. Г., Якубсон К. И. Нейтронный активационный анализ в геологии и геофизике. М., «Наука», 1972, стр. 165.
41. Богатиков Б. Ф., Денисова Л. А., Марунина Н. И., Чернова А. II. Тр. Моск, ин-та тонкой хим. технол., 4, 12 (1974).
42. Богатырев И. О., Заборенко К. В. Радиохимия, 10, 250 (1968).
43. Божеволънов Е. А. Люминесцентный анализ неорганических веществ. М., «Химия», 1966, стр. 149, 260.
44. Б ойко В. К. Бюл. ВИМС, № 10 (222), 3 (1961).
45. Бокач-Полгар Э., Курц-Чики И. Ж. аналит. химии, 18, 1206 (1963).
46. Бокач-Полгар Э., Курц-Чики И. Ж. аналпт. химии, 21, 558 (1966).
47. Бондаренко С. К., Кислов Б. И. Ж. аналит. химии, 30, 1179 (1975).
48. Бондаренко С. К., Любимов В. Ф. Сб. «Материалы III Межотраслевого совещания по методам получения и анализа ферритовых, сегнето- п пьезоэлектрических материалов и сырья для них», ч. 1. Донецк, изд. ВНИИ реактивэлектрон, 1970, стр. 250.
49. Бондаренко С. К., Любимов В. Ф., Кислов Б. И., Уманец Т. И., Щедрина Л. П. Сб. «Методы получения и анализа ферритовых, сегнето-, пьезоэлектрических и конденсаторных материалов и сырья для них». Харьков, изд. ВНИИ монокристаллов, 1975, стр. 145.
50. Боич-Осмоловская К. С. Тр. Ин-та геол. Арктики, 149, 92 (1967).
51. Боич-Осмоловская К. С. Уч. зап. НИИ геол. Арктики. Региональная геология, вып. 2, 179 (1964).
52. Борзов В. Л., Есина Н. К., Шарыгина И.Н.Сб. «Материалы II Межотраслевого совещания по методам получения и анализа ферритовых материалов и сырья для них», ч. 2. М., изд. ИРЕА, 1969, стр. 53.
53. Борк В. А., Швыркова Л. А., Ким Л. Б. Ж. аналит. химии, 25, 1079 (1970).
54. Боровик С. А. Докл. АН СССР, 38, 270 (1943).
55. Боровик С. А., Боровик-Романова Т. ф Михайлова Г. В., Павленко Л. И. Зав. лаб., 19, 1200 (1953).
56. Боровик-Романова Т. Ф. Спектрально-аналитическое определение
183
щелочных и щелочноземельных элементов (в водах, растениях, почвах и породах). М.. Изд-во АН СССР, 1956, стр. 120, 127.
57. Боровик-Романова Т. Ф. Спектрально-аналитическое определение щелочных и щелочноземельных элементов (в водах, растениях, почвах и породах). М., «Наука», 1965.
58. Боровик-Романова Т. Ф., Беляев Ю- И., Куценко Ю. И., Павленко Е. И., Савинова Е. И., Фарафонов М. М- Спектральное определение редких и рассеянных элементов. М., Изд-во АН СССР, 1962.
59. Боровик-Романова Т. Ф., Королев В. В., Куценко Ю. И. Ж. аналит. химии, 9, 265 (1954).
60. Боровик-Романова Т. Ф., Королев В. В-, Куценко Ю. И., Фарафонов: М. М. Изв. АН СССР. Сер. физ., 19, 196 (1955).
61. Боувн Г., Гиббонс Д. Радиоактивационный анализ. М., Атомиздат, 1968.
62. Браун Т., Максим И., Галацеану И. Ж. аналит. химии, 14, 542 (1959).
63. Брей Д. М. Сб. «Редкие элементы в изверженных горных породах». М., ИЛ, 1952, стр. 109.
64. Брицке М. Э. Анализ металлургических продуктов методом эмиссионной фотометрии пламени. М., «Металлургия», 1969.
65. БудевскийО., Краснобаева И. Докл. Болг. АН, 13, 67 (1960).
66. Буколов И. Е., Астахов К. В., Зимин В. И., Таиров В. С. Ж. неорган. химии, 7, 1577 (1962).
67. Букреев Ю. Ф. Тр. Тамбовского ин-та хим. машиностроения, вып. 2, 84 (1968).
68. Бурков В- В., Подпорина Е. К. Стронций. Минералогия, геохимия и главные типы месторождений. (Труды Ин-та минерал., геохимии и кристаллохимии редких элементов. Вып. 12). М., Изд-во АН СССР, 1962.
69. Бурксер Е. СБурксер В. В- Тр. I сессии Комиссии по определению абсолютного возраста геологических формаций. АН СССР. М., Изд-во АН СССР, 1954, стр. 30.
70. Бурриелъ-Марти Ф., Рамирес-Муньос X. Фотометрия пламени. М., ИЛ, 1962.
71. Бусев А. И., Типцова В. Г., Иванов В. М. Практическое руководство по аналитической химии редких элементов. М., «Наука», 1966, стр. 383.
72. БыковВ. П., Сорокин И. В. Зав. лаб., 27, 1371 (1961).
73. Быковский И. И., Калистратова А. В., Курчатов В-В., ЧижовВ.А.^ Юзвук Ю. А. Сборник работ по некоторым вопросам дозиметрии и радиометрии ионизирующих излучений, вып. 2. М., Госатомиздат, 1961, стр. 192.|
74. Вайвадс Я. К. Сб. «Активационный анализ». Рига, «Зинатне», 1976, стр. 122.
75. Вайнштейн Э. Е., Лебедев В. И. Геохимия, 1961, 362.
76. Василевская А. Е., Кальян Г. А. Сб. «Спектральный анализ в геологии» (Материалы IV семинара по спектральному анализу для работников организаций Министерства геологии СССР). М., изд. ВИМС, 1971, стр. 70.
77. Васильев В. В., Григорьев Н. И. Практическое руководство по качественному химическому полумикроанализу. Изд-во ЛГУ, 1966, стр. 264.
78 Васюкевич В. А., Гончаров С. Ф. Тр. ВНИИ ж.-д. транспорта, вып. 251, 4 (1963).
79. Васютинский А. И., Могилевская В. И. Ж. аналит. химии, 22, 1895 (1967).
80. Вахобова Р. У., Валитова И. X., Кавардакова О. А., Вахобов А. В-Сб. «Успехи полярографии с накоплением». Изд-во Томского гос. ун-та, 1973, стр. 179.
81. Вдовенко В. М. Труды П-й Международной конференции по мирному использованию атомной энергии, Женева, 1958. Доклады советских ученых, т. IV. М., Атомиздат, 1959, стр. 41.
82. Венедиктова Р. В, Ильинская Т. А., Чернышева Л. И- Сб. «Радиохими-
184
ческий анализ продуктов деления». М.—Л., Изд-во АН СССР, 1960, стр. 63.
S3. Виноградов А. П. Геохимия редких и рассеянных химических элементов в почвах. М., Изд-во АН СССР, 1950.
84. Виноградов А. П. Геохимия, 1971, 261.
85. Виноградова 3. А. Гидробиологический журнал, 1, 12 (1965).
86. Воздвиженская С. А., Горбенко Ф. П. Органические реагенты в аналитической химии. Тезисы докладов IV Всесоюзной конференции, ч. 1. Киев, «Наукова^ думка», 1976, стр. 32.
87. Войтко И. И., Митрофанова О. Г. Зав. лаб. 42, 662 (1976).
88. Воробьев Г. Г. Сб. «Метеоритика», вып. 20. М., Изд-во АН СССР, 1961, стр. 185.
89. Воробьев Т. И., Рыкова Г. А., Штернина Э. Б. Ж. неорган. химии, 15, 2644 (1970).
90. Галицина Э. И., Курганова В. И. Сб. «Новые методы анализа химического состава подземных вод». М., изд. Мин-ва геологии СССР, 1967, стр. 110.
91. Галкина Л. Л. Радиохимия, 9, 116 (1967).
92. Герасимовский В. И., Беляев Ю. И. Геохимия, 1963, 1111.
93. Герасимовский В. И., Лебедев В. И. Геохимия, 1958, 553.
94. Герлинг Э. К., Ященко М. Л., Левский Л. К., Овчинникова Г. В. Геохимия, 1958, 535.
95. Гиллебранд В. Ф., Ленделъ Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., «Химия», 1966.
96. Гильберт Э. Н., Артюхин П. И. Изв. СО АН СССР. Сер. хим., вып. 2, 90 (1967).
97. Главин Г. Г. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты», ч. 1. Харьков, изд. ВНИИ монокристаллов, 1971, стр. 52.
98. Главин Г. Г., Карпов Ю.А., Олжатаев Б. А. Зав. лаб., 35, 172 (1969).
99. Глонти Г. Г. Ж. аналит. химии, 19, 637 (1964).
100. Голутвина М. М., Казакова Т. А., Николаев Ю. М., Меркелова Н.В. Медицинская радиология, 7, 62 (1962).
101. Горбенко Ф. П. Автореферат докторской диссертации. Киевский гос. ун-т, 1970.
102. Горбенко Ф. П. Доклады VII Конференции заводских и производственных лабораторий. Алма-Ата, «Наука», 1964, стр. 50.
103. Горбенко Ф. П. Сб. «Материалы II Межотраслевого совещания по методам получения и анализа ферритовых материалов и сырья для них», ч. 2. М., изд. ИРЕА, 1969, стр. 99.
104. Горбенко Ф. П. Сб. «Методы анализа химических реактивов и препаратов», вып. 14. М., изд. ИРЕА, 1967, стр. 34.
105. Горбенко Ф. И. Сб. «Химическая промышленность», вып. 1. Киев, изд. Укр.НИИНТИ, 1966, стр. 22.
106. Горбенко Ф. П-, Даиомко В. М., Надежда А. А., Дунаевская К. А. Авт. свид. СССР 171658 (1964); Вюл. изобр., № 11 (1965).
107. Горбенко Ф. П., Киселева Н. Е., Лапицкая Е. В., Целинский Ю. К. Сб. «Методы анализа химических реактивов и препаратов», вып. 14. М., изд, ИРЕА, 1967, стр. 76.
108. Горбенко Ф. П., Кучкина Е. Д. Авт. свид. СССР 194073 (1965); Бюл. изобр., №8 (1967).
109. Горбенко Ф. П-, Кучкина Е. Д. Сб. «Методы анализа химических реактивов и препаратов», вып. 18. М., изд. ИРЕА, 1971, стр. 160.
110. Горбенко Ф- П., Кучкина Е. Д., Олевинский М. И. Ж. аналит. химии, 23, 1301 (1968).
111. Горбенко Ф. П., Кучкина Е. Д., Олевинский М. И. Радиохимия, 12, 661 (1970).
112. Горбенко Ф- П., Лапицкая Е. В. Ж. аналит. химии, 23, 1139 (1968); Зав. лаб., 34, 105 (1968).
113. Горбенко Ф- П., Лапицкая Е. В. Сб. «Аналитическая химия и экстрак
185
ционные процессы». Киев, «Наукова думка», 1970, стр. 40, 44; Сб. «Методы анализа химических реактивов и препаратов», вып. 17. М., изд. ИРЕА, 1971, стр. 176, 227.
114. Горбенко Ф. Л., Лапицкая Е. В., Дзиомко В. М., Дунаевская К. А.г Воздвиженская С. А., Надежда А. А., Калинина К. Е. Сб. «Реактивы и особо чистые вещества», вып. 37. М., изд. ИРЕА, 1975, стр. 5.
115. Горбенко Ф. П., Лапицкая Е. В-, Целинский Ю. К. Сб. «Методы анализа химических реактивов и препаратов», вып. 14. М., изд. ИРЕА, 1967, стр. 80.
116. Горбенко Ф. П., Надежда А. А. Ж. аналит. химии, 24, 671 (1969); Укр. хим. ж., 34, 625 (1968).
117. Горбенко Ф. Л., Надежда А. А., Воздвиженская С. А., Дехович Э. И. Ж. аналит. химии, 30, 1484 (1975).(
117а. Горбенко Ф. П., Надежда А. А., Воздвиженская С. А. Ж. аналит. химии, 31, 1898 (1976).
118. Горбенко Ф. Л., Надежда А. А., Воздвиженская С. А., Лошкарева Н. И. Сб. «Методы получения и анализа ферритов, сегнето-, пьезоэлектрических и конденсаторных материалов и сырья для них». Харьков, изд. ВНИИ монокристаллов, 1975, стр. 164.
119. Горбенко Ф. Л., Сачко В. В-, Лапицкая Е. В., Надежда А. А. Ж. прикл. химии, 42, 2212 (1969).
120. Горбенко Ф. Л., Цилинский IO. К., Лапицкая Е. В., Кучкина Е. Д.г Киселева Н. Е. Тр. ВНИИ хим. реактивов и особо чистых хим. веществ-(ИРЕА), вып. 29, 92 (1966).
121. Горелов И. И., Бабич В. А. Ж. неорган. химии, 16, 902 (1971).
122. Горелов И. П-, Колесова М. X. Ж. неорган. химии, 14, 2687 (1969).
123. Горохова Л. ВДятловаН.М., Григорьев А. И. Органические реагенты в аналитической химии. Тезисы докладов III Всесоюзной конференции. М., изд. ГЕОХИ АН СССР, 1971, стр. 272.
124. Гусев С. И., Кумов В. И., Битовт 3. А. Ж. аналит. химии, 15, 746.' (1960).
125. Даймонд Р. М., ТакД. Г. Экстракция неорганических соединений. М., Госхимиздат, 1962.
126. Дегтярев Ю. Н. Радиохимия, 7, 733 (1965).
127. Дегтярева О. Ф., Островская М. Ф. Ж. аналит. химии, 18, 245 (1963).
128. Дегтярева О. Ф., Островская М. Ф. Зав. лаб., 30, 174 (1964).
129. Дедков IO. М., Макарова В. Л., Винокурова В. А., Чащихина М. В.г Саввин С. Б. Ж. аналит. химии, 20, 440 (1965).
130. Дзиомко В. М. Ж. Всес. хим. о-ва им. Д. И. Менделеева, 6, 108 (1961).
131. ДибобесИ .К ., Лебедев В. И., Е. 7/. Гигиена и санитария, № 11,
42 (1962).
132. ДракинС.И., Карапетъянц М. X., Ербанова Л. Н., Курмалиева Р. X., Лантухова Л. В., Соколов В. Б. Труды I Конференции по аналитической химии неводных растворов и их физпко-хпмическим свойствам, ч. 2. М., изд. Моск, хим.-технол. ин-та им. Д. И. Менделеева, 1968, стр. 52.
133. Дракин С. И., Карапетъянц М. X., Курмалиева Р. X. Ж. неорган. химии, 14, 2697 (1969).
134. Дятлова Н. М., Темкина В. Я., Колпакова И. Д. Комплексоны. М., «Химия», 1970.
135. Егоров Ю. В., Пушкарев В. В-, Ткаченко Е. В. Ж. аналит. химии, 6, 505, 508 (1961).
136. Егоров Ю. В., Пушкарев В. В., Ткаченко Е. В. Радиохимия, 3, 87 (1961).
137. Есиков А. Д., Бесчастнова Г. С., Яковлев Г. II. Бюл. Комис, по определению абсолютного возраста геол, формаций. АН СССР, вып. 5, 76, 82 (1962).
138. Есиков А. Д., Бесчастнова Г. С., Яковлев Г. Н. Изв. АН СССР, Сер. геол., № 12, 69 (1959).
139. Есиков А. Д., Каленчук Г. Е. Сб. «Химический анализ минералов и их химический состав». М., «Наука», 1964, стр. 3.
186
140. Жеенбаев Ж. Изв. выучеб, заведений. Физика, № 3, 103 (1960).
141. Живописцев В. П., Пятосин Л. П. Уч. зап. Перм. ун-та, № 159, 254 (1966),
142, Жиглинский А. Г., Зайдель А. И., Кунд Г. Г. Геохимия, 1962, 976.
143. Жиглинский А. Г., Кунд Г. Г. Оптика и спектроскопия, 7, 836 (1959).
444. Жиглинский А. Г., Кунд Г. Г., Хлопина Т. И. Ж. прикл. спектроскоп., 10, 205 (1969).
445. Жинка Л., Сиртеш Л. Радиохимия, 12, 774 (1970).
146. Житков Р. Д., Кедринский И. А. Тр. Ленингр. технол. ин-та им. Ленсовета, 48, 197 (1958).
147. Жуков А. И., Баранов Г. П., Плясунов П. В. Ж. неорган. химии, 7, 1452 (1962)
148. Заборенко К. Б., Богатырев И. О., Малыгина Н. Л. Радиохимия, 8, 352 (1966).
149. Зайдель А. Н. Основы спектрального анализа. М., «Недра», 1965, стр. 271.
150. Зайдель А. И., Ж иглинский А. Г., Кунд Г. Г. Бюл. Комис, по определению абсолютного возраста геол, формаций. АН СССР, вып. 5, 60 [1962).
151. ЗайделъА.Н., Жиглинский А. Г., ЧайкоЮ. Чехословацкий физический журнал, 8, 530 (1958).
152. Зайдель А.Н., Калитиевский Н. И., Липис Л. В., Чайка М. П. Эмиссионный спектральный анализ атомных материалов. М.— Л., Физматгиз, 1960, стр. 578.
453. Зайцев Е. И., Курочкин С. С., Днепровский И. С., Марголин Е. М. Зав. лаб., 40, 1339 (1974).
154. Зайцев Е. И., Марголин Е. М., Полыця О. П., Панькин В. В., Щекин К. И. Ж. аналит. химии, 29, 668 (1974).
155. Зайцев Е. И., Шинкина М. А. Авт. свид. СССР 177147 (1964); Бюл. изобр., № 24 (1965).
156. Зацепина Л. Н., Сапарлыев Ч., Павлоцкая Ф. И. Гигиена и санитария, № 10, 68 (1966).
157. Зверева М. И., Леонова Л. А. Сб. «Вопросы аналитической химии минеральных веществ». Изд-во ЛГУ, 1966, стр. 141.
158. ЗиминаН.М., Вережникова Н. С., Берестнева Т. И. Тр. лаборатории ионообменных процессов и сорбции. Н.-и. физ.-хим. ин-т Воронежского гос. ун-та, вып. 1, 55 (1966).
159. Зиновьева В. К. Сб. «Определение загрязнений биосферы продуктами ядерных испытаний». М., Изд-во АН СССР, 1959, стр. 155.
460. Зиновьева В. К., Жилкина М. И., Шведов В. П., Яковлева Г. В. Радиохимия, 1, 613 (1959).
161. Золотавин В. Л., Букреев Ю. Ф., Мувгин В. И., Пророк М. М. Тр. Ин-та химии Уральск, науч, центр. АН СССР, вып. 17, 159 (1970).
162. Золотавин В. Л., Пономарева J1. К. Радиохимия, 2, 104 (1960).
163. Золотов Ю. А. Экстракция внутрикомплексных соединений. М-, «Наука», 1968.
164. Золотов Ю. А., ЛамбревВ. Г. Радиохимия, 8, 627 (1966).
165. Золотов Ю. А., Ламбрев В. Г., Чмутова М. К., Сизоненко И. Т. Докл. АН СССР, 165, 117 (1965).
166. Золотовицкая Э. С., Яковенко Е.И. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты», ч. 2. Харьков, изд. ВНИИ монокристаллов, 1971, стр. 23.
167. ЗыринН.Г., Обухов А. И., БелицинаГ. Д. Сб. «Микроэлементы и естественная радиоактивность почв». Изд-во Ростов, гос. ун-та, 1962, стр. 242.
168. ИвановВ.И., ЛеншинаН.Я. Тр. Комиссии по аналит. химии. АН СССР, 11, 418 (1960).
169. ИвановВ.И., ЛеншинаН. Я., Иванова В. С. Изв. АН СССР. Сер. хим., № 4, 518 (1957).
170. Иванов И. Л., Козырева Н. А., Зав. лаб., 31, 566 (1965).
187
171. Иванов И. И., Козырева И. А. Тр. ВНИИ хим. реактивов и особо чистых хим. веществ (ИРЕА), вып. 28, 137 (1966).
172. Иванова Л. М. Радиохимия, 9, 622 (1967).
173. ИвановаЛ. М., Жилкина М. И. Сб. «Определение загрязнений биосферы продуктами ядерных испытаний». М., Изд-во АН СССР, 1959, стр. 168.
174. Иванова Л. М., Шведов В. И. Радиохимия, 5, 185 (1963).
175. Идзава М., Цубота X., Касаи А. Бунсэки кагаку, 10, 1230 (1961); РЖХим, 1962, 10Д29.
176. ИдзноЭ., Суда К. Бунсэки кагаку, 13, 62 (1964); РЖХим, 1964, 23Г62.
177. ИжакИ. Г., Изотова И. В. Вести, техн, и эконом, информ. НИИ техн.-эконом. исслед. Гос. ком-та хим. и нефт. пром-сти при Госплане СССР, вып. 10, 47 (1963).
178. Иида, Ямасаки. Нихон кагаку дзасси, 75, 189 (1954); РЖХим, 1957, 1203.
179. Икрами Д. Д-, Парамзин А. С. Изв. АН ТаджССР. Сер. физ.-мат. и геол.-хим. наук, № 2, 66 (1970).
180. Имаи X. Нихон кагаку дзасси, 82, 1176 (1961); РЖХим, 1962, 16Д40.
181. Инарида М-, С имаму pa A. Radioisotopes 20, 12 (1971); РЖХим, 1971, 13Б578.
182. Кабачник М. И., Дятлова И. М., Медведь Т. Я., Медынцев В. В., Поликарпов Ю. М., Царева 3. И- Изв. АН СССР. Сер. хим., № 9, 2058! (1968).
183. Кабачник М. И., Ластовский Р. Л., Медведь Т. Я., Медынцев В. В.г Колпакова И. Д., Дятлова И. М Докл. АН СССР, 177, 582 (1967).
184. Кавамура С- Бунсэки кагаку, 10, 975 (1961); РЖХим, 1962, 8Б282.
185. Калинин А. И., Кузнецов Р. А., Моисеев В. В., Соколова М. И. Сб. «Радиохимические методы определения микроэлементов». М.— Л., «Наука», 1965, стр. 180.
186. Канаев И. А., Дорофеева И. И., Трофимов Н.В.Сб. «Физико-химические методы анализа металлов и сплавов». М., ОНТИ, 1969, стр. 80.
187. Карабаш А- Г., Бондаренко Л. С., Морозова Г. Г., Пейзулаев Ш. И. Ж. аналит. химии, 15, 623 (1960).
188. Карабаш А. Г., Пейзулаев Ш. И- Сб. «Получение изотопов. Мощные гаммаустановки. Радиометрия и дозиметрия». М., Изд-во АН СССР, 1958, стр. 36.
189. Карабаш А. Г., Пейзулаев Ш. И., Слюсарева Р. Л., Смирнова-Аверина И- И., Самсонова 3. И. и др. Физический сборник (Материалы X Всесоюзного совещания по спектроскопии, ч. 2. Атомная спектроскопия), вып. 4 (9). Изд-во Львов. гос. ун-та, 1958, стр. 556.
190. Карнаухов А. С., Мизера М., ПалаушР. Ж. аналит. химии, 15, 502 (1960).
191. Карпенко Л. И., Фадеева Л. А., Шевченко Л. Д. Ж. аналит химии, 30, 1330 (1975).
192. Катыхин Т. С. Ж. аналит химии, 27, 849 (1972).
193. Кацура Т. Бунсэки кагаку, 10, 1207 (1967); РЖХим, 1962, 11Д46.
194. Кипиани Р., Гвоздек К., Крупенникова А. Тр. Ин-та защиты растений ГрузССР, 14, 275 (1961).
195. Ким Гун Он, Хан Дон Эк, Кан Дзюн Сик. Пунсок хвахак, 8, 17 (1970); РЖХим, 1971, 12Г101.
196. Кириллов А. И., Алхименкова Г. И. Зав. лаб., 31, 57 (1965).
197. Кист А. А., Лобанов Е. М., Свиридова А. И., Хатамов Ш. Сб. «Нейтронно-активационный анализ». Ташкент, «Фан», 1971, стр. 95.
198. Коблянский А. Г. Ж. общей химии, 24, 18 (1954).
199. Кодири С., Старчик Л. П., Зав. лаб., 36, 191 (1970).
200. Кодочигов П. И., Глазунов М. П-, Меднис И. В., Спицын В. И. Сб. «Нейтронно-активационный анализ». Рига, «Зинатне», 1966, стр. 115.
201. Козин А. Н-, Коростелева И. А-, Юрова В. П. Тр. Куйбышев. НИИ нефт. пром-сти, вып. 39, 227 (1968).
202. Колесников И. И. Сб. «Методы анализа руд Кольского полуострова». Апатиты, Кольский филиал АН СССР, 1970, стр. 57.
188
203. Колосов И. В. Изв. Тимирязевск. с.-х. акад., № 1, 212 (1968).
204. Кольцов Г. В. Тр. Ин-та биол. внутр, вод. АН СССР, вып. 18 (21), 205 (1968).
205. Канторович А. Э., Минина Л. ДАнтаков Р. М. Тр. Сиб. НИИ геол., геофиз. и минеральн. сырья, вып. 14, 142 (1961).|
206. Кон Хо Ын, Пак Чен Хва, Ли Сун Га. Пунсок хвахак, 4, 78 (1965); РЖХим, 1967, 5Г66.
207. Копейкин Ю. А., Загарских Т. И. Сб. «Микроэлементы в почвах, водах и организмах Восточной Сибири и Дальнего Востока и их роль в жизни растений, животных и человека». Улан-Удэ. Изд. Бурятского компл. НИИ СО АН СССР, 1961, стр. 258.
208. Коренман И. М. Микрокристаллоскопия. М., Госхимиздат, 1955, стр. 144.
209. Корлисс Ч., Бозман К. Вероятности переходов и силы осцилляторов 70 элементов. М., «Мир», 1968.
210. Корпусов Г. В., Озиранер С. Радиоактивный стронций. М., Госатомиз-дат, 1961.
211. Корчемная Е. К., Наумова В. И-, Ермаков А. Н. Ж. аналит. химии, 24, 342 (1969).
212. Крешков А. П., Кузнецов В. В. Зав. лаб., 34, 134 (1968).
213. Крешков А. Л., Кузнецов В. В. Ж. аналит. химии, 25, 49 (1970).
214. Крешков А. П., Яровенко А. Н-, Саюшкина Е. Н., Зеленина Л. И. Изв. высш, учебн. заведений. Химия и хим. технол., 8, 196 (1965).
215. Криницкая Л. В., Киреева А. Ю., Дятлова Н. М., Колпакова И. Д., Бихман Б. И. Органические реагенты в аналитической химии. Тезисы докладов III Всесоюзной конференции. М., изд. ГЕОХИ АН СССР, 1971, стр. 269.
216. Кручкова Е. С., Пухова В. М., Трубникова Л. И. Изв. высш, учебн. заведений. Химия и хим. технол., 17, 614 (1974).
217. Крылов В. Н., Гедеонов Л. И., Раков Н. А., Трофимов А. М. Радиохимия, 15, 654 (1973).
218. Крюкова Т- А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М., Госхимиздат, 1959, стр. 226.
219. Ксандопуло Г. И., Щербов Д. П. Зав. лаб., 24, 1432 (1958).
220. Ксандопуло Г. И., Щербов Д. П. Сб. «Химические, физико-химические и спектральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961, стр. 63.
221. Кузнецов В. В. Тезисы докладов научно-технической конференции Моск. хим.-технол. ин-та им. Д. И. Менделеева. М., изд. МХТИ 1969, стр. 36.
222. Кузнецов В. И. Зав. лаб., 14, 545 (1948).
223. Кузнецов В. И. Сб. «Органические реагенты в неорганическом анализе». М., «Наука», 1969, стр. 17.
224. Кумов В. И., Битовт 3. А., Песис А. С. Ж. неорган. химии, 3, 1181 (1958).
225. Кунин Л. Л., Маликова Е- Д., Чапыжников Б. А. Определение кислорода, углерода, азота и водорода в щелочных и щелочно-земельных металлах. М., Атомиздат, 1972.
226. Курокава К. Нихон кагаку дзасси, 89, 1076 (1968); РЖХим, 1969, 19Г65.
227. Курокава К. Нихон кагаку дзасси, 88, 188 (1967); РЖХим, 1968,11Г41.
228. Куртасова Л. 3. Сб. «Спектральный анализ в геологии» (Материалы IV семинара по спектральному анализу для работников организаций Министерства геологии СССР). М., изд. ВИМС, 1971, стр. 133.
229. Кучкина Е. Д. Автореферат кандидатской диссертации. Донецкий гос. ун-т, 1970.
230. Кучкина Е- Д-, Литвинова Г. В., Сораченко Л. Т., Горбенко Ф. П. Сб. «Методы получения и анализа ферритов, сегнето-, пьезоэлектрических и конденсаторных материалов и сырья для них». Харьков, изд. ВНИИ монокристаллов, 1975, стр. 158.
189
231. Лаврухина А. К., Малышев В. В., Родин С. С. Ж. Всес. хим. о-ва им. Д. И. Менделеева, 8, 227 (1963).
232. Лаврухина А. К., Малышева В. Г., Лавлоцкая Ф. Л. Радиохимический анализ. М., Изд-во АН СССР, 1963, стр. 168.
233. Лакомкин И. Г. Тр. Ленингр. технол. ин-та им. Ленсовета, № 35, 34 (1956).
234. Лапицкая Е. В. Автореферат кандидатской диссертации. Днепропетровский гос. ун-т, 1971.
235. Лапицкая Е. В., Горбенко Ф. Л. Радиохимия, 10, 90 (1968).
236. Лапицкая Е. В., Кучкина Е. Д., Горбенко Ф. П. Ж. неорган. химии, 13, 2774 (1968).
237. Лебедев В. И. Ж. аналит. химии, 14, 283 (1959).
238. Левин В. И., Курчатова Л. И., Малинин А. В. Авт. свид. СССР 225343 (1966); Бюл. изобр., № 27 (1968).
239. Левшин Б. А. Тр. Упр. геол. Сов. Мин. ТуркмССР, вып. 7, 397 (1972).
240. Липчински А., Кулев И. Годишник Висш. хим.-технол. ин-та (Бургас), 2, 43 (1965); РЖХим, 1967, 11Г78.
241. Лисовский И. Л., Смахтин Л. А., Винник М- М., Захарова С. Н. Хим. пром-сть, № 3, 235 (1971).
242. Литовченко Г. Д. Шипицын С. А. Изв. АН СССР. Сер. физ., 23, 1156 (1959).
243. Личев В., Динков Д., Ташкова А. Научн. тр. НИИ винарска и пивоварка пром-ст — София, 10, 7 (1968); РЖХим, 1970, 16Р341.
244. Лобанов Е. М., Мингалиев Г. Г., Хуснутдинов Р. И. Сб. «Активационный анализ элементного состава геологических объектов». Ташкент, «Фан», 1967, стр. 64.
245. Логинова Л. Г. Бюл. науч.-техн, информ. Мин-во геол, и охраны недр СССР, № 5—6 (39—40), 127 (1962)
246. Логинова Л. Г., Галицына Э. И. Сб. «Новые методы анализа химического состава подземных вод». М., изд. ВНИИ гидрогеол. и инженерн. геологии, 1967, стр. 100.
247. Лукин А. М., Зеличенок С. Л., Чернышева Т. В. Ж. аналит. химии, 19, 1513 (1964).
248. Лукин А. М., Смирнова К. А., Чернышева Т. В. Ассортимент реактивов на стронций и барий (обзорная информация). М., изд. НИИТЭХим, 1972, стр. 17.
249. Лукин А. М., Чернышева Т. В. Тр. ВНИИ хим. реактивов и особо чистых хим. веществ (ИРЕА), вып. 33, 170 (1971).
250. Львов Б. В. Атомно-абсорбционный спектральный анализ. М., «Наука», 1966.
251. Маки М. Бунсэки кагаку, 4, 156, 217, 302 (1955); РЖХим, 1957, 1197.
252. Малинина В. И., Кудямов Б. Я. Новости нефт. техн. Геология, № 12, 37 (1958).
253. Малюшко Л. Д. Тр. Всес. н.-и. геол, пн-та, 146, 140 1966).
254. Манжелий Л. С., Бланк А. Б., Булгакова А. М., ЛикитинаЛ. М., Мартынчеико Л. Л. Сб. «Методы анализа галогенидов шёлочных и щелочноземельных металлов высокой чистоты», ч. 2. Харьков, изд. ВНИИ монокристаллов, 1971, стр. 120.
255. Маслов И. А., Лукницкий В. А. Справочник по нейтронному активационному анализу. Л., «Наука», 1971.
256. Мацуда С., Кавамото X., Ямагата Л. Бунсэки кагаку, 14, 1061 (1965); РЖХим, 1966, 13Г180.
257. Методы определения и анализа редких элементов. М., Изд-во АН СССР, 1961, стр. 67, 125.
258. Методы радиохимического анализа. Женева, Всемирная организация здравоохранения «Медицина», 1967, стр. 55, 63, 69.
259. Методы радиохимического анализа. Доклад объединенного комитета экспертов Всемирной организации здравоохранения, Продовольственной и Сельскохозяйственной организации при ООН. М., Атомиздат, 1962, стр. 44.
190
260. Миллер А. Д. Труды Межведомственного совещения по гидрогеохи-мическому методу поисков рудных месторождений, 1960. Изд-во Томского гос. ун-та, 1962, стр. 212.
261. Миллер В. Б., Нейман М. Б., Сазонов Л. А. Ж. аналит. химии, 7, 269 (1952).
262. Миллер 10. М., Попонова Р. В., Устинов В. И. Сб. «Химические реактивы и препараты», вып. 27. М., изд. ИРЕА, 1965, стр. 103.
263. Милованов Г. И., Белякова Л. Т. Требования промышленности к качеству сырья, вып. 70. М., Госгеолтехиздат, 1960.
264. Минц И. М., Былинкина А. А. Сб. «Материалы Конференции по итогам научных исследований. Ин-т общ. и коммун, гигиены АМН СССР». М., изд. АМН СССР, 1965, стр. 41.
265. Мокиенко В. Ф., Лапашева Т. И., Митрофанов В. 3. Тр. Волгогр. НИИ нефт. и газ. пром-сти, вып. 8, 101 (1965).
266. Морачевский Ю.В., Новиков А. И. Тр. Комиссии по аналит. химии. АН СССР, 9 (12), 121 (1958).
267. Морачевский Ю. В., Новиков А. И. Уч. зап. ЛГУ, № 272, 112 (1959).
268. Мори И. Якугаку дзасси, 85, 486, 679, 686 (1965); РЖХим, 1966, ЗГ15, 4Г26, 4Г27.
269. Мори И. Бунсэки кагаку, 16, 1320 (1967); РЖХим, 1968, 20Г49.
270. Москвин Л. Н. Радиохимия, 6, ПО (1964).
271. Москвина М. М., Харцизов А.Д. Radioisotopy, 6, 819 (1965).
272. Мосулишвили Л. М., Кучава П. Е., Гинтури Э. Н. Сообщения АН ГрузССР, 59, 93 (1970).
273. Мудрецов А. И. Ж. неорганич. химип, 16, 1199 (1971).
274. Муся С., Мунэмори М., Хосогаки Р. Бунсэки кагаку, 19, 1618 (1970); РЖХим, 1971, 13Г109.
275. Навтанович М. Л., Сутырин 10. Е., Черняк А. С. Сб. «Исследования в области химип и технологии минеральных солей и окислов». М.— Л., «Наука», 1965, стр. 300.
276. Надежда А. А. Автореферат кандидатской диссертации. Днепропетровский гос. ун-т, 1969.
277. Надежда А. А., Иванова И. П-, Горбенко Ф. П., Лошкарева Н. И. Сб. «Материалы IV Межотраслевой конференции по методам получения и анализа ферритовых, сегнето-, пьезоэлектрических и конденсаторных материалов и сырья для них». М., изд. ИРЕА, 1973, стр. 211.
277а Надежда А. А., Иванова К. П., Горбенко Ф. П., Маслиева ф. И. Укр. хим. ж., 42, 760 (1976).
278. Нагоя когё гидзюцу сикэнсе хококу, 13, 83 (1964); РЖХим, 1965, 10Б593.
279. Назаренко В. А., Полуактов 11. С. Полумикрохимический анализ минералов и руд. М., Госхимиздат, 1950, стр. 132.
280. Назин А. Г., Левин В. И., Голутвина М. М. Сб. «Методы получения радиоактивных препаратов». М., Госатомиздат, 1962, стр. 118.
281. Нарбутт К. И., Беспалова И. Д., Зав. лаб., 24, 617 (1958).
282. НвуенВан Быу, Жиглинский А. Г., Туркин 10. И. Ж. прикл. спектроскоп., 9, 193 (1968).
283. Нестерова Ю. С-, Арапова Г. А. Сб. «Химический анализ и формулы минералов». М., «Наука», 1969, стр. 24.
284. Писи Т., Фудвивара И. Кёто дайгаку когаку кэнкюсё ихо, № 16, 38 (1959); РЖХим, 1960, 95525.
285. Новиков А. И. Изв. СО АН СССР. Сер. хим. вып. 3, 64 (1965).
286. Новиков А. И. Труды Ташкентской конференции по мирному использованию атомной энергии, т. 2. Ташкент, Изд-во АН УзбССР, I960,, стр. 349.
287. Новиков А. И., Егорова Л. А., Веселаго Л. С. Докл. АН ТаджССР, И, 33 (1968).
288. Новиков А. И., Старовойт И. А. Докл. АН ТаджССР, 8, 25, (1965).
289. Номура Ц., Пакагава Т. Бунсэки кагаку, 19, 397 (1970); РЖХим,. 1970, 21Г38.
290. Ноткина М. А., Добкина Б. М. Зав. лаб., 26, 1126 (1960).
191
291. Ноткина М. А., Добкина Б. М. Науч. тр. Гиредмет, 3, 155 (1961).
292. Ноткина М.А., Солодовник С. М. Зав. лаб., 23, 569 (1957).
293. Ноткина М. А., Солодовник С. М. Физический сборник. (Материалы X Всесоюзного совещания по спектроскопии, ч. 2. Атомная спектроскопия), вып. 4 (9). Изд-во Львов, гос. ун-та, 1958, стр. 341.
294. Огнева Э. Я., Огнев В. Р., Райхбаум Я. Д. Зав. лаб., 34, 1450 (1968).
295. Орлов Н. А., Львов Б. В. XVII Всесоюзный съезд по спектроскопии. Тезисы докладов. Атомный спектральный анализ. Минск, «Наука и техника», 1971, стр. 102.
296. Орлов Н. А., Победоносцев В. А., Савельев Ю. А. Ж. прикл. спектроскоп., 24, 214 (1976).
297. Орлова Е. И. Труды Всесоюзной конференции по медицинской радиологии (Вопросы гигиены и дозиметрии). М., Медгиз, 1957, стр. 190.
298. Основы металлургии, т. 3. М., Госна^техметаллургиздат, 1963, стр. 471.
299. Отмахова 3. И., Чащина О. В., Слезко Н. И. Зав. лаб., 35, 685 (1969).
300. Павлоцкая Ф. И. Сб. «Физико-химические методы исследования почв». М., «Наука», 1966, стр. 69.
301. Павлоцкая Ф.И. Труды научно-методического совещания ученых социалистических стран по применению изотопов и излучений в сельском хозяйстве. ВАСХНИЛ им. В. И. Ленина. Совет по использованию атомной энергии в сельском хозяйстве. М., изд. ВНИЭСХ, 1961, стр. 86.
302. Павлоцкая Ф. И., Федосеев Г. А., Бабичева Е. В., Зацепина Л. Н., Родионова Л. М., Тюрюканова Э. Б. Радиохимические методы определения 90Sr в природных объектах. М., изд. ГЕОХИ АН СССР, 1963.
303. Павлоцкая Ф. И., Федосеев Г. А., Бабичева Е. В., Зацепина Л. Н., Тюрюканова Э. Б. Почвоведение, № 2, 105 (1964).
304. Павлюченко М. М., Филонов Б. О. Сборник научных работ ИОНХ АН БССР, вып. 1. Минск, изд. АН БССР, 1960, стр. 27.
305. Паничев Н. А., Туркин Ю. И. Ж. прикл. спектроскоп., 12, 213 (1970).
306. Пантелеева Л. И., Занько А. А. Науч. зап. Львов, политехи, ин-та, вып. 29, 101 (1955).
307. Патин С. А. Сб. «Гидрофизические и гидрохимические исследования». Киев, «Наукова думка», 1965, стр. 68.
308. Певцов Г. А., Манова Т. Г. Ж. аналит. химии, 23, 96 (1968).
309. Певцов Г. А., Манова Т. Г. Сб. «Химические реактивы и препараты», вып. 29. М., изд. ИРЕА, 1966, стр. 7.
310. Певцов Г. А., Манова Т. Г. Сб. «Методы анализа химических реактивов и препаратов», вып. 8. М., изд. ИРЕА, 1964, стр. 66.
311. Певцов Г. А., Манова Т. Г. Там же, стр. 45.
312. Певцов Г. А., Манова Т. Г., Красильщик В. 3. Сб. «Методы анализа химических реактивов и препаратов», вып. 16. М., «Химия», 1969, стр. 74.
313. Певцов Г. А., Манова Т. Г., Рагинская Л. К. Лебединская К. Л. Там же, стр. 83.
314. Певцов Г. А., Рагинская Л. К., Манова Т. Г., Красильщик В. 3. Зав. лаб., 35, 1073 (1969).
315. Певцов Г. А., Рагинская Л. К., Манова Т. Г., Красильщик В. 3. Сб. «Химические реактивы и препараты», вып. 32. М., изд. ИРЕА, 1970, стр. 150.
316. Пейзулаев Ш. И., Карабаш А. Г., Крауз Л. С-, Костарева Ф. А. и др. Зав. лаб., 24, 723 (1958).
317. Пенчев Н. П., Панова Д. Хр. Изв. хим. ин-та Болгар. АН, 3, 53 (1955).
318. Петрова Т. В., Саввин С. В. Ж. аналит. химии, 24, 490 (1969).
319. Петрова Т. В.,тСаввин С. Б., Джераян Т. Г. Ж. аналит. химии, 30, 918 (1975).
320. Петрова Т. В., Саввин С. Б., Хакимходжаев И. Ж. апалит, химии, 25, 2110 (1970).
192
321. Петрова Т. В., Хакимходжаев Н., Саввин С. Б. Изв. АН СССР. Сер. хим., № 2, 259 (1970).
322. Перрин Д. Органические аналитические реагенты. М., «Мир», 1967, стр. 327.
323. Пилипенко А. Т., Алтаев И. М. Тр. науч.-техн. о-ва черн. металлургии. Укр. республ. правление (Киев), 4, 67 (1956).
324. Пих М, С., Чередниченко И. Ф., Целинский Ю. К. Сб. «Методы анализа химических реактивов и препаратов», вып. 14. М., изд. ИРЕА, 1967, стр. 85.
325. Поздняков А. А., Лаврухина А. К. Тр. Комиссии по аналит. химии. АН СССР, 9(12), 161 (1958).
326. Поливанова Н. Г., Мошкович Г. Н., Богданова Н. Н., Карасева И. А. Тр. Ленингр. н.-и. и проект, ин-та основн. хим. пром-сти, вып. 17, 58 (1975).
327. Полуэктов Н. С. Методы анализа по фотометрии пламени. М., «Химия», 1967.
328. Полуэктов Н. С. Экспрессные методы анализа при помощи фотометрии пламени в цветной металлургии. М., Металлургиздат, 1958.
329. Полуэктов Н. С., Бельтюкова С. В. Ж. аналит. химии, 25, 2106 (1970).
330. Полуэктов Н. С., Виткун Р. А. Укр. хим. ж., 26, 648 (1960).
331. Полуэктов Н. С., Виткун Р. А., Овчар Л. А. Ж. аналит. химии, 15, 264 (1960).
332. Полуэктов Н. С., Мищенко В. Т. Сб. «Методы определения и анализа редких элементов». М., Изд.-во АН СССР, 1961, стр. 108.
333. Полуэктов Н. С., Никонова М. П., Лейдерман Ц. А., Лауэр Р. С. Ж. аналит. химии, 12, 699 (1957).
334. Полуэктов Н. С., Попова С. Б. Ж. аналит. химии, 15, 437 (1960).
335. Полуэктов Н. С., Попова С. Б., Овчар Л. А. Там же, стр. 131.
336. Портнов А. И. Ж. общей химии, 18, 605 (1948).
337. Потапова С. А. Радиохимия, 5, 110 (1963).
337а. Посторонко А. И., Горбенко Ф. П. Техническая и экономическая информация. Серия «Методы анализа и контроля производства в химической промышленности», № 4, М., изд. НИИТЭХим, 1974, стр. 57.
338. Починок X. Н. Ж. прикл. химии, 5, 1078 (1932).
339. Прокопчук Ю. 3., Брежнева Н. Е., Корпусов Г. В. Радиохимия, 13, 245 (1971).
340. Прокопчук Ю. 3., Брежнева Н. Е., Корпусов Г. В., Прохорова Н.П. Радиохимия, 15, 42 (1973).
341. Прокопчук Ю. 3., Корпусов Г. В-, Брежнева Н. Е. Радиохимия, Там же, стр. 35.
342. Прудников Е. Д. Изв. высш, учебн. завед. Химия и хим. технол., 14, 1331 (1971).
343. Прудников Е. Д. Изв. высш, учебн. завед. Химия и хим. технол. 18, 186 (1975).
344. Прудников Е. Д. Ж. прикл. спектроскоп., 14, 145 (1971).
345. Прудников Е. Д., Ж. аналит. химии, 27, 2129, 2327 (1972).
346. Прудников Е. Д. Ж. прикл. спектроскоп., 24, 571 (1976).
347. Прудников Е. Д., Шапкина Ю. С. Ж. аналит. химии, 26, 710 (1971).
348. Прудников Е. Д., Шапкина Ю. С. Ж. аналит. химии, 28, 1055 (1973).
349. Прудников Е. Д., Шапкина Ю. С. Ж. аналит. химии, 31, 274, (1976).
350. Прудников Е.Д., Шапкина Ю. С., Семов М. Л. Изв. высш, учебн.
завед. Химия и хим. технол., 14, 196 (1971).
351. Пршибил Р. Комплексоны в химическом анализе. М., ИЛ, 1960, стр. 317, 345.
352. Пушинов Ю. В., Черкесов А. И. Сб. «Физико-химические исследования природных сорбентов и ряда аналитических систем», вып. 1. Изд-во Саратовск. гос. ун-та, 1966, стр. 91.
353. Пушкарев В. В., Багрецов В. Ф., Пузако В. Д. Радиохимия, 6, 120 (1964).
7 Н. С. Полуэктов и др.
193
354. Реакции и реактивы для качественного анализа неорганических соединений. М.— Л., Госнаучтехиздат, 1950, стр. 177.
355. Росоловский В. Я. Сакк Ж. Т. Ж. неорган. химии, 15, 2640 (1970).
356. Руднев Н. А., Пустовалова М. Н., Малофеева Г. И. Ж. аналит. химии, 25, 1085 (1970).
357. Русанов А. К. Основы количественного спектрального анализа руд и минералов. М., «Недра», 1971, стр. 272, 275, 301, 333, 336.
358. Русанов А. К., Алексеева В. М., Хитрое В. Г. Количественное спектральное определение редких и рассеянных элементов в рудах и минералах. М., Госгеолтехиздат, 1960.
359. Ручик А. С., Старостина Л. С. Тр. Гос. НИИ горно-хим. сырья, вып. 4, 242 (1958.)
360. Рысс И. Г., Нилус Э. Л. Химическая технология. Республиканский межведомственный научно-технический сборник, вып. 1. Изд-во Харьковского гос. ун-та, 1965, стр. 42.
361. Рязанов И. А., Айдаров Т. К. Ж. прикл. спектроскоп., 21, 5 (1974).
362. Саввин С. Б. Органические реагенты группы арсеназо III. М., Ато-миздат, 1971, стр. 149, 213, 318.
363. Саввин С. Б., Акимова Т. Г., Дедкова В. П. Органические реагенты для определения Ва2+ и SO^~. М., «Наука», 1971, стр. 135.
364. Саввин С. Б., Дедков Ю. М. Ж. аналит. химии, 19, 21 (1964).
365. Саввин С. Б., Дедков Ю. М., Макарова В. П. Ж. аналит. химии, 17, 43 (1962).
366. Саввин С. Б., Петрова Т.В. Ж. аналит. химии, 24, 177 (1969).
367. Саввин С. Б., Петрова Т.В., Хакимходжаев Н. Авт. свид. СССР 244695 (1967); Бюл. изобр., № 18 (1969).
368. Саввин С. Б., Петрова Т. В., Хакимходжаев И. Ж. аналит. химии, 24, 1460 (1969).
369. Седнев М. П., Старобинец Г. Л., Акулович А. М. Ж. аналит. хи-, мии, 21, 23 (1966).
370. Седых Э. М. Стекло и керамика, № 4, 46 (1971).
371. Селиванова Н. М., Зубова Г. А. Ж. аналит. химии, 12, 466 (1957).
372. Селинов И. П. Изотопы. Справочные таблицы, т. 1. М., «Наука», 1970.
373. Сендел Е. Колориметрические методы определения следов металлов. М., «Мир», 1964, стр. 336.
374. Сенявин М. М., Никита И. Г. Сб. «Радиохимические методы определения микроэлементов». М.— Л., «Наука», 1965 стр. 118.
375. Середа Г. А., Бобовникова Ц. И., Егоров В. В., Жигаловская Т. Н.г Махонько Э. П. Гидрохимические материалы, 43, 24 (1967).
376. Середа Г. А., Бобовникова Ц. И., Жигаловская Т. Н., Егоров В. В.у Махонько Э. П. Гидрохимические материалы, 41, 6 (1966).
377. Си Чжи-вэнъ, Тан Цзо-хуа, Ван Цзин-тан. Вести. Сычуаньск. ун-та, сер. естеств. и., № 5, 53, (1959); РЖХим, 1960, 69149.
378. Сигэмацу Ц., Осио Т. Доитай то хосясэн, 4, 105 (1961); РЖХим, 1963, 10Б371.
379. Сигэмацу Ц., Табуси М., Мураками Т. Нихон сио гаккайси, 17 294 (1964), РЖХим, 1965, 8Л80.
380. Сигэмацу Ц., Уэсуги К., Мураками Т. Бунсэки кагаку, 13, 1032 (1964); РЖХим, 1965, 13Г159.
381. Сизоненко Н. Т., Беленко Л. Э. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты», ч. 2. Харьков, изд. ВНИИ монокристаллов, 1971, стр. 202.
382. Силантьев А.Н., Чумичев В. Б., Вакуловский С. М. Тр. Ин-та эк-сперим. метеорологии. Гл. упр. гидрометеорологической службы при Сов. Мин. СССР, вып. 2, 15 (1970).
383. Силантьев А. И., Чумичев В. Б., Рослый Е.И. Там же, стр. 5.
384. Симатори К. Ригаку дэнки дзянару, 7, 265 (1965); РЖХим, 1966, 18Г35.)
194
385. Синицын Б. В., Бараков С. М., Баранова М. К., Бардин В. А., За-демидко Г. А., Получение стронция (Обзор литературы). М., изд. Ги-редмета, 1972, стр. 14.
386. Синочкин Ю. Д., Перумов Д. А., Патин С. А., Ажажа Э. Г. Радиохимия, 4, 198 (1962).
387. Славин В. Атомно-абсорбционная спектроскопия. Л., «Химия», 1971.
388. Смирнова И. С., Круглова Л. А., Казак В. Г., Дубинин В. Г., XI Всесоюзное совещание по рентгеновской спектроскопии. Ростов-на-Дону. Тезисы докладов. Л., 1975, стр. 96.
389. Смолин Д. Д., Тихонова Л. И., Рытова А. А. Ж. общей химии, 32, 2418 (1962).
390. Соляник С. К., Гитис Э. Б., Завьялова Н. Д. Тр. Н.-и. и проектн. ин-та основной химии, 32, 63 (1974).
391. Сорокина Н. И., Голубева В. М. Тр. ЦНИИ черной металлургии, вып. 37, 98 (1964).
392. Старик И. Е., Александрук В. М. Докл. АН СССР, 159, 1055 (1964).
393. Старобинец Г. Л., Седнев М. П., Дубовик Т. Л. Тр. Комиссии по аналит. химии. АН СССР, 15, 323 (1965).
394. Стары И. Экстракция хелатов. М., «Мир», 1966, стр. 307.
395. Степанова Н. Д., Степанов А. В. Ж. прикл. химии, 42, 1670 (1969).
396. Стефановий Б.,ЯньиЙТ. Гласник хем. друшт. (Београд), 20, 569 (1955): 21, 125 (1956).
397. Столяров К. П. Методы микрохимического анализа. Изд-во ЛГУ, 1960.
398. Столярова И. А., Акельева А. С. Тр. Всес. н.-и. геол, ин-та, 125, 179 (1966).
399. Столярова И. А., Мойжес И. Б. Там же, стр. 157.
400. Столярова И. А., Шувалова Н. И. Информ, сб. Всес. н.-и. геол, ин-та, № 51, 89 (1961).
401. Страдомский В. Б., Гончарова С. Н. Гидрохимические материалы, 41, 28 (1966).
402. Стромберг А. Г., Захарова Э. А. Сб. «Современные методы анализа материалов». М., Металлургия, 1969, стр. 91.
403. Сулайманкулов К., Ногоев К., Рысмендеев К., Токмергенова Л. Сб. «Исследование взаимодействия мочевины с неорганическими соединениями». Фрунзе, «Илим», 1964, стр. 15.
404. Такамото С. Нихон кагаку дзасси, 81, 1539 (I960): РЖХим, 1961, 9Д41.
405. Такитани С., Судзуки М., Фудзита И., Ходзуми К. Бунсэки кагаку, 14, 597 (1965); РЖХим, 1966, 13Г53.
406. Такитани С., Хата Й., Судзуки М. Бунсэки кагаку, 18, 626 (1969); РЖХим, 1970, ЗГ69.
407. Тамура Д., Мацусима Й. Якугаку дзасси, 82, 1569 (1962); РЖХим, 1964, 12В54.
408. Танака М. Нихон кагаку дзасси, 85, 117, 119 (1964); РЖХим, 1964, 22Г48, 22Г49.
409. Танака Г., Томикава А., Кавамура X., Ояги Й. Нихон кагаку дзасси, 89, 175 (1968); РЖХим, 1969, 2Г74.
410. Тананаев Н. А. Капельный метод. М.— Л., Госхимиздат, 1954, стр. 60.
411. Тананаев И. В., Терешин Г. С., Сокол В. И. Ж. неорган. химии, 11, 2279 (1966).
412. Тарковская И. А., Горбенко Ф. П. Ж. аналит. химии, 20, 1185 (1965).
413. Тарковская И. А., Горбенко Ф. ПЕмельянов В. Б., Олевинский М. И. Тр. Комиссии по аналит. химии. АН СССР, 15, 336 (1965).
414. Тарковская И. А., Шевченко С. И. Тр. по химии и хим. технол. (Горький), вып. 3 (24), 49 (1969).
415. Терентьев А. П., Литвин К. И., Рухадзе Е. Г. Ж. аналит. химии, 14, 288 (1959).
416. Терентьев А. П., Рухадзе Е. Г., Литвин К. И. Ж. аналит. химии. И, 55 (1956).
417. Тиба М. Бунсэки кагаку, 14, 1161 (1965); РЖХим, 1966, 15Г50.
7*
195
418. Тихомиров В. И., Кузнецова А. А., Ваторовская Э. Д. Радиохимия, 6, 182 (1964).
419. Тихонова Л. И. Ж. неорган. химии, 13, 2687 (1968).
420. Тихонова Л. И. Ж. неорган. химии, 14, 2368 (1969).
421. Толмачев В. И., Ломакина Г. Г. Ж. физ. химии, 33, 2302 (1959).
422. Толмачев В. Н., Ломако Л. А., Мирошник Л. В. Высокомолекул. соединения. Б. Кратк. сообщения, 10, 905 (1968).
423. Участника 3. Б. Бум. пром-сть, № 7, 10 (1954).
424. Уэсуги К., Табуси М., Мураками Т. Бунсэки кагаку, 13, 440 (1964); РЖХим, 1965, 9Г136.
425. Фабрикова Е. А., Баум Л. И. Сб. «Новые методические исследования по анализу редкометальных минералов, руд и горных пород». М., «Наука», 1970, стр. 55.
426. Фабрикова Е.А., Исаева А. Г. Тр. Ин-та минерал., геохим., кристаллохим. редких элементов АН СССР, вып. 18, 175 (1963).
427. Файглъ Ф. Капельный анализ. М., Госхимтехиздат, 1933, стр. 208.
428. Феръянчич Ю. А. Тр. Центр, н.-и. горноразвед. ин-та, вып. 77, 227 (1967).
429. Фор Г., Пауэлл Дж. Изотопы стронция в геологии. М., «Мир», 1974-
430. Франц, пат. 1538528 (1968); РЖХим, 1969, 18Г69П.
431. Фудвинага Т., Кояма М., ЦурубоС. Нихон кагаку дзасси, 84, 128 (1963); РЖХим, 1964, 6Б317.
432. Фудвита Т., Сигамацу Ц. Бунсэки кагаку, 19, 893 (1970); РЖХим, 1970, 24Г199.
433. Хамагути X. Бунсэки кики, 7, 703 (1969); РЖХим, 1970, 13Г167.
434. Хёринг Р. Хелатообразующие ионообменники. М., «Мир», 1971, стр. 239.
435. Хивенс О. Лазеры на твердых веществах. М., «Мир», 1969.
436. Хисано К., Ояма К. Бунсэки кагаку, 20, 228 (1971); РЖХим, 1971, • 18Г143.
437. Худякова Т. А., Востоков В. М. Тр. по химии и хим. технол. (Горький), вып. 2 (20), 101 (1968).
438. Худякова Т. А., Мешкова Л. А. Изв. высш, учебн. завед. Химия и хим. технол., 13, 1576 (1970).
439. Целинский Ю. К., Киселева Н. Е., Лапицкая Е. В., Кучкина Е. Д., Сб. «Методы анализа химических реактивов и препаратов», вып. 14. М., изд. ИРЕА, 1967, стр. 101.
440. Церковницкая И. А., Боровая Н. С., Бахвалова М. Н. Сб. «Методы количественного определения элементов». Изд-во ЛГУ, 1964, стр. 84.
441. Цинь Цицзун, Чэнь Цзун-фан, Пань Шунъ-кан. Юаньцзынэн, № 9, 854 (1964); РЖХим, 1965, 17Г30.
442. Цубаки И. Бунсэки кагаку, 12, 849 (1963); РЖХим, 1964, 22Г101.
443. Челап М. Б. Гласник хем. друшт. (Београд), 21, 225 (1956).
444. Черкашина Т. В., Салтыкова А. М., Петрова Е. И., Горянская Г. Р., Луговская В. И., Блохина Д. И. Ж. прикл. спектроскоп. 9, 508 (1968).
445. Чжан Чжу-цвюнъ. Хуасюэ сюэбао, 31, 547 (1965); РЖХим, 1966, 14Г45.
446. Чуенко Л. И. Информ, сб. Всес. н.-и. геол, ин-та, № 51, 113 (1961).
447. Чуенко Л. И. Сб. «Методы химического анализа минерального сырья», вып. 5. М., Госгеолтехиздат, 1959, стр. 35.
448. Чуенко Л. И. Тр. Всес. н.-и. геол, ин-та, 125, 66 (1966).
449. Чуенко Л. И., Фрейде М. В. Информ, сб. Всес. н.-и. геол, ин-та, № 3, 128 (1956).
450. Чупахин М. С., Крючкова О. И., Рамендик Г. И. Аналитические возможности искровой масс-спектрометрии. М., Атомиздат, 1972.
451. Шабурова В. П., Юделевич И. Г., Серякова И. В., Золотов Ю. А. Ж. аналит. химии, 31, 255 (1976).
452. Шамаев В. И., Гарбаускас Г. К., Кондратенко А. А., Мартынов А. М. Тр. Моск, хим.-технол. ин-та им. Д. И. Менделеева, вып. 62, 256 (1969).
196
453. Шарм Г. Методы аналитической химии. Количественный анализ, ч. 1 и 2. М., «Химия», 1969.
454. Шахова Н. В., Алимарин И. П., Золотов Ю. А. Докл. АН СССР, 152, 884 (1963).
455. Ш ахтахтинский Г. Б., Асланов Г. А., Джафаров Г. А. Азерб. хим. ж., № 3, 78 (1969).
456, Шварценбах Г., Флашка Г. Комплекснометрическое титрование. М., «Химия», 1970.
457. Шведов В. Л., Анкудинов Е. Л., Бунин Б. Г., Максимова А. М., Леонова Л. М. Радиохимия, 4, 110 (1962).
458. Шведов В. Л., Жилкина М. И., Зиновьева В. К. Радиохимия, 1, 109 (1959).
459. Шведов В. Л., Жилкина М. Л., Зиновьева В. В., Леонова Л. М., Макарова Т. Л., Анкудинов Е. Л., Бунин Б. Г. Исследование содержания 90Sr в зерновых культурах (урожай 1958 г.). М., Изд-во АН СССР, 1960.
460. Шведов В. Л., Леонов В. Л. Ж. аналит. химии, 31, 385 (1976).
461. Шведов В. Л., Макарова Т. Л., Леонова Л. М., Павлова Н. А. Радиохимия, 1, 616 (1959).
462. Шведов В. Л., Степанов А. В., Горский Н. И. Радиохимия, 5, 690 (1963).
463. Шведов В. Л., Тэн Тон, Степанов А. В. Ж. аналит. химии, 15, 16 (1960).
464. Шведов В. П., Фомин В. В. Радиохимия, 11, 565 (1969).
465. Шевченко В. Б., Смелов В. С. Сб. «Экстракция. Теория, применение, аппаратура», вып. 2. М., Госатомиздат, 1962, стр. 257.
466. Шевченко В. Б., Шмидт В. С. Радиохимия, 3, 121 (1961).
467. Шевченко В. В., Несанелис М. 3. Сб. «Монокристаллы и техника», вып. 7. Харьков, изд. ВНИИ монокристаллов, 1972, стр. 164.
468. Шевченко Ф. Д., Гаевська Н. Ш., Byaina Л. О., Агеев В. А. В1сник Кшвського ун-ту, сер. xiM., № 11, 28 (1970).
469. Шевченко Ф. Д., Byaina Л. О. В1сник Кшвського ун-ту, сер. ф!з. та xiM, № 7, 138 (1967).
470. Шевченко Ф. Д., Вузина Л. А. Укр. хим. ж., 33, 991 (1967).
471. Шевченко Ф. Д., Вузина Л. А. Укр. хим. ж., 34, 499 (1968).
472. Шевченко Ф. Д., Вузша Л. О., Висоцъка Г. О. В1сник Кшвського унту, сер. xiM., № 9, 31 (1968).
473. Шеллард Э. Количественная хроматография на бумаге и в тонком слое. М., «Мир», 1971.
474. Шемет В. В., Малыгина Л. В., Луфт Б. Д., Милявский Ю. С., Павельева О. X. Ж. аналит. химии, 27, 2341 (1972).
475. Шестов И. Н., Шурубор А. В., Тверъе Ф. М. Нефтегазовая геология и геофизика. Научи.-техн, сб., № 8, 33 (1965).
476. Шнайдерман С. Я. Изв. Киев, политехи, ин-та, 14, 140 (1954).
477. Шакина Н. Т., Тимофеев Е. Ф., Новикова В. А. Зав. лаб., 39, 169 (1973).
478. Штепа Л. Л. Сб. «Материалы IV Межотраслевой конференции по методам получения и анализа ферритовых, сегнето-, пьезоэлектрических и конденсаторных материалов и сырья для них». М., изд. ИРЕА, 1973. стр. 194.
479. Штефек М., Вирш М., Райс И. Ж. аналит. химии, 31, 1364 (1976).
480. Щебетковский В. Н., Егоров Ю. М. Радиохимия, 10, 83 (1968).
481. Щелков Е. Е. Изв. АН КиргССР. Сер. естеств. и техн, наук, 6, № 3, 89 (1964).
482. Электромиграционный метод в физико-химических и радиохимических исследованиях. М., Атомиздат, 1971.
483. Эфендиев Г. X., Маков Н. Н., Александров Р. А. Азерб. хим. ж., № 2, 93 (1967).
484. Яньи/i Т., Челап М. Б., Зарубица Л., РадоновиЬ Л. Д. Гласник хем. друшт. (Београд), 29, 201 (1963).
197
485. Ясуда К. Бунсэки кагаку, 17, 289 (1968); РЖХим, 1969, 2Г42.
486. Abbey S., Lee N. J., Bouvier J. L. Pap. Geol. Surv. Can., N 19, 26 (1974).
487. Abd el Raheem A. A., Osman F. A. Z. anal. Chem., 169, 328 (1958).
488. Absolon K. Acta geol. et geogr. Univ. Comenianae. Geol., N 6, 293 (1961).
489. Adam J., Ettre K., Gergely G., Varadi P. F. Magy. kem. folyoirat, 62, 223 (1956).
490. Adams P. B., Passmore W. 0. Anal. Chem., 38, 630 (1966).
491. Ahrens L. H. Bull. Geol. Mag., 60, 217 (1949).
492. Ahrens L. H. Spectrochim. acta, 4, 302 (1951).
493. Ahrens L. H., Taylor S. R. Spectrochemical analysis. London, Addison— Wesley Co., 1961, p. 206, 408,
494. Akaza I. Bull. Chem. Soc. Jap., 39, 971 (1966); РЖХим, 1967, 8B2.
495. Aldous К. M., Dagnail R. M., Ebdon L. C., West T. S. Anal. chim. acta, 50, 335 (1970).
496. Aldrich L. T., Herzog L. F., Holyk W- K., Whiting F. B., Ahrens L. H . Phys. Rev., 89, 631 (1953).
497. Aldrich L. T., Wetherill G- W., Davis G. L. Geochim. et cosmochim. acta, 10, 238 (1956).
498. Allison J. B., Becker R. S. J. Phys. Chem., 67, 2675 (1963).
499. Almassy G., Dezso J., Szell T. Magy.kem folyoirat, 60, 373 (1954).
500. Algyier R. Parfum. mod., 45, N 33, 54 (1953); РЖХим, 1953, 9150.
501. Alvarez Gonzalez F., Roca Adell M.t Fernandez Cellini R. An. Real. soc. esp. fis. у qyim., B54, 278 (1958).
502. Amos M. D., Willis J. B. Spectrochim. acta, 22, 1325 (1966).
503. A nderegg G. Experientia, 20, Suppl. N9, 75 (1964); РЖХим, 1966, 14Б564.
504. A nderegg G. L., Eplattenier F., Schwarzenbach G. Helv. chim. acta, 46. 1400 (1963).
505. Andersen N. R.t HumeD. jV. Anal, chim., acta, 40, 207 (1968).
506. Ando A., Fuwa K., Vallee B. Anal. Chem. 42, 818 (1970).
507. Angyal S. J., Davies К. P. Chem. Communs N10, 500 (1971); РЖХим, 1971, 20B53.
508. ArgieroL., Manfredini S., Palmas G. Chim. e ind. (Ital.) 43, 896 (1961).
509. Argiero L., Manfredini S., Palmas G. Minerva nucl., 3, 421 (1959).
510. Arnikar H. J., Mehta О. P. Current Sci., 28, 400 (1959).
511. Arribas J. S. Inform, quim. anal., 15, 161 (1961); РЖХим, 1962, 19Д34,
512. Arribas J. S., Alvarez В. M. L. Inform, quim. anal., 17, 145 (1963); РЖХим, 1964, 19Г1.
513. Arribas J. S., Alvarez В. M. L., Moro G. R- An. Real soc. esp. fis. у quim., B57, 581 (1961); РЖХим, 1962, 16Д38.
514. Avni R. Spectrochim. acta, B23, 619 (1968).
515. Avni R., Bonkobza A. Appl. Spectrosc., 23, 483 (1969).
516. Baetsle L., Hays D. J. Inorg. Nucl. Chem. 21, 133 (1961 —1962).
517. Bagliano G., Ossicini L., Lederer M. J. Chromatogr., 21, 471 (1966).
518. Bailey B. W., Aldous К. M. 16 Colloq. Spectroscop. Int. Heidelberg, 1971, Vorabdr. Bd. 2, London, 1971, p. 338; РЖХим, 1974, 13Г29.
519. Bailey R. A., Yaffe L. Can. J. Chem., 38, 1871 (1960).
520. Baker J. R., Pearson K. N. Anal. chim. acta, 50, 255 (1970).
521. Ballada J., Jeanmaire L. Rapp. CEA, NR-3653 (1968); Anal, abstr., 18, 2756 (1970).
522. Ballczo H., Doppler G. Z. anal. Chem., 151, 16 (1956).
523. Ballczo H., Doppler G. Z. anal. Chem., 152, 321 (1956).
524. Ballczo H., Schenk W. Mikrochim. acta, 1954, 163.
525. Barcza L., Mester O. Ann. Univ. sci. budapest. Sec. chim., 4, 33 (1962); РЖХим, 1963, 13Г59.
526. Bark L. S. Mikrochim. acta, 1958, 117.
527. Barnabas T., Badve M. G., Barnabas J. Anal. chim. acta, 12, 542 (1955).
528. Barreto H. S. R., Barreto R. C. R. J. Chromatogr., 4, 153 (1960).
198
529. Barry Wh., Beer Z. de, Hourlier P. Publ., Groupem. avancem. meth, spectrogr., 1961, p. 415; РЖХим, 1962, 22Д98.
530. Bascunan A., Cetina R., Comez-Lara J. J. Inorg. Nucl. Chem., 33, 1013 (1971).
531. Bassett J., Leton G. B., Vogel A. J. Analyst, 92, 279 (1967).
532. Bastion R., Weberling R., Palilla F. Anal. Chem., 29, 1795 (1957).
533. Baum M. Radex Rdsch. N 3, 158 (1964); РЖХим, 1965, 20Г112.
534. Beccaluva L., Venturelli G. Atom. Absorpt. Newslett., 10, 50 (1971).
535. Bek F., JanouSkovd J-, Moldan B. Atom. Absorpt. Newslett., 13, 47 (1974).
536. Bek F., Janouikovd J., Moldan В. Chem.listy, 66, 867 (1972).
537. Belcher С. B., Brooks К. A. AnaE chim. acta, 29, 202 (1963).
538. Benes J. Collect. Czech. Chem. Communs, 35, 591 (1970).
539. Benei J., Kyrs M. Collect. Czech. Chem. Communs, 31, 4405 (1966).
540. Bennett W. E., Skoylin D. O. Anal. Chem., 38, 518 (1966).
541. Berak L., Munich J. Collect. Czech. Chem. Communs, 26, 276 (1961).
542. BergE. W., Herrera N. M. Anal. chim. acta, 60, 117 (1972).
543. Bergamini C., Rovai A. Anal. chim. acta, 15, 43 (1956).
544. Berkey E., Sweeney G. G., Hickam W. Nucl. Appl., 5, 344 (1968); РЖХим, 1969, 16Г126.
545. Bernhard H., Grass F. Mikrochim. acta, 1£66, 426.
546. Bezdekova A., Budisinsky B. Collect. Czech. Chem. Communs, 30, 818 (1965).
547. Bheemasankara Rao Ch., Vonkateswarlu V. Z. anal. Chem., 198, 183 (1963).
548. В iagi L., Pirani R., Simboli G. Mineral, et petrogr. acta, 9, 163 (1963).
549. Biheller J. H. Atompraxis, 12, 465 (1966).
550. Billings G. K., Angina E. E. Bull. Can. Petrol. Geol., 13, 529 (1965).
551. Billings G. K., Harriss R. С. Тех. J. Sci., 17, 129 (1965); РЖХим, 1966, 5И173.
552. Billon J. P. Meth. phys. anal., 6, 231 (1970).
553. Birks F. T. Atom. Energy Res. Establ. [Rept.], N 1177, 1, Appendix I, II (1953); РЖХим, 1955, 16538.
554. Birks F. T. Spectrochim. acta, 8, 167 (1956).
555. Blacdel W- S., Olsen E. D., Вuchanan R. F. Anal. Chem., 32, 1866 (1960).
556. Blake C. A., Baes C. F., Brown К. B. Ind. and Eng. Chem., 50, 1763 (1958); РЖХим, 1959, 39182.
557. Blasius E., Brozio B. J. Chromatogr., 18, 572 (1965).
558. Blo^eld E., Levi H. W., Schiewer E. Ber. Hahn-Meitner-Inst.-Kern-forsch., Berlin, N 69, 40 (1968); РЖХим, 1969, 4Л25.
559. Bobtelsky M., Kerles S. Bull. Soc. chim. France, 1955, 328.
560. Bodner R. L., Hendricker D. G. Inorg. Chem., 9, 1255 (1970).
561. Bogs U., Gbthe W. Mikrochim. acta, 1963, 931.
562. Вokacs-Polgar E., Kurcz-Csiky J. Nature, 204, 1057 (1964).
563. Bolomej R. A., Wish L. J. Amer. Chem. Soc., 72, 4483 (1950).
564. Boni A. L. Anal. Chem., 32, 599 (1960).
565. BonigG. Omagiuacad. prof. Raluca Ripan. Bucuresti, Editura Academiei RSR, 1966, p. 157.
566. Bonnin A. Bull. Soc. chim. France, 1956, 1563.
567. Bosch-Reig F-, Burriel-Marti F. Inform, quim. anal, pura apl. Ind., 22, 207 (1968).
568. Bouquiaux J. J., Gillard J. H. Anal. chim. acta, 30, 273 (1964).
569. Bouquiaux J. J., Gillard-Baruh J. H.C. Radiochim. acta (BRD), 9, 153 (1968).
570. Bouquiaux J. J., Gillard-Baruh J. H. Radiochim. acta (BRD), 14, 39 (1970).
571. Bouilloux G- Bull. Soc. chim. France, 1954, 1347.
572. Bovy R., Duyckaerls G. Anal. chim. acta, 11, 134 (1954).
573. Bowen H. J. M. Adv. Sci., 12, 585 (1956).
574. Bowen H- J. M., Cawse P. A. Atom. Energy Res. Establ. [Rept.] N R2925, 1959; РЖХим, 1959, 86241.
199
-’575. Brandt P. J., RietB. T. Anal. Chem., 38, 1790 (1966).
576. Bray L. A. Nucl. Sci. and Eng., 20, 362 (1964).
-577. Breault E. A. J. Assoc. Offic. Agric. Chem., 48, 719 (1965).
578. Brenan C., LovattJ. H., Smith H. Nature, 211, 68 (1966).
579. Brinkman U. A., Kingma G. Z. anal. Chem., 249, 16 (1970).
-580. Broadbank R. W. C., Dhabanandana S., Harding R. D. Analyst, 85, 365 (1960).
581. Brunfelt A. O., Steinnes E. Anal. chim. acta, 48, 13 (1969).
582. Bruninx E. J. Inorg. Nucl. Chem., 23, 317 (1961—1962).
583. Brusset H., Madaule-Aubry F., Brunet M. Bull. Soc. chim. France, 1967, 1973.
584. Bruyne P. J. Inorg. Nucl. Chem., 32, 346 (1970).
585. Bryant E. 4., Sattizahn J. E., Warreu B. Anal. Chem., 31, 334 (1959).
586. Bryant F. J., Chamberlian A. C. MorganA., Spicer C. S. J. Nucl. Energy, 6, 22 (1957).
587. Buchtela K. Leisigang M. Radiochim. acta, 1, 208 (1963).
588. Budesinsky B. Collect. Czech. Chem. Communs, 32, 1688 (1967).
589. В udSsinsky B., Haas K., Bezdekova A. Ibid., p. 1528.
590. BudSsinsky B., Urzalova D. Taianta, 13, 1217 (1966).
591. BudSsinsky B., Urzalova D., Bezdekova A. Acta chim. Acad. sci. hung., 52, 37 (1967); РЖХим, 1967, 18Г98.
592. В ud$ s insky B., West T. S. Analyst, 94, 182 (1969).
593. Budesinsky B., West T. S. Taianta, 16, 399 (1969).
594. Bundy W. S., Pavlik А. Пат. США 3065052 (1962); РЖХим, 1964, 21Л57П.
595. Burk W. Kernenergie, 3, 1204 (1960).
596. Burns R. E., Moore R. L., Platt A. M., Swift W. H. Chem. Eng. Progr. Symp. Ser., 60, 1 (1964); РЖХим, 1965, ЗЛЗО.
597. Burriel-Marti F., Alvarez H. C. Inform, quim. anal., 15, N 6, 151 (1961); РЖХим, 1962, 19Д116.
598. Burriel-Marti F., Alvarez H. C. Inform, quim. anal., 16, N 3, 68; N 6, 159 (1962); РЖХим, 1963, 9Г188, 23Г15.
599. Burriel-Marti F., Ramirez M. J. Rexach M. de Lizarduy M. L. Rev. cienc., apl., 12, 16 (1958); РЖХим, 1958, 77323.
600. Burton J. D., Love R. M., Mercer E. R. Analyst, 91, 739 (1966).
601. Butler F. E. Anal. Chem., 35, 2069 (1963).
602. Camozzo S. Chimica, 30, 113 (1954).
603. Campbell M. H. Anal. Chem., 37, 252 (1965).
604. Canic V. D., Petrovic S. M. Z. anal. Chem., 211, 321 (1965).
605. Capacho-Delgado L., Manning D. C. Analyst, 92, 553 (1967).
606. Carlsqvist B., Dyrssen D. Acta chem. scand., 19, 1293 (1965).
607. Carr R. A. Limnol. and Oceanogr., 15, 318 (1970); РЖХим, 1971, 1Г208.
608. Carron J.—P., Hennion J., Nicole J., Tridot G. Chim. anal., 50 (9), 455 (1968).
609. Ceccetti G., Cotta-Rasumino F., Stacchini A. Metallurgia ital., 61, 405 (1966); РЖХим, 1970, 8Г99.
610. Celsi S. A. Bol. Soc. quim. Peru, 25, 73 (1959); РЖХим, 1960, 56683.
611. Cerrai E.t Ghersini G. J. Chromatogr., 13, 211 (1964).
612. Cerrai E.t Ghersini G. J. Chromatogr., 15, 236 (1964).
613. Cerrai E., Ghersini G. J. Chromatogr., 18, 124 (1965).
614. Cerrai E., Pelati L., Schreiber B., Tviulzi C. Energ. nucl., 10, 45 (1963).
615. Cerrai E., Testa C. J. Chromatogr., 7, 112 (1962).
616. Cerrai E., Testa C., Triulzi C. Energ. nucl., 9, 377 (1962).
617. Cervellati 4., Cicognani G. Minerva nucl., 5, 48 (1961).
618. Chakrabarti C. L., Katyal Mohan. Anal. Chem., 43, 1302 (1971).
619. Chalmers R. A., DickD. M., Anal. chim. acta, 31, 520 (1964).
620. Chalmers R. 4., Telling G. Mikrochim. acta, 1967, 1126.
621. Champion К. P., Taylor J. C., Whittem R. N. Anal. Chem., 38, 169 (1966).
622. Champion К. P., Whittem R- N. Analyst, 93, 550 (1968).
200
623. Chandra R. J. Chem. Educ., 39, 397 (1962).
624. Chandra U. Govt. India Atom. Energy Commis. [Rept], N BARC-454 (1970); РЖХим, 1970, 19И329.
625. Chandrasekaran V. R., Lalit B. Y., Proc. Nucl. and Radiat. Chem, Symp. Bombay, 1964, Bombay, 1965, p. 57.
626. Chandrasekaran V. R., Lalit B. Y. Proc. Nucl. and Radiat. Chem.
Symp. Walteir, 1966, Delhi, 1966, p. 20; РЖБиохим, 1967, 17Ф101.
627. Chappell B. W., Compston W-, Arriens P. A., Vernon M. J. Geochim. cosmochim. acta, 33, 1002 (1969).
628. ChasanD. E., Norwitz G. Appl. Spectrosc., 24, 283 (1970).
629. Chasan D. E., Norwitz G. Taianta, 18, 499 (1971).
630. Chatterjee R., Dey-Arun K. Microchem. J., 10, 171; 11, 437 (1966).
631. Chatterjee, R-, Dey-Arun K. Microchem. J., 12, 151 (1967).
632. Cherry K. D., Hobbs J. В. M., Erlank A. J., Willis J. P., Can. Spectrosc., 15, 3, 16 (1970).
633. Chesterikoff A. Bull. Soc. franp. miner, et cristallogr., 93, 357 (1970); РЖХим, 1970, 24Г197.
634. Chow T. J., Tompson T. G., Anal. Chem., 27, 18 (1955).
635. Chowdhury A. N., Das A. K., Z. anal. Chem., 261, 126 (1972).
636. Christian G. D., Feldman F. J. Appl. Spectrosc., 25, 660 (1971).
637. Christensen H. T. Taianta, 13, 1203 (1966).
638. Christova R., llkova P. Anal. chim. acta 33, 434 (1965).
639. Christova R., Kruschevska A: Anal. chim. acta, 36, 392 (1966).
640. Chwastowska J., Rozanska В. Chem. anal. (PRL), 21, 85 (1976).
641. Claisse F. Can. Spectrosc., 12, 20 (1967).
642. Claisse F. Rev. Groupem. avancem. meth, spectrogr. jan.-mars, 1967, 10; РЖХим, 1968, 2Г31.
643. Cohen P., Pardo G., Wormser G. Energ. nucl., 1, 89 (1957).
644. Collin R. L., Anal. Chem., 33, 605 (1961).
645. Collins A. G. Producers Monthly, 26, 24 (1962); РЖХим, 1963, 15Г81.
646. Cooley C. R., Richardson G. A. Chem. Eng. Progr. Symp. Ser., 60, 22 (1964); РЖХим, 1965, 19Л27.
647. Cotta-Ramusino F., Intonti R. Metallurgia ital., 59, 645 (1967); РЖХим, 1968, 8Г155.
648. Cotterill J. C. Atom. Energy Res. Establ. [Rept], N AM-46 (1959); РЖХим, 1960, 34555.
649. Cotterill J. C., Res. Group U. K. Atom. Energy Auth., N AERE-AM94, (1963); РЖХим, 1964, 16Г128.
650. Covello M., Ciampa G., Mauna F. Rend. Accad. sci. fis. e mat. Soc. naz. sci. lett. ed arti Napoli, 35, 292 (1968—1969); РЖХим, 1970, 7Г50.
651. Cowgill U. M. Appl. Spectrosc., 22, 415 (1968).
652. Creger C. R-, Ansari M. N. A., Conch J. R., Colvin L. B. Int. J. Appl. Radiat. and Isotop., 18 71, (1967); РЖБиохим, 1967, 19Ф60.
653. Crouthamel С. E., Fudge A. J. J. Inorg. Nucl. Chem., 5, 240 (1958).
654. Cvjeticanin D., Milic N. Bull. «В. Kidric» Nucl. Sci., 15, 73 (1964); РЖХим, 1965, 23Б446.
655. Cvjeticanin N. J. Chromatogr., 32, 384 (1968).
656. CzibolyC., Zsinka L., Szirtes L. Magy. kern, lapja, 24, 470 (1969).
657. Dalton J. C., Welch G. A. Anal. chim. acta, 15, 317 (1956).
658. Dankwortt P W., Eisenbrand J. Lumineszenz-Analyseimfiltriertenultra-violetten Licht, 7 Aufl. Leipzig, 1964.
659. - Das H. A ., Hoede D., Macke J. F., Zonderhuis J. Radiochem. and Radio-anal. Lett.,4, 171 (1970).
660. D'atta S. K. Z. anal. Chem., 148, 259, 267 (1955).
661. David D. J., Analyst, 87, 576 (1962).
662. David D. J. Analyst, 94, 884 (1969).
663. David D. J. Nature, 187, 1109 (1960).
664. David D. J. Rev. Univ. ind. Santander, 4, 207 (1962).
665. Davis P. S. Nature, 183, 674 (1959).
666. Dawson I. B., Ellis D. I. Spectrochim. acta, A23, 565 (1967).
201
667. Deamer D. W-, MeekD. W., Cornwell D. G. J. Lipid Res., 8, 2551 (1937); РЖХим, 1967, 22Б1003.
668. Debras J., Voinovitch I. A. Chem. anal., 3, 302 (1958).
669. Debras-Guedon J., Voinovitch I. 4. Bull. Soc. franf. ceram., N 46, 75 (1960).
670. Debras-Guedon J., Voinovitch I. A. Compt. rend., 248, 77, 3421 (1959).
671. Dejardin G., Mesnard G., Uzan R., Caband B. Compt. rend., 255, 1712 (1962).
672f Descube J., Rogues N., Roussebet F., Girard M. L. Ann. biol. clin., 25, 1011 (1967).
673. Diamond] J. J. Anal. Chem., 27, 913 (1955).
674. Diaz-Guerra J. P., Capdevila P. C. An. Real. soc. esp. fis. у quim., B64, 937 (1968).; Anal. Abstr., 18, 2060 (1970).
675. Dickinson G. W., Fassel V. A. Anal. Chem., 41, 1021 (1969).
676. Dietze H.-J., Lildke W. Exp. Techn. Phys., 17, 289 (1969).
677. Dijk H., Ijssoling F. P., Loman H. Anal. chim. acta, 27, 563 (1962).
678. Dinnin J. Geol. Surv. Profess. Paper, N 424-D, 392 (1961).
679. Dixon K. Analyst, 83, 362 (1958).
680. Dobrzanski B., Glinski J., Magierski J., Malicki M. Agrochimica, 10, 259 (1966).
681. Dojlido J. Pr. Inst. gosp. wod., 5, 5 (1969); РЖХим, 1969, 24Г154.
682. Dorado E. Rev. plast, 7, 311 (1956);
683. Dorado B., Boucher Y. Farmacognosia, 17, 15 (1957); РЖХим, 1957, 44823.
684. Drekopf K., Winzen W. Brennstoff-Chemie, 38, 208 (1957).
685. Druding L. F. Anal. Chem., 35, 1582 (1963).
686. Dubuouoy C., Guillaumont R., Bouissieres G. Radiochim. acta, 8, 49 (1967).
687. Dupuis М.-C., Dupuis M., Le Nagard MMichot H. Rapp. CEA, N 2712, ; 8 (1965); РЖХим, 1965, 23Б420.
688. Dupuy C., Savoyant L. 3rd. Int. Congr. Atom. Absorpt. and Atom. Fluoresc. Spectrom. (Paris, 1971), v. 1. London, 1973, p. 381; РЖХим, 1974, 13Г172.
689. Diising W., Kunze H. Ъ. anal. Chem., 162, 345 (1958).
690. Duyckaerts G., Lejeune R, J. Chromatogr., 3, 58 (1960).
691. Dybczynski R. Proc. 3rd. Anal. Chem. Conf. Budapest, v. I, 1970, p. 35; РЖХим, 1971, 4Г55.
692. DyckR., Veleker T. J. Anal. Chem., 31, 1640 (1959).
693. DyrssenD. Acta chem. scand., 11, 1277 (1957).
694. Dyrssen D. Acta chem. scand., 15, 1614 (1961).
695. Dyrssen D. Svensk. kem. tidskr., 67, 311 (1955).
696. Dyrssen D., Ekberg S. Acta chem. scand., 13, 1909 (1959).
697. Dyrssen D., LiemD. H. Acta chem. scand., 14, 1100 (1960).
698. Eakins J.D., Gomm P. A. Health Phys., 12, 1557 (1968).
699. Edge R. A., Brooks R- R-, Ahrens L. H., Amdurer S. Geochim. cosmo-chim. acta, 15, 337 (1959).
700. EichoffJ. Appl. Spectrosc. 14, 3 (1960).
701. Elbeih I. I. M., Abon-Elnaga M. A. Anal. chim. acta, 17, 397 (1957).
702. Eljers L. A., Hallbach P. F., Velten R. J. Anal. Chem., 36, 540 (1964).
703. ElvingP.J., Van Atta R.E. Anal. Chem., 22,1375 (1950).
704. Elwell W. T., Gidley J. A. F. Atomic-absorption spectrophotometry. Oxford, Pergamon Press, 1966.
705. Emi K., Toei K., Wada T., Miyata H., Harada K. J. Chem. Soc. Jap., Pure Chem. Sec., 78, 979 (1957).
706. Erlenmeyer H., Hahn H., Sorkin E. Helv. chim. acta, 34, 1419 (1951).
707. Eulitz G. Nukleonik, 2, 85 (1960).
708. Evans G. H., Strain H. H. Anal. Chem., 28, 1560 (1956).
709. Fairbairn H. W., Ahrens L. H., Gorfinkl L. G. Geochim. cosmochim. acta, 3, 34 (1953).
202
710. Fairbairn H. W-, Hurley P. M., P inson W- H. Geochim. cosmochim. acta, 23, 135’ (1961).
711. Fassel V. 4., Kniseley R. N. Anal. Chem., 46, iilOA (1974).
712. Faucherre J. Bull.! Soc. chim. France, 1953, 900.
713. Faure G., Hurley P- M. J. Petrology, 4, 31 (1963).
714. Faure G., Hurley P. M., Fairbairn H. W. J. Geophys. Res., 68, 2323 (1963).
715. Fernando Q. Anal. chim. acta, 12, 432 (1955).
716. Filby R. H., Haller W. 4., Shah K. R. J. Radioanal. Chem., 5, 277 (1970).
717. Firsching F. H. Taianta, 2, 326 (1959).
718. Fishman M. J., Downs S. C. Geol. Surv. Water-Supply Paper, N 1540-C, 23 (1966).
719. Fbldvari-Vogl M., Kliburszky B. Acta geol., 7, 1 (1961).
720. Foreman H., Roberts M. B. Taianta, 9, 559 (1962).
721. Fornaseri M., Grandi L. Geochim. cosmochim. acta, 19, 218 (1960).
722. Fornaseri M., Grandi L. Metallurgia ital., 53, 243 (1961).
723. FonargeJ- Anal. chim. acta, 12, 342 (1955); 18, 225 (1958).
724. Francis С. V. Microchem. J., 7, 375 (1963).
725. Francois M., Jezequel M. Bull, trim., CEBEDEAU, N 50, 269 (1960); РЖХим, 1962, 4И323.
726. Frei R- W., Stillman H. Mikrochim. acta, 1970, 184.
727. Fresenius В Chem. Labor, und Betrieb, 4, 299 (1953).
728. Fresenius R-, Jander G. Handbuch der analytischen Chemie. Teil II. Qualitative Nachweisverfahren, Bd. II. Elementeder Zweiten Gruppe. Berlin. Springer Verlag, 1955, S. 77, 300.
729. Fritz J. S., Garralda В. В. Anal. Chem., 36, 737 (1964).
730. Fritz J. S., Garralda В. B. Taianta, 10, 91 (1963).
731. Fritz J. S., Garralda В. В., Karraker S. K. Anal. Chem., 33, 882 (1961).
732. Fritz J. S., Karraker S. K. Anal. Chem., 32, 957 (1960).
733. Fritz J. S., Peters M. A. Taianta, 16, 575 (1969).
734. Fritz J. S., Sickafoose J. P., Schmitt M- A. Anal. Chem., 41, 1954 (1969).
735. Fritz J. S., Waki H., Garralda В. В. Anal. Chem., 36, 900 (1964).
736. Fuchs H., Schindler R- Z. angew. Geol., 10, 267 (1964).
737. Fujita T., Shigematsu T. Jap. Analyst, 19, 893 (1970); Atom. Absorpt. Abstr. 1970 925.
738. Fujiwara S., Nagashima K. Anal. Chem., 38, 1464 (1966).
739. Fulton 4., Butler L. R- P. Spectrosc. Lett., 1, 317 (1968).
740. Gabis V- Bull. Soc. frang. miner, et cristallogr., 84, 273 (1961); РЖХим, 1962, 15Д98.
741. Gagliardi E., Khadem-Awal M. Mikrochim. acta, 1969, 397.
742. Gagliardi E., Likussar W. Microchim. acta, 1965, 765.
743. Gagliardi E., Raber H. Monats. Chem., 93, 360 (1962).
744. GalleO. K. Appl. Spectrosc., 25, 664 (1971).
745. Gamot E., Philibert J., Vialette Y. Colloq. nat. CNRS, N 923, 278 (1970); РЖХим, 1970, 23Г120.
746. Garda A. Inform. Comis. nac. energ. atom., N 205, 19 (1967); РЖХим, 1968, 21Б722.
747. Garcia A., A gyirre F. Scientia, 28, 22 (1961).
748. Garcia C. A. Rev. Fac. Farmac. у bioquim., 29, 22 (1967).
749. Garcia-RamosL. A., Montagut В. M. Afinidad, 24, 187 (1967), РЖХим, 1968, 8Г72.
750. Gazert F., Potzl K., Reiter R- Atomkernenergie, 7, 106 (1962).
751. Gazith M., Zangen M. Isr. J. Chem., 8, 76 (1970); РЖХим, 1971, 8Г58.
752. Gersonde K. Anal. Biochem., 25, 459 (1968).
753. Gertner A., Kodenja J-, Pavisic D., Grdinic V. Acta pharm. jugosl., 21, N 3, 125 (1971).
754. Getoff N. Osterr. Chem. Ztg, 62, 334 (1961).
755. Getoff N., Bildstein H. Osterr. Chem. Ztg, 66, N 3, 72 (1965).
203
756. GhoseA.K., Dey-Arun К. Analyst, 95, 698 (1970).
757. Chose A. K., Dey-Arun K. Z. anal. Chem., 234, 185 (1968).
758. Girardi F., Pietra R. Anal. Chem., 35, 173 (1963).
759. Girardi F., Pietra R. Anal. chim. acta, 30, 188 (1964).
760. Glascock R. F., Bryant D. T. W., Radioisotopes and Radiat. Dairy ; Sci. and Technol. Vienna, 1966, p. 85.
761. Gloyna E. F., Bhagat S. K., Felsing W. A. J. Water Pollut. Control.
Federat., 35, 893 (1963).
762. Gmelins Handbuch der anorganischen Chemie. System-Nummer 29. Strontium. Erganzungsband. Weinheim, Verlag Chemie, 1960.
763. Gobin R. Vide, 9, 56 (1954).
764. Goddard D. R., Nwankwo S. I. J. Chem. Soc., A, 1371 (1967).
765. Goheen M. W., Robinson R. J. Anal. chim. acta, 30, 234 (1964).
766. Goldin A. S., Velten R. J., Feishkorn G. W. Anal. Chem., 31, 1490 (1959).
767. Goldman M., Anderson R. P. Anal. Chem., 37, 718 (1965).
768. Golightly D. W., Mossotti V. G., Phillips W. C. Spectrosc. Lett., 2, 43 (1969).
769. Gomez Coedo 4., Dorado Lopez M. T., Jimenez Seco J. L. Rev. met. CLNIM, 7, 390 (1971); РЖХим, 1972, 9Г146.
770. Gordon С. M., Larson R. E. Radiochem. and Radioanal. Lett., 5, 369 (1970).
771. Gordon L., Firsching F. H. Anal. Chem., 26, 759 (1954).
772. Gordon L., Reimer C., BurttB. P. Ibid., p. 842.
773. Gorski N. Z. Naturforsch., 24b, 402 (1969).
7, 74. Goto H. Sci. Repts Tohoku Imper. Univ. Ser. I, 29, 204, 287 (1940).
775. Goto H., Suzuki S., Saito M. J. Chem. Soc. Jap., Pure Chem. Sec., 81, 578 (I960); РЖХим, 1961, 4Б295.
776. Goto H., Suzuki S., Saito M. Sci. Rept Res. Inst. Tohoku Univ., A14, 269 (1962); РЖХим, 1964, 14Б386.
777. Gotte H., Petze D. Z. Elektrochem., 58, 636 (1954).
778. Gottschalk G., Dehmel P. Techn.-wiss. Abhandl. Osram-Ges., 8, 37 (1963).
779. Govindaraju K. Colloq. nat. CNRS, N 923, 133 (1970).
780. Govindaraju K. Publ. Groupem. avancem. meth, spectrogr., N 4 319
(1963).
781. Govindaraju K., L'homel N. Atom. Absorpt. Newslett., 11, 115 (1972).
782. Grabowski R. J., Unice R- C. Anal. Chem., 30, 1374 (1958).
783. Graham E. R. Soil Sci., 88, 11 (1959).
784. Greenhalgh R., Riley J. P., Tongudai M. Anal. chim. acta, 36, 439 (1966).
,785. Gregorowiez Z., Olejniczenko M. Chem. anal. (PRL), 13, 877 (1968).
786. Griepink B., Terlouw J. К., Kreuk C. W. Mikrochim. acta, 1965, 1011.
787. Grail P., Grass F., Buchtela K. J. Radioanal. Chem., 1, 179 (1968).
788. Gross D. J. Chromatogr., 10, 221 (1963); Nature, 180, 596 (1957).
789. Guedon J., Debras-Guedon J., Volnovitch J. Chim. anal., 43, 267 (1961).
790. GundlachH. Z. anal. Chem., 171, 9 (1959).
791. Gunn E. L. Anal., Chem., 33, 921 (1961).
792. GunninkR., Cobble J. W- Phys. Rev., 115, 1247 (1959).
793. Gupta В. P., Dutt Yag, Singh R. P. Indian J. Chem., 5, 322 (1967).
794. Gutsche B., Herrmann R. Z. anal. Chem., 269, 260 (1974).
795. Hadzistelios I., Papadopoulou C. Taianta, 16, 337 (1969).
796. Hajjty J. Geol. Surv. Water-Supply Paper, N 150-A, IV, 9 (1960).
797. Hagivara K. Bull. Govt Ind. Res. Inst., Osaka, 17, 184 (1966); РЖХим, 1967, 22Г61.
798. Hahn R. В. J. Chem. Educ., 30, 349 (1953).
799. Hahn R. B., Backer R. O., Nucleonics, 14, 90 (1956).
800. Hahn R. B., Backer C., Backer R- Anal. chim. acta, 9, 223 (1953).
891. Hahn R. B., Joseph P. T., Salciccioli G. G. Taianta, 11, 1073 (1964).
802. Hahn R. В., Straub С. P. J. Amer. Water Works Assoc., 47, 335 (1955).
803. Hahn-Weinheimer P., Ackermann H. Z. anal. Chem., 194, 81 (1963).
804. Haindberger L., SanchezS. Mikrochim. acta, 1962, 760.
805. Hamaguchi H., Ikeda N., Iwasa A. Radioisotopes, 19, 377 (1964).
204
806. Hamaguchi H-, Ikeda N. Kawachima T. Jap. Analyst, 7, 243 (1958): РЖХим, 1959, 14492.
807. Hamaguchi H., Опита N., Watanabe T., Kuroda R. Nature, 211, 1295 (1966).
808. Hantabal E., fcoftik M., Rusek V-, Trnovec T. Chem. zvesti, 18, 203 (1964).
809. Harley J. H. J. Assoc. Offic. Agric. Chem., 46, 371 (1963).
810. Harrison G. E. Nature, 182, 792 (1958).
811. Hartkamp H., Specker H. Z. anal. Chem., 152, 107 (1956).
'812. Harvey С. O., Murray K. Z. H. Analyst, 83, 136 (1958).
813. Harzgorf C. Z. anal. Chem., 262, 167 (1972).
814. Hashmi M. H., Shahid M. A., AyazA. A. Chughtal F. R., Nasin H., Adil A. S. Anal. Chem., 38, 1554 (1966).
815. Haskin L., Vandenbosch R. Phys. Rev., 123, 184 (1961).
816. Hawley J- E., Mac Donald G. Geochim. cosmochim. acta, 10, 197 (1956).
817. Hayatsu A., Farguhar R. M. J. Sci. Instrum., 44, 141 (1967).
818. Headridge J. B., Magee R. J. Taianta, 1, 117 (1958).
819. Hedge С. E„ Walthall F. G. Science, 140, 1214 (1963).
820. Heffernan B. J. Metallurgia, 83, 205 (1971).
821. Heier K. S., Taylor S. R. Geochim. cosmochim. acta, 15, 284 (1959).
822. Heinrich H., Lange J. Fortschr. der Mineral., 50, 35 (1972).
823. Heisig G. B., Pollard F. H. Anal. chim. acta, 16, 234 (1957).
824. Heitner-Wirguin C., Albu A. Taianta, 9, 79 (1962).
825. Henry A. T. Analyst, 89, 255 (1964).
826. Herak M. J., Vuficic N., Jagodic V. Microchim. acta, 1969, 16.
827. Hering R., Heinrich L., Brauer E. Wiss. Z. Padagog. Inst. Gustrow. F. Biol.-Chem.-Polytechn., 4, 53 (1965—1966); РЖХим, 1967, 16Г130.
828. Herr W- Proceedings Symposium on Radioactivation Analysis (Vienna, 1959). London, Butterworth, 1960.
829. Herrmann A. J., Hoffmann R. O. Monatsber. Dtsch. Akad. Wiss. Berlin, 1, 690 (1959).
830. Herrmann G., S trassmann F. Z. Naturforsch., 10a, 146 (1955).
831. Herrmann R., Alkemade C. Th. J. Flammenphotometrie. 2 Aufl. Berlin, Springer, 1960.
832. Hershey H. C., Mitchell R. D., Webb W. H. J. Inorg. Nucl. Chem., 28, 645 (1966).
833. Hertyk W-, Gregorowicz Z. Chem. anal. (PRL), 11, 1105 (1966).
834. Herzog L. F., Pinson W. H., Hurley P. M. Amer. J. Sci., 258, 191 (1960).
835. Hesselbarth H. Z. anal. Chem., 248, 167 (1969).
836. Hetzel H. F. Mikrochim. acta, 1971, 54.
837. Heyden M., Kopfermann H. Z. Phys., 93, 9 (1935).
838. Heyden M., Kopfermann H. Z. Phys., 108, 232 (1938).
839. Heyndryckx P., Gouyon M. A. Meth. phys. anal., 5, 204 (1969).
840. Higuchi H., Tomura K., Takahashi H., Опита N. Anal. chim. acta. 44, 431 (1969).
841. Higuchi H., Tomura K., Takahashi H., Опита N., Hamaguchi H. U. S. Dep. Commer. Nat. Bur. Stand. Spec. Publ., N 312/1, 334 (1969).
842. Hildon M. 4., Sully G. R. Anal. chim. acta, 53, 192 (1971).
843. Hilton D- A., ReedD- Mikrochim. acta, 1966, 508.
844. Hingorani S. B. Chandrasekaran V. R- Proc. Nucl. and Radiat. Chem. Symp. Bombay, 1964, Bombay, 1965, p. 66.
845. Hircq B. 3rd Int. Congr. Atom. Absorp. and Atom. Fluoresc. spectrom. (Paris, 1971), v. 1. London, 1973, p. 199; РЖХим, 1974, 12Г30.
846. Hlasivcova N., Novak J. Silikaty, 13, 157 (1969).
847. Hofer A. Z. anal. Chem., 249, 115 (1970).
848. Hoff R. W., Hollander J. M., Michel M. C. J. Inorg. Nucl. Chem., 18, 1 (1961).
849. Hooper P. R. Anal. Chem., 36, 1271 (1964).
850. Horak J. Scr. Fac. sci. natur. UJEP brun. Chem., 4, 9 (1975).
851. Horak J. Spisy Prirodoved. fak. Univ. Brne, N 1, 19 (1968).
205
852. Horner D.E., Crouse D. J-, Brown К. B., Weaver B. Nucl. Sci. and Eng., 17, 234 (1963).
853. Horr C.A. Geol. Surv. Bull., N 1496-C, I—IV, 33 (1962).
854. Horsford P. M., Ilbery P. L. T. Austral. J. Sci., 23, 298 (1961).
855. Hovorka V., Holzbecher Z. Collect. Czech. Chem. Communs, 14, 186 (1949).
856. Howard J. M. H. Chem. and. Ind., N 17, 688 (1967).
857. Huffman C., Mesik J. D. Appl. Spectrosc., 21, 125 (1967).
858. Hummel R- W., Smales A. A. Analyst, 81, 110 (1956).
859. Hunter G. J., Mitchell V- F. Res. Group. U. K. Atom. Energy Auth., N AERE-AM99 (1964); РЖХим, 1965, 11Б481.
860. Hunter G. J., Perkins M- Res. Group. U. K. Atom. Energy Auth., N AERE-AM3 (1963); РЖХим, 1963, 14Б385.
861. Hybbinette A. Sven. Kem. Tid., 55, 151 (1943).
862. Ibbett R. D. Analyst, 92, 417 (1967).
863. Ichikuni M. Ann. Inst, hydrol. et climatol., 30, 79 (1961).
864. Ikeda N., Akaishi J., Ono Y. Radioisotopes, 8, 233 (1959).
865. Ikeda S. J. Chem. Soc. lap., Pure Chem. Sec., 78, 1428, 1431 (1957); РЖХим, 1958, 81281.
866. Inoue Y. Bull. Chem. Soc. Jap., 36, 1324 (1963); РЖХим, 1964, 14Б839.
867. Intonti R., Stacchini A. Metallurgia ital., 58, 335 (1966).
868. Intonti R-, Stacchini A. Spectrochim. acta, B23, 437 (1968).
869. Irving H., Silva J. J. Chem. Soc., 1963, 945, 1144.
870. Irving H., Stacey M. H. J. Chem. Soc., 1961, 2019.
871. Ishibashi M., Shigematsu T., Ishida T. Bull. Inst. Chem. Res. Kyoto Univ., Suppl. issue, 1954, 60; РЖХим, 1957, 37931.
872. Ishibashi M., Tsutsui T. Jap. Analyst, 5, 437 (1956); Anal. Abstr., 1957, 1455.
873. Ishimori T., Watanade K., Nakamura E. Bull. Chem. Soc. Jap., 33, 636 (1960); РЖХим, 1961, 6Б349.
874. Jackson W- M., Gleason G. I., Hammons P. J. Anal. Chem., 42, 1242 (1970).
875. J ansen G., Richardson G. L., Platt A . M., Bray L. А. Пат. США 3154500 (1964); РЖХим, 1966, ЗЛ24П.
876. Jasinski T., Smagowski H- Chem. anal. (PRL), 14, 829 (1969).
877. Jaskolska H. Chem. anal. (PRL), 14, 285 (1969).
878. JasmundK., Seek H. A. Beitr. Mineral, and Petrogr., 10, 275 (1964).
879. Jeanmaire L., Patti F., BullierD. Rapp. CEA, N 2844 (1965); РЖХим, 1966, 11И215.
880. JeczalikA. Przegl. geol., N 8, 382 (1955).
881. Jedlewska A. Chem. anal. (PRL), 7, 381 (1962).
882. Jervis R. E., Wong К. У. Nucl. Activat. Techn. Life Sci. Vienna, 1967, p. 137.
883. Joensuu O., Suhr N. H. Appl. Spectrosc. 16, 101 (1962).
884. Johnson J. O., Edwards K. W- Geol. Surv. Water-Supply Paper, N 1696-E (1967).
885. Johnson W. C. Anal. Chem., 38, 954 (1966).
886. Jokl V., Undeutsch M., Majer J. J. Chromatogr., 26, 208 (1967).
887. Jones W. F. Mikrochim. acta, 1967, 1004.
888. Jonsson G. AE-Repts, N 163 (1964).
889. Jule H. P. Anal. Chem., 38, 818 (1966).
890. Jury R. V., Webb M. S., Webb R. J. Anal. chim. acta, 22, 145 (1960).
891. Kabasawa У., Tanimura T., Tamura Z. Jap. Analyst, 23, 89 (1974); РЖХим, 1974, 19Г58.
892. Kahn H. Z. J. Metals, 18, 1101 (1966).
893. Kailman S. Anal. Chem., 20, 449 (1948).
894. Kalra A. K. Mikrochim. acta, 1969, 396.
895. Kalvoda R. Anal. chim. acta, 18, 132 (1958).
896. Kar K. R., Singh G., Jain S. C. Mikrochim. acta, 1968, 614.
897. Kaufmann W. Pharmazie, 22, 667 (1967).
206
898. Kawamura S.\ Izawa M. Bull. Chem. Soc. Jap., 39, 624 (1966); РЖХим, 1967, 23Б549. \
899. Kawashima T. Int. J. Appl. Radiat. and Isotop., 20, 806 (1969).
900. Khalifa H. Z. Anal. Chem., 159, 410 (1958).
901. Khalifa H. Z. an»l. Chem., 203, 161 (1964).
902. Khalifa H., Khater M. M. J. Chem. U.A.R., 10, 123 (1967).
903. Khan M. M. J. Phys. Chem., 66, 10 (1962).
904. KhanM. M., TagdiM. A. E. J. Amer., Chem. Soc., 84, 3037 (1962).
905. Kiba T., Konetani M- Bull. Chem. Soc. Jap., 31, 1013 (1958); РЖХим, 1959, 85200.
906. Kiba T., Miura A., Sugioka Y. Bull. Chem. Soc. Jap., 36, 663 (1963); РЖХим, 1964, 2Б326.
907. Kiba T., MizukamiS. Bull. Chem. Soc. Jap., 31, 1007 (1958); РЖХим, 1959 85203
908. Kiba T., Ohashi S., Minabe T. Bull. Chem. Soc. Jap., 28, 443 (1955); РЖХим, 1956, 21899.
909. Kiba T., Ohashi S., Toda S. Bull. Chem. Soc. Jap., 29, 745 (1956); РЖХим, 1957, 50641.
910. Kick H. Z. anal. Chem., 163, 252 (1958).
911. Kieste W., Hecht F. Radiochim. acta, 3, 48 (1964).
912. Kimura K., Bull. Chem. Soc. Jap., 33, 1038 (1960); РЖХим, 1961, 11Б330.
913. Kinsman D. J., Holland H. D. Geochim. cosmochim. acta, 33, 1 (1969).
914. Kiso Y. J. Sci. Hiroshima Univ., Ser. A, Div. 2, 27, 23 (1963); РЖХим, 1965, 9B34.
915. Kleinmann J-, Svoboda V. Anal. Chem., 41, 1029 (1969).
916. Klotzer D., Levi H. W. Radiochim. acta, 6, 81 (1966).
917. Knafft J. Radioisotopy, 7, 131 (1966).
918. Knapstein H. Z. anal. Chem., 1975, 255 (1960).
919. Kobatake T., Iwachido T., T&el K. Taianta, 14, 607 (1967).
920. Koch G. Kerntechnik, 7, 394 (1965).
921. Koch M. Sb. Vysoke skoly zemed. Вгпё, A, 179 (1960).
922. Kocheva L. L., Doychinova V. Докл. Волг. АН, 28, 1649 (1975).
923. Koda Y. J. Inorg. Nucl. Chem., 25, 733 (1963).
924. Koda Y. Radioisotopes, 12, 363 (1963).
925. Kodama H., Brydon J. E., Stone В. C. Geochim. cosmochim. acta, 31, 649 1967).
926. Kohn B. Chem. zvesti, 23, 721 (1970).
927. Koirtyohann S. P., Pickett E. E. Anal. Chem., 38, 585 (1966).
928. Koirtyohann S. P., Pickett E. E. Spectrochim. acta, B23, 673 (1968).
929. KolarikZ.,Hefnd J., Moravec A. J. Inorg. Nucl. Chem., 29, 1279 (1967).
930. Kolarik Z., Pankova H. Collect. Czech. Chem. Communs, 27, 166 (1962).
931. Kolarik Z., Pankova H. Collect. Czech. Chem. Communs, 29, 2023 (1964).
932. Kolthoff I. M., ElvingP. I. Treatise on analytical chemistry, v. 4, N. Y., J. Wiley, 1966, p. 11.
933. KonigK. H., DemelK. J. Chromatogr., 39, 101 (1969).
934. Konopac J. Collect. Czech. Chem. Communs, 30, 2558 (1965).
935. Kooi J. Anal. Chem., 30, 532 (1958).
936. Kooi J- Progr. Nucl. Energy, 1, 351 (1959.)
937. Kopp J. FKroner R. C. Appl. Spectrosc., 19, 155 (1965).
938. KorblJ., Vydra F. Chem. listy, 51, 1457 (1957).
939. Korkisch J., Hazan I. Taianta, 11, 523, 1157 (1964).
940. Koster H. M. Contribs Mineral, and. Petrol., 12, 168 (1966).
941. Kraus K. A., Nelson F. Пат. США 3188169 (1965); РЖХим, 1966, 15Г64.
942. Krause H. Z. anal, chem., 128, 98 (1948).
943. Krepelka J., Kasak F. Jad. energ., 12, 368 (1966); РЖХим, 1967, 6И300.
944. Krleza F. Croat, chem. acta, 30, 231 (1958—1959).
945. Kroner R. C., Kopp J. F- A. Analysis Instrum., v. 4. N. Y., 1967, p. 91.
207.
946. Krtil J. J. Inorg. Nucl. Chem., 22, 247 (1961).
947. Krtil J., Rais J. J. Radional. Chem., 27, 47 (1975).
948. Kundra S. K., Thompson L. C. J. Inorg. Nucl. Chem., 30, 1847 (1968).
949. Kunzendorj H. J. Radioanal. Chem., 9, 311 (1971).
950. Kuroda R., Arino H. Taianta, 11, 343 (1964).
951. Kurodz R., Oquma K. Anal. Chem., 39, 1003 (1967).
952. Kyrs M., Michalukovd V., Benes J., Kadlecovd L. Collect. Czech. Chem. Commons, 34, 1709 (1969).
953. KyrsM., Podesva S. Пат. ЧССР, 104661 (1962); РЖХим, 1964, ЗЛ11П.
954. Lange G., Herrmann G., Strassmann F. I. Inorg. Nucl. Chem., 4, 146 (1957).
955. LaugE. P., Mikalis A., Bollinger H. M., Dinitrojf J. M. J. Assoc. Offic. Agric. Chem., 46, 752 (1963).
956. Laxmi S., Prakash Som, Prakash Satya. Z. phys. Chem. (RRD), 61, 247 (1968).
957. Lazzarini E, Int. J. Appl. Radiat. and Isotop. 16, 443 (1965).
958. Lehn J. M., Sauvage J. P. Chem. Communs, N 9, 440 (1971).
959. Lerner M. W., Reiman W. Anal. chim. acta, 13, 439 (1955).
960. Lesigang-Buchtela M. Mickrochim. acta, 1969, 1027.
961. Lesigang M., Hecht F. Mikrochim. acta, 1964, 508.
962. Levi M. C., Danon J. J. Chromatogr., 6, 269 (1961).
963. Lewandowski 4., Szczepaniak W. Prace Komis, mat-przyrodn. Poznan, towarz. przyjaciol nauk, 12, 3 (1965).
964. Leyden D. E., Whidby I. E. Anal. chim. acta, 42, 271 (1968).
965. Liebman A. M. Anal. Chem., 34, 1370 (1962).
966. Lieser К. H., Bastian J., Hecker А. В. H. Z. anal. Chem., 228, 98 (1967).
967. Lieser К. H., Bastian J., Hecker A. В. H., Hild W. J. Inorg. Nucl. Chem., 29, 815 (1967).
968. Lieser К. H., Bernhard H. Z. anal. Chem., 219, 401 (1966).
969. Lieser К. H., Hiibenthal K. Z. anal. Chem., 207, 5 (1965).
970. Ligten J. W. L., Velthuyzen H. Microchim. acta, 1964, 759.
971. Lingane J. J. J. Electroanal. Chem., 15, 211 (1967).
972. Loley F., Malissa H., Anal. chim. acta, 34, 278 (1966).
973. Lotrian J., Johann in-Gilles A. Spectrochim. acta, B24, 479 (1969).
974. LoveridgeB. A. Atom. Energy Res. EstabL [Rept], N C/M 380, (1959> РЖХим, 1960, 72453.
975. Loreridge B. A., Webster R. K., Morgan J. W., Thomas A. M., Smales A. A. Anal. chim. acta, 23,154 (1960).
976. Luca C., Armeanu St. An. Univ. Bucuresti. Ser. sti. natur. Chim., 14, 121 (1965).
977. Lucchesi C. A. Anal. Chem., 29, 370 (1957).
978. Lucchesi P. J., Lewin S. Z., Vance J. E. Anal. Chem., 26, 521 (1954).
979. LuisP. Mikrochim. acta., 1959, 536.
980. Luis P. Mikrochim. acta, 1961, 529.
981. Luke C. L. Anal. Chem., 28, 1443 (1956).
982. Luke C. L., Campbell M. E. Anal. Chem., 26, 1778 (1954).
983. Lumb R. F., Martell 4. E. J. Phys. Chem., 57, 690 (1953).
984. Lyle S. J., Nair F. C. Taianta, 16, 813 (1969).
985. Macasek F., Cech R. Chem. zvesti, 19, 107 (1965).
986. Mach M. Chem. Prumysl., 8, 303 (1958).
987. Madi J. J. Inorg. Nucl. Chem., 22, 169 (1961—1962).
988. Magee R. J., Headridge J. B. Analyst, 80, 785 (1955).
989. Mageru Y., Weisbuch H., Gradinaru M., Radulescu M., Coman J. An. stiint Univ. Iasi, 11, Ser lb, 121 (1966).
990. Magill W. 4., SvehlaG. Z. anal. Chem., 270, 177 (1974).
991. Magno P. J., Knowles F. E. Anal. Chem., 37, 1112 (1965).
992. Maivicini A., Vido L. Secondo colloq. Franco-Ital. fis. sanitar. Salug-gia, 1961, Roma, 1966, p. 19.
993. Maier J., Dvofakovd E., Nagyovd M. Chem. zvesti, 20, 313 (1966).
994. Major T., Gado P. Magy. kern, folyoirat., 74, 196 (1968).
208
995. Major W. JY e. a. Trans. Amer. Nucl. Soc., 10, 71 (1967).
996. MajumdarS- Л-, De Anil K. Anal. chim. acta, 24, 356 (1961).
997. Majumdar S. Singh В. R. Anal. chim. acta, 20, 275 (1959).
998. Manning D. C., Fernandez F. Atom. Absorpt. Newslett., 9, 65 (1970)-
999. Marchand B. Strahlentherapie, 116, 614 (1961).
1000. Marcu G- Stud. Univ Babes-Bolyai, Ser. chem., 12, 139 (1967).
1001. Marcu G., Botar A. Rev. roum. chim., 12, 645 (1967).
1002. Marcu G-, Botar A. Stud. Univ. Babes-Bolyai. Ser. chem., 12, 11 (1967).
1003. Marcu G., Ghizdavu L. Stud. Univ. Babes-Bolyai. Ser. chem., 14, 39 (1969).
1004. Marcu G., Sacelean V. Stud. Univ. Babes-Bolyai. Ser. chem., 13, 89 (1968).
1005. Marcu G-, Tomus M., Solea M. Ibid., p. 1519.
1006. Marenco A. e. a. Chim. anal., 50, 133 (1968).
1007. Marhol M. Z. anal. Chem., 231, 265 (1967).
1008. Markl P-, Markl I., Hecht F. Mikrocnim. acta, 1968, 1.
1009. Marro F., Pelati L. Rend. 1st. lombardo sci. e lett. Sci. mat., fis., chim. e geol., Л95, 215 (1961); РЖХим, 1963, 24Г81.
1010. Marsh R. H., Hahn R. B. Anal. Chem., 40, 641 (1968).
1011. Martin A., Quintin M. Colloq. nat. CNRS, N 923, 187 (1970); РЖХим,
1970, 23Г138.
1012. Maruta T., Takeuchi T. Jap. Analyst, 22, 602 (1973); Atom. Absorpt. Abstr., 5, 683 (1973).
1013. Massa V., Susplugas P., Salabert J. Trav. Soc. pharm. Montpellier, 34, 175 (1974); РЖХим, 1975, 1Г162.
1014. Massart D. L. J. Radioanal. Chem., 4, 265 (1970).
1015. Mathias 4., Warhurst E. Trans. Faraday Soc., 58, 942, 948,953 (1962).
1016. Matsui M. Bull. Chem. Soc. Jap., 39, 581 (1966); РЖХим, 1967, 23Б551.
1017. Mavrodineanu R., Boiteux H. L’analyse spectrale quantitative par la flamme, Paris, 1954.
1018. MaydanD., ToicherJ., ZeidenbergZ. Isr. Atom. Energy Commis. [Repts], N 619 (1961); РЖХим, 1962, 13Б292.
1019. Mazza L., Sardo L. Gazz. chim. ital., 93, 1155 (1963).
1020. Mazza L., Sardo L., Frache R. Gazz. chim. ital., 95, 599 (1965).
1021. Mazza L., Sardo L., Frache R., Dadone A. Ibid., p. 610.
1022. Me Dowell W- J., Coleman C. F. J. Inorg. Nucl. Chem., 25, 234 (1963).
1023. McDowell W. J-, Coleman C. F. J. Inorg. Nucl. Chem., 27, 1117 (1965).
1024. Me Dowell W. J., Coleman C. F. J. Inorg. Nucl. Chem., 28, 1083 (1966).
1025. McDowell W- J., Harmon H. D. J. Inorg. Nucl. Chem., 31, 1473 (1969).
1026. Me Gee W- W-, Winefordner J. D. Anal. chim. acta, 37, 429 (1967).
1027. Me Intyre R. T. e. a. Anal. Chem., 28, 1316 (1956).
1028. Me Millan J. W- Res. Group U. K. Atom. Energy Auth., N AERE-AM92 (1963).
1029. Me Naughton G. S., Woodward R. N. New Zealand J. Sci., 4, 523 (1961).
1030. Medlint J. H. e. a. Atom. Absorpt. Newslett., 8, 25 (1969).
1031. Mellichamp J. W- Anal. Chem., 26, 977 (1954).
1032. Merritt W. F. Can. J. Chem., 36, 425 (1958).
1033. Michalewska M., Kazimierczyk S. Chem. anal. (PRL), 16, 573 (1971).
1034. Mikulski J., Stronski I. J. Chromatogr., 17, 197 (1965).
1035. Mikulski J., Stronski I. Nukleonika, 7, 769 (1962).
1036. Miller С. C., Magee R. J. I. Chem. Soc., 1951, 3183.
1037. Mills A. A. Can J. Chem., 42, 73 (1964).
1038. Milton G. M., Grummitt W. E. Can. J. Chem., 35, 541 (1957).
1039. Minami E., Honda M., Sasaki Y. Bull. Chem. Soc. Jap., 31, 372 (1958);
РЖХим, 1960, 829.
1040. Miro M., Sutton D. C. Appl. Spectrosc., 24, 220 (1970).
1041. Mirza M. Y. Anal. chim. acta, 40, 229, 235 (1968).
1042. Mirza M. Y. Radiochim. acta (BRD), 8, 216 (1967).
1043. Mirza M. Y. Riso Rept, N 159 (1967); РЖХим, 1968, 12Б511.
1044. Mirza M. Y., Aziz A. Int. J. Appl. Radiat. and Isotop. 20, 135 (1969).
209
1045. Mirza M. У., SaniA. R. Int. J. Appl. Radiat. and Isotop., 18, 437 (1967).
1046. Misumi S., Taketatsu T. J. Inorg. Nucl. Chem., 20,127 (1961).
1047. Miyake У., Saruhashi K., Katsuragi Y., Kanazawa T., Sugimura Y. Recent research Fields of the hydrosphere, atmosphere and nucleare geochemistry. Tokyo, Kenkyusha, 1964, p. 127; РЖХим, 1966, 24И313.
1048. Modreanu F. Naturwissenschaften, 45, 310 (1958).
1049. Modreanu F., Fisel S., Carpov 4. Naturwissenschaften, 44, 615 (1957).
1050. Moenke 4., Moenke-Blankenburg L., Quillfeldt W. Mikrochim. acta, Suppl., 1968, 221.
1051. Mogno У., Kinindai T. C. r. Acad, sci., C267, 1373 (1968).
1052. Moldan B., MikSovsky M. Collect. Czech. Chem. Communs, 36, 1673 (1971).
1053. Molnar F., Horvath A., Khalkin V. A. J. Chromatogr., 26, 215 (1967).
1054. Molt E. L. Chem. weekbl., 60, 245 (1964).
1055. MontfordB., Cribbs S. C. Atom. Absorpt. Newslett., 8, 77 (1969).
1056. Montgomery H. A. C. Analyst, 85, 524 (1960).
1057. Moor С. E., Lalli M. M., Anderson K. L., Brady J. L., Me Lafferty J. J. Anal. chim. acta, 15, 1 (1956).
1058. Moore С. E. Atomic Energy Levels, v. II (24Cr — 41Nb). Washington, CNRS, 1952, p. 189, 467.
1059. Moore F. L. Anal. Chem., 37, 1235 (1965).
1060. Moore L. J., Moody J. R., Barnes I. L., Cramlich J. W-, Murphy T. J., Paulsen P. J., Shields W. R- Anal. Chem., 45, 2384 (1973).
1061. Morgan G. B. e. a. Water Works Eng., 116, 33, 53 (1963).
1062. Moriquchi У., Miyazaki M., Ueno K. Bull. Chem. Soc. Jap., 41, 1344 (1968); РЖХим, 1969, 6B144.
1063. Morrison G. H., KashubaA. T. Anal. Chem., 41, 1842 (1969).
1064. Mossotti V. G., Duggan M. Appl. Optics, 7, 1325 (1968).
1065. Mukherji A. K., Dey 4. K. Chim. anal., 39, 148 (1957).
1066. Murugaiyan P. e. a. Taianta,15, 1119 (1968).
1067. Muzzarelli R. A. 4., Bate L. C. Taianta, 12, 823 (1965).
1068. Myers A. T., Havens R. G., Dunton P. J. Geol. Surv. Bull., N 1084— 1, 207 (1961).
1069. Nagasaki C., T6el K. Jap. Analyst, 21, 87 (1972); РЖХим, 1972, 17Г75.
1070. Nagase У., Matsumoto U., Izumi T. Annu. Proc. Gifu Coll. Pharm., N 7, 53, 69 (1957); РЖХим, 1958, 77177.
1071. Nakanishi S., Miyoshi K. New Nippon Electrotechn. Rev., 6, 103 (1971); РЖХим, 1972, 2Г64.
1072. Nakano S. Bull. Chem. Soc. Jap., 29, 219 (1956); РЖХим, 1957, 19483.
1073. Nakano S. J. Chem. Soc. Jap., 73, 912 (1952).
1074. Ndsanen R., Uusitalo E. Acta chem. Scand, 8, 835 (1954).
1075. Nascutiu T. Stud. .?i cere. chim. Acad. (RSR), 13, 1025 (1965).
1076. Natelson S., Sheid B. Anal. Chem., 33, 396 (1961).
1077. Navrdtil O., Kolafik Z. Collect. Czech. Chem. Communs, 26, 3009 (1961).
1078. Navrdtil O., Kotas J. Collect. Czech. Chem. Communs, 30, 1824 (1965).
1079. Naylor P. M. Develop, and Eng. Group U. K. Atom. Energy Auth. [Rept], N 317(W) (1961); РЖХим, 1962, 9Б336.
1080. Nelson F., Kraus K. J. Amer. Chem. Soc., 77, 801 (1955).
1081. Nelson F., Michelson D. C. J. Chromatogr., 25, 414 (1966).
1082. Nemoda D. S. Bull. Inst. Nucl. Sci., 9, 95 (1959).
1083. Neumann G. M. Taianta, 19, 1202 (1972).
1084. Niepel J. Z. Lebensmitteluntersuch. und Forsch., 128, 144 (1965).
1085. Nordwall H. J., Citteus G. J. Res. Group U. K. Atom. Energy Auth., N AERE-R4493, (1961); РЖХим, 1964, 23Л30.
1086. Nordwall H. J., Healy T. V. Англ. пат. 1079901 (1967); РЖХим, 1968, 17Л8П.
1087. Norwitz G. Analyst, 90, 554 (1965).
1088. Noshkin V. E., Mott N. S. Taianta, 14, 45 (1967).
1089. Novak V. Sb. ved. pr. VSHT Pardubice, 1963, 253.
1090. Novdk J., DobidSovd L. Sklar a keramik, 18, 138 (1968).
210
1091. Novdk V., Dvofdkovd E., Majer J. Chem. zvesti, 23, 161 (1969).
1092. Novak V., Dvofdkovd E., Svicerkovd M., Majer J. Ibid., 330.
1093. Novak V., Tockstein A. Collect. Czech. Chem. Communs, 27, 41 (1962).
1094. Nozaki T. J. Chem. Soc. Jap., Pure Chem. Sec., 80, 1278 (1959); РЖХим, 1960, 73052.
1095. Oesper R. E., Fulmer R. E. Anal. Chem., 25, 908 (1953).
1096. Ogura T., Tarutani T., Misumi S. Mem. Fac. Sci. Kyushu Univ., C5, N 4, 129 (1964).
1097. Okac 4., Pech J. Collect. Czech. Chem. Communs, 13, 400, 514 (1948).
1098. O' Laughlin J. W., Karnin G. J., Berner D. L., Banks С. V. Anal. Chem., 36, 2110 (1964).
1099. Ongaro D., Calapaj G. G. Minerva nucl., 8, 273 (1964).
1100. Orr P. B. Trans. Amer. Nucl. Soc., 8, 334 (1965).
1101. Osmond R. G. D., Healy C., Marshall G. F. Analyst, 86, 616 (1961).
1102. Ostacoli G. e. a. Ric. sci., 38, 318 (1968); РЖХим, 1969, 7B158.
1103. Ott R. Monatsh. Chem., 92, 1 (1961).
1104. Owers M. J. Atom. Energy Res. Establ. [Rept], N R-3010 (1959); РЖХим, 1960, 72427.
1105. Owers M. J., Evett T. W. Analyst, 89, 544 (1964).
1106. Palacios O., Brody T.A., Martinez A.M. Rev. mexicana fis., 8, 27 (1959); РЖХим, 1960, 21399.
1107. Parker A. Chem. Geol., 4, 445 (1969).
1108. Parry E. P., Dollman G. W. Anal. Chem., 36, 1783 (1964).
1109. Parsons M. L., Me Elfresh P. M. Appl. Spectrosc., 26, 472 (1972).
1110. Pascal P. Nouveau traite de chimie minerale, v. 4. Paris, Masson, 1958, p. 546.
1111. Patti F. Bull. Soc. chim. France, 1970, 4145; Anal, abstr., 21, 2752 (1971).
1112. Pattnaik R., Pani S. J. Indian Chem. Soc., 38, 896 (1961).
1113. Pauwels M., Glfbels R., Hoste J. Anal. chim. acta, 36, 210 (1966).
1114. Pauwels M., Gifbels R., Hoste J. Anal. chim. acta, 42, 213 (1968).
1115. Pavlikovd E., Kvapil M., Weiss D. Rudy, 10, N 4 (1962).
1116. Pedersen C. J. Proc. 10th Int. Conf. Coordinat. Chem., Tokyo-Nikko,
1967. Abstrs Papers. Tokyo, Chem. Soc. Jap., 1967, p. 264; РЖХим, 1968, 14B142.
1117. Peltier S., Duval C. Anal. chim. acta, 1, 355 (1947).
1118. Pentscheff N. P., Nonowa D. Chr. Докл. Болгар. АН, 7, № 1, 45 (1954).
1119. Peppard D. F., Mason G. W., McCarty S., Johnson F. D. J. Inorg. Nucl. Chem., 24, 321 (1962).
1120. Peppard D. F. e. a. J. Inorg. Nucl. Chem., 5, 141 (1957).
1121. Perdue H. D., Conover A., Sawley N., Anderson R. Anal. Chem., 40, 1773 (1968).
1122. Pereira de Brito D. Rev. IBPT., N 10, 22 (1958); РЖХим, 1960, 42350.
1123. Peter G. Atompraxis, 13, 116 (1967).
1124. Petho A. Bol. geol. у minero, 80, N 5, 62 (1969).
1125. Petit J. M., Grubis B., Michard J. Bull. Soc. chim. France, 1966, 2717, 2720; РЖБиохим, 1967, 16Ф74.
1126. Petrow H. G. Anal. Chem., 37, 584 (1965).
1127. Petruska J. A. e. a. Can. J. Phys., 33, 640 (1955).
1128. Petruska J. A. e. a. Can. J. Phys., 33, 693 (1955).
1129. Pfeil E. Chem. Labor, und Betrieb, 5, 418 (1954).
1130. Pjeil E., Friedrich A., Wachsmann Th. Z. anal. Chem., 158, 429 (1957).
1131. Pickett E. E., Koirtyohann S. P. Spectrochim. acta, B23, 235 (1968).
1132. Pinson W. H., Herzog L. F., Fairbairn H. W., Cormier R. F. Geochim. cosmochim. acta, 14, 331 (1958).
1133. Pinta M. 17-e Congr. G. A. M. S. Paris, 1954; p. 153; РЖХим, 1957, 69113.
1134. Pinta M., Riandey C. Meth. phys. anal., 5, 76 (1969).
1135. Pivonkovd M., Kyrs M. J. Inorg. Nucl. Chem., 31, 175 (1969).
1136. Plocek L. Sklar a keram., 6, 145 (1956).
211
1137. Plocek L. Ibid., p. 171.
1138. Podobnik B., Dular M., Korosin J. Mikrochim. acta, 1965, 1073.
1139. Podobnik В., Korosin J., Kosta L. Z. anal, chem., 218, 184 (1966).
1140. Polacek J. Symp. pracov. bansk. prflm. Pribram, Sek. N. s. l.,s. a., 1972, p. 203; РЖХим, 1974, 17Г192.
1141. Pollard F. H., Martin J. V. Analyst, 81, 348 (1956).
1142. Pollard F. H., Me Omie J. F. TV., Martin J. V. Ibid., p. 353.
1143. Pollard F. H., Nickless G., Spincer D. J. Chromatogr., 11, 542 (1963).
1144. Pbllmann G. Diss. Dokt. Naturwiss. Fak. allg. wiss. Techn. Univ. Munchen, 1972; РЖХим, 1974, 1Г184.
1145. Poonia N. S. Mikrochim. acta, 1969, 211.
1146. Poonia N. S., Sahni N. S. Indian J. Chem., 5, 203 (1967).
1147. Porter C., Cahill D., Schneider B., Robbins P., Perry W., Kahn B. Anal. Chem., 33, 1306 (1961).
1148. Porter C. R., Kahn B., Carter M. W., Rehnberg G. L., Pepper E. W. Environ. Sci. and Technol., 1, 745 (1967).
1149. Poskanzer A. U., Foreman В. M. J. Inorg. Nucl. Chem., 16, 323 (1961).
1150. Possidoni de Albinati J. F. An. Asoc. quim. argent., 47, 275 (1959).
1151. Possidoni de Albinati J. F., Capaccioli J. H. An. Asoc. quim. argent., 42, 115 (1954).
1152. Povondra P., Sulcek Z. Chem. listy, 56, 918 (1962).
1153. Povondra P., Sulcek Z., Pfibil R., Stangl R. Taianta, 8, 705 (1961).
1154. PraSilovi J., Sebesta F. J. Chromatogr., 14, 555 (1964).
1155. Price N. B., Angell G. R. Anal. Chem., 40, 660 (1968).
1156. Price W. J. Analytical Atomic Absorption Spectrometry. London-New York — Rheine, Heyden, & Son Ltd., 1974.
1157. Priscott В. H. J. Polarogr. Soc., 11, N 2, 18 (1965).
1158. Pro M. J., Mathers 4. P. J. Assoc. Off. Agric. Chem., 39, 225 (1956).
1159. Prod. Group. U. K. Atom. Energy Auth., N 84 (W) (1960); РЖХим, 1961, 6Д89.
1160. Prod. Group U. K. Atom. Energy Auth., N 99 (W) (1960); РЖХим, 1961, 17Б286.
1161. Prod. Group U. K. Atom. Energy Auth., N 155 (W) (1960); РЖХим, 1962, 12Б315.
1162. Prod. Group U. K. Atom. Energy Auth., N 157(S) (1963); РЖХим, 1964, 4Г153.
1163. Prod. Group. U. K. Atom. Energy Auth., N 583(W) (1964); РЖХим, 1965, 2Г83.
1164. Puymbroeck S. van, Borght O. Anal. chim. acta, 57, 441 (1971).
1165. Quentin К. E. Z. Lebensmitteluntersuch. und Forsch., 99, 85 (1954).
1166. Qureshi M., Akthar T. Z. anal. Chem., 227, 89 (1967).
1167. Qureshi M., Israili A. H. Anal. chim. acta, 41, 523 (1968).
1168. Qureshi M., Khan M. A. Taianta, 13, 117 (1966).
1169. Qureshi M., Nabi S. A. J. Chem. Soc., A, 1971, 139.
1170. Qureshi M., Rathore H. C., Kumar R. J. Chem. Soc., A, 1970, 1986.
1171. Raag V., Berlin E. P., Longobucco R. J. Advances Electron Tube Techn.. Oxford—London—New York—Paris, Pergamon Press, 1963, p. 249.
1172. Rains T. C., Zittel H. E., Ferguson M..Anal. Chem., 34, 778 (1962).
1173. Rats J., Rodesva S., Kyrs M. Anal. chim. acta, 36, 90 (1966).
1174. Randa Z. e. a. J. Radioanal. Chem., 14, 437 (1973).
1175. Randow F. Atompraxis, 14, 462, 466, 494 (1968).
1176. Rane A. T., Bhatki K. S. Anal. Chem., 38, 1598 (1966).
1177. Rane A. T., Phatak V.B. Proc. Nucl. and Radiat. Chem. Symp., Poona, s. 1., s. a., 1967, p. 211.
1178. Rann C. S., Hambly A. N. Anal. Chem., 37, 879 (1965).
1179, Rao S. R., Khan 4. 4., Kamath P. R. Proc. Nucl. and Radiat. Chem. Symp., Bombay, 1964, Bombay, 1965, p. 199.
1180. Ray S. В. C., Chauhau U. P..S. Indian J. Chem., 3, 542 (1965).
1181. Rayner L.W. W. Mikrochim. acta, 1970, 193.
1182. Reed L. L., Myers M. N., Sabol W. W. Taianta, 6, 215 (1960).
212
1183. Reedijk J. Recueil trav. chim., 88, 499 (1969).
1184. Reese К. M. Anal. Chem., 42, 26A (1970).
1185. Regan J. G. T., Tyler J. F. C. Analyst, 101, 32 (1976).
1186. Reissig H. Kernenergie, 4, 440 (1961).
1187. Retief D. H., Cleaton-J ones P. E., Turkstra J. De Wet W. J. Arch. Oral. Biol., 16, 1257 (1971); РЖХим, 1972, 7Ф138.
1188. Rhodes D. TV., Me Henry J. R., Amos L. L. Пат. США 3032497 (1962); РЖХим, 1963, 20Л25П.
1189. Rlepe W., Kaiser H. Z. anal. Chem., 223, 321 (1966).
1190. Riley J. R., Tonqudai M. Chem. Geol., 2, 263 (1967).
1191. Ritze F., Kasperek K. Elektromedizina, 12, 236 (1967).
1192. Roberts W. M. B. Nature, 183, 887 (1959).
1193. Roca M., Capdevila C. An. quim. Real. Soc. esp. fis. у quim., 65, 31 (1969).
1194. Rokosz A., Stepak R. Chem. anal. (PRL), 20, 525 (1975).
1195. Rooke J. M., Fisher A. M. Geochim. cosmochim. acta, 26, 335 (1962).
1196. Roos J. T. H., Price W. J. Analyst, 90, 89 (1969).
1197. Rotger K. Z. anal. Chem., 227, 321 (1967).
1198. Rozycki C., Lachowicz E. Chem. anal. (PRL), 15, 255 (1970).
1199. Rubeska I. Collect. Czech. Chem. Communs, 30, 1292 (1965).
1200. Rubeska I., Moldan B. Anal. chim. acta, 37, 421 (1967).
1201. Riidisiile A. Nachweis, Bestimmung und Trennung der chemischen Ele-mente Bd. 6. Bern, Paul Haupt, 1923, S. 728, 740, 741.
1202. Rupp ,4. F. Chemistry, 29, 34 (1956).
1203. Russell D. S. e. a. Anal. chim. acta, 25 , 81 (1961).
1204. Rydberg J. Acta chem. Scand., 14, 157 (1960).
1205. Saas A., Genevey C., Grauby A. Methodes d’extraction de separation et de dosage du strontium dans les sols., Saclay. CEA (France), 1968, p. 13, 61.
1206. Saas A., Grauby A. Colloq. nat. CNRS, N 923, 197 (1970).
1207. Saito N. Jap. Sci. Monthly, 10, 452 (1957); РЖХим, 1958, 60098.
1208. Salomon J., Vance J., Baase W. A. Anal. Chem., 41, 1864 (1969).
1209. Salutsky M. L., Kirby H. W. Anal. Chem.. 27, 567 (1955).
1210. Samsahl K. AE-Repts, AE N 168, 1964; РЖБиохим, 1966, 2Ф93.
1211. Samsahl K. Sci. and Techn. Aerospace Repts, 1, 1151 (1963).
1212. Sanchez В. P. Rapp. CEA, N 3753 (1969).
1213. Sanchez В. P., Burriel M. F. Quim. anal, (pura у apl.), 29, 349 (1975).
1214. Sanchez В. P., May S., Burriel M. F. Inform, quim. anal., 24, 106 (1970).
1215. Sastri V. S., Chakrabarti Ch. L., Willis D. E. Taianta, 16, 1093 (1969).
1216. Scharrer K., Heilenz S. Atompraxis, 5, 170 (1959).
1217. Scheidhauer J., Messaingulral L. Rapp. CEA, N 1701 (1960).
1218. Schmid A., Zipf K. Biochem. Z., 331, 144 (1959).
1219. Schmitt D. H., Fritz J. S. Taianta, 15, 515 (1968).
1220. Schneider R., Santopadre G. Igiene mod., 55, 177 (1962).
1221. Schoenfeld I. Isr. Atom. Energy Commis. |Repts], N 1016, 20 (1965); РЖХим, 1966, ЗЛ31.
1222. Schoenfeld I. Isr. J. Chem., 3, 47 (1965); РЖХим, 1966, 1Г202.
1223. Schow D. M. Bull. Geol. Soc. Amer., 65, 1151 (1954).
1224. Schroeder H. J. Chromatogr., 6, 361 (1961).
1225. Schulte К. E., Henke G., Tfan K. S. Z. anal. Chem., 252, 358 (1970).
1226. Schulz W. W., Mendel J; E., Richardson G. L. Ind. Eng. Chem. Process Design and Develop., 2, 134 (1963).
1227. Schulze W., Scheffler M. Z. anal. Chem., 226, 395 (1967).
1228. Schumacher E., Triedli W. Helv. chim. acta, 43, 1706 (1960).
1229. Schumacher E., Streiff H. J. Helv. chim. acta, 40, 221, 228, 234 (1957).
1229a. Schumann G. Radiostrontium. Symposion des «Sonderausschuss Ra-dioaktivitat». Miinchen, Verlag Gersbach und Sohn, 1961, S. 15.
1230. Schwander H. Schweiz, miner, und petrogr. Mitt., 40, 289 (1960).
1231. Schwarzenbach G., Gysling H. Helv. chim. acta, 32, 1314 (1949).
1232. Seidler H., Hdrtig M. Nahrung, 10, 359 (1966).
213
1233. Seiler H., Artz К., Erlenmeyer II. Helv. chim. acta, 39, 783 (1956).
1234. Seijas-Rivas M. J. An. Fac. farm, у bioquim., 7, 607 (1956).
1235. Sekine T., Komatsu Yu. Jap. Analyst, 24, 94 (1975); РЖХим, 1975, 19Г16.
1236. Senegacnik M., Paljk S. Z. anal. Chem., 224, 375 (1966).
1237. Senegacnik M., Paljk S. Z. anal. Chem., 232, 409 (1967).
1238. Senegacnik M., Paljk S. Z. anal. Chem., 244, 375 (1969).
1239. Senegacnik M., Paljk S., Juznic K. Z. anal. Chem., 233, 81 (1968).
1240. Senegacnik M., Paljk S., Kristan J. Z. anal. Chem., 249, 39 (1970).
1241. Serbanescu A. e. a. Rev. chim. (RSR), 26, 863 (1975).
1242. Seyb К. E., Herrmann G. Z. Elecktrochem., 64, 1065 (1960).
1243. Shaw D. M. Can. Mineral., 6, 467 (1960).
1244. Shima M., Honda M. Geochim. cosmochim. acta, 31, 1995 (1967).
1245. Shiozaki M., Seto Y., Higano R. J. Oceanogr. Soc. Jap 20, 7 (1964)-РЖХим, 1965, 17E128.
1246. Shipman W. H. Anal. Chem., 38, 1175 (1966).
1247. Shukla S. K., Lederer M. J. Chromatogr., 9, 255 (1962).
1248. Sillen L. G., Martell A. E. Stability Constants. (Special Publication N 17 and 25). London—Oxford, Alden Press, 1964 and 1971.
1249. Singhal К. C., Banerjee В. K. Technology (India), 6, 193 (1970).
1250. Singhal К. C. e. a. Technology (India), 5, 117 (1968).
1251. Singhal S. P., Jain B. D. Current Sci., 28, 324 (1959).
1252. Sistkova N. V., Kolafik Z., Barta K., Pankova H. J. Inorg. Nucl Chem 30, 1595 (1968).
1253. Sistkova N. V., Kolarik Z., Chotivka V. J. Inorg. Nucl. Chem 32 637 (1970).
1254. Sistkovd. N. V. e. a. J. Inorg. Nucl. Chem., 31/2911 (1969).
1255. Skafi M., Liser К. H. Z. anal. Chem., 251, 177 (1970).
1256. Skougstad M. W. Geol. Surv. Water-Supply Paper, N 1496-B, 19 (1961)
1257. Skurnik-Sartg S. e. a. Taianta, 16, 1488 (1969).
1258. Slater L. J. inorg. Nucl. Chem., 31, 851 (1969).
1259. Smit J., Robb W. J. Inorg. Nucl. Chem., 26, 509 (1964).
1260. Sommer G. Z. anal. Chem., 151, 336 (1956).
1261. Sorantin H. Mikrochim. acta, 1966, 957.
1262. Souliotis A. G., Belkas E. P., Grimanis A. P. Analyst, 92, 300 (1967).
1263. Stanley C., Kruger P. Nucleonics, 14, 114, 116 (1956).
1264. Starke R. Freiberger. Forschungsh., N 150 (1964).
1265. Starke R. Riihlicke D. Bergakademie, 13, 505 (1961).
1266. Stary J. Anal. chim. acta, 28, 132 (1963).
1267. Steigmann A. J. Soc. Chem. Ind., 66, 353 (1947).
1268. Stein P. C. Anal. Chem., 34, 352 (1962).
1269. Stephens R. Taianta, 20, 765 (1973).
1270. Stephens R., West T. S. Spectrochim. acta, B27, 515 (1972).
1271. Steeppier M. L. anal. Chem., 250, 237 (1970).
1272. Stoeppler M., Cremer M., May K. Radiostrontiumanalytik, I. Abtren-nung und Bestimmung von 89Sr und 80Sr aus Graphitproben und Leach-losungen, Arbeitsvorschriften, Kernforschungsanlage Julich, 1971.
1273. Stoeppler M., May K. J. Radioanal. Chem., 32, 485 (1976).
1274. Stone R. G. Analyst, 88, 56 (1963).
1275. Strajelda F. Collect. Czech. Chem. Communs, 28, 3345 (1963).
1276. Strajelda F. Collect. Czech. Chem. Communs, 30, 2320 (1965).
1277. Strasheim A. e. a. J. S. Afr. Chem. Inst., 20, 25 (1967).
1278. Strelow F. W. E. Anal. Chem., 32, 1185 (1960).
1279. Strelow F. W. E. Anal. Chem., 40, 928 (1968).
1280. Strelow F. W. E., Boshoff M. D. Taianta, 18, 983 (1971).
1281. Strelow F. W. E. e. a. J. S. Afr. Chem. Inst., 21, 93 (1968).
1282. Strelow F. W. E., Weinert С. H. S. Taianta, 17, 1 (1970).
1283. Strelow F. W. E., Zyl C. R., Nolte C. R. Anal. chim. acta, 40, 145 (1968)..
1284. Strong A. B., Rehnberg L. G., Moss U. R. Taianta, 15, 73 (1968).
1285. Stronsky I., Zielinski A. Nucleonika, 9, 801 (1964).
214
1286. Sugihara T. T. е. a. Anal. Chem., 31, 44 (1959).
1287. Sulcek Z., Povondra P., Stangl R. Taianta, 9, 647 (1962).
1288. Sunderman D. N., Townley C. W. The Radiochemistry of Barium, Calcium and Strontium. Nucl, Sci. Ser., U. S. Atom. Energy Commis. Washington, NAS-NS3010, 1960, p. 73.
1289. Sunderman D. N., Meinke W. W. Anal. Chem., 29, 1578 (1957).
1290. Sundkvist G. Acta chem. scand., 15, 1485 (1961).
1291. Surak J. G., Martinovich R. J. J. Chem. Educ., 32, 95 (1955).
1292. Susi6 M. V., Maksimovic Z. B. Bull. «В. Kidric» Inst. Nucl. Sci., 14, 135 (1963).
1293. Suzuki Y. Int. J. Appl. Radiat. and Isotop., 15, 599 (1964).
1294. Suzuki H., Losni E., Oheshi K. J. Chem. Soc. Jap., 13, 132 (1960).
1295. Svoboda V., Chromy V. Taianta, 12, 431 (1965).
1296. Svoboda V., Kleinmann I. Anal. Chem., 40, 1534 (1968).
1297. Swarberg J. E. e. a. Anal. Chem., 42, 1828 (1970).
1298. Szarvas P., Koronddn I., Raisz I. Magy. kem. folyoirat, 72, 441 (1966).
1299. Szmytoivna M., Latour J. Prace Komis, farmac. Poznan towarz. przyja-ciol nauk, 1, 31 (1962).
1300. Taddeussi A., Barbieri M. Metallurgia ital., 58, 281 (1966).
1301. Talmni I. K., Agarwal R. P. Anal. chim. acta, 9, 216 (1953).
1302. Takamoto S. J. Chem. Soc. Jap., Pure Chem. Sec., 81, 915 (1960); РЖХим, 1961, ЗД56.
1303. Tali V. Eesti Pollumaj. Acad, teaduslike toode kogumik, N 9, 176 (1959).
1304. Talvitie N. A., Demint R. J. Anal. Chem., 37, 1605 (1965).
1305. Taussky H. H. Microchem. J., 7, 89 (1963).
1306. Taylor A. E., Paige H. H. Anal. Chem.. 27, 282 (1955).
1307. Taylor R. W., Boulogne A. R. Health Phys., 15, 25 (1968).
1308. Tenoutasse N., De Dander A. Silicates Ind., 37, 13 (1972).
1309. Teree T. M., Cohn S. II. J. Nucl. Med., 7, 848 (1966).
1310 Tesarik B. Chem. prum., 20, 181 (1970).
1311. Testa C., Santori G. Energ. nucl. Milano, 17, 320 (1970).
1312. Theis M. Radex Rdsch., N 1, 333 (1955).
1313. Thompson G., Bankston D. C. Spectrochim. acta, B24, 335 (1969).
1314. Thompson J. K., Wilson C. L. Mikrochim. acta, 1957, 334.
1315. Tillu M, M. Current Sci., 24, 45 (1955).
1316. Timar J. Elelmiszervizsg. kozl., 8, 174 (1962).
1317. Titze II. J. Chromatogr., 20, 193 (1965).
1318. Tockstein A., Novak V. Mikrochim. acta, 1962, 142.
1319. Tdei K., Kobatake T. Taianta, 13, 1194 (1966).
1320. Tompsett S. L. Proc. Assoc. Clin. Biochem., 5, 125 (1968).
1321. Tristram D. R., Phillips C. S. G. J. Chem. Soc., 1955, 580.
1322. Trnka J. Collect. Czech. Chem. Communs, 25, 2232 (1960).
1323. TsubotaH. Bull. Chem. Soc. Jap., 36, 1545 (1963); РЖХим, 1965, 2Б411.
1324. Tsubota II., Kitano Y. Bull. Chem. Soc. Jap., 33, 770 (1960); РЖХим, 1961, 4Д30.
1325. Tsuchitani Y., Tomlto Y., Ueno K. Taianta, 9, 1023 (1962).
1326. Tsuyoshl N., Genkichi N. Jap. Analyst, 16, 1314 (1967); Anal. Abstr., 16, 1118 (1969).
1327. Tucholka-Szmeja B. Chem. anal. (PRL), 1, 255 (1956).
1328. Turekian К. K., Gast P. W., Kulp J. L. Spectrochim. Acta, 9, 40 (1957).
1329. Turekian К. K., Wedepohl К. H. Bull. Geol. Soc. Amer., 72, 175 (1961).
1330. Turekian К. K. e. a. Antarct. Journal US, 2, 186 (1967).
1331. Ullmann J. Spisy pfirodoved. fak. Univ. Brne, N 1, 45 (1968).
1332. Uman G. A. Anal. Chem., 37, 287 (1965).
1333. Umland F., Meckenstock K.-U. Z. anal. Chem., 165 161 (1959).
1334. Umland F., Meckenstock K.-U. Z. anal. Chem., 176, 96 (1960).
1335. Uzumasa Y., Nasu Y., Seo T. J. Chem. Soc. Jap., Pure Chem. Sec., 81, 430 (1960); РЖХим, 1960, 84418.
1336. Vafnstein E. E., Lebedev V. I. Chim. anal., 43, 398 (1961).
1337. Vancompernalle G. Pedologic, 11, 277 (1961).
215
1338. Van Uitert Le Grand G., Fernelius W. C., Douglas В. E. J. Amer. Chem. Soc., 75, 2736 (1953).
1339. Vaughan A., Townshend A. Anal. chim. acta, 43, 134 (1968).
1340. Veal D. J., Armstrong F. E. Trans. Amer. Nucl. Soc., 11, 95 (1968).
1341. Verbeek F., Thun H. Anal. chim. acta, 33, 378 (1965).
1342. Visapda A. Tekn. kem. aikakauslehti, 20, 146 (1963).
1343. Vlacil F. Collect. Czech. Chem. Communs, 26, 658 (1961).
1344. Vitek B„ Reif 4. Atompraxis, 11, 369 (1965).
1345. Voinovltch I. A., Vilnat J. Publ. groupem. avancem. meth, spectrogr., 8, 11 (1961).
1346. Vorlicek J. Rudy, 15, 373 (1967).
1347. Vorlicek J., Vydra F. Collect. Czech. Chem. Communs, 31,2510 (1966).
1348. Vries G. e. a. Anal. chim. acta, 21, 568 (1959).
1349. Wald M., Schonfeld. T. Monatsch. Chem., 90, 189 (1959).
1350. Wdnke U. 16 Colloq. Spectrosc. Int. Heidelberg, 1971. Hauptvortr. und Ber., London, 1972, S. 232; РЖХим, 1974, 13Г176.
1351. Wdnke H., Konig H. Z. Nalurforsch., 14a, 929 (1959).
1352. Warren J. M., Spencer H. Clin. chim. acta, 38, 435 (1972); РЖХим, 1972, 22Ф110.
1353. Wearer B. J. Inorg. Nucl. Chem., 30, 2233 (1968).
1354. Webb M. S. Atom. Energy Res. Establ. [Rept], N AM15 (1959).
1355. Webb W. H., Hershey H. C., Mitchell R. D. Пат. США 3381164 (1968); РЖХим, 1969, 16Л215.
1356. Wedepohl К. H. Z. anal. Chem., 180, 246 (1961).
1357. Weis H. V., Shipman W. H. Anal. Chem., 29, 1764 (1957).
1358. Wetsz H., Klockow D., Fritsche U. Z. anal. Chem., 244, 289 (1969).
1359. Welford G. A., Chiotis E. L. Anal. chim. acta., 31, 376 (1964).
1360. Welford G. A. e. a. Taianta, 5, 168 (1960).
1361. Wendlandt W. W., Iftikhar All S., Stembridge С. H. Anal. chim. acta, 31, 501 (1964).
1362. Wenger P., Cassimatis D. Helv. chim. acta, 45 , 783 (1962).
1363. Wenger P. E. e. a. Pharm. acta helv., 37, 472 (1962).
1364. Werner G., Hanning R., Dedek W., Holzapfel H. Proc. 3rd. Anal. Chem. Conf., Budapest, 1970, v. 1. 1970, p. 75.
1365. West P. W., Mukherfi A. K. Anal. Chem., 31, 947 (1959).
1366. Wheelwright E. J., Bray L. A., Roberts F. P., Moore R. L. Пат. США 3173757 (1965); РЖХим, 1966, 16Л28П.
1367. Whitehead A. В., Heady H. H. Appl. Spectrosc., 22, 7 (1968).
1368. Wiles D. M., Tomlinson R. H. Can. J. Phys., 33, 133 (1955).
1369. Wilkins D. H. Taianta, 2, 277 (1959).
1370. Wilkinson P. R. e. a. Anal. Chem., 26, 767 (1954).
1371. Willard H. H., Goodspeed E. W. Ind. and Eng. Chem., Anal. Ed., 8, 414 (1936).
1372. Willis J. B. e. a. Spectrochim. acta, B24, 157 (1969).
1373. Winkler R., Sansoni B., Starke K. Chem. Zvesti, 21, 571 (1967).
1374. Wish L. Anal. Chem., 33, 53 (1961).
1375. Witzmann H., Oeser W., Piesche L. Reinstoffe Wissenschaft und Technik. Berlin, 1963, S. 199; РЖХим, 1964, 5B16.
1376. Wolter R., Bahn U. Schriftenr. Ver. Wasser-Boden und Lufthyg. Ber-lin/Dahlem, N 33, 93 (1970).
1377. Woodward C. Atom. Absorpt. Newslett., 8, 121 (1969).
1378. Yofe J,, Avni R., Stiller M. Anal. chim. acta, 28, 331 (1963).
1379. Yusaf M. e. a. Collect. Czech. Chem. Communs, 40, 3645 (1975).
1380. Zaremba J. Chem. anal. (PRL), 7, 244 (1962).
1381. Zeigler M., Rittner W. Z. anal. Chem., 211, 170 (1965).
1382. Zelinkova M. Ceskosl. biol., 2, 110 (1953).
1383. Zeltmeisl M. J., Haworth D. T. J. Chromatogr., 30, 637 (1967).
1384. Zenkt M. Anal. chim. acta, 83, 267 (1976).
1385. Zima S., Giacintov P. J. Radioanal. Chem., 7, 19 (1971).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азо-азокси БН 55
Азо-азокси ФМП 55
Азокрасители как реагенты на стронций 21, 22
Активационный метод ИЗ
определение стронция в
биологических материалах 114, 116
воде ИЗ, 115, 116 кристаллах галогенидов щелочных металлов 114
Ализарин S 26
Алкалиметрический метод определения стронция 84
Алюминия окись 73
Амперометрическое титрование 87
Арсенатно-иодометрический метод определения стронция 84
Атомно-абсорбционная спектрофотометрия 108
в анализе алюминиевых сплавов 134 биологических материалов 133 вод 125, 126 минералов, горных пород, руд 127, 131
солей и других соединений 136, 137, 145
стекла 137, 142
цемента 137, 141
используемые пламена 108 метод импульсного испарения 110 устранение помех 109
2,7-бис-Азопроизводные хромот-
роповой кислоты 21, 22
1,5-5пс-(ДикарбоксиметиламинОме-тил)-2,6-диоксинафталин 27, 28
3,5-бис-(Дикарбоксиметиламино-метил) -4,4' -диокси-7н/>анс-сти ль-бен 27, 28
3,3'-бис-(2-Окси-3-карбоксинафтила-зо)-фенолфталеин 22
3,3'-бис-(Хромотропазо)фенолфтале-ин 22
Виолуровая кислота 27, 70
Вода
содержание стронция 9 определение стронция 122
Галогениды стронция, анализ 144
Горные породы, содержание стронция 9
Гравиметрические методы определения стронция 79
в виде
карбоната 81 оксалата 82 сульфата, метод возникающих реагентов 80, 81
тартрата 82
в водах 124 минералах 127 рассолах и рапе нефтяных промыслов 124
окислах металлов 136 рудах 127
2,2'-Диаминодиэтиловый эфир 17
Диацетил оксимсалицилальгидразон 18
Диацетилоксимсалицилоилгидразон
Дибензоилметан 17, 18
5,7-Дибром-8-оксихинолин 17, 18,
26,153
Ди-н-бутилфосфорная кислота 51
Дигексиловый эфир фенилсульфо-ниламидофосфорной кислоты 53
3,5-Диметилпикриновая кислота 20
Диметилсульфоназо III 89
Динонилнафталинсульфокислота 59
Ди-2-этилгексилфосфорная кислота 51, 73, 74
fJ-Изопропилтрополон 51
Изотопного состава стронция определение
спектральное НО масс-спектральное 117
Изотопы стронция 7,8, 158
Инверсионная полярография 95
Ионообменное отделение стронция использование растворов комплексообразующих веществ 64 неорганические ионообменные материалы 67
применение органических растворителей 63
с помощью
217
катионитов 61
анионитов 65
Карбоксинитразо 91
Качественные реакции 19
микрокристаллоскопические 28, 29 осаждения в водных растворах с реагентами
3,5-диметилпикриновой кислотой 20
карбонатами 19, 20
2-окси-4-нитрохалконом 20 оксалатами 20
органическими реагентами 20, 21 пикролоновой кислотой 20 сульфатами 20 фосфатами 20 хроматами 20
флуоресцентные с
1,5-бис-(дикарбоксиметилами-нометил)-2,6-диоксинафталином 27
3,5'-бис-(дикарбоксиметилами-нометил)-4,4'-диокси-транс-стильбеном 27
1-ди-(карбоксиметил)-аминоме-тил-2-окси-З-нафтойной кислотой 27
куркумином 27, 28
морином 27, 28 флуорексоном 27, 28 цветные с
2,7-бис-азопроизводными хромотроповой кислоты 21, 23
3,3'-бис-(2-окси-3-карбоксинаф-тилазо)-фенолфталеином 22
о-крезолфталеинкомплексоном
мурексидом 22
2-оксинафталин (1-'азо-2)-нафта-лин-1-сульфокислотой 22
родизонатом натрия 22
9,10-фенантренхинон-2-сульфо-кислотой 22
эриохром черным Т 22, 25
цветные реакции с использованием экстракции органическими растворителями 26
Карбонат стронция 19, 20, 35
Кверцетин 26
Комплексные соединения стронция 17
Кондуктометрическое титрование 87 Коэффициенты распределения щелочноземельных металлов между ионообменником и элюентом 61, 68
о-Крезолфталеинкомплексон 22, 24
Купферон 54
Куркумин 28
218
Литосфера, содержание стронция 9-Локализация зон стронция на хроматограмме 70 электрофореграмме 75
Масс-спектрометрия 117 искровые источники 117 метод изотопного разбавления 118-наложение линий ионов 119
8-Меркаптохинолин 59
Металлический стронций
анализ на примеси 143, 144, 149 получение 11
Метилизобутилкетон 50
Метод изотопного разбавления 118 определение кислорода во фториде стронция 144 инфракрасной спектроскопии для определения нитрата и сульфата стронция в смеси с соответствующими солями бария 93
Минералы стронция 10
Молибдат циркония 68
Морин 27, 28
Мурексид 22, 92
Нафтеновые кислоты 58
Нитробензол 49
Нитрохромазо 22, 88
Обнаружение
зон стронция на бумажной хроматограмме 27, 70
по окрашиванию пламени 28 с помощью метода дифференциального термического анализа 30
1-Оксиантрахинон 26, 54
2-Оксинафталин-(Г-азо-2)-нафталин-
1-сульфокислота 22
2-Окси-4'-нитрохалкон 20
8-Оксихинолин 53, 92
Определение
бария в смешанных титанатах бария и стронция 156
воды в иодиде стронция 147 европия в иодиде стронция 153 кадмия в смешанных титанатах ба-
рия и стронция 152
кислорода в
металлическом стронции 143
фториде стронция 147
нитрата в смеси карбонатов кальция, стронция и бария 148
основного|вещества в иодиде стронция 154
примеси металлов в стронции и его соединениях 144, 149
примеси неметаллов в стронции и его препаратах 144 радиоактивного стронция 157
в биологических материалах 174 водах 168 воздухе, осадках 171 почвах 172
продуктах деления и облученных препаратах 179 составляющих в ниобатах кальция, стронция, бария 155 стронция в
биологических материалах 133 водах 122 внутрикомплексных соединениях 156
горных породах, рудах, минералах 126
двуокиси марганца 136, 137 закиси-окиси урана 138 металлах и сплавах 134 окиси алюминия 137 окиси магния 138 окислах металлов 137 органических соединениях и материалах 137 перекиси бария 139 почвах 126 солях и других соединениях металлов 135 стеклах 142 трехокиси вольфрама 137 уране и его соединениях 135 цементах 141
Определение стронция методами активационным ИЗ атомно-абсорбционным 108 гравиметрическим в виде карбоната 81 оксалата 82 сульфата 80 тартрата 82 масс-спектральным 117 полярографическим и другими электрохимическими 94 спектрографическим 95, 110 спектрофотометрическим с диметилсульфоназо III 89 карбоксинитразо 91 о-крезолфталеинкомплексоном 91 мурексидом92 8-оксихинолином 92 родизоновой кислотой 93 хлорфосфоназо III 90 эриохром серым 3BL-cynpa 92 титриметрическим 83 титрования амперометрического 87 комплексонометрического 86,156
кондуктометрического 87 спектрополяриметрического 88 спектрофотометрического 86
эмиссионным пламеннофотометрическим 103
сцинтилляционным 129 Ортаниловый С 88 Отделение стронция методами
извлечения нитрата органическими растворителями 33
ацетоном 33
этанолом 34
этанолом в смеси с эфиром 33 осаждения в виде
карбоната 35
нитрата 31
оксалата 38
оксалата в присутствии этанола 39
родизоната 40
сульфата 36
фосфата 40
хромата 39
осаждения посторонних элементов 42, 43
бария в виде хлорида и сульфата 43, 44
бария в виде хромата 44
кальция в виде ферроцианида 46
соосаждения с
гидроокисью железа 41 гидроокисью титана 42 двуокисью марганца 42 нитратом бария 34 нитратом свинца 34 оксалатом кальция 39 сульфатом бария 38 сульфатом свинца 38
фокусирующего ионного обмена 76 хроматографии
ионообменной 60
с помощью катионинов 61 анионитов 65
распределительной 69 на бумаге 69
тонкослойной 72
с обращенными фазами 73 экстракции 47
алкилфосфорными кислотами 51
в виде р-дикетонатов 50 иодида 47 перхлората 47 полииодида 49 роданида 47 купфероната 54
нафтеновыми кислотами 58 8-оксихинолината 53
219
посторонних элементов 59 рутения 59 сурьмы 60 тория 59
трибутилфосфатом 54, 55 экстрагентом АТ 55
АФТ 57
БТ-1 58
электродиализа 78 электрофореза 74, 167 Отделение стронция
от бария осаждением
хлорида и сульфата бария 43 хромата бария 44, 45, 46
от кальция
ионообменнойхроматографиейЭО в виде
карбоната 35
нитрата 31
в присутствии ацетона 33 этанола 34 оксалата 38 родизоната 40 сульфата 36 хромата 39
осаждением ферроцианида кальция 46
Отделение 80Sr от 80Y методами ионного обмена 165 тонкослойной хроматографии 167 экстракции 160 электрофореза 167
Перекись стронция 15
Переработка стронциевых руд 10 Пикролоновая кислота 20 Пирогаллол 22, 26
Пламеннофотометрический метод определения стронция в
водах 124
горных породах и минералах 127, 129
кальций-стронций-бариевом нио-бате 155
почвах 127
рудах 127
уране 135
Подвижность ионов щелочноземельных металлов на бумаге 74
Полярографический метод 94 инверсионный с накоплением 95 потенциал полуволны восстановления 94
Применение стронция 12 Продукты деления 179
Разнолигандный комплекс стронция с 8-оксихинолином и 1-оксиантрахиноном 53, 54
Распределительная хроматография 69 колоночная 73 на бумаге 69 с обращенными фазами 73 тонкослойная 72 сорбенты 73
Распространение в природе 7, 9 Рентгенофлуоресцентный метод влияние состава образца 121 определение стронция в биологических материалах 134 водах 126 осадочных породах 127, 132 стронций-бариевых титанатах 154
характеристика рентгеновского излучения стронция 120
Родамин С 26
Родизоновая кислота 25, 93 Рубидиево-стронциевый метод определения возраста горных пород 117
Свойства стронция 13
Силикагель 73
Содержание стронция в воде 8, 9 глубоководных осадках 9 земной коре 8 литосфере 9 лунном грунте 9
Соединения стронция 14
Спектральное определение изотопного состава ПО
Спектрографический эмиссионный (дуговой и искровой) метод определения стронция в водах 123, 124 горных породах, минералах 127 металлическом молибдене 134 стронций-кальций-висмутовом титанате 154 фториде стронция 151
Спектрофотометрический метод 88 определение стронция в водах 122, 124 почвах 127 рудах 127 хлориде бария 136
Теноилтрифторацетон 48, 50 Титриметрические методы 83 арсенатно-иодометрический 84 комплексонометрический 84, 85 кондуктометрический 84, 87 оксалатный 83, 84 потенциометрический 86 спектрофотометрический 85
Тонкослойная хроматография 72 9,10-Фенантренхинон-2-сульфокис-лота 22
220
Фенилантраниловая кислота 26 1-Фенил-3-метил-4-бензоилпиразо-лон-5 50
1 -Фе ни л-З-метил-4-ка пр и лпира зо-лон-5 50
Фенолфталеина производные 91
Ферроцианид калия 46
Физические и химические свойства стронция 13
Флуорексон 27
Флуоресцентные реакции 27, 28
Фотометрические методы с
диметилсульфоназо III 89
карбоксинитразо 91
мурексидом 92
нитроортаниловым С 88
производными фенолфталеина 91 родизоновой кислотой 93 хлорфосфоназо III 90
Хелант БТ 58
Хлорфосфоназо III 90, 127
Хроматография на бумаге 69
Цветные реакции стронция 21
Целлюлоза 73
Циркония молибдат 69
Циркония фосфат 69
Экстрагент АТ 55,57
Экстрагент АФТ 55, 57
Экстрагент БТ-1 58
Электродиализ 78
Электролиз 78
Электрофорез 74
локализация зон стронция 75
электролиты для отделения стронция от других элементов 76
Эмиссионный спектрографический
метод 95
буферные смеси 99
возможные помехи 100
линии спектра 98
определение в
водах 123
горных породах 127,128
других объектах 136—139
с предварительным химическим обогащением 149
с применением автоматического фотометрирования 151
Эмиссионная фотометрия пламени 103
анионный эффект 105
влияние катионов металлов 106
влияние состава раствора 104
ионизация атомов стронция в пламени 107
применение в анализе вод 124, 125
горных пород 127,129
других объектов 136—140
Эриохром черный Т 25
ОГЛАВЛЕНИЕ
От редколлегии ................................................. 3
Предисловие..................................................... 5
Глава I
Физико-химическая и химико-аналитическая характеристики стронция и его соединений.................................. ............. 7
Общие сведения ................................................. 7
Краткие исторические данные.................................. 7
Нахождение стронция в природе............................. 7
Способы переработки минералов.............................. 10
Получение стронция ..................................... И
Применение стронция........................................ 12
Физические и химические свойства стронция........................ 13
Соединения стронция.............................................. 14
Неорганические соединения стронция......................... 14
Органические соединения стронция........................... 17
Глава II
Качественное обнаружение стронция............................ 19
Реакции осаждения.............................................. 19
Цветные реакции................................................ 21
Реакции с использованием экстракции органическими растворителями ...................................................... 26
Флуоресцентные реакции..................................... . 27
Микрокристаллоскопические реакции.............................. 28
Окрашивание пламени........................................... 28
Другие методы.................................................. 30
Глава III
Методы отделения стронция от сопутствующих элементов........... 31
Методы осаждения ............................................. 31
Методы, основанные на осаждении стронция................... 31
Методы, основанные на осаждении посторонних элементов ... 42
Экстракционные методы.......................................... 47
Методы, основанные на экстракции стронция.................. 47
Методы, основанные на экстракции посторонних элементов . . 59
Хроматографические методы...................................... 60
Ионообменная хроматография................................. 60
Распределительная хроматография............................ 69
222
Электрофорез ................................................... '4
Электродиализ .................................................. 78
Электролиз .................................................... 78
Глава IV
Количественное определение стронция............................ 79
Химические методы анализа...................................... 79
Гравиметрические методы..................................... 79
Титриметрические методы.................................... 83
Физико-химические методы анализа............................... 88
Спектрофотометрические методы.............................. 88
Электрохимические методы................................... 94
Физические методы анализа...................................... 95
Спектральные методы........................................ 95
Активационный метод........................................ И8
Масс-спектрометрический метод............................... И7
Рентгенофлуоресцентный метод................................ И9
Глава V
Определение стронция в природных и промышленных объектах . . . 122
Анализ вод...................................................... 122
Анализ минералов, горных пород, руд и почв...................... 126
Анализ биологических материалов............................... 133
Анализ металлов и сплавов..................................... 134
Анализ других объектов........................................ 135
Глава VI
Анализ стронция и его соединений.............................. 143
Определение примеси элементов в стронции и его соединениях . . . 143
Определение неметаллов.................................... 143
Определение металлов...................................... 149
Анализ препаратов, содержащих стронций........................ 153
Глава VII
Определение радиоактивного стронция........................... 157
Общие сведения................................................ 157
Отделение 90Y от 90Sr......................................... 160
Экстракционные методы..................................... 160
Хроматографические методы................................. 165
Электрофоретические и другие методы....................... 167
Определение стронция в различных объектах..................... 168
Анализ вод................................................ 168
Анализ воздуха, осадков .................................. 171
Анализ почв............................................... 172
Анализ биологических материалов........................... 174
Анализ продуктов деления и облученных препаратов.......... 179
Литература................................................ 182
Предметный указатель.......................................... 217
Аналитическая химия стронция. Полуэктов Н. С., Мищев. и о В. Т., Кононенко Л. И., Бельтюкова С. В. М., «Наука», 1978, стр. 223.
В книге рассматриваются основные химические свойства стронция, методы его идентификации, отделения и химические, физические и физико-химические методы определения, в том числе в различных природных и промышленных материалах. Обсуждены вопросы определения радиоактивного стронция.
Книга рассчитана на широкий круг химиков-аналитиков и научных работников, а также на преподавателей, аспирантов и студентов жузов.
Таблиц 51. Библ. 1385 назв.
Николай Сергеевич Полуэктов, Валентин Трофимович Мищенко, Людмила Ивановна Кононенко, Светлана Вадимовна Бельтюкова
Аналитическая химия стронция
Серия: «Аналитическая химия элементов»
Утверждено к печати ордена Ленина Институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор Л. В. Рузайкина
Редакторы издательства Р. А. Баранова, Л. Г. Чекалова Художественный редактор С. А. Литвак
Технический редактор П. С. Лашина Корректоры М. М. Баранова, Л. И.Харитонова
ИБ № 7001
Сдано в набор 16.05.78.
Подписано к печати 12.12.78.
Т-18173. Формат 60х90>/1»
Бумага типографская № 1
Гарнитура обыкновенная Печать высокая
Усл. печ. л. 14. Уч.-изд. л. 15,3
Тираж 2200 экз. Тип. зак. 506 Цена 1 р. 90 к.
Издательство «Наука»
117485, Москва, В-485, Профсоюзная ул., 94а 2-я типография издательства «Наука»
121099, Москва, Г-99, Шубинсний пер., 10