/






















































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Text
Кт)
и
термостойки*
полимеры
КгУ. Бюллер
Тепло-
и „
термостойкие
полимеры
Перевод с немецкого
Н. В. АФАНАСЬЕВА и Г. М. ЦЕЙТЛИНА
Под редакцией
Я. С. ВЫГОДСКОГО
МОСКВА «ХИМИЯ» 1984
Spezialplaste
von
DR. KONRAD ULRICH BUHLER
Leipzig
AKADEMIE-VERLAG • BERLIN
1978
6П7.55
Б 982
УДК 678
Бюллер К.-У.
Тепло- и термостойкие полимеры; Пер. с нем./
Под ред. Я. С. Выгодского. — М.: Химия, 1984 —
1056 с, ил.
Монография охватывает широкий круг вопросов, касающихся
синтеза, строения, структуры, свойств и применения тепло- и термостойких
полимеров с высокими физнко-механическими, электроизоляционными и
оптическими характеристиками. В ней рассмотрено более 150 классов
алифатических, циклоалифатических, ароматических, карбо- и
гетероциклических полимеров. Содержится обширный справочный материал.
Монография предназначена для инженерно-технических и научных
работников, занятых получением, переработкой и применением
полимерных материалов, а также преподавателей, аспирантов и студентов
химико технологических вузов.
1056 с, 143 табл., 301 рис., 2855 литературных ссылок.
_ 2803090000-096 псал
Б 050 @1)-84 %-84
© Akademie-Verlag Berlin, I978
Издательство «Химия>, 1984 г.
СОДЕРЖАНИЕ
Предисловие к русскому изданию 14
Предисловие к немецкому изданию . . - - 15
1. Перспективы развития производства пластмасс 16
2. Строение и особенности структуры полимеров с повышенной
термостойкостью ¦. 24
2.1. Определение понятий «теплостойкость» и «термостойкость» ... 24
2.2. Теплостойкость 25
2.2.1. Температура плавления 25
2.2.2. Температура стеклования 30
2.3. Термостойкость 31
3. Методы определения термостойкости полимеров .40
3.1. Экспресс-методы 41
3.1.1, Методы изучения химических процессов, протекающих при
деструкции полимеров 41
.1.1. Термогравиметрический анализ 41
Л.2. Изотеннскопия 42
.1.3. Поглощение кислорода 42
3.1.2. Методы определения температур плавления и стеклования . , . . 43
.2.1. Дифференциально-термический анализ 43
.2.2. Определение температуры стеклования 44
.2.3. Определение температуры размягчения 44
3.
3.
3.
3.
3.
3-
3.2. Длительные испытания полимеров на термостойкость 45
Литература 46
4. Термостойкие полиолефины и фторсодержащие полиолефины . .... 46
4. К Полибутен-1 46
4.1.1, Получение и свойства бутена-1 47
4.1.2. Стереоспецифическая полимеризация бутена-1 49
4Л.З. Физико-химические свойства полибутена-1 . " 51
4.1.3.1. Структура 51
4.1.3.2. Кристалличность 52
4.1.3.3. Свойства растворов 55
4.1.4. Переработка изотактического полнбутена-1 56
4.1.5. Эксплуатационные свойства изотактического полнбутена-1 . . . . 58
4.1.5.1. Теплофизические свойства 58
4.1.5.2. Физико-механические свойства 59
4.1.5.3. Электрические свойства -ч .... 62
4.1.5.4. Химическая стойкость и коррозионное растрескивание 62
4.1.5.5. Токсичность 63
4.1.6. Основные области применения полибутена-1 63
4.1.7. Модификация полибутена-1 65
4.1.8. Сополимеризация и привитая сополимеризация 65
4.1.9. Воздействие ионизирующего излучения 66
4.2. Полп-4-метилпентен-1 66
4.2.1. 4-Метилпентен-1 66
4.2.2. Полимеризация 4-метилпентена-1 68
4.2.3. Физико-химические свойства поли-4-метилпентена-1 71
4.2.3.1. Структура 71
4.2.3.2. Растворимость 71
4.2.3.3. Термостойкость 71
4.2.3.4. Радиационная стойкость 72
4.2.4. Переработка поли-4-метилпентена-1 73
4.2.4.1. Литье под давлением 73
4.2.4.2. Экструзия 74
4.2.4.3. Формование с раздувом 75
4.2.4.4. Пленки и пенопласты • 75
4.2.4.5. Прессование - • 75
4.2.4.6. Термоформование пленок и листов 75
4.2.4.7. Поверхностная обработка изделий 76
4.2.5. Эксплуатационные свойства поли-4-метилпентена-1 76
4.2.5.1. Теплофнзические свойства 76
4.2.5.2. Физико-механические свойства 77
4.2.5.3. Электрические свойства 78
4.2.5.4. Оптические свойства 78
4.2.5.5. Химическая стойкость , .... 79
4.2.5.6. Длительная стойкость и стойкость к старению , . . . . ... 80
4.2.6. Волокна из поли-4-метилпентена-1 80
4.27. Применение поли-4-метилпентена-1 82
4.2.7.1. Лабораторная и медицинская техника . . - 82
4.2.7.2. Электротехника 82
4.2.7.3. Упаковочные материалы 83
4.2.8. Сополимеризация и привитая сополимеризация 85
4.3. Поливинилциклогексан . . ¦ . , 85
4.3.1. Винилциклогексан 85
4.3.2. Получение поливинилциклогекеана 86
4.3.2.1. Ионно-координационная полимеризация винилциклогексана .... 87
4.3.2.2. Полимеризация винилциклогексана в присутствии оксида хрома(III)
и оксида алюминия , . . 88
4.3.2.3. Катионная полимеризация винилциклогексана 89
4.3.2.4. Гидрирование полистирола до поливииилциклогексана 90
4.3.3. Физико-химические свойства изотактического поливинилциклогекеана 90
4.3.3.1. Кристалличность 90
4.3.3.2. Термостойкость 91
4.3.3.3. Растворимость и свойства растворов •. 93
4.3.4. Переработка изотактического поливинилциклогекеана 93
4.3.5. Свойства и применение изотактического поливинилциклогекеана . . 94
4.4. Поливинилфторид , 95
4.4.1. Винилфторид 96
4.4.2. Полимеризация винилфторида 98
4.4.2.1. Радикальная полимеризация 99
4.4.2.2. Ионно-координационная полимеризация 101
4.4.3. Физико-химические свойства поливинилфторида 101
4.4.3.1. Структура 101
4.4.3.2. Растворимость и свойства растворов 102
4.4.4. Переработка поливинилфторида 1,02
4.4.5. Эксплуатационные свойства поливинилфторида ........ 104
4.4.5.1. Теплофизические свойства . • 104
4.4.5.2. Физико-механические свойства 105
4.4.5.3. Электрические свойства . 106
4.4.5.4. Химическая стойкость 107
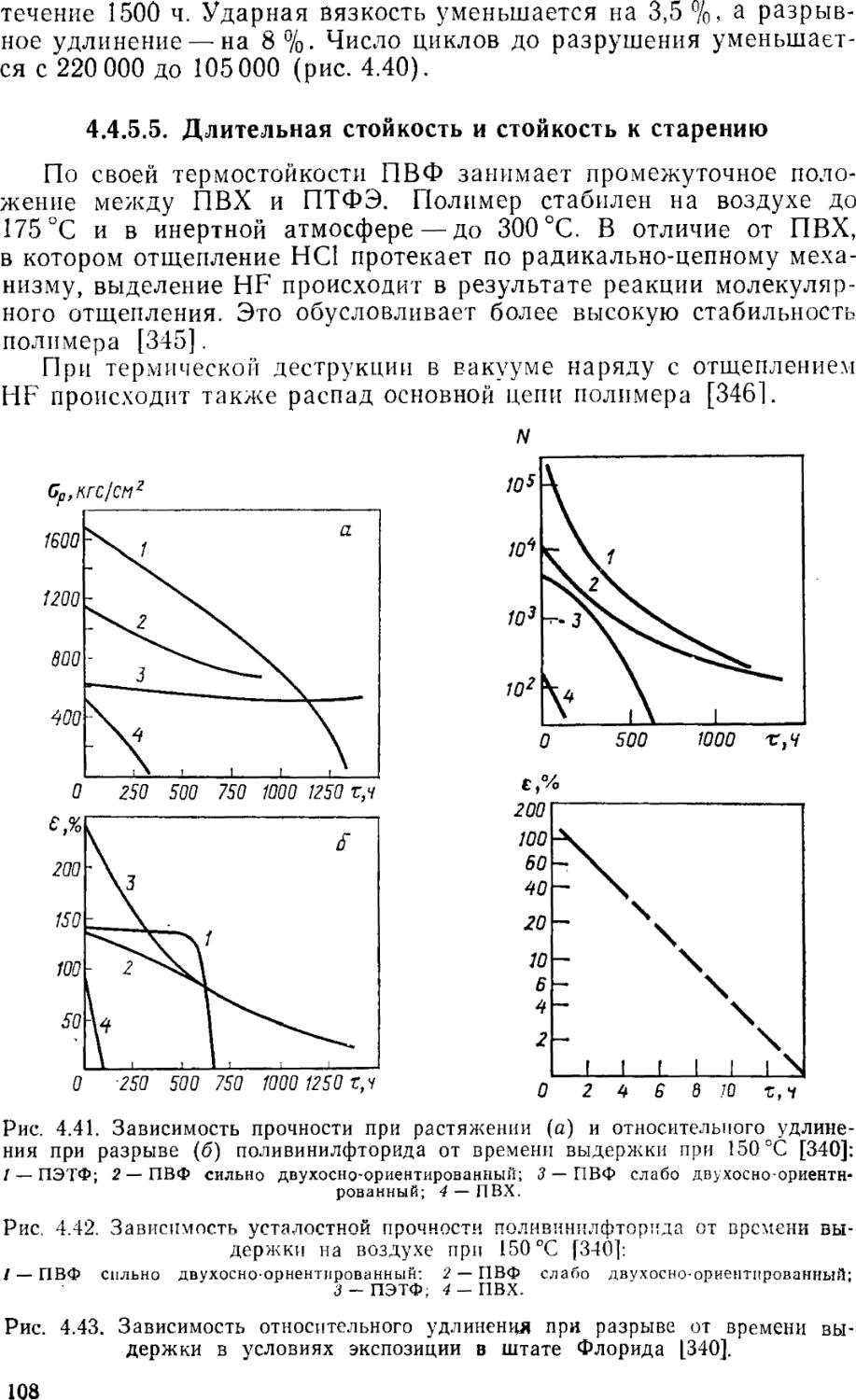
4.4.5.5. Длительная стойкость и стойкость к старению 108
4.4.5.6. Радиационная стойкость 109
4.4.6. Применение поливинилфторида . . 109
4.4.7. Сополимеризация и привитая сополимеризация 111
4.5. Поливинилиденфторид , 112
4.5.1. Винилиденфторид .... . 113
4.5.2. Полимеризация винилиденфторида 114
4.5.3. Физико-химические свойства поливинилидеифторида 115
4.5.3.1. Структура .115
4.5.3.2. Кристалличность 116
4.5.3.3. Свойства растворов . 118
6
1 1 S
4.5.3.4. Термическая и радиационная стойкость l:Q
4.5.4. Переработка поливинилиденфторидз ^
4.5.4.1. Экструзия lV
4.5.4.2. Литье под давлением
4.5.4.3. Прессование и литьевое прессование
4.5.4.4. Нанесение покрытий
4.5.4.5. Формование волокон
4.5.5. Эксплуатационные свойства поливинилиденфторпда
4.5.5.1. Физико-механические свойства . • . .
4.5.5.2. Электрические свойства 124
4.5.5.3. Химическая стойкость 12^
4.5.6. Применение поливинилидеифторида . . ........... 126
4.5.6Л. Электротехника и электроника 127
4.5.6.2. Химическое машиностроение 127
4.5.6.3. Другие области применения 128
4.5.7. Полимерные композиции на основе поливинилиденфторпда и мета-
крилатов 128
4.5.7.1. Совместимость 128
4.5.7.2. Свойства полимерных композиций на основе поливииплиденфторида
и полиметилметакрилата 129
4.5.8. Сополимеры винилиденфторида « . 130
4.5.8.1. Сополимеры винилиденфторида с гексафторпропиленом 130
4.5.8.2. Сополимеры трифторхлорэтилена с винилидеифторидом 131
Литература 131
5. Линейные полимеры с ароматическими карбоциклами в цепи 141
5.1. Полифенплены 142
5.1.1. Получение полифениленов 143
5.1.1.1. Реакция сочетания галогенсодержащих ароматических
углеводородов 143
5.1.1.2, Окислительная дегндрополиконденсация ароматических соединений . 145
5.1.1 3. Дегидрирование полициклогексадиена-1,3 148
5.1.1.4. Полимеризация по реакции Дильса — Альдера 149
5.1.1.5. Радикальное арилирование ароматических углеводородов
5.1.1.6. Электролитическая полимеризация
5.1.1.7. Циклополимеризация ненасыщенных алифатических п
ароматических соединений
5.1.1.8. Дегидратация фенолов
5.1.2. Физико-химические свойства полифениленов ^ . .
5.1.2.1. Кристалличность
5.1.2.2. Температура плавления
5.1.2.3. Растворимость ?
5.1.2.4. Стойкость к термической и термоокислительной деструкции , . . . 154
5.1.3. Эксплуатационные свойства материалов и переработка полифенилена 157
5.1.3.1. Ненаполненные формовочные материалы на основе полифеиилена . '57
5.1.3.2. Материалы на основе полифенилена 158
5.2. Поли-п-ксилилен 161
5.2.1. Получение поли-п-ксилилена 162
5.2.1.1. Пиролиз «-ксилола в вакууме 162
5.2.1.2. Реакция Вюрца — Фиттига с использованием дигалоген-гс-ксилолов 163
5.2.1.3. Диссоциация четвертичных аммониевых или фосфониевых
производных л-ксилола 164
5.2.1.4. Изомеризационная полимеризация л-замещенных производных
стирола 165
5.2.1.5. Изомеризационная полимеризация диазопроизводных л-ксилола . • 165
5.2Л.6. Гидрирование поли-л-ксилилидена 166
5.2.2. Промышленное получение поли-л-ксилилена 166
5.2.3. Физико-химические свойства поли-л-ксилилена 168
5.2.3.1. Кристалличность 168
5.2.3.2. Растворимость 169
5.2.3.3. Термическая и радиационная стойкость 170
5.2.4. Эксплуатационные свойства поли-л-ксилиленов I72
5.2.4.1. Теплофизические свойства 172
5.2.4.2. Физико-мсданические свойства 173
5.2.4.3. Электрические свойства 174
5.2.4.4. Химическая стойкость 174
5.2.5. Применение поли-л-ксилиленов 175
5.2.6. Модификация поли-п-ксилиленов 175
5.2.7. Сополимеры л-ксилилена 176
5.2.8. Политетрафтор-л-ксилилен 177
5.3. Поликсилилидены 178
5.3.1. Получение поликсилилиденов 179
5.3.1.1. Взаимодействие ароматических диальдегидов с бнс(трифенилфосфо-
ний)ксилилендигалогенидами по Виттигу 179
Конденсация ксилиленбис(диэтилового эфира фосфоновой кислоты)
с ароматическими диальдегидами 179
Конденсация по Кневенагелю ароматических диальдегидов с
ароматическими диацетонитрилами 180
Дегидрогалогенироваиие ксилилеидигалогенидов 181
Конденсация бис(диметилсульфоиийтетрафторборатов) 182
Кислотнокаталитическое разложение ароматических бисдиазоалкапов 182
Реакции дегалогеиирования 182
Свойства поликснлилиденов 183
Полибеизилы 184
Получение полибеизилов 185
Конденсация беизилгалогенидов по реакции Фриделя — Крафтса . . 185
Конденсация простых диэфиров л-ксилиленгликоля с ароматическими
углеводородами по Фрнделю — Крафтсу 186
Конденсация п-ксилилеидигалогеиидов или дизфиров л-ксилиленгли-
коля с фенолами по Фриделю—Крафтсу 187
Дегидрирование полиметилеицнклогексана ¦ . 187
Физико-химические свойства полибеизилов 187
Переработка и эксплуатационные свойства материалов на основе
полибенэилов 188
5.5. Полназометииы 190
5.5.1. Полишиффовы основания 191
5.5.2. Поликетанилы 195
5.5.3. Полнальдазины 196
5.5-4. Полнкетазины 197
5.6. Просты^ ароматические полиэфиры и полиариленсульфоноксиды . - 198
5.6.1. Полифениленоксиды 199
5.6.1 Л. Получение полифениленоксида 199
5.6.1.2. Промышленное производство поли-2,6-диметил-1,4-фениленоксида . 205
5.6.1.3. Сополимеры 2,6-диметил-1г4-феиилоксида 206
5.6.1.4. Физико-механические свойства полифениленоксида 208
5.6.1.5. Переработка поли-2,6-диметил-1,4-фениленоксида и его композиций
с полистиролом 217
Эксплуатационные свойства поли-2,6-диметил-1,4-фенилеиоксида и
его композиций с полистиролом .............. 223
Применение полидиметилфениленоксида и его смесей с полистиролом 230
Сополимеры и привитые сополимеры 231
Модификация поли-2,6-днметил-1,4-фениленоксида ..¦•¦•• 234
Полиметилендифенилоксид 235
Получение полиметилеидифенилоксида 235
Получение композиций на основе полиметилендифенилоксида . . 235
Свойства и применение полиметилендифенилоксида 236
Полигидроксизфир 237
Получение полигидроксизфира 237
Физико-химические свойства полигидроксизфиров 238
Переработка полигидроксиэфира на основе дифеиилолпропана и эпи-
хлоргидрнна 241
5.6.3.4. Свойства полигидроксиэфира на основе дифенилолпропана и эпи-
хлоргидрина 243
5.6.3.5. Применение полигидроксиэфира на основе днфенилолпропана и эпи-
хлоргидрина - - 244
5.6.4. Полиариленсульфоноксиды - - 245
5.6.4.1. Получение полиариленсульфоноксидов . 245
5.6.4.2. Физико-химические свойства полиариленсульфоноксидов ..... 250
5.6.4.3. Переработка полиариленсульфоноксидов 262
5.6.4.4. Эксплуатационные свойства ароматических полиариленсульфоноксп-
5.6.4.5. Применение полиариленсульфоноксидов 272
Литература 274
5.7. Ароматические полисульфиды 284
5.7.1. Получение ароматических полисульфидов 285
5.7.1.1. Электрофильное присоединение серы или ее галогенидов к
ароматическим соединениям 285
5.7.1.2. Реакция замещения галогенсодержащих ароматических соединений . 286
5.7.2. Физико-химические свойства полифенилеисульфидов 289
5.7.2.1. Температура плавления и кристалличность 289
5.7.2.2. Растворимость 290.
5.7.2.3. Термостойкость 290
5.7.3. Переработка поли-я-фениленсульфида . 292
5.7.3.1. Формованный материал 292
5.7.3.2. Покрытия . . . . . . . . 292
5.7.3.3. Разветвленный полифениленсульфид 293
5.7.4. Эксплуатационные свойства поли-я-фениленсульфида 293
5.7.5. Применение поли-я-фениленсульфида 295
5.8. Ароматические сложные полиэфиры (полиарилаты) . 296
5.8Л. Получение полиарилатов 297
5.8.1.1. Переэтерификация зфиров дикарбоновых кислот диолами .... 297
5.8.1.2. Переэтерификация диэфиров ароматических диолов дикарбоиовыми
кислотами ... . - 297
5.8.1.3. Поликонденсация дихлорангидридов ароматических дикарбоновых
кислот с щелочными солями бисфенилов на границе раздела фаз . 297
5.8.1.4. Поликонденсация ароматических диолов с дихлорангидридами
ароматических дикарбоновых кислот в высококипящем растворителе ¦ „ 298
5.8.2. Строение и свойства ароматических полиэфиров ....... 299
5.8.3. Промышленные ароматические полиэфиры 343
5.8.3.1. Поли-я-оксибензойная кислота 343
5.8.3.2. Поли-1,4-циклогексилендиметилентерефталат 347
5.8.3.3. Полиэтиленоксибензоат 349
5.8.3.4. Полибутилентерефталат 349
5.8.4. Ароматические полисульфонаты 350
5.8.4.1. Получение 350
5.8.4.2. Физико-химические свойства 351
5.8.4.3. Переработка и эксплуатационные свойства 355
5.9. Ароматические полиангидриды 356
5.9.1. Получение 356
5.9.2. Свойства 357
5.10. Полициклоамиды 361
5.10.1. Алициклические полиамиды 361
5.10.2. Алифатически-ароматические полиамиды 384
5.10.2.1. Получение 384
5.10.2.2. Физико-химические свойства 386
5.10.2.3. Промышленные алифатически-ароматические полиамиды ..... 390
5.10.3. Ароматические полиамиды (полиарамиды) 394
5.10.3.1. Получение 394
5.10.3.2. Физико-химические свойства 418
5.10.3.3. Переработка и свойства ароматических полиамидов 427
5.10.4. Гетероциклические сополиамиды 434
5.10.4.1. Получение ,.,..,.,...,,, 434
9
5.10.4.2, Свойства 435
5.10.5. Ароматические полиамидогидразпды 453
5.10.5.1. Получение 453
5.10.5.2. Физико-химические свойства 456
5.10.5.3. Переработка и эксплуатационные свойства . 457
Литература 461
6. Гетероциклические цепные полимеры . . 469
6.1. Цепные полимеры с пятичленным гетероциклом и двумя гетероато-
мами в цикле 470
6.1.1. Полипиразолы 470
6Л.1.1. Получение полипиразолов 470
6.1.1.2. Свойства полипиразолов 482
6Л.2. Полигидантоины 483
6.1.2.1. Получение полигидантоинов 433
6.1.2.2. Физико-механические свойства 485
6.1.2.3. Переработка, свойства и применение полигидантоинов 487
6.1.3. Полиоксазолы и полиазооксазолы 493
6.1.4. Полиоксазолидоны 494
6Л.4Л. Поаучение поли-2-оксазолидонов 494
6.1.4.2. Физико-химические свойства полиоксазолидоиов 496
6.1.4.3. Переработка и эксплуатационные свойства поли-2-оксазолидонов 497
6.1.5. Политиазолы 497
6.1.5.1. Получение политиазолов 501
6.1.5.2. Физико-химические свойства политиазолов 501
6.1.5.3. Политиазольные волокна 506
6.1.5.4. Политиазольная пленка . . . ...... „ 507
6.2. Циклоцепые полимеры с тремя гетероатомами в пятичленном ге-
тероцнкле 508
6.2.1. Полнтриазолы 508
6.2.1.1. Получение политриазолов 508
6.2.1.2. Физико-химические свойства ароматических поли-4-фенил-1,2,4-три-
азолов 523
6.2.L3, Переработка и эксплуатационные свойства ароматических поли-4-фе-
нил-1,2,4-триазолов . 523
6.2.2. Полиоксадиазолы 525
6.2.2.1. Получение поли-1 Д4-оксадиазолов t . 525
6.2.2.2. Получение поли-1,2,4-оксадиазолов 529
6.2,2.3- Физико-химические свойства полиоксадиазолоЕ 531
6.2.2.4. Поли-1Д4-оксадиазольные волокна 546
6.2.3. Политиадиазолы 549
6.2.3.1. Получение политиадиазолов , . 549
6.2.3.2. Физико-химические свойства политиадиазолов 551
6.2.3.3. Политиадиазольные волокна 555
6.3. Гетероциклические полимеры с четырьмя гетероатомами в
пятичленном цикле 555
6.3.1. Политетразолы 555
6.3.1.1. Полибистетразолы 555
6.3.1.2. Полиаминотетразолы 557
6.3.1.3. Свойства полятетразолов 557
6.3.2. Комплексы политерефталонлоксалоилбисамидгидразонов с
металлами 557
6.4. Полимеры с шестичленными гетероцикламн 559
6.4.1. Полипиразины 559
6.4.2. Полидикеллшперазипы 559
6.4.2.1. Получение полидикетопиперазинов 559
6.4.2.2. Свойства полидикетопиперазинов 565
6.4.3. Политриазины - 565
6.4.3.1. Получение политриазинов 566
6.4.3.2. Физико-химические свойств-a политриазинов ..,.,,.,., 570
10
G.4,3-3. Переработка и эксплуатационные свойства политриазинов .... 581
Литература %8\
7, Полибензгетероциклические полимеры ?85
7.К Полиимиды б85
7.1.1. Ароматические и алифатические полиимиды 589
7.1.1.1. Получение 589
7.1 Л.2. Физико-химические свойства алифатических и ароматических поли-
имидоз * ¦ • 692
7.1.1.3. Полиимидные пленки
7ЛЛ.4. Полиимидные волокна 725
7.1 Л.5. Полиимиды для склеивания металлов 729
7ЛЛ.6. Полиимидные лаки 731
7Л.1.7. Полиимидные связующие 733
7.1.1.8. Полиимидные пресс-композиции 737
7Л.1.9. Переработка прессованием и пресс-литьем 744
7Л.1Л0. Полиимидные пенопласты 745
7Л.2. Полиимиды с гетероциклическими звеньями 772
7Л.2Л. Получение полиимидов с гетероциклами 772
7.1.2.2. Свойства полиимидов с гетероциклами 777
7Л.З. Пол нами доимиды 798
7Л.ЗЛ. Получение полиамидоимидов 799
7Л.3.2. Физико-химические свойства полиамидоимидов 808
7Л.З.З. Полиамидоимидиые волокна 810
7Л.ЗА Полиамидоимидные пленки 811
7.1.3.5. Кабельная изоляция я покрытия на основе полиамидоимндоз . . . 812
7.1.3.6. Полиамидоимидные связующие 813
7.1.3.7. Полиамидоимидные пресс-массы 815
7.1.4. Полиэфироимиды 816
7.1.4.1. Получение полиэфироимидов 816
7.1.4.2. Физико-химические свойства полиэфироимидов 821
7.1.4.3. Кабельная изоляция на полиэфироимидиой основе 835
7.1.4.4. Полиэфироимидные пленки 835
7Л .5. Полисульфоимиды 835
Литература 839
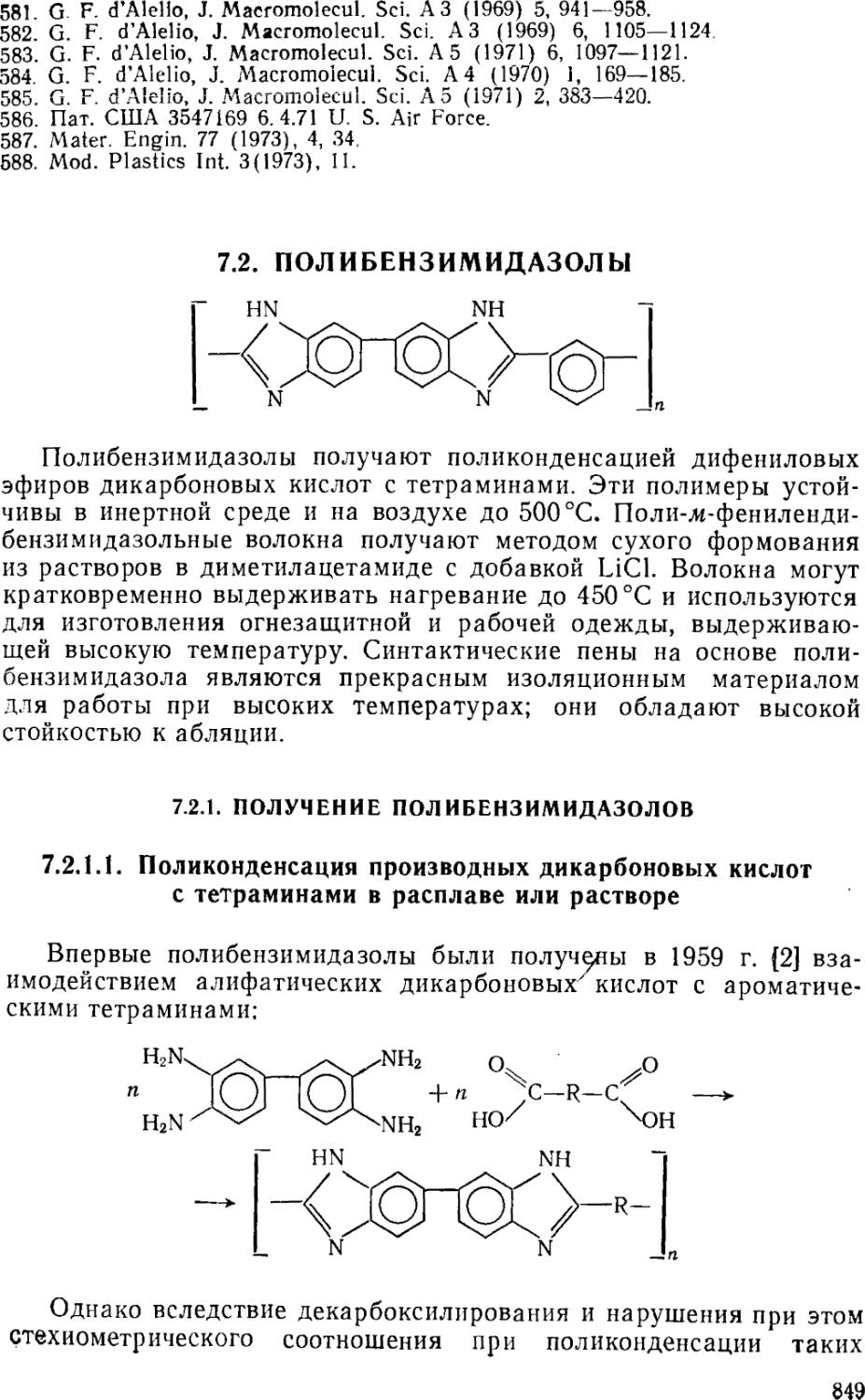
7.2, Полибензимидазолы 849
7.2.1. Получение полибензимндазолов 849
7.2ЛЛ. Поликонденсация производных дикарбоиовых кислот с тетраминами
в расплаве или растворе 849
7.2Л.2. Поликонденсация диальдегидов с тетраминами 875
7.2Л.З. Взаимодействие диацетильиых производных ароматических
соединений с тетрамииами 876
7.2.1.4. Термическая циклизация поли-(о-ацетамидо)амидов 876
7.2.1.5. Поликонденсация ароматических я-бис(гидроксиимииохлорметиль-
ных) соединений с ароматическими диаминами 877
7.2Л.6. Поликонденсация с использованием соединений, содержащих имида-
зольные циклы 878
7.2Л.7. Промышленные способы получения поли-2,2'-ж-фенилен-5,5'-ди6ензи-
мидазола 879
7.2.2, Физико-химические свойства полибензимидазолов 879
7.2.2Л. Температурные переходы 879
7.2.2.2. Растворимость и свойства растворов 880
7.2.2.3. Термостойкость 881
7.2.3. Поли-ж-фенилендибензимидазольные волокна 887
7.2.3.1. Получение 887
7.2.3.2. Свойства 889
7.2.3.3. Применение 890
7.2.4, Синтактические полибензимидазольные пенопласты 891
11
7.2.5. Полибензимидазольные клеи для металлов
7.2.6. Полибензимидазольные стеклопластики
7.2.7. Полибензимидазольные пленки
7.2.8. Мембраны и корпуса аккумуляторов из поли~2,2'-октаметилен-5,5'-
дибензимидазола
7.3. Полибензоксазолы ..*.,.
7.3.1. Получение полибензоксазолов 897
7.3.1.1. Поликонденсация бис-о-аминофенолов с производными днкарбоно-
вых кислот в расплаве 898
7.3.1.2. Высокотемпературная поликонденсация в растворе 898
7.3.1.3. Двухстадийная поликонденсация с промежуточным образованием
полигидрокси- или полиметоксиамидов - 898
7.3.2. Физико-химические свойства полибензоксазолов ........ 901
7.3.2.1. Температура размягчения 901
7.3.2.2. Растворимость .... 919
7.3.2.3. Термостойкость 919
7.3.3. Полибензоксазольные пленки 921
7.3.4. Полибензоксазольные волокна 925
7.4. Полибензтиазолы 926
7.4.1. Получение полибензтиазолов 926
7.4.2. Физико-химические свойства полибензтиазолов 929
7.4.3. Переработка и свойства полибензтиазолов 929
7.5. Полихиноксалины * * . 933
7.5.1. Получение полихиноксалииов 933
7.5.1.1. Поликонденсация 6исA,2-дикар6онильных соединений) с бис(о-диа-
минамн) в расплаве 957
7.5.1.2. Поликонденсация бис-A,2-дикарбонильных соединений) с
ароматическими бис(о-диаминами) - 958
7.5.2. Физико-химические свойства полихиноксалннов 960
7.5.2.1. Температура стеклования 960
7.5.2.2. Растворимость 960
7.5.2.3. Термостойкость 961
7.5.3. Полихиноксалиновые пленки и волокна 964
7.5.4. Связующие на основе полихиноксалинов 964
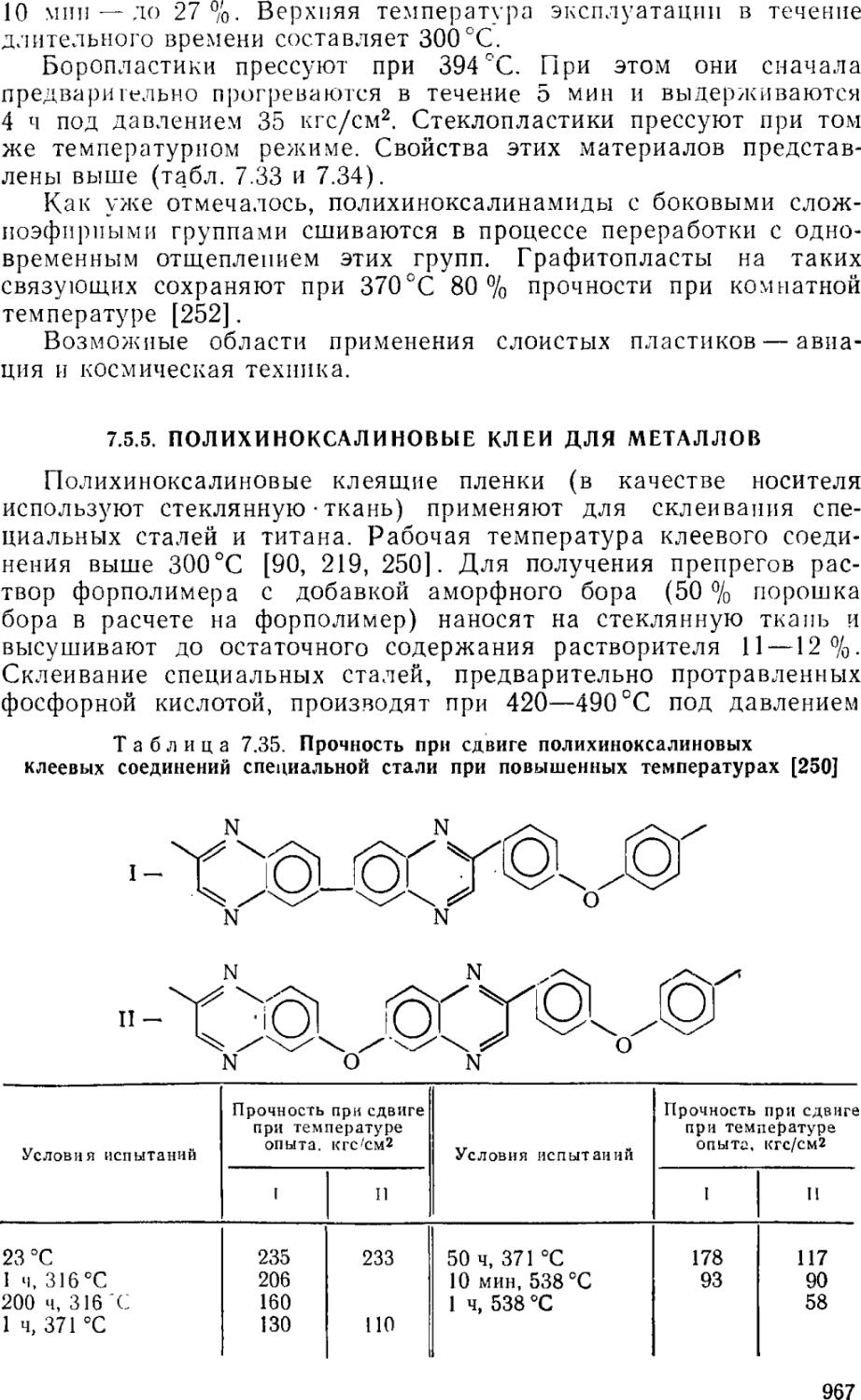
7.5.5. Полихиноксалиновые клеи для металлов . • . . . 967
7.6. Полихиназолоны . . . 968
7.6.1, Получение полихиназолонов 968
7.6.1.1, Незамещенные полихиназолоны 968
7.6.1.2. Получение поли-3-метил- или поли-З-фенилхиназолонов-4 .... 969
7.6.2, Физико-химические свойству полихиназолонов , . - 971
7.6.3, Переработка и свойства полихиназолонов 980
7.7. Полихиназолиндионы 981
7.7.1. Получение полихиназолиндионов 981
7.7.1.1. Синтез полимеров из ароматических бис-(о-аминокарбоновых)
кислот и диизоцианатов 981
7.7.1.2. Взаимодействие бис(оксазиндионов) с диаминами 984
¦ 7.7.2. Свойства полихиназолиндионов . ....•., 984
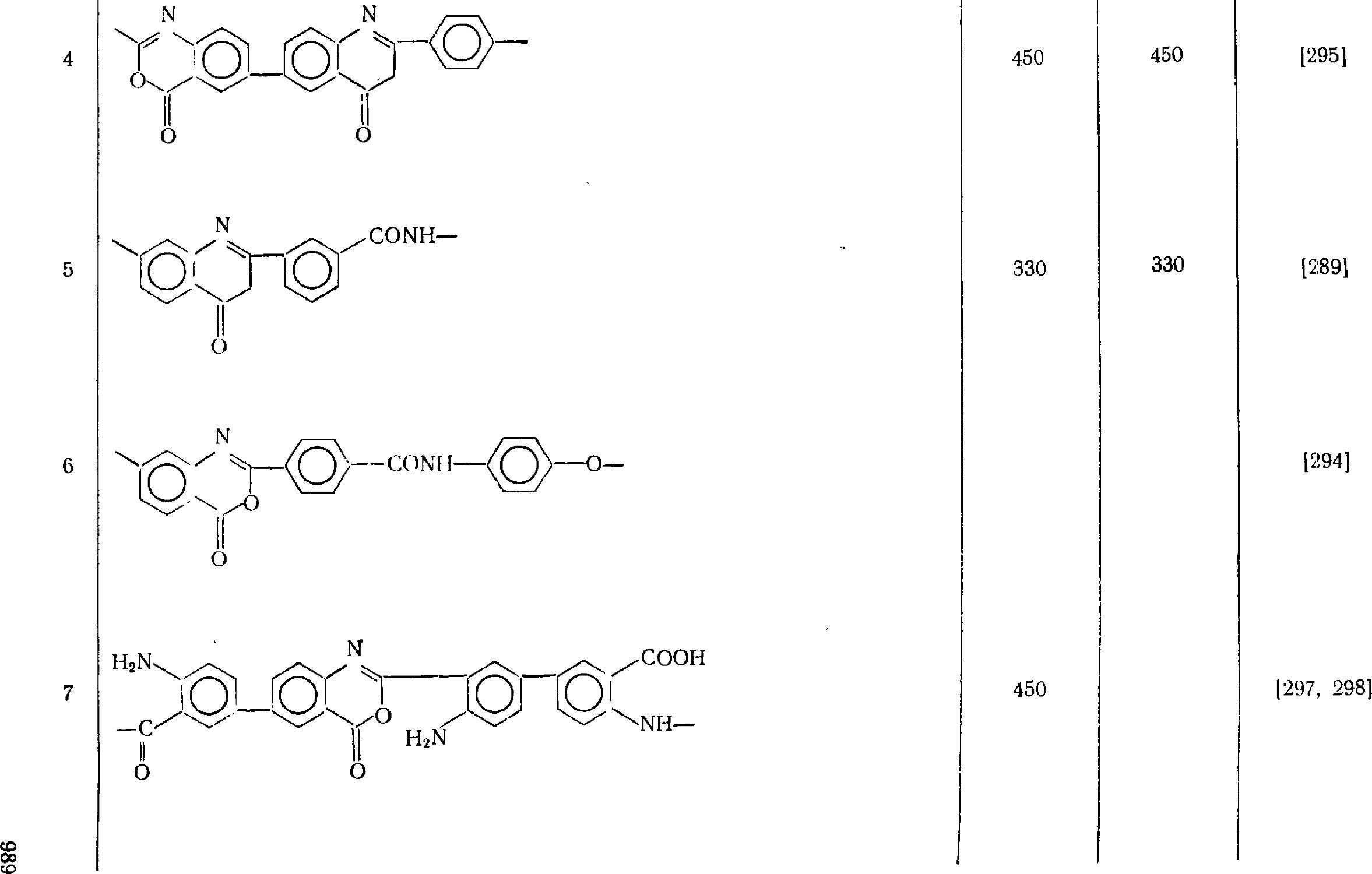
7.8. Полибензоксазиноны • 986
7.8.1. Получение полибензоксазинонов 986
7.8.1.1. Поликонденсация бис(о-аминокарбоновых) кислот с дихлорангидри-
дами кислот 986
7.8.1.2. Взаимодействие бис-о-аминофенолов с диглиоксалевыми кислотами . 990
7.8.2. Свойства полибензсксазинонов 990
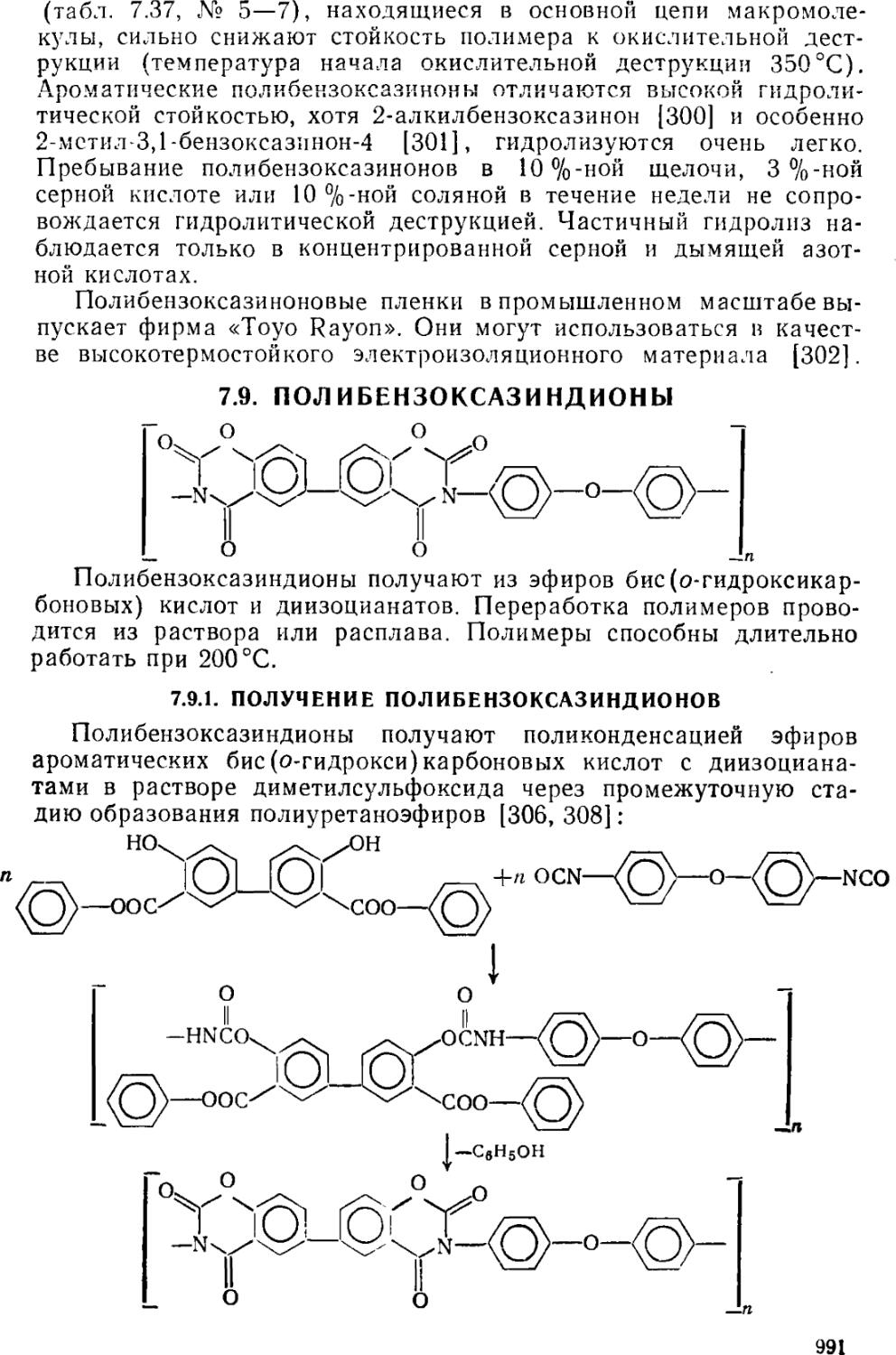
7.9. Полибензоксазиндионы 991
7.9.1, Получение полибензоксазиндионов 991
7.9.2. Свойства полибензоксазиндионов •..•.. 992
Литература щ . 992
12
8. Лестничные полимеры • - • ¦ 998
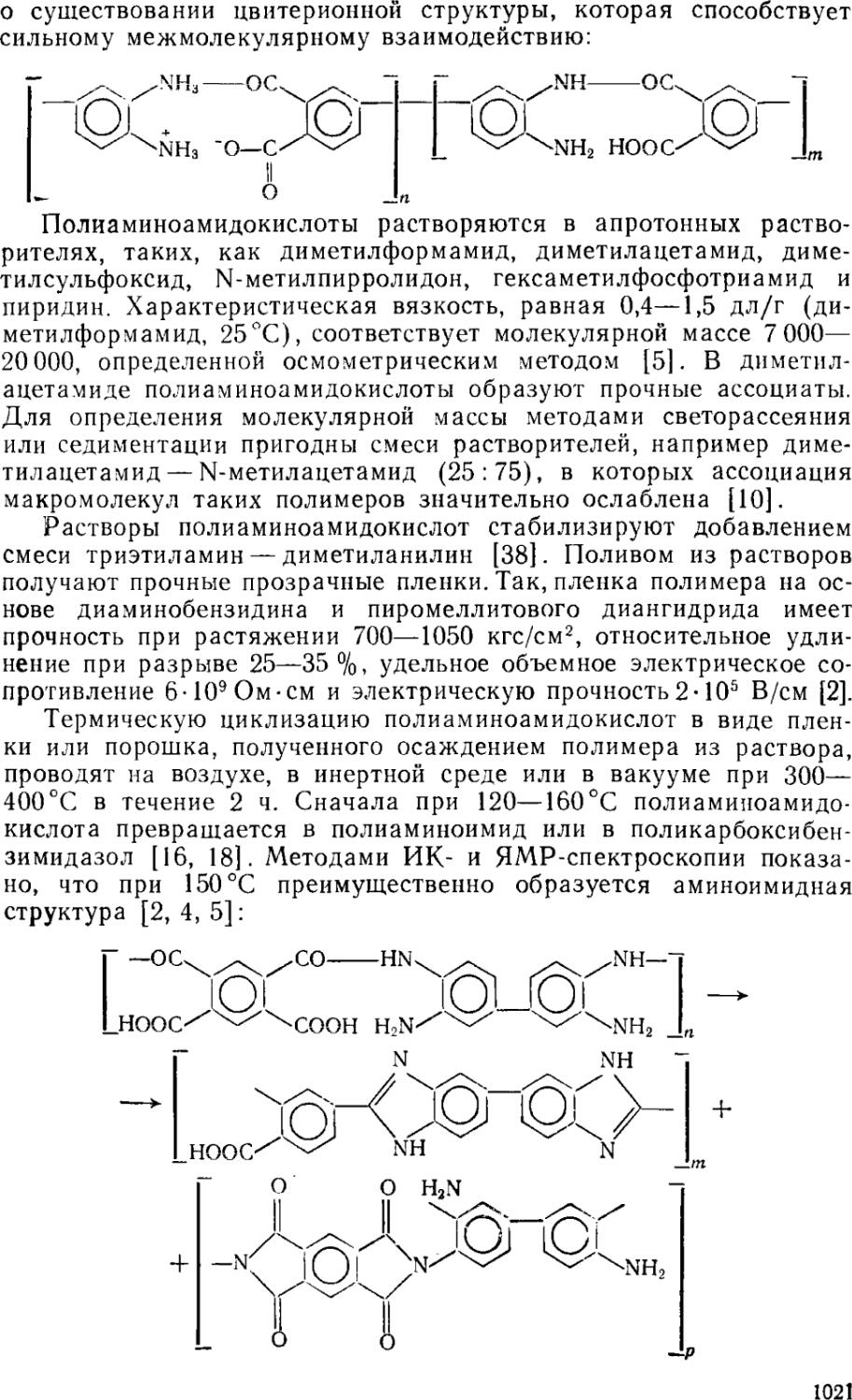
Kl, Пирроны
8.1.1. Получение пирронов •
8.1.1.1. Двухстадийная поликонденсация • 999
8.1.1.2. Одностадийная поликонденсация в растворе Ю22
8.1.1.3. Поликонденсация в расплаве - * '022
8.1.2. Физико-химические свойства пирронов 1°22
8.1.2.1. Температура размягчения '°23
8.1.2.2. Растворимость 1023
8.1.2.3. Термостойкость 1024
8.1.3. Переработка пирронов ¦ Ю26
8.1.4. Свойства материалов из пирронов 1027
8.1.5. Применение пирронов 1 28
8.2. Полиимидазобензфенантролины « Ю29
8.2.1. Получение полиимидазобензфенантролинов 1033
8.2.2. Физико-химические свойства полиимидазобензфенантропинов . . . 1034
8.2.2Л. Отношение к растворителям , 1035
8.2.2.2. Термостойкость 1036
8.2.3. Полибензимидазофенантролиновые волокна 1037
8.2.4. Полибензимидазофенаитролиновая пленка . , . . « 1039
8.3. Полиизоиндолохиназолиндионы 1039
8.3.1. Получение полиизоиндолохиназолиндионов 1040
8.3.2. Свойства полиизоиндолохиназолиндионов 1044
8 4. Политетразапирены 1044
8.4.1. Получение политетразапиренов 1044
Литература 1046
Предметный указатель 1049
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Предлагаемая советскому читателю книга профессора К.-У.
Бюллера (ГДР) в немецком издании буквально называется
«Специальные полимеры» или «Полимеры со специальными
свойствами». Однако практически в ней идет речь преимущественно о
тепло- и (или) термостойких полимерах. Это подтверждают и
первые общие главы книги, в которых анализируются сами понятия
термостойкости и теплостойкости, даются методы их оценки, а
также описываются способы управления этими важнейшими
свойствами. Да и большая часть книги посвящена полимерам,
особенностью которых является возможность их эксплуатации при
повышенных температурах. Естественно, что при этом отдельные
полимеры характеризуются и другими специфическими свойствами,
например электрофизическими, оптическими, химическими. Такой
широкий спектр свойств тепло- и термостойких полимеров создает
поистине неограниченные возможности для их практического
применения. Весьма наглядным в этом плане является пример
ароматических линейных полиимидов, которые могут эксплуатироваться
при температурах до 300—400°С, сохраняя ценные свойства и при
криогенных температурах вплоть до температуры жидкого гелия.
Необходимо отметить, что появившаяся в конце 50-х годов в
результате возросших требований современной техники химия
тепло- и термостойких полимеров очень быстро превратилась в
один из важнейших разделов химии высокомолекулярных
соединений. Для синтеза таких полимеров потребовалась разработка
новых эффективных методов получения исходных мономеров.
Большое число оригинальных исследований привело к созданию
интересных способов синтеза таких полимеров, а также методов
их переработки в различные материалы и изделия.
Немаловажным представляется и экономический аспект проблемы тепло- и
термостойких полимеров, их конкурентоспособность с другими
типами полимерных и неполимерных материалов.
Все эти вопросы подробно рассмотрены в книге К.-У. Бюллера,
причем последовательность представления различных классов
полимеров и однотипность построения отдельных разделов (синтез
мономеров, полимеров, их свойства, переработка, области
применения) позволяют читателю самому провести сопоставительный
анализ описываемых полимеров и выбрать те из них, которые
наиболее подходят для данной области практического использования.
Немалую пользу при этом оказывает громадный табличный
материал, приведенный в книге, а также обширная библиография,
насчитывающая около 3 тыс. ссылок. Большой удельный вес в
анализируемом материале и библиографии занимают работы
советских ученых. Однако в списке литературы, цитируемой в
немецком издании, фигурирует фамилия только первого автора. При
подготовке русского издания библиография осталась в прежнем
виде, поскольку перечисление всего авторского коллектива
повлекло бы за собой существенное увеличение и без того большого
14
объема монографии. По этой же причине пришлось отказаться от
заманчивой перспективы дополнить русское издание книги
списком литературы, появившейся после 1973 г. Можно лишь
отметить, что такая библиография была бы не меньше списка
литературы, имеющегося в книге, хотя принципиально новых
полимеров за этот период появилось очень немного.
Мы полагаем, что книга К.-У. Бюллера в первую очередь
заинтересует тех читателей, которые занимаются практическим
использованием полимерных материалов. Несомненно, она
окажется полезной и для научных работников, сфера деятельности
которых — синтез и исследование полимеров.
Я. С. Выгодский
ПРЕДИСЛОВИЕ К НЕМЕЦКОМУ ИЗДАНИЮ
Успехи научных исследований и разработок, освоение новых
технологических процессов в нефтехимии, интенсификация и
оптимизация существующих технологических процессов переработки
полимерных материалов, широкое применение их в самых
различных областях промышленности, сельском хозяйстве, медицине
привели к значительному увеличению масштабов производства
пластических масс. В 1955—1960 гг. даже сформировалось
понятие «массовые», или крупнотоннажные, полимеры. В дальнейшем,
однако, оказалось, что эксплуатационные показатели многих
полимеров не удовлетворяют все возрастающим требованиям.
Например, многим отраслям промышленности потребовались
неметаллические материалы с хорошими механическими свойствами при
температурах свыше 300 °С. Это поставило новые задачи перед
химиками, работающими в области синтеза полимеров, а также перед
технологами, занимающимися переработкой пластмасс в изделия.
Нужно было получить такие высокомолекулярные соединения
(может быть в небольших количествах), которые по многим своим
показателям отличались от сегодняшних крупнотоннажных
полимеров. В дальнейшем для обозначения этих полимеров стали
использовать термин «полимеры со специальными свойствами», или
«специальные пластмассы».
Но известно, что одно дело получить какой-то результат или
синтезировать продукт один раз и совсем другое дело получить
воспроизводимые результаты или организовать выпуск продукта
в промышленном масштабе. Это в полной мере относится и к
полимерам со специальными свойствами. Поэтому, для того чтобы
производство этих полимеров стало экономичным, надо наладить
их получение в большом количестве и по возможности расширить
области применения.
Пусть эта книга поможет решению поставленной задачи, пусть
она также подтвердит слова Вильгельма Оствальда, который
сказал в 1910 г., что из всех энергий самой ценной является энергия
творческого духа и она должна меньше растрачиваться попусту.
Профессор К. Тиниус
1. ПЕРСПЕКТИВЫ РАЗВИТИЯ
ПРОИЗВОДСТВА ПЛАСТМАСС
Широкое внедрение синтетических материалов во все отрасли
народного хозяйства является существенным элементом научно-
технической революции, которая охватывает все промышленно
развитые страны. В этом процессе решающую роль играет
развитие производства полимеров с улучшенными эксплуатационными
свойствами. В результате замены традиционных материалов
пластмассами стал возможен переход от процессов изготовления
изделий отдельными партиями к поточному их производству. Это
позволило значительно повысить производительность труда, понизить
себестоимость продукции и существенно сократить затраты
рабочего времени. При этом главное требование заключается в
разработке типового ассортимента полимеров, которые по своим
эксплуатационным свойствам были бы, по крайней мере, равноценны
традиционным материалам.
Наряду с эффективностью применения пластмасс возникает
проблема получения полимеров с особыми свойствами, которые
позволяют совершенно по-новому подойти к решению вопросов,
касающихся придания формы, конструирования изделий и их
использования.
По сравнению с другими материалами производство
полимерных материалов за последние 20 лет развивалось очень высокими
темпами. В то время как объем производства чугуна удваивается
за 11 лет, цемента—за 9—10 лет и алюминия—за 7—8 лет,
удвоение объема производства пластмасс происходит примерно за 5 лет.
Согласно прогнозам в 1983 г. полимеры должны были выйти на
первое место по объему производства.
Примерно из 33 млн. т общего объема производства полимеров
в 1971 г. на крупнотоннажные полимеры, такие, как полиэтилен,
поливинилхлорид и полистирол, а также на продукты их
модификации приходилось примерно 63%. Из оставшегося количества
приблизительно 32 % составляли конструкционные пластмассы,
такие, как полипропилен, алифатические полиамиды, полиэфиры,
полиуретаны, амино- и фенопласты, полиметакрилаты,
поликарбонаты и эпоксидные смолы.
Остальные полимеры представляли собой так называемые
полимеры с особыми физическими, или специальными, свойствами.
Эти полимеры благодаря своему строению обладают более
высокой термостойкостью при длительном температурном воздействии
или другими специфическими свойствами, которые не могут быть
достигнуты в результате модификации крупнотоннажных или
конструкционных полимеров. Примерами таких полимеров,
применяющихся более десятилетия в качестве термостойких
конструкционных материалов, являются политетрафторэтилен, кремнийор-
ганические полимеры и полиформальдегиды. С 1960 г. началось
производство еще около 40 новых типов полимеров со
специальными свойствами (табл. 1.1). Наряду с термостойкими полиме-
16
Таблица 1.1. Развитие производства пластмасс
(синтетических полимеров)
*ства (год)
Начало
пронзво;
1901
1914
1915
1928
1929
^930
Озо
1930
ГЭЗО
1930
1930
1931
1931
1931
1934
1936
1937
1937
1937
Полимер
Фенолоформальде-
гидные смолы
Поливиниловый
эфир монохлорук-
сусной кислоты
Диметилбута
диеновый каучук
Полиметилметакри-
лат
Мочевиноформаль-
дегидные смолы
Тиополимеры
(полисульфидный
каучук)
Полиакрилат
Поливинилацетат
Полистирол
Полиакрилонитрил
Поливинилхлорид
Полихлорбутадиен
Полиизобутилен
Полиэтиленоксид
Политетрафторэтилен
Поливиниловый
эфир
Синтетический
каучук *
Поливиннлкарбазол
Полиамид 6,6
Первый изготовитель
«Blumer»
(Германия)
«Chem. Fabrik
Griesheim»
(Германия)
«Bayer A. G.»
(Германия)
«Rohm und Haas»
(Германия)
«IG Ludwigsthafen»
«Thiocol Co.»
(США)
«IG Ludwigsthafen»
«Hoechst»
«IG Ludwigshafen»
«IG Ludwigshafen»
«IG Ludwigshafen»
«Du Pont»
«IG Ludwigshafen»
«IG Ludwigshafen»
«IG Ludwigshafen»
«IG Ludwigshafen»
«IG Schkopan»
«IG Ludwigshafen»
«Du Pont»
i маркэ
Торгова!
Акрилоид
Каурит
Тиокол
Лувикан
Автор и
получен!
Байер,
1872
Кондаков,
1Q00
I cTUU
Рем,
1901
Гольд-
ШМИДТ,
1877
Байер,
1926
Кас-
пари,
1873
Клатте,
Симон,
1 Q QQ
Рено,
1QQQ
I OOO
Каро-
зерс?
1925
Бутлеров,
1873
Шлоф-
фер,
1930
Висли-
цениус,
1 Я7Я
1о/o
Хофман,
1909
Каро-
зерс,
1931
о
о
к
со s
X « „ S
ч So g
Максима
темпера!
длнтелы
эксплуат
125
50
75
65
40
65
65-8С
75
60
60
30
230
60—а-
170
80
I- о ?^пнотоннажное,
С. В. Лебедева освоено в
^ каучука по методу
Продолжение табл. 1.1
год)
детва (
Нач
npoi
1938
1938
1939
1939
1939
1939
1947
1953
1954
1958
1959
1959
1960
1960
1961
Полимер
Полиэтиленимин
Поливинилиденхло-
рид
Полиэтилен низкой
плотности
Поливинилпирроли-
дон
Поли-е-капроамид
Полиуретаны
Полиэтилентерефта-
лат
Полиэтилен
высокой плотности
Полиамид 11
Поликарбонат
Полипропилен
Полиформальдегид
Пентон [поли-3,31-
(бисхлорметокси)
пропиленоксид]
Поливинилфторид
Полиимиды
Первый изготовитель
«IG Ludwigshafen»
«BASF Dow Chem.»
ICI
«IG Ludwigshafen»
«IG Ludwigshafen»
«IG Wolfen»
ICI
«Hoechst»
«Aquitaine-
Organico»
«Bayer AG»
«Montecatini»
«Du Pont»
«Hercules Powder»
«Du Pont»
«Dynamit Nobel»
«Diamond
Shamrock»
«Du Pont»
«Monsanto»
«American
Cyanamide»
«Quantum Inc.»
«Dixon Co.»
«General Electric»
ICI
та
я марк
гова
Тор
Диофан,
Саран
Коллидон
Десмодур
Рильсан
Макролон
Моплен
Дельрин
Пентон
Тедлар
PVF ЕР
L90
Дал вор X
6500
Каптон,
Веспель,
Пайэр ML
Скайбонд,
AFR XPI
Квантед
Мелдин
Джемон
Х-13
Полиамид
1«
к х
а?
о >>
3°
Рено,
1838
Фоусетт,
Хопфф,
1 uoO
Габриель,
1899
Байер,
1935
Каро-
зерс,
1932
Циглер,
1953
Айнхорн,
Натта,
1954
Бутлеров
1872
Фартинг,
1951
Штарк-
везер,
1932
X
Бод-
жерт,
1908
о
альная
ггура
>ной
цЦ
70
65
80
130
90
80
135
120
100
100
140
260
Продолжение табл. IJ
1
с:
ю
il
II
1961
1961
1962
1962
1964
1964
1964
1964
1964
1964
1965
Полимер
Поли-л*-фениленизо-
фталамид
Поли-1,4-циклогек-
силенднметилентс-
рефталат
Полиоксиэфир
Поливинилиденфто-
рпд
Поли-4-метилпен-
тен-1
Полибепзимидазол
Полиамидоимид
Поли-2,6-диметил-
1,4-фениленоксид
Полисульфоноксид
Полиамид 12
Алициклический по*
лнамид из
пробковой кислоты и 1,4-
бис(аминометил)
циклогексана
Первый изготовитель
СССР
«Du Pont»
СССР
«Eastman Kodak»
«Union Carbide»
«Bakelite Ltd.»
«Pennsalt Co.»
«Diamond
Shamrock»
«Kureha Chem.»
ICI
СССР
«Narmco»
«Celanese»
«Westinghouse»
«Schramm»
(ФРГ)
«Rhone-Poulenc»
«Hitachi»
«General Electric»
AKU
(Нидерланды)
СССР
«Mitsubishi
Edogawa»
«Union Carbide»
«Uniroyal»
(Великобритания)
«Huls»
«Emser Werk AG»
«Tennessee
Eastman»
i марка
гова*
Top
Аримид,
ПМ 67,
ПАК-1,
ДФО, СТП-1
Номекс
Фенилон
Кодель-П,
Тенайт
PCDT
Фенокси
Кинар
Дальвор
DS
KF (КФ)
ТРХ
Аймидайт
Полимер
Аманнм
Феногарант
Кермель,
Родефталь
HI-400
РРО
Норил,
Арнокс
(сополимер)
Арилокс
Бакелит
Р 1700
Юкардел
Арилон
(сополимер)
Вестамид L
Гриламид
L
Q2
ор и
учен!
Il
Дюпон,
1948
Натта,
1955
Бринкер,
1959
Фрош,
1 СкЛО
1У4/
Хантер,
1911
Ы
S w й a1
' Man
тем
ДЛИ
эксп
230—
250
ПО
80
150
130
315
240
210
100—
140
150
70
110
19
Продолжение табл. IJ
хства (год)
Начало
произво;
1965
1965
1965
1966
1966
1966
1966
1966
1966
1967
1967
1967
1968
1968
1968
Полимер
Полиметилендифе-
нилоксид
Поли-п-ксилилен
Полибутен-1
Полиамид 7
Полиамид 9
Полиэфиримид
Полн-^-ксилилен-
адипамид
Полигексаметилен-
терефталамид
Полифениленсуль-
фид
Политриметилгекса-
метилентерефтал-
амид
Полиариленсуль-
фоноксид
Полифенилен
Полиимидазобензо-
фенантролин
Полигидантоины
Полиэтнленоксибен-
зоат
Первый изготовитель
«Westinghouse
Electric»
«Union Carbide»
«Petro-Chem. Со.»
«Huls»
СССР
СССР
«General Electric»
«Dr. Beck and Co.»
(ФРГ)
«Dr. Herberts»
(ФРГ)
«Schenectady»
(США)
«Du Pont»
«Celanese»
«Dow Chem.»
«Phillips Petroleum»
«Dynamit Nobel»
«3 M Co.»
ICI
«Eitel Mc Cullough
Inc.»
(США)
«Monsanto»
«Celanese»
«Bayer AG»
«Nippon Rayon»
i марка
Торговая
Дорнл
Парилен
Бу-Таф
Вестолен
ВТ
1711
Энант
Пел аргон
Аймидекс
Теребек
Алловес
Изомид
MXD-6-Fib-
ге
Найлон-бТ
Resin QX
4735
Райтои
Трогамид
Т
Астрел
Бранд
360
Полимер
380
Эймак 221
ВВВ-во-
локна
Резистофол
I Do 4089,
LK 4025,
LK 2217
Полиэфир
A-Tell
2«
Автор и
получет
Шварц,
Натта,
1954
Дженев-
рес,
1898
Кадби,
1963
Ризе,
1872
в я х
Максима
те м пер ai
длителы
250
80
30
190
90
100
180-
260
80-
100
260
250
300
200
75
Продояжение табл. /./
ГОД)
детва (
х?
1968
1969
1970
1970
1970
1970
1971
1971
1971
1972
1972
Полимер
Алициклический
полиамид из декан-
дикарбоновой
кислоты и бис(/г-амн-
ноциклогекенл) мета-
па
Полнбеизоксазинон
Полибензоксазин-
дион
Поли-/г-бензамид(?)
Полиамид 4
Полиамид 3
Политерефталоил-
оксалилбисамидра-
зон
Поли-/г-оксибензой-
ная кислота
Полиоксибензил
Полибензоксазол
Полибутилентсре-
фталат
Первый изготовитель
«Du Pont»
«Toyo Rayon»
«Bayer AGx>
«Du Pont»
«Alrac»
(США)
«Hochst»
AKZO
(Нидерланды)
«Carborundum Co.»
«Midland Silicones»
СССР
«General Electric»
«Eastman Kodak»
Ci
a
вд
ргова
о
H
Квана
IDO 4051
Волокно В
PRD-49
Энкатерм
Эконол
Хайлок
210
Метолон
Валокс
Тенайт
РМТ
и
тор и
лучен
йода,
Фишер,
1910
Брин-
кер,
1959
и
о
г^ а _ s
г; >^ О ^
^j оЭ »Q сч
5|^
* ? = 5
НО
300
200
250
Негорючий
300
230
300
45
Примечание. Для периода с 1901 до I960 г. (крупнотоннажные полимеры,
конструкционные полимерные материалы) приводятся первый изготовитель и одно торговое
наименование, для периода с I960 "до 1972 г.— первый изготовитель в перечне промышленных
производств.
рами получены полимеры с полупроводниковыми или фотохром-
ными свойствами, а также полимеры, применяющиеся в
фармакологии или в ракетных топливах.
Полимеры со специальными свойствами имеют высокую
стоимость: если 1 кг крупнотоннажных полимеров стоит 2—4 марки,
1 кг конструкционных полимерных материалов — 4—10 марок, то
1 кг выпускаемых в промышленном масштабе полимеров со
специальными свойствами стоит 10 000 марок и выше.
Основным направлением при разработке новых полимеров для
замены традиционных материалов является модификация
крупнотоннажных и конструкционных полимеров. Улучшение эксплуата-
ционных свойств полимеров осуществляется либо путем изменения
условий синтеза, либо усовершенствованием процессов их перс-
работки.
Изменение условий проведения реакции достигается
использованием сомономеров при сополнмсрпзации, привитой сополимери-
зацией, а также полимераналогнчнымн превращениями. Так,
теплостойкость поливиннлхлорида повышается при снижении
температуры полимеризации в результате статистического включения в
основную цепь звеньев бутена-1 в ходе сополимеризации или
посредством дополнительного хлорирования. На стадии переработки
возможна модификация свойств материалов путем введения в
композицию волокнистых или порошкообразных сферических
наполнителей для получения армированных полимерных материалов,
смешением с низкомолекулярными или высокомолекулярными
соединениями для получения пластифицированных материалов или
полимерных композиций или окислительным либо радиационно-
химическим сшиванием. Повышение теплостойкости полнвинил-
хлорида на стадии переработки достигается введением в
композицию стеклянных волокон или путем сшивания
пластифицированных полимеров. Ударную вязкость поливинилхлорида можно
повысить проведением привитой сополимеризации с бутадиеном,
введением хлорированного полиэтилена или путем сшивания
пластифицированных полимеров.
Техническая реализация способов получения полимеров с
заданными размерами путем модификации свойств
крупнотоннажных и в меньшей степени конструкционных полимеров дает
возможность значительно повысить темпы развития и эффективность
процесса общественного воспроизводства.
Согласно прогнозам до 2000 г. крупнотоннажные полимеры
полиэтилен, поливинилхлорид и полистирол, а также некоторые
конструкционные пластмассы останутся основными заменителями
таких традиционных материалов, как древесина, кожа и металлы.
Хотя начало промышленного производства этих полимеров
относится к 30-м и 40-м годам нашего столетия (см. табл. 1.1), часть из
них продолжают использовать для получения изделий общего
назначения, несмотря на четырехкратное увеличение объема
производства синтетических полимеров за последние десять лет. С
момента появления на мировом рынке полипропилена в 1959 г. ни
один новый полимер не достиг такого объема промышленного
производства, чтобы его можно было включить в группу
конструкционных пластмасс. Вряд ли можно ожидать, что какой-нибудь
средний по объему производства полимер перейдет в группу
крупнотоннажных полимеров.
Верхняя температура длительной эксплуатации
крупнотоннажных полимеров лежит ниже 100 °С, причем путем модификации
можно лишь незначительно повысить эту температуру.
Верхнюю температуру длительной эксплуатации около 150°С
имеют лишь некоторые представители конструкционных пластмасс,
такие, как поликарбонат, ненасыщенные полиэфиры, сетчатые по-
22
лиуретаны, полиамиды, а также реактопласты на основе феноло-
и мочевиноформальдегидных смол.
Необходимость создания полимеров с верхней температурой
длительной эксплуатации выше 150°С была вызвана развитием
электротехники, электроники, самолетостроения и космонавтики.
Прогресс в электротехнической промышленности заключается
в миниатюризации сопротивлений, конденсаторов,
электродвигателей при сохранении их мощности. Это потребовало повышения
рабочих температур. Температуры длительной эксплуатации
полимеров, применяемых для изготовления электрической изоляции,
фольги, деталей включателей и кожухов, должны превышать
200 °С.
Развитие современной микроэлектроники было бы немыслимо
без создания специальных полимеров, обладающих высокой
эластичностью и стабильностью размеров при температурах до 300°С.
Например, только благодаря наличию полиимидов стало
возможным рентабельное изготовление гибких печатных схем,
устойчивых в ваннах для пайки, температуростойких плоских
многоканальных кабелей и гибких интегральных схем. Хотя применение
пластмасс в электронике по сравнению с общим их выпуском
невелико, именно эта область имеет решающее значение при
осуществлении научно-технической революции во всей химической
промышленности. Комплексная автоматизация и
совершенствование систем управления производственными процессами
обусловливают постоянное ужесточение требований к современной
электронике.
Переход к сверхзвуковым скоростям, наблюдающийся в
настоящее время в гражданской авиации, также предъявляет
совершенно новые требования к материалам. При скоростях, равных
удвоенной скорости звука, температура на внешних поверхностях
достигает 150 °С, а при трехкратном превышении скорости
звука — 300°С. Количество применяемых термостойких
пропитывающих составов при изготовлении элементов слоистых
конструкций крыльев и пультов сверхзвуковых пассажирских самолетов
при трехкратном превышении скорости звука составляет 2,9 т для
одного самолета [1].
Еще более жесткие (хотя и при кратковременной
эксплуатации) требования к материалам предъявляются космической
техникой. Тепловая защита летательных аппаратов должна
противостоять кратковременному действию температур, равных 5000°С.
Это достигается применением полимеров, которые при пиролизе
приобретают структуру, обеспечивающую минимальную
теплопроводность и достаточную прочность.
Верхнюю температуру длительной эксплуатации, равную 150°С,
пока что нельзя повысить при использовании недорогих
модифицированных крупнотоннажных полимеров, так как термостойкость
полимеров примерно пропорциональна значениям энергии
химических связей в цепи. Для получения термостойких полимеров
необходим переход от полимеров с С—С-связями (энергия связи
23
58,6 ккал), к гетерополимерам с более высокой энергией связи,
такими, как —С—О— G0 ккал), —C = N— (94 ккал) или
—С=С— A00 ккал). Термостойкость повышается также за счет
того, что эти связи в циклических полимерах находятся в
сопряжении. При использовании ароматических циклов получаются (по
крайней мере, на первый взгляд) термостойкие полимеры более
сложной структуры.
Полимеры, выпускаемые с 1960 г. (см. табл. 1.1), относятся
исключительно к высокотермостойким. Поскольку большая часть
новых полимеров имеет стратегическое значение, расходы на их
создание во многих случаях поступают из государственного
бюджета. Так, например, в США получение полибензимидазолов
(раздел 7.2) финансировалось исключительно авиационной
промышленностью США. За последние десять лет число полимеров со
специальными свойствами значительно увеличилось. Однако по
сравнению с числом научных исследований в области
термостойких полимеров или имеющимся на сегодняшний день объемом
информации их промышленное производство сравнительно невелико.
Одной из причин этого является необходимость создания новой
технологии переработки указанных полимеров, поскольку лишь
незначительная часть их может перерабатываться в изделия
обычными методами, применяемыми для переработки термопластов.
Каждый из полимеров со специальными свойствами, как
правило, применяется лишь в одной или нескольких областях.
2. СТРОЕНИЕ
И ОСОБЕННОСТИ СТРУКТУРЫ ПОЛИМЕРОВ
С ПОВЫШЕННОЙ ТЕРМОСТОЙКОСТЬЮ
2.1. ОПРЕДЕЛЕНИЕ ПОНЯТИЙ «ТЕПЛОСТОЙКОСТЬ»
И «ТЕРМОСТОЙКОСТЬ»
Понятие «термостойкость полимерных материалов» используется
в литературе неоднозначно. С одной стороны, оно характеризует
температурный интервал плавления или температуру размягчения
пластмасс, с другой стороны, это понятие используется в качестве
характеристики верхней предельной температуры, при которой в
определенных условиях и при заданном времени выдержки не
происходит существенных изменений механических или
электрических свойств полимеров. Время и условия выдержки
устанавливаются с учетом требований данной конкретной области
применения.
Экстремальные условия работы конструкций в космическом
пространстве требуют от материалов, используемых в космической
технике, примерного постоянства свойств: в течение 2000 ч при
24
250 °C, 500 ч при 350°С, 100 ч при 500°С и 2 мин при 1000°С.
В электротехнике для специальной изоляции требуется
постоянство свойств в течение 25 000 ч при 250°С.
Тепло- и термостойкость полимеров связаны с их химическим
строением и определяются физическими (температура плавления
и температура стеклования) и химическими (стойкость к
термической, термоокислительной и гидролитической деструкции)
факторами. При кратковременном тепловом воздействии свойства
материалов часто определяются исключительно влиянием физических
факторов. В случае длительной термостойкости решающими в
значительной степени являются химические факторы. Отсюда
следует, что термостойкость полимеров представляет собой величину,
зависящую от времени.
2.2. ТЕПЛОСТОЙКОСТЬ
К термостойким полимерам относятся как
частично-кристаллические, так и аморфные полимеры.
Частично-кристаллические полимеры имеют достаточно четкую
температуру плавления. Верхняя температура длительной
эксплуатации этих полимеров ограничена температурой плавления,
которая лежит выше температуры стеклования аморфных полимеров.
Поскольку в этой области аморфная часть не находится больше
в стеклообразном состоянии, то механическая прочность
полимеров определяется долей нерасплавившейся кристаллической фазы.
Температура плавления является также минимальной
температурой переработки термопластичных материалов.
Для аморфных полимеров температура стеклования является
верхней границей области применения. Вблизи температуры
стеклования происходит переход из стеклообразного в
высокоэластическое состояние, при котором для аморфных полимеров
наблюдается резкое падение прочности.
Определение верхней температуры эксплуатации полимеров из
данных по температурам плавления или стеклования носит,
однако, только ориентировочный характер и позволяет судить о
поведении материалов при кратковременном действии температур.
Для оценки длительной стойкости решающими являются прежде
всего химические факторы B.3).
2.2.1. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
Температура плавления определяется термодинамически как
отношение энтальпии плавления АЯПл к энтропии плавления
Строго говоря, это соотношение справедливо только для
соединений данного полихмергомологического ряда и при постоянном
давлении. В реальных условиях фазовый переход
частично-кристаллических полимеров происходит не в определенной точке, а
25
в некотором температурном интервале, величина которого ллвисит
от степени кристалличности.
Высокую температуру плавления можно ожидать при высоком
значении энтальпии плавления и (или) низкой энтропии плавления.
Энтропия плавления, связанная с конформацпонной подвижностью
цепей, находится в прямой зависимости от структуры
макромолекул: высокая жесткость neuen и симметрия в пределах цепи
приводят к малым значениям AS и тем самым к высоким
температурам плавления.
В соотношении Т„л = ДЯпл/Д5Пл энтропия плавления является
определяющим фактором. Путем сравнения значений «мольной»
энтальпии плавления и энтропии плавления для ряда полимеров
Годемари [2] было показано, что между температурой плавления
и энтальпией плавления, отнесенной к молекулярной массе
мономерного звена, нет никакой взаимосвязи (табл. 2.1): введение
ароматических циклов в полиэфиры приводит к повышению
температуры плавления, хотя энтальпия плавления остается
практически неизменной. Полидекаметилентерефталат плавится при
138 °С, полидекаметиленадипииат — при 79,5 °С. Отношение
АЯпл/Мол. масса составляет для обоих полиэфиров 36 кал/г.
Полиамиды имеют значительно более высокие температуры
плавления, чем соответствующие полиэфиры, хотя при этом и не
наблюдается существенных различий в значениях энтальпии плав-
Та блица 2.1. Энтальпия и энтропия плавления полимеров [2]
Полимер
гиг
°С
кал/моль
Д#пл/Мол.
масса,
кал/моль
AS,
(ка л/моль -°С)
ДА/связь,
(кал/моль-дО
Полипропилен изотак-
тический
Полистирол изотакти-
ческий
Полидекаметиленсеба-
цинат
Полидекаметиленади-
пинат
Полидекаметилентерефталат
Политетраметилентере-
фталат
Политетраметиленизо-
фталат
Полиэтилентерефталат
Полидекаметиленсеба-
цинамид
Полигексаметилеиади-
пннамид
Поликапроамид
Полиэтиленоксид
Полиакрилонитрил
Политрифторхлорэтилен
Политетрафторэтилен
176
239
80
79,5
138
230
152,5
267
216
267
225
66
317
210
335
2 600
2 000
12 000
Л 0 200
11000
7 600
10 100
5 500
8 300
10 300
5 100
1980
1200
1200
1 460
62,0
19,2
35,0
36,0
36,0
33,0
45,0
28,1
24,5
45,0
45,0
45,0
23,0
10,3
14,6
5,78
3,90
34,0
29,0
27,0
15,1
23,7
10,2
17,0
19,1
10,3
5,35
2,0
2,49
2,9
2,90
1,95
1,55
1,60
1,91
1,9
3,0
1,7
0,77
1,36
1,46
2,68
1,0
1,25
1,45
26
ления. Полидекаметиленсебацинат плавится при 80 °С, мольная
энтальпия плавления его составляет 35 кал/г. Для полидекамети-
леисебацинамида соответствующие величины равны 216 °С и
24,5 кал/г.
В то же время на основании данных, приведенных в табл. 2.1
можно сделать вывод, что полимеры, у которых энтропия
плавления, отнесенная к числу звеньев в цепи, имеет меньшее значение,
как правило, обладают более высокой температурой плавления.
В частности, энтропия плавления, конформация цепей, структура
и температура плавления полимера связаны следующим
соотношением:
Энтропия плавления равна разности между значениями энтропии
полимера в твердом состоянии и в расплаве при температуре
плавления.
Низкая энтропия плавления и, следовательно, высокая
температура плавления полимера наблюдаются при малой S\ или
большой 5s. В принципе низкая энтропия плавления возможна в том
случае, когда степень упорядоченности расплавленного полимера
незначительно отличается от степени упорядоченности твердого
полимера, т. е. если в расплаве сохраняется максимальная
степень упорядоченности. Полиолефины имеют в расплавленном
состоянии, как правило, большую энтропию и, следовательно,
низкие температуры плавления, так как в результате высокой
подвижности цепей в расплаве возможно большое число конформации
макромолекул. Количество возможных конформации цепей в
расплаве и энтропия понижаются, если вследствие сильного меж- и
внутримолекулярного взаимодействия в расплаве сохраняется
высокая степень упорядоченности. Конформации свернутых
макромолекул определяются главным образом внутримолекулярными
ван-дер-ваальсовыми силами притяжения и отталкивания
заместителей соседних атомов цепи и ориентацией связей соседних
звеньев цепи. Потенциальный барьер, который нужно преодолеть
при свободном вращении вокруг простой связи, тем выше, чем
меньше длина связи между атомами и чем больше сила
отталкивания заместителей в цепи.
Уменьшение длины С—С-связи в полифторуглеводородах
происходит вследствие высокой электроотрицательности атома фтора.
Гране-конфигурации углеродных цепей полиэтилена при
замещении атомов водорода фтором превращаются из планарной
зигзагообразной формы в слабоспиралевидную форму. Эффект
повышения жесткости цепей способствует повышению температуры
плавления со 138°С (полиэтилен) до 327°С (политетрафторэтилен).
При введении объемистых заместителей конформации цепей
изменяются и переходят из ступенчатого транс-положения в гош-ио-
ложение. Повышение жесткости цепей тем сильнее, чем объемистее
заместители и чем более упорядоченно они расположены
(табл. 2.2). Замещение атомов водорода в* цепи полиэтилена на
27
Таблица 2.2. Строение звена и температура плавления полиолефинов
Звено
-—СН2—СН2—
—СН2—СН(СНз) —
—СН2—СН(СН2СН3) —
—СН2—СН (СН2) 2CH3—
—СН2—СНСНзСН (СН3J—
—СН2—СН(СН2)зСН3—
—СН2—СН—
1
Полимер
Полиэтилен
Полипропилен
Полибутен-1
Полннентен-1
Поли-4-метилпентен-1
Полигексен-1
Полнвинилциклогексан
Температура
плавления,
°С
138
198-212
125-140
130
230
55
выше 360
изопропиленовые группы приводит к повышению температуры
плавления со 138°С (полиэтилен) до 230°С (поли-4-метилпен-
тен-1). Введение в боковую цепь объемистых циклогексановых
циклов повышает температуру плавления (для поливинилцикло-
гексана она равна 360°С).
Замещение в молекуле полиэтилена атомов водорода на
линейные гибкие боковые цепи снижает, однако, температуру
плавления.
Полигексен-1 плавится при 55 °С, в то время как полиэтилен
плавится при 138°С.
Увеличение жесткости цепей линейных полимеров достигается
введением в основную цепь карбо- или гетероциклов.
Максимальная жесткость получается в том случае, когда ароматические ядра
копланарны и сопряжены. Линейный поли-л-фенилен
представляет собой неплавкий нерастворимый и
высококристаллический продукт; в то же время высокомолекулярный поли-л*-фе-
нилен еще растворим. Влияние структурной изомерии хорошо
иллюстрируется рис. 2.1.
Термодинамически линейный поли-л-фенилен представляет
собой идеальный термостойкий полимерный материал. Однако
практическая реализация его термодинамических характеристик
сильно затруднена. Синтез высокомолекулярного поли-гс-фенилена
трудно воспроизводим. Его нерастворимость и неплавкость
исключают возможность использования традиционных способов
переработки в изделия. Необходимый компромисс между высокой
температурой плавления и перерабатываемостью полимера достигает
путем введения шарнирных групп между ароматическими
кольцами, таких, как —О—, —СО—, —NH—, —N=N—, —S— или
—SO2—. При повышении гибкости цепей и снижении степени
кристалличности получаются плавкие и растворимые полимеры.
Ароматические простые полиэфиры, полисульфоноксиды, поли-
28
сульфиды, сложные полиэфиры и полиамиды уже более пяти лет
применяются в качестве термостойких полимеров специального
назначения.
Такое же влияние оказывает введение объемистых боковых
цепей (конкретные примеры приводятся ниже при рассмотрении
отдельных классов полимеров). .
Уменьшение числа возможных конформаций цепей и,
следовательно, повышение их жесткости и температуры плавления
полимера можно достигнуть также сшиванием молекулярных цепей.
Температурная граница применения сильносшитых полимеров
определяется их стойкостью к термоокислительной деструкции.
Часто сетчатые полимеры, хотя и являются довольно формоустой-
чивыми, имеют очень большую жесткость и хрупкость, что
ограничивает их практическое применение. Этого недостатка лишены
лестничные полимеры, построенные подобным образом. Такие
лестничные полимеры, как полихиноксалины G.5) или полиими-
доазобензофенантролины (8.2) представляют собой определенным
образом сшитые линейные полимеры, у которых в каждом
мономерном звене сшиваются только две молекулярные цепочки:
Линейный полимер
(две соседние цепи)
Лестничный полимер
Лестничный полихиноксалин
При хороших механических свойствах и высокой стойкости к
термоокислительной деструкции лестничные полимеры являются
неплавкими продуктами.
Следующим фактором, влияющим на
степень упорядоченности в расплавах
полимеров и тем самым на энтропию плавления
и температуру плавления является
межмолекулярное взаимодействие (ван-дер-
ваальсовы силы, водородные связи),
которое характеризуется энергией когезии.
Энергия когезии и температура плавления
повышаются с увеличением длины цепи. Эта
зависимость сильнее проявляется в области
низких значений молекулярных масс. По
Бильмейеру [5], температура плавления
Рис. 2.!. Зависимость температуры плавления
анкетированных п неаннелнрованных пелиароматнческих
углеводородов от числа углеродных атомов [3, 4].
и средняя молекулярная масса связаны следующим
соотношением:
1 аЛ-Ь
пл
Мол. масса
где а и b — эмпирические константы.
Незначительная энтропия плавления полиамидов обусловлена
тем, что полиамиды даже в расплавленном состоянии сохраняют
кристалличность. Превращение триклинной кристаллической
модификации в гексагональную с повышенным числом степеней
свободы происходит выше температуры плавления, так что энтропия
увеличивается уже в этой области.
Когезионные силы и температуры плавления зависят также от
симметрии элементарных звеньев в цепи. Чем выше симметрия,
тем более прочное взаимодействие возникает между цепями и тем
более плотную упаковку они имеют в кристалле. Принцип
симметрии цепей был использован Престоном при создании
«упорядоченных» ароматических сополиамидов для получения
высокоплавких продуктов. Упорядоченные сополиамиды с ж-фениленовыми
группами плавятся вследствие их высокой симметрии при
температуре примерно на 100° выше, чем соответствующие
статистические сополиамиды на основе одних и тех же исходных веществ.
2.2.2. ТЕМПЕРАТУРА СТЕКЛОВАНИЯ
Температура стеклования в противоположность температуре
плавления термодинамически неопределима. Температура
стеклования высокомолекулярных соединений характеризует переход из
стеклообразного (твердого — хрупкого) в высокоэластическое
состояние. Этот переход осуществляется в температурном интервале
от 5 до 20°; температура стеклования зависит от условий
испытаний, т. е. от скорости охлаждения полимера. Для аморфных
полимеров температура стеклования является верхней границей
области применения.
Таблица 2.3. Строение звена и температурные переходы полимеров [7]
Звено
—-СН2—СНг—
—СН2—СН(СНз)—
—СН2-С(СНзJ~
—СН2—СН2—О—
—СН2—СН(СНз)—О—
—СН2—С(СНзJ—О—
—СН2—СНСНгСНз—
-СНг-СНС(СН3)з—
Полимер
Полиэтилен
Полипропилен изотак-
тический
Полиизобутилен
Полиэтиленоксид
Полипропиленоксид
изотактический
Полиизобутиленоксид
Полибутен-I изотакти-
Т1 J"l j-4* i^ Ж Ж Тш
ческии
Поли-3,3-диметилбу-
тен-1
Гпл- °С
144
170
44
67
70
170
300
>320
тс, °с
-20
-15
-73
-67
-75
g
<50
<60
Тс/Тил
0,61
0,58
0,63
0,61
0,58
0,61
<0,56
<0,56
Взаимосоязь между температурой стеклования и молекулярной
структурой полимера такая же, как и в случае температуры
плавления (табл. 2.3). Температура стеклования повышается с
увеличением внутренней жесткости цепи и межмолекулярного
взаимодействия. Шарнирные группы в полимерной цепи, которые
способствуют сегментальной подвижности, снижают температуру
стеклования. Введение объемных группировок, а также увеличение
ориентации или кристаллизации повышают температуру
стеклования.
Взаимосвязь между температурой стеклования и температурой
плавления может быть выражена в виде эмпирического
соотношения, предложенного Бименом [6] :
i = const « 0.58 — 0,78
По другим данным [7], это отношение равно 0,56—0,63 (см.
табл. 2.3).
2.3. ТЕРМОСТОЙКОСТЬ
Термостойкость полимеров определяется стойкостью связей в
макромолекуле при повышенных температурах. Поскольку
прочностные свойства полимеров зависят от их молекулярной массы,
то ее уменьшение вследствие термической, термоокислительной
или гидролитической деструкции цепи приводит к характерному
снижению механической прочности. Стойкость химических связей
представляет собой верхний уровень колебательной энергии,
который может занимать молекула без разрыва связей. Так как
колебательная энергия увеличивается с повышением температуры,
то термостойкость полимеров можно охарактеризовать энергией
диссоциации связей в макромолекуле.
Из сопоставления значений энергии диссоциации различных
химических связей в макромолекулах (табл. 2.4) можно сделать
следующие выводы.
1. По сравнению с такими связями, как Si—Si- или Р—Р-,
С—С-связь обладает наибольшей энергией диссоциации. Поэтому
наибольшую стойкость можно ожидать у полимера с С—С-связями.
2. Значение энергий диссоциации связей между углеродными
и другими атомами превышают лишь значения энергии диссоциации
Р—О-, Si—О-, В—N- и В—О-связей (см. табл. 2.4). Из
неорганических термостойких полимеров в промышленном масштабе
производится лишь полимерный фосфонитрилхлорид (полидихлорфосфа-
зен) [8].
3. Из полимеров с С—С-связями наибольшую стойкость имеют
карбоциклические ароматические соединения. Стойкость
алифатических С—С-связей увеличивается при замене водорода у
углеродных атомов атомами фтора.
Термостойкость полимеров повышается, если они состоят не
из отдельных цепей, а из двух соединенных между собой цепочек
(лестничные полимеры). Термическая диссоциация отдельных
связей еще не приводит в данном случае к снижению молекулярной
31
Таблица
Тнп связи
N—N
Si—Si
Р-Р
Si—S
С—С
s—s
Р—H
с—s
С—N
Si—C
Si—H
С—H
с—о
С—Cl
P—0
N—H
Si—Cl
С—С
С—Cl
S—H
C-H
B-C
с—с
C-H
C—H
c-c
C-H
C—H
C—H
B—N
Si—0
0—H
c-c
C—H
C—F
c=c
B--0
c—s
Si-F
C—F
c=o
C-.C
2.4. Энергия диссоциации химически»
Стандартное вещество
Гидразин
I
У
С$Н5—СНд—С }]л
Нитромет;
Силаны
С6Н5СН2—H
Диэтиловый эфир
В алифатических соединениях
В алифатических соединениях
В ароматических соединениях
СС1з—H
Третичный
Вторичный
Гексафторэтан
Метай
В ароматических соединениях
CF3—H
Силиконы
В ароматических соединениях
Ацетилен
CF4
н2с=сн2
В(ОСН3)з
CeFe
Кетоны
Ацетилен
; связей при 25°С
Энергия диссоциации,
ккал/моль
37
45
53
61
63
63
63
66
68
70
74
77.5
79
80-81
82
84
85
83-85
86
87
89
89
89
89
94
94
97
98-102
102
103
104
106
110
120
121
121
125
126
129
143
145
174
200
массы и ухудшению свойств материала. Этот, так называемый
«принцип многократных связей» далее будет рассмотрен
подробнее.
Использование энергии диссоциации в качестве основного
параметра при выборе структурных элементов для создания
полимеров повышенной химической термостойкости ограничивается
следующими факторами.
1. Термостойкость, которую должен иметь полимер в
соответствии со значениями энергии диссоциации, не достигается, так как
в большинстве случаев окислительная деструкция начинается ниже
температуры, при которой происходило бы разрушение цепей
согласно энергии диссоциации. Окисление происходит при действии
кислорода воздуха или озона и часто протекает как цепная
реакция. Склонность полимера к окислению можно понизить путем
исключения таких легкоокисляемых структур, как алифатические
С—Н-связи, и посредством создания плотных непористых
поверхностей.
2. Разрушение молекулярных цепей поликонденсационных
полимеров обусловливается наряду с термическим распадом связей
гидролизом, протекающим при повышенных температурах. Вода,
вызывающая гидролиз, может диффундировать в полимер извне
или образовываться непосредственно в полимере при повышенных
температурах вследствие реакций перегруппировки, таких, как
циклизация (см., например, механизм деструкции полиимидов).
Скорость гидролиза зависит от химического строения и
морфологии полимера, которая в свою очередь определяет проницаемость.
3. Термостойкость полимеров, содержащих связи с различной
энергией диссоциации, не прямо пропорциональна энергии
диссоциации наиболее слабых связей в цепи. Термическое разрушение
связей происходит в том случае, если в некоторой области цепи
локализуется значительная кинетическая энергия
микроброуновского движения. Соответственно максвелловское распределение
энергии при данной температуре определяется числом связей в
полимерной цепи с кинетической энергией, превышающей среднее
значение и достаточной для разрыва цепей. У полимеров,
содержащих в макромолекуле связи с различными значениями энергии
диссоциации, с увеличением доли стабильных связей уменьшается
вероятность концентрации максимума энергии, лежащего выше
среднего значения кинетической энергии, на связях с
незначительной энергией диссоциации [9].
4. При анализе термостойкости следует остановиться также на
стерических факторах. Термическая деструкция полигалогеноле-
финов протекает с отщеплением галогенводорода чаше всего по
механизму цепных реакций. Поли-3,3-бисхлорметилпропиленоксид
(«Пентон»)
СН2С1
—СН2—С—СН2—О—
Н2С1
—П
2 Зак. 1S 33
в соответствии со своим строением (алифатические С—С-, С—CI-,
С—Н- и С—О-связи) должен обладать термостойкостью,
примерно равной термостойкости ПВХ. Однако этот полимер
устойчив к длительному нагреву до 150°С, поскольку в
противоположность ПВХ из-за стерических затруднений отщепление НС1 по
цепному механизму становится невозможным.
5. Наряду с энергией основных связей определенный вклад в
термостойкость полимеров вносят также водородные связи, впн-
дер-ваальсовы силы, дисперсионные силы.
6. Вследствие обменного взаимодействия карбоцпкличеекпх и
гетероциклических ароматических ядер в цепи возникает
дополнительная резонансная стабилизация, которая не учитывается
энергией диссоциации. В противоположность поли-я-ксилнлену
E.2.3.3) в поли-д-ксилилидене E.3.2) бензольные ядра могут
вступать в сопряжение через алифатические двойные связи:
n
CH=CH—(( )>—CH= I Поли-я-ксилилиден
СН2—СН2—(х/У)—СН2— ] Поли-я-ксил плен
СН=]
Лп
Результатом дополнительной резонансной стабилизации является
повышенная термостойкость. В инертной атмосфере деструкция
поли-я-ксилилидена начинается при 405 °С, в то время как
температура начала разложения поли-/г-ксилилена составляет 323 °С.
7. Структура полимеров не является совершенно идентичной
структуре основного мономерного звена. Виниловые полимеры в
отличие от мономерных звеньев содержат в цепи разветвления и
двойные связи. Строение концевых звеньев определяется наличием
инициатора, растворителя и примесей. Реакция циклизации у поли-
гетероциклов протекает во многих случаях не полностью, поэтому
такие полимеры содержат ряд фрагментов с незначительной
термостойкостью. Именно указанные отклонения от идеального
строения основного мономерного звена и являются вследствие более
низких значений энергии диссоциации исходными точками
(местами) начала термической деструкции.
Увеличение термостойкости полимеров происходит прежде
всего при переходе от полиолефинов или полпхлоролефинов к по-
лифторолефинам. Вследствие высокой электроотрицательности
атома фтора размеры С—С-связи уменьшаются. Это влечет за
собой, во-первых, вытягивание и повышение жесткости цепей,
связанные со значительным повышением температуры плавления но
сравнению с соответствующими полихлоролефинами, во-вторых,
повышение энергии диссоциации С—С-связи с 85 до 97 ккал/моль.
Частичная или полная замена склонных к окислению С—Н-связей
с энергией диссоциации 94 ккал/моль на С—F-связи с энергией
диссоциации 121 ккал/моль обусловливает повышенную стойкость
фторполимеров к термоокислительной деструкции. Отщепление
фтористого водорода от поливннилфторида D.4.5), аналогичного
34
ПВХ, протекает не по цепному механизму. Политетрафторэтилен
производится в промышленном масштабе с 1946 г. и благодаря
устойчивости к длительному нагреву до 260°С сделался одним из
первых термостойких полимеров специального назначения. Из
прочих полифторолефннов стали выпускаться в 1960 г. поливинил-
фторид D.4) и в 1961 г. поливинилиденфторид D.5).
На основании рассмотренных данных о строении
высокоплавких и стойких к термической или термоокислительной деструкции
полимеров можно сделать вывод о том, что карбоциклические
полиароматические углеводороды должны обладать более высокой
термостойкостью, чем алифатические полимеры, включая поли-
фторолефины. Вследствие высокой жесткости цепей
ароматические полимерные углеводороды имеют очень высокую
температуру плавления или вообще являются неплавкими продуктами.
Энергия диссоциации ароматических С—С-евязей составляет
120 ккал/моль, что примерно на 30 ккал/моль больше энергии
диссоциации связей С—С в алифатических соединениях, которая
близка энергии диссоциации С—Н-связей в ароматических
углеводородах (ED = 102 ккал/моль по сравнению с 89—94 ккал/моль
для третичных или вторичных алифатических С—Н-связей).
Атомы водорода в ароматических соединениях в
противоположность алифатическим не могут отщепляться под действием
свободных радикалов (термоокислительная деструкция). Как следует
из значений энергии диссоциации, в карбоциклических
ароматических углеводородах С—Н-связи разрушаются раньше С—С-связей.
Дальнейшее повышение термической стойкости можно осуществить
посредством замены слабых С—Н-связей С—F-связями, например
поли-я-ксилилен E.2) — политетрафтор-м-ксилилен E.2.8).
Несмотря на высокую термостойкость, карбоциклические
полиароматические углеводороды, такие, как полифенилены E.1),
до сих пор имеют лишь второстепенное техническое значение,
поскольку не решена проблема переработки неплавких и
нерастворимых полимеров.
Оптимальное соответствие между термостойкостью, перераба-
тываемостью и свойствами полимерных материалов достигается
для карбоциклических ароматических полимеров посредством
введения таких атомов или атомных группировок между
ароматическими кольцами, как —О—, —СО—, —NH—, —S—, —SO2—. Это
приводит к снижению сопряжения между ароматическими
кольцами и повышению гибкости цепей макромолекул. Полимеры,
построенные таким образом, плавки и растворимы в полярных
растворителях. Гибкие связи имеют достаточную стойкость к
термической и термоокислительной деструкции, поэтому термостойкость
ароматических полимерных углеводородов большей частью при
этом не ухудшается. Из значений энергии диссоциации (см.
табл. 2.4) и температур разложения ароматических двухъядерных
соединений как модельных соединений для ароматических
полимеров с алифатическими мостиковыми группами (табл. 2.5) можно
сделать вывод, что при создании термостойких полимеров для
2*
1 35
Таблица 2.5. Температуры разложения ароматических
двухъядерных соединений
Соединение
разл1
510
428
422
320
483
427
420
370
соединения между собой карбоциклических ароматических ядер
пригодны прежде всего простые эфирные, сульфо-, кето-, метиле-
новые, амидные, сложные эфирные, и сульфидные группы. Такие
недостатки подобных полимеров, как низкая температура
плавления и пониженная термическая и термоокислительная стойкость
по сравнению с исходными карбоциклическими ароматическими
полимерными углеводородами компенсируются лучшей их пере-
рабатываемостью. Полимеры, построенные по этому структурному
принципу, — ароматические простые полиэфиры, полисульфоны,
поликарбонаты, полиамиды, сложные полиэфиры (полиарилаты)
полисульфиды и поли-д-ксилилен в первой половине 60-х годов
стали выпускаться в промышленном масштабе (см. табл. 1.1).
Они в основном перерабатываются в изделия традиционными
методами (в термопластичном состоянии или из раствора).
Термостойкость (длительный нагрев) указанных карбоциклических
ароматических полимеров с алифатическими мостиковыми группами
колеблется от 100 до 250 °С и не может превышать 300 °С.
Более высокую термостойкость по сравнению с
карбоциклическими ароматическими полимерами имеют гетероциклические
ароматические полимеры. Из сопоставления значений температуры
разложения карбоциклических ароматических углеводородов и
соответствующих гетероциклических ароматических систем
(табл. 2.6) видно, что гетероциклические ароматические
углеводороды обладают более высокой термостойкостью. С увеличением
количества введенных гетероатомов в ароматические системы
число наиболее слабых С—Н-связей снижается. Другим
преимуществом данного класса полимеров по сравнению с
карбоциклическими полимерами является простота их синтеза. Реакция
образования гетероциклов может одновременно служить для связи
между собой различных полифункциональных мономеров (цикло-
поликонденсация). В отличие от получения незамещенных
карбоциклических ароматических полимерных углеводородов синтез
указанных полимеров включает несколько стадии. Например,
36
полиоксадиазол F.2.2) получается по следующей схеме:
- -о
Ъг
\
NH—
О
V©
,o>
N
|-нс1
\Н—
-и2о
О
N N
J
Полигидразид
(растворим в
органических
растворителях,
Гпл=350-400°С)
Полиоксадиазол (нерастворим в
органических растворителях, не
плавится)
п
Ступенчатый механизм реакции позволяет перерабатывать
полимер, образующийся на промежуточной стадии, традиционными
методами, поскольку вследствие повышенной гибкости цепей
(реакция циклизации еще не завершилась) он растворяется и легче
плавится, чем конечный продукт с замкнутыми гетероциклами.
Переход в нерастворимый и высокоплавкий конечный продукт
осуществляется в сформованном материале. Длительная термостойкость
полигетероциклов лежит в интервале температур 200—300°С. Из
полимеров этого класса в промышленном масштабе
выпускаются лишь полиоксадиазолы F.2.2) и полигидантоины F.1.2) (см.
табл. 1.1).
Таблица 2.6. Температуры разложения ароматических карбо-
и гетероциклов
Карбоцик-шческое
разл'
°С
Гетероциклическое
соединение
Т
разл'
°С
Гетероциклическое
соединение
разл*
OC
600
630
520
N-
NH
N
600—620
620-650
620-650
N
О
620-650
650
630
87
При использовании в реакции полнциклнзации соответствую-
щих тетрафункциональных мономеров получают лестничные
полимеры:
О
-_CONH
СООН HaN^^^^^NHs'
О
Полиаминоамидокцслота
—н2о
—н2о
Полиамииоимид
О О
Полиимидазопнрролон
—П
Длительная термостойкость полибензгегероциклов лестничного
строения колеблется от 300 до 500 °С. Их неплавкость является
следствием высокой жесткости цепей. Наличие незначительного
числа атомов водорода обусловливает высокую стойкость
полимеров к термоокислительной деструкции. Высокая термостойкость
обеспечивается также высокими энергиями диссоциации внутри
ароматических, полностью сопряженных систем, значения которых
превышают ПО ккал/моль. Повышенную термостойкость
полимерам лестничного строения придает также реализуемый в них
принцип многократных связей. Разрушение лестничного строения и,
следовательно, изменение свойств полимеров этого класса
происходит лишь при разрыве определенной комбинации связей в
макромолекуле. На примере лестничных полимеров, состоящих из
конденсированных шестичленных циклов, можно показать сле-
38
дующие комбинации связей, необходимые для разрушения
макромолекул:
Тесслер [11] на основе подобных моделей, состоящих из шес-
тичленных конденсированных циклов, выполнил статистический
расчет процесса деструкции по методу Монте-Карло с
использованием ЭВМ. При установлении ряда допущений (все
макромолекулы имеют одинаковую длину цепей; все связи в цепях имеют,
несмотря на их положение, одинаковую прочность; скорость
деструкции пропорциональна общему числу связей) были
определены зависимости отношения среднечисловой молекулярной массы
к исходной молекулярной массе как функции времени (рис. 2.2).
Если взять за основу идентичные реакции разложения, то можно
отметить существенные различия между лестничными и линейными
полимерами в отношении изменения молекулярной массы в
процессе деструкции. В полимерах с простыми цепями с самого начала
и до окончания процесса деструкции происходит резкое падение
молекулярной массы. Для лестничного полимера при деструкции
вначале наблюдается индукционный период, характеризующийся
незначительным изменением молекулярной м_ассы. Однако после
завершения периода индукции отношение Мя/Л1я0 уменьшаемся
намного медленнее по сравнению с линейным полимером.
Промышленное получение лестничных полимеров сопряжено с
рядом трудностей, связанных, например, с неколичественным
протеканием циклизации и деструкцией на отдельных стадиях
процесса конденсации. Несмотря на возможное применение этих
полимеров для получения абляционных и невоспламеняющихся
текстильных материалов, лишь один лестничный полимер — полибен-
Рис. 2.2. Статистические
данные о деструкции
лестничных, полулестничных и
линейных полимеров
(расчеты выполнены по методу
Монте-Карло) [11]:
/ — лестничный полимер
(замкнутые двойные цепи); 2 — по-
лулестиичный полимер (одна
одинарная связь на 24 цикла);
3 — полулестиичиый полимер
(одна одинарная связь на 12
циклов); 4 —линейный полимер.
о
0,05 OJQ 0,15 0м20 0,25 - К t
39
зпмпдазобензофенантролин («ВВВ-волокна») (8.2.3) —
выпускается в опытно промышленном масштабе.
Лестничные полимеры, в которых лестничные сегменты
соединены друг с другом с помощью одинарных простых связей,
называются полулестничными полимерами («блок-лестничные»
полимеры):
^ ^^ ^Ч^ ^S^ ^*\^ > "S. л"Ч. j^
Лестничный полимер
(схематически)
Полулестничныи
полимер
(схематически)
Бензгетероциклические полулестничные полимеры (гл. 7)
получают и перерабатывают со значительно меньшими затратами,
чем лестничные полимеры. Согласно расчетам Тесслера (см.
рис. 2) на основе строения полимера, занимающего
промежуточное положение между линейными и лестничными полимерами,
можно предсказать его поведение при деструкции. Как видно из
рис. 2.2, по термостойкости полулестничный полимер с одной
простой связью на 12 циклов (фрагменты из двух конденсированных
шестичленных циклов) значительно превосходит 'линейные
полимеры. Деструкция полулестничного полимера с одной простой
связью на 24 цикла (фрагменты из четырехконденсированных
шестичленных циклов) протекает с индукционным периодом.
Соединение лестничных сегментов простыми связями способствует
повышению гибкости цепей. Некоторые полулестничные полимеры
являются плавкими продуктами. Их длительная термостойкость
лежит в области 200—500 °С. Такие полулестничные полимеры, как
полиимиды G.1.1), полиэфиримиды G.1.4), полиамидоимиды
G.1.3) и полибензимидазолы G.2), выпускаются в промышленном
масштабе уже с середины 60-х годов (см. табл. 1.1), другие
полимеры этого типа находятся еще на стадии разработки.
3. МЕТОДЫ ОПРЕДЕЛЕНИЯ ТЕРМОСТОЙКОСТИ
ПОЛИМЕРОВ
Для оценки термостойкости полимеров применяются экспресс-
методы, позволяющие охарактеризовать температурные
зависимости различных химических превращений и физических свойств,
и длительные испытания, служащие для установления верхней
температуры длительной эксплуатации по отношению к
определенным свойствам материалов.
Поскольку данные, полученные с помощью экспресс-методов
(например, термограв1шетрического и
дифференциально-термического анализа), дают лишь ориентировочную оценку термостойко-
40
сти, для получения сведении о верхней температуре длительной
эксплуатации полимеров необходимо иметь результаты
длительных испытаний. Многие процессы деструкции протекают во
времени и поэтому не регистрируются экспресс-методами.
3.1. ЭКСПРЕСС-МЕТОДЫ
Химические процессы деструкции полимеров в зависимости от
температуры можно контролировать с помощью термографимет-
рии, изотенископии, по поглощению кислорода и методом
дифференциально-термического анализа.
Для неплавких полимеров температура начала разложения
является предельной температурой, выше которой происходят
скачкообразные изменения свойств материалов. '
Теплостойкость полимеров, которые размягчаются ниже
температуры разложения, характеризуется температурой стеклования.
Температурный интервал плавления позволяет оценить
наименьшую температуру, при которой возможна переработка полимера
в изделия традиционными методами и в случае
частично-кристаллических полимеров определить верхнюю температуру
длительной эксплуатации.
Температура стеклования определяется по скачку на графиках
температурных зависимостей удельного объема, удельной
теплоемкости, коэффициента теплопроводности, диэлектрической
проницаемости, показателя преломления или модуля кручения. В случае,
аморфного полимера температура стеклования позволяет
определить верхнюю границу теплостойкости. Для определения
температурного интервала стеклования или размягчения используются
инструментальные методы — методы определения
теплостойкости по Вика или Мартенсу и деформационной теплостойкости.
3.1.1. МЕТОДЫ ИЗУЧЕНИЯ ХИМИЧЕСКИХ ПРОЦЕССОВ,
ПРОТЕКАЮЩИХ ПРИ ДЕСТРУКЦИИ ПОЛИМЕРОВ
3.1.1.1. Термогравиметрический анализ
Термогравиметрический анализ заключается в нагревании
полимера при заданном градиенте температур в вакууме, инертной
атмосфере, на воздухе или в кислороде и при одновременной
регистрации потери массы образца в зависимости от температуры
(рис. 3.1). В том случае, если не происходит обратимых
процессов выделений влаги или отщепления низкомолекулярных
соединений в результате циклизации, температура начала потери массы
образца характеризует начало разложения полимера. Скорость
нагрева выбирается, как правило, в пределах от 1 до 5 °С/мин.
Испытания проводятся в изотермическом режиме [для
нахождения констант скоростей при заданной температуре
определяются скорости разложения (рис. 3.2)]. При определении
изотермических потерь массы можно одновременно анализировать и
41
m, % от асх.
100
200 400 600 800 T,°C
200 400 600 Г,MUH
Рис. 3.1. Кривые динамического термогравиметрического анализа полипиромел-
литимида 4,4'-днаминодифенилоксида в среде гелия, ДГ = 3 °/мин [21].
Рис. 3.2. Кривые изотермического термогравнметрического анализа полиппромел-
литимида 4,4'-диаминодифенилоксида на воздухе [21].
регистрировать мольное соотношение различных газообразных
продуктов деструкции методами газовой хроматографии или масс-
спектрометрии.
Разновидностью этого метода является дифференциальный
термогравиметрический анализ, при котором с включением RC-
цепей посредством электронного дифференцирования импульсов
непосредственно получается дифференциальная кривая.
Максимумы на дифференциальных кривых характеризуют температуры
максимальной скорости деструкции.
Недостатком термогравиметрического анализа является то
обстоятельство, что потеря массы, обусловленная отщеплением
газообразных продуктов деструкции, в отдельных случаях может
компенсироваться увеличением массы при протекании процессов
окислительной деструкции. Кроме того, при
термогравиметрическом анализе при высоких температурах не регистрируются реакции
гидролиза, сопровождающиеся образованием большого количеств
ва фрагментов разрушенных макромолекул.
3.1.1.2. Изотенископия
Метод изотенископии заключается в измерении давления
газов, образующихся при нагревании исследуемого образца в
замкнутой системе или вакууме [12, 13]. Температура, при которой
начинается изотермическое изменение давления, называется
температурой начала разложения. Метод используется для изучения
как полимеров, так и-низкомолекулярных жидких модельных
соединений, что позволяет проводить сравнительную оценку
стойкости высокомолекулярных и низкомолекулярных соединений с
одинаковым строением мономерного звена.
3.1.1.3. Поглощение кислорода
Метод поглощения кислорода полимером заключается в
нагревании полимера и кислорода в замкнутой системе и
одновременной регистрации волюмометрическим или манометрическим
методами количества поглощенного кислорода [14] (рис. 3.3).
Поскольку термоокислительная деструкция полимеров начинается
43
Рис. 3.3. Оценка стойкости полпвшшлцнклогексана к
термоокислительной деструкции (по количеству
поглощенного кислорода А при 180—220 °С) [22].
Д-Ю^моль О2 fr полимера
32V
80 Г, nun
обычно при более низких температурах, чем
термическое разложение или
термодинамические превращения, то определение
поглощения кислорода является существенным
дополнением к данным
термогравиметрического анализа или изотенископии при
изучении химических процессов, протекающих при деструкции
полимеров.
3.1.2. МЕТОДЫ ОПРЕДЕЛЕНИЯ ТЕМПЕРАТУР ПЛАВЛЕНИЯ
И СТЕКЛОВАНИЯ
3.1.2.1. Дифференциально-термический анализ
Наряду с такими общепринятыми методами, как определение
температуры плавления по Кофлеру или с помощью микроскопа,
гемпературный интервал плавления термостойких полимеров
можно определять методом дифференциально-термического анализа.
Навеску исследуемого полимера нагревают в печи вместе с
инертным веществом (эталонный образец). Скорость нагрева
выбирается так, чтобы обеспечить линейный характер повышения
температуры. Разность между температурой исследуемого образца
и эталонного вещества измеряется с помощью измерительного
зонда с термочувствительным элементом и автоматически
записывается как функция температуры (рис. 3.4). Полученные
эндотермические или экзотермические пики соответствуют тому интервалу
температур, при которых в полимерах протекают физические
превращения или химические реакции. Поэтому дифференциально-
термический анализ позволяет одновременно с регистрацией
температурного интервала плавления и температуры стеклования
установить температурный интервал, в котором в зависимости от
природы среды протекают различные процессы деструкции. Пу«
тем сопоставления полученных данных с данными о составе
газов пиролиза, определенном в результате термогравиметрического
анализа в изотермическом режиме при соответствующей
температуре C.1.1.1), можно сделать вывод о механизме процесса
деструкции.
Рнс. 3.4. Кривые
дифференциально-термического анализа поли-1,4-циклогексилендиме-
тилентерефталата, d «= 1,197 г/см3, содер-
жание граяс-изомера 70 % [23]:
/ — температура стеклования (88 °С); 2~
температура кристаллизации A33 °С); 3 — температура
плавления {293°С).
SQ 1QQ 15Q Z№ 250 300t,°C
43
3.1.2.2. Определение температуры стеклования
Температурные зависимости ряда физических характеристик
полимеров, таких, как удельный объем, удельная теплоемкость,
показатель преломления, коэффициент теплопроводности,
диэлектрическая проницаемость, модуль кручения, носят линейный
характер, который изменяется скачкообразно в области
температуры стеклования. Этот скачок функции свойство — температура не
для всех перечисленных характеристик данного полимера
находится при одинаковой температуре, поэтому для температуры
стеклования следует указывать метод ее определения.
Обычно температуру стеклования определяют по данным
измерении крутильных колебаний. В качестве исследуемого образца
для испытаний на кручение используют стандартные пленочные
полоски. Этот метод применим и для полимеров, которые не
перерабатываются общепринятыми для термопластов способами.
Образцы для испытаний получают осаждением полимера на
проволочной сетке или полимеризацией непосредственно на ней [15].
Полученная кривая кручения после резкого падения при
температуре размягчения позволяет оценить теплостойкость полимерных
материалов при кратковременном нагружении.
3.1.2.3. Определение температуры размягчения
Поведение полимерных материалов при размягчении в
технике характеризуется не температурой стеклования, а
температурой, при которой в определенных условиях реализуется заданная
деформация прессованных или литьевых стандартных брусков,
изготовленных из исследуемого полимера. Общепринятыми
унифицированными методами испытаний являются определение
теплостойкости по Вика и по Мартенсу, а также определение
«температуры допустимой деформации», «деформационной теплостойкости»
и «температуры нулевой прочности».
Для оценки теплостойкости по Вика [16] стальную
иглу-пуансон цилиндрической формы сечением 1 мм2, нагруженную 5 или
1 кгс, помещают на горизонтальную поверхность образца и при
скорости нагрева 50°С/ч определяют температуру, при которой
игла-пуансон вдавливается в образец на глубину 1 мм.
При определении теплостойкости по Мартенсу [17] образец
в виде бруса прямоугольного сечения стандартных размеров
нагружают усилием, при котором изгибающее напряжение постоянно
и равно 50 кгс/см2, и нагревают со скоростью 50 °С/ч. Заданное
отклонение рычага на приборе соответствует теплостойкости
полимеров по Мартенсу.
Для аморфных полимеров отклонение значений
теплостойкости по Вика от температуры стеклования составляет примерно
5—10 град, значения теплостойкости по Мартенсу лежат
примерно на 20—25 град ниже температуры стеклования.
«Температура допустимой деформации», или «деформационная
теплостойкость» [18], представляет собой температуру, при кото-
44
рои образец для испытаний в виде стержня под приложенным к
его концу изгибающем напряжении 18,48 кгс/см2 или 4,62 кгс/см2
и скорости нагрева 2 °С/мин изгибается на 0,254 мм.
Деформационная теплостойкость примерно на 5—10 °С ниже
соответствующей температуры стеклования.
3.2. ДЛИТЕЛЬНЫЕ ИСПЫТАНИЯ ПОЛИМЕРОВ
НА ТЕРМОСТОЙКОСТЬ
При оценке длительной термостойкости полимеров время
проведения испытаний составляет по меньшей мере 8—12 мес.
Параметры, по которым определяется длительная термостойкость,
выбираются в зависимости от областей применения.
При использовании полимеров в качестве конструкционных
материалов, предназначенных для работы при высоких
температурах, ударная вязкость и относительное удлинение при разрыве
являются теми критическими характеристиками, которые прежде
всего изменяются при тепловом старении. Изменение
механических показателей определяют как при температуре теплового
старения, так и при комнатной температуре после теплового
старения. Чаще всего изменение свойств оценивают в процентном
отношении в зависимости от времени хранения.
Длительная термостойкость комбинированных материалов
оценивается обычно по прочности при изгибе. При склеивании
металлов тепловое старение прежде всего влияет на прочность
клеевого соединения на сдвиг.
При использовании полимеров в виде пленок и при нанесении
защитных покрытий оценивают изменение физико-механических и
диэлектрических свойств, а также прочность на истирание после
теплового старения. Особые критерии для оценки свойств при
испытании электроизоляционных лаков рассмотрены в гл. «Поли-
эфироимиды» G.1.4).
Для волокон определяется влияние теплового старения на
прочность, удлинение и модуль.
Длительная термостойкость полимеров
характеризуется температурой, которая не
вызывает при продолжительном тепловом
воздействии существенных изменений
свойств материалов (снижение
показателей, измеренных при комнатной
температуре, составляет примерно 10 % по
сравнению с исходным значением). Эта величина
приводится, как правило, для 25 000 ч
(примерно 3 года). Она определяется путем
Часы
100000
50000
Рис. 3.5. Оценка стойкости полиамидной A) и
полиэфирной B) изоляции проводов к длительному иа-
греву (путем графической экстраполяции) [20].
Годы
10
4
2
WQ 22Q 2601^
12
2
Сутки
20
Ю
6
ч
2-
Часы
15
10
4S
экстраполяции результатов испытании, полученных в течение
8—12 мес. (рис. 3.5) [19].
Другим параметром длительной термостойкости является
период полураспада, или время выдержки, при котором
соответствующие показатели снижаются на 50 % от их исходного значения.
Литература к главам 1—3
1. Mater. Engng. 65 A967) 6, 20—22.
2. G. de Gaudemaris, Publ. Inst. Francais du Petrole 20 A971).
3. F. Wallenberger, Angew. Chem. 76 A964) 11, 484-494.
4. J. Joncs, Chem. in Britain 6 A970) 6, 251—259.
5. Ф. Бильмсйер. Введение в химию полимеров, Издатинлнт, 1959.
6. R. Beaman, J. Polymer Sei. 9 A952), 470—472.
7. H. Elias, Schweizer Aren, angew. Wiss. Techn. 33 A967) 5, 134—142.
8. Chem. Engng. 77 A970) 12, 65—66.
9. J. Stille, J. Polymer Sei. В 4 A966), 791—793.
10. F. Wellenberger, Polymer Engn. Sei. A966) 10, 369.
11. M. Tessler, J. Polymer Sei. А 14 A966), 2521—2532.
12. С. Doyle, WADC Techn. Report 59—95 A959) WPAFB.
13. C. Doyle, WADD Techn. Report 60—283 A960) WPAFB.
14. E. Bobalek, J. appl. Polymer Sei. 25 A959), 210—218.
15. J. Gillham, J. appl. Polymer Sei. 10 A966), 159—169.
16. TGL 17274, DIN 53460, VDE 0302/111. 43, ASTM D 1525-85-T.
17. TGL 14071, DIN 53458, DIN 53462.
18. ASTM-D 648-56, DIN 53461.
19. DIN 46432, Blatt 2.
20. B. v. Bornhaupt, Kunststoffe 61 A971) 1, 46—55.
21. C. Sroog, J. Polymer Sei. А 3 A965), 1373—1390.
22. E. Зинин, Пластмассы A970) 1, 32—34.
23. R. Schulken, J. Polymer Sei. С 6 A964), 17—29.
4. ТЕРМОСТОЙКИЕ ПОЛИОЛЕФИНЫ
И ФТОРСОДЕРЖАЩИЕ ПОЛИОЛЕФИНЫ
4.1. ПОЛИБУТЕН-1
-—сн—сн2—-
[—СН— СН2—1
СН2СНа \п
В основе процесса получения изотактического полибутена-1,
выпускаемого в промышленном масштабе с 1965 г., лежат работы
Натта по стереоспецифической полимеризации высших сс-олефи-
нов. Его преимущества перед полиэтиленом и полипропиленом
заключаются прежде всего в весьма незначительной
температурной зависимости механических показателей и крайне
незначительной склонности к ползучести и образованию трещин вследствие
внутренних напряжений. Основные области применения полибуте-
на-1—производство труб, пленок, емкостей, кабельной изоляции
и пенопластов.
46
4.1.1. ПОЛУЧЕНИЕ И СВОЙСТВА БУТЕНА-1
Бутен-1 при комнатной температуре и нормальном давлении
представляет собой бесцветный газ. Физико-химические свойства
бутена-1 приведены ниже:
Тил (на воздухе при давлении насыщенного пара), °С —185,35
Ткип G60 мм рт. ст.), °С • -6,26
dTldP G60 мм рт. ст.) 0,0337
Плотность при 25 °С, г/см3 0,0588
Температура вспышки, °С —80
Предел взрывоопасиости на воздухе при давлении 760 мм рт. ст. и
температуре 20 СС, % (объемн.)
верхняя граница 9,3
нижняя граница 1,6
Температура вспышки по YDE 0173, °С 445
ГКр 146,4
Ркр 39,7^
Теплота парообразования при 25 °С, ккал/моль - . . 4,87
Теплота испарения вблизи точки кипения, ккал/моль .Г .... 5,238
Свободная энергия образования, ккал/моль 17,09
Энтропия, кал/(моль-°С) 73,04
Удельная теплоемкость при 20 °С, ккал/(кг-ьС) 0,543
Энтальпия полимеризации при 25 °С, ккал/моль —20,7
В промышленности бутен-1 получают дегидратацией я-бута-
нола, каталитическим дегидрированием бутана или димеризацией
этилена. При получении бутена дегидратацией я-бутанола
последний сначала превращают в эфир стеариновой кислоты, при
кипячении которой образуется чистый я-бутен-1. Днмеризация
этилена в бутен-1 протекает на каталитической системе кобальт —
активный уголь. При температуре 50 °С, давлении 50 кгс/см2 и
50%-ном превращении получают я-бутен с выходом 75%,
основную часть которого составляет бутен-1 [1—3]. Если в качестве
носителя катализатора применяется металлический кобальт, то
димеризация может протекать в интервале температур 25—30 °С
14].
Каталитическое дегидрирование фракции С4 в я-бутены при
переработке нефти протекает на катализаторе Сг2Оз/А12Оз при
500—600СС. Чисто термическое дегидрирование замещенных
углеводородов выше С3 невозможно вследствие энергетических
затруднений, так как энергия связи С—С на 30 ккал меньше
энергии связи С—Н. При дегидрировании я-бутана на каталитической
системе Сг2Оз/А12Оз обычно получают смесь бутенов [5].
Бутен-1 получается так же, как промежуточный продукт, при
термическом и каталитическом крекинге. При пиролизе высших
углеводородов (легкая фракция бензина, дизельное топливо)
я-бутен содержится в составе газов крекинга в количестве от 0,5
до 4%. В табл. 4.1 приведены данные о содержании я-бутена
в продуктах пиролиза при получении этилена в зависимости от
типа сырья а условий реакции,
47
Таблица 4.1. Содержание н-бутена в газе пиролиза
при получении этилена
Сырье
Легкий бензин *
Лигроин *
Сырая нефть *
Ромашкинская нефть с различными
пределами кипения
90—120 °С
90—120°С
180—210 °С
180—210 °С
240—270 °С
240—270 °С
Легкая бензинолигроиновая
фракция (за один проход)
Температура
реакции,
вС
775
830
825
850
730
760
720
790
720
790
720
790
Выход
м-бутена,
% (масс,)
3,7
1,3
0,6
1,6
2,4
0,8
3,0
2,3
2,6
2,05
2,7
1,8
3,4
Литературный
источник
[6]
[6]
[6]
[6]
[6]
[в]
m
[7]
[7]
[7]
[71
[7]
[8]
* По способу Лурги.
Другим источником «-бутена являются отходящие газы,
образующиеся при атмосферной перегонке нефти, а также при рифор-
минге и крекинге. В зависимости от условий процесса и состава
нефти доля фракций С4 составляет в среднем 4—5 % (от массы
нефти). Содержание w-бутена во фракции С4 достигает при этом
около 20 %.
Разделение фракции С4 при каталитическом дегидрировании
или при каталитическом или термическом крекинге требует
многочисленных операций. Характеристики фракции С4 приведены
в табл. 4.2. При разделении смеси вначале абсорбцией умеренно
концентрированной серной кислотой отделяют изобутен. Бутен-1
и бутен-2 разделяют перегонкой. Насыщенные углеводороды мож-
Таблица 4.2. Характеристика углеводородов С4 [9]
Углеводород
Изобутан
Изобутен
к-Бутен-1
Бутадиен-1,3
и-Бутан
траис-Бутен-2
цис-Бутен-2
Молекулярная
масса
58,12
56,10
56,10
54,09
58,12
56,10
56,10
Температура
кипения,
°С
-11,73
—6.90
—6,26
—4,41
-0,50
0,88
3,72
Температура
затвердевания,
°С
-159,6
-140,35
-185,35
— 108,9
-138,3
-105,4
-138,9
48
но разделять экстрактивной перегонкой с водным фурфуролом,
ацетоном или ацетонитрилом. По Клею [10], возможно также
непосредственное выделение бутена-1 из фракции С4 методами
экстрактивной перегонки.
Основными примесями е техническом бутене-1 являются бутан,
бутен-2, изобутен и следы ацетилена, диена, воды, оксида и
диоксида углерода, кислорода, а также сернистых соединений. В то
время как бутан, бутен-2 и изобутен инертны по отношению к
катализатору и при полимеризации вызывают лишь снижение
выхода продукта, вода, метанол, кислород и соединения серы
приводят к отравлению каталитической системы, и их необходимо
удалить перед процессом полимеризации. При повышенных
температурах и е присутствии катализатора очень легко протекает
изомеризация бутена-1 в бутен-2.
Бутен-1 в небольших количествах нетоксичен, при
повышенных концентрациях оказывает анестезирующее действие.
4.1.2. СТЕРЕОСПЕЦИФИЧЕСКАЯ ПОЛИМЕРИЗАЦИЯ БУТЕНА-1
До открытия стереоспецифнческой полимеризации были
известны лишь высоковязкие маслообразные или аморфные
продукты полимеризации бутена-1, которые получались радикальной
или катионной полимеризацией [И]. Применение этих продуктов
ограничивалось производством клеев и кабельной
промышленностью.
Изотактический высококристаллический полибутен-1 был
получен вскоре после открытия Натта [12, 13] в 1954 г. стереоспе-
цифической полимеризации пропилена. Высококристаллический
полибутен-1 (ПБ) был впервые выпущен в промышленном
масштабе в ФРГ в 1965 г. под марками Вестолен БТ и Петротекс и
в США под маркой Бу-Туф. Фирма «Petro Тех Chemical Со.»
в дальнейшем прекратила его изготовление. В 1970 г. началось
опытное производство ПБ фирмой «Mobil Chem. Corp.» (США)
преимущественно для производства упаковочного материала.
К 1971 г. производство полибутена Вестолен БТ имело мощнасть
12 000 т в год [13]. В 1972 г. фирмой «Nichiya Chemicals»
(дочернее предприятие «Nippon Oil and Fats Go.») было организовано
производство мощностью 5600 т в год.
Изотактический полибутен-1 получается полимеризацией при
низком давлении бутена-1 в присутствии стереоспецифических
катализаторов Циглера — Натта. По технологии и типу
каталитических систем процесс получения полибутена-1 аналогичен
процессу полимеризации пропилена. В то же время каталитические
системы, предназначенные для полимеризации этилена при низком
давлении, при полимеризации бутена-1 или неактивны
(производные циклопентадиенилтитана + алюминииалкил), или они изоме-
ризуют бутен-1 в бутен-2 [например, СгС13 + АЦСгНвЬ].
Типичные двух- и трехкомпонентные системы для полимеризации
бутена-1 приведены ниже;
49
Литературно
источник
TiCU—Al(C2HsJCl [15,16]
TiCla—AI(C2H5JCI [17-191
VCU—Al(C2Hs)a [21]
WC16—A1(C2H5K [22]
TiCI3—А1(С2Н5)з—А1(С2Н5JС1 [23]
TiCI3—AI(C2H5K—NaCl [24]
TiCI3—AI (C2H5K-CuSCH3 [29]
TiCIa—Al (C2H5) 3—VOCI3 [30]
TiCI4—Al (C2H5) 3-C6H5NO2 [25]
TiCU—AI (C2H5) 3—SnCI2 [26]
TiCU—А!(С4Нэ)з—Sn(C4H9KH [31]
VCI3-AI (C12H25K-As (CH3) 3 [27]
CrCh—Al (C4H9JH — ТГФ — технический углерод [28]
В качестве соединений переходных металлов применяют гало-
гениды п оксигалогениды титана, ванадия и вольфрама.
Использование трехкомпонентной системы позволяет увеличить выход
полимера заданной тактичности. Роль составляющих в
трехкомпонентной каталитической системе [14] заключается в ускорении
превращения алюминийалкилов в алюминийгалогендиалкилы,
являющиеся активными центрами трехкомпонентной каталитической
системы.
При полимеризации бутена-1 на катализаторах Циглера —
Натта наряду с изотактическим полимером образуются два других
стереоизомера — атактический и стереоблочный:
H C2H5 H C2H5 H H H C2H5
..._ ^ ^ 1~Г""
H H H H H C2HS H H
Атактический полибутен-1
H C2H5 H C2H5 H C2H5 H C2H5
II II II II
С—С С—С С—С С—С
II II II II
нн нн нн нн
Изотактический полнбутен-1
Н С2Н5 H С2Н5 H С2Н5 H С2Н5 H H С2Н5 H С2Н5
С—С С-С С—С С—С С—С С С—С
II II II II II I II
НН НН НН НН H С2Н5 H H H
Стереоблочный полибутен-1
Степень тактичности полимера определяется природой
каталитической системы. Согласно Натта [21], имеется
непосредственная связь между структурой нерастворимого каталитического
комплекса и долей изотактических фрагментов в полимере.
Степень тактичности уменьшается по мере того, как реакционный
продукт из многокомпонентной каталитической системы
становится аморфным и легко диспергируемым. Кристаллические катали-
69
тнческие системы позволяют получать высококрпсталлические
изотактические полимеры. В присутствии растворимых
катализаторов получается атактический продукт. Другими факторами,
существенно влияющими на тактичность синтезируемого ПБ, являются
склонность к старению данной каталитической системы, а также
введение добавок (например, пиридина) к каталитическому
комплексу. ПБ с повышенной стереорегулярностью (эфирорастворимая
часть <С2°/о) получается при добавках в качестве активатора ди-
этилалюминийгидрида [32].
Для получения полнбутена-1 с высокой степенью тактичности
вместо циглеровской системы можно использовать двухкомпонент-
ную систему цпнкалкнл или кадмийалкил/трихлорид титана [33].
Бутен-2 из-за стернческих препятствий полимеризуется с
трудом и изомеризуется в бутен-1. Несмотря на то что равновесная
концентрация бутена-1 при этом составляет всего 3—5%,
вследствие высокой скорости полимеризации на каталитической системе
Циглера — Натта происходит количественное превращение буте-
на-2 в высокомолекулярный полибутен-1 [39].
Условия стереоспецифической полимеризации бутена-1 близки
использующимся при получении изотактического полипропилена.
Реакцию проводят в растворе инертных углеводородов, таких, как
гептан, октан, циклогексан, петролейный эфир, бензол или толуол,
при атмосферном или пониженном д? влении. Однако вследствие
малого давления паров бутена-1 возможна полимеризация в
массе в присутствии пропилена [37]. Соотношение металлалкил/сое-
динение переходного металла для двухкомпонентной системы
находится в пределах от 5:1 до 2:1. Полимеризация проводится
в интервале температур 20—180 °С [38], Мономеры и
растворители не должны содержать влаги и соединений с активным
водородом. Дезактивирование катализатора протекает также под
влиянием серосодержащих соединений, кислорода и оксида углерода.
Молекулярную массу можно регулировать введением водорода
[34], винилгалогенидов [35] или цинкалкилов [36].
Полимеризацию обрывают дезактивацией каталитической
системы спиртами или смесями спирт—НС1. Полимер отмывают от
остатков катализатора растворением в углеводородах типа
гептана при 80°С или диспергированием в воде [42, 43].
Полимеризация бутена-1 в газовой фазе описана Ракусом [40].
Наполненный ПБ может быть получен непосредственно
полимеризацией на катализаторах Циглера — Натта в присутствии
инертных наполнителей [41].
4.1.8. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИБУТЕНА-1
4.1.3.1. Структура
Получающийся полимеризацией на катализаторах Циглера —
Натта полибутен-1 представляет собой смесь стереоизомеров,
состоящую из атактического, изотактического и стереоблочного
51
полимеров (ilx строение описано выше). Оценка тактичности и сте-
реорегулярности может проводиться методом ИК-спектроскопии.
Полосы поглощения при 1208, 1098, 1059, 1028, 922, 849 и 816 см-1
характеризуют степень кристалличности; для расплава и раствора
полимера в CS2 они отсутствуют. Интенсивность полос при 1220,
972 и 917 см-1 пропорциональна стереорегулярности полимера
[44]. Другой метод определения тактичности основан на
экстрагировании полимера диэтиловым эфиром. Изотактическая часть и
полимерные цепи с длинными стереоблокамн не растворяются, в то
время как атактическая часть и стереоблочный полимер с
короткими сегментами диэтиловым эфиром вымываются [45, 46].
4.1.3.2. Кристалличность
Изотактический ПБ существует по меньшей мере в трех
полиморфных кристаллических модификациях (табл. 4.3). В отличие
от других полиолефинов эти три модификации легко выделить
в чистом виде.
При кристаллизации изотактического ПБ из расплава сначала
образуются термодинамически нестабильные кристаллы
модификации II. Образование этой модификации происходит с высоким
отрицательным температурным коэффициентом [51]. Период по-
Таблица 4.3. Кристаллографические характеристики
изотактического полибутена-1 [13, 47—50]
Характеристика
Модификация
I
Ромбоэдрическая
17,7
17,7
6,5
6,5
3,
0,95
130—140
1450±50
3,6±0.3
135,5
Из
модификации II
Тетрагональная
14,89
14,89
20,87
6,8
Из
Р4
0,89
120-126
1500±300
3,8±0,7
124,0
Из расплава
III
Ромбическая
12,49
8,96
7,6
0,90
90—96
1550=Ы50
4,1 ±0,3
106,5
Из раствора
Элементарная ячейка
Размеры элементарной
ячейки, A
а
b
с
Период идентичности, А
Конформация цепи
Пространственная группа
Плотность, г/см3
Температурный интервал
плавления, СС
Энтальпия плавления,
кал/моль
Энтропия плавления,
кал/ (моль -°С)
Разновесная температура
плавления, °С
Стабильность при 20 °С
Образование модификации
52
Рис. 4.1. Изменение периода фазового полупревращения ^
модификация II — модификация I для сополимера буте- jqqqq
на-1 с пропиленом [54].
1QQQ г
лупревращения при переходе расплава в
модификацию II составляет 380 мин при 140 °С
и 2 мин при 90°С.
Термодинамически нестабильная
модификация II со временем необратимо переходит
в модификацию I. Этот переход в большой 0 2 4 6 с,%(пол)
степени зависит от температуры. При
температурах выше 20 °С и ниже 80 °С модификационные изменения
протекают медленно, максимальная скорость достигается при
комнатной температуре [52]. Скорость превращения зависит также от
молекулярной массы, тактичности, приложенных внешних
нагрузок, добавок и компонентов сополимеризации.
Высокомолекулярный ПБ с повышенным содержанием атактической или стереоб-
лочной фазы превращается медленнее, чем полимер с высокой
степенью стереорегулярности, и низкой молекулярной массой [53].
Скорость перехода для экструдированного образца полимера с
молекулярной массой 4000 в 100 раз выше скорости перехода в
исходном образце [54]. Давление и приложенная нагрузка, а также
остатки катализаторов ускоряют фазовый переход [55]. Добавка
нзопропанола или эфира уксусной кислоты приводит к увеличению
скорости перехода [56]. Сополимеры бутена-1 с короткоцепными
олефинами имеют небольшие изотактические блоки в полимере,
что также ускоряет фазовый переход в термодинамически
стабильную модификацию I.
На рис. 4.1 показан период полупревращения модификации II
в модификацию I для сополимеров бутена-1 с пропиленом в
зависимости от содержания пропилена в сополимере. Напротив, для
сополимера с длинноцепочными а-олефинами или разветвленными
олефинами скорости превращения сильно снижаются вследствие
стерическпх эффектов [54] :
Содержание Период
в сополимере, полупреврцце-
% (мол.) ння II-I, сут
Октен-1 1,5 105
Додецен-1 3,0 125
4-Метилпентен-1 7,0 51
При содержании в сополимере 7 % (мол.) 4-метилпентана-1
скорость перехода уменьшается в 50 раз [54]. Аналогично изотакти-
ческому полипропилену ПБ имеет стабильную модификацию I,
соответствующую конформации Згспираль (рис. 4.2). В
ромбоэдрической элементарной ячейке молекулы упакованы более плотно,
чем в нестабильной модификации II. Переход тетрагональной
формы II в гексагональную форму I связан с увеличением
плотности, твердости, вязкости и прочности материалов (рис. 4.3).
53
Третью кристаллическую модификацию изотактического ПБ
можно получить из раствора, например осаждением раствора ПБ
в бензоле или декалине, растворением в амилацетате при 90—
120°С и кристаллизацией при температурах ниже 58°С [47] или
при получении пленок поливом из раствора в тетрахлориде
углерода либо толуоле [59]. Модификация III стабильна при
комнатной температуре. В расплаве или при высоких температурах
происходит ее переход в модификацию II [49, 60]. Для всех трех
модификаций характерна тонколамелярная структура полимерных
кристаллов.
Размер сферолитов, образующихся из расплава
кристаллизующегося ПБ, равен примерно 100 мкм.
В случае сополимеров с пропиленом
получаются сферолиты очень малых
размеров [61].
Кристалличность ПБ можно
определить также по плотности [62].
Содержание кристаллической фазы
рассчитывается по формуле
¦—[«-
где da — плотность аморфного ПБ при 25 °С,
равная 0,868 г/см3; dc — плотность
кристаллической модификации I при 25 °С, равная
0,950 г/см3; d — плотность образца при 25 °С
Температура стеклования ПБ
линейно возрастает с увеличением
степени кристалличности. Так, при
увеличении степени кристалличности с 43
до 74 % температура стеклования
повышается со 128 до 132 °С [62].
Рис. 4.2. Пространственная модель
модификации I изотактического полибутена-1 [57].
Рис. 4.3. Зависимость твердости по Шору A) и
прочности при растяжении B) вблизи предела
текучести от продолжительности хранения
кристаллизующегося расплава прн комнатной
температуре (полиморфное превращение
полибутена-1 из тетрагональной формы II в
стабильную гексагональную форму I) [58],
200
-юо
70
50
, шла/70 D
Y
\ \
GpyKCC
2
m-
! 1
54
4.1.3.3. Свойства растворов
Растворимость изотактического ПБ в данном растворителе и
критическая температура растворения зависят не только от
химического строения и средней молекулярной массы, но и от степени
упорядоченности ПБ. При экстрагировании кипящим диэтило-
вым эфиром можно разделить атактическую и изотактическую
части.
Изотактический ПБ растворим при повышенных температурах
в ароматических углеводородах, таких, как бензол, толуол,
декалин, тетралин, и в хлорированных углеводородах, таких, как
а-хлорнафталин или хлороформ. 6-Температура растворения в
анизоле атактического ПБ равна 82,6°С и изотактического ПБ 89,1 °С
[63]. При охлаждении раствора изотактического ПБ до комнатной
температуры выпадает осадок. Так, при охлаждении толуольного
раствора полимера в течение 24 ч до 40СС происходит фазовое
разделение [63].
Для фракционирования изотактического ПБ применяют
системы лигроин — этанол [64] и циклогексанон — циклогексанол с
гликолем C:1) при П5СС.
Определение молекулярно-массового распределения методом
гель-проникающей хроматографиии описано в работах [65, 66].
Для предотвращения деструкции ПБ в растворе при повышенных
температурах используются антиоксиданты, такие, как 4-метил-2,6-
цп-трет-бутил фенол.
Коэффициенты уравнения Марка — Хаувинка — Куна для вис-
козиметрического определения молекулярной массы приведены для
ряда растворителей в табл. 4.4; данные о размерах невозмущенного
клубка ПБ в растворах приведены в табл. 4.5.
Таблица 4.4 Коэффициенты в уравнении Марка — Хаувинка — Куна
[к]] = КМа для полибутена-1 ([ц\ измеряется в см3/г)
Полимер
Атактиче-
ский
Изотактический
Растворитель
Бензол
Этилцикло-
гексан
Анизол
Гептан
»
Этилцикло-
гексан
Нонан
Декалин
пература,
°С
30
70
86,2
35
60
70
80
115
2,24-10~2
0,73- 10~2
12,3-10-'
0,47-10
1,5 -10~2
0,73 • 10~2
0,58- 10~2
0,95. 10
а
0,72
0,80
0,50
0,80
0,69
0,80
0,80
0,73
Молекулярная
масса
3-102-5.103
4- 104—1,3 - 106
1- 105—1,3- 106
4-104-9- 105
4-104-9-105
8-104-9-105
1-103—9-Ю5
4- 10*—9 • 105
Литературный
источник
[67]
[63J
[63]
[64]
[64]
[63]
[63]
164]
Таблица 4.5. Невозмущенные размеры клубка полибутена-1 [63]
Показатель
Температура, °С
SIMf ¦ 103, A
К • Ю5, 100 мл/г
Го/М'^.Ю3, A
гв/М[!2-\0\ А
Метод
Изотактический
полибутен-1
анизол — этил-
циклогексан
70
1234=5
775+25
427
183±0,05
Определение
в Э-растворителе
Изотактический
полибутен-1
н-ноиан
80
520+50
1290+90
427
3,0±0,25
Светорассеяние
Атактический
полибутен-1
н-нонан
35
590+50
1180±70
427
2,76+0,20
Светорассеяние
Примечание. S—Z-среднее значение радиуса вращения; /( — константа вязкости;
г —расстояние между концами цепи (экспериментальное значение); г —расстояние между
концами цепн (рассчитано по длине связи и валентному углу).
4.1.4. ПЕРЕРАБОТКА ИЗОТАКТИЧЕСКОГО ПОЛИБУТЕНА-1
Полибутен-l можно перерабатывать на обычных машинах для
переработки термопластов методами экструзии, литья под
давлением, ротационным литьем, каландрованием или прессованием.
Хорошая перерабатываемость, в частности, методом ротационного
литья обусловлена более низкой вязкостью расплава по сравнению
с другими полиолефинами, что объясняется подвижностью боковых
групп. Высокомолекулярный ПБ перерабатывается значительно
легче, чем полиэтилен или полипропилен такой же молекулярной
массы. Полибутен с Mw = 3-106 имеет при 190°С такой же
показатель текучести расплава, как ПЭ с Mw = 18 • 104 [53]. Кривая
зависимости текучести расплава от температуры для ПБ значительно
круче, чем для ПЭ, и смещена (по сравнению с полипропиленом)
в область более низких температур.
При производстве труб из ПБ методом экструзии применяют
шнеки такой же геометрии, как для ПЭНП, со степенью сжатия
от 3: 1 до 4: 1. Температура по зонам от загрузочной воронки до
сопла составляет 140—165СС. Вследствие усадки материала
необходимо увеличить внутренний диаметр сопла на 10—15 % по
отношению к калибровочным размерам. При этом намотка на барабан
должна быть очень слабой, так как при превращении в стабильную
модификацию I в течение 3—7 сут происходит дальнейшая усадка
на 1—2 %. Поэтому резку трубчатых изделий можно производить
не ранее чем через 1 сут после экструзии [68].
Переработку ПБ литьем под давлением можно осуществлять на
том же оборудовании, что и переработку ПЭ при температурах
200—290°С. Вследствие пониженной теплопроводности требуется
более длительное охлаждение его в форме, чем полиэтилена и nq-
лйпропилепа. Время вылеживания литьевых изделий до их
использования должно составлять примерно 7 сут для того, чтобы
произошло качественное превращение в кристаллическую форму I и
сформировались оптимальные прочностные свойства.
Для ротационного литья емкостей применяется ПБ с
показателем текучести расплава, равным примерно 20. При сравнимой
толщине стенок продолжительность цикла почти такая же, как и для
ПЭВП. Вследствие незначительной скорости кристаллизации
изделия из ПБ, полученные ротационным литьем, не имеют остаточных
напряжений. При показателе текучести расплава, равном 20,
изделия имеют модуль упругости при растяжении около 1700 кгс/см2
[58].
Для получения волокон из ПБ используются сополимеры с 10—
20 % этилена или пропилена. При переходе полимера в
кристаллическую форму I прочность волокна может достигать 940 кгс/см2.
В качестве порообразователей для получения вспененного ПБ
применяют смеси из азодикарбонамида, 2,5-диметил-2,5-пероксп-
гексана, серы и триаллилцианурата [73] или непрореагировавший
бутен-1 [74]. Эластичные пенопласты, содержащие 3% талька,
применяются для изготовления изоляции [75, 76].
Тонкие пленки для упаковки получают путем полива из
раствора ПБ в изоамилацетате при 66°С [77].
ПБ с неорганическими и органическими наполнителями хорошо
деформируются без заметного снижения механических показателен
[79]. Графит используют как усиливающий наполнитель [80].
Полибутен-1 не совмещается с ПЭНП, но может
перерабатываться в смеси с ПЭВП и полипропиленом.
Заготовки из ПБ можно сваривать, склеивать и резать. Для
соединения стыкуемых деталей используют сварку нагретым газом,
нагретым инструментом, за счет трения, токами высокой частоты
и ультразвуком [69]. Сварка пластин при нагревании
осуществляется по режиму:
Температура нагревательного элемента, СС 250U0
Давление на нагревательный элемент,
кгс/см2 0,5
Время нагрева, с 30—40
Давление прижима при сварке, кгс/см2 1—2
Время охлаждения под давлением, с . . Около 90
Оптимальное соотношение прочности при растяжении
сваренных и исходных материалов («фактор сварки») составляет 0,65—
0,70. Длительные испытания показали очень высокий срок службы
сваренных труб из ПБ. На трубопроводах для горячего
водоснабжения, работающих под давлением, со сварными фитингами для
соединения труб (диаметр трубы 32 мм), эксплуатировавшихся
зимой в течение 3 лет при температуре воды 60—80СС и давлении
3 кгс/см2, не было обнаружено никаких повреждений [70].
При склеивании ПБ синтетическими клеями поверхности
стыкуемых деталей следует предварительно обработать хромовой
смесью из бихромата калия или натрия и серной кислоты.
57
Таблица 4.6 Технологические режимы обработки резанием
полибутена-1 [69]
Технологи ческая
операция
Строгание
Фрезерование
Точение
Сверление
8,5 мм
23 мм
Резка
ножовкой
дисковой пилой
ленточной пилой
Скорость,
м/мин
5-35
15-70
5-100
5
14
55—130 ход/мин
1000-2000
500-1500
Скорость
подачн,
мм/об
0,2
0,2
0,1-0,8
0,1—0,3
0,1—0,3
0,1 мм/зуб
0,3 мм/зуб
0,1 мм/зуб
Задний
угол,
град
10-15
10-15
10—15
—
—
30—40
30-35
30-35
Угол
зажима,
град
10-15
10-15
10-15
25-30 *
25-30*
5-8
10-12
8-10
• Угол наклона спирали.
В отдельных случаях, например при соединении труб с трубными
муфтами, можно использовать специальные клеи без
предварительной обработки [71,72].
Технологические режимы процессов обработки полибутена-1
резанием приведены в табл. 4.6.
Материал заготовки, подвергаемой обработке, должен
находиться в стабильной модификации I. Если в полимере содержится
нестабильная модификация II, то после механической обработки
в заготовке появляется усадка, снижающая точность размеров
изделий.
4.1.5. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА
ИЗОТАКТИЧЕСКОГО ПОЛИБУТЕНА-1
4.1.5.1. Теплофизические свойства
Основные показатели теплофизических свойств ПБ приведены
ниже:
Температура, °С
плавления 124—130
размягчения 120—126
хрупкости —15
стеклования —45
Теплостойкость по Вика, °С 96
Термический коэффициент линейного
расширения, град-' 1,5 • 10~4
Удельная теплоемкость, кал/(г-°С) . . 0,49
Коэффициент теплопроводности, ккал/(м X
X ч-°С) 0,20
Плотность, г/см3 . 0,915
Водопоглощение за 24 ч (пленка
толщиной 0,3 мм), % 0,01-0,026
Деформационная теплостойкость при
4,64 кгс/см2, °С 155
58
По температуре плавления, составляющей 124—130 С, ПБ
можно расположить между ПЭНД и ПЭВД. Его плотность, равная
0,915 г/см3, несколько ниже, чем плотность ПЭНП, и близка к
плотности нзотактического полипропилена.
Негорючие рецептуры на основе ПБ можно получить путем
введения 3—7 % оксида сурьмы, 4—8 % фторобората аммония, 2—4 %
твердого парафина и 3 % гексахлорэтана. Продолжительность
горения материала на основе ПБ, полученного по этой рецептуре,
всего 0,5—0,9 с после воспламенения [81].
4.1.5.2. Физико-механические свойства
Преимуществами ПБ по сравнению с другими полиолефинами,
выпускаемыми промышленностью, является малая ползучесть,
незначительная склонность к образованию трещин вследствие
возникновения остаточных напряжений, а также небольшая
зависимость прочности при растяжении от температуры.
Физико-механические свойства изотактического ПБ
определяются наряду с типом кристаллической структуры (при обычных
условиях модификации I) средней молекулярной массой и
тактичностью, а также предварительной тепловой и механической
обработкой. Медленное охлаждение и ориентация кристаллитов
способствуют повышению степени упорядоченности, и благодаря
образованию сферолитов — улучшению механических свойств. Основные
показатели физико-механических свойств ПБ приведены ниже:
Показатель текучести расплава при 5,0 кгс, г/10 мин . . , 0,5
Предел текучести при растяжении, кгс/см2 150—250
Прочность при растяжении, кгс/см2 250—400
Относительное удлинение при разрыве, % 300—400
Ударная вязкость Не разрушается
Предельное изгибающее напряжение, кгс/см2 150—250
Модуль упругости при изгибе, кгс/см2 800—2000
Модуль эластичности, кгс/см2 5000—9000
Твердость при вдавливании шарика в течение 60 с, кгс/см2 250—450
Твердость по Шору, шкала D 60—68
Модуль упругости при сжатии, кгс/см2 2180
Остаточное удлинение при разрыве, % 16
Ударная вязкость образцов с надрезом, кгс-см/см2
20 °С Не разрушается
0°С 25
-10 °С 25
-20 °С 16
Поведение ПБ при растяжении заметно отличается от
характера деформации полипропилена и полиэтилена. В то время как
у ПП и ПЭ после превышения предела текучести резко
ослабляется зависимость прочности при растяжении от относительного
удлинения (на кривой появляется плато, соответствующее уровню
напряжения, при котором материал течет), у ПБ прочность
69
¦ Р, кгс/см2.
200Ы
X, ,
2
1 1 1
3
1 1
0 50 100150 200250 300 350 ?,%
Рис. 4.4. Деформационно-прочностные кривые различных полимеров [58]:
/ —ПЭВД; 2 — ПП; 3 — полибутен-1.
Рис. 4.5. Температурная зависимость прочности при растяжении полиолефинов,
полученных ионно-координационной полимеризацией [53]:
/ — ПЭ; 2 — ПП; 3 — полибутен-1.
Рис. 4.6. Кривые ползучести различных полимеров при растяжении под
нагрузкой 150 кгс/см2 при 23 °С [53]:
/ —ПЭ; 2 — ПП; 3 — полнбутен-1.
возрастает с увеличением относительного удлинения вплоть до
разрушения образца (рис. 4.4).
Результаты исследований двойного лучепреломления
полиолефинов свидетельствуют о том, что выше предела текучести при
растяжении при одинаковом удлинении у ПБ наблюдается более
высокая степень ориентации по сравнению с полиэтиленом и
полипропиленом [82]. При разрушении образца происходит стягивание
ориентированных областей, как если бы в этих зонах была
аккумулирована упругая энергия. Такое поведение объясняется
присутствием свернутых макромолекул внутри кристаллических областей,
которые способствуют более широкому распределению напряжений
внутри образца [62].
Повышенная прочность при растяжении ПБ по сравнению
с этим показателем для ПЭНП и полипропилена обусловлена ори-
ентационным эффектом. При степени кристалличности 50—60 %
прочность ПБ при растяжении составляет 300—350 кгс/см2 по
сравнению со 140—200 кгс/см2 у ПЭНП и полипропилена.
Такие прочностные характеристики, как прочность при
растяжении и модуль упругости зависят от тепловой предыстории и
степени кристалличности образца. Быстрое охлаждение расплава,
способствующее образованию более мелких кристаллитов, приводит
к повышенным значениям прочности. Прочность при растяжении
ПБ снижается с повышением температуры меньше, чем у ПЭ или
ПП (рис. 4.5). Это объясняется незначительным уменьшением
степени кристалличности при повышении температуры по сравнению
со степенью кристалличности ПЭ и ПП. Такой же характер
температурной зависимости имеет модуль растяжения и сдвига. В
области от 0 до 80 °С прочность при растяжении с повышением
температуры снижается менее интенсивно, чем в области выше 80 °С,
60
Рис. 4.7. Предел усталости полибутена-1
при различном удлинении образцов при
23 (о), 60 (б) и 100 °С (в) [54].
Ю
6
/о
10 '
103
, Y
10-
2-
¦ —
I
"(Г
"¦————,
——-—.
— ——
- —
1 т
/
Г
t
^ ^т ^т ^а
t
ю
Ю'
to'
Особенностью ПБ является
очень незначительная склонность
его к ползучести, т. е. к развитию
пластических деформаций под
нагрузкой при температурах ниже
температуры размягчения. Ре- f
зультаты долговременных
испытаний ПБ на растяжение по ргКГС/см2
DIN 53444 показаны на рис. 4.6.
Хотя ПБ при комнатной темпе- ^
ратуре под действием растяги- б
вающего напряжения 150 кгс/см2
имеет большее начальное удлине- 2
ние (8%), чем ПЭ D%),
нарастание пластических деформаций
у ПЭ или ПП с увеличением
продолжительности нагружения
происходит намного быстрее, чем у
ПБ. После 500 ч нагружения от- /"
носительное удлинение образцов
ПБ составляет 13 % по сравне- Р,кгс/см2
нию с 800 % у ПЭ и 700 % у ПП. Ю2
Такое поведение ПБ под нагруз- 6
кой связано с наличием большого
числа свернутых молекул в
кристаллических областях. Боковые Ю
этильные группы создают
дополнительные энергетические
барьеры, которые противодействуют
скольжению цепей относительно
друг друга. Для конструктора
очень важным показателем
является, как правило, предел текучести при 2%-ном удлинении
(рис. 4.7). Это значение удлинения вполне может быть принято
при расчете и конструировании сосудов и трубопроводов из ПБ.
Формовочные материалы на основе ПБ обладают высокой
эластичностью. У ПБ и ПП способность восстанавливать размеры и
форму одинакова.
Вязкость ПБ при температурах выше температуры замерзания
имеет тот же порядок величины, что и вязкость ПЭВП.
ПБ хорошо совмещается с наполнителями без заметного
снижения прочностных показателей. Физико-механические показатели
ПБ, наполненного различным количеством технического углерода,
приведены в табл. 4,7,
2 =;
2-
i
1Q
61
Таблица 47 Физико-механические свойства полибутена-1,
наполненного техническим углеродом [83]
Показатель
Плотность, г/см3
Прочность при растяжении, кгс/см2
Предел текучести при растяжении,
кгс/см2
Модуль упругости при изгибе,
кгс/см2
Относительное удлинение при
разрыве, %
Твердость по Шору, шкала D
Показатель текучести расплава,
г/10 мин
Термический коэффициент линейного
расширения, град-'
Содержание технического углерода, %
0
0,912
270
155
1800
350
65
0,5
15- 10~
3
0,915
270
155
1800
350
65
0,5
15-Ю-5
23
1,02
180
170
5300
100
69
0,1
15 - 10~5
4.1.5.3. Электрические свойства
Полибутен-1, так же как ПЭ и ПП, является отличным
электроизоляционным материалом. Электрические характеристики
полибутена-1 не отличаются от соответствующих показателей для прочих
полиолефинов:
Диэлектрическая проницаемость при 102—107 Гц 2,2—2,
Тангенс угла диэлектрических потерь 7-10~*
Удельное объемное электрическое сопротивление, Ом-см .... 1018
Удельное поверхностное электрическое сопротивление, Ом ... 1018
Электрическая прочность кВ/мм 70
4.1.5.4. Химическая стойкость и коррозионное растрескивание
ПБ при комнатной температуре имеет высокую стойкость к
действию разбавленных неорганических кислот, оснований, солей и
большинства алифатических органических растворителей.
Механические характеристики ПБ снижаются в кислотах, являющихся
окислителями, вследствие химической деструкции и в
ароматических и хлорированных углеводородах в результате набухания
полимера. После 6 мес. выдержки при 23 °С в концентрированной и
разбавленной соляной кислоте, фосфорной кислоте, серной кислоте,
а также в 20 %-ном растворе едкого натра снижения прочности
материала не обнаружено. В концентрированной азотной кислоте
потеря прочности составляет 40 %. При одинаковых условиях
прочность не изменяется при выдержке ПБ в воде, метаноле, этаноле,
ацетоне, минеральных маслах, 36 %-ном растворе формальдегида,
а также в 3 %-ном пероксиде водорода. При выдержке в бензоле
прочность снижается до 78 %• В диэтиловом эфире и силиконовом
масле прочность повышается до 105 % [58]. ПБ незначительно на-
62
бухает в днэтиловом эфире, изоокгане, бензоле, толуоле и тетра-
гидронафталине как при 23 °С, так и при 60 °С [84].
Полибутен-1 обладает отличной стойкостью к коррозионному
растрескиванию, что обусловлено «внутренним отжигом» при
превращении в кристаллическую модификацию I. Появления трещин
на образцах вследствие возникновения остаточных напряжений
после 2000 ч выдержки в спиртах и моющих средствах не
наблюдалось [53].
4.1.5.5. Токсичность
Полибутен-1 с токсикологической точки зрения является
безвредным соединением. Добавление в корм крысам 1—10 % ПБ
в течение 6 мес. не вызывало у них никаких изменений [85].
4.1.6. ОСНОВНЫЕ ОБЛАСТИ ПРИМЕНЕНИЯ ПОЛИБУТЕНА-1
Материалы на основе ПБ используются в тех областях, где
требуется сочетание высокой трещиностойкости, незначительной
склонности к ползучести и низких температурных коэффициентов
механических характеристик.
Трубы из полибутена-1 применяются прежде всего для
изготовления хозяйственно-питьевых и оросительных водопроводов,
отопительных систем в жилых домах, а также трубопроводов для
транспортирования природного газа и химически агрессивных сред. Эти
трубы при длительном нагружении высокими давлениями и при
повышенных температурах обладают значительно более высокой
длительной прочностью и незначительной склонностью к
коррозионному растрескиванию, чем трубы из ПЭ или полипропилена.
Срок службы труб диаметром 32 мм и толщиной стенки 2,9 мм при
температуре испытаний 80°С и тангенциальном напряжении а,
равном 100 кгс/см2, для ПЭНП и ПП составляет меньше 1 ч, в то
время как для ПБ он достигает 1500 ч [53]. Трубы из ПБ имеют
высокую стойкость к колебаниям температуры. Трубы из ПБ,
полностью заполненные водой, охлаждали до —60 °С и
размораживали в течение 120 ч пять раз. При этом диаметр труб увеличивался
на 0,5%, кроме этого, никаких изменений не происходило [68].
Трубы из ПБ, а также сополимеров бутена с 5 % этилена [87]
пригодны при длительных условиях эксплуатации для всех тепло-
проводящих систем внутри жилых зданий. Трубы из ПБ
применяются в системах водяного отопления при изготовлении
отопительных панелей в полу помещения для нагревания поверхности
пола до 28 °С. Трубы могут быть непосредственно уложены с
катушек под бесшовный (сплошной) или мозаичный пол [86].
Оригинальная технология для укладки хозяйственно-питьевых
водопроводов из ПБ была применена в Финляндии [88].
Трубопроводы, предназначенные для работы под давлением 8,5 кгс/см2,
были уложены зимой над замерзшим озером, сварены и после
оттаивания ледяного покрова погружены с бетонными кольцами на дно
озера.
63
и,
80
40
0
/а
- ^
->?
(-^
1
\
Рис. 4.8. Зависимость усадки полнбутспа-1 от
температуры [58]:
/ — ориентированный ПЭ; 2 — полибутен-1;
3 — радиационно-сшитый ПЭ.
70 90 110 ПО I,°C
Трубы из ПБ имеют отличную стойкость к механическому
царапанию [68]. В обогатительной установке более одного года
успешно использовали трубы из ПБ диаметром 100 мм для
транспортирования водных дисперсий песка с острыми кромками (содержание
твердого вещества 5%) при скорости потока 5 м/с. В тех же
условиях стальные трубы с твердостью по Бринеллю 180 должны были
заменяться через каждые 2 мес.
Высокая прочность при действии внутреннего давления в
сочетании с высокой химической стойкостью позволяет широко
использовать трубы из ПБ в химической промышленности. Для
транспортирования 30%-ной отработанной серной кислоты применяли
трубопроводы из ПБ при 60 °С и давлении 4 кгс/см2 [89].
Хорошая адгезия к различным материалам делает ПБ
пригодным для кэширования бумаги и текстильных изделий, а также
алюминия.
Большегрузные контейнеры, изготовленные из ПБ методом
ротационного литья, отличаются незначительной ползучестью,
высокой прочностью и трещиностойкостью, а также коррозионной
стойкостью к действию агрессивных сред. Они применяются в
химической промышленности (складские емкости для хранения,
небольшие напорные емкости) и в агротехнике (силосные башни для
хранения корма, емкости для растворов удобрений) [58].
Пленки из ПБ, полученные экструзией с раздувом, имеют ряд
преимуществ по сравнению с полиэтиленовыми пленками — они
обладают более высокой прочностью, незначительной
проницаемостью и большей стойкостью к коррозии. Они применяются в
основном для изготовлениия сборников отходов одноразового
пользования [58]. В подвесной клети мешок из ПБ размером 500 X
X 150X0,03 мм может быть нагружен 70 кг воды. Для мешка из
ПЭНП тех же размеров толщиной 0,075 мм предельная нагрузка
составляет всего 30—40 кг воды. Кроме того, прозрачные и стойкие
к кипячению пленки из ПБ применяются для упаковки промыш*
ленных товаров и пищевых продуктов.
Для специальных упаковочных целей изготавливают
ориентированные термоусаживающиеся пленки из ПБ различной
жесткости [58]. Прочность при растяжении таких пленок составляет
100—120 кгс/см2. Усадка пленок происходит при температурах от
100 до 180°С. Зависимость усадки от температуры для полибу-
тена-1 и полиэтилена показана на рис. 4.8. Нижняя граница усадки,
составляющая 30 % при 95 °С, обеспечивает пригодность пленки из
ПБ для упаковки пищевых продуктов.
64
Хорошая совместимость ПБ с наполнителями и значительное
улучшение механических свойств позволяют использовать эти
рецептуры в качестве материалов для покрытия полов.
Смеси ПБ с битумом пригодны б качестве связующего для
морозостойких дорожных покрытий [90].
Кабельные оболочки из ПБ имеют при хороших
диэлектрических характеристиках незначительную жесткость и высокую тре-
щиностойкость.
4.1.7. МОДИФИКАЦИЯ ПОЛИБУТЕНА-1
Полибутен-1 можно хлорировать в растворе и в твердой фазе.
Для хлорирования в растворе применяются такие растворители,
как метиленхлорид, тетрахлорид углерода или дихлорбензол. При
хлорировании стереоблочных полимеров при 50 °С 28%-ное
содержание хлора достигалось за 150 мин [91]. С увеличением
содержания хлора уменьшается степень кристалличности полимера. ПБ с
содержанием хлора более 30 % — полностью аморфный полимер [92].
Сульфохлорирование ПБ инициируется с помощью радикальных
инициаторов или облучением [93]. Сульфирование ПБ проводится
в гексане концентрированной серной кислотой при 0°С [94]. Его
можно осуществлять также путем взаимодействия ПБ с
расплавленной серой при температурах от 150 до 200 °С. Сульфированный
ПБ используется в качестве присадок к смазочным маслам [95].
4.1.8. СОПОЛИМЕРИЗАЦИЯ И ПРИВИТАЯ СОПОЛИМЕРИЗАЦИЯ
Степень кристалличности сополимеров, полученных
статистической сополимеризацией бутена-1 с этиленом, уменьшается с
увеличением содержания этилена. Сополимеры бутена, содержащие 20 %
этилена, являются полностью аморфными. С понижением степени
кристалличности связано уменьшение плотности и повышение
проницаемости для газов и паров, а также увеличение эластичности и
гибкости полимера [96]. Сополимеры с пентеном-I и 3-метилбуте-
ном имеют кристаллическую структуру.
Таблица 4.8. Параметры процесса сополимеризации бутена-1
(г,) с олефинами
0,15±0,05
0,019
0,019
—
0,16
8,5
0,227
Сомономер
Бутен-2
Этилен
»
З-Метил-
бутен-1
Пропилен
31 ±0,7
29,5
29,6
3,25
3,60
0,013
4,39
Г, °С
-20
0-75
130-220
60
0-75
Условия
Катализаторы Фриде-
ля — Крафтса
Катализаторы Циглера
То же
Р = 1000 кгс/см2
Р = 100 мм рт. ст.
Катализаторы Циглера
То же
Литературный
ИСТОЧНИ1-
[И]
[102]
[32]
[1031
104л
;ю5
[32]
8 Зак. 1В
65
Параметры процесса сополимеризации бутена-1 с бутеном-2 и
другими олефннами приведены в табл. 4.8.
Привитые сополимеры ПБ были получены прививкой стирола,
винилхлорида [100] и акрилонитрила [101] к пероксндированному
полимеру.
4.1.9. ВОЗДЕЙСТВИЕ ИОНИЗИРУЮЩЕГО ИЗЛУЧЕНИЯ
При действии на аморфный и кристаллический ПБ унзлучения
F0Со) или пучка электронов с дозами 54—200 Мрад происходит
его сшивание. Содержание растворимой фазы в бензоле при 70 °С
составляет примерно 17% [106]. При облучении улучами при
—196 °С и дозе 30 Мрад происходит деструкция полимера [107].
Исследования структуры радикалов облученного ПБ различной
кристаллической модификации были проведены Хукадой [107].
4.2. ПОЛИ-4-МЕТИЛПЕНТЕН-1
-— сн2—сн—
СНз
сн2—сн.
Поли-4-метилпентен-1 получается стереоспецифической
полимеризацией и выпускается в промышленном масштабе, начиная
с 1968 г.
Полн-4-метилпентен-1 отличается от других полиолефинов,
производимых промышленностью, высокой температурой
размягчения B40°С), высокой прозрачностью и очень низкой плотностью
Он применяется в основном для получения прозрачной упаковки,
стерилизуемых медицинских изделий одноразового пользования и
химической посуды.
4.2.1. 4-МЕТИЛПЕНТЕН-1
4-Метилпентен-1 синтезируют из оксида углерода и водорода
по методу Фишера — Тропша [108], из пзомасляного альдегида и
ацетона (с получением 4-метилпентанола) с последующей
дегидратацией в присутствии оксида алюминия при 275—400 °С [109—111]
или каталитической димеризацией пропилена.
В промышленности 4-метилпентен-1 получают исключительно
последним способом. Пропилен образуется в качестве побочного
продукта при получении этилена. Технический и экономический
интерес представляет переработка пропилена в димеры, такие, как
изопрен и 4-метилпентен-1, а также получение полимеров на их
основе.
Димеризация пропилена в 4-метилпентен-1 была впервые
проведена в 1961 г. Шраммом [112] при использовании в качестве
катализаторов щелочных металлов. По Шоу [113], механизм реакции
на первой стадии заключается во взаимодействии пропилена с ще-
лочным металлом с образованием гидрида щелочного металла и
алкила щелочного металла:
СНз—СН=СН2 + 2К ч=* (СН3—СН=СН)"К+ + КН
Резонансно-стабилизированный аллильный анион реагирует
с пропиленом с образованием изогексильного аниона [114]:
(СН2=--СН-=СН2)"К+ + СНз—СН=СН2 —> /СН2—СН—СН2—СН \"К+
V СНз СН2/
На последней стадии реакции алкил калия регенерируется, и
образуется 4-метилпентен:
К+/СН2—СН—СН2—СН=СН2\" + СНз—СН=СН2 + К —>
\ СНз /
СНз—СН—СН2—СН=СН2 + (СН=СН—СНз)"К+
С
КН
Нз
В качестве катализаторов используются металлический калий,
рубидий или цезий (в виде суспензий в гептане или на носителях,
таких, как А12О3, MgO—КгСОз, К2О, технический углерод), гидрид
калия или алкил калия [115—128].
Фирма «British Petroleum Со.» в Грангемоуте (Шотландия)
осуществила каталитическую димеризацию пропилена с получением
4-метилпентена-1 при 135—165°С и давлении 40—60 кгс/см2 на
установке мощностью 2000 т в год. При общем превращении за
один проход, составляющем 30%, выход 4-метилпентена-1,
пересчитанный на прореагировавший пропилен, достигает 80—90 %
[129]. Технологическая схема установки для получения 4-метил-
пентена-1 показана на рис. 4.9.
Установка состоит из двух основных частей: реактора и ректификационных
колонн для разделения углеводородов. Свежий пропилеи смешивается в
смесителе с не вступившим в реакцию оборотным пропиленом и после нагревания до
температуры реакции под давлением подается в реактор. Затем реакционная
смесь поступает в колонну, в которой иепрореагировавший пропилен отделяется
от изомеров гексеиа и возвращается в процесс. В следующей колоиие
углеводороды С4 и С5 отделяются от изомеров гексена. Гексеновая фракция разделяется
в отдельной высокоэффективной колонне: 4-метилпеитеи-1 {Ткнп = 53,88 °С)
отводится из верхней части колой- , „
Рециркуляция пропилена легкие пентен-1
углеводороды , *-
ны, тяжелолетучие изомеры: гек-
сен-1 (Гкип = 60,70°С), трансА-
метилпентен-2 (Укип = 58,55°С)
и цмс-4-метилпеитеи-2 (Гкип =
= 56,30 °С).
Рис. 4.9. Технологическая схема
каталитической димеризации
пропилена с образованием 4-метил-
пентеиа-1 [129]:
/ — смесительный резервуар; 2 —
реактор; 3 — сепаратор пропилена: 4 —
сепаратор фракции С«; 5 — дистилляцн-
онная колонна,
3*
ШЩ шщш угтШомОы ¦
67
собираются в отстойнике. По этому способу можно получить 4-метилпентеи-1
с чистотой 97,4%. Примеси состоят из гра«с-4-метилпентена-2A,5 %), цис-4-ые-
тилпентена-2 @,9%), гексена-1 @,1%) и легких углеводородов @,1%).
Физико-химические свойства 4-метилпентена-1 приведены ниже:
Tnq (воздух 760 мм рт. ст,), °С ... —153,63
Ткпа G60'мм рт. ст.), °С 53,88
dT/dp G60 мм рт. ст.) 0,04
Плотность при 25 °С, г/см3 0,6594
1,3799
Температура вспышки (по ASTM D 286-30),
°С 305
Теплоемкость Ср при 25°С, кал/(моль-°С) 30,23
Теплота сгорания при 25 °С и постоянном
давлении, ккал/моль 962,56
В присутствии катализаторов 4-метилпентен-1 легко изомери-
зуется в 4-метилпентен-2 или 2-метилпентен-2 [130—134], который
является промежуточным продуктом при синтезе изопрена:
СН2=С—СН2—СН2—СНз =<=>: СН2=С—СН=СН2 + СН4
СНз СНз
Каталитическая димеризация 4-метилпентена-1 в присутствии
алюминийалкилов приводит к образованию 2-изобутил-6-метилгеп-
тена-1:
СНз
АШз I
2СН2=СН—СН2-СН—СНз =*=* СН2=С-СН2—СН2—СН2—СН
СНз СН2СН(СНзJ СНз
4.2.2. ПОЛИМЕРИЗАЦИЯ 4-МЕТИЛПЕНТЕНА-1
Поли-4-метилпентен-1 (ПМП) был впервые получен Натта
в 1955 г. в присутствии каталитической системы Циглера диэтил-
алюминийхлорид — трихлорид титана при атмосферном давлении
в гептане. Дальнейшее развитие работ в области синтеза ПМП
осуществлялось большей частью фирмами ICI, «Montecatini»,
«Hochst», «Toyo Rayon», «Du Pont» и «British Petroleum Co.»
прежде всего благодаря сравнительно широкой доступности 4-ме-
тилпентена-1.
В 1964 г. в Англии была пущена первая полупромышленная
установка для получения ПМП мощностью 37 т/год [136]. В 1968 г.
появилась вторая установка — мощностью 2000 т/год («ТРХ»).
Организовано производство ПМП в Канаде и США. В СССР
опытное производство ПМП было начато в 1970 г.
В то время как при ионно-координационной полимеризации
4-метилпентена-1 с использованием катализаторов Циглера — Нят-
та образуются продукты с высокой степенью тактичности:
СН—СН2—СН—СН2
(СНзЬСНСНа СНаСН(СНэЬ
68
при катионной полимеризации в гептане при 0—60 С в
присутствии кислот Льюиса, таких, как А1С13, TiCI3, или систем,
аналогичных катализаторам Циглера— Натта, например TiCl3 —
— А1(СаН5)С12, частично происходит миграция водорода, которая
приводит к получению продуктов полимеризации с диметилбутано-
выми фрагментами в цепи:
—СН2—СНа—СНг—С(СНзJ—
ЯМР — спектроскопические исследования структуры полимеров
4-метилпентена-1 в зависимости от использованной каталитической
системы были проведены Гудричем [135]. Наибольшее количество
диметилбутановых фрагментов (примерно 70%) получается при
использовании в качестве катализатора этилалюминийдихлорида.
При применении трихлорида алюминия продукт полимеризации
содержит 46 % диметилбутановых фрагментов. При катионной
полимеризации 4-метилпентена-1 получаются низкомолекулярные
продукты дегтеобразной или высоковязкой консистенции.
В промышленности при получении ПМП используются
катализаторы Циглера — Натта и аналогичные им системы, такие, как
триэтилалюминий — трихлорид титана [137], диэтилалюминийхло-
рид —трихлорид титана [135, 138—140], триизобутилалюминий —
трихлорид титана [141], трихлорид титана — диэтилалюминий-
сульфат — трихлорид фосфора [142], трихлорид ванадия —
триизобутилалюминий [141, 143, 144] или тетрахлорид титана — тетраде-
циллитий — алюминий [145, 146]. Тактичность, молекулярно-мас-
совое распределение и выход полученного ПМП определяются
температурой полимеризации, природой и составом каталитической
системы, концентрацией катализатора, временем проведения
реакции, а также дипольным моментом среды, в которой она протекает.
При использовании каталитической системы алюминийалкил —
трихлорид ванадия в соотношении 2 : 1 происходит увеличение
молекулярной массы образующегося ПМП в следующем ряду [144];
триэтилалюминий < три-«-децилалюминий <С
триизобутилалюминий <С диэтилалюминийхлорид.
В системе трихлорид ванадия — триэтилалюминий с
повышением температуры полимеризации и концентрации мономера
молекулярная масса образующегося ПМП уменьшается. С
увеличением времени реакции до 2 ч молекулярная масса сначала
повышается, а затем остается постоянной. При мольном соотношении
компонентов каталитической системы
триэтилалюминий—трихлорид ванадия, равном 2:1, молекулярная масса полимера не
зависит от концентрации катализатора. При мольном соотношении,
большем чем 2:1, происходит уменьшение молекулярной массы с
увеличением содержания триэтилалюминия в каталитической системе.
Если полимеризация 4-метилпентена-1 проводится в смеси
растворителей, состоящей из н-гептана и галогенированных
ароматических углеводородов, в присутствии трихлорида титана — диэтил-
алюминийхлорида, то степень тактичности ПМП возрастает с
увеличением дипольного момента смеси растворителей [139] (табл. 4.9)\
69
Таблица 4.9. Влияние дипольного момента смеси
растворителей на выход и степень тактичности
поли-4-метилпентена-1 [139]
Условия полимеризации: 4-метилпеитен-1 @,25 моль); T1CI3 F,3 ммоль);
25 ммоль); растворитель II — н-гептан E0 мл)
Полимер
Дипольный
момент [1
Выход, %
Степень
тактичности, % *
о-дихлор-
беизол
2,33
88
91,8
-и-фторто-
луол
1,67
80
85,0
Растворитель I A0
-и-хлор-
толуо 1
1,77
84
88,8
хлорбензол
1,55
•
84
87,5
мл)
фтор-
бензол
1,45
73
84,0
КСИЛОЛ
0,39
77
84,0
к-геп-
таи
0
73
81,0
• Степень тактичности определяли по выходу нерастворимой фракции в к-гептане (экст*
ракция в течение 24 ч прн 98 °С).
Это связано с незначительной сольватацией каталитических систем
Циглера — Натта растворителями с малыми дипольными
моментами.
В системе тетрахлорид титана — тетрадециллитий алюминия
молекулярная масса ПМП резко снижается, когда мольное
соотношение указанных компонентов каталитической системы становится
больше 2 : 1 (рис. 4.10). Считают, что получающиеся в процессе
полимеризации низкомолекулярные аморфные сополимеры состоят
из атактических блоков, образующихся в результате катионной
полимеризации, и изотактических блоков — продуктов ионно-коорди-
национной полимеризации [145].
Другая возможность регулирования молекулярной массы при
ионно-координационной полимеризации 4-метилпентена-1
заключается в добавке водорода [140]. Остатки катализатора, которые
оказывают чрезвычайно большое влияние на оптические свойства
ПМП, удаляют обработкой полученного полимера спиртовой НС1
и этилацетатом [147].
Из 4-метилпентена-2 в присутствии катализаторов Циглера —
Натта, таких, как А1(С2Н5)з—TiCl3—СгС13, также получается
поли-4-метилпентен-1, поскольку происходит промежуточная
изомеризация в 4-метилпентен-1 [148, 149].
0 12
TiCl4-Al (Си, H
Рис. 4.10. Зависимость характеристической
вязкости стереорегуляриого поли-4-метилпентеча-1 от
мольного соотношения компонентов
каталитической системы [145].
70
4.2.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
ПОЛИ-4-МЕТИЛПЕНТЕНА-1
4.2.3.1. Структура
При полимеризации 4-метилпентена-1 в присутствии
катализаторов Циглера — Натта образуются полимеры с преимущественно
изотактической структурой. Атактическую фракцию можно
выделить путем экстракции кипящим гептаном, циклогексаном или ди-
этиловым эфиром. В зависимости от природы используемой
каталитической системы содержание атактической фракции колеблется
от 5 до 25% [139, 145].
Кристаллическая фаза в изотактическом ПМП составляет
примерно 40 % по данным рентгеноструктурного анализа или около
30 % —по данным калориметрического метода. Путем термической
обработки можно повысить степени кристалличности примерно
до 60% [150].
Изотактический ПМП кристаллизуется в тетрагональную
систему: размеры элементарной ячейки составляют а = Ь = 18,66 А,
с=13,80А [I5I, 152]. Плотность кристаллической фазы равна
0,828 г/см3, аморфной — 0,838 г/см3 [154]. Согласно Фрэнку [152],
боковые группы, расположенные снаружи молекулярной спирали,
встраиваются в свободные пространства спирали другой молекулы.
При максимально возможном сближении макромолекулярных
цепей достигается состояние, характеризующееся малым запасом
энергии. В результате измерений теплоемкости полимера при
температурах от —200 до 260°С был сделан вывод о существовании
двух полиморфных модификаций, которые являются стабильными
при температурах выше 172°С.
ПМП при 50—90 °С кристаллизуется из раствора ксилол —
амилацетат при их соотношении 1 : 1 в кристаллы ламелярного
типа, кривизна которых увеличивается с уменьшением
температуры кристаллизации [153].
4.2.3.2. Растворимость
Изотактический ПМП растворим при повышенных
температурах в ароматических и хлорированных ароматических
углеводородах. Атактические полимеры растворяются в кипящем гексане,
циклогексане или простых эфирах. Среднее значение
молекулярной массы можно определить по данным измерения
характеристической вязкости раствора в декалине A00 мл/г) при 135°С по
уравнению [155]:
4<81
4.2.3.3. Термостойкость
ПМП обладает более высокой термической стойкостью, чем
полиэтилен или полипропилен. Его разложение начинается в
инертной атмосфере при 360 °С и на воздухе — при 155 °С [157].
71
О
10
Q
Рис. 4.11. Кривые термической деструкции поли-4-метилпентена-1 в вакууме [156].
Рис. 4.12. Зависимость характеристической вязкости стабилизированного и неста-
билизированного полн-4-метилпентена-1 в вакууме от продолжительности
термической деструкции при 3O2°C [156]:
/fe-ПМП, стабилизированный 0,2% дигидроантрацена; 2 — ПМП, стабилизированный
О,27о топанола; 3 — нестабилизнрованный ПМП.
Энергия активации термической деструкции в вакууме составляет
53,5 ккал/моль. На рис. 4.11 показана зависимость потери массы
от времени пиролиза при температурах от 291 до 341 °С. При
времени пиролиза 180 мин и 300 °С летучие продукты разложения
имеют следующий состав [156]: изобутен — 57,5%, пропан — 33%,
изобутан — 3 % ; 4-метилпентен-1 — 2,42 %, изопентан — 2,2 %, 2,3-
диметилбутан — 1,26 %, w-пентан — 0,26 % и пропилен — 0,14 %.
Содержание мономера в летучих продуктах не превышает 2,42%.
Жидкие продукты деструкции состоят из разветвленных
насыщенных и ненасыщенных углеводородов Си—Ci8.
Деструкция протекает по радикально-цепному механизму и
характеризуется очень большой длиной кинетической цепи.
Водород отрывается преимущественно от третичного, но частично и
от вторичного атомов углерода при первичном углеродном
радикале. Ослабление углеродной цепи приводит к выделению
летучих продуктов. Кривая снижения молекулярной массы ПМП при
деструкции в вакууме на начальной стадии носит крутой
характер, однако затем зависимость становится прямолинейной со
слабым наклоном (рис. 4.12). Начальное резкое уменьшение
молекулярной массы связано с наличием статистически распределенных
слабых мест в цепи. На первой стадии деструкции происходит
перегруппировка этих слабых мест (например, двойных связей)
с образованием структур повышенной термической стойкости.
4.2.3.4. Радиационная стойкость
При облучении ПМП у-лучами или быстрыми электронами на
воздухе вследствие отщепления водорода и протекания
окислительных процессов происходит разложение макромолекул. При
облучении в вакууме происходит сшивание и образование
двойных связей [158]. Строение радикалов облученного ПМП иссле-
72
довали Кузомото [159], Чарльз [160] и Пинкертон [161]. В
процессе облучения при —173°С вначале образуются радикалы
следующей структуры
СН(С4Н9)—СН2—СН— СН2—СН(С4Н9)
которые при нагревании до —30 °С превращаются в аллильные
радикалы [159] :
СН(С4Н9)—СН—СН=СН(С4Н9)
4.2.4. ПЕРЕРАБОТКА ПОЛИ-4-МЕТИЛПЕНТЕНА-1
ПМП в чистом виде не применяется в технике вследствие
хрупкости и недостаточной прозрачности. Выпускаемый в
промышленности полимер всегда содержит небольшое количество
а-олефинов. ПМП поставляется в виде гранулята или сыпучего
порошка. Переработка его в изделия осуществляется экструзией,
литьем под давлением, формованием из расплава с раздувом и
прессованием на обычных машинах для переработки термопластов.
Особенности и режимы переработки ПМП определяются высокой
температурой плавления полимера, равной 232°С, узким
температурным интервалом плавления, низкой вязкостью и тиксотропией
расплава, которая является следствием гибкости цепей полимера,
обусловленной наличием изопропильных боковых групп. Из-за
ограниченной стойкости полимера к термоокислительной
деструкции его переработку во избежании охрупчивания и изменения
цвета материала нельзя проводить при длительных временах
выдержки при высоких температурах. Температура начала
разложения чистого ПМП на воздухе составляет примерно 154°С [157].
Путем введения стабилизаторов, таких, как дифенил-п-фенилен-
диамин, фенил-Р-нафтиламин, ионол или дигидроантрацен, можно
сместить температуру начала окисления в область 220—240 °С (см.
также рис. 4.12). При переработке в изделия методами,
используемыми для термопластов, наиболее предпочтительным является
интервал температур 250—300°С [162].
4.2.4.1. Литье под давлением
ПМП может перерабатываться в изделия на всех современных
литьевых машинах. Как правило, температура переработки
колеблется от 270 до 300 °С, температура литьевой формы должна
составлять примерно 70°С. Усадка в форме линейно увеличивается
с возрастанием толщины стенки и составляет 1,5—3,0%; при
неблагоприятных условиях могут получаться и более высокие ее
значения. На рис. 4.13 приведена зависимость усадки от
температуры цилиндра. Усадка изделия в форме в направлении течения
расплава выше, чем в направлении, поперечном направлению
потока [163]. Вследствие более высокого значения энтальпии и
малой плотности @,813 г/см3) оптимальное значение объема
впрыска составляет примерно 60 % от данного значения для поли-
73
о
260 2вО 300 320 t,C
ZOO
250 300 t;G
Рис. 4.13. Зависимость усадки изделий из поли-4-метилпентена-1 от температуры
цилиндра [163]:
1— в направлении течения; 2 — в поперечном направлении.
Рис. 4.14. Оценка текучести различных полимеров в литьевой форме со
спиралевидным каналом [164]:
/—ПЭНД (ПТР-2); 2 — ПП; 3 — полн-4-метнлпентен-1.
стирола. Объемная скорость пластикации такая же, как и для
ПЭВП. На рис. 4.14 показана сильная температурная зависимость
вязкости расплава ПМП (в сравнении с другими полиолефинами),
построенная путем оценки текучести в литьевой форме со
спиралевидным каналом. Малая вязкость расплава ПМП в случае
поршневых машин приводит к потере материала через сопло, если
входное отверстие формующей головки не является двойным
коническим.
Расплав ПМП при течении ведет себя как псевдопластичные
тела. По сравнению с ПММА, ПЭ и полипропиленом происходит
значительно более сильное уменьшение вязкости расплава с
ростом напряжения сдвига (рис. 4.15).
4.2.4.2. Экструзия
Хорошие результаты при экструзии ПМП достигаются при
использовании шнеков с высокой степенью сжатия, (L/D = 20),
наиболее пригодными являются двухшнековые экструдеры. Скоростной
напор может быть повышен за счет дополнительного применения
делительных дисков с отверстиями и
решеток. Зона декомпрессии не является
необходимой.
При экструзии труб диаметром более
15 мм применяется калибровка изделий
как при избыточном, так и при
пониженном давлении; при меньших диаметрах
Рис. 4.15. Зависимость вязкости расплава
полимеров от напряжения сдвига [164]:
1 — полн-4-метнлпентен-1; 2 — ПЭНД: 3 — ПММА; 4-^
полипропилен; 5 — ГШХ.
74
трубы достаточно использовать калибрующие насадки. Для
отдельных зон экструдера применяются следующие температуры
[164]:
Температура вблизи загрузочной воронки, °С 235
Температура на конце шнека, °С ..«,,. 260
Температура экструзионной головки, °С . . . 320
Необходимо исключить провисание трубы (вплоть до намотки).
Вследствие высокой теплостойкости и отличных диэлектрических
свойств ПМП может применяться в качестве изоляции для
проводов и кабелей. Во избежание образования воздушных включений
в виде пустот и пузырей следует соблюдать определенные условия
охлаждения. Высокие скорости намотки достигаются при
температуре на конце шнека 280 °С и в экструзионной головке 320 °С.
4.2.4.3. Формование с раздувом
Формование полых изделий методом экструзии с раздувом
осуществляется при температуре расплава, равной 275°С.
Температурный профиль вдоль шнека должен поддерживаться в
пределах 240—270°С. Во избежание образования следов течения и
линий стыка расплава на изделии применяются спиралевидные
рассекатели (торпедо). Оптимальная прозрачность изделий
достигается при температуре экструзионной головки 290 °С. Этим
методом изготавливаются полые изделия объемом до 2 л [164].
4.2.4.4. Пленки и пенопласты
Экструзия пленок через щелевые головки осуществляется при
высоких частотах вращения шнека и температуре расплава 300°С.
Тонкие пленки пригодны для кэширования бумаги [164].
Вспененные материалы на основе ПМП можно получать путем
добавления смесей дивинилбензола (пероксида) с вспенивающим
агентом и нагревания под давлением до 200 °С. Сетчатые
пенопласты обладают высокой стабильностью размеров вплоть до
140°С [167].
4.2.4.5. Прессование
Для специальных целей при изготовлении толстых пластин и
блоков используют метод прессования. Температура плит при
прессовании поддерживается в интервале 290—300 °С.
4.2.4.6. Термоформование пленок и листов
Формование изделий из пленок и листов осуществляется при
тех же режимах переработки, что и в случае полипропилена.
Трудности осуществления технологических процессов переработки
связаны с узким температурным интервалом плавления ПМП. Глу-
75
бокая вытяжка под вакуумом применяется только для тонких
пленок. В случае толстых листов необходим предварительный их
подогрев в течение 2—3 мин при 190—200 °С. Формование изделий
осуществляется в интервале 230—240 °С.
4.2.4.7. Поверхностная обработка изделий
Так же как и для полипропилена, перед нанесением печатного
рисунка, окрашиванием и металлизацией поверхность
формованных изделий из ПМП подвергают предварительной обработке
химическими реактивами. Наиболее пригодны для этой цели
хромовая смесь, подкисленная раствором перманганата, или
специальные составы, используемые для нанесения окунанием первого
покровного слоя [166].
4.2.5. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА
ПОЛИ-4-МЕТИЛПЕНТЕНА-1
Как уже отмечалось, ПМП в чистом виде в технике не
применяется.
4.2.5.1. Теплофизические свойства
Основные теплофизические свойства ПМП приведены ниже:
Температура, °С
стеклования 29
плавления 228—335 .
хрупкости 38
Теплостойкость по Вика, °С 179
Термический коэффициент линейного
расширения, 1/°С 11,7-10*
Показатель преломления 1,456
Удельная теплоемкость, кал/(г-°С) . . . 0,52
Коэффициент теплопровод- Теплота образования,
ности, кал/(см-с-°С) . . 4 - 10~4 кал/моль 2850±450
Горючесть, см/мин ... 2,5 Энтропия образования,
Теплота плавления, ккал/ кал/моль 2500
элементарное звено . . 4,71 Газопроницаемость при
Плотность, г/см3 .... 0,813 25 °С, см3/см2/с/см/см рт.
Водопоглощение за 24 ч ст.
@,3 мм пленки) .... 0,01 О2 27-Ю-10
N2 6,5 • Ю-10
Высокая температура плавления B32°С) является следствием
объемной структуры изотактических макромолекул с боковыми
третичными бутильными группами. Теплостойкость по Вика,
равная 179 °С, значительно выше, чем у обычных термопластов
(полипропилен и полистирол 90 °С, ПВХ 82 °С, ПММА 110°С). Это
позволяет стерилизовать формованные изделия из ПМП
перегретым паром. Относительно высокие значения термического
коэффициента линейного расширения обусловлены низкой температу-
76
рой стеклования B9 С). При добавлении 40% атактического
ПМП температура хрупкости снижается с 38 до —8°С [168].
Плотность ПМП приближается к теоретически возможной для
термопластов. Газопроницаемость ПМП значительно выше, чем
у других полиолефинов. По отношению к водяным парам она
составляет при 38°С и 90% относительной влажности воздуха
(95—107) г/см3-24 ч (для пленки толщиной 0,025 мм).
4.2.5.2. Физико-механические свойства
По физико-механическим свойствам ПМП во многом
аналогичен полипропилену.
Ниже приведены физико-механические свойства изотактиче-
ского поли-4-метилпентена-1:
Прочность при растяжении, кгс/см2 280
Относительное удлинение при разрыве, % '5
Модуль упругости при растяжении, кгс/см2 15 000
Твердость по Роквеллу 67—74
Твердость по Шору . 80
Модуль упругости при изгибе, кгс/см2 12 000
Ударная вязкость образца с надрезом (по Изоду), кгс-см/см2 . . 0,28—0,44
Прочность термопластов при растяжении, как правило,
уменьшается при повышении температуры; изменение прочности
определяется средней молекулярной массой и степенью кристалличности
полимера. Для ПМП влияние температуры на прочность при
растяжении выражено сильнее, чем для полипропилена или
полистирола (рис. 4.16). Это объясняется изменениями, происходящими
в кристаллической фазе, и явлениями, связанными с ростом сфе-
ролитов.
Модуль упругости при растяжении, равный 15 000 кгс/см2 (при
20°С) несколько выше, чем у полипропилена A2 000 кгс/см2).
При температурах выше 150°С ПМП жестче, чем полипропилен
и поликарбонат, при комнатной температуре —наоборот
(рис. 4.17). Ударная вязкость образца с надрезом (по Изоду),
равная 0,28—0,44 кгс -см/см2, ниже, чем у таких кристаллических
полимеров, как полиамид 6,6 @,55 кгс см/см2) или
полиформальдегид @,66—0,77 кгс-см/см2) [163].
Склонность к ползучести при 20 °С
под растягивающей нагрузкой 50
кгс/см2 при времени выдержки менее
6 сут у ПМП менее выражена, чем
Рис. 4.16. Зависимость прочности при растяже-
нии различных полимеров от температуры
[169]:
1 — ПС; 2 — ПП; 3 — поли-4-метилпентен-1. 0 25
150 t, ff
о 10 m2 ю3 io* ю5 ю6 io7z,c
Зависимость динамического модуля упругости различных полимеров от
температуры [163]:
/ — ПС; 2 —ПК; 3 — поли-4-метшшентен; 4 — ПП; 5 — ПЭНД.
Рис. 4.18. Кривые ползучести различных полимеров при растяжении под
нагрузкой 50 кгс/см2 [163]:
/ — ПЭВД; 2 — ПП; 3 — поли-4-метилпеитеи-1.
у полипропилена. При более длительных временах действия
нагрузки ползучесть ПМП больше, чем у полипропилена (рис. 4.18).
4.2.5.3. Электрические свойства
ПМП по своим электрическим свойствам незначительно
отличается от других полиолефинов. Его диэлектрическая
проницаемость B.12) ниже, чем у ПП B,25) и ПЭНП B,28). Частотная
зависимость тангенса угла диэлектрических потерь и
температурная зависимость тангенса угла диэлектрических потерь при
различных частотах приведены на рис. 4.19 и 4.20. Основные
электрические свойства ПМП приведены ниже:
Диэлектрическая проницаемость при
102—106 Гц 2,12
Удельное объемное электрическое
сопротивление, Ом-см 1018
Электрическая прочность, кВ/мм ... 28
Дугостойкость Подвержен разложению
без коксования
4.2.5.4. Оптические свойства
При определенных условиях охлаждения расплава можно
получить ПМП с прозрачностью, равной 90 °/о, т. е. такой же, как
для ПММА (92%). Однако в отличие от аморфных полимеров
при повышенных значениях толщины стенки фасонных деталей
появляется помутнение материала.
Отличная прозрачность ПМП объясняется незначительной
разницей плотностей аморфных и кристаллических областей в
полимере. Разность значений плотности составляет при температуре
78
HO'3 -
420 -80 -<tO 0 40 80 120 160 t°G
Рис. 4.19. Частотная зависимость тантенса угла диэлектрических потерь для
различных полимеров [163]:
1 — поли-4-метилпентен-1; 2 — ПТФЭ; 3 — ПЭНД.
Рис. 4.20. Температурная зависимость тангенса угла диэлектрических потерь
поли-4-метилпентеиа-1 при частотах от 32 до 31 600 Гц [163].
плавления 7%. При 60°С обе фазы имеют одинаковые значения
плотности, при более высоких температурах плотность аморфных
областей выше плотности кристаллических областей. При
неблагоприятных условиях при охлаждении расплава полимера на
поверхности сферолитов и между ними образуются микрополости,
обусловливающие получение непрозрачных продуктов. Более
высокую прозрачность имеют изделия из сополимеров 4-метилпен-
тена-1 с незначительным количеством пентена-1 или гексена-1
[136]. Поэтому в промышленном ПМП всегда содержится немного
сомономеров.
Прозрачность ПМП можно улучшить введением структурообра-
зователей, которые в процессе кристаллизации способствуют росту
концентрации сферолитов и уменьшению их размеров [170]. В
качестве структурообразователей используются кристаллические по-
лиолефины с более высокой температурой плавления по сравнению
с ПМП, например изотактический поли-З-метилбутен-1 (ТПп =
= 310°С), изотактический поли-З-метилпентен-1 (Гпл >• 350сС)
и изотактический поли-4,4-диметилбутен-1 (Тпл > 350°С). При
1 %-ной концентрации полимеры, сохраняющие кристалличность в
расплаве ПМП, в процессе охлаждения расплава инициируют
процесс структурообразования.
4.2.5.5. Химическая стойкость
ПМП имеет такую же стойкость к действию химических
агентов, как полиэтилен или полипропилен. Химическая деструкция
полимера происходит при воздействии окисляющих сред, таких,
как хлор, бром, концентрированная азотная кислота или олеум.
Ароматические или хлорированные углеводороды, а также
сложные эфиры вызывают при комнатной температуре набухание
полимера. Ниже приведены данные о набухании нзотактического поли-
79
4-метилпентена-1 в ароматических и хлорированных
углеводородах при 25°С [171]:
Бензол
Толуол
Ксилол
Тетрахлорид углерода
Метиленхлорид . . .
21,6
26,3
24,2
70,7
20,0
1-Хлорбутан . 33,4
1-Хлоргексан 25,9
1-Хлороктан ....... 19,7
1-Хлордекан 16,3
Электронно-микроскопические исследования структуры набухшего
ПМП и их механические свойства были проведены Хейзом [171].
Набухание происходит в межламеллярных или аморфных
областях.
Поверхностно-активные вещества вызывают коррозионное
растрескивание ПМП. Однако стойкость его к образованию трещин
выше, чем у других прозрачных полимеров, таких, как ПММА
или ПС.
4.2.5.6. Длительная стойкость и стойкость к старению
Из-за наличия третичных атомов углерода в основной и
боковых цепях ПМП весьма подвержен термоокислительной и
фотохимической деструкции. Путем введения стабилизаторов (см.
разд.4.2.4 и рис. 4.12) процесс деструкции замедляется. На рис. 4.21
приведена температурная зависимость долговечности ПМП,
характеризующейся температурно-временной зависимостью
снижения прочности при растяжении на 50 % от исходного значения.
При 200 °С уменьшение прочности на 50% наступает после 25 ч,
при 130°С — после 1 ч.
Облучение стабилизированного ПМП электронами или у-лу-
чами в дозах, используемых для стерилизации, не оказывает
влияния на физико-механические свойства материала.
4.2.6. ВОЛОКНА ИЗ ПОЛИ-4-МЕТИЛПЕНТЕНА-1
Средняя молекулярная масса ПМП, полученного ионно-коор-
динационной полимеризацией, слишком велика для проведения
формования волокна из расплава. Путем термической деструкции
в высоком вакууме при 270—280°С
можно понизить среднюю
молекулярную массу ПМП без ухудшения
свойств полимера. При этом
степень кристалличности ПМП и
структура изотактических полимер-
Рис 4.21. Снижение прочности при
растяжении поли-4-метилпентена-1 на 50 % от
исходного значения [163].
100
Ю
10
/О-
F, r(денье
50
о m во. m m 2001°9с
Рис. 4.22. Зависимость модуля упругости
волокон из поли-4-метилпентена-1 от температуры
[172]:
/ —ПП; 5 —ПМН высокой степени кристалличности;
3 — ПМП низкой степени кристалличности.
ных цепей остаются неизменными [165].
Перед началом формования аморфная
часть полимера отделяется путем
экстракции в петролейном эфире, цикло-
гексане, диэтиловом эфире или ацетоне. Процесс формования
волокна из расплава ПМП может осуществляться со скоростью до
500 м/мин. Полученные нити, однако, из-за высокой степени
ориентации, которая достигается в процессе формования, не могут
подвергаться дополнительной вытяжке. Наилучшие результаты
достигаются в том случае, если в процессе формования удается избежать
вытяжки полученного волокна и она проводится отдельно.
Разрывная прочность волокна при этом во много раз выше прочности
волокон, полученных по первому методу. В табл. 4.10 приведены
физико-механические характеристики волокон из ПМП, полученных
формованием из расплава при различных молекулярной массе и
температурах формования.
Усадка волокон из ПМП в воде составляет 9%, на воздухе
при 150°С— 13% и при 200°С — 21 % [172].
На рис. 4.22 сопоставлены температурные зависимости модуля
упругости двух образцов волокон из ПМП с различной степенью
кристалличности. Модуль упругости ПМП больше, чем
полипропилена, лишь при температурах выше 140 °С.
Таблица 4.10. Физико-механические свойства волокон
из поли-4-метилпентена-1 [165]
Показатель
Характеристическая вязкость
в декалине при 135°С,
100 мл/г
Кратность вытяжки
Температура вытяжки, °С
Прочность при растяжении,
г/деиье
Относительное удлинение при
разрыве, °/о
Модуль упругости при
растяжении, г/депье
300
2,38
8
175
2,8
23
30
Температура формования, °С
279
1,85
12
155
3,7
27
29
282
1,35
13
140
2,4
26
17
275
1,90
6,5
155
3,6
26
33
250
1,24
12
155
5,1
25
35
81
4.2.7. ПРИМЕНЕНИЕ ПОЛИ-4-МЕТИЛПЕНТЕНА-1
Удачное сочетание высокой температуры плавления и
прозрачности, низкой плотности, а также хороших диэлектрических и
физико-механических свойств ПМП открывает широкие
перспективы его применения. Относительно высокая стоимость,
составляющая 9 марок за 1 кг, ограничивает это применение лишь
специальными областями, в которых невозможно использование
крупнотоннажных полимеров.
4.2.7.1. Лабораторная и медицинская техника
Высокая теплостойкость, прозрачность и химическая стойкость,
а также физиологическая инертность позволяют использовать
ПМП для различных целей. В промышленном масштабе из него
изготавливают бюретки, мерные цилиндры, колбы, стаканы и
другую химическую посуду, которая благодаря запасу прочности при
высокой теплостойкости и прозрачности имеет ряд преимуществ
по сравнению со стеклянной посудой [173]. Для фильтрования
гелей под давлением изготавливают колонки диаметром 385 мм
и толщиной стенки 6—15 мм [174]. Из ПМП делают также
смотровые стекла и трубопроводы в узлах приборов. При окраске
полиэфирных волокон при температурах ванны от 140 °С обычные
стальные бобины заменяют бобинами из ПМП.
Физиологическая инертность и возможность стерилизации
ПМП горячим паром позволяют широко использовать его в
медицинской технике. В основном ПМП применяется для изготовления
медицинских шприцев и канюлей, соединительных деталей,
измерительных сосудов, а также емкостей для медикаментов и
стерилизованной воды. Стерилизация может проводиться при
температурах вплоть до 160 °С; 40-кратная стерилизация изделий из ПМП
при 134 °С не изменяет свойств материала [163].
4.2.7.2. Электротехника
Хотя вследствие чувствительности ПМП к действию УФ-лучей
его нельзя использовать на открытом воздухе, благодаря отличной
прозрачности и высокой теплостойкости ПМП можно применять
для изготовления интенсивных источников света, отражателей
ламп и рефлекторов.
В электротехнической промышленности применение ПМП
основано на его высокой теплостойкости, хороших электрических
свойствах и незначительном водопоглощении. Использование
ПМП позволяет заполнить пробел между полиолефинами с
низкими температурами размягчения и термостойкими полимерами,
такими, как ПТФЭ. Основными областями применения ПМП
являются изоляция силовых кабелей, производство печатных схем
32
и коаксиальных кабелей в высокочастотной технике, которые
подвержены действию высоких температур (при запаивании,
герметизации), ПМП используется также в качестве изоляции в тех
случаях, хчогдз необходим визуальный контроль электропроводки
и монтажа.
4.2.7.3. Упаковочные материалы
Применение ПМП для упаковки основано на его
физиологической безвредности, высокой теплостойкости и прозрачности, а
также на хорошей проницаемости по отношению к кислороду и
диоксиду углерода.
Непористые пленки используются для упаковки овощей и
свежего мяса.
Из ПМП изготавливают тару и упаковку одноразового
пользования для готовой пищи. Блюда в упаковке из ПМП можно
нагревать с помощью высокочастотных или инфракрасных
обогревателей, а также в любой домашней духовке до 200 °С. Для более
высоких температур применяются дублированные пленки из ПМП
и бумаги. Основное их достоинство заключается в том, что готовое
блюдо сразу после нагревания в упаковке из ПМП можно подать
на стол. Бутыли и канистры из ПМП совершенно прозрачны, при
заполнении горячими жидкостями они не меняют формы и
размеров [175].
4.2.8. СОПОЛИМЕРИЗАЦИЯ И ПРИВИТАЯ СОПОЛИМЕРИЗАЦИЯ
Гомополимеры поли-4-метилпентена-1 с трудом растворяются
в большинстве растворителей и поэтому не могут
перерабатываться из раствора в волокна или пленки.
Для повышения растворимости ПМП Кэмпбелл [165] провел
сополимеризацию 4-метилпентена-1 с высшими а-олефинами. В
результате сополимеризации мономера с 16 % пентена-1 температура
плавления полимера снижается до 170 °С. Сополимеры с 20%
гексена-1 имеют температуру плавления 195 °С; они растворимы в
циклогексане с образованием растворов вплоть до 20%-ной
концентрации и могут хорошо перерабатываться в пленки методом
полива из раствора. Сополимеры с содержанием гексена-1 более 25 %
обладают каучукоподобными свойствами. Константы
сополимеризации равны Г] (гексен) = 5,4; г2 D-метилпентен-1) = 0,62 [178].
Смеси мономеров 4-метилпентен-1—гексен-1 в соотношении 80:20
или 90:10 могут полимеризоваться до образования высоковязких
растворов, из которых без отделения катализатора методом полива
непосредственно получают пленки. Сополимеры с 10 % гексена-1
плавятся при 205 °С.
Сополимеры с 16 % октена-1 легко растворяются в хлороформе,
циклогексане, тетрахлориде углерода, горячем тетрагидрофуране
и горячем ксилоле. Температура плавления их составляет
188 °С.
Сополимеры 4-метилпентена-1 с 16% октадецена-1 умеренно
растворяются при комнатной температуре в циклогексане, хлоро-
83
Таблица 4.11. Физико-механнческие
свойства сополимеров
4-метилпентена-1 [129]
Сополикер
Высокомолекулярный пол
пометил пентен-1
Сополимер мстилпентена с де-
ценом-1 E,4%)
Сополимер 4-метнлпентена-1 с
деценом-1 F,7%)
Сополимер 4-метнлпентена-1 с
генсеком-1 F,3%)
Сополимер 4-метилпентена-1 с
гексеном-1 A2%)
Сополимер 4-метилпентена-1 с
деценом-1 (8,5%)
Сополимер 4-метилпснтеиа-1 с
гексеном-1 A2%)
Прочность
при 1
20 °С
288
203
169
232
134
246
225
растяжении.
кгс/см2
50 °С
140
98
91
98
70
119
112
100 °С
70
63
63
63
49
84
63
Ударная вязкость
по Изоду,
КГС-СМ/СМ2
0 °С
5,4
3,3
3,3
3,3
3,3
6,5
5,4
20 "С
7,5
4,3
35,5
5,4
54
10,3
18,0
40 °С
25
54
54
37
54
43
—
стойкость
по
Вика,
°С
195
115
105
138
100
МО
100
Модуль
упругости
при
изгибе,
кгс/см2
17 000
6 300
3 500
7 200
2 800
—
5 900
форме, тетрахлориде углерода, но легко растворяются в кипящих
циклогексане, хлороформе, тетрахлориде углерода и ксилоле.
Температура плавления указанных сополимеров составляет
примерно 182 °С.
Тройной сополимеризацией 4-метилпентена-1 с гексеном-1 и
дивинилбензолом в циклогексане получены растворимые терполи-
меры, которые при нагревании до 225 °С могут сшиваться по
свободным винильным группам [165]. Температуры размягчения и
физико-механические свойства сополимеров 4-метилпентена-1 с
высшими а-олефинами приведены в табл. 4.11.
Сополимеры 4-метилпентена-1 со стиролом были получены
Андерсоном [176], Кренделем [157] и Кисейным [177] в
присутствии катализаторов Циглера — Натта. Константы сополимеризации
при 45 °С составляют г\ = 3,67 ± 0,22 D-метилпентен-1) и г2 =
= 0,89 + 0,05 (стирол) [176]. Сополимеры обладают более
высокой термостойкостью по сравнению с ПМП. При введении в
качестве сомономера 5—9 % стирола температура, при которой в
инертной атмосфере сополимер разлагается на 10%, равна 395 °С,
т. е. она выше, чем у соответствующих гомополимеров (у ПМП
384 °С; у ПС 345 °С) [157].
Радиационно-химическая прививка стирола к ПМП в области
температур от 23 до 85 °С изучалась Уилсоном [180, 181]. Хотя
равновесная степень прививки стирола при комнатной
температуре, составляющая 19,6%, значительно выше, чем в случае
полипропилена (8,8%) или полиэтилена D,5%), из трех
перечисленных полимеров ПМП имеет наименьшую скорость прививки. Это
объясняется повышенной скоростью обрыва цепей в данной
системе.
84
4.3. ПОЛИВИНИЛЦИКЛОГЕКСАН
—СН— СН2— '
[-СН—СН2—-|
цикло-СъНц \п
Изотактический поливинилциклогексан получается ионно-коор-
динационной полимеризацией винилциклогексана или
гидрированием изотактического полистирола.
Исходный винилциклогексан образуется при термическом
разложении циклогексилэтилацетата.
Поливинилциклогексан можно перерабатывать в изделия при
температуре около 330 °С обычными традиционными методами пе-
раработки термопластов. Верхняя температура длительной
эксплуатации на воздухе равна примерно 220 °С.
Полимер был получен в Институте нефтехимического синтеза
АН СССР и до сих пор выпускается опытными партиями.
Поливинилциклогексан в интервале температур от —160 °С до
220 °С и диапазоне частот от 50 до 1010 Гц обладает отличными
диэлектрическими свойствами. До настоящего времени он является
единственным высокочастотным изоляционным материалом,
который можно применять до 200 °С.
4.3.1. ВИНИЛЦИКЛОГЕКСАН
Винилциклогексан можно получать термическим разложением
(пиролизом) циклогексилэтилацетата, димеризацией бутадиена, де-
гидрогалогенированием производных этилциклогексана и другими
способами.
Пиролиз циклогексилэтилацетата протекает с 80 %-ным
выходом винилциклогексана [182—184]:
500 °С
СН2—СН2—О—СО—СНз > СН=СН2
I
Циклогексилэтилацетат получается при взаимодействии цикло-
гексилэтилового спирта с ацетангидридом. Циклогексилэтиловый
спирт в качестве исходного продукта может быть получен
гидрированием фенилэтилового спирта или по реакции Гриньяра.
Гидрирование фенилэтилов,ого спирта происходит в присутствии
скелетного никелевого катализатора [185] или по Сабатье [186].
Гидрирование в присутствии никеля Ренея проводится при 160°С
и давлении 100 кгс/см2 в этаноле, используемом в качестве
растворителя. Наилучшие результаты получают при 10%-ной
концентрации катализатора, пересчитанной на фенилэтиловый спирт, и
54%-ной концентрации этанола в реакционной смеси. Суммарный
85
выход винилциклогексана составляет 30—35 % [187]:
На/Ni tCH3COJO
од—сн2он > сн2—сн2он
I I
СвН5 цикло-СвНи
—> СН2—СН2—О—СО—СНЭ
Пиролиз
СН2—СН2-О—СО-СНз -.. ап > СН=СН2
| 500 С
Циклогексилэтиловый спирт по реакции Гриньяра получают
взаимодействием сухого газообразного этиленоксида с циклогек-
силмагнийхлоридом в диэтиловом эфире [228, 229]:
Mg (CH2JO
Cl > Mg—Cl > СН2—СН2—ОН
i I I
цикло-CeMw цикло-СвНи цикло-С6Ни
Димеризация бутадиена с получением винилциклогексена
проводится в присутствии оксида хрома и оксида алюминия или
фосфорной кислоты и силикагеля в качестве катализаторов при 400 °С.
Селективное гидрирование полученного винилциклогексена при
комнатной температуре (катализатор платина — активный уголь)
приводит к получению винилциклогексана [230].
Винилциклогексан из 1-хлор-2-циклогексилэтана путем
отщепления хлористого водорода был впервые получен Егоровой [231]
в 1910 г. Хорошие выходы винилциклогексана достигались также
при нагревании 1-иод-2-циклогексилэтана в хинолине [183].
В 1929 г. Штаудингер [188] получил винилциклогексан
пиролизом гидрированного полистирола при 350—400 °С.
Винилциклогексан получается также пиролизом ацетилированного
нафтенового спирта при 350—450 °С [189].
Физические свойства винилциклогексана приведены ниже
[183]:
Температура кипения, °С
при 749 мм рт. ст 130—131
при 15 мм рт. ст 83—85
Плотность при 20 °С, г/см3 0,8060
20
Показатель преломления nD 1,4470
Молекулярная рефракция 36,41
4.3.2. ПОЛУЧЕНИЕ ПОЛИВИНИЛЦИКЛОГЕКСАНА
Поливинилциклогексан (ПВЦГ) был впервые получен в
1929 г. Штаудингером путем гидрирования полистирола.
Полимеризация винилциклогексана осуществляется в присутствии
комплексных катализаторов, катализаторов, состоящих из оксидов
хрома и алюминия и кислот Льюиса. Винилциклогексан не поли-
86
меризуется по радикальному механизму с образованием аллиль-
ных радикалов. В 1939 г. Шорыгин [190] термической
полимеризацией получил очень низкомолекулярные продукты.
4.3.2.1. Ионно-координационная полимеризация
винилциклогексана
Винилциклогексан полимеризуется с выходом примерно 50 %
с образованием кристаллического ПВЦГ в присутствии
каталитических систем, таких, как триизобутилалюминий — тетрахлорид
титана [187, 191 —195], триэтилалюминий—тетрахлорид титана
[196], триэтилалюминий—трихлорид титана [197],
триизобутилалюминий— трихлорид титана [198], диэтилалюминийхлорид —
трихлорид титана [199], триэтилалюминий — трихлорид ванадия
[200], этил-бис-гексаметилдисилазоалюминий — тетрахлорид
титана [201], трехкомпонентные системы с N, О, Р, As или S в качестве
комплексообразующего элемента [202—206] или четырехкомпо-
нентные системы [207] в алифатических или ароматических
углеводородах, используемых в качестве растворителя. При ионно-ко-
ординационной полимеризации выход винилциклогексана более
50 % не достигается, так как в процессе полимеризации
происходит частичная изомеризация винилциклогексана в этилиденцикло-
гексан [187, 208]:
СН=СН2
При использовании в качестве катализатора системы
триизобутилалюминий— тетрахлорид титана 48% винилциклогексана
полимернзуется, 25 % изомеризуется в этилиденциклогексан и
9%—в аллилциклогексан; 18% винилциклогексана остается не-
лрореагировавшим. В присутствии каталитических систем с малым
содержанием активных соединений алюминия, например с три-
этилалюминием, изомеризация протекает примерно на 15 %.
Полимеризация этилиденциклогексана на катализаторах Циглера —
Натта не протекает. Винилциклогексан не изомеризуется при
контакте мономера лишь с одним из компонентов каталитической
системы, например с тетрахлоридом титана или триизобутилалю-
минием.
Степень упорядоченности ПВЦГ определяется природой
растворителя, используемого при ионно-координационной
полимеризации. В плохих растворителях для ПВЦГ, каким является,
например, н-гептан, образуются высококристаллические продукты, в
хороших растворителях, например в бензоле или циклогексане,
получаются аморфные продукты.
При полимеризации винилциклогексана в гептане с
использованием каталитической системы триизобутилалюминий —
тетрахлорид титана оптимальным является соотношение Al(i— С4Н9)з —
— TiCl4 = 1 : 1. Отклонения от эквимольного соотношения
приводят к снижению молекулярной массы (рис. 4.23).
87
А,моль/(г-ч)
12 3
[А10ш-С4Нэ)з]
[TiCI4]
¦мо/ttftm
w
во
60
40
20
AMi«
- ^s
f^rii
-1 ^
e^iB
—*2
^* 3
0 20 MO 60 80 x,muh
32
16
8
la - U /0
**—"^
1
0 20 40 т,мин
Рис. 4.23. Зависимость молекулярной массы поливинилциклогексенат полученного
ионно-координационной полимеризацией, от мольного соотношения компонентов
каталитической системы (полимеризация в гептане при 80 °С, концентрация
катализатора 1 %) [187].
Рис. 4.24. Зависимость скорости ионно-координационной полимеризации винил-
циклогексана от мольного соотношения компонентов каталитической системы
[полимеризация в присутствии А1(нзо-С4Н9)з—TiCl4 в гептане при 80 °С,
концентрация катализатора I %] [187].
Рис. 4.25. Зависимость скорости ионно-координационной полимеризации винил-
циклогексана от концентрации катализатора [полимеризация в присутствии
АЦизо^НэЬ—TiCl4 с соотношением компонентов 1:1 в гептане при 80 °С]
[187].
При повышении концентрации триизобутилалюминия скорость
реакции уменьшается (рис. 4.24) [187]. Оптимальная температура
полимеризации, при которой достигаются наибольшие глубина
полимеризации и молекулярная масса, составляет примерно 80 °С.
Увеличение концентрации катализатора приводит к повышению
скорости полимеризации и снижению молекулярной массы
(рис. 4.25 и 4.26).
Скорость полимеризации и молекулярная масса
увеличиваются с возрастанием концентрации ВЦГ в растворителе (гептане)
(рис. 4.27 и 4.28).
Сополимеры винилциклогексана с пропиленом были получены
в присутствии каталитической системы трихлорид титана — ди-
этилалюминийхлорид [232]. Константы сополимеризации
составляют г\ (пропилен) = 0,049; гг (винилциклогексан) = 80.
4.3.2.2. Полимеризация винилциклогексана в присутствии
оксида хрома (III) и оксида алюминия
Высококристаллический поливинилциклогексан получается
полимеризацией винилциклогексана на носителе оксид хрома(III) —
оксид алюминия в растворе гептана. При 80°С удалось получить
ПВЦГ с характеристической вязкостью в циклогексане, равной 0,5
A00 мл/г) [209, 210].
Так же как и при полимеризации в присутствии катализаторов
Циглера, происходит частичная изомеризация винилциклогексана
в этилиденциклогексан [211]. Если катализатор оксид хрома(III)
с оксидом алюминия модифицировать триизобутилалюминием, то
88
A y моль} (г-
"Ту
20 40 60
Рис. 4.26. Зависимость характеристической вязкости поливинилциклогексана,
полученного ионно-координационной полимеризацией, от концентрации
катализатора [полимеризация в присутствии А1(изо-С4Н9)з—TiCl4 с соотношением
компонентов 1 : 1 в гептане при 80 °С] [187].
Рис. 4.27. Зависимость характеристической вязкости поливинилциклогексана,
полученного ионно-координационной полимеризацией, от концентрации мономера
в растворителе [полимеризация в присутствии А1(ызо-С4Н9)з—TiCU с
соотношением компонентов 1 : 1 в гептане при 80 °С] [187].
Рис. 4.28. Зависимость скорости ионно-координационной полимеризации
винилциклогексана от концентрации мономера в растворителе (полимеризация в
присутствии А1(изо-С4Н9)з—TiCl4 с соотношением компонентов 1 : 1 в гептане при
80 °С) [187].
выход полимера можно увеличить вдвое. Максимальный выход при
полимеризации винилциклогексана на оксиде хрома (III) с оксидом
алюминия составляет примерно 20%.
4.3.2.3. Катионная полимеризация винилциклогексана
Винилциклогексан полимеризуется в присутствии кислот
Льюиса, таких, как хлорид или бромид алюминия, в растворе этилен-
хлорида в интервале температур от —144 до 7°С с образованием
аморфных полимеров, молекулярная масса которых составляет
2000—10 500 [196, 199,212].
На основании данных ИК- и ЯМР-спектроскопии можно
сделать вывод о том, что в процессе роста цепи при полимеризации
на катализаторах Фриделя — Крафтса происходит
внутримолекулярный перенос водорода. Третичные карбониевые ионы,
образующиеся при переносе водорода, обусловливают получение
полимеров с циклогексилиденовыми группами:
Кат
Н2С=СН
4R—CH-,—СН
С- Но С-л
Медленно
•СН2 СН2
Подобным образом протекает и полимеризация 3-метил буте-
на-1 и винилцпклопентана.
4.3.2.4. Гидрирование полистирола
до поливинилциклогексана
Другой метод получения ПВЦГ заключается в полимерана-
логичном гидрировании полистирола при 170°С и давлении
170 кгс/см2 в циклогексане с использованием активного никеля в
качестве катализатора [188, 213—215]. В этих условиях
деструкция протекает очень незначительно, содержание двойных связей
в полимере составляет менее 0,2 % [216]. Температура
стеклования гидрированного полистирола, равная 150Х, примерно на
15° выше, чем температура стеклования высококристаллического
изотактического поливинилциклогексана [217].
4.3.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
ИЗОТАКТИЧЕСКОГО ПОЛИВИНИЛЦИКЛОГЕКСАНА
4.3.3.1. Кристалличность
Согласно Натта [218], изотактический ПВЦГ, полученный
ионно-координационной полимеризацией в плохих растворителях
или гидрированием изотактического ПС, образует
тетрагональную элементарную ячейку с размерами а = Ь = 21,9 А, с — 6,50 A.
Цепи располагаются в виде четверных спиралей с
пространственной группой I4i/a. Элементарная ячейка содержит 16
мономерных звеньев. Каждая спираль с правой винтовой резьбой
окружена четырьмя спиралями с левой резьбой. Схема упаковки
молекул в изотактическом ПВЦГ показана на рис. 4.29. Ноетер
приводит следующие размеры тетрагональных элементарных
ячеек: а = Ь = 20,48 A, с — 44,58 A. Согласно приведенной схеме
элементарная ячейка содержит 96 мономерных звеньев в четырех
спиралях по 24 звена в каждой [219].
По данным ИК- [196] и ЯМР-спектроскопии [220] можно
сделать вывод о том, что
ПВЦГ также содержит
незначительное количество
макромолекул, в которых
мономерные звенья
соединены по типу «голова к
голове».
Рис. 4.29. Схема упаковки
молекул изотактического
поливинилциклогексана [218].
90
4.3.3.2. Термостойкость
Изотактический ПВЦГ устойчив в вакууме до 380 °С и на
воздухе— до 230°С [221,222]. Стойкость ПВЦГ зависит от степени
тактичности и возрастает в следующем ряду в зависимости от типа
каталитических систем, использующихся при полимеризации: ди-
этилалюминийхлорид — трихлорид титана < триизобутилалюми-
ний — тетрахлорид титана < триизобутилалюминий — трихлорид
титана. Кривые ДТА и ТГА для поливинилциклогексана в вакууме
и на воздухе, а также для стабилизированного ПВЦГ на воздухе
показаны на рис. 4.30. При нагревании в вакууме на кривых
появляются два эндотермических пика. Первый пик при температуре
328 °С отвечает температуре плавления, второй пик при
температуре 380 °С — разложению полимера. Энергия активации процесса
термической деструкции в интервале температур от 390 и 420 °С
равна 56,5 ккал/моль [221].
При нагревании ПВЦГ на воздухе на кривых ниже температуры
плавления появляются два эндотермических пика с максимумами
при температурах 235 и 310 °С, соответствующими процессам
окислительной деструкции полимера. Энергия активации процесса
термоокислительной деструкции на воздухе в температурном
интервале 240—320 °С составляет примерно 31 ккал/моль [221].
Кривые поглощения кислорода изотактическим ПВЦГ (рис.. 4.31)
имеют S-образную форму с характерным индукционным периодом,
который при повышении температуры смещается в область более
коротких времен.
Основными продуктами разложения при термоокислительной
деструкции ПВЦГ при температурах ниже 200 °С являются вода
AT
50
300 400 t;C
по t;c ,
Рис. 4.30. Кривые дифференциально-термического (а) и термогравиметрического
(&) анализа изотактического поливинилциклогексана [222]:
/ — в вакууме; 2— на воздухе; 3 — стабилизированный ПВЦГ на воздухе.
Рис. 4.31. Кривые поглощения кислорода изотактическим поливинилциклогекса-
ном [222].
91
и циклогексанон. При температурах выше 250 °С выделяются
также формальдегид и ацетальдегид. В ИК-спектрах ПВЦГ,
подвергнутого термоокислительной деструкции, появляются полосы,
соответствующие альдегидным группам [223]. Разложение ПВЦГ на
воздухе при температуре 310 °С сопровождается снижением
содержания углерода с 87,24 до 81,26%, водорода с 12,73 до 11,78% и
кислорода — до 6,93 %. По поглощению кислорода и анализу
выделяющихся продуктов разложения можно сделать вывод о том,
что термоокислительная деструкция ПВЦГ протекает по
классическому радикально-цепному механизму [222]. При разложении
гидропероксидов выделяется вода. Наиболее вероятным является
следующий механизм образования циклогексанона:
—сн—сн2—¦
о2
•• * СН СНд ¦ * *
—О
—OH
СН—СН2—
о.
CH—CH2—
При более высоких температурах происходит изомеризация пе-
роксидных радикалов:
—СН—СН2
При разрыве углеродных цепей могут также образовываться
карбонильные группы:
цикло-С6Ни
—CH—CH2—CH—CH2—CH—CH2— *••
! I
О« цикло-С6Нп
Us
• • * —CH—CH2
I
цикло-Се,Нц
\
X—СН2—СН—СН2
02
4.3.3.3. Растворимость и свойства растворов
Изотактический ПВЦГ растворим при комнатной температуре
в цпклогексане, трихлорэтилене и хлороформе. При повышенных
температурах он растворяется в ароматических углеводородах,
таких, как "бензол, толуол, ксилол, ароматические хлоруглеводороды,
а также в декалине и тетралине [193, 199, 209, 210]. Температура
помутнения для растворов ПВЦГ в хлорированных ароматических
углеводородах равна для хлорбензола—10 °С, для о-дихлорбен-
зола 35 °С и для трихлорбензола 40 °С [197]. В пеитане и простых
эфирах ПВЦГ растворяется лишь частично. Фракционирование
ПВЦГ можно проводить экстрагируя его бензолом при различных
температурах [192,197]. Характеристическая вязкость ПВЦГ в
циклогексане при 25 °С, равная 1,18 A00 мл/г), соответствует
молекулярной массе 300 000 (определена методом светорассеяния)
[187].
Параметр взаимодействия Хаггинса % при 37 °С для растворов
в толуоле равен 0,465, второй вириальныйкоэффициент — 3,3 моль-
• см3-г~2-104. Незначительные различия в значениях параметра
растворимости по сравнению с изотактическим ПС такой же
молекулярной массы обусловлены тем, что толуол является «лучшим»
растворителем для ПС, чем для циклоалифатического поливинил-
цикл огексана [215].
4.3.4. ПЕРЕРАБОТКА ИЗОТАКТИЧЕСКОГО
ПОЛИВИНИЛЦИКЛОГЕКСАНА
ПВЦГ можно перерабатывать в изделия обычными методами,
используемыми для переработки термопластов. Литье под
давлением проводится [224] при температурах цилиндра 325—330 °С
и температурах литьевой формы 150—200 °С. При обычных
температурах переработки стабилизации ПВЦГ не требуется, если
полученные изделия эксплуатируются при температурах ниже 120 °С.
При получении изделий, применяемых при температурах порядка
200 °С, рекомендуется вводить в полимер стабилизаторы, такие, как
антрацен, триалкилфосфит [222], замещенные фенолы, вторичные
ароматические амины, олово- или кремнийорганические
соединения [224].
Из ПВЦГ можно вырабатывать волокна мокрым формованием
из раствора с последующей непосредственной горячей вытяжкой
подсушенного волокна [225]. Для формования волокна пригодны
5—8 %-ные растворы полимера в хлороформе, трихлорэтилене
(вязкость 30—140 Пз) или циклогексане [226], для осадительной
ванны используется метанол или изопропанол. Отдельные нити
нагреваются до 70—80 °С для удаления растворителя и
вытягиваются при 150—270 °С в 5—7 раз. Оптимальный температурный
интервал для ориентации волокон составляет 175—190°С> Волокна
имеют разрывную прочность 3,5 г/денье и относительное
удлинение при разрыве более 35 %. Применение волокон при повышен-
93
ных температурах ограничивается тем, что при 95 °С наблюдается
30%-ная потеря прочности, а при 150°С прочность уменьшается
на 80 % по сравнению с исходным значением при комнатной
температуре. При формовании волокон из расплава или сухим
методом из хлороформа или трихлорэтилена получаются лишь
хрупкие короткие волокна.
4.3.5. СВОЙСТВА И ПРИМЕНЕНИЕ ИЗОТАКТИЧЕСКОГО
ПОЛИВИНИЛЦИКЛОГЕКСАНА
ПВЦГ является единственным полимером, который можно
перерабатывать в изделия обычными для термопластов методами и
применять в качестве высокочастотного диэлектрика вплоть до
200 °С. Физико-механические, теплофизические и электрические
свойства изотактического ПВЦГ приведены ниже [193, 215, 217,
223, 224]:
Температура, °С
плавления 325—330
стеклования 118 (калориметрия)
128 (крутильный
маятник)
Плотность, г/см3
при 25 °С 0,9467
при 90 °С 0,9272
Теплостойкость по Вика, °С 330—350
Показатель преломления п^ , 1,51 — 1,56
—dp/dT, г/(смэ-°С) 4,5-Ю-4
Прочность при растяжении, кгс/см2 170—200
Ударная вязкость, кгс-см/см2 5—8
Прочность при изгибе, кгс/см2 200—230
Диэлектрическая проницаемость при —160 4-220 °С и 50—
1010 Гц 2,1-2,4
Тангенс угла диэлектрических потерь при —10ч-220°С и
50—10'° Гц E-8). Ю-4
Объемное электрическое сопротивление при 20—150 °С,
Ом-см 1015-1018
Температура стеклования изотактического ПВЦГ, равная
118°С, примерно на 20° выше, чем температура стеклования атак-
тического ПВЦГ [227].
Диэлектрические свойства ПВЦГ в температурном интервале
от — 180 до 160 °С почти не изменяются.
По своим диэлектрическим свойствам ПВЦГ превосходит
поликарбонат. Температурные зависимости диэлектрической
проницаемости и тангенса угла диэлектрических потерь в интервале частот
50 Гц—1 МГц приведены на рис. 4.32. Диэлектрическая
проницаемость в области температур стеклования от 100 до 120°С
изменяется; с повышением частоты изменения диэлектрической
проницаемости смещаются в область более высоких температур [223]. Выше
и ниже температуры стеклования диэлектрическая проницаемость
слабо зависит от температуры. В стеклообразном состоянии
температурный коэффициент Ае/е составляет примерно 0,2%, в
высокоэластическом состоянии примерно — 0,6 %. Этот параметр
• ¦ • ¦ L ^
| 1 1 1 ! 1
2,5
г i
1
f .? 3 4
1 1
f 5
| |
-160-140-120 -WO -80 -60 -40--20 0 20 40 60 80 100 120 Ш t,°C
Рис. 4.32. Температурная зависимость диэлектрической проницаемости е и
тангенса угла диэлектрических потерь tg o изотактического поливинилциклогексана
при различных частотах [223]:
] _ 50 Гц; 2 — 400 Гц; 3 — 1000 Гц; 4 — 5000 Гц; 5 — 100 кГц; 6 — 500 кГц; 7 — I МГц.
почти вдвое меньше, чем у ПС или ПТФЭ. Диэлектрическая
проницаемость в интервале температур от — 160 до 200 °С изменяется
максимум на 2 %•
Характер температурных зависимостей диэлектрической
проницаемости ПВЦГ, а также тангенса угла диэлектрических потерь
объясняется исключительно типом дипольной поляризации ПВЦГ.
Выше и ниже температуры стеклования на оба указанных
параметра оказывает влияние лишь тепловое движение макромолекул.
Удельное объемное электрическое сопротивление в интервале
температур 20—150°С составляет 1015— 1018 Ом/см.
4.4. ПОЛИВИНИЛФТОРИД
[—СН2—CHF—]п
Винилфторид получается в промышленном масштабе
присоединением фтористого водорода к ацетилену или пиролизом 1,1-ди-
фторэтана.
Радикальная полимеризация мономера осуществляется
преимущественно в суспензии при 40—150°С и давлении до 1000 кгс/см2.
Температура плавления ПВФ составляет примерно 230 °С.
Пленки из ПВФ получают чаще всего из раствора или дисперсий.
Для переработки в изделия методами, используемыми для
термопластов, при температурах выше 200 °С применяют образцы
полимера средней вязкости или модифицированный ПВФ.
Эксплуатационные свойства ПВФ сильно зависят от степени ориентации
материала.
ПВФ отличается высокой стойкостью к атмосферным
воздействиям, термическому старению и агрессивным средам. Разрывное
удлинение после 10 лет атмосферного старения в субтропическом
климате уменьшается лишь на 50 % ¦ Верхняя температура длитель-
95
ной эксплуатации составляет около 140 СС. В основном ПВФ ис-»
пользуется в качестве покрытий в строительстве.
ПВФ производится в промышленном масштабе с 1963 г.
4.4.1. ВИНИЛФТОРИД
Винилфторид был впервые получен в 1901 г. [233,234] путем
отщепления брома от дибромфторэтилена цинком
SbF3 Zn
2Вг— СН2—CHBr2 > Вг—СН2— CHF2 + Br—CH2—CHBrF *¦ 2CH2=CHF
и взаимодействием 1,1-дифтор-2-иодэтана с реактивом Гриньяра
[235]:
Ce45MgBr
F2CH— CH2I > CH2=CHF
В промышленности винилфторид получают по реакции
присоединения фтористого водорода к ацетилену или пиролитическим
отщеплением фтористого водорода от фторированных алканов.
При проведении реакции присоединения фтористого водорода
к ацетилену при 20 °С и давлении 10 кгс/см2 без катализатора
конверсия ацетилена составляет примерно 15 % [236]. При
использовании в качестве катилазатора активного угля,
пропитанного цианидом меди (I) или цианидом калия, при температуре
реакции 160 °С получают винилфторид примерно с 50 %-ным
выходом. В качестве побочного продукта образуется 1,1-дифторэтан
[237, 238]. Оксиды хрома и соли хромовой кислоты на носителе —
древесном угле могут использоваться как катализаторы в
интервале температур 200—400 °С [239]. При использовании оксидов
цинка выход винилфторида при 300 °С составляет примерно 70 %.
Около 10 % ацетилена остается непрореагировавшим, в виде
побочного продукта выделяется до 20 % дифторэтана [240]. При
использовании в качестве катализатора активного оксида алюминия
[241] или трифторида алюминия [242] и температуре реакции
300 °С 1,1-дифторэтан, образующийся в качестве побочного
продукта, при возвращении его в процесс может также
взаимодействовать с избыточным ацетиленом, давая винилфторид. 96 %-ная
степень превращения при продолжительности реакции 8 ч и 120°С
достигается при применении активного технического углерода,
пропитанного раствором трифторацетата ртути [243]. В качестве
катализаторов реакции присоединения фтористого водорода к
ацетилену также запатентованы соли кадмия—активный уголь
[244], трифторид алюминия — графит [245], трифторид
алюминия— оксид алюминия [246] и сульфат алюминия [247].
Другой промышленный способ получения винилфторида также
основан на использовании ацетилена. При присоединении к нему
двух молей фтористого водорода сначала образуется
1,1-дифторэтан [248], из которого при термокаталитическом отщеплении
фтористого водорода получается винилфторид:
2HF Т, Кат
HCs=CH > F2CH—СН3 —> CHF=CH2
Некатализируемый пиролиз 1,1-дпфторэтапа приводит к
получению винилфторида с хорошими выходами лишь при температуре
выше 800 °С [249]. При использовании таких катализаторов, как
ТЮ.2, ZrO2 или KF на активном угле, пиролиз протекает с
достаточной скоростью уже при 400—600 СС [250]. При каталитическом
дегидрофторированпи с использованием системы сульфат
алюминия— активный уголь в качестве катализатора при 250—400 °С
выход винилфторида составляет 30—40 % [251]. Применение
каталитической системы хлорид палладия — оксид алюминия при
300 °С позволяет повысить конверсию до 70 % и выше [252].
Винилфторид также получается пиролизом 1,2-дифторэтана,
1,1-хлорфторэтана и 1-фтор-2-хлорэтана. При проведении
пиролиза 1,2-дифторэтана при 200—400°С в качестве катализатора
используют сульфат кальция, гранулированный алюминий или
активный уголь [253]. 1,1-Хлорфторэтан пиролизуется при 600—
750 °С, образуя винилфторнд с максимальным выходом 50%
[254]. При взаимодействии 1,1-хлорфторэтана с натронной
известью при 400 °С винилфторид получается с выходом около 60%
[255].
Почти количественный выход достигается при использовании
порошкообразной меди или хлорида меди (I) при 500—800 °С
[256, 257], никеля или сплавов никеля при 400—700 °С [258] или
системы хлорид палладия — оксид алюминия [259].
При пиролизе 1-фтор-2-хлорэтана в присутствии
каталитического количества брома при 450—600 °С винилфторид образуется с
количественным выходом [260].
Другие способы получения винилфторида основаны на
взаимодействии фтористого водорода с винилхлоридом. При
непосредственном взаимодействии в газовой фазе при 200—500 °С в
присутствии каталитической системы оксид хрома — алюминий [261], или
хлорид палладия — оксид алюминия [262] винилфторид
образуется с выходом, не превышающим 40 %. При двухстадийном способе,
заключающемся во взаимодействии винилхлорида с фтористым
водородом в присутствии сильных кислот при 0 °С с получением
1,1-хлорфторэтана и 1,1-дифторэтана с последующим пиролизом
реакционной смеси при 400—800 °С, винилфторид получают
с 85%-ным выходом [263].
При комнатной температуре винилфторид представляет собой
бесцветный газ. Физико-химические свойства винилфторида
приведены ниже:
Температура, °С
кипения —72,2
плавления —160,5
Плотность при температуре кипения, г/см3 .... 0,853
Дипольный момент, Д 1,42±0,01
Потенциал ионизации, эВ 10,4
Критическая температура, °С 54,7
Критическое давление, кгс/см2 53,4
Теплота образования при 25 °С, ккал/моль .... 28
Давление пара при 21 "С, кгс/см2 . 25,2
4 Зак. 18 97
Теплоемкость, кал/(г-0С)
при —28.9 °С . , . . . . 0,484 (жидкий)
0,224 (газ, 1 кгс/см2)
при 4,4 °С 0,510 (жидкий)
0,241 (газ, 1 кгс/см2)
при 27,7 СС 0,650 (жидкий)
0,252 (газ, 1 кгс/см2)
Предел взрывасмости на воздухе, % (объемн.) . . . 2,6—21,7
Температура вспышки, СС 460±5
Винилфторпд практически нерастворим в воде, очень мало
растворяется в спирте и ацетоне.
Винил фторид обладает очень незначительной токсичностью. При
60—80%-ной концентрации он оказывает слабое, полностью
обратимое наркотическое действие.
Для получения высококачественного полимера решающее
значение имеет чистота винилфторида. Недопустимо, чтобы
содержание в мономере ацетилена превышало 5-10~4% и кислорода —
20-10~4%. Фтористый водород удаляется из мономера
пропусканием его через абсорбционные колонны с гидроксидом натрия.
Примеси ацетилена удаляют промывкой в аммиачном растворе
хлорида меди (I) или в N-метилпирролидоне [264], пропусканием
через оксид меди — карбид кремния при 250—400 °С [265] или
путем каталитического превращения в винилхлорид с последующим
отделением перегонкой [266]. Путем дистилляции под давлением
при температурах от —50 до —25 °С содержание кислорода
уменьшается до допустимого уровня.
4.4.2. ПОЛИМЕРИЗАЦИЯ ВИНИЛФТОРИДА
Поливинилфторид (ПВФ) был впервые получен в 1932 г. [267]
методом радикальной полимеризации мономера под высоким
давлением в толуольном растворе.
В процессе полимеризации винилфторид ведет себя, скорее, как
этилен, чем как прочие винилгалогениды. Так как радикальная
полимеризация чаще всего проводится при температурах выше
критической температуры мономера E7,4 °С), то для достижения
достаточно высоких скорости полимеризации и молекулярной массы
необходимо создание высокого парциального давления мономера.
Полимеризация винилфторида может осуществляться также на
каталитических системах Циглера — Натта.
В промышленности полимеризацию проводят в водной
суспензии или эмульсии для того, чтобы получить полимер в виде,
пригодном для дальнейшей переработки.
ПВФ выпускается в промышленном масштабе фирмой «Du
Pont» с 1963 г. (пленки Теслар или Тедлар). Фирма «Dynamit
Nobel» изготавливает порошкообразный поливинилфторид под
названием PVF EPL 90. Для нанесения покрытий используется
модифицированный ПВФ Далвор X 6500, выпускаемый фирмой «Diamond
Shamrock» (США). В 1968 г. потребление ПВФ составило 100 т.
Стоимость его в зависимости от рецептуры колеблется от 10 до
20 марок за 1 кг.
98
4.4.2.1. Радикальная полимеризация
Радикальная полимеризация винилфторида в водной суспензии
проводится при температурах от 40 до 150°С и давлениях до
1000 кгс/см2. В качестве инициаторов используют пероксиды, азо-
соеднпения, а также борорганические соединения.
Впервые полимеризация в полупромышленном масштабе была
осуществлена фирмой «Du Pont» в конце 40-х годов в суспензии
при использовании в качестве инициатора бензоилпероксида при
85 °С и давлении 900 кгс/см2. Продолжительность реакции
составляла 6—10 ч [268—271]. При использовании азосоединений и при
более низких температурах полимеризацию можно проводить при
давлениях от 25 до 100 кгс/см2 с хорошими выходами.
Суспензионную полимеризацию при температурах реакции ниже
100 °С можно инициировать бензоилпероксидом [268—272],
персульфатом аммония [272, 273], додеканоилпероксидом [274],
бис(перфторацетил)пероксидом [276], бис(трифторметил)перокси-
дом [277], диизопропилпероксидикарбонатом [275, 279], динитри-
лом азобисизомасляной кислоты [268, 272, 275], азобисизобутил-
амидингидрохлоридом [272, 278], азобистриметилвалеронитрилом
[272], азобисдиметилвалеронитрилом [272], комплексами триалки-
ла бора с аммиаком [274, 280—282] и с пероксидом водорода
[275, 283, 284], а также алкилами цинка, кадмия и бериллия [285,
286]. При температурах полимеризации выше 100 °С в качестве
инициаторов можно использовать диэтил- или ди-трет-бутилпер-
оксид [272].
С повышением температуры полимеризации уменьшается
молекулярная масса ПВФ и увеличивается степень его разветвленности
[272]. Влияние давления на выход и молекулярную массу ПВФ,
полученного суспензионной полимеризацией при использовании
в качестве инициатора 0,2 % бензоилпероксида, при различных
температурах полимеризации показано на рис. 4.33 и 4.34. Средняя
молекулярная масса и выход полимера при заданной концентрации
инициатора увеличиваются с возрастанием давления.
На скорость полимеризации сильно влияют кислород воздуха
и ацетилен, который присутствует в мономере в качестве примеси.
2500
Рис. 4.33. Зависимость 2000
выхода суспензионного
полнвинилфторида от fSOO
давления [272].
Рис. 4.34. Зависимость
молекулярной массы
суспензионного поливинил-
фторида от давления
[272].
4*
Г}(аДМФ),дл/г
1000-
500-
Щ
Р,КГС/СПг Q 2QQ
Р,кгфпг
99
Рис. 4.35. Зависимость молекулярной массы
суспензионного поливиннлфторнда от
концентрации агента передачи цепи [272]:
/ — метанол; 2 — изопропиловый спирт; 3 — 1,3-днок.
солан.
о
ю
цепей >
При содержании кислорода 135-
• 10—4 %, давлении 900 кгс/см2 и
температуре 80—100°С полимеризация в
присутствии 0,2 % бензоилпероксида
ускоряется, но при концентрации
кислорода 500-Ю-4 % она оказывается
ингибированной. В идентичных
условиях ацетилен при концентрации 0,1 %
также оказывает ускоряющее
действие на полимеризацию, причем с почти количественным
выходом образуются сетчатые нерастворимые полимеры. В
количестве 2% ацетилен уже ингибирует полимеризацию. 1,1-Ди-
фторэтан как примесь не оказывает влияния на
полимеризацию.
Среднюю молекулярную массу ПВФ можно определить при
введении агентов передачи цепи. На рис. 4.35 показано влияние
концентрации метанола, изопропанола и диоксолана на
характеристическую вязкость образующегося ПВФ. При проведении
полимеризации в бензоле снижаются средняя молекулярная масса и
термостойкость ПВФ [272].
В качестве стабилизаторов дисперсии при суспензионной
полимеризации ПВФ используют лаурилсульфат магния [287],
метилцеллюлозу [279] и додецилбензолсульфонат натрия
[279].
Описана непрерывная суспензионная полимеризация
винилфторида в присутствии низкомолекулярных олефинов как регуляторов
длины цепи [288], протекающая в две стадии: 60—90°С/100—
600 кгс/см2 и 90—140 °С/280—900 кгс/см2 [289].
При эмульсионной полимеризации винилфторида обычные
эмульгаторы нельзя использовать из-за очень высокой степени
передачи кинетической цепи мономеров. Для этой цели пригодны
перфторированные карбоновые кислоты с семью-восемью
углеродными атомами. В отличие от других способов полимеризации при
эмульсионной полимеризации винилфторида, проводимой при
давлениях ниже 50 кгс/см2, могут достигаться высокие степени
превращения [290, 291].
Радиационно-химическое радикальное инициирование
полимеризации винилфторида в суспензии или эмульсии позволяет проводить
процесс при более низких температурах и высоких скоростях
полимеризации [292—303]. ПВФ, полученный при низких температурах
радиационно-химическим способом, характеризуется более
высокой температурой стеклования и повышенной термостойкостью по
сравнению с обычными полимерами.
100
4.4.2.2. Ионно-координационная полимеризация
При цонно-координацпонной полимеризации втшилфторида
используют гомогенные каталитические системы, состоящие из
растворимых, не содержащих галогенов солей ванадия и моно- или
диалюминийалкилов, таких, как ванадплацетплацетонат—моно-
хлоралюминийэтилат в растворе в метиленхлориде или ДМФ
1286]. Средняя молекулярная масса полимеров, однако, очень
невелика, по стереорегулярности такие полимеры близки к ПВФ,
полученному радикальной полимеризацией. На гетерогенных
каталитических системах, состоящих из алюминийалкилов и галоге-
нидов титана или ванадия конверсия очень незначительна.
4.4.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИВИНИЛФТОРИДА
4.4.3.1. Структура
По данным ИК-спектроскопии и спектрам комбинационного
рассеяния, промышленный ПВФ имеет атактическую структуру [304].
Степень кристалличности и температура плавления ПВФ сильно
зависят от температуры полимеризации (табл. 4.12). С повышением
температуры полимеризации от 0 до 180°С температура плавления
ПВФ снижается с 230 до 165°С [286, 305], что объясняется
увеличением содержания звеньев, соединенных по типу «голова к
голове».
По данным ЯМР A9 F)-спектроскопии [306, 307], число звеньев,
соединенных по типу «голова к голове», в интервале температур
полимеризации от 25 до 189°С увеличивается с 26 до 32%.
Поскольку по плотности упаковки атомы фтора близки к
атомам водорода, ПВФ имеет относительно высокую степень
кристалличности, достигающую 40—50 %-
Кристаллический ПВФ относится к орторомбической
кристаллической системе с пространственной группой Ст2т [308]. Размеры
элементарной ячейки равны а = 8,57 А, Ь = 4,95 А, с = 2,52 А.
Макромолекула ПВФ, так же как и ПЭ, представляет собой
плоскую зигзагообразную цепь. В элементарной ячейке содержатся два
Таблица 4.12. Температура плавления и степень
кристалличности поливинилфторида, полученного
при различных температурах [286]
Температура
полимеризации,
°С
180
85
40
30
Температура
плавания.
°С
164
197—205
205-215
218-225
Степень
кристалличности,
%
37
44
45
Температура
полимеризации,
°С
20
10
0
Температура
плавления,
°С
222-230
220—235
225-235
Степень
кристалличности,
%
45
48
50
101
мономерных звена [309]. Галайк [310], напротив, пришел к
выводу, что кристаллическая структура ПВФ имеет вид
гексагональной кристаллической решетки с размерами элементарной ячейки
а = Ь = 4,93 A и с = 2,53 А.
4.4.3.2. Растворимость и свойства растворов
ПВФ при температуре ниже 100 °С не растворяется в известных
растворителях. При температурах выше 100 °С он растворяется
в N-замещенных аминах или амидах, таких, как N-ацетилпипери-
дин, N-метил-М-изопропилпирролидон, 2-пирролидон, 5-метил-2-
пирролидон, в динитрилах, кетонах, тетраметиленсульфоне, диме-
тилсульфоксиде, диметилсульфонамиде, лактонах, таких, как Y-бу-
тиролактон, тетраметиленмочевине и триэтилфосфате. При
охлаждении растворов происходит гелеобразование.
Для формования пленок методом полива наиболее пригодны
тетраметилмочевина и диметилсульфонамид [272].
При определении средней молекулярной массы ПВФ вискози-
метрическим, седиментационным и осмометрическим методами
в качестве растворителей использовали диметилформамид, диме-
тилацетамид и -у-бутиролактон [272, 3.11]. Для подавления
межмолекулярного взаимодействия наиболее эффективной оказывается
добавка хлорида лития @,1 н. LiCl в ДМФ).
Для продуктов, выпускаемых в промышленном масштабе с
молекулярной массой 76 000—234 000, справедливо следующее
соотношение:
[г\] = 6,42 ¦ 10Mj85 (ДМФ/LiCl, 90 °С)
Для константы седиментации можно записать:
So = 0.0929ЛГ0*40 (ДМФ/LiCl, 100 °С)
Размер невозмущенного клубка- макромолекул А—^гЦМ
составляет 0,786 [311].
Отклонения при вискозиметрическом определении молекулярной
массы связаны с образованием ассоциатов и разветвлением цепей.
4.4.4. ПЕРЕРАБОТКА ПОЛИВИНИЛФТОРИДА
ПВФ перерабатывается в изделия преимущественно из
растворов и дисперсий, но наряду с этим применяются методы,
используемые для переработки термопластов, такие, как прессование, ка-
ландрование, экструзия или литье под давлением.
Переработка высоковязких марок ПВФ методами,
используемыми для получения изделий из термопластов, осуществляется при
температурах выше 200 °С и предполагает надежную стабилизацию
полимера акцепторами кислот и с помощью радикальных ловушек
с тем, чтобы предотвратить термическую деструкцию при его
переработке. В качестве стабилизаторов используются моноэфиры
малеиновой или фумаровой кислот [312], фосфиты, соли бария и
102
кадмия высших жирных кислот [313], монолаурпловый эфир
глицерина [314], терпены и дисульфиды [315], фенолы, например
4,4'-бутилиден-бисB-т/?ег-бутил-5-метилфенол) [316]. По данным
[317], оптимальной рецептурой для внутренней и внешней
стабилизации являются составы, содержащие 0,25—0,75 масс. ч. триде-
цилфосфпта, 0,25—0,75 дипентаэритритола, 0,25—0,75 ди-трег-бу-
тил-я-крезола и 0,05 масс. ч. воска. Добавка SiC>2 или А12Оз к
пигментированным рецептурам оказывает каталитическое действие на
термическую деструкцию ПВФ. Термостойкость ПВФ, полученного
в присутствии борорганических соединений в качестве
инициаторов, повышается при обработке аммиаком, анилином или ацетами-
дом [318].
Для переработки литьем под давлением наиболее пригодны
марки ПВФ средней вязкости (полученные полимеризацией в
водном метаноле) с температурой размягчения примерно 155 °С [272].
Высокомолекулярные марки ПВФ могут перерабатываться этим
методом только при введении в них больших количеств
пластификаторов и стабилизаторов. В качестве пластификаторов используют
теломеры винилфторида с трифторметилиодидом [324] или тело-
меры трифторхлорэтилена [325]. Другие возможности улучшения
перерабатываемости ПВФ методами, используемыми для
термопластов, заключаются во введении на стадии полимеризации
небольших количеств винилацетата [336, 337] или в приготовлении
составов с поливинилацетатом [336], поливинилпиридином [338],
АБС-сополимерами [339] или сополимерами этилена с винилацета-
том или метилметакрилата с этилметакрилатом [339]. Получение
пленок или нанесение покрытий осуществляется методом полива
из раствора, формованием из дисперсий, а также экструзией или
каландрованием.
При отливе пленок ПВФ частично или полностью растворяют
при 130°С в растворителях, таких, как ДМФ, и 8— 10^%-ные
растворы выливают на бесконечную, нагретую до 120—130 °С стальную
ленту. На этой установке могут использоваться также органозоли
в инертных растворителях, содержащих 20—85 % ПВФ [326—
331]. Покрытия можно наносить по методу «катушка —
покрытие» или литьевым способом. Внутреннюю облицовку производят
окунанием или обливанием. При использовании водных дисперсий
необходимо добавление таких растворителей, как тетраметилмо-
чевина. гтг>лч
При экструзии пленок из высокомолекулярных образцов ПВФ
целесообразно использовать в качестве добавок, понижающих
вязкость расплава и улучшающих технологические свойства
перерабатываемого материала, 15—30% высококипящих инертных
растворителей, таких, как ТМС. НМП. пропилкарбонат, циклогексанон
или Y-бутиролактон [333, 3341. Кроме того, методом экструзии
можно перерабатывать в пленки дисперсии полимера в высоко-
кипящих растворителях. Без добавок растворителей пленки могут
быть получены экструзией из модифицированного ПВФ типа
Далвор (Diamond Shamrock Corp.). Столингс [317] приводит
ЮЗ
условия переработки полимера на экструдерах со степенью сжатия
от 2,2 : ] до 3 : 1 и шнеках с L/D от 20 : 1 до 24 : 1 в виде
температурного профиля от зоны I до зоны IV, составляющего 130—215 °С.
ПВФ средней вязкости с коэффициентом Фикентчера 60—70,
стабилизированный 1 % стеарата бария или кадмия и 0,2 % бисфе-
нола А, можно перерабатывать в прозрачные пленки толщиной
0,05 мм на четырехвалковых L-образных каландрах при
температурах вальцевания 196, 210, 218 и 203°С [332].
При нанесении порошкообразных покрытий из
модифицированного ПВФ на стальные листы необходим предварительный нагрев
листов до 330 °С [317].
Пленки и покрытия из ПВФ, предназначенные для наружного
использования, стабилизируют бензофеноном [319—322] или
различными триазолами [323].
4.4.5. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА ПОЛИВИНИЛФТОРИДА
Поливинилфторид сочетает в себе отличную атмосферостойкость
с высокой термо- и химической стойкостью, а также с хорошими
физико-механическими и электрическими характеристиками.
4.4.5.1. Теплофизические свойства
Ниже приведены основные теплофизические характеристики по-
ливинилфторида [340,341]:
Температура, °С
плавления 190—210
нулевой прочности
хрупкости
Плотность, г/см3 ....
Показатель преломления .
25
"D
Теплота плавления, ккал/
элементарное звено . .
Влагопоглощение, % • ¦
Поверхностное натяжение,
дин/см
Проницаемость (толщина
пленки 0,025 мм), г/100
м2/ч
Н2О D0°С) . . . .
О2 B5 °С)
N2 B5 °С) . . . .
СО2 B5 °С) . . . .
Около 300
— 180
1,38—1,55
1,45
1,80
0,5
28
190
0,3
0,02
1,3
Усадка сильно двухосно-
ориентированных пленок, %
°С
°С
130
150
170°С
180
190
°С
°С
Усадка слабо двухосио-
ориеитированных пленок
130°С
150 °С
170°С
180 °С
190°С
3
6
21
30
40
0
0
1
2
20
Температура плавления полимера уменьшается с повышением
температуры полимеризации.
Пленки из ПВФ в значительной степени проницаемы по
отношению к видимому и ультрафиолетовому свету. Прозрачность
пленок толщиной 0,025 мм составляет в области от 400 нм до 1,4 мкм
более 90 % и в области 230—400 нм — 80 %. В области ниже 200 нм
прозрачность, однако, сильно ухудшается [340]. С высокой прони-
104
цаемостью по отношению к УФ-лучам связана и отличная атмо-
сферостойкость ПВФ.
В ориентированных пленках показатель преломления зависит
от угла падаюшего света к направлению вытяжки. Показатель
преломления в направлении вытяжки равен 1,464 и по толщине
пленки— 1,444. Степень ориентации влияет также на оптическое
двойное лучепреломление пленок из ПВФ. В сильно ориентированных
пленках оно равно 0,0186, в слабо ориентированных пленках —
0,006. Термическая усадка увеличивается с возрастанием степени
ориентации ПВФ.
Проницаемость пленок из ПВФ по отношению к воде и
органическим средам незначительна.
Формованные материалы на основе ПВФ проявляют
самозатухающие свойства.
4.4.5.2. Физико-механические свойства
Вследствие ориентации при вытяжке кристаллических областей
физико-механические свойства ПВФ сильно зависят от степени
ориентации пленок. Сильное двухосное растяжение пленок
приводит к значительно более высоким значениям прочности при
растяжении, ударной вязкости, сопротивлению продавливанию и
меньшей усталостной прочности при изгибе. Напротив, образцы с
незначительной ориентацией обладают более высокими
деформируемостью и прочностью при разрыве.
Из-за высоких степени кристалличности и температуры
плавления ПВФ температурная зависимость физико-механических свойств
до 140°С выражена относительно слабо. На рис. 4.36 сопоставлены
температурные зависимости прочности при растяжении,
относительного удлинения и модуля упругости при растяжении двух
пленок из ПВФ различной степени ориентации, полиэтилентерефта-
лата и ПВФ. По числу изгибов, достигающему 230000, ПВФ близок
к ПЭНП.
Следует отметить высокую износостойкость, равную 4,8 (для
слабо ориентированной пленки) или 3,4 (для сильно двухосно-
ориентированной пленки) (ASTM D 523 = 53 Т). Соответствующие
бр,кгс/см2
2000
^ ^. .- , 10000
/Шок \ ^ .„„ ^^ / 1 W 5000
1000
500
0
10O t, С
100 t?C
50
100 t,°C
Рис. 4.36. Зависимость прочности при растяжении (а), относительного удлинения
при разрыве (б) и модуля при растяжении (в) образцов поливинилфторида от
температуры [340]:
I — ЛВФ сильно двухосно-ориентированный; 2 — ПВХ слабо двухосно-орнентированный;
5-ПВХ; 4-ПЭТФ.
105
Рис. 4.37. Частотная
зависимость тангенса угла
диэлектрических потерь
(а) и диэлектрической
р проницаемости (б) пле-
f Гц нок из поливинилфтори-
1 да [340].
средние показатели для обычных лаковых пленок колеблются от
0,5 до 1.
Механическая дисперсия кристаллического ПВФ в широком
температурном интервале была исследована Кавасаки [342].
Прочность при растяжении, кгс/см2 . -
Относительное удлинение при разрыве, %
Модуль упругости при растяжении, кгс/см2
Сопротивление продавливанию, кгс/см2
Прочность при изгибе, число циклов
при 23 °С
при —17 °С
Ударная вязкость, кгс-см/0,025 мм . .
Коэффициент износа
Износостойкость, мин/мм
Сильно двухосно-
ориентнрованная
пленка
1400
110
20 000
2,80
230 000
100 000
5,7
0,15
3,4
Слабо двухосноориен
тированная пленка
670
260
17 000
1,05
10 000
2
0,24
4,8
4.4.5.3. Электрические свойства
Пленки из ПВФ обладают высокой диэлектрической
проницаемостью G—9) и хорошей электрической прочностью; тангенс угла
диэлектрических потерь их составляет 0,03—0,06.
125 tt°G
50 75 100 125 150 t,°G
Рис. 4.38. Температурная зависимость диэлектрической проницаемости (а) и
тангенса угла диэлектрических потерь (б) пленок из поливпнилфторида [340]:
/~ПВФ сильно двухосно-ориентированный; 2—ПВФ слабо двухоспо ориентированный;
S - ПЭТФ.
106
10
25 50
WO !2St,°C
Рис. 4.39. Температурная зависимость удельного
объемного электрического сопротивления пленок
из поливинилфторида [340]:
/ — ПВФ сильно лвухосно-орнентировонный: 2 — ГЖФ
слабо двухосно-ориентнрованный; 3 — ПЭТФ.
Температурная и частотная
зависимости электрических показателей ПВФ
выражены относительно слабо. В
сочетании с высокой тепло- и химической
стойкостью это открывает широкие
перспективы применения ПВФ в
электротехнической промышленности.
На рис. 4.37 приведена частотная зависимость диэлектрической
проницаемости и тангенса угла диэлектрических потерь. На рис. 4.38
и 4.39 представлены температурные зависимости диэлектрической
проницаемости, тангенса угла диэлектрических потерь и удельного
объемного электрического сопротивления. Зависимость тангенса
угла диэлектрических потерь, определенного при 105 Гц и 80 °С,
имеет четко выраженный минимум.
Электрическая прочность в зависимости от толщины пленки
колеблется от 2000 до 5000 В/0,025 мм.
Поляризованный ПВФ проявляет пьезоэлектрические свойства
[343].
Механизм подвижности носителей зарядов в ПВФ в
зависимости от температуры был изучен Мак-Глю [344]. В интервале
температур от 120 до 20 °С подвижность носителей зарядов
уменьшается на пять порядков.
4.4.5.4. Химическая стойкость
Пленки из ПВФ обладают высокой стойкостью к
неокислительным кислотам, основаниям и органическим растворителям.
При выдержке пленок ПВФ в 10 %-ной НС1 или в 10%-ной
NaOH при 60 °С в течение 7 сут не отмечалось изменений прочности
при растяжении или ударной вязкости. Механические показатели
сохраняются практически неизменными даже после двухчасового
кипячения пленок ПВФ в 10%-ной НС1, 10 %-ном NaOH, тетра-
хлориде углерода, бензоле, ацетоне или метилэтилкетоне [340].
Вследствие отличной стойкости к гидролизу механические
свойства ПВФ очень незначительно изменяются даже при выдержке
его в водяных парах при 100 °С. Прочность и модуль упругости при
растяжении остаются при этом неизменными после выдержки в
N
з-ю5
Рис. 4.40. Зависимость усталостной
прочности поливинилфторида от времени выдерж- 1-Ю5
ки 1з водяном паре при 100 °С [340].
107
течение 1500 ч. Ударная вязкость уменьшается на 3,5 %, а
разрывное удлинение — на 8 %. Число циклов до разрушения
уменьшается с 220 000 до 105000 (рис. 4.40).
4.4.5.5. Длительная стойкость и стойкость к старению
По своей термостойкости ПВФ занимает промежуточное
положение между ПВХ и ПТФЭ. Полимер стабилен на воздухе до
175 °С и в инертной атмосфере — до 300 °С. В отличие от ПВХ,
в котором отщепление НС1 протекает по радикально-цепному
механизму, выделение HF происходит в результате реакции
молекулярного отщепления. Это обусловливает более высокую стабильность
полимера [345].
При термической деструкции в вакууме наряду с отщеплением
HF происходит также распад основной цепи полимера [3461.
N
О 250 500 750 1000 1250 г, y
€,%
200
150
0 250 500 750 Ю00 1250 Г, y
W*
W*
W3
W2
V
0
200
100
60
40
20
W
6
4
2
1
500 Ю00 r,V
\
\
\
\
\
1 1 1 I I \
2 4 ? 8 W
Рис. 4.41. Зависимость прочности при растяжении (а) и относительного
удлинения при разрыве (б) поливинилфторида от времени выдержки при 150 °С [340]:
/ —ПЭТФ; 2 — ПВФ сильно двухосно-ориентированный; 3 — ПВФ слабо двухосно-ориентн-
рованный; 4 — ПВХ.
Рис. 4.42. Зависимость усталостной прочности поливинилфторттда от времени
выдержки на воздухе при 150 °С [340]:
/ — ПВФ сильно двухосно-орнентнрованный: 2 — ПВФ слабо двухосноориентпрованный;
3-ПЭТФ; 4-ПВХ.
Рис. 4.43. Зависимость относительного удлинения при разрыве от времени
выдержки в условиях экспозиции в штате Флорида [340].
108
Термоокислнтельная деструкция протекает относительно
медленно вплоть до температуры около 175 °С. Хотя после 1000 ч
старения на воздухе при 150 °С пленки темнеют, прочностные
характеристики полимера сохраняются на высоком уровне, На рис. 4.41
показано влияние продолжительности выдержки на воздухе при
150 °С на прочность при растяжении и относительное удлинение
при разрыве, а па рис. 4.42 — на прочность при знакопеременном
изгибе.
При атмосферных воздействиях пленки из ПВФ
характеризуются чрезвычайно длительным "сроком службы и способностью
сохранять большую часть своей первоначальной прозрачности и
прочности. После 10 лет атмосферного воздействия в штате Флорида
относительное удлинение при разрыве уменьшилось лишь на 50 %.
Как при естественном, так и при искусственном атмосферном
воздействии логарифм величины относительного удлинения при
разрыве линейно понижается с увеличением времени выдержки
(рис. 4.43). Минимальный срок службы покрытий из ПВФ
составляет приблизительно 30 лет.
Эрозия поверхности пленок, вызванная естественным старением,
также является незначительной. После двух лет атмосферного
воздействия (штат Флорида) износостойкость по ASTM D 658-44
ориентированных пленок снижается с 3,4 до 2,9 мин/мм [340]. Даже
после 10 лет атмосферного воздействия пленки еще могут быть
растянуты на холоду.
4.4.5.6. Радиационная стойкость
Стойкость ПВФ к ионизирующему излучению выше, чем
у ПТФЭ. В то время как физико-механические показатели ПТФЭ
ухудшаются уже при дозе облучения 2 Мрад, для ПВФ необходима
доза облучения 32 Мрад [347].
Радиационно-химический выход Ghf по фтористому водороду
при облучении дозой в 100 Мрад на воздухе или в инертной
атмосфере составляет 54—75. ПВФ, облученный дозой 100 Мрад, при
воздействии на него кислот сразу приобретает черный цвет и
становится хрупким [348].
4.4.6. ПРИМЕНЕНИЕ ПОЛИВИНИЛФТОРИДА
В основном ПВФ используют в качестве защитных покрытии
в строительстве, для упаковки пищевых продуктов и в качестве
пленочных прослоек при переработке реактопластов.
Покрытия из ПВФ имеют чрезвычайно высокие адгезию,
эластичность и способность к глубокой вытяжке, а также великолепную
атмосферостойкость, стойкость к растворителям и химическим
реагентам. ПВФ применяется для покрытия изделий из стали,
алюминия, стекла, твердой древесины, а также полимерных пленок и
пенопластов на основе ПВХ, АБС-пластиков, полистирола,
полиэфиров и полиуретанов. Металлы и полимеры с ПВФ покрытиями
могут хорошо деформироваться, так как ПВФ обладает значитель-
109
но более высокими гибкостью и деформируемостью по сравнению
с подложкой [349]. Адгезионную прочность пленок из ПВФ можно
повысить путем их модификации сополимерами метилметакрилата
(98%) и глицидилметакрилата B%) [350]. Сборные
строительные элементы конструкций из алюминиевых листов в форме тра-
пецни с нанесенной на них пигментированной пленкой из ПВФ
использовали в СССР как декоративные элементы фасадов зданий
[352].
Детали приборов, покрытые ПВФ, имеют примерно такую же
коррозионную стойкость, что и детали с керамическими
покрытиями, но расходы на их нанесение составляют лишь 25% от
расходов на нанесение защитных керамических покрытий.
Стойкость к атмосферным воздействиям пленок из ПВХ, АБС-
пластиков и полиэфиров с ПВФ покрытием повышается по
сравнению с исходными во много раз. Листы из фенопластов пли
полиэфиров, армированных стеклянным волокном, покрытые ПВФ,
сохраняют блеск поверхностей в 6 раз дольше, чем материалы без
ПВФ покрытий [361]. Листы с ПВФ покрытием используются в
качестве декоративной облицовки стен в кухнях, ваннах, больницах,
кабинах самолетов. Они имеют более высокую стойкость, и их
можно чистить органическими растворителями.
Для облицовки стен в больницах хорошо зарекомендовали себя
комбинированные материалы, состоящие из текстильной подложки
с тонким слоем ПВФ. Стойкие к царапанию и химически стойкие
покрытия препятствуют появлению и размножению бактерий.
Брызги иода или крови, попавшие на покрытия, легко удаляются с их
поверхности сильным моющим средством или растворителем [353].
Материалы на основе специальных стекол с ПВФ покрытием
применяются в космической технике [354].
Разметка улиц, выполненная из пигментированных АБС-пласти-
ков с ПВФ покрытием, служит в несколько раз дольше.
Пленки из ПВФ вследствие их термической стойкости
используются для упаковки пищевых продуктов и пленочных прослоек
при переработке реактопластов. Упаковки из ПВФ можно
нагревать как в печах с высокочастотным обогревом, так и на обычных
кухонных плитах. Мясо в упаковке из ПВФ можно замораживать
и варить.
В качестве пленочных прослоек ПВФ используют при
переработке фенольных и эпоксидных смол, силиконов, полиэфиров. Тол-
шина пленок колеблется от 12 до 25 мкм. Преимущества этих
пленок по сравнению с аналогичными материалами на основе
полиэфиров заключаются в том, что толщину пленок из ПВФ можно
уменьшить до 25 % и за счет повышения температуры переработки
до 205 °С увеличить скорость технологического процесса. Пленки
из ПВФ являются единственным материалом, который можно
использовать в качестве пленочной прослойки в системах
закаливания, содержащих фторид бора [341].
ПВФ применяется в электронике для получения тонкой
коррозионно-стойкой изоляции с высокими электрической прочностью и
по
гибкостью, а также для покрытия медной фольги в печатных
схемах. Для изготовления комбинированных материалов на основе
меди и ПВФ используется пленка, которая с одной стороны
покрывается адгезивом (поливинилбутиралем). придающим ей
самоклеящие свойства [355].
Сенсибилизированные пленки из ПВФ могут применяться в
пожарной сигнализации и инфракрасных кристаллических
детекторах [356].
4.4.7. СОПОЛИМЕРИЗАЦИЯ И ПРИВИТАЯ СОПОЛИМЕРИЗАЦИЯ
Радикальную сополимеризацию винилфторида, так же как и
гомополимеризацию, можно осуществлять в массе, растворе,
эмульсии или суспензии. Сомономеры, используемые для сополпмериза-
ции с винилфторидом, перечислены ниже:
Сомономер
Акриламид
Акрилонитрил
Этилен
а-Ацетаминоакрил-
амид
Дициклопентадиен
Дифтордихлорэтилен
Дифторизобутилен
Гексафторпропилен
Гексафторциклобу-
тен
Изобутилен
Диэтиловый эфир
малеиновой кислоты
Перфторпропилен
Пропилен
Стирол
Тетрафторэтилен
1,1,2,4-Тетрафтор
бутадиен-1,3
Литературный
источник
[395]
[369, 370, 395]
[357, 365, 366-370]
[391]
[365
394
404
375
[370
[398]
[395, 399]
[357, 369, 401]
[396, 397]
[369]
[357, 368-374]
[402]
Сомономер
Трифторэтилен
Трифторбутадиен
Трифторхлорэтилен
Винилацетат
Винилбутират
Винилхлорид
Винилиденхлорид
Винилиденфторид
Винилкарбонат
Винилпропионат
а-Цианакриламид
Литературный
источник
[382, 384]
[403]
[369, 382,
387, 393]
[336, 337, 358,
370, 376-381]
[379, 395]
[357, 369, 370,
372, 375, 380,
385-390, 405
406, 414, 415]
[369]
[364, 369, 371,
379, 382, 383]
[400]
[379, 380]
[364]
В табл. 4.13 приведены константы сополимеризации некоторых
систем.
Сополимеры с винилхлоридом растворимы в ДМФ, ТГФ,
фурфуроле и дихлорбензоле и пригодны для получения волокон
методом сухого формования или формования из раствора [357—359].
Их свето- и термостойкость возрастают с увеличением содержания
в сополимере винилфторида [406]. Сополимеры с 30%-ным
содержанием винилфторида устойчивы до 200 °С н выше [414]. Винил-
фторид может сополимеризоваться с Еннилхлоридом в присутствии
смеси катализаторов, состоящей из тетраизопропилтитана и алю-
минийалкилов, при 30 °С [385].
Сополимеры с изобутиленом или перфторпропплсном обладают
высокими гибкостью и прочностью и примерно такой же тепло- и
термостойкостью, как и гомополимер [357, 360, 361].
Тройные сополимеры с винилхлоридом и тетрафторэтнленом
Ш
Таблица 4ЛЗ. Константы сополимеризации винилфторида
с различными сомономерами [286, 363, 364]
Сомономер (г2)
Этилен
Винилхлорнд
Тетрафторэтилен
Хлортрифторэтилен
Акрилоннтрил
Винилацетат
Винилиденфторид
т, °с
30
30
30
25
30
80
30
30
25
0,3
0,05
0,27
0,27
0,18
0,8
0,001
0,16
5,5
1,7
11,0
0,05
0,05
0,06
1,2
24
2,9
0,17
0,51
0,55
0,013
0,013
0,011
0,96
0,024
0,46
0,935
растворяются в ацетоне, этилацетате, метилэтилкетоне и в гало-
гензамещенных углеводородах [362].
Привитые сополимеры стирол — ПВФ [407], метилметакри-
лат — ПВФ [407—409] и винилацетат — ПВФ [407] обладают
более высокой адгезионной прочностью по сравнению с гомополи-
мером.
Радиационно-химической прививкой мономера винилфторида
к природным и синтетическим полимерам, таким, как целлюлоза,
триацетат целлюлозы, ПВХ, ПС, ПММА [410, 411] или полиамид
1412], можно понизить растворимость или степень набухания
полимеров и повысить их термостойкость.
Хлорирование ПВФ лучше всего проводить в суспензии тетра-
хлорида углерода при комнатной температуре [413]. Вплоть до
40—50%-ного содержания хлора реакция протекает быстрее, чем
при хлорировании ПВХ. При продолжительном проведении реакции
достигается максимальное содержание хлора, равное 62%. По
данным ЯМР-спектроскопии, хлорирование СНг- и CHF-rpynn
вплоть до содержания хлора около 50 % протекает с одинаковой
скоростью. С увеличением содержания хлора повышается
растворимость ПВФ, снижаются теплостойкость по Вика и степень
кристалличности. ПВФ, содержащий 30 °/о хлора, растворим при
комнатной температуре в ДМФ. При 60 %-ном содержании хлора
полимер растворяется в тетрахлориде углерода. Хлорированный
ПВФ, содержащий 40 % хлора, является полностью аморфным.
При содержании хлора 20 % теплостойкость по Вика уменьшается
от 160 до 125 °С.
4.5. ПОЛИВИНИЛИДЕНФТОРИД
[~CH2-CF2-]„
Винилиденфторид получают в промышленном масштабе
термическим дегидрогалогенированием 1-хлор-1,1-дифторэтана.
Полимеризацию проводят преимущественно в суспензии или эмульсии
в присутствии радикальных инициаторов.
\\%
Поливинилиденфторид можно перерабатывать на обычных
машинах для переработки пластмасс при 210—280 °С.
По сравнению с другими термопластичными фторполимерами
поливинилиденфторид обладает большей механической прочностью,
более высокой износостойкостью и незначительной ползучестью.
Его теплостойкость составляет около 150 °С.
Поливинилиденфторид имеет отличную стойкость к растворителям и агрессивным
средам при повышенных температурах, а также ионизирующему
излучению.
Поливинилиденфторид применяется в основном в
электротехнике и электронике, в химическом машиностроении, строительстве
и для упаковки,
4.5.I. ВИНИЛИДЕНФТОРИД
Винилиденфторид получают из фторалканов термическим де-
гидрохлорированием, пиролизом в присутствии алифатических
углеводородов или хлора либо дегалогенированием.
Исходными соединениями для термического дегидрогалогениро-
вания являются 1,1-дифтор-1-хлорэтан [415—424], 1,1,1-трифторме-
тан [425, 426], 1Д-дифтор-2-хлорэтан [420], 1,1,1-бромдифторэтан
[427] и фторциклоалканы, например 1-метил-2,3-дифторциклобутан
[428, 429]. Дегидрогалогенирование проводят при температурах от
600 до 1500 °С и в течение от 30 до 0,3 с. В присутствии
катализаторов, таких, как платина, родий, никель, медь или фторид магния,
процесс проводят при 400—700 °С. При термическом дегидрохлори-
ровании 1,1-дифтор-1-хлорэтана при 850 °С степень превращения
достигает 97 %.
Пиролиз смесей фторалканов и алифатических углеводородов,
таких, как трифторметан — этилен [430, 431], трифторхлорметан
или трифторбромметан—метан [432], дифторхлорметан — метан
[433], дифторхлорметан — метилхлорид [434, 435], а также
пиролиз фторалканов и хлора, например 1,1-дифторэтан — хлор [436]
или дифтордихлорметан — хлор [437], протекают в аналогичных
условиях.
Дегалогенирование фторалканов, таких, как 1,2-дихлор-1Д-дн-
фторэтан [438,440] или 1,1,1-трифтор-2-хлор-2,2-дибромметан [439],
проводят в присутствии цинка или амальгамы натрия.
Промышленное применение нашел в настоящее время способ
термического дегидрогалогенирования. Используемые в качестве
исходного сырья фторалканы Сг производятся холодильной
промышленностью. Исходным сырьем для 1-хлор-1,1-дифторэтана,
который применяется преимущественно для произоодства винилиден-
фторида, служат 1,1,1-трихлорэтан, винилиденфторид или
ацетилен:
СНз—СС13 + 2HF —> СНз—CC1F2 + 2НС1
CH2=CC12 + 2HF —>¦ СНз—CC1F2 + HC1
с 12
г-CHFj, + СНз—CC1F2 + HC1
Винилпделфторид представляет собой бесцветный нетоксичный
газ. Ниже приведены физико-химические свойства винилиденфто-
рида:
Температура, °С
плавления —144
кипения —82
Плотность (жидкий, 23,6 °С), г/см3 0,617
Критическая температура, °С 30,1
Критическое давление, кгс/см2 . 45,2
Критическая плотность, г/см3 0,417
Теплота образования, ккал/моль —80±8
Удельная теплоемкость СР при 25 °С, кал/(моль-°С) 14,37
Теплота испарения B5 °С), кал/(г-моль) 980
Пределы взрываемости на воздухе, % (объемн.) 5,8—20,3
4.5.2. ПОЛИМЕРИЗАЦИЯ ВИНИЛИДЕНФТОРИДА
Винилиденфторид полимеризуется по радикальному механизму.
Впервые процесс получения поливинилиденфторида был описан
в 1948 г. в одном из патентов фирмы «Дюпон» [441].
Полимеризацию проводят, как правило, выше критической
температуры мономеров C0,1 °С) в массе, суспензии, эмульсии или
в растворе.
Полимеризацию в массе проводят при 60—120°С и давлении
от 30 до 300 кгс/см2 [441, 442]. Высокие давления необходимы для
достижения достаточно высокой молекулярной массы (порядка
100 000). Полимеризацию инициируют пероксидами, такими, как
ди-т/?ег-бутилперрксид [443], трет-бутилпероксиизобутират [444],
боралкилы [445], или у-излучеиием [446, 447].
При суспензионной полимеризации в качестве инициаторов
используют ди-трег-бутилпероксид [448, 449] и боралкилы [450, 451].
Эмульсионную полимеризацию винилиденфторида применяют
главным образом при получении паст и латексов для нанесения
покрытий [452—454]. В присутствии перкарбонатов, таких, как ди-
изопропилпероксидикарбонат [455, 456] или трет-бут ил пер оке и изо-
пропилкарбонат [457], эмульсионная полимеризация протекает
с высоким выходом и ниже критической температуры. Как и в слу*
чае других фторированных мономеров, наиболее эффективными
эмульгаторами являются перфорированные соединения, такие, как
перфторкаприлат аммония [458].
Путем радиационного инициирования полимеризации в растворе
при низких температурах можно получать полимеры с
температурой плавления выше 175°С и молекулярной массой более 300 000
[459—462].
В патентах фирмы «Dow Chemical» описано инициирование
полимеризации винилиденфторида катализаторами Циглера — Натта
[463, 464].
Поливимилиденфторид впервые в опытно-промышленном
масштабе был получен фирмой «Pennwalt Chemical Corp.» (США)
в 1960 г. под названием «Кинар». В 1962 г. была создана
полупромышленная установка мощностью несколько сот тонн в год, и
Ш
в 1965 г, в Келверт Сити была пущена промышленная установка
для получения мономера и полимера. Кинар выпускается в виде
порошка с насыпной плотностью 0,5 г/см3 и в виде гранулята с
насыпной плотностью 1 г/см3. Стоимость его составляет около
8 дол /к г.
В 1970 г. фирма «Diamond Shamrock Corp.» (Огайо, США)
выпустила новую марку поливинилиденфторида, полученного
суспензионной полимеризацией, под названием «Далвор DS».
В том же году в Японии фирмой «Kureha Chemical Со.» была
пущена промышленная установка мощностью 250 т в год [465].
Эта фирма поставляет полимер, применяемый для нанесения
покрытий и для получения изделий методом экструзии («Дайкин»),
а также пленок толщиной до 9 мкм. Стоимость полимера
составляет 5 йен за 1 кг.
Гранулированный поливинилиденфторид, перерабатываемый
литьем под давлением и экструзией, производится с 1973 г. фирмой
«Dynamit Nobel» под названием «Дифлор 2000» [555, 556].
4.5.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
ПОЛИВИНИЛИДЕНФТОРИДА
4.5.3.1. Структура
По данным ЯМР (J9F)-спектроскопии [466—468], выпускаемый
в промышленности поливинилиденфторид, содержит наряду с
обычным присоединением мономерных звеньев по типу «голова к
хвосту» примерно 10 % мономерных единиц, соединенных между собой
по Timv «голова к голове»:
СН2 CF2 CF2 СН2
Основные полосы в ЯМР A9F)-спектре показаны на рис. 4.44. Доля
присоединений «голова к голове» составляет в выпускаемых в
промышленном масштабе полимерах марки Кинар 301 10,4 % и
Дайкин 8,7 % [469]. Доля присоединений типа «голова к голове»
уменьшается только при радиационно-химической полимеризации
винилиденфторида в твердом состоянии при температуре жидкого
азота [461]. В полимерах, полученных при —78 С, содержание
присоединений типа «голова к голове» такое же, как и в продуктах,
выпускаемых в промышленном масштабе.
2 """ 2 """
22
Рис. 4.44. ЯМР (Р19)-спектр I \j? "CF2 -2
поливинилиденфторида II ~
E6,4 МГц, 0,25%-ный рас- I CH2-CH2-CF2-CH2-CF2-
твор ПВДФ в диметилацет- I \ f
амиде при комнатной тем-
115
4.5.3.2. Кристалличность
Кристалличность порошкообразного и неориентированного по-
ливинилиденфторида, определенная рентгеноструктурным методом,
составляет приблизительно 40—50 % [470].
Полнвинилиденфторид существует в трех кристаллических
формах, двух моноклинных (а и у) и одной орторомбической
модификации (?). Относительная доля различных кристаллических форм
зависит от условий получения полимера, а также от термической и
механической предыстории.
В а-модификацни, или фазе II, по Натта [471], макромолекулы
имеют ступенчатую конформацию (транс-гош-транс-гош'-располо-
жение) [472—474].
Молекула образует 2гспираль, причем дипольный момент С—F-
связи направлен параллельно оси цепи [475, 476].
Размеры элементарных ячеек составляют а — 9,64 А, Ь = 4,64 А,
с = 5,02 А, ? = 91°5' [477] или а = 4.96 A, Ь = 9,64 А, с = 4,62 А
и ? = 90° [479].
ИК-спектры а- и ?-модификации ПВДФ показаны на рис. 4.45,
Относительная доля а-модификации определялась по полосе
836 см-1 [478].
При радиационно-химической полимеризации в газовой фазе
[470] образуется преимущественно а-модификация.
Кристаллизация из расплава при атмосферном давлении независимо от
скорости охлаждения приводит к получению кристаллитов
исключительно а-модификации. Кристаллизация осуществляется при
120—130°С [480, 481]. Из таких растворителей, как хлорбензол,
ДМФ, ацетон, смеси ацетон — амилацетат C:1), поливинилиден-
фторид также кристаллизуется в а-модификацию [477, 482].
Конформации, образующиеся при растворении поливинилиден-
фторида, зависят от сил межмолекулярного взаимодействия в
растворе. При растворении поливинилиденфторида в диоксане, ТГФ
циклогексаноне или амилацетате а-модификация превращается
в ?-модификацию.
?-Модификация образуется также путем ориентации
поливинилиденфторида при 50—60 °С или кристаллизацией из расплава при
давлениях выше 5000 кгс/см2. В кристаллической ?-модификации,
или в фазе I, по Натта [471], макромолекулы
поливинилиденфторида расположены в виде плоских
z* ° зигзагообразных цепей. Дипольный
""Ч /"——ч ~^Л 70 момент молекулы направлен перпен-
\ J \Г\ I а дикулярно оси цепи. Элементарная
I/ V ^/ . ячейка ?-кристаллов является орто-
80
Рис. 4 45. ИК-спектры а- и ?-кристаллических
модификаций поливинилиденфторида:
а — кристаллическая а-модификацня (из 0,1%-ного
10 раствора в смеси хлорбензола с ДЧФ>: б —
кристаллическая ^-модификация (из 0,[%-ного рас-
v>,CM-' S50 500 4S0 40Q твора в дмсо).
U6
ромбической и принадлежит к пространственной группе Ст2т. Ди-
польные моменты двух цепей в элементарной ячейке направлены
параллельно друг другу вдоль Ь-оси [472; 473, 476. 483].
Плотность кристаллов ?-модификации. определенная рентгено-
структуриым методом, составляет 2,0 г/см3 [484]. Относительная
доля ?-модификацин определяется по полосе 852 см-1 [478]. При
инициировании полимеризации винилиденфторида химическими
соединениями преимущественно образуется кристаллическая ?-мо-
дификация [470]. Образование кристаллов ?-модификации из
расплава происходит лишь при давлениях выше 5000 кгс/см2 [485,
486]. При давлении ниже 5000 кгс/см2 из расплава
кристаллизуются смеси а- и ?-модификации [470, 476].
?-Модификация образуется также при механической ориентации
пленок а-модификации при 50—60 °С [473, 483, 487]. Термическая
обработка при 150°С под нагрузкой способствует дальнейшему
повышению степени ориентации ?-кристаллов. Образцы, содержащие
высокоориентированные ?-кристаллы, могут выдерживать нагрев
до 160°С без каких-либо изменений [480].
При использовании в качестве растворителей диоксана, ТГФ,
циклогексанона, амилацетата или диметилсульфоксида образуется
?-модификация [477,482,488].
При растворении ?-модификации в ДМФ или смеси
амилацетат—ацетон A:3) происходит ее превращение в а-модификацию.
При нагревании в расплаве выше 200 °С ?-модификация
превращается в а-модификацию [481].
Третья кристаллическая модификация поливинилиденфторида
выделяется при охлаждении из 0,3 %-ных растворов в смеси
хлорбензол— ДМФ (9 : 1). Размеры моноклинной элементарной ячейки
этой ^-модификации составляют а =8,75 А, Ь =5,14 A, с = 4,88 А
и ? = 91° [4911. Незначительную стабильность ?- и ^-модификаций
(формы I и III) по сравнению с а-модификацией (форма II) Хасе-
гава [479] объясняет стерическими эффектами и
электростатическим дипольным взаимодействием.
Поливинилиденфторидные пленки вследствие высокого
содержания в них кристаллической фазы обладают относительно
низкой B0%) прозрачностью. Содержание кристаллической фазы
можно снизить путем обработки порошка полимера растворами
неорганических солей в полярных органических растворителях,
например NaCl—СН3ОН. Степень прозрачности тонких пленок,
обработанных 0,3 % NaCl, достигает 68 % [489, 490].
При облучении 7"лУчами дозой 109 рад. степень
кристалличности порошкообразного поливинилиденфторида снижается с 40—
50 до 13—20%, при этом уменьшается плотность упаковки в
кристаллических областях [470].
Наилучшая перерабатываемость поливинилиденфторида типа
Далвор и КФ обусловлена более низкой средней молекулярной
массой, более высокой степенью кристалличности и
незначительной степенью разветвленности [478, 492].
117
Рис. 4.46. Вискозпметрическое определение
молекулярной массы поливиннлиденфторнда в днме-
тилформамиде при температуре 30 °С [478].
4.5.3.3. Свойства растворов
70000 80000 ЮОООО 150000 f1„
ных эфирах, кетонах и
Поливинилиденфторид растворим в
сильно полярных растворителях,
сложаминах, таких, как амилацетат, ацетон,
циклогексанон, диоксан, ТГФ, хлорбензол, ДМФ, ДМАА и ДМСО.
Исследования молекулярно-массового распределения различных
типов поливинилиденфторида методом гель-проникающей
хроматографии в циклогексаноне при 100 °С были проведены Столингсом
[478]. Соотношение между молекулярной массой, определенной
осмометрическим методом, и характеристической вязкостью в ДМФ
при 30 °С приведено на рис. 4.46.
4.5.3.4. Термическая и радиационная стойкость
При нагревании поливинилиденфторида в инертной среде или
на воздухе выше 250 °С постепенно меняется окраска полимера и
происходит линейная потеря массы вследствие отщепления
фтористого водорода (рис. 4.47) [493—495]. Изменение окраски
обусловливается образованием сопряженных структур, таких, как
—CH=CF—CH=CF—. Даже при сильном изменении окраски
полимера снижения характеристической вязкости и ухудшения
физико-механических свойств полимера не происходит. Только
относительное удлинение несколько уменьшается вследствие образования
сетчатых структур.
Потеря массы после 60 мин нагревания в вакууме при 375 °С
равняется 74 % [496].
Максимальная температура длительной эксплуатации
поливинилиденфторида составляет 150 °С.
Интересно, что поливинилиденфторид
обладает высокой стойкостью к облучению
7-лучами и быстрыми электронами. Как
правило, поливинилидены при облучении де-
структируется по основной цепи, поскольку
в ней возникают высокие стерические
напряжения. В противоположность обычным
винилиденовым полимерам вследствие
небольшого объема фторзаместителей
зигзагообразная конформация не напряжена,
так что при облучении поливинилиден-
20-
О
300 SOQ 700
Рис. 4.47. Кривые деструкции поливинилиденфторида
на воздухе (/) и в среде азота B) (ДТ — 5 °С/мин)
[494].
118
Рис. 4.48. Зависимость прочности при
растяжении (а) и относительного
удлинения при разрыве (б) поливинилфтори-
да A) и поливинилиденфторида B) от
дозы облучения [438].
фторида вместо разрушения
цепей происходит их сшивание с
образованием сетчатых структур
[497].
Отношение GAeCTP : осш при
облучении 7-лучами в вакууме
составляет 0,18—0,21 [496].
Зависимость прочности при
растяжении и относительного
удлинения при разрыве
поливинилиденфторида от дозы облучения
показана на рис. 4.48.
60 Доза,Прад
4.5.4. ПЕРЕРАБОТКА ПОЛИВИНИЛИДЕНФТОРИДА
Поливинилиденфторид в отличие от других фторсодержащих
полимеров легче всего перерабатывается в изделия литьем под
давлением и экструзией. Переработку в термопластичном состоянии
можно проводить на обычных машинах для переработки
термопластов. Вследствие относительно высокой вязкости расплава при
переработке необходима реализация незначительных скоростей
деформации, что вполне возможно, поскольку поливинилиденфторид
стабилен при 250 °С в течение более 1 ч. Зависимость вязкости
расплава от скорости сдвига для двух промышленных марок
поливинилиденфторида (Кинар и КФ) приведена на рис. 4.49.
Переработка его ротационным литьем, вихревым напылением,
газопламенным напылением или в плазме невозможна из-за высокой
вязкости расплава.
4.5.4.1. Экструзия
Поливинилиденфторид перерабатывается методом экструзии
в трубы, профильно-погонажные изделия, пленки и оболочки
кабелей. Температура переработки в зависимости от вязкости расплава
Рис. 4.49. Зависимость вязкости расплава поливи-
ннлиденфторида при 220—310 °С от скорости
сдвига [492].
/О3
Ю
того или иного типа полимера колеблется от 210 до 290 С. Все
соприкасающиеся с расплавом детали в экструдере, а также головка
и шнек должны быть изготовлены из высококачественной или
хромированной инструментальной стали, отношение L/D для шнека
должно составлять от 15 : 1 до 20: 1. Экструдирование пленок из
поливинилиденфторида с молекулярной массой 82 000, согласно
Столингсу [478], осуществляется по режиму: зона 1 :245°С; зона 2:
250 °С; зона 3: 260 °С; зона 4: 260 °С; головка экструдера: 270 °С;
частота вращения шнека 40 об/мин. Температура переработки не
должна превышать 290°С, поскольку в противном случае под
действием сдвиговых напряжений происходит быстрое отщепление
фтористого водорода и коррозия основных частей экструдера.
4.5.4.2. Литье под давлением
Поливинилиденфторид может перерабатываться в изделия как
на червячных, так и на поршневых литьевых машинах. Цилиндр,
торпеда, шнек или поршень и литьевая головка должны быть
изготовлены из высококачественной стали или хромированы. В
зависимости от типа полимера рекомендуется поддерживать
температуру цилиндра от 215 до 260 °С и температуру литьевой формы
от 20 до 50 °С [478, 492, 499, 500].
Наибольшая прочность изделий получается при литье под
давлением на нижних температурных границах (рис. 4.50).
Кристаллический поливинилиденфторид имеет довольно
высокую усадку (рис. 4.51).
При толщине материала более 10 мм в изделии появляются
пустоты. В этом случае следует работать на верхней
температурной границе при высоких давлениях литья. Пустоты в
формованном изделии можно устранить также путем нагревания полимера
на 10 °С выше температуры плавления и медленного охлаждения,
при этом образуются микрокристаллы размером около 1 мкм [501].
Сушка поливинилиденфторида перед литьем под давлением не
производится.
4.5.4.3. Прессование и литьевое прессование
Методом прессования получают детали простой конфигурации,
а также заготовки в виде блоков, предназначенные для
дальнейшей механической обработки резанием. Предварительно
подогретый гранулят нагревают в прессе до 210 °С и прессуют под
давлением 70—700 кгс/см2. Выдержка материала в прессе при этой
температуре должна составлять минимум 5 мин. Прессованное изделие
можно извлечь из пресса при 100 °С [499].
При изготовлении относительно небольшой партии изделий или
деталей сложной конфигурации предпочтение следует отдать
литьевому прессованию, а не литью под давлением [499, 500].
Расплавленный поливинилиденфторид прессуют под давлением
70 кгс/см2 в пресс-форме, нагретой до 190—200 °С. После
охлаждения отпрессованное изделие может быть извлечено из пресс-формы
при температуре 105 °С.
120
U,°c
270
-5'50-
I70
///////////////////////У/////
I I
J L
250 260 27Q 280 Ь,С
200 350 500 650 600 950 WO Р,кгс/см'
Рнс. 4.50. Влияние температуры переработки на прочность литьевых изделий
из поливинплнденфторида [478]:
Температура, СС
Зона 1
Зона 2
Зона 3
260
246
218
274
260
232
288
260
232
Рис. 4.51. Влияние давления литья на усадку изделий из
поливинилиденфторида при различных температурах цилиндра [492].
4.5.4.4. Нанесение покрытий
Очень тонкие покрытия, толщиной от 0,1 до 0,5 мм, можно
наносить на поверхности деталей вихревым напылением или
ротационным способом [478, 492]. Металлические детали
предварительно нагревают до 260—400 °С, наносят поливинилиденфторид
и для создания однородной, непористой пленки снова расплавляют
при 400 СС.
Тонкие пленочные покрытия получают поливом из раствора
в ДМАА поливинилиденфторида типа ЕМ при комнатной
температуре [502]. Адгезионную прочность пленок из
поливинилиденфторида к металлическим, полимерным или деревянным поверхностям
можно повысить путем предварительной обработки пленки водным
раствором щелочи [503].
Покрытия из поливинилиденфторида наносят из раствора или
дисперсии [492, 500]. При использовании 25 %-ных растворов
поливинилиденфторида в ДМАА толщина наносимых пленок не
должна превышать 0,07 мм, так как в противном случае в пленке
остается растворитель (удаляемый при 230—250 °С).
Для нанесения покрытий из поливинилиденфторида из
дисперсий применяют пропиленкарбонат — метилгексилкетон или диме-
тилфталат — диизобутилкетон. При нанесении покрытий из
дисперсий растворитель удерживается в них в очень незначительной
степени. Это позволяет наносить пленки толщиной до 0,2 мм за одну
технологическую операцию.
121
4.5.4.5. Формование волокон
Поливинилиденфторид можно перерабатывать в волокна
методами мокрого формования и формования из расплава [478, 480,
504]. Мокрое формование проводят из 27 %-ного раствора поливи-
нилиденфторида в ДМАА при 55 °С. Осадительной ванной служит
смесь 55 % ДМАА и 45 % Н2О. Уже через 1 с после прохождения
через осадительную ванну происходит кристаллизация нити в ?-мо-
дификацию. Мокрым формованием получают волокна прочностью
455 кгс/см2, формованием из расплава — максимум 730 кгс/см2.
Нити из поливинилиденфторида со средней молекулярной массой
88000, полученные формованием из расплава, после шестикратной
вытяжки при 40 °С имеют прочность при растяжении 3,7 г/денье и
относительное удлинение при разрыве 18%-
4.5.5. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА ПОЛИВИНИЛИДЕНФТОРИДА
Поливинилиденфторид при тщательной его обработке
представляет собой почти прозрачный полимер с высоким поверхностным
блеском. Температурный интервал его применения колеблется от
—60 до 150 °С.
В табл. 4.14 приведены теплофизические характеристики двух
промышленных типов поливинилиденфторида.
По сравнению с другими термопластичными фторполимерами
поливинилиденфторид обладает большой механической прочностью,
более высокой износостойкостью и значительно меньшей
ползучестью. Это объясняется очень плотной упаковкой, обусловленной
Таблица 4.14. Теплофизические свойства
поливинилиденфторида [478, 492, 494, 500, 505]
Показатель
Температура, °С
плавления
стеклования
хрупкости
Теплостойкость по Вика, °С
Деформационная теплостойкость, °С
при 4,64 кгс/см2
при 18,6 кгс/см2
Плотность, г/см3
20
Показатель преломления nD
Удельная теплоемкость, кал/(г-°С)
Термический коэффициент линейного
расширения, 1/°С
Коэффициент теплопроводности при 25—100°С,
кал/(см-ч-°С)
Проницаемость для паров воды @,025 мм
пленки), г/24 ч-м2
Вязкость расплава, Пз
Усадка, см/см
Водопоглощение, %
Кинар
(«Pennwalb)
170
-46
-62
—
.150
91
1,76
1,42
0,33
1,5-Ю-6
1.0
1,0
104-105
0,02
0,04
КФ
(«Kureha»l
—
-20
165
150
98
1,76-1,77
1,42
0,33
1,2- 10"е
—
1О3-1О4
0,02-0,03
0,03
122
Таблица 4.1
5. Физико-механические свойства
поливинилиденфторида [478, 492, 494, 500, 505]
Показатель
Прочность при растяжении,
кгс/см2
при 25 °С
при 100°С
Относительное удлинение при
разрыве, %
при 25 °С
при 100°С
Предел текучести при
растяжении, кгс/см2
при 25 °С
при 100 °С
Прочность при сжатии при
25 °С, кгс/см2
Модуль упругости при
растяжении при 25 °С, кгс/см2
Модуль упругости при
изгибе при 25 °С, кгс/см2
Модуль упругости при
сжатии при 25 °С, кгс/см2
Ударная вязкость (по Изоду),
кгс- см/см2
Ударная вязкость образца с
надрезом (по Изоду), кгс-
•см/см2
Износостойкость (Taber—Cs—
15; 0,5 кгс), мг/1000 циклов
Твердость по Шору, шкала D
Твердость по Роквеллу
Коэффициент трения
скольжения по стали
Кии ар
(«Fennwall)*
492
352
300
400
389
176
703
8 437
14 060
8 437
162
16,2
17,6
80
—
0,14-0,17
Далвор
(«Diamond
Shamrock»)
436-583
—
70-100
—
464-485
—
—
15 300-17 000
—
—
—
—
—
—
—
КФ
(«Kureha»)
500-600
300-400
200—300
300-400
500-600
200-300
900—1 000
12 000—14 000
16 000—18 000
12 000-14 000
—
10-20
7,1 (нагрузка
1 кгс)
—
110
—
регулярной структурой полимера и близкими объемами боковых
атомов H и F. Вследствие того, что атомы фтора и водорода
имеют примерно одинаковые радиусы, между соседними цепями
могут образовываться псевдоводородные связи, т. е. происходит
физическое сшивание цепей [494]:
F H F H
I I I I
С—С—С—С
I I I I
F H F H
• * ¦ ¦
H F H F
I I I I
¦ • • —С—С—С—С— • • •
I I ! I
H F H F
4.5.5.1. Физико-механические свойства
Физико-механические свойства поливинилиденфторида типа
Кинар, Далвор и КФ приведены в табл. 4.15. Водопоглощение
123
о
Рис. 4.52. Кривые ползучести
поливинилиденфторида КФ под нагрузкой 140 A) и 240 B) кгс/см2
[492].
полимера настолько незначительно, что
оно не оказывает никакого влияния на
его физико-механические свойства. По-
ливинилиденфторид отличается высокой
стойкостью к упругим деформациям под нагрузкой (рис. 4.52).
Стойкость к старению поливинилиденфторида сохраняется в
течение 20 лет [506]. После двухлетнего старения в промышленной
атмосфере относительное удлинение при разрыве снижается на
14 %, а прочность при растяжении увеличивается на 4 %. Высокая
стойкость полнвинилнденфторида к естественному старению
объясняется тем, что поливинилиденфторид как в видимой, так и в
ультрафиолетовой области обладает очень незначительным
поглощением [494].
Окислительная коррозия полимера затрудняется вследствие
высокой энергии С — F-связей.
4.5.5.2. Электрические свойства
Вследствие симметричного расположения атомов фтора в по-
ливинилиденфториде можно было бы ожидать очень малые
значения тангенса угла диэлектрических потерь и диэлектрической
проницаемости. Однако эти показатели (табл. 4.16) необычайно
высоки: 8—10 и 0,04—0,05 соответственно. Это связано с
постоянным дипольным моментом, равным 2,1 Д, у плоской зигзагообраз-
Та блица 4.16. Электрические свойства поливинилидеифторида
[478, 492, 494, 500, 505]
Показатель
Диэлектрическая
проницаемость
при 60 Гц
при 103 Гц
при 106 Гц
при 109 Гц
Тангенс угла диэлектрических
потерь
при 60 Гц
при 103 Гц
при 106 Гц
при 109 Гц
Удельное объемное
электрическое сопротивление, Ом-см
Электрическая прочность
(кратковременно), В/см
Кинар
(«PennwalW
8,40
7,72
6,43
2,98
0,0497
0,0191
0,159
05П
2- 10м
260 @,32 см)
1280 @,02 см)
Дальвор
(«Diamond
Shamrock»)
8-9
0,02
0,22
4- 1014-1- 1015
КФ
(«Kureha»)
10,1
9,5
7.5
0,05
0,02
0,22
C-6) • 1014
4000 @,035 мм)
124
е-
15
10
о-700 «Гц
о-10 кГц
с-1кГц
о -то гц
о-ЮГц
-50
-tga-10'2
50
100
150 t,°C
о-100 кГц
о-10 КГЦ
с-1 кГц
о-100 ГЦ
9-Ю Гц-
D
Рис. 4.53. Температурная зависимость диэлектрической проницаемости (а) и
тангенса угла диэлектрических потерь (б) для поливинилиденфторида [507].
ной конформации. Угол между атомами фтора составляет менее
180° [478].
Температурные зависимости диэлектрической проницаемости
и тангенса угла диэлектрических потерь в интервале температур
от —50 до 50 °С приведены на рис. 4.53.
Изучению диэлектрической релаксации поливинилиденфторида
посвящены многочисленные исследования [472, 507—514].
Аналогично другим частично-кристаллическим полимерам в поливинили-
денфториде протекают релаксационные процессы, которые в
порядке уменьшения соответствующих температур обозначаются
буквами а, ? и у. а-Переход G0°С/110 Гц или 130°С/100 кГц)
ориентированного поливинилиденфторида зависит от толщины ла-
мелей и числа дефектных мест в кристаллических областях и связан
с процессами молекулярной подвижности в этих областях.
Термодинамические параметры процесса равны: АН — 24 ккал/моль
125
или AS = 26 ккал/моль [472, 507]. ?-Переход (—40°С/110 Гц;
0°С/100 кГц) приписывается микроброуновскому движению
основных цепей в аморфных областях. Активационные параметры
равны ЛЯ270 = 22 ккал/моль или AS270 = 47 ккал/моль. у-Релаксация
проявляется при температуре —70 °С и частоте 110 Гц и сводится
к локальным осциллирующим колебаниям застеклованных
основных цепей. Поведение поливинилиденфтормда в условиях
диэлектрической релаксации очень сильно зависит от типа
кристаллической модификации и степени ориентации образцов.
Исследования пироэлектрических и пьезоэлектрических свойств
поливпнилиденфторида были проведены Накамурой [515], Глас-
сом [516], Фукаотой [517], Луонго [518], Мак-Фи [519] и
Бергманом [520]. Эти свойства ПВДФ являются следствием
перестройки ступенчатой гранс-гош-конформации в плоскую
зигзагообразную конформацию, при которой диполи расположены под
прямым углом к поверхности пленки. Пьезоэлектрическое
поведение поливинилиденфторида обусловлено сегнетоэлектрическими
свойствами кристаллической ?-модификации.
4.5.5.3. Химическая стойкость
Поливинилиденфторид стоек к большинству агрессивных
химических сред и огранических растворителей. Деструкция
макромолекул протекает лишь в первичных аминах при повышенных
температурах. Поливинилиденфторид сульфируется дымящей
серной кислотой. В ряде полярных растворителей происходит
размягчение полимера вследствие его набухания.
После 30 сут выдержки поливинилиденфторида в агрессивных
химических средах при 50 °С прочность при растяжении
уменьшается [494]: в 95 %-ной H2SO4 на 1%, в дымящей HNO3 на
13%, в ледяной уксусной кислоте на 25 %, в 35 %-ной НС1 на
1 %, в диэтиламине на 24 %, в сухом хлоре при 25 °С на 3%, в
броме при 25 °С на 7% ив 90 %-ной Н2О2 при 25 °С на 24%,
в 50 %-ном NaOH и NH4OH не изменяется. После 30 сут
выдержки поливинилиденфторида в ацетофеноне, пиридине или
амилацетате при 50 °С относительное удлинение при разрыве не
изменяется [478]. После выдержки ПВДФ в 95 %-ной H2SO4 при 50 °С
в течение 30 сут происходит уменьшение относительного
удлинения при разрыве на 10%, в циклогексаноне, наоборот,
относительное удлинение при разрыве увеличивается на 10 %.
Поливинилиденфторид при 100 °С стоек к хлористому
водороду и диоксиду серы. Образования трещин вследствие остаточных
напряжений в органических растворителях не происходит [492].
4.5.6. ПРИМЕНЕНИЕ ПОЛИВИНИЛИДЕНФТОРИДА
Основными областями применения поливинилиденфторида
являются электротехническая промышленность, химическое
машиностроение, строительство и упаковка.
Поливинилиденфторид представляет собой трудновоспламеняю-
щийся полимер, он затухает вне пламени.
126
4.5.6.1. Электротехника и электроника
Поливинилиденфторид представляет собой
высококачественный изоляционный материал, предназначенный для работы в
жестких условиях, при которых требуется высокая термостойкость,
повышенные изоляционные характеристики и высокое
сопротивление пробою. При диаметре провода 0,36 мм и толщине
изоляционного слоя 0,15 мм удельное объемное электрическое
сопротивление изоляции после выдержки в воде в течение 1 ч при
20 °С составляет 3,5-1015, а при 80 °С — 1,3- 10м Ом-см.
Соответствующие значения при диаметре провода 0,6 мм и толщине
изоляционного слоя 0,3 мм равны 7-Ю14 или 1,3-1012 Ом-см [492].
Поливинилиденфторид обладает отличным сопротивлением
пробою. При радиусе заострения 0,08 мм пробой пленки толщиной
0,20 мм (тип КФ) при нагрузке 8 кгс происходит через 24 ч и при
нагрузке 9 кгс через 1 ч. В аналогичных условиях пробой
политетрафторэтилена происходит под нагрузкой 3 кгс за 1 ч и
полиамида 6,10 при нагрузке 6 кгс за 1 ч.
Изоляция проводов, изготовленная из поливинилиденфтори-
да, хорошо зарекомендовала себя при работе в условиях
космических полетов. Однако высокое значение тангенса угла
диэлектрических потерь ограничивает его применение в качестве
высокочастотной изоляции.
Усадочные шланги из поливинилиденфторида превосходят
другие усадочные материалы по прочности на истирание. Радиацион-
но-химически сшитый поливинилиденфторид пригоден для
изготовления кабельных соединительных муфт [521].
В электронике поливинилиденфторид используется как
перспективный электретный материал. Получение электретов
осуществляется путем двухосной вытяжки пленок из
поливинилиденфторида при 50—170 °С на 220—400 %• Такой материал, переведенный
в ?-модификацию, нагревается под нагрузкой и для
образования электрета подвергается действию поля напряжением 320—
1000 кВ/см [522]. В качестве электретных материалов пригодны
также сополимеры винилиденфторида с тетрафторэтиленом, ви-
нилфторидом или трифторхлорэтиленом [523]. Стабильность
поверхностного заряда, равного 10—20 эл.-ст.ед./сма,
экстраполировалась до 10 лет. Электреты из поливинилиденфторида могут
применяться вплоть до 130 °С, а также в присутствии масел и
растворителей [492, 524].
Поливинилиденфторид обладает свойствами сегнетоэлектри-
ческих кристаллов и может использоваться в качестве
пироэлектрических детекторов для электромагнитного излучения [519].
Чувствительность выпускаемого в промышленности
поливинилиденфторида для длины волны 10,6 мкм составляет 17 В/Вт при
100 Гц [516].
4.5.6.2. Химическое машиностроение
В химической промышленности поливинилиденфторид
применяется для изготовления трубопроводов, деталей насосов, уплот-
127
нителей, вентилей и для облицовки резервуаров, автоклавов и
скрубберов.
Поливинилиденфторид может применяться для изготовления
труб, предназначенных для транспортирования хлора, диоксида
хлора, 60 %-ной серной кислоты и 50 %-ного раствора едкого
натра, эксплуатируемых при температурах до 150 °С. Не было
обнаружено никаких изменений в насосах и мешалках для 40 %-ного
раствора хлорида железа (III) при 120 °С и в диафрагмах
насосов для 98 %-ного влажного хлористого водорода после 6 мес.
работы. Краны из поливинилиденфторида для 21 %-ной соляной
кислоты могут применяться до 100°С, а для 30 %-ного раствора
хлората натрия — до 40°С [505]. Для компрессоров,
предназначенных для перекачки хлоргексафторида, поливинилиденфторид
является единственным пригодным материалом.
Сульфированием поливинилиденфторида дымящей серной
кислотой с предварительным набуханием можно изготавливать
ионообменные мембраны с высокой гибкостью и низким электрическим
сопротивлением [525]. Микропористые мембраны из
поливинилиденфторида [526] и мембраны, покрытые силиконовым каучуком
[527], применяют для очистки воды.
Обработкой поливинилиденфторида аммиаком, аминами или
щелочными металлами в автоклавах при 300—500 °С получают
мембраны с углеродной основой, которые пригодны для селективного
выделения пентана и циклогексана из углеводородных смесей [528].
4.5.6.3. Другие области применения
Стальные или алюминиевые листы, покрытые ПВДФ,
применяют в строительстве для облицовки фасадов. Лакокрасочные
покрытия на основе дисперсий ПВДФ служат для защиты древесины
или кирп-ичной кладки от атмосферной коррозии. Особо важными
достоинствами ПВДФ являются высокая стойкость к царапанию и
прочность на истирание, а также высокая стойкость к старению.
ПВДФ при его применении для наружной защиты устойчив в
течение 20—30 лет.
Упаковочные пленки на основе ПВДФ можно использовать
в пищевой промышленности. Они поддаются горячему тиснению и
свариванию при 185°С. Кроме того, ПВДФ применяется при
изготовлении стерилизуемой упаковки в фармацевтической
промышленности и медицинского инструмента [478].
4.5.7. ПОЛИМЕРНЫЕ КОМПОЗИЦИИ
НА ОСНОВЕ ПОЛИВИНИЛИДЕНФТОРИДА И МЕТАКРИЛАТОВ
4.5.7.1. Совместимость
Полиметилметакрилат (ПММА) и полиэтилметакрилат (ПЭМА)
при переработке методами, используемыми для термопластов,
образуют с ПВДФ совместимые смеси [529]. Гомогенные смеси
получают путем смешения ПВДФ с метилметакрилатом при нагревании
до 40 °С [530].
128
пр,%
-rry
/и
30
40
'50
\
t
o
\
0,4 0,8 1,2
<р, ой. доли
О 0,4 0,3 1,2
ipnB49. ой.дош
Рис. 4.54. Зависимость температуры стеклования композиции поливинилиденфто-
рид — полиметакрилат от ее состава [529]:
с — композиция с ПММА; б — композиция с ПЭМА.
Рис. 4.55. Зависимость прозрачности (Пр) композиции поливинилиденфторид —
полиметилметакрилат от ее состава [529].
Совместимость подтверждается прозрачностью полимерных
композиций в широком интервале составов. Такие композиции
имеют одну температуру стеклования, которая лежит между
значениями температур стеклования соответствующих гомополимеров.
Температура стеклования полимерной композиции линейно зависит от
объемной доли компонентов смеси (рис. 4.54).
Кристаллический ПВДФ хорошо растворяется в расплаве
аморфного ПММА. Раствор аморфного ПВДФ оказывает
пластифицирующее действие на ПММА. Однако при содержании ПВДФ
выше 33 % происходит кристаллизация ПВДФ компонента. При
таком соотношении компонентов наступает резкое ухудшение
прозрачности полимерной композиции (рис. 4.55).
Композиции на основе ПВДФ и ПЭМА представляют собой
аморфные системы вплоть до эквимольного соотношения
компонентов смеси. Композиции с высшими полиалкилметакрнлатами
негомогенны.
4.5.7.2. Свойства полимерных композиций на основе
поливинилиденфторида и полиметилметакрилата
Прочность и модуль упругости при растяжении уменьшаются
с увеличением содержания ПВДФ в композиции (рис. 4.5G).
Компоненты смеси в композиции при с .^.^-з^гс/см2
одинаковых условиях облучения УФ-луча- р>
ми обладают такими же свойствами, как и боо
гомополимеры. Поскольку ПВДФ в компо-
wo-
Рис. 4.56. Зависимость физико-механических свойств ?00
композиции поливинилиденфторид — полиметилметакри-
от ее состава [529]:
129
— прочность при растяжении; 2 — модуль упругости при
растяжении.
5 Зак. 18
Зиции оказывает пластифицирующее действие и вязкость расплава
композиции значительно не повышается при увеличении
молекулярной массы ПММА, можно путем удвоения его молекулярной
массы получить композиции, стойкость которых к действию УФ-
лучей будет в 2 раза выше.
4.5.8. СОПОЛИМЕРЫ ВИНИЛИДЕНФТОРИДА
Из сополимеров винилиденфторида техническое значение имеют
пока сополимеры с гексафторпропиленом и хлортрифторэтиленом.
Сополимеры винилиденфторида с гексафторпропиленом
изготавливаются фирмой «Дюпон» под названием «Витон А» или
«Витон В» и фирмой «3 M Со» — под названием «Флуорел».
Сополимеры хлортрифторэтилена с винилиденфторидом выпускаются
под названиями «Эластомер 3700» и «Кел Ф» («Kellogg»).
Оба сополимера представляют собой термостойкие эластомеры,
обладающие отличной химической стойкостью.
4.5.8.1. Сополимеры винилиденфторида
с гексафторпропиленом
Сополимеризация винилиденфторида с гексафторпропиленом
проводится в эмульсии в присутствии
окислительно-восстановительных систем [531—535]. Получаемый полимер представляет собой
белый порошок со средней молекулярной массой около 150 000. При
его вулканизации под действием кислотных акцепторов происходит
отщепление фтористого водорода и образование (за счет радикалов
или двойных связей) полимерной сетки.
Для получения типовой рецептуры 100 масс. ч. сополимера,
20 масс. ч. оксида магния, 15 масс. ч. технического углерода и
1 масс. ч. гексаметнленднаминкарбамата перемешивают в течение
1 ч при 150 °С и выдерживают 24 ч при 200 °С [536—539].
Вулканизацию сополимеров с ацильными или сложноэфирными
концевыми группами проводит также хлорангидридами карбоновых
кислот или пентаэритритом [540].
Термостойкость сополимеров винилиденфторида с
гексафторпропиленом составляет около 200°С, и кратковременно их можно
использовать до 300 °С. Потеря массы вплоть до 320 °С составляет
лишь 1 %. Энергия активации процесса термической деструкции
равна 23 ккал/моль [541]. Интенсивное разложение сополимеров
начинается при температурах выше 340 °С.
Ниже приведены свойства сополимера Флюорел в невулканизо-
ванном и вулканизованном состоянии [531]:
Исходный сополимер
Содержание фтора, % 60
Плотность, г/см3 1.85
Твердость по Шору, шкала А 40
Вязкость по Муни ML A+4) 100°С 135
130
Вулканизованный сополимер
Прочность при растяжении,' кгс/см2 161
Прочность при 100%-ним удлинении, кгс/см2 22
Твердость по Шорз% шкала А 320
Износостойкость (Taber H, 22; нагрузка 1 кгс), мг/1000 циклов 31
Диэлектрическая проницаемость при 100 кГц 11,4
Тангенс угла диэлектрических потерь при 100 кГц 0,0125
Электрическая прочность, кВ/мм 25
Удельное объемное электрическое сопротивление, Ом-см
при 50%-ной отн. влажности 2 • 1013
при 90%-ной отн. влажности 1,5 • 1013
Вулканизат представляет собой негорючий продукт, устойчивый
к тошшвам, растворителям, смазкам и гидравлическим маслам.
Он обладает отличной стойкостью при старении и нечувствителен
к озону [542—544].
Сополимеры винилиденфторида с гексафторпропиленом
применяются для изготовления уплотнителей, манжет, шлангов,
штампованных деталей, для пропитки защитной одежды и в качестве
фрикционного материала для дисковых тормозов [545, 546]. Детали
из сополимера марки «Флуорел» использовали при создании
космического корабля «Аполлон» [547].
4.5.8.2. Сополимеры трифторхлорэтилена
с винилиденфторидом
Сополимеры трифторхлорэтилена с винилиденфторидом
получаются также эмульсионной полимеризацией [548—550].
Вулканизация сополимера проводится в присутствии гексаметилендиамин-
карбамата, оксида цинка и фосфита свинца при 180°С под
давлением с последующей термической обработкой при 180°С [551.
553]. Эластомер стабилен при температуре 200 °С. При 250—300 °С
энергия активации дегидрохлорирования и дегидрофторирования
в вакууме составляет примерно 29 и 34 ккал/моль. Интенсивно
сополимер разлагается лишь при температуре около 300 °С [552].
Сополимеры трифторхлорэтилена с винилиденфторидом имеют
относительное удлинение при разрыве 300—600%. Они устойчивы
к действию таких растворителей, как алифатические углеводороды,
хлорпроизводные углеводородов, спирты, а также гидравлические
жидкости и смазочные масла. Во фторуглеводородах, эфирах ди-
карбоновых и фосфорных кислот сополимеры набухают. Эластомер
представляет собой негорючий продукт, устойчивый к действию
озона.
Применение сополимера ограничивается его высокой
ползучестью.
Промышленные продукты Кел Ф 5500 или Эластомер 3700
содержат 50 % и 70 % винилиденфторида соответственно.
Литература к главе 4
1. Пат. США 2380385 10.7.45 Shell Development.
2. Пат. США 2581228 A950) Phillips Petroleum.
3. Пат. США 2606940 12. 8. 52, Phillips Petroleum.
б*
4. Пат, СССР 272300 3. 6. 70.
5. К. Hochmuth, Chem. Engng. Progr. 44 A948), 421—430.
6. E. Herpers, Gaswarme internat. 16 A967), 289.
7. M. Таниевский, Нефтехимия, 6 A966), 1, 63—66.
8. Р. Spitz, Oil Gas J. 66 A968), 108—111.
9. F. Schlimper. Chem. Technol. 21 A969) 9, 543.
10. Пат. США 3235741 15. 2. 66 Phillips Petroleum.
11. R. Meier, J. ehem. Soc. A950), 3656—3671.
12. G. Natta, Chim. e Ind. 38 A956), 124.
13. G. Natta, Makromolekulare Chem. 21 A956), 240—244.
Kunststoffe A971) 7, 347.
14. A. Zambielli, Chim. Ind. (Milano) 44 A962), 529.
15. Акцент, заявка 1910482 17. 9. 70. Chem. Werke Huls.
16. Пат. США 2964510 9.4.56 Esso.
17. Англ. пат. 827464 3. 2. 60 Destillers Co.
18. Фр. пат. 1596024 24.7.70 Chem. Werke Huls.
19. Пат ФРГ 1495350 11.3.71 Chem. Werke Huls.
20. Пат. США 2936302 10. 5. 60 Petro Тех.
21. Пат. США 3197452 27.7.65 Montecatini.
22. Пат. США 3047551 31. 7. 62 Sun Oil.
23. Пат. ФРГ 1058736 4.6.59 Hochst.
24. Ит. пат. 564233 12.6.57 Montecatini.
25. Англ. пат. 851583 19. 10.60 Bataafse Petroleum.
26. Пат. США 3190866 22. 6.65 Petro Тех.
27. Пат. США 3072629 8. I. 63 Eastman Kodak.
28. Пат. США 3202644 24. 8.65 Cabot Corp.
29. Акцепт, заявка 1814643 9.7.70 Ube Ind.
30. Опубл. заявка 1948623 1.4.71 Huls.
31. Акцепт, заявка Японии 7214450 28.4.72 Jap Bur. Ind. Technol.
32. G. Natta, J. Polymer Sei. 51 A961), 387—398.
33. J. Boor, J. Polymer Sei. С I A963), 237.
34. Ит. пат. 554013 l. 5. 57 Montecatini.
35. Белы. пат. 626642 28.6. 63 Montecatini.
36. G. Natta, Chim. Ind. (Milano) 43 A961), 871—874.
37. Аигл. пат. 909081 24. 10.62 Montecatini.
38. Акцепт, заявка 1910482 17.9.70 Huls.
39. T. Otsu, J. Polymer Sei. A 14 A966), 1579—1593-
40. J. Rakus, J. Polymer Sei. В 4 A966), 467—468.
41. Фр. пат. 1534598 26. 7. 68 Huls.
42. Фр. пат. 146240 24. 2. 65 Huls.
43. Англ. пат. 1153781 29. 5.69 Petro Тех.
44. A. Nishioka, Chem. High Polymers (Japan) 19 A962) 211, 667—671.
45. E. Beati, Makromolekulare Chem. 61 A963), 104—115.
46. G. Natta, Chim. Ind. (Milano) 37 A955), 927—932.
47.'V. Holland, J. Polymer Sei. В 2 A964), 519—521.
48. V. Holland, J. Appl. Phys. 35 A964), 3241—3246.
49. H. Holden, J. Polymer Sei. С 6 A964), 209—211.
50. F. Danusso, Makromolekulare Chem. 61 A963), 139—156
51. J. Boor, J. Polymer Sei. AI A963), 59—84.
52. F. Danusso, Makromolekulare Chem. 88 A965), 149—158.
53. J. Plemikowski, Kunststoffe 55 A965), 431—437.
54. J. Fpglia, Appl. Polymer Symp. 11 A969), 1—18.
55. R. Hughes, J. Polymer Sei. С 6 A964), 43—54.
56. F. Herre, «Wandel in der chemischen Technik», Munchen 1963.
57. G. Natta. Nuove Cimiento A0) 15 A957). 59—65
58. C. Lindegren, Polymer Engin. Sei. 10 A970) 3, 163—169,
59. C. Champitt, J. Polymer Sei. С 6 A964), 43—54.
60. С. Geacintov, J. Polymer Sei. С 6 A964), 197—208,
61. Англ. пат. 1084953 31.7.64 ICI.
62. С. Rohn. Mod. Plastics A971) 3.
63. W. Krigbaum, J. phys. Chem. 65 A961), 1984.
132
64. S Stivala, J. appl. Polymer Sei. 7 A963), 97—102.
65 W. Ring, Makromolekulare Chem. 103 A967), 83—90.
66 J Cazes, J. chem. Educ. 47 A970). A 505—514.
67. R. Endo, Bu!!, chem. Soc. (Japan) 37 A964), 950.
68. S. Pregun, Papers presentent the SPE 28th Antec, New York Ш70.
69. .Г. Zimmermann, Plastverarbeiter 20 A969), 4, 245—252.
70. Modem Plastics 44 A967) 11, 54.
71. Акцепт, заявка 1258530 A968) Huls.
72. Акцепт, заявка 1287240 16. 1.69 Huls.
73. Фр. пат. 1600531 4.9.70 Huls.
74. Опубл. заявка 2005966 26.8.71 BASF.
75. Опубл. заявка I778I03 29. 7. 71 BASF.
76. Англ. пат. 1252522 3. И. 71 BASF.
77. Акцепт, заявка Японии 7138689 15. 11.71 Kuraray.
78. Фр. пат. 1534598 26.7.68 Huls.
79. Т. Reed, Plast, Technol. 12 A966), 2, 38.
80. Пат. США 3433742 18. 3. 69 Petro Тех.
81. Опубл. заявка 2063670 1.7.71 Mobil Oil.
82. S. Hoshino, J. Polymer Sei. 58 A962), 185.
83. Mod. Plastics 43 A965), 94.
84. G. Binder, Werkstoffe und Korrosion 19 A968), 120—236.
85. G. Bornmann, Arch. Toxikologie (Berlin) 23 A968), 240—244.
86. Kunststoff — Berater 17 A972), 3, 156.
87. Акцепт, заявка 2012913 8. 10. 70 Mobil Oil.
88. Kunststoffe 61 A971), 467.
89. D. Bollmann, Plastverarbeiter 20 A969) 7, 485—493.
90. Опубл. заявка 2043864 9. 3. 72 Huls.
91. Ит. лат. 582004 15.9.58 Montecatini.
92. G. Natta, Chem. Ind. (Milano) 37 A955), 927.
93. Ит. пат. 564806 2. 7. 57 Montecatini.
94. Акцепт, заявка Японии 7138563 13.11.71 Furukawa Chem. Ind.
95. Пат. США 3631019 28. 12.71 Standard Oil.
96. Опубл. заявка 2058015 3.6.71 Mobil Oil.
97. A. Turner, Polymer 7 A966), 23—30.
98. H. Sobue, J. chem. Soc. (Japan), Ind. chem. Sect. 68 A965), 700—702.
99. J. Brown, J. Polymer Sei. A 1 10 A972) 7, 1997—2004.
100. G. Natta, J. Polymer Sei. 34 A959), 685—698.
101. Пат. США 3141862 21. 7. 64 Esso Research.
102. G. Natta, Chim. Ind. (Milano) 41 A959), 764.
103. L. Bogetich, J. Polymer Sei. 61 A962), 3—8.
104. F. Edgecombe, Nature 198 A963), 1085.
105. A. Ketlev, J. Polymer Sei. В 1 A963), 121 — 124.
106. J. Mark, J. Amer. Chem. Soc. 87 A965), 1423—1429.
107. K. Hukada, J. chem Soc. (Japan), Ind. chem. Sect. 67 A964) 12, 2163—2167.
108. R. Koch, Ber. Reichsamt Wirtschaftsausbau Pruf-Nr. 43 (P. В. 52003) 93.
109. Англ. пат. 1233020 26. 5. 71 ICI.
ПО. Англ. лат. 1225559 17.3.71 ICI.
111. J. Henne, J. Amer. chem. Soc. 66 A944), 1650—1658.
112. Пат. США 2986588 30. 5. 61 California Research.
113. A. Shaw, J. org. Chem. 30 A965), 3286—3289.
114. J. Wilkes, J. org. Chem. 32 A967), 3231—3233.
115. Англ. пат. 914144 28. 12.61 British Petroleum.
116. Англ. пат. 933253 8.8.63 British Petroleum.
117. Англ. пат. 945967 8. 1.64 British Petroleum.
118. Англ. пат. 1003576 8.9.65 British Petroleum.
119. Англ. лат. 1005609 22.9.65 British Petroleum.
120. Англ. лат. 1066113 19.8.67 British Petroleum.
121. Фр. пат. 1356267 7.4.64 Ethyl Corp.
122. Фр. пат. 1377787 6. 11 64 British Petroleum.
123. Фр. лат. 1364460 19.6.64 Vereinigte Glanzstoff-Fabriken,
124. Фр. лат. 1357252 3.4.64 British Petroleum.
133
125. Пат. США 3175020 23-3.65 California Research.
126. Нидерл. пат. 6509224 9.3.66 British Petroleum.
127. Нидерл. пат. 6402598 14.9.64 Goodyear.
128. Белы. пат. 613324 15.2.62 Goodyear.
129. J. Hambling, Rubber Plast. Age 49 A968), 3, 224—227.
130. Англ. пат. 934449 21.8.63 British Petroleum.
131. Англ. пат. 934450 21.8.63 British Petroleum.
132. Англ. пат. 991705 A965) British Petroleum.
133. Англ. пат. 1002894 A965) British Petroleum.
134. Англ. пат. 1007325 A965) British Petroleum.
135. J. Goodrich, J. Polymer Sei. В 2 A964) 4, 353—357.
136. H. Raine, Appl. Polymer Symp. 11 A969), 39—45.
137. Y. Atarashi, Chem. Higt Polymer (Japan) 21 A964) 228, 264—269.
138. Kunststoffe 57 A967) 9, 731.
139. J. di Pietro, Polymer Engin. Sei. 7 A967) 1, 35—37.
140. Пат. СССР 328098 2. 2. 72.
141. У. Хо, Изв. АН СССР, Сер. Хим. A970), 6, 1391—1395.
142. Акцепт, заявка Японии 7120818 11.6.71 Toray Ind.
143. P. Tait, Kinet. Mech. Polyreactions, Int. Symp. Macromol. Chem. Prepr.
A969) 2, 259—265.
144. J. Me Kenzie, Polymer 13 A972) 11, 510—514.
145. T. Campbell, J. appl. Polymer Sei. l A959), 78—83.
146. Пат. США 2721189 18. 10. 55 Du Pont.
147. Акцепт, заявка Японии 7105155 8.2.71 Toyo Rayon.
148. N. Phung, Bull. Soc. Chim. France A967) 10, 3618—3626.
149. Англ. пат. 1027758 27.5.66 Inst. Franc, du Petrole.
150. F. Karash, Polymer 8 A967) 10, 547—560.
151. G. Natta, Makromolekulare Chem. 35 A960), 93.
152. F. Frank, Phil. Mag. 4 A959), 200.
153. F. Khoury, J. Res. Nat. Bur. Stand. A 76 A972), 3, 225—252.
154. J. Griffith, J. Polymer Sei. 44 (I960), 389—396.
155. A. Hoffmann, Papers presented at the IUPAC Symp. Macromol. Chem., Paris
1963.
156. L. Reginato, Makromolekulare Chem. 132 A970), 113—134.
157. У. Хо, Пласт, массы A972) 3, 57—59.
158. D. Goodhead, J. Polymer Sei. А2 A971), 6, 999—1024.
159. N. Kusomoto, Rep. Progr. Polymer Phys. (Japan) 8 A965) 315.
160. M. Charles, J. Polymer Sei. А 2 10 A972) 8, 1609—1610.
161. D. Pinkerton, Aust. J. Chem. 24 A971) 1, 183—185.
162. А. Буният-заде, Пласт, массы A972) 4, 33—35.
163. «TPX Methylpentene Polymers», Firmenschrift, ICI.
164. M. Day, Plastics & Polymers A968) 4, 101—108.
165. T. Campbell, J. appl. Polymer Sei. 5 A961) 4, 184—190.
166. Patentanm. 1254933 23. 11.67 Dynamit Nobel.
167. Акцепт, заявка Японии 7117581 15.5.71 Today Ind.
168. Акцепт, заявка Японии 7214712 2.5.72 Furukawa Electric.
169. R. Isaacson, J. appl. Polymer Sei. 8 A964), 2789—2799.
170. Англ. пат. 1085914 4. 10.67 ICI.
171. Y. Hase, Polymer J. 2 A971) 5, 560—580.
172. G. Bryant, Textile Res. J. 37 A967) 7, 552—563.
173. K. Euler, Kunststoffe — Plastics A971) 2, 51—53.
174. Plastnyheterna A971) 2, 5.
175. Англ. пат. 1249322 13. 10.71 ICI.
176. J. Anderson, J. Polymer Sei. 56 A962). 391—402.
177. О. Киссии, Eur. Polymer J. 8 A972), 3, 487—499.
178. Yuji Atarashi, Kogyo Kagaku Zasshi 68 A969). 2487.
179. J. Kubik, Petrochemia 11 A971) 6, 151—156.
180. J. Wilson, J. macromol. Sei. A 6 A972) 2, 391—402.
181. J. Wilson, J. Macromol. Sei. A 5 A971) 4, 777—792.
182. Р. Левина, Науч. труды Моск. ун-та, 7 A950), 241.
134
183 Р. Левина, ЖОХ, 4 A934), 9, 1250.
184 А. Платэ, ЖОХ, 20 A950), 472.
185. Р. Левина, ЖОХ, 7 A937), 353.
186. Р. Розанов ЖОХ, A929) 10, 2309.
187 А Перельман, Пласт, мяссь* A964) 8, 3—6.
188 H Staudinger, Бег. 62 A929), 2406—2411.
189 Пат СССР 308994 9. 7. 71,
190, П, Шорыгин, ЖОХ, 9 A939), 945.
191 Т. Campbell, J. appl, Polymer Sei. 1 A959), 73—83.
192. С Overberger, J. Polymer Sei. 44 A960), 483—491,
193. А. Топчиев, J. Polymer Sei, 52 A961) 157, 199—204.
194. Фр, пат. 1581067 12,9.69. В Америк.
195. Англ. пат,1212558 18. 11.70. В Америк.
196. J, Kennedy, J. Polymer Sei. A 2 A964), 5029—5040.
197. G. Parravano, J. Polymer Sei, 3 A965) 12, 4029—4046.
198. Б. Крепцель, Km, Mech. Polyreactions, Int. Symp. macromol, Chem., Preprints
A969) 2, 281—284.
199. A. Ketly, J, Polymer Sei. A 2 A964), 4461—4474.
200. К. Dunham, J. Polymer Sei. A 1 A963), 751—762.
201. В. Клейнер, Высокомол. соед,, Б13 A971), 1, 63—66.
202. Пат, США 2958688 1. 11,60 Eastman Kodak.
203. Пат. США 2969345 24. 1.61 Eastman Kodak.
204. Пат. США 2969346 24. 1. 61 Eastman Kodak.
205. Пат. США 2973348 28. 2. 61 Eastman Kodak.
206. Пат. США 30263311 20.2.62 Eastman Kodak.
207. Пат. США 2972607 21.2.61 Eastman Kodak.
208. В. Клейнер, Пласт, массы A967) 4, 3—7.
209. А. Топчиев, J, Polymer Sei. 53 A961), 195—202,
210. А. Топчиев, ДАН СССР, 130 A960) 2, 344—345.
211. А. Топчиев, Нефтехимия, 3 A963), 74.
212. Н. Прищепа, Усп. хим., 35 A966), вып. 11, 1986—1998.
213. Н. Staudinger, Ber. 62 A929), 263.
214. A. Steinhofer, DBP 1131885 A962).
215. G. Taylor, J. Polymer Sei. В 6 A968), 699—705.
216. R, Kelchner, J. Polymer Sei. А 2 8 A970), 799—806.
217. A. Abe, J. Polymer Sei. В 7 A969), 427—435,
218. G. Natta, Makromolekulare Chem. 33 A959), 247—248.
219. H. Noether, J. Polymer Sei. С 16 A967), 725—753.
220. В, Каргин, Усп. хим., 35 A966), Вып. 6, 1006—1029.
221. В. Клейиер. Пласт, массы A971) 11, 48—50.
222. Е. Зинин, Пласт, массы A970) 1, 32—34.
223. С. Барсамян, Пласт, массы A968) 3, 13—14.
224. Е. Зинин, Труды Моск. хим,-технолог, института им. Д. И, Менделеева
A969), 61, 233—236.
225. S, Jamison, Text, Res, J, 40 A970) 10, 955—966.
226. Пат. СССР 318643 28. 10, 71, M, Зверев.
227. В, Талыков, Высокомол. соед., AI4 A972) 2, 432—439.
228. R. Adams, J, Amer. chem. Soc. 48 A926), 1901.
229. В. Егорова, ЖОХ, 43 A910), 1116,
230. Я. Паушкин, Нефтехимия, 1 A961), 1, 60.
231. В. Егорова, Ж. Русск. физ.-хим. р_бше?таа, 43 A910), 116.
232. Ю. Киссин, Высокомол. соед., А14 A972) 1, 3—8.
233. F. Swarts, Bull. Acad. roy. Belg. A901), 383.
234. F. Swarts, Bull. Acad. roy. Belg. A909), 728.
235. F. Swarts, Bull. Soc. chim. France 4 A919) 25, 145.
236. A. Grosse, J. Amer, chem. Soc. 64 A942), 2289.
237. A. Newkirk, J. Amer, chem. Soc. 68 A946), 2467,
238. Пат. США 2626963 27. 1, 53 Union Carbide.
239. Пат. США 2899000 23. 6. 59 Du Pont.
240. Пат. США 2716142 23. 8. 55 Du Pont.
241. Пат. США 2634300 7. 4. 53 Phillips Petroleum.
135
242. Нидерл. пат. 6405140 11. 11.64 Diamond Alkali.
243. Ит. пат. 846005 1.7.69 ittontecatini.
244. Фр. пат. 1410317 10.9.65 Soc. Electro-Chim.
245. Пат. СССР 174622 7.9.65.
246. Фр. пат. 1325750 3. 5. 63 St. Gobain.
247. Пат. США 2716143 23.8.55 Du Pont.
248. Пат. США 2425991 19.8.47 Du Pont.
249. D. Sianesi, Chim. e Ind. A968), 619.
250. Акцепт, заявка 1210799 17.2.66 Continental Oii.
251. Пат. США 2695320 23. 11.54 Monsanto.
252. Акцепт, заявка Японии 7211729 12.4.72 Electro Chem. Ind.
253. Белы. пат. 633525 12. 12. 63 Montecatini.
254. Фр. пат. 1399051 14.5.65 Daikin Kogyo.
255. G. Pritchard, J. phys. Chem. 68 A964) 7, 1786—1792.
256. Англ. пат. 921254 20. 3. 63 Union Carbide.
257. Акцепт, заявка 1175225 6. 8. 64 Union Carbide.
258. Акцепт, заявка Японии 7121607 18.6.71 Electro Chem. Ind.
259. Акцепт, заявка Японии 7211728 12. 4.72 Electro Chem. Ind.
260. Пат. США 3621067 16. 11.71 Atlantic Richfield.
261. Англ. пат. 1020716 23.2.66 Montecatini.
262. Акцепт, заявка Японии 7211726 12.4.72 Electro Chem Ind.
263. Пат. США 2678884 30. 3. 54 С. Thomas.
264. Фр. пат. 1324408 19.4.63 St. Gobain.
265. J. Krests, Chem. Prumysl 16 A966), 94—102.
266. Пат. США 2889378 2. 6. 59 Allied Chem.
267. H. Starkweather, J. Amer. chem. Soc. 56 A934), 1870.
268. Пат. США 2419008 15.4.47 Du Pont.
269. Пат. США 2461523 15. 2. 49 Du Pont.
270. Пат. США 2419009 15. 4. 47 Du Pont.
271. Пат. США 2419010 15.4.47 Du Pont.
272. G. Kalb, J. appl. Polumer Sei. 4 A960), 10, 55—61.
273. Акцепт, заявка Японии 7108869 5.3.71 Kuraray.
274. К. Kresta, Chem. Prumysl 16 A966) 5, 282—286.
275. А. Гуфаров, Узбекский химич. ж. 16 A972) 3, 59—61.
276. Пат. США 2792423 17. 5. 57 Du Pont.
277. Пат. США 3069404 18. 12.62 Du Pont.
278. Англ. пат. 905879 12.9.62 Du Pont.
279. Акцепт, заявка Японии 7109347 9,3.71 Electro Chem Ind.
280. Белы. пат. 646722 19. 10.64 Montecatini.
281. Белы. пат. 635081 18. 11.63 Montecatini.
282. Пат. США 3112298 26. 11.63 Union Carbide.
283. Акцепт, заявка Японии 7109592 11.3.71 Onoda Cernent.
284. Пат. США 3645998 29. 2. 72 Onoda Cernent.
285. G. Caporiccio, Chim. e Ind. 52 A970) 1, 28—36.
286. D. Sianesi, J. Polymer Sei. A 1 6 A968) 1, 335—352.
287. Фр. пат. 1388525 5.2.65 Diamond Alkali.
288. Пат. США 3265678 A966) Du Pont.
289. Англ. пат. 1077728 A967) Du Pont.
290. Англ. пат. 1161958 A968) J. Frielink.
291. Пат. США 3573242 30.3.71 Phillips Petroleu:n.
292. Белы. пат. 615568 13.4.62 Continental Oil.
293. Англ. пат. 919683 27.2.63 Continental Oil.
294. Фр. пат. 1394584 2.4.65 Daikin Kogyo.
295. P. Manno, A. С. S. Polymer Preprints 4 A963), 1, 79—89.
296. P. Manno, Nucleonics 22 A964) 6, 64—67.
297. P, Manno, Nucleonics 22 A964) 9, 72—76.
298. E. Волкова, ДАН СССР, 167 A966) 5, 1057—1060.
299. X. Усманов, Высокомол. соед. 5 A963) 8, 1277,
300. Kh. Usmanov, J. Polymer Sei. AI 9 A971), 1779-1783.
301. Л. Губарева, Известия ВУЗов, Хим. и хим. технол. 14 A971) 8 1252-
1255.
136
A Yulchibaev, Kinet. Mech. Polyreactions, Int. Symp. Macromol. Chem.
Prepr. 3 A969), 315—319.
H Nishimura, Kogyu Kagaku Zasshi 74 A971) 34, 489—494.
J Konig, J. Polymer Sei. A24 A966) 3, 401—404.
С. Wilson, J. Polymer Sei. С A965) 8. 97.
F Weigert, Organ, magn. Resonance 3 A971), 373—377.
J. Lando, J. Polymer Sei. A 4 A966), 941—951.
G Natta, Atti Acad. Nazi. Lincei, Rend. Classe Sei. Fis. Nat. 31 A961), 350.
R. Golike, J. Polymer Sei. 42 A960), 583—584.
311 M Wallach, J. Polymer Sei. A 1 4 A966), 2667—2674.
312. Пат. США 2457035 21. 12.48 Monsanto.
313. Опубл. заявка 2035259 20. 1.72 Dynamit Nobel.
314. Пат. США 2476606 19.7.49 Du Pont.
315. Англ. пат. 898182 6. 6. 62 Du Pont.
316. Акцепт, заявка Японии 7205417 16.2.72 Electro Chem. Ind.
317. J. Stellings, J. appl. Polymer Sei. 14 A970), 2, 461—470.
318. Акцепт, заявка Японии 7205416 16. 2.72 Onoda Cernent.
Пат. США 3297462 10. 1.67 Du Pont.
Пат США 3288880 29. 11.66 Du Pont.
Пат. США 2917402 15. 12. 59 Du Pont.
Пат. США 2970066 31. 1. 61 Du Pont.
Пат. США 3018269 23. 1.62 Du Pont.
T. Dougherty, J. Amer. chem. Soc. 86 A964), 460.
Пат. США 2820772 21. 1. 58 3 M Co.
Пат. США 679761 23. 8. 57 Du Pont.
Пат. США 555794 28. 12.55 Du Pont.
Patentanm. 1154939 26.9.63 Du Pont.
Patentanm. 1153885 5.9.63 Du Pont.
330. Англ. пат. 837839 A960) Du Pont.
331. Англ. пат. 862853 15. 3. 61 Du Pont.
332. Пат. ФРГ 2032825 5. 1. 72 Dynamit Nobel.
Пат. США 2953818 27. 9. 60 Du Pont.
Англ. пат. 845634 24. 8. 60 Du Pont.
Пат. США 3096229 2. 7. 63 Du Pont.
Акцепт, заявка Японии 7119863 3. 6.71 Kuraray.
337. Опубл. заявка 2058823 31. 5. 72 Dynamit Nobel.
338. Пат. США 3627854 14. 12.71 Phillips Petroleum.
339. Опубл. заявка 2035446 20. 1.72 Dynamit Nobel.
340. V. Simril, J. appl. Polymer Sei. 4 A960) 10, 62—68.
341. D. Jazgi, Plast, mod. Elast. 20 A968) 3, 68—70.
342. К. Kawasaki, J. Polymer Sei. A2 9 A971) 11, 2095—2097.
343. E. Fukada, J. appl. Phys. 11 A972), 1, 36—40.
344. A. Me Glue, Polymer 13 A972) 8. 371—378.
345. A. Patrick, Adv. Fluorine Chem. 2 A961), 1.
346. W. Hart, J Res. Nat. Bureau Stand. 51 A953), 327.
347. M. Bro, J. appl. Polymer Sei. 6 A962), 456—460.
348. L. Epstein, Nat. Acad. Sei. Nat. Council Publ. 842 A961), 141—146.
349. VDE-Ztg. 108 A966), 4, 125—128.
350. Белы. пат. 620723 28. 1.63 Du Pont.
351. Пат. США 3676290 11. 6. 72 Westinghouse Electric.
352. Kunststoffechnik 9 A970) 11,409.
353. Kunststoffe 60 A970) 8, 601.
354. Modem Plastics 46 A963) 8, 90—91.
355. Пат. США 3677882 18. 7. 72 General Electric.
356. Kunststoff-Berater A972) 5, 369.
357. Белы. пат. 635081 18. 11.63 Montecatini.
358. Англ. пат. 592113 8.9.47 ICI.
359. Акцепт, заявка Японии 613995 25.4.61 Daiwa Spinning.
360. Белы. пат. 645617 24. 10.64 Diamond Alkali.
361. Пат. США 2823200 11.2.58 Monsanto,
137
362. Пат. США 2409948 22. 10. 46 Du Pont.
363. Y. Imanishi, Makromolekulare Chem. 70 A964), 77.
364. G. Natta, J. Polymer Sei. A3 A965), 4263—4278.
365. Белы. пат. 645961 16.7.64 Union Carbide.
366. Пат. США 2497323 14. 2. 50 Du Pont
367 R. Burkhart, J. Polymer Sei. A 1 A963), 1137—1145.
368. Англ. пат. 583482 19. 12.46 ICI.
369. Фр. пат. 1478785 28.4.67 Montecatini.
370. D. Sianesi, Chim. Ind. (Milano) 52 A970) I, 37—42.
371. Ит. пат. 704235 20.4.66 Montecatini.
372. Ит. пат. 704906 27. 4. 66 Montecatini.
373. Пат. США 2479367 16.8.49 Du Pont.
374. Пат. США 2468664 26. 4. 49 Du Pont.
375. D. Sianesi, Chim. Ind. (Milano) 52 A970) 2, 139—145.
376. Пат. США 2792423 14. 5. 57 Union Carbide.
377. Пат. США 2499097 28. 2. 50 Du Pont.
378. Пат. США 2406717 27.8.46 Monsanto.
379. Белы. пат. 611530 29. 12.61 Continental Oil.
380. Англ. пат. 946050 8. 1.64 Continental Oil.
381. Пат. ФРГ 1238204 6.4.57 Du Pont.
382. Пат. ФРГ 1086433 4.9.60 Hoechst.
383. Пат. США 2468054 26. 4. 49 Du Pont.
384. Пат. США 2456255 14. 12. 48 Du Pont.
385. Белы. пат. 618320 17.9.62 Montecatini.
386. Белы. пат. 562433 16. 5. 58 Solvay.
387. Пат. США 2866721 30. 12.58 3 M Со.
388. Пат. США 2362960 14. 11.44 Monsanto.
389. Акцепт, заявка Японии 613995 25.4.61 Daiwa Spinning.
390. Пат. США 2917473 15. 12.59 Union Carbide.
391. Пат. США 2592248 8. 4. 52 Eastman Kodak.
392. Пат. США 2579061 18. 12.51 Du Pont.
393. Англ. пат. 593605 21. 10.47 Du Pont.
394. Англ. пат. 608807 21.9.48 Du Pont.
395. Пат. США 2917494 15. 12.59 Monsanto.
396. Белы. пат. 562701 27.4.58 Solvay.
397. Англ. пат. 949752 19. 2. 64 Du Pont.
398. Белы. пат. 645617 24.9.64 Diamond Alkali.
399. Пат. США 2823200 11.2.58 Monsanto.
400. Пат. США 2847401 12. 8. 58 Monsanto.
401. Пат. ФРГ 1114279 9. 7.60 Kulzer & Со.
402. Пат. США 2992211 11.7.61 3 M Со.
403. Пат. США 2986556 30. 5. 61 3 M Со.
404. Пат. США 2956988 18. 10. 60 Du Pont.
405. Пат. ФРГ 850668 6. 9. 52 Bayer.
406. О. Кхаликова, Науч. труды Ташкентск. уни-та A970), 379, 86—92 105—
ПО.
407. Т. Латнпов, Науч. труды Ташкенского уни-та A969) 349, 3—10.
408. А. Юльчибаев, Науч. труды Ташкентск. уни-та A969), 349, 11 — 16
409. G. Goutiere, Compt. Rend. Acad. Sei. 257 A963), 3, 674—676.
410. Kh. Usmanov, J. Polymer Sei. A 1 9 A971) 5, 1459—1462.
411. Kh Usmanov, Kinet. Mech. Polyreactions, Int. Symp. Macromol. Chem. Prepr.
A969), 4, 251-255.
412. С. Юльчибаев, Узб. хим. ж., 15 A971) 5, 75—76.
413. R. Bacskai, J. Polymer Sei. A l 9 A971), 991—1004.
414. L. Weintraub, Chem. Ind. (London) 48 A965), 1963.
415. Пат. США 2627529 3. 2. 53 Secony Vacuum Oil,
416. Пат. США 2401987 A946) Du Pont.
417. Пат. США 2551573 8.5.51 Du Pont.
418. Пат. США 2628989 17.2.53 Allied Chem.
419. Англ. пат. 732269 22.6.55 Du Pont.
420. Пат. США 2774799 18. 12.56 Kellogg.
m
421 Пат ФРГ 1068695 A958) Hochst.
422. Англ. пат. 823998 19. 11.59 3 M Со.
423 Акцепт, заявка 1906206 8. 10.70 Fluorwerke Donna.
424. Опубл. заявка 2044370 23. 3. 72 Bayer.
425. Пат. США 3188356 8.6.65 Permwait.
426 Фр. пат. 1330146 21.6.63 Stauffer Chem.
427. Фр. пат. 1337360 13.9.63 St. Gobain.
428. Пат. США 2733278 31.1.56 Du Pont.
429. Англ. пат. 778750 10. 7. 57 Du Pont.
430. Пат. США 3047637 31. 7. 62 Dow Chem.
431. Акцепт, заявка Японии 22453 5. 10. 65 Kureha Chem.
432. Пат. США 3089910 14.5.63 Dow Chem.
433. Пат. США 2687440 24. 8. 54 Dow Chem.
434. Пат. США 3073870 15. 1. 63 Dow Chem.
435. H Madai, J. prakt. Chem. 19 A963), 202—205.
436. Пат США 2722558 1. 11.55 Dow Chem.
437. Пат. США 2723296 8. 11. 55 Dow Chem.
438. Пат. США 2401897 11.5. 46 Du Pont.
439. Пат. ФРГ 952713 A956).
440. G. Bachmann, Ind. Engin. Chem. 41 A949) 1, 70—72.
441. Пат. США 2435537 3. 2. 48 Du Pont.
442. Пат. США 2635093 A950) AUled Chem.
443. Patentanm. 1167537 9.4.64 Pennwalt.
444. Акцепт, заявка Японии 71.09476 10. 3. 71 Daikin Kogyo.
445. Белы. пат. 562433 16.5.58 Solvay.
446. Пат. США 2865824 23. 12. 58 Allied Chem.
447. С. Малкевич, Высокомол. соед., Б 10 A968) 12, 881—882
448. Patentanm. 1167532 9.4.64 Pennwalt.
449. Опубл. заявка 2141796 2.3.72 Pennwalt.
450. Англ. пат. 1004172 A965) Solvay.
451. Белы. пат. 562701 27.5.58 Solvay.
452. Опубл. заявка 2063248 24.6.71 Diamond Shamrock.
453. Акцепт, заявка Японии 7041596 26. 12.70 Kureha.
454. Пат. США 3475396 A969) Diamond Shamrock.
455. Акцепт, заявка Японии 7114465 17.4.71 Kureha.
456. Фр. пат. 1419741 3. 12.65 Kureha.
457. Акцепт, заявка Японии 7120820 11.6. 71 Kureha.
458. Опубл. заявка 2213135 28.9.72 Diamond Shamrock.
459. Англ. пат. 1188889 A970) Asahi Glass.
460. Акцепт, заявка Японии 7114667 20.4.71 Kureha.
461. W. Doll, J. appl. Polymer Sei. 14 A970) 7, 1767—1773.
462. E. Волкова, ДАН СССР, 167 A966), 5, 1057—1059.
463. Фр. пат. 1566523 A969) Dow Chem.
464. Пат. США 3380977 A968) Dow Chem.
465. Japan Plastics Age 8A970) 2, 14.
466. R. Naylor, J. Polymer Sei. 44 A960), 1—7.
467. С. Wilson, J. Polymer Sei. С 8 A965), 97—112.
468. С. Wilson, J. Polymer Sei. A 1 A963), 1305—1310.
469. S. Yano, J. Polymer Sei. A2 8 A970), 1057—1072.
470. E. Гальперин, Высокомол. соед., А 12 A970) 8, 1880—1885.
471. G. Natta, J. Polymer Sei. A3 A965), 4263—4278.
472. H. Katukami, J. Polymer Sei. A2 8 A970), 1177—1186.
473. J. Lando, J. Polymer Sei. A l 4 A966), 941—951.
474. W. Doll, J. macromol. Sei. В 4 A970), 309—322.
475. E. Гальперин, Высокомол. соед., 7 A965), 933—938.
476. W. Doll, J. macromol., Sei. В 2 A968) 2, 205—218.
477. К. Okuda, J. Polymer Sei. В 5 A967), 465—468.
478. J. Stallings, Polymer Engin. Sei. 11 A971) 6, 507—512.
479. R. Hasegawa, Polymer J. 3 A972) 5, 491—610.
480. R. Teulings, J. Polymer Sei. В 6 A968), 441—445.
481. S. Enomoto, J. Polymer Sei. A2 6 A968), 861—869.
482. Л. Тарутина, Высокомол. соед., Б 12 A970) 10, 780—782
483. К. Sakaoku, J. macromol. Sei. В I A967), 401—412.
484. Е. Baer, J. macromol. Sei. В 1 A967), 309—320.
485. W. Doll, J. macromol. Sei. В 4 A970) 4, 889—896.
486. С. Берлянт, Высокомол. соед., Б 13 A971) 1, 37—40.
487. Б. Космынин, Высокомол. соед., А 14 A972) 6, 1365—1370.
488. G. Cortili, Spectrochim. Acta 23 А A967), 285, 2216.
489. Опубл. заявка 2130370 5. 1. 72 Kureha.
490. Акцепт, заявка Японии 7053941 20.7.70 Kureha.
491. Е. Гальперин, Высокомол. соед., Б 14 A972) I, 37—38.
492. Y. Hoshi, Mod. Plastics A971) 1.
493. J. Сох, J. appl. Polymer Sei. 8 A964) 6, 2935—2961.
494. W. Barnhart, SPE Techn. Papers 17th Ann. Techn. Conf. 1961, 8—7, Pt. 1.
495. H. Ishii, Kobunshi Kagaku 27 A970) 307, 858—864.
496. L. Wall, J. Polymer Sei. A 14 A966) 2, 349—365.
497. G. Sands, ACS Polymer Preprints 6 A965) 2, 987—991.
498. R. Timmermann, J. appl. Polymer Sei. 6 A962) 22, 456—460.
499. A. Dukert, SPE Techn. Papers A962) 8, 23—3 I8th Ann. Meeting.
500. British Plastics 34 A961) 9, 473—475.
501. Акцепт, заявка Японии 7102918 25. 1. 71 Kureha.
502. Акцепт, заявка Японии 7114536 19.4.71 Kureha.
503. Акцепт, заявка Японии 7111996 27.3.71 Y. Iketa.
504. J. Stallings, J. appl. Polymer Sei. 14 A970), 461—470.
505. F. Ingraham, Ind. Engin. Chem. 56 A964) 9, 53—55.
506. Chem. Engin. News 44 A966) 35, 38.
507. S. Yano, J. Polymer Sei. A28 A970), 1057—1072.
508. T. Wentinik, J. appl. Phys. 32 A961), 1063—1069.
509. С. Кабин, Высокомол. соед., 3 A961) 4, 618—623.
510. Y. Ishida, Kolloid-Ztg. 200 A964), 48—54.
511. A. Peterlin, Kolloid-Ztg. 203 A965), 68—75.
512. A. Katukami, Kobunshi Kagaku 26 A969), 83.
513. H. Sasabe, J. Polymer Sei. A 27 A969), 1405—1414.
514. N. Koizeuni, J. Polymer Sei. В 7 A969), 815—820.
515. К. Nakamura, J. Polymer Sei. A29 A971), 161—173.
516. A. Glass, J. appl. Physics 42 A971) 13, 5219—5222.
517. E. Fukaota, Polymer J. 2 A971) 5, 656—662.
518. J. Luongo, J. Polymer Sei. A 2 10 A972) 6, 1119—1123.
519. J. Mc Fee, IEEE Trans. Sonics Ultrason. 19 A972) 2, 305—313
520. J. Bergman, ACS Polymer Preprints 12 A971) 1, 689—693.
521. W. Fullhase, Schwei?en und Schneiden 20 A968) 4, 84—85.
522. Опубл. заявка 2147992 4.5.72 Kureha.
523. Опубл. заявка 2159861 8.6.72 Kureha.
524. К- Shimada, Japan J. appl. Physics 11 A972) 6, 910.
525. G. Richards, J. appl. Polymer Sei. 8 A964) 5, 2269—2280.
526. Пат. США 3642668 15.2.72 Polaroid Corp.
527. Пат. США 3620895 16. 11.71 Polaroid Corp.
528. Опубл. заявка 2038915 18.2.71 Kureha.
529. J. Noland, Adv. Chem. Ser. 99 A971) 2, 15—28.
530. Опубл. заявка 2141617 24.2.72 Kyreha.
531. Revue Prod. chim. 64 A961) 1289, 419—421.
532. Англ. пат. 823974 18. 11. 59 3 M Co.
533. Пат. США 3051677 A962) Du Pont.
534. Пат. США 3023187 27. 2. 1962 3 M Co.
535. Опубл. заявка 2161861 29.6.72 t)u Pont.
536. J. Smith, J. appl. Polymer Sei. 5 A961), 460—467.
537. A. Moran, Ind. Engin. Chem. 51 A959), 831.
538. R. Radcliff, Techn. Rdsch. (Bern) 53 A961) 9, 9—11
539. D. Stivers, Ind. Engin. Chem., Prod. Res. Dev. 3 A964) I, 61—67.
540. D. Rice, ACS Polymer Preprints 12 A971) 1, 396—402.
541. T. Дегтева, Высокомол. соед., 7 A965), 1198—1202.
542. A. Moran, Kunststoffe — Plastics 8 A961) 1, 50—51.
140
543. A Moran, Gummi — Asbest 13 A960) 12, 1006—1018.
544 N Luijendijk, Rev. gen. Caoutchouc Plast 43 A966) 7—8, 981—988.
545. Chem. Ind. 20 A968), 751.
546 W. Kadi, Kautschuk —Gummi —Kunststoffe 20 A967) 12, 729—733.
547. Kunststoffe —Plastics A971) 6,248.
548. Пат. США 2468054 A949) Du Pont.
549 Пат. США 2738343 A956) Kellogg.
550. Пат. США 2752331 A956) Kellogg.
551. W. Robbe, Rubber Age 82 A957) 2, 286.
552. T. Дегтева, Высокомол. соед., 3 A961) 5, 671—678.
553. G. le Guellec, Rev. gen. Caoutchouc 38 A961) 7—8, 1110—1122.
554. U. Sandhof, Kunststoffe 60 A970) 5, 327—334.
555. Kunststoff-Berater A974) 2, 82.
556. Plastverarbeiter 25 A974) 3, 178.
5. ЛИНЕЙНЫЕ ПОЛИМЕРЫ С АРОМАТИЧЕСКИМИ
КАРБОЦИКЛАМИ В ЦЕПИ
Линейные полимеры с ароматическими карбоциклами в цепи
наряду с высшими полиолефинами и фторсодержащие полиоле-
фины нашли практическое применение уже в начале 60-х годов.
Полиэтилентерефталат и поликарбонат, являющиеся одними из
первых представителей этого класса, получают в промышленном
масштабе соответственно с 1946 и 1958 гг.
Ниже приведены данные о структуре и верхней температурной
границе практического использования ряда линейных полимеров
с ароматическими карбоциклами в цепи:
Г_сн2—
Полифенилен B50 °С)
СНг
Поли-гс-ксилплен A00 °Q)
~]
он _„
Полишдроксибензил B00 °С)
/
СНз
-*«
Простой ароматический
цолиэфир B00 °С)
Полифеннлен-
сульфид B20 °С)
Полисульфонэфир A80-260 °С)
Полиметилендифеннлоксид B50 °С)
Подиарилат C00 °С)
141
СНз
СН3 О _„
Поликарбонат A35 °С)
\/^ \/ -Ire
Полиамид B30 °С)
В зависимости от атомов или групп атомов, соединяющих карбо-
циклы, а также ориентации циклов в цепи верхняя температура
длительной работоспособности полимеров колеблется от 100 до
250 °С. Повышенная термостойкость полимеров этого класса по
сравнению с термостойкостью линейных алифатических полимеров
является следствием высокой жесткости цепи, а также повышенной
энергии разложения ароматических С—С- или С—Н-связей.
Поли-п-фенилен, характеризующийся наиболее высокой
температурой длительной работоспособности (около 250°С),
представляет собой в силу особенностей своей структуры (непосредственное
соединение n-фениленовых циклов) неплавкий и нерастворимый
продукт, вследствие этого, трудно поддающийся переработке в
изделия.
Оптимальное соотношение между термостойкостью и
эксплуатационными свойствами материалов наблюдается для полимеров,
в которых ароматические циклы соединяются такими атомами и
группами атомов, как —О—, —СО—, —NH—, —S— или —SO2—.
Линейные ароматические полимеры, построенные по этому
принципу, такие, как полифениленоксиды, полиарилаты,
поликарбонаты или полиариленсульфоноксиды хотя и обладают по
сравнению с поли-л-фениленом пониженной термостойкостью, однако
вследствие повышенной гибкости цепей могут перерабатываться
в изделия из раствора или расплава.
Среди вновь создаваемых термостойких полимерных
конструкционных материалов эти полимеры занимают значительное место.
5.1. ПОЛИФЕНИЛЕНЫ
[-©-L
Полифенилены можно получить конденсацией ароматических
галогенсодержащих соединений, окислительной дегидрополикон-
денсацией ароматических углеводородов, циклоприсоединением
бисацетиленов к бисциклопентадиенонам или радикальным арили-
рованием ароматических углеводородов.
Высокомолекулярный поли-/г-фенилен является неплавким и
нерастворимым продуктом. Его термическое разложение з
инертной среде начинается при 550 °С и на воздухе — при 450 °С
142
В промышленности полифенилен получают окислительной ка-
тионной полимеризацией бензола, а также при взаимодействии
терфенилов с ж-бензолдисульфохлоридим.
" Поли-л-фенилен, полученный полимеризацией бензола, может
перерабатываться в изделия спеканием при 400 °С и давлении
14 000 кгс/см2.
Низкомолекулярные растворимые полифенилены из терфенила и
бензолдисульфохлорида используются для получения
высокотермостойких и весьма стойких к действию внешних факторов
композиционных материалов с асбестовыми или углеродными волокнами.
Они применяются для изготовления элементов конструкций,
работающих при высоких температурах (при 400°С в течение 100 ч)
в присутствии горячих агрессивных сред.
5.1.1. ПОЛУЧЕНИЕ ПОЛИФЕНИЛЕНОВ
5.1.1.1. Реакция сочетания галогенсодержащих
ароматических углеводородов
Полифенилены можно получать из ароматических
галогенсодержащих соединений по реакциям Ульмана, Вюрца — Фиттига, путем
окислительной конденсации с реактивами Гриньяра или литийаро-
матнческими углеводородами.
Реакция Ульмана. Реакция Ульмана используется прежде всего
для получения замещенных полифениленов, например полиметил-
фенилена, полинитрофенилена или полиперфторфенилена. В
оптимальных условиях можно получать полимеры с молекулярной
массой до 300 000 [1]. Конденсация дигалогенсодержащих соединений
с порошком меди осуществляется в отсутствие растворителей или
в полярных растворителях при температурах от 100 до 300 °С:
—CuX2
Jrt
Наряду с порошкообразной медью в реакции можно
использовать также серебро [2—4] или ртуть [5].
Полагают [6, 7], что конденсация по Ульману протекает по
механизму образования неустойчивых медьорганических соединений
и промежуточных радикальных продуктов:
:
CuX
Полиперфторфенилен был получен конденсацией по Ульману
из 1,4-дибромперфторбензола, 1,4-дииодперфторбензола или 1,4-ди-
хлорперфторбензола [8—14].
143
При конденсации 3,3'-диметил-4,4'-дииоддифенила образуются
полимеры, растворимые в бензоле, с молекулярной массой
примерно 1000 [15]:
HaCv
ni—
\ /
/ \L^/ l 220-270
Высокомолекулярные продукты с молекулярной массой 300 000
можно выделить при использовании а-метилнафталина в качестве
растворителя [1, 16].
Конденсация дииоддинитробифенила в ДМФ при 120—140°С
приводит к получению высокомолекулярного нерастворимого
продукта желтого цвета [17]:
-О-О)-1
Си/ДМФ
I-
120-140 °C
v /
n
Незамещенный поли-я-фенилен получают взаимодействием
я-дибромбензола с активированной порошкообразной медью в
бензоле при 250 °С [18].
Реакция Вюрца — Фиттига. По реакции Вюрца — Фиттига по-
лифенилен был получен впервые в Риге в 1872 г.
Na или Са(ОНJ
>
Диоксан
Поли-tt- пли поли-ж-фенилен с молекулярной массой до 3000
был получен из п~ или ж-дибромбензола в диэтиловом эфире
[19,20].
По этой реакции получают также метилзамещенные поли-ж-фе-
нилен [21] и поли-я-фенилен [1, 16].
Синтез политетрафтор-я-фенилена из тетрафтордибром- или
тетрафтордииодбензола не удается осуществить из-за близкой
реакционной способности атомов фтора и брома или иода [8].
Синтез полифенилена по реакции Вюрца — Фиттига ускоряется
в присутствии соединений с активным водородом [22].
Окислительная конденсация с использованием реактивов
Гриньяра. Этот метод применяется для синтеза замещенных или
соединенных в ж-положении полифениленов. По этому способу
образуются более высокомолекулярные продукты, чем по реакции
Вюрца — Фиттига.
Полиперфторфенилен и полиперфтортолуилен [9, 23, 24]
получают из соответствующих монохлор- или монобромсоединений
в ТГФ при взаимодействии с реактивом Гриньяра при —30°С и
разложением при 64 °С. Реакция протекает путем нуклеофильного
замещения атома фтора в ароматическом кольце под действием
144
реактива Гриньяра:
ТГФ
«F—Л ))—MgX
-r-
64 "С
L_ F
/ \
F J
-f MgFX
n
Поли-л*-фенилен образуется в результате получения реактива
Гриньяра с ж-дибромбензолом и разложением его в присутствии
хлорида железа(III) или хлорида кобальта(П) [25].
Окислительная конденсация с литий ароматическими
соединениями. Дилитийарилен можно получать из дибромбензола при
обработке его амальгамой натрия и последующим взаимодействием
с литием [26] :
Na/Hg
В присутствии галогенндов переходных металлов (например,
FeCl2, CoCl2, NiCl2 или МпС12) в результате конденсации
образуются полифенилены [26—32].
Механизм конденсации заключается в образовании
промежуточных медьароматических соединений. Получаемые при их
разложении свободные радикалы рекомбинируют с образованием
полифенил енов:
CuCL
Cl
-LiCl
CuCl
CuCl
-2 cuci
5.1.1.2. Окислительная дегидрополиконденсация
ароматических соединений
Бензол, толуол, галогенпроизводные бензола и многоядерные
ароматические углеводороды, например бифенил и терфенил, при
35 °С в присутствии кислот Льюиса, сокатализатора и окисляющего
агента образуют полифенилены:
СиС12; А1С1з/Н2О Г
—CuCl; HCI
Эта реакция была детально изучена в начале 60-х годов Кова-
чичем [33—48].
В качестве кислот Льюиса применяют А1С13, А1Вг3, FeCl3, SbCl5,
M0CI5 или МоОС13. Катализаторами служат Н2О, H2S, НСООН и
145
нитроэтан. Из окисляющих агентов используют диоксид марганца,
диоксид свинца, хлоранил, я-бензохинон, диоксид азота, три-
оксид азота, нитробензол, хлорид пли бромид меди(П). Пентахло-
рид молибдена или хлорид железа одновременно выполняют
функции кислоты Льюиса и окислителя.
По механизму эта реакция напоминает катионную
полимеризацию олефинов. В системе бензол —А1С13—СиС12—Н2О протекают
следующие элементарные стадии реакции [34]:
Н2О + А1С1
Инициирование
[Н2О • А1С13] ч=^ Н+[А1С13ОН]"
+ H
Рост цепи.
(алкилирование)
Al CL
Окисление
Алкилирование
и окисление
Обрыв цепи
Инициирование происходит за счет комплексов Льюиса и
образовавшегося бензониевого иона (сг-комплекс). При присоединении
комплекса к молекуле бензола возникает новый замещенный бен-
зониевый ион. Окисление циклогексадиеновых фрагментов
ароматических структур препятствует обратимости реакции роста цепи.
Обрыв цепи наступает вследствие отрыва протона.
Выход полимера зависит от соотношения кислоты Льюиса и
окисляющего агента. В системе А1С1з—СиС12—Н2О при
соотношении А1С13—СиС12 выше 2: 1 достигается приблизительно
количественный выход. При незначительном избытке А1С13 выход
уменьшается примерно до 70 %. Это приводит к образованию комплекса
бензол
[38]:
—А1С13—CuCI2 A:1:1) в качестве побочного продукта
Cl СеН5 Cl
«Cl=--Al=Cl—Си—С1=А1—С1—
А.
сен
6П5
Cl
Молекулярная масса образующегося полифенилена определяется
температурой полимеризации, концентрацией мономера и
природой растворителя. Она уменьшается в следующем ряду
растворителей, используемых в процессе полимеризации: бензол > тетра-
хлорид олова > тетрахлорид титана > /г-дихлорбензол > 1,2,4-
трихлорбензол > о-дихлорбензол. Снижение молекулярной массы
происходит также при повышении температуры реакции и
уменьшении концентрации бензола.
Недостаток этого способа заключается в образовании
конденсированных многоядерных систем, ветвлении цепей и внедрении
галогенов в полимер. Разветвления и конденсированные
многоядерные ароматические соединения образуются по механизму о-алки-
лирования:
70
н
U/ -AlCb
=\ CuCl,
H
-ИС1
Циклизация
H
Внедрение атомов хлора является следствием рекомбинации
концевых групп растущего полимерного катиона с ионами хлора:
Н—
чо>-
Аг
СГ
+ >¦ h—
Аг
H—
1,
'—Cl
147
Содержание хлора в полимере зависит от типа каталитической
системы. При использовании системы А1С*13—СиС12 и температуре
реакции 35 СС в образующемся полифенилене содержится 2 %
хлора. Высокое содержание хлора наблюдается при использовании
в качестве окисляющего агента МпО2 или РЬО.
Окислительная дегидрополиконденсация применима также к
таким галогенпроизводным бензола, как хлорбензол [46, 49], фтор-
бензол [46], бромбензол [46], толуол [48, 49], этилбензол [50],
кумол [50], ксилол [49, 51], многоядерным ароматическим
углеводородам, например нафталину (или антрацену) [52], фенолам,
самому фенолу [53], крезолу [54], пирокатехину, резорцину и
гидрохинону [53], приводя к образованию соответствующих
замещенных полифениленов. В таких же условиях замещенные полифени-
лены получают, исходя из аминофенолов [49, 55] и ацетилбензола
[56].
Поли-я-фенилены, как правило, неплавки и нерастворимы
[57—59] и вследствие этого не могут перерабатываться в изделия
общепринятыми методами.
Статистическое распределение о-, м- и я-фениленовых ядер в
цепи, а также высокая степень разветвленности препятствуют
плотной упаковке молекул, способствуют уменьшению кристалличности
и приводят вследствие этого к получению растворимых и плавких
полифениленов.
Низкомолекулярные плавкие полифенилены можно получать
в расплаве окислительной дегидрополиконденсацией смеси бифени-
ла, о- и ж-терфенилов или трифенилбензола либо системы фенол —
бифенил [38, 60—63]. Для обеспечения механической циркуляции
реакционной смеси вязкость снижают введением избытка А1С13.
Снижение вязкости обусловлено низкой температурой плавления
комплексов А1С13 с ароматическими углеводородами. При 130—
150°С и времени реакции 6—10 ч достигается 30—40 %-ный выход
полифенилена с молекулярной массой 1800—2500. Выше 170 °С
получаются неплавкие продукты [64]. Эти низкомолекулярные по-
лифенилены с температурой плавления 300—400 °С могут
перерабатываться с добавкой отвердителей известными методами
получения изделий из реактопластов.
5.1.1.3. Дегидрирование полициклогексадиена-1,3
Ионно-координационной полимеризацией циклогексадиена-1,3
в присутствии катализаторов Циглера, например триизобутил-
алюминий — тетрахлорид титана, получается с хорошими выходами
полициклогексадиен-1,3, представляющий собой соединенные в
«-положении циклы, с молекулярной массой 10000. Ароматизация
путем обработки хлоранилом в кипящем ксилоле или галогениро-
вание с последующим пиролизом при 300—380 °С приводят к
получению поли-гс-фенилена с высокими выходами [65—69] :
O(U30-C4H9KAl/TiCI4 Г / \ ! Ароматизация Г //-^ 1
* L -\J-l —=*-* HQH.
148
При катионной полимеризации циклогексадиена-1,3 образуются
1,2- и 1,4-замещенные соединения, что приводит при ароматизации
к получению в полифенилене звеньев, соединенных в положении
1г2н 1,4 [68]:
О -=* [-ОН
, -1Н]
m
/
5.1.1.4. Полимеризация по реакции Дильса — Альдера
1,4-Циклоприсоединение бисциклопентадиенонов или биспиро-
нов к диенофилам с тройными связами позволяет получать с
высокими выходами высокомолекулярные плавкие, хорошо
растворимые полифенилены [70—80, 126]:
п
Синтез фенилзамещенных полифениленов проводят в таких
ароматических растворителях, как 1,2,4-трихлорбензол или сс-хлор-
нафталин при 180—260 °С, при этом достигается молекулярная
масса около 30 000—100 000.
Этот метод был использован также для получения поли-я-фе-
нилена из я-фениленбиспирона и я-диэтинилбензола [80, 126] :
О
пО=? ,)—(( У>—// Wd
о
—со?
Нерастворимый полимер желтого цвета содержит 90 % поли-
я-фенилена. Одновременное образование я- и И!-фениленовых
149
групп протекает по следующему механизму [136]:
О + НС==С
90% 10%
-СО
5.1.1.5. Радикальное арилирование ароматических
углеводородов
Получение полифениленов по механизму свободнорадикально-
го арилирования осуществляется при пиролизе ароматических
углеводородов, а также при термическом разложении
бифункциональных ароматических пероксидов, диазосоединений, металлор-
ганических соединений, иодониевых соединений, карбоновых
кислот и сульфохлоридов в присутствии ароматических
углеводородов.
При пиролизе бифенила при 400 °С [81] или смеси бензол —
бифенил в присутствии соединений серы при 650—950 °С [82]
образуются с хорошими выходами воскообразные полифенилены.
Образование твердых органических остатков при пиролизе
бензола в глиняных трубах было описано Бертло [83] еще в 1866 г.
При разложении моно-, ди- и тринитробензолов в присутствии
ароматических углеводородов при 400—600 °С получаются лишь
относительно низкомолекулярные полифенилены [84].
Бисдиазосоединения разлагаются в присутствии соединений
меди при 400 °С до полифениленов, которые, однако, содержат в
цепи азогруппы [18, 85—8]
pHSH*
<-<o>-<^'
где
150
— H, —COOH; m<Cln.
Среди методов радикального арилирования для синтеза поли-
фениленов практическое значение имеет лишь разложение
ароматических дисульфохлоридов в присутствии терфенилов [89—92].
Путем регулируемого присоединения расплавленного ж-бензол-
дисульфохлорида к расплавленной смеси терфенилов при
температуре 300—310 °С можно получить как олигомерные, так и
высокомолекулярные полифенилены:
Cl—SO.
SO2—Cl
зоо °с
— 2SO2. —2НС1
SO2-C1
-2SO2. -2HC1
и т. д.
Добавки CU2CI2 или FeCb оказывают каталитическое действие.
За ходом реакции можно следить по выделяющимся диоксиду
серы и хлористому водороду [89]. Для описания процесса
конденсации предложен следующий механизм реакции:
Ar—SCL— Cl
Ar*+
+ «Cl
АГ +
Cl
HC1
•so2a
HCI +
Суммарная реакция записывается в виде:
ArSO2Cl + (СЪ—R —> Ar—(Tj) + S02 + HC1
Реакция арилирования с дисульфохлоридами применима лишь
для ароматических углеводородов с малой реакционной
способностью. Арилирование алкилбензолов или фенолов приводит к
образованию в качестве побочных продуктов смеси хлорированных
ароматических углеводородов и полисульфонов.
Растворимые плавкие низкомолекулярные полифенилены,
полученные взаимодействием терфенилов с ж-бензолдисульфохлори-
дом, могут перерабатываться обычными методами получения
изделий из реактопластов.
5.1.1.6. Электролитическая полимеризация
Полифенилены образуются при электролизе таких
ароматических углеводородов, как бензол, толуол, ксилол, триметилбензол
и мезитилен, в присутствии кислот Льюиса [93—97]. Высокие
выходы продуктов были достигнуты при электролизе комплексов
бензола с галогенводородными кислотами и галогенидами
алюминия вида
С6Н6 • НХ • А1Х3 или С6Н6 • НХ • А12Хе
с использованием платины в качестве измерительного электрода
и системы Си — СигСЬ в качестве эталонного электрода [98]. По
свойствам полученные полимеры аналогичны полимерам,
синтезированным окислительной дегидрополиконденсацией.
Высокомолекулярный полифенилен образуется также при
электролитическом окислении бензола в присутствии таких акцепторов
аротонов, как ацетонитрил [99].
5.1.1.7. Циклополимеризация ненасыщенных алифатических
и ароматических соединений
Разветвленные полифенилены типа
__ п
можно получить полициклотримеризацией диэтинилароматических
соединений, например диэтинилбензола или диэтинилнафталина,
в присутствии комплексов кобальта с триалкилфосфитом или
катализаторов Фишера, состоящих из тетрахлорида титана, три-
хлорида алюминия и алюминия [100—103].
152
5.1.1.8. Дегидратация фенолов
Полифенилены с незначительным содержанием гидроксильных
групп образуются с 18—32 %-ным выходом восстановлением
фенолов хлоридом цинка при повышенных температурах [104—107].
Полигидроксифенилены получают дегидратацией дифенолов
хлоридом цинка при температурах от 200 до 400 °С.
5.1.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИФЕНИЛЕНОВ
В зависимости от метода синтеза полифенилены содержат в
определенных количествах функциональные группы, а также
дефектные звенья в макромолекуле. Об идеальной структуре поли-
/г-фенилена или поли-ж-фенилена можно говорить лишь
приблизительно. Свойства полифениленов поэтому сильно зависят от
метода получения. Полифенилены, полученные окислительной дегид-
рополиконденсацией E.1.1.2) или дегидрированием полициклогек-
саднена-1,3 E.1.1.3), имеют коричневую окраску. Светлые
продукты образуются при циклоприсоединении по Дильсу — Альдеру
E.1.1.4).
5.1.2.1. Кристалличность
Поли-/г-фенилены, полученные окислительной дегидрополикон-
денсацией E.1.1.2) или дегидрированием полициклогексадиенов
E.1.1.3), являются кристаллическими. Полимеризация по
Дильсу— Альдеру бисциклопентадиенонов или биспиронов с бисаце-
тиленами E.1.1.4), а также синтез Вюрца — Фиттига приводят
к образованию аморфных полифениленов.
Степень кристалличности поли-/г-фениленов, полученных
окислительной дегидрополиконденсацией, можно повысить
прессованием при температурах выше 150 °С [47]. Это можно объяснить
влиянием температуры на процесс отщепления относительно
больших атомов хлора от полимерных цепей (примерно один Cl
на 20 фениленовых звеньев).
Орторомбическая элементарная ячейка поли-п-фенилена
состоит из двух проходящих через элементарные ячейки сегментов
цепи на одно фениленовое звено. Размеры элементарной ячейки
равны а = 7,81 A, b = 5,53 А и с = 4,20 А (ось молекул). Длина
элементарного звена составляет 4,20—4,30 А [47].
5.1.2.2. Температура плавления
Зависимость температуры плавления олигомерных фениленов
от числа соединенных ядер и вида соединений приведена на
рис. 5.1. Наиболее сильная зависимость температуры плавления
от числа соединенных бензольных колеи наблюдается в случае
соединения в «-положении. Поли-я-фенилены с восемью
соединенными фенильными ядрами уже нерастворимы.
Пониженная температура плавления низкомолекулярных
полифениленов объясняется замещением бензольных ядер, а также
153
100-
Рис. 5.1. Зависимость температуры плавления олигофе-
иилснов от числа бензольных колец, соединенных в о-
(/), м-B) и п-C)-положениях.
чередованием м- и /i-положений при их
соединении.
Фенилзамещенные полифенилены,
полученные циклоприсоединением бисциклопентадие-
нонов к бисацетиленам E.1.1.4), являются
плавкими продуктами. Форполимеры с
молекулярной массой 1775, которые получены
взаимодействием терфенилов с ж-бензолди-
сульфохлоридом, размягчаются при 204 °С
Энтальпия и энтропия образования поли-я-фенилена при
температуре 250 К равны —34,5 ккал/моль и 18,2 кал/(моль-°С)
соответственно [108].
5.1.2.3. Растворимость
Линейный поли-л-фенилен нерастворим в известных
органических растворителях, а также в НС1 и КОН. В горячей
концентрированной серной кислоте, а также в дымящей азотной кислоте
наблюдается постепенное растворение при одновременном
сульфировании или нитровании соответственно.
Напротив, на основе высокомолекулярных фенилзамещенных
полифениленов, соединенных в м- и /г-положениях, которые
синтезированы путем 1,4-циклоприсоединения E.1.1.4), можно получить
растворы в бензоле или хлороформе до 15%-ной концентрации
[76, 80].
Низкомолекулярные полифенилены, соединенные в о-, м- и
л-положениях, которые получены взаимодействием л*-бензолдисуль-
фохлорида с терфенилами E.1.1.5), растворяются в хлороформе,
дихлорэтане и бензоле.
Молекулярная масса фенилзамещенных полифениленов E.1.1.4),
определенная осмометрическим методом, составляет 40 000—100000
[76]. Для низкомолекулярных поли-/г-фениленов молекулярную
массу можно определить также по данным УФ-спектроскопии
[109—111]. Установлена [112] связь между длиной волны
максимального поглощения % и степенью полимеризации Р:
2,08-Ю3
- 0,634 ¦ cos
+ 1)
5.1.2.4. Стойкость к термической и термоокислительной
деструкции
Поли-л-фенилены стабильны примерно до 550°С в инертной
среде и до 450°С — на воздухе. Суммарные потери массы, по
данным ТГА, при нагреве р инертной среде до 850 °С колеблются от
154
10 до 15% [ИЗ, 114]. Исключение составляют поли-гс-фенилены,
которые получают разложением диазониевых соединений E.1.1.5).
Из-за наличия в цепи азогрупп разложение полимера в инертной
среде начинается уже при 350 °С [85].
В ряду бензол — бифенил-гс-терфенил-гс-кватерфенил
температура разложения уменьшается с увеличением молекулярной
массы от 600 до 470°С (воздух). Повышенная стабильность олигомер-
ных я-фениленов по сравнению с высокомолекулярным поли-гс-фе-
ниленом объясняется структурной нерегулярностью (разнозвен-
ностью) высокомолекулярных продуктов [115].
Полифенилены, представляющие собой статистические смеси
о-, м- и я-изомеров, которые получают взаимодействием Л1-бен-
золдисульфохлорида с терфенилами E.1.1.5), стабильны в
инертной среде до 500 °С. Потеря массы при нагревании до 900 °С (ТГА)
составляет 25 % [63].
Фенилзамещенные полифенилены E.1.1.4) стабильны в
инертной среде и на воздухе примерно до 530 °С (рис. 5.2). В то время
как на воздухе при температуре выше 530 °С происходит очень
быстрое разложение, в среде азота до 800 °С потеря массы
составляет примерно 30% [73, 79, 80]. Из сравнения ИК-спектров
пиролизованного фенилзамещенного полифенилена и исходного
полимера следует, что потеря массы обусловлена отщеплением
боковых фенильных групп [116].
После 100 ч термического старения на воздухе при 350 °С
заметных потерь массы не наблюдается.
Политетрафторфенилены по термостойкости не отличаются от
своих нефторированных аналогов [117].
Стабильность монозамещенных поли-я-фениленов уменьшается
в следующем ряду заместителей [45] :
—H > —Cl > —Br > —NH2 > —NO2
Поли->г-фенилены по термической стойкости весьма близки к
поли-гс-фениленам [118].
100 100 300 400 500 600 t,°C
Рис. 5.2. Кривые термической
деструкции фенилзамещенного полифенилена в
азоте (/) и на воздухе B) [116].
Рис. 5.3. Зависимость скорости образования летучих продуктов разложения при
пиролизе поли-/г-фенилена от температуры [119].
155
Основными продуктами деструкции поли-я-фенилсна при
660°С являются водород A моль/звено), метан @,1 моль/звено),
бензол @,03 моль/звено) и нафталин @,01 моль/звено) [119].
Зависимость скоростей образования летучих продуктов разложения
от температуры деструкции приведена на рис. 5.3. Дегидрохлори-
рование при 450 °С обусловлено наличием хлора, содержащегося
в полимере при получении его окислительной дегидрополиконден-
сацией. Водород выделяется при образовании из линейного поли-
я-фенилена полианнелированных ароматических углеводородов;
500-600 °С
—н-
550-700 °С
п *-
>700 °С
п *-
-*т
156
При разрыве дифенильных связей, а также при расщеплении
ароматических циклов выделяются летучие углеводороды:
-с2н2
С9Н5
н.
н.
2СН4
Пиролитический остаток наряду с углеродом содержит также
незначительные количества бифенила, /ьтерфенила и я-кватерфе-
нила [34].
5.I.3. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА МАТЕРИАЛОВ
И ПЕРЕРАБОТКА ПОЛИФЕНИЛЕНА
5.1.3.1. Ненаполненные формовочные материалы
на основе полифенилена
Высокомолекулярный неплавкий поли-я-фенилен производится
в США с 1967 г. фирмой «Eitel — McCullough Inc.»
окислительной дегидрополиконденсацией бензола E.1.1.2) и выпускается под
торговой маркой «Эймак 221» [120].
Методом спекания, называемым также изостатическим
прессованием, при 400 °С и давлении 14 000 кгс/см2 из поли-л-фени-
лена получают заготовки, из которых путем механической
обработки могут быть изготовлены изделия [42, 121].
Свойства полифениленовых образцов, полученных спеканием,
приведены ниже [120].
Плотность, г/см3 1,23
Прочность, кгс/см2
при растяжении 140—280
при сжатии 703
Относительное удлинение при разрыве, % • ¦ " 8—12
Модуль упругости, кгс/см2 21000—31000
Термический коэффициент линейного расширения
(при 20—600 °С) а-105, 1/°С 3-4
Твердость по Шору, шкала D 60—80
Удельное объемное электрическое сопротивление,
Ом-см Ю15
Электрическая прочность В/мм ....... 6@—830
Поли-я-фенилен обладает великолепной стойкостью к
ионизирующему излучению [42]. После облучения дозой 895 Мрад
157
tg o Рис, 5.4. Зависимость диэлектрической
проницаемости (У) и тангенса угла диэлектрических
потерь B) фенилзамещенного полифенилена
от температуры при 100 кГц [80].
уменьшения прочности
отпрессованных изделий не отмечалось.
Поли-л-фенилен, полученный
окислительной дегидрополиконденсацией
при температурах выше 150 °С E.1.1.2),
обладает полупроводниковыми и фотополупроводниковыми
свойствами [122, 123]. Температурные зависимости диэлектрических
характеристик фенилзамещенного полифенилена представлены на
рис. 5.4.
5.1.3.2. Материалы на основе полифенилена
Материалы на основе полифенилена, модифицированного п-кси-
лиленгликолем. Композиционные материалы на основе
полифенилена и стеклянных или графитовых волокон можно получать
пропиткой армирующих волокон растворами низкомолекулярных по-
лифениленов [получены окислительной дегидрополиконденсацией
терфенилов E.1.1.2)], модифицированных л-ксилиленгликолем, и
отверждением препрегов при 200—280 °С [64].
При нагревании /г-ксилиленгликоля с я-толуолсульфокислотой
вначале образуется растворимый в хлороформе теломер с четырьмя
ксилиленгликолевыми звеньями:
но—сн2—(Tj)—сн2—он
но—сн2
но-сн2
но-сн
сн2—он
—(Q)—сн2—о—сн2—(Q)—сн2—
он
но-сн2
\—сн2-о-сн2
¦Q
сн2-он
НО—СН2—(С ))—СН2—ОН
—SO3H
НзС
,_SO2-O-[-CH
Взаимодействие теломера с низкомолекулярным полифениленом
в таких растворителях, как хлороформ, трихлорэтилен или
хлорбензол, при температуре от 70 до 80 °С приводит к получению
поЛифенилена, отверждающегося при нагревании:
НзС—<|^—5О2~О-Г-СН2—((^—CH2~O-j ~s°2—<0)—
НО—Г—СН2 (Q) CH2-O-1-SO2
II
— CHa + НзС—
SO3H
СН2—
,_о-[-сн,
*
\
—О—1—
СН2—О—1—SO
СНз
н3с—
so- +н2о
—(iQ)—СН2О—|—СН2—/Q\—СН2—О—I — SO2—
Н-ш
Модификацию полифенилена можно осуществлять также
нагреванием смеси полифенилен — я-ксилиленгликоль — «-толуолсуль-
фокислота в хлорированных растворителях.
Для получения композиционных материалов стеклянные или
углеродные ткани или ровницу пропитывают раствором
модифицированного полифенилена и растворитель удаляют. Прессование
термореактивных препрегов проводят при 175—200 °С под
давлением 70—150 кгс/см2 с последующим постепенным их нагреванием
до 280 °С в среде азота.
В ходе термической обработки происходит образование
поперечных связей с немодифицированными сегментами полифениле-
новых цепей:
\
>200 °С
о
сн2—/
но—|—сн
)—сн2-о
—(( ))—СНэ
Слоистые материалы такого типа обладают отличными
абляционными свойствами.
159
Отверждение полифенилена м-бензолдисульфохлоридом.
Низкомолекулярные растворимые и плавкие полифенилены
производятся в промышленности с 1968 г. фирмой «Monsanto» путем
взаимодействия терфенилов с ж-бензолдисульфохлоридом [124, 125].
Аналогичную реакцию используют для получения слоистых
пластиков и наполненных пластмасс [89].
Низкомолекулярный полифенилен вместе с
ж-бензолдисульфохлоридом растворяют в хлороформе, дихлорэтане или бензоле и
после добавления Си2С1г в качестве катализатора используют для
пропитки высокотермостойких подложек. После удаления
растворителя при 50°С их прессуют при давлении 35—70 кгс/см2 и
температуре 300 °С. Дальнейшая термическая обработка полученных
заготовок осуществляется в течение 24 ч при 275, 300 и 325 °С.
Ниже приведены физико-механические свойстна композиционных материалов
на основе полифениленов [I — полифенилен D8—52%), армированный
асбестовым волокном; II — полифенилен C0—50%), армированный 45—55%
графитового волокна] [89]:
! II
Плотность, г/см3 1,4—1.7 1,4—1,5
Прочность, кгс/см2
при растяжении
при 25 °С 1400—1900
при 200 °С 1000-1400
при 300 °С 700—1000
при изгибе
при 25 °С 2100-2600 6330
при 250 °С — 3870
при 300 °С 1760—2100 —
при сдвиге — 210—315
Модуль упругости при изгибе, кгс/см2 . . . A,25—1,75) 105 A,4—1,7) 10е
Прочность при изгибе после старения в течение
2000 ч при 300 °С, кгс/см2
при 25 °С — 3500
при 300 °С — 1900
Время выдержки, в течение которого происходит
снижение прочности при изгибе до 350 кгс/см2, ч
при 400 °С 30-150
при 350 °С 200-400
при 300 °С 1000-1500
при 240 °С . - 7 000-15 000
при 200 °С 6—12 лет
Материалы на основе полифенилена кратковременно A00 ч)
работают под нагрузкой вплоть до 400°С. Их термостойкость
составляет 240 °С.
Для материалов на основе полифенилена, армированных
углеродными волокнами, характерна чрезвычайно высокая стойкость
к действию растворителей, кислот и оснований при повышенных
температурах. Воздействие на них оказывают лишь горячие
кислоты— окислители, например азотная кислота, или пиридин.
Данные об изменении прочности при изгибе (в %) компози-
в течение 2000 ч в различных агрессивных средах приведены ниже
ционных материалов на основе полифенилена после их выдержки
160
[] полнфенилен D8—52%), армированный асбестовым
волокном; II —- полнфепилен C0—50%), армированный графитовым
волокном] [89]:
H2SO4, КОНЦ.
H2SO4, 50%-ная
HNO3, конц.
НШ3, 50%-пая
НС1. конц.
НС1, 7,5%-ная
HF, 40%-на я
NaOH, 75%-ный
NaOH, 50%-ный
NaOH, 20%-ный
NaOH, 50%-нып
NaCl, 20%-ный
NaOCl, 15,6%-ный
Н2О2, 100%-ный
Диметилформамнд
Пиридин
Водяной пар
Бензол
Хлороформ
Петролейный эфир
Моторное масло
25
90
25
90
90
25
90
90
90
25
90
25
90
140
25
25
25
90
90
100
80
61
90
150
1
-79 C50 ч)
-87 D40 ч)
-60
-24
0
+0,5
+49
11
+4,4
+2,0
-6,0
-3,4
— 100
-7,7
-59
-1,9
+ 12
-0,3
+ 11,1
+6,5
+ 17
+ П
+ 14,5
-5,2
+8
-15
—69
-3,4
+30
Благодаря высоким теплостойкости и химической стойкости
в сочетании с хорошими механическими свойствами материалы на
основе полифенилена являются идеальными конструкционными
материалами, которые могут использоваться при высоких
температурах и в контакте с агрессивными средами. Материалы на
основе полифенилена, армированные графитовыми волокнами,
обладают такими же значениями коэффициента трения, как и
аналогичные материалы на основе эпоксидных или полиимидных
связующих, но значительно лучшими адгезионными свойствами к
металлическим поверхностям. В химической промышленности их
применяют для изготовления деталей насосов, мешалок и других
элементов конструкций, т. е. там, где требуется сочетание высокой
прочности со стойкостью к агрессивным средам при повышенных
температурах.
Благодаря отличным абляционным свойствам (материалы на
основе полифениленов и асбестового наполнителя превосходят
аналогичные материалы на основе эпоксидных смол) их можно
использовать для изготовления элементов реактивных двигателей.
5.2, ПОЛИ-л-КСИЛИЛЕН
Поли-л-ксилилен получают в промышленности пиролизом
ди-л-ксилилена и полимеризацией образующеюся при этом гс-кси-
лилена на охлаждаемой поверхности. Полимеризация и формование
6 Зак. 18 161
изделий проводят в Одну Технологическую стадию. Верхняя
температура длительной эксплуатации поли-/г-ксилилена в
инертной среде и на воздухе составляет 220 и 100°С соответственно.
Полимер имеет хорошие диэлектрические свойства, высокую
стойкость к растворителям, стабильность размеров и крайне
незначительную проницаемость.
Поли-/г-ксилилен применяется для покрытия металлов в
электронике и.для защиты их от коррозии.
Поли-я-ксплилен и галогенпроизводные поли-/г-ксилилена были
получены фирмой «Union Carbide» с 1965 г. в
опытно-промышленном и с 1970 г. — в промышленном масштабе. Эти продукты
выпускаются под торговыми марками «Парилен N» (поли-/г-ксилилен),
«Парилен С» (полихлор-я-ксилилен) и «Парилен D» (полидихлор-
я-ксилилен).
Стоимость поли-/г-ксилилена составила в 1970 г. 500 дол/кг.
5.2.1. ПОЛУЧЕНИЕ ПОЛИ-л-КСИЛИЛЕНА
5.2.1.1. Пиролиз я-ксилола в вакууме
Пиролизом я-ксилола в вакууме при 700—1150°С/1—5 мм рт. ст.
получают я-ксилилен с максимальным выходом 25%, который
сразу же полимеризуется с образованием смеси полимеров и оли-
гомеров [127—129]:
При введении полученного пиролизом я-ксилилена в
растворитель при —78 °С с последующим нагреванием образуются
растворимые высокомолекулярные продукты с молекулярной массой до
200000 [130, 131]. Ингибиторы не влияют на процесс
полимеризации [133]. По Шварцу [132, 134], при пиролизе я-ксилола вначале
образуются /г-ксилильные радикалы, при диспропорционировании
которых получаются «-ксилол и я-ксилилен:
н. +
2Н3С—^QN—СН2
Будучи очень активным, я-ксилилен быстро полимеризуется:
лН2С=/ )=СН2 —> Г—СН2—О—СН2—1
\—/ L vrt/ Jn
Хотя я-ксилилен вследствие его высокой химической активности
можно рассматривать как псевдорадикал, полимеризация в инерт-
162
ных растворителях при —78 С ускоряется в присутствии кислот
Льюиса [135].
Промышленное получение поли-л-ксилилена пиролизом ди-я-
ксилилена в вакууме подробно рассмотрено в разд. 5,2,2.
5.2.1.2. Реакция Вюрца —Фиттига с использованием
дигалоген-л-ксилолов
Конденсация дигалоген-я-ксилолов в присутствии
восстановителей проводится в инертных растворителях при температурах
ниже 25°С:
/ZZZ\ Восстановление
пх-сн2(Q)
Г—сн2—(Cj)—сн2—1
В качестве восстановителей применяют щелочные металлы
[136—141] и металлорганические соединения, например динатрий-
тетрафенилэтан [142], натрийнафталин [143] и литийметил [144].
При использовании переходных металлов и их солей, например
хлорида хрома (II) [145, 146], железа, никеля, кобальта [147]
или хлорида меди (I) [148], восстановление происходит в водной
суспензии.
Восстановление дйгалоген-я-ксилолов может осуществляться
электролитическим способом:
—СН2—X -^- Г—
nX-CH2—(|Q>)—СН2—X -^- Г—СН2—(JQ^>—СН2—
Для электролиза а, а'-дихлор-я-ксилола или а, а'-дибром-а-кси-
лола на ртутных электродах катодный потенциал должен
составлять 1,2В. Максимальный выход по току равен 96% [149].
Механизм полимеризации включает отщепление галогенидионов
посредством переноса заряда двух электронов с образованием ксилил-
аниона. При дальнейшем отщеплении в а'-положении образуется
я-ксилилен, который затем полимеризуется:
х—сн2—<rv>—сн2—x + 2e"—> х—сн2—(( )>—(
х—сн2—(СЪ—сн2 •
лН2С==/ >==СН2 —>
Отщепление и полимеризация протекают на катоде или в
растворе. На аноде происходит окисление галогенидионов до
галогенов. При использовании три- или тетрагалогензамещенных я-кси-
лолов можно получать соответствующие галогенсодержащие
полимеры.
За исключением поли-2-хлор-л-ксилилена, полимеры,
синтезированные этим методом, имеют сетчатое строение,
о* 163
По аналогичному механизму, включающему i ,6-дегидрогалоге-
нирование, поли-я-ксилилены можно получить поликонденсацией
а-галоген-я-ксилола в присутствии оснований [150].
5.2.1.3. Диссоциация четвертичных аммониевых
или фосфониевых производных п-ксилола
Разложением четвертичных аммониевых или фосфониевых
производных я-ксилолов горячим раствором гидроксидов натрия
получают в качестве промежуточного продукта я-ксилилен, который
тут же полимеризуется [151, 152]:
NaOH
Полимеризация
—НгО; —NaCl; —N(CH3K
[_cH2_0-cH2-]rt
Образующийся поли-я-ксилилен отличается от поли-я-ксилиле-
нов, полученных другими методами, хорошей растворимостью
[153].
Технический интерес может представить электролитический
метод проведения данной реакции:
СНз СНз
' + /7=г\ | + 2е-
НзС—N—СН2—(ГУ)—СН2—N—СНз —^-> 2(CH3KN +
I V^ I
¦<-> Н2С={ >=
В качестве растворителей при электролизе пригодны ДМФ,
ДМСО, пропиленкарбонат или тетраметиленсульфон. Обрыв цепи
при полимеризации я-ксилилена происходит за счет отрыва
водорода от молекул растворителя. При этом растворитель
восстанавливается [154].
Таким методом можно нанести на алюминий, используемый в
качестве катода, прочно сцепляющееся покрытие толщиной до
1 мкм. Вблизи этого слоя процесс полимеризации
затормаживается, так как покрытие катода образующимся поли-я-ксилиле-
ном препятствует переносу катионов.
Для электролитического нанесения поли-гг-кснлиленовых
покрытий пригодны производные ксилола, например га-метилбензил-
триметиламмонийнитрат, л-ксилиленбистриметиламмонийнитрат
или я-ксилиленбистрифенилфосфонийхлорид,
164
5.2.1.4. Изомеризационная полимеризация /г-замещенных
производных стирола
При наличии в «-положении у стирола фенилацетонитрильной
группы наблюдается почти полная ионизация водорода при
третичном атоме углерода заместителя и перегруппировка в я-ксили-
леновую структуру. Образующиеся промежуточные производные
n-ксилилена полимеризуются по свободно-радикальному цепному
механизму [155]:
CN
«-на
-сн=сн2
CN
С \ ( jj—СН=СН2
с6н
6П5
CN
==/ >=CH—СНз
C6HS
CN
_ CeHe
СНз
В качестве катализаторов для перегруппировки можно
использовать грет-бутилат калия или трег-бутиловый спирт. В ходе
реакции между ионизированными и неионизированными формами
я-бензилцианостирола образуются олигомеры, которые снижают
выход замещенного поли-я-ксилилена.
Подобный механизм лежит в основе полимеризации а.а'-дйа-
рил-1,4-хинодиметана до замещенных поли-я-ксилиленов [156] :
С6Н5
5.2.1.5. Изомеризационная полимеризация
диазопроизводных я-ксилола
Диазо-п-ксилол в присутствии эфирата трифторида бора не
образует я-метилфенилзамещенный полиметилен, а подвергается изо-
меризационной полимеризации с получением поли-/г-ксилилена
[157]:
+ - ВРз/Диэтиловый эфир
-CH=N=N
—N2
[-сн2—<О)—сн2-]
165
5.2.1.6. Гидрирование поли-л-ксилилидена
Поли-я-ксилилиден E.3) в результате полимераналогичных
превращений можно восстанавливать этилатом натрия в жидком
аммиаке до поли-/г-ксилилена [158]:
Na/NH3
5.2.2. ПРОМЫШЛЕННОЕ ПОЛУЧЕНИЕ ПОЛИ-л-КСИЛИЛЕНА
В промышленности для получения поли-я-ксилилена в качестве
исходного продукта для пиролиза применяются димеры /г-ксилола
и ди-я-ксилилен. В противоположность пиролизу я-ксилолов при
пиролизе димеров можно получать неструктурированные продукты
с хорошими эксплуатационными свойствами [159—169].
Ди-я-ксилилен (ТПЛ = 284°С) получается путем пиролиза
/г-ксилола в присутствии водяного пара при атмосферном
давлении:
НзС—
—СНз
900
Ди-м-ксилилен очищают перекристаллизацией из ксилола.
Выход л-ксилола составляет примерно 60 %.
Таким методом получается дихлорди-я-ксилилен (исходный
продукт для синтеза полихлор-/г-ксилилена) :
В результате хлорирования я-ксилилена образуется смесь
изомеров дихлорди-я-ксилилена. Так как при пиролизе этой смеси
образуется исключительно хлор-я-ксилилен, то разделения смеси
изомеров не требуется. Отделение дихлорди-я-ксилилена (ГКип
185°С/0,04 мм рт. ст.) от полихлорированных димеров я-ксили-
лена осуществляется перегонкой в глубоком вакууме.
Процесс получения поли-/г-ксилилена из ди-я-ксилилена состоит
из трех стадий: выпаривания димера при 250°С/1 мм рт. ст.,
пиролиза при 600°С/0,5 мм рт. ст. и полимеризации образующегося
166
сап
Рис. 5.5. Технологическая схема получения поли-д-ксилилена [170]:
I _ испаритель B50 °С, 1 мм рт. ст.); 2 — пиролизатор F80 °С; 0,6 мм рт. ст.); 3 — сепара«
тор B5 °С, 0,1 мм рт. ст.); 4 — низкотемпературная ловушка (—70 С); 5 — вакуумный
насос @,01 мм рт. ст.).
в качестве промежуточного продукта я-ксилилена на охлаждаемых
поверхностях при температуре ниже 30°С/0Д мм рт. ст. (рис. 5.5):
—сн2
Этот способ применяется также для получения замещенных поли-
rz-ксилиленов, например поли-3-хлор-гс-ксилилена и поли-3,6-ди-
хлор-я-ксилилена.
На первой стадии испаряют твердый димер в глубоком вакууме
при 250 °С [170]. Ди-гс-ксилилен превращается во время пиролиза
при 680°С/0,5 мм рт. ст. количественно в две молекулы гс-ксили-
лена вследствие разрыва обеих метилен-метиленовых связей.
Образование гс-ксилилена является следствием высокого напряжения
цикла димера и относительно высокой стабильности гс-ксилилена.
При одинаковых значениях времени контакта степень
превращения ди-я-ксилилена в rz-ксилилен составляет при 400°С — 6%, при
500 °С — 50 % и при 680 °С — 100 % -
На третьей стадии процесса происходят одновременно
конденсация паров гс-ксилилена на охлажденных до 25 °С поверхностях
и полимеризация. Для я-ксилилена критическая температура
конденсации, выше которой полимеризация сильно замедляется,
равна примерно 30 °С. Для замещенных гс-ксилиленов это значение
лежит в области более высоких температур: для 2-метил-гс-ксили-
лена 60 °С, для 2-этил-гс-ксилилена 90 °С, для 2-хлор-гс-ксилилена
90 °С и для дихлор-гс-ксилилена 130°С. При конденсации на
охлаждаемых поверхностях на промежуточных стадиях образуются
сначала бирадикалы. На стадии роста цепи происходит
присоединение молекул я-ксилилена к обоим концам цепи, имеющим не-
спаренные электроны. Поскольку полимеризация не
сопровождается реакциями обрыва цепи, то молекулярная масса полимеров
достигает 500 000. Выход продуктов полимеризации увеличивается
167
100
во
60
АО
20
0
—
—
—
—
|
-40
\
V
V
-20 0 20 40 60 t,°C
Рис. 5.6. Зависимость выхода поли-я-ксилилена от
температуры полимеризации [172].
с понижением температуры и достигает
при —50°С 100% (рис. 5.6).
Молекулярная масса образующегося поли-гс-кси-
лилена в области температур от —50 до
+50 °С практически не зависит от
температуры полимеризации [172]. В све-
жеполученном поли-д-ксилилене методом ЭПР показано наличие
5-10~4 моля свободных радикалов на звено.
Для нанесения покрытий используются камеры размером до
35X100 см. Процесс конденсации на охлаждаемых поверхностях
напоминает, в известном смысле, вакуумное напыление металлов.
В противоположность металлизации в вакууме (Р = 10~5 мм рт. ст.)
нанесение я-ксилилена осуществляется при Р = 0,1 мм рт. ст. При
этих условиях длина свободного пробега молекул я-ксилилена
в газовой фазе составляет примерно 1 мм и субстрат наносится
равномерно со всех сторон. Скорость наращивания слоя 4-хлор-я-
ксилилена составляет примерно 5 мкм/мин, я-ксилилен полимери-
зуется немного медленнее.
В промышленности фирма «Union Carbide» осуществляет
нанесение поли-гс-ксилиленового покрытия или только поставляет ди-
меры, а потребители по технологии, полученной по лицензии
(около 10 000 дол.), сами наносят покрытия [171]. Стоимость
установки для нанесения покрытий составляет от 10 000 до 20 000 дол.
Стоимость поли-д-ксилиленового покрытия толщиною 0,01 мм
равна примерно 0,25—0,35 цент/см2.
Пленки из поли-гс-ксилилена получают полимеризацией на
стеклянных пластинках, предварительно обработанных
смазывающими веществами (выделенными из раствора галогенидами
щелочных металлов). Полимерные пленки могут быть сняты после
удаления (путем растворения) смазывающих веществ.
5.2.3. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИ-я-КСИЛИЛЕНА
5.2.3.1. Кристалличность
Кристаллизация поли-я-ксилилена происходит при
температурах ниже температуры стеклования полимера F0—80 °С). Так как
макромолекулы вследствие их незначительной подвижности не
способны к кристаллизации, то кристаллизация поли-д-ксилилена
может осуществляться лишь в процессе полимеризации [172—175].
При полимеризации гс-ксилилена при температурах от —17 до
30 °С образуется поли-я-ксилилен в виде моноклинных
кристаллов а-модификации, причем процессы полимеризации при
одновременном складывании цепей и кристаллизации складчатых
сегментов цепей (длиной приблизительно 80 А) протекают
последовательно. В а-модификации ароматические кольца располагаются
168
параллельно друг другу, угол между плоскостью бензольных
колец и метиленовыми группами равен 90°. Элементарная ячейка
ct-модификации включает два звена. Размеры элементарных ячеек
моноклинной «-модификации составляют а = 5,92 A, 6=10,64 A
и с (ось цепи) = 6,55 A, ? — 134,7° [176]. Теоретическая
плотность составляет 1,185 г/см3. Эти размеры элементарных ячеек
были определены Лизером [177] и Вундерлихом [178].
В интервале температур 220—260 °С а-кристаллы необратимо
превращаются в тригональную ?-модификацию. Эта
кристаллическая модификация образуется также при температурах
полимеризации ниже —78 °С. В области температур от —196 до —78 °С
вследствие низкой скорости полимеризации процессы
полимеризации и кристаллизации протекают одновременно. Размеры
элементарных ячеек тригональной ?-модификации составляют а =
= 20,52 А, с (ось цепи) = 6,58 А [179]. Элементарная ячейка
содержит 16 звеньев, рассчитанное значение плотности составляет
1,153 г/см3. До а — ?-перехода при температурах 220—260°С
?-модификация метастабильна. Этот переход сопровождается
сильным охрупчиванием полимера.
При 270 °С наблюдается обратимое смектическое превращение
?-модификации (?i — ?2-nepexoA).
Из таких растворителей, как а-хлорнафталин, поли-я-ксилилен
кристаллизуется преимущественно в а-модификацию с
незначительной долей ?-кристаллитов [172, 176, 180].
5.2.3.2. Растворимость
Поли-п-ксилилен и полидихлор-я-ксилилен нерастворимы до
температур ниже 200°С в известных растворителях. При
температурах выше 270 °С они растворяются в ароматических и
хлорированных ароматических углеводородах, например в бензилбензоате,
метилбензилфеноле, бромнафталине, а также в хлорированном би-
фениле. При охлаждении растворов ниже 200 °С полимер
осаждается в виде геля.
Поли-п-ксилилен, полученный из четвертичных аммониевых
производных я-ксилола E.2.1.3) или реакцией Вюрца E.2.1.2),
вследствие низкой молекулярной массы растворяется лучше, чем
полимер, полученный пиролизом я-ксилола или ди-гс-ксилилена.
Вискозиметрические исследования растворов поли-я-ксилилена
были проведены Шефгеном [181, 182] и Елинеком [183].
Приведенная вязкость образцов промышленного поли-я-ксилилена в
бензилбензоате при 298°С под N2 равна 1,30 A00 мл/г) при
концентрации 0,065 г/100 мл.
Поли-3-хлор-гс-ксилилен и поли-3-бром-я-ксилилен обладают
повышенной растворимостью. Вязкость их растворов в хлорнафта-
лине может быть измерена при 150 °С.
Замещение алкилами приводит к значительному улучшению
растворимости поли-я-ксилиленов. Поли-4-метил-гс-ксилилен
растворим в нагретом толуоле, а поли-м-бутил-я-ксилилен растворим
даже при комнатной температуре в хлороформе [169].
169
5.2.3.3. Термическая и радиационная стойкость
Применение поли-л-ксилилена при повышенных температурах
ограничивается стабильностью относительно слабых дибензиль-
ных групп в полимерных цепях. По термостойкости поли-л-ксили-
лен уступает поли-л-фенилену [202]. Верхняя температура
длительной работоспособности поли-л-ксилилена и полихлор-я-ксили-
лена, полученного пиролизом в вакууме ди-м-ксилилена или ди-
хлор-ди-/г-ксилилена, в инертной среде составляет 220 °С, на
воздухе для полихлор-/г-ксилилена эта температура равна 130 °С,
для поли-/г-ксилилена составляет 100°С [170].
При деструкции в вакууме разрушение полимера происходит
вначале по слабым местам в цепи, таким, как разветвления.
Вследствие выделения летучих продуктов деструкции ускоряется
процесс разрыва цепей по закону случая. Параллельно с этим
протекают реакции, сопровождающиеся отщеплением водорода и
образованием сопряженных структур [183]. При пиролизе поли-л-
ксилилена в вакууме при 515 °С выделяются также газообразные
продукты: водород, метан, этилен и этан (рис. 5.7). В качестве
нелетучих продуктов деструкции образуются толуол, /г-ксилилен,
/г-этилтолуол, 1,2-ди-я-толилэтан и 4,4'-диметилстильбен. Согласно
кинетическим исследованиям Елинека [184], /г-ксилилен
отщепляется только после протекания определенного индукционного
периода. /г-Ксилилен образуется путем деструкции и гидрирования
олигоксилиленов, которые в свою очередь являются продуктами
термических превращений. Жидкие продукты — толуол и м-этил-
толуол образуются в результате протекания процесса деструкции
по следующему механизму:
НзС
НзС—/Q) + Н3С-СН2
—сн3
H
—сн3
Этилен и этан выделяются при деструкции в вакууме на
следующих стадиях реакции:
СНз + НзС-СНэ
170
О 20 40 60 80 WO 120 т,мин
Рис. 5.7. Зависимость выхода газообразных А'Ю~3, М0/!ь/з6ено
продуктов разложения при пиролизе поли- .__
я-ксилилена в вакууме при 515°Сотвре- /ии
мени выдержки [184]:
I — метан; 2 — водород; 3 — этилен; 4 — этан.
Энергия активации термической
деструкции поли-/г-ксилилена в
вакууме составляет 45 ккал/моль.
Термостойкость поли-/г-ксилиле-
на, полученного пиролизом ди-/г-
ксилилена или /г-ксилола, выше по сравнению с аналогичными
полимерами, которые получены по реакции Вюрца E.2.1.2) или
из четвертичных аммониевых производных м-ксилола E.2.1.3).
Термическое разложение полимеров, полученных пиролизом,
начинается при 323°С, полимеров — продуктов реакции Вюрца —
при 245°С и полимеров, полученных из /i-ксилолтриметиламмо-
нийхлорида, — при 302 °С.
При термоокислительной деструкции поли-/г-ксилилена в
области температур 420—480 °С появляются также СО, СО2 и Н2О.
Пероксиды, образующиеся в начале деструкции, способствуют
разрушению полимерных цепей с образованием промежуточных
продуктов— альдегидов, кетонов и кислот, которые затем декарбони-
лируются или декарбоксилируются:
00.
—CH2-CH2-(Q)-
|о2
о2
1Н
ООН
О
I!
-сн + • он + • сн2-
(CjS
¦-©
+ СО
о
-с
о2
о
н2о
(О)—с—°"h0H
Термоокислительная деструкция поли-я-ксилилена, полученного
пиролизом ди-/г-ксилилена или /г-ксилола, начинается на воздухе
при 300 °С. При 580 °С потеря массы составляет 85%. Деструкция
поли-/г-ксилилена, полученного по реакции Вюрца и из /г-ксилол-
триметиламмонийхлорида, начинается на воздухе при 220 и 265 °С
соответственно.
171
ТТоли-л-ксилилены характеризуются высокой стойкостью к
действию ионизирующего излучения (в вакууме). Прочность при
растяжении и электрические свойства не меняются при облучении
их у-лучами дозой 10 Мрад. Облучение на воздухе приводит к
интенсивному разложению и большой хрупкости полимера [170].
5.2.4. ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА ПОЛИ-га-КСИЛИЛЕНОВ
Поли-я-ксилилены отличаются главным образом
незначительной проницаемостью, хорошими диэлектрическими свойствами
в очень широком температурном интервале, высокой стабильностью
размеров и стойкостью к действию растворителей. Недостатком их
является невысокая температура длительной эксплуатации на
воздухе из-за незначительной стойкости к окислительной деструкции.
5.2.4.1. Теплофизические свойства
Данные об основных теплофизических свойствах поли-я-ксили-
лена, поли-хлор-/г-ксилилена и полидихлор-/г-ксилилена приведены
в табл. 5.1.
По проницаемости водяных паров полихлор-/г-ксшшлен B0 см3-
•см/см2-с-см рт. ст. -1010) аналогичен ПЭВП и лучше таких
обычно применяемых материалов с хорошими защитными свойствами
Таблица 5.1. Теплофизические свойства поли-л-ксилиленов [170, 185, 186]
ТТЛ т/ л ri л фл тт г__
показатель
Температура, °С
плавления
стеклования
Термический коэффициент
линейного расширения а-105, 1/°С
Термический коэффициент
объемного расширения, ?-lO~4 см3/(г/°С)
Коэффициент теплопроводности
Я • 10~4, кал/(см • с • °С)
Удельная теплоемкость, кал/(г¦ °С)
Плотность, г/см3
Показатель преломления nD
Водопоглощение (в течение 24 ч),
Газопроницаемость пленки
толщиной 0,025 мкм при 23 °С, см3-см/
/(см2-с-см рт. ст.-Ю10)
о2
N2 .
Не
н2
со2
нао
Поли-rz-кси-
лилен
405
60-70
6,9
4,3
3
0,20
1,10-1,12
¦ 1,661
0,01
0,24
0,047
2,5
3,2
1,3
57
Полихлор-
гг-ксилилен
280
80-100
3,5
0,17
1,29
1,639
0,06
0,030
0,0036
0,99
0,66
0,046
20
Полидихлор-
п-ксилилен
300
110
Таблица 5.2. Физико-механические свойства
поли-л-ксилиленов [170, 186, 187]
1 9 ^Ч VJ* гъ ^\ rt Т* ^% Tf т_
.¦юкэззтелъ
Прочность при растяжении, кгс/см2
Относительное удлинение при
разрыве, %
Модуль упругости при изгибе,
кгс/см2
Модуль упругости при растяжении,
кгс/см2
при 23°С
при 200°С
Прочность при растяжении
(волокна), г/деньс
Модуль упругости при растяжении
(волокна), г/денье
Твердость по Роквеллу
Износостойкость (Taber CS 17; I кгс)
П0ЛН-/1-КС11-
лклен
630
200
24 500
24 600
17 600
3,5
36
85
8,8
Нолнхлор-
п-ксилилея
910
200
28 000
28 000
17 600
80
22,5
Полндихлор-
«-к си л и лея
400
5—10
28 000
по отношению к водяному пару, как ПВДХ, ПЭТФ и ПТФЭ.
Проницаемость таких газов, как О2, N2, Не или СО2, примерно
такая же, как у ПЭТФ [185].
5.2.4.2. Физико-механические свойства
Поли-п-ксилилены обладают отличной стабильностью размеров
при повышенной влажности воздуха. При выдержке полихлор-я-
ксилиленовых пленок при 40 °С и относительной влажности
воздуха 95% изменение размеров составляет лишь 0,25-10~5 см/см.
При тех же условиях изменение размеров поликарбоната равно
0,70-Ю-5 см/см, а ПЭТФ 3,0-10 см/см [186].
Поли-л-ксилилены вплоть до температур около —165 °С
сохраняют эластичность (табл. 5.2). Пленки из полихлор-/г-ксилилена
толщиной 0,05 мм можно изгибать на 180° при —165 °С шесть раз.
В тех же условиях пленки из ПЭ ломаются после трех, из ПЭТФ
после двух и из ПТФЭ уже после первого изгиба на 180°.
Изменение прочности при растяжении поли-я-ксилиленовых
пленок после теплового старения при температурах от 100 до 175 °С
показано на рис. 5.8. 6,кгс/см2
Термической обработкой поли-п-ксили- р' ¦ -
ленов можно повысить их твердость и
прочность. Это обусловлено увеличением
степени кристалличности и плотности материала.
Рис 5.8. Зависимость прочности при растяжении поли-
л-ксилилена при 100—175°С от времени выдержки
[186].
800
600
400
200
—
—
m
—
<
"с
i
<
\
150\
1
1
\\
\\
120\ \
1оо 4ч
I I
0 0,1 1 Ю Ю0 т,ч
173
Рис. 5.9. Зависимость пробивного напряжения по-
ли-л-кснлиленов от толщины пленки [170]:
/ — полихлор-я-ксилилен; 2 — поли-л-ксилилен.
5.2.4.3. Электрические свойства
Данные о важнейших электрических
свойствах поли-д-ксилиленов приведены
в табл. 5.3.
Характеристики поли-д-ксилилена в
значительной степени аналогичны
соответствующим показателям для ПТФЭ. Поли-/г-ксилилены при
использовании в электронике можно наносить на элементы
конструкций любых размеров в виде очень тонких полимерных
покрытий. При толщине покрытия менее 5 мкм электрическая
прочность полихлор-я-ксилилена выше, чем у полп-л-ксилнлена. На
рис. 5.9 приведена зависимость пробивного напряжения от толщины
пленки поли-я-ксилилена и полихлор-/г-ксилилена.
Снижение электрической прочности после 100 000 ч теплового
старения при 130 °С для полихлор-«-ксилилена составляет 20%.
5.2.4.4. Химическая стойкость
Поли-я-ксилилен, полихлор-л-ксилилен и полидихлор-/г-ксилилен
при температурах ниже 150°С нерастворимы в известных
органических растворителях. Полихлор-д-ксилилен и поли-п-ксилилен
в хлорнафталине растворяются при 175 и 265 °С соответственно.
При комнатной температуре эти полимеры набухают в
ароматических и галогенароматических соединениях.
Таблица 5.3. Электрические свойства поли-л-ксилиленов [170]
Показатель
Диэлектрическая проницаемость
при 60 Гц
при 103 Гц
при 106 Гц
Тангенс угла диэлектрических по-
ТРПК
при 60 Гц
при 103 Гц
при 106 Гц
Удельное поверхностное
электрическое сопротивление, Ом
при 50%-ной отн. влажности
при 90%-ной отн. влажности
Удельное объемное электрическое
сопротивление, Ом-см
Электрическая прочность, кВ/мм
Поли-п-кснли-
лен
2,65
2,65
2,65
0,0002
0,0002
0,0006
МО13
9.10м
1,4-1017
260
Полнхлор-
п-ксилилен
3,15
3,10
2,95
0,020
0,019
0,013
МО14
7-Ю11
8,8-1016
145
Полидихлор-
п-кснлилея
2,82
0,003
174
Образования трещин вследствие внутренних напряжений при
контакте с моющими средствами не обнаружено.
Поли-я-ксилилен устойчив в концентрированной серной кислоте
и 5 н. азотной кислоте.
5.2.5. ПРИМЕНЕНИЕ ПОЛИ-л-КСИЛИЛЕНОВ
Основными областями применения поли-я-ксилиленов является
электротехника и электроника.
Полимеры с такими же хорошими диэлектрическими
свойствами, как, например, полистирол, имеют значительно худшие
защитные свойства по отношению к водяному пару, что не позволяет
наносить гомогенные покрытия на детали.
Печатные и интегральные схемы защищают от действия влаги
и агрессивных сред путем нанесения покрытий толщиной 0,02 мм
из поли-я-ксилилена или полихлор-п-ксилилена. В
полупроводниковой технике поли-п-ксилилен и полихлор-п-ксилилен
используются для пассивации поверхности, а также для защиты от коррозии.
В слоях толщиной 0,4 мкм микропоры отсутствуют. Герметизация
не влияет на электрические характеристики полупроводников.
Использование полихлор-п-ксилилена между пластинами
конденсаторов позволяет при той же емкости уменьшить их размеры
на 20 % по сравнению с конденсаторами с прослойками из
полистирола. Конденсаторы с полихлор-п-ксилиленовой изоляцией могут
длительно эксплуатироваться вплоть до 170 °С [188].
Возможность получения очень тонких однородных пленок с
незначительной газопроницаемостью и высоким пропусканием
мягкого и ультрамягкого рентгеновского излучения позволяет
применять поли-п-ксилилен для изготовления окошек счетчиков
рентгеновского излучения [185].
Мембраны из поли-п-ксилилена можно использовать при
изготовлении детекторов микрометеоритов.
Высокое пропускание в видимой области (90 % у поли-п-ксили-
лена, 95 % У полихлор-п-ксилилена) делает поли-п-ксилилены
незаменимыми материалами для осветления поверхности оптических
линз.
Металлы, чувствительные к влаге, например литий, могут
сохраняться ограниченное время благодаря поли-п-ксилиленовым
покрытиям [189—191].
При нанесении покрытий на теплообменники повышаются их
коррозионная стойкость и производительность, так как водяная
пленка на поверхности быстро переходит в капли. При наличии
достаточных мощностей для нанесения покрытий установки для
опреснения морской воды можно оснащать теплообменниками с по-
ли-п-ксилилеиовыми покрытиями.
5.2.6. МОДИФИКАЦИЯ ПОЛИ-л-КСИЛИЛЕНОВ
Для поли-п-ксилилена характерны те же реакции замещения,
что и для поли-я-ксилола.
175
В присутствии ионов серебра б качестве катализатора
происходит сульфирование полимера концентрированной серной
кислотой. Сульфированный полимер содержит одну сульфогруппу на
звено. Поли-я-ксилиленсульфокислота очень сильно набухает в
воде или разбавленных щелочах, однако в раствор не переходит
[192].
При нитровании поли-я-ксилилена концентрированной азотной
кислотой при 50 °С образуется полидинитро-я-ксилилен.
Нитрованный полимер обладает примерно такой же чувствительностью
к ударному воздействию, как и тетранитрат пентаэритрита и той
же бризантностью, что и тринитротолуол [187].
Хлорирование сульфурилхлорндом при облучении УФ-лучами
протекает преимущественно по этиленовым мостикам между
ароматическими кольцами [129].
При добавлении серы поли-я-ксилилен вулканизуется [193].
Привитые сополимеры поли-я-ксилилена с виниловыми
мономерами можно получать окислением поли-я-ксилилена до пер-
оксидных соединений и прививкой в эмульсии [194].
Привитую сополимеризацию можно проводить также путем
аминирования хлорметилированных поли-я-ксилиленов.
Инициирование протекает по следующему механизму [195]:
-сн2-0-сн2- _ ^ _ +тсу
/
/~~Нн Cl
Г )
сн2 . _
Vh2
п
Этим методом к поли-/г-ксил плену были привиты стирол, метил-
метакрилат, акрилонитрил, акриловая кислота, метакриловая
кислота и акриламид.
5.2.7. СОПОЛИМЕРЫ л-КСИЛИЛЕНА
При сополимеризации я-ксилилена с алкнл- пли галогенпроиз-
водными я-ксилилеиа ниже критической температуры конденсации
мономеров образуются статистические сополимеры, которые по
свойствам занимают промежуточное положение между
соответствующими гомополимерами [127, 129, 187]. Напротив, если
проводить сополимеризацию производных п-ксилилена при
температурах, лежащих между критическими температурами конденсации
обоих мономеров, то происходит лишь полимеризация мономера
с более низкой критической температурой конденсации [196].
Сополимеры с олефинами получаются в том случае, если
раствор я-ксилилена при —78 °С добавляется порциями к нагретому
до 100°С раствору мономера. При температуре —78°С сополиме-
ризация с ненасыщенными мономерами не протекает вследствие
значительно более высокой реакционной способности д-ксилилена
'[131],
176
При сополимеризации я-ксилиленов с диоксидом серы
образуются соответствующие полисульфоны [197]:
:=( \=сн2 + nso2 —> Г—сн2—(О)—сн2—so2—1
\—/ L \N—/ An
Эти сополимеры нерастворимы в растворителях и могут быть
отпрессованы между плитами, нагретыми до 200 °С, при давлении
500 кгс/см2.
Чередующиеся сополимеры получают также с трихлоридом
фосфора, используемым в качестве сомономера [198]:
пН2С=/\=СН2 + яРС13 —> \—СН2—/QN—CH2-PCI3—I
При полнмераналогнчных превращениях, обусловленных соль-
волизом, получаются соответствующие фосфоновые кислоты, хлор-
аигидриды фосфоновых кислот или их сложные эфиры.
Были получены сополимеры с нитробензолом, n-HHTpo3O-N,N-
диметиланилином, а-нитрозонафтолом, хлоранилом и малеиновым
ангидридом [131].
При пропускании кислорода через раствор n-ксилилена при
—78 °С образуется пероксид поли-п-ксилилена:
пО2 + «Н2С=/ \=СН2
Г—О—СН2—(CjS—СН2—О—1
Пероксид поли-гг-ксилилена в сухом виде разлагается со
взрывом [199].
5.2.8. ПОЛИТЕТРАФТОР-ЛМ(СШ1ИЛЕН
В ряду производных n-ксилилена политетрафтор-п-ксилилен
занимает особое место вследствие высоких температуры плавления и
теплостойкости.
Политетрафтор-гс-ксилилен получается по аналогии с поли-п-
ксплнленом пиролизом фторированных производных ксилилена
с одновременной полимеризацией. При пиролизе цнклоди-а,а,а',а'-
тетрафтор-я-ксилилена образуется полимер с выходом 72 % при
те.х?пературе пиролиза 720—730 °С и температуре конденсационной
камеры—35°С [200]:
п
F2C ^Q) CF
=( )=CF2
FoC—(( Y)—CF
\77
По другому способу политетрафтор-я-ксилилен получают
пиролизом а,а'-бис(алкилсульфонил)-а,а,а'а'-тетрафтор-/г-ксилилена:
CF2—SO2—R CF2
—2SO2; —2R
[-CF2—@)—CF
CF2—SO2—R
CF2
Ниже приведены свойства политетрафтор-я-ксилилена [200] :
Диэлектрическая
проницаемость при 60 Гц .... 2,36
Тангенс угла
диэлектрических потерь 0,0008
Удельное объемное
электрическое сопротивление,
.Ом-см 1,3-1014
Электрическая прочность,
кВ/мм 250
Температура, °С
плавления 500
стеклования .... 90
Плотность, г/см3 1,506
Прочность при растяжении,
кгс/см2 435
Относительное удлинение
при разрыве, % .... 120
Модуль упругости при
растяжении, кгс/см2 . . 25 300
Физико-механические показатели примерно такие же, как и
у поли-/г-ксилилена. В отличие от поли-п-ксилилена политетрафтор-
я-ксилилен обладает хорошей свето- и теплостойкостью. Пленки из
политетрафтор-п-ксилилена остаются эластичными после облучения
солнечными лучами (инсоляции) в течение 3600 ч. После 3000 ч
выдержки в термошкафу при 250 °С прочность при растяжении
увеличивается с 460 до 750 кгс/см2, а относительное удлинение при
разрыве уменьшается со 120 до 5 %. Возрастание прочностных
характеристик обусловлено повышением степени кристалличности
полимера при термической обработке.
После 3000 ч выдержки при 250 °С происходит незначительное
ухудшение электрических свойств. Тангенс угла диэлектрических
потерь увеличивается с 0,0008 до 0,0009 и электрическая прочность
уменьшается с 248 до 148 кВ/мм.
Политетрафтор-п-ксилилен нерастворим в известных
органических растворителях. В концентрированном растворе едкого натра
и концентрированной азотной кислоте он разлагается.
Полигексафтор-я-ксилилен можно получить пиролизом 4-три-
фторметил-2,3,5,6-тетрафторфенилацетата [201].
5.3. ПОЛИКСИЛИЛИДЕНЫ
HO>-CR=CR-L
Поликсилилидены получают конденсацией дикарбонильных
соединений с реакционноспособными производными ксилола,
разложением замещенных бисдиазоалканов, дегалогенированием либо
дегидрогалогенированием галогензамещенных производных
ксилола.
178
Некоторые замещенные поликсилилидены являются
растворимыми полимерами с температурой плавления 180—250 °С.
Термическая деструкция начинается при температурах выше
400 СС.
Благодаря высокому темновому сопротивлению и хорошей
фотопроводимости замещенные поликсилилидены используются в
качестве полупроводниковых материалов в электрофотографии.
5.3.1. ПОЛУЧЕНИЕ ПОЛИКСИЛИЛИДЕНОВ
5.3.1.1. Взаимодействие ароматических диальдегидов
с бис(трифенилфосфоний)ксилилендигалогенидами
по Виттигу
При действии алкоголята лития бис(трифеннлфосфоний)ксили-
лендигалогенпды превращаются в бис(трифенилфосфораны),
которые сразу же вступают в реакцию поликонденсации с диальдеги-
дами с образованием поликсилилиденов [203—210]:
2Вг"
MeOR
—MeBr
ОН С
-О-
-сно
-О=Р(СвН5)з
Этот метод используется также для получения незамещенных
поликсилилиденов с м- и о-фениленовыми группами.
Поликсилилидены, полученные по способу Виттига, имеют молекулярную
массу около 1500 и содержат довольно большое количество цис-вшп-
леновых групп.
5.3.1.2. Конденсация ксилиленбис(диэтилового эфира
фосфоновой кислоты) с ароматическими диальдегидами
Поликонденсация проводится в присутствии г/?ег-бутилата
калия в таких растворителях, как толуол или ДМФ.
Многоступенчатая реакция протекает через образование ?-гидргксифосфорных
соединений, которые перегруппировываются при выделении
179
анионов фосфорной кислоты в соответствующие поликсилилидены
[211—213]:
С2Н5ОЧ || / У
>-СН2—(О)—сн2-
о
о
ОС2Н5
онс—(( ))—сно
О
QS-c»
сн-
,/
ОС2Н6
о
В отличие от полимеров, полученных по реакции Виттига,
данные поли-я-ксилилидены содержат практически только транс-вини-
леновые группы. В качестве исходных мономеров могут
использоваться такие химически инертные кетоны, как терефталофенон и
изофталофенон [214]. Бис(дифенилфосфиноксиды), используемые
в качестве мономеров, получаются по реакции Арбузова из
сложных этиловых эфиров дифенилфосфиновой кислоты и
соответствующих дигалогенксилиленов [213].
5.3.1.3. Конденсация по Кнёвенагелю ароматических
диальдегидов с ароматическими диацетонитрилами
При конденсации в присутствии алкоголята натрия в
спиртовом растворе получают полиариленцианоэтенилены [215—219]:
Н /7^\ C2H5ONa
пН2С—(
При конденсации о-диальдегидов получаются циклические
продукты:
CN
СНО NC—СН
СНО NC—СН2
NaOR
>¦
An
180
Такую же активность, как дпацетонптрил при конденсации с ди-
альдегидамп, имеют замещенные в ядро диннтроксилнлены [220] :
NO,
NO2
Yj
сн=с н
-сн=сн—
NO2
5.3.1.4. Дегидрогалогенирование ксилилендигалогенидов
По аналогии с методами Хараша при обработке м- или д-за-
мещенного ксилилендихлорпда амидом натрия или алюминия
в жидком аммиаке получают полнксилилидены с выходами,
близкими к количественным. Карбанионы, образующиеся на первой
стадии реакции, алкилируются по галогенметильным группам с
образованием из двух молекул одной и перегруппировываются путем
?-отщепления в поликсилилиден [158, 221—224]:
X—СН2
—вн
х—сн2
-^ X—СН2
х—сн2-
сн-х
сн2—х
х_сн2—(g>—сн=сн
Гсн—/Q)—сн=сн
—X"
СН2—X
-сн2—х -
В"
Г=
В полярных растворителях, например ДМФ или ДМСО,
внутримолекулярное дегидрогалогенирование протекает с образованием
co-галогенхинодиметана, который полимеризуется в поли-а-галоген-
ксилилен и при действии оснований дегидрогалогенируется с
получением поликсилилидена [150, 209,225, 226]:
х-сн
>—сн2-х
н2с
х-сн
сн-х
СН—X
181
в
По этому способу можно получать также замещенные в ядро
поликсилилидены.
5.3.1.5. Конденсация бис(диметилсульфонийтетрафторборатов)
Нагреванием п- или о-замещенных ксилолбис(диметилсульфо-
кийтетрафторборатов) в водном растворе щелочи получаются
поликсилилидены с хорошими выходами [227, 228]. Реакция
протекает через образование промежуточных сульфонийилидов и хино-
диметановых структур.
5.3.1.6. Кислотно-каталитическое разложение
ароматических бисдиазоалканов
Дифенилзамещенные поликсилилидены можно получить по
Сметсу [229] путем поликонденсации ароматических
бисдиазоалканов, катализируемой кислотами:
N2C—|
¦CNS
-N2
По этому способу можно синтезировать полимеры с молекуляр*
ной массой до 5000.
5.3.1.7. Реакции дегалогенирования
Путем дегалогенирования в присутствии металлоалкиленов из
я-бис(дибромметил)бензола [230], гс-бром-а,р-фтор-р-хлорстирола
и /г-бром-а-хлор-р-дихлорстирола [231, 232], а также хлорзамещен-
ных дисульфонийпроизводных «-ксилола [233] можно получать
соответствующие поликсилилидены, например
CF=CF—Cl
C4H9Li
182
5.3.2. СВОЙСТВА ПОЛИКСИЛИЛИДЕНОВ
Все описанные выше поликсилилидены представляют собой
относительно низкомолекулярные продукты. Наиболее высокие
молекулярные массы (около 5000) были достигнуты при
конденсации ксилиленбис(диэтилового эфира фосфоновой кислоты) E.3.1.2)
или при кислотно-каталитическом разложении ароматических бис-
диазоалканов E.3.1.6).
Температура плавления, а также растворимость незамещенных
поликсилилиденов определяются долей цмс-виниленовых групп,
а также типом замещения фениленовых групп. Незамещенный по-
ли-гс-ксилилиден — неплавкий и нерастворимый продукт.
Поликсилилидены, в которых соседние фениленовые группы соединены
в о,о-, Oyti- и ж,/г-гюложениях, нерастворимы и неплавки, если связь
осуществляется через транс-вини леновые группы (получение по
5.3.1.2). При содержании в цепи цмс-виниленовых групп —
получение полимеров по реакции Виттига E.3.1.1) — образуются плавкие
растворимые поликсилилидены. Если у виниленовых групп имеются
боковые фениленовые группы (получение по 5.3.1.2 или
соответственно 5.3.1.6), то поликсилилидены растворяются в обычных
растворителях, например бензоле, хлороформе и диоксане, и плавятся
в интервале температур 180—250 С. Полиариленцианоэтилен,
содержащий только /г-замещенные фениленовые группы, не плавится
и не растворяется, в то же время ж-замещенные аналоги
представляют собой плавкие и растворимые продукты.
Незамещенный поли-/г-ксилилиден в инертной среде и на
воздухе стабилен примерно до 400 °С в то время как на воздухе при
температуре выше 400 °С наблюдаются интенсивные потери массы,
которые при 525°С достигают 100 %, в инертной среде масса
остатка при 900 °С составляет 55 % [158].
Поли-сс-циано-/г-ксилилиден стабилен в инертной среде и на
воздухе вплоть до 500 °С. При термическом старении при 500 °С
потери массы составляют после 50 мин 15 % и после 100 мин
25 %. Из анализа ИК-спектров образцов, обработанных при 550 °С,
следует, что цианогруппы циклизуются частично в иминоинденовые
или соответственно в хинолиновые группы [216] :
NH
\
чсн_/о\
Влияние структуры на спектральные характеристики, а также
электропроводность и фотоэлектрическая проводимость
поликсилилиденов были исследованы Херхольдом [209, 213, 218, 234].
183
Максимумы поглощения поли-я-ксилилидена определяются типом
замещения фениленовых колец, природой заместителей в фениль-
ных ядрах и виниленовых группах, а также долей цис-виниленовых
групп. Максимум поглощения для незамещенного поли-я-ксилили-
дена находится примерно при 2350 см-1, полимер имеет желтую
окраску. Введение о- или >г-фениленовых групп прерывает цепь
сопряжения, что приводит к гипсохромному сдвигу. Поли-л*- или
поли-о-ксилилидены являются бесцветными продуктами. Наличие
в «-фениленовых кольцах таких сильных электронодонорных
заместителей, как метоксигруппы в 2,5-положенни, вызывает батохром-
иый сдвиг от желтого к темно-красному цвету. Незначительный
батохромный сдвиг к оранжевому цвету происходит при введении
цианогрупп в виниленовые группы. Часть цис-виниленовых групп
в цепи обусловливает незначительный гипсохромный сдвиг в связи
с уширением полосы поглощения к коротким длинам волн.
Поликсилилидены характеризуются очень высоким темновым
сопротивлением при комнатной температуре. Энергия активации
темновой проводимости Ed в соответствии с уравнением
при замещении в фениленовых и виниленовых группах уменьшается
от начального значения ED = 2,45 эВ, характерного для поли-гс-
ксилилидепа, до Ed = 1,94 эВ—для поли-гс-2,6-диметокси-ос-циан-
окснлилпдена.
Электропроводность поликсилилиденов сильно увеличивается
при экспонировании, при этом наблюдается ярко выраженный
внутренний фотоэффект с малыми временами нарастания и затухания.
Без введения примесей фотопроводимость сильно смещается в
видимую область спектра. К батохромному сдвигу длинноволновой
границы фотопроводимости приводит введение в фениленовые
группы в качестве заместителей метоксигрупп или цианогрупп в
виниленовые. Максимум фотопроводимости на длинноволновой
границе поглощения находится при 500—600 нм. Максимум полосы
поглощения совпадает с максимумом фотопроводимости.
Высокое темновое сопротивление и хорошая фотопроводимость
замещенных поликсилилиденов положены в основу успешного
использования их в качестве светочувствительных
(фотоэлектрических) полупроводниковых материалов, пригодных для целей
электрофотографии. В этой области поликсилилидены могут заменить
такие высокоомные неорганические полупроводники, как оксид
цинка или селен.
5.4. ПОЛИБЕНЗИЛЫ
Полибензилы получают поликонденсацией бензилгалогенидов
или я-ксилилендигалогенидов, либо простых диэфиров п-ксилилен-
гликоля с ароматическими углеводородами по Фриделю — Крафтсу.
134
Линейные полибензилы являются аморфными веществами. Их
гомологи — полнизопропплидены стабильны на воздухе вплоть до
450°С и обладают хорошими эксплуатационными свойствами при
температурах до 150 °С.
Форполимеры иа основе фенолов и простых диметиловых эфи-
ров п-ксилиленгликолей используют в промышленности в качестве
связующих при получении композиционных материалов. Отверж-
денные материалы на основе полиоксибензила и стеклянного
волокна могут длительно эксплуатироваться до 200—250°С. Они
применяются в электротехнике и химическом машиностроении для
изготовления элементов конструкций, подвергающихся большим
тепловым и механическим нарушениям.
5.4.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗИЛОВ
5.4.1.1. Кондесация бензил галогенидов
по реакции Фриделя — Крафтса
Полибензил был впервые получен в 1885 г. Фриделем [234] при
действии хлорида алюминия на бензилхлорид:
AlCla Г
В дальнейшем реакцию Фриделя — Крафтса с бензилгалогени-
дами изучали Ингольд [235], Якобсон [236], Бецци [237], Шринер
[238], Валэнтайн [239], Кеннеди [240], Браун [241] и Хаас [242],
которые показали, что конденсация протекает по различным
положениям ароматического ядра. Образующиеся полибензилы
являются сильно разветвленными низкомолекулярными аморфными
полимерами с температурой размягчения 70—80 °С.
Линейный замещенный полибензил получается нагреванием
хлорметилдурола при 100 °С в присутствии Fe2O3 [242]:
С3Н4 /СН
тт //Y4 pu Г-1 Fe2°3
n H—\\)/—CH2C1
НзС
"НзС
п
Бисхлорметилированные ароматические углеводороды
конденсируются с ароматическими соединениями в присутствии тетра-
хлорида олова с образованием линейных олигомерных бензилов,
которые при избытке бисхлорметилированного компонента
способны отверждаться с получением сетчатых полимеров [243—247] :
—Q)?+ «cich2—<jQ>—ch2ci
?
ClCHa— (О/ СНгС1
An SaCli
Сетчатый полибензил
185
1,4-БисB-хлоризопропил) бензол конденсируется с ароматиче
скими углеводородами под влиянием кислот Льюиса при комнат
ной температуре с образованием полимеров, содержащих изопро
пиленовые и индановые группировки [248, 249] :
СНз СНэ
2п Cl—С (Си С—Cl + п (CS
I v^y I W—>v
СНз
ч
\Г.Н—
^СН
СНз
СНз
СНз
СНз
CH3J
п
Димеризация 1,4-бисB-хлоризопропил) бензола с образованием
индановой структуры подавляется использованием
соответствующих катализаторов, а именно тритилгексафторарсената — хлорида
алюминия — нитробензола, и проведением реакции при
температуре ниже —20 °С в хлорированных растворителях, например ме-
тилхлориде, метиленхлориде или хлорбензоле. В этих условиях
образуются высокомолекулярные полиариленизопропилидены с
температурой стеклования 130—300 °С и хорошими
эксплуатационными свойствами [249] :
СНз
СНя
-НС1
СНз
СНз "я
5.4.1.2. Конденсация простых диэфиров п-ксилиленгликоля
с ароматическими углеводородами по Фриделю — Крафтсу
Диэфиры /г-ксилиленгликоля конденсируются с ароматическими
углеводородами в присутствии тетрахлорида олова с отщеплением
спирта и образованием олигомерных метилароматических
углеводородов [250, 251]:
н,с-о-сн2—
—сн2-о-сн3
°, SnQ4
186
Отверждение олигомеров с образованием сетчатых структур
происходит в присутствии таких эффективных катализаторов Фри-
деля— Крафтса, как хлорид железа(III) [252].
5.4.1.3. Конденсация я-ксилилендигалогенидов или диэфиров
/г-ксилиленгликоля с фенолами по Фриделю — Крафтсу
Ксилилендигалогениды или диэфиры ксилиленгликоля
конденсируются с фенолами в присутствии катализаторов Фриделя —
Крафтса с образованием полигидроксифенилиденов [253—254]:
ОН
/Р^\ f, SnCl4
НзС—О—СН2—(Г))—СН2—О—СНз -
ГН- ' fil
—СН
Вначале под влиянием кислот Льюиса ксилилендигалогениды
или диэфиры ксилиленгликоля ионизируются, затем происходит
электрофильное замещение в феноле образующимися ионами кар-
бония.
Олигомерные гидроксифенилидены этого типа изготавливаются
в промышленном масштабе с 1969 г. фирмой «Midland Silicones»
(Barry, Англия) под маркой «Ксилок 210» в качестве связующих
для композиционных материалов. Отверждение олигомеров
(молекулярная масса 600—800), нанесенных на армирующее волокно,
происходит при нагревании с гексаметилентетрамином [253]. При
этом образуются сначала диметиленаминовые мостики, которые
при повышенных температурах превращаются в стабильные мети-
леновые и азометиновые группы. Особенности переработки таких
связующих рассмотрены в 5.4.3.
5.4.1.4. Дегидрирование полиметиленциклогексана
Полиметиленциклогексан, синтезированный циклополимериза-
цией гексадиена-1,6, можно каталитически дегидрировать в
присутствии палладия или платины, а также дегидрировать
химическим путем при действии перхлората калия, получая растворимые
полибензилы [255] :
5.4.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИБЕНЗИЛОВ
Полибензилы, полученные конденсацией бензилгалогенидов по
Фриделю — Крафтсу E.4.1.1), представляют собой сильно
разветвленные аморфные полимеры, которые размягчаются при 70—
187
80 С [242]. Они растворимы б ароматических и хлорированных
растворителях, таких, как бензол, толуол, тетрахлорид углерода
и хлороформ, но нерастворимы в метаноле, петролейном эфире
и ацетоне.
Полиариленизопропилидены, полученные поликонденсацией по
Фриделю — Крафтсу при температурах ниже —20 °С E.4.1.1),
имеют температуру стеклования от 130 до 300 °С [249].
Прозрачные полимеры стабильны в инертной среде до 500 °С и на воздухе
до 450 °С. Они растворяются в метилхлориде, метиленхлориде,
хлорбензоле и о-дихлорбензоле.
5.4.3. ПЕРЕРАБОТКА И ЭКСПЛУАТАЦИОННЫЕ СВОЙСТВА
МАТЕРИАЛОВ НА ОСНОВЕ ПОЛИБЕНЗИЛОВ
Полиариленизопропилидены, полученные поликонденсацией по
Фриделю — Крафтсу, могут перерабатываться из растворов в
хлорбензоле или дихлорбензоле в прозрачные термостойкие
пленки. Свойства образцов полиариленизопропилиденов на основе
1,4-бисB-хлоризопропил)бензола и дифенилоксида приведены
ниже [249]:
СНз
СНзп
-©>—Ш-Ш-
Температура стеклования,
°С
Прочность ' при
растяжении, кгс/см2
Относительное удлинение
при разрыве, % . . . .
Модуль упругости при
растяжении, кгс/см2 ....
152
810
20-30
25 300
СНз
Модуль упругости при
изгибе, кгс/см2
при 0°С
при 25 °С
при 50 °С
при 100°С . . . .
при 125°С . . . .
при 150°С . . . .
37 100
35 000
33 500
30 400
28 800
20 700
Олигомерные продукты конденсации фенолов с диметиловыми
эфирами ксилиленгликоля E.4.1.3) выпускаются в
промышленности фирмой «Midland Silicones» под маркой «Ксилок 210» в
качестве связующих для композиционных материалов. Слоистые
пластики на их основе получают пропиткой армирующих волокон
60 %-ным раствором полигидроксибензила, предварительным
отверждением при 120—130 °С, прессованием препрегов при 150—
175°С и давлении 7—70 кгс/см2 и последующей термообработкой
при температурах, лежащих в интервале 175—250 °С [256—258].
В качестве армирующих материалов используют
преимущественно стеклянные, асбестовые и углеродные волокна. Свойства
полимерной матрицы можно варьировать в широком интервале
введением дифенолов, алкилфенолов, нафталина, дифенилоксида
или дифенилсульфона.
Материалы на основе полигидроксилбензилов и стеклянных
волокон аналогичны по термостойкости полиимидным материалам.
Прочность при изгибе снижается на 50 % только после 2000 ч вы-
188
Таблица 5.4. Свойства композиционных материалов на основе
полигидроксибензила [256]
Показатель
Армирование
стеклотканью
Армирование
асбестовой
тканью
Армирование
6Э% (объеми.)
углеродных
волокон
Плотность, г/см3
Прочность, кгс/см2
при растяжении
при 25°С
при 250 °С
при изгибе
Прочность при сдвиге (в
горизонтальном направлении), кгс/см2
при 25 °С
при 230°С
Модуль упругости, гкс/см2
при растяжении
при изгибе
Ударная вязкость по Изоду,
кгс-см/см2
Твердость по Роквеллу
Горючесть
Электрическая прочность, кВ/мм
Диэлектрическая проницаемость в
сухом состоянии при 1 МГц
Тангенс угла диэлектрических
потерь в сухом состоянии при 1 МГц
1,77
4420
3066
3,8- 105
105
M 120
Самогасящийся
28—34
4,77
0,0107
1,65
1357
1913
1,4- 105
19,7
R 120
Самогасящийся
1,48
1720
821
492
1,41 ¦ 105
держки при 250 °С, а при 300 °С такое уменьшение прочности
наблюдается через 300 ч. Свойства композиционных материалов
на основе полигидроксибензилов приведены в табл. 5.4.
Прочность при растяжении стекловолокнистых материалов, равная
4430 кгс/см2, уменьшается при 250 °С на 30 %. Материалы
устойчивы при комнатной температуре к действию большинства
органических растворителей, кислот и щелочей. При 90 °С в
присутствии кислот и оснований механические характеристики
ухудшаются. Материалы на основе полигидроксибензилов и стеклянных
волокон обладают исключительно высокой стойкостью к
ионизирующему излучению. При облучении у-лучами дозой 4000 Мрад
прочность при изгибе повышается на 11 %, а модуль упругости
при изгибе — на 1 %. Композиционные материалы на основе
сетчатых полигидроксибензилов и стеклянных волокон применяются
в химическом машиностроении и электротехнике (детали насосов
для транспортирования смесей хлорсилан — бензол не
подвергаются коррозии после 9 мес. эксплуатации). Они являются
идеальными материалами для подшипников в ядерной технике. В
электротехнике они находят применение при изготовлении
переключателей высокого напряжения, трансформаторов и печатных
схем.
189
Заготовки на основе армированных асбестовыми волокнами
полигидроксибензилов лучше обрабатываются резанием, чем стек-
ловолокнистые материалы. По сравнению с асбонаполненными фе-
нолоальдегидными смолами рабочая температура элементов
конструкций в химическом машиностроении (соединительные фланцы,
фиттинги для трубопроводов или погружаемые трубы) из этих
материалов может быть повышена со 175 до примерно 250 °С.
Асбонаполненные материалы используются также в качестве
антифрикционных материалов для подшипников, работающих без
смазки до 200 °С.
Материалы на основе полигидроксибензилов и углеродных
волокон используются для изготовления элементов конструкций в
авиации и космонавтике, работающих при повышенных
температурах под нагрузкой.
Литьевые полигидроксибензилы, наполненные асбестовой
мукой, обладают такими же реологическими характеристиками и
поведением при отверждении, как и аналогичные материалы на
основе литьевых фенолоальдегидных смол. Долговечность (при
250 °С) формованных изделий, полученных литьем под давлением
или литьевым прессованием, в 8 раз больше, чем
соответствующих материалов на основе фенолоальдегидных смол.
Г
6.5. ПОЛИАЗОМЕТИНЫ
=С R—\О)~С R=N-Ar-N=
Полиазометины получаются поликонденсацией ароматических
дикарбонилсодержащих соединений с ароматическими диаминами
или обменными реакциями.
Эти полимеры перерабатываются почти исключительно по
рабочему циклу, предложенному Д'Алелио [259].
Сопряженные полиазометины стабильны на воздухе до 500 °С
и в инертной среде вплоть до 1000 °С. Радиационно-химическая
деструкция наступает при дозах облучения, равных 200 Мрад
(у-излучение) или 1000 Мрад (облучение электронами).
Сетчатые полишиффовы основания и полиазины представляют
интерес как коксующиеся абляционные материалы.
К ароматическим полиазометинам относятся полишиффовы
основания, поликетанилы, полиальдазины и поликетазины общей
формулы
[-R-CR'-N-]*
Необычайная стабильность этих полимеров может быть
обусловлена тем, что низкомолекулярные азометины, такие, как
С6Н5—CH=N—С6Н5 или С6Н5—N=CH—СвН4—GH=N—С6Н5
перегоняются без разложения при атмосферном давлении/
Полишиффовы основания (R = Н) и поликетанилы (R = ал-
кил; арил) представляют собой продукты поликонденсацин аро-
190
матических гс-дикарбонилсодержащйх соединений с ароматически
ми /i-диаминами или продукты гомополиконденсации
ароматических л-аминокарбонилсодержащих соединений:
г R R 1 Г
L=C—Ar— C=N—Ar'—N= J L
=N—Ar—C=
При взаимодействии соответствующих ароматических /2-дикар-
бонилсодержащих соединений с гидразином получают полнальда-
зины (R = Н) или поликетазины (R = арил, алкил):
R R -I
=С—Ar— C=N—N=in
5.5.1. ПОЛИШИФФОВЫ ОСНОВАНИЯ
=CH—(ГУ)—CH=N—R—N=
L V4—•/ An
Полимерные шиффовы основания получают поликонденсацией
ароматических диальдегидов с ароматическими диаминами или
ароматическими дитиоизоцианатами, а также путем обменной реакции
Шиффа под действием оснований (альдегидного, аминного,
двойного обмена).
Прямой конденсацией ароматических диальдегидов с
диаминами Марвел впервые в 1957 г. получил полимерные шиффовы
основания [260—262]:
?OHC—<( ))—CHO + rcH2N—<( )>—NH
—н2о
Поликонденсацию можно проводить в таких растворителях, как
бензол или толуол, при непрерывной отгонке азеотропной воды
[263—268] или в расплаве [264]. По этому методу максимальная
степень полимеризации равна 10.
Почти количественные выходы достигаются при проведении
поликонденсации в растворе в бензилиденанилине или в анилине
и бензальдегиде [269].
Наряду с диальдегидами при поликонденсации с
ароматическими диаминами или дибензилиденарилендиаминами могут
использоваться диацетали [270, 271]:
4 y
п VH—Ar'—Crc +/zH2N—Ar"—NH2
~ROH
—^ [=CH—Ar'—CH=N—Ar"— N=]ft
Конденсация диальдегидов с ароматическими
дитиоизоцианатами имеет то преимущество по сравнению с диаминами, что
191
реакция необратима, причем единственным низкомолекулярным
продуктом реакции является газообразный диоксид серы [272,273] :
/^чЧ /s~^\ Пиперидин
понс—(ГУ)—сно + no=s=N—<^Q)—n=s=o —_S02 >
Поликонденсация, катилизируемая пиперидином, протекает
через образование комплекса основания ампнного типа с дитиоизо-
цианатом, который, присоединяясь к альдегиду с отщеплением
катализатора и диоксида серы, дает полиазометин:
—(( ))— N—S
-О-
¦СНО
+
H
"¦ \O)~"N=CH~
H H
Уменьшение реакционной способности амино- или альдегидных
групп при прямой поликонденсации диаминов и диальдегидов
компенсируется также использованием при синтезе полиазометинов
обменной реакции Шиффа под влиянием оснований. Аминные
компоненты в основаниях Шиффа могут быть количественно заменены
другими аминами, если летучесть отщепляющегося амина выше,
чем летучесть амина, который должен входить в состав
полимерного шиффового основания [264, 274, 275] :
nH2N—Ar—NH2 + лС6Н5—N=CH—Ar'—CH=N—C?H5
Bn - 1)C6H5—NH2 + C6HSN[=CH—Ar'—CH=N—Ar—N=]nH2
Аналогично протекает обмен под влиянием альдегидов [264,
275]:
/Ю=СН—Ar—СН=О + пСвНь—CH=N—Ar'—N=CH—C6H5
ii
B/г ~ 1)С6Н5—СНО + С6Н5—CH[=N—Ar'—N=CH—Ar—CH=]nO
При реакции двойного обмена могут быть заменены
альдегидные и аминные функции двух различных шиффовых оснований
192
[264, 276] :
raC6H5№=CH—Ar'—CH=N- C6H5 + «С6Н5—CH=N~Ar~N-=CH—C6H5
li
Bл- l)CeH5CH=N—C6H5 + С6Н5—N[=CH—Ar'—CH=N—Ar—N=]„CHC6H5
Наряду с альдегидами в реакции обмена можно применять
соответствующие ацетали [271]:
m(ROJ=CH—Ar'—СН—(ORJ + пАт—CH=N—Ar"—N^CH—Ar
I
Bй - l)ArCH(ORJ + (ROJ[=CH—Ar'—CH=N—Ar"—N=]„CH—Ar
Обменные реакции проводят в растворе или расплаве. Наиболее
высокие молекулярные массы полимеров достигаются при
проведении реакции в расплаве.
Сетчатые полишиффовы основания получаются при
использовании трифункциональных компонентов (трикарбонильных
соединений, триаминов) [275, 277].
Для гомополиконденсации ароматических аминоальдегидов в
настоящее время используют лишь изомерные аминобензальде-
гиды [264]. м- и /г-Аминобензальдегиды можно получать лишь в
виде тримеров, которые в бензилиденанилине образуют теломеры.
Поликонденсация о-аминобензальдегида осуществляется в
расплаве при 300 °С.
Свойства полишиффовых оснований определяются строением,
методом синтеза, а также их последующей термической
обработкой.
Полишиффовы основания, которые состоят только из
ароматических колец, соединенных в ^-положении азометиновыми
группами, например иоли-гс-ксилилиден-гс-бензидин
представляют собой высокосопряженные системы и имеют при
достаточной молекулярной массе глубокую черную окраску.
Сопряжение нарушают путем введения между ароматическими ядрами
диаминов, таких групп атомов, как —(СН2L—. Атомы или группы
атомов, такие, как—О—, —S—, —NH—, —SO2— или —СН2—,
в известной мере принимают участие в образование цепи
сопряжения («псевдосопряженные полишиффовы основания»).
Снижение сопряжения происходит также при наличии ароматических
колец, соединенных в ж-положении [276].
При переходе от поли-п-ксилилиден-/г-фенилендиамина (I) к
поли-м-ксилилиден-ж-фенилендиамину (II)
(О
7 Зак. 18 1Q3
,CH=NV
(П)
-¦rt
при степени полимеризации 6 происходит батохромный сдвиг
максимума полосы поглощения в УФ-спектре с 317 до 253 мкм.
Несопряженные полишиффовы основания, которые получаются
поликонденсацией в растворе или реакцией двойного обмена,
представляют собой продукты от белого до кремового цвета,
стабильные в инертной среде до 300 °С и на воздухе — до 250 °С [264].
Поликонденсация в растворе сопряженных или
«псевдосопряженных» диаминов с терефталевым альдегидом приводит в итоге
к получению низкомолекулярных хрупких неплавких полимеров
желтого цвета. Те же мономеры дают возможность при проведении
поликонденсации в расплаве получать высокосопряженные
полишиффовы основания черного цвета.
Метод двойного обмена, имеющий лучшую воспроизводимость,
чем поликонденсация в расплаве, приводит к образованию
плавких пластичных полимеров, при нагревании которых выше 250 СС
происходит усиление окраски от желтого до оранжево-коричнево-
черного цвета. Усиление окраски и нерастворимость продуктов
реакции черного цвета связаны с интенсивным протеканием
процесса конденсации при удлинении цепи сопряжения.
Термостойкость полишиффовых оснований, полученных
реакцией поликонденсации в расплаве, сильно зависит от
максимальной температуры проведения реакции. Вплоть до 400°С
поликонденсация протекает не полностью, хотя эти полишиффовы
основания (за исключением несопряженных полимеров с
алифатическими мостиковыми группами) стабильны на воздухе до 480 °С и в
инертной среде — до 550 СС. При максимальной температуре
поликонденсации около 600 СС температура разложения на воздухе
полимеров колеблется от 500 до 550 °С. Потеря массы в инертной
среде составляет при 700 °С 5 % и при 1176°С 18 % [264].
При температурах поликонденсации выше 600 °С происходит
отщепление водорода. Это приводит к образованию поперечных
связей между соседними цепями
-н2
=N—(Q—N=CH—
?
—CH=
194
и к внутримолекулярным перегруппировкам в термически
стабильные фенантридиновые и фенантролиновые структуры:
гот
N
_н2
[=сн—<Q>~CH=N—(Q>—N=],, ~
N=
— П
Если максимальная температура конденсации составляет
И7б°С, то образующиеся полимеры стабильны в инертной среде
до 1100 °С и на воздухе приблизительно до 500 °С.
Сшитые полишиффовы основания, полученные при
использовании трифункциональных мономеров, дают при пиролизе высокие
выходы графита и вследствие этого пригодны для использования
в качестве абляционных материалов [275, 277]. Выход графита
пропорционален плотности полимерной сетки.
Наличие в полишиффовых основаниях системы сопряжения
приводит к делокализации энергии у- или электронного
излучения. Термограммы полишиффовых оснований остаются
неизменными после облучения 7-лУчами дозой 200 Мрад или облучения
электронами дозой 1000 Мрад в инертной среде или на воздухе
[278].
5.5.2. ПОЛИКЕТАНИЛЫ
Поликонденсация ароматических диаминов с ароматическими
дикетонами с образованием поликетанилов
»H2N-<Q>—NH2 + „H3C-C-(Q)-C-CH3 —
о
о
L CH3 СНз
протекает значительно медленнее, чем соответствующие реакции
с ароматическими диальдегидами [259].
7*
195
В процессе поликонденсации в растворе ароматических
диаминов с /г-диацетилбензолом образуются нерастворимые олигомеры
черного цвета со средней степенью полимеризации от 3 до 6.
При конденсации в расплаве с дикетонами в отличие от
альдегидов сначала образуются плавкие продукты поликонденсации
черного цвета, которые становятся неплавкими лишь при более
высоких степенях превращения. Процесс поликонденсации в
растворе смеси анилин—ацетофенон или С6Н5 — С(СНз) =N — C6Hs
ускоряется в присутствии каталитических количеств кислот
Льюиса, например хлорида цинка. Аналогично, но с более высокой
скоростью, чем при получении соответствующих полишиффовых
оснований, протекает реакция обмена в расплаве. Пониженная
вязкость расплава поликетанилов по сравнению с полишиффовы-
ми основаниями объясняется наличием боковых алифатических
групп. По термостойкости поликетанилы близки к полученным в
аналогичных условиях полишиффовым основаниям.
Гомополиконденсацию изомерных аминоацетофенонов с
получением кетанилов типа
/Г
СНз
-*п
можно осуществлять в расплаве при 200—300 °С в присутствии
хлорида цинка. Самой незначительной реакционной способностью
обладает /г-аминобензофенон. В растворе поликонденсация не про-
ТвКЭбТ
5.5.3. ПОЛИАЛЬДАЗИНЫ
[=СН—Ar—CH=N—N=]n
Можно предположить, что полиальдазины имеют такую же
термостойкость, как полишиффовы основания, поскольку
сопряжение ароматических колец в них не прерывается вследствие
наличия в цепи азогрупп.
Полиальдазины были впервые получены в 1950 г. Марвелом
^[262] поликонденсацией в растворе ароматических диальдегидов
с гидразином:
nOHC—Ar—СНО + «H2N~NH2 —-* [=СН—Ar—CH=N—N=]„
Низкомолекулярные продукты желтого цвета не
растворяются и не плавятся.
Поликонденсацией в расплаве гидразина с ацеталями или
реакцией обмена с альдегидами или аминами также получают
низкомолекулярные неплавкие продукты [279].
Высокомолекулярные полиальдазины были синтезированы
методом двойного обмена при температурах ниже температуры
разложения мономерных азинов, используемых для их синтеза.
Плавкие полиальдазины черного цвета получают в бензилиден-
анилине поликонденсацией ароматических диальдегидов с
гидразином [263] или реакцией двойного обмена [279],
196
Полиальдазины имеют относительно невысокую
термостойкость. При температурах выше 175°С начинается выделение
азота, которое протекает очень интенсивно в области 370—425°С и
количественно — при 700 °С. При этом начинается превращение
полиальдазинов в полимеры полистильбеновой структуры,
которые в свою очередь конденсируются при 700—1176°С с
отщеплением водорода и образованием полиароматических систем [279] :
Г=СН—(О)—CH=N-N=CH—(О^~ CH=N-N=CH—(Cj)—CH=N-N=]
L \^./ v^*v W/ Jn
—N2
—сн=сн
—сн=1
сн=_„
О превращении соответствующих мономеров дибензилиденазина
в стильбены с отщеплением азота
сообщалось Куртиусом еще в 1889 г. [280].
Благодаря возможности превращения в полиароматические
системы полиальдазины представляют интерес для космонавтики
как карбонизующиеся абляционные материалы.
5.5.4. ПОЛИКЕТАЗИНЫ
Поликонденсацией ароматических дикетонов с гидразином
в растворе получают низкомолекулярные поликетазины желтого
цвета [279]:
иНзС—С—{СЪ—С—СНз + nH2N—NH2 >
II
о
«
о
СНэ
СНа
шп
При взаимодействии с альдегидами по реакции двойного
обмена получают продукты от коричневого до черного цвета,
которые на промежуточной стадии являются плавкими соединениями.
По теплофизическим свойствам поликетазины близки к полиаль-
дазинам.
197
5.6. ПРОСТЫЕ АРОМАТИЧЕСКИЕ ПОЛИЭФИРЫ
И ПОЛИАРИЛЕНСУЛЬФОНОКСИДЫ
Из ароматических карбоциклических цепных полимеров, в
которых ароматические ядра связаны шарнирными атомами или
группами атомов, большое значение как конструкционные
материалы имеют простые полиэфиры и полисульфоноксиды.
Поли-2,6-диметил-1,4-фениленоксид
-(О
/
N
СНз
\
сн3
производится в четырех странах в количестве 50 тыс. т в год.
В смеси с полистиролом его используют как термостойкий
конструкционный материал для длительной работы в интервале
температур 120—200 DC.
Полиметилендифенилоксид
представляет собой термостойкий олигомер, используемый как
электроизоляционный лак или связующее для длительной
эксплуатации до 250 °С.
Полигидроксиэфир типа
СНз
ОН
используют для покрытия металлов или в качестве «сухого» клея
для склеивания металлов. Область длительной работоспособности
находится в интервале температур от —60 до 80 °С. В
промышленности полисульфоноксиды получают из бисфенола А и ди-
хлордифенилсульфона
СНз
СНЭ
п
а также из ароматических дисульфохлоридов и многоядерных
ароматических соединений:
Эти термопластичные конструкционные материалы применяются
в электротехнике, электронике и самолетостроении. Полисульфон-
19а
Оксиды с изопропйлиденовыми группами в цепи используют при
температурах до 150°С, а соответствующие полностью
ароматические полимеры — до 260 °С.
5.6.1. ПОЛЙФЕНИЛЕНОКСИДЫ
Полифениленоксиды получают из галогенфенолятов по Ульма^
ну, гомоконденсацией ароматических диазооксидов или
окислительной дегидрополиконденсацией фенолов. В промышленном
масштабе с 1964 г. производится поли-2,6-диметил-1,4-фенилен-
оксид окислительной дегидрополиконденсацией 2,6-диметилфено-
ла в присутствии медноаммиачного комплекса. Переработку
термопласта осуществляют в интервале температур 250—300 °С. По-
лифениленоксид и его смеси с полистиролом применяются как
термостойкие конструкционные материалы в электротехнике,
электронике, автомобилестроении. Полифениленоксид термостабилен в
процессе длительной эксплуатации при 210 °С, а его смеси с
полистиролом— при 100—145 °С. В смеси с полистиролом его
используют в качестве материала конструкционного назначения.
5.6.1.1. Получение полифениленоксида
Конденсация галогенфенолятов по Ульману. Гюнтер [281]
впервые получил полифениленоксид разложением серебряной
соли галогенфенола:
R
\
—AgX
-°-\Q
о/
n
Аналогично протекает превращение 2,4,4,6-тетрагалогензамещен*
ного циклогексадиенона в присутствии ртути [282] :
Hg
^Вг
вп
\
Br J
п
Из галогенфенолятов щелочных металлов в присутствии в
качестве катализатора медной бронзы Гольден [283] получил ряд
ароматических полиэфиров, таких, как поли-4-хлор-1,4-фениленоксид,
1UJ
поли-2,4-дибром-1,4-фениленоксид и поли-2,3,4,6-тетрахлорфени-
леноксид. Молекулярная масса полимеров составляет 2000, они
размягчаются в интервале 180—250°С.
Полифениленоксид с молекулярной массой до 10 000 получают
конденсацией /г-бромфенолятов щелочных металлов в
растворителях (нитробензоле или пиридине) при ПО—200°С в присутствии
меди или ее солей в качестве катализатора [284, 285].
Конденсация по Ульману галогенфенолятов представляет
собой типичную реакцию нуклеофильного ароматического
замещения, при которой образуются соли Cu+I, играющие роль
катализатора. Реакционная способность галогенфенолятов в ряду
заместителей понижается в следующей последовательности:
I — Вг > Cl !S> F [286]. Высокомолекулярные полифениленокси-
ды образуются при поликонденсации галогенфенолов в щелочной
среде в присутствии таких окислителей, как РЬО2 или K3[Fe(CNN]
[287, 288]:
R
-°н
ОН", Кат
—НХ
\
R
П
Для достижения высоких степеней превращения концентрация
катализатора должна составлять до 10 % (мол.). Таким образом,
получены полимеры из 2,6-диметилфенола, 2,6-дипропилфенола,
2-пропил-6-фенилфенола и др. Для конденсации несимметрично
замещенных галогенфенолов в качестве окислителей можно
использовать пероксиды и персульфаты [290]. По Прайсу [287],
конденсация протекает по радикальному механизму путем
окислительного отщепления галогена:
R
XR
-О'
Предполагается, что на промежуточной стадии образуется хи-
нолэфир.
Разложение бензол-1.4-диазооксидов. При фотохимическом
или термическом разложении бензол-1,4 диазооксидов образуются
феноксидные бирадикалы или цвиттер-ионы, которые рекомбини-
руются в полиэфиры [291, 292] :
200
4
°-\о>
г
-*n
Однако вследствие побочных реакций, например взаимодействия
с растворителем, этим методом получают только
низкомолекулярные полиэфиры.
Окислительная дегидрополиконденсация фенолов. В 1956—
1962 гг. Хэй [293, 294] предложил получать полиэфиры из 2,6-ди-
замещенных фенолов окислительной дегидрополиконденсацией в
присутствии в качестве катализатора комплекса меди с аминами:
«О—он+т°>
СцС]/Пиридин
R
—(С У;—о—
R
n
Этот метод до сих пор является единственным промышленным
способом получения линейных высокомолекулярных
ароматических простых полиэфиров. С 1964 г. он используется фирмой
«General Electric» для получения поли-2,6-диметил-1,4-фениленоксида.
Процесс протекает по ступенчатому механизму путем
окислительной радикальной конденсации. В качестве мономеров для
синтеза линейных высокомолекулярных полиэфиров по способу Хэя
могут применяться только такие производные фенола, которые
содержат заместители в положениях 2 и 6, с размером не больше
определенного, такие производные легко окисляются.
Окислительная дегидрополиконденсация самого фенола приводит к
образованию смолоподобного сшитого продукта [294]. Наоборот, о-крезол
в ацетонитриле или смеси нитробензол — толуол A:3) в
присутствии комплекса медь — пиколин с хорошим выходом превращается
в поли-2-метил-1,4-фениленоксид [295, 296]. Фенолы, используемые
для получения полимеров, приведены в табл. 5.5. Если оба о-заме-
стителя содержат изопропильные или грег-бутильные группы, то
окислительная конденсация подавляется и процесс протекает с
образованием новой С—С-связи в тетраалкилдифенохиноне:
R
он + о2
CuCl/Пиридин
R
*=( ) +2Н2О
R
201
Таблица 5.5. Окислительная дегидрополиконденсация
2,6-дизамещенных фенолов [297]
;—ОН
R
СНз
СНз
СНэ
СНз
СНз
СНз
СНз
R'
СНз
СН2СН3
СН(СНзJ
С(СНзK
QH5
ОСНз
Cl
Основной
продукт
реакции
полиэфир
+
+
+
+
+
+
тетраал-
кнлдифе-
нохинон
+
R
СН2СН3
СН(СН3J
С(СН3)з
Cl
NO2
ОСНэ
R'
СН2СН3
СН(СН3J
С(СН3)з
Cl
NO2
ОСНэ
Основной
продукт
реакции
полиэфир
+
+
тетраал-
килдифе-
нохинон
+
Заместители, повышающие окислительный потенциал фенолов,
замедляют окислительную конденсацию, поэтому реакцию
следует проводить при более высоких температурах. Окислительная
дегидрополиконденсация 2,6-диметилфенола протекает энергично
уже при комнатной температуре; 2-хлор-6-метилфенол реагирует
при 60 °С; для 2,6-дихлорфенола требуется более высокая
температура; 2,6-динитрофенол не вступает в реакцию в условиях
окислительной поликонденсации. Катализаторами для такого процесса
служат системы, включающие соли Си1+ и амины, которые в
присутствии кислорода воздуха превращаются в основные комплексы
Си2+. Из аминов наиболее эффективны производные пиридина.
Алифатические амины пригодны только в том случае, если
содержат по крайней мере один короткоцепной заместитель. В
патентной литературе наряду с системой Си — амин приводятся
также системы с Мп, Со и Pd.
Ниже перечислены каталитические системы, которые можно
использовать для окислительной дегидрополиконденсации
фенолов:
Система
Соль одновалентной меди — амины
Соль одновалентной меди — пиридин
Соль одновалентной меди—пиридин — ацетонитрил
Комплекс медь—поливинилпиридин
CuCl — 2-пиридинон, Ы-метил-2-пиридинон, валеролак-
там
СигО — амнн
CuS — амин
Двухвалентная медная соль органической кислоты—.
амин
Литературный источник
[298-307, 360-362]
[308-311]
[312, 313]
[314, 315]
[316]
[317, 318]
[319, 321]
322-324]
202
Система Литературный источник
Карбонат меди — иод — третичный диамин [325]
Медь — соль двухвалентной меди — кетон — амин [328, 329]
Медь- соль двухвалентной меди—спирт—амин [326, 327]
Медный порошок — FeC!« — пиридин .. . [330]
Соль двухвалентного марганца — амин [331, 332]
Соль двухвалентного марганца — метилат натрия [333, 334]
Ацетилацетонат трехвалентного марганца [335]
Дицнклопентадиенилмарганец [336]
Комплекс кобальт — амин [337, 338]
Ацетат кобальта — N-пропилиденаминофенол [339]
Палладий — оксид алюминия [340]
PdCl2-Fe(NO3K [341]
V2O5 — фосфорная кислота — амин [342J
Комплекс железо — порфирин + соединения Си, Со, Мп [343]
или Ni
Каталитическую активность системы Си — амин можно
повысить добавлением бромидов щелочных и щелочноземельных
металлов [347]. Использование в качестве катализаторов РЮ2,
активированной МпО2 [344], AgO [345] или Ks[Fe(CNN] [346],
приводит к образованию низкомолекулярных полиэфиров с
высоким содержанием тетраалкилдифенохиноновых фрагментов. При
катализе окислительной дегидрополиконденсации фенолов солью
одновалентной меди с амином активным является комплекс Си2+.
Для стадии инициирования предложен следующий механизм:
H H Cl
АгОЧ У°К > /
/ \ / X Медленно у \ ^r *
(Ру)/ Cl Т)Аг (Ру)з ОТ ОАр
H H
l
+2ArOH
-2ArO
ArOv Cl (PyJ HO Cl (PyJ
>< >< 7^ X X
(Py)/ V \>H BblCTf° (PyJ Cl XOH
Лимитирующей стадией реакции является окисление комплекса
Си — фенол — амин. Для получения высокомолекулярных
полиэфиров соотношение амин: Cd должно быть возможно более высоким.
С уменьшением соотношения амин: Си, а также с увеличением
размера аминного лиганда возрастает выход тетраалкилдифенохи-
нона как побочного продукта и уменьшается молекулярная масса
образующегося полиэфира [349, 350]. Если двухъядерный
комплекс меди содержит два аминных лиганда на атом меди, то в
результате переноса электронов легко генерируются арилоксира-
дикалы и становится возможной реакция образования С—О-связи.
2U3
При более низком соотношении амин: медь, например один
пиридиновый лиганд на атом меди в двухъядерном комплексе,
выделение образовавшегося арилоксирадикала затруднено. Хинонный
кислород с атомом одновалентной меди, координационно-связы-
вающим арилоксирадикал, способствует реакции образования
С—С-связи, приводящей к диалкилдифенохиноновой структуре
[348]:
АГО ^О ^Ру
Са2+ CiF -
2АгОН|-2Н2О
Cu2t tu2
Медленно I О2
2*
+ о
При взаимодействии между собой арилоксирадикалов сначала
образуются олигомерные радикалы, которые затем претерпевают
дальнейшее превращение через промежуточную стадию получения
хинонэфиров [351]:
О.
Зависимость количества поглощенного кислорода и
характеристической вязкости полифениленоксида от продолжительности реак-
204
0,2
Рис. 5.10. Зависимость количества поглощенного кис- Уо2,%(уоя)
лорода и характеристической вязкости про окисли- ^г^у - * "у
тельной дегидрополиконденсации 2,6-диметилфенола 8д„ -$$
от продолжительности реакции [297].
ции представлена на рис. 5.10. Характери- J
стическая вязкость становится выше 0,1 при го
поглощении 97 % и более кислорода.
Скорость окислительной дегидрополи- ku 80 12а шг>тн
конденсации не зависит от концентрации
мономера и имеет первый порядок по катализатору [348,352,353].
Кинетика окислительной дегидрополиконденсации 2,6-диалкилфе-
нола отвечает механизму реакции Михаэля — Ментена для энзи-
матических систем которые имеют нулевой порядок по субстрату
и первый порядок по энзиму.
5.6.1.2. Промышленное производство
поли-2,6-диметил-1,4-фениленоксида
2,6-Диметилфенол, являющийся исходным мономером для
синтеза полифениленоксида, получают из крезольной фракции
каменноугольной смолы или алкилированием о-крезола метанолом.
Извлечение иг,я-крезольной фракции — продукта
высокотемпературного коксования каменного угля — производят дистилляцией или
экстракцией [354]. При перегонке лг,я-крезольной фракции 2,6-ди-
метилфенол отбирают в виде первой фракции или азеотропа с
водой. При охлаждении прозрачного крезольного раствора 2,6-диме-
тилфенол кристаллизуется или его извлекают экстракцией
бензолом. Для противоточной экстракции из крезольной смеси
используют систему метанол — парафиновое масло [355].
При получении 2,6-диме .илфенола метилированием крезола
смесь о-крезол — метанол в соотношении 1:1,5 пропускают при
350 °С над активированным силикатом алюминия [356]. В
продуктах пиролиза фенолоформальдегидных полимеров также
содержится 2,6-диметилфенол (т. пл. 49 °С, т. кип. 212 °С). Условия реакции
и основные представления о механизме окислительной
поликонденсации 2,6-диметилфенола описаны ниже. В промышленности
реакцию проводят ступенчато при пропускании воздуха через раствор
2,6-диметилфенола в пиридине при комнатной температуре в
присутствии солей одновалентной меди. Полимер в пиридине не
растворяется, в процессе поликонденсации выпадает из раствора.
Средняя молекулярная масса промышленного полифениленоксида
составляет 20 000—30000. Бесцветный продукт отмывают от
катализатора, обрабатывая его такими соединениями, как тиомочевина
и тиосемикарбазид, образующими с ним комплексы.
Светопроницаемость полифениленоксида может быть повышена обработкой
его раствора в восстановительной среде цинком или алюминием
[364, 365]. Образующийся в качестве побочного продукта тетра-
метилдифенохинон выделяют растворением полимера в толуоле,
205
пропусканием НС1 с последующим отделением динатриевой соли
[363].
Молекулярная масса полимера, получающегося при
окислительной поликонденсации, регулируется добавлением циклических
бифункциональных соединений, содержащих кислород, серу или азот
[366]. Наличие небольшого количества воды не препятствует
окислительной поликонденсации 2,6-диметилфенола в присутствии смеси
соль одновалентной меди — пиридин. До мольного соотношения
пиридин: вода, равного 10:1, реакция ускоряется; при соотношении
1:1 — происходит обрыв. Наличие небольшого количества щелочи
не влияет на скорость протекания реакции в присутствии воды и
метанола. Насыщенные углеводороды и полярные растворители,
такие, как нитробензол, ингибируют эту реакцию [367].
Окислительную дигидрополиконденсацию 2,6-диметилфенола
можно проводить в эмульсии с применением в качестве
катализатора солей одновалентной меди с поливинилпиридином в смеси
вода—бензол. В щелочной среде основным продуктом реакции
является полифениленоксид, а в кислой среде — тетраметилдифе-
нохинон [368].
Процесс окислительной дегидрополиконденсации
2,6-диметилфенола разработан в исследовательской лаборатории «General
Electric». Дальнейшие исследования проводились совместно с
голландской фирмой «N. U. Kunstzijde Union» (AKU), Arnheim.
В США аналогичное производство было организовано в 1964 г. под
названием РРО. В 1966 г. начался выпуск модифицированных
полистиролом материалов норил и арнокс, в 1968 г. — материала,
наполненного стеклянным волокном. Объем производства в 1970 г.
составил 30 000 т в «General Electric» и 10 000 т —в AKU [357].
В 1975 г. объем производства в Голландии составил 20 000 т [358].
Цена 1 кг РРО составляет 11 и 1 кг Норил 6 марок ФРГ. Почти
50 % стоимости приходится на 2,6-диметилфенол. В СССР полимер
производится под названием «Арилокс» [359]. В 1970 г. японская
фирма «Mitsubisi Edogawa» начала производство
модифицированного РРО. Окислительной дегидрополиконденсацией смеси
2,6-диметилфенола с 2-метил-6-аллилфенолом получен полиэфир Родок
[фирма «Rocketdyne» (США).]
5.6.1.3. Сополимеры 2,6-диметил-1,4-фенил оксида
Свойства полифениленоксида могут быть реализованы только
в специальных областях применения. Это обусловило
необходимость тщательного изучения характеристик его композиций с
различными термопластами (табл. 5.6). Такие композиции были
получены главным образом гомогенизацией в термопластичном
состоянии или растворением обоих полимеров, окислительной
дегидрополиконденсацией 2,6-диметилфенола в присутствии
растворенного компонента, например второго мономера, полимеризующегося
по радикальному механизму, или радикальной полимеризацией
винилового мономера в присутствии растворенного полифенилен-
206
Таблица 5.6. Полимерные смеси,
содержащие поли-2,6-фениленоксид
Второй компонент
Свойства смеси
Литератур
ный источник
Материалы получены гомогенизацией в термопластичном состоянии
Тройной сополимер этилена»
пропилена и дициклопеитадиена
Сополимер этилена с винилаце-
татом
Сополимер этилена с этилакри-
латом
Кумароно-инденовая смола
Кумароновая смола
Сополимер метилметакрилата со
стиролом
Сополимер ос-метилстирола с
акрилонитрилом
Сополимер стирола с бутадиеном
Сополимер винилфторида с гек-
сафторпропиленом
Полистирол
Полисилоксан
Поликарбонат
Полигидроксиэфир
Сополимер стирола с циклопен-
тадиеном
Модифицированный каучуком
полистирол
Сополимер стирола, этилена и
силоксана
Пресс-композиция с 5%
сополимера
Деформационная
теплостойкость 190 °С
Прочность при растяжении
682 кгс/см2
Ударная вязкость образцов с
надрезом, 22 кгс* см/см2
Пресс-композиция с 10%
смолы
Модуль упругости при изгибе
27 800 кгс/см2
Прочность при растяжении
870 кгс/см2
Лучшая перерабатываемость
То же
Лучшая перерабатываемость и
повышенная ударная вязкость
Пониженная горючесть
Повышенная эластичность
Повышенная адгезия
Повышенные удлинение и
ударная вязкость
Повышенная ударная вязкость
[369]
[370]
[370]
[371]
[371]
[372]
[372]
[373-375]
[376]
[377, 378]
[379]
380]
381]
371]
[382, 383]
[384]
Материалы получены растворением одного или обоих компонентов
Сополимер стирола с бутадиеном
Смесь полистирола с каучуком
[385]
[386]
Материалы получены полимеризацией мономера
в присутствии растворенного полифениленоксида
Стирол
Замещенный в ядро стирол
[388-391]
[387]
Продолжение
Второй компонент
Свойства смеси
Литературный источник
Материалы получены окислительной дегидрополиконденсацией 2,6-диметилфенола
в присутствии второго компонента, находящегося в растворе
Тронной сополимер этилена,
диена и пропилена
Тройной сополимер этилена,
пропилена и иорборнепа
Каучук
Бутадиенстирольный каучук
[392J
[393]
[394]
[394]
оксида. Из многочисленных композиций, описанных в патентах,
в промышленности получают смесь полифениленоксида с
полистиролом блочной полимеризацией стирола в присутствии
растворенного полифениленоксида или смешением обоих компонентов в
термопластичном состоянии («General Flectric»). Этот продукт
выпускается под торговым названием «Норил». Под влиянием
полистирола термостойкость понижается до 100—145°С, однако масса
перерабатывается гораздо легче, чем чистый простой полиэфир.
К этому следует добавить и экономические соображения: 1 кг
чистого полимера стоит 11 марок, тогда как его смесь с
полистиролом — только 6 марок ФРГ. Наряду со стандартным типом Норил
731 выпускается смесь с полистиролом Норил SEI с
самозатухающими свойствами, композиция, наполненная 20 % (Норил GFN2)
и 30 % (Норил GFN3) стеклянного волокна, композиция для
гальваники Норил PN235, а также Норил FN215, представляющая
собой самозатухающий, вспененный полупродукт или формовочный
материал [626]. Переработка и свойства этих материалов
рассмотрены в смеси с полимерами на основе чистого
полифениленоксида. Эта смесь является недорогим пластиком конструкционного
назначения A кг стоит 6 марок ФРГ). Темпы роста производства
этих пластмасс выше, чем всех известных до сих пор
термопластов.
5.6.1.4. Физико-механические свойства полифениленоксида
Температуры плавления, стеклования и кристалличность. Поли-
1,4-фениленоксид плавится при 260°С. Наличие в структуре
атомов галогенов или алкильных групп приводит к понижению точки
плавления. Выше 300 °С плавятся только полм-2,6-дихлор-; 2,6-ди-
метокси- и 2,3,5,6-тетрабром-1,4-фениленоксиды (табл. 5.7).
Термодинамические характеристики полифениленоксидов, содержащих
различные заместители, приведены в табл. 5.8. Замещение в
бензольном ядре приводит к затруднению вращения вокруг простой
эфирной связи. Это повышает температуру стеклования поли-2,6-
диметилфениленоксида на 117СС, а поли-2,6-дифенил-1,4-фенилен.-
208
оксида — на 130°С по сравнению с поли-1,4-фениленоксидом,
температура стеклования которого составляет 90 °С [395]. Энтальпия
плавления поли-2,6-диметилфениленоксида равна 6—20 кал/г
[396—399].
В процессе синтеза полифениленоксид выпадает из раствора;
его степень кристалличности 40—50%. Однако полимер,
образующийся при охлаждении расплава, имеет аморфную структуру. При
добавлении 0,005—0,5 % таких структурообразователей, как
изатин, индиготин, антрахинон и 2-меркаптобензтиазол, скорость
кристаллизации расплава достигает 0,001 мл/мин [400]. Степень
кристалличности полифениленоксида, выделяющегося из раствора,
зависит от концентрации и природы растворителя [399]. В а-хлор-
нафталине при концентрации полимера выше 50 % кристаллизации
не происходит; она протекает при более низкой его концентрации.
Размеры элементарной ячейки по [409] а = 8,45 A, b = 6,02 A,
у = 90°, а по [410] а = b = 11,76 А, с = 16,83 А (тетрагональная
элементарная ячейка). Данные электронно-микроскопических
исследований гедритных кристаллов полифениленоксида приведены
в [399, 411]. Молекулярная подвижность поли-2,6-диметилфенилен-
оксида, поли-2,6-дифенилфениленоксида, поли-2-метил-6-фенилфе-
ниленоксида и поли-1,4-фениленоксида исследована
релаксационным методом [408, 412] с учетом характера связи фенил —
кислород. Структуру макромолекулы полифениленоксида можно пред-
Таблица 5.7. Температуры плавления и разложения
(температура начала разложения в инертной атмосфере)
полифениленоксидов [401—407]
Полифенилеиоксид
Поли-2,3,5,6-тетрафторфениленоксид
Поли-2-изопропилиден-6-фенил-1,4-фениленоксид
Поли-2-аллил-6-фенил-1,4-фениленоксид
Поли-2-изопропилиден-6-метил-1,4-фениленоксид
Поли-2-аллил-6-метил-1,4-фениленоксид
Поли-2,6-диаллил-1,4-фениленоксид
Поли-2,6-диэтил-1,4-фенилеиоксид
Поли-2,3,5,6-тетрахлор-1,4-фениленоксид
Поли-2-метил-6-грег-бутил-1,4-фениленоксид
Поли-2,6-дибром-1,4-фениленоксид
Поли-2,3,6-трихлор-1,4-фенилеиоксид
Поли-2,6-диизопропил-1,4-фениленоксид
Поли-2-бром-1,4-фениленоксид
Поли-2,6-ди-грег-бутил-1,4-фениленоксид
Поли-1,4-фениленоксид
Поли-2,6-диметил-1,4-фениленоксид
Поли-2,5-дихлор-1,4-фениленоксид
Поли-2-хлор-1,4-фениленоксид
Поли-2,6-дихлор-1,4-фениленоксид
Поли-2,6-диметокси-1,4-фениленоксид
Поли-2,3,5,6-тетрабром-1,4-фенилвноксид
Поли-2,6-Дифенил-1,4-фениленоксид
т
°С
98
116
117
117
117
160
180
192
217
220
222
225
240
246
262
262
265
270
300
300
300
484
т
разл»
UC
280
220
280
260—290
360
ставить как последовательность жестких 2,6-диметилоксифещ
новых фрагментов длиной 5,34 А [413]:
СН
Диамагнитная восприимчивость полифениленоксида при
степенях полимеризации до 350 линейно зависит от молекулярной
массы. Найденная из этой зависимости константа
внутримолекулярного взаимодействия более чем на 10 % выше, чем у полиэтилена
и полиэтиленокснда [414]. Смесь полифениленоксида и
полистирола лишь в ограниченном числе случаев имеет область
совместимости. В этой области по данным механической или
диэлектрической релаксации и калориметрии с достоверностью можно
определить температуру стеклования [415—417].
На рис. 5.11 представлена зависимость температуры
стеклования от содержания полифениленоксида в его смеси с
полистиролом. Сравнительные исследования механической и диэлектрической
Таблица 5.8. Термодинамические характеристики
полифениленоксидов [395, 408]
Полимер
/СН3 -
Vh, .
/ 3
—<Q>—о-
\с«н5 .
/CBHS -
\С6Н5 .
п
п
п
гс. °с
90
207
180
220
кал/°С
на звено
2,76
3,45
4,11
Гпл, °С
262
262
474
Степень
кристалличности,
72
40
52
0,69
0,90
0,69
AS.1
(кал/моль-°С)
3,50
2,18
3,85
210
Рис. 5.11. Температура стеклования смеси поли-2,6- Тс , С
диметил-1,4-фениленоксида с полистиролом:
I _ определение диэлектрическим методом ПОП Гц); 2~ 220
определение' колориметрическим методом.
180
релаксации показали [416J, что
гомогенная фаза образована областью, содержа- /40
щей не только сегменты, но и области,
обогащенные полифениленоксидом и полисти- до
ролом. В целом смесь тем не менее, доста- 0 2S 50
точно однородна, чтобы можно было
заметить отличие ее температуры стеклования от Тс исходных
компонентов. При гомогенизации в термопластичном состоянии при
280—330 °С вязкость расплава полистирола значительно ниже, чем
полифениленоксида. Поэтому сначала полистирол образует
непрерывную фазу, в то время как полифениленоксид существует
в виде дисперсной фазы. В результате взаимной диффузии
происходит смешение, но при охлаждении остается фаза, обогащенная
полифениленоксидом, которая диспергируется в полистирольной
матрице. Высокоэластическое состояние смеси полимеров может
быть предсказано по температуре стеклования и свободным
объемам компонентов [417].
Растворимость. Изучена растворимость только поли-2,6-диме-
тил-1,4-фениленоксида. Он растворяется в ароматических
углеводородах, таких, как бензол, толуол, ксилол, и в хлорированных
углеводородах, таких, как метиленхлорид, хлороформ, хлорбензол,
дихлорбензол; не растворяется в алифатических углеводородах,
таких, как н-гексан, циклогексан, а также в спиртах, кетонах,
сложных эфирах. Растворы в бензоле и толуоле, полученные при 40 °С
и при комнатной температуре, стабильны. При 0°С через
несколько часов и при —5°С мгновенно раствор мутнеет [418]. При
растворении в метиленхлориде прозрачные растворы получают при
концентрации полимера до 20%, но при хранении происходит
почти количественное осаждение его [419]. Время до помутнения
раствора тем меньше, чем выше концентрация полимера в
растворе и больше его молекулярная масса; оно составляет 2 мин при
2%-ной концентрации раствора и 1 ч —при 1 %-ной. Такое же
соотношение наблюдается для растворов в бромхлорметане и ме-
тиленбромиде. Это объясняется тем, что такие растворители, как
метиленхлорид, образуют комплекс B: 1 в расчете на звено поли-
2,6-диметилен-1,4-фениленоксида). Комплекс нестабилен и легко
разрушается под действием бензола, хлороформа, 900-кратного
избытка метиленхлорида или при нагревании суспензии.
По-видимому, метиленхлорид располагается в дефектных местах спиралей
полимера, вследствие чего происходит его кристаллизация и
осаждение из раствора.
Термодинамические исследования системы полидиметилфени-
леноксид—метиленхлорид показали, что константа Хаггинса в
интервале температур 48—75 °С составляет 0,46—0,50. Энтальпия
2П
растворения ДЯ = 1404 кал на звено [410]. Кристаллизация также
протекает при использовании в качестве растворителей сс-пинена
[409] и декалина [410]. Фазовое состояние системы полимер—¦
толуол — этанол описано в [425].
Для определения молекулярно-массового распределения
полимера методом фракционного осаждения можно использовать
системы: дихлорэтилен — нитрометан [287], хлороформ — метанол
[418], хлороформ — этанол — этиленгликоль [420], толуол —
метанол [418]. Хорошие результаты достигаются также при дробном
осаждении из 0,4 %-ного раствора в диоксане при нагревании:
первая фракция выделяется при 65°С [421].
Гель-проникающая хроматография поли-2,6-диметил-1,4-фенп-
леноксида описана в [422], поли-2,6-дифенил-1,4-фепиленоксида —
в [423]. Из смесей поли-2,6-диметилфениленоксида с
полистиролом или полисилоксаном полифениленоксидная составляющая
может быть проэкстрагирована метиленхлоридом [424]. Для виско-
зиметрического определения молекулярной массы пригодны
бензол, толуол, хлороформ, хлорбензол и тетрахлорид углерода [422,
426, 427]. Константы в уравнении Марка — Хаувинка для
различных простых ароматических полиэфиров приведены в табл. 5.9.
Изучена вязкость растворов поли-2,6-диметил-1,4-фениленоксида в
толуоле и о-дихлорбензоле в интервале температур 20—80 °С
[427]. Метод светорассеяния использован для определения
молекулярной массы поли-2,6-диметил-1,4-фениленоксида [418, 422] и
поли-2,6-дифенил-1,4-фениленоксида [423]. Инкремент показателя
преломления dn/dc B5 °С, 546 мкм) составляет для поли-2,6-ди-
метил-1,4-фениленоксида 0,124 мл/г в хлороформе, 0,118 мг/л в
толуоле и 0,114 мл/г в бензоле.
Таблица 5.9. Константы в уравнении Марка — Хаувинка
для вискозиметрического определения молекулярной массы
полифениленоксидов [422, 423, 426, 428, 429]
Полимер
Растворитель
Температура,
°С
-10*
Размеры
см/(г-моль)
ратурный
источник
Поли-2,6-диме-
тил-1,4-фенилен-
оксид
Поли-2-метил-
6-фенил-1,4-фени-
леноксид
Поли-2,6-дифенил-
1,4- фенил еноксид
Бензол
Толуол
Хлорбензол
Тетрахлорид
углерода
Хлороформ
Бензол
Хлороформ
25
25
25
90
25
25
25
25
50
25
2,60
2,85
3,78
5,14
4,63
7,55
3,86
3,494
3,705
3,56
0,69
0,68
0,66
0,63
0,64
0,585
0,70
0,609
0,638
0,62
(830±5)-10
(835±10)-10
(845±20)-10
— и
— 11
— и
422
426
426
422
426
426
428
423
423
429
212
80
60
20
0
—
—
—
200
1
1 1
400 600
Г 1
800 1000 t?C
Рис. 5.12. Кривая термической деструкции по-
ли-2,6-диметил-1,4-феипленоксида [432].
Химическая и фотохимическая
деструкция. Несмотря на высокую
температуру плавления, по
термостойкости полифениленоксид аналогичен
сложным ароматическим полиэфирах^,
поликарбонатам или полиангидридам,
уступая в этом отношении ароматическим полиамидам. В табл. 5.7
приведены температуры начала разложения в инертной атмосфере
некоторых полифениленоксидов. Температура начала разложения
поли-1,4-фениленоксида колеблется от 360 до 390°С.
Увеличение числа заместителей в ароматическом ядре
приводит к снижению термостойкости, которая уменьшается в
следующем ряду: полифениленоксид > поли-2,6-дихлорфениленоксид >
> поли-2,6-дибромфениленоксид > политетрахлорфениленоксид.
Промышленный поли-2,6-диметил-1,4-фениленоксид в инертной
атмосфере устойчив до 350 °С. На воздухе окислительная деструкция
начинается примерно при 125°С. В инертной среде деструкция
этого полимера протекает в две стадии [119, 430—432]. При
температуре выше 360 °С в качестве летучего продукта выделяется
с большой скоростью фенол (рис. 5.12). При температуре выше
393°С выделяются следующие продукты [в % (масс.) в расчете
на загруженный полимер]: фенол — 0,2; о-крезол — 5,4; ж-кре-
зол — 0,4; 2,6-диметилфенол — 24,4; 2,5-диметилфенол — 3,2; 2,3-ди-
метилфенол — 9,0; 3,5-диметилфенол — 0,3 [119]. При температуре
выше 450 °С выделяются такие продукты деструкции, как вода,
водород, метан, диоксид углерода. Выделение воды является
следствием конденсации образующихся фенольных фрагментов цепи
с получением полициклических соединений. Метан образуется за
счет отщепления метильных групп от фенольных фрагментов. Выше
500 °С полианнелирование многоядерных ароматических
соединений сопровождается выделением водорода. Согласно [119],
термическая деструкция поли-2,6-диметил-1,4-фениленоксида может быть
описана схемой, представленной на рис. 5.13.
Стойкость этого полимера к термоокислительной деструкции
зависит от условий синтеза, т. е. природы катализатора,
растворителя, продолжительности реакции и концентрации пиридина в
смеси растворителей. Чем меньше продолжительность процесса и
ниже концентрация пиридина в смеси растворителей, тем выше
термостойкость полимера [437]. Термоокислительная и
фотохимическая деструкция полимера сопровождается его
структурированием с выделением СО2, СО, азота (из остатков катализатора) и
водорода [431, 433]. Чувствительность поли-2,6-диметил-1,4-фени-
леноксида к окислению объясняется наличием в нем гидроперокси-
дов, образующихся на стадии синтеза, которые выполняют функ-
21
Рис. 5.13. Механизм термической деструкции поли-2,6-диметил-1,4-фениленсксида
[119]-
ции активных центров в процессе деструкции. Нагревание
полимера на воздухе при 125°С сопровождается потерей массы с
выделением СО2, СО, N2, H2. Потеря массы в ходе дальнейшего
нагревания в течение 1200 ч компенсируется, как это видно из
рис. 5.14, за счет окисления метильных групп. Наличие сшитого
продукта в полимере обнаруживается после экспозиции в течение
1000 ч [434].
При термоокислении при 260°С 2,6-диметилбензолфенилового
эфира, который моделирует полимер, выделяется 4-метилксантон,
214
100 200 500 1000 2000 5000 tt v
ПО 100 80 60 25t,G
Рис. 5Л4. Изменение массы пленки в зависимости от продолжительности
экспозиции при термоокислительной деструкции поли-2,6-диметил-1,4-фениленоксида на
воздухе при 125°С [434].
Рис. 5Л5. Зависимость времени поглощения кислорода A0 мл/г полимера) поли*
2,6-диметил-1,4-фениленоксидом и его смесями с полистиролом [415]:
I — термостабилизированная смесь полиднметилфениленоксида с полистиролом; 2 —
негорючая смесь пол идиметнл фен ил ен оксида с полистиролом; 3 — нестабилизированные полидиме»
тилфениленоксид и его смесь с полистиролом.
2-гидрокси-З-метилбензофенон, 2-фенокси-З-метилбензальдегид,
2-фенокси-З-метилбензойная кислота, 2-фенокси-З-метилбензило-
вый спирт и 2-феиокси-3-метилбензил-2-фенокси-3-метилбензоат
[436].
Нестабилизированный поли-2,6-диметил-1,4-фениленоксид и его
смесь с полистиролом не различаются по стойкости к
термоокислительной деструкции (рис. 5.15), для них характерно одинаковое
поглощение кислорода в интервале температур 60—140 °С [415].
Более высокой стойкостью отличаются негорючие смеси полимера
с полистиролом, а также их смеси со стабилизатором. В
противоположность чистому поли-2,6-диметил-1,4-фениленоксиду его смеси
с полистиролом не структурируются: в процессе термоокисления
происходит только деструкция. Это объясняется тем, что не может
происходить рекомбинация радикалов полифениленоксида.
В качестве термостабилизаторов для поли-2,6-диметил-1,4-фе-
ниленоксида и его сополимеров с полистиролом предпочтительны
системы, включающие 2-меркаптобензимидазол и фосфиты. Ниже
перечислены наиболее известные стабилизаторы для поли-2,6-ди-
метилфениленоксида и его сополимеров с полистиролом:
Литературный
источник
Алканоламин — фосфит — сульфид
2-Меркаптобензтиазол — фосфит
т/эет-Бутилфенол — фосфит — тиопропионат — дикарбамат . . .
Сульфид кадмия — тридсцилфосфит
2-Меркаптобензтиазол—?-нафтилфенилендиамин
2-Меркаптобензимидазол — Ы-феиилэтил-2,5-диметоксианилин . .
а-Нафтол
Оловоорганические стабилизаторы — трихлорид фосфора — димст-
оксихлорфосфин
Этилендиамин — фталимид
[445]
[449
[450
[438
[447
[448
[444]
[446]
[451]
[452]
215
О 200 400 600 800 ЮОО т,ч
Рис. 5.16. Зависимость содержания золь- (/) и
гель-фракции B) от продолжительности
облучения для пленки толщиной 12 мкм при
фотоокислительной деструкции и поли-2,6-диметил-1,4-фе-
ниленоксида [435].
Поглощение 10 мл кислорода на 1 г
сополимера на основе 2,6-диметил-1,4-
фениленоксида и стирола происходит
р рд
при 125°С за 19 ч. Добавление в качестве стабилизатора 0,5%
тридецилфосфита с сульфидом кадмия в тех же условиях
увеличивает это значение до 118 ч [438]. Повышение стабильности поли-
2,6-диметил-1,4-фениленоксида достигается также сополиконденса-
цией с такими соединениями, как 2,6-дибромфенол [440] или 2-ме-
тил-б-метилтиометилфенол [439], или взаимодействием концевых
гидроксильных групп со стиролом [441,442] или акриламидом [443].
Фотоокисление поли-2,6-диметил-1,4-фениленоксида при
облучении УФ-лампой сопровождается деструкцией или
структурированием полимера. С увеличением продолжительности облучения
содержание гель-фракции достигает 65% (рис. 5.16). При этом
молекулярная масса неструктурированной части полимера
уменьшается. После УФ-облучения полимера на воздухе в течение 2 ч
молекулярная масса сохраняется на уровне 93 % и в течение
100 ч — на уровне 30% от исходного значения [435]. Для
процессов, связанных с отщеплением СО и СО2, предложен
следующий механизм [415]:
о2 rh
RCH2 > RCH2O—О *- RCH2OOH
(О)
RCOOH ¦* RCHO
СО2 RH+CO
J
RH
RCH2OH
Структурирование происходит за счет рекомбинации метилено-
вых радикалов:
2--СН-
о
!
o
•er
CH2OCH2
5.6.1.5. Переработка поли-2,6-ДИметил-1,4-фениленоксида
и его композиций с полистиролом
Текучесть. Зависимость вязкости расплава композиций поли-
фениленоксида с полистиролом от скорости сдвига и аналогичная
зависимость для поликарбоната и АБС-пластика при
температурах их переработки показаны на рис. 5.17 [453]. Поведение
полимеров при различных температурах расплава показано на
рис. 5.18. Зависимость вязкости расплава от температуры для
смеси поли-2,6-диметил-1,4-фениленоксида с полистиролом носит
более ярко выраженный характер, чем для поликарбоната и АБС-
пластика (рис. 5.19). Относительно более сильная зависимость
вязкости расплава от градиента сдвига, температуры расплава
позволяет легко регулировать характеристики текучести, определяющие
процесс переработки полимера. Наполненные стеклянным
волокном композиции качественно ведут себя так же, как и ненапол-
ненные. Зависимость вязкости расплава от температуры для смеси
поли-2,6-диметил-1,4-фениленоксида с полистиролом, ненаполнен-
ной и наполненной 30 % стеклянного волокна, при двух
различных градиентах сдвига показана на рис. 5.20.
Экструзия. Поли-2,6-диметил-1,4-фениленоксид и его смеси
с полистиролом, ненаполненные и наполненные стеклянным
волокном, перерабатываются экструзией в прутки, трубы, полые
изделия, пленки и листы. Наилучшие результаты достигаются в том
случае, если перед переработкой стеклянное волокно пропитать
17—23%-ным раствором полифениленоксида в смеси
растворитель— нерастворитель, например хлороформ— изопропиловый
1Q3
о
10
Рис. 5.17. Кривые текучести смеси поли-2,6-диметил-1,4-фениленоксида с
полистиролом, поликарбоната и АБС-пластика [453]:
/ — АБС-пластик, 204 °С; 2 — смесь полимеров типа «Норил», 282 °С; 3—поликарбонат, 287 °С.
Рис. 5.18. Кривые текучести смеси поли-2,6-диметил-1,4-фенилеиоксида с
полистиролом при различных температурах расплава [453].
217
10
315 2O2 253 226 2m t°C
10
Рис. 5.19. Кривые текучести смеси поли-2,6-диметил-1,4-фениленоксида с
полистиролом типа «Норил» при различных обратных температурах расплава [453]:
1 — смесь полидиметилфенилеиоксида с полистиролом, 10 с; 2 — АБС-пластик, 10 с^^З-
поликарбонат, 10 с" ; 4 — поликарбонат, 1000 с"; 5 — смесь полндиметнлфеннленоксида
полистиролом, 1000 с~]; 6 — АБС-пластик, 1000 с.
Рис. 5.20. Кривые текучести смеси поли-2,6-диметил-1,4-фениленоксида с
полистиролом при различных обратных температурах расплава [454]:
1 — смесь поли-2,6-диметил-1,4-фениленоксида с полистиролом с 30% стеклянного волокна;
2 — иеиаполнеииая смесь поли-2,6-диметил-1,4-фениленоксида с полистиролом.
спирт [455, 456], или силанами [457]. Если экструдер не имеет
зоны дегазации, то полимер перед экструзией следует высушить
при 110°С Поскольку в процессе экструзии не образуются
агрессивные газы, то необходимость применения антикоррозионной
арматуры отпадает [458]. Чистый поли-2,6-диметил-1,4-фениленоксид
Таблица 5.10. Экструзия смеси поли-2,6-диметил-
1,4-фениленоксида с полистиролом [461]
Характеристики экструдера
и режимы переработки
Диаметр мундштука,
мм
Ширина зазора сопла,
мм
Диаметр дорна, мм
Шнек и зона конуса
Остаточное давление в
зоне дегазации»
мм рт. ст.
Диаметр для
калибровки, мм
Температура цилиндра,
°С
1-я зона
2-я зона
3-я зона
4-я зона
Адаптер, °С
Изготовление |
плнт
3,3
труб
34,5
27,9
Не
охлаждаются
711
260
260
260
260
257
711
27,9
316
332
316
295
254
1
Характеристики экструдера
и режимы переработки
1
Температура сопла, °С
сзади
слева
в середине
спереди
справа
Температура расплава,
°С
Давление расплава,
кгс/см2
Частота вращения
шнека, об/мин
Температура масла в
охлаждающих валках,
°С
1-е валки
2-е валки
3-й валки
Скорость приема, м/мин
Производительность,
кг/ч
Изготовление
плит
275
271
275
275
155-162
48
160
163
ПО
0,4
55
труб
254
249
297
282
98-105
24
45
218
Таблица 5.IL Переработка литьем под давлением
поли-2,6-диметил-1,4-фениленоксида, его смесей
с полистиролом и композиций, наполненных стеклянным
волокном [454, 458]
Режим переработки
Температура формы, °С
Температура цилиндра, СС
зона загрузки
зона сжатия
зона питания
Температура сопла, OC
Давление впрыска, кгс/см2
Скорость впрыска
Частота вращения шпека,
об/мии
Время цикла, с
ПФО
90
300
340
350
350
1000-1500
30
Ненаполнен-
ная смесь
80
250
260
270
260
1000-1500
В ы со
30
Смесь, наполненная
стеклянным волокном
20%
80
277
282
290
290
1220
к а я
73
30
30%
80
290
290
290
290
1220
73
30
перерабатывается при следующих температурах: 1-я зона 275—
285 °С, 2-я зона 300—315 °С, 3-я зона 290—300 °С, 4-я зона 280—
290 °С и в головке 245—265 °С. Температура переработки смесей
с полистиролом лежит на 40° ниже, чем для чистого полимера.
Шнек должен иметь стенки с равномерным шагом и одинаковой
глубины. Длина шнека должна составлять 24 диаметра [460].
Условия переработки смесей с полистиролом приведены в табл. 5.10
[461] для экструдера диаметром 3'/2" с длиной шнека 28D
(экструдер для труб) или диаметром 2!/2" с длиной 24D и зоной
дегазации (экструдер для плит). Плиты принимаются на два
охлаждающих валка и правятся третьим.
Литье под давлением. Поли-2,6-диметил-1,4-фениленоксид и его
смеси с полистиролом могут перерабатываться на обычных
плунжерных литьевых машинах или на литьевых машинах со шнеко-
вой пластикацией. Для чистого полимера температура переработки
на плунжерных литьевых машинах колеблется от 330 до 360 °С,
а на шнек-машинах — от 300 до 350°С [462]. Температура
переработки сополимеров с полистиролом на 40° ниже.
Шнек-машины более удобны для переработки, чем плунжерные
литьевые машины. Важнейшие параметры для литья под
давлением полифениленоксида, его сополимеров с полистиролом и
наполненных стеклянным волокном композиций приведены в
табл. 5.11.
Полифениленоксид гигроскопичен, и перед литьем его
необходимо подсушивать при ПО—120°С. Целесообразно использование
закрытой воронки предварительного подогрева, в которой
материал сразу после сушки смешивается с наполнителем. В шнеко-
вых литьевых машинах применяют обычные шнеки. Сопло должно
быть возможно более коротким при минимальном сечении 5, мм.
213
В направлении от воронки к соплу оптимальный температурный
градиент составляет всего 50°. Высокая температура стеклования
полифениленоксида обусловливает необходимость литья с высокой
скоростью при давлении 1000—1500 кгс/см2. Поверхностный
глянец на литьевых изделиях достигается регулированием
температуры формы: при температуре формы 80—90°С получают изделия
с матовой поверхностью, выше 100 °С — с поверхностью с высоким
глянцем. Сопла с клапаном можно использовать лишь при
высоких температурах расплава. Выдержка под давлением необходима
только в случае толстых стержневых литников. Вследствие
быстрого застывания подпитка составляет 50—60 % давления при
литье, что обеспечивает получение изделий с невысокими
остаточными напряжениями. Потеря давления при впрыске может быть
уменьшена за счет применения сопел с отверстиями диаметром
3,5 мм [460].
Рекомендуют литниковые втулки с минимальным диаметром
5 мм, которому должны соответствовать определенные размеры
сечения разводящих каналов. Впускной литник должен находиться
в том месте изделия, где толщина стенок составляет от 1,9 до
3,8 мм; длина впускного литника должна быть минимальной.
Продолжительность цикла при изготовлении изделий с толщиной
стенок 0,8; 1,6 и 6,3 мм составляет соответственно 15, 30 и 60 с.
Продолжительность цикла для наполненных стеклянным волокном
композиций может быть на 30 % меньше. Истирание в шнеке,
цилиндре и форме несколько выше, чем для ненаполненных
материалов. Изменение температуры, давления и ширины каналов для
смеси полидиметилфениленоксида с полистиролом такое же, как
и для ненаполненных материалов. На рис. 5.21 показана
зависимость пути течения от давления для наполненной и ненаполнен-
ной смеси полидиметилфениленоксида с полистиролом при
различном сечении каналов, а на рис. 5.22 — зависимость пути течения
от толщины стенок изделия при различных температурах массы.
Усадка уменьшается с повышением температуры и давления
как для наполненных, так и для ненаполненных композиций. На
рис. 5.23 показана зависимость
усадки от давления для разнотол-
щинных изделий.
Глубокая вытяжка под вакуумом.
Листы из
полидиметилфениленоксида могут быть получены
глубокой вытяжкой под вакуумом в
Рис. 5.21. Зависимость пути течения от
давления для различных сечений каналов при
литье под давлением смеси
полидиметилфениленоксида с полистиролом (температура
смеси 290 °С; температура формы 93 °СЛ
[454]:
. ненаполнзнная смесь, смесь,
40O . ?OQ. 1200 P}Krcjcti* наполненная 30% стеклянного вдлокна.
0
220
l>Cf1 j_
Б0
50
30
го
10
0 0fi 1,0 1,5 2? 2,5 a,MM
Толщина стенки
"мм
_L
700' 800 900 1000 Р,кгс/спг
Рис. 5.22. Зависимость пути течения от толщины стенок формуемого изделия при
различных температурах при литье под давлением смеси поли-2,6-диметил-1,4-фе-
ниленоксида с полистиролом (температура формы 93 °С, давление 1050 кгс/см2)
[454].
Рис. 5.23. Зависимость усадки от давления (температура формы 93 °С,
температура массы 290 °С) при литье под давлением смеси поли-2,6-диметил-1,4-фенилен-
оксида с полистиролом [454]:
^———— —ненаполненная смесь; смесь, наполненная 30% стеклянного волокна.
интервале температур 150—160 °С [463]. Листы толщиной более
2 мм должны предварительно нагреваться с двух сторон в течение
20—30 с для того, чтобы избежать перегрева и сильного
потемнения материала. Вследствие высокой температуры размягчения
необходимо небольшое время охлаждения (8—10 с). Изделия можно
извлекать из формы при 130—140 °С.
Тонкие листы и пленка. Тонкие листы получают каландрова-
нием смесей полидиметилфениленоксида с другими полимерами.
Смесь, содержащая 5 % тройного сополимера этилена, пропилена
и диена, может быть переработана в тонкий лист каландрованием
при 290 °С за 7 мин [392, 393]. В СССР пленку получают методом
полива из раствора полидиметилфениленоксида в хлороформе
[359]; прочность пленки возрастает с увеличением молекулярной
массы в интервале 24 000—50 000 с 480 до 690 кгс/см2. В качестве
растворителя для полива из раствора пригодны также тетрахлор-
этан и пентахлорэтан [464].
Улучшение поверхности. В отличие от изделий, получаемых
из других полимеров, изделия из полидиметилфениленоксида
могут изменять глянец в зависимости от режима переработки: при
низких температурах переработки получают матовую поверхность,
при высоких температурах достигается отличный глянец.
Металлизацию полидиметилфениленоксида проводят в вакууме или
гальванизацией окунанием [465]. Для лакирования используют
алкидные, акрилатные и эпоксидные лаки. Нанесение печати
возможно тиснением или трафаретным методом.
Сваривание и склеивание. Изделия из
полидиметилфениленоксида можно соединять между собой свариванием ультразвуком,
зеркальным методом, методом горячей ванны, ротационным
221
методом и свариванием электрическим сопротивлением. Ниже
приведены типичные режимы сваривания различными методами:
Ультразвуковой Зеркальный Ротационный
Давление, кгс/см2 1,75-8,8 — 20
Время, с 0,3-2 20 -
Температура поверхности, °С . . — 260—288 —
Скорость вращения, м/мин ... — — 360
Для склеивания можно применять такие растворители, как ди-
хлорэтилен, толуол, хлороформ, смесь дихлорэтилена с тетрахло-
ридом углерода. Наилучшие результаты получаются при
использовании 10 %-ных растворов. Продолжительность цикла,
включающая нанесение, соединение и выдержку под давлением, составляет
10—15 с. Прочность при растяжении при этом достигает
315 кгс/см2. Склеивание полидиметилфениленоксида с другими
материалами осуществляют с помощью каучуковых, эпоксидных и
силиконовых клеев.
Удаление стружки. Для удаления заусенцев можно
использовать обычные станки. Для обработки полидиметилфениленоксида,
наполненного стеклянным волокном, следует пользоваться
вольфрам-карбидным инструментом. Для сверления применяют сверла
с углом резания 5° и боковым углом резца 118°. Число оборотов
при сечении 12 мм составляет 400 об/мин, а для больших сверл —
325 об/мин [466]. Режимы механической обработки изделий,
полученных из смеси полидиметилфениленоксида с полистиролом,
приведены в табл. 5.12.
Для фрезерования применяют фрезы с углом резания 15° и
четырьмя нарезками; частота вращения должна составлять
325 об/мин. В качестве смазки следует использовать силиконовые
масла, так как при применении обычных смазок происходит
набухание полимера. Удаление заусенцев с изделий, изготовленных из
ненаполненных композиций, рекомендуется проводить при 120°С.
В случае композиций, наполненных стеклянным волокном,
нагревания не требуется.
Таблица 5. 12. Удаление стружки с изделий, полученных
из смеси полидиметилфениленоксида с полистиролом [466]
Толщина
материала, мм
Ниже 6
6-25
25-40
Выше 40
Скорость ленточной
пилы, м/мнн
10-12
8-10
6-8
3-5
Скорость
циркульной пнлы*, м/мии
Ниже 32
Ниже 32
Ниже 30
Ниже 25
Характеристика пилы,
зуб/см
ленточная
пнла
5-7
5-6
5-6
4-5
циркульная
СО СО СО
• Частота вращения циркульной, пнлы составляет 1220 об/мин.
222
Таблица 5.13. Теплофизические характеристики
полидиметилфениленоксида, его смесей с полистиролом
и композиций, наполненных стеклянным волокном
Показатель
Температура стеклования, °С
Плотность, г/см3
Показатель преломления
Деформационная
теплостойкость, °С
при 4,6 кгс/см2
при 18,6 кгс/см2
при 28,0 кгс/см2
Удельная теплоемкость,
кал/(г-°С)
Коэффициент
теплопроводности, ккал/(м-ч-°С)
Термический коэффициент ли-
иейного расширения при
-29~66°С, 1/°С
Усадка, %
Температура длительной
эксплуатации на воздухе, °С
Температура хрупкости, °С
ПФО
220
1,06
1,575
180-191
0,40
0,160
5,2-10~5
130
-170
ПФО + ПС
155
1,06
1,633
138
130
0,32
0,186
5,9- 10~5
0,5-0,7
105
ПФО + ПС
B0%
волокна)
1,21
147
144
142
0,143
3,6- 10~5
0,3-0,4
120
ПФО + ПС
C0%
волокна)
156
152
148
0,136
2,5- 10~5
0,2-0,3
5.6.1.6. Эксплуатационные свойства поли-2,6-диметил-
1,4-фениленоксида и его композиций с полистиролом
Теплофизические свойства. Важнейшие теплофизические
свойства полидиметилфениленоксида, его смесей с полистиролом и
наполненных композиций приведены в табл. 5.13. Термостойкость
полидиметилфениленоксида составляет 190 °С, а его смесей с
полистиролом около 140°С. Это значение близко термостойкости
полиформальдегида и поликарбоната. При наполнении
стеклянным волокном термостойкость повышается максимум на 20°.
Термический коэффициент линейного расширения
полидиметилфениленоксида на 10 % ниже, чем у поликарбоната и значительно
меньше, чем у полиформальдегида и АБС-пластика, и мало зависит от
температуры. Наполненные стеклянным волокном смеси
полидиметилфениленоксида с полистиролом похожи в этом отношении на
металлы [467] :
а. 1/°С
Полиформальдегид 8,1 - 10—5
АБС-пластик F—9)-Ю-5
Поликарбонат 6,6 • 10*~5
Полидиметилфеннлоноксид ... 5,2 • 10~5
Смесь полидиметилфениленоксида с
полистиролом 5,9 - 10~5
Фенольная смола C,0—4,4) • 10~5
Смесь полидиметилфениленоксида с
полистиролом, наполненная 2О°/о
стеклянного волокна 3,6-10~5
223
Алюминии 2,7 • Ю-8
Цинк 2,6-Ю-3
Смесь полидиметилфенилеиоксида с
полистиролом, наполненная 30% стеклянного
волокна 2,5- 10~s
Латунь 2,0-Ю-5
Высококачественная сталь 1,0 - 10—5
Следствием низкого значения коэффициента линейного
расширения является малая усадка @,3—0,4 %) Для композиций,
наполненных стекловолокном, что позволяет получать изделия с высокой
точностью размеров при литье.
Физико-механические свойства. Полидиметилфениленоксид
отличается от других пластмасс тем, что его физико-механические
свойства мало изменяются в широком температурном интервале.
Физико-механические свойства полидиметилфениленоксида, его
Таблица 5.14 Физико-механические свойства
полидиметилфениленоксида, его смесей с полистиролом
и композиций, наполненных стеклянным волокном
Показатель
Прочность при растяжении,
к г с/см2
при 23°С
при 93 °С
Относительное удлинение при
разрыве, %
прн 23 °С
при 93°С
Модуль упругости при
растяжении, кгс/см2
при 23°С
прн 93 °С
Прочность при изгибе, кгс/см2
при — 18°С
прн 23°С
при 93 °С
Модуль упругости при
изгибе, кгс/см2
при —18 °С
при 23°С
прн 93°С
Ударная вязкость образца с
надрезом, кгс-см/см2
Твердость прн вдавливании
шарика, кгс/см2
Прочность прн сжатии при
10%-ной деформации, B3 °С),
кгс/см2
Твердость по Роквеллу
Ползучесть при растяжении
C00 ч, 140 кгс/см2), %
Коэффициент трения
ПФО
775
420
80
26 000
23 000
25 000
9,5
1200
118-120
0,70
0,18—0,23
ПФО + ПС
655—675
450
20—30
30-40
25 000
16 150
1 120
950
525
27 000
25 300
8 300
7,7
1 250
1 150
119
0,70
ПФО + ПС
B0%
волокна)
1020
750
4—6
4-6
65 000
52 750
1600
1 450
950
61000
52 000
41500
8,6
1240
106
0,33
ПФО + ПС
C0%
волокна)
1200
1 020
4—6
4—6
93 500
90 000
1750
1550
1275
81 000
73 100
70 300
1260
108
0,20
224
Е-10~3,кгс/сп2
40
Рис. 5.24. Зависимость прочности при растяжении различных полимеров от
температуры [468]:
/ — полнднметллфеинленокснд; 2 — смесь полидиметилфеннленоксида с полистиролом; 3 —
поликарбонат; 4 — полиформальдегид.
Рис. 5.25. Зависимость модуля упругости при растяжении различных полимеров
от температуры [467]:
1 — полндиметнлфениленоксид; 2— полисульфон; 3 — поликарбонат; 4 — Норил; 5 — поли-
ацеталь (гомополимер); 6 — полиамид 6,6.
смесей с полистиролом и композиций, наполненных стеклянным
волокном, приведены в табл. 5.14. Прочность при растяжении
смесей полидиметилфениленоксида с полистиролом несколько ниже,
чем у чистого полимера, но значительно выше, чем у полиамида
6,6, АБС-пластика или поликарбоната. При высоких температурах
прочность сохраняется на достаточно высоком уровне. На рис. 5.24
показана зависимость прочности при растяжении от температуры
для ряда полимеров. Отношение прочности при растяжении к
плотности у полидиметилфениленоксида на 25 % выше, чем у
поликарбоната или алюминия, и на 40 % выше, чем у
полиформальдегида или цинка. Модуль упругости при растяжении мало
зависит от колебаний температуры и влажности. Ниже 100°С
жесткость полидиметилфениленоксида и его смесей с полистиролом
примерно одинакова. Однако при температуре выше 100°С
жесткость резко уменьшается. Вместе с тем чистый полидиметилфе-
нпленоксид до 200°С имеет очень хорошие характеристики. На
рис. 5.25 показана зависимость модуля упругости при растяжении
от температуры для ряда полимеров. По прочности и модулю
упругости при растяжении полидиметилфениленокспд превосходят
только металлы.
Полидиметилфениленоксид и его смеси с полистиролом
отличаются от других термопластов е %
сравнительно низкой хладотеку- /°
честью, что обусловливает
высокую стабильность размеров мате-
2,0
1,5
Рис. 5.26. Кривые ползучести различных
полимеров под нагрузкой 141 кгс/см2 при
23 °С [467]:
1 — АБС-пластик; 2 — полиамид 6,6; 3 — поли*
ацеталь; 4 — Норил; 5 — поликарбонат.
8 Зак, 18
0,5
1 1
1 2
3
4
-
5
1 1
IQQ ZQQ 3QQ № 5QQ xt4
225
D WO 200 300 400 t,Y
Рис. 5.27. Кривые ползучести смесей полидиметнлфеннленокснда с полистиролом,
наполненных стеклянным волокном, при различных температурах и нагрузках
[454]:
/ —-211 кгс/см2, 116 °С; 2 — 352 кгс/см2, 82 °С; 5 — 352 кгс/см2, 23 °С; 4 — 211 кгс/см2, 23 °С.
Рис. 5.28, Зависимость ударной вязкости различных полимеров от температуры
[468]:
I — поликарбонат; 2 — полндиметилфениленоксид: 3 — смесь полиднметилфениленоксида со
со стиролом; 4 — полиформальдегид.
риалов. Поэтому изделия из полидиметилфениленоксида являются
прекрасными заменителями металлов [469]. На рис. 5.26 показано
изменение ползучести при растяжении смесей
полидиметилфениленоксида с полистиролом и других полимеров. Зависимость
ползучести стеклонаполненных материалов от времени, температуры
и приложенной нагрузки представлена на рис. 5.27.
Изменение ударной вязкости по Изоду в температурном
интервале от —45 до 25°С показано на рис. 5.28. Выше 25°С ударная
вязкость изменяется мало. Полидиметилфениленоксид применяется
там, где необходимо, чтобы соответствующая вязкость
сохранялась вплоть до низких температур. Стойкость к истиранию для
этого полимера сравнима с поликарбонатом.
Электрические свойства. Электрические свойства
полидиметилфениленоксида не меняются в широком интервале температур и
Тут
100
BQ
60
20
0
ус/!.
-
-
-
ed.
V
[
300 400
Рис. Б.29. Зависимость тангенса угла диэлектрических потерь
полидиметилфениленоксида от продолжительности выдержки в воде при 10* Гц [470].
Рнс. 5,30. Зависимость электрической прочности пленки из смеси
полидиметилфениленоксида с полистиролом от ее толщины [471].
Рис. 5,31. Стойкость к токам утечки смеси полидиметилфенилеиоксида с
полистиролом (Норил КВ25) по VDE 0303 [460]:
1 — 0,1%-ный раствор NHX1 + 0,5% иекаля; 2— 0,1%-иый раствор NH4Clf ao DIN 53480.
226
частот и не зависят от влажности. Как видно из табл. 5.15
различия между диэлектрическими характеристиками
полидиметилфениленоксида и его смесей с полистиролом незначительны. По
сравнению с другими конструкционными материалами полидиме-
тилфенилснокснд имеет очень низкий ig о, который не меняется
в широком интервале температур и частот. Влажность оказывает
на него небольшое влияние (рис. 5.29). Пробивное напряжение
тонких пленок из смеси полидиметилфениленоксида с
полистиролом составляет около 1 кВ. На рис. 5.30 показана зависимость
пробивного напряжения от толщины пленки. Стойкость к токам
утечки смесей полидиметилфениленоксида с полистиролом
позволяет использовать их в производстве электробытовых приборов
(рис. 5.31).
Таблица 5.15. Электрические свойства
полидиметилфениленоксида, его смесей с полистиролом
и композиций, наполненных стеклянным волокном
Показатель
Диэлектрическая
проницаемость
23 °С, 50%-ная Отн.
влажность, 50 Гц — 1 МГц
23 °С, сухой материал,
50 Гц — 1 МГц
66 °С, 50 Гц — 1 МГц
121 °С, 50 Гц—1 МГц
Тангенс угла диэлектрических
потерь tgo-104
23°С, 50%-ная отн.
влажность
50 Гц
1 кГц
1 МГц
23 °С, сухой материал
50 Гц
1 кГц
1 МГц
66 °С
50 Гц
1 кГц
1 МГц
121 °С:
50 Гц
1 кГц
1 МГц
Удельное объемное
электрическое сопротивление, Ом-см
23 °С, сухой материал
23°С, 50%-ная отн.
влажность
66 °С
121 °С
ПФО
2,56
10"
ПФО + ПС
2,68-2,69
2,68-2,69
2,66—2,67
2,66
6,6
5,8
14
4,0
7,5
6,3
10
10"
8,6-1016
1.5- 101в
ПФО + ПС
B0%
волокна)
2,95-2,98
2,93-2,96
2,93-2,96
2,91-2,95
16
14
17
15
15
15
21
16
18
47
24
23
1,6 - 1016
3,5-1016
1,2.10"
5,6-1015
ПФО + ПС
C0%
волокна)
3,11-3,15
3,10-3,13
3,10-3,14
3,08-3,14
20
18
21
18
17
18
29
21
22
59
34
29
3,9 • 101в
З.О-Ю18
6,6-1015
9,4-1015
227
Таблица 5.16. Влияние продолжительности выдержки в воде
на свойства наполненной 30% стеклянного волокна смеси
полидиметилфениленоксида с полистиролом [454]
Условия
23 °С, 50%-ная отн.
влажность
24 °С, в воде
Кипящая вода
Время
выдержки
24 Ч
7 сут
24 ч
7 сут
24 ч
7 сут
Увеличение массы,
%
+0,03
+0,03
+0,06
+0,12
+0,28
+0,28
Увеличение размеров,
%
+0,01
+0,01
+0,01
+0,01
+0,01
+0,01
Химическая стойкость. Полндиметилфениленокснд обладает
еысокой гидролитической стойкостью, особенно к действию
кипящей воды, и незначительным водопоглощеннем, что обеспечивает
высокую стабильность размеров. При стерилизации медицинских
инструментов паром при 135°С их эксплуатационные
характеристики не ухудшаются. В табл. 5.16 показано изменение массы и
размеров изделий из смесей полидиметилфениленоксида с
полистиролом при выдержке их в воде при различных условиях, а в
табл. 5.17 — изменение физико-механических свойств при
многократном воздействии горячего пара в условиях стерилизации при
135°С.
Полимер имеет хорошую стойкость к действию кислот,
щелочей, углеводородов, спиртов и моющих средств (табл. 5.18).
Многократное погружение в стиральный раствор в течение 400 ч при
90°С не сопровождается изменением массы полимера и его
окраски. Недостатком полидиметилфениленоксида является
растворимость в ароматических и хлорированных углеводородах, таких,
как бензол, толуол, дихлорэтилен, трихлорэтилен и хлороформ, а
также появление усталостных трещин при действии
ароматических, циклоалифатических и хлорированных растворителей.
Образование усталостных трещин при изгибающем напряжении
420 кгс/см2 при контакте с гептаном при 26 °С наблюдается для
смесей полидиметилфениленоксида с полистиролом, наполненных
стеклянным волокном, через 7 сут; без нагрузки при контакте с
Таблица 5.17. Влияние обработки горячим паром на свойства
композиций из смеси полидиметнлфениленоксида
с полистиролом, наполненных стеклянным волокном [454]
Стерилизация паром при
135 °С, число циклов
0
25
50
Прочность
при изгибе»
кгс/см2
1575
1 175
1 165
Модуль упругости
при изгибе кгс/см2
73 100
70 300
70 300
Ударная вязкость
образцов с надрезом,
кгс-см/см2
10
8,5
9,1
228
Таблица 5.18. Химическая стойкость
полидиметилфениленоксида [472]
Выдержка 7 сут при 23 °С
Вода
Серная кислота
3%-ная
30%-ная
Соляная кислота
10%-ная
38%-ная
Азотная кислота
Уксусная кислота
Плавиковая кислота,
48%-пая
Увеличение
массы, %
0,10
0,10
0,08
0.10
0,93
0,11
0,13
2,08
Выдержка 7 гут при 23 °С
Щелочной раствор
1%-ный
10%-ный
Аммиак, 10%-пый
Хлорид натрия, 10%-ный
Этанол
100%-иый
50%-ный
Гептан
Увеличение
массы» %
+0.Н
+0,09
+0,15
+0,09
+0.45
+0,34
+0,39
этиленгликолем при 86 °С — через 3 сут и при изгибающем
напряжении 420 кгс/см2 при контакте с масляной кислотой, маисовым
маслом, гептаном и метанолом при 86°С — через 3 сут.
Неориентированная полидиметилфениленоксидная пленка при контакте с
эфиром или «-бутилгалогенидами покрывается трещинами. Двух-
осно-ориентированная пленка при контакте с метанолом, этанолом,
ксилолом, «-бутилгалогенидами, диэтиловым и дифениловым эфи-
рами растрескивается под нагрузкой [473].
Горючесть. Полидиметилфениленоксид и его смеси с
полистиролом в отсутствие специальных добавок горючи. Смесь полимеров,
полученная фирмой «General Electric» типа SE, отвечает
требованиям группы 1 стандарта 94 Underwrites Laboratories: при
поджигании лопатки толщиной 1,6 мм на бунзеновской горелке
затухание наступает через 25 с без образования капель. Для придания
негорючести к полидиметилфениленоксиду добавляют 0,1—5%
красного фосфора [474].
Атмосферостойкость. За 3 года пребывания смеси полидиметил-
фениленоксида с полистиролом на воздухе при 90 °С
физико-механические характеристики уменьшаются на 50 %. У наполненных
стеклянным волокном образцов 50%-ное снижение прочности
происходит за то же время при 110°С. Действие УФ-лучей приводит
к пожелтению полимера на глубину 50 мкм без изменения физико-
механических свойств [460].
Влияние облучения УФ-лучами на свойства полимера изучалось
Сидху [435]. Текучесть полидиметилфениленоксида сильно
ухудшается при облучении на воздухе. Уже после 15 ч облучения
пленки толщиной 50 мкм относительное удлинение при разрыве
снижается с 131 до 3%. Прочность при растяжении после 300 ч
облучения равна 75 % от исходного значения. Ударная вязкость
образцов с надрезом после облучения в течение 100 ч составляет
80%, а после 1000 ч — 72% от исходного значения. Тангенс угла
Диэлектрических потерь уже через 10 ч облучения скачкообразно
229
возрастает с 10~2 до 10 . Это связано с образованием в полимере
карбонильных и гидроксильных групп. Основной максимум
диэлектрической дисперсии в процессе облучения смещается в
область более высоких температур.
5.6.1.7. Применение полидиметилфениленоксида
и его смесей с полистиролом
Полидиметилфениленоксид и его смеси с полистиролом
используют главным образом для изготовления деталей в
электротехнике и электронике, деталей арматуры, оптических и медицинских
приборов, автомобилей.
Смесь полидиметилфениленоксида с полистиролом применяют
в тех случаях, когда требования к формоусгойчивостм не столь
жестки, а полимер должен иметь высокую текучесть. Кроме того,
применение смеси полимеров более экономично: цена 1 кг
полидиметилфениленоксида 11 марок ФРГ, а его смеси с
полистиролом— 6 марок ФРГ. Для достижения более высокой прочности
используют стеклонаполненную композицию. В электротехнике
полидиметилфениленоксид и его смеси с полистиролом применяют
для изготовления выключателей приборов бытового назначения,
многоконтактных выключателей электронных приборов,
программных выключателей, мощных выключателей распределительных
щитов, сердечников катушек, реле и деталей телевизоров, таких,
как переключатели программ. Ткань, пропитанная полидиметил-
фениленоксидом, служит изоляционным материалом для
наполненных маслом кабелей высокого напряжения [475].
Благодаря хорошим физико-механическим свойствам,
стабильности размеров и стойкости к моющим средствам
полидиметилфениленоксид является идеальным материалом для изготовления
деталей стиральных машин, узлов моечных автоматов, таких, как
колеса с лопастями, вентилей, расходомеров и насосов для
щелоков. Наполненные стеклянным волокном смеси
полидиметилфениленоксида с полистиролом не разрушаются промывными
водами при 90°С. Высокая стабильность размеров обеспечивает
применение этих композиций для изготовления регуляторов
давления воды [477]. Бытовые кипятильники для воды,
изготовленные из смеси полидиметилфениленоксида с полистиролом литьем
под давлением, имеют тепловой коэффициент полезного действия
95 % по сравнению с 12,5 % для обычных моделей из металла.
Стоимость таких изделий на 30 % ниже стоимости традиционных
кипятильников; продолжительность эксплуатации оценивается в
10 лет [476]. Фирма «Fa Duropipe» выпускает трубы, работающие
под давлением горячей воды 7 кгс/см2 при 100°С; их стойкость
оценивается в 25 лет [460].
Высокая стойкость к истиранию и гидролизу позволяет исполь-
вовать наполненную стеклянным волокном смесь
полидиметилфениленоксида с полистиролом в производстве двигателей
корабельных насосов [467]. Через 2000 ч работы износ составил мак-
230
симально 0,02 мм по сравнению с 0,03 мм для изделий из латуни.
Стоимость насосов для холодной воды составляет лишь 30 %
их стоимости при изготовлении из латуни.
Хирургические инструменты из полидиметилфениленоксида
великолепно стерилизуются горячим паром (лучше, чем аналогичные
изделия из нержавеющей стали).
Пористый полидиметилфениленоксид без покрытия применяется
в качестве носителя в хроматографии. При использовании в
качестве газа-носителя азота рабочая температура может достигать
435°С, что позволяет разделить олигомеры этиленгликоля и поли-
этиленгликоль [478, 479].
В оптике из смесей полидиметнлфениленоксида с полистиролом
получают детали камер и корпуса проекционных аппаратов. В
производстве точных приборов их используют при изготовлении
печатной техники, электробритв, оборудования для сушки волос. В
автомобильной промышленности полифениленоксид применяют везде,
где необходимы высокая прочность и точность размеров в
широком температурном интервале [480]. Полифениленоксид
применяют для изготовления контактных соединений, изоляторов,
цоколей ламп, термостойких деталей для нагревательных приборов и
кондиционеров, украшений с матовой поверхностью, деталей
холодильников, прежде всего металлизированных наружных частей.
Благодаря высокой термостойкости полифениленокснда эти детали
можно получать методом гальванизации [465].
Высокая стабильность размеров и хорошая перерабатывае-
мость при очень узких допусках позволяют применять
полифениленоксид в производстве уличных и спортивных наручных
часов.
Пленку и пенопласты из полифениленоксида используют в
качестве термоизоляционного материалу для алюминиевых емкостей
или сосудов, работающих под давлением, против термоудара,
например, при контакте с жидким водородом [481, 482].
5.6.1.8. Сополимеры и привитые сополимеры
Описано получение сополимеров 2,6-диметилфенола с 2-метил-
6-трет-бутилфенолом, 2,6*изопропилфенолом, 2-метил-6-фенилфено-
лом и 2,6-дифенилфенолом [483]. Структура сополимера зависит
от реакционной способности 2,6-дизамещенного фенола в реакции
окислительной дегидрополиконденсации. При сополиконденсации
2,6-диметилфенола с 2,6-дифенилфенолом образуется
статистический сополимер [484]. При окислительной
дегидрополиконденсации 2,6-дифенилфенола в присутствии поли-2,6-диметилфенилен-
оксида получают статистический сополимер, тогда как при
подобной поликонденсации 2,6-диметилфенола в присутствии поли-2,6-
дифенилфениленоксида — блок-сополимер. Это объясняется низкой
реакционной способностью 2,6-дифенилфенола. Образование
статистического сополимера обусловлено более быстрым протеканием
обменной реакции мономера А (например, дифенилфенола)
231
с замещенным поли-1,4-фениленоксндом на основе мономера В
(например, 2,6-дпметилфенола) по сравнению с окислительной
поликонденсацией мономера А [485] :
ОН
он
ft]
о
Н3С
сн.
он
kl >k2
Реакция обмена протекает через промежуточную стадию
образования нестабильного хинонокеталя. При этом происходит
превращение арилоксимономера из одного радикала в другой:
О
о
о
о
о
о
232
Обменные реакции ускоряются с повышением температуры и
протекают при низкой концентрации аминов, так как повышение
температуры способствует диссоциации кеталя. По этому
механизму из поли-2,6-диметил-1,4-фениленоксида и поли-2,6-дифенил-
1,4-фениленоксида в присутствии тетраметилдиаминобутан —
бромида меди образуется блок-сополимер. Статистические и блок-
сополнмеры с эквимольным соотношением 2,6-диметилфенокси- и
2,6-дифенилфенокси-звеньев являются аморфными GС = 226°С).
Взаимодействием поли-2,6-диметил-1,4-фениленоксида с фенолами
в присутствии таких инициаторов, как 3,3/,5,5'-тетрафенил-4,4/-ди-
фенохинон, пероксид бензоила или трег-бутиловый эфир надбен-
зойной кислоты, при 100—180°С по аналогичному механизму
получают низкомолекулярный полигидроксиэфир [486] :
СНз
\
н—
СНз
R
Получение блок-сополимера с полиацеталями возможно путем
механохимического взаимодействия обоих гомополимеров [487].
Сополимеры, содержащие полиамидные, простые полиэфирные,
полисилоксановые и поликарбонатные блоки в цепи, образуются
при взаимодействии соответствующих мономеров с олигомерными
фениленоксидами, содержащими концевые гидроксильные группы
[.488, 489]. Прививка стирола к полидиметнлфеннленоксиду
233
осуществляется катионной полимеризацией мономера в растворе
полимера [490]. Другой метод получения привитых сополимеров
состоит во взаимодействии литийсодержащих поли-2,6-диметил- или
поли-2,б-дифенил-1,4-фениленоксидов с такими винильными
мономерами, как метилметакрилат и стирол [491—494]. При
добавлении бутиллития к 2%-ному раствору полимера в тетрагидрофу-
ране при продолжительности реакции до 1 ч деструкции не
происходит.
5.6.1.9. Модификация поли-2,6-диметил-1,4-фениленоксида
Полидмметилфениленоксид, так же как 2,6-диметилфенол,
нитруется, галогенируется и сульфируется. Сульфирование проводят
смесью нитроалкана, например нитрометана с хлорсульфоновой
кислотой, при — 20~100°С [495, 496]. Пленки сульфированного
полимера, отлитые из раствора в метаноле, метаноле —
хлороформе или метаноле — изопропаноле — хлороформе, используют в
качестве катионообменных мембран для обратного осмоса
отработанной воды [497, 498]. Водопроницаемость мембран из
сульфированного полидиметилфениленоксида в 8—10 раз выше, чем у
гомогенных мембран на основе ацетата целлюлозы или
удерживании 95% соли в ходе извлечения хлорида натрия из 1 %-ного
раствора. Для хлорирования полидиметилфениленоксида в ядро
в качестве растворителя используют тетрахлорид углерода, а
катализатора— FeCb- Хлорирование в боковую цепь при облучении
УФ-лучами сопровождается частичным хлорированием в ядро
([499, 500]. Хлорирование осложняется сильным снижением
молекулярной массы и термостойкости. При 26 %-ном содержании
хлора температура начала деструкции на воздухе составляет 184 °С
(хлорированный в ядро полимер) и 174 °С (хлорированный в
боковую цепь полимер).
Эпоксидированием сополимера 2,6-диметилфенола с аллилфе-
нолом получен полимер с эпоксигруппами в цепи [501—503].
Удлинение цепи полидиметилфениленоксида возможно при
взаимодействии полимера с бис(л-хлорфенил)сульфоном, дигалогенан-
гидридами или диизоцианатами в растворе [504]. Для
структурирования полидиметилфениленоксида используют его
функциональные производные, которые сшивают либо в процессе переработки
из расплава, либо при взаимодействии с бифункциональными
соединениями [505, 506].
Полидиметилфениленоксид с гидроксильными группами
получают окислительной дегидрополиконденсацией в присутствии гид-
роксидов щелочных или щелочноземельных металлов [507] ; под
влиянием формальдегида или дикарбоновых кислот в присутствии
кислот Льюиса полимер сшивается [508, 509]. В качестве
сшивающих агентов для полидиметилфениленоксида пригодны также
я-малеидобензоилхлорид или Г^-B-хлорэтил) -тетрагидронафтал-
имид [510].
5.6.2. ПОЛИМЕТИЛЕНДИФЕНИЛОКСИД
Олигомерные простые полиэфиры используют в качестве
пропитывающего электроизоляционного лака. Термически отвержден-
ный полимер может быть использован при температурах до 250°С.
Полиметилендифенилоксид выпускается в промышленном
масштабе с 1965 г. под названием «Дорил» фирмой «Westinghouse
Micarta Division» (Хэмптон, США) [511, 512].
5.6.2.1. Получение полиметилендифенилоксида
Исходным продуктом для получения
полиметилендифенилоксида (ПМФО) служит дифенилоксид, который хлорметилированием
превращается в смесь гомологов и изомеров с различным
количеством хлора. Олигомеры получают двумя методами. По первому
методу гомополимеры получают из хлорметилированного продукта
с метилатом натрия, который превращается в соответствующее
метоксисоединение из которого в присутствии катализаторов Фри-
деля — Крафтса, таких, как FeCb (около 0,008% в расчете на
мономер), образуются олигомеры со степенью полимеризации около
9 [513]. По второму методу хлорметилированный дифенилоксид
взаимодействует с фенолом:
С1Н2С СН2С1
—HC1
,0H
Вследствие различной функциональности исходных продуктов
образующиеся олигомеры представляют собой сильно разветвленные
соединения с молекулярной массой 500—1300 и температурой
размягчения 55—115°С. При взаимодействии с формальдегидом
получаются термореактивные продукты.
5.6.2.2. Получение композиций
на основе полиметилендифенилоксида
Гомополимер растворяется в ароматических растворителях,
например в толуоле. Сополимеры с фенолом растворяются в
смесях растворитель — нерастворитель, например этанол — толуол
A : 1). Жизнеспособность этого раствора 6 мес. Достоинством
связующих на основе ПМФО является длительное пребывание в
235
стадии В. Препрсги, клеевые пленки и гибкие ленты сохраняются
в течение многих лет. Ниже 110°С отверждения не происходит.
В стадии В при содержании сухого остатка до 60 % растворы
ПМФО имеют относительно низкую вязкость, так что в них можно
вводить до 30 % наполнителя. Аналогично фенольным смолам"
слоистые пластики на основе ПМФО прессуют при 170 °С под
давлением 50—70 кгс/см2; дополнительную обработку проводят
при 350°С. Композиции, наполненные асбестовым и стеклянным
волокном, прессуют при 170 °С и давлении 70 кгс/см2 в течение
15—17 мин.
5.6.2.3. Свойства и применение лолиметилендифенилоксида
Отвержденный ПМФО инертен по отношению к алифатическим,
хлорированным и ароматическим углеводородам. Он не
разрушается под действием кислот и щелочей. Температура длительной
эксплуатации составляет 250 °С A0 000 ч); кратковременно
полимер выдерживает нагревание до 300 °С [514]. ПМФО
используют в качестве электроизоляционного лака для катушек,
конденсаторов, сопротивлений, электродвигателей и
трансформаторов. Вследствие того что полимер может находиться в течение
длительного времени в стадии В, его можно наносить на провода
и после намотки проводить отверждение непосредственно на
катушке.
Слоистые пластики по термостойкости лишь незначительно
уступают материалам на силиконовых и полиимидных связующих.
Потеря массы при старении на воздухе при 260°С составляет
2,1 % через 100 ч и 7,8 % через 500 ч. Прочность при изгибе и
электрическая прочность при этой температуре практически не
меняются в течение 1000 ч. Полиметиленднфенилоксид используют
в электротехнике в качестве основы печатных схем, для изоляции
слоев обмотки в трансформаторах. Для трубок с толщиной стенки
3,1 мм электрическая прочность составляет 360 kB/мм, прочность
при сжатии 650 кгс/см2 и диэлектрическая проницаемость 4,6.
Показатели свойств изделий из наполненных стеклянным и
асбестовым волокном композиций на основе ПМФО связующего
представлены ниже [512]:
Плотность, г/см3
Прочность при изгибе, кгс/см2
Прочность при сжатии, кгс/см2
Усадка, %
Водопоглощение, B5 °С, 24 ч), %
Электрическая прочность, кВ/мм
Удельное поверхностное электрическое
сопротивление, Ом
сухой образец ... . ...
после выдержки в течение 24 ч в воде . .
Тангенс угла диэлектрических потерь . . . .
36
Стеклянное
волокно
1,76-1,85
1000-1300
1800-2100
0,35-0,50
0,10-0,16
13-16
2-Ю12
2- 1010
0,01-0,015
Асбестовое
ВОЛОКНО
1,85-1,95
900—1000
0,70-0,90
0,10-0,16
13-15
2- 10"
2- 109
0,05-0.08
5.6.3. ПОЛИГИДРОКСИЭФИР
—OCH2CHCH2O—
он
Полигидроксиэфир является термопластичным полимером,
получаемым взаимодействием двухатомных фенолов с эпихлоргид-
рином. В отличие от эпоксидных смол, получаемых из тех же
исходных соединений, полигидроксиэфир имеет большую
молекулярную массу и не содержит эпоксигрупп. В термопластичном состоянии
его можно перерабатывать в прутки, пленку, различные детали.
Изделия из полигидроксиэфира имеют низкую
газопроницаемость, высокую жесткость и хорошие физико-механические
свойства в температурном- интервале от —60 до 80 °С. Основная область
применения—покрытия по металлу и клеи для металла (твердые
клеи). Для покрытий используют также и растворы.
Полигидроксиэфир под торговым названием «фенокси» выпускают с 1962 г.
из эпихлоргидрина и дифенилолпропана фирма «Union Carbide»
и с 1963 г. фирма «Bakelite». В 1968 г. цена 1 кг составляла 2 дол.
(США).
5.6.3.1. Получение полигидроксиэфира
Синтез термопластичного полигидроксиэфира протекает
аналогично синтезу эпоксидных смол путем полиприсоединення с
последующим отщеплением НС1 на промежуточной стадии:
он + сн2—ch-ch2ci
о
СНз
С—О—ОСН2СНСН2С1
ОН"
сн.
он
NaOH
—fia Cl
—н2о
СНз
НО—( У)—С— ( ))—|
1
СНз
О
—о—
|он'
СНз
I
-с—((у)—осн2снсн2-
I Ч^/ I
СНз ОН
237
При взаимодействии эпоксидной группы с фенольным гидро-
ксилом сначала образуется хлоргидрин. После дегидрохлорирова-
ния происходит полиприсоединение. Полиприсоединение должно
проводиться в таких условиях, чтобы исключить образование
сильно разветвленных или сшитых структур рследствие участия
вторичных алифатических гидроксильных групп в процессе
полиприсоединения на глубоких стадиях превращения. При синтезе
низкомолекулярных полигидроксиэфиров для подавления образования
разветвлений процесс следует проводить в мягких условиях или
в присутствии избытка эпихлоргидрина. Высокомолекулярные
полимеры получают в две стадии. На первой ста/щи за счет избытка
эпихлоргидрина образуется относительно низкомолекулярный
продукт, например, rc/i'-диглицидиловый эфир дифенилолпропана.
После удаления избытка реагента в присутствии
соответствующего катализатора, например моногидрата гидроксида лития или
гидроксида бензилтриметиламмония, проводят реакцию с чистым
дифенилолпропаном. При проведении реакций в таких
растворителях, как метилэтилкетон, диоксан, тетрагидрофуран или смесь
толуол — бутанол F0:40), молекулярная масса полигидроксиэфира
может достигать 120000.
Для придания полимеру технологических свойств, характерных
для термопласта, в процессе синтеза в систему добавляют
смешивающийся с водой растворитель; при добавлении избытка воды
происходит выделение полигидроксиэфира. Полимер, который
перерабатывается экструзией или литьем под давлением, имеет
молекулярную массу 30 000—40 000. Продукт с высоким выходом
при 80 °С получается при проведении процесса в течение 60 ч
[515]. На второй стадии реакции (без растворителя) образуется
полимер с молекулярной массой 5000 [516]. При добавлении на
этой стадии других бисфенолов получаются чередующиеся
сополимеры [517]. Этерификацией вторичных гидроксильных групп
кислотами, их ангидридами или хлорангидридами можно повысить
эластичность и понизить горючесть полигидроксиэфира.
Самозатухающий полимер получается при введении дифенилфосфиниль-
ных и хлорацетатных групп. При добавлении эфиров тиоуксусной
кислоты в присутствии пероксидов или при длительном контакте
с кислородом происходит сшивание полимера [518]. В случае
применения бифункциональных компонентов, например фенольных
смол с метилольными группами, эпоксидных смол или
полиуретанов [519], образуются термореактивные полимеры.
5.6.3.2. Физико-химические свойства полигидроксиэфиров
Температура стеклования. Температура стеклования зависит
от природы фенольного остатка в цепи. Промышленный полигид-
роксиэфир на основе дифенилолпропана и эпихлоргидрина имеет
температуру стеклования 100°С. Наличие между фенильными
ядрами полярных, например сульфоновых, групп повышает
температуру стеклования за счет образования водородных связей с гид-
роксильными группами. Повышение температуры стеклования про-
238
исходит и за счет присутствия алициклических мостиков, которые
увеличивают жесткость цепи вследствие ограничения вращения
ароматических ядер [519]. Этерификация гидроксильных групп
полигидроксиэфиров сопровождается понижением температуры
стеклования на 20—30 °С [518]. Выше температуры стеклования
полигидроксиэфиры представляют собой эластомеры с низкой
прочностью. Ниже приведены температуры стеклования
различных полигидроксиэфиров [519]:
гс. -с
—о—(Cj)—о—сн2—сн—сн2— 60
он
СНз
I
—С-СН2—СН—(Г))—О—СН2—СН-СН2- 75
| | \^7 |
СНз СНз ОН
—°—О—СН2—U^>—О-СН2—СН-СН2- 80
он
С\ СНз
—°—О—с—Ua>—О-СН2-СН—СН2- 85
СНз ОН
-О—(ГУ)—СН—/PS\—О-СН2—СН-СН2- 95
СН(СН3J ОН
СНз
—°—(С))—с—(СЪ—О-СН2-СН—СН2- 100
СНз ОН
С\ СНз /CI
/Г^)—С—/ГЪ—О—СН2—СН—СН2— 115
Cl/ СНз \ci 0Н
СНз
-О—(Г))—С—(Г))—О—СН2—СН—СШ— 115
W// | \\~•/ I
он
СНз
^ "(Ъ—О-СН2-СН-СН2— 120
I
он
н3с снэ
c, ec
О—СН2—СН—СН2 — 135
ОН
¦'—О—СН2—СН—СН2— 140
ОН
—О—СН2—СН—СН2- 155
ОН
-о-
СНз
Q)—О—СН2-СН-СН2— 175
он
СН(СНзJ
Растворимость. Полигидроксиэфир растворяется в полярных
растворителях, таких, как эфиры, кетоны, простые циклические
эфиры и хлорированные углеводороды. Термодинамически
хорошие растворители располагаются в следующий ряд: диметилформ-
амид > метилэтилкетон >> метоксиэтанол > диоксан >> циклогек-
санон >> хлороформ. Хорошие осадители располагаются в
следующей последовательности: бензол < тетралин < толуол <С тетра-
хлорид углерода -< бутанол < гексан [520].
Для фракционирования полигидроксиэфира используют элю-
энтный метод [521], метод Бейкера — Вильямса [522],
гель-проникающую хроматографию [523]. В случае элюэнтного метода в
качестве носителя используют песок, обработанный триметилхлор-
силаном, для того чтобы избежать необратимой сорбции полимера
в процессе фракционирования. В качестве элюента используют
смесь хлороформа с гексаном при 31 °С. По методу Бейкера —
Вильямса фракционирование проводят при температурном
градиенте 30—70 °С с использованием в качестве элюента смеси
хлороформ— тетрагидрофуран (от 1 : 3 до 3 : 7). Средняя
молекулярная масса промышленного полигидроксиэфира из днфенилолпро-
пана и эпихлоргидрина составляет 15 000—45 000. По данным
фракционирования, полидисперсность колеблется от 1,5 до 3,5
[521]. Для определения средней молекулярной массы используют
методы светорассеяния, ультрацентрифугирования и
вискозиметрию. Светорассеяние проводят в хлороформе и тетрагидрофуране
при 25 °С; соответствующие инкременты показателя преломления
240
Таблица 5.19. Коэффициенты в уравнении Марка — Хаувиика
для полигидроксиэфира из дифенилолпропана
и эпихлоргидрина [520, 521]
Растворитель
Тетрагидрофуран
Циклогексанои
Хлороформ
Дноксан
Температура, °С
25
30
30
30
/(¦ю-4
1,48
2,86
2,53
2,02
а
0,765
0,714
0,691
0t743
Растворитель
Диметилформ-
амид
Толуол —
тетрагидрофуран*
B:1)
Температура, °с
30
30
/ЫО
1,165
16,3
а
0,797
0,50
* 6—растворитель.
составляют 0,172 и 0,221 мл/г [521]. Для седиментации в
ультрацентрифуге при 30 °С используют смесь толуол — тетрагидрофуран
A:2). Константа седиментации связана с молекулярной массой
уравнением: 50 = 0,0165-M0-51 [520]. В табл. 5.19 приведены
значения коэффициентов в уравнении Марка — Хаувинка.
На графике зависимости логарифма характеристической
вязкости от среднемассовой молекулярной массы при Mw больше
70 000 наблюдается отклонение от теоретической прямой, которое
свидетельствует о том, что при таких молекулярных массах
существуют длииноцепные разветвления [521]. Другим фактором,
свидетельствующим о появлении длинноцепных разветвлений,
является изменение в этой области физико-механических свойств:
Молекулярная масса
45000 54000 76000
расплава B20 °С,
Показатель текучести
3 кгс/см2), г/мин . .
Температура стеклования, °С
Относительное удлинение при разрыве, %
Ударная вязкость по Изоду, кгс-см/см2 .
0.880
90
7-20
60
0,330
85
20
50
0,068
100-105
60-150
133
5.6.3.3. Переработка полигидроксиэфира
на основе дифенилолпропана и эпихлоргидрина
Текучесть. Переработку полигидроксиэфира на основе
дифенилолпропана и эпихлоргидрина проводят из расплава в интервале
температур 220—280 °С. Зависимость вязкости расплава от
скорости сдвига показана на рис. 5.32, а. Вязкость расплава
полигидроксиэфира зависит от температуры гораздо сильнее, чем
вязкость полистирола и полиэтилена (рис. 5.32,6). Это позволяет
легко регулировать вязкость расплава, изменяя температуру
переработки.
Экструзия. Экструзию полигидроксиэфира проводят при 150—
200°С и давлении 70—775 кгс/см2. Так же как и в Случае
полиэтилена и полиамида, при переработке полигидроксиэфира
841
/¦яг
2-Ю6
1-Юв
2-Ю5
НО5
\3
\
O
F
150
200
250 t?G
Рис. 5.32. Кривые текучести полигидрокси-
эфира [524]:
а — вязкость расплава в зависимости от скорости
сдвига (длина сопла 0,1—25 мм, диаметр 2,1 мм);
6 — вязкость расплава в зависимости от
температуры; / — ударопрочный полистирол, 100 с~ ;
2 — полиэтилен низкого давления, 100 с" ; 3 —
полигидроксиэфир иа основе дифеннлолпропана
и эпнхлоргидрина, 10 с~';3' —то же, 100 с.
необходимо применять шнеки с
высоким отношением сжатия.
Эластические свойства полигидроксиэфира
при низких температурах позволяют
получать из него пленки методом
раздува [525].
Литье под давлением. Для литья
под давлением используют
обычные литьевые машины шпекового
типа со шнековой пластикацией и
плунжерного типа; температура
220—275 °С, давление 700—1400
кгс/см2, оптимальная температура
пресс-формы 60 °С [524].
Покрытия. Покрытия из
полигидроксиэфира наносят в кипящем
слое или из раствора [526]. При нанесении из раствора
некоторые затруднения возникают из-за невысокой скорости
испарения полярных растворителей. Для покрытия толщиной 10 мкм
продолжительность сушки составляет 1 ч. Ускорение сушки
достигается использованием смеси растворителей или наполнителей.
Покрытия, отверждающиеся при комнатных температурах,
получают с применением изоцианатов и производных триазина.
Отверждающиеся при нагревании рецептуры содержат в качестве от-
вердителя мочевино-, феноло- или меламиноформальдегидные
смолы.
Склеивание полигидроксиэфиром металлических и
неметаллических изделий. Для склеивания используют пленочный полигидр-
оксиэфирный твердый клей. Пленку наносят между
склеиваемыми частями и кратковременно нагревают под давлением
15 кгс/см2. Температура нагрева при склеивании металлов в
течение нескольких секунд колеблется от 315 до 345 °С. При 260 °С
продолжительность склеивания составляет минуты, а при 190 °С —
0,5 ч. Неметаллические изделия склеивают при 150—200°С.
Толщина клеевого слоя для непористых поверхностей равна 10—
100 мкм. Прочность клеевого шва при сдвиге составляет 250 кгс'см2
[527]. Склеиваемые поверхности должны быть предварительно
обработаны растворителем и слегка опескоструены. Наилучшие
результаты достигаются при травлении поверхности хромовой
смесью.
5.6.3.4. Свойства полигидроксиэфира
на основе дифенилолпропана и эпихлоргидрина
Свойства полигидроксиэфира на основе дифенилолпропана и
эпихлоргидрина приведены ниже [527, 531, 532]:
Температура стеклования, °С 100
Плотность, г/см3 1,17—1,18
Показатель преломления 1,5978
Прочность при растяжении, кгс/см2 640
Относительное удлинение при, разрыве, % 50—100
Модуль упругости при растяжении, кгс/см2 27 000
Прочность при сжатии, кгс/см2 730—840
Модуль упругости при сжатии, кгс/см2 22 500—24 000
Прочность при изгибе, кгс/см2 . 1 000
Модуль упругости при изгибе, кгс/см2 29 000
Ударная вязкость, кгс-см/см2 65
образцов с надрезом 13
Твердость по Роквсллу 123
Деформационная теплостойкость под нагрузкой 4,7 кгс/см2,
°С 90
Коэффициент теплопроводности, А,-10~4, кал/(см-с-°С) ... 4,2
Термический коэффициент расширения, 1/°С
от -30 до 65 °С 3,2 • 10-в
от-65 до 65 °С 3,8-10-°
Усадка, % 0,40 Водопоглощение за 24 ч,
Электрическая прочность, % 0,13
кВ/мм 20,8 Газопроницаемость плен-
Удельное объемное элек- ки толщиной 24 мкм,
трическое сопротивление, см3/25 мкм/24 ч/см2
Ом-см 5-Ю3 кислород 1
Диэлектрическая проницае- диоксид углерода . . 35
мость водяной пар .... 0,6
при 60 Гц 4,1 Удельная теплоемкость,
при 1 МГц .... 3,8 кал/(г-°С) 0,4
Тангенс угла диэлектриче- Горючесть Медлен-
ских потерь но заго-
при 60 Гц 0,0012 рается
при 1 МГц .... 0,030
По таким физико-механическим свойствам, как прочность при
растяжении, твердость, жесткость, ударная вязкость и удлинение,
полигидроксиэфир близок поликарбонату.
На рис. 5.33 показаны кривые ползучести различных
полимеров. Несмотря на относительно высокое начальное значение, за
3000 ч удлинение возрастает незначительно. Этерификация
полигидроксиэфира длннноцепными кислотами, такими, как
стеариновая, приводит к трехкратному увеличению относительного
удлинения при разрыве при сильном уменьшении прочности. Этерифи-
кация бензойной кислотой приводит к увеличению прочности,
однако относительное удлинение при разрыве при этом уменьшается
на 4% [529]. Физико-механические свойства сильно зависят от
молекулярной массы полимера (рис. 5.34). При увеличении
молекулярной массы с 30 000 до 80 000 ударная вязкость
возрастает в 5 раз. В температурном интервале от —60 до 80 °С физико-
механические свойства относительно мало зависят от температуры.
При приближении к температуре стеклования происходит резкое их
243
1UQUQ
2000
0 40000 вОООО 120000 Hw
Рис. 5.33. Кривые ползучести различных полимеров при 23 °С [528]:
/ — полигпдрооксиэфир иа основе дифенилолпропана н эпихлоргидрина, 210 кгс/см2; 2 —
поликарбонат, 280 кгс/см2; 3 — полиформальдегид, 210 кгс/см2.
Рис. 5.34. Зависимость ударной вязкости полигидроксиэфира от средней
молекулярной массы [519].
ухудшение. Модуль упругости при изгибе при 80 °С сохраняет
70 % от значения при комнатной температуре по сравнению с
50 % для полиформальдегида и АБС-пластика и 30 % Для
полипропилена [530].
По электрическим свойствам полигидроксиэфиры аналогичны
отвержденным эпоксидным смолам. Однако при температурах,
близких температуре стеклования (выше 80 °С), наблюдается
резкое ухудшение электрических характеристик.
Полимер имеет исключительно низкую, близкую к полиэтилен-
терефталату газопроницаемость для кислорода и диоксида
углерода. Этерификация вторичных гидроксильных групп приводит к
повышению проницаемости [518]. Химическая стойкость и
стойкость к действию погодных факторов хуже, чем у поликарбоната.
Полигидроксиэфир стоек к действию 10 %-ного гидроксида натрия,
10 %-ной серной кислоты, 10 %-ной азотной кислоты, 10 %-ного
аммиака, глицерина, минеральных и растительных масел.
Полимер набухает и даже растворяется в бензине, кетонах, сложных
эфирах, ароматических хлорированных углеводородах. При эте-
рификации вторичных гидроксильных групп происходит
увеличение стойкости полимера к действию полярных растворителей.
Двухосно-ориентированные пленки склонны к растрескиванию под
нагрузкой только при контакте с диэтиловым эфиром и
хлорбензолом [473]. Воздействие внешних погодных факторов приводит к
пожелтению и появлению хрупкости. Термическая деструкция
незначительна до 200 °С. Этерификация полигидроксиэфира
вызывает снижение эластичности при одновременном улучшении
химической и термостойкости.
5.6.3.5. Применение полигидроксиэфира
на основе дифенилолпропана и эпихлоргидрина
Полимер может эксплуатироваться при температурах до 80 °С.
Он используется главным образом в качестве твердого клея и в
виде покрытий. Его применяют для склеивания стали, алюминия,
244
латуни, древесины, бумаги, тканей, полистирола, полиакрплатов,
стекла, керамики и асбеста. Особенно ценным является
возможность склеивания им металлических труб. Высокая прочность при
сдвиге (до 250 кгс/см2) позволяет склеивать элементы
строительных конструкций. Полигидроксиэфир пригоден в качестве
связующего для ферритовых кристаллов к магнитофонным лентам. Из-за
низкой атмосферостойкости он применяется в лаковой технике
только как грунтовка по металлам, но хорошо совмещается с
лаками на основе алкидных, эпоксидных смол и хлоркаучука,
используемых для декоративных покрытий. Особенно пригодны
растворы полимера для высоконаполненных цинком грунтов, так
как металлический компонент в силу высокой полярности
раствора, находится в суспензии. Коррозионно-стойкие покрытия
облегчают последующее сваривание металлических деталей. Лаки
используют также для лакирования консервных банок. Благодаря
высокой стабильности размеров изделия, полученные литьем под
давлением, применяют в электротехнике и приборостроении [533].
5.6.4. ПОЛИАРИЛЕНСУЛЬФОНОКСИДЫ
Полиариленсульфоноксиды получают взаимодействием галоге-
нированных диарилсульфонов с фенолятами металлов или
конденсаций по Фриделю—Крафтсу хлорсульфированных простых
ароматических эфиров. Полиариленсульфоноксиды с
алифатическими группами в основной цепи отличаются стойкостью в
процессе длительного термостарения при 150 °С:
СНз
СНз
—О
п
Для аналогичных чисто ароматических полисульфоноксидов эта
температура составляет 180—260 °С:
Переработка осуществляется обычными методами, применяемыми
для термопластов. Основными областями применения таких
полимеров является электротехника, электроника и самолетостроение.
Полиариленсульфоноксиды и их композиции с АБС-пластиками
в промышленности производятся с 1965 г.
5.6.4.1. Получение полиариленсульфоноксидов
Полиариленсульфоноксиды с алифатическими группами в
основной цепи. Полисульфоны на основе бисфенолов с центральным
углеродным атомом между ароматическими ядрами получают
поликонденсацией бисфенолятов щелочных металлов с галогени-
рованными дифенилсульфонами в таких безводных полярных
245
апротонных растворителях, как дпметилсульфоксид, диметнлформ-
амид или сульфолаи(тетрагидротиофен-1,1-диоксид) в интервале
температур 130—250 СС [535,540,541]:
130-250 °C
—Cl —
Скорость нуклеофильного замещения галогена, связанного с
ароматическим ядром, фенолят-ионом зависит от основности
фенолята и электроотрицательности заместителей в дигалогенсодер-
жащем соединении. Полисульфон на основе дифенилолпропана и
дихлордифенилсульфона выпускается с 1964 г. фирмой «Union
Carbide» под названием «Бакелит Р 1700» (литьевая масса) и
«Бакелит Р 3500» (экструзионная масса). В 1970 г. объем
производства составил 13 000 т. Цена 1 кг полимера составляет 13
марок ФРГ. Наполненные 30 и 40 % стеклянного волокна литьевые
композиции ZNPGF 1006 или ZNPGF 1008 рекомендованы для
процессов с жидким азотом.
В сочетании с АБС-пластиками фирма «Uniroyab выпускает
продукт марки «Арилон». Полисульфоноксид, совмещенный с
сополимером стирола с акрилонитрилом, выпускается фирмой
«Union Carbide» под маркой «Юкардел Р 4174». С 1973 г. эта
фирма выпускает полисульфон ВХ L627. Такой полисульфон
получают из дифенилолпропана и дихлордифенилсульфона при
150—160 °С: дифенилолпропан растворяют в смеси хлорбензол —
диметилсульфоксид C:1) при 60—80 °С и вытесняют воздух
инертным газом. После добавления стехиометрического
количества 50%-ного раствора NaOH образуются две фазы: верхний
слой — хлорбензол и нижний слой — раствор натриевой соли
дифенилолпропана в водном диметилсульфоксиде. После азеотропной
отгонки воды с хлорбензолом при 140 °С натриевая соль
дифенилолпропана выпадает из раствора в осадок и вновь растворяется
после удаления избытка азеотропа при 155—160°С.
Поликонденсацию проводят введением стехиометрического количества
дихлордифенилсульфона E0%-ный раствор в хлорбензоле) с
последующей отгонкой растворителя при 160 °С. Для достижения
необходимой молекулярной массы смесь выдерживают при 150—
160 °С. Температура не должна опускаться ниже 150 °С во
избежание выделения полисульфона на стенках реакционного сосуда.
При температурах выше 160 °С разлагается растворитель и
происходит структурирование поликонденсата. По достижении
необходимой молекулярной массы поликонденсацию прекращают,
вводя метиленхлорид, при этом происходит этерификации
концевых фенольных групп. После охлаждения раствора полимера и
разбавления хлорбензолом выделяющуюся поваренную соль
отфильтровывают, осаждают полимер в этаноле и после отделения
высушивают при 135°С; выход 90—98 %. Промышленный полимер
246
имеет молекулярную массу 30 000-60 000 [535-539]. Для
достижения высокой молекулярной массы необходимо очень точное
соблюдение стехиометрического соотношения.
Влияние состава реакционной смеси на приведенную вязкость
образующегося полисульфона показано на рис. 5.35, а. В
присутствии воды одновременно с поликонденсацией протекает гидролиз
дихлордифенилсульфона с образованием натриевой соли. Поэтому
молекулярная масса зависит от содержания воды в системе, это
связано с нарушением стехиометрии реагирующих веществ.
Влияние количества воды на приведенную вязкость полимера
показано на рис. 5.35,6. В незначительной степени происходит
также и гомополиконденсация натриевой соли днфенплолпропана.
Образующийся в результате гидролиза NaOH вызывает
деструкцию полисульфона за счет атаки по активированным простым
эфирным связям, находящимся в /г-положенпн к сульфогруппе
[540].
Нерастворимость Li-, Ca- и Mg-солей дифеннлолпропана в ди-
метилсульфоксиде делает их непригодными для синтеза. В то
время как реакционная способность дихлор-, дибром- и дииодпроиз-
водных дифенилсульфона примерно одинакова, в сравнимых
условиях скорость реакции с дифторпроизводными значительно выше.
Методами ИК- и ЯМР-спектроскопии было показано, что
поликонденсация сопровождается образованием связей в положении
1,4; о- и ^-соединение ароматических ядер отсутствует [535].
Нуклеофильное замещение хлора происходит по
бимолекулярному механизму. Ускоряющее действие диметилсульфоксида в
ходе поликонденсации объясняется преимущественной сольватацией
катионов. Материал Арилон фирмы «Uniroyal» на 48—60 %
состоит из полиариленсульфоноксида и на 40—52 % из АБС-пласти-
ка. Полисульфон получают из дихлордифенилсульфона и смеси
бисD-гидроксифенил)-/г-диизопропилбензола с дифенилолпропа-
ном. Поликонденсацию калиевых солей проводят в смеси сульфо-
лан — бензол. Смешение полимера с АБС-пластиком осуществляют
в смесителе в течение 2 мин при 238 °С с последующей экструзией
при 190 °С [542—545]. Известны также композиции с полиэтиленом«
Рис. 5.35. Влияние
соотношения реагентов
(а) н содержания
воды в днфенилолпропа-
не (б) на
молекулярную массу
полисульфона, полученного
поликонденсацией
дихлордифенилсульфона
с натриевой солью
дифёнилолпропана
[535J.
96 98 100 102 Ш
Л W~Na -со/»! моль] моль
О 1 2 S 4 5 6 ?
,НаО -Na - co/ibj no/rbjno/гь
247
полипропиленом [546], каучуком [547—549], винилсодержащим
привитым полипентенамером [550], полиамидом [551],
поликарбонатом [553] и полиамидоимидом [552].
Полностью ароматический полисульфоноксид. Полностью
ароматический полисульфоноксид, в котором ароматические ядра
связаны только кислородными и сульфоновыми группами,
получается по реакции Фриделя — Крафтса из ароматических сульфо-
галогенидов
Кат п /j^~^\
[so2—(Q)—о
?
или из ароматических я,я'-дисульфогалогенидов и многоядерных
ароматических соединений [554, 561]
nClSO2
Введение сомономеров дает возможность получать сополимеры с
желаемыми технологическими и потребительскими свойствами.
Сополимеры, в которых сульфогруппы расположены между
дифенильными и дифенилоксидньши группами, выпускаются с
1967 г. в промышленном масштабе фирмой «Minnesota Mining and
Manufacturing Company C M Со.)» под названием «Астрел
Бранд 360» (экструзионный и литьевой сорта, ГС = 288°С) и 380
(для покрытий, Тс = 310°С). Цена 1 кг экструзионного полимера
50 дол. США. Температура длительной эксплуатации 260°С.
С 1972 г. фирма ICI производит легко перерабатывающийся поли-
ариленсульфоноксид, в котором, как полагают, дифенилоксидные
звенья связаны только сульфоновыми группами. Температура
длительной эксплуатации этого полимера 180 °С [555, 556].
Взаимодействие ароматических дисульфохлоридов с двухъядер-
ными ароматическими соединениями или поликонденсацию ди-
арилмоносульфохлоридов проводят в таких растворителях, как
нитробензол, ^-хлорнитробензол, тетрахлорэтилен, диметилсуль-
фон, тетраметплсульфон, ди-п-толуолсульфон, в интервале
температур 100—250 °С. Концентрация реагентов составляет 30—50%
[557—560]. Растворитель должен обладать достаточно высокой
растворяющей способностью по отношению к полисульфону и
быть инертным к электрофильной атаке сульфохлоридом. В
качестве катализаторов Фриделя — Крафтса применяют FeCb,
SbCU, MoCU, 1пС13 или трифторметансульфокислоту в
количестве 0,1—1 % (мол.). При поликонденсации в нитробензоле при
130°С приведенная вязкость A %-ный раствор в диметилацет-
амиде при 25 °С) через 20 ч составляет 0,3—0,4 A00 мл/г) [561,
562]. Наличие сульфогрупп дезактивирует оба соседних
ароматических ядра и препятствует дальнейшему замещению. В качестве
сомономера для дисульфохлорида используют соединения не ме-
248
нее чем с двумя ароматическими ядрами. Бензол как реагент инги-
бирует дальнейшую поликонденсацию. Дезактивирующий эффект
сульфогруппы приводит исключительно к моносульфированию в
ароматическом ядре. При стехиометрическом соотношении
реагентов и оптимальной температуре образующийся полисульфон не
имеет разветвлений и не содержит гель-фракции.
Ниже перечислены мономеры, пригодные для получения
полностью ароматических полисульфонов по методу Фриделя — Крафт-
са [562]:
C1-SO,—(Q
so2-ci
212-213
131-132
219—220
С/ so2-
ci-so,—\О)~
(_)) 69,5-70,5
140-141
,O>-<O)-
CH,
SO-C1
SO2—Cl
95-96
115-116
158,5-160
249
S02—Cl
128-129
Ароматические полисульфоны можно синтезировать также го-
мополиконденсацией солей щелочных металлов ароматических
л-гидрокси-л-галогензамещенных моносульфонов [564, 565]:
При нагревании полимеров с 1—2 % дифенилоксид-4,4'-дисуль-
фохлорида при 200—340 °С происходит их структурирование [566,
567]. Полимеры структурируются также при облучении быстрыми
электронами и у-лучами дозой 50 Мрад в присутствии 1 % серы
при 300 °С. Текучесть структурированного продукта
обнаруживается через 24 ч при 270 °С, в то время как несшитый полимер при
этой температуре проявляет пластическое течение уже через
10 мин [568].
5.6.4.2. Физико-химические свойства полиариленсульфоноксидов
Температура стеклования. Полиариленсульфоноксиды
представляют собой аморфные полимеры с температурой стеклования выше
170°С. Температура стеклования зависит от соотношения простых
эфирных и сульфосвязей в элементарном звене, характера
соединения ароматических ядер, а также от строения и содержания
алифатических групп в цепи полимера.
В табл. 5.20 полиариленсульфоноксиды расположены в порядке
повышения температуры стеклования. Температура стеклования
полимеров, содержащих только л-замещенные фениленовые
группы, соединенные между собой простыми эфирными и сульфоно-
выми связями, повышается с увеличением количества сульфоно-
вых групп и доли непосредственно связанных между собой фе-
ниленовых циклов в элементарном звене полимера. Для звена,
содержащего четыре связанных в «-положении фениленовых цикла,
наблюдается следующая зависимость между химическим
строением и температурой стеклования:
Соотношение сульф о новые/простые эфирные
связи 1:3 1:2 1:1 2: 1
Температура стеклования, °С 180 210 220 275
Температура стеклования поли-л-дифенилсульфона (две суль-
фогруппы на четыре фениленовых цикла) выше 400°С.
Температура стеклования полимера на основе дихлордифенилсульфона и
бисфенолов с центральным углеродным атомом между ароматичен
250
скими ядрами типа
п
повышается с увеличением количества и объема заместителей. По-
лисульфоноксиды, в которых центральный углеродный атом
входит в состав цикла, находящегося в боковой цепи, имеют
температуру стеклования 200—300 °С [569].
В полисульфоноксидах общей формулы:
температура стеклования повышается в следующем ряду:
СН3 СНз СНз CF3
А=-СН2— < —С— < —С— < —С—
< —С- <
NH <
Наличие хлора или метильных групп в фенильном ядре
приводит к повышению температуры стеклования.
Растворимость. Полиариленсульфоноксиды на основе дихлор-
дифенилсульфона и дифенилолпропана при комнатной температуре
растворяются в кетонах, например в ацетофеноне и циклогекса-
ноне, хлорированных углеводородах, таких, как метиленхлорид,
хлороформ, тетрахлорэтан, циклических эфирах типа диоксана и
тетрагидрофурана, полярных ароматических углеводородах —
хлорбензоле и дихлорбензоле, диметилформамиде, диметилацетамиде
и N-метилпирролидоне. В процессе выдержки растворов таких
полимеров в некоторых растворителях происходит их помутнение
за счет выпадения кристаллических олигосульфонов.
Высокомолекулярные полиариленсульфоноксиды аморфны [535]. Полностью
ароматические полимеры растворяются в диметилформамиде,
диметилацетамиде, диметилсульфоксиде, 7~бутиролактоне, лькрезоле,
N-метилпирролидоне, анилине и пиридине [561]. Осадительной
251
Таблица 5.20. Температуры стеклования ароматических полисульфоноксидов
Строение звена
с> °С
Компонент
Арилона
Полисульфон
Бакелит UCC
175
180 (HDT)*
180
180
190
200
СНз
I
СН(СНз)
я
СНз /
СНз
Компонент
промышленного полисульфона
фирмы ICI
200
205
205
205
205
210
220
175
230
_я
Продолжение табл. 5.20
Строение звена
Гс, °С
SO2—
—О
СНз
—S02—(( )>—'
so2—i
СНз
СНз'
/
. СНз /
СНз СНз
-°-o-so=-<o)
—П
230
230
235
250
260 (HDT)
-о—(Г Y>—so.
*о
-SO
-©-¦
о
260 (HDT)
[669J
265 (HDT)
[569]
275
275 (HDT)
[561]
[569]
290
[561J
Продолжение табл. 5.20
Строение звена
те -с
Литературный
источник
295 (HDT)
300 (HDT)
360 (HDT)
>400
[569]
[572, 575]
[56Ц
[561]
[576]
* Деформационная теплостойкость.
системой для дробного фракционирования полисульфона на основе
дифенилолпропана и дихлордифенилсульфона служит смесь
хлороформ— метанол. Фракционирование проводят, используя 0,7%-
ный раствор в диметилсульфокснде постепенным понижением
температуры, начиная с 80 °С, до разделения фаз [570].
Молекулярно-массовое распределение исследовано методом
гель-проникающей хроматографии [570, 571]. Для полимера из
дпхлорднфенилсульфона и дифенилолпропана получены следующие
уравнения для вискозиметрического определения молекулярной
массы:
Диметилсульфоксид, 105,5 °С [г\] = 0,145 • Мо>5°
Диметилформамид, 25 °С [rj] = 0,103 • M0-55
Тетрагидрофуран, 25 °С [ц] = 0,079 ¦ Л10>58
Хлороформ, 25 °С [т]1 = 0,024 • M0-72
Для определения вязкости полностью ароматических полисуль-
фоноксидов пригодны диметилформамиди диметилацетамид [561].
В случае диметилформамида (при 25°С) характеристическая
вязкость и среднечисловая молекулярная масса связаны уравнением
[т]] = 0,033-Л!0-65 (мл/г).
Для промышленного полимера дихлордифенилсульфона и
дифенилолпропана среднемассовая и среднечисловая молекулярные
массы, определенные светорассеянием или осмометрией,
составляют соответственно 40 000 и 20 400 [570]. Для раствора этого
полимера в диметилсульфоксиде 9-температура равна 105,5°С.
Размеры клубка составляют д/(гоJ/М = 802 4= 20 - 10~и см (по
данным вискозиметрии, при 9-температуре) или д/(гоJ/-М = 796 ± 20Х
X Ю~п см (по данным вискозиметрии, в различных растворителях
и среднемассовой молекулярной массе). Константа Хаггинса %
имеет следующие значения [570]:
Растворитель Хлороформ Тетрагидрофу- Диметилформа- Диметилсульф-
ран мид оксид
Температура, 25,0 25,0 25,0 105,5
°С
X 0,376 0,468 0,480 0,500
Молекулярные параметры для полностью ароматического
полисульфона имеют следующие значения: ^/(гоJ/М = 753 ± 20 • 10~и
см; В = 1,3-10-27 см3-г-2 моль2; % — 0,45 [577].
Стойкость к термической и термоокислительной деструкции.
Полиариленсульфоноксиды представляют собой термопласты с
высокой стойкостью к окислительной деструкции. Сульфогруппа
образует с соседними ароматическими ядрами сопряженную систему
с высокой степенью резонансной стабилизации. Энергия
распределяется по всей молекуле и не приводит к разрыву цепи.
Нагревание полисульфона на основе дихлордифенилсульфона и
дифенилолпропана в вакууме выше 380 °С сопровождается
структурированием и деструкцией полимера. Независимо от природы газовой
среды интенсивная потеря массы происходит между 500 и 550 °С
9 Зак. 18 257
О ЮО 300 500 700 t°C
50"
400 500 500 700 800 9D0 t;°C
5 10
15 20 25t,4
5
Рисг 5.36. Кривые термической деструкции полисульфона на основе дихлордифе-
нилсульфона и бисфенола А на воздухе A) и в азоте B) (АГ = 10°С/мин)
[579].
Рис. 5.37 Кривые изотермической деструкции полисульфона на основе дифенилол-
пропана прн 380 °С [580]:
а — содержание геля в зависимости от продолжительности деструкции при 380 "С; б —
характеристическая вязкость в зависимости от продолжительности деструкции при 380 "С.
Рис. 5.38. Кривые изотермической деструкции полисульфона на основе дихлор-
дифенилсульфона и дифенилолпропана [582]:
а — выход газов в зависимости от температуры деструкции (/— феиол; 2 —бензол; 3 —
замещенный длфениловын эфир; 4 — толуол; 5 — дифениловый эфир; 6 — л-крезол; 7-*
п-этилфеиол);
б — потери массы в зависимости от продолжительности термообработки при 393—493 X.
(рис. 5.36). При нагревании полимера в инертной атмосфере до
800 °С коксовый остаток составляет 30—40 % [579].
При нагревании полисульфона из дихлордифенилсульфона и
дифенилолпропана в изотермических условиях при 380°С гель-
фракция появляется через 3 ч и спустя 20 ч составляет 75 %
(рис. 5.37, а). В аналогичных условиях молекулярная масса золь-
фракции возрастает до точки гелеобразования, затем начинает
снижаться (рис. 5.37,6). Основным продуктом деструкции в вакууме
25S
до 400 °С является SO2. В макромолекуле такого полимера
наименьшую энергию связи (в ккал/моль) имеет связь С—S [579].
148
П
Ниже 500°С в газообразных продуктах распада присутствуют
также метан и фенол. На рис. 5.38 показано изменение массы по-
лисульфона на основе дихлордифенилсульфона и дифенилолпро-
пана при деструкции в интервале температур 393—493 °С в
инертной атмосфере и при интенсивности выделения продуктов распада
в зависимости от температуры деструкции. Метан в основном
образуется в результате гомолитического распада слабых связей
в изопропилиденовых группах. Выше 500 °С выделяются водород,
сероводород, оксид углерода, бензол, толуол, /г-крезол, /г-этилфе-
нол, дифениловый эфир и замещенный продукт на его основе. При
600 °С количество выделившегося SO2 составляет уже 34 % от
теории. Наличие следов влаги ускоряет распад ароматических
простых эфирных связей [581]. Энергия активации процесса
термической деструкции составляет 73 ккал/моль [583].
Деструкция полисульфона начинается с отщепления SO2 с
образованием бирадикала следующего строения:
СНз
Дальнейший распад происходит по схеме, приведенной на с. 260
[582].
Полисульфон на основе дихлордифенилсульфона и дифенилол-
пропана устойчив к окислению на воздухе и в кислороде до 140 °С
[584]. Поглощение кислорода за 9000 ч при 140 °С составляет
3 см3 О2. Потеря массы за 7000 ч при 125°С на воздухе
составляет менее 0,2 % ¦ Структурирования в этих условиях не
происходит. В условиях термического старения при 180° С на воздухе
наблюдается линейная зависимость молекулярной массы от времени
(рис. 5.39). Полностью ароматический полимер, не содержащий
Рис. 5.39. Зависимость вязкости раствора
полисульфона на основе дихлордифенилсульфона
дифенилолпропана от времени
термообработки при 180 °С [585].
9*
и
I
он
СНз
СНэ
i i
ОН
1
он
СНз
. СНз
ОН
сн
он
С2Н
2П5
I
он
Нз
I
сн
с2н.
1
СНз
! 1
он
(а
I
35
90
85
80
\370°C
J L
0 100 200 300400 500 600
J L
0
•400 800 1200 r.v
Рис. 5.40. Термогравиметрические кривые полисульфоиа (Полимер 360) [586]:
/ — в азоте; 2 — на воздухе.
Рис. 5.41. Кривые термической деструкции чисто ароматических полисульфонокси-
дов (Полимер 360) [561].
в цепи алифатических групп (Полимеры 360 и 380), независимо от
стехиометрического состава по термическим свойствам аналогичен
полисульфону на основе дихлордифенилсульфона и дифенилолпро-
пана. Независимо от типа газовой среды сильное уменьшение
массы наблюдается при 500 °С (рис. 5.40).
На рис. 5.41 приведены результаты термостарения при 260, 315
и 370 °С. После термообработки в течение 1000 ч потеря массы
при 260°С составляет 1—2% и при 315°С — 4—8%. При 370°С
15%-ное уменьшение массы происходит уже через 100 ч [561].
Основными продуктами деструкции полностью ароматических по-
лисульфоноксидов при 475°С являются SO2, дифенил, дифенилок-
сид и фенол. В небольших количествах образуются также бензол
и дибензофуран. Изучалась термическая деструкция и сшитого
полисульфоноксида [563].
Радиационная стойкость. По сравнению с другими полимерами
полиариленсульфоноксиды отличает высокая стойкость к действию
ионизирующих излучений. При облучении быстрыми электронами
молекулярная масса возрастает вплоть до дозы 80Мрад,
отвечающей точке гелеобразования [587]. Деструкция и структурирование
протекают параллельно. После дозы 80Мрад молекулярная масса
зольфракции постоянно уменьшается (рис. 5.42). При дозе выше
200Мрад содержание гель-фракции G0%) остается постоянным.
При дозе 400Мрад полисульфон окрашивается в коричневый цвет
[588]. Квантовые выходы
деструкции и сшивания
полисульфоиа на основе дифени-
лолпропана и дихлордифе-
Рис. 5.42. Зависимость
молекулярной массы золь-фракции поли-
сульфона на основе
дихлордифенилсульфона и дифенилолпропана
от дозы облучения [588].
100 200 300 Доэа,Мрад
261
нилсульфона при дозах 100—500 Мрад имеют близкие значения:
для деструкции 0,08 и для сшивания 0,16. Квантовый выход
газообразных продуктов облучения (ф-103) составляет: водород — 57,
метан — 17, СО — 11, СО2 — 5, SO2 — 4, этан — 4, COS — 1 и
этилен— 0,5 [588]. Возможно, что высокая радиационная стойкость
определяет то, что значительная доля осколков, образующихся при
радиолизе, обусловливает сшивание полимера:
СНз
СН,
СНз
сн4
-cm
Возникающие радикалы рекомбинируют с возникновением
поперечных связей. Методом ЭПР показано, что в полисульфоне на
основе дихлордифенилсульфона и дифенилолпропана после
облучения ^-лучами большая доля образовавшихся радикалов должна
участвовать во вторичных реакциях с атомом водорода, который
при радиолизе отщепляется от изопропилиденовой группы [589].
5.6.4.3. Переработка полиариленсульфоноксидов
Текучесть. Вязкость расплава полностью ароматического поли-
сульфона типа «Полимер 360» значительно выше, чем полисуль-
фона на основе дихлордифенилсульфона и дифенилолпропана и
других конструкционных пластиков. Зависимость вязкости
расплава ароматического полисульфона, полидиметилфениленоксида
и поликарбоната от температуры показана на рис. 5.43, а. При
повышении температуры на 25°С вязкость уменьшается на один
порядок [590]. Существенно меняется также и скорость сдвига
полностью ароматических полисульфонов (рис. 5.43,6). Определены
вязкости расплавов фракций полисульфона из
дихлордифенилсульфона н дифенилолпропана [571]. Даже с введением поправки,
учитывающей зависимость температуры стеклования от среднечис-
ловой молекулярной массы, в двойных логарифмических
координатах вязкость расплава — молекулярная масса наблюдается
отклонение от теоретической прямой (рис. 5.44). Тем не менее
композиции полисульфона с АБС-пластиком даже при обычных
температурах переработки характеризуются хорошей текучестью. При
скорости сдвига более 10 с-1 вязкость расплава этой композиции
262
300 350 Ц00 t,°C
Рис. о.43. Кривые текучести ароматических
полнариленсульфоноксидов [590]:
а — зависимость вязкости от температуры
расплава; 6 — зависимость вязкости от градиента сдай-'
гг; i — пол!!карбокат: 2 = пплн-2.6-диметилфени-
.теноксид; 3 — полпсульфон дихлордифенилсульфо-
на и дифен«лолпропана: 4 — чнсто ароматический
полпсульфон.
при 232 °С ниже, чем у
поликарбоната или смеси полидиметилфенилен-
оксида с полистиролом (рис. 5.45).
Экструзия. Полиариленсульфон-
оксиды гигроскопичны, поэтому
непосредственно перед переработкой
их подсушивают. Наиболее
целесообразно нагревать полимер в
термошкафах с циркуляцией воздуха
при толщине слоя до 25 мм при
150 °С в течение 12 ч или при
200 °С — 6 ч. Во избежание
повторного увлажнения материала
загрузочная воронка экструдера должна быть нагрета до 200 °С.
Переработка влажного материала сопровождается образованием в
изделиях пузырей и трещин с одновременной коррозией цилиндра
и шнека. Для стабилизации полисульфонов используют фосфаты
или фосфиты щелочных или щелочноземельных металлов, а также
оксид цинка [592, 593].
В табл. 5.21 приведены условия экструзии полиариленсульфон-
оксидов. Полностью ароматические полисульфоны перерабатывают
при температуре на 80 °С выше температуры стеклования [561].
Для этих полимеров (например, Полимер 360) необходимо
использовать шнеки с глубокой нарезкой и отношением сжатия
2,5:1. Переработка при 350—400 °С проводится при верхней
2-70° -
О -
Рис. 5.44. Кривая текучести полиариленсульфоноксида дифенилолпропана и ди-
хлордкфенилсульфона при 240 °С и низкой скорости сдвига [571].
Рис. 5.45. Кривые текучести расплавов полимеров при 232 °С [591]:
I — смесь полиариленсульфоноксида с АБС-пластиком; 2 — полидиметнлфениленокснд с
полистиролом; 3 — полистирол.
253
Таблица 5.2!. Экструзия полиариленсульфоноксидов [586. 596]
Параметр
Отношение сжатия
Температура цилиндра, °С
1-я зона
2-я зона
3-я зона
Температура сопла, °С
Частота вращения шнека, об/мин
Давление расплава, кгс/см2
ПОЛНОСТЬЮ
ароматический
полимер типа 360
2,5: 1
230-265
260-315
315-340
340—410
Полнсульфон на
основе
дихлордифенилсульфона и
дифенилолпропана
2: 1-3: 1
300
315
332
343
25
105
температурной границе, допустимой для обычных экструдеров. Экс-
трудеры для таких аномально-высоких температур должны иметь
цилиндр с индукционным обогревом и большее, чем обычное,
расстояние между цилиндром и приводом [594]. Обогреваемая зона
представляет собой охлаждаемую водой медную трубчатую
спираль, через которую пропускают индукционный ток. Из-за высокой
вязкости расплава при экструзии требуются большие усилия
сдвига. Щелевая насадка не должна иметь дробильных пластин
и других приспособлений, препятствующих течению. Для
улучшения перерабатываемости полностью ароматических полисульфонов
добавляют 5 % дифенилсульфона [595].
Температура цилиндра при экструзии полисульфона из
дихлордифенилсульфона и дифенилолпропана составляет 315—370 °С.
Для кабельной изоляции необходима температура 410 °С. Для
изготовления пленки экструзией через насадку с широкой щелью
используют шнеки длиной 25 D и плоско нарезанной зоной сжатия.
Вытяжка пленки производится путем отжига на охлаждающих
вальцах. Ориентируют пленку при 200 °С [585].
Литье под давлением. Режимы литья полисульфонов и их
смесей с АБС-пластиком приведены в табл. 5.22.
Полностью ароматические полисульфоны типа «Полимер 360»
перерабатывают литьем под давлением 2800 кгс/см2 и
температуре 430 °С.
Распределительные каналы, так же как при литье
поликарбоната, должны быть как можно более короткими [590]. Полисуль-
фон на основе дихлордифенилсульфона и дифенилолпропана может
перерабатываться как на плунжерных, так и на литьевых
машинах со шнековой пластикацией. Для изделий с толщиной стенок
до 2,5 мм температура формы может быть ниже 100 °С. Сложные
изделия с длинными литниками формуют при температуре формы
до 160°С [597]. Для переработки материалов типа «Арилон»
можно использовать обычные шнеки или шнеки с мелкой нарезкой.
Отношение сжатия не превышает 2,6:1. Изделия с высоким
поверхностным глянцем получают из предварительно подсушенного
материала.
264
Прессование. Полностью ароматические полисульфоны
необходимо подсушивать перед прессованием в течение 3 ч при 260 °С.
Пресс-форму и пресс предварительно нагревают до температур
соответственно 315 и 380 °С, нагретый высушенный материал
загружают в форму, поддерживая давление на урове 140 кгс/см2 до
начала плавления полимера, сопровождающегося понижением
давления. При плавлении давление составляет 105 кгс/см2. В точке,
в которой вследствие термического расширения расплава
давление возрастает, его снижают до 70 кгс/см2 и массу охлаждают.
Отпрессованные изделия извлекают из формы при 55°С и быстро
охлаждают холодной водой [586]. Полисульфон из дихлордифе-
нилсульфона и дифенилолпропана может быть отпрессован при
температурах 220—275 °С.
Пленки и покрытия. Пленки из полимера на основе дихлор-
дифенилсульфона и дифенилолпропана, полученные поливом из
раствора в диметилформамиде, имеют прочность при растяжении
570 кгс/см2. При поливе из раствора при 100°С прочность при
растяжении составляет 480 кгс/см2 [600]. Двухосная вытяжка
приводит к повышению прочности до 1200 кгс/см2 [601]. В США в
промышленности пленки получают методом полива из раствора.
Пленка может легко наноситься на стальную ленту. Трудности
обусловлены медленной фильтрацией раствора и удалением
остатков растворителя. Покрытия на основе полностью ароматических
полимеров типа «Полимер 380» можно получать напылением из
раствора в смеси N-метилпирролидон — толуол F0:40). Покрытия
имеют исключительно высокие твердость и стойкость к
истиранию— 0,03 г/1000 циклов (Taber CS, нагрузка 17,1 кг) [561]. Для
получения покрытий на основе полисульфона и дихлордифенил-
сульфона и дифенилолпропана полимер используют в виде
порошка, пленки или раствора. Кэширование металлических лент
проводят полисульфоновой пленкой при 370 °С под давлением
5,5 кгс/см2. Порошок наносят на носитель, нагретый до 260 °С.
Таблица 5.22. Литье под давлением полисульфоиов
[590, 591, 596]
Параметр
Температура формы, °С
Температура цилиндра, °С
зона загрузки
зона сжатия
зона питания
Температура сопла, еС
Давление впрыска, кгс/см2
Частота вращения шнека,
об/мин
Продолжительность цикла, с
Полностью
ароматический
полисульфон
260
382
412
390
1470
76
42
Полисульфон
из дихлордифе-
нилсульфона и
дифенилолпро-
паиа
93
354
354
360
360
980
51
67
Смесь полисуль-
фоиа и АБС-
пластнка
70-93
243
260
270
265
Сварка и склеивание. Полностью ароматические полиеульфоны
сваривают ультразвуком или склеивают с помощью таю^х
растворителей, как диметилформамид и N-метилпирролидон. Полимер из
дихлордифенилсульфона и дифенилолпропана сваривается при
370 °С за 10 с с применением тефлоновых пресс-форм. Перед
свариванием соединяемые детали должны высушиваться в течение
3—6 ч при 120 °С для удаления влаги. Для склеивания
используют 5%-ные растворы в метиленхлориде. Смеси полисульфона
с АБС-пластиком (типа «Арилон») сваривают ультразвуком. Для
склеивания используют раствор полимера в метилэтилкетоне или
метиленхлориде.
Обработка поверхности. Полисульфоноксиды на основе
дихлордифенилсульфона и дифенилолпропана, а также его смеси с АБС-
пластиком могут подвергаться гальванической металлизации с
получением покрытия такого же качества, как и в случае чистого
АБС-пластика. Для нанесения покрытий пригодны лаки на основе
полиакрилатов, нитрата целлюлозы и алкидных смол. В качестве
пигментов применяют диоксид титана, технический углерод или
органические красители @,005—0,02%).
5.6.4.4. Эксплуатационные свойства ароматических
полиариленсульфоноксидов
Полиариленсульфоноксиды являются термопластичными
конструкционными материалами, сочетающими высокую прочность,
жесткость, твердость и ударную вязкость в широком
температурном интервале с хорошими диэлектрическими свойствами и
отличной химической стойкостью.
Теплофизические свойства. Критерием оценки длительной
работоспособности служит температура длительной формоустойчи-
вости (HDT)*. Чисто ароматические полимеры, содержащие
дифенильные группировки (типа «Полимер 360» и ICI), имеют
более высокую жесткость, чем полимеры с дифенилизопропилидено-
выми группами в цепи. Поэтому HDT для первых B75 °С) на 100°
выше, чем для последних A75°С). За счет увеличения
концентрации алифатических углеродных атомов в смесях полисульфон —
АБС-пластик (типа «Арилон») HDT составляет 150 °С. По
теплопроводности, термическому коэффициенту линейного расширения
и усадке полиеульфоны не отличаются от ряда крупнотоннажных
полимеров (табл. 5.23).
Физико-механические свойства. Полиариленсульфоноксиды
являются высокотермостойкими конструкционными материалами с
с высокой прочностью, жесткостью, твердостью н ударной
вязкостью (табл. 5.24).
По отношению прочность: плотность полиарнленсульфоноксиды
принадлежат к той же группе материалов, что и сталь, алюминий,
поликарбонат и полиамид. Хорошие физико-механические свой-
* Аналогична деформационной теплостойкости,
266
Таблица 5.23. Теплофизические свойства ароматических
полиариленсульфоноксидов [585, 586, 602, 603]
А - чиото ароматический полисульфон типа «Полимер 360»
? _ цисте- ароматический полисульфои типа IL.I
С - поллсульфон на основе дихлордифеиилсульфона и дифеиилолпропана типа «Бакелит 1700>
D — смесь полимеров полисульфон АБС типа «Арилон»
Показатель
Плотность, г/см3
Температура плавления, °С
Температура стеклования, °С
Показатель текучести расплава
C50 °С, 3 кгс/см2), г/мин
Показатель преломления
Коэффициент теплопроводности,
ккал/(м-ч-°С)
Термический коэффициент линейного
расширения а-106, 1/°С
Деформационная теплостойкость при
17 кгс/см2, °С
Усадка, %
Водопоглощсние за 24 ч при 23 °С,
Горючесть
Температура разложения (при
нагрузке I кгс), °С
Газопроницаемость пленки
толщиной 50 мкм, см3/(м2-ат-24 ч)
кислород
азот
диоксид углерода
А
1,36
288
0,164
47
275
0,80
1,4
343
в
1,37
230
55
303
0,70
0,43
с
1,24-1,25
194-202
195
0,25-0,40
1,633
0,223
31
175
0,76
0,22
Замозатухаюший
-
230
40
95
D
114
150
Таблица 5.24. Физико-механические свойства полисульфонов [586, 602, 603]
Условные обозначения А, В, С, Д — см. в табл. 5.23.
Показатель
Прочность при растяжении, кгс/см2
Относительное удлинение при
разрыве, %
Модуль упругости при растяжении,
кгс/см2
Прочность лри изгибе, кгс/см2
Модуль упругости при изгибе,
кгс/см2
Прочность при сжатии, кгс/см2
Ударная вязкость образцов с
надрезом, кгс-см/см2
Модуль упругости при сжатии,
кгс/см2
Твердость по Роквеллу
Износостойкость A кгс), мг/1000
ЦИКЛОВ
А
920
10
26 000
1210
27 800
1260
10
24 000
М110
40
в
840
24 400
1290
25 700
3,4
М88
С
760
50-100
25 200
1080
27 300
1295
6,9
26 000
М69
20
D
525
20 000
770
19 000
43
Р117
267
Till!
О 50 100 150 200 250 t,°C
О 50 100 150 ZOO 250 t,*C
Рис. 5.46. Температурная зависимость модуля упругости при изгибе полиарилен-
сульфоноксида [586, 609]:
/ — полностью ароматический полисульфоноксид; 2 — полисульфон с дихлордифенилсульфо«
ном и дифенилолпропаиом.
Рис. 5.47. Зависимость прочности при растяжении полисульфона от температуры
[586, 5911:
/ — полиостью ароматический полнсульфои; 2 — смесь полиариленсульфоиа с АВС-пластиком.
ства сохраняются в широком интервале температур [604—608],
который для чисто ароматических полимеров равен 240—280°С;
для продуктов взаимодействия дихлордифенилсульфона с дифени-
лолпропаном составляет 100—175°С, для смесей полисульфона
с АБС-пластиком до 150°С. Последние материалы имеют самую
высокую ударную вязкость (образцов с надрезом) среди
термостойких конструкционных полимеров D3 кгс-см/см2). Модуль
упругости при изгибе почти не изменяется в температурном интервале
0—130°С (рис. 5.46). Выше 130°С модуль упругости при изгибе
для продукта взаимодействия дихлордифенилсульфона с дифени-
лолпропаном резко уменьшается, в то время как для чисто
ароматического полимера до 270 °С сохраняется почти линейная
зависимость.
На рис. 5.47 показана зависимость прочности при растяжении
от температуры чисто ароматических полисульфонов, продуктов
реакции дихлордифенилсульфона с дифенилолпропаном и смесей
полисульфонов с АБС-пластиком. Последние при 93°С сохраняют
50 % прочности при комнатной температуре, а у чисто
ароматических полимеров это значение достигается только при 215°С.
Изменение относительно удлинения при разрыве у полисульфона на
г основе дихлордифенилсульфона
и дифенилолпропана показано на
рис. 5.48. Полиариленсульфонок-
600\- X \ сиды отличаются высокой
стойкостью при термостарении при вы-
4оо\- /S^*~^ \ U7°n соких температурах (рис. 5.49, а).
200-
о
268
б-,кгс/см
800
Рис. 5.48. Деформационно-прочностные
кривые полкариленсульфоноксида ди-
хлордифенилсульфона и дифенилолпропа-
7 8 1,% на при различных температурах [609].
300
200
a
.—-^
-
1 t 1
200
^^
260°C
i i i i
°G
12G
80
40
5
1
\2Ю
i i
°c
490
*
—
170
i
WC
I
100
гусут
50
150
250
350
Рис. 5.49. Зависимость прочности при растяжении от продолжительности
экспозиции [586, 610]:
а — термостарение полиариленсульфоноксида; б — старение полисульфона на основе дифе-
нилолпропана.
Для чисто ароматического полисульфона через 4000 ч
термообработки на воздухе при 200 °С прочность при растяжении еще не
изменяется; при 260 °С сохраняется 50% прочности [586].
Прочность не изменяется также через 1000 ч нагревания при 150 °С
в атмосфере насыщенных водяных паров. Модуль упругости при
изгибе не изменяется за 1100 ч нагревания на воздухе при 300°С
[611]. На рис. 5.49,6 показано изменение прочности при
растяжении для продукта взаимодействия дихлордифенилсульфона с дифе-
нилолпропаном. Через 6 мес нагревания на воздухе при 110°С
прочность при растяжении уменьшается на 10 и удлинение на
5% [584].
Ароматические полисульфоны отличаются высокой
устойчивостью к ползучести (рис. 5.50, а). После года выдержки
полисульфона из дихлордифенилсульфона и дифенилолпропана под
нагрузкой 210 кгс/см2 суммарное удлинение составляет около 1 %, что
вдвое меньше, чем у полиформальдегида в тех же условиях.
Ползучесть полимеров остается низкой и при высоких температурах.
Полисульфон из дихлордифенилсульфона и дифенилолпропана
спустя год после выдержки при 100 °С под нагрузкой 210 кгс/см2
100 200 300 Ш 500 600 trf4
2000 Ш0 6000 8000
Рис. 5.50. Кривые ползучести различных полимеров:
О — при 23 °С и различных нагрузках G — чисто ароматический полисульфон; 2—полисулъ«
фон дихлорднфенилсульфона и дифенилолпролаиа; 3 — смесь полисульфона с АБОпласти«
ком) [591, 612];
О « при 100 °С и нагрузке 210 кгс/см2 (/ — полисульфон на основе днхлордифенилсульфона
и дифенилолпропана; 2 — поликарбонат) [613].
269
удлиняется менее чем на 2 % (рис. 5.50,6). Это значение для
смесей полисульфонов с АБС-пластика-ми через 300 ч при 70 °С под
нагрузкой 70 кгс/см2 или при 2*3 °С под нагрузкой 140 кгс/см2
составляет около 1 % [614].
Полиариленсульфоноксиды отличаются высокой стойкостью к
действию ионизирующих излучений. При облучении у-лучами
прочность при растяжении и относительное удлинение при разрыве не
изменяются при дозе 100 Мрад [586, 588]. Выше этой дозы
облучения удлинение начинает медленно уменьшаться для полимера
на основе днхлордифенилсульфона и дифенилолпропана, достигает
минимума и при дозе 200 Мрад снова составляет 90 % от
исходного значения. До 200 Мрад модуль с увеличением дозы линейно
возрастает с 25000 до 28 000 кгс/см2. В то же время полисульфоны
отличаются незначительной стойкостью к облучению УФ-лучами
на воздухе. В пленках из чисто ароматического полисульфона
через 300 ч облучения обнаруживаются сильные изменения
поверхностной структуры, появляется хрупкость. Порошкообразный
продукт, растворимый в 5 %-ном гидроксиде натрия, может быть
отмыт с поверхности ацетоном [611]. Под действием УФ-лучей
полимер на основе дихлордифенилсульфона и дифенилолпропана
становится жестким, его прочность при растяжении и относительное
удлинение при разрыве уменьшаются [615].
Электрические свойства. Хорошие электрические свойства по-
лиарленсульфоноксидов (табл. 5.25) не ухудшаются при
нагревании до 200 °С.
Для чисто ароматических полисульфонатов в интервале
0—270 °С диэлектрическая проницаемость остается практически
постоянной. Тангенс угла диэлектрических потерь не изменяется до
Таблица 525. Электрические свойства полисульфонов [586, 602, 603]
Условные обозначения А, В, С см. в табл. 5.23
Показатель
Диэлектрическая проницаемость
при 60 Гц
при 106 Гц
при 109 Гц
при 106 Гц A75 °С)
Тангенс угла диэлектрических
потерь
при 60 Гц
при 106 Гц
при 109 Гц
при 106 Гц A75 °С)
Электрическая прочность, В/мм
Удельное электрическое
сопротивление
объемное, Ом-см
поверхностное, Ом
Дугостойкость, с
А
9,94
3.24
3,26
3,24
0,003
0,001
0,001
14 000
3.2- 1016
6,2 • 1015
67
в
3,5
0,006
1017
с
3,14
3,10
2,75
0,008
0,0056
0,003
16 700
5- 1016
3- 1016
122
270
Рис. 5.51. Зависимость диэлектрических
свойств чисто ароматических полисуль-
фонов от температуры [5861:
lt 2 — диэлектрическая проницаемость; 3, 4,
*5 — тангенс угла диэлектрических потерь.
180°С, а при более высоких
температурах резко возрастает
(рис. 5.51). Влажность практиче-
ски не влияет на диэлектрические
свойства полисульфона на основе
дихлордифенилсульфона и дифе-
[6] Д
50 т
рфуф ф
нилолпропана [613]. Диэлектрическая проницаемость чисто
ароматического полисульфона увеличивается вследствие высокого водо-
поглощения с 4,0 до 4,75 A00 Гц). Электрическая прочность с
повышением температры изменяется мало. Диэлектрическая
проницаемость и tgo сильно зависят от влажности воздуха (рис. 5.52).
Зависимость tgo от частоты для полимера на основе
дихлордифенилсульфона и дифенилолпропана выражены менее ярко, чем в
случае поликарбоната или полиэтилентерефталата (рис. 5.53).
Пробивное напряжение пленки толщиной 0,3 мм из полисульфона,
дихлордифенилсульфона и дифенилолпропана за 540 дней при
150°С увеличивается на 1,3% и через ПО дней при 210°С на
5,2% [610].
Химическая стойкость. Полиариленсульфоноксиды стойки к
действию большинства агрессивных сред (в том числе и при
нагревании). Медленная деструкция протекает под действием окисляющих
концентрированных кислот. В полярных растворителях, за
исключением спиртов, происходит их набухание. Увеличение массы по-
лиариленсульфоноксидов в неорганических и органических средах
показано в табл. 5.26. Появление усталостных трещин в случае
чисто ароматических полисульфонов происходит при контакте
во во у>,%
7
7Ог 10* W6
W
Рис. 5.52. Зависимость диэлектрических свойств чисто ароматических
полисульфонов от влажности воздуха:
/ — диэлектрическая проницаемость, 2 — тангенс угла диэлектрических потерь.
Рис. 5.53. Зависимость тангенса угла диэлектрических потерь ароматического
полисульфона на основе дихлордифенилсульфона и дифенилолпропана от частоты
[5851":
t — полиариленсульфоноксид; 2 — поликарбонат; 3 — полиэтилентерефталат.
271
Таблица 5.26. Химическая стойкость
полиариленсульфоноксидов [586, 616]
Среда
Серная кислота
Соляная кнслота
Плавиковая кислота
Пероксид водорода
Уксусная кислота
Метанол
Этанол
Реактивное топливо
Раствор гидроксида натрия
Ацетон
Трихлорэтилен
Трансформаторное масло
Силиконовое масло
i\un цеп 1 f «
ПИЯ %
40
10
10
20
10
10
100
20
10
Изменение
полиостью
ароматический полисульфон
(тиоа «Полимер 360»,
выдержка 30 сут
прн 25 °С)
2,2
—
5,8
1,0
4,0
11,0
0,6 (т. кип.)
1,4 (т. кип.)
25,4
20,2
0,5 (80 °С)
0,8 F5 °С)
массы, %
на основе полисуль-
фоидихлордифенил-
сульфона и дифеиил-
олпропана (типа
«Бакелит 1700»,
выдержка 7 сут прн
25 °С)
0,36
-0,42
—0,52
0,08
0,05
—
с кетонами, например ацетоном или циклогексаноном. Двухосно-
ориентированные пленки из полимера на основе дихлордифенил-
сульфона и дифенилолпропана при контакте с метанолом, этанолом,
тетрахлоридом углерода, «-бутилгалогенидами, хлорбензолом, ди-
этиловым эфиром, ди-«-амиловым эфиром, дифенилоксидом и
ацетоном образуют спиралевидные усталостные трещины [473].
Растрескивание смесей полисульфонов с АБС-пластиками под
нагрузкой происходит при воздействии этанола, пропанола, этилен-
гликоля, тетрахлорида углерода.
5.6.4.5. Применение полиариленсульфоноксидов
Полиариленсульфоноксиды используют главным образом в
самолетостроении, производстве бытовых электроприборов,
упаковочных материалов, в электротехнике. Применение специальных
типов полисульфонов определяется температурой длительной
эксплуатации и их стоимостью. Чисто ароматический полисульфон
типа «Полимер 360» (применяется до 260°С) стоит 160 марок
ФРГ за 1 кг; полисульфон на основе дихлордифенилсульфона и
дифенилолпропана — 10 марок-за 1 кг (применяется до 150°С).
Чисто ароматический полисульфон может использоваться в таких
областях, где требуется высокая термостойкость и где ранее
применяли только керамику, реактопласты или отливки из цинка,
бронзы, свинца. Поскольку полимер является термопластом, из
272
него можно изготовлять сложные изделия без дополнительной
механической обработки литьем под давлением.
Применение их в электротехнике позволяет повысить
температуру эксплуатации электротехнических деталей. Полисульфоны
применяют для изготовления движущихся частей реле, катушек,
электрических клемм, деталей потенциометров, выключателей,
проводящих клемм, печатных схем, цоколей трубок, корпусов
инструментов, аккумуляторных батарей для никель-кадмиевых
элементов [617], кабельной изоляции для высокотермостойких
клеящих электроизоляционных лент. Низкий tgO при высоких частотах
позволяет использовать их в качестве конденсаторной изоляции
в производстве телевизоров в виде пленки толщиной 4—6 мкм.
Такие конденсаторы имеют повышенную проводимость и более
высокие температуры эксплуатации, чем конденсаторы из
поликарбоната или диэлектрики из полиэтилентерефталата [618].
Электроизоляционная пленка производится в Швейцарии в
промышленных масштабах на экструдерах с широкой щелевой насадкой
[598, 599].
Набухающие в воде пленки из полисульфонов используют в
качестве мембран для топливных элементов [619, 620].
Сульфированный полисульфон пригоден для изготовления полупроницаемых
мембран для обессоливания морской воды обратным осмосом.
Сульфирование полимера на основе дихлордифенилсульфона и ди-
фенилолпропана проводят хлорсульфоновой кислотой в растворе
1,2-дихлорэтана. Мембрана толщиной 150 мкм с 0,1—0,2 мэкв
сульфогрупп на 1 г при пропускании 900 л/м2 в сутки
задерживает 84,4 % соли [624]. Самозатухающие свойства, высокая
термоизоляционная способность (особенно металлизованных в вакууме
пленок) и отсутствие запаха при деструкции позволили
использовать полиариленсульфоноксиды в авиастроении для внутренней
облицовки кабин, в измерительных приборах, защитных
шлемах. В производстве бытовых приборов полисульфоны применяют
для изготовления приспособлений разматывающих машин для
пряжи, стиральных машин (барабанов, баков, деталей насосов).
Пленки из полисульфонов на основе дихлордифенилсульфона и ди-
фенилолпропана в США используют в качестве
высококачественного упаковочного материала для пищевых продуктов. Пищевую
посуду из полисульфонов можно подвергать как горячей
стерилизации, так и глубокому охлаждению. Устойчивость при
стерилизации горячим паром делает возможным применение полисульфонов
в производстве медицинских инструментов. Из полисульфонов
получают высокотермостойкие покрытия; их применяют для
склеивания металлов. При склеивании стали клеем из полимера на
основе дихлордифенилсульфона и дпфенилолпропана прочность при
растяжении в зависимости от температуры изменяется следующим
образом [625]:
Температура, °С —55 25 155 205
Прочность при растяжении, кгс/см2 . . 230 246 155 36,5
273
Прочность при сдвиге клеевого соединения
высококачественной стали может быть повышена путем введения в полисульфон
фенольных гидроксильных групп. Полисульфон, содержащий 0,2 %
фенольных гидроксильных групп, обеспечивает прочность при
сдвиге клеевого соединения высококачественной стали 814 кгс/см2
при 150°С [622]. При склеивании бериллия, легированного медью,
клеевое соединение прекрасно выдерживает действие топлива для
двигателей, масла и морской воды в интервале температур от —50
до 200 °С [621]. Высокая термостойкость полимеров на основе ди-
хлордифенилсульфона и дифенилолпропана делает их пригодными
для использования в ксерокопировальной технике [585]. Высокая
рабочая температура таких полисульфонов обеспечивает
получение копий с оптимальной четкостью, резкостью контуров и хорошей
ретушируемостью. В химическом машиностроении полисульфон
используют в качестве материала для фильтрующих элементов
[623].
Литература к главе 5, разделы 5.1.—5.6
1. S. Claesson, Makromolekulare Chem. 7 A951), 46—61.
2. R. Pummerer, Chem. Ber. 64 A931), 2477—2486.
3. R. Pummerer, Chem. Ber. 57 A924), 747—750.
4. M. Dunkel, Chem. Ber. 57 A924), 739—746.
5. T. Cohen, Chem. Commun. A967), 451.
6. A. Levin, Tetrahedron A965), 4531—4536.
7. P. Gragerov, 2, org. Khim. 5 A969), 3—8.
8. M. Hellmann, J. Amer. chem. Soc. 77 A955), 3650—3658
9. W. Pummer, Polymer Preprints 7 A966), 1071—1076.
10. E. Nield, J. chem. Soc. A959), 166—171.
11. W. Pummer, J. Res. Natl. Bur. Stand. 63 A A959), 167.
12. Пат. США 3046313 24. 7. 62 US Dept. of Navy.
13. Англ. пат. 1052482.
14. W. Postelnak, Ind. Engin. Chem. A958), 50 1602.
15. W. Kern, Angew. Chem. 62 A950), 337—338.
16. R. Gehm, Acta chem. Scand. 5 A951), 270—282.
17. W. Kern, Makromolekulare Chem. 77 A964), 90—113.
18. A. Berlin, J. Polymer Sei. 55 A961), 621—628.
19. F. Riese, Ann. 164 A872), 161.
20. G. Golkschmidt, Monatsh. Chem. 7 A886), 40.
21. W. Fuchs, Ber. 55 A922), 738—745.
22. E. Hoess, Picatinny Arsenal Techn. Rep. 3236-AD-467450 A950).
23. E. Fear, Sei. Techn. Aerospace Rept. 3 A965) 10, 1619.
24. G. Brooke, J. chem. Soc. A964), 729—733.
25. Пат. США 3320183, 16.5.67 General Electric.
26. H. Winkler, J. org. Chem. 28 A963), 1733—1740.
27. G. Wittig, Ber. 90 A957), 875—892.
28. M. Jozefowicz, Bull. Soc. chim. France A963), 2036—2066.
29. В. Богомольный, Изв. АН СССР, сер. хим., 1961, 1912.
30. W. v. Heitz, Makromolekulare Chem. 98 A966), 29—41.
31. M. Kharash, J. Amer. chem. Soc. 65 A943), 498—500
32. R. Barcel, С R. Acad. Sei. 253 A961), 1801—1808.
33. P. Kovacic, J. Polymer Sei. 47 (I960) 149, 45—54,
34. P. Kovacic, J. Amer. chem. Soc. 85 A965) 4, 454—458
35. P. Kovacic, Tetrahedron 11 A962), 467—469.
36. P. Kovacic, J. org. Chem. 28 A963) 7, 1864—1867.
37. P. Kovacic, J. org. Chem. 28 A963) 4, 468—472.
38. P. Kovacic, J. org. Chem. 29 A964) 1, 100—104.
39. P. Kovacic, J. Polymer Sei. A2 A964), 1193—1203,
274
40 P. Kovacic, J. or g. Chem 29 A964), 2416—2420.
41 P Kovacic, J. org. Chem. 30 A965), 3176—3181.
42. P. Kovacic, J. Polymer Sei. A3 A965), 4297—4298.
43. P. Kovacic, J. Polymer Sei. A4 A966) 1, 5—28.
44. P. Kovacic, J. Polymer Sei. A 14 A966) 6, 1445—1462.
45 P. Kovacic, J. org. Chem. 31 A966) 8, 2467—2470.
46. P. Kovacic, J. Polymer Sei. A 15 A967) 5, 945—964.
47. P. Kovacic, J. appl. Polymer Sei. 12 A968) 7, 1735—1743.
48. P. Kovacic, J. Polymer Sei. A 17 A969), 111—125.
49 M. Kuwata, Mem. Fac. Engin. Hiroshima Univ. 2 A955), 55—67.
50 P. Kovacic, J. Amer. chem. Soc. 82 A960), 1917—1923.
5t. P. Kovacic, J. org. Chem. 26 A961), 762—765.
52. A. Берлин, Высокомол. соед., А 14, 1972, 7, 1591 — 1595.
53. G. Ehlcrs, IUPAC Symp. Macromolecul. Chem. Tokyo 1966.
54. M. Larson, Inorg. Chem. 5 A966), 801—809.
55. B. Ellis, J. Polymer Sei. A ПО A972), 553—568.
56. Пат. СССР 302022, 30. 11.71.
57. G. Hall, J. chem. Soc. A966), 2043—2047.
58. D. Packham. Chem. Commun. A965), 207—215.
59. G. Ostrum, Chem. Commun. A966), 53—60.
60. Пат. США 3678006, 18.7.72 Hughes Aircraft.
61. Пат. США 3595811, 27.7.71 Hughes Aircraft.
62. Пат. США 3582498 1. 6. 71 Hughes Aircraft.
63. N. Bilow, J. macromolecul. Sei. А 1 A967), 183—198.
64. N. Bilow, J. macromolecul. Sei. A3 A969), 3, 501—525.
65. С Marvel, J. Amer. chem. Soc. 41 A959), 448—452.
66. F. Dawans, J. Polymer Sei. A 2 A964), 3277—3295.
67. С. Marvel, J. Polymer Sei. A3 A965), 1553—1565.
68. D. Frey, J. Polymer Sei. A 1 A963), 2057—2065.
69. Нидерл. пат. 6402650 A964) Inst. Franc, du Petrole.
70. J. Stille. J. Polymer Sei. В 4 A966), 791—793.
71. J Stille, J. Polymer Sei. A 15 A967), 2721—2729.
72. J. Stille, Macromolecules 2 A969), 85—93.
73. J. Stille, J. Polymer Sei. В 7 A969) 7, 525—527.
74. J. Stille, J. macromolecul. Sei. A3 A969), 1043—1058.
75. J. Stille, Advan. Chem. Ser. 91 A969), 625.
76. J. Stille, Macromolecules 4 A971) 4, 515—517.
77. W. Reid, Naturwissenschaften 53 A966), 306—308.
78. W. Reid, Angew. Chem. 80 A968), 932—942.
79. W. Wrasidlo, J. Polymer Sei. В 7 A969), 519—523.
80. J. Stille, Makromolekulare Chem. 154 A972), 49—61.
81. D. de Halas, Nucl. Sei. Abstr. 17 A963) 5 '3113.
82. Пат. США 1976468, 1934.
83. M. Berthelot, Bull. Soc. chim. France 6 A866), 268.
84. E. Fields, J. Amer. chem. Soc. 89 A967), 724—725.
85. В. Парини, Изв. АН СССР, сер. хим., 1958, 12, 1499.
86. А. Berlin, J. Polymer Sei. 55 A961), 675—682.
87. J. Panaiotov, Compt. rend. Acad. Sei. Bulg. 21 A968) 455.
88. Яп. пат. 7226680, 18. 7. 72.
89. G. Ensor, Brit. Polymer J. 2 A970), 264—269.
90. G. Ehlers, J. Polymer Sei. A 15 A867), 1802—1804.
91. J. Jones, J. macromolecul. Sci.-Revs. 2 A968) 2, 303—318.
92. Опубл. заявка ФРГ 2159392 13.7.72 Gulf Oil Canada.
93. A. Shepard, J. Polymer Sei. 4 A966), 511—518.
94. Англ. пат. 1136699 A968) Esso Research.
95. Пат. США 343769, 8. 4. 69 Esso Research.
96. Фр. пат. 1534181 A968) Esso Research.
97. Пат. США 3437570, 8. 4. 69 Esso Research.
98. N. Wisdom, Abstr. N. Y. Etectr. Soc. 5. 5. 69.
99. T. Osa, J. Amer. chem. Soc. 91 A969) 3994—4002.
100. В. Коршак, Высокомол. соед., Б 13, 1971, 12, 873—875.
275
lui. Пат. СССР 309606, 1. 10.71.
102. Опубл. заявка ФРГ 2058250 8.6.72 V. Korshak.
103. W. Bracke, J. Polymer Sei. A i 10 A972) 7, 2097—2107.
Ш4. Y. Paushkin, Magyar Kern. Lap. 22 A967), 122—125.
105. Я. Паушкин, Высокомол. соед., А 9, 1967, 1293—1297.
106. Y. Paushkin, J. Polymer Sei. С 16 A967), 2615—2624.
107 Я. Паушкии, Высокомол. соед., Б 12, 1969, 5, 376—378.
108. И. Рабинович, Докл. АН СССР, 200, 1971, 4, 890—892.
109. М. Dewar, J. ehem. Suc. A952), 3544—3550.
ПО. A. Dawydow, Z. exp. Teor. Fiz. 18 A948), 515—521.
111. H. Suzuki, Bull. chem. Soc. (Japan) 33 A960), 109—121.
112. W. Kuhn, Helv. chim. Acta 31 A948), 1780—1788.
113. A. Берлин, Изв. АН СССР, 1959, 1674—1677.
114. D. Vincent, J. macromolecul. Sei. A3 A969) 3, 485.
115. В. Коршак, Высокомол. соед., Б 13, 1971, 9, 695—699.
116. J. Stille, J. Polymer Sei. D5 A971), 385—430.
117. J. Cotter, J. appl. Polymer Sei. 12 A968), 2481—2490.
118. G. Brown, J. chem. Engin. Data 6 A961), 125.
119. E. Fitzer, Ber. Dtsch. keram. Ges. 48 A971) 4, 157—165.
120. Eimac 221 Bull., Eitel Mc Cullough Inc., San Carlos, USA.
121. Пат. ФРГ 1178529, 24. 10.64 BASF.
122. F. Beck, Ber. Bunsenges. phys. Chem. 68 A964), 558—567.
123. Пат. ФРГ il78529, 24.9.64 BASF.
124. Rubber & Plastics Age A969), 197.
125. Europa-Chemie 18 A969), 6.
126. J. Stille, Macromolecules 5 A972) 5, 541—546.
127. M. Szwarc, J. Polymer Sei. 6 A951), 319—329.
128. M. Szwarc, J. Amer. chem. Soc. 73 A951), 1379.
129. M. Kaufman, J. Polymer Sei. 13 A954), 3—12.
130. L. Errede, J. Amer. chem. Soc. 79 A957), 4952—4955.
131. L. Errede, J. Amer. chem. Soc. 82 (I960), 2, 436—439.
132. L. Errede, J. Amer. chem. Soc. 82 (i960), 5218—5223.
133. Патентная заявка ФРГ 1103030, 23.3.61.
134. M. Szwarc, Discussions Faraday Soc. 2 A947), 46.
135. L. Errede, J. Amer. chem. Soc. 82 (i960) 19, 5224—5227.
136. A. Farthing, Nature 164 A949), 915.
137. A. Ваншейдт, Высокомол. соед., 2, i960, 9, 1383—1390.
138. A. Wanscheidt, J. Polymer Sei. 52 A961) 157, 179—188.
139. J. Golden, J. chem. Soc. A961), 1604—1610.
140. А. Ваншейдт, Высокомол. соед., 2, I960, 12, 1818—1823.
141. F. Dammont, J. Polymer Sei. В 1 A963) 6, 339—340.
142. Патентная заявка 1090429 6. 10.60 Phrix-Werke.
143. S. Okabe, J. chem. Soc. (Japan), Ind. chem. Sect. 70 A967) 7, 1243—1246.
144. J. Moritani, J. ehem. Soc. (Japan), Ind. chem. Sect. 68 A965) 2, 296—299.
145. I. Hoyt, ACS Polymer Preprints 5 A964) 2, 680—687.
146. H. Lunk, J. Polymer Sei. A 3 A965) 8, 2983—2990.
147. К. Sisido, J. Polymer Sei. A 1 A963) 6, 2101—2107.
148. H. Doorenbos, ACS Polymer Preprints 10 A969) 2, 1351 —1352.
149. H. Gilch, J. Polymer Sei. A 1, 4 A966), 6, 1351—1357.
150. H. Gilch, J. Polymer Sei. A 1, 4 A966), 6, 1337—1349.
151. В. Кравцов, Высокомол. соед., 9, 1967, 3, 550—555.
152. L. Taylor, J. Polymer Sei. В 1 A963), 3, 117—119.
153. Англ. пат. 807196, 7. 1.59 Du Pont.
154. S. Ross, J. appl. Polymer Sei. 11 A967) 7, 1209—1215.
155. D. Kluiber, J. org. Chem. 30 A965), 2037—2041.
156. H. Hefter, Chem. Ber. 104 A971), 2581—2587.
157. П. Буренко, Высокомол. соед., 5, 1963, 11, 1597—1602.
158. D. Hoeg, J. Polymer Sei. В 2 A964) 7, 697—701.
159. Пат. США 3117168 7. 1.64 Union Carbide.
160. Пат. США 3149175 A964) Union Carbide.
161. Пат. США 3155712 3. 11.64 Union Carbide.
276
162 Англ. пат. 883939 A961) Union Carbide.
163 Англ. пат. 883840 A961) Union Carbide.
164 Аргл пат. 833941 A961) Union Carbide.
165. Акцепт, заявка ФР'Г 1085673, 21.7.60 Union Carbide.
166. W. uorham, J. Polymer bei. A 1 4 A966), 3027—3039.
167. L. Errede, Quart. Rev. (London) 12 A958), 301—312.
168. W. Gorhani, ACS Polymer Preprints 6 A965) 1, 73—83.
169. W. Gorham, J. Polymer Sei. A 1 4 A966) 12, 3027—3039.
170. «Parylene for Electronics», Firmenschrift, Union Carbide, 1970.
171. Modern Plastics 8 A970), 22—23.
172. S. Kubo, J. Polymer Sei. 10 A972) 10, 1949—1966.
173. S. Kubo, Makromolekulare Chem. 157 A972), 299—302.
174. S. Kubo, J. appl. Phys. 42 A971> 12, 4558—4565.
175. S. Kubo, J. appl. Phys. 42 A971) 12, 4565—4570.
176. S. Kubo, Makromolekulare Chem. 162 A972), 1—7.
177. G. Lieser, Dissertation Naturw. Fakultat Mainz 1971.
178. B. Wunderlich, J. appl. Phys. 42 A971), 4565—4569.
179. W. Niegisch, J. appl. Phys. 37 A966) 11, 4041—4046.
180. A. Kajnera, Kolloid-Z. 224 A968) 2, 124—128.
181. J. Schaefgen, J. Polymer Sei. 15 A955), 19—29.
182. J. Schaefgen, J. Polymer Sei. 41 A959), 133—141.
183. H. Jellinek, J. Polymer Sei. A 1 8 A970) 9, 2517—2534.
184. H. Jellinek, J. Polymer Sei. A 1 9 A971), 2605—2615.
185. M. Spivack, Rev. Scientific Instrum. 41 A970) 11, 1614—1616.
186. «Bakelite Parylene», Firmenschrift, Union Carbide 1965.
187. L. Auspos, J. Polymer Sei. 15 A955), 19—29.
188. M. Spivack, J. electrochem. Soc. 116 A969) 11, 1592—1594. .
189. S. Pitts, Sei. Techn. Aerospace Rep. 7 A969) 17, 3184.
190. Пат. США 176625 1.3.62 Union Carbide.
191. Patentanm. 1216255 12.5.66 Union Carbide.
192. R. Coriey, J. Polymer Sei. 13 A954), 137—142.
193. Ю. Юкельсон, Изв. высшей школы, хим. и хим. технология, 8, 1965, 6.
194. Г. Колесников, Высокомол. соед, Б 10, 1968, 3, 182—185.
195. Г. Колесников, Высокомол. соед., А 11, 1969, 12, 2643—2651.
196. Пат. США 3342754 A966) Union Carbide.
197. W. Barb, J. Polymer Sei. 10 A953), 49—62.
198. L. Errede, ACS Polymer Preprints 1 A959) 2, 293—299.
199. L. Errede, J. Amer. chem. Soc. 79 A957), 6507—6510.
200. S. Chow, J. appl. Polymer Sei. 13 A969) 11, 2325—2332.
201. Пат. США 3626032 7. 12.71. U. S. Dept. of Navy.
202. А. Калашник, Высокомол. соед., 8, 1966, 3, 526—531.
203. G. Drefahl, Chem. Ber. 91 A958), 1274—1280.
204. G. Drefahl, Chem. Ber. 94 A961), 907—914.
205. R. Me Donald, J. Amer. chem. Soc. 82 A960) 17, 4669—4671.
206. A. Ваншейдт, Высокомол. соед., 5, 1963, 6, 805—810.
207. Г. Лапицкий, Высокомол. соед., А 9, 1967, 1274—1279.
208. G. Manecke, Makromolekulare Chem. 129 A969), 183—202.
209. H. Horhold, Makromolekulare Chem., 131 A970), 105—132.
210. G. Kossmehl, Makromolekulare Chem. 131 A970), 37—54.
211. G. Drefahl, Mitteilungsblatt Chem. Ges. DDR 12 A965), 140.
212. Пат. Дании 51436 5. 11.66 G. Drefahl.
213. H. Horhold, Z. Chem. 12 A972), 41—52.
214. Пат. Дании 84272 A972) H. Horhold.
215. W. Funke, Makromolekulare Chem. 74 A964), 71—91.
216. R. Lenz, J. org. Chem. 25 A960) 5, 813—817.
217. Пат. США 3336261, 1966.
218. H. Horhold, Plaste + Kautschuk 17 A970) 2, 84—88.
219. J. Gole, С. R. Hebd. Seances Acad Sei. 259 A964), 3022—3024
220. Пат. США 3422071, 14. 1. 69 U. S. Air Force.
221. E. Zidaroff, Naturwiss, 52 A965) 1, 13.
222. W. Dunnavant, J. Polymer Sei. A3 A965) 10, 3649—3653.
277
223. Пат. США 3110687, 12. 11.63 3 M Co.
224. R. Wade, J. Polymer Sei. В 5 A967), 565—567.
225. Пат. Дании 64889 A968) H. Horhold.
226. Пат. Дании 91567 20. 7. 72 H. Horhold.
227. M. Kanbe, J. chem. Soc. (Japan), Ind. chem. Sect. 71 A968) 8. 1276—1280.
228. M. Kanbe, J. Polymer Sei. A 1 6 A968) 4, 1058—1060.
229. G. Smets, J. Polymer Sei. A 1 7 A969), 3313—3328.
230. К. Ouchi, Aust, J. chem. 19 A966), 333—335.
231. E. Панов, Изв. АН СССР, сер. хим., 190, 1970, 122—126.
232. M. Ballester, J. Amer. chem. Soc. 88 A966), 957—968.
233. Пат. США 3532643, 6. 10. 70 Dow Chem.
234 С. Friedel, Bull. Soc. chim. France 43 A885), 53.
235. С Ingold, J. chem. Soc. A928), 2249.
236. R. Jacobson, J. Amer. chem. Soc. 54 A932), 1513—1518.
237. S. Bezzi, Gazz. chim. Ital. 66 A936), 491.
238. R. Shriner, J. org. Chem. 6 A941), 305—318.
239. L. Valentine, J. chem. Soc. A956), 4768—4779.
240 J. Kennedy, J. macromolecul. Sei. 1 A966), 541—553.
241. H. Brown, J. Amer. chem. Soc. 75 A953), 6285—6292.
242. H. Haas, J. Polymer Sei. 15 A955), 503—514.
243. L. Phillips, Transactions J. Plast. Inst. 32 A964), 298.
244. Пат. США 2870098 A965) Shell.
245 Пат. США 3405091 A965) Westinghouse Electric.
246. J. Doedens, Ind. Engin. Chem. 53 A961), 59—62.
247. A. Ваншейдт, Высокомол. соед., 4, 1962, 9, 1303—1309.
248. Ю. Митин, Докл. АН СССР, 115, 1957, 97—99.
249. A Fritz, J. Polymer Sei. A 1 10 A972) 8, 2365—2378.
250. Англ. пат. 1094181 6. 12.67 Royal Air Force.
251. Англ. пат. 1099123 A968) Midland Silicones.
252. Англ. пат. 1127122 A969) Midland Silicones.
253. Англ. пат. 1150203 A969) Midland Silicones.
254. G. Harris, Brit. Plastics 42 A969) 10, 108—113.
255. С. Marvel, J. Amer. chem. Soc. 80 A958), 1740—1744.
256. G. Harris, Werkstoffe und Korrosion 22 A971) 3, 227—234.
257. G. Harris, Brit. Plastics 42 A969) 10, 108—113.
258. G. Harris, Brit. Polymer J. A970) 11, 270—276.
259. G. F. d'Alelio, J. macromolecul. Sei. C3 A969), 1, 105—234.
260. С Marvel, J. Amer. chem. Soc. 79 A957), 6000—6002.
261. С Marvel, J. Amer. chem. Soc. 80 A958), 832—835.
262. С Marvel, J. Amer. chem. Soc. 81 A959), 2668—2670.
263. G. F. d'Alelio, J. macromolecul. Sei. A 1 A967) 7, 1251—1258.
264. G. F. d'Alelio, J. macromolecul. Sei. AI A967) 7, 1161—1249.
265. J. Akitt, Makromolekulare Chem. 56 A962), 195—199.
266. A. Topchiev, J. Polymer Sei. С 1 A963), 1305—1313.
267. S. Stivalla, J. Polymer Sei. В2 A964), 943—946.
268. R. Me Donald, J. org. Chem. 24 A959), 1246—1251.
269. G. F. d'Alelio, J. macromolecul. Sei. A 1 A967) 7, 1321—1330.
270. G. F. d'Alelio, J. macromolecul. Sei. AI A967) 7, 1279—1298.
271. G. F. d'Alelio, J. macromolecul. Sei. A 2 A968) 2, 285—333.
272. Пат. Дании 76970, 12. 11.70.
273. H. Horhold, Z. Chemie 12 A972) 2, 41—52.
274. G. F. d'Alelio, J. macromolecul. Sei. A 1 A967) 7, 1259—1278.
275. G. F. d'Alelio, J. macromolecul. Sei. A 2 A968) 6, 1235—1259.
276. G. F. d'Alelio, J. macromolecul. Sei. AI A967) 7, 1299—1320.
277. G. F. d'Alelio, J. macromolecul. Soi. A 2 A968), 6, 1223—1234.
278. G. F. d'Alelio, J. macromolecul. Sei. AI A967) 7, 1331—1364.
279. G. F. d'Alelio, J. macromolecul. Sei. A 2 A968) 5, 979—1043.
280. T. Curtius, J. prakt. Chem., Ser. II, 39 A889), 45.
281. W. Hunter, J. Amer. chem. Soc. 33 A911), 194.
282. W. Hunter, J. Amer. chem. Soc. 43 A921), 131—135.
283. J. Golden, Soc. ehem. Ind (London) Monogr. 13 A961), 231.
278
284. G Brown, Polymer Preprits 5 A963) 2, 39.
285 G Brown, Polymer Preprints 6 A964) 1, 195.
286 H Weingarten, J. org. Chem. 29 A964), 977—978, 3624—3626.
287 С Price, J. Amer. chem. Soc. 82 A960), 3632—3634.
288 С Price, j. Poiymer Sei. 61 A962), S. 28.
289 С Price, J. Polymer Sei. 61 A962), 135—141.
290 Пат. США 3236807, 22. 2. 66.
29 t. О. Sus, Ann. 598 A956), 123.
292 M. Dewar, J. chem. Soc. A958), 917—922.
293. A. Hay, J. Amer. chem. Soc. 8t. A959), 6335—6336.
294. A. Hay, J. Polymer Sei. 58 A962), 581—589.
295. В. Гуцалюк, Изв. АН Казахской ССР, хим., 21, 1971, 4, 59—63.
296 Англ. пат. 1255590, 1. 12. 71.
297. А. Hay, Anvanc. Polymer Sei. 4 A967), 496—527.
298. Пат. США 3642699 15.2.72 General Electric.
299. Опубл. заявка ФРГ 2011710 1. 10.70 General Electric.
300. Акцепт, заявка ФРГ 2011705 1.10.70 General Electric.
301. Пат. СССР 339561, 24.5.71.
302. Пат. Японии 7211513 11.4.72 Sumitomo Electric.
303 Пат. Японии 7211512 11.4.72. Sumitomo Electric.
304. Пат. СССР 326196, 19. 1.72.
305. Пат. СССР 275407, 3. 7. 70.
306. Пат. Японии 7128423 A971) Mitsui Petrochem.
307. Пат. Японии 7231718 15.8.72 Kanegafuchi Chem. Ind.
308. Пат. США 3306874 A962) General Electric.
309. Англ. пат. 930993 A958) General Electric.
310. Акцепт, заявка Японии 7203989, 3.2.72. Daicell.
311. Акцепт, заявка Японии 7022153, 27.7.70 Today Ind.
312. Акцепт, заявка Японии 7102265, 20. 1.71 Тогау Ltd.
313. Опубл. заявка ФРГ 2012443 14. 10.71 A. Pravednikov.
314. Е. Tsuchida, Nippon Kagaku Kaishi 8 A972) 7, 1313—1316.
315. E. Tsuchida, Nippon Kagaku Zasshi 7 A971) 11, 2370—2374.
316. Акцепт, заявка Японии 7210072 25.3.72 Kenegaiuchi Cbem. Ind.
317. Акцепт, заявка Японии 7109596 11.3.71 Mitsui Petrochem. Ind.
318. Акцепт, заявка Японии 7122934 30.7.71 Mitsui Petrochem. ind.
319. Опубл. заявка ФРГ 2155303 22.6.72 Japan Synthetic Rubber.
320. Опубл. заявка ФРГ 2160419 13.7.72 Mitsubishi.
321. Акцепт, заявка Японии 7111835 26.3.71 Тогау Ind.
322. Пат. СССР 297655, 11.3.71.
323. Пат. СССР 317683, 19. 10.71.
324. Пат. СССР 295781, 12.2.71.
325. Опубл. заявка ФРГ 2217161 2. 11.72 Mitsubishi Gas.
326. Пат. СССР 335257, 11.4.72.
327. Пат. СССР 328132, 2.2.72.
328. Пат. СССР 341814, 14.6.72.
329. Пат. СССР 321527, 19. 11.72.
330. Акцепт, заявка Японии 723195Ы6. 8. 72 Тогау Ind.
331. Опубл. заявка ФРГ 2061116 24.6.71 Sumitomo Chem.
332. Акцепт, заявка Японии 7205111 14.2.72 Sumitomo Chem.
333. Акцепт, заявка Японии 7200619 8. 1.71 Sumitomo Chem.
334. Акцепт, заявка Японии 7040305 17. 12. 70 Sumitomo Chem.
335. Акцепт, заявка Японии 7114469 17.4.71 Sumitomo Chem.
336. Акцепт, заявка Японии 7041235 24. 12.70 Mitsui Chem.
337. Акцепт, заявка Японии 7023555 7. 8. 70 Kanegafuchi Ind.
338. Опубл. заявка ФРГ 2114101 5. 10.72 Hoechst.
339. Акцепт, заявка Японии 7232836 22. 8. 72 Kanegafuchi Chem. Ind.
340. Акцепт, заявка Японии 7203746 1.2.72 Mitsubishi.
341. Опубл. заявка ФРГ 2057267 27.5.71 Atlas Chem. Ind.
342. Акцепт/заявка Японии 7121741 19.6.71 Mitsubishi.
343. Акцепт, заявка Японии 7232837 22. 8. 72 Asahi Dow Ltd.
344. E. Me Nelis, J. org. Chem. 31 A936), 1255-1259,
279
345. В. Lindegren. Acta ehem. Scand. 14 (I960). 1203-1210.
346. G. Нау nes, J. chem. Soc. A956), 2823—2831.
347. D. O. S. 2228071 21. 12.72 General Electric.
348. С Price, Macromolecules 4 A971), 363—369.
349. G. Endres, J. org. Chem. 28 A963), 1300—1305.
350. G. Endres, J. Polymer Sei. 58 A962), 593—598.
351. H. Finkbeiner, SPE Trans. 2 A962), 112—119.
352. E. Tsuchida, Makromolekulare Chem. 151 A972), 221—234.
353. B. Brooks, British Polymer J. 2 A970) 4, 193—196.
354. E. Boerle, Brennstoff-Chemie 30 A949), 392.
355. Пат. США 2789146 A954).
356. Англ. пат. 717588 A951) ICI.
357. Plast, mod. Elast. 22 A970) 1, 22.
358. Chem. Horizons (Overseas Rep.) 16.4.70, 63.
359. B. Judkin, Plast Massy A972) 3, 63—64.
360. Пат. СССР 214801, 30.9.69.
361. Пат. СССР 267074, 1.4.70.
362. Пат. СССР 238781, 23. 10.69.
363. Пат. США 3637593 25. 1.72 General Electric.
364. Пат. США 3573254 30.3.71 General Electric.
365. Акцепт, заявка Японии 7121222 15.6.71 Sumitomo Chem. Ind.
366. Пат. СССР 298610 16.3.71.
367. Y. Kamiya, Nippon Kagaku Kaishi A972) 12, 2379—2384
368. E. Tsuchida, Nippon Kagaku Kaishi A972) 12, 2416—2420
369. Опубл. заявка ФРГ 2062044 24.6.71 Sumitomo Ltd.
370. Опубл. заявка ФРГ 2119371 4. 11.71 General Electric.
371. Пат. США 3639499 1.2.72 General Electric.
371а. Опубл. заявка ФРГ 2136747 27. 1.72 Uniroyal.
372. Акцепт, заявка Японии 7127871 13.8.71 Sumitomo Chem
373. Пат. США 3660531 2. 5.72 Uniroyal.
374. Опубл. заявка ФРГ 2211005 16. 11.72 General Electric.
375. Опубл. заявка ФРГ 2211006 16.11.72 General Electric.
376. Фр. пат. 2086141 4.2.72 Uniroyal.
377. Нидерл. пат. 6517152 Uniroyal.
378. Нидерл. пат. 6517240 Uniroyal.
379. Пат. США 3063872 13. 11.62 General Electric.
380. Англ. пат. 1110195 General Electric.
381. Пат. США 3631126 28. 12.71 Uniroyal.
382. Опубл. заявка ФРГ 2136838 2.3.72 General Electric.
383. Опубл. заявка ФРГ 2136837 24.2.72 General Electric.
384. Опубл. заявка ФРГ 2138470 24.2.72 General Electric.
385. Акцепт, заявка ФРГ 2000118 16.7.70 General Electric.
386. Опубл. заявка ФРГ 2111043 30.9.71 General Electric.
387. Опубл. заявка ФРГ 2057107 3.6.71 Sumitomo.
388. Нидерл. пат. 6500032.
389. Пат. США 3356761.
390. Пат. США 3383435.
391. Пат. США 3384682.
392. Опубл. заявка ФРГ 2061115 16.6.71 Sumitomo Ltd.
393. Пат. США 3658949 25. 4. 72 Subitomo Ltd.
394. Опубл. заявка ФРГ 2107935 23.9.71 General Electric.
395. W. Wrasidlo, J. Polymer Sei. A 2 10 A972) 9, 1719—1729.
396. F. Karasz, J. Polymer Sei. B3 A965) 7, 561—563.
397. F. Karasz, J. Polymer Sei. A2 6 A968) 6, 1141 —1148.
398. A. Shultz, J. Polymer Sei. A27 A969), 1577—1583.
399. J. Barrales-Rienda, Kolioid-Z. 244 (I971), 317—323.
400. A. Packer, J. Polymer Sei. В A961) 6, 435—441.
401. Б. Маличенко, Успехи химии, 40, 1971, 3, 547—571.
402. С. Priee, J. Polymer Sei. 49 A961), 267—272.
403. А. Нау, Polymer Preprints 2 A961) 2, 319.
404. А. Нау, J. Polymer Sei. B3 A965), 887—889.
280
405 Фр пат. 1322152 A963).
406 J Сох J. appl. Polymer Sei. 9 A965), 513—522.
407 J Lancaster, J. appl. Polymer Sei. 9 A965). 1955—1971.
408 A Eibenberg. J. Polymer Sei. С 35 A971), 129—149.
409. W. Butte. J. Polymer Sei. 61 A962), S 28— S 29.
410 E Magre, 2nd Microsymposium on the structure of organic solid, Prag 1968.
411 A Savolainen, Acta chem. Scand. 26 A972) 2, 846—847.
412 S de Petris, J. Polymer Sei. A 2 10 A972) 8, 2365—2378.
413 F. Karasz, J. appl. Phys. 41 A970) 11, 4357—4360.
414 F Balta-Calleja, J. Macr. Sei. В 6 A972) 2, 387—396.
415. P Kelleher, Polymer Engin. Sei. 10 A970) 1, 38—42.
416. W. Me Knight, Advan. Chem. Ser. 99 A971) 3, 29—41.
417. W. Me Knight, J. Polymer Sei. A2 10 A972), 1639—1655.
418. J. Barrales-Rienda, European Polymer J. 3 A967) 4, 535—550.
419. A. Factor, J. Polymer Sei. В 7 A969), 205—209.
420. E. Turska, Polimery 16 A971) 6, 269—272.
421. M. Bethell, Dissertation, Univ. Manchester 1968.
422. P. Akers, Polymer 9 A968) 11, 575—584.
423. A. Shultz, J. Polymer Sei. A2 10 A972), 273—282.
424. Пат. США 3644227 22. 2. 72 General Electric.
425. P. van Emmerik, J. Polymer Sei. С 38 A972), 73—8b.
426. D. Pepper, J. Polymer Sei. В A966) 4, 939—941.
427. J. Barrales-Rienda, Ann. Real Soc. Esp. Fis. Quim Ser. В 66 A970) 9+10,
767—777.
428. D. White, J. Polymer Sei. A 1 10 A972) 6, 1565—1578.
429. D. White, J. Polymer Sei. A 1 8 A970) 6, 1427—1438.
430. A. Faktor, J. Polymer Sei. A 1 7 A969) 1, 363—377.
431. J. Penczek, Polimery 13 A972), 121—123.
432. E. Fitzer, High Temperatuies—High Pressures 3 A971), 53—63.
433. Z. Slama, Chem. Prumysl A970), 270—273.
434. P. Kelleher, J. appl. Polymer Sei. 11 A967), 1, 137—144.
435. A. Sidhu, Z. Werkstofftechnik 2 A971) 6, 298—304.
436. R. Jerussi, J. Polymer Sei. A 1 9 A971), 2009—2029.
437. A. Kim. Plast. Massy A972) 3, 10—12.
438. Опубл. заявка ФРГ 2120154 16. 11.71 General Electric
439. Акцепт, заявка ФРГ 1961603 9.7.70 General Electric.
440. Пат. СССР 306145, 11.6.71.
441. Акцепт, заявка Японии 7040551 19. 12. 70 Sumitomo.
442. Акцепт, заявка Японии 7132427 21.9.71 Sumitomo.
443. Акцепт, заявка Японии 7102837 23. 1.71 Sumitomo.
444. Пат. СССР 311936, 19.8.71.
445. Опубл. заявка ФРГ 2123415 23. 11. 72 A. Katchman.
446. Акцепт, заявка Японии 7128184 16.8.71 Sumitomo.
447. E. Dragowska, Polimery 17 A972) 3, 131—135.
448. Пат. СССР 328151, 2.2.72.
449. Акцепт, заявка Японии 7124782 16.7.71 Sumitomo.
450. Акцепт., заявка Японии 7142029—7142033 П. 12. 71 Sumitomo.
451. Акцепт, заявка Японии 7134624 11. 10.71 T. Takemura.
452. Пат. США 3563934 16.2.71 General Electric.
453. Е. Jaskot, SPE J. 24 A968) 7, 33—36.
454. Kunststoffe 59 A969) 10, 663—666.
455. Фр. пат. 1484306 A964) General Electric.
456. Нидерл. пат. 6609492 General Electiic.
457. Пат. США 3708455 2. 1. 73 Asahi Dow.
458. Kunststoffe 57 A967) 5, 366.
459. J. Barrales-Rienda, Rov. Plast. Mod. 139 A968), 3—7.
460. J. Bussink, Kunststoffe 61 A972) 10, 649—652.
461. Kunststoffe 59 A969), 26.
462. J. Barrales-Rienda, Rev. Plast. Mod. 132 A967), 458—461.
463. Brit. Plastics 41 A968) 10, 90—94.
464. Акцепт, заявка Японии 7125628 23.7.71 Hitachi Chem.
28
465. Kunststoffe-Plastics 19 A972) 10, 401.
466. M. Silverman, Mod. P^stics 45 A968) 9, 128—130.
467. Gummi - Asbest — Kunststoffe 23 A970) 4, 420—423.
468. Brit. Plastics 39 A966) 8, 441—443.
469. Brit. Plastics A971) 12, 53—55.
470. D. Flashman, Rubber & Plastics Age 47 A966) 4, 383—386.
471. J. Barrales-Rienda, Rev. Plast. Mod. A971), 28—42.
472. R. Steinbuch, Kunststoffe-Plastics 12 A965) 3, 149—155.
473. M. Macnulty, J. Mater. Sei. 6 A971), 1070—1075.
474. Пат. США 3663654, 16. 5. 72.
475. Опубл. заявка ФРГ 2123291 25. 11.71 AKZO N. V.
476 Kunststoffe-Plastics 18 A971) 5,204.
477. Kunststoffe-Plastics 10 A972) 2, 63.
478. R. van Wigh, Advan, Chromatogr. Proc. Int. Symp. 6th 1970, 122—124.
479. R. van Wigh, Column Chromatogr. int. Symp. Separ. Methods 5th 1969, 254—
256.
480. Kunststoff-Rundschau 16 A969) 8, 483—484.
481. J. Knobbout, Sei. Techn. Aerospace Rep. 7 A969) 22. 4206.
482. J. Knobbout, Sei. Techn. Aerospace Rep. 8 A970) 3, 485.
483. A. Hay, ACS Polymer Preprints 10 A969), 92—96.
484. Пат. США 3639337, 1.2.72 Generai Electric.
485. G. Cooper, Advan. Chem. Ser. 99 A971) 27, 431—450.
486. D. White, J. Polymer Sei. A 1 9 A971) 3, 663—675.
487. J. Fejgin, J. Polymer Sei. В 4 A966), 615—622.
488. Нидерл. пат. 6413958 General Electric.
489. Пат. США 3668273, 6. 6. 72 General Electric.
490. Акцепт, заявка Японии 7201210 13. 1.72 Sumitomo.
491. A. Chalk, J. Polymer Sei. A 1 7 A969), 691—705.
492. A. Chalk, J. Polymer Sei. A 1 9 A971 >, 3067—3069.
493. A. Chalk, J. Polymer Sei. A 1 7 A969), 1359—1369.
494. A. Chalk, J. Polymer Sei. A 1 7 A969), 2537—2545.
495. Пат. США 3631130 28. 12.71 General Electric.
496. Пат. США 3259592 5. 7. 66 General Electric.
497. С. Plummer, U. S. Office Saline Water Res. Develop. Progr. Rep. A970), 551.
498. S. Kimura, Ind. Engin. Prod. Res. Develop. 10 A971) 3, 335—339.
499. W. Schoene, Angew. Makr. Chem. 7 A969), 15—28.
500. A. Savolainen, Suomi Kemistilehti В 44 A971), 143—145.
501. К. Tsou, J. Polymer Sei. A2 A964), 4425—4431.
502. H. Hoyt, J. appl. Polymer Sei. 8 A964), 1633—1641.
503. R. Neville, J. appl. Polymer Sei. 12 A968), 607—618.
504. Пат. США 3703564, 21. 11.72 General Electric.
505. Англ. пат. 1071768 A967) General Electric.
506. Пат. США 3375298 A967) General Electric.
507. Пат. США 3396146 A967) General Electric.
508. Пат. США 3275225 A967) General Electric.
509. Пат. США 3406147 A967) General Electric.
510. Опубл. заявка ФРГ 2057272 25.5.72 General Electric
511. Kunststoffe 61 A971) 7, 481.
512. D. Sassano, 28th Techn. Conf. Pap. A970), 498—502.
513. Белы. пат. 632201 A963) Westinghouse.
514. L. Mortillaro, Mat. Plast. Elast. 32 A966) 12, 1257—1259.
515. Аигл. пат. 980509 A965) Shell.
516. Пат. США 3006892, 27. 1.59, CIBA.
517. N. Reinking, J. appl. Polymer Sei. 7 A963), 2145—2152.
518. N. Reinking, J. appl. Polymer Sei. 7 A963), 2153—2160.
519. Пат. США 3177090, 6.4.65, Shell.
520. J. Brzezinski, Polimery 14 A969) 3, 117—120.
521. G. Myers, J. Polymer Sei. А 2 A964), 2631—2645.
522. J. Brzezinski, Polimery 11 A966), 423—429.
523. R. Miles, SPI 20th Ann. Meeting RIP Div., 1965.
524. R. Norwalk, Plastics Technol. 2 A963), 34—39.
282
525 Packaging News (London) 10A963) 10, 25—26.
526 Пат США 3177090 A965) R. Baynes.
527. Mat. Design. Engin. 56 A962) 7, 12.
528. R. Bugel, ASTM Symp. Atlantic City, 27. 6. 63.
529 «phenoxy» Bulletin 10181. Union Carbide.
53O. Brit. Plastics 40 A962) 3, 172.
531 Gummi —Asbest —Kunststoffe 17 A969) 2, 165—166.
532. Mod. Plastics 40 A962) 3, 169—172.
533. Rubber & Plastics Age 45 A964) 1, 62—63.
534. M. Sauers, Reg. Techn. Conf. SPI, Acron Sect. 1970, 1—6.
535. R. Johnson, J. Polymer Sei. A 15 A967) 9, 2375—2398.
536. Пат. США 2822351 A958) Union Carbide.
537. Фр. пат. 1475231 A965) Union Carbide.
538. Патентная заявка Франции 2015676 30.4. 70 Bayer.
539. Фр. пат. 2050554 7.5.71 Union Carbide.
540. R. Johnson, J. Polymer Sei. A 15 A967) 9, 2415—2427.
541. Нидерл. пат. 6408130 Union Carbide.
542. Пат. США 3554972 12. 1.71 Uniroyal.
543. Опубл. заявка ФРГ 2025467 10. 12. 70 Uniroyal.
544. Фр. пат. 2056205 18. 6. 71 Uniroyal.
545. Фр. пат. 2090204 18. 2. 72 Uniroyal.
546. Опубл. заявка ФРГ 2122734, 2. 12.71 ICI.
547. Опубл. заявка ФРГ 2122735, 2. 12.71 ICI.
548. Опубл. заявка ФРГ 2035143 28. 1.71 Uniroyal.
549. Опубл. заявка ФРГ 2035148 28. 1.71 Uniroyal.
550. Опубл. заявка ФРГ 2041532 2.3. 71 Bayer.
551. Опубл. заявка ФРГ 2122735, 26. 11.71 ICI.
552. Пат. США 3658938 25. 4. 72 Union Carbide.
553. Опубл. заявка ФРГ 2051890 6.5.71 Borg-Warner Со.
554. М. Cudby, Polymer 6 A965), 589.
555. Plastverarbeiter 23 A972) 12, 856.
556. Kunststoffe 63 A973) 2, 122.
557. Опубл. заявка ФРГ 2038167 11.2.71 ICI.
558. Опубл. заявка ФРГ 2038166, 11.2.71 ICI.
559. M. Cudby, J. Polymer Sei. С 2 A969) 22, 747—760.
560. В. Jennings, J. Polymer Sei. С 16 A967) 2, 715—724.
561. H. Vogel, J. Polymer Sei. A 18 A970), 2035—2047.
562. Опубл. заявка ФРГ 2200502, 20. 7. 72 ICI.
563. В. Сергеев, Высокомол. соед., А 11, 1969, 12, 2636—2642.
564. Опубл. заявка ФРГ 2038168 11.2.71 J. Rose.
Б65. Англ. пат. 1265145, 1.3.72 ICI.
666. Пат. США 3579475 18. 5. 71 М. Jones.
567. Опубл. заявка ФРГ 2139535, 10.2.71 ICI.
568. Опубл. заявка ФРГ 2163920, 13.7.72 ICI.
569. С. Виноградова, Высокомол. соед., А 14, 1972, 12, 2545—2552.
570. G. Allen, European Polymer J. 5 A969), 319—334.
571. N. Mills, J. Macromolecul. Sei. В 4 A970) 4, 863—876.
572. Пласт, массы, 1971, 7, 45—46.
573. С. Либина, Пласт, массы, 1972, 1, 23—25.
574. Пат. СССР 295438, 1. 10.71.
575. Пат. СССР 301073, 11.6.71.
576. W. Daly, J. Polymer Sei. В 10 A972) 7, 519—525.
577. G. Allen, European Polymer J. 6 A970), 1635—1641.
578. M. Акутин, Труды Моск. хим.-техн. ин-та им. Д. И. Менделеева 1970 66
212-214. ' '
579. W. Haie, J. Polymer Sei. A 1 5 A967) 9, 2399—2414.
580. A. Davis, Makromolekulare Chem. 128 A969), 242—25L
581. Ж. Левантовская Высокомол. соед., А 13, 1971, 1, 8—15.
582. H. Tarnowiecki, Mitt. Oesterr. Kunststoofinstitut 24 A970) 6 296—298
583. T. Hsueh, Anal. Calor. Proc. Symp. 2nd 1970, 103—112.
584. P. Kelleher, J. appl. Polymer Sei. 12 A968), 1199—1208.
283
585. H. Classer Kunststoffe 61 A971) 4, 232—234.
586. R. Bringer, Applied Polymer Symp. 11 A969), 189—208.
587. Опубл. заявка ФРГ 2163930 13.7.72 ICI.
588. A. Davis, Makromolekulare Chem. 129 A969), 63—72.
589. A. Lyons, Makromolekulare Chem. 157 A972), 103—109.
590. R. Burns, Plastics Design & Processing A972), 4.
591. P. Byra, SPI Reg. Techn. Conf., Acron Sect., Sept. 1970, Techn. Papers 10—16.
592. Опубл. заявка ФРГ 2211758, 10.9.71 ICI.
593. Опубл. заявка ФРГ 2221969 7. 12. 72 Union Carbide.
594. Kunststoff-Technik 10 A971) 7, 260.
595. Опубл. заявка ФРГ 2129163, 16. 12.71 ICI.
596. H. Bassett, Plastics Techn. A965) 10, 49—51.
597. H. Bassett, Gummi — Asbest — Kunststoffe 19 A966) 5, 551—553.
598. Kunststoff-Berater A972) II, 582.
599. Kunststoff-Plastics A972) 12, 502.
600. Опубл. заявка ФРГ 2141913 24.2.72 Rhone-Poulenc.
601. Акцепт, заявка Японии 7113839 13.4.71 Mitsubishi Rayon.
602. В. Benson, SPE-J. 23 A967) 7, 31—35.
603. G. Morneau, Mod. Plastics 47 A970) 1, 150—152.
604. J. Huml, Plast. Hmoty Kaucuk 7 A970) 4, 102—106.
605. M. Russo, Materie Plast. Elast. 34 A968) 2, 157—159.
606. Kunststofftechnik 7 A968) 10,362—363.
607. В. Лапшин, Пласт, массы, 1, 1967, 74—78.
608. F. Riehl, Hydrocarbon Processing 46 A967) 3, 161—164.
609. Firmenschrift Bulletin J-27000, 036-6, Union Carbide, 1965.
610. T. Bugel, SPE-J. 24 A968) 3, 52—55.
611. W. Alvino, J. appl. Polymer Sei. 15 A971), 2521—2537.
612. Mod. Plastics A965), 87—89.
613. R. Walten, Mod. Plast. Encycl. 1970/71, 220.
614. Brit. Plastics 41 A968) 11, 122.
615. И. Хухрева, Высокомол. соед., А 14, 1972, 5, 1122—1126.
616. Mater. Design. 5 A969), 89—91, 196.
617. L. Scala, NASA-CR-93 194, QR-6.
618. Пат. США 3264536.
619. Пат. США 3632404 4. 1.72 Amicon.
621. Е. Maganiello, Adhesives Age 14 A971) 3, 24—27.
622. Опубл. заявка ФРГ 2130407 23. 12.71 ICI.
623. Фр. пат. 1591737 12. 6. 70 Dynamit Nobel.
624. Опубл. заявка ФРГ 2225283 14. 12. 72 Rhone-Poulenc.
625. Firmenschrift Union Carbide, Bulletin 5-1416 A965).
626. Kunststoff-Berater A973) 6, 454—455.
627. Plastverarbeiter 25 A974) 2, 114.
5.7. АРОМАТИЧЕСКИЕ ПОЛИСУЛЬФИДЫ
Ароматические полисульфиды получают путем электрофильного
присоединения серы или ее галогенидов к ароматическим
соединениям, гомополиконденсацией я-галогентиофенолятов или
взаимодействием ароматических дигалогенидов с серой и карбонатом
натрия. В промышленности поли-/г-фениленсульфид получают
с 1968 г. поликонденсацией в N-метилпирролидоне /г-дихлорбензола
с сульфидом натрия. При 315—370 °С из полимера можно получить
покрытия или изделия литьем под давлением. Температура
длительной эксплуатации поли-я-фениленсульфида 180—260 °С. Основ-
284
ной областью его применения являются термостойкие и коррозион-
ностойкие покрытия металлов, стеклонаполненные литьевые
изделия для электротехники.
5.7.1. ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ ПОЛИСУЛЬФИДОВ
5.7.1.1. Электрофильное присоединение серы
или ее галогенидов к ароматическим соединениям
Электрофильное присоединение серы или ее галогенидов,
таких, как S2C12, SC12, к бензолу, алкилбензолам или многоядерным
ароматическим соединениям, таким, как нафталин, антрацен или
фенантрен, в присутствии катализаторов Фриделя — Крафтса типа
А1С13, FeCl3 или серной кислоты сопровождается образованием
(в различных соотношениях) разветвленного полисульфида и
серосодержащих гетероциклических соединений как побочных
продуктов:
Кат
+ SX
[-04.
где X = Cl, Cl2.
Полисульфид с температурой плавления 295 °С был впервые
получен в 1898 г. [1] взаимодействием бензола и серы в
присутствии А1С1з- Аналогичные продукты были синтезированы в 1909 г.
конденсацией тиофенола [2] в присутствии А1СЬ и в 1910 г.
конденсацией тиофенола в концентрированной серной кислоте [3].
Взаимодействием тиофенола с тионилхлоридом получены
низкомолекулярные полисульфиды [4].
Ароматические полисульфиды общей формулы
_п
образуются при взаимодействии ж-ксилола с серой в присутствии
А1С13 при 80—90 °С [7]. Полимеры, содержащие до 90% серы,
получают нагреванием ароматических соединений с серой в
концентрированной серной кислоте [8]. Аморфные полисульфиды
с температурой плавлени-я 100—110°С образуются при
взаимодействии нафталина, антрацена или фенантрена с S2C12 в
хлороформе или тетрахлорнде углерода в присутствии висмута или сурьмы
(в реакции с S2C12 образуется in situ катализатор Фриделя —
Крафтса) [5]. Дальнейшее нагревание этого полимера выше
90 °С приводит к получению высокомолекулярного продукта [6].
Линейный высокомолекулярный полисульфидоэфир
285
синтезирован из дифенилоксида и SC12 в хлороформе с
применением в качестве катализатора Fe — SCh. В аналогичных условиях
в присутствии S2C12 с S2C12 — Fe или S2C12 — FeCb образуется
аморфный полимер. Взаимодействие дифенилоксида с S2C12 или
SC12 в присутствии А1С13 в хлороформе приводит к получению
гетероциклических соединений:
Ароматические полисульфиды можно получать также по
реакции Зандмейера между бисдиазотированными ароматическими
соединениями и Na2S [10]:
Г—/ON—S—1
5.7.1.2. Реакция замещения галогенсодержащих
ароматических соединений
Ароматические полисульфиды были получены нуклеофильным
замещением ароматических галогенидов [11—13] в конце 40-х
годов. При взаимодействии /г-дихлорбензола с серой и безводным
карбонатом натрия при 300—340 °С образуется полисульфид
с температурой плавления 285—350СС и молекулярной массой
9000—17 000:
Cl—(Q)—CI + 3S + 2Na2CO3 —>
—> Г—^QN—S—1 + 2NaCl + 2CO2 + Na2S2O2
В аналогичных условиях при использовании о- и ж-дихлорбензо-
лов получаются только олигомерные продукты [10]. Недостатком
синтеза на основе n-дихлорбензола является образование
разветвленных макромолекул. Механизм этого процесса изучен Ленцем
114]. Согласно [15], на стадии инициирования образуются
радикалы серы (их концентрация в расплавленной сере при 300 °С
составляет 10~3 моль/л), которые реагируют с дихлорбензолом:
+ Cl или f ) +н
286
Взаимодействие карбонатных ионов с серосодержащими
радикалами приводит к образованию тиофенолятных ионов:
Ci—<O/—s** + Na2CO3 —> Ci—Hj)—S"Na+ + NaOS*-, + CO2
—Cl—(Q)—Cl + Na2CO3 —> Cl—(Cji) Cl + NaOSx_i + CO2
\ / \ /
Тиофенолятные ионы реагируют с дихлорбензолом:
CI— <O>~S— (Q)—Cl + NaCl
Cl—/Q)—Cl + Cl— (Q>—C1
S"Na+
\
S—{{_))—Cl + NaCl
Рост цепи сопровождается образованием линейных и
разветвленных продуктов. В ходе поликонденсации сера реагирует с
карбонатом натрия с образованием сульфида натрия, взаимодействие
которого с хлором, содержащимся в концевой группе
макромолекулы, протекает с получением тиофенолятных групп:
Na2S
—> Na+S~—<ГУ>—S—<TJ)—••¦ + NaCl
Конденсация сопровождается увеличением молекулярной массы:
—NaCl
Разветвленные продукты получены также при использовании
в качестве исходного мономера /г-хлорбензойной кислоты [46].
При использовании /г-хлорфенола синтезированы полифенилен-
сульфиды с гидроксифенильными и циклогексадиеновыми
группами в цепи. На основе /г-хлортолуола получен полимер с фени-
ленсульфидными, бензилсульфидными и 6-метил-1,3-фениленсуль-
фидными группами в цепи. Строго линейные полимеры получены
287
гомополнконденсацией солен щелочных металлов л-галогентиофе-
нола в твердой фазе или растворе [14, 16, 17]:
Температура плавления л-галогентиофенолятов щелочных
металлов колеблется от 200 до 400 °С;
гпл- сс гпл« °с
IC6H4SNa 220 BrC6H4SNa 315
BrC6H4SLi 325 BrC6H4SK 325
ClC6H4SLi 330 ClC6H4SNa 375
Поликоиденсацию в твердой фазе проводят при температуре
на 20—30° ниже температуры плавления полимера. Для
получения линейных полимеров необходимы чистые мономеры; при этом
ниже 300 °С реакция протекает в течение нескольких суток. По
этому способу фирма «Dow Chemical» получила полифениленсуль-
фид с температурой плавления 270—315 °С и температурой
стеклования 150 °С под маркой «Экспериментальная смола QX 4375.1»
[18].
Поликонденсация л-галогентиофенолятов щелочных металлов-
в растворе таких растворителей, как пиридин или хинолин,
протекает в 50—100 раз быстрее, чем в твердой фазе. Выход за 6 ч
при 250 °С при использовании пиридина составляет в случае хлор-
замещенного тиофеиолята 85 %, а фторзамещенного 91 %.
Реакционная способность уменьшается в ряду галогенов I > Вг > F >
> Cl и щелочных металлов Li > Na > К. Этот способ получения
поли-л-фениленсульфнда (ПФС) был усовершенствован [19, 20],
и в 1968 г. фирма «Philips Petroleum» организовала его
производство под названием «Райтон». Литьевой полимер выпускается под
маркой «Райтон R6» (ненаполненный) и «Райтон R4»
(наполненный 40% стеклянного волокна) [423]. В 1973 г. его объем
производства увеличился на 2700 т в год.
В 1969 г. цена 1 кг полимера составила 1,5 дол. США.
Промышленный способ производства этого полимера основан
на применении л-дихлорбензола и Na2S-9H2O. Поликонденсацию
проводят в гексаметилфосфотриамиде или N-метилпирролидоне
в инертной среде при температуре выше 125°С.
Высокомолекулярный полимер получают, возвращая 30—50 %
низкомолекулярного ПФС в реактор [21, 22]. Используемый в качестве
растворителя N-метилпирролидон регенерируется путем экстракции смесью
метиленхлорид — бензол [23] или метиленхлорид — хлороформ —
тетрахлорэтан [24]. Низкомолекулярный продукт можно
получать этим способом, используя в качестве регулятора роста цепи
хлорбензол.
Применяя смесь дихлорбензола с дихлоранилином,
синтезируют термореактивные сополимеры, отверждаемые дикарбоновыми
кислотами. Замещенные полифениленсульфиды такие, как поли-
2-метил-1,4-фениленсульфид (т. пл. 60—100 °С), поли-2,6-диметил-
фениленсульфид (т. пл. 160—180°С), получены [25] поликонден-
288
сациеи в растворе литиевых и натриевых солеи алкилзамещенных
я-галогентнофенолов с применением в качестве катализатора
меди. При взаимодействии 4,4/-димеркаптодифенила или 4,4'-ди-
меркаптодифенилоксида с 4,4'-дихлорд1тфеиилсульфоном по
аналогии с полиарилеисульфоноксидами получают полисульфонтиоэфи-
ры или полисульфоны с простыми эфирными пли тиоэфирными
связями [26].
Линейные ароматические полпсульфиды с простыми
эфирными, метпленовымн или изопропплидеиовыми группами в цепи
образуются при поликонденсации в растворе бнстпофенолятов
щелочных металлов с ароматическими 1,4-дигалогенидами в N-ме-
тилпирролндоне пли дпметплсульфоксиде при 200—210 °С [27],
например
СНз
п Na S—
О>
-SNa + «Cl-
О
-Cl
—NaCl
СНз
СНз
СНз
п
5.7.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
ПОЛИФЕНИЛЕНСУЛЬФИДОВ
5.7.2.1. Температура плавления и кристалличность
Линейные поли-я-фениленсульфиды представляют собой
преимущественно кристаллические полимеры с температурой
плавления кристаллической фазы 287 °С и температурой стеклования
150 СС. Наличие заместителей в фенильном ядре, таких, как гид-
роксп-, амино- или метнльиые группы, приводит к понижению
температуры плавления полимера более чем на 100° [25,46].
Кристаллическая структура ориентированного поли-/г-фениленсульфи-
да аналогична структуре поли-я-фениленоксида [30]. Орторомбн-
ческая элементарная ячейка включает четыре мономерных
остатка. При этом она пронизывает две макромолекулярные цепи: одна
макромолекула проходит через центр ячейки, а вторая под
некоторым углом. Ячейка имеет следующие размеры: а = 8,67 А; Ъ =
= 5,61 A, с =10,26 А; пространственная группа Pbcn — D2h.
Атом серы, входящий в цепь, которая имеет форму зигзага,
располагается в плоскости A00). Плоскости фениленовых циклов
чередуются под углом 45 или —45° к этой плоскости. Так же как
в алифатических серосодержащих полимерах, атом серы
расположен под углом 110 с:
10 Зек. 18
239
777,%
F/7/9
UU
80
60
20
- \
\
i 1
s. /
i i
1
0
Рис. 5.54. Кривые деструкции поли-гс-фенилен-
сульфида на воздухе A) и в среде азота {2}
[47].
Кристаллографическая плотность
поли-гс-фениленсульфида составляет
1,43 г/см3, тогда как
экспериментально определенные значения плотности
200 Ш 600 воо t,°c аморфНОГО и поликристаллического
полимеров—соответственно 1,32 и 1,35 г/см3. В спектре
механической релаксации поли-/г-фениленсульфида имеется пик при
температуре ниже — 200 °С и при —108 °С [45]. По аналогии с поли-я-фе-
ниленоксидом ?-релаксация при —108 °С объясняется
затруднением вращения вокруг оси —S—С6Н4—S—. Энергия активации
крутильных колебаний составляет 11+2 ккал/моль.
5.7.2.2. Растворимость
Ниже 175 °С линейные поли-п-фенилен-
сульфиды не растворяются в органических
растворителях. Выше 175°С они
растворяются в хлорированных ароматических и
алифатических углеводородах,
ароматических простых эфирах и кетонах. Вязкость
измеряют при 250 °С в растворе дифенил-
оксида. Характеристическая вязкость 0,166
A00 мл/г) соответствует среднечисловой
молекулярной массе 40 000.
5.7.2.3. Термостойкость
Линейные поли-/г-фениленсульфиды ста-
бильны на воздухе и в инертной атмосфере
до 420 °С. Наличие в фенильном цикле
заместителей, таких, как гидрокси-, амино-
или метильная группа, понижают
температуру начала разложения до 270—300°С
[46]. Кривые ТГА для поли-п-фениленсуль-
фида, полученного из л-бромтиофенолята,
до 540 °С совпадают при нагревании в азоте
и на воздухе; потеря массы до этой
температуры составляет около 45 % (рис. 5.54).
При нагревании до 800 °С потеря
массы на воздухе составляет 85 %, в азоте
60% [47].
0 12 3
m,%
VU
80
60
40
20
- S
-
1
— i
1
/U3175*y
/
f
/
^396,5°
i i i i
7 2 3
Рис. 5.55. Кривые изотермического ТГА поли-л-
фениленсульфида [29]:
о — деструкция в вакууме; б — деструкция в азоте.
290
Изучено поведение поли-л-фениленсульфида при
изотермическом нагревании в вакууме и кислороде [29]. При деструкции
в вакууме количественное разложение с образованием летучих
продуктов происходит через б ч при 442 °С (рис. 5.55). При
термообработке в кислороде автоускорение разложения больше, чем
в вакууме. Скорость деструкции при 350 °С в вакууме 0,005 %/мин,
в кислороде 0,009 %/мин. Летучими продуктами деструкции в
вакууме полимера, полученного из гс-бромтиофенолята, в интервале
температур 20—450°С являются Н2 (8,5%), СН4 @,4%), Н2О
A,4%), СО A,0%); H2S (84,5%); СО2 A,1%); SO2 A,8); С6Н6
A,3%) [47]. Содержание водорода в газообразных продуктах
деструкции сильно возрастает при повышении температуры и при
550—620 °С достигает 61 %. Невысокое содержание кислорода
в исходном продукте A %) ответственно за образование СО, СО2,
SO2 и Н2О. Жидкие и Боскоподобные продукты деструкции состоят
из следующих соединений:
ИК-спектральный анализ твердых продуктов деструкции
показывает, что при термообработке происходит конденсация фениле-
новых циклов по о-положению. При 620 °С остаток содержит
только Уз исходной серы. Полимер состоит из сшитых фениленовых
и бензтиофеновых циклических продуктов:
Деструкция линейного поли-гс-фениленсульфида в инертной
среде включает следующие стадии [47] :
-н2
-O-s-O
291
-o>-s-
—H2S
\o>-sH-^--O + H2S
v_^ —...
Разрыв связей углерод — сера и отщепление водорода от фе-
ниленовых циклов приводят к образованию продуктов деструкции
с концевыми фенильными и меркаптофенильными группами.
Сероводород образуется в результате разрыва меркаптановой связи
и взаимодействия с избыточным водородом. Рекомбинация
радикалов внутри цепи приводит к структурированию полимера.
5.7.3. ПЕРЕРАБОТКА ПОЛИ-я-ФЕНИЛЕНСУЛЬФИДА
Хотя при температуре выше 287 °С происходит сшивание
линейного поли-д-фениленсульфида, его можно перерабатывать, как
термопласт. Вязкость расплава при 303°С составляет 7-Ю4 —
— 2-Ю6 Пз.
5.7.3.1. Формованный материал
Поскольку формованный материал из ПФС при комнатной
температуре является хрупким, для переработки используют
наполненные стеклянным волокном, асбестом, техническим
углеродом и оксидом железа композиции [43]. Прессование проводят
под давлением 70—140 кгс/см2 при температуре формы 315—
370 °С. В случае толстостенных изделий необходимо применение
более высоких давлений [32]. При термообработке пресс-изделий
при 100—300 °С в течение 3—12 ч происходит улучшение
прочности [31]. Для литья под давлением используют экструдеры со
шнековой пластикацией. Экструзия ПФС проводится при 370 °С
(температура цилиндра 315—370 °С и температура формы 65—
120°С). Для предотвращения окрашивания ПФС в процессе
переработки используют термостабнлизаторы в количестве (до 1 %),
такие, как фенилфосфиновая кислота и диоктилфосфит [33]. Для
получения пенопластов дисперсию ПФС и термически стойкого
наполнителя в воде или растворителях сначала подвергают
предварительному прессованию с последующим окончательным
отверждением при 250—500 °С [34, 35].
5.7.3.2. Покрытия
Покрытия на основе ПФС получают из дисперсий, порошка
или растворов [32, 36]. Перед покрытием металлы подвергают
поверхностной обработке. Для обезжиривания поверхности
титана, алюминия и бронзы достаточно пескоструйной обработки«
292
-o>-s-
—H2S
\o>-sH-^--O + H2S
v_^ —...
Разрыв связей углерод — сера и отщепление водорода от фе-
ниленовых циклов приводят к образованию продуктов деструкции
с концевыми фенильными и меркаптофенильными группами.
Сероводород образуется в результате разрыва меркаптановой связи
и взаимодействия с избыточным водородом. Рекомбинация
радикалов внутри цепи приводит к структурированию полимера.
5.7.3. ПЕРЕРАБОТКА ПОЛИ-я-ФЕНИЛЕНСУЛЬФИДА
Хотя при температуре выше 287 °С происходит сшивание
линейного поли-д-фениленсульфида, его можно перерабатывать, как
термопласт. Вязкость расплава при 303°С составляет 7-Ю4 —
— 2-Ю6 Пз.
5.7.3.1. Формованный материал
Поскольку формованный материал из ПФС при комнатной
температуре является хрупким, для переработки используют
наполненные стеклянным волокном, асбестом, техническим
углеродом и оксидом железа композиции [43]. Прессование проводят
под давлением 70—140 кгс/см2 при температуре формы 315—
370 °С. В случае толстостенных изделий необходимо применение
более высоких давлений [32]. При термообработке пресс-изделий
при 100—300 °С в течение 3—12 ч происходит улучшение
прочности [31]. Для литья под давлением используют экструдеры со
шнековой пластикацией. Экструзия ПФС проводится при 370 °С
(температура цилиндра 315—370 °С и температура формы 65—
120°С). Для предотвращения окрашивания ПФС в процессе
переработки используют термостабнлизаторы в количестве (до 1 %),
такие, как фенилфосфиновая кислота и диоктилфосфит [33]. Для
получения пенопластов дисперсию ПФС и термически стойкого
наполнителя в воде или растворителях сначала подвергают
предварительному прессованию с последующим окончательным
отверждением при 250—500 °С [34, 35].
5.7.3.2. Покрытия
Покрытия на основе ПФС получают из дисперсий, порошка
или растворов [32, 36]. Перед покрытием металлы подвергают
поверхностной обработке. Для обезжиривания поверхности
титана, алюминия и бронзы достаточно пескоструйной обработки«
292
Таблица 5.27. Физико-механические, теплофизические
и электрические свойства полифениленсульфида [28, 36]
Показатель
Лииейиий ПФС
ненапол-
ненный
40%
стеклянного
волокна
Разветвленный ПФС
ненапол-
ненный
30%
стеклянного
волокна
асбестовое
волокно
растяжении,
Плотность, г/см3
Прочность при
кгс/см2
при 20 °С
при 120°С
при 200°С
Относительное удлинение при
разрыве, %
при 20 °С
при 120°С
Прочность при изгибе, кгс/см2
Модуль упругости при изгибе,
кгс/см2
при 20°С
при 120°С
при 230°С
Модуль упругости при
сжатии, кгс/см2
Ударная вязкость образцов с
надрезом, кгс-см/см2
при 20°С
при 150°С
Твердость по Шору
Температура стеклования, °С
Теплостойкость по Вика, °С
Деформационная
теплостойкость (при 18,5 кгс), °С
Верхняя температура
эксплуатации, °С
Термический коэффициент
линейного расширения ос-105,
/°С
коэффициент
расширения
/
Термический
объемного
?-10\ см3/(г>°С)
ниже 150°С
выше 150 °С
Водопоглощение, %
Усадка за 3 ч при 205 DC, %
Диэлектрическая постоянная
при 103 Гц
при 106 Гц
Тангенс угла диэлектрических
потерь
при 103 Гц
при 106 Гц
Удельное объемное
электрическое сопротивление, Ом-см
Электрическая прочность,
kB/mm
1,34
770
330
3
1400
42 000
1,6
5,4
86
150
137
260
0,93
5,68
3,1
3,26
0,0Q04
0,0007
23,5
1,64
1470
330
3
2 600
154 000
42 000
4,3
9,8
92
218
260
3,8
0,0037
0,0066
19,6
1,33
550—700
2-4
28 000
105 000
101
180
3
0,008
2.8—3,0
2,5-1016
21,5
780
520
1
5
1 ПО
76 000
23 000
87
205
800
160
I
5
870
61 000
10 000
90
294
Еиэ ,кгс/см
160000
100000
50000
100 150 ZOO t°C
О 50 100 150 200 250 t°C
Рис. 5.56. Зависимость прочности при растяжении поли-я-фенилснсулъфида от
температуры [28]:
/ — полн-л-фениленсульфид с 40% стеклянного волокна; 2 — ненаполненный поли-я-фенилен-
сульфнд.
Рис. 5.57. Зависимость модуля упругости при изгибе поли-я-фениленсульфида от
температуры [28].
Ползучесть ПФС при комнатной температуре исключительно
мала. Хорошие физико-механические свойства в течение многих
месяцев термостарения на воздухе остаются на достаточно
высоком уровне. ПФС на воздухе не горит. Кислородный индекс
составляет 44 % по сравнению с 47 % для ПВХ [28]. ПФС
отличает высокая стойкость к действию растворителей и агрессивных
сред. Ниже 175°С органические растворители вообще не
действуют на ПФС. Выше 175 °С они растворяются в ароматических
углеводородах, ароматических простых эфир ах и кетонах. После
выдержки в течение 24 ч в углеводородах, тетрахлориде углерода,
спиртах, кетонах, таких органических кислотах, как уксусная и
муравьиная кислота, 10 %-ной азотной, 37 %-иой соляной
кислотах, 30 %-ном гидрокснде натрия, неорганических солях, при
93°С прочность практически не изменяется; 10%-ное уменьшение
прочности при 93 °С происходит в пиридине, ацетонитриле и
растворе карбоната натрия. В тех же условиях при контакте с трн-
хлорэтиленом прочность снижается на 30 %, в пшохлорите
натрия— на 50 °/о. Деструкция полимера за счет окисления
сульфидных связей при 93 °С за 24 ч происходит количественно в бромной
воде, царской водке или 96 %-ной серной кислоте.
5.7.5. ПРИМЕНЕНИЕ ПОЛИ-п-ФЕНИЛЕНСУЛЬФИДА
ПФС используют для получения высокотермостойких
покрытий слоистых пластиков и литьевых изделий, которые сочетают
хорошую стабильность размеров при высоких температурах с
высокой химической стойкостью [32, 36, 38—41]. Линейные ПФС
после отверждения имеют высокую адгезию к стеклу,
керамическим материалам, алюминию, титану, бронзе и стали, хотя
полимер не содержит функциональных групп, которые образуют
химические связи с носителем. После нанесения ПФС на стеклянные
пластины отделение полимерного слоя возможно только при вы-
295
сокотемпературном выплавлении. Покрытия устойчивы к действию
оснований и кислот, кроме кислот-окислителей, например
концентрированных серной и азотной кислот. Эти покрытия устойчивы на
воздухе многие месяцы при 230 °С и многие недели при 315°С.
Литьевые детали и слоистые пластики используют в
электротехнике вследствие их негорючести и высокой термостойкости.
Литьевые изделия из ПФС, наполненного техническим углеродом,
применяют в качестве нагревательных элементов. Электрическое
сопротивление возрастает с увеличением содержания технического
углерода в композиции [42].
Запатентован [44] способ получения штырей, соединительных
трубок и мундштуков для литья под давлением ПФС. Композиции
с углеродными волокнами характеризуются хорошими
абляционными свойствами.
5.8. АРОМАТИЧЕСКИЕ СЛОЖНЫЕ ПОЛИЭФИРЫ
(ПОЛИАРИЛАТЫ)
Хотя полнэтилентерефталат как алпфатпчески-ароматический
полиэфир производится с 1947 г. и по объему производства
A млн. т. в год) может быть отнесен к многотоннажным
полимерам, другие алифатическо-ароматические и чисто
ароматические полиэфиры, производство которых началось после 1960 г.,
в силу небольшого объема производства относятся к специальным
пластикам.
Поли-я-оксибензойная кислота E.8.3.1) представляет собой
исключительно жесткий конструкционный материал, устойчивый в
условиях длительной эксплуатации при 300°С. Перерабатывается
ковкой, спеканием в плазме. Поли-1,4-циклогексилендиметилентере-
фталат E.8.3.2)
Г—О-СН3— / \— СН.ОСО—<^)— СО—1
используется как волокнообразующий полимер с повышенной
гидролитической стойкостью. Покрытия по металлам имеют хорошую
адгезию, высокие ударную вязкость и эластичность. Волокна из
полиэтиленокенбензоата по характеристикам напоминают шелк,
гидролитически они более устойчивы, чем полнэтилентерефталат
Г—осн2сн2о—/Q)—со~]
Полибутилентерефталат применяют для изготовления литьевых
изделий с точными размерами, которые предпочитают литьевым
изделиям из латуни
296
Г-О(СН2LОСО— <О>—- C0~]rt
5.8.1. ПОЛУЧЕНИЕ ПОЛИАРИЛАТОВ
5.8.1.1. Переэтерификация эфиров
дикарбоновых кислот д иолам и
Переэтерификация применяется в основном при получении алп-
фатическо-ароматических полиэфиров, но ее используют также при
синтезе чисто ароматических полиэфиров:
/iHOROH + п R"OOCR'COOR" *=? [—OROOCR'CO-]*
Процесс катализируется такими слабыми основаниями, как
гидроксиды и алкоголяты щелочноземельных металлов,
комплексными алкоголятами типа NaHTi(OC4H9N или CaTi(OC4H9N
[146, 159].
5.8.1.2. Переэтерификация диэфиров ароматических диолов
дикарбоновыми кислотами
Диэфнры двухатомных фенолов (главным образом диацетаты)
реагируют с дикарбоновыми кислотами с получением алифатиче-
ско-ароматических [166, 167] и чисто ароматических полиэфиров
[77, 165]:
пНзССОО—<О)—СН2—(Q)—ОСОСНз+/гНООС—\Q)~ СОЭН
PSV-oco—^Г))—со-]
i-снзсоон
Эти полимеры синтезируют также гомополиконденсацией [52,
53]:
пНСОС—(^f)—ОСОСНз ~ > Г-СО—/ГУ)—0—1
\^У -CHgCOOH L \^v7 Jn
Поликонденсацию проводят в расплаве либо в высококипящем
инертном растворителе. В качестве катализаторов используют
магний, цинк, ацетаты Na и Zn, бутилат Ti, /г-толуолсульфокислоту и
оксид сурьмы. Выделение уксусной кислоты происходит при 210 °С.
Для получения высокомолекулярных полимеров процесс проводят
в расплаве при пониженном давлении или в твердой фазе при
нагревании до 300 °С. Диацетаты о,о'-бисфенолов и бнсфенолов
с объемистыми заместителями вследствие пространственных
затруднений не реагируют с дикарбоновыми кислотами [167].
5.8.1.3. Поликонденсация дихлорангидридов ароматических
дикарбоновых кислот с щелочными солями бисфенолов
на границе раздела фаз
При растворении бисфеиолов в водных растворах щелочей
происходит образование соответствующих фенолятных дианионов,
которые реагируют с дихлорангидрндами дикарбоновых кислот в ра-
297
створе не смешивающихся с водой растворителей. Процесс
протекает при интенсивном перемешивании:
СНз
«NaO (( Y) С (( "Л ONa + ttClOC—(( )) СОС1
—NaCl
СНз
СНз
_п
Процесс представляет классическую реакцию Шоттен —
Баумана в применении к бифункциональным соединениям. Диффузион-
но-контролируемый процесс протекает на границе раздела фаз.
Растворителем для дихлорангидридов кислот и одновременно для
образующегося полимера служат хлорированные углеводороды,
такие, как метиленхлорид, дихлорэтан или трихлорэтан [92, 125, 160,
161]. Во избежание гидролиза дихлорангидридов процесс проводят
при 0—25 °С. Образование фенолятов катализируется небольшими
количествами четвертичных аммониевых, фосфониевых или сульфо-
ниевых солей [77, 124, 162]. Реакционная способность бисфенолов
возрастает с увеличением их кислотности [164]. Поликонденсация
на границе раздела фаз в противоположность процессу полиэтери-
фикации является неравновесным процессом. Кроме того, при этом
не протекают межмолекулярные обменные реакции. Следствием
такого течения процесса является необычное молекулярно-массо-
вое распределение [115].
5.8.1.4. Поликонденсация ароматических диолов
с дихлорангидридами ароматических дикарбоновых кислот
в высококилящем растворителе
Взаимодействие ароматических и алифатических диолов с
числом атомов углерода более пяти с дихлорангидридами кислот
сопровождается образованием высокомолекулярных полиэфиров с
хорошим выходом [78, 79, 125, 163]:
«HOROH + иСЮС—(С))—СОС1 —> —OROCO
Г
В зависимости от реакционной способности исходных веществ
процесс проводят при температурах от 150 до 300 °С. В качестве
растворителей используют высококипящпе галогенсодержащие
ароматические соединения и полиароматические соединения, которые
при комнатных температурах не могут служить растворителями,
а при высоких температурах являются растворителями
образующихся полиэфиров такие, как хлорбензол, о-дихлорбензол, ж-тер-
фенил, галогенированные бифенилы, дифенилоксид и нафталин.
Выделяющийся хлористый водород удаляется из реакционной
298
смеси током инертного газа. Скорость взаимодействия бисфенолов,
имеющих два заместителя при центральном атоме углерода, с те-
рефталоилдихлоридом зависит от возникающего индукционного
эффекта
i
о—н
и уменьшается в следующем ряду [88] :
CF3 ^f H СНз H СНз
I I I I I I
—С— > —С— > —С— > —С— > —С— > —С—
СНз
CF3
H
Молекулярная масса образующихся полимеров зависит от
реакционной способности реагентов. Реакционная способность хлоран-
гидридов кислот снижается в ряду [111]:
С ЮС—(Cj)—C0C1 > СЮС—,-Ач—СОС1 >
> С10С—
./(
ZJ
-COCI
Этим способом нельзя получить полиэфиры из алифатических
диолов с числом атомов углерода меньше шести вследствие
протекания побочной реакции образования простых эфирных связей и
катализируемой кислотами перегруппировки.
5.8.2. СТРОЕНИЕ И СВОЙСТВА АРОМАТИЧЕСКИХ ПОЛИЭФИРОВ
В табл. 5.28 приведены свойства 180 ароматических полиэфиров,
классифицированных следующим образом:
№ 1—4 (группа 1) полиэфиры на основе ароматических оксикарбоновых
кислот
(группа 2) полиэфиры на основе двухатомных фенолов и
ароматических дикарбоновых кислот
(группа 3) полиэфиры на основе многоядерных неконденсированных
диолов и ароматических дикарбоновых кислот
№ 56—92 (группа 4) полиэфиры на основе бисфенолов с заместителями при
центральном углеродном атоме и ароматических дикарбоновых
кислот
№ 93—119 (группа 5) полиэфиры на основе бисфенолов, содержащие
циклические заместители при центральном углеродном атоме, и
ароматических дикарбоновых кислот.
№ 5—19
№ 20-55
299
№ 120—132 (группа 6) полиэфиры на основе конденсированных многоядерных
ароматических диолов и ароматических дикарбоновых кислот
№ 133-— 150 (группа 7) полиэфиры на основе бисфенолов и алифатических
дикарбоновых кислот
№ 151 —179 (группа 8) полиэфиры на основе алифатических диолов и
ароматических днкарбоновых кислот
В табл. 5.28 в графе «Температура стеклования» иногда
приводится Гразм (температура размягчения). Вследствие структурных
особенностей число полиэфиров с температурой стеклования выше
300°С весьма ограниченно. Для повышения температуры
стеклования требуются максимальное днпольное взаимодействие, сильное
межмолекулярное взаимодействие, высокая симметрия молекул,
резонанс между ароматическими ядрами и сложноэфнрной группой
и заторможенность движения макромолекул.
Эти условия лучше всего выполняются для чисто ароматических
симметричных полиэфиров, которые содержат только
незамещенные фенпленовые и сложноэфирные группы. Ряд этих полиэфиров
имеет температуру плавления, превышающую температуру их
разложения.
Замещение бензольных или нафталиновых колец галогенами
или боковыми алифатическими группами приводит к снижению Тс
(группа 2, № 7—9). Отношение Тс: ТПл для чисто ароматических
полиэфиров составляет 0,68—0,89 [85].
Полиэфиры на основе бисфенолов с различными заместителями
при центральном углеродном атоме и ароматических дикарбоновых
кислот синтезированы Коршаком п Виноградовой с сотр. [73, 77—
83, 87—90, 94—98, 101, 104—108, ПО, 111, 114—125, 129, 158, 162,
164). Эти полимеры размягчаются при 250—300 °С (группы 3—5),
перерабатываются экструзией и литьем под давлением и
используются как высокотермостойкие конструкционные материалы и
электроизоляционные пленки [173].
Полиэфиры на основе бисфенолов с монозамещенным
центральным атомом углерода размягчаются при температуре около
250°С, с дизамещенным центральным атомом углерода
(группа 4)—в интервале 300—350°С. Наиболее высокие температуры
размягчения среди полимеров этого класса имеют эфиры, в
которых центральный углеродный атом между ароматическими ядрами
входит в состав боковой циклической группы (группа 5) *.
Алифатическо-ароматическне полимеры (группы 7 и 8) плавятся
при 100—300 °С. Наиболее высокую кристалличность и вместе с тем
лучшую волокнообразующую способность имеют полиэфиры с
симметрично построенными цепями. Температура плавления их
уменьшается с увеличением длины алифатических участков. В
полиэфирах терефталевой кислоты и линейных алифатических диолов
температуры плавления зигзагообразно изменяются при увеличении
числа метиленовых групп в остатке гликоля (рис. 5.58). Структур-
* Полимеры с такими группами было предложено называть нардовыми (от
латинского слова «cardo» — петля), так как такие группы можно рассматривать
как петли по отношению к основной цепи макромолекулы. — Прим. ред.
300
200
100
о
В
П
О 100 200 300 400 500 600 t?С
Рис. 5.58. Зависимость температуры плавления алнфатнческо-ароматнческих
полиэфиров на основе терсфталевой кислоты и линейных алифатических гликолем
от числа мстиленовых групп в глпкопе.
Рис. 5.59. Кривые термической деструкции ароматических полиэфиров в азоте
AT = 10 "С/мин [169]:
Cl
2 —
L-0-.IQN-co-J
ОСНз
ОСНз
4 —
-О—(С})—(СН2JСО-
п
ные различия приводят к тому, что определенное
рентгенографически звено полимера в случае полиэфиров с четным числом
метиленовых групп в диоле соответствует элементарному звену,
в то время как для продуктов с нечетным числом метиленовых
групп эта величина соответствует двум элементарным звеньям
[93, 168].
Алифатическо-ароматические полиэфиры на основе дифенилди-
карбоновой кислоты (№ 152) и нафталин-2,6-дикарбоновой
кислоты (№ 164) вследствие ярко выраженного ароматического
характера имеют более высокие температуры плавления, чем
соответствующие политерефталаты.
В полимерах на основе ароматических дикарбоновых кислот
с карбоксильными группами, связанными с бензольными ядрами
через метиленовые группы, кристалличность и температура
плавления (№ 159, 160) значительно ниже, чем у полиэфиров на основе
чисто ароматических дикарбоновых кислот (№ 151, 152).
Политерефталаты всегда имеют более высокие степень кристалличности и
температуру плавления, чем соответствующие полиизофталаты. Это
обусловлено большей симметрией в случае соединения фениле-
новых групп в n-положении. Наиболее высокой термической ста-
301
Таблица 5.28. Температура стеклования, плавления и разложения ароматических полиэфиров
Элементарное звено
(-
100 мл
Ж
творите
Рас
О
-Г
в
m
га
о
Е-,
и
о
г*
m
га
о.
о
зл.
воздух
i
X
-ератур
очник
— И
чё
Группа L Полиэфиры на основе ароматических оксикарбоновых кислот
-^
(Т/-С-
¦ А
—оч
II
о
-Оч .</
-О-(СН,J-
О
80
185—205
220—225
550
380
[48-61]
[52]
[62]
[63—72]
Группа 2. Полиэфиры на основе двухатомных фенолов и ароматических дикарбоновых кислот
-°-о
-с-;О)-с-
o о
> 500*
[73]
-о—(С IV-
—О"
-о-с. /-. .с-
о l^Jj о
Cl
ю
11
12
о
№
CI
Cl
\
о \С1А
НзС,
-С—
0 \сн3 °
о
о
h
о
о
о
>450*
360
> 360
305
395
355
360
13
14
15
Элементарное звено
СН3
-о—('(У)—о-с—(С})—с—^О)—с-
О СНз О
с
s
S
1,03
0,56
Растворитель
Крезол
Крезол
о
о
s
га
* о.
о
320
340*
350—370 *
Продолжение
о
о
и
о
О.
и
о
U
табл. 5.28
Литературный
источник
175, 84]
[82]
[83]
16
UCO
о
17
о—с—-,
о
F\
18
/ V о
F/ \F
F\_/F
19
0,80
0,67
с6н5он
С2С Uri2
Крезол
I !
340-360 *
270 :
> 400
90U
со
o=n
о
"со
38
s
сд
00
164
со
ел
00
СД
со
СП
о
§
Г
Г
о
о
о
I
о
H
Й
-с
¦g
п
о
100 мл/г
Растворитель
71 ГГ* Y °С
с \ рэзм/
¦ разл,
(в Ni), °C
5
¦ разл
(иа воздухе), °С
Литературный
источник
24
о -— -с- — о
25
26
27
-0-<
28
29
сн2
О
О
-о—(П)—ся2—(( )>—о-с.
О-СвНп
-С—.
СН3
Л о
СНз
о
1,26
1,18
1,04
0,13
Крезол
ж-Крезол
ж-Крезол
345*
150
ПО
360
348
300
254
450-470
400
375
480
410
[821
[87-89]
[85, 89]
[85]
[90]
[91]
8oe
со
о
II
—s-
II
о
о
о
о
ii
о
о
II
-S-
о
о
1-о—(( У)—s— (
II
о
о
о
О IVJJ О
II
о
с—
II
о
O-СвНц
о
о
-с—
e
о
о
0,38
0,11
л*-Крезол
л*-Крезол
249
170
400*
>400
331
246
325
>400
435
405
415
oie
46
47
48
49
50
51
52
о_сч
UEOJ A
_O-(CH2J-
-О-(СН2K-О—(О)— N=N
о
-О—(СН2J—О—С—<
II
о
-О-(СН2K-О-С—{( ))—с—
о
0,5
—о
—о—(( ))—с—(( ))—с—(Г ))—¦
0,53
II
о
il
о
il
о
о
о
—с—
II
О
—с—
А
СНС13
Крезол
240—260
255-270
145-180
350
430
400
270
350
340
305
Продолжение табл. 5.28
Элементарное звено
g
53
54
о
-на
О
.с—(Г))—0-с
II \N-v ii
о
о
"
420
350
[98]
[98]
55
|—О—С
О
250
[981
Группа 4. Полиэфиры на основе бисфенолов с заместителями при центральном углеродном атоме
и ароматических оикарбоновых кислот
56
—О-
СС13
О
о
270
[99]
57
58
59
60
61
О О
l
-о—a iv-си-
о
о
СНз
I
С—(
СНз
СНз
о о
-О—
о l W J о
1,18
0,70
0,60
0,80
снсь
СНС13
Крезол
СНС13
280
240
250-260
350
280
419
397
[87-89,
97]
[89, 97]
[90]
[73,
87-89,
100]
[73, 89,
101]
Продолжение табл. 5.28
Элементарное звено
s
S
5 S
2
S.
62
63
64
65
[102]
0,38
Крезол
230
300
270
420-440
424
[103, 90]
[87]
[82]
66a
666
—О
67
68
69
70
—о
320—330
360
200-250*
350'
410
[80]
430
(81]
438
[104]
[105]
[105]
[87—89,
106, 107]
Продолжение табл. 5.28
Элементарное звено
ч
8
71
72
73
74
75
С—
-сн-
о
0,60
0,45
CHCh
Крезол
290
400—415 *
300
424
450
[190]
240—250
[89, 106|
[901
[87, 108]
[Ю91
[91, 103,
110-113]
СНз
с/ сн3 \С1
СНз
77
Н3С
Н3
СН3
78
СН
79
—О
(СН2K
СНз
СНз
СН3
О
-о-с-
\ А
(СН2).
снв
-с—
II
о
I1
о
о
0,57
Фенол
C2H2CI4
156
НО
160
350-365
190-200
190— 205
[83]
00
IT]], 100 мл/г
Растворитель
T °G
), -С
разл.
(на воздухе), °С
s
Литературный
источник
СНз
о о
84
CH3
С—
О IWJ О
сн.
85
о
86
CF3
C
о о
0,57
0,48
0,70
0,70
Крезол
снсь
Крезол
СНС13
296*
265*
330*
310*
446
434
[87-89,
1П]
[89]
[90]
187-89]
o=n
o-n
о
ел
00
w
о
i
О
X
n
]], 100 мл/г
Растворитель
со
о
о
со
о
о
Т (Т* Y °С
с V разы)
1 разл.
<на воздухе), °С
Литературный
источник
t 90,
СО
-О
о
о
91
—О
92
Ю
0,48
0,34
0,50
СНС la
СНСЬ
Крезол
300
270
300
[87-891
[89]
[90]
Продолжение табл. 5,28
Элементарное звено
л/г
§
тель
a
О
g
я
а
m
я
^°
о
о
m
1ЭЛ.
о
к
п
рны(
тер a
гочн
Группа 5. Полиэфиры на основе бисфенолов* содержащих циклические заместители при центральном углеродном атоме,
и ароматических дикарбоновых кислот
93
94
С-
0,80
0,46
СНС13
СНС13
330
270
330—350
375
360
[73,77,89,
П5, П6]
[73,77,89,
114, 117]
[95]
96
с—
-с
«
о
97
с—
II
О
с
О
i
98
C
—<(Q)—(С2В4Н10)—^>—С-
e
e
99
-(С2В,оН1О)
О
0,57
1,2
2,4
Крезол
C2H2CI4
снсь
290—305
340
355—360
340
310
[95]
[82]
420
[80]
420
400
[81]
8
OU
/
о
со
CD
S
о
s
I
[i|l, 100 мл/г
П
О
П
*
Растворитель
°c
T °C
l
о
СИ
г разл.
(в Ns), °С
1 разл.
(иа воздухе), °С
Литературный
источник
103
a
—C6H5
104
i—O-C—(( ))—G-
o
105
c—
106
с-
0,75
0,7^
Крезол
Крезол
360-380
290
340*
{83]
360
375
[116, 120,
121]
[1011
458
[87, 108J
о
со
, 100 мл/г
п
о
о
о
Растворитель
i
*
СО
со
о
Y
'пл'
4
О
). °G
ё
о
разл,
(на воздухе*. °С
<?)
Литературный
источник
110
111
П2
113
ох ют д
0,41
2,2
0,71
1,14
Крезол
Крезол
Крезол
Крезол
305-315*
325—360*
310—340 *
370-380
Продолжение табл. 5.28
Элементарное звено
g
PI О
ьГз
3
я
о.
ц
U4
с—
1,36
Крезол
1
115
116
0,85
Крезол
0,98
» C2H2Cl4
315-350
[98, 122]
370-400
[122]
360-390
[831
117
О
118
119
г-о-с-ЧПУ-с-
POL
^ II
о
0,90
(с2в4н10)—а ;>—с-
1Д
С2Н2С14
снсь
380—395
350
265
420
180]
410
410
[81]
[1171
Элементарное звено
л/г
g
гель
зори'
я
о.
и
о
раз*
/7 родолжение
и
о
Ч
и
о
g
CQ
n
и
о
"ЗГ
л.
озду
n ш
табл. 5.28
а
г
о.
&|
S.5
Лите
исто
120
121
122
Группа 6. Полиэфиры на основе конденсированных многоядерных ароматических диолов
и ароматических дикарбоновых кислот
>500
о
350
>500
[1241
[124]
[125]
123
>500
О IWJ О
124
>500
V-c
О IWJ О
125
—О
320
>500
C
О
127
—О.
> 500
Шгт
со
о
w
п
S
г»
я
H
ta
•О
S
h], 100 мл/г
Растворитель
s
c\ разм/
со
о
to
о
°G
i
о
to
о
to
о
1 разл.
(иа воздухе). °С
СО
CD
Литературный
источник
—о
182
О
200-235
300
133
134
135
136
Группа 7, Полиэфиры на основе бисфенолов и алифатических дикарбоновых кислот
О
о
С—(СН2M— С—
А А
108
96
57
315
230
192
294
310
304
329
n—n—n о—n—n
0=0
O-.C.
O-o
[t\], 100 МЛ/Г
Растворитель
с\ разм^
Т °С
¦'пл*
со
5
ю
00
со
), °С
л разл.
(на воздухе), °С
Литературный
источник
I
о
0,36
0,50
Крезол
Крезол
120
80
100
145-170*
98
300
OC?
[Т)]( 100 мл/г
Растворитель
(т*
V р
Y °c
со
о
Т °С
2
разл.
(на воздухеК °С
Литературный
источник
a 48
149
.150
Y-, _/
—О
°—С"(СН2)8
о
о
A
'C—(СЪ O-C—(CH2)8-C-
II \^/ II II
0,35
0,70
Крезол
Крезол
220
150
312
Группа 8, Полиэфиры на основе алифатических диолов и ароматических дикарбоновых кислот
151
152
153
-О-(СН2J-О-С—
6
-С—
il
о
c—
о
о
о
о
73-79
265 ;
355
340
СЛ
СЛ
о u
O 0
o=n o=c
S S '4 Гу-°
O=0
170
220
420
145
145
t (t* Y °c
гразл. (в N2), °C
гразл.
(на воздухе), °С
], ЮО мл/г
Растворитель
Литературный
источник
LZZ
22Z
99S
2LZ
OOS
091
zei
08
96
О
II
—э-
o
-э—о—Чгнэ)-о—
о
о
-э—(S)—э-о-е(гнэ)-
i
гнэ)-
о о
-Э—(О/—HN—ЧгНЭ)—HN (О/ Э-О—г(гНЭ)-0—
о
II
—э
о
о
—s—Чгнэ)—s—<Q)—э—о—г(гнэ)-о—
-г(гнэ)—о—
-е(гнэ)—о—
о о
—э—гнэ—(Cj)—(О)—гнэ — э—о—Чгнэ)—о—
о
II
—э—ен:
гнэ—э—о—г(г
гнэ
I
991
99 I
69I
I9I
09 I
691
o*e
3
СО
3
[1\]. 100 мл/г
Растворитель
<45
<45
S
<45
32
54
34
°G
(в №). »С
320
320
320
т
-'разл.
(на воздухе), °С
CuOO
00
СО
ю
Литературный
источник
173
174
175
176
177
178
179
-O-(CH2)8-O-C—|VVj
—О—( СН2)! о—О—С
-о- сн2—/^)—сн2—о-с—(Г^)—с—
—О—(СН2)9—О—С—
—О—(СН2)ю—О—С
-Ph— СНзНС
усн2—оч усн2—оч
^ ^С^ ^СНСН2—(
\N—сн2/
^ 2 N^c/ 2 ^с
\о-сн/ \о-сн/
)-С
о
о-с
II
<35
<25
56
250-290 *
;—С—0-
о
I I
1220—240*
С—О—
II
о
95-102
125-130
305
бильностью отличается поли-я-оксибензойная кислота (№ 1).
Интенсивное уменьшение массы в инертной среде наблюдается при
температуре выше 550 °С (рис. 5.59). Полпарилаты ароматических
дикарбоновых кислот и бисфенолов с двумя заместителями при
центральном атоме углерода (группы 4 и 5) в инертной среде
устойчивы до 400 °С. Термическое и термоокислнтельное старение
полимера сопровождается в интервале температур 250—275 °С
увеличением молекулярной массы, а выше 300 °С — образованием
нерастворимой части. Выше 350 °С параллельно протекают деструкция и
структурирование полимера [116]. Газообразными продуктами
деструкции полиарилатов в условиях термического и
термоокислительного разложения являются СО2, СО и Н2 [120]. Количество
СО2 в газообразных продуктах деструкции в 2—3 раза выше, чем
СО, и в 200 раз выше, чем Н2 [116]. При термообработке полиари-
лата (№ 104) в вакууме при 500 °С за 4 ч выделяется 14,46%
газообразных продуктов деструкции, которые содержат СО2—10,9 %,
СО—3,56 %, Н2—0,002 %. Энергии активации соответствующих
продуктов составляют 25,5; 30,7; 31,0 ккал/моль. Процесс
термоокисления в аналогичных условиях сопровождается выделением
9,5 % СО2, 5,7 % СО и 0,02 % Н2; соответствующие энергии
активации составляют 23; 19,2 и 24,2 ккал/моль. Стойкость
полиарилатов повышают введением в макромолекулы ингибирующих групп,
например гидроксильных, в о-положении к карбонильной группе
или атомов фосфора в основную полимерную цепь. При нагревании
полиарилатов в присутствии катализаторов Фриделя — Крафтса,
таких, как А1С13, TiCl4, SnCl4, ZnCh, или при облучении УФ-лучами
в растворе происходит перегруппировка Фриса с образованием ок~
сибензофеноновой структуры [117]:
—ОС
п
п
В результате такой перегруппировки температура начала
деструкции повышается на 40°, а энергия активации — на 12 ккал/моль.
Полиарилаты, содержащие серу (№ 162) или фосфор (№ 67, 69,
167, 169, 171, 176), получают сополиконденсацней с использованием
соответствующих мономеров. Термостойкость повышается за счет
введения в цепь полиэфира карборановых циклов (№ 32, 33, 66, 98,
99, 107, 108, 117, 118). Изучены полидисперсность и
гидродинамические свойства полимеров в зависимости от условий синтеза [115,
139, 170—172]. У полиарилатов на основе бпс-2,2-C-хлор-4-гидр-
342
оксифенил) пропана методом ЯКР С показано наличие
поворотных изомеров
Cl Cl Cl
О— —О
и
СНз
соотношение которых зависит от типа растворителя,
использованного при низкотемпературной поликонденсации [110]. Наличием
этих поворотных изомеров объясняют зависимость
гидродинамических свойств полимеров от условий синтеза.
5.8.3. ПРОМЫШЛЕННЫЕ АРОМАТИЧЕСКИЕ ПОЛИЭФИРЫ
В то время как объем производства полиэтилентерефталата со
ставляет 1 млн. в год, другие чисто ароматические и алифатическо-
ароматпческпе полимеры, также, как поли-о-оксибензойная
кислота, полп-1,4-циклогексилендиметилентерефталат, полиэтиленокси-
бензоат и полибутплентерефталат, выпускаются до сих пор в отно
сительно небольших количествах. В табл. 5.29 приведены свойства
некоторых промышленных полиэфиров, данные об объеме произ-«
водства которых отсутствуют.
5.8.3.1. Поли-л-оксибензойная кислота
Поли-п-оксибензойную кислоту получают гомополиконденсацией
n-гидроксибензойной кислоты с блокированной карбоксильной
группой или соответствующего хлорангидрида:
Переработка такого неплавкого полимера производится
методом ударного прессования (ковка), спеканием или в плазме.
Верхняя температура длительной эксплуатации полимера 300°С.
Благодаря очень высоким жесткости и теплопроводности поли-п-окси*
бензойную кислоту используют в качестве конструкционного
материала. Основные области применения этого полимера—электро«
техника и самосмазывающиеся подшипники.
Получение. Основная трудность при синтезе поли-п-оксибензой-
ной кислоты заключается в возможности декарбоксилирования
кислоты в процессе получения высокомолекулярного полимера.
Тетрамер я-гидроксибензойной кислоты описан еще Фишером
[174]. Из-за сравнительно низкой реакционной способности фе-
нольного гидроксила гомополиконденсацию /г-гидроксибензойной
кислоты необходимо проводить при температурах выше 150°С.
Однако в этих условиях мономер декарбоксилируется.
Высокомолекулярные полимеры получают с использованием я-ацетоксибен-
343
зойной кислоты [5,8.1.2] [50, 52] или хлорангидрида /г-гидрокси-
бензойной кислоты [51, 57].
Из феиилового эфира /г-гидроксибензойной кислоты
высокомолекулярный полимер получается с высоким выходом в присутствии
бутилтитаната в перхлоролигофенилене при нагревании в токе
азота в течение 4 ч при 170—190 °С и 10 ч при 340—360 °С [58].
При получении полимера через хлорангидрид кислоты натриевую
или калиевую соль /г-гидроксибензойной кислоты нагревают в
инертном растворителе, таком, как полихлордифенил, в
присутствии SOC12, РСЬ, SOBr2, РОСЬ или РС15 в инертной среде в
течение 5 ч при 80 °С и после отгонки растворителя реакцию
продолжают при 300 °С [51]. Поли-/г-оксибензойную кислоту с 1971 г.
выпускает в опытно-промышленном масштабе под названием
«Эконол» фирма «Carborundum» (США). После доведения объема
производства до 220 т в год стоимость продукта должна снизиться
с 300 марок ФРГ за 1 кг до 50 марок. Мономер легко получается
карбоксилированием фенола.
Физико-химические свойства. Поли-я-оксибензойная кислота
является линейным высококристаллическим полимером. При 550°С
в инертной среде и на воздухе полимер разлагается, не плавясь
[55]. Ниже 500 °С разложение протекает очень медленно. Потеря
массы за 1 ч термообработки при 460 °С на воздухе составляет
3%, при 400°С —0,5% и при 320°С —0,06%. Основными
продуктами разложения в вакууме при 500—565°С являются СО, СОг и
фенол. Остаток имеет практически полифениленовую структуру.
Для деструкции полимера предложен следующий механизм [56]:
Lc-o—.. —
Il = \^У II ^У 5 J
0 0 0 0 0
со + но—(Qs—с—о—(С» + со» +
о
,o
>—<( )> + нс—
о
Энергия активации процесса до отщепления 30 % летучих
продуктов деструкции составляет 59,6 ккал/моль. Высокая
упорядоченность полимера сохраняется до температуры 425 °С. Поли-л-
оксибензойная кислота не растворяется в известных
растворителях, набухает в хлорированном дифениле лишь выше 300 °С.
Промышленный полимер — «Эконол» имеет среднечисловую
молекулярную массу 8000—12 000 (по концевым группам).
Переработка. Поли-я-оксибензойная кислота выше 430 °С
проявляет способность к пластическому течению аналогично
металлам. Переработка осуществляется методами, используемыми
344
Таблица 5.29. Свойства промышленных
ароматических полиэфиров
Условные обозначения: ПЭТФ — полиэтилентерефталат; ПЭОБ — поли-
этиленоксибензоат; ПБТФ —полибутилентерефталат: ПЦДТ — поли-1,4-цикло-
гексилендиметилентерефталат; ПОБ — поли-/г-оксибензойная кислота; ПА1 — по-
лиарилат бисD-гидроксифенил)флуорсна и терефталевон кислоты; ПАП — поли-
арилат 2,2~бисC,5-дихлор-4-гидроксифенил) пропана и смеси терефталевои и изо-
фталевой кислот A:1)
Показатель
ПЭТФ
1,37
740
50—300
1650
1 170
28 500
Не
ломается
4
1,2-2,0
73—79
188
130
7
255-258
0,4
3,3
3,2
3,1
2 • 10"*
5-ю-;
2-10
4- 10*
300
ПЭОБ
1,34
580
15-25
220-250
ПБТФ
1,31
15
1500
844
22 000
Не
ломается
4
0,6-2,0
36-49
178
150
6
220-225
0,4
3,91
3,86
3,74
6.10"'
3-10-
1 • 10~2
5-1015
ПЦДТ
1,226
45
2 800
165
305
0,3
3,18
2-10
" ПО6
752
70 300
0,02
3,8
2 • 10~4
ыо15
160
ПА1
1,46
15
300
360
3,3
4-10
1 • 1015
Плотность, г/см3
Прочность при
растяжении, кгс/см2
Относительное
удлинение при
разрыве, %
Твердость по
шарику, кгс/см2
Прочность при
изгибе, кгс/см2
Модуль упругости
при изгибе, кгс/см2
Ударная вязкость
Ударная вязкость
образцов с надрезом,
кгс-см/см2
Усадка, %
Температура
стеклования, СС
Теплостойкость по
Вика, °С
Температура
длительной
эксплуатации, °С
Термический
коэффициент линейного
расширения a-105,
1/°С
/С
Температура
плавления, °С
Водопоглощение, %
Диэлектрическая
проницаемость
при 60 Гц
при 103 Гц
при 10б Гц
Тангенс угла
диэлектрических потерь
при 60 Гц
при 103 Гц
при I06 Гц
Удельное объемное
электрическое
сопротивление, Ом-см
Электрическая
прочность, кВ/мм
обычно для металлов—ударным прессованием (ковкой),
спеканием и в плазме.
Ударное прессование проводится при энергии от 1000 до
10000 кгс-м. Материал нагревают в форме до 155—260°С и
подвергают ударному формованию. За счет высокой кинетической
энергии силы сдвига вызывают внутреннее нагревание материала,
которое и приводит к течению полимера. В случае непрерывного
процесса продолжительность цикла составляет 6—10 с.
Формованные изделия не имеют микропустот; плотность 1,50 г/см3.
Этим методом можно перерабатывать и наполненный
полимер.
Спекание проводят при 430 °С под давлением 420—840 кгс/см2.
В зависимости от плотности время формования колеблется от
нескольких минут до I ч. В противоположность ударному
прессованию при этом методе получают изделия с некоторой
пористостью.
При переработке в плазменной струе порошок полимера
вводится в плазму ионизированного гелия или аргона при
3000°С и с высокой скоростью напыляется на поверхность.
Этим методом можно нанести покрытие толщиной 0,01—5 мм
на титан, алюминий или сталь. Микропористость покрытий
можно устранить полированием. Теплопроводность поли-л-окси-
бензойной кислоты в 3—5 раз выше, чем у других полимеров.
Такой полимер может легко обрабатываться механическим
способом.
Эксплуатационные свойства. Полиоксибензойная кислота как
высокотермостойкий конструкционный материал с исключительно
высокой жесткостью пригодна для длительной эксплуатации
при температурах до 300°С (см. табл. 5.29). Высокий модуль
связан с высокой кристалличностью полимеров. Относительно
невысокая прочность при изгибе, равная 752 кгс/см2, может быть
увеличена до 1120 кгс/см2 введением в качестве наполнителя
АЬОз. Хорошие диэлектрические свойства в сочетании с очень
высокой теплопроводностью делают полиоксибензойную кислоту
идеальным конструкционным материалом для электроники.
Поскольку силы сдвига вызывают локальное течение, полимер
обладает самосмазывающими свойствами. Он инертен ко всем
органическим растворителям при комнатной температуре. При
контакте с хлорированным дифенилом выше 320 °С полимер
набухает. Полиоксибензойная кислота устойчива в холодных
концентрированных кислотах и основаниях, но разрушается в них
при нагревании.
Применение. Поли-я-оксибензойную кислоту используют в
электротехнике и электронике для высокотермостойких печатных
схем, корпусов диодов и транзисторов, концевых выключателей,
цоколей и зажимных пластин. Наполненный титанатом бария
полимер применяют как высокочастотный изолятор.
Самосмазывающая способность полимера, которую варьируют введением
графита, нитрида бора, сульфида молибдена или политетра-
346
фторэтилена, обусловливает применение полиоксибензойной
кислоты для изготовления подшипников, втулок и уплотнителей.
Наполненный карбидом кремния материал используется в
производстве юрмозных и шлифовальных дисков. Полимер
используют также для получения приводных устройств, насосов и труб
для эксплуатации в агрессивных средах, а также деталей
текстильных машин, работающих под большой нагрузкой.
5.8.3.2. Поли-1,4-циклогексилендиметилентерефталат
Поли-1,4-циклогексилендиметилентерефталат (ПЦДТ)
получают при взаимодействии диметилтерефталата с дигидроксиме-
тилциклогексаном в расплаве. Дигидроксиметилциклогексан
синтезируют гидрированием диметилтерефталата в две стадии.
Полимер отличается от полиэтилентерефталата значительно более
высокой гидролитической стойкостью. Волокно на основе поли-
1,4-циклогексилендиметилентерефталата выпускают с 1961 г., а
порошок для покрытий — с 1972 г.
Получение. Полупродуктом для обоих исходных компонентов
является я-ксилол. Диол получают гидрированием
диметилтерефталата в две стадии [151, 153]:
НзСООС—(О/—СООСНз ->¦ НзСООС—( )—СООСНз
y-y pd \ /
СиСгО2
—> носн2—' у—сн2он
Для получения волокнообразующего полимера используют
более высокоплавкий транс-изомер (т. пл. 67 °С).
Поликонденсация диметилтерефталата с дигидроксиметилциклогексаном
протекает аналогично процессу образования полиэтилентерефталата
в расплаве при 220—240 °С в токе инертного газа в присутствии
NaHTi(OC4HgN. Метанол, выделяющийся в результате переэте-
рификации, отгоняют при 270°С/1 мм рт. ст. [152].
Физико-химические свойства. Температура плавления и
плотность ПЦДТ определяются содержанием в полиэфире транс-1,4-
диметилциклогексановых звеньев. ПЦДТ на основе цис-1,4-гидр-
оксиметилциклогексана плавится при 257 °С. Температура
плавления полимера, в синтезе которого использовали 1,4-дигидроксиме-
тилциклогексан, содержащий 80 или 100 % транс-изомера,
составляет 300 или 318 °С. Плотность ПЦДТ, полученного из мономера,
содержащего 100 % tjwc-дигидроксиметилциклогексана, 1,208 г/см3,
а содержащего 100% транс-изомера -— 1,190 г/см3 [143]. ПЦДТ
после формования волокна аморфен; кристализация происходит
при ориентации волокна. Цис- и транс-изомеры ПЦДТ разли-
347
чаются также по размерам триклинной элементарной
ячейки [140]:
Транс-Шип а = 6.37A, b = 6.63A, с = 14.2A, а = 89,4, ? — 47,1, Y =
tfuc-ПЦДТ a = 6,02A, b = 6.01A, с = 13.7A, а = 89,1, ? = 53,1. \*
Длина элементарного звена граяс-ПЦДТ 154 А, а цис-ПЩХТ
145 Л. ПЦДТ с содержанием г/кшс-изомера 70 % растворяется
при комнатной температуре в феноле, крезоле, л-хлорфеноле,
сильных кислотах типа трифтор- и трихлоруксусной кислот;
при нагревании выше 100 °С — дифенилоксиде и хинолине.
Светорассеяние изучено для растворов полимера в
трихлоруксусной кислоте, а вискозиметрия — для растворов в смеси фенол —
тетрахлорэтан F0:40) [141]. Из-за заторможенного вращения в
цепи ПЦДТ размеры клубка у него в 2 раза больше, чем у поли-
этилентерефталата такой же молекулярной массы.
Волокна. Волокна из ПЦДТ получают обычным формованием
из расплава. С целью кристаллизации волокно ориентируют при
100—200 °С и в течение 2 мин термообрабатывают при 175—
220 °С. Для сухих и влажных волокон прочность при растяжении
колеблется от 2,5 до 4,0 г/денье, относительное удлинение при
разрыве — от 20 до 35%. Волокна из ПЦДТ обладают высокой
эластичностью при растяжении. При нагревании до 250 °С
прочность при растяжении снижается почти линейно до 35 % от
исходного значения, в то же время относительное удлинение при
разрыве возрастает вдвое. После 1680 ч выдержки в термошкафу
с принудительной циркуляцией воздуха при 160°С прочность
при растяжении уменьшается на 40 % и относительное удлинение
при разрыве—на 8%. По сравнению с полиэтилентерефталатом
он имеет более высокую гидролитическую стойкость, которая
обусловлена экранирующим влиянием циклогексановых колец на
сложноэфирную связь. При кипячении ПЦДТ в течение 3 ч в
воде прочность и масса не уменьшаются, в то время как у поли-
этилентерефталата в тех же условиях масса уменьшается на
20% и прочность при растяжении—на 43% [142]. После 24 ч
выдержки в концентрированной серной кислоте при 50 °С
прочность при растяжении и относительное удлинение при разрыве
ПЦДТ уменьшается вдвое; полиэтилентерефталат в тех же
условиях разрушается полностью. Волокна из ПЦДТ используют
для изготовления обивки для мебели, спальных мешков, шалей
и подушек. В промышленности волокна из ПЦДТ выпускаются
фирмой «Eastman-Kodak» под названием «Кодел II».
Покрытия. С 1972 г. фирма «Eastman-Chemical» выпускает со-
поли-1,4-циклогексилендиметилентереизофталат в виде порошка
для покрытий под названием «Тенайт PCDT». Полимер наносят
на холодную подложку в электростатическом поле с
минимальной толщиной слоя 75 мкм [154, 155]. На металлах без
адгезионного слоя это покрытие отличается хорошей адгезией, глянцем,
ударной вязкостью и эластичностью. В процессе нанесения поро*
шок не окрашивается. Покрытия на основе ПЦДТ используют
в производстве металлической мебели, арматуры, деталей машин
и предметов домашнего обихода.
5.8.3.3. Полиэтиленоксибензоат
Полупродуктами для производства полиэтиленоксибензоата
Г—ОСН2СН2О—<Y^j)—СО—1
служат /г-оксибензойная кислота и этиленоксид.
Полимер отличается от полиэтилентерефталата более
высокой гидролитической стойкостью. Формованием из расплава из
него получают высококристаллическое шелкоподобное волокно
(т. пл. 220—225 °С), которое отлично окрашивается [63—72].
При вытяжке полиэтиленоксибензойного волокна выше
температуры стеклования (80°С) происходит переход из а-модификации
в ?-модификацию с зигзагообразной конформацией [67]. Как в
сухом, так и во влажном состоянии прочность при растяжении
составляет 4,0—5,3 г/денье и относительное удлинение при
разрыве 15—25%. Модуль упругости равен 33—75 г/денье. Волокно
из полиэтиленоксибензоата используют в смесевых волокнах в
сочетании с полиэтилентерефталатом или полиолефином.
Креповые ткани с хорошими текстильными свойствами получают путем
обработки ориентированного волокна щелоком при 100—200 °С.
Полиэтиленоксибензоат выпускается фирмой «Nippon Rayon» под
названием «Полиэстер» с 1968 г. в количестве 1000 т в год. Цена
1 кг полиэтиленоксибензоата составляет 17,6 дол. США, что вдвое
дешевле натурального шелка.
5.8.3.4. Полибутилентерефталат
Г-о(Сн2L-осо—<О)—со-1
Полибутилентерефталат (ПБТФ) является
высококристаллическим термопластичным полимером конструкционного
назначения с температурой плавления 227 °С и температурой
стеклования 36—49 °С [128, 130, 133—138]. При переработке экструзией
температура цилиндра должна быть 275—290 °С, отношение
сжатия шиека 1:3—1:4. При литье под давлением рекомендуется
температура цилиндра 250—270°С и температура формы 30 °С.
Перед переработкой гранулят подсушивают при 75—90 °С.
Кристаллизация в форме протекает очень быстро, так что можно
проводить переработку с коротким циклом. Высокая текучесть
полимера дает возможность получать литьевые изделия с
длинными или сложными литьевыми каналами.
Свойства полибутилентерефталата приведены в табл. 5.29.
Хорошие прочностные свойства ПБТФ сочетаются с высокой
349
ударной вязкостью при низких температурах и высокой
износостойкостью. Коэффициент трения по стали составляет 0,13. По
проницаемости для паров воды ПБТФ занимает промежуточное
положение между полиэтиленом и полиамидом. Недостатком
ПБТФ является низкая температура стеклования. Уже выше
50 °С при высоких нагрузках полимер деформируется. Этот
недостаток может быть нивелирован за счет наполнения материала
стеклянным волокном.
ПБТФ и наполненные стеклянным волокном композиции
применяют для литьевых изделий с точными размерами, такими,
как зубчатые колеса, ролики скольжения, подшипники, лопасти
вентиляторов, детали для насосов и вентилей, пишущие и
счетные устройства и предметы домашнего обихода. ПБТФ может
заменить отливки из латуни. Из ПБТФ получают прозрачные
пленки, которые могут быть подвергнуты сильной вытяжке и
двухосной ориентации. В США ПБТФ используют как
высококачественный упаковочный материал для пищевых продуктов.
Производят этот полимер в ФРГ фирмы «BASF» (Ультрадур),
«Farbwerke Hohst» (Хостадур) и в США «Celanese Corporation»
(Селанекс), «Eastman-Kodak» (Тенайт РТМТ) и «General
Electric» (Валокс). Цена 1 кг Валокс в 1972 г. составляла 1,5 дол.
США.
5.8.4. АРОМАТИЧЕСКИЕ ПОЛИСУЛЬФОНАТЫ
Полисульфонаты отличаются от полиэфиров наличием в мак«
ромолекуле сульфоновой группы вместо карбонильной:
L о
Полиэфир
О О
II
L О
о
Полису льфонат
я
Полисульфонаты получают взаимодействием ароматических ди-
сульфохлоридов с бисфенолятами. Температура размягчения
полимеров колеблется от 120 до 250°С, на воздухе они стабильны
до 350 °С. Полимеры перерабатывают из раствора и
прессованием. Возможные области использования материалов —
электроизоляционная пленка и температуростойкие фотоматериалы.
5.8.4.1. Получение
Ароматические полисульфонаты получают из ароматических
дисульфохлоридов и дифенолятов или из /г-гидроксисульфохло-
ридов в расплаве [183], в растворе [175, 176] или на границе
35U
раздела фаз [177—179, 181, 184—188]:
SO2CI + мНО—
В качестве катализаторов процесса используют
четвертичные аммониевые и сульфониевые соединения. Поликонденсацию
проводят в растворе циклических кетонов или амидов кислот,
таких, как N-метилпирролидон, диметилацетамид и гексаметил-
фосфотриамид при температуре от —40 до 0°С. В случае
поликонденсации на границе раздела фаз дисульфохлорид растворяют
в ароматических углеводородах или метиленхлориде и к нему
добавляют бисфенол в виде раствора в водной щелочи, который
содержит катализатор. Взаимодействие протекает при
температурах от 25 до 70 °С. Необходимым условием образования
высокомолекулярных продуктов является высокая чистота исходных
дисульфохлоридов.
Сополимеры с чередующимися сложноэфирными и сульфо-
натными группами в цепи получают из бисфенолов и л-хлорфор-
мил-л'-сульфохлоридов:
СНз
/Fn\ t /^\ I /^\ NaOH
лСЮС-
\^У
—с
-SO2C1 + лНО—
s-o—
п
Сополимеры с сульфонатными и карбонатными группами в
цепи синтезируют фосгенированием полисульфонатов,
содержащих гидроксильные группы [180].
5.8.4.2. Физико-химические свойства
В табл. 5.30 приведены температуры размягчения, плавления
и разложения полисульфонатов. Эти полимеры представляют
собой прозрачные аморфные полимеры, кристаллизующиеся из
раствора. При термообработке увеличения упорядоченности не
происходит. Температура размягчения ароматических
полисульфонатов лежит между 120 и 250 °С. Формоустойчивость при
нагревании ниже, чем у соответствующих полиэфиров: полисульфо-
нат на основе ж-фенилендисульфохлорида и дифенилолпропана
(№ 4) размягчается при температурах от 95 до 138 °С в то время
как температура стеклования соответствующего полиэфира изофта-
левой кислоты и дифенилолпропана равна 180°С. Ароматические
полисульфонаты растворяются в хлорированных углеводородах
351
Таблица 5.30. Температуры размягчения, плавления и разложения ароматических полисульфонатов
Элементарное звено
100 мл/г
Растворитель
Гразм- °с
разл
Литературный
источник
so2—(^Q>—о-
—so2
so2—о
СНз
—s
0,14
ДМФА
150—160
335
145-200
0,36
0,62
ТГФ
ДМФА
0,20
ДМФА
95-138
250—260
160-165
170—180
185—205
365
290
[177]
[175, 176]
[178]
[178]
[177}
[179}
[177]
10
п
12
13
0,37
— SO,
о
— SO
2^
СНз
|
СНз
0,31
-so,—<( ))—сн2—:( )>— so2-o— Y ))—сн2—ff )>—o-
СНз
—so2
\oji"O'
-SO2-O— ( У)—C—;
Нз
0,27
СНз
СНз
СНз
СНз
о
Нэ
0г07
ДМФА
ДМФА
ТГФ
снсь
270-280
200-210
122
220
174—176
185-215
310
305
360
|177]
[178]
[177]
[178]
Ц80]
[182J
2^/v/
15
16
Продолжение табл. 5.30
14
Элементарное звено
100 мл/г
Растворитель
'разм' ^
Т
разл
(в N2>
°С
Литературный
источник
[181]
СНз
ОСНз
ОСНз
2^ ^. ^so2
\Г\\
LsJJ
\
—о—/(у).—сн=сн—с—сн=-сн—(С)\—о—
о
СНз
ОСН;
[181J
СНз
\
о
-so2
[183]
—SO2—Ar- SO2—О— (СН2J—N—(CH2J— О—
I
(СН2J
RO—P—OR
II
о
Рис. 5.60. Кривые термической деструкции
ароматических полисульфонатов на воздухе
Д7* 6°С/ [178]:
1 —
-so,—,7-vi—с—
типа метиленхлорида, дихлорэтана, 1,1,2-трихлорэтана,
хлорбензола; в тетрагидрофуране, диметилформамиде, диметилацетамиде
и гексаметилфосфотриамиде. Характеристическая вязкость 0,42
A00 мл/г) полимера № 5 в диметилформамиде при 30 °С
соответствует средней молекулярной массе 50 000, определенной
светорассеянием [177].
В [178] приведены данные по молекулярно-массовому
распределению полимера, найденному гель-проникающей хроматографией
в тетрагидрофуране. Полисульфонаты стабильны на воздухе и в
инертной атмосфере до 350 °С (рис. 5.60). Ароматические группы
различного строения не оказывают значительного влияния на
термостойкость; верхняя граница определяется стабильностью суль-
фонатной связи; расплав полимера быстро разлагается при 400 °С
с количественным выделением SO2. При дальнейшем повышении
температуры (выше 500 °С) разложение замедляется, потеря
массы при 900°С составляет 50% [178]. Ароматические
полисульфонаты имеют отличную гидролитическую стойкость. Молекулярная
масса не меняется после 24 ч выдержки в 10 %-ной серной
кислоте и 10 %-ном гидроксиде натрия. При 80—85 °С в аналогичных
условиях молекулярная масса даже несколько возрастает [177].
5.8.4.3. Переработка и эксплуатационные свойства
Ароматические полисульфонаты перерабатывают из раствора
(в волокна и пленки) или прессованием. Пленки, полученные
поливом из раствора, имеют прочность при растяжении 600—
900 кгс/см2, относительное удлинение при разрыве 2—6 % и
модуль упругости при растяжении 24О0О—32 0ОО кгс/см2 [179].
Прочность увеличивается при вытяжке. Максимальная
температура эксплуатации полисульфонатов составляет 175 СС. Возможные
области применения пленок и пресс-изделий — электротехника и
автомобилестроение. Сополимеры со светочувствительными
группами в цепи получают поликонденсацией дисульфохлоридов с бис-
фенолами и дизинилальацетоном или дизинилальциклопентано-
ном [181]. Полисульфонаты, структурирующиеся под действием
облучения, пригодны в качестве термостойкого фотоматериала и
для изготовления печатных плат.
5.9. АРОМАТИЧЕСКИЕ ПОЛИАНГИДРИДЫ
Ароматические полиангидриды получают путем термического
отщепления ацетангидрида от диацетилированных ароматических
дикарбоновых кислот. Температура плавления лежит в интервале
200—400 °С. Волокна, полученные из расплава, имеют хорошую
прочность и гидролитически стабильны.
5.9.1. ПОЛУЧЕНИЕ
Ароматические полиангидриды получают ацетилированием
ароматических дикарбоновых кислот и термическим отщеплением
ацетангидрида от образующегося в качестве промежуточного продукта
смешанного ангидрида [189—209, 211]:
НООС—<^_Д)—СООН+ 2СН3СО—О—СОСНз
—2Сн3соон
/гНзСОС—О-СО ^Q^ СО—О—СОСНз Смешанный ангидрид
1 (СН3СОJО
НзСОС—Г—ОСО (Cj) СО—1—ОСОСНз Полнтерефталевый
[<«-!>,
ангндрнд
Вначале при взаимодействии при 160 °С ароматических
дикарбоновых кислот с ацетангидридом получают смешанный ангидрид.
Нагревание диацетата при 160—200 °С приводит к
низкомолекулярному форполимеру с выделением ацетангидрида.
Высокомолекулярный полимер образуется при нагревании форполимера в
интервале температур 220—280 °С/1 мм рт. ст. при дальнейшем
выделении ацетангидрида.
356
Рис. 5.61. Зависимость температуры
плавления сополиангидрида тиодипропионовой и
терефталевой кислот от содержания
терефталевой кислоты [198].
Рис. 5.62. Кривые гидролиза алифатических и ароматических полиангндридоз
при выдержке в 1 н. растворе NaOH при комнатной температуре [190]:
/ _ [_СО(СН2)8СО—О— ]„:
2- Г_СО—(Г^\—О-(СН2K-О
же, кристаллический;
3-Г—СО—/О\—О—СН2—О—[{ \)—СО—
у—СО—О— , аморфный; Г - то
- Г—СО—
L
, аморфный;
—СО—О-(СН2J-О—
]
5.9.2. СВОЙСТВА
Ароматические полиангидриды представляют собой
кристаллические полимеры с хорошими волокнообразующими свойствами и
высокой гидролитической стойкостью. В табл. 5.31 приведены
температуры плавления и стеклования ароматических полиангидридов.
Наиболее высокую температуру плавления (около 400°С) имеет
политерефталевый ангидрид. Введение в макромолекулу мети-
леновых, простых эфирных и тиоэфирных групп приводит к
увеличению эластичности и понижению температуры плавления на
100—250 °С [191,211]. Полиангидриды с пятичленными гетероцик-
лами и метиленовыми группами в цепи (№ 26—29) плавятся в
интервале 60—120 °С [197]. Сополиангидриды на основе
терефталевой кислоты и дикарбоновых кислот с пятичленными гетероцикла-
ми (№ 30—34) плавятся при температуре тем более высокой, чем
больше содержание терефталевой кислоты в сополимере (рис. 5.61).
Сополимер с амидными связями в цепи вследствие образования
межмолекулярных водородных связей имеет более высокую
температуру плавления (№ 35) [199]. Кристаллический
полиангидрид можно аморфизовать путем быстрого охлаждения расплава
[190].
В отличие от алифатических полиангидридов, полученных еще
Карозерсом [210], ароматические полимеры отличаются
необычайно высокой гидролитической стабильностью (рис. 5.62). После вы-
357
Таблица o.31. Температура стеклования и плавления
ароматических полиангидридов
Элементарное звено
Литературный
источник
8
10
11
12
13
С—
II
О
с
II
о
'Г))—С-О
\^У II
О
с-о-
II
о
c
II
о
)—C-0-
II
о
СНз
— С ( )/
II С^7
о
с
о
-с-
о
"|
о
СНз О
^~\ ( )/ С—О—
<^У |j
|j
о
о-
(сн,J—(Q >—с-о
V^ II
о
||
о
-с—(Q
II ^=^
о
-с
II
о
-с-
о
-с-
I!
O
о
—(сн2M—<О)~ с-°-
о
—о—сн2—о—{Г))—с—о-
\~/ II
о
—(СН2J—О—(О/—с—°"
о
fi
о
Q
о
ii
Q
140
122
>400
259
290—296
46
39
84
45
95
75
238-240
332
>340
215
257-263
118
220
206-208
267
192-194
[189, 190]
[189,190]
[189, 190]
[189, 190]
[189, 190]
[189, 190]
[189, 190]
[189, 190]
[189,190]
[189,190]
[189,190]
[189, 190]
[189,1901
Продолжение табл. 5.31
14
/
-с—(( ) ;—о-(сн,),-о-
II
о
/
ОСН
/
ОСН3
о
ОСНэ
15
16
17
18
19
А
-с-
II
о
/ОСНз
-((У)—О-(СН2L-О-
/
о
ОСНз
—с—о—
«
о
с—сн2
4
—сн2~с-о-
—С-СНг-0—(C^S О-СНа— С-О—
II
о
70
65
105
СНз
-с—сн2—о—О—с—
-сн2—с^о—
20
21
22
О СНз
-С-(СН2J—S—(CH2J—S—(СН2J—С—О—
А А
-С—СН2—S—(СН2J— С —СН2—С—О—
II к
о о
С—(СН2J—S—СН2—/Г^)—CH2-S—(СН2J—С-О—
и \^/ «
о о
208-210
162—175
160-172
158
152
196—202
75
83
91
23
24
—С—CH2-S—СН2
А
СН2—S-C—О-
—С—(СН2J—S—(СН2J—С—О—
e
A
88
55
[189, 190]
[189,190]
[189.190]
[189. 190]
[189, 190]
[189, 190]
[2П1
[211]
[211]
[211]
[211]
359
Продолжение табл. 5Я1
Элементарное звено
Лтл- °С
25
—С—(СН2J—S-
U
о
26
-С-(СН2J-
А
)—S—(СН2J—С—О-
II
о
—(СН2J—С—О—
о
27
-С-(СН2J—II J—(CH2)»-C—О-
II s II
о о
28
—С—(СН2J-
II
О
N
СН3
-(СН2J—С—О
II
о
29
—С—(СН2J—к J—(СН2J—С-О-
II ^^ W
II о II
о о
30
о
-С— О— С-
II I!
о о
31
32
33
-с
II
о
II о
О
-С—О—С-
II II
о о
:—с-о—
II
о
)—С—О—
о
-с-
II
о
о
N
—с-
А , о
СНз
-с—о—с-
e A
-с—о—с-
II II
о
<3)—с-о-
—С-О-
о
34
N
СНя
-r°-r®-i-°-
II II
о о
о
35
-с
о
)_C-NH-CH2-NH^C—(О)—
45-55
67
78
128
85
260
285
250
280
220
325-330
О
-С—О-
360
держки в 1 н. растворе NaOH в течение 150 ч гидролизованная
часть ароматического полиангидрида составляет менее 20%.
Гидролитическая стойкость кристаллических полиангидридов вдвое
выше, чем аморфных [190]. Из расплава ароматических
полиангидридов можно получить кристаллические волокна. По
физико-механическим свойствам (прочность при растяжении 40 кгс/мм2,
относительное удлинение при разрыве 17,2%) эти волокна аналогичны
волокнам из ароматических полиамидов и сложных полиэфиров
[189].
5.10. ПОЛИЦИКЛОАМИДЫ
Кристалличность полиамидов с циклическими группировками
в цепи зависит от содержания амидных связей. Стоимость
исходных веществ (диаминов и дикарбановых кислот), получаемых из
продуктов нефтехимии, находится на обычном уровне. Алицикли-
ческие и алпфатически-ароматические полиамиды многих типов
производятся в промышленном масштабе с середины 60-х годов.
Они устойчивы к гидролизу и применяются в качестве
высокотермостойких полимеров конструкционного назначения,
перерабатываемых обычными способами. Из чисто ароматических
полиамидов (полиарамидов) в промышленности выпускается только поли-
л*-фениленизофталамид. Верхняя температура длительной
эксплуатации этого полимера равна около 230СС; он используется в
качестве волокна для огнезащитной одежды, высокотермостойких
газовых фильтров и электроизоляции. Пресс-массы на основе поли-ж-
фениленизофталамида перерабатываются при 320—330 °С. В
опытном масштабе выпускаются регулярные сополиамиды с карбо- и
гетероциклами в цепи. Из регулярных карбоциклических
ароматических сополиамидов вырабатываются высокомодульные волокна.
Они обладают более высокой термостойкостью, чем
соответствующие статистические сополиамиды. Регулярные сополимеры с
гетероциклическими звеньями в цепи лучше растворяются, поэтому их
легче перерабатывать. Их стойкость к термоокислительной
деструкции выше, чем у карбоциклических ароматических полиамидов.
5.I0.I. АЛИЦИКЛИЧЕСКИЕ ПОЛИАМИДЫ
Алициклические полиамиды получают теми же методами, что и
алифатические. Полиамиды из алициклических аминокарбоновых
кислот (табл. 5.32, № 1—3) неплавки, разлагаются при 490 °С [212].
Полимеры алифатических дикарбоновых кислот и алициклических
диаминов плавятся при температуре на 50 °С выше, чем полиамиды
на основе соответствующих нециклических мономеров
алифатического ряда с таким же числом углеродных атомов (№ 4—7). В США
начиная с 1965 г. фирма «Tennessee Eastman» выпускает алицикли-
ческий полиамид пробковой кислоты и 1,4-бис (аминометил)цикло-
гексана под маркой Q2, а с 1968 г. фирма «Du Pont» — полимер на
основе декандикарбоновой кислоты и бис (я-аминоциклогексил)
метана под названием «Квиана» (объем производства 16 000 т в год,
цена 1 кг 15 дол. США),
361
Таблица 5.32. Температура стеклования, плавлении и разложения алифатически-ароматических
и циклоалифатических полиамидов
1
2
3
4
5
€
7
Элементарное звено
—С С/ NH—
О
—С—Г"'44]—NH—
О ^-^
—С-СН2 ( \—NH—
С2Н5
—С—(СН2J—СН—(СН2O—CO-NH—/ \—СН2—/UN—NH—
О
-С-(СН2),о—С—NH—/ \—СН2 / \—NH-
II H ^ ' > '
О О
—С—(СНа)в—С—NH—СН2 ( S—CH2-NH—
II II v '
О О
-C—(CH2L—C—NH-CH2—/ \—CH2-NH—
II II \ /
II , II N—'
0 0
°c
130
8'i
296
разл'
490
490
490
Литературный
источник
[212]
[212]
[212]
[213]
Квиана
[214,215]
[216,215]
10
11
12
13
14
15
—С
CH2—NH—
НзС
—с—с—N7 :n-
Ai
СНз
C—(CH2L—C—NH
II II
II
О
II
О
—С— (СН2L—С—NPk
А
А
NH—
NH-
СНз
NH—
NH—
—С—(СН2L—С—NH4 ,MH—
А А к
-C-(CH2L-C-NH—
|| ||
о о
SO
)—NH—
160
400
376
340
250
179
№
16
17
18
19
20
21
Элементарное звено
О
—С—(СН2L—С—NH^ ^~ Л
A A Qfc
—С—(СН2L—С—NH—CH2 ^ ^ ^СН2—NH—
II II 1 f\\
о о ILJJ
—С—(СН2)С—С—NH {(У) NH—
II II vr^/
О О
—С—(СН2N—С—NH. .. ^NH—
II II irV
—С—(СН2)б—С—NHv ,NH—
о о /^~S\
—С—(СН2N—С—NH NH—
4 АЛ_Д
Продолжение i
т
с*
°С
141
115
"с
243
340
196
V
150
табл. 5.32
Литературный
источник
[223)
[222]
[221]
[221]
[221]
[2241
22
23
24
25
26
27
28
6—С—NH>
.NH-
— С—(СН2)8—С—NH—(())—NH-
A i ^
-С— (СН2)„— С—NB. .- /NH—
II II 4As/
О О
—С—(CH2)8-C—NH4 /NH—
II \—/
О
II
°
-C-(CH2)8—С—NH—(Г})—S02—(П)—NH-
ii il ^/ \^y
о о
—С— (СН2)8—С—
A A
о
f
сн5
.NH—
—С-(СН2)8—C-NH—СН2—((у)—GH2—NH-
II II \^У
о о
B24)
319
184
125
133
268
[221}
[221]
12211
[220]
[223]
[225]
Продолжение табл. 5.32
29
30
31
32
33
34
Элементарное звено
—С—(СН2)8—С—NH— СН2^ -. /CH2— NH—
А А ©
—С—(СНа)
II
0
II N
О
-с—(С)
А ^
5-С- NH. /^ ^. JSIH-
А "О.Ж
С
Г> С—NH—<CH2)a—NH—
/ 11
О
))—C~NH-(CH2K—NH-
О
У)—С—NH—(СН2L— NH—
/ il
/ \\
О
Г> С— NH—(CH2M—NH— '
1
220
°С
193
455
399
436
353
Граэл'
Литературный
источник
[222]
[132!
[132J
11321
[132}
36
о
—С
II
о
—С-
II
о
—с-
4
С—NH—(СН2)б—NH—
II
О
—С—NH^(CH2O-NH-
II
о
, у—С—NH—(СН2)8—NH—
J/ II
О
,—С—NH— СН2ч /(СН2)з—NH-
о
С—(С^))—C-NH—(СН2I2—NH-
II W>Y II
О
О
40
41
42
-С—(О/ C-N-(CH2M-N-
н \^/ || | |
О О СНз СНз
—NH-(CH2L— NH-
II
О
I!
о
.C-NH—(CH2N—NH—
ТУГ 4
149
142
371
350
341
186
260
250
198
400
'a?
•JOD
Продолжение табл. 5.32
№
43
44
45
Элементарное звено
—С. ^ /С—NH—(СН2)8—NH—
А@°
—С. /. .С—NH—(СН2I2—NH—
о iyj о
123
Гпл>
°с
184
Гразл-
Литературный
источник
[221]
[228]
[228]
—С—Ar—C-NH— (CH2)l2—NH— Аг=-
А А
N
46
47
—С—< QJ\—С—NH—(CH2N—NH-
i/ A
CV
Cl
—С <( ))—C-NH-<CH2)„—NH-
II
О
290
340
341
349
[226]
[226]
-48
49
.50
51
—С
II
о
С—NH-(CH2N—NH—
275
334
[2261
Br
y
—С—(Cy>—С—NH— (CH2N—NH—
О
Br
i
340
342
[226]
A
.C—NH—(CH2)e—NH—
230—240
1
о
С—NH—(СН2)в—NH—
220-23C
Продолжение табл. 5.32
52
53
-54
55
Элементарное звено
О
II
—С\ .-s. ^Р>. ^. /С—NH— (CH2)e—NH—
О
и
_с^ !
»^X4>^C-NH-(CH,),-NH-
Су
О
й
f j
о ILJJ .
V ^\ /C-NH-(CH2)o—NH—
о
1
Q ]
°G
134
133
124
128
°G
М>азл'
o^
Литературный
источник
[230]
[230]
[230]
[230]
56
57
58
59
-ОС СО—NH—(CH2L—NH—
-ОС СО—NH—(CH2)e—NH-
d-i-b
\^У
-ОС СО—NH—(CH2)8—NH—
—ОС СО—NH— (CH2)„—NH-
0 \
128
[230]
122
[230)
120
[230)
93
[230)
»
60
61
62
63
Элементарное звено
—ОС СО—NH—(СН2)ю—NH—
с
о ^
о
4
3
3 —(Cj)—С—NH—tCH2L—NH-
-у
3
1 '7=Л
э—; ГУ)—с—NH—(сн2N—NH—
3
э—(О")—C-NH—(CH2)8-NH-
П родолжение
°с
85
195
190
175
Гщг
°с
V
габл. 5.5^
Литературный
источник
[230]
[2301
[230]
[230)
О
64
—С-
А
о
175
65
—С <
II
О
О
Р—(Г^—C-NH-(CH2)io—NH-
о
149
66
О
о
СНз О
NH-(CH2L-NH-
184
О
67
-о
II
о
Р —( ТУ)—С—NH—(СН2N—NH—
сн, о
182
68
С
о
СНэ О
142
со
Продолжение табл. 5.32
69
70
71
71а
72
73
74
о
-С (()
1 ч~
О
1 у
О
II
СНз
О
СНз
\—so2—<^
1)—so2—<C
n—so2—(Q
Элементарное звено
)—C-NH-(CH2)9-NH-
0
WC-NH-(CH2IO-NH_
Л \ г* MU1 /^*IJ \ \Ti-I
i\ С* МЫ Г*\-Л С\А \Ti-T
1 / \-t 1\П V— Г12 vil 1\П—*
~~ 0 CH,
J)—C-NH—(CH2L-MH-
0
У)—C—NH—(CH2N—NH—
—/ II
0
^)/—C—NH—(CH2I2-NH—
~^ 0
°C
140
140
^пл-
°C
380
335
358
310
T a л1
Литературный
источник
|230|
[230]
[231]
f231|
[231]
[231]
B28)
75
76
c-СИ
С-
А
0-
-so,
)—с-
А
НзС
СН3
77
78
79
II
о
с
II
(О)
А ^ А
>380
CHj
> 400
430
СН3
350
Н3С
350
[231]
[231]
[231]
[226,
232, 233]
, [232, 233]
[232]
Продолжение табл. 5.32
s\
?2
$3
34
S5
f?
0
О
h -
j) y—
J
Нз
T>—с—
к,
0
Элементарное звено
/ \N_
Cv /СНз
1зКнз
O-CN-
Q_(ch2J_Qn-
Госс
>400
> 400
>400
160
T
435
340
V
380
460
440
Литературный
источник
[234, 235]
[234]
[234]
[236]
[236]
86
87
88
С—/О' C~
о
i
О
89
90
91
О
—Сч
II
О
— Сч
II
о
—(СН2K
II
о
II
о
I '
НзС
:—N
\
:—N
.С—N
il
О
N—
СНз
СНз
— (СН2J
(СН2K—¦
340
280
315
290
250
160
445
Na
92
93
94
95
96
Элементарное эвен
—ОСч /с°—\ V—
<o>
—ОС .СО—NT \—
О Ни,
СН3,
—OCv .СО—N^ \—
/qN \СНз
-ОС, CO-N^~N—(СН2J—{^~)N-
—^*^\ /СО—N у—(СН2)з—( .N—
П родолжение
°СС
325
350
350
"с
215
215
V
га<5л. 5.52
Литературный
источник
[232]
[232]
[232]
[236]
[236]
<97
'98
99
100
101
302
103
О
-С—
о
-с-
II
о
-Cs
II
о
-с»
II
о
о
о
о
о
¦к>
N-
о
о
—(СН,),—/~~^N_
-HS
о Г|/ о
о
Cl-
Со
-si
CD
320
270
210
170
455
300
275
455
270
250
370
460
465
455
450
390
[236]
[236]
[236]
[236]
[236]
[236]
|226]
Продолжение табл. 5.32
»,
104
105
106
107
Элементарное звено
оС1/ о
,Вг
О О ^^
/Вг
CI
rc.
°с
°С
370
360
360
255
т
разл-
390
390
377
Литературный
источник
[226]
[226]
[226]
[236]
СК
108
( II
\ci°
320
400
[236]
109
-Cs
о
C—N? V-
300
380
ПО
X
N N
СХ- хх- хх
NOS
[241]
111
—N
r
-c-
II
о
>500
400
[237]
[238]
Продолжение табл. 5.23
N°
112
113
114
115
116
Элементарное звено
\'Х^'^Х-С—NH— (CH2N—NH—
-c-(ch2J-n; ;n-(chsJ-c-n; ;n-
A w A V-/
о о
II . Il
~S —(Г))—S—NH—(CH2J—NH—
fi \V>y il
II v f II
О О
о о
II 11
— S—r<^ s—NH—(CH2J—NH-
o о
II ¦ ¦— . . Il
—S (( jj (( jy S—NH—(CH2J—NH^~
О O
°c
V
290
220
290
250
302
Литературный
нсточиик
B391
[2401
[242J
[2421
[2421
о
117
265
1242]
° (Q)—S-NH-(CH2N-NH-
О
118
О
— S-
о
о
S—NH—(CH2J—NH-
A
380
[242]
о
119
y
290
[242]
О
-NH—(CH2J—NH—
о
120
—S—(Ту)—
II ^У
О
II
S-NH-(CH2N—NH-
О
288
|242]
о о
121
1243]
Полимер Q2 используется исключительно как термостойкий,
устойчивый к гидролизу пластик конструкционного назначения.
Перерабатывается обычными методами, принятыми для
термопластов. Вязкость расплава при 320°С при скорости сдвига 30 с1
составляет 106 сП и при 110 с-1 4,4-105 сП. Температура при литье
под давлением колеблется от 325 до 345 °С [214].
Эксплуатационные свойства этих полимеров и стеклонаполненных материалов на
их основе приведены в табл. 5.33.
Таблица 5.33. Свойства алициклических полиамидов
на основе 1,4-бисD-амннометил)циклогексана н пробковой кислоты [214]
Показатель
Плотность, г/см3
Температура стеклования, °С
Температура плавления, °С
Прочность при растяжении, кгс/см2
Относительное удлинение при разрыве, %
Модуль упругости при растяжении, кгс/см2
Ударная вязкость, кгс-см/см2
Твердость по Роквеллу
Температура длительной эксплуатации при нагрузке
18,5 кгс/см2, °С
Тангенс угла диэлектрических потерь
при 20 °С
при 100°С
Ненапол-
ненный
полимер
1,12
86
296
840
30
26 000
3,1
110
90
0,02
0,07
Полимер,
наполненный
30%
стеклянного волокна
(термообра-
ботаниый)
1400
5
63 000
3,5
116
290
В^отличие от полиамида 6 и полиамида 6,6 полиамид из
пробковой кислоты и 1,4-бис(аминометил)циклогексана имеет гораздо
более низкое водопоглощение. В кипящей воде за 24 ч водопогло-
щение не превышает 3,5 %, модуль упругости при изгибе при этом
не меняется. Полиамид 6 в тех же условиях имеет водопоглощение
9 %, модуль уменьшается на 18 %. За 14 сут пребывания алицик-
лического полиамида при 110°С и 100 %-ной влажности воздуха
или за 8 недель воздействия моющих средств при 70 °С состояние
поверхности и прочность при изгибе остаются неизменными. Али-
циклические полиамиды используют в тех случаях, когда
требуются ^более высокие, чем для полиамида 6 или полиамида 6,6,
влагостойкость при высоких температурах, например для изготовления
деталей стиральных и посудомоечных машин, фотографических и
электроприборов.
5.10.2. АЛИФАТИЧЕСКИ-АРОМАТИЧЕСКИЕ ПОЛИАМИДЫ
5.10.2.1. Получение
Способ получения смешанных алифатически-ароматических
полиамидов определяется реакционной способностью исходных
веществ, их температурой разложения, а также температурой плав-
384
ления и разложения образующихся полимеров. В табл. 5.32
приведены температуры стеклования, плавления и разложения таких
полимеров.
Полиоксиамиды. Из-за высокой температуры плавления (около
400 °С) полиоксиамиды нельзя получать поликонденсацией в
расплаве. Высокомолекулярные полимеры образуются при
поликонденсации в растворе дифенилтиооксалата с соответствующим
диамином при 130—200 °С [218] или поликонденсацией оксалилхло-
рида с диаминами на границе раздела фаз [244, 245].
Полиамиды алифатических дикарбоновых кислот и
ароматических диаминов (табл. 5.32, № 12—30). Поликонденсацией в
расплаве можно получать не очень высокоплавкие полиамиды,
исходя из стабильных в расплавленном состоянии диаминов с
достаточно высокой реакционной способностью (№ 13,19,24) или из
ароматических диаминов с алифатическими группами между
аминогруппами и ароматическим ядром, например из м- или /г-ксили-
лендиамина (№ 17, 28, 29) [222, 246—248]. Вследствие низкой
реакционной способности ароматических аминогрупп
высокомолекулярные алифатическо-ароматические полиамиды на основе гс-фенилен-
диамина, диаминодифенилсульфона или диамиподифенилоксида
могут быть получены только низкотемпературной
поликонденсацией в растворе при использовании в качестве кислотных
компонентов хлорангидридов алифатических дикарбоновых кислот [249].
Полиамиды с диметилбензольными группами могут быть получены
из гс-диацетоксипроизводных и алифатических динитрилов [250]:
/СН3
/7=гч h2so4/hcooh
Н3ОСОСОСН2—(( ))—СН2ОСОСН3 + NC(CH2NCN ->
/СН3
—СН2—^Q\—CH2NHCO(CH2NCONH—
НзС
п
Полиамиды терефталевой кислоты и алифатических диаминов
(табл. 5.32, № 31—40). Политерефталамиды алифатических
диаминов получают, как и чисто ароматические полиамиды,
низкотемпературной поликонденсацией на границе раздела фаз E.10.3.1)
[132] или в растворе [251, 252]. Поликонденсация в расплаве
пригодна только при получений низкоплавких полиамидов на
основе диаминов, содержащих более восьми атомов углерода в
молекуле. Политерефталамиды диаминов с числом атомов углерода
от двух до шести при температурах плавления 350—450 °С заметно
деструктируют. Полигексаметилентерефталамид (№ 35)
получается в твердой фазе при нагревании соли A:1) ниже температуры
плавления [253].
13 Зак. 18 385
Полиизофталамиды и поли-о-фталамиды. Полиамиды изофта-
левой кислоты и алифатических диаминов, отличающиеся
пониженной температурой плавления и хорошей растворимостью, могут
быть получены с хорошим выходом в расплаве или
низкотемпературной поликонденсацией в растворе. Поскольку при
использовании в реакции с первичными диаминами о-фталевой кислоты
образуются имиды, то применяют вторичные диамины или производные
ппперазина. Предпочтительно такие полимеры получать полнкон-
денсацией в растворе.
Полиамиды фосфор- или серосодержащих дикарбоновых кислот.
Полиамиды фосфорсодержащих ароматических дикарбоновых
кислот и алифатических диаминов (табл. 5.32, № 52—60),
отличающиеся хорошей растворимостью и относительно низкой
температурой плавления, могут быть получены как низкотемпературной
поликонденсацией на границе раздела фаз и в растворе, так и
поликонденсацией в расплаве [230]. Синтез полимеров, исходя из
дихлорангидрида бисD-карбоксифенил)сульфона и алифатических
диаминов или производных пиперазина (№ 71—77), проводят на
границе раздела фаз [231].
Полиамиды ароматических дикарбоновых кислот и дипроизвод-
ных дипиперидила (табл. 5.32, № 78—ПО). Поликонденсация ди-
хлорангидридов ароматических дикарбоновых кислот с
производными пиперазина или дипиперидила осуществляется на границе
раздела фаз в системе хлороформ — вода или низкотемпературной
поликонденсацией в хлороформе [232, 234, 236, 252].
Низкомолекулярные полипиперидиламиды получают также в расплаве. В
противоположность алифатическим диаминам производные
пиперазина при взаимодействии с ортофталевой кислотой образуют
высокоплавкие полимеры.
5.10.2.2. Физико-химические свойства
Наличие в цепи полиамидов ароматических групп способствует
повышению жесткости, температуры стеклования и
теплостойкости полимеров. Температура стеклования поли-ж-фениленадипа-
мида на 80° выше, чем у соответствующего ему по числу атомов
углерода линейного полигексаметиленадипамида. В отличие от
аналогичных сложных полиэфиров в алифатическо-ароматических
полиамидах не наблюдается отчетливая связь между температурой
стеклования и температурой плавления. Это объясняется
одновременным влиянием симметрии цепи, сопряжения карбонильной
группы с ароматическим ядром и затруднением свободного
вращения на прочность водородных связей и кристалличность.
Температура плавления полиамидов с я-фениленовыми
группами в цепи на 70° выше, чем у соответствующих им линейных
полимеров на основе гексаметилендиамина. Так, поли-п-фенилен-
адипамид (табл. 5.32, № 12) плавится при 340 °С, а его линейный
аналог с тем же числом углеродных атомов — полигексаметилен-
адипамид при 265 °С. Напротив, температура плавления поли-лг-
фениленаднпамида (№ 13) 250 °С равна примерно температуре
386
плавления соответствующего алифатического полиамида. Поли-о
фениленизофталамид (№ 14) плавится при 179 °С, что значительно
ниже, чем у соответствующего линейного полиамида. Подобные
соотношения наблюдаются также в случае полиамидов
ароматических дикарбоновых кислот и алифатических диаминов. Поли-
гексаметилентерефталамид плавится при 370 °С (№ 35), что на
140° выше, чем у соответствующего ему полиамида на основе
гексаметилендиамина и пробковой кислоты (температура
плавления 232 °С). Полигексаметиленизофталамид плавится на 30° ниже,
чем полигексаметиленамид на основе пробковой кислоты.
Значительно более низкие температуры плавления алифатически-арома-
тических полиамидов с ж- или о-фениленовыми звеньями по
сравнению с подобными продуктами с /г-фениленовыми группами
обусловлены тем, что сегменты, соединенные в /г-положении, больше
способствуют межмолекулярному взаимодействию амидных групп
и образованию водородных связей.
Различие в температурах плавления алифатическо-аромати-
ческих полиамидов с одинаковым числом атомов углерода в звене,
но с разным положением фениленовых групп, например
Г — HNHaC^^. ^CH2NHCO(CH2LCO— T
Тпя = 243 °С
пл
Г—NH(CH2NNHCCk^ ^CO—-1
пл
198°С
объясняется сопряжением карбонильной группы с ароматическим
ядром. В полиизофталамиде карбонильная группа сопряжена с
бензольным кольцом. Ограничение вращения молекулярных
сегментов препятствует образованию водородных связей. Это
обусловливает понижение температуры плавления и кристалличности по
сравнению с изомерными амидами адипиновой кислоты. Алифати-
ческо-ароматические полиамиды на основе бисD-карбоксифенил)-
сульфона (№ 71—77) вследствие наличия сульфоновой группы,
обусловливающей повышение гибкости цепи, плавятся ниже, чем
соответствующие политерефталамиды. Внутри гомологического
ряда закономерности изменения температуры плавления
полиамидов ароматических дикарбоновых кислот и алифатических
диаминов (например, № 31—37) такие же, как и для алифатических
полиамидов. Температура плавления полиамидов с одинаковыми
остатками диамина уменьшается в следующем ряду в зависимости
от строения остатка дикарбоновой кислоты:
-о- « -о- > -<о
13* 387
о-
СНЭ
У полиамидов алифатических дикарбоновых кислот и
ароматических диаминов температура плавления уменьшается в
следующем ряду в зависимости от строения остатков диаминового
компонента:
-СН2
—СН2-
—Н2С
Полиамиды ароматических дикарбоновых кислот и N, N'-
диметилзамещенных алифатических диаминов (№ 40 и 41)
плавятся при температуре на 100° ниже, чем соответствующие
незамещенные аналоги. В то время как алифатическо-ароматические
полиамиды на основе незамещенных алифатических диаминов
растворяются только в концентрированной серной и трифторуксус-
ной кислотах, полимеры, содержащие N-метилзамещепные амид-
ные группы, растворяются также в хлорированных растворителях
или кетонах [132]:
Среди алифатическо-ароматических полиамидов полиоксиами-
ды и полиамиды с пиперазиновыми и дипиперидильными
фрагментами в цепи занимают особое место. По высокой температуре
3OO
плавления (около 400 С) и хорошей растворимости в N-метилпир-
ролидоне или тстраметилмочевине полиокснамиды блинке к чисто
ароматическим полиамидам, чем алифатнческо-ароматнческне
полиамиды длинноцепных дикарбоновых кислот [218].
Полиамиды с пиперазиновыми и дипииерндильнымн группами
в цепи (№ 78—ПО) представляют собой также особый класс и
могут быть отнесены к N-алкилированным алифатическо-арома-
тическим полиамидам. Поскольку у этих полимеров невозможно
образование водородных связей, они отлично растворяются в
хлороформе или смеси хлороформ — метанол [232, 236]. В случае
полимеров с пиперазиновыми и дипиперидильными группами
отсутствие водородных связей, влияющих на температуру плавления,
компенсируется высокой жесткостью гетероциклических групп.
Пиперазиновые полиамиды размягчаются с разложением при
300—350°С. Дипиперидилполиамиды плавятся между 200 и 400°С.
Продукты взаимодействия тере-, изо- и ортофталевых кислот с
одними и теми же пиперазиновыми производными имеют близкие
температуры размягчения в отличие от аналогичных полимеров на
основе соответствующих линейных диаминов. Термическая
деструкция алифатическо-ароматических полиамидов начинается около
400 °С, а термоокислительная — при 350 °С. До 600 СС выделяются
летучие продукты, количество которых соответствует
разложению 90% полимера. Такое поведение алифатическо-аромат1
полиамидов определяется прочностью алифатическ^ "
Несмотря на высокие температуры плавления этих -:.;:;::;.; ,
применение ограничивается температурами ниже 200°С.
Механизм термической деструкции полиамидов ароматических
дикарбоновых кислот и алифатических диаминов исследован в
[228]. Энергия активации термической деструкции в зависимости
от природы дикарбоновой кислоты колеблется в пределах 10—
28 ккал/моль. Вследствие смещения электронов за счет влияния
бензольного ядра происходит первичный акт деструкции, связанный
с разрывом связи N—С;
I
С—М—|—СН2— СН,—
Возникающие при этом радикалы
—/ГЪ—CpNH+CH2CH2 —> (Г ))—CONH + СН2СН2
распадаются или рекомбинируют в соответствии со схемой:
(S)—CONH + CH2CH2
СН2СН2 + СН2СН2 —> СН2СН2СНгСНг
389
/Q)—CONHNHCO—/ON—
В результате протекания вторичных реакций образуются
термически устойчивые конечные продукты:
(( У)—CONH
NCO или <( ))—CN+H2O
<g>-CN + H2O-. -
CONHCH2CH2—
—(С )>—COOHH-NHs
Н2О
—>¦ CH2CH2NH2 + • • ¦ —(Cj/—СООН
2 CH2NH2 —> CH2NHCH2 \- NH3
2- • • —^QN—СООН —* (Tj)—СО—<^N + CO2 + H2O
2 (Q\—NCO —> -.—<Q|)—N=C=N—<1Q> + CO
5.10.2.3. Промышленные алифатическо-ароматические
полиамиды
В промышленности алифатическо-ароматические „полиамиды
производятся в ограниченном объеме. В США фирма «Du Pont»
выпускает в опытном масштабе поли-ж-ксилиленадипамид
(волокно МХ-6) и фирма «Celanese» — полигексаметилентерефтал-
амид (найлон-бТ). С 1967 г. фирма «Dynamit NoBeb (ФРГ)
выпускает термостойкий конструкционный полимер политриметил-
гексаметилентерефталамид под маркой «Трогамид Т».
Волокна. Свойства волокон на основе полигексаметилентере-
фталамида и поли-л*-ксилиленадипамида, а также полиамида 6,6
приведены в табл. 5.34. Волокно из поли-л*-ксилиленадипамида,
имеющего относительно низкую температуру плавления B43°С)(
может быть получено формованием из расплава [247]. Недостатком
этого волокна является высокая чувствительность к влаге;
температура стеклования и модуль упругости у него выше, чем у
полиамида 6,6. Из полигексаметилентерефталамида волокно
получают методом мокрого формования из раствора в
концентрированной серной кислоте [247] или сухим методом из трифторуксусной
кислоты [132]. Наряду с хорошей теплостойкостью и стабильностью
рамеров полигексаметилентерефталамид отличается низкой
способностью к образованию вмятин, вследствие чего он очень ценен
для использования в производстве шинного корда.
Материалы. Аморфный алифатически-ароматический полимер
терефталевой кислоты и 2,4,4-триметилгексаметилендиамина вы-
390
Таблица 5.34. Свойства алифатическо-ароматических
полиамидных волокон [247J
Показатель
Полигекоа-
мегнлен гере-
фталамнд
Полч-лг-
KCil.ill.iCH-
адипамнд
I So л иге кг с,
мегилеи-
адипамил
Плотность, г/см3
Температура стеклования, °С
Температура плавления, °С
Водопоглощение, %
Прочность волокна, г/денье
Относительное удлинение прн разрыве, %
Модуль упругости, г/денье
Остаточная прочность после выдержки в
течение 5 ч, %
при 150°С
при 185°С
при 220°С
1,21
180
370
4,5
4,5
35
45
100
100
60
90
243
4,3
29
50
1,14
60
265
4,5
4,9
57
40
85
40
0
пускаемый под маркой «Трогамид Т», легко перерабатывается,
СНз
—HN—СН2СНСН2—С—СН2СН2—NHOC
—СО-
сн,
сн3
п
имеет отличную прозрачность и высокие ударную вязкость и
стабильность размеров в условиях повышенных температур и
влажности. Наполненный стеклянным волокном материал
выпускается под маркой «Трогамид Т 935». Полимер перерабатывают
экструзией, литьем под давлением и выдуванием. Перед
переработкой гранулы полимера подсушивают при ПО—120 °С. Для
экструзии используют короткие шнеки с отношением сжатия
1:1,5—1:2,5, температурное поле от воронки до сопла должно иметь
температуру 250—280 °С [254]. Литье под давлением осуществляют
при 250—320 °С и давлении впрыска 1300 кгс/см2, частота
вращения шнека 20—60 об/мин, температура формы 70—90 °С.
Вследствие высокой вязкости расплава работают с открытыми соплами
(без обратного клапана). Усадка отлитых деталей при 100—130°С
составляет 0,02—0,03%- Для переработки выдуванием применяют
выдувные машины со шнековой пластикацией (длина шнека 15—
20 D). Лучшие результаты достигаются при использовании трех-
зонных шнеков со степенью сжатия от 1,5 до 2,5. Температурный
режим от воронки до формы следующий: 240-250-255-255-245-245 °С.
Поли-2,4,4-триметилгексаметилентерефталамид представляет собой
жесткий полимер конструкционного назначения с высокими
ударной вязкостью и теплостойкостью. Ниже приведены
физико-механические и теплофизические свойства полимера [254]:
Предел текучести, кгс/см2
при —40 °С
при 23 DC
при 77 °С
при 93 °С
1 184
691
462
360
391
Относительное удлинение при рязрыве %
при —40 °С 9,5
при 23 °С 8,8
Прочность при растяжении, кгс'см2
при -40 °С 896
при 23 °С 685
при 77 °С 535
при 93 °С 500
Относительное удлинение при разрыве, %
при — 40 °С 134
при 23 °С 132
Модуль упругости при растяжении, кгс/см2
при — 40 °С 38 669
при 23 °С 28 474
при 77 °С 26 717
при 93 X 20 881
Предел прочности при изгибе при удлинении 5 %,
кгс/см2
при —40 °С 1 467
при 23 °С 932
при 77 °С 721
при 93 X 658
Модуль упругости при изгибе, кгс'см2
при —40 °С ' 38 985
при 23 °С 27 139
при 77 °С 22 780
при 93 °С 22 217
Ударная вязкость, кг см/см2
при 20 СС Не
разрушается
при — 50 °С 60
образцов с надрезом
при 20°С 10-15
при — 50 °С 3-5
Твердость при вдавливании шарика через 60 с,
кгс/см2 1 250
Прочность при сжатии, кгс/см2
I % деформации при сжатии 238
10 % деформации при сжатии 1 035
Модуль упругости при сжатии, кгс/см2 23 834
Твердость по Роквеллу, шкала M ........ 93
Износостойкость, мг/1000 циклов 21
Водопоглощение, % 4
Плотность, г/см3 1,12
Показатель преломления 1,566
Температура стеклования, °С 148
Теплостойкость по Вика, СС . . . . ....... 145
Теплостойкость по Мартенсу, °С .......... 100
Температура длительной эксплуатации, СС
при нагрузке 4,6 кгс 140
при нагрузке 18,5 кгс 130
Удельная теплоемкость, кал/г-°С) 0,35
Коэффициент теплопроводности. ккал'(\г ч X) ... 0,18
Усадка, % . . . . ' 0,5
Термический коэффициент линейного расширения,
1/°С 51 - Ю-6
Горючесть
Самозатухающий
Светопроницаемость, % 85—90
Есд, кгс/см*
10*
s
103
s
102
5
10
•
-*„
*^m m
—
S'
l"
-Л.
0,3
0,6
Ofi
0,2
О
0 40 80 120
Рис. 5.63. Зависимость модуля упругости при
сдвиге и логарифма декремента затухания поли-2,4,4-
триметилгексаметилентерефталамида от
температуры [254].
б,кгс/см2
700
600
500
то
300
200
то
о
J
n
n
i
У
]
y
<
01 5 1 510 5102 5103 %9ч
Рис. 5.64. Изменение длительной прочности при растяжении поли-2,4,4-триме-
тилгексаметилентерефталамида на воздухе при постоянной нагрузке [254];
а — 20 °С; б — 20 °С и условный предел упругости 2%; в — 100 °С и условный предел
упругости 2%.
В отличие от других полиамидов аморфный поли-2,4,4-триме-
тилгексаметилентерефталамид характеризуется отличной
прозрачностью даже в толстых слоях. Пластик горюч, однако пламя не
распространяется, и при извлечении из пламени горение
прекращается. Продукт представляет собой термопластичный полимер.
Модуль упругости до 93 °С сильно зависит от температуры. Из
зависимости модуля упругости при сдвиге от температуры следует,
что изделия до 130 °С сохраняют высокую формоустойчивость
(рис. 5.63).
На рис. 5.64 показано изменение прочности при растяжении от
времени пребывания под постоянной нагрузкой при 20 и 100°С.
Поли-2,4,4-триметилгексаметилентерефталамид имеет хорошие
электроизоляционные свойства. Ниже приведены электрические
свойства поли-2,4,4-триметилгексаметилентерефталамида [254]:
Удельное поверхностное электрическое
сопротивление, Ом
в нормальных условиях >5-1013
после 24 ч выдержки в воде > 1 • 10[3
Удельное объемное электрическое
сопротивление, Ом см
в нормальных условиях >5-1014
после 4 ч выдержки при 80 %-ной отн.
влажности . >5*1014
Диэлектрическая проницаемость
в нормальных условиях
при 1 кГц 3,5
при 1 МГц 3,1
после 7 сут. выдержки при 100 %-ной
отн. влажности
при 1 кГц 3,9
при 1 МГц 3,4
393
Тангенс угла диэлектрических потерь
сухого продукта
в нормальных условиях
при 1 кГц 0,028
при 1 МГц 0,022
после 7 сут выдержки при 200% -ной
отн. влажности
при 1 кГц 0,030
при 1 МГц 0,030
Электрическая прочность (пластина 1 мм),
кВ/см 250
Стойкость к скользящим разрядам (D1N
53480), кА 3
Во влажной атмосфере изменение диэлектрических свойств по-
ли-2,4,4-триметилгексаметилентерефталамида незначительно.
Равновесное содержание влаги при 20 °С и 65%-ной относительной
влажности составляет 3%. Поли-2,4,4-триметилгексаметилентере-
фталамид инертен к. большинству алифатических и ароматических
углеводородов, сложных эфиров, кетонов, хлорированных
углеводородов, таких, как трихлорэтилен и тетрахлорид углерода, а
также к разбавленным минеральным кислотам. При выдержке в этих
средах не происходит образования трещин на поверхности
изделий. В некоторых кетонах, хлорированных углеводородах и
алифатических спиртах полимер набухает. Полимер устойчив к действию
горячей воды до 80 °С, выше этой температуры наблюдаются
ухудшение механических свойств и потеря прозрачности. Поли-2,4,4-
триметилгексаметилентерефталамид растворяется в феноле,
муравьиной кислоте, концентрированной серной кислоте, диметил-
формамйде и смеси хлороформ — метанол 80 : 20 % (объемн.).
Основные области применения алифатическо-ароматического
полиамида терефталевой кислоты и 2,4,4-триметилгексаметилен-
диамина — детали измерительных приборов для газов и жидкостей,
регуляторы давления [255], прозрачные корпуса и выключатели в
электротехнике и защитные покрытия для осветительных приборов.
В вычислительной технике и точных приборах полиамид
используют для изготовления зубчатых колес и зубчатых передач. Его
используют также в производстве защитных устройств, корпусов
печатных машин и крышек доильных аппаратов Такое применение
поли-2,4,4-триметилгексаметилентерефталамида объясняется
сочетанием в нем термостойкости и стабильности размеров, присущих
поликарбонату, с высокой прозрачностью, присущей полиметил-
мета кр ил ату.
5.10.3. АРОМАТИЧЕСКИЕ ПОЛИАМИДЫ (ПОЛИАРАМИДЫ)
5.10.3.1. Получение
Температуры стеклования, плавления и разложения
ароматических полиамидов приведены в табл. 5.35.
Полиамиды ароматических аминокарбоновых кислот (№ 1—5).
Высокомолекулярные полимеры из аминокарбоновых кислот мож-
394
но получить только через их хлорангидриды:
H2N>. -. .СООН H2N. . .СОС1 ^
\,<>-Сх SOCI2 \^>у^ Эфир; НС]
—IIC1; -
^ ^^ ^ Основание
—H Cl
Поликонденсацию проводят в присутствии акцепторов
хлористого водорода, таких, как триэтиламин или пиридин, в
растворителях типа гексаметилфосфотриамида, диметилацетамида, тетра-
метилмочевины или хлороформа [257, 260, 261] или на границе
раздела фаз [259, 266, 321].
Ароматические полиамиды дикарбоновых кислот и обычных
ароматических диаминов. Поликонденсация в расплаве для
получения ароматических полиамидов не применяется, так как
исходные диамины обладают относительно низкой реакционной
способностью, а образующиеся полиамиды чаще всего при температурах
расплава разлагаются. Высокомолекулярные полимеры получают
низкотемпературной поликонденсацией ароматических диаминов и
дихлорангидридов дикарбоновых кислот на границе раздела фаз
или в растворе. При поликонденсации на границе раздела фаз
диамин растворяют в водной щелочи B моля щелочи на моль
диамина) и хлорангидрид в таком растворителе, как бензол, цикло-
гексанон, тетрагидрофуран, тетрахлорид углерода, и проводят
процесс при интенсивном перемешивании и температуре 0—20 °С
[275, 280, 282, 284, 290, 322—326]. Концентрация хлорангидридов
лежит в пределах 0,05—0,5, а диаминов 0,05—0,2 моль/л.
Межфазная поликонденсация протекает с очень высокой скоростью, при
этом образуется полимер с почти количественным выходом.
Взаимодействие происходит на границе раздела фаз или в пограничной
области органического растворителя. Гидролиз хлорангидридов
вследствие их плохой растворимости в водной фазе протекает в
незначительной степени. Для получения высокомолекулярного
полимера мольное соотношение диамина и дихлорангидрида должно
быть 0,93:0,90 [275]. При выборе растворителя очень важно
набухание в нем полиамида.
Высокомолекулярные полиамиды получают также
эмульсионной поликонденсацией; процесс протекает с высокой скоростью
[296, 308]. При этом в отличие от поликонденсации на границе
раздела фаз полимер образуется в органической фазе, так как диамин
почти полностью находится в ней, тогда как нейтрализация
выделяющегося хлористого водорода протекает в водной фазе. Выход
и молекулярная масса полимера определяются коэффициентом
распределения диамина, составом органической фазы, поверхностным
натяжением, степенью дисперсности и степенью набухания
образующегося полиамида в органической фазе. Поликонденсация
м-фенилендиамина с изофталоилхлоридом в системе
тетрагидрофуран— вода—ЫагСОз протекает количественно в течение 1 мин.
395
Таблица 5.35. Температуры стеклования, плавления и разложения ароматических полиамидов
1
2
3
4
Б
6
7
Элементарное звено
\ТГ_т // \\ /"•
1\П ""¦ ( ( ) /' Vj '
О
-NH-rQj-C-
^S-sj II
^ 0
-NH OC—
\ /
0)
—NH—\Cj) \O)—C—
О
_NH__Q_o-0_c-
0
—С—(См—C—NH—(C)) NH— '
il ^/ ii Х^У
0 0
о о \x-
286
503
424
> 500
500
> 400
^разл
(в N2). °C
500
650
520
(на
воздухе), °c
450
Литературный
источник
[259-265]
[256, 257]
[258]
[266]
[267]
[226, 268,
272—277,
419]
[268,
270—272]
10
-c-
o
о
С
II
о
и
12
13
14
О
О
о
¦С—NH NH—
о W
о
C-NH NH
II I I
о I I
о
о
О
—a ))—nh—
О
о
~i~NH~<O/~i~<O)~NH~ Сульфон т
о
о
380
295
> 350
340
> 400
380
482
484
430
392
430
405
Элементарное звено
15
16
17
18
—С—(
О
О
о
-с-/C>-с-
II "^У II
H;
NH
\
о
о
О
СНз
СНз
¦NH-
СН3
»
Z—NH
19
NH—
Продолжение табл. 5.35
Т
разл
(в N2), °C
Т
разл
(на
воздухе), °С
Литературный
источник
325
365
365
380
[223]
[270]
[270]
[224. 228J
320
[269, 312]
ОСН3
20
21
22
23
24
-с—<
II
о
C
о
V
ЮСНз
-С— ( ))—C-NH— ( )>—СН=СН <( )>—NH-
II
о
-с—(
А
о
о
-C-(C}>-C-NH-\(O,
о о „
уСНз
-N=N—(С))—NH"
25
26
-С-
II
О
-С
II
о
!1
о
о
-С—NH
II
О
С—NH
II
О
СНз
СНз
> 200
> 360
320
360
> 400
Продолжение табл. 5,35
27
28
29
30
-с—{С
О
—с—/Г^
II Y4*-^
II
О
-с—(П*
о
\_C_N
о
D-C-NI
A
О
Элементарное звено
с6н5
Н-|ЯI ff)]-NH-
с
о
о
H (CjS—Р—<^Q)—NH—
^пл'
305
290
390
290
(в N2). OC
^ 350
т
разл
(на
воздухе), °С
350
Литературн1лй
нсточннк
[289]
[289]
[223]
31
32
33
34
~c-O-^NH-CH2-<O)-'
2—NH—
О
-сч
II
о
-Cv
г
о
-с.
о
35
36
II
о
II
О
о
СНз
С—NH—(<ГЪ—NH—
||
О
II
О
С—NH NH—
NH—
СНз
С—NH
II
II
о
NH—
.NH—
225
> 400
> 400
365
240
352
330
350
[250]
[268, 272,
275, 293]
[268,272,279
290—293,
296—311]
[268, 269]
[290]
[270]
37
1
о
^C-NH-
II
О
—(^5)—NH—
350
[290]
Элементарное авено
38
39
40
41
—С
/
42
43
-с.
II
о
О
о
,-C—NH-
II
О
NH—
Продолжение табл. 5.35
тс.-с
т °с
2пл* ^
раз л
(в N2). °С
Т
разл
(на
воздухе), вС
Литературный
источник
300
> 400
365
380
380
[224]
[270]
[290, 293]
[224]
[290]
[312]
44
45
С.
А
С*.
II
О
46
47
48
О
A
49
50
О
X—NH
О
О
.С—NH-
II
О
о
~ R=-s- -о-сн2-о. -со-
- Сульфон I
о
СН2—(( ))—NH-
СНз
Н3
—С>». /\ ^С—
II |ГЛ1 II
о iyj о
—СО ОС—NH-
\_/
НзСО
-NH—
о-
\—NH—
СНз
СН,
/
ОСНз
—NH
315
400
400
375
240
420
[293, 295]
[295]
[281, 282,
294,295,425]
[282, 290,
293]
[282, 290]
[313]
[269]
Продолжение табл. 5.35
№
51
52
53
54
55
56
-ОС
N
<
-ОС,
<
о
-с-
°с
-с-
°с
-с—
осг
Элементарное звено
ОС —NH NH_
Сот
Cl
-<о
\
)—С—NH—((^)
О
)—С—NH—(Г^)
О
—С—NH—( Q
о
N—NH—
f
о
>— S—E)—NH-
о
СНз
)— С—(Cj)—NH-
СНз
Гпл* °С
200
185
305
340
360
360
т
разл
(в N2J, °C
404
406
418
т
разл
(на
воздухе), °С
Литературный
ИСТОЧНИК
[269]
[269]
[270]
[226]
[226]
Г226]
57
58
59
60
61
62
Cl
\
о \
Cl
Cl
-С—(Г))—C-NH-
II N»' II
о V, о
Cl
\
о \с,о
Вг,
о
-S —
I!
О
СНз
СНз
NH—
Л
II
о
C-NH—(СЪ—NH-
?
о
Bi
\
-С—<|
II
о о
Bi\
л
о о
о
СНз
I
СИ,
355
360
360
330
360
360
404
398
418
412
401
383
Продолжение табл. 5.35
гразл
N2). °G
разл
(ца
воздухе) °С
340
360
340
394
395
353
380
№
Элементарное звено
в
\
63
\Вг°
Вг
64
II
о \Вго
65
66
СНз
II
о
о
OT
—СО СО NH NH—
67
§п§)
—со со—ь
68
69
70
71
72
О
с-
II
о
73
74
NH—
о
-С—( Ъ—О— Ъ—C-NH
о
О
.NH—
О
450
400
>450
450
380
355
376
378
380
380
367
Продолжение табл. 5.35
№
Элементарное звеио
Гразл
(в N2) °
T
раз л
(на
воздухе), °G
О
1Ъ
76
77
—С
О
О
—с—
И
о
'—Л /—\ /—\ II
—(ГУ)—О—(Г))—C-NH—(ГУ)—S
78
- С— '
79
il
о
и
о
О
О
—с—NH—;
о
||
О
С—:f^—С—NH NH—
NH-
|
о
340
400
>400
356
380
380
380
80
-с—i
II
о
81
—С-
II
О
О
U
82
h
С—NH-
II
О
—NH-
83
-с—,
II
о
-С—NH-
O
i—NH—
UC
^о
*о
> 400
> 400
330
> 400
470
480
360
460
Продолжение табл. 5.35
Элементарное звено
гс,-с
т °с
Т
разл
(в N2), °С
разл
(на
воздухе), °С
Литературный
источник
84
[316]
—С—vA )/—CF<2—ч
О
85
О
86
о
о
о
-С—NH—R—NH— R =
О
©-с-
р-Ю,
с--
о
NH—
F
260
249
F\ /
жг -я
V
[230)
[230]
87
о
168
88
II
О
О
Л-
-С—
239
89
О
СНз О
-NH—
271
90
О
СНз О
240
Продолжение табл. 5.35
№
Элементарное звено
rc, °c
разл
(в N2), °C
разл
(на
воздухе), °С
Литературный
источник
О
91
-nh ш-
О
СНэ
О
О
92
93
94
05
и
О
| «
СНэ О
NH-
о
о
A
~NH?—гЯ\,—
о
-С
О
II
;—Р
C
A
170
230
320
224
222
[230]
[230]
[223]
[223]
[223]
96
97
О
—С
II
о
-NH
А
¦NU—
-С—,
II
О
к У А
-NH>. л ^NH-
-СНз
98
-СО OCNH—(Г ))—NH—
10 1 Х" J
-Р—
I
СНз
99
—СО OCNH>,
О I
NH—
100 i
—СО OCNHv^^^^NH—
CH
СНз
164
220
162
150
145
Продолжение табл. 5.35
10t
102
1Q3
104
Элементарное звено
II ^^/"^N4
A Q
-Л
II "^
i
с
\ N
0
0
/C—NH—f^si—P jA|—NH—
] A A«
)
\C-NH—(Qy-(Qj—ий-
0
^C—NH NH—
II 1
V
rc. °c
400
разл
(в N2), °C
380
380
T
разл
(на
воздухе), QC
Литературный
источник
C17]
C18]
1224]
B24]
105
106
107
108
109
ПО
415
О
0
и
\\
О
о
II
II
—s
II
1!
О
-с
II
II
о
0
О
-nh—(О/ сн>—;О>—NH-
о
о
—NH>
о
I
365
400
400
274
155
340
340
[318]
[318]
[318]
[320]
[320]
[412]
В зависимости от механизма образования двух фаз и скорости
перемешивания молекулярная масса пол и-лг-фениленизофтал амида
изменяется в пределах A,3—22,2)-104 и полидисперсность Mw/Mn—
— 1,04 — 2,3. При поликонденсации в растворе в качестве
растворителей преимущественно используют диметилацетамид, гекса-
метилфосфотриамид или смеси с акцептором хлористого водорода,
такие, как метиленхлорид — триэтиламин, диоксан — пиридин [251,
270, 327, 328].
За исключением полиамидов с /г-фениленовыми группами,
другие ароматические полиамиды остаются в растворе при
использовании в синтезе растворителей амидного типа или растворителей
с добавками неорганических солей, таких, как хлориды лития или
кальция, повышающих степень сольватации. Образующиеся
растворы могут непосредственно применяться для формования волокон.
Поликонденсацией в диметилацетамиде фирма «Du Pont» с 1961 г.
производит ароматический полиамид на основе изофталоилхлорида
и ж-фенилендиамина (Номекс). Большая часть выделяющегося
НС1 в конце процесса-находится в связанном виде в растворителе
и отделяется в виде хлорида аммония после обработки аммиаком,
а кепрореагировавший хлоргидрат диметилацетамида
нейтрализуется Са(ОНJ. Образующийся при этом хлорид кальция
повышает сольватирующую способность диметилацетамида.
Поли-;и-фениленизофталамид был получен и в нерастворителе
типа циклогексанона, ацетонитрила, бензонитрила или
нитробензола в присутствии в качестве акцептора НС1 третичных аминов
[297]. Так же, как при эмульсионной поликонденсации,
молекулярная масса полимера зависит от порядка и скорости смешения
реагентов.
Сополиамиды регулярного строения. Модификация полиамидов
возможна за счет использования в качестве диаминных
компонентов блоков с предварительно образованными амидными связями
и концевыми аминогруппами:
H2N
п
Подобные полимеры, синтезированные Престоном, могут
содержать м- и /г-фениленовые фрагменты в любом соотношении.
Престон назвал эти полимеры регулярными (упорядоченными). Такие
полиамиды и полиамиды на основе обычных диаминов и хлоран-
гидридов кислот синтезируют на границе раздела фаз [329—332]
и в растворе [283,333—336]. Даже при очень высоком содержании
/г-фениленовых групп полимеры можно получить в растворе, так
как кристаллизация продуктов затруднена в силу сложной приро-
416
ды элементарного звена полимера. Аналогичным образом можно
синтезировать полимеры, содержащие в структуре азогруппы [333]
или сложноэфирныи связи [337].
Для синтеза диампнных блоков важна симметрия. Диамины
типа
получают из фенилендиамина и нитробензоилхлорида с
последующим восстановлением нитрогруппы. Трехъядерные диамины,
содержащие в середине молекулы остатки изомерных фталевых
кислот
образуются из фталоилхлорида и большого избытка диамина [332]j
или из фталоилхлорида и нитроанилина с последующим
восстановлением [338].
Элегантный синтез многоядерных ароматических или бензгете-
роциклических диаминов с центральной азогруппой
осуществляется окислительной димеризацией асимметричных ароматических
диаминов с различными по основности аминогруппами [333]:
H2N—(( V/—CO-NH—(Г I)—NH2 2—^
CuVCu«
H2N—s'QN—CONH—(Q)—N=N—((Ql)—NHCO—(f?)\—NH2
Поликонденсация с образованием полиэфироамидов
регулярного строения является саморегулирующейся, так как
аминогруппы имеют гораздо более высокую реакционную способность, чем
фенольные гидроксилы [337]:
Образующийся в качестве промежуточного продукта дифенилол-
амид реагирует со следующей молекулой дихлорангидрида дикар-
боновой кислоты с образованием регулярного полиэфироамида.
14 За*, la 417
5.10.3.2. Физико-химические свойства
Температуры стеклования и плавления. Температуры
стеклования, плавления и разложения ароматических полиамидов (за
исключением сополиамидов) приведены в табл. 5.35. Для многих
полиамидов наблюдаются аномальные значения температуры
плавления, что связано с термической деструкцией таких полимеров
вблизи температуры плавления. Температура плавления в первую
очередь определяется ориентацией феннленовых групп в цепи.
Полиамиды с феппленовыми группами, соединенными
преимущественно в /г-положении, например поли-/г-амш:обензойная кислота (№ 1,
табл. 5.35) пли поли-/г-фенилентерефталамид (№ б, табл. 5.35),
являются исключительно высокоплавкими полимерами (т. пл. около
500 °С). Наличие в структуре м- и особенно о-фепиленовых групп
приводит к снижению температуры плавления. Так, температура
плавления полимера с преимущественно о-фениленовыми группами
лежит около 185°С (№ 52, табл. 5.35). Такие низкие значения
температур плавления в случае о-фениленовых групп объясняются
затруднением вращения и неполнотой образования водородных
связей.
В зависимости от типа связей в остатках диамина и дикарбоно-
вой кислоты по уменьшению температуры плавления полимеры
можно расположить в следующий ряд: п, п >• м, п> м, м> о,
п > о, ж > п, о > м, о > о,о (полиамиды № б, 7, 32, 8, 50, 33, 34,
51, 52). Ароматические полиамиды с я,я'-дифенильными, n,nf-
дифенилоксидными, /г,я-дифенилметановыми, /г,/г'-дифенилсулъфо-
новыми фрагментами в цепи плавятся в том же температурном
интервале, что и полимеры с ж-фениленовыми связями. В ряду
диаминов температура плавления политерефталамидов снижается
следующим образом:
41Я
о
Полимеры на основе .w-фенилендиамина и различных дикар-
боновых кислот по температуре плавления можно расположить,
в ряд:
Ck
eu
> -Q>- > -o-
V,
» \
СНз
Для интересных в практическом отношении ароматических
полиамидов возможность варьирования температуры плавления выше
400 °С ограничена только двумя мономерами. Статистические
сополимеры на основе трех компонентов плавятся вследствие
асимметрии цепи при более низкой температуре, чем соответствующие го-
мополимеры,
14*
419
Таблица 5.36. Температуры стеклования, плавления и разложения регулярных ароматических сополиамидов [329,334]
Элементарное звено
toi.
дл/г
Растворитель
ПЛ'
разл1
°С
—NB
-NH—
Л—NH4
О
С—NH>
/С—NH—
II
О
С—NH—i
О
О
о
NH—
NH—С
NH—С
NH—Сч
О
NH—С ;
II
о
NH—(
ЛЧН—С.
li
О
,NH-C—
II
О
NH-C
II
^NH-C—(
О
О
—NB—(Г^)—С- NB—(Cj)—NH—С /Г^—NH—C
M
о
о
II
о
c—
II
о
с—
о
о
c
1,48
1,92
ЦЗО
1,31
1,27
1,42
0,99
1,98
ДМАА-
ДМАА -
ДМАА-
ДМАА-
ДМАА-
ДМАА-
ДМАА —
-LiCl
-LiCl
-LiCl
-LiCl
-LiCl
-LiCl
LiCl
H2SO4
290
305
295
335
410
450
460
467
475
490
480
555
450
485
Таблица 5.37. Температуры плавления н разложения
ограниченно регулярных сополиамндов [339]
Элементарное звено
—NH
6
о
1,90
0,19
2,80
2,70
0,75
3,45
290
400
405
495
490
450
405
440
440
450
525
Метод, предложенный Престоном и основанный на использовав
нии диаминов, содержащих амидные связи E.10.3.1.3), открывает
широкие возможности для получения ароматических полиамидов
с сбалансированными свойствами, такими, как термостойкость,
кристалличность, температура стеклования и модуль упругости
[329, 334, 336, 340—342]. В табл. 5.36 приведены физические
свойства ряда полиамидов, различающихся только характером связей
между фениленовыми группами. Из данных этой таблицы особенно
наглядно видно различие в температурах плавления при наличии
в звене п—я-связей. При переходе от полимеров с ж-фениленовыми
группами к я-фениленовым группам температура плавления
повышается скачкообразно с 410 до 555 °С. О регулярном строении
сополимера свидетельствует температура стеклования, которая на
60—110° выше, чем у соответствующего ему статистического
сополимера с ^-фениленовыми группами в цепи (см. табл. 5.36) [334].
Сополимеры, полученные из предварительно синтезированных
асимметричных диаминов с амидными связями, плавятся так же,
421
как статистические сополимеры. Престон назвал эти полимеры
«ограниченно-регулярными» [339]. В табл. 5.37 приведены данные
об ограниченно-регулярных полиамидах на основе диаминобенз-
анилида и изо- или терефталевой кислоты. Полимеры этой
группы с лг-фениленовыми группами плавятся при температуре на
110°С ниже, чем соответствующие им регулярные сополимеры
(табл. 5.36).
Структура и кристалличность. Ароматические полиамиды имеют
аморфную структуру. Однако известно много примеров, когда в
результате термообработки при температуре около 300 °С или
горячей вытяжке соответствующего волокна полимеры становятся
частично-кристаллическими [341, 343]. Термообработанный поли-/г-
фенилентерефталамид принадлежит к орторомбической
кристаллической системе с пространственной группой D\a [344] и
следующими размерами элементарной ячейки: а = 22,8 А, Ъ = 5,5 А,
с = 8,1 А. Фениленовые группы расположены параллельно друг
другу, а амидная группа выступает из плоскости. Это приводит
к структурированию полиамидов за счет возникновения
водородных связей. В отличие от поли-я-фенилентерефталамида
ароматические полиамиды с ж-фениленовыми группами в цепи имеют
«волнообразное» расположение фениленовых циклов [345]. Вдоль оси
волокна, чередуясь, расположены бензольные ядра под углом 10
и 20° к оси. Амидные группы располагаются почти
перпендикулярно плоскости бензольных колец, вращение которых
обусловливает низкую симметрию ароматических ж-полиамидов и
вследствие этого незначительную упорядоченность, повышенную
растворимость и пониженную температуру плавления.
Растворимость. Так же как температура плавления и теплофи-
зические свойства, растворимость ароматических полиамидов
зависит от ориентации фениленовых групп, природы мостиковых
атомов и групп в полимерах, полученных на основе многоядерных
дикарбоновых кислот и диаминов, а в случае сополимеров — от
регулярности строения.
Растворимость ароматических полиамидов ухудшается с
увеличением содержания /г-фениленовых групп. Полимеры, полностью
состоящие из фениленовых групп, соединенных в я-положении,
растворяются только в сильных кислотах, таких, как
концентрированная серная и трифторуксусная кислоты. Полиамиды с ж-фени-
леновыми фрагментами растворяются также, в диметилформамиде,
диметилацетамиде, диметилсульфоксиде. В таком порядке сольва-
тирующая способность растворителей понижается. Последняя,
также как стабильность растворов, повышается при добавлении
неорганических солей типа хлорида лития и кальция. Процесс
растворения ароматических полиамидов в системе диметиламид
карбоновой кислоты — галогенид лития представляет собой
конкурентную реакцию амидной группы карбоновой кислоты полиамида
и карбонильной группы амидного растворителя за
координирование с атомом лития. Блокирование мест, акцептирующих
водородную связь в полиамиде, вследствие координации с литием может
422
привести к образованию новых водородных связей с растворителем
[345]:
Li
/ \
I? О R О О
\ ^ Li \ <? ^
С СН
I I
N N
W
D O
\ S
с
|
N
С
N
нс^ /сн3
I чсн3
H
С
I
N
зС/~
В противоположность поли-гс-фенилентерефталамиду
регулярные сополимеры, содержащие только n-фениленовые группы, при
синтезе в смеси диметилацетамида с хлоридом лития находятся
в растворе [334]. В результате кристаллизации при переработке
ароматических сополиамидов в пленки и волокна их растворимость
ухудшается. Затрудненная кристаллизация из раствора в процессе
синтеза объясняется сложным строением элементарного звена
регулярных ароматических сополиамидов. Растворимость
полиамидов с n-фениленовыми группами улучшается при наличии в
диамине пли дикарбоновой кислоте мостиковых групп типа С(СНз)г>
СН2, СО, SO2, PO(C6H5) и РО(СНз) между ароматическими
ядрами (№ 13, 14, 26, 31, 55, 61, 62, 64, 65, 78, 79, 85—100, 107). Эти
полимеры растворяются также в диметилформамиде и ж-крезоле.
Подробное исследование растворов поли-ж-фениленизофталамида
(№ 33) проведено в [301, 302]. Предложены следующие уравнения,
связывающие молекулярную массу и характеристические вязкости
или константы седиментации
диметилформамид, 25 °С
= 5,5- 10~4iM0*74
So=l,9.1OM0-44
диметилформамид +0,25 г LiCl на 100 мл, 25 °С
50 = 2,8 • 10~2М°-39
серная кислота, 96 %-ная, 25 °С
= 1,2 .1(Г3М0'67
Теплота растворения в диметилформамиде А// = —36 кал/г и
в диметилформамиде с добавкой 0,25 г LiCl на 100 мл АН =
= —38 кал/г. В табл. 5.38 приведены размеры клубка и константы
423
/00
J0O 400 5U0 Ш 700 t,C
Рис. 5.65. Кривые термической
деструкции полиамидов в вакууме [269].
Номера на кривых соответствуют номерам
полимеров в табл. 5.35.
взаимодействия поли-ж-фенилен-
изофталамида в растворе диме-
тилформамида и серной
кислоты.
Растворы поли-/г-бечзамида в
диметилацетамиде (№ 1) при
5%-ной концентрации являются
жидкокристаллическими [262].
Концентрационная зависимость вязкости имеет максимум при 5 %.
Оптическая анизотропия и дихроизм в области концентраций 5—
10 % объясняется параллельной укладкой молекулярных цепей.
Термическая и термоокислительная деструкция. Стойкость
ароматических полиамидов к термической и термоокислительной
деструкции определяется ориентацией фениленовых циклов в цепи,
природой мостиковых атомов и групп атомов в полимерах,
полученных из многоядерных дикарбоновых кислот и диаминов, а для
сополиамидов—регулярностью их строения [269, 276, 293, 315,
346]. Ароматические полиамиды с о-фениленовыми группами в
инертной среде разлагаются уже при 250 °С. Температура начала
деструкции полиамидов с м- и /г-фениленовыми группами
колеблется от 350 до 500 °С. При этом стойкость возрастает в ряду м, м <;
< м, п < п7п.
На рис. 5.65 приведены значения потери массы для девяти
полиамидов в вакууме. Сополимеры регулярного строения (см.
табл. 5.36) значительно стабильнее соответствующих нерегулярных
полиамидов [329, 347]. В зависимости от строения заместителей и
других групп по убыванию стабильности в условиях термической
Таблица 5.38. Размеры клубка и константы взаимодействия
поли-лс-фениленизофталамида [302J
M
см. 10
-8
CM-IQ-8
Длина
сегмента,
Коэффициент
Гщльден-
брацдта
X
Константа
Хаггннса
1,3
1,2
1,4
0,9
1,8
Диметил форм амид
1,3 0,7 35
2,8
Диметилформамид + 0,25 г LiCl иа 100 мл
1,2
1,4
29
Серная кислота, 96%-ная
0,6 | - | - | 39
2,6
8,0
0,1
-0,2
424
деструкции в интертнои среде полиамиды можно расположить в
ряд:
\сн3
-<О>—2-<О>- ~
-Qr- >
о
СНз
r
СНз
Температура разложения на воздухе ароматических
полиамидов с /г-фениленовыми группами лежит в интервале 380—450 °С.
В зависимости от характера групп в звене полиамида стабиль*
ность на воздухе уменьшается в следующем ряду:
СНз
\
/
СНз
Нз
425
O)-N=C=N
I
-N=C: 4-
Внсококонденсиро-
ванная карбо-гете-
роциклическая
система
HCN Ч-
ОУ-СО-1-NH^O
OVCONH —
OVCONH2 -z
+
Высококонденси-
рованыая
ароматическая
система
Высококонденсированнад
CH4 + ароматичеекая
система
426
Ароматические полисульфонамиды (№ 108, 109) стабильны на
воздухе и в инертной среде до 350°С. При 600°С потеря массы
составляет на воздухе 95 % и в вакууме 40% [320].
Полимеры № 14 н 110 выпускаются в СССР в
полупромышленном масштабе под марками 4Т и СИ [412]. При деструкции
ароматического полиамида, содержащего только м- и я-фениленовые
циклы и амидные группы, выделяются СО2, СО, Н2О, HCN, метан,
а также следы воды, бензола, толуола, анилина и бензонитрила.
В твердом остатке обнаружены фенилендиамин, аминобензанилид
и олигомеры [276, 285, 304, 346, 347, 349]. При 375 °С до 85 %
летучих продуктов деструкции составляет СО2. В интервале
температур 375—450 °С содержание СО2 в газообразных продуктах
деструкции уменьшается, а количество СО достигает
максимального значения (около 45%). В области температур 450—550 °С
образуется до 65 % водорода и 12 % HCN.
Деструкция ароматических полиамидов начинается с разрыва
связи между ароматическим кольцом и NH-группой [347]# За счет
отрыва водорода от фениленовых групп образуется концевая амид-
ная группа, дегидратация которой приводит к получению нитрила.
В свою очередь отщепляющаяся вода гидролизует амидную связь
с образованием концевых амино- и карбоксильных групп.
Основной продукт деструкции при низких температурах ССЬ, который
выделяется вследствие декарбоксилирования таких карбоксильных
групп. Для процесса образования летучих продуктов деструкции
полиамидов, содержащих только фениленовые и амидные группы,
предложен следующий механизм (см. с. 426) [347].
Энергия активации деструкции ароматических полиамидов
составляет 38—50 ккал/моль в инертной среде и 30—50 ккал/моль
на воздухе [276]. Энергия активации и температура разложения
(см. табл. 5.36) при термоокислении ароматических полиамидов
заметно ниже, чем при деструкции в инертной среде. Проведение
окисления в среде 18О показало, что в СО2 и Н2О, образующихся
в результате деструкции, этот изотоп не содержится [350]#
Следовательно, в ходе деструкции в присутствии кислорода окисление не
происходит. Ускорение термической деструкции в присутствии
кислорода связано с тем, что он как парамагнетик увеличивает
вероятность перехода ароматического полиамида в триплетное состояние.
5.10.3.3. Переработка и свойства
ароматических полиамидов
Поли-ж-фениленизофталамидные волокна. Поли-ж-фениленизо-
фталамидные нити или волокна выпускаются в промышленном
масштабе в США фирмой «Du Pont» под названием «Номекс» и
в СССР под названием «фенилон». Исследовательские работы в
этой области проводились в США с конца 40-х годов. Выбор для
промышленного производства именно этого волокна связан с
удачным сочетанием его хорошей перерабатываемое™ и сравнительно
высокой стойкости к термоокислительной деструкции. Более термо-
427
стойкий полиамид с я-фениленовыми фрагментами в цепи нельзя
переработать из растворов в органических растворителях, например
в диметилформамиде и диметилацетамиде. Для формования
волокна из поли-ун-фениленизофталамида используеот реакционные
растворы полимера в диметилацетамиде с добавкой LiCl или растворы
полиамида в диметилформамиде с LiCl.
В случае сухого формования раствор поли-ж-фениленизофтал-
амида в смеси 95% диметилформамида и 5% LiCl при 128 °С
впрыскивают в прядильную шахту при 225 °С (горячий воздух).
Скорость вытяжки конечного волокна составляет 80 м/мин. После
вытягивания в 4—5 раз волокна кипятят в воде для удаления LIC1
[327]. При формовании из раствора используют растворы в
диметилацетамиде с 2,2 % СаСЬ. Осадительной ванной служит 53 %
водный раствор СаС12 при 72 °С [292]. Кристаллизация аморфного
волокна осуществляется при вытяжке в парах в течение 1 ч под
давлением 7 кгс/см2 [282] или термообработкой ориентированного
волокна 1—3 с при 300—350 °С [334, 351, 352]. Поли-ж-фенилен-
изофталамидное волокно обладает длительной термостойкостью
до 230 °С. Физико-механические свойства поли-ж-фениленизофтала-
мидных волокон приведены ниже [353, 354]:
Феннлон Номекс
Прочность при растяжении, г/денье
при 20 °С, 65%-ная отн. влажность. . 5,0 6,5
при 20 °С, мокрое 4,1
при 250 °С 3,1
при 300 °С 3,1 1,2
Относительное удлинение при разрыве, %
при 20°С, 65%-ная отн. влажность. . 16—20 28
при 20 °С, мокрое 14
при 250 °С 22
при 300 °С 23
Начальный модуль упругости, г/денье
при 20 °С . г ИЗ
при 250 °С 63
при 300 °С 75
Прочность при растяжении при 300 °С составляет еще 40 % от
исходного значения (рис. 5.66), относительное удлинение при
разрыве и начальный модуль упругости — 82 или 55 % от исходных
значений соответственно.
После 100 ч выдержки волокна в сухом воздухе при 300 °С
остаточная прочность составляет 55 % (рис. 5.67). Волокно
обладает высокой стойкостью к кислотам и щелочам даже при
высоких температурах [355]. За 100 ч выдержки при комнатной
температуре в 10 %-ной H2SO4, 70 %-ной H2SO4, 10 %-ной HNO3, 10
о/оной NaOH и 28 %-ной NH4OH прочность при растяжении не
меняется. Прочность при растяжении снижается на 50 % через 100 ч
выдержки в 70 %-ной HNO3 или 35 %-ной НС1 при комнатной
температуре или через 8 ч —в 10 %-ной НС1, 70 %-ной H2SO4 или
10 %-ной NaOH при 95 °С. Действие водяного пара при 155 °С
через 100 ч приводит к уменьшению прочности на 40 % и через
1000 ч на 70 %•
428
Рис. 5.66. Зависимость
остаточной прочности при
растяжении поли-лг-фенилен-
изофталамидных волокон о г
темпсратуры [353].
Рис. 5.67. Зависимость
остаточной прочности при
растяжении поли-льфенилен-
изофталамидпых волокон от
продолжительности
термообработки при 250 и 300°С
[353].
Q 20 40 60 60
100 200 3001°0
Поли-лг-фениленизофталамидные волокна обладают очень
высокой стойкостью к действию ионизирующих излучений (табл. 5.39).
При дозе около 1000 Мрад прочность и относительное удлинение
практически не изменяются.
При контакте с открытым пламенем волокно горит, однако вне
пламени гаснет без образования капель. Стойкость к возгоранию
можно увеличить отжигом ароматического полиамидного волокна
при 250 °С с последующим нагреванием в течение 1 ч на воздухе
при 425 °С [356], Окрашенное в черный цвет волокно негорюче, но
имеет более низкие физико-механические показатели.
Предполагается, что при высокотемпературной обработке происходит
отщепление водорода и сшивание полимера.
Содержание влаги до 4 % благоприятно влияет на текстильные
свойства волокна. Влага удаляется при нагревании в интервале
температур 50—150 °С. С помощью оксида трития было показано,
что значительно ниже температуры стеклования поли-лг-фенилен-
изофталамида водообмен происходит с высокой скоростью [345].
Ароматические полиамиды применяют для изготовления
огнезащитной одежды, шин и приводных ремней, для
электроизоляционных тканей, для фильтрации газов и горячего воздуха.
По термостойкости волокна на основе поли-ж-фениленизо-
фталамида уступают полисульфонамидным. Так, на основе
продукта взаимодействия терефталевой кислоты с бис(л-аминофе-
нил)сульфоном (см. табл. 5.35, № 14) из раствора в диметил-
формамиде получено волокно с верхней температурой
эксплуатации 300 °С. С 1969 г. это волокно производится на пилотной
установке ВНИИВ в СССР под названием «сульфон Т» [425].
Таблица 5.39. Радиационная стойкость
поли-ж-фениленизофталамидиого волокна [299, 303]
Доза, Мрад
0
8-107
Прочность
при растя-
женин,
г/денье
5,92
5,85
Относительное
удлинение
при разрыве,
%
11,2
12,5
Доза, Мрад
1,9.10е
1,4-109
Прочность
при
растяжении,
г/денье
5,74
5,90
Относительное
удлинение
при разрыве,
°/о
12,1
11,7
429
Бумага из поли-Л!-фениленизофталамида. Фирмой «Du Pont»
методом спекания [357] из коротких волокон Номекс и
измельченного пленочного материала получена высокотеплостойкая
бумага, применяемая в качестве изоляционного материала.
Непористый материал имеет плотность 0,7—1,0 г/см3. При толщине
бумаги 0,5 мм прочность при растяжении составляет 30 кгс/см2,
относительное удлинение при разрыве 19 %, усадка при 285 °С
0,3% [358]. При 200 °С удлинение не изменяется, уменьшение
прочности при растяжении при 225°С составляет 32%.
Удельное объемное электрическое сопротивление не зависит от
толщины бумаги и составляет 1,3* 1016 Ом-см; при 250 °С оно
снижается до 1011 Ом-см. Диэлектрическая проницаемость в интервале
температур 0—200°С при частоте 102—10" Гц изменяется в
пределах 2,8—3,4. Электрическая прочность при 250 °С составляет
95 % от исходного значения. Верхняя температура длительной
эксплуатации (снижение прочности при растяжении за 10 лет
на 50%) равна 220 °С. Покрытая полиамидом бумага может
подвергаться горячей сварке. Бумагу используют главным
образом в качестве изоляции в электродвигателях и
трансформаторах.
Пресс-массы на основе поли-л«-фениленизофталамида. Поли-уИ-
фениленизофталамид типа фенилона можно перерабатывать
прессованием C20—330°С, 15—20 мин). Переработка литьем под
давлением и экструзией невозможна вследствие высокой
вязкости расплава E-107Пз) [306,359].
Поскольку при 330 °С протекает деструкция полимера, для
переработки прессованием в состав композиции следует вводить
стабилизаторы — фосфор- и борорганические соединения, а
также бисфенолы и алкилфенолы [304]. Если переработку проводят
при температурах, близких к температуре стеклования, то пресс-
изделия обладают неудовлетворительными
физико-механическими свойствами. Введение 1 % бисфенола приводит к
уменьшению нерастворимой части в процессе термоокислення на воздухе
при 330 °С в 2 раза. Другая возможность стабилизации
заключается в использовании в синтезе поли-ж-фениленизофталамида
бромизофталевой кислоты [298]. Введение в сополимер 0,025—
0,05 % (мол.) кислоты уменьшает поглощение кислорода при
340 °С вдвое. Наличие атомов галогена в цепи не влияет на
термоокислительную деструкцию полимера. Кристаллизация
происходит при 340 °С за 40 мин. Этот процесс можно существенно
ускорить добавлением структурообразователей, например оксида
титана. Скорость окисления кристаллического полимера вдвое
меньше, чем аморфного.
На стр. 431 приведены свойства изделий из поли-ж-фениленизо-
фталамида в интервале температур от — 70 до 250 °С [306, 307].
При 250 °С прочность при растяжении составляет 630 кгс/см2,
или 61 % от исходной. За 100 ч при 270 °С прочность и
удлинение практически не изменяются. После выдержки изделий в воде
430
Плотность, г/см3 1,33—1,36
Температура стеклования,
°С \ ш 270-280
Температура хрупкости, °С —70
Температура плавления
кристаллической фазы, °С . 430
Теплостойкость по Вика,
°С 270
Теплопроводность, ккал/(м-
•ч-°С)
при 20 °С 0,16
при 50 °С 0,17
при 100 °С 0,20
при 150 °С 0,22
при 200 °С 0,24
Удельная теплоемкость,
ккал/(кг-°С)
при 20 °С 0,34
при 50 °С 0,39
при 100 °С 0,46
при 150 °С 0,52
при 200 °С 0,58
Прочность при растяжении,
кгс/см2
при —70 °С 1 270
при 20°С . . . . 1 030
при 120 °С 818
при 250 °С 630
Относительное удлинение
при разрыве, %
при — 70 °С 5
при 20 °С 5
при 120 °С 5,8
при 250 °С 6,6
Прочность сжатия, кгс/см2 . 3 200
Модуль упругости при
сжатии, кгс/см2 44 000
Твердость по Бринеллю,
кгс/мм2 34
Ударная вязкость, кгс-
•см/см2 20-35
Удельное объемное
электрическое сопротивление,
Ом см
при 20 °С 2 - 10!*
при 200 °С 2- 1С14
при 250 °С 6.10'2
Электрическая прочность,
кВ/мм
при 20 °С 65
при 200 °С 22
при 250 °С 19
при комнатной температуре в течение 10 сут прочность при
растяжении снижается на 11 % и относительное удлинение при
разрыве на 4 %. Пресс-изделия из поли-лг-фениленизофталамида
стойки к действию алифатических углеводородов,
трансформаторного масла, концентрированной соляной, 98% -ной уксусной
кислоты и 40%-ного раствора КОН. Однако после 165 ч
выдержки в 70 %-ной серной или 57 %-ной азотной кислоте при
комнатной температуре прочность при растяжении понижается
соответственно на 50 и 30 % • Д° 250 °С удельное объемное и
поверхностное электрическое сопротивление почти не изменяются,
электрическая прочность при этом уменьшается до 30 % от исходного
значения. По антифрикционным свойствам поли-лг-фениленизо-
фталамид превосходит бронзу [309]. При введении 40% графита
температура переработки понижается до 280 °С [360]. Изделия
из такой композиции имеют прочность при изгибе 387 кгс/см2 и
модуль упругости при изгибе 79 400 кгс/см2.
Высокомодульное волокно. Наряду с поли-л^-фениленизофтала-
мидом как исходным продуктом для получения волокон и пресс-
изделпй, для изготовления высокомодульных волокон большое
значение приобрели и другие ароматические полиамиды с я-фени-
леновыми группами (табл. 5.40). Среди этих полимеров
полиамиды, содержащие только л-фениленовые группы, имеют
наибольшую плотность упаковки, высокие степени ориентации и
кристалличности. Это позволяет получать материалы, отличающиеся по-
431
»в*
§3
AS
о
§d
'g
О
о о
Прочность
при растяжении
г/денье
GO
00
Относительное
удлинение
прн разрыве, %
ff)
¦M
О
О
Модуль
упругости, г/денье
мимо высокой термостойкости высокими модулем (в 3—б раз
выше) и прочностью (в 2—3 раза выше) по сравнению с поли-л*-
фенилен-изофтал амидом. При этом модуль полиамидов,
содержащих /г-фениленовые фрагменты, выше, чем для стеклянного
волокна C2,6 г/денье) и стальной проволоки B80 г/денье).
Рассчитанные по прочности связей значения модуля упругости
составляют Для поли-/г-бензамида и поли-«-фенилентерефталами,да
1500 г/денье [420]. Как видно из табл. 5.40, найденные значения
составляют соответственно 1040 и 770 г/денье.
По сравнению с волокнами на основе ароматических
полиамидов с «-замещенными фениленовыми группами стеклянные
волокна имеют низкую износостойкость, существенно
различающиеся термические коэффициенты линейного расширения волокна
и полимерной матрицы, ограниченную совместимость и высокую
плотность. Недостатком органических высокомодульных волокон
является анизотропия физико-механических свойств.
Поли-я-бензамид получают поликонденсацией хлоргидрата п-
аминобензоилхлорида в растворе хлорида лития в тетраметилмо-
чевине или диметилацетамиде; осадительной ванной служит вода
(в процессе формования) [260, 261, 361, 362, 413, 414]. Требуемая
вязкость прядильного раствора достигается его термической
обработкой. Поли-я-бензамид выпускается в США фирмой «Du Pont»
под названием «Файбр В» или «Кевлар». Объем его
производства (преимущественно для шинного корда) составил в 1977 г.
25 000 т. Поли-я-фенилентерефталамид в виде нагретого до
80—100 °С раствора в серной кислоте осаждается в
охлажденную до 1 °С воду. Волокна промывают водой при 50°С и
подвергают ориентации при 250 °С [363, 415]. Еще в 1972 г. фирма «Du
Pont» довела производство органических высокомодульных
волокон марки PRD-49 до 50 000 т [416,417]. Волокно представляет
собой, вероятно, модифицированный поли-п-бензамид или смесь
поли-«-бензамида с поли-п-фенилентерефталамидом. Прочность
при растяжении его волокна почти в 2 раза выше B5000 кгс/см2),
чем у полиамида 6,6; относительное удлинение при разрыве 2%.
Модуль упругости A30 000 кгс/см2) почти в 20 раз выше, чем у
полиамида 6,6, и вдвое больше, чем у стеклянного волокна.
Плотность A,45 г/см3) на 40 % ниже, чем у стеклянного
волокна.
Это волокно в виде ткани, лент, ровницы, рубленых прядей
используют в авиации, ракетной и космической технике,
машиностроении, электронике и при изготовлении спорттоваров. В
авиации высокомодульное волокно PRD-49 используют как
армирующий наполнитель с эпоксидными, фенольными, полиэфирными и
полинмидными связующими для изготовления несущих
конструкционных элементов, например облицовки соединения крыла с
корпусом самолета и винта с держателем, обтекателей, элементов
облицовки стен кабины, а также конструкционных элементов,
испытывающих высокие нагрузки, например роторов
вертолетов,
433
Композиции с эпоксидными смолами при 175 °С сохраняют
еще 73 % исходной прочности при комнатной температуре;
модуль упругости при этой температуре не изменяется [418]. Цена
1 кг волокна составляет 15—200 долл. США. Несмотря на то что
высокомодульное PRD волокно значительно дороже, чем
обычное стеклянное волокно, его использование в композиционных
материалах позволяет понизить массу конструкционных
элементов и, следовательно, получить выигрыш в удельной прочности
по сравнению с алюминием и материалами, армированными
стеклянным волокном. Уменьшение массы самолета сопровождается
увеличением дальности полета и полезной нагрузки и
уменьшением расхода горючего. Применение PRD-49 для изготовления
высоконапряженных конструкционных элементов ограничено
низкой прочностью при сжатии.
Хорошие результаты достигаются при использовании
композиций с углеродными и борными волокнами. Основные области
применения—производство спорттоваров (теннисных ракеток,
лыж, удилищ и клюшек для гольфа). В СССР получено
высокотермостойкое волокно вниивлон с модулем 930 г/денье [421].
Высокомодульное волокно на основе полиамида с /г-фрагмен-
тами в цепи вырабатывается в СССР на пилотной установке
ВНИИВ под названием «терлон» [424]. Оно получается методом
мокрого формования из раствора полимера в концентрированной
серной кислоте. Волокно используется для армирования
полимеров и изготовления технических тканей, работающих при
высоких температурах.
5.10.4. ГЕТЕРОЦИКЛИЧЕСКИЕ СОПОЛИАМИДЫ
Ароматические полиамиды с гетероциклами в цепи имеют
лучшую растворимость по сравнению с ароматическими
полиамидами с карбоциклическими группами.
В табл. 5.41 приведены температуры плавления и разложения
сополиамидов с имидазолидоновыми (№ 1 и 2), гидантоиновыми
(№ 3—8), тиазольными (№ 9—19), оксадиазольными (№ 20—
31), тиадиазольными (№ 32), триазольными (№ 33,34), бензи-
мидазольными (№ 35—37), бензоксазольными (№ 38—46), бенз-
тиазольными (№ 47), хиназолоновыми (№ 48—50), хиназолин-
дионовыми (№ 51,52), бензоксазиноновыми (№ 53,54), бензокса-
зиндионовыми (№ 55), феноксасульфидными (№ 56), фенокса-
сульфоновыми (№ 57), фенантридиновьши (№ 58) и карборано-
выми циклами в цепи полиамида. Полиамиды с имидными
циклами— полиамидоимиды — описаны в 7.1.3.
5.10.4.1. Получение
По аналогии с синтезом сополимеров с карбоциклическими
ароматическими группами полиамиды с гетероциклическими
звеньями в цепи синтезируют в растворе взаимодействием дихлоран-
434
гидридов дикарбоновых кислот с диаминами, которые уже
содержат в молекуле гетероцикл [341, 343, 364, 365, 369, 372, 379, 389,
392]:
N N
NH2
ЯС1ОС—
NHOC—
СОС1
СО-
Получение полимеров с высокой молекулярной массой
обеспечивается добавлением к растворителям (диметилацетамид, гек-
саметилфосфотриамид, N-метилпирролидон) хлоридов лития или
кальция. Раствор сополимера непосредственно из реактора
поликонденсации поступает на формование волокна. Для синтеза
сополимеров пригоден также метод, предусматривающий
использование в качестве исходных соединений продуктов, содержащих
амидные группы [377, 390, 406—409]:
CONH
NHOC
соо-0
Н2 N
H2N
NH2
CONH
n
5.10.4.2. Свойства
Ароматические полиамиды с гетероциклами в цепи в таких
растворителях, как диметилацетамид, диметилформамид, гекса-
метилфосфотриамид, N-метилпирролидон, растворяются с
добавкой солей LiCl или СаС12. Сополимеры, содержащие в цепи
также "простые эфирные и сульфоновые группы, растворяются в этих
растворителях без добавления солей. В отличие от сополимеров
с карбоциклическими группировками аналогичные продукты с
гетероциклами в цепи имеют преимущественно кристаллическую
структуру. Температура плавления сополимеров регулярного
строения, содержащих битиазольные (№ 9—13), тиазольные (№ 18)
или бензоксазольные (№ 38—46) звенья, лежит около 500 °С.
435
Таблица 5.41. Температуры плавления и разложения гетероциклических полиамидов
1
2
3
4
л
-NH—({
о \-
-С—(Г
О
о-
1—f°
. 1 1
-г0
1
^N—NH—С—<^QN R = —H,—СНз
О
,со—
О
R=—\О/—СН2 (О/ • [Qi • (О/
^NH—
j
>—о—А
T °C
A разл1
300
Литературный
источник
[364]
[364]
[363]
[366]
[365]
СНз
о-
-€>-¦
282
НзС СНз
_NH—(( )) О
q^ НзС СНз
—СО—[Tj]—CO-NH
Os
СНз
НзС СН;
о
о
[365]
[367]
Продолжение табл. 5.41
—n;
10
11
—СО frjj СО—NH
-CQ-
„со—
'—(( )>—со-
О
540
535
575
490
485
[369]
[369,
370-372]
[369]
12
13
14
15
16
—NH
—_
N
-—NH—(^
ГН]
-NH— С Y) —,—N N—n— X Y>— NH-G— С Y> -S02—
¦со—
-NH.
-NH-®-
Jl
о
о
с—
535
520
340
525
475
490
475
540
540
520
475
C69]
[369]
C69J
I.369J
Продолжение табл. 5.41
Т °С
ггтт»
Гразл. °
Литературный
источник
18
19
—NH—R-
—NH
20
21
22
с—
II
о
-R
О
345
470
380
400
465
490
450
[365]
[340, 343]
[374, 375]
[374, 375]
[375]
23
N N
О
N
24
25
N N
26
—NH-
N N
О
N N
C
О
i
27
28
N N
N N
О О
NH—С—(СН2L—С-
e I
) NH-C-(CH2)8-C-
II II
О О
400
515
465
>350
>350
>350
480
440
300
300
300
[375]
[340, 343
276]
[340, 343
[373]
[373]
[373]
Продолжение табл. 5.41
№
29
N N -.
—NH—R—II J—R'—NH—C—[Г)!—С—
0 « ^ II
0 0
Tnn- 'C
1 разл* ^
Литературный
источник
[365]
R= —
ск
/Cl
30
31
/
Cl
Cl
Cl
\
—NH.
—NH—CH2—
N N
IL 1
XX
o
о
370
350
490
[276, 343,
370]
[376]
32
—NH—R-^SnT ^R
R =
'-C—,-Я^—С—
il KJJ il
о ^-^ о
33
34 I
—NH-
NH—С
II
450
[3651
455
[343]
[365]
N
n^\r'-nh-c—(QJ—c
/\
P
N
^ 35
со
N
\co—
[378]
38
40
Продолжение табл. 5.41
№
36
37
N
^У HN
N
370
Гразл* *G
410
Лнтературный
источник
[343]
[377]
NH-C
О
/
О
jy-
N
(СН2)8
О
о
N
о
-NH—С.
A
380
390
[3811
[381]
41
42
43
—NH
—NH
44
45
46
—NB
47
NH— С— (СН2)8~С—
А А
Ш— С—(СН2N—С—
11 II
о о
—С—(Q) C~
NH—С—(СН2)8—С—
I! II
О О
N
с—
II
о
530
> 500
350
170
234
180
455
530
450
450
375
450
475
Продолжение табл. 5AI
°С
Т °С
разл'
Литературный
источник
48
R-NH-C
[365]
49
50
о
—NH
¦ -<о>-о-(о
о
R =
—C2H5, —СНз
о
о
[365]
о
—NH
51
521
—NH 1
NH
О
,R—NH— C-
If
О
о
«--
N
NH—С
53
—NH
-о-
-N
О
О
54
N
О
\
[365]
1365]
О О
|384]
[383]
Продолжение табл. 5AI
58
59
60
61
62
63
CD
/Cl
R =
—NH>
N
Ar— C—
О
—С—С2В4Н! о—С—NH
О
О
—С—C2B4Hio—С—NH—(Г))—;
II II \^У
о о
NH—
—С-С2В4Н10—С—NH (Y ))—CH2-
It II
О О
—С—С2ВюН,о—С—NH 1
А 4
;—NH—
.—NH—
С
—С—C2B10Hi0— С—NH— (СН2N—NH—
!1 II
о о
7V = 350
= 480
7V = 300
420
400
400
400
400
230
400
Таб лица 5.42. Физико-механические свойства гетероциклических ароматических полиамидов
Элементарное звено
Прочность
при
растяжении,
г/денье
Относительное
удлинение
при
разрыве,
Модуль
упругости,
г/денье
—NH—I
N N
N-
riLJ
У с о
о о
О
N N
г
О
) NH—С
II
О
6,7
7.8
4>3
11,0
6,6
4,5
114
168
—NH.
-NHv
О
о
N
О
N
N
N
О
Ч
N
о
II
N
N
,ш—с
ЮГ о
9
3,5
5,4
2,2
2,5
5,5
6,0
7,8
154
56
[365]
[364]
[379]
[379]
бр,г/'деньег
бр,г/'денье
7
О 100 200 300 400 SQOt,°C
О 40 80 ПО
Рис. 5.68. Зависимость прочности при растяжении (J) и относительного
удлинения при разрыве B) полибитиазоламидного волокна (№ 10) от температуры
[3401.
Рис. 5.69. Кривые термической деструкции на воздухе полиамидов с оксадиа-
зольными и тиазольными циклами в цепи [340];
N N
1 —
о
о -I
п
N N
3-
_NH_(O>— Ц
Наличие в структуре мостиковых простых эфирных или сульфо-
новых групп приводит к снижению температуры плавления.
Способность к переработке, термическая стабильность и
жесткость делают потенциально возможным использование
полиамидов с гетероциклами в цепи для изготовления
высокотермостойких и высокомодульных волокон [404]. Они могут
перерабатываться методом сухого и мокрого формования. При сухом
формовании должна быть предусмотрена предварительная стадия
экстрагирования остатков соли, содержащейся в
поликонденсационном растворе, для получения волокна с оптимальными
физико-механическими и термическими свойствами. Полимеры,
содержащие гетероциклические звенья, при комнатной температуре
имеют хорошие физико-механические свойства (табл. 5.42).
Остаточная прочность при высоких температурах и стойкость к
термоокислительной деструкции этих полимеров выше, чем
полиамидов с карбоциклами в цепи. Полибензтиазольные волокна с амид-
ными группами отличает высокая термическая стойкость при
действии кратковременных A—5 мин) нагрузок. Остаточная
прочность при 450 °С составляет 28% исходной прочности (рис. 5.68).
Светостойкость и стойкость к окислительной деструкции поли-
452
бензтиазоламидных волокон, однако, невелика. Уже после 2 ч
нагревания на воздухе при 300 °С происходит уменьшение
прочности при растяжении на 44 %, относительного удлинения при
разрыве на 52 % и модуля упругости на 83 % [372].
В качестве стабилизаторов для полибензтиазоламидов
используются соли Си11 и Си1, например ацетат Си11. После 2 ч
нагревания на воздухе при 300 °С при содержании ацетата меди 1,8%
остаточная прочность при растяжении составляет 74 %,
относительное удлинение при разрыве — 93 % и модуль упругости —
88% [371].
Наиболее высокую стойкость на воздухе имеют
гетероциклические полиамиды с феноксасульфидными и хиназолиндионовыми
группировками (соответственно полимеры № 56 и № 51,52). Так,
через 500 ч нагревания на воздухе при 250°С волокна на
основе полимера с феноксасульфидными группами в цепи имеют
остаточную прочность 90% [364], а полиамиды с
хиназолиндионовыми циклами — 50 % [365]. На рис. 5.69 показало изменение
прочности в процессе термообработки для полиамидов с оксадиазоль-
ными и тиазольными группами. Более высокая стойкость к
окислительной деструкции полиамидов с гетероциклическими
группами по сравнению с полимерами с карбоциклическими звеньями
объясняется меньшим содержанием нестойких к окислению амид-
ных групп [340]. Полибензимидазоламиды более пригодны для
склеивания специальных сталей, чем полибензимидазолы [377].
Прочность при сдвиге после 100 ч нагревания на воздухе при
300 °С клеевого шва при склеивании специальных сталей
составляет 1,6—1,8 кгс/см2. Свойства полиамидов с имидными
циклами приведены в 7.1.3.
5.10.5. АРОМАТИЧЕСКИЕ ПОЛИАМИДОГИДРАЗИДЫ
Ароматические полиамидогидразиды
Г-HN—<1Q|)—CONHNHOC—(Q)—CO-j
в промышленном масштабе производятся фирмой «Monsanto».
Полиамидогидразидные высокомодульные волокна пригодны для
изготовления наружных слоев корда, специальных защитных
тканей, в качестве наполнителей в высоконагружаемых изделиях.
5.10.5.1. Получение
Регулярные полиамидогидразиды. Стереорегулярные
полиамидогидразиды получают из диаминов с симметрично
расположенными гидразидными группами и дихлорангидридов ароматических
453
дикарбоновых кислот [392].
H2N
п
CONHNHOC
CONHNHCO
—2HCI
COCI
--HN
CONHNHOC
CONHNHOC
Поликонденсацию проводят в растворе в диметилацетамиде в
присутствии 5% хлорида лития при —10—0°С. Симметричные
диаминогидразиды получают взаимодействием нитробензоилхло-
рида с гидразидом изофталевой или терефталевой кислоты (или
их дихлорангидридов) либо реакцией дихлорангидридов с
гидразидом нитробензойной кислоты с последующим восстановлением
образующихся динитросоединений [392] :
H2NNHCO
CONHNHa
CONHNHa
O2N ;
-COCI
ГЛЛ, CONHNHOC-
-COCI
"©
-CONHNHOC-
"NOS
I"
H2N-
CNHNHC
A
A
,—CNHNHC-
II II
О О
,—NH2
Диаминодигидразид с д-фениленовыми группами получают
также прямым взаимодействием 1 моля терефталоилхлорида с 2
молями /г-аминобензгидразида [410]. Температура плавления
симметричных диаминов в зависимости от орие-нтации трех фени-
леновых групп находится в пределах 150—300 °С.
Полиамидогидразиды с ограниченной регулярностью. При
одностадийном взаимодействии /г-аминобензгидразида с терефталоил-
454
хлоридом могут возникнуть два типа регулярных структур:
или
[—HN—i(})—С—NHNHC ((у,—С—1
о о о J
[—NHNHC—(С^)—NHC—/Г^)—С—1
о о о Jn
присоединение «голова к хвосту»;
[— HN— (О\)—CNHNHC—(С))—CNHNHC—(С))—NHC—(С))—С-1
^у il il ^У i il у^у il <^/ и
О О О О О 0-1
присоединение «голова к голове» и «хвост к хвосту».
Если к раствору терефталоилхлорида в тетрагидрофуране
добавить раствор /i-аминобензгидразида в диметилацетамиде при
0°С, то поликонденсация будет протекать в режиме
саморегулирования. Образующиеся in situ симметричные диамины,
чередующиеся в положении «голова к голове» и «хвост к хвосту», с
избытком терефталоилхлорида дают полиамидогидразид [393—395,
4101:
H2NNHOC П. )\ NH2
H2N—(Су)—CNHNH2 + CfC—(С))—CCI ->
NC^7 |j II \^У j| -HGl
0 0 0
—»- «H2N—(Cy)—CNHNHC—<(Y>—CNHNHC—((y)—NH2
Х\^У I! Il \^r/ II H \^У
\\ | ||
О О О О
C1C (( )) CCI
о ^ о
_HN—/(^—CNHNHC—(())—CNHNHC—(П- NHC—('Г^—С-
V^7 j| || X4^/ || || X^V || ^ ||
о о о о о о _п
Высокая селективность процесса поликонденсации и
регулярность полиамидогидразидов обусловлены более высокой
реакционной способностью гидразидных групп /i-аминобензгидразида в
реакции с хлорангидридами по сравнению с аминогруппами. При
высокой скорости перемешивания структурная неоднородность
полимера незначительна (реакция избытка дихлорангидрида дикарбо-
новой кислоты с аминогруппой /i-аминобензгидразида). Если,
напротив, твердый терефталоилхлорид вводят в раствор /г-аминобенз-
гидразида в диметилацетамиде, то процесс поликонденсации почти
перестает зависеть от различия в реакционной способности амино-
и гидразидных групп и начинает диффузионно контролироваться
455
поверхностью терефталоилхлорида. Селективность реакции и
регулярность образующегося при этом полиамидогидразида очень
низки [394]. Структура олигомеров, образующихся при
взаимодействии терефталоилхлорнда с я-амннобензгидразидом в различных
условиях, исследована методом ЯМР [395]. Престон [404] получил
сополимеры, которые наряду с амино- и гидразидными группами
содержат в цепи оксадиазольные и бензоксазольные циклы.
5.10.5.2. Физико-химические свойства
Ароматические полиамидогидразиды не имеют четкой
температуры плавления, так как в интервале температур 250—300 °С
происходит их циклодегидратация с образованием полиоксадиазо-
лов. Волокна и пленки, полученные из раствора, имеют
желтоватый цвет и степень кристалличности 20—30 %. Горячая вытяжка
при 300 °С позволяет увеличить степень кристалличности до 90 %.
В пленках, приготовленных поливом из раствора, молекулярные
цепи предпочтительно ориентированы к плоскости пленки.
Псевдогексагональная элементарная ячейка полиамидогндразида на
основе терефталоилхлорида и я-аминобензгидразида включает два
элементарных звена и имеет следующие размеры: а — 8,5 А; Ь —
= 4,9 А, с (ось цепи) = 29,6 А, теоретическая плотность составляет
1,51 г/см3 [397, 398].
Полностью «упорядоченные» (регулярные)
полиамидогидразиды с высоким содержанием я-фениленовых групп растворяются в
N-метилпирролидоне и диметилацетамиде с добавкой 5 % хлорида
лития. Полностью упорядоченные полимеры с преобладанием м-фе-
ниленовых групп растворяются в диметилацетамиде,
N-метилпирролидоне и диметилсульфоксиде [392], Полиамидогидразиды с
ограниченной регулярностью, синтезированные из
терефталоилхлорида и я-аминобензгидразида, обладают более высокой
растворимостью в смеси диметилацетамид — LiCl, чем полностью
упорядоченные [393].
Растворимость полиамидогидразидов улучшают введением в их
структуру оксадиазольных и бензоксазольных фрагментов [404].
Изучены свойства растворов полностью упорядоченных
полиамидогидразидов на основе терефталоилхлорида и я-аминобензгидра-
зида [399]. Благодаря наличию я-фениленовых фрагментов в поли-
гидразидной цепи макромолекулы в растворе образуют сильно
проницаемые клубки. Для раствора в диметилсульфоксиде при
25 °С уравнение Марка—Хаувинка—Куна имеет вид:
[Л] = 6,15 - Ю-5^1'06
При средних значениях молекулярной массы
характеристическая вязкость имеет очень большую величину. Так, при
характеристической вязкости 9,0 (дл/г) молекулярная масса составляет
всего 73 000. Разрушение или агрегация не происходят при
измерении вязкости в интервале [i\] = 3—8 (дл/г). Размеры клубка
г2/М » 10"8А растворенной макромолекулы следует отнести за счет
456
необычно низкой гибкости цепи. Для светорассеяния используют
зеленый свет E46 1 А), так как при применении голубого света
флуоресценция зависит от концентрации раствора. В растворе
диметилсульфоксида при 25°С инкремент показания
преломления dn/dc = 0,166 см3/г н внриальный коэффициент Ь = около
К)-3 см3 -моль-г-1.
5.10.5.3. Переработка и эксплуатационные свойства
Волокна. Ограниченно-регулярные полнамидогидразиды могут
быть переработаны в волокна мокрым формованием из раствора
в смеси диметилацетамид — LiCl или сухим формованием из
раствора в диметилсульфоксиде. Волокна с оптимальными свойствами
получают мокрым формованием с последующей горячей вытяжкой
при 300 СС с фактором вытяжки 1,1—2,0 [393]. В табл. 5.43
приведены свойства таких волокон на основе пяти изомерных поли-
амидогидразидов ограниченной регулярности с различным
содержанием я-фениленовых групп в цепи. Прочность при растяжении
и модуль упругости увеличиваются с повышением содержания этих
групп, а относительное удлинение уменьшается. Так, модуль
упругости увеличивается с 85 до 650 г/денье при переходе от полимера,
в котором соотношение п- и u-фениленовых групп составляет 1:1
к полимеру, содержащему только /г-фениленовые фрагменты.
Физико-механические свойства, кристалличность, плотность нитей из
полностью упорядоченных полнамидогидразидов с п-фениленовы-
ми группами примерно такие же, как и у «ограниченно-упорядочен-
Е, г/денье
Рис. 5.70. Зависимость модуля упругости высокомодульных органических волокон
(элементарных нитей) от относительного удлинения при разрыве [396].
Рис. 5.71. Деформациотю-прочпостмые кривые полиамидогпдразпдных,
текстильных и неорганических высокомодульных волокон [396]:
/ — графитовое волокно; 2 — полнамидогидразидное волокно PABH-T(G); 3 — стеклянное
волокно S: 4 — стальная нить; 5 — полнамидогидразмдное волокно РАВН-Т(Т); 6 — полнэтн-
лентерефталатное волокно; 7 — волокно из полиамида E,6; S — полнампдогидразндное
волокно РАВН-Т(Е); 9 — волокно из полиамида 6,6; /0 — стеклянное волокно Е-
457
Таблица 5.43. Физико-мехаиические свойства волокон* из ароматических полиамидогидразидов
ограниченной регулярности после горячей вытяжки [393, 396]
Элементарное звено
Прочность
при
растяжении,
г/д.енье
5,4
6,0
6,7
10,5
15,0
Относительное
удлинение
при
раз
рыве,
18,5
9,6
3,3
4,8
3,0
.С—NH—NH—С
и »
оо
С—NH—NH—
о
—NH (С)) С—NH— NH—С.
—NH—(Г))—С—NH—NH—С
V^ 11 il
о
—NH—(?))—C—NH—NH—С
v^/ II II
0 0
л
о
NH—NH—С
4
МН— С (Г*)) С—
» ^^ il
о о
NH
С
°
—NH—NH—С /())—NH—С /ГУ) С—
1 ^^ « ^^ II
il rr^T
о Iwj o
o
II
o
C—NH—NH—С
II II
0 0
—NH
-NH
—C-NH-NH—С
II II
0 0
C-NH-NH—С
||
0
||
0
NH—С А
О
JA
'—' II
О
85
114
289
323
650
* Олытное волокно Х-500 фирмы «Monsanto».
ного» (регулярного) полимера на основе терефталоилхлорида и
п-аминобензгидразида [393].
Однако ограниченно-регулярный полиамидогидразид лучше
растворяется и потому легче формуется.
Деформационно-прочностные характеристики органических высокомодульных волокон в
отличие от показателей высокомодульных неорганических волокон
можно регулировать в широких пределах путем варьирования
условий формования одних и тех же полимеров. Между модулем
упругости и относительным удлинением при разрыве (рис. 5.70)
существует следующая зависимость M = 1000 D~0-58 (г/депье) [396,
405].
Фирма «Monsanto» на основе высокоупорядоченного полимера
из терефталоилхлорида и м-аминобензгидразида выпускает три
типа волокна РАВН-ТХ-500 с различными значениями модуля
упругости и относительного удлинения (табл. 5.44). На рис. 5.71
приведены деформационно-прочностные характеристики для
различных органических и неорганических волокон. Волокно типа
РАВН-Т(Е) (модуль упругости 90—150 г/денье и относительное
удлинение при разрыве 22—40%) особенно целесообразно
применять там, где высокую кинетическую энергию необходимо гасить
за счет высокого удлинения материала, как, например, в
регуляторах режима работы самолета, защитных шлемах и экранах [403].
Волокно типа РАВН-Т(Т) используют в производстве шинного
корда [402]_ Возможность варьирования свойств волокна для
специальных типов корда, в частности его теплостойкости и
текстильных характеристик, делает полиамидогидразид идеальным
волокном для использования в шинах специального назначения. Поли-
амидогидразидное волокно РАВН-Т(9) в 4—5 раз прочнее
стального, равного ему по массе, и имеет вдвое большую жесткость.
Волокно этого типа служит армирующим наполнителем для
полимеров. При 300 °С у полиамидогидразидного волокна отсутствует
усадка; при 200 °С относительное удлинение почти не меняется.
Таблица 5.44. Свойства полиамидогидразидных
высокомодульных волокон [396]
Показатель
РАВН-Т(Т^
1,44
0,3
30
>390
3,3
390
72-81
15-20
2 250-2 700
10 500-12 000
C2-39)- 104
0,1
PABH-T(G)
1,47
0,57
85-93
525
2,0
525
135-153
3-4
5 850-7 200
19 700-22 000
(84—105) ¦ 106
0,1
РАВН-Т(Е)
45-54
20—40
810-1 350
6 000-7 000
A-2) • 1059
0,1
Плотность, г/см2
Двойное лучепреломление Дп
Степень кристалличности, %
Температура стеклования, °С
Водопоглощение при 25 °С, %
Температура начала разложения, °С
Прочность при растяжении, г/текс
Относительное удлинение при
разрыве, %
Модуль упругости, г/текс
Прочность при растяжении, кгс/см2
Модуль упругости, кгс/см2
Усадка при 400°С, %
459
G20
wu
60
Pfl
I
ex.)
a
i ^
WO
200 t,°C
Рис. 5.72. Зависимость
прочности (а) и модуля
упругости (б) ттолиами-
догндразидных волокон
типа PABH-T(G) от
температуры [396].
При 100 °С модуль упругости не меняется, а при 200° уменьшается
на 50% (рис 5.72).
Способы получения и свойства ткани и пряжи на основе поли-
амидогидразидных волокон описаны в [400, 401]. Они отличаются
высокой стойкостью к термической деструкции. При нагревании на
воздухе при 200 °С в течение 10 сут прочность не меняется.
Уменьшение прочности после 20 сут составляет 16 %. После 3 сут
нагревания на воздухе 300 °С происходит уменьшение прочности при
растяжении на 26% и модуля упругости на 54% [396]. При этом
происходит циклодегидратация гидразидных фрагментов с
образованием полиоксадиазоламидов. Полиамидогидразидные волокна
непригодны для длительной работы на солнечном свету.
Облучение УФ-лучами сопровождается быстрым снижением прочности
[396]. Эти волокна устойчивы при комнатной температуре к
действию 10%-ной серной кислоты и 10%-ного раствора аммиака; при
Таблица 5.45. Физико-механические свойства пластмасс
на эпоксидной смэле и полигидразидных
или стеклянных волокнах [400]
Показатель
Содержание волокна, %
Плотность, г/см3
Прочность при изгибе, кгс/см2
при 25 °С
при 121 °С
Moдvль упругости при изгибе, кгс/см2
при 25 °С
при 121 °С
Удельный модуль упругости при изгибе, км
Прочность при растяжении, кгс/см2
при 25 °С
при 121 °С
Модуль упругости при растяжении, кгс/см2
при 23 °С
при 121 °С
Удельный модуль упругости при растяжении, км
Прочность при сжатии, кгс/см2
при 25 °С
при 121 СС
Модуль упругости при сжатии, кгс/см2
при 85°С
при 121 °С
Удельный модуль упругости при сжатии, км
Мсжслойная прочность при сдвиге, кгс/см2
PABH-TfG) —
эпоксидная
смола
60,0
1,301
3 656
1434
260 000
154 700
1567
3 584
1 336
281 000
70 300
2 162
1 406
914
189 800
175 800
1 460
211
Стеклянное
волокно К —
эпоксидная
смола
55,0
1,799
5 765
4 520
239 000
211 000
1328
3 937
2 882
225 000
77 300
1 250
4 429
1 898
246 100
217 900
1 366
302
460
Таблица 5.46. Электрические свойства слоистых пластиков
на основе полиамндогидразидяого волокна типа PABH-T(G)
или стеклянного волокна марки Е и эпоксидного связующего [396]
Показатель
Диэлектрическая проницаемость
при 102 Гц
при 106 Гц
при 8-Ю9 Гц
Тангенс угла диэлектрических потерь
при 102 Гц
при 106 Гц
при 8-Ю9 Гц
Электрическая прочность при 1000 В/с, В/мм
Удельное объемное электрическое сопротивление,
Ом см
Дугостойкость, с
Волокно
PABH-T(G)
5,20
4,38
3,40
0.028
0,029
0,010
19 600
1,13- 1016
125
Стеклянное
волокно
5,66
5,18
4,46
0,012
0,011
0,009
19 520
2,48 • 1015
182
90 °С наблюдается значительное уменьшение прочности [393].
Выдержка в воде при 90 СС приводит к снижению прочности при
растяжении волокна РАВН-Т(9) на 60%, относительного удлинения
при разрыве на 86% и модуля упругости на 76%. За 2 сут
кипячения в воде прочность и модуль не меняются, удлинение
составляет 83 % от исходного значения. После 15 мин действия пара
высокого давления прочность при растяжении снижается на 72%;
модуль упругости при этом не меняется.
Армированные пластмассы на основе полиамидогидразидных
волокон. На основе полиамидогидразидной ровницы и тканей и
эпоксидных, полиэфирных, полиимидных, фенолоформальдегидных
и меламиноформальдегидных связующих можно получать
армированные пластики [400, 401]. Преимущество полнамидогидразидных
волокон перед стеклянными состоит в более высокой
износостойкости, невысокой чувствительности к влаге, хорошей
совместимости с полимерным связующим, а также в более высокой прочности
и жесткости в расчете на единицу массы. При одинаковой массе
прочность армированных пластиков на основе
полиамидогидразидных волокон выше. В табл. 5.45 и 5.46 приведены
физико-механические и электрические свойства пластиков на основе эпоксидного
связующего, наполненного полиамидогидразидным и стеклянным
волокнами.
Литература к главе 5, разделы 5.7—5.10
1. H. Genvresse, Bull. Soc. chim. France 17 A898), 599.
2. J. Deuss, Rec. Trav. chim. 28 A909), 136.
3. T. Hilditch, J. chem. Soc. 97 A910), 2579.
4 H. Tasher, J. chcm. Soc. 131 A928), 1141.
5. Z. Binenfeld, Bull. chim. France A961), 679—683.
6. Нидерл. пат. 6410567 12.5.65 Institut za Kemihska, Technoloska i Metalurska
Istrazivanja
7. И. Юкель, Высокомолек. соед., Б 10, 1968, 116.
461
8. Нидерл. пат. 6507380 13. 12.65 Soc. Nat. Patrole Aquitaine.
9. T. Fuyisawa, J. Polymer Sei. В 8 (!970), 19—24
10. M. Schmidt, Diplomarbeit, Inst, org. Chemie Munchen 1959.
11. A. Macallum, J. org. Chem. 13 A948). 154—159.
12. Пат. США 2513188 27. 6. 1950 A. Macallum.
13. Пат. США 2538941 23. 1.51 A. Macallum.
14. R Lenz, J. Po.ymer Sei. 41 A959), 333-358.
15. D. Gardner, J. Amer. chem. Soc. 78 A956), 3279—3288.
16. R. Lenz, J Polymer Sei. 58 A962), 351—367.
17. R. Lenz, J. Polymer Sei. 43 (I960), 167—181.
18. M. Russo, Materie Plast. Elast. 34 A968) 1. 61—64.
19. Пат. США 3354129 21. 11 67 Phillips Petroleum.
20. Фр. пат. 1437406 A967) Phillips Petroleum.
21. Пат. США 3607843 21. 10.71 Phillips Petroleum.
22. Опубл. заявка ФРГ 2026655 5. 1.72 Phillips Petroleum.
23. Пат. США 3697487 10. 10.72 Phillips Petroleum.
24. Пат. США 3687907 29. 8. 72 Phillips Petroleum.
25. S. Tsunawaki, J. Polymer Sei. A 2 A969), 1511.
26. L Baron, Makromolekulare Chem. 140 A970), 83—89.
27. Англ. пат. 1160666 A967).
28. R. Jones, Hydrocarbon Processing A972) 11, 89—91.
29. N. Christopher, J. appl. Polymer Sei. 12 A968) 4, 863—870.
30. B. Tabor, European Polymer J. A971), 1127—1133.
31. Пат. США 3562199 9.2.71 Phillips Petroleum.
32. Mod. Plastics Encyclop. 1971/72, 89—93.
33. Опубл. заявка ФРГ 2131996 5. 1.72 Phillips Petroleum.
34. Пат. США 3677994 18. 7.72 Phillips Petroleum.
35. Пат. США 3592783 13.7.71 Phillips Petroleum.
36. Mat. plast. Elast. 35 A969) 7, 946—947
37. Пат. США 3616186 26. 10. 71 Phillips Petroleum.
38. Белы. пат. 645262 A964) Dow Chem.
39. Белы. пат. 644106 A965) Dow Chem.
40. Фр. пат. 1386040 A965) Dow Chem.
41. Фр. пат. 1386042 A965) Dow Chem.
42. Пат. США 3521214 21.7.70 Phillips Petroleum.
43. Опубл. заявка ФРГ 2102512 19.8.71 Phillips Petroleum.
44. Пат. США 3695276 3. 10. 72 H. Coale.
45. A. Eisenberg, J. Polymer Sei. С 35 A971), 129—149.
46. В. Hortung, IUPAC-Symposium Helsinki 1972.
47. G. Ehlers, J. Polymer Sei. A 1 7 A969) 10, 2955—2967.
48. Акцепт, заявка Японии 7214767 2. 5. 72 Yunichika Co.
49. Акцепт, заявка Японии 7217411 22.5.72 Asahi Chem.
50. Опубл. заявка ФРГ 2164473 27. 7. 72 Sumitomo Cnem.
51. Опубл. заявка ФРГ 2165015 27.6. 72 Japan Soda.
52. R. Gilkey, J. appl. Polymer Sei. 2 A959), 198—202.
53. J. Economy, Polymer Preprints 11 A970) 1, 332.
54. Kunststoff-Berater 16 A971) 4, 285.
55. J. Economy, SAMPE-J. 6 A970) 6, 21—27.
56. H. Jellinek, J. Polymer Sei. A 1 10 A972) 6, 1719—1728.
57. T. Kametani, Yakugaku Zasshi 80 A960). 1188.
58. Акцепт, заявка ФРГ 2025516 3. 12. 70.
59. Chem. Engin 77 A970) 6, 31.
60. Kunststoffe 6! A971) 1, 36.
61. SPE J. 26 A970) 4, 33—34.
62. R. Anschutz, Ber. В 52 A919), 1860—1875.
63. Акцепт, заявка Японии 7202513 24. 1.72 Yunichika Co.
64. Акцепт, заявка Японии 7219172 1.6.72 Polyather Polyester Development.
65. Акцепт, заявка Японии 7214051 26. 4.72 Kanebo Co.
66. Акцепт, заявка Японии 7214765 2.3. 72 Kanebo Со.
67. M. Kuroishi, Nippon Kagaku Kaishi 8 A972) 7, 2181—2190
68. Фр. пат. 2107677 9. 6. 72 Yunichika Co.
462
69 Акцепт, заявка Японии 7218889 31.5. 72 Yunichika Со.
70 Chem. Engin. News 46 A968), 14.
7!. Пат. США 2471023 A945) ICI.
72 Англ. пат. 579462 6.8.46 Calico Printers.
73. В. Коршак, Пласт, массы, 2, 1961, 1, 9—13.
74 Фр. пат. 1163702 A958).
75. Кан. пат. 603582 A960).
76. Кан. пат. 674714 A963).
77. В Коршак, Высокомолек. соед., 4, 1962, 3, 339—344.
78. В. Коршак, Высокомолек. соед., 4, 1962, 987—994.
79. В. Коршак, Высокомолек. соед., 5, 1963, 799—804.
80. В. Коршак, Высокомолек. соед., А 13, 1971, 4, 848—853.
81. В. Коршак, Высокомолек. соед., А 14, 1972, 6, 1306—1312.
82. В. Коршак, Высокомолек. соед., А 8, 1966, 3, 548—552.
83. С Виноградова, Высокомолек. соед., Б 13, 1971, 9, 681—685.
84. Белы. пат. 563173 A957).
85. G. Ehlers, J. Polymer Sei. A 1 7 A969) 12, 3413—3415.
86. Англ. пат. 1079516 A967).
87. В. Коршак, Высокомолек. соед., А 7, 1965, 10, 1813—1817.
88. С. Виноградова, ДАН СССР, 164, 1965, 563—566.
89. В. Коршак, ДАН СССР, 156, 1964, 880—883.
90. В. Коршак, Высокомолек. соед., А 7, 1965, 10, 1689—1691.
91. R. Ismail, Angew. Makromolekul. Chem. 8 A969) 70, 99—116.
92. W. Eareckson, J. Polymer Sei 40 A959), 399—406.
93. R. Wilfong, J. Polymer Sei. 54 A961), 385—410.
94. В. Коршак, Высокомолек. соед., 1, 1959, 834.
95. В. Коршак, ДАН СССР, 177, 1967, 1, 120—133.
96. С. Виноградова, Высокомолек. соед., 7, 1965, 12, 2052—2056.
97. В. Панкратов, Изв. АН СССР, сер. хим., 1965, 342—348.
98. С. Виноградова, Изв. АН СССР, сер. хим., 1969, 2554—2560.
99. Z. Jedlinski, J. Polymer Sei. A 1 7 A969) 9, 2587—2604.
100. Акцепт, заявка Японии 7213784 25.4.72 Hitachi Electric.
101. В. Коршак, Изв. АН СССР, сер. хим., 1967, 747—752.
102. Англ. пат. 1141719 A969) S. Evans.
103. К. Goldammer, Vysokomol. Soedin. 10 A968) 4, 821—825.
104. В. Коршак, Высокомолек. соед., 4, 1962, 982—986.
105. Д. Израилов, Пласт, массы, 9, 1966, 13—16.
106. В. Коршак, Изв. АН СССР, сер. хим., 1965, 1649—1654.
107. С. Карапетян, Высокомолек. соед., 6, 1964, 1550—1554.
108. Пат. СССР 170662 A965).
109. Пат. США 3573250 30.3.71 Allied Chem.
ПО. V. Korshak, Polymer Letters В 10 A972) 6, 429—431.
111. В. Коршак, Высокомолек. соед., А 14, 1972, 6, 1267—1272.
112. Опубл. заявка ФРГ 1951551 22.4.71 Dynamit Nobel.
113. R. Ismail, Angew. Makromolekul. Chem. 15 A971) 212, 157—188.
114. В. Коршак, Хим. волокна, 3, 1965, 16—19.
115. Л. Дубровина, Высокомолек. соед., А 12, 1970, 6, 1308—1314.
116. V. Rode, J. Polymer Sei. С 16 A968) 6, 3255—3263.
117. V. Korshak, J. Polymer Sei. A 1 7 A969) 1, 157—172.
118. Ю. Васильева, Высокомолек. соед., А 14, 1972, 10, 2041—2046.
119. В. Панкратов, Изв. АН СССР, сер. хим., 1965, 1286—1288.
120. В. Родэ, Высокомолек. соед., А 7, 1965, 9, 1614—1616.
121. С. Виноградова, ДАН СССР, 181, 1968, 6, 1393—1396.
122. С. Виноградова, Высокомолек. соед., 11, 1969, 1, 27—34.
123. С. Виноградова, Изв. АН СССР, сер. хим., 4, 1969, 931—938.
124. В. Коршак, Высокомолек. соед., 4, 1962, 345—350.
125. В. Коршак, Успехи химии, 29, 1960, 569—628.
126. Z. Jedlinski, Kinet. Mech. Polyreact. IUPAC Symp. 1969, 113—125
127. H. Weyland, European Polymer J. A970) 6, 1339—1346.
128. M. Gilbert, Polymer 13 A972) 7, 327—332.
129. С. Виноградова, Высокомолек. соед., 1, 1959, 357.
463
130. G. Farrow, Makromolekulare Chem. 38 A970), 147—158.
131. A Thompson, Trans. Faraday Soc. Ш A956), 1383—1397.
132. W. Eareckson, J. Polymer Sei. 40 A959), 343—358.
133. Chem. Engin. 78 A971) 15, 64.
134. D. Asmus, Kunststoffe 62 A972) 10, 635—637.
135. Опубл. заявка ФРГ 2224088 14. 12.72 ICI.
136. A. Baron, SPE Techn. Papers Reg. Techn. Conf. California Sect. 27.3.72.
137. R. Boyer, Rubber Chem. Technol. 36 A963), 1303.
138. Kunststoffe 62 A972) 4, 208.
139. В. Меньшов, Высокомолек. соед., А 14, 1972, 8, 1766—1773.
140. С. Воуе, J. Polymer Sei. 55 A961), 275—284.
141. L. Moore, 137th ACS Nat. Meeting, Cleveland 1960.
142. E. Martin, Mod. Textiles Mag. 40 A959) 2, 43.
143. E. Martin, Textile Res. J. 32 A962), 619.
144. Пат. США 2917549 15. 12.59 Eastman Kodak.
145. Акцепт, заявка Японии 7236276 12.9.72 Kuraray Co.
146. С. Kibler, J. Polymer Sei. А 2 A964), 2115—2125.
147. Англ. пат. 1024487 A966) Du Pont.
148. Акцепт, заявка Японии 7218895 31. 5. 72 Kanebo Со.
149. Акцепт, заявка Японии 7214766 2. 3. 72 Kanebo Со.
150. Chem. Engin. 78 A971) 15, 64.
151. Англ. пат. 879264 A961) Eastman Kodak. '
152. Пат. США 2901466 A959) Eastman Kodak.
153. Пат. США 2105664 Du Pont.
154. Kunststoffe 62 A972) 11,581.
155. Kunststoffe 63 A973) 2, 122.
156. А. Микитаев, Высокомолек. соед., А 14, 1972, 9, 1936—1941.
157. Англ. пат. 968390 A964).
158. В. Коршак, Высокомолек. соед., 1, 1959, 839.
159. J. Smith, J. Polymer Sei. A 1 4 A966), 1851—1859.
160. A. Conix, Ind. Engin. Chem. 51 A959), 147—150.
161. P. Morgan, J. Polymer Sei. A 2 A964), 437—459.
162. В. Коршак, Высокомолек. соед., 2, 1960, 1475—1480.
163. G. Schweitzer, J. Polymer Sei. 24 A957), 33—41.
164. С. Виноградова, Высокомолек. соед., Б 14, 1972, 6, 457—462.
165. А. Conix, Ind. Chim. Belg. 22 A957), 1457—1462.
166. M. Levine, J. Polymer Sei. 28 A958), 179—184.
167. S. Temin, J. org. Chem. 26 A961), 2518—2521.
168. С Fuller, J. chem. Educat. 20 A963), 3.
169. G. Ehlers, Papers presented at the 12th Canadian High Polymer Forum
A964).
170. В. Коршак, Изв. АН СССР, сер. хим, 1972, 6, 1409—1410.
171. Ю. Васильева, Высокомолек. соед., А 13, 1971, 7, 1475—1483.
172. S. Pavlova, J. Polymer Sei. С 16 A967) 5, 2649—2658.
173. Л. Радивилова, Пластмассы, 1970, 10, 39—41.
174. Е. Fischer, Ann. 372 A910), 32.
175. Пат. США 3656862, 23.2.71 Phillips Petroleum.
176. Пат. США 3549595, 22. 12.70 Phillips Petroleum.
177. G. Ehlers, J. Polymer Sei. A 2 A964) 3, 1051—1056.
178. J. Work, J. Polymer Sei. A 1 6 A968) 7, 2022—2030.
179. A. Conix, Angew. Chem. 72 A960) 3, 116—117.
180. W. Firth, J. Polymer Sei. В 10 A972) 8, 637—641.
181. J. Arcesi, J. appl. Polymer Sei. 15 A971) 3, 513—518.
182. J. Wilson, J. Polymer Sei. A 110 A972) 4, 1157—1164.
183. Е. Кузнецов, Труды Казанского хим-.-техн- ин-та, 40, 1969, 2, 351—358.
184. Англ. пат. 1015202 A965) Borg-Warner Со.
185. Пат. США 3236808 A966) Borg-Warner Со.
186. Пат. США 3236809 A966) Borg-Warner Со.
187. Англ. пат. 916660 A963) Gevaert Photo.
188. Англ. пат. 974342 A964) Gevaert Photo.
189. A. Conix, Makromolekulare Chem. 24 A957), 76—79.
464
190 A Coni.x, J. Polymer Sei. 29 A958), 343—353.
191 N Yoda, Makromolekulare Chem. 32 A959), 1—12.
192* N' Yoda, Kogyo Kagaku Zasshi 65 A962), 667.
193 N Yoda, Bull* Chem. Soc. (Japan) 32 A959), 1120.
194. N. Yoda, Kobunshi Kagaku 19 A962), 490.
195 N Yoda, Kobunshi Kagaku 19 A962), 553.
196. N. Yoda, Kobunshi Kagaku 19 A962), 613.
197. N. Yoda, Makromolekulare Chem. 55 A962), 174—190.
198 N. Yoda, Makromolekulare Chem. 56 A962), 10—35.
199. N. Yoda, J. Polymer Sei. AI A963), 1323—1338.
200. Белы. пат. 552044 Gevaert Photo.
201. Белы. пат. 553897 Gevaert Photo.
202. Белы. пат. 552538 Gevaert Photo.
203. Белы. пат. 840846 Gevaert Photo.
204. Англ. пат. 840847 Gevaert Photo.
205. Акцепт, заявка Японии 614841 A961) Toyo Rayon.
206. Акцепт, заявка Японии 6210944 A962) Nippon Celluloid.
207. Англ. пат. 838986 ICI.
208. Англ. пат. 968715 ICI.
209. Англ. пат. 998356 ICI.
210. W. Carothers, J. Amer. chem. Soc. 54 A932), 1569—1579; 1579-1587.
211. N. Yoda, Makromolekulare Chem. 56 A962), 36—54.
212. R. Muromova, Vysokomol. Soedin. 5 A963), 10, 1473—1478.
213. Опубл. заявка ФРГ 2034541 20. 1.72 Bayer.
214. M. Watson, SPE-J. A965) 5, 475—479.
215. Б. Жубанов, Изв. АН КазССР, сер. хим., 22, 1972, 4, 53—63.
216. Т. Шеин, Высокомолек.соед., 7, 1965, 8, 1447—1451.
217. J. Schaeigen, J. Polymer Sei. 40 A959), 377—387.
218. H. Hall, J. Polymer Sei. В l A963) 6, 277—278.
219. J. Preston, Polymer Preprints 6 A965) 2, 761—772.
220. G. Jarrett, Brit. Polymer J. 2 A970) 5, 229—232.
221. H. Hopff, Makromolekulare Chem. 47 A961), 93—113.
222. E. Carlston, Ind. Engin. Chem. 49 A957), 1239—1240.
223. Т. Медведь, Высокомолек. соед., 5, 1963, 1309—1314.
224. Г. Колесников, Высокомолек. соед., А 1 4, 1972, 3, 608—612.
225. Англ. пат. 506125 Du Pont.
226. G. Roques, Compt. rend. С 264 A967), 63—66, 178—180.
227. Опубл. заявка ФРГ 2064573 20. 7. 72 Hochst.
228. Г. Колесников, Высокомолек. соед., А 1 2, 1970, 1, 177—181.
229. R. Beaman, J. Polymer Sei. 40 A959), 329—336.
230. Т. Фрунзе, Высокомолек. соед., 1, 1959, 670.
231. С. Stevens, J. Polymer Sei. 40 A959), 359—366.
232. M. Katz, J. Polymer Sei. 40 A959), 337—342.
233. S. Bruck, Polymer 7 A966) 5, 231—241.
234. W. Morgan, J. Polymer Sei. A 2 A964), 181—224.
235. S. Bruck, Polymer Preprints 8 A967) 2, 1181—1189.
236. J. Preston, J. Polymer Sei. A l 8 A970), 1841—1850.
237. Т. Соколова, Высокомолек. соед., Б 11, 1969, 6, 451—453.
238. А. Волошина, Высокомолек. соед., Б 9, 1967, 9, 683—684.
239. R. Deanin, J. Polymer Sei. A 1 6 A968) l, 235—240.
240. M. Sumoto, J. chem. Soc. (Japan), Ind. chem. Sect. 68 A965) 10, 1989—1994.
241. В. Саржевская, Украинск. хим. жур., 35, 1969, 4, 390—392.
242. S. Sundet, J. Polymer Sei. 40 A959), 389—397.
243. Пат. США 3595834 27. 7. 71 Pennwalt Co.
244. Л. Соколов, Высокомолек. соед., 2, 1960, 1744.
245. Л. Соколов, Высокомолек. соед., 4, 1962, 1817—1821.
246. R. Mekherjea, J. Polymer Sei. AI 10 A972) 5, 1547—1552.
247. В. Sprague, Textile Res. J. 35 A965) 11, 999.
248. N. Yoda, J. Polymer Sei. A 2 A964), 253-255.
249. Пат. США 3287323 A966) Du Pont.
465
250. L. Lee, J. Polymer Sei. A 1 9 A971) 2, 557—561.
251. P. Morgan, J. Polymer Sei. С 1 A963), 1075—1096.
252. P. Morgan, J. Polymer Sei. A 1 2 A964), 2693-2703.
253. Пат. США 3232909 A966) Celanese.
254. «Trogamid T» Firmenschrift Dynamit Nobel 1972.
255. Plastverarbeiter 23 A972) 12, 855.
256. Пат. США 3203933 A965) Monsanto.
257. А. Семенова, Хим. волокна, 1969, 5, 2—4.
258. N. Boyer, Technikas Apskats 31 A961), 13—20.
259. Пат. США 3225011 A965) Monsanto.
260. Пат. США 3637606 25. 1.72 Du Pont.
261. Англ. пат. 1262002 2. 2. 72 Du Pont.
262. В. Калмыкова, Высокомолек. соед., Б 13, 1971, 10, 707—708.
263. Пат. США 3595951 17.7.71 Du Pont.
264. Пат. США 3591673 6.7.71 Du Pont.
265. Пат. США 3699085 17. 10.72 Du Pont.
266. Пат. США 3240758 A966) Monsanto.
267. Акцепт, заявка ФРГ 1951077 29.4.71 Bayer.
268. Г. Кузнецов, Высокомолек. соед., А 11, 1969, 7, 1491 — 1500.
269. R. Dine-Hart, J. Polymer Sei. В 2 A964), 369—373.
270. Пат. США 3063966 A962) Du Pont.
271. В. Савинов, Хим. волокна, 1965, 4, 22—25.
272. Е. Краснов, Высокомолек. соед., 8, 1966, 3, 380—386.
273. А. Федоров, Высокомолек. соед., Б И, 1969, 2, 129—132.
274. В. Савинов, Высокомолек. соед., Б 10, 1968, 2, 111—114.
275. H. Mark, J. Polymer Sei. 61 A962), S 49—S 53.
276. К. Калашник, Высокомолек. соед., А 1 4, 1972, 6, 1396— 1402.
277. Опубл. заявка ФРГ 2219646 28.4.71 Du Pont.
278. О. Федотова, Высокомолек. соед., Б 10, 1968, 6, 461—465.
279. 3. Приходько, Хим. волокна, 22, 1971, 19—21.
280. Пат. США 2831834 A958) Du Pont.
281. А. Щетннин, Хим. волокна, 1968, 5, 50—52.
282. Пат. США 3094511 A963) Du Pont.
283. J. Holsten, Appl. Polymer Symp. 9 A969), 63—74.
284. Л. Соколов, Высокомолек. соед., 2, I960, 693—710.
285. А. Калашник, Высокомолек. соед., Б 14, 1972, 5, 346—348.
286. Пат. США 3697478 10. 10. 72 Monsanto.
287. Пат. США 3660361 2.5.72 Monsanto.
288. Г. Колесников, Высокомолек. соед., Б 9, 1967, 11, 819—821.
289. С. Хорьков, Высокомолек. соед., А 11, 1969, 9, 2061—2066.
290. Пат. США 3006899 A961) Du Pont.
291. Пат. США 3642706 15. 2. 72 Montecatini.
292. Опубл. заявка ФРГ 2037254 3. 2. 72 Hochst.
293. В. Беляков, Высокомолек. соед., А 12, 1970, 3, 610—619.
294. А. Щетинин, Хим. волокна, 1968, 1, 22—25.
295. Англ. пат. 1201531 5.8.70 ICI.
296. И. Некрасов, Высокомолек. соед., А 1 4, 1972, 4, 789—793.
297. С. Медведь, Высокомолек. соед., А 1 4, 1972, 6, 1313—1319.
298. С. Рафиков, Высокомолек. соед., А 13, 1971, 10, 2324—2331.
299. P. Opt, Textile Res. J. 37 A967) 12, 1050—1055.
300. J. Preston, Faserforschg. Textiltechn. 22 A971) 3, 153—162.
301. В. Захаров, Хим.волокна, 1968, 1, 16—18.
302. И. Некрасов, Высокомолек. соед., A3, 1971, 8, 1707—1715.
303. J. Ross, Appl. Polymer Symp. 9 A969), 23—47.
304. M. Акутин, Пласт, массы, 1970, 12, 57—59.
305. Хим. волокна, 1968, 6, 72.
306. Л. Соколов, Пласт, массы, 1967, 9, 21—23.
307. А. Кузнецов, Высокомолек. соед., 1965, 7, 1592—1596.
308. Л. Соколов, ДАН АН СССР, 158, 1964, 1139-1148.
309. А. Трофимович, Вестник машиностроения, 50, 1970, 2, 50—51.
466
310 В Коршак, Механика полимеров, 1969, 2, 201—206.
311. L. Johnson, J. appl. Polymer Sei. 13 A069) 9, 1825-1832.
312 Г Колесников, Высокомолек. соед., Б 12, 1970, 11, 839—841.
313 И Елин, Высокомолек. соед., А 14, 1972, 9, 1964 1969.
314. L. Starr, J. Poiyrner Sel A 1 4 A966) 12, 304!—3046.
315. S. Nishizaki, J. Polymer Sei. A l 6 A968) 6, 1769—1773.
316 Б. Маличенко, Высокомолек. соед.. А 13. 1971. 4, 96G—970.
317. Опубл. заявка ФРГ 2061774 13.7.72 Hochst.
318. А. Терентьев, Высокомолек. соед., 5, 1963, 837—841.
319. Пат. США 3591559 6.7.71 Du Pont.
320. Y. Imai, J. Polymer Sei. A 1 10 A972) 8, 2257—2264.
321. Англ. пат. 901159 A962) Du Pont.
322. E. Wittbecker, J. Polymer Sei. 40 A959), 289—297.
323. О. Федотова, Высокомолек. соед., 2, 1960, 1020—1025.
324. Л. Туреикий, Высокомолек. соед., 3, 1961, 1449—1,455.
325. О. Федотова, Высокомолек. соед., 3, 1961, 1524—1527.
326. О, Федотова, Высокомолек. соед, 3. 1961, 1528—1534.
327. Англ. пат. 871581 A961) Du Pont.
328. Пат. ФРГ 1243820 A960) Du Pont.
329. J. Preston, J. Polymer Sei. A 1 4 A966). 2093—2105.
330. Пат. США 3240760 A966) Monsanto.
331. Пат. США 3242213 A966) Monsanto.
332. Пат. США 3049518 A962) Du Pont.
333. H. Bach, Polymer Preprints A972).
334. J. Preston, J. Polymer Sei. A 1 4 A966), 529—539.
335. Пат. США 3232910 A966) Monsanlo
336. J. Preston, J. Polymer Sei. В 2 A964) 1171—1174.
337. J. Preston, J. Polymer Sei. A 1 8 A970) 11. 3135-3144.
338. Пат. США 2001526 A935) Du Pont.
339. J Preston, J. Polymer Sei. В 4 A966) 12, 1033—1038.
340. J. Preston, Faserforschg. und Te.xtiltechn. 22 A971) 3, 153—162.
341. J. Preston. J. Polymer Sei. С 19 A967), 7-27.
342. J Preston, J. Polymer Sei. С 1 A971), 664—671
343. J. Preston, J. Polymer Sei. В 4 A966V 267—272.
344. H Morawetz, Proc. Natl. Acad. Sei. 49 A963), 789—794.
345 H Herlinger, Textilpraxis A972), 181-183; 243—346.
346. E. Краснов, Высокомолек. соед., 8, 1966, 3, 380—386.
347. G Ehlers, J. Polymer Sei. A 1 8 A970), 3511—3527.
348. E Краснов, Высокомолек. соед., 8, 1966, 1970—1975.
349. L. Scala, J. appl. Polymer Sei. 12 A968), 2339—2357.
350. A. Блюменфельд, Высокомолек. соед., Б 14, 1972, 6, 403.
351. Англ. пат. 877885 A961) Du Pont.
352. J. Weiss, J Polymer Sei. С 19 A967), 29—39.
353. Хим. волокна, I968, 6, 72.
354. «Nomex» Firmenschrift NP-33 Du Pont A964).
355. «HT 1-Faser» Firmenschrift Du Pont 1961.
356. S. Hirsch, Appl. Polymer Symp. 9 A969), 187—194.
357. Пат. США 2999788 A961) Du Pont,
358. Nomex-Papiere, Firmenschrift Du Pont 1965.
359. А. Долженков, Докл. Моск. ин-та инженеров сельского хоз-ва 7, 1971 4
177—185. ' '
360. Опубл. заявка ФРГ 2162159 6.7.72 Celanese Со.
361. Фр. пат. 1526745 A968) Du Pont.
362. Белы. пат. 726050 A969) Du Pont.
363. Опубл. заявка ФРГ 2219646 28.4.71 Du Pont.
364. H. Kunzel, Makromolekulare Chem. 138 A970). 223—250.
365 H, Kunzel, Makromolekulare Chem. 130 A969), 103—144.
366. Фр. пат. 2038701 8. 1.71.
367. Опубл. заявка ФРГ 2009741 30.9.71 Bayer.
368. В. Sillion, Compt. rend. С 1972, 274 B2),* 1795—1798.
467
369. J. Preston, J. Polymer Sei. С 23 A968), 441—448.
370. Пат. США 3576769 27. 4. 71 .Monsanto.
371. H. Bach. Appl. Polymer Symp. 9 A969), 177—185.
372. H. Bach, Appl. Polymer Symp. 9 A969), 167—176.
373. M. Bruma, Rev. Roumaine Chim. 13 A968) 8, 1069—1073.
374. B. Culbertson, J. Polymer Sei. В 5 A967) 9, 807—812.
375. В. Коршак, ДАН СССР, 180, 1968, 5, 1105—1108.
376. К. Saotome, Makr. Chem. 100 A967), 91—99.
377. Franz. Patentanm. 68652 7.7.66 Inst. Franc, du Petrole.
378. Акцепт, заявка Японии 7127819 12.8.71 Teijin Ltd.
379. J. Preston, Appl. Polymer Symp. 9 A969), 75—88.
380. W. de Winter, Int. Symp. Macromol. Chem. 1UPAC, Kinet. Mech. Holyreac-
tions A969) I, 35—39.
381. R. Hirohashi, Polymer 11 A970) 6, 297—308.
382. Y. Imazu, Yuki Gosei Kagaku Kyokai Shi 29 A971) 7, 717—720.
383. Г. Кудрявцев, Высокомолек. соед., Б 10, 1968, 4, 295—298.
384. Фр. пат. 1601083 18.9.70 Inst. Franc, du Petrole.
385. Опубл. заявка ФРГ 1946789 25.3.71 Bayer.
386. О. Федотова, Высокомолек. соед., Б 11, 1969, 1, 53—55.
387. В. Коршак, Высокомолек. соед., А 14, 1972, 8, 1761—1765.
388. Y. Iwakura, Makromolekulare Chem. 77 A964), 41—50.
389. W. de Winter, J. Polymer Sei. A 1 8 A970), 1955—1966.
390. G. Rabilloud, Bull. Soc. chim. France A966), 926—932.
391. В. Коршак, Высокомолек. соед., А 14, 1972, 7, 1557—1564.
392. J. Preston, J. macromoleciil. Sei. A7 A973), 1, 45—65.
393. J. Preston, J. macromolecul. Sei. A 7 A973) 1, 67—98.
394. R. Morrison, J. macromolecul. Sei. A 7 A973) I, 99—118.
395. J. Randall, J. macromolecul. Sei. A 7 A973) 1, 119—133.
396. W. Black, J. macromolecul. Sei. A 7 A973) 1, 137—171.
397. V. Holland, J. macroinolecul. Sei A 7 A973) 1, 173—182.
398. R. Miller, J. macromolecul. Sei. A 7 A973) 1, 183—186.
399. J. Burke, J. macromolecul Sei. A 7 A973) 1, 187—200.
400. M. Lillyquist, J. macromolecul. Sei. A 7 A973) 1, 203—227.
401. D. Zaukelies, J. macromolecul. Sei. A 7 A973) 1, 229—279.
402. G. Raumann, J. macromolecul. Sei. A 7 A973) 1, 281—293.
403. R. Laible, J. macromolecul. Sei. A 7 A973) 1, 295—322.
404. J. Preston, J. macromolecul. Sei. A 7 A973) 1, 325—348.
405. W Black, J. macromolecul. Sei. A 7 A973) 1, 3—41.
406. Белы. пат. 660339 A965) Monsanto.
407. Пат. США 3376269 A968) Monsanto.
408. Пат. США 3484407 A969) Monsanto.
409. Белы. пат. 709304 A968) Monsanto.
410. Пат. США 3584046 A971) Monsanto.
411. L. Frost, J. Polymer Sei. A 16 A968), 215—233.
412. Э. Телешов, Пласт, массы, 1973, 2, 3—8.
413. Пат. США 3719642 6.3.73 Du Pont.
414. Пат. США 3595951 17.8.71 Du Pont.
415. Опубл. заявка ФРГ 2219703 9. 11.72 Du Pont.
416. Kunststoffe 63 A973) 2, 106.
417- Kunststoff-Berater 18 A973) 1, 46—48.
418. M. Hanson, NASA Techn. Note 1972, NASA-TN D 7120.
419. А. Федоров, Высокомолек. соед., Б 15, 1973, 1, 74—77
420. G. Fielding, Textile Res. J. 41 A971), 861.
421. Хим. волокна, 13, 1971, 1, 76.
422. J. Burke, J. Polymer Sei. A 2 A969), 1—25.
423. Europlastics Monthly 46 A973) 7, 77; 80.
424. Хим. волокна, 1972, 6, 20—21.
425. Хим. волокна, 1969, 4, 78.
6 ГЕТЕРОЦИКЛИЧЕСКИЕ ЦЕПНЫЕ ПОЛИМЕРЫ
Карбоциклические цепные полимеры, состоящие из
ароматических колец, например полифенилен E.1), имеют пока
ограниченное техническое применение вследствие трудностей, связанных
с их получением и переработкой в изделия. Термостойкость
полимеров с ароматическими гетероциклами в цепи (тиадиазольный,
оксадиазольный или триазиновый) всегда выше, чем у
ароматических, карбоциклических цепных полимеров, так как с
увеличением числа гетероатомов в ароматической системе уменьшается
количество слабых С—Н-связей. Ниже приведены некоторые ге-
тероциклы, представляющие наибольший интерес:
О
N
I
Пиразол
О N
XX
о
Гидантоин
N г,
N г,
О"
Оксазол
N N
Оксазолидон
N N
Х XX X)
Изоксазол
Тиазол
NH
Триазод
о
Оксадиазол
N M
?
-N'
Тиадиазол
N
Тетразол
N
Пиразин
Дикетопнперазин
Триазнн
В противоположность карбоциклическим полимерам при
получении полигетероциклических полимеров образование гетероцикла
может сопровождаться одновременным протеканием реакции роста
цепи полимера (циклополиконденсация). Это дает возможность
при ступенчатом проведении процесса осуществлять синтез
полимеров с чередующимися гетеро- и карбоциклическими звеньями,
не содержащими между циклами атомы или группы атомов,
которые являются слабыми местами в макромолекуле. В большинстве
случаев образование гетероциклоцепных полимеров протекает
через промежуточную стадию получения полимера с открытой
цепью, который на второй стадии превращается в конечный
циклический продукт. Благодаря этому можно проводить переработку
растворимых и плавких форполимеров (замыкание цикла еще не
произошло) непрерывным методом. Формирование гетероциклов
происходит в процессе формования.
Термостойкость гетероциклических полимеров определяется
энергией резонанса гетероцикла, сопряжением гетеро- и карбо-
469
циклических колец, а также содержанием карбоциклических
циклов и групп, соединяющих циклы. Самую высокую термостойкость
имеют политиадиазолы, поли-1,3,4-оксадпазолы и поли-4-фенил-
1,2,4-триазолы. Для полимеров, в которых гетероцикл чередуется
с я-фениленом, температура начала разложения в инертной среде
составляет 400—500°С, а на воздухе 340—400 °С. Полигидантоины
выпускаются в промышленном масштабе с 1968 г. Они
применяются в качестве электроизоляционного лака и
электроизоляционной пленки с температурой длительной работоспособности
около 200 °С. Политиадиазолы, полиоксадиазолы и поли-4-фенил-
1,2,4-триазолы пригодны для изготовления термостойких волокон
с температурой длительной работоспособности около 300 °С.
Сшитые политриазины до температуры 260°С обладают хорошей
адгезией и эластическими свойствами.
6.1. ЦЕПНЫЕ ПОЛИМЕРЫ С ПЯТИЧЛЕННЫМ
ГЕТЕРОЦИКЛОМ И ДВУМЯ ГЕТЕРОАТОМАМИ
В ЦИКЛЕ
6.1.1. ПОЛИПИРАЗОЛЫ
N
п
Полипиразолы получают поликонденсацией бис-р-дикетонов
с дигидразидами или диполярным полиприсоединением диэтиниль-
ных соединений к дитетразолам. Ароматические полипиразолы
устойчивы на воздухе и в инертной среде до 450 °С.
6.1.1.1. Получение полипиразолов
Строение, температура размягчения и температура
разложения полипиразолов приведены в табл. 6.1.
Полипиразолы. Незамещенные 1,3-полипиразолы (№ 1,2)
получены Стиллом [25] методом диполярного полиприсоединения
циэтинильных соединений к гс-фенилен-3,3-дисиднону:
N—О—N
470
5-Фенил- или 5-метилзамещенные 1,3-полипиразолы (№ 5—13,
16—18) получают взаимодействием дипиразолов с доступными
хлорзнгидридами дикарбоновых кислот [32, 39]:
п
R'-
R-
n ^C-
.0
чл
—H Cl
, NH
N
¦R'-
N
СО—R"—
R"—СО— _
n
Подобные полимеры могут быть получены поликонденсацией
бис^-дикетонов с дигидразинами кислот с образованием в
качестве промежуточных продуктов линейных полигидразонов [27
29—31, 33]:
nR'—С—СН2—С—R—С—СН2—С—R' + «NH2—NH— R"—NH—NH2
I! Il il II
о о о о
о о
II II
R'— С—СН2— С—R—С—СН2—С—R'
II II
—HN—N N—NH—R"— -I
Полигидразои
-R-
—Н2О
п
N
N
R"-
R'
n
Полипиразол
—Н2О
Для полипиразолов возможны две изомерные формы:
R'-
-R-
N
I
N
И
п
R'-
N
N
R"-
-»ri
При сильной енолизации бис-р-дикетонов наиболее вероятно
1,3-присоединение [29].
3.5-Диметилзамещенные 1,4-полипиразолы. 3,5-Диметилзаме-
щенные 1,4-полипиразолы (№ 25—37) получают из дигидрази-
471
Таблица 6.1. Температуры размягчения и разложения полипиразолов
№
Элементарное звено
разм'
Т
разл
(в N2),
°С
Т
разл
(на
воздухе).
Литературный
источник
— А )>—N—N
N—N—
N—N—
U-^AJ
—R-N—N N—N—
—Я—N—N
Fe
. R =
N—N—
460
460
420
420
[25, 26]
[25]
[26]
- -o-°-<D>-. -O-s°'-<a-
[27]
F\._/ F\_/
p/ \p p/ \p
ч
СНз НзС
N— CO(CH2LCO—N
СНз
Л_
j ^N-CO(CH2NCO-N^
,СН3 НзС
;n— co(ch2Lco-n
\
S=ti
N=
СНэ
НзС
,
¦ 'N—CO(CH2LCO—N'
СНз
H3C
\
10
— CO(CH2LCO—
H3C
N=
—сн2—сн2—(
У3
^J—CO(CH2NCO—N(
100-120
210—220
218-225
118—124
Продолжение табл. 6.1
Элементарное звено
Т °С
разм'
разл
Т
разл
(на
воздухе),
°С
11
НзС
\
СНз
N—СО— R— СО— N |
N N=
п
12
13
^R
НзС
\
СО—К
СНз
N—СО—R
НзС
\
14
,CH3
Ы—со
НзС
\
(S)—(сн2J
-С(СН2N-,
CH2-t —
122—140
260-280
•• -(СН2)8-
[29. 31]
[29, 301
15
ctr
-CO-R-CO-N
N-
R
Fe
16
HsCr
17
N— СО—(СН2L— СО—i/
N~\(CH2).
\
\-CO—(CH2)*—CO-N^
L=N N=
18
[—СО—(СН2L— СО—
/CeHs
19
100—120
240—250
218—225
260-280
Продолжение табл. 6.1
Элементарное звено
т
разм'
Т
разл
В N2).
°С
Т
разл
(на
воздухе),
°С
20
21
23
-(СН2N-
СНз
N—
НзС"
F\_/
/'"V
156—174
141-146
350
НзС
24
/
Л,
сн»
N—
"СНз
25
НзС,
-его:
,j=n
/
\
НзС
\
26
N
СНз
N—
НзС
,
(
27
Н3
eut
НзС'
Л
/СНз
сн.
НзС'
262-265
120-132
180-190
13Ц
450
[35]
500
[34]
[31]
[34]
Продолжение табл. 6.I
»
29
30
31
32
НзС.
—NH—(CH2N—NHCO—N^
N=kc
vr~CH2~
НзС,
-NCpCHa
^СНз
Н3СЧ
>=(— CH2-
N=
^CH3
-0-сн2-
НзС"
^—^
Элементарное звено
/CH,
:н2 сн2-_.
^N—СО—
'rj ri C*^
In—*~4j\J—i^j~jnl4 v^V_/ "
/CH3
) CH2 i=\
V—СО—(СН2L—СО—
НзС/
^-сн. _/СНз
"^—/ )n-co-(ch2L-co-
НзС^
Гразм* °С
208—215
230—300
228-230
245-256
Т
разл
(в N2).
разл
(иа воз-
д^хе),
Литературный
источник
[31]
[36]
[36]
136]
33
34
35
36
НзС
—N:
СНз
-СНз
i—CONH— (CH2N-NHCO-
Н3С
\
У
СНз
—n:
НзС
-N
'N—CONH^(CH2N—NHCO—
-СНз НзС"
—CONH
чСНз
—К
н3сч
(
N
—сн2—т=-
НзС"
¦NHCO—
СНз
/СНз
— CONH—,
NHCO-
СНз
37
38
СНз
N=1
-СНз
1 U U
NH NH
220-235
230—246
230—276
260—272
210-254
350—374
Лродоло/ссние табл. в Л
»
39
40
41
42
43
44
Xj
NH
\
\h
Vy
\/
CH3
A \
NH
-V
NH
NH
-[QJ—сн2-
-Г)—сн2—
Элементарнее звене
ОТ
440
>500
разл
(в N2)f
°С
450
450
т
разл
(иа
воздухе),
°С
450
400
Литературный
источник
[37]
[37]
[37]
[37]
[42]
[42]
дов и тетраазетилалканов [35]
п
0
II
rSf^ 2 НзС~с\
yj +/гн3с-с/СН
о
Н3С^
-LOl-^ЛсНз НзС
о
0
1
-сн.
—н2о
-СНз
или поликонденсацией метилзамещенных бис-р-дикетонов с ди-
гидразинами [31]:
О СНз О О СНз О
[| I II II I II
«НзС—С—СН—С—R—С—СН—С—СНз + «H2N—NH—R'—NH—NH2 w
—Н2О
II
N
R'—
П
3,4-Полипиразолы. 3,4-Полипиразолы (№ 38—42) получают ди-
полярным полиприсоединением [37]:
rtN2CH—R—CHN2
^C—R'—C^
NH
NH
3,5-Полипиразолы. 3,5-Полипиразолы получают с высоким
выходом диполярным полиприсоединением диэтинильных соединений
к замещенным тетразолам при 200°С в растворе тетралина
(№ 43—50) [25, 39] :
п
(СУ) N—N4 /—А А!—N ¦ i
—С=СН
N—N
N—N—
16 Зак. 18
481
Аналогичные полимеры получаются из этинилтетразолов [40]:
К
-in
Незамещенные 3,5-полипиразолы можно синтезировать из поли-
диэтинилбензола и гидразина в пиридине [42]:
N—NH
L "^ in
N2H4
сн2—
п
сочетанием
Полидиэтинилбензол получают окислительным
[43, 44].
6.1.1.2. Свойства полипиразолов
Полипиразолы представляют собой порошки от желтого до
коричневого цвета.
Различают 1,3-полипиразолы (№ 1—20), где R = —Н, —СН3—
—С6Н5A); 1,4-полипиразолы (№21—37), где R = — CH3(II);
3,4-полипиразолы (№ 38—42) (III); 3,5-полипиразолы (№43—
50) (IV).
II
N
N
Xf LA
N NH
II
NH
III
NH
IV
Температуры размягчения и начала разложения
полипиразолов приведены выше (см. табл. 6.1). Полипиразолы, полученные
поликонденсацией в растворе,
растворяются в полярных
растворителях, таких, как диметилформамид,
диметилацетамид, диметилсульфо-
ксид, гексаметилфосфотриамид,
крезол и тетрахлорэтан. Полимеры,
синтезированные в расплаве, имеют
ограниченную растворимость вслед-
го-
о
200 300 400 500 Б0О 700 t,
C
N-
-N
Рис. 6.1. Кривые термической деструкции
полипиразола (табл. 6.2, № 46) и политри-
азола [39]:
/. 4 — на воздухе; 2, 3 — в азоте.
С6Н5 СвН5
I 1
N N N N N
482
ствие частичного структурирования. Полибиспиразолы
нерастворимы в полярных растворителях и концентрированных
кислотах.
Температуры размягчения и термическая стойкость почти не
зависят от изомерного состава полипиразолов. Деструкция
полностью ароматических полипиразолов начинается ниже их
температур размягчения. Температура размягчения полипиразолов,
в ароматических ядрах которых имеются метильные группы,
колеблется от 150 до 250 °С. В этой же области размягчаются алифа-
тическо-ароматические полипиразолы, в то время как
алифатические полимеры размягчаются при температуре около 100 °С.
Термостойкость полипиразолов лежит в тех же пределах, что и
политриазолов (рис. 6.1). Ароматические полипиразолы на воздухе
и в азоте устойчивы до 470°С. Выше этой температуры на воздухе
происходит быстрое разложение, в то время как в инертной среде
этот процесс протекает довольно медленно. При 800°С в инертной
среде остаток ароматических полипиразолов составляет около
60%- Деструкция алифатическо-ароматических полимеров этого
класса начинается при температуре выше 350 °С. Пленки из 3,5-
полипиразолов получают поливом из раствора в диметилформа-
миде на поверхность ртути. Прочность при растяжении пленки из
полипиразола № 43 составляет 1400 кгс/см2. Высокая прочность
объясняется наличием сильных водородных связей между
макромолекулами [42] :
N—NH N—NH
HN—N HN—N
-R
О
6.1.2. ПОЛИГИДАНТОИНЫ
О
-о-
Ч
О
Полигидантоины в промышленности получают взаимодействием
диизоцианатов с бисглицидиловыми эфирами. Ароматические
полигидантоины не плавятся и разлагаются при 300 °С. Они
могут длительно работать при 200 °С. В промышленном масштабе
лаки и пленки из полигидантоинов получены в 1968 г.
Изготовление электроизоляционной пленки и нанесение кабельной изоляции
проводят из раствора в метиленхлориде или крезоле.
6.1.2.1. Получение полигидантоинов
Полигидантоипы получают взаимодействием диизоцианатов
с бисглицидиловыми эфирами или нитрилами, а также
взаимодействием диизоциаиатов с синильной кислотой или полиприсое-
16*
динением диаминов или диизоцианатов к эфирам малеиновой или
фумаровой кислоты.
Взаимодействие диизоцианатов с бисглицидиловыми эфирами.
Для синтеза полимеров [46—59] использована известная в
органической химии реакция, описанная Велером [45] в 1911 г.:
пС2Н5ОСОСН2Ш
NHCH2COOC2H5
+ OCN
сн2
NCO
[
—iV-Vi—NCONH
I
\
CH2COOC2H5
о
—CH2—^Qj)—NHCON—1
C2H5OOGCH2J„
I-
C2H5OH
О
п
Синтез осуществляют в расплаве или в растворе. В расплаве
процесс проводят при 200 °С, используя в качестве катализатора
триэтилендиамин. Образующийся при этом форполимер —
замещенный полиуреидоэфир циклизуется при 250°С. Для получения
полигидантоинов с более высокой молекулярной массой
используют либо скрытые диизоцианаты, либо фенилуретаны [48]. При
синтезе в растворе в качестве растворителя применяют фенол или
хлорированные углеводороды, такие, как метиленхлорид и
хлороформ [46, 51, 53].
Для получения на первой стадии достаточно
высокомолекулярных полиуреидоэфиров синтез проводят при 50°С. Фенол и
крезол являются не только растворителями, но и
высокоактивными катализаторами. Превращение на первой стадии должно
быть количественным, так как спирт, выделяющийся при
циклизации, реагирует с изоцианатом с образованием уретанов [53|.
Форполимеры — полиуреидоэфиры хорошо растворяются во
многих органических растворителях, за исключением алифатических
углеводородов и спирта. В присутствии химических
дегидратирующих агентов циклизация проходит при 50°С. При термической
циклизации в растворе выше 150 °С наряду с циклизацией
протекает деструкция форполимера [46].
Используемые в качестве мономеров бисглицидиловые эфиры
получают взаимодействием ароматических диаминов с хлоруксус-
484
ной кислотой в присутствии таких оснований, как карбонат
кальция:
H2N
.—СН2—(Q;—NH2 + 2CiCR2COOCH3
CH2
NHCR2COOCH3
Cy
CH3OOCCR2HN
Их получают также конденсацией формальдегида с эфирами
ct-анилинокарбоновых кислот, синтезируемых по методу Штрекера
из анилина [53] :
NH2 NHCR2CN NHCR2COOC2H5
CR2O — HCN
C2H5OH, H2O
CH2
CH2O
-н2о
NHCR2COOC2H5
—> C2H5OOCCR2HN—
При этом одновременно образуется трисглицидиловый эфир,
который используют для получения сшитых полигидантоинов:
c2h5ooccr2hn
СН2
сн2—,;Q)—nhcr2cooc2h5
—NHCR2COOC2H5
В промышленном масштабе полигидантоины до сих пор полу-,
чают только в крезоле. Образующийся раствор используется для
нанесения кабельной изоляции [51].
В реакции с диизоцианатами вместо бисглицидиловых эфиров
применяют соответствующие динитрилы [60] :
п
Г
L
NCONH
CH2CN
Н2СО
+ «OCN (Q) СН2
gCH2—(^)~NHCON-1
NC-CH2J„
Образующийся в качестве промежуточного продукта иминоги-
дантоин в результате гидролиза превращается в гидантоин:
О О
п
Н2О
О
О
485
Взаимодействие эфиров малеиновой и фумаровой кислот с
диаминами и диизоцианатами
2ROOCChh=CHCOOR + NH2(CH2)*NH2 —>
ROOCCHNH(CH2 )XNHCHCOOR
I
ROOCCHa
I
CH2COOR
OCN-
-©—*-©
-NCO
CH2COOR
N-(CH2)*-N
CH2COOR
-N—
Поскольку ароматические диамины обладают низкой
реакционной способностью, этот способ пригоден для получения прежде
всего алифатическо-ароматических полигидантоинов [53].
Синтез полииминогидантоинов из диизоцианатов и синильной
кислоты. Взаимодействие диизоцианатов с дикарбамоилцианами-
дами в растворителях основного характера или гомополиконденса-
ция последних [61] приводит к образованию поли-5-иминогидан-
тоинов:
О О
rtNCCONHRNHCOCN
«OCNR'NCO
Пиридин
—R—N^\N R' N^\N—
n
ИЛИ
nNCCONHRNHCOCN
Пиридин
—HCN
О
L HN-
n
Дикарбамоилцианамиды, используемые в качестве мономеров,
получают из диизоцианатов и синильной кислоты:
OCNRNCC4 2HCN —> NCCONHRNHCOCN
Полигидантоины, полученные поликонденсацией, имеют более
низкую молекулярную массу.
В табл. 6.2 показано строение элементарных звеньев ряда
полигидантоинов и полииминогидантоинов.
6.1.2.2. Физико-механические свойства
Полигидантоины растворяются в феноле, крезоле, метиленхло-
риде и серной кислоте; набухают или частично растворяются в
таких сильнополярных растворителях, как диметилформамид, диме-
тилацетамид, диметилсульфоксид и N-метилпирролидон,
486
300 400 t,C
Рис. 6.2. Кривые
термической деструкции поли-
гидантоинов в аргоне (а)
и на воздухе {6} (цифры
на кривых соответствуют
номерам полимеров,
перечисленных в табл. 6.2)
[48].
Ароматические полигидантоины (№ 3—12) разлагаются ниже
температуры плавления; полииминогидантоин (№ 13) плавится
при 250°С, а аналогичные алифатические соединения — при 100°С.
Разложение ароматических полигидантоинов в инертной среде
начинается при 320СС, а на воздухе —при 300°С (рис. 6.2).
При 400 °С потеря массы относительно невелика.
Термоокислительную стойкость полигидантоинов можно повысить добавлением
аминов, фосфитов и многоатомных фенолов [57]. В течение
длительного времени ароматические полигидантоины термостабильны
при 200 °С.
6.1.2.3. Переработка, свойства и применение полигидантоинов
Полигидантоины используются главным образом для получения
термостойкой проволочной изоляции и изготовления
электроизоляционной пленки (из растворов полимеров в крезоле или метилен-
хлориде). Раствор форполимера применяется в качестве
связующего для стеклопластиков.
Проволочная изоляция. ПолигиДантоиновую проволочную
изоляцию получают из 20—35 %-ных растворов полимера в крезоле
или метиленхлориде. Вязкость лаковых растворов регулируют
путем разбавления их ароматическими углеводородами, такими, как
толуол или ксилол. Оптимальные изоляционные свойства имеют
полимеры с молекулярной массой выше 10 000 [51].
Верхний предел термостойкости ароматических
полигидантоинов, оцениваемой по электрической прочности, составляет 200°С
(по DIN 46453). До 250 °С диэлектрические свойства
полигидантоинов практически не изменяются. Стойкость к действию
растворителей можно повысить, добавляя 0,3—1 % таких соединений, как
ацетилацетонат железа или бутилат титана. При тепловом ударе
(изгибание проволоки с изоляцией при температуре эксперимента)
трещины появляются при температуре выше 280 °С.
Преимуществом полигидантоинов по сравнению с другими теплостойкими
электроизоляционными лаками являются отличная перерабатывае-
Мость и хорошая совместимость с другими лаками, используемыми
для проволочной изоляции. Такие лаки выпускает фирма «Bayer
AG» под названиями «Резистерм РН20» и «Резистерм РН10». Лак
для проволочной изоляции выпускается в виде 30%-ного раствора
в крезоле, который стабилен при хранении. Вязкость 15%-ного
раствора лака в крезоле при 20 °С с pH = 20 составляет 2000 сП,
а лака с pH = 10 — 6000 сП.
487
Таблица 6.2. Строение элементарных звеньев некоторых полигидантоинов и полииминогидантоинов
1
2
3
4
Элементарное звено
О
II
—(СН2N—N^NS
J—
О
-(СН2)б—N-^N
J
О
||
<
HUrf
L
1
О
II
0
II
_
""OTT
0 0
Литератур ный
источник
[48, 54]
[54]
[46]
[48. 54]
о
о
148]
о 9
-nK^n—r—n^^n—
E41
о о
NN
[47. 48]
О О
СН2—(
r— о —м^^м—
148]
Продолжение табл. 6Л
Элементарное звено
Литературный
источник
о
о
n-
о-
О-
X
О
О
10
,=-© ¦
H-
\^7
ш
о
о
11
СНз
НзС СНз
[38]
[46]
[55]
12
13
о о
N II
о о
—R—NKNM R' N^^N—
О^ ^NH HI
R =-—(СН2L—, — (CH2)g— ,
R/ = _(СН2L— —(СН2N—
— (СН2L—, —(СН2N—,
R =— а )>—^н2—¦;
R' = -(СН,),—.
OJ
—(СН2L—¦ —(СН2N—.
1501
[611
jn* I 1 11 1 1
-700-50 u 50 100 150 200 t^C
-50 0 50 100 150 200 t,C
50
ioo
-50 0 50 100 150 200 t°C
Рис, 6.3. Зависимость диэлектрической проницаемости (а), тангенса угла
диэлектрических потерь (б) и удельного объемного электрического сопротивления (в)
полигидантоинов от температуры [62].
Рис. 6.4. Деформационно-прочностные кривые полигидаптоиновой пленки [62].
Электроизоляционная пленка. Полигидантоиновые пленки
получают поливом из раствора в метиленхлориде. Пленки отличаются
хорошими диэлектрическими свойствами при 260 °С и высоким
электрическим сопротивлением. Ниже приведены показатели
свойств полигидантоиновых электроизоляционных пленок [64] :
Плотность, г/см3 ....... 1,27
Прочность при растяжении, кгс/см2 1000
Относительное удлинение при растяжении, % 100
Водопоглощение (за 24 ч), % 4,5
Термостойкость, ЭС 160
Формоустойчивость, °С 270
Электрическая прочность (пленка 40 мм), кВ/мм . . . 200
Удельное объемное электрическое сопротивление, Ом см
сухая пленка 4-Ю16
после выдержки в течение 24 ч при 95 %-ной отн.
влажности , 2- 1016
Диэлектрическая проницаемость при 50 Гц 3,3
Тангенс угла диэлектрических потерь при 50 Гц .... 1,5 - 10 —3
Стойкость к тлеющему разряду, мин 2000
На рис. 6.3 показано изменение диэлектрической проницаемости,
тангенса угла диэлектрических потерь и удельного объемного
электрического сопротивления от температуры. На рис. 6.4 и 6.5
представлена диаграмма удлинения при растяжении (для 20 °С)
и температурная зависимость прочности при растяжении. На
^гс/см2 Рис- 6-6 приведена зависимость е и
tgo от частоты.
Рис. 6.5. Зависимость прочности при
растяжении полигидантоиновой пленки от темпе-
0 50 100 150 200 t,°C ратуры [62].
492
ю m2 ю3 ю* m5 юе f,ru ., , с t
ю ю2 ю3 ю* ю5 m5 f,ru
Рис. о.б. Зависимость диэлектрической проницаемости (а) и тангенса угла
диэлектрических потерь (б) полигидантоиновой пленки от частоты [62].
Полигидантоиновые электроизоляционные пленки устойчивы
к действию разбавленных кислот и щелочей, пропиточных смол,
углеводородов, таких, как бензол, циклогексан, спиртов и эфиров,
таких, как этилацетат. Они сильно набухают и даже растворяются
в ацетонитриле, ацетилацетоне, циклогексаноне, о-дихлорбензоле,
диоксане, диметилформамиде, уксусном ангидриде, крезоле, мети-
ленхлориде, три- и тетрахлорэтане, трихлорэтилене и
концентрированной серной кислоте [64].
Полигидантоиновую пленку можно склеивать с помощью таких
растворителей, как метиленхлорид. Для склеивания с металлами
используют двухкомпонентную композицию на основе
полиуретанов или эпоксидных полимеров. Сваривание осуществляется
термоимпульсным методом при температуре выше 300 °С. Пленка
обладает хорошей термостойкостью, что позволяет проводить глубокую
вытяжку при высоких температурах; может длительно
эксплуатироваться при 160 °С. Она используется в качестве пазовой изоляции
электродвигателей, кроющих лаков для трансформаторов и
катушек [62]. Металлизированные медью пленки используют для
получения печатных плат. Пленки можно сваривать при 260 °С [63].
Самоклеющаяся лента на основе полигидантоина пригодна для
защиты печатных плат. Полигидантоиновая электроизоляционная
пленка марок Шо-4089 и «Резистофол» выпускается фирмой
«Bayer AG». Самоклеющаяся лента с отверждающимся при
нагревании клеевым слоем, а также свариваемые образцы на основе
этой пленки производятся фирмой CMC в Дюссельдорфе.
Связующее для стеклопластиков. Связующее наносят в виде
раствора форполимера на стеклянное волокно. Циклизацию
проводят при 150°С. Верхняя температурная граница применения
стеклопластика в условиях длительной эксплуатации 200—220 °С.
Через 100 ч термостарения на воздухе прочность при изгибе
стеклопластика составляет 3300 кгс/см2.
6.1.3. ПОЛИОКСАЗОЛЫ И ПОЛИИЗООКСАЗОЛЫ
Полиизооксазолы получаются аналогично 3,5-пиразолам при
взаимодействии полидиэтинилбепзола с гидроксиламином [42,
68—70] :
Г N—О Т
—iUj—с=с-с=с- +«h2noh —-> —iLJj—сн2—
*" in L ^ J/t
493
Синтез полиднэтинилбензола проводят окислительной
поликонденсацией [43, 44]. При взаимодействии бис-/г-цианбензонитрилоксида
с 1,4-диэтинилбензолом образуется неплавящийся до 500 °С
полимер с чередующимися 1,4-фениленовыми и 3,5-изооксазольными
звеньями [232] :
(Г))—CN~>0
-O-feCH
-О-
О
о
О
Полиизооксазолы с метиленовыми мостиками в цепи устойчивы
на воздухе до 280 °С и в ииертной среде до 320 °С. Температура
разложения полимеров лежит ниже их температуры плавления.
Прочность пленки на основе полиизооксазола, содержащего фени-
леновые, изооксазольные и метиленовые звенья, составляет
650 кгс/см2 [42]. Полиоксазолы, имеющие строение
устойчивы в инертной среде до 400 °С [67].
6.1.4. ПОЛИОКСАЗОЛИДОНЫ
-N/4
-CHaO ,
о-
О'
-о
IQj
,—OCH,—
Поли-2-оксазолидоны получают из диглицидиловых эфиров и
диизоцианатов или диуретанов, а также дегидрохлорированием
полиуретанов с хлорметильными боковыми группами. Температура
размягчения ароматических поли-2-оксазолидонов составляет 170—
200 °С; они термически устойчивы до 300 °С. Перерабатываются
прессованием при 200 °С. Могут длительно эксплуатироваться при
150°С. Прочность при сдвиге клеевого соединения высокосортной
стали с помощью полиоксазолидонов можно повысить путем
последующей термообработки.
6.1.4.1. Получение поли-2-оксазолидонов
Циклоприсоединение диглицидиловых эфиров к диизоцианатам.
Поли-2-оксазолидоны получают 1,3-циклоприсоединением диглици-
днловых эфиров к диизоцианатам с использованием в качестве
катализаторов четвертичных галогенидов аммония, хлорида и бу-
тилата лития в полярных растворителях типа дихлорбензола или
494
диметилформамида [15—20]:
uOCXRNCO
[.
«НаС CHR'CH—СН2 Y k^N R N\/ - -„
О О
При 140—160 °С в диметилформамиде даже без катализаторов
образуются полиоксазолидоны [18]. В случае использования бути-
лата лития происходит образование уретан-аниона, который
реагирует с эпоксидной группой [20] :
... — '())—NCO —> (у.—МНСООС4Н9
C4H9OH
л
—NCOOC4H9
С4Н9ОН
••¦—(( h—NCOOC4H9 + НаС СНСЫ2О—;( );—
V
о"
!
/СН2СНСН2О—
_с4н9о-
соос4н9
Взаимодействие диглицидиловых эфиров и диуретанов. Эта
реакция протекает в диметилформамиде в присутствии триэтилами-
на, пиридина или тетраметиламмонийиодида с образованием
растворимых полиоксазолидонов [21, 22]:
rtROCONHR'NHCOOR Г /^N—R'—
I
Г /^N—R'—N^X. p„ Л
I I ji
пН2С CHR"CH-GH2 L Ю О* -„
Поскольку полимеры плохо растворяются, в этих условиях можно
получить только относительно низкомолекулярные продукты.
Дегидрохлорирование полиуретанов с боковыми хлорметиль-
ными группами. Полиуретаны с хлорметильнымп группами
получаются при взаимодействии бисхлоргидринов с диизоцианатамп
495
в анизоле в присутствии дибутилдилаурата олова в качестве
катализатора [23] :
nClCH2CHCH2OROCH2CHCH2CI + nOCNR'NCO —>
I I
ОН ОН
СН2С1 СН2С1
I I
—OCHCH2OROCH2CHOCNHR'NHC—
О
п
В диметилформамиде в присутствии метилата Na происходит
циклизация:
СН2С1 СН2С1
I I
—OCHCH2OROCH2CHOCNHR'NHC
II II
О О -!„
—CH2OROCH2 ^M
Скорость циклизации зависит от структуры полиуретана. Так,
полиуретан на основе дифенилметандиизоцианата "циклизуется при
комнатной температуре меньше чем за минуту. Этим методом
можно получить полиоксазолидоны с характеристической вязкостью
в диметилформамиде до 0,5 A00 мл/г).
6.1.4.2. Физико-химические свойства полиоксазолидонов
Строение звена и температуры размягчения наиболее известных
полиоксазолидонов приведены в табл. 6.3. Ароматические
полиоксазолидоны аморфны [15]. Они существуют в двух изомерных
формах, наличие которых подтверждают данные ЯМР-спектро-
скопии:
и
L 1
-о
Поли-2-оксазолидоны хорошо растворяются в диметилформамиде,
диметилсульфоксиде и крезоле, умеренно растворимы в метилен-
хлориде и хлороформе [15, 21]. В ароматических углеводородах,
спиртах и эфирах не растворяются. Температура размягчения по-
ли-2-оксазолидонов прежде всего зависит от строения исходных
уретанов [21]. Ароматические полиоксазолидоны размягчаются
в интервале температур 170—200 °С, алифатическо-ароматиче-
ские — в интервале 80—110 °С? алифатические полимеры предстаз-
wo
Рис. 6.7. Кривые термической деструкции поли-2-оксазолидонов на воздухе
(цифры на кривых соответствуют номерам полимеров, перечисленных в табл. 6.3)
П51:
ляют собой вязкие масла. Большинство ароматических полиоксазо-
лидонов стабильно на воздухе и в инертной атмосфере до 300 °С
(рис. 6.7).
6.1.4.3. Переработка и эксплуатационные свойства поли-2-
оксазолидонов
Поли-2-оксазолидоны могут перерабатываться в изделия
прессованием при 200 °С и давлении 70 кгс/см2. Изделия могут длительно
эксплуатироваться при 140—170°С [20]. Физико-механические
свойства поли-2-оксазолидонов приведены ниже (строение
полимеров соответствует данным табл. 6.3) [20] :
Полимер
Прочность при растяжении, кгс/см2 667 562
Относительное удлинение при разрыве, % 6,6 3,9
Прочность при изгибе, кгс/см2 1 050 1 170
Модуль при изгибе, кгс/см2 29 000 34 500
Ударная вязкость, кгс-см/см2 11 20
образца с надрезом 2,5 2,8
Деформационная теплостойкость при 18.5 кгс/см2. °С , . . . 143 176
Прочность при сдвиге клеевого шва при склеивании
высокосортных сталей с помощью полиоксазолидонов повышается при
последующем нагревании и в течение 2 ч при 100°С [19]. Так, например,
клей, состоящий из 5 масс. ч. диглицидилового эфира дифенила,
2 масс. ч. полиуретана с активными изоцианатными группами,
1,8 масс. ч. диаминодифенилметана, 0,8 масс, ч. тетраметиламмо-
нийиодида, имеет прочность при сдвиге 182 кгс/см2 при —195 °С,
158 при 25 °С, 83 при 121 °С и 18 кгс/см2 при 204 °С.
6.1.5. ПОЛИТИАЗОЛЫ
497
Таблица 6.3. Строение звена и температуры размягчения полноксазолидонов
Элементарное звено
Т-П1-
дл/г
Растворитель
разм'
°С
Литературный
источник
*N—, -Я^,—N^V"CH2O—l^v—OCH2-
ад-^-ош,-
ОСН2—
—СН2О
сн2—
СНз О*
OpO-i
S—СН20—(Ръ—ОСН
d У1
0,16
0,18
0,39
0,12
ДМФ
ДМФ
Крезол
ДМФ
165
235
170
177
170
275
[21]
[21]
[21]
[21. 23]
[21]
[21]
СНз
о
N^ СН2О ,
о—Чо
СН3
I
—сн2снсн2—с—сн2сн2—n
СНз СНз 0J-
.СН2О (<^> ОСНг—
сн2о
—осн2—
СНз
СНз
СНз
СНз
осн2—
осн2—
0,41
0,09
0,П
0,09
0,51
0,10
0,11
0,41
0,13
0,25
ДМФ
ДМФ
ДМФ
ДМФ
Крезол
ДМФ
ДМФ
ДМФ
ДМФ
ДМФ
195
123
130
192
188
125
150
170
134
90
100—
112
[15, 17]
A9, 20]
[21]
[21, 23J
11В. 21]
[15, 20]
[15]
[15]
Продолжение табл. 6.3
lTll,
дл/г
0,12
0,30
0.20
0,21
Растворитель
ДМФ
Крезол
ДМФ
Масло
т
разм'
°с
83
88
86
160
157
Литературный
источник
о-
-N—сн2снсн2о—.-^—осн2-
^_ он
—('О)—СН2—
о-
—СН2О(СН2LОСН2-
L ОХ
СН2О(СН2LОСН2—
°—^о -"^сн3 о^—°
—сн2
о-
о-
|
О
N— (CH2N—N
СН2О(СН2LОСН2—
о-
ю о-
-о
СНз
о-
¦N-
-к
о
¦СНз
Н3С СНз
СНз
-с—
СНз
[24]
[15, 23]
[231
[23]
СНз
-с—4Q>—0СН2-
СНз
[15]
15}
[151
—ОСН2-
Политиазолы получают поликонденсацией а-галогенкетонов
с дитиоамидами. Алифатическо-аромэтические политиазолы
пригодны для изготовления волокон, они устойчивы к действию УФ-лу-
чей, обладают химической стойкостью, но имеют низкую
теплостойкость. Пленки из политиазолов характеризуются хорошими
диэлектрическими свойствами.
6.1.5.1. Получение политиазолов
Политиазолы получены впервые Эрленмейером [1—7] в 1944 г.
взаимодействием а-галогенкетонов с дитиоамидами:
«BrCH2CORCOCH2Br + /zH2NCR'CNH2 —
BrCH2C— — R
[
—H20; —НВг
и il
S S
N N
-CNH2
Рядом исследователей синтезированы высокомолекулярные
полимеры [8—13]. Поликонденсацию проводят в таких растворителях,
как диметилформамид, ацетон и уксусная кислота, при 30—
150°С. Для синтеза полибитиазолов используют а-галогенкетоны
и дитиооксамидьг.
BrCH2CORCOCH2Br г N N
U w
H2NC—CNH2 I J J i
Vl
Jl I
S S
t
6.1.5.2. Физико-химические свойства политиазолов
Политиазолы — сильно окрашенные кристаллические
полимеры. Данные об их строении, температурах плавления и
разложения приведены в табл. 6.4.
Алифатическо-ароматические политиазолы плавятся при 230—
500 °С (№ 1—6). Температура плавления их понижается с
увеличением числа мети леновых групп в цепи. Температура
разложения ароматических политиазолов, состоящих из чередующихся
ароматических и тиазольных циклов (№ 7—19), лежит ниже и к
температуры плавления. Политиазолы, в цепи которых находятся
мостичные атомы и группы, например —О—, —СН2—, плавятся
при 350—450 °С. Полиаминотиазолы плавятся выше 460 °С с
разложением (№ 21—26). При 500 °С ароматические политиазолы
(№ 7—19) ограниченно стабильны.
На рис. 6.8 приведены результаты термогравиметрического
анализа политиазолов и политетрафторэтилена.
Алифатическо-ароматические политиазолы разлагаются на воздухе при 350—500 °С.
Стабильность их уменьшается с увеличением числа метиленовых
групп в цепи. Полиаминотиазолы (№ 21—26) стабильны на воз-
501
Таблица 6.4. Температура плавления (размягчения) и разложения политиазолов
1
2
3
4
5
6
Элементарное звено
N N
r R'-r V-
1
!
R = -(CH2L-, — (СН2L-, -(СН2L-, —fCH2O— -(СН2)9- -(СН2O-
R'=-(CH2L—, -, — (СН2O— -(СН2L- -(СН2L-, -(СН2O—
N , ч N
ь
!
\
M о
о
N /^^ N
~~(CH2)—f Х
\ "( ( ) / m(\ ^^\
N у—v N
—(СН,L—f ^~\C/—(Iх ^^
s ' -—s
N у . N
-(сн2)8—f \-\Or-f ^1—
s
1
v ' M ^
Ь
/—\ N /—\ /—\ N
s-
J
T °C
500
265
250
265
Уразм = 226
T °C
разл1
>500
290
487
493
440
Литературный
источник
[7]
[И]
[11]
[3, 8, l I
12]
[12]
[Ю]
N ,—ч
-rv®
N
N ,—ч N
10
N ,—ч N
-О-
N
N
N
13
IN
> 500
240-250
>500
^разм = 368
>500
> 500
> 500
>5UO
>500
500
400
[П, 13]
[3, 8, 11,
13]
[H]
[11]
[8, 11]
[101
Продолжение табл. 6.4
14
15
16
17
18
19
N j—,
S
о
Vvo>-
о
N y—y
—fAl
N
—(СН2)б~
-О-
Элементарное звено
N
S
N
i
^=^ fi I
с
о
-л N
S
>520
>520
>520
440
>520
220-240
разл' L
>500
(в N2)
Литературный
источник
[13]
[13]
[13]
[13]
[13]
[13]
20
21
22
23
24
25
26
27
28
-соон ноос
460
460
460
460
460
460
> 500
(в N2)
350
400
390
400
440
405
500
20
О
400 450
550 t°C
Рис. 6.8. Кривые термической деструкции по-
лнтиазолов в азоте: № 8 и 7 (в табл. 6.4):
/ — иолитиаэол № 8 (табл. 6.4); 2 — полиамид HT-};
3 — политпазол № 7 (табл. 6.4); 4 —
политетрафторэтилен [11].
духе до 350 °С.
Алифатическо-ароматические политиазолы растворяются в
серной кислоте, бензолсульфо- и три-
фторуксусной кислотах и ведут себя в
растворе как полиэлектролиты [11].
Исследовать светорассеивание
окрашенных и флуоресцирующих
растворов невозможно.
Политиазолы с 2,2-дифенильными группами в цепи (№ 9, 14,
15) растворяются в диметилформамиде, диметилацетамиде и тетра-
гидрофуране. Алифатическо-ароматические политиазолы образуют
с серной кислотой, бензол- и нафталинсульфокислотами
кристаллические соли, которые растворяются в нитробензоле и этиленгли-
коле.
Полиаминотиазолы с гс-фениленовыми группами растворяются
в концентрированной соляной кислоте, а с ж-фениленовыми —
в серной и уксусной кислотах, диметилформамиде, диметилсуль-
фоксиде, ж-крезоле.
6.1.5.3. Политиазольные волокна
Алифатическо-ароматические политиазолы пригодны для
изготовления волокон из расплава, из раствора и сухим формованием.
Волокна отличаются высокой химической стойкостью и
устойчивостью к действию УФ-лучей. Волокна на основе алифатических
дитиоамидов не пригодны для работы при высоких температурах
[[И, 12].
Политетраметиленфенилентиазол
N
[¦
Pjj'денье
2,0
можно формовать из расплава в атмосфере азота при 287 °С. Сухое
формование проводят из 15%-ного
раствора в муравьиной кислоте в
токе горячего воздуха при 115—
120 °С. При мокром формовании из
раствора в муравьиной кислоте оса-
дительной ванной служит вода.
В табл. 6.5 приведены
характеристики волокна, полученного различ*
40 50 еа%
Рис. 6.9. Деформационно-прочностные
кривые политетраметиленфенилентиазольных
волокон при мокром формовании (/) н
формовании из расплава B) [12].
5UI
Таблица 6.5. Характеристики
политетраметиленфенилентиазольных волокон [12]
Показа1ель
Плотность, г/см3
Температура плавления, °С
Кратность вытяжки
Прочность при растяжении» г/денье
Относительное удлинение при разрыве, %
Модуль при 4% -ном удлинении, г/денье
Температура нулевой прочности, °С
Формование
из расплава
1,33
242
3X8
1,9
64
24,7
-
Сухое
формование
1,33
242
2,5 X 80
1,5
37
17,7
240
ными методами. Волокна из расплава имеют более высокую
жесткость. На рис. 6.9 показано изменение относительного
удлинения при разрыве политиазольных волокон. В нерастянутом
состоянии C5,5 денье) прочность составляет 0,7 г/денье и удлинение
129 %. С увеличением молекулярной массы политиазола прочность
возрастает. Политиазольное волокно отличается высокой
гидролитической стойкостью. Выдержка в течение 24 ч в 40 %-ном растворе
щелочи или 20%-ном растворе серной кислоты не влияет на
прочность, модуль и удлинение при растяжении. После 24 ч пребывания
в 40%-ной серной кислоте прочность при растяжении понижается
на 68 % •
6.1.5.4. Политиазольная пленка
Из раствора алифатическо-ароматических и ароматических по-
литиазолов может быть получена пленка, отличающаяся высокой
прочностью и хорошими диэлектрическими свойствами. Показатели
свойств политиазольной пленки приведены в табл. 6.6.
Таблица 6.6. Показатели свойств политиазольной пленки [10]
Показатель
Полимер
№ 13
(см. табл. 6.4)
Температура размягчения, °С
Прочность при растяжении, кгс/см2
Относительное удлинение при разрыве,%
Диэлектрическая проницаемость при 1000 Гц
Удельное объемное электрическое
сопротивление, Ом см
Температура разложения, °Q
226
255
13
5,8
3,7- 1012
320
368
283
7
3,6
8- 1015
400
607
6.2, ЦИКЛОЦЕПНЫЕ ПОЛИМЕРЫ С ТРЕМЯ
ГЕТЕРОАТОМАМИ В ПЯТИЧЛЕННОМ ЦИКЛЕ
6.2.1. ПОЛИТРИАЗОЛЫ
- N N
NH
—П
6.2.1.1. Получение политриазолов
Политриазолы получают в полифосфорной кислоте
взаимодействием полигидразидов или полиоксадиазолов с гидразином.
Полимеры с триазольным циклом (табл. 6.7) можно разбить на пять
групп:
N N
¦R—
R—
NH2
N N
V
-R—
-N N
П
N
—N N
II II
¦N
Незамещенные поли-1,2,4-трназолы (№ 1, 2)
Поли-4-амино-1,2,4-триазолы (№ 3—26)
Алкил (арил) замещенные в положение 4 поли-1,2.4-
триазолы (№ 27—32)
Поли-1-фенил-1,2,4-триазолы (№ 33—41)
Поли-!ДЗ-триазолы (№ 42—48)
R—
Из перечисленных полимеров практическое применение для
изготовления высокотермостойких волокон и пленок нашли поли-4-фе-
нил-1,2,4-триазолы (№ 27—29).
Поли-4-фенил-1,2,4-триазолы. Получение поли-4-фенил-1,2,4-
триазолов основано на использовании реакции, предложенной
Хьюсгеном [95] для синтеза замещенных триазолов, путем
конденсации высокомолекулярных полигидразидов с ароматическими
508
аминами в полифосфорной кислоте [83, 84] :
HN—NH HN—NH
N
СеН
6
сен
6П5
Количественное превращение достигается при нагревании в
течение 3 ч при 260 °С, 24 ч при 204 °С или 140 ч при 175 ГС. При
взаимодействии анилина с поли-n- или поли-ж-фениленгидразидом
оптимальная концентрация анилина составляет 0,44 моль на 150 г
полифосфорной кислоты. Предполагается, что при поликонденсации
сначала образуется аддукт полигидразида с полифосфорной
кислотой [83]:
О _ о О
НО—
О О
II II
,—с с—
О о
II /0Н || /ОН
—Р—
но
• I
о о
ОН
—i Г\\— С С- + -О- Р
?/он
HN—NH
-"+ О" II ¦ - "So-
^-^ HN—N
Ниже 200°С анилин присоединяется к углеродному атому,
связанному с фосфором, с последующим отщеплением фосфорного
остатка и циклизацией. При температуре выше 200 °С происходит
раскрытие карбонильной связи с образованием фосфор —
кислород с последующей протонизацией оксадиазольного цикла. Анилин
атакует углеродный атом оксадиазольного цикла и расщепляет
связь углерод — кислород. С отщеплением протона происходит
циклодегидратация и образование поли-4-фенил-1,2,4-триазола.
При концентрации анилина 0,44 моль на 150 г полифосфорной
кислоты полигидразид не деструктирует. Анилин реагирует с
полифосфорной кислотой с образованием соли. Уменьшение
концентрации анилина ниже 0,44 моль на 150 г кислоты приводит к
гидролизу полигидразида за счет избытка полифосфорной кислоты
(рис. 6.10). При увеличении концентрации анилина сверх этого
количества происходит деструкция полимера вследствие переамиди-
рования.
Анилин может непосредственно реагировать с полиоксадиазо-
лом F.2.2), политриазол может быть получен в одну стадию в
полифосфорной кислоте из дикарбоновых кислот, гидразинсульфата
и анилина [86, 89, 96, 97] :
N N 1 Г N N 1
О
+
п
1Q
N
I
509
Таблица 6.7. Температуры стеклования, плавления и разложения некоторых политриазолов
Элементарное звено
Гпл-
т
разл
(BgN2).
т
разл
(на
воздухе),
°С
180—240
400
400
320
320
N N
N
I
NH2
N N
N
I
NH2
N N
N
NH2
N N
N
NH2
N N
N
NH2
-(CH2J-
¦(СН2)з-
-(CH2L-
-(CH2M-
"(CH2N~
36Q
137-141
300
260
300-315
Ns
10
11
12
13
Элементарное звено
M M
г
_l
1
\ /
N
i
!) (CH2O—
NH2
M M
N
i
4
J—(CH,)r-
NH2
M KT
11
_l
ч J—(CH,)r-
NH2
N m
J~(CH2I0-
i
Tc-
°C
°C
218-225
225-260
245
П родолженпе
7
разл
(в N2),
°C
разл
(на воз-
Духе),
QG
табл. 6.7
Литературный
источник
[76]
[76]
[77]
[77]
N N
N
NH2
N N
(CH2J0-
N N
N
NH2
N N
V
NH2
N N
N
i
NH2
-(CH2K"S-(CH2K-
-(CH2L-S-(CH2L—
225—228
210—213
150-155
160-165
19
20
21
22
Элементарное звено
\т м
-1
Y
J—(CH2M-S-(CH2M-
NH2
Y
J—(CH2K-SO2-(CH2K-
NH2
M M"
г*
. J—(CH2L-SO2-(CH2L-
N
1
NH2
N N
Г и
v J—(CH2N-SO2—(CH2)S-
NH
1
°C
206—208
208—210
258-260
220-225
Продолжение
разл
(в N2),
°G
T
разл
(на
воздухе),
°G
табл, 67
Литера
турный
источник
[79]
[79, 80]
[79, 80],
[79, 80J
23
N N
V
NH2
-(CH2J-O(CH2)*O-(CH2J-
N N
—СН
NH2
N N
25
N
NH2
N N
J ОСН2—(Q) СН2О-
26
N
NH2
-®-
(CH2J—(С ) )—(СН2JО-
N
27
[81]
[82]
120
[82]
180
[82J
512
Продолжение табл. 67
Элементарное звено
ТТЛ1
Т
раз л
<в N2),
Т
разл
(на
воздухе).
Литературный
нсточник
N N
28
N N
29
30
31
AJ-
N
СНз
N N
N N
512
400
[83. 85]
187]
[86]
188]
32
33
34
35
N N N
N-
-N
(CF2);
о
N
-N
N
CF2)
250
365
130
340
225
320
1891
460
[39]
370
[39]
480
[39]
Продолжение табл. 6.7
Элементарное звено
с
°С
разл
разл
(на воз-
Духе),
36
37
39
F F
N N 4 A N N
ii i ^ / h i
200
210
225
200
300
255
300
450
440
470
450
40
41
42
43
44
45
C6H5
N n
CeH5 CeH5
N N F\ /F N N
—H2 С
ч _
N4 /N—CH2
—Н2С
>=1
N N
N
=N N
/N-CH
JL
1—R—
R=-(CH2N- -(CH2N-, -(CH2N-, -(CH2)e-
R' = — (CH2J—, —(CH2L—, —(CH2M—, — O(CH2JO-
CH2OCOCH2—N
140
125
270
265
450
470
170—200
[391
[39],
[90]
[91]
[92}
[91J
Продолжение табл. 6.7
№
Элементарное звеио
T
пл-
°G
разл
(в N2),
71
разл
(на воз-
духе).
46
I >
R-N
H
47
48
49
R'=-(CH2J-, _(CH2L—, -(CH2L-, -(CHj)e—, -(CH2M-
342
500
490
Рис. 6.10. Зависимость молекулярной массы t)np (8 НСООН с С=0,5%\дл/г
политриазола от концентрации анилина в рас- /000
творе поли-л«-(л)-фениленгидразида в
полифосфорной кислоте [83]. 800 —
Поли-4-феинлен-1,2,4-триазолы могут
получаться также взаимодействием 4-Н-
1,2,4-политриазолов с гидразидами циан-
карбоновых кислот [98], конденсацией в
расплаве гидразида терефталевой
кислоты с нитрилом той же кислоты [72],
по реакции ж(/г)-фениленбистриазолов с Ь^М'-дифенилизофтали-
мидоилхлоридом [99]. В последнем случае из-за высокой
чувствительности к влаге имидоилхлоридов получаются
низкомолекулярные продукты:
3 5 7 9
G} моль/150 г к-ты
Пиролиз
—H Cl; —N2
/zHN
N N
N
N n
C6HS
4-Метилзамещенные поли-1,2,4-триазолы можно получать
циклизацией полифенилен-Ы-метилазиламидразонов в вакууме при
250 °С [88]:
NH-
О'
сн3
NH
N N
-ОА
HN-
-NH
СНз
п
N N n
Поли-1-фенил-1,2,4-триазолы. Поли-1 -фенил- 1,2,4-триазолы
(№ 33—41) получают 1,3-биполярным присоединением динитрилов
521
к 2,2'-дифенил-5,5/-ж(гс)-фенилентетразолам при 180°С дод давле»
чием [39]:
«NCRCN
R = _(CF2)a- —(( ))—,
F3C
Полимеры растворяются в хлорбензоле и трихлорбензоле, но
не растворяются в концентрированных кислотах. Температура
стеклования находится в пределах 125—250°С, температура
плавления равна 255—365 °С. Термоокислительная деструкция на
воздухе протекает при 370 °С (№ 34) или 480 °С (№ 35).
Поли-4-амино-1,2,4-триазолы. Поли-4-амино-1,2,4-триазолы
(№ 3—26) образуются из тетразинов или при взаимодействии поли-
гидразидов F.2.2.1) с гидразином. При реакции безводного
гидразина с дииминоэфирами при 60 °С сначала образуются поли-1,2-ди-
гидротетразины, которые в присутствии НС1 превращаются в поли-
4-амино-1,2,4-триазол [82, 231]:
2nH2N—NH2
Диэтиловый
эфир
20%-ная НС1
Ароматические дигидразиды или полигидразиды F.2.2.1) при
взаимодействии с гидразином под давлением при 300 °С
превращаются в поли-4-амино-1,2,4-триазолы [100]:
522
HN—NH HN—
I I ^ I I
i i О в и
О О X^ О О
N N
2H2NNH2
_п
N N п
-ОА
NH2
п
Алифатические поли-4-амино-1,2,4-триазолы (№ 5—26)
плавятся при 150—250 °С. Температура плавления ароматических поли-
триазолов выше температуры разложения. Поли-4-амино-1,2,4-
триазолы с температурой разложения на воздухе 320 °С и в
инертной среде 400 °С гораздо менее термостойки, чем 4-фенилзамещен-
ные полимеры (№ 27—29).
Поли-1,2.3-триазолы. Поли-1,2,3-триазолы (№ 42—48)
получают циклоприсоединением /?Ж-диазидов к диэтинильным
соединениям [92, 93]:
—<О)—(О)—N3
-Jft
Их можно получить также гомоциклоприсоединением ш-этинил-
ш'-азидов [90, 91]. Полимер из чередующихся 1,2,3-триазольных и
/i-фениленовых циклов (№ 47) стабилен на воздухе до 500 °С.
6.2.1.2. Физико-химические свойства ароматических
поли-4-фенил-1,2,4-триазолов
Ароматические поли-4-фенил-1,2,4-триазолы образуют
стабильные растворы в муравьиной кислоте до концентрации 35%.
Характеристическая вязкость в муравьиной кислоте 1,79 или 2,56 (дл/г)
соответствует среднемировой молекулярной массе 13 600 или
26 700 [83]. Для поли-л*(и)-фенилен-4-фенил-1,2,4-триазола
деструкция в азоте начинается при 512°С; область, в которой происходит
в основном разрушение полимера, лежит в интервале температур
515—575 °С. Остаток при 900 °С составляет 58%.
Термоокислительная деструкция на воздухе начинается при 400 °С.
6.2.1.3. Переработка и эксплуатационные свойства
ароматических поли-4-фенил-1,2,4-триазолов
Из растворов ароматических поли-4-фенил-1,2,4-триазолов
получают термостойкие пленки и волокна. Волокна можно
вырабатывать сухим и мокрым формованием. Волокно, полученное по сухому
523
Таблица 6.8 Характеристики поли^-фенил-^г^-триазольных
волокон [87]
N—— N N N ~
Показатель
Прочность при растяжении кгс/см2
Относительное удлинение при
разрыве, %
Модуль, г/денье
Усадка, %
Температура,
°С
25
1 ПО
200
300
25
100
200
300
25
100
200
300
300
Сухое
формование
1,98
1,76
0,90
0,19
24,3
19,0
23,6
33,5
38,1
37,22
20,34
40
Мокрое
формование
3,08
2,62
1,69
0,95
8,4
6,9
7,5
12,7
86
74
55
12
10
способу из растворов в муравьиной кислоте, имеет прочность при
растяжении 0,8 г/денье. После трехкратной вытяжки волокна при
300—350 °С прочность увеличивается до 2 г/денье [87]. Волокно
с повышенными прочностью и термостойкостью получают мокрым
формованием из 30%-ного раствора в муравьиной кислоте в 1 %-
ный раствор NaOH. Такое волокно сохраняет 47% при 250 СС и
31 % при 300 °С исходной прочности. Повышенная прочность
волокна объясняется большей ориентацией макромолекул волокнообра-
зующего полимера. Недостатком поли-4-фенил-1,2,4-триазольного
волокна является низкая стойкость к УФ-лучам. В табл. 6.8 при-
Таблица 6.9. Физико-механические свойства
поли-4-фенил~1,2,4-триазольных пленок [87]
Показатель
Плотность, г/см3
Температура стеклования, °С
Прочность при растяжении, кгс/см2
Относительное удлинение при разрыве, %
Неотож-
женная
пленка
1,23
2,65
738
6,0
Пленка,
отожженная
в течение 1 ч
при 100 °С
280
647
13,0
524
ведены характеристики политриазольных волокон, полученных
разными способами.
Поливом из растворов поли-4-фенил-1,2,4-триазола в
муравьиной кислоте получают пленки, которые обладают хорошими
диэлектрическими свойствами. Термостойкость пленок можно
повысить путем отжига при 290 °С (табл. 6.9). Покрытия на алюминии
и высококачественных сталях в широком температурном интервале
имеют хорошие прочность при сдвиге и эластичность.
6.2.2. ПОЛИОКСАДИАЗОЛЫ
N N
О
—П
Поли-1,3,4-оксадиазолы получаются при взаимодействии
производных дикарбоновых кислот с гидразином или дигидразидами
дикарбоновых кислот. Полиоксадиазольное волокно вырабатывают
мокрым формованием из олеума или термической циклизацией
полигидразидных волокон. Теплостойкость волокна около 300 °С.
Поли-1,2,4-оксадиазолы имеют более низкую теплостойкость, чем
поли-1,3,4-оксадиазолы.
6.2.2.1. Получение поли-1,3,4-оксадиазолов
Поли-1,3,4,-оксадпазолы получают в две стадии—через
промежуточное образование полигидразида (форполимера) и
последующую его термическую циклизацию — или в одну стадию — в
полифосфорной кислоте либо олеуме. Поли-1,3,4-оксадиазолы можно
получать также циклизацией поли-Г^-ациламидгидразонов, которые
синтезируют 1,3-биполярным присоединением хлорангидридов
кислот к производным тетразолов, так же как и при нагревании по-
лиоксазолидинов.
Циклодегидратация полигидразидов. Полигидразиды
образуются при взаимодействии производных дикарбоновых кислот с
гидразином или дигидразидами: '
CIOC—jQpCOC1 + H2NNHC—jQj-CNHNH
C—jQj-C
f/~\~—CNHNHC—f/~\i—CNHNHC—
У? A e У1 e A
Поликонденсацию проводят в расплаве в инертных
растворителях, таких, как нитробензол или ксилол, при 170—200 °С [166—
169] (при этом образуются относительно низкомолекулярные
продукты), в полярных растворителях, таких, как гексаметилфос-
фотриамид, диметилформамид и диметилацетамид или N-метил-
пирролидон, с добавкой 5 % LiCl при низких температурах (при
525
этом образуются высокомолекулярные полимеры с хорошими во-
локнообразующимп свойствами) [156, 168, 170, 171].
Алифатические полигидразиды плавятся при 200—300 °С, смешанные алифа-
тическо-ароматические — при 250—300 °С, ароматические—в
интервале температур 350—400 °С. Все эти полимеры, за исключением
алифатических полигидразидов, растворяются в диметилсульф-
оксиде. Полигидразиды служат исходными продуктами для
получения волокна. Циклизация гидразидов в оксадиазолы описана еще
в 1900 г. [172, 173], однако для получения полимеров эта реакция
была применена лишь в 1960 г. [147, 174]:
Г-CNHNHC—(f^,—CNHNHC—(f^{—1 -77+
II « ICJJ » il IUJ -Н2°
L о о \-' о о ^^ }п
N N N N
-QrKoJ-\Qr^J~-
n
Циклодегидратация полигидразидов происходит
преимущественно в вакууме в твердой фазе (с получением пленок, порошка
и волокон) [151, 152, 159, 161, 175—177], в инертной среде или
в растворителях — при высокой температуре в присутствии
дегидратирующих средств, таких, как серная и я-толуолсульфокислота,
фосфорилхлорид, хлорангидриды и ангидриды карбоновых кислот
[149]. Циклизация сопровождается ухудшением растворимости.
Процесс может контролироваться ИК-спектроскопически [138]
по понижению интенсивности полос гидразидных групп (—С=0
1650 см-1; NH—3300—3400 см-1) и возрастанию интенсивности
полос поглощения, отвечающих колебаниям оксадиазольного цикла
(—C=N— 1550 см-1, 1,3,4-оксадиазольный цикл — 970 см-1).
Скорость циклизации в твердой фазе зависит от химического строения
и конформации молекул полигидразида [176]. С увеличением
содержания в макромолекуле полигидразида циклических звеньев
повышаются жесткость и упорядоченность макромолекул.
Понижение подвижности макроцепей обусловливает уменьшение скорости
термической циклизации. Средние значения энтальпии и энтропии
циклизации составляют: Д# = 49 — 53 ккал/моль и AS* = 14 —
— 17 ккал/моль соответственно. Для ароматического
полигидразида с 1,3-фениленовыми звеньями эти значения гораздо выше
(Д# = 62 ккал/моль и AS* =34 ккал/моль).
Различие в скорости циклизации ароматических полиоксадиа-
золов обусловлено также различием в межмолекулярном
взаимодействии, которое в процессе формирования гетероцикла
приходится преодолевать в первую очередь. Скорость циклизации
уменьшается и вследствие ориентации цепей полигидразидов при вытяжке.
В неориентированных полигидразидах цепь существует в складчатой
(цисоидной) форме, которая предпочтительно и подвергается
циклизации. Ориентированные полигидразидные волокна находятся
526
преимущественно в вытянутой (трансоидной) форме, которая имеет
меньшую реакционную способность:
о о о
Сч
Складчатая цепь Ориентированная цепь
Скорость термической циклизации можно увеличить, используя
кислые катализаторы, например, в присутствии бензолсульфокис-
лоты она возрастает в 15 раз [151].
Одностадийная циклополиконденсация дигидразидов дикарбо-
новых кислот или производных дикарбоновых кислот с гидразином.
Алифатические и ароматические поли-1,3,4-оксадиазолы могут
получаться в одну стадию из дигидразидов дикарбоновых кислот или
гидразинсульфата и дикарбоновых кислот, их диамидов или дини-
трилов с использованием в качестве растворителя и
дегидратирующего средства полифосфорной кислоты или олеума [128, 131, 132,
140, 143—145, 148, 153, 178—187]:
N—N
H2NNHC—,74,—CNHNH2
il {LJJ и
ПФК
H2NNH2 • H2SO4 + X—fZ-V,—X— ^^J °
О
Jj_
n
X = —COOH, —CONH2, —<
Для получения полиоксадиазолов олеум более удобен, чем
полифосфорная кислота. В полифосфорной кислоте реакция
завершается при 140—180 °С за 1—8 ч, в то время как в олеуме процесс
с достаточной скоростью идет при 80—120 °С. Впервые
взаимодействие карбоновых кислот с гидразинсульфатом с целью получения
полиоксадиазолов осуществил в полифосфорной кислоте Ноегебау-
эр в 1959 г. [188]. Молекулярная масса полимера при
одностадийном синтезе в полифосфорной кислоте достигает 3-Ю5.
Молекулярная масса зависит от концентрации мономеров и содержания Р2О5
в растворе. Поли-1,3,4-оксадиазол остается в растворе и может
быть непосредственно использован для получения волокна мокрым
способом [153].
Циклизация поли-^ациламидгидразонов. Поли-Ы-ациламид-
гидразоны могут быть получены поликонденсацией в растворе
дигидразидов с дииминоэфирами [125, 155, 191, 192]:
«Н5С2О—С—(Г^—С—ОС2Н5 + ftH2NNHC (A,—CNHNH2 —. н >
II О И II О II -с2н5он
NH ^^ NH О — О
—fA\—С—NHNH—С—Гр>Г.—С—NHNH—С—1
lUJ 11 11 Lv^J 11 11
^^ NH О ^' О NHjft
527
или из диамидгидразонов и дихлорангидридов дикарбоновых
кислот [162, 193]:
/iH2NNH— С
II
NH
С—NHNH2 + rtClOC—17^|—COC1
NH
—HCI
HNNH—С—Г
C—NHNH—С ,У-Ч,—С—
II II
NH О
"ОТ
n
Поли-1,3,4-оксадиазол образуется при нагревании с внутримолеку
лярным отщеплением аммиака:
—HNNH—С—'
О -
—NH3
п
1,3-Биполярное присоединение хлорангидридрв кислот к про-
шодным тетразолов. Этим методом поли-1,3,4-оксадиазолы были
первые получены в 1960 г. [147, 194]:
HN—N
>
—NH
—\ I +»сюс—Q—coci
\N==N *^=/
HN—N
>
=N'
N N
о
-COC1
Молекулярная масса синтезированных полимеров оказалась
недостаточной для получения пленок и волокон. Подобным образом
происходит присоединение диизоцианатов к дитетразолам [195].
Термические превращения полиоксазолидинов. Путем полимер-
аналогичных превращений полигидразидов с муравьиной кислотой
в полярных растворителях получают поли-1,3,4-оксазолидины,
которые при нагревании с выделением формальдегида и воды
превращаются в соответствующие поли-1 Д4-оксадиазолы [154]:
О
Ш-Cll
—Н2О. —СН2О
_©:
N N
О
6.2.2.2. Получение поли-1,2,4-оксадиазолов
Впервые поли-1,2,4-оксадиазолы получены Блумстромом [133]
в 1962 г. циклизацией поли-о-ациламиноксимов. Их можно
синтезировать также 1,3-биполярным присоединением нитроксидов к
нитрилам.
Циклизация поли-О-ациламиноксимов. Поли-О-ациламинокси-
мы получают поликонденсацией в растворе диаминоксимов с ди-
карбоновыми кислотами, эфирами и тиоэфирами дикарбоновых
кислот. В качестве растворителей используют гексаметилфосфотри-
амид, диметилсульфоксид или N-метилпирролидон; синтез может
быть проведен и на границе раздела фаз [196—198] :
C=NOH + пСЮС—(Г^)—О—(|Q^—COC1
?
—HC1
л f \ f
NH2
NH2 NH2 О О
Во избежание побочных реакций, например образования поли-
амидоксима или сшивания по NH2-rpynnaM,
nHON=C—(О)—C=NOH -f пСЮС—<Q)—°—(\j)—COCI —*
NH2 NH2
—>¦ Г—CNHC—(СУ)—CNHC—(())—°—Uj)—1
il l N—' и il ^^-у >—'
L О NOH HON О Jn
ИЛИ
NH NH О
C=O C=O
—n
реакцию проводят при низких температурах [199].
Поли-О-ациламиноксим не растворяется в органических
растворителях. Циклизация в случае образования поли-1,2,4-оксадиазола
протекает в более мягких условиях, чем при получении поли-1,3,4-
оксадиазолов из полигидразидов. Замыкание цикла происходит
при нагревании порошкообразного форполимера в вакууме при
200 "С или в полифосфорной кислоте, диметилсульфоксиде и N-ме-
тилпирролидоне при 180—220 С [199]:
NH2
C
NH2
О
О
N
V
п
Механизм циклизации заключается в нуклеофильной атаке
аминогруппой карбонильного атома углерода, при этом образуется
промежуточный комплекс, который после миграции ацильной
группы дегидратируется;
.о
(II
¦C=N—О—С-
\т—ОН
о
о
II
N—С—
II.
С—NH-OHH
.N— О
Циклизация ускоряется в присутствии дегидратирующих агентов,
например уксусного ангидрида [134].
1.3-Биполярное присоединение нитроксидов к нитрилам
rtCX-NC
nNC-
-CN->0
-CN
N -O
Циклоприсоединение нитроксидов к нитрилам происходит при
низких температурах в сильно разбавленных растворах и протекает
по катионному механизму [200—203]. Аналогичным образом под
действием солнечного света протекает полимеризация /г-цианбензо-
нитроксида [136]:
NC—
CN->O
-f- NC
Av
+ NC
530
\
О
—CN~>O
6.2.2.3. Физико-химические свойства полиоксадиазолов
Температуры размягчения и плавления. Полиоксадиазолы —
соединения с высокой степенью кристалличности, от белого до
желтого цвета. Температуры размягчения, плавления и разложения
полиоксадиазолов приведены в табл. 6.10.
Температуры размягчения поли-1,2,4- и поли-1,3,4-оксадиазолов
имеют близкие значения и повышаются по мере увеличения
жесткости цепи. Алифатические поли-1,3,4-оксадиазолы (№ 13—23)
размягчаются в интервале температур 100—250 °С, плавятся выше
300 °С с разложением. Циклоалифатические поли-1,3,4-оксадиазолы
(№ 24—27) плавятся при 200—300°С, алифатическо-ароматиче-
ские —при 300—400 °С (№ 29—33). Ароматические
полиоксадиазолы разлагаются ниже температуры плавления. Температура
размягчения их лежит в пределах 300—450 °С.
Растворимости С увеличением жесткости цепи макромолекул
поли-1,3,4-оксадиазолов растворимость ухудшается. Алифатические
полиоксадиазолы с 8—10 метиленовыми группами между оксадиа-
зольными циклами растворяются в серной и муравьиной кислотах,
диметилсульфоксиде, диметилформамиде, диметилацетамиде, N-ме-
тилпирролидоне, бензиловом спирте и дихлорэтане [140].
Циклоалифатические полиоксадиазолы (№ 24—27) растворяются в
серной, муравьиной и дихлоруксусной кислотах; ароматические —
в серной, полифосфорной и трифторуксусной кислотах, в
муравьиной и дихлоруксусной кислотах, феноле и крезоле они лишь
набухают. Карбонильная и простая эфирная группы между фениль-
ными ядрами придают ароматическим полиоксадиазолам
растворимость в диметилсульфоксиде и диметилформамиде. Исследованы
[128, 131, 132] свойства растворов поли-2,5-D'4"-дифенилфталид)-
1,3,4-оксадиазола, полученного одностадийной полициклоконденса-
цией в полифосфорной кислоте (№ 54).
Установлена зависимость между характеристической вязкостью
и молекулярной массой:
Зависимость константы седиментации от молекулярной массы
описывается уравнением
•ль °с
*0(ДМАА)
= 7,2
10-15М°'3
531
Таблица 6.10. Температуры размягчения, плавления и разложения некоторых полиоксадиазолов
Элементарное звено
т
разм*
т
разл1
N О N О
_(СН»L—1^ ^i—(СН2L—1!^ ^
N N
N О N О
-(СН2L—\ ^— (СН2)8-
N
-(СН»)в
N О
N
N О
-(СН2)8—
N
-©-
N
N О
350
212
350
350
>400
>400
10
11
12
13
N—О N О
N N-
N
О N N О
N N
N-—N
—CH=CH—^ ^—СН=СН—I
о о
N N
-СН=СН—IL J*—СН=СН—II J—(СН2) " \
N N N N
J— (СН2)8—1^
о о
N N
=r_«
N
—c==c
N N
О
N N
-(CH2L—^ J (CH2L-
O O
245
320
>400
390
180—210
230
310
275
Ц331
[137,
138]
[139]
[121]
[123]
[123]
[140-
142]
Продолжение табл. 6J0
Элементарное звено
Г
разм*
°С
>250
> 150
> 150
100-150
115
У
350
300
разл*
°С
380 (N2)
290
300
Литературный
источник
14
15
16
17
18
19
N N N N
-(CH2L—l J— (CH2O
О "О
N N N N
N N
-(СН2N—4^ J—(СН2N^
о о
-(СН2M—^ J— (CH2M-
о
N N
N N N N
-(СН2O
о
о
N N N N
-(CH2)O—1!^ JJ—(СН2)8—JL
о о
N N N N
—(СН.)В—l J— (СН2)9—II
[141]
[140]
[140]
[140]
[140—
144]
[140, 143,
144]
-21
23
25
26
N N N N
(СН2)ю
О
N N N N
-(CH2)I2—l!^ J—(CH2)l2—II J-
o о
N N N N
'(CH2)I5-
N N
О
N N
-(CH2JU—^ J— (CH2J0
О O
120
120
112-115
118-120
>350
280-300
200
390
390
390
11«.
143]
[1401
[140J
[140J
[145]
[145]
[145]
27
28
29
30
31
Элементарное звено
N N N
ММ M
-R—Ц. J— R'—IL
0 0
R=-(CH2),_8-,
R'= — СНг— S—CH2—,
R = —CH2—S—CH2—,
R' =—CH2-SO2—CH2—
NT NT
-(CH2L—l x
0
¦4
N N
n i
—(CH2O—L y
0
N N
-(CH2)a—1^ x
0
J_
U
M
L
—CH2—S—CH2—,
—CH2—S—CH2—,
I N
J—
—C-H2—0 O2—CjH2—, —(Cjrl2)i 8—
T
1 разм(
°c
—CH2— S— CH2—, —CH2—S02—CH2—
—CH2—S02—CH2—
, —CH2—S02—CH2—
M NT
>
N N
ii ii
N N
u
J-
Продолжение i
250
> 400
350
T
1 разл1
°C
390
314
372
"абл. 6.10
Литературный
источник
[145]
[146]
[141, 142,
147]
[147]
[141,
1421
32
33
34
35
о
N N N N
o
36
37
о о
N N N N
О LWJ О
170
300
>400
>400
> 400
447
450 •
275
450
[Н1,
1471
[116, 138,
1481
[116, 138,
141, 142,
147—153J
[141,
1421
[130, 141,
142, 148,
150, 154,
155]
Продолжение табл. 6.1
Элементарное звено
разл-
°G
38
O3H
39
N N
о"
N NC\
N-
O3H
N—N
_^J I
N N
40,
N N
N N
41
440
>400
460
N N
N N
430
380
440
370
470
430
300
430 ;
[147]
[138, L47,
L57]
[138, L41
L57]
[137,
L38]
Ц57]
Продолжение табл, 6.10
Элементарное звено
1 разм-
разл*
°С
N N
R = — (CH2)a— — (CH2L—, -(СН2)8—, —
сГ
390
400
51
52
53
N N
N N
54
О1 ЮГ V
390
340
340
Продолжение табл. 6.10
55
56
57
58
59
Элементарное авено
.. N N ^v N N
-См
N
л
ONO
J_
N 0 ^^ 0
/^ N N N N
-IM
N
V
J-
/^ N N N N
^M
N
N
'-©-«-O-V-
M N 1
разм-
°C
400
350
V
T
разл*
°G
Литературный
источник
[163]
{138,
147]
[138, 141
162]
[138]
[141]
о
СНз
I
N
60
N N
-Nv^N—CH2-
CH2—
N
61
о
N N N
62
i U
о s о'
N N N
63
N N
N N
64
—(О)—V JL-ch2-c2b10h10~ch2—1^
^=^ о о
>464
390
464
200-230
Ц381
[1641
1165}
во
60
40
20
WO
Рис. 6.11.
300 500 700 t,"C
О
200 400 600 t,°C
Кривые термической деструкции ароматических поли-1,3,4-оксадиазо-
лов в вакууме г'"п'1
N N
арома!
[138]:
1-
2 —
-V. У
L
N N N N 1
п
Рис. 6.12. Кривые термической деструкции полиизофталоилгидразида (а) и поли-
л!-фенилен-1,3,4-оксадиэзола {б) в вакууме [176].
Изучено строение цепи сорбционными методами [129]. Наличие
в цепи полиоксадиазолов таких объемистых заместителей, как
фталидная группа, приводит к уменьшению плотности упаковки,
широкому распределению пор в полимере и снижению
механической прочности.
Термостойкость. Термостойкость поли-1,3,4-оксадиазолов
примерно на 150° выше, чем поли-1,2,4-оксадиазолов. Расчет я-элек-
тронных систем незамещенных 1,2,4- и 1,3,4-оксадиазольных циклов
показывает, что 1,3,4-оксадиазол значительно более стабилен [137].
Отмечается [194], что 1,3,4-оксадиазольный цикл по спектральным
характеристикам и распределению электронной плотности
эквивалентен /г-фениленовой структуре. При пиролизе поли-1,3,4-окса-
диазолов на воздухе, в инертной атмосфере и в вакууме получаются
близкие результаты. Температуры начала разложения
полиоксадиазолов приведены в табл. 6.10. Самой высокой термостойкостью
обладают полимеры, в которых оксадиазольные циклы связаны
карбоциклическими ароматическими циклами. Деструкция таких
полимеров начинается выше 450 °С (№ 34—42, рис. 6.11). До
100 °С наблюдается незначительная потеря массы, которая связана
с удалением сорбированной влаги. В температурной области 100—
450 °С потери массы не происходит. Выше 450 °С в инертной
атмосфере и на воздухе наблюдается быстрая деструкция полимеров,
544
А, моль/осн. моль
Рис. 6.13. Кривые дифференциально-термического анализа поли-ж-фенилен-1,3,4-
оксадиазола в азоте [176].
Рис. 6.14. Зависимость состава газов от температуры пиролиза полиоксадиазола
№ 36 (табл. 118) в изотермических условиях [137]:
/ — N2; 2 — NO; 3 — СО2; 4 — СО; 5 — О2; 6 — (CNJ; 7 — Н2.
причем скорость деструкции максимальна в интервале температур
460—560°С. Остаток поли-1,3,4-оксадиазолов после нагревания до
700°С составляет 45—55%- Нагревание поли-/г-фенилен-1,3,4-окса-
диазола в течение 2 ч при 450 °С на воздухе сопровождается
образованием хрупкого продукта черного цвета, ИК-спектр которого
идентичен исходному полимеру [150]. О термической стойкости
поли-я-фенилен-1,3,4-оксадиазола и соответствующего полигидра-
зида дает представление рис. 6.12; участок кривой, отвечающий
деструкции полигидразида, соответствует температурному
интервалу начала деструкции полиоксадиазола, т. е. в ходе ТГА поли-
изофталгидразид частично циклизуется. Данные ДТА поли-лг-фе-
нилен-1,3,4-оксадиазола представлены на рис. 6.13.
В продуктах пиролиза поли-1,3,4-оксадиазола обнаружены N2,
N0, СО2, СО, О2, (CNJ и Н2 [137]. Зависимость состава летучих
продуктов деструкции в вакууме от температуры для
полиоксадиазола № 36 представлена на рис. 6.14. Остаток после деструкции
состоит из динитрила дикарбоксидифенилоксида, дифенилоксида,
дифенила, бензонитрила, бензола и сшитого карбонизованного
продукта.
Наиболее слабой связью в 1,3,4-оксадиазольном цикле является
связь С—О. По данным Родэ [137], деструкция начинается с
расщепления сначала С—О—С-связи, а затем С = 1М-связи:
N N
О'
N N
LUT
HC-O-
CN + О2 + СО2 -Ь СО
H2NNH— С—]/~С,—С—NHNH2+N2
Разрушение С—О—С-связи сопровождается образованием О2 и
СО, которые, реагируя между собой, дают СО2. Содержание азота
18 Зак. 18
545
40h
20
кость полиоксадназолов,
Рис. 6.15 Кривые термической деструкции поли-
1,2,4-оксадиазола (/) и гто/ш-1,3,4-оксадиазола [2)
в вакууме [138].
в летучих продуктах деструкции
соответствует его наличию в C = N-rpynne.
Введение в ароматические поли-1,3,4-
оксадиазолы простых эфирных групп
приводит к снижению их термостойкости
с 450 до 430 °С (№ 44—47). Термостой-
в которых оксадиазольные циклы
связаны непосредственно друг с другом (№ 36, 45), примерно на
150° ниже, чем аналогичных полимеров, содержащих оксадиазоль-
ные звенья, соединенные между собой ароматическими карбоцик-
лами (№ 37, 47). Алифатическо-ароматические поли-1,3,4-оксади-
азолы разлагаются при 320—370°С (№ 29—33). Начало
разложения соответствующих алифатических полимеров лежит в
температурном интервале 270—300 °С (№ 13—23); циклоалифатиче-
ские (№ 24—26)—устойчивы до 390°С.
Термостойкость полп-1,2,4-оксадиазолов значительно ниже, чем
поли-1,3,4-оксадиазолов, что хорошо видно из рис. 6.15.
Разложение на воздухе начинается уже при 300 °С. В том же
температурном интервале полимер разлагается и в инертной атмосфере.
Газообразные продукты пиролиза содержат О2, СО, СО2, N2, NO, (CNJ.
В составе низкомолекулярных жидких и твердых продуктов
деструкции поли-1,2,4-оксадиазола № 8 обнаружены динитрил ди-
карбоксиднфеннлоксида, бензонитрил, фенилизоцианат, дифенил,
дифениловый эфир и бензол [137]. При деструкции происходит
одновременный разрыв связей С—О, N—О и N—С. Образование
СО2 и NO2 является следствием окисления выделяющимся
кислородом СО и NO [137, 204]. Наличие в продуктах деструкции бензо-
нитрила и фенилизоцианата свидетельствует о том, что
деструктивные реакции являются обратными 1,3-биполярному присоединению
[199]:
ShcN+''
6.2.2.4. Поли-1,3,4-оксадиазольные волокна
Полп-1,3,4-оксадиазольные волокна получают термической
дегидратацией соответствующих полигидразидов в волокне или
формованием из растворов полп-1,3,4-оксадназолов в олеуме (при
одностадийном синтезе ароматических полимеров). Предельная
температура длительной эксплуатации ароматических поли-1,3,4-
оксадиазолов составляет примерно 300 °С. Полиоксадиазольные
546
Таблица 6.11. Превращение полигидразидкых волокон
в полиоксадиазольные [151]
1'ежим циклизации
Кратность вытяжки
Бензол сульфокислота,
1% (мол.) в расчете
на полигидразид
2 ч при 250°С
2 ч при 265°С
16 ч при 300°С
х 1,85 при 380 °С
х 1,28 при 430 °С
волокна в течение многих лет выпускаются на пилотной установке;
промышленное производство их отсутствует.
Термическая дегидратация полигидразидов в волокне. Поли-
гидразидные волокна могут быть получены сухим формованием из
растворов в гексаметилфосфотриамиде, диметилацетамиде, диме-
тилсульфоксиде или N-метилпирролидоне [152, 177]. По патенту
фирмы «Du Pont» [126] готовят 10 %-пый раствор полигидразида
в диметилсульфоксиде с добавкой 5—12% LiCl при 90°С; волокно
формуют при 110°С в противотоке нагретого до 260°С азота. Поли-
гидразид из дихлорангидрида терефталевой кислоты и изофталди-
гидразида имеет прочность при растяжении 6 г/денье,
относительное удлинение при разрыве 8%, модуль 151 г/денье. Прочность
ароматических полигидразидных волокон заметно снижается при
температуре выше 200 °С. Ориентированные (кристаллические)
волокна получают предварительной вытяжкой на 200 % при 300 °С
и последующей вытяжкой на 120% ПРИ 340 °С. Термическая
циклизация при 290°С завершается за 160 ч [152, 177]. При
циклизации в присутствии кислых катализаторов, таких, как бензолсуль-
фокислота, процесс ускоряется в 15 раз [151].
В табл. 6.11 приведены оптимальные условия превращения
полигидразидных волокон в полиоксадиазольные.
Формование из олеума ароматических поли-1,3,4-оксадиазолов.
Конденсация производных дикарбоновых кислот с гидразинсуль-
фатом в олеуме приводит к образованию поли-1,3,4-оксадиазолов,
10 %-ные растворы которых могут быть непосредственно
использованы для мокрого формования [120, 153, 187, 205—207]. Осади-
тельной ванной служит 40—50 %-ная серная кислота с добавкой
неорганических солей, например сульфата аммония, хлорида и
нитрата цинка, сульфата железа и магния, или разбавленные
серная, фосфорная или соляная кислоты также с добавкой солей.
Сформованное волокно после 4—5-кратной вытяжки промывают
теплой водой и высушивают. Формованием из растворов
концентрированной серной кислоты начиная с 1971 г. во Всесоюзном научно-
исследовательском институте искусственного волокна на пилотной
установке выпускается полиоксадиазольное волокно оксалон [238].
Свойства волокон на основе ароматических поли-1,3,4-оксадиа-
золов. Оптимальными свойствами обладают волокна, полученные
из поли-.м(/1)-фенилен-1,3,4-оксадиазолов [151 —153]. В табл. 6.12
приведены характеристики полиоксадиазолыюго волокна, получен-
18* 647
P, r/денье
Б
Рис. 6.16.
Деформационно -
прочностные кривые по-
ли-1,3,4-окса д и а -
зольных {1) и по-
ли-л!-фенилен и з о -
фталамидных
волокон B) [153].
Рис. 6.17.
Зависимость
физико-механических свойств
поли-лг-(я) - феии-
лен- 1,3,4-оксадиа-
зольных волокон
(% от их значения
при 23 °С) от
температуры -[177].
кого различным способом. Диаграмма изменения прочности при
растяжении (рис. 6.16) показывает, что полиоксадиазольное
волокно ведет себя аналогично полиамидному. Прочность во
влажном состоянии составляет 88 % прочности сухого волокна.
Ароматические полиоксадиазольные волокна отличает высокая
термостойкость (рис. 6.17). При 200 °С происходит лишь незначительная
потеря прочности. При 300 °С сохраняется 60% исходной прочности.
Выше 400°С наблюдается карбонизация волокна без плавления.
Поли-1,3,4-оксадиазольное волокно обладает высокой
стабильностью размеров. Усадка после 30 мин старения в среде сухого
воздуха при 250 °С составляет 1,4%; через 30 мин выдержки
в воде при 130°С—1,5%. На рис. 6.18 показано изменение
прочности полиоксадиазольного волокна в процессе старения.
Сохранение 50% прочности при 400 °С наблюдается через 30 ч выдержки
при этой температуре, а при 300°С — через 700 ч. Остаточная
прочность 1 г/денье в условиях термостарения при 400 °С достигается
через 48 ч, а при 300 °С — через 100 ч.
Таблица 6.12 Характеристики поли-ж(я)-фенилен-1,3,4-оксадиазольных
волокон [151, 153]
Показатель
Циклизация
полигид-
разидных
волокон
Формо
вание
из олеума
Показатель
Циклизация
полигид
разидных
волокон
Формование
из олеума
Прочность при
разрыве, г/денье
при 23°С
при 200°С
при 400°С
Относительное
удлинение при
разрыве, %
при 23°С
при 200°С
8,0
4,9
1.6
10,0
5,6
4,1
3,2
9,7
15,0
Модуль, г/денье
при 23°С
при 200°С
при 400 °С
Водопоглощение,
%
Плотность, г/см3
227
166
63
1,41
114
4,5
1,41
54S
Gptr( денье
2 -
0
1
a
1
, г/денье
' P,г/денье
5\
20
40
200 Ш 600 800 z,i
u 5 ?,%
Рис. 6.18. Зависимость прочности при растяжении поли-ж-(п)-фениленоксадиа-
зольных волокон при термическом старении на воздухе [151]:
а — при 400 °С; б — при 300 °С.
Рис. 6.19. Деформационно-прочностные кривые при термическом старении
полила- (гс)-фенилен-1,3,4-оксадиазольного волокна [153]:
/ — нетермообработанный образец; 2 — на воздухе при 300 °С в течение 30 мин; 3 — в
водяном паре при 130 °С в течение 30 мин.
Изменение прочности при растяжении полиоксадиазольного
волокна в условиях термостарения показано на рис. 6.19.
Ароматические поли-1,3.4-оксадиазольные волокна устойчивы в
разбавленных кислотах и щелочах. При кипячении в течение 24 ч в 10 %-ной
серной кислоте прочность снижается на 75%. а в 10 %-ной
щелочи—на 85%. Стойкость к УФ-лучам примерно такая же, как
у ароматических полиамидов. Облучение в течение 20 ч в федомет-
ре понижает прочность при растяжении на 33 % и относительное
удлинение при разрыве на 66 %.
Полиоксадиазольное волокно с хорошими окрашиваемостью и
ионообменными свойствами получают путем сульфирования
полимера [119]. Фосфор- [208] и галогенсодержащие [115] полиокса-
диазолы обладают способностью к самозатуханию.
6.2.3. ПОЛИТИАДИАЗОЛЫ
N N
Политиадиазолы получают термической циклизацией полиди-
тиагидразидов или полиоксатиагидразидов. Полимеры стабильны
на воздухе до 400° и плавятся выше 375 °С. Политиадиазольные
волокна изготавливают сухим формованием форполимеров с
вытяжкой при 275 °С.
6.2.3.1. Получение политиадиазолов
Политиадиазолы могут быть получены поликонденсацией в
растворе ароматических тетратианатов с ароматическими дитиагидра-
зидами и дйгидразидами; дихлорангидридов дикарбоновых кислот
549
-—S—CH3 H2N—NH—(
-C-NH-NH2 H3C-S~C-
s s
-c— s—сн3 ci—c-
o о
B)
-HAN-NH2
ГМФА
(t)
C)
-C—NHNH—C—
(I (I _,
D)
E)
I Р2О5/ПиридИн I
-С—NH—NH2 NH2-NH— C«
3 S
ГМФА
дмсо
(e)
-C-NH—NH2
3
ГМФА
il.'
—c-
II
?—NH—NH—C—(Q^-C—NH—NH—
s s
i Политиагид{>азид
— C-
11
S
-C—NH—NH-
O о
Полн оксати агидразиду
-С—NH—NH—C-
ii
S
-IUO
г1 Л
с ароматическими дитиагидразидами; термической гомополикон-
денсацией ароматических дитиагидразидов и взаимодействием
ароматических полигидразидов с пентасульфидом фосфора с
последующей циклизацией образующихся в качестве промежуточных
продуктов полидитиагидразидов или полиоксатиагидразидов (см.
с. 550).
Циклизация полидитиагидразидов или полиоксатиагидразидов
протекает с отщеплением соответственно H2S или воды в
значительно более мягких условиях, чем при замыкании оксадиазольного
цикла [101, 102]. Так, образование поли-1,3,4-оксадиазолов при
250 °С протекает в течение продолжительного времени (несколько
часов), в то время как политиадиазольный цикл замыкается за
несколько минут. Для получения политиадиазольного волокна или
пленки предпочитают использовать полиоксатиагидразид.
Взаимодействие полиоксадиазолов с P2S5 в кипящем пиридине позволяет
за 30 мин получить полимер, хорошо растворимый в диметилацет-
амиде и пиридине. Высокоупорядоченный сополимер, в цепи
которого содержатся тиадиазольные, оксадиазольные и фениленовые
фрагменты, получен по реакции 4,4/-A,3,4-тиадиазол-2,5-диил)ди-
бензоилхлорида с изофталоилгидразидом через промежуточную
стадию образования форполимера [102]:
nClC—/(у)
II <^У/
N N
II
О
СС1 + пН2ШНС
11 1
О О
CNHNH2
11
О
N N
6.2.3.2. Физико-химические свойства политиадиазолов
Политиадиазолы — кристаллические полимеры от желтого цвета
до цвета охры. Плавятся политиадиазолы, за небольшими
исключениями, при температуре выше 375 °С. Некоторые их свойства
приведены в табл. 6.13.
По стойкости к термоокислительной деструкции
политиадиазолы близки к аналогичным полиоксадиазолам. Ароматические
политиадиазолы разлагаются на воздухе примерно при 450°С,
алифатические и ароматическо-алифатические — при 380 °С. Скорость
деструкции максимальна в интервале температур 400—600OC;
остаток после деструкции до 700 °С составляет около 50 %.
Ароматические политиадиазолы растворяются только в сильных кислотах,
таких, как серная и метансульфокислота (в которой растворяются
551
1
2
3
4
5
Таблица 6.13. Характеристическая вязкость [rj], температуры плавления и разложения
Элементарное звено
S S
-ifV-f Vf V
g g
/—\ S S
S S
^3^ N N L^J N N
n/ I\ 14 1ч IN
Дл/г
0,14
0,14
0,50
0,60
0,26
T , °C
300
>375
>375
>375
300
политиадиазолов
разл
(на воздухе),
°С
360
360
450
460
450 V
Литературный
источник
[104]
[103, 104]
[101, 102,
[103, 104]
[104]
N
i-
N
N N 4 *> N N
N
Cl
Cl
\
N
CK
C1
s s
N N k<^>j N N
10
N N
J.24
0,56
0.28
0,28
> 375
> 375
> 375
>375
450
456
450
440
i
Ц04]
[104]
[104]
[104]
1.103]
Продолжение табл, 6.13
ni].
дл/г
0,70
0,93
0,35
гпл
>
>
>
>
>
•с
375
375
375
375
375
T
разл
(на воздухе),
Литератур*
ный
источник
380
380
360
360
381
[104]
[104]
1104]
1104]
[104]
Элементарное звено
11
12
,13
И
-(сн2L—f ^ri—(сн2L—
11 II II II
NN NN
15
N N
Таблица 6.14. Характеристики политиадиаэольных волокон [101, 102]
Показатель
Поли-
тиа-
диазол
Сополимер
тиади-
аэола
и окса-
диазола
Показатель
Поли
тиа-
диазол
Сополимер
тиадн-
азола
и окса-
диазола
Прочность при
растяжении, г/денье
Относительное
удлинение при разрыве, %
3,5
14
1,3
3,0
Модуль, г/денье
Температура плавления,
°С
Температура
разложения на воздухе, °С
78
375
450
46
464
464
все полимеры, перечисленные в табл. 6.13, кроме № 10 и 11).
Гидролиза полимеров не происходит при воздействии на политиа-
диазолы 20 %-ной щелочи и 20 %-ной серной кислоты.
6.2.3.3. Политиадиазольные волокна
Политиадиазольные волокна получают формованием из
растворов полиоксатиагидразидов в пиридине с последующей термической
циклизацией. Оптимальные свойства имеет волокно, однократно
вытянутое на стадии получения полиоксатиагидразида; его
циклизация происходит за несколько минут. Политиадиазольное волокно
негорюче. Характеристики политиадиазольных волокон приведены
в табл. 6.14. Волокно на основе поли-и(п)-фенилен-1,3,4-оксадиа-
зола сохраняет 92 % исходной прочности после нагревания в
течение 144 ч на воздухе при 300 °С.
6.3. ГЕТЕРОЦИКЛИЧЕСКИЕ ПОЛИМЕРЫ С ЧЕТЫРЬМЯ
ГЕТЕРОАТОМАМИ В ПЯТИЧЛЕННОМ ЦИКЛЕ
6.3.1. ПОЛИТЕТРАЗОЛЫ
6.3.1.1. Полибистетразолы
"-R-
¦N—R'—N-
I I
N
/N
N
Полибистетразолы получают поликонденсацией динатриевых
солей бистетразолов с дигалогенидами
С1(СН2JС1 + Na-
I.
-Na
—NaCl
N
N
-(сн,ь-
¦N-O-N
N
N
ббб
Таблица 6.15. Температура разложения политетразолов
Элементарное звено
N=N
N
N=N
N
N=N
N
О.
N
N
N—N
N
_cho—/r^s—сн
N=N
N
СНзО
i
N
ОСНз
О
-NH-
г
N
8
CH,
^^
NH—n N—
%:
—(CH2N-~NH-
N
240
[105]
240
[105]
270
[105]
120
>300
[105]
[105]
245
[105]
240
[105]
240
[105]
125
[105]
556
или при взаимодействии полиамидов с HN3 в присутствии РС15
[105]
[.-HN(CH2NNHCO(CH2LCO—]„
HN3 _
-(CH2N-N-
-(СН2L-
-N—
N
N
_/2
6.3.1.2. Полиаминотетразолы
Полиаминотетразолы получают взаимодействием поликарбоди-
имидов с HN3 в среде хлорированных углеводородов:
Поликарбодиимиды получают поликонденсацией диизоцианатов
в присутствии 3-метил-1-фенил-3-фосфоленоксида-1 [105].
6.3.1.3. Свойства политетразолов
Температуры разложения политетразолов приведены в табл. 6.15.
Полимеры растворяются только в концентрированных кислотах,
таких, как серная и соляная.
6.3.2. КОМПЛЕКСЫ ПОЛИТЕРЕФТАЛОИЛОКСАЛОИЛБИСАМИД-
РАЗОНОВ С МЕТАЛЛАМИ
Me NH2
N С N
N С N—С—
О
I I II
H2N Me О
Комплексы политереф|талоилоксалоилбисамидразонов с
металлами представляют собой псевдогетероциклическую систему с пя-
тичленным гетероциклом. Волокна на основе таких полимерных
хелатных комплексов выдерживают нагревание при 1500°С в
течение 20 с.
Исходными продуктами для получения таких полимеров служат
терефталоилхлорид и оксамидразон, синтезируемый из гидразина и
дициана:
=C—C=N
+
H2N-NH2
H2N—NH
NH-NH2
Г
V-c'
557
Поликонденсацией на границе раздела фаз 15%-ный водный
раствор NaCl — гептан при комнатной температуре получают поли-
оксамидразон [236, 237] :
n
l/
г
.
нСЮС
COCI
—iici
—HN—N.
M—NH— C-
с-с/
Hiink \nh?
о
II
Политерефталоилоксалоилбисамидразон нерастворим в
органических растворителях. Полиоксамидразон в водных растворах гидр-
оксидов щелочных металлов окрашивается в темно-красный цвет.
Растворимость повышается с увеличением атомной массы
щелочного металла. В присутствии ионов Ме2+ при комнатной
температуре быстро образуется хелатный металлокомплекс политерефта-
лоилоксалоилбисамидразона. Ионы металлов входят в состав
молекулы в эквимольном количестве:
I— с
L Ч) h2i
—с
^
—N4
HoN^
^NH2
.N-
\
;-<S>-
Me
2 +
4/
Me
(O
-C—N>
II
О
H-
6
N
j-c—
H2N
-N N-
-¦Me
/\
H
или
c-
I
с
N
Me
N
О
l
N
Хелаты олова и циркония окрашены в желтый цвет, висмута и
свинца — в красный, меди — зеленый, цинка и кадмия —
оранжевый, кальция — коричневый и железа — в черный цвет. Волокна
таких металлокомплексных полимеров получают формованием
щелочных растворов полиоксамидразонов в кислотную осадительную
ванну и непосредственно после образования нитей проводят хела-
тирование.
Гомогенные бикомпонентные волокна можно получить при
совместном формовании с ксантогенатом целлюлозы. В процессе
осаждения металлокомплексных волокон при высоких
температурах происходит ступенчатое превращение макромолекул, сопро-
558
вождающееся отщеплением воды и азота, и образование остатка,
состоящего из оксида металла и углерода. Кислородный индекс ме-
таллокомплексных волокон равен 0,52 и значительно превышает
кислородный индекс других негорючих волокон (полибензимида-
зола 0,41; поли-ж-фениленизофталамида 0,28; полиакрилонитрила
0,18). При температуре пламени 1500°С волокно разрушается за
20 с. С 1972 г. негорючие металлокомплексные политерефталоил-
оксалоилбисамидразонные волокна выпускает фирма AKZO
(Голландия) под торговым названием «Энкатерм».
6.4. ПОЛИМЕРЫ С ШЕСТИЧЛЕННЫМИ ГЕТЕРОЦИКЛАМИ
6.4.1. ПОЛИПИРАЗИНЫ
4
N V\
Полипиразины получают циклополиконденсацией бис-я,я'-(а-га-
логенацетил) ароматических соединений с аммиаком в диметил-
формамиде или в диметилацетамиде при 60—70°С [234] или в
расплаве [233]:
R 1
R—СНС Ar ССН—R
I II il I
X О ОХ
N
-Ar—"
N
п
где
= -H, -(СН2M-СНз, -(СН2I3-СН3;
Ar =
Незамещенные полипиразины (R = H) растворяются в сильных
кислотах — серной, фосфорной или муравьиной—с добавкой пер-
оксида водорода. Увеличение растворимости полимеров в
присутствии Н2О2 связано с образованием полярных N-оксидных групп
в цепи. Алкилированные полипиразины [R = СН3—(CH2)s— или
СН3—(СНгЬз—] растворяются в пиридине, СС14, муравьиной и
серной кислотах. В инертной среде термическая деструкция таких
полимеров начинается выше 400°С.
—N;
о
л
6.4.2. ПОЛИДИКЕТОПИПЕРАЗИНЫ
О
N—R—
—П
—N
N-R-
—П
6.4.2.1. Получение полидикетопиперазинов
Некоторые характеристики этих полимеров представлены
в табл. 6.16. Поли-2,5-дикетопиперазины с 1,4-расположением
559
1
2
3
T
О
О
о
0
а б л и ц а 6.16.
"(ОТ
^--^сн,
-О—
Характеристическая вязкость и температура разложения полидикетопиперазинов
Элементарное звено
¦-©-
l-nl-
дл/г
0.21
0,18
0,33
Растворитель
Д и хлор-
уксусная
кислота
То же
*
Т °С
раз л'
в
азоте
340
340
280
на
воздухе
300
300
300
Литературный
источник
[ПО]
[ПО]
[ПО]
о
/
О
_N' \j_R-_p
О
О
о
f
4NR'-
о
«--(Ob
—(СН2J, —(Cfb),—.
0,18
280
300
ЦЮ1
345
[106]
[106]
R' =
-(CH,)-,
¦ -\O)
(CH2N-
'S
7
S
9
—NHOCHs-
-NHOCH2-
—NHOCHa-
HN-
>
0
HN-
>
Элементарное звено
О
/ NHC^
)—CH2ONHCNH fP^I^ II
«NH » ^W\ °
О
)—CH2ONHCNH—/Q)—CH2—(С]
-NH II ^—' ^—
О
)—CH2ONHCNH(CH2)eNHC—
-NH II 4
\J
[41.
дл/г
0,07
0Л2
0,13
Продолжение табл. 6.16
Растворитель
ДМФА
ДМФА
ДМФА
в
азоте
на
воз-
Духе
234
267
212
Литературный
источник
[107.
108]
[107.
108]
[107,
108]
10
11
12
13
HN
(
О
-О(СН2J—( >—(CH2JOCNH
о
О
HN-
-О(СН2J—(
-NHC—
II
О
О
о
HN
-O(CH2J—( )—(CH2JOCNH(CH2NNHC
>-NH A
о
О
//
HN
—NH(CH2L—( )—(CH2LNHC(CH2LC-
Ут> о о
0,27
0,13
0.20
0,21
ДМФА
ДМФА
ж-Кре-
зол
Три-
фтор-
этанол
60
40
210
162
188
235
1107,
108]
[107,
1081
[107,
108]
[109]
связей (№ 1—4) получают поликонденсацией бис-2-хлорацетамидов
ароматических диаминов в диметилацетамиде или фенилглицидило-
вом эфире при 110—150°С в присутствии эпоксисоединений в
качестве акцепторов НС1 и четвертичных галогенидов аммония,
например триэтилбензиламмонийхлорида в качестве катализатора.
Поли-2,6-дикетопиперазины № 5 и 6 получают из диангидридов
общей формулы Оч ,NRN4 ,0 и диаминов с образо-
\—СО/
ванием на первой стадии полиамидокислот, которые циклизуются
в смеси этилацетат — пиридин при 110°С [106]:
О
О
л О.
\
/
о
«H2N—(^)—О—
О О
-NHCCH2N—(СЪ—NCH2CNH—
I ^ I
СНаСООН СНаСООН
О
—п
_п
Поли-2,5-дикетопиперазины № 7—13 получают, используя 2,5-
дикетопиперазин с функциональными группами в положении 3,6,
например 3,6-бис(аминометил)-2,5-дикетопиперазин или 3,6-бис-
(?-гидроксиэтил) -2,5-дикетопиперазин. Процесс проводят в диме-
тилсульфоксиде при 50—60 °С [107—109]:
H2NCH2—( }—СН2 + OCN-
о
—(Ту)—О—(QS—NCO
—NHCH2—( у—CH2NHCONH-
NH
h
561
Напротив, полимеризация моиомерных 2,5-дикетопиперазинов как
циклических лактамов протекает по катионному механизму с
раскрытием цикла и образованием полиглицнна [111 —113].
6.4.2.2. Свойства полидикетопиперазинов
Полидикетопиперазины, в которых пиперазиновые циклы
связаны только ароматическими или алифатическими группировками,
растворяются в органических кислотах — муравьиной, дихлор-,
трихлор- и трифторуксусной, но не растворяются в полярных
растворителях, таких, как диметилацетамид, диметилформамид и ди-
метилсульфоксид.
Термическая деструкция в инертной среде начинается при
300 °С. Полиднкетопиперазины с.амидными связями в цепи
растворяются в полярных растворителях, например в диметилформамиде
и ж-крезоле. Полимер, полученный на основе О-3,6-бис(аминоокси-
метил)-2,5-дикетопиперазина, оптически активен; он разлагается
при 250 °С.
6.4.3. ПОЛИТРИАЗИНЫ
Политриазииы получают поликонденсацией производных триа-
зина с бифункциональными соединениями, полициклотримериза-
цией динитрилов или дицианатов, циклизацией замещенных дини-
трилов.
Густосетчатые полиэфир-1,3,5-триазины пригодны для
изготовления конструкционных гибких элементов, используемых в
авиационной и космической технике, к которым предъявляются
жесткие требования в отношении стойкости к облучению и термической
стойкости.
п
Поли-6-фенил-1,2,4-триазин
п
может быть использован в качестве клеев и термостойких
связующих. Полимер выдерживает длительное нагревание при 260 UC.
565
6.4.3.1. Получение политриазинов
Поли-1,2,4-триазины. Поли-1,2:4-трпазипы получают циклополи-
конденсацией ароматических дигидразидинов с ароматическими
диглиоксалями или дибензилами [209, 210, 229, 230] :
nH2NNHC—II J—CNHNH2 +
I! N «
NH NH
О О
Циклополиконденсация протекает при комнатной температуре
в растворителях типа лг-крезол, пиридин, диметилформамид, диме-
тилацетамид, диметилсульфоксид. Нагревание в инертной среде
при 300 °С сопровождается увеличением характеристической
вязкости (в ,м-крезоле) с 0,6—0,9 до 1,3 A00 мл/г). Полимер состоит
из изомерных звеньев следующего строения:
СеН
6П5
R—
Поли-1,3,5-триазины. Поли-1,3,5-триазины получают
поликонденсацией производных триазина с бифункциональными
соединениями, полициклизацией нитрилов и полициклотримеризациеи ди-
нитрилов и дицианатов.
При действии на цианурхлорид аминов, фенолов или спиртов
по мере замещения реакционная способность незамещенных
атомов хлора понижается, что облегчает ступенчатое проведение
реакции с нуклеофильными агентами. Полиэфиртриазины,
полученные методом межфазной поликонденсации [216, 217, 233] при
взаимодействии цианурхлорида с натриевыми солями фенолов,
представляют собой продукты, строение которых сильно зависит
от продолжительности контакта на поверхности раздела. Длитель-
566
ный контакт приводит к получению сильно разветвленных
продуктов.
Линейные политриазины можно синтезировать из 2-фенил- или
2-пилеридил-4,6-дихлортриазинов, получаемых замещением атома
хлора в цианурхлориде при взаимодействии его с фенолом или
пиридином [214, 215, 224]:
п f \ +яНО—
|
С
., N4 Na2CO3
-С—(С У,—ОН >
-HCJ
СНз
-O-
с6н5
Наиболее высокую молекулярную массу имеет полимер,
полученный в растворителе при 180—200 °С. Поликонденсацня на
границе раздела фаз вследствие низкой реакционной способности
атомов хлора в производных триазина приводит к образованию
низкомолекулярных продуктов. Высокомолекулярные
политриазины получаются при поликонденсации в растворителе 2-замещен-
ного 4,6-дихлортриазина с диаминами [214, 215, 219, 220, 224]:
п
I
N
+«H2NArNH2
Cl
-НС]
—^ j)—NHArNH—
N
—П
Высокомолекулярный полимер получается при взаимодействии
2-«-бутокси-4,6-дихлор-1,3,5-триазина с 4,4/-диаминодифениловым
эфиром [[х\] = 1,15 дл/г при 30°С в НСООН] при проведении
поликонденсации в N-метилпирролидоне при 30—140СС в
присутствии карбоната лития в качестве акцептора кислоты. В отсутствие
акцептора соответствующий поли-1,3,5-триазин образуется с
отщеплением бутоксигрупп [219, 220]:
OC4HS
п
+ nH2N-
—О
667
LiiCO3
OC4H9
N
О
N
Полииминотриазины получают поликонденсацией в расплаве
2-замещенных 4,6-диалкиламино- или диариламино-1,3,5-триазинов
с 2,6-дихлортриазинами или их эфирами [221, 222]:
п
C6
К Л
N
Ce
с6н5
6н4~
Полимеры представляют собой низкомолекулярньте хрупкие
продукты. Линейные полистирилтриазины образуются при
взаимодействии 2,6-диметш1-4-фенш1-1Д5-триазинов с диальдегидами [213]:
п
+ пОСН-
h2so4
3
-2Н2О
с6н5
=нс
сн=сн
N
сн=
В концентрированной серной кислоте максимальная степень
поликонденсации при 25—60 °С составляет 11. При температуре
выше 100 °С частично протекает сульфирование. Использование
2,4,6-триметилтриазинов приводит к образованию сшитых
продуктов. Структурированные полимеры получаются также при исполь-
зевании ароматических [225] или перфорированных
алифатических диамидинов [226, 227) :
HN
п
у
NH
N
—rt
Наряду со свободными амидинами применяют соответствующие
эфиры карбоновых кислот [225]:
п
HsCaOOCHN/ \ f NNHCOOC2HS
170-190 вС
ОН
—п
Ароматические динитрилы в присутствии в качестве
катализаторов циклотримеризации хлорсульфоновой кислоты, аминов или
металлов образуют сшитые полимеры с 1,3,5-триазиновыми
циклами в цепи [217, 228]:
Исходные динитрилы легко получаются из соответствующих
диаминов по реакции Зандмейера. При циклотримеризации в
массе реакция протекает с индукционным эффектом. Скорость
полимеризации прямо пропорциональна концентрации катализатора [217].
При использовании третичных аминов с увеличением их
основности скорость процесса возрастает. Наиболее высокую
каталитическую активность проявляют циклические амины. По каталитической
активности эти амины можно расположить в ряд: изохинолин >
> пиридин > хинолин > трет-амииы. Способность к циклотриме-
ризации у ароматических дициановых эфиров выше, чем у арома-
56у
тических ди нитрилов [217] :
3NC0—(( ))—С—(( ))—OCN
I
С
Нз
Процесс протекает в присутствии как кислых, так и основных
катализаторов
6.4.3.2. Физико-химические свойства политриазинов
Некоторые свойства политриазинов приведены в табл. 6.17.
Ароматические поли-1,2,4-триазины, за исключением линейных
политриазиновых эфиров (№ 12—20) и полиаминотриазинов
(№ 21—33), не плавятся. Для фенилзамещенных поли-1,2,4-три-
азинов температура стеклования колеблется от 200 до 260°С, а
для полииминотриазинов (№ 34—39)—от 180 до 360 °С.
Полимеры, содержащие в цепи остатки пиридина (№ 1—6), имеют по
сравнению с другими поли-1,2,4-триазинами исключительно
высокую растворимость. Они образуют 30%-ные растворы в
хлороформе, сиж-тетрахлорэтане или смеси лг-крезола с толуолом A:4).
В диметнлформ амиде, диметилацетамиде и N-метилпирролидоне
концентрация растворов не превышает 1 °/о [209, 210, 229, 230].
Линейные поли-1,3,5-триазины (№ 12—20) растворяются в анилине,
гексаметилфосфотриамиде, N-метилпирролидоне, бензилбензоате,
хлорнафталине, нитробензоле и бензиловом спирте [214, 215, 224];
алкоксипроизводные полиамино-1,3,5-триазинов хорошо,
растворяются в N-метилпирролидоне, муравьиной кислоте, диметилсульф^
оксиде, ж-крезоле и серной кислоте; поли-1Д5-триазиноны
растворяются только в концентрированной серной кислоте [219, 220].
570
Высокая резонансная энергия 1,3,5-триазинового цикла (82
ккал/моль) обеспечивает высокую термостойкость полимеров с три-
азиновым циклом. Так, 2,4,6-трифенпл-1,3,5-триазин перегоняется
при атмосферном давлении па воздухе при 460 °С без разложения.
Линейные полиэфир-1,3,5-триазины (№ 12—20) стабильны в
инертной среде до 300 °С. Полиимино-1,3,5-триазины разлагаются
на воздухе при 350 °С, а в инертной среде —при 400 °С [221]. При
нагревании алкоксизамещенных поли-1,3,5-триазинов (№ 25—28)
при 300 °С на воздухе происходит отщепление алкила и
превращение в соответствующие поли-1,3,5-триазиноны (№ 29—32) [219, 220]:
О(СН2)ХСН3"
-к J-R-
N
>300 °С
п
о
—IL J—
-R-
N
п
Поли-1,3,5-триазиноны стабильны на воздухе до 350 °С и в
инертной среде до 400 °С. Полимеры с виниленовыми группами
разлагаются при температуре выше 380 °С [212]. Разложения
сшитых полиэфиртриазинов (№ 20) на воздухе начинается при 375 °С.
Объемистые заместители в боковой цепи, например флуореновые,
оказывают экранирующее действие в процессах гидролитического
и термоокислительного разложения [217].
При деструкции в инертной среде коксовый остаток при 900 °С
составляет 55—70 %¦ Термодеструкция полиэфиртриазина (№ 20)
протекает как реакция первого порядка. Основными продуктами
деструкции являются СО2, СО, Н2, фенол и производные триазина.
Реакция разложения инициируется гидролитическим расщеплением
эфирных связей и сопровождается распадом триазинового цикла
[217]. Поли-6-фенил-1,2,4-триазины (№ 1—6) стабильны в
инертной среде до 350 °С и на воздухе до 375 °С.
На основании данных ДТА и ТГА можно предположить [211],
что в инертной среде между 305 и 465 °С происходит
экзотермический распад N—N-связи триазинового цикла с образованием бен-
зонитрила:
N
^-
N
С6Н
—'А--—С—N=C—
N+
6П5
На второй стадии происходит рекомбинация промежуточного
продукта с замыканием триазинового цикла:
N
.—С—N=C— -
11
N
671
N
ir
N
N
N
N
N
N
N
Таблица 6.17. Некоторые свойства политриазинов
Элементарное звено
N
N
N
N
С6Н5
N ХС6Ы3
N
с6н5
с
°С
260
205
223
разл
пл
°С
370
375
320
355
разл
(на воз-
духе),
395
390
320
380
C6H5
N
N
с6н5
,N
N
с6н5
CeH5
N
N
?—S-
C6H5
N
—CH=CH—/Q)—CH=CH-
365
360
220
250
380
365
380
380
Продолжение табл. 6J7
разл
разл
(на воз-
315—320
355—385
440
340
400
380
Элементарное звено
ОСНз
t J.—сн=сн—^qN—сн=сн-
GH=CH—(( ))—CH=GH—
—сн=сн
N
C6H5
:h=gh-
GaH5
свн5
СНз
14
N
СбН5
15
16
17
18
N
270—310
355-385
295—305
315—320
290-310
340-350
400
320
365
365
Продолжение табл. 6.17
Элементарное звено
nji
°C
разл
(в N2),
°G
разл
(на воз-
духе).
19
285-310
395
OR-
20
350—410
c6h5
21
N
585
495
с6н5
22
N=
225
NH N
о
N
23
—NH—(( ) —(Г ))—NH-
470
460
0
N
C6H5
24
I— NH— l^ J NH-
400
410
N
N
OC4H9
25
Продолжение табл. б.17
26
27
28
29
Элементарное звено
(
ЭС4Н9
;CL,«_@-©-nh-
ОС4Н9
С
)C4H9
0
NJI
°C
•c1
(в N2),
°C
(иа
воздухе),
°C
300
230
300
Литературный
источник
[219]
[219]
[219]
[220)
CD
о
X
—NH
N
О
NH—
31
—а У)—сп,
¦NH—
0
32
N
—(С Г?—NH-
33
N
С6Н5
34
Oi
SO
N
250
185
400
400
350
230
350
[219]
[219]
[219]
[221]
[221]
Продолжение табл. 6.17
T
°C
T
раз л
(в N2J,
°C
раз л
(иа воз-
177-182
365
185
250
355
404
Элементарное эзено
-N—к J—N-
N N
C6H5
4
N
СНз
C6H5
N
Cl
t <^\ ^
Выше 480 °С протекает эндотермический распад остатка с
выделением HCN, аммиака, следов воды и дпциана. При температурах
от 690 до 760 °С происходит отщепление водорода и превращение
продукта в высококонденсированную ароматическую систему.
Температура длительной эксплуатации поли-6-фенил-1,2,4-триазинов на
воздухе составляет 260°С. Максимальная потеря массы при
нагревании на воздухе в течение 50 ч при 290°С составляет 13%.
6.4.3.3. Переработка и эксплуатационные свойства
политриазинов
Поли-6-фенил-1,2,4-триазины (№ 1—6) можно использовать в
качестве связующих и клеев, отличающихся высокой
термостойкостью [229, 230]. Титановое клеевое соединение, полученное на
основе полифенилтриазина, армированного стеклянной тканью при
287°С и давлении 2,5 кгс/см2, при комнатной температуре имеет
прочность при растяжении 175 кгс/см2. После 2000 ч нагревания
на воздухе при 260 °С прочность при растяжении составляет
147 кгс/см2.
Пленка, полученная поливом из раствора в ж-крезоле,
сохраняет хорошую гибкость после кипячения в течение 10 ч в 40 %-ном
растворе гидроксида калия. Поли-1,3,5-триазины с перфторалкиль-
ными группами представляют собой эластомеры, которые могут
применяться при температурах до 430 °С [227].
Для получения формованных изделий из полиэфир-1,3,5-триази-
нов (№ 20) используют технологию, применяющуюся при
переработке каучуков. В отличие от других термостойких полимеров
сшитые полиэфиртриазины имеют аномальновысокую ударную
вязкость; их твердость по Бринеллю 1000—1700 кгс/см2, модуль
упругости 30 000—50 000 кгс/см2, относительное удлинение при разрыве
3—6%. За счет свободных цианатных групп сшитые
полиэфиртриазины обладают высокой адгезией к различным металлам. Из
таких полимеров можно изготавливать стойкие к облучению,
термостойкие, гибкие конструкционные изделия и клен для воздушного
и наземного транспорта [217], Из бутоксплнрованных поли-1,3,5-
триазинов (№ 25—28) можно формовать эластичные пленки из
растворов в N-метилпирролидоне с добавкой Lid или из H2SO4.
Хрупкие волокна получают формованием из растворов в
N-метилпирролидоне с LiCl в осадительную ванну диметилформамид —
вода [219, 220].
Литература к главе 6
1. Н. Erlenmeyer, Helv. chim. Acta 27 A944), 489—493.
2. II. Erlenmeyer, Helv. chim. Acta 27 A944), 947—948.
3. H. Erlenmeyer, Helv. chim. Acta 27 A944), 969—970.
4. H. Erlenmeyer, Helv. chim. Acta 28 A945), 1281—1282.
5 H. Erlenmeyer, Helv. chim Acta 29 A946), 275—279.
6. H. Erlenmeyer, Helv. chim. Acta 29 A946), 1080-1084.
7. H. Erlenmeyer, Helv. chim. Acta 29 A946), 1924—1929.
8. J. Mulvaney, J. org. Chemistry 26 A961), 95—97.
9. Y. Inoue, Kogyo Kagaku Zasshi 63 A960), 1014.
10. J. Craven, J. Polymer Sei. B3 A965), 35—37,
58 L
11. W. Sheehan, J. Polymer Sei. A3 A955) 4, 1443—1462.
12. W. Shcehan, J. appl. Polymer Sei. 9 A965) 4, 1445—1471.
13. D. Longone, J. Polymer Sei. A3 A965) 9, 3117—3130.
14. В. Patnaik, J. Polymer Sei. A l 7 A969), 12, 3407—3412.
15. W. Whitmore, J. Polymer Sei. A l 8 A970) 10, 2759—2773.
16. G. Sperenza, J. org. Chem. 23 A958). 1922—1924.
17. S Sandler, J. appl. Polymer Sei. 9 A965), 1994—1996.
18. S. Sandler, J. Polymer Sei. 5 A967) 6, 1481 — 1485.
19. S. Sandler, J. appl. Polymer Sei. 13 A969) 12, 2699—2703.
20. R. Dileone, J. Polymer Sei. A 18 A970) 3, 609—615.
21. Y lwakura, J. Polymer Sei. A 1 4 A966) 4, 751—760.
22. Y. lwakura, J. org. Chem. 29 A964), 379—382.
23. Y. lwakura, J. Polymer Sei. A 1 5 A967) 8, 1865—1880.
24. 3. Пазенко, Высокомолек. соед., сер. А, 1970, 7, 41—47.
25. J. Stille, J. Polymer Sei. В 4 A966) 5, 329—331.
26. E. Neuse, Chem. & Ind. A965), 2005.
27. E. Neuse, Macromolecules 1 A968) 2, 171 — 178.
28. E. Кронгауз, Высокомолек. соед., А II, 1969, I, 48—56.
29. V. Korshak, J. Polymer Sei. A3 A965) 7, 2425—2439.
30. В. Коршак, ДАН СССР, 149, 1963, 3, 602—605.
31. В. Коршак, Высокомолек. соед., 6, 1964, 7, 1195—1202.
32. В. Коршак, Высокомолек. соед., 6, 1964, 6, 1087—1091.
33. В. Коршак, Изв. АН СССР, сер. хим., 1964, 1281—1288.
34. Е. Kronhaus, ISGS A967), 880.
35. J. Schaefer, J. Polymer Sei. В 3 A965) 2, 95—98.
36. В. Коршак, Высокомолек. соед., 67 1964, 6, 1078—1086.
37. В. Коршак, ДАН СССР, 152, 1963, 5, 1108—И10.
38. J. Stille, Makromolekulare Chem. 154 A972), 49—61.
39. J. Stille, J. Polymer Sei. A 1 7 A969) 9, 2493—2505.
40. J. Stille, Macromolecules 2 A969) 5, 465—468.
41. Опубл. заявка ФРГ 2219002 26. 10. 72 Labofina S. A.
42. W."Bracke, J. Polymer Sei. A l 10 A972) 4, 983—992.
43. А. Нау, J. Polymer Sei. 7 A969), 1625—1634.
44. D. White, Polymer Preprints 12 A971) 1, 155.
45. M Wheeler, J. Amer. chem. Soc. 45 A911), 383.
46. Y. Imai, J. Polymer Sei. A 1 5 A967) 9, 2289—2296.
47. В. Sillion, J. Polymer Sei. A4 A966) 11, 2903—2906.
48. R. Salle, Bull. Soc. chim. France A968) 8, 3378—3386.
49. Белы. пат. 678286 A966) Bayer.
50. L. Stoicescu-Crivetz, Rev. Roumaine chim. 15 A970) 1 129—134
51. W. Dunwald, Farbe und Lack 75 A969) 12, 1157—1160.
52. Акцепт, заявка ФРГ 1230568 A966) Bayer.
53. R. Merten, Angew. Chem. 83 A971) 10, 339-347.
54. В. Трезвов, Высокомолек. соед., Б 13, 1971, 11. 832-837,
55. Опубл. заявка ФРГ 1952396, 20.4.71 Bayer.
56. Пат. США 3397253 A965) Bayer.
57. Белы. пат. 700041 A966) Bayer.
58. Пат. США 3448170 A966) Bayer.
59. Опубл заявка ФРГ 1543616 A966) Bayer.
60. H. Kunzel, Makromolekulare Chem. 130 'A969), 103—144.
61. A. Oku, Makromolekulare Chem.. 78 A964), 186—193.
62. «Elektroisolierfolie-Versuchsprodukt I Do 4089», vorlaufiges Merkblatt Baver
AG.
63. Kunststoff-Berater A972) 12, 643—644.
64. «Verarbeitungsrichtlinien fur Resistherm PH 10», Firmenschrift Bayer AG,
65. «Verarbeitungsrichtlinien fur Resistherm PH 20» Firmenschrift Baver
66. W. Dunwald, Kunststoffe 62 A972) 6, 347—350.
67. T. Shono, J. Polymer Sei. В 5 A968) 11, 1001—1005.
68. Фр. пат. 2072502 29. 10.71 Labofina S. A.
69. Опубл. заявка ФРГ 2059239 2. 12. 71 Labofina S. A,
70. Англ- пат. 1275177 24. 5. 72 Labofina S. A,
682
71. Пат. США 2512599 A950) H. Bates.
72. H. Weidinger, Ber. 96 A963), 1064—1070.
73. В. Коршак, Изв. ЛН СССР, сер. хим.. 11, 1968, 2663.
74 J Stille, J. Polymer Sei. A3 A965) !2, 4284—4286.
75. J. Fisher, J. appl. Chem. 4 A954). 212—219.
76. M. Школнпа, Диссертация, ИНЭОС АН СССР, 1959.
77. J. Fisher, Brit. P. 693172 A959).
78. Пат. США 2615862 A952).
79. Англ. пат. 676785 A952) J. Fisher.
80. Пат. США 2395642 A941) W. Pritchard.
81. Т. Lieser, Ber. 84 A951), 4—12.
82. L. Stoicescu-Crivetz, J. Polymer Sei. С 22 A969), 761—771.
83. J. Hulsten, J. Polymer Sei. A3 A965) 11, 3905—3917.
84. А. Дьяченко, Высокомолек. соед., А 9, 1967 10, 2231—2235.
85. Акцепт, заявка Японии 715226 7. 2. 71 Teijin Ltd.
86. В. Коршак, Высокомолек. соед., А 11, 1969, 1, 7—10.
87. J. Hulsten, J. Polymer Sei. С 19 A967), 77—88.
88. J. Sundquist, Makromolekulare Chem. 134 A970), 287—298.
89. E. Кронгауз, Высокомолек. соед., Б 10, 1968, 2, 108—11L
90. E. Johnson, J. Polymer Sei. В 4 A966) 12, 977—979.
91. M. Baldwin, J. Polymer Sei. В 5 A967) 9, 803—806.
92. W. Gilliams, Makromolekulare Chem. 128 A969), 263—271.
93. E. Кронгауз, Высокомолек. соед., Б 9, 1967, 8, 563.
94. Р. Hergenrother, J. Polymer. Sei A 1 9 A971), 2377—2393.
95. R. Huisgen, Tetrahedron 11 (i960), 241—251.
96. В. Коршак, Высокомолек. соед., All, 1969, 1, 69—72.
97. В. Коршак, Высокомолек. соед., А 9, 1967, 10, 2231—2235.
98. Белы. пат. 628618 2.2.63 BASF.
99. С. Marvel, Makromolekulare Chem. 44—46 A961), 388—397.
100. J. Holsten, J. Polymer Sei. A3 A965) 12, 4284—4286.
101. A. Frazer, J. Polymer Sei. С 19 A967), 65—101.
102. A. Frazer, Appl. Polymer Symp. 9 A969), 119—131.
103. В. Коршак, Высокомолек. соед., Б 10, 1968, 3, 157—159.
104. A. Frazer, Air Force Material Laboratory Report AFML-TR-65-221 A965).
105. E. Dyer, J. Polymer Sei. А 1 6 A968) 4, 729—742.
106. Y. Imai, Kobunshi Kagaku 29 A972) 9, 610—614.
107. V. Crescenzi, Makromolekulare Chem. 141 A971), 199—210.
108. С Crescenzi, Chim. Ind. (Milano) 52 A970) 11, 1103—1109.
109. V. Crescenzi, Makromolekulare Chem. 120 A968), 220—224.
110. Y. Iwakura, J. Polymer Sei. А 1 6 A968), 1097—1108.
111. H. Elias, Makromolekulare Chem. 144 A971), 213—221.
112. H. Elias, Makromolekulare Chem. 144 A971), 183—192.
113. H. Elias, Makromolekulare Chem. 144 A971), 193—212
114. Пат. ГДР 83464 20.7.71 A. Wende.
115. Опубл. заявка ФРГ 2223624 16. 11.72 ICI.
116. Fr. Demande 2111913 13.7.72 Inventa.
117. T. Yoshida, Kogyo Kagaku Zasshi 74 A971) 9, 1945—1948.
118. Пат. СССР 332111, 14.3.72.
119. T. Кравченко, Хим. волокна, 14, 1971, 3, 15—16.
120. Акцепт, заявка Японии 7137914 8. 11.71 Furukawa Electric.
121. И. Шопов, Высокомолек. соед., Б 14, 1972, 5, 325—327.
122. Пат. СССР 317685, 19. 10. 71.
123. Пат. США 3632560 4. 1.72 Union Carbide.
124. Пат. США 3629198 21. 12.71.
125. Акцепт, заявка Японии, 7103191, 26. 1.71.
126. Пат. США 3642707 15.2.71 Du Pont.
127. Пат. СССР 349697, 4.9.72.
128. С. Виноградова, Высокомолек. соед., А 14, 1972, 4, 915—919.
129. Г. Береснева, Высокомолек. соед., А 14, 1972, 4, 731—734.
130. В. Родэ, ДАН СССР, 171, 1966, 2, 355—358.
131. С. Павлова, Высокомолек. соед., А 13, 1971, 12, 2643—2652.
583
132. Г. Тимофеева, Высокомолек. соед, А 13, 1971, 12, 2653—2660.
133. Пат. США 3044994 17.7.62 D. Bloomstrom.
134 W Funde. Makromolekulare Chcrn. 99 A966), 1—8.
135 S. Hong. Daehan Hevahak Hwoejie 15 A971) 3, 121 — 125,
136 M. Akiyama. J. Polymer Sei. В 4 A966) 5, 305—308.
137. V. Rode, J. Polymer Sei. A 1 6 A968) 5, 1351—1365.
138. V. Korshak, J. Polymer Sei. С 16 A967), 2635—2647.
139. N. Yoda, Makromolekulare Chem. 108 A967), 170—181.
140. Y. hvakura, Makromolekulare Chem. 94 A966), 103—113.
141. F. Wallenberger, J. Polymer Sei. A 2 A964), 1157—1169.
142. M. Sarasohn^J. Polymer Sei. A 14 A966) 7, 1649—1664.
143. T. Unishj, J. Polymer Sei. В 2 A964) 2, 237—239.
144. T. Unishi, J. Polymer Sei. A 1 3 A965) 9, 3191—3198.
145. Y. Iwakura, Makromolekulare Chem. 95 A966), 248—260.
146. В. Мухина, Высокомолек, соед., сер. Б, 1967, 284—289.
147. Е. Abshire, Makromolekulare Chem. 44 A961). 388—39
148. V. Iwakura, J. Polymer Sei. A3 A965) 1, 45—54.
149. С. Overberger, Science 141 A963), 176.
150. Y. Iwakura, Makromolekulare Chem. 104 A967), 66—76.
151. A. Frazer, Appl. Polymer Symposia 9 A969), 89—106.
152. A. Frazer, J. Polymer Sei. С 19 A967), 89—94.
153. Y. imai, J. appl. Polymer Sei. 14 A970), 225—239
154. Д. Frazer, J. Polymer Sei. A 2 A964), 1181—1183
155. Kuh. пат. 67091, 1963.
156. В. Коршак, Высокомолек. соед., А 9, 1967, 87—91.
157. Е. Кронгауз, Высокомолек. соед., А 9, 1967, 1, 87—91.
158.'В Коршак, Высокомолек. соед., А 13, 1971, 7, 1557—1563.
159. Пат. США 3567698 2. 3. 71 U. S. Dept. of Navy.
160. С. Виноградова, Высокомолек. соед.. А 13. 1971, 7, 1507—1515
IG1 В. Коршак, Высокомолек. соед.. ДАН СССР, 197, 1971, 3. 597—600,
162. Р. Hergenrother, Polymer Preprints 10 A969) 2, 772—778.
163. Y. Iwakura, Makromolekulare Chem. 108 A967), 160—I69.
154. G. Kossmehl, Makromolekulare Chem. 123 A969), 233—244
165. W. Memeger, Appl. Polymer Symp. 9 A969), 119—131.
166. J. Fisher. Chem. Ind. (London) 1952, 244.
167 Пат. США 2512633 A960) J. Fisher.
168. A. Frazer, J. Polymer Sei. A 2 A964), 1147—1156.
160. Пат. США 2615862 A952) Celanese.
170. A. Frazer, J. Polymer Sei. A2 A964), 1137—1145.
171. В. Коршак, Изв. АН СССР, сер. хим., 1965, 726—728.
172. R. Stolle, J. prakt Chem. 68 A903) 2. 30.
173. N. Pellizzare, Atti Acad. Lincei Rend. 8 A899) 1, 328.
174. R. Huisgen, Tetrahedron 11 (I960), 241—251.
175. A. Frazer, J. Polymer Sei. A 2 A964), 1157—1169.
176. A. Frazer, J. Polymer Sei. A 1 4 A966) 7, 1649—1664.
177. A. Frazer, J. Polymer Sei. A 2 A964), 1171 — 1179.
178. E Кронгауз, Высокомолек. соед., А 12, 1970, 1. 135—139.
179. Акцепт, заявка Японии 7041030 23. 12.70 Teijin Ltd.
180. Акцепт, заявка Японии 7039831 15. 12. 70 Teijin Ltd.
181. Акцепт, заявка Японии 7016718 10.6.70 Teijin Ltd.
182. Акцепт, заявка Японии 7016719 10.6.70 Teijin Ltd.
183. К. Корнев, Ж. гетероциклич. соед,, 1960, 353.
184. Пат. СССР 272552, 3. 6. 70.
185. Акцепт, заявка Японии 7109598 11.3.71 Fujikura Cable Works.
186. Акцепт, заявка Японии 7121903 22.6.71 Teijin Ltd.
187. Акцепт, заявка Японии 7115512 26.4.71 Furukawa Electric.
188. Акцепт, заявка Японии 256946 A959) W. Neugebauer.
189. M. Saga, Kogyo Kagaku Zasshi 70 A967), 584.
190. J. Kranz, Chem. Ber. 96 A963), 1049—1058.
191. Акцепт, заявка Японии 7103191, 26. 1.71.
192. Белы. пат. 628775, 1963.
584
193. M. Saga, J. Polymer Sei. В 4 A966) 11, 869—873.
194. R. Huisgen, Tetrahedron 11 A960), 241—251.
195 M Bruina, Rev. Roumaine Chim. 14 A969) 5, 659—665.
196 G Quesnel. С R Acad- Sei. С 272 A971) 17, 1499—1502.
197. В. Коршак, ДАН СССР, 166, 1966, 2, 356—360.
198. T. Shono, Kogyo Kagaku Zasshi 70 A967), 1250.
199. N. Dokoshi. Makromolekulare Chem. 108 A967), 170—1/1.
200. R Huisgen, Tetrahedron Letters 17 A961), 587.
201 С Overberger, J. Polymer Sei. B3 A965), 735—738.
202. J. Cotter, Chem. Commun. A966), 336—337.
203. H. Livingstoti, J. Polymer Sei. Л 1 7 A969) 12, 3434—3436.
204. В. Родэ, ДАН СССР, 176, 1967, 5, 1089—1092.
205. Акцепт, заявка Японии 7105537 10.2.71 Furukawa Electric.
206. Акцепт, заявка Японии 7105539 10.2.71 Furukawa Electric.
207. Акцепт, заявка Японии 7105539 10.2.71 Furukawa Electric.
208. S. Копуа, Kogakuin Daigaku Kenkyu Hokoku 31 A972), 162—166.
209. P. Hergenrother, J. macromoiecui. Sei. A 5 A972) 2, 365—382.
210. P. Hergenrother, J. Polymer Sei. A 1 7 A969) 3, 945—957.
211. W. Wrasidlo, Makromolekules 3 A970) 5, 548—555.
212. E. Greth, Makromolekulare Chem. 125 A969), 33—41.
213. E. Greth, Makromolekulare Chem. 123 A969), 203—222.
214. R. Audebert, J. Polymer Sei. С 16 A968), 3245—3254.
215. R. Audebert, Bull. Soc. chim. France A970) 2, 606—611.
216. L. Picklesimer, J. Polymer Sei. A 3 A965), 2673—2684.
217. V Pankratov, Plenarvortrag Hochpolymerentagung Berlin 1972.
218. I. Gavat, Rev. Roumaine 12 A967) 2, 131 — 137.
219. J Sundquist, Dissertation Helsinki 1971.
220. J. Sundquist, Angew. makromolekulare Chem. 14 A970). 203—212.
221. G. Fhlers, J. Polymer Sei A 2 A964), 4989—5003.
222. G. Ehlers, J. Polymer Sei. A 1 4 A966), 1645—1646.
223. Пат. США 3297639 A967) L Picklesimer.
224. R. Audebert, Compt. rend. Acad. Sei. 258 A964), 4749—4751.
225. E. Greth, Makromolekulare Chem. 125 A969), 24—32.
226. E. Doriman, Rubber Chem. Technol. 39 A966), 1175.
227. Chem. Week A964) 2, 35.
228. D Anderson. J. Polymer Sei. A 14 A966), 1689—1702.
229. P. Hergenrother, Report D 180-12911-1 Boeing Res. Lab. 1971.
230. P. Hergenrother, SAMPE-J. 3 A971) 1, 1—16.
231. L. Stoicescu-Crivetz, Rev. Roum. Chim. 11 A966), 1127—1131.
232. С Overberger, J. Polymer Sei. B3 A965), 735—738.
233. R. Biehle, J. Polymer Sei. AI 10 A972) 10, 2919—2923.
234. J. Higgins, Macromolecules 2 A969) 5, 558—559.
235. Фр. пат. 2088889 11.2.72 Esso Research.
236. D. v. Krevelen, Angew. Makr. Chem. 22 A972), 133—157.
237. Англ. пат. 705592 A966), 713132 A967), 712798 A967) 748357 П9691
748358 A969), 748359 A969), псе Akzo Rcs.
238. Хим. волокна, 1971, 2, 64.
7. ПОЛИБЕНЗГЕТЕРОЦИКЛИЧЕСКИЕ ПОЛИМЕРЫ
7.1. ПОЛИИМИДЫ
Полибензгетероциклические полимеры по своему строению
занимают промежуточное положение между
полигетероциклическими полимерами и лестничными полимерами (гл. 8). В литературе
применительно к этим полимерам используются термины «блок-
585
лестничные» или «полулестничные полимеры»:
W W
—X/ \-Cs ^>>/ ^Х—Аг—
•©"©
или
X
z
X
j
VY—Ar—
Полибснзгетероциклические (полулестничные) полимеры
отличаются от обычных полиароматических и
полигетероциклических полимеров (гл. 5 и 6) повышенной радиационной и
термической стойкостью, что следует из величин энергий стабилизации
B. 3). Высокая энергия диссоциации внутри сопряженных бенз-
гетероциклических цепей также обусловливает высокую
термостойкость полимеров этого типа. В этом случае, если в
замкнутую бициклическую систему в качестве гетероатомов W, X, Y, Z
входят О, S, N, NH, то вследствие уменьшения количества
водородных атомов стойкость к окислительной деструкции полимеров
возрастает. Температура начала их разложения, по данным ТГА,
в инертной среде находится в интервале 400—700 °С.
Термостойкость полимеров с одинаковыми арильными
радикалами возрастает в следующем ряду: полибензоксазиндионы << по-
либензоксазино <С полихиназолоны <С полибензимидазолы <С по-
лихиноксалины <С полиимиды <С полихиназолиндионы<Сполибенз-
оксазолы <z поли-Ы-фенилбензимидазолы << полибензтиазолы.
По данным ТГА, при нагревании полимеров в изотермических
условиях на воздухе при 370 °С полимеры по увеличению
стойкости к окислительной деструкции располагаются в ряд:
полибензимидазолы <: поли-Ы-фенилбензймидазолы << полибензтиазолы <
полихиноксалины <С полибензоксазолы <С полиимиды.
Синтез полибензгетероциклических полимеров осуществляется
циклополиконденсацией из ди- или тетрафункциональных
мономеров через стадию промежуточного образования растворимых и
плавких продуктов. Полигетероциклоцепные полимеры, состоящие
из гетероциклов, связанных телько фениленовыми радикалами,
отличает высокая жесткость цепи; они в большинстве случаев
неплавки и нерастворимы, температура стеклования находится в
пределах 400—600 °С. Их переработка осуществляется на стадии
плавкого и растворимого линейного форполимера. Циклизацию
проводят в готовых изделиях или полупродуктах. Наличие
«шарнирных» атомов и групп в структуре макромолекулы, таких, как
О, S, SO2, СО, СН2, приводит к снижению теплостойкости и
увеличению гибкости:
лОХ
п
586
Эти полимеры можно перерабатывать как обычные
термопласты. Производство полигетероариленов было начато в 1961 г.
С тех пор объем производства и ассортимент выпускаемых
продуктов неуклонно возрастают. Среди производимых в промышленном
масштабе циклоцепных полимеров наиболее важными являются
полиимиды
—N1
N—Ar—
п
объем производства которых в 1971 г. составил 300 т. Они
выпускаются в виде пресс-изделий, пленок, волокон, связующих, пено-
пластов с длительной работоспособностью до 260 °С.
Полимеризацией бисмалеимидов получают сшитые полимеры
яналогичной структуры, которые могут длительно
эксплуатироваться при 220—250 °С. Температуру длительной эксплуатации по-
лиимидов можно повысить до 300 °С, используя сополимеры,
содержащие наряду с имидными оксадиазольные и бензоксазольные
фрагменты (см. 7.1.2). Полиампдопмиды лучше перерабатываются,
чем гомополимеры (см. 7.1.3)
N—Ar—NH—
п
Армированные пластики, пленки, волокна и кабельную
изоляцию на их основе можно эксплуатировать в течение
продолжительного времени при 200—240 °С.
Полиэфироимиды используют предпочтительно для получения
кабельной изоляции, длительно работающей при 190 °С
N—Ar —
Полибензимидазолы (см. 7.2) в виде волокон или связующих
выдерживают кратковременный нагрев до 450 °С. Синтактические
687
полибензнмидазольиые пснопласты обладают отличной
абляционной стойкостью.
— П
Полибензоксазолы используют при склеивании металлов и для
изготовления электроизоляционных пленок, длительно работающих
N
О
R
N
О
Ar —
п
при температурах до 300 °С.
Полихиноксалпны (см. 7.5) применяют для изготовления
пленок,
N N
R
ю:
N
N
R
п
покрытий, связующих и клеев для склеивания металлов,
длительно эксплуатируемых до 300 °С.
Электроизоляционные пленки из полибензоксазинонов (см. 7.8)
пригодны для длительной работы до температуры 250 °С.
N
N
У
Аг-
-0
п
Из полибензоксазиндионов (см. 7.9) можно формовать
различные изделия, пленки и покрытия, верхний температурный предел
О
о
п
длгьельной эксплуатации которых составляет 230 °С.
Полисульфимиды G.1.5), полибензтиазолы G.4), полихиназо-
лоны G.6) и полихиназолиндионы G.7) по своим свойствам
аналогичны другим полигетероциклоцепным полимерам. Сведения об
их промышленном производстве отсутствуют.
588
I.I.I. АРОМАТИЧЕСКИЕ И АЛИФАТИЧЕСКИЕ ПОЛИИМИДЫ
Ароматические полиимиды (пилиаримиды) в общем объеме
производства высокотермостойких полимеров специального
назначения занимают первое место. Их производство уже в 1971 г.
составляло 300 т в год. Ароматические полиимиды получают через
промежуточную стадию образования полнамидокислот или ари-
ленбисмалеимидов с последующим превращением в полиимид
непосредственно в процессе изготовления формованных изделий,
пленок, электроизоляции, волокон и пенопластов. Длительная
эксплуатация ароматических полиимидов возможна до температуры
260°С. Кратковременное нагревание такие полимеры
выдерживают до 480 °С. Ароматические полиимиды негорючи и в широком
температурном интервале имеют хорошие физико-механические и
диэлектрические свойства. Они устойчивы при комнатной
температуре к действию растворителей и концентрированных кислот, за
исключением серной и азотной. Главные направления
использования ароматических нолиимидов—электроника, электротехника и
авиация.
7.1.1.1. Получение
Полиимиды были впервые получены Боджертом и Реншоу
[114] в 1908 г. полико^денсацией эфиров или ангидрида 4-амино-
фталевой кислоты. Высокомолекулярные продукты были
синтезированы в 1955 г. путем двухстадийной поликонденсации пиромел-
литового диангидрида с диаминами [53, 249—252]. Широкие
исследования в этой области проводят в США фирмы «Westinghouse
Electric», «Whittacer Со», «Monsanto», «Du Ponst», в Англии —
«Royal Aircraft». В СССР глубокие исследования в этом
направлении выполнены В. В. Коршаком, С. В. Виноградовой
с сотр. и М. М. Котоном с сотр. (Институты элементооргани-
ческих и высокомолекулярных соединений АН СССР).
Первые технические продукты, появившиеся на рынке, —
электроизоляционные лаки и пленки — были произведены фирмой «Du
Pont». Выпускаемые в настоящее время продукты представлены
в табл. 7.1. Температуры плавления, размягчения и разложения
полиимидов, синтезированных в настоящее время, приведены в
табл. 7.2.
Мономеры для получения полиимидов. Диангидриды
ароматических тетракарбоновых кислот, такие как пиромеллитовый диан-
гидрид и диангидрид дифенилоксидтетракарбоновой кислоты
получают сложным путем.
В промышленности пиромеллитовый диангидрид получают
окислением тетраалкилбензолов, таких как триметилизопропилбензол,
диметилдиизопропилбензол, диэтилдиметилбензол или метилтри-
этилбензол. Окисление проводят действием азотной кислоты или
перманганата калия в жидкой фазе либо газофазным окислением
на воздухе при 450 °С в присутствии пентаоксида ванадия [406].
Полученный продукт очищают перекристаллизацией из ацетан-
589
Страна
США
США
США
США
США
США
США
США
США
США
США
США
США
США
Таблиц
Торговое название
Каптон H
Пленка H
Каптон ИГ
Каптон НВ
Волокно НТ-1, PRD-14
PI-4707
PI-2501
PI-9622
Р 105 А
Р 13 N
Монсанто 710
Скайбонд 700
Скайбонд 703
Панралан
Пайр ML
ЕМ 34
WRD-9371
HRH-324
UHU QO7
Имидайт 1832
а 7.1. Торговые названия
Изготовитель
«Du Pont»
«Du Pont»
«Du Pont»
«Du Pont»
«Du Pont»
«CIBA-Geigy»
«CIBA-Geigy»
«Monsanto»
«Monsanto»
«Du Pont»
«Du Pont»
«Bloomingdale Cyana-
mid»
«Narmco»
и фирмы-изготовители полиимидов
Материал
Электроизоляционная пленка
Пленка, сваривающаяся при
нагревании
Двухслойная пленка
Волокно
Связующее для слоистых
пластиков
То же
Пресс-ком позиция
Связующее для слоистых
пластиков
То же
Связующее для слоистых
пластиков
Электроизоляционный лак
Клеевая пленка (носитель
стеклянная ткань)
Ткань, пропитанная
связующим
Элементы сотовой структуры
Связующее для слоистых
пластиков
Состав
Полипмид на оспопе пиро-
меллитового диангидрида и
диаминодифенилового эфира
Тот же полиимид, покрытый
сополимером этилена с тетра-
фторэтиленом
Полиимид —
политетрафторэтилен
Низкомолекулярный
полиимид с концевыми норборне-
новыми группами
Полиамидокислота на основе
диэфира бензофенонтетракар-
боновой кислоты и
ароматических диаминов
Полиимид — борное волокна
Метлбонд 840
PI-5505
PI-5515
Квонтад 159
Скайбоид RI-7271
Полиимид/XPI
182
NR50
Poly-X
Джемон 2010
Полиимид 2080
Джемон 2012
Джемон 3010
Мелднн
Веспел SP1
Веспел SP5
Веспел SP21
Веспел SP211
Веспел SP31
Аримид ПМ-1, ПМ-2,
ПМ-4
Аримид
«Narmco»
«Quantum Inc.»
«Monsanto»
«Cyanamid»
«Du Ponb
«Du Pont»
«Raychem»
«General Electric»
«Upjohn»
«General Electric»
«General Electric»
«Duxon Со»
«Du Pont»
«Du Pont»
«Du Pont»
«Du Pont»
«Du Pont»
Клеевой полимер
То же
Порошок для получения пе-
нопластов
Пресс-композиция
Пресс-литьевая композиция
Пресс-композиция
То же
Пресс-композиция,
наполненная 25% стеклянного
волокна
Волокно
Связующее для слоистых
пластиков
Пресс-композиция
Пресс-композиция,
наполненная 15% графита и 25%
стеклянного волокна
Пресс-композиция,
наполненная 60% стеклянного
волокна
Стержни, пластины
Формованные изделия
Наполненные материалы
То же
Полнимидиая пленка
Волокно
Алифатически-ароматический
полиимид
Термопластичный полиимид
Ароматический полиимид
Наполнитель — коротковолок-
нистое стеклянное волокно
30% (объемн.)
40% графита
15% графита
10% политетрафторэтилена
95% дисульфида молибдена
Продолжение табл. 7Л
Страна
СССР
СССР
СССР
Франция
Англия
Торговое название
ПАК-1, Аримид ПМ
СП-3, СП-6
СТ-1
ПМ-67
Аримид ДФО
Керимид 601
Кинел 5504
Кинел 5514
Кинел 5515
Кинел 5505
Кинел 5508
Кинел 5517
Кинел 5518
M 33 и MN 3
Компаунд VO 1011
Компаунд VO 5502
IP 380
IPA 380
QX-13
Изготовитель
«Rhone-Pouienc»
«Yorkshire Chem. Ltd.»
ICI (до 1971 г.)
Материал
Связующее для слоистых
пластиков
То же
Клеевой полимер
Пресс-композиция
Пресс-литьевая композиция
Связующее для слоистых
пластиков
Наполненная
пресс-композиция
Пресс-литьевая композиция
Пресс-композиция
То же
»
»
Литьевая композиция
Связующее для слоистых
пластиков
Клеевой полимер
Клеевая пленка для
склеивания металлов
Связующее для слоистых
пластиков, пресс-композиция
Состав
Полиамидокислота на основе
пиромеллитового диангид-
рида и диаминодифенилового
эфира
Полиимид на основе диангид-
рида дифенилоксидтетракар-
боновой кислоты и диамино-
ТТ ? I /Г4НЪ S*\ II Г] П f\ Л^ Л f I ft Л
дифснилоксида
Бисмалеимид
Наполнитель — стеклянное
волокно
То же
Наполнитель — графит
Наполнитель — графит и
дисульфид молибдена
Политетр афторэтилен
Бисмалеимид
Наполнитель — графит и
асбест
Бисмалеимид
Эфир со-аминодикарбоновой
кислоты
гидрида, комплексообразованием с анизолом или вакуумной
сублимацией.
Исходным продуктом для синтеза бисC,4-дикарбоксидифени-
лового эфира) является о-ксилол, из которого бромированием с
последующим аминированием и диазотированием получают 3.4-
ксиленол:
СНэ СНэ СН3 СНз
СНа
—НВг
СН3
NH3
>¦
—НВг
:SX"CH" NaNO2
Н +
СН3
Вг
NH2 ОН
Образующийся 3,4-ксиленол при взаимодействии с бромксилолом
дает тетраметилдифенилоксид [253]:
О
ОН Вг
CHS
Н3С
>
—HBr
СНэ
При окислении перманганатом калия получается соответствующая
тетракарбоновая кислота [254].
Двухстадийная поликонденсация производных тетракарбоновых
кислот с диаминами. При получении полиимидов на первой стадии
происходит образование полиамидокислот:
О О
—(( ))—NHS
—((у)—о—((У)—
п
На второй стадии протекает циклодегидратация этих полиами
докислот с превращением их в полиимиды:
—HNOC. ^ ^СООН
—N;
"^CONH—/Q\—О ''О)—
о о
п
II
о
»-<O>->-<O)-
—п
593
Таблица 7.2. Температуры размягчения, плавления и разложения полиимидов
Элементарное звено
1 разм1
Гразл
(в N2),
т
раз л
(на воз-
Духе),
о
—N:
о
N—
(СН2J-
:N~(CH2)e-
о
135
400
170
о о
—N;
\
Л
N—R—
R =
О О
N-R-
О
О. Cl Ci ,0
—n:
с/'
ci
:N-(CH,b
а\
200—300
400
Тпл = 228-280
332
Uродолжение табл. 7.2
№
Элементарное звено
разм1
°С
Г
разл
(в N2i.
Т
разл
(на
воздухе),
Оч Cl Cl
;n-(ch2)8-
Cl Cl ^V)
Ox Cl Cl /O
>-(CH2)9-
O^ Cl CI
4
Cl
-N.
o" ci ci
Ov Cl
10
o^ ci Ci
N-(CH2)n-
= 201-247
Гпл = 200-216
330
о
11
—n:
12
13
N—(CH2JSO2(CH2J—
О
C6HS
//
14
265-325
—R— R = -(CH2N-,
Продолжение табл. 7.2
№
15
16
17
О
/^
—N
\r-
О
Y
—N
О
>
—N
A
J-N—L
Ф
Элементарное звено
о
N—Ar—
?j-(OH,b
"разм*
разл
<в Na),
°G
Т
разл
(на
воздухе),
420
Литературный
источник
[50]
[134] '
[186, 207]
18
О
420
[186, 207]
410
[186, 207]
-20
485
[186, 2071
'21
420
[186, 207]
Продолжение табл. 7.2
Элементарное звено
разм'
т
разл
(в N2),
Т
разл
(на воз-
духе),
°С
Литературный
источник
О
О
—N
Y
О
•—(СН2K-
420
420
450
[186, 207]
[186, 207]
[186, 207]
[40, 169,
239]
26
27
28
29
О О
с; n~(ch2J-
о
о о
Л /N~(CH2N~
о
о о
-n; g n-(Ch2I0-
o о
о о
Y
О
«-о
"V-
1
205
420
125
400
78
420
Продолжение табл. 7.2
T
разл
(в N2),
°C
(на воз-
Духе),
ос
360
370
280
290
170
о о
Элементарное звено
О О
-CH2(CF,LCHa-
О О
СНз
о о
о о
35
СНз
ОТ >-(СН2)з-С-(СН2K
СНз
о о
о о
36
350
135
135
л - 320
330
Продолжение табл. 7.2
Элементарное звено
О
37
-(CH2)s
о
о
о
38
iOi
39
о
О
Л
о
о
т
т/ y/~\y \nch2chch2o
он
—ОСН2СНСН2
разМ'
°с
по
л = 325
Гпл = 310
разл
(в N2),
410
разл
(на
воздухе),
О О
40
—n:
i
NCH2CHCH2C
ОСОСНз
) ОСНгСНСНг—
ОСОСНз
= 146
395
о о
41
о
4
NCH2CHCH2O—Г/^Vi-
он ^
)СН2СНСН2—
он
= 200
380
о о
42
l
2f)СН2СНСН9—
ОСОСНэ ^-^ ОСОСНз
117
400
о о
43 ! —n;
\
N-(CH2N-
о
300
Элементарное звено
44
45
46
47
—(СН2),-
QI >-(СН2N-
(СН2)8"
Продолжение табл. 7.2
разм*
°С
'разл
vb N2),
раз л
(на
воздухе),
280
210
190
О
43
о
49
О
о
50
Г0П"°~Пр
^ О О ^^
о
о
о
51
-о-с—|
о о
270
440
450
415
420
Продолжение табл. 7,2
разм'
°С
раз л
(в N2),
°С
разл
(на воз-
Духе),
°С
310
270
205
О
52
о
53
о
54
so
Элементарное звено
о
] >'-(СН2)в-
о
о
N-(CH2)8-
l
ю
о
55
—N
56
57
NH2 О
58
195
500
4C0
450
1511
11, 2, 150,
165, 208,
2251
[152]
[1, % 208,
225]
Продолжение табл. 72
Элементарное звено
раэм'
(в N2),
т
разл
(на
воздухе),
°С
59
о
о
60
о
61
-< IOI X4OMQ
490
330
о о
Щ
CI
62
or>
\
,ci
[208]
II il
о о
О О
63
eu
XI CL
ci
/
[208]
Cl
О О
F, ,FF,
64
О
О
[208]
О KO3S SO3K
65
300
1199]
Продолжение табл. 7.2
Элементарное звено
разм*
°С
Гразл
разл
(на воз-
духе).
Литературный
источник
66
67
68
О
ШСНз
N(CH3J
NHCH,
OI ХОНОУ-
N(CH3),
[6]
[197, 247]
[197, 247,
248]
о o
69
[187, 197,
247]
N
HN
HN—C6H5
70
71
18]
500
450
[198]
О
[198]
Продолжение табл. 7.2
Элементарное звено
разм1
°С
разл
разл
(иа
воздухе),
73
-К 1О1 >-*
74
\
[208]
[208]
R=-]
C
О О О
о о
75
о о
О
о
о
77
о
о
о
¦<jco;
о
500
[7]
500
171
[3. 5, 1421
Продолжение табл. 7.2
Элементарное звено
1 разм'
Т
'разл
(в N2),
т
разл
(на
воздухе),
Литератур*
иый
источник
78
79
[3, 5, 149]
[9, Ю, 66,
208]
О^ О \>
400
[И]
О
81
—n:
500
[67]
о о
[240]
83
[12]
Продолжение табл. 7.2
Элементарное звено
разл
(на
воздухе),
°С
О
—n:
k^
H"N—C6H5
490
86
О
484
О
87
О
[208]
88
89
о
л
О
О
260
[14, 111,
142, 143]
[14J
Продолжение табл. 7,2
Элементарное звено
разы1 '
•С
^разл
(на воз-
Литературный
источник
90
О
91
92
NH— C6HS
—с6н5
[61]
[187]
[61]
93
94
95
96
NH—С6Н,
Г„„ = 460
410
Продолжение табл. 7.2
Элементарное звено
размф
'разл
(в N2),
°С
разл
(на
воздухе),
UC
Литературный
источник
97
98
99
360
415
320
47O
[151
[15J
A851
100
—N'
101
102
103
326
480
116, 185]
[229]
260
[13]
450
[13]
¦о
Продолжение табл. 7.2
Элементарное звено
1 разм*
°С
разл
(B.N2).
т
разл
(на воз-
Литературный
источник
104
105
Каптон («Du Pont»)
Аримид ПМ (СССР)
525
490
400
[1-4,
17-19, 57,
82, 88, 92,
99, 104, 119,
122, 135,
137, 139,
149, 153,
158, 162,
163, 165,
166, 172,
183, 193,
200-202,
208, 212]
[65, 208J
а об
340
360
[155, 1791
107
470
[2, 5, 146,
1C5, 200,
208]
108
500
[200, 208]
109
[208]
Продолжение табл. 7J2
Элементарное звено
разм*
°С
Гразл
(на воз-
Литературный
источник
О
о
345
430
300
[2, 163,
208]
12]
[1791
113
—N
114
R
-< oi >-«-
О О
115
450
A371
[97, 144]
CH3
\
SO3H
410
[80, 166,
200, 208)
Продолжение табл. 7J2
Элеиентарцое аввно
'разы*
°С
Гразл
(на воз-
Духе),
°С
116
117
118
О
О
463
375
375
230
119
325
[1791
О О
120
400
320
[I, 2, 193,
2261
121
СНз
355
[179, 203)
о о
122
CF3
r
CF3
[208]
Продолжение табл. 72
№
Элементарное звено
разм'
°С
Гразл
(на воз-
123
124
125
490
445
480
126
—n;
127
О О
128
о
о
400
Ц7Э]
430
[22, 23, 25]
123]
Продолжение табл. 7.2
разм*
°С
т
разл
разл
(на воз-
)
515
385
520
440
129
130
о
131
Элементарное звено
000
(NH2J
6н6
о о
132
-< JOI >-О-СН2-сн>-О-
470
200
о
133
Г
300
О
134
480
О О
135
-Опт >-<о:
Продолжение табл. 7.2
Элементарное звено
разм'
°С
разл
(в N2),
Г
разл
(на воз-
Духе),
°С
Литературный
источник
136
137
138
[22]
[133]
[230]
0 0 0
139
-N
О О
I40
о
о о
N
141
—n;
N
о о
о
142
1
о
!!
о
[83]
[31]
350
[29, 30, 407]
300
[3, 5, 142,
149]
Продолжение табл. 7.2
разм'
°C
т
разл
(в N2),
°с
т
разл
(на воз-
ДУохе,,
320
375
340
№
О
ИЗ
о
О
144
145
Элементарное звено
1!
о
о
n-ch,—^ч—
146
О
147
О
о
148
-< толпе >
415
[21, 151]
425
[21, 151]
430-480
[3, 5, 142]
Элементарное звено
разм'
т
разл
разл
(на
воздухе).
О
149
130
151
152
-< lOnOi XO.
240
153
—N;
420
154
—N
О
155
о
270
470
О
156
—n:
250
Продолжение табл. 7.2
Элементарное звено
разм*
°С
гразл
(в N2).
Г
разл
(на воз-
дувхе).
О
157
158
159
265
305
230
160
-N
161
162
163
440
[163]
290
[3]
285
430
320
[21, 163]
430
[163]
Продолжение табл. 7,2
Элементарное звено
разм1
Т
раз л
(в N2).
°С
разл
(на
воздухе),
С
Литературный
источник
О
164
375
[21, 151]
165
385
[21]
о
о
166
405
500
О
167
—N'
\
г
О
385
О
168
-N
l
О
!!
I
; (CF2K (
150—160
380
[184]
Продолжение табл. 7.2
Элементарное звеио
разм1
Т
разл
(в N2),
°С
¦* разл
(на воз-
)
О
169
il
170
171
О
—к
о
о
О
О
о
155-160
230
195
о
172
173
О
174
О
175
—N
О
О
О
255
л = 430
210
250
340
Продолжение табл. 7.2
Элементарное звено
разм,
DG
Т
разл
(в N2),
°С
Т
разл
(на воз
духе),
°С
176
о
О
177
—N'
О
178
О
О
i
-СН2—,y-Vl—СН:
—сн2-
500
230
340
о
179
О
II
S'il
О
о
440
450
О
О
180
-N'
согг
О
О
О
X
О
181
—N
г
О
О
О
290
Продолжение табл. 7а
Элементарное звено
разм-
°С
разл
(в Na).
°С
разл
(на воз-
Литературный
источник
Скайбонд 700 («Monsanto»)
О О
182
183
184
О
О
О
[70]
[87, 208, 209]
[208]
о
185
О
О
186
О
187
о
о
О
.N—I
О
260—270
260-270
375
470
Продолжение табл. 7.2
Элементарное звено
разм1
°С
Т
разл
(в N2),
°С
т
раз л
(на
воздухе),
°С
188
189
N
380
[21]
[62]
190
1151]
О
О
191
—N
¦/ I /^л1
О
О
у
470
[21]
О
О
192
—n:
N-
270
[141
Продолжение табл. 7.2
Элементарное звено
разм*
°С
Т
раз л
(в N2).
°С
разл
(на воз*
духе),
°С
Литературный
источник
гез
о
.N
I
го-
C6H5
о
о
194
II
-< ion-°mDi >
v
i
195
—N
475
[36]
[2U]
[208]
о
196
О
CF3
|_
I
CF3
CF3
CF3
[38]
О
197
О
CF3
С—i
CF3
О
[208]
О
О
198
DJ V
!ес
345
[15]
о
Продолжение табл. 72
Элементарное звено
Т
раз л
(в N2),
°С
Т
разл
(на
воздухе).
°С
199
О
200
-К
о
о
\
о
I
о
О
320
370
о
201
,N—fVSi—S—j
о
202
—N.
Y
о
\>
315
305
о
о
203
—n:
Q) \*-сн2—г^
сн2-
о
;o
I
240
Продолжение табл. 7.2
Элементарное звено
разм1
Т
разл
(в N2),
разл
(на воз-
дуохе),
О
204
< I
Y
205
о
О
о
\> o
;о
О
С
NH
О
415
410
О
О
206
—N
\
о
О
425
[15]
О
207
СНз
'Si—(
СНз
О
>-€
€к
О
О
208
Ci !
ел
-о \
Л
О
л
А
[1511
282
420
Продолжение Tuo.i. 7.2
Элементарное звено
' разм-
раял
N2).
(на
воздухе К
О
209
—N'
I
О
210
о
о
211
О
о
о
о
О
270
300
270
470
470
О
О
212
О
О
О
О
213
О
О
C
214
215
II
О
О
О
:оьн
О
О
230—240
190—200
230—240
260—265
470
440
440
1
Продолжение табл. 7..
Кя
Элементарное звено
разм1
°С
Т
разл
(в N2),
°С
разл
(на
воздухе),
°С
О
216
О
217
—Ny
О
218
О
Л
О
О
130—140
175-180
215
480
520
430
450
о
219
О
220
-< от
I
о
о
221
о
о
—rrVr— (CF2K
О
о
о
о
о
222
-N'
¦(CF2O—|
О
163
155
180
Тип = 332
175
ГПл = 330
440
460
470
380
390
[184]
[184]
[184]
[184]
Прооолжение табл. 7.2
Элементарное звено
1 разм-
°С
разл
(В N2).
°С
Г
разл
1на воз
духе).
"С
223
О
224
0
225
130
по
180
= ззо
430
о
о
226
-N'
": (СF2) 16 -
,—,^чГ"\
227
-К jQj °
228
229
о
N-
О
О
350
230
290
200
390
о
о
Продолжение табл. 7.2
Элементарное звено
разм'
°С
Т
разл
(в N2).
°С
1 разл
(на воз-
о
230
231
О
О
О
о
232
—N
о
х
О
| О ,7-У:
О
о
215
202
О
233
О
о
234
[601
[921
О
О
235
208
450
[12, 13, 204]
Продолжение табл. 7.2
Элементарное звено
разм'
Т
раэл
(в N2>.
°С
Т
разл
(на
воздухе),
°С
Cl
Cl
о
о
l
о
о
о
l
Cl
Cl
СНз
СНз
\
Cl
202
450
194
о
Cl
239
N-
IQ
Cl
l
СНз
-г©
СНз
163-167
385
[419]
О
С
240
N Г^\! N
CI CI
Cl
сн, /
О
Cl
222
405
1419]
О
241
СНз
[419]
Продолжение табл. 7.2
Элементарное звено
разм'
°С
Т
разл
(в N2),
°С
Т
разл
(на
воздухе),
Литературный
источник
242
Cl
Cl
243
О
244
II
о
о
I
О
СНз
)—с
СНз
174
187-192
184
400
[419]
330-370
[419]
330
[419]
О
Cl
246
О
о
CI
—(( )>—CHS
(
о
о
247
| 173-175
О
о-
СНз
-с
СН
200—215
О
,C1 Cl
ce ^
СНз
Cl
С1
\
CI
о
Cl Br
Вг
СНз /
Вг
СНз \
Br
330-360
335
350
[419]
[419]
[419]
Полиамидокислоты получают главным образом введением
тщательно очищенных твердых днангидридов тетракарбоновых кислот
в раствор диаминов в отсутствие влаги и кислорода при
температуре около 50 °С. В качестве апротонных растворителей
используют диметилформамид, 1^,М-днэтилформамид, Ы,1Ч-диэтилацет-
амид, М.М-диметилоксиацетамид, N-метилпирролидон, Ы-ацетил-2-
пкрролидои, пиридин, гексаметилфосфотриамид, диметилсульф-
оксид, диметилсульфон или тетраметиленсульфон [2, 121, 216, 225,
226, 255, 262, 263].
Эти растворители образуют с диангидридами активные
комплексы. В диметилсульфоксиде полиамидокислоты получаются с
более высокой молекулярной массой, чем в диметилформамиде или
диметилацетамиде, что объясняется его более высокой
полярностью. При добавлении диангидрида молекулярная масса
полимера непрерывно растет, достигая максимального значения при экви-
мольном соотношении реагирующих веществ. При использовании
смеси двух диангидридов [222] или двух диаминов получаются
сополимеры полиампдокнслот [223]. Поскольку свойства пол ними-
дов зависят от степени полимеризации полиамидокислот, следует
тщательно соблюдать условия синтеза. Нагревание массы выше
50°С сопровождается частичным превращением в полнимид и
приводит к деструкции полиамидокислоты за счет выделяющейся при
этом воды. Гидролитическая деструкция полиамидокислот при
температурах поликонденсации выше оптимальной приводит к
образованию солей [209] ;
О
II
С—СГ H3N
-ОН (ДМАА)
I
v
При частичной имидизации отщепляющаяся вода находится в
реакционной системе в виде гидроксильных ионов, а амидный
растворитель комплексно связывает ионы водорода. Нагревание
массы выше 100°С сопровождается, кроме того, выпадением
низкомолекулярных продуктов в осадок. Оба процесса приводят к
снижению молекулярной массы полиамидокислот и, как следствие, к
ухудшению свойств полиимидов. Только при применении в
качестве растворителя ж-крезола можно проводить реакцию при
температурах до 160 °С [256].
Соблюдение указанной выше последовательности загрузки
реагирующих веществ является фактором, оказывающим
решающее влияние на поликонденсационный процесс. Полимеры с
наиболее высокой молекулярной массой образуются при введении в
раствор диамина твердого диангидрида в количестве, на 0,4—0,5 %
(мол.) превышающем эквимольное. Если оба компонента добав-
670
ляют в растворенном виде, то молекулярная масса полпампдокнс-
лоты будет тем меньше, чем меньше концентрация растворов.
Высокомолекулярные полпаыидокнелоты могут быть пол\чигы
при введении и растворитель гомогенной эквимольнои смеси твор-
дого дпангидрпда и твердого диамина [209]. Различие между
гетерогенным и гомогенным методами синтеза заключается в том,
что в первом случае аминолиз и гидролиз представляют собой
последовательные реакции, в то время как при гомогенном
(растворном) методе смешения оба процесса протекают
одновременно [257]:
О
kA, H2NRNH2
Соотношение констант скоростей реакций k,\/kr составляет
около 5. Реакционная способность диаминов по отношению к днан-
гидридам возрастает в следующем ряду [266]: гексаметнленди-
амнн > декаметилендпампн > 4>4/-дпаминодифенплметан > 4,4Г-
дпаминодифенилоксид > w-фенилендиамин > л/-фенилендиамин >>
> льтолуилендиамин > 4,4/-диамино-3,3/-диметилдифенилметан >
> 4,4/-диаминодифенилсульфон.
Синтез полиамидокислот представляет собой бимолекулярную
реакцию ацилирования амина. В результате нуклеофильной атаки
аминогруппы происходит раскрытие ангидридного цикла [267].
Кинетические исследования [200] показали, что в процессе
синтеза полиамидокислот сначала образуются моноацнлнрованные
диамины. Увеличение молекулярной массы происходит за счет их ди-
мерпзации:
О
О + H2NRNH2
А
671
При введении 78 % (мол.) диангидрида к рассчитанному
количеству диамина, находящемуся в растворе, устанавливается
равновесие [257, 267]. Не вступивший в реакцию диамин [22% (мол.)]
реагирует с карбоксильными группами полиамидокислоты с
образованием соли:
h'n—О
X0NH—О—
При нагревании происходит дальнейшее превращение:
CONH-
—н2о
Для модельных систем, включающих ароматические амины и фта-
левый или пиромеллитовый ангидриды, получены константы
равновесия [243].
672
Полиамидокислоты существуют не только в виде 1,4-диамидов,
но в известной мере и в виде 1,3-диамндных структур:
HNOC
CONHR—"
1,4-Днамид
1,3-Диамнд
Синтез полиамидокислот сопровождается изменением окраски.
Окрашенные кристаллы удалось выделить из разбавленных
растворов в тетрагидрофуране. Стабильность этих соединений,
имеющих характер комплексов с переносом заряда, уменьшается с
увеличением основности использованных растворителей [257]*.
Для получения полиимидных пленок с оптимальными
свойствами необходимо, чтобы на стадии синтеза
полиамидокислотьГполучался продукт со степенью полимеризации около 100. Как
видно из рис. 7.1, прочность при растяжении полиимидной пленки на
основе пиромеллитового диангидрида и диаминодифенилоксида
сильно возрастает при увеличении среднемассовой молекулярной
массы соответствующей полиамидокислоты с 20 000 до 130 000.
При дальнейшем увеличении молекулярной массы этот показатель
меняется незначительно. В интервале значений 80 000—200 000
относительное удлинение при разрыве возрастает линейно.
Максимум прочности при растяжении достигается при более
низком значении среднечисловой молекулярной массы, чем
удлинение. Этот пример лишний раз свидетельствует о том значении,
которое имеет соблюдение условий синтеза полиамидокислоты
(чистота растворителя и исходных веществ, температура реакции,
стехиометрия) для получения полиимидов с оптимальными
эксплуатационными свойствами.
В то время как
полиамидокислоты на основе пиромеллитового бр,кгс/см2
ароматических 1500
диангидрида и
диаминов в процессе синтеза в
амидных растворителях образуют
гомогенные растворы, продукт
взаимодействия с
алифатическими диаминами выпадает из
раствора; растворение происходит
только после интенсивного пере-
Рис. 7.1. Зависимость
физико-механических свойств пленки на основе полипиро-
меллитимидодифенилоксида от
молекулярной массы исходной
полиамидокислоты:
а — изменение прочности при растяжении и
относительного удлинения при разрыве в
зависимости от средиемассовой молекулярной
массы; б —изменение прочности при
растяжении и относительного удлинения при
разрыве в зависимости от средиечисловой
молекулярной массы [259].
22 Эак. 18
бр, кгс/см*
1500
1QQQ-
500-
О 2W 4-ЯГ Мп
1'1052-1Q5Mw 0
W
2-10* 4-10* M
п.
673
100 г,сут
0
20 z,cym
Рис. 7.2.
Зависимость приведенной
вязкости раствора
полиамидокислоты
на основе пнро-
меллитового диан-
гпдрида и 4,4'-ди-
а минодифеиилмета-
на от
продолжительности
выдержки:
а— 10%-ный раствор
[257]; б — раствор
в диметилацетамиде
при 23 °С [255].
мешивания в течение многих часов. Скорость растворения этого
осадка возрастает, если в процессе синтеза к раствору
алифатического диамина добавлять диангидрид в виде порошка [262, 263]
Осаждение порошкообразной полиамидокислоты из раствора
происходит в случае взаимодействия ароматических диангидридов с
ароматическими аминами при проведении синтеза в тетрагидро-
фуране [115] .Полиамидокислоты из ароматических диангидридов
и ароматических диаминов являются сильно растворимыми
полиэлектролитами [264]. Стабильность их растворов уменьшается с
повышением температуры и содержания воды и понижением
концентрации раствора. Поскольку растворы полиамидокислот
непосредственно используются для переработки в пленки, волокна,
кабельную изоляцию и полупродукты, их стабильность имеет
большое практическое значение.
На рис. 7.2 показано изменение вязкости 10 %-ного раствора
полиамидокислоты на основе пиромеллитового диангидрида и диа-
минодифенилметана в диметилформамиде при 0 и 20 °С. В то
время как при хранении этого раствора при 0 °С его вязкость
зависит от времени линейно, при 20 °С в начальный период
наблюдается очень сильное уменьшение вязкости. На рис. 7.2, б
показано изменение вязкости растворов полиамидокислоты при хранении
в зависимости от концентрации. В результате гидролиза и
взаимодействия с растворителем происходит деструкция
полиамидокислоты. Гидролиз происходит за счет воды, выделяющейся при
частичной имидизации в процессе хранения, а также влаги,
вносимой исходными веществами и растворителем [205]. Полиамидо-
кислота в виде 10 %-ного раствора при 35°С после 200 ч
хранения имеет степень имидизации 20 %. Вследствие гидролиза
образуются соли аминов:
соон
н2о
—HNOC
СООН
НООС^ ^ ^СОО" HaN—
- дмфа
^CONH— 20 °с
В результате сольватации ионов растворителем деструктиро-
вавшиеся участки макромолекулы отходят друг от друга, что и
674
приводит к уменьшению вязкости. Одновременно со снижением
яязкости наблюдается изменение окраски раствора из-за
фотоокисления концевых аминогрупп [257].
Зависимость реакций деструкции от концентрации растворов
полиамидокислот определяется обменными процессами с
растворителем:
+ НССЩСНзЬ
HCONH-
Концентрированные растворы полиамидокислот при —15 °С в
отсутствие влаги могут храниться до 6 месяцев. Исключение
представляют растворы полиамидокислоты на основе пиромеллитового
диангидрида и м- или n-фенилендиаминов, в которых уже через
несколько суток образуется гель. Наряду с этим растворы
полимеров на основе диаминобензофенона и диангидридов тетракарбо-
новых кислот в N-метилпирролидоне отличает более высокая
стабильность: вязкость их не меняется после 300 ч хранения при
100 °С или 1000 ч при 80 °С [80].
Превращение полиамидокислот в полиимиды. Циклодегидрата-
ция полиамидокислот в полиимиды осуществляется либо при
нагревании в интервале температур 150—300 °С, либо при комнатной
температуре в присутствии дегидратирующих агентов (химическая
имидизация). Общая схема двухстадийного процесса синтеза поли-
имидов из диангидридов и диаминов приведена на с. 676 [257].
Циклизацию пленки на основе полиамидокислоты можно
провести в течение 2—3 ч путем окунания ее в раствор агента дигид-
ратацпи, в качестве которого используют ангидриды кислот,
например уксусный, пропионовый, масляный, валериановый,
бензойный ангидриды и ангидрид нафталинкарбоновой кислоты,
применяемые вместе с такими основаниями, как 4-изопропилпиридин,
N-диметилбензиламин, изохинолин, 4-бензилпиридин, N-диметил-
додециламин, триметиламин, триэтилендиамин, N-метилморфолнн,
диэтилциклогексиламин, диметилциклогексиламин, 4-бензоилпири-
дин, 2,4,6-коллидин, никотиновая кислота, пиридин, а также кете-
ны и дикетены [ 79, 130, 215, 217, 221, 268]. Применение таких
дегидратирующих агентов, как РСЦ, трифторацетангидрид, N,N'-
дицпклогексилкарбодиимнд, приводит преимущественно к
образованию пзонмпдного цикла [260, 261, 270]. Полннзонмнды
представляют собой желтые или оранжевые порошки, превращающнеся
22* 675
rtooa
хоон
соон
l,W,H2O
ноос.
соо
оос 'соон
H3N—Ar—NH3
нооо^-соон
ноос^соон
—Ar-NO2
Неактивные
компоненты
Комплекс
ноо
О
¦Ar—NH—С
ноос
XOQH
с-кнч
о
РС1
л
Полиамидокислота
-Ar—
л
OaN—Ar—
Неактивные
компоненты
-Ar-N
-HO-C. XOOH
HOOC ^C
o"o
—Ar—N
N—
п
676
при нагревании в соответствующие полиимиды:
о
о:
но—
II
_—Ar—N
N—Ar—~
С—ОН
-*-
о "С,
ДМФА
РС13
п
О
Термическую циклодегидратацию можно проводить в растворе
при 80—150 °С (однако более предпочтительным является
проведение процесса в твердой фазе при 250—300 °С), причем толщина
слоя циклизуемой полиамидокислоты в виде порошка-, волокна или
пленки не должна превышать 200 мкм [273, 274].
Порошкообразный полимер с размером частиц 20—40 мкм получают
диспергированием растворов полиамидокислот с последующей сушкой в
токе инертного газа при 250—300 °С [125].
Процесс имидизации протекает одновременно с процессом
формования изделий, который рассмотрен ниже. Скорость процесса
имидизации в растворе зависит от температуры, природы
растворителя и химического строения полиамидокислоты. Полиамидо-
кислота из пиромеллитового диангидрида и ж-фениленднамина
циклизуется в растворе диметилформамида при комнатной
температуре исключительно медленно (после выдержки в течение 12 ч
при 25°С циклизация не обнаруживается) [258]. При 80°С цик-
лодегидратация в той же системе практически завершается за
11 мин. Имидизация является реакцией первого порядка.
Константа скорости этой реакции при 80°С составляет 3,87-10~3 с1.
Имидизация в растворе сопровождается протеканием побочной
реакции образования солей полиамидокислот, которые, отщепляя воду,
превращаются в разветвленные полиамиды и далее в линейные
полиимиды:
—н2о
$77
Контроль за ходом циклодегидратации полнамидокислот
осуществляется по изменению интенсивности характеристических
полос поглощения имидных групп (при 1380 и 1780 см-1) в ИК-спек-
трах полимера [2, 123, 276, 277]. Имидизация полиамидокислоты
из пиромеллитового диангидрида и 4,4/-диаминодифенилметана в
диметилформамиде при 150°С происходит за 10 мин почти
количественно. В то же время для получения того же самого полиими-
да из полиамидокислоты, синтезированной в тетрагидрофуране,
необходимо многочасовое нагревание в твердой фазе при 180 °С
[257]. Полиамидокислота из пиромеллитового диангидрида и диа-
минодифенилоксида циклизуется почти количественно за 45 мин
при 220 °С [135]. Через 20 мин при 100 °С степень циклизации
составляет 22 %; длительное нагревание при этой температуре
сопровождается уменьшением молекулярной массы [107].
Циклизация протекает легче, если полимер содержит группы,
способные образовывать сопряженные связи. Кинетика процесса
имидизации изучена рядом авторов [200, 204, 279, 408]. При ими-
дизации в твердой фазе энергия активации непрерывно растет, а
константа скорости уменьшается. Это обусловлено
пространственными затруднениями (наиболее реакционноспособные группы цик-
лизуются в первую очередь), а также увеличением жесткости
цепи. Так, энергия активации циклодегидратации
полиамидокислоты из пиромеллитового диангидрида и диаминодифенилоксида
возрастает с 23 до 30 ккал/моль, в то время как абсолютное
значение констант скорости уменьшается больше чем на порядок.
В толстых пленках скорость реакции имидизации лимитируется
диффузией воды из реакционной зоны. В случае термической
циклодегидратации содержание изоимидных звеньев в полимере
составляет 1—3% (мол.) [272]. Концентрация этих групп может быть
определена ИК-спектроскопическим методом по соотношению ин-
тенсивностей /io.95//9,85-
Хотя при 300 °С процесс имидизации протекает практически
количественно, тем не менее кратковременная
высокотемпературная термообработка приводит к существенному изменению
физико-механических характеристик. Это объясняется изменением сте
пени кристалличности полимера и структурированием полиимидг
[135, 275]. Предполагается, что при высоких температурах
—СО—N<^ в цикле постоянно разрываются и рекомбинируют. Пр*
этом возможна не только внутримолекулярная рекомбинация (
образованием исходного гетероцикла, но и межмолекулярная;
678
со
>< >'-
со
X
со-
\
\
N—R'—
или
со
хсо
1
N—R'—
N-R'—
X
со
\
\
N-R—
R
/R\co-
При температуре выше 300 °С методом ЭПР в иолиимидах
обнаруживается высокая концентрация свободных радикалов
[280], одновременно в ИК-спектре уменьшается интенсивность
полосы при 1780 см (С=О имидного цикла) [275]. Сшиванне
макромолекул вызывает возрастание модуля упругости и повышение
температуры размягчения при высокотемпературной
термообработке полиимидов *.
Поликонденсация в спиртовом растворе диэфиров тетракарбо-
новых кислот с диаминами при комнатной температуре,
приводящая к образованию солей, также протекает в две стадии [111,
218—220, 249, 282]. При проведении реакции в смеси
растворителей, содержащей амидный растворитель, например N-метилпирро-
лидон, циклообразование происходит через промежуточную
стадию получения полиэфирамида:
О О
* Изменение свойств полиимидов при высокотемпературной обработке
может быть обусловлено повышением их молекулярной массы в результате
твердофазной поликонденсации по концевым амино- и ангидридным группам. —Прим.
ред.
679
Преимуществом такого метода является то, что-в отличие от
полиамидокислот промежуточные продукты—соли аминов
гидролитически гораздо более стойки. Форполимеры используют главным
образом в качестве связующих для армированных пластиков,
обеспечивающих получение низковязких растворов с высоким
содержанием сухого остатка [278,290]. При циклизации выделяется
азеотропная, сравнительно легко удаляющаяся смесь
растворителя, спирта и воды. На основе аминодикарбоновых кислот, в
которых одна из карбоксильных групп в о-положении этерифицирова-
на, фирмой «Rhone-Poulenc» под маркой ТР 380 выпускаются
самоконденсирующиеся клеевые составы [281]:
II
с.н -A-v""
—П
Исходные продукты для них получают на основе о-ксилола и
л-нитробензоилхлорида:
Гидролитическую стойкость полиамидокислот повышают путем
блокирования свободных окарбоксильных групп вторичными
аминами [172] :
О О
(С2Н5)!ЫОС-х/\ ХОСТ
О)
iO ХЛ \ NT 'ГЛСЛС*^ N/* 4\PONT//^ 14 \
680
(C2H5JNCOX ^ ^COOH
socl2
Поликонденсация таких соединений с ароматическими
диаминами приводит к образованию полиамидокислот с молекулярной
массой до 50 000, СООН-группы которых блокированы
аминогруппами:
—H CI
n
Такие полиамидокислоты отличаются более высокой
гидролитической стойкостью и низкой скоростью циклизации при
температурах ниже 200 °С. Молекулярная масса форполимера в виде
0,5 %-ного раствора в диметилацетамиде, содержащего 1 % воды,
не изменяется в течение 500 ч. Имидизация проходит
количественно при 200 °С за 10 ч.
Полиэфироамиды получены поликонденсацией диэфирдихлор-
ангидридов тетракарбоновых кислот с ароматическими диаминами
[283]. Полиамидокислоты, блокированные аминогруппами,
синтезированы в растворе N-метилпирролидона из Ы,№-бис(этоксикар-
бонил)пиромеллитимида и диаминов [137]; термическая
циклизация протекает с отщеплением этилуретана;
О О
681
Циклизация полицианамидов. При взаимодействии дихлоран-
гидридов ароматических дициандикарбоновых кислот с
ароматическими диаминами в амидном растворителе, таком, как N-метил-
пирролидон, при 0°С образуются полицианамиды, которые при
нагревании превращаются в полииминоимиды [156, 233, 331]:
/\ /-СОС1
LCl
n
n
В отличие от полиамидокислот в этом случае получаются
стабильные промежуточные продукты синтеза. Особенностью
процесса является то, что при циклизации не выделяются какие-либо
летучие продукты.
Двухстадийная поликонденсация в расплаве тетракарбоновых
кислот или их диэфиров с диаминами. Способ применим лишь к
полиимидам, которые размягчаются при температуре ниже
температуры циклизации. Плавкие полиимиды получают на основе
-циромеллитового диангидрида и линейных алифатических
диаминов с числом атомов углерода не менее восьми или разветвленных
диаминов с числом атомов углерода больше семи (табл. 7.2,
№ 34—42).
Такие же полимеры получены из циклоалифатических
тетракарбоновых кислот и алифатических или ароматических диаминов
(№ 6—11, 19, 21). Синтез этих плавких полиимидов описан в
[249, 252, 284, 285].
При нагревании тетракарбоновых кислот или их диэфиров с
диаминами при ПО—140°С образуются промежуточные
продукты— соли, которые на второй стадии при нагревании до 250—
300°С отщепляют воду и превращаются в полиимид [53, 96, 251]:
НООС
.соон
П0-140°С
—FbN "OOC
СОО" H3N(CH2)X—
->¦
250-300 °С
-СН3ОН; — Н2О
п
682
п
Для получения чисто ароматических полиимидов данный
способ непригоден, так как уже при малых степенях превращения
образуются неплавкие продукты, что препятствует синтезу
высокомолекулярных полигетероариленов.
Одностадийный метод синтеза поликонденсацией при высоких
температурах ароматических тетракарбоновых кислот с диаминами.
При поликонденсации тетракарбоновых кислот или их диангидри-
дов с диаминами в среде высококипящих органических
растворителей, таких, как нитробензол, бензонитрил, тетраметиленсульфон
или гликоль, в инертной атмосфере при 195—210°С полиимид
образуется в одну стадию [23, 108, 193, 286, 287]. Молекулярная
масса полимера в ходе синтеза регулируется добавкой
растворителей, например N-метилпирролидона, циклотетраметиленсульфона
или следов воды в реакционную массу. Такой раствор полимера
может быть использован непосредственно для склеивания,
получения волокон и покрытий. Аналогично (в одну стадию) получают
полиимпды при взаимодействии дитиоангидрида пиромеллитовой
кислоты с диаминами в диметилацетамиде при 140—150 °С с
отщеплением сероводорода [181].
Взаимодействие ароматических диимидов с дигалогенидами.
Поликонденсация ароматических диимидов или соответствующих
солей щелочных металлов с алифатическими дигалогенидами или
ароматическими бисхлорметильными соединениями в
диметилацетамиде при 70—140 °С ускоряется при использовании в качестве
катализаторов таких оснований, как третичные амины и КгСОз
[288, 289] :
ttHN
—ch2ci
-HCI
о
о
—N:
\
-сн,
сн>-
1 I
683
С ароматическими дигалогенидами реакция не протекает [288,
289]. Исходя из бисгалогенметилдисилоксанов, получены полиими-
ды, содержащие в цепи силоксановые группы.
Реакции ароматических диангидридов с диизоцианатами или
диизотиоцианатами. Об образовании имидов из ангидридов карбо-
новых кислот и изоцианатов известно еще со второй половины
прошлого столетия [295, 296]. При взаимодействии ортофталевой
кислоты с фенилизоцианатом в присутствии пиридина или
хлорной кислоты при 180°С получен N-фенилфталимид [298, 299]. Эта
реакция использована для синтеза полиимидов [294, 300]:
—Jl
Ее проводят в пиридиновом растворе или расплаве.
Предполагается, что процесс протекает через промежуточную стадию
образования полимеров с семичленным гетероциклом, который
превращается в имидный с отщеплением СО2 [294, 300]:
О
-со2
NR—
При проведении этого синтеза в пиридине добавление воды
вплоть до эквимольного соотношения вода — диизоцианат
ускоряет реакцию. При дальнейшем увеличении содержания воды в
системе скорость процесса понижается. При мольном соотношении
вода: изоцианат = 0,056 скорость реакции повышается в 2 раза
[150]. Роль воды в этом процессе заключается в промежуточном
образовании замещенной мочевины, которая собственно и
реагирует с диангидридами. Этот способ синтеза защищен рядом
патентов [128, 291—294, 301].
Взаимодействие оснований Шиффа с диангидридами. Шиффовы
основания, полученные взаимодействием диаминов с кетонами или
альдегидами, при поликонденсации с ароматическими диангидри-
684
дами в апротонных растворителях образуют полиимиды [330]
u О
R
/
4 -V*
п
Полибисмалеимиды. Впервые сшитые полиимиды были
получены радикальной полимеризацией бисмалеимидов в 1948 г. [314].
Бисмалеимиды получают взаимодействием малеииового ангидрида
с ароматическими или алифатическими диаминами:
О
NH2RNH2
i
НС
-CONHRNHCOCH
НС—СООН НООС—СН -Н2°
О
N—R—N
1
Полимеризация бисмалеимидов с образованием сшитых поли-
имидов осуществляется в расплаве или в растворителе при
температуре выше 200°С под влиянием инициаторов радикального
типа [315]:
О О - о О
-R-K
I
\j-R- К
О
п
При взаимодействии бисмалеимидов с диаминами образуются
также линейные полималеимиды с низкой термостойкостью и проч-
ностью [303, 304] :
—R"-NH2
NH— R' — NH—
Сшитые полиимиды с оптимальной термостойкостью
получаются при сочетании обоих процессов путем взаимодействия
различных количеств диаминов с бисмалеимидами или полимеризацией
бисмалеимидов в присутствии линейных полимеров [58, 59, 92,
100, 101, 103, 168, 235, 305, 311, 316].
Преимуществом этого метода по сравнению с синтезом поли-
имидов из диангидридов и диаминов является то, что
полимеризация с образованием полимеров сшитой структуры не
сопровождается выделением низкомолекулярных летучих продуктов. Это дает
возможность перерабатывать такие полимеры прессованием или
литьевым прессованием с образованием непористых изделий. Фор-
полимеры этого типа выпускаются в промышленном масштабе с
1969 г. фирмой «Rhone-Poulenc» под торговым названием «Кинел»
в качестве лаковых смол, пресс- и пресс-литьевых композиций
[164, 316, 317]. Полимеры с повышенной термостойкостью
получают в том случае, когда исходные бисмалеимиды содержат
предварительно синтезированные ароматические имидные группы [109,
306,312]:
N—
—R—N:
N—
О
—R—N
Эти низкомолекулярные продукты с малеимидными концевыми
группами полимеризуются в процессе переработки с образованием
высокотермостойких сшитых полиимидов. Если необходимо
повысить жизнеспособность композиции, процесс отверждения можно
ингибировать добавлением гидрохинона [236]. Наличие
заместителей при двойной связи бисмалеимидов снижает их реакционную
686
способность. Ряд соединений, например N.N'-декаметиленбисди-
хлормалеимид, при облучении в растворе в присутствии бензофе-
нона подвергается фотополимеризации [213, 318] (табл. 7.2,
№ 6—10):
П [j n—(сна)ю—
CL
ci ci
-(CH2)r0—>
J "l
Высокомолекулярные полималеимидоэфиры могут быть
получены при взаимодействии бисхлормалеимидов, содержащих в
молекуле алифатические или ароматические группы между имид-
мымм циклами, с бисфенолами в таких апротонных растворителях,
как диметилсульфоксид или диметилформамид, при комнатной
температуре в присутствии неорганических оснований, например
СаО или К2СО3, или каталитических количеств триэтиламина
[419,420]:
Cl
о
о
CI Ц
о о
+ «но-
О
СНз
СНз
он
К2СО3; ДМФА
О
Уу-Q^Q
п
В противоположность сшитым полиимидам линейные полимале-
нмидэфиры (табл. 7.2, № 39) растворяются в хлорированных
углеводородах и полярных растворителях. Температура их
стеклования 165—220 °С, они стабильны на воздухе до 300 °С. По реакции
Дильса — Альдера бисмалеимиды реагируют с диенами при
высоких температурах [322—326] или под действием света и
сенсибилизаторов [186, 207] с образованием полициклических полннми-
дов.
687
Высокомолекулярный линейный полиимид с температурой
стеклования 360 °С образуется при взаимодействии 2,5-диметил-3,4-ди-
фенилбутадиена с М^/-4,4'-C,3/-диметилдифенил)бисмалеимидом
[320] :
П + п СН.—СН=С—С=СН—СН,
N—
п
При фотохимическом взаимодействии бензола с бисмалеимидами
образуется полиимид, ограниченно растворимый вследствие
протекания реакций сшивания [186, 207]:
+ n N-R-N
(с6н5Jсо
О
N-R-
Полимеризация олигоимидов с концевыми норборненовыми
группами. Предложен способ получения олигоимидов с концевыми
норборненовыми группами, которые при температуре выше 260 °С
образуют сшитые полимеры [68, 168, 196, 307—310, 313].
Полимеры этого типа выпускаются в промышленном масштабе начиная с
1970 г. фирмой «США GEIGY» (США). Исходными
соединениями для их синтеза служат малеиновын ангидрид, циклопентадиен,
диаминодифенилметан и диангидрид бензофенонтетракарбоновой
кислоты. Сначала конденсацией в диметилформамиде получают
олигоамидокислоту, которая превращается в порошкообразный
олигоимид распылительной сушкой. При нагревании в сушилках с
циркуляцией воздуха при 200 X получают олигоимид с
концевыми норборненовыми группами с молекулярной массой 1300.
Прессование этого продукта при температуре выше 260°С
сопровождается отверждением полимера. При этом протекает процесс,
обратный реакции Дильса — Альдера, с отщеплением циклопента-
диена и образованием олигоимнда. Форполимер в результате со-
полимеризации с циклопентадиеном образует неплавкий сшитый
полимер:
688
о
О 4-H2N-
О
о
соон
о
о
о
о
О | О
о о о
<c^ct5rc-NH-
|200°С
-cH24OhCH2
о
о о
290 °С Низкомолекулярный полиимир
N
N
XI
О
Достоинством этого метода синтеза является отсутствие
выделения летучих продуктов, что дает возможность получать
пустотелые изделия по обычной для пенопластов технологии.
Полиприсоединение дивинильных соединений к диимидам. Этот
процесс протекает в присутствии оснований [422] :
О
nHN
N—CH2CH2SO2CH2CH2—
—п
Полиприсоединение проводят в таких растворителях, как
пиридин, диметилсульфоксид, диметилформамид, в среде азота при
20—110°С с использованием в качестве катализатора трет-бутя-
лата Na. Ароматические амины, такие, как N-фенил-р-нафтиламин,
ингибируют радикальную полимеризацию дивинилсульфона.
Процесс начинается с миграции водорода и может быть представлен
следующей схемой:
О О О О
нв
DI >н
нв
1-в-
о
HN i О ' xn-ch2ch2so2ch=ch2 + в"
690
Полимеризация замещенных лактамов с раскрытием цикла.
Линейные алифатические полиимиды образуются при
полимеризации ?-карбоксизамещенных лактамов, например ?-карбоксиметил-
капролактама, ?-карбоксиметилвалеролактама, ?-карбоксивалеро-
лактама, ?-карбоксикапролактама, путем их нагревания выше
температуры плавления A50—170°С). Синтез таких полимеров
описан в [98, 169, 326, 327]:
/СН2— СН2ч -
/ XNH 200 °с Г уСН2—СОч
н,с: I —7г> —не; ;n(ch2K-
L ХСН2—СОХ
П
СН2СООН
?-Карбоксиметил-
капролактам
По ли-2,6-д ноксо- \ ,4-пипе-
рицинилтриметилен
Карбоксиметилкапролактам получают следующим образом [328,
329] : _
* NH / \NH Г/ \NH / \NH"
со \ ^-со I \ ^со \
НООС—СН—COOR
В процессе полимеризации ?-карбоксилактамов образуется би-
циклическая система, которая через димерный промежуточный
комплекс и димер с иминолактонной структурой
перегруппировывается в полиимид [169]:
H2N —
691
Полимераналогичные превращения замещенных полиимидов.
Полиимиды, полученные на основе диаминов, содержащих
заместители в о-положенни к аминогруппе, при температуре выше
350 °С претерпевают химические превращения, связанные с
реакциями этих заместителей с имидным циклом [120, 197].
Полиимиды с алкокси- и гидроксигруппами при температуре около 350°С
выделяют СО2 и превращаются в полибензоксазол (см. 7.3):
t
-со2
Полиимиды с о-анилиновыми группами при 340—430 °С в
вакууме почти количественно превращаются в N-замещенные поли-
бензимидазолы (см. 7.2):
NH—С6Н5
О
370 °С
>-
—со2
С6Н5
. ^г <s~Z>-
Поли-о-аминоимиды в аналогичных условиях превращаются в
полиимидазопирролоны (пирроны) :
t
—н2о
N
7.1.1.2. Физико-химические свойства алифатических
и ароматических полиимидов
Полиимиды представляют собой частично-кристаллические
полимеры от желтого до темно-красного цвета с высокими
температурой размягчения и термостойкостью. Физико-механические
свойства, термостойкость и эксплуатационные свойства этих
продуктов определяются строением макромолекул и внутри- и
межмолекулярным взаимодействием сегментов цепи, наличием (или
отсутствием) связей, придающих гибкость макромолекулам, и долей
алифатических атомов углерода по отношению к ароматическим
и циклоалифатическим элементам цепи.
692
Полиимиды, содержащие в макромолекулах алициклические
или алифатические фрагменты, а также N-замещенные линейные
полималеимпды [146], в большинстве случаев плавятся и
размягчаются при температуре ниже 200СС (табл. 7.2, № 1—3, 5—11,
17—19, 25—28). Полиимиды, полученные из диангидридов
ароматических тетракарбоновых кислот и алифатических диаминов
(№ 30—55), размягчаются в интервале температур 200—300°С;
плавкими являются полимеры на основе пиромеллитового диан-
гидрнда и алифатических диаминов с числом атомов углерода
больше семи. Плотность составляет 1,30—1,36 г/см3.
Классификация ароматических полиимидов предложена в
работах [3, 12]. В полимерах № 56—82 (табл. 7.2) имидные циклы
связаны только с ароматическими циклами (группа I по
указанной классификации). Эти продукты отличаются исключительно
высокой жесткостью, они нерастворимы и разлагаются при
температуре выше 500°С без размягчения и плавления. Полимеры
№ 83—103, полученные из ароматических диангидридов,
содержащих «шарнирные» атомы и группы атомов, такие, как —О—,
—S—, —SO2, —СО—, —СН2—, —С(СН3)г—, относятся к группе II.
Эти полимеры размягчаются при температурах около 400°С.
Ароматические полиимиды № 104—154 содержат «шарнирные» атомы
и группы в диамине. Они относятся к группе III и отличаются от
полимеров групп I и II более высокой эластичностью,
обусловленной возможностью вращения вокруг связей в остатке амина.
Температура их размягчения лежит выше 350°С. Полимеры № 155—
236 являются ароматическими полиимидами, содержащими
шарнирные группы как в остатке кислотного, так и аминного
компонентов (группа IV). Это высокоэластичные продукты с
температурой размягчения 200—350°С. Полиимиды группы IV плавятся,
формуются, как термопласты, и растворяются в полярных
растворителях. Ароматические полиимиды — жесткие твердые полимеры,
прочность которых A000—2000 кгс/см2) мало зависит от
строения; модуль упругости зависит от числа шарнирных атомов и
уменьшается последовательно от полимеров группы I к
полимерам группы IV, в то время как удлинение изменяется в обратной
зависимости. Окраска полиимидов определяется сопряжением
имидного цикла с ароматическими ядрами и зависит от наличия
атомов и групп атомов между ними [332]. Полипиромеллитимиды
диаминодифенилсульфона имеют желтоватую окраску, аминоди-
фенилоксида — желтую, диаминодифенилсульфида —
темно-красную. Плотность ароматических полиимидов колеблется от 1,32 до
1,50 г/см3.
Температурные переходы и физико-механические свойства.
У большинства ароматических полиимидов деструкция начинается
до достижения температуры фазовых переходов. Полиимиды с
повышенной подвижностью макромолекул, обусловленной наличием
«шарнирных» групп или пониженной плотностью упаковки, при
наличии в боковой цепи объемистых групп размягчаются.
Сравнение теплофизических и физико-механических свойств полиимидов
693
Таблица 7.3. Физико-механические свойства ароматических полиимидов [1, 3—5]
Гру
Элементарное звено
f i
-
Прочность
при растяжении,
кгс/см2
-195°С
20 °С
400 °С
2 000
1 000
1500
2 300
Относительное
удлинение
при разрыве, %
- 195 °С
20 °С
400 °С
Модуль
упругости,
КГС/СМ2
20 °С
15
120 000
75 000
70 000
о
II
—N;
О
О
о
— N
1500
1400
1 500
5
9
4
Элементарное звено
0
III
-©--о-
о
О
о о
Продолжение табл. 7.3
Прочность
прн растяжении,
кгс/см2
-195 °С
20 °С
400 °С
Относительное
удлинение
при разрыве, %
-195°С
20е С
400 °С
Модуль
упругости,
кгс/см2
20 °С
3 500
3 000
2 020
2 000
400
400
40
30
100
130
120
140
35 000
32 000
о
IV
о
С
О
о
о
О
2 500
2 ООП
100
30 000
1 500
50
27 000
1300
90
24 000
о
предполагает, однако, примерно одинаковые условия синтеза и
переработки. На понижение температуры размягчения и
ухудшение физико-механических свойств влияют степень циклизации
полимера и условия его имидизации, которые обусловливают
наличие пустот в полиимидах. В ароматических полиимидах группы I,
содержащих только имидные и ароматические циклы, например
о
—N'
циклические группы копланарны, следствием чего являются
высокая плотность упаковки полимеров и жесткость макромолекул.
Полимеры этого типа нерастворимы, очень хрупки и не имеют
фазовых переходов ниже температуры разложения. Модуль
упругости при комнатной температуре составляет около 105 кгс/см2,
при 400 °С— 104—105 кгс/см2 (табл. 7.3).
Высокие значения модуля упругости обусловлены сильными
внутри- и межмолекулярными взаимодействиями. При нагревании
при 400 °С в течение 15 мин модуль упругости возрастает на
10—40%. Несмотря на высокую жесткость макромолекул,
полиимиды группы I под напряжением выше 800 кгс/см2 проявляют
известную текучесть.
Полиимиды группы II, содержащие заместители («шарниры»)
между ароматическими ядрами, такие, как О, S, СО, SO2, CH2,
—N;
до-®-
О
С(СН3Ь по свойствам близки к полимерам группы I. Высокая
жесткость имидных циклов мешает проявлению эластичности за
счет атомов и групп, находящихся в диангидридном компоненте.
Полимеры этой группы, за небольшими исключениями, не
размягчаются (табл. 7.3, № 83—103). Прочность при растяжении и
модуль упругости полимеров группы II несколько ниже, чем у поли-
имидов группы I. Полиимиды группы II размягчаются в том
случае, когда плотность их упаковки понижается за счет наличия
между ароматическими ядрами диангидрида таких групп, как
фталидные (табл. 7.3, № 96—100). Некоторая эластичность
проявляется и при наличии в аминном компоненте ж-фениленовых
фрагментов (табл. 7.3, № 84, 89, 102).
698
Весьма сильно отличаются по свойствам полиимиды группы III,
в которых в остатке аминного компонента имеются «шарнирные»
атомы (табл. 7.3, № 104—154). например
о
—N
обеспечивающие внутримолекулярное вращение в остатках
диамина, приводящее к ослаблению межмолекулярного
взаимодействия и уменьшению плотности упаковки. Полимеры этой группы в
широком температурном интервале обладают повышенными
эластичностью, прочностью и вязкостью и имеют ярко выраженную
область размягчения. К этой группе полимеров относятся
полиимиды, производимые в промышленном масштабе, — Аримид ПМ и
ПАК-1 (СССР) и Каптон («Du Pont», США). Температура
размягчения продуктов, полученных на основе пиромеллитового диан-
гидрида и различных диаминов, изменяется в следующей
последовательности:
—S—.
699
СН3
г—о—r?V
IQT-TQ
^он
При комнатной температуре прочность при растяжении
составляет около 2000 кг/см2, относительное удлинение при разрыве
100—150 %, модуль упругости 30000—35000 кгс/см2 (см. табл. 7.3).
Полиимиды группы III имеют высокую эластичность и при низких
температурах: при —195 °С их относительное удлинение при
разрыве составляет 30—60 %. Эластичность увеличивается при
возрастании числа «шарнирных» атомов в остатке диамина. С
повышением температуры до 400 °С модуль упругости уменьшается с
30 000—35 000 до 100—500 кгс/см2. Длительная выдержка при
400 °С (больше 20 мин) приводит к увеличению модуля упругости
до 2000—5000 кгс/см2 за счет сшивания полимеров, легкость
протекания которого связана с их повышенной эластичностью. После
термической обработки при 400 °С полиимиды перестают
размягчаться. Исследования механической релаксации полиимидов на
основе пиромеллитового диангидрида и диаминодифенилового
эфира в интервале температур 200—400 °С показало наличие трех
вторичных релаксационных переходов с максимумами потерь при
121, 204 и 357 °С [349, 350]. Основной релаксационный процесс
начинается при 287 °С.
Повышенной эластичностью и узким интервалом температур
размягчения характеризуются полиимиды группы IV (табл. 7.3,
№ 155—236) на основе диангидридов и диаминов, содержащих
«шарнирные» группы, например:
—О—(( У)—
Выше температуры размягчения возможна необратимая,
остаточная деформация. Некоторые полимеры группы IV плавятся. Тем-
700
пература размягчения зависит от числа и строения «шарнирных»
атомов и групп, обеспечивающих вращение макромолекулярных
сегментов. Замена л-фенилсновых фрагментов в цепи на л-фениле-
новые сопровождается снижением температуры размягчения По
лнимиды на основе диангидрида дифенилокситетракарбоновой
кислоты в зависимости от строения остатка аминного компонента
по уменьшению температуры размягчения располагаются в
следующий ряд: "
> -€к /©- > -<0\ /О- >
(CFa)a—<^S^_
701
нилоксида и различных диангидридов в зависимости от строения
использованного ангидрида располагаются в ряд, в котором
температура размягчения снижается с 345 до 175 °С:
> к )Г<СРЛ-0^>
Четко выраженную температуру плавления, лежащую ниже
температуры разложения полимеров, имеют только полиимиды
группы IV, в которых не менее двух атомов кислорода входит в
состав диамина и один эфирный мостик — в состав ангидрида
(табл. 7.3, № 172, 229, 231, 235, 236) или кислородный мостик —
в диамин и длинная перфторметиленовая цепочка — в диангидрид
(табл. 7.3, № 220, 221). Подобные полиимиды можно
перерабатывать компрессионным или литьевым прессованием. Прочность
при растяжении при комнатной температуре составляет 1000—
2000 кгс/см2, относительное удлинение при разрыве 15—100% и
модуль упругости 20 000—30000 кгс/см2. Как и в случае полиими-
дов группы III, сшивание полимеров, протекающее выше 350°С
оказывает лишь незначительное влияние на их свойства. С
увеличением продолжительности термообработки эластичность полиими-
дов группы IV снижается.
Кристалличность. Полиимид из пиромеллитового диангидридг
и n-фенилендиамина кристаллизуется непосредственно в ходе ими
дизации. Полиимиды групп III и IV, содержащие в цепи макро
молекулы «шарнирные» атомы, характеризуются незначительной
702
степенью упорядоченности, которая возрастает при термообработке
[2, 112]. Скорость кристаллизации повышается при воздействии
паров растворителей, таких, как диметилформамнд, при высоких
температурах. Из данных рентгеноструктурного анализа и ИК-
спектроскопии [202, 333] следует, что при растяжении пленки и
вытяжке волокна степень кристалличности увеличивается,
способствуя возрастанию прочности при растяжении и модуля
упругости.
Растворимость и свойства растворов. Вследствие сильного
межмолекулярного взаимодействия большинство ароматических
полиимидов не растворяется в органических растворителях и
разбавленных кислотах. Они растворяются с разложением только в
дымящей азотной кислоте, а некоторые из них и в концентрированной
серной кислоте [2]. Для измерения вязкости используют
хлориды мышьяка или сурьмы или их смеси. При этом происходит
лишь незначительная деструкция полимеров [335, 341].
Межмолекулярное взаимодействие можно существенно ослабить, вводя
в боковую цепь полимеров в составе ангидрида (табл. 7.3, № 96—
98, 198—205) и/или амина (табл. 7.3, № 125—130, 146, 147, 162—
167, 179, 187—191) объемистые группировки, такие, как фталид-
ные или флуореновые. Плотность упаковки понижается, и
полимер приобретает растворимость в органических растворителях,
таких, как диметилформамид, диметилацетамид и тетрахлорэтан
[15, 62, 334]. Растворимость полиимидов с объемистыми
заместителями в аминном остатке увеличивается в следующем ряду диан-
гидридов, использованных при их синтезе [334].
—SO
—О—|
Растворимость полиимидов можно также увеличить введением
в диамин в о-положение к аминогруппе заместителей с гидрокси-
и алкоксигруппами:
ю < юла < о
С(СНзJ и т. д.
Такие полиимиды растворяются в диметилформамиде, диметил-
ацетамиде, N-метилпирролидоне, тетрагидрофуране и циклотетра-
метиленсульфоне [129, 179, 203]. В том случае, когда Х =
= C6H5NH, полимеры растворимы в диметилформамиде, диметил-
сульфоксиде, диметилацетамиде и нитробензоле [61, 187, 247].
Полиимиды из ароматических диангидридов и алифатических
диаминов (табл. 7.3, № 30—55) растворяются при комнатной
температуре в диметилформамиде, jw-крезоле и тетрахлорэтане [51].
703
Полимеры на основе диангидрида циклопентаитетракарбоновон
кислоты и ароматических диаминов растворимы в воде [ПО].
Поскольку ароматические полиимиды ограниченно растворяются в
органических растворителях, исследования о фракционировании
и молекулярных массах, результаты которых представлены
(табл. 7.4), весьма немногочисленны. Свойства растворов
полимеров, полученных из пиромеллитового диангидрида и 3,3'-бисD-
аминофсиил)фталида, изучены методами [23, 338, 339]
светорассеяния, седиментации, вискозиметрии и электронной микроскопии
(табл. 7.2, № 125). Инкремент показателя преломления dn/dC
в растворе в диметилформамиде при 25 °С составляет 0,231 г на
100 мл. Фракционирование в системе нитробензол—дихлорэтан
возможно при температуре 50—60 °С. Полиимид, полученный
одностадийной и двухстадийной полициклоконденсацией, имеет
молекулярную массу 125 000 и 20 000 соответственно.
Молекулярно-массовое распределение и гидродинамические
свойства полипиромеллитимида 2,2-бисC-амино-4-гидроксифенил)-
пропана исследованы [203] методом экстракции при 20 °С между
двумя несмешивающимися жидкостями с использованием диме-
тилформамида в качестве растворителя и смеси хлороформ —
гептан в качестве осадителя. Зависимость константы седиментации от
молекулярной массы выражается уравнением
So = 9,3 • 10~2iW0'57
Полидисперсность полимера, полученного в одну и две стадии,
Mw/Mn равна 1,9 и 1,3 соответственно. Для большинства
полимеров возможно определение молекулярной массы только для поли-
амидокислот, поскольку в типичных для полиимидов
растворителях— азотной и серной кислотах — полимер разлагается.
Циклизация полиамидокислот сопровождается лишь незначительным
изменением молекулярной массы [337].
При изучении свойств растворов полиамидокислоты на основе
пиромеллитового диангидрида и диаминодифенилового эфира в
диметилацетамиде методом светорассеяния наблюдается
подавление полиэлектролитного эффекта при добавлении бромида лития
[336]. Инкремент показателя преломления составляет 0,210 г на
100 мл. При осмометрических измерениях равновесие
устанавливается через 7—10 ч. В зависимости от метода синтеза Мп
изменяется от 13 000 до 55000, a Mw — от 9 900 до 226 000.
Характеристическая вязкость полимера приведена в табл. 7.4. Значения
экспоненты, равные 0,78—0,8, характерны для сольватированного
проницаемого клубка. Уравнение, связывающее константу
седиментации и молекулярную массу, имеет следующий вид:
So = 2,70 • 10~2М0>39
Размеры невозмущенного клубка макромолекулы
полиамидокислоты (го/М)Чя = 0,848; стерический фактор 1,24. Исследование
растворов полиамидокислоты на основе пиромеллитового
диангидрида и бензидина в диметилацетамиде при 25 °С методами свето-
704
рассеяния и вискозиметрии показало [191, 192], что полиэлектро-
лптнып эффект полностью подавляется при добавлении 0,1 м/л
LiBr. Инкремент показателя преломления в этой системе dn/dC
составляет 0,265 мл/г. Диаграмма Зимма свидетельствует о
линейной угловой и концентрационной зависимости величины КсДО.
Значение второго внриального коэффициента для МШ=2'1О-4 —
— Ы0~6 колеблется от 36-10~4 до 27,6-10~4 моль-мл-г~2.
Гидродинамические размеры клубка составляют 574 A, плотность
клубка 0,0041 г/мл. Относительно большие размеры невозмущенного
клубка {yh2iM= l,054A) свидетельствуют о том, что полиамидо-
кислота в растворе существует в виде весьма протекаемых
клубков. Это обусловливается высокой жесткостью макромолекул по-
лиамидокислоты и сольватацией их с растворителем. Инкремент
показателя преломления для растворов полиамидокислоты,
полученной из пиромеллитового диангидрида и бензидина, в чистом
диметилацетамиде при 25 °С составляет dn/dC = 0,375. Вследствие
диссоциации карбоксильных групп в системах, не содержащих LiBr,
наблюдается более заметное расширение клубка, чем в растворах
с LiBr. Полиамидокислота с Мш = 75 000 в растворе диметилацет-
амида с LiBr имеет [т]] = 2,24 дл/г, тогда как в чистом
диметилацетамиде [т]]=2,65 дл/г. В системах, содержащих LiBr, с
увеличением разбавления наблюдается возрастание оптической
анизотропии. Это приводит к увеличению соотношения осей в
эллипсоиде. При добавлении триэтиламина к разбавленным растворам
полиамидокислоты происходит сильное возрастание вязкости.
Расширение клубка указывает на электростатическое отталкивание
одноименно заряженных ионов, которые образуются в результате
переноса протона:
—СООН + N(C2H5K ч=^ —СОО" + Ш(С2Н5K
Стойкость к термической и термоокислительной деструкции.
Стойкость полиимидов к термической и термоокислительной
деструкции определяются строением звена полиимидов (числом и
типом простых связей в цепи), строением концевых групп,
концентрацией нециклизованных звеньев, степенью сшивания, молекуляр-
но-массовым распределением, степенью кристалличности и
морфологией. Выше были приведены температуры начала разложения
полиимидов в инертной среде и на воздухе.
Полиостью ароматические полиимиды стабильны в инертной
-реде до 480 °С. «Шарнирные» группы, находящиеся между
ароматическими ядрами, вызывают снижение термостойкости.
Разложение на воздухе обычно начинается на 10—50° ниже, чем в
инертной среде, и проходит с гораздо более высокой скоростью. Из
цанных ДТА следует, что при деструкции на воздухе полипиро-
меллитимида и диаминодифенилоксида (табл. 7.2, № 104, Каптон,
А.римид ПМ) количественное разложение при 680 °С происходит
: образованием летучих продуктов, а в среде сухого гелия в
интерзале температур 500—700 °С потеря массы составляет только 40 °/о
23 Зак. 18 706
Таблица 7.4. Вискозиметрическая молекулярная масса полиимидов и полиамидокислот
In] = /с - ма
Элементарное звено
С
1
) О
II
о с
)
СНз
1
Растворитель
Диметилформамид
Диметилформамид
Диметилацетамид
Диметилсульфоксид
Г, °С
25
20
20
20
/МО4
32,83
1,68
2,85
13,1
а
0,516
0,83
0,78
1,11
Литературный
источник
[23, 338,
339]
-
[203]
[203]
[203]
о
4
<rCH>
у^СН2
О
О
ж-Крезод
Диметилацетамид
Диметилацетамид-
LiBr
Диметилацетамид -
LiBr
25
25
25
25
4,30
1,85
2,38
3,40
0,647
0,80
0,78
0,78
[169, 210]
[336, 337]
[340]
[1911
300 500 700 t,cC
Рис. 7.3. Кривые термической деструкции полипиро-
меллитимиддифенилоксида на воздухе A) ив
среде гелия B) [278].
(рис. 7.3). Алифатическо-ароматические
полиимиды разлагаются на воздухе ив
инертной среде между 200 и 300 °С (табл. 7.2,
№ 30—55). Полиимиды на основе алици-
клических диангидридов стабильны до
420 °С (табл. 7.2, № 16—24). Среди
ароматических полиимидов наиболее высокой
термостойкостью характеризуются такие
полимеры, которые состоят только из ароматических и имидных
циклов (полиимиды группы I, № 56—82). Так, полипиромеллитимид
д-фенилендиамина в инертной среде устойчив до 500 °С, а на
воздухе— до 450 °С. Внутри этой группы термостойкость снижается
в следующем ряду:
/^ Q ,
>
о-
-п-
О
Стойкость полипиромеллитимида бензидина к
термоокислительной деструкции увеличивается при введении в ароматическое
ядро одного или двух атомов хлора (в аминном компоненте), а
использование октафторбензидина приводит к уменьшению
стабильности. Полипиромеллитимид разлагается в инертной среде при
температуре ниже 400 °С при наличии в аминном остатке гидро-
кси- или метоксигрупп [23]. д-Замещенные структуры
характеризуются более высокой устойчивостью, чем ж-структуры (№ 56,58).
Стабилизация полиимидов достигается за счет введения в
макромолекулы атомов галогенов в составе ангидридного компонента
[64, 105, 245[. При этом повышается как термическая, так и
окислительная стойкость полиимидов. Введение в полиимиды в амин-
ный и/или в ангидридный компонент «шарнирных» атомов и
групп сопровождается понижением термостойкости (полимеры
групп II—IV, № 82—236). Для полиимидов на основе диамино-
дифенилового эфира и различных диангидридов стабильность
уменьшается в следующей последовательности:
,—(CF2K—,
, (CF2N .
708
N
Для полипиромеллитимидов диаминов с «шарнирными»
атомами или группами термостойкость уменьшается в зависимости от
природы мостиковой группы в следующем ряду (группа III):
-О— > —S— > -СН2 (СН2J— > -NH— > —СН— >
> —SO,— > С
(СНзJ
На рис. 7.4 представлены данные термогравиметрического
анализа полипиромеллитимидов групп I и III в среде гелия. На
рис. 7.5 приведены результаты изотермического ТГА в вакууме
полипиромеллитимида диаминодифенилового эфира (№ 104). При
термической деструкции полипиромеллитимидов с различными
«шарнирными» атомами между /г-связанными фениленовыми
группами стабильность на воздухе уменьшается в следующей
последовательности [208]:
—s- > -so2— > —сн2— > -со— > —so— > —о- > \:(сн3J
Из этого вытекает, что стойкость к термоокислительной дест-
100 200 300 400 500 600 700 8QQ t,°C
1
S ?,ч
Рис. 7.4. Кривые термической деструкции полипиромеллитнмидов на основе
диаминов общей формулы H2NRNH2 в гелии [255]:
/ — Н-4,4'-Дифенил-; 2 — Н-1,3-фенилен-; 3 — Н-4,4'-дифенилокспд-; 4 — Н-4,4'-днфеннлметан-.
Рис. 7,5. Кривые изотермической деструкции полипиромеллитимиддифенилоксида
[278].
709
/00- 300 500 700 t°C
Рис. 7.6. Кривые термической деструкции
ароматических полипиромеллитимидов с боковыми
циклическими группировками на воздухе A, 2) и в гелии C,4):
1, 3 — полимер на основе 3,3-бисD'-аминофеннл)фталида;
2, 4— иа основе 9,9-бисD'-амннофенил)флуорена [23].
рукции непосредственно связана с
электроотрицательностью «шарнирных» атомов.
Наличие между фенильными ядрами
как в ангидридном, так и в аминном
компоненте перфторалкильных групп приводит к
понижению температуры размягчения полиимидов, тем не менее
эти полимеры стабильны в инертной среде до 450 °С, а на воздухе
до 400 °С (табл. 7.2 № 212, 226) [117].
Важным фактором, влияющим на термостойкость полиимидов,
является жесткость макромолекул. Полиимиды, содержащие в
макромолекуле боковые циклические группы, центральный атом
которых связан с основной цепью, отличаются более высокой
термостойкостью, чем аналогичные полимеры, в которых углеродный
атом циклической группы не входит в основную цепь. Полипиро-
меллитимиды различных диаминов с боковыми объемистыми
группировками в порядке понижения термостойкости
располагаются в следующий ряд:
о
На рис. 7.6 приведены данные о потере массы полипиромел-
литимидами'3,3-бисD/-аминофенил)фталида и 9,9-бисD'-аминофе-
нил)флуорена в гелии и на воздухе.
Наряду с химическим строением повторяющегося звена на
термостойкость полиимида влияет и способ получения полимеров.
Низкомолекулярные фракции, содержащие полимеры с
фенильными и ангидридными групп-ами, оказывают стабилизирующее
действие при термоокислительной деструкции [162]. Полимеры,
полученные химической (каталитической) циклизацией, более
стабильны, чем аналогичные продукты, синтезированные термической
имидизацией, что объясняется более высоким содержание изо-
7IQ
имидных звеньев в первых полимерах. Полиимиды с концевыми
фенильными или двумя ангидридными группами обладают более
высокой стойкостью к тсрмоокислнтелыюй деструкции, чем поли-
меры с концевыми амндными или ацетамидными группами.
Наличие влаги при деструкции в инертной среде приводит к
увеличению потери массы за счет гидролиза. Термоокислительная
деструкция протекает легче во влажной атмосфере [162]. Сшитые
полиимиды имеют более высокую стойкость к термоокислительной
деструкции. Синтез сшитых полиимидов осуществляется за счет
использования на стадии получения полиамидокислот
многоосновных карбоновых кислот [237] или диаминов с карбоксильными
[234] или ацетильными группами [343]. При этом сшивание
полимеров протекает в процессе имидизации. Дополнительное
сшивание полиимидов может осуществляться с помощью дигидрази-
дов [238]. Полиимиды, полученные полимеризацией продуктов
взаимодействия малеинового ангидрида с ароматическими
диаминами, например диаминодифенилметаном, обладают более
низкой стойкостью к термической и окислительной деструкции, чем
соответствующие полипиромеллитимиды (рис. 7.7). Начало
уменьшения массы у полимеров как на воздухе, так и в среде азота
наблюдается при 390 °С [212]. Термическая деструкция полибис-
малеимида диаминодифенилметана протекает в обеих средах при
500 °С приблизительно одинаково (потеря массы составляет около
30%)- При 700 °С на воздухе продукт полностью разлагается с
образованием летучих соединений, в то время как в азоте остаток
при этой температуре составляет около 50%. Полииминоимиды
общей формулы
—N
N—Ar—
NH
имеют более высокую стойкость к термоокислительной деструкции,
чем соответствующие полиимиды [156]. Деструкция полипиромел-
литимида 4,4'-диаминодифенилоксида на воздухе протекает при
400 °С, в то время как соответствующий полииминоимид (Аг =
= С6Н4— О—СеН4) стабилен на воздухе до 450°С.
Рис. 7.7. Кривые
термической
деструкции полибис-
малеимида на
основе
диаминодифенилметана на
воздухе (а) и в среде
азота (б) [212].
711
Табл
0
II
и
—к \С
VA:
Y
S
о
-к \C
II
s
0
II
-ал
j
(
D
1
II
(
A
ица
7.5
С
с
|
)
о
А
Y
Л
H
Згт
^ п
VJ
. Состав
[%
(мол.)] гелеобразных продуктов разложения
Элементарное звено
(
1
-об
Y
Л
(
1
-об
р
- s—(С))—
N—/П>—S—O—
Г, °G
20-400
400—450
450—550
550—620
20—400
400—450
450-550
550-620
20—400
400—450
450—550
550-620
20-400
400—450
450-550
550—620
CO2
78,0
43,7
11,1
1,0
82,1
41,0
24,7
2,4
37,7
20,7
1,1
64,3
26,5
19,1
1,3
ароматических полиамидов
со
19,7
53,0
46,3
23,6
14,5
45,9
49,2
18,0
60,1
52,3
21,3
27,8
60,3
48,6
12,8
HCN
0,2
0,2
4,3
3,7
0,5
1,0
4,8
9,2
0,3
2,0
3,0
0,4
0,9
4,4
4,9
н2о
0,5
—
0,4
0,3
0,4
0,8
0,2
0,4
0,8
0,6
0,7
1,0
0,5
0,5
0,7
0,4
0,4
5,6
8,6
0,7
0,1
1,1
6,4
0,1
3,8
9,0
0,4
0,2
2.4
8,4
н2
1,0
2,6
32,1
61,6
0,4
1,2
11,4
55,0
0,8
20,2
64,7
0,3
2,2
19,7
66,3
в вакууме
свнв
0,2
1,0
0,2
1,2
0,2
0,2
0,4
0,2
0,2
0,4
0,2
0,4
0,5
0,3
cs2
—
—
—
—
—
0,1
0,1
—
—
—
—
0,1
[347]
со
—
—
—
0,3
3,8
1,6
0,2
—
—
—
0,6
2,6
0,9
0,1
H2S
—
—
—
1,1
5,9
6,5
7,9
—
—
5,2
6,4
3,9
5,2
Летучие продукты деструкции полиимидов в вакууме состоят
главным образом из СО и CO2j a также СН4, синильной кислоты,
воды, водорода, бензола, аммиака и бензонитрила (табл. 7.5).
Серосодержащие полиимиды отщепляют, кроме того, H2S, COS, CS
[163, 166, 171, 182, 272, 344, 345, 347, 348]. Содержание СО2 в
газообразных продуктах деструкции уменьшается с повышением
температуры, содержание СО проходит через максимум при
температуре около 450 °С, а водород выделяется в количестве более
5% (мол.) при температуре выше 500 °С. Коксовый остаток при
600 °С составляет 40—60 % от массы исходного полимера и
состоит из аминов, фенолов и производных фталимида. Дальнейшее
повышение температуры приводит к образованию высококонден-
сированных полиароматических продуктов. Предложен следующий
механизм образования полупроводниковых полимеров,
получающихся при нагревании в вакууме полипиромеллитимида диамино-
дифенилоксида [345]:
-N
О
600 С
700вС '
О
еоо
При 620°С остаток составляет около 65 %• При повышении
температуры до 800 °С масса остается неизменной, а плотность
увеличивается до 1,65 г/см3. Ненасыщенные связи в высококонденсиро-
ванной ароматической системе придают коксовому остатку
полупроводниковые свойства. Нет единой точки зрения на механизм
термической деструкции ароматических полиимидов в вакууме и
образования СО2. Ряд авторов [182, 344] полагает, что
выделение СО2 обусловлено разложением нециклизованных звеньев
в полимере:
713
I
—co2
CONH—Q)—
H2O
I
+ 2CO2
Диоксид углерода выделяется уже при 300 °С. Энергия активации
этого процесса составляет 20 ккал/моль, т. е. она того же порядка,
что и для гидролитической деструкции ароматических полиамидов
A5—20 ккал/моль). Образование СО является следствием гомо-
литического распада имидных циклов. Энергия активации этого
процесса составляет 50—60 ккал/моль [182]. Некоторые
исследователи [139, 272] полагают, что выделение СОг связано с
разложением в полиимидах изоимидных групп. В ароматических
полиимидах существует подвижное равновесие между имидными
(содержание около 98%) и изоимидными структурами, которое с
повышением температуры смещается в сторону образования
имидных структур:
N—R—'
N—R—
АН = —10 ккал/моль
Наличие СО2 в продуктах деструкции относят за счет
разложения изоимидных звеньев, которые постоянно появляются в ходе
процесса. Однако Элерс [347] полагает, что СО2 образуется
вследствие димеризации макромолекул с концевыми изоцианатными
группами, появляющимися в ходе деструкции:
П
714
п
—NCO+nOCN—
Оксид углерода образуется при распаде бензоильного радикала:
[AJ
Кинетические исследования деструкции полипиромеллитимидов
и соответствующих модельных соединений позволили предложить
[166, 171] внутримолекулярный механизм превращений, связанных
с отщеплением СО2, через переходное изомерное ионное состояние:
ю:Ой-€>>
,=N-(Q
соа
Реакции, приводящие к отщеплению СО и СОг, являются
конкурирующими. Константы скорости выделения СО и СОг при
600 °С из фталимида, использованного в качестве модельного
соединения, составляют соответственно 5,3«Ю-5 и 2,12* 10 с [171].
Наличие азометиновых групп в твердых остатках после деструкции
ароматических полиимидов доказано методом ЭПР [153]. Энергия
активации процесса термоокислительной (воздух) и термической
(вакуум) деструкции полипиромеллитимида диаминодифенилокси-
да составляет 33 и 74 ккал/моль соответственно [345]. С ростом
концентрации кислорода скорость термоокислительной деструкции
увеличивается [162, 346]. На рис. 7.8 приведены данные,
позволяющие сравнить скорость деструкции полиимида на воздухе и в
вакууме. Выделение при деструкции на воздухе летучих
продуктов при 485 °С завершается за 5 ч [344].
Гидролитическая стойкость. При действии на ароматические
полиимиды при высоких температурах воды, водяных паров или
щелочных растворов они деструктируются до полиамидокислот, а
при длительном воздействии — до соответствующих диаминов и
тетракарбоновых кислот. Полимеры устойчивы к действию воды
716
Рис. 7.8. Изменение скорости
выделения летучих продуктов
при термической деструкции по-
лнппромеллитимида диамино-
дифенилоксида [344]:
а — на воздухе; б — в вакууме.
при комнатной
температуре. Пленки полиимидов
при кипячении в воде в
течение нескольких
недель или месяцев
утрачивают эластичность [2].
В растворе при температуре выше 150°С ароматические
полиимиды гидролизуются очень быстро, при этом самую низкую
гидролитическую стойкость имеют полипиромеллитимиды [151]. По
гидролитической стойкости полиимиды в зависимости от строения
диангидрида можно расположить в следующий ряд:
w,
0,7
0,5
0,5
0,4
0,2
0,1
0
%'MUH
4a5°C^\ a
/475 \
20 40 ВО m.%
2,4
2,0
I?
1,2
0,8
0,4
0
Vq-MUH
y—4 BOf°r **
- \
Vs.
~ 590 \\
| | ^^S^-
Ю 20 30m,%
На гидролитическую стойкость полиимидов большое влияние
оказывает также физическая структура полимеров в твердом
состоянии. Кристаллические полиимиды дифенилоксидтетракарбоно-
вой кислоты не разлагаются при нагревании в воде. При действии
растворов щелочей на полиимидные пленки имидные циклы
гидролизуются с образованием солей полиамидокислот [352] и
обесцвечиванием пленок:
О
п
п
Металлические соли полиамидокислот растворяются в воде,
аммиаке и разбавленных (< 1 н.) щелочах, но не растворяются
в полярных органических растворителях и концентрированных
щелочах. При обработке солей полиамидокислот соляной кислотой
образуются полиамидокислоты, что сопровождается усадкой пле-
716
нок. Гидролитическая активность гидроксидов щелочных металлов
уменьшается в следующем ряду:
КОН > NaOH > LiOH
В насыщенных растворах гидроксида бария при комнатной
температуре гидролиз не протекает. Щелочной гидролиз
ароматических полиимидов происходит уже при комнатных температурах и
сопровождается медленной деструкцией образовавшейся соли по-
лиамидокислоты. При гидролизе полипиромеллитимида диамино-
дифенилоксида в 4 н. КОН при 100 °С через 20—25 ч выделяется
90—95% исходных мономеров [93].
Для идентификации соединении, использованных для синтеза
полиимидов, пригодна реакция Инга — Маиске. Имид реагирует с
гидразином с образованием промежуточного продукта,
распадающегося на гидразид и амин [91]. Гидразинолиз полиимидов гидра-
зингидратом протекает количественно уже при комнатной
температуре:
О N—R О
N2H4
N—R >
н2о
Из полимера образуется соответственно дигидразид кислоты и
диамин [351]:
7.1.1.3. Полиимидные пленки
Получение. Формование пленок из реакционных растворов по-
лиамидокислот проводят методом полива. Наиболее высокие
физико-механические показатели пленок достигаются в том случае,
когда после полива растворитель удаляется при возможно более
низкой температуре A00—150 °С) и циклизация проводится путем
кратковременного нагрева при 400°С [257].
717
О гидролитической нестойкости полиамидокислот в растворе
уже говорилось выше. Растворы полнамндокнслот сразу же после
синтеза следует перерабатывать или хранить при температуре
ниже О °С. Для полива в большинстве случаев используют
растворы в диметилформамиде или диметилацетамиде. Вследствие
ассоциации ионизованных карбоксильных групп полиамидокислот
пленки, полученные поливом из раствора, содержат до 20 %
растворителя, который пластифицирует их, придавая им высокие
эластичность и прочность. Из водно-тетрагидрофурановой смеси
пленки можно отливать только после добавления хлороформа, так как
в противном случае испарение растворителя сопровождается
выпадением осадка.
Свойства полиимидных пленок зависят не только от
молекулярной массы полиамидокислот, как об этом уже говорилось, но
и от условий имндизации, т. е. от вязкости раствора, толщины
пленки и скорости нагревания. Предельная толщина пленки,
которая может быть получена методом полива, составляет 0,1 мм.
При формовании более толстых пленок имидизация происходит
быстрее, чем удаление растворителя, и полимер осаждается в
процессе пленкообразования. В случае химической имидизации этих
трудностей удается избежать. В процессе получения пленки
поливом имидизация вследствие двухстадийного механизма протекает
через стадию образования промежуточного хрупкого продукта
[257];
H3N—Ar—~—NHCO
СООН
—Ar—
H3N-Ar
—N1
—Ar— —N
- НООС
Эластичная соль
поли-
амидкар-
боновой
кислоты
СООН
Хрупкая соль
полиимидкарбо-
иовон кислоты
Эластичный
полиимид
На рис. 7.9 показано, как меняется в процессе имидизации
эластичность пленок полиамидкарбоновых кислот различной мо-
718
Рис. 7.9. Зависимость эластичности поли-
имидной пленки от условий имидизации и
молекулярной массы исходной полиамидо-
кислоты [257]:
/ — ннзкомолекулярный полимер G00); 2 —
полимер со средней молекулярной массой; 3 —
высокомолекулярный полимер.
Гт} |-"C ^Т ^ | 1 |
0 WO 200 300 t,°C
лекулярной массы. На основе полиамидокислот с т]пр < 0,2 нельзя
получить эластичные пленки, так как на завершающей стадии
процесса имидизации при 400 °С протекают деструктивные
реакции, связанные с окислением по аминогруппам или
взаимодействием с остатками растворителя. Для полиимидных пленок с
хорошими механическими свойствами средняя степень полимеризации
должна быть около 100. Хорошие свойства пленки объясняются
тем, что в процессе формования сразу же после испарения
растворителя, находясь еще на стадии полиамидокислоты, она
отделяется от подложки, при 150—180 °С пленка дает усадку, и при
300—400 °С хрупкая пленка превращается в эластичный полиимид.
Свойства. Фирма «Du Pont» выпускает полиимидную пленку на
основе продукта взаимодействия пиромеллитового диангидрида с
диаминодифенилоксидом в промышленном масштабе под
названием «Каптон Н» или «Н-пленка». Свариваемые многослойные
пленки на основе «Каптона Н» и сополимеров этилена с тетра-
фторэтиленом или политетрафторэтилена выпускаются под
названиями «Каптон HF» и «Кантон НВ». В СССР пленка на основе
ПМДЛ и/или дифенилоксидтетракарбоновой кислоты и диамино-
дифенилоксида выпускается под названием «Аримид ПМ 1»,
«Аримид ПМ 2» и «Аримид ПМ 4».
Пленка полипиромеллитимида диаминодифенилоксида
прозрачна. Тонкие пленки окрашелы в золотисто-желтый, толстые —
в коричневый цвет. Их отличают высокая термо- и хемостойкость,
хорошие механические и электрические свойства, а также
устойчивость к действию ионизирующих излучений [353]. Свойства
пленок полипиромеллитимида диаминодифенилоксида приведены
ниже [354, 356]:
1,42
385
Плотность, г/см3
Температура стеклования,
°С
Термостойкость, °С (время,
в течение которого при
экспозиции на воздухе
удлинение достигает 1 %)
кратковременно . . .
1 неделя
1 месяц
1 год
10 лет 240—260
Температура нулевой проч-
480
340
300
275
II ОСТИ.
Термический коэффициент
линейного расширения от
—14 до 38 °С, 1/°С . . . .
815
2,0- 10"
Коэффициент
теплопроводности, кал/(смс-°С) . . . 3,5-10"
Усадка, %
при 200 °С 0,2
при 250 °С 0,3
при 300 °С 0,5
при 400 °С 3,5
Водопоглощение при 23 °С
и 20—80%-ной отн.
влажности, см/(см-%) 2,2'10
Прочность при растяжении,
кгс/см2
при —195 °С 2 460
при 25 °С 1 750
при 200 °С 1 190
при 300 °С 980
при 450 °С 280
—S
Тангенс угла
диэлектрических потерь прн 1 кГц
при 25 °С 0,003
при 200 °С 0,002
Электрическая прочность,
кВ/мм 280
Удельное электрическое
сопротивление объемное, Ом-
•см
при 25 °С 1019
при 200 °С 1014
поверхностное, Ом . . 1016
Относительное удлинение
при разрыве, %
при —195 °С 2
при 25 °С 70
при 200 °С 90
при 300 "С ПО
при 450 °С ПО
Модуль упругости при
растяжении, кгс/см2
при —195 °С 35 800
при 25 °С 30 300
при 200 °С 18 300
Диэлектрическая
проницаемость при 1 кГц
при 25 °С 3,5
при 200 °С 3,0
Длительная эксплуатация пленок возможна при 240—260 °С,
кратковременно они выдерживают температуру до 480 °С.
Пленка остается гибкой при температуре кипения гелия D К),
сохраняя при этом 50 % от значения относительного удлинения при
комнатной температуре. Физико-механические свойства мало
изменяются до 250° (рис. 7.10). При 500°С прочность при
растяжении полиимидной пленки вдвое больше, чем у полиэтиленовой
пленки при комнатной температуре. Этот показатель за 1000 ч при
300 °С на воздухе уменьшается лишь наполовину. Нулевая
прочность наблюдается при 815°С, что на 300° выше, чем у алюминия.
При 250 °С за 1000 ч полимер сохраняет 90 % прочности [75].
Вытяжка при 500 °С в течение 60 с полиимидной пленки,
обработанной триизоцианатотрифенилтиофосфатом, приводит к сильному
уменьшению усадки ее в процессе высокотемпературной
эксплуатации [412].
Полипиромеллитимид диаминодифенилоксида является
превосходным полимером для изготовления пленок
электроизоляционного назначения, длительно работающих при высоких температурах.
Благодаря высоким механическим характеристикам при
высокотемпературном воздействии полиимидная пленка может заменять
гораздо более толстую изоляцию из стекла, керамики и слюды.
Дугостойкость полимера составляет 230 с. Стойкость к действию
коронарного разряда обеспечивается путем нанесения покрытия из
алюминиевого порошка [82]. Такая пленка устойчива в течение
500 ч к коронарному разряду 500 В. По диэлектрическим
показателям прн комнатной температуре она аналогична полиэтиленте-
рефталатной пленке. Диэлектрическая проницаемость и тангенс
угла диэлектрических потерь мало зависят от частоты (рис. 7.11).
Температурные зависимости пробивного напряжения и удельного
Ор,кгс/см2 рнс 7 10 Зависимость
относительного удлинения при
разрыве (а) и прочности
при растяжении (б) полипи-
ромеллитимидной пленки
на основе диаминодифеиил-
-2020 50 100 ПО t,°C -2020 60 100 /40 180 t.°C оксида от температуры [355].
100
90
80
70
а
— ^У
^ f*i I I
2000
1500
1000
Таблица 7.6. Химическая стойкость пленки полипиромеллитимида
диаминодифенилоксида [358]
Среда
Бензол
Толуол
Метанол
Ацетон
NaOH, 10%-ный
Уксусная кислота
(ледяная)
п-Крезол
Трансформаторное
масло
Вода
pH 1
pH 4,2
pH 7
pH 8,9
pH 10
Условия выдержки
а среде
} 1 год при 23 °С
1
1
5 сут при 23 °С
36 сут при И0°С
22 сут при 200 °С
180 сут при 150 °С
14 сут при Ю0°С
14 сут при 100 °С
70 сут при 100 °С
14 сут при 100°С
4 сут при 100 °С
Модуль
упругости
при
растяжении
Прочность
при
растяжении
Удлинение
при
разрыве
% от исходного значения
100
97
140
160
100
94
100
67
Разрушаете
102
102
100
100
100
100
100
100
85
100
100
65
65
65
65
60
82
66
73
62
я
62
77
ЮО
30
30
30
20
10
объемного электрического сопротивления приведены на рис. 7.12
и 7.13, а на рис. 7.14 представлено изменение удельного объемного
электрического сопротивления в условиях термостарения на
воздухе при 300 °С. Зависимость диэлектрических свойств полиимидов
от степени имидизации изучалась в [74]. Полиимидная пленка
загорается только при прямом контакте с открытым пламенем,
т. е. обладает самозатухающими свойствами.
Пленка полипиромеллитимида диаминодифенилоксида
обладает отличной стойкостью к действию органических растворителей,
но довольно низкой стойкостью к действию кипящей воды
(табл. 7.6). После 5 сут пребывания в дистиллированной воде
при 100 °С прочность при растяжении уменьшается с 1850 до
1000 кгс/см2, а относительное удлинение при разрыве — с 62 до
6,3% [366]. Прочность гидролизованной полиимидной пленки
tg 6
10'
Ю
5 ~~
25 50 75 100 125 150 175 t,°C
Рис. 7.11. Зависимость диэлектрической проницаемости (/) и тангенса угла
диэлектрических потерь B) полипиромеллитнмидной пленки на основе
диаминодифенилоксида от частоты [355].
Рис. 7.12. Зависимость пробивного напряжения пиромеллитимндиой пленки на
основе днамшюднфенилокепда от температуры (толщина пленки 0,0025 мм,
градиент напряжения 500 В/с) [355].
721
10" -
10
12
50 100 150 200 250 tfC
z, недели
Рис. 7.13
Зависимость удельного
объемного электро-
сопро тивления
пленки полипиро-
меллитимида диа-
минодифенилок с и-
да от температуры
[355].
Рис. 7.14.
Зависимость удельного
объемного электро-
сопро тивления
пленки полипиро-
меллитимида диа-
минодифеиилокси-
да от
продолжительности
нагревания на воздухе при
300 °С [357].
можно почти полностью восстановить путем термообработки. Так,
если пленку, подвергшуюся гидролизу, нагревать в течение 80 ч
яри 224 °С или 2 ч при 310 °С, то прочность при растяжении
увеличивается с 1000 до 1700—1800 кгс/см2, а относительное
удлинение при разрыве с 6,3 до 38,8 или 50,7 % [158].
В процессе термообработки вновь возникают амидные связи,
распавшиеся в результате термогидролиза. При этом происходит
ацилирование концевых аминогрупп одной деструктированной
цепи ангидридными группами другой. Регенерированная таким
образом полиимидная пленка обладает более высокой гидролитической
стойкостью, чем пленка, не подвергавшаяся гидролизу. В
щелочных средах и при воздействии гидразина происходит интенсивное
разрушение полимера.
Полиимиды мало устойчивы к действию УФ-излучения.
Материалы становятся хрупкими уже после 6-месячной выдержки на
солнце. Полипиромеллитимид диаминодифенилоксида
характеризуется высокой радиационной стойкостью [359]. Хотя окраска по-
лиимидных пленок усиливается уже при дозе выше 108 рад,
физико-механические и электрические свойства при этом практически
не изменяются. При облучении полиимидной пленки у~лУчами
дозой 4-Ю10 рад в вакууме прочность при растяжении составляет
90 % первоначального значения, а относительное удлинение при
разрыве — только 20% от исходной величины, равной 65%.
Кислород воздуха ускоряет радиолиз этого полимера [95]. В
результате облучения улучами на воздухе прочность при растяжении
составляет 50, а удлинения— 10 % от исходного значения. В то же
время при облучении на воздухе дозой 109 рад термостойкость
[244] и электрические свойства изменяются незначительно [367];
Облучение электронами дозой 1010 рад не приводит к изменению
диэлектрических свойств и эластичности пленок [2]. Полистирол
в этих условиях становится совершенно хрупким. Облучение в
течение 40 сут в ядерном реакторе тепловыми нейтронами при плот-
722
ности потока I013 нейтронов (см2-с) сопровождается появлением
легкой окраски, ио физико-механические свойства при этом не
изменяются [356]. Проницаемость полиимидной пленки по
отношению к водороду после облучения дозой 4 -10'° рад
увеличивается в 10 раз.
Применение. Полиимидная пленка используется в
электротехнике и электронике. В электротехнике полиимидную пленку
применяют для изоляции обмотки электродвигателей и
трансформаторов, в качестве диэлектриков в конденсаторах. По сравнению
с обычной изоляцией полиимидная пленка негорюча, обладает
высокой термостойкостью (при эксплуатации в течение 20 000 ч
при 250°С не происходит пробоя), хорошими
физико-механическими н электрическими свойствами в интервале температур от
-—2E9 до 250 °С. Кроме того, при ее использовании достигается
большая экономия в массе и объеме по сравнению с применением
традиционных материалов, таких, как слюда, стекло, керамика.
Быстрый отвод тепла препятствует перегреву. Экономически
целесообразно использование полиимидной изоляции для проводов,
движущихся частей двигателей постоянного тока, локомотивов
сверхскоростных поездов [214]. За счет увеличения сечения
медного провода с полиимидной изоляцией без изменения размера
мощность электродвигателей увеличивается на 12%. При этом
стоимость 1 кВт мощности двигателя понижается на 8—9%. При
сохранении сечения провода и мощности двигателя длину обмотки
можно сократить на 7 %, что снижает стоимость на 3 %.
Пленка, покрытая сополимером этилена с тетрафторэтиленом,
при 260—280°С сваривается и при этой температуре может каши-
роваться на другие субстраты. Подобная изоляция после
нагревания до 450 °С прочно связывается с поверхностью металла.
В электронике полиимиды используют при изготовлении
многоканальных кабелей, гибких печатных схем, многопозиционных
выключателей. Полиимид является единственным эластичным
материалом, выдерживающим высокие температуры, развивающиеся
при пайке: при 260—300 °С в течение нескольких минут не
наблюдается повреждений. Нанесение медного слоя производят
гальванизацией или путем кэширования тонкими медными пленками
в присутствии специальных полиуретановых клеев. Ниже
приведены показатели свойств полиимидной пленки, кашированной
медью [360] :
Толщина пленки полиимида с медной фольгой, мкм 75
Толщина медной фольги, мкм 35
Стойкость к припаиванию по MIL-P-13949 при 260 °С . . . . Отсутствие
пузырей
Прочность при отслаивании медной пленки за 20 с при 260 °С,
кгс/25 мм > 3
Поверхностное сопротивление по MIL-P-13949 (при 35 °С,
4 сут, 90%-ная отн. влажность), Ом Ю12
Внутреннее сопротивление (в тех же условиях), Ом . . . Ю14
Электрическая прочность, кВ/0,04 мм 5,5
Диэлектрическая проницаемость при 1 МГц 4,1
Тангенс угла диэлектрических потерь при 1 МГц 130 • 10~~
723
При нанесении медного слоя гальваническим способом сначала
происходит образование шершавой поверхности пленки, затем
с помощью вакуума напыляется тонкий хромовый слой и слой
золота толщиной 2—ЗА. Металлизирование медью проводится в
сернокислотной ванне. Благодаря использованию плоских полиимид-
ных многоканальных кабелей в высшей степени экономично
решается задача соединения электронных приборов и заменяется
«лес» проводов. Высокая плотность проводов в интегральных
многопозиционных выключателях требует получения проводниковых
соединений между отдельными слоями с помощью полнимидных
носителей.
Механическое сверление отверстий слишком неточно и
неэкономично, так как в современных цепях выключения расстояние
между отверстиями составляет 1—2 мм и диаметр их не
превышает 0,2—0,3 мм. Полиимиды можно обработать гидразином. За
счет селективного травления можно получить капиллярные
отверстия для соединения компенсаторов выключения [351, 361]. На
металлизированную медью полиимидную пленку сначала наносят
светочувствительный слой, а затем соответствующий фотонегатив.
Этим достигается определенное маскирование медного слоя.
Только в точках, в которых установлено выключение отдельных
плоскостей, медь существует в свободном виде и протравливается
FeCl3. Таким образом можно получить светочувствительные
пленки. При погружении на 5 мин в нагретый до 45—50 °С раствор гид-
разипгидрата (пленка толщиной 0,02 мм) в полиимидном слое
протравливаются отверстия, которые необходимы для соединения
компенсаторов выключения. После снятия медного слоя поли-
имидная пленка подвергается повторной металлизации. При этом
достигается не только покрытие цепей, но и соединение их между
собой. Этот метод дает возможность развиваться дальше процессу
микроминиатюризации гибких интегральных многопозиционных
цепей схем.
Склеивание компенсаторов выключения производится клеями
горячего отверждения при небольших давлении и температуре
160"С. Высокая стабильность размеров обеспечивает применение
ее в электронной технике.
Самоклеящиеся полиимидные пленки могут быть получены
путем поверхностного гидролиза пленки 10%-ным раствором
щелочи с последующим нанесением слоя тройного сополимера акри-
лонитрила, бутилакрилата и метакриловой кислоты [57]. В
результате пиролиза полиимидной пленки в вакууме при 800°С
образуется органический полупроводник с удельным объемным
электрическим сопротивлением 5-Ю2 Ом-см, который можно
использовать в широком температурном интервале [358, 362].
Из металлизированной полиимидной пленки можно
изготавливать защитные жилеты для авиапассажиров, используемые при
пожарах в самолетах [363]. Покрытая алюминием полиимидная
пленка была использована в конструкции космического корабля
Аполлон для защиты двигательной группы от воздействия солнр«-
724
ного излучения и тепла. Теплопроводность пленки можно
увеличить, получая комплексы полимеров с нонами металлов [113, 127].
При содержании в пленке 5 % ионов металлов диэлектрические
свойства остаются такими же, как и у пемодпфицированпых
полимеров, удельное объемное электрическое сопротивление при этом
уменьшается на 20% [194]. Высокая эластичность полиимидпой
пленки при низких температурах дает возможность применять ее
для обкладки сосудов для хранения жидкого водорода [364].
В ядерной технике также широко используют полиимидную пленку
[175].
7.1.1.4. Полиимидные волокна
Получение. Полиимидные волокна могут быть получены из
полнамидокислот как сухим, так и мокрым формованием в
воду. Использование сухого формования возможно благодаря
достаточно высокой стабильности физико-механических свойств
волокна, подвергаемого термической имидизации. Волокно на основе
полипиромеллитимида диаминодифенилоксида получают из 20—
30 %-ного раствора в диметилформамиде, диметилацетамиде, ди-
метилсульфоксиде или N-метилпирролидоне в инертной среде при
температуре ниже 65 °С с последующей термической
циклизацией при 200—250 °С. При 300—350 °С волокно подвергается
дополнительной вытяжке на 150—200 % и, наконец, термофиксации
при 400°С. В неориентированном состоянии волокно имеет
прочность при растяжении 2 г/денье и относительное удлинение при
разрыве 50—80%. После вытяжки разрывная прочность
возрастает до 7 г/денье, а удлинение уменьшается до 12—14 % [368—
370, 372,421].
При мокром формовании в качестве осадительной ванны
используют воду или смесь диметилформамид — вода B0:80).
В процессе мокрого формования волокна возможна циклизация
в полнимид путем добавления в прядильный раствор таких
катализаторов циклодегидратации, как пиридин или амины, а также
введением в осадительную ванну ацетангидрида [371]. Кинетику
процесса циклизации волокна изучают по абсорбции фенола из
концентрированных водных растворов [145].
Свойства. Ароматические полиимидные волокна выпускаются
фирмой «Du Pont» начиная с 1968 г. под названием НТ-1 или
PRD-14, а в СССР с 1972 г. производится Аримид [423]. Физико-
механические свойства различных ароматических полиимидных
волокон приведены в табл. 7.7.
Прочность ароматических полиимидных волокон в известной
мере зависит от скорости нагружения разрывной машины:
увеличение скорости приводит к получению завышенных значений
прочности (рис. 7.15). Прочность при растяжении ароматических
полиимидных волокон уменьшается с повышением температуры
(рис. 7.16); волокно с исходной прочностью 3 г/денье при 300°С
сохраняет 44% прочности при комнатной температуре [373].
Аналогичным образом зависит от температуры и модуль упругости
725
Таблица 7.7. Физико-механические свойства ароматических полиимидных волокон
Элементарное звено
Прочность
прн растяжении,
г/денье
U
Относительное
удлинение
при разрыве, %
8
Модуль
упругости,
г/денье
о
¦ ui У-о
О
2,2
22
43
6,9
4,3
13
72
47
[421]
[373]
о о
О О
о о
О
о о
—N'
о
S
сн'-
9,3
5,7
3,4
1
5,2
4,8
5
14,5
26
2,5
4
300
53
65
260 '
190
[3741
[421]
[42Ц
i, г/денье
1Б ?,%
200
400
Рис. 7.15. Зависимость прочностных свойств волокна полипиромеллитимида ди-
аминодифенилоксида от скорости нагружеиия [373]:
1 — 100%/мин при 2J °С; 2 — ]0%/мин при 2J °С; 3 — 100%/мин при 300 "С.
Рис. 7.16. Зависимость прочности при растяжении волокна полипиромеллитимида
диаминодифенилоксида от температуры [373].
Рис. 7.17. Зависимость модуля упругости волокна из полипиромеллитимида диа-
мииодифенилоксида от температуры [373].
(рис. 7.17). Путем экстраполяции к нулевой прочности удалось
установить, что предельной температурой прочности является
560 °С, а для модуля упругости — 620 °С.
Это дает основание считать, что волокна полипиромеллитимида
диаминодифенилоксида могут кратковременно выдерживать
температуру 550 °С.
При нагревании волокна на воздухе при 283°С прочность при
растяжении резко понижается только в начальный период. Через
300 ч прочность практически перестает изменяться (рис. 7.18).
Уменьшение прочности при растяжении до 1 г/денье происходит
при термообработке на воздухе при 400 СС в течение 10 ч, при
333 °С — 500 ч, при 283 °С — 1000 ч. Уменьшение механической
прочности полиимидного волокна при термообработке ниже 425 °С
на воздухе связано с хрупкостью, развивающейся за счет
сшивания полимера. Характеристическая вязкость полиимидов возрастает
с увеличением времени термообработки вплоть до точки гелеобра-
зования. При нагревании в среде кислорода прочность уменьшается
в гораздо большей степени, чем на воздухе или в инертной среде.
Ароматические полиимидные волокна обладают отличной
стабильностью размеров. Усадка в токе сухого воздуха при 400 СС
Таблица 7.8. Химическая стойкость ароматических
полиимидных волокон при 100 СС [373]
Агент
H2SO4
НС1
HF
NaOH
Концентрация,
%
10
10
10
0,4
Время
выдержки,
ч
260
260
150
4
Уменьшение
прочности,
%
52
71
36
57
Агент
Н2О
NaOCl
C.H,CON(CH3J
Концентрация,
%
0,5
Время
выдержки,
ч
1400
150
150
(при
166 °С)
Уменьшение
прочности,
%
4
37
—
728
Рис. 7.18. Зависимость прочности при растяже- бр,г/денье
нии полиимидиого волокна от продолжительно- 7
сти нагревания на воздухе при 283 °С [373].
составляет менее 2%. При 300 СС
усадки не обнаруживается.
Полиимидные волокна негорючи.
В пламени бутана материал без
воспламенения или размягчения пироли-
зуется с медленным образованием хрупкого углеродного остатка.
В табл. 7.8 приведены сведения о химической стойкости полиимид-
ных волокон.В органических растворителях волокно не набухает и
не растворяется. Только диметилацетамид вызывает его
незначительную усадку, не влияет на прочность и кристалличность.
Длительное кипячение в воде сопровождается небольшим изменением
прочности при длительной экспозиции и незначительной усадкой.
Волокно устойчиво к действию разбавленных кислот, но
разрушается в концентрированных кислотах. В разбавленных водных
растворах щелочей также происходит гидролиз. Влияние окисляющих
или щелочных моющих средств незначительно.
Ароматические полиимидные волокна, так же как
ароматические полиамидные, отличаются высокой стойкостью к действию
ионизирующих излучений [188]. Прочность пряжи B00 денье, 60
нитей) полиимида на основе пиромеллитового диангидрида и ди-
аминодифенилоксида при исходном значении 6,57 г/денье после
облучения у-лучами дозой 9-Ю5 или 1,6-107 рад составляет
соответственно 6,80 или 6,47 г/денье. После облучения дозой 1,6-107 рад
удлинение уменьшается с 12,8 до 11,8%. При облучении указанной
выше дозой при 425 °С прочность после облучения составляет 91 %
от исходной.
Применение. Полиимидные волокна используют для
изготовления негорючей одежды, технических тканей, применяющихся в тех
случаях, когда требуется высокая термостойкость. Из них шьют
одежду для пожарников и летчиков. Ткани применяют для
фильтрования горячих газов, в качестве термо-и электроизоляции. По-
лиимидная ткань служит для изготовления корда,
применяющегося для получения специальных шин и приводных ремней,
выдерживающих высокие нагрузки. Рубленое волокно используют
в качестве наполнителя в высокотермостойких композициях на
основе полиимида и других полимеров. Ткань обладает
прозрачностью для электромагнитных волн, что обеспечило ее применение
в конструкциях антенных обтекателей.
7.1.1.5. Полиимиды для склеивания металлов
Полиамидокислоты в виде растворов, паст и клеящей пленки
(подложка — стеклянная ткань) используют в качестве
высокотермостойких клеев для металлов. Товарные продукты выпускают
729
под названием СТ-1 (СССР), «Керимид» («Rhone-Poulenc»),
«Квантед-159» (Quantum Inc., USA), FM-34 («American Cyana-
mid»), «Метлбонд-840» («Narmco»), DP 5505 («Du Pont»), IPA-380
(«Rhone-Poulenc»).
Для улучшения адгезии рекомендуется поверхность металла
обезжиривать и протравливать. Для травления высокосортных
сталей применяют раствор из 35 % НС1, 85 % Н3РО4, 60 % HF;
титан протравливают смесью NaF — CrO3 — H2SO4.
Для получения клеящей пленки концентрированный
высоковязкий раствор полиамидокислоты (иногда с диспергированным
в нем наполнителем) наносят на стеклянную ткань и подвергают
в заданном режиме термообработке A ч при Ю0°С, 15 мин при
150, 200, 250 °С и 5 мин —при 300 °С). Для нанесения на ткань
полимерного слоя в количестве около 40% необходимо трех-,
четырехкратное повторение операции [70]. В процессе склеивания
температура должна превышать температуру стеклования, но быть
ниже температуры разложения полиимида. Для этих полимеров
температурная область переработки ограниченна. В большинстве
случаев склеивание проводят в течение 5 мин при 400—450 °С и
давлении 140 кгс/см2 [404].
В качестве аминного компонента для адгезивных полиимидов
на основе диангидрида бензофенонтетракарбоновой кислоты
наилучшие результаты получаются при использовании ж-фениленди-
амина. Применение таких диаминов, как диаминоацетанилид, ди-
аминодифенилметан, диаминобензанилид или диаминодифенил-
оксид, приводит к образованию полиимидов с низкой текучестью и
плохой адгезией. В том случае, если в процессе получения
полиамидокислоты на основе диангидрида бензофенонтетракарбоновой
кислоты и ж-фенилендиамина концевые группы блокируют /г-ами-
ноацетанилидом, прочность при растяжении клеевого шва
склеенной высокосортной стали повышается на 75 % при 287 °С. Однако
блокирование концевых групп фталевым ангидридом
сопровождается лишь незначительным увеличением прочности при сдвиге
[143, 404]. Более высокое содержание монофункционального
компонента в клеевой рецептуре приводит к снижению молекулярной
массы и сильному уменьшению прочности.
Ниже приведены рецептуры полиимидных клеев,
представляющие наибольший интерес:
а) ж-фенилендиамин — 89 масс, ч., диангидрид
бензофенонтетракарбоновой кислоты — 99,5 масс, ч., я-аминоацетанилид —
4 масс, ч и фталевый ангидрид— 1 масс. ч.
б) ж-фенилендиамин — 96 масс, ч., диангидрид
бензофенонтетракарбоновой кислоты—100 масс. ч. и л-аминоацетанилид—
4 масс. ч.
Клеевые соединения высокосортных сталей с помощью
клеящей пленки с основой из стеклянной ткани имеют прочность при
сдвиге 210 и 140 кгс/см2 при комнатной температуре и 287°С
соответственно. Как видно из рис. 7.25, через 1000 ч термообработки
при 315 °С прочность клеевого соединения еще составляет 50%
730
исходной прочности. Понижение прочности при сдвиге до
56—70 кгс/см2 при 371 °С происходит через 40 ч. при 343 °С
—через 100 ч и при 315 °С — через 400—450 ч. Клеевые композиции,
содержащие в качестве наполнителя порошки металлов или
оксидов, имеют такую же прочность при сдвиге, как и ненаполненные
композиции (за исключением композиций с такими
наполнителями, как As2Ss и AS2S4). Наиболее подходящей основой для
клеев, используемых для склеивания титана, служат полибензимида-
зольные сополимеры (см. 7.1.2), нанесенные на стеклянную ткань
[404]. Хотя исходная прочность при сдвиге титанового соединения
почти в 2 раза ниже, чем в случае гомополиимида, в процессе
термостарения она изменяется значительно меньше. Через 1000 ч при
315 °С прочность при сдвиге клеевого соединения на основе поли-
бензимидазолимида составляет 56 кгс/см2.
Наиболее важной областью использования полиимидных клеев
является авиация, где их применяют при изготовлении сотовых
конструкций с наружным слоем из титана и высокосортной стали
[405]. Подобные клеевые соединения в конструкциях
сверхзвуковых самолетов могут эксплуатироваться до 30 000 тыс. ч при
450 °С. Шлифовальные круги на основе полиимида E7% поли-
имида и 43 % алмаза) в качестве связующего имеют
преимущество по сравнению с кругами на основе алмаз-фенолоформальде-
гидного олигомера, так как вследствие более высокой
термостойкости первые круги дают возможность работать при более
высоких скоростях [73, 132, 410].
7.1.1.6. Полиимидные лаки
Электроизоляционные лаки и лаки горячей сушки на основе
полиимидов выпускают под торговыми названиями ПАК-1
(СССР), Родефтал, Керимид («Rhone-Poulenc»), P-13N (TRW,
Inc. Systems Group, USA), Пайр ML («Du Pont») и AI-220
(«Anaconda Wire Co. Cable»). Из-за перегрузок и дефектов
изоляция проводов электродвигателей и катушек более, чем другие
элементы конструкций, подвержена термоударам, что опасно при
их эксплуатации. Поэтому изоляция для обмотки
электродвигателей является одной из основных областей применения
термостойких полимеров. Лаки для проводов можно наносить путем горячей
сушки растворов полиамидокислот Пайр ML или ариленбисмале-
имидов Родефтал. В качестве растворителей для лаков на основе
полиамидокислот наряду с диметилформамидом и диметилацет-
амидом используют смесь N-метилпирролидон — /г-ксилол, которая
отличается меньшей токсичностью, обеспечивает большую
стабильность растворов и лучшую перерабатываемость. За один
рабочий ход может быть нанесен слой толщиной 5 мкм, при этом
достигается полное удаление растворителя и воды, образующейся
в процессе имидизации при горячей сушке A мин, 400 °С).
Нанесение толстых слоев требует многослойного лакирования.
Электроизоляционный лак Пайр MZ представляет собой рас-
гвор полиамидокислоты на основе пиромеллитового диангидрида
731
f, Y
50000
рцппп
?QUUU
10000
5000
2000
1000
500
200
то
50
20
10
V \
_ \
\
\
\
\
—
Годы
\ _
\ -
\
V
\
—
\_
ь
з
2
Месяиы
12
10
6
2
Сутки
30
20
10
6
4
2
Часы
15
10
200 240 280 t°C
Рис. 7.19. Термостабильность полиимидной (/)
и полиэтилентерефталатной B) проволочной
изоляции по DIN 46432 [378].
и диаминодифенилоксида в смеси
N-метилпирролидон — диметилацетамид
[360]. Жаростойкость полученной на
этой основе проволочной изоляции
составляет 400 мин против 60 мин по
сравнению с эпоксидной изоляцией и
сохраняется на 99% при 300 °С [378].
На рис. 7.19 представлены данные о
термостойкости полиимидной и
полиэфирной проволочной изоляции.
Продолжительность эксплуатации изоляции из по-
лиимида при 240 °С составляет 25 000 ч
(около трех лет), такое же время поли-
этилентерефталатная изоляция
выдерживает только при 210 °С. Недостатком
растворов полиамидокислот является их
кислотный характер и нестабильность при
хранении. Для хранения этих лаков
необходима аппаратура из высококачественной стали. Низкая
прочность к истиранию полиимидной изоляции проводов требует
предельно точной установки напряжения на обмотке
электродвигателей. Проволочную изоляцию на основе ариленбисмалеимидов
наносят из растворов (смесь ацетон — ацетофенон) с
последующей горячей сушкой при 260 °С [316].
Проволочная полиимидная изоляция имеет электрическую
прочность 95 kB/мм; удельное объемное электрическое
сопротивление ее изменяется от 1,5-1018 (сухая) до 2,5*1015 Ом-см через
24 ч пребывания в воде. Лаки отличаются высокой адгезионной
прочностью. При 200 °С она составляет 10 кгс (по UTE-C-26-937),
через 240 ч термостарения при 300 °С — 4 кгс.
Полиимидная проволочная изоляция применяется в тех
случаях, когда при перегрузках наблюдаются перегревы (станки,
локомотивы), когда требуется высокая надежность (генераторы,
холодильные агрегаты), когда благодаря миниатюризации можно
переходить на более высокие температуры, при которых
оборудование должно в течение длительного времени работать.
Полиимидные лаки горячего отверждения используют в
качестве коррозионно-стойких, термостойких, стойких к действию
УФ-лучей покрытий сверхзвуковых самолетов, а также в виде
покрытий на политетрафторэтиленовой пленке для повышения ее
стойкости в процессе старения. Для полиимидного эмаль-лака,
содержащего алюминиевый пигмент (соотношение пигмент : связую-
щее = 2:1), при нанесении на высококачественную сталь через
100 ч экспозиции при 426 °С потеря массы составляет только 30%
[397].
732
При покрытии конденсаторной жести диэлектрическим слоем
ариленбисмалеимид наносят на субстрат методом вакуумного
испарения и проводят радиационно-химическую трехмерную
полимеризацию [398].
Полиимиды с сульфогруппами используют в качестве мембран
для электродиализа (катионообменники). Металлические
электроды покрывают путем нанесения полиамидокислот, содержащих
в макромолекулах сульфогруппы, с последующей имидизацией
[97]. Нанесение полиимидного слоя на крупногабаритные изделия
осуществляют методом напыления в электростатическом поле,
используя для этой цели полиамидокислоты [87].
7.1.1.7. Полиимидные связующие
При совмещении полиимидов с такими наполнителями, как
стеклянные ткани, маты, ровница, стальные, борные и углеродные
волокна, получают конструкционные материалы,
характеризующиеся высокой термостойкостью, хорошими
физико-механическими и электроизоляционными свойствами [159, 167, 173, 176, 205,
242, 387—389]. Технические полиамидокислоты, используемые для
пропитки, выпускают под названием ПАК-1 (СССР), «Скайбонд
700» и «Скайбонд 703» («Monsanto»), PJ2501 и «Пиралан» («Du
Pont»), «Имидайт 1832» («Narmco») и «X 13-Полиимид» (ICI).
Пропиточные составы на основе ариленбисмалеимидов выпускают
под названиями «Керимид» и «Родефталь» («Rhone-Poulenc») и
P13N (TRW Inc. Systems Groop, США). Клеящая пленка
(носитель— стеклянная ткань) производится «Bloomingdale Cyanamid»
под маркой F-34.
Раствором полиамидокислоты пропитывают стеклянную ткань,
получая активный препрег (метод «живого сшивания») с
последующей термообработкой или в одну стадию приготавливают
композицию, подвергая ее затем формованию (метод «мертвого
сшивания») [360]. Во многих случаях используют металлические
порошки или SiO2 [411]. Формование препрегов осуществляется
в вакуумном мешке или прессованием, а также в автоклавах
[176]. Прессование композиции полиимидный форполимер —
наполнитель связано с геометрией формующего агрегата с
программированным регулированием температуры и давления. После
загрузки масса нагревается до 180°С и при контактном давлении
выдерживается 30 мин. В течение 4 ч давление повышается до
4 кгс/см2 и затем при той же температуре A80°С) доводится до
140 кгс/см2. Для прессования печатных схем достаточно давления
5 кгс/см2. Для завершения процесса циклизации проводится
термообработка при каждой из температур 204, 232, 260, 288 и 370°С
в течение 4 ч.
При переработке методом формования в вакуумном мешке
препрег в формующем агрегате накрывается пористой тканью или
воздухопроницаемой пленкой и после вакуумирования нагревается
1 ч до 130 °С, 30 мин при 130°С и 2 ч при 180 °С. Готовое изделие
733
ьиии
4000
2000
0
l
400
800
- —
5 1
1200
|
Z,4
Рис. 7.20. Зависимость прочности при изгибе поли-
имидного стеклопластика от продолжительности
нагревания при 300°С [360]:
/ — метод «живого сшивания» ; 2 — метод «мертвого
сшивания».
после охлаждения под вакуумом при
температуре до 65 °С извлекают из формы
и подвергают термообработке по 2 ч при
205 и 230 °С и по 4 ч при 260, 290, 320 °С
[176, 360, 386].
В случае метода «мертвого сшивания» препрег предварительно
нагревают для удаления остатков растворителя и воды,
выделяющейся в ходе циклизации. Прессование проводят при температуре
на 20° выше температуры стеклования полимера. Так, полипиро-
меллитимид диаминодифенилоксида прессуют при 410 °С. Хорошая
теплостойкость достигается, однако, при использовании метода
«живого сшивания». Композиции, пропитанные раствором арилен-
бисмалеимида, прессуют после удаления растворителя при 100°С
и давлении 15 кгс/см2. В течение 1 ч повышают температуру до
250 °С и прессуют при этой температуре 10 мин. После
охлаждения слоистый пластик, извлеченный из формы, нагревают 24—48 ч
при 200 °С [85, 316]. Из пропитанного волокна методом намотки
без давления можно изготовить трубы [205].
В случае получения слоистых пластиков на основе графитового
и борного волокон рекомендуется сначала проводить
предварительную, а затем окончательную пропитку волокна, после чего
осуществлять термообработку [391]. Для поверхностной
обработки стеклянной ткани в качестве аппретов используют аминоси-
ланы и комплексные соединения хрома [205]. Улучшение адгезии
достигается также путем использования полиимидов,
модифицированных эпоксидированным полифениленоксидом [409]. Верхняя
температура длительной эксплуатации стеклопластиков на поли-
имидном связующем составляет 250 СС; кратковременно они
выдерживают нагрузки до 400 °С. В специальных конструкциях
стабильность обеспечивается в течение 2000 ч при 300 °С [76]. При
использовании полибисмалеимидного связующего верхняя
температура эксплуатации составляет 230 °С.
Свойства слоистых пластиков зависят от объема, который
в конструкции занимают поры. Полиимидные связующие,
полученные из полиамидокислот, вследствие высокой пористости
(объем пор составляет до 20—25 %) имеют при комнатной
температуре относительно низкую прочность при изгибе. Однако
ценность этих материалов проявляется при высоких температурах, что
видно из данных табл. 7.9. При изменении пор от 24 до 9 %
прочность при изгибе при комнатной температуре возрастает линейно
от 3850 до 5700 кгс/см2 [392, 393]. Прочность полиимидных
стеклопластиков резко уменьшается только после длительного
нагревания при 300 °С (рис. 7.20). Существенное снижение прочности
наблюдается после выдержки в течение 3000 ч при 275 °С. Это
734
объясняется недостаточной термической стойкостью шлихтовки
стеклянной ткани [390].
Полиимидные слоистые пластики, которые в процессе
изготовления при нагревании связующего образуют межмолекулярные
амидные связи (сшиваются), имеют более высокую
термостойкость на воздухе, чем пластики на немодифицированном полними-
де [343]. Ниже приведены наиболее важные рецептуры для
получения модифицированных полиамндокислот:
Таблица 7.9. Свойства полиимидных связующих
[173, 316, 360, 394]
Показатель
Прочность при изгибе, кгс/см2
при 25 °С
при 280 °С
Модуль упругости при изгибе,
кгс/см2
при 25 °С
при 280 °С
Прочность при растяжении,
кгс/см2
Прочность при сдвиге, кгс/см2
при 25 °С
при 280 °С
Диэлектрическая
проницаемость
при 1,5 МГц
при 600 МГц
после 24 ч пребывания в
воде
при 1,5 МГц
при 600 МГц
Тангенс угла диэлектрических
потерь
при 1,5 МГц
при 600 МГц
после 24 ч пребывания в
воде
при 1,5 МГц
при 600 МГц
Удельное электрическое
сопротивление, Ом
поверхностное
через 4 ч при 70 °С
через 24 ч при 20 °С
объемное, Ом-см
Электрическая прочность,
кВ/мм
Водопоглощение, %
Плотность, г/см3
Полиариленбнсмалеимид
стеклопластик
D0% связующего)
6 400
5 100
280 000
210 000
3 500
520
4,70
5,40
з
12-10
— 3
16-10
1,3-10м
22
2,05
4 000
3,42
3,84
3,65
4,11
56-10
31 • 10
87 • 10"
53.10"'
1014
1013
1.16
Ароматический полиимид
графито-
пласт
7 700
5 700
1 780 000
1 680 000
521
402
1,53
боропласт
13 600
11800
1 860 000
1 470 000
700
420
1.94
735
GU3 ,
4000
3000
2000
1000
140-10
200 400 600 800 Z,4
10'
m4 ios ios ю;
Рис. 7.21. Изменение прочности при изгибе в процессе отверждения полиимидного
связующего в стеклопластике при 315 °С (содержание связующего 35%) [343]:
1 — неотвержденное связующее; 2 — отвержденное (рецептура а); 3 — отоержденчое
(рецептура б).
Рис. 7.22. Зависимость тангенса угла диэлектрических потерь (/) и
диэлектрической проницаемости B) полиимидных стеклопластиков от частоты.
а) ж-фенилендиамин — 50 масс, ч., диаминоацетанилид —
25 масс, ч., диаминобензойная кислота—25 масс, ч., диангидрид
бензофенонтетракарбоновой кислоты—100 масс, ч.;
б) ж-фенилендиамин— 50 масс, ч., я-диаминофеноксиацетани-
лид — 35 масс, ч., диаминобензойная кислота —15 масс. ч. и
диангидрид бензофенонтетракарбоновой кислоты—ПО масс. ч.
Пластики на основе отвержденного сшитого связующего
(содержание 35%) через 100 ч термостарения при 315°С имеют
прочность при изгибе на воздухе 3375 кгс/см2 по сравнению
с 1690 кгс/см2 для пластика с неотверждающимся связующим.
Однако через 1000 ч старения прочность при изгибе в обоих
случаях снижается до 845 кгс/см2 (рис. 7.21). Ползучесть
стеклопластиков на основе полиимидного связующего проявляется только
в случае, если термическая нагрузка превышает температуру
размягчения или при температуре испытания происходит
окислительная либо гидролитическая деструкция [167, 205]. Ускорения
ползучести не происходит при выдержке однонаправленного
стеклопластика на основе полипиромеллитимида при 250 °С и полибис-
малеимида при 230 °С в течение 1000 ч при изгибающем
напряжении 1000 кгс/см2. Использование графитовых волокон не приводит
к увеличению времени, в течение которого при высоких
температурах прочность сохраняется неизменной. Диэлектрические свойстза
полиимидных стеклопластиков в широком интервале температур
л частот не зависят от этих параметров (рис. 7.22).
Полиимидные армированные пластики применяются для
изготовления высокопрочных элементов в электронике и как
конструкционный материал в скоростной авиации и ракетостроении.
Наиболее важной областью применения является изготовление
обтекателей антенн сверхзвуковых самолетов. Только фирма «Соп-
vair» произвела свыше 900 таких конструкций (Radome). Для
защиты от коррозии они имеют керамическое покрытие [395].
В электронике армированные пластики используются в
производстве компьютеров и в приборах средств связи, к которым предъяв-
736
ляются жесткие требования для обеспечения надежностей работы
при высоких температурах [81, 396].
В авиастроении полинмидные стеклопластики применяют для
изготовления элементов крыльев и фюзеляжа (в виде сотовых
конструкций). Получение полупродуктов с сотовой структурой для
сэндвичевых конструкций производится путем пропитки
стеклянных матов с гексагональной структурой ячейки и последующей
термообработки. Сотовые конструкции на полиимидной основе
выпускаются под названиями HRH-324 и HRH-327 фирмой «Hexcell
Corp. С» (США). Цена 28,3 дм3 составляет 400—500 дол. США,
что значительно ниже стоимости аналогичных конструкций из
высокосортной стали и титана, имеющих такую же прочность [399,
400]. За 10000 ч при 204°С модуль упругости при сдвиге поли-
имидных сотовых конструкций практически не меняется [394].
В случае использования полиимидных сотовых конструкций
в авиастроении их покрывают бериллиевой жестью, для
приклеивания которой применяют полиимидную клеящую пленку
(носитель— стеклянная ткань) [401]. При строительстве первых
американских сверхзвуковых транспортных самолетов для получения
этих сотовых конструкций было израсходовано 2600 кг полиими/т
общей стоимостью 3,6 млн. дол. [402, 403].
7.1.1.8. Полиимидные пресс-композиции
При получении формованных изделий изполиимидов групп 1—3
прессованием происходит термическая циклизация частично-ими-
дизоваиной полиамндокислоты или термическая полимеризация
ариленбисмалеимидов либо олигомерных полиимидов с концевыми
норборненовыми группами. Переработка пресс-композиций на
основе ариленбисмалеимидов или биснорборнилолигоимидов
(соответственно Кинел и P13N) осуществляется по обычной
технологии, в то время как прессование порошков на основе частично-
имидизованной полиамидокислоты требует методики, сходной
с принятой в порошковой металлургии (Vespel).
Пресс-массы на основе бисмалеимидов выпускаются с 1970 г.
фирмой «Rhone Poulenc» под марками «Кинел M 33» и «Кинел
MN3» (ненаполненные), «Кинел 5504» и «Кинел 5514»
(армированные стеклянным волокном). Композиция для литьевого
прессования находится в стадии испытаний. Перед прессованием табле-
тированную композицию предварительно подогревают в печах
с высокочастотным нагревом и прессуют при 220—250 °С и
давлении 100—300 кгс/см2. При загрузке массы в предварительно
нагретую до 250 °С форму время прессования составляет 2 мин на
1 мм толщины стенки изделия. Изделия выгружают горячими и
подвергают термообработке в термошкафу в течение 24 ч при
250 °С [316, 317]. При получении опытных образцов композиций,
армированных стеклянным волокном, литьевое прессование
проводят при 200 °С и давлении 300—800 кгс/см2. Свойства пресо-И8-
делий приведены в табл. 7.10. Прессованные изделия на ogh<"'B^
24 Зак. 18 737
Таблица 7.10. Свойства
Показатель
Материал типа Веспел
ненапол-
ненный
графита
с 15%
графита
и [0%
тефлона
с 15%
MoS2
Полибисма-
леимид
ненаполиен-
иый
типа МЗЗ
Плотность, г/см3
Температура длительной
эксплуатации,°С
Теплопроводность,
кал/(см • с • °С)
Усадка после
формования, %
Водопоглощение за 24 ч,
%
Прочность при разрыве,
кгс/см2
при 25 °С
при 250 °С
при 316°С
Относительное
удлинение при разрыве, %
при 25 °С
при 250°С
Модуль упругости при
изгибе, кгс/см2
при 25 °С
при 250 °С
при 316 °С
Прочность при изгибе,
кгс/см2
при 25 °С
при 250 °С
при 316 °С
Прочность при сжатии,
кгс/см2
при 25 °С
при 150°С
при 250°С
Ударная вязкость, кгс.
см/см2 образца с
надрезом
Твердость по Роквеллу
Диэлектрическая
проницаемость при 1 МГц
Тангенс угла
диэлектрических потерь при 1 МГц
1,43
243
0,32
910
465
370
7-9
6-8
31500
20 000
18 000
1200
630
2 800
2 100
1400
52
Е 45—48
3,4
0,005
1,51
630
420
350
4—6
3-5
38 000
26 000
23 000
1050
560
2 250
1500
900
2,6
Е 32-44
7,6
0,004
1,55
420
3—4
32 000
710
1300
1050
Е 5-25
1,60
820
460
350
6-8
5-7
35 000
1350
Е 40—55
1,30
38 000
28 000
1400
500
M 127
3,3
738
полиимидных
пресс-изделий
Полибисмзлеимид (наполненный
стеклянным волокном)
Кииел 5504
Кинел 5514
масса
для
литьевого
прессования
коэффициент уплотнения
8-8,5
1,90
12,4- 10~4
0,20
0,20
1900
250 000
180 000
75 000
3 500
2500
1400
2 350
1300
92
М. 125
4,7
0,009
4,5-5
1,70
8,5 • 10~*
0,20
0,75
450
131000
105 000
1500
1250
2 450
33
M 126
4,5
0,017
3,5
1,60
80 000
75 000
1300
1 100
8,1
Полиимид с концевыми
норборненовыми группами типа
Р13 N
ненапол-
нениый
1,33
выше
300
0,12
0,40
510
400
1,4
1,8
33 500
22 000
780
480
2 600
4,33
0,006
с 50%
(объемн.)
O1U2
1,75
выше
300
0,70
0,30
550
350
0,6
0,8
9 000
850
730
2 400
I 200
7,5
5,03
0,001
40 %
(объемн.)
стеклянного
волокна
1,83
выше
300
0,08
1,8
1000
655
0,8
0,6
15 500
12 500
2 600
2 350
2 300
1500
49
6,0
0,006
Полиимид
Джемон
ЗОЮ с 60%
стеклянного
волокна
0,20
2 000
1690
270 000
206 000
2 950
4,84
0,009
24*
739
Показатель
Материал типа Веспел
ненапол-
нениый
с 15%
графита
с \Ь%
графнта
и 10%
тефлона
с 15%
MoS2
Полнбнсма-
леимид
ненаполнен-
ный
типа МЗЗ
Удельное электрическое
сопротивление
объемное, Ом см
поверхностное, Ом
Электрическая
прочность, кВ/мм
Коэффициент трения без
смазки на воздухе
1OIG-1O17
Ю15—10'6
0,29
1015
0,24
наполненных стеклянным волокном полибисмалеимидов обладают
самозатухающими свойствами. Длительная эксплуатация их
возможна при температуре до 250 °С. Через 8000 ч при 200 °С
прочность при изгибе уменьшается с 2650 до 1500 кгс/см2, модуль
упругости при изгибе —с 130 000 до 105 000 кгс/см2.
Пресс-изделия стойки к действию разбавленных кислот, алифатических и
ароматических углеводородов, а также спиртов, простых и
сложных эфиров. Разрушение происходит под действием водных
растворов щелочей при высоких температурах. Изделия на основе
прессованных полибисмалеимидов применяют в авиастроении для
изготовления деталей реактивных двигателей, кромок несущих
крыльев, обтекателей. В электротехнической промышленности из
них изготавливают штекеры, врезные шпонки. Фирма «General
Electric» выпускает термореактивную и пресс-массу под
названием «Джемон»; Джемон 2010 и ЗОЮ содержат в качестве
наполнителя 25 и 60 % стеклянного волокна. Джемон 2012 —
пресс-масса, наполненная 15 % графита. Прессование проводят при 170—
250 СС и давлении 140—420 кгс/см2. Протекающий при этом
процесс полимеризации не сопровождается выделением
низкомолекулярных продуктов [190, 379]. Из табл. 7.10 видно, что при 260°С
прочность при растяжении уменьшается на 14%, а относительное
удлинение при разрыве — на 10%. Цена 1 кг Джемона ЗОЮ
составляет 7,6 дол. США.
Олигоимиды с концевыми норборненовыми группами
перерабатываются по технологии, аналогичной технологии переработки
пресс-масс на основе бисмалеимидов. Композиции этого типа
с 1972 г, выпускаются фирмой «Ciba Geigy» под маркой Р 13N.
740
Продолжение табл. 7АО
1 iолибисм п л епм ид (на» алией н ый
стеклянным волокном)
Кинел 5504
Кинел 5514
масса
для
литьевого
прессования
коэффициент уплотнения
8-8,5
9. Ю15
22 _
4,5-5
1U16
1U14
18
3,5
Полиимнд с концевыми
нороирненивымн группами типа
Р13 N
неиапол-
менный
ю16
4- 1014
15
с 50«
(объеми.)
SiO2
1U'5
3- 1014
16,3
40%
(объемн.)
стеклянного
волокна
5- 1U15
2- 1014
14,2
Полинмид
Джемон
ЗОЮ с 60%
стеклянного
волокна
9-Ю15
3- 10i5
50
Прессование при температуре 270—315 °С и давлении 35—
210 кгс/см2 не сопровождается выделением летучих продуктов
деструкции. Для завершения процесса необходима выдержка при
нагревании до 1 ч [196]. Термостойкость изделий из таких пресс-
композиций не отличается от термостойкости полиостью
ароматических полиимидов. Диэлектрические свойства мало зависят от
температуры. Изделия на основе олигоимидов с концевыми нор-
борненовыми группами применяют в авиационной и
электротехнической промышленности.
Прессование порошков на основе полнамидокислот требует
специальной технологии и поэтому производится самими
производителями. Прецизионные прессованные изделия на основе
полиимидов выпускаются фирмой «Du Pont» под названием «Веспел».
Ненаполненные материалы — марки SP-1, SP-21 и SP-22 —
представляют собой композиции, содержащие 15 и 40 % графита;
SP-31 содержит 15% MoS2, a SP-5 —30% (объемн.) коротково-
локнистого стеклянного волокна [424] - В состав Веспел 211
входит 15% графита и 10% политетрафторэтилена [375]. Для
прессования частично-имидизованных полиамидокислот необходимы
продукты, совершенно не содержащие растворителя. Использование
полимеров, осажденных из растворов при двухстадийном
синтезе, связано с рядом трудностей, обусловленных наличием неэкст-
рагирующихся остатков растворителя. Подходящие
порошкообразные полна ми докислоты могут быть получены методом осадитель-
ной поликонденсации в смеси тетрагидрофуран — диоксан. Оба
растворителя хорошо растворяют диамин и диангидрид тетракар-
боновой кислоты, являясь нерастворителями образующейся поли-
741
Р, кгс/см г
1000
•800
Р, кгс/см2
OQQ
600 h
Рис. 7.23. Зависимость
относительного
удлинения при разрыве
от напряжения для
полиимидных
изделий, полученных
прессованием [376]:
а — ненаполнениые
полимеры; б —полимеры,
наполненные 25% графита.
амидокислоты. Высокая летучесть растворителей обеспечивает
легкость сушки осажденного продукта [257]. Для получения
полимера смесь диамина и диангидрида при перемешивании можно
добавлять к тетрагидрофурану. Растворение, поликонденсация и
осаждение следуют быстро друг за другом. В результате
образуется полиамидокислота, которая после экстракции эфиром и
сушки в вакууме измельчается в порошок. Перед прессованием нена-
полненный или наполненный минеральным наполнителем полимер
подвергают термообработке при 180°С в течение 30 мин [86,
126]. В ходе прессования порошок полиамидокислоты под
давлением 35 кгс/см2 нагревают до 200—220 °С, в течение 1 мин
давление повышают до 70—140 кгс/см2 и затем еще за 10 мин давление
увеличивают до 280—420 кгс/см2. После извлечения из
пресс-формы образец нагревают до 250°С в течение нескольких часов для
завершения процесса имидизации. Во избежание коробления
изделий из-за выделяющихся летучих продуктов окончательную
термообработку проводят в прессе под давлением 35 кгс/см2.
В одном процессе можно совместить синтез и переработку
полимера, нагревая смесь диангидрид—диамин в пресс-форме до
410°С при медленном повышении давления [94]. Для
промышленного производства втулок и вкладышей подшипников предпочитают
технологию, напоминающую порошковую металлургию [176].
Для этого частично-имидизованный полимер таблетируется в
прессе, ему придается заданная'форма, после чего в печи проводится
окончательное нагревание. Формованные изделия сложной
конфигурации подвергают дополнительной обработке, Возрастающая
пластичность полиимидов при увеличении гидростатического
давления облегчает их холодное формование в заготовку. Профили,
полученные экструзией под давлением 6000 кгс/см2, имеют
гладкую блестящую поверхность [180].
Свойства изделий, полученных спеканием, представлены в
табл. 7.10. Длительная работоспособность изделий составляет
300°С; кратковременно они выдерживают нагрузки до 500°С. Не-
наполненный материал имеет более высокую прочность при
растяжении, однако прочность на смятие выше у подшипников для
изделий на основе пластиков, наполненных графитом или M0S2.
Изделия из полиимидных композиций, содержащих 15% графита,
длительно работают при 300°С. Они могут использоваться для
изготовления подшипников и уплотнений, не требующих смазки и
742
Е,кгс/см2
5D000
4U0Q0
30000
20000
10000
200
150
100
SO
О 100 200
I
l4^ 1
0 200 400 600 800
Рис. 7.24. Зависимость модуля упругости при изгибе от температуры [376] для
полиимидных изделий:
/ — ненаполненные полимеры; 2 — полимеры, наполненные 25% графита.
Рис. 7.25. Зависимость прочности при сдвиге полиимидного клеевого соединения
от продолжительности термостарения при 315 °С (толщина клеевого соединения
1,6 мм, связующее соответствует рецептуре «а» в разделе 7.1.1.5 [404]:
/ — ненаполненный полимер; 2 — 5% As2S; 3—5% As2S-; 4 — 5% TiO2.
выдерживающих высокие нагрузки. При кратковременном нагру-
жении изделия работают до 100 ч при 350°С. Пластики,
наполненные графитом и политетрафторэтиленом, имеют прекрасные
антифрикционные свойства (при трении скольжения), однако
механическая прочность их ниже, чем у ненаполненных материалов [95].
На рис. 7.23 показано изменение прочностных свойств
ненаполненных и наполненных графитом пластиков на основе частично-
имидизованной полиамидокислоты, изделия из которых получены
прессованием. Модуль упругости при изгибе почти линейно
уменьшается с повышением температуры (рис. 7.24). На рис. 7.25
показано изменение прочности при сдвиге наполненного и ненаполнен-
ного клеевого соединения в условиях длительного старения при
315°С [404].
Данные о химической стойкости ненаполненных полиимидных
формованных изделий приведены в табл. 7.11. Тангенс угла
диэлектрических потерь при 50 Гц составляет 0,0015 при 23СС и
0,0815 при 264°С. Влияние различных факторов на трение
скольжения полиимидов изучено в работе [170]. Коэффициент трения
скольжения полиимидов не изменяется до 130 °С. При температуре
Таблица 7.11. Химическая стойкость изделий
из ненаполненных полиимидов [278]
Агент
HNO3
HNO3
HC1
NaOH
СО
CX
Я v^
° я
15
70
38
5
CXo
о
E я
<u >;
23
23
23
23
3"
о
о*
СО
720
120
120
120
о о
а рт
н о
0 и ^
66
37
71
56
Агент
N2O4 (газ)
н2о
тра-
О as
«К
О х
«о
S *
Н ь
23
23
100
100
№
S
о
о.
СО
120
720
400
1000
и
Se
о о
H о
О ci,
О н«
60
43
48
44
743
скользящей поверхности выше 150 С коэффицент трения
скольжения уменьшается на 25 % по сравнению с исходным значением,
оставаясь затем неизменным до 300 °С. В этом отношении поли-
имиды отличаются от всех термопластичных материалов. У поли-
метилметакрилата и полиамидов наблюдается постоянное
увеличение коэффициента трения скольжения с температурой. Поли-
имидные самосмазывающпеся подшипники имеют p-V =
= 120(кгс/см2) • (м/с). Износ при 400°С очень низкий. При
кратковременных нагрузках полиимидные подшипники могут применяться
вплоть до 540 °С. Для самосмазывающихся подшипников в
композицию рекомендуют добавлять политетрафторэтилен, графит или
MoS2 [424].
Прецизионные детали из наполненного полиимида применяют
в авто- и авиастроении для уплотнений, не требующих смазки,
седел клапанов и подшипников [131]. В турбинах фирмы «Rolls-
Royce» более 100 полиимидных вкладышей подшипников в
механизме привода работают без смазки в интервале температур от
—60 до 150°С [377]. В приводе двигателя «Боинга-747»
использованы полиимидные уплотнения-прокладки для различных
компрессоров. В пишущих машинках, калькуляторах и компьютерах по-
лиимиды используют для изготовления подшипников, роликов
скользящих и ведущих шин. Применение полиимидных деталей
в автомобилестроении для изготовления подшипников, уплотнений
и втулок экономически целесообразно, так как более высокие
затраты компенсируются более длительной непрерывной работой
этих изделий по сравнению с изделиями, изготовленными из
традиционных материалов.
7.1.1.9. Переработка прессованием и пресс-литьем
Полиимиды с мостиковыми группами в остатке ангидридного и
аминного компонентов (№ 155—236, табл. 7.2) и алифатическо-
ароматические полимеры (№ 30—55) в противоположность
ароматическим полиамидам групп 1—3 (№ 56—154) могут
перерабатываться прессованием и пресс-литьем как обычные термопласты.
Композиции этого типа выпускаются под названиями «Полиимид
XPI 182» («Cyanamid Co.»), Poly-X («Raychem»), NR-50 («Du
Pont»), ПМ-67 (СССР), «Полиимид ДФО СП-3, «Полиимид ДФО»
СП-6» (СССР) и Х-13 PI (ICI) [381,412,587].
Эти материалы перерабатывают при температурах выше
300°С. Вязкость расплава ароматического полиимида на основе
дифенилоксидтетракарбоновой кислоты и диаминодифенилоксида
(полиимид ДФО) составляет 104 Пз при 380°С [52] и алифати-
ческо-аромэтического полипиромеллитимида нонаметилендиамина
3-Ю6 Пз при 340°С [53]. Формование ДФО проводят при
370—390°С и давлении 500—2000 кгс/см2. Отпрессованные таким
образом изделия отличаются высокой термостойкостью (табл. 7.12).
Нагревание в течение 1 ч при 380 °С не приводит к изменению
прочностных и деформационных свойств; после 3 ч нагревания при этой
744
Таблица 7.12. Свойства полиимидных пресс-изделий,
полученных по обычной технологии [52, 379]
Т Tfii/- a a 51 то rt 1»
Прочность при растяжении, кгс/см2
при—150 °С
при 20 °С
при 200 °С
Относительное удлинение при разрыве, %
при — 150°С
при 20 °С
при 200 °С
Модуль упругости, кгс-/см2
при 20 °С
при 200°С
Ударная вязкость, кгс • см/см2
Диэлектрическая проницаемость при 50 Гц
Тангенс угла диэлектрических потерь
Электрическая прочность, кВ/мм
Удельное объемное электрическое
сопротивление, Ом-см
Водопоглощение, %
при 20 СС
при 100 °С
Полип иромел-
литимид
гексаметнлен-
диамина
1200
700
60
ПО
32 000
12 000
50—80
Полнимид на основе
днангидрмда
дифеннлоксидтетра-
карбоновой кислоты
и днаминоднфенил-
оксида
2 500
1500
800
40
80
120
32 000
12 000
1,0
0,002
100—200
1017
—
0,5-0,8
температуре прочность снижается на 10 %, а удлинение — на 50 %'.
Уменьшение массы при нагревании при 210°С в течение 3000 ч
составляет менее 0,05%. Выше 400 °С начинается образование
трехмерной полимерной сетки. Диэлектрические свойства мало зависят
от температуры. Прессованные изделия из ДФО характеризуются
высокой стойкостью к действию излучений. После облучения \~ЛУ~
чами дозой 10 000 Мрад прочность при растяжении и модуль
упругости не изменяются, относительное удлинение при разрыве
уменьшается на 20 %.
Ненаполненные и наполненные композиции на основе полиими-
дов этого типа используют для изготовления изделий авиационной
техники, таких, как самосмазывающиеся подшипники, уплотнения
[380], детали компрессоров и кулачки. В машиностроении и
электротехнической промышленности их используют для получения
конструкций, перекрытий и корпусов, выдерживающих большие
нагрузки [33, 381].
7.1.1.10. Полиимидные пенопласты
Полиимидные пенопласты сохраняют стабильность размеров в
широком температурном интервале, они негорючи и обладают
хорошими термо- и звукоизоляционными свойствами. Первые
пенопласты, описанные в патенте фирмы «Du Pont» в 1966 г.,
получали нагнетанием (под давлением) воздуха в сшитый полимер [413].
745
Фирма «Monsanto» выпускает с 1970 г. полиимидный
пенообразователь Скайбонд R1-7271 для получения материалов плотностью
0,016—0,3 г/см3 [176, 382—384, 414—417]. Предварительное
вспенивание полиамидокислоты проводят при 175 °С. Образующаяся
при этом оранжевая хрупкая пена при 300 °С за несколько минут
окрашивается в желтый цвет. Для количественной имидизации
время выдержки должно составлять 4—16 ч при 300 °С. Пено-
пласты, полученные на основе продуктов взаимодействия диангид-
ридов с ароматическими диизоцианатами, имеют низкую
теплостойкость.
Таблица 7.13. Свойства полиимидных пеиопластов [140, 383]
Показатель
А м- W ?\ Ы 4 U 1 V* ml л. и
Плотность, г/см3
Прочность при растяжении, кгс/см2
Относительное удлинение при
разрыве, %
Модуль упругости при растяжении,
кгс/см2
Прочность при сжатии, кгс/см2
на 5%
на 10%
на 25%
Модуль упругости при сжатии,
кгс/см2
при 20 °С
при 371 °С
Давление, необходимое для 10% -ио-
го сжатия, кгс/см2
при 93 °С
при 204 °С
при 232 °С
при 288 °С
при 316 СС
Термический коэффициент линейного
расширения при 20—300 °С, 1/°С
Поглощение света при
максимальном дымообразовании в ходе
пиролиза, %
Коэффициент теплопроводности, кал/
(смс°С)
при 22 °С
при 371 °(
Диэлектрическая проницаемость при
8,5 Гц
при 25 °С
при 148°С
при 303 °С
Тангенс угла диэлектрических
потерь при 8,5 Гц
при 25 СС
прн 148 °С
при 303 'С
Синтактические
пены
0,37
31,9
1,8
6450
3230
1420
2-10-5
2,39 • 10~*
3,10- 10
Пеиопласты
с открытыми i
0,107
0,98
1,48
2,81
1,40
1,40
0,84
0,70
0,14
0,215
6,60
9,49
16,52
8,43
7,03
6,32
2,46
0,14
3
торами
0,328
24,9
42,9
76,2
37,9
33,7
16,9
15,5
0,70
3
1,41
1,36
1,38
0,0062
0,0014
0,0019
746
Рис. 7.26. Зависимость прочности при растяжении Р,%{отисх.)
полннмидных пенопластов от температуры [140];
1 — гц-итактнчрская пснэ; 2 — гтенз с открытыми порзмз;. д
60\-
Синтактические пены на полиимидной
основе получены Рэндом [140]. Полиимид-
пый форполимер в N-метилпирролидоне 20
A5%-пый раствор) смешивают с полыми 100 150 200 250 зоо t/c
стеклянными микросферами и смесью
ацетангидрида с 4-бензоилпиридином в качестве
дегидратирующего средства. Циклизацию проводят в пресс-форме при 66 °С.
Композицию, состоящую из полиимида и полых стеклянных
шариков, нагревают в реакторе в среде инертного газа до 316 °С для
удаления растворителя, при этом полимер сшивается.
Физико-механические свойства полиимидных пенопластов с открытыми
порами приведены в табл. 7.13.
Пенопласт с открытыми порами и плотностью 0,016 г/см3
отличается высокой плотностью. Пены плотностью выше 0,10 г/см3
представляют собой твердые продукты, характеризующиеся
высокой стабильностью размеров [383]. Пены плотностью 0,012 г/см3
сохраняют сжимаемость и гибкость до —256 СС. Уменьшение
массы полиимидного пенопласта плотностью 0,009 г/см3 при
выдержке в течение 370 ч при 371 °С па воздухе составляет 30%.
Стойкость к термоокислительной деструкции уменьшается с
увеличением плотности пенопласта.
По свойствам синтактические полиимидные пенопласты
занимают промежуточное положение между пенополиуретанами и
керамическими или графитовыми пеноматериалами. По сравнению
с аналогичными пенами с открытыми порами они. имеют
значительно более высокие прочность и термостойкость, так как в этих
материалах полиимидная матрица играет только роль
связующего [140]. Свойства синтактических пен с соотношением
стеклянных сфер и полиимида, равным 60:40, приведены в табл. 7.13
и на рис. 7.26.
Пенополиимиды с открытыми порами используют в авиации
в качестве заполнителей в элементах сотовых конструкций, а
также как тепло- и звукоизоляционный материал [385]. Их
негорючесть и незначительное дымообразование в процессе пиролиза
являются их большим преимуществом по сравнению с обычными
пенопластами. Светопроницаемость через 120 с пиролиза пенопо-
лиимида с открытыми порами при плотности 0,2 г/см3 в
дымогарной камере составляет более 97% [383]. Высокие диэлектрические
характеристики пенопластов с открытыми порами обеспечили их
применение в радарной технике в качестве высокочастотных
изоляционных материалов. Применение негорючих, тепло- и
звукоизоляционных материалов в строительстве ограничено
специальными областями.
Синтактические пены применяют как термостойкий
теплоизоляционный материал на транспорте. Пенопласты этого типа обла-
747
Таблица 7.14. Температуры плавления (размягчения) и разложения полиимидов с гетероциклами
Элементарное звено
разл
(в N2),
разл
(на
воздухе),
—N,
О О
w
О О
N N
520
500
520
500
N N
>35О
—к
—n:
N
> 350
335
380
Продолжение табл. 7.14
Элементарное звено
раз л
* N?).
Т
раз л
(на воз-
2ь
Литературный
источник
N N
10
360
A951
380
[1951
360
[195}
11
12
13
14
О
Y
О
:n-R
N
II II
N N
-R-
R = арил, алкил; R' =
-(CH,)*-
О
¦N
О
О
о
О
О
О
O
[440]
[440]
485
500
[425, 426,
428]
[426]
Продолжение табл. 7,14
[Элементарное звено
(в Na).
разл
(на воз-
Литературный
источник
о
о
О
о
о
16
II
r-pOO
о о
Л 7
[426]
[426]
[426]
19
20
.SS
T
A Й
R = алкил, арил
, -(CHi)e-, -(СН2).
О Q
—n:
о о
N
N
[44<Ч
[4461
400
[28, 174)
Продолжение табл. 7.14
Элементарное звено
•'S1
Т
разл
UN,,.
т
разл
(иа воз-
о о
о
565
550
45O
510
24
25
26
27
О
о о
-< тот >
II 1
О
N N
О О
>
о
N N
О
DJ
575
625
590
575
570
625
575
Продолжение табл. 7.14
Элементарное звено
разл
(в N2).
•С
разл
(на воз-
о о
28
о о
29
30
О
о о
555
580
280
580
565
о о
31
32
33
О О
34
N
N
\
О
N
О
О
О
N
N
575
570
520
475
595
570
585
425
Продолжение табл. 7.14
35
36
37
0 0
II
i ,
О (
II
-Oo"
О (
(
1
Э <
1
i (
( K
i
Элементарное
N
о
о ч—^
звено
N
-C
N
5>-VSor
о
N
V
280
(в N2),
°C
570
T
разл
(на
воздухе),
°G
Литературный
источник
[432]
[432]
[432]
о о
38
4N-
о
о
о
580
570
о
39
-< юг >
О
[432]
О
40
<pQz®
N
О
N
R'
V
-О-, —СН2-, -С(СН3J-, [Q]
[438]
s
Продолжение табл. 7A4
Элементарное звено
1 разл
,bN2).
разл
(на воз-
духе),
41
42
43
О
О
N
о
N
560
305
610
570
570
625
44
45
46
47
N
N
N
О
N
590
555
520
280
560
550
550
Продолжение табл. 7.14
о
52
53
О
54
570
595
615
570
615
[430, 444]
[432]
N32]
Продолжение табл. 7.14
Элементарное звено
л пл1
°С
(в N2),
разл
(на воз-
Духе),
55
-N
О
56
О
57
N
ч
N
о
О
560
350
550
575
58
О
[454]
о о
59
N
о
500
[165, 43D
о о
N
60
ЧХОГ>
HN
540
550
[430]
6]
О
575
575
500
[427, 451]
Продолжение табл. 7.14
Элементарное звено
°С
Гразл
(в N2),
°С
^разл
(на воз-
>
Литературный
источник
62
о о
N
N
63
N
64
N
NH
530
510
480
D511
510
145Ц
N
NH
N
66
о
О
О
67
68
500
1451]
500
500
[451]
[165]
510
[451]
Продолжение табл. 7.14
71
480 [451]
72
О
О
""Y
HN
o
о
73
с—i
il
о
NH
О
О
О
74
—N
N
NH HN
О
о
N
N
О
75
о
^ HN
NH
NH
°-о-
О
О
500 | 1.451]
Продолжение табл. 7.14
Элементарное звено
раз л
(в N2».
разл
(на воз-
oyni
о
О
570
о
X = —S—
о
575
605
575
79 |
Si О О
80
О О
81
О О
о о
о
О-
о
-он-
о
—(( ))—О—
—о
[433]
СН,
СНэ
\_
1433]
о
О
[433]
)—о—
дают исключительно высокой способностью поглощать
кинетическую энергию при мгновенных нагрузках и быстро восстанавливать
свою форму [140].
7.1.2. ПОЛИИМИДЫ С ГЕТЕРОЦИКЛИЧЕСКИМИ ЗВЕНЬЯМИ
Получение сополимеров, содержащих наряду с имидным
другие гетероциклы, дает возможность существенно улучшить
многие их свойства (например, стойкость к окислительной деструкции,
физико-механические свойства, перерабатываемость). Большая
часть исследований в этой области относится к синтезу.полиокса-
диазол- и полибензоксазолимидов (табл. 7.14). Наиболее
значительный вклад в развитие направления, связанного с созданием
сополигетероариленов, внесли Коршак (ИНЭОС АН СССР),
Престон (США) и Годемари (Институт нефти, Франция). Волокна
на основе полиоксадиазолимидов и полибензоксазолимидов в
США выпускаются на пилотной установке фирмой «Chemstrand».
Верхняя температура длительной эксплуатации лежит около
300 °С.
7.1.2.1. Получение полиимидов с гетероциклами
Поликонденсация ароматических диангидридов с
гетероциклическими диаминами. Полиимиды с гетероциклическими звеньями
в цепи синтезируют двухстадийным способом с получением на
первой стадии в растворе полиамидокислоты путем
взаимодействия диангидридов тетракарбоновых кислот с ароматическими
диаминами, в структуре которых уже содержатся готовые
гетероциклы. Промежуточный продукт при нагревании или в результате
химической имидизации превращается в соответствующий поли-
гетероарилен. Процесс может быть представлен следующей
схемой:
О О
N
Полибензоксазоламидокислота
О
о
—п
Лолибензоксаэолимид
772
Этим методом были получены полиоксадиазолимиды [425, 427,
428], политиазолимиды [427, 429], политриазолимиды [427, 429],
полмбензоксазолимиды [430—432]. полибензтиазолимиды [430—
432], полибензимидазолимиды [426, 427, 429, 430], а также поли-
имиды, содержащие в цепи сульфоэфирные групппы [433].
Синтез диаминов с бензоксазольными группами описан в [435, 436],
а с бензтиазольными — в [436]. Сополимеры получают также го-
мополиконденсацией ангидридов ароматических ы-амино-о-дикар-
боновых кислот, содержащих в молекуле гетероциклы [437]:
nH2N
—н2о
-_п
Применение диаминов, содержащих гетероциклы, для
получения сополимеров имеет то преимущество по сравнению с другими
методами синтеза, что в этом случае циклизация количественно
протекает при температуре ниже 300 °С. Полиоксадиазолы, поли-
бензоксазолы, полибензимидазолы могут быть получены через
стадию образования соответствующего форполимера, однако в
инертной среде даже при 350 °С не удается добиться
количественной циклодегидратации. При синтезе же полибензтиазолов
промежуточные продукты выделить не удается [432].
Синтез сополимеров из исходных соединений, содержащих имид-
ные циклы. Сополимеры с гетероциклами можно получать лг
основе исходных продуктов, в состав которых входят имидные
циклы например [440]:
nH2NNHCR—N
II
О
—Rr.NHNH2+ nClOC—(())—СОС1
у—у —нс\
773
о
о
—CONHNHCOR—
о
j— RCONHNHCO
о
о
N N
-in
Полиоксадиазолимиды можно синтезировать также на основе
бистетразолимидов через промежуточную стадию образования по-
литетразол имидов:
О О
HN— N.
N—NH
:r—n
ClCOR'COCl | —НС1
О О
1-
N— N— COR'CO-
\NsasN
i-
О
N N
О
N N
-R-N
_!rt
Синтез сополимеров из исходных соединений, не содержащих
гетероциклы. В этом случае в качестве промежуточного продукта
получаются полиамидокислоты, содержащие кроме СООН-групп
и другие функциональные группы. Поэтому в ходе циклизации
наряду с имидизацией происходит замыкание и другого гетероцикла.
774
Полиоксадиазолимиды могут быть синтезированы гомополи-
конденсацией ангидридов ш-аминодикарбоновых кислот,
содержащих гидразидные группы [437], или поликонденсацией гидразида
гс-амннобензойной кислоты с дихлорангидрпдами дпкарбоновых
кислот [453], например
H2N
Q>—coNHNHa +
1
—coNHNHoc—an—conhnhco—<
—coci
0—25 °C
0—25 °C
—UN-
-CONHNHOC—
—CONHNHCO—'
NHCO
HOOC
—H2O 150—170 °C
—nQN—CONHNHCO—^Q)—CONHNHCO—
>I70°C
N N
NH2
CO-
cooh n
N—
n
Циклизация полиамидокислот с гидразидными фрагментами
протекает при 150—170°С с образованием полиимидогидразида,
который превращается в полиоксадиазолимид при температуре
около 200°С [453]. Полибензоксазолимиды получены Коршаком
775
с сотр. [438] и Токаревым [439] совместной поликонденсациеи
бис-о-аминофеиолов, диангидридов тетракарбоиовых кислот и
хлорангидридов дикарбоновых кислот:
О о
H2N
! R 1
О + rcCIOCR'COCl
1
-—HN,
NHOC
НО НООС
CONH
(/~^) NHCOR'CO—
соон он
—П
R—¦
Докоши [454] предложил способ синтеза, при котором
гетероциклическая группа (бензоксазол, бензимидазол, бензтиазол или
бензоксазинон) содержится в диангидридном компоненте. При
взаимодействии ангидрида тримеллитовой кислоты с о-дизаме-
щенными ароматическими диаминами при низких температурах
образуется диангидрид. Для конденсации используют диамины,
содержащие в качестве о-заместителей NH2, ОН, SH или СНО-
группы, которые при 200—280 °С участвуют в процессе
формования другого гетероцикла:
<40рС
776
7.1.2.2. Свойства полиимидов с гетероциклами
Полиимиды, содержащие различные гетероциклы, отличаются
высокой степенью упорядоченности; это соединения от желтого
до коричневатого цвета, не имеющие четкой точки плавления.
Эндотермический пик на кривой ДТА, соответствующий
«плавлению», совпадает с температурой разложения полимеров,
приведенной в табл. 7.14, которая охватывает область от 280 до 600 °С. По
влиянию химического строения остальных фрагментов на свойства
такие полиимиды не отличаются от карбоциклических
ароматических полипмидов: полимеры с «-замещенными ароматическими
ядрами имеют более высокую температуру «плавления», чем м-за-
мещенные полимеры; группы, способствующие увеличению
гибкости, находящиеся между циклами, снижают температуру
размягчения. При температуре разложения 580—625 °С в инертной среде
и вакууме самую высокую термостойкость из всех полимеров,
содержащих кроме имидного другой гетероцикл, имеют полибен-
зоксазолимиды (табл. 7.14, № 24—51). Термостойкость в инертной
среде и вакууме уменьшается в следующем ряду: полибензоксазо-
лимнд >> полибензтиазолимнд > поли-1,3,4-оксадиазолимид > по-
либензнмидазолимид ;> карбоциклические ароматические
полиимиды > поли-4-фенил-1,2,4-триазолимид. В ином порядке эти
полимеры выстраиваются по уменьшению стойкости к
термоокислению: полибензоксазолимид > полибензтиазолимид > поли-1,3,4-
оксадиазолимнд > карбоциклические ароматические полиимиды >
поли-4-фенил-1,2,4-триазолимид >• полибензимидазолимид > по-
лнтназолимид.
В противоположность ароматическим полиамидам с
гетероциклическими группами E.10.4) термостойкость внутри отдельных
групп подобных сополиимидов изменяется мало. Полиимиды с
гетероциклическими звеньями в цепи, имеющие низкую температуру
плавления (температура перехода, связанная с плавлением, в
области температур 280—350 °С, табл. 7.14, № 43,48, 57), устойчивы в
вакууме до 500°С. В то же время низкоплавкие гомополиимиды
начинают деструктироваться лишь немногим выше их температуры
плавления [443]. Полибензоксазолимиды и полибензтиазолнмпды
выдерживают кратковременный нагрев до 500 °С. Бензоксазольные звенья
начинают разлагаться при температуре около 550 °С, в то время как
для нмндных циклов этот процесс протекает уже при 500 °С [141].
777
q Таблица 7.15. Физико-механические свойства волокон из полиимндов с гетероцикламн в основной цепи [425, 430, 442, 453]
Элементарное звено
Прочность
при
растяжении,
г/деиье
Относительное
удлинение
при разрыве,
Модуль
упругости,
г /денье
< loi xo
о
о
О
5,2
2,1
1,4
0,8
3,9
1,0
3,5
6,1
6,0
3,7
1.4
ПО
44
58
31
19,1
18,0
100
42
3,4
164
о
о
—N
о
' "Ч
N N
О
6,3
1,4
0,7
0,4
9,7
9,7
7,8
3,1
122
11
О О
О
N
-iuf y о-
о
О
2,8
25
130
О О
6
N
-OsO-iotv
О
N
О
О
4,8
2,4
1,6
1,1
5,1
10,2
6,8
4,2
168
57
67
Продолжение табл. 7.15
Элементарное звено
Прочность
при
растяжении,
г/денье
О
о
О
о
О
о
о
о
Относительное
удлинение
при разрыве,
О
о
О
о
О
о
О
О
Модуль
упругости,
г/деиье
О
о
о
см
о
§
СП
о
о
о
о
8
5,2
2,0
1.1
4,0
5,0
3.6
6,4
2,0
1.5
5,8
14,9
15,7
о
О
О
—N
IQL
\
у
N
4,7
1.8
1,7
0,6
19,3
22.6
30,5
О
148
63
32
203
65
32
57
28
15
О Ю 20 30 10 50 т,сут
Рис. 7.27. Зависимость прочности при растяжении волокон из карбо- и
гетероциклических полиимидов от продолжительности термостарения на воздухе при
300 °С П77];
О О
3 —
__ п
Полиимиды с гетероциклическими звеньями не растворяются
в чистых органических растворителях. Полиоксадиазолимиды
растворяются в N-метилпирролидоне, диметилацетамиде, диметил-
формамиде с добавками LiCl [447]. Сополимеры с ариленсульфо-
ноксидными блоками в цепи (табл. 7.14, № 79—81) хорошо
растворяются в диметилформа-миде, диметилацетамиде, гексаметил-
фосфотриамиде [433]. Физико-механические свойства волокон на
основе полиимидов с гетероциклическими звеньями в цепи
приведены в табл. 7.15. Самой высокой прочностью на воздухе при
растяжении при 500°С A,7 г/денье) обладает полибензоксазолимид-
ное (табл. 7.14, № 38) волокно.
На рис. 7.27 сопоставляется прочность полибензоксазолимид-
ных, полиоксадиазолимидных и ароматических полиимидных
волокон при термическом старении на воздухе при 300°С. Симметрия
гетероциклических диаминов не оказывает влияния на термостой-
7O1
Таблица 7.16. Температуры размягчения, плавления и разложения в азоте и на воздухе полиамидоимидов
Элементарное звено
разм*
(в N2)
разл
(на воз-
Литературный
источник
О
ч
о
о
<
420
i168, 479]
460
[479]
360
[464,468,
479]
о
о
о
I!
l
Полимер AJ-10 («Amoco»)
О
NH—C-
II
О
о
)T
410-420
1468]
350
300
|461,468t
476, 479]
465]
Продолжение табл. 7.16
№
Элементарное звено
разм1
°С
Т
разл1
boN2)
т
разл
(на
воздухе),
°С
О
<-<O>-NH-?
il
о
f
|—С—NH—((у)—NH—С-
« ^у ii
о о
о
о
-NH— С—j
II
О
: С—NH-
II
О
i—NH—C-
II
О
о
о
О
500
460
420
450
430
320
о
10
О
о
11
О
о
о
12
-о-
О
О
13
200
120
300
300
300
300
[480]
[480]
[480]
[480]
П родолжение табл. 7ЛЬ
Элементарное звено
размр
т
разл'
(в N2),
разл
(на воз-
духе>,
°С
Литературный
источник
о
о >-о-сн.-<о -
О
О
о—с4н9
О—СбН5
о
О-С4Н9
О— С6Н5
180
150
250
300
[480]
[480]
[497]
о
17
M—(CH2)e—NH—С—СН2-
II
О
65
о
о
о
18
^' У
N
-О-
N
о
о
о
о
о
о
19
ГоГ>-
С—NH-
II
О
о
>360
400
> 360
420
О
О
20
1оп-ын-
-©-
О
О
> 360
410
Продолжение табл. 7.16
Элементарное звено
разл
т
разл
(на воз-
дух?).
Литературный
источник
о
II
285
> 360
400
[189]
380
[462]
400
[478,484]
о
о
24
д
о
о
25
26
о
27
О
I С—NH-
(I
О
NH—С—
II
о
о
о
О
о
380
[189,478]
О
О
> 360 I 500
NH—С—
> 360 | 410
450
420
[478]
! [478, 484]
[484]
о
Продолжение табл. у .16
Элементарное звено
Г
раз м
шг
разл
\в
разл
(на
воздухе*.
°С
Литературный
источник
о
о
С—NH— (СН2)в— NH—СО—
368
О
311
-NH—С—
II
О
I
300
[478]
[455]
425
14551
31
32
33
34
О
О
О
О
о
о
О
О
о
I
о
о
о
о
370
480
[455]
310
440
[455]
307
324
470
-NH—С—
II
О
| 500 | 450
-NH—С—
II
О
[455]
[455]
CD
Продолжение табл. 7.16
Элементарное звено
разм*
°С
пл1
°С
разл
(в N2>,
Т
разл
(на воз-
Духе),
Литературный
нсточник
35
36
37
о
о
II
\
N _
О
о
о
Л^-с-кн-
^
О
о
о
355
425
350 440
о
280
355
420
О
[484]
[484]
[458, 462,
472—475,
489]
I I I
38
39
40
41
о
о
О
о
о
о
о
СН,
il
о
-NH—C-
II
О
—мн—с-
II
о
О
350
1462]
390
[478]
420
'478]
360
[478]
Продолжение табл. 7.16
Элементарное звено
разм1
"С
Т
разл
(На
воздухе),
°С
42
N-(CH2N-]
1
С—NH—(СН2)б—NH—С—
II II
О О
о
о
43
J—NH—С—((_))—C-NH-
o ^ о
О
44
—NH—
280
310
290
о
45
—NH— С—(СН2)«—С—NH—
46
240
1512]
500
420
S463]
47
500
420
1463]
Продолжение табл. 7Л6
Элементарное эаено
разм1
°С
разл
(в N2),
(на
воздухе),
°С
43
—n:
49
:n-r-
N N
NH—С—
О
300
350
50
N N
a
О
51
О
52
N N
О
N N
380
[467]
370
[4671
380
D83]
кость полимеров на их основе, однако физико-механические
свойства полиимидов на основе асимметричных диаминов (по
определению Престона, «сополиимиды с ограниченной
упорядоченностью») значительно хуже, чем у продуктов на основе
гетероциклических диаминов симметричного строения (по Престону —
«полностью упорядоченные гетероциклические сополимеры» [430]).
Ниже приведены физико-механические свойства полибензокса-
золимидной пленки, полученной методом полива (табл. 7.14,
№ 50) [454]:
Тангенс угла
диэлектрических потерь
при 1 кГц
при 22
при 180
при 1 Мгц
при 22 °С
при 180°С
Электрическая прочность,
кВ/мм
Удельное электрическое
сопротивление
объемное, Ом см
при 22 °С 2
при 180 °С 7
Прочность при растяжении,
кгс/см2
при • 22 °С
при 200 °С
Относительное удлинение
при разрыве, %
при 22 °С
при 200 °С
Модуль упругости при
растяжении
при 22 °С, кгс/см2 . . .
Прочность при
многократном изгибе
при 22 °С, циклы . . .
Плотность при 25 °С, кг/см3
Диэлектрическая
проницаемость при 1 кГц
при 22 СС
при 180°С
при 1 МГц
при 22 °С
при 180°С
1800
900
21
48
35 300
30 000
1,41
3,4
3,1
3,3
3,0
°С
°С
0,0038
0,0041
0,0068
0,0033
210
101а
ю17
поверхностное при
22 °С, Ом
Химическая стойкость, %,
после выдержки
в воде B4 ч при 22 °С)
в 10%-иой щелочи E ч
при кипячении) . . .
в конц. H2SO< B4 ч
при 30°С)
9-10'5
0,9
17
3.5
7.1.3. ПОЛИАМИДОИМИДЫ
Полиамидоимиды представляют собой сополимеры, сочетающие
свойства полимеров этих двух классов:
Г—ОС—гЯ\Ч—CONH-
NH—
—П
-in
798
Они получаются поликонденсацией ароматических поликарбоновых
кислот и диаминов, диизоцианатов или дигидразндов. Верхний
температурный предел эксплуатации в условиях длительного
нагревания составляет 240°С. В промышленном масштабе их выпускают
с 1964 г. Основные области использования: связующие для
армированных пластиков, кабельная, изоляция, волокна и пленки.
7.1.3.1. Получение полиамидоимидов
Впервые полиамидоимиды были получены в 1942 г.
взаимодействием диаминов с трикарбоновыми кислотами [517]. Однако
интенсивные исследования в этой области в СССР, США, Франции и
Японии начались только в 1960 г. Их промышленное производство
начато в 1964 г. с выпуска фирмой «Атосо» продукта Полимер
A J.
Основные характеристики полиамидоимидов приведены в
табл. 7.16, названия технических продуктов, производимых в
настоящее время, приведены в табл. 7.17.
В качестве исходных веществ для синтеза этих полимеров
применяют полиамины и поликарбоновые кислоты, в состав которых
входят амидные или нмидные группы. Процесс проводят, как
правило, в апротониых биполярных растворителях, таких, как диме-
тилформамид, диметилацетамид, диметилсульфоксид, N-метилпир-
ролидон, при комнатной температуре. Образующаяся в качестве
промежуточного продукта полиамидокислота характеризуется
гораздо более высокой стойкостью растворов, чем форполимер,
получаемый при синтезе полиимидов. Процесс циклизации не
отличается от имидизации обычных полнамидокислот — его проводят
Таблица 7.17. Торговые марки полиамидоимидов
и их фирмы-изготовители
Страна
США
США
Франция
ФРГ
Япония
Торговая марка
Полимер AJ-10
Полимер AJ-11
Торлои 3000
Торлон 4000
Аманим
Арамидил
Кермел 201, 203, 233?
234,
Родефтал 310,311
Родефтал 101, 200
Феногараит
HJ-400, HJ-401, HJ-200
Фирма-изготовитель
«Amoco Chemicals»
«Westinghouse»
«Rhone Poulenc»
То же
«Schramm»
«Hitachi»
Применение
Электроизоляционные лаки
Клеи
Пластики
Плеики
Клеи
Волокна
Электроизоляционные лаки
Лаки горячего
отверждения
Лаки
Кабельная изоляция
Лаки горячей
сушки
79Э
либо химической циклодегидратацией, либо нагреванием при 200—
280 °С.
Поликонденсация хлорангидридов и ангидридов трикарбоновых
кислот с диаминами. Поликонденсацию ароматических диаминов
с хлорангидридами ангидридов ароматических трикарбоновых
кислот, главным образом хлорангидридоангидридом тримеллитовой
кислоты [459, 473—475, 514—516, 518—524], проводят в растворе
диметилацетамида, N-метилпирролидона, диоксана, нитробензола
или бензонитрила при 0—40°С или на границе раздела двух фаз
метилэтилкетон — вода [473—475].
—CH
-NH,
—HGi
Cj)—СН2—(Q)—NH~
-н2о
п
-oc^
—CH
—NH-
Эта схема иллюстрирует процесс получения полиамидоимида
марки «Полимер AJ-10», выпускаемого фирмой «Атосо».
Использование таких акцепторов НС1, как триэтиламин или карбонат
калия, позволяет получать высокомолекулярные полиамидокислоты.
Межфазная поликонденсация, протекающая с высокой скоростью,
приводит к образованию высокомолекулярных, легко
выделяющихся из раствора полиамидокислот. Кроме того, достоинством этого
метода является возможность применения менее чистых
исходных веществ [473, 475].
Использование л-аминобензойной кислоты или изофталоилхло-
рида позволяет варьировать в любых пределах соотношение амид-
ных и имидных звеньев [492, 513]. Сшитые полиамидоимиды
получают хлорированием полиамидокислот SOC12, PCI или РСЦ с
последующим взаимодействием с диаминами [498]. При добавлении
к раствору полиамидоимида в N-метилпирролидоне раствора диан-
гидрида тетракарбоновой кислоты в осадок практически мгновенно
выпадает гелеобразный продукт. Если этот гель нагреть до 100—
120 °С, то получится стабильный раствор, из которого можно
формовать эластичные пленки [476]. Схема получения подобных поли-
800
имидов приведена ниже:
—NHCO—
о
У
X
о
\
о
11
-Метилпирролидон
О
IЮОС
GONH—
CONH
,-снг-(О)-
_H2N—.
— п
NHOC
о
—П
Взаимодействие ангидридов трикарбоновых кислот с диизоци-
анатами. Поликонденсация ангидридов ароматических
трикарбоновых кислот с ароматическими диизоцианатами сопровождается
выделением СОг и приводит к получению полиамидоимида в одну
стадию [458, 468, 470—472, 486, 494, 532—534, 543—545] :
О
СН2—/QN—NCO
-со2
СНо—
NH—
Процесс проводят в таких растворителях, как диметилформ-
амид, диметилацетамид, N-метилпирролидон, нитробензол, бензо-
нитрил или ацетофенон. Из-за высокой реакционной способности
26 Зак 16
801
изоцианатов к реагентам и растворителям предъявляются очень
жесткие требования по чистоте. Содержание влаги в реакционной
смеси должно быть менее 0,05%. Раствор постепенно нагревают
до 160—180 °С и по достижении необходимой вязкости реакцию
прекращают. Скорость выделения СО2 достигает максимума при
125—130 °С и уменьшается до нуля при 140—180 °С. Реакционно-
способные изоцианатные группы количественно реагируют за 1ч
при 120°С [468]. Полученный раствор полимера стабилен при
комнатной температуре и может быть использован для
непосредственной переработки в пленки, волокно и кабельную изоляцию.
Наиболее высокомолекулярные продукты получают при проведении
процесса при 140 °С в нитробензоле с использованием в качестве
катализатора триэтилендиамина [472].
Частичная замена трикарбоновои кислоты на диангидрид тетра-
карбоновой, или хлорангидрид дикарбоновой кислоты [458], или
я-аминобензойную кислоту [494, 503], а также диизоцианатов на
диамины позволяет варьировать соотношение амидных н имидных
групп, а следовательно, и свойства сополимеров [488, 501]. При
получении электроизоляционных лаков применяют избыток диизо-
цианата. В процессе горячей сушки наряду с увеличением
молекулярной массы происходит структурирование полимера за счет ци-
клотримеризации по свободным изоцианатным группам.
Поликонденсация с использованием мономеров, содержащих
амидные группы. При взаимодействии диангидридов тетракарбоно-
вых кислот с дигидразидами ароматических дикарбоновых кислот
[512, 527—529, 541] или амидогидразидами [530, 531] получаются
полнамидоимиды с хорошим выходом:
О
О -Н nH2NNHOCRCONHNH2
i i
rHOOC^ ^^ ^CONHNHOCRCONHNH—
L —OC
I -H°°
NNHOCRCONH—
—ft
Поликонденсацию проводят в биполярных апротонных
растворителях (диметилформамид, диметилацетамид или диметилсульф-
оксид). Циклизация количественно протекает за 15 мин при 200—
280 °С и остаточном давлении 2 мм рт. ст. Трудность представляет
802
получение высокомолекулярного полимера. Таким
были синтезированы полиимидомочевины [542]:
О О
же способом
—П
Другой способ основан на использовании диаминов,
содержащих в своем составе амидные связи, которые при взаимодействии
с диангидридом в растворе образуют сначала полиамидокислоту,
которую термической или химической циклодегидратацией
превращают в полиамидоимид [460, 514, 515] :
О О
;о + nU2N—
CONH-
NHG
—NH2
_ нею
Cv^ /ч. /CONH
Ж
I
CONH—Г^ч|—NHCO
,—NH—
GOOH
—o:
J-H.0
о
— П
В качестве диаминов используют диаминобензанилиды [459],
олигоамиды с концевыми аминогруппами, диамины с азогруппами,
полученные окислительной конденсацией [485], амины,
содержащие, наряду с амидными группами также гетероциклические
26*
803
группы [525]. Применение гидразидов аминокарбоновых кислот
с частичной заменой тетракарбоновых кислот на дихлорангид-
риды дикарбоновых кислот позволяет получать полиамидокис-
лоты с гидразидными группами, которые при термической или
химической циклодегидратации превращаются в полиамидоимиды
с оксадиазольными звеньями в цепи [457]. Применение диангид-
рида бензофенонтетракарбоновой кислоты приводит к получению
разветвленных или сшитых полимеров с С=Ы-связями. Синтез
полиамидоимидов возможен и при использовании в качестве
кислотных компонентов диангидридов с амидными группами в цепи:
О О
О
[
ноос>
-NHOC-
—N
CONHArNHC
Такие диангидриды получают взаимодействием ангидрида три*
меллитовой кислоты с диацетильными производными диаминов
[116, 509,526]:
О О
СООН
НзСОСШАгШСОСН3 + О
I-
СООН
СНзСООН
CONHArNHOC
804
Поликонденсация с использованием мономеров, содержащих
имидные циклы. Дикарбоновые кислоты или их хлорангидриды,
содержащие имидные циклы, взаимодействуют с аминами или ди-
изоцианатами с образованием полиамидоимидов [189, 478, 495] :
о
HOOCR—N^ [Qj ^N—RCOOH + H2NArNH2
—OCR—N
H2O
N—RCONHArNH—
О
При использовании полифункциональных аминов получают
сшитые полимеры с высокой термостойкостью [459] :
N-R-N
CONH—,
N—R-
Применение в качестве кислотного компонента дихлорангид-
рида следующего строения
СЮС
СОС1
805
приводит к получению полиамидоимидов с повышенной
растворимостью [455].
При взаимодействии диангидридов, полученных из трикарбо-
новых кислот, с /г-аминобензойной кислотой образуются дикарбо-
новые кислоты с имидными циклами:
соон
НООС—
соон
о
Реакции с диизоцианатами приводят к получению
полиамидоимидов с ангидридными группами в цепи [496, 505]. При
взаимодействии имида тримеллитовой кислоты с диизоцианатами
образуется полиамидоимид (в одну стадию) [504].
В синтезе полиамидоимидов можно также применять диамины,
содержащие имидные циклы. Из подобного диамина и дихлоран-
гидрида дикарбоновой кислоты при 25—40 °С в диметилацетамиде
в присутствии пиридина или пропиленоксида получен
полиамидоимид [463]:
/tH2N—гЯ^|—NHC
N—fpN|—NH2 + rtClOCRCOCI
I
—HN—i
NHCORCO—
—я
806
Взаимодействие имида тримеллитовои кислоты с диацетильны-
ми производными диаминов приводит к получению бисимидов [484]:
О
соон
+ H3COCNH
ноос
ШСОСНз
\
I-
СНзСООН
CONH
NHCO
NH
Поликонденсация таких диимидов и диаминов в ж-крезоле или
смеси ж-крезол — диметилацетамид при 150—180 °С протекает в
течение 8—24 ч с отщеплением аммиака, при этом получается поли-
амидоимид с выходом 30—70 % •
nHN
CONH— R'— NHC
NH -f /iH2NRNH2
CONH—R'—NHCO
NH3
CONH2 -i
CONH—J,
-R-n;
CONH—R'—NHCO—I
n
807
Полиамидоимиды получают и реакцией диимидазолидов,
содержащих имидные циклы, с диаминами [481] :
о * о
N
п
N—СО—'
СО—N
H2NRNH2 130 °С
—ОС
-< IQ
-CONHRNH—
-In
7.1.3.2. Физико-химические свойства полиамидоимидов
Полиамидоимиды представляют собой порошки желтого цвета
плотностью 1,2—1,6 г/см3. Температуры размягчения и плавления,
растворимость и термическая стойкость полиамидоимидов зависят
от соотношения амидных и имидных звеньев, порядка их
чередования в макромолекуле, содержания алифатических и «шарнирных»
групп в цепи.
Полимеры с л-фениленовыми фрагментами размягчаются при
300—400°С. Наличие в боковой цепи алкоксигрупп и ароксигрупп
(табл. 7.16, № 13—15) обусловливает резкое понижение
температуры размягчения. Полиамидоимиды, содержащие в цепи только
фталимидные, амидные и /i-фениленовые фрагменты, плавятся и
разлагаются при температуре выше 360 °С (табл. 7.16, № 18—20,
23, 26). При наличии в цепи мостиковых групп типа О, SO2, CH2,
С(СНзЬ температура размягчения лежит ниже 350 °С.
В отличие от ароматических полиимидов полиамидоимиды
растворяются в полярных растворителях, таких, как диметилформа-
мид, диметилацетамид, диметилсульфоксид, N-метилпирролидон,
иногда с добавкой LiCl. Полиамидоимиды с мостиковыми гибкими
группами или другими гетероциклическими звеньями в цепи между
ароматическими ядрами растворяются также в фенолах и тетра-
гидрофуране [355, 507], а полученные из о-замещенных
диаминов— в хлороформе и диоксане (табл. 7.16, № 13—15). Такие
растворы в противоположность тем, которые получаются при синтезе
форполимеров для полиимидов G.1.1.3—7.1.1.7), стабильны при
хранении при комнатной температуре в течение 6 мес.
По термостойкости полиамидоимиды занимают промежуточное
положение между ароматическими полиимидами и ароматическими
полиамидами. Температура начала разложения полиамидоимидов
808
200 400 600 800t,°C
10000 Z.4
Рис. 7.28. Кривые термической деструкции полиимида (/) и полиамидоимида
B) в среде азота [358]:
О О
2-
—п
у
-< о
о
Рис. 7.29. Кривые термической деструкции полиамидоимида на основе тримелли-
тового ангидрида и диаминодифенилметана на воздухе [537J.
в зависимости от химического строения в инертной среде находится
между 400 и 500 °С, а на воздухе составляет 350—450 °С.
Сополимеры алифатических диаминов на воздухе разлагаются уже при
300°С [512, 535]. Более низкая термостойкость полиамидоимидов
по сравнению с ароматическими полиимидами, как это видно из
рис. 7.28, связана с меньшей жесткостью цепи, меньшей
симметрией структурных элементов и относительно высокой
подверженностью амидных групп окислению. Термостойкость их прямо
пропорциональна содержанию в полиамидоимидах имидных циклов
[458].
На рис. 7.29 приведены данные изотермического ТГА полиими-
дов на основе ангидрида тримеллйтовой кислоты и 4,4'-диаминоди-
фенилметана на воздухе при 205 и 260 °С. По убыванию
термической и термоокислительной устойчивости полиамидоимидов в
зависимости от строения мостиковых групп их можно расположить
в следующий ряд [536]: имид > —О— > —S— > —SO2— >
> _NH~ > —СН2— > —С(СН3J— -
Введение в структуру сульфоновых и оксадиазольных групп
способствует повышению термостойкости. Полиамидоимид на основе
бис (Ы-4-фенилен-4'-хлорформилфталимид)сульфона имеет темпе-
809
ратуру разложения 450 °С и стабилен на воздухе при 400 °С в
течение 6 ч (табл. 7.16, № 34). Термическая деструкция полиамидоими-
дов начинается с относительно слабых амидных связей. В составе
продуктов деструкции полиамидоимида с простыми эфирными
связями в цепи (табл. 7.16, № 11) при 400 °С обнаружены СО2 —
83,54%, СбНе —9,6%, СО —6,8% и Н2 — 0,6% [480].
7.1.3.3. Полиамидоимидные волокна
Полиамидоимидные волокна с длительной работоспособностью
до 260 °С выпускаются с 1971 г. фирмой «Rhone Poulenc»
(Франция) под названием «Кермел». Исследовательские работы в этом
направлении были начаты в 1963 г. При взаимодействии
ангидрида тримеллитовой кислоты, полученного из триметилбензола, с ди-
изоцианатами, преимущественно с дифенилметандиизоцианатом, и
диаминами в диметилформамиде и диметилацетамиде получают
полиамидоимид [468, 539, 540]. Из профильтрованного раствора
можно формовать волокно сухим или мокрым методом.
Сухое формования проводят в обогреваемой вертикальной
колонне. Растворитель нагревается током горячего газа выше
температуры кипения, испаряется и регенерируется. Сушка волокна
проводится при 240°С и остаточном давлении 1—50 мм рт. ст. [510].
Осадительной ванной при мокром формовании служит смесь
растворитель — вода, например N-метилпирролидон — вода C0 :70)
[508]. Полученное в начале гель-волокно вытягивается в 1,5 раза,
промывается и высушивается. Для повышения степени
упорядоченности волокно подвергают вытяжке в 4—7 раз при 380 °С [508,
510]. Фирма «Rhone Poulenc» выпускает волокна Кермел 201, 203
и 233, а также штапельное волокно (Кермел 234). Прочность при
растяжении волокна составляет 25 и 60 г/текс, относительное
удлинение при разрыве 10—20 %, модуль упругости 500—900 г/текс.
В табл. 7.18 приведены данные о свойствах полиамидоимидного
волокна Кермел. Волокно разлагается на воздухе при температуре
выше 380 °С. Потеря массы через 2 ч при 350 °С на воздухе
составляет 5% и при 380°С — 8—10%- Полиамидные волокна с имид-
ными циклами в цепи имеют длительную температуру
эксплуатации при 260 °С. Остаточная прочность после 1000 ч работы при этой
температуре составляет 80 % для Кермел 201 и 40 % для Кермел
203.
Так же как полиамиды, полиамидоимиды нестойки к действию
УФ-лучей. Они негорючи, при контакте с открытым пламенем на
воздухе только коксуются, причем без выделения токсичных газов.
Кислородный индекс (минимальное количество кислорода, при
котором материал воспламеняется) составляет 21 %. В нейтральных
средах полиамидоимидные волокна устойчивы к гидролизу. Через
50 ч пребывания в воде при 160°С потеря прочности волокна
Кермел 203 составляет 35%. Вследствие недостаточной
гидролитической стойкости имидных циклов полиамидоимидные волокна
разрушаются в щелочных средах, особенно при нагревании.
610
Таблица 7.18. Свойства полиамидоимидных волокон [468, 540]
Показатель
Непрерывное
марок
201
1,39
50-60
30
25
45-50
45—50
45-50
35-40
8-10
960
2,1
0,29
0
0
ВОЛОКНО
203
45-50
18
40
20
12
2.2
0,2
Рубленое
волокно
марки 231
1,34
40-45
20
15
20-25
20
10-15
5
16-20
500
3,4
0,30
0,4
0,2
Плотность, г/см3
Прочность при растяжении, г/текс
при 177 °С
при 260 °С
при 177 °С через 300 ч
при 177 °С через 1000 ч
при 260 °С через 30 ч
при 260 °С через 1000 ч
Относительное удлинение при разрыве, °/о
Начальный модуль, г/текс
Водопоглощение при 22 °С и 65%-ной отн.
влажности, %
Удельная теплоемкость при 40 °С, кал/
/(г-°С)
Усадка, %
10 мин на воздухе, 150 °С
5 мин в воде, 100 °С
Основными направлениями использования полиамидоимидных
волокон является применение их для отделки кабин самолетов,
в фильтрах для разделения горячих газов [491] или
расплавленного свинца, в виде волокон для упрочнения бумаги
электроизоляционного назначения или абляционных материалов. Из них
изготавливают защитную одежду для пожарников и летчиков,
спецодежду для работающих в стекольном производстве, металлургии,
нефтехимии. Окрашивание полиамидоимидных волокон можно
проводить при высокой температуре в присутствии сшивающих
агентов. Поликонденсацией в растворе пиромеллитового диангид-
рида, гидразида n-аминобензойной кислоты и терефталоилхлорида
[457] получают полиамидокислоту с гидразидными группами, из
которой вырабатывают волокно мокрым формованием (осадитель-
ная ванна диметилацетамид — роданид кальция или смесь диме-
тилацетамид — этанол — глицерин). В результате термической или
химической циклодегидратации получают термостойкие волокна
с оксадиазольными циклами в цепи. С 1972 г. фирма «Teijin»
(Япония) начала промышленное производство полиамидоимидных
волокон.
7.1.3.4. Полиамидоимидные пленки
Эти пленки получают поливом из растворов полиамидокислоты
или полиамидоимида. Оптимальные свойства достигаются при
условии ориентации и термообработки выше 300 °С [463, 489, 500, 529,
533]. Физико-механические свойства опытного продукта фирмы
«Hitachi» приведены ниже [482]:
811
Плотность, г/см3
Прочность при растяжении,
кгс/см2
при 23'
при 100
150 °С
°С
°С
при
при 200°С
при 230°С
Относительное
при разрыве, %
при 23°"
при 100
при 150°С
при 200СС
при 230°С
удлинение
С
°С
1,36
1500
1200
840
500
300
14
11
12
32
50
Модуль упругости при
растяжении при 23 °С, кгс/см2
Диэлектрическая
проницаемость при 20—240 °С
Тангенс угла
диэлектрических потерь при 0—220 СС
Удельное объемное
электрическое сопротивление,
Ом-см
при 23
при 100
при 150°С
при 200°С
при 230°С
°С
°С
22 600
3,7-4,4
0,01
1018
3-Ю15
2 ¦ 10м
1013
ю12
Прочность при растяжении пленки при нагревании ее до 230 °С
уменьшается с 1500 до 300 кгс/см2 и сохраняется на этом уровне
в течение 20 сут при этой температуре [482]. Диэлектрическая
проницаемость и тангенс угла диэлектрических потерь в интервале
температур 20—220 °С остаются практически неизменными. Поли-
амидоимидная пленка отличается высокой стойкостью к действию
ионизирующих излучений. После облучения у"лУчами дозой
1000 Мрад прочность при растяжении уменьшается на 10%,
относительное удлинение при разрыве с 14 до 9,5%- Стойкость пленки
из сополимера к действию УФ-излучения ниже, чем у полиимидной
пленки. Для пленки полиамидоимида тримеллитовой кислоты и ди-
аминодифенилоксида после 6000 ч облучения УФ-лучами
(расстояние от источника до пленки 10 см) исходная прочность уменьшается
на 50% и относительное удлинение при разрыве —на 20% [464].
Тангенс угла диэлектрических потерь после 4000 ч облучения
увеличивается с 0,001 до 0,013. Под воздействием погодных факторов
хрупкость у полиамидоимидных пленок появляется гораздо раньше,
чем у полиимидных. В то же время наличие амидных связей в
макромолекуле обеспечивает более высокую адгезию к меди, чем у по-
лиимида. Повышению адгезии способствует каширование пленки
полиамидом 6,6 [493]. Нежелательную адгезию к поверхности
стекла и металлов можно понизить добавлением фосфорных
соединений, например трифенилфосфата [490].
Возможные области применения полиамидоимидных пленок
следующие: обмоточная изоляция для трансформаторов,
двигателей и генераторов, приборы атомной техники, отделочный
материал для воздушного и наземного транспорта. Полиамидоимиды
с сульфокислотными группами пригодны для получения ионооб-
менников [499].
7.1.3.5. Кабельная изоляция и покрытия
на основе полиамидоимидов
Полиамидные электроизоляционные лаки выпускаются под
марками: «Полимер AJ» («Amoco», США), «Феногарант 615/79»
(«Schramm»), HJ 400 или 401 («Hitachi») и «Родефтал 310» или
«Родефтал 311» («Rhone Poulenc»). В качестве растворителя для
812
Рис. 7.30. Кривые предельной температуры длительной ра- А,ч
ботоспособности кабельной изоляции [358]: 20000
1 — полиэфир Пзогел 200; ? — полиамидоимид AJ-10: 3 — полиимид 4г,г,пп
Пайр ML. luuuu
изготовления лака для кабельной изоляции пред-
почитают N-метилпирролидон. При взаимодей- ,'000~
ствии тримеллитовой кислоты с диаминами
получают растворы полиамидокислот, стабильность юо
их значительно выше, чем у соответствующих
полиамидокислот, из которых получают поли- t80 260
имиды. Имидизация происходит в процессе
горячей сушки.
Растворы полиамидокислот, полученных взаимодействием
ангидрида тримеллитовой кислоты с диизоцианатами, могут быть
непосредственно использованы для наложения кабельной
изоляции [503]. Модифицированные лактамами полиамидоимиды [546]
используют в качестве лаков для кабельной изоляции в виде
растворов в ж-крезоле. Полиамидоимидная кабельная изоляция
наряду с высокой термостойкостью B20 °С) имеет также хорошие
диэлектрические свойства, высокие влагостойкость, стойкость
к истиранию и действию мгновенных температурных перегрузок.
Кривые длительной работоспособности кабельной изоляции на
основе полиамидоимидов, полиимидов и полиэфиров показаны на
рис. 7.30. При термостарении в течение 20 000 ч предельная
верхняя температура для полиамидоимидной и полиимидной
кабельной изоляции составляет 240 и 245 °С соответственно.
Диэлектрическая проницаемость при 1 кГц полиамидоимидной
кабельной изоляции составляет 3,8 и 4,5 соответственно при 25 и
200°С, а тангенс угла диэлектрических потерь для этих же
температур 0,007 и 0,010. Электрическая прочность сухой изоляции
180 kB/мм и влажной 160 kB/мм. После 42 сут термостарения на
воздухе при 300 °С электрическая прочность, измеренная при
комнатной температуре, составляет 82 % от исходной.
Модифицированная лактамом кабельная изоляция может работать при
нагревании до 200 °С [477].
В многослойных лаковых покрытиях предпочтительно
использовать полиамидоимидный лак в качестве наружного слоя. Поли-
амидоимидные лаки горячего отверждения производят фирмы
«Westinghouse» (Арамидил), «Hitachi» (HJ-200) и «Rhone Poulenc»
(Родефтал 101 и 200). Отверждение при 250°С завершается за
15 мин. Лаковая пленка имеет исключительно высокую адгезию
к стали, меди, легированному алюминию и стеклу и может
выдерживать нагревание до 400 °С [466].
7.1.3.6. Полиамидоимидные связующие
Для получения слоистых пластиков на полиамидоимидном
связующем применяют растворы полиамидокислот [360, 456, 459,
538]. Промежуточные продукты синтеза полиамидоимидов хорошо
813
Р,кгс/см*
2000
500
Рис. 7.31. Зависимость прочности при изгибе стеклопластиков на основе
различного связующего от продолжительности термостарения при 315 °С [547]:
J — полимер на основе .и-аминобензойной кислоты, л-феннлендиамина н пнромеллнтового
днангидрида; 2 — полипнромеллнтимнд диаминодифеннлсульфида.
Рис. 7.32 Кривые изотермической деструкции полиамидоимидов при 325 °С [547]:
/ — полнамидоимпд Amoco AJ-10; 2 — полнамндонмид Amoco AJ-11.
растворяются в диметилформамиде, диметилацетамиде, N-метил-
пирролидоне, диметилсульфоксиде и хинолине. Эти растворы
могут содержать до 40 % ароматических и эфирных растворителей и
до 60—80 % кетонов, например метилэтилкетона, ацетона или цик-
логексанона. В отличие от полиимидных форполимеров в случае
полиамидоимидных промежуточных продуктов при той же
концентрации растворы имеют значительно более низкую вязкость.
Кроме того, эти растворы значительно более стабильны.
Для получения стекловолокнистых препрегов применяют метод
окунания с использованием низковязких растворов, содержащих
20—30 % сухого остатка. Применение метода намазывания
требует высоковязких растворов с содержанием сухого остатка
35—45%- Переработка препрегов осуществляется прессованием,
методом вакуумного мешка и резинового мешка под давлением.
Перед прессованием препрег высушивают при 150°С в течение
10—20 мин и выдерживают в прессе 20 мин при 200 °С под
давлением 50—100 кгс/см2. Для достижения оптимальных свойств
слоистые пластики на полиамидоимидном связующем подвергают
термообработке, постепенно повышая температуру от 200 до
300°С. Физико-механические свойства стеклопластиков приведены
в табл. 7.19 и на рис. 7.31. На рис. 7.32 показана зависимость
прочности при изгибе от продолжительности нагревания связую-
Таблица 7.19. Свойства стеклопластиков на основе
полиамидоимидиого связующего [176]
Показатель
Прочность при растяжении, кгс/см2
Относительное удлинение при разрыве, %
Модуль упругости при изгибе, кгс/см2
Прочность при изгибе, кгс/см2
Твердость по Роквеллу, шкала Е
Метод
прессования
3 900
1,84
230 000
5 200
65
Метод резинового
мешка
под давлением
3 800
1,76
210 000
3 900
65
814
щего на основе полиамидоимида ж-аминобензойной кислоты, я-фе-
нилендиамина и циромеллитового диангидрида.
Использование термореактивных полиамидоимидных
связующих дает возможность получить материалы, отличающиеся более
высокими прочностными свойствами при повышенных
температурах [459].
7.1.3.7. Полиамидоимидные пресс-массы
Полиамидоимидные пресс-массы и пластики на их основе
выпускает фирма «Amoco» (США). Прессование проводят при
300—330 °С под давлением 250—350 кгс/см2 [176, 463, 588]. Этим
методом получают пластины толщиной 3—б мм и размером 65 X
X 450 мм. Свойства наполненных и ненаполненных
полиамидоимидных пластиков приведены в табл. 7.20. Предельная верхняя
Таблица 7.20. Свойства полиамидоимидных пластиков [176]
Показатель
Плотность, г/см3
Прочность при
растяжении, кгс/см2
при 23 °С
при 150 °С
при 260 °С
Относительное
удлинение при разрыве
при 23 °С, %
Модуль упругости
при изгибе, кгс/см2
при 23 °С
при 150 °С
при 260 °С
Прочность при изгибе,
кгс/см2
при 23 °С
прп 150сС
при 260 °С
Прочность при сжатии
при 23 °С, кгс/см2
Ударная вязкость
при 23 °С, кгс- см/см2
Твердость по Роквеллу,
шкала Е
Верхняя предельная
температура
длительной работоспособности,
Диэлектрическая
проницаемость
Тангенс угла
диэлектрических потерь
Водопо! лощение, %
Нена-
полнен-
ный
пластик
1,41
930
820
625
2,5
49 000
38 000
31500
1650
1 300
990
2 450
3,8
104
295
3,8
0,001
0,28
ПТФЭ
1,46
600
430
150
2,0
40 000
31000
27 000
920
770
410
2 150
3,4
81
280
3,8
0,002
0,27
Пластики, наполненные
¦
графитом
1,45
910
710
365
3,4
49 500
39 000
35 000
1550
1090
540
2 400
4,3
98
295 .
0,27
ПТФЭ
и
графитом
1,65
620
430
210
2,2
42 000
34 000
29 000
1050
850
435
2 600
4,3
76
295
0,26
силикатом
Са
1,58
920
780
460
2,3
81000
84 000
33 000
1700
1220
705
2 900
3,4.
101
290
3,9
0,008
0,30
AI
1,60
920
830
280
3,0
55 000
47 000
34 000
1550
1340
555
2 600
4,8
74
300
0,22
815
температура длительной работоспособности изделий составляет
около 310 °С. При 260 °С прессованные изделия сохраняют еще
67 % прочности при растяжении и 60 % при изгибе (от значения
при комнатной температуре).
Прессованные полиамидные изделия используют в
производстве пишущих машинок, компьютеров и деталей арматуры,
которая выдерживает большие нагрузки.
7.1.4. ПОЛИЭФИРОИМИДЫ
Полиэфироимиды получают из диангидридов, содержащих
эфирные группы, в сочетании с диаминами, применяя в процессе
этерификации мономеры с имидными или имидообразующими
группами. Верхняя температурная граница длительной
работоспособности полиэфироимидов равна 190°С. Используются они
главным образом в качестве лаков для кабельной изоляции.
Промышленное производство полиэфироимидов было начато в 1966 г.
7.1.4.1. Получение полиэфироимидов
Поликонденсация диангидридов тетракарбоновых кислот,
содержащих эфирные группы с диаминами. Процесс проводят по
обычной для полиимидов двухстадийной схеме [548—550, 562,
570—572, 577] :
О о
;О + nH2NRNH2
Ol—С—О—R'—О—С
J II II
О О
Полнамидокислоты получают поликонденсацией в растворе
N-метилпирролидона, диметилформамида, диметилацетамида, ди-
метилсульфоксида или ж-крезола. Термическую циклизацию
проводят, например, для лаковой пленки по следующему режихму: 1 ч
при 100°С, 1 ч при 200°С и 1—2 ч при 240°С [548]. Наибольшая
скорость имидизации наблюдается в температурном интервале
180—210 °С [573].
816
ИК-спектроскопические, дилатометрические, термомеханические
и рентгеноскопические исследования показали, что в процессе ими-
дизации сначала происходит форкристаллизация, обусловленная
уменьшением подвижности структурных элементов и повышением
упорядоченности макромолекул. Кристаллизация молекулярных
сегментов происходит при температуре выше 220 °С [574]. Синтез
диангидридов из ангидридов трикарбоновых кислот и диацетатов
бисфенолов протекает по схеме
О -Ь СНзСОО—R—ОСОСНз + О
О
—СНзСООН
соон
Переэтерификацию проводят в хлорированных растворителях
при 270—320 °С. Выход составляет 60—90%. По некоторым
данным, эта реакция протекает через промежуточную стадию
образования четырехчленного цикла [548]:
О
г- Л
О-Н O-C
817
Другой способ получения диангидридов, содержащих сложно-
эфирные группы, состоит во взаимодействии 4-хлорформил-о-фта-
левого ангидрида с бисфенолами в присутствии акцепторов
хлористого водорода (например, пиридина):
СОС1
НО—R—ОН +
О
COCJ
Температура плавления таких соединений колеблется от 150 до
300 °С.
Поликонденсация мономеров, содержащих имидные циклы.
В этом случае полиэфироимиды получают при взаимодействии ди-
карбоновых кислот или диолов, содержащих имидные циклы,
соответственно с диолами или полиэфирами с концевыми гидроксиль-
ными группами или дикарбоновыми кислотами и их производными
[477, 551]. Аналогичным образом полиэфироимиды синтезируют из
гидроксиалкилпроизводных имида тримеллитовой кислоты. Так же
как и полиамидоимиды, полиэфироимиды можно получить в м-
крезоле или смеси л^-крезол — диметилацетамид при 120—180 °С
из диимидов, содержащих эфирные группы, и диаминов [484]:
NH + NH2RNH2
—NH3
818
Диимиды с эфирными группами получают реакцией имида
тримеллитовой кислоты с диацетатами бисфенолов
о
\
QJ ^NH + НзССО—R—ОС—СНз +
НООС-""
*• HN
-СНзСООН ч
или переэтерификацией эфира имида тримеллитовой кислоты гли-
колями
СОСНз
—СН3ОН
о
Получение мономеров, содержащих имидные группы.В
промышленности полиэфиронмиды получают из имидов, образующихся
непосредственно в присутствии компонентов, необходимых для
синтеза полиэфиров [378, 552—556, 566—567, 569]. Для этих целей
чаще всего используют ангидрид тримеллитовой кислоты, пиромел-
литовый диангидрид и диангидрид бензофенонтетракарбоновой
кислоты, диаминодифенилметан, я-фенилендиамин, /г-аминобензой-
ную кислоту, аминоэтанол, аминоуксусную кислоту, терефталевую
и итаконовую кислоты, фенилиндандикарбоновую кислоту, бензо-
фенондикарбоновую кислоту, этиленгликоль, глицерин, трис(гидр-
оксиэтил)изоцианурат. В условиях этерификации B00—280 °С)"
имидизация происходит чрезвычайно быстро, в то время как
относительно медленно протекающий процесс поликонденсации
замедляется выделяющейся в ходе имидизации водой. При добавлении
819
в состав исходных продуктов ароматических дигидроксисоединений
получают полиэфироимиды с высокой устойчивостью к тлению
[557].
—он
НО(СН2J
—О(СН2JОН
Так как при синтезе полиэфиров с использованием в качестве
кислотного компонента самой терефталевой кислоты такие диолы
не применяют, более высокая реакционная способность
компонентов в процессе получения полиэфироимидов объясняется
повышенной кислотностью дикарбоновых кислот, содержащих имидные
группы [378]. Если диамин используется в избытке, образуется
сополимер, содержащий эфирные, амидные связи и имидные гетеро-
циклы. При избытке три- и тетракарбоновых кислот получаются
разветвленные и сшитые полимеры. В том случае, когда применяют
структурирующие агенты с симметрично расположенными
функциональными группами, полиэфироимиды для изоляции проводов
получаются повышенной термостойкости [558, 559]. В
промышленности для их производства чаще всего используются трисB-гидро-
ксиэтил)цианураты. Наряду с этим часто применяют пиромелли-
товую, тримезиновую, меллитовую кислоты и симметричные нафта-
линкарбоновые кислоты. Полиэфироимид, полученный при
использовании трисB-гидроксиэтил)изоцианурата, имеет следующее
строение:
0 о н2с-с
СООСНг
о—
Структурирование в полиэфироимидной лаковой пленке можно
осуществить в процессе ее горячей сушки, используя такие
катализаторы переэтерификации, как ацетат цинка и трибутилорто-
титанат [551, 568].
Аппараты для синтеза полиэфироимидов должны обеспечивать
быстрый подвод тепла в условиях быстро меняющейся температуры
820
Рис. 7.33. Кривые
термической
деструкции при
различной
температуре в среде азота
(а) и при
различной
продолжительности выдержки
на воздухе при
240 °С (б) [564];
1 — поли-л-фенилен-
терефталат; 2 — поли-
эфироимид № 8
(табл. 7.21); 3 — по-
липнромеллитимид
диаминодифенил-
окснда.
0 200 400 t,°C
200 ¦ Х,Ч
до 260 °С, а колонны — эффективный отвод выделяющейся воды,
препятствуя одновременно потерям гликоля. Должны быть
предусмотрены загрузочные устройства для дозировки твердых
продуктов, создания вакуума, контроля степени заполнения с учетом
изменения скорости перемешивания и температурных перепадов [553].
7.1.4.2. Физико-химические свойства полиэфироимидов
Полиэфироимиды окрашены в слегка желтый цвет. В отличие
от ароматических полиимидов они очень легко кристаллизуются.
Температуры размягчения, плавления и разложения линейных
полиэфироимидов приведены в табл. 7.21.
Полиэфироимиды, содержащие в цепи только фталимидные,
фениленовые и эфирные группы, разлагаются при температуре
выше 400 °С, но ниже температуры плавления (табл. 7.21, № 3,
4, 6, 20). При наличии в цепи «шарнирных» групп типа О, S, SO2,
СН2 полимеры плавятся в интервале температур 350—430 °С; их
температура размягчения колеблется от 250 до 300 °С. В случае
модификации политерефталатами в зависимости от степени
структурирования они размягчаются при 320—400°С. Линейные
полиэфироимиды с алифатическими группами в цепи (табл. 7.21, № 13,
14,30—37) плавятся в интервале температур 125—300 °С.
Линейные полиэфироимиды хорошо растворяются в полярных
растворителях типа диметилформамида и диметилацетамида, ди-
метилсульфоксида и N-метилпирролидона. Эти растворы стабильны
при комнатной температуре до 6 мес. Как видно из рис. 7.33, по
термостойкости полиэфироимиды занимают промежуточное
положение между ароматическими полиимидами и ароматическими
полиэфирами. Линейные ароматические полиэфироимиды стабильны
на воздухе до 350 °С и в инертной среде до 400 °С. Стойкость к
термоокислительной деструкции в зависимости от природы
ароматических групп в основной цепи уменьшается в следующем ряду
[573]:
821
Таблица 7.21. Температуры размягчения, плавления и разложения полиэфиримидов
Элементарное звено
раэм1
т
разл
B„N2),
разл
(на
воздухе),
О
>360
>500
395
о
О
О
о
о
)—о-с-
II
о
гаг>
соон
> 500
> 500
о
о
>500
о
р
ОСНз
> 500
Продолжение 'табл. 7.21
Элементарное звено
разм'
-•С
Т
разл
"gN2>,
т
разл
<на воз-
)
О
- с—о—(\01)—о—с
о о
о
10
• 310
>360
>500
430
>500
410
360
360
О
11
12
13
14
О
W
о
290
>355
>500
320
300
405
350
[484,572,
5481
[541,
573-575]
400
[484]
385
[484,570]
Продолжение табл. 7.2I
Элементарное ввено
разм'
т
раз л
(в N2>,
°С
разл
(на воз-
Литературный
источник
О
15
16
17
270 360
335
[571,575]
[571,575]
290
[571,575]
18
О
[571,575]
19
21
О О II
II II _s*^s\
с-о о-с—<г\\ \
1548]
> 500
[548]
235
[548]
Продолжение табл. 7.21
О
23
24
>500
>500
[548]
[548]
> 5001
26
27
28
Продолжение табл. 7.21
Элементарное звено
разм'
Г
разл
Гразл
(на воз-
«5g).
29
30
31
N-(CH2J
О
350
410
250
225
390
380
32
О
к
О
О
О
200
400
О
о
33
С—О-(СН2J—О—С
о
о
155
380
О
34
—N
с-о-(сн2J-о-с
Il il
Il
165
415
Продолжение табл. 7,21
Элементарное звено
разм1
Т
раз л
(в N2),
разл
(на
воздухе),
О
35
36
—N'
О
А
37
О
—n:
о
о
С—О—(СН2J—О—(СН2J—О—С
о
С—О— (СН2J—О—(СН2J—'
130
140
380
400
125
375
/77,%
200 400 600
Рис. 7.34. Кривые термической деструкции полпэфироимидов при различной
температуре в среде азота:
О О
—N
iQi
,—с—о
О
-о—с—,
о
Y
О
—п
Полиэфироимиды с алифатическими группами в цепи имеют
i инертной среде примерно такую же температуру разложения,
сак и полностью ароматические полимеры, однако при температуре
зыше 450°С скорость их разложения намного выше, чем у послед-
1их (рис. 7.34). Лаковые пленки на основе сшитого полиэфиро-
шида терефталевой кислоты, глицерина, этиленгликоля,
ангидрида тримеллитовой кислоты и диаминодифенилметана на воздухе
тачинают интенсивно разлагаться выше 380 °С. Эта температура
ia 30° превышает температуру процесса отверждения лаковой
тленки [576]. Полиэфироимиды трехмерной структуры, полученные
*заимодействием полнамидокислоты с 4,4/-диB-гидроксиэтокси)-
Шнафтилом-1,Г с последующей имидизацией [566] стабильны на
юздухе и в среде азота до 480 °С.
—N
CONHR—
COO(CH2J—
27 Зак. 18
аЗЗ
7.1.4.3. Кабельная изоляция на полиэфироимидной
основе
Полиэфироимиды имеют верхнюю температуру длительной
работоспособности около 190 °С, что значительно ниже, чем у
ароматических полиимидов, однако технология получения кабельной
изоляции на их основе не связана с теми трудностями, которые
возникают при переработке полиимидов через растворы полиамидо-
кислоты G.1.1.6). Лаковые растворы выпускаются в имидизован-
ной форме. Эти растворы в крезоле, N-метилпирролидоне, диме-
тилацетамиде, диметилформамиде практически неограниченно
стабильны. Процесс горячей сушки происходит в температурном
интервале 255—350 °С. Методы отверждения подобных лаковых
полимеров рассматривались выше. Кабельная изоляция на
полиэфироимидной основе имеет высокую прочность при истирании
в сочетании с хорошими механическими и электрическими
свойствами [360, 378, 552, 560—563]. Промышленные марки полиэфиро-
имидов приведены в табл. 7.22.
Диэлектрическая проницаемость в широком интервале от 60 до
106 Гц практически не зависит от частоты. При повышении
температуры от 20 до 250 °С она возрастает с 3,0 до 4,!. В этом
температурном интервале тангенс угла диэлектрических потерь
изменяется от 0,005 до 0,025 при 104 Гц. Кабельная изоляция
становится хрупкой при температуре выше 350 °С.
Электроизоляционные лаки характеризуются высокой
стойкостью к мгновенным нагрузкам и тлению. Стойкость к
воздействию мгновенных нагрузок определяется путем измерения числа
витков вокруг изолируемого провода до отделения изоляционного
слоя, после чего подсчитывается количество трещин на их
поверхности. Верхняя температурная граница до образования трещин
для полиэтилентерефталата за 15 мин экспозиции при трех витках
составляет 150°С. Полиэфироимиды ведут себя так же, но при
однократном изгибе при 180—200 °С. Для оценки устойчивости к
тлению определяют время, в течение которого происходит короткое
Та
Страна
США
ФРГ
блица 7.22.
Торговое
название
Имидекс
Изомид
Теребек
Аллобек
Полиэфироимидные лаки
Фкрма-изготовитель
«General Electric»
«Schenectady»
«Dr. Beck and Со,»
«Dn Herberts and Со,»
для кабельной изоляции
Исходные соединения
Диаминодифенилоксид, диан-
гидрид на основе тримеллито-
вого ангидрида и диацетокси-
бензола
Полиэфир, пиромеллитовый
ангидрид, трис(гидроксиэтил)-
изоцианурат
Полиэфир, диамины,
пиромеллитовый ангидрид, тримелли*
товьш ангидрид
834
замыкание при напряжении переменного тока 220 В и постоянной
нагрузке. Модификация полиэфироимидов фенолами дает
возможность увеличить устойчивость к тлению в 5 раз. При термодеструк-
цин таких модифицированных полимеров на поверхности
изолируемого провода образуется продукт разложения, обладающий
высокой адгезионной прочностью и в течение длительного времени
обеспечивающий хорошую изоляцию [378]. Устойчивость к тлению
увеличивается также при сшивании симметричными
полифункциональными соединениями, например трисB-гидроксиэтил)изо-
циануратом. При этом значительно повышается и термостойкость.
Для оценки теплостойкости изоляционного слоя определяется некий
показатель, который устанавливается при нагревании изоляции под
нагрузкой. Для полиэфироимидной изоляции при 300 °С в
зависимости от степени структурирования этот показатель равен 30—
70 %¦ В тех же условиях для полиэтилентерефталата он составляет
лишь 10 %.
Однако в результате структурирования увеличивается и
чувствительность изоляции к действию мгновенных нагрузок, особенно
при знакопеременных тепловых ударах. Этот недостаток удается
ликвидировать путем нанесения поверхностного слоя на основе
полиамидоимидного связующего. Оптимальный состав рецептуры
лаков изоляционного назначения (соединения, образующие имид-
ные группы, сшивающий агент, фенольные соединения)
устанавливается экспериментально. Полиэфироамидимиды в отличие от по-
лиэфнримидов обладают относительно низкой способностью к
испарению в процессе нанесения лакового слоя, что связано с
большей склонностью к образованию водородных связей [378, 565].
7.1.4.4. Полиэфироимидные пленки
Полиэфироимидные пленки получают поливом из растворов
полимеров или форполимеров в N-метилпирролидоне, диметилформ-
амиде и диметилацетамиде, ж-крезоле. Верхняя температурная
граница длительной работоспособности полиэфироимидной пленки
составляет 190 °С. Свойства пленок приведены в табл. 7.23.
Основные области применения пленок — обмоточная изоляция и
печатные схемы.
7.1.5. ПОЛИСУЛЬФОИМИДЫ
Полисульфоимиды описаны в ряде работ [580—586]. Их
отличает от полиимидов наличие в имидном цикле вместо одной
карбонильной группы сульфогруппы:
0 0 О О
0 О
Полиамид
п
^n
27*
835
Таблица 7.23. Физико-механические свойства полиэфироимиднои пленки
Элементарное звено
Прочность
при
растяжении,
КГС/СМ2
20 °С
200 °С
Относительное
удлинение
при разрыве,
20 °С
200 °С
Модуль
упругости,
КГС/СМ2
Литературный
ИСТОЧНИК
20 °С
200 °С
1550
775
16
[548]
1 120
450
15
32 00Q
17 900
[548]
о
о
||
О
СН3
СНз
)—о—
о
О
1050|
О-
990
450
425
700
450
14
6
4
23
21
О
3—С
I0 900
38 200
25 301
3 600
13 40С
1548J
1548J
10 70 [548]
Гомополиконденсация б-аминосахарина в расплаве или в раст
воре, например в диметилформамиде, я-крезоле, хлорфеноле, ни
трофеноле или полифосфорной кислоте, приводит к образована
растворимого форполимера, циклизация которого сопровождаете
получением полимера с преимущественно псевдосахариновыми
индазольными звеньями:
H2N
SO2NH2-
Таблица 7.24. Температура разложения полисульфоимидов
Идеализированное строение
элементарного звена
Литературный
источник
650
[585]
500
[5841
[584]
838
Гомополимеры с высоким содержанием сульфоимидных звеньев
получают циклополиконденсацией 4-амино-2-сульфампдобензоата
или ангидрида 4-амино-2-сульфобензойной кислоты.
Поликонденсация «лг-бис-сахарпна» с бензидшюм или фенилендиамином
приводит к получению полимера с преимущественно сульфоимидными
звеньями в цепи. Полностью циклизованный полисульфоимид —
¦неплавкий и нерастворимый продукт, в то время как форполимер
растворяется в диметилацетамиде. Гомополимер стабилен на
воздухе и в среде азота до 650 °С, а сополимер — до 500 °С
{табл. 7.24).
Литература к главе 7, разделы 7.1—7. К5
1. S. Nishizaki, J. ehem. Soc. (Japan), Ind. ehem. Sect. 66 A963) 3, 382—386.
2. E. Sroog, J. Polymer Sei. A3 A965) 4, 1373—1390.
3. А. Рудаков, ДАН СССР, 172, 1967, 899—902.
4. А. Рудаков, ДАН СССР, 174, 1967, 6, 1352—1355.
5. М. Котон, Хим. волокна, 1969, 4, 15—22.
6. И. Кардаш, Высокомолек. соед., Б 9, 1967, 12. 873—876.
7. К. Мозгова, Пласт, массы, 1968, 10, 14—15.
8. А. Русанов, Изв. АН СССР, сер хим., 1968, 7, 1654.
9. 3. Плонка, Высокомолек. соед., А 7, 1966, 12, 2177—2178.
10. А. Дулов, Высокомолек. соед., А12, 1970, 2, 401—408.
11. О. Goins, J. Polymer Sei. В 6 A968) 11, 821—825.
12. А. Рудаков, Высокомолек. соед., А12, 1970, 3, 641—648.
13. А. Анненкова, Пласт, массы, 1969, 8, 41—43.
14. Н. Адрова, Высокомолек. соед., Б9, 1967, 1, 22—23.
15. С. Виноградова, Высокомолек. соед., 13, 1971, 7, 1507—1516.
16. Г. Колесников, Высокомолек. соед., А12, 1970, 2, 317—322.
17. D. Hummel, Farbe & Lack 74 A968) 1, 11—18.
18. К- Власова, Высокомолек. соед., А13, 1971, 3, 603—607.
19. Ш. Туйчиев, Высокомолек. соед., Б13, 1971, 7, 1463—1467.
20. S. Nishizaki, J. ehem. Soc. (Japan), Ind. ehem. sect. 68 A965) 9, 1756—
1761.
21. Г. Слоннмскнй, ДАН СССР, 182, 1968, 4, 851—854.
С. Виноградова, Высокомолек. соед., АИ, 1969, 12, 2725—2740.
22. Н. Адрова, Высокомолек. соед., Б10, 1968, 5, 354—356.
С. Виноградова, Высокомолек. соед., А9, 1967, 5, 1091—1098
23. В. Родэ, Высокомолек. соед., АИ, 1969, 7, 1617—1621.
С. Виноградова, Изв. АН СССР, 10, 1967, 2267—2274.
24. Г. Слонимский, ДАН СССР, 1967, 10, 2267—2274.
25. П. Грибкова, Высокомолек. соед., AI2, 1970, 1, 220—228.
26. Е. Краснов, Хим. волокна, 1970, 40—48.
27. S. Nishizaki, Sei. Techn. Aerospace Rep. 4 A966), 16, ЗОН.
28. S. Hirsch, Chem. Engin. News 45 A967) 41, 48.
29. С. Павлова, Изв. АН СССР, сер. хим., 8, 1968, 1900—1902.
30. J. Rose, ACS Polymer Preprints 11 A970) 1, 339—346.
31. Г. Колесников, Высокомолек. соед., 8, 1966, 1440—1444.
32. О. Goins, J. Polymer Sei. В 6 A968) 11, 821—825.
33. А. Рудаков, Пласт, массы, 1967, 9, 26—29.
34. Г. Колесников, Высокомолек. соед., Б10, 1968, 10, 742—745.
35. Пат. СССР 257010, 11. 11.69.
36. Фр. пат. 1539074, 5.8.68.
37. M Котон, ДАН СССР, 177, 1967, 3, 579—581.
38. Пат. США 3356648 A967) F. Rogers.
39. H. Reimschuessel, Macromolecules 2 A969) 6, 567—569.
40. H. Reimschuessd, J. Polymer Sei. В 4 A966) 12, 953—954.
839
41. T Asahara, J. chcm. Soc. (Japan), Ind. ehem. Sect. 71 A968) 6, 918—922.
42 T Asahara, J. ehern. Soc. (Japan), Ind. ehem. Sect. 72 A969) 8, 1923—1925.
43. M. Russo, J. Polymer Sei. Л 1 A969) 12, 3337—3349.
44. T. Asahara, J. ehem. Soc. (Japan), Ind. ehem. Sect. 70 A967) 12, 2388—
2392.
45. А. Рудаков; Высокомолек. спел., А9, 1967, 9, 1985—1988.
46. В. Кудрявцев, Высокомолек. соед., А 9, 1967.
47. В. Кудрявцев, ЖПХ, 41, 1968, 8, 1825—1829.
48. Е. Федорова, Высокомолек. соед., Б 10, 1968, 2, 119—123.
49. J. Stille, J. Polymer Sei. A3 A965) 6, 2397—2399.
50. R. Oda, J. Polymer Sei. В 7 A969), 1, 11—15.
51. С. Виноградова, Высокомолек. соед., AI3, 1971, 5, 1146—1150
52. M. Koton, Plaste u. Kautschuk 14 A967) 10, 730—734.
53. Пат. США. 2710853 14.6.55 Du Pont.
54. Акцепт, заявка Японии 7041396 25. 12. 70 Teijin Ltd.
55. Y. Iwakura, J. Polymer Sei. A 17 A969) 2, 597—602.
56. J. Heller, J. Polymer Sei. В 6 A968) 3, 153—157.
57. Пат. США 3728150 17.4.73 Du Pont.
58. Fr. Demande 2142743 9. 3. 73 Rhone-Poulenc.
59. Опубл. заявка 2246026 29. 3.73 Union Carbide.
60. Fr. Demande 2142716 9.3.73 Inst. Franc, du Petrole.
61. Пат. США 3705870, 12.12.72. CIBA.
62. Пат. США 3705869, 12. 12.72. CIBA.
63. Акцепт, заявка Японии 7134989 19. 10.71 Toray Ind.
64. Пат. СССР 319618, 2. 11.71.
65. С. Харьков, Хим. волокна, 1971, 289—304.
66. Пат. США 3705875 12. 12.72 U. S. Air Force.
67. Пат. США 3702318 7. 11. 72 U. S. Air Force.
68. H. Lubowitz, ACS Polymer Preprints 12 A971) 1, 329—334.
69. R. Kray, ACS Div. org. Coating Plast Chem. Pap. 31 A971) 1, 569—577.
70. Пат. США 3700649 24. 10. 72 Int. Harvester Со.
71. Акцепт, заявка Японии 7219707 5.7.72 Toray Ind.
72. G. Saksaganskii, Elektrofiz. App. 9 A971), 57—59.
73. C. Jepson, Plast. Mod. Elast. 20 A968) 7, 81—83.
74. Г. Лущейкин, Пласт, массы, 1972, 8, 28—31.
75. H. Баюшкина, Стабилизация полимерных материалов и изделий из них,
1971, 2, 60—64.
76. И. Давыдова, Стабилизация полимерных материалов и изделий из них, 1.971,
2, 64—69.
77. Акцепт, заявка Японии 7214503, 1.5.72 Toray Ind.
78. Акцепт, заявка Японии 7219615 5.6.72 Toray Ind.
79. Англ. пат. 1283077 26.7.72 Du Pont.
80. Опубл. заявка ФРГ 2061360 15. 6.72 BASF.
81. M. Stitch, Ann. Rel. Phys. Symp. A971) 9, 86—92.
82. Акцепт, заявка Японии 7137432 4. 11.71 Toray Ind.
83. Опубл. заявка ФРГ 2055211 25.5.72 Batteile Inst.
84. Пат. США 3666723 30. 5. 72 CIBA-GEIGY.
85. Опубл. заявка ФРГ 2152972 4. 5. 72 Rhone-Poulenc.
86. Акцепт, заявка Япония 7128647 20.8.71 Matsuhita Electric.
87. Пат. США 3652355 28. 3. 72 General Electric.
88. Пат. СССР 329188, 9. 2. 72.
89. Акцепт, заявка Японии 7123916 8.7.71 Toray Ind.
90. Акцепт, заявка Япр.нии 7123917 8.7.71 Toray Ind.
91. H. Ing, J. chem. Soc. A926), 2348.
92. Опубл. заявка ФРГ 2113063 7.10.71 General Electric.
93. К. Власова, Пласт, массы, 1971, 11, 56—57.
94. Пат. СССР 318597, 28. 10.71.
95. M. Hanson, SAMPE Techn. Conf. A971), 31—38.
96. Пат. США 3624050 30. 11.71 J. Strickrodt.
97. Фр. пат. 2050251 7.5.71 Inst. Franc, du Petrole.
98. H. Reimschuessel, J. Polymer Sei. В 4 A966), 12, 953—954.
99. Г. Лущейкин, Высокомолек. соед., Б14, 1972, 1, 53—56.
100. Акцепт, заявка Японии 7137732 6. 11.71 Toray Ind.
101. Акцепт, заявка Японии 7137730 6. 11.71 Toray Ind.
102. Акцепт, заявка Японни 7129500 27.8.71 Japan Polyurethane.
103. Акцепт, заявка Японии 7137734 6. 11.71 Torav Ind.
104. Пат. США 3616177 26. 10. 71 С. Gumermon.
105. Пат. СССР 314777, 21.9. 7i.
106. Опубл. заявка ФРГ 2118388 28. 10.71 D. Dixon.
107. К. Власова, Пласт, массы, 1971, 10, 24—25.
108. Пат. США 3607838 21.9.71 General Electric.
109. Опубл. заявка ФРГ 1954526 3.6.71 TRW Inc.
ПО. Акцепт, заявка Японии 7106793 20.2.71 Hitachi Chemicals.
111. К. Власова, Пласт, массы, 1971, 9, 15—17.
112. А. Смирнова, Синтез, свойства и применение полимеров, тезисы, 15, 196J
146—149.
113. М. Котои, ДАН СССР, 100, 1971, 1, 118—121.
114. M. Bogert, J. Amer. chem. Soc. 30 A908), 1135—1144.
115. Акцепт, заявка Японии 7127821 12.8.71 Toray Ind.
116. Опубл. заявка ФРГ 2016778 15.4. 71 Nat. Res. Dev. Co.
117. J. Critchley, Gt. Brit. Roy. Aircr. Estab. Techn. Rep. A970), TR 70178.
118. Опубл. заявка ФРГ 2103500, 22.7.71.
119. Б. Гинзберг, Высокомолек. соед., А13, 1971, 7, 1463—1467.
120. А. Праведников, Высокомолек. соед., А K, 1971, 2, 425—436.
121. Пат. США 3179634 20. 4. 65 Du Pont.
122. M. Бессонов, Высокомолек. соед., Б 13,1971, 7, 509—514.
123. К. Власова, Пласт, массы, 1971, 8, 65—66.
124. Акцепт, заявка Японии 7116906 10.5.71 Kureha Chem.
125. Пат. СССР 294843, 4.2.71.
126. Акцепт, заявка Японии 7041H6, 23. 12. 70.
127. Пат. СССР 295782, 12.2.71.
128. Фр. пат. 1591043, 29.5.70.
129. Акцепт, заявка Японии 7117145 12.5.71 Showa Electric.
130. Пат. США 3600361 17.8.71 Du Pont.
131. Пат. ФРГ 1956461 15.7.71 V. Korshak.
132. Пат. США 3585013 15.6.71 Du Pont.
133. Акцепт, заявка Японии 7009393, 4.4.70.
134. Опубл. заявка ФРГ 2004380 5.8.71 BASF.
135. И. Гугель, Хим. волокна, 1971, 4. 75—76.
136. Sei. Techn. Aerospace Rep. 8 A970) 1, 49.
137. Y. Imai, J. Polymer Sei. В 8 A970), 8, 555—558.
138. А. Рудаков, Высокомолек. соед., Б 13, 1971, 7, 542—547.
139. Н. Анненкова, Высокомолек. соед., Б 13, 1971, 3, 201—205.
140. Р. Rand, SPE techn. Papers A972).
14). В. Коршак, Высокомолек. соед., А 14, 1972, 7, 1528—1533.
142. А. Якубович, Высокомолек. соед., А 14, 1972, 10, 2174—2182.
143. Р. Hergenrother, Boeing Scientific Res. Lab. D 180-12911-1.
144. A. Праведников, Высокомолек. соед., А 14, 1972, 10, 2148—2154.
145. H. Михайлов, Хим. волокна, 14, 1972, 5, 51—53.
146. Кудрявцев, Хим. волокна, 14, 1972, 2, 39—41.
147. Н. Block, Polymer 13 A972) 11, 527—535.
148. Г. Колесников, Пласт, массы, 1970, 12, 20—23.
149. Н. Адрова, Пласт, массы, 1972, 11, 64—65.
150. Р. Carleton, J. appl. Polymer Sei. 16 A972) 11, 2983—2989.
151. В. Коршак, Высокомолек. соед., А14, 1972, 9, 1924—1928.
152. А. Праведников, Высокомолек. соед., Б14, 1972, 3, 185—188.
153. Л. Бикбулатова, Высокомолек. соед.. Б14, 1972, 6, 469—472.
154. Н. Зуберман, Высокомолек. соед., Б14, 1972, 7, 541—542.
155. В. Коршак, Высокомолек. соед., Б14, 1972, 8, 592—595.
156. Ю. Васильева, ДАН СССР, 201, 1971, 4, 850—853.
157. М. Котон, Высокомолек. соед., Б14, 1972, 12, 900—911.
158. R. de Iasi, J. appl. Polymer Sei. 16 A972) 11, 2909—2919,
841
159. W. Scheck, SAMPE Quart 3 A972) 4, 22—29.
160. J. Barton, Polymer 11 A970), 212—221.
161. Kunststoff-Berater 2 A970), 212—220.
162. R. Dine-Hart, Brit. Polymer J. 3 A971), 163—168.
163. E. Краснов, Высокомопек. соед., А12, 1970, 4, 873—884.
164. M. Girard, Verre text. plast. renf. 4 A970I0, 2205—2210.
165. Л. Кикурина, Высокомолек. соед., Б12, 1970, 10, 2205—2210.
166. D. Bishop, J. appl. Polymer Sei. 14 A970) 2, 345—354.
167. G. Menges, Plastverarbeiter 21 A970) 5, 1—8.
168. Фр. пат. 1572798 A969) TRW Inc.
169. Trans. New York Acad. Sei. Ser. II 33 A971) 2, 219—230.
170. G. Erhard, Kunststoffe 62 A972) 1, 2—9.
171. T. Johnston, J. macromol. Sei. A3 A969) 6, 1161—1182.
172. P. Delvings, J. Polymer Sei. В 8 A970) 1, 29—35.
173. К. Rogers, Composites l A970) 5, 286—289.
174. S. Hirsch, J. Polymer Sei. A 17 A969) 1, 15—22.
175. Kunststoff-Berater A972) 6, 427.
176. R. Fabian, Materials Engin. Sei. A971) 8, 26—31.
177. J. Preston, Faserforschg. Textiltechn. 22 A971) 3, 153—162.
178. J. Preston, Chem. Technol. 1 A971) 11, 664—671.
179. В. Азаров, Пласт, массы, 1971, 7, 37—38.
180. К. Рае, J. Polymer Sei. В 10 A972) 7, 531—535.
181. Y. Imai, J. Polymer Sei. 10 A972), 7, 2091—2096.
182. A. Телешова, Высокомолек. соед., А 13, 1971, 10, 2309—2315.
183. В. Евстафьев, Высокомолек. соед., А 13, 1971, И, 2565—2571.
184. J. Critchley, J. Polymer Sei. A 1 !0 A972) 6, 1798—1825.
185. О. Федотова, Высокомолек. соед., А 14, 1972, 6, 1256—1266.
186. N. Kardush, J. Polymer Sei. A 1 10 A972) 4, 1093—1096.
187. В. Коршак, Высокомолек. соед., А 14, 1972, 3, 686—694.
188. P. Opt, Textile res. J. 37 A967) 12, 1050—1055.
189. J. Preston, J. Polymer Sei. Al A972) 5, 1377—1389.
190. Mater. Engin. 71 A970) 3, 45.
191. B. Vollmert, Angew. Makromol. Chem. 23 A972), 117—139.
192. A. Horvath, Angew. Makromol. Chem. 23 A973), 141—156.
193. В. Коршак, Высокомолек. соед., Б 13, 1971, 8, 603—605.
194. M. Котон, ДАН СССР, 200, 1971, 118—121.
195. Н. Адрова, Высокомолек. соед., В 13, 1971, 11, 824—827.
196. S. Kaplan, SPI Techn. Conf. Febr. 1972, Techn. Papers.
197. H. Малегина, Высокомолек. соед., А 13, 1971, 12, 2800—2808.
198. Э. Телешов, Высокомолек. соед., А 14, 1972, 1, 150—155.
199. Ю. Васильева, Высокомолек. соед., А 14, 1972, 1, 143—149.
200. М. Котон, Высокомолек. соед., А 13, 1971, 6, 1348—1357.
201. Ш. Туйчиев, Высокомолек. соед., А 13, 1971, 7, 1463—1467.
202. В. Пшеницина, Высокомолек. соед., А 14, 1972, 3, 628—632.
203. Г. Тимофеева Высокомолек. соед., А 13, 1971, 10, 2348—2353.
204. Л. Лайус, Высокомолек. соед., А 13, 1971, 9, 2006—2010
205. R. Kleinholz, Gummi — Asbest — Kunststoffe A972) 3, 204—214
206. L. Lee, J. Polymer Sei. A l 9 A971), 3169—3174.
207. Y. Musa, J. Polymer Sei. A 1 A972), 2, 319—327.
208. R. Dine-Hart, Makromolekulare Chem. 153 A972), 237—254.
209. E. Johnson, J. appl. Polymer Sei. 15 A971 , 2825—2839.
210. H. Reimschuessel, J. Polymer Sei. A 1 9 A971), 3071—3073
211. J. de Abajo, Angew. Makr. Chem 19 A971), 121 — 134.
212. P. Bailly, Chim. Ind. (Paris) 104 A971) 21, 2683—2689
213. G. Smets, J. prakt. Chem. 313 A971) 3, 546—560.
214. Kunststoffe — Plastics A972) 5, 191.
215. Пат. США 3179630 20.4.65 Du Pont.
216. Пат. США 3179634 20.4.65 Du Pont.
217. Пат. США 3541057 17. 1.70 Du Pont.
218. Пат. США 324213G 22.3.66 Du Pont.
219. Пат. США 3376260 2. 4.68 Du Pont.
842
»20 Пат США 3391120 2.7.68 Du Pont.
I21 Пат США 3249588 3. 5. 66 Du Pont
!22 Пат США 3264250 2. 8. 66 Du Pont.
!23' Пат США 3424718 28. 1.69 Du Pont.
»24 Пат США 3342897 19.9.67 Du Pont.
!25 Пат США 3179633 20.4.65 Du Pont.
!26 Пат США 3179631 20.4.65 Du Pont.
J27 Пат США 3410875 12. 11.68 General Electric.
>28 Пат США 3440215 22. 4. 69 General Electric.
»29 Пат. США 3476705 4. 11. 69 3 M Со.
>30. Пат. США 3455879 15.7.69 Du Pont.
!31 Пат США 3536666 27. 10. 70 3 M Со.
J32 Пат США 3282897 1. 11.66 Du Pont.
!33. Пат. США 3516967 23.6. 70 Du Pont.
!34. Пат. США 3533997 13. 10. 70 Du Pont.
!35. Пат США 3380964 30. 4. 68 Rhodiaceta.
!36. Пат США 3533996 13. 10. 70 Rhodiaceta.
!37. Пат. США 3492270 27. 1. 70 Du Pont.
:38 Пат. США 3436372 1. 4. 69 Du Pont.
!39. Пат. США 3542744 24. 11. 70 H. Reimschuessel.
!40. Пат. США 3207728 21. 9.65 Du Pont.
!41. Пат. США 3502625 24.3.70 Eastman Kodak.
!42. С. Browning, J. Composites Mater. 4 A970), 390—403.
!43. А. Праведников, Высокомолек. соед., А 13, 1971, 8, 1863—1869,
!44. J. Zurukowska-Orszdk, Polimcry-Tworzywa 15 A970) 7, 344 449.
!45. С. Рафиков, Высокомолек. соед., Б 12, 1970, 3, 234—237.
!46. Опубл. заявка ФРГ 2223803 22. 2. 73 Westinghouse Electric.
!47, Пат. СССР 296785, 2.3.71.
!48. Фр. пат. 2087170 4. 2.72 Inst. Franc, du Petrole.
!49. Пат. США 2731447 A956) Du Pont.
:50. Пат. США 2712543 A955) Du Pont.
!51. Пат. США 2867609 6. 1. 59 Du Pont.
:52. Пат. США 2880230 A959) Du Pont.
!53. S. Marvel, J. Amer. chem. Soc. 80 A958), 1197—1199.
154. Пласт, массы, 9, 1967, 26—29.
155. С. Sroog, J. Polymer Sei. С 16 A967) 2, 1191—1209.
!56. Пат. США 3277043 A966) Du Pont.
157. R. Dine-Hart, J. appl. Polymer Sei. H A967) 5, 609—627.
!58. W. Wrasidlo, ACS Polymer Preprints 5 A964) 1, 141 — 146.
!59. M. Wallach, J. Polymer Sei. A 2 A968) 5, 953—960.
!60. Пат. США 3179632 20. 4. 65 Du Pont.
»61. Пат. США 3261811 A966) Du Pont.
!62. Англ. пат. 898651 18.9.59 Du Pont.
I63. Кан. пат. 659328 12.3.63 Du Pont.
!64. G. Bower, J. Polymer Sei. A 1 A963), 3135—3150.
I65. L. Frost, J. appl. Polymer Sei. 8 A964) 3, 1039—1051.
!66. С. Закощиков, Пластмассы, 1966, 4, 9—11.
I67. Р. Hergenrother, NARMCO-Report DDC-AD-602679, Narmco Research and
Development, April 1964.
!68. Англ. пат. 803347—803349 A959) Du Pont.
!69. Белы. пат. 649336, 649337 A967) Du Pont.
270. Англ. пат. 941158 A966) Du Pont.
271. Г. Колесников, Высокомолек. соед., Б 11, 1969, 8, 617—620.
272. F. Gay, J. Polymer Sei. A l 6 A968) 7, 1935—1943.
273. Кан. пат. 645073 A962) Du Pont
274. Пат. США 3073785 A963) Du Pont.
275. A. Рудаков, ДАН СССР, 161, 1965, 3, 617—619.
276. Е. Федорова, Высокомолек. соед., Б 10, 1968, 4, 273—277.
277. Л. Лайюш, Высокомолек. соед., А 9, 1967, 10, 2185—2192.
278. Пат. США 3190856 22. 6. 65 Shawingan Со.
279. J. Kreuz, J. Polymer Sei. A 1 4 A966) 9, 2607—2616.
843
280. А. Болдырев, ДАН СССР 163, 1965, 5, 1143—1146.
281. Фр. пат. 144875 A968) Rhone-Poulenc.
282. S. Nishizaki, Kogyo Kagaku Zasshi 73 A970) 8, 1873—1875.
283 С. Виноградова, Высокомолек. соед., А 13, 1971, 5, 1190—1198.
284. I. rabushi, Kogyo Kagaku Zasshi 67 A964) 7, 1084—1086.
285. Пат. США 2900369 A959) Du Pont.
286. С. Виноградова. Пзв АН СССР, сер. хим., 6, 1968, 1405—1407.
287. С. Виноградова, Кинетика и механизм полиреакции, тезисы симпозиума ПО
макромолекуляриой химии, Будапешт, 1969, 1, 53—57.
288. S. Nishizaki, Kogyo Kagaku Zasshi 68 A965) 2, 383—387.
289. S. Nishizaki, Kogyo Kagaku Zasshi 68 A965) 3, 574—576.
290. Пат. США 3347808 A967) Monsanto.
291. Фр. пат. 1473600 6.7 67 Hitachi Chem. Co.
292. Фр. пат. 1485931 16.5.67 Societe Rhodiaceta.
293. 1501198 2. 10. 67 Mobil Oil Со.
294. R. Myers, ACS Polymer Preprints 10 A969) 1, 186—192
295. A. Wurtz, Ann. 42 A854) 3, 54.
296. F. Gumpert, J. prakt. Chem. 31 A885), 119.
297 F. Gumpert, J. prakt. Chem. 33 A885), 293.
298. L. Otvos, Tetrahedron Letters 15 A960), 322—326.
299. С. Hurd, J. org. Chem. 24 A959), 388—392.
300. R. Myers, J. Polymer Sei. A 1 7 A969) 10, 2757—2762.
301. Англ. пат. 1105437 6.3.68 Soc. Rhodiaceta.
302. P. Tawney, J. org. Chem. 26 A961), 15—21.
303. T. Sheremeteva, J. Polymer Sei. С 16 A967), 1631—1646.
304. P. Kovacic, J. Amer, chem Soc. 81 A959), 1187—1190.
305. Белы. пат. 718016 A969) Rhone-Poulenc.
306. Нидерл. пат. 6818601 A969) TRW Inc.
307. Нидерл. пат. 6813940 A969) TRW Inc.
308. Нидерл. пат. 6809244 A969) TRW Inc.
309. Нидерл пат. 6809665 A969) TRW Inc.
310. Нидерл. пат. 6901939 A969) TRW Inc.
311. Фр. пат. 1555564 A969) Rhone-Poulenc.
312. Фр. пат. 1537135 A969) Rhone-Poulenc
313. Фр. пат. 1581983 A969) TRW Inc.
314. Пат. США 2444536 A948) Du Pont.
315. M. Yamada, Chem. High Polymers (Tokyo) 26 A969) 290, 401—407.
316. Kunststoffe 60 A970) 6, 401—402.
317. Kunststoffe 62 A972) 7, 424—426.
318. R. Кап, Tetrahedron 24 A968) 7, 3085—3093.
319 J. Stille, J. org. Chem. 26 A961), 4026—4029.
320. Пат. США 2890206 A959) Union Carbide.
321. Пат. США 2890207 A959) Union Carbide.
322. J. Stille, J. Polymer Sei. A 2 A964) 3, 1487—1491.
323. J. Stille, J. Polymer Sei. A3 A965) 6, 2397—2399.
324. J. Stille, Macromolecules 1 A968) 5, 463—464.
325. J. Krimmel, ACS 146th Meeting 1964, Abstracts.
326. H. Reimschuessel, J. Polymer Sei. В 4 A966) 12, 953—954.
327. H. Reimschuessel. Macromolecules 2 A969) 6, 567—569.
328. H. Reimschuessel, J. Heterocycl. Chem. 1 A964), 193.
329. H. Reimschuessel, J. org. Chem. 34 A9G9), 959.
330. Фр. пат. 1576862 23.6.69 Bayer AG.
331. Англ. пат. 1259641.
332. Б. Биксон. Высокомолек. соед., 12, А 1970, 1, 69—73.
333. М. Котон, ЖПХ, 38, 1965, 12, 2728—2734.
334 С. Виноградова, Высокомолек. соед., А 11, 1969, 12, 2725—2740
335. W. Collins, J. Polymer Sei. B3 A965), 81—82.
336. M Wallach, J. Polymer Sei. A2 5 A967), 653—662.
337. M. Wallach, ACS Polymer Preprints 8 A967) 2,1170.
338. С. Павлова, Высокомолек. соед., А 12, 1970, 56—72.
339. С. Павлова, Высокомолек. соед., Б 10, 1968, 6, 398.
844
340 M. Wallach, ACS Polymer Priprints 6 A905) 1, 53—63.
341 H Szymanski, J. Polymer Sei. B3 A965), 81—82.
342 T Asahara, J. ehem. Soc. (Japan), Ind. chem Sect. 72 A969) 6, 1389—1391.
343. G Br>wer: .T Polymer Sei. A 1 A968) 4, 877—887.
344 S. Bruck, ACS Polymer Preprints 5 A964) 1. 148.
345 S. Bruck, Polymer *5 A964) 9,435—446.
346. L. Wall, Sei. Techn. Aerospace Rep. 7 A969) 7, 1110.
347. G. Ehiers, J. Polymer Sei. A 1 8 A970), 3511—3527.
348. J. Heacock, SPE Trans. A965) 4, 105—110.
349. E. Butta, J. appl. Polymer Sei. 13 A969) 6, 1073—1081.
350. D. Kline, J. appl. Polymer Sei. 12 A968) 3, 593—604.
351 J. Jones, J. Polymer Sei. С 2 A969) 22, 773—784.
352. S Nishizaki, Kogyo Kagaku Zasshi 69 A966) 7, 1393—1395.
353. L. Amborski, ACS Polymer Priprints 4 A963) 1,175—182.
354. Machine Design 36 A964), 232
355. W. Tatum, 5th Electrical Insulation Conf., Chicago Proc. A963) I, 1—4
356. «Kapton», Firmenschrift Du Pont 1965.
357. J. Jones, Chem. Ind. (London) 1962, 1686—1688.
358. «Polyimide Technology» Firmenschrift Hughes Aircraft, 1965.
359. П. Фефелов, Механика полимеров, 6, 1969, Uli —1112.
360. W. Grunsteidl, Kunststoffe 58 A968) 11, 739—744.
361. Англ. пат. 50985 A966) J. Jones.
362. S. Bruck, Polymer 6 A965), 6, 319—332.
363. H Reynolds, Mater. Engin. 67 A968) 4, 94—95.
3S4. J. Hoggatt, Sei. Techn. Aerospace Rep. 4 A966) 15, 2855.
365. W. Parkinson, Report ORNL 4229 A967).
366. R. de Iasi, J. appl. Polymer Sei. 15 A971), 2965—2974.
367. P. Belloc, Rev. gen. Electricite 78 A969) 1, 50.
368. Англ. пат. 1038738 A966) Du Pont.
369. Нидерл. пат. 6504004 A964) Du Pont.
370. Фр. пат. 1432038 A966) Du Pont.
371. Пат. США 2900369 A959) Du Pont.
372. В. Рудаков, Хим. волокна, i966, 5, 20—23.
373. R. Irwin, J. Polymer Sei. С 19 A967), 41—48.
374. M. Котон, Вестник АН СССР, 36, 1966, 8, 56—63.
375. Plastverarbeiter 21 A970) 8, 758.
376. Firmenschrift Westinghouse Rep. A967) 7, AD 608002.
377. Kunststoffe A971) 10, 936.
378. B. v. Bornhaupt, Kunststoffe 61 A971) 1, 46—56.
379. Modern Plastics 47 A970) 3, 55.
380. Chem. Engin. 77 A970) 11, 97.
381. H. Щерба, Вестник машиностроения, 51, 1971, 4 43—45.
382. Plastverarbeiter 21 A970) 6,595.
383. G. Kline, Modern Plastics 47 A970) 7, 120—122.
384. W. Farrissey, ACS Polymer Preprints 9 A968) 2, 1581 — 1586
385. Пат. США 3542703 24. 11. 70 Monsanto.
386. V. Chase, SPI 23rd Techn. Conf 6.2. 1968.
387. R. Copeland, SPI Reinf. Plast. Сотр. 23rd Ann. Conf. 1968, 3 С 1—9
388. R. Pride. SPI Reinf. Plast. Сотр. 23rd Ann. Conf. 1968, 17 С 1—8
389. J. Freeman, SPE Trans. 5 A965) 2 75—63.
390. J. Courtright, Plast. Design. Processing 7 A967), 10.
391. E. Burns, Nation. SAMPE Teohn. Conf. Papers A969) 9
392. Bull. Nr. 5042 В Firmenschrift Monsanto.
393. J. Petker, SPI Reinf. Plast. Сотр. Div. 23rd Ann. Techn. Conf 1968 17-B
1-17-B 6.
394. P. Giuliani, Publ. Inst. Franc. Petr. 20 A971), 79—109.
395. Chem. Week 14. 1. 1967 S 21.
396. J. Schiller, SPI Techn. Conf. Papers 13 A967), 905—910.
397. Firmenschrift, Hughes Aircraft 1965.
398. Опубл. заявка ФРГ 2015080 28.3. 70 General Electric.
399. A. Marshall, Mat. Engin. 70 A969) 3, 42—43.
845
400. A. Marshall, SPI 25th Ann. Western Sect Conf. .Mai 1968.
401. R. Kantner, Adhesives Age 12 A969) 11, 24—32.
402. B. Pascuzzi, Adhcsivps Age 14 A971) 2, 26—33
403. Adhasion 10 A968), 448—449.
404. H. Burgman, J. appl. Polymer Sei. 12 A968), 805—829.
405. J. Mahon, Sei. Techn. Aerospace Rep. 7 A969) 7, 1158.
406. E. Бабин, ЖПХ, 1973, 3, 598—604.
407. В. Коршак, Тр. Моск. хим-техн. ин-та, 1972, 70, 172—174.
408. С. Виноградова, Тр. Моск. хим-техн. ин-та, 1972, 70, 168—171.
409. Т. Размахнина, Тр. Моск. хим-техн. ин-та, 1972, 70, 215—217.
410. Фр. пат. 2136181 9.2.73 Du Pont.
411. Пат. США 3700538 24. 10.72 U. S. Aeronaulics a Space Admin.
412. Фр. пат. 2109447 30.6.72 С. N. E. S.
413. Пат. США 3249561 3. 5. 66 Du Pont.
414. Пат. США 3483144 9. 12.69 Monsanto.
415. Пат. США 3520837 21.7.70 Monsanto.
416. Пат. США 3554935 12. 1.71 Monsanto.
417. Пат. США 3554939 12. 1.71 Monsanto.
418. W. Farrissey, J. appl. Polymer Sei. 14 A970), 1093—1101.
419. H Relies, J. Polymer Sei., Polymer Chem. Ed 11 A973) 3, 561—571.
420. H. Relies, J. org. Chem. 37 A972), 3637—3645.
421. Англ. пат. 903271 A962) Du Pont.
422. M Russo, J. Polymer Sei. 7 A969) 12, 3337—3349.
423. Plastverarbieter 24 A973) 9, 579.
424. Kunststoffe 63 A973) 5, 342.
425. J. Preston, Appi. Polymer Symp. 9 A969), 107—117.
426. J. Frost, J. Polymer Sei. А Гб A968) 1, 215—233.
427. J. Preston, J. Polymer Sei. A l 5 A967), 2429—2438.
428. J. Preston, Polymer Preprints 9 A968) 2, 1143—1149.
429. J. Preston, Polymer Preprints 6 A965) 2, 757—760.
430. J. Preston, J. Polymer Sei. A 1 7 A969), 10, 3027—3037
431. J. Preston, Appl. Polymer Sei. 9 A967), 145—158
432. J. Preston, J. Polymer Sei. 7 A969), 283—296.
433. Фр. пат. 1561519 28. 3.69 Bayer.
434. J. Preston, J. Heterocycuc Chem. 2 A965), 441—448.
435. P. Hergenrother, J. Polymer Sei. A3 A965), 1665—1674.
436. J. Preston, J. Heterocyclic Chem. 5 A968), 269—275.
437. Фр. пат. 2031817 20. 11.70 Inst. Franc, du Petrole.
438. В. Коршак, Высокомолек. соед., А 13 1971, 4, 971—976.
439. А. Токарев. Хим. волокна, 5, 1970, 20—22.
440. Акцепт, заявка Японии 7102110 19. 1.71 Showa Electric.
441. Акцепт, заявка Японии 7102111 19. 1.71 Showa Electric.
442. J. Preston, Appl. Polymer Symp. 9 A969), 145—158.
443. J. Preston, J. Polymer Sei. A 14 A966), 529—539.
444. Y. Imai, Chem. High Polymers (Japan) 27 A970) 305, 664—660
445. Опубл. заявка ФРГ 18U107 11.6. 70 Bayer.
446. Акцепт, заявка Японии 7121656 19.6.71 Furukawa Electric.
447. M. Bruma, Rev. Roumaine Chjm. 16 A971) 6, 935—940.
448. Акцепт, заявка Японии 7127820 12.8.71 Toray Ind.
449. Акцепт, заявка Японии 710840 1.2.71 Toray Ind.
450. Акцепт, заявка Японии 7024791 18.8.70 Toray Ltd.
451. M. Котон, Высокомолек. соед., А 14, 1972, 9, 2034—2041.
452. Акцепт, заявка Японии 7117904 19.5.71 Toray Ind.
453. J. Preston, J. Polymer Sei., Polymer Chem. 10 A972) 11, 3373-3386.
454. N. Dokoshi, J. Polymer Sei. A 1 8 A970), 8, 2197—2217.
455. W. Wrasidlo, J. Polymer Sei. A 1.7 A969) 7, 1589—1598.
456. В. Bol ton, Polymer Engin. Sei. 6 A966), 3, 227—230.
457. A. Токарев, Хим. волокна, 1970, 5, 20.
458. S: Terney, J. Polymer Sei. A 1 8 A970), 683—692.
459. Л. Чудина, Пластмассы, 1970, 8, 12—16.
460. L. Frost, J. Polymer Sei. A 1 A963), 3135—3150.
846
461. Фр. пат. 1386617 A964) Ariioco.
462 Г Колесников, Высокомолек. соед., А 10, 1968, 8, 1845—1851.
463. W. Alvino, J. Polymer Sei. А 1 9 A971), 2209—2224.
464. VV. Alvino, J. appi. Polymer Sei. 15 A971), 2123—2140.
465. В. Родэ, Высокомолек.'соед., А 12. 1970.7. 1566—1573.
46G. Paint Technology 35 A971) 6, 46—47.
467. H. Адрова, Высокомолек. соед., Б 13, 1971, 12, 900—902.
468. J. Chambion, Chimie & Industrie, Uenie chim. 106 A973) 7, 453-458.
469. Пат. США 3453236 1. 7. 69 Ashland О il Co.
470. Пат. США 3541038 17. 11.70 Hitachi Ltd.
471. Пат. США 3518230 30.6. 70 Schenectady Chem.
472. Y. Imai, Kobunshi Kagaku 29 A972) 323, 182—185.
473 H. Uchiyama, Chem. High. Polymers (Japan) 28 A971) 309, 73—79.
474. H. Uchiyama, Chem. High Polymers (Japan) 27 A970) 305, 667—669.
475. H. Uchiyama, Chem. High. Polymers (Japan) 28 A971) 309, 80—84.
476. D: Kruh, Polymer Leiters 11 A973), 2, 139—146.
477. W. Dunwald,'Kunststoffe 62 A972) 6, 347—350.
478. С. Рафиков, Высокомолек. соед., А 14, 1972, 10, 2201—2204.
479. Б. Жубанов, Высокомолек. соед., Б 13, 1971, 8, 618—621.
480. О. Федотова, Высокомолек. соед., А 13, 1971, 8, 1886—1893.
481. F. Hayano, J. Polymer Sei. А 1 10 A972) 4, 1263—1266.
482. M. Nakano, Hitachi Review 20 Nr. 12, 503—509.
483. M. Bruma, Rev. Roumaine Chim. 16 A971) 10, 1591—1599.
484. S. Bade, Angew. Makr. Chemie 21 A972), 65—77.
485. Пат. США 3597392 3. 8. 71 Monsanto.
486. Акцепт, заявка Японии 7221592 17.7.72 Toray Ind.
487. Акцепт, заявка Японии 7225475 11.6.72 Toray Ind.
488. Акцепт, заявка Японии 7225478 11.6.72 Toray Ind.
489. Акцепт, заявка Японии 7237707 22. 10. 72 Toray Ind.
490. Англ. пат. 1309501 14.3.72 Toray Ind.
491. Опубл заявка ФРГ 2239707 22.2.73 Rhone Poulenc.
492. Заявка Яп. 7217856 11.9.72 Toray Ind.
493. Яп. заявка 7230782 9. 11.72 Toray Ind.
494. Яп. заявка 7232835 22.8.72 Toray Ind.
495. Яп. заявка 7230312 7. 8. 72 Teijin'Ltd.
496. Яп. заявка 7230311 7.8.72 Toray Ind.
497. J. Adduci, ACS Polymer Preprints 12 A971) 1, 611—614.
498. Яп. заявка 7220394 17.6.72 Toray Ind.
499. Фр. пат. 2105446 2.7.72 Inst. Franc, du Petrole.
500. Опубл. заявка ФРГ 2155678 25.5.72 General Electric.
501. Яп. заявка 7211836 13.4.72 Toray Ind.
502. Яп. -э*явка 7207468 2. 3. 72 Toray Ind.
503. Яп. заявка 7203988 3. 2. 72 Toray Ind.
504. Акцепт, заявка Японии 7141394 7. 12.71 К. Tanaka.
505. H. Адрова, Высокомолек. соед, Б 13, 1971, 10, 761—763.
506. Опубл. заявка ФРГ 2124884 2. 12.71 Allied Chem.
507. Опубл. заявка ФРГ 2029078 9. 9. 71 M. Suzuki.
508. Опубл. заявка ФРГ 2106704 26. 8. 71 Rhodiaceta.
509. Акцепт, заявка Японии 7127178 6.8.71 Toray Ind.
510. Фр. пат. 2045566 9. 4. 71 Rhodiaceta.
511. Акцепт, заявка Японии 7116909 10.5.71 Teijin Ltd.
512. Y. Imai, Makromolekulare Chem. 94 A966), 114—123.
513. Акцепт, заявка Японии 7116908 10.5.71 Teijin Ltd.
514 Пат. США 3049518 A962) Du Pont.
515. Пат. США 3179635 A965) Westinghouse Electric.
516. Пат. США 3260691 A966) Monsanto.
517. Пат. США 2421024 A942) С. Frosch.
518. T. Shono, J. chem. Soc. (Japan), Ind. chem. Sect. 71 A968) 10, 1738—1741.
519. T. Shono, J. chem. Soc. (Japan), Ind. ehem. Sect. 72 A969) б' 1392—1394*
520. С. Рафиков, Высокомолек. соед., Б 11, 1969, 3, 165 — 169.
521. Y. Imai, Chem. high Polymers (Japan) 27 A970) 305, 670-672.
347
522. Y. Imai, Kobunshi Kagaku 28 A971) 309, 73—79.
523. Акцепт, заявка Японии 7115573 25.4.71 Teijin Ltd.
524. Фр. пат. 1386617 A964) Standard Oil.
525. T. Unishi, Kogyo Kagaku Zasshi 74 A971) 1, 121—124.
526. Пат. США 3182073 A965) D. Loncrini.
527. M. Котон, Хим. волокна, 1969, 4, 15—22.
528. M. Hasegawa, J. chem. Soc. (Japan), Ind. ehem. Sect. 70 A967) 12, 2392—
2395
529. M. Котон, Высокомолек. соед., Б 11, 1969, 7, 507—509.
530. В. Sillion, Bull. Soc. chim. France 5 A967), 4296—4298.
531. Y. Imai, Chlym. High Polymers 27 A970) 302, 384—393.
532. Y. Imai, Chem. High Polymers 27 A970) 305, 667—669.
533. Опубл. заявка ФРГ 2043994 11.3.71 Rhone-Poulenc.
534. Фр. пат. 1577256, 1575839, 1566393 A965) Rhone Poulenc.
535. В. Коршак, Высокомолек. соед., Б 9, 1967. 10, 736.
536. H. Freeman, SPE Trans. 2 A962), 216—221.
537. «Bulletin AI Polymer», Firmenschrift, Amoco Chem., 1965.
538. Kunststoff-Berater 7 A962) 6, 385.
539. R. Pigeon, Textilia 47 A971) 7, 35—39.
540. J. Corbiere, Publ. Inst. Franc, du Petrole 20 A971), 111—133.
541. T. Unishi. J. Polymer Sei. B3 A965) 8, 679—383.
542. T. Unishi, Kogyo Kagaku Zasshi 68 A965) 11, 2275—2277.
543. Англ. пат. 1058236 A963), 1041868 A964), 1045395 A965) Bayer AG
544. Пат. ФРГ 1266427 A963) Bayer AG.
545. Белы. пат. 719083 A968) Bayer AG.
546. Опубл. заявка ФРГ 1770202 A965) Bayer AG.
547. J. Freeman, SPE Transactions A965) 4, 75—83.
548. D. Loncrini, J. Polymer Sei. A 1 4 A966), 1531 — 1541.
549. M. Котон, Хим. волокна, 1969, 4, 15—22.
550. Пат. США 3182073 A965) General Electric.
551. Англ. пат. 973377 A964) Dr. Beck & Co.
552. К. Schmidt, Electrotechn. Ztg. В 15 A963) 21, 603-607.
553. D. Lindenberg, Farbe & Lack 76 A970) 4, 365—369.
554. Опубл. заявка ФРГ 1494452, 1494454, 1520061, 1520068 Dr. Herberts & Co.
555. Англ. пат. 973377 A964) Dr. Beck & Co.
556. Oesterr. P. 245701 Chem. Werke Witten.
557. Пат. ФРГ 1285653 Dr. Herberts & So.
558. Опубл. заявка ФРГ 1640423 Dr. Herberts & Co.
559. Англ. пат. 1082181 Schenectady Chem.
560. P. Novak, Kunststoff-Rdschau A965) 12, 120.
561. H. Адрова, Высокомолек. соед., сер. Б, 1968, 137—139,
562. К. Власова, Пластмассы, 1969, 2, 25—26.
563. В. Таликов, Высокомолек. соед., А 11, 1969, 6, 1303—1308.
564. D. Loncrini, Proc. 6th Electrical Insulation Conf. A965).
565. Опубл. заявка ФРГ 1494457 Dr. Herberts & Co.
566. Z. Jedlinski, Thermal Analysis, Vol. 3, Proc. Third ICTA Davos 1971, 197—
203.
567. Яп. заявка 7234490 21. 11.72 Teijin Ltd.
568. Пат. США 2985624 A961) Anaconda WLre & Cable.
569. Опубл. заявка ФРГ 2052330 27.4.72 Cella Lackfabrik.
570. Е. Гуцалюк, Изв. АН Каз. ССР, сер. хим., 21, 1971, 5, 71—74.
471. Н. Адрова, Высокомолек. соед., Б 13, 1971, 10, 764—767.
572. Е. Гуцалюк, Изв. АН Каз. ССР, сер. хим., 21, 1971, 6, 50—55.
573. М. Котон, Высокомолек. соед., А 15, 1973, 2, 310—313.
574. Н. Адрова, Высокомолек, соед., А 14, 1972, 10, 2166—2173.
575. А. Рудаков, Высокомолек. соед., Б 13, 1971, 10, 764—767.
576. Р. Penczek, Farbe u. Lack 78 A972) 1, 37—42.
577. Пат. США 3274159 20. 9. 66 Union Carbide.
578. Пат-США 3377321 9.4.68 Standard Oil.
579. В. Vollmert, Kunststoffe 56 A966) 10, 680—694.
580. G. F. d'Alelio, J. Macromolecul, Sei. A3 A969) 5, 927—940.
848
581 G F. d'Alello, J. Maeromolecul. Sei. A3 A969) 5, 941—958.
582 G F d'Alelio, J. Macromolecul. Sei. A3 A969) 6, 1105—1124
583 G. F. d'Alelio, J. Macromolecul. Sei. A 5 A971) 6, 1097—1121.
584 G F. d'Alelio, J. Macromolecul. Sei. A4 A970) 1, 169—185.
585. G. F. d'Alelio, J. Macromolecul. Sei. A 5 A971) 2, 383—420.
586 Пат. США 3547169 6.4.71 U. S. Air Force.
587. Mater. Engin. 77 A973), 4, 34.
688. Mod. Plastics Int. 3A973), II.
7.2. ПОЛИБЕНЗИМИДАЗОЛЫ
HN NH
Полибензимидазолы получают поликонденсацией дифениловых
эфиров дикарбоновых кислот с тетраминами. Эти полимеры
устойчивы в инертной среде и на воздухе до 500 °С. Поли-ж-фениленди-
бензимидазольные волокна получают методом сухого формования
из растворов в диметилацетамиде с добавкой LiCl. Волокна могут
кратковременно выдерживать нагревание до 450 °С и используются
для изготовления огнезащитной и рабочей одежды,
выдерживающей высокую температуру. Синтактические пены на основе поли-
бензимидазола являются прекрасным изоляционным материалом
для работы при высоких температурах; они обладают высокой
стойкостью к абляции.
7.2.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗИМИДАЗОЛОВ
7.2.1.1. Поликонденсация производных дикарбоновых кислот
с тетраминами в расплаве или растворе
Впервые полибензимидазолы были получены в 1959 г. {2]
взаимодействием алифатических дикарбоновых кислот с
ароматическими тетраминами:
H2N.
п
H2N
О
Ч)Н
—R-
N
—п
Однако вследствие декарбоксилирования и нарушения при этом
стехиометрического соотношения при поликонденсации таких
849
мономеров образуются только низкомолекулярные продукты. В
основе метода лежит реакция, открытая в 1878 г., по которой
взаимодействием о-фенилендиамина с муравьиной кислотой был получен
бензимидазол [1].
При использовании хлорангидридов кислот реакция протекает
очень быстро и сопровождается "образованием солей. В случае
метиловых эфиров кислот протекает побочная реакция метилирования
аминогрупп, приводящая к нарушению соотношения реагирующих
веществ [3].
Наиболее подходящими для получения полибензимидазолов
оказались дифениловые эфиры дикарбоновых кислот. Синтез
проводят при стехиометрическом соотношении тетраминов и дифени-
ловых эфиров дикарбоновых кислот в инертной среде при 250 —
300 °С и пониженном давлении с получением на первой стадии
форполимера, который в измельченном виде в твердом состоянии
при 400°С в высоком вакууме превращается в
высокомолекулярный полибензимидазол [4, 111]. Вторую стадию проводят также в
токе инертного газа при атмосферном давлении [5]. Образующиеся
при этом полимеры отличаются низким содержанием гельфракции
и повышенной растворимостью.
Промышленный способ получения поли-ж-фенилендибензими-
дазола рассмотрен в разделе 7.2.1.7. Масс-спектрометрический
анализ летучих соединений, выделяющихся в ходе
поликонденсации в расплаве, показал, что они содержат только воду и фенол,
отщепляющиеся одновременно [21]. На первой стадии процесса
при 300 °С выделяется половина стехиометрического количества
воды и практически весь фенол. При 400 °С количество
выделившейся воды достигает второго максимума, что обусловлено
дальнейшей поликонденсацией форполимера, образовавшегося на
первой стадии процесса. Состав полимера после первой стадии
процесса не отвечает поли-лг-фениленбисбензимидазолу C20H12N4, a
соответствует строению частично гидратированного продукта [22—24]:
NH
NH3
Полигидроксиимидазолин
-NHCO—^A^—
Полиамид
По данным ИК-спектроскопии [21], преобладают гидроксиими
дазолиновые группы. Исходя из этого, Фогель [4] предложил сле
дующий механизм процесса:
он
350
О
ОН
Q)—NH—С
n:-
"h.
-H2O
Поликонденсация протекает аналогично процессу омыления
полиэфиров и щелочного гидролиза амидов. Аддукт, образующийся на
первой стадии, стабилизируется за счет отщепления фенола.
В пользу такого механизма образования полибензимидазолов
могут служить и данные о деструкции полимера [23].
Другой механизм образования полибензимидазолов предложен
Левиным [25] и Врасидло [26]. Они предполагают, что на
промежуточной стадии образуется альдоль, дегидратация которого
приводит к получению основания Шиффа, превращающегося при
отщеплении фенола в бензимидазол:
NH2
+н2о
Поликонденсацию проводят также в растворе фенола, крезола,
днметилформамида, диметилацетамида, диметилсульфоксида, N-ме-
тилпирролидона или полифосфорной кислоты. При использовании
фенола эквимольные количества тетрамина и дифенилового эфира
дикарбоновой кислоты нагревают в среде инертного газа (во
избежание окисления тетрамииа) при непрерывном удалении фенола
до образования твердого продукта. Для превращения в
высокомолекулярный продукт измельченный форполимер прогревают в
вакууме при 350—400 °С. Применение в качестве растворителя ди-
метилформамида и диметилацетамида предпочтительно в тех
случаях, когда полимерный раствор предлагается использовать в
качестве связующего. Молекулярная масса полимеров, образующихся
в растворе, вследствие протекания реакции переамидирования
меньше, чем при синтезе их в расплаве [89].
851
Таблица 7.25. Температуры размягчения и разложения полибензимидазолов
Элементарное звено
разм1
разл
(в N2),
°С
разл
(на
воздухе),
N
HN
N
HN
N
N
NH
450
Тил = 490
590
450
550
420
10
V
NH
N
О
HN
H
Имидайт 850 («Narmco»)
N
N
N
4NH
N
HN
N
О
390
> 400
540
600
480
500
500
[4, 121]
[4, 81, 78, 79,
83-85, 90, 91,
93, 97, 100, 105,
107, 110, 112, 114,
115, 11.9, 125]
[4]
[41
[961
Продолжение табл. 7.25
11
12
13
14
N
N
N
HN
N
HN
Элементарное звено
N
N
N
NH
N
rv
•О
/ 'Ш
N
N
• о
^1
N
¦uc>-
г
разм*
"С
Гразд
(в N2).
550
71
раз л
(на воз-
Д?се>"
Литературный
источник
[58]
[103]
[58]
[4]
15
16
17
18
19
N
N
H
N
On
HN
N
N
Q
N
HN
N
NH
N
<
HN
>
NH
200
400
500
430
465
500
Продолжение табл. 7.25
разл'
°С
Гразл
(в N2i,
т
1 разл
(на воз-
Литературный
источник
460
400
N
HN
22
Элементарное звено
N
HN
СНз
23
24
25
26
27
28
N
N
N N
Ч_.^Л L
N
-О
N
HN
N
N
СНз СНз
NH
N
СНз СН
N
О
NH
V-(CH2)a-
N
N
H
\—CH=CH—
H
N
N
/ ^«i-1
с
HN
(СН2),
530
Гпл = 420-450
= 470
275, Гпл = 420
585
450
600-700
[80]
[27]
1130)
[4, 60]
[3J
[30, 60]
Продолжение табл, 7.25
Т
разл'
т
разл
J разл
(на
воздухе),
°С
Тпл =: 450
410
350
410
350
N
N
Элементарное звено
V—(СН2L-
H
N
CF2L-
N
0
N
N
/
HN
NH
N
л
NH
N
(CH2K-S-(CH2K-
CH3 СНз
[Qj (Q) 4—(CH2K-si-o-si-(CH2K
NH СНз СНз
234t
Гпл = 470
220
400—410
380,
Тпл = 500
350
490
Продолжение табл. 7.25
Элементарное звено
Т
разл1
°С
Т
разл
(в N2),
°С
Т
разл
(на воз-
у>
Литературный
40
41
42
43
44
N
HN
N
HN
CH2 N R
N CH2 N
CH2 N
О
N CH2 N
\—(СН2L-
'jQ \ y—(CH2K~S-(CH2K
360
390
490
[65]
[64, 65, 86]
[64, 65]
{66]
[63]
N
О
HN
N
о—(Г IV-
NH
225
260-275
380-400
Продолжение табл. 7.25
№
Элементарное звено
разл1
°С
Г
разл
(в N2),
°С
Г
разл
(на
воздухе).
R"
51
52
53
<0-so'
54
N
N
S02
N
Ж
>
NH
N
О
SO2
N
О
О
55
56
57
58
59
N
v
HN
N
N
HN
N
О О N
H
N О О N
NH
О О N
N N
и-
N N
N
О
NH
N
4
NH
Гпл=510
[67]
[67]
[67]
Тпл > 400
500
[80]
520
[80]
Продолжение табл. 7.25
№
60
61
62
63
N
-о
\ /
HN
N
-o
HN
0'
HN
HN
HN
HN
U
O)
X
IQ
0
M M
11 J
0
>^
N
OC)
CH2
CH2
Элементарное эвено
N 1
^NH ^4^
NH
NH
NH
lOl
разл1
°C
Тпл = 480
500
T
разл
(в N2),
°C
480
500
T
разл
(иа воз-
духе),
400
Литературный
источник
[80]
[ИЗ]
[64, 65]
[68, 69]
00
S? 64
HN
О NH
OrUC
NH
HN О
65
C6H5
N
66
N
N
А.
67
C6H5
Гпл = 450
300
510
460
480
Продолжение табл. 7.25
CeH5
N
Элементарное звено
N
Ч
N
С,Н»
с.н.
с6н5
разл-
°С
л разл
(на воз-
Литературный
источник
[70]
[701
[70J
СНз
N
\
N
—(CF2K-
N
C
F2]
171]
[62]
Продолжение табл. 7.25
Элементарное звено
1 разл1
•С
т
разл
Гразл
(на
воздухе),
°С
СНз СНз /СНз
74
75
CHav СНз СНз /СНз
76
77
345
335
320
350
450
410
415
450
78
79
СНзч СНз СНз /СНз
N | СНз I N
80
О
/
81
380
340
245
230
480-490
450
410
395
[72]
[72]
[72]
[72]
Продолжение табл. 7.25
Элементарное звено
разл1
Т
'разл
(в N2),
°С
т
разл
(на воз-
N
N
N
СНз
N
СН2 ! N
Ж
N
210
400
85
86
87
N
HN
N
H
N
HN
и
Г®
N R N
J
HN
О
•— R = —CH2
88
NH—С—R—
Hi
>500
500-560
700
600*
500
450-500
I122I
[871
[106, 128]
[9, 731
Продолжение табл. 7.25
Элементарное звено
разл1
°С
т
разл
(в N2),
°С
Т
разл
(на воз-
Духе),
°С
N
NH
N
H
N
H
-NH— С—R— С—NH-
O О
j_C~NH—(CH2N—NH—С—(СН2)8
II 11
О О
(СН2)8—С—О—(СН2N—О—С—(СН2)8-
NH О О
NH
NH-C—(( ))—
О
430
Высокомолекулярные полибензимидазолы можно получить в
одну стадию в полифосфорной кислоте A16%) при 130—250 °С
[б—ц? 107]. Во избежание окисления вместо самих тетраминов
используют их хлоргидраты, которые при температуре выше 150 °С
отщепляют НС1 и поэтому при температуре синтеза в
полифосфорной кислоте существуют в виде свободных оснований. Таким же
образом из ароматических бис(о-анилиноаминов) и дифениловых
эфиров дикарбоновых кислот получают термостойкие поли-N-
фенилбензимидазолы (табл. 7.25, № 65—71) [3, 62, 70, 71, 78, 89,
100, 102, 127]:
с6н5осо—/QV-cooc6h5 -
свн5он
—н2о
П
Циклизация промежуточных о-замещенных полиамидов
протекает в две стадии [78] :
= 14 ккал/моль
ДЯ = 9 ккал/моль
п
873
Ниже приведены термодинамические константы этого процесса:
Температура циклизации, К
298 400 500
Н° 0,9 22 23
S° 0,4 56,4 58,6
G° +0,8 —0,6 -6,3
Степень циклизации поли-1М-фенилбензимидазолов,
определенная по теплотам сгорания [78] и ИК-спектральным методом [89],
составляет 90—95%. Поликонденсацией ароматических
«полиаминов» с дифениловыми эфирами дикарбоновых кислот в расплаве
были получены полимеры с боковыми бензимидазольными
группами [90]:
«H2N—r^^^NH-<^^-<f:^V-NH-I^^---NH2 +
—H2O
-CeH5OH
При использовании диангидридов тетракарбоновых кислот
образуются полибензоиленбензимидазолы лестничной структуры [12]:
H2N
О
JLn
Подробнее полимеры этого класса рассмотрены в гл. 8.
§74
Полибензимидазолы образуются в одну стадию при
взаимодействии тетраминов ароматического ряда с динитрилами
ароматических дикарбоновых кислот в присутствии аммонийных солей,
например хлорида аммония или сульфата аммония, или алкокси-
соединений [13, 14, 93, 97, 112]:
NH2
Реакцию проводят в твердой фазе при 200—400 °С в среде
инертного газа или в вакууме. Описан синтез полибензимидазолов
из тетраминов и иминоэфиров дикарбоновых кислот [16, 17]:
H2Ns
ЛМН2
-NH3; -CeH5OH
7.2.1.2. Поликонденсация диальдегидов с тетраминами
При взаимодействии тетраминов с бисульфитными
производными диальдегидов в растворе диметилацетамида, диметилформ-
амида, диметилсульфоксида или N-метилпирролидона с
практически количественным выходом образуются полибензимидазолы
[14, 18,115]:
H2N
п
H2N
I
ОН
СН— SO3Na
ОН
—NaHSOs;
—Н2О
п
875
7.2.1.3. Взаимодействие диацетильных производных
ароматических соединений с тетраминами
Поликонденсацию проводят в две стадии [19, 20]:
+ ttCH3CO (О)—СОСНз
*NH2
-н2о|
250 °С
N=?~O~?-
I
СНз
I
Н3С
NH2
H2N
-in
п
На первой стадии в расплаве образуются полиазометины,
которые при 300 °С отщепляют метан и превращаются в полибензими-
дазолы.
7.2.1.4. Термическая циклизация поли-(о-ацетамидо)амидов
При взаимодействии 4,4'-'Диацетамидо-3,3'-Диаминодифенил-
окснда с дихлорангидридами дикарбоновых кислот образуются
поли (о-ацетамидо) амиды [15]:
О
H2N
NHCOCH3 + «C1OCRCOC1
—НС1
О
Н3С С—NH—/Q>\—'
—COHN
_«
Термическая циклизация полимера, полученного с использовав
нием алифатических дикарбоновых кислот, например адипиновой
87fi
кислоты, протекает с отщеплением уксусной кислоты:
N
NHCOCH3
NHCO(CH2b— J„
t
—CH3COOH
Иная картина наблюдается при использовании в качестве аци-
лирующего агента дихлорангидрида ароматической дикарбоновои
кислоты. На первой стадии процесса циклизации выделяется вода
и образуется поли-К-ацетилбеизимидазол. При дальнейшем
повышении температуры происходит отщепление СО и образование
поли-Г^-метилбензимидазола:
NHCOCHs
NHCO—rA|—
—п
t
-СО
п
7.2.1.5. Поликонденсация ароматических я-бис-
(гидроксииминохлорметильных) соединений
с ароматическими диаминами
Поликонденсацию таких соединений с ароматическими
диаминами проводят в растворе диметилацетамида или диметилформ-
амида в присутствии акцепторов НС1 при 20—240 °С, получая на
первой стадии растворимые полнамидоксимы, которые в
присутствии дегидратирующих средств, таких, как пиридин и /или /г-то-
луолсульфохлорид, превращаются в полибензимидазолы [126]:
HON
п
ч
NOH
I
—HG1
877
7.2.1.6. Поликонденсация с использованием соединений,
содержащих имидазольные циклы
При конденсации тетраминов с нитрокарбоновыми кислотами
с последующим восстановлением продуктов реакции образуются
диамины с бензимидазольными гетероциклами:
^\ H2N
НООС—|V-V|—NO2
^^ HoN
H2N-
NH2
Взаимодействие этих соединений с диизоцианатами приводит
к образованию полибензимидазолмочевин [95], а с дихлорангид-
ридами дикарбоновых кислот — полибензимидазоламидов (табл.
7.25, № 81). В результате поликонденсации 2,2'-бис(о-аминофе-
нил^-б^'-дибензимидазола с дихлорангидридами дикарбоповых
кислот с одновременной циклизацией получают полибензимидазо-
хиназолины [132, 133]:
878
7.2.1.7. Промышленные способы получения пол и-2,2'-ж-
фенилен-5,5'-дибензимидазола
Доступность исходных продуктов и наличие подходящих
растворителей позволило создать промышленное производство поли-
бензимидазола на основе диаминобензидина и дифенилизофтала-
та. Последний получают из фенола и изофталоилхлорида при
130°С {Тпп = 77—78°С). Синтез диаминобензидина основан на
нитровании предварительно ацилированного бензидина с
последующим восстановлением динитросоединения железом [30].
Канцерогенная активность бензидина требует особых мер
предосторожности при работе с ним. Так как ароматические амины легко
окисляются, синтез проводят в инертной среде. Для очистки от
следов железа тетрамин перекристаллизовывают из кипящей
воды в присутствии активированного угля. Бесцветные кристаллы
плавятся при 176—178°С [5]. Для получения полибензимидазола
примерно стехиометрические количества диаминобензидина и ди-
фенилизофталата нагревают в токе азота в реакторе и
перемешивают в течение 1,5 ч при 290 °С. По достижении необходимой
вязкости мешалку выключают, и форполимер вспенивается за счет
выделения фенола и воды. Объемная пена после измельчения
в шаровой мельнице поступает в другой реактор, в котором
порошок нагревается в токе азота 3 ч при 385 °С. Выделяющийся
фенол, вызывая набухание полимера, способствует ускорению
реакции, протекающей в твердой фазе. Образующийся поли-2,2Чи-
фенилен-5,5'-дибензимидазол имеет характеристическую вязкость
в концентрированной серной кислоте 0,72—1,00 дл/г. Дифенил-
изофталат вводят с небольшим избытком @,2—0,5%), чтобы
компенсировать его частичный унос вместе с удаляемой из сферы
реакции водой [5]. Избыток диаминобензидина приводит к получению
низкомолекулярного продукта, в то время как при избытке дифе-
нилизофталата образуется высокомолекулярный полимер,
значительная часть которого представляет собой трудно фильтрующийся
гель.
7.2.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИБЕНЗИМИДАЗОЛОВ
Полибензимидазолы представляют собой продукты от желтого
до оранжевого цвета; плотность их составляет 1,2—1,4 г/см3.
Некоторые их свойства приведены в табл. 7.25.
7.2.2.1. Температурные переходы
Незамещенные полностью ароматические полибензимидазолы,
в которых отсутствуют «шарнирные» группы и атомы в нуклео-
фильном компоненте, а электрофильный компонент состоит из
я-замещенных или конденсированных ароматических ядер,
разлагаются йри температуре ниже температуры размягчения или
плавления (табл. 7.25, № 2, 6, 13—17). Полимеры, содержащие
m
«шарнирные» атомы или группы в остатке кислоты или тетрами-
на, размягчаются в интервале температур 250—450°С.
Длительная термообработка полибензимидазолов в высоком вакууме
приводит к повышению температуры размягчения на 20—50° [86].
Температура размягчения полимера тем ниже, чем выше в нем
содержание групп, повышающих гибкость макромолекулы. Поли-
бензимидазолы с двумя или более простыми эфирными связями
в звене (табл. 7.25, № 55—57) плавятся в интервале
температур 300—500 °С. Плавкие продукты образуются и в том случае,
если в состав тетрамина и/или кислотного компонента входят
оксадиазольные или фурановые циклы (табл. 7.25, № 9, 23,
58—60). Полибензимидазолы с алифатическими звеньями в цепи
(табл. 7.25, № 4, 26—35, 43) размягчаются в интервале
температур 230—280°С, температура их плавления составляет 420—
470 °С.
Поли-Ы-алкил(арил)бензимидазолы из-за уменьшения вклада
водородных связей и наличия объемистых заместителей в боковой
цепи имеют низкую степень упорядоченности и четко выраженную
температуру размягчения. Самая высокая степень кристалличности
у полибензимидазолов на основе тетраминобензола и
ароматических гс-дикарбоновых кислот (максимальная жесткость
макромолекулы, высокая упорядоченность за счет водородных связей).
7.2.2.2. Растворимость и свойства растворов
Ароматические полибензимидазолы растворяются в
концентрированных кислотах, таких, как серная, муравьиная и метансульфо-
кислоты. В апротонных растворителях (диметилформамид, диме-
тилацетамид, диметилсульфоксид, N-метилпирролидон, гексаметил-
фосфотриамид) они растворяются только с добавкой хлорида
лития. После растворения поли-ж-фенилендибензимидазола в диме-
тилацетамиде через 1—3 сут при комнатной температуре
наблюдается гелеобразование. Это происходит за счет кристаллизации
полимера с одновременной сольватацией [5]. При повышении
температуры до 140 °С полимер снова переходит в раствор. Растворимость
ароматических полибензимидазолов тем хуже, чем выше симмет-
тричность и жесткость макромолекулы.
Полимеры, содержащие в своем составе остатки дикарбоно-
вых кислот, в которых карбоксильные группы находятся в
«-положении, всегда значительно хуже растворяются, чем
соответствующие полибензимидазолы с м- и о-замещенными
ароматическими ядрами. Растворимость улучшается при наличии
«шарнирных» групп и атомов как в остатке а минного, так и кислотного
компонента.
Полимеры, содержащие в обоих исходных соединениях
простые эфирные связи, растворяются без добавки хлорида лития
в диметилформамиде, диметилацетамиде, диметилсульфоксиде и
метилпирролидоне, а также в тетрахлорэтане [89].
Повышенной растворимостью отличаются цнапэтилированные
полибензимидазолы, полученные при обработке полимера акрило-
нитрилом [119]. Полибензимидазолы на основе ароматических
дикарбоновых кислот с объемистыми заместителями в боковой
цепи растворяются лучше, чем незамещенные продукты [117].
Полибензимидазолы с алифатическими фрагментами в цепи
(табл. 7.25, № 4, 26—35, 43) лучше растворяются, чем
ароматические. При этом растворимость улучшается с увеличением
длины алифатической цепи. В качестве растворителей можно
использовать муравьиную, серную и метансульфокислоты, лькрезол,
диметилформамид, диметилацетамид, диметилсульфоксид, N-ме-
тилпирролидон и циклотетраметиленсульфон. Однако, когда число
метиленовых групп в алифатическом фрагменте превышает 20,
полимеры становятся нерастворимыми. Методом дробного
осаждения в системе диметилацетамид — гексан при 61 °С поли-лг-фе-
ниленбензимидазол (табл. 7.25, № 7) с характеристической
вязкостью 0,78 дл/г в диметилацетамиде был расфракционирован на
шесть фракций, среднечисловая молекулярная масса которых
находится в интервале 11030—42 040 [5, 81]. Для проведения
экстракционного фракционирования в диметилацетамиде необходимо,
чтобы полимер растворялся в интервале температур 70—240 °С
[5,81].
При изучении молекулярно-массового распределения поли-лс-
феиилендибензимидазола седиментационным методом в
ультрацентрифуге выведена следующая зависимость между [г|] и
молекулярной массой [56]: [г\] =1,736-10~3 М0-63 дл/г
(диметилацетамид, 30°С). Значение коэффициента а = 0,63 указывает на то,
что для данного полимера диметилацетамид при 30°С является
термодинамически плохим растворителем [81].
Полибензимидазолы образуют относительно стабильные соль-
ваты и гидраты. Характеристическая вязкость полибепзимидазола
в диметилацетамиде, содержащем 2,27 % воды, выше, чем в
безводном диметилацетамиде. В интервале температур 10—135 °С
характеристическая вязкость уменьшается с 0,806 до 0,626 дл/г, при
этом константа Хаггинса возрастает с 0,371 до 0,50. Отсюда,
следует, что с повышением температуры диметилацетамид становится
более плохим растворителем. Термодинамически более хорошим
растворителем для поли-ж-фенилендибензимидазола
диметилацетамид оказывается в присутствии хлорида лития.
7.2,2.3. Термостойкость
Незамещенные ароматические полибензимидазолы начинают
терять массу при 500 °С. В инертной среде при температурах выше
500 °С скорость выделения летучих продуктов деструкции
относительно мала, в то время как на воздухе из-за наличия весьма
неустойчивых N—Н-связей этот процесс протекает очень быстро
(рис 7.35).
Стойкость к термоокислительной деструкции незамещенных
полибензимидазолов примерно такая же, как и у сополимера
о
200
WO 600
Q 200 400 600 800 О?
Рис. 7.35. Кривые термической деструкции полибензимидазолов при различной
температуре на воздухе A, 4) и в среде азота {2, 3) [4]:
HN NH
HN
1
N N ^^
(СН2L-
Рис. 7.36. Кривые термической деструкции полибензимидазолов с перфтормети-
леновыми группами при различной температуре [3]:
HN NH
7-
2-
(CF2),
тетрафторэтилена с гексафторпропиленом [78]. Температуры
начала разложения полибензимидазолов приведены в табл. 7.25. По-
либензимидазолы с алифатическими группами (табл. 7.25, № 4,
26—35, 43) имеют более низкую термостойкость, чем полностью
ароматические полимеры. В инертной среде разложение начинается
при 450—470 °С, а на воздухе — около 350 °С (см. рис. 7.35).
Разложение начинается по метиленовым группам основной цепи. Замена
СН2 (табл. 7.25, № 29, 32, 34, 71, 73) на CF2-rpynnbi
сопровождается уменьшением термостойкости (рис. 7.36). Деструкция в этом
случае начинается с отщепления HF за счет водорода имидазоль-
ного цикла и фтора дифторметиленовой группы.
882
О 200 4QQ 600 800 t,°C
Рис. 7.37. Кривая термической деструкции поли-2,2'-вшшлнден-5,5'-дибенз1]мпда-
зола при различной температуре [3].
Рис. 7.38. Кривые термической деструкции полибензимпдазолов при различной
температуре в среде азота [3]:
HN
о
Полибензимидазолы с ненасыщенными алифатическими
группами (табл. 7.25, № 27) имеют более высокую термостойкость за
счет сопряжения двойной связи с ароматической системой
(рис. 7.37). Полимеры с алициклическими группами в основной
цепи по термостойкости занимают промежуточное положение
между алифатическими и ароматическими полибензимидазолами
(табл. 7.25, № 30, 33). Термостойкость уменьшается в следующем
ряду:
^ j ^» г плит. » \ :> /' г и I wi ¦ _ ^
V
>— ~>тран
> транс.
>
По термостойкости полибензимидазолы ферроцендикарбоновой
кислоты (№ 20) аналогичны алифатическим полибепзимидазолам.
Термостойкость полибензимидазолов с нафталиновыми
группами в основной цепи зависит от изомерии использованных нафта-
линдикарбоновых кислот (№ 13—16). Свойства
полибензимидазолов 2,6-нафталиндикарбоновых кислот такие же, как и у других
ароматических полибензимидазолов. На основе же 1,2- и 1,7-наф-
талиндикарбоновых кислот получают полимеры, которые по
термостойкости занимают промежуточное положение между
алифатическими и ароматическими полибензимидазолами (рис. 7.38).
При введении в структуру мостиковых групп термостойкость
понижается в следующей последовательности: О > СН2 > SO2.
883
100 200 300 400
О 200 400 600 800 t,°C
Рис. 7.39. Кривые термической деструкции полибензимидазолов при различной
температуре в среде азота (/, 5) и на воздухе B, 4) [3]:
C
1.2-
-< ж
3,4-
Рис. 7.40. Кривые термической деструкции полиборбензимидазолов при
различной температуре в азоте [3]:
1 —
—В
2-
NH
NH
NH
NH
n
п
Полибензимидазолы с N-алкилзамещенными группами имеют
несколько более высокую стойкость к термической и окислительной
деструкции (№ 72—85).
Полибензимидазолы с N-фенилзамещенными циклами имеют
значительно более высокую стойкость к термической и термоокис-
лнтельной деструкции, что особенно проявляется в условиях тер-
мостарения (рис. 7.39). При замене группы ,С в имидазольном
цикле на бор (боримидазолы) или —Р=О (№ 64)
термостабильность не повышается, но улучшается растворимость полимера
(рис. 7.40). Введение в макромолекулы полибензимидазолов крем-
нийорганнческпх групп в остатках кислотного компонента (№ 24,
834
37) не приводит к увеличению термостойкости и вызывает
понижение температуры размягчения [27]. Полибепзоилендибензимида-
золы (№ 85—87) благодаря частично-лестничной структуре имеют
более высокую термостойкость, чем обычные полибензимидазолы.
Полимеры этого типа рассмотрены в гл. 8.
В результате структурирования полибензимидазолов за счет
взаимодействия с формальдегидом с последующей
термообработкой термостойкость повышается на 30—50° [28]:
сн2о
n
Механизм деструкции поли-2,2/-Л1-фенилен-5,5/-дибензимидазо-
ла изучали методами масс-спектрометрии, ДТА, пиролитической
газовой хроматографии и ИК-спектроскопии [23, 29]. Оказалось,
что ниже 500°С выделяется только вода (табл. 7.26). Выше 550°С
среди летучих продуктов деструкции обнаружены HCN, NH3, H2
и СО2, свидетельствующие о распаде гетероцикла. Из данных
Таблица 7.26. Состав остатка поли-ж-фенилендибензимидазола, полученного
при деструкции при различных температурах [23]
Температура
пиролиза,
°С
25
319
487
586
606
Масса
остатка,
"°
100
99,0
98,3
96,8
94,7
с, %
73,46
73,77
73,66
73,60
75,49
и. %
4,28
4,02
4,13
3,94
3,18
N, %
11,12
11,90
12,05
11,34
11,61
Температура
пиролиза,
°С
670
730
817
916
Масса
остатка.
%
89,5
85,4
81,3
77,1
с. %
77,12
77,84
77,82
80,58
н, %
2,75
1,81
1,52
1,07
N. %
10,35
5,41
3,74
2,50
885
-c-O-
элементного анализа твердого остатка можно сделать вывод о
карбонизации бензимидазольных циклов в процессе деструкции.
На основании этого предложен следующий механизм деструкции
[23]:
NH _ NH
\ ^^ Н2О
N ^/ NH
0)
На первом этапе при температуре ниже 550°С происходит
медленный гидролиз имидазольных циклов в гидроксиимидазолин.
Сольватированная и абсорбированная вода удерживается в поли-
бензимидазоле даже при термообработке его в вакууме при
400 °С. За счет раскрытия гидроксиимидазолинового цикла
образуется таутомерная амидная структура. Ухудшение растворимости
полибензимидазолов при термообработке в интервале температур
450—500 °С свидетельствует о том, что одновременно происходит
структурирование полибензимидазолов за счет обменных реакций
по амидным группам и реакций конденсации. Расщепление амид-
ных связей при 550—625 °С протекает через имидный таутомер
с образованием аминофенильных радикалов, бензонитрильных
групп и гидроксирадикалов:
B)
При разложении бензонитрила выделяется HCN, и образуется
карбонизованный остаток:
HCN + Карбонизованный остаток
Рекомбинация о-фенильных радикалов приводит к получению
2,2'-диаминодифенилов, которые с отщеплением аммиака
превращаются в карбазольные или антраценовые группировки:
—nh3
886
Образование СО можно объяснить следующим образом:
NH2
fi00.c
+ СО +
D)
Рекомбинация радикалов, образующихся при 600—800°С,
сопровождается выделением водорода и приводит к получению
лестничных структур:
E)
Предложена несколько иная схема процесса деструкции поли-
бензимидазолов с образованием в качестве промежуточного
продукта бензина [23] :
NH
XN
:NH
О
Бензин
:NH + H2 —> NH3
HN ОН
'NH + O
ИЛИ
NH
Hgr
:NH +
+
а
1
OH
-C=NH
Карбонизованный остаток -f СО + NH3
I
|-н2о
XONH2
7.2.3. ПОЛИ-л-ФЕНИЛЕНДИБЕНЗИМИДАЗОЛЬНЫЕ ВОЛОКНА
7.2.3.1. Получение
Производство этих волокон по заказу министерства обороны
США организовала фирма «Celanese». Пилотная установка в
Саммите (штат Нью-Джерси) работает по следующей схеме [5].
887
бр , г/денье
6
Q
0
100
200
Рис. 7.41. Зависимость прочности при растяжении (а) и относительного
удлинения при разрыве (б) поли-.и-фенилендибензимидазольных волокон от
температуры:
1 — исходный материал; 2 — высокоориентироваиные волокна; 3 — кристаллические
волокна [31].
Поли-ж-фенилендибензимидазол растворяют в диметилацет-
амиде при 130 °С под давлением 5,5 кгс/см2. Во избежание
фазового расслоения при хранении в раствор добавляют 2 % хлорида
лития, который в процессе формования волокна легко отмывается
водой. Для стабилизации раствора можно использовать также
хлорид цинка, N-метилморфолин, тетраметиленсульфон, триэтил-
амин или триэтаноламин. Для предотвращения
структурирования за счет окисления растворение проводят в среде азота. После
фильтрации под давлением получают 25%-ный раствор полибен-
зимидазола, вязкость которого составляет 3000 Пз при 25 °С.
Формование нагретого до 150°С раствора проводят в среде горячего
азота с содержанием кислорода менее 2,5%. Нагретая до 180°С
фильера находится в головке колонки с электрическим обогревом,
нижняя часть которой «промывается» азотом, нагретым до 240 °С.
После приемного устройства в колонне с паровым обогревом
волокно вытягивается на 6 % [99]. Затем нить на бобине отмывается
горячей водой от следов диметилацетамида и хлорида лития и
сушится в течение 48 ч. Прочность при растяжении волокна
составляет 1,7 г/денье, относительное удлинение при разрыве 115% и
модуль упругости 44 г/денье. Волокно после формования
подвергается дополнительной вытяжке в горячих печах при 520 °С при
кратности вытяжки 2:1. Такое дополнительно ориентированное
волокно имеет прочность при растяжении 4,9 г/денье,
относительное удлинение при разрыве 24 %, модуль упругости 105 г/денье.
Степень кристалличности волокна можно повысить обработкой его
смесью фенол : вода A:1) под давлением. После 2 ч выдержки
волокна в автоклаве при 250 °С удлинение увеличивается с 18,4 до
48 %, прочность при растяжении понижается с 4,46 до 2,52 г/денье.
Мокрое формование проводится из 20—25%-ного раствора поли-
л-фениленднбензимидазола в диметилацетамиде с 2 % хлорида
лития в осаднтельную ванну из этнленгликоля или глицерина
[114].
7.2.3.2. Свойства
Отличительной особенностью полибепзимидазольного волокна
являются его исключительно высокая термостойкость, негорючесть
на воздухе и хорошие текстильные свойства [32]. Полн-л-фени-
лендибензимидазольное волокно может использоваться до
температуры 450 °С. Нагревание волокна от комнатной температуры до
100°С сопровождается сначала увеличением прочности за счет
удаления сорбированной воды, затем ее постепенным снижением
(рис. 7.41,6). При температуре выше 200 °С сильно возрастает
удлинение. Исключением являются высокоориентированные волокна.
Наличие NH-групп в бензимидазольном цикле делает полимер
довольно чувствительным к окислению при высоких температурах.
На рис. 7.42 приведены данные о влиянии термостарения на
прочность полибензимидазольного и ароматического полиамидного
волокон. Усадку волокна при нагревании можно понизить путем
его обработки раствором хлорсульфоновой кислоты в фосфорил-
хлориде с последующими нейтрализацией и термообработкой. По-
ли-ж-фенилендибензимидазольное волокно довольно чувствительно
к свету (рис. 7.43). При длительной эксплуатации на свету
необходима его стабилизация. Напротив, волокно весьма устойчиво
к Y-излучению. После облучения дозой 8,4-107 или 1,2-109 рад
прочность при растяжении снижается с 5,19 до 4,34 или 4,13 г/денье;
относительное удлинение при разрыве снижается с 20,3 до 20,1 или
17,2 о/о [34,91].
Высокотермостойкое полибензимидазольное волокно весьма
чувствительно при высоких температурах к действию влаги и
химических агентов. На рис. 7.44 приведена диаграмма растяжения
полибензимидазольного волокна в условиях различной ориентации
и влажности. Ориентация и кристаллизация волокна не улучшают
его влагостойкость. Пребывание волокна в агрессивных средах
р, % (от асх.)
бр,%(отисх.)
90
ВО
70
ВО
50
40
30
20
10
0
--
—
—
—
—
—
I I I
\
\
\
\
i i x
10 20 40 ВО 100 200 г,Ч
50-
60 г,ч
Рис. 7.42. Зависимость прочности при растяжении термостойких волокон от
продолжительности термообработки при 305 °С [33]:
/ — поли-л-фенилендибензимидазольное волокно; 2 — поли-л*-фениленизофталамидное волокно
Рис. 7.43. Зависимость прочности при растяжении поли-л!-фенилендибензимида-
зольного волокна от продолжительности облучения ксеноновой лампой [33].
889
бр, rjденье
Б
,г/денье
О
5 10 15 20 Z5 ?,%
Рис. 7.44. Зависимость
прочностных свойств по-
ли-.и-феннленбензимида-
зольного волокна
различной степени
упорядоченности от влажности
воздуха [31]:
а — аморфное волокно; б —
высокоориентированное кри-
сталлическое волокно; / —
15%-ная относительная
влажность; 2 — 65%-ная; 3 —
100%- ная.
Ю ?,%
приводит к уменьшению прочности и сильной усадке (табл. 7.27).
Высокая усадка обусловлена сольватацией, приводящей к образо^
ванию межмолекулярных связей [33]. Кристаллизация поли-ж-фе*
нилендибензимидазольного волокна путем его вытяжки в 88 %-ной
муравьиной кислоте и последующее повторное формование не
отражаются на усадке, так как кристаллы уже существуют в сольвати-
рованной форме. При выдержке волокна в агрессивных средах
прочность уменьшается сравнительно мало. Значительно меньше
сведений имеется о волокнах на основе других полибензимидазо-
лов. Поли-2,2'-октаметилен-5,5'-дибензимидазольное волокно
(табл. 7.25, № 35) с прочностью при растяжении 7,2 г/денье,
относительным удлинением при разрыве 13 % и модулем упругости
150 г/денье может быть получено из растворов в муравьиной
кислоте [134].
7.2.3.3. Применение
Высокие термостойкость и негорючесть поли-^-фенилендибен-
зимидазольного волокна определили его использование главным
образом в наземном и воздушном транспорте. Хорошие текстиль-
Таблица 7.27. Химическая стойкость поли-ж-фенилендибензимидазольного
волокна [33]
Среда
нсоон
H2SO4
HNO3
о н
88
88
10
10
60
70
70
10
10
70
*
га
&
ер а ту
с
Е
20
20
20
20
60
20
95
20
20
20
W
CQ
И
О
16
192
16
192
102
96
8
16
192
16
ЯЭ'
Q
О
57
59
37,7
40,1
45,9
43,2
35
32,5
35
40,1
к
га д
В" О
о о
н а
и а>
о °
48,1
48,3
86,2
79,1
55,6
82,7
75
56,1
69,9
68,9
Среда
HN03
на
HF
NaOH
NH4OH
&
=г
к «
О g
70
35
35
10
48
10
10
40
50
28
ЯЗ'
&
ер a ту
«и
Но
20
20
20
95
20
20
95
20
60
20
2
а
CQ Ш 5*
192
16
192
8
96
96
8
96
102
96
сз
^t
43,2
30,6
30,2
33,5
31,6
11,8
14,2
6,6
18,8
22,4
№
точна
f- О
О с
50,1
76,5
78,1
73
79,6
87,8
91,4
83,1
51,2
95,4
890
ные свойства и отличное качество при носке (водопоглощение
14,4%) сделали этот материал весьма ценным для изготовления
одежды для летчиков и защитной одежды для работающих в
условиях высоких температур. Хорошей окрашиваемостью катион-
ными красителями характеризуются полибензимидазольные
волокна, подвергнутые первоначальному сульфоалкилированию
[110]. Ткань из полибензимидазольного волокна воспламеняется
только после 2 мин выдержки при 900 СС [79]. В чистом кислороде
воспламенение происходит только под давлением 1,35 кгс/см2.
Особый показатель для волокна, используемого на транспорте, —
самозатухаемость достигается путем бромирования (увеличение
массы на 80%) или фосфорилирования [83]. Химическая
модификация при этом не сопровождается уменьшением
термостойкости. В ткани на основе немодифицированного полибензимидазола
зона пламени от горелки с температурой 980—1200 °С
распространяется только на 10 %• При выдержке в течение 3 с образование
пузырей на поверхности еще не наблюдается [36].
Полибензимидазольные волокна и ткани применяются также для изготовления
тормозных парашютов для сверхзвуковых самолетов и
космической техники, мешков для парашютов и спасательных средств,
предохранительных сеток, высокотемпературных газовых
фильтров.
7.2.4. СИНТАКТИЧЕСКИЕ ПОЛИБЕНЗИМИДАЗОЛЬНЫЕ ПЕНОПЛАСТЫ
Низкая плотность и высокая термостойкость обусловили
использование поли-ж-фенилендибензимидазольных пенопластов в
качестве прекрасного изоляционного материала для ядерных
реакторов и топливных элементов, а также абляционного
теплозащитного слоя в космической технике [42, 43, 82]. Пеноматериалы
получают путем сплавления низкомолекулярных поли-ж-фенилен-
дибензимидазолов, со степенью полимеризации 2—3 и больше 3
в присутствии 10—15 % углеродных волокон или волокон на
основе оксида алюминия и диоксида кремния и 10—20 %
микропористых сфер из диоксида кремния или фенольного полимера. Смесь
нагревают в течение 20 мин при 260°С и 10 мин при 315 °С.
Наполнитель обеспечивает равномерность газовыделения, легко
контролируемую на второй стадии синтеза, и образование пенопласта
с однородной пористой структурой. Пенопласты, полученные без
наполнителя, имеют большие пустоты за счет локального
газовыделения и характеризуются хрупкостью. Пенопласт формуют в
металлических формах при 315 °С с последующей термообработкой
при 450сС. Верхняя предельная температура длительной
работоспособности поли-ж-фенилендибензимидазольного пенопласта,
наполненного 20% полых сфер из SiO2 и 15% волокна на основе
А12О3—SiO2, составляет 315 СС [41]. Физико-механические свойства
синтактических поли-ж-фенилендибензимидазольных пенопластов,
состоящих из 65 % полимера, 20 % микрополых сфер из SiC>2,
15% волокна AI2O3—SiO2, приведены ниже [44, 82]:
891
1,82
1,91
2,40
0,0046
Диэлектрическая
проницаемость при 9,3 Гц
при 24 °С
при 540 °С
при 1000°С
Тангенс угла
диэлектрических потерь при 9,3 Гц
при 24 °С
при 540 °С 0,0182
при 1000 °С 0,0423
Коэффициент
теплопроводности при 120—260 °С,
ккал/(м-ч-°С) 0,05-0,1
Водопоглощение, % ... 34
Максимально допустимая
температура эксплуатации
под нагрузкой, °С 315
Кажущаяся плотность,
г/см3 0,4-0,6
Размер пор, мкм 30—300
Прочность при сжатии,
кгс/см2
при 24 °С 255
при 315 °С 200
при 540 °С 54
Модуль упругости при
сжатии, кгс/см2
при 24 °С 17 000
при 315 °С 2 500
Прочность при растяжении,
кгс/см2
при 24 °С 90
при 315 °С 80
при 540 °С 20
Особая ценность синтактических полибензимидазольных
пластов заключается в возможности их легкой переработки вместе
с наполнителем, причем коксование сопровождается образованием
графитированного продукта с высоким выходом. Этот продую
имеет высокоориентированную структуру и характеризуется мини
мальной усадкой. Коксовый остаток обладает высокой
отражательной способностью [45]. Абляционные свойства полибензимидазо
лов можно улучшить путем химического (через образование
пероксидных групп в диаминобензидине) или термического
структурирования. Блокирование аминогрупп в имидазольном цикле
сопровождается понижением склонности полимеров к окислительной де
струкции. При термической обработке полибензимидазолов i
инертной среде до 800 °С со скоростью нагревания 3°С/мин обра
зуется сильно сшитый черный продукт, представляющий собой по
липиробензимидазол с эмпирической формулой C47H14N8. Тепло
проводность такого пирополимера ниже, .чем графита, вследствие
наличия в структуре атомов азота. Синтактические полибензими
дазольные пенопласты в промышленном масштабе были получень
в 1968 г. фирмой «Whittaker» в Сан-Диего (США) и получили на
звание «Имидайт SA» (термо- и электроизоляционный материал
наполненный 20 % SiO2 и 15 % А12О3—SiO2) и «Имидайт PC:
(абляционный материал, наполненный 20 % микросфер на основ*
фенольного полимера и 15% углеродного волокна).
7.2.5. ПОЛИБЕНЗИМИДАЗОЛЬНЫЕ КЛЕИ ДЛЯ МЕТАЛЛОВ
Полибензнмидазольные клеи на основе поли-ти-фенилендибен-
зимидазола используют в авиационной и космической технике длу
склеивания конструкций из бериллиевой стали и легированногс
титана, находящихся под большими напряжениями [47, 48, 82]
Клеи представляют собой растворы олигобензимидазола со
степенью полимеризации 3—4, содержащие диспергированный
алюминиевый порошок или стеклянное волокно; соотношение наполни-
892
Рис. 7.45. Зависимость прочности при сдвиге бсд,кгс/см
стального соединения, склеенного поли-л^фенилен-
бензимидазолышм клеем, от условий отверждения
[46]:
1 — нагревание при 260 "С; 2 — нагревание при 287 "С;
$ — нагревание при 315 °С; 4 — нагревание при 315 °С
с последующим дополнительным прогревом при 370 °С inn\,
в токе азота.
100
50
100 200 300 t,C
тель : связующее = 1:1. Наполнитель понижает термический
коэффициент расширения и уменьшает усадку. Наилучшие прочностные
характеристики достигаются при использовании в качестве
наполнителя стеклянного волокна; для улучшения адгезии вводят поли*
имид. При склеивании специальных сталей прочность клеевого
шва при растяжении составляет 210 кгс/см2. Для увеличения
стойкости к окислению, особенно при склеивании легированных сталей,
в качестве антиоксиданта применяют As(AsS4) [49].
Как видно из рис. 7.45, условия отверждения оказывают
большое влияние на прочность клеевого соединения. Оптимальные
свойства достигаются при нагревании в течение 1 ч при 220 °С и
1 ч при 315 °С под давлением 150 кгс/см2. Дополнительная
термообработка в сухом азоте (нагревание до 370ЪС в течение 45 мин
и выдержка при этой температуре 1 ч) позволяет существенно
повысить прочность клеевого шва. При малых поверхностях
склеивания наилучшие результаты достигаются при давлении 2 кгс/см2.
Ограниченность применения полибензимидазольных клеев
обусловлена пористостью клеевого шва, так как процесс склеивания
сопровождается выделением фенола и воды. Это связано с
большими трудностями, особенно при больших поверхностях
склеивания.
Вместе с тем достоинством поли-ж-фенилендибензимидазольного
клея является его способность кратковременно выдерживать
большие нагрузки (рис. 7.46). На рис. 7.47 показана зависимость
прочности клеевого шва от продолжительности эксплуатации при
высоких температурах. При 205 °С прочность при растяжении за
1000 ч практически не меняется, а при 260 °С она медленно
уменьшается. Прочность при сдвиге при пониженных температурах
даже выше, чем при комнатной. При —196 °С она составляет 338,
бсд,КГС/СМ2
250\
200 300 t,°C
о
Рис. 7.46. Зависимость прочности при бдвиге стального соединения, склеенного
поли-л^фениленбензимидазольным клеем, от температуры [51].
Рис. 7.47. Зависимость прочности при сдвиге поли-иг-фениленбензимидазольного
клеевого шва от продолжительности эксплуатации при 205 A) и 260 °С B).
893
я при —253 С—400 кгс/см2 [47]. Топливо и гидравлические
жидкости не влияют на прочность клеевых соединений. После 2 ч
пребывания в кипящей воде прочность уменьшается на 30% [52].
Олиго-льфенилендибензимидазол для склеивания металлов
выпускают в США с 1964 г. (фирма «Whittaker») под названием
«Имидайт 850» и «Имидайт 1850». Стоимость 1 кг этого клея
в 1971 г. составляла 200 дол.
7.2.6. ПОЛИБЕНЗИМИДАЗОЛЬНЫЕ СТЕКЛОПЛАСТИКИ
Для пропитки стеклянной ткани или ровницы используют оли-
гобензимидазолы со степенью полимеризации 3—4.
В качестве аппрета, улучшающего адгезию, используют а-бром-
толилсилан, карбоксифенилсилан, гидроксилированные алифати-
ческо-ароматические полибензимидазолы [94].
Связующее наносят на ткань из расплава или раствора.
Раствор олигобензимидазола в диметилацетамиде, диметилформами-
де или N-метилпирролидоне многократно наносят на ткань до
получения определенного содержания связующего и в течение
30 мин высушивают при 120°С. Остатки растворителя удаляют
в вакууме при 160 °С. Недостаток этого метода нанесения
связующего на ткань состоит в том, что олигобензимидазол
взаимодействует с растворителем. Поэтому прочность пластиков при таком
способе нанесения связующего на ткань меньше, чем при
пропитке расплавом полибензимидазола. В последнем случае можно
получить пластик с более высоким содержанием
полибензимидазола [82].
Для нанесения связующего из расплава ткань с нанесенным на
нее порошком полимера пропускается через нож, нагретый выше
температуры плавления олигобензимидазола.
Переработку препрегов осуществляют контактным
прессованием, нагревая пакет в токе инертного газа до 370°С под
давлением 15 кгс/см2 с выдержкой при этой температуре в течение 3 ч.
Отформованное изделие охлаждается в прессе под давлением
до 100°С, после чего в инертной среде выдерживается
последовательно при 315, 345, 370 и 400 °С в течение 24 ч и еще 8 ч при
425 °С.
бр,КГС/СМ*
10000
8000
6000
4000V-
2000
¦из,кгс/сп<
10000
8000
6000
4000
2000
-200 0 200 400
- 2
t
f !
5
v
,кгс/см2
40000
30000
20000
10000
-200
0 200 400 t,cC
Рис. 7.48. Зависимость прочностных (/) и деформационных B) свойств поли-
ж-фениленбензимидазольного стеклопластика при растяжении (а) и изгибе (б)
(толщина образца Змм, содержание связующего 19 %, продолжительность ВЫ'
держки при данной температуре 30 мин) от температуры [51].
Cai, кгс/см*
SQOO
6000
4000
ZOOO
U3
40000
30UO0
20000
10000
SOO 750 т,ч
рис. 7.49. Зависимость модуля упругости (/) и прочности при изгибе B) поли-
бензимидазольного стеклопластика от продолжительности термостарения при
260°С (толщина образца 3 мм, содержание связующего 19%) [51].
Рис. 7.50. Зависимость диэлектрической проницаемости (/) н тангенса угла
диэлектрических потерь B) полибензимидазольного стеклопластика от температуры
(толщина 3 мм, содержание связующего 19 %, продолжительность выдержки при
данной температуре 30 мин, частота 9,37 МГц) [51].
Дальнейшее улучшение свойств наблюдается при нагревании
готового изделия на воздухе в течение 3 ч при 370 °С. При этом
за счет окисления происходит частичное структурирование
полимера. Армированные пластики, полученные из расплава при
толщине 3 мм и содержании поли-лг-фенилендибензимидазола 40%»
имеют следующие показатели: прочность при изгибе 6300—
8000 кгс/см2, прочность при сжатии 3850—4600 кгс/см2, прочность
при растяжении 5300—6000 кгс/см2. Модуль упругости при всех
типах нагрузок составляет 316 000 — 385 000 кгс/см2. Прочностные
свойства при кратковременной нагрузке сохраняются вплоть до
высоких температур (рис. 7.48). После нагревания в течение
30 мин при 425 °С пластик с 20% поли-ж-фенилеидибензимида:
зольного связующего сохраняет 75 % прочности при растяжении,
50 % прочности при сжатии и 33 % прочности при изгибе. При
выдержке в течение ПО ч при 315°С прочность при растяжении
уменьшается на 54%, прочность при сжатии — на 24% и
прочность при изгибе — на 30% [54]. В результате термообработки
в течение 1 ч при 540 °С прочность при растяжении снижается на
16,5%, а модуль упругости при растяжении — на 50%. Изменение
прочностных характеристик полибензимидазольного
стеклопластика при термостарении при 260 °С показано на рис. 7.49.
Прочность остается неизменной при выдержке 4000 ч при
175°С. Электроизоляционные характеристики поли-м-фенилендн-
бензимидазольных стеклопластиков находятся на том же уровне,
что и для обычных стеклопластиков. Однако они очень незначи
тельно изменяются с температурой (рис. 7.50). Такие материалы
применяют в авиации (например, детали сопел). Олиго-ж-фени-
лендибензимидазольные связующие выпускает в США фирма
«Whittaker» под торговой маркой «Имидайт 850».
7.2.7. ПОЛИБЕНЗИМИДАЗОЛЬНЫЕ ПЛЕНКИ
Пленки получают поливом из раствора в муравьиной кислоте
[89], диметилсульфоксиде [3] или диметилацетамиде с добавкой
хлорида лития. Прочность при растяжении составляет 680—
890
1400 кгс/см2, относительное удлинение при разрыве 5—30% [89].
При 200 °С прочностные характеристики сохраняются на уровне
70 % от исходного значения. Диэлектрическая проницаемость
4—5, тангенс угла диэлектрических потерь D—4,7)-10~3 и
удельное объемное электрическое сопротивление 1012 Ом-см.
Пленки из ароматических полибензимидазолов пригодны в
качестве высокотемпературной изоляции обмотки электродвигателей
[89]. Поли-ж-фенилендибензимидазольные мембраны [125], так
же как и мембраны из поли-М-фенилбензимидазоламида [124],
с успехом используются для обратного осмоса. Полибензимида-
зольные пленки благодаря наличию системы полисопряжения
обладают полупроводниковыми свойствами [55].
7.2.8. МЕМБРАНЫ И КОРПУСА АККУМУЛЯТОРОВ
ИЗ ПОЛИ-2,2-ОКТАМЕТИЛЕН-5,5'-ДИБЕНЗИМИДАЗОЛА
Из всех алифатическо-ароматических полибензилимидазолов
до недавнего времени нашел практическое применение только по-
ли-2,2'-октаметилен-5,5'-дибензимидазол.
Полиоктаметилендибензимидазол 3,3'-диаминобензидина и се-
бациновой кислоты (табл. 7.25, № 35) получают в
полифосфорной кислоте при 100—235 °С. Температура стеклования полимера
275 °С, температура начала разложения на воздухе 395°С и в
среде гелия 410 °С. При формовании пленки поливом из раствора
в муравьиной кислоте образуются высокоориентированные
кристаллиты. Нагревание в интервале температур 120—140°С
сопровождается аморфизацией полимера [37, 38]. Прочность при
растяжении этой пленки после выдержки в течение 5 сут в 40%-ном
растворе едкого кали возрастает с 520 до 580 кгс/см2 [11]. Как
электроизоляционный материал полимер может использоваться
вплоть до 200°С [401. Полиоктаметилендибензимидазол
характеризуется высокими прочностными свойствами при низких
температурах. Так, ударная вязкость образца с надрезом при 250 и
— 196°С составляет 8,5 и 5,0 кгс-см/см2, а прочность при сжатии
2000 и 6000 кгс/см2 соответственно. Полимер используется для
изготовления стерилизуемых корпусов шприцев, в качестве
разделительных мембран для AI — Zn-элемеитов, которые применяются
в космической технике (при исследовании возможных форм
существования живых существ на других планетах все детали
космического корабля должны быть тщательно стерилизованы) [39].
Для обеспечения возможности переработки полиоктаметилен-
дибензимидазола литьем под давлением необходимо блокировать
концевые группы полимера монокарбоновыми кислотами или
о-диаминами. Обычно применяемые в Ag — Zn-элементах
целлюлозные мембраны разрушаются при стерилизации в присутствии
электролитов. Полиоктаметилендибензимидазольные мембраны
устойчивы при нагревании в 40%-ном растворе КОН,
насыщенном Hg2O. Однако, в силу того что полимер не смачивается
электролитом, объемное электрическое сопротивление слишком велико
896
для применения их в качестве разделительных мембран. Этот
недостаток можно устранить путем цианэтнлирования полиоктамети-
леиднбензимидазола с последующим омылением продукта
реакции [11]:
(СН2)8~
H2C=CIICN
CH3ONa
Димстилацетамнд
(CH2)e-
кон
NH
CH2CH2CN
(CH2)8-
СНаСНзСОО" К*
Карбоксиэтилированный полимер после 36 ч пребывания
в 40%-ном растворе КОН при 145°С имеет прочность при
растяжении 485 в сухом состоянии и 140 кгс/см2 при поглощении 90 %
электролита; удельное объемное электрическое сопротивление
составляет 1340 Ом-см.
7.3. ПОЛИБЕНЗОКСАЗОЛЫ
п
Полибензоксазолы получают поликонденсацией бис-о-аминофе-
нолов с дикарбоновыми кислотами или их производными. Волокна
и пленки формуют из промежуточного поли (о-гидрокси)амида.
Температура длительной работоспособности полибензоксазолов, не
содержащих алифатических звеньев, составляет 300°С.
Применяются они главным образом как высокотермостойкие
электроизоляционные материалы и клеи для металлов.
7.3.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗОКСАЗОЛОВ
Получение полибензоксазолов основано на реакции, которую
в 1876 г. обнаружил Ладенбург [155] при сплавлении о-амино-
фенола с бензоилхлоридом. Общее уравнение реакции можно
29 Зак. 18
897
предстапить следующим образом:
О
г©
п
Полибепзоксазолы получают в две стадии через
промежуточное образование поли(о-гидроксн) амида или в одну стадию в
растворе. В СССР получают поли (о-метокси)амид на основе продукта
взаимодействия З.З'-диметокаМ^'-диамннодпфенилметана и изо-
фталевой кислоты под названием «метолон». Полиметоксиамид
в процессе термической переработки в клеи, пленки, формованные
изделия превращается в сшитый полибензоксазол [189].
7.3.1.1. Поликонденсация бис-о-аминофенолов
с производными дикарбоновых кислот в расплаве
Впервые поликонденсациеп в расплаве полибензоксазолы были
получены в 1959 г. [136, 137]. Высокомолекулярные
полибензоксазолы образуются при нагревании в инертной среде эквимольных
количеств бис-о-аминофенолов с днкарбоновымн кислотами или
их дифениловыми эфирами при 250—300°С с выделением воды
или фенола. Продолжительность реакции составляет 5—30 ч [11,
138, 139].
Привлекательность данного метода связана с простотой
аппаратурного оформления процесса и отсутствием пизкомолекулярной
фракции и корродирующих низкомолекулярных продуктов.
7.3.1.2. Высокотемпературная поликонденсация
в растворе
Высокомолекулярные полибензоксазолы можно получить гомо-
поликонденсацией о-аминогидроксикарбоповых кислот или поли-
конденсацпей ароматических дикарбоновых кислот с хлоргидра-
тами бис-о-амииофенолов в полифосфориой кислоте [6, 131,
141 —142, 144]. В феноле или крезоле образуются полимеры
небольшой молекулярной массы.
7.3.1.3. Двухстадийная поликонденсация с промежуточным
образованием полигидрокси- или полиметоксиамидов
В этом случае сначала взаимодействием бис-о-аминофенолов
и дикарбоновых кислот или производных тех и других соединений
в растворе получают соответствующие о-замещенные полиамиды,
которые затем при нагревании превращаются в полибензоксазолы
[Il, 16, 138—140, 143, 145—152, 182, 184]:
OR
R'HN-
R"\ M'"
o-«
^NHCR'
n
На первом этапе реакцию проводят при температурах от —10
до 20°С в апротонных растворителях (диметилформамид, диме-
тнлацетамид, N-метплпнрролидон, тетраметнленсульфон). В ди-
метилацетамиде реакция затруднена из-за протекания побочного
процесса переамидирования [182]. Нагревание раствора полигидр-
оксиамида в диметилацетамнде в течение 2 ч при 100°С приводит
к снижению относительной вязкости на 10%. Сульфолан и N-ме-
тилпмрролндон являются инертными растворителями в данном
процессе. Наиболее высокомолекулярные полигидроксиамиды
образуются при добавлении к раствору в сульфолане (концентрация
мономеров 1 моль/л) через 45 мин после начала реакции диметпл-
ацетамида или тетраметилмочевины в качестве акцепторов НС1
D моля на моль дихлорангидрида дикарбоновой кислоты).
Сложность технического оформления этого процесса связана с
корродирующим действием выделяющегося НС1. При применении
незамещенных бис-о-аминофенолов и днкарбоновых кислот возможно
образование разветвленных или сшитых продуктов.
Этого можно избежать, применяя N- или О-ацилировапиые
бнс-о-аминофенолы [153, 154] или иминоэфиры днкарбоновых
кислот [16]. В последнем случае поликонденсацию с бис-о-амино-
феиолами проводят в растворе диметилацетамида или N-метпл-
пнрролидона при 100—190°С. Полн-о-гидрокси- или поли-о-мет-
оксиамиды, образующиеся на первой стадии процесса,
растворяются в 96 %-ной H2SO4, N-метнлпирролидоне, диметилсульфоксиде,
днметилацетамиде, гексаметнлфосфотриамиде и диметплформамиде
с добавкой хлорида лития. Растворы могут быть непосредственно
использованы для формования пленок или волокон. Прочность
при растяжении полигидрокснамидной пленки на основе 3,3'-ди-
амино^Д'-дигндроксидифенила и пзофталевоп кпслотт.т составляет
1600 кгс/ем2, относительное удлинение при разрыве 30—50 °/о
[150].
29*
899
Термическая циклизация промежуточного о-замещенного
полиамида проводится в твердой фазе (пленка, волокно) в среде
инертного газа или вакууме при 220—300 °С. Скорость циклизации
можно контролировать по изменению интенсивности полосы
поглощения бензоксазола при 1620 см~1 [138]. Процесс циклизации
осложняется гидролизом амидных связей и уменьшением
молекулярной массы на 5—10% [147]. Однако это уменьшение
компенсируется реакцией макромолекул по концевым функциональным
группам на заключительной стадии циклизации. Скорость
циклизации зависит от гибкости о-замещенного (гидрокси- или метокси-
полиамида); энергия активации этого процесса зависит от
строения дикарбоновой кислоты, изменяясь при взаимодействии с ди-
оксибензидином в порядке возрастания в следующей
последовательности [147] :
При термической циклизации линейных поли (о-гидрокси)
амидов в инертной среде образуются линейные полибензоксазолы.
Термообработка полиметоксиамидов, напротив, сопровождается
структурированием.
Линейные полибензоксазолы сшиваются на воздухе при 400 °С
[179J. Сшитые полимеры могут быть также получены на основе
структурированных поли (о-гидрокси) амидов, содержащих двойные
связи [156—158]. Двухстадийная гомополиконденсация гидрокси-
аминобензойной кислоты приводит к образованию
низкомолекулярных продуктов. Полимеры с высокой молекулярной массой могут
быть получены только косвенным методом через промежуточную
стадию образования полиэфироамидов [139]:
H2N
.соон
SOC12
5-'m
Полибензоксазолнмиды синтезированы из днкарбоновых кислот,
содержащих в своем составе имидные звенья, и бис-о-аминофено-
лов с термической циклизацией промежуточно образующихся поли-
900
о-гидроксиамидоимидов при 375 °С [183]:
О
С1С
п
COCI
—2HG1
о
СО—
—2Н2О
—п
о
—П
7.3.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИБЕНЗОКСАЗОЛОВ
Полибензоксазолы представляют собой аморфные вещества от
желтого до оранжевого цвета. Температуры их размягчения и
разложения приведены в табл. 7.28. В основу классификации поли-
бензоксазолов положено строение исходных бис-о-аминофенолов.
Продукты их взаимодействия с ароматическими дикарбоновыми
кислотами в табл. 7.28 даны под № 1—38; с алифатическими —
№ 39—66, сополимеры —№ 67—78.
7.3.2.1. Температура размягчения
Чистоароматические полибензоксазолы, содержащие в своем
составе только бензоксазольные и фениленовые звенья,
разлагаются при температуре выше 500 СС. Температура разложения их
лежит ниже температур плавления или размягчения. Полимеры,
содержащие «шарнирные» группы типа О, СН2, СО или SO2
в остатках бис-о-аминофенола или дикарбоновой кислоты, размяг-
901
Таблица 7,28, Температура размягчения и разложения полибензоксазолов
Элементарное звено
разм'
разл
(на
воздухе),
Т
разл
<в N8).
Литературный
источник
о
N
N
О
о
о
N
о
N
О
N
530
425
520
450
500
500
450
[139, 161]
[139, 161]
[177]
[138, 139,
142, 147, 160,
162, 184.
187]
[138, 139,
143, 147, 148,
150, 160, 162,
178, 184]
о
о
N
N
[139]
Cl
8
10
11
CD
370
285
[139]
[113, 147,
160, 178,
188]
[143]
[143]
[178, 180]
Продолжение табл. 7.28
Элементарное звено
разм'
Т
разл
(на воз-
Духе),
°С
Т
разл
(в N2),
Литературный
источник
О
N
о
N
о
О
N
О
О
280
270
500
450
420
[178]
[84, 162]
[140. 162]
[163]
[142]
17
18
19
о
N
N
О
N
V
N
20
о
¦Q
so2
SO;
о
N
N
О
N
О
21
N
22
СО
о
ел
240
520
[1421
410
[140, 149,
150]
420
[140, 149,
182, 184]
[160]
[143]
[НО, 182]
о
СП
23
N СО
О
N СН2
24
О
Метолон (СССР)
25
N СН,
О
N
СНЯ
26
N
О
N
' \
О
N
N
Л
о
Продолжение табл. 7,28
Элементарное звено
255
500
500
Литературный
источник
[135]
[159, 179,
189]
[176]
[163, 164]
N
500
[163. 164,
1811
СНз
сня
N
О
N
300
[159, 163.
164]
СН3
400
1163. 164]
О
180
[163, 164]
о
1165]
Продолжение табл. 7.28
Элементарное звено
разм'
°С
разл
(на
воздухе),
°С
Г
разл
(в N2),
Литературный
источник
N
О
-О-
300
320
350
[159, 163,
164]
A59, 163
164]
[159, 163,
164}
N
О
N
О
350
400
[159, 163,
164]
400
[159, 163,
164]
О
N
О
N
350
[159, 163,
164]
/7родолжение табл. 7.28
Элементарное зветю
разы
"С
разл
(на
воздухе),
° С
разл
(в N2),
—СН?—СНо—
— х = 2,4
сп=сн—
о
N
О
N
О
О
N
О
N
О
N
(СН2L—
(СН2N-
(СН8)в—
250
520
300
О
СО
46
47
48
49
50
N
О
N
N
О
N
о
о
N
о
N
—/ .УЛ1
О
N
О
N
N
О
N
О
О
^ \
N
О
=г 4,
-(СН2)8-
—(CF,)S— (П)~
250
350
300
345
300
[161
[168]
[162]
[11]
[167]
[142]
Ko
51
52
со
53
54
N
-с
V
N
-<;
N
/ V
V
N
V
СНз
^ I
^^ СНз
СНз
СНз
-. 1
(бгН
^^ СНз
СНз
тбгЬ
с
^ СНз
:н2
Or
г
СНз
Элементарное звено
N
'Ч
\ /
0
N
'Ч
V
N
'Ч
0
N
^ /
0
»—СНз—0—СНз—
>—(сн2L-
>—(СН2N-
> сн2—о—сн2—
т
разм'
°с
Продолжение табл. 7.28
т
разл
(иа воз-
«2»).
т
разл
(в N2),
°С
250
420
420
200
Литературный
источник
[163, 164]
[164]
[166]
[163, 164]
N
СНз
N
О I ьпз I О
СНз СНз
400
[103, 164,
181]
N
СНз
N
О | ^лз | О
СНз СНз
300
[163, 164]
N
r
О ! СНз I О
Cl CI
220
[163, 164]
N
СНз
//
О
N
V
i СН2—О—СН2—
200
[159, 163,
164]
Продолжение табл. 7.28
Элементарное звено
разм*
°С
Т
раз л
(на
воздухе),
d
Т
разл
(в N2).
(СН2L-
СН,
—(СН,)б—
о
(CH2)S-
350
350
о
V
N
о
1 Ъ—(сн?;б-
330
A661
N
О
Y
о
N
сн,—о—сн2—
300
[159, 163.
1641
N
(СН2L
400
[159, 164,
165]
N
о
(СН2N-
400
[163, 1641
QC@
N
Продолжение табл. 7.28
Элементарное звеио
разм*
°С
Т
разл
(на воз-
Духе),
Т
разл
В N2).
N
О
N
О
N
СНз
СНз
(СН2)8-
N
О
N
V
О
N
О
СНз
525
525
450
450
300
N
N
N
V
N
О
N
V
сн3
N
о
О
N
О
о
N N
N
N
SO2 N
Ч
N
320
1159, 163,
L64]
300
[159, 163,
164]
520
[173]
[169, 184]
[140]
Продолжение табл. 7.28
Элементарное звено
разм'
°С
т
разл
^на
воздухе),
разл
(в N2.)
°С
75
76
77
78
О
550
550
чаются в интервале температур 250—400 С. Температура
размягчения полибензоксазолов, полученных на основе дикарбоновых
кислот Сб—С8, колеблется от 250 до 280 °С.
7.3.2.2. Растворимость
Большинство ароматических полибензоксазолов растворяется
только в полифосфорной или концентрированной серной кислотах.
Наличие в составе полимеров «шарнирных» атомов и групп
увеличивает их растворимость. Полимеры на основе бисC-амино-4-гидр-
оксифенил)сульфона и ароматических дикарбоновых кислот
растворяются также в диметилацетамиде, диметилформамиде, N-ме-
тилпирролидоне и диметнлсульфоксиде [184]. При введении в
полимер объемистых заместителей между фенильными ядрами в
остатках бис-о-аминофенолов получаются полимеры, растворимые
(кроме указанных растворителей) в крезоле и хлороформе (с изо-
пропенильной группой — №26—30; фенилэтильной — №32—34;
дифенилметилыюй — № 35; флуореновой — № 36—38).
Растворимость полибензоксазолов строения
возрастает по мере увеличения объема заместителей при
центральном углеродном атоме, находящемся между фенильными
ядрами [159]. Полибензоксазолы на основе алифатических
дикарбоновых кислот растворяются в лг-крезоле и муравьиной кислоте.
7.3.2.3. Термостойкость
Ароматические полибензоксазолы (табл. 7.28, № 1—38)
стабильны на воздухе до 450 °С, а в инертной среде до 500 °С.
Наиболее высокую термостойкость имеют полимеры, в которых
содержатся карбоциклические ароматические и бензоксазольные звенья
(№ 1, 2, 4, 5, 7, 13, 14). Температура начала разложения их как
в инертной среде, так и на воздухе лежит выше 450°С (по
данным ТГА). Полимер на основе 3,3'-диоксибензидпна и терефта-
левой кислоты (№ 4) разлагается на воздухе при температуре
выше 500°С. Полибензоксазолы, в которых между фенильными
ядрами находится замещенная метиленовая группа (№ 24—38)
919
80
Рис. 7.51. Кривые термической
деструкции ароматических полибензоксазолов
при различной температуре на воздухе
[1431:
1 — полимер № 4; 2 — полимер № 9; 3 — по-
лпиер № 20; 4 — полимер № 10; 5 — полимер
№ 21 (табл. 7.28).
разлагаются при более низких
температурах, чем полностью
ароматические полимеры;
температура их разложения на
воздухе колеблется от 300 до 450 °С.
Уменьшение термостойкости в
этой группе тем больше, чем больше объем заместителей при
центральном углеродном атоме. Термостойкость понижается и при
наличии между фенильными ядрами групп SO2— и —О— (№ 16—
22) (рис. 7.51). Температура начала разложения полимеров, в
которых «шарнирные» группы находятся в кислотном остатке, в
инертной среде составляет 400—450°С (табл. 7.28, № 3, 8—12).
Полимеры, в которых «шарнирные» группы находятся как в
остатке аминного, так и кислотного компонентов, начинают разлагаться
при 350 °С.
Такие заместители в ароматическом ядре, как Cl и СНз, сильно
понижают термостойкость, что хорошо видно из данных табл. 7.28
(ср. № 28—30 и № 26, 27). Полибензоксазолимиды (№ 75—78) в
инертной среде стабильны до 550°С. Полимеры с алифатическими
звеньями в цепи (№ 39—66) разлагаются на воздухе при 250—
400 °С.
Изменение массы поли-п-фенилендибензоксазола в
изотермических условиях на воздухе в интервале температур 462—646°С
показано на рис. 7.52. Энергия активации процесса термической
деструкции ароматических полибензоксазолов на основе бис(о-
аминофенолов) и терефталевой кислоты на воздухе достигает 58 ±
± 3 ккал/моль. Ароматические полибензоксазолы с «шарнирными»
группами в остатках как электрофильного, так и нуклеофиль-
ного компонентов имеют энергию активации деструкции около
40 ккал/моль [160].
По аналогии с термическим разложением полибензимидазолов
G.2.2.3) деструкция полибензоксазолов представляет собой
сочетание гомолитических и гетеролитических процессов [ 160]. Основными
летучими продуктами разложения поли-я-фенилендибензоксазола
являются СО, СО2, Н2, HCN, бензол, метан, NH4CN, NH4HCO3.
При 200°С в результате доциклизации выделяется вода. При более
высоких температурах выделяющаяся вода вызывает
гидролитическое расщепление бензоксазольных циклов, а образующиеся в
результате этого амидные связи разрушаются далее, приводя к
получению осколков макромолекул с концевыми карбоксильными и
аминогруппами. Разложение последних приводит к выделению
920
in,
20
1B
12
8
а
P*i i
2
""^^—о
I !
40 ВО 120 т,мин
О 40 80 т,мин
гп,%
.5
100
Рис. 7.52. Кривые изотермической деструкции поли-л-
фенилендибензоксазола на воздухе [160]:
а- 1 — 577 °С; 2 — 590; 3 — 605; 4 — 617; 5 — 628 °С;
6-1 — 565 °С: 2 — 581; 3-600; 4-617; 5 — 636; 6 — 646 °С;
в - / - 531 °С; 2 ~ 553; 3 - 569; 4 — 591;
г - 1 - 462 °С; 2 - 481; ,i — 500; 4—531 'С.
СО2 и NH3. Вследствие протекания
радикальных процессов образуются метан, бензол и
СО. Наличие в продуктах деструкции солей
является следствием протекания вторичных
реакций. В остатке после термообработки
полимеров при 615°С [75% (масс.)] бензокса-
зольные циклы ИК-спектральным методом не
обнаруживаются. Предполагается, что при на-
т.мин гревании происходит структурирование поли*
п-фенпленбепзоксазола [84, 160]:
\
-<ск .а
-ЧГ ))—c-N
С—N
О
\
С—N
N
О,
-®-< ж -с
о
С—N
/
\
7.3.3. ПОЛИБЕНЗОКСАЗОЛЬНЫЕ ПЛЕНКИ
Полибензоксазольные пленки получают поливом из растворов в
диметилформамиде, диметилацетамиде, диметилсульфоксиде, N-ме-
тилпирролидоне или гексаметилфосфотриамиде поли (о-гидрокси)-
921
Таблица 7.29 Физико-механические свойства полибензоксазольной
из раствора [143, 150]
пленки, полученной поливом
№
Элементарное звено
Прочность
при растяжении,
Относительное
удлинение
при разрыве,
%
Модуль упругости
при растяжении,
КГС/СМ2
N
N
О
о
N
о
900-1 000
I 580
1250
2,6
31000
8,4
43 000
10,9 32 000
N
О
N
О
N S02 N
о
О
N SO2 N
О
О
N SO2 N
Гот
о
о
1 120
7,1
2G 000
1 200
12
210
12,1
32 000
1 120
10,9
23 000
или поли (о-метокси) амидов, синтезированных по методике,
описанной в 7.3.1.3, с последующей циклизацией при 220—300 СС в
вакууме или инертной среде [143, 149, 150].
Прямая переработка из растворов возможна исключительно для
полибензоксазолов, в которых фениленовые группы разделены
замещенными метиленовыми мостиками, а в остатке дикарбоновой
кислоты содержатся «шарнирные» группы (например, О или S):
СНз
п
Подобные полибензоксазолы растворяются в ж-крезоле и
хлороформе. Физико-механические свойства полибензоксазольных
пленок приведены в табл. 7.29. По свойствам при высоких
температурах полибензоксазолы близки к полиимидам [183].
Пленки линейных ароматических полибензоксазолов при 150 °С
сохраняют 50 и 40 % прочности при комнатной температуре
соответственно. Термической обработкой на воздухе полибензоксазола
на основе бис (З-гидрокси-4-аминофенил) метана и изофталевой
кислоты получают сшитый полимер, сохраняющий 40 % исходной
прочности при 400°С. Отличную термостойкость имеют также
сшитые полибензоксазолы, полученные термической циклизацией
пленок поли (о-метокси) амидов [179]. К увеличению прочности
пленок, полученных из растворов форполимеров, приводит
вытяжка их в процессе термической циклизации [188]. Полибензокса-
зольная пленка имеет высокую стойкость к термоокислительной
деструкции. При термообработке на воздухе при 300 °С в течение
24 и 72 ч пленка сохраняет 80 и 72 % исходной прочности
соответственно. Прочность пленки, выдержанной 9 ч при 400 °С,
составляет при этой температуре 500 кгс/см2. Выдержка полимера в
течение 10 сут при 400°С в вакууме не отражается на прочностных
и деформационных свойствах [150]. Пленки из полибензоксазолов
обладают хорошей гидролитической стойкостью. После выдержки
в кипящей воде в течение 120 и 240 ч прочность при растяжении
составляет 90 и 75 % от исходной прочности. Прочность и
удлинение пленки практически не меняются после 4,5 ч обработки
кипящими 10 %-ным едким натром или серной кислотой.
Характеристическая вязкость полибензоксазола на основе 3,3'-диамино-4,4'-ди-
гидроксидифенилпропана и изофталевой кислоты после выдержки
в течение 24 ч в 30 %-ной H2SO4 при 100°С и в 20%-ной НС1 при
60 СС увеличивается на 4 и 3—6% соответственно [181].
В результате облучения ароматического полибензоксазола
Y-лучами дозой 500 Мрад прочность уменьшается на 90%.
Диэлектрическая проницаемость ароматических полибензоксазолов
в интервале температур от —150 до 300°С составляет 3,4—3,5
[178]. Тангенс угла диэлектрических потерь при —30 СС проходит
924
О 50 100 150 ZOO t,°C
Рис. 7.53. Зависимость объемного электрического
сопротивления полибензоксазольной пленки от
температуры [143]:
/ — полимер №5: 2— полимер №8 (табл. 7-28); 3 -^
полиимидная Н-пленка.
через максимум, а в интервале
температур 50—210 °С практически не
меняется B,5-10~3 — 2,8-10~3). Электрическая
прочность пленки из ароматических по-
либензоксазолов составляет 180—250
kB/мм. Через сутки работы пленки
электрическая прочность, измеренная при
300 °С, снижается до 24 kB/мм. Зависимость удельного объемного
электрического сопротивления пленки от температуры показана
на рис. 7.53.
Полибензоксазольная пленка медленно горит только в
контакте с открытым пламенем. Пленка используется в
электротехнической промышленности в качестве изоляции обмоток
электродвигателей.
7.3.4. ПОЛИБЕНЗОКСАЗОЛЬНЫЕ ВОЛОКНА
Полибензоксазольные или полибензоксазолимидные волокна
изготавливают мокрым формованием форполимера, полученного
поликонденсацией в растворе с последующей термической
циклизацией при 220—270°С [174, 175]. Обычно используют 15%-ные
растворы поли (о-гидрокси) амидов в диметилацетамиде и воду
в качестве осадителя. При формовании волокон из
высокомолекулярных полимеров необходимо использовать мягкую осадительную
ванну, например 40—70%-ные растворы диметилацетамида или
диметилформамида в воде. Сформованное в таких водных
растворах поли (о-гидрокси) амидное волокно обладает повышенной
способностью к вытяжке вследствие пластифицирующего действия
остатков растворителя. Прочность при растяжении при комнатной
температуре волокна полибензоксазола, имеющего строение
О О
N
N
О
о
О
составляет 5,2 кг/денье, относительное удлинение при разрыве
4 % и модуль упругости 146 г/денье [183]. При 300 и 400 °С
прочность при растяжении составляет соответственно 2 и 1,1 г/денье,
относительное удлинение при разрыве 5 и 3,6 %» модуль упругости
63 и 32 г/денье. Прочность, удлинение и модуль волокна, термо-
обработашюго на воздухе в течение 14 сут при 300°С, составляют
соответственно 2,1 г/денье, 3,1 % и 87 г/денье.
Полибензоксазолимидные волокна характеризуются высокой светостойкостью.
925
7.4. ПОЛИБЕНЗТИАЗОЛЫ
s s
Полибензтиазолы
карбоновых кислот
4-аминофталимидом.
на воздухе до 500 °С
зующем способны к
до 280 °С.
N
получают из бис-о-аминотиофенолов и ди-
или взаимодействием толуидинов с серой и
Ароматические полибензтиазолы устойчивы
. Стеклопластики на полибензтиазольном свя-
длительной эксплуатации при температурах
7.4.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗТИАЗОЛОВ
Полибензтиазолы впервые получены Киприановым [190] в
1962 г. гомополиконденсацией в расплаве З-меркапто-4-аминобен-
зойной кислоты. Однако высокомолекулярные полибензтиазолы
удалось синтезировать только двухстадийным методом через
промежуточное образование полисульфидов или полиамидов.
Поликонденсация бис(о-аминотиофенолов) с дихлорангидридамп дн-
карбоновых кислот в растворе амидных растворителей (например,
N-метилпирролидон или диметилацетамид) в присутствии
акцепторов хлористого водорода, таких, как пиридин, триалкил-
амин или ацетат Na, проводится при —5 — 0°С; в результате
образуются полиамиды или полисульфиды [191, 192, 202]:
SH С ЮС
СОС1
1
1
—n+m
926
Циклизация таких полимеров в пленках, полученных поливом
из растворов в крезоле или муравьиной кислоте, проводится при
200—300°С. Полибензтназолы получают также в одну стадию
в полифосфорной кислоте при 200—250 °С. В качестве электро-
фильного компонента используют свободные дикарбоновые
кислоты, их хлорангидриды, нитрилы, амиды или а ми дины [129, 162,
192, 193]:
fl
,NH,
X=—COOH, -COOR, -CONH2, —CN, —COC1, —С
На основании данных ИК-спектроскопни предложен
следующий механизм образования полибензтиазолов [193]:
l\n,2
О
—н2о
—свн5он
927
Интересный способ получения полибензтиазолов предложен
французскими учеными, которые синтезировали такие полигетеро-
арнлены взаимодействием бис(о-аминотиофенолов) с диглиокса-
лиловыми соединениями. В результате реакции З.З'-дитиобензи-
дина с ароматическими бисглиоксалевыми кислотами или их эфн-
рами в ж-крезоле или диоксане при 100—150°С сначала
образуются полибензтиазолины, которые при 250—400°С превращаются
в полибензтиазолы [197] :
H,N
О О
NH2
SH
-f- n
H5C
2OC-
II
0
-c
il
0
С2Н5ОН I Диоксан, 100 °С
—СО2|.«-Крезол, 150 °С
— П
В полифосфорпой кислоте при 100—200 °С в качестве
промежуточных продуктов образуются полибензтиазиноны, которые, де-
карбонилируясь при 250—400°С, дают полибензтиазолы:
+ n C2H5OC—С
о о
—(О)—°—(О)—с-сос2н5
|
оо
I
п
I-1
со
N
—п
В основу промьттттленного способа получения полибензтиазолов
может быть положена реакция толуидина, серы и 4-аминофтали-
923
Рис. 7.54. Кривые термической деструкции т,%
поли-2,6-бензоксазола A) и поли-2,6-бенз- ЮО
тиазола B) при различной температуре на
воздухе [129|. ' " во
мида [194—196]. При этом вначале 60
образуется плавкий ц растворимый
форполимер, который циклизуется *о
в процессе переработки при 95—
155°С. 20
100 200 300 400 500 t°G
7.4.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИБЕНЗТИАЗОЛОВ
Полибензтиазолы — аморфные полимеры от желтого до
оранжевого цвета. Ароматические полибензтиазолы растворяются
только в концентрированной серной и полифосфорной кислотах.
Полибензтиазолы, полученные с использованием алифатических
дикарбоновых кислот, растворяются также в муравьиной, трифтор-
уксусной кислотах и крезоле. Температура стеклования их лежит
в интервале 250—500 СС (табл. 7.30). Полимеры плавятся выше
температуры разложения.
Как видно из рис. 7.54, по термостойкости полибензтиазолы
близки полибензоксазолам и полибензимидазолам. Полимеры,
содержащие только тиазольные и фениленовые циклы, разлагаются
на воздухе при 500—600°С и в инертной среде —при 700 °С
(табл. 7.30). Выше 600 °С происходит дегидрирование и
структурирование полимеров. Содержание водорода в ароматических по-
либензтиазолах после нагревания на воздухе при 816°С
составляет около 45% от исходного значения [193]. Полибензтиазолы
на основе алифатических дикарбоновых кислот разлагаются на
воздухе при 300—400 °С.
7.4.3. ПЕРЕРАБОТКА И СВОЙСТВА ПОЛИБЕНЗТИАЗОЛОВ
Стеклопластики на основе полибензтиазолов могут быть
получены путем пропитки наполнителя раствором форполимера в ди-
метилформамиде или диметилацетамиде с последующей
термической циклодегидратацией в процессе прессования [196].
Прочность и модуль упругости при растяжении полибензтиазольного
стеклопластика при комнатной температуре достигают 4000 и
250 000 кгс/см2 соответственно. Прочность и модуль упругости при
растяжении стеклопластика, выдержанного в течение 300 ч на
воздухе при 315°С, составляют соответственно 56 и 78% от
исходных; после 6-часовой термообработки при 425 °С эти
показатели составляют соответственно 52 и 76 % [196].
Полибензтиазольные стеклопластики длительное время
сохраняют свои свойства при высоких температурах и по стойкости
к термоокислительной деструкции превосходят аналогичные поли-
бензимидазольные материалы G.2.6). При 288°С при разрывном
напряжении 2600 кгс/см2 разрушение происходит через 1000 ч
30 Зак. 18
Таблица 7.30. Температуры стеклования и разложения полибензтиазолов
Элементарное звено
Т
разл
Т
разл
(на
воздухе),
вС
Литературный
источник
> 450
650
680
520
[129. 190, 192]
540
[192]
550
[162, 193, 202]
600
[162, 191, 193,
202]
\
N
N
233
[193, 197]
[1931
S HN NH
[2031
О
\
LQ
\
[198]
g
Продолжение табл. 7.30
разл
(в N2).
°С
т
разл
(на воз-
Литературный
источник
238
450
430
120Ц
[11, 162]
[20Ц
[200]
[1991
Элементарное звено
—(SF2)«-
Пластики на основе алифатическо-ароматических полибензти-
азолов, например полиоктаметилендибензтиазола (табл. 7.30,
№ 11), могут быть получены прессованием при 300°С под
давлением 70 кгс/см2 [11].
7.5. ПОЛИХИНОКСАЛИНЫ
N N ,—ч
п
Полихиноксалины получают поликонденсацией бисA,2-дикар-
бонильных) соединений и бис(о-диаминов). Ароматические
полихиноксалины устойчивы на воздухе до 550 °С. Полимеры
используют для изготовления пленок, волокон; их применяют в качестве
связующих и клеев, которые работают при температурах до 300°С.
Они могут применяться для изготовления покрытий, клеев для
металлов, электроизоляции, связующих для материалов,
используемых в авиационной промышленности.
7.5.1. ПОЛУЧЕНИЕ ПОЛИХИНОКСАЛИНОВ
Полихиноксалины получают в расплаве или растворе по схеме
—(Г))—С=О
H — полихиноксалин
СеН5 — полифенилхииоксалин
Хиноксалины были синтезированы из 1,2-дикарбонильных
соединений и о-диаминов в 1884 г. [256, 257]. Применительно к
полимерам эта реакция впервые была использована в 1961 г. [258].
Работы по синтезу и исследованию этих полигетероариленов
ведутся в США и во Франции *.
Другой вариант синтеза полихиноксалинов основан на
взаимодействии а-бромацетильных производных ароматических
соединений с бис(о-ди.аминами) с промежуточным образованием поли-
аминокетонов [241].
Полихиноксалинимиды синтезируют из диаминов, содержащих
в своем составе хиноксалиновые циклы. Аналогичным образом из
дибензилов, содержащих амидные группы, получают полихин-
оксалинамиды (табл. 7.31, № 56—68) [251, 252]. Их можно
* Д также в СССР и ГДР. — Прим. ред,
933
Таблица 7.31. Температуры стеклования и разложения полихиноксалинов
Элементарное звено
Т
разл
(в N2).
°С
Т
разл
(на
воздухе),
°С
Литературный
источник
N
N
x ;
v :
N
N
350
305
280
575
640
565
550
600
535
430
515
530
625
[208, 218, 224,
230, 241, 2501
[2171
[210, 218, 230,
250, 255]
[90, 217-219,
210, 224, 250,
2551
[218, 250]
N
10
11
195
530
510
[218, 250]
213
133
560
520
535
510
[218, 250]
[218, 250]
520
450
580
450
540
520
[217, 224]
[217]
[210]
Продолжение табл. 7.31
12
13
14
15
16
Элементарное звено
N N
N N к
X Ж Ж )-O-s-0-
NON
N N ^^
N N
'с-
°с
270
т
разл
(BeN2),
540
т
разл
(на воз-
510
460
420
500
460
Литературный
источник
[210, 217, 219,
224, 246]
[207, 246]
[207, 246]
[207, 246]
[207, 246]
17
N
18
SO
N
19
N
N
sSO2 N
N
21
212
450
12201
430
[220, 246]
440
[207, 246]
420
[207, 246]
465
[220]
Продолжение табл. 7.31
22
23
24
25
N
V ^^ Ч^ ^***4.
N
V. ^ *Ч.
Y^
N
Y^
CsHs N
N
V >rf Ч
N
N
N
Элементарное злено
N
-(QV-
218
254
317
т
разл
в°с2'
Г
разл
(на
воздухе),
440
520
520
540
Литературный
источник
[220]
[2Щ
[211]
[212, 213, 224,
241, 244, 245,
248, 250]
26
27
28
29
30
сбн5-
N
С6Н5
N
N
i)e-
320
285
260
550
550
450
540
530
520
445
[212, 213, 224,
2501
[210, 255]
[90, 210, 250,
255]
[250]
[209]
Продолжение табл. 7.ol
Элементарное звено
•?
разл
«в N2b
°G
T
разл
(на
воздухе).
°G
Литературный
источник
N
298
253
268
550
450
515
520
530
510
445
[212, 213, 224,
250]
[212, 213, 224.
250]
[210]
[210, 224, 246,
250, 255]
[209]
S7
39
N
^C6H5
N
40
С6Н5
а?
41
Ю5
258
243
490
490
490
520
510
510
[220, 246, 250]
[220, 246, 250]
[220, 24а]
[220, 250]
[220, 250]
[220]
Продолжение табл. 731
Элементарное звено
Т
разл
Т
разл
(на UO3-
духе),
Литературный
источник
"
oi
N
О
о о
217
260
550
530
[242]
[240, 243, 247J
[215, 240, 243,
247]
45
46
47
48
230
N—(
N
Н3
-сн2
N
-сн2
о
о о
274
254
290
535
515
С6Нб
О
<Ж?До;
Л
4
О
[215, 240, 243]
[232]
[214, 240, 243,
247]
[215, 240, 243,
247]
Продолжение табл. 7.3I
[214, 240, 243,
247]
[215, 240, 243,
247]
52
53
300
240
540
с6н;
о
54
255
525
О
О
[215, 240, 243,
247]
[214, 240, 247,
243]
[215, 240, 243,
247]
N
|
О
55
255
310
О О
[215, 240, 243,
247 J
C6H5'
Продолжение табл. 7.31
Элементарное звено
°С
раз л
(в N2).
°С
Г
разл
(на
воздухе),
О
N
N
О
О
—NH—<
CONH
NH—
279
456
432
340
60
61
62
63
64
266
448
N
N
NON
298
429
N
N
ХЖ
H>-NH-<
О L WJ О
325
CONH
N
N
N
—NH—С—
О
A
N
228
:—NH—(( ))—
442
SO2 N
C6H5
О О
278
429
^6^5 N SQ2 N ^бНб
о LvJJ о
Продолжение табл. 7.31
Элементарное звено
Т
разл
Т
разл
(на воз-
N
N
О
^/^¦^
с6н5
¦ч.
N
шж
с6н5-
о
о
.С
CONH—
247
253
:—nh—(Г ))—
460
435
350
69
N О
70
=- -О-
О
О N
NO
71
II
жххххж
О N ! NO
Продолжение табл. 7.31
Элементарное звено
т
разл
(в N2),
"С
Т
разл
(на воз-
Духе),
N
О N
О—
О
N0 S ON
toi XjQKJQ^XM
О S ON
N О
N О
NH N
450
430
N NH
76
77
78
79
SQ
81
R — —j
450
450
450
500
500
500
400
400
400
Продолжение табл. 7.3}
Элементарное звено
разл
(в NZ),
°С
разл
(на воз-
духр).
NH N
NH N
NH NH N
NH NH N
МН N
NH N
400
380
86
87
720
700
N NH NH
N О
N
89
90
91
92
«о
520
460
1254]
[254]
[234, 235]
[236]
[225]
[226]
[206]
Продолжение табл. 7JSI
Элементарное звено
разл
BeN2).
(на воз-
Н3С СНз
N \/ N
N /\ N
НзС СНз
N
N
NH N
H
400
680
390
460
400
N [Oi N
N ^ N
•98
100
101
О N
700
500
[2491
700
500
12491
683
460
B041
500
400
[2211
[2231
Продолжение табл. 7.31
Элементарное звено
разл
oN2),
т
разл
(на воз<
духе),
ON
О
О
NO
о»
ON NO
Н3С СНз
N
N
N /\
НзС СНз
ON
N О
1
жхжх:®
JON NO
х
NO
N
NO
NH
ХЖХЖ!
N N NH О
400
400
200
синтезировать также из диаминов с хиноксалиновыми циклами и
дихлорангидридов дикарбоновых кислот [242]. Лестничные поли-
хиноксалины получают из циклических тетракетонов и
ароматических тетраминов [204—206, 249, 253, 254] :
H2N
—Я
7.5.1.1. Поликонденсация бисA,2-дикарбонильных)
соединений с бис(о-диаминами) в расплаве
Эквимольные смеси диглиоксалей или дибензилов с тетрамина-
ми нагревают сначала в среде инертного газа при 180—200 °С,
затем в вакууме, при этом температуру ступенчато повышают до
400 °С [204, 208, 209, 210]. Поликонденсация сопровождается
затвердеванием реакционной массы. Вследствие отклонения от экви-
мольности за счет частичной сублимации исходных веществ при
этрм образуются сшитые полихиноксалины. Производные глиок-
саля, используемые в качестве мономеров для синтеза полигетеро-
ариленов, получают окислением диацетильных соединений
ароматического ряда диоксидом селена:
О
U
M [1
о о
Ароматические диглиоксали плавятся в интервале температур
100—160°С. Температура плавления дибензилов составляет 60—
160 °С. Синтез ароматических бис(о-диаминов) описывается
следующей схемой [209] :
(сн3соJо
HNO3
NHCOCHa >
CH3GONH—¦
CH3CONH
Н2О
ЫНСОСНз >
957
Ароматические тетрамины плавятся в интервале температур
110—270°С; они относятся к числу очень канцерогенных веществ.
7.5.1.2. Поликонденсация бисA,2-дикарбонильных)
соединений с ароматическими бис(о-диаминами)
Поликонденсацию диглиоксалей или дибензилов с
ароматическими бис(о-диаминами) проводят в растворе диметилформамида,
диметилацетамида, гексаметилфосфотриамида, N-метилпирроли«
дона, диметиланилина, л*-крезола или полифосфорной кислоты
в интервале температур от —10 до 250°С [90, 206—208, 211—220,
250, 255]. Взаимодействие тетраминов с диглиоксалями в растворе
при 250 °С приводит к образованию промежуточных полимеров,
которые циклизуют в виде пленок в вакууме при 350—400 °С до
полихиноксалинов:
H2N
О—с=° -
—.П
Ароматические дибензилы обладают гораздо более высокой
реакционной способностью, чем соответствующие диглиоксали:
H—С- С—,
u
Диглиоксаль
с-с-н
il и
оо
il il О в и
оо \^ о о
Дибеизил
Полифенилхиноксалины (в одну стадию) образуются при
взаимодействии тетраминов с дибензилами в растворе при
температурах до 200 °С. На молекулярную массу полимера очень сильное
влияние оказывает порядок введения в реакцию исходных
веществ. Наиболее высокомолекулярные полифенилхиноксалины
получаются при добавлении электрофильного компонента в твердом
виде или в виде дисперсии в соответствующем растворителе к
дисперсии или раствору тетрамина. При смешении растворов обоих
компонентов образуются сшитые не полностью циклизованные по-
956
лимеры с низкой термостойкостью:
R-C
UM
N
—C=N
I
O=C— R
Такую межмолекулярную реакцию удается подавить,
поддерживая низкой концентрацию тетракетона в реакционной среде,
добавлением его в виде суспензии или порошка. Тогда реакционно-
способные карбонильные группы реагируют внутримолекулярно
с образованием гетероцикла, давая линейные полифенилхинокса-
лнны.
Незамещенные полихиноксалины в промышленном масштабе
получают в ж-крезоле [250]: 20 %-ную дисперсию бис(/г-феннлен-
глноксальгидрат)оксида в течение 3 мин вводят в 20%-ный
раствор З.З', 4,4/-тетраминодифенилоксида в ж-крезоле и
реакционную смесь выдерживают 4 ч при 75 °С при постоянном
перемешивании. Раствор полимера в ж-крезоле стабилен при комнатной
температуре в течение 90 сут и может быть использован
непосредственно для получения пленок, волокон и препрегов.
В процессе взаимодействия дикарбонильных соединений с тетр-
аминами образуется смесь изомеров:
О О
R
"r
lO
V^«
N
2,2'-Изомер
N
—m
959
Соотношение между 2,2'- и 3,3'-изомерами определяется
природой радикала (R — Н, СбН5) и реакционной способностью
мономеров. Незамещенные полихиноксалины (R = H) существуют
главным образом в виде 2,2'-изомера. Взаимодействие диглиокса-
лей с тетраминами, нуклеофильность которых относительно
высока, например с тетраминодифенилоксидом, приводит к
образованию в основном 3,3'-изомера (табл. 7.31, № 12). В полифенил-
хиноксалинах (табл. 7.31, № 23—41) соотношение изомеров зависит
от реакционной способности карбонильных групп дибензилов.
В табл. 7.31 все полимеры независимо от изомерного состава
показаны в виде изомеров строения 2,2'.
7.5.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИХИНОКСАЛИНОВ
В зависимости от структуры и степени сопряжения в цепи
полихиноксалины имеют окраску от желтого и красного до черного
цвета. Температура плавления их лежит выше температуры
начала разложения. Полимеры, содержащие только хиноксалиновые
и бензольные циклы, имеют кристаллическую структуру;
наличие шарнирных групп приводит к аморфизации полихинокса-
линов. Температуры стеклования и разложения полимеров
приведены в табл. 7.31 ('№ 1—22 — незамещенные полимеры; № 23—
41—полифенилхиноксалины; № 42—55 — полихиноксалинимиды;
№ 56—68 — полихиноксалинамиды; № 69—89 — сополимеры;
№ 90—105 — полихиноксалины лестничной структуры).
7.5.2.1. Температура стеклования
Температура стеклования чисто ароматических нефенилирован-
ных полихиноксалинов колеблется от 300 до 350°С (табл. 7.31,
№ 1—3, 5); полимеров, в состав которых входят «шарнирные»
группы (например, —О—, —S—, —SO2—, —СО—), — от 150 — до
300 °С (№ 4—8, 10—22), температура стеклования полифенил-
хиноксалинов (№ 23—41) на 10—35° ниже, чем у их нефенилиро-
ванных аналогов. Температуры стеклования полихиноксалиними-
дов (№ 42—55) находятся в пределах от 200 до 300 °С;
полихиноксалинамиды размягчаются при 230—280°С.
7.5.2.2. Растворимость
Растворы полимеров имеют такую же окраску, как и сами
полихиноксалины. Ароматические нефенилированные
полихиноксалины (табл. 7.31, № 1—3,5) растворяются в концентрированной
серной кислоте и частично в гексаметилфосфотриамиде. Наличие
«шарнирных» групп в макромолекуле обеспечивает растворимость
в гексаметилфосфотриамиде, феноле и л*-крезоле; причем
растворимость улучшается с увеличением содержания этих групп в
полимере [207, 220]. Фенилированные полимеры растворяются гораздо
960
лучше, чем незамещенные полихиноксалины. Растворителями для
них служат серная кислота, фенол, ж-крезол, дихлорбензол, тетра-
хлорэтан, хлороформ, сесквигидрат гексафторацетона, гексафтор-
изопропанол, нитробензол и пиридин. Для полифенилхиноксали-
нов с «шарнирными» группами можно получить растворы до
30 %-ной концентрации [211, 212, 220].
Гидродинамические свойства растворов полихнноксалинов в
м-крезоле изучены в [244]. Сополимеры, содержащие наряду с
хиноксалиновым имидный гетероцикл и амидные связи,
растворяются в тех же растворителях, что и полихиноксалины [214, 2151.
Лестничные полихиноксалины (№ 90—105) в силу высокой
жесткости цепей растворимы ограниченно. Лестничные полимеры
с тиазиновыми и дигидропиразиновыми звеньями (№ 96, 100)
растворяются в серной и метансульфокнелоте [221, 222]; при
наличии диоксиновых групп (№ 101 —105) — только частично в
концентрированной серной кислоте.
7.5.2.3. Термостойкость
Наличие боковых фенильных групп способствует увеличению
стойкости полихиноксалинов к окислительной деструкции, а
«шарнирные» группы понижают их термостойкость. Чисто
ароматические незамещенные полихиноксалины (№ 1—3,5) стабильны на
воздухе до 520°С и в инертной среде до 580°С. При наличии
«шарнирных» групп (№ 4, 6—22) полимеры устойчивы в инертной
атмосфере до 500—550 СС и на воздухе до 450—500 °С. Стойкость
полифенилхиноксалинов на 20—30° выше, чем соответствующих
незамещенных полимеров (рис. 7.55 и 7.56). Влияние «шарнир-
400
500
0
120 160 Т,ч
Рис. 7.55. Кривые термической деструкции полихиноксалинов при различной
температуре на воздухе и в среде гелия [210]:
/ — полихиноксалин № 34; 2 — полихиноксалин № 12 (табл 7.31); сплошные линии — на
воздухе; пунктирные — в гелии.
Рис. 7.56. Кривые термической деструкции полихиноксалинов при различной
температуре на воздухе [210]:
/ — полихиноксалиь Ni 34; 2~ полихииоксалнн № 12 (табл. 7.31); сплошные линии — при 316;
пунктирные — при 371 °С.
31 Зак 18
961
100
80
60
40
20
Pue. 7.57. Кривые 430 термической деструкции по-
лифенилхиноксалннов на воздухе [224]:
1 — полимер № 25; 2 — полимер Kt 31; 3 — полимер №34
(табл. 7.31).
ных» групп на термостойкость наглядно
иллюстрируется рис. 7.57.
Наиболее уязвимым местом при
окислительной деструкции незамещенных поли-
хиноксалинов является атом водорода пи-
0 50 100 150 т,ч
- - - - - - г'1 г -
разинового цикла [224], который первым подвергается
окислению:
t
Медленно
N
N
N
N
о—о.
[01
Быстро
Пероксидный радикал, образовавшийся в результате окисления,
реагирует с атомом водорода пиразинового кольца, превращаясь
в относительно более стабильный продукт — гидропероксид:
Реакцией, лимитирующей скорость деструкции, является
распад гидропероксида с образованием
резонансно-стабилизированного продукта хиноидной структуры:
О—ОН
loi
Медленно
962
Деструкция фенилхиноксалинов, не содержащих «шарнирных»
атомов и групп, протекает в три стадии [224]. Сначала между
500 и 640 °С разрушается пиразиновый цикл и отщепляются
гетероциклические фрагменты:
С6Н5СНЭ
Среди летучих продуктов деструкции при 640°С полимера
№ 26 (табл. 7.31) обнаружено 4,7 % СН4, 5,4 % Н2О, 15,9 % HCN,
3,5% СО2, 20,3% (CNJ, 19,7% бензола, 18,2% толуола и
1,8% бензонитрила. Стабильный азотсодержащий остаток при
640—690°С дегидрируется:
C20H10N3 —>¦ C20H4N3 +
При пиролизе при 1300—1400 °С происходит конденсация с
образованием ароматических систем, в которых на два атома азота
приходится 20 атомов углерода.
Полифенилхиноксалины с простыми эфирными связями между
бензольными ароматическими ядрами (№ 31—33) при
термоокислении разрушаются по мостиковому кислородному атому:
N
Фенильные и феноксидные радикалы реагируют с кислородом
с образованием пероксидных радикалов, которые с отрывом фе-
нильной боковой группы превращаются в хиноидные структуры:
[О]
о
Кинетика процесса деструкции изучена в [245].
31*
963
Полихиноксалинамиды (№ 56—67) разлагаются в инертной
среде при 320—460СС. Термостойкость зависит,от изомерии дикар-
боновой кислоты [251]. Полихиноксалинамид с сложноэфирными
группами (№ 68) при температуре выше 200°С сшивается с
отщеплением эфирных групп. Такой сшитый полихиноксалинамид
начинает разлагаться на воздухе при температуре выше 480°С
[252]. В соответствии с той ролью, которая отводится
полисопряженным системам B.3), следует ожидать, что лестничные поли-
хиноксалины должны иметь более высокую термостойкость, чем
их нелестничные аналоги. Действительно, лестничные полимеры
№ 94 и 97—99 в инертной среде устойчивы до 700 °С и на воздухе
до 500 °С. Относительно низкая термостойкость лестничных поли-
хиноксалинов, приведенных в табл. 7.31, может быть вызвана
недостаточной степенью циклизации этих полимеров.
7.5.3. ПОЛИХИНОКСАЛИНОВЫЕ ПЛЕНКИ И ВОЛОКНА
Полихиноксалиновые пленки получают поливом из растворов
полимеров в крезоле [212, 250]. Растворитель удаляется за 18 ч
при 70°С в сушильных шкафах с принудительной циркуляцией
воздуха, и сформировавшаяся пленка нагревается в вакууме в
течение 6 ч при 200°С и 1 ч при 300°С для завершения циклизации
и удаления остатков растворителя. Свойства пленок различных
полихиноксалинов приведены в табл. 7.32. Пленки окрашены в
желтый цвет. Они могут применяться в качестве
высокотермостойкой электроизоляции. В циклизованном состоянии полихин-
оксалины сохраняют некоторую термопластичность, которая дает
возможность прессовать из них при 420—480 °С пластики [219].
Полихиноксалиновые волокна могут быть получены мокрым
формованием из крезольных растворов в воду [250]. Волокна из
полиC,3'-/г-фенилен-6,6'-ди-2-фенилхиноксалина) (см. табл. 7.31,
№ 25) при комнатной температуре имеют прочность при
растяжении 6,5 г/денье, относительное удлинение при разрыве 3,2 % и
модуль упругости 60 г/денье.
7.5.4. СВЯЗУЮЩИЕ НА ОСНОВЕ ПОЛИХИНОКСАЛИНОВ
Для получения стекло- и боропластиков на основе
полихиноксалинов используют растворы форполимеров. При выдержке
20 %-ного раствора полимера в -и-крезоле в течение 3 мес.
молекулярная масса его остается неизменной.
Препреги на основе стеклянного волокна получают путем
окунания в 15—20%-ные растворы форполимера в ж-крезоле с
последующей сушкой на воздухе в течение 1 ч при 130 °С в
термошкафах [219, 250]. Препреги сохраняют способность к
переработке в течение 4 мес. Преимущество препрегов этого типа по
сравнению с аналогичными материалами на основе других
термостойких гетероциклических полимеров заключается в их
исключительной способности к переработке из высококонцентрированных
964
Таблица 7.32. Свойства полихиноксалиновых пленок
N N
N
III-
N CfiH
6П5
Показатель
Прочность при растяжении, кгс/смг
при 23 °С
при 177 °С
Относительное удлинение при
разрыве, %
при 23 °С
при 177 °С
Модуль упругости при растяжении,
кгс/см2
при 23 °С
при 177 °С
Диэлектрическая проницаемость при
1 кГц
при 25 °С
при 200 °С
Тангенс угла диэлектрических
потерь при 1 кГц
при 25 °С
при 300 °С
j
|2J2|
660
17
4,6
0,02
0,07
п
1250J
1 260
980
4,5
21
28 000
19 700
ш
12оО|
1 190
850
8,5
25
26 700
16 900
[255|
811
5,5
3,46
3,22
растворов (небольшое содержание растворителя). Это
объясняется тем, что выше 400°С полимер проявляет термопластические
свойства, а остатки крезола выполняют функцию пластификатора.
Переработка препрегов осуществляется при 370 °С. Давление
в процессе прессования зависит от содержания остаточного
растворителя. При содержании 0,8 % растворителя давление равно
35 кгс/см2, а при 2—3% — 14 кгс/см2. Последующую
термообработку проводят в инертной среде в общей сложности в течение
40 ч в интервале температур от 205 до 400 °С. В процессе термо-
965
Таблица 7.33. Физико-механические свойства
полихиноксалиновых стеклопластиков [250]
I-
II —
Показатель
Плотность, г/см3
Содержание
связующего, %
Пористость, %
Прочность при изгибе
при 23 °С, кгс/см2
1 ч, 371 °С
50 ч, 371 °С
10 мин, 538 °С
I
1,92
24,8
4,0
7 800
4 200
2 500
2 100
П
1,71
33,1
8,6
7 000
4 300
3 100
1200
Показатель
Модуль упругости при
изгибе при 23 °С, кгс/см2
1 ч, 371 °С
50 ч,371 °С
10 мин, 538 °С
I
342 000
245 000
184 000
225 000
и
256 000
172 000
176 000
154 000
обработки выделяется 0,8 % летучих, состоящих из 0,3 % воды и
0,5 % крезола.
Физико-механические свойства стеклопластиков на основе по-
лихиноксалинового связующего приведены в табл. 7.33.
Физико-механические свойства боропластиков на основе поли-
хиноксалинового связующего I после выдержки в течение 3 мин
при температуре испытаний приведены з табл. 7.34.
Как видно из табл. 7.33, стеклопластик на основе поли-2,2'-
D,4/г-оксидифенилен)-6/,6-оксидихиноксалина при комнатной
температуре имеет прочность при изгибе 7800 кгс/см2 и модуль
упругости 342 000 кгс/см2. При 371 °С через 1ч и 50 ч соответственно
эти значения уменьшаются до 50 и 32%, а при 538 °С через
Таблица 7.34. Физико-механические свойства
полихиноксалиновых боропластиков [219]
Пока та гель
Прочность при изгибе,
кгс/см2
Модуль упругости при
изгибе ?10~4, кгс/см2
Температура, °С
23
17 780
297
133
13 850
255
177
14 480
263,6
212
12 650
260
316
15 540
170
361
16 940
85
966
10 мин — до 27%. Верхняя температура эксплуатации в течение
длительного времени составляет 300°С.
Боропластики прессуют при 394 °С. При этом они сначала
предварительно прогреваются в течение о мин и выдерживаются
4 ч под давлением 35 кгс/см2. Стеклопластики прессуют при том
же температурном режиме. Свойства этих материалов
представлены выше (табл. 7.33 и 7.34).
Как уже отмечалось, полихиноксалинамиды с боковыми слож-
ноэфирпыми группами сшиваются в процессе переработки с
одновременным отщеплением этих групп. Графитопласты на таких
связующих сохраняют при 370°С 80% прочности при комнатной
температуре [252].
Возможные области применения слоистых пластиков —
авиация и космическая техника.
7.5.5. ПОЛИХИНОКСАЛИНОВЫЕ КЛЕИ ДЛЯ МЕТАЛЛОВ
Полихиноксалиновые клеящие пленки (в качестве носителя
используют стеклянную-ткань) применяют для склеивания
специальных сталей и титана. Рабочая температура клеевого
соединения выше 300°С [90, 219, 250]. Для получения препрегов
раствор форполимера с добавкой аморфного бора E0 % порошка
бора в расчете на форполимер) наносят на стеклянную ткань и
высушивают до остаточного содержания растворителя 11 —12%.
Склеивание специальных сталей, предварительно протравленных
фосфорной кислотой, производят при 420—490 °С под давлением
Таблица 7.35. Прочность при сдвиге полихиноксалиновых
клеевых соединений специальной стали при повышенных температурах [250]
I-
II-
Условия испытаний
23 °С
I ч, 316°С
200 ч, 316'С
1 ч, 371 °С
N
О
Прочность при сдвиге
при температуре
опыта. кгс'смЗ
I
235
206
160
130
n
233
ПО
N
Условия испытаний
50 ч, 371 °С
10 мин, 538 °С
1 ч, 538 °С
Прочность при сдвиге
при температуре
опыта, кгс/см2
I
178
93
II
117
90
58
967
15 кгс/см2 в среде азота за 2—3 ч. Значения прочности при сдвиге
клеевого шва на полихиноксалиновой основе приведены в табл. 7.35.
Выше температуры стеклования полимера B70 или 280°С) при
длительном нагревании происходит его разрушение вследствие
пластической деформации. Термообработка полимера и клеевого
соединения выше 400°С приводит к сильному ухудшению
термопластичности. Ползучесть при 230°С под нагрузкой 70 кгс/см2 не
проявляется в течение 60 сут.
Перед склеиванием титановую жесть обрабатывают раствором
форполимера в крезоле, высушивают в среде азота при 400°С и
склеивают в автоклаве под давлением. По этой же технологии
изготавливают титановые сотовые конструкции. Такие клеи
применяют в авиации и космической технике.
7.6. Полихиназолоны
о О
Полихиназолоны отличаются от полибензимидазолов наличием
дополнительной карбонильной группы в гетероцикле:
О О
HN NH JI JI
к N N *
Они получаются взаимодействием ароматических бис(о-амино-
амидов) дикарбоновых кислот с дихлорангидридами дикарбоно-
вых кислот или поликонденсацией бисбензоксазинонов с
диаминами.
Термическая деструкция полифенилхиназолонов в
окислительной среде начинается при температуре выше 425 °С. Полиметилхин-
азолоны устойчивы на воздухе до 325 °С и в инертной среде —до
400 °С. Полихиназолоны могут перерабатываться из раствора
в пленки и волокна. Их длительная работоспособность достигает
200° С. Эти полимеры могут применяться для изготовления
электроизоляционной пленки.
7.6.1. ПОЛУЧЕНИЕ ПОЛИХИНАЗОЛОНОВ
7.6.1.1. Незамещенные полихиназолоны
Незамещенные полимеры получают из бис(о-аминоамидов)
дикарбоновых кислот и дихлорангидридов дикарбоновых кислот
968
[260,261]:
_„
На первой стадии в растворителях амидного типа (диметил-
формамид, диметилацетамид, N-метилпирролидон, гексаметилфос-
фотриамид) получают при —20-^60°С в присутствии хлорида
лития линейные полимеры, представляющие собой полиамиды
с о-амидными звеньями, циклизация которых в твердой фазе
осуществляется в вакууме при 250—400 °С.
7.6.1.2. Получение поли-3-метил- или
поли-З-фенилхиназолонов-4
Линейные полихиназолоны получают поликонденсацией в
растворе или в расплаве 3-метил- или 3-фенилзамещенных бисбенз-
оксазинонов с ароматическими диаминами [262—268]:
= _CH3, —С6Н5
96S
Г] уд 1С
0,6
0,4
0,2
Рис. 7.58. Зависимость молекулярной массы
(%Д/С, 0,5 г в 100 мл крезола, 25 °С) продукта
поликонденсации бисB-метилбензоксазинона) с
З^'-диаминобензанилидом в га-хлорфеиоле от
продолжительности реакции [265]:
1 — 25 "С; 2—150; 3 — 180 °С.
0 1 2 3 Т,7
Исходные бисбензоксазиноны получают расщеплением по
Гофману ароматических диимидов с последующими ацилированием и
циклодегидратацией продуктов реакции [263, 266]:
о
НООС
соон
rcoci
О
НООС
Поликонденсация в растворе протекает при температурах 150—
200 °С в инертной среде в таких полярных растворителях, как ди-
метилформамид, диметилацетамид, N-метилпирролидон, диметил-
сульфоксид, /г-хлорфенол или м-крезол [262, 265, 267, 268].
Эффективным растворителем является /г-хлорфенол. Молекулярная
масса образующегося полимера уменьшается с повышением
температуры синтеза, что хорошо видно из рис. 7.58. Наиболее
высокомолекулярные продукты образуются при 10%-ном мольном
избытке бисбензоксазинонов, который необходим для компенсации
их потерь за счет гидролиза, вызываемого водой, выделяющейся в
ходе процесса [268]. Поликонденсация фенилзамещенных
бисбензоксазинонов с диаминами протекает гораздо медленнее, чем
соответствующих метилзамещенных соединений [266].
При нагревании полихпназолонов, полученных в растворе, в
твердой фазе в вакууме при 200—300 °С молекулярная масса
полимеров увеличивается. Этн полимеры не растворяются в
органических растворителях, но растворяются в концентрированной
серной кислоте [262, 265].
В полифосфорной кислоте при 160—200 °С получаются
низкомолекулярные полимеры [268].
Поликонденсация в расплаве позволяет получать
высокомолекулярные полигетероарилены, но с относительно низкой степенью
циклизации [266, 267]. При проведении синтеза в полифосфорной
кислоте можно исходить не из бнсбензоксазинонов, а из проме-
97U
жуточных продуктов их получения — о-ацетилампнокарбоновых
кислот [268]:
НООС СН2
I
7.6.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПОЛИХИНАЗОЛОНОВ
В табл. 7.36 представлены свойства некоторых полимеров,
разделенных на три группы:
0 0 0 0
\T—R—
N ' N
Незамещенный полихиназолон (№№ 1-3)
О
N N
Полпметилхкназолон (№JVs 4-17)
N
Полифенилхиназолон № 18-22
Полихиназолоны, содержащие только бензольные и
гетероциклические группы, деструктируются ниже температуры плавления.
Полиметилхиназолоны, имеющие «шарнирные» группы как в
остатке диамина, так и бисбензоксазинона, размягчаются в
интервале температур 420—490 °С [265]. Полимеры, в остаток аминного
компонента которых входят сульфоновые, простые эфирные и гид-
антоиновые звенья, плавятся при 230—320 °С [264].
Температура стеклования полиметилхиназолонов с
«шарнирными» группами в цепи колеблется от 210 до 240 °С [265]. Поли-
метилхиназолоны гораздо лучше растворимы, чем полифенилхина-
золоны (ср. № 4—16 и 18—22 в табл. 7.36). Полифенилхиназо-
лоны с «шарнирными» группами в остатке диамина растворяются
только в я-хлорфеноле и дихлоруксусной кислоте, в то время как
полимеры с боковыми метильными группами растворяются также
971
Таблица 7.36. Температуры плавления н раэложення полихинаэолонов
Элементарное звено
(в N2),
°С
(на
воздухе),
°С
о
J!
N
О
Qu-o-
N \^=^
J J
О
H3C
N N
400
400
400
380
380
[260]
350
[260]
400
R-—
НзС
425
О
N
о
N
О
СН3
—Ar—N\/N
—Аг—
> 300
290-320
325
[262|
/
[264]
[264]
Продолжение табл. 7.36
Элементарное звено
разл
BQN2),
Т
разл
(на
воздухе),
о
220—270
СНз
СНз
[264]
I \x_>v
J
10
11
О
N
СНэ
о
-о—(( )>—< )>—о—л у,—
О
сНз
о о
сн2
-N^\A
H.C-V
N
258-305
475
350
485
Продолжение табл. 7.36
№
12
13
14
15
О
н3с N ^
о
НзС N
о
о
сн2
rCHt
Элементарное звено
О
^ N ХС
О
^ N ХС
О
ЮСС'
о
Нз
Нз
V
450
473
470
445
^разл
(В N2^.
°С
490
480
460
420
т
разл
(на
воздухе),
Литературный
источник
[265, 274]
[265]
[265, 269]
[265, 269]
О
О
сн
16
НзС
С—NH-
О
440
415
[265, 269]
о
О
CH2
17
A
N
N
CF3
[272]
18
О
N
[270, 271]
19
500
[268]
Продолжение табл. 7.36
Элементарное звено
т
разл
(в N2).
т
разл
<на воз*
дуй).
Литературный
источник
20
R =
С6Н.
О
N
О
l/R-
N CeHs
О
л
N
>420
> 300
23
N N
II II
N N
N
II
N
R—
24
N
N
X = —, — 0— —S—,
R—
—SO2— —CH2—
25
N СНз
Y
О
Y
430-450
550
[273]
[237-239]
[275]
200
400
Рис. 7.59. Зависимость от температуры
потери массы полихиназолонов [267]:
1,3 — полимеры № 18; 2, 4 — полимеры № 20
(табл. 7.36); 3, 4 — на аоздухе; /. 2 — в аргоне.
в крезоле, бензиловом спирте,
диметилформамиде, диметилацет-
амиде, диметилсульфоксиде и
N-метилпирролидоне [266, 267J.
500 t,°c Дальнейшего улучшения
растворимости можно добиться, вводя в
состав полимера сульфоновые, простые эфирные и гидантоиновые
группы [264].
Полиметилхиназолон, полученный поликонденсацией в
растворе и термообработанный в твердой фазе при 250—400 °С,
растворяется только в концентрированной серной кислоте [265]. По-
лифенилхиназолоны обладают более высокой стойкостью к
термической и термоокислительной деструкции, чем соответствующие
незамещенные и метилзамещенные продукты. Температура начала
разложения полифенилхиназолонов в инертной среде и на воздухе
составляет 425 °С (рис. 7.59). Незамещенные продукты
разлагаются в инертной среде при 425 °С и на воздухе при 350—380°С
[260].
Метилзамещенные полимеры в инертной среде по
термостойкости не отличаются от других полихиназолонов, тогда как на
воздухе термоокислительная деструкция протекает уже при 325 °С.
В аргоне 20 %-ная потеря массы достигается при 550 °С, а на
воздухе— при 450 °С [262]. Через 100 ч термостарения на воздухе
при 300 °С потеря массы составляет 50% [263].
7.6.3. ПЕРЕРАБОТКА И СВОЙСТВА ПОЛИХИНАЗОЛОНОВ
Из 15 %-ных растворов полихиназолонов в я-хлорфеноле
поливом можно получить прочные высокотермостойкие пленки. Пленка,
приготовленная из полимера № 4 (см.
табл. 7.36), имеет при комнатной
температуре прочность при растяжении
ИЗО кгс/см2 и относительное удлинение
при разрыве 14 % [267].
Хорошая растворимость полимеров
особенно с сульфоновыми, простыми
эфирными и гидантоиновыми группами
в цепи, позволяет использовать их для
получения электроизоляционных пленок
ig pv [ом-см]
16
1,6 Ifi 2,0 2,2 1JT-1O3
Рис. 7.60. Зависимость удельного объемного
электрического сопротивления полиметилхиназолоно-
вой пленки от температуры [265]:
/—полимер № 16; 2 — полимер № 12; 3 — полимер № 11
(табл. 7.36).
980
и высокотермостойких волокон [264]. Пленка на основе подобного
полиметилхиназолона (табл. 7.36, № 7) характеризуется
следующими диэлектрическими показателями:
Диэлектрическая проницаемость при 800 Гц
Тангенс угла диэлектрических потерь при 800 Гц
Удельное объемное электрическое сопротивление,
Ом-см
При 20 "С
3,1
0,0017
При 200 "С
3,0
0,0020
3 - 10'6
3-1013
На рис. 7.60 показана температурная зависимость удельного
объемного электрического сопротивления ряда полиметилхиназо-
лонов.
7.7. Полихиназолиндионы
О
О
Полихиназолиндионы получают из ароматических бис(о-амино-
карбоновых кислот) и диизоцианатов или ароматических
диаминов и бис(оксазиндионов). Ароматические полихиназолиндионы
устойчивы в инертной среде до 560 °С и на воздухе до 520 °С.
Переработка их возможна на стадии форполимеров, так как они
не плавятся и растворяются только в концентрированных азотной
и серной кислотах. Эти полимеры можно применять для
изготовления электроизоляционной пленки, клеев для металлов и
связующих для армированных пластиков. Их температура
эксплуатации колеблется от 350 до 400 °С.
7.7.1. ПОЛУЧЕНИЕ ПОЛИХИНАЗОЛИНДИОНОВ
7.7.1.1. Синтез полимеров из ароматических
бис-(о-аминокарбоновых) кислот
и диизоцианатов
В этом случае в качестве промежуточных продуктов
образуются полиуреидокислоты. Их циклизация протекает через
промежуточную стадию образования полииминобензоксазинонов [276—
981
281]:
нею с.
п
соон
rtO=C=N'—\Q/—N—C=0
i
HOOC
-HNCHN-^
II
О
о
COOH
^NHCNH—(Г)
II \^'
О
Полиурендокарбоновая
кислота
n
l-
i1! -H2O
Полииминобензоксазинон
Полихиназолиндион
Вместо бис(о-аминокарбоновых кислот) можно применять их
динитрилы [285]. Взаимодействие диизоцианатов с бис(о-амино-
карбоновыми кислотами) осуществляется в расплаве, растворе
или суспензии. В расплаве процесс проводится в инертной среде
при 160 °С и стехиометрическом соотношении реагирующих
веществ. Дополнительное нагревание при 230—350°С приводит к
дальнейшему увеличению молекулярной массы [276].
Поликонденсацию в растворе проводят в таких растворителях,
как диметилацетамид, N-метилпирролидон или циклотетрамети-
ленсульфон, при низких температурах в присутствии аминов в
качестве катализаторов или в полифосфорной кислоте при 100—
160 °С. При этом твердый диизоцианат вводят в раствор диамино-
дикарбоновой кислоты и катализатора. Катализаторами
низкотемпературной поликонденсации служат пиридин, триэтилендиамин,
триэтиламин. Оловоорганические соединения не обладают
каталитической активностью. Каталитическая активность возрастает по
мере увеличения основности аминов; наиболее эффективными ка-
982
Afrff00nA МЩ.Н2 SO4
100
, г/100мЛ WH4.H
Рис. 7.61. Зависимость характеристической
вязкости (/) и растворимости B) продукта
поликонденсацин 4,4'-дифенилметандиизо-
цианата с бисантраниловой кислотой в
полифосфорной кислоте от концентрации
мономеров (а), продолжительности
реакции (б) и температуры (б) [277]:
а — соотношение мономеров 1:1, концентрация
мономеров 10%, продолжительность реакции 5 ч;
б — соотношение мономеров 1: I,
продолжительность реакции 5—20 ч, температура 150—160 °С;
в — соотношение мономеров 1:1, концентрация
мономеров 5%, температура 150—160 °С.
тализаторами являются третичные амины. Оптимальное
количество хлоргидрата триэтиламина составляет 2 моля на 1 моль ди-
изоцианата. Наиболее высокомолекулярный полимер получают
при 20°С [278]. В полифосфорной кислоте высокомолекулярные
полимеры получают в сравнительно мягких условиях (рис. 7.61):
оптимальная температура составляет 140—160 °С, концентрация
реагентов не выше 5 % (при более высоких концентрациях
образуются сшитые продукты), продолжительность реакции 5 ч [276,
277].
Суспензионную поликонденсацию проводят в разбавленной
щелочи. Образующиеся при этом полиуреидокарбоновые кислоты
имеют характеристическую вязкость 0,8 дл/г (в H2SO4) [276].
Форполимеры растворяются в N-метилпирролидоне, диметил-
ацетамиде и диметилсульфоксиде с добавкой LiCl, a также в
концентрированной серной кислоте. Судя по растворимости полиуре-
идокарбоновых кислот, можно заключить, что они имеют частич-
но-сшитую структуру за счет взаимодействия карбоксильных и
изоцианатных групп п в известной мере циклизованы. Циклоде-
гидратация форполимеров в порошке или пленке при 160—180°С
протекает с образованием полииминобензоксазинона
(характеристическая полоса поглощения 1775 см-1). Внутримолекулярное
превращение последних в полихиназолиндиокы (полосы
СО-группы при 1720 и 1660 см-1) происходит при 230—250 °С. При этой
температуре сублимируются амины, образующиеся в результате
гидролиза изоцианатов, и низкомолекулярные продукты
поликонденсации.
983
7.7.1.2. Взаимодействие бис(оксазиндионов) с диаминами
Аналогично получению полихиназолонов из ароматических бис-
оксазиндионов и ароматических диаминов образуются полихии-
азолиндионы [282]:
О О
—н2о
-in
При использовании три- или тетраминов получают сшитые
полихиназолиндионы или высококонденсированные системы. Вместо
бис(оксазиндионов) можно использовать ароматические бис(о-
алкилуретано)хлорангидриды дикарбоновых кислот [283, 286]:
О
A-ci
ос + - ™-<О-
NH2
—H Cl
—CH3OH
_л
7.7.2. СВОЙСТВА ПОЛИХИНАЗОЛИНДИОНОВ
Полихиназолиндионы представляют собой аморфные продукты
от желтого до коричневого цвета. Ароматические
полихиназолиндионы разлагаются ниже температуры плавления. Они
нерастворимы в органических растворителях; при комнатной температуре
984
растворяются в дымящей азотной кислоте, при нагревании — в
концентрированной серной кислоте. Термосюйкость их находится
на уровне [ермостойкости полиимидов, полибензимидазолов и по-
либензоксазолов.
Ароматические полихиназолиндионы устойчивы на воздухе до
520 °С и в инертной среде до
560°С (рис. 7.62). ту%
Полихиназолиндионы стойки wo
к действию растворов кислот и
щелочей. Переработка
полимеров может быть осуществлена
только на стадии промежуточных
полиуреидокарбоновых кислот.
Полихиназолиндионы могут быть
использованы в качестве
высокотемпературной электроизоля- 60
ции (кабельная изоляция и
пленка), связующих для слоистых
пластиков и клеев для металлов,
длительно работающих при 350—
400 °С.
Я0-
80-
50-
300 %0 500 600
800 t,°C
Рис. 7.62. Кривые термической деструкции полихиназолиндионов при различной
температуре [276]:
2, 3, 4, 6 ~ на воздухе; 1,5 — в среде азота;
NH
К 4—
о
NH
NH
Jn
NH
NH
2.5-
985
7.8. ПОЛИБЕНЗОКСАЗИНОНЫ
Полибенз-3,1-оксазиноны-4 получают поликонденсацией
ароматических бнс(о-аминокарбоновых) кислот с дихлорангидридами
дикарбоновых кислот. Они характеризуются исключительно
высокой гидролитической стойкостью. Полимеры устойчивы на
воздухе до 400 °С и в инертной среде до 450 °С. Полибензоксазиноны
могут быть использованы для изготовления высокотермостойкой
электроизоляционной пленки.
7.8.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗОКСАЗИНОНОВ
7.8.1.1. Поликонденсация бис(о-аминокарбоновых)
кислот с дихлорангидридами кислот
Первая стадия процесса—образование полиамидокарбоновых
кислот — проводится в растворе или на границе раздела фаз
[268, 287—295] при взаимодействии ароматических бис(о-амино-
карбоновых кислот с дихлорангидридами ароматических
дикарбоновых кислот или алициклическими дикетенами [295]. Циклоде-
гидратация осуществляется при нагревании или в присутствии
дегидратирующих веществ:
+ л сюс
ноосх
coci
1
Полиамидокарбоновая
кислота
п
Полибензоксазинон
О возможности получения производных бензоксазинонов
взаимодействием аминокарбоновых кислот с хлорангидридами карбо-
новых кислот сообщалось еще в 1883 г. [296].
Поликонденсация проводится при 20—40°С в N-метилпирроли-
доне, диметилацетамиде, гексаметилфосфотриамиде, N-метилкап-
986
ролактаме, полифосфорной кислоте [287, 288] или пиридине [289].
Высокомолекулярные полимеры образуются при добавлении
твердого дихлорангидрида к раствору диаминодикарбоновой кислоты
в инертной среде. Эффективными растворителями являются N-ме-
тилиирролидон и N-метилкапролактам [287]. Следы воды
вызывают резкое снижение молекулярной массы образующейся поли-
амидокислоты. Так, при взаимодействии дикарбоксибензидина
с изофталоилхлоридом в N-метилпирролидоне при эквимольном
соотношении реагентов характеристическая вязкость в реакционной
среде при увеличении содержания воды с 0,001 до 3 % снижается
с 1,28 до 0,35 дл/г. Более высокомолекулярные продукты
образуются при использовании дихлорангидрида в количестве, на 4 %
(мол.) превышающем эквимольное, что необходимо для
компенсации потерь этого реагента в результате протекания побочных
реакций.
Для межфазной поликонденсации применяют систему
хлороформ— вода — сода [268]. Диаминодикарбоновая кислота
растворяется в воде в присутствии соды. После растворения кислоты к
раствору при 5°С добавляют раствор хлорангидрида в
хлороформе. Полнамидокислота образуется в солевой форме, из которой
ее выделяют в виде свободной кислоты путем подкисления НО.
Ароматические полиамидокислоты растворяются в диметил-
формамиде, диметилацетамиде, диметилсульфоксиде,
N-метилпирролидоне, N-метилкапролактаме, серной и полифосфорной
кислотах. Эти растворы являются исключительно гидролитически
стойкими. Через 10 сут хранения раствора в серной кислоте или
N-метилпирролидоне характеристическая вязкость практически не
меняется [287].
Циклодегидратация пленки или порошка полиамидокислоты
протекает выше 200°С и может контролироваться по появлению
в ИК-спектрах полосы при 1760 см-1, отвечающий колебаниям
карбонильной группы беизоксазинонового цикла. Циклизация при
360°С протекает количественно [288, 295, 297]. Термическая
циклизация ускоряется при добавлении 0,1—5% фосфорорганических
соединений [290]. Химическая циклодегидратация может
протекать в растворе полиамидокислоты в N-метилпирролидоне под
действием ацетангидрида при 55 °С [268] или в результате
обработки пленки смесью ацетангидрид — пиридин с последующим
нагреванием при 130—180 °С [287].
Гомополиконденсация ароматических бис(о-аминокарбоновых)
кислот при 150—160°С в полифосфорной кислоте протекает по
схеме [297, 298] :
H2N
Таблица 7.37. Температура разложения полибензоксазинонов
Элементарное звено
разл
(в N2). °C
разл
(на воздухе),
°С
Лнтературный
источник
N
О
N
¦"V
О
сн2
о
т
500
550
360
450
350
[290, 299, 304]
[287, 288, 291,
303-305]
[268]
г
N
О
о
il
450
450
[2951
N
CONH—
О
—CONH-
о
330
330
[2891
[294]
H2Nk
О
соон
O
450
B97, 298]
7.8.1.2. Взаимодействие бис-о-аминофенолов
с диглиоксалевыми кислотами
Полибензоксазиноны получают также циклополиконденсацией
бис-о-аминофенолов с диглиоксалевыми кислотами [299] :
+ п но-с-с—/Г))—с-с-он —>
О О 0 0
N
UUO
_о о о
7.8.2. СВОЙСТВА ПОЛИБЕНЗОКСАЗИНОНОВ
Различие между полибензоксазолами и полибензоксазинонами
формально заключается в наличии в гетероцикле дополнительной
карбонильной группы:
NN NN
О
Полибензоксаэол
о о
Полибензоксазинон
Ароматические полибензоксазиноны неплавки и благодаря
сопряжению окрашены в желтый или коричневый цвет. Полимеры,
полученные из ароматических бис (о-аминокарбоновых) кислот и
хлорангидридов циклоалифатических дикарбоновых кислот,
бесцветны [295]. Ароматические полибензоксазиноны растворяются
только в дымящей азотной и концентрированной серной кислотах.
По термостойкости полибензоксазиноны близки к полибенз-
оксазолам (табл. 7.37). Уменьшение
массы начинается при 450 °С в
инертной среде и при 400 °С на воздухе
(рис. 7.63). При 900°С уменьшение
массы составляет на воздухе 30 % и в
инертной среде 24%. Метиленовые
(табл. 7.37, № 3) или амидные группы
700 t,C
Рис. 7.63. Кривые термической деструкции по-
либензоксазинонов и соответствующих поли-
амидокислот при различной температуре [288]:
/, 3 — в азоте; 2, 4 — на воздухе;
1.2-
3, 4
990
(табл. 7.37, № 5—7), находящиеся в основной цепи
макромолекулы, сильно снижают стойкость полимера к окислительной
деструкции (температура начала окислительной деструкции 350°С).
Ароматические полибекзоксазиноны отличаются высокой
гидролитической стойкостью, хотя 2-алкилбензоксазинон [300] и особенно
2^мстил-3,1-бензоксазннон-4 [301], гидролизуются очень легко.
Пребывание полибензоксазинонов в 10%-ной щелочи, 3 %-ной
серной кислоте или 10%-ной соляной в течение недели не
сопровождается гидролитической деструкцией. Частичный гидролиз
наблюдается только в концентрированной серной и дымящей
азотной кислотах.
Полибензоксазиноновые пленки в промышленном масштабе
выпускает фирма «Тоуо Rayon». Они могут использоваться в
качестве высокотермостойкого электроизоляционного материала [302].
7.9. ПОЛИБЕНЗОКСАЗИНДИОНЫ
°
о
—л
Полибензоксазиндионы получают из эфиров бис(о-гидроксикар-
боновых) кислот и диизоцианатов. Переработка полимеров
проводится из раствора или расплава. Полимеры способны длительно
работать при 200 °С.
7.9.1. ПОЛУЧЕНИЕ ПОЛИБЕНЗОКСАЗИНДИОНОВ
Полибензоксазиндионы получают поликонденсацией эфиров
ароматических бис(о-гидрокси)карбоновых кислот с диизоциана-
тами в растворе диметилсульфоксида через промежуточную
стадию образования полиуретаноэфиров [306, 308] :
NCO
991
Полибензоксазиндионы выпускаются с 1971 г. фирмой «Bayer»
в виде опытных партий по цене 100—200 марок ФРГ за кг.
Взаимодействие бис-о-гидроксинитрилов с диизоцианатами
сопровождается образованием полииминобензоксазиндионов [309]:
—п
7.9.2. СВОЙСТВА ПОЛИБЕНЗОКСАЗИНДИОНОВ
В противоположность полибензоксазинонам
полибензоксазиндионы плавятся и растворяются. Температура плавления
ароматических полимеров находится в интервале 300—400 °С. Термическая
деструкция происходит при температурах выше 400°С.
Полибензоксазиндионы растворяются в хлорированных углеводородах,
например метиленхлориде и трихлорэтилене, а также в диметил-
формамиде, диметилацетамиде, N-метилпирролидоне и крезоле.
Переработка в пленки и волокна осуществляется из растворов.
Полимеры можно перерабатывать литьем под давлением при
360—380 °С [310].
Физико-механические свойства полимеров мало меняются в
интервале температур от —180 до 300°С. Кратковременно их можно
эксплуатировать при температурах до 360°С.
Полибензоксазиндионы могут длительно работать при 180—230°С. Водопоглощение
полимеров составляет около 6 % • Полибензоксазиндионы
устойчивы к действию концентрированных кислот и разбавленных
щелочей. Концентрированные растворы щелочей вызывают их
деструкцию.
Литература к главе 7, разделы 7.2—7.9
1. Е. Wundt, Ber. Il A878), 826.
2. Пат. США 2895948 21. 7. 59 Du Pont.
3. Е. Marvel, SPE J. 20 A964), 220.
4. H. Vogel, J. Polymer Sei. 50 A961) 154, 511—539.
5. A. Conciatori, J. Polymer Sei. С 19 A967), 49—64.
6. Y. Iwakura, J. Polymer Sei A 2 A964) 6, 2605—2615.
7. Y. Iwakura, Makromolekulare Chem. 77 A964), 41—50.
8. Фр. пат. 1363757 A963) ТеЩп Ltd.
9. J. Horikawa, Chem. High Polymers (Japan), 24 A967) 267, 501—504.
10. S. Inoue, Makromolekulare Chem. 95 A966), 236—247.
11. F. Trischter, J. appl. Polymer Sei. 11 A967) 7, 1325—1331.
12. J. Colson, J. Polymer Sei. A t 4 A966), 59—70.
13. D. Packham, Polymer 10 A969) 12, 923—931.
14. Опубл. заявка ФРГ 1943499 4.3.71 BASF.
15. В. Коршак, Высокомолек. соед., А И, 1969, I, 35—42.
992
16. Р. Гитина, Высокомолек. соед., 8, 1966, 9, 1535—1538.
17 M. Saga, J. ehem. Soc. (Japan), Ind. ehem. Sect. 70 A967) 4 581-588.
18. J. Higgins, J. Polymer Sei A ! 8 A970), 1, 171 — 177.
19. D. Gray, ACS Polymer Preprints 8 A967) 2, 1138—1148.
20. D. Gray, Macromolecules 1 A968) 6, 473—478.
21. D. Gray, J. Macromolecul. Sei. 1 A A967) 3, 395—411.
22. А. Изынеев, Изв. АН СССР, сер. хим., 11, 1963, 2019—2023.
23. G. Shulman, J. Macromolecul. Sei. A 1 A967) 3, 413—428.
24. В. Коршак, Изв. АН СССР, сер. хим., 1963, 10, 1828—1836.
25. H. Levine, Report AFML-TR-64-365/1 A964).
26. W. Wrasidlo, J. Polymei Sei. A 2 A964) 11, 4795—4808.
27. J. Mulvaney, J. Polymer Sei. 50 A961), 541—547.
28. В. Родз, Изв. АН СССР, сер. хим., 11, 1968, 2662—2663.
29. G. Shulman, ACS Polymer Preprints 6 A965) 2, 773—777.
30. H. Vogel, J. Polymer Sei. A 1 A963), 1531—1541.
31. R. Singleton, J. Polymer Sei. С 19 A967), 65—78.
32. R. Singleton, ACS Polymer Preprints 9 A968) 2, 1158—1164.
33. R. Singleton, Appl. Polymer Symp. 9 A969), 133—144.
34. J. Ross, Appl. Polymer Symp. 9 A969), 23—47.
35. J. Ross, Textile Res. J. 41 A971) 2, 146—153.
36. Mater. Engin. 71 A970) 4, 19—20.
37. S. Frenkel, J. Polymer Sei С 2 A969) 22, 813—825.
38. Б. Гинзбург, Высокомолек. соед., А 9. 1967, 11, 2385—2391.
39. Sei. Techn. Aerospace Rep. 6 A968) 12, 1895.
40. Л. Лайус, Пластмассы, 8, 1965, 34—38.
41. А. Мс Leod, Mod. Plast. Encycl. 1970/71, 244.
42. В. Marks, ACS Div. org. Coating Plast. Chem., 155th Meeting 28.1.1968,
Techn. Papers.
43. R. Dickey, Sei. Techn. Aerospace Rep. 6 A968) 13, 2088.
44. Mod. Plast. Encycl. 1969/70, 261.
45. R. Dickey, J. Macromolecul. Sei. A3 A969), 4, 573—584.
46. Firmenschrift Whittacker Co., 1965.
47. S. Litvak, Adhesives Age 11 A968) I, 17—24.
48. S. Litvak, Adhesives Age 11 A968) 2, 24—28.
49. D. Flom, ACS Polymer Preprints 8 A967) 2, 1190—1202.
50. J. Hill, Adhesives Age 9 A966) 7, 32.
51. «Imidite 850 and 1850», Firmenschrift Narmco Res. Co., 1964.
52. J. Licari, Prod. Engin. 7. 12. 1964.
53. E. Plueddemann, SPI Reinf. Plast. Div., 22nd Ann. Techn. Conf. Proc. A967),
9-A1-9-17-10.
54. A. Mackay, Mod. Plast. 43 A966), 5, 149—152.
55. В. Касаточкин, Высокомолек. соед., 7, 1965, 7, 1147—1153.
56. D. Wiff, Sei. Techn. Aerospace Rep. 7 A969) 23, 4332.
57. T. Narayan, J. Polymer Sei. A 15 A967) 5, 1113—1118.
58. С. Marvel, J. Polymer Sei. 2 A964) 6, 2559—2569.
59. T. Фрунзе, Высокомолек. соед., 6, 1964, 7, 1251 — 1255.
60. H. Адрова, Высокомолек. соед., 7, 1965, 2, 305—307.
61. F. Trischler, J. appl. Polymer Sei. 13 A969), 1, 101—106.
62. Р. Гитина, Высокомолек. соед., Б 9, 1967, 6, 447—452.
63. Фр. пат. 1570033 6.6.69.
64. Т. Фрунзе. Высокомолек. соед., 7, 1965, 2, 285—289.
65. И. Маммарова, Высокомолек. соед., 8, 1966, 5, 777—782.
66. А. Изынеев, Высокомолек. соед., 7, 1965, 2, 280—286.
67. В. Коршак, Изв. АН СССР, сер. хим., 9, 1968, 2143—2144
68. Н. Адрова, ДАН СССР, сер. хим., 158, 1964, 130—137.
69. Н. Адрова, ДАН СССР, сер. хим., 158, 1966, 18—26.
70. Г. Цейтлин, Высокомолек. соед., 11, 1969, 1, 22—26.
71. М. Тепляков, Высокомолек. соед., Б 9, 1967, 10, 767—769.
72. М. Тепляков, Высокомолек. соед., АН, 1969, 8, 1858—1862.
73. G. Rabilloud, Bull. Soc. chim. France 32 A966), 926—932.
74. Y. Iwakura, Makromolekulare Chem. 77 A964), 41—50.
32 зак. 18 993
75. Акцепт, заявка Японии 7105716 12.2.71 Teihin Ltd.
76. Т. Фрунзе, Высокомолек. соед., 6, 1964, 5, 901—905.
77. А. Изынеев, Изв. АН СССР, сер. хим., 11, 1964, 2104—2106.
78. R. Philips, J. Polymer Sei. В 2 A964) 1, 47-49.
79. W. Freeston, J. Fire & Ftarnmability 2 A971) 1, 57—76.
80. J. Preston, J. Polymer Sei. A 5 A967), 2429—2438,
81. T. Helmiak, ACS Polymer Preprints 11 A970) 1, 291—296.
82. Kunststoff-Berater 2 A970), 118-120.
83. S. Tenun, Report NAS 98811 A970).
84. В. Коршак, Пластмассы, 1972, 2, 40—42.
85. H. Herlinger, Textilpraxis A972) 3/4, 181 — 183, 243—246.
86. В. Коршак, Высокомолек. соед., А 14, 1972, 11, 2329—2334.
87. В. Коршак, Высокомолек. соед., А 14, 1972, 12, 2647—2652.
88. И. Рабинович, Высокомолек. соед., А 15, 1973, 2, 360—366.
89. А. Праведников, Доклад на конгрессе по полимерам, г. Карл-Марксштадт,
1973.
90. Р. Hergenrother, Report D180-12911-1 A971) Boeing Scientific Research Lab.
91. P. Opt, Textile Res. J. 37 A967) 12, 1050—1055.
92. Y. Hsu, Gaofenzi Tongxun 7 A965) 4, 252—256.
93. Опубл. заявка ФРГ 2047933 13.5.71 Kuraray Co.
94. Пат. США 3578644 11.5.71 Whittaker Co.
95. Акцепт, заявка Японии 7114474 17.4.71 Mabushita Electric.
96. Пат. СССР 293016, 15. 1.71.
97. Опубл. заявка ФРГ 2042623 22.4.71 Kuraray Со.
98. Акцепт, заявка Японии 7106631 19.2.71 Teijin Ltd.
99. Пат. США 3584104 8. 6. 71 Celanese.
100. Пат. СССР 299520, 26.3.71.
101. В. Marks, Report NASA-CR-1723 A971).
102. Акцепт, заявка Японии 7115514 26.4.71 Teijin Ltd.
103. В. Коршак, Кинетика и механизм полиреакции, Международный конгресс
по макромолекулярной химии, Будапешт, препринты, 1969, I, 59—63.
104. Пат. США 3598767 10.8.71 U. S. Dept. of Navy.
105. Акцепт, заявка Японии 7117312 24.4.71 Matsushita Electric.
106. В. Коршак, ДАН СССР, 198, 1971, 4, 841—843.
107. Акцепт, заявка Японии 7127825 12.8.71 Matsushita Electric.
108. И. Рабинович, ДАН СССР, 198, 1971, 3, 597—600.
109. Опубл. заявка ФРГ 2121179 11. П. 70 R. Кгау.
110. Пат. США 3629479 21. 12.71 J. Szita.
111. В. Коршак, Пластмассы, 1971, 7, 8—12.
112. Пат. США 3655632 11.4.72 Kuraray Со.
ИЗ. Пат. США 3620998 16. 11.71 Research Со.
114. Пат. США 3619453 9. 11.71 Celanese.
116. Пат. США 3630972 28. 12.71 Research Со.
116. R. Yokota, Kobunshi Kagaku 29 A972) 6, 428—481.
117. Пат. СССР 337387, 5. 5. 72.
118. К- Sidman, Report AD 748248 A971).
119. Яп. заявка 7223448 12.10.72 Kuraray.
120. Пат. СССР 350805, 13. 9. 72.
121. Акцепт, заявка Японии 7232769 21.8.72 Kuraray.
122. Пат. США 3714129 30. 1.73 Ashland Oll.
123. Пат. СССР 364644, 28. 12. 72.
124. Опубл. заявка ФРГ 2244908 29.3.73 Teijin Ltd.
125. Пат. США 3699038 22. 4. 70 Celanese.
126. Пат. США 3729453 24. 4. 73 Celanese.
127. Д. Тугуши, Тр. Моск. хим.-техн. ин-та, 1972, 70, 182—184.
128. В. Коршак, Тр. Моск. хим.-техн. ин-та, 1972, 70, 175—177
129. Y. Imai, Makromolekulare Chem. 83 A965), 179—187
130. J. Green, J. Macromolecut. Sei. A 1 A967), 135
131. H. Kovacs, J. Polymer Sei. A 1 6 A968), 2103—2112
132. M. Saga, J. Polymer Sei. A 1 8 A970), 2265—2274
133. Пат. США 3503929 A970) 3 M Co.
994
134. Б. Гинзбург. Высокомолек. соед., 8, 1966, 278—281.
135 В. KoDinaK, Пластмассы. 1970, 12, 35—42.
136. Пат. США 2904537 A9591 Du Pont.
137 Англ. пат. 811758 A959) Du Pont.
138. T. Kubcta, J Polymer Sei В 2 A964) 6. 655—659.
139. W. Моуег, J. Polymer Sei A3 A965). 2107—2121.
140. А Якубович, Высокомолек. соед., 9 1967. 8. 1782—1786.
141 Y. Iwakura, Makromolekulare Chem. 83 A965), 167—178.
142. Y (mai. Makromolekulare Chem. 95 A966), 236—247.
143. А. Якубович, Высокомолек. соед., 9, 1967, 9, 1973—1984.
144. Y Iwakura, Makromolekulare Chem. 77 A964). 33—44.
145. А. Якубович, ДАН СССР, 159, 1964. 630—637
146. А. Якубович, Высокомолек. соед., 8, 1966, 2, 272—277.
147. А. Якубович, Высокомолек. соед., 9, 1967, 9, 1914—1919.
148. А. Якубович, Высокомолек. соед., сер. Б, 1968, 11, 859—861.
149. Г. Браз. Высокомолек. соед., 12, 1970, 6. 1396—1402.
150. А. Якубович. Пластмассы. 1970, 9, 17—19.
151. Пат. СССР 280835, 3.9.70.
152. Пат. СССР 297291, 11.6.71.
153. Акцепт, заявка Японии 15995 A968).
154. Акцепт, заявка Японии 15995 A968).
155. А. Ladenburg, Berichte 9 A876). 1524.
156. Акцепт, заявка Японии 698238. 1969.
157. Фр. пат. 1548308 6.2.68 Batelle Inst.
158. Фр. пат. 1548309 6.2.68 Batelle Tnst.
159. А. Павлов, Высокомолек. соед., All, 1969, 1, 11—15.
160. И. Кардаш, Высокомолек. соед., All. 1969. 9. 1996—2001.
161. Y. Tmai, Makromolekulare Chem. 84 A965), 179—187.
162. Y. Tmai, Makromolekulare Chem. 83 A965). 167—178.
163. А. Павлов, Изв, АН СССР, сер. хим., 1965, 1912—1913.
164. А. Павлов, Высокомолек, соед., 8, 1966, 1599—1603.
165. Пат. США 3563950, 16.2.71.
166. А. Павлов ДАН СССР, 163, 1965. 116—119.
167. Р Гитина, Высокомолек. соед., Б 9, 1967, 7, 498—499.
168. Пат. США 3560438 2. 2. 71 Dow Chem.
169. А. Якубович, 15 Всесоюзная конференция по высокомолек,. соед., Москва,
из-во Наука, 1965.
170. А. Якубович. Высокомолек. соед., 13. 1971, 994—1002.
171. V. Preston, .1. Polvmer Sei. А 1 7 A969) 1, 283—296.
172. V. Preston, Appi Polvmer Svmn 9 (]Q69). 145—157.
173. B. Culbertson, Л Polymer Sei. В 4 A966). 249—253.
174. Ю. Андрющенко, Хим. волокна, 1968, 3, 10—13.
175. Ю. Андрющенко. Хим. волокна, 1968, 12, 923.
176. Пат. СССР 361185, 7. 12. 72.
177. В Евстафьев, Высокомолек. соед., А 13, 1971, 11, 2565—2571.
178. В. Воищев, Пластмассы, 1972, 8. 39—41.
179. И. Елин, Пластмассы, 1973, 2, 39—41.
180. А. Якубович. Высокомолек. соед., А 14, 1972, 10, 2174—2182.
181. А. Павлов, Пластмассы, 1972, 4, 4041.
182. А. Якубович, Высокомолек. соед., А 14, 1972, 8, 1822—1832.
183. Л. Preston, Л. Polymer Sei. А 1 10 A972). 1377—1389.
184. Г. Браз, Высокомолек. соед., 8, 1966. 2. 272—277.
185. Акцепт, заявка Японии 7132520 22.9.71 Toray Ind.
186. Пат. США 3671491 20.6.72 Celanese.
187. Акцепт, заявка Японии 7201223 13. 1.72 Furukawa Electric.
188. Акцепт, заявка Японии 7121960 22.6.71 Toray Ind.
189. Э. Телешов, Пластмассы, 1973, 2, 3—8,
190. А. Киприянов ЖОХ, 32, 1962, 4040—4047.
191. Р. Hergenrother, Л. Polymer Sei. A4 A966) 9, 2341—2347.
192. Р. Hergenrother, J. Polymer Sei. A l 6 A968) 10, 2939—2943.
32* 995
193. P. Hergenrother, J. Polymer Sei. A3 A965) 5, 1665—1674.
194. Пат. США 3047543 A962) A. Morton.
195. Пат. США 3397187 A968) W. Месит.
196. Kuntstoff-Barater A970) 2, 117—120.
197. G. Rabilloud, Makromolekulare Chem. 146 A971), 215—226.
198. Пат. СССР 303335, 13.5.71.
199. Опубл. заявка ФРГ 2138830 24.2.72 Aquitaine-Organico.
200. Фр. пат. 2067512 24.9.71 Inst. Franc, du Petrole.
201. Пат. США 3560438 2. 2. 71 Dow Chem.
202. В. Коршак, Высокомолек. соед., Б 9, 1967, 12, 870—873.
203. Пат. СССР 276405, 14. 7. 70.
204. J. Stille, J. Polymer Sei. В 4 A966) 9, 665—667.
205. J. Stille, J. Polymer Sei. В 5 A967) 11, 989—992.
206. J. Stille, J. Polymer Sei. В 4 A966), 39—41.
207. J. Stille, J. Polymer Sei. A4 A966), 551—562.
208. J. Stille, J. Polymer Sei. Л 2 A964) 9, 3867—3875.
209. P. Hergenrother, J. Polymer Sei. A 16 A968) 11, 3170—3173.
210. P. Hergenrother, J. Polymer Sei. A l 5 A967) 6, 1453—1466.
211. W. Wrasidlo, J. Polymer Sei. В 8 A970) 2, 69—74.
212. W. Wrasidlo, J. Polymer Sei. A l 7 A969) 12, 3393—3405.
213. W. Wrasidlo, J. Polymer Sei. В 7 A969) 4, 281—286.
214. J. Augl, J. Polymer Sei. A 1 8 A970) 11, 3145—3153.
215. J. Augl, J. Polymer Sei. A l 9 A971), 1343—1350.
216. N. Popov, J. Polymer Sei. A 1 7 A969) 7, 1803—1814.
217. J. Stille, J. Polymer Sei. A3 A965) 3, 1013—1030.
218. P. Hergenrother, Macromolecules 3 A970) 4, 387—393.
219. P. Hergenrother, J. appl. Polymer Sei. 14 A970) 4, 1037—1048.
220. W. Wrasidlo, Macromolecules 3 A970) 5. 544—547.
221. M. Okada, J. Polymer Sei. A 1 6 A968) 5, 1259—1271.
222. A. Banihashemi, J. Polymer Sei. A 8 A970) 11, 3211—3224.
223. R. Wolf, J. Polymer Sei. A 1 7 A969) 9, 2481—2491.
224. W. Wrasidlo, J. Polymer Sei. A 1 8 A970) 5, 1107—1130.
225. Опубл. заявка ФРГ 1944796, 11.3.71.
226. F. de Schryver, J. Polymer Sei. А 1 5 A967), 545—552.
227. Пат. США 3563917 16.2.71 С. Marvel.
228. Пат. США 3620997 16. 11.71 Research Co.
229. R. Wolf, J. Polymer Sei. A 1 6 A968), 1503—1514.
230. В. Sillion, J. Polymer Sei. В 2 A964) 2, 203—207.
231. J. Stille, J. Polymer Sei. В 2 A964) 2, 209—211.
232. А. Берлин, Ж. ВХО им. Менделеева, 11, 1966, 5, 591—592.
233. H. Jadamus, J. Polymer Sei. A 1 4 A966) 11, 2831—2842.
234. I. Sopov, J. Polymer Sei. A l 7 A969) 7, 1803—1814.
235. И. Шопов, Высокомолек. соед., Б 9, 1967, 6, 415—416.
236. Акцепт, заявка Японии 7040552 16. 12.70 Toyo Rayon.
237. Акцепт, заявка Японии 7127823 12.8.71 Furukawa Electric.
238. Акцепт, заявка Японии 7127826 11. 8. 71 Furukawa Electric.
239. Пат. СССР 304276, 25.5.71, Inst. Heterorg. Verbinde.
240. Пат. США 3726899 10.4.73 U. S. Dept. of Navy.
241. W. Lee, ACS Polymer Preprints 12 A971) 2, 217—222.
242. Пат. США 3630994 28. 12. 71 Ashland Oil.
243. Пат. США 3642700 15.2. 72 J. Augl.
244. G. Hagnauer, Nuova Chim. 49 A973) 1, 59—63.
245. N. Burningham, ACS Div. Org. Coatings Plast. Chem Pap. 31 A971) 1,
101 — 112.
246. Пат США 3661850 9. 5. 72 J. Stille.
247. Пат. США 3654226 4. 4. 72 U. S. Dept. of Navy.
248. W. Wrasidlo, ACS Div. Org. Coatings Plast. Chem. 30 A970) 2 97—104
249. F. Arnold, J. Polymer Sei. В 7 A969) 10, 749—753.
250. P. Hergenrother, J. Macromolecul. Sei. С 6 A971) 1, 1—28.
251. J. Duffy, J. Polymer Sei. A 1 10 A972) 4, 1123—L131.
996
252. J. Duffy, J. Polymer Sei., Polymer Letters 11 A973) 1, 29—32.
253. J. Higgins, J. Polymer Sei. В 10 A972) 4, 301—303.
254. J. Higgins, J. Polymer Sei. A 1 JO A972) 6. 1609—1615.
255- X. Раубах. Высокомолек. соед.. А 15. 1973, 2. 367—371.
256. V Korner,' Ber. 17 A884), 573.
257. О. Hinsberg, Ber. 17 A884), 318.
258. W. Sorenson, Polymer Preprints 2 A961) 2, 226—231.
259. B. Sillion, BuM. Soc. Chim. France A964), 1793—1800.
260. G. Rabilloud, Makromolekulare Chem. 125 A969), 264—282.
261. Акцепт, заявка Японии 6931350 15. 12.69 Toyo Rayon.
262. В. Sillion, Bull. Soc. chim. France A965). 171—176. '
263. J. Serlin, J. Polymer Sei. 60 A962) 169, S 59—S 63.
264. E. Radimann, Makromolekulare Chem. 145 A971), 21—38.
265. A. Fukami, Mitsubishi Denki Lab. Rep. 12 A972) 1—3, 37—48.
266. B. Sillion, Compt. rend. Acad. Sei. С 265 A967), 1234—1236.
267. В. Sillion, J. Polymer Sei. С 22 A969) 2, 827—848.
268. P. Schlack, An gew. Makr. Chem. 15 A971), 25—36.
269. A. Fukami, J. chem. Soc. (Japan), Ind. chem. Sect. 73 A970) 6, 1239—1243.
270. Фр. пат. 1523133 A966) Inst. Francais du Petrole.
271. Фр. пат. 1423631 A963) Inst. Francais du Petrole.
272. Акцепт, заявка Японии 7223196 28.6.72 Toray Ind.
273. В. Коршак, ДАН СССР, 196, 1971, 6, 1357—1360.
274. Фр. пат. 1603128 30.4.71 Inst. Francais du Petrole.
275. Акцепт, заявка Японии 7112746 1.4.71 Toray Ind.
276. N. Yod a. J. Polymer Sei. В 4 A966), 1, 11—16.
277. M. Kurihara, J. Polymer Sei. A 1 5 A967) 7, 1765—1781.
278. M. Kurihara, J. Polymer Sei. A 1 5 A967) 10, 2523—2534.
279. Акцепт, заявка Японии 7008993 1.4. 70 Toyo Rayon.
280. Акцепт, заявка Японии 6433304, 6434599, 6434603 A964) Toyo Rayon.
281. Акцепт, заявка Японии 6927677 7. 11.69 Toyo Rayon.
282. Акцепт, заявка Японии 7125011 19.7.71 Toray Ind.
283. Пат. СССР 312856, 31.8.71.
284. Акцепт, заявка Японии 7041032 23. 12. 70 Toyo Rayon.
285. Пат. США 3674749 4. 7. 72 Du Pont.
286. Пат. СССР 328139, 2. 2. 72.
287. К. Ikeda, J. Polymer Sei. 5 A967) 9, 2359—2374.
288. N. Yoda, J. Polymer Sei. В 4 A966) 8, 551—556.
289. R. Salle, Compt. rend. Acad. Sei. С 267 A968), 1213—1216.
290. Акцепт, заявка Японии 6920635 4. 9. 69 Toyo Rayon.
291. M. Kurihara, J. Macromolecul. Sei. A 1 A967) 6, 1069—1087.
292. Акцепт, заявка Японии 7126872 4.8.71 Toray Ind.
293. Акцепт, заявка Японии 7001631 20. 1.70 Toyo Rayon.
294. Фр. пат. 2010603 20. 2. 70 Bayer AG.
295. Y. Hagiwara, Makromolekulare Chem. 126 A969), 48—55.
296. P. Friedlander, Ber. 16 A883), 2277.
297. M. Kurihara, Makromolekulare Chem. 105 A967), 84—94.
298. M. Kurihara, Makromolekulare Chem. 107 A967) 112—123.
299. Фр. пат. 2041423 29. 1.71 Inst. Francais du Petrole.
300. D. Zentmeyer, J. org. Chem. 14 A949), 967—981.
301. M. Bogert, J. Amer. chem. Soc. 33 A911), 951.
302. Materie plast. Elestomeri 35 A969) 7, 948.
303. Акцепт, заявка Японии 7032944 23. 10. 70 Toray Ind.
304. Акцепт, заявка Японии 7032629 21. 10.70 Toray Ind.
305. G. Rabilloud, Makromolekulare Chem. 108 A967), 18—51.
306. L. Bottenbruch, Deutsche Farbenzeitschrift 25 A971) 12, 668
307. L. Bottenbruch, 179. DECHEMA Kolloquium 7. 10. 1971.
308. Пат. Южно-Афр. Респ. 68037, 1968.
309. Пат. США 3657186 18. 4. 72 Du Pont.
310. «Polybenzoxazindione», Firmenschrift, Bayer AG.
8. ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ
В этой главе рассмотрены полимеры, которые содержат в
элементарном звене не менее трех конденсированных циклов
Пирроны
Q
ПС У©
Полиимидазобензофена втролины
Полиизоиндолохина-
золиндионы
Политетразапирены
Если конденсированная циклическая система связана простой
связью, лестничная структура нарушается (полулестничный, блок-
лестничный полимеры)*. В случае двоесвязанных (двутяжевых)
конденсированных систем образуются истинно лестничные
полимеры. Лестничные полихиноксалины были описаны ранее G.5).
Лестничные полимеры получают циклополиконденсацией тет-
рафункциональных мономеров. Такие полимеры отличаются очень
высокой термостойкостью. Связь между строением и свойствами
лестничных полимеров рассмотрена в гл. 2. Высокая жесткость
этих полимеров обусловливает высокую температуру стеклования
(> 300°С) и их неплавкость. Температура разложения на воздухе
составляет около 500 СС и в инертной среде 600СС.
Высокая стойкость к термо- и термоокислительной деструкции,
а также реакционная стойкость лестничных полимеров
обусловлены тем, что благодаря своеобразной структуре таких полимеров
деструкция происходит только при одновременном разрыве
нескольких связей в макромолекуле. Кроме того, в системе с
конденсированными циклами высокая полисопряженность и низкая
концентрация периферийных водородных атомов обусловливают до-
* Английский термин «Step-Ladder».
полнительный выигрыш в термостойкости. Однако на практике
принцип лестничного строения молекулы реализуется не
полностью вследствие незавершенности циклизации. Хотя лестничные
полимеры довольно широко используются в космической и
ядерной технике как абляционно-стойкие конструкционные элементы,
а также для изготовления негорючих текстильных материалов,
в промышленном масштабе производится только полибисбензи-
мидазобензофенантролин (волокно ВВВ).
8.1. ПИРРОНЫ
Полибензоилендибензимидазолы (пирроны, полиимидазопйр-
ролоны) получают взаимодействием диангидридов тетракарбоно-
вых кислот с ароматическими тетраминами. Пирроны устойчивы
на воздухе до 450 °С, отличаются высокими химической и
радиационной стойкостью. Пленки получают поливом из растворов фор-
полимеров с последующей термической циклизацией.
Формованные изделия можно получить высокотемпературным прессованием
полигетероарилена или форполимера, а также непосредственно из
смеси мономеров. Верхняя температура длительной эксплуатации
изделий из пирронов составляет 300°С. Пирроны могут быть
использованы для изготовления волокон, полупроницаемых мембран,
сепараторов в аккумуляторах или в качестве полупроводников.
Синтез и исследование пирронов проводится главным образом
в ИНЭОС АН СССР и в исследовательском центре Лэнгли, штат
Вирджиния (США). ;
8.1.1. ПОЛУЧЕНИЕ ПИРРОНОВ
Поликонденсацию ароматических тетраминов с производными
тетракарбоновых кислот проводят многостадийно или в одну
стадию в полифосфорной кислоте, расплаве или твердой фазе.
Температуры размягчения и разложения пирронов приведены в табл. 8.1.
8.1.1.1. Двухстадийная поликонденсация
В этом случае на первой стадии в растворе получают поли-
аминоамидокислоту, которая на второй стадии в процессе
термической циклодегидратации в твердой фазе превращается в поли-
аминоимид или поликарбоксибензимидазол, а затем в пиррон.
999
Таблица 8.1. Температуры размягчения и разложения пирронов
Эдемеятарное звено
'разм'
°С
разл
(в Na К
°С
разл
(на
воздухе).
о
Л,
О
II N
.^nT^ S
JQL-1 s
'J 1
JQ
'J
О
/
СН2—N>
СН-/ \сНо }
400
О
О Cl
N-
о-
400
[3-14,
47, 49,
64]
[46]
[56]
595
540
13,5,7, 8,
18-20,
47, 48]
Продолжение табл. 8.1
Элементарное звено
разм1
Г
рвзл
(в N2),
т
разл
(на воз-
)
N
N
О
N
О
О
о
I
so2—
о
N
N
118, 21,
47]
N
т
410
[47]
N О ||
0_
380
555
[48, 52,
61. 72]
N О ||
i
N
;soi
Продолжение табл. 8.1
Элементарное звено
разм'
разл
(в N2),
Т
разл
»на воз-
350
18
N SO2
va ^
N
N
19
l
N
О
20
О
О
I!
.0—
о
21
CH2 —
N
600
350
400
330
500
460
480
[47]
[51]
[51]
[511
Продолжение таСг. 8.1
Элементарное звено
разм'
°G
Т arin
разл
(boN8).
Т
разл
(на
воздухе),
°С
N
N
N
О—
340
390
430
420
450
¦N>
l
О
л
N-
N
O
540
5, 7]
о
'26
tuCi
N
о
о
О
N
о
370
[5, 7, 47]
400
[47]
28
[17]
Продолжение табл. 8.1
Элементарное звено
разм1
°С
Т
разл
(и №>¦
°С
т
разл
1на
воздухе),
°С
Литературный
источник
N
I
N
О
N
360
340
500
615
410
470
505
[47, 48]
[47, 48]
[47]
о
32
-N
о
430
147]
О
33
О
N
О
350
530
[16, 48]
О
34
N 0 0
N
305
[48]
N
35
)—с.
"N
О
л»
N
460
[47]
Продолжение табл. 8.1
Элементарное звено
разм'
°С
Т
разл
(в N2),
°С
Т
разл
(на
воздухе),
N
N
Q
I
N
II
о
о
N
о
C-Ccf^-
А
N
470
32Q
460
N
39
т
40
О
41
N
¦N
I
42
~ч
&
О
о—а ))—
380
330
330
300
540
116, 48]
M8J
116, 48]
[48]
Продолжение табл. 8.I
Элементарное звено
разм'
°С
разл
(в N2),
°
т
разл
(на воз-
духе),
О
о
I
480
375
400
46
[<33, 69]
О О
47
so.
[25]
О
О
48
[24]
49
[55]
N
Продолжение табл. 8Л
N
53
N
О
N
54
-N'
55
N
N
А
N
и N
N
[271
[271
600
450
[451
Такой процесс может быть представлен следующей схемой:
о
о
./Ч-Яа-'Ч
H2N,
i
20 °С
- —ос
Полиамидоамино-
кислота
NH—
180 °С, Ю-3 мм рт. ст.
НООС
N
Поликарбоксибензимидазол
—П
Полиамиионмид
I 300-350°С, 10~3 мм рт. ст.
О
Пиррои
Поликонденсация в растворе может проводиться в амидных
растворителях при низких или при высоких температурах. Однако
наиболее важным является взаимодействие диангидридов тетра-
карбоновых кислот с ароматическими тетраминами при —20-г-
Ч-50°С в среде таких растворителей, как диметилформамид, ди-
метилацетамид, диметилсульфоксид, N-метилпирролидон или гек*
Ю16
саметилфосфотриамид, приводящее к получению полиаминоамидо-
кислот [2, 3, 6, 9, 11, 14, 16—21, 27—37, 47, 51, 72].
Раствор диангидрида при перемешивании добавляют к
раствору тетрамина. Основная трудность при проведении реакции
заключается в том, что диангидридьт ароматических тетрякарбоновых
кислот плохо растворяются в амидных растворителях, а наличие
нерастворенного диангидрида приводит к гелеобразованию. Геле-
образование наступает также при изменении порядка загрузки
компонентов, т. е. при добавлении тетрамина к раствору
диангидрида [5, 9].
Реакционная способность диангидридов тетракарбоновых
кислот и их тенденция к гелеобразованию уменьшаются в следующей
последовательности [47] :
о
;о >
О >
(слабое сшивание)
О
(отсутствие сшивания)
О
О
О
(отсутствие сшивания)
О О
(отсутствие сшивания\
,25 вС
Для получения высокомолекулярных продуктов с [т]]дмаа=
1,0—1,5 дл/г необходима высокая степень чистоты исходных
1017
мономеров и 1—2%-ный избыток диангидрида. В этом случае
избыточный диангидрид реагирует со свободными аминогруппами
с образованием имидных мостиков [5]:
о
о
NH OCv
ноос
—н2о
соон
Учитывая высокую скорость окисления таких ароматических
тетраминов, как тетраминобензол, удобнее к суспензии хлоргид-
рата тетрамина добавлять раствор диангидрида и проводить про-
1018
цесс в присутствии акцепторов НС1 (третичных аминов) [5]
О О
«< IoC)o+n
4СГ
R3N
-In
В этом случае реакция протекает значительно медленнее,
продолжительность ее достигает 20 ч. Для ускорения процесса
получения полиаминоамидокислот можно использовать диэфироди-
хлорангидриды тетракарбоновых кислот:
•4СГ
_n
Акцепторы НС1 в этом случае не применяются. Можно
синтезировать пирроны, используя прием, которым обычно пользуются при
получении полиамидов. Для этого сначала взаимодействием тетр-
аминов с диэфиром тетракарбоновой кислоты получают соль,
которая выкристаллизовывается из реакционной смеси [12]:
;о + 4С4н9он
ноос
XOOH HOOCs
+
о
II
СОС4Н9
1019
0
11
II
0—CN
HeOC-"
M
A
\ /
<o>
0
II
/Q- OC4H9
V-C—0"
il
0
H2N
В диметилацетамиде эти соли дают линейный полимер. При
строгом соблюдении эквимольного соотношения реагентов гелеобразо-
ваыие не происходит. Из соли A:1) можно получать пиррон в
одну стадию в твердой фазе при 300°С [49]., Другой путь,
способствующий предотвращению гелеобразования в ходе синтеза поли-
аминоамидокислоты при наличии электрофильного компонента,
заключается в блокировании о-аминогрупп путем ацетилирования
[22]:
О О
О
NHCOCHa
СООН
1—NHCO—;
СО—
СООН
_п
Ацетамидные группы не реагируют с ангидридами, исключая
возможность образования сшитого полимера.
Реакционную способность аминогрупп в тетраминах можно
значительно уменьшить введением хлора в о-положение к
аминогруппам. Тетрамины этого типа, например, З^-дихлор-о^'-диами-
нобензидин
Cl
реагируя с диангидридами, не дают геля даже при избытке
электрофильного компонента в реакционной среде [46].
Полиаминоамидокислота, образующаяся в качестве,
промежуточного продукта, имеет аморфную структуру и либо плавится
выше 300 °С, либо вообще не плавится [47]. Это свидетельствует
1020
о существовании цвитерионнои структуры, которая способствует
сильному межмолекулярному взаимодействию:
Полиаминоамидокислоты растворяются в апротонных
растворителях, таких, как диметилформамид, диметилацетамид, диме-
тилсульфоксид, N-метилпирролидон, гексаметилфосфотриамид и
пиридин. Характеристическая вязкость, равная 0,4—1,5 дл/г
(диметилформамид, 25°С), соответствует молекулярной массе 7 000—
20 000, определенной осмометрическим методом [5]. В диметил-
ацетамиде полиаминоамидокислоты образуют прочные ассоциаты.
Для определения молекулярной массы методами светорассеяния
или седиментации пригодны смеси растворителей, например
диметилацетамид— N-метилацетамид B5:75), в которых ассоциация
макромолекул таких полимеров значительно ослаблена [10].
Растворы полиаминоамидокислот стабилизируют добавлением
смеси триэтиламин — диметиланилин [38]. Поливом из растворов
получают прочные прозрачные пленки. Так, пленка полимера на
основе диаминобензидина и пиромеллитового диангидрида имеет
прочность при растяжении 700—1050 кгс/см2, относительное
удлинение при разрыве 25—35%, удельное объемное электрическое
сопротивление 6*109Ом-см и электрическую прочность 2-105 В/см [2].
Термическую циклизацию полиаминоамидокислот в виде
пленки или порошка, полученного осаждением полимера из раствора,
проводят на воздухе, в инертной среде или в вакууме при 300—
400°С в течение 2 ч. Сначала при 120—160°С полиаминоамидо-
кислота превращается в полиаминоимид или в поликарбоксибен-
зимидазол [16, 18]. Методами ИК- и ЯМР-спектроскопии
показано, что при 150°С преимущественно образуется аминоимидная
структура [2, 4, 5]:
O-JQ
1021
Поликонденсация ароматических диангидридов и тетраминов
в N-метилпирролидоне, диметилацетамиде, диметилсульфоксиде,
пиперидине, пиридине, этиленгликоле и расплавленном феноле
при 150—240 °С приводит к образованию полиаминоимида или по-
ликарбоксибензимидазола [3, 39, 40]. Эти продукты не
растворяются в органических растворителях и растворяются только в
концентрированной серной кислоте. Циклодегидратация полиами-
ноамидокислот до пирронов при 300—400 °С в большинстве
случаев происходит в процессе их переработки в изделия (8.1.3).
Если в рецептуре часть тетрамина заменить на ароматический
диамин, то в аналогичных условиях получатся полипирронимиды
[69].
8.1.1.2. Одностадийная поликонденсация в растворе
Пирроны в одну стадию образуются при взаимодействии
диангидридов тетракарбоновых кислот с ароматическими тетраминами
в полифосфорной кислоте при 180—220 °С [3, 26, 41, 64]. Вместо
склонных к окислению тетраминов предпочтительно использовать
их хлоргидраты. В противоположность низкотемпературной
поликонденсации при высокотемпературном синтезе в полифосфорной
кислоте избыток диангидрида вызывает понижение молекулярной
массы. Этим методом получают высокомолекулярные пирроны
с характеристической вязкостью до 1,8 дл/г в серной кислоте.
В результате взаимодействия пирронов с такими нуклеофильными
компонентами, как о-аминофенол или о-аминотиофенол, в
полифосфорной кислоте образуются полибензимидазолы, содержащие
в о-положении боковые бензазольные группы, растворимые в
органических растворителях [73, 74].
8.1.1.3. Поликондеисация в расплаве
Пирроны образуются в одну стадию при постепенном
нагревании смеси диангидрида и тетрамина в интервале температур 100—
450 °С [3]. Для получения высокомолекулярных полимеров
необходимо, однако, проведение процесса под высоким давлением.
В этом случае возможно совмещение в одном процессе синтеза и
переработки полимера в изделия [13] (8.1.3).
8.1.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПИРРОНОВ
Пирроны представляют собой сильно окрашенные полимеры,
плотность которых колеблется от 1,30 до 1,40 г/см3. В табл. 8.1
приведены температуры размягчения и разложения пирронов.
Полимеры № 1—3 представляют собой истинно лестничные
полимеры, в то время как в полимерах № 4—42 пирроновые звенья
соединены ординарными связями; полимеры № 43—55 содержат
в своем составе наряду с пирроновыми, также другие группы. На
основании данных ИК-спектроскопии (положение полосы С=О
группы) показано, что пирроны существуют в двух изомерных фор-
1022
мах [4] :
цнс-Бислактамная структура
гранс-Бислактамиая структура
Методами пиролитической газовой хроматографии и масс-спек-
трометрического анализа было установлено, что в пирронах на
основе пиромеллитового диангидрида каждое третье, а в пирронах
на основе диангидрида бензофенонтетракарбоновой кислоты
каждое четвертое звено остается нециклизованным [62]. По теплотам
сгорания полиаминоамидокислоты и соответствующего пиррона на
основе диангидрида дифенилоксидтетракарбоновой кислоты и тет-
раминодифенилоксида (№ 13) была рассчитана степень
циклизации, оказавшаяся равной 83%. Вследствие наличия системы
сопряжения пирроны сильно окрашены (от желтого до оранжевого
цвета). Полимер № 1, полученный из пиромеллитового
диангидрида и тетраминобензола, окрашен в черный цвет.
8.1.2.1. Температура размягчения
Для большинства пирронов температура разложения
находится ниже температуры размягчения или плавления. Пирроны, в
которых имидазопирролоновые звенья разделены «шарнирными»
атомами или группами (О, СО, SO2, СН2), размягчаются в
интервале температур 300—400 °С. Температура размягчения
понижается с увеличением числа этих групп в звене полимера.
8.1.2.2. Растворимость
Растворимость пирронов зависит от химического строения
полимера и способа его получения.
Лестничные пирроны № 1—3 нерастворимы так же, как и
большинство других пирронов, полученных термической циклизацией
пленок высокомолекулярных полиаминоамидокислот, что
обусловлено их частично-сшитой структурой. Пирроны, в макромолекулах
которых содержатся «шарнирные» группы, связывающие
имидазопирролоновые звенья, а также полимеры, полученные в
полифосфорной кислоте и расплаве, растворяются в серной кислоте,
1023
m,
юо
во
60
40
20
—
-
-
-
f Г
XV 2
\
\
\ 1
200 400 500 800 t^
20 40 Т,
Рис. 8.1. Кривые тер-
мической деструкции
пиррона на основе пи-
ромеллитового диан-
гндрида и диамино-
бензиднна при
различной температуре [9]:
/ — на воздухе; ? — в
среде азота.
Рис. 8.2. Кривые
изотермической
деструкции пиррона на
основе пиромеллитового
диангидрида и ди-
аминобензидина в
вакууме [9].
диметилформамиде, диметилацетамиде и диметилсульфоксиде.
Пирроны на основе трифенилфосфиноксидтетракарбоновой
кислоты (№ 19—22), которые получены химической циклизацией поли-
аминоамидокислот с последующей термической циклизацией поли-
аминоимидов в нитробензоле при 200 °С, растворяются в смеси
тетрахлорэтан — фенол, муравьиной и трифторуксусной кислотах
[51].
8J-2.3. Термостойкость
По стойкости к термической и термоокислительной деструкции
пирроны близки к полиамидам. Они устойчивы на воздухе до
450°С и в инертной среде до 500 °С. Деструкция в инертной среде
протекает, однако, значительно медленнее, чем на воздухе
(рис. 8.1). Относительно низкая термостойкость объясняется
наличием нециклизованных звеньев, по которым и начинается
деструкция. Стойкость к термической деструкции в зависимости от
строения диангидрида изменяется в следующей последовательности:
О
ш Стойкость к термоокислительной деструкции возрастает с
увеличением содержания карбонильных групп в элементарном звене
макромолекулы. Наиболее высокой стойкостью к
термоокислительной деструкции на воздухе характеризуется полимер на осно-
1024
ве диангидрида бензофенонтетракарбоновой кислоты и тетрамино-
бензофенона (№ 25). Уменьшение массы пиррона (табл. 8.1, № 4)
в изотермических условиях показано на рис. 8.2. Газообразные
продукты пиролиза пирронов в вакууме состоят из СО, СО2, воды
и небольших количеств аммиака, дициана и водорода [7, 50, 52].
Ниже 400 °С в результате доциклизации выделяется почти только
одна вода. В состав газообразных продуктов деструкции полимера
на основе диангидрида дифенилоксидтетракарбоновой кислоты и
тетраминодифенилоксида, общее количество которых составляет
2,28 %, входят 2 % СО, 0,20 % СО2 и 0,04 % Н2 (температура
пиролиза 500°С). При 600°С выделяется 8,15% летучих,
содержащих 5,96% СО, 1,78 СО2 и 0,41 Н2 [52]. Выше 550°С в ИК-спект-
рах пирронов увеличивается интенсивность частот, характерных
для бензимидазольного цикла, что связано с участием во
вторичных реакциях (выше 480°С) карбонильных групп пирронов [7].
Энергия активации процесса термической деструкции пирронов
составляет около 30 ккал/моль [8, 20, 52].
Механизм термической деструкции пирронов аналогичен
механизму деструкции полиимидов и полибензимидазолов [50, 52].
Вода, выделяющаяся в результате дополнительной циклизации
при температурах до 390 °С, вызывает гидролитическое
расщепление связи .N—СО:
ОХ ЮН:
Главным продуктом, образующимся в результате деструкции,
является СО, выделяющийся при распаде имидазопирролонового
цикла:
^г1
СО +
Карбонизированный остаток
33 Зак. 10
1025
8.1.3. ПЕРЕРАБОТКА ПИРРОНОВ
Пленки, связующие для армированных пластиков и покрытия,
получаются так же, как и в случае полиимидов (путем
использования полиаминоамидокислот с последующей циклизацией
непосредственно в изделиях) [14, 22, 42]. При формовании
стеклопластиков стеклянную ткань пропитывают раствором форполимера.
Сушка пропитанной ткани проводится при 150 °С с последующим
прессованием при 325 °С под давлением 35 кгс/см2. В качестве
связующего используют также полиимидазопирролонимиды [65].
Прессованные изделия получают, применяя смеси диангидридов
и тетраминов, исходя из форполимеров или пирронов. В
последнем случае прессование проводится при высоких
температурах.
Прессование пирронов аналогично прессованию керамических
материалов. В ходе предварительного формования мономеров
используют пресс-формы, покрытые графитом, обеспечивающие
удаление летучих продуктов реакции [13]. Смесь мономеров
нагревают под давлением 280 кгс/см2 при скорости нагревания 5 °С/мин
до 450 °С и выдерживают прессуемую массу при этой температуре
в течение 1 ч. После охлаждения изделия извлекают из формы.
В ходе прессования масса расплавляется, одновременно протекает
процесс поликонденсации. Главная трудность при прессовании
заключается в необходимости удаления воды из твердого продукта.
При слишком высокой скорости повышения температуры и
давления получаются пористые изделия, в то время как при низкой
скорости образуются материалы низкой плотности. Этот метод
не обеспечивает хорошие физико-механические свойства изделий
и высокую плотность. Удовлетворительные результаты
достигаются только в случае использования солей мономеров A:1) [49].
Прессование, исходя из промежуточных полимеров (полиамино-
имид, поликарбоксибензимидазол или полиаминоамидокислота),
проводят сначала при 260 °С и давлении 70 кгс/см2, причем
давление дается после разогрева массы до заданной температуры.
Затем температуру повышают до 325 °С, а давление сбрасывают до
нуля, что обеспечивает удаление летучих. И наконец, температуру
повышают до 450 °С. Переработка пирронов прессованием
осуществляется при 500°С [6].
Предварительно таблетированный порошок нагревают до
400 °С и загружают в формы, облицованные стеклянной тканью.
Массу под давлением 35—70 кгс/см2 нагревают до 475 °С, после
чего давление и температуру устанавливают в пределах 350—
700 кгс/см2 и 520—535 °С. Цикл горячего прессования составляет
20—60" мин. Изделие выдерживают 1—5 мин при максимальной
температуре и после охлаждения под давлением до 250°С
извлекают из формы. В ходе прессования материал при нагревании до
350°С сначала расширяется, а при дальнейшем повышении
температуры до 475°С постепенно уплотняется. При 475—500°С
материал приобретает пластичность, и давление снижается.
1026
Полученные таким образом изделия в отличие от изделий,
отпрессованных из порошка, полностью гомогенны и изотропны. За
время цикла прессования потеря массы составляет от 10 до 20 %.
Пластичность пирронов при 500°С является следствием разрыва
ковалентных связей лестничного полимера с последующим
возникновением новой лестничной структуры. Этим можно объяснить
появление в пресс-изделиях маслянистых продуктов деструкции,
которые частично состоят из бензонитрила.
8.1.4. СВОЙСТВА МАТЕРИАЛОВ ИЗ ПИРРОНОВ
Лестничная графитоподобная структура пирронов
обусловливает их уникальную химическую, термическую и радиационную
стойкость.
В зависимости от химического строения полимера прочность
при растяжении колеблется от 700 до 1 600 кгс/см2 (пленка,
полученная поливом, с последующей термической циклизацией),
относительное удлинение при разрыве — от 2 до 5% и модуль
упругости— от 35000 до 70 000 кгс/см2. Ниже приведены свойства
материалов из пиррона на основе пиромеллитового диангидрида и
диаминобензидина [5, 6, 14] :
2,5
35 000-70 000
1,25-1,36
70-90
Пленки и
прессованные изделия
Прочность при
растяжении, кгс/см2 1100-1600*
700-1 100**
Относительное удлинение
при разрыве, %
Модуль упругости при
растяжении кгс/см2
Плотность, г/ем3
Твердость по Роквеллу,
шкала В
Термический
коэффициент линейного
расширения, 1/°С
при 35—200 °С
при 200—420 °С
Удельное объемное
электрическое сопротивление,
Ом-см
Электрическая
прочность, кВ/мм
Количество
абсорбированной влаги, %
Степень набухания
высушенной пленки в воде, %
18
36
10
10
-в
-в
101в
1000
7—12
1
Стеклопластик иа
основе пирроиа
№ 5
Содержание связующего,
%
Прочность при изгибе,
кгс/см2
при 25 СС
при 316°С
Модуль упругости при
изгибе, кгс/см2
при 25 °С
при 316°С
Потеря массы при 325 °С,
%
при старении в
течение 24 ч
при старении в
течение ПО ч
36
3 500
3 450
190 000
190 000
6,5
27,1
* Пленка получена поливом из раствора форполимера с последующей термической ци«
клизацией.
•• Изделие получено из мономеров методом прессования.
Прессование не обеспечивает получение изделий с такой
прочностью, которая достигается при приготовлении пленки поливом
из раствора форполимера с последующей термической циклиза-
33*
1027
цией. Прочность такой пленки не зависит от того, в каких
условиях проводилась циклодегидратация: в вакууме, инертной среде
или на воздухе [5]. При высоких температурах прочность пленки
остается достаточно высокой. При 100 °С прочность при
растяжении составляет 93 % и модуль упругости 88 %, при 200 °С —
соответственно 68 и 67 % от исходного значения. Относительное
удлинение при разрыве в этом температурном интервале
возрастает на 30 %.
По прочности пирроновые стеклопластики находятся на уровне
полиимидных стеклопластиков [46]. Прочность и модуль
упругости при изгибе до 300 °С практически не меняются.
Высокотермостойкие клеи для металлов получены путем
обработки пирронов аммиаком при 200 °С [44]. Склеивание алюминия
с помощью таких полибензимидазоламидов проводят за 1 мин
при 400 °С под давлением 14 кгс/см2.
Пирроновые пенопласты отличаются высокой стойкостью к
абляции [68] и исключительно высокой радиационной стойкостью.
При облучении пирроновой пленки ^излучением дозой 21 Мрад
прочность при растяжении увеличивается на 32%, и при дозе
58 Мрад она достигает исходного значения; модуль претерпевает
подобные же изменения. При облучении дозой 5000 Мрад
электроизоляционные свойства изменяются незначительно [43].
Пирроны имеют очень высокую стойкость к различным
химическим агентам. Прочность при растяжении пленки толщиной
0,025 мм за 1 ч при 250 °С в 96 % -ной серной кислоте и 2 н.
NaOH составляет соответственно 76 и 58 % от исходного
значения, а модуль упругости — 97 и 64 % • ¦
Пирроны обладают фотополупроводниковыми свойствами.
Максимальная чувствительность пиррона на основе пиромеллито-
вого диангидрида и диаминобензидина проявляется при толщине
пленки 0,610 мкм, а на основе диангидрида бензофенонтетракар-
боновой кислоты и диаминобензидина — 0,55 мкм [11].
Отношение фототок: темновой ток составляет 300. В интервале 102—105
В/см существует линейная зависимость фототока от напряжения.
Проводимость пирронов, инициированная электронным потоком,
изучена в [43].
8.1.5. ПРИМЕНЕНИЕ ПИРРОНОВ
Пирроны могут применяться в качестве конструкционных
материалов в авиации и космической технике, в атомной
промышленности, для изготовления батарейных сепараторов, щелочных
элементов и полупроницаемых мембран для очистки морской
воды. В последнем случае они действуют аналогично
неорганическим молекулярным ситам [14].
В процессе термической циклизации полиаминоамидокислот за
счет выделяющейся воды образуются поры, размер которых
обеспечивает диффузию через них воды, но недостаточен для
проникновения солей. Именно наличием в мембранах таких пустот
объясняется высокое водопоглощение без набухания. Число пере-
1028
Рис. 8.3. Зависимость коэффициента
диффузии воды и поваренной соли через пирроно-
вую мембрану из пиромеллитового диангид-
рида и диаминобензидина от
продолжительности прямого осмоса (толщина пленки
2,5 мкм) [14]:
I — диффузия
воды, мл • 10
NaCl, мг/мл.
— 2
2 — диффузия
40-
носа (константа мембраны),
выражающее отношение количества
проникающей воды к давлению, для
полимера на основе
пиромеллитового диангидрида и
диаминобензидина в случае 0,5 %-ного раствора
хлорида натрия при давлении
105 кгс/см2 составляет 9,8-10~8
г/(см2-с-атм). Фильтрация воды и
диффузия хлорида натрия через
мембрану пиррона из пиромеллитового диангидрида и
диаминобензидина при прямом осмосе показаны на рис. 8.3.
Поскольку пирроны обладают фотополупроводниковыми
свойствами, их можно применять в электрофотографии [11].
8.2. ПОЛИИМИДАЗОБЕНЗФЕНАНТРОЛИНЫ
Полибисбензимидазобенз-
фенантролин (полимер ВВВ)
N
Полиимидазобензфенантролины (полинафтоиленбензимидазо-
лы) получают взаимодействием 1,4,5,8-нафталинтетракарбоновой
кислоты или тетракарбоновых кислот с конденсированными
ароматическими ядрами с аналогичным расположением
карбоксильных групп и ароматических тетраминов. От пирронов их отличает
только наличие в структуре пяти- и шестичленного гетероцикла
вместо двух пятичленных.
Полимеры этого типа устойчивы на воздухе до 500 СС и в
инертной среде до 600°С. Из растворов в серной кислоте поли-
имидазобензфенантролина на основе нафталинтетракарбоновой
кислоты и диаминобензидина мокрым формованием в воду могут
быть получены волокна, которые кратковременно выдерживают
нагревание до 800 °С и длительно работают при 300 °С. Полиими-
дазобензфенантролиновые волокна производятся на пилотной
установке начиная с 1969 г. и используются исключительно для
военной и космической техники.
1029
Таблица 8.2. Строение н температура разложения полннмндазобензфенантролинов
Элементарное звено
Гразл
т
'разл
(на
воздухе),
°С
600
600
600
450
500
550
600
600
450
400
500
Продолжение табл. 8,2
9
10
11
V5
чя
О
\
N
О
\
^ \
N
0
/ ч
o—<
0
\n
N
g
Элементарное звено
У
0
N
= —, —0—, —CH2—
T
разл
разл
(на
воздухе),
°С
400
Литературный
источник
[106]
[107, 108]
[71, 109]
8.2.1. ПОЛУЧЕНИЕ ПОЛИИМИДАЗОБЕНЗФЕНАНТРОЛИНОВ
Эти полимеры получают одностадийной поликонденсацией в
полифосфорной кислоте [26, 71, 93—95, 99]:
НООС
п
НООС—
—соон
-<0>~~С00н "н
N
—н2о
Эквимольные количества тетракарбоновой кислоты и тетрами-
на нагревают в полифосфорной кислоте в течение 7 ч при 100 °С
и 20 ч при 190—200 °С. В противоположность аналогичным по
строению полибензоиленбензимидазолам (пирронам) полимеры
типа ВВВ (полибисбензимидазобензфенантролины) имеют
линейное, неразветвленное строение и сохраняют растворимость в
полифосфорной кислоте в течение всего процесса. Это чрезвычайно
важно для формирования структуры полигетероарилена,
поскольку тетракарбоновая кислота при 200 °С растворяется
относительно плохо [93].
Дальнейшая термообработка приводит к увеличению
молекулярной массы образовавшегося полимера. Характеристическая
вязкость полибензимидазобензфенантролинов (например,
полимера № 7, табл. 8.2) возрастает после 114 ч нагревания при 165—
180 °С с 0,49 до 0,60 дл/г, а после 4—5 ч при 350—400 °С она
достигает 1,01 дл/г [91].
При температурах до 200 °С реакция протекает через две
промежуточные стадии:
НООС—(( V,—
НООС—(( У,—СООН
Полна миноамидо-
кислота
—п
1033
Полиамипоимид
—H2O
Полнбеизимидазобензфенаитролин
Поликонденсация тетраминов и тетракарбоновых кислот в
расплаве [26, 91] и в растворе, исходя из 1,4-диамино-5,8-ДИкарбокси-
нафталина или аналогичных диаминодикарбоновых кислот с
подобным расположением аминогрупп, приводит к образованию
низкомолекулярных полимеров:
H2N
СООН
п
H2N
—н2о
-П
В случае многостадийного синтеза подобного синтезу пирро-
нов (8.1.1.1) в диметилформамиде или диметилсульфоксиде
проводят первую стадию процесса [88]. Во избежание
предварительного структурирования полиаминоамидокислот на этом этапе
необходимо получать полимер с относительно невысокой
молекулярной массой, что достигается добавлением в раствор
неорганических солей типа КС1, NaCl, СаС12, KBr, KI. В
диметилсульфоксиде в ряду катионов активность уменьшается в следующей
последовательности: К+> Li+^s Na+> Ca2+. Для анионов
активность уменьшается в следующем ряду: Cl" > В г > 1~.
8.2.2. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
ПОЛИИМИДАЗОБЕНЗФЕНАНТРОЛИНОВ
Эти полимеры имеют окраску от глубоко коричневого до
черного цвета; они разлагаются при 500—600 °С, не плавясь и не
размягчаясь. Температуры разложения полимеров приведены в табл.
8.2. Исходя из строения элементарного звена, все полибензимида-
зобензфенантролины можно разделить на шесть типов изомеров:
1034
Все полимеры имеют низкую степень кристалличности [90].
Исследование релаксации механических напряжений полимера
№ 2 (BBL) показало, что максимум потерь наблюдается при
—50°С/1 Гц, а полимера № 7 (ВВВ) при — 120°С/1 Гц [86, 87].
Более низкое положение максимума в последнем случае
объясняется несколько более высокой эластичностью за счет
присутствия в макромолекуле ординарных связей, которых нет у полимера
№ 2.
8.2.2.1. Отношение к растворителям
Полиимидазобензфенантролины в противоположность пирро-
нам растворяются в концентрированной серной, полифосфорной
1035
и бензойной кислотах, а также в диметилсульфате. Растворы
полимеров окрашены в глубокий красный цвет [91, 92]. Полимеры
этого типа растворяются также в водных растворах
восстановителей, например сульфид Na—NaOH — пиридин. При этом
происходит восстановление карбонильных групп практически без
изменения молекулярной массы [90]. Растворимость полиимидазобенз-
фенантролинов с ординарными связями (полимер ВВВ № 7),
соединяющими имидазофенантролиновые блоки, растворяются
значительно лучше, чем чисто лестничные полимеры (полимер
BBL № 1). Из полимера ВВВ 1 %-ный раствор в
концентрированной серной кислоте можно приготовить, перемешивая
реакционную массу в течение 2—3 сут при комнатной температуре. Из
полимера BBL 0,2 %-ный раствор в аналогичных условиях
получается только через 2 недели [91].
Исследование растворов полимера ВВВ [90] показало, что
дробным осаждением из метансульфокислоты и серной кислоты
соответственно метанолом и водой не удается разделить продукт
на фракции, различающиеся по молекулярным массам.
Хорошие результаты получаются при применении
гель-проникающей хроматографии при использовании в качестве носителя
пористой кремневой кислоты, а в качестве элюента — серной
кислоты.
Для определения молекулярной массы седиментацией в
ультрацентрифуге служит водный раствор смеси сульфита Na и NaOH
с пиридином. Сильная абсорбция растворов ВВВ в области 4350—
5460 A делает возможным их исследование только в случае
применения лазерного источника (изучение растворов
светорассеянием) с длиной волны 6328 А. Коэффициент экстинкции растворов
ВВВ в метансульфокислоте при этой длине волны равен
1,25-103 см3/(г-см).
Для вискозиметрических исследований пригодны растворы
полимеров в 96 %-ной серной кислоте или метансульфокислоте.
Константа Хаггинса составляет 0,3—0,5. Экспонента а в уравнении
Марка — Хаувинка — Куна равна 1, что свидетельствует о
высокой жесткости макромолекул, существующих в форме сильно
проницаемого клубка. То, что в растворах ВВВ в 92 %-ной серной, а
для высокомолекулярных полимеров даже в 96 %-ной серной
кислоте константа Хаггинса превышает 1, а также вид угловой
зависимости при светорассеянии, свидетельствует об ассоциации
макромолекул. Для нефракционированного ВВВ при
характеристической вязкости в метансульфокислоте 2,66 дл/г и среднечисловой
молекулярной массе 97 300 среднеквадратичный радиус инерции
<S2> =6,36-Ю-12 см2.
8.2.2.2. Термостойкость
Как отмечалось, полиимидазобензфенантролины устойчивы в
инертной среде до 600 °С и на воздухе до 500 °С (рис. 8.4).
Старение ВВВ на воздухе при 315°С сопровождается 10 %-ной потерей
юзе
200 ?Ю
100150200 300 500 800 Т,ч
Рис. 8.4. Кривые термической деструкции полибисимидазофенантролина при
различной температуре в среде азота [91].
Рис. 8.5. Кривые термической деструкции полибензимидазофенантролина при
315 °С на воздухе [91].
массы за 1500 ч (рис. 8.5). Такая же потеря массы при 400 °С
наблюдается за 7—8 ч. Первый максимум, соответствующий
выделению газообразных продуктов при нагревании полимеров ВВВ
(табл. 8.2, № 7) в вакууме, приходится на 720 °С. Основную часть
продуктов составляют СО [40 % (мол.)] и водород [31,5 % (мол.)].
Выделение СО является следствием разрушения шестичленного
цикла. При 900 °С (второй максимум) в состав газов пиролиза
входит 46,9% (мол.) N2; 31,7% Н2; 6% Н2О; 4,9% HCN; 2,7%
(мол.) акрилонитрила и небольшие количества ацетилена,
пропилена, фенола, ацетонитрила и бутадиена [93]. При этой
температуре декарбонилирование сопровождается превращением гетеро-
иикла:
—П
8.2.3. ПОЛИБЕНЗИМИДАЗОФЕНАНТРОЛИНОВЫЕ ВОЛОКНА
Хорошая растворимость полимера ВВВ, синтезированного из
нафталинтетракарбоновой кислоты и диаминобензидина,
обусловила возможность его использования для изготовления
высокотермостойкого волокна, которое получают путем формования 2—5 %-
ного раствора полибензимидазобензфенантролина в 97%-ной
серной кислоте в осадительную ванну, представляющую собой 70%-
ную серную кислоту. Зависимость вязкости растворов при 30 °С
от концентрации показана на рис. 8.6.
Для переработки в волокно методом осаждения в воду
пригоден ВВВ с характеристической вязкостью 2,4—3,5 дл/г в 97%-ной
1037
и
10
8
e
2
Ж
—
г
•
^¦IOOOO
\> 20000
Рис. 8.6. Зависимость вязкости раствора в 97 %-ной
H2SO4 при формовании полибензимидазофенантролино-
вого волокна осаждением в водную ванну от
характеристической вязкости и концентрации полимера
(температура 30 X) [95].
серной кислоте, вязкость прядильного
раствора в которой должна быть около 4000 Пз
[93—95]. После коагуляции полимер промы-
вают разбавленной щелочью, горячей водой
¦0,51,0 1,5 2,0 1ц},щг и высушивают при 60°С. Кроме того, для
промывки можно использовать также амид-
ные растворители, например диметилформамид, гексаметилфос-
фотриамид и диметилацетамид, а также водные растворы дитио-
натов [98].
В неориентированном состоянии волокно из ВВВ в
зависимости от молекулярной массы имеет прочность при растяжении
1,25—1,40 г/денье и относительное удлинение при разрыве 60—
70 %. При горячей вытяжке прочность можно увеличить в 3 раза.
На рис. 8.7 показаны зависимости прочности и удлинения от
степени ориентации и температуры процесса. Оптимальные
результаты достигаются при 525—575 °С (продолжительность обработки
4 с) и степени вытяжки 2,2:1. При этом волокно имеет прочность
при растяжении 5,2 г/денье и относительное удлинение при
разрыве 5%.
Волокно ВВВ характеризуется значительно более высокой
термостойкостью, чем аналогичное волокно из ароматических
полиамидов и полибензимидазолов (рис. 8.8). При 700 °С сохраняется
50% исходной прочности; термообработка на воздухе при 360 °С
в течение 30 ч приводит к снижению прочности на 50 %. Волокно
ВВВ имеет исключительно высокую стойкость к действию УФ-
излучения. После 400 ч облучения в федеометре прочность,
удлинение и модуль практически не меняются. Термостойкость
волокна можно повысить, проводя его термообработку при 500—700 °С
и кратковременно при 1200 °С, в ходе которой происходит декар-
550 t,°C
150 250 350 450 550
Рис. 8.7. Зависимость прочности при растяжении (а) и относительного удлинения
при разрыве (б) полибензимидазофенантролинового волокна от степени вытяжки
при различной температуре [94]:
О — 1,31—1,32; О— 1,40-1,42; Д - 1,51-1,55; D - 1,58.
1038
Рис. 8.8. Зависимость прочности при растяже- бр,% (от
80
нии высокотермостойких волокон от
температуры (время экспозиции образца при
температуре испытаний 1 мин) [95]:
/ — поли я-фениленизофталамид Номекс: 2 — поли-
бензимидазол; 3 — полибисбеизимидазобензфенаитро-
лин В ВВ.
60-
40-
200
бонилирование шестичленного гетеро- 20
цикла [100]. Декарбонилированное
волокно кратковременно выдерживает
нагревание до 900 °С.
Полибензимидазобензфенантролиновые волокна выпускаются
на пилотной установке фирмой «Celanese» (США) начиная с
1969 г. Такое волокно применяется только для военных целей и в
космической технике.
8.2.4. ПОЛИБЕНЗИМИДАЗОФЕНАНТРОЛИНОВАЯ ПЛЕНКА
Пленку можно получить поливом из раствора полимера в ме-
тансульфокислоте или диметилсульфате, а также путем
вальцевания при высоких градиентах сдвига, причем сильное
межмолекулярное взаимодействие ослабляется за счет усилий при сдвиге в
твердом состоянии [90].
Литьевые пленки из полимера № 2 (см. табл. 8.2)
чувствительны к действию высоких температур, кипящей воды и кипящего
1 н. раствора NaOH. Логарифмическая вязкость этого полимера
(С — 0,0625 дл/г) в метансульфокислоте при 25 °С уменьшается
за 45 ч термостарения на воздухе при 260 °С с 3,67 до 2,96. После
42 ч кипячения в воде ее значение составляет 2,32 и после 15 ч в
1 н. NaOH—1,71. Одновременно возрастает жесткость материала,
которая проявляется в смещении максимума механических потерь
(8.2.2) в область более высоких температур. Понижение
молекулярной массы и хрупкость являются следствием разрушения не-
циклизованных звеньев полимера BBL, в результате чего
нарушается лестничная структура, и полимер в известной мере
приобретает гибкость. Термоокисление и гидролитическая деструкция
приводят к образованию жестких осколков цепи с лестничной
структурой [87]. Абляционные свойства полибисбензимидазобенз-
фенантролинов можно улучшить, добавляя ж-фениленбисмале-
имид и 4,4'-дифенилметанбисмалеимид [97].
8.3. ПОЛИИЗОИНДОЛОХИНАЗОЛИНДИОНЫ
о
-П
Полиизоиндолохиназолиндионы получают из диамидов
ароматических бис(о-аминов)дикарбоновых кислот и диангидридов
ароматических тетракарбоновых кислот. Полимеры устойчивы в инерт-
1039
ной среде до 490 °С и на воздухе до 430 °С. Полимеры неплавки,
поэтому их перерабатывают на стадии промежуточной полнамидо-
кислоты.
8.3.1. ПОЛУЧЕНИЕ ПОЛИИЗОИНДОЛОХИНАЗОЛИНДИОНОВ
Как отмечено выше, их получают взаимодействием амидов
бис(о-амино)дикарбоновых кислот с диангидридами тетракарбо-
новых кислот [75—80]. Процесс протекает через промежуточную
стадию образования соответствующей полиамидокислоты, которая
при термообработке или в присутствии дегидратирующих средств
через полиимидоамид превращается в изомерный полиизоиндоло-
хиназолиндион, изомеризующийся далее в стабильный полиизоин-
долохиназолин-10,12-дион (промежуточный изомер имеет
карбоксильные группы в положении 5 и. 11).
Первая стадия осуществляется в обычных растворителях апро-
тонного типа, таких, как диметилформамид, диметилацетамид,
N-метилпирролидон и диметилсульфоксид. Процесс проводят при
комнатной температуре в присутствии 2—3 % LiCl (для
улучшения растворимости исходного диамида). Наиболее высокая
молекулярная масса ([rj] > 3 дл/г) достигается при добавлении
растворителя к однородной смеси исходных веществ. Оптимальная
концентрация мономеров не превышает 15 %. Форполимер
растворяется во всех растворителях, которые применяются для синтеза.
Такой раствор может быть использован для получения пленки
поливом.
Поликонденсация в полифосфорной кислоте осложнена
гидролизом амидных групп. Поликонденсации в расплаве препятствует
высокая температура плавления исходного диаминодиамида
(Тпл > 300 °С). Циклизацию проводят в твердом состоянии в
изделии, используя форполимер в виде порошка или пленки, в
присутствии дициклогексилкарбодиимида или смеси ацетангидрид-
пиридин в качестве дегидратирующих агентов [76] или путем
нагревания в вакууме или инертной среде [75]. За первые 2 ч при
150—220 °С происходит образование полиамидоимида, который
О О
H2NCO
¦
H2NC<
Полиамидкарбоновая кислота
150-220 *С
J
1040
N—
Полиимидоамид
I
—П.
I 250-420 e(
О О
о о
II JI II II
???
N
N
230-240 °С
—п
Полиизоиидолохиназолин-10,12-днон
230-240 °С
О
N N
-oe
Полиизонидолохииазолии-5,11-дион
Амиды дикарбоновых кислот с о-расположением аминогрупп получают
из соответствующих дикарбоновых кислот по схеме [771:
н3с—(О/—S°2C1
Na2CO3, H2O
НзС—(Г )>—
^.
—СНз
CeHe, РС1з
н3с
C1OG
SO2NH
H2NOC
—(Q)—СНз
. NH3
NHSO2
1041
Таблица 3.3. Температура разложения полиизоиндолохиназолиидионов
Элементарное звено
Т
разл
(в N2),
О О
N
W
о о о о
N
О О 0 0
[77]
490
432
[75-78]
[79]
о о
х
О О
180]
0 0
0 0
450
432
[76, 77]
О О
О О
II
о
N
455
430
[76, 77]
О
о»
100
во
60
40
20
400 600 t,°C
тывают
Рис. 8.9. Кривые термической деструкции полнизоиндолхин-
азолиндионов при различной температуре [75]:
г —полимер Лв 2; 3, 4 — полимер № 3 (табл, 8.3); 2, 4 у*
на воздухе; /. 3 — в среде азота.
в последующие 30 мин при 250 °С превращается
в полиизоиндолохиназолиндион: или
взаимодействием таких диаминодикарбоновых кислот с
фосгеном в концентрированной соляной кислоте
получают бисоксазиндионы, которые обраба-
аммиаком [81]:
H2Nv^ ^^ ^-4. ^NH2 HCl
NH2
8.3.2. СВОЙСТВА ПОЛИИЗОИНДОЛОХИНАЗОЛИНДИОНОВ
Температуры разложения некоторых полиизоиндолохиназолин-
дионов приведены в табл. 8.3.
Полиизоиндолохиназолиндионы разлагаются ниже
температуры плавления. Они растворяются только в концентрированной
серной и дымящей азотной кислотах. Относительно низкая
термостойкость лестничных полимеров объясняется неполнотой
циклизации (рис. 8.9). Деструкция в инертной среде начинается при
490 °С и на воздухе при 430 °С. Полиизоиндолохиназолиндионы
гидролитически стойки.
8.4. ПОЛИТЕТРАЗАПИРЕНЫ
i>Q
Политетразапирены получают взаимодействием тетраминонаф-
талина с производными ароматических дикарбоновых кислот.
Полимеры неплавки, устойчивы на воздухе до 340 °С и в инертной
среде до 410 °С.
8.4.1.ПОЛУЧЕНИЕ ПОЛИТЕТРАЗАПИРЕНОВ
Эти полимеры получают взаимодействием 1,4,5,8-тетрамино-
нафталина с дихлорангидридами или дифениловыми эфирами
ароматических дикарбоновых кислот [85]. По аналогии с получе-
1044
нием полибензимидазолов сначала при 150 °С образуется
промежуточный полимер, который при 250—300 °С циклизуется:
п
H2N—(rS)—NH
H2N—(Q/ NH
|-
н2о
NH2 ОСбН5
I—свн5он
HN—(( ))—
=о-<о>-
_rt
Полимеры, образующиеся на первой стадии реакции,
растворяются в бензоле, хлороформе, диметилформамиде, диметилаце-
тамиде, диметилсульфоксиде. Полигетероарилен — политетразапи-
рен — растворяется частично в концентрированной серной кислоте.
Политетразапирены представляют собой неплавкие порошки,
которые вследствие наличия полисопряженных конденсированных
циклических систем имеют окраску от коричневого до черного
цвета.
Полимеры этого типа менее стойки, чем соответствующие по-
либензимидазолы (рис. 8.10). В инертной среде потеря массы
начинается при температуре 410°С, а на воздухе — при 340 °С.
Масса остатка в инертной среде при 900 °С составляет около 75 %; на
воздухе при 750 °С полимер деструктируется полностью. Свойства
политетразапиренов приведены в
табл. 8.4.
Политетразапиреновая пленка
может быть получена поливом из
раствора форполимера с последующей
термической циклизацией.
Диэлектрическая проницаемость при 100 МГц
составляет 3—4, удельное объемное
электрическое сопротивление 1013 —
1015 Ом-см.
m, %
100
80-
60-
40-
Рис. 8.10. Кривые термической деструкции по- 20
литетразапиренов при различной температуре
[85]:
/, 3 — полимер № 1; 2, 4 — полимер № 3
(табл. 8.4); 3, 4 — на воздухе; 1, 2— в среде азота.
200 400 600 800 t,vC
1045
Таблица 8.4. Температура разложения политетраэапиреиов [85]
JVs
Элементарное звено
Г
разл
(в N2),
°С
4 разл
(на воздухе)
«С
410
410
370
340
347
270
Литература к главе 8
1. V. Bell, Polymer Preprints 6 A965), 747—752.
2. V. Bell, J. Polymer Sei. В З A965) 12, 977—984.
3. F. Damans, J. Polymer Sei. A3 A965), 3549—3571.
4. J. Colson, J. Polymer Sei. A 1 A966) 59—70.
5. V. Bell, J. Polymer Sei. A 5 A967) 12, 3043—3060.
6. С. Johnson, Polymer Engin. Sei. 10 A970) 6, 340—344.
7. R. Jewell, J. Macromolecul. Sei. A3 A969) 6, 1147—U59.
8. R. Jewell, J. appl. Polymer Sei. 12 A968), 1137—1145.
9. А. Праведников, ДАН СССР, 172, 1967, 6, 1347—1349.
10. G. Berry, J. Polymer Sei. A28 A970) 12, 2089—2094.
11. P. Rencroit, J. Polymer Sei. С 30 A970), 261—270.
12. V. Bell, J. Polymer Sei. В 5 A967) 10, 941—946.
13. P. Morgan, J. Polymer Sei. В 7 A969) 6, 437—442.
14. H. Scott, J. Polymer Sei. В 8 A970) 8, 563—571.
15. H. Priee, SPE techn. Papers 15 A969), 617—621.
16. Л. Лайус, Высокомолек. соед., А 12, 1970, 8, 1834—1840.
17. Б. Жубанов, Труды ин-та химических наук АН Каз. СССР, 1970, 28, 118-
122.
18. В. Коршак, ДАН СССР, 182, 1968, 6, 1327—1330.
19. М. Котон, Хим. волокна, 1969, 4, 15—22.
20. Э. Телешов, Высокомолек. соед., А 20, 1968, 2, 422—428.
21. Пат. СССР 257012, II. 11.69.
22. L. Frost, J. Polymer Sei. A I 9 A971), 1045—1070.
23. G. D'Alelio, J. Macromolecul. Sei. A 2 A968) 6, 1275—1280.
24. W. Dunnavant, J. Polymer Sei. В 6 A968) 1, 49—52.
25. Акцепт, заявка Японии 7036040, 17. 11.70.
26. J. Szita, J. Polymer Sei. A 19 A971) 3, 691—700.
27. P. Dutt, J. Polymer Sei. A I 8 A970), 3225—3234.
28. Акцепт, заявка Японии 7015990, 3.6.70 Тогау IncL
29. Акцепт, заявка Японии 7122937 30.6.71 Тогау Ind/
30. Акцепт, заявка Японии 7127822 12.8.71 Тогау Ind.
1046
31. Пат. СССР 268645, 10.4.70.
32. Пат. СССР 295779, 12.2.71.
33. Пат. СССР 293011, 15. 1.71.
34. Пат. СССР 275388, 3. 7. 70.
35. Пат. СССР 308031, 1.7.71.
36. А. Берлин, Высокомолек. соед., А 12, 1970, 938—947.
37. V. Bell, J. appl. Polymer Sei. 14 A970) 9, 2385—2398.
38. Пат. США 3640960 8. 2. 72.
39. H. Priee, SPE techn. Papers 15 A969), 617—623.
40. B. Sillion, Compt. rend. Acad. Sei. С262 A966), 926—932.
41. J. Szita, J. Polymer Sei. A 1 9 A971) 2, 415—421.
42. W. Glover, Polymer Preprints 7 A966), 819—823.
43. P. Rencroft, J. appl. Polymer Sei. 14 A970), 1361—1372.
44. Пат. США 3640956 8. 2. 72 Du Pont.
45. F. Arnold, J. Polymer Sei. A 1 8 A970) 8, 2079—2081.
46. G. Bower, J. appl. Polymer Sei. 16 A972), 345—352.
47. В. Коршак, Высокомолек. соед., А 14, 1972, I, 186—201.
48. А. Рудаков, Высокомолек соед., А 14, 1972, 1, 169—176.
49. Р. Morgan, J. appl. Polymer Sei. 16 A972), 2029—2050.
50. С. Павлова, ДАН СССР, 206, 1972, 2, 359—363.
51. Г. Колесников, Высокомолек. соед., А 14, 1972, 10, 2065—2078.
52. В. Родэ, Высокомолек. соед., Б 13, 1971, 10, 732—736.
53. J. Heller, J. Polymer Sei. В 6 A968) 3, 153—157.
54. А. Праведников, Высокомолек. соед., Б 14, 1972, 3, 185—188.
55. R. Murphy, J. Polymer Sei. В 6 A968), 3, 241—245.
56. D. Packham, Polymer 10 A969) 12,923—931.
57. R. Evers, J. Polymer Sei. А I 9 A971) 2, 543—551.
58. Б. Лиогоньский, Высокомолек. соед., Б 10, 1968, 9, 678—682.
59. Б. Лиогоньский, Изв. АН СССР, сер. хим., 5, 1966, 945.
60. Г. Белова, Высокомолек. соед., А 10, 1968, 7, 1561—1569.
61. И. Рабинович, Высокомолек. соед., А 15, 1972, 2, 360—366.
62. G. Sykes, SPI techn. Papers 19 A973), 610—612.
63. Акцепт, заявка Японии 7204946 10.2.72 Тогау Ind.
64. Пат. СССР 339558, 24. 5. 72.
65. Акцепт, заявка Японии 7142319 14. 12.71 Тогау Ind.
66. Акцепт, заявка Японии 7208234 9. 3. 72 Тогау Ind.
67. Пат. США 3657190 18.4.72 Avco Со.
68. А. Мс Lain, NASA Techn. Note A972), D-6711.
69. В. Коршак, Труды Бурятского ии-та естественных наук, СО АН СССР, 1971,
10, 78—86.
70. Е. Краснов, Высокомолек. соед., Б 13, 1971, 7, 431—435.
71. А. Берлин, Высокомолек. соед., Б 13, 1971, 3, 198—201.
72. И. Рабинович, ДАН СССР, 198, 1971, 3, 597—600.
73. В. Коршак, Кинетика и механизм полиреакций, Международный
симпозиум по макромолекулярной химии, препринты, 1969, 1, 59—63.
74. Пат. СССР 312857, 31.8.71.
75. M. Kurihara, Macromolecules 3 A970) 6, 722—727.
76. G. Rabilloud, Compt. rend. Acad. Sei. С 263 A966), 862—865.
77. G. Rabilloud, Makromolekulare Chem. 108 A967), 18—51.
78. В. Коршак ДАН СССР, 200, 1971, 1, 114—117.
79. M. Kurihara, J. Polymer Sei. В 6 A968) 12, 875—882.
80. Акцепт, заявка Японии 6928517 22. 11.69 Тогау Ind.
81. Акцепт, заявка Японии 7039247 10. 12.70 Тогау Ind.
82. В. Коршак, Труды Моск. хим.-техн. ин-та, 1972, 70, 178—181.
83. Опубл. заявка ФРГ 2153602 27.4.72 Westinghouse Electric
84. Пат. СССР 291937, 6. 1. 71.
85. F. Davvans, J. Polymer Sei. A 2 A964) 11, 5005—5016.
86. J. Gillham, J. appl. Polymer Sei. 16 A972) 8, 2153—2155.
87. J. Powell, J. appl. Polymer Sei. A973).
88. С. Белых, Высокомолек. ссед., А 14, 1972, 5, 1215—1220.
89. F. Arnold, J. Polymer Sei. В 6 A968) 11, 815—819.
10 7
90. G. Berry, J. Macromolecul. Sei. A3 A969) 6, 1125—1146.
91. R. van Deusen, J. Polymer Sei. A I 6 A968) 7, 1177—1193.
92. R. van Deusen, J. Polymer Sei. В 4 A966) 3, 211—214.
93. W. Gibbs, J. Macromolecul. Sei. A 2 A968) 7, 1291—1302.
94. W. Gloor. Appl. Polymer Symp. 6 A967), 151—163.
95. W. Gloor, Appl. Polymer Symp. 9 A969), 159—166.
96. Пат. США 3562380 9. 2. 71 Celanese.
97. Б. Лиогонький, Труды Лит. АН, Б, 1972, 4, 123—130.
98. Пат. США 3620999 16. 11.71 С. Marvel.
99. F. Arnold, J. Polymer Sei. A 1 8 A970) 8, 2079—2089.
100. Пат. США 3575941, 10.4.71.
101. Пат. США 3523151 A971) Celanese.
102. Пат. США 3574171 6.4.71 Celanese.
103. Пат. США 3574170,6.4.71 Celanese.
104. А. Берлин, Изв. АН СССР, сер. хим., 1966, 945.
105. А. Берлин, Высокомолек. соед., А 9, 1967, 1936—1945.
106. А. Берлин, Высокомолек. соед., Б 10, 1968, 678.
107. Б. Лиогонькнй, Высокомолек. соед., Б 10, 1968, 574.
108. Г. Шамраев, Высокомолек. соед., А 12, 1970, 401—408.
109. А. Берлин, Высокомолек. соед., А 13, 1971, 198—207.
ПО. F. Arnold, Macromolecules 2 A969), 497—504.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абляционные материалы 190, 195
АБС-пластик 217, 218, 223, 244, 247,
262, 263
Алифатически-ароматические
полиамиды 384, 386 ел.
Алифатически-ароматические
полиэфиры 296, 297 ел.
Алициклические полиамиды 361 ел.,
384
Аллобек 834
«Аманим» 799
«Амоко» 814
Арамидил 799, 813
«Арилокс» 206
«Арилон» 246, 247, 264, 267
Аримид 591, 592, 699, 705, 719, 725
«Арнокс» 206
Ароматические полиамидогидразиды
453' ел.
Ароматические полиамиды 394 ел.,
430
Ароматические полиангидриды 356,
357
Ароматические полисульфиды 284 ел.
«Астрел бранд 360» 248
«Бакелит» 246, 252, 267, 272
Блок-лестничные полимеры 586, 998
Боропластики 964, 966
«Бу-Туф» 20, 49
«Валокс» 350
Веспел 591, 737, 740, 741
Вестолеи БТ 49
«Вииивлон» 434
Волокна
борные 434, 590
В 432
ВВВ 40, 999, 1029, 1037
высокомолекулярные 431, 434, 459
высокоориентированные 888
графитовые 736
кристаллические 888
МХ-6 390
ориентированные 547
полиамидные 390, 427, 548
полиамидогидразидные 456, 157,
461
полиамидоимидные 799, 810
полиаминотетразоловые 557
полиангидридные 361
полибензимидазольные 849
полибензимидазофенантролино-
вые 1037
полибензоксазиндиоиовые 992
полибензоксазолимидные 772
полибензоксазольные 897, 925
из полибутена-1 57
поливинилциклогексаиовые 93
Волокна
полигидразидные 526, 547
пилиимидные 725, 778 ел.
из поли-4-метилпентена-1 80 ел.
полиоксадиазолимидные 772
полиоксадиазольные 546
полисульфонатные 355
политиациазольные 555, 1054
политиазольные 506, 1054
политриазиновые 581
политриазольные 523
полифенилендибензимидазольные
887 ел.
полихиназолонойые 968, 980
полихиноксалиновые 933, 964
из поли-1,4-циклогексилендиме-
тилентерефталата 347
полиэтиленоксибензоатные 296
РАВН 457, 459—461
PRD 433, 434
углеродные 434
Вспененные материалы 75
Гетероциклические цепные полимеры
469 ел.
Графит 195
Графитопласты 967, 1027
«Дайкин» 115
«Далвор» ЮЗ, 115, 117, 123, 124
Деструкция
пиронов 1024
полиазометинов 190
полиамидов 389, 424, 452, 809,
814
полиамидокислот 670, 764
полиарилатов 301, 342
полиариленсульфоноксидов 257
полибензимидазолов 881 ел.
полибензоксазиндионов 992
полибензоксазинонов 990
полибензоксазолов 920, 921
поливинилиденфторида 118
поливинилфторида 103, 108
поливинилциклогексана 91, 92
полигалогенолефинов 33
полигидантоинов 487
полидикетопиперазина 565
полиизоиндолохиназолиндионов
1044
полиимидазобензфенантролинов
1037
полиимидов 692, 693, 705, 709.
поли-п-ксилена 170
поликсилилидеиов 179
поли-4-метилпеитена-1 80
полиоксадиазолов 544, 545
полиоксазолидонов 496
полипиразолов 482
1049
Деструкция
полисульфонатов 355
политетразапиренов 1045
политиадиазолов 551
политриазииов 571
политриазолов 522, 523
полифениленов 142, 154—156
полифениленоксидов 213
полифениленсульфидов 290
полихиназолонов 968, 971, 980
полихиназолиндионов 984
полихиноксалинов 961—964
полиэфироимидов 821, 833
Джемонд 591, 740
«Дифлор 2000» 115
Жесткость 29
«Живое сшивание» 733
полимеризация
157
Изогол 813
Изомеризационная
165
Изомид 834
Изостатическое прессование
Изотенископия 42
«Имидайт» 591, 733, 892, 894, 895
"Имидекс" 834
Имидизация 670, 677, 681
Инсоляция 178
Каландрование 221
Кантон 590, 699, 705, 719
Карбоциклические цепные полимеры
142 ел.
Кардовые полимеры 300
Катализаторы Циглера — Натта 68
Квантад 591, 730
Кватерфенил 155, 157
«Квиана» 361
«Кевлар» 433
«Керимид» 592, 730, 731, 733
«Кермел» 799, 810
«Кинар» 114, 115, 119, 122—124
«Кинел» 592, 686, 737
Кислородный индекс 295
Клеи 237, 244, 799
пирроновые 1028
из полибутена-1 57
полибензимидазольные 892
полибензоксазольные 897
полиимидные 729
политриазиновые 581
полихиназолиндионовые 981, 985
полихиноксалиновые 933, 967
«Кодел» 348
Композиционные материалы 128 ел.,
158, 189, 217, 223, 230, 235, 236,
247, 262, 292 ел., 350, 734, 737
«Ксилок 210» 187, 188
КФ 117, 119, 122—124, 131 ,
1050
Лаки
полиамидоимидные 799
полигидантоиновые 483, 487
полиимидные 731
полиэфироимидные 834
Латексы 114
Лестничные полимеры 29, 31, 38, 40,
585, 961, 964, 998 ел.
Литье под давлением
полибензоксазиндионов 992
полибутена-1 56
поливинилиденфторида 120
поливинилфторида 103
полигидроксиэфиров 238, 242
поли-2,6-диметил-1,4-фениленок-
сида 219
поли-4-метилпентена-1 73
полипропилена 74
полисульфонов 264
полиэтилена 74
«Мелдин» 591
«Мертвое сшивание» 733
«Метлбонд» 591, 730
Методы
поглощения кислорода 42
Фишера — Тропша 66
экспрессные 41
«Метолон» 898
Модификация 21, 22, 65
Молекулярно-массовое распределение
69, 257, 357, 704, 881
«Монсанто-710» 590
Набухание 79
Найлон-бТ 390
«Номекс» 416, 427, 428, 430
«Норил» 206, 208, 217, 218, 225
Обрыв цепи 146
«Оксалон» 547
Ориентация 60
Отверждение 160
«Пайр» 590, 731, 812
Пасты 114
Парилеиы 162
Пенопласты 75, 231, 292
пирроиовые 1028
полиимидные 745 "ел.
синтактические 588, 747, 849, 891
Пентон 33
Перегруппировка Фриса 342
Переработка
полибензилов 188
полигидроксиэфиров 238, 241
полиимидов 744
полиоксазолидонов 497
политриазинов 581
политриазолов 523
полифенилена 157
полифениленоксидов 217
Петротекс 49
«Пиралои» 733
Пирролоны 692
Пирроны 988, 999 ел., 1022 ел.
Пленки
двухслойная 590
клеевая 590
пнрроновые 999, 1026—1028
полнамидогидразидные 456
полиамидоимидные 799, 811
полиаминоамидокислотные 1021
полибензимидазольные 895
полибензимидазофенантролино-
вые 1039
полибензоксазиндионовые 992
полибензоксазиноновые 991
полибензоксазольные 897, 921,
922
из полибутена-1 57, 64
поливинилиденфторидные 115,
117, 119
поливини лфторидные 95, 102,
104, 106
полигидантоиновые 483, 492, 1053
полигидроксиэфирные 244
полиимидные 673, 717
из поли-п-ксилиленов 168, 173
из поли-4-метилпентена-1 75
полиоксазолидоновые 1053
полисульфонатные 355
полисульфоновые 265, 273
политетразапиреновая 1045
нз политетрафтор-п-ксилилена 178
политиазольные 507
политриазиновые 581
политриазольные 523
полифениленоксидные 220, 231
полихиназолнндионовые 981
полихиназолоновые 968, 980
полихиноксалиновые 933, 964, 965
полиэфироимидные 835, 836 ел,
самоклеящиеся 724
Покрытия
полиамидоимидные 812 ел.
поливинилиденфторидные 114,
115, 121
поливинилфторидные 104, 109
полигидроксиэфнрные 242, 244
- из поли-л-ксилилена 168, 174
пирроновые 1026
полисульфоновые 265
полифеннленсульфидные 292
из поли- 1,4-циклогексилендимети-
лентерефталата 347
Ползучесть 60, 77, 78, 113, 225, 226,
243, 269, 294
Полиазометины 190 ел.
Полиазооксазолы 493
Полнамидогидразиды см.
Ароматические полиамидогидразиды
Полиакрнлаты 128, 129
Полиальдазины 190, 191, 196 ел.
Полиамидоимиды 434, 587, 772 ел.,
799 ел., 808 ел., 815 ел.
Полиамидокислоты 591, 593, 670, 675,
717, 986, 1040
Полиаминоамидокислоты 38, 999,
1016, 1023
Полиаминоимиды 38, 999, 1016, 1019—
1021, 1034
Полиаминотиазолы 501
Полиаминотетразолы 557
Полиамиды 225, 850, 926
Полиарамиды 361, 394 ел.
Полиарнлаты 141, 297 ел.
Полиариленизопропилидены 188
Полиариленсульфоноксиды 198,
245, ел., 262 ел., 272 ел.
Полиацетали 225, 233
Поли-О-ациламиноксимы 529
Поли-л-бензамид 433
Полибензгетероциклические полимеры
585 ел.
Полибензилы 184 ел.
Полнбензимидазолы 587, 849 ел.,
875 ел. 894—896, 999, 1016,
1021, 1022
Полибензоилиденбензимидазолы 999
Полибензоксазиндионы 588, 991 ел.
Полибензоксазиноны 588, 986 ел., 990
Полибензоксазолимиды 772, 777, 900,
925
Полибеизоксазолы 588, 897 ел,, 901,
922, 990
Полибензтиазолимиды 777
Полибензтиазолы 588, 926 ел.
Полибисбензимидазобензофенантро-
лин 999, 1029
Полибисмалеимиды 685
Полибистетразолы 555
Поли-3,3-бисхлорметилпропиленоксид
33
Полибутен-1 46, 49 ел.
Полибутилентерефталат 296, 349 ел.
Поливинилиденфторид 112 ел., 126 ел
Поливинилфторид 95 ел., 109 ел.
Поливинилциклогексан 43, 85 ел.,
93 ел.
Полигексаметиленадипамид 391
Полигексаметиленизофталамид 387
Полигексаметилентерефталамид 390.
391
Полигексафтор-/г-ксилилен 178
Полигидантоины 470, 471, 483 ел.
Полигидразиды 37, 509, 522, 525
полимераналогичные превращения
528
Полнгидразоны 527
Полигидроксиамиды 898
Полигидроксибензил 141
Полигидроксинмидазолин 850
Полигидроксифенилидены 187
Полигидроксиэфиры 198, 233, 237 ел,
Полиизооксазолы 494
1051
Полиизофталамид 387
Поли-2,4-дибромЛ,4-фениленоксид 200
Полидикетопиперазины 559
Поли-2,6-диметил-164-фениленоксид
198, 201, 205, 214, 234
Полидитиагидразиды 549, 551
Полидихлор-л-ксилилен 162, 169, 172,
174
Полидиэтинилбензол 482, 494
Полиизоимиды 675
Полиизоиндолохиназолиндионы 998
Полиизоиндолохиназолины 1039 ел.
Полиизопропилидены 185
Полимидазобензфенантролииы 998,
1029 ел.
Полимидазопирролоиы 38, 692, 999,
1026
Полиимидгидразиды 775
Полиимиды 585, 587, 738 ел., 772 ел.
Полииминобензоксазиноны 981, 982
Полииминогидантоины 486» 488
Полииминоимиды 711
Полииминотриазины 586, 571
Поликарбодиимиды 557
Поликарбонат 142, 217, 218, 223, 225,
226, 243, 244, 263, 269, 271
Поликетазины 190, 191, 197 ел.
Поликетанилы 190, 195 ел.
Поли-л*-ксилиленадипамид 390, 391
Поли-л-ксилилены 34, 141, 161 ел.,
168 ел.
Поликсилилидены 34, 178 сл„ 183 ел.
Поли-я-ксилилиден-я-бензидин 193
Поли-ж-ксилилиден-ж-фениленди-
амин 193
Поли-я-ксилилиден-я-фенилендиамин
193
Полималеимидоэфиры 687
«Полимер 380» 265
«Полимер 360» 261, 262, 264, 267, 272
Полимерные композиции 128 ел.
Полиметилендифенилоксид 141, 235 ел.
Поли-4-метилпентен-1 66 ел.
Полиметилфенилен 142
Поли-2-метил-1,4-фениленоксид 201
Полиметилхиназолоиы 968, 971
Полиметоксиамиды 898
Полииитрофенилен 142
Полиоксадиазолимиды 772, 774
Полиоксадиазолы 470, 525 ел.
Полиоксазолидены 494, 496
Полиоксазолы 493
Полиоксамидразон 558
Полиоксатиагидразиды 549, 551
Полиоксиамиды 385, 389
Поли-я-оксибензойная кислота 296,
342, 343 ел.
Полиоксидиазол 37
Полиоктаметилендибензимидазол 896
Полиолефины 46 ел., 1049
Полиперфторфенилен 143, 144
Полипиразины 559
1052
Полипиразолы 470
Полипиромеллитимид 42, 708, 719
Полипирронимиды 1022
Полиприсоединение 690
Полисопряженные системы 964, 990,
998, 1023
Полистирилтриазины 568
Полистирол 90, 227, 242, 263
Полисульфидимиды 588
Полисульфиды 926
Полисульфидоэфир 285
Полисульфоимиды 835
Полисульфонамиды 427
Полисульфонаты 350 ел.
Полисульфоноксиды 198, 245, 248
Полисульфоны 141, 177
Политетразапиреиы 998, 1044
Политетразолы 555
Политетраметиленфенилентиазол 506
Политетрафтор-л-ксилилен 177
Политетрафторфенилены 155
Политетрафторэтилен 501, 590, 815
Поли-2Д4,6-тетрахлорфеннленоксид
200
Политиадиазолы 470т 549 ел.
Политиазолы 497, 501 ел.
Политриазиноны 571
Политриазины 470, 565 ел.
Политриазолы 470, 508 ел.
Политриметилгексаметилентерефта-
ламид 390, 393
Полиуреидокислоты 981, 982
Полиуреидоэфир 484
Полиуретаноэфиры 991
Поли-л*-фениленадипамид 386
Поли-л(-фенилеиизофталамид 361, 387,
424, 427 ел.
Полифенилеиоксиды 199 ел., 208 ел.,
230 ел.
Полифениленсульфиды 141,286,292 ел.
Полифеиилены 28, 141, 142 ел.
Полифеиилентерефталамид 433
Полифеиилхиноксалииы 933, 958
Полиформальдегид 222, 226, 244
Полифосфонитрилхлорид 31
Полифторолефииы 1050
Полихииазолиндионы 588, 981 ел.
Полихиназолоны 588, 968 ел.
Полихиноксалинамиды 967
Полихиноксалииы 29, 588, 933 ел.,
960 ел.
Полихлор-п-ксилилеи 162 166, 172'
174
Поли-4-хлор-1,4-фениленоксид 200
Полицианамиды 682
Полициклоамиды 361 ел.
Полициклогексадиен-1,3 148
Поли-1,4-циклогексилендиметиленте-
рефталат 43, 296, 347 ел.
Политиффовы основания 190 ел.
«Полиэстер» 349
Полиэтилен 242, 247
Полиэтиленоксибензоат 296, 349 ел.
Полиэтилентерефталат 271, 196
Полиэфироамиды 417, 587, 679, 681,
816 ел., 834 сл„ 900
Полиэфиртриазины 566, 571, 581
Полиэфиры 141, 297, 350
Полулестничный полимер 40, 586, 998
Порообразователи 57
Прессование
поливинилиденфторида 121
поли-4-метнлпентена-1 75
полисульфонов 265
Препреги 964
Привитая сополнмеризация 65, 83,
111
«Принцип многократных связей» 33
Прозрачность 78, 104, 117
Простые ароматические полиэфиры
198 ел., 201
Прочность
полиамидов 393, 424, 432
полиамидоимидов 814, 815
полибензилов 188
полибутена-1 60, 64
поливинилиденфторида 113, 119,
299
поливинилфторида 105
поливинилхлорида 105
полиимидов 700, 702, 743
поли-/г-ксилилена 173
поли-4-метилпентена-1 77
полипропилена 77
полистирола 77
полисульфонов 268
полифениленоксидов 225
полифениленсульфидов 299
полиэтилентерефталата 105
«Псевдосопряженные полишиффовы
основания» 193
Разветвленные полимеры 287, 293,
899
Райралан 590
Райтон 288, 293
Радиационная стойкость .
полиамидов 429
полиарилеисульфоноксидов 261
полибензоксазолов 924
полибутена-1 66
поливинилиденфторида 118
поливинилфторида 109, 119
полиимндов 722
поли-/г-ксилилеиа 170
поли-4-метилпентена-1 80
полифениленоксидов 216
винилфторида 99
Реакция
Велера 484
Вюрца — Фиттига 144, 163
Виттига 179
Гофмана 970
Дильса — Альдера 149, 687
Реакция
Инга — Манске 717
Кнёвенагеля 180
нуклеофильного замещения 286
обмена 192, 193, 232
переэтерификации 297
сочетания 143
Шиффа 191, 192
Шоттен — Баумана 298
Штрекера 485
Ульмана 143, 199, 200
Фриделя — Крафтса 184—187, 248
«Резистерм» 487
«Резистофол» 493
Родефтал 731, 733, 799, 812
«Родок» 206
Рост цепи 146, 287
Ротационное литье 57
Сварка 57, 221
Связующее 493, 581
пирроновое 1026
полиамидоимидное 813
полибензимидазольное 894
полимидное 733
полихиназолиндионовое 981, 985
полихинооксалиновое 933, 964
«Селанекс» 350
Скайбонд 590, 733, 746
Склеивание 221, 242, 1028
Совместимость 128
Сополимеризация 65, 83, 111
тройная 84, 112
Сополимеры
амидов 416, 422, 434 ел.
бутена-1 65, 66
винилиденфторида 130, 131
винилфторида 112
винилциклогексана 89
с гетероциклами 772 ел.
2,б-диметил-1,4-фениленоксида
206, 215, 217 ел.
2,6-диметилфеиола 231
/z-ксилилена 176
4-метилпентена-1 83, 84
привитые 231, 233
Сопряженные полиазометниы 190
Спекание 157
Стабилизаторы 80, 102
Стеклопластики 964, 966, 1027
«Сульфон Т» 430
Сшивание 29, 119, 216, 261, 292, 711,
899, 900, 924, 957
Тактичность 69
Твердофазная поликонденсация 679
Теломеры 192
Температура нулевой прочности 44
Температура плавления 24, 25 ел,,
29
полиамидов 362 ел., 386, 387, 396,
418, 421, 436
йолиамидогидразидов 466
1053
Температура плавления
полиамидоимидов 782 ел., 808,
822 ел.
полиангидридов 356, 358 ел.
полибензоксазнидионов 992
полнбутеиа-1 59
полибутилентерефталата 349
поливинилфторида 101, 104, 114,
122
поливинцлциклогексаиа 94
полиимидов 594, 693, 748 ел.
поликсилилиденов 183
поли-4-метилпентена-1 76
полиоксадиазолов 531, 532 ел.
полисульфидов 285, 286, 288, 289
полисульфонатов 351, 352 ел.
политетрафтор-п-ксилилена 178
политиадиазолов 552 ел.
политиазолов 501, 502
политриазинов 572 ел.
политриазолов 510
полифеиилена 153
полифениленоксидов 208
полихиназолонов 972 ел.
Температура разложения 36, 37, 42
пирроиов 1000 ел.
полиамидов 362 ел., 396, 420, 421,
436
полиамидоимндов 782 ел., 822 ел.
полибензимидазолов 852 ел.
полибензоксазиионов 988
полибензоксазолов 902 ел.
полибентиазолов 930 ел.
полидикетопиперазинов 560 ел.
полиизоиндохиназолины 1042
полиимидазобензфенаитролинов
1030 ел.
полиимидов 594, 749 ел.
полипиразолов 470, 472
полисульфоимидов 838
полисульфоиатов 351, 352 ел.
политетразапиреиов 1046
политетразолов 556
политиадиазолов 552 ел.
политиазолов 501, 502
политриазииов 572 ел.
политриазолов 510
полихиноксалинов 934 ел.
Температура размягчения 24
определение 44
пирронов 1000 ел., 1023
полиамидоимидов 782 ел., 808,
822 ел.
полиарилатов 300, 302 ел.
полибензилов 185, 187
полибензимидазолов 852 ел.
полибензоксазолов 902 ел.
полиимидов 594, 693, 748
полиоксадиазолов 531, 532 ел.
голиоксазолидонов 496, 498
полипиразолов 470, 472
полисульфонатов 351, 352 ел.
1054
Температура размягчения
политиазолов 501, 502
полифениленоксидов 209
полихиназолоиов 971, 972 ел.
Температура стеклования 30, 41,
302 ел.
определение 44
полиамидов 362 ел., 386, 395, 418,
421
полиангидридов 358 ел.
полиарилатов 300, 302 ел.
полиариленсульфоноксидов 250
полибензтиазолов 930 ел.
полибутилентерефталата 350
поливинилиденфторида 122, 129
полнвиннлциклогексана 94
полигидроксиэфиров 238, 239,
241, 243
полиимидов 687, 688
поли-4-метилпеитена-1 76
полисульфидов 288
полисульфоноксидов 252 ел.
политриазииов 572 ел,
политриазолов 510
полифенилеиоксидов 208, 211,223
полихииоксалинов 934 ел., 960
полихиназолоиов 971, 972 ел.
«Теиайт» 348, 350
Теплостойкость 24, 41
по Вика 44
деформационная 44
по Мартенсу 44
Теплофизические свойства 104
полиамидов 391
полиариленсульфоноксидов 266,
267
полибутена-1 58
поливииилиденфторида 122
поливинилциклогексана 94
поли-/г-ксилиленов 172
поли-4-метилпентена-1 76
полифениленоксидов 223
полифеииленсульфидов 295
Теребек 834
«Терлон» 434
Термостойкость 24, 31
гетероциклических полимеров 469
длительные испытания 45 ел.
методы определения 40 ел.
пирронов 1024
полиамидов 429
полиамидоимидов 808
полиариленсульфонокендов 257
полибензимидазолов 881
полибензоксазолов 919
поливинидиденфторида 118, 124
поливинилфторида 95, 108
поливииилциклогексаиа 91
полиизоиидолхиназолиидионов
1044
полиимидазобензфенантролинов
1036
Термостойкость
полиимидов 686, 708
поли-л-ксилилеиов 170, 174
поли-4-метилпеитена-1 80
полиоксадиазолов 544, 546
политиадиазолов 551
полифспилснов 154
полифениленоксидов 213
полифениленсульфидов 290
полихиноксалинов 961
полишиффовых оснований 194
Терполимеры 84, 112
Терфенил 155, 157
Торлон 799
«Трогамид» 390, 391
«Ультрадур» 350
Уравнение Марка — Хаувинка — Куна
55, 456
Усадка 64, 73, 74, 120, 220, 224
Фактор сварки 57
«Фенилон» 427, 428, 430
Феногарант 799, 812
«Фенокси» 237
Физико-механические свойства
полиамидов 391
полнамидогидразидов 459, 460
полиамидоимидов 814, 815
полна риленсульфоноксидов 266,
267
полибензоксазиндионов 992
полибензоксалов 901, 924
полибензтиазолов 929 ел.
полибутена-1 59
поливинилиденфторида 123
поливинилфторида 105
поливинилциклогексаиа 94
полигидроксиэфира 243, 244
полиимидов 693, 694 ел., 726
поли-л-ксилилена 173
поли-4-метилпентена-! 77 ел.
полифениленоксидов 224 ел.
полифениленсульфидов 294 ел.
полихиназолонов 980
полихиноксалинов 965 ел.
Формование
поливинилиденфторида 122
поливинилфторида 102
поливинилциклогексаиа 93
полиимидов 725
поли-4-метилпентена-1 75, 81
полисульфонатов 355
политиадиазолов 555
политиазолов 506
политриазолов 523
полифениленсульфида 292
поли-1,4-циклогексилендиметилен-
терефталата 348
полиэтиленоксибензоата 349
фотопроводимость 184
Фторсодержащие полиолефины 46 ел.
Химическая стойкость
пирронов 1023, 1028
полиариленсульфоноксидов 271
полибензимидазрлов 890
полибензоксазипдиопов 992
полибензоксазипопов 991
полибепзоксазолов 919
поливинилиденфторида 126
поливинилфторида 107
полигидроксиэфира 244
полиизоинл^лхнназолиндионов
1044
полиимидов 715, 716, 721, 728,
743
поли-/г-ксилиленов 174
поли-4-метилпентена-1 79
политетразапиренов 1045
полифениленоксидов 228
полихиназолиндиоиов 985
полихиноксалинов 960
Хладотекучесть 225
«Хостадур» 350
Цвитер-ионы 1021
Циклизация 34, 39, 147, 675, 678,
680—682, 704, 717, 775, 816,
873, 876, 900, 927, 958, 981,
987, 1021, 1040.
Циклодегидратация 593, 675, 677,
678, 929, 987, 1022, 1028
Циклополикоиденсация 527, 566, 586,
998
Циклополимеризация 152
Шиффовы основания 684, 851
«Эймак 221» 157
«Эконол» 345
Эксплуатационные свойства
полиамидогидразидов 457
полиариленсульфоноксидов 266
полибензилов 188
поливинилиденфторида 122 ел.
поливииилфторида 104 ел.
поли-л-ксилиленов 172
поли-4-метилпеитена-! 76 ел.
полиоксазолиденов 497
цолиоксибензойной кислоты 346
полисульфонатов 355
полисульфоноксидов 248
политриазолов 523
политриазинов 581
полифенилена 157 ел.
полифениленоксидов 223
полифениленсульфидов 293
Экструзия
полиарилеисульфоноксидов 262
полибутена-1 56, 64
поливинилденфторида 119
поливинилфторида 103
1055
Экструзия
полигидроксиэфиров 238, 241
поли-2,6-диметил-1,4-фенилено-
ксид 217
поли-4-метилеиа-1 74
полифенилеисульфидов 292
Эластомер 3700 131
Электреты 127
Электрические свойства
полиамидов 392, 393
полиамидогидразидов 461
полиамидоимидов 815
полибеизоксазолов 925
поливинилиденфторида 124
поливииилфторида 106
поливинилциклогексана 94
полигидроксиэфира 244
Электрические свойства к
полиимидов 736, 740
поли-л-ксилиленов 174
поли-4-метилпентена-1 79
полисульфонов 270, 271
политетразапиренов 1045
политетрафторэтилена 79
полифеииленоксидов 226 ел.
полифеииленсульфидов 294
полиэтилена 79
Электролитическая полимеризации
152
Электропроводность 184
«Энкатерм» 559
Эффект полиэлектролитиый 704; 7f
«Юкардел Р 4174» 246
Конрад-Ульрих Бюллер
ТЕПЛО- И ТЕРМОСТОЙКИЕ
ПОЛИМЕРЫ
Редактор А. А. Рогайлина
Художник Е. В. Бекетов
Художественный редактор К. К- Федоров
Технические редакторы Л* А* Молодцова, О* В. Тюрина,
С* Ю. Титова
Корректор Т* В. Смирнова
ИБ
1294
Сдано в наб. 09.0L84. Подп„ в печ. 10.07.84. Формат бумаги
60><90Yie. Бумага Кн. журн. Гарнитура литературная. Печать
высока*. Усл. печ. л. 66* Усл. кр* ютт. 66. Уч.-изд. л. 72,55.
Тираж 4500 зкз. Заказ № 18. Цена 5 р. 10 к. Изд. № 2403.
Ордена „Знак Почета*1 издательство „Химия",
107076» Москва. Стромынка» 21/2.
Ленинградская типография № 2 головное предприятие ордена
Трудового Красного Знамени Ленинградского объединения
«Техническая книга» им. Евгении Соколовой Союзполиграфпрома
при Государственном комитете СССР по делам издательств,
полиграфии а книжной торговли. 198052, г. Ленинград, Л-52,
Измайловский проспект, 29.