/





















































































































































































































































































































































































































































































Author: Глушко В.П.
Tags: тепло термодинамика химия физика математическая физика издательство наука химическая термодинамика
Year: 1981
Similar




Text
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ВЫСОКИХ ТЕМПЕРАТУР
ГОСУДАРСТВЕННЫЙ ИНСТИТУТ ПРИКЛАДНОЙ ХИМИИ
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
СПРАВОЧНОЕ ИЗДАНИЕ
В ЧЕТЫРЕХ ТОМАХ
Издание третье, переработанное и расширенное
Согласовано с Государственной службой
стандартных справочных данных
РЕДАКЦИОННАЯ КОЛЛЕГИЯ:
В. П. ГЛУШКО (ответственный редактор),
Л.-З^ГУРВИЧ (зам. ответственного редактора),
Г. А. 6ЕРГМАН, И. В. ВЕЙЦ, В.А.МЕДВЕДЕВ,
Г. А. ХАЧКУРУЗОВ, В. С. ЮНГМАН v
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1981
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
Том III
ЭЛЕМЕНТЫ
В, Al, Ga, In, Tl, Be, Mg, Ca, Sr, Ba
И ИХ СОЕДИНЕНИЯ
Книга 1
ВЫЧИСЛЕНИЕ
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1981
УДК 536+541Л
Термодинамические свойства индивидуальных веществ. Справочное издание в 4-х т.
/Л. В. Гурвич, И. В. Вейц, В. А. Медведев и др. — 3-е изд., перераб. и расширен.—
Т. III. Кн. 1 —М., Наука, 1981.—472 с.
Издание состоит из четырех томов, каждый том — из двух книг: в первой описано вычисление
таблиц термодинамических свойств, во второй книге помещены соответствующие таблицы.
В первой книге III тома изложены результаты критического выбора постоянных
(молекулярных констант, энтальпий образования и фазовых переходов, теплоемкости, энергий диссоциации
и т. п.), необходимых для расчета термодинамических свойств соединений В, Al, Ga, In, Tl, Be,
Mg, Ca, Sr и Ва, описаны расчеты таблиц термодинамических свойств и дана оценка их
достоверности для каждого вещества. Для ряда газов рассмотрены свойства при повышенных давлениях.
Л. В. ГУРВИЧ
И. В. ВЕЙЦ
В.А.МЕДВЕДЕВ
В. С. ЮНГМАН
Г. А. БЕРГМАН
В.Ф.БАЙБУЗ
В. С. ИОРИШ
А.В.ГУСАРОВ
АВТОРЫ
С. И. ГОРБОВ
И. И. НАЗАРЕНКО
В. Я. ЛЕОНИДОВ
В. Г. РЯБОВА
О.В.ДОРОФЕЕВА
Е. Л. ОСИНА
М.Е.ЕФИМОВ
Е.А. ШЕНЯВСКАЯ
П. И. ТОЛМАЧ
М.Ф. МОСКОВСКАЯ
Ю. С. ХОДЕЕВ
В. Ю. ЗИЦЕРМАН
Л. Н. ГОРОХОВ
Н. Э. ХАНДАМИРОВА
И. В. СИДОРОВА
М. С. ДЕМИДОВА
20503-345
055 @2)-81
Подписное издание 2602040600
© Издательство «Наука», 1981 г.
Эта книга не может быть полностью или частично
воспроизведена или размножена, введена в
информационно-поисковую систему или передана
но линиям связи в любой форме или любыми
средствами (в том числе электронными
устройствами и на магнитных носителях информации)
без письменного разрешения издательства
«Наука».
ОГЛАВЛЕНИЕ
Предисловие к III тому 6
Глава 20. Бор и его соединения 8
Глава 21. Алюминий и его соединения 92
Глава 22. Галлий и его соединения 169
Глава 23. Индий и его соединения . 189
Глава 24. Таллий и его соединения 211
Глава 25. Бериллий и его соединения 230
Глава 26. Магний и его соединения 266
Глава 27. Кальций и его соединения 301
Глава 28. Стронций и его соединения 341
Глава 29. Барий и его соединения 379
ПРИЛОЖЕНИЯ
Приложение 3. Термодинамические свойства газов при повышенном
давлении 420
Приложение 5. Термодинамические свойства водных ионов в
стандартном состоянии 423
Литература ....,,. W\
ПРЕДИСЛОВИЕ К III ТОМУ
В III томе настоящего издания рассматриваются термодинамические свойства 10 элементов (В,
Al, Ga, In, Tl, Be, Mg, Ca, Sr и Ва) и их соединений с кислородом, водородом, галогенами,
азотом и серой. Галлий, индий и таллий впервые включены в справочник, семь других элементов
представлены существенно большим числом соединений по сравнению с предыдущими изданиями.
Всего в III томе рассматриваются 297 веществ. Во второй книге тома приведены таблицы
термодинамических свойств 93 веществ в конденсированном состоянии и таблицы
термодинамических свойств 273 газов в стандартном состоянии. Приблизительно для 80 газов и веществ в
конденсированном состоянии таблицы термодинамических свойств публикуются впервые.
Материалы первой книги III тома изложены в 10 главах, каждая из которых содержит
сведения о соединениях одного элемента. Расположение материалов в главах первой книги и
соответствующих таблиц во второй книге третьего тома следует стандартному термохимическому
порядку (см. рис. 1 в первой книге первого тома). Помимо десяти глав, первая книга
настоящего тома содержит два Приложения. В одном из них, являющемся продолжением Приложения 3,
включенного в два первых тома справочника, содержатся данные, позволяющие учесть
отклонения термодинамических свойств ряда газов при высоких давлениях от их свойств в стандартном
состоянии. В Приложении 5 представлены вспомогательные данные о термохимических свойствах
некоторых ионов в водных растворах, использованные при подготовке третьего тома справочника.
В текстах глав и Приложений первой книги III тома нумерация таблиц, рисунков и
уравнений поглавная, так же как в двух первых томах; нумерация таблиц термодинамических свойств,
включенных во вторую книгу тома, сплошная и является продолжением нумерации таблиц
вторых книг I и II томов.
Во второй книге III тома, помимо ее основной части, включающей 343 таблицы
термодинамических свойств, имеется Дополнение, в котором представлены 19 таблиц термодинамических
свойств веществ в конденсированном состоянии, а также 4 таблицы свойств газов. В Дополнение,
в частности, включены таблицы свойств неравновесных модификаций кристаллических веществ
и веществ в стеклообразном состоянии. Кроме того, в Дополнении приведены две таблицы с ви-
риальными коэффициентами и критическими постоянными для 21 вещества, рассматриваемых
в III томе.
При расчетах таблиц термодинамических свойств использовались методики, изложенные в
предыдущих томах, главным образом в трех методических главах тома I. В текстах глав третьего тома
даны ссылки на соответствующие уравнения или разделы этих методических глав, использованные
при обработке экспериментальных данных, оценках постоянных и расчетах таблиц
термодинамических свойств.
Подготовка материалов III тома в основном была завершена к середине 1978 г., при этом была
использована литература, опубликованная по 1977 г. включительно. Более поздние публикации
учитывались, как правило, в тех случаях, когда полученные в них данные могли позволить
уточнить расчеты соответствующих таблиц термодинамических свойств.
Основная авторская работа по III тому выполнена Л. В. Гурвичем, И. В. Вейц, В. А.
Медведевым, В. С. Юнгманом и Г. А. Бергманом. Кроме того, выбор молекулярных постоянных и
подготовку соответствующих текстов проводили В. G. Иориш, И. И. Назаренко, О. В.
Дорофеева, В. Г. Рябова, Е. Л. Осина, Е. А. Шенявская при участии С. М. Толмачева, В. В. Ка-
спарова, Ю. М. Ефремова, расчеты термодинамических функций газов — В. С. Иориш,
С. И. Горбов, И. И. Назаренко, О. В. Дорофеева, Е. Л. Осина, М. С. Демидова, выбор
постоянных и расчеты термодинамических свойств веществ в конденсированном состоянии —
П. И. Толмач при участии А. Я. Якобсона и В. Н. Вдовина, выбор термохимических
величин— В. Я. Леонидов, А. В. Гусаров, М. Е. Ефимов, М. Ф. Московская, В. Г. Рябова,
Е. А. Шенявская, Ю. G. Ходеев, Л. Н. Горохов, Н. Э. Хандамирова, И.В.Сидорова приучастии
6
Предисловие к Ш тому
A. Г. Ефимовой, О. М. Гайсинской и А. М. Емельянова. Приложение 3 подготовлено
B. Ф. Байбуз и В. Ю. Зицерманом при участии В. В. Гоголевой, Приложение 5 — В. А.
Медведевым.
Основная техническая работа, связанная с проведением расчетов, подготовкой материалов,
оформлением рукописи, составлением списка литературы, выполнена Е. С. Маевской, А. И.
Варшавской, В. С. Шмелевой, И. Г. Байбуз, Н. Р. Симагиной, С. А. Равинской.
Общее редактирование тома проведено В. П. Глушко и Л. В. Гурвичем, глав 20, 22, 25—29
и второй книги — В. С. Юнгманом, разделов по молекулярным постоянным и глав 21, 23, 24 —
И. В. Вейц, разделов по термохимическим постоянным — В. А. Медведевым. Предварительное
редактирование разделов по термодинамическим свойствам веществ в конденсированном
состоянии — Г. А. Бергманом.
При подготовке материалов третьего тома ряд вопросов был обсужден с ведущими советскими
ж зарубежными специалистами. Так, выбор значений ключевых термохимических величин для
соединений бора, алюминия, бериллия, магния, кальция, стронция и бария был выполнен в
рамках деятельности соответствующей Рабочей группы КОДАТА—MGHG. Дискуссии с членами этой
группы, в первую очередь с докторами Д. Д. Уагманом и В. Эвансом (НЁС США) и профессором
Ж. Дровартом (Бельгия), а также с докторами И. Л. Ходаковским (ГЕОХИ АН СССР), М. Чей-
зом (группа JANAF) и Д. Хилденбрандом (Стэнфордский исследовательский институт)
способствовали подготовке этого тома. Редколлегия признательна также профессорам А. А.
Мальцеву и В. П. Спиридонову, кандидатам химических наук Е. 3. Засорину, Н. Ф. Степанову и
А. И. Дементьеву (Химфак МГУ) и другим советским и зарубежным ученым за информацию
о результатах выполненных ими с сотрудниками экспериментальных и теоретических
исследований. Особой благодарности заслуживает эффективная помощь, оказанная авторскому
коллективу при подготовке этого тома Отделом вычислительной техники ИВТАН (руководитель
Г. П. Малюжонок).
Глава 20
БОР И ЕГО СОЕДИНЕНИЯ
В (к, ж), В (аморф.), В, В+, В2, ВО, ВО", ВО2, ВОг, В2О, В3О2, В2О3 (к, ж), В2О3 (стекл.), В,О3,
ВН, ВН2, ВН3, В2Нв, НВО, ВОН, НВО2 (к, ж), НВО2, НВОН, В (ОНJ, Н„ВОН, НВ (ОНJ,
Н3ВО3 (к, ж), Н3ВО3, В2(ОНL, Н3В3О3, H3B3Oe, BF, BF2, BF2, BF3, BF4, B2F4, FBO, F2BO, F3B3O3,
BHF, BH,F, BHF2, FBOH, FB (OHJ, F2BOH, BCI, BC12, BCI3, B2CI4, C1BO, C12BO, C13B3O3, BHC1,
BH2G1, BHC12, C1BOH, C1B (OHJ, С12ВОН, BFCI, BF2CI, BFC12, F9GIB3O3, FCI2B3O3, BHFC1, BBr,
BBr2, BBr3, BI, BI2, BI3, BS, BS2, B2S, B2S2, B2S3 (к, ж), B2S3, BN (к, ж), BN, BH3NHS, B3N3H6,
ВС, ВС2, В2С, В4С (к)
В настоящей главе рассматриваются термодинамические свойства 77 веществ — бора и его
соединений с кислородом, водородом, галоидами, серой, азотом и углеродом. Для семи веществ в
справочнике приведены свойства в конденсированном состоянии, для остальных веществ — в
газообразном состоянии. Для элементарного бора и окиси бора, помимо их свойств в кристаллическом
состоянии, представлены свойства в аморфном и стеклообразном состояниях. Термодинамические
свойства четырех газов (В, В+, ВО, ВО2) рассчитаны до 10000К, остальных — до 6000 К. Для
шести газов в Приложении 3 рассмотрены данные, учитывающие отклонения их свойств при
повышенных давлениях от свойств в стандартном состоянии.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов бора
(Вл=Ю,81) и других элементов. Благодаря тому, что атомные массы изотопов 10В и Х1В существенно
отличаются друг от друга и содержание менее распространенного изотопа 10В значительно,
различие молекулярных постоянных изотопических модификаций соединений бора существенно
влияет на термодинамические свойства природной смеси изотопов. Поэтому во всех случаях, когда
молекулярные постоянные были известны для разных модификаций или их различие могло быть
оценено, а учет этих различий позволял уточнить расчет термодинамических функций,
соответствующие данные принимались во внимание в расчетах, что отмечено в конкретных текстах.
В отличие от таблиц JANAF [1543, 1544] в справочнике не рассматриваются многие известные
положительно заряженные ионы, типа ВО+, НВО + и BFJ. Потенциалы ионизации
соответствующих нейтральных молекул высоки, и образованием таких ионов в равновесных условиях при
температурах до 6000 К можно несомненно пренебречь.
В (к, ж). Термодинамические свойства кристаллического и жидкого бора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 530.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 20.1. За стандартное состояние бора в интервале от Т=0 до Тт принята, как и в других
справочниках, р-ромбоэдрическая (высокотемпературная) модификация 1. При Т < 298,15 К
термодинамические функции вычислены по результатам измерений теплоемкости Р-бора
(чистотой 99,9%) в работе Богданова и др. [40] A6—280 К) и кристаллического бора с неидентифици-
рованной структурой в работе Джонстона и др. [1590] A7—308 К). Для Т <С 50 К приняты дан_
ные [40], а в интервале 50—280 К проведено усреднение данных [40] и [1590]; точность
выбранных значений теплоемкости бора при Т <^ 100 К оценивается в 10%, а при Т ^> 100 К —-в 1%2_
Погрешности принятых значений S° B98,15 К) и П° B98,15 К)—11° @), совпадающих с рекомен.
1 Литературные данные о полиморфизме бора неполны и противоречивы (описано более 10 полиморфных
модификаций [366]). Образование некоторых модификаций связано, по-видимому, с термической предысторией
образцов, дефицитом атомов В в решетке [2408], а также с наличием примесей [2857]. Согласно [2408]
«-ромбоэдрический бор, устойчивый при сравнительно низких температурах, при нагревании до ~1643 К превращается
в промежуточную ^'-модификацию, при —1863 К — в C"-модификацию, а при 1913 К — в||3-ромбоэдрическую
модификацию (энтальпии превращений неизвестны). Образующаяся при кристаллизации жидкого бора р-ромбо-
эдрическая модификация легко закаливается до низких температур; термодинамические свойства этой
модификации определены достаточно надежно.
2 Расхождения между теплоемкостями бора, определенными в [40] и [1590], велики и достигают 30% при Т <
<50 К и 2—5% при Т > 50 К. Близость значений S° B98,15 К) и Н° B98,15 К)—Н° @), вычисленных по данным
[40] и [1590], объясняется случайным совпадением (кривые теплоемкости по данным этих работ несколько рая
пересекаются).
Бор и его соединения
В (к, ж)
Т а б л и
Ц
Вещество
В
В
В2Оз
В203
НВО2
НзВОз
B2S3
BN
В4С
a q° /у\—
= 10,694.
а 20.1.
Принятые значения
в твердом
Состояние
к1, гекс.
ж
аморф.
к1, гекс.
ж
стекл.
к1, куб.
ж
к, трикл.
ж
К, MOHOKJ
ж
к, гекс.
к, гекс.
ж
к, гекс.
а+ЪТ—сг1
Ю
оо-
го ^
кДжХ
Хмоль
Ш 1,222
—
1,318
9,301
—
10,30
Т) 8,46
13,393
¦ —
I. 17,2
—
2,628
—
—
5,611
r-2+dT2+eT3
термодинамических величии
и жидком
к
ю
со"
го
о
Дж • К-1 •
5,90
—
6,53
53,97
—
80,5
49,0
—
88,743
—
100
—
14,81
—
—
27,11
(в Дж-К
состояниях
?Г
оо"
го
¦ CS1
о а.
МОЛЬ"
11,09
—
11,95
62,76
—
62,97
54,71
—
81,34
—
111,72
—
19,71
—
—
53,09
¦МОЛЬ
Коэффициенты
НИИ
а
14,924
31,4
16,050
64,141
127,047
22,309
37,24
105
65,982
180
98,95
146
18,732
51,635
67
105,922
для с;
Ь-103
16,171
—
10,013
64,643
—
154,440
58,58
—
109,620
—
72,43
—
38,886
—
—
0,507
[ для бора и его соединении
в уравне-
(Г)а
С • Ю-6
7,208 б
—
6,296
18,359
31,38
4,786
—.
—
15,397
—
7,841
—
8,456 в
68,731
—
47,944 г
). б <М0в=—6,496, е-109=1
Интервал
температуры
К
298,15—2348
2348—6000
298,15—2000
298,15-723
723—3200
298,15-723
298,15—509
509—1500
298,15-444,1
444,1-800
298,15—840
840—2000
298,15—1200
1200—3240
3240—3500
298,15—2700
или
Т
К
2348
—
—
723
—
—
509
—
444,1
—
840
—
—
3240
—
2700
053. в (М08=—12,464.
\1гН
или
Д""
кДжХ
Хмоль
50,2
—
—
24,56
—
—
14,3
—
22,3
—
48,5
—
—
81
—
—
г<М06=
дованными КОДАТА—МСНС [864], оцениваются в 0,08 Дж^К^-моль и 0,008 кДж-моль
соответственно. В интервале 298,15—2348 К для С (В, к) принято уравнение (см. табл. 20.1),
выведенное совместной обработкой результатов измерений энтальпии бора в работах Мак-До-
налда и Сталла [1991] C33—1668К, примесей в образце не более 0,2%), Уайза и др. [2862] (821 —
1103 К, 2 образца, фазовый состав которых не был достаточно исследован) и Стаута и др. [26241
A820—2218 К, образец содержал 99,96% В). Точность описания данных [1991] и [2624]
составляет 0,4%, а данных [2862] — 1,1%. Температура плавления бора 2348+50 К принята по
результатам измерений Кимпела и Мосса [1708], которые учли влияние примесей на температуру
плавления. С принятым значением в пределах погрешности согласуются также результаты определений
[1458] B365 К), [2624] B343 К), [1940] B315+20 К) и [911, 912] B270-2350 К). Определенные
ранее более высокие значения в работах [2486] (>2420 К), [859] B423 К) и [334] B453—2543 К)
не подтвердились. ЗцЦчение энтальпии плавления бора, 50,2 + 1,7 кДж-моль, принято по
результатам единственного экспериментального измерения [2624]. Для С° (В, ж) принято значение
31,4 Дж-К~1-моль, полученное оценкой в справочнике [1503]. Результаты оценок [1543] и [2452]
составили 30,5 и 31,4 Дж-К~1-моль~1 соответственно. Погрешности вычисленных значений Ф° (Т)
при 298,15, 1000, 3000 и 6000 К оцениваются в 0,08, 0,2, 1,5 и 5 Дж-К~1-моль~1 соответственно.
Термодинамические функции В (к), приведенные в табл. 530, незначительно отличаются от
вычисленных в справочниках [105, 1503, 1543] (в пределах 0,4 Дж-К-моль в значениях Ф° (Т)
до 2348 К). Для В (ж) соответствующие расхождения увеличиваются и достигают 3,5 и 7 ДжХ
хК~1-моль~1 при 3000 и 5000 К соответственно. Эти расхождения обусловлены в основном тем,
что в справочниках [105, 1503, 1543] приняты оцененные значения энтальпии плавления бора.
В настоящем справочнике за стандартное состояние бора принят кристаллический бор (|3-ром-
боэдрическая модификация) и в соответствии с этим
AfH°(p-B, к)==0.
9
В (аморф.)
Глава 20
Давление пара бора В (к)=В (г) вычислено с использованием значения энтальпии
сублимации бора
Д//°(В, к, 0) —560 + 5 кДж-моль,
принятого на основании данных, приведенных в табл. 20.2. Анализ работ, результаты которых
представлены в таблице, показывает, что наиболее надежные измерения выполнены в работах Аки-
шина и др. [3], Пола и Маргрейва [2198], Робсона и Гиллеса [2353], Хилденбранда и Холла
[1415], Мара и Бедфорда [1941], а также Стормса и Мюллера [2622] (приведенные погрешности
характеризуют воспроизводимость измерений). По данным работы [2622] энтальпия сублимации
бора может быть рассчитана только по уравнению II закона термодинамики. Точность найденной
таким образом величины сравнима с точностью расчета по III закону термодинамики, поскольку
исследование было выполнено в широком интервале температур, и благодаря использованию новой
техники измерений в экспериментах была достигнута высокая точность определения температуры.
Принятая величина является средневзвешенной из указанных работ, ее погрешность учитывает
неточность термодинамических функций В (к).
Таблица 20.2. Результаты определений Д,Я° (В, к, 0) (в кДж-моль)
Авторы
Метод
А,Н° (В, к, 0)
II закон
III закон
Сирей, Майерс, 1957 [2486] Эффузионный, В (к)=В (г), 2115-2413 К, 9 то- 609,3+82 574,9±4,1
чек
Акишин и др., 1959 [3] Масс-спектрометрический, В (к)=В (г), 1765— 515,8+20 568,0+1,4
2233 К, 75 точек
Шиссель, Вильяме, 1959 [2457] Масс-спектрометрический, В (к)=В (г) 540+20а
Приселков и др., 1960 [338] Эффузионный, В (к)=В (г). 1651—1764 К, — 413—521б
24 точки
Ферхаген, Дроварт, 1962 [2765] Масс-спектрометрический,
1. В (к)=В (г), 1760—2440 К, 46 точек 535 в 536 в
2. 0,25В4С (к)=В (г)+0,25С (к) 1730-2400 К, — 531 в
38 точек
Алкок, Гривзон, 1962 [492] Эффузионный, В (к)=В (г), 8 точек 557,5+28 581,4±0,5
Шиссель, Трулсон, 1962 [2456] Масс-спектрометрический, В (к)=В (г), 4 серии 550+20 а —
измерений, Гср= 1962—2121 К
Пол, Маргрейв, 1963 [2198] Испарение с открытой поверхности, В (к)=В (г), 549,2+17 565,3+0,6
1781—2152 К, 11 точек
Робсон, Гиллес, 1964 [2353] . Эффузионный, 0,25В4С (к)=В (г)+0,25С (к), 597+20 556,8+1,1
2184—2522 К, 16 точек
Хилденбранд, Холл, 1964 [1415] Торзионно-эффузионный, 0,25В4С (к)=В (г)+2 554+13 559,2+0,3
+0.25С (к), 2341—2615 К, 43 точки
Map, Бедфорд, 1976 [1941] Торзионно-эффузионный, В (к)=В (г), 2086— 586+19 556,0+0,5
2251 К, 27 точек
Торзионный (испарение с открытой поверхности), 496+42 556,2+0,8
2099—2253 К, 14 точек
Масс-спектрометрический, 1823—2085 К, 15 точек 540,0±5,8 —
Стормс, Мюллер, 1977 [2622] Масс-спектрометрический, В (к)=В (г), 1721— 569,8+0,8 —
2118 К, 136 точек
а Детали эксперимента и расчетов не приведены.
6 Результаты расчета сильно зависят от величины отношения площади испарения к площади эффузионного отв ерстия,
увеличиваясь с уменьшением этого отношения.
в Результаты измерений приведены только в виде графика.
В (аморф.). Термодинамические свойства аморфного бора 1 при температурах 100—2000 К
приведены в табл. Д.41.
1 Состояние аморфного бора следует отличать от состояния гипотетического стекловидного бора. Аморфный бор
характеризуется неупорядоченностью икосаэдрических группировок (В12), которые имеются во всех
модификациях кристаллического бора.
10
Бор и его соединения В (г)
Значения постоянных, принятых для расчета термодинамических функций В (аморф.),
представлены в табл. 20.1. При Т <С 298,15 К расчет проводился по результатам измерений
теплоемкости В (аморф.) в интервале 18—308 К, проведенных Джонстоном и др. [1590] на образце,
приготовленном термическим разложением диборана чистотой 99,8% при 700° С. Кривая С°р (Т) имеет
минимум при —30 К; экстраполяция ниже 18 К приводит к значению S° A8 К)—S° @) =
=0,05 Дж-К^-моль. Погрешности принятых по [1590] значений S° B98,15 К) — S° @) х
и Н°- B98,15 К) — Н° @) оцениваются в 0,2 Дж-К-моль~1 и 0,04 кДж-моль соответственно.
В интервале 2»8,15—2000 К для теплоемкости В (аморф.) принято уравнение (см. табл. 20.1),
выведенное по результатам измерений энтальпии в работе Уайза и др. [2862] E10—1234 К).
Точность значений С° (Т), вычисленных по этому уравнению, оценивается в 1%Цпри 1000 К и 5% при
1500 К. Данные Магнуса и Данца [1909] B98—1174 К) по энтальпии образца В (аморф.) чистотой
97% ненадежны, они лежат на 10% ниже данных [2862] и не принимались во внимание при
вычислении термодинамических функций. Расчет термодинамических функций В (аморф.) проводился
до 2000 К, поскольку при нагревании (в вакууме) до температуры около 2000 К происходит его
переход в кристаллический бор (^-ромбоэдрическую модификацию).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются (без учета
остаточной энтропии) в 0,2, 1 и 3 Дж-К~1-моль; пренебрежение остаточной энтропией
увеличивает погрешность, по-видимому, на 1—2 Дж-К~1-моль~1. Термодинамические функции В (аморф.),
приведенные в табл. Д.41, практически совпадают с рассчитанными в справочнике Халтгрина
[1503] B98-1200 К).
Одной из особенностей термохимии соединений бора является то, что большое число
исследований выполнено с аморфным бором, в особенности с бором, полученным при пиролизе диборана.
Для обеспечения возможности сравнения этих данных с более поздними, которые были получены
при работе с кристаллическим бором, необходима энтальпия образования В (аморф.). В настоящем
справочнике принята энтальпия образования
AfH°(B, аморф., 298,15 К) = 4,4 ± 1,0 кДж • моль.
Это значение было получено сопоставлением результатов измерений энтальпий образования
В2О3 (стекл.), В2Н6 (г), ВС13 (г) и BN (к) из бора в кристаллическом и в аморфном состояниях
(в кДж-моль):
ДуЯ° B98,15 К) д ДО(В
(из кристалличе- 1 298 lif К)*"
ского бора) ' '
—1254,9+1,4 4,6 + 1,3
36,6+2,0 4,3 + 1,8
—404,5+1,3 3,6 + 2,4
—250,5 + 1,5 5,1+2,5
Обсуждение приведенных величин см. в соответствующих текстах; расчет энтальпии
образования аморфного бора четырьмя независимыми путями приводит к хорошо согласующимся
величинам.
В (г). Термодинамические свойства газообразного бора в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 531.
Уровни энергии атома В, использованные в расчете термодинамических функций, приведены
в табл. 20.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
Is22s22p и Is22s2p2. Энергия расщепления основного состояния 2Р принята по результатам
измерений Эдлена и др. [1010], подтвержденным в работе Брауна и др. [770] и уточняющим прежние
данные [1313, 2053]. Энергии подуровней квартетного состояния *Р были измерены впервые Рой-
гом и Тонделло [2357]. Энергии остальных состояний приняты по данным работ [770, 1010, 1232],
практически совпадающими рекомендацией Мур [2053]. Состояния Is22s2p2 2P и 2р3 *S, 2P, 2D
лежат выше ионизационного предела и не включены в таблицу.
В2О
в2н
BCL
BN
Вещество
з (стекл.)
6 (Г)
> (г)
(к)
ДуЯ" B98,15
(из аморфного
—1264,0+2
28,1 + 3
-408,1 + 2
—255,6+2
К)
бора)
,0
,0
,0
,0
1 Термодинамические функции В (аморф.) в настоящем справочнике вычислены без учета остаточной энтропии ,
величина которой неизвестна. Келли и Кинг [1658] оценили 5° (В, аморф., 0) = 1,05±0,40 Дж-К^-моль
без указания метода оценки.
И
В+(г)
Глава 20
Таблица 20.3. Уровни энергии атомов В и В+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Состояние
Ti
СМ
Pi
В Is22s22p 2P1/z 0 2
*Р3/! 15,25 4
Is22s2j2. ¦ 4Р1/з2 28870,0 2
4Р3/* 28875,0 4
4Р5/ 28881,3 6
Is22s2p2 *D ' 47857 10
Is22s2,p2 2S 63560,6 2
Атом
Электронная
конфигурация
Состояние
СМ
Pi
В+ ls-2s2 J5 0 1
Is22s2p 3P0 37336,7 1
аР: 37342,4 3
3Р2 37358,3 5
Is22s2p !P 73396,6 3
Is22;J 3P0 98912,6 1
8Pi 98921,8 3
3Р2 98934,3 5
Термодинамические функции В (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии атома В. Погрешности
вычисленных значений Ф° (Т) во всем интервале температур не превышают 0,02 Дж-К~1-моль
и обусловлены главным образом неточностью принятого значения R; в S° (T) и, в особенности,
в С°(Т) при Т^>8000К погрешности увеличиваются из-за пренебрежения ридберговскими
состояниями и могут достигать 0,1 и 1 Дж-К~1-моль в значениях S0 A0 000 К) и С A0 000 К)
соответственно.
Ранее таблицы термодинамических функций В (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106], в таблицах JANAF [1543] и справочниках [1044, 1493, 1904, 1974],
а также в работах: [1436] до 10 000 К, [1749] до 8000 К, [2855] до 5000 К, [2632] до 3000 К и
[1639, 2838] до 2000 К. Результаты всех расчетов, за исключением [1749, 2632],
удовлетворительно согласуются с вычисленными в настоящем справочнике значениями термодинамических
функций; максимальные расхождения в Ф° (Т) составляют 0,02—0,03 Дж-К~1-моль~1. В случае
расчета [1749] расхождения в Ф° (Т) превышают 0,06 Дж-К~1-моль~1, что обусловлено большим
отличием выбранной величины R.
Энтальпия образования В (г)
A//°;(B, г, 0) = 560 +--5 кДж-моль
соответствует принятой в справочнике величине &3Н° (Bs к, 0) (см. В (к, ж)).
В+(г). Термодинамические свойства газообразного положительного иона бора в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 532.
Уровни энергии иона В+, использованные в расчете термодинамических функций, приведены
в табл. 20.3, в которой даны валентные состояния с энергиями ниже 100000 см, относящиеся
к электронным конфигурациям Is22s2, Is22s2p и Is22/?2. Энергии этих состояний приняты по
результатам спектральных измерений Ольме [2158а], который впервые определил энергии триплетных
состояний (см. [2053]).
Термодинамические функции В+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
значений во всем интервале температур не превышают 0,02 Дж-К-моль~1 и обусловлены главным
образом неточностью принятого значения R.
Ранее таблицы термодинамических функций В+ (г) вычислялись в справочнике [1543] до
6000 К, а также в работах [1436] до 10 000 К и [1246] до 50 000 К. Результаты этих расчетов
удовлетворительно согласуются между собой и с данными настоящего справочника (максимальные
расхождения составляют 0,02—0,03 Дж-К-моль~1).
Константа равновесия реакции В+(г)+е (г) = В (г) вычислена с использованием значения
A,i?0 @) = —800,638 + 0,001 кДж-моль, соответствующего потенциалу ионизации атома В
10 (В)=66 928,1+0,1 см~1=800,638+0,001 кДж-моль,
12
Бор и его соединения
В2(г)
принятому по результатам измерений границы ридберговской серии npF (re=4-J-ll) в
спектре В в работе Эдлена и др. [1010]. В работе [770] получено практически совпадающее
значение. Принятому значению-/0 соответствует энтальпия образования иона В+, равная
+й кДж-моль
В2 (г). Термодинамические свойства газообразного дибора в стандартном состоянии в интервале
температур 100—6000 К приведены в табл. 533.
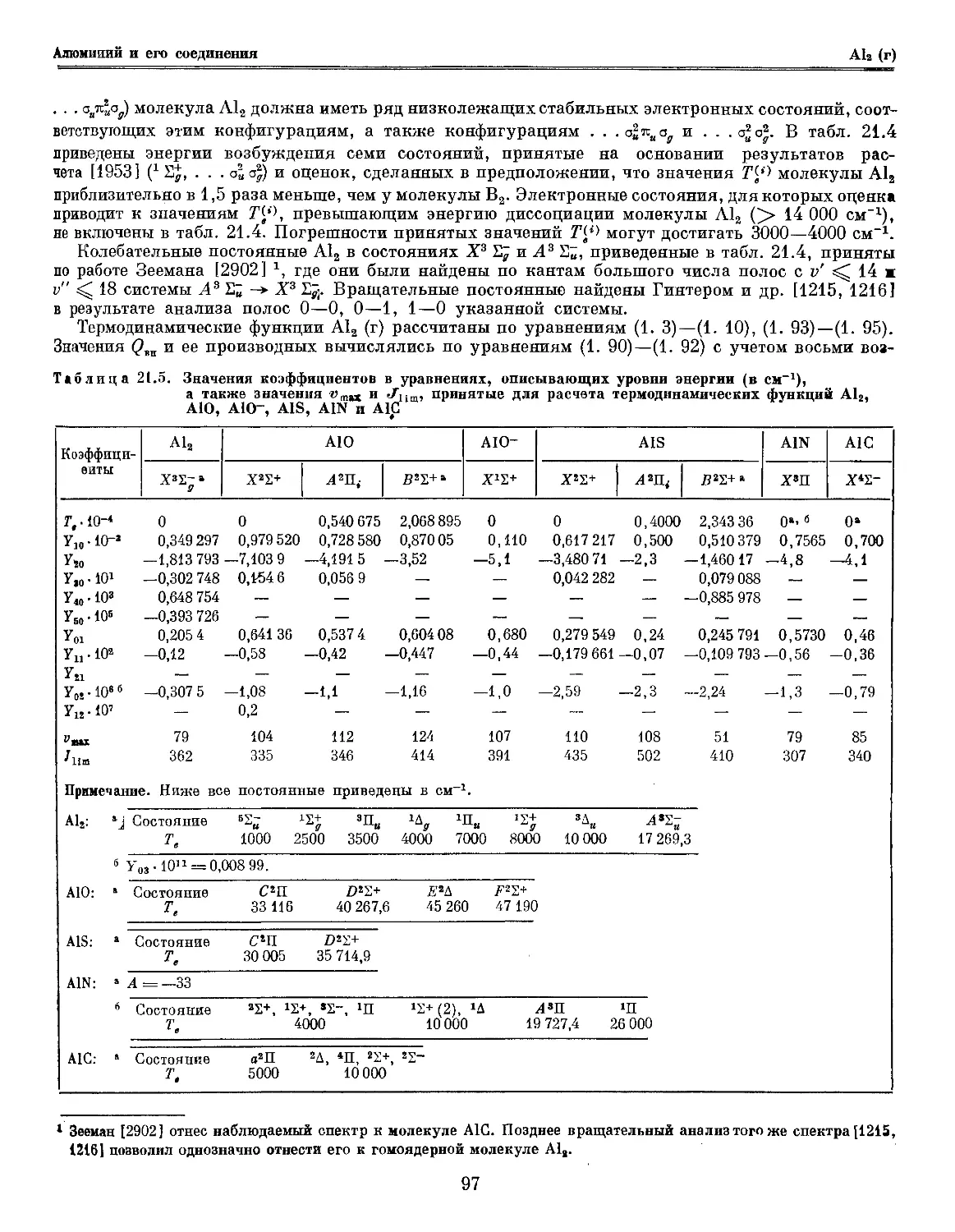
Таблица 23.4. Принятые значения
Молекула
Состояние
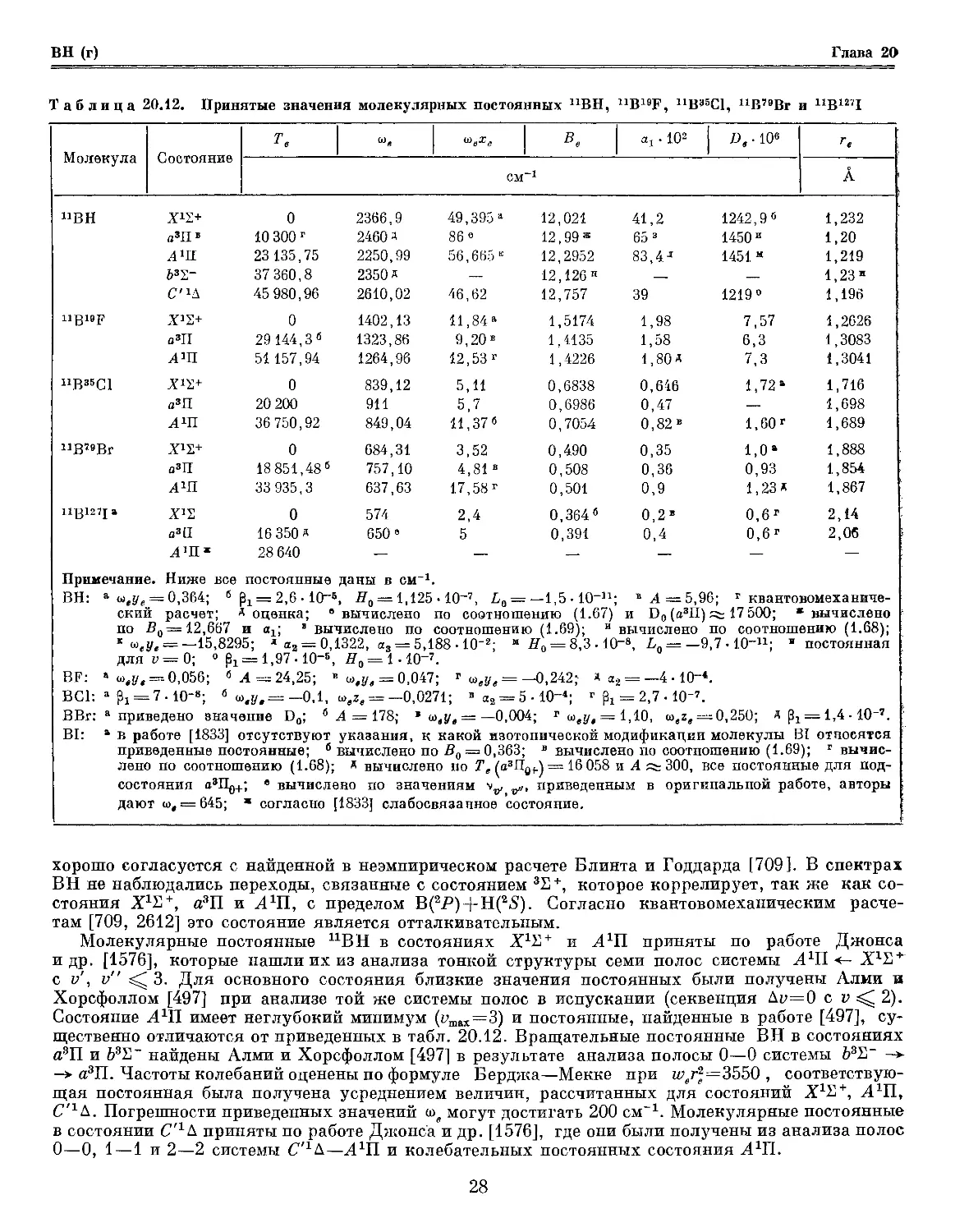
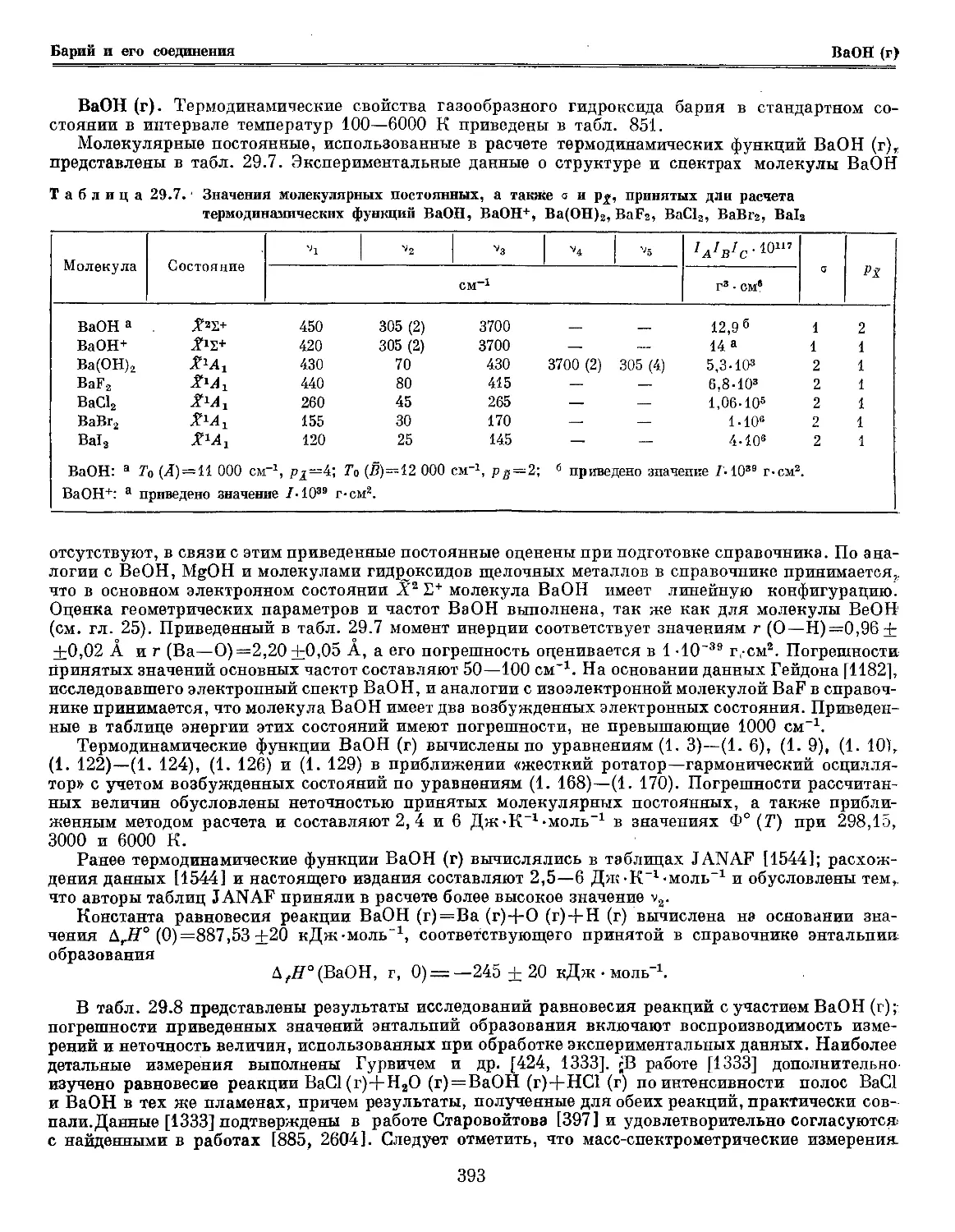
молекулярных постоянных !132, nBlsO
nB2a Х3Ъ~ 0 1051
я52- 1300 1204
АЧ- 15 000 1104
В*2~ 30 573,4 937,
ngi6Q X2S+ 0а 1885
Amt 23 958,85" 1260
В22+ 43174,0г 1281
СгП. 55 346,1 Д 1315
ВО" А'Ч:+ 0 1900 а
nB32S x2T+ 0 1180
АЧ1{ 16 042,5б 753
52Е+ 36 223 770
Cm 38 983,5 892
ngi4]\j ХЧ1-а О6 1514
Cm' 27 850 1317
UB12C X*S-a 0 991
Примечание. Ниже все постоянные даны в
В2: а Молекула имеет еще 17 электронных
Состояние 3ПЯ 1Ду
Те 5000 6000
Состояние Щд ъ^g
Те 30 000 31000
6 вычислено по соотношению A.68).
ВО: а т = 6,5-Ю-3; б ^ = — 2-Ю-8; в А =
ВО-;а Оценка; б вычислено по соотношений:
г вычислено по соотношению A. 69); Д
ш
3 9
9
16
4 2
69 И,
70 И
69 10
,3 11
11
17 6
35 4
4
64 6
6 12
15 14
6
СМ.
,35
8
,0
,6
810
157
66
,1
5
31"
62
0
74
3
9
6б
состояний с
10 000
ъц
33 000
—122,26;
> A.67) и
вычислено
Ве
СМ
1,212
1,28
1,25
1,160
1,7820
1,4132
1,5171
1,484
1,80в
0,79489
0,62291
0,6335
0,7060
1,666
1,555
1,069
энергиями меньше
1S+ 3Д 3S'
г т
Do
по
14 000 19 000
здм в2-
34 000 35 000
= 2,5-Ю-2; Д А =
1
1
1
1
1
1
2
1
1
0
0
0
0
2
1
1
ВО", nBS2S
• 102
,4
,1
,7
,1
66
96
,10
,7
,5Г
605
592
47»
70 е
5
0
0в
40 000:
12-
21 000
38 000
t6,4.
= 79 500; в вычислено по
соотношению A. 68)
BS: а ш^уе==—4-10~3; 6 А = —332,15; в вычислено по соотношению A.69);
A.68).
BN: а Молекула имеет еще 8 электронных состояний
Состояние А3~Е+ аЧ1 j33?~
,Те 4400 4800 5600
6 А = —30.
ВС: а Молекула имеет еще 15 электронных
Состояние а2П &2Д, АЧ1 c2S~, d-
Те 7300 11600 13 500
Состояние г2Д /2?~ С4?~
7200 i
6
5
6
7
6
7
8
8
6
1
1
1
1
8
8
5
11B14N ,
¦10°
,4
,8б
,4б
,0
,326
,09
,5
,3
,5Д
,4
,7
,53
,74 г
,1
,7
г
22 000
3S+
39 000
i ЧВ12С
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
ге
А
,590
,546
,565
,625
,2043
,3524
,3053
,3196
,20а
,6091
,8177
,8026
,7076
,281
,326
,665
26 000
соотношению A. 38);
г вычислено
с энергиями меньше 40000:
с*А
2 000 14 000 24 000
состояний с
18 000
от е
L
2С
Гв 33000 35 000 41000 42 000 47 000
6 вычислено по соотношению A.67) и Do = 37 400
по соотношению A.68).
•
энергиями меньше
?42- /6П, ?22+, /
) 300 28 000
39 000
50 000:
,2]
I
1 вычислено по соотношению
A-
по соотношению
69); г вычислено
13
В2 (г) Глава 20
Молекулярные постоянные ПВ2, использованные для расчета термодинамических функций,
приведены в табл. 20.4. Эти постоянные получены в результате спектральных исследований [974,
1243] и квантовомеханических расчетов [319, 507, 508, 646, 851, 996а, 1106, 1739, 1785, 1864,
2104, 2170, 2525, 2640, 2641, 2740]. На основании анализа электронного спектра поглощения
молекул UB2, изолированных в матрице инертного газа [1243], а также по данным прецизионного
теоретического расчета [996а] принимается, что основное состояние В2 принадлежит к типу
SL~ (конфигурация . . .2d^i^). Согласно квантовомеханическим расчетам [646, 996а] молекула
В2 имеет 42 устойчивых возбужденных состояния с энергиями ниже 60000 см, коррелирующих
с валентными состояниями атомов, однако только состояние B3L~ (конфигурация . . .2ам2^3ог)
наблюдалось экспериментально [974, 1243]. В таблице приведены энергии возбуждения 19 не-
наблюдавшихся состояний с Те < 40000 см по данным расчетов [646, 996а]. Погрешность
значений Те для состояний а5Е~ и А32>~ не превышает 500 см [996а], а для остальных состояний
может достигать 2000—3000 см. Молекулярные постоянные В2 в состояниях Х32~ и \В3?~
получены Дугласом и Герцбергом [974] из колебательного анализа восьми полос секвенций Др=0
и +1 (i/ ^ 4, v' <1 3) и вращательного анализа полос 0—0, 1—0 и 1—1 системы 53S~ -> Х31Г.
Постоянные В2 в состояниях а5И~ и А3Ъ~ приняты по результатам квантовомеханического
расчета [996а]. Погрешности значений ше и Ве в этих состояниях могут достигать 100 и 0,05 см
соответственно.
Термодинамические функции В2 (г) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом 20 возбужденных электронных состояний. Величины Qmx. вр состояний X31~ и а5 Е~ находились
непосредственным суммированием по колебательным уровням энергии и интегрированием по /
с помощью соотношений типа A. 82). В расчете учитывались колебательно-вращательные уровни
со значениями / ^ /шах,»! величины /шаХ;0 находились из условий A. 81). Энергии уровней
вычислялись по уравнениям A. 61) и A. 65) со значениями Ykl, полученными для природной смеси
изотопов 10В и ПВ по формулам A. 66) на основании молекулярных постоянных 11В2 из табл. 20.4,
причем для обеспечения выполнения условий A. 50) и A. 59) в формулу для уровней энергии
добавлены члены с Y30 и Y03. Соответствующие значения Yk!, а также ушах и /цт для состояний
Х3Е~ и а5Е~ приведены в табл. 20.5. Мультиплетность этих состояний учитывалась
статистическими весами 3 и 5 соответственно. Для остальных возбужденных электронных состояний
принималось, что (?й.„ = (лМ <?SbP-
Погрешности вычисленных термодинамических функций В2 (г) обусловлены главным образом
неточностью энергии возбуждения и молекулярных постоянных состояний а5 ?-, а также, при
высоких температурах, отсутствием надежных данных об энергиях других низколежащих состояний
и погрешностью принятого значения энергии диссоциации В2. При температурах до 2000 К
особенно существенны погрешности в значениях С°, составляющие при 298,15 К около 3 Дж-К X
Хмоль. Общие погрешности в значениях Ф° (Т) составляют 1, 3 и 4 Дж-К~1-моль при 298,15,
3000 и 6000 К соответственно.
Ранее термодинамические функции В2 (г) вычислялись в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF [1543], а также в справочниках [977, 1493, 1904, 1974]
и в работах [1072, 2463, 2855]. Во всех расчетах не учитывались низколежащие возбужденные
электронные состояния (за исключением В3!,- в расчетах [105, 106, 1072, 1974, 2463]), что и
обусловило приблизительно одинаковые расхождения данных табл. 533 со всеми перечисленными
расчетами, которые составляют 5 и 10 Дж-К~1-моль~1 в значениях Ф° C000 К) и S° C000 К) и
достигают 9 и 14 Дж-К^-моль в значениях Ф° F000 К) и S0 F000 К).
Константа равновесия реакции В2 (г)=2В (г) вычислена с использованием принятой в
справочнике энергии диссоциации
ЬгН° @) = Do (В2) = 290 ± 15 кДж • моль.
Эта величина основана на результатах масс-спектрометрического изучения равновесия
реакции В (г)+В (к)=В2 (г) при 2330 К в работе Ферхагена и Дроварта [2765]. Ее погрешность
определяется прежде всего неточностью измерения константы равновесия A3 кДж-моль), а также
погрешностями энтальпии сублимации бора и термодинамических функций В2 (г) и В (к).
Принятой энергии диссоциации соответствует
&fH°(B2, г, 0) = 830 + 18 кДж • моль.
14
Бор и его соединения
ВО (г)
Таблица 20.5. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения vms,% и Jim, принятые для расчета термодинамических функций В,,
ВО, ВО-, BS, BN и ВС 2
Коэффициенты
Те'
Ую
У»о
^40
у5о
Уо1
Ун
Уи
Уо2
у12
V
J 03
¦'lim
ю-4 о
ю-3 1,
Ю-1 —0,
101 —0,
103
106
1,
102 —1,
10s —6,
107
1010 0,
!
Е
С«2~
060323
913042
669102
—
—
234346
438896
—
64
.—.
357190
40
L87
Примечание. Ниже все
В„:
ВО:
BS:
BN:
ВС:
а Состояние
Г.
Состояние
^е
а Состояние
а Состояние
Те
а ^ = _30;
6 Состояние
те
а Состояние
те
Состояние
Те
Зпм
5000
Зп„
26 000
!2
-*•
*
0,1300 0
1,213055 1,
—0,949500 —1,
0,244385 —0,
—
—
1,304 1,
—1,12 — 1,
—
—6,02 —6,
-0,
0,277917 —0,
115
261 ;
постоянные даны
1Д in Ч;
6000 10 000
30 000 30 573
АШ{ В2?+
ВО
во-
№
0
89551 1,900
17848 —1,1
21 —
— —
— —
8012 1,80
69 —1,5
— —
46 -6,5
2 —
06 —
67 85
261 284
В СМ".
h 1Ц+ ЗД Л 3V—
7 ^gi "м ^ 11
14 000 15 000
4 31000 33 000
С2П
23 958,85 43174 55 346,1
Л2
2+ СЧ1
36 223 38 983,5
Л32+
4400
аШ
7300
12Д
33 000
аЧ1 BsYr
4800 5600
Ь2Д, АЧ1 c2S
6'Е+ с:Д d1!!
0
1
—0
—0
1
—4
0
—0
—1
BS
ST2S+
A
1
,186565 0
,579708 —0
,894759 —0
,18679 0
,07052
,805233 0
,616847 —0
—
,437 —1
—
109
334
19 000 21000
34 000 35 000
f
7200 12 000 14 000
-, d22+ е2П В
11600 13 500 18 000 20 300
;22~ C4S~
35 000 41 000
/LП ?4S+
42 000 47 000
2Ща
,60425
,758232
,467784
,005901
,049880
,631015
,603592
—
,745
—
100
298
SJIg
22 000
з?+
38 000
BN
0
1,5244
—1,246
,
1,688
—2,55
—8,3
_
—
61
227
39 000
е1Ъ+ СЧ1 /Щ
24 000 27 850 39 000
/en, ff2S+j
28 000
ВС
0
0,990
—0,66
1,07
—1,0
-5,0
_
—
75
249
ВО (г). Термодинамические свойства газообразного оксида бора в стандартном состоянии в
интервале температур 1000—10 000 К приведены в табл. 534.
В табл. 20.4 представлены молекулярные постоянные 1гВО, принятые в справочнике на
основании анализа результатов исследований электронных спектров этой молекулы. В спектрах ВО
наблюдались 4 системы полос, связанных с переходами между четырьмя электронными
состояниями: 42П*^12Е+ [995, 1555, 1559, 2076, 2450, 2696], 5223+-*Х2?+ [1555, 1800, 2076],
Б2?+-н>. A2U [2076] и С2П -> Х2?+ [207, 234, 848]. Помимо этих состояний, данные для которых
приведены в табл. 20.4, молекула ВО должна обладать связанными квартетными состояниями
DП, 4Е и 4Д), коррелирующими, так же как состояние Х2Е+, с пределом В BР)+О CР). Однако,
как показали квантовомеханические расчеты Мичелса и др. [2018а], эти состояния имеют энергии
свыше 40 000 см и не рассматриваются в справочнике.
В работе Мальцева и Катаева [233] в спектре испускания разряда в парах В2О3 в области
15
ВО (г) Глава 20
2500 А была обнаружена группа полос, отнесенная к переходу из состояния 22+ с энергией
~40 000 см. Поскольку анализ структуры полос этой системы в работе [233] не проводился,
а последующие квантовомеханические расчеты (см. [2018а]) показали, что молекула ВО в области
энергий до 50 000 см имеет только два связанных состояния с симметрией 2Е + (Х2?+ и 52Б+),
это состояние не включено в табл. 20.4. Следует отметить, что энергия верхнего состояния
перехода, обнаруженного в работе [233], практически совпадает с энергией состояния а*?+, найденной
в расчете Мичелса и др. [2018а].
Постоянные молекулы Х1В1вО в состояниях Х2?+, Л2П и БаБ+, приведенные в табл. 20.4,
получены Лагерквистом и др. [1800] в результате анализа вращательной структуры девяти полос
системы 522+ -*¦ Х22+ (v' ^ 3, v" ^ 4 ) и исследований систем полос Л2П->Х22+, 522+->
->X2S + и i?2X!+ -> А2Пв работах Дженкинса [1555, 1559] и Милликена [2076]. Они хорошо
согласуются с данными других авторов, в частности Данна и Хаисона [995]. Постоянные ВО в
состоянии С2П приняты по работе Мальцева и др. [234], в которой был выполнен анализ
вращательной структуры семи полос системы С2П—Х2? + с v' ^ 2 в спектре молекул 10ВО и 11ВО.
, Термодинамические функции ВО (г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBB и ее производных вычислялись по уравнениям A. 90)^-
A.92) с учетом трех возбужденных электронных состояний и в предположении, что (>W вр =
=z(PilPx)Q[mi m- Статистическая сумма по колебательно-вращательным уровням энергии
состояния Х2И + и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням энергии и интегрированием по значениям / с помощью
уравнений типа A. 82); мультиплетность уровней учитывалась статистическим весом 2. В расчет
были включены все уровни энергии этого состояния с / ^ /шах.я последние находились из
условий A. 81). Колебательно-вращательные уровни энергии состояния Х22 + вычислялись по
уравнениям A. 64а) и A. 65). Значения коэффициентов Ykl в этих уравнениях для изотопической
модификации ВО, соответствующей природной смеси изотопов бора, вычисленные по соотношениям
A. 66) и принятым постоянным молекулы 11ВО, приведены в табл. 20.5; коэффициент Y03 введен
для обеспечения условия A. 59). В таблице 20.5 приведены также значения ушах и /Ит.
Постоянные молекулы ВО в состоянии Х2Е + известны с удовлетворительной точностью, а
электронные состояния, не учитывавшиеся в расчете, имеют высокие энергии. Поэтому погрешности
табулированных термодинамических функций ВО (г) при температурах до 3000 К не превышают
0,02 Дж-К-моль~1 в значениях Ф° (Т) и S° (T) и обусловлены в основном неточностью
фундаментальных постоянных. При более высоких температурах становятся существенными
погрешности из-за отсутствия экспериментальных данных о высоких колебательно-вращательных уровнях
состояния Х2И+, применения приближенной методики для учета вклада возбужденных
электронных состояний и пренебрежения рядом валентных состояний, общие погрешности в значениях
Ф° (Т) при 6000 и 10 000 К оцениваются в 0,1 и 0,3 Дж-К^-молъ.
Термодинамические функции ВО (г) вычислялись ранее в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF [1543], справочниках [977, 1904, 1974], в работах
[1044, 1493, 1947, 2854]. Результаты расчетов работ [977, 1493, 1904] и ряда других отличаются
от данных табл. 534 из-за использования приближенных методик и пренебрежения возбужденными
состояниями ВО, расхождения достигают 0,7 и 3 Дж-К~1-моль в значениях Ф° (Т) и С°р (Т).
Наилучшее согласие (в пределах 0,03—0,07 Дж-К-моль в значениях Ф° (Т) и S° (T)) имеет
место с предыдущим изданием [105], расхождения с [1543] не превышают 0,07, 0,4 и
1,5 Дж-К^-моль в значениях Ф° (Т), S0 (Т) и С°р (Т).
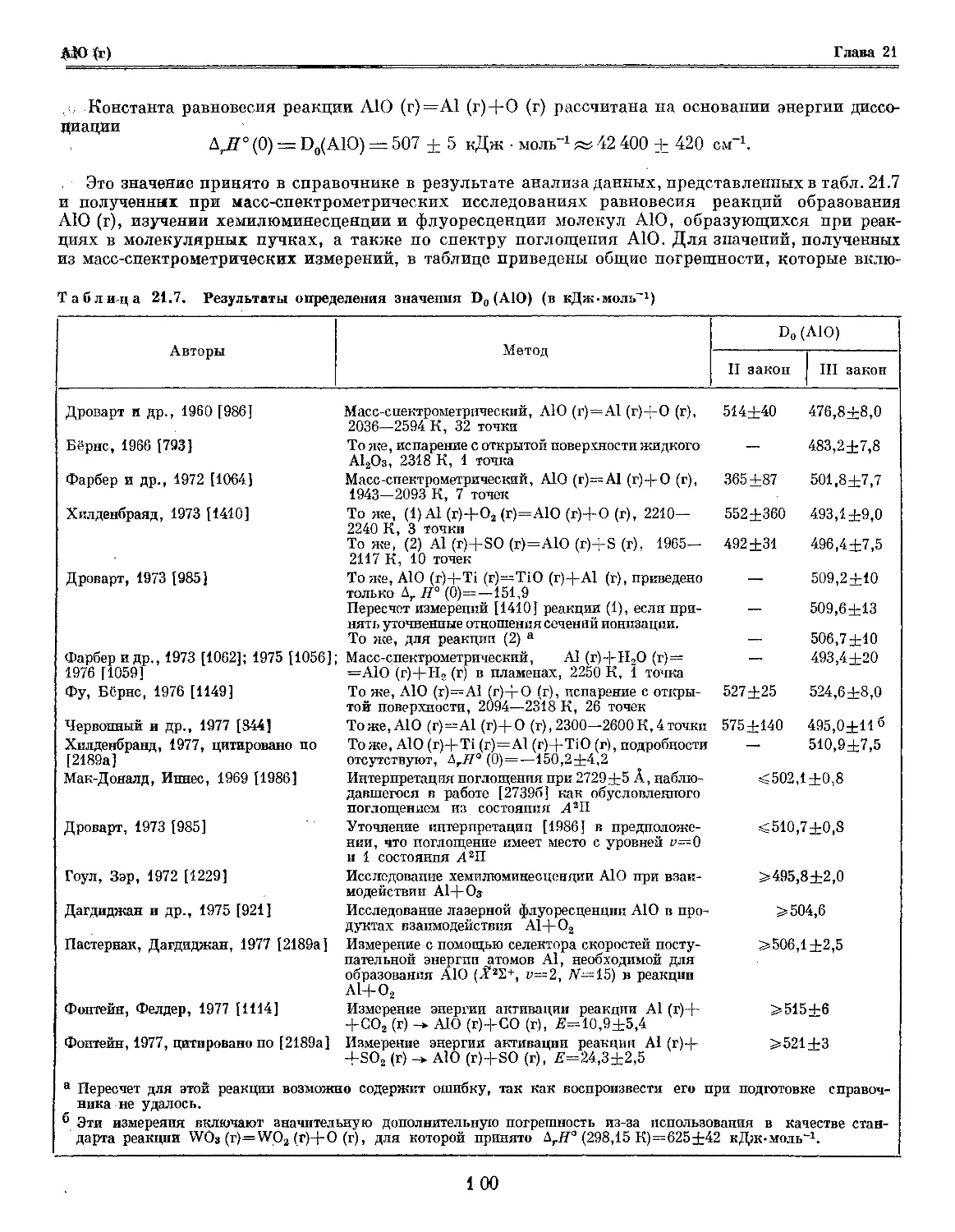
Константа равновесия реакции ВО (г)=В (г)+О (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (ВО) = 800+10 кДж • моль «* 66 900 + 850 см.
Это значение основано на результатах масс-спектрометрических исследований обменных изо-
молекулярных реакций в работах Дроварта и др. [880, 989, 2745]. Авторы этих работ измерили
константы равновесия реакций ВО с UO, UO2, ScO, YO, LaO, реакций SiO с UO, UO2, LaO,
реакций ScO с GeO и др. Полученные данные позволяют вычислить шесть значений энергии
диссоциации ВО, среднее из которых составляет 800,8+7,8 кДж-моль, причем эти значения
зависят только от энергий диссоциации SiO и GeO. Погрешность принятого значения учитывает
погрешности энергий диссоциации этих молекул (см. т. 2), сечений ионизации и термодинамических
функций компонентов реакций.
16
Бор и его соединения
ВО- (г)
Близкие, но менее точные значения, подтверждающие сделанный выбор, были найдены по
результатам изучения процессов испарения смесей В+ВеО и B+MgO [2485, 2486],
пересчитанным в работе [2585] G85+40 кДж-моль), фотометрических исследований борсодержащих
пламен 11161] (801+30 кДж-моль) и масс-спектрометрического исследования равновесия реакции
0,5В2О2=ВО [696] G99+18 кДж-моль).
Принятому значению энергии диссоциации соответствует
г, 0) = 6,783 + 11 кДж
ВО" (г). Термодинамические свойства газообразного отрицательного иона оксида бора в
стандартном состоянии при температурах 298,15—6000 К приведены в табл. 535.
В табл. 20.4 приведены молекулярные постоянные ВО", принятые в справочнике. Спектр ВО"
до настоящего времени не был получен, поэтому принятые постоянные основаны на оценках и
результатах квантовомеханического расчета Саммерса и Тиррелла [2637а]. Так же как изоэлектрон-
ные молекулы СО и BF, ион ВО" должен иметь основное состояние ХХ2+ (конфигурация . . .1тс45о2)
и электронные состояния с высокими энергиями возбуждения. Молекула СО и ион СО+, несмотря
на значительное различие в энергиях диссоциации, имеют близкие межъядерные расстояния и
частоты колебаний (см. т. 2, табл. 15.4). На основании этого и постоянных молекулы ВО, для ВО"
были приняты значения (вв=1900 +100 см и ге=1,20+0,03 А. Близкое, хотя и более высокое
значение ге было получено в неэмпирическом квантовомеханическом расчете [2637а].
Термодинамические функции ВО" (г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95) без учета возбужденных электронных состояний. Статистическая сумма
по колебательно-вращательным уровням энергии состояния Хг^+ и ее производные находились
непосредственным суммированием по колебательным уровням и интегрированием по / с помощью
уравнений типа A.82). Колебательно-вращательные уровни энергии вычислялись по уравнениям
A. 65) и A. 62), коэффициенты Ykl в этих уравнениях тождественны молекулярным постоянным
О б 204 В /^/
( ) ( ), ффц kl р р
ВО", приведенным в табл. 20.4. В расчете учитывались все уровни с значениями
A 81) С /
/„
, рд р
последние определялись из условий A. 81). Соответствующие значения ут
Y б 205
р
Ykl представлены в табл. 20.5.
и коэффициентов
рд
Погрешности рассчитанных термодинамических функций ВО" (г) обусловлены отсутствием
экспериментальных данных о молекулярных постоянных этого иона и составляют 1, 2 и 3 ДжХ
хК^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Ранее таблица термодинамических функций ВО" (г) в литературе не публиковалась; в работе
[2598] были рассчитаны значения Ф' (Т) для !Г=1600-^-2100 К, которые в пределах 0,25 ДжХ
X К-моль
согласуются с данными табл. 535.
Таблица 20.6. Значения молекулярных постоянных (в см) в состоянии
термодинамических функций ВО4а (о = 2, р% = Щ.
^ , принятые для расчета
Постоянная
uB02
10ВО2
vj 1051,9 1052,8
v2B) 448,8 6 465,3 6
v3 1301,6 1350,2
Постоянная
Г*11
ж12
ж13
ж22
х23
a Учитывались также следующие состояния:
X4L . TQ = 150,1 см, а = 2, ,- = 2;
АШи, * ГО = 18291,6 см, о = 2, Р2 = 4;
ВЧХ, То = 24 508,0 см-1, с = 2, ps = 2.
"ВО2
—3,87
—0,13
—22,39
1,03
—3,26
—10
6 Вместо
10ВО2
—3,87
0,49
-21,96
1,02
—3,46
—10
Постоянная
Во
ох
а2
аз
W
"ВО2
10ВО2
0,3283 0,3283
0,0021 0,0021
—0,0018 —0,0018
0,0029 0,0029
31,43 32,66
цважды вырожденной частоты v2 в расчетах
испольвовались невырожденные значения v^ = 409,4 см,
v|==440,4
A0ВО2)
v j = 488
("ВО2)
см-1 (nBO2), vf=423,3
ДЛЯ СОСТОЯНИЯ jP2II
2 см (nBO2), vf = 456
цля состояния -^*Пу1 .
см, v2' = 456,5 см
и vf = 440,4 см,
5 см-1^^ 507,3 см
17
ВОг (г) Глава 20
Константа равновесия реакции ВО" (г)=В (г)+О (г)+е (г) вычислена с использованием
значения Ь.ГН° @)=1092 +11 кДж-моль, соответствующего принятому в справочнике сродству ВО
Ао (ВО) = —292+5 кДж-моль.
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакции
ВО+С1~=ВО"+С1, выполненном Сривастава и др. [2598] A623—2100 К, 9 точек). По этим
данным было найдено Д//° @)=56,7+2,2 кДж-моль (III) и 89,9+6,0 кДж-моль (II). Расчеты
по уравнению III закона показали небольшой систематический ход вычисляемых величин (с
ростом температуры от 1623 до 2100 К величина AJJ0 уменьшается на 9 кДж-моль). Енсен[1566}
при исследовании пламен с добавками бора нашел значения констант равновесия реакций
НВО2+е=ВО"+ОН и BO.j+H2 = BO~+H2O при 2475 К. Расчет по уравнению III закона и этим
измерениям приводит к значениям t\rH° @)=354 и 216 кДж-моль соответственно. Этим
величинам соответствуют значения сродства ВО к электрону, равные —251+30 и —236 + 40 кДж-моль.
Однако автор работы [1566] высказывает сомнение в том, что в условиях измерений достигалось
равновесие изучавшихся реакций, и отмечает, что истинное значение сродства ВО к электрону
должно быть более отрицательным. Таким образом, данные [1566] не противоречат результатам
работы [2598].
Принятому значению сродства к электрону соответствуют
kfH° (ВО", г, 0) = — 285,217 + 12 кДж . моль,
Do (ВО") = 950,8 + 13 кДж • моль ж 79 500 + 1100 см.
ВО2 (г). Термодинамические свойства газообразного диоксида бора в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 536.
Молекулярные постоянные ВО2, использованные для расчета термодинамических функций,
приведены в табл. 20.6. В справочнике принято, что в основном электронном состоянии -Х'2П?
молекула ВО2 имеет линейную структуру симметрии Deo*- Интерпретация электронного спектра
испускания ВО2 в работах Коряжкина и др. [181, 182, 183] и ИК-спектра молекул ВО3,
изолированных в матрице, в работах Серебренникова и Мальцева [379, 380] основана на угловой
структуре ВО2. Однако анализ этих работ и сравнение с другими исследованиями электронного спектра
[1575], ИК-спектра [2583] и спектров лазерной флуоресценции [960, 2410, 2411, 2573а] не
позволяют согласиться с выводами этих авторов. Наиболее исчерпывающая информация о
молекулярных постоянных ВО3 получена Расселом и др. [2410] при исследовании с высоким разрешением
спектра лазерной флуоресценции ВО2. Авторы [2410] при вычислении
колебательно-вращательных уровней учитывали ангармоничность колебаний, колебательно-вращательное взаимодействие,
эффект Реннера—Теллера, резонанс Ферми, спин-орбитальное взаимодействие. Полученные
в работе [2410] молекулярные постоянные 10ВО2 и UBO2 в основном электронном состоянии
приведены в табл. 20.6. Принятому значению Во соответствует г (В—О) = 1,267 А. Поскольку
авторам [2410] удалось получить лишь значение ш3+3а;зз=1336,1 см, постоянная х33 была оценена
в справочнике по соотношению D0=a>2/4a>,:r()—coe/2 в предположении, что частота
антисимметричного валентного колебания ш3 связана с энергией связи ВО—О 1. Энергия подсостояния Х^П^
принята равной постоянной спин-орбитального расщепления А =—150,1 см, найденной в работе
[2410]. Согласно данным Джонса [1575] молекула ВО2 имеет возбужденные состояния А*Пи и
№• ?*, энергии которых приведены в табл. 20.6.
Термодинамические функции ВО2 (г) вычислены по уравнениям A.3)—A.6), A.9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом поправок на ангармоничность колебаний и взаимодействие колебаний и вращения
по уравнениям A. 131)—A. 133), резонанс Ферми — по уравнениям A. 153)—A. 155), возбуж-
1 Основные частоты ВО2, определенные в работе [2410], согласуются с полученными в работах [960, 1575, 241:1,
2573а, 2583], но противоречат данным Коряжкина и др. ?181, 182, 183] из электронного спектра испускания
и Серебренникова и Мальцева [379, 380] из ИК-спектра ВО2 в матрице, которые получили для частоты vs
существенно более высокие значения A840 и 2081 см соответственно). Однако результаты работ [181, 182, 183, 379,
380] не представляются убедительными.
18
Бор и его соединения
ВО, (г)
денные состояния Л2Ии и i?2?* — по уравнениям A. 168)—A. 170), т. е. при условии, что
колебательно-вращательная статсумма в этих состояниях и в основном состоянии одинакова. Для
приближенного учета эффекта Реннера—Теллера (е0ш2=—86,4 [2410]) в состоянии ^2П„ вместо
одной дважды вырожденной частоты v2 при вычислении термодинамических функций
использовались невырожденные значения v^ и v& в состояниях Z2ng3/2 и Z2II^i/s (см. табл. 20.6). Расщепление
основного электронного состояния учитывалось по уравнениям A.171)—A.173), если принять
нодсостояние Х2П^/, возбужденным состоянием, обладающим отличными от основного состояния
частотами v* и v* (остальные постоянные, в том числе постоянные ангармоничности, колебательно-
вращательного взаимодействия и резонанса Ферми, приняты такими же, как в основном
состоянии). Общие погрешности рассчитанных функций обусловлены приближенным характером
расчета и недостаточно точной и полной информацией о молекулярных постоянных ВО2 и
оцениваются в 0,1, 0,2, 0,5 и 1 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000, 6000 и 10 000 К.
Пренебрежение поправкой на центробежное растяжение не приводит к заметному увеличению
погрешностей рассчитанных величин.
Термодинамические функции ВО2 (г) вычислялись в предыдущем издании справочника [105],
таблицах JANAF [1543] и работе [1974] в приближении «жесткий ротатор—гармонический
осциллятор» по молекулярным постоянным, определенным в работах [1575, 2573а]. Некоторое
отличие в принятых молекулярных постоянных, а также учет в настоящем справочнике
ангармоничности колебаний, колебательно-вращательного взаимодействия, резонанса Ферми и эффекта
Реннера—Теллера приводит к расхождениям с результатами расчетов [105, 1543], которые
достигают 1 Дж'К~1-моль в значениях Ф° (Т). В расчете [1974] пренебрежение
спин-орбитальным расщеплением основного состояния ВО2 привело к заметным ошибкам в термодинамических
функциях при низких температурах; расхождение с данными табл. 536 составляют 2,2 Дж-КХ
Хмоль и 0,7 кДж-моль в значениях Ф° B98,15 К) и Н° B98,15 К)-Я° @).
Константа равновесия реакции ВО2 (г)=В (г)+2О (г) вычислена с использованием значения
ЬГН° @)=1378,566+21 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0)= — 325 + 20 кДж
моль
Эта величина основана на результатах исследований, представленных в табл. 20.7.
Погрешности приведенных значений характеризуют воспроизводимость измерений и неточность величин,
использованных в расчетах. Принятая величина является средневзвешенной из данных,
приведенных в таблице, причем наибольший вес придавался работам [2598, 2747], для которых имеет место
хорошее соответствие расчетов по II и III законам термодинамики.
Таблица 20.7. Результаты определений Д^Я0 (ВО2, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (BOjj, г, 0)
II закон
III закон
Каскан, Милликен, 1960 [1632] Фотометрическое исследование борсодержащих —352 —
пламен, ОН (г)+НВО, (г)=ВО2 (г)+Н2О (г),
1500—2200 К, 4 точки" ,
Мальцев и др., 1961 [237, 248] Фотометрический, В2О3 (ж)+1/2 О2 (г)=2ВО2 —318±20 —
(г), средняя температура 1750 К ;
Каскан и др., 1961 [1631] То же, 1660—1950 К, 4 точки —328+20 —
Русин, Татевский, 1965 [351, 352] Фотометрия продуктов взрыва в сферической —432+70 —288+30
бомбе, ОН (г)+НВО2 (г)=ВО2 (г)+Н„О (г),
2971 и 3186 К
Соммер, 1963 [2580] Масс-спектрометрический — —318а
Уй и др., 1971 [2598, 2747] Масс-спектрометрический, ОН (г)+НВО2 (г)= —330+25 —328±15
=ВО2 (г)+Н„О (г), 1160—1400 К, 7 точек
Макаров, Никитин, 1976 [230] Электронный удар, МВО2 (г)=М+ (г)+ —273±35
+ВО2(г)+е (г), где M=Li, Na, К, Rb, Сз
а В работе [2580] приведена только полученная величина.
19
ВОа- (г)
Глава 20
ВО2 (г). Термодинамические функции газообразного отрицательного иона диоксида бора в
стандартном состоянии в интервале температур 298,15 — 6000 К приведены в табл. 537.
Молекулярные постоянные ВО^, использованные для расчета термодинамических функций,
приведены в табл. 20.8. Экспериментально изучены ИК-спектры иона ВО^ в решетке кристаллов
галогенидов щелочных металлов [935, 936,1444, 2037, 2062, 2548, 2549, 2550, 2758] и ИК-спектры
Таблица 20.8. Значения молекулярных постоянных, а также аи р%, принятые для расчета термодинамических
функций ВОJ, В2О, В2Оа, НВО, ВОН, FBO, C1BO, BS2, B2S, B2S2, BC2 и В2С
Молекула
Состояние
BOj Х*Ц
в2о хщ
В2О2 Х^Ц
НВО Х^+
ВОН 1М'
FBO XW
сто ^ls+
BS2 1т,%л
B2S Х1^
B2S2 J?l?+
"DP F^2Y+ a
JJ\-J2 -Л ^
Ь2Ь ^ -^
ВОН: а Приведено значение
BS2: a xmg v>, Г0=440 см
BC2: a Л2Я, Г0=13 000см-1
В2С: а А12, Го=15 000 см-1
v2
1050 600 B)
1000 200 B)
2100 650
2850 754 B)
1050 1250
900 502 B)
673 404 B)
510 120 B)
650 100 B)
1300 400
1100 200 B)
1000 200 B)
1А1ВТС ¦ 10-"' rs ¦ см».
, о=2, рхч=2; АЧ1и, 1
, о=1, Р1=4.
0=1, Р1=1.
CM"
2000
1100
1899
1817
3600
2081
1958
1015
700
1215
2000
1700
V= 13 776 см-
_
—
420 B) 213 B)
— —
— —
—. —
— —
_ —
— —
350 B) 180 B)
— —
— —
-\ e=2, Pi=4; 5*2+
/ • 10'»
Г-CM2
8,3
6^3
25,4
2,1
0,4 a
9,0
16,8
31
11
68
6,5
6,8
, 7V=24 072
a
2
2
2
1
1
1
1
2
2
2
1
2
CM,
Pi
1
1
1
1 ¦
1
1
1
2
1
1
2
1
o=2,
газообразных метаборатов щелочных металлов [232, 437, 438, 780, 2496]. Молекулы метаборатов
щелочных металлов изучены также методом газовой электронографии [10, 122, 124, 125].
Близость основных частот группы ВО2 в этих молекулах к частотам иона ВО^ в решетке кристаллов
галогенидов щелочных металлов позволяет предположить, что молекулы метаборатов относятся
к ионному тину. Исходя из этого, молекулярные постоянные BOJ оценены в настоящем
справочнике на основе имеющихся экспериментальных данных для метаборатов щелочных металлов. Для
ВО7 в основном электронном состоянии г 2* принимается линейная структура симметрии D ^д
с величиной г (В—О)=1,25+0,03 А, как в метаборатах щелочных металлов [10, 122, 124, 125].
Погрешность приведенного в табл. 20.8 значения / составляет 4-Ю0 г-см2. Основные частоты
ВО7, приведенные в таблице, представляют собой округленные значения частот, наблюдаемых
в ИК-спектрах ионов ВО^ в решетках кристаллов галогенидов щелочных металлов и в ИК-спект-
рах метаборатов щелочных металлов (см. выше). Погрешности в значениях \ и v2 оцениваются
в 50 см, v3 — 100 см. Можно ожидать, что возбужденные электронные состояния ВО^ лежат
в области высоких энергий, поэтому они в настоящем справочнике не рассматриваются.
Термодинамические функции ВО^" (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и составляют около 1, 2 и 3 Дж-К~1-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К (в том числе из-за неточности молекулярных
постоянных 0,5, 1 и 1,5 Дж-К~1-моль).
Термодинамические функции BOJ (г) вычислялись ранее в справочнике [1543].
Незначительное отличие оцененных в [1543] молекулярных постоянных приводит к расхождениям с
настоящим изданием, которые достигают 0,7 и 1 Дж-К
6000 К.
20
•моль в значениях Ф° (Т) и S° (T) при
Бор и его соединения В2О (г), В2О2 (г)
Константа равновесия реакции ВО" (г) =В (г)+20 (г)+е (г) вычислена с использованием
значения ДД/0 @) =1738,566+25 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д//° (ВО-, г, 0) = — 685 + 25 кДж • моль.
Принятая величина является средней из величин —663+20 и —703+25 кДж -моль", полученных
масс-спектрометрическим методом Сривастава и др. [2598] (ВО2+С1"=ВО~+С1, 1623—2100 Ks
10 точек, АГН° @)=49 кДж-моль-1 (II) и 10,7+3,0 кДж-моль-1 (III)) и Енсеном [1566] (см. также
[1565]) (НВО2+е=Н+ВО~, 2475 К, Дг#° @)=73+15 кДж-моль-1 (III)).
Принятой энтальпии образования соответствует сродство к электрону ВО2
Ао (ВО2)=—360+25 кДж-моль-1.
В2О (г). Термодинамические свойства газообразного оксида дибора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 538.
Молекулярные постоянные В3О, использованные для расчета термодинамических функций,
приведены в табл. 20.8. Экспериментальные данные о строении и спектре В2О в литературе
отсутствуют. В настоящем справочнике на основании результатов квантовомеханических расчетов [109 б]
принимается, что в основном электронном состоянии X1?* молекула В2О имеет линейную
структуру симметрии -Ош4. Для расчета момента инерции принято г (В—О) =1,32+0,05 А, как в
молекуле В2О3. Погрешность / составляет около 5-10~40 г-см2. Основные частоты В2О рассчитаны
в приближении валентного силового поля по валентным силовым постоянным фрагмента В—О—В
молекулы В2О3 и в предположении /в//г=0,01. Погрешности в значениях vx и v3 оцениваются
в 100 см~х, v2 — 50 см. Возбужденные электронные Состояния ВаО в настоящем справочнике
не рассматриваются.
Термодинамические функции В2О (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных, в частности, принятой структуры этой молекулы, пренебрежением возбужденными
электронными состояниями и приближенным характером расчета; они составляют около 5* 7
и 10 Дж-К-^моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Термодинамические функции В2О (г) вычислялись ранее в справочнике JANAF [1543]. Расчет
выполнен для нелинейной структуры (J? В—О—В =150°) со значениями частот, существенно
большими, чем принятые в настоящем справочнике. Соответствующие расхождения
термодинамических функций достигают 13 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции В2О (г)=2В (г)+О (г) вычислена с использованием принятого
в справочнике значения Д^0 @) =1100+100 кДж-моль, оцененного сопоставлением энергий
связей в молекулах ВО, ПВО и НВО2.
Принятому значению соответствует
Д/Д°(В2О, г, 0) = 266,783 + 100 кДж • моль.
В2О2 (г). Термодинамические свойства газообразного диоксида дибора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 539.
Молекулярные постоянные В2О2, использованные для расчета термодинамических функций,
приведены в табл. 20.8. В литературе отсутствуют экспериментальные данные о структуре и
молекулярных постоянных В2О2, за исключением основных частот v3 и v6, наблюдавшихся в ИК-спек-
трах [281, 367а, 3676, 2583, 2831, 2843, 2846]. По аналогии с изоэлектронной молекулой G2N2
и в согласии с изотопным сдвигом v3 в ИК-спектре молекул В2О2, изолированных в матрице из Аг
[3676, 2583], в настоящем справочнике принимается, что в основном электронном состоянии х??
молекула В2О2 имеет линейную структуру симметрии D coh1. Приведенный в таблице момент
инерции молекулы В2О2 вычислен по структурным параметрам г (В—О) =1,22+0,03 А и г (В—В) =
=1,70+0,05 А, оцененным сравнением с молекулами В2О3, B2F4 и В2С14. Его погрешность состав-
1 Линейное строение В2О2 предполагается также в работах [1530, 2583, 2843]. Мэлони и др. [1929] в результате
анализа энергий связей приняли для В2О2 нелинейную несимметричную структуру. Однако позже в работе [2644 ]
было показано, что имеющихся термохимических данных недостаточно для установления строения молекулы.
21
BaOg (к, ж)
Глава 20
ляет 1,3 -Ю9 г-см2. Основные частоты v3 и v6 молекулы 11В2О2 приняты по данным [3676, 2583];
их погрешности с учетом матричного и изотопного сдвигов оцениваются в 20 и 5 см соответственно.
Остальные частоты, приведенные в таблице, оценены в результате расчета в приближении
валентного силового поля, если полагать силовую постоянную /в—в такой же, как в молекуле B2F4.
Погрешности в частотах vlt v2 и v4 оцениваются примерно в 10% от их величин. Оцененные таким
образом основные частоты В2О2 близки к вычисленным в работах [1530, 2583, 2843] с
использованием несколько отличающихся силовых постоянных. Возбужденные электронные состояния В2О2,
которые, как можно ожидать по аналогии с C2N2, лежат в области высоких энергий, в настоящем
справочнике не принимаются во внимание.
Термодинамические функции В2О2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)s
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и составляют около 2, 6 и 10 Дж-К^-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К (в том числе из-за неточности молекулярных
постоянных 1—3 Дж-К^-моль).
Термодинамические функции В2О2 (г) вычислялись ранее в справочниках [105, 1543, 1904,
1974], а также в работах [1044, 1929, 2843, 2854, 2855]. Отличие молекулярных постоянных,
принятых в этих расчетах, от оцененных в настоящем справочнике приводит к расхождениям в
термодинамических функциях, которые достигают в значениях Ф° (Т): 6—9 Дж-К^-моль для [105,
1543, 1974], 1 Дж-К^-моль-1 для [1904, 2854] и 13-15 Дж-К^-моль-1 для [1044, 1929, 2843,
2855].
Константа равновесия реакции В2О2 (г)=2В (г)+2О (г) вычислена с использованием
значения АГН° @) =2077,566+14 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH° (В2О2> г, 0) = — 464 ± 10 кДж • моль.
Принятая величина основана на исследованиях равновесий, представленных в табл. 20.9.
Помимо указанных в таблице погрешностей воспроизводимости были также учтены погрешности,
связанные с неточностью термодинамических функций G нДж-моль при 1500 К). Принятое
Таблица 20.9. Результаты определений АуН° (В2О2, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (В2О2, г, 0)
11 закон
III закон
Инграм и др., 1956 [1530] Масс-спектрометрический, 2/з В (к)+ —453+100 —449,3+5,2
+2/зВ2О3 (ж)=В2О2 (г), 1305—1500 К, 3 точки
То же, а/3В(к)+2/»В2О3(г)=ВаО2(г) —452±10 —458,8±6,0
Сирей, Майерс, 1957 [2486] Эффузионный, 2В (K)+2MgO (к)=В2О2 (г)+ — —461,3
+2Mg (г), 1375 К, 1 точка а
Шир, 1958 [2449] Эффузионный, 2/зВ (K)+a/sB2O3 (ж)=В2О2 (г), —507+70 —466,4+1,2
1340—1410 К, 4 точки
Торзиошшй, та же реакция, 1294—1457 К, —512±10 —464,8+1,6
14 точек
Рентцепис и др., 1960 [2327] Эффузионный, С (к)+В2О2 (г)=1/3В2О3 (ж)+ — -457,4+5,1
+4/зВ (к)+СО (г), 1603—1656 К, 5 точек
Соммер, 1963 [2580] Не указан —464
а В работе проведены 6 измерений в интервале 1337—1436 К. Однако сами авторы указывают, что условия были
достаточно близки к равновесным только при 1375 К, когда исходная смесь была спрессована и эффузионная камера
имела минимальное отверстие.
в справочнике значение является средневзвешенным из приведенных в таблице величин (за
исключением выпадающего значения —449,3+5,2 кДж-моль [1530]), причем больший вес придавался
результатам работы Шира [2449].
В2О3 (к, ж). Термодинамические свойства кристаллического и жидкого триоксида дибора
в стандартном состоянии при температурах 100—3200 К приведены в табл. 540.
22
Бор и его соединения В2О3 (к, ж)
Значения постоянных, принятых для расчета термодинамических функций, приведены
в табл. 20.1. В справочнике за стандартное состояние В2О3 (к) принята гексагональная
модификация [1332], устойчивая от Т=0 до Ттх.
При Т < 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости образца В2О3 чистотой 99,9%, выполненных с точностью ~0,5% Керром и др. [1674] A8—
297 К). Экстраполяция данных [1674] ниже 18 К дает 5° A8 К) =0,1 Дж-К^-моль-1.
Погрешности принятых значений S° B98,15 К) и Н° B98,15 К) — Н° @), которые совпадают с
рекомендованными КОДАТА—MGHC [864], оцениваются в 0,3 Дж -К -моль и 0,04 кДж -моль
соответственно. Данные Келли [1654] по Ср (В2О3, к) в интервале 53—295 К в пределах погрешности
удовлетворительно согласуются с данными [1674]. В интервале 298,15—723 К для С ВаО3, к)
принято уравнение (см. табл. 20.1), выведенное на основании результатов измерений Шмидт [457]
в интервале 303—706 К с точностью ~0,5% на образце, содержавшем 0,1 % влаги. С данными [457]
в пределах 1 % согласуются результаты измерений энтальпии В2О3 (к) в работе Саутарда [2588 J
E70—720 К). Температура плавления В2О3 G23+1 К) принята по результатам тщательных
измерений на чистых образцах (примеси менее 0,1%), выполненных Шмидт [457] и Крачеком и др.
11769]. Близкое значение получено Доноуэ и Хаббардом [972] G24 К). Значения температуры
плавления В2О3, найденные в работах [1983, 2328, 2667], менее надежны. Энтальпия
плавления В2О3 B4,56+0,15 кДж -моль-1) принята по данным Шмидт [457]; с этим значением
удовлетворительно согласуются значения АтН°, вычисляемые из энтальпий растворения кристаллического
и стеклообразного В2О3 (см. ниже) с последующим пересчетом к температуре плавления В2О3.
Для С° (ВгО3, ж) принято уравнение (см. табл. 20.1), выведенное на основании совместной
обработки данных Шмидт [457 ] по теплоемкости В2О3 (ж) E80—910 К) и данных по энтальпии В2О3 (ж)Ё
полученных Красовицкой и др. [198] A015—2154 К), Саузардом [2588] C81—1777 К), Шпильрай-
ном и др. [461] G60-2210 К) и Воскресенской и др. (см. [105], с. 729) (800—1400 К).
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,3, 0,8, 2 и 4 Дж-К^-моль соответственно. Термодинамические функции В2О3 (к),
приведенные в табл. 540, практически совпадают с вычисленными в других справочных изданиях [105,
1503, 1544, 1656]. Для В2О3 (ж) расхождения увеличиваются с ростом температуры, достигая
3 Дж-К^-моль в значении Ф° C000 К), что обусловлено уточнением данных по Дш#°, а также
по теплоемкости и энтальпии В2О3 (ж) в работах [457, 461 ].
В справочнике принято значение энтальпии образования
Д//°(В2О3, к, 298,15 К) = —1273,5 + 1,4 кДж.моль,
рекомендованное КОДАТА—МСНС [864] на основании совместного анализа результатов большой
группы измерений энтальпий реакций, включающих В2О3, Н3ВО3, BF3 и т. д. (см. [864]). Основное
значение для выбора рекомендованной величины имели измерения энтальпии сгорания В2О3 (к)
во фторе, выполненные Джонсоном и Хаббардом [1578].
Давление пара В2О3 (к, ж) =В2О3 (г) вычислено с использованием принятой в справочнике
энтальпии сублимации
Д.Я ° @) = 433 + 8 кДж • моль,
основанной на приведенных в табл. 20.10 измерениях давления пара. Приведенные в таблице
погрешности отражают только воспроизводимость измерений.. Дополнительно необходимо учесть
неточность термодинамических функций В2О3 в газообразном и конденсированном состояниях,
что приводит к погрешности в &JI° (Г), равной 8, 12 и 15 кДж-моль для величин, полученных
из измерений при 1500, 2000 и 2500 К соответственно. Особенно сильно неточность функций
сказывается на высокотемпературных измерениях [1258]. Приведенная в таблице погрешность расчета
А,Н° @) по III закону из масс-спектрометрических измерений [285] включает оцененную
погрешность сечений ионизации. При пересчетах данных работ [35, 277, 417, 2592] по рекомендации [2851
были исключены низкотемпературные точки, в которых возможно образование заметных
количеств НВО2 (г). По этой же причине привели к завышенным значениям давлений пара результаты
измерений методом протока [16, 39, 868], в которых не были приняты специальные меры по тща-
При высоких давлениях ( > 65 МПа) и температурах (около 1400 К) устойчива ромбическая модификация BaOs
[2269].
23
(стекл.)
Глава 20
Таблица 20.10. Результаты определений Д8ДС (В2Оз, к, 0) (в кДж-моль
Авторы
Шпейзер и др., 1950 [2592]
Несмеянов, Фирсова, 1960 [35, 277,
4171
Саулен и др., 1955 [2586]
Сирен, Майерс, 1957 [2486]
Шир, 1957 [2448]
Уайт и др., 1960 [2843]
Меши и др., 1960 [2009]
Никитин, Акишин, 1962 [285]
Уайт и др., 1961 [2845]
Бюхлер, Берковиц-Маттук, 1963 [777
Хилденбранд и др., 1963 [1416]
Блэкберн, Бюхлер, 1965 [696]
Грин, Маргрейв, 1966 [1258]
Фарбер, Фриш, 1969 [1051]
Метод
Эффузионный, 1449—1642 К, 15 точек
Эффузионный, 1402—1515 К, 8 точек
Протока, 1567—1808 К, 5 точек
Эффузионный, 1501—1566 К, 6 точек
Эффузионный, 1406—1626 К, 13 точек
Торзионный, 1414—1621 К, 19 точек
Эффузионный, 1409—1610 К, 6 точек
Спектрофотометрический, —1400—1800 К
Масс-спектрометрический, 1323—1451 К,
4 точки
Масс-спектрометрический, 1288—1529 К,
331 точка
Эффузионный, ~1400 К, 1 точка
Масс-спектрометрический, 1220—1641 К,
16 точек
Масс-спектрометрический, -—1200—1440 К,
19 точек
Торзионный, 1450—1584 К, 24 точки
Масс-спектрометрический, —1180—1550 К
Протока, 1956—2094 К, 4 точки
Точек кипения, 1949—2419 К, 12 точек
Масс-спектрометрический, —1320 К
\„Н° (ВгО
II закон
385 ±27
380±40
292+100
440±100
416+16
415 ±8
373 ±50
370±15
378 ±56
400±4
—
395±5
436±3
438±10
430 ±2
372 ±90
397 ±14
415±5
з, к, 0)
III закон
430+2
429 ±2
424+13
426±2
440+1
439 ±1
432±6
—
—
449 ±8
429
—
—
432±0,3
—
444 ±4
444 ±3
—
тельной осушке газа-носителя и измерительной аппаратуры (см. [2586]). Результаты расчетов
по уравнению II закона по данным работ [696, 777,1416 ] оказались в удовлетворительном согласии
с расчетами по уравнению III закона. Принятое значение является средним из величин, найденных
по методу III закона и обработки по методу II закона данных, полученных в этих трех работах.
С этим значением согласуются результаты измерений [35, 277, 417, 2486, 2586, 2845].
В2О3 (стекл.). Термодинамические свойства стеклообразного триоксида дибора при
температурах 100—723 К приведены в табл. Д.42.
Значения постоянных, принятых для расчета термодинамических функций, приведены
в табл. 20.1. При Т < 298,15 К термодинамические функции В2О3 (стекл.) вычислены по
результатам измерений теплоемкости в работе Турдакина и Тарасова [407а] E9,6—295 К, точность
—0,5%). Экстраполяция ниже 60 К выполнена по линейному закону и приводит к S° F0 К)—
—S°@)=16,2Дж-К-1-моль-1.Погрешностивычисленныхзначений S° B98,15 К) и Н° B98,15 К)—
Н° @) оцениваются в 1,3 Дж-К-моль~1 и 0,1 нДж-моль" соответственно. Остаточная
энтропия В2О3 (стекл., 0) =7,9+1,0 Дж-К^-моль была вычислена сравнением энтропии В2О3 (ж)
в точке плавления из данных по &mS° (В2О3) и значениям S° (В2О3, к, Тт) и S° (B2O3, стекл.,
Тт)—S° (B2O3, стекл., 0), вычисленным в настоящем справочнике. Принятое значение S° (B2O3,
стекл., 298,15 К) близко к рекомендованному в справочнике [406] (80,8+1,3 Дж-К^-моль).
В справочниках [1658] и [1543] приводятся более низкие значения G7,8+2,0 и 78,4 Дж-К~хХ
Хмоль), поскольку в этих изданиях принимались заниженные значения Д„Д° (В2О3). Уравнение
для С (В2О3, стекл.) в интервале 298,15—723 К выведено по результатам измерений энтальпии
в работе Саузарда [2588] C82-1777 К).
Погрешности вычисленных значений Ф° (Т) для В2О3 (стекл.) в интервале 298,15—723 К
оцениваются в 1,0—1,5 Дж-К~1-моль и обусловлены в основном неточностью расчета остаточной
энтропии.
Термодинамические функции В2О3 (стекл.), приведенные в табл. Д.42, отличаются от
приведенных в таблицах JANAF [1543] примерно на 3 Дж-К^-моль в значениях Ф° (Т) и S° (T)
(см. выше).
24
Бор и его соединения
В2О3 (г)
В справочнике принята энтальпия образования
Д/Я°(ВаО8, стекл., 298,15 К)=—1254,9 ±1,4 кДж
моль
полученная на основании принятой величины АуН° (В2О3, к, 298,15 К) и значения энтальпии
перехода кристаллического В2О3 в стеклообразный AtrH° B98,15 К) =18,6+0,3 кДж-моль.
Последнее основано на измерениях энтальпии растворения в воде A8,58+0,25 кДж -моль) и
сожжения во фторе A8,54+4,2 кДж-моль) образцов В2О3 (к) и В2О3 (стекл.) в работе Джонсона и
Хаббарда [1578]. Близкие значения при аналогичных измерениях энтальпии растворения в воде
были получены Саузардом [2588] и Фазолино [1069] A8,24 и 18,12 кДж-моль).
Многочисленные измерения энтальпии сгорания В (аморф.) в О2 и энтальпии ряда других
реакций (гидролиз и разложение В2Н6, гидролиз ВС13 и т. д.) приводят к величине энтальпии
образования В2О3 (стекл.) из В (аморф.) (—1264,0+2,0 кДж-моль), удовлетворительно согласующейся
с принятой выше величиной. Библиографию соответствующих работ см. в справочниках [105, 406].
В2О3 (г). Термодинамические свойства газообразного триоксида дибора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 541.
Молекулярные постоянные, использованные для расчета термодинамических функций В2О3г
приведены в табл. 20.11. Несмотря на многочисленные исследования структуры и спектров
молекулы В2О3, некоторые детали ее строения и сведения о ее частотах колебаний остаются пока не
выясненными. Впервые к выводу об угловой F-образной структуре этой молекулы пришли Акишин и
Спиридонов [8], которые в результате электронографического исследования определили
межатомные расстояния В=О и В—О и -^ В—О—В =95°. В более детальном электронографическом
Таблица 20.11.
Значения молекулярных постоянных, а также а и р%, принятые для расчета
термодинамических функций В2О3, В(ОНJ, Н2ВОН, F2BOH, B2C14, C12BOH, B2S*
Молекула
В2Оз
В(ОНJ
Н2ВОН
F2BOH
В2С14
С12В0Н
B2S»
a Fo=63(
Состоя-
V2
Х}Аг 2100 1050
1Ы' 3600 3600
2ХА' 3600 2800
Х*А' 3600 1450
iMi 1122 401
2х А1 3600 1250
2х А х 1314 650
Зсм, /пр=1,33-10-38 г-см
490 85
1500 1200
2700 1600
1250 1050
176 — а
1050 1000
120 70
ем-
500
1100
1250
1000
728
550
125
=2.
1
2050
900
1100
700
289
530
1314
1120
540
1050
500
917B)
450
650
V8
490
600
1000
480
512B)
350
130
520
600
600
450
104B)
270
135
ХЮ11'
г3 • см'
2-10*
60
4
1,113-10»
5,4-105
1,2-10*
8,7-10*
а
2
1
1
1
4
1
2
РХ
1
2
1
1
1
1
1
исследовании Ежова и др. [123, 125] были получены близкие значения расстояний В=О и В—О,
но существенно больший -^ В—О—В =132°. Позднее Спиридонов и Засорин [25956] провели новое
электронографическое исследование с использованием более совершенной экспериментальной
техники и методики расшифровки электронограмм, причем в ходе структурного анализа вновь были
рассмотрены различные модели молекулы В2О3. Наилучшее согласие с экспериментальными
данными было получено для F-образной модели симметрии C2t с параметрами г (В=0) =1,219 А,
г (В—О) =1,323 А и -4 В—О—В =137°. Спиридонов, Гершиков и др. [87а, 2595а] обработали те же
экспериментальные данные по новой методике совместного структурного и колебательного
анализа, в которой осуществляется параметризация молекулярной составляющей интенсивности
на структурные параметры и силовые постоянные. В результате выполненного анализа в этой
работе были определены 5 структурных параметров и 11 силовых постоянных (последние были
использованы для расчета частот колебаний), причем наилучшее согласие с экспериментом было
достигнуто для неплоской и несколько искаженной F-образной структуры симметрии С2: г(В=О) =
=1,190+0,005 А, г (В—О) =1,337+0,009 А, ^ В-О—В=135,8±5,7°, -$ О—В—0=187,6+4,9°,
s =9,2+6,0 (угол выхода связи В = О из плоскости В—О—В). Найденные таким образом значения
25
В2О3 (г) Глава 20
г (В=О), г (В—О) и ^ В—О—В близки к полученным в работах [123, 125, 25956], а
обнаруженные авторами неплоскостность молекулы В2О3 и нелинейность фрагментов О=В—О нельзя
признать существенными при учете погрешностей определения этих параметров.
Следует отметить, что найденные Спиридоновым, Гершиковым и др. [87а, 2595а] структурные
параметры соответствуют равновесной конфигурации молекулы В2О3, в то время как во всех
остальных исследованиях определялись «эффективные» значения этих параметров, усредненные для
большой совокупности колебательных состояний, которые заселены в условиях электронографического
эксперимента (~1600 К). Неэмпирический квантовомеханический расчет, выполненный Рамбиди
и др. [2288а], привел к равновесной конфигурации молекулы В2О3, близкой к найденной в работах
[87а, 2595а] (^ В—О—В=139°, г(В-О)=1,24 А, г(В=О)=1,36 А, -? О-В-О=175°) х.
Приведенное в табл. 20.11 произведение главных моментов инерции молекулы В2О3 в
состоянии Х1А1 рассчитано по эффективным значениям структурных параметров, найденным в работе
Спиридонова и Засорина [25956], что позволяет косвенно учесть зависимость этих
параметров от колебательного возбуждения. Погрешность принятого значения IaIbIc оценивается
в 5-Ю-115 г3-см6.
В ИК-спектре паров триоксида дибора [237, 283, 404, 981, 2843, 2846] однозначно была
интерпретирована только одна полоса в области ~2000 см, отнесенная к валентным колебаниям В = О
(vx и v6). В работах [283, 2843, 2846] наблюдались также полосы в области 1300 и 740 см,
отнесенные авторами этих работ к колебаниям связи В—О (v7) и к деформационному колебанию О=В—О
(v8), однако последующие исследования спектров молекул В2О3, изолированных в матрицах
из аргона, показали, что обе полосыг по-видимому, не принадлежат этой молекуле.
Наиболее полные исследования спектров молекул В2О3, изолированных в матрицах из аргона,
были выполнены Мальцевым с сотрудниками. В работах [378а, 2493а, 24936] в ИК-сцектре 11В216О3
ими наблюдались полосы в области 2062, 518, 489 и 73 см и слабая полоса при 462 см; в работах
[380а, 3806 ] в спектре КР была зарегистрирована только одна полоса при 1062 см. Изучение изо*
топического сдвига в этих полосах и условий их появления в ИК-спектрах матриц при изменении
условий напыления матриц привели Серебренникова и др. [24936] к заключению, что полоса
2062 см принадлежит антисимметричному колебанию связи B=O(v6), а полоса 1062 см —
симметричному колебанию связи В—О (v2). Поскольку и в ИК-спектре и в спектре КР в каждой
из этих областей наблюдалось только по одной полосе молекул uB2leO3 и 10В216О3, авторы [378а,
2493а, 24936] пришли к выводу о совпадении частот симметричного и антисимметричного
колебаний этих связей (vlT v6 и v2, v7 соответственно). Частоты 73, 462, 489 и 518 см были отнесены в этих
работах к деформационным колебаниям В—О—В (v4), О=В—О (v3 и v8) и внеплоскостному
колебанию О=В—О (v9). Неактивная в ИК-спектрах частота внеплоскостного колебания О—В—О (v6)
в работах [367а, 24936] оценивается равной 500 см. Результаты более ранних исследований
ИК-спектров В2О3 в матрицах, полученные в работах Соммера и др. [2582, 2583] и Ветлнера и
Варна [2831], в общем согласуются с данными [378а, 380а, 3806, 2493а, 24936], за исключением
того, что эти авторы отнесли к симметричному и антисимметричному колебаниям связи В—О
в молекуле nB2leO3 (v2 и v7) частоты 729 и 1239 см. Варьирование условий напыления матриц и
тщательное удаление следов воды из образцов триоксида дибора позволило Серебренникову и др.
[378а, 2493а, 24936] показать, что эти две частоты, по-видимому, принадлежат борным кислотам
или полимерным молекулам окиси бора. Следует отметить, что в ИК-спектре молекул триоксида
дибора, содержащих по 50% изотопов 10В и UB, в работах [378а, 2493а, 24936] в области 2100 см
были зарегистрированы только те полосы B061 и 2128 см), что и в спектрах чистых молекул
AГВ216О3 и 10В216О3 соответственно), хотя можно было ожидать появления новых частот,
принадлежащих молекуле 11В10В16О3. Наоборот, при замене изотопа 16О на изотоп 18О в ИК-спектрах
появлялось больше полос, чем это следовало ожидать для молекулы ВаО3 с симметрией С2,. Непонятно
1 Р-образная структура молекулы В2О8 подтверждается изучением отклонения молекулярного пучка в
неоднородном электрическом поле [1613], а также квантовомеханическим расчетом [845]. Тейлор [2671] рассмотрел
применение правила произведений Теллера—Редлиха к интерпретации ИК-спектров изотопзамещенных
молекул триоксида дибора для семи наиболее вероятных моделей этой молекулы; он показал, что наблюдавшееся
отношение частот несовместимо с бипирамидальной и линейной структурами B8OS, и пришел к выводу, что
наиболее вероятной является F-образная структура симметрии Сг„.
26
Бор и его соединения ВН (г)
также отсутствие в спектре КР полос в области 2000 см. Таким образом, исследования спектров
молекулы В2О3 не позволяют сделать однозначный выбор значений основных частот этой молекулы.
Результаты исследований спектров молекул В2О3 в работах [378а, 380а, 3806, 2493а, 24936]
получили качественное подтверждение в работах Спиридонова, Гершикова и др. [87а, 2595а],
которые из анализа электронографических данных (см. выше) определили 11 постоянных силового
поля этой молекулы и рассчитали все 9 ее основных частот (v1=2134+63, v2=1024+49, v3=493 +
±18, v4=98+9, v5=777±127, v6=2031+51, v7=H44±40, vg=558±19 и. v,=703±121 см). Если
учесть, что приведенные выше спектральные данные относятся к молекуле 11В2О3 и, кроме того,
могут содержать неточности из-за матричных сдвигов, а данные [87а, 2595а] получены для
свободных молекул, соответствующих природной смеси изотопов бора, данные [87а, 2595а] и [378а,
380а, 3806, 2493а, 24936] удовлетворительно согласуются между собой, за исключением частот
\ и v,.
В табл. 20.11 для основных частот молекулы В2О3 приняты усредненные и округленные
значения, основанные на совокупности данных [87а, 2595а и 378а, 380а, 3806, 2493а, 24936];
погрешности приведенных основных частот оцениваются в 50 см для \, v2) vg) v7 и v9, 100 см в v5,
20 см в v3 и v8 и 15 см в v4.
Возбужденные электронные состояния ВаО3 в справочнике не рассматриваются, поскольку кван-
товомеханический расчет [340а] показал, что их энергии превышают 35 000 см.
Термодинамические функции В2О3 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета и составляют 4, 8 и 12 Дж- К~1-моль~1 в значениях Ф° (Т)
и S° (T) при 298,15, 3000 и 6000 К соответственно, в том числе из-за неточности принятых
значений постоянных 2—6 Дж- К-моль.
Термодинамические функции В2О3 (г) вычислялись ранее в справочниках [105, 106, 1493, 1543,
1544, 1904, 1946, 1974], а также в работах [1044, 1045, 2094, 2583, 2831, 2843, 2855].
Термодинамические функции В2О3 (г), вычисленные во всех этих работах, меньше приведенных в табл. 541,
причем расхождения из-за использования в соответствующих расчетах отличающихся (более
высоких) значений основных частот частично компенсируются выбором других структурных
параметров этой молекулы. Наилучшее согласие с данными настоящего справочника имеет место для
таблиц JANAF [1543, 1544], соответствующие расхождения не превышают 1—3 Дж-К~1-моль
в значениях Ф° (Т) и S° (T). Расхождения в этих же функциях составляют 6—16 Дж-К-моль~1
для предыдущего издания настоящего справочника [105] и 1—4 Дж-К-моль~1 для справочника
Мак-Брайд и др. [1974]. В справочниках [106,1493,1904,1946 ] в расчетах была принята бипирами-
дальная структура молекулы В2О3, в связи с чем соответствующие данные на 35—40 Дж- К- моль
меньше приведенных в табл. 541. Расхождения с данными [1044, 2843, 2855] составляют 25—
30 Дж-К~1-моль~1, с работой [2831] — около 5 Дж-К~1-моль; в остальных работах расчеты
были выполнены для ограниченного интервала температур и расхождения с данными табл. 541
имеют тот же характер.
Константа равновесия реакции В2Ог(г)=2В (г)+30 (г) вычислена с использованием значения
Дг#° @) =2694,683+13 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации В2О3 (к). Этим величинам также соответствует
AfH°(B2Os, г, 0)= —834,334 + 8,1 кДж • моль.
ВН (г). Термодинамические свойства газообразного гидрида бора в стандартном состоянии при
температурах 100—6000 К приведены в табл. 542.
Молекулярные постоянные, принятые на основании анализа результатов исследований
электронного спектра 11ВН и квантовомеханических расчетов, представлены в табл. 20.12. В спектре
ВН наблюдалось 16 электронных переходов, в том числе системы полос .4 41 ** X1 Е+ [497, 1576,
1880, 1881, 2113, 2195, 2696, 2698], В1^ -> XXZ+ [623, 1576], ВЪ+-+АЧ1 [973], Ь»Е- -> asH
[497, 832, 1881], С"ХД -+Am [1576], С1^ <- ХХ2 + [623], (р-Я*-+АШ |973, 1576] и переходы
в 9 синглетных ридберговских состояний с энергиями свыше 55000 см, изученные в спектре
поглощения ВН в работе [623]. Интеркомбинационные переходы в спектре ВН не наблюдались и
энергия состояния а3П принята по результатам неэмпирического квантовомеханического расчета
Штерна и Калдора [2612]; ее погрешность, оцененная из сравнения рассчитанных и известных из
экспериментов энергий возбуждения состояний .4 41, С"ХД и В1^*, не превышает 1500 см. Она
27
ВН(г)
Глава 20
Таблица 20.12. Принятые значения молекулярных постоянных "ВН, nB19F, "B35C1, nB79Br и
Молекула
"ВН
HB19F
"В35С1
пВ"Вг
11B127J
Состояние
хъ+
о3Пв
Am
63s-
л»П
х1^
а3П
441
а3П
441
а X1!!
a3 Q
4Ш*
Примечание. Ниже все
ВН: а
BF: »
ВС1: »
ВВг: а
BI: »
«,?„ = 0,364; 6
те
0
10 300г
23 135,75
37 360,8
45 980,96
0
29 144,3б
51157,94
0
20 200
36 750,92
0
18 851,486
33 935,3
0
16 350 л
28 640
постоянные
3, = 2,6 • 10
ский расчет; л оценка; "
по Ва= 12,667
2366,9
2460*
2250,99
2350 Д
2610,02
1402,13
1323,86
1264,96
839,12
911
849,04
684,31
757,10
637,63
574
650"
—
даны в см
, Яо = 1,125
вычислено
и ot; ' вычислено по
" <о,у, =—15,8295; s а4 = 0,
для d = 0; о р!
<».у. = 0,056; б
Pi = 7 • Ю-8; ° и
= 1,97.10-6,
4=24,25; ¦
.У. = -0.1.
приведено значение Do; б
в работе [1833] отсутствуют
приведенные постоянные; б
лено по соотношению A.68);
состояния оаП0+
дают и, = 645;
о
49,
86
56
46
11
9,
12
5
5
И
3
4
17
2
5
395
в
665
—
62
84 а
20 в
53 г
И
7
37 «
52
81в
58 г
4
—
• Ю-7, L
в.
СМ
12,021
12,99 =
к 12,2952
12,126н
12,757
1,5174
1,4135
1,4226
0,6838
0,6986
0,7054
0,490
0,508
0,501
0,3646
0,391
—
,„ = —1,5.10-»;
по соотношению A.67) ]
41
65
83
39
1
1,
1
0
0
0
0
0
0
0
0
в
1 '.
•102
2
98
58
80 л
646
47
82 в
35
36
9
2"
,4
—
D, ¦ 10е
1242,9б
1450 й
1451»
1219°
7,57
6,3
7,3
1,72»
—
1,60'
1,0»
0,93
1,23л
0,6г
0,6г
—
1,
1
1,
1,
1
1
1
1
1
1
1
1
1
1
2
2
г.
А
232
20
219
23»
196
2626
3083
3041
716
698
689
888
854
867
,14
,06
—
4 = 5,96; г квантовомеханиче-
30 (а3П)
соотношению A.69); и вычислено
L322, а3 = 5,188.10-2;
Яо=1.10-'
ш,у, = 0,047
г
ш,ге = — 0,0271;
А =178; » ш,у, =
указания, к
* #О = 8,3-10-
= —0,242; л а2 =
= 5 ¦ 10-*; г рх =
= _0,004; г ш,у. = 1
\ 1
, == g
= —4 • 10-*
= 2,
до,
7 • 10-'.
какой изотопической модификации
вычислено по Во -
л вычислено по
= 0,
Те
; е вычислено по значениям vt
я согласно
1833] слабосвязанное
% 17 500; ж вычислено
по соотношению
A.68);
,7 • 10""; ж постоянная
= 0,250; л р1 =
молекулы BI
363; " вычислено по соотношению A.69);
а3Пд+) = 16 058 и
у ту,, приведенным ъ
состояние.
& 300, все постоянные
= 1,
4 • 10-'.
относятся
г
вычис-
для под-
оригинальной работе,
авторы
хорошо согласуется с найденной в неэмпирическом расчете Блинта и Годдарда [709]. В спектрах
ВН не наблюдались переходы, связанные с состоянием 3И+, которое коррелирует, так же как
состояния X1!)*, а3П и АХИ, с пределом ВBРL-НB5). Согласно квантовомеханическим
расчетам [709, 2612] это состояние является отталкивательным.
Молекулярные постоянные ПВН в состояниях ZXS+ и А1!! приняты по работе Джонса
и др. [1576], которые нашли их из анализа тонкой структуры семи полос системы АгИ <- Х1!!"
с v', v" ^ 3. Для основного состояния близкие значения постоянных были получены Алми и
Хорсфоллом [497] при анализе той же системы полос в испускании (секвенция Дь>=0 с v ^ 2).
Состояние АЩ имеет неглубокий минимум (ушк=3) и постоянные, найденные в работе [497],
существенно отличаются от приведенных в табл. 20.12. Вращательные постоянные ВН в состояниях
а3П и &3?~ найдены Алми и Хорсфоллом [497] в результате анализа полосы 0—0 системы Ь3Е~ ->
—> а3П. Частоты колебаний оценены по формуле Берджа—Мекке при u>er^=3550 ,
соответствующая постоянная была получена усреднением величин, рассчитанных для состояний Х1И+, ЛХП,
CaA. Погрешности приведенных значений wg могут достигать 200 см. Молекулярные постоянные
в состоянии С'1 А приняты по работе Джонса и др. [1576], где они были получены из анализа полос
0—0, 1—-1 и 2—2 системы CaA—АгП и колебательных постоянных состояния А1!!.
28
Таблица 20.13. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см), а также значений vmix и i/1Im,
принятые для расчета термодинамических функций ВН, BF, BC1, ВВг и BI
и
-§
Коэффициенты
т, ¦ ю-*
Yw • Ю-8
v20 • 10-1
Y30 ¦ 101
У40 • Ю2
У01
У11 • 101
Y21 • 102
Y31 ¦ 10»
02 ^
Y12 • 10*
Y03 • 10'
Уо* • 10"
(o0 = D9)-10-4
a2 • 10*
a3 ¦ 10'
"шах
Aim
ВН
Х12+
0
2,369135
—4,988 040
0,517 470
—1,905 1
12,043 6
—4,209 7
0,312
—0,256
—124,67
0,261
1,125
—1,768
2,905 216
0,523 803
1,139173
0,167 766
22
68
АШ*
2,313 649
2,253 55
-5,688 2
-15,814
—
12,3141
—8,365
13,26
—52,08
-145,5
—
0,834
—9,77
0,591662
4,398 627
4,481725
4,202 309
3
35
Примечание. Ниже все постоянные даны в см
ВН: а Соотояние
т.
BF: » Состояние
г.
а'П Ь3Б"
10 300 37 360
С'1Д
,8 45 980,96
о»П АШ
29 144,3 511
57,94
;
0
1,
—1,
0,
—0,
1,
-о,
о,
-о,
BF
410 265
198 830
594
017 65
535 5
2055
006 04
—
775
—
—
—
—
—
—
—
108
285
ВС1: »
ВВг: а
ВС1
*iS+*
0
0,844127
-0,533 660
0,137 450
—0,001136
0,6913
—0,067 014
0,001543
—
—0,176
0,000 72
—
—
—
—
—
—
166
360
Состояние а3П
Т, 20 200
Состояние Am
Тв 33 935
0
0
—0
-0
-0
0
—0
—0
,3
X1SH-
,689 309
,357 041
,003 183
,002 693
,49719
,035 8
—
—
,103
—
—
—
—
—
—
—
121
359
4Ш
36 750,92
ВВг
1
0
-0
-0
0
—0
-0
,885148
,762 6
,488
,040 88
—
,5155
,036 8
—
—
,096
—
—
—
—
—
—
—
27
251
0
0
-0
—0
0
—0
0
-0
Х12+
,578 641
,238 476
,011439
—
,3712
,041
,000 73
—
,062
—
—
—
—
—
—,
—
111
451
BI
1
0
-0
—0
0
—0
-0
а'П
,635 0
,654 794
,480120
,283 424
—
,397 6
,041
—
—
,062
—
—
—
—
—
—
—
47
294
ВН (г) Глава 20
Термодинамические функции ВН (г) были рассчитаны по соотношениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Q2m и ее производных находились по уравнениям A. 90)—A. 92)
с учетом четырех возбужденных электронных состояний. Составляющие состояний а3П, 63Е~ и СпА
вычислялись в предположении, что (?(*>д> вр = (pjpx) Q(.fJ_ вр. Значения QifJ^, Qif^ вр и их
производные находились непосредственным суммированием по колебательно-вращательным уровням
этих состояний с помощью соотношений A. 73)—A. 75). В расчете учитывались все уровни обоих
состояний, ограниченные их предельными кривыми диссоциации. Энергии уровней вычислялись
по уравнениям A. 65) и A. 60), вырождение уровней состояния А1!! учитывалось статистическим
весом. Коэффициенты Ykl в уравнениях A. 65) и A. 60), пересчитанные для изотопической
модификации ВН, соответствующей природной смеси изотопов бора, с помощью соотношений A. 66) и
постоянных молекулы ПВН, приведены в табл. 20.13. Для обеспечения условий A. 51) и A. 59)
в уравнения для уровней энергии состояния ХХ2+ введены коэффициенты Ys0, Yi0 и У04.
В табл. 20.13 приведены также значения итйж, /,jm и коэффициентов в уравнениях,
аппроксимирующих предельные кривые диссоциации состояний Хх?+ и АгП (см. т. 1, кн. 1, с. 41).
Погрешности рассчитанных термодинамических функций ВН (г) при низких температурах
обусловлены главным образом неточностью универсальных постоянных, выше 2000 К становятся
заметными погрешности из-за отсутствия экспериментальных данных об энергии состояния а3П
и колебательно-вращательных уровней состояния Z1E+ с v ^> 3 и большими значениями /.
Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,02, 0,3 и 1,5 Дж-К-^моль.
Термодинамические функции ВН (г) вычислялись ранее в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF [1543], справочниках [977, 1904, 1974] и работах [1044,
1336, 1493, 1819, 1946, 2463, 2796, 2855]. В большинстве расчетов не учитывались возбужденные
состояния этой молекулы, а в [105] — для энергии состояния а3И было принято существенно
завышенное значение. Все расчеты были выполнены с использованием приближенных методик, что
благодаря невысокой энергии диссоциации ВН должно было привести к значительным ошибкам.
Однако эти ошибки скомпенсированы пренебрежением возбужденными состояниями.
Расхождения данных [977, 1336, 1543, 1904, 1974] и табл. 542 достигают 3,5, И и 15 Дж-К^-моль
в значениях Ф° (Т), S° (Т) и С°р (Т), проходя в последней функции через максимум в области
4000—5000 К. Расхождения с данными предыдущего издания несколько меньше (доЗ и9 Дж- К-1Х
Хмоль) в значениях Ф° (Т) и S° (T)).
Константа равновесия реакции ВН (г)=Н (г)+В (г) была вычислена на основании энергии
диссоциации
АгН° @) = Do (ВН) = 27 800 + 400 см да 333 ± 5 кДж • моль.
Эта величина была получена на основании данных о предиссоциации при вращении молекулы ВН
на четырех колебательных уровнях (у=0-г-3) состояния А1!!, которое имеет общий диссоциацион-
ный предел с состоянием Хх?+. Предиссоциация была изучена в работах Алми и Хорсфолла [497],
Герцберга и Мунди [1396] и Джонса и др. [1576]. Авторы [1396] из данных по предиссоциации
показали, что потенциальная кривая состояния А1!! имеет максимум и построение
предельной кривой диссоциации (вырождающейся в этом случае в прямую) позволяет определить
только энергию максимума относительно состояния Z1S+, v=0. Наиболее полный анализ
предиссоциации, выполненный в работе Джонса и др. [1576], дал для этой величины значение
28 850+150 см.
Расчет потенциальной кривой состояния АШ для определения величины барьера над диссоциа-
ционным пределом В BР)+Н BS) проводился в ряде работ. Наиболее точными из них
представляются полуэмпирический расчет Харли [1517] и неэмпирический расчет Блинта и Годдарда [709]>
которые привели к хорошо согласующимся значениям 1250 см и 1210+160 см. Однако Харли
отметил, что полученное им значение является верхним пределом этой величины и рекомендовал
для нее 970+325 см. Принятое в справочнике округленное значение Do (ВН) соответствует
барьеру в 1050см. В пределах погрешностей принятое значение согласуется с рекомендациями
авторов [1576] и [709], а также с результатами неэмпирического квантовомеханического расчета Do (BH)>
в работе Майера и Росмуса [2015].
Вильсон и Мак-Ги [2859] выполнили масс-спектрометрические измерения потенциалов'появле-
ния В+ и ВН+ из ВН3, их данным соответствует Do (BH)=350 кДж-моль (в расчете
использовано значение 10 (ВН)=9,77 эВ [623]). Исследования равновесия реакции бора с водородом»
с образованием ВН, выполненные Стеком и др. [2609а] (масс-спектрометрический метод, 2100—
30
Бор и его соединения
ВН2 (г)
2400 К), приводят к значению Do (ВН)=325Ч;20 кДж-моль. Результаты этих работ хорошо
согласуются с принятой величиной, которой соответствует
A/ff°(BH, г, 0) = 443,034 + 7,0 кДж ¦ моль.
ВН2 (г). Термодинамические свойства газообразного дигидрида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 543.
Молекулярные постоянные ВН2, использованные при расчете термодинамических функций,
приведены в табл. 20.14. Молекула ВН2 в основном электронном состоянии Х2АХ имеет
симметричную нелинейную структуру (группа симметрии С2Л- Структурные параметры ВН2 в этом
состоянии, г (В—Н)=1,18+0,01 Ан^ Н—В—Н=131 +1°, были вычислены Герцбергом и Джонсом
[1395] на основании анализа структуры полос системы А*Вг -*¦ S2A1 в электронном спектре
поглощения ВН/. В пределах погрешностей эти результаты согласуются с теоретическими расчетами,
выполненными в различных приближениях [609, 647, 712, 730, 773, 1395, 1492, 2737].
Приведенное в таблице значение IaIbIc вычислено по этим структурным параметрам, его погрешность не
превышает 1 • 10~120 г3-см6.
Таблица 20.14. Значения молекулярных постоянных, а также о и р%, принятые для расчета
термодинамических функций ВН2, BF2, BFj, BHF, ВС12, BHCI, BFC1, ВВг2, В13
Молекула
вн2
BF2
BFj
BHF
BC12
BHCI
BFC1
BBr2
BI2
BH2: a
BHF: a
BC12: a
BHCI: a
Состояние
%XAX
XMxa
J?M' a
J?M 'a
Pi,»
A*BU Тй =
A*A", To =
A2Blt To =
А*А", Го =
2650
1200
1200
1300
780
750
1320
640
540
= 5150 см, о =
= 15 300 см-1, o =
= 15 300 cM-i, a =
= 10000 см, c =
= 2,
= 1,
= 2,
= 1,
CM
1030
480
600
1000
270
900
365
160
110
J»I =
Pl =
Pl =
Pl =
2.
2.
2.
2.
2800
1350
1150
2750
850
2750
830
720
630
BFC1:
BBr2:
BI2:
1,
4,
4,
8,
8,
3,
1,
7,
4,
a ^M
a J[2.?
а ?2В
г3 • см6
2 ¦ Ю-2
З-lOi
5-101
2•lO-i
8-Ю2
5
9-102
7.10»
0-10*
\ Г0 = 20000
ь Г„==10000
ь го = юооо
a
2
2
о
1
2
1
1
2
2
СМ, о =
CM, a =
СМ~1, О =
PS
2
2
1
2
2
2
2
2
2
1, ,1=2.
1, РХ=2.
Колебательный спектр ВН2 экспериментально не изучался. Принятое значение основной
частоты деформационного колебания v2 оценено Герцбергом [87] на основании данных по
инерционному дефекту. Основные частоты симметричного (vx) и антисимметричного (vs) валентных
колебаний вычислены в настоящем справочнике по уравнениям поля валентных сил с силовой
постоянной связи В—Н, равной соответствующей постоянной в молекуле ВН3 (/г=4,08.105 дин-см)
[1617]. Погрешности найденных таким образом частот не превышают 100 см. Исследование
электронного спектра ВН2 [1395] показывает, что молекула ВН2 имеет невысокое электронное
возбужденное состояние AiB1 (см. табл. 20.14). В этом состоянии молекула имеет линейную структуру
(симметрия Дед) с межатомным расстоянием г (В—Н) = 1,17+0,01 А.
и# Термодинамические функции ВИ2 (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
1 Приведенные структурные параметры соответствуют вращательным постоянным А000=41,649, Bw0='4,241 в
Сш= 6,001 см.
31
ВН3 (г)
Глава 20
A.130) с учетом возбужденного электронного состояния A2Bt по уравнениям A.168)—A.170).
Погрешности рассчитанных термодинамических функций обусловлены главным образом
отсутствием экспериментальных данных о частотах колебаний молекулы ВН2 и приближенным
характером расчета. Суммарные погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К не
превышают 0,3, 1 и 2 Дж-К~1.моль.
Ранее термодинамические функции ВН2 (г) вычисляли авторы справочника JANAF [1543]
для линейной модели молекулы. Значения Ф° (Т) и S° (T), приведенные в табл. 543, на 7—
13 Дж.К~1.моль~1 превышают значения, вычисленные в [1543].
Константа равновесия реакции ВН2 (г)=В (г)+2Н (г) вычислена с использованием значения
^,Н° @)=697,068 +25 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^^ВНа, г, 0) = 295±25 кДж • моль.
Эта величина основана на измеренных Вильсоном и Мак-Ги [2859] потенциалах появления ряда
ионов из молекулы В2Н8 и измеренном Гангули и Мак-Ги [1166] потенциале появления ВЩ" из
ВН3СО. Эти исследования привели к совпадающему значению hfH° (ВЩ, г, 0)=1183+10 кДжХ
Хмель". Потенциал ионизации 10 (ВН2)=888+22 кДж-моль был вычислен по предложенному
в [2859] циклу. Потенциалы появления ВЩ были также измерены в более ранних и менее точных
работах [1073, 1761, 1762].
ВН3 (г). Термодинамические свойства газообразного тригидрида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 544. Щ Щ
Молекулярные постоянные ВН3, использованные при расчете термодинамических функций,
приведены в табл. 20.15. Экспериментальные данные о структуре молекулы ВН3 отсутствуют. На ос-
Т а б лиц а 20.15. Значения молекулярных постоянных и о в основном электронном состоянии (р% = 1),
принятые для расчета термодинамических функций BHg, BF3, BFj", BH2F, BHF2, BC13,
BH2C1, BHC12, BF2C1, BFC12, BHFCI, BBr3, BI3
Молекула
BH3
UBF3
"BF,
BFj-
BH2F
UBHF2
i°BHF2
nBCl3
«BC13
BH2C1
"BHC12
l0BHCl2
"BFaCl
«BFjCl
"BFC12
1OBFC12
BHFCI
nBBr3
1()BBr3
"BI,
10BI3
Состояние
1Ы[
11A[
?lA[
Xх A x
X1A1
1M,
JTMJ
11A[
XlAt
1^1
iMj
X1A1
%lA1
21Al
X1A1
Xi A'
Ж1 А [
Ж1А[
Ж1А[
&A[
2623
888
888
770
2700
1162
1193
471
471
2700
740
762
1242
1286
1312
1355
2620
279
279
190
190
v2
1125
691,22
719,28
. 360B)
1180
2619
2619
454,9
474,3
720
2617
2617
696
701
565
572
670
375
390
305
320
CM
2808 B)
1453,72B)
1505,25B)
1080 C)
1600
545
555
954,2B)
993,7B)
1600
287
289
427
427
271
271
370
819 B)
856 B)
704 B)
737 B)
1605 B)
479,36B)
481,13 B)
530 C)
2800
1402
1451
256 B)
256 B)
2800
1089
1100
1421
1470
993
1031
1270
151 B)
151 B)
100B)
100 B)
v
_
—
—
—
1050
1200
1190
—
930
892
892
366
366
338
338
1050
—
—
—
v
_
—
—
—
1030
924
945
—
990
784
795
604
629
524
547
860
—
—
—
/,7B/C.1O117
¦ex S3 ty
Г* • CM6
8,5.10-2
1,068.10s
1,072-103
5,1 -103
2,6
8,232-101
7,908-101
t 3,805 • 10*
1,07-101
1,38-103
1,31 -103
1 3,8-103
1 1,24-10*
3,2-10*
1
> 7,25-105
| 5,7.10*
О
6
6
6
12
2
2
2
6
6
2
2
2
2
2
2
2
1
6
6
6
6
32
Бор и его соединения ВН3 (г)
новании теоретических расчетов [534, 804, 1191, 2173, 2174, 2643] в настоящем справочнике
принимается, что молекула ВН3 в основном состоянии Х1А[ плоская (симметрия Dah) и
характеризуется величиной г (В—Н)=1,18+0,01 А. Погрешность значения IaIbIc, вычисленного по этим
данным и приведенного в таблице, не превышает 1-10~119 г3-см6. Принятые значения основных частот
ВН3 получены Калдором и Портером [1617] при исследовании инфракрасного спектра поглощения
продуктов пиролиза ВН3СО, изолированного в матрице из Аг. Отнесение частот подтверждено
согласием вычисленного и наблюдаемого изотопического смещения по бору и водороду и условиями
появления спектра. Частота симметричного колебания \, неактивная в ИК-спектре, вычислена
авторами [1617] по уравнениям поля валентных сил с использованием силовых постоянных /г=
=4,08-105 дин-см, /а/г2=0,46-105 дин-см и /s/>2=0,59-105 дин-см, отвечающих
экспериментально наблюдаемым частотам. С учетом возможного матричного сдвига погрешности
наблюдаемых частот оцениваются в 10 см. Погрешность в \ может достигать 100 см г. Сведения о
возбужденных электронных состояниях молекулы ВН3 отсутствуют.
Термодинамические функции ВН3 (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A.122)—A.124), A.128),
{1.130). Погрешности рассчитанных величин обусловлены главным образом приближенным
характером расчета. Суммарные погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
составляют 0,5, 2 и 3 Дж-К~1-моль~1; погрешности, связанные с неточностью принятых молекулярных
постоянных, не превышают 0,5 Дж-К~1-моль.
Ранее термодинамические функции ВН3 (г) вычислялись в таблицах JANAF [1543], Мак-Брайд
и др. [1974], а также Уилкинсом и др. [2855] (Т ^ 5000 К), и, в более узком интервале
температур, Юшиным [473] (Т ^ 1300 К) и Эндрюсом и Пиментелом [523] (Т ^ 2000 К). Все расчеты
выполнены по оцененным молекулярным постоянным. Значения Ф° (Т), вычисленные в этих
работах, систематически превышают приведенные в табл. 544. Соответствующие расхождения
достигают 0,8 Дж-К^-моль [1543, 1974], 1,2 Дж-К^-моль [473], 4,8 Дж-К^-моль [2855] и
6,7 Дж-К^-моль [523].
Константа равновесия реакции ВН3 (г)=В (г)+ЗН (г) вычислена с использованием значения
АгНа @)=1112,165 +11 кДж- моль, соответствующего принятой в справочнике энтальпии
образования
г, 298,15 К) = 92 + 10 кДж - моль.
Эта величина основана на приведенных в табл. 20.16 данных.
Из таблицы видно, что результаты определений энтальпии образования ВН3 противоречивы.
Кинетические исследования [622, 791, 816, 1074, 1288] приводят к величине 92 кДж-моль.
Исследования равновесий [629а, 2536, 2605] и измерения потенциалов появления методом
электронного удара [1166, 1167, 2859] дают 148 кДж-моль. Исследование равновесия реакции бора с
водородом [109] могло привести к неверному результату, так как при масс-спектрометрическом
исследовании аналогичной реакции водорода с карбидом бора в работе [2609а] было обнаружено, что
равновесие ВН3 с твердой фазой не достигается, причем концентрации ВН3 намного превышают
равновесные.
Противоречие между данными, полученными разными методами, могло бы быть объяснено
существованием у молекулы ВН3 возбужденного состояния с энергией около 50 кДж-моль
(~4000 см), которое оказывается заселенным в условиях высокотемпературных реакций и
измерений методом электронного удара. Оно может также объясняться наличием активационного
барьера при разрыве связей. Однако имеющиеся данные, в том числе исследования процесса
диссоциации В2Н6 в работах [629а, 2605], не дают оснований для таких предположений. Таким
образом, имеющиеся в настоящее время данные не позволяют сделать достаточно обоснованный
выбор энтальпии образования ВН3. Поскольку трудно предположить, чтобы кинетические
исследования энергий связей ВН3—СО и ВН3 — ВН3 имели ошибки в 60 и 110 кДж-моль
соответственно, в справочнике отдано предпочтение величине, вычисленной из кинетических измерений»
1 Частоты ВН3 ранее неоднократно оценивались разными авторами на основании данных о частотах бороводоро-
дов и СН3 [523, 1171, 1846, 2064, 2338, 2507]. Однако оцененные величины существенно отличаются от
экспериментально наблюдаемых и имеют большой разброс.
33
ВяНв (г)
Глава 20
Таблица 20.16. Результаты определений
(ВН3, г, 298,15 К) (в кДж-моль ')
Авторы
Метод
Д,#°(ВНз, г, 298,15 К)
II вакон III закон
Бауэр, 1956 [622]
Карабедион, Беясов, 1964 [816]
Фелнер, Коски, 1965 [1074]
Гротевольд и др. 1966 [1288]
Барг, Фу Юан Шин, 1966 [791]
Зинке и др., 1964 [2536]
Бейлис и др., 1966 [629а, 2605]
Девятых, Юшин, 1969 [109]
Коски и др., 1964 [1073, 1762]
Вильсон, Мак-Ги, 1967 [2859]
Гангули, Мак-Ги, 1969 [1166, 1167]
Анализ данных восьми работ по кинетике реакций 78
с участием ВН3; D(BH3—ВН3)=Н9
Анализ данных по кинетике пиролиза ВНзСО; 85—98
D(BH3—ВНз)=134—160
Исследование кинетики пиролиза ВН3СО; D(BHg— 97 ±10
СО)=97±10
Исследование кинетики пиролиза ВНзСО; D(BHa— 90±10
СО)=90±10
Спектрофотометрическое исследование равнове- 92 ±5
сия реакции 2BH3PF3=B2He+2PF3 и кинетики
реакции BH3PF3=BH3+PF3; D(BH3—BH3)=
=147 ±10
Масс-спектрометрическое исследование равнове- — 157
сия В2Нв=2ВН3, 773-925 К, D(BH3—ВНз)=278
То же, 650—800 К, 5 точек а, D(BH3—ВН3)=262 — 149
Исследование равновесия реакции В (аморф.)+ — 87
+8/2Н2=ВНз методом протока, 1300 К, 1 точка
Электронный удар, D(BH3—ВН3)=172 104
Электронный удар с использованием моноэнерге- 148±10
тических электронов, расчет тремя независимыми
путями D(BH3—ВН3)=260±15
То же, ВНзСО=ВН3+СО; D(BH3—CO)=148±20 148±20
а Значения констант равновесия вычислены по проведенным в [629а, 2605] интенсивностям ионных токов.
которые лучше проанализированы в литературе, и выбор энтальпии образования ВН3 в других
справочных изданиях и обзорных работах также основывается на них.
В2Н6 (г). Термодинамические свойства газообразного диборана в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 545.
Молекулярные постоянные В2Н6) использованные для расчета термодинамических функций,,
приведены в табл. 20.17. Спектроскопические [520, 1804, 1885, 2270], электронографические [605,
1377] исследования, а также квантовомеханические расчеты [792, 925, 1148, 1203, 1388, 2079?
показывают, что молекула В2Н6 имеет две мостиковые водородные связи между атомами бора и
относится к точечной группе симметрии Dn- Произведение главных моментов инерции 1А1Б1сг
приведенное в таблице, рассчитано по вращательным постоянным Ло=2,655 69, #„=0,606 463
и С0=0,557 279 см, найденным для uB2He из анализа вращательной структуры ИК-полос Ла-
ферти и др. [1804] г. Этим значениям вращательных постоянных соответствуют структурные
параметры: г (В. . .В)=1,763 А, г(В-Н)=1,201 А, г (В-НЫОСТ)=1,320 A, J* Н-В-Н=Ш,0%
^ НМО1!Т—В—НМОО1=96,2°, которые находятся в хорошем согласии с электронографическими
данными [605, 1377]. Колебательные спектры молекулы В2Н6 исследовались в газообразном [520,.
824,1140,1640, 1804, 1885, 2270, 2272, 2560, 2616, 2670, 2823], жидком [515, 1804, 1885] и твердом
[1804, 1885] состояниях как при низком, так и при высоком [520, 1804, 2272, 2560] разрешении!
В настоящем справочнике частоты vx—v4, vn и v15 приняты на основании данных Каррейра и др.
[824] по спектру комбинационного рассеяния газообразного В2Н6, при этом отнесение частоты
v3 принято по данным [520, 640, 1885, 2560, 2670, 2823] (авторы [824] рекомендовали для этой
частоты значение 1317 см). Частота v6 найдена Лордом и Нильсеном [1885] в спектре
комбинационного рассеяния твердого В2Н6. Частоты v13 и v17 получены из ИК-спектра газообразного В2Б№
[1640J. Частоты v6, v7 и v12 приняты по результатам исследования ИК-спектра в работе Смита в
1 Кроме вращательных постоянных Аа, Во, Сй в работе [1804] приведены постоянные центробежного растяжения:;
/)у=1,17-10"?, DJK=l,97-i0~- и DK=i,97-10"^ см, полученные в приближении симметричного волчка.
34
Пор и его соединения
В2Нв (г)
Таблица 20.17.
Значения молекулярных постоянных и а в основном электронном состоянии (Рх=1),
принятые для расчета термодинамических функций В2Нв, Н3Вз06, Р2С1Вз0з, ГС12Вз0з
и B3N3He
Молекула
B2He
НзВзОв
ргС1В3О3
FC12B3O3
B3N3He
Молекула
ванв
НзВзОв
F2C1B3O3
FC12B3O3
B3N3He
v2
v.
v.
vn
4,2
см
2530,2 2112,2
3356 1239
1435 1374
1443 1242
3488 2545
v,,
-и
1183
819
1166
1103
942
790
595
847
832
857
-.в
833,1
1473
788
783
1300
v»
1768 850 2608,?
1196 478 833
472 380 312
434 358 158
1195 782 917,
v,.
55 800
745
1464
1410
5 719
"0
369,3
200
1413
1352
394
см *
1905 972,7
1350 B) 939 B)
576 457
483 394
1465 B) 1406 B)
1025
1147 B)
195
233
1096 B)
2519,65
462 B)
700
680
990 B)
1601
280 B)
700
450
518 B)
1173,8 —
833 B) 650 B)
520 200
450 150
987 B) 787 B)
215 B)
200
200
280 B)
_
—
150
150
—
2597
3200 B)
1267
1197
3486 B)
• 10117
r3 • сив
24,44
l,8-105
4,5-105
1.10»
8350
915
1397 B)
950
762
2520 B)
4
3
2
2
6
Милса [2560], причем две последние рассчитаны из составных частот J. Значения основных частот
v8, vie (Лафферти и др. [1804]), v10 (Прингл и Мейнцер [2272]) и v14, v18 (Андерсон и Баркер [520])
приняты по началам соответствующих полос, для v9 принято значение, основанное на оценке
авторов [2560]. Погрешности принятых частот составляют менее 1 см для v5, vg, v10, v14, v16 и vlg;
до 10 см для Vj— v4, vn, v12, v15 и v17; 15—30 см для ve, v7 и v13 и 100 см для v9. Сведения о
возбужденных электронных состояниях В2Нв в литературе отсутствуют.
Термодинамические функции В2Н6 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
{1. 130) без учета возбужденных электронных состояний. Погрешности вычисленных величин
обусловлены в основном приближенным характером расчета и составляют 0,5, 6 и 10 Дж-К~хХ
Хмоль в значениях Ф° (Г) при 298,15, 3000 и 6000 К. Погрешности, связанные с неточностью
постоянных, не превышают 0,1—1 Дж-К-моль~1.
Ранее термодинамические функции В2Н6 (г) рассчитывались в справочнике JANAF [1543],
а также работах Сандарама [2638] (до 2000 К) и Визи и Сивина [2780] (до 1000 К). Расхождение
результатов расчета [1543] с данными, приведенными в табл. 545, не превышает 0,7 Дж- К- моль
в значении Ф° F000 К), расхождения с данными [2780] составляют 1,3 Дж- К- моль в значении
Ф° A000 К).
Константа равновесия реакции В2И6 (г)=2В (г)+6Н (г) вычислена с использованием значения
Д^0 @) =2363,779 +10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(В2Нв, к, 298,15 К) = 36,6 ±2,0 кДж • моль.
Эта величина основана на приведенных в табл. 20.18 результатах измерений и является
средневзвешенной из данных работ [1307, 1308, 2275, 2538]. Величина энтальпии образования В2Нв
яз В (аморф.), полученная в работе [2274], хорошо согласуется с принятыми в настоящем
справочнике значениями энтальпии образования В2Н6 из В (к) и энтальпии перехода В (к) в В (аморф.).
В работе [1310] образующийся в реакции В (аморф.) мог отличаться от полученного пиролизом
В2Нв в проточной системе (см. [2274]).
1 В работе [2560] найдены также постоянные Кориолиса для ряда полос.
HBO (г) Глава 20
Таблица 20.18. Результаты определений А/Н° (В2Н6, г, 298,15 К) (в кДж-моль)
Авторы
Метод
Н° (В2Н6, г,
298,15 К)
Рот, Бергер, 1937B376] Измерение энтальпий гидролиза диборана и растворе- 22+9
ния аморфной окиси бора
Прозен и др., 1959 [2275] Измерение энтальпии реакции В2Н0 (г)+6Н2О (ж)= 35,4+3,0
=2Н3ВО3 (р-р 1000Н„О)+6Н2 (г), АГН° B98Д5 К)=
=—466,3+2,3
Ганн, Грин, 1960 [1307, 1308] То же, Лг Н° B98,15 К)=— 468,4+0,7 37,5+2,0
Скиннер и др., 1961 [2538] То же, Н3ВО3 (р-р 500Н„О), Дг 11° B98,15 К)= 35,6+3,0
=-466,5+2,1
Прозен и др., 1958 [2274] Измерение энтальпии термического разложения дибо- 28,1+3,0 а
рана В2Нц (г)=2В (аморф.)+ЗН2 (г) в проточной
системе
Ганн, Грин, 1961 [1310] То же, взрыв в присутствии БЬНз 21 а
Мак-Кой, Бауэр, 1956 [1979] Измерение энтальпии реакции 0,5В2Нв (г)+ 23
+(CH3KN (r) = (CH3KNBH3 (к) и др. данные
Ганн, 1965 [1307] То же 31
а Энтальпия образования В,Нб из аморфного бора.
НВО (г). Термодинамические свойства газообразного оксид-гидрида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 546.
Молекулярные постоянные НВО, использованные для расчета термодинамических функций,
приведены в табл. 20.8. Как показывают результаты квантовомеханических расчетов Томсона
и Уишарта [2691], Дилла и др. [954], Зюбиной и др. [137а], а также спектрального исследования
Лори и Портера [1888], молекула НВО в основном электронном состоянии Хг^+ имеет линейное
строение. Приведенный в таблице момент инерции соответствует структурным параметрам
г (В—Н)=1,18+0,02 А (как в ВН3) и г (В=О) = 1,20+0,02 А (как в ВО). Последние
удовлетворительно согласуются с результатами квантовомеханических расчетов [137а, 954, 2691]. Погрешность
значения / составляет 1-100 г-см2. Значения основных частот в табл. 20.8 рекомендованы для
Н"ВО Лори и Портером [1888], которые исследовали ИК-спектр Н"ВО, Н10ВО, DUBO и D10BO
в матрице из Аг. Не наблюдавшаяся в спектре НПВО частота vx рассчитана авторами [1888] по
силовой постоянной /b-d- Погрешность \ оценивается в 100 см, a v2 и v3 — в 10 см.
Согласно [Ю9б] молекула НВО имеет три триплетных возбужденных состояния с энергиями
30 000-40 000 см.
Термодинамические функции НВО (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
значений молекулярных постоянных, а также приближенным характером расчета и составляют
0,5, 2 и 3 Дж-К~1-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности из-за
неточности молекулярных постоянных не превышают 1 Дж-К-моль.
Ранее таблицы термодинамических функций НВО (г) рассчитывались во 2-м издании
настоящего справочника [105] и в справочнике JANAF [1544]. Расчет [105] выполнен по оцененным
значениям молекулярных постоянных, расхождения данных [105] и табл. 546 составляют 0,1 —
2,5 Дж-К-моль~1 в значениях Ф° (Т). В расчете [1544] учтены три триплетных состояния НВО,
•соответствующие расхождения с данными табл. 546 не превышают 0,8 Дж-К-моль.-
Константа равновесия реакции НВО (г)=Н (r)+B (г)+О (г) вычислена с использованием
значения &ГН° @)=1210,817 +25 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д//0(НВО, г, 0) = —188 + 25 кДж • моль.
Эта величина основана на результатах масс-спектрометрического измерения равновесия D2 (г) +
+В2О3 (ж)=БВО (r)-j-DBO2 (г), проведенного Фарбером и Фриш [1051] F серий опытов при
Tev=1260 К, константы равновесия в публикации не приведены). Авторы [1051] по II закону
36
Бор и его соединения ВОН (г), НВОг (к, ж)
нашли Дг#° A260 К)=457,7+6,3 кДж-моль, чему соответствует ДД° @)=511,9 кДж-моль'1
и приведенное выше значение kfH° (HBO, г, 0).
ВОН (г). Термодинамические свойства газообразного моногидроксида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 547.
Молекулярные постоянные ВОН, использованные для расчета термодинамических функций,
приведены в табл. 20.8. Экспериментальные данные о спектре и структуре ВОН в литературе
отсутствуют. Согласно квантовомеханическому расчету Зюбиной и др. [137а] молекула ВОН имеет
нелинейную структуру симметрии Са с геометрическими параметрами -$ В—О—Н=125+10°г
г (В—О) = 1,30+0,03 А и г (О—Н)=0,96+0,02 А. Приведенное в табл. 20.8 произведение
моментов инерции, соответствующее этим параметрам, имеет погрешность 1-1018 г3-см6. Для частоты
Vj (В—О) молекулы ВОН принято значение аналогичной частоты НВО2, а для va (В—О—Н) и
v3 (О—Н) — соответствующие характеристические частоты. Погрешности частот va и v2 могут
достигать 10—15%.
Термодинамические функции ВОН (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных, а также приближенным характером расчета и составляют 1,
2 и 3 Дж- К- моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К, в том числе 1—2 Дж- К- моль
из-за неточности молекулярных постоянных.
Таблица термодинамических функций ВОН (г) публикуется впервые.
Константа равновесия реакции ВОН (г)=В (г)+О (г)+Н (г) вычислена с
использованием значения &ГН° @)=1042,817+30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д//°(ВОН, г, 0) = —20 + 30 кДж-моль.
Принятая величина основана на разности энтальпий образования НВО и ВОН, вычисленной
в работе Зюбиной и др. [137а] (см. НВО).
НВО2 (к, ж). Термодинамические свойства кристаллической и жидкой метаборной кислоты
в стандартном состоянии в интервале температур 298,15—1500 К приведены в табл. Д. 43.
Значения постоянных, использованных для расчета термодинамических функций,
представлены в табл. 20.1. В справочнике за стандартное состояние НВО2 (к) принята кубическая
модификация у-НВО2 [2891а], устойчивая в интервале от Т=0 до Тт х. В литературе отсутствуют
какие-либо экспериментальные данные о теплоемкости и энтальпии НВО2 (к, ж). Оцененные
значения термодинамических функций при 298,15 К, приведенные в табл. 20.1, а также уравнение для
теплоемкости НВО2 (к) в интервале 298,15—509 К заимствованы из таблиц JANAF [1543].
Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К) — Н° @) составляют 5 Дж-IT1-моль
и 0,8 кДж-моль соответственно, а величин теплоемкости при Т ^> 298,15 К — приблизительно
10%. Температура плавления E09+1 К) принята по данным Крачека и др. [1769], полученным
для образца f-HBO2, содержащего 79,40% В2О3 при 79,45% по стехиометрии. Равновесное
давление водяного пара в точке плавления согласно [1769] равно 7,0 атм. Энтальпия плавления A4,3 +
+3,0 кДж-моль) и теплоемкость НВО2 (ж) A05+20 Дж-К~1-моль~1) оценены по простой
аддитивной схеме как полусумма соответствующих величин для Н2О и В2О3.
Погрешности вычисленных значений Ф° (Т) при 298,15, 500, 1000 и 1500 К оцениваются в 4,
6, 18 и 25 Дж-К~1-моль соответственно. Термодинамические функции НВО2 (к), приведенные
в табл. Д. 43 и в таблицах JANAF [1543], практически совпадают. Таблица термодинамических
функций НВО2 (ж) в литературе отсутствует.
В справочнике принята энтальпия образования
а, к, куб., 298,15 К) = —804,6 + 1,0 кДж • моль.
1 Кроме y-HBO2 известны' еще две менее стабильные модификации — ромбическая (а) и моноклинная (р) с
температурами плавления 449,15+0,2 К и 474,05+0,5 К соответственно [387, 409, 1769, 2199]. Температуры
превращения различных модификаций НВО2 неизвестны, за исключением температуры монотропного перехода
а -> р D33 К [2662]).
37
НВОа (г)
Глава 20
Принятое значение для термодинамически устойчивой кубической модификации основано на
измеренных Килди и Прозеном [1705] энтальпиях растворения у-НВО2 и Н3ВО3 в щелочи 1.
НВО2 (г). Термодинамические свойства газообразной метаборной кислоты в стандартном
состоянии в интервале температур 100—-6000 К приведены в табл. 548.
Молекулярные постоянные НВО2, использованные для расчета термодинамических функций,
приведены в табл. 20.19. Экспериментальные данные о строении молекулы НВО2 в литературе
Таблица 20.19. Значения молекулярных постоянных, а также о и р^, принятые для расчета
термодинамических функций НВО2, НВОН, F2BO, FBOH, С12ВО, С1ВОН
Молекула
нво2
НВОН
F.jBO
FBOH
СЦВО
С1ВОН
Состояние
I'M'
I'M'
Х2В2
Х*А'
3700
3600
1369
3600
1300
3600
205в
2750
856
1300
550
1250
^4
см-1
1050 1250
1250 1050
491 700
1250 1050
270 530
1050 830
570
1000
1450
500
1000
370
600
400
500
250
370
250
U*в1 с ¦ 1°117
г3 • см'
9,7
1,5
1.10»
45
11,7.10»
1,9.10*
а
1
1
2
1
2
1
Pi
1
2
2
2
2
2
отсутствуют. По аналогии с метаборатами щелочных металлов, структура которых исследована
методом газовой электронографии [9, 10, 122, 124, 125], в справочнике принимается, что в
основном электронном состоянии ХА' молекула НВО2 имеет угловую структуру симметрии С, с
линейной группировкой ОБО. Приведенное в таблице значение 1д1в1с вычислено по оцененным
структурным параметрам г (В=О) = 1,22+0,03 А, г (В—О) = 1,32+0,03 А, г (О—Н) =0,96+0,02 Аи
-^ Н—О—В = 115+10°. Оценка была выполнена на основании соответствующих величин в
молекулах В2О3, F2BOH и метаборатов щелочных металлов. Погрешность значения IaIbIo составляет
5-10-118 г3-смв.
В исследованиях ИК-спектров газообразной НВО2 в работах [232, 280, 1348, 2844] были
определены частоты \ и v2, приведенные в табл. 20.19. Отнесение Уайтом и др. [2844] полосы 1420 см
к частоте v3 вызвало сомнение у авторов последующих работ [232, 280, 1348], поэтому для этой
частоты, а также частот v5 и v6 приняты значения, оцененные по частотам метаборатов щелочных
металлов [437, 438, 2496] и молекулы В2О3. Частота деформационного колебания В—О—Н, (v4)
принята такой же, как в других молекулах, содержащих группу ВОН. Погрешности v3 и v4
оцениваются в 100 см, остальных частот — в 50 см. Можно ожидать, что возбужденные
электронные состояния НВО2 лежат в области высоких энергий, и в справочнике они не рассматриваются 2.
1 В работе [1705] были также измерены энтальпии растворения {!- и а-модификаций НВО2 в воде ив
щелочи,откуда были найдены их энтальпии образования —794,8±1,0 кДж-моль и —789,4±1,0 кДж-моль
соответственно. Близкие значения (—794,6±1,2 и —789,2±1,2 кДж-моль) были найдены Соколовой и др. [387].
В более ранних калориметрических исследованиях [1704, 2378, 2601] были получены менее точные
результаты, в основном из-за недостаточной чистоты исходных образцов.
Результаты исследований равновесия диссоциации Н3ВО3(к)={3-НВО2(к)+Н2О (г) [1200, 2006, 2661,2662]
и Н3ВО3 (к)=а-НВО2 (к)+Н20 (г) [679, 1847, 2661, 2662, 2680] были проанализированы Эвансом и др.
[1045а] и привели к значениям —795,7 и —789 кДж-моль, которые хорошо согласуются с измерениями
[387,1705], но уступают им по точности. На основании этих данных в справочнике в дальнейших расчетах
принимаются энтальпии образования
к, монокл., 298,15 К) = —794,7 + 1,0 кДж • моль,
Д^0 (а-НВО2, к, ромб., 298,15 К) = —789,3 + 1,0 кДж • моль.
2 После завершения работы над этим разделом Секачев [367а] исследовал ИК-спектр продуктов взаимодействия
молекул В2О3 и Н20, изолированных в матрице из аргона, и отнес три полосы D94, 2058 и 3733 см) к частотам
vB, v2 и vj молекулы HUB16O2. Расчет термодинамических функций НВО2 (г) на основании этих частот приводит
к величинам, согласующимся с данными табл. 548 в пределах 0,4—1,7 Дж-К^-моль в значениях Ф° (Г) и
1,1—1,8 Дж-К^-моль в значениях 5° (Г).
38
Бор и его соединения
НВОН (г)
Термодинамические функции НВО2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и составляют около 2, 5 и 8 Дж-К«моль"х
s значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Термодинамические функции НВО2 (г) вычислялись ранее в справочниках [105, 1543, 1904,
1974], а также в работах [1044, 2844, 2855]. Расхождения результатов этих расчетов и настоящего
издания не превышают 3—4 Дж-К-моль в значениях Ф°(Т) и обусловлены главным образом
отличием принятых значений основных частот.
Константа равновесия реакции НВО2 (г)=Н (г)+В (г)+20 (г) вычислена с использованием
значенияДг#° @)=1829,600+11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
^^НВО г, 0) = —560+10 кДж-моль,
основанной на данных, представленных в табл. 20.20.
Таблица 20.20. Результаты определений А.уН° (НВО2, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (HBOS, г, 0)
II закон
III закон
Штаккельберг п др., 1937 [2601 ] Протока, НВ02(к)=НВО2(г), 433 и 453 К, 2 точки —687 —689
Абрикосов и др., 1960 [16] Протока, VaBjjOs (ж)+1/!Н20 (г)=НВО2 (г), —545 —583
1113—1637 К, 7 точек. Данные представлены в
виде уравнения
Меши и др., 1960 [2009] Масс-спектрометрический, V2B2O3 (ж)+1/2Н2О (г)= —553±6 —555±8
=НВО2(г), 1061—1451 К, 10 точек
Тоже, V2B2O3 (r)+V2H2O (г)=НВО2 (г), 1323— —536±100 —555±10
1451 К, 4 точки
Уайт и др., 1960 [2844] Измерения интенсивности ИК-спектра испуска- —564±10 —
ния, V2B2O3 (ж)+1/2В2О (tf=DBO2 (г), 1250—
1450 К
Рандалл, Маргрейв, 1960 [2290] Протока, V2B2OS (ж)+х/гН,0 (г)=НВО2 (г), — —560±10
1273 К, 1 точка
Фарбер и др., 1960—1962 [1046а] Эффузионный,1/2В2О3(ж^+1/2Н2О(г)=НВО2(г), —570±10 —579±10
1071—1325 К
То же, 1143 К, 1 точка — —573
Та же реакция, протока, 1070—1323 К, —624±40 —561 ±10
То же, 1148 К, 1 точка — —563
Фарбер, Блауэр, 1962 [1049] Протока в условиях молекулярного течения, — 563±10
1/2ВгО3(ж)+1/2Н2О(гM=НВО2 (г), 1148 К.З точки
Соммер, 1963 [2580] Та же реакция, масс-спектрометрический — —572а
Рандалл, 1968 [2289а] Та же реакция, протока — —560а
Фарбер, Фриш, 1969 [1051] Масс-спектрометрический, ВаОз (ж)+В20 (г)= —552±10 —
=2DBO2(r), 1160-1405 К, 6 точек
а В этих работах не приведены измеренные константы равновесия, укачаны только вычисленные по ним
значения энтальпии образования.
Приведенные в таблице погрешности включают воспроизводимость измерений, неточности
термодинамических функций, сечений ионизации и использованных в расчетах термохимических
величин. Принятое значение является средневзвешенным из вычисленных по уравнению III
закона из данных [1046а, 1049, 2009, 2290] и по уравнению II закона из данных [1051, 2844].
Результаты измерений в работах [16, 2601] и, частично, данные эффузионных измерений в работе [1046а],
приводят к выпадающим значениям, и поэтому они не принимались во внимание.
НВОН (г). Термодинамические свойства газообразного гидрид-гидроксида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 549.
39
В(ОНJ (г) Глава 20
Молекулярные постоянные, использованные для расчета термодинамических функций НВОН (r)f
приведены в табл. 20.19. Структура и спектры молекулы НВОН не исследовались. Произведение
моментов инерции IaIbIc в состоянии Х2А', приведенное в таблице, вычислено для плоской
структуры этой молекулы (симметрия Cs) по структурным параметрам, оцененным сравнением с
молекулами В(ОНJ, ВН2 и BHF: -? Н-В-О=125+5°, *$ В—О—Н=115+10° (трансположение),.
г (В—О)=1,34+0,03 А, г (В—Н)=1,18+0,05 А, г (О—Н)=0,96 +0,02 А. Погрешность значения
IaIbIc составляет 0,5-10~117 г3-см6. Принятые значения частот валентных колебаний О—Н (vi),
В—Н (v2), В—О (v4) и деформационных колебаний В—О—Н (v3) и Н—В—О (v5) оценены по
соответствующим частотам молекул BHF, BFOH, ВСЮН, В(ОНJ. Частота v6 крутильного
колебания ОН-группы рассчитана по высоте барьера вращения E000+2000 см), принятого таким же,.
как в молекуле Н2ВОН. Погрешности частот v2 и v5 оцениваются в 200 см, остальных — в 100 см.
Сведений о возбужденных электронных состояниях НВОН в литературе нет, по аналогии с
молекулой BHF можно предполагать, что их энергии превышают 15 000 см.
Термодинамические функции НВОН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных, а также приближенным
характером расчета и составляют 3, 5 и 8 Дж-К-моль в значениях Ф° (Т) при 298,15, 300О
и 6000 К (в том числе 1,5—3 Дж-К~1-моль из-за неточности молекулярных постоянных).
Таблица термодинамических функций НВОН (г) публикуется впервые.
Константа равновесия реакции НВОН (г)=В (г)+2Н (г)+О (г) вычислена с использованием
принятого в справочнике значения
ДД° @) = 1305 + 25 кДж ¦ моль.
Это значение было оценено в предположении о равенстве средних энергий связей В—Н ш
В-ОН в молекулах НВОН, ВН3 и В(ОНJ.
Принятой величине соответствует
Д^Я0 (НВОН, г, 0) = —66,149 + 25 кДж • моль.
В(ОНJ (г). Термодинамические свойства газообразного дигидроксида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 550.
Молекулярные постоянные В(ОНJ, использованные для расчета термодинамических функций,,
приведены в табл. 20.11.
В литературе отсутствуют сведения об экспериментальных измерениях или квантовохимиче-
ских расчетах молекулярных постоянных В(ОНJ. В настоящем справочнике оценки этих
постоянных сделаны в предположении, что молекула В(ОНJ представляет собой фрагмент молекулы
Н3ВО3 (плоская структура симметрии Cs) и имеет дублетное основное электронное состояние 2А'.
Структурные параметры В(ОНJ, использованные для расчета IaIbIc, перенесены из В(ОНK:
г (В-О) = 1,34+0,03 А, г (О—Н)=0,96+0,02 А, ^ В-О—Н=115+10° и ^ О-В-О = 120+5°.
Погрешность вычисленного значения IaIbIc может достигать 1-1018 г3>см6. В качестве основных
частот В(ОНJ взяты округленные значения соответствующих основных частот Н3ВО3: валентные
колебания О—Н (vx и v2), валентные колебания В—О (v3h vg), деформационные колебания В—О—Н
(v4 и v5), деформационное колебание О—В—О (v7) и крутильные колебания О—Н (v8 и v9).
Погрешности двух последних частот могут достигать 150 см, для v7 — 50 см и для остальных-
частот 100 см.
Термодинамические функции В(ОНJ (г) вычислены по формулам A.3)—A.6), A.9), A.10),.
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью
принятых значений молекулярных постоянных, а также приближенным характером расчета (в том числе-
учетом внутренних вращений как гармонических колебаний) и составляют 4,10 и 15 Дж- К- моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее таблица термодинамических функций В(ОНJ (г) была рассчитана в справочнике JANAF
[1543]. Оценка молекулярных постоянных В(ОНJ в [1543] была сделана в предположении
структуры симметрии C2h с линейным фрагментом О—В—О. Вследствие этого расхождения
40
Бор и его соединения НгВОН (г>
между результатами расчета [1543] и данными табл. 550 составляют 10—15 Дж-К-моль
в значениях Ф° (Т) и S° (T).
Константа равновесия реакции В(ОНJ (г)=В (г)+2О (г)+2Н (г) вычислена с использованием
значения ДГЯ° @)=1915,634+40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования:
bfH° (В (ОНJ, г, 0) = —430 ± 40 кДж ¦ моль,
оцененной сопоставлением средних энергий связей в молекулах В(ОНJ, В(ОНK, BF2, BF3, ВС12
и ВС13.
Н2ВОН (г). Термодинамические свойства газообразного дигидрид-гидроксида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 551.
Молекулярные постоянные, использованные для расчета термодинамических функций
Н3ВОН (г), приведены в табл. 20.11. Структура и спектры молекулы Н2ВОН не исследовались.
Произведение моментов инерции IaIbIo в состоянии ХгА', приведенное в таблице, вычислено для
плоской структуры этой молекулы (симметрия Са) для структурных параметров -$ Н—В—0 =
= 4 Н-В-Н=120+3°, ^ В-О—Н=115+5°, г (В—О) = 1,33±0,03 А, г (В—Н)=1,18+0,05 А
и г (О—Н) =0,96+0,02 А. Эти параметры были приняты на основании сопоставления результатов-
двух неэмпирических расчетов строения Н2ВОН, выполненных Диллом и др. [954] (транс —
4 Н—В—0=121,8°, цис — J* Н-В-О=117,2°, ^ Н—В—Н=121°, ^ В-О—Н=112,3°,
Г(В-О)=1,334 А, транс — г (В—Н) = 1,165 А, цис — г (В—Н) = 1,163 А, г(О-Н)=0,984 А,.
барьер вращения ОН-группы F0=5036 см) и Гропеном и Иохансеном [1272] (-$ В—О—Н=114°,
r(B—O)=l,33 A hV0=5735 см при фиксированных значениях ^ Н—В—О = -$ Н—В—Н=120°,
г (В—Н)=1,20 А, г (О—Н)=0,95А). Погрешность приведенного значения IaIbIo составляет
1-10~117г3-см6. Принятые значения частот валентных колебаний О—Н (\), [В—Н (v2, v3) и
В—О (v7), деформационных колебаний Н—В—Н (v4), Н—В—О (ve), В—О—Н (v6) и зонтичного-
колебания (vg) были оценены на основании соответствующих частот молекул BH2F, F2BOH и
В(ОНK, принятых в настоящем справочнике. Частота v9 крутильного колебания ОН-группы
соответствует высоте барьера внутреннего вращения Fo=5000+1000 см, принятого по результатам
расчетов [954, 1272]. Погрешности значений v2, v3, v4) v6 оцениваются в 200 см", v9 — в 150 см,
остальных частот — в 100 см. Сведения о возбужденных электронных состояниях Н2ВОН
отсутствуют, по аналогии с молекулами ВН3 и В(ОНK можно предполагать, что они имеют высокие
энергии.
Термодинамические функции Н2ВОН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета и составляют 4, 6 и 10 Дж-К-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К (из них 2—4 Дж-К-моль из-за неточности молекулярных
постоянных).
Таблица термодинамических функций Н2ВОН (г) публикуется впервые.
Константа равновесия реакции Н2ВОН (г)=В (г)+ЗН (г)+О (г) вычислена с использованием
значения &ГН° @)=1736,885 +11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(H2BOH, г, 0) = —282 + 10 кДж • моль.
Принятая величина основана на исследовании равновесия реакции 1/3В2Н6 (г)+1/3В(ОНK (к) =
=Н2ВОН (г), C00 К, 1 точка), выполненного Портером и Гуптой |2255а]. В этом исследовании
газообразный диборан помещался в реакционный сосуд с большим избытком борной кислоты.
Смесь выдерживалась в течение нескольких недель и затем манометрически измерялось
равновесное давление диборана и масс-спектрометрическим методом измерялось отношение интенсивностей
ионных токов НаВОН+ и В2Нд в равновесной смеси. Погрешность принятой величины оценена
с учетом погрешности измерений, термодинамических функций и энтальпий образования В2Н6 (г)
и В(ОНK (к).
Оценка в предположении о равенстве средних энергий связей В—Н и В—ОН в молекулах
Н2ВОН, ВН3 и В(ОНK приводит к менее отрицательной величине —267+15 кДж-моль. Авторы
41
ИВ (ОНJ (г)
Глава 20
работы [2255а] полагают, что эта разность может быть объяснена бблыпей стабильностью
несимметричной молекулы Н2В0Н по сравнению с симметричными ВН3 и В(ОНK. Однако в случае
НВ(ОНJ разность составила только 4 ккал/моль, что на наш взгляд не подтверждает это
объяснение.
НВ@НJ (г). -Термодинамические свойства газообразного гидрид-дигидроксида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 552.
Молекулярные постоянные, использованные для расчета термодинамических функций НВ(ОНJ,
приведены в табл. 20.21. Произведение моментов инерции IjJbIg в состоянии %хА' (симметрия С,)
Таблица 20.21.
Значения молекулярных постоянных и я в основном электронном состоянии (p_g=i),
принятые для расчета термодинамических функций НВ(ОНJ, Н3ВОз, В2(ОНL, Н3В3Оз,
BaF4, F3B3O3, FB(OHJ, CI3B3O3, С1В(ОНJ и BHSNH3
Молекула
НВ(ОНJ
Н3ВОз
В2(ОН)«
НзВзОз
B2F4
F3B3O3
FB(OHJ
С1зВ3Оз
С1В(ОНJ
BHsNHs
Молекула
НВ(ОНJ
НзВО8
В2(ОН),
НзВзОз
B2F4
F3B3O3
FB(OHJ
ClsBsO,
С1В@НJ
BHaNHs
В2(ОНL :
BjF. : •
BH3NH3 : '
v3 1 v4
CM
3600 3600 2600 1500
3600 1080 880 625
3600D) 1400 1320B) 1200B)
2616 906 800 1477
1404 673 318 а
1372 826 495 1416
3600 3600 1500 1300
1037 807 333 1379
3600 3600 1500 1200
968 3337 2340 1343
V,
v10 I vu
1
v12
CM
900 600 600 540
540B) 600B) — —
600D) 575 400 300B)
1389 B) 1213 B) 990 B) 530 B)
1375 150 1151,2 541
1373B) 964B) 462 316B)
500 700 600 600
390B) 150B) 664 150
370 610 600 600
1608 B) 1301 B) 1186 B) 603 B)
1 F0=0, /ир=0,4.10-88 г-см*. om=l, nm=2.
1200
600
1150
1197
1368
646
1200
375
1100
1052
Vl3
—
150
1050 B)
—
630B)
—
480 B)
—
—
»Fo=147cm-i, /пр=0,394-10-*8 г-см2, om=2, nm=2.
1 Fe=675 см, /np=0,027-10-38 г-см*, om=3, л
m=3.
1200
3600 B)
1100 B)
918
326
711
1100
1345 B)
900
a
v14
_
—
a
343 B)
—
185 B)
—
150 B)
—
—
1100
1490 B)
700
380
657
170
900
1183 B)
700
3386 B)
IA1BIC ¦ 10
Г3 • CM*
105,55
11.102
2,14-10*
5820
2,55-10*
2,1-10»
1,1.10»
1,9.10s
3,6-103
24
""8
900
1200 B)
700
2620 B)
380
1436 B)
540
760 B)
540
2415 B)
117
0
1
3
2
6
4
6
1
6
1
3
вычислено по вращательным постоянным Ао, Во и Со, найденным Кавашима и др. [1644а] при ис-
•следовании микроволнового спектра НиВ(ОН)я (Л0=2,0511A24) см, ?0=0,342 941 (9) см,
€¦„=0,293 694 (9) см) и Н10В(ОНJ D0=2,1093 B25) см, ?0=0,342903 (9) см, Со=
=0,294933 (9) см), после пересчета этих вращательных постоянных на природную смесь
изотопов бора. Кавашима и др. [1644а] на основании этих данных и результатов исследования
молекул FjBOH и FB(OHJ, приняли, что в молекуле НВ(ОНJ углы вокруг атома бора равны 120°,
-4 В—О—Н=114,1°, г (В-О)=1,344 А, г (В—Н)-1,189 А и г (О—Н)=0,941 А. Спектры моле-
жулы НВ(ОНJ не исследовались. Принятые значения частот колебаний О—Н (vx и va), В—О
42
Жор и его соединения Н3ВО3 (к, ж)
(v4 и vg) и В—Н (v3), деформационных колебаний О—В—О (v12), В—О—Н (v5, v7) и Н—В—О (ve),
зонтичного колебания (v9) и крутильных колебаний О—Н групп (vj0 и vu) оценены по
соответствующим частотам колебаний молекул В(ОНK, FB(OHJ и BHF2, принятым в настоящем справочнике.
Погрешности частот v10 и vn оцениваются — в 150 см, \—v5 и v7—v9 — в 100 см, v6 — в 200 см,
v12 — в 50 см. Сведения о возбужденных электронных состояниях НВ(ОНJ отсутствуют, но
можно предполагать, что их энергии достаточно высоки.
Термодинамические функции НВ(ОНJ (г) вычислены по уравнениям A. 3)—A. 6), A. 9),
<1. 10), A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических
•функций обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета и составляют 6, 10 и 15 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000
и 6000 К (в том числе 1—5 Дж-К~1-моль~1 из-за неточности молекулярных постоянных).
Таблица термодинамических функций НВ(ОНJ (г) публикуется впервые.
Константа равновесия реакции НВ(ОНJ(г)=В (г)+ЗН(г)+2О (г) вычислена с использованием
значения ДГЯ° @) =2335,668 +11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(HBe(OHJ, г, 0) = —634 + 10 кДж-моль.
Эта величина получена в результате обработки данных исследования равновесия реакции
Л/6В2Н6 (г)+2/3В(ОНK (к)=НВ(ОНJ (г), C00 К, 1 точка), выполненного Портером и Гуптой
|2255а] (см. текст по Н2ВОН). Оценка в предположении о равенстве средних энергий связей В—Н
л В—ОН в молекулах НВ(ОНJ, ВН3 и В(ОНK приводит к совпадающей величине — 630+
+ 15 кДж-моль.
Н3ВО3 (к, ж). Термодинамические свойства кристаллической и жидкой ортоборной кислоты
в стандартном состоянии при температурах 100—800 К приведены в табл. Д. 44.
Значения постоянных, использованных для расчета термодинамических функций,
представлены в табл. 20.1. В настоящем справочнике за стандартное состояние Ы3ВО3 (к) принята триклин-
ная модификация [2891], устойчивая в интервале от Т=0 до Тт х. При Т <^ 298,15 К
термодинамические функции вычислены по результатам измерений теплоемкости образца Н3ВО3 (к)
¦с содержанием примесей не более 0,05%, выполненных Джонстоном и Керром [1599] A7—301 К)
•с точностью ~0,5% при Т > 50 К и ~1 % при более низких температурах. Экстраполяция
теплоемкости при Т < 17 К приводит к значению S° A7 К)=0,75 Дж-К~1-моль. Аномальное
изменение кривизны зависимости теплоемкости от температуры вблизи 80 К связано с разупорядоче-
нием водородных связей в Н3ВО3. Погрешность принятых по [1599] значений S0 B98,15 К) и
Н° B98,15 К) — Н° @) (см. табл. 20.1) оценивается в 0,5 Дж-К^-моль и 0,06 кДж-моль
•соответственно.
Экспериментальные данные о теплоемкости и энтальпии Н3ВО3 (к, ж) при Т > 298,15 К,
а также об энтальпии плавления Н3ВО3 в литературе отсутствуют. Для теплоемкости в интервале
298,15—444,1 К используется уравнение, аппроксимирующее принятое в табл. 20.1 значение
€'р B98,15 К) и значения С° (Т) при более высоких температурах, оцененные в справочнике
f 1543] сравнением теплоемкости Н3ВО3 и составляющих ее окислов (В2О3 и Н2О). Температура
плавления D44,1 +0,3 К) принята по данным Крачека и др. [1769], полученным для весьма
чистого образца Н3ВО3. Равновесное давление Н2О в точке плавления определено в этой работе
равным 4,0 атм. Близкие значения Тт (в пределах 442,7—443,7 К) были получены в работах [2000,
-2601]; найденное в [654] значение 454 К завышено. Энтальпия плавления B2,3+4,0 кДж-моль)
и теплоемкость Н3ВО3 (ж) A80+30 Дж-К'^моль) оценены, как и для НВО2 (к, ж), по аддитив-
дой схеме.
Погрешности вычисленных значений Ф° (Г) при 298,15, 500 и 800 К оцениваются в 0,3, 4 и
13 Дж-К'моль соответственно. Термодинамические функции Н3ВО3 (к), приведенные
в табл. Д.44 и в таблицах JANAF [1543], практически совпадают. Таблица термодинамических
«функций НЭВО3 (ж) публикуется впервые.
Согласно данным [734] при высоких давлениях образуются ромбическая (р > 42 000 атм) и тетрагональная
модификация (р > 57 000 атм).
43
Глава 2ft>
В настоящем справочнике принята энтальпия образования
Д/Я°(Н,ВО8, к, 298,15 К) = —1094,9 + 0,8 кДж • моль,
основанная на энтальпии образования В2О3 (к), измерениях энтальпии растворения В2О3 (к)
в воде в работах Джонсона, Хаббарда [1578], Саузарда [2588] и Фазолино [1069] и измерениях
энтальпии растворения Н3ВО3 в работах Смиско, Мейсон [2543], Ван Артсдалена, Андерсона
[2752], Килди, Прозена [1705], Фазолино [1069] и Девиной и др. [108а]. Анализ этих данных
был выполнен при подготовке рекомендаций КОДАТА—MCHG [864]. Гуд и др. [1230] измерили
энтальпию сгорания бора в кислороде в присутствии плавиковой кислоты и энтальпию
растворения Н3ВО3 (к) в плавиковой кислоте. Из этого термохимического цикла было вычислено значение-
энтальпии образования, равное — 1094,4+1,0 кДж-моль, прекрасно согласующееся с
принятой величиной.
Н3ВО3 (г). Термодинамические свойства газообразной ортоборной кислоты в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 553.
Молекулярные постоянные Н3ВО3, использованные для расчета термодинамических функций,
приведены в табл. 20.21. Структурные параметры были оценены на основании данных о строении
Н3ВО3 в кристаллическом состоянии и в растворах [892, 893, 2890, 2891], а также данных о
строении молекулы F2BOH (см. ниже). Для Н3ВО3 в основном электронном состоянии 1А' принята
плоская конфигурация (симметрия C3h) с параметрами «^ О—В—О=120° ,^ В—О—Н = 115+10°,
г (В—О)=1,34+0,03 А, г (О—Н) =0,96 + 0,02 А. Соответствующее значение Ц1в1о (см. таблицу)
имеет погрешность около 2-Ю15 г3-см6. Измерения основных частот Н3ВО3 в кристаллическом
состоянии и в растворах проводились во многих работах [215, 381, 506а, 678, 755, 827, 998, 1001af
1237, 1402а, 1481а, 1612а, 1775а, 1784а, 1967а, 2029а, 20386, 2230, 2494, 2761а, 2778, 2779], и
только в одном неопубликованном исследовании Грина и др. согласно справочнику [1543] был
получен ИК-спектр паров Н3ВО3, в котором наблюдались три сильные C220, 1490, 1199 см)
и две слабые (881 и 669 см) полосы. Наиболее полное исследование ИК-спектра пленок, а также-
спектра КР порошков и растворов Н3ВО3, D3BO3 и Н310ВО3 выполнено Дюригом и др. [998].
В этой работе сделано отнесение основных частот Н3ВО3 в конденсированном состоянии в
предположении о наличии значительных межмолекулярных взаимодействий (водородных связей).
Наибольшее влияние эти взаимодействия оказывают на частоты, связанные с колебаниями, в которых
участвуют атомы Н: валентные колебания О—Н (vx и v6) и крутильные колебания группы В—О—Н
(v5 и v10). Для двух последних в работе [998] найдены значения свыше 800 см, чему соответствует
барьер внутреннего вращения около 8000 см. Следует ожидать, что в свободной молекуле Н3ВОз:
барьер должен быть меньше. Так, квантовомеханические расчеты молекул Н2ВОН [954, 1107а]
и F2BOH (см. ниже) позволяют предполагать, что он составляет 4000 +2000 см. Значения
крутильных частот F00+150 см), приведенные в табл. 20.21, соответствуют этой величине. Для \ и ve
выбрано характеристическое значение C600+100 см), хотя в спектрах конденсированной Н3ВОз;
из-за наличия водородных связей эти частоты равны примерно 3200 см. Значения остальных
основных частот Н3ВО3 (деформационное колебание В—О—Н, v2 и v8; валентное колебание В—О,.
v3 и v7; зонтичное колебание ВО3, v4; деформационное колебание О—В—О, v9) приняты по
рекомендации Дюрига и др. [998] (согласующейся в основном с результатами других работ). Погрешности
этих частот не превышают 20 см. Следует отметить, что из упомянутых выше полос в ИК-спектре
паров Н3ВО3 две можно с уверенностью отнести к v7 и v8.
Термодинамические функции Н3ВО3 (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных значений обусловлены приблизительно в равной мере
неточностью принятых молекулярных постоянных и приближенным характером расчета (в том числе
учетом внутренних вращений в приближении гармонического осциллятора). Эти погрешности
могут достигать 6, 12 и 20 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции Н3ВО3 (г) рассчитывались во 2-м издании настоящего»
справочника [105], в справочниках JANAF [1543], Мак-Брайд и др. [1974] и ряде других работ-
[1044, 1904, 1946, 2428, 2838, 2855]. Расхождения результатов различных расчетов обусловлены
главным образом различием вычисления составляющей внутреннего вращения (за исключением
расчетов [1904, 2855], содержащих, по-видимому, ошибки, и расчетов [1946, 2428, 2838], в
которых не приведены использованные молекулярные постоянные, что затрудняет анализ их резуль-
44
Вор и его соединения
В2 (ОНL (г)
татов). Расчет термодинамических функций Н3ВО3 (г) в предыдущем издании справочника [105]
выполнен в предположении заторможенного внутреннего вращения групп О—Н с очень низким
барьером. Это приводит к расхождениям с данными настоящего издания, достигающим около
-38 Дж-К~1-моль~1 в значениях Ф° (Т). Приблизительно такие же расхождения имеют место со
-справочником JANAF [1543], где принято свободное вращение этих групп. С расчетами Мак-
Брайд и др. [1974] и Эванса [1044] расхождения достигают 17 Дж-К~1-моль в значениях
Ф° F000 К), так как эти авторы частоту крутильного колебания v12 приняли равной около 200 см.
Константа равновесия реакции Н3ВО3 (г)=ЗН (г)+В (г)+30 (г) вычислена с использованием
значения ДГЯ° @) =2940,451+11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я°(Н8ВО81 г, 0) = —992 + 10 кДж.моль.
Эта величина основана на хорошо согласующихся между собой результатах исследований
равновесия реакции Н3ВО3 (к)=Н3ВО3 (г) в работах [324, 1550а, 2601] (см. табл. 20.22).
Измерения проведены при низких температурах, и погрешности в энтальпии реакции из-за неточности
термодинамических функций не превышают 2 кДж-моль. Расчеты по результатам масс-спектро-
метрических измерений [2009] носят приближенный характер.
Таблица 20.22. Результаты определении Ayff° (Н3ВОз, г, 0) (в кДж-моль г)
Авторы
Метод
AfH°(EsBO3, г, 0)
II закон
III закон
Штакельберг и др., 1937 [2601] Протока, Н3ВО3 (к)=Н3ВО3 (г), 382—413 К, —978±7 —992±1
6 точек
То же, НВО2 (к)+Н2О (г)=Н3ВО3 (г), 415— —993+7 —1006+3
453 К, 10 точек
То же, НзВОз (г)=НВО2 (г)+Н2О (г), 433 и 453 К, — —901 +60
2 точки
Джальмес и Галак, 1937 [1550а] Протока, Н3ВО3 (к)=Н.,ВО3 (г), 377—413 К, —985±5 —992±1
24 точки
Меши и др., 1960 [2009] а Масс-спектрометрический, 1/2В2О3 (ж)+3/2Н2О (г)= — —1085
=Н3ВОз (г), 1451 К, 1 точка
То же, V2B2O3 (г)+3/2НаО (г)=Н3ВО3 (г) - -1086
То же, Н3ВО3 (г)=НВО2 (г)+Н2О (г) — —1091
Рандалл, 1968 [2289а] Протока, ^^Оз (ж)+3/2Н2О (г)=Н3ВО8 (г), — —982
экспериментальные данные не приведены
Петропавловский и Торочешников, Протока, HSBO3 (к)=Н3ВОз (г), 383—415 К, дан- —1003 —992±2
1969 [324] ные представлены в виде уравнения
а Значения констант равновесия приближенно оценены по интенсивностям ионных токов, приведенным в
работе [2009],
В2 (ОНL (г). Термодинамические свойства тетрагидроксида дибора в стандартном состоянии
;в интервале температур 100—6000 К приведены в табл. 554.
Молекулярные постоянные В2(ОНL, использованные для расчета термодинамических функций,
приведены в табл. 20.21. Экспериментальные данные о строении молекулы В2(ОНL в литературе
отсутствуют. В справочнике принимается, что В2(ОНL имеет плоское строение и состоит из двух
трупп В(ОНJ (симметрия C2h). Величина г (В—В)=1,70+0,07 А принята такой же, как в
молекуле B2F4, а остальные параметры, как в молекуле В(ОНJ. Величина IaIbIo, приведенная
в табл. 20.21, имеет погрешность около 4-101* г3-см8. Единственное исследование спектра В2(ОНL
выполнено Никитиным и др. [284]. Авторы [284] получили ИК-спектр суспензии порошка
В2(ОНL и B2(ODL в области 400—4000 см и выполнили отнесение ряда основных частот.
В табл. 20.21 по данным [284] приведены частоты антисимметричных валентных колебаний
В—О (v3) и деформационного колебания О—В—О (v10). Полоса 1150 см отнесена к симметричному
валентному колебанию В—О (v5), хотя авторы [284] приписали ее валентному колебанию В—В.
45
(г) Глава 2»
Для последнего (v2) принято значение, близкое к соответствующей частоте молекулы B2F4. Также
по аналогии с B2F4 приняты значения основных частот v7 и vu (веерные колебания В(ОНJ), \
(симметричное валентное колебание В—О) и v12, v13 (деформационные колебания О—В—О).
Значения частот валентных колебаний О—Н (v2), деформационных колебаний В—О—Н (v4 и ve) в
крутильных колебаний О—Н (v9) приняты такими же, как в молекулах Н3ВО3 и В(ОНJ. Наконец,
крутильное колебание вокруг связи В—В, поскольку соответствующая частота по аналогии с
молекулами B2F4 и В2С14 должна быть очень низкой, рассматривается в настоящем справочнике
как свободное внутреннее вращение. Значение /пр для группы В(ОНJ, вычисленное по принятым:
структурным параметрам, приведено в таблице, его погрешность составляет 1-Ю9 г-см2.
Погрешности принятых значений основных частот составляют 150 см'1 для v9, 100 см для vx—ve и 50 см
для остальных. Мультиплетность основного электронного состояния принята равной 1. Сведения
о возбужденных электронных состояниях В2(ОНL отсутствуют.
Термодинамические функции В2(ОНL (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130), без учета возбужденных электронных состояний; составляющие свободного внутреннего,
вращения рассчитывались по уравнениям A. 164)—A. 165). Погрешности вычисленных таким
образом функций определяются как неточностью молекулярных постоянных, так и приближенным
характером расчета (в том числе учетом заторможенных внутренних вращений О—Н как
гармонических колебаний) и составляют 8, 15 и 25 Дж-К-моль в значениях Ф° (Т) при 298,15,3000*
и 6000 К.
Ранее термодинамические функции В2(ОНL (г) рассчитывались в справочнике JANAF [1543)
до 6000 К в предположении свободного вращения вокруг связи В—В. Расхождения результатов,
расчета [1543] с данными, приведенными в табл. 554, достигают 14 Дж-К-моль в значении
Ф° F000 К) и объясняются главным образом различием основных частот В2(ОНL (в справочнике-
[1543] для крутильных частот О—Н принято заниженное значение 200 см).
Константа равновесия реакции В2(ОНL (г)=2В (г)+4О (г)+4Н (г) вычислена с
использованием значения Дг#° @) =4221,268+100 кДж-моль, соответствующего принятой в справочнике-
энтальпии образования
г, 0) = —1250 ±100 кДж-моль.
Эта величина оценена в предположении о равенстве средних энергий связи В—В в молекулах
Вг(ОНL, B2F4 и В2С14.
Н3В3Оз (г). Термодинамические свойства газообразного бороксина в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 555.
Молекулярные постоянные Н3В3О3, использованные для расчета термодинамических функций,,
приведены в табл. 20.21. Электронографическое исследование паров Н3В3О3, выполненное Чангом
и др. [835], показало, что эта молекула имеет плоское циклическое строение (симметрия D3h) с
параметрами г (В—Н) = 1,192+0,017 А, г (В—О)=1,3758+0,0021 A, ^O-B-O = ->iB-O—В =
=120+0,04° и «$Н—В—О=120°. Соответствующее значение IaIbIc, приведенное в таблице,
имеет погрешность 1-Ю18 г3-см6. Инфракрасный спектр Н3В3О3 исследовался в гэзоеой фазе-
[282, 1267, 1837а, 1838, 2806] и в матрице из Аг [1616]. В работе Гримма и др. [1267] изучены
различные изотопные модификации Н3В3О3 и выполнен расчет колебательного спектра. На
основании экспериментальных данных [1267], находящихся в хорошем согласии с результатами работ
[282, 1616, 1837а, 1838, 2806], в табл. 20.21 приведены основные частоты v6, v8—v12, активные-
в ИК-спектре. Погрешности этих значений составляют около 10 см. Для остальных частот
приняты значения, рекомендованные Калдором и Портером [1616] из данных по составным полосам
и по теоретическому расчету. Их погрешности могут достигать 30 см. Сведения о возбужденных
электронных состояниях Н3В3О3 в литературе отсутствуют.
Термодинамические функции Н3В3О3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных величин определяются неточностью молекулярных
постоянных, приближенным характером расчета, а также пренебрежением различием молекулярных
постоянных изотопических модификаций Н3В3О3; они составляют 4, 10 и 15 Дж-К-моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности, обусловленные неточностью
молекулярных постоянных, не превышают 1—2 Дж-К~1-моль~1.
46
Бор и его соединения Н3В3Ов (г>
Ранее термодинамические функции Н3В3О3 (г) вычислялись в справочнике JANAF [1543].
Расхождения в значениях Ф° (Т), табулированных в [1543J и в настоящем издании справочник»
достигают 17 Дж-К~1-моль~1. Они обусловлены тем, что выбор значений основных частот в [1543];
был сделан некорректно и противоречит рекомендациям [1267, 1616].
Константа равновесия реакции Н3В3О3 (г)=ЗН (г)+ЗВ (г)+ЗО (г) вычислена с использование»*
значения А,Л° @) =4260,451 +18 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^ЧНзВзОз, г, 0) = —1192 ± 10 кДж • моль.
Шолетт и Портер [2512] масс-спектрометрическим методом исследовали равновесие реакцию
В2О3 (ж)+В (к)+3/2Н2 (г)=Н3В3О3 (г) A290—1481 К, 10 точек), этим данным соответствуют
значения &.fH° (Н3В3О3, г, 0) =—1219+85 кДж-моль (II закон) и —1294+20 кДж-моль (III
закон). Портер и Гупта [2255] исследовали равновесие реакции Н3В3О3 (г)=1/аВ2Нв (г)+В2О3 (к>
в статических условиях, парциальные давления компонентов реакции определялись масс-спектро-
метрически. Измерения были выполнены при 258, 300 и 348 К и привели к значениям Д^Я°(Н3В3О3,-
г, 0) =—1217+130 кДж-моль (II закон) и —1189+11 кДж-моль (III закон). В этой же работе
была исследована обратная реакция C00 К, 9 измерений), bfH° (Н3В8О3, г, 0) =—1191 +6 кДжХ
Хмоль, а также аналогичная реакция со стеклообразным В2О3 C00 К, 5 измерений),.
A^°(H3B3O3, г, 0)==—1195+6 кДж-моль. Указанные погрешности учитывают
воспроизводимость измерений и неточность термодинамических функций.
Принятое значение основано на данных работы [2255], где искомая величина была получена*
тремя независимыми путями. Авторы работы [2512] отмечают, что их измерения могли быть
искажены в результате того, что термодинамические свойства компонентов в смеси В2О3 (ж)+В (к)»
заметно отличаются от свойств соответствующих чистых компонентов.
Н3В3О6 (г). Термодинамические свойства газообразного гексоксид-тригидрида трибора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 556.
Молекулярные постоянные Н3В3О6, использованные для расчета термодинамических функций,,
приведены в табл. 20.17. В литературе отсутствуют экспериментальные данные о молекулярных
постоянных Н3В3Ов в парах. По аналогии с молекулой Н3В3О3 (см. выше), а также на основании
данных рентгеноструктурного анализа орторомбической модификации метаборной кислоты,,
характеризующейся образованием кристаллического тримера Н3В8О6 [2211], в справочнике
принимается, что молекула Н3В3О6 в основном электронном состоянии ХА' имеет плоскую
циклическую структуру симметрии CSh со структурными параметрами: г (В—О)=1,38+0,02 А (в цикле)
и 1,34+0,02 А (вне цикла), г (О—Н) =0,96+0,02 А, ^В—О—В=-?Ю—В—О=120°, ^В-О-Н=
=115+10°. Произведение моментов инерции, вычисленное по этим параметрам (см. таблицу),
имеет погрешность около 3-1013 г3-смв. Основные частоты Н3В3О6 приняты по данным Парсона
[2189], исследовавшего колебательные спектры Н3В3О6 (к). Эти частоты существенно отличаются
от оцененных Уайтом и др. [2844] из данных по спектрам молекул (RBOK, но согласуются с
соответствующими частотами, принятыми в настоящем справочнике для аналогичных молекул.
Погрешности в частотах могут составлять примерно 10—15% от их величины. Возбужденные
электронные состояния молекулы Н3В3О6 в справочнике не рассматриваются.
Термодинамические функции Н3В3О6 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), 1. 10),
A. 122)—A. 124), A. 130) и A. 128) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета и составляют около 8, 15 и 25 Дж-К-моль*
в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно (в том числе из-за неточности
молекулярных постоянных 1—3 Дж-К-моль во всем интервале температур).
Термодинамические функции Н3В3О6 (г) вычислялись ранее в справочниках [1543, 1974] и*
в работе Уайта и др. [2844] по молекулярным постоянным, оцененным авторами последнего
расчета. Различие главным образом основных частот приводит к расхождениям с данными
настоящего издания, достигающим для [1543, 1974] 21 Дж-К~1-моль в значениях Ф° (Т).
Константа равновесия реакции Н3В3Ов (г)=ЗН (г)+ЗВ (г)+6О (г) вычислена с
использованием значения Д^Г3 @) =6073,8 +27 кДж'Моль, соответствующего принятой в справочнике
энтальпии образования
Д^0 (Н3В3Ов, г, 0) = —2265 + 25 кДж • моль.
47
BF (г) Глава 20
Принятая величина является средней из рассчитанных по результатам масс-спектрометриче-
ского исследования равновесия реакции В2О3 (ж)+Н3ВО3 (г)=Н3В3О6 (г), выполненного Мепш и
Чапкой [2009] (—2253+30 кДж-моль, 1451 К, 1 точка) и равновесия реакции ЗВ2О3 (ж)+
+ЗН2О (г)=2Н3В3О6 (г), которое было исследовано методом протока Рэндаллом и Маргрейвом
[2290] (—2197+170 кДж-моль по II закону и —2265+25 кДж-моль по III закону, 1000—
1273 К, 3 точки) и Блауэром и Фарбером [700] (—2275+30 кДж'Моль, 1147 К, 1 точка).
BF (г). Термодинамические свойства газообразного фторида бора в стандартном состоянии
при температурах 100—6000 К приведены в табл. 557.
Молекулярные постоянные BF, принятые в справочнике на основании результатов
исследований электронных спектров, представлены в табл. 20.12. В спектре BF экспериментально
наблюдались переходы, связанные с двадцатью двумя возбужденными состояниями [828], в том числе
переходы 441 *t Х^+ [602, 828, 848, 849, 918, 1775, 2024, 2162, 2352, 2830] и а3П—XXZ+ [1834].
Так же как у молекул других галогенидов группы Ша, состояния Х*?+, а3П и АгН молекулы BF
коррелируют с диссоциационным пределом В BP)+F BP3i2)- Все остальные наблюдавшиеся
состояния BF, согласно данным Катона и Дугласа [828], являются состояниями ридберговского
типа; они расположены существенно выше 50 000 см и в настоящем справочнике не
рассматриваются.
Колебательные постоянные BF в состояниях Х1^* и 441 определялись на основании анализа
колебательной структуры системы 441—ХХБ+ в ряде работ [828, 848, 849, 2024, 2162].
Приведенные в табл. 20.12 колебательные постоянные приняты по работе Онака [2162] и получены по
началам 13 полос этой системы (v' ^ 5, 0 ^ v" ^ 6). В пределах точности измерений они хорошо
описывают положения начал 16 полос этой системы, измеренных Катоном и Дугласом [828].
Вращательные постоянные состояния Хг% + молекулы UB19F определялись в работах [809,
828, 848, 1889, 2162] и удовлетворительно согласуются между собой. Приведенные в табл. 20.12
вращательные постоянные в состояниях Х11>+ и 441 получены Катоном и Дугласом [828] в
результате анализа полос 0—0, 1—0, 2—0, 3—0 системы А1!!—Хг1>+ и данных Онака [2162].
Значение постоянной Во (X1!!"), найденное авторами [828], хорошо согласуется с полученным Лова-
сом и Джонсоном [1889] из микроволнового спектра BF (Бо=1,507 234 8 см).
Колебательные и вращательные постоянные 11B19F в состоянии а3П приняты по работе Барроу
и др. [602], которые выполнили анализ структуры шести полос системы Ь32+—а3И и полосы 0—0
системы c3S+—а?П. Значение Те (а3Щ получено Лебретоном и др. [1834] при исследовании
интеркомбинационной системы а3П—Хх?+.
Термодинамические функции BF (г), приведенные в табл. 557, вычислены по соотношениям
A. 3)—A. 6) A. 9), A. 10), A. 93)—A. 95). Значения <?вн и ее производных рассчитывались по
уравнениям A. 90)—A. 92) с учетом двух возбужденных состояний и в предположении Q\*Q\ вр =
={pJPx) .Qwx.ep • Величина (?S.BP и ее производные по температуре для основного электронного
состояния вычислялись непосредственным суммированием по колебательны^ уровням и
интегрированием по / с помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии
с значениями / ^/ша1> с, где/шах>, находились из условий A.81). Колебательно-вращательные
уровни всех трех состояний вычислялись по уравнениям A. 62), A. 65). В расчетах
использовались значения коэффициентов Ykl, а также vm&x и /Ит, полученные для природной смеси изотопов
бора при помощи соотношений A. 66) и постоянных молекулы 11B19F. Соответствующие величины
приведены в табл. 20.13. Коэффициенты Yi0 и У21 определены из условий A. 50) и A. 57). Мульти-
плетность и вырождение уровней в состояниях а3П и 441 учитывались статистическими весами
6 и 2 соответственно.
Постоянные молекулы BF известны с достаточной точностью, и погрешности вычисленных
термодинамических функций BF (г) вплоть до 3000—4000 К обусловлены лишь неточностью
фундаментальных постоянных, при более высоких температурах становится существенным отсутствие
экспериментальных данных об энергии высоких колебательно-вращательных уровней состояния
ХХЕ+. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,02, 0,03 и
0,1 Дж-К-^моль.
Термодинамические функции BF (г) ранее вычислялись в предыдущих изданиях настоящего
справочника [105, 106], а также в справочниках [1543], [1974] и работах [499, 1493, 1904, 2796,
2855]. За исключением работ [2796, 2855] (Ф° (Т) до Т ^ 5000 К) все расчеты выполнены до
6000 К.
48
Бор и его соединения
BF2 (г)
Результаты всех расчетов, кроме [1493, 2796], удовлетворительно согласуются с данными
табл. 557, расхождения не превышают 0,2 и 0,7 Дж-К-моль в значениях Ф° (Т) и S° (T),
несмотря на пренебрежение состоянием а3П и использование приближенных методов расчета. Это
обусловлено большими величинами энергий возбужденных состояний, энергии диссоциации и
постоянных ше и Ве молекулы BF. Наилучшее согласие имеет место с данными [105, 106]
(расхождения не превышают 0,1 и 0,3 Дж-К-моль). Расхождения с работой [1493] достигают 0,8
и 2,2 Дж'К~1'Моль, а с работой [2796], где были использованы неверные постоянные BF,
16 Дж-К^-моль.
Константа равновесия реакции BF (r)=B (r)+F (г) вычислена с использованием значения
дгЯ° @)=D0 (BF)=742,275 +11 кДж-моль=62 050+1000 см, соответствующего принятой
в справочнике энтальпии образования
„••¦ bfH°(B?, г, 0) = —105 + 10 кДж.моль.
Принятая величина основана на приведенных в табл. 20.23 данных. Приведенные в таблице
погрешности включают воспроизводимость измерений, а также неточность термодинамических
функций и сечений ионизации, использованных в расчетах термохимических величин. Как видно
из таблицы, в работах Хилденбранда были получены значения, практически совпадающие в
пределах их погрешностей. Принятая величина является средней из этих данных.
Таблица 20.23. Результаты
Авторы
определений AfH° (BF, г, 0) (в кДж-моль *)
Метод
ДуЯ° (BF, г, 0)
II закон
III закон
Гросс и др., 1963 (см. [1543]) Протока, 2/3В (k)+VsBFs (r)=BF (г) — —120
Блауэр и др., 1964 [704] То же, 1307—1505 К, 2 точки, значения констант —НО —125
экстраполированы к бесконечно-большой
поверхности бора
Хилденбранд, Мурад, 1965 [1420] Масс-слектрометрический, BF3 (г)+2Са (г)= —126+80 —107+12
=BF (r)+2CaF (г), 1583—1734 К, 15 точек
То же, BF3(r)+Ca(r)=BF(r)+CaF2(r), 1583— —114+40 —106±10
1734 К, 15 точек
Хилденбранд, 1968 [1406] То же, A1F3 (r)+BF (r)=AlF (r)+BF3 (г), 1320— — —114+10
1400 К, 4 точки
Дайблер, Листон, 1968 [950] Фотоионизационный, B2F*=BF3+BF, Дг#° @)= —91+12
=211±10
В работе [2597] было выполнено масс-спектрометрическое исследование реакции BF3 (г)+
+2В (k)=3BF (г) A477—1650 К, 5 точек). Вычисленная по методу II закона и измеренным в этой
работе ионным токам величина ДГЯ° @) = —368+290 кДж-моль имеет большую погрешность и
приводит к нереальному значению kfH° (BF)=—470 кДж-моль. Возможно, что в этой работе не
достигалось равновесие между конденсированной и газообразной фазами.
BF2 (г). Термодинамические свойства газообразного дифторида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 558.
Молекулярные постоянные, использованные для расчета термодинамических функций BFa,
приведены в табл. 20.14. Прямые экспериментальные данные о структуре молекулы BF2
отсутствуют. В справочнике принимается, что в основном электронном состоянии %?АХ молекула BF2
имеет нелинейную симметричную структуру (симметрия Civ). Предположение о подобной
структуре подтверждается результатами исследований спектра электронно-спинового резонанса [796,
2114, 2893] и многочисленными теоретическими расчетами (см., например, [774, 1492, 1975, 2249,
2690]). Приведенное в табл. 20.14 произведение главных моментов инерции вычислено в
предположении, что молекулы BF2h BF3 имеют одинаковые структурные параметры: -^ F—В—F=120+10°
и г (В—F)=l,31 +0,03 А. Результаты теоретических расчетов и исследования спектра ЭПР в
пределах погрешностей согласуются с этими величинами. Погрешность в значении IaIbIc оценива-
49
BF2~ (г) Глава 20
ется в 1 • 10~116 г3-см6. Приведенные в табл. 20.14 основные частоты вычислены по уравнениям
поля валентных сил и силовым постоянным, принятым равными соответствующим постоянным
BF3 *. Погрешности в оцененных таким образом частотах могут достигать 10—15%. Теоретические
расчеты [2689, 2690] показывают, что первое возбужденное состояние BF2 AiB1 имеет энергию
25 800+5000 см2.
Термодинамические функции BF2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128 ), A. 130)
без учета возбужденных электронных состояний. Погрешности рассчитанных термодинамических
функций составляют 1, 2 и 3 Дж-К~1-моль~1 в значениях Ф° (Г) при Г=298,15, 3000 и 6000 К и
обусловлены неточностью принятых молекулярных постоянных A—1,5 Дж-К~1-моль~1), а также
приближенным характером расчета.
Ранее термодинамические функции BF2 (г) вычислялись в предыдущих изданиях настоящего
справочника [105, 106], в справочнике JANAF [1543], Мак-Брайд и др. [1974] и Уилкинсом
[2855]. Расхождения в функциях, приведенных в табл. 558 и вычисленных в работах [105, 106,
1543, 1974], не превышают 0,7—1,3 Дж-К~1-моль в значениях Ф° (Т) и связаны с некоторым
отличием в принятых значениях частот и структурных параметров. В работе [2855] для BF2
принята линейная структура и существенно иные молекулярные постоянные. Расхождения между
результатами этого расчета и значениями функций, приведенными в табл. 558, составляют 25—
27 Дж-К^-моль'1.
Константа равновесия реакции BF2 (r) = B (r)+2F (г) вычислена с использованием значения
к,Л° @) = 1216,55+11 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
i\fH° (BF2> г, 0) = —502 + 10 кДж • моль.
Принятая величина основана на фотоионизационных измерениях Дайблера и Листона [950],
из которых для реакции B2F4+F=BF3+BF2 было получено Дг#° @) =—277+5 кДж-моль.
Заметно отличающаяся величина &fH° (BF2, г, 0) =—589+20 кДж-моль (III закон) была
вычислена по результатам масс-спектрометрического исследования равновесия реакции BF3 (r)+BF (r) =
=2BF2 (г) в работе Сривастава и Фарбера [2597] A477—1650 К, 5 точек). Выполненные в этой же
работе Сривастава и Фарбера измерения константы равновесия реакции BF3 (r)+2B (k)=3BF (г)
привели к ошибочным результатам (см. выше) 3.
BF^ (г). Термодинамические свойства газообразного отрицательного иона дифторида бора
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 559.
Спектр и структура иона BF^ экспериментально не исследовались. По аналоги с изоэлектрон-
ной молекулой CF2 в справочнике принимается, что в основном электронном состоянии Х1А1 ион
BF" имеет нелинейную структуру (симметрия C2v); с этим выводом согласуется квантовомеханиче-
ский расчет, выполненный Рябовым и др. [354]. Приведенное в табл. 20.14 произведение главных
моментов инерции в состоянии Х1А1 вычислено при г (В—F) = l,30+0,05 А и -$F—В—F=
=105+15°. Расчет [354] дал для угла значения в пределах 90—100°. Погрешность значения
IaIbIc может достигать 1-Ю16 г3-см6. Основные частоты BF" оценены на основании сравнения
соответствующих частот молекул CF2 и BF2, их погрешности в значениях \ и v3 оцениваются
в 100 см, в значении v2 — 60 см. По аналогии с изоэлектронной молекулой CF2 можно ожидать,
что BF^ имеет связанное триплетное состояние а?Вх с энергией 15 000—20 000 см.
Термодинамические функции BF~ (г) рассчитаны в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130) с учетом только основного электронного состояния Х1А1. Погрешности функций опреде,-
1 /г=6,62; /гг=1,1 и /а/г2=0,6 (в 106 дин-см).
2 При разряде через пары BF3 при низком давлении [1774], а также при возбуждении паров BF3 электронным
ударом [1399] наблюдался спектр испускания, отнесенный к молекуле BF2, однако анализ структуры полос,
расположенных в области 2100—3700 А, не проводился, и отнесение полос к молекуле BF2 является лишь
предположительным.
3 В масс-спектрометрическом исследовании Лау и Хилденбранда [1822а], опубликованном после завершения
подготовки настоящего справочника, было получено значение —502+15 кДж-моль, подтверждающее
сделанный выбор.
50
Бор и его соединения BF3 (г)
ляются неточностью оцененных молекулярных постоянных, пренебрежением возбужденными
состояниями и приближенным характером расчета, они могут достигать 2, 4 и 6 Дж-К-моль~1
в значениях Ф° (Т) при 298,15, 3000 и 6000_К.
Ранее термодинамические функции BF2 (г) вычисляли авторы справочника JANAF [1544].
Расхождения данных [1544] и табл. 559 не превышают 1, 3 и 4 Дж>К~1-моль~1 в значениях Ф° (Т)
и 5° (Т).
Константа равновесия реакции BF2 (r)=B (r)+2F (r)-j-e (г) вычислена с использованием
значения Дг.йГ0 @)=1444,55+15 кДж-моль, соответствующего принятому в справочнике сродству
BF2 к электрону
А о (BF2) = — 228+10 кДж-моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции BF~ (r)-f-
+F (r)=BF2 (r)+F~(r), выполненном Сривастава и др. [2600] A383—1700 К, 6 точек,
AJt0 @)=—140+20 кДж-моль по II закону и —99,7+4,0 кДж-моль по III закону).
Принятому сродству к электрону соответствует
A^O(BF", г, 0) = —730 + 14 кДж.ыопь.
BF3 (г). Термодинамические свойства газообразного трифторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 560.
Молекулярные постоянные BF3 (г), использованные для расчета термодинамических функций,
представлены в табл. 20.15. Результаты спектроскопических [238, 516, 558, 559, 771, 772, 993,
1138, 1159, 1206, 1207, 1208, 1209, 1852, 1853, 1866, 1994, 2140, 2754, 2868, 2886] и электронографи-
ческих [736, 1752, 1782, 1855] исследований, а также измерения дипольного момента BF3 [1874,
2117] и теоретические расчеты [274, 531, 533, 1104, 1240, 1841 ] свидетельствуют о том, что в
основном электронном состоянии ЁгА[ молекула BF3 имеет плоскую структуру (симметрия D3h).
Приведенные в табл. 20.15 произведения главных моментов инерции IaIbIc вычислены по
вращательным постоянным 50(nBF3)=0,34505 +0,0005 см и ?0A0BF3)=0,344 74+0,000 5 см, полученным
Гином и др. [1208] в результате анализа тонкой структуры полос \ в ИК-спектре поглощения
uBF3h10BF3. Этим постоянным соответствуют межатомные расстояния г0 (В—F) = 1,3096+0,001 А
и г0 (В—F)=l,3090+0,001 А соответственно для 10BF3 и nBF3. С принятыми значениями г (В—F)
прекрасно согласуются результаты электронографических измерений [1752, 1782] A,311 + 0,001 А),
а также значения г (В—F) =1,3110 А и 1,3102 А, вычисленные на основании анализа
вращательного спектра комбинационного рассеяния UBF3 [1138] и обертона 2v3 в ИК-спектре BF8 [771].
Для частоты колебания \ в табл. 20.15 приведены значения, полученные в работе [2886] при
анализе спектра КР газообразного BF3. Значения частот v2, v3 и \ измерялись многими авторами
(см. [238, 772, 993, 1206, 1207, 1208, 1852, 1866, 1994, 2140, 2754]) и в пределах 1 см согласуются
между собой. В табл. 20.15 для этих частот приняты значения, полученные в работах [772, 1208,
2140, 2868] (v2), [238, 772, 993, 1852, 1866, 2754, 2868] (v3) и [772, 993, 1207, 1852, 1994, 2868] (v4).
Погрешность принятых частот \, va и v4 не превышает 1 см, частоты v3 _ 5 см (с учетом
возможного резонансного смещения) х. В работах [772, 993, 1207, 1208, 1209, 1956, 2043, 2288, 2868]
на основании результатов исследований основных и комбинационных полос й анализа
изотопических сдвигов были сделаны попытки определить постоянные ангармоничности и взаимодействия
колебаний с вращением. Однако имеющихся данных недостаточно для определения полного набора
этих постоянных.
Исследование электронного спектра [1948] показало, что первое возбужденное состояние
BF3 расположено на 45 000—50 000 см выше основного состояния.
Термодинамические функции BF3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122)—A. 124), A.128), A.130).
Расчет проводился с учетом отличия молекулярных постоянных разных изотопных модификаций
BF3. Погрешности рассчитанных термодинамических функций составляют 0,1,3 и 5'Дж-К-моль~1
в значениях Ф° (Т) при 298,15, 3000 и 6000 К и обусловлены главным образом приближенным
1 Гин и др. [1206] получили для невозмущенной резонансом частоты v3 значения 1502,5 гЬО^'см и 1449 ±3 см
(для J1BF3 и *°BF3) и постоянные кориолисова взаимодействия.
51
B,Ft~ (r) Глава 20
характером расчета, погрешности из-за неточности молекулярных постоянных малы и не
превышают 0,1 Дж-К-мояь~1.
Измерения теплоемкости BF3, выполненные Эйккеном и Шредером [1041] (см. [2594]) и
Стрелковым и др. (см. [105, с. 727]), приводят к значениям S° B98,15 К) =254,4+2,5 и 253,8+2,5 Джх
хК^-моль, которые хорошо согласуются с величиной, приведенной в табл. 560.
Ранее таблицы термодинамических функций BF3 (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106], авторами справочника JANAF [1543], Мадером [1904], Мак-
Брайдом и др. [1974], Эвансом [1044], Хаффом и др. [1493] и в работах [505, 1312, 1539, 1856,
2594, 2905] (до температур 1000—2500 К). Результаты всех расчетов (значения Ф° и S°), кроме
[2092, 2855], в пределах 0,3 Дж-К~1-моль~1 согласуются с приведенными в табл. 560. Расхожде-
дия с данными [2092, 2855] достигают 1 —1,5 Дж-К~1-моль~1 из-за ошибок, допущенных в этих
расчетах.
Константа равновесия реакции BF3 (r)=B (r)+3F (г) вычислена с использованием значения
Аг#° @) = 1924,966 +5,1 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^H°(BF г, 298,15 К) = -1135,95 + 0,80 кДж • моль.
Принятая величина рекомендована КО ДАТА—МСНС [864] в результате совместного анализа
•большой группы измерений энтальпий реакций, включающих BF3, В2О3, Н3ВО3 и т. д. (см. [864])
и является оптимальной для всей совокупности данных. Однако основное значение для выбора
этой величины имели работы Джонсона и др. [1577], Гросса и др. [1285] и Домальского,
Армстронга [968], в которых были измерены энтальпии сгорания кристаллического бора во фторе,
а также измерения энтальпии сгорания бора в кислороде в присутствии плавиковой кислоты
Гудом и Монссон [1230] и энтальпии растворения BF3 в плавиковой кислоте Ганном [1307].
BFJ (г). Термодинамические свойства газообразного отрицательного иона тетрафторида бора
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 561.
,. Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 20.15. Экспериментальные данные о молекулярных постоянных свободного иона BFJ
в литературе отсутствуют. В соответствии с теоретическими расчетами [1104, 1840а] но аналогии
со структурой изоэлектронной молекулы CF4, а также в предположении, что при переходе в
свободное состояние из соединений типа XBF4, где X — щелочной металл, структура BFJ мало
изменяется, в настоящем справочнике принято, что ион BF^ в основном электронном состоянии Х1А1
имеет тетраэдрическое строение (симметрия Td). Приведенная в табл. 20.15 величина IaIbIc
рассчитана для межатомного расстояния г (В—F) = l,43+0,05 А, найденного в работах [2204, 2885]
в результате исследований структуры молекул XBF4 (X=Na, К, Rb, Cs и NH4) методом ЯМР.
С этим значением в пределах погрешностей согласуются результаты рентгеноструктурных
исследований ионных кристаллов RbBF4, NH4BF4 [1445], NaBF4 [643] и KBF4 [644] 1. Погрешность
в произведении главных моментов инерции BF" может достигать 1- 1014г3-см6. Приведенные в табл.
20.15 основные частоты приняты по результатам исследований ИК-спектра и спектра КР водных
растворов соединений типа XBF4 (X — щелочной металл) [617,1236], в которых расщепление частот,
обусловленное взаимодействием иона BFJ с окружением, минимально. С принятыми значениями в
пределах погрешностей согласуются частоты, найденные при изучении колебательных спектров
кристаллических соединений XBF4 [615, 721, 722, 884, 1015, 1837, 2283, 2677] и их расплавов при
различных температурах [616, 2282]. Погрешности приведенных значений составляют 80, 40, 100 и
50 см соответственно для vlt v2, v3 и v4. По аналогии с молекулой CF4 можно предполагать, что BFJ
не имеет низкорасположенных электронных состояний.
Термодинамические функции BF^ (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A.122)—A. 124), A. 128), A. 130).
Погрешности вычисленных значений определяются неточностью принятых молекулярных
постоянных и приближенным характером расчета и составляют 2, 6 и 8 Дж- К- моль в значениях Ф° (Т)
при 298,15, 3000 й 6000 К, из которых 2—4 Дж-К~1-моль — из-за неточности постоянных.
Термодинамические функции BFJ (г) публикуются впервые.
1 Неэмпирический расчет дает для г (В—F), значение 1,39 А [1104], а расчет в приближении GNDO/2 — 1,5Q A
11840а].
52
Бор и его соединения
Константа равновесия реакции BF~ (r)=B (r)+4F (г)-(-е (г) вычислена с использованием
значения &ГН° @) =2619,1+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^0 (BF-, г, 0) = —1750 + 40 кДж • моль.
Принятая величина основана на данных работ [505, 2600J. Альтшуллер 1505] вычислил энергию
кристаллической решетки KBF4 (к) F36+30 кДж-моль), откуда из цикла Борна—Габера было
получено Д^#° (BF~, г, 0) =—1755+40 кДж-моль. В этом расчете было использовано значение
L;H° (KBF4, к, 0) =—1882 кДж-моль по данным справочника [2787]. Сривастава и др. [2600]
масс-спектрометрическим методом исследовали реакцию BF3(r)+BF~ (r)=BF~(r)+BF (г) A523—.
1703 К, 3 точки). Однако ион BF~ в масс-спектре не был обнаружен, и для расчета значения
константы равновесия был использован порог чувствительности прибора. Из этих данных по
уравнению III закона был найден предел &ГН° @) J> 5+25 кДж-моль, которому соответствует
значение Д/#° (BF~, г, 0) > —1753+30 кДж-моль-1.
B2F4 (г). Термодинамические свойства газообразного тетрафторида дибора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 562.
Молекулярные постоянные, использованные для расчета термодинамических функций B2F4r
приведены в табл. 20.21. Спектроскопические [1000, 1089] и электронографические [924]
исследования, а также квантовомеханический расчет [894] показывают, что молекула B2F4 имеет структуру,,
соответствующую симметрии ZJA x и обладает заторможенным внутренним вращением вокруг связи
В—В. Приведенные в табл. 20.21 значения произведения главных моментов инерции и величины
/др соответствуют структурным параметрам: г (В—F)=l,317+0,003 А, г (В—В) = 1,720+0,006 А и
«3F—В—F=117,3+0,3°, полученным в электронографическом исследовании Даниэлсона и др.
[924]. Величина потенциального барьера У0=147+60 см также принята по работе [924].
Значения основных частот \, v2) v3, v6, ve, vg и v10 приняты по работе Дюрига и др. [1000],
которые исследовали спектр КР и ИК-спектр газообразного B2F4. Значения v7, v9, vu и v12 найдены в
работе Гейлеса и Селфа [1185] из инфракрасного спектра газообразного B2F4. Частота v4 оценена
Даниэлсоном и др. по величине потенциального барьера. Погрешности частот составляют
от 1 до 5 см. Сведения о возбужденных электронных состояниях B2F4 в литературе отсутствуют.
Термодинамические функции B2F4 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124)т A. 128),
A. 130) с учетом заторможенного внутреннего вращения по уравнениям A. 160)—A. 162).
Погрешности вычисленных величин определяются как неточностью молекулярных постоянных, так и
приближенным характером расчета и составляют 2, 6 и 10 Дж-К^-моль в значениях Ф° (Т)\
при 298,15, 3000 и 6000 К, в том числе из-за неточности постоянных ~1 Дж-К~1-моль~1.
Ранее термодинамические функции B2F4 (г) вычислялись в таблицах JANAF [1543] в
предположении о свободном вращении вокруг связи В—В. Расхождения результатов расчета [1543] с
данными, приведенными в табл. 562, составляют от 2 до 9 Дж-К-моль в значениях Ф° (Т); они
обусловлены тем, что в таблицах JANAF приняты неверные значения частот v8 и v10, а барьер
внутреннего вращения принят равным нулю.
Константа равновесия реакции B2F4 (г)=2В (r)+4F (г) вычислена с использованием значения
ДД° @) =2864,696+12 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^Н°(В.2?^ г, 298,15 К) = —1438 ± 7 кДж • моль.
Принятая величина основана на измерении теплового эффекта реакции B2F4 (r)-f-Gl2 (r) =
=0,92 BF3 (г)+0,46 BF2C1 (г)+0,32 BFC12 (г)+0,30 ВС13 (г) в работе Ганна и Грина [13091
(ДГЯ° B98,15 К) = —244,3+4,2 кДж-моль). Состав продуктов реакции предполагался
равновесным и устанавливался соответствующими расчетами.
1 Известны исследования спектров [1185, 2145], а также квантовомеханические расчеты [1104, 1303, 1487Jr
в которых для молекул B2F4 в газообразном состоянии принималась структура, соответствующая симметрии
?>ы- Однако авторы работ [1000, 1089] показали, что этот вывод был ошибочным, и в настоящем издании для
B2F4 принята симметрия D2h.
53
FBO (г), F2BO (г) Глава 20
FBO (г). Термодинамические свойства газообразного оксид-фторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 563.
Молекулярные постоянные, использованные для расчета термодинамических функций FBO,
приведены в табл. 20.8. Исследование ИК-спектра молекул FBO в матрицах из Ne и Аг,
выполненное Снелсоном [2569], и квантовохимический расчет Томсона [2687] показали, что молекула FBO
имеет линейную структуру. Момент инерции в состоянии ZXS+ рассчитан по структурным
параметрам: г (В—F)=1,31+0,03 А (перенесено из BF3), r (В=О)=1,20+0,03 А (перенесено из ВО),
его погрешность составляет 0,5-10~39 г-см2. Близкие значения межатомных расстояний были
рассчитаны в работе [2687] (г (В—F)=l,26 А и г (В=О) = 1,19 А). Для частот v2 и v3, соответствующих
деформационному колебанию и колебанию связи В=О, приняты значения, найденные Снелсоном
[2569] в ИК-спектре молекул FUBO, изолированных в матрице, из Ne. He наблюдавшаяся в спектре
частота \ вычислена по формулам поля валентных сил, если принять силовую постоянную /в—f
равной соответствующей постоянной в молекуле BF3 (/в—f=6,6-105 дин-см). Погрешности
принятых значений частот оцениваются в 100 см для \ и 10—15 см для v2 и v3 1.
Термодинамические функции FBO (г) вычислены по уравнениям A.3)—A.6), A.9), A.10), A.122)—
A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический осциллятор».
Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
значений молекулярных постоянных, а также приближенным характером расчета и составляют 0,5,
2 и 3 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000К, из них 0,5—1 Дж-К'^моль
из-за неточности молекулярных постоянных.
Ранее термодинамические функции FBO (г) рассчитывались во 2-м издании настоящего
справочника [105], таблицах J ANAF [1543] и справочнике Мак-Брайд и др. [1974], а также Уилкинсом
и др. [2855] (Т ^ 5000 К), в справочнике [305] воспроизведена таблица из 11543]. Во всех
расчетах использованы оцененные значения молекулярных постоянных. Расхождения между данными'
табл. 563 и расчетов [105, 1974, 2855] достигают 2—4 Дж-К-моль~1 в значениях Ф° (Т),
расхождения с данными [1543] не превышают 0,5 Дж-К~1-моль.
Константа равновесия реакции FBO (r)=F (г)+В (г)+О (г) вычислена с использованием
значения ДГЯ° @)=1477,058+11 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —593+ 10 кДж • моль.
Принятая величина является средневзвешенной из результатов работ [1048, 1049, 1429].
Фарбер [1048] методом протока при молекулярном течении исследовал реакцию В2О3 (ж)+ВР3(г)=
=3FBO (г) (999—1253 К, 19 точек). Низкое общее давление позволило свести к минимуму
количество F3B3O3 в равновесной смеси. Из этих измерений было найдено &fH° (FBO, г, 0) = —664 +
+22 кДж-моль (II закон) и —595+10 кДж-моль (III закон). Из аналогичных измерений Фар-
бера и Блауэра [1049] A054—1253 К, 15 точек) были получены значения —593+50 кДж-моль
(II закон) и —589+8 кДж-моль (III закон). Хилденбранд и др. [1429] в масс-спектр ометриче-
ском исследовании равновесия реакции F3B3O3 (r)=3FBO (г) (980—1230 К, 10 точек) измерили
интенсивности ионных токов компонентов этой реакции; этим данным соответствует ^fH° (FBO,
г, 0) = —613+25 кДж-моль (II закон). Погрешности всех величин приведены с учетом
воспроизводимости измерений и неточности термодинамических функций.
F2BO (г). Термодинамические свойства газообразного оксид-дифторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 564.
Молекулярные постоянные, использованные для расчета термодинамических функций F2BO (г),
приведены в табл. 20.19. Анализ вращательной структуры полос в электронном спектре молекулы
F2BO показал, что в основном состоянии Х2В2 она имеет плоское строение (симметрия С2р).
Приведенное в табл. 20.19. произведение моментов инерции вычислено по структурным параметрам
г (В—О)=1,40+0,05 А, г (В—F)=1,30+0,05 А и ^ F—В—F=126+5°, найденным Матыосом
[1960] при исследовании электронного спектра F2BO по значениям вращательной постоянной
Соо... трех изотопических модификаций этой молекулы. Погрешность в IaIbIc составляет Зх
X 10~115 г3- см6. Основные частоты vl5 v2 и v3, приведенные в табл. 20.19, приняты по данным Матьюса
1 Снелсоы [2569], приняв /B_F =8- 10е дин-см, рассчитал vx=964 см~1.
54
Бор и его соединения F3B3O3 (г)
и Иннеса [1962], полученным при исследовании электронного спектра F2UBO, их погрешности
оцениваются 1—2 см'1. Остальные частоты оценены из сравнения с молекулой BF3; погрешности
этих частот могут составлять до 10% от их величины. Первое возбужденное состояние А2А2
молекулы F2BO согласно работе [1962] имеет энергию Т0=П 171 см и в справочнике не
рассматривается.
Термодинамические функции F2BO (г) вычислены по уравнениям A. 3)— A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета и составляют около 1, 4 и 6 Дж-К~1-моль~1
в значениях Ф° (Т) при 298,15, 3000 и 6000 К (в том числе из-за неточности постоянных 1 —
2 Дж-К^-моль).
Термодинамические функции F2BO (г) вычислялись ранее в справочнике JANAF [1543],
данные [1543] и табл. 564 согласуются в значениях Ф° (Т) в пределах 0,2 Дж-К~1-моль~1.
Константа равновесия реакции F2BO (r) = B (r)+2F (г) + О (г) вычислена с использованием
значения ДГЯ° @)=1791,333 +60 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^^ВО, г, 0) = —830 + 60 кДж-моль-1.
Принятая величина оценена в предположении, что D0(F2B—O)=D0 (OB—О), Do (F2BO—H)=
=D0 (HO—H) и Do (FaBO—H)=D0 (OBO—H). Эти оценки приводят к значениям LfH° @),
равным —830, —808 и —851 кДж-моль соответственно. Первые два соотношения были
предложены в таблицах JANAF [1543].
F3B3O3 (г). Термодинамические свойства газообразного трифторбороксина в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 565.
Молекулярные постоянные, использованные для расчета термодинамических функций F3B3O3,
приведены в табл. 20.21. Экспериментальные данные о структуре FgB3O3 в литературе отсутствуют.
Структурные параметры F3B3O3 были оценены на основании предположения, что эта молекула,
так же как молекула Н3В3О3, имеет плоскую кольцевую структуру (симметрия DSh), и
использования соответствующих данных для молекул Н3В3О3 и BF3. Произведение моментов инерции
вычислено приг(В—О)=1,38 +0,02 А, г (В—F) = l,31 +0,02 А и^ В—О—В = ^ О—В—О = -$ О—В—F=
=120°, его погрешность составляет 3-1013 г3-см6. Инфракрасный спектр F3B3O3 исследовался
в работах [1102, 1816, 2741]. В работе Латимера и Девлина [1817] на основании оцененного
силового поля молекулы выполнен расчет колебательного спектра F3B3O3. В работе [1102]
наблюдались только 4 частоты в спектре F3B3O3 (к), а в работе [2741] — две области поглощения в ИК-
•спектре F3B3O3 (г). В табл. 20.21 для основных частот, активных в ИК-спектре (v2, ve, v8—v12),
приняты значения, полученные Латимером и Девлином [1816] для твердых пленок F3B3O3.
Значения частот \, v3, v4 и v5 рассчитаны в работе [1817], а для частот v7t v13! v14 приняты величины,
оцененные в [1543] на основании сравнения со сходственными соединениями В3О8(ОНK, Н3В3О3 и их
дейтерированными производными. Погрешности частот оцениваются в 15 см в значениях vu
и v12 и от 20 до 40 см в v2, ve, vg—v10. Погрешности частот, значения которых рассчитаны или
оценены, могут составлять 10—15%. Сведения о возбужденных электронных состояниях в литературе
отсутствуют.
Термодинамические функции F3B3O3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных таким образом функций определяются как неточностью
молекулярных постоянных, так и приближенным характером расчета и составляют 4, 12 и
20 Дж'К-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К. Погрешности из-за неточности
постоянных могут достигать 4—5 Дж-К-моль~1.
Ранее таблицы термодинамических функций F3B3O3 (г) рассчитывались в JANAF [1543] и
справочнике Мак-Брайд и др. [1974]. Расхождения результатов этих расчетов с
приведенными в табл. 565, составляют от 4 до 11 Дж-К~1-моль~1 в значениях Ф° (Г) и обусловлены тем,
что в обоих расчетах были использованы главным образом оцененные значения основных частот
молекулы F3B3O3.
55
BHF (г)
Глава 20
Константа равновесия реакции F3B3O3 (г)=ЗВ (г)+ЗО (r)+3F (г) вычислена с использованием
значения АГЯ° @)=5026,174+19 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH°(FsB3O3, г, 0) = —2374 + 12 кДж • моль.
Принятая величина основана на приведенных в табл. 20.24 результатах исследований
равновесия реакции В2О3 (к, ж)+ВГ3 (r)=F3B3O3 (г). Она является средней из значений, полученных
по уравнению III закона, а также найденного по уравнению II закона из данных [2253]. Средняя
Т а б л и
Фишер
Фарбер,
Портер
Фарбер
ц а 20.24. Результаты
Авторы
и др., 1961 [1101]
1962 [1047, 1054]
и др., 1962 [2253]
Блауэр, 1962 [1049]
Хилденбранд и др., 1963 [1429]
определений А^Н° (F3B3O3, г, 0) (в кДж-мол!
Метод
Измерение интенсивностей ИК-спектров,
700—1000 К
Протока, 330—454 К, 6 точек
То же, 1000 К, 1 точка
Масс-спектрометрический, 412—974 К, 7 точе»
Метод молекулярного потока, 999—1054 К, 5
точек
Протока, 598—1103 К, 14 точек
II закон
—2351
—2368±3
—
. -2384±3
—2255+200
—2360 ±2
*
3В3О3, г, 0)
III закон
—2376 + 23
—2383+3
—2406
—
-2360+5
—2373+9
величина совпадает с рассчитанной по наиболее детальным измерениям, выполненным в работе
[1429]. Приведенные в табл. 20.24 погрешности отражают только воспроизводимость измерений
и неточность других термохимических величин, использованных в расчетах; неточность
термодинамических функций приводит к дополнительной погрешности в энтальпии образования
F кДж-моль для измерений, выполненных при 1000 К). При оценке погрешности принятой
величины учтено расхождение результатов измерений в различных работах.
BHF (г). Термодинамические свойства газообразного гидридфторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 566.
Молекулярные постоянные, использованные для расчета термодинамических функций ВHF (г),
приведены в табл. 20.14. Экспериментальные данные о структуре и спектре BHF отсутствуют,
поэтому молекулярные постоянные были оценены. По аналогии с симметричными молекулами
ВХ2, где Х=Н, F, G1, Вг, I в справочнике принимается, что молекула BHF в основном
электронном состоянии Х2А' имеет структуру с ^ Н—В—F=120+10° (симметрия Cs). Это предположение
подтверждается квантовомеханическим расчетом Бротчи и Томпсона [767], которые вне
зависимости от выбора базисных волновых функций получили для угла Н—В—F значения 121—122°,
а также аналогией с изоэлектронными молекулами НСО и НВО". Приведенное в табл. 20.14
произведение главных моментов инерции вычислено для г (В—Н)=1,18+0,02 А и г (В—F)=l,31 +
Jr0,03 А, оцененных по соответствующим расстояниям в молекулах ВН2, BF2 и BHF2.
Погрешность в IaIbIc не превосходит 2-1018 г3-см6. Основные частоты молекулы BHF, приведенные
в табл. 20.14, вычислены по силовым постоянным, принятым близкими к соответствующим
постоянным в молекулах ВХ2 и ВХ3: /в_н=4,1, /b-f=6,6 и /0t/r1r2=0,53 (в 105 дин-см). Погрешности
принятых значений частот BHF составляют 10—15%. Энергия возбуждения состояния А2А"
была оценена как средняя из энергий возбуждения состояний А2В± в молекулах ВН2 и BF2.
Практически к тому же значению A3 170 см) приводит квантовомеханический расчет Бротчи и
Томпсона [767]. Погрешность принятого значения То может достигать 5000 см.
Термодинамические функции BHF (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Вклад возбужденного электронного состояния А2А" учитывался по уравнениям A. 168)—
A.170). Погрешности рассчитанных термодинамических функций составляют 1,2 и 3 Дж- К- моль
в значениях Ф° (Т) и при 298,15, 3000 и 6000 К и обусловлены в основном неточностью оцененных
значений молекулярных постоянных.
56
Бор и его соединения BH2F (г), BHF2 (г)
Термодинамические функции BHF (г) публикуются впервые.
Константа равновесия реакции BHF (г)=В (г)+Н (r)-j-F (г) вычислена с использованием
значения АГН° @)=1208,309 +20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A//°(BHF, г, 0) = —355 + 20 кДж • моль.
Принятая величина была оценена в предположении о равенстве средних энергий связей в
молекулах BHF, ВН2 и BF2.
BH2F (г). Термодинамические свойства газообразного дигидрид-фторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 567.
Экспериментальные данные о спектре и структуре молекулы BH2F в литературе отсутствуют.
Молекулярные постоянные, использованные для расчета термодинамических функций BH2F (г)
и приведенные в табл. 20.15, оценены при подготовке настоящего справочника. По аналогии с
симметричными молекулами ВХ3, где Х=Н, F, C1, Вг, I, и с молекулами BHF2 и ВНС12 в справочнике
принимается, что в основном электронном состоянии Х1А1 молекула BH2F имеет плоскую
структуру (симметрия С2в). Произведение главных моментов инерции, приведенное в табл. 20.15,
вычислено в предположении, что все углы равны 120+10°, а межатомные расстояния г (В—Н) =
=1,18+0,02 А, г (В—F)=l,31+0,03 А были оценены по межатомным расстояниям в молекулах
ВН3 и BF3. Погрешность в IaIbIc не превосходит 6-10~118 г3-см6. Основные частоты BH2F,
приведенные в табл. 20.15, оценены сравнением с частотами ВН3 и BF3, а также в результате расчета
по уравнениям поля валентных сил в предположении, что силовые постоянные BH2F равны
соответствующим силовым постоянным молекул ВН3 и BF3 г. Частоты BHF2 и ВНС12, оцененные
аналогичным образом, согласуются с экспериментальными данными в пределах 10—15%, поэтому
можно предполагать, что погрешности в принятых значениях частот имеют тот же порядок.
Термодинамические функции BH2F (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122) —A. 124), A. 128),
A.13(д, с учетом, только основного электронного состояния. Погрешности рассчитанных
термодинамических функций составляют 1, 2 и 4 "Дж-"?^"ч-молъ~ч в звачипш:*.^ ""у!4) -а^ъч1^^>,<>§§§^
и 6000 К и обусловлены неточностью оцененных значений постоянных A—2 Дж-К~1-моль) и
приближенным характером расчета.
Таблица термодинамических функций BH2F (г) публикуется впервые.
Константа равновесия реакции BH2F (г)=В (г)+2Н (r)+F (г) вычислена с использованием
значения ДГЯ° @)=1389,343+21 кДж-моль", соответствующего принятой в справочнике
энтальпии образования
t\fH°(BK2F, г, 0) = —320 + 20 кДж • моль.
Эта величина была оценена на основании предположения о равенстве средних энергий связей
в молекулах BH2F, BH3 и BF3. Поскольку для BHF2 рассчитанное таким образом значение меньше
по абсолютной величине, чем найденное экспериментально, в оценку для BH2F была внесена
соответствующая поправка (—6 кДж-моль).
BHF2 (г). Термодинамические свойства газообразного гидрид-дифторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 568.
Молекулярные постоянные, использованные при расчете термодинамических функций BHF2 (г) *
приведены в табл. 20.15. Исследование микроволнового спектра изотопзамещенных BHF2 по бору
и водороду, выполненное Касуйя и др. [1634], свидетельствует о том, что в основном электронном
состоянии Х1А1 молекула BHF2 имеет плоскую структуру (симметрия С2„). Приведенные
в табл. 20.15 произведения главных моментов инерции UBHF2 и 10BHF2 вычислены по
вращательным постоянным А 0=248,487 972 и 257,5923707, 50=35,0133558 и 35,00871921, Со=
=30,826 75445 и 30,759 578 68 см (соответственно для UBHF2 и 10BHF2), найденным в работе
1 Деформационная силовая постоянная/хву принималась равной среднему арифметическому из силовых
постоянных /хвх и /yby1 a постоянная внеплоскостного деформационного колебания f? оценивалась по соотноше-
нгао B/д (ВХ3)+/Д (BY3))/3.
57
FBOH (г) Глава 20
[1634] х. Им отвечают структурные параметры г (В—Н)=1,189+0,010 А, г (В—F)=l,311 +
+0,005 Ai4 F—В—F=118,3+l°. Приведенные в табл. 20.15 основные частоты получены в
работах [1897, 2205, 2206, 2258], в которых исследовались инфракрасные спектры поглощения
газообразных UBHF2 и 10BHF2. Значения частот, измеренные и рассчитанные в [1897, 2205, 2206,
2258] 2, согласуются в пределах 5 см. Исключением является частота \, соответствующая
деформационному колебанию Н—В—F. Эта частота наблюдалась только в работах Переса и Бека
[2205, 2206] в виде слабого плеча на полосе, соответствующей колебанию vx nBHF2, и ее отнесение
сомнительно. Поскольку, однако, оценки, выполненные различными методами [1897,-2205, 2206,
2258], приводят для частоты v5 к значениям, изменяющимся в пределах от 1000 до 1350 см, в
справочнике для нее с погрешностью 200 см принимаются величины, полученные в [2205, 2206].
Сведения о возбужденных электронных состояниях BHFa в литературе отсутствуют.
Термодинамические функции BHF2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Расчет проводился с учетом отличия молекулярных постоянных двух изотопических
модификаций BHF2. Погрешности рассчитанных таким образом термодинамических функций
составляют 0,3, 2 и 4 Дж-К'^моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К и обусловлены
главным образом приближенным методом расчета.
Ранее термодинамические функции BHF2 (г) вычисляли авторы справочника JANAF [1543],
а также Рао и др. [2296], Варриер и др. [2797] и Нагараян [2096] до температур 1000—2000 К.
Данные [1543] и табл. 568 согласуются в пределах 0,25 Дж-К~1-моль~1 в значениях Ф° (Т) и
S° {T).
Константа равновесия реакции BHF2 (r)=B (r)+H (r)+2F (г) вычислена с использованием
значения Ь.,Н° @)=1665,584+8,6 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH° (BHF2, г, 0) = —735 + 7 кДж • моль.
Принятая величина является средневзвешенной из полученных по результатам исследования
равновесия реакции 1/e B2He (r)+2/3 BF3 (r)=HBF2 (г) в работах Портера и Уазона [2258]
(изучение интенсивности полос В2Н6 и BF3 в ИК-спектре продуктов реакции, Т=296и 360 К, Д^#° (BHF2,
г, 0)=—740 кДж-моль (II) и —734,7+6,5 кДж-моль (III)) и Дазо и Монжо [933] (равновесие
в разделительной колонке, 297 К, — 737,7 кДж-моль (III)).
FBOH (г). Термодинамические свойства газообразного гидроксид-фторида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 569.
Молекулярные постоянные FBOH, использованные для расчета термодинамических функций,
приведены в табл. 20.19. В литературе отсутствуют сведения об экспериментальных или
теоретических исследованиях молекулярных постоянных FBOH. В справочнике принимается, что эта
молекула в дублетном основном электронном состоянии Х2А' имеет плоское строение симметрии Cs.
Структурные параметры FBOH, на основании которых рассчитана величинаIaIbIc (cm. табл. 20.19),
оценены по аналогии с молекулами В(ОНJ и BF2 г (В—F)=l,31 +0,02 А, г (В—О)=1,34+0,03 А,
г (О-Н) =0,96+0,02 А, ^ F—В—0=120+5° и -? В—О-Н=115+10° (трансположение).
Погрешность IaIbIc составляет 5-Ю17 г3-см6. Основные частоты плоских колебаний \ (валентное
колебание О—Н), v2 (валентное колебание В—F), v3 (деформационное колебание В—О—Н), v4
(валентное колебание В—О) и v5 (деформационное колебание F—В—О) оценены по аналогии с
молекулами BF2, B(OHJ, НВО2 и ВОН. Частота крутильного колебания v6 группы ОН принята по
оцененному барьеру внутреннего вращения (F0=2400+700 см, как в молекуле F2BOH) при
п=1 и /цр=1,04-10~40 г-см2. Погрешности принятых значений vx—v4 оцениваются в 100 см,
a v5 и v6 — в 50 см.
По аналогии с молекулой BF2 можно предполагать, что нижнее возбужденное состояние FBOH
имеет энергию около 20 000 см, поэтому оно не рассматривается в справочнике.
Термодинамические функции FBOH (г) вычислены по формулам A.3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический осцилля-
1 Авторы [1634] нашли также постоянные центробежного растяжения BHF2.
2 В работе [897] отнесение наблюдавшихся частот не проводилось. В работе [1892] выполнен лишь
предварительный анализ, исправленный впоследствии авторами [1897].
58
Бор и его соединения FB (ОНJ (г), F2BOH (г)
тор» без учета возбужденных электронных состояний. Погрешности вычисленных значений
обусловлены неточностью принятых значений молекулярных постоянных, а также ^приближенным
характером расчета, в том числе учетом заторможенного внутреннего вращения как
гармонического колебания, и составляют 3, 5 и 8 Дж- К- моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К
(из которых 1—2 Дж-К-моль~1 из-за неточности молекулярных постоянных).
Таблица термодинамических функций FBOH (г) публикуется впервые.
Константа равновесия реакции FBOH (r)=F (г)+В (г)+О (г)+Н (г) вычислена с
использованием значения Дг#° @)=1565,092+30 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
A^^FBOH, г, 0) = —465 + 30 кДж.моль,
«цененной в предположении о равенстве средних энергий связей В—F и В—ОН в молекулах FBOH,
В(ОНJ и BF2.
FB(OHJ(r). Термодинамические свойства газообразного дигидроксид-фторида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 570.
Молекулярные постоянные FB(OHJ, использованные для расчета термодинамических
функций, приведены в табл. 20.21. В литературе отсутствуют сведения об экспериментальных или
теоретических исследованиях молекулярных постоянных FB(OHJ. В справочнике для этой молекулы
в электронном состоянии ХгА' принимается плоское строение (симметрия С,) и структурные
параметры из молекул BF3 и Н3ВО3: г (В—О) = 1,34+0,03 А, г (В—F)=1,31+0,02 А, г (О—Н) =
=0,96+0,02 А, J$ О-В-О = ^с F-B-O=120+3°, -^ В—О-Н=115+10°. Соответствующее
значение IaIbIc (см. табл. 20.21) может иметь погрешность 2-10~115 г3-см6 г. Основные частоты
FB(OHJ v1 и v2 (валентные колебания О—Н), v3 и v7 (валентные колебания В—О), v5 и v6
(деформационные колебания В—О—Н), vg (деформационное колебание О—В—О), vn и v12 (крутильные
колебания О—Н) перенесены из молекулы Н3ВО3. Основная частота v4 (валентное колебание В—F)
оценена сравнением частот аналогичного колебания в молекулах BFC12, BHFG1 и BH2F, а частоты
"V, (деформационное колебание О—В—F) и v10 (зонтичное колебание) оценены по частотам
молекулы BF3. Погрешности принятых значений vu и v12 составляют 150 см, v8—v10 — 50 см и
остальных частот — 100 см. Сведения о возбужденных электронных состояниях FB(OHJ
отсутствуют.
Термодинамические функции FB(OHJ (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных значений обусловлены неточностью принятых значений
молекулярных постоянных, а также приближенным характером расчета (в том числе учетом
заторможенных внутренних вращений как гармонических колебаний) и составляют 6, 10 и
15 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К (из них 1—3 Дж-К^-моль
из-за неточности молекулярных постоянных).
Таблица термодинамических функций FB(OHJ (г) публикуется впервые.
Константа равновесия реакции FB(OHJ (r)=F (г)+В (г)+20 (г)+2Н (г) вычислена с
использованием значения ЬГН° @) =2602,909 +21 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
Д^" (FB(OHJ, г, 0) = —1040^+ 20 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей В—F и В—ОН в
молекулах FB(OHJ, BF3 и Н3ВО3.
F2BOH (г). Термодинамические свойства газообразного гидроксид-дифторида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 571.
Молекулярные постоянные F2BOH, использованные для расчета термодинамических функций,
представлены в табл. 20.11. Величина IaIbIo соответствует вращательным постоянным F2UBOH,
Кавашима и др. [16446] при исследовании микроволнового спектра определили вращательные постоянные
F^B (ОНJ, Fl0B (ОНJ, FUB (ODJ и Fl0B (ODJ. Этим постоянным соответствуют структурные параметры и
произведение моментов инерции FB(OHJ /А/д/с=1154-10~117 г3-см6, хорошо согласующиеся с оцененными
в настоящем справочнике.
59
ВС1 (г) Глава 20
найденным Такео и Керлом [2650] в результате исследования микроволнового спектра различных
изотопных модификаций F2BOH. В работе [2650] было показано, что эта молекула имеет плоское-
строение (симметрия С,) с параметрами г (В—F)=1,32+0,02 А, г (В—О) = 1,34+0,02 А, г (О—Н) =
=0,94+0,01 А, ^ F-B—F=U8+1°, ^ F-B-0 (цисположение) = 122,8+1° и J$ В—О—Н=
=114,1 + 1°. Экспериментальных данных об основных частотах F2BOH нет. Оценки \ (валентное
колебание О—Н), vg (деформационное колебание В—О—Н) и v4 (валентное колебание В—О) сделаны,
как и для FBOH, по постоянным молекул НВО2 и ВОН. Частоты v2h v6 (валентное колебание В—F),
v6 и v7 (деформационное колебание F—В—О) и vg (деформационное колебание F—В—F) оценены
сравнением соответствующих частот BF3 и Н3ВО3. Квантовомеханический расчет [1842] барьера,
внутреннего вращения в молекуле F2BOH дал величину ~2400 см, что сравнимо с результатами
расчета Fo для молекулы Н2ВОН (см. также Н3ВО3). Эта величина представляется заниженной,,
однако отсутствие других данных не дает оснований для ее пересмотра. В настоящем справочнике
частота крутильного колебания v9 принимается по приведенному выше значению Vo, поскольку
расчет термодинамических функций через Vo при такой неопределенности не представляется
целесообразным. Погрешность значений v9 может достигать 150 см", погрешности \—v6 могут
составлять 100 см, v6—v8 — до 50 см. Статистический вес основного электронного состояния F2BOH
принят равным 1. Сведения о возбужденных электронных состояниях молекулы F2BOH
отсутствуют.
Термодинамические функции F2BOH (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью
оцененных значений основных частот, а также приближенным характером расчета (в том числе учетом:
заторможенного внутреннего вращения в приближении гармонического осциллятора), они
составляют 4, 6 и 10 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К (из них 1 —
2 Дж-К-моль из-за неточности основных частот).
Ранее таблица термодинамических функций F2BOH (г) рассчитывалась в справочнике JANAF'
[1543] по оцененным значениям всех молекулярных постоянных. Расхождения между значениями
Ф° (Т), вычисленными в настоящем издании справочника и в [1543], достигают 9 Дж-К~1-моль~1'
и объясняются в основном различием в оценке ряда основных частот.
Константа равновесия реакции F2BOH (r)=2F (г)+В (г)+О (г)+Н (г) вычислена с
использованием значения Д^0 @) =2263,367+16 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
A^°|(F2BOH, г, 0) = —1086 + 15 кДж • моль,
оцененной в предположении о равенстве средних энергий связей В—F и В—ОН в молекулах.
F2BOH, BF3 и Н3ВО3.
ВС1 (г). Термодинамические свойства газообразного хлорида бора в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 572.
Молекулярные постоянные ВС1, принятые в справочнике на основании анализа результатов^
исследований электронных спектров, представлены в табл. 20.12. В спектре ВС1 экспериментально'
изучены переходы 4Ш ** ХХ2+ [1394, 1569, 1836, 2020, 2021, 2178, 2696] и а3П -* ХгЕ + Ц832а,
1836]. Аналогично другим галогенидам группы IIIA, основное состояние Х1?+ молекулы ВС1
с электронной конфигурацией. . . а2а2тг4а2 и нижние возбужденные состояния А1!! и а3П,
соответствующие электронной конфигурации. . . о2 о2 л4 аи, коррелируют с диссоциационным пределом
В BР)+С1 BР%). В спектрах ВС1 не наблюдались переходы, связанные с другими валентными
состояниями этой молекулы; на основании данных для молекулы BF можно предполагать, что-
они имеют энергии не ниже 40 000 см".
Приведенные в табл. 20.12 колебательные постоянные ВС1 в состояниях ХгБ+ и А1!! найдены
Герцбергом и Хашли [1394] по (Акантам 28 полос системы A1U—X1L+ (z/, v" <C 8). Эти
постоянные хорошо описывают полосы с v" ^ 10, полученные позднее в работе Паннетье и др. [2178].
Вращательные постоянные в состояниях Х1!!* и АЩ также приняты по работе [1394], где они
получены из анализа вращательной структуры полос с у', d" ^ 6. Значения этих постоянных были-
подтверждены исследованием Паннетье и др. [2178]. Колебательные и вращательные постоянные-
ВС1 в состоянии а3П получены Лебретоном и др. [1836] по кантам полос 0—0, 1—1 и 2—2 и
вращательной структуре полос 0—0 и 1—1 системы а3П—XXS+.
60
Вор и его соединения ВСЬ (г)
Термодинамические функции BG1 (г) вычислены по соотношениям A.3) — A. 6), A. 9), A. 10),
¦{1.93)—A. 95). Значения Qsn и ее производных рассчитывались по уравнениям A. 90) — A. 92)
с учетом двух возбужденных состояний в предположении, что Q^ ^^(pJpA QW .
Статистическая сумма по колебательно-вращательным уровням состояния X'1S+ и ее производные
находились непосредственным суммированием по колебательным уровням и интегрированием по
значениям / с помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии с
значениями /=<^/Шах vj гДе Лпах „ определялись из условий A. 81). Колебательно-вращательные уровни
состояния Х1% + вычислялись по уравнениям A. 65), A. 62). Значения коэффициентов Ykl в этих
уравнениях для изотопической модификации ВС1, соответствующей природной смеси изотопов
бора и хлора, полученные по соотношениям A. 66) и постоянным молекулы 11В35С1, приведены
в табл. 20.13 вместе с величинами vimx и /Иш. Коэффициенты Y30 и У40 определены из условий A. 50),
коэффициент Y%x —¦ из условий A. 57). Мультиплетность и вырождение уровней в состояниях
а?П и А1!! учитывались статистическими весами 6 и 2 соответственно.
Погрешности вычисленных термодинамических функций BG1 (г) при Т ^ 3000 К определяются
практически лишь неточностью фундаментальных постоянных, при более высоких температурах
начинает сказываться отсутствие экспериментальных данных об энергии высоких колебательно-
вращательных уровней основного состояния, а также пренебрежение ненаблюдавшимися
возбужденными состояниями молекулы ВС1. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
составляют 0,02, 0,03 и 0,1 Дж-К~1-моль соответственно.
Термодинамические функции ВС1 (г) вычислялись в предыдущих изданиях справочника
1105, 106], справочниках [1543, 1904, 1974] и работе [499]. В большинстве расчетов не
учитывались возбужденные состояния ВС1 и расхождения с данными табл. 572 достигают 0,6 и
2,7 Дж-К~1-моль в значениях Ф° (Г) и 5° (Г). Расхождения с предыдущим изданием [105],
где были учтены два возбужденных состояния, существенно меньше и не превышают 0,1 и
0,6 Дж-К~1-моль~1 соответственно.
Константа равновесия реакции ВС1 (г)=В (г)+С1 (г) вычислена с использованием
значения ДД" @)=D0 (BC1) =569,620+11 кДж-моль?^47 610+950 см, соответствующего принятой
в справочнике энтальпии образования
Д//° (ВС1, г, 0) = 110 + 10 кДж . моль^1.
Принятое значение основано на исследовании равновесия реакции 2В (к)+ВС13 (г)=ЗВС1 (г),
проведенном методом протока A373—1573 К, 8 точек) Креневым и др. [199, 200, 201]. Из данных
этой работы было вычислено ^/Ho (ВС1, г, 0)=109+8 кДж-моль (III) и 113+17 кДж-моль (II).
Менее точное значение 135+40 кДж-моль было получено по уравнению II закона и результатам
масс-спектрометрического исследования этой же реакции, выполненного Сривастава и Фарбером
|2597] A513—1813 К, 7 точек). Линейная экстраполяция колебательных уровней состояния
X1S+ и экстраполяция уровней состояния АЧ1, выполненная в работе [589], приводят к
значениям 406 кДж-моль-1 C3 900 см) и 533+25 кДж-моль-1 D4 500+2000 см).
ВС12 (г). Термодинамические свойства газообразного дихлорида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 573.
Молекулярные постоянные, использованные для расчета термодинамических функций ВС12 (г),
приведены в табл. 20.14. Экспериментальные данные о структуре молекулы ВС12 в литературе
отсутствуют. Так же как и для BF2, в справочнике принимается, что в основном электронном
состоянии Х2Аг молекула ВС12 имеет нелинейную симметричную структуру (симметрия С2„) с
геометрическими параметрами, близкими к соответствующим параметрам ВС13: *4 С1—В—С1=120 +
+10°, г (В—С1)=1,74+0,04 А. Произведение моментов инерции ВС12, приведенное в табл. 20.14,
вычислено по этим параметрам, его погрешность составляет 2-1015 г3-см8. Приведенные
в табл. 20.14 частоты вычислены по уравнениям поля валентных сил, если принять силовые
постоянные ВС12 равными соответствующим постоянным ВС13 х. Погрешности в оцененных таким
образом величинах могут достигать 10—15%. Следует отметить, что Дессо и др. [946, 947]
получили спектр люминесценции, отнесенный ими к радикалу ВС12. Предварительный анализ
колебательной структуры привел к значению v2 более низкому, чем принятое в табл. 20.14 B00 см).
¦ /г=3,23; /гг=0,7 и /а/г3=0,3 (в 10» -дин-см)-.
61
ВС13 (г) Глава 2»
Однако авторы [946] отметили, что для проведения корректного анализа необходимо получить
спектр с более высоким разрешением. Приведенная в таблице энергия первого возбужденного
состояния А2В1 принята на основании теоретического расчета Хасти и Маргрейва [1367] с учетом
того, что для молекулы ВС1 аналогичный расчет приводит к заниженному значению энергии во&-
буждения состояния А1!!. Погрешность принятой величины не превышает 3000 см.
Термодинамические функции ВС12 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A. 9), A.10), A.122)—A. 124), A. 128), A. 130)
с учетом возбужденного состояния А2Вг по уравнениям A. 168)—A. 170). Погрешности
рассчитанных термодинамических функций составляют 1, 3 и 4 Дж-К-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К и обусловлены неточностью принятых значений молекулярных
постоянных A—2 Дж-К~1-моль~1) и приближенным характером расчета.
Ранее термодинамические функции ВС12 (г) вычислялись в предыдущих изданиях настоящего
справочника [105, 106], справочниках JANAF [1544] и Мак-Брайд и др. [1974]. Во всех
расчетах, кроме [1544], не учитывалось возбужденное электронное состояние. Расхождения величин
Ф° {Т), приведенных в табл. 573 и вычисленных в [105], [1544] и [1974], достигают 1,3, 3,2 и
3,6 Дж-К~1-моль соответственно и связаны с некоторым отличием в оценках молекулярных
постоянных ВС12.
Константа равновесия реакции ВС12 (г)=В (г)+2С1 (г) вычислена с использованием значения
A^ff0 @) =861,240+12 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
&.fH°(BCl2, г, 0) = —62+11 кДж-моль.
Эта величина основана на данных, приведенных в табл. 20.25, их погрешности учитывают
воспроизводимость измерений и неточности других постоянных, использованных в расчетах.
Принятое значение основывается на фотоионизационных измерениях [951], результаты которых
Таблица 20.25. Результаты определений А*Н° (ВС12, г, 0) (в кДж-моль)
Авторы
Метод
Ayff^BCla, г, 0)
И закон
III закон
Русин, Татевский, 1969 [353] Метод взрыва в сферической бомбе, 10 опытов — —77
Дайблер, Уокер, 1969 [951] Фотоионизационный, В2С14+С1=ВС1з+ВС12, —62+11
Дг#°@)=-94,6+3,0
Сривастава, Фарбер, 1971 [2597] Масс-спектрометрический, ВС13+ВС1=2ВС12, —90+14 — 99+10 а
1513—1813 К, 7 точек
а В работе [2597] константы равновесия вычислены по соотношению ионных токов. Однако ионы BClt
образуются не только при ионизации ВС12, но и при диссоциативной ионизации ВС1з [951, 2167], что должно было
привести к завышенным (по абсолютной величине) значениям Ду#° (ВС12).
подтверждаются более ранними измерениями, выполненными методом электронного удара [729,
1761, 2167], методом фотоионизации [1952] и методом [фотоэлектронной спектроскопии [729].
ВС13 (г). Термодинамические свойства газообразного трихлорида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 574.
Молекулярные постоянные, использованные для расчета термодинамических функций ВС13 (г),,
приведены в табл. 20.15. Электронографические измерения [1259, 1752, 1855, 2412, 2593],
исследования колебательно-вращательных спектров [516, 1816, 1852, 1866, 1883, 2484, 2868] и
теоретический расчет [274] показывают, что молекула ВС13 в основном электронном состоянии ХгА'ь
имеет форму равностороннего треугольника с атомом бора в центре, и относится к точечной группе
симметрии Z)за- Приведенное в табл. 20.15 произведение главных моментов инерции вычислено для*
межатомного расстояния г (В—С1)=1,7387+0,0044 А, найденного Канака и др. [1752] в
результате электронографического исследования. Близкие значения межъядерного расстояния В—CL
62
Бор и его соединения
ВС13 (г)
были получены Райаном [2412] A,7340 А) и Леви и Брокуэем [1855] A,73 А). Погрешность
величины IaIbIc не превышает 6-Ю15 г3-см6. Значения основных частот молекул 11ВС13 и 10ВС13,
приведенные в табл. 20.15, приняты по рекомендации Волфе и Хамфри [2868], которые
исследовали на приборе с высоким разрешением инфракрасные спектры газообразных тригалогенидов
бора и рассчитали соответствующие силовые постоянные. Сравнение вычисленных по этим
постоянным и наблюдаемых экспериментально частот молекул смешанных галогенидов бора показало, что
рекомендуемые ими частоты ВС13 являются достаточно точными. Значение \ совпадает с
полученным Скраби и Лачером [2484] и Линдеманом и Вильсоном [1866], значения v2 и v3 — с
полученными в работе [1866] и Левиным и Абрамовитцем [1852]. Погрешности принятых частот не
превышают 1 см х.
Исследования электронного спектра ВС13 [1948, 2234] показывают, что энергия первого
возбужденного состояния составляет 48 200 см, поэтому оно не рассматривается в справочнике.
Термодинамические функции ВС13 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Расчет проводился с учетом отличия частот колебаний разных изотопных модификаций
молекул ВС13 (по бору) и произведения их моментов инерции (по хлору). Погрешности
рассчитанных термодинамических функций составляют 0,7, 4 и 6 Дж-К~1-моль в значениях Ф° (Т) при
298,15, 3000 и 6000 К и обусловлены главным образом приближенным характером расчета.
Погрешности из-за неточности молекулярных постоянных не превышают 0,6 Дж-К~1-моль.
Термодинамические функции ВС13 (г) вычислялись в предыдущих изданиях справочника
[105, 106], справочниках [1543, 1974], а также в работах [1312, 1539, 1856, 2092, 2594, 2787,
2838, 2905] для Т ^ 1000-^-2500 К. Все расчеты выполнены по молекулярным постоянным,
близким к принятым в настоящем справочнике. Расхождения в функциях, вычисленных в
перечисленных выше справочных изданиях и приведенных в табл. 574, не превышают 0,3—0,8 Дж- К- моль
в значениях Ф° (Т) во всем интервале температур и обусловлены незначительной разницей в
принятых частотах колебаний и межъядерных расстояниях.
Таблица 20.26. Результаты определений Ду?Г° (ВС13, г, 298,15 К) (в кДжмоль)
Авторы
Трост, Отфийе,
1876 [2732, 2733]
Джонсон и др.,
1959 [1589]
Гальченко и др.
1960 [80, 83, 85
Лаубенгайер,
Метод
Калориметрические
измерения энтальпии
хлорирования аморфного
бора и энтальпии
гидролиза ВС1з (ж)
Калориметрическое
измерение энтальпии
хлорирования аморфного
бора в проточном
калориметре
, То же, с
использованием микропечи
Измерение энтальпии
Сере, 1945 [1824] гидролиза BCJ3 (ж)
а Энтальпия образования из аморфного
6 В результаты
(см. [1308]).
Д,Я°(ВС1„ г,В
298,15 К)
—404 а
—407,9 ±3,2 а
—408,4±3,2 а
—403,9
бора.
Авторы
Капустинский,
Самойлов, 1952
[150]
Скиннер, Смит,
1953 [2539]
Фазолино, 1965
[1069]
Ганн, Грин,
1960 [1308]
Лачер и др.,
1952 [1788]
измерений внесена поправка, учитывающая энтальпию
Метод
Измерение энтальпии
гидролиза ВС1з (ж)
То же
То же
То же, измерена также
энтальпия смешивания
растворов НзВОз и НС1
Калориметрическое
определение энтальпии
хлорирования дибора-
на при 80° С
смешивания растворов
AfH° (BC13, г,
298,15 К)
—377,4+5,4
-404,1 ±2,0 6
—403,5±2,0б
—404,5±1,3
-423+20
НзВОз и НС1
1 Молекулярные постоянные ВС13, приведенные в табл. 20.15, относятся к природной смеси изотопов хлора.
Коумфорд и др. [875] проанализировали изотопный эффект, обусловленный атомами звС1и37С1, на частоте vs
в ИК-спектре ВС13, замороженного в Аг матрице, и показали, что он мал. Лбвешпусс [1883] наблюдал
соответствующий изотопный эффект в спектре комбинационного рассеяния газообразного ВС13 и получил для частоты
vx значения от 461,8 см (nB37Cls) до 474,5 см (UBS*C1,).
63
В2С14(г), С1В0 (г) Глава 20
Константа равновесия реакции ВС13 (г)=В (г)+ЗС1 (г) вычислена с использованием значения
Дг#° @)=1322,334 +5,2 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
kfH°(BCls, г, 298,15 К) = — 404,5 + 1,3 кДж • моль-1.
Эта величина основана на результатах измерений, приведенных в табл. 20.26.
Наиболее точное значение энтальпии образования ВС13 (г) из кристаллического бора, принятое
в справочнике, было вычислено по данным работы Ганна и Грина [1308]. Близкие значения были
также получены по результатам измерений авторов [1069] и [2539]. Для энтальпии образования
ВС13 (г) из аморфного бора по данным работ [80, 83, 85, 1589] можно принять —408,1 +
+2,0 кДж-моль.
В2С14 (г). Термодинамические свойства газообразного тетрахлорида дибора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 575.
Молекулярные постоянные, использованные для расчета термодинамических функций В2С14,
приведены в табл. 20.11. Спектроскопические [637, 999, 1871, 1935, 2145] и электронографические
исследования [2413] показали, что в основном электронном состоянии Х1А1 молекула В2С14
принадлежит к точечной группе симметрии D2d- Приведенные в табл. 20.11 произведение главных
моментов инерции и величина 1лр соответствуют структурным параметрам: г (В—С1) = 1,75+0,01 А,
г (В—В) = 1,70+0,07 AM С1—В—С1=118,65+0,66°, найденным Райаном и Хедбергом [2413]
в электронографическом исследовании В2С14. Колебательные спектры молекулы В2С14
исследовались в работах [999, 1598, 1871, 1935, 2145]. Приведенные в таблице основные частоты приняты
по данным Дюрига и др. [999], которые исследовали инфракрасный спектр газообразного и спектр
комбинационного рассеяния жидкого В2С14. Величина потенциального барьера F0=630+35 см
и частота v4 найдены Джонсом и Райаном [1598] при изучении спектра КР газообразного В2С14.
Погрешности принятых частот составляют от 1 см в значениях v2) v4—v6 до 5 см в v7. Сведения
о возбужденных электронных состояниях В2С14 в литературе отсутствуют.
Термодинамические функции В2С14 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
{1. 130) с учетом заторможенного внутреннего вращения по уравнениям A. 160)—A. 162).
Погрешности вычисленных величин определяются неточностью молекулярных постоянных и
приближенным характером расчета и составляют 2, 9 и 12 Дж-К-моль в значении Ф° (Т) при
298,15, 3000 и 6000 К. Погрешности из-за неточности молекулярных постоянных не превышают
1—2 Дж-К^-моль-1.
Ранее термодинамические функции В2С14 (г) рассчитывались в таблицах JANAF [1543] в
предположении о заторможенном вращении вокруг связи В—В. Расхождение результатов расчета
[1543] с данными, приведенными в табл. 575, составляют от 7 до 13 Дж-К~1-моль~1 в значениях
Ф° (Г), они обусловлены главным образом тем, что авторы [1543] использовали неверные значения
двух основных частот этой молекулы.
Константа равновесия реакции В2С14 (г)=2В (г)+4С1 (г) вычислена с использованием значения
ДГЯ° @) =2089,264+14 кДж-моль, соответствующего принятой в справочнике энтальпии
•образования
Д/Г^С^, г, 298,15 К) = —490+J10 кДж . моль.
Эта величина является суммой энтальпии образования В2С14 (ж) и энтальпии ее испарения.
Ганн и др. [1311] измерили энтальпию реакции В2С14 (ж)+С12 (г)=2ВС13 (г), Дг#° B98,15 К) =
=—284,5+8,4 кДж-моль, чему соответствует ДД°(В2С14, ж, 298,15 К) =—524,9 +
+8,8 кДж-моль". Данным о давлении пара В2С14 (ж), измеренным Эрри и др. [2744] B10—
234 К, 7 точек), при расчете по III закону соответствует Д,Д° (В2С14, ж, 298,15 К)=35,0 +
+4,0 кДж-моль. В расчете были использованы сведения о теплоемкости В2С14 (ж), полученные
Линевским и Уортиком [1872].
С1ВО (г). Термодинамические свойства газообразного оксид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 576.
Молекулярные постоянные, использованные в расчете термодинамических функций С1ВО (г),
представлены в табл. 20.8. Исследование ИК-спектра молекул С1ВО в матрице из Аг, выполненное
64
Бор н его соединения С12ВО (г)
Снелсоном [2570], свидетельствует, что в основном электронном состоянии Хх?+ они имеют
линейную структуру. Приведенный в таблице момент инерции соответствует значениям г (В—С1)=1,74 +
±0,05 А (перенесено из ВС13) и г (В—О)=1,20±0,03 А (перенесено из ВО), его погрешность
составляет 1 • 10~39 г-см2. Для основных частот приняты значения, отнесенные Снелсоном [2570]
к молекуле 38CIUBO, их погрешности за счет матричных сдвигов оцениваются в 10 см.
Термодинамические функции G1BO (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных величин обусловлены неточностью принятых молекулярных
постоянных, в том числе пренебрежением различием частот колебаний разных изотопических
модификаций молекулы С1ВО, а также приближенным характером расчета, они составляют 1, 2
и 4 Дж- К- моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К (из которых 0,5—1 Дж- К, мояь
из-за неточности молекулярных постоянных).
Ранее таблицы термодинамических функций С1ВО (г) были вычислены в справочниках JANAF
[15431 и Мак-Брайд и др. [1974] по оцененным значениям молекулярных постоянных.
Расхождения в термодинамических функциях, приведенных в табл. 576 и в справочниках [1543, 1974],
обусловлены главным образом различиями в принятых значениях v2, они достигают 0,4 ж
2,1 Дж-К-моль~1 в значениях Ф° (Т).
Константа равновесия реакции С1ВО (г)=В (г)+С1 (г)+О (г) вычислена с использованием
значения Д^0 @)=1245,403+11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —319 + 10 кДж-моль-1.
Эта величина основывается на работах Блауэра и Фарбера [701] и Блауэра и др. [703], в
которых эффузионным методом была исследована реакция V3B2O3 (ж)-Ь1/8ВС13 (г)=С1ВО (г). По
данным работы [701] A234—1389 К, 5 точек) были найдены значения энтальпии образования,
равные — 310+100 кДж-моль (II закон) и —319±5 кДж-моль (III закон), а по данным работы
[703] A279 К, 2 измерения) —319+15 кДж-моль (III закон).
С12ВО (г). Термодинамические свойства газообразного оксид-дихлорида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 577.
Молекулярные постоянные, использованные для расчета термодинамических функций С12ВО,
цриведены в табл. 20.19. В литературе отсутствуют экспериментальные данные о молекулярных
постоянных С12ВО. По аналогии с FaBO в справочнике принимается, что в основном электронном
состоянии %гВг молекула С12ВО имеет плоскую структуру симметрии С^. Приведенное в табл. 20.19
произведение моментов инерции вычислено по структурным параметрам г (В—О)=1,40 +0,05 А,
г (В—G1)=1,74+0,05 А и -? С1—В—С1=126+5°, оцененным по соответствующим параметрам
молекул F8BO и ВС13, его погрешность составляет 3-Ю14 г3-см*. Основные частоты молекулы
С1аВО, приведенные в табл. 20.19, оценены из сравнения с молекулами F2BO и C12BF, их
погрешности оцениваются в 10—15%. Возбужденные электронные состояния С12ВО, по-видимому, так же
как у F2BO, имеют высокие энергии и не рассматриваются в справочнике.
Термодинамические функции С12ВО (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 1J4), A. 128), A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета, они составляют 1, 5 и 7 Дж-К-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К, в том числе из-за неточности молекулярных постоянных
1—3 Дж-К-^моль.
Таблица термодинамических функций С12ВО (г) публикуется впервые.
Константа равновесия реакции С12ВО (г)=В (г)+2С1 (г)+О (г) вычислена с использованием
значения Д^Р @)=1406,023+60 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
ДЯ" (С12ВО, г, 0) = —360 + 60 кДж • моль.
Эта величина была оценена сопоставлением энергий связей в молекулах С12ВО, ВО», С1.ВОН,
Н,0 н НВО2 (см. FjBO).
65
С13В3О3 (г), ВНС1 (г) Глава 2»
С13В3О3 (г). Термодинамические свойства газообразного трихлорбороксина в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 578.
Молекулярные постоянные, использованные для расчета термодинамических функций С13В3О3,
приведены в табл. 20.21. Экспериментальные данные о структуре С13В3О3 в литературе отсутствуют.
Приведенная величина IaIbIc вычислена для плоской кольцевой молекулы (группы симметрии
D3h) с г (В—О)=1,38±0,02 А, г (В-С1)=1,74 +0,03 А, ^ В-О-В = ^ О—В-0 = -3 О—В—С1=
=120°, ее погрешность составляет 2-10~112 г3-смв. Структурные параметры С13В3О3 оценены на
основании соответствующих параметров молекул Н3ВаО3 и ВС13 (см. выше). Спектры молекулы
С13В3О3 в газовой фазе до настоящего времени не исследовались. Изучен ИК-спектр поглощения
этой молекулы в твердой фазе (в стабилизированных пленках) [1816] и спектр комбинационного
рассеяния раствора в ВС13 [1238]. Приведенные в табл. 20.21 частоты v6—-v9 и vu, активные в ИК-
спектре, найдены в работе Латимера и Девлина [1816], частоты \— v3 и v10, активные в КР-спектре—
в работе Губо и Келлера [1238]. Значения неактивных частот v4 и v5 рассчитаны Латимером и Дев-
лином [18П] по найденному ими набору силовых постоянных потенциала Юри—Бредли. Частота»
неплоского деформационного колебания О—В—Cl, v12, принята равной частоте плоского
деформационного колебания О—В—Cl, v1Ol частоты v13 и v14 оценены по соответствующим частотам ко-
^й'ажжй.ъ^й^.«^^.?й.^\з^>8^з"й^з^)з^)з-^OTpesnuObTHтгриаятыхчастот составляют от W до 40 см
для частот \—>vu, а для частот v4, v5, v12—v14 10—30% от их величины. Сведения о возбужденных
электродных состояниях С13В3О8 в литературе отсутствуют.
Термодинамические функции С13В3О3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных функций определяются как неточностью молекулярных
постоянных, так и приближенным характером расчета и могут достигать 8, 15 и 25 Дж-К~1-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К, из них из-за неточности молекулярных постоянных
6—10 Дж-К^-моль-1.
Ранее таблицы термодинамических функций С13В3О3(г) рассчитывались в таблицах JANAF[1543J
и Мак-Брайд и др. [1974]. Расхождения данных, приведенных в табл. 578, с результатами этих
расчетов составляют 4—16 Дж-К~1-моль~1 в значениях Ф° (Т) и обусловлены тем, что оба расчета
были выполнены до опубликования работ J1816, 1817J.
Константа равновесия реакции С13В3О3 (г)=ЗВ (г)+ЗС1 (г)+30 (г) вычислена с использованием
значения Дг#° @) =4409,209+21 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
^fHo(C]aBsOs, г, 0) = —1630+15 кДж-моль.
Принятая величина основывается на исследованиях равновесия реакции В2О3 (ж)+ВС13 (г) =
=С13В3О3 (г) в работах [699, 701, 2255]. Блауэр и Фарбер [699] провели исследования методом
протока E36—825 К, 6 точек), этим данным соответствует &fH° (С13В3О3, г, 0) =—1640 +
+80 кДж-моль (II закон) и —1640+7 кДж-моль (III закон). Из эффузионных измерений этих
авторов [701 ] A234—1309 К, 3 точки) было найдено ДуЯ0 (С13В3О3, г, 0) =—1623+100 нДж-моль
(II закон) и —1616+10 кДж-моль (III закон). Портер и Гупта [2255] масс-спектрометрическим
методом (800—1200 К, 13 точек) измерили интенсивности ионных токов компонентов приведенной
реакции, этим данным соответствует Д^Я0 (С13В3О3, г, 0) =—1571+20 кДж-моль (II закон).
В работе [2255] была также определена константа равновесия реакции Н3В3О3 (г)+ЗНС1 (г) =
=С13В3О3 (г)+ЗН2 (г) при 1150 К, по которой было найдено Д, Н° (С13В3О3, г, 0) =—1500 +
+20 кДж-моль". Принятое в справочнике значение основано на работах Блауэра и Фарбера
[699, 701], результаты которых, включая обработку по II закону, находятся в хорошем
соответствии. Приведенные погрешности учитывают неточность термодинамических функций участников
реакций (в случае измерений при высоких температурах это основной источник ошибок),
использованных в расчетах термохимических величин и сечений ионизации, а также воспроизводимость
измерений.
ВНС1 (г). Термодинамические свойства газообразного гидрид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 579.
Молекулярные постоянные ВНС1, использованные для расчета термодинамических функций
и приведенные в табл. 20.14, оценены так же, как соответствующие постоянные BHF (см. выше).
66
Бор и его соединения ВН2С1 (г), ВНС12 (г)
Произведение главных моментов инерции в Х2А' состоянии рассчитано по структурным
параметрам г (В—Н)=1,18 ±0,02 А, г (В—С1)=1,74+0,04 А, *$ Н—В—С1=120+10°. Основные частоты
вычислены по силовым постоянным /в—н=4,1, /в—01=3,2 и /a/rjr2=0,39 (в 10Б дин-см).
Погрешность приведенного значения 1а1Ыс составляет 1-Ю17 г3-см8, основных частот — 10—15%,
величины То (Л2А") — 5000 см.
Термодинамические функции BHG1 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом вклада возбужденного электронного состояния А2А" по уравнениям A. 168)—
A. 170). Погрешности рассчитанных термодинамических функций составляют 1, 2 и
4 Дж-К~1-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К, они обусловлены в основном
неточностью оцененных молекулярных постоянных (в особенности в значениях S° (T) и С" (Т)) и
в меньшей степени приближенным характером расчета.
Таблица термодинамических функций ВНС1 (г) публикуется впервые.
Константа равновесия реакции ВНС1 (г)=В (г)+Н (r)-f-Cl (г) вычислена с использованием
значения Ь.,Н° @)=780,654+21 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^0 (BHQ, г, 0) = 115 ±>20 кДж • моль.
Эта величина была оценена в предположении о равенстве средних энергий связей в молекулах
ВНС1, ВН2 и ВС12.
ВН2С1 (г). Термодинамические свойства газообразного дигидрид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К, приведены в табл. 580.
Экспериментальные данные о спектре и структуре молекулы ВН2С1 в литературе отсутствуют.
Молекулярные постоянные ВН2С1, использованные при расчете термодинамических функций и
приведенные в табл. 20.15, оценены так же, как молекулярные постоянные BH2F, см. выше.
Значение IaIbIc рассчитано по структурным параметрам г (В—Н)=1,18+0 02 А, г (В—С1)=1,74+.
+0,04 Аи^ Н—В—Н=-$ С1—В—Н=120+10°, его погрешность не превосходит 2-1017 г3-смв,
погрешности в частотах составляют 10—15%. Так же как у молекулы ВС13, .возбужденные
электронные состояния должны иметь большие энергии.
Термодинамические функции ВН2С1 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом только основного ХгАг состояния. Погрешности в функциях обусловлены
отсутствием экспериментальных данных о молекулярных постоянных и приближенным характером
расчёта, они достигают 1, 3 и 5 Дж-К'^моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций ВН2С1 (г) публикуется впервые.
Константа равновесия реакции ВН2С1 (г)=В (г)+2Н (г)-|-С1 (г) вычислена с использованием
значения Д^0 @) = 1188,688+21 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Af//°5(BH2C1, г, 0) = —77 + 20 кДж • моль.
Эта величина была получена на основании предположения о равенстве средних энергий связей
в молекулах ВН2С1, ВН3 и ВС13 с учетом поправки (—6 кДж-моль"), оцененной из сравнения
полученных таким же образом значений AjH° для ВНС12 и BHF2 с найденными экспериментально.
ВНС12 (г). Термодинамические свойства газообразного гидрид-дихлорида бора в стандартном
состоянии в интервале температур 100—6000 К, приведены в табл. 581.
Молекулярные постоянные, использованные для расчета термодинамических функций ВНС12 (г),
приведены в табл. 20.15. Структура ВНС12 экспериментально не исследовалась. В согласии с-ре-
зультатами исследования тонкой структуры ряда полос в инфракрасном спектре ВНС12 [1893,
1896] и по аналогии с BHF2 в справочнике принимается, что в основном электронном состоянии
¦XхАг молекула ВНС12 имеет плоскую структуру симметрии C2v. Произведение главных моментов
инерции, приведенное в табл. 20.15, вычислено по оцененным структурным параметрам г (В—Н)=*
67
ВНСЬ (г)
Глава 20
=1,18±0,02 А, г (В—С1)=1,74 ±0,04 Аи^ Н—В— Cl=-? C1—В—С1=120+10° К Его
погрешность не превышает 2-10~115 г3-смв. ИК-спектр ВНС12 исследовался в работах [611, 612, 1893,
1894, 1896, 1933 J. Наиболее тщательное изучение спектра ВНС12 выполнено Линдсом и др. [612,
1893, 1896], которые в спектре газообразного BHG12 в области 500—2800 см наблюдали пять
частот UBHG12 и четыре частоты 10ВНС12 [612], а также исследовали спектр молекул ВНС12,
изолированных в матрице из Аг [611]. В табл. 20.15 приведены значения основных частот молекул
10ВНС12 и "ВНСЦ, найденные в работе [612]2. Частоты v1( v2, v4, v5, v6 наблюдались
экспериментально, их погрешности не превышают 1 см. Частота v3 вычислена, так как она расположена вне
исследованной экспериментально области спектра, ее погрешность оценивается равной 10%.
Сведения о возбужденных электронных состояниях ВНС12 в литературе отсутствуют, но по
аналогии с молекулами BXS можно предполагать, что они имеют высокие энергии.
Термодинамические функции ВНС12 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом различия частот разных изотопных модификаций ВНС12. Погрешности
рассчитанных величин при низких температурах в основном обусловлены отсутствием экспериментальных
данных о структурных параметрах ВНС12, при высоких температурах — приближенным методом
расчета и составляют 1, 3 и 5 Дж-К-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К,
погрешности из-за неточности молекулярных постоянных не превышают 1—2 Дж-К-моль~1.
Ранее таблицы термодинамических функций ВНС12 (г) вычисляли авторы справочника JANAF
[1543], а также Линде и Басе [1895] и Нагараян [2096] соответственно до Т < 1200 иГ< 2000 К.
Все расчеты выполнены по молекулярным постоянным, близким к принятым в настоящем
справочнике. Расхождения между термодинамическими функциями, приведенными в табл. 581 и
вычисленными в [1543] и [1895], не превосходят 0,1 Дж-К-моль~1 в значениях Ф° (Т) и 5° (Г).
Расхождения с данными [2096] достигают 0,5 Дж-К-моль~1.
Константа равновесия реакции ВНС1а (г)=В (r)+H(r)-f-2Gl (г) вычислена с использованием
значения kJHa @)=1263,174+5,4 кДж-моль, соответствующего принятой в справочнике
энтальпии образование
^Н°(ВЕС\2, г, 0) = —247,9 + 2,0 кДж • моль.
Эта величина основана на результатах исследований равновесия двух реакций: A) 6ВНС12 (г) =
=4ВС13 (г)+В2Н4 (г) и B) ВС13(г)-ЬН2(г)=ВНС12(г)+НС1 (г), представленных в табл. 20.27.
Таблица 20.27. Результаты
Авторы
Линде, Басе, 1964 [1895]
Мюриб и др., 1965 [2083]
Атвуд, Шелтон, 1970 [544]
Мук и др., (см. [1544])
эпределений h.fH° (BHC12, г, 0) (в кДж-моль)
Метод
Статический, реакция A), 298 К
То же, 2 точки, 273 и 298 К
То же, реакция B), 893—1023 К, значения К" (Т)
в работе не приводятся
Протока, реакция B), 1173—1573 К, 21 точка,
данные представлены в виде уравнения
Реакция B), 1073-1275 К.
Д/Я°(ВНС12, г, 0)
II закон
III закон
— —247,2±1,7
—249,2 —248,1 ±1,5
—249,2 —
-271 ±8 -247,3±5,0
-253±13 -250,6±3,7
Погрешности, приведенные в таблице, вычислены с учетом воспроизводимости измерений,
погрешностей термодинамических функций и термохимических величин компонентов исследованных
реакций. Расчеты энтальпии образования ВНС1а по уравнению III закона приводят к величинам,
1 Линде и Басе [1896], приняв для г (В—С1)=1,75 А, нашли в результате анализа тонкой структуры полосы v2
значения г (В—Н)=1,13±0,2 А, -4 С1—В—С1=119,7±3°. Исследование структуры полосы ve, связанной
с внеплоскостным колебанием [1893], привело к значениям А = 1,57±0,05 см и А— 5=1,47±0,05 см,
которые не противоречат результатам работы [1896].
s Мандирола я Вестеркамп [1933] предложили иное отнесение частот, ошибочность которого была показана Линд-
сом в работе [1894].
68
Бор и его соединения С1ВОН (г), С1В(ОВJ (г)
совпадающим в пределах их погрешностей. Принятое значение является средневзвешенным из них;
его погрешность оценена с учетом погрешностей отдельных измерений и их сходимости.
С1ВОН (г). Термодинамические свойства газообразного гидроксид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 582.
Молекулярные постоянные СШОН, использованные для расчета термодинамических функций,
приведены в табл. 20.19.
В литературе отсутствуют сведения об исследованиях молекулярных постоянных СШОН.
В справочнике принимается, что в основном электронном состоянии %?А' молекула СШОН, как
и FBOH, имеет плоское строение симметрии С„. Структурные параметры СШОН, на основании
которых выполнен расчет IaIbIc (см. таблицу), оценены по аналогии с молекулами ВС12 и В(ОНJ:
г (В— С1)=1,74+0,02 А, г (В—О) = 1,34±0,03 А, г (О—Н)=0,96+0,02 А, «$ С1-В—О=120 +
+5° и Jj В—О—Н=115+10° (трансположение). Погрешность IaIbIc составляет 2-Ю16 г3«смв.
Оценка основных частот \ (валентное колебание О—Н), v2 (деформационное колебание В—О—Н)
и v3 (валентное колебание В—О) выполнена, как и для FBOH, по аналогии с молекулами НВО2
и ВОН. Частота v4 (валентное колебание В—С1) принята такой же, как в молекуле BFC1. Частота
v6 (деформационное колебание О—В—С1) оценена сравнением деформационных частот в ВС12,
В(ОНJ и BFC1. Частота ve крутильного колебания принята такой же, как в FBOH. Погрешности
в значениях \—v4 могут составлять 100 см, остальных частот — 50 см. Тан же как молекулы
BHF и BFC1, молекула СШОН должна иметь возбужденное электронное состояние с энергией
от 7000 до 15 000 см, ввиду отсутствия экспериментальных данных о молекулярных постоянных
СШОН это состояние не представлено в табл. 20.19.
Термодинамические функции СШОН (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности вычисленных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета (в том числе учетом заторможенного внутреннего вращения
в приближении гармонического осциллятора) и составляют 3, 5 и 8 Дж-К~1-моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К (из которых 1—2 Дж> К- моль из-за неточности молекулярных
постоянных).
Таблица термодинамических функций СШОН (г) публикуется впервые.
Константа равновесия реакции СШОН (г)=С1 (г)+В (г)+О (г)+Н (г) вычислена с
использованием значения Д^ЕГ0 @)=1387,437+25 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^У0 (С1ВОН, г, 0) = —245 + 25 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей В—С1 и В—ОН в
молекулах СШОН, В(ОНJ и ВС12.
С1В(ОНJ (г). Термодинамические свойства газообразного дигидроксид-хлорида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 583.
Молекулярные постоянные С1В(ОНJ, использованные для расчета термодинамических
функций, приведены в табл. 20.21. В литературе отсутствуют сведения об исследованиях молекулярных
постоянных СШ(ОНJ. В справочнике принимается, что в основном электронном состоянии ХгА'
эта молекула имеет плоское строение симметрии С, со структурными параметрами, перенесенными
изВС13 и Н3ВО3: г(В-О)=1,34+0,03 А, г (В-С1)=1,74+0,02 А, г(О-Н)=0,96+0,02 А,
^ О—В—О = -^С1—В—0=120+3° и-?В—О—Н=115+10°. Погрешность вычисленного по этим
параметрам значения IaIbIc (см. таблицу) составляет 5'101В г3-см6. Основные частоты \ и v2
(валентные колебания О—Н), v3 и ve (валентные колебания В—О), v4 и v5 (деформационные
колебания В—О—Н), v8 (деформационное колебание О—В—О), vu и v12 (крутильные колебания О—Н)
перенесены из молекулы Н3ВО3. Основная частота v7 (валентное колебание В—С1) оценена
сравнением частот аналогичного колебания в молекулах BF2C1, BH2C1 и BHFC1. Частоты v9
(деформационное колебание О—В—С1) и v10 («зонтичное» колебание) приняты по данным для BF2C1 в
предположении, что замена атома F на изоэлектронную группу ОН не изменяет существенно величины
соответствующих частот. Погрешности принятых значений основных частот могут достигать 150 см
для крутильных колебаний (vn и v12), 100 см для \— ve и около 50 см для остальных частот.
69
CIjBOH (г), BFC1 (г) Глава 20
Термодинамические функции С1В(ОНJ (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных значений обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета (в том числе учетом заторможенного
внутреннего вращения в приближении гармонического осциллятора) и составляют 6, 10 и
15 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К (из которых 1—3 Дж-К^-моль
из-за неточности молекулярных постоянных).
Таблица термодинамических функций С1В(ОНJ (г) публикуется впервые.
Константа равновесия реакции С1В(ОНJ (г)=С1 (г)+В (г)+2О (г)+2Н (г) вычислена с
использованием значения ?±ГН° @) =2401,254+21 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
ЬХН° (С1В(ОН)а, г, 0) = —796 ± 20 кДж • моль,
оцененной в предположении о равенстве средних энергий связей В—С1 и В—ОН в молекулах
С1В(ОНJ, ВС13 и Н3ВО3.
С12ВОН (г). Термодинамические свойства газообразного гидроксид-дихлорида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 584.
Молекулярные постоянные С12ВОН, использованные для расчета термодинамических функций,
приведены в табл. 20.11. Сведения об исследованиях молекулярных постоянных С12ВОН в
литературе отсутствуют. По аналогии с молекулой F2BOH (см. выше) в настоящем справочнике
принимается, что С12ВОН имеет в основном электронном состоянии ХХА' плоскую структуру симметрии
С, с параметрами г (В—О)=1,34 +0,03 А, г (В—G1)=1,74+0,02 А, г (О—Я) =0,96+0,02 А,
J? С1—В—С1=120+2°, -3 G1—В—0=120+3° и ^ В—О—Н=115+10°, оцененными по
соответствующим величинам в молекулах Н3ВО3, ВС13 и F2BOH. Погрешность вычисленного по этим
параметрам значения IaIbIc (см. таблицу) составляет 1-Ю"14 г3-см6. Оценки основных частот \
(валентное колебание О—Н), v2 (деформационное колебание В—О—Н) и v3 (валентное колебание
В—О) сделаны по молекулам НВО2 и ВОН. Частоты v4 и v. (валентные колебания В—Gl), v6
(зонтичное колебание), v7 (деформационное колебание С1—В—О) и vg (деформационное колебание
С1—В—G1) оценены сравнением соответствующих частот ВС13, Н3ВО3, а также BFGla. Частота v9
крутильного колебания ОН принята такой же, как в молекуле F2BOH. Погрешности принятых
значений vx—v4 составляют около 100 см, v5—v8 — 50 см и v9 —¦ 150 см.
Термодинамические функции С12ВОН (г) вычислены по формулам A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью
принятых значений молекулярных постоянных, а также приближенным характером расчета (в том
числе учетом заторможенного внутреннего вращения в приближении гармонического осциллятора)
и составляют 4, 6 и 10 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К (из них 1 —
2 Дж-К-моль из-за неточности молекулярных постоянных).
Таблица термодинамических функций С12ВОН (г) публикуется впервые.
Константа равновесия реакции С12ВОН (г)=2С1 (г)+В (г)+О (г)+Н (г) вычислена с
использованием значения АГН° @)=1862,057 +16 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
A^^CLjBOH, г, 0) = —600 + 15 кДж . моль,
оцененной в предположении о равенстве средних энергий связей В—С1 и В—ОН в молекулах
С12ВОН, ВС13 и Н3ВО3.
BFC1 (г). Термодинамические свойства газообразного фторид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К, приведены в табл. 585.
Структура и спектры молекулы BFG1 экспериментально не исследовались. Молекулярные
постоянные BFC1, приведенные в табл. 20.14, оценены так же, как для молекулы BHF (см. выше).
Произведение главных моментов инерции в состоянии Х2А' вычислено по постоянным г (В—F) =
=1,31+0,03 А, г (В—С1) =1,74+0,04 А и ^ F-B—С1=120+10°, его погрешность равна 5х
Х10~116 г3-см8. Основные частоты вычислены по силовым постоянным /в—р=6,6, /в—ci=3,2 ш
70
Бор и его соединения BF2C1 (г)
/в/Г]Г2=0,46 (в 105 дин-см), погрешности принятых значений частот не превышают 10—15%,
погрешность в То (Л2А") составляет 5000 см.
Термодинамические функции BFC1 (г) рассчитаны в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом возбужденного электронного состояния А2А" по уравнениям A. 168)—A. 170).
Погрешности в термодинамических функциях BFC1 (г) составляют 1, 3 и 5 Дж-К~1-моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К и обусловлены в основном неточностью оцененных
молекулярных постоянных A—2 Дж-К>моль) и в меньшей степени приближенным характером
расчета.
Ранее термодинамические функции BFC1 (г) вычисляли авторы справочника JANAF [1543],
Мак-Брайд и др. [1974] и Эванс [1044]. Функции, приведенные в табл. 585 и в [1543], отличаются
не более, чем на 1 Дж-К-моль~1 в значениях Ф° (Т) и S° (T). Расхождения с функциями,
приведенными в [1974] и [1044], достигают 2—3 Дне-К-моль.
Константа равновесия реакции BFG1 (г)=В (r)+F (г)+С1 (г) вычислена с использованием
значения Дг Н° @)=1037,895 +11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH°(BFGl, г, 0) = —281 + 10 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании р^новесия реакции
BF2 (r)+BCl2 (r)=2BFCl (г), выполненном Сривастава и Фарбером [2597] A523—1800 К, 7
точек, расчет по III закону приводит к значению —281+10 кДж-моль, по II закону — к
значению — 226+15 кДж-моль, где погрешности учитывают неточность термодинамических величин
и сечений ионизации, использованных в расчетах, а также воспроизводимость измерений).
Совпадающее значение было получено в предположении о равенстве средних энергий связей в
молекулах BFC1, BF2 и ВС12.
BF2C1 (г). Термодинамические свойства газообразного дифторид-хлорида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 586.
Молекулярные постоянные, использованные для расчета термодинамических функций BF2C1,
приведены в табл. 20.15. Структура молекулы В F2C1 экспериментально не исследовалась. По
аналогии с BHF2 и симметричными галогенидами бора ВХ3 в настоящем справочнике принимается, что
в основном электронном состоянии ХгАг молекула BF2C1 имеет плоскую структуру симметрии
С2о. С такой структурой согласуются результаты исследований инфракрасного спектра BF2C1,
выполненных Линдеманом и Вильсоном [1866] и Волфе и Хамфри [2868]. Произведение главных
моментов инерции, приведенное в табл. 20.15, вычислено по постоянным г (В—F)=l,31+0,03 A,
г (В—С1)=1,74 +0,04 Аи^ F—В—С1=-4 F—В—F = 120 +5°, равным соответствующим значениям
в молекулах BF3 и ВС13. Его погрешность составляет 5-Ю15 г3-см6. Основные частоты молекул
nBF2Cl и 1OBF2C1, найденные при исследованиях инфракрасного спектра газообразного BF2C1
в области 300—1500 см в работах [1866, 2868], совпадают. Они приведены в табл. 20.15, их
погрешности не превышают 1 см. Сведения о возбужденных электронных состояниях BF2C1 в
литературе отсутствуют.
Термодинамические функции BF2C1 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A.122)—A. 124), A. 128), A. 130).
Расчет проводился с учетом различия частот разных изотопных модификаций BF2C1. Погрешности
вычисленных величин обусловлены в основном приближенным характером расчета и в меньшей
степени неточностью структурных параметров этой молекулы. Погрешности в значениях Ф° (Т)
при 298,15, 3000 и 6000 К составляют 1, 3 и 5 Дж-К~1-моль, погрешности из-за неточности
постоянных не превышают 1—2 Дж-К-моль.
Ранее термодинамические функции BF2C1 (г) вычисляли авторы справочника JANAF [1543],
Эванс [1044] и Нагараян [2091] (Т ^ 1000 К). Данные табл. 586 и [1543] совпадают в пределах
0,13 Дж-К~1-моль~1 в значениях Ф° (Т) и S° (T). В работе [2091] допущены ошибки и
расхождения составляют 2 Дж-К~1-моль~1 в значениях Ф° (Г) и 5° (Т) уже при 298,15 К.
Константа равновесия реакции BF2C1 (r)=B (r)+2F (г)+С1 (г) вычислена с использованием
значения &ГН° @)=1719,856 +7,1 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
l, г, 298,15 К) = —888 + 5 кДж • моль.
71
BFCIjj (г), FaClB3O3 (г) Глава 20
Принятая величина основана на исследованиях равновесия реакции 2BF3 (г)+ВС13 (г) —
=3BF2C1 (г), выполненных масс-спектрометрическим методом Портером и др. [2253] C50—600К,,
измерялась зависимость интенсивности ионных токов от температуры, Д^Я° (BF2C1, г, 298,15 К)=
=—890,1+1,0 кДж-моль (II)) и Сривастава и Фарбером [2597] B98|К, f Д^0 (BF2C1, rr
2&8,15К)=— 886,7 кДж-моль (III)). В работах Хиггинса и др. [1404] и Ганна, Санборна [1312]
методом ИК-спектроскопии было исследовано равновесие реакции BF3 (r)+BCl3(r)=BF2Cl (г) +
+BFC12 (г). Из этих измерений было найдено в хорошем соответствии с результатами работ [22531
и [2597], что сумма энтальпии образования BF2C1 и BFC12 составляет — 1533,0 кДж-моль.
BFC12 (г). Термодинамические свойства газообразного фторид-дихлорида бора в стандартном
состоянии в интервале температур 100—6000 К, приведены в табл. 587.
Молекулярные постоянные, использованные для расчета термодинамических функций BFC12 (г),.
приведены в табл. 20.15. Прямые экспериментальные данные о структуре BFC12 в литературе
отсутствуют. На основании результатов исследований инфракрасного спектра BFC12 [1866, 2868}
и по аналогии с другими молекулами ВХУа ч справочнике принимается, что молекула BFC12
в основном электронном состоянии Х1А1 имеет плоскую структуру (симметрия CZv). Приведенное
в табл. 20.15 произведение главных моментов инерции вычислено по структурным параметрам
г (В—F) = 1,31 +0,03 А, г (В-С1)=1,74+0,04 А, ^ F-B-C1=^ С1-В-С1=120+5°, принятым
равными соответствующим параметрам в молекулах BF3 и ВС13. Погрешность значения Idslc
не превышает 2-1014 г3-см6. Основные частоты, приведенные в табл. 20.15, найдены Волфе и
Хамфри [2868] при исследовании инфракрасного спектра газообразного BFC12 в области 300—
1500 см. Частоты vl5 v4, v6 наблюдались непосредственно, они совпадают с измеренными в работе-
Линдемана и Вильсона [1866]. Частоты v2, v3 и vg были вычислены по составным частотам и
обертонам, отнесение которых выполнено в работе [2868]. В пределах 1—15 см они согласуются с
частотами, оцененными Линдеманом и Вильсоном [1866]. Погрешности в \, v4 и ve не превышают
1 см, погрешности в va, v3 и v6 могут достигать 10 см. Сведения о возбужденных электронных
состояниях BFC12 в литературе отсутствуют.
Термодинамические функции газообразного BFC12 вычислялись в приближении «жесткий
ротатор — гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A. 122)—A. 124),.
A. 128), A. 130) с учетом только основного электронного состояния. В расчете учитывалось
различие молекулярных постоянных изотопических модификаций BFC12. Погрешности вычисленных
функций составляют 1, 4 и 6 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К, они
обусловлены в основном приближенным характером расчета. Погрешности из-за неточности
молекулярных постоянных не превосходят 1—2 Дж-К~1-моль в значениях Ф° (Т).
Ранее таблицы термодинамических функций BFC12 (г) вычисляли авторы справочника JANAF
[1543], Эванс [1044] и Нагараян [2091] до Г<1000К. Данные табл. 587 и [1543] совпадают
в пределах 0,2 Дж-К-моль~1 в значениях Ф°(Т) и S° (T). В расчете [2091] допущена ошибка,
и расхождения с данными табл. 587 достигают 1,5 Дж-К-моль уже при 298,15 К.
Константа равновесия реакции BFC12 (г)=В (r)+F (г)+2С1 (г) вычислена с использованием
значения А^0 @)=1517,934+7,1 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(BFC12, г, 298,15 К) = —643 ± 5 кДж • моль.
Принятая величина является средней из вычисленных по результатам исследований равновесия
реакции BF3 (г)+2ВС13 (r)=3BFCl2 (г) в работах Портера и др. [2253] C50-600 К, bfH° (BFC12,
г, 298,15 К) = -645,4+1,0 кДж-моль (И)) и Сривастава и Фарбера [2597] B98 К, Д/Я° (BFCl2r
г, 298,15 К) =—640,9 кДж-моль (III)). Эти измерения подтверждаются также работами Хиггинса
и др. [1404] и Ганна и Санборна [1312] (см. текст по BF2C1).
F2C1B3O3 (г). Термодинамические свойства газообразного дифторхлорбороксина в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 588.
Молекулярные постоянные, использованные для расчета термодинамических функций F2C1B3O3,
приведены в табл. 20.17. Экспериментальные данные о структурных параметрах F2C1B3O3 в
литературе отсутствуют. Произведение моментов инерции вычислено для плоской кольцевой структуры
(симметрия Civ) по структурным параметрам, оцененным на основании соответствующих данных
в молекулах LHSB3O3, BF3 и ВС13; г (В-О)-1,38±0,02 А, г (B-F)=l,31+0,03 А, г (В-С1) =
72
Бор и его соединения FC12B3O3 (г)
=1,74+0,04 А, ^В-О-В = -§ О—В-О = -§ О—B-F=<$ О—В-С1=120°; его погрешность
составляет 5-10~113 г3-см6. Основные частоты v2, v4, v8, v10—v12, v14, vie, v18 приняты по работе Лати-
мера и Девлина [1817], в которой исследован ИК-спектр поглощения этой молекулы в твердой
фазе (в стабилизированных пленках). Для частот vX) v3, v5—v7, v9, v13, v15 приняты величины,
рассчитанные Латимером и Девлином по найденному ими набору силовых постоянных, частоты
v17, v19, v20, v21 оценены по соответствующим частотам колебаний в молекулах F3B3O3, C13B3O3.
Погрешности частот, наблюдавшихся в работе [1817], оцениваются в 10—40 см, погрешности
рассчитанных и оцененных частот — в 10—30% от их величины. Сведения о возбужденных
электронных состояниях F2G1B3O3 в литературе отсутствуют.
Термодинамические функции F2C1B3O3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных функций определяются неточностью молекулярных
постоянных и приближенным характером расчета, они составляют 5, 15, 20 Дж-К~1-моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К, в том числе из-за неточности постоянных 4—8 Дж-К^-моль.
Таблица термодинамических функций F2C1B3O3 (г) публикуется впервые.
Константа равновесия реакции F2G1B3O3 (r)=3B (r)+2F (r)-j-Cl (г)+30 (г) вычислена с
использованием значения Д,_<ЕГ° @)=4819,519+21 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
HfHol(F2ClB3O3, г, 0) = —2125J+20 кДж-моль.
Эта величина была оценена в предположении о равенстве средних энергий связей в молекулах
F2C1B3O3, F3B3O3 и С13В3О3.
FC12B3O3 (г). Термодинамические свойства газообразного фтордихлорбороксина в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 589.
Молекулярные постоянные, использованные для расчета термодинамических функций FC12B3O3,
приведены в табл. 20.17. Экспериментальные данные о структурных параметрах FC12B3O3 в
литературе отсутствуют. Произведение моментов инерции вычислено для плоской структуры этой
молекулы (симметрия С2г) по структурным параметрам, оцененным на основании соответствующих
постоянных в молекулах Н3В3О3, BF3 и ВС13 : г (В—O)=l,38+0,02 A, jr (В—F)=l,31+0,03 А,
г (В—С1)=1,74 +0,04 А, -ЗВ—О—В = -$О—В—О=-?О—В—F=-$O—В—С1=120°, его
погрешность составляет 1-10~112 г3-см6. Основные частоты v2, v4_v6, v10—v14, v16, v17 приведены
в табл. 20.17 по работе Латимера и Девлина [1817], которые исследовали ИК-спектр поглощения
этой молекулы в твердой фазе (в стабилизированных пленках). Для частот v1? v3, v,— v9, v15 приняты
значения, рассчитанные Латимером и Девлином по найденному ими набору силовых постоянных,
частоты v18—,val оценены по соответствующим частотам колебаний в молекулах F8B3O3 и С13В3О3.
Погрешности частот, наблюдавшихся в работе [1817], составляют 10—40 см, остальных частот —
Ю—30% от их величины. Сведения о возбужденных электронных состояниях в литературе
отсутствуют.
Термодинамические функции FC12B3O3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A.130). Погрешности вычисленных таким образом функций определяются неточностью
молекулярных постоянных и приближенным характером расчета и составляют 5, 15 и 20 Дж-К-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К, из них из-за неточности молекулярных постоянных
4-8 Дж-К'^моль.
Таблица термодинамических функций FC12B3O3 (г) публикуется впервые.
Константа равновесия реакции FC12B3O3 (г)=ЗВ (r)+F (г)+2С1 (г)+30 (г) вычислена с
использованием значения ДГЯ° @) =4613,864+21 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
^Н° (FC12B3O3> г, 0) = —1877 + 20 кДж • моль.
Эта величина была оценена в предположении о равенстве средних энергий связей в молекулах
FC12B3O3) F3B3O3 и С13В8О3.
73
BHFCI (г), BBr (г) =========== Глава 20
BHFC1 (г). Термодинамические свойства газообразного гидрид-фторид-хлорида бора в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 590.
Экспериментальные данные о спектре и структуре молекулы BHFC1 в литературе отсутствуют.
Приведенные в табл. 20.15 молекулярные постоянные BHFC1 оценены так же, как и
молекулярные постоянные BH2F (см. выше), с учетом измеренных экспериментально частот молекул BHF2
и ВНС12. Приведенное в табл. 20.15 произведение главных моментов инерции вычислено по
структурным параметрам г (В—Н)=1,18±0,02А, г (В—F)=1,31 ±0,03 А, г(В—С1)=1,74+0,04 А и
-$ X—В—У=120 +10°. Погрешность в IaIbIc оценивается в 6-10~118 г3-см6, погрешности в
частотах не превышают 10—15%.
Термодинамические функции BHFG1 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A. 10), A. 122)—A. 124), A. 128), A. 130)
€ учетом только основного электронного состояния ХгА'. Погрешности в термодинамических
функциях BHFG1, обусловленные неточностью оцененных молекулярных постоянных и
приближенным характером расчета, составляют 1, 3 и 5 Дж-К-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К.
Таблица термодинамических функций BHFG1 (г) публикуется впервые.
Константа равновесия реакции BHFC1 (г)=В (г)+Н (r)+F (г)+С1 (г) вычислена с
использованием значения &ГН° @) =1452,929 +21 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Aff^BHFCl г, 0) = —480 ± 20 кДж • моль,
оцененной в предположении о равенстве средних энергий связей в молекулах BHFCI, BH3, BF3
и ВС13.
ВВг (г). Термодинамические свойства газообразного бромида бора в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 591.
Молекулярные постоянные ВВг, принятые в справочнике на основании результатов
исследования электронных спектров, представлены в табл. 20.12. В спектре ВВг наблюдались переходы
А1!!—ХХЕ+ [2020, 2021, 2025, 2363] и а3П—Х^ + [1832а, 1835]. Также как у других галогенидов
элементов группы IIIA все три состояния, соответствующие конфигурациям электронной оболочки
.. .тс4а2 и ... тс4ая, коррелируют с диссоциационным пределом В BР)-\-Вт B-Р=/2). Молекула ВВг
должна обладать рядом связанных валентных электронных состояний с энергиями свыше
30 000 см, однако сведения о них отсутствуют и они не рассматриваются в справочнике.
Приведенные в табл. 20.12 колебательные постоянные молекулы uB79Br в состояниях X1? + и А1!!
найдены Розенталером [2363] по кантам полос v ^ 4, v" ^ 5. Для основного состояния они хорошо
согласуются с результатами анализа колебательной структуры системы полос а3П—Х^+ (г/<^6,
и'^ 7) в работах Лебретона и др. [1832а, 1835]. Колебательные постоянные в состоянии а3П
приняты по данным [1832а, 1835]. Вращательные постоянные в состояниях X1 2+ и А1!! приняты
по работе [2363], где был выполнен анализ структуры полос 0—0 C ветви) и 1—1 (одна ветвь)
перехода А1!!—Xxti+. Вращательные постоянные в состоянии а3П найдены в работах [1832а,
1835] из анализа вращательной структуры полос 0—0, 1—0 и 2—2, при этом для состояния XlS*
были получены значения, совпадающие с данными [2363].
Термодинамические функции ВВг (г) были вычислены по соотношениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производных по температуре рассчитывались по
уравнениям A. 90)—A. 92) с учетом двух возбужденных электронных состояний. Статистические суммы
по уровням энергии состояний Х1^* и а3П и их производные находились непосредственным
суммированием по колебательным уровням и интегрированием по / с помощью уравнений типа A. 82),
для состояния А1!! принималось, что Q^l. ^ — (Р^УР^) Qifx.sp колебательно-вращательные уровни
состояний Хх?+ и а3П вычислялись по уравнениям A. 65), A. 62), в расчетах учитывались все
уровни с значениями / ^ /Шах, «! гДе Jmax, „ находились из условий A. 81). Значения
коэффициентов Ykl для молекулы ВВг, соответствующей природной смеси изотопов бора и брома, полученные
по постоянным иВ79Вг и соотношениям A. 66), приведены в табл. 20.13. Коэффициенты Ys0 и У40
определены с учетом условий A. 50). Мультиплетность и вырождение вращательных уровней в
состояниях а3П и АгП учитывались статистическими весами 6 и 2 соответственно.
При невысоких температурах основные погрешности рассчитанных термодинамических функций
обусловлены неточностью фундаментальных постоянных и вращательной постоянной ВВг, при
74
Бор и его соединения ВВг2 (г), ВВг3 (г)
более высоких температурах становятся существенными отсутствие экспериментальных данных
о высоких колебательно-вращательных уровнях состояния Хг%+, а также пренебрежение высокими
электронными состояниями. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут
достигать 0,03, 0,05 и 0,3 Дж-К~1-моль~1 соответственно.
Ранее термодинамические функции ВВг (г) вычислялись в таблицах JANAF [1543J и в работе
[499] (до 6000 К). Расхождения данных табл. 591 и этих работ обусловлены различием
молекулярных постоянных, методов расчета и пренебрежением возбужденными состояниями ВВг в [499,
1543]. Эти расхождения достигают 0,5 и 1,2 Дж-К -моль в значениях Ф° (Т) и S° (T).
Константа равновесия реакции ВВг (г)=В (г)+Вг (г) вычислена по принятой в справочнике
энергии диссоциации
АгН° @) = Do (ВВг) = 37 000 ± 1000 см «* 443 ± 12 кДж • моль.
Эта величина принята на основании короткой экстраполяции колебательных уровней
состояния Л 41 по постоянным, приведенным в табл. 20.12 (vmB,x=8, To-\-Go (8)=37 120 см), последний
наблюдавшийся колебательный уровень (у=4) имеет энергию ~36100 см. Потенциальная кривая
состояния А1!! может иметь максимум величиной в 500—1000 см, что учитывается погрешностью,
приписанной принятому значению. Последнему соответствует
A^°(BBr, г, 0) = 234,923 + 13 кДжТьюль.
ВВг2 (г). Термодинамические свойства газообразного дибромида бора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 592.
Экспериментальные данные о структуре и спектре молекулы ВВг2 отсутствуют, поэтому
в табл. 20.14 приведены молекулярные постоянные, оцененные при подготовке справочника на
основании постоянных молекулы ВВг3. Приведенное в табл. 20.14 произведение главных моментов
инерции в электронном состоянии Х2АХ вычислено для симметричной нелинейной структуры
(симметрия С.и) о ^ Вт— В— Bi=120+10° в. г (В—Вт}=1,89+0,04 А.. Погрешность Г АЩс составляет
2-1014 г^-см6. Основные частоты, приведенные в табл. 20.14, вычислены по силовым постоянным,
принятым равными соответствующим постоянным ВВг3 \ их погрешности не превышают 10—15%.
Оценка энергии возбуждения состояния А2В± по аналогии с соответствующими состояниями
молекул BF2 и ВС12 приводит к значению То (Л2В1) = 10 000+5000 см 2.
Термодинамические функции ВВг2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом возбужденного состояния Л2В± по уравнениям A. 168)—A. 170). Погрешности
рассчитанных термодинамических функций ВВг2 (г) обусловлены главным образом неточностью оцененных
молекулярных постоянных, а также приближенным характером расчета и могут достигать 2, 4
и 6 Дж- К- моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции ВВга (г) вычислялись в таблицах JANAF [1543] по близким
значениям молекулярных постоянных, но без учета возбужденного электронного состояния.
Расхождения данных табл. 592 и [1543] не превосходят 1 Дж-К~1-моль в значениях Ф° (Т).
Константа равновесия реакции ВВг2 (г)=В (г)+2Вг (г) вычислена с использованием принятой
в справочнике величины
i\rH° @) = 695 + 25 кДж • моль,
оцененной сопоставлением энергий связей в молекулах ВХ, ВХ2 и ВХ3, где X=Br, Cl, F. Принятой
величине соответствует
Д//° (ВВг2, г, 0) = 100,846 + 25 кДж • моль.
ВВг3 (г). Термодинамические свойства газообразного трибромида бора в стандартном состоянии
в интервале температур 100—6000 К, приведены в табл. 593.
Молекулярные постоянные, использованные при расчете термодинамических функций ВВг3 (г),
приведены в табл. 20.15. Результаты электронографических измерений [1751, 1855] и исследова-
1 /г=2,55; /гг=0,55 и /а/г2=0,25 (в 105
* Как и в случае BF3, при возбуждении паров ВВг3 электронным ударом в спектре флуоресценции в
ультрафиолетовой области наблюдаются полосы, которые, возможно, связаны с образованием молекул ВВг2 [1399].
75
BI (г) Глава 20
ний колебательно-вращательных спектров [506, 516, 1088, 1239, 1852, 1866, 2789, 2833, 2868}
показывают, что молекула ВВг3 в основном %}А\ состоянии имеет плоскую симметричную
структуру (симметрия D3h). Приведенное в табл. 20.15 произведение главных моментов инерции
вычислено для межатомного расстояния т (В—Вг) = 1,893+0,005 А и 4 Вг—В—Вг=120°,
полученных Канака и др. [1751 ] при электронографическом исследовании ВВг3. Его погрешность
составляет 1-Ю13 г3-смв. ИК-спектр и спектр КР ВВг3 исследовались неоднократно как в жидком
[506, 516, 1088, 1239, 1866, 2789], так и в газообразном [1852, 1866, 2243, 2868] состояниях.
Приведенное в табл. 20.15 значение \ найдено Линдеманом и Вильсоном [1866] в спектре КР жидкого-
ВВг3. Частоты v3 и v4 приняты по результатам измерений Линдемана и Вильсона [1866], Левина
и Абрамовица [1852] и Волфе и Хамфри [2868] в ИК-спектре газообразных 10ВВг3 и иВВг8. Для
частоты v2 в табл. 20.15 приведены значения, близкие к средним из найденных в ИК-спектре
газообразного ВВг3 в работах Волфе и Хамфри [2868] и Помпосиело и др. [2243]. Погрешности в
значениях vx, v3 и v4 не превышают 1—3 см, погрешность v2 может достигать 10 см. В пределах
указанных погрешностей принятые частоты удовлетворительно согласуются с найденными в других
исследованиях спектров жидкого и газообразного ВВг3. Исследования электронного спектра ВВг9
показали, что первое возбужденное состояние имеет энергию свыше 40 000 см [1948, 2234].
Термодинамические функции ВВг3 (г) рассчитаны в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A.122)—A. 124), A. 128), A. 130)
без учета возбужденных электронных состояний. Расчет проводился с учетом отличия
молекулярных постоянных разных изотопических модификаций ВВг3. Погрешности вычисленных
термодинамических функций составляют 1, 5 и 7 Дж- К~1-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К
и обусловлены главным образом приближенным характером расчета; погрешности из-за неточности
молекулярных постоянных не превышают 0,8 Дж-К~1-моль~1.
Ранее термодинамические функции ВВг3 (г) вычислялись в таблицах JANAF [1543], Эвансом
[1044] и Якешем и Папоушеком [1539] до Т^ 1500 К. Расхождения данных табл. 593 ж этих
расчетов не превышают 0,2 Дж-К~1-моль~1.
Константа равновесия реакции ВВг3 (г)=В (г)+ЗВг (г) вычислена с использованием значения
Р @)=1096,769 +5,8 кДж-моль, соответствующего принятой энтальпии образования
^Н°(ВВт3, г, 298,15 К) = —205,3 + 3,0 кДж • моль.
Эта величина получена как сумма энтальпий образования и испарения ВВг3 (ж). Выбор
энтальпии образования ВВг3 (ж) основывается на измерениях энтальпии гидролиза, выполненных Бертло
[671] (—228 кДж-моль), Поландом [2239] (—234 кДж-моль) и Скиннером и Смитом [2540].
Работа [2540] привела к наиболее точному значению Д^0 (ВВг3, ж, 298,15 К) = —240,2 +
+3,0 кДж-моль. Энтальпия испарения Д„Я° (ВВг3, ж, 298,15 К) =34,9 +0,8 кДж-моль была
вычислена как средняя из расчетов по II и III законам и измерениям давления пара в работах
Барбера и др. [577] (метод изотенископа, 273—363 К, Д„Я° B98,15 К) =35,1 кДж-моль (II)
и 34,4 кДж-моль (III)) и Нисельсона, Петрусевич [287] C08—383 К, ДД° B98,15 К) =
=35,7 кДж-моль (И) и 34,5 кДж-моль (III)). Близкие значения были найдены по менее точным
измерениям Стока и Кусса [2618] (статический метод, 223—363 К, Д„# B98,15 К) =
=33,6 кДж-моль (II) и 34,4 кДж-моль (Ш)). В расчете были использованы оцененные
термодинамические функции ВВг3 (к, ж) [578]. Поскольку измерения производились при низких
температурах, точность расчетов по II и III законам термодинамики предполагалась одинаковой.
BI (г). Термодинамические свойства газообразного иодида бора в стандартном состоянии прн
температуре 100—6000 К приведены в табл. 594.
Спектр молекулы BI (г) исследовался только в двух работах [751,1833], авторы которых отнесли
наблюдавшиеся группы полос к переходам а3П -> Хх?> + и Л1П *- Х1!!*. Следует отметить,
что Лебретон и др. [1833] показали, что полосы, отождествленные авторами [751] с переходом
АгП—ZXS+, в действительности принадлежат молекуле ВС1. Так же как у молекул других гало-
генидов группы IIIA, состояния а3П и А1!! должны иметь общий диссоциационный предел с
состоянием Х1?+ и соответствовать конфигурации электронной оболочки . . ,п*'ап. Другие возбужденные
состояния, по-видимому, имеют энергии свыше 35 000 см, и не рассматриваются в справочнике.
Колебательные и вращательные постоянные BI в состояниях Ха?+ и а3П, приведенные
в табл. 20.12, приняты по работе Лебретона и др. [1833 ]. Авторы [1833 ] выполнили приближенный
76
Жор и его соединения ВЬ (г)
анализ вращательной структуры полос 1—0 и 0—0 перехода а3По+ — Х1Е+, а также выделили
канты 12 полос этого перехода (v" <I 9, v' <I 7) и <?-кант полосы 0—0 перехода а3^—Ха2+.
Точность постоянных, приведенных в таблице, невелика и погрешности в значениях шв и Ве могут
.достигать 2—5 см и 0,003 см соответственно. Следует отметить, что в работе [1833] отсутствуют
указания на то, какой изотопической модификации BI принадлежат найденные постоянные. Спектр
•был получен при работе с BIS, содержащим природную смесь изотопов бора, это позволяет отнести
лринятые постоянные к молекуле nBmI.
В работе [1833 ] в области 28 650 см в спектре поглощения иодида бора наблюдались две
диффузные полосы, отнесенные к переходу АХП <- ХХ2+. Энергия состояния АХП принята на
основании этого отнесения. Анализ данных для молекул других галогенидов бора позволяет
предполагать, что это состояние является слабосвязанным.
Термодинамические функции BI (г) были вычислены по соотношениям A. 3)—A. 6), A. 9),
<A. 10), A. 93)—A. 95). Значения ()„„ и ее производных по температуре рассчитывались по
уравнениям A. 76)—A. 78) с учетом только одного возбужденного состояния (а3П) в предположении,
что Q™lm вр = QQifl вр- Величина (?км# вр и ее производные для состояния X1!,* находились по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным и интегрированием
ло вращательным уровням энергии с помощью уравнений типа A. 82). В расчетах учитывались все
уровни энергии с значениями / ^ /шм> „, где /шах „ определялось из условий A. 81). Колебательно-
вращательные уровни состояния ХХЕ+ вычислялись по уравнениям A. 65), A.62). В табл. 20.13
приведены значения коэффициентов Ykl в этих уравнениях, а также величины vnMX и /1Ш.
Коэффициенты Y30 и У21 получены с учетом условий A. 50) и A. 57), в последнем случае при В1Ы=0.
Коэффициенты Ykl, приведенные в табл. 20.13, получены по соотношениям A. 66) для
изотопической модификации молекул BI, соответствующей природной смеси изотопов бора, на основании
предположения, что принятые в табл. 20.12 постоянные принадлежат молекуле nBmI.
Основные погрешности рассчитанных термодинамических функций BI (г) обусловлены низкой
точностью известных молекулярных постоянных и фактическим отсутствием данных о
колебательно-вращательных уровнях состояния XXS+, энергии диссоциации BI и возбужденных
электронных состояниях этой молекулы. Они могут достигать 0,3, 1 и 2 Дж-К~1-моль~1 в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Ранее термодинамические функции BI (г) вычислялись в таблицах JANAF [1543] по оцененным
значениям постоянных и без учета возбужденных электронных состояний. Расхождения между
данными [1543] и табл. 594 достигают 2, 6 и 10 Дж-К~1-моль~1 при 6000 К в значениях Ф° (Г),
JS° (Т) и С\ (Т) соответственно.
Константа равновесия реакции BI (г)=В (г)+1 (г) рассчитана с использованием принятого
в справочнике значения энергии диссоциации
ДД° @) = Do (BI) = 29 500 + 1000 см = 353 + 12 кДж • моль.
Эта величина принята на основании сопоставления энергий диссоциации других галогенидов
я тригалогенидов бора, а также данных Лебретона и др. [1833 ] об энергии состояния АгИ молекулы
BI (см. выше). Поскольку состояния Х1Е+иЛ1П имеют общий диссоциационный предел, энергия
•состояния АЩ является нижним пределом для энергии диссоциации BI.
Гринбер и др. [94, 95] па основании измерений убыли веса твердой фазы определили константу
равновесия реакции 2ВР (к)+В13 (г)=ЗВ1 (г)+Р2 (г) в интервале 1348—1573 К. Приведенному
в этой работе уравнению соответствуют значения Do (В1)=640 кДж-моль (II закон) и 511 кДжХ
Хмоль (III закон). По-видимому, эти измерения или их интерпретация содержат ошибки, так как
полученные величины превышают Do (ВВг).
Принятой энергии диссоциации соответствует
^^"(BI, г, 0) = 314,163 + 13 кДж.моль.
В12 (г). Термодинамические свойства газообразного дииодида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 595.
Экспериментальные данные о структуре и спектре В12 в литературе отсутствуют, поэтому
молекулярные постоянные, приведенные в табл. 20.14, были оценены на основании аналогии с
другими молекулами МХ2, где М=В, Al, a X=H, F, C1. Приведенное в табл. 20.14 произведение
главных моментов инерции вычислено в предположении, что в электронном состоянии ХгАх молекула
77
В13 (г) Глава 20
В12 имеет симметричную угловую структуру (симметрия С2„) ,|^jl—В—I = 120 +10° и г (В—I)=2,12 +
+0,05 А(как в В13). Погрешность в IaIbIc составляет 1-1013 г3-смв. Основные частоты,
приведенные в табл. 20.14, вычислены по силовым постоянным, принятым равными соответствующим
постоянным В13 х. Погрешности в частотах не превышают 10—15%. По аналогии с другими
молекулами ВХ2 (Х=Н, F, C1) принимается, что молекула В12 имеет возбужденное электронное
состояние А2Вг с энергией 10 000+5000 см.
Термодинамические функции В12 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом возбужденного электронного состояния АгВх по уравнениям A. 168)—A. 170).
Погрешности рассчитанных таким образом термодинамических функций В12 (г) составляют 2, 5 и 8 ДжХ
XК-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К, они обусловлены как неточностью,
оцененных молекулярных постоянных (соответственно 1,5—4 Дж-К-моль), так и
приближенным характером расчета.
Ранее термодинамические функции В12 (г) вычислялись в таблицах JANAF [1543].
Расхождения данных табл. 595 и [1543] не превосходят 3 Дж-К-моль в значениях Ф° (Т) и связаны
с отличием в принятых молекулярных постоянных В12, а также с тем, что авторы [1543] не
учитывали в расчете состояние А2Вг.
Константа равновесия реакции В12 (г)=В (г)+21 (г) вычислена с использованием принятого
в справочнике значения
Дг#° @) = 545 ± 25 кДж • моль.
Эта величина была оценена сопоставлением энергий связей в молекулах ВХ, ВХ2 и ВХ3, где
X=F, C1, I.
Принятой величине соответствует
/\fH° (BI2, г, 0) — 229,326 + 25 кДж • моль.
В13 (г). Термодинамические свойства газообразного трииодида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 596.
Молекулярные постоянные, использованные для расчета термодинамических функций В13 (г),
приведены в табл. 20.15. Электронографические измерения [1614] показывают, что в основном
электронном состоянии ^А^ молекула В13, так же как другие молекулы тригалогенидов бора,
имеет плоскую симметричную структуру (симметрия DSh). Приведенное в табл. 20.15 значение
IaIbIc вычислено по данным Какубари и др. [1614] г (В—1)=2,118 +0,005 А. Погрешность в МвРо
составляет 1 • 10~112 г3- см6. ИК-спектр В13 в различных агрегатных состояниях исследовали Вентинк
и Тьенсуу [2833], Левин и Абрамовиц [1852], Финч и др. [1088] и Волфе и Хамфри [2868].
Приведенные в табл. 20.15 значения частот v2, vg и \ найдены Волфе и Хамфри |2868], частоты v2 и vs
непосредственно наблюдались в ИК-спектре газообразного В13, а частота v4 оценена из
наблюдаемых обертонов. Частота \ принята по рекомендации Вентинка и Тьенсуу [2833], которые оценили
значение \ из комбинационных полос, наблюдаемых в ИК-Спектре газообразного В13. Погрешности
принятых значений частот оцениваются в 5 см для v2, v3 и v4 и 10 см — для vx. В пределах
указанных погрешностей с принятыми значениями удовлетворительно согласуются частоты,
полученные в работах [1088, 1852]. Исследование электронного спектра газообразного В13, выполненное
Мария и др. [1948], показало, что первое возбужденное состояние имеет энергию —29 000 см.
Термодинамические функции В13 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом только основного состояния Х1А'1. Расчет проводился с учетом отличия частот колебаний
разных изотопных модификаций В13. Погрешности рассчитанных термодинамических функций
обусловлены главным образом приближенным характером расчета и в меньшей степени —
неточностью молекулярных постоянных. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
составляют 2, 5 и 8 Дж-К-моль~1, в том числе до 1 Дж-К-моль из-за неточности
молекулярных постоянных.
Ранее термодинамические функции В13 (г) вычислялись в таблицах JANAF [1543], Эвансом
[1044] и в работах [1085, 1539, 2093] до Т < 1000 К. Данные [1543] и [1044] согласуются
/,= 1,95; /гг=0,38 и /«/»*=0,17 (в 106 дин-аГ*).
78
Бор и его соединения BS (г)
с табл. 596 в пределах 2 и 0,6 Дж-К-моль~1, расхождения обусловлены использованием в этих
расчётах менее точных молекулярных постоянных В13.
Константа равновесия реакции В13 (г)=В (г)+3 I (г) вычислена с использованием значения
АДГ0 @) =856,006 +6,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH° (В1„, г, 298,15 К) = 21,4 + 4,0 кДж • моль.
Принятое значение основано на измерениях энтальпии образования и сублимации В13 (к).
Финч и др. [1086 ] измерили энтальпию гидролиза В13 (к), которая приводит к значению kfH° (В13,
к, 298,15 К) = — 42,6+3,5 кДж-моль. Энтальпия испарения Дс#° (В13, ж, 401 К) =45,8 +
+0,5 кДж-моль была вычислена по уравнению It закона на основании измерения давления пара
методом изотенископа C36—480 К, 9 точек) в работе Андерсона и Белза [513]. Ей соответствует
ДД°(В13, к, 298,15 К)=64,0+1,7 кДж-моль, если принять Дт#° (В13, к, 322,9 К) = 12,9 +
+0,8 кДж-моль и оцененные теплоемкости по данным [2169].
BS (г). Термодинамические свойства газообразного сульфида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 597.
Молекулярные постоянные 11B32S, принятые на основании спектральных исследований [180,
641, 1985, 2531, 2900, 2901], приведены в табл. 20.4. В спектрах молекулы BS наблюдались 7
систем полос, связывающие 8 нижних электронных состояний: A2U(—Х2?+ [2531, 2900, 2901],
С2ПГ-Х2? + [2900, 2901], ?2? + -* y!2IIJ1985, 2901], 2Д-> ^12П; [180, 1985], 2Е+ -> 2П, [1985],
?2?+ -> Х2?+ [641] и Е21> + -> А2И4 [641]. Помимо этих восьми состояний молекула BS по
аналогии с изоэлектронной молекулой CN должна обладать рядом квартетных состояний с энергией
30 000—40 000 см. В табл. 20.4 приведены данные для четырех нижних электронных состояний
с энергией до 40 000 см, остальные состояния в справочнике не рассматриваются.
Колебательные постоянные ше и wexe BS в состоянии Х2? + приняты по данным Зеемана [2900,
2901 ], полученным по кантам полос cv" ^ 4 систем А2П{ ->• Х2?+ и С2ПГ—Х2?+. Постоянная <оеув
добавлена согласно Мак-Доналду и Иннесу [1985] для правильного описания уровней с v" =
=19—22, переходы на которые были идентифицированы в системе -42П4—Х2?+. Вращательные
постоянные состояния Х2? + получены Зееманом [2901] из анализа полос 3—0, 4—0, 5—0 и 2—1
системы ^42П,-—Х2?+. Колебательные постоянные состояния АЧТ{ получены Зееманом [29011
по кантам полос системы А2П{—Х2? + с и' ^ 5 и хорошо описывают уровни с г/ ^ 11, переходы
с которых были зарегистрированы в работе [2531 ]. Вращательные постоянные и постоянные
мультиплетного расщепления состояния ^42П^ приняты по результатам анализа четырех полос
3_0, 4—0, 5—0 и 2—1 системы -42П,—Х2?+ [2901 ] и полос 0—0 систем Б2Е—А2П{ и
2Д_42П,. [1985]. Колебательные постоянные состояния В2 ?+ найдены Коряжкиным и
Мальцевым [180*] по кантам полос ci;'^4 системы _В2?—A2U(, а вращательные постоянные определены
из анализа 0—0 полосы той же системы в работе [1985]. Молекулярные постоянные в состоянии
С2ПГ приняты по работе Зеемана [2901].
Термодинамические функции BS (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBS и ее производные вычислялись по уравнениям A. 90)—A. 92) с
учетом трех возбужденных электронных состояний и при условии, что для состояний 2?2Е+ и С2НГ
QW в = (pjpx) (?S вр- Колебательно-вращательные статистические суммы состояний Х2Е+ и
АЧ1{ и производные находились непосредственным суммированием по колебательным уровням и
интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались все
уровни состояний Х2?+ и А2П( со значениями / <J /шах, „, последние определялись по
соотношениям A. 81). Энергия колебательно-вращательных уровней рассчитывалась по уравнениям A. 65)
и A. 62). Коэффициенты Ykl в этих уравнениях, приведенные в табл. 20.5, получены для
изотопической модификации BS, соответствующей природной смеси изотопов бора и серы, по
соотношениям A. 66) и молекулярным постоянным из табл. 20.4. Для выполнения условий A. 51) введены
постоянные У40 и У50 для состояний Х2?+ и постоянные У30 и У40 для состояния ^42П;. В табл. 20.5
представлены также значения итЛ1 и /lim. Мультиплетность и вырождение состояний Х2?+, А2И4
учитывались статистическими весами 2 и 4 соответственно.
Погрешности рассчитанных термодинамических функций при температурах до 3000 К
определяются главным образом неточностью фундаментальных постоянных и не превышают 0,02—
0,04 Дж-К-Моль в значениях Ф° (Т) и S° (T). При более высоких температурах становятся
79
Глава 20
существенными погрешности, обусловленные пренебрежением более высокими электронными
состояниями, в том числе квартетными состояниями, приближенным способом учета состояний
?2?+ и С2ПГ, а также неточностью экстраполяции колебательно-вращательных уровней основного
состояния. Погрешность в значении Ф° F000 К) может достигать 0,3 Дж-К~1-моль.
Ранее термодинамические функции BS (г) вычислялись в справочниках JANAF [1543], Мак-
Брайда и др. [1974], Эвансом [1044] и в справочнике [977]. Все расчеты проводились по близким
значениям молекулярных постоянных, и их результаты практически совпадают с приведенным
в табл. 597 для Т <^ 3000 К. При 6000 К расхождения данных [1543] и табл. 597 достигают 1 и
4 Дж-К^-моль в значениях Ф° (Т) и S° (T).
Константа равновесия реакции BS (r)=B (r)+S (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг//° @) = D0(BS) = 575 ± 30 кДж • моль = 48 000 + 2500 см'1.
Эта величина основана на приведенных в табл. 20.28 результатах масс-спектрометрических
исследований равновесия реакций в газовой фазе. Приведенные в таблице погрешности включают
Таблица 20.28. Результаты определений Do (BS) (в кДж-моль)
Авторы
Гингерич, 1970
[1204]
Метод
Масс-сдектромет-
ричеокий, CS+B—
=BS+C, 2230—
2330 К, 5 точек
То же,
GeS+B=BS+Ce,
2057-2344 К, 8
точек
D0(BS)
II закон
III закон
— 542+8
' — 604+14
Авторы
Уй, Дроварт,
1970 [2745]
Метод
Масс-спектромет-
рический YS+B=
=» BS+Y, 2236-
2482 К, 21 точка
То же,
YS+BO=BS+YO,
2236—2482 К,
21 точка
D0(BS)
II закон
III закон
565+21 582+16
567+23 568±21
воспроизводимость результатов измерений, неточность сечений ионизации, термодинамических
функций и энергий диссоциации компонентов исследованных реакций. В расчетах были
использованы значения Do (CeS)=565+13 кДж-моль, Do (YS)=527+13 кДж-моль, Do (YO)=718±
+11 кДж-моль, рекомендованные в справочнике [406], и значение Do (CS)=710+1 кДж-моль
^см. гл. 15). Принятая величина основана на результатах измерений авторов [2745], которые
получили хорошее согласие расчетов по II и III законам для двух независимых реакций. Данные [1204],
опубликованные в виде краткого сообщения, приводят к плохо согласующимся между собой
величинам. При оценке погрешности принятого значения учтено расхождение результатов измерений
в работах [2745] и [1204].
Принятой энергии диссоциации соответствует энтальпия образования
&fH° (BS, г, 0) = 259,785 + 30 кДж • моль.
BS3 (г). Термодинамические свойства газообразного дисульфида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 598.
Молекулярные постоянные, использованные для расчета термодинамических функций BS2 (г),
приведены в табл. 20.8. Исследования электронных и ИК-спектров поглощения [179, 762]
показали, что в основном Х2Ид и возбужденном А*Пи состояниях молекула BS2 имеет линейную
структуру (симметрия Da,k). Приведенный в табл. 20.8 момент инерции вычислен для значения
г (В—S)=1,70+0,05 А, оцененного на основании сравнения межатомных расстояний в молекулах
окислов и сульфидов бора. Погрешность величины / составляет 2-Ю9 г-см2. Основные частоты,
приведенные в табл. 20.8, приняты по результатам исследования электронного и ИК-спектра
поглощения 11BS2 в матрице из Ne в работе [762]. Погрешности частот, наблюдавшихся в спектре
<Vj и v3 в состоянии ХгШд и \, v2, vs в состоянии ^42ПЫ), с учетом матричного сдвига оцениваются
в 10 см; погрешность значения v2, оцененного из соотношения
80
— в 20 см
-1
Бор ¦ его соединения B*S (г)
Постоянная спин-орбитального взаимодействия в состоянии Х2Ид и энергии возбужденных
состояний также приняты по данным [762].
Термодинамические функции BS2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический осциллятор».
Подсостояние -Х^Пу, рассматривалось как возбужденное состояние BS2, составляющие этого под-
состояния и состояний АгЛи и Б??и вычислялись по уравнениям A. 168)—A 170) без учета
различия постоянных в этих и основном Х2Пз/2 состояниях. Погрешности рассчитанных значений
обусловлены неточностью принятых молекулярных постоянных, пренебрежением различием
постоянных изотопических модификаций молекулы BS2, а также приближенным характером
расчета и оцениваются в 3, 6 и 8 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К (из
которых 2—3 Дж-К-моль из-за неточности молекулярных постоянных).
Таблица термодинамических функций BSa (г) публикуется впервые.
Константа равновесия реакции BS2 (r)=B (r)+2S (г) вычислялась с использованием принятой
в справочнике энергии атомизации
АГН° @) = 1060 ± 20 кДж ¦ моль.
Принятое значение основано на масс-спектрометрическом исследовании равновесия реакций
2CeS+B=BS2+2Ce и 2BS=BS2+B, выполненном Гингиричем [1204] B279—2319 К, по 3 точки).
Из этих измерений энтальпия атомизации BS2 была найдена равной 1079+25 кДж-моль и 1060 +
+22 кДж-моль соответственно. В работах Соммера и др. [2580, 2581] энтальпия образования
BSj (г) была определена в масс-спектрометрических исследованиях (973—1173 К). В этих работах
практически отсутствует описание эксперимента, а для энтальпии образования BS2 (г) приводятся
значения 103 и 54+42 кДж-моль. Этим величинам соответствуют значения энтальпии
атомизации, равные 1006 и 1055 кДж-моль.
Измерения давления насыщенного и перегретого пара над BS2 [92] и B2S3 [91] не могут быть
использованы для вычисления энтальпии образования газообразных BS2 и B2S3, поскольку в
условиях экспериментов состав пара должен был быть существенно более сложным (см. [1014, 1256]),
чем'это предполагали авторы [91, 92].
Принятой энтальпии атомизации соответствует энтальпия образования
AfH° (BS2, г, 0) = 49,570 + 21 кДж • моль.
B3S (г). Термодинамические свойства газообразного сульфида дибора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 599.
Молекулярные постоянные, использованные для расчета термодинамических функций B2S,
приведены в табл. 20.8. Экспериментальные данные о строении и спектрах B2S в литературе
отсутствуют. В справочнике по аналогии с В2О принято, что в основном электронном состоянии
Х1^} молекула B2S имеет линейную структуру симметрии Dcoa- Приведенный в табл. 20.8 момент
инерции вычислен для значения г (В—S)=1,72 +0,05 А, оцененного из сравнения расстояний В—О
и В—S в различных окислах и сульфидах бора. Погрешность в / составляет около 1 • 10~зэ г-см2.
Основные частоты B2S оценены из сравнения результатов расчета этих величин в приближении
поля валентных сил (силовые постоянные B2S были оценены по силовым постоянным молекул ВО,
BS и В2О) с соответствующими частотами,,принятыми в справочнике для других соединений бора.
Погрешности в значениях \ и v3 оцениваются в 100 см, v2 — в 20 см. Так же как В2О, молекула
BSS может обладать возбужденными состояниями с низкими энергиями, однако сведения о них
отсутствуют, и они в справочнике не рассматриваются.
Термодинамические функции B2S (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью оцененных молекулярных постоянных, неопределенностью структуры
B^S, отсутствием сведений о'возбужденных электронных состояниях и приближенным характером
расчета и составляют 5, 8 и2 Дж-К^-моль в Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций B2S (г) публикуется впервые.
Константа равновесия реакции B2S (r)=2B (r)+S (г) вычислена с использованием принятого
в справочнике значения
ДГЯ° @) = 800 + 100 кДж • моль.
81
B2S2 (r), B2S3(k, ж) Глава 20
Эта величина была оценена сопоставлением энергий связей молекул BS, B2S3, ВО, В2О3, В2О.
Принятому значению соответствует
AfH°(B2S, г, 0) = 594,785+100 кДж • моль.
BjS2 (г). Термодинамические свойства газообразного дисульфида дибора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 600.
Молекулярные постоянные, использованные для расчета термодинамических функций B2S2,
приведены в табл. 20.8. В литературе отсутствуют экспериментальные данные о молекулярных
постоянных B2S2, за исключением частоты антисимметричного валентного колебания v3,
наблюдавшейся в ИК-спектрах [762, 2581 ]. В справочнике по аналогии с ВаО2 принято, что в основном
электронном состоянии ^SJ молекула B2S2 имеет линейную структуру симметрии D mi. Приведенный
в табл. 20.8 момент инерции вычислен по оцененным структурным параметрам: г (В—В)=1,70 +
+0,05 А и г (B=S) = l,62+0,05 А (г (В—В) принято таким же, как в В2О2, а г (B=S) оценено из
сравнения расстояний В=О и B=S в различных окислах и сульфидах бора). Погрешность момента
инерции составляет 4-109 г-см2. Основная частота v3 принята по результатам исследования
ИК-спектра 11B2S2 в газовой фазе [2581]; ее погрешность оценивается в 30 см. Остальные
частоты B2S2, приведенные в табл. 20.8, являются средними между результатами двух оценок: из
сопоставления основных частот молекул ВО, BS и В2О2 и в предположении, что отношение всех
частот молекул B2S2 и В2О2 такое же, как отношение v3 (B2S2)/v3 (B2O2). Погрешности частот \,
v2, v4 и. vb оцениваются в 10—15% от их величин. Предполагается, что возбужденные электронные
состояния B2S2 лежат в области высоких значений энергии и не рассматриваются в справочнике.
Термодинамические функции B2S2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и составляют около 4, 8 и 12 Дж-К~1-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К (из которых из-за неточности молекулярных постоянных
2—5 Дж-К^-моль-1).
Таблица термодинамических функций B2S2 (г) публикуется впервые.
Константа равновесия реакции B2S2 (r)=2B (r)-j-2S (г) вычислена с использованием значения
Д^й @)=1534,57+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(B2S2, г, 0) = 135 + 50 кДж • моль.
Принятая величина основана на работе Соммера [2580]. В работе не сообщаются детали
эксперимента, однако в более ранней работе Соммера и др. [2581 ] сообщается о масс-спектрометрическом
исследовании системы ZnS (к)+В (к), в парах над которой был обнаружен ион B2S2. Следует
отметить, что в [2581] приводится значительно более низкое значение AfH° (B2S2, г, 0), равное
75 кДж-моль. В справочнике отдано предпочтение более позднему сообщению [2580].
B2S3 (к, ж). Термодинамические свойства кристаллического и жидкого трисульфида дибора
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. Д. 45.
Значения постоянных, использованных для расчета термодинамических функций,
представлены в табл. 20.1. В справочнике за стандартное состояние B2SS (к, ж) в интервале от Г=0 до-
температуры плавления принята моноклинная модификация со сложной слоистой структурой [842^
952а]. В литературе отсутствуют какие-либо данные о термодинамических свойствах B2S3 (к, ж)г
за исключением температуры и энтальпии плавления. Значения термодинамических функций при
298,15 К, приведенные в табл. 20.1, оценены с использованием различных эмпирических методов;
погрешность принятых значений S° B98,15 К) и Н° B98,15 К)—Н° @) оценивается в 8 Дж-К~1Х
Хмель" и 2 кДж-моль соответственно. Для теплоемкости B2S3 (к) в интервале 298,15—840 К
принято уравнение, выведенное методом Ландия [214], принимая температуру плавления BgSg,
равной 840+10 К по данным Чен и Гиллес [842, 843]. Результаты более ранних исследований [254Г
2047] приводили к резко заниженным значениям этой величины E83 и 690 К). Энтальпия
плавления D8,5+7 кДж-моль) была определена в работе [843] как разность энтальпии сублимации
и испарения B2S3, вычисленных по данным о давлении пара B2S3 (к) F60—840 К) и B2S3 (ж) (842—
938 К). Теплоемкость B2S3 (ж) A46 +20 Дж-К~1-моль~1) оценена по теплоемкости B2OS (ж) и А12О3 (ж).
82
Бор и его соединения B2S3 (г)
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 8, 15 и
30 Дж-К-моль~1 соответственно. Расхождения термодинамических функций B2S3 (к, ж),
приведенных в табл. Д. 45 и в справочниках [578, 2031] B98—1100 К), достигают 8 Дж'К~1-моль~1
в значениях Ф° (Т) и обусловлены различием оценок теплоемкости и величины S0 B98,15 К).
В справочнике принята энтальпия образования трисульфида дибора
A^c|(B2S?t к, 298,15 К) = —243 + 3 кДж^моль.
Эта величина основана на измеренной Ария и др. [20, 264, 265 ] энтальпии реакции 2В (аморф.)+
+3S (k)=B2S3 (к), Дг#° B98,15 К)=—252,1 +2,3 кДж- моль. В выполненных[еще в прошлом веке
измерениях Сабатье [2418, 2419] были найдены энтальпия гидролиза B2S3 и энтальпия его реакции
с раствором иода, которые приводят для энтальпии образования B2S3 (к) к значениям —309 и
—315 кДж-моль.
В справочнике принята энтальпия сублимации трисульфида дибора
A,H°(B2S3, к, 0) = 258 ± 15 кДж.моль.
Это значение основано на масс-спектрометрических измерениях давления пара B2S3 (к),
выполненных Чен и Гиллесом [843] F60—938 К, 82 точки, Д8#° @)=236+35 кДж-моль (II) и
258+14 (III)). В более ранней работе Грина и Гиллеса [1256] масс-спектрометрическим
методом F33—722 К) были измерены интенсивности ионных токов, которым при расчете по
уравнению II закона соответствует значение 160 +20 кДж- моль. В кратком сообщении Соммера [2580]
(см. также [2581 ]) сообщается о масс-спектрометрическом определении величины А^Н° (B2S3,
Г) 0)=42 кДж-моль, которой соответствует As#° (B2S3, к, 0)=286 кДж-моль.
B2S3 (г). Термодинамические свойства газообразного трисульфида дибора в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 601.
Молекулярные постоянные, использованные для расчета термодинамических функций B2S3,
приведены в табл. 20.11. Результаты электронографического исследования Акишиным и
Спиридоновым [7] структуры молекул B2S3 (г (В—S) = 1,81 +0,02 А, г (B=S)=l,65+0,03 A, J$B—S—В =
=96+5°), так же как и первоначального исследования этими же авторами структуры В2О3,
по-видимому, являются неточными. В справочнике по аналогии с В2О3 принимается, что в основном
электронном состоянии хАг молекула B2S3 имеет угловую У-образную структуру симметрии Сйв.
Приведенное в табл. 20.11 произведение моментов инерции вычислено на основе следующих
структурных параметров: г (B=S)=l,62+0,05 А, г (В—S)=l,72+0,05 А и ^В—S—В=130+10°.
Длина связи В—S оценена из сравнения величин г (В—О) и г (В—S), принятых в справочнике для
различных кислородных и серных соединений бора; -^В—S—В оценен из сравнения с В2О3.
Погрешность в IaIbIc составляет около 5-Ю13 г3-см6.
Из анализа результатов исследования ИК-спектров B2S3 [762, 1257, 2581] можно заключить,
что надежно определена только частота v6 антисимметричного валентного колебания связи B=S.
В справочнике частота v6 принята из анализа ИК-спектра паров nB2S3 [2581 ]. Остальные частоты
B2S3, приведенные в табл. 20.11, оценены из сравнения с частотами В2О, ВО2, В2О3 и B2S, BS2.
Погрешность в значении v6 оценивается в 50 см, остальных частот — в 15—20% от их величин.
Возбужденные электронные состояния B2SS в справочнике не рассматриваются.
Термодинамические функции B2S3 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и составляют около 10, 15 и 20 Дж-К~1-моль~1
в значениях Ф° (Т) при 298,15, 3000 и 6000 К (из которых из-за неточности молекулярных
постоянных 8—12 Дж-К-моль во всем интервале температур).
Термодинамические функции B2S3 (г) вычислялись ранее в работе Нагараяна [2094] (до 1000 К),
значительные расхождения данных [2094] и табл. 601, достигающие 23 Дж-К-моль~1 в
значениях Ф° (Г), обусловлены различием постоянных, использованных в расчетах, а также ошибками
в расчете [2094].
83
BN (к, ,ж) • Глава 2»
Константа равновесия реакции B2S8 (r)=2B (r)+3S (г) вычислена с использованием значения
А^0 @)=1930,871 +18 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации B2S3 (к). Этим величинам также соответствует
A^^B^s, г, 0) = 13,484 + 15 кДж • моль.
BN (к, ж). Термодинамические свойства кристаллического и жидкого нитрида бора в
стандартном состоянии при температурах 100—3500 К приведены в табл. 602.
Значения постоянных, принятых для расчета термодинамических функций, представлены
в табл. 20.1. В справочнике за стандартное состояние BN (к) в интервале 0—3240 К принята
гексагональная (графитоподобная) модификация *. При Т <^ 298,15 К термодинамические функции
вычислены по результатам измерений теплоемкости, выполненных Узструмом и Биллом [см. 28381
E-300 К). Погрешность значений S° B98,15 К) и Я° B98,15 К)—Я° @), принятых по [2838]
(см. табл. 20.1), оценивается в 0,17 Дж-К~1-моль и 0,025 кДж-моль соответственно. Измерения
теплоемкосш в работе [1005] A9—301 К) ненадежны ввиду наличия в исследованном образце
значительных примесей магнетита и не учитывались в расчете. Уравнения для теплоемкости BN (к)
в интервалах 298—1200 К и 1200—3240 К (см. табл. 20.1) выведены на основании согласующихся
между собой данных по энтальпии, полученных в работах Мак-Доналда и Сталла [1990] B98—
1683 К) и Магнуса и Данца [1908] B98—1174 К), и измерений теплоемкости в интервале 1300—
2200 К, выполненных Профетом и Сталлом [2273]. Точность принятых значений теплоемкости
оценивается в 1% при 1000 К, 3% при 2000 К и 10% при 3000 К. Температура плавления C240 +
+200 К) определена Уенторфом [2834] при изучении ее зависимости от давления C0—80 кбар).
Близкие значения Тт (~3273 К) были получены ранее в работах [812, 1145]. Энтальпия плавления
BN (81 +25 кДж-моль) оценена по энтропии плавления BN, принятой равной 25 Дж-К-моль~1.
Теплоемкость BN (ж) оценена по соотношению Ср=33,5/г Дж-К^-моль-^см. т. 1,с. 79), ее
погрешность может достигать 15 Дж-К~1'Моль~1.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3500 К оцениваются
в 0,1, 0,4, 1 и 4 Дж-К~1-моль соответственно. Расхождения между термодинамическими
функциями BN (к), приведенными в табл. 602 и в справочниках [105, 1543], не превышают 1 Дж-КХ
Хмель в значениях Ф° (Т). Для BN (ж) соответствующие расхождения растут с повышением
температуры и достигают 4Дж-К~1-моль~1 при 3500 К.
Константа равновесия реакции BN(k)=B (r)+N (г) вычислена с использованием значения
ДГЯ° @)=1278,39+5,2 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH0(BN, к, 298,15 К) = — 250,5 + 1,5 кДж • моль.
Принятая величина основана на данных, приведенных в табл. 20.29. Их погрешности
учитывают воспроизводимость измерений и неточность использованных в расчетах величин.
Результаты измерений энтальпии образования нитрида бора из кристаллического бора
находятся в хорошем соответствии. Наиболее точные измерения с хорошо охарактеризованным
образцом были проведены Вайсом и др. [2863], и этой работе придается наибольший вес при вычислении
принятой средневзвешенной величины. Энтальпия образования нитрида бора из аморфного бора
была измерена Гальченко и др. [82, 83]. Для дальнейших расчетов принимается полученное в этой
работе значение —255, 6+2,0 кДж-моль, которое находится в удовлетворительном соответствии
е результатами исследования равновесия диссоциации BN (к). Из данных по исследованию
равновесия диссоциации следует, что коэффициент испарения азота из нитрида бора составляет
6-Ю [984, 1414]. В соответствующих расчетах были использованы термодинамические функции
кристаллического бора, что вносит некоторую дополнительную погрешность в результаты, так как
в действительности в равновесии, видимо, принимает участие бор в аморфной или
микрокристаллической форме. Однако вносимая этим погрешность невелика, так как термодинамические
функции кристаллического и аморфного бора достаточно близки (хотя остается неясным вопрос о
возможной остаточной энтропии аморфного бора, см. В (аморф.)) и при нагревании происходит
дальнейшая кристаллизация аморфного бора. Помимо приведенных в табл. 20.29 данных исследования
диссоциации BN были проведены также в работах [44, 1448, 1887, 2436].
При высоких давлениях устойчивы две модификации BN — гексагональная (структурный тип вюртцита) и
кубическая (структурный тип сфалерита).
84
Бор и его соединения
BN(r)
Таблица 20.29. Результаты
Авторы
определений AfH° (BN,k, 298,15 К) (в кДж-
Метод
моль)
A/H°{BN, к, 298,15 К)
II закон
III закон
Шиссель, Вильяме, 1959 [2457] Исследование равновесия реакции 2BN (к)= — —251 а
=2В (aMop(J>.)+N2 (г) масс-спектрометрическим
методом
Дрегер и др., 1962 [984} То же методом испарения с поверхности A421— — —251+8 а
2031 К)
Хилденбранд, Холл, 1963 [1414] То же торзионным методом A630—ШОК), —254+10 а — 251,4+2,0 а
29 точек6
By, 1971 [2870] То же масс-спектрометрическим методом A500 247,7±2,0 а —
1800 К)
Дворкин и др., 1954 [1005] Измерение энтальпии сгорания BN в кисло- —250,1+3,0
роде
Гальченко и др., 1960 [82, 831 Измерение энтальпии реакции В (аморф.)+ —255,6+2,0 а
+V2N2(r)= BN(k)
Томпсон, Зинке, 1961 (см. [1543]) Измерение энтальпии реакции BN (K)+NF3(r)= —251,5+8,0
=BF3 (r)+N2 (г)
Вайс и др., 1966 [2863] Измерение энтальпии сгорания BN во фторе —256,3+1,5
Гросс и др., 1967 [1285] То же —253,2±1,5
а Энтальпия образования BN (к) из аморфного бора.
" Измерения производились прл разных эффузионных отверстиях. Равновесные давления получены
экстраполяцией.
BN (г). Термодинамические свойства газообразного нитрида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 603.
В табл. 20.4 приведены молекулярные постоянные nB14N, полученные в результате
спектральных исследований [975, 2066, 2696], в которых наблюдались две системы полос С3П ** Х3Л4 и
41 ->1?, и квантовомеханических расчетов [1957, 2003, 2042, 2104, 2767]. На основании анализа
электронного спектра поглощения BN в матрице Ne и Аг [2066], а также теоретических
расчетов [1957, 2003, 2767] в справочнике принимается, что молекула BN имеет основное состояние
XaUi (конфигурация . . .4o2lu35a). Помимо возбужденного состояния С3П (конфигурация
. . .4о1тт:35о2) молекула BN имеет ряд низколежащих состояний, соответствующих той же
конфигурации (ЧТ), а также конфигурациям . . .4о5а1л4 C I+, i?+), . . .4o2lrc4 (i?+) и . . А°Чп25<г*
(8 2", *Д,1 ?+). Энергии возбуждения этих состояний приняты на основании квантовомеханического
расчета [2003] (ASH+, аЧ1, В3 Е" и Ь1!") и сравнительных оценок с изоэлектроиными молекулами
С2 и BeO (c*A, &Т,+, еЧ,+ и /2П) *. Погрешности значений Т. могут достигать 2000—3000 см.
Колебательные постоянные состояний Х3П( и С3П, приведенные в табл. 20.4, определены Дугласом
и Герцбергом [975] по кантам 10 полос системы С3П -*¦ X3U.{ (z/, v" ^ 3), вращательные
постоянные найдены авторами[975] из анализа структуры Р-ветвей полос @—0), A—0) и @—1) с
погрешностью 0,05 см. Постоянная спин-орбитального взаимодействия А основного состояния Х3П^
экспериментально не получена и принята приблизительно равной соответствующей постоянной
в молекуле A1N, поскольку в молекулах ВеО и MgO они практически совпадают. Для остальных
состояний приведенные в табл. 20.4 частоты колебаний и вращательные постоянные оценены на
основании значений г„, полученных из квантовомеханических расчетов [2003, 2767], и приняты
одинаковыми для изоконфигурационных состояний.
Термодинамические функции BN (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qss и ее производные вычислялись по уравнениям A. 90)—A. 92) с уче-
1 Результаты нового квантовомеханического расчета низколежащих электронных состояний молекулы BNJ6e3
учета корреляции, проведенного Хаитом и Барановским [419а], удовлетворительно согласуются с данными
работы [2003]. Значения Ф° G"), вычисленные по данным работы [419а], согласуются в пределах 0,6 Дж«К"*-
•моль'1 с приведенными в табл. 603.
85
BH3NH3 (г) Глава 20
том девяти возбужденных электронных состояний в предположении, что Q^H Бр = (р{1рЛ Qi*l вр.
Колебательно-вращательная статистическая сумма состояния XaHf и ее производные находились
непосредственным суммированием по колебательным уровням и интегрированием по значениям /
с помощью уравнений типа A. 82). В расчете учитывались все уровни состояния Х3Н( со
значениями / ^ /Шах, »> последние находились из условий A. 81). Энергия колебательно-вращательных
уровней рассчитывалась по уравнениям A. 65), A. 43), A. 62). Постоянная спин-орбитального
расщепления А, коэффициенты Ykl, а также значение ршах и /Ит приведены в табл. 20.5. Постоянные
Yk! были получены для изотопической модификации, соответствующей природной смеси изотопов
В и N, по формулам A. 66) и постоянным молекулы UB14N из табл. 20.4.
Погрешности рассчитанных термодинамических функций обусловлены отсутствием
экспериментальных данных об энергиях возбуждения и молекулярных постоянных низколежащих
возбужденных состояний, неточностью колебательных и вращательных постоянных основного
состояния, а также отсутствием экспериментальных данных об энергии диссоциации молекулы
BN. Суммарная погрешность в значениях Ф° (Т) может достигать 0,2, 2 и 4 Дж-К~1-моль при
298,15, 3000 и 6000 К соответственно.
Таблицы термодинамических функций BN (г) ранее вычислялись в предыдущем издании
настоящего справочника [105], таблицах JANAF [1543], справочниках Мак-Брайд и др. [1974], Дугласа
и Бекетта [977], Мадера [1904], Эвансом [1044] и в работе Уилкинса и др. [2855]. Все расчеты
проводились по молекулярным постоянным, близким к принятым в табл. 20.4. Возбужденные
электронные состояния либо не учитывались вообще [977], [1904], [2855], либо учитывались
только частично (С3П в [105], [1974] и С3П, х ? и 1П в [1543]). Этим обусловлены расхождения
результатов перечисленных расчетов и данных табл. 603, достигающие 3 и 7 Дж -К -моль в
значениях Ф° F000 К) и S° F000 К) соответственно.
Константа равновесия реакции BN (г)=В (r)+N (г) вычислена на основании значения энергии
диссоциации
Дг#°. @) = Do (BN) = 540 ± 100 кДж • моль да 45 000 + 8000 см.
Эта величина была получена линейной экстраполяцией колебательных уровней основного
состояния по постоянным, приведенным в табл. 20.4. Ей соответствует
?>.fH°(BN, г, 0) = 490,818 + 100 кДж • моль.
BH3NH3 (г). Термодинамические свойства газообразного аммин-борана в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 604.
Молекулярные постоянные, использованные для расчета термодинамических функций
BH3NH3 (г), приведены в табл. 20.21. В справочнике принято, что в основном электронном
состоянии Ё1А1 молекула BH3NH3 имеет структуру симметрии С3г, с внутренним вращением вокруг
связи В—N. Структура симметрии С3„ установлена в результате исследования кристаллического
BH3NH3 [1495, 1875]. В предположении симметрии С3„ интерпретированы колебательные спектры
твердого BH3NH3 [1239a, 2669] и молекул BH3NH3, изолированных в матрице [2557]; этаноподоб-
ная структура BH3NH3 подтверждается также квантовомеханическими расчетами [532, 552, 927а,
948, 954, 1148, 11496, 1234, 1455а, 2046, 2172, 2222, 2741а, 2759, 2760]. Приведенные в табл. 20.21
произведение главных моментов инерции BH3NH3 и приведенный момент инерции волчка
вычислены по структурным параметрам г (В—N) = 1,657 А, г (В—H) = l,162 A, r (N—Н)=1,032 А,
-$N—В—Н=104,2°, -^>В—N—Н=111,7°, полученным в наиболее строгом неэмпирическом
расчете Дилла и др. [954]. Сравнение этих значений со структурными параметрами молекулы
(CH3KNBH3, определенными в исследовании микроволнового спектра [998а], а также параметрами
кристаллического BH3NH3 [1495, 1875] и газообразных и кристаллических производных
BH3NH3 [420а] позволяет заключить, что погрешности рассчитанных в [954] структурных
параметров не превышают 0,05 А для длин связей и 5° для валентных углов. Соответствующие
погрешности в значениях IaIbIc и 1щ оцениваются 5 -1017 г3 -см8 и 1-10"1 г -см2.
Для барьера внутреннего вращения молекулы BH3NH3 в квантовомеханических расчетах [532,
552, 954, 1234, 1455а, 2046, 2172, 2741а, 2759] получены значения в интервале 500—1150 см.
Единственным неэмпирическим расчетом, где проводилась оптимизация как всех геометрических
параметров, так и барьера внутреннего вращения, является работа [954], в которой найдено Vg=
86
Бор и его соединения Bg N3He (r)
=675 см. В лучшем из неэмпирических расчетов с фиксированной геометрией вычислено
У0=1070 см [2759], однако авторы [954] указывают, что для этана в аналогичном расчете с жесткой
геометрией было получено F0=1280 см, тогда как при оптимизации геометрии значение Vo
снизилось до 1060 см и оказалось близким к экспериментальной величине A040 см). Поэтому в
справочнике для барьера внутреннего вращения принято значение, рассчитанное в работе Дилла
я др. [954]. Его погрешность оценивается в 175 см.
Исследованы ИК-спектры и спектры КР твердого BH3NH3 [1239a], раствора BH3NHS [2669]
и молекулы BH3NH3 и ее изотопзамещенных, изолированных в аргоновой матрице [2557]. Для ряда
частот, наблюдавшихся в работах [1239а, 2557, 2669], имеются значительные расхождения, что
может быть обусловлено полимеризацией молекул в твердом й жидком состояниях. В табл. 20.21
представлены основные частоты молекул BH3NH3, найденные Смитом и др. [2557] из ИК-спектра
молекулы UBH3NH3, изолированной в аргоновой матрице. Погрешности в их значениях с учетом
матричного сдвига оцениваются в 10—20 см. Предполагается, что возбужденные электронные
состояния молекулы BH3NH3 лежат в области высоких энергий и в справочнике не рассматриваются.
Термодинамические функции BH3NH3 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом внутреннего вращения по уравнениям A. 160)—A. 162). Погрешности рассчитанных
значений обусловлены неточностью принятых молекулярных постоянных и приближенным
характером расчета и составляют около 2, 6 и 12 Дж-К~1-моль в значениях Ф° (Г) при 298,15, 3000
и 6000 К (из которых из-за неточности постоянных около 2 Дж-К-моль во всем интервале
температур).
Таблица термодинамических функций BH3NH3 (г) публикуется впервые.
Константа равновесия реакции BH3NH3 (r)=B (г)+6Н (r)+N (г) вычислена с использованием
значения ДГЯ° @)=2423,740 +16 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 298,15К) = —115 + 15 кДж-моль.
Принятая величина основана на энтальпии образования кристаллического аммин-борана
"(—175+10 кДж-моль), найденной при измерении энтальпии сгорания в работе Шаулова и
др. [442], и оцененной энтальпии сублимации F0+10 кДж-моль"). Энтальпия образования
BH3NH3 (г) оценивалась ранее в работах [532, 7726, 1979].
B3N3H6 (г). Термодинамические свойства газообразного боразола в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 605.
Молекулярные постоянные, использованные для расчета термодинамических функций
B3N3H6 (г), приведены в табл. 20.17. На основании результатов исследований спектров [708, 899,
1500,1616, 1630, 2139, 2271, 2388а, 2521, 2590, 2808], теоретических расчетов [3466, 530а], а также
по аналогии с молекулами С6Н6 и В3О3Н3 для основного электронного состояния 1А[ молекулы
B3N3He в справочнике принята плоская структура симметрии D3h x. Приведенное в табл. 20.17
произведение моментов инерции IaIbIc соответствует структурным параметрам г (В—N) =
=1,435 ±0,005 A, r(B-H) = l,260+0,015A, r(N-H)=l,050 +0,015 A, ^N-B-N = ^ B-N-B=
= -$ Н—В—N = -^c Н—N—В=120+5° (как в бензоле и бороксине), полученным Харшбаргером
и др. [1357] при электронографическом исследовании B3N3H6. Изучены колебательные спектры
газообразного [899, 1616, 1630, 2139, 2271, 2288а, 2521] и жидкого [899, 2139] боразола, а также
молекул B3N3H6, изолированных в матрицах [1616]. В работах [346а, 708, 899, 1500, 2521]
проведен расчет частот колебаний B3N3He. Интерпретация спектров B3N3H6, предложенная в
работах [899, 2271, 2521, 2590, 2808], затем была изменена авторами [1616, 1630, 2139, 2388а].
Основные частоты, найденные в наиболее полных исследованиях [1616, 2139, 2388а], достаточно
хорошо согласуются между собой, за исключением частоты v16. В табл. 20.17 для частот vx—v4,
/v18—v20 приняты значения, полученные Калдором и Портером [1616] при исследовании спектров
Экспериментальным данным, полученным в электронографическом исследовании Харшбаргера и др. [13571f
наилучшим образом соответствуют две неплоские структуры боразола симметрии C3v и С2. Однако обе они не
согласуются с результатами спектроскопических исследований B3N3He {708, 899, 1500, 1616, 1630, 2139, 2271,
2388а, 2521, 2590, 2808]. Возможно, что в электронографическом эксперименте наблюдается эффективная струи,
тура, искаженная внутримолекулярными колебаниями.
87
ВС (г) Глава 20
КР газообразного B3N3He. Значения частот v8—v17 найдены Нидензу и др. [2139] при изучении
ИК-спектра B3N3H6 х. Частоты v5, v6, v7 неактивны в колебательных спектрах B8NsHe, значения
v, и v7 были определены Калдором и Портером [1616] из анализа составных частот, значение v&
жолучено Рихтером и Свердловым [346а] в результате расчета силового поля этой молекулы. По-
ррешности принятых значений частот оцениваются в 200 см для v6, 50 см для ve и v7, 30 см для
vM, 5 см для vx—v4, v9—v13, v15—v20 и 1 см для v8. Возбужденные электронные состояния B3N3He
согласно данным [668, 1615] лежат выше 50 000 см и в связи с этим в справочнике не
рассматриваются.
Термодинамические функции B3N3He (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных функций определяются главным образом приближенным
характером расчета и неточностью молекулярных постоянных, они составляют 1, 12и20 Дж-К~хХ
Хмель в значениях Ф° (Г) при 298,15, 3000 и 6000 К, в том числе 0,2—1 Дж-К^-моль из-за
неточности молекулярных постоянных.
Ранее термодинамические функции B3N3He (г) вычислялись в таблицах JANAF [1543], а также
в работах Крофорда и Эдсалла [899] до 1000 К и Рихтера и Свердлова [346а]. Данные [1543J
¦ табл. 605 согласуются в пределах 0,1—2 Дж-К-моль в значениях Ф° (Т), имеющиеся
расхождения обусловлены использованием в [1543] неточных значений частот по работе [899]. В
расчете [899], по-видимому, допущены ошибки и расхождения с данными табл. 605 достигают
19 Дж-К^-моль.
Константа равновесия реакции B3N3He (r)=3B (r)+3N (г)+6Н (г) вычислена с
использованием значения Д^0 @) =4874,806+20 кДж-моль, соответствующего принятой в справочник»
энтальпии образования
(B8N3H6, г, 298,15 К) = —512' ± 13 кДж • моль.
Принятая величина основана на измерениях энтальпии сгорания B3N3He (ж) в работе Кмлди
ж др. [1703], которым соответствует Д^Л^В^Ыв, ж, 298,15 К)=—542,6+13 кДж-моль,
а также на измерениях давления пара над B3N3H6 (ж) в работах Стока и др. [2619, 2620] и Виберга
н др. [2848, 2849, 2850]. Сток и Поланд [2619] провели измерения в интервале 197—217 К A9 точек,
ДгЯ°=30,2±0,6 кДж-моль), Сток и др. [2620] — в интервале 223—273 К G точек, Д,#°=
=34,3+7,0 кДж-моль), Вибер и др. [2848, 2849, 2850] — в интервале 219—324 К A4 точек,
А,Д°=30,1 +0,6 кДж-моль). Энтальпии испарения были вычислены по уравнениям II закона
термодинамики и пересчитаны к 298,15 К с помощью оцененной величины ДСр=50 Дж -К -моль.
Принятая величина вычислена по значению Д,Я° (B3N3H6, ж, 298,15 К)=30,2+0,6 кДж-моль.
ВС (г). Термодинамические свойства газообразного карбида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 606.
Спектры молекулы ВС экспериментально не исследовались. Приведенные в табл. 20.4
молекулярные постоянные ВС приняты на основании квантовомеханического расчета Куба и Орн [17631
н оценок. Теоретическое исследование ВС, выполненное с учетом конфигурационного
взаимодействия [1763], показало, что основным электронным состоянием этой молекулы является состояние
X* Е" (конфигурация. . . 5 о In2). Согласно расчету [1763] молекула ВС имеет 18 связанных
возбужденных состояний, коррелирующих с валентными состояниями атомов В и С. В табл. 20.4
приведены данные для 15 состояний, имеющих энергии ниже 50 000 см. Погрешность принятых
значений Т, может достигать 3000—5000 см. Приведенные в таблице значения г, и о># найдены
авторами [1763], их погрешности не превышают 10 и 30% соответственно.
Термодинамические функции ВС (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производные вычислялись по соотношениям A. 90)—A. 92)
е учетом 15 возбужденных состояний, в предположении, что Qiil.Bg—{Pi/px)Qmi.sf Статистическая
сумма по колебательно-вращательным уровням энергии состояния X* ?" и ее производные
находились по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по / с помощью соотношений типа A. 82). В расчете учитывались все ко-
1 Значение vle, найденное Калдором и Портером A068 см *) [1616], отличается от приведенного в табл. 20.17
(990 см) и основанного на данных [2139, 2388а].
88
Бор и его соединения ВСз (г)
лебательно-вращателыше уровни состояния X* 2" со значениями /^ /шах, „ последние находились
по формуле A. 81). Уровни энергии вычислялись по уравнениям A. 62) и A. 65) с
коэффициентами Ykl, идентичными с постоянными из табл. 20.4 и приведенными вместе со значениями и^^
ж /Иш в табл. 20.5. Мультиплетность состояния X4S" учитывалась статистическим весом 4.
Погрешности рассчитанных термодинамических функций обусловлены главным образом
отсутствием экспериментальных данных о молекулярных постоянных ВС, а также неопределенностью
энергий возбуждения низколежащих электронных состояний. Общие погрешности в значениях
Ф° (Т) можно оценить в 2, 4 и 6 Дж-К-моль при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции ВС (г) вычислялись в таблицах JANAF [1543] по оцененным
молекулярным постоянным, существенно отличающимся от принятых в настоящем справочнике,
и без учета возбужденных электронных состояний. Расхождения данных [1543] и табл. 606
составляют 3—9иЗ—15 Дж-К-моль в значениях Ф° (Г) и S° (T) соответственно. Значения
Ф° (Т) в интервале 2000—2300 К, рассчитанные авторами [2769], отличаются от данных табл. 606
примерно на 4 Дж-К-моль из-за различия в принятых молекулярных постоянных.
Константа равновесия реакции ВС (г)=В (г)+С (г) вычислена с использованием принятой
в справочнике энергии диссоциации
[Дг#°!@) = Do (ВС) = 450 ± 30 кДж. моль ^37*400]+|2500 см,
основанной на масс-спектрометрическом исследовании равновесия реакции ВС (г)=В (г)+С (г),
выполненном Верхагеном и др. [2768, 2769]. Полученное в этом исследовании значение константы
равновесия при 2400 К было исправлено с учетом принятых в настоящее время сечений ионизации.
Погрешность принята по оценке авторов работы [2768, 2769].
Принятой энергии диссоциации соответствует
Д/П(ВС, г, 0)= 821,185 + 30 кДж • моль.
ВС2 (г). Термодинамические свойства газообразного дикарбида бора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 607.
Молекулярные постоянные ВС2, принятые для расчета термодинамических функций,
приведены в табл. 20.8. На основании исследования спектра ЭПР молекул ВС2, изолированных й матрице
инертного газа [1005а], и квантовомеханического расчета [2686а] принимается, что в основном
электронном состоянии X2 Е+ (конфигурация. . .1 тс47 а1) эта молекула имеет линейную
несимметричную структуру (симметрия СЮр)- Приведенное в табл. 20.8 значение момента инерции вычислено
по структурным параметрам г (В—С)=1,36+0,05 А и г (С—С)=1,26+0,05 А, полученным Том-
соном [2686а] из расчета в приближении Хартри—Фока. Погрешность величины / не превышает
1-109 г-см2. Частоты валентных колебаний vx и v3 оценены по силовым постоянным /В_С=7Х
Х105 дин-см, /с_с=11 -105 дин-см, принятым на основании сопоставления соответствующих
величин в молекулах В2, С2 и С3. Погрешности значений vx и v3 могут достигать 200 см.
Надежная оценка частоты v2 затруднительна. По аналогии с молекулой С3 следует ожидать, что
деформационное колебание имеет низкую частоту (около 100 см) и высокую положительную
ангармоничность. Приведенное в табл. 20.8 значение v2 учитывает эту аналогию, его погрешность может
составлять 50%. Энергия возбуждения состояния у12П (конфигурация . . . 1 тт;37 о2) принята по
расчету [2686а], ее погрешность оценивается в 4000 см.
Термодинамические функции ВС2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9) A. 10), A. 122)—A. 124), A. 126),
A. 129) с учетом одного возбужденного состояния по уравнениям A. 168)—A. 170). Погрешности
табулированных величин обусловлены главным образом неточностью принятых значений
молекулярных постоянных (в особенности частоты деформационного колебания), а также
приближенным методом расчета и могут достигать 8, 12 и 15 Дж-К-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К соответственно.
Ранее таблица термодинамических функций ВС2 (г) не публиковалась. Рассчитанные в
работе [2769] значения Ф° (Т) в интервале 2000—2300 К отличаются от приведенных в табл. 607 на
6,3 Дж-К-моль из-за различия в оценке молекулярных постоянных.
89
В2С(г), В4С (к) Глава 20
Константа равновесия реакции ВС2 (г)=В (г)+2С (г) рассчитана с использованием принятой
.в справочнике энтальпии атомизации
V7° @) = 1200 ± 25 кДж • моль.
Принятая величина была найдена по результатам масс-спектрометрического исследования
равновесия реакции ВС2 (г)=В (г)+2С (к) B083—2344 К, 5 точек), выполненного Верхагеном и др. [2768,
2769]. Значения, рассчитанные по уравнениям II и III законов, равны соответственно 1222 +
+85 кДж-моль и 1201+25 кДж-моль. Приведенные погрешности учитывают
воспроизводимость измерений и неточность сечений ионизации и термодинамических функций, использованных
в расчетах.
Принятой энтальпии атомизации соответствует
Д/Я°(ВС2, г, 0) = 782,37 + 25 кДж • моль.
В2С (г). Термодинамические свойства газообразного карбида дибора в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 608.
Спектры и структура молекулы В2С экспериментально не исследовались. Молекулярные
постоянные, принятые для расчета термодинамических функций и приведенные в табл. 20.8,
основаны на результатах квантовомеханического расчета Томсона [2686а]. В справочнике
принимается, что молекула В2С в основном электронном состоянии X1 SJ (конфигурация . . . 1 п^4о|) имеет
линейную симметричную структуру (симметрия D^t)- Несимметричный изомер ВВС согласно
[2686а] имеет более высокую энергию и рассматривается как возбужденное электронное
состояние г Е этой молекулы. Погрешность энергии возбуждения, приведенной в табл. 20.8, не
превышает 4000 см. Момент инерции, приведенный в табл. 20.8, вычислен на основании величины
г (В—С)=1,38 +0,05 А, полученной в расчете [2686а], его погрешность не превышает 1 -10~39 г -см2.
Частоты валентных колебаний vx и v3 оценены по силовой постоянной /в_с=7-105 дин-см,
принятой на основании сопоставления соответствующих величин в молекулах В2, С2 и С3.
Погрешности значений vx и v3 не превышают 200 см. Деформационная частота v2 оценена так же, как
в молекуле ВС2, и ее погрешность может достигать 100 см.
Термодинамические функции В2С (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124) и A. 126),
A. 129) с учетом только одного возбужденного состояния по уравнениям A. 168)—A. 170).
Погрешности табулированных величин обусловлены главным образом неточностью принятых
значений молекулярных постоянных (в особенности частоты деформационного колебания), а также
приближенным методом расчета и могут достигать 8, 12 и 15 Дж-К-моль~1 в значениях Ф° (Т)
при 298,15, 3000 и 6000 К соответственно.
Ранее таблица термодинамических функций В2С (г) не публиковалась. Рассчитанные в
работе [2769] значения Ф° (Г) в интервале 2000—2300 К отличаются от приведенных в табл. 608
примерно на 2 Дж-К~1-моль, что обусловлено различием в оценке молекулярных постоянных.
Константа равновесия реакции В2С (г)=2В (г)+С (г) вычислена с использованием принятой
в справочнике энтальпии атомизации
ДгЯ° @) = 1060 + 30 кДж • моль.
Принятая величина была получена по результатам исследования равновесия реакции В2С (г) =
=В (г)+В (к)+С (к), выполненного Верхагеном и др. [2768, 2769] масс-спектрометрическим
методом B249—2344 К, 4 точки). Обработка этих данных по уравнениям II и III закона приводит
к значениям 1094 и 1059+28 кДж-моль соответственно. Погрешность учитывает не только
воспроизводимость измерений, но и неточность использованных термодинамических функций
(свыше 20 кДж-моль) и сечений ионизации.
Принятой энтальпии атомизации соответствует
bfH°{B2C, г, 0) = 771,185 ± 30 кДж • моль.
В4С (к). Термодинамические свойства кристаллического карбида тетрабора в стандартном
состоянии при температурах 100—2700 К приведены в табл. 609.
90
Бор и его соединения В4С (к)
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 20.1. В справочнике за стандартное состояние В4С (к) в интервале 0—2700 К принимается
гексагональная (ромбоэдрическая) модификация [250] х. При Т <[ 298,15 К термодинамические
функции вычислены по результатам измерений теплоемкости, проведенных Келли [1654] E4—
294 К) для образца В4С, содержащего примесь 4% свободного углерода. Точность этих измерений
теплоемкости оценивается в 0,5%. Экстраполяция теплоемкости к Т=0 приводит к S° E3 К) =
=0,20 Дж-К~1-моль~1. Погрешности принимаемых по [1654] значений S° B98,15 К) и
Д° B98,15 К)— Н° @) (см. табл. 20.1) составляют 0,13 Дж^К^-моль и 0,020 кДж-моль
соответственно. В интервале 298,15—2700 К для теплоемкости В4С принято уравнение (см. табл. 20.1),
полученное совместной обработкой методом Шомейта результатов измерений энтальпии В4С (к)
в работах Кинга [1710] D30—1726 К, 28 измерений, образец В4С, содержащий 4% свободного
углерода) и Шейндлина и др. [451] A702—2574 К, 10 измерений, образец В4С, содержащий 3,3%
свободного углерода и 2,3% примесей окислов). Согласно этому уравнению теплоемкость В4С (к)
круто растет в интервале 298—800 К, имеет почти линейную зависимость от температуры в
интершале 1000—2000 К, а при Т >• 2000 К вновь наблюдается ускоренный рост теплоемкости. Точность
принятых значений теплоемкости оценивается в 2% при 1000 К, 5 % — при 2000 К и 8% при 2500 К.
Принятое в справочнике значение температуры плавления В4С B700+50 К) является средним из
"результатов определений этой константы в работах Шейндлина и др. 1451] B713 К), Киффера и
др. [1698] B723 К), Долова и др. [964а] B743 К) и Вийяр 127851 B633 К). Ввиду инконгруэнтного
характера плавления В4С расчет термодинамических функций проводился только для В4С (к) 2.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2700 К оцениваются в 0,2, 1 и
5 Дж-К~1-моль~1 соответственно. Различия между термодинамическими функциями В4С (к),
приведенными в табл. 609 и таблицах JANAF [1543], растут с увеличением температуры и достигают
4 Дж-К -моль в значении Ф° B700 К), это объясняется тем, что в настоящем справочнике
учтены данные работы [451].
В справочнике принята энтальпия образования карбида бора
Д//° (В4С, к, 298,15 К) = —62 + 6 кДж • моль".
Принятая величина основана на измерениях энтальпии сгорания карбида бора во фторе (До-
мальский, Армстронг [969], kfH° (В4222С, к, 298,15 К) = —71,5+14 кДж-моль) и в хлоре (Галь-
ченко и др. [80, 84], ДуЯ° (В4233С, к, 298,15 К) = —58,2+17 кДж-моль), а также измерений
энтальпии растворения карбида бора, бора и графита в расплаве состава MnOi6NiOi4Bo 02 (Хонг,
Клеппа [1475а], LfH° (В4017С, к, 298,15 К) = —61,9+6 кДж-моль) 3. Приближенная
экстраполяция этих величин к стехиометрическому составу дает —67,7+15 кДж-моль [969], —55,0 +
+18 кДж-моль [80, 84] и —61,6+6 кДж-моль [1475а]. Принятая величина является
средневзвешенной из этих значений. Измерения энтальпии сгорания карбида бора в кислороде,
выполненные Смитом и др. [2546], привели к значению энтальпии образования —45 кДж-моль,
точность которого ограничивается низкой степенью сгорания карбида бора и недостаточной
надежностью химических анализов.
Исследования испарения карбида бора приводят к результатам, менее надежным по сравнению
с термохимическими. Эти работы использованы в справочнике при выборе энтальпии сублимации
бора.
Константа равновесия реакции В4С (к)=4В (г)+С (г) вычислена с использованием значения
Дг#° @) = 3012,858 + 21 кДж • моль,
«оответствующего принятой энтальпии образования В4С (к).
* По данным [496] область гомогенности ромбоэдрической модификации В4С лежит при комнатной температуре
в пределах составов В4С—В6,5С (или В12С3—В13С2). При высоких температурах область гомогенности
расширяется за счет изоморфного замещения атомов бора атомами углерода в икосаэдрических группах В12 [250]. Кроме
В4С известны [2237] карбиды бора с небольшим содержанием углерода (В8С, В49С3, В2БС и В81С).
1 В таблицах JANAF [1543] температура плавления В4С принята по [964а] B743 К), оценена энтальпия
плавления (Дт#=105 кДж-моль), теплоемкость В4С (ж) A36 Дж • К • моль) и вычислены термодинамические
функции В4С (ж) до 3500 К.
•» В работе [1475а] при пересчете результатов измерений от 1320 К к стандартной [температуре, была допущена
«шибка.
Глава 21
АЛЮМИНИЙ И ЕГО СОЕДИНЕНИЯ
А1 (к, ж), AI, А1+, А12, АЮ, А1О", АЮ2, АЮ;, ALO, А12О2, А12О3 (к, ж), у-А12О3 (к), А12О3,
А1Н, АШ2, А1Н3 (к), АШ3, АЮН, НАЮ, НАЮ2, А1(ОНJ, А1@НK (к), А1@НK, A1F, AlF2r
AIF2, A1F3 (к), A1F3, AlFj, AIaF6, FA1O, F2A1O, A1HF, A1H2F, AIHF2, FA10H, FA1(OHJ,
F2AIOH, A1C1, A1C12, AlClg (к, ж), A1C13, A12C16, C1A1O, C12A1O, A1HC1, A1H2C1, A1HC12, С1АЮН,
C1A1 (OHJ, C12A1OH, A1FC1, A1F2C1, A1FC12, A1HFC1, AlBr, AlBr2, AlBr3 (к, ж), А1Вг3, Al2Br6,
AH, A1I2, A1I3 (к, ж), A1I3, AI2Ie, AIS, A1S2, A12S, AI2S2, A12S3 (к, ж), A1N (к, ж), A1N, AlCr
A1C2, A12C2, A14C3 (к)
В настоящей главе рассматриваются термодинамические свойства 66 веществ — алюминия и его
соединений с кислородом, водородом, галоидами, серой, азотом и углеродом. Для 11 веществ
в справочнике представлены свойства в конденсированном состоянии, причем для А1Н3, А1(ОНK,
А1С13, АШг3 и АП3 в ограниченном интервале температур, поскольку АШ3 разлагается при
нагревании, а остальные вещества имеют низкие критические температуры, см. ниже. Для А12О3 (к)
помимо свойств в стандартном состоянии дополнительно рассмотрены свойства метастабильной
модификации -f-Al2Os. Для 64 веществ приводятся данные об их свойствах в газообразном
состоянии, причем для Al, A1+ н АЮ — при температурах до 10 000 К, а для всех остальных — до 6000 К.
В Приложении 3 рассмотрены данные, позволяющие учитывать отклонения термодинамических
свойств хлорида, бромида и иодида алюминия при повышенных давлениях от свойств в
стандартном состоянии; для этих же газов, а также для алюминия даны критические постоянные.
В отличие от таблиц JANAF [1543, 1544] в справочнике не рассматривается ряд известных
положительно заряженных газообразных ионов, таких, как АЮ+, A1FJ и другие; потенциалы
ионизации соответствующих нейтральных соединений достаточно велики, а их энергии диссоциации
(атомизации) малы, поэтому эти соединения в равновесных условиях должны диссоциировать до»
начала их заметной ионизации.
Следует отметить, что для многих соединений, рассматриваемых в главе 21, сведения о
постоянных, необходимых для расчета их термодинамических свойств, в литературе частично или
полностью отсутствуют, а в ряде случаев имеющиеся данные очень сомнительны. Расчеты
соответствующих таблиц термодинамических свойств основаны на приближенных оценках молекулярных
и термохимических постоянных, которые для ряда соединений алюминия, в частности с О ¦ S,
малонадежны из-за отсутствия аналогии в строении однотипных соединений бора и алюминия.
А1 (к, ж). Термодинамические свойства кристаллического и жидкого алюминия в стандартном-
состоянии при температурах 100—4500 К приведены в табл. 610.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние А1 (к) в интервале от Т=0 до температуры
плавления 933,61 К принята кубическая модификация (структурный тип Си) х. При Т < 298,15 К
термодинамические функции алюминия вычислены по результатам измерений теплоемкости,
выполненных Джиоком и Мидсом [1197] A5—300 К) и Бергом [659] B,7—20 К). Данные работ [2223?
@,1—4 К), [959] A,2—4,2 К) и [1924] E5—296 К) удовлетворительно согласуются с выбранными»
значениями С°р (А1, к); результаты работ [913, 1265, 1477, 1748, 2115] менее надежны и не
принимались во внимание. Принятые значения S° B98,15 К) и Н° B98,15 К)—Н° @) совпадают с
рекомендованными КОДАТА—МСНС [864]; их погрешности оцениваются в 0,08 Дж-К-моль~1 ш
0,010 кДж-моль соответственно. В интервале 298,15—933,52 К для теплоемкости А1 (к) принята
уравнение (см. табл. 21.1), полученное Буйко и Дэвисом [801а] на основании обработки данных
по энтальпии и теплоемкости алюминия [548, 765, 1830, 1987, 2155, 2742, 2871]. Это уравнение-
аппроксимирует экспериментальные данные с точностью 0,5 % до 700 К и 1 % до 900 К. Температура
плавления алюминия (933,61 +0,10 К) принята как вторичная постоянная точка МПТШ-68 [255а }
и основана на прецизионных измерениях, проведенных Мозером и др. [2068] с помощью газового
термометра, ж является наиболее точным из многочисленных измерений Tm (A1) (библиографию-
При 1,16 К у А1 (к) имеется переход в сверхпроводящее состояние. Согласно данным [2397] при р > 200 кбар
образуется новая модификация алюминия (по-видимому, гексагональная).
92
Алнжиннй и его соединения
А1 (к, ж
Таблица 21.1. Принятые значения термодинамических величин для алюминия ж его соединений
в твердом и жидком состояниях
Вещество
[AI
А12О3
А1гО3
АШз
А1(ОИ)з
A1F3
A1C1S
AlBi-з
Alls
A12S3
A1N
AI4C3
Состояние
к, куб
ж
к1, гекс. (а)
(корунд)
к1, гекс.
к1, гекс.
ж
к, тетр. (y)
к, гекс. (а)
к1, монокл. (y
(гиббсит)
к11, гекс.
к1, куб.
к, монокл.
ж
к, монокл.
ж
к, гекс.
ж
к11, гекс. (а)
к1, гекс. (y)
ж
к, гекс.
к, гекс.
ж
к, гекс. ¦
2"
ш
?з 1
кДжХ
X моль
4,565
—
10,016
,
—
10,3
5,44
12,46
11,62
—
16,98
—
21,92
—
22,3
—
18,632
—
3,87
—
16,47
Примечание. С°0=а-\-ЪТ—гГ+й
«<М06=
16,167. б оИО6
= —39,75?
3,15 К)
оэ
CS1
о
Дж • К
28,35
—
50,92
—
—
53,3
30,04
68,44
66,50
—
109,29
—
180,25
—
190
—
116,86
_
—
20,17
—
88,95
3,15 К)
оз
CVI
о С
СО
• МОЛЬ
24,35
—
79,03
—
—
82,72
40,21
91,97
75,12 -
—
91,13
—
100,56
—
98,9
—
105,06
—
—
30,09
—
116,78
ГЧ-еГ'ЧвДж-К-1
,?.1О9—{
5,539.
Коэффициенты в
уравнении
а
27,527
31,75
97,056
122,679
107. 571
162,9
95,584
49,182
56,024
-26,262
92,628
64,936
125,5
148,41
125,0
70,626
121,0
127,790
100,2
160
45,94
37,342
67,0
148,74
•МОЛЬ^.
Для Ср(Г)
6-Ю3
с • Ю-5
—8,046 1,966а
— —
39,020 26,363
6,878 50,237
16,569 —
— —
69,508 26,92б
24,761 14,538
168,866 12,803
590,444 в —
9,085 8,819
87,864 —
— —
-426,94г —
— —
94,809 —
— —
20,258 25,574
36,48 —
— —
3,347 14,979
7,866 —
— —
33,46 37,279
Интервал
температуры
К
298,15—933,61
933,52—4500
298,15—500
500—1200
1200—2327
2327—5400
298,15—2300
298,15-500
298,15-500
298,15-728
728—2100
298,15-465,7
465,7—600
298,15-371,16
371,16-700
298,15-461,47
461,47-900
298,15—1273
1273—1373
1373-4500
298,15-1800
1800—2700
2700—4700
298,15—2500
Tir или
т
К
933,61
—
.
—.
2327
_
—
—
.
728
—
465,7
—
371,16
—
461,47
—
1273
1373
—
2700
—
2500
в d-10e=—1036,437, f 109=659.372
г <М0«=893,70.
или \,Я
кДжХ1
10,7
—
;
111,4
0,565
35,4
11,25
—
15,9
—
0
66
68
—
см. в [406]). Энтальпия плавления A0,7+0,1 кДж-моль) принята по данным [801а, 1987], она
согласуется с принятым в справочнике уравнением для С°р (А1, к) и данными для энтальпии А1 (ж)
и не противоречит менее точным измерениям Л,ИЯ в работах [547, 548, 846, 2155, 2517, 2595, 2865].
Значение теплоемкости жидкого алюминия C1,76+0,50 Дж-К-моль) принято на основании
измерений энтальпии А1 (ж), проведенных Мак-Доналдом [1987] в широком интервале температур
(941—1647 К). Результаты других измерений энтальпии А1 (ж) в работах [2871] (до 1273 К), [548]
(до 1036 К) и [2397] (до 1425 К) приводят к менее надежным значениям, лежащим в пределах
29—31 Дж-К^-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 4500 К оцениваются в 0,05,
0,2, 3 и 5 Дж-К'-моль~1 соответственно. Термодинамические функции А1 (к, ж), приведенные
в табл. 610, незначительно (в пределах 0,2 Дж -К -моль для Ф° (Т)) отличаются от данных
справочников [105, 1503, 1543].
Давление пара алюминия А1 (к, ж)=А1 (г) вычислено на основании значения b.JI0 @) = Д,Я°
(А1, к, 0)=327,346+4,0 кДж-моль, соответствующего принятой в справочнике энтальпии суб-
лимацин
Д,7Г (А1, к, 298,15 К) = 329,7 ± 4,0 кДж • моль.
¦ 93
А1(г)
Глава 21
Эта величина основана на результатах измерений давления пара алюминия, приведенных
в табл. 21.2, где в качестве погрешностей дана воспроизводимость измерений. Как видно из таблицы^
Таблица 21.2. Результаты определений Д,?Г° (А1, к, 298,15 К) (в кДж-моль")
Авторы
Фаркаш, 1931 [1065]
Баур, Бруннер, 1934 [626]
Микулинский, Умова, 1948 [257]
Брюэр, Сирей, 1951 [746]
Портер и др., 1955 [2256]
Джонсон и др., 1956 [1586]
Приселков и др., 1959 [337]
Ильшнер, Хумберт, 1960 [1527]
Фикалора и др., 1968 [1077]
Кулифеев, Ухлинов, 1968 [208]
Рао, Мотцфельдт, 1970 [2292]
Поттер и др., 1970 [2261]
Бодров, Николаев, 1974 [42]
Метод
Эффузионный, 1476 К, 1 точка
Точек кипения, 1734—2237 К, 4 точки
Эффузионный, 1773 К, 1 точка
Эффузионный, ячейка из карбида тантала,
1383—1460 К, 4 точки
То же, ячейка из окиси бериллия, 1410—
1468 К, 6 точек
Масс-спектрометрический, по зависимости
интенсивности ионного тока от Т
\Н° (А1,
II закон
_
348
—
597±380
333±90
325 ±12
Масс-спектрометрический, поверхностная иони- 327,4 ± 1,7
зация, по зависимости интенсивности ионного
тока от Т (средние температуры в разных сериях
опытов составляют 1270—1418 К)
Эффузионный, 1273—1473 К, 11 точек
Испарение с открытой поверхности в атмосфере
Аг, 1273—1573 К
Масс-спектрометрический, по зависимости
интенсивности ионного тока от температуры
Эффузионный, 1383—1572 К, 11 точек
Эффузионный, 1556 К, 1 точка
Торзионный, 66 точек, интервал температуры
не указан
Атомно-адсорбционный, 1318—1548 К, 9 точе*
328±28
315
322
338+20
—
331 ±6
с 347 ±40
к, 298,15 К)
III закон
324,6
312,0+6,0 ;
317,4 \
316±12
329,2±1,1
—
—
314,7 ±1,5
— !
—
312,4±0,9
332,7
332,5±1,3
319,3+1,6
измерения давления пара алюминия приводят к значениям энтальпии сублимации,
неудовлетворительно согласующимся между собой. Принятое значение является средневзвешенным из работ
[746 х, 1586, 2261, 2292]. При этом больший вес придавался работе Поттера и др. [2261], в которой
была использована совершенная, неоднократно проверенная методика и получено хорошее
согласие между расчетами по II и III законам термодинамики. Принятая величина совпадает с
рекомендацией КО ДАТА—МСНС [864]. Погрешность принятой величины отражает главным образом
несогласованность результатов различных измерений. Погрешность в энтальпии сублимации,
связанная с неточностью термодинамических функций, не превышает 0,7 кДж-моль.
А1 (г). Термодинамические свойства газообразного алюминия в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 611.
Уровни энергии атома А1, использованные в расчете термодинамических функций, приведены
в табл. 21.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. . .3s23p, . . .3s3p2 и . . .3s*3d и имеющие энергии ниже ионизационного предела. Энергии
состояний приняты по результатам спектрального исследования Эрикссона и Исберга [1037]. Эти данные
практически совпадают с рекомендацией Мур [2053].
Термодинамические функции А1 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т <^ 6000 К не превышают 0,02 Дж-К~1-моль~1 и обусловлены, главным образом,
неточностью принятого значения R; при более высоких температурах погрешности в S° (T) и
С°р (Т) увеличиваются из-за неучета ридберговских состояний и могут достигать 0,2 и 2 Дж-К~хХ
Хмель в значениях S° A0 000 К) и С°р A0 000 К) соответственно.
Ранее таблицы термодинамических функций А1 (г) рассчитывались в предыдущих изданиях
1 Опыты с ячейкой из окиси бериллия.
94
Алюминий в его соединения
А1+ (г»
Таблица 21.3. Уровни энергии атомов А1 и А1+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Состояние
Ti
СМ
Pi
А1 . . .3s*3p *Р1/г 0 2
2Ps/[ 112,061 4
. . .3s3p* *P*U 29 020,41 2
*Р^ 29066,96 4
4Р5/* 29142,78 6
. . .3s23d 2ЯЭ/' 32 435,435 4
2?>5/* 32 436,778 6
Атом
Электронная
конфигурация
Состояние
Ti
СМ
Pi
А1+ . . .3s2 !? 0 1
. . .3s3p *P0 37 392,0 1
»/»! 37 453,8 3
аР2 37 579,3 5
. . .3s3p ip 59 849,7 3
. . .Зр2 W 85479,0 5
• • .Зр2 *Р0 94084,5 1
*РХ 94146,8 3
3Р2 94 267,7 5
. . .3s3i здз 95546,8 7
3D2 95 547,9 5
aD1 95 548,8 3
настоящего справочника [105, 106] и в справочниках [1493, 1543, 1904, 1974] до 6000 К, а также
в работах Хилзенрата [1436] до 10000 К, Каца и Маргрейва [1639] до 2000К, Хейзе и Ви-
ланда [1384] до 1500 К, Хаффа [1493] до 3000 К и Кольского и Гилмера [1749] до 8000 К. При
Т ^ 3000 К результаты этих расчетов (за исключением ошибочного расчета [1384])
удовлетворительно согласуются с данными настоящего справочника; соответствующие расхождения не
превышают 0,02 Дж-К~1-моль~1. В интервале 3000^ Т <1 6000 К расхождения в значениях С°р (Т}
возрастают и достигают 1 Дж-К-моль~1. При Т > 6000 К становятся заметнымиЦтакже
расхождения в Ф° (Т) и S° (T), обусловленные учетом ридберговских состояний в расчетах [1436, 1749.]-
Энтальпия образования А1 (г)
bf&° (A1, г, 298,15 К) = 329,7 ± 4,0 кДж . моль
принята по рекомендации КОДАТА—МСНС [864] (см. А1 (к, ж)).
А1+ (г). Термодинамические свойства газообразного положительного иона алюминия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 612.
Уровни энергии иона А1+, использованные в расчете термодинамических функций, приведены
в табл. 21.3, в которой даны валентные состояния с энергиями ниже 100 000 см, относящиеся
к электронным конфигурациям . . .ЗД . . .3s3p, . . .3sSdn . . .Зр2 и лежащие ниже ионизационного,
предела А1+. Энергии этих состояний приняты по рекомендации Мур [2053].
Термодинамические функции А1+ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)„
A. 22)—A. 25). Значения Qai и ее производных находились непосредственным суммированием ш*
уровням энергии. Погрешности вычисленных значений термодинамических функций во всем
интервале температур не превышают 0,03 Дж-К-моль и обусловлены главным образом
неточностью принятого значения R.
Ранее таблицы термодинамических функций А1+ (г) вычислялись в предыдущем издании
справочника [105] и в справочнике JANAF [1543], а также в работах Хилзенрата [1436] до 10 000 К ж
Грина и др. [1246] до 50 000 К. Результаты этих расчетов согласуются с данными настоящего-
справочника в пределах 0,03 Дж-К~1-моль.
Константа равновесия реакции А1 +(г)+е (г) =А1 (г) вычислена на основании значения ДГЯ° @)=
=—577,538+0,001 кДж-моль, соответствующего потенциалу ионизации атома алюминия
/0(А1) = 48 278,37 + 0,02 см = 577,538+ 0,001 кДж-моль,
принятому по результатам измерений границ ридберговских серий в спектре А1 в работах Эрикс-
сона и Исберга [1037, 1038]. Ему соответствует также
bfH° (АГ, г, 0) = 904,884 + 4,0 кДж • моль.
95
АЬ (г)
Глава 21
А12 (г). Термодинамические свойства газообразного диалюминия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 613.
В табл. 21.4 приведены молекулярные постоянные А12, принятые на основании спектральных
исследований [1215, 1216, 2902], квантовомеханического расчета [1953] и оценок. В спектре
испускания А12 наблюдался единственный переход А3 Е; ->• X3 Е^ [1215, 1216, 2902]. Квантовомехани-
ческий расчет в приближении Хартри—Фока с учетом корреляционной энергии [1953] подтвердил,
что основным электронным состоянием является состояние Xs EJ (конфигурация. . . о|те|), и
показал, что помимо экспериментально исследованного возбужденного состояния А3 Е^ (конфигурация
Таблица 21.4. Принятые значения молекулярных постоянных 27А12,
27А112С
"КЯ г\ гг а те \г т]
27А12
27А11вО
«А11вО-
27A132S
«A1UN
2'A112G
а
О.
Vh^Uw ^ у } п Г1 У1, Р
Г
Х*2~* 0
Л82- 17 269,3
Х22+ 0а
ЛЧ1, 5 406,7»
В22+ 20 688,95
Ст( 33116 я
#22+ 40 267,6
Я2Д 45 260в
^2Х+ 47 190 *
Хг2+ 0
Х2Б+ 0
А*ЩЛ 4 000а
В22+ 23 380,69 Д
С*П4 30 005 Д
Z>22+ 35 714.9Д
Х3Па О6
Л'П 19 727.4Д
Х42-а 0
со
350,01
278,80
979,23
728,5
870,05
856
817,47
—
—
1100 а
617,36
500а
511,17
529,25»
430,0 е
756,5В
—
700 б
Примечание. Ниже все постоянные даны в см.
А12: а
б
АЮ: а
А1О-: а
AIS: а
A1N: *
А1С: •
Состояние 52- XSJ 3J
1 1Д
Г, 1000 2500 3500 4000
ш,у, = —0,0105; » сову„ = -
т = 1. Ю-2; « р1==2. Ю-8;
шению A.69); в вычислено
"приведено значение То, А
0,0104.
со х
в.
см-1
2,022б 0,2054
0,831» 0,190 7
6,97 0,64136
4,15 0,537 4*
3,52 0,604 08
6 0,6011
4,795» 0,565 92
- 0,493 5*
— 0,508 8*
5,26 0,68в
3,42 0,279 9
2,16 0,24»
1,52 0,2461
— 0,25
— 0,2402
4,8" 0,573 0
— 0,5811 я
4,1» 0,46 *
1пм гц здя
7000 8000 10000
в А = —128,9; г вычислено по Во
по соотношению A.68); ж А =76;
= -64,8; *
приведена постоянная
27А11вО, 27AIleO-, 27AI32S,
ах • 103
1,2
1,3
5,80
4,2Д
4,47
4,5*
4,6-
—
4,4'
1,8
0,7»
1,1
0,7»
3,6»
5,6
—
3,6Д
D
0
0
1
1
1
,-10»
3075
3875
08б
2»
16
Iе
1
1
2
1
0
0
0
0
0
1
0
1
,9*
2"
к
26
22 г
225
23 г
Зг
Зг
—
8*
= 0,5353; Д вычислено по
•»,?,=»-0,1107;
для v = 0.
оценка; 6 вычислено по соотношению A.67), принимая Do = 58500; B вычислено
A.38); г вычислено по соотношению A
оценка; 6 вычислено по соотношению
.69); Д вычислено по соотношению A.
A.67) и значению О0(АЩ) % 27 000; в
68)
н <х2 =
27AI14N,
г.
А
2,467
2,560
1,6179
1,767
1,6670
1,671
1,723
1,844"
1,816Д
1,57»
2,0275
2,19
2,1623
2,145
2,1887
1,791
1,779»
2,10б
соотно-
-5.10-»;
по соотношению
вычисленс
ношению A-69); г вычислено по соотношению A-68); Д приведено значение
значение &GU.
/2
молекула имеет еще 8 низколежащих электронных состояний:
*S+B) и ХД с энергией 10000 и »П с Те
и значениям AGl^= 746,9 и
чение для v = 0.
Do = 30 000;
= 26 000. 4 = —33;
32+, Щ 32~ и :
» вычислено по
г вычислено по соотношению A.68)
молекула имеет 5 низколежащих электронных состояний: о2П с 7^ = 5000
с энергией 10000; 6 оценка; в вычислено по соотношению A
лено по соотношению A.38
A.68).
; х вычислено по соотношению A.
.67) и DO(A1G) =
2+
по соот-
То; • приведено
с энергией 4000
соотношению A.67^
; '
приведено зна.
и 2Д, *П, 2
= 30 000; "
69); * вычислено
2- и г2+
вычис-
по соотношению
96
Алюминий и его соединения
А12 (г)
. • • <уг*оу) молекула А12 должна иметь ряд низколежащих стабильных электронных состояний,
соответствующих этим конфигурациям, а также конфигурациям . . ¦ <%ъиад и . . . а^а2. В табл. 21.4
приведены энергии возбуждения семи состояний, принятые на основании результатов
расчета [1953] (L SJ, . . . a2uaf) и оценок, сделанных в предположении, что значения fw молекулы Als
приблизительно в 1,5 раза меньше, чем у молекулы В2. Электронные состояния, для которых оценка
приводит к значениям TW, превышающим энергию диссоциации молекулы А12 О 14 000 см),
не включены в табл. 21.4. Погрешности принятых значений TW могут достигать 3000—4000 см.
Колебательные постоянные А12 в состояниях X3 EJ и А3 Е;, приведенные в табл. 21.4, приняты
по работе Зеемана [2902] х, где они были найдены по кантам большого числа полос с v' ^ 14 ж
v" ^ 18 системы А3 ?п -*¦ X3 Ejj- Вращательные постоянные найдены Гинтером и др. [1215, 1216]
в результате анализа полос 0—0, 0—1, 1—0 указанной системы.
Термодинамические функции А12 (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения Qin и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом восьми воа-
Таблица 21.5. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения vma и <Тиш, принятые для расчета термодинамических функции А12,
А1О, А1О-, A1S, AIN и А1С
Коэффициенты
А1г
9
г, • ю-* о
Ую • Ю-3 0,349 297
ySo —1,813 793
У,о • 101 _о,ЗО2 748
Ум • 10' 0,648 754
у60. Ю5 _0,393 726
Уо1 0,205 4
уп. юг _0д2
Уи
Уо. • 10е б —0,307 5
У18-10'
"я* 79
Лл 362
0
0,979 520
—7,103 9
0,164 6
—
—
0,641 36
—0,58
—
—1,08
0,2
104
335
АЮ
B*2+»
0,540 675 2,068 895
0,728 580 0,87005
—4,1915 —3,52
0,056 9 —
— —
— —
0,537 4 0,604 08
—0,42 —0,447
— —
—1,1 —1,16
— —
112 124
346 414
Примечание. Ниже все постоянные приведены в см~]
Als: aj Состояние
Те
52~
У+ ЗТТ 1Д 1
^а и» "г
А1О-
Х12+
0
0,110
—5,1
—
—
—
0,680
—0,44
—
—1,0
—
107
391
1000 2500 3500 4000 7000 80О(
s yos. 1011 = 0,008 99.
А10: * Состояние
Те
A1S: * Состояние
Те
A1N: *Л=— 33
0 Состояние
т.
А1С: а Состояние
Т.
33 116
СЧ1
30 005
*2+, х?-
?22+ ?2д
F2H+
40 267,6 45 260 47 190
Д*2+
35 714,9
ь sj;- 1Ц 4+ B)
1Д
4000 10000 IS
5000
2Д 4Д 2J+ 22-
10000
Х*2+
0
0,617 217
—3,480 71
0,042 282
—
—
0,279 549
—0,179 661
—
—2,59
—
110
435
8Да
) 10 000
A1S
Лт*
0,4000
0,500
—2,3
—
—
—
0,24
-0,07
—
—2,3
—
108
502
17 269
41
) 727,4 26 000
2,343 36
0,510379
—1,460 17
0,079 088
—0,885 978
—
0,245 791
AIN
хт
о»-б
0,7565
-4,8
—
—
—
0,5730
-0,109 793-0,56
—
—2,24
—
51
410
3
—
—1,3
—
79
307
А1С
0»
0,700
4,1
—
—
—
0,46
-0,36
—
—0,79
—
85
340
1 Зееиан [2902] отнес наблюдаемый спектр к молекуле А1С. Позднее вращательный анализ того же спектра [1215,
1216] позволил однозначно отнести его к гомоядерной молекуле А1а.
97
А10 (г) Гаава 21
бужденных электронных состояний, приведенных в табл. 21.5, в предположении, что р
=(PtlPx) Qifj.w Величина <?км?вр находилась непосредственным суммированием по колебательным
уровням и интегрированием по / с помощью уравнений типа A.82). В расчете учитывались все
уровни состояния X3 EJ со значениями /^ JmaXf „; последние находились по соотношению A. 81).
Мультиплетность основного состояния учитывалась статистическим весом 3. Энергия колебательно-
вращательных уровней рассчитывалась по уравнениям A. 62) и A. 65) с коэффициентами Ykl,
приведенными в табл. 21.5 вместе со значениями vmax и /Иш. Значения Ykl получены по постоянным
из табл. 21.4 с учетом условий A. 50) и A. 59), для обеспечения которых были добавлены члены,
с Г40, Y6o и Y03.
Погрешности вычисленных термодинамических функций определяются главным образом
неопределенностью в числе и энергиях возбуждения низколежащих электронных состояний, о
которых отсутствуют какие-либо экспериментальные данные. При температурах, превышающих 3000 К,
становятся существенными погрешности, связанные с неточностью энергии диссоциации А12.
Общие погрешности в значениях Ф° (Т) при Т ^ 6000 К могут достигать 6—8 Дж-К~1-моль~1.
Ранее таблицы термодинамических функций А12 (г) вычислялись в справочниках [105, 106,.
977, 1974], работах [2463] (<?„ до 6000 К), [2609] (Т < 2200 К) и [2855] (до 5000 К). Во всех
расчетах не учитывались возбужденные состояния А12, кром'е того, в работах [977, 1974, 2855]
принималось, что основное состояние А12 является синглетным х Е? состоянием, и использовались,,
так же как в [105 и 106],, оцененные значения молекулярных постоянных. Все это предопределило
большие расхождения данных табл. 613 с результатами всех предыдущих расчетов, достигающие'
для [977, 1974, 2855] 8—9 Дж-К^-моль-1 при 298,15 К и 17—25 Дж-К^-моль при 6000 К:
в значениях Ф° (Т) и 5° (Т).
Константа равновесия реакции А12 (г)=2А1 (г) вычислена с использованием принятой в
справочнике энергии диссоциации
АгН° @) = DO(A12) = 165 + 20 кДж • моль« 13 800 ± 1500 см.
Эта величина является средневзвешенной из найденных в масс-спектрометрических
исследованиях, которые представлены в табл. 21.6. Указанные в таблице погрешности учитывают воспроиз-
Таблица 21.6. Результаты определений Do (А12) (в кДж • моль)
Авторы
Метод
DO(A12)
Уй, Дроварт, 1971 [2746] А12 (г) = 2А1 (г), ~ 1700 К 163 + 20 (III)
Стерне, Коль, 1973 [2609] AlAu (г) + А1 (г) = А12 (г) + Аи (г), 1921—2032 К, ДГЯ° @) = 184+14 (III) 159 + 22 (III)
2А1Аи(г) = А12(г) + Аи2(г), 1921—2032 К, ArH° @) = 287 + 14 (III) 173 + 38A11)
Ш а тиль он и др., 1975 А12 (г) = 2А1 (г), 1632—1795 К 170+21A11)
[839]
Гугги и др., 1975 [1304] А12 (г) = 2А1 (г), детали эксперимента не указаны 172
водимость измерений, неточность использованных в расчетах термохимических величин, сечений'
ионизации и термодинамических функций. При обработке данных [2609] были использованы
значения Do (AlAu)=343+17 кДж-моль и Do (Au2)=227 +11 кДж-моль [406].
Принятой энергии диссоциации соответствует
A,ff° (Ala, г, 0) = 489,692 + 20 кДж • моль.
А1О (г). Термодинамические свойства газообразного оксида алюминия в стандартном
состоянии при температурах 100—10 000 К приведены в табл. 614.
Молекулярные постоянные АЮ, принятые в справочнике на основании результатов
исследований электронного спектра и теоретических расчетов этой молекулы, представлены в табл. 21.4.
В электронном спектре АЮ наблюдались девять систем полос Л2П<** X2 Е+ |1730, 2364а], В2 Е+ spt
2 Е+ [6366, 1230а, 1799,1996а, 2242а, 2361а, 23616, 2392а, 2491а], СЩ^Х2 Е+[636а, 867а, 1230а„
98
Алюминий и его соединения А1О (г)
1921а, 1986а, 2739а], ?>2Е+^Х2?+ [1773а, 2531а, 2534а, 25346,] Л«Е+**ЛЧ1, [867а, 1986,
2739а], Е2А^А*П; [867а, 1986, 2534а, 27396], ЯЕ+^АШ, [2532а], С2П(->В2Л+ [1230а,
2739а], D2 2+ -> В2 Е+ [22646]. Так же как молекула ВО, молекула А1О может обладать
квартетными состояниями 4П и 4 2. Скамп [2443в] на основании квантовомеханического расчета считает
их отталкивательными состояниями, а Колб и др. [1748а] высказали предположение, что молекула
АЮ в этих состояниях может образовываться при реакциях атомов А1 с молекулами О3.
Молекулярные постоянные АЮ в состояниях X2 Е+ и В2 2+, приведенные в табл. 21.4, получены
Лагерквистом и др. [1799] в результате исследования вращательной структуры восьми полос
соответствующего перехода (i/ ^ 3, v" ^ 5). Принятые значения колебательных постоянных хорошо
согласуются с результатами анализа системы В2Л+—Х2?+ по кантам полос (i/ ^ 12, v" ^ 11)
в работах [6366, 2392а]. Принятые значения вращательных постоянных близки к найденным из
анализа структуры полос той же системы в работе Сена [2491а] и системы D2L+—Х2? + в работе
Сингха [2531а]. Молекулярные постоянные в состоянии A2Hf приняты по работе Мак-Доналда
и Иннеса [1986], которые определили значения колебательных постоянных по кантам полос
системы D2 Е+—.<42П,. (у" ^ 3) и провели анализ двух подполос 0—0 этой системы. Последний
удовлетворительно согласуется с анализом полосы 0—0 системы ?2Д—Л2П,-, выполненным в той же
работе, однако при изучении вращательной структуры двух подполос 0—0 системы ^2Б+—A2Iif
Сингх [2532а] получил слегка отличающиеся значения. Приведенные в табл. 21.4 вращательные
постоянные АЮ в этом состоянии основаны на величине Во, усредненной из данных [1986] для двух
подсостояний.
Постоянные АЮ в состояниях С2П^, D2 Е+, Е2Ь. и F2 Е+, приведенные в табл. 21.4, приняты по
данным Майю и Бекара [1921а], Сингха, Саксены [25346] и Сингха [2531а], Мак-Доналда и
Иннеса [1986а] и Сингха [2532а] соответственно.
Термодинамические функции АЮ (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBB и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом шести
возбужденных электронных состояний, причем составляющие состояний С2П^ ZJ?+, РДиР?+
находились в предположении, что (^ вр = (pjpx) <2S вр- Статистические суммы по колебательно-
вращательным уровням энергии состояний X2 Е+, А2И4, В2 Е+ и их производные рассчитывались
по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по значениям / с помощью уравнений типа A. 82). В расчетах учитывались все
уровни энергии со значениями /^/mMl!>, где последние определялись по формуле A. 81);
в табл. 21.5 приведены соответствующие значения vmu% и /1Ш для каждого из этих состояний.
Колебательно-вращательные уровни энергии состояний X2 ?+, А2П{ и В2 Е+ вычислялись по
уравнениям A. 65), A. 62) и A. 42) — последнее для состояния ^42П^. Значения коэффициентов YM
в этих уравнениях приведены в табл. 21.5; коэффициенты Yk0 были получены из постоянных,
включенных в табл. 21.4 с учетом условий A. 50). Вырождение уровней состояния у!2П^ и мультиплет-
ность уровней в состояниях X2 Е+ и В2 ?+ учитывались в расчетах статистическим весом 2.
• Погрешности рассчитанных термодинамических функций АЮ (г) при температурах до 3000 К
обусловлены главным образом неточностью принятых фундаментальных постоянных и не
превышают 0,02—0,03 Дж-К-моль~1. При более высоких температурах начинает сказываться
отсутствие надежных данных о колебательно-вращательных постоянных АЮ в состоянии А211{, а также
экспериментальных данных о колебательно-вращательных уровнях энергии состояния X2 И+
с г; > 10. Выше 5000 К становится существенной неполнота данных о возбужденных электронных
состояниях этой молекулы. Погрешности в значениях Ф° (Т) при 6000 и 10 000 К оцениваются
в 0,1 и 0,3 Дж-К^-моль.
Термодинамические функции АЮ (г) раньше вычислялись в предыдущих изданиях
справочника [105, 106], таблицах JANAF [1543, 1544], справочниках [1493, 1904, 1974]'и работах [745,
977, 986, 1235, 2796]. Расхождения данных табл. 614 с результатами всех расчетов, кроме [1543,
1544], весьма значительны, так как их авторам было неизвестно о существовании у молекулы
АЮ электронного состояния с низкой энергией возбуждения. Эти расхождения становятся
заметными при Т > 1000 К и при высоких температурах достигают 4—6 Дж>К-моль~1 в значениях
S° (Т) и Ср (Т). Расхождения с данными [1544] не превышают 0,2, 0,6 и 0,8 Дж-К-моль в
значениях Ф° (Т), S° (Т) и С (Т). Авторы [1544] учли в расчете И возбужденных состояний АЮ,
в том числе пять не наблюдавшихся экспериментально; однако имеющиеся расхождения
обусловлены не этой причиной, а использованием авторами [1544] приближенных методик расчета.
99
ты
Глава 21
,; Константа равновесия реакции АЮ (г)=А1 (г)+0 (г) рассчитана на основании энергии
диссоциации
Дг//° @) = D0(A10) = 507 + 5 кДж • моль «* 42 400 + 420 см.
Это значение принято в справочнике в результате анализа данных, представленных в табл. 21.7
и полученных при масс-спектрометрических исследованиях равновесия реакций образования
АЮ (г), изучении хемилюминесценции и флуоресценции молекул АЮ, образующихся при
реакциях в молекулярных пучках, а также по спектру поглощения АЮ. Для значений, полученных
из масс-спектрометрических измерений, в таблице приведены общие погрешности, которые вклю-
Табли-ца 21.7. Результаты определения значения Do (A1O) (в кДж-моль)
Авторы
Метод
Do (АЮ)
И закон III закон
Масс-спектрометрический, АЮ (г)=А1 (г)+О (г), 514+40 476,8±8,0
2036—2594 К, 32 точки
Тоже, испарение с открытой поверхности жидкого — 483,2±7,8
А12Оз, 2318 К, 1 точка
Масс-спектрометрический, АЮ (г)=А1 (г)+О (г), 365+87 501,8±7,7
1943—2093 К, 7 точек
То же, A) А1 (г)+О2 (г)=АЮ (г)+О (г), 2210— 552±360 493,1±9,0
2240 К, 3 точки
То же, B) Al (r)+SO (г)=АЮ (r)+S (г), 1965— 492±31 496,4±7,5
2117 К, 10 точек
То же, АЮ (r)+Ti (г)=ТЮ (г)+А1 (г), приведено — 509,2±10
только ДгЯ°@)=—151,9
Пересчет измерений [1410] реакции A), если при- — 509,6±13
нять уточненные отношения сечений ионизации.
То же, для реакции B) а — 506,7±10
Фарбер и др., 1973 [1062]; 1975 [1056]; Масс-спектрометрический, A3 (г)+Н2О (г)= — 493,4±20
Дроварт и др., 1960 [986]
Берне, 1966 [793]
Фарбер и др., 1972 [1064]
Хилденбранд, 1973 [1410]
Дроварт, 1973 [985]
1976 [1059]
Фу, Берне, 1976 [1149]
=АЮ (г)+Н, (г) в пламенах, 2250 К, 1 точка
То же, АЮ (г)=А1 (г)+О (г), испарение с откры- 527±25
той поверхности, 2094—2318 К, 26 точек
Тоже, АЮ (г)=А1 (г)+О (г), 2300—2600К,4точки 575±140 495,0±11б
Тоже, АЮ (r)+Ti (г)=А1 (r)+TiO (г), подробности
отсутствуют, АГН° @)=—150,2±4,2
Интерпретация поглощения при 2729+5 А,
наблюдавшегося в работе [27396] как обусловленного
поглощением из состояния А%К
Уточнение интерпретации [1986] в
предположении, что поглощение имеет место с уровней v=0
и 1 состояния АШ
Исследование хемилюминесценции АЮ при
взаимодействии А1+Оа
Исследование лазерной флуоресценции АЮ в
продуктах взаимодействия А1+О2
Пастернак, Дагдиджан, 1977 [2189а] Измерение с помощью селектора скоростей
поступательной энергии атомов А1, необходимой для
образования АЮ (^22+, у=2, N=15) в реакции
А1+О2
Фонтейн, Фелдер, 1977 [1114] Измерение энергии активации реакции А1 (г)+
+СО2 (г) -> АЮ (г)+СО (г), Я=10,9±5,4
Фонтейн, 1977, цитировано по [2189а] Измерение энергии активации реакции А1 (г)+
+SOa (г) -> АЮ (r)+SO (г), ?=24,3+2,5
Червонный и др., 1977 [844]
Хилденбранд, 1977, цитировано по
[2189а]
Мак-Доналд, Иннес, 1969 [1986]
Дроварт, 1973 [985]
Гоул, Зэр, 1972 [1229]
Дагдиджан и др., 1975 [921]
524,6±8,0
495,0±11 б
510,9±7,5
«502,1 ±0,8
<510,7±0,8
>495,8±2,0
>504,6
» 506,1 ±2,5
>521±3
а Пересчет для этой реакции возможно содержит ошибку, так как воспроизвести его при подготовке
справочника не удалось.
б Эти измерения включают значительную дополнительную погрешность из-за использования в качестве
стандарта реакции WO3 (r)=WO2 (г)+О (г), для которой принято ДГЯ° B98,15 К)=625±42 кДж-моль-1.
100
Алюминий и его соединения АЮ (г)
чают воспроизводимость измерений, неточность сечений ионизации и использованных в расчетах
термодинамических функций и термохимических величин. Для остальных измерений, позволив-1-
ших определить только границу возможных значений, приведены величины, характеризующие
только воспроизводимость измерений. При выборе принятой величины не учитывались масс-
спектрометрические исследования скорости испарения с открытой поверхности [793, 1149], старые
и впоследствии пересмотренные измерения Дроварта и др. [986], измерения Фарбера и др. [1056,
1059, 1062], которые были выполнены только при одной температуре, а также данные Червонного
и др. [844], которые включают значительную погрешность, связанную с неудачным выбором
стандарта. В таблице не представлены результаты спектрофотометрических исследований равновесия
образования АЮ в пламенах [98, ИЗО, 1567, 2137] и разряде [1131]. Данные .[98], по-видимому,
относятся не к молекуле АЮ, а к другим соединениям алюминия (АЮН), а данные [ИЗО, 1567,
2137] содержат большие ошибки из-за отсутствия достоверных сведений о вероятностях
электронных переходов в этой молекуле.
Принятое значение основано на обработке по III закону результатов масс-спектрометрических
измерений [985, 1064, 1410] и данных Хилденбранда, приведенных в [2189а], а также
интерпретации данных по спектру поглощения АЮ авторами работ [985, 1986], исследованиях хемилюминес-
ценции АЮ в работах [921, 1229, 2189а] и кинетическом исследовании [1114]. Результаты всех
этих исследований, выполненных разными методами, находятся в хорошем согласии. Следует
отметить, что данные, полученные в работах [844, 1056, 1059, 1062, 1131], в пределах погрешностей
их определения также не противоречат принятому значению Do (АЮ). Ему соответетвует
AfH° (АЮ, г, 0) = 67,129 '+ 6,4 кДж • моль.
АЮ~ (г). Термодинамические свойства газообразного отрицательного иона оксида алюминия
в стандартном состоянии для температур 298,15—6000 К приведены в табл. 615. |
В литературе отсутствуют сведения об экспериментальных исследованиях спектров АЮ~|,
поэтому приведенные в табл. 21.4 молекулярные постоянные основаны на приближенных оценках1
АЮ" изоэлектронен молекулам SiO, CS, A1F и ВС1; так же как эти молекулы, ион АЮ" должей
иметь основное состояние -41+, соответствующее конфигзтрации электронной оболочки . . .п*а2'.
По аналогии с молекулами SiO и A1F можно предполагать, что даже нижнее возбужденное
состояние АЮ" (состояние 3П с конфигурацией. . .тг4атс) имеет высокую энергию (~25 000 см или выше)
и расположено вблизи ионизационного предела этого иона. Поэтому возбужденные состояния
АЮ" в справочнике не рассматриваются. i
Присоединение одного электрона на связывающую а-орбиталь молекулы АЮ приводит к резкому
упрочнению связи А1—О, энергия разрыва которой возрастает примерно на 16 000 см. По|
видимому, это сопровождается уменьшением межъядерного расстояния и частоты колебания, Kof
торые у АЮ" должны иметь значения промежуточные между соответствующими величинами
у молекул АЮ и SiO. На основании этих соображений в справочнике для последующих расчетов
приняты значения <о#=1100+100 см и г,=1,57+0,04 А, см. табл. 21.4. •
Термодинамические функции АЮ" (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95)i
Значения QBK и ее производных вычислялись без учета возбужденных электронных состояний
АЮ", в предположении, что Qtv—Q^. вр- Статистическая сумма по колебательно-вращательным
уровням энергии состояния ХгЬ+ и ее производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по значениям v и интегрированием по значениям / с помощью
уравнений типа A. 82). В расчете учитывались все уровни со значениями / ^ Jmas, v-> последние
определялись из условий A. 81); в табл. 21.5 приведены соответствующие значения vm&% и JUmi
Уровни энергии состояния Хг^+ вычислялись по уравнениям A. 65) и A. 62); коэффициенты YHf
в этих уравнениях, приведенные в табл. 21.5, тождественны постоянным из табл. 21.4.
Погрешности рассчитанных термодинамических функций АЮ" (г) при Т ^ 1000 К обусловлены
главным образом неточностью оценки величины г„, при более высоких температурах становятся
существенными погрешности из-за неточности оценок других постоянных АЮ"; пренебрежение
возбужденными электронными состояниями АЮ" практически не влияет на точность расчета
термодинамических функций. Погрешности рассчитанных значений Ф° (Т) при 298,15, 3000 и 6000 К
оцениваются в 0,5, 1 и 2 Дж-К~1-моль~1.
Термодинамические функции АЮ" (г) ранее вычислялись в таблицах JANAF [1544] и в работе
[2599] (значения Ф° (Г) для 2000—2300 К). Расхождения данных [1544] и табл. 615 составляют
101
AIO, (г)
Глава 21
до 2—3 Дж-К-моль~1 в значениях Ф° (Г) и S° (T) из-за различий в оценке молекулярных
постоянных АЮ" и до 3,5Дж- К~1«моль в значении С°р F000 К) из-за того, что в [1544] приближенно
^ 3П Р 2599 17 Д К1 1
д ,Д р ( ) , р
вклад состояния а3П. Расхождения с данными [2599] составляют —1,7 Дне» К* моль
Ю О
Константа равновесия реакции АЮ" (г)=А1 (г)+О (г)+е (г) вычислена с использованием
значения Д^йГ @)=841,000±11 кДж-моль, соответствующего принятому в справочнике сродству
к йлектрону
Ао (АЮ) = — 334+10 кДж-моль.
Эта величина получена из результатов масс-спектрометрического исследования равновесия
реакции АЮ (г)+С1" (г)=АЮ" (г)+С1 (г), выполненного Сривастава и др. [2599] B080—2222 К,
5 точек). Обработка данных этих авторов по уравнениям II и III законов приводит к значениям
tLfH0 @) для исследованной ими реакции, равным —34+100 и 14,7+5,0 кДж-моль. Принятая
величина основана на последнем значении. Ей также соответствуют
г, 0) = —266,871 + 12 кДж-моль,
699,8 + 12 кДж • моль т 58 500 + 1000 см.
АЮ4 (г). Термодинамические свойства газообразного диоксида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 616.
Молекулярные постоянные, использованные для расчета термодинамических функций АЮ2,
приведены в табл. 21.8. Спектр и структура газообразного диоксида алюминия экспериментально
Таблица 21.8.
Значения молекулярных постоянных, а также j и р?, принятые для расчета термодинамических
функций АЮ2, АЮ2", А12О, А1ОН, НА1О, FA1O, С1А1О, A1S2, A12S и А1С2
Молекула
Состояние
А1О2 2%Af
А1Ог- I1?
А12О 1Щ
А1ОН A'JS
НАЮ 2^
FA1O Х1Ъ
сшо 2хг
A1S2 2*Af
A12S &Ц
А1Сг Х«Е
А1О2: а А*Вг, Го=7000 см, а
A1S2: a 1SB2, Го=Ю000 см,
1096
750
600
750
1900
740
490
600
450
800
=2, pi=2;
а—Z, pj—г
V2
v8
СМ"
/. 1039
г • см2
496 550 1,9-1026
400B) 1000 14,5
110B) 994 26,5
320B) 3600 5,7
500 B) 1050 5,2
330 B) 1148 . 15,3
250 B) 1094 27
350 350 3,3-10» б
80 B) 600 40,7
150 B) 1900 14
^ приведено значение /А/в/с-10117 г*-см*
6 приведено значение IjJbIq ¦ 1011' г*-см8.
а
2
2
2
1
1
1
1
2
2
1
РЖ
2
1
1 -
1
1
1
1
2
1
2
не исследовались. Методом матричной изоляции изучены ИК-спектры продуктов реакции атомов
А1 (а также Ga, In и Т1) с молекулами О2 [304а, 3046, 1091, 2904а]. Рассмотрение относительных
интенсивностей полос при изменении концентраций реагирующих веществ, разбавлении матрицы
и изменении ее температуры, а также анализ изотопического сдвига полос позволили авторам этих
работ выделить полосы, относящиеся к молекулам МО2. Эти полосы интерпретированы как
валентные колебания связей О—О и М—О, сходные с аналогичными колебаниями в треугольных сильно
ионизированных молекулах диоксидов щелочных металлов М+Ог, см. [522]. На основании этих
данных в справочнике для АЮ2 в основном электронном состоянии Х2А2 принята циклическая
треугольная структура симметрии С2„. Приведенное в табл. 21.8 произведение моментов инерции
вычислено по оцененным структурным параметрам: г (О—О)=1,35 +0,05 А и г (А1—О)=1,9 +0,1 А.
Для г (О—О) принято значение, равное среднему между величиной, оцененной для диоксидов
щелочных металлов A,32 А, см., например, [493а, 521, 5226, 1288а]), и межатомным расстоянием
102
Алюминий и его соединения
АЮ2 (г)
в молекуле Ог A,37 А, см. т. 1). Сравнение величин г (М—О) в молекулах МОН и оцененных
значений г (М—О) в молекулах МО2, где М—атомы щелочных металлов[493а, 521—521в, 5226, 1288а,
2164а], показывает, что в диоксидах это расстояние в среднем на 0,2 А больше, чем в
соответствующих гидроксидах. На основании этого в справочнике для АЮ2 принято значение г (А1—О)
примерно на 0,2 А большее, чем в молекуле А12О. [Погрешность величины IaIbIg составляет
6.10-iie гз.смв#
Частоты v1=v (О—О) и v2=vomi (A1—О), приведенные в табл. 21.8, приняты по работе Осина
и др. [304а, 3046], в которой исследован ИК-спектр продуктов реакции А1+О2, изолированных
в аргоновой матрице. Не наблюдавшаяся в спектре частота v3=vacilM (A1—О) оценена
Серебренниковым и др. [379а ] из анализа нормальных колебаний молекулы АЮ2 по найденным в работе [304а ]
частотам \ и v2 и закономерностям в силовых постоянных диоксидов металлов группы IIIA,
наблюдавшимся в работе [2904а]. Погрешность частоты v3 оценивается в 150 см, частот \ и vg
¦с учетом возможного матричного сдвига — в 30 см. В работе [1091 ] к молекуле АЮ2
ориентировочно были отнесены несколько отличающиеся значения частот A178, 717 и 525 см),
наблюдавшиеся в ИК-спектре продуктов распыления А12О3 в аргоновой матрице. По аналогии с молекулой
LiOa, которая согласно теоретическим расчетам [493а, 1288а, 2164а] имеет возбужденное
электронное состояние 2Z?2 с энергией 5000—8000 см, в справочнике принято, что молекула АЮг
имеет возбужденное электронное состояние Л2Вг с энергией 7000+3000 см.
¦Термодинамические функции АЮ2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом возбужденного состояния Л2В2 по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены главным образом неточностью принятых молекулярных
постоянных АЮ2, в том числе структуры этой молекулы, и в меньшей степени приближенным характером
расчета; они составляют от 1,5 до 5 Дж-К-моль в значениях Ф° (Т) при температурах 298,15—
6000 К.
Термодинамические функции АЮ2 (г) вычислялись ранее в справочнике JANAF [1543, 1544]
в предположении о линейной структуре этой молекулы. Соответствующие расхождения данных
[1544] и табл. 616 наиболее существенны при низких температурах, они составляют 13, 6 и 4 ДжХ
хК^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Константа равновесия реакции АЮ2 (г)=А1 (г)+20 (г) вычислена с использованием принятой
в справочнике энтальпии атомизации
Дг/7° @) = 900 + 30 кДж • моль.
Таблица 21.9. Результаты определений ABf22"° (А1О2, г, 0) (в кДж-моль)
Авторы
Фарбер и др., 1971 [1063]
Фарбер и др., 1972 [1064]
Фарбер и др., 1973—1976 [1056,
1059, 1062]
Пол, 1976 [2197]
Червонный и др., 1977 [844]
Жо, Варне, 1980 [1444а]
Метод
Масс-спектрометрический, А1О» (г)+А1 (г)=
=2А1О (г), 1663—19S3 К, 7 точек
То же, 1943—2093 К, 7 точек
Масс-спектрометрическое исследование
равновесий в пламенах, АЮ (г)+Н2О (г)=А1О, (г)+Н, (г),
2250 К, 1 точка
Масс-спектрометрический, испарение с открытой
поверхности, АЮ2 (г)+А1 (г)=2АЮ (г), 2450 К,
1 точка
Масс-спектрометрический, А1О2 (г)+А1 (г)=
=2АЮ (г), 2600К, 1 точка
Масс-спектрометрический, АЮ» (г)=А1 (г)+2О (г)
ДгЯа B98 К)=907±25 " ,
Дв,Я° (АЮ2, г, 0)
II закон
III закон
1077±100 996±30
1000±22 995 ±30
— 980±30
— <945±30
— <910±30
- 898±25а
а В данные [1444а] внесена поправка, обусловленная тем, что в этой работе использовались термодинамические
функции из таблиц JANAF.
103
A10J (г), AUO (г) Глава 21
Результаты масс-спектрометрических исследований энтальпии атомизации АЮ2 (г) приведены
в табл. 21.9; погрешности соответствующих величин в основном обусловлены неточностью
термодинамических функций АЮ2 (г). В работах [844, 2197] в масс-спектрах не удалось обнаружить
жон АЮг, что позволило оценить только верхний предел энтальпии атомизации АЮ2. Как видно
жз таблицы, измерения Фарбера и др. [1056, 1059, 1062—1064] привели к существенно более
высоким величинам по сравнению с измерениями Пола [2197], Червонного и др. [844] и Хо и Барнса,
[1444а]. Сривастава и Фарбер в обзоре [2597а] отмечают, что в работах [844] и [2197]
использовались вольфрамовые эффузионные ячейки, способные восстанавливать окислы алюминия и понижать
концентрацию АЮ2 в паре. Однако в работе [1444а] этот источник ошибок был устранен. Данные
[1056, 1059, 1062—1064] вызывают сомнение, так как в этих работах ион с массой 59,
интерпретированный как A10J, наблюдался на аппаратуре, существенно менее чувствительной, чем
использованная авторами работ [844, 2197]. Дополнительные замечания по этим работам Фарбера и др.
см. в статье [1630а].
Принятая величина основана на хорошо согласующихся результатах измерений [1444а] и
[844, 2197], ей соответствует энтальпия образования
Д^0 (АЮу г, 0) = —79,088 + 30 кДж • моль*
(г). Термодинамические свойства газообразного отрицательного иона диоксида алюминия
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 617.
Молекулярные постоянные, использованные для расчета термодинамических функций AlOi,
приведены в табл. 21.8. В литературе отсутствуют экспериментальные данные о молекулярных
постоянных свободного иона АЮг *• В справочнике принимается, что в основном электронном
состоянии ХгИ ион АЮг имеет линейную структуру симметрии Dmh. Приведенный в табл. 21.8
момент инерции АЮг вычислен для значения г (А1—О)=1,65 +0,1 А, оцененного на основании
сравнения межъядерных расстояний в молекулах ВО, ВОг и АЮ. Его погрешность составляет
2-Ю9 г-см2. Основные частоты АЮг оценены на основе принятых в справочнике частот колеба-
жий молекул НВО2, ВОг и НАЮ2. Погрешности в значениях \ и vg оцениваются в 100 см, v2 —
в 50 см. Сведения о возбужденных электронных состояниях АЮг в литературе отсутствуют;
ио аналогии с изоэлектронной молекулой SiO2 в справочнике принимается, что их энергии
превышают 20 000 см.
Термодинамические функции АЮг (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью оценки молекулярных постоянных, пренебрежением возбужденными
состояниями и приближенным характером расчета; они составляют 4, 6 и 8 Дж-К-моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Термодинамические функции АЮг (г) вычислялись ранее в справочнике JANAF [1544].
Расхождения данных табл. 617 и [1544] не превышают 0,2 Дж-К~1-моль~1 в значениях Ф° (Т).
Константа равновесия реакции АЮг (г)=А1 (г)+20 (г)+е (г) вычислена с использованием
значения Дг Н° @) =1305+60 кДж-моль, соответствующего принятому в справочнике сродству
к электрону
А0 (АЮа) = —405+50 кДж-моль.
Эта величина вычислена по результатам масс-спектрометрического исследования равновесия
реакции АЮ2 (г)+-СГ (г)=А10г (г)+С1 (г), выполненного Сривастава и др. [2599] B088—2222 К,
5 точек, A^f°=—34+100 кДж-моль по II закону и —56,6+2,5 кДж-моль по III закону).
Основной вклад в погрешность принятой величины вносит неточность термодинамических функций
АЮ2 и АЮг и возможные ошибки определения концентрации этих компонентов (см. АЮ2).
Принятой величине соответствует
A^°J(A10", г, 0) = —484,088 + 60 кДж.моль.
А12О (г). Термодинамические свойства газообразного оксида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 618.
1 В работах [175, 823, 2052] исследованы ИК-спектры и спектры КР алюминатных ионов в щелочных растворах,
где образуются преимущественно ионы Al(OH)j.
104
Алюминий и его соединения ЛЬО (г)
Молекулярные постоянные, использованные для расчета термодинамических функций А12О,
приведены в табл. 21.8. Экспериментальные данные о строении и частотах колебаний А12О
противоречивы. Иванов и др. [138] на основании электронографического исследования пришли к выводу,
что молекула А12О имеет нелинейную структуру с -^ А1—О— А1=141 +5°. Позднее Толмачев
и Рамбиди [2708] показали, что эта величина при ее определении из электронографических
измерений зависит от величины частоты деформационного колебания, и для низких значений v2 угол будет
близким к 180°. Линевский и др. [1873] вИК-спектре А12О, изолированного в матрицах из Аг
и Кг, наблюдали две полосы, отнесенные к частотам \ и v3j что позволило им сделать вывод о
нелинейной структуре молекулы и на основе изотопных сдвигов частоты v3 оценить -^ А1—О—А1=
=145+10°. Позднее Найт, Велтнер [1730] и Линч и др. [1891 ] показали, что отнесение полосы
715 см в работе [1873] к частоте vx молекулы А12О было ошибочным. А Хинчклифф и Огден [1439]
на примере Ga2O и 1п2О пришли к выводу, что оценка валентных углов в этих молекулах по
отношению основных частот их изотопных модификаций дает только нижний предел величины угла
М—О—М, а учет поправки на ангармоничность увеличивает ее почти до 180°. Мальцев и Шевель
ков [239, 240] в ИК-спектре газообразного А12О наблюдали только полосу, связанную с колебанием
v3, но отметили, что отсутствие частоты va не может рассматриваться как доказательство линейной
структуры А12О, так как соответствующий переход может быть малоинтенсивным. Изучение
отклонения молекулярного пучка в электрическом поле привело авторов [784] к выводу, что для А12О
наиболее вероятной является линейная структура. В пользу линейной структуры А1аО
свидетельствует также неэмпирический квантовомеханический расчет Вагнера [2788].
На основании изложенного в справочнике принято, что в основном электронном состоянии
Х1^ молекула А12О имеет линейную структуру симметрии Dmh. Приведенный в табл. 21.8 момент
инерции вычислен для значения г (А1—О)=1,72 +0,01 А, найденного при электронографическом
исследовании Толмачевым и Рамбиди [2708]. Это значение хорошо согласуется с результатами
теоретического расчета [2788]. Погрешность в значении / оценивается в 3-109 г-см2.
ИК-спектр А12О исследовался как в газообразном состоянии [239, 240, 1873], так и в
низкотемпературных матрицах [304а, 321, 1091, 1873, 1927, 2568], однако надежно определена только
частота антисимметричного валентного колебания v3. Поскольку полосы, наблюдаемые в ИК-
спектре газообразного А12О, широкие, приведенное в табл. 21.8 значение v3 принято по результатам
исследований ИК-спектра в матрице из Аг [321, 1091, 1873, 2568]; с ним в пределах погрешности
согласуется значение, полученное при исследовании спектра в паре (950 см). Частоты vx и v2,
приведенные в табл. 21.8, вычислены по уравнениям поля валентных сил в предположении, что
/„.=0,25/,. и /а/г2=0,005/г; эти значения \ и v2 в пределах погрешностей совпадают с результатами
расчета [2788 ] и с оценкой, выполненной Снелсоном [2568 ] на основании сравнения с молекулой
Li2O *. Погрешности в значениях vx, v2 и v3 оцениваются в 100, 30 и 40 см соответственно.
Возбужденные электронные состояния А12О в справочнике не рассматриваются, так как
исследования электронных спектров [1091, 1873, 1986] показали, что они лежат выше 25 000 см.
Термодинамические функции А12О (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены в основном неточностью оцененных
молекулярных постоянных, в частности структуры этой молекулы, и приближенным характером
расчета; они могут достигать 6—12 Дж-К~1-моль в значениях Ф° (Т) в интервале 100—6000 К.
Термодинамические функции А12О (г) вычислялись ранее в справочниках [105, 106, 977, 1544,
1974], а также в работах [447, 2095, 2609, 2855] (в последних трех до 2000 К). Во всех расчетах,
кроме [1544], для А12О принята нелинейная структура (^ А1—О—А1 = 110-^-145°) и
использовались оцененные молекулярные постоянные, существенно отличающиеся от приведенных
в табл. 21.8. Расхождения результатов этих расчетов с данными табл. 618 достигают 5—8
Дж-К^-моль в значениях Ф° (Г) для [477, 1543, 2095, 2609] и 10—14 Дж-К-^моль для
[105, 977, 1974, 2855]. Расхождения данных [1544] и табл. 618 не превышают 1 Дж-К-моль
в значениях Ф° (Т).
1 В работах [321,1873, 1927, 2568] к частоте симметричного колебания vt была отнесена полоса 715 см *, а в работе
[1927] с деформационным колебанием v2 связывалась полоса с максимумом при 503 см". Однако впоследствии
в работах [1730, 1891] было показано, что эти полосы принадлежат колебаниям полимерных молекул.
105
А1аО2 (г)
Глава 21
Константа равновесия реакции А120 (г)=2А1 (г)+О (г) вычислена с использованием принятой
в справочнике энтальпии атомизации
ДгЯ° @) = 1045 ± 25 кДж • моль.
Эта величина основана на приведенных в табл. 21.10 данных, погрешности которых учитывают
воспроизводимость соответствующих измерений, неточность сечений ионизации и
термодинамических функций. Наиболее надежные измерения выполнены в работах [209, 746, 942, 986, 1064,
Таблица 21.10. Результаты
Авторы
определений Дв/2Г (А12О, г, 0) (в кДж-моль)
Метод
AaiH° (АЦО, г, 0)
II закон
III закон
Брюэр, Серей, 1951 [746] Эффузионный,*/3А1 (г)-Р/зЛ12О3 (к)=А1.0 (г), 1097+40 1055+16
1466—1853 К, 13 точек
Портер и др., 1955 [2256] Масс-спектрометрический, 4/зА1 (к, ж)+ 1066+22 —
-р/зА12О,(к)=А1*О (Г), Гср=1750 К
Дроварт и др., 1960 [986] Масс-спектрометрический, А12О (г)=2А1 (г)+О(г), 1195+45 1016±22
2036—2594 К, 30 точек
Фарбер и др., 1972 [1064] То же, 1943-2093 К, 7 точек 901 ±100 1029±20
Хилденбранд, 1973 [1410] То же, 2104-2256 К, 9 точек 1063+27 1063±25
Червонный и др., 1977 [844] То же, 2300—2600 К, 4 точки 1227+150 1089±40
Берне, 1966 [793] Масс-спектрометрический, испарение е открытой — 1004±25
поверхности, 2318 К, 1 точка
Де Мария и др., 1968 [942] Масс-спектрометрический, 4/3А1 (r)+l/3Al20s (к)= 1043+10 1042±16
=А12О (г), 1313—1511 К, 16 точек
Фарбер и др., 1973 [1062] То же, 1833—2240 К, 8 точек 1060±33 —
Кулифеев, Ухлинов, 1969 [209] Эффузионный, 4/*А1 (ж)+1/зА12О3 (к)=А120 (г), 1036+25 1057+16
1443—1576 К, 8 точек
Рао, Мотпфельдт, 1970 [2292] То же, 1556 К, 1 точка — 1031±17
Томпсон, 1973 [2685] Масс-спектрометрический, 4/3А1 (ж)+1/«А1а03 (к)= 1058+25 1051+16
=А12О (г), 1438—1580 К, 17 точек
Фу, Берне, 1976 [1149] Масс-спектрометрический, испарение с открытой 1047+20 1077+25
поверхности, А1аО (г)=2А1 (г)+О (г), 2049—
2318 К, 26 точек
1410, 2292, 2685], среднее из которых принято в справочнике. Погрешность принятой величины
оценена с учетом воспроизводимости измерений A5 кДж-моль) и погрешностей, связанных с
неточностью термодинамических функций A0—18 кДж-моль).
Принятой энтальпии атомизации соответствует энтальпия образования
Д^0 (А1аО, г, 0) = —143,525 + 26 кДж.моль.
А12О2 (г). Термодинамические свойства газообразного диоксида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 619.
Молекулярные постоянные, использованные для расчета термодинамических функций А12О2,
приведены в табл. 21.11. В литературе отсутствуют экспериментальные данные о структуре А12О2.
По аналогии с Li2O2 [521, 2881] в справочнике принимается, что в основном электронном состоянии
ХгАд молекула А12О2 имеет ромбическую структуру симметрии D2h (Vh) x. Приведенное в табл. 21.11
произведение моментов инерции А12О2 вычислено по структурным параметрам г (А1—О)=1,8 +
1 На основании оцененных энергетических характеристик линейной и циклической моделей Дроварт и др. [986]
также рекомендовали для А1аО2 ромбическую структуру. Малони и др. [1929], основываясь на расчетах
энергий связей, приняли для А12О2 угловую несимметричную структуру; однако позже Сивернд [2644] предложил
более правильную схему расчета, которая привела для ромбической структуры к хорошему согласию с
экспериментальными данными. Циклическое строение А12О2 предполагают также авторы исследования ИК-спектра
молекул А12О2, изолированных в матрице [1091,1949].
106
Алюминий н его соединения
А1аО2 (г)
Таблица 21.11. Значения молекулярных постоянных, а также s и p_g, принятые для расчета
термодинамических функций А12О2, НА1О2, F2A1O, FA1OH, С12АЮ, С1А1ОН, A12S2 и А12С2
Молекула
Состояние
ALO2 lM,
HA1O2 X*A'
F-jAlO J?2B2
FA1OH X*A'
CljAlO %2Вг
С1АЮН IU'
AlaS2 ^1-4»
A12C2 *»2+
700
3700
900
750
850
750
500
2000
v2
500
1000
700
3600
450
3600
350
500
А12Сг: а Приведено значение /-1039 г-см2.
v3
44,
CM
700 80
750 1200
260 300
900 1200
150 220
520 1200
500 70
1000 200 B)
500 -
380
950
250
600
200
350
100 B)
900
400
260
150
200
150
650
—
We-1»1"
Г» • CM*
20-10*
29
41-102
39-101
38-10»
15-102
19-10»
54 a
4
1
2
1
2
1
4
2
PX
1
1
2
2
2
2
1
1
+0,1 А и -^1 О—А1—0=50+10°, оцененным из сравнения со значениями аналогичных параметров
для А1Оа, ЫО2 [493а, 521, 521в, 1288а, 2164а] и 1Л2О2 [521, 2881]. Погрешность величины 1л1в1с
составляет 1-Ю14 г3-см6. Приведенные в табл. 21.11 частоты колебаний \—v4 и v6 оценены на
¦основании сравнения с частотами колебаний АЮ2, LiO2 [522] и Li2Oa [2881]. Частота v5 принята
по результатам исследований ИК-спектров поглощения изолированных в матрицах продуктов,
образующихся при добавлении кислорода к системе А1+А12О [1949], и продуктов распыления
А12О3 [1091]. Погрешности в частотах v1—v4 и ve оцениваются в 20% от их величин, в частоте v5 —
в 30 см. Возбужденные электронные состояния А12О2 в справочнике не рассматривались ввиду
грубости оценок постоянных в основном состоянии.
Термодинамические функции А12О2 (г) вычислены по уравнениям A. 3)—A. 6), A.9), A.10),
A. 122)—A. 124), A. 130) и A. 128) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью принятой молекулярной структуры А12О2, оценкой молекулярных
постоянных и приближенным характером расчета; они могут достигать 5, 8 и 12 Дж-К~1.моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Термодинамические функции А1аО2 (г) вычислялись ранее в справочниках [977, 1543, 1544,
1974] ив работах [986,1929]. Дуглас и Бекетт [977] принимали линейную структуру О=А1—А1=О,
а Малони и др. [1929] — нелинейную структуру А1—О—А1=О. Расхождения
термодинамических функций, вычисленных в работах [977, 1929] и приведенных в табл. 619, достигают
соответственно 20 и 8 Дж-К~1.моль~1 в значениях Ф° (Т). В справочниках JANAF [1543, 1544], Мак-
Брайд и др. [1974] и в работе Дроварта и др. [986], так же как и в настоящем справочнике,
принималась ромбическая структура А12О2; расхождения достигают 13, 4 и 20 Дж-К~1-моль~1 в
значениях Ф° (Т) соответственно для [986, 1544, 1974] и обусловлены различиями в оценке
молекулярных постоянных.
Константа равновесия реакции А12О2 (г)=2А1 (г)+20 (г) вычислена с использованием
принятого в справочнике значения энтальпии атомизации
Дг#° @) = 1550 ± 50 кДж • моль.
Это значение является средним из вычисленных по уравнению III закона и результатам масс-
спектрометрических измерений равновесия реакции А12О2 (г)=2А1 (г)+2О (г), выполненных
Дровартом и др. [986] и Фарбером и др. [1064]. В работе [986] измерения проводились в
интервале 2281—2594 К A3 точек), откуда для энтальпии атомизации А12О2 получено ДД0 @)=858 +
+200 кДж-моль (II закон) и 1493+20 кДж-моль" (III закон). В работе [1064] исследования
проводились в интервале 1943—2090 К E точек), им соответствуют ДД0 @)=1343 +180 кДж- моль
(II закон) и 1580+20 кДж-моль (III закон). Приведенные погрешности включают
воспроизводимость измерений, неточность сечений ионизации и термодинамических функций.
107
AljO3 (к, ж) Глава 21
Менее точные значения 1582+40 кДж-моль" и 1608+30 кДж-моль были найдены по
уравнению III закона из данных, полученных в аналогичных измерениях Червонного и др. [844] и
исследовании испарения с открытой поверхности, выполненном Фу и Бёрнсом [1149].
Принятой энтальпии атомизации соответствует
AfH° (А12О2, г, 0) == —401,742 + 50 кДж • моль.
А12О3 (к, ж). Термодинамические свойства кристаллического и жидкого триоксида диалюминия
в стандартном состоянии при температурах 100—5400 К приведены в табл. 620.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 21.1.В справочнике за стандартное состояние А12О3 (к) в интервале 0—2327 К принимается
корунд — а-модификация А12О3, имеющая ромбоэдрическую решетку1. При Г<^298,15К
термодинамические функции вычислены по измерениям теплоемкости А12О3, проведенным в НБС США [11521
A3—300 К) с точностью ~0,1% при Т ^> 90 К и 0,5—1% при более низких температурах. В
указанных пределах эти данные согласуются с измерениями теплоемкости А12О3, проведенными
Фьюгейтом и Свенсоном [1149а] в интервале 2—25 К.
Расчет по данным [1152] и [1149а] приводит к значениям S° B98,15 К) и Н° B98,15 К)—Н° @),.
представленным в табл. 21.1 и принятым КОДАТА—МСНС [864] в качестве ключевых величин.
Их погрешности составляют 0,10 Дж-К-моль и 0,02 кДж-моль. Менее точные данные па
теплоемкости а-А12О3 ниже 298,15 К получены в ряде других работ; эти измерения проводились,
как правило, с целью проверки калориметров по теплоемкости корунда, используемого как
стандартное вещество в низкотемпературной калориметрии.
В интервале 298,15—1200 К наиболее надежные данные по энтальпии и теплоемкости А13О$
получены в ряде работ НБС США [1152, 1210, 1212]. На основании этих исследований в работе
[1152] вычислена таблица термодинамических функций А12О3 до 1200 К; эти данные
рекомендованы в качестве стандарта теплоемкости и энтальпии А1аО3. Выполненные в ряде последующих
работ [145, 458, 2790, 2836, 2837] измерения энтальпии и теплоемкости а-А12О3 согласуются с
рекомендованными в [1152] с высокой точностью. Принятые в настоящем справочнике (см. табл. 21.1)
уравнения для теплоемкости А12О3 в интервалах 298,15—500 К и 500—1200 К аппроксимируют
данные [1152] с точностью ~0,3% для теплоемкости и ~0,05% для энтальпии и энтропии.
При Т > 1200 К расхождения между результатами измерений энтальпии А12О3 (к) [128, 145, 162,
163, 433, 452] увеличиваются и приводят к неопределенности до 0,5% в данных по энтальпии А12Оз.
при 2000 К и до 3% в значениях теплоемкости при 2000 К. Принятое в табл. 21.1 уравнение для
теплоемкости А12О3 в интервале 1200—2327 К основано на данных Кантор и др. [145].
Температура плавления A12OS B327 +4 К) принята как вторичная реперная точка МПТШ-68. Энтальпия
плавления А12О3 A11,4+3,0 кДж-моль) и значение теплоемкости А12О3 (ж) A62,9+5,0 Дж-К X
Хмоль) приняты на основании измерений энтальпии жидкого А12О3 в работах Шпильрайна
и др. [25156] (до 3190 К) и Кантор и др. [145] (до 2476 К).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 5000 К оцениваются
в 0,04, 0,3, 2 и 10 Дж-К~1-моль~1 соответственно. Термодинамические функции А12О3 (к), при-
1 Корунд — единственная кристаллическая модификация А12О8, термодинамически стабильная во всем интервале
температур от Т—0 до температуры плавления. Разновидности корунда—рубин и сапфир—имеют такую же
структуру и отличаются от корунда лишь окраской, обусловленной примесями. А12О3 (к) имеет несколько мета-
стабильных модификаций — у, 8, х, р, -ц, х, 6 и {3. (Обзор сведений об условиях их получения, структуре,
наличии примесей и устойчивости см.: Торопов и др. [406в, вып. 2, с. 19—34]). Данные о термодинамических
свойствах метастабильных модификаций Al2Og практически отсутствуют (кроме энтальпий образования). Это
связано не только с трудностями приготовления чистых образцов, их идентификации и химического анализа,
но и со сложными взаимными переходами, температуры которых зависят от термической истории образца,
примесей, дисперсности, дефектности структуры и т. д. Среди метастабильных модификаций А12О3 наиболее
важное практическое значение имеет ^-модификация, существующая в нескольких разновидностях. 7-А12О3 имеет
структуру типа шпинели дефектного типа (тетрагональная сингония) и плотность в пределах 3,3—3,4 г-смГ3.
Он образуется при дегидратации гидратных форм триоксида диалюминия (в первую очередь бёмита, АЮОН)
при температурах 250—600° С и содержит структурно связанную воду в количестве до 1—2% (иногда и другие
примеси). Различные формы у-А12О3 при нагревании устойчивы до температур 1300—1900 К и превращаются
в корунд (а-А12О3). В качестве промежуточных модификаций между у- и а-А12О3 могут образовываться 5гг в-
и х-модификации А12О8. О термодинамических свойствах ^-А12О3 см. ниже.
108
Алюшшнй и его соединения Т-А1зО3 (к)
веденные в табл. 620, незначительно отличаются от данных справочников [105, 1544]. Для жидкого
А12О3 соответствующие расхождения увеличиваются с ростом температуры (до 1 Дж-К-моль
в значении Ф° D000 К)), что обусловлено использованием в настоящем справочнике данных Шпиль-
райна и др. [25156] по энтальпии А12О3 (ж).
Константа равноЕесия реакции А12О3 (к)=2А1 (г)+ЗО (г) вычислена х с использованием
значения ДГД"° @) =3058,604+8,1 кДж-моль", соответствующего принятой в справочнике энтальпии
образования корунда
^Н° (а-А1аО8, к, 298,15|К) = —1675,7 + 1,3 кДж • моль.
Эта величина основана на совпадающих результатах измерений энтальпии сгорания алюминия,
выполненных Ма [1911] и Холли и Хубером [1461]. Менее точные величины получены этим же
методом Шнейдером, Гаттовым [2460] (—1682+8 кДж-моль"), Снайдером, Зельтцем [2574]
(—1670 кДж-моль-1), а также в более ранних измерениях [674, 2001, 2057, 2385, 2386, 2388, 2804].
Принятое значение совпадает с рекомендацией КО ДАТА—МСНС [864], оно подтверждается
данными по энтальпиям растворения и дегидратации А1(ОНK (к) и АЮОН (к) (см. Ходаковский
и др. [424а]).
Измерения ЭДС реакций с участием триоксида диалюминия приводят к величинам, заметно
различающимся между собой: —1649 к ж-моль [251а], —1641+5 кДж-моль [1195],
-1668 кДж- моль-1 [2613], —1681 нДж-моль [2692а], —1664 нДж-моль [2327а], -1665 +
+7 кДж-моль [2730а], —1686 кДж-моль [2685а]. Эти измерения существенно уступают по
точности данным, полученным в работах Ма [1911] и Холли и Хубера [1461], и, по-видимому,
содержат систематические ошибки.
у-А12О3 (к). Термодинамические свойства -f-модификации триоксида диалюминия при
температурах 298,15—2300 К приведены в табл. Д. 46.
Значения постоянных., принятые для расчета термодинамических функций, приведены
в табл. 21.1. За стандартное состояние у-А13О3 в справочнике принята тетрагональная
модификация (структурный тип искаженной шпинели) 2.
Экспериментальные данные по термодинамическим свойствам 1"-А12О3 в литературе отсутствуют
(за исключением данных по энтальпии образования). В справочнике термодинамические функции
у-А12О3 воспроизводят данные из таблиц JANAF [1544].
Значение S° B98,15 К) оценено с учетом данных о термодинамической стабильности у-А12О8
по отношению к а-А12О3; точность этого значения 8 Дж-К-моль~1. Теплоемкость "f-Al2O3 при
Т > 298,15 К принята на 4,7% большей, чем теплоемкость а-А12О3, на основании сравнения
данных по энтальпии образца А12О3 марки ТА-600 в интервале 1003—1177 К, определенных в работе
Марчидана и др. [1944а], с данными по энтальпии <х-А12О3. Согласно [1544] образец А12О3 марки
ТА-600 близок по составу к у-А12О3. Уравнение для теплоемкости у-А12О3 в интервале 298,15—
2300 К (см. табл. 21.1) выведено методом Шомейта по табулированным в [1544] значениям
энтальпии -f-Al2O3. Величина Н° B98,15 К)—Н° @) оценена при подготовке настоящего справочника, ее
погрешность составляет 0,3 кДж-моль. Данные по температуре плавления у-А12О3 отсутствуют,
за исключением оценки в [1544] B290 К). Погрешности табулированных значений Ф° (Т) при
298,15, 1000 и 2000 К оцениваются в 5, 7 и 10 Дж-К-моль~1 соответственно.
Принятое в справочнике значение энтальпии образования
&fH° (-{-А}2Оа, к, 298,15 К) = —1657 + 5 кДж • моль
основано на измерениях энтальпии перехода ^-модификации А12О3 в а-модификацию, результаты
которых приведены в табл. 21.12.
1 В связи с отсутствием достоверных данных о существовании А12О3 в газообразном состоянии и невозможностью
расчета хотя бы относительно надежных значений его термодинамических свойств в табл. 620 приводится не
парциальное давление этого газа над А12О3 (к, ж), а константа равновесия диссоциации А12О3 (к, ж) на одноатомные
газы. Это обеспечивает возможность корректного учета образования А12ОЯ (к, ж) в процессах, протекающих
в любых системах, содержащих алюминий и кислород, без использования данных для А12О3 (г). Грубая оценка
роли А12О3 (г) в таких системах может быть выполнена отдельно на основании данных, приведенных в табл. 621.
1 О полиморфизме А12О3 см. с. 108; для f-Al2O3 известно несколько кристаллических форм [406в]: плотная
модификация y-A12O3 с примесью 0,4% Li2O, мелкодисперсная форма y-A12O3 с примесью структурно связанной воды
(до 1—2%), модификация y'-A12O3 с неупорядоченными дефектами (при —1200 К f'-AljO, переходит в y-
109
АМ>з (г)
Глава 21
Таблица 21.12. Результаты определений энтальпии перехода у-А12Оз -> а-А12Оз (в кДж-моль")
Авторы
Рот, Тройч, 1936 [2385а]
Келли и др., 1946 (см. [1543])
Йококава, Клеппа, 1964 [2885а]
Ямада и др. (см. [1544])
Костомаров, Рай, 1963 [1762а]
Мукайбо и др., 1969 [2071а]
Джани, Мак-Ферсон, 1973 [1167а]
Фан Зуан и др., 1974 [2222а]
Метод
Калориметрический, сожжение смеси т-А12Оз
с парафиновым маслом
Калориметрический, растворение А1 (к) и
Т-А12Оз (к) в щелочи
Калориметрический, растворение у- и «-А12Оз
в расплаве, Г=978 К
Калориметрический, А12Оз-Н2О (к)=г-А12Оз (к)+
+Н20 (г), 773 К
ДТА, Г=1473К
ДТА, Г=978К
ДТА, Т=1473 К
ДТА, Т=1370К
ДТА, Г=1203К
ДТА, Г=1303К
Д/гЯ (Г)
-33
-65
—22,2
—18
—46
—22,2
—33,5
—24
-37,9
—28,5
Д,ГЯ B98,15 К)
—33
—65
—18,8
—16
—39
—18,8
—26,8
—18
—32,9
—32,8
Приведенные в табл. 21.12 величины имеют широкий разброс. Калориметрические измерения
в общем случае более точны, чем измерения методом дифференциального термического анализа-
Из калориметрических работ наиболее точны измерения Иококавы и Клешш [2885а]. Эта работа
выгодно отличается от исследований Рота и Тройча [2385а] и Келли и др. (см. [1543]), в частности,
тем, что найденное в ней значение не является малой разностью двух больших величин. Приняв
тая величина основана на измерениях Иококавы и Клеппы [2885а]. Близкие величины получены
в работах [1167а, 2071а] методом ДТА и в калориметрических измерениях Ямады и др. (см. [1544]).
При оценке погрешности принятого значения учитывалось плохое соответствие результатов
измерений, которое может быть связано с существованием нескольких форм f-модификации А12О3 (см.
выше).
А12О3 (г). Термодинамические свойства газообразного триоксида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 621.
Молекулярные постоянные, использованные для расчета термодинамических функций А12О3,
приведены в табл. 21.13. Экспериментальные данные о строении и спектрах А12О3 в литературе от-
Таблица 21.13. Значения молекулярных постоянных, а также а и рх, принятые для расчета
термодинамических функций А12О3, А1(ОНJ, AI(OHK, FA1(OHJ, F,A1OH, CIA1(OHJ,
С12АЮН
Молекула
А1203
А1@НJ
А](ОНK
FA1(OHJ
F2A1OH
С1А1(ОНJ
С12АЮН
Состояние
^1
iMj 900
Ж2 А' 650
Ж1 А' 3400
Ж1 А' 650
ЖХА' 750
Ж1 А' 650
Ж1 А' 750
V2
700
900
650
900
3600
900
3600
v3
300
3400
1050
3500
800
3500
450
Ч
70
3400
270
3500
950
3500
600
400
250
300
850
1200
500
1200
v8
""в
СМ
900 700 300
1050 1150 300
400
300
3400B) 900B) 280BI150B)
280 1050 1150
250 260 300
280 1050 1150
200 150 220
250
250
200
250
—
300 B)
300
—
260
—
•1
V12
— —
— —
300 30©
— —
300 300
— —
'aVc-ю1»
Г3-СМ6
19 • 103
50 • 101
55 • 102
50 • 102
44 • 102
i5 • 103
41 • 10s
a
2
1
3
1
1
1
1
Px
1
2
1
1
1
1
1
сутствуют. По аналогии с В2О3 в справочнике принимается, что в основном электронном состоянии
%гАх молекула А12О3 имеет F-образную структуру симметрии С2с. Приведенное в табл. 21.13.
произведение моментов инерции рассчитано по структурным параметрам г (А1=О)=1,65+0,05 А,
г (А1—О)=1,75 +0,05 А, -? A1—О—А1=135 ±20°, оцененным из сравнениях В2О3 и с длинами свя-
110
Алюминий и его соединения А1Н (г)
зей В—О и А1—О, принятыми в справочнике для различных кислородных соединений бора и
алюминия. Погрешность в IaIbIo составляет 1-1013 г3-см6. Основные частоты колебаний
молекулы А12О3 оценены на основании сравнения с соответствующими частотами молекулы В2О3 и
других кислородных соединений бора и алюминия. Погрешности в частотах оцениваются в 20—•
30% от их величин. Возбужденные электронные состояния А12О3 неизвестны и в справочнике Н&
рассматриваются.
Термодинамические функции А12О3 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета и могут достигать 15—20 Дж-К-моль~1 в
значениях Ф° (Г); обоснованная оценка их величины в настоящее время невозможна.
Термодинамические функции А12О3 (г) вычислялись ранее Мадером [1904J для бипирамидаль-
ной модели молекулы с существенно отличными от принятых в табл. 21.13 молекулярными
постоянными; расхождения в значениях Ф° (Г), приведенных в табл. 621 и вычисленных в [1904],
достигают 16 Дж- К-моль.
Константа равновесия реакции А12О3 (г)=2А1 (г)+30 (г) вычислена с использованием принятой,
в справочнике энтальпии атомизации
Дг#° @) = 1940 + 100 кДж • моль.
Хо и Варне [1444а] в результате масс-спектрометрического исследования состава насыщенных
паров над А12О3 (к) не обнаружили в масс-спектре ион А12Оз и пришли к выводу, что парциальное
давление А12О3 меньше 4-10"3 атм при 2318 К. Это приводит для энергии атомизации А12О3 (г)
к значению <^ 1943 кДж-моль. Значительно большая величина B-10~5 атм при 2600 К), которой
соответствует baiH° @)=2215 кДж-моль, была оценена Фарбером и др. [1064, 2797а]. Оценка
в предположении, что А^Я° (Al2O3)=2D0 (АЮ) + Да(Я° (А12О), дает 2060 кДж-моль. Эту
величину следует рассматривать только как верхнюю границу возможных значений, поскольку ана-.
логичная оценка для В2О3 (г) приводит к величине, практически совпадающей с принятой в
справочнике, а устойчивость В2О3 относительно других окислов бора значительно выше, чем это можно
ожидать из сравнения А12О3 с другими газообразными окислами алюминия. Таким образом, эта
оценка подтверждает значение, вычисленное по данным работы [1444а], которое принимается
в справочнике.
Принятой энтальпии атомизации соответствует
bfH°(Al2Os, г, 0) =—544,959 ± 100 кДж • моль.
А1Н (г). Термодинамические свойства газообразного гидрида алюминия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 622.
Молекулярные постоянные А1Н, принятые в справочнике на основании результатов
исследований электронного спектра и теоретических расчетов этой молекулы, представлены в табл. 21.14.
В электронном спектре А1Н наблюдались восемь систем полос: .Л 41 ** Х1Е+ [581, 610, 652, 1036,
1065, 1066, 1471, 1474, 1488, 1507, 1518, 1718, 2063, 2775, 2903], Сх?+ -+Х1!.* [648, 650, 652,
1241, 1676, 1678, 2143], D^+ ** Хх?+ [650, 652, 1676, 1678, 2143], ElU ** ХХЕ+ [1798, 2143],
C^-» 441 [650, 651, 652,1471], ЕгП -> .441 [1470, 1798], Ь3?" -* а3П [832, 1466, 1467],
с3П -»¦ а3П [1469, 1717]. Квантовомеханические расчеты [1266, 2203а] показали, что, кроме восьми
электронных состояний, переходы между которыми наблюдались в спектре, молекула А1Н имеет
еще несколько связанных состояний с энергиями от 43 000 до 51 000 см CЕ+, 3П и Б?~).
В табл. 21.14 включены только четыре нижних валентных электронных состояния АШ. Состояния
Х^+, а3И и АгП имеют общий диссоциационный предел А1 BР)-\-Л BS) и соответствуют
конфигурациям электронной оболочки . . .5а2 и . . .5о2тг. Состояние fes?~ соответствует конфигурации
. . .2л2. Более высокие состояния, имеющие энергии свыше 42 000 см, согласно [1266] имеют
характер ридберговских состояний.
Приведенные в табл. 21.14 постоянные АШ в состоянии XXS+ и вращательные постоянные в
состоянии А41 найдены в работе Зеемана и Риттера [2903] из анализа вращательной структуры
девяти полос системы .441 ** Х1?+ (и" ^ 4, v'=0 и 1). Молекула АШ в состоянии А1!! имеет
только два связанных колебательных уровня ж согласно результатам анализа иредиссоциацни
в этом состоянии максимум на потенциальной кржвой Уо (г)- Колебательные постоянные АШ
111
АШ (г)
Глава 21
Таблица 21.14. Принятые значения молекулярных постоянных А1Н, A1F, AlCl, AlBr и АН
Молекула
Состояние
"А1Н X12+
а3пг
АЧ1
Ъ3 2-
"AFOF X12+
а3П
лт
Ь32+
27А136С1 X12+
а3П
Ь3Ъ
"А1'вВг X12+
а3П
,441
!7А1Ш1 X1 S+
а3П
Примечание. Ниже все
А1Н: а шеуе=0,2389; б <
До=6,7О4 и ах; е
= -22,86; к р1 =
_ 8 1-10~9.
У.
0
15 400
23 660,25
41603
0
27 239,4 в
43 947
44 813,2
0
24 594
38 254
43 590
0
23 776,3 в
35 879,5
0
22 089,5 "
1682,563
1763
1341,08
1810
801,52
830,3
804,69
786,37
481,3
524,35
449,96
350 г
378,0
411,25
297,2
316,1
337,4
постоянные даны в см
х2=1,6М0-3;
вычислено по
в Pi=5,73
29,09 a
47
91,84 я
65
4,658
4,6
6,3525
7,64 e
1,95
2,175
4,37 B
—
1,28
1,85
6,40 е
1,0
2,0
ю-6, pa
соотношению A,69);
в.
CM
6,390 66
6,911 Д
6,386 88
6,759 *
0,552 479
0,557 18
Д 0,556 5
0,562 80
0,243 942
0,250
0,259
0,226 «
0,159 204
0,164 8
0,155 5
0,117 700
0,124 0
=2,82-10-';
ж приведено
= —4,9957-10; л приведена постоянная для
A1F: * а2=1,72-10-5, а3=— 4,7-Ю-8
=0,004845; е <о„
А1С1: » о2= 4,7-Ю-», а3 =
постоянная при v
AlBr: * о2=2,03-10-6; б
Уе = —0,009;
= 5,7-Ю-9; 6
= 0.
6 Pi=l,
- «2=-2
5-10-'; *
• 10.
h= -5,3-10-", Ht=
Pi = 2,06-Ю0; Я,= —1
числено по соотношению A.68); " ^еув-
A1I: » o2 = l,048-10-6,
оз=2,46-10-9;
,12.10-";
= —0,527
6 Pi= 9,57-10-", .
40=47,6; г р
-4,46-Ю-14;
' 4% 130; г
* о3 = —1
aj • 10*
D. ¦ Ю7
1858,1 б 3570,6 в
4140 е 4000 ж
7322,9 4701,4 *
— 4360 *
8 49,841 а 10,46 б
46,8 9,81 г
55 11
45,7 ж 11,8
3 16,111 3а 2,503б
20 —
60 —
— —
8,604 48 а 1,128 5 б
9 г ja
21,6 * 1,83 !
5,585 9 а 0,652 12 б
8,3 0,44
г 4=40,4; * вычислено по
г»
А
1,647 8
1,584
1,648 3
1,602 х
1,654 36
1,647 4
1,648 5
1,639 1
2,130 11
2,10
2,07
2,21 Д
2,294 80
2,255
2,322
2,537 09
2,472
значениям
значение Do; Яо = — 8,35-10"9; и a>,jr,=
у=0; * приведено значение
Do; ffo=
!=-2,57-10-»; Д о>,у.=0,01779, шйгй=
в шеУ,= —0,216; Г оценка; Д
вычислено по соотношению A
,75-10-*; 3 Pi= 1,4.10-*, р2 =
(У, = 4,59-Ю-15; вЛ = 200.
приведена
.69); я вы-
= 1,3-10-».
в состоянии Л-Ч! найдены Хароном [1518] на основании величины Д&д и значений ДС?у„ Л^/.
для молекулы АШ. Постоянные АШ в состояниях а3П и й3Е" приняты по работам Холста [1466,
1467], который выполнил анализ структуры полосы 0—0 в системе Ь3?~ -» а3П, и Чаллакомбе
л Алми [832]. Поскольку в спектре АШ не наблюдались интеркомбинационные переходы, для
энергии состояния а3П и колебательных постоянных в этом состоянии приняты значения, полученные
в теоретическом расчете Пелиссье и Мальрьё [2203а]. Соответствующее значение Тв находится в
хорошем согласии с энергиями состояния а3П у молекул ВН, GaH и InH, принятыми в справочнике;
погрешность этой величины, по-видимому, не превышает 4000 см.
Термодинамические функции АШ (г) вычислены по соотношениям A. 3)—A. 10), A. 93)—
A. 95). Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом трех
возбужденных состояний; статистические суммы по колебательно-вращательным уровням энергии
состояний ХХ11+, а3П, ЛХП, Ь32~ и их производные находились по соотношениям A. 76)—A. 78)
непосредственным суммированием по уровням энергии. В расчете учитывались все колебательно-
вращательные уровни, ограниченные предельными кривыми диссоциации этих состояний (для
состояния А1!! — предельной прямой диссоциации). Энергии уровней состояний Хх?+ и а3П
вычислялись по уравнениям A. 65) и A. 61), состояний А1!! и Ь3?~ — по уравнениям A. 65) и A. 60);
мультиплетность и вырождение уровней состояний а3П, А1!! и 63Е" учитывались
статистическими весами. Значения коэффициентов Ykl в уравнениях, аппроксимирующих
колебательно-вращательные уровни, коэффициентов а( в уравнениях, аппроксимирующих предельные кривые
диссоциации, а также величин vma% и /1!т приведены в табл. 21.15. Коэффициенты У40 и Y31 для
состояния ХХЕ+ определены из условий A. 50) и A. 57) соответственно, коэффициенты Yoa и YOi для
состояний Хх?+ и а3П и коэффициенты Уоз для состояний ^41 и Ь3Е~ — из условий A. 59).
112
Таблица 21.15.
Значения коэффициентов в уравнениях, описывающих уровни энергии (в см'1), а также значения vmtx и Jim,
принятые для расчета термодинамических функций А1Н, A1F, А1С1, АШг и АН
Коэффициенты
Г..10-* 0
Ую-10"» 1,683 036
Ум-Ю -2,948 315
У.о-101 3,587 2
У,о-10» 1,204 36
Ум-10» -
Ув1 6,39185
Yu-iffl —18,82
У21.10» 2,767 9
У«.10« -1,495 94
Ум.10« —357,6
Уц.10' 57,3
Уи.10' —2,82
Уо8'Ю« 19,946
Ум-10й -4,140 23
(оо-В,).10-* 2,463 419
Ol 0,303 465
Oj-104 0,481570 7
в,-10» 0,333160 3
iw 25
/иш 85
А1Н
о»П
1,540 0
2,763
-8,7
—
—
—
6,911 1
-41,4
—
—
—400,0
—
—
—8,35
-2,821
0,923 413
0,785 246
3,077 697
5,526 608
9
49
Примечание. Ниже приведены значения анергий
A1F: а в»П, Г,=27 239,4; АШ,
А1С1: а а*П, Г,=24 594; .441,
AlBr: a а»П, Г,=23 776,3.
All: ¦ am, «Г,=22 089,5.
441
2,366 025
1,341 08
-9,184
-228,6
—
—
6,386 88
-73,229
—
—
—470,14
-4995,7
—
34,607 1
—
0,097 0
3,971 22
1,548 623
81,173 58
1
25
воабуждения в
Г,=43 947; 6s 2+, Г,=44 813,2.
Г,=38 254; 6*S
, Г,=43 590.
Ь»2-
4,160 3
1,810
-6,9
—
—
—
6,930 6
-34,3
—
—
—436,0
—
—
-8,1
—
1,212 813
0,639 633
2,256 237
2,642 916
12
152
см.
A1F
0
0,801 311
—0,459 3
—0,065 435
0,021 606
-0,676 7
0,552 479 8
-0,498 41
0,017 2
-0,000 47
-1,046
—0,015
—
—
—
—
—
—
—
179
451
А1С1
0
0,480 230
—0,201 705
0,041 206
0,000 447
—
0,242 491 6
-0,159 678
0,004 64
-0,000 056
-0,247 3
—0,005 22
—
—
—
—
—
—
—
218
571
АШг
0
0,378 101
-0,130 483
0,014 843
—
0,159 204
-0,086 045
0,020 3
—
-0,112 85
0,002 06
—
—0,112
—
—
261
649
АН
0
0,316 118
—0,100 803
0,009 268
,
0,117 699 8
—0,055 859
0,000 002 5
,
—0,065 212
0,000 957
0,000 004 6
227
678
АШг (г) Глава 21
При температурах до 2000 К основные погрешности вычисленных термодинамических функций
АШ (г) обусловлены неточностью принятых фундаментальных постоянных. При более высоких
температурах становятся существенными погрешности из-за отсутствия экспериментальных данных
об энергии состояния а3П, а также пренебрежения состояниями с энергиями выше 42 000 см»
Погрешности в значениях Ф°(Г)при298,15,3000и6000К составляют 0,02, 0,1 и0,5 Дж-К-^моль.
Термодинамические функции АШ (г) ранее вычислялись в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF/[1543], работах [977, 1336, 1819, 1904, 1974, 2463, 2796,
2855]. Наилучшее согласие с данными табл. 622 (в пределах 0,2 и 1 Дж-К~1-моль~1 в значениях
Ф° (Т) и S° (T) соответственно) имеет место для расчетов [105, 106], в которых учтен вклад
возбужденных состояний. Расхождения с данными [1543, 1974, 1904, 1336] достигают 1,3, 4 и 5,5 ДжХ
XК-моль в значениях Ф° (Т), S° (Т) и С° (Т) и обусловлены использованием в этих расчетах
приближенной методики и пренебрежением вкладом состояния а3П.
Константа равновесия реакции АШ (г)=А1 (г)+Н (г) вычислена на основании энергии
диссоциации этой молекулы
ДД°@) = О0(АШ) = 24 600 + 150 см ^294,3 + 1,8 кДж • моль.
Эта величина принята в справочнике на основании данных по предиссоциации при вращении
молекул АШ и AID, полученных в работах [5736, 1036, 1396, 1471, 1474, 1505, 1506, 2775J.
Наиболее тщательные измерения, выполненные Холстом и Хультеном [1474], показали, что обрыв
вращательной структуры в состоянии АгИ наблюдается у АШ при i;=0, /=20 и v=l, /=9, а у АШ
при v=0, /=31, у=1, /=23 и v=2, / = 10. Анализ этих данных привел авторов [1396, 1505, 1506]
к выводу, что потенциальная кривая состояния А1!! для невращающейся молекулы имеет максимум.
По предельным кривым диссоциации состояния АгП молекул АШ и АШ, построенным по
данным [1474], Герцберг и Мунди [1396] нашли, что энергия максимума потенциальной кривой
этого состояния равна соответственно 25 610 и 25 775 см над минимумом состояния Х1!!".
Различие в этой величине для двух изотопических модификаций обусловлено туннельным эффектом,
приводящим к диссоциации дейтерида и особенно гидрида при энергиях, меньших энергии
максимумов на эффективных потенциальных кривых вращающихся молекул. Благодаря этому эффекту
значение, полученное для дейтерида, ближе к энергии максимума на потенциальной кривой
невращающейся молекулы, а полученное для гидрида — к энергии предела диссоциации. На
основании этих данных Герцберг и Мунди [1396] приняли De (АШ) <1 25 610 см.
Высота максимума на потенциальной кривой состояния А1!! оценена Харли [1517] равной
1200+400 см в предположении, что она примерно такая же, как у молекулы ВН (см. выше).
Эта величина вместе с энергией максимума, найденной в работе [1396] из данных для AID, приводит
к значению De (A1H)=24 600+400 см.1
В 1979 г. Балтаян и Неделек [5736] исследовали время жизни молекул АШ и AID на предис-
социированных уровнях состояния Аг11 ж смогли определить форму максимума на потенциальной
кривой этого состояния. Из этих данных авторы [5736] нашли De (A1H)=25 490+80 см.
Практически совпадающее значение B5 435 см) получено Майером и Росмуссм [2015] в результате кван-
товомеханического расчета. Принятое в справочнике значение основано на величинах, найденных
в работах [5736, 1396, 2015], и значении G @, Х1?+)=834 см; ему соответствует
A^°(A1H, г, 0) = 249,08 + 4,4 кДж • моль.
А1Н2 (г). Термодинамические свойства газообразного дигидрида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 623.
Молекулярные постоянные, положенные в основу расчета термодинамических функций АШ3 (г),
приведены в табл. 21.16. Исследование электронного спектра АШ3 (см. [87]), а также
теоретический анализ [2098] показали, что в основном электронном состоянии Ж2Аг молекула А1Н2 имеет
симметричную нелинейную структуру (группа симметрии С2„) с межатомным расстоянием
г (А1—Н)=1,59+0,02 А и ^ Н—А1—И=119+5°. Этим структурным параметрам соответствует
произведение главных моментов инерции А1Н2, приведенное в табл. 21.16. Его погрешность не
1 Харли получил более низкое значение De (АШ), приняв для энергии максимума данные авторов [1396],
относящиеся к АШ.
114
Алюминий и его соединения
А1Н2 (г)
Таблица 21.16. Значения молекулярных постоянных, о и р% в основном электронном состоянии, принятые
для расчета термодинамических функций АШ2, A1F2, AlF-2, A1HF,A1C12, АШС1, AlFCl
А1Вг2, и АП2
Молекула
Состояние
СМ
Г3-СМ6
о
PZ
АШ2 i'Mj» 1900 850 2000 0,11 2 2
A1F2 1*А^ 800 230 930 31-Ю1 2 2
AlFj iMj 850 300 900 32-101 2 1
AlHF 1*A"- 1100 500 1900 8,5 1 2
A1C12 ^Мг» 480 140 600 53-102 2 2
A1H2: a Л2^, Го=10 000 см, о = 2, Pl=2.
A1F2: a Л2]?!, Го = 21 600 см, о = 2, рА = 2.
A1HF: а Л2^", Г0 = 14 000 см, о = 1, рл = 2.
А1С12: а АгВг, Г0 = 15 300 см, о = 2, Pj = 2.
Молекула
Состояние
СМ
г3-см6
а
Pt
А1НС1 i44'a 800 420 1900 40 12
AlFCl lW* 880 190 520 13-102 х 2
А1Вг2 l2i!3 360 95 490 48-Ю3 2 2
АП2 ^2Лга 290 70 410 23-10* 2 2
АШС1: а ЛМ", Г0=10 000 см~', 0 = 1,^ = 2.
A1FC1: * Л2Л", Г0=18 000 cir», 0 = 1,^ = 2.
А1Вг2: а Л2ЯЬ Г0 = 10 000 см-', 0 = 2,^ = 2.
АП2: а Л2В3, Г0=10 000 см-\ 0 = 2,^ = 2.
превышает 1-1019 г3-см". В электронном спектре АШ2 проанализирована только одна полоса и
частоты колебаний получены не были. Приведенные в табл. 21.16 частоты А1Н2 вычислены по урав-
¦„-.-.„ п 't i,4 i ъ\ лп ~ - соответствующей
постоянной в АШ3- При этом отношение ]ri\Tjr")=l\) оказывается таким же, как в молекулах ВН и ВН
Погрешности оцененных таким образом основных частот АШ2 могут достигать 15 20% 2 3*
Согласно Герцбергу [87], в спектре АШ2 наблюдалась только одна, частично предис'социиро-
ванная полоса, соответствующая переходу на уровень состояния А*ВХ с энергией около 15 200 см
который расположен вблизи диссоциационного предела этого состояния. Оценка энергии состояния
T(AiB АШ2) Г0(А»П, А1Н)
А2Вг по соотношению
ПРИВ°ДИТ к значению 5600 см.
Теоретичеi, ВН2) Т0(АЩ, ВН)
ский расчет в приближении CNDO, выполненный Чарски и Малеком [825], дал явно завышенную
величину Г0=18 000-^20 000 см. Значение, приведенное в табл. 21.16, принято как округленное
среднее из этих величин; его погрешность оценивается в 5000 см.
Термодинамические функции А1Н2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
вклада возбужденного электронного состояния А2Вг по уравнениям A. 168)—A. 170), т. е. без учета
различия структуры АШ2 в основном и возбужденном состояниях. Погрешности'рассчитанных
термодинамических функций оцениваются в 2, 4 и 6 Дж-К-моль~1 в значениях! Ф° (Т) шш
298,15, 3000 и 6000 К. Они обусловлены в основном неточностью оцененных значений
молекулярных постоянных, в частности энергии состояния А2Вг, и в меньшей степени приближенным
характером расчета.
Таблица термодинамических функций АШ2 (г) публикуется впервые.
Константа равновесия реакции АШ2 (г)=А1 (г)+2Н (г) вычислена с использованием значения
ДД° @)=509,414+60 кДж-моль, соответствующего принятой в справочнике энтальпии
образования '
t\fH° (АШ2, г, 0) = 250 + 60 кДж • моль.
Эта величина оценена сопоставлением энергий связей в молекулах АШ, A1F, A1F А1С1 и А1С1
Оценка с использованием данных для ВН и ВН2 приводит к значению LfH° (A1H2, г, bWl45 кДжХ
Хмоль, что, соответствует значительно большей термодинамической стабильности А1Н М
Такая оценка представляется необоснованной, так как в отличие от бора для алюминия не
характерно существование стабильных газообразных соединений с водородом.
115
(к), A1H, (г) Глава 21
АШ3 (к). Термодинамические свойства кристаллического тригидрида алюминия в стандартном
состоянии при температурах 100—500 К приведены в табл. Д.47.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние А1Н3 (к) принята гексагональная
модификация (<х-АШ3), относящаяся к структурному типу A1F3 [767а, 2738а] Ч
Термодинамические функции А1Н3 (к) при Т < 298,15 К вычислены по результатам измерений
теплоемкости, проведенных Зинке и др. [2537] A5—308 К) на образце, содержащем 94,3% АШ3,
¦ 1,9% 1ЛАШ4, 1,9% А1аО3, 1,6% LiCl и 0,3% эфира. Результаты измерений исправлялись на
содержание примесей. Точность измерений теплоемкости при Т > 50 К равна 1,0—1,5%. Ввиду
значительного разброса экспериментальных данных при Т < 30 К величина S° C0 К) =0,10 ДжХ
X К-моль вычислена по уравнению Дебая. Погрешности принятых по [2537] значений S0
B98,15 К) и Н° B98,15 К)—Н° @) оцениваются в 0,4 Дж-К^-моль и 0,08 кДж-моль
соответственно. При Т > 298,15 К для теплоемкости АШ3 (к) принято уравнение, выведенное по
значениям С; B98,15 К)=40,21 Дж-К^-моль [2537], С°„ D00 К)=50 Дж-К^.моль и С°р F00 К) =
=60 Дж-К-моль"; последние оценены на основании сравнения экспериментальных данных по
теплоемкости дигидридов щелочноземельных металлов и А1Н3. В литературе отсутствуют данные
о температуре плавления А1Н3. Разложение наиболее стабильной модификации (<х-АШ3)
происходит при 380—440К и зависит от скорости нагрева. Данные по энтальпии образования и энтропии
А1Н3 (к) при 298,15 К приводят к значению Ду(?=46 кДж-моль, которое соответствует
равновесному давлению водорода в 2,6-105 атм (при Г=298,15 К).
Погрешности вычисленных значений Ф° (Т) при 298,15 и 500 К оцениваются в 0,3 и 1 ДжХ
XК-моль соответственно. Расхождения между термодинамическими функциями А1Н3 (к),
приведенными в табл. Д.47 и в справочнике [578] B98—400 К), составляют около 1 Дж-К~1Х
Хмоль в значении Ф° D00 К) из-за различия оценок теплоемкости АШ3 (к).
В справочнике принята энтальпия образования тригидрида алюминия
, к, гекс, 298,15 К) = —11,4 ± 1,0 кДж • моль.
Эта величина основана на совпадающих результатах измерений энтальпии разложения АШ3 (к),
выполненных Зинке и др. [2537J и Кирпичевым и др. [166 ]. Измерения в этих работах проводились
в калориметрической бомбе с электрическим нагревателем для разложения образца [2537] и
в сканирующем калориметре [166]. Близкое значение —8,5 кДж-моль получено Клауди
и др. [857а] с использованием калориметра Кальве.
А1Н3 (г). Термодинамические свойства газообразного тригидрида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 624.
Молекулярные постоянные, использованные для расчета термодинамических функций А1Н3 (г),
приведены в табл. 21.17. Экспериментальные данные о спектре и структуре молекулы А1Н3 в
литературе отсутствуют. По аналогии с ВН3 и тригалогенидами бора и алюминия, а также с учетом
результатов теоретических расчетов [45, 489, 2575], в справочнике принимается, что в основном
электронном состоянии Х1А[ молекула АШ3 имеет плоскую структуру симметрии D3h. Произведение
главных моментов инерции, приведенное в табл. 21.17, вычислено для межатомного расстояния
г (А1-^Н)=1,59+0,03 А, оцененного на основании сравнения величины г (В—Н) в ВН8 и ВН и
г (А1—Н) в А1Н. Теоретические расчеты [489, 2575] дали для г (А1—Н) те же значения A,59 и
1,598 А). Погрешность в величине IaIbIc составляет 4«10~119 г3-смв. Основные частоты А1Н3,
приведенные в табл. 21.17, рассчитаны по формулам поля валентных сил и оцененным значениям
силовых постоянных. Силовые постоянные АШ3 оценивались двумя—тремя способами, каждая на
основании соответствующих постоянных молекул А1Н2, ВН3, А1НС1а, A1F3 и А1С13 и соотношений
постоянных fr/fx и frlf в этих молекулах. Принятые значения основных частот являются средними
из полученных таким образом, их погрешности с учетом соответствующих расхождений
оцениваются в 100 см для vlt v, и 200 см для v2, v4.
Согласно данным Брауэра ж др. [767а] существует семь различных модификаций^ А1Н, (a, a, J9, у, I, b я «),
образующихся в зависимости от методов получения и наличия примесей. При нагревании большинство
модификаций превращается в «-А1Н,.
116
Алюминий и его соединения
АЮН (г)
Таблица 21.17. Значения молекулярных постоянных и о в основном электронной состояния (р<г=1),
принятые для расчета термодинамических функций АШз, AIF3, A1F4~, A1H.F, A1HF2*
A1C1S, A1H2C1, A1HC12, A1F2C1, A1FC12, A1HFC1, AlBr3 и Alls
Молекула
Состояние
АШз ^А[
A1F3 I'M;
AlFr XlAx
A1H2F -f1A i
A1HF2 IM,
А1С13 ^А[
АШ2С1 I'M,
АШС12 JPM!
A1F2C1 -?Мг
A1FC12 I'M,
A1HFC1 I'M,
AlBr3 I'M J
Alls I'M!
1900
690
650
1950
800
375
1950
481
820
450
1950
228
156
700
297
220 B)
800
1950
180
500
1968
500
850
500
155
135
V3
CM
2000 B) 1000 B)
935 B) 263 B)
800 C) 320 C)
1000 2000
260 900
610 B) 150 B)
1000 2000
150 579
250 900
150 610
600 800
510 B) 93 B)
425 B) 64 B)
— —
— —
700 750
700 550
_
650 700
655 472
200 260
200 220
400 500
— —
— —
/Al?/c.10"'
r3 • см6
0,51
3,98-10»
1,39-10"
2,2-101
4,6-102
1,05-1O5
1-102
6,8-103
1,3-104
3,3-104
1,8.10s
1,9-106
1,36-107
О
6
6
12
2
2
6 ,
2
2
2
2
1
6
6
Термодинамические функции А1Н3 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности рассчитанных величин обусловлены до»
3000 К главным образом неточностью оцененных молекулярных постоянных, при более высоких
температурах становится существенным приближенный характер расчета. Погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 1, 4 и 6 Дж^К^-моль.
Таблица термодинамических функций АШ3 (г) публикуется впервые.
Константа равновесия реакции А1Н3 (г)=А1 (г)+ЗН (г) вычислена с использованием значения
Дг#с @)=760,448 +60|кДж -моль, соответствующего принятой в сиравочнике энтальпии
образования
Д/Я°(А1Н8, г, 0) = 215 + 60 кДж • моль.
Эта величина оценена сопоставлением энергий связей в молекулах АШ, A1F, A1F3, A1C1 и A1CI,-
Оценка, основанная на сопоставлении с энергиями связей в ВН и ВН3, приводит к величине-
&,Н° (АШ3)»—7 кДж-моль, что соответствует существенно более высокой термодинамической
стабильности АШ3 (г). Такая оценка представляется менее оправданной, так как в отличие от
бора для алюминия не характерно образование водородных соединений и характер связей в этих
соединениях, по-видимому, иной.
АЮН (г). Термодинамические свойства газообразного гидроксида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 625. ;
Молекулярные постоянные, использованные для расчета термодинамических функций АЮН,
приведены в табл. 21.8. Экспериментальные данные о спектре и структуре молекулы АЮН
отсутствуют. Неэмпирический расчет потенциальной поверхности, выполненный Зюбиной и др. [13761,
показал, что в основном электронном состоянии Х*Е + молекула АЮН имеет линейную структуру
симметрии Ссог- Приведенное в табл. 21.8 значение главного момента инерции рассчитано для
линейной структуры и оцененных межатомных расстояний г (А1—О)=1,72+0,05 А 1 (как в А12О) и
г (О—Н) =0,96 ±0,02 А (характеристическое значение). Погрешность момента инерции не
превышает 3-10"*° г-см2. Значения основных частот приняты на основании оценок. Частота vx (A1—О)
оценена из сопоставления значения <ве молекулы A1F с частотой 764 см, вычисленной для АЮН
по силовой постоянной /ai-Oi перенесенной из молекулы А1гО. Частота деформационного колеба-
1 В теоретическом расчете [1376] для г (А1—О) получено значение 1,66 А.
117
НА10 (г)
Глава 21
ния оценена по соответствующим частотам гидроксидов щелочных металлов. Для частоты
колебания группы О—Н принято характеристическое значение. Погрешности в принятых значениях
частот могут достигать 100, 50 и 100 см соответственно. Молекула АЮН имеет изомер НАШ;
согласно теоретическому расчету [1376] структура этого изомера линейна, а энергия его превышает
энергию АЮН приблизительно на 18 000 см.
Термодинамические функции АЮН (г) вычислены по уравнениям A. 3)—A. 10), A. 122) —
A. 124), A. 126), A. 129). Погрешности вычисленных термодинамических функций обусловлены
главным образом неточностью оцененных значений молекулярных постоянных, в том числе
структуры молекулы АЮН, и в меньшей степени приближенным характером расчета; они составляют 4,
10 и 16 Дж-К^-моль-1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции АЮН (г) рассчитывались в справочнике JANAF [1543]
и в работе Уилкинса и др. [2855] (Т ^ 5000 К). Расхождения между термодинамическими
функциями, приведенными в табл. 625 и вычисленными в [1543] и [2855] , достигают 23 и 10 Дж-К~1Х
Хмоль в значениях Ф° (Т) соответственно. Они обусловлены главным образом разницей в оценке
частоты деформационного колебания и структурных параметров этой молекулы.
Константа равновесия реакции АЮН (г)=А1 (г)+О (г)+Н (г) вычислена с использованием
значения АГН° @) =980,163+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —190 ± 50 кДж • моль.
Результаты измерений энтальпии образования АЮН (г) приведены в табл. 21.18. Соответствующие
погрешности учитывают воспроизводимость измерений, погрешности использованных в расчетах
Таблица 21.18. Результаты определений AfH° (АЮН, г, 0) (в кДж-моль)
Авторы
Метод
Д/Я°(АЮН, г, 0)
II закон
III закон
Фарбер и др., 1967 [1051а] Масс-спектрометрический, VsA^ (к)+1/8А1 (г)+ —179±10 —
(см. [1543]) а +1/2D2(r)=AlOD (г), три серии, средние
температуры 1980, 2000 и 2060 К
То же, 1/зА12Оз (к)+4/3А1 (r)+D2O (r)=2A10D (г) —182±12 —
Молекулярного потока, А12Оз (к)+2Н2 (г)= — —163+.30
=2АЮН (г)+Н2О (г)
Фарбер и др., 1973 [1062] Масс-спектрометрический, 1/зА12О3(к)+1/зА1(ж)+ —189±15 —245±25
+V2D2 (r)=A10D (г), 1833—2240 К, 2 серии,
8 точек'
ToHte,Al2O(r)+D2O(r)=2AlOD(r),1898-2240K, —183+.20 —170±25
4 точки
Фарбер и др., 1975 [1056] Масс-спектрометрическое исследование равнове- — —130+30
сий в пламенах, АЮ (г)+Н2 (г)=А1ОН (г)+Н (г),
2250 К, 1 точка
Тоже, А1(г)+Н2О (г)=-АЮН (г)+Н (г), 2250 К, — —116+30
1 точка
а В [1543] отсутствуют первичные экспериментальные данные [1051а]. Приведенные в таблице значения A^ff3
пересчитаны с учетом разности термодинамических функций АЮН, принятых5в [1543] и в настоящем справочнике.
При 2000 К различие в термодинамических функциях изменяет энтальпию реакции примерно на 50 кДж на один
моль АЮН.
величин, а также учитывают оценку точности измерений авторами рассматриваемых работ.
Как видно из таблицы, результаты исследований энтальпии образования АЮН (г) плохо
согласуются между собой. В справочнике принято среднее из значений, рассчитанных по III закону по
данным работ [1051а, 1062]. Принятая величина не противоречит данным по энергиям связей
A1F, A1G1, А1(ОНJ, BF, BG1 и ВОН.
НАЮ (г). Термодинамические свойства газообразного оксид-гидрида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 626.
118
Алюминий и его соединения НАЮг (г)
Молекулярные постоянные, использованные для расчета термодинамических функций НАЮ,
приведены в табл. 21.8. Спектр и структура НАЮ экспериментально не исследовались. На
основании неэмпирического расчета Зюбиной и др. [1376 ] в справочнике принимается, что молекула НАЮ
в основном электронном состоянии Хг^+ имеет линейную структуру (симметрия С^). Приведенное
в табл. 21.8 значение главного момента инерции вычислено для межатомных расстояний г (А1—Н) =
=1,59+0,03 Лиг (А1—О) = 1,62+0,03 А, принятых равными соответствующим значениям в
молекулах А1Н3 и А1О х. Погрешность значения / не превышает 0,3-10~39 г-см2. Приведенные
в табл. 21.8 частоты колебаний рассчитаны по уравнениям поля валентных сил и оцененным
значениям силовых постоянных. Постоянная /ai—о принята такой же, как в молекулах FA1O и С1АЮ
F,6-105 дин-см), постоянная /ai-h перенесена из А1Н3 B,1-105 дин-см), деформационная
постоянная /а/гх-г3 принята равной @,11 +0,03)-105 дин-см на основании предположения, что
отношение /м—х//а в НАЮ такое же, как в молекулах HBO, FBO, G1BO. Погрешности принятых
значений составляют 200, 70 и 100 см в \, v2 и v3.
Термодинамические функции НАЮ (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический осциллятор»;
погрешности рассчитанных функций обусловлены неточностью принятых значений молекулярных
постоянных, а также приближенным характером расчета и составляют 2, 4 и 6 Дж- К-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции НАЮ (г) вычислялись в справочнике JANAF [1543],
Дугласом и Бекеттом [977], Мадером [1904] и Уилкинсом и др. |2855] (Т <! 5000 К). Все расчеты
были выполнены по оцененным значениям молекулярных постоянных. Термодинамические
функции, вычисленные в работах [977, 1904, 2855], меньше приведенных в табл. 626 на 1—8,5 ДжХ
XК-моль в значениях Ф° (Г); расхождение с данными [1543] также лежит в этих пределах.
Константа равновесия реакции НАЮ (г)=Н (г)+А1 (г)+О (г) вычислена с использованием
значения ДГЯ° @)=770,163+70 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я°(НА1О, г, 0) = 20|+70 кДж • моль.
Эта величина основана на разности энергий НАЮ и АЮН (см. табл. 21.8) и принятом значении
ДД° (АЮН, г).
НА1О2 (г). Термодинамические свойства газообразной метаалюминиевой кислоты в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 627.
Молекулярные постоянные, использованные для расчета термодинамических функций НАЮ2,
приведены в табл. 21.11. В литературе отсутствуют экспериментальные данные о структуре и
частотах колебаний НА1Оа. В справочнике по аналогии с НВО2 принято, что в основном электронном
состоянии ХгА' молекула НАЮ2 имеет угловую структуру симметрии Са с линейной группировкой
ОАЮ а. Приведенное в табл. 21.11 произведение моментов инерции молекулы НАЮ2 вычислено
для структурных параметров: г (А1=О)=1,62+0,05 А, г (А1—О)=1,72+0,05 А, г (О—Н)=0,96 +
+0,021А и -=$ Н—О—А1=115+10°. Длины связей А1=О и А1—О оценены из сравнения с
молекулами ВО, В2О, НВО2, АЮ и А1аО; г (ОН) и -^ Н—О—А1 приняты такими же, как в молекуле НВО2.
Погрешность в значении IjJbIo составляет 5-Ю17 г3-см6. Основные частоты НАЮ2 оценены из
сопоставления со значениями частот аналогичных колебаний в молекулах FA1O, АЮН, НВО2.
Погрешности в значениях v1? v2 и \ оцениваются в 200 см, v3 и v6 — в 100 см и, v5 — в 75 см.
Возбужденные электронные состояния НАЮ2 в справочнике не рассматриваются, в предположении,
что их энергии превышают 20 000 см.
Термодинамические функции НАЮ2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных термодинамических функций обусловлены главным образом
неточностью оценки молекулярных постоянных, в частности структуры НАЮ2, и приближенным
характером расчета; они составляют 8—15 Дж-К-моль в значениях Ф° (Т) в интервале
температур 298,15-6000 К.
• Расчет [1376] дал несколько меньшие значения межатомных расстояний, равные 1,54 и 1,59 А соответственно,
2 Нельзя также исключить, что молекула НАЮ2 имеет циклическую структуру.
119
А1@НJ (г), А1 (ОН), (к) Глава 21
Термодинамические функции НАЮ2 (г) вычислялись ранее в справочниках [1543, 1904].
Расхождения данных табл. 627 и этих расчетов не превышают 1 Дж'К»моль в значениях Ф° (Г)
и обусловлены некоторыми различиями в оценке постоянных НАЮ2.
Константа равновесия реакции НАЮ2 (г)=Н (г)+А1 (г)+20 (г) вычислена с использованием
значения к^Д0 @)=1386,946+60 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
0 г, 0) = —350 +.60 кДж-моль-1.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции
АЮ (г)+Н2О (г)=НАЮ2 (г)+Н (г) в пламенах, выполненном Фарбером и Сриваставой [1056г
1059]. По отношению интенсивностей ионных токов авторы работы [1056] нашли ДГС B250 К) =
=86+25 кДж-моль, которому и соответствует принятое значение. Поскольку найденная в
работе [1056] энтальпия образования АЮН (г) существенно отличается от значения, принятого
в справочнике (см. выше), погрешность приведенного значения Д^.ЕР|(НАЮа, г) увеличена по
сравнению с ее оценкой в [1056].
А1(ОНJ (г). Термодинамические функции газообразного дигидроксида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 628.
Молекулярные постоянные, использованные для расчета термодинамических функций А1(ОНJ),
приведены в табл. 21.13. В литературе отсутствуют экспериментальные данные и квантовомехани-
ческие расчеты, связанные с исследованием структуры и частот колебаний молекулы А1(ОНJ.
В справочнике структурные параметры А1(ОНJ оценены в предположении, что эта молекула
представляет фрагмент молекулы А1(ОНK. Произведение главных моментов инерции в основном
электронном состоянии Х2А' вычислено с учетом плоской структуры симметрии С, молекулы А1(ОНJ.
Значения межатомных расстояний и углов перенесены из молекулы А1(ОНK, при этом принималась
во внимание некоторая аналогия между молекулами А1(ОНJ и A1F2, A1C12. Погрешность значения
IjJbIc, учитывающая неточность оценки структурных параметров, составляет 2-Ю15 г3-см*.
Основные частоты колебаний А1(ОНJ, приведенные в табл. 21.13, оценены на основании сравнения
с частотами А1(ОНK и A1F2. Погрешности принятых значений частот составляют 20—25% от их
величин.
Термодинамические функции А1(ОНJ (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных величин обусловлены неточностью оценки молекулярных постоянных,
а также приближенным характером расчета; они составляют 6, 12 и 20 Дж- К-моль в значениях
Ф° (Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций А1(ОНJ (г) публикуется впервые.
Константа равновесия реакции А1(ОНJ (г)=А1 (г)+2Н (г)+20 (г) вычислена с
использованием значения ДГЯ° @)=1887,98+30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —635|+30 кДж.моль.
Эта величина основана на результатах исследования равновесия реакции А1 (г)+2Н2О (г) =
=А1(ОНJ (г)+2Н (г), выполненного Енсеном и Джонсом [1567] методом спектрофотометрии
пламен B280—2640 К, 8 пламен, Д^0 @)=—666+45 кДж-моль по II закону и —636±30ькДжХ
Хмоль — по III закону). Погрешность принятого значения включает воспроизводимость
измерений и неточность величин, использованных при обработке экспериментальных данных.
А1(ОН)з (к). Термодинамические свойства кристаллического тригидроксида алюминия в
стандартном состоянии при температурах 100—500 К приведены в табл. Д.48.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние А1(ОНK ^принята гиббсит-моноклинная
Т-модификация, термодинамически стабильная в интервале 0—500 К [2414а]г. Термодинамические
* Выше 500 К гиббсит становится неустойчивым по отношению к продуктам его разложения (диаспор АЮОН в
Н2О). Для А1(ОНK (к), кроме гиббсита, известны также метастабильные модификации: триклинный т'-г
моноклинный байерит (а) и триклинный нордстандит (см. [406]).
120
Алюминий и его соединения AI @НK (г)
функции гиббсита при Т < 298,15 К вычислены на основании измерений теплоемкости образца
гиббсита, содержащего 0,5% примесей (Na2O — 0,35%, окислов железа 0,13% и СО2 — 0,01 %),.
выполненных Хемингуэем и др. [13856] в интервале температур 13,5—298 К. Точность этих
измерений, проведенных в адиабатическом калориметре, оценивается в 0,3%. Экстраполяция
теплоемкости ниже 13,5 К приводит к значению S° A3,5 К) =0,40 Дж-К~1-моль. Погрешности
принятых по [13856] значений S° B98,15 К) и Н° B98,15 К)—Н° @) (см. табл. 21.1) оцениваются
в 0,2 Дж-К~1«моль~1 и 0,03 кДж-моль соответственно. Менее точные данные по теплоемкости
гиббсита, полученные в работе [25126] E3—297 К), не принимались во внимание при вычислении,
термодинамических функций. В интервале 298,15—500 К для теплоемкости А1(ОНK принято
уравнение (см. табл. 21.1), выведенное Ходаковским и др. [424а] по результатам измерений
теплоемкости гиббсита в адиабатическом калориметре B40—380 К) и в дифференциальном сканирующем
калориметре C40—480 К) в работе [13856]. Погрешности значений теплоемкости, рассчитанных
по этому уравнению, не превышают 1% при 480 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 400 и 500 К оцениваются в 0,1, 0,2
и 0,5 Дж-К-моль соответственно.
Термодинамические функции А1(ОНK (к), приведенные в табл. Д.48 и в справочнике [578],
отличаются на 1,5 Дж-К-моль~1 в значениях Ф° (Т) из-за использования данных Хемингуэя
и др. в настоящем издании.
В справочнике принята энтальпия образования тригидроксида алюминия
\AfH°\(Al(OH)s, к, гиббсит, 298,15"К) = —1293,5 ±1,2 кДж • моль.
Эта величина является средней из данных Хемингуэя и др. [13856] (—1293,1 +1,2 кДж-моль,
измерения энтальпии растворения А1 и А1(ОН)8 в плавиковой кислоте), Гросса и Хеймана [1275а]
(—1294,2+1,4 кДж-моль, измерения энтальпии растворения А1С13 и А1(ОН)8 в плавиковой
кислоте) и Ходаковского и др. [424а] (—1293,3+1,3 кДж-моль, измерения растворимости гиббсита.
и бемита в щелочных растворах). Измерения энтальпии растворения А1С13-6Н2О и А1(ОНK в
плавиковой кислоте в работе [575а] привели к заметно меньшей величине —1282 кДж-моль.
А1(ОНK (г). Термодинамические свойства газообразного тригидроксида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 629.
Молекулярные постоянные, использованные для расчета термодинамических функций А1(ОНK^
приведены в табл. 21.13. Экспериментальные данные о спектре и структуре молекулы А1(ОН)»
в литературе отсутствуют, поэтому структурные параметры и частоты колебаний А1(ОНK были
оценены. Приведенное в табл. 21.13. произведение моментов инерции в состоянии %ХА' вычислена
для плоской структуры молекулы симметрии C3h с -$ О—А1—0=120+3°, -4> А1—О—Н=115+10°,
г (О—Н)=0,96+0,02 А (перенесены из В(ОНK) и г (А1—О)=1,78+0,05 А (оценено сравнением
с АЮ, ВО и В(ОНK). Погрешность значения IaIbIc составляет 1-1O~1U г3-см6. Частоты колебаний
О—Н \ и ve приняты несколько большими, чем в В(ОНK. Частоты колебаний А1—О v2 и v7
деформационного колебания О—А1—О f*8 и зонтичного колебания v4 оценены с учетом близости свойств
группы ОН и атома фтора из сопоставления соответствующих частот В(ОНK, BF3 и A1F3. Для
частот деформационных колебаний А1—О—Н v3 и v9 приняты значения несколько меньшие, чем в
молекуле В(ОНK. Частоты крутильных колебаний ОН групп v5 и v10 оценены из сравнения с
величиной vKpyx молекулы A1F2OH. Погрешности принятых значений vx и ve оцениваются в 200 см~\
частот v2, v3, v5, v7) v9 и v10 — в 100 см и частот v4 и vg — в 50 см.
Термодинамические функции А1(ОНK (г) вычислены по уравнениям A. 3)—A. 10), A. 122)'—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор»; их
погрешности обусловлены главным образом неточностью оценки молекулярных постоянных,
а также приближенным характером расчета и могут достигать 8, 20 и 30 Дж- К- моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций А1(ОНK (г) публикуется впервые.
Константа равновесия реакции А1(ОНK (г)=А1 (г)+30 (г)-(-ЗН (г) вычислена с использованием
значения ДД^0 @)=2715,797 ±100 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH° (А](ОНK, г, 0) = —1000 ± 100 кДж ¦ моль.
121
A1F (г) Глава 21
Эта величина оценена сопоставлением средних энергий связей в молекулах АЮН, A1F, A1F3,
A1G1 и А1С18.
A1F (г). Термодинамические свойства газообразного фторида алюминия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 630.
Молекулярные постоянные, принятые в справочнике на основании анализа результатов
исследований электронного и вращательного спектров A1F, приведены в табл. 21.14. В электронном
спектре A1F экспериментально наблюдались переходы между 16 синглетными и триплетными
состояниями а3П -> Х1^ [1759, 2364, 2365], АХП <* Х*2+ [599, 600, 2106, 2107, 2108, 2109, 2284,
2354, 2364, 2365, 2391], BXZ+ ^ Х1^ [600, 604, 2109, 2110, 2392], CLE+ ^ XXS+ [600, 604, 2109,
2392 ], ЕЧ1 «- X12+, F'u *- X1 S+, Я1 Е+ <- Xх Е+ [600, 604, 2392], С1 ?+ <* АХП [600, 2106, 2107,
210S], Dlb -* Am, FlU -> АХП, G1^ -+ Am [600, 2108], b3 S+ <* a?U. [600, 961, 962, 2354, 2364,
2391], c32+ ** am [600, 601, 961, 962, 1757, 2364], d32+ -* asU [600, 601, 1757], /3П -> а3П [600,
1757], c3?+<^&3?+ [601, 961, 962, 1757, 2364], fll->b32,+, e3E+->53S+ и #3 E+ -> 63 S+ [600,
601, 1757]. Так же как у других галогенидов элементов группы IIIА, основное состояние Хг2+
молекулы A1F с электронной конфигурацией . . .тс4а2 и нижние возбужденные состояния А1!! и
а3П, соответствующие электронной конфигурации . . .тс4атс, коррелируют с диссоциационным
пределом Al Ci-*)+F BP3i,)- Кроме того, молекула A1F имеет еще два связанных валентных состояния
Ь3И+ и В1Е+, а все остальные связанные состояния, по-видимому, являются ридберговскими
состояниями [1757, 2565а]. В табл. 21.14 представлены данные только для трех возбужденных
состояний, энергии которых не превышают 50 000 см.
Приведенные в табл. 21.14 колебательные постоянные молекулы A1F в состоянии X1S+
получены Барроу и др. [599] на основании анализа системы Am -*¦ X1!,* по (?-кантам 40 полос (г/,
v" ^ 16). Они хорошо согласуются с результатами анализа различных систем полос в
работах [2107, 2354, 2391]. Вращательные постоянные в состоянии Х*?+ приняты по работе Вайса
и др. [2874], которые исследовали вращательные переходы с / ^ 14, i;^4b микроволновом
спектре A1F. Они определялись также в результате анализа тонкой структуры полос различных
систем [599, 600, 1759, 2110], а также при изучении микроволнового спектра в работах [1452,
1860, 1862]. Колебательные постоянные A1F в состоянии А1П приняты по данным Барроу
и др. [599], вращательные постоянные — по результатам исследований систем С1Е+—АгП,
-ОХД— Am, Fm—Am, GlL+—Am, проведенных Ноде и Хьюго [2106, 2108]. Колебательные и
вращательные постоянные состояния а3П найдены Коппа с соавторами [1759], состояния &3Е+ —
в работе Барроу и др. [600].
Термодинамические функции A1F (г) вычислены по уравнениям A. 3)—A.10), A. 93)—A.95).
Значения QBn и ее производных по температуре рассчитывались по уравнениям A. 90)—A. 92)
с учетом трех возбужденных состояний в предположении, что (?(*>. вр = [р^Рх) Qif*. вР- Величина
Qxo2.*t Для основного состояния ХХЕ+ и ее производные по температуре вычислялись по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью уравнения типа A. 82). В расчетах
учитывались все уровни энергии со значениями / ^/maI, „, где /шах> „ находились из условий A. 81).
Колебательно-вращательные уровни состояния Х2Е+ вычислялись по уравнениям A. 65), A. 62).
Значения коэффициентов Ykl, а также величин ушах и /Ит, использованные в расчетах, приведены
в табл. 21.15. Коэффициенты Y30, Yi0 и Уб0 определялись из условий A. 50). Мультиплетность и
вырождение вращательных уровней возбужденных состояний учитывались статистическими
весами.
Молекулярные постоянные A1F в состоянии XXS+ известны с хорошей точностью, и при
температурах до 3000—4000 К погрешности в термодинамических функциях A1F (г) определяются
лишь неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль~1. При более
высоких температурах начинает сказываться отсутствие экспериментальных данных об энергии
высоких колебательно-вращательных уровней основного состояния. Однако соответствующие
погрешности невелики и не превышают 0,05 Дж-К~1-моль~1 в значении Ф° F000 К).
Ранее термодинамические функции A1F (г) вычислялись в предыдущих изданиях настоящего
справочника [105, 106], в справочнике JANAF [1544], справочных изданиях [977, 1904, 1974,
2805] и работе [499]. За исключением [2805] (Т <! 5000 К), все расчеты выполнены до 6000 К.
Наилучшее согласие данных табл. 630 имеет место с расчетом [105], в котором учтено состояние
а*П. и применен модифицированный метод Гордона—Барнес; соответствующие расхождения не
122
Алюминий и его соединения
AlFs (г)
превышают 0,04 Дж-К-моль в значениях Ф° (Т) и S0 (Г). Расхождения с данными
американских справочных изданий [977, 1904, 1974, 2805], в которых не учитывалось состояние а3П и
использовались более грубые методы расчета, достигают 0,25, 1 и 3,5 Дж-К-моль~1 в значениях
Ф° (Т), S° (Т) и С°р (Т) при высоких температурах; расхождения с таблицами [1544],
обусловленные практически только использованием в JANAF приближенного метода расчета, не превышают
0,2, 0,5 и 0,6 Дж-К-моль~1 в значениях этих функций.
Константа равновесия реакции A1F (г)=А1 (r)-f-F (г) вычислена с использованием значения
дгЯ° @)=D0 (A1F)=670,021 +5,0 кДж-моль-
в справочнике энтальпии образования
000 +400 см, соответствующего принятой
A//°(A1F, г, 0) = —265,4+|3,0 кДж-моль.
Эта величина основана на приведенных в табл. 21.19 результатах исследований равновесия
реакции V3 A1F3 (к)+2/3А1 (к, ж)=А№ (г). Приведенные в табл. 21.19 погрешности включают
воспроизводимость измерений, неточность термодинамических функций и погрешность энтальпии
Таблица 21.19. Результаты определений А/Н° (AIF, г, 0) (в кДж-моль)
Авторы
Гросс и др., 1948 [1275]
Блекбёрн, 1955 (см. [1544])
Баймаков, 1957 [26]
Уитт, Барроу, 1959 [2864]
Портер, 1960 [2252]
Семенкович, 1960 [376]
Хилденбранд, 1963 (см. [1544])
Мацусима и др., 1964 [1968]
Лайд, 1965 [1862]
Ко и др., 1965 [1733]
Хилденбранд, Серд, 1965 [1425]
Метод
Статический с отбором проб, 1193 К, 1 точка
Эффузионный, 828—1008 К, 12 точек
Точек кипения, 1287—1349 К, 5 точек
Торзионный, 838—922 К а
Эффузионный, 933 К, 1 точка
Переноса, 1100—1500 К6
Торзионный, 882—931 К, 8 точек
То же, 856—932 К, 8 точек
То же, 866—931 К, 8 точек
То же, 867—927 К, 6 точек
ДТА, 1250—1330 К, 23 точки
Оценка по температурной границе появления
микроволнового спектра A1F
Переноса, 1199—1348 К, 10 точек
Масс-сиектрометрический, BeF2 (г)+2А1 (к)=
-=Ве (k)+2A1F (г), 819-916 К, 8 точек
То же, торзионный, 897—926 К, три серии, 25 точек
а Результаты измерений представлены в виде графика.
6 Результаты измерений представлены в виде уравнения.
II закон
_
—309 ±20
—
—230
—
—266
—277 ±8
—264 ±6
—262±2
—264±6
—263
—
—253
—268+11
—268+30
A1F, г, 0)
III закон
—253
—269 ±4
—252+11
—260,6
—262,9
—265,2 4-3,0
—266,7±2,4
—265,4+2,4
—265,6+2,0
—265,9+2,0
—262,9±15
—261 ±8
—260,6+4,0
—
—273+13
образования A1F3 (к). Как видно из таблицы, результаты измерений совпадают в пределах их
погрешностей. Наиболее полные исследования проведены Хилденбрандом в 1963 г. (результаты
этой работы известны только по справочнику [1544]). В хорошем согласии с ними находятся
измерения, выполненные Семенковичем [376]. Для этих двух работ характерно хорошее соответствие
расчетов по II и III законам. Несколько меньшие величины получены в работах Мацусима
и др. [1968] и Ко и др. [1733]. Принятая величина является средневзвешенной из результатов
этих четырех работ.
Относительно короткая экстраполяция (примерно на 6000 см) колебательных уровней
состояния А1!!, проведенная Барроу и др. [599], приводит к значению Do (A1F)=58 400+1500 см.
Расхождения между этой величиной и принятым значением позволяют предполагать наличие
максимума на потенциальной кривой состояния АХИ с высотой 2400+1600 см.
AIF2 (г). Термодинамические свойства газообразного дифторида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 631.
123
AlFj (г) Глава 2t,
Спектр и структура молекулы A1F2 экспериментально не исследовались. Молекулярные
постоянные A1F2, приведенные в табл. 21.16, оценены при подготовке настоящего справочника.
По аналогии с другими молекулами дигалогенидов элементов подгруппы IIIA, а также принимая
во внимание результаты теоретического расчета Хасти и Маргрейва [1367], в справочнике
принимается, что в основном электронном состоянии Х2Аг молекула A1F2 имеет нелинейную
симметричную структуру (симметрия С2„). Произведение главных моментов инерции, приведенное
в табл. 21.16, вычислено по структурным параметрам г (А1—F)=1,63+0,03 А и 2$. F—Al—F=
=120+10°, принятым по аналогии с соответствующими параметрами A1F3. Погрешность значения
IaIbIc составляет 1-1015 г3-см6. Приведенные в табл. 21.16 частоты колебаний A1F2 вычислены
по силовым постоянным A1F3 х; их погрешности могут достигать 10—15%. Энергия возбуждения
состояния Л2Вг оценена по результатам теоретического расчета [1367], скорректированным
сравнением рассчитанных и измеренных энергий возбуждения двухатомных молекул галогенидов А1
и В. Погрешность значения То (Л25Х), приведенного в табл. 21.16, оценивается в 5000 см.
Термодинамические функции A1F2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом вклада
возбужденного электронного состояния A2BX по уравнениям A. 168)—A. 170). Погрешности:
термодинамических функций составляют 1, 3 и 5 Дж-К~1-моль~1 в значениях Ф° (Т) при 298,15,
3000 и 6000 К. При Т ^ 2000 К они обусловлены неточностью оцененных значений молекулярных
постоянных, при более высоких температурах сказывается приближенный характер расчета.
Термодинамические функции A1F2 (г) ранее вычислялись в предыдущих изданиях
справочника [105, 106] и таблицах JANAF [1544], справочниках [977, 1904], а также работе Уилкинса
и др. [2855] (Т <^ 5000 К). Все расчеты выполнены по оцененным молекулярным постоянным в,
за исключением [1544], с учетом только основного электронного состояния A1F2. Расхождения
в значениях Ф° (Т), приведенных в табл. 631 и вычисленных в [105, 1544, 1904], не превосходят
соответственно 1,1, 0,4 и 0,2 Дж-К~1-моль в значениях Ф° (Т). В расчете [2855], очевидно,
допущена ошибка, так как при использовании близких молекулярных постоянных расхождения
достигают 6 Дж-К-моль в значениях Ф° (Т).
Константа равновесия реакции A1F2 (г)=А1 (r)+2F (г) вычислена с использованием значения
Д^Г3 @)=1151,896+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(AlF2, г, 0) = —670 + 40 кДж • моль.
Результаты измерений энтальпии образования A1F2 (г) представлены в табл. 21.20.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неопределенность в сечениях
ионизации и погрешности термодинамических функций. Полученные в работах [1052, 2748]
значения энтальпии образования представляются ошибочными. Если Д^0 (A1F2, г) < —750 кДжХ
Хмоль, то в условиях масс-спектрометрических измерений Портера [2252] при 933 К
парциальное давление A1F2 должно быть почти на два порядка больше, чем парциальное давление A1F,.
а в работе [2252] показано, что оно существенно меньше парциального давления A1F. Принятая,
величина А^Н° (A1F2) является средней из данных работ [1020, 1021] и величины —680 кДжХ
Хмоль, вычисленной из данных [1052] при подготовке настоящего справочника (см. таблицу)..
Погрешность оценена с учетом противоречивости имеющихся данных.
j (г). Термодинамические свойства газообразного отрицательного иона дифторида алюмж-
ния в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 632.
Спектр и структура A1FJ экспериментально не исследовались. По аналогии с изоэлектронжш
молекулой SiF2 в справочнике принимается, что AlFi" в основном электронном состоянии ХХА^_
имеет нелинейную структуру симметрии С2с с межатомным расстоянием г (Al—F)=1,60+0,06 А
и углом -^ F—Al—F=100+10°. Этим структурным параметрам соответствует произведение
главных моментов инерции, приведенное в табл. 21.16. Его погрешность оценивается в 1-1015 г3-см*.
Приведенные в табл. 21.16 основные частоты оценены на основании сравнения основных частот
молекул A1F2 и SiF2, их погрешности могут достигать 20%. По аналогии с SiF2 можно ожидать, что»
AIF2 имеет стабильное возбужденное состояние а3В1 с энергией 20 000 см или больше.
* /г=4,9, /„=0,3 н /„«=0,17 (в 10» дин-см).
124
Алюминий и его соединения
A1F3 (к)
Таблица 21.20. Результаты определений AfH° (AlFt, г, 0) (в кДж-моль)
Авторы
Элерт, Маргрейв, 1964 [1020,1021]
Фарбер, Харрис, 1971 [1052]
Уй и др., 1972 [2748]
Метод
Электронный удар
Масс-спектрометрический, - 2A1F2 (г)—AIF3 (r)+
+A1F (г), 1243—1301 К (константы равновесия
не приведены, ДГЯ° взято по [1543])
Масс-спектрометрический, 2A1F (г)=А1 (r)+AlF« (г
1408-1639 К, 5 точек
То же, 1598 К, 1 точка
Масс-спектрометрический, A1F2 (r)+AlClt (r)=
=A1F2C1 (г)+А1С1 (г) и A1F2 (г)+А1С1, (г)=
=A1FC12 (r)+AlF(r), 1491 К, по 1 точке
Масс-спектрометрцческий, 2A1F (г)=А1 (г)+
+A1F2 (г), 1453—1675 К, 7 точек
* Результаты измерений зависят от энергии электронов.
* „ Do (AlCU)
получено при подготовке настоящего справочника в предположении, что ^с TXTj^V
DO(A1CI2) DO(A1C13)
или D0(A1Fj) ~ D0(AIF3) -
= 0,718.
II закон
A1F2, г, 0)
III закон
—685±90
—836 ±60
, —765±15
—730±30
Do (A1C1)
— DO(A1F)
—639+16
—787 ±10 а
—751 а
—680 б
—756 ±10
1),/4J
Термодинамические функции A1F2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
* учетом только основного электронного состояния. Погрешности в функциях при температурах
до 3000 К определяются в основном неточностью оценки молекулярных постоянных, при более
высоких температурах становятся существенными погрешности из-за приближенного характера
расчета. Погрешности в Ф°(Г) при 298,15, 3000 и 6000 К составляют 2, 4 и 6 Дж-К~1-моль.
Ранее термодинамические функции AIF2 (г) вычислялись в таблицах JANAF [1544].
Расхождения данных [1544] и табл. 632 составляют около 1 Дж-К~1-моль и связаны с тем, что авторы
[1544] приняли несколько отличающиеся значения постоянных AlFj и учли два
возбужденных электронных состояния с энергиями 25 000 и 40 000 см.
Константа равновесия реакции A1F2 (г)=А1 (r)+2F (т)-\-е (г) вычислена с использованием
значения ДДГ° @)=1289,896 ±45 кДж-моль, соответствующего принятому в справочнике
сродству к электрону
Ао (A1F2) =—138+20 кДж-моль.
Эта величина основана на масс-спектрометрическом 'исследовании равновесия реакции
AlFj (r)+F" (r)=F (r)+AlF2i(r) A705—1900 К, 3 точки), выполненном Сривастава и др. [2600].
Вычисленная из этих данных энтальпия реакции A12,8 ±5,0 кДж'Моль, расчет по III закону)
равна разности сродства к электрону A1F2 и F. Значительно менее точное исследование реакции
A1F, (г)-(-е (r)=AlF2 (r)+F (г) методом электронного удара, выполненное Петти и др. [2221],
приводит к значению AQ (A1F2) ^ —83 кДж-моль, которое не противоречит результатам
исследования авторов [2600]. Погрешность принятого значения увеличена по сравнению с
оценкой в [2600], ему соответствует
A^^AIF", г, 0) = —808 ±540 кДж ¦ моль.
A1FS (к). Термодинамические свойства кристаллического трифторида алюминия в стандартном
¦состоянии при температурах 100—2100 К приведены в табл. 633.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние A1F3 (к) в интервале 0—728 К принимается
гексагональная (ромбоэдрическая) модификация со слоистой структурой, а при Т >• 728 К —
кубическая модификация. Термодинамические функции A1F3 (к) при Т •< 298,15 К вычислены на
основании данных о теплоемкости, полученных Кингом [1711] E4—296 К) на образце чистотой
125
A1F3 (к)
Глава 2t
99,85% A1F3. Экстраполяция теплоемкости к 7=0 дала 5° E1 К)=2,34 Дж-К^-моль [1711].
Погрешности принятых по [1711] значений S° B98,15 К) и Н° B98,15 К)—Я0 @) оцениваются
в 0,4 Дж-К-моль~1 и 0,04 кДж-моль соответственно. В интервале 298,15—728 К для
теплоемкости A1F3 (к) принято уравнение (см. табл. 21.1), выведенное Дугласом и Дитмарсом [978]
по результатам 40 измерений энтальпии образца A1F3 чистоты 99,91% с точностью ~0,1%.
Измерения энтальпии A1F3(k), выполненные в работах [2152] D01 — 1401 К) и [229] D40—1060 К),
менее точны и не учитывались. Температура G28+1 К) и энтальпия @,565+0,050 кДж-моль)
полиморфного превращения приняты на основании измерений [978]. В работе [2152] найдено
близкое значение температуры превращения G27 К); данные других авторов [891, 1131, 2676]
менее надежны и приводят к значениям Ttr в пределах 713—733 К. Для теплоемкости
высокотемпературной модификации A1F3 принято уравнение (см. табл. 21.1), выведенное по результатам
18 измерений энтальпии в интервале 728—1123 К [978]. Точность аппроксимации значений
теплоемкости этим уравнением оценивается в ~1%. Результаты измерений [2152 и 229] менее точны,
хотя в пределах их погрешностей не противоречат данным [978]. Термодинамические функции
A1F3 (к) вычислялись до температуры 2100 К, при которой давление пара A1F3 превышает 100 атм.
Температура плавления (тройная точка A1F3), вычисленная экстраполяцией данных по диаграмме
плавкости в системе A1F3—NaF3, равна ~2500 К при давлении 3-104 атм [2029г]. Энтальпия
плавления A1F3 A12 кДж-моль) вычислена Хонгом и Клеппой [1475] по результатам
термохимических измерений энтальпий смешения A1F3 (к) с Li3AlF6 (ж) и NasAlF6 (ж) при ~1300 К *.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 1500 и 2000 К оцениваются
в 0,2, 0,4, 0,8 и 4 Дж-К-моль~1 соответственно.
Термодинамические функции A1F3 (к), приведенные в табл. 633 и таблицах JANAF [1543],
совпадают.
В справочнике принимается энтальпия образования
bfH°(A\F3, к, 298,15 К) = —1510,4 + 1,3 кДж • моль.
Это значение, совпадающее с рекомендацией КО ДАТА—МСНС [864], основано на наиболее
точных измерениях, выполненных Рудзитисом и др. [2402]. С принятым значением в пределах
погрешностей согласуются результаты измерений [170, 967, 1281, 1283, 1401, 1950, 2658],
представленные в табл. 21.21.
Таблица 21.21. Результаты определений &.,Н° (AIF3, к, 298,15 К) (в кДж-мтепГ1)
Авторы
Гросс и др., 1954 [1281, 1283]
Колесов и др., 1961 [170]
Домальский. Армстронг, 1965 [967]
Рудзитис и др., 1967 [24021
Хойес, Эган, 1966 [1401]; Маркин и
др., 1967 [1950]; Танака и др., 1971
[2658]
Метод
Измерение энтальпии реакции 3/2PbF2 (к)+А1 (к)=
= АЩ (к)+8/2РЬ (к)
То же
Измерение энтальпии сгорания алюминия (в смеси
с тефлоном) во фторе
Измерение энтальпии сгорания алюминия во фторе
Измерения ЭДС реакции 1/2М«(к)+1/зА1Рз(к'1=
=VsMgF2 (k)+V3A1 (к), 873 К
а Приведенные погрешности характеризуют воспроизводимость измерений.
Д.,#О(А1Р„ к,
298, 15 К) »
—1510+7
-1507+7
— 1507,9+6,7
-1510,4+1,3
—1507,3 (III)
Давление пара трифторида алюминия A1F3 (k)=A1F3 (г) рассчитано с использованием принятой
в справочнике энтальпии сублимации
Дг#° @) = AsH° (A1F3, к, 0) = 298,0 + 4,0 кДж ¦ моль.
Эта величина основана на результатах измерений давления пара над A1F3 (к), приведенных
в табл. 21.22; погрешности в таблице отражают только воспроизводимость измерений. Неточность
термодинамических функций, использованных в расчетах, приводит к погрешности в энтальпии
1 Данные по Тт ж АтН для A1F3 в расчетах термодинамических функций не использовались.
126
Алюминий и его соединения
A1F3
Таблица 21.22. Результаты определений Д8Я° (AIF3, к, 0) (в кДж-моль)
Авторы
Ольбрпх, 1928 [2158]
Руфф, Ле Буше, 1934 [2407,
Нарышкин, 1939 [270]
Евсеев и др., 1959 [116]
Уитт, Барроу, 1959 [2864]
Ко и др., 1965 [1733]
Ерохип и др., 1967 [126]
Бюхлер и др., 1967 [781]
К раус, Дуглас, 1968 [1772]
Сошш, Поляченок, 1973 [391
Метод
Протока, 1371—1567 К, 9 точек
1832] Эффузионный, 1367—1524 К, 9 точек
Протока, 1108—1273 К, 3 точки
Протока, 980—1123 К, 12 точек
Тораионный, 955 и 1063 К, 2 точки
Протока, 1027—1184 К, 23 точки, данные
приведены в виде уравнения
Масс-спектрометрический, 893—1165 К, данные
представлены в виде уравнения
Масс-спектрометрический, 951—1077 К, по
интенсивности ионного тока
Протока, 1194—1258 К, 8 точек
, 392] Точек кипения, 1328—1464 К, 9 точек
ASH°
II закон
353+30
364 ±30
265+900
337+8
302
200
297
294±5
299,9+1,5
305+18
(A1F,, к, 0)
III закон
302,9+1,5
298,2+2,0
301+14
299,0+1,0
299,0+2,0
298,2+2,0
299,3+3,0
298,0+0,1
298,3 ±0,5
сублимации в 3,5 кДж-моль х. При обработке экспериментальных данных учитывалось
образование молекул AlaFe (их доля составляет 0,2% при 1000 К и 6% при 1500 К). Испарение A1F3
исследовалось также в работах [380в, 1275, 2012, 2567], однако эти измерения менее надежны и, в
частности, не могут быть пересчитаны с учетом сложного состава пара. Принятое в справочнике
значение является средневзвешенным из работ [116, 126, 391, 392, 1733, 1772, 2864]. При этом
наибольший вес придавался работе Крауса и Дугласа [1772], данные которых имеют хорошую
воспроизводимость и согласие результатов обработки по II и III законам.
A1F3 (г). Термодинамические свойства газообразного трифторида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 634.
Молекулярные постоянные, использованные для расчета термодинамических функций A1F3 (г),
приведены в табл. 21.17.На основании результатов электронографического исследования,
выполненного Акишииым и др. [4], и теоретического расчета [874] в справочнике принимается, что
молекула A1F3 в основном электронном состоянии Х1А[ имеет плоскую структуру симметрии Dsh.
Исследования спектров молекулы A1F3 подтверждают эту структуру. Приведенное в табл. 21.17
произведение главных моментов инерции вычислено для межатомного расстояния г (Al—F) =
=1,63+0,01 А, найденного в работе [4]. Погрешность значения IaIWc не превышает 2-1015 г3-см6.
Частоты колебаний v2, v3 и v4, приведенные в табл. 21.17, получены Бюхлером и др. |781] при
исследовании ИК-спектра газообразного A1F3. С принятыми значениями в пределах погрешностей
их определения согласуются частоты, измеренные Мак-Кори и др. [1977] в спектре газообразного
A1F3 и Снелсоном [2565а], Линевским [1868] и Янгом и Шерком [2880] в ИК-спектре молекул
A1F3, изолированных в низкотемпературных матрицах из Ar, Ne, Кг, Хе. Частота \ в спектре
A1F3 не наблюдалась. Приведенное в табл. 21.17 значение рассчитано для поля валентных сил
по силовым постоянным, вычисленным из частот У3 и. v4 и в предположении, что отношение frr/fr
в A1F3 такое же, как в молекуле А1С13. Значения vl5 близкие к принятому, оценены в работах
[2565а, 390] Ч Погрешности в значениях v2—v4 оцениваются в 10—20 см", погрешность в \
может достигать 10%. Сведения о возбужденных электронных состояниях A1F3 в литературе
отсутствуют, но можно предполагать, что их энергия превышает 25 000 см.
Термодинамические функции A1F3 (г) рассчитаны в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности вычисленных таким образом
термодинамических функций составляют 2, 3 и 5 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000 и
6000 К и обусловлены в основном приближенным характером расчета; погрешности из-за
неточности молекулярных постоянных не превышают 1 Дж-К"'1-моль.
1 Бюхлер и др. [781] получили в результате оценки явно завышенпое значение Vj=800 см х.
127
A1F4~ (г) Глава 21
Ранее термодинамические функции A1F3 (г) рассчитывались в предыдущих изданиях
настоящего справочника [105, 106], в справочниках JANAF [1543], Мак-Брайд и др. [1974], Мадера
[1904], Дугласа и Бекетта [977] и работе Уилкинса и др. [2855] (Г <; 5000 К). Все расчеты
выполнены по оцененным значениям молекулярных постоянных (за исключением расчета [1543]).
Расхождения в функциях, приведенных в табл. 634 и вычисленных в [105, 106, 977, 1904, 1974,
2855], составляют 1—3,3 Дж- К~1-моль в значениях Фс (Г) и обусловлены различием в принятых
молекулярных постоянных. Расхождения с функциями, вычисленными в [1543], не превышают
0,5 Дж'К-моль~1 в значениях Ф° (Г) и объясняются тем, что в [1543] для частоты \ принято
более низкое значение.
Константа равновесия реакции A1F3 (г)=А1 (r)+3F (г) вычислена с использованием значения
Д^0 @)=1765,389+5,8 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации A1F3 (к). Этим величинам также соответствует
AfH°\(AlFs, г, 0) = —1206,218 + 4,2 кДж моль.
A1F" (г). Термодинамические свойства газообразного отрицательного иона тетрафторида
алюминия в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 635.
Спектр и структура свободного иона AlFjj экспериментально не исследовались. Молекулярные
постоянные, использованные для расчета термодинамических функций и приведенные в табл. 21.17,
ОЦенеНЫ» По анаЛОГИИ С BFJ И ИЗОЭЛеКТрОННОЙ МОЛекуЛвЙ SiF4 в справочнике принимается, что
A1FJ в основном электронном состоянии Х1Аг имеет структуру правильного тетраэдра
(симметрия Td). Произведение главных моментов инерции, приведенное в табл. 21.17, соответствует
межатомному расстоянию г (А1—F) = 1,69+0,05 А. Это значение получено Спиридоновым и Ерохиным
[396] и Вайда и др. [2749] в результате электронографического исследования структуры
газообразных NaAlF4 и KA1F4 соответственно. Авторы [396, 2749] нашли, что в указанных молекулах
межатомные расстояния г (А1—FBHeiIIH) и г (А1—FM00T) равны между собой и, следовательно, тетра-
эдрическая структура аниона A1FJ остается жесткой и не деформируется в результате
взаимодействия с атомами щелочных металлов. Погрешность IaIbIc не превышает 3-1014 г3-см6. Частоты
колебаний AlF^, приведенные в табл. 21.17, являются средними из частот, полученных при
исследовании ИК-спектров поглощения молекул фтор алюминатов щелочных металлов MA1F4 (M=Li,
Na, К, Rb, Cs), изолированных в матрицах благородных газов [915а, 1482а, 1495а] и спектров
КР фторалюминатов, образующихся в расплавах смесей A1F3—MF (M=Na, К) [639, 1199, 2302,
2414]. Частоты, отнесенные к колебаниям иона A1FJ в конденсированном состоянии [639, 1199,
2302, 2414] и в соединениях, изолированных в низкотемпературных матрицах [915а, 1482а, 1495а],
отличаются не более чем на 5 %. Учитывая возможное изменение частот колебаний при переходе
к свободному иону A1FJ, погрешности в значениях v<5 приведенных в табл. 21.17, составляют 10%
¦от их величин. По аналогии с изоэлектронной молекулой SiF4 в справочнике принимается, что
A1FJ не имеет электронных состояний с низкими энергиями возбуждения.
Термодинамические функции AlFj (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130).
Погрешности функций определяются неточностью принятых молекулярных постоянных и приближенным
характером расчета; при высоких температурах преобладают последние. Погрешности в
значениях Ф° (Г) составляют 2, 7 и 10 Дж-К-^моль при 298,15, 3000 и 6000 К.
Ранее термодинамические функции A1FJ вычисляли авторы справочника JANAF [1544].
Расхождения в функциях, вычисленных в [1544] и приведенных в табл. 635, не превосходят 0,5 ДжХ
хК~1-моль~1 в значениях Ф° (Т).
Константа равновесия реакции A1FJ" (г)=А1 (r)+4F (т)-\-е (г) вычислена с использованием
значения Д^" @) =2566,446 +40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —1930 + 40 кДж • моль.
Эта величина основана на масс-спектрометрических измерениях Гусарова и др. 1107а] и Сри-
вастава и др. [2600]. В работе[107а ] при 1020 К измерены константы равновесия реакций AlFi" (r)-f-
+2KF (r)=KF2 (r)+KAlF4 (г) и KFa (r)=KF (k)+F" (г). Комбинируя эти данные и используя
АД° (KF, к, 0)=242 кДж-моль, для реакции AlFjf (r)+KF (r)=KAlF4 (r)+F~ (г) получено
@)=127кДж-моль. Этой величине соответствует Ду#° (A1FJ, г, 0) =—1925+40кДж-мояь~1
128
Алюминий и его соединения
AI,F, (г)
(в расчете использованы значения термодинамических функций и AyfT°L(KAlF4l г, 0) =—1873 кДжХ
Хмоль, принятые авторами [107а] по литературным данным).
Сриваставаидр. [2600] измерили константу равновесия реакпиж2А1Р2 (r)+AlF2 (г)=2А1Р (г)+
+A1FJ (г) в интервале 1130—1485 К, 7 точек. Отсюда по уравнению III закона найдено ?±,Л° @) =
=—160+60 кДж-моль, чему соответствует Д^Я0 (AlFj, г, 0) =—1780+120 кДж-моль.
Основным источником погрешности этой величины является низкая точность использованного в расчете
значения Д^Я0 (A1F2, г, 0). Однако можно провести расчет энтальпии образования AlFi по данным
[2600], используя результаты исследования реакции 2A1F (г)=А1 (r)+AlFa (г), Д^0 @)=102+_
+10 кДж-моль, выполненного этими же авторами в работе [2748] (см. текст но A1F2).
Комбинация этих данных дает для реакции 2A1F (г) + AlFi" (г)=2А1 (r)+AlFj (г) величину Д^0 @) =
=44+60 кДж'Моль, чему соответствует Д^Я° (AlFj, г, 0)=—1954+70 кДж-моль. Возможно,
что в работах [2600, 2748] имеются какие-то систематические ошибки, взаимно компенсирующие
друг друга, что приводит к хорошему,-, огласию значений ДЛ70 (A1FJ, г), вычисленных по данным
1107а] и [2600, 2748]. 2
Al2Fe (г). Термодинамические свойства газообразного гексафторида диалюминия в стандартном
состоянии в интервале 100—6000 К приведены в табл. 636.
Молекулярные постоянные, использованные для расчета термодинамических функций AlaFe,
приведены в табл. 21.23. Экспериментальные данные о структурных параметрах A12F6 в литературе
Таблица 21.23. Значения молекулярных постоянных, принятые для расчета термодинамических функций
A12F,, А12С16, А12Вг„ и А121в в основной электронном состоянии Х1А.д (о=4, p%=i)
Молекула
A12F6
А1гСЦ
Al2Bre
Al2Ie
ух
830
523
410
340
600
336
203
145
370
217
139
93
V4
200
99
59
42
90
36
25
20
630
438
350
310
210
165
114
82
v8
995
625
530
415
340
230
140
90
vt.
см
60
19
18
13
v»
970
612
485
405
va
260
117
76
54
""is
600
420
350
291
Yu
230
142
90
64
VI.
150
77
70
54
VI.
805
484
387
320
v»
575
320
198
140
VI.
300
176
111
81
lAhl
rs.
3,0-
5,7.
8,9
5,9
3M»
10*
10*
• 10»
10"
отсутствуют. Произведение моментов инерции Al2Fe вычислено для мостиковой модели симметрии
Du с r(Al-FBHeinH)=l,63±0,05 А, г (Al-F^)=1,80 ±0,05 А, ^ FBHeHIII-Al-FMiemH=120±10ol
-? FMoot —Al—FMOOI=80±10°. Значения межатомного расстояния г (А1—FmemB) и угла -? Fmtm—
—Al—Fmmn перенесены из молекулы A1F8. Значение г (Al—FM0CI) принято, исходя из соотношения
г (А1—Хвнвшя)/г (А1—Хмоот) и разности г (А1—ХИОС1)—г (А1—Хвжешн), имеющих достаточно
постоянные величины в ряду молекул А12С1в, А12Вгв и А121в. Значение угла -^ FU0llT—Al—Fx0eT оценено
сравнением с аналогичным углом в молекуле A12F2 (см. [388]) г. Погрешность IaIbIc составляет
1'10~ш г3-см6. В табл. 21.23 для частот колебаний, активных в ИК-спектрах (vg, v9) v13) v16, v17,
v18), приняты величины, предложенные Снелсоном [2565а] на основании результатов исследования
ИК-спектра A12F6 в Ne-, Ar- и Kr-матрицах. Для остальных частот приведены значения,
рекомендованные в справочнике JANAF [1543]. Эти частоты оценены по частотам молекул А12С16, А1аВгв,
А121в, причем для проверки правильности оценки использовано сравнение вычисленного значения
энтропии с полученным Краусом и Дугласом [1772] в результате калориметрических измерений 2.
Погрешности принятых значений частот составляют 100 см в vlt v2, vu, 50 см в v8, ve, v8, vUf
25 см в v4, v7, v9> v12— v15, v17, 15 см в v8, v10 и v18. Сведения о возбужденных электронных
состояниях AlaF6 в литературе отсутствуют.
Термодинамические функции A12F6 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
1 Кертисс [914а] получил в результате квантовомеханического расчета этой молекулы близкие значения
структурных параметров Al8Fe, за исключением величины г (А1—FKOOT)=1,72 A.
2 Сивин [915] рассчитал основные частоты Al2Fe на основании оцененного силового поля молекулы. Однако
рассчитанные им значения не удовлетворяют ожидаемому закономерному изменению частот в ряду молекул
Al2Fe, А1аС1„, А12Вгв и А1а1в.
129
FA10 (г) Глава 2t
A. 130). Погрешности термодинамических функций при низких температурах определяются в
основном неточностью оцененных значений молекулярных постоянных, при увеличении температуры
преобладающими становятся погрешности из-за приближенного характера расчета. Погрешности
в значениях Ф° (Т) при 298,15, 3000, 6000 К составляют 5, 15, 25 Днс-К^-моль.
Ранее термодинамические функции Al2Fe (г) рассчитывались в справочнике JANAF [1543].
Расхождения результатов этого расчета с данными, приведенными в табл. 636, не превышают
0,5 Дж-К^-моль.
Константа равновесия реакции A12F6 (г)=2А1 (r)+6F (г) вычислена с использованием значения
ДГЯ° @)=3735,342+17 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/ff°(AlaFe, г, 0) = —2617 ±15 кДж • моль.
Эта величина основана на масс-спектрометрических исследованиях зависимости интенсивности
ионных токов A1FJ и A1FJ, образующихся из молекулы A1F3, и ионного тока Al2Fg, образующегося
из A12F6. Эти данные позволяют вычислить Do (A1F3—A1F3). Бюхлер [781] провел две серии
измерений в интервале 971—1077 К. В этой работе указаны только значения энтальпии испарения
A1F3 и A12F6, вычисленные по II закону, разность их дает Do=213 кДж-моль. В масс-спектро-
метрическом исследовании Ерохина и др. [126] проведена калибровка аппаратуры методом
изотермического испарения, что позволило вычислить парциальные давления компонентов и
определить энергию диссоциации как по II B15 кДж-моль), так и по III законам B04,3+10 кДжХ
Хмоль). Принятая в справочнике величина основывается на обработке по III закону данных
[126], полученных в интервале температур 893—1165 К. При оценке ее погрешности учтена
неточность термодинамических функций, приводящая к погрешности в энергии диссоциации в 12 кДж X
Хмоль.
FA1O (г). Термодинамические свойства газообразного оксид-фторида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 637.
Молекулярные постоянные, использованные для расчета термодинамических функций FA1O,
приведены в табл. 21.8. Структура молекулы FA1O экспериментально не исследовалась. По
аналогии с другими оксид-галогенидами алюминия и бора в справочнике принимается, что в основном
электронном состоянии X1 2+ молекула FA1O имеет линейную структуру симметрии CaV. С такой
структурой согласуются результаты исследования ИК-спектра молекул FA1O, изолированных
в матрице из Аг [24686]. Приведенное в табл. 21.8 значение главного момента инерции вычислено
для межатомных расстояний г (А1—F)=l,63+0,03 А и г (А1=О)=1,62+0,03 А, принятых
равными соответствующим расстояниям в молекулах A1F3 и АЮ. Погрешность значения / составляет
1-10~39 г-см2. Приведенные в табл. 21.8 основные частоты \ и v3 найдены Шнёкелем при
исследовании ИК-спектра FA1O [24686]. Значение частоты деформационного колебания рассчитано по
оцененной силовой постоянной, полученной в предположении, что у FA1O /ai—f//k^20+5, как
это имеет место для аналогичных постоянных молекул HBO, FBO, С1ВО и ВгВО *. Частоты 368
и 1022 см, наблюдавшиеся Снелсоном [2571 ] в ИК-спектре в матрицах и приписанные им
молекуле FA1O, принадлежат, как показал Шнёкель, молекулам SiF4. Погрешности принятых
значений \ и v3 оцениваются в 10 см, частоты v2 — в 50 см.
Термодинамические функции FA1O (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124) и A. 129) в приближении «жесткий ротатор—гармонический осциллятор». Погрешности
термодинамических функций при температурах до 1000 К обусловлены в основном неточностью
принятых значений молекулярных постоянных, с ростом температуры увеличиваются погрешности
из-за приближенного характера расчета. Погрешности в значениях Ф° (Т) составляют 2, 5 и 7 Дж X
хК-^моль при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций FA1O (г) рассчитывались авторами справочника
JANAF [1544], Мак-Брайд и др. [1974], Мадером [1904], Дугласом и Бекеттом [977], до 5000 К
Галкиным [305] и Уилкинсом и др. [2855]. Все расчеты, за исключением [1544], выполнены по
оцененным молекулярным постоянным. Расхождения данных табл. 637 и справочника JANAF
[1544] составляют 1—1,5 Дж-К-моль в значениях Ф° (Т) и S° (T) и обусловлены различием
принятых в расчетах постоянных FA1O. Вычисленные в [305, 977, 1904, 2855] термодинамические
1 Соответствующие силовые постоянные равны (в 105 дин-см 1): /^j_p=5,01, /A]_O=6,62, /„/^2=0,25.
130
Алюминий в его соединения F2AIO (г)
функции меньше приведенных в табл. 637 на 1—4 Дж-К~1-моль~1 в значениях Ф° (Т) в основном
из-за использования завышенного значения; V
Константа равновесия реакции FA1O (г)=А1 (r)-fF (г)+О (г) вычислена с использованием
значения Д^0 @)=1221,404 +30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
АуНЦ?АЮ, г, 0) = —570 + 30 кДж • моль.
Эта величина основана на результатах измерений, выполненных Фарбером и Петерсеном [1053]
и Фарбером и Сривастава [1056]. В работе [1053] эффузионным методом в условиях молекулярного
потока исследована реакция А12О8 (k)+A1F3 (r)=3FA10 (г) B200—2225 К, 8 точек); результатам
измерений соответствует AjH° (FA1O, г, 0) =—586+10 кДж-моль (III закон). При масс-спектро-
метрическом исследовании в пламенах [1056] изучены реакции A1F (г)+АЮ (r)=FA10 (г)+
+А1 (г) B250 К, 1 точка, AfH° (FA1O, г, 0) =—568+20 кДж-моль, III закон) и A1F (г)+Н2О (г) =
=FA1O (г)+Н2 (г) B250 К, 1 точка, AfH° (FA1O, г, 0) =—554+20 кДж-моль"\ III закон).
Принятое значение является средним из данных этих работ. Сривастава и Фарбер [2596] провели
также масс-спектрометрическое исследование продуктов реакции A1F (г)+А12О (r)=FA10 (г)+
-{-2А1 (г) A540—1923 К, 8 точек). В этой работе получены только величины, пропорциональные
константе равновесия. При расчете по уравнению II закона они приводят к значению АуН" (FA1O,
г, 0)==—635+35 кДж-моль, которое существенно отличается от принятого.
F2A1O (г). Термодинамические свойства газообразного оксид-дифторида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 638.
Молекулярные постоянные, использованные для расчета термодинамических функций F2A1O,
приведены в табл. 21.11. В литературе отсутствуют экспериментальные данные о спектре и
структуре молекулы F2A1O. По аналогии с F2BO в справочнике принимается, что в основном
электронном состоянии %2В2 молекула F2A1O имеет плоскую структуру симметрии C2v. Приведенное
в табл. 21.11 произведение главных моментов инерции F2A1O вычислено по оцененным структурным
параметрам: г (А1-О)=1,80+0,05 А, г (А1—F)=l,63±0,05 A, ^ F—A1—F=120+10°. Длина
связи А1—F принята такой же, как в молекуле A1F3, а величина г (А1—О) оценена из сравнения
расстояний В—О и А1—О в молекулах ВО, В2О, F2BO и АЮ, А12О. Погрешность в IaIbIc
составляет 1-1014 г3-см6. Частоты колебаний v2—ve F2A1O, приведенные в табл. 21.11, оценены на
основании сравнения с частотами молекулы A1F3, частота \ (А1=О) — на основании сравнения
v (B=O), v (В—F) и v (Al—F) в молекулах F2BO и F2A1O. Погрешности в оцененных таким образом
частотах не превышают 15—20%. Сведения о возбужденных электронных состояниях F2A1O
отсутствуют; по аналогии с молекулой F2BO можно предполагать, что молекула F2A1O имеет
возбужденные электронные состояния с энергией 10 000—20 000 см.
Термодинамические функции F2A1O (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью оцененных молекулярных постоянных и приближенным характером
расчета и составляют 3, 6 и 8 Дж- К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции F2A1O (г) рассчитывали авторы справочника JANAF [1544].
Расхождения в данных табл. 638 и [1544] составляют 3 и 5 Дж-К-моль в значениях Ф° (Т)
и S° (T) и связаны с различием в оценке молекулярных постоянных F2A1O и, в меньшей степени,
тем, что в расчете [1544] учитывались возбужденные состояния fi*Bx (Г0=10 000 см) и В2В
(Г0=20 000 см).
Константа равновесия реакции F2A1O (г)=А1 (r)-f-O (r)+2F (г) вычислена с использованием
значения Д^0 @)=1828,679+50 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH°(F2A\O, г, 0) = —1100Й+50 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции A1F (г)+
+FA1O (r)=F2A10 (г)+А1 (г), проведенного Уй и др. [2748] с использованием двойной эффузион-
ной камеры. A450—1675 К, 7 точек). Расчет по уравнению III закона дает для энтальпии
образования — 1102+35 кДж-моль'1, по II закону 1078+35 кДж-моль.
131
AlHF(r), AlHgF (г) Глава 21
A1HF (г). Термодинамические свойства газообразного гидрид-фторида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 639.
Спектр и структура A1HF экспериментально не изучались. Приведенные в табл. 21.16
молекулярные постоянные, использованные для расчета термодинамических функций A1HF (г), оценены
при подготовке настоящего справочника. По аналогии с симметричными молекулами А1Х2, а также
учитывая теоретические предсказания Уолша [2793], принимается, что молекула A1HF в основном
электронном состоянии Ё%А' имеет нелинейную структуру с углом -^ Н—А1—F=120+10°
(точечная группа симметрии С,). Приведенное в табл. 21.16 произведение главных моментов инерции
вычислено, исходя из значений для межатомных расстояний г (А1—Н)=1,59 +0,03 А и г (Al—F) =
=1,63+0,05 А, равных соответствующим величинам в симметричных молекулах А1Х2 и А1Х3
(Х=Н, F). Погрешность значения IaIbIg оценивается в 3-10~ш г3-см6.
Частоты колебаний, приведенные в табл. 21.16, вычислены по силовым постоянным, принятым
близкими силовым постоянным симметричных молекул А1Х2 г. Погрешности в оцененных
значениях основных частот не превышают 10—15%. По аналогии с трехатомными молекулами дигалоге-
нидов и дигидридов элементов подгруппы IIIA можно ожидать, что молекулы типа A1XY, где
X, Y=H, F, С1, должны иметь стабильные возбужденные электронные состояния со сравнительно
невысокой энергией возбуждения. Квантовомеханический расчет электронных состояний BHF,
выполненный Бротчи и Томпсоном [767], привел к значению То (Л2А"), близкому к среднему из
энергий возбуждения состояний А2ВХ у молекул ВН2 и BF2 (см. табл. 20.13). Значение То (А2А")
для A1HF, приведенное в табл. 21.16, оценено в этом же предположении; его погрешность может
достигать 6000 см.
Термодинамические функции A1HF (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
вклада возбужденного электронного состояния А2А" по уравнениям A. 168)—A. 170).
Погрешности термодинамических функций обусловлены неточностью оцененных молекулярных
постоянных и приближенным характером расчета. Погрешности в значениях Ф° (Т) составляют 1, 3 и
5 Дж-К^-моль при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1HF (г) публикуется впервые.
Константа равновесия реакции A1HF (г)=А1 (г)+Н (r)+F (г) вычислена с использованием
значения ДГЯ° @) =830,655+40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH° (A1HF, г, 0) = —210 ± 40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
A1HF, А1Н2 и A1F,.
A1H2F (г). Термодинамические свойства газообразного дигидрид-фторида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 640.
Спектр и структура молекулы A1H2F экспериментально не исследовались. Молекулярные
постоянные, использованные для расчета термодинамических функций A1H2F (см. табл. 21.17)
оценены при подготовке настоящего справочника. По аналогии с симметричными молекулами А1Х3
принимается, что в основном электронном состоянии Х1А1 молекула A1H2F имеет плоскую
структуру с углами ^ Н—А1—Н=^ H—Al—F=120+10° (точечная группа симметрии С2(]) и
межатомными расстояниями, равными соответствующим расстояниям в молекулах А1Н3 и A1F3: г (А1—Н) =
=1,59+0,03 А и г (Al—F)=l,63+0,05 А. Этим структурным параметрам соответствует
произведение главных моментов инерции, приведенное в табл. 21.17. Его погрешность оценивается
в 5-Ю17 г3-см6. Приведенные в табл. 21.17 частоты колебаний оценены из сравнения значений
частот А1Н3, A1F3, A1C13 и АШС12. Погрешности частот va, v5 и ve оцениваются в 100 см, частот
\, v3 и v4 — в 200 см.
Термодинамические функции A1H2F (г) рассчитаны в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности рассчитанных величин обусловлены до
2000 К в основном отсутствием экспериментальных данных об основных частотах и структурных
1 /ai-f=5'0' /ai-h=2'1- />1Г2=О,19 (в 106 дин-см).
132
Алюминий и его соединения AlHF2(r), FA10H (г)
параметрах молекулы AlHaF; при более высоких температурах основным источником ошибок
является приближенная методика расчета. Погрешности в значениях Ф° (Т) составляют 1, 5 и 7 ДжХ
хК^-моль при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1H2F публикуется впервые.
Константа равновесия реакции A1H2F (г)=А1 (г)+2Н (r)+F (г) вычислена с использованием
значения ДДГ0 @)=1096,689 +40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я° (AlHaF, г, 0) = —260 + 40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах A1H2F,
АШ3 и AJF,.
A1HF2 (г). Термодинамические свойства газообразного гидрид-дифторида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 641.
Спектр и структура молекулы A1HF2 экспериментально не исследовались. Молекулярные
постоянные A1HF2, приведенные в табл. 21.17 и использованные для расчета термодинамических
функций, оценены таким же образом, как постоянные A1H2F (см. выше). Произведение главных
моментов инерции в электронном состоянии Х1А1 соответствует структурным параметрам г (А1—
— Н)=1,59±0,03А, r(Al—F) = l,63+0,05A и-$Н-А1—F=-3F—Al-F=120±10°. Погрешность
значения IaIbIo составляет 1-1015 г3-см6. Погрешности в принятых значениях основных частот
оцениваются в 50 см для v3 и ve, 100 см для \, \ и v6 и 200 см для v2.
Термодинамические функции A1HF2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности в их значениях ниже 1000 К обусловлены
в основном неточностью оцененных молекулярных постоянных, а при более высоких
температурах— применением приближенного метода расчета. Они составляют 2, 5 и 8 Дж-К-моль~1
в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1HF2 (г) публикуется впервые.
Константа равновесия реакции A1HF2 (г)=А1 (г)+Н (r)+2F (г) вычислена с использованием
значения А^0 @)=1427,930+25 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH°(AmF2, г, 0) = —730 ±25 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах AlHFa,
АШ3 и A1F3.
FA1OH (г). Термодинамические свойства газообразного гидроксид-фторида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 642.
Молекулярные постоянные, использованные для расчета термодинамических функций FA1OH,
приведены в табл. 21.11. Экспериментальные данные о спектре и структуре молекулы FA1OH
в литературе отсутствуют. Произведение моментов инерции в электронном состоянии %2А'
вычислено для плоской молекулы симметрии С, с -? F—A1—0=120+10°, -? А1—О—Н=1,15+10°
(трансположение), г (А1—F) = l,64+0,05 А, г (А1—О)=1,78+0,05 А, г (О—Н)=0,96+0,02 А.
Все структурные параметры FA1OH оценены сопоставлением с соответствующими структурными
параметрами A1F2 и А1 (ОНJ. Погрешность значения IaIbIo составляет 1 -10~116 г3-см6. Значения
частот v1 = vA1_0, v2 = v0_H и v4 = vA1_o_H перенесены из НАЮ2. Частота v3 = vA1_p принята близкой
к va?-f в A1F2. Частота v5 = vQ_A1_p оценена сравнением соответствующих деформационных частот
A1F2 и А1 (ОНJ. Частота крутильного колебания ОН-группы v6 рассчитана так же, как для F2BOH
(см. соответствующий текст). При этом 7^,,, для FA1OH принято равным 1,13-10~*° г-см2, п=1,
а высота барьера вращения ОН-группы Уо=700+350 см перенесена из F2A1OH. Погрешности
принятых частот vx—v6 оцениваются в 20% от их величин. Молекула FA1OH, так же как молекулы
A1F2 и А1С12 по-видимому, должна иметь возбужденное электронное состояние с энергией 10 000—
20 000 см; ввиду того что все постоянные FA1OH приняты на основании грубых оценок, это
состояние не включено в табл. 21.11.
Термодинамические функции FA1OH (г) вычислены по формулам A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении модели «жесткий ротатор—гармонический осциллятор»;
133
FAl(OHJ(r), F2A10H (г) Глава 21
вклад возбужденных электронных состояний не учитывался. Погрешности вычисленных
термодинамических функций обусловлены отсутствием экспериментальных данных о молекулярных
постоянных этого соединения, включая его структуру, а также приближенным характером расчета;
они составляют 4, 7 и 10 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций FA1OH (г) публикуется впервые.
Константа равновесия реакции FA1OH (г) =А1 (r)+F (г)+О (г)+Н (г) вычислена с
использованием значения Д,Л0 @)=1517,438 +40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —6503+^40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах FA1OH,
A1F2 и А1 (ОН),.
FA1 (ОНJ (г). Термодинамические свойства газообразного дигидроксид-фторида алюминия
в стандартном состоянии в интервале температур 100—6000 К приведены в табл. 643.
Молекулярные постоянные, использованные для расчета термодинамических функций
FA1 (ОНJ, приведены в табл. 21.13. Спектр и структура FA1 (ОНJ экспериментально не
исследовались. Произведение главных моментов инерции в электронном состоянии ХМ.' вычислено для
плоской молекулы симметрии Св со следующими структурными параметрами, перенесенными из
A1F3 и А1 (ОНK: -? О—А1—О = -? F—А1-О=120+10°, *$ А1-О-Н=115+10°, г (А1—F) =
=1,63+0,05 А, г (А1—О)=1,78 +0,05 А, г (О—Н) =0,96+0,02 А. Погрешность значения 1АГв1с
составляет 1-10~114 г3-см6. Частоты v3, v4 валентных колебаний связей О—Н приняты несколько
большими, чем в А1 (ОНK, так как в FA1 (ОНJ водородные связи должны быть слабее. Частоты
v1 = vAailo, v2 = vXci™o, v6 = v0_A1_0, v7 = vlY-o-н, v8 = v\c!™0-h и крутильные частоты vu и v12
перенесены из А1 (ОНK. Частота vg=vA1_P оценена по величине vA1_p в молекулах A1FC12, A1H2F,
A1HFG1. Учитывая близость масс и изоэлектронность группы ОН и атома F, частоты v9=v0_ai-f
и vlo=v3№T приняты примерно такими же, как в молекуле A1F3. Погрешности принятых значений
частот оцениваются в 15—20%.
Термодинамические функции FA1 (ОНJ (г) вычислены по формулам A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осщллятор».
Погрешности вычисленных термодинамических функций обусловлены приближенной оценкой
значений молекулярных постоянных, в том числе структурных параметров FA1 (ОНJ, а также
приближенным характером расчета и составляют 6, 12 и 20 Дж- К- моль в значениях Ф° (Г) при 298,15,
3000 и 6000 К.
Таблица термодинамических функций FA1 (ОНJ (г) публикуется впервые.
Константа равновесия реакции FA1 (ОНJ (г)=А1 (r)+F (г)+20 (г)+2Н (г) вычислена с
использованием значения &ГН° @) =2400,255+70 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
Д7Я°(FA1(OHJ, г, 0) = —1070 ±70 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
FA1 (ОН),, A1F3 и А1 (ОН),.
F2A1OH (г). Термодинамические свойства газообразного гидроксид-дифторида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 644.
Молекулярные постоянные, использованные для расчета термодинамических функций F2A1OH,
приведены в табл. 21.13. Поскольку спектр и структура молекулы F2A1OH экспериментально
не исследовались, все молекулярные постоянные оценены. Произведение главных моментов
инерции в электронном состоянии Х1А' соответствует плоской структуре молекулы симметрии С,
с углами J$ F—А1—F=120+10°, ^ F—А1—0=120+10°, -? А1—О—Н=115+10° (приняты
по аналогии с F2BOH) и межатомными расстояниями г (А1—О)=1,78+0,05 А, г (О—Н)=0,96 +
+0,02 А (перенесены из А1(ОНK) и г (А1—F)=l,63+0,05 А (перенесено из A1F3). Погрешность
значения 1А1в1о составляет 1 • 10~1М г3 • см6. Значения основных частот v1 = vA1_0, va = v0_H
и vs — vai-o-h перенесены из НАЮ2 и АЮН. Частоты v3 = va7-f, \ = vaT-f, v6 = vF_A1_0, v7=vF_Al_p>
134
Алюминий и его соединения А1С1 (г)
v8=v3oht оценены сравнением с соответствующими частотами A1F3 и А1@НK. Частота
крутильного колебания ОН группы v9 оценена так же, как в F2BOH (см. соответствующий текст), причем
высота барьера вращения F0=700+350 см принята на основании значений Vo, рассчитанных
для НоВОН E040 см [9543, 5740 см [1272]), F2BOH B400+200 см [1842]) и Н2АЮН
A520 см [1273]). Приведенный момент инерции равен 1,33-10~40 г-см2. Погрешности принятых
значений частот оцениваются в 15—20%.
Термодинамические функции F2A1OH (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
значений молекулярных постоянных, в частности структурных параметров F2A1OH, а также
приближенным характером расчета; они составляют 6, 10 и 15 Дж-К-моль~1 в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Таблица термодинамических функций F2A1OH (г) публикуется впервые.
Константа равновесия реакции F2A1OH (г)=А1 (r)-f-2F (r)+O (г)+Н (г) вычислена с
использованием значения Дг//° @) =2079,713+40 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
Д^ЕГ^АЮН, г, 0) = —1135 + 40 кДж.моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
F2A1OH, A1F3 и А1(ОНK.
AIC1 (г). Термодинамические свойства газообразного хлорида алюминия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 645.
Молекулярные постоянные A1G1, принятые в справочнике на основании анализа результатов
исследований электронного и вращательного спектров этой молекулы, приведены в табл. 21.14.
В электронном спектре A1G1 наблюдались переходы между состояниями АгИ <^ Х1^* [680, 8406,
1468, 1472, 1473, 1569, 1918], а3И -+ Хг%+ и 63? -> а3П [2502], а также шесть систем полос,
расположенных в вакуумном ультрафиолете и, по-видимому, связанных с переходами из ридбергов-
ских электронных состояний А1С1 в основное состояние [2316]. Поскольку энергии возбуждения
этих состояний превышают 50 000 см [2316], они не рассматриваются в справочнике.
Аналогично другим галогенидам элементов группы IIIA основное состояние X1S+ и состояния
й3П ii'11 молекулы A1G1 коррелируют с атомными состояниями А1BР)+С1 B.Ps/2).
Колебательные постоянные А1С1 в состояниях XXS+ и А1!!, найденные в работах [680, 1472,
1473, 1918], хорошо согласуются между собой. Принятые значения этих постоянных А135С1
получены Бхадури и Фаулером [680] в результате анализа колебательной структуры системы А1!!—
Х1?+ по (J-кантам (i/ ^ 9, у" ^ 13). Вращательные постоянные А135С1 в состоянии Хг?+,
приведенные в табл. 21.14, получены Вайсом и Горди [2873] при исследовании в микроволновом
спектре этой молекулы вращательных переходов, соответствующих значениям / ^ 19, v ^ 8.
Они хорошо согласуются как с результатами исследования Лайда [1862], так и с данными,
полученными из анализа вращательной структуры ряда полос системы А1!!—Хх?+ [8406, 1472, 1473].
Вращательные постоянные в состоянии Л 41 приняты по работам Холста [1468, 1473]. Принятые
значения колебательных и вращательных постоянных А1С1 в состояниях а3П и Ь3Е найдены
Шарма [2502] на основании исследований систем полос а3П—ХХ2+ и 63S—а3П и дополнительных
оценок.
Термодинамические функции А1С1 (г) вычислялись по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения (?вя и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом
трех возбужденных состояний в предположении, что (?<?>. вр = (pjpx) Qif]. BV- Статистическая сумма
по колебательно-вращательным уровням состояния Хг2+ и ее производные вычислялись по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82). В
расчетах учитывались все уровни энергии со значениями / ^ /шах, „> где /Шах, „ находились из
условия A. 81);.соответствующие значения vmai и /Пш приведены в табл. 21.15.
Колебательно-вращательные уровни состояния Х1^* вычислялись по уравнениям A. 65), A. 62). Значения
коэффициентов Ykl в этих уравнениях для изотопической модификации молекул А1С1, соответствующей
природной смеси изотопов хлора, полученные по отношениям A. 66) и постоянным из табл. 21.14,
135
A1CI (г)
Глава 2f
приведены в табл. 21.15; коэффициенты Ys0, Yie определялись с учетом условий A. 50). Мульти-
плетность и вырождение вращательных уровней возбужденных электронных состояний
учитывались соответствующими статистическими весами.
Молекулярные постоянные А1С1 в состоянии X1 S+ известны с хорошей точностью, поэтому
погрешности рассчитанных термодинамических функций при Т ^ 3 000 К обусловлены лишь
неточностью основных физических постоянных; при более высоких температурах становится
существенным отсутствие экспериментальных данных об энергии высоких
колебательно-вращательных уровней состояния ZXS+. Пренебрежение электронными состояниями с энергиями
свыше 50 000 см практически не влияет на точность рассчитанных величин; погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 0,02, 0,03 и 0,1 Днс-К^-моль.
Ранее термодинамические функции А1С1 (г) вычислялись в предыдущих изданиях
справочника [105, 106], в справочниках [977, 1543, 1904, 1974] и работах [499, 1124, 1235, 1384].
Все расчеты выполнены до 6000 К, за исключением [1124] (Т < 2400 К) и [1384] (Т < 1500 К).
Данные, приведенные в табл. 645 и большинстве указанных справочных изданий, согласуются
в пределах 0,3 и 1 Дж-К-моль в значениях Ф° (Т) и S° (T); имеющиеся расхождения
обусловлены главным образом использованием в этих изданиях приближенных методов расчета. В
справочнике НБС США [977] не учитывались возбужденные электронные состояния А1С1, в связи
с чем расхождения с этим изданием достигают почти 4 Дж-К-моль~1 в значениях Ср F000 К).
Константа равновесия реакции А1С1 (г)=А1 (г)+С1 (г) вычислена с использованием значения
LJS0 @)=D0 (A1C1) =498,166 +5,0 кДж-моль?»^ 640+400 см, соответствующего принятой
в справочнике энтальпии образования
А/Я°(А1а, г, 0) = — 51,2 ±[3,0 кДж моль.
Выбор этой величины основан на исследованиях равновесия реакции 2А1 (ж)+А1С13 (г) =
—ЗА1С1 (г), результаты которых приведены в табл. 21.24. Приведенные в таблице погрешности
учитывают воспроизводимость измерений и погрешность используемого в расчете значения
LjH° (A1C13, г). Неточность термодинамических функций увеличивает погрешность еще на
2 кДж'Моль. Для выбора значения Д^#° (А1С1, г), помимо приведенных в табл. 21.24 данных,
использованы результаты исследования равновесия реакций А1 (ж)+КС1 (к)=А1С1 (г)+К (г)
Таблица 21.24. Результаты
Авторы
определений KfH° (A1C1, г, 0) (в нДж-моль)
Метод
Д/Я°(А1С1, г, 0)
II закон
III закон
Гросс и др., 1948 [1275] Протока, 1295—1275 К, 7 точек — 80±100 —44,1±2,5
Расселл и др., 1951 [2409] Протока, 1500 К, 1 точка — —50
Хаймгартнер, 1952 [1382] Протока, 1170—1383 К, 9 точек — —52,5±3,5
Семенкович, 1960 [375] Протока, 1273—1473 К, 6 точек —70±3 —57,3±2,5
Кикути и др., 1964 [1701] Протока, 1273—1523 Ка —85 —57,0±3,5
Танабе и др., 1964 [2657] Протока, 1173—1573 Ка —65 —62,8+2,0
Фриш и др., 1965 [1146] Эффузионный, 931—1034 К, 9 точек —58±3 —45,9±2,0
Гинсберг, Спарвальд, 1965 [1214] Протока, 1423 и 1500 К, 2 точки —67 —48,8±4,0
Рао, Дадапе, 1966 [2293] Протока, 1125—1425 К, 11 точек —51,1 ±5 —51,2±2,0
Митани, Нагаи, 1967 [2036] Протока, 1273—1473 Ка —71 —56,5±2,5
Бандоравалла, Алтекар, 1968 [574] Протока, 1273—1473 К, 6 точек —54±30 —48,1±3,5
а Экспериментальные данные даны только в виде уравнения.
A023 К, 3 измерения) и А1 (jK)+NaCl (к, ж)=А1С1 (r)+Na (г) A023 К, 3 измерения и 1243 К,
5 измерений), выполненного Гроссом и др. [1275] статическим методом. Из этих данных по III
закону рассчитаны значения Д/#°(А1С1, г ,0), равные —54,7,-54,9 и —54,2 кДж-моль
соответственно. При проведении этих расчетов учтено образование в парах димеров К2С12 и Na2Cl2.
Как видно из табл. 21.24, результаты определений величины bfH° (A1C1)
неудовлетворительно согласуются между собой. Из всех рассмотренных работ наиболее надежной представля-
136
Алюминий и его соединения . А1С13 (г)
ется работа Рао и Дадапе [2293], где измерения проведены в широком интервале температур
и для которой имеет место хорошее согласие между величинами, вычисленными по II и III
законам. Принятое в справочнике значение основывается на этой работе. Следует отметить, что
средневзвешенное из данных табл. 21.24 (за исключением крайних выпадающих величин) и
исследований равновесий с КС1 и NaCl [1275] составляет —51,5 кДж-моль, что совпадает с
данными [2293].
Экстраполяция (примерно на 2000 см) колебательных уровней состояния АХИ приводит
к значению DO(A1C1) та 43 500+600 см. Это дозволяет предполагать, что потенциальная кривая
состояния АгП у А1С1 имеет максимум высотой около 1900 ±700 см.
А1С12 (г). Термодинамические свойства газообразного дихлорида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 646.
Экспериментальные данные о спектре и структуре молекулы А1С12 в литературе отсутствуют.
Молекулярные постоянные, приведенные в табл. 21.16, оценены. По аналогии с другими
молекулами дигалогенидов элементов подгруппы IIIA, а также учитывая результаты теоретического
расчета Хасти и Маргрейва [1367] и оценок, выполненных ранее (см. [106, 1382]), в справочнике
принимается, что в электронном состоянии X2At молекула А1С12 имеет угловую симметричную
структуру (симметрия С2„). Произведение главных моментов инерции, приведенное в табл. 21.16,
вычислено по структурным параметрам г (А1—С1)=2,06+0,05 А и -§ С1—А1—С1=120+10°,
принятым равными соответствующим величинам в молекуле А1С13. Погрешность значения IaIbIg
может достигать 1-1014 г3-см8. Приведенные в табл. 21.16 частоты колебаний А1С12 вычислены
по силовым постоянным (/г=2,6, /гг=0,15, /а/г2=0,10, в 105 дин-см), перенесенным из А1С13.
Погрешности в рассчитанных значениях частот оцениваются в 10—15%. Энергия возбуждения
состояния А*ВХ принята на основании теоретического расчета [1367], скорректированного
сравнением рассчитанных в [1367] и известных из экспериментальных данных энергий возбужденных
электронных состояний двухатомных галогенидов бора и алюминия. Погрешность в значении
Тй (A2Bj), приведенном в табл. 21.16, может достигать 4000 см.
Термодинамические функции А1С12 (г) вычислялись в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом вклада возбужденного электронного состояния A~2Bt по уравнениям A. 168)—A. 170).
Погрешности рассчитанных термодинамических функций А1С12 (г) определяются в основном
неточностью оцененных молекулярных постоянных; при высоких температурах существенными
становятся погрешности, обусловленные приближенным характером расчета. Погрешности
в значениях Ф° (Т) составляют 1, 3 и 6 Дж-К^-моль при 298,15, 3000 и 6000 К.
Ранее термодинамические функции' А1С12 (г) вычислялись в справочниках [105, 106, 977,
1544, 1904]. Все расчеты выполнены по оцененным молекулярным постоянным. Данные табл. 646
и справочника [1544] согласуются в пределах 0,7 Дж-К-моль в значениях Ф° (Т) и S° (Г).
Расхождения между данными [105, 106, 977 и 1904] и табл. 646 достигают 2,8 Дж-К-моль
в значениях Ф° (Т) и S° (T) и обусловлены некоторым различием в оценке структурных
параметров и частот колебаний.
Константа равновесия реакции А1С12 (г)=А1 (г)+2С1 (г) вычислена с использованием
значения А,Л° @) =856,586+40 кДж-моль, соответствующего принятой в справочнике энтальпиж
образования ^,?
AfH° (А1С12, г, 0) = —290 + 40 кДж • моль.
Результаты измерений энтальпии образования А1С12 (г) представлены в табл. 21.25. Для"ра-
бот [115, 831 ] приведены значения, полученные их авторами, так как пересчет соответствующих
данных оказался затруднительным. Погрешности всех данных приняты по оценке авторов
исследований и, очевидно, они занижены. Несмотря на то что в работе Блума и Босмана [710]
получено хорошее соответствие результатов расчетов по II и III законам (авторы [710] вычислили
парциальные давления компонентов реакции только для одной температуры), найденная величина
—365 кДж-моль представляется ошибочной, так как она приводит к неоправданно высокой
стабильности А1С12 1. Вызывает сомнение также величина —318 кДж-моль [115]/так как она
1 Если принять значение А/Я° (А1С12, г), найденное в работе [710], парциальное давление А1С12 [в.условиях
опытов по изучению реакции 2А1 (ж)+А1С13 (г)=ЗА1С1 (г)[(см. выше А1С1 (г)) должно составлять десятки
атмосфер.
137
А1С13 (к, ж)
Глава 21
Таблица 21.25. Результаты определений Д/#° (А1С12, г, 0) (в кДж-моль *)
Авторы
Метод
Д/Я°(А1С12, г, 0)
II закон
III закон
Чей и др., 1967 [831] Протока, исследовалась система А1 (ж)+А1С1з (г), — —276+13
800—1000 К, учитывалось образование А1С1, А1С12.
А1С13, А12С14, А12С1в
Долгов, 1970 [115] Пересчет результатов исследований системы А1 (ж)-}- — —318+13
+А1С13 (г), в работах [831,1382, 1700, 2293, 2675],
учитывалось образование А1С1, А1С12, А1С1з, А12С1в
Фарбер, Харрис, 1971 [1050,1052] Масс-спектрометрический, А1С13 (г)+А1С1 (г)= —395+75 —293+15
=2А1С12(г), 1150—1430 К, 5 точек
Масс-спектрометрический, A1F» (г)+А1С1» (г)= — —283 а
=A1F2C1 (г)+А1С1 (г) и A1F2 (r)+AlCl,(r)=AlFCl2
(r)+AlF (г), 1491 К, 1 точка
Блум, Босман, 1975 [710] Протока, А12Оз (к)+4НС1 (г)+Н2 (г) =2А1С12 (г)+ —363+13 —
+ЗН2О (г), 1230—1500 К, 5 точек
То же, 1500 К, 1 точка — —365+4
DO(A1C12) DO(A1C1)
а Величина получена при подготовке справочника в предположении, что д (AIF ) == D (AIF) =0,743 или
DO(A1C12) DO(A1C13) п„,„ ¦ ого
DO(A1F2) - DO(A1F3) ~v'nb-
приводит к тому, что в условиях рассмотренных выше опытов по исследованию равновесия
реакции с образованием A1G1 (г) основным газообразным компонентом становится А1С12, а его
давление превысит давление А1С1 на порядок. Результаты расчетов, выполненных авторами
работ [831] и [115], приводят к различным значениям AfH° (A1C12, г) в связи с тем, что в одной
из них ([831]) в качестве предполагаемого компонента учитывался А1аС14, а в другой ([115]) это
соединение не рассматривалось. Поскольку в настоящее время нет экспериментальных данных,
подтверждающих или опровергающих существование в исследуемом интервале температур
заметных количеств A12G14, эти работы позволяют лишь утверждать, что энтальпия образования
А1С12 должна лежать между —276 и —318 кДж-моль.
Принятая в справочнике величина ^fH° (А1С12, г) основывается на результатах измерений
Фарбера и Харриса [1050, 1052]. Эта величина не противоречит существующему в настоящее
время представлению о том, что основными газообразными компонентами системы А1 (ж) +
+А1С13 (г) являются А1С13 и А1С1, а А1С12 не может образовываться в количествах, много
больших A1G1. Погрешность принятой величины увеличена по сравнению с рекомендацией авторов
[1050, 1052] с учетом противоречивости имеющихся данных.
А1С13 (к, ж). Термодинамические свойства кристаллического и жидкого трихлорида алюминия
в стандартном состоянии при температурах 100—600 К приведены в табл. Д.49 *.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние А1С13 (к) в интервале 0—465,7 К принята
моноклинная модификация со слоистой решеткой 2.
Термодинамические функции А1С13 (к) при Т <^ 298,15 К вычислены по результатам
измерений теплоемкости в работе Джастиса [1607] A3—310 К), проведенных с точностью —10% при
13 К и 0,5% при Т > 30 К. Экстраполяция теплоемкости ниже 13 К по уравнению Дебая
привела к значению 5° A3 К)=0,36 Дж-К-моль. Погрешности принятых по [1607] значений
S° B98,15 К) и Н° B98,15 К)-Н° @) оцениваются в 0,5 Дж-К^-моль и 0,08 кДж-моль'1
соответственно. Результаты измерений теплоемкости А1С13 [1094] (90—273 К) занижены в сред-
1 Критическая температура А1С13 равна 625,65 К (см. табл. Д. 65).
2 Согласно [372] у А1С13 при 413 К имеется фазовый переход, связанный с переходом из одной политипной
модификации в другую (тип кристаллической структуры не меняется). При расчете термодинамических функций
А1С13 (к) этот переход не учитывался, поскольку энтальпия перехода неизвестна и, по-видимому, мала.
138
Алюминий и его соединения А1С13 (г)
нем на 1,5% и не учитывались при расчете функций. В интервале 298,15—465,7 К для
теплоемкости А1С13 (к) принято уравнение (см. табл. 21.1), рекомендуемое в [1543] на основании
измерений энтальпии в работах Фишера [1094] B73—461 К), неопубликованной работе Мак-До-
налда B98—465 К) (см. [1543]) и с учетом данных Джастиса [1607]. Температура плавления
А1С13 D65,7+0,2 К) принята по данным Фостера [1122] D65,7 К) и Смитса и др. [2561] D65,8 К);
результаты других, менее точных измерений [112, 288, 687, 1482, 1668, 2515, 2730] приводят
к значениям Гт=463 -f- 467 К. Энтальпия плавления А1С13 C5,4+0,8 кДж'Моль) и
теплоемкость А1С13 (ж) A25,5+3,0 Дж-К~1-моль~1) приняты по рекомендации [1543], основанной
на измерениях энтальпии A1CL, (ж) в работах Фишера [1094] D60—504 К) и Мак-Доналда D60—
493 К) (см. выше). Давление пара в точке плавления А1С13 равно 2,3 атм. Расчет
термодинамических функций А1С13 (ж) проводился до 600 К, поскольку критическая температура А1С13
равна 625 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 600 К оцениваются в 0,3, 1,5
и 2,5 Дж-К~1-моль соответственно. В интервале температур 100 <J T ^ 600 К
термодинамические функции А1С13 (к, ж), приведенные в табл. Д. 49 и в справочнике JANAF [1543]
совпадают г.
В настоящем справочнике принята энтальпия образования трихлорида алюминия
Д/Я°(А1а8, к, 298,15 К) ==—705,1 + 1,2 кДж • моль,
¦основанная на величинах, полученных Гроссом и Хейманом [1277] при измерении энтальпии
сгорания алюминия в хлоре (—706,3+0,7 кДж-моль) и Кофлиным [888, 889] при измерении
энтальпии растворения А1 и А1С13 в соляной кислоте (—704,0+0,6 кДж-моль). Измерения
энтальпии растворения алюминия [685, 2330, 2332, 2387, 2887] и трихлорида алюминия [108,
327, 620, 671, 1724, 1726, 2377, 2380] в более старых работах уступают по точности измерениям
[888, 889] и проводились при различающихся температурах и концентрациях, что затрудняет
их использование для расчета энтальпии образования А1С13 (к). Семененко и др. [373] провели
исследования, аналогичные проведенным в работах [888, 889]. Однако для энтальпии
растворения А1С13 (к) они нашли величину, отличающуюся примерно на 10 кДж-моль, и получили
kfH° (А1С13, к, 298,15 К) =—714,5 кДж-моль. Хорошее соответствие результатов работ [888,
889] и [1277] показывает, что в работе [373], вероятно, имеется систематическая ошибка,
связанная с реакцией трихлорида алюминия с соляной кислотой. Очевидно, содержат ошибки также
измерения энтальпии сгорания алюминия в хлоре, выполненные в работе [2520].
А1С13 (г). Термодинамические свойства газообразного трихлорида алюминия в стандартном
•состоянии в интервале температур 100—6000 К приведены в табл. 647.
Молекулярные постоянные, использованные для расчета термодинамических функций А1С13 (г),
приведены в табл. 21.17. Электронографическое исследование пара А1С13, выполненное Засори-
ным и Рамбиди [134], результаты исследования ИК-спектра и спектра КР газообразного А1С13
[368, 636, 1727] и молекул А1С13, изолированных в матрицах [320, 633, 634, 1850, 2244, 2467,
2511], а также квантовомеханический расчет ab initio, проведенный Со и Ричардсом [2578],
однозначно показывают, что молекула А1С13 в основном электронном состоянии ХгА' имеет
плоскую симметричную структуру (группа симметрии D3h) 2. Приведенное в табл. 21.17
произведение главных моментов инерции вычислено по значению г (А1—С1) =2,06 +0,01 А, найденному
Засориным и Рамбиди [134] 3. Погрешность значения IaIbIg не превышает 3-Ю14 г3-смв.
Приведенные в табл. 21.17 частоты \, v3 и v4 найдены в результате исследования ИК-спектра
1 В таблицах JANAF [1543] термодинамические функции А1С13(ж) рассчитаны до 1500 К, т. е. значительно выше,
чем ГЕр.
2 Заключение Лесицкого и Шерка [1850] о пирамидальной структуре А1С13, основанное на результатах
исследования ИК-спектра в матрице, неверно. В работах [633, 634, 2467, 2511] показано, что авторы [1850], помимо
спектра А1С13, наблюдали в области частоты vx спектры комплексов А1С13Х2, где X — соединение азота.
3 Повторное электронографическое исследование структуры А1С13 с использованием более совершенной
аппаратуры и методики, выполненное Засориным и др. [2595в], дало г (А1—С1)=2,068+0,004 А и -4 С1—А1—С1=
= 119,39 ±0,42°.
139
AUCl, (г)
Глава 21-
^v3) [1727] и спектра КР (v1? v4) [636] газообразного А1С13 х. В пределах возможных матричных
сдвигов с этими значениями согласуются частоты в спектрах молекул А1С13, изолированных
в матрицах [320, 368, 633, 634, 2467, 2511]. Частота v2 наблюдалась только в ИК-спектре
молекул, изолированных в матрицах. Ее значения варьируются от 174 [320, 368] до 183 см [1850,.
2511]. Приведенное в табл. 21.17 значение является округленным средним. Погрешности в
принятых частотах оцениваются равными 10—15 см. Сведения о возбужденных электронных
состояниях А1С13 в литературе отсутствуют; по аналогии с галогенрдами бора в справочнике
принимается, что их энергии превышают 20 000 см.
Термодинамические функции А1С13 (г) вычислялись в приближении «жесткий ротатор-
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130>
без учета возбужденных электронных состояний. Погрешности термодинамических функций
обусловлены в основном использованием приближенного метода расчета и в меньшей степени
неточностью молекулярных постоянных. Они составляют 1, 5 и 8 Дж-К • моль в Ф° (У)
при 298,15, 3000 и 6000 К, в том числе из-за неточности постоянных менее 1 Дж-К~1-моль.
Ранее термодинамические функции А1С13 (г) вычислялись в предыдущих изданиях настоящего-
справочника [105, 106], таблицах JANAF [1543], справочниках [977, 1856, 1904, 1974] и
работах [1124, 1384] до Т < 1000 К. Расхождения данных табл. 647 и JANAF [1543] не превышают
0,3 Дж-К-моль в значениях Ф° (Т) и S° (T). Максимальные расхождения между
результатами расчетов, выполненных в [105, 977, 1856, 1904, 1974] и приведенных в табл. 647, составляют
0,3—2,2 Дж-К-моль в значениях Ф° (Т) и обусловлены различием в принятых значениях
частот и межатомных расстояний.
Константа равновесия реакции А1С13 (г)
0 @I26850672 Д1
р
^ @)=1268,506+7,2
образования
А1 (г)+ЗС1 (г) вычислена с использованием значения
й
р 3 () () ()
кДж-моль, соответствующего принятой в справочнике энтальпии
г, 0) = —582,3 + 6,0 кДж • моль
Эта величина вычислена на основании принятой в справочнике энтальпии образования
А12С16 (г) и энтальпии диссоциации А1гС1в (г)=2А1С13 (г), результаты измерения которой
приведены в табл. 21.26. Как видно из таблицы, эти измерения находятся в хорошем соответствии.
Таблица 21.26. Результаты определений DO(AIC13—A1C13) (в кДж-моль)
Авторы
Фишер и др., 1932 [1100]
Смите, Мейеринг, 1938 [2561]
Фриланд, Сталл, 1967 [2784]
Поляченок и др., 1976 [331]
Метод
Статический (изотенископа), 605—944 К, 20
точек
Статический, 669—816 К, 6 точек
Статический-, 513—823 К, данные приведены
в виде уравнения
Статический, 650—1100 К', 43 точки, данные
приведены в виде уравнения
Do (A1C13-A1C13)
II закон
133,5±5,0
122,9±7,6
121,5
128,0
III закон
128,3±0,7
128,3 ±0,4
128,1 ±1,4
127,6±0,4
Для последующих расчетов в справочнике используется среднее значение Do (А1С13—А1С13) =
=128,1+12 кДж-моль, погрешность которого учитывает не только воспроизводимость
измерений (см. табл. 21.26), но и неточность термодинамических функций.
А12С16 (г). Термодинамические свойства газообразного гексахлорида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 648.
Молекулярные постоянные, использованные для расчета термодинамических функций А12С1»,
приведены в табл. 21.23. Исследования спектров [633, 636, 1727, 1877, 1951], а также электроно-
1 Поскольку полосы газообрааного AlClj в спектре КР, полученные при высоких температурах, довольно
широкие, не исключено, что частота vx=392+394 см~а, наблюдавшаяся в спектрах КР в матрицах [633, 6341, ближе
к ее истинному значению.
140
Алюминий и его соединения С1А1О (г)
графические исследования [4, 133, 756, 2175, 2506] показывают, что молекула А12С16 в основном
электронном состоянии ХхАд имеет мостиковую структуру и принадлежит к точечной группе
•симметрии D2h. Произведение моментов инерции вычислено по структурным параметрам
г (А1-С1внешн) = 2,065 + 0,003 А, г (А1-С1М00Г) = 2,252 + 0,003 А, ^ С1внвшн-А1-С1внешн = 123,4 +
+ 1,6°, -^ С15Ю0Т—А1—С1МООТ = 91,0 + 1,6°, найденным Шеном в результате электронографического
исследования [2506]. Погрешность значения IaIbIo составляет 2-1012 г3»см6. Колебательные
спектры молекулы А12С1в исследовались в газовой [636, 1727, 1951] и жидкой фазах [323, 1193]
и в матрицах [320, 633, 1877, 2467]. Приведенные в табл. 21.23 частоты, активные в ИК-спектре
А12С16, приняты по работам Клемперера [1727] (vg, v13, v16), Перова и др. [320] (v14, v18) и Битти
и др. [633] (v1? v17). Частоты, активные в спектрах КР, найдены Битти и Хордером [636] (v2, v3,
V4> V7> vu> vi2) vie)' Марони и др. [1951] (v6) и Першиной и Раскиным [323] (v5, v10) x. Для
частоты vg, не наблюдавшейся экспериментально, принято среднее значение между величинами,
оцененными авторами работ [1951, 2165]. Погрешности экспериментально наблюдаемых частот
•составляют 1—10 см, погрешность в v9 может достигать 50 см. Сведения о возбужденных
электронных состояниях А12С16 в литературе отсутствуют.
Термодинамические функции А12С16 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
{1. 130); их погрешности составляют 8, 20 и 30 Дж-К~1-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К и определяются в основном приближенным характером расчета. Погрешности,
обусловленные неточностью молекулярных постоянных, не превышают 3 Дж-К-моль~1.
Ранее термодинамические функции А12С1в (г) рассчитывались в справочниках JANAF [1543],
Дугласа и Бекетта [977], Льюиса и др. [1856] (до 3000 К), а также в работах Венкатесварлу
и Нагараяна [2764] (Т < 1000 К) и Сивина и Тёрсет [917] (до 3000 К). Данные в табл. 648 и
и [1543 ] согласуются в пределах 3 Дж-К^-моль в значениях Ф° (Т) и S° (Г). В работах [917, 977,
1856, 2764] для многих частот использованы неверно отнесенные или неправильно оцененные
величины, поэтому рассчитанные значения термодинамических функций существенно (на 16—
.20 Дж-К~1-моль в значениях Ф° (Т)) отличаются от вычисленных в настоящем справочнике.
Константа равновесия реакции А12С16 (г)=2А1 (г)+6С1 (г) вычислена с использованием
значения кгН° @) =2665,112+9,4 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH° (А12С16, г, 0) = —1292,7+ 5,0 кДж . моль.
Эта величина основана на энтальпиях образования и сублимации А1С13 (к) с образованием
А12С1в (г). Последняя получена на основании представленных в табл. 21.27 результатов
измерений давления паров трихлорида алюминия. Эти измерения проводились в области температур
и давлений, при которых диссоциация А12С16 (г) с образованием А1С13 (г) практически
отсутствует. Как видно из таблицы, результаты обработки всех измерений по уравнению III закона
находятся в хорошем согласии, за исключением работы [1714], где давление пара достигало
десятков атмосфер и не учитывалось отклонение от законов идеального газа. Приведенные
в табл. 21.27 погрешности отражают только воспроизводимость измерений. Неточность
термодинамических функций приводит к дополнительной погрешности, равной 5 кДж-моль. Для
последующих расчетов в справочнике для энтальпии сублимации трихлорида алюминия с
образованием А12С16 (г) используется значение 114,8+5,0 кДж-моль.
С1А1О (г). Термодинамические свойства газообразного оксид-хлорида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 649.
Молекулярные постоянные, использованные для расчета термодинамических функций С1А1О,
приведены в табл. 21.8. Структура С1А1О экспериментально не исследовалась. По аналогии
с другими оксидами алюминия и бора принято, что молекула С1А1О в основном электронном
состоянии ХХ2+ линейна (симметрия CaV). Приведенное в табл. 21.8 значение главного момента
4 В 1980 г. Транквипь и Фуасье [2724а] измерили в матрицах инертных газов ИК- и КР-спектры А12С16 и
предложили несколько иную интерпретацию спектров, чем принятая в настоящем справочнике. Однако расхождения
термодинамических функций, рассчитанных по данным работы [2724а] и приведенных в справочнике, не
превышают в значениях Ф° (Т) 2 Дж-К^-моль.
141
C12AJO (г)
Глава 2f
Таблица 21.27. Результаты измерений энтальпии сублимации 2А1СЬ (к)=А12С1б (г) (в кДж-моль 1)
Авторы
Тредуэлл, Теребези, 1932 [2730]
Фишер и др., 1932 [1100]
Смите и др., 1938 [2561, 2563]
Нарышкин, 1939 [271]
Такахаши, 1954 [2648]
Данн, Грегори, 1958 [996]
Нисельсон, 1965 [288]
Денисова, Баскова, 1969 [110],
Кинг, Сигмиллер, 1971 [1714]
Виола и др., 1977 [2776]
Метод
Статический, 389—476 К, 14 точек
Статический, 395—450 К, 14 точек
Протока, 392—450 К, 9 точек
Статический, 421—482 К, 19 точек
Статический, 427—466 К, 12 точек, данные
представлены в виде уравнения
390—423 К, 5 точек
Эффузионный, 298—322 К, данные
представлены в виде уравнения
Эбулиоскопический, 471—506 К, 19 точек
Статический, 468—606 К, 26 точек
Измерение плотности пара, 474—569 К, 24 точки
То же, 462-511 К, 55 точек
Статический (изотенископа), 387—466 К, 48
точек
То же, 456—530 К, 80 точек
Д„Я°
II закон
117,5±4,0
117,9+1,2
116,2±4,5
103+10
98
172
125
117,4+0,6
115,7±0,6
127,7+0,4
121,2+0,6
116,9+0,4
116,1+0,2
@)
III закон
114,7±0,2
115,0+0,1
115,1+0,1
114,6±0,1
И4,7±0,7
115,1
115,9+0,5
114,5+0,1
114,1 ±0,1
110,2+0,4
111,2±0,1
115,0+0,1
114,5+0,1
инерции вычислено по оцененным структурным параметрам г (А1—С1) =2,06 +0,05 А и г (А1=
=О)=1,62+0,03 А, которые приняты такими же, как в молекулах А1С13 и АЮ. Погрешность
значения / составляет 3 • 10~39 г • см2. Частоты vi==va1_ci и v3==vai—о найдены Шнёкелем [24686}
в ИК-спектре молекул 35С1А116О, изолированных в матрице из аргона. Деформационная частота
рассчитана так же, как для FA1O *. Погрешности принятых частот оцениваются в 10, 50
и 10 см соответственно.
Термодинамические функции G1A1O (г) вычислялись по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический осциллятор» без
учета возбужденных электронных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета;
они составляют 2, 6 и 8 Дж-К-^моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К, в том
числе 2—4 Дж-К-моль~1 из-за неточности молекулярных постоянных.
Ранее таблицы термодинамических функций С1А1О (г) вычислялись в справочниках JANAF
[1543], Мак-Брайд и др. [1974], Мадера [1904] и Дугласа и Бекетта [977] по оцененным
значениям молекулярных постоянных. Расхождения в функциях, приведенных в табл. 649 и
рассчитанных в перечисленных выше работах, составляют 1—3,6 Дж-К~1-моль~1 в значениях
Ф° (Т); они обусловлены в основном различием в принятых значениях частоты деформационного
колебания и межатомного расстояния г (А1—С1).
Константа равновесия реакции С1А1О (г)=А1 (г)+С1 (г)-(-О (г) вычислена с использованием
значения Ari/° @)=1043,749+30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH° (G1 АЮ, г, 0) = — 350 + 30 кДж • моль.
Результаты измерений энтальпии образования С1А1О (г) представлены в табл. 21.28.
Приведенные в таблице погрешности включают воспроизводимость данных, погрешности
термодинамических функций и погрешности использованных в расчетах термохимических величин.
Термодинамические функции компонентов, не рассматриваемых в настоящем издании, взяты из
справочника Барина и др. [578], а термохимические величины — из справочника [406]. Принятое
значение энтальпии образования С1А1О (г) является средневзвешенным из результатов обработки
экспериментальных данных по III закону.
С12А1О (г). Термодинамические свойства газообразного оксид-дихлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 650.
1 Соответствующие силовые постоянные равны (в 108 дин-см): /А1_С1=2,92, /А1_О=6,62, fJr1r2=0,i5±0,Q5.
142
Алюминий и его соединения
А1НС1 (г)
Таблица 21.28. Результаты
Авторы
эпределений АуН° (С1А1О, г, 0) (в кДж-моль)
Метод
Д/Я°(С1АЮ, г, 0)
II закон
III закон
Гринбаум и др., 1964 [1248а] Эф&узионный в условиях молекулярного потока, — —340+20
VsAlaOs (ж)+1/зА1С13 (г)=С1А1О(г), 2400 К, 6
измерений
Рао, Дадапе, 1970 [2292а] Протока, 6№О(к)+4А1С1з(г)=ША1„О4(к)+5ШС12 -387+60 —281+50
(г)+2С1А1О (г), 1269—1446 К, 10 точек
Сривастава, Фарбер и др.. 1971 Масс-спектрометрический,А12О(г)+С1(г)=С1А1О(г)+ —316+40 —360+25
[2596] ' +А1(г), 1473—1600 К, 5 точек
Атчайя, Дадапе, 1971 [543а] Протока, La2O3 (к)+'/2А12С1в (r)=2LaOCl (к)+ —378+50 —388+40
+С1А1О (г), 622—793 К, 7 точек
Протока, La»O3 (к^+А1С13 (r)=2LaOCl (к)+С1А1О (г), —290+50 —335+40
836—899 К, точек
Молекулярные постоянные, использованные для расчета термодинамических функций С12АЮ,
приведены в табл. 21.11. В литературе отсутствуют экспериментальные данные о спектре и
структуре С12АЮ. По аналогии с F2BO в справочнике принимается, что в основном электронном
состоянии Х2В2 молекула С12АЮ имеет плоскую структуру симметрии C2v. Приведенное в табл. 21.11
произведение моментов инерции С12АЮ вычислено по структурным параметрам г (А1—О) = 1,80 +
+0,05 А, г (А1—С1)=2,06+0,05 Аи^ С1—А1—С1=120+10°, оцененным на основании сравнения
с молекулами F2A1O и А1С13. Погрешность значения IjJbIc составляет 1-10~113 г3-см8. Основные
частоты v2—vg C12A1O, приведенные в табл. 21.11, оценены сравнением с частотами
соответствующих колебаний молекулы C12A1F, частота v1(Al—О) принята близкой соответствующей
частоте молекулы F2A1O. Погрешности принятых значений частот оцениваются в 20%.
Сведения о возбужденных электронных состояниях С12АЮ в литературе отсутствуют; по аналогии
с молекулой F2BO можно предполагать, что в области 10 000—20 000 см расположены два
возбужденных состояния С12АЮ.
Термодинамические функции С12АЮ (г) вычислены по уравнениям A. 3)—A. 6), A.9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены главным образом неточностью оцененных молекулярных постоянных и в меньшей степени
приближенным характером расчета; они составляют 4, 8 и 12 Дж-К~1-моль~1 в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Таблица термодинамических функций С12АЮ (г) публикуется впервые.
Константа равновесия реакции С12АЮ (г)=А1 (г)+О (г)+2С1 (г) вычислена с использованием
значения Д,.//0 @) = 1563,369+60 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
&fH°(Cl2AlO, г, 0) = —750 + 60 кДж-моль.
Эта величина оценена в предположении, что Do (Cl2AlO)/D0 (ClAlO)=D0 (F2A10)/D0 (FA1O).
A1HC1 (г). Термодинамические свойства газообразного гидрид-хлорида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 651.
Молекулярные постоянные А1НС1, использованные для расчета термодинамических функций,
приведены в табл. 21.16. Спектр и структура А1НС1 экспериментально не исследовались. По
аналогии с другими молекулами A1XY в справочнике принимается, что в основном электронном
состоянии Х2А' молекула А1НС1 имеет угловую структуру симметрии Cs. Молекулярные постоянные,
приведенные в табл. 21.16, оценены так же, как молекулярные постоянные A1HF (см. выше).
Погрешность в значении 1А1в1о, вычисленном по структурным параметрам г (А1—Н)=1,59 +
+0,03 А, г (А1—С1) =2,06+0,05 Аи^ Н—А1—С1=120+10° составляет МО16 г3-см6.
Погрешности в значениях частот оцениваются в 10—15% х, а погрешность величины То (Л2А") —
в 5000 см.
1 Частоты рассчитаны по силовым постоянным (в 105 дин-см) /дт-сг^^б, /д]_н=2,1 и /а/г]Г2= 0,15.
143
А1НаС1(г), A1HCU (г) Глава 21
Термодинамические функции А1НС1 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
возбужденного состояния Л2А" по уравнениям A. 168)—A. 170). Погрешности рассчитанных
термодинамических функций обусловлены неточностью оцененных молекулярных постоянных и (выше
3000 К) приближенным характером расчета; они составляют 1, 4 и 6 Дж-К~1-моль~1 в значениях
Ф°(Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1HG1 (г) публикуется впервые.
Константа равновесия реакции A1HG1 (г)=А1 (г)+Н (г)+С1 (г) вычислена с использованием
значения Дг#° @) =683,000+40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^0 (А1НСЗ, г, 0) = —20 + 40 кДж. моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах A1HG1,
А1Н2 и А1С1,.
АШ2С1 (г). Термодинамические свойства газообразного дигидрид-хлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 652.
Экспериментальные данные о спектре и строении молекулы А1Н2С1 в литературе отсутствуют.
На основании сравнения с симметричными молекулами А1Х3 в справочнике принимается, что в
основном электронном состоянии Х1Аг молекула А1Н2С1 имеет плоскую структуру симметрии С2в.
Приведенные в табл. 21.17 молекулярные постоянные А1Н2С1 оценены так же, как постоянные
A1H2F. Произведение главных моментов инерции соответствует структурным параметрам
г(А1—Н)=1,59+0,03 А, г (А1—G1) =2,06+0,05 А и J$ H—A1—Н = -$ Н—А1-С1=120+10°;
его погрешность составляет 5-1016 г3-см8. Погрешности в значениях частот, оцененных по
частотам молекул АШ3, А1С13 и А1НС12, могут достигать 15—20%.
Термодинамические функции А1Н2С1 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A.130) с учетом
только основного электронного состояния. Погрешности табулированных величин составляют
1, 5 и 7 Дж- К- моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К; они обусловлены неточностью
оценки молекулярных постоянных и (выше 1000 К) приближенным характером расчета.
Таблица термодинамических функций А1Н2С1 (г) публикуется впервые.
Константа равновесия реакции АШ2С1 (г)=А1 (г)+2Н (г)+С1 (г) вычислена с использованием
значения Дг#° @) =929,034 ±40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^^АШД, г, 0) = — 50 ± 40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах А1Н4С1,
АШ3 и А1С13.
А1НС12 (г). Термодинамические свойства газообразного гидрид-дихлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 653.
Структура молекулы А1НС12 экспериментально не исследовалась. На основании сравнения
с симметричными молекулами А1Х3 в справочнике принимается, что в основном электронном
состоянии XхАх она имеет плоскую структуру симметрии С2„. Приведенное в табл. 21.17
значение IaIbIc рассчитано по оцененным структурным параметрам г (А1—Н)=1,59+0,03 А,
г (А1—Cl)=2,06+0,05 A, J$ Н—А1—С1=^ С1—А1—С1=120+10°, его погрешность равна
1 -10~114 г3-см6. Основные частоты, приведенные в табл. 21.17, кроме значения v3, найдены Шнё-
келем [2468а] при исследовании ИК-спектра молекул А1НС12, образующихся в матрице из аргона
в результате реакции продуктов фотолиза молекул А1С13 с НС1. Их погрешности оцениваются
в 10—15 см. Частота v3=vci-Ai-0b не наблюдавшаяся экспериментально, принята равной
соответствующей частоте в молекуле А1С13, ее погрешность оценивается в 30 см.
Термодинамические функции АШС12 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122) — A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности рассчитанных величин обусловлены
главным образом приближенным методом расчета и (ниже 1000 К) неточностью молекулярных
постоянных; они составляют 2, 6 и 8 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
144
Алюминий и его соединения CIA1OH (г),С1А1 (ОН)г (г)
Таблица термодинамических функций А1НС12 (г) публикуется впервые.
Константа равновесия реакции А1НС12 (г)=А1 (г)+Н (г)+2С1 (г) вычислена с использованием
значения Дг#° @)=1097,620+25 кДж-моль", соответствующего принятой в справочнике
энтальпии образования
г, 0) = —315 + 25 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
AlHClj, А1Н3 и А1С13.
С1А1ОН (г). Термодинамические свойства газообразного гидроксид-хлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 654.
Молекулярные постоянные, использованные для расчета термодинамических функций G1A1OH,
приведены в табл. 21.11. Спектр и структура молекулы С1АЮН экспериментально не
исследовались. Произведение главных моментов инерции в основном состоянии Х2А' вычислено для плоской
молекулы симметрии С, с -$¦ G1—А1—0=120+10°, *$ А1—О—Н=115+10е (трансположение),
г(А1—Cl)=2,06+0,05 A, r (A1—О)=1,78 +0,05 А, г (О—Н)=0,96+0,02 А. Эти структурные
параметры оценены на основании соответствующих параметров молекул А1С12 и А1(ОНJ.
Погрешность значения 1А1в1с составляет 5-10~115г3-см6. Значения частот vx=vA1_0, v2 = v0_H hv4=vAi_0_h
перенесены из молекулы НАЮ2. Частота v3=vA1_G1 принята примерно равной этой частоте в
молекуле A1FG1. Частота v5=v0_A1_Gl оценена по соответствующим основным частотам А1С12, А1(ОНJ
и с учетом близости свойств группы ОН и атома фтора, A1FG1. Частота крутильного колебания
ОН-грушш vg принята равной соответствующей частоте в A1FOH. Погрешности приведенных
в табл. 21.11 значений основных частот оцениваются в 20%. Молекула С1А1ОН, так же как
молекулы A1F2, A1FG1 и А1С12, по-видимому, имеет возбужденное электронное состояние в области
10 000—20 000 см; ввиду грубой оценки всех постоянных G1A1OH это состояние не включено
в табл. 21.11.
Термодинамические функции С1А1ОН (г) вычислены по уравнениям A.3)—A. 10), A. 122)—
A. 124), A. 128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор» без
учета возбужденных состояний. Погрешности функций обусловлены грубой оценкой принятых
значений молекулярных постоянных, а также приближенным характером расчета и составляют
4, 7 и 12 Дж-К^-моль'1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций С1А1ОН (г) публикуется впервые.
Константа равновесия реакции С1А1ОН (г)=А1 (г)+С1 (r)-f-0 (г)+Н (г) вычислена с
использованием значения Д^0 @) =1369,783 ±40 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
Д/Я°(С1АЮН, г, 0) = —460J+ 40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
С1А1ОН, А1С1Я и А1(ОНJ.
С1А1(ОНJ (г). Термодинамические свойства газообразного дигидроксид-хлорида алюминия
в стандартном состоянии в интервале температур 100—6000 К приведены в табл. 655.
Молекулярные постоянные, использованные для расчета термодинамических функций
С1А1(ОНJ, приведены ь габл. 21.13. Спектр и структура молекулы С1А1(ОНJ экспериментально
не исследовались. Произведение моментов инерции в электронном состоянии ХХА' вычислено
для плоской молекулы симметрии С, со структурными параметрами, перенесенными из молекул
А1С1, и А1(ОН)8: ^ О-А1-О = ^С1-А1-О=120+10°, ^ А1-О-Н=115+10°, г (А1—О) =
=1,78+0,05]А, r(Al—G1) =2,06 + 0,06 А, г (О—Н)=0,96 + 0,02|А. Погрешность приведенного значения
IJbIc составляет 5-Ю14 г3-см8. Частоты валентных колебаний связей О—Н (v3, v4) приняты
немного большими, чем в А1(ОНK, так как в С1А1(ОНJ водородные связи, по-видимому, слабее.
Частоты v1 = vAT-o, v2 = v\c1™o, ve = v0_A1_0, v7 = vab1'Lo-h, v8 = vaTJLo_h и частоты крутильных колебаний
vii и vi2 перенесены из А1(ОНK. Частота v5=<vAl_G1 оценена по соответствующим частотам молекул
A1F2C1, A1H2C1 и A1HFC1. Учитывая близость масс и изоэлектронность группы ОН и атома F,
частоты ve=vo_Ai_Gi и vio=v30Ht приняты близкими к значениям этих частот в молекуле A1F2C1.
Погрешности принятых значений оцениваются в 15—25%.
145
С1аАЮН (г), AIFC1 (г) Глава 21
Термодинамические функции С1А1(ОНJ (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных величин обусловлены грубой оценкой молекулярных постоянных,
а также приближенным характером расчета и составляют 8, 15 и 25 Дж-К~1-моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К (в том числе 5—6 Дж-К~1-моль из-за неточности молекулярных
постоянных).
Таблица термодинамических функций С1А1(ОНJ (г) публикуется впервые.
Константа равновесия реакции С1А1(ОНJ (г)=А1 (г)+С1 (г)+20 (г)+2Н (г) вычислена с
использованием значения &ГН° @) =2232,600+70 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
A^°(G1A](OHJ, г, 0) = —860 ±70 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
С1А1(ОНJ, А1С13 и А1(ОНK.
С12АЮН (г). Термодинамические свойства газообразного гидроксид-дихлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 656.
Молекулярные постоянные, использованные для расчета термодинамических
функций С12АЮН, приведены в табл. 21.13. Спектр и структура молекулы С12АЮН
экспериментально не исследовались. Приведенное в табл. 21.13 произведение главных моментов
инерции в состоянии XхА' соответствует плоской структуре молекулы симметрии С„ со
структурными параметрами, оцененными на основании сравнения с параметрами А1(ОЫK. А1С13 и F2BOH
и равными -? С1—А1—С1=->[ С1—А1—0=120+10°, ^ А1—О—Н=115±10°, г (А1—О) = 1,78 +
+0,05 А, г (О—Н)=0,96 +0,02 А и г(А1—С1)=2,06 +0,06 А. Погрешность значения IaIbIp составляет
5 • 10~114 г3 • см6. Значения частот v1 = vA1_0, v2 = vQ_H и V5 —vai—о—н пеРенесены из молекул НАЮ2
и АЮН. Частоты v3 = vA^LCi, v4 = vaT-съ \ = ^i-ai-o' v? — vgi-ai-ci' vs = v»oht оценены по
соответствующим частотам молекул A1G13, A1(OHK и с учетом близости свойств группы ОН и атома фтора,
молекулы A1FC12. Величина крутильной частоты.v9 принята такой же, как в молекуле F2A1OH.
Погрешности принятых частот vx—vn оцениваются в 15—25% от их величин.
Термодинамические функции С12АЮН (г) вычислены по формулам A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении модели «жесткий ротатор—гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены грубой оценкой
молекулярных постоянных, а также приближенным характером расчета и составляют 6, 12
и 20 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций С12АЮН (г) публикуется впервые.
Константа равновесия реакции С12АЮН=А1 (г)+2С1 (г)+О (г)+Н (г) вычислена с
использованием значения &ГН° @)=1749,403+40 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
Д^Л0 (CLjAlOH, г, 0) = —720 ±40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
С12АЮН, А1С1, и А1(ОН),.
AIFCI (г). Термодинамические свойства газообразного фторид-хлорида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 657.
Спектр и структура молекулы A1FC1 экспериментально не исследовались. По аналогии с
другими молекулами дигалогенидов бора и алюминия в справочнике принимается, что в основном
электронном состоянии Х2А' молекула A1FC1 имеет структуру симметрии Са. Молекулярные
постоянные A1FC1, использованные для расчета термодинамических функций и приведенные в табл. 21.16,
оценены так же, как постоянные молекулы A1HF (см. выше). Погрешность величины IaIbIc,
вычисленной по оцененным структурным параметрам г (А1—F)=1,63+0,04 А, г (А1—С1)=2,06 +
+ 0,06 Аи4 F—А1—С1=120+10°, не превышает 5«10~115 г3-см6. Погрешности в значениях
основных частот составляют 10—15%, погрешность величины То (А2А") оценивается в 5000 см.
146
Алюминий в его соединения A1F2C1 (г), AlFCb (г)
Термодинамические функции A1FC1 (г) вычислены в приближении «жесткий ротатор—гармо*
нический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
вклада возбужденного электронного состояния Л2 А" по уравнениям A.168)—A.170). Погрешности
рассчитанных термодинамических функций в основном обусловлены неточностью молекулярных
постоянных, а также приближенным характером расчета (выше 1000 К); они составляют 1, 4* и
6 Дж-К-^моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции A1FC1 (г) вычисляли авторы таблиц JANAF [1544] в
приближении «жесткий ротатор—гармонический осциллятор» с учетом двух дублетных
возбужденных состояний; результаты обоих расчетов согласуются в пределах 1 Дж-К-моль~1 в значениях
Ф° (Г) и S° (T).
Константа равновесия реакции A1FC1 (г)=А1 (r)+F (г)+С1 (г) вычислена с использованием
значения ЬГН° @) = 1004,241 +40 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^0 (A1FC1, г, 0) = —480 + 40 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах A1FC1,
A1F2 и А1С12.
A1F2CI (г). Термодинамические свойства газообразного дифторид-хлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 658.
Экспериментальные данные о спектре и строении молекулы A1F2C1 в литературе отсутствуют.
Приведенные в табл. 21.17 молекулярные постоянные A1F2C1, использованные для расчета
термодинамических функций, оценены по постоянным молекул A1F3 и А1С13. В основном состоянии
%гАг молекула A1F2C1 должна иметь плоскую структуру симметрии С2„. Произведение главных
моментов инерции соответствует межатомным расстояниям г (А1—F) = 1,63+0,04 А, г (А1—С1) =
=2,06 +0,06 А и'углам *$?—А1—С1=^ F—A1—F = 120+10°. Погрешность значения IaIbIc не
превышает 3-1014 г3-см6. Погрешности оцененных частот составляют 10—15% от их величины.
Близкие значения постоянных A1F2C1 были получены ранее в результате оценок авторами [1544],
а также Соломоником и др. [389].
Термодинамические функции A1F2C1 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A. 122) — A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности рассчитанных величин составляют 2,
5 и 8 Дж- К- моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К. При температурах ниже 1000 К
они обусловлены в основном неточностью молекулярных постоянных, при более высоких
температурах становятся существенными погрешности из-за применения приближенного метода расчета.
Ранее термодинамические функции A1F2G1 (г) вычислялись в справочниках [977, 1544, 1904],
а также Соломоником и др. [389] (Т < 6000 К). Данные табл. 658 и расчетов [1544, 389]
согласуются в пределах 0,3 Дж-К-моль. Расхождения с данными [977, 1904] достигают
1—2 Дж-К^-моль.
Константа равновесия реакции A1F2C1 (г)=А1 (r)-f-2F (r)-fCl (г) вычислена с использованием
значения Дг#° @)=1597,516+8 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я°(А1Р2С1, г, 0) = —996 + 7 кДж • моль.
Эта величина основана на исследовании испарения A1F3 (к) в присутствии А1С13 (г),
проведенном Краусом и Дугласом [1772а] методом протока A196—1257 К, 8 точек). В расчетах авторы
учитывали образование A1F2C1, A1FC12 и смешанных молекул Al2FnCl6_B и нашли, что для реакции
2/3А1Р3(г)+г/3А1С13(г)=А1Р2С1 (г) Дг#°A225 К)=2+4 нДж-моль. Погрешность принятого
значения учитывает неточность измерений и термодинамических величин, использованных в
расчетах.
A1FC12 (г). Термодинамические свойства газообразного фторид-дихлорида алюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 659.
Спектр и структура A1FC12 экспериментально не исследовались. Молекулярные постоянные
A1FC12, использованные для расчета термодинамических функций и приведенные в табл. 21.17,
оценены по постоянным молекул A1F3 и A1C1S. Приведенное в табл. 21.17 произведение главных мо-
147
AIHFC1 (г), AlBr (г)| Глава 2$
ментов инерции молекулы A1FC12 в состоянии Х1А1 рассчитано для плоской структуры симметрии
С2„ с параметрами г (A1-F)=1,63 +0,04 А, г (A1-G1) =2,06+0,05 А, ^ F-A1-C1=^ Gl—A1—С1-=
=120+10°. Погрешность значения IaIbIc не превышает 5-10~114 г3-см6. Погрешности оцененных
значений основных частот составляют 10—15%. Близкие значения молекулярных постоянных
A1FC12 оценены авторами [1544] и Соломоником и др. [389].
Термодинамические функции A1FG12 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128) и A. 130) с учетом
только основного электронного состояния; их погрешности обусловлены главным образом
приближенным характером расчета и (ниже 1000 К) неточностью оцененных молекулярных
постоянных. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К составляют 2, 6 и 10 Дж-К-
• моль.
Ранее термодинамические функции A1FG13 (г) вычисляли авторы справочных изданий [977,
1544, 1904] и Соломоник и др. [389] (Г ^ 6000 К). Термодинамические функции, приведенные
в табл. 659 и справочнике JANAF [1544], совпадают в пределах 0,05 Дж-К~1-мояь~1.
Расхождения с данными [389, 1904] не превышают 1—2 Дж-К'Моль~1.
Константа равновесия реакции A1FG12 (г)=А1 (r)+F (г)+2С1 (г) вычислена с использованием
значения ДгЯ°@)=>1432,861+8 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я° (AlFCla, г, 0) == —789 + 7 кДж • моль.
Эта величина основана на выполненном Краусом и Дугласом [1772а] исследовании
испарения A1F3 (к) в присутствии А1С13 (г) (метод протока, 1196—1257 К, 8 точек). Из этих данных
авторы нашли для реакции V3A1F3 (г)+2/3А1С13 (r)=AlFCl2 (г) значение Д^0 A225 К)=2+4 кДж-
-моль. Погрешность принятой величины учитывает неточность измерений, а также
термодинамических величин, использованных в расчетах.
A1HFC1 (г). Термодинамические свойства газообразного гидрид-фторид-хлорида алюминия
в стандартном состоянии в интервале температур 100—6000 К приведены в табл. 660.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 21.17. Спектры и структура A1HFC1 не исследовались, и все постоянные оценены
на основании данных для молекул типа А1Х3 и A1XY2, где X, Y=H, F, Gl. Приведенное в табл.
21.17 значение IiIbIc рассчитано для плоской структуры молекулы A1HFC1 (точечная группа Cs)
с межатомными расстояниями г (А1—Н)=1,59+0,03 А, г (А1—F)=l,63+0,04 А, г (А1—С1) =
=2,06+0,05 А и углами J} Н—А1—F=-$ F—Al—Cl = ^ H—Al—Gl=120+10°. Погрешность
произведения главных моментов инерции составляет 5-Ю15 г3-см6. Частоты колебаний, приведенные
в таблице 21.17, оценены по частотам молекул А1Х3 и A1XY2; их погрешности составляют 15—20%.
Термодинамические функции A1HFG1 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с
учетом только основного электронного состояния. Погрешности рассчитанных величин обусловлены
приближенным методом расчета и неточностью оценки молекулярных постоянных; они составляют
3, 10 и 12 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1HFG1 (г) публикуется впервые.
Константа равновесия реакции A1HFG1 (г)=А1 (г)+Н (r)-f-F (г)+С1 (г) вычислена с
использованием значения ДГЯ° @)=1265,275 +25 кДж-моль, соответствующего принятой в
справочнике энтальпии образования
г, 0) = —525 + 25 кДж • моль.
Эта величина оценена в предположении о равенстве средних энергий связей в молекулах
A1HFG1, АШ3, A1F3 и А1С13.
AlBr (г). Термодинамические свойства газообразного бромида алюминия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 661.
Молекулярные постоянные А179Вг, принятые в справочнике на основании результатов
исследований электронного и вращательного спектров этой молекулы, представлены в табл. 21.14.
В электронном спектре AlBr наблюдались две системы полос: а3По+, a3nt -> Х1Е+ [1805, 2021,
2503] и ЛЧ1 ** XXS+ [900, 1483, 1560, 1561, 1919, 2021, 2022, 2285, 2285а]. Помимо этих трех со-
148
Алюминия и его соединения AlBr (i)
стояний, молекула AlBr может иметь другие связанные состояния в области 40 000—50 000 см,
бблыпая часть которых должна быть ридберговскими, и все они не рассматриваются в справочнике.
Состояние А1П и подсостояние а3Пси- коррелируют с диссоциационным пределом AlBiJa/2)+BrBP5/j),
остальные компоненты состояния а3П, по-видимому, имеют общий диссоциационный предел с
состоянием ХХЕ+.
Колебательные постоянные в состояниях ХХЕ+ и АгП, приведенные в табл. 21.14, найдены
Яннергреном |1560, 1561] по (?-кантам полос системы 4Ч1-ГЕ+ (г/ ^ 3, v" <; 7). Рам [2285,
2285а] при исследовании системы полос А1!!—Х1Е+ (г/ ^ 3, v" <С 6) рассчитал значения
колебательных постоянных ш"е, и>ех"е, и/, ч>ех'е и <о,у'е, близкие к принятым в справочнике, однако для
состояния ХХИ+ он дополнительно получил постоянную <*>tye=—0,06 см. Благодаря большому
отрицательному значению юеув эти постоянные приводят к быстрому схождению колебательных
уровней состояния ХХ2+ при энергии, не достигающей 25% от величины Do (AlBr). Поэтому
в справочнике отдано предпочтение постоянным, найденным Яннергреном г.
Вращательные постоянные 27А179Вг в состоянии X1 Е+ приняты по работе Вайса и Горди [2873 ],
которые исследовали вращательные переходы с v <J! 5, / <J 27 в микроволновом спектре этой
молекулы. Близкие, но существенно менее точные значения получены Яннергреном [1561] и Рамом
[2285а] при исследовании вращательной структуры ряда полос системы А1!!—Хх?+.
Вращательные постоянные AlBr в состоянии А1!!, приведенные в табл. 21.14, найдены Яннергреном [1561],
они несколько отличаются (на 0,0004 см в значении Bt) от соответствующих данных [2285а].
Следует отметить, что состояние А1!! у молекулы AlBr является слабосвязанным; в спектрах
наблюдались переходы только на уровни с v ^ 3, причем на уровнях v=2 и 3 имеет место предиссо1
циация (см. [2285]).
Приведенные в табл. 21.14 колебательные постоянные AlBr в состоянии «3П приняты по работе
Шарма [2503], который рассчитал их по кантам полос двух.подсистем (i>'-<^ 12). Вращательные
постоянные основаны на результатах анализа полосы 0—0 в обеих подсистемах, выполненного
Лакшминараяна и Харанахом [1805].
Термодинамические функции AlBr (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения Qn и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом одного
возбужденного состояния (а3П) в предположении, что (?jjj)# 1?> = 6()№вр>. Статистическая суммй
по колебательно-вращательным уровням энергии состояния Х1!!* и ее производные находились
по уравнениям A. 73)—A. 75) непосредственным суммированием по значениям v и
интегрированием по значениям / с помощью уравнений типаA. 82). В расчете учитывались все уровни
состояния Х1Е+ со значениями / ^ /шах „, последние определялись из соотношений A. 81);
соответствующие значения уша1 и /Ит приведены в табл. 21.15. Колебательно-вращательные уровни
состояния X1 ?+ рассчитывались по уравнениям A.65) и A.62); в табл. 21.15 приведены значения
коэффициентов YM в этих уравнениях, они получены для изотопической модификации, соответствующей
природной смеси изотопов брома, по постоянным, принятым для молекулы 27А179Вг, и уравнениям
A. 66); коэффициенты Yk0, включая Yso, найдены с учетом условий A. 50).
При температурах до 1000 К погрешности рассчитанных термодинамических функций AlBr (г)
обусловлены главным образом неточностью фундаментальных постоянных, принятых в
справочнике. При более высоких температурах становятся существенными неточность известных
колебательных постоянных в состоянии ХХЕ+, отсутствие экспериментальных данных об уровнях этого
состояния с энергией больше 3000 см, а также приближенный учет вклада состояния а3П.
Пренебрежение вкладом слабосвязанного состояния А1!!, а также других ненаблюдавшихся
электронных состояний вносит существенно меньшие ошибки в значения Ф° (Т) и S° (T). Погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,03, 0,1 и 0,3 Дж-К-моль.
Термодинамические функции AlBr (г) ранее вычислялись в таблицах JANAF [1543] и работе
Альтмана [499]; расхождения данных [1543] и табл. 661 достигают 0,1, 1 и 5 Дж-К~1-моль
В работе [2285а] приведены волновые числа начал семи полос, позволяющие рассчитать соответствующие
значения ДФ/г—Дб'/2. Эти значения существенно отличаются от рассчитанных Рамом по кантам полос и
незакономерно изменяются с увеличением квантового числа v. По-видимому, точность определения этих величин в
работе [2285а] экстраполяцией значений волновых чисел линий ф-ветвей от /%40 к /=0 невелика;
соответствующие значения плохо описываются колебательными постоянными, найденными автором [2285, 2285а], и
значительно лучше постоянными, принятыми в табл. 21.14.
149
А1Вг2 (г) Глава 21
в значениях Ф° (Т), S° (Т) и С°р (Т) и обусловлены использованием в таблицах [1543] менее точной
методики расчета и отличающихся молекулярных постоянных АШг, а также пренебрежением
вклада состояния аЧ1. В работе [499], по-видимому, допущены ошибки в расчетах, и расхождения
достигают 3—4 Дж-К~1-моль~1 в значениях Ф° (Т) и S° (T).
Константа равновесия реакции АШг (г)=А1 (г)+Вг (г) вычислена с использованием принятой
в справочнике энергии диссоциации
АгН° @) = D0(AlBr) = 432 + 6 кДж • моль« 36 100 + 500 см.
Это значение получено как средневзвешенное из результатов обработки измерений равновесия
реакции образования АШг (г) в работах [90, 374, 1286а]. Семенкович |374] исследовал
равновесие реакции NaBr (ж)+А1 (ж)=А1Вг (r)+Na (г) A точка, 1175 К) газокинетическим методом.
Пересчет данных [374] с учетом образования в парах иодида натрия молекул Na2I2 и Nal привел
к значению Do (AlBr) =437+10 кДж-моль. Горохов и др. [90] исследовали реакцию NaBr (г)+
+А1 (г)=А1Вг (r)+Na (г) масс-спектрометрическим методом A251—1389 К, 35 точек); эти
измерения дают значение 442+12 кДж-моль (III закон). Детальное изучение равновесия 2А1 (ж) +
+АШг3 (г)=ЗА1Вг (г) выполнено Гроссом и Левиным [1286а] методом протока A015—1568 К,
12 точек). Обработка данных [1286а] привела к значениям Do (А1Вг)=429+5 [кДж-моль (III
закон) и 418+7 кДж-моль (II закон).
Рам [2885, 2885а] при исследовании спектра испускания АШг обнаружил предиссоциацию
на уровнях и=2 и 3 состояния Л 41 при /=93 и 67 соответственно. Автор [2885, 2885а]
интерпретировал эту предиссоциацию как обусловленную взаимодействием состояния Л 41 с отталкиватель-
ной ветвью другого слабосвязанного состояния, коррелирующего с диссоциационным пределом
AlBPVs)+Br (Чр*/г), и рекомендовал Do (А1Вг)=37 000+200 см (с-442,5+2,5 кДж-моль-1).
Как видно из приведенных величин, это значение несовместимо с результатами наиболее точных
измерений равновесия образования АШг (г), полученными Гроссом и Левиным. Поскольку трудно
допустить наличие такой систематической ошибки в данных [1286а], а исследование предиссоциа-
ции в состоянии АЩ может дать завышенное значение Do (AlBr), если на потенциальной кривой
состояния, вызывающего предиссоциацию, имеется максимум или это состояние является оттал-
кивательным, в справочнике отдано предпочтение величине, основанной на исследованиях
равновесия реакций с участием АШг (г).
Принятому значению t соответствует
г, 0) = 13,269 + 7,2 кДж • моль.
А1Вг2 (г). Термодинамические свойства газообразного дибромида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 662.
Экспериментальные данные о структуре и спектре молекулы А1Вга в литературе отсутствуют.
Молекулярные постоянные, приведенные в табл. 21.16, оценены при подготовке справочника.
По аналогии с другими молекулами дигалогенидов бора и алюминия принято, что в состоянии
ХгАх молекула А1Вг2 имеет угловую структуру симметрии Civ. Произведение главных моментов
инерции, приведенное в табл. 21.16, вычислено в предположении, что -$ Вг—А1—Вг=120+10°,
г (А1—Вг) =2,22 +0,05 А (как в А1Вг3). Погрешность значения IJbIg оценивается в 2-1013 г3- см6.
Частоты колебаний оценены по силовым постоянным молекулы AlBrg1. Погрешности приведенных
частот оцениваются в 15—20%. Энергия возбуждения состояния АгВх оценена по данным для
других дигалогенидов бора и алюминия; ее погрешность составляет 5000 см.
Термодинамическиэ функции А1Вг2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
возбужденного электронного состояния по уравнениям A. 168)—A. 170). Погрешности
рассчитанных термодинамических функций обусловлены главным образом неточностью молекулярных
постоянных, в том числе энергии возбуждения состояния А2ВХ, а также приближенным характером
расчета. Общие погрешности в значениях Ф° (Т) могут достигать 2, 4 и 8 Дж-К~1-моль при
298,15, 3000 и 6000 К.
Таблица термодинамических функций АШг2 (г) публикуется впервые.
1 Выло пршшто (в 10е дпи-см): /,.= 2,19, /гг=0,13 и /„/^=0,08.
150
Алюминий и его соединения АШг3 (к, ж),А1Вг3 (г)
Константа равновесия реакции А1Вг2 (г)=А1 (г)+2Вг (г) вычислена с использованием
значения Д^0 @)=723,192+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^Н°(А1Вт2, г, 0) = —160 ±50 кДж.моль.
Эта величина оценена сопоставлением энергий связей в молекулах АШг, АШг2, А1Вг8 и в
соответствующих соединениях алюминия с фтором и хлором.
А1Вг3 (к, ж). Термодинамические свойства кристаллического и жидкого трибромида алюминия
в стандартном состоянии при температурах 100—700 К приведены в табл. Д.50 г.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 21.1. В справочнике за стандартное состояние АШг3 (к) в интервале температур 0—371,16 К
принята моноклинная модификация [2326а]. Термодинамические функции АШг3 (к) при
Г< 298,15 К вычислены по значениям теплоемкости, измеренным Уэббом и др. [2824] A1,6—
308,6 К) на образце, содержащем не менее 99,9% А1Вг3. Точность измерений при Т > 30 К
оценивается в 0,5%. Экстраполяция теплоемкости ниже 12 К по уравнению C°P=3D F1,3/Г)-Ь
+ 3? (92,4/7)+?" A73/Г) [2824] приводит к значению S0 A2 К) =4,02 Дж- К- моль. Погрешности
принятых по [2824] значений S° B98,15 К) и Я° B98,15 К)—#°@) оцениваютсяв ^ОДж-К^-моль
и0,1 кДж- моль^соответственно. Уравнение для теплоемкости А1Вг3(к)в интервале298,15—371,16 К
(см. табл. 21.1) выведено на основании данных [2824] по теплоемкости B98—308,6 К) и
результатов измерений энтальпии в работе Фишера [1094] B73—367 К). Температура плавления
C71,16 ±0,10 К) найдена Тонстэдом [2692]; менее точные измерения [131, 485, 687, 691, 1094,
1533,1547, 1668,1726] приводят к значениям Тт, лежащим в интервале 369,6—371,1 К. Энтальпия
плавления A1,25 ±0,80 кДж-моль) принята по согласующимся между собой калориметрическим
измерениям Фишера [1094] и криоскопическим определениям Тонстэда [2692]. Значение
теплоемкости А1Вг3 (ж) A25,0+3,0 Дж> К* моль) основано на результатах измерений энтальпии,
проведенных Фишером [1094] в сравнительно небольшом интервале температур C74—406 К).
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 700 К оцениваются в 0,6, 2 и
6 Дж-К-моль~1 соответственно.
Расхождения между термодинамическими функциями А1Вг3 (к, ж), приведенными в табл.
Д. 50 и в справочнике [1544], не превышают 0,5 Дж-К>моль в значениях Ф° (Т) при
Т < 700 К 2.
В справочнике принята энтальпия образования
A^°(AlBr3, к, 298,15К) = —511,5 + 1,0 кДж.моль.
Результаты исследований энтальпии образования А1Вг3 (к) приведены в табл. 21.29. Как
видно из этой таблицы, данные большинства работ расходятся значительно больше, чем
это можно было бы объяснить приписанными им погрешностями. Вероятно, это является
следствием высокой гигроскопичности А1С13 и А1Вг3 и недостаточно точно замкнутых термохимических
циклов в работах [328, 526, 1025, 1726]. Методически наиболее совершенной является работа
Гросса и др. [1274, 1286]. Совпадающее в пределах погрешностей вначение получено Ефимовым
и др. [126а, 1017а]. В этих исследованиях использованы образцы высокой чистоты, приняты
особые меры по предохранению образцов от влаги и исследованные реакции составляли строго
замкнутый цикл. Принятая величина является средневзвешенной из измерений, выполненных
Гроссом и др. [1274, 1286] и Ефимовым и др. [126а, 1017а].
А1Вг3 (г). Термодинамические свойства газообразного трибромида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 663.
Экспериментально структура молекулы А1Вг3 не исследовалась. По аналогии с другими
молекулами тригалогенидов бора и алюминия в справочнике принимается, что в состоянии ДМ/
молекула А1Вг3 имеет плоскую симметричную структуру (симметрия DSh). С такой структурой
1 Критическая температура А1Вг3 равна 763 К (см. табл. Д.65).
2 В таблицах JANAF [1544] термодинамические функции А1Вг8 (к, ж) рассчитаны до 1500 К, хотя для этого
вещества Г1р=763±2К.
151
А1Вг3 (г)
Глава 21
Таблица 21.29. Результаты определений Д/-ЕГ° (А1Вг3, к, 298,15 К) (в кДжмоль)
Авторы
Метод
AfH° (А1Вг3, к,
298,15 К)
Плотников, Якубсон, 1938 [328]
—515+5
Измерение энтальпии растворения АШгз (к)+
+3000 Н2О(ж)=А1Вг8-3000 Н2О(р-р), ДгЯеB88К)=
=-382,7±4,0 а
Измерение энтальпии растворения А1Вг3 и А1С1з в со- —509+5
ляной кислоте, АШгз (к)+ЗС1~ (ад)=А1С1з (к)+
+ЗВг" (ад), АГН° B98 К) =—59,6+4,0
То же, ДГЯ° B98 К)=-38,3±3,3 —530±5
Измерение энтальпии реакций: А1 (к)+3/2Вг2 (ж)= —511,12±0,84
=А1Вг8 (р-р в Вг2), ДгЯ°B98К)=-501,79+0,33;
АШгз (к)+Вгг (ж)=А1Вгз (р-р в Вг2), ДГЯ° B98 К)=
= 9,33±0,08
Измерение энтальпии растворения AlClj и АШгз в ще- —496,7 ±3,0
лочи (—353,1 ±1,9 и —424,5±1,8 соответственно)
Измерение энтальпий рчстворения AlBrj (к), А1С13 (к), —512,9±1,9
КВг (к) и KG1 (к) в соляной кислоте, А1С1з (к)+
+ЗКВг (к)=А1Вг8 (к)+ЗКС1 (к), ДГЯ° B98 К)-=
=63,4±1,3
а Менее точные значения энтальпии этой реакции были найдены в ныне устаревших работах [108, 671].
Клемм, Танке, 1931 [1726]
Эли, Уотте, 1954 [1025]
Гросс и др., 1971 [1274, 1286]
Энтони и др., 1973 [526]
Ефимов и др., 1979 [126а, 1017а]
согласуются результаты спектральных исследований [320, 368, 636, 2244, 2468]. В справочнике
принято значение г (А1—Вг) =2,22 +0,03 А, оцененное на основании данных Шена [2506] о длине
внешней связи А1—Вг в молекуле А12Вгв (см. ниже) и заключения, сделанного Засориньш я Рам-
биди [134], о том, что длины внешних связей М—С1 в молекулах M2Cle (М=А1, Fe) совпадают в
пределах ошибки эксперимента с межатомными расстояниями в молекулах MCI31. Соответствующее
этому значению г (А1—Вг) произведение главных моментов инерции, приведенное в табл. 21.17,
имеет погрешность 3-10~113 г3-см6.
Приведенные в таблице значения частот симметричного валентного колебания \ и
деформационного колебания \ получены Битти и Хордером [636 ] при исследовании спектра КР
газообразного А1Вг3. Близкие значения v4 получены в работах [320, 2244] при исследованиях ИК-спектра
молекул А1Вг3, изолированных в матрицах из Хе и Аг, а также ИК-спектра газообразного А1Вг,
[368]. Частота антисимметричного валентного колебания v3 наблюдалась только в ИК-спектре
молекул А1Вг3, изолированных в матрицах из Хе [320] и Аг [2244, 2468], в которых получены
хорошо согласующиеся значения. Погрешности в значениях \ и v4 не превышают 5 см,
погрешность в v3 из-за возможного матричного сдвига может достигать 10—15 см 2.
Принятое значение частоты внеплоскостного колебания v2 найдено Понгом и др. [2244, 2245а]
при исследовании ИК-спектра молекул AlBrs, изолированных в матрице из Аг. Селиванов и
Мальцев [368] в ИК-спектре паров А1Вг3 в области 107 см и Перов и др. [320] в ИК-спектре молекул
А1Вг3 в матрице из Аг в области 109 см наблюдали полосу, отнесенную ими к частоте va этой
молекулы. Однако это отнесение, по-видимому, ошибочно, так как Понг и др. в повторном
исследовании [2245а] специально отметили отсутствие полос в области 107—109 см. Кроме того,
найденному в работах [320, 368] значению v2 соответствует аномально высокое для этого класса
молекул отношение силовых постоянных /г и / , равное 40. В молекулах A1F3 и А1С13 оно равно 20,
в тригалогенидах бора —10, а данным [2245а] соответствует ~20 3. Погрешность принятого
значения v2 оценивается в 10 см. Сведения о возбужденных состояниях А1Вг3 в литературе
отсутствуют; по аналогии с другими тригалогенидами бора и алюминия можно предполагать, что их
энергии превышают 20 000 см.
1 В пределах погрешностей принятое значение согласуется с рекомендованным для А1Вг3 Засориным и Рам-
биди [134] B,20 ±0,02 А) на основании данных для А1аВгв.
а Отнесение в работе [636] к частоте v3 слабой широкой полосы при 360 см оказалось ошибочным.
• В справочнике JANAF [1544] имеется ссылка на неопубликованную работу Шерка и Лукаса, в которой для
v2 при исследовании ИК-спектра А1Вг8 в матрице из Аг получено значение 162 см.
152
Алюминий и его соединения АЬВг6 (г)
Термодинамические функции А1Вг3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130) с учетом
только основного электронного состояния. Погрешности рассчитанных величин составляют 2,
7 и 10 Дж-К-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К и обусловлены в основном
приближенным характером расчета; погрешности из-за неточности молекулярных постоянных,
не превышают 1—2 Дж-К-моль~1.
Термодинамические функции АШг3 (г) вычислялись ранее в справочнике JANAF [1544],
а также в работах Уэбба и др. [2824] (Т < 3000 К) и Юшина и др. [474а] (Т < 1500 К).
Расхождения данных табл. 663 и [1544, 2824] не превышают 0,4 Дж- К-моль в значениях Ф° (Т) и S° (Т)
и обусловлены главным образом различием в оцененной величине г (А1—Вг).
Константа равновесия реакции А1Вг3 (г)=А1 (г)+ЗВг (г) вычислена с использованием
значения &ГН° @) = 1067,115+8,9 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
\fH° (А]Вг3, г, 0) = —386 ± 8 кДж • моль.
Эта величина основана на измерениях равновесия реакции А12Вг6 (г)=2А1Вг3 (г), выполненных
Фишером и др. [1100] (статический метод и измерение плотности пара, 613—854 К, 11 точек).
Результатам измерений [1100] соответствуют значения энтальпии диссоциации А12Вг6, равные 125,0 +
±0,5 кДж-моль (III закон) и 121,5 ±4,5 кДж-моль (II закон), где погрешности
характеризуют только воспроизводимость измерений. Близкие значения получены аналогичным методом
в работе Суворова и др. [401] E13—693 К, 127,2 кДж-моль (III закон) и 112 кДж-моль (II
закон)), однако эта работа опубликована в сокращенном виде, что не позволило провести ее
детальную обработку. Погрешность принятого значения Д^Я° (А1Вг3, г) учитывает неточность
термодинамических функций компонентов реакции, энтальпии образования А14Вгв (г) и воспроизводимость
измерений.
А12Вгб (г). Термодинамические свойства газообразного гексабромида диалюминия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 664.
Молекулярные постоянные, использованные для расчета термодинамических функций А12Вгв (г)г
приведены в табл. 21.23. Электронографические исследования А1аВгв [4, 2175, 2506],.
а также исследования спектров [320, 482, 635, 636] показывают, что молекула А12Вг6 в состоянии
ХХА имеет мостиковую структуру и принадлежит к точечной группе симметрии Dih.
Произведение моментов инерции вычислено по структурным параметрам г(А1—Вгмешн) = 2,222+ 0,005 А,.
/¦(А1-ВгИ0С1НК) = 2,414 ±0,007 А, ДВгвяеши-А1-Вг1нешн= 122,8 ±3,3°, Д Вгяов„-А1-Вгив01И11 =
=92,3+0,9°, найденным Шеном [2506] в результате электронографического исследования.
Погрешность значения IaIbIo составляет 4-Ю11 г3-смв. Колебательные спектры молекулы А12Вг»,
исследовались в газовой [320, 636], жидкой [322, 482, 1194, 2362] и твердой фазах [322, 635, 2824],
а также в матрице [320]. В указанных работах получены согласующиеся наборы частот колебаний.
8 табл. 21.23 для частот v2—v4, v7, v12 приняты величины, полученные в спектре КР паров Битти
и Хордером [636]. Значения частот v1? ve, vu, v14, v17, v18 найдены Битти и др. [635] в результате
исследования ИК-спектров и спектров КР А12Вгв в твердой и жидкой фазах. Для частот v8, v13,
vie приняты величины, полученные в газовой фазе Перовым и др. [320]. Частоты \, v9, v10 и v15.
в спектрах А12Вгв не наблюдались. Частота v5 рассчитана с использованием среднего значения
отношений v< (Al2Bre)/v< (А12С1в)=0,7+0,1. Значение v15 оценено на основании значений
соответствующей частоты в молекулах А12С1„ и А121„1. Для частоты v9 принято среднее значение
междувеличинами, рассчитанными авторами работ [483, 635]. Частота v10 рассчитана Битти и др. [635].
Погрешности принятых значений составляют от 2—5 см для частот, наблюдавшихся в спектрах
паров, и до 20—30 см для остальных частот. Сведения о возбужденных электронных состояниях
А12Вгв в литературе отсутствуют.
Термодинамические функции А12Вгв (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
1 В работе [635] к частоте v16 предположительно отнесена полоса 79 см х, однако авторы [635] отмечают, что эта
полоса может относиться к колебаниям решетки. Кроме того, это значение превышает частоту v16=77 см
в молекуле А12С1е.
153
АИ (г) Глава 21
A. 130). Погрешности рассчитанных значений определяются приближенным характером расчета
и при умеренных температурах неточностью ряда частот колебаний. Они оцениваются в 10, 25
и 40 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции А12Вгв (г) рассчитывались в справочнике [1544], а также
в работе Уэбба и др. [2824] до 3000 К. Расхождения данных, приведенных в табл. 664 и
справочнике [1544], составляют 10—11 Дж-К-моль в значениях Ф° (Г) и 5° (Т) и объясняются
главным образом различием в принятых значениях частот v5, v9, v10 и v15.
Константа равновесия реакции А12Вг8 (г)=2А1 (г)+6Вг (г) вычислена с использованием
значения ДГЯ° @) =2259,23 +11 кДж-моль, соответствующего принятой в справочнике энтальпии
•образования
^fH°{A[2Bv6, г, 0) = —897 + 7 кДж ¦ моль.
Эта величина вычислена как сумма энтальпии образования А1Вг3 (к) (см. выше) и энтальпии
«сублимации 2А1Вг3 (к)=А12Вг6 (г) Д,Яо@)=87+7 кДж-моль, являющейся средневзвешенной
из значений, приведенных в табл. 21.30 и найденных по результатам измерений давления пара
А12Вг„ (г) над А1Вг3 (к, ж).
Таблица 21.30. Результаты определений энтальпии реакции 2А1Вг8 (к, ж)=А12Вг„ (г) (в кДж-моль)
Автор
Фишер и др., 1932 [1100]
Данн, Грегори, 1958 [996]
Джонсон и др., 1968 [1581]
Зульцманн, 1974 [2637]
Метод
Иаотенископа, точек кипения, весовой метод
Руффа и переноса, 392—523 К, 27 точек
Эффузпонный, 273—310 К, данные
представлены в виде уравнения
Точек кипения, 536—639 К, 15 точек
То же, 639—761 К, 14 точек
Статический, 302—335 К, 13 точек
Д„Я° @)
II закон
91,5±0,6
87,7
95,4±0,4
103,3±0,5
94,7±1,5
III закон
86,6±0,2
87,7
84,9 ±0,3
81,7±0,8
88,7±0,2
Приведенные в таблице погрешности отражают только воспроизводимость измерений. При
вычислении средневзвешенной были учтены погрешности из-за неточности термодинамических
функций (около 7 кДж-моль при 500 К) и отклонение результатов данной работы от средней величины.
•Средневзвешенное оказалось близким к результатам наиболее полного исследования,
выполненного Фишером и др. [1100].
АН (г). Термодинамические свойства газообразного иодида алюминия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 665.
В табл. 21.14 представлены молекулярные постоянные АИ, принятые на основании
результатов исследований электронного и вращательного спектров этой молекулы. В электронном спектре
АИ наблюдались две системы полос: АШ +* X1?,* [1955а, 2022] и а3П -> ХХЛ+ [1806, 1955а, 2020,
2021]. Так же как у других галогенидов элементов группы IIIA, основное Х1^ и возбужденные
а3П и А1!! состояния молекулы АИ коррелируют с диссоциационными пределами AlBPi/2, з/2)+
+1BР>/2). Сравнение с другими молекулами этого класса позволяет предполагать, что молекула
АН должна иметь помимо состояний, приведенных в табл. 21.14, другие связанные состояния в
области 30 000—50 000 см, главным образом состояния ридберговского типа; в справочнике эти
состояния не рассматриваются.
Вращательные постоянные АИ в состоянии X12+, приведенные в табл. 21.14, получены Вайсом
и Горди [2872, 2873] при исследовании микроволнового спектра АН (у ^ 14, / <; 39). С этими
постоянными согласуются в пределах погрешности их определения менее точные значения,
найденные из анализа вращательной структуры полос системы а3П ->¦ X1 ?+ в работе [1806].
Вращательные постоянные АН в состоянии а3П приняты по работе [1955а], в которой выполнен анализ
структуры полос 0—0, 0—1, 1—0, 1—2, 2—0, 2—1 подсистемы а3П0—XXL+ и полос 0—0 и 1—1
подсистемы а3Пх—ХХЕ+ с использованием значений вращательных постоянных в
найденных Вайсом и Горди [2873].
154
Алюминий и его соединения АН (г)
Колебательные постоянные АН в состояниях X1 ?+ и а3^ определялись в работах [1955а,
2021 ] и хорошо согласуются между собой. Приведенные в табл. 21.14 значения постоянных найдены
Мартиным и Барроу [1955а] в результате анализа колебательной структуры переходов
а3По+— ХХЕ+ (г/< 9, v" < 7) и аЧ^—Х1^ (i/ < 11, v" < 9), проведенного по началам полос,
волновые числа которых были определены по кантам соответствующих полос и результатам
анализа вращательной структуры.
Переход А1!!—Xх ?+, характерный для всех галогенидов элементов группы IIIA, в случае
молекулы АН представляет собой диффузное поглощение в области )Д 3426—3175 А [1955а, 2022].
Это свидетельствует о том, что у АН состояние А1!! является нестабильным или слабосвязанным.
Термодинамические функции АН (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения (?,„ и ее производных по температуре рассчитывались по уравнениям A. 90—1. 92) с
учетом возбужденного состояния а3П в предположении, что <?^# вр = 6()№ вр. Статистическая сумма
по колебательно-вращательным уровням энергии состояния X1 S+ и ее производные по температуре
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням по уравнениям типа A.82). В расчете
учитывались все уровни со значениями / ^ Jma.it гДе «Лши, ¦> находились из условий A. 81).
Колебательно-вращательные уровни энергии состояния ХХ2+ вычислялись по уравнениям A.62),
A. 65). Значения коэффициентов Ykl в этих уравнениях, а также величин #„„ и /1Ш приведены
в табл. 21.15; коэффициенты Yk0 получены из постоянных, принятых в табл. 21.14, с учетом
условий A. 50).
Молекулярные постоянные АН в состоянии Х1^* известны с хорошей точностью, поэтому
погрешности рассчитанных термодинамических функций при температурах вплоть до 2000—3000 К
определяются только неточностью фундаментальных постоянных. При более высоких
температурах становятся существенными погрешности из-за отсутствия экспериментальных данных об
энергии колебательно-вращательных уровней этого состояния cd^ 20, а также из-за пренебрежения
ненаблюдавшимися состояниями с энергиями более 30 000 см. Погрешности в значениях Ф° (Т)
при 298,15, 3000 и 6000 К составляют 0,02, 0,05, и 0,2 Дж-К^-моль.
Термодинамические функции АН (г) ранее вычислялись в таблицах JANAF [1543];
расхождения данных [1543] и настоящего справочника достигают 0,5, 2 и 5 Дж-К-моль~1 в значениях
Ф° (Т), S0 (Т)иС°р(Т) и обусловлены тем, что авторы[1543] использовали более грубую методику ине
учитывали состояние а8П.
Константа равновесия реакции АН (г)=А1 (г)+1 (г) вычислена по принятой в справочнике
энергии диссоциации
ДгЯ° @) = DO(A1I) = 30 590 ±150 см« 366 + 2 кДж • моль.
Это значение получено Мартиным и Барроу [1955а] на основании построения потенциальной
кривой отталкивательного состояния АХП по данным о максимумах поглощения, наблюдавшихся
в диффузном спектре АИ, соответствующем переходу Л 41 <- X1!,*. Полученная таким образом
зависимость V (г) для области 2,30 ^ г ^ 2,80 А была экстраполирована кг->юи после
внесения поправки, учитывающей распределение молекул АН в условиях эксперимента по
вращательным уровням состояния ХХ1,+, привела к указанной выше величине.
Принятое значение находится в хорошем согласии с результатами исследований равновесия
реакций с участием АН (г) в работах [367, 377, 1413], но превосходит их по точности. Сандулова
и др. [367] исследовали равновесие реакции 2А1 (ж) + АИ 3 (г)=3 All (г) статическим методом (988—
1193 К, 10 точек); полученным в этой работе данным соответствуют значения Do (All)=395+25
(II закон) и 378±10 кДж-моль (III закон). Приведенные погрешности здесь и ниже учитывают
воспроизводимость измерений, неточность термодинамических функций и термохимических
величин, использованных в расчетах. Семенкович [377] исследовал реакцию А1 (ж)+К1 (г)=АП (г) +
+К (г) газокинетическим методом при 1205 К. Обработка его измерений с учетом присутствия
в парах иодида калия молекул KI и К212 привела к значению Do (All)=354+15 кДж-моль.
Хилденбранд и Клейншмидт [1413, 17166] исследовали равновесие реакции Sr (г)+АП(г) =
=SrI (г)+А1 (г) масс-спектрометрическим методом. Эти измерения использованы для
выбора значения Do (Sri), данные для которого менее надежны.
Принятой энергии диссоциации соответствует
bfH° @) = 68,509 + 4,5 кДж • моль.
155
АП2 (г), АП3 (к, ж) Глава 2t
АИ2 (г). Термодинамические свойства газообразного дииодида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 666.
Спектр и структура АП2 экспериментально не исследовались. Молекулярные постоянные АП2,.
приведенные в табл. 21.16, оценены. По аналогии с другими молекулами дигалогенидов бора и
алюминия в справочнике принято, что в электронном состоянии Х2Аг молекула АПа имеет угловую-
структуру симметрии С2с. Произведение главных моментов инерции, приведенное в табл. 21.16,
отвечает оцененным структурным параметрам ^ I—А1—1 = 120+10°, г (А1—I)=2,45+0,05 А.
Его погрешность составляет 1-1012 г3-см6. Частоты колебаний рассчитаны по силовым
постоянным молекулы АП31, их погрешности могут достигать 15—20%. Энергия возбуждения состояния
ЛгВ1 оценена по данным для молекул ВХ2, A1F2 и А1С12, ее погрешность достигает 5000 см.
Термодинамические функции АП2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A. 122)—A. 124), A. 128), A. 130) с учетом
возбужденного электронного состояния по уравнениям A. 168)—A. 170). Погрешности
рассчитанных величин обусловлены неточностью оцененных молекулярных постоянных, в том числе
энергии состояния Л2Ви а также приближенным характером расчета; погрешности в значениях
Ф° (Т) составляют 3, 5 и 10 Дж-К^-моль при 298,15, 3000 и 6000 К.
Таблица термодинамических функций АП2 (г) публикуется впервые.
Константа равновесия реакции АП2 (г)=А1 (г)+21 (г) вычислена с использованием значения
Дг/Г° @)=591,672+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —50 + 50 кДж • моль.
Эта величина оценена сопоставлением энергий связей в молекулах АН, АП2, АП3 и
аналогичных соединениях алюминия с фтором и хлором.
А113 (к, ж). Термодинамические свойства кристаллического и жидкого трииодида алюмжния
в стандартном состоянии ири температурах 298,15—900 К приведены в табл. Д. 51 а.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние АП3 (к) в интервале 0—461,47 К принята
гексагональная модификация. Экспериментальные данные о теплоемкости АН3 (к) при
Т <С 298,15 К в литературе отсутствуют; в работе Фишера и др. [1100] измерена энтальпия А113
в интервале 273—480 К. Результаты этих измерений позволяют рекомендовать значение
С; B98,15 К)=98,9±1,0 Дж-К^-моль. Энтропия АН3 (к) при 298,15 К, приведенная в табл.
21.1, вычислена исходя из величины S0 B98,15 К) для А1216 (г) и энтропии сублимации А121, (к)
при 298,15 К (см. ниже); погрешность принятого значения S° B98,15 К) для АИ3 (к) оценивается
в 15 Дж-К-моль~1. Величина Н° B98,15 К) — Н° @) оценена с использованием уравнения
Cf=D E7,8/7)+2Я A10/Г)+? (Ш/Т)+Е A404/Г), выведенного с учетом принятых выше
значений Ср B98,15 К) и S° B98,15 К). Погрешность Я° B98,15 К)—Я0 @) не превышает 1 кДж-
• моль. Для теплоемкости АП3 (к) в интервале 298,15—461,47 К принято уравнение (см. табл.
21.1), полученное на основании результатов измерений энтальпии, выполненных Фишером и др.
[1100]. Погрешность значений теплоемкости АП3, рассчитанных по этому уравнению, оценивается
в ~1%. Температура плавления D61,47+0,05 К) принята по результатам измерений Тонстэда
[2692], проведенных для образца, содержащего > 99,99% АП3. Значения Тт, полученные Молем
и др. [2050, 2051] (—452,7 К), ошибочны, по-видимому, вследствие значительного содержания
примесей в исследованных образцах. Энтальпия плавления A5,9+0,5 кДж-моль) и значение
теплоемкости АН3 (ж) A21,0+3,0 Дж'К-моль~1) приняты по результатам измерений Фишера
и др. [1100] в интервале 273—480 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 900 К оцениваются в 8, 10
и 15 Дж-К-моль соответственно. Термодинамические функции АП3 (к, ж), приведенные в табл.
Д. 51 и таблицах JANAF [1543], практически совпадают3.
1 Использованы значения (в, 106 дин-см l): /r=l,63, /rr=0,096 и fJi^—QfiQ.
2 Критическая температура АП3 равна 983 К (см. табл. Д.65).
* В [1543] табулированы свойства АП3 (к, ж) до 2000 К, т. е. существенно выше критической температуры.
156
.Адншвшш и его соединения
А113 (г)
Принятая в справочнике энтальпия образования
Д//°(А113) к, 298,15 К) = —302,9 + 2,0 кДж • моль
¦основана на результатах измерений, приведенных в табл. 21.31; их погрешности оценены при
подготовке справочника. Как видно, эти исследования плохо согласуются между собой, так как
расхождения значительно превышают погрешности измерений в отдельных работах. Измерения
энтальпии растворения АП3 (к) в воде [671, 934] приводят к близким величинам, однако они
содержат систематическую ошибку из-за невозможности надежного расчета поправки на энтальпию
Таблица 21.31. Результаты определений AfH° (Mis, к, 298,15 К) (в кДЖ'Модь")
Авторы
Метод
Д,Я° (АП3, г,
298,15 К)
Бертло, 1878 [671]
Клемм, Танке, 1931 [1726]
Дир, Зли, 1954 [934] (см. также
[1025])
Корбетт, Грегори, 1954 [881]
Энтони и др., 1973 [526]
Ефимов и др., 1979 [1017а]
Измерение энтальпии растворения Alls (к) в воде, ДГЯ" —322
(гаН2О, 298 К)=—386,3
Измерение энтальпии растворения АНз и A1CU в соля- —295,4±3,0
ной кислоте АНз (к)+ЗС1~ (а?)=А1С13 (к)+31~ (aq),
АГН" B98 К)=— 79,3±3,0
Измерение энтальпии растворения АНз (к) в воде, —321
ДГЯ° (о>Н20, 298 К)=—387,4±6,3
Исследование равновесия реакции Alls (к)+ЗНС1 (г)= —305,0±5,0
=А1С1з (к)+ЗШ (г) статическим методом, 298 К, 1 точка,
ДГЯ°B98К)=42,7+3,8
Измерение энтальпии растворения АНз и А1С1з в раство- —282,3+2,8
ре NaOH, Alls (K)+3NaCl G759 Н2О)=А1С13 (к)+ 3NaI
E239 Н2О), ДГЯ° B98 К)=90,8±2,5
Измерение энтальпий растворения АП3 (к), AlClj (к), —302,9±2,0
KI (к) и КС1 (к) в соляной кислоте, А1С13 (к)+ЗК1 (к)=
=АПз (к)+ЗКС1 (к), ДГЯ° B98 К)=80,1 ±1,4
разведения полученных растворов до бесконечного разведения (эта поправка принята по
справочнику [2787] равной —5 кДж-моль). В работах [1726] и [526] получена хорошая
воспроизводимость измерений, однако в них возможны погрешности, связанные с высокой гигроскопичностью
А1С13 (к) и АП3 (к) и с недостаточно точно сходящимися термохимическими циклами. Отметим,
что работа Энтони и др. [526] привела также к заниженному значению энтальпии образования
АШг3 (см. выше). Принятое в справочнике значение основывается на измерениях Ефимова и др.
[1017а], в которых были использованы образцы высокой чистоты, приняты меры по устранению
возможности их гидролиза и исследованные реакции составляли замкнутый цикл. Исследование
равновесия обменной реакции с участием АП3 (к) [881] привело к близкому, но менее точному
значению энтальпии образования АП3 (к).
АП3 (г). Термодинамические свойства газообразного трииодида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 667.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 21.17. Результаты электронографических исследований [4, 2506] показывают, что
молекула АП3 в состоянии Х1А'1 имеет плоскую структуру симметрии D3h. Произведение главных
моментов инерции, приведенное в табл. 21.17, вычислено для г (А1—I) =2,449+0,013 А,
найденного Шеном [2506] при электронографическом исследовании пара A1IS. Несколько менее
точное значение 2,44+0,02 А получено Акишиным и др. [4]. Погрешность в IaIbIo составляет
5-1012 г3-см6. Частоты колебаний \ и v4, приведенные в табл. 21.17, получены Битти и Хорде-
ром [636] при исследовании спектра КР газообразного АП3 при температуре ~ 700° С. Частота
v3 экспериментально не наблюдалась. Оценки ее величины на основании частот и силовых
постоянных, а также их отношений для молекул других тригалогенидов бора и алюминия дают для v3
значение 425+30 см, которое и принимается в настоящем справочнике *. Приведенная в табл.
* Близкие значения получены в результате оценок в работах [320, 390].
157
А1а16 (г) Глава 21
21.17 частота v2 оценена на основании предположения, что у АП3 отношение силовых постоянных
/г и / имеет такую же величину, как у молекул A1F3, A1C13 и А1Вг3 (~ 20) (см. с. 152).
Селиванов и Мальцев [368] при исследовании ИК-спектра паров АП3 отнесли к частоте
v2 полосу в области 77 см. По-видимому, это отнесение, так же как для А1Вг3 (см. выше),
является ошибочным. Частоте v2=77 см соответствует отношение /г//„ ~ 56, которое представляется
неправдоподобно большим для молекул этого класса; она также приводит к соотношению / < /а,
в то время как для всех изученных тригалогенидов бора, алюминия, галлия и индия справедливо
условие / ]> /я. Погрешности в значениях \ и \ могут составлять 5—10 см, в v2—20 см.
Сведения о возбужденных электронных состояниях АН3 в литературе отсутствуют; можно
предполагать, что их энергии превышают 15 000—20 000 см.
Термодинамические функции АП3 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128), A. 130).
Погрешности в значениях Ф° (Т) составляют 3, 9 и 12 Дж-К^-моль при 298,15, 3000 и 6000 К и
обусловлены в основном приближенным характером расчета, а также отсутствием надежных
экспериментальных данных о частотах v2 и v3.
Ранее термодинамические функции вычислялись в справочнике JANAF [1543], а также Юши-
ным и др. [474а] (Т <^ 1500 К). Расхождения данных [1543] и табл. 667 достигают 8—12 ДжХ
X К-моль в значениях Ф° (Т) и S°(T). Они обусловлены различием в выборе основных частот
АП3, который был сделан авторами [1543] до публикации работы [636]. Расчет [474а] согласуется
с настоящим изданием в пределах 3 Дж-К~1-моль.
Константа равновесия реакции АП3 (г)=А1 (г)+31 (г) вычислена с использованием значения
Д,^0 @) =834,835+6,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/гГв(АП8, г, 0) = —186 + 5 кДж • моль.
Эта величина основана на измерениях, представленных в табл. 21.32. Она является
средневзвешенной из приведенных результатов расчетов по уравнению III закона термодинамики.
Приведенные в таблице погрешности включают погрешности термодинамических функций,
воспроизводимость измерений и погрешности использованных в расчетах энтальпий образования.
Таблица 21.32. Результаты определений hfH° (А11з, г, 0) (в кДж-моль)
Фишер и
Грегори,
Авторы
др., 1932 [1100]
1977 [1260]
Метод
Метод переноса и измерение плотности пара,
А121в (г)=2А11з (г), 482-578 К, 8 точек
То же, Alls (к)=АП3 (г)
Статический и измерение плотности пара, А12Те, (г}=
=2АП3 (г), 614—750 К, 14 точек
Спектрофотометрический, АНз (к)=А11з (г), 420 К,
два измерения
II закон
—190±10
—194±6
—187+10
—192
(АП3, г, 0) ..vr
III закон
-182+10
—192±6
—180±10
-186+8
А1а16 (г). Термодинамические свойства газообразного гексаиодида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 668.
Молекулярные постоянные, использованные для расчета термодинамических функций А121в,
приведены в табл. 21.23. Электронографические исследования А121в [2175, 2506], а также
исследования спектров [482, 635, 636] показывают, что молекула А1316 имеет мостиковую структуру
и принадлежит к точечной группе симметрии Dih. Произведение моментов инерции вычислено
для значений г (А1-1внешн) = 2,449 + 0,013 А, г (А1-1К0„) = 2,634+0,030 А, Д1внешн_А1-1внетя =
= 115,0+7,4°, Д1МОСТ—А1—1МОСТ== 99,6 + 4,5°, найденных в работе Шена [2506]. Погрешность
IaIbIc составляет 1-1009 г3-см". Колебательные спектры молекулы А121„ исследовались в газовой
[636], жидкой [482, 635, 1568] и твердой [635] фазах. В табл. 21.23 для частот v2— v4, v7, v12
приняты величины, полученные в спектре КР паров А121в Битти и Хордером [636]. Значения \, vgf
158
Алюминий и его соединения • A1S (г)-
vu, v13—v18 найдены Битти и др. [635] при исследовании ИК-спектра и спектра КР А1216 в твердой
и жидкой фазах. Частоты v5, v6, v9, v10 экспериментально не наблюдались. Принятая частота v5
рассчитана по среднему значению отношения v<(Al2I6)/vf(Al2Cl6)=0,5+0,l. Для частот v6, v9
в табл. 21.23 приведены средние значения из величин, рассчитанных авторами работ [483, 635],
для v10 — рассчитанное Битти и др. [635]. Погрешности принятых значений оцениваются в 5 см
для частот, наблюдавшихся в газовой фазе, и 10—25 см для остальных частот. Сведения о
возбужденных электронных состояниях А1216 в литературе отсутствуют.
Термодинамические функции А121е (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных термодинамических функций определяются как неточностью
молекулярных постоянных, так и приближенным характером расчета и составляют 12, 25
и 40 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции А121в (г) рассчитывались в справочнике JANAF [1543].
Расхождения результатов расчета [1543] с данными, приведенными в табл. 668, составляют
5 Дж-К~1-моль~1 для всего интервала температур и объясняются некоторым различием в
принятых значениях молекулярных постоянных.
Константа равновесия реакции А121в (г)=2А1 (г)+61 (г) вычислена с использованием значения
Дг#° @) = 1775,67+14 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д//°(А1216, г, 0) ——478 + 12 кДж • моль.
Эта величина основана на энтальпии сублимации 2АП3 (к)=А1216 (г), рассчитанной по III
закону из данных работы Фишера и др. [1100] (метод переноса, 483—578 К, 8 точек) и равной
Д,Я° @)=123,5+12 кДж-моль. Основным источником погрешности в значении энтальпии
сублимации является неточность термодинамических функций компонентов реакции. Близкая
величина, 121 кДж-моль (III), получена по данным работы Грегори [1260] (статический метод,
спектрофотометрическое определение концентраций, 420 К, 2 измерения).
A1S (г). Термодинамические свойства газообразного сульфида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 669.
Молекулярные постоянные 27A132S, принятые на основании спектральных исследований
[241, 1777, 1827, 1900] и оценок, приведены в табл. 21.4. В спектрах молекулы A1S наблюдались
три системы полос, связанные с переходами между четырьмя электронными состояниями:
В2Л+—Х22+ [1777, 1827, 1900], С2П—Х2?+ [241, 1777] и Z>2?+-X2?+ [241 ] х. По аналогии
с изоэлектронными молекулами BS, АЮ, CN молекула A1S должна иметь низколежащее
стабильное состояние 2П<) не обнаруженное в экспериментальных исследованиях, а также ряд
квартетных состояний с энергиями около 25 000—35 000 см. В табл. 21.4 приведены молекулярные
постоянные для пяти электронных состояний с энергией до 40 000 см.
Молекулярные постоянные в состояниях X2L+ и j52?+ приняты по результатам анализа
колебательной (полосы с г/ <^ 11 и v" <С 10) и вращательной (полосы 0—0, 0—1, 0—2, 1—0,
2—0, 3—0) структуры системы 2?2И+—Х2Е+, проведенного Лавенди и др. [1827]; они хорошо
согласуются с менее точными данными [1900]. Энергия возбуждения состояния -42П,. оценена
по данным для молекулы АЮ и соотношению A. 89). Погрешность величины Те, по-видимому,
не превышает 1000 см. Значения ш, и Ве для состояния А2П{ оценены на основании
предположения, что эти постоянные, как и у молекулы АЮ, приблизительно на 15—20% меньше
соответствующих величин в основном состоянии. Погрешность принятых значений ш, и Ве может
достигать 50 см и 0,02 см соответственно. Значение Д&д состояния С2П (см. табл. 21.4)
вычислено по таблице кантов полос с v ^ 1 системы С2П—X2 ?+, приведенной в работе Мальцева
и др. [241]. Вращательная постоянная Во найдена Кронеквистом и Лагерквистом [1777] из
приближенного анализа полосы 0—0 той же системы. Молекулярные постоянные в состоянии D21)+
приняты по данным Мальцева и др. [241], полученным из анализа полос 0—0, 0—2, 1—О
и 1-1 системы Д2?+-Х2?+.
Термодинамические функции A1S (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBH и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом четырех
1 Для электронных состояний A1S принята символика, аналогичная современной символике состояний
она отличается от использованной в оригинальных работах.
159
AlSa (г) Глава 21
возбужденных электронных состояний, причем для состояний С2Л и D2 Е+ принималось, что
Qv№.tjf=={PilPx\QMi.mf Колебательно-вращательные статистические еуммы. состояний Ха?+,
А2П4, В2 ?+ и их производные находились непосредственным суммированием по колебательным
уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В расчете
учитывались все уровни этих состояний со значениями / <^ /Шах, v> последние находились из
условий A. 81). Энергия колебательно-вращательных уровней рассчитывалась по уравнениям A. 65)
и A. 62). Коэффициенты Ykl, полученные для изотопической модификации A1S, соответствующей
природной смеси изотопов, по соотношениям A. 66) и данным табл. 21.4, приведены вместе
со значениями ушах и /1Ш в табл. 21.5. Для обеспечения выполнения условий A. 51) введены
постоянные У30 для состояния Х2?+ и Ys0 и У40 для состояния 5а2+. Мультиплетность и
вырождение уровней состояний Х2Е+, АгИ( и B2L+ учитывались статистическими весами 2, 4 и 2
соответственно.
Погрешности рассчитанных термодинамических функций при температурах до 3000 К
определяются главным образом неточностью принятых значений энергии возбуждения и
молекулярных постоянных состояния А2И(. При более высоких температурах к ним добавляются
погрешности, обусловленные пренебрежением ненаблюдавшимися квартетными состояниями,
приближенным способом учета части электронных состояний, а также неточностью экстраполяции
колебательно-вращательных уровней основного состояния. Общие погрешности в значениях
Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,1, 1 и 3 Дж-К-моль соответственно.
Ранее термодинамические функции A1S (г) вычислялись в справочнике JANAF [1543] и
работе Папоушека [2181]. Расхождения практически совпадающих между собой данных [1543]
и [2181 ] с приведенными в табл. 669 обусловлены пренебрежением в этих работах
возбужденными электронными состояниями A1S; они достигают 10 и 16 Дж-К~1-моль"'1 в значениях
Ф° F000 К) и S0 F000 К) соответственно.
Константа равновесия реакции A1S (г)=А1 (r)+S (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#' @) = Do (A1S) = 368 + 6 кДж • моль ж 30 800 + 500 см.
Это значение основано на масс-спектрометрических исследованиях равновесия реакций с
участием A1S (г). Фикалора и др. [1077] исследовали реакцию Al (r)-j-1/3S2 (r)=AlS (г) A316—
1515 К, 10 точек), а Уй и Дроварт [2746] — изомолекулярную реакцию A1S (r)+S (г)=А1 (г)+
+S2 (г) A504—1763 К, результаты измерений приведены в виде уравнения). Результатам этих
измерений соответствуют значения Do (A1S), равные соответственно 404 и 384 кДж-моль при
расчете по II закону и 365,7+6 и 369,8+6 кДж-моль при расчете по III закону1. Принятое
значение является средним из двух последних величин. Его погрешность определяется в
основном неточностью сечений ионизации F кДж-моль).
Принятой энергии диссоциации соответствует
Д^" (A1S, г, 0) = 234,131 + 7,2 кДж • моль.
A1S2 (г). Термодинамические свойства газообразного дисульфида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 670.
Молекулярные постоянные, использованные для расчета термодинамических функций A1S2,
приведены в табл. 21.8. В литературе отсутствуют экспериментальные данные о структуре
и спектрах молекулы A1S2. По аналогии с молекулой АЮ2 в справочнике принимается, что в
основном электронном состоянии Х2А2 молекула A1S2 имеет циклическую треугольную структуру
симметрии С2„. Это согласуется с циклической структурой молекул дисульфидов щелочных
металлов, установленной в работе [2851а]. Приведенное в табл. 21.8 произведение моментов
инерции вычислено для оцененных структурных параметров г (А1—S)=2,3+0,l А и г (S—S)=l,95 +
+0,05 А. Для г (S—S) принято значение, равное межатомному расстоянию в молекуле S2
(см. т. 1). Значение г (Al—S) оценено из сравнения межатомных расстояний, принятых в
справочнике для соединений алюминия с кислородом и серой. Погрешность значения IaIbIc составляет
МО14 г3-см6.
1 При обработке данных [1077] учтена разница в энтальпии сублимации алюминия, использованной в [1077]
при калибровке измерений и принятой в настоящем справочнике.
160
Алюминий и его соединения AUS (г)
Частота v1==v (S—S) молекулы A1S2 принята равной частоте колебания иона S? (см. т. 1).
Частоты колебаний связей А1—S v2 и v3 оценены из сравнения с соответствующими частотами
молекул АЮг, АЮ и A1S. Погрешность принятых частот оценивается в 15—20%. По аналогии
с АЮ2 предполагается, что молекула A1S2 имеет низколежащее возбужденное электронное
состояние А2В%. Погрешность в его значении оценивается в 5000 см.
Термодинамические функции A1S2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом возбужденного состояния А2В2 по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены главным образом неточностью оценки структуры и
молекулярных постоянных A1S2 и в меньшей степени приближенным методом расчета; они составляют 4,
8 и 12 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A1S2 (г) публикуется впервые.
Константа равновесия реакции A1S2 (г)=А1 (r)+2S (г) вычислена с использованием принятой
в справочнике энтальпии атомизации
Дг#° @) = 630 ± 50 кДж • моль.
Эта величина оценена сопоставлением энтальпий атомизации сульфидов и окислов алюминия
и бора; ей соответствует
kH° (A1S2, г, 0) = 246,916 ± 50 кДж-моль.
A12S (г). Термодинамические свойства газообразного сульфида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 671.
Молекулярные постоянные, использованные для расчета термодинамических функций A12S,
приведены в табл. 21.8. Экспериментальные данные о строении A12S в литературе отсутствуют.
В справочнике по аналогии с А12О принято, что в основном электронном состоянии J?1!^
молекула A12S имеет линейную структуру симметрии jDco*. Приведенный в табл. 21.8 момент инерции
вычислен для значения г (Al—S)=2,13+0,05 А, оцененного в результате сравнения принятых
в справочнике расстояний А1—О и Al—S в различных соединениях алюминия с кислородом
в серой. Погрешность в / не превышает 2-Ю9 г-см2.
Мальцев и Шевельков [239, 448] в ИК-спектре газообразного A12S наблюдали полосу 430 см,
которую они предположительно отнесли к частоте v3. Поскольку данные этой работы вызывают
ряд сомнений, в справочнике основные частоты A12S оценены из сравнения результатов расчета
в приближении валентного силового поля (силовые постоянные A12S оценены по силовым
постоянным АЮ, A1S и А12О) с соответствующими частотами, полученными для других серных
и кислородных соединений алюминия. Погрешности в значениях vlf v2 и v3 оцениваются в 100,
25 и 100 см соответственно. Ввиду грубой оценки постоянных молекулы A12S в основном
состоянии ее возбужденные электронные состояния в справочнике не рассматриваются.
Термодинамические функции A12S (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткийротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оценки молекулярных
постоянных, включая структуру молекулы A12S, и использованием приближенного метода
расчета; они составляют 6, 10 и 15 Дж-К -моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A12S (г) публикуется впервые. Значения Ф° (Т) для
температур 1200—1800 К вычислялись в работе Уй и Дроварта [2746]. Несмотря на
существенные расхождения в принятой структуре и оценке постоянных A12S, данные табл. 671 и
работы [2746] согласуются в пределах 2 Дж-К-моль~1.
Константа равновесия реакции A12S (г)=2А1 (r)+S (г) вычислена с использованием принятой
в настоящем справочнике энтальпии атомизации
Дг#° @) = 700 ± 15 кДж моль.
Это значение является средним из полученных по уравнению III закона из результатов масс-
спектрометрических исследований [1077, 2746]. В работе Фикалора и др. [1077] исследована
реакция A12S (г)=2А1 (r)-j-1/2S2 (г) A316—1515 К, 8 точек). Результатам этих измерений
соответствуют значения энтальпии атомизации A12S, равные 649 кДж-моль (II закон) и 699,8 ±
±8,0 кДж-моль (III закон). В расчетах учтена разность значения энтальпии сублимации
161
AljS2(r) Глава 21
алюминия, использованного в [1077] при калибровке, и принятого в настоящем справочнике.
Уй и Дроварт [2746] исследовали равновесие изомолекулярной реакции A12S (r)+3S (г)=2А1 (г)+
+2S2 (г) A539—1763 К, результаты измерений представлены в виде уравнения), откуда найдены
энтальпии атомизации 720 кДж-моль (II закон) и 703,2+6,0 кДж-моль (III закон).
Приведенные в тексте погрешности учитывают разброс экспериментальных данных и неопределенность
сечений ионизации F кДж-моль). При выборе рекомендованной величины учтена неточность
термодинамических функций A12S (г), приводящая к погрешности в 12 кДж-моль в энтальпии
атомизации.
Принятой энтальпии атомизации соответствует энтальпия образования
bfH°(A\2S, г, 0) = 229,477 ±17 нДж-моль.
AIaS2 (г). Термодинамические свойства газообразного дисульфида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 672.
Молекулярные постоянные, использованные для расчета термодинамических функций A12S2,
приведены в табл. 21.11. Экспериментальные данные о строении и частотах колебаний A12S2
в литературе отсутствуют. В справочнике принимается, что по аналогии с А12О2 молекула A12S2
в основном электронном состоянии ХгАв имеет плоскую ромбическую структуру симметрии
Dih (Vh). Такая структура предсказывается как одна из наиболее вероятных для молекулы
Li2S2 [1627a] г. Приведенное в табл. 21.11 произведение моментов инерции A12S2 вычислено по
структурным параметрам г (А1—S) =2,2+0,1 А, -$ S—Al—S=60+10°, оцененным из сравнения
со структурными параметрами молекул A1S2, АЮа, А12О2, LiO2 [521, 521в], 1Л2О2 [521, 2881],
Li2S2 [1627а]. Погрешность значения IaIbIc составляет 1-1018 г3-см6. Основные частоты A12S2
оценены из сравнения с частотами молекул А12О2, АЮ и A1S. Погрешности частот могут
достигать 20% от их величин. Возбужденные электронные состояния A12S2 в справочнике не
рассматриваются ввиду грубости оценок постоянных в основном состоянии.
Термодинамические функции A12S2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью оценки молекулярных
постоянных, включая структуру молекулы A12S2, и приближенным характером расчета; они
составляют 8, 12 и 16 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций A12S2 (г) публикуется впервые. В работе [2746]
рассчитаны значения Ф° (Т) для температур 1200—1800 К; из-за различий в принятой структуре
и оценке постоянных расхождения данных табл. 672 и этой работы достигают 16 Дж-К~1-моль.
Константа равновесия реакции A12S2 (r)=2Al (r)+2S (г) вычислена с использованием
принятой в справочнике энтальпии атомизации
ДгЯ° @) = 1100 ± 30 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия
изомолекулярной реакции A12S2 (r)+4S (г)=2А1 (r)-j-3S2 (г), проведенном Уй и Дровартом [2746] A565—
1763 К, результаты измерений представлены в виде уравнения). По данным этой работы
вычислены значения энтальпии атомизации 1121 кДж-моль (II закон) и 1093+20 кДж-моль (III
закон). При вычислении погрешности последней величины учтены воспроизводимость измерений,
неточность сечений ионизации F кДж-моль) и термодинамических функций A6 кДж-моль).
Равновесие реакций в системе A1+S исследовали также Фикалора и др. [1077]. Для A1S и A12S
результаты работ [1077] и [2746] оказались в полном соответствии. Однако исследование
реакции A12S2 (г)=2А1 (r)-j-2S (г) A316—1515 К, 10 точек) приводит по данным [1077] к
величинам 1053 кДж-моль (II закон) и 1301+25 кДж-моль" (III закон) 2, значительно отличающимся
от данных [2746]. Причины этого расхождения для A12S2 неясны. В справочнике отдано
предпочтение работе [2746 ], так как полученное в ней значение лучше согласуется с аналогичными данными
1 Као [1627а] выполнил ab initio расчет Li2S2 и показал, что минимальной энергии отвечает неплоская
циклическая структура симметрии С2„, а энергия плоской структуры превышает ее всего на 600 см.
2 При вычислении этой величины учтена разность значения энтальпии сублимации алюминия, использованного
в [1077] при калибровке и принятого в настоящем справочнике.
162
Алюминий и его соединения A12S3(k,~5k)
для окислов алюминия. Кроме того, в работе [2746], в отличие от [1077], исследована изомоле-
кулярная реакция, что уменьшает возможность ошибок, связанных с калибровкой.
Принятой энтальпии атомизации соответствует энтальпия образования
bfH°(Al2S2, г, 0) = 104,262 + 31 кДж • моль.
A12S3 (к, ж). Термодинамические свойства кристаллического и жидкого трисульфида диалюми-
ния в стандартном состоянии при температурах 100—4500 К приведены в табл. 673.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 21.1. В справочнике за стандартное состояние A12S3 (к) в интервале 0—1273 К принята
а-гексагональная модификация, а в интервале 1273—1373 К — у-ромбоэдрическая модификация
(структурный тип корунда) [1105] *.
Термодинамические функции A12S3 (к) при Т <^ 298,15 К вычислены по результатам измерений
теплоемкости, выполненным Ко и др. [1736] A2—311 К) для образца чистотой —99,8% A12S8
(образец a-Al2S3 с небольшой примесью у-модификации и кубической модификации A12S3).
Погрешности измерений теплоемкости составляют по данным [1736] 5% при —12 К, 0,2%
в интервале 20—50 К и 0,1% при Т > 50 К. Экстраполяция ниже 12 К приводит к S0 A2 К) =
=0,10 Дж-К^-моль. Погрешности принятых значений 5° B98,15 К) и Н° B98,15 К)— Н° @)
оцениваются в 0,20 Дж-К~1-моль~1 и 0,04 кДж-моль соответственно.
Экспериментальные данные по энтальпии и теплоемкости A12S3 при Т > 298,15 К в литературе
отсутствуют. Уравнение для теплоемкости a-A!2S3 (см. табл. 21.1) выведено на основании
экспериментального значения Ср B98,15 К)=105,06 Дж-К-моль [1736] и двух значений
С?(800К)=140 Дж-К^-моль и С; A273 К)=152 Дж-К'^моль, оцененных с
использованием экспериментальных данных по теплоемкости А12О3 (к). Температура полиморфного
превращения A12S3 принята по данным Флао [1105], а температура плавления — по согласующимся
между собой данным [684, 1105, 1741]. Ввиду отсутствия экспериментальных данных энтальпия
превращения A12S3 принята равной 0. Энтальпия плавления A12S3 F6+15 кДж-моль) оценена
в предположении, что энтропия плавления A12S3 равна энтропии плавления А12О3 [30а].
Теплоемкости y-Al2S3 и A12S3 (ж) оценены с учетом данных по теплоемкости А12О3.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются
в 0,2, 6, 18 и 35 Дж-К-моль~1 соответственно. Расхождения между термодинамическими
функциями A12S3 (к, ж), приведенными в табл. 673 и в справочнике [578] B98—1800 К), достигают
5 Дж-К~1*моль~1 в значениях Ф° (Г), главным образом из-за того, что в настоящем справочнике
учтены данные [1736] по теплоемкости A12S3 при низких температурах.
Энтальпия образования трисульфида диалюминия принята равной
A/flr0(AlaS8, к, 298,15 К) = —648,5 + 4,0 кДж • моль.
Эта величина основана на результатах измерений энтальпии растворения сульфида
алюминия и А1С13-6Н2О в соляной кислоте, выполненных Ко и др. [1736]. При обработке
измерений [1736] использовано значение энтальпии образования &уН° (А1С13-6Н2О, к, 298,15 К)=
= —2691,6+1,3 кДж-моль, вычисленное по данным, полученным в работе Кофлина [889].
Менее точные значения kj,H° (A12S3) получены в работах Сабатье [2417] (—586 кДж»моль,
измерение энтальпии гидролиза сульфида алюминия), Коршунова [177] (—513 кДж-моль,
измерение энтальпии реакции сгорания алюминия в сере) и Капустинского и Голутвина [148]
(—723 кДж-моль, измерение энтальпии реакции сгорания алюминия в сере).
Константа равновесия реакции A12S3 (к, ж)=2А1 (r)-j-3S (г) вычислена с использованием
значения
ДгЯ° @) = 2123,815 + 8,9 кДж • моль,
соответствующего принятой в справочнике энтальпии образования A12SS (к).
1 В литературе имеются указания на существование {3-AlgS8, образующегося в присутствии карбида алюминия
(гексагональная модификация, структурный тип вюртцита) [1105], а также двух модификаций высокого
давления — кубической (дефектная структура шпинели) [24436] и тетрагональной (структурный тип p-In2Sa) [972а].
163
A1N(k, ж) Глава 21
A1N (к, ж). Термодинамические свойства кристаллического и жидкого нитрида алюминия
в стандартном состоянии при температурах 100—4000 К приведены в табл. 674.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 21.1. В справочнике за стандартное состояние A1N (к) в интервале от Г=0 до температуры
плавления 2700 К принята гексагональная модификация (структурный тип вюртцита) х.
При Т < 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости A1N (к), выполненным Ма и др. [1915] E3—297 К, 98% A1N) и Демиденко и др. [109а]
E4—330 К, 99,9% A1N), которые согласуются между собой в пределах их погрешности @,3—
0,5%). Экстраполяция теплоемкости приводит к значению S0 E3 К) =0,35 Дж>К-моль~1.
Погрешности принятых значений S° B98,15 К) и Н° B98,15 К) — Н° @) оцениваются
в 0,08 Дж-К-моль~1 и 0,013 кДж-моль соответственно. В интервале 298,15—1800 К для
теплоемкости A1N (к) принято уравнение (см. табл. 21.1), полученное в работе Ма и др. [1915]
на основании измерений энтальпии в интервале 298—1800 К с точностью 0,9 %. Измерения
энтальпии A1N, проведенные в ограниченных интервалах температур Сато [2439] B73—871 К)
и Мезаки и др. [2015а] B98—1113 К), менее точны и не учитывались, хотя в пределах их
погрешности они согласуются с данными [1915]. В интервале 1800—2700 К для теплоемкости
A1N (к) принято уравнение, рассчитанное по значениям С°р A800 К)=51,5 Дж-К-моль~х [19151
и С], B700 К) =58,6 Дж-К~1-моль~1 (оценка). Принятое значение температуры плавления
B700+200 К) основано на данных Реннера [23266], согласно которым при нагревании A1N
в атмосфере азота под давлением до 2670 К не наблюдается плавления. Менее надежные
измерения Фридериха и Ситтига [1145] дали Гш=2500 К. Энтальпия плавления A1N F8+25 кДжХ
Хмоль); оценена с использованием приближенного значения Д„,5'О=25 Дж-К~1-моль.
Теплоемкость жидкого A1N F7+15 Дж-К~1-моль) оценена как равная 33,5- п Дж-К-моль.
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 3000 и 4000 К оцениваются
в 0,05, 0,4, 3 и 6 Дж-К-моль~1 соответственно. Расхождения в термодинамических функциях
A1N (к, ж), приведенных в табл. 674 и вычисленных в справочниках [105, 1543], не превышают
2 Дж-К-моль в значениях Ф° (Т).
Константа равновесия реакции A1N (к)=А1 (r)+N (г) вычислена2 с использованием значения
Д,ЛУ° @)=1112, 135+5,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Ь,Н° (A1N, к, 298,15 К) = —319,0 + 3,0 кДж • моль.
Таблица 21.33. Результаты определений LfH° (A1N, к, 298,15 К) (в кДж-моль)
Автор
Метод
Фихтер, Енни, 1922 [1079] Измерение энтальпии сгорания A1N (к) в
кислороде
Май др., 1961 [1915] Тоже
Нейман и др., 1932 [2126] Измерение энтальпии сгорания алюминия в азоте
Нёйгебауэр, Маргрейв, 1957 [2118] То же
Прескотт, Хинке, 1928 [2268] Исследование равновесия реакции 2A1N (к)+
+3/2С (k)=V2A1,C3 (k)+N2 (г), 1774—1909 К,
14 точек
Хилденбранд, Холл, 1963 [1414] Исследование равновесия реакции 2A1N (к)=
=2А1 (r)+N2 (т) торзионным методом, 1776—
1972 К, 29 точек
Д/Я° (A1N
II закон
—303
, к, 298,15 К)
III закон
—316,4±1,7
—24(
—319
—319+30
—328±25
)
,9±0,8
—315±10
—312,6±1,0
1 При высоких давлениях Верещагин и др. [63а] установили существование другой кристаллической
модификации A1N.
* Для A1N (к) в табл. 674 вместо значений логарифма давления насыщенного пара A1N приведены значения
логарифма константы равновесия реакции диссоциации на одноатомные газы. Это сделано в связи с низким
содержанием молекул A1N в насыщенном паре, а также низкой точностью рассчитанных термодинамических
свойств АШ(г).
164
Алюминий и его соединения АШ (г)
Эта величина является средневзвешенной из приведенных в табл. 21.33 результатов
калориметрических измерений [1915, 2118]. Близкие значения получены в работе [2268] при
исследовании равновесия реакции нитрида алюминия с углеродом и в работе [1414] при исследовании
испарения нитрида алюминия. В работе [1414] измерения проводились при разных диаметрах
эффузионных отверстий и равновесные давления рассчитывались экстраполяцией найденных
таким образом величин. Для коэффициента испарения получено значение 2-Ю. Испарение A1N
изучалось также в работах [44, 697, 984, 1202, 1449, 2457, 2870], а равновесие А12О3 (к)+ЗС (к)+
+N2 (г)=2АШ (к)+ЗСО (г) — в работе [1127] (см. также [2434, 2435]), однако полученные
в них значения менее точны, в частности из-за неравновесных условий испарения A1N (к).
АШ (г). Термодинамические свойства газообразного нитрида алюминия в стандартном
состоянии в ийтервале температур 100—6000 К приведены в табл. 675.
В спектре A1N наблюдалась только одна система полос А3И -> Х3П [2523]. Приведенные
в табл. 21.4 молекулярные постоянные A1N основаны на результатах этого исследования и
приближенных оценках. По аналогии с молекулой BN в справочнике принимается, что АШ имеет
основное состояние Х3Л{ (конфигурация . . .а2тс3а) и ряд низколежащих электронных
состояний, соответствующих той же конфигурации (ХП), а также конфигурациям . ..ол4о C?+, ХЕ+),
. . .о2тс4 ^S"), . . .а2 л2 о2 C2~, 1Д, гИ+) и . . .отс3с2 CП, ХП). Энергии возбуждения этих состояний
оценены сравнением с энергиями соответствующих состояний молекулы BN, причем для групп
состояний C?+, 12+, 32", Ш) и (*Д, 1E+, 1Е+) приняты средние энергии возбуждения г.
Погрешность оцененных таким образом значений может достигать 3000—5000 см. Вращательные
постоянные АШ в состояниях Х3И и А3Н и значение Д&/2 в основном состоянии приняты по работе
Симмонса и Мак-Доналда [2523], которые выполнили анализ структуры полос 0—0 и 0—1
системы А3П -» Х3П. Остальные постоянные основного состояния, приведенные в табл. 21.4,
оценены при подготовке справочника.
Термодинамические функции A1N (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения Qm и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом
девяти возбужденных электронных состояний в предположении, что Qiil.3t = (PilPx)IQioJ.ir
Статистическая сумма по колебательно-вращательным уровням состояния Х3П и ее производные
находились непосредственным суммированием по колебательным уровням и интегрированием
по значениям / с помощью уравнений типа A. 82). В расчете учитывались все уровни
состояния Х3Л со значениями /^ Jmax, *>> последние находились из условий A. 81). Энергия
колебательно-вращательных уровней рассчитывалась по уравнениям A. 65), A. 43) и A. 62).
Постоянная спин-орбитального расщепления А и коэффициенты Ykl, идентичные соответствующим
постоянным из табл. 21.4, а также значения vmx и /1Im приведены в табл. 21.5.
Погрешности рассчитанных таким образом термодинамических функций обусловлены
отсутствием экспериментальных данных об энергиях возбуждения и молекулярных постоянных низко-
лежащих электронных состояний, а также неточностью колебательных и вращательных
постоянных основного состояния молекулы АШ. Погрешности в значениях Ф° (Т) могут достигать
2, 5 и 8 Дж-К'^моль при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций A1N (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106] и справочнике JANAF[1543]. В расчетах [105, 106] не
учитывались возбужденные состояния A1N; расхождения данных [105, 106] и табл. 675
увеличиваются с ростом температуры и достигают 10 и 14 Дж-К-моль в значениях Ф° F000 К)
и 5° F000 К) соответственно. В таблицах [1543] за основное состояние АШ принято состояние 12,
поэтому значительные расхождения данных [1543] и настоящего издания имеют место при всех
температурах и достигают 22 и 26 Дж-К~1-моль при 6000 К в значениях Ф° (Г) и 5° (Т).
Константа равновесия реакции A1N (r)=Al (r)+N (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (A1N) = 360 + 40 кДж . моль« 30 000 + 3300 см.
Это значение оценено Гингеричем [1205а] на основании измеренной им энергии диссоциации
изоэлектронной молекулы ВР.
1 В 1979 г. опубликованы результаты неэмпирического расчета молекулы АШ [22036], подтвердившие тип
основного электронного состояния Х3П и правильность оценки средней энергии возбуждения для состояний 12+,
2" и Ш.
165
А(С(г), Aid frj Глава 21
Ему соответствует энтальпия образования
Д/Г(АШ, г, 0) = 438,164 + 40 кДж • моль.
А1С (г). Термодинамические свойства газообразного карбида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 676.
В литературе отсутствуют сведения об экспериментальных исследованиях спектров А1С или
теоретических расчетах постоянных этой молекулы. В табл. 21.4 приведены молекулярные
постоянные A1G, принятые на основании оценок. По аналогии с молекулой ВС принимается, что
молекула A1G имеет основное электронное состояние X4 S" (конфигурация . . . ол2) и ряд низко-
лежащих возбужденных электронных состояний. В табл. 21.4 приведены значения энергий
возбуждения пяти первых состояний, оцененные по данным для молекулы ВС. Погрешности
оценок могут достигать 5000 см. Приведенные в табл. 21.4 значения ге и а>в оценены путем
сопоставления соответствующих величин в молекулах В2, А12, С2 и ВС. Погрешности в
значениях гв и сое могут достигать 0,2 А и 200 см.
Термодинамические функции А1С (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значение QBS и ее производных вычислялись по соотношениям A. 90)—A. 92) с учетом
пяти возбужденных состояний в предположении, что Qlil.Bs==(PilPx)Qifl.Br Статистическая
сумма по колебательным и вращательным уровням энергии состояния X* ХГ и ее производные
находились по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по / с помощью соотношений типа A. 82). В расчете учитывались
все колебательно-вращательные уровни состояния Х*?+ со значениями / ^ /щах, „! последние
находились по формуле A. 81). Уровни энергии вычислялись по уравнениям A.' 65) и A. 62)
с коэффициентами Ун, идентичными с постоянными из табл. 21.4 и приведенными вместе со
значениями ишях и /Ит в табл. 21.5. Мультиплетность X4 2~ учитывалась статистическим весом.
Погрешности рассчитанных термодинамических функций А1С (г) обусловлены главным
образом неточностью оценок молекулярных постоянных А1С в основном электронном {[состоянии
и энергий возбуждения низколежащих электронных состояний; они могут достигать 6, 10 и
15 Дж-К^-моль ~х в значениях Ф° {Т) при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций А1С (г) вычислялись авторами справочника
JANAF [1543]. Расхождения данных [1543] и табл. 676 достигают 8 и 15 Дж- К-моль в
значениях Ф° F000 К) и S° F000 К) соответственно и обусловлены отличающейся оценкой
постоянных и пренебрежением возбужденными электронными состояниями А1С в расчете [1543].
Константа равновесия реакции А1С (г)=А1 (г)+С (г) вычислена на основании принятой
энергии диссоциации
Дг#° @) = Do (A1C) = 360 + 50 кДж • моль «* 30 000 + 4000 см.
Эта величина оценена из сопоставления энергий диссоциации молекул В2, С2, А12 и ВС.
Принятому значению соответствует
A^°(A1C, г, 0)^678,531 + 50 кДж • моль.
А1С2 (г). Термодинамические свойства газообразного дикарбида алюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 677.
Молекулярные постоянные А1С2, принятые для расчета термодинамических функций,
приведены в табл. 21.8. По аналогии с молекулой ВС2 принимается, что молекула А1С2 в основном
электронном состоянии X2 2+ имеет линейную несимметричную структуру (точечная группа С^).
Приведенное в табл. 21.8 значение момента инерции вычислено по структурным параметрам
r (A1—С)=1,8+0,2 А и г (С—С)—1,26+0,05 А, оцененным на основании данных о
молекулах А12, С2 и ВС2. Погрешность величины / составляет 3-10~39 г-см2. Частоты валентных
колебаний vx и v3 оценены по силовым постоянным / ... р.=5>108 дин-см и / ,„ „,=
,, ,„, . Jr(Al—d) "• ¦<r(G—С)
—11 -11)° дин-см , принятым на основании сопоставления соответствующих величин в
молекулах А12, С2 и С3. Погрешности значений vx и v3 составляют 200 см. Деформационная частота v2
оценена по силовой постоянной /ж/г1г2=0,025-105 дин-см, которая соответствует значению v2
в молекуле ВС2. Погрешность значения v2 оценивается в 80 см.
166
Алюминий и его соединения ' Л1,0„ (г), А14С3 (к)
Термодинамические функции А1С2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 126) и A. 129).
Погрешности вычисленных термодинамических функций обусловлены главным образом неточностью
принятых молекулярных постоянных, отсутствием сведений о возбужденных состояниях, а также
приближенным методом расчета и составляют 10—25 Дж-К-моль в значениях Ф°(Т) во всем
интервале температур.
Таблица термодинамических функций А1С2 (г) публикуется впервые.
Константа равновесия реакции А1С2 (г)=А1 (г)+2С (г) вычислена с использованием значения
ДГЯ° @)=1069,716+35 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(AlC2, г, 0) = 680 + 35 кДж • моль.
Эта величина является средней из вычисленных по уравнению III закона из результатов
масс-спектрометрических исследований реакции А1 (г)+2С (графит)=А1С2 (г). Чапка и Берко-
виц [853] исследовали это равновесие при 2100 К, результатам их исследования соответствует
&fH° @) =672+15 кДж-моль. Стерне и Коль [2608а] провели измерения в интервале 2082—
2171 К E точек); этим измерениям соответствует Ь.^Н° @)=691 +14 кДж-моль. Неточность
термодинамических функций А1С2 (г) приводит к погрешности в энтальпии образования в 25—
30 кДж-моль.
А12С2 (г). Термодинамические свойства газообразного дикарбида диалюминия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 678.
Молекулярные постоянные А12С2, принятые для расчета термодинамических функций,
приведены в табл. 21.11. В настоящем справочнике принимается, что молекула А12С2 в основном
электронном состоянии XX2J имеет линейную симметричную структуру (точечная группа D гаА).
Приведенное в табл. 21.11 значение момента инерции вычислено по структурным параметрам,
принятым равными соответствующим параметрам молекулы А1С2. Погрешность величины / может
достигать 2-Ю8 г-см2. Частоты колебаний А12С2, приведенные в табл. 21.11, оценены по
уравнениям поля валентных сил при тех же значениях силовых постоянных, что и в молекуле А1С2.
Погрешности принятых значений vl5 v2, v3, v4 и v5 могут достигать 200, 100, 200, 100 и 50 см
соответственно.
Термодинамические функции А12С2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A. 122)—A. 124), A. 126) и A. 129).
Погрешности рассчитанных термодинамических функций обусловлены главным образом неточностью
оценки молекулярных постоянных, в том числе структуры А12С2, и приближенным характером
расчета; они составляют 15—25 Дж*К~1-моль~1 в значениях Ф° (Т) во всем интервале температур.
Таблица термодинамических функций А12С2 (г) публикуется впервые.
Константа равновесия реакции А12С2 (г)=2А1 (г)+2С (г) вычислена с использованием
значения ДГЛ° @)=1517,062 +40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfE°(Al2C2, г, 0) = 560+ 40 кДж • моль.
Это значение является средним из величин, вычисленных по результатам
масс-спектрометрических исследований системы А1+С. Чапка и Берковиц [853] исследовали реакции 2А1 (г) +
+2G (графит) =А12С2 (г) и А12 (г)+2С (графит) =А12С2 (г) при 2100 К. Из этих измерений
вычислены значения Д^Я0 (А12С2, г, 0) = 552+10 кДж • моль и 549+21 кДж • моль. Стерне
и Коль [2608а] исследовали вторую из этих реакций в интервале 2051—2171 К (9 точек).
Обработка данных [2608а] по уравнению III закона дает 572+22 кДж-моль. Приведенные
погрешности для работы [853] учитывают неточность сечений ионизации и использованных в расчетах
термохимических величин, для работы [2608а] учтена также воспроизводимость измерений.
Неточность термодинамических функций А12С2 (г) приводит к дополнительной погрешности
в принятом значении энтальпии образования, равной 30—35 кДж-моль.
А14С3 (к). Термодинамические свойства кристаллического трикарбида тетраалюминия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 679.
167
А14С, (к) Глава 21
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 21.1. В справочнике за стандартное состояние А14С3 (к) в интервале 0—2500 К принята
гексагональная (ромбоэдрическая) модификация г.
Термодинамические функции А14С3 (к) при Т < 298,15 К вычислены по результатам
измерений теплоемкости, выполненных Фурукавой и др. [1153] A8—380 К) на образце состава 94,8%
А14С3, 1% А1, 1% С, 1,3% A1N, 2,2% А12О3 и 0,06% Fe. В экспериментальные значения
теплоемкости внесена поправка на содержание примесей. Точность измерений теплоемкости при
Т > 50 К следует оценить в 0,5%. Экстраполяция теплоемкости ниже 18 К приводит к
значению S° A8 К)=0,05 Дж-К^-моль-1. Погрешности принятых по [1153] _значений S° B98,15ДО
и Н° B98,15 К)—Я0 @) оцениваются в 0,5 Дж- К- моль и 0,05 кДж- моль соответственно. В
интервале 298,15—2500 К для теплоемкости А14С3 (к) принято уравнение, найденное методом Шо-
мейта в результате совместной обработки измерений энтальпии в работах Дугласа и др. [979]
B73—1173 К) и Бинфорда и др. [693] B98—1774 К). При обработке данным обеих работ
приписывалась погрешность, равная 1%, а результаты двух выпадающих измерений в работе [6931
при 1277 и 1672 К не учитывались. При выводе уравнения для теплоемкости не учитывались
также измерения энтальпии А14С3 (к), выполненные Сато [2437] для образца, содержащего
75% А14С3. Температура инконгруэнтного плавления А14С3 B500+100 К) принята по данным
Руффа и Еллинека [2406], которые показали, что А14С8 (к) плавится с разложением на графи»
и раствор углерода в расплавленном А1.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2500 К оцениваются в 0,3,
2 и 8 Дж«К-моль~1 соответственно. Расхождения между термодинамическими функциями
А14С3 (к), приведенными в табл. 679 и в справочнике^[1543], растут с увеличением температуры,,
достигая 7 Дж-К^-моль в значении Ф° B500 К). В справочнике [578] B98—1800 К) принято
неверное значение стандартной энтропии А14С3 (к)' A05 Дж-К-моль вместо 88,95 Дж-К^Х
Хмоль).
Константа равновесия реакции А14С3 (к)=4А1 (г)+ЗС (г) вычислена с использованием значе--
ния Д^Г* @)=3644,899+16 кДж-моль, соответствующего принятой в справочнике энталыига
образования
b.fH° (А14С3, к, 298,15 К) = —206,9 ± 2,0 кДж • моль.
Это значение является средневзвешенным из измерений Кинг и Армстронга [1715] (—208,6 ±
±5,0 кДж'Моль) и Блачника и др. [695] (—206,6+2,1 кДж-моль). Кинг и Армстронг [1715]
сжигали в кислороде карбид алюминия и провели тщательный анализ продуктов сгорания,
в том числе установили, что основным продуктом сгорания являются 8-А12О3 (от 47 до 77%)
и а-А12О3. Для энтальпии перехода 8- в а-модификацию было принято значение —11,3±
+1,7 кДж-моль. Бланчик и др. [695] измерили энтальпию растворения А14С3 и А1 в соляной
кислоте и нашли Д^0 B98,15 К)=—17,2+2,1 кДж-моль для реакции А14С3 (к)+6Н2 (г)=
=4А1 (к)+ЗСН4 (г). Более ранние измерения энтальпии сгорания А14С3 в кислороде [673, 675,
1624, 1914, 2001, 2371, 2866 2] привели к недостаточно точным значениям из-за низкой чистоты
исходных образцов и недостаточно полного анализа продуктов сгорания. Лучшей из этих работ
является работа Ма [1914], в которой получена энтальпия образования —224+8 кДж-моль.
Исследования равновесия реакций с участием карбида алюминия [811, 853, 1268, 2010, 2235,
2267, 2268] не приводят к надежным значениям энтальпии образования. Наиболее близкие
результаты к приведенным выше термохимическим величинам получены Кемпбеллом [811]
(—215 кДж-моль, измерение активности алюминия, 1193 К, 13 измерений), Планте, Шрейе-
ром [2235] (—220+17 кДж-моль, эффузионный метод, 0,25 А14С3 (к)=А1 (г)+0,75 С (та),
1651—1924 К, 12 измерений), Меши и Серей [2010] (—254 кДж'МОль, эффузионный метод,
1651—1965 К, 5 точек; —188 кДж-моль, торзионный метод, 1500—1850 К, 56 точек).
31 Ai4Cg — наиболее устойчивый карбид алюминия в конденсированном состоянии в системе AI—С, диаграмма
состояния которой изучена недостаточно полно. В ряде работ (см. [67, 626, 997]) указывается на существованше
других карбидов алюминия: А18С2, А13С, А12(С2)8.
2 Результаты этих измерений пересмотрены Ефименко (см. [1715 ]).]
Глава 22
ГАЛЛИЙ И ЕГО СОЕДИНЕНИЯ
Ga (к, ж), Ga, Ga+, GaO, Ga2O, Ga2O3 (к, ж), GaH, GaOH, GaF, GaF2, GaF3 (к), GaF3, GaCl,GaCl2,
GaCI3 (к, ж), GaCI3, Ga2Cl6
В настоящей главе рассматриваются 14 веществ — галлий и его соединения с кислородом,
водородом, фтором и хлором. Для четырех веществ приведены данные о термодинамических свойствах
в конденсированном состоянии и для 13 веществ — в газообразном состоянии.
Термодинамические функции газообразных галлия и его иона рассчитаны для температур до 10 000 К, для
остальных газов — до 6000 К. Для галлия в Приложении 3 приведены сведения о его
критических постоянных.
Термодинамические свойства всех веществ рассчитаны для природной изотопической смеси
галлия (атомный вес 69,72) и других элементов. Однако разница в молекулярных постоянных
изотопических модификаций молекул учитывалась только для GaCl, так как для других
молекул отсутствуют соответствующие данные, а влияние изотопического эффекта на результаты
расчетов термодинамических функций несущественно.
Ga (к, ж). Термодинамические свойства кристаллического и жидкого галлия в стандартном
состоянии при температурах 100—4000 К приведены в табл. 680.
Значения постоянных, использованные для расчета термодинамических функций,
приведены в табл. 22.1. В справочнике за стандартное состояние Ga (к) принимается ромбическая
Т а б л в
Вещество
Ga
Ga2O3
GaFs
GaCl3
ца 22.1. Принятые
значения
в кристаллической
Состояние
К
ю
о .
сц 1
кДжХ
X моль
к, ромб. 5,572
ж —
к, монокл. 14,55
ж —
к, гекс. 16,7
к, трикл. 20,0
ж —
К
ю
оо
оз
о
со
Дж-К
40,81
,—
84,94
—
96
134
—
термодинамических величин для галлия и его соединений]
и жидкой состояниях
И
§8
• МОЛЬ
26,07
—
92,13
—
89,0
96,0
—
Коэффициенты i
НИИ ДЛЯ Ср
а
а
26,069
114,398
160,0
77,837
22,661
109,0
Примечание. C°p=a+bT—cT 2+dT2 (в Дж-К^-моль).
Ga: a Табличные данные; 6 d-
Юв=0,241.
6.10»
_
—0,301
14,962
—
37,442
245,98
—
уравне-
(Т)
с • Ю-5
_
—2,367 5
23,759
—
—
—
—
Интервал
температур
К
298,15-302,92
302,92—4000
298,15—2080
2080—4000
298,15—2000
298,15—351
351 —1000
Т
1 m
К
302,92
—
2080
—
—
351
—
д н
кДжХ
X моль
5,565
—
100
—
—
11,13
—
модификация *. При Т < 298,15 К термодинамические функции Ga (к) вычислены по
результатам измерений теплоемкости в работах Адамса и др. [484] A5—300 К). Эйхенауэра и Шульце
[1022а] F—21 К) и Зайделя и Кисома [2490а] @,35—4,4 К). Эти данные приводят к значениям
5° B98,15 К) и Н° B98,15 К)—Я0 @) (см. табл. 22.1), погрешности которых оцениваются
в 0,2 Дж-К»моль и 0,02 кДж-моль соответственно. Измерения низкотемпературной
теплоемкости Ga (к) в работах [671а, 859а, 2384] менее точны и не учитывались в расчете. Согласно
данным Буатара и др. [719а] B94,45—302,75 К), полученным для образца галлия высокой
чистоты (99,9999%) с использованием калориметра Кальве, теплоемкость твердого галлия резко
возрастает в интервале —2° вблизи температуры плавления. К сожалению, точность этих изме-
1 При 1,09 К ромбическая модификация переходит в сверхпроводящее состояние [2490а]. Кроме ромбической
модификации для галлия известны две фазы высокого давления, а также две метастабильные модификации [4066].
169
Ga (к, ж)
Глава 22
рений невысока (в интервале 294—299 К данные [719а] на ~3% ниже данных Адамса
и др. [484]), а результаты определения теплоемкости при двух наиболее высоких температурах
C02,45 и 302,75 К) вызывают сомнение в их достоверности и не были использованы в настоящем
справочнике. Скорректированные данные [719а] были использованы для расчета
термодинамических функций в интервале 298,15—302,92 К. Температура плавления галлия C02,92+0,01К)
принята по прецизионным данным Ло и Шлотта [18226]; с ними хорошо согласуются результаты
измерений Роззера и Гоффмана [2354а] C02,93+0,05 К) (ссылки на другие измерения см. в
справочнике [406]). Энтальпия плавления галлия E,565+0,020 кДж-моль") принята по данным
Адамса и др. [484], исправленным на аномальное возрастание теплоемкости Ga (к) вблизи точки
плавления [719а] (величина поправки составляет —0,020 кДж-моль). Другие измерения
(ссылки см. в справочнике [406]) менее точны. Для теплоемкости жидкого галлия принято
уравнение (см. табл. 22.1), выведенное методом Шомейта на основании теплоемкости Ga (ж) в точке
плавления B8,58+0,17 Дж-К-моль) [719а], данных по энтальпии жидкого галлия,
полученных Резухиной и Кочетковой [191а] E88—985 К) и рассчитанных по результатам измерений
теплоемкости галлия, выполненных Адамсом и др. [484] C07—323 К), Ченом и Турнбул-
лом (8416) C50—600 К) и Банчила [27а] A100—1600 К), а также трех значений энтальпии
галлия (при 2000, 2500 и 3000 К), найденных экстраполяцией данных [27а].
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 3000 и 4000 К оцениваются
в 0,1, 1, 3 и 4 Дж-К-моль~1 соответственно. Расхождения между термодинамическими
функциями Ga (ж), приведенными в табл. 680 и в справочниках [1503, 2632], достигающие
2 Дж-К-моль~1 в значениях Ф° B500 К), вызваны уточнением теплоемкости жидкого галлия
в работе [27а].
В справочнике за стандартное состояние галлия принимается Ga (к, ромб.) и в соответствии
с этим
AfH°(Ga, к, ромб.) = 0.
Давление пара галлия в реакции Ga (к, ж) = &а (г) вычислено с использованием энтальпии
сублимации
AsH°(Ga, к, 0) = 271,0 + 4,0 кДж . моль.
В табл. 22.2 представлены результаты измерений этой величины. Принятое значение является
средневзвешенным из данных [491, 1903, 2080, 2591]. В работах [97, 537а, 793, 988] приведены
только энтальпии сублимации, вычисленные по II закону термодинамики, и отсутствуют сведения
о давлении пара, что не позволило провести их обработку по III закону. В работе [249] измерения
Таблица 22.2. Результаты определений h,H° (Ga, к, 0) (в кДж-моль")
Авторы
Хартек, 1928 [1359]
Спейзер, Джонстон, 1953 [2591]
Дроварт, Хониг, 1957 [988]
Гурвич, 1960 [97]
Кохран, Фостер, 1962 [862]
Мунир, Сирей, 1964 [2080]
Алкок и др., 1965 [491]
Мекар и др., 1966 [1903]
Берне, 1966 [793]
Артур, 1967 [537а]
Матерн и др., 1969 [249]
Метод
Эффузионный, 1198—1391 К, 18 точек
Эффузионный, 1230—1518 К, 19 точек
Масс-спектрометрический, 1035—1320 К
Обработка измерений аномальной дисперсии света
в парах галлия в работе [305а], ТСр=1360 К
Эффузионный, 1179—1383 К, 6 точек
Торзионно-эффузионный, 1204—1603 К, 104 точки
Эффузионный, 1281—1437 К, 9 точек
Протока, 1173—1273 К, 3 точки
Эффузионный, 1196—1472 К, 9 точек
Масс-спектрометрический, Гср=1132 К, 11 точек
Масс-спектрометрический, 900—1200 К, 33 точки
Эффузионный, 1081—1164 К, 17 точек,
радиоактивный образец
1090—1185 К, 11 точек, нерадиоактивный образец
1062—1213 К, 19 точек, нерадиоактивный образец
Д8Я°(
II закон
308 ±24
297+22
251 ±17
245 ±4
274±10
266+2
269+14
256±131
264±11
266±9
274±13
262±22
248±19
252 ±22
Ga, к, 0)
III закон
277,6±1,2
270,9 ±1,3
—
—
288,7±1,0
273,5±0,3
270,9+0,5
272,0±1,9
269,3±0,5
—
—
259,7 ±0,5
259,9 ±0,5
266,8±0,7
170
Галлий и его соединения
Ga (г), Ga+ (г)
проводились с радиоактивным и обычным образцами и была обнаружена зависимость давления
от размера эффузионного отверстия, что противоречит данным других авторов, в частности [1903].
Приведенные в таблице погрешности отражают только воспроизводимость измерений.
Погрешность принятой величины оценена с учетом расхождения результатов указанных выше работ и
неточности термодинамических функций Ga (ж), которой соответствует погрешность в энтальпии
сублимации около 3 кДж-моль.
Ga (г). Термодинамические свойства газообразного галлия в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 681.
Уровни энергии атома Ga, использованные в расчете термодинамических функций, приведены
в табл. 22.3, в которой даны валентные состояния Ga, относящиеся к электронным конфигурациям
. . .4s24p, . . .4s24J, . . .4s24/, . . .4s4p2 и расположенные ниже ионизационного предела. Энергии
состояний . . As2Al приняты по результатам экспериментального исследования Йоханссона и Ли-
Таблица 22.3. Уровни энергии атомов Ga и Ga+, принятые для расчета термодинамических функций
Атом
Электрон-
гурация
Состояние
Ga .. As2ip 2Pw
2Ps/2
...4s24d Ю3/г
2%
...4sV 4Р1Д
4/4
Т{
СМ'
0
826,19
34 781,66
34 787,85
37 972
38 338
38 913
Pi
2
4
4
6
2
4
6
Атом
Ga
Ga+
Электронная
конфигурация
.. .4s24/
.. .4s2
...isip
... isip
Состояние
гр
iS
spo
*Pi
3 p
1 p
Ti
CM
41454,18
0
47 370
47 816
48 750
70 700
Pi
14
1
1
3
5
3
цена [1572], в котором удалось существенно уточнить энергию состояния . . .4s24/ BF) по
сравнению с величиной, рекомендованной Мур [2053]. Энергии подуровней состояния . . . 4s4/>2 (*F)
приняты по данным [2053]. Состояния . . .4s4p4d BZ>) и . . .4s4p4/ BF), по-видимому, лежат выше
ионизационного предела Ga и не учитывались в расчете.
Термодинамические функции Ga (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
термодинамических функций при Т <d 4000 К не превышают 0,02 Дж-К-моль~1, что обусловлено только
неточностью принятого значения R; при более высоких температурах погрешности в S0 (Т) и Ср (Т)
увеличиваются из-за пренебрежения ридберговскими состояниями и могут достигать 0,7 ДжХ
ХК^-моль в значении 5° A0 000 К).
Ранее таблицы термодинамических функций Ga (г) вычислялись в работах Гурвича [97] до
3500 К, Каца и Маргрейва [1639] до 2000 К, Кольского и др. [1749] до 8000 К, Хилзенрата [1436]
до 10 000 К, а также в справочнике Сталла и Зинке [2632] до 3000 К. Результаты этих расчетов
при Т <^ 4000 К совпадают в пределах 0,02 Дж- К"-моль (за исключением расчета [1749]) с
приведенными в настоящем справочнике. Расчет [1749] выполнен с неточными значениями
физических постоянных, что приводит к ошибкам около 0,05 Дж-К~1-моль~1 даже при низких
температурах. При Т >¦ 4000 К результаты расчета [1436] отличаются от данных настоящего справочника
из-за различия в ограничении статсуммы по электронным состояниям; расхождения в Ф° (Т)
составляют около 0,2 Дж-К~1-моль при 10 000 К.
Энтальпия образования Ga (г)
AfH° (Ga, г, 0) = 271,0 ± 4,0 кДж • моль
соответствует принятой в справочнике величине A,ff° (Ga, к, 0).
Ga+ (г). Термодинамические свойства газообразного положительного иона галлия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 682.
171
GaO (г) Глава 22
Уровни энергии иона Ga+, использованные в расчете термодинамических функций, приведены
в табл. 22.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. . .4s2 и . . .4s4p. Энергии этих состояний приняты по рекомендации Мур [2053]. Валентные
состояния, относящиеся к конфигурациям . . . isAd, . . . 4s4/ и ... 4р2, имеют энергии свыше
100 000 см и не учитывались в расчете.
Термодинамические функции Ga+ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),,
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
значений во всем интервале температур не превышают 0,02 Дж-К~1>моль и обусловлены
главным образом неточностью принятого значения R.
Ранее таблицы термодинамических функций Ga+ (г) вычислялись в работах Грина и др. [12461
до 50 000 К и Хилзенрата и др. [1436] до 10 000 К. Вычисленные в этих работах значения
совпадают с данными настоящего справочника в пределах указанной выше погрешности.
Константа равновесия реакции Ga+ (г)+е (r) = Ga (г) вычислена с использованием значения
Дгйго@)=—578,845+0,001 кДж-моль, соответствующего потенциалу ионизации атома галлия
/0 (Ga)=48 387,63±0,05 см=578,845+0,001 кДж-моль.
Это значение принято на основании экстраполяции ридберговской серии . . As2nf BF) (n=A-^-8)
по измерениям Йоханссона и Лицена |1572]. В работе [316] найдено практически совпадающее
значение. Принятому значению /0 (Ga) соответствует .
A,#°(Ga+, г, 0) = 849,845 + 4,0 кДж • моль.
GaO (г). Термодинамические свойства газообразного оксида галлия в стандартном сеетеянии
при температурах 100—6000 К приведены в табл. 683.
Молекулярные постоянные e9Ga160, принятые на основании результатов исследований
электронного спектра этой молекулы, приведены в табл. 22.4. В спектре испускания GaO наблюдались
три системы полос в областях 3900—4200 А, 3430—3485 А и 4450—4460 А. До настоящего времен»
выполнен анализ только первой из этих трех систем, принадлежащей переходу Z?2?+ -» Х2? +
[101, 304а, 443а, 1301, 1485, 2305, 2306, 2491, 2875а], две другие, по аналогии с спектральными
переходами молекулы АЮ, были отнесены авторами [304а, 443а] к переходам С2П ->• Х*? + в
J52E+ -»¦ A2TLt соответственно. Молекула GaO может иметь и другие связанные валентные
электронные состояния, но их энергии должны превышать 30 000 см и они, так же как состояние C2Ut
не представлены в табл. 22.4.
Приведенные в табл. 22.4 значения колебательных постоянных молекулы 69Ga160 в состояниях
Х2Е + и 52Е + приняты по работе Ядава и др. [2875а], которые нашли их по кантам 26 полос,
принадлежащих пяти секвенциям (и" ^8, v' ^ 7) перехода Б22+ -> Х2?+. Анализ колебательной
структуры этой системы полос был подтвержден в работе [2875а ] хорошим согласием измеренных
и рассчитанных кантов полос молекулы 71Gaie0. Принятые значения колебательных постоянных
хорошо согласуются также с найденными ранее в работах [101, 2306]. Принятые в табл. 22.4
значения вращательных постоянных основаны на результатах анализа тонкой структуры полос
0—0 и 1—0 этой системы в спектрах молекул 69Ga160 и 71GaleO, выполненного Ядава и др. [2875а].
Анализ тонкой структуры полосы 0—0 в работе «[2306] содержал ошибки и привел к несколько
отличающемуся значению 5q=0,4271 cm, а анализ полос 0—0 и 0—1 в работе [304а] не дал
определенных результатов.
Приведенная в табл. 22.4 энергия состояния Л2П; соответствует предположению, что система
полос в области 4460 А принадлежит переходу Z?2? + ->¦ А2П{, ее погрешность оценивается
в 1500 см.
Термодинамические функции GaO (г) были вычислены по уравнениям A. 3)—A. 10), A. 93)—
A.95). Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A.92) с учетом
состояний -42П,. и Б2?+ и в предположении, что Q\j>\. щ= (PilPx) Qmx. вР-
Колебательно-вращательная статистическая сумма состояния Х2?+ и ее производные вычислялись по уравнениям A. 73)—
A. 75) непосредственным суммированием по колебательным уровняем и интегрированием по
вращательным уровням энергии с помощью соотношений типа A. 82). В расчетах учитывались вс&
уровни энергии с значениями / ^ /„,„, последние находились из условий A. 81). Колебательно-
вращательные уровни состояния Х2Е+ вычислялись по уравнениям A. 65), A. 62), мультиплет-
ность уровней учитывалась статистическим весом. Значения коэффициентов Ykl в этих уравнениях
172
Галлий и его соединения
GaO (rl
Таблица 22.4. Молекулярные постоянные GaO, GaH, GaF, GaCI
Молекула
Состояние
69Ga160 X2S+
А2П{ 3
B2S+ 25
MGaH X*S+
а3П0- 17
а3П0+ 17
а3П1 17
o3ll2 17
Am 23
eeGa19F X12+
а3П0+ 33
a3lli 33
Am 47
eeGa35Cl X12+
а3П0+ 29
в*Щ 29
Am —40
0
500 Д
699,7
0
331,7
337,08
622,01
903
919
0
105,5
427,8
365,7
0
524,1
855,7
261
ше
766,84
—
761,54
1604,52
1640,54
1640,54
1631,17
1631,17
1180 м
622,2
663,02
662,1
542,35
365,0
395,8
395,1
120 в
Примечание. Ниже все постоянные даны в смг1.
GaO: a wtyt = 0,02) рассчитано на
г рассчитано по соотношению A
GaH: а сзд,=0,36; б «2=—5-10; в
постоянная для v=0; ж
л pi = 2,2.10~6; p2 = 5-l
О;
и соответствующей постоянно!
GaF: a а2 = 1,02-Ю; б ч>,у,-
A.68); Д («^ = -0,43;
Ве
СМ
6,21 а
—
3,13 е
28,77 а
62,72 г
62,72
58,22 и
58,22
—
3,2
2,18 б
1,45 Д
9,55 е
1,1
2,5
2,5
—
основании значения Во
.68); Д опенка
Pi=—6-10~в,
3.= -М(
0,436 3 °
—
0,409 9
6,137
6,359
6,394
6,687
6,811 в
5,14 н
0,359 516
0,371 0
0,372 0
0,358
0,149 904
0,157 8
0,156 9
—
аг ¦ 102
0,41 в
—
0,15
18,1 б
22 Д
27,6 ж
30,6 к
—
—
0 0,286 42 а
0,30 в
0,302
0,535 ж
5 0,079 35 а
—
—
= 0,434 18; в рассчитано
=-0,03,
4.Ю-2; 3 Pi=— 7,5-Ю-5; ра=1,45
м рассчитано
GaD.
D,-
0,56
—
0,5
337 в
243 е
262 3
448,5
620 е
400 н
0,5
0,46
0,47
0,62
10" '
Ге
А
г 1,725
—
г 1,778
1,663
1,634
1,629
* 1,593
1,579 е
1,817 е
1,774369
г 1,746 7
г 1,744 4
1,778
0,1 б 2,201 689
—
—
—
2,146
2,152
—
по соотношению A.69);
=—6,195; Д а2=— 5,8-
•10; и ш„у,=-
по соотношению A.68); н рассчитанс
= —0,3; в рассчитано по соотношению
у. = -0,6; ж
GaCI: a а2 = 1,8-10; б рассчитано по соотношению
аг==—3
A.68);
•ю-4.
~7»4t7 *
Ю; е приведена
к О2=—3,85-Ю-2;
> по соотношению A.66)
A.69); г рассчитано
в приведено приближенное
по соотношению
значение АСуа-
приведены в табл. 22.5, коэффициенты Yk0 получены по принятым колебательным постоянным
GaO (см. табл. 22. 4), с учетом условий A. 50), в той же таблице приведены значения ушах и /lim.
Погрешности рассчитанных термодинамических функций при умеренных температурах
обусловлены в основном отсутствием экспериментальных данных об энергии состояния А2П4, при
высоких температурах становятся существенными пренебрежение электронными состояними с
энергиями свыше 25 000 см, а также ограниченность данных о колебательно-вращательных уровнях
•состояния X2 2+ и отсутствие таких данных для состояния А2И{. Погрешности в значениях Ф° (Т)
при 298,15, 3000 и 6000 К оцениваются в 0,05, 3 и 5 Дж-К^-моль.
Термодинамические функции GaO (г) ранее вычислялись в работах Бруэра и Розенблатта
G45] (Ф° (Т) при Т < 3000 К), Разинаса и др. [2305] (Т < 3000 К) и Барнса [793] (для
Т ^ 2500 К). Расхождения данных [745] и табл. 683 не превышают 1 Дж-К-моль'1 и
обусловлены тем, что авторы [745] не учли возбужденные электронные состояния GaO.
Существенные расхождения данных работ [2305] (до 9 Дж • К~1-моль~1) и [793] (до 16 Дж X
ХК • моль) с табл. 683 не могут быть объяснены иначе, как ошибками в расчетах в этих работах.
Константа равновесия реакции GaO (r) = Ga (г)+О (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДД° @) = Do (GaO) = 370 + 20 кДж • моль я* 31 000 + 1500 см.
Эта величина основывается на результатах масс-спектрометрических исследований равновесия
«реакции GaO (r) = Ga (г)+О (г), выполненных Барнсом [793] (Г=2068 К, 4 точки) и Фу, Барн-
173
Ga2O (г)
Глава 22
Таблица 22.5.
Коэффициенты в уравнениях, описывающих уровни энергии (в см"), а также значения
vmax и е/цщ, принятые для расчета термодинамических функций GaO, GaH, GaF и GaCl
Коэффициенты
г,-ю-*
У,о-1О-8
Уго-Ю
Узо-Ю
У40-Ю4
у01
Уц-Ю2
У21-10*
У02-Юв
Уи-10»
У22-Ю5
Уоз-Ю12
Уо4-Ю»
01
аг104
аз-Ю8
ушах
Jlim
Примечание.
GaO: a Те(Ат
GaO
0
0,765 924
—0,619 309
0,199 880
-0,326 920
0,435 264
-0,408 541
—
—0,577 341
—
—
-2,235 099
—
—
—
—
—
98
348
Ниже все величины
) = 3500, Г, (В* 2+) =
GaH
Х*2+
0
1,604 87
—2,911 22
4,873 46
-159,183
6,137
—18,1
—5,0
-337,0
—0,6
0,1
18505,6
-0,5
2,334 52
0,301 684
0,371 399
0,415 893
25
85
даны в см.
= 25 699,7. GaF:
GaCl:
а3П
1,762 201
1,631 17
—5,822
-74,7
—
6,687
—30,6
—385,0
-448,5
2,2
-5,0
30 081,1
—
0,572 321
1,909 921
4,621 832
1,137 238
5
45
a Ге(а3П) = 33 427,
a Те (а3П) = 29 855
GaF
*'2+а
0
0,622 654
-0,336 032
0,144 504
—0,372 990
0,359 516
-0,286 42
0,102
-0,50
—
—
-0,359 558
—
—
—.
—
—
169
518
S.
,7.
GaCl
0
0,362 688
—0,110 119
0,012 054
—0,002 949
0,147 972
—0,077 820
0,017 539
—0,097 438
—
—
-0,018 889
—
—
__
—
287
722
сом [1149] (Г=2318 К, 1 точка). Обработка данных этих работ приводит к значениям 362
и 380 кДж-моль.
Погрешность рекомендованной величины оценена с учетом расхождения результатов
измерений, неточности использованных в расчетах термодинамических функций и сечений ионизации.
Принятому значению соответствует
A/Fe(GaO, г, 0) = 147,783 + 20 кДж • моль.
Ga2O (г). Термодинамические свойства газообразного оксида дигаллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 684.
Молекулярные постоянные Ga2O, использованные для расчета термодинамических функций,
приведены в табл. 22.6. В настоящее время не существует однозначной интерпретации
экспериментальных данных о строении и частотах колебаний Ga2O. Рамбиди и Толмачев [341] при электроно-
графическом исследовании паров Ga2O установили, что средней в условиях эксперимента
(Г=1300 К) является нелинейная конфигурация этой молекулы с -? Ga—О—Ga=140+10°.
Однако позднее теми же авторами [2708] было показано, что в случае низкой частоты
деформационного колебания равновесной структуре Ga2O может отвечать -^ Ga—О—Ga=150—180°.
Мальцев и Шевельков [240] в ИК-спектре газообразного Ga2O наблюдали только одну полосу,
отнесенную к частоте v3; правда, авторы отметили, что отсутствие в спектре частоты vx нельзя
рассматривать как доказательство линейной структуры Ga2O, так как эта полоса может быть
малоинтенсивной. Хинчклифф и Огден [1439] в ИК-спектре молекул Ga2O, изолированных в матрице
из N2, наряду с интенсивной полосой v3 наблюдали слабую полосу 471 см, отнесенную ими к
частоте \ г. Поводом для такого отнесения послужило изучение зависимости интенсивности полос
В матрице из аргона наблюдалась только полоса vs.
174
Галлий и его соединения
Ga2O (г)
от температуры, хотя подтвердить это количественно из-за их слабой интенсивности не удалось.
По той же причине оказалось невозможным изучить изотопные сдвиги этой полосы по кислороду.
Поэтому отнесение vx Хинчклифф и Огден рассматривают как предварительное. Из отношения
значений частоты v3 в спектрах разных изотопических модификаций молекулы Ga2O авторы [1439J
оценили -3 Ga—О—Ga=142°, но показали, что учет ангармоничности колебаний может
увеличить угол до —170°.
На основании результатов электронографического исследования [341, 2708] и интерпретации
ИК-спектров молекул Ga2O, изолированных в матрицах [1439], в справочнике принято, что в ос-
Таблица 22.6.
Значения молекулярных постоянных, а также о и j)|, принятые для расчета
термодинамических функций Ga2O, GaOH, GaF2, GaF3, GaCL, GaCb, и Ga2Cl6
Молекула
Ga2O
GaOH
GaF2
GaF3
GaCl2
GaCb
Ga2Cl6
GaOH: a
GaF2: a
GaCl2: a
Электронное
состояние
2>A
2* A?
XlA
2*Af
vx
472
600
720
700
410
381
416
приведено значение /•
A'Bi, 7o=15C
J2Bb TO=1OC
00 см
H0 cm
200
310 B)
160
200
110
145
324
1039 г-см2.
, o=2, Pl=2
, o=2, Pl=2
v3
CM
809
3600
760
759,2 B) 190
450
457 B) 128
168 89
Ga2Cle: a
•
v5
B) -
B) -
30
v7=120,
vii=472,
vi5=70,
—
—
—
—
—
310 a
v8=477,
Vl2=113,
vw=400,
We-
Г3 • CM
4,3.10s
8,1 a
9,5-102
7-103
Ц.ЮЗ
12-104
8,7-10»
v9=202,
vi3=310,
v17=280,
10"'
б
G
2
1
2
6
2
6
4
vio=17,
Vl4=120,
Pi
1
1
2
1
2
1
1
vi8=155 (в см).
новном электронном состоянии ХгА молекула Ga2O имеет нелинейную структуру симметрии С2с.
Приведенное в табл. 22.6 произведение моментов инерции вычислено для значений г (Ga—О) =
= 1,82+0,03 Аи^ Ga—О—Ga=140+10°, найденных Толмачевым и Рамбиди [2708], его
погрешность составляет 2-10~114 г3.см6.
ИК-спектр Ga2O исследовался как в газообразном состоянии [239, 240], так и в
низкотемпературных матрицах [304а, 321, 1437, 1439, 1927], однако надежно определена только частота
антисимметричного валентного колебания v3. Поскольку в ИК-спектре паров Ga2O наблюдаются
широкие полосы, приведенное в табл. 22.6 значение v3 принято по результатам исследования Хинчк-
лиффом и Огденом [1439] ИК-спектра Ga2O в матрице из N2. По результатам той же работы
принято значение vx. Частота \ вычислена по уравнениям поля валентных сил в предположении, что
fjr2=0,05 fr x. Погрешность в частотах оценивается в 50 см. Возбужденные электронные
состояния Ga2O в справочнике не рассматриваются, так как согласно предварительным результатам
исследования электронного спектра (см. [446]) их энергии превышают 25 000 см.
Термодинамические функции Ga2O (г) были вычислены по уравнениям A. 3)—A. 6), A. 9)т
A. 10), A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор —гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных, включая неопределенность структуры Ga2O, а также приближенным
характером расчета; они могут составлять 3—8 Дж-К~1-моль в значениях Ф° (Т) в интервале 100—
6000 К.
Термодинамические функции Ga2O (г) вычислялись ранее до 2000 К в работах Шевелькова
и Мальцева [447], Баттаба и др. [618], а также для нескольких температур Кохраном и Фостером
1 Маковецкий и др. [1927] к частотам vx и v2 отнесли полосы 596 и 417см \ наблюдаемые в ИК-спектре иопещп
Ga2O, изолированных в матрице из аргона. Однако в работе [1438] было показано, что эти полосы не
принадлежат молекуле GaaO. Ошибочным оказалось также отнесение Мальцевым и Шевельковым [239] полосы 420 см~*
к частоте vx. Эта полоса, как признали сами авторы позднее, обусловлена, по-видимому, загрязнениями [240].
175
Gas03 (к, ж)
Глава 22
[862]. Данные [447] и табл. 684 согласуются в пределах 0,5 Дж-К~1-моль~1, расхождения
результатов расчета [618] с данными табл. 684 достигают 3 Дне-К-моль.
Константа равновесия реакции Ga2O (r)=2Ga (г)+О (г) вычислена с использованием значения
ЬГН° @) =880,783 +14 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^ ° (Ga2O, г, 0) = —92 + 12 кДж • моль.
Эта величина основана на приведенных в табл. 22.7 данных, полученных при исследованиях
равновесия реакций с участием Ga2O (г); погрешности приведенных значений отражают только
воспроизводимость измерений. В случае работы [618] в качестве погрешности приведена оценка
авторов этой работы. В ней из одних и тех же измерений (при двух отношениях Н2 : Н2О) были
Таблица 22.7. Результаты определений ДдЕГ° (Ga2O, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (Ga2O, г, 0)
II закон
III закон
Кохран, Фостер, 1962 [862] Эффузионный, 2Ga (>K)+SiO, (K)=Ga,0 (г)+ —131+50 —97,1+4,0
+SiO (г), 1179—1380 К, 6 точек
Эффузионный, 2Ga(Ht)+MgO (K)=Ga2O (г)+ —110+60 —77,1+4,0
+Mg(r), 1179—1383 К, 5 точек
Фрош, Тёрмонд. 1962 [1147] Протока, 4/sGa (ж)+1/з0а20з (K)=Ga2O (г), 1073— —103+25 —95,0±1,5
1273 К, 4 точки
Берне, 1966 [793] Масс-спектрометрический, Ga2O (r)=2Ga (г)+ — —72,5±2,5
+О (г), 2068 К, 4 измерения
Щукарев и др., 1969 [470] Масс-спектрометрический, Ga2O3 (к)—Ga2O (г)+ 2+70 —135+8 а
+О2(г), 1728—1841 К, 5 точек
Масс-спектрометрический, */8Ga (aO+VsGajOs (к)= —72+20 —99,2±3,0
=GaaO (г), 1013—1103 К, 7 точек
Баттат и др., 1974 [618] Протока, 2Ga (ж)+Н20 (r)=Ga2O (г)+Н2 (г), —79 —88,3±8
1186—1324 К, данные представлены в виде
уравнения]
Чаплыгин, 1975 [432] Протока, 1208—1291 К, 2Ga (ж)+Н20 (г)= —113+40 —91,9±1,0
=Ga2O (г)+Н2 (г), 20 точек
а Авторы [470] принимали без какого-либо обоснования, что коэффициент испарения составляет 0,15. Если
принять, что коэффициент испарения равен единице, то эти измерения приводят к значению —83 кДж-моль.
вычислены константы равновесия реакций с образованием GaaO (г) и GaOH (г), так как в условиях
этих измерений оба этих вещества образовывались в сравнимых количествах. Принятая величина
является средней из данных [432, 470, 618, 862, 1147]. При этом не были учтены измерения Кох-
рана, Фостера [862] с MgO (к) и измерения Бёрнса [793], приводящие к выпадающим значениям.
По данным работы Щукарева и др. [470] принималась величина, пересчитанная с учетом
коэффициента испарения, равного единице. При оценке погрешности были учтены неточность
термодинамических функций (соответствующая погрешность 10 кДж-моль в энтальпии образования) и
расхождения результатов измерений в разных работах.
Ga2O3 (к, ж). Термодинамические свойства кристаллического и жидкого триоксида дигаллия
в стандартном состоянии при температурах 100—4000 К приведены в табл. 685.
Значения постоянных, принятых для расчета термодинамических функций, приведены в табл.
22.1. В справочнике за стандартное состояние Ga2O3 (к) до Г=2080 К принята моноклинная
модификация (P-Ga2O3), относящаяся к структурному типу 6-А12О3 [1745а] и являющаяся единственной
стабильной модификацией GaaO3 г. Термодинамические функции Ga2O3 (к) при Т <^ 298,15 К
1 По данным [829, 2398] кроме ^-модификации существуют четыре метастабильные модификации Ga2O3:
гексагональная а (структурный тип <х-А12О3), кубическая у (шпинельная структура), кубическая S (структурный тип
Т12О3, С-тип) и «-модификация низкой симметрии. Температуры монотропных переходов равны 773 К (8 -> е
у -> а), 898 К (а -> р), 923 К (у -* 0) и 1143 К (* -> р). . .,
176
Галлий и его соединения
GaH (г))
вычислены по результатам измерений теплоемкости образца Ga2O3 чистотой 99,9%, выполненных
Кингом [1712] E4—296 К). При Т < 54 К были использованы данные по теплоемкостям менее
чистого образца Ga2O3 (—98,7%), полученные Адамсом и Джонстоном [484] A6—301 К); в
интервале 54—296 К эти данные лежат в среднем на 1% ниже данных Кинга [1712]. Погрешности
принятых значений S° B98,15 К) и Н° B98,15 К)—Н° @) оцениваются в 0,4 Дж-К^-моль
и 0,04 кДж-моль соответственно. В интервале 298,15—2080 К принято уравнение для
теплоемкости (см. табл. 22.1), выведенное совместной обработкой по методу Шомейта
результатов измерений энтальпии [3-Ga2O3, проведенных в работах Миллза [2030] B98—
1800К), Цагарейшвили и Гвелесиани [428] C69—1596 К) и Панкраца и Келли [2177]
C96—1800 К). При обработке точность измерений [2030] оценивалась в 0,5%, [428] — в 1%
и [2177] — в 2%. Температура плавления Ga2O3 B080 + 20 К) принята на основании согласующихся
между собой данных Macao и др. [2041] B082+20 К) и Шнейдера и др. [2466] B068+20 К).
Значения температуры плавления, найденные в работах [2398] A998 К), [2802] B013 К) и [829]
B173 К), менее надежны. Энтальпия плавления |3-Ga2O3 A00+20 кДж-моль) и теплоемкость
Ga2O3 (ж) A60+25 Дж-К~1-моль~1) приняты равными соответствующим величинам для А12О3.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000, 3000 и 4000 К оцениваются
в 0,3,1, 4, 10 и 15 Дж- К- моль соответственно. Расхождения этих значений с данными
справочника [578] составляют не более 0,3 Дж- К-моль.
В справочнике принято значение энтальпии образования
Д/Г (Ga2O3, к, 298,15 К) — —1091,0 + 3,0 кДж • моль,
основанное на приведенных в табл. 22.8 результатах измерений. Данные, полученные методом
ЭДС [518, 1635, 1728] и в термохимических исследованиях [77, 1912], находятся в хорошем
соответствии. Принятая величина является средневзвешенной из найденных в этих работах.
Таблица 22.8. Результаты определений A/fi"° (Ga2O3, к, 298,15 К) (в кДж-моль)
Авторы
Кляйндинст, Стевенсон, 1972 [1728
Смит, Чаттерджи, 1973 [2556]
Катояма, Казука, 1973 [1635]
Андерсон, Донахыо, 1977 [518]
Рот, Веккер, 1932 [2373]
Клемм, Шник, 1936 [1725]
Гаджиев, Шарифов, 1961 [77]
Ма, 1962 [1912]
Метод
ЭДС ячейки с твердым электролитом, 873—
1273 К, 18 точек, 2 образца
То же, 823—1073 К, результаты измерений
представлены в виде уравнения
То же, 1083—1303 К, результаты измерений
представлены в виде уравнения
То же, 777—1091 К, 13 точек
Калориметрическое измерение энтальпии
сгорания Ga, 3 опыта, сжигание с
вспомогательным веществом
То же, 2 опыта, сжигание с вспомогательным
веществом
То же, 4 опыта
То же, 5 опытов
Д/Я° (Ga2O3, к, 298,15 К)
II закон
III закон
—1055+10 —1088,0+3,0
—1082 —1065,8
—1097 —1088,9+3,0
—1110±5 -1089,2+3,0
-1070
—1083,7+8,0
—1085,7±6,0
—1092,2+1,5
Константа равновесия реакции Ga2O3 (к, ж)=2йа (г)+30 (г) вычислена на основании
значения ДГЯ° @) =2363,732+8,5 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования Ga2O3 (к), Ga (г) и О (г).
GaH (г). Термодинамические свойства газообразного гидрида галлия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 686.
Молекулярные постоянные GaH, принятые в справочнике на основании результатов
исследований электронного спектра этой молекулы, представлены в табл. 22.4. В спектрах GaH
наблюдались 5 систем полос: ^По-^Х1^ [2263, 2264], а3П0+ ** Хх?+ [1220, 2122], а^^Х1^
[1220, 2119, 2122], а3П2 -> Хх?+ [1220] и 4Ш ^ Хх?+ [1220, 1778, 2122]. Эти состояния связаны
с электронными конфигурациями . . .о2о2 (Х1?+) и . . .о2атс и коррелируют с атомами галлия
177
GaH (г) Глава 22
и водорода в основных состояниях: Ga BРу)+Я BS) (Хх?+, о3П0-, а3^) и Ga BРз,2)+Н BS)
(а3П0+, а3П2, А1!!). Со вторым пределом должно также коррелировать состояние 3 Е+, которое не
наблюдалось в спектрах и, так же как у молекул ВН и А1Н, вероятно, является отталкиватель-
ным состоянием. По аналогии с этими же молекулами можно предполагать, что молекула GaH
должна иметь связанные валентные и ридберговские состояния с энергиями свыше 35 000—
40 000 см. По-видимому, переходы в эти состояния наблюдал Гартон [1173] в спе
поглощения GaH в области 2160—2400 А. Эти состояния не включены в табл. 22.4.
Молекулярные постоянные GaH в состояниях Х1Е+, а3П0+, а 3ПХ и а3П2 приняты по работе Гин-
тера и Иннеса [1220], которые выполнили анализ тонкой структуры семи полос перехода а3П0+—
ХХЕ+ (г/, v" <^ 3), семи полос перехода а3^—ХХБ+ (v', v" <[ 3) и полосы 0—0 перехода а3П2—
X1 И+. Принятые вращательные постоянные в состояниях а3^ и а3П2 вычислены как средние
из данных [1220] для двух компонент (+ и —), колебательные постоянные в состоянии а3П2
приняты такими же, как в состоянии a3nx. Молекулярные постоянные в состоянии а3П0- основываются
на результатах анализа структуры полос 0—0, 1—0, 1—1, 2—1 и 2—2 перехода а3П0—ХХЕ+
в работах Пойнера и др. [2263, 2264]. Поскольку значения Д&/2 и ДСт»/,, рассчитанные по данным
авторов [1220, 2263, 2264] для состояний «3П0- и а3П0+, близки, а для состояния а3П0+ была
определена также величина Д??»/„ для обоих состояний приняты одинаковые значения колебательных
постоянных по данным [1220]. Приведенные в табл. 22.4 значения вращательных постоянных
в состоянии а3По- рассчитаны из данных для у=0, 1 и 2, найденных Пойнером и др. [2264].
Состояние .4 41 молекулы GaH является слабосвязанным. В спектре GaH наблюдались полосы
с сильно размытой структурой, связанные с переходом с уровня у=0 этого состояния.
Вращательная структура аналогичных полос в спектре GaD предиссоциирована меньше и полоса 0—1 была
проанализирована Кронквистом и др. [1778]. Приведенные в табл. 22.4 вращательные постоянные
GaH в этом состоянии рассчитаны по данным [1778] для GaD и по соотношениям A. 66).
Термодинамические функции GaH (г), приведенные в табл. 686, рассчитаны по уравнениям
A. 3)—A. 10), A. 93)—A. 95). Значения <2ВЯ и ее производных вычислялись по уравнениям A. 90)—
A. 92) с учетом возбужденного состояния а3И (расщепление этого состояния на подсостоя-
ния а3П0-, а3П0+, а3Пх и а3П2 во внимание не принималось). Статистические суммы по колебательно-
вращательным уровням энергии состояний X1S+ и а3П и их производные находились
непосредственным суммированием по уровням энергии каждого состояния с помощью соотношений A. 76)—
A. 78). В расчетах учитывались все колебательно-вращательные уровни, ограниченные
предельными кривыми диссоциации состояний Х1Е+ и а3П, энергия уровней вычислялась по уравнениям
A. 65), A. 60) для состояния ХХЕ+ и по уравнениям A. 65), A. 61) для состояния а3П, мультиплет-
ность и вырождение уровней состояния а3П учитывались статистическим весом 6. Значения
коэффициентов Ykl в уравнениях A. 65), использованные в расчетах, приведены в табл. 22.5;
коэффициенты F40, Y03 и YOi для состояния Z1E+ получены из условий A. 50) и" A. 59), коэффициенты
Ykl для состояния а3П основаны на молекулярных постоянных GaH в подсостоянии я3^,
коэффициент Y03 введен для обеспечения условий A.59).
При Т <^ 1500 К основные погрешности в термодинамических-функциях GaH (г) обусловлены
неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль~1. При более
высоких температурах начинают сказываться погрешности из-за отсутствия экспериментальных
данных о колебательно-вращательных уровнях энергии с v ^> 3, а выше 4000 К — из-за
пренебрежения высокими электронными состояниями. Погрешности из-за пренебрежения мульти-
плетным расщеплением подуровней состояния а3П и вкладом состояния А1!! не существенны
во всем интервале температур. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
оцениваются в 0,02, 0,04 и 0,08 Дж-К-^моль.
Таблица термодинамических функций GaH (г) публикуется впервые. В работе Шнейдера
[2463 ] были рассчитаны значения QES и In QBK для GaH (г) и определены коэффициенты полиномов,
аппроксимирующих эти величины для У=1000-^-5300 К. Соответствующие значения Ф° (Т)
отличаются от данных табл. 686 на 0,6—0,8 Дж-К~1-моль.
Константа равновесия реакции GaH (r) = Ga (г)+Н (г) вычислена на основании энергии
диссоциации
ДгЯ° @) = Do (GaH) = 271,6 ± 2,5 кДж . моль« 22 700 ± 200 см.
Эта величина была принята в справочнике в результате анализагимеющихся спектральных данных.
Исследования предиссоциации GaH и GaD в состоянии .441 в работах Гинтера, Иннеса [1220}
178
Галлий и его соединения
GaOH (г)
и Кронквиста и др. [1778] показали, что потенциальная кривая этого состояния у невращающейся
молекулы имеет максимум, а уровень v—О G700«^23 900 см) лежит выше соответствующего диссо-
циационного предела Ga BА/2)+Н BS). Из этого следует, что Do (GaH)^22 900 см. Энергия
последнего наблюдавшегося колебательного уровня состояния а3Пх составляет T0Q-\-G0 C)«
^21 500 см. Короткая экстраполяция уровней этого состояния (vmitt=5) дает для его предела
значение 22 920 см. Если принять, что погрешность экстраполяции не превышает 30% от ее
величины, то D0(GaH) ^ 22 500 см. В справочнике принято значение, среднее между двумя
пределами. Ему соответствует
A//°(GaH, г, 0)== 215,434 +[4,8 кДж-моль
GaOH (г). Термодинамические свойства газообразного гидроксида галлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 687.
Молекулярные постоянные GaOH, использованные в расчете термодинамических функций,
приведены в табл. 22.6. Спектр и структура GaOH экспериментально не исследовались. По
аналогии с молекулой АЮН и молекулами гидроксидов щелочных металлов в справочнике
принимается, что в основном электронном состоянии X1 ? молекула GaOH имеет линейную структуру
(симметрия Ссо„)- Приведенный в табл. 22.6 момент инерции соответствует структурным
параметрам г (Ga—О)=1,82+0,05 А (как в Ga2O) и г (О—Н)=0,96+0,02 А, его погрешность составляет
1-Ю9 г-см2. Частота vx (валентное колебание Ga—О) оценена путем сопоставления значения
шв (GaF)=622,2 см с величиной vx, вычисленной по силовой постоянной, перенесенной из
молекулы Ga2O E60 см). Частота; v3 оценена по частотам деформационных колебаний молекул
гидроксидов щелочных металлов. Для v3 принято ее характеристическое значение. Погрешности частот
vi> V2 и V3 оцениваются в 50—100 см. По аналогии с молекулами двухатомных галогенидов
галлия можно предполагать, что энергии возбужденных состояний GaOH превышают 30 000 см.
Термодинамические функции GaOH (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 126) и A.129)-'В приближении «жесткий ротатор"—гармонический осциллятор».
Погрешности вычисленных значений обусловлены неточностью молекулярных постоянных, а также
приближенным характером расчета и составляют 1, 3 и 5 Дж- К-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Ранее таблица термодинамических функций GaOH (г) вычислялась в работе Баттата и др.
[618] для Т <; 2000 К. Расхождения между результатами расчета [618] и данными настоящего
справочника достигают 4,7*Дж-К-моль~1 в значении S° B000 К), главным образом из-за разной
оценки постоянных.
Таблица 22.9. Результаты определений Д/Н (GaOH, г, 0) (в кДж-моль)
Авторы
Метод
bfH°(GaOE, г, 0)
II закон
III закон
Гурвич, Вейц, 1958 [90, 98] Исследование равновесия реакции Ga (г)+ — —141
+ ОН (г) = GaOH (г) в пламенах, 3070 К (оценка)
Булевич, Сагден, 1958 [790] Спектрофотометрия пламен H2+O2+N2, Ga —117+20 —137±40
(r)+H2O(r)=GaOH (г)+Н (г), 1770—2450 К,
16 точек
Гурвич и др., 1969 [100, Спектрофотометрия пламен H2+O2+N2, —90+28 —132,6+4,6
101, 1334] Ga (г)+НаО (r>=GaOH (г)+Н (г), 1670—
2200 К, 12 точек
Масс-спектрометрический, Ga2O (г)+Н2О (г)= —149+50 —132,6+10
=2GaOH (г), 938—1071 К, 7 точек
¦Келли, Падли, 1971 [1665] Спектрофотометрия пламен Н2+О2+СОо и —125,3±4,2 —123,4±4,2
H2+O2+N2, Ga (г)+Н2О (r)=GaOH »+
+V2H2(r), 1950—2750 К, 32 точки
Баттат и др., 1974 [618] Протока, Ga (ж)+Н20 (г)= GaOH (r)+V2H2(г), —180 —157±17
1186—1324 К, 11 точек, данные представлены
в виде уравнения
179
GaF (г) Глава 22
Константа равновесия реакции GaOH (r) = Ga (г)+О (г)+Н (г) вычислена с использованием
значения hrH° @) =858,817 +11 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A,#°(GaOH, г, 0) = —125 + 10 кДж • моль.
В табл. 22.9 представлены результаты исследований равновесия реакций с участием GaOH (г),
в качестве погрешностей рассчитанных величин дана только воспроизводимость измерений.
Наиболее полные и удовлетворительно согласующиеся между собой результаты были получены в
работах [100, 101, 1334] и [1665] и на них основана принятая величина. Неточность использованных
в расчетах термодинамических функций приводит к дополнительной погрешности в энтальпии
образования около 7 кДж-моль. Авторы работы [618] из измерений скорости уноса галлия (при
двух отношениях Н2 : Н2О) попытались определить одновременно константы равновесия реакций
образования GaOH (г) и Ga2O (г), так как в условиях опытов оба вещества образовывались в
сравнимых количествах. При этом, однако, погрешность определения энтальпий образования обоих
веществ оказывается значительной.
GaF (г). Термодинамические свойства газообразного фторида галлия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 688.
Молекулярные постоянные 89Ga19F, принятые в справочнике на основании результатов
исследований электронного и. вращательного спектров этой молекулы, приведены в табл. 22.4.В
электронном спектре GaF наблюдались переходы а3П0+**Х12+ [589, 596, 598, 1452, 2829, 2830],
а^^Х1!* [589, 598, 2829, 2830] и i1n*iI1E+ [598, 2829, 2830]. Подобно другим галоге-
нидам элементов группы III А, все три состояния молекулы GaF, включая ненаблюдавшиеся
компоненты состояния 3П, коррелируют с атомами в основных состояниях, причем состояние X1 ?+
должно коррелировать с пределом Ga BPi/2)+F (а-?Ч)> а состояние ЛЧ1 — с пределом Ga B/\)+
+F Bi\). Можно предполагать, что не наблюдавшиеся в спектрах другие связанные состояния
GaF имеют энергии около 50 000 см и выше; в справочнике они не рассматриваются.
Приведенные в табл. 22.4 колебательные постоянные GaF найдены Барроу и др. [596] на
основании полученных ими ранее [598] данных о кантах полос систем а3П0+—ХХ2+ A6 полос,
v" < 3, v' < 3), (Л^-Х1 ?+ A6 полос v" < 5, v' < 6) и АШ—Х1 ?+ B2 полосы, v" < 7, v' < 5),
а также измерений начал полос 0—0, 0—1, 1—2, 2—3 системы а3Па—X1 ?+ и полосы 0—0 системы
а3П0+—Хх?+. Эти значения постоянных хорошо согласуются с полученными в более ранних
работах [2829, 2830]. Вращательные постоянные GaF в состоянии Х12+ приняты по работе Хоэфта
и др. [1452], в которой исследовался микроволновый спектр этой молекулы, включавший
переходы су^Зи/^2. Близкие значения были получены в работе [596J из анализа вращательной
структуры ряда полос. Вращательные постоянные в состояниях а3П и А1!! основаны на данных
[596] [(см. [1491а]).
Термодинамические функции GaF (г) были рассчитаны по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения (?вн и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом
одного возбужденного состояния а3П и в предположении, что Qm?al = 6()№ вр. Энергия возбуждения
состояния а3И была принята равной энергии подсостояния а3Пх. Статистическая сумма по
колебательно-вращательным уровням энергии состояния Х1!!" и ее производные находились по
уравнениям A.73)—A-75) непосредственным суммированием по v и интегрированием по J с
помощью соотношений типа A. 82). В расчете учитывались все уровни энергии с значениями / ^ /maI,
последние определялись из условий A. 81). Колебательно-вращательные уровни энергии
состояния ХХБ+ вычислялись по уравнениям A. 65) и A. 64а), значения коэффициентов Yhl в этих
уравнениях, а также величин утах и /1т приведены в табл. 22.5. Значения коэффициентов Yk0 были
уточнены из условий A. 50), коэффициент У03 введен для обеспечения условий A. 59).
Постоянные молекулы GaF в состоянии X1 ?+ известны с удовлетворительной точностью,
поэтому при температурах до 1500—2000 К погрешности рассчитанных термодинамических функций
определяются неточностью фундаментальных постоянных и не превышают 0,02 Дж-К-моль.
При более высоких температурах становятся существенными отсутствие экспериментальных
данных о большей части колебательно-вращательных уровней этого состояния, а также применение
приближенной методики для учета вклада состояния а3П. Пренебрежение в расчете состоянием
А1!! ж другими электронными состояниями практически не влияет на точность результатов
расчета. Погрешности в значениях Ф° (Т) при 3000 и 6000 К составляют 0,05 и 0,3 Дж- К- моль.
180
Галлий и его соединения GaF2 (г), GaFs (к)
Термодинамические функции GaF (г) ранее вычислялись в справочнике [305] для Т ^ 5000 К.
Результаты этого расчета и данные табл. 688 отличаются из-за использования авторами [305]
приближенной мэтодики и пренебрежения состоянием а3П; расхождения достигают 1,2, 2,8 и
3,7 Дж-К~1-дголь~1 в значениях Ф° (Т), S° (Г) и С"р (Т) при высоких температурах.
Константа равновесия реакции GaF (r) = Ga (r)-f-F (г) вычислена с использованием принятого
в справочнике значения энергии диссоциации
АгН° @) = Do (GaF) = 580 ± 10 кДж • моль я» 48 500 + 850 см.
Эта величина основана на исследовании равновесия реакции CaF (r) + Ga (r)=Ca (r)+GaF (г),
выполненном масс-спектрометрическим методом Мурадом и др. [2082] A358—1456 К, 5 точек).
Расчет по II закону из этих данных дает D0(GaF)=572 +30 кДж- моль, расчет по III закону •—
значение 579+10 кДж-моль (погрешность значения Дг#° принята по рекомендации авторов [2082]).
Короткая экстраполяция колебательных уровней состояния А1!! (примерно на 1500 см)
приводит к энергии диссоциационного предела этого состояния, равной около 51 150 см над
уровнем v=0, /=0 состояния Х1!4" и величине D0(GaF)=50 300+500 см. Расхождение между
этим значением и полученным из данных [2082], по-видимому, обусловлено существованием
максимума на потенциальной кривой состояния АгИ (см. [598, 2082]), высота которого в соответствии
с приведенными данными составляет 1500+900 см.
Принятой энергии диссоциации соответствует также
bfH°(GaV, г, 0) = —231,725 +И кДж • моль.
GaF2 (г). Термодинамические свойства газообразного дифторида галлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 689.
Молекулярные постоянные GaF2, использованные для расчета термодинамических функций,
приведены в табл. 22.6. В литературе отсутствуют какие-либо экспериментальные данные о
геометрических параметрах и частотах колебаний GaF2. В справочнике принимается, что в основном
электронном состоянии Х2АХ молекула GaF2 имеет симметричную нелинейную структуру (группа
симметрии С2о) с параметрами г (Ga—F)=l,80+0,05 А (как в GaF3) и -$ F—Ga—F=120+5°
(как в ВН2, BF2, А1Н2). Погрешность вычисленного значения IaIbIc составляет 5-10~115 г3-см6.
Основные частоты GaF2, принятые в справочнике, вычислены по уравнениям поля валентных сил
и силовым постоянным молекулы GaF3, соответствующим ее частотам, приведенным в табл. 22.6
(/г=4,84-10в, /гг=0,28-105, /а/г2=0,10-105 дин-см). Погрешности принятых частот GaF2
составляют 10—15%. По аналогии с A1F2 можно предполагать, что у молекулы GaF2 имеется
электронное состояние А2Вг с энергией возбуждения 15 000+5000 см".
Термодинамические функции GaF2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом возбужденного электронного состояния АгВ1 по уравнениям A. 168)—A. 170).
Погрешности вычисленных значений определяются в основном неточностью оценки
молекулярных постоянных, а также (при высоких температурах) приближенным характером расчета,
погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 2, 3 и 5 Дж-К^-моль.
Таблица термодинамических функций GaF2 (г) публикуется впервые.
Константа равновесия реакции GaF2 (r) = Ga (r)+2F (г) вычислена с использованием
принятого в справочнике значения
Д Д° @) = 960 + 50 кДж • моль.
Эта величина оценена по энтальпиям атомизации молекул GaF, GaF3, A1F, A1F2 и A1F3,
принятым в справочнике, ей соответствует
Д//° (GaF2, г, 0) = —534,450 + 50 кДж • моль.
GaF3 (к). Термодинамические свойства кристаллического трифторида галлия в стандартном
состоянии при температурах 298,15—2000 К приведены в табл. 690.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 22.1. В справочнике за стандартное состояние GaF3 (к) принята гексагональная модифи-
181
GaFs (г) Глава 22
нация (структурный тип VF3 [740а]). В литературе отсутствуют сведения о термодинамических
свойствах GaF3 (к). Значения S° B98,15 К) и Н° B98,15 К)—Я0 @), приведенные в табл. 22.1;
оценены по различным эмпирическим методикам (см. [216, 427, 814а]) с использованием данных
для A1F3 (к) и GeF3 (к); точность принятых значений оценивается в 8 Дж-К~1-моль~1 и 1,5 кДж-
•моль соответственно. В интервале 298,15—1500 К для теплоемкости GaF3 (к) принято уравнение
(см. табл. 22.1), выведенное по двум значениям С°р (GaF3, к) при Г=298,15 и 1500 К, которые были
оценены путем сравнения экспериментальных данных по теплоемкости A1F3 (к) и LaF3 (к).
Погрешности в значениях С°р (GaF3, к), вычисленных по этому уравнению, могут достигать 10%.
В литературе отсутствуют экспериментальные данные о температуре плавления GaF3. Сравнение
известных температур плавления A1F3 B523 К) и InF3 A445 К) позволяет сделать вывод, что
температура плавления GaF3 должна быть выше 1500 К; при этой температуре давление
насыщенного пара GaF3 составляет около 2,4 атм.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 8, 10 и
20 Дж-К-моль~1 соответственно.
Таблица термодинамических функций GaF3 (к) публикуется впервые.
Принятое в справочнике значение
-1
AfH°(GaF3, к, 298,15 К) = —1175 + 25 кДж • моль
основано на исследованиях равновесия реакции 4/3GaF3 (K)+SiO2 (стекл.)=2/3Са203 (K)+Si F4(r),
выполненных Сониным [391 ] статическим методом G53 К, 1 точка). Равновесие этой реакции
устанавливалось медленно, что могло привести к существенным неточностям. Погрешность, связанная
с неточностью термодинамических функций GaF3 (к), составляет около 10 кДж-моль.
Давление пара трифторида галлия GaF3 (к, ж) = ОаР3 (г) было вычислено с использованием
энтальпии сублимации
Asff°(GaF3, к, 0) = 255±15 кДж • моль.
Эта величина принята на основании результатов измерений давления пара, приведенных
в табл. 22.10. Во всех работах экспериментальные данные представлены только в виде уравнений.
Таблица 22.10. Результаты определении АеЕГ (GaFa, к, 0) (i
Авторы
Фезер и др., 1972 [1071а]
Жегульская и др., 1972 [130а]
Сидоров, Жегульская, 1975 [2517а]
Сонин, 1975 [391]
Метод
i кДж-моль *)
Масс-спектрометрический и торзионный,
714-1015 К
Масс-спектрометрический, 805—940 К
Масс-спектрометрический, 800—920 К
Статический, 1080—1241 К
Д,Я° (GaF
II закон
246
260
252
271
s, к, 0)
III закон
256,3
247,5
256,3
258,4
Как было показано (см., например, [130а]), количество димерных молекул в парах над трифтори-
дом галлия не превышает 1%, и не принималось во внимание. Как видно из таблицы, данные всех
авторов удовлетворительно согласуются между собою. В справочнике рекомендовано среднее
значение. Неточность термодинамических функций приводит к погрешности в энтальпии сублимации
в 10 кДж- моль для измерений, выполненных при 1000 К.
GaF3 (г). Термодинамические свойства газообразного трифторида галлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 691.
Молекулярные постоянные, использованные для расчета термодинамических функций GaF3,
приведены в табл. 22.6. Электронографическое исследование [2а] и исследование ИК-спектра
GaF3 [1365]|показали, что эта молекула в основном электронном состоянии ХгА имеет плоскую
структуру (группа симметрии D3h). Произведение моментов инерции IaIbIo вычислено для г (Ga—
—F)=1,80 +0,1 А, оцененного на основании сравнения величины межатомного расстояния М—F
в молекулах лтипа MF и MF3, гдеМ=В Al, Ga, In, Sc, Y, La. Это сравнение показало, что соответ-
182
Галлий и его соединения GaCl (r)
ствующие величины отличаются не более, чем на 0,1 А. В электронографическом исследовании
Акишин и др. [2а] нашли г (Ga—F)=l,88+0,02 А, что на 0,11 А больше re (Ga—F); по-видимому,
в измерениях [2а] имела место систематическая ошибка. Погрешность значения IaIbIc не
превышает 3-10~114 г3-см6. Хести и др. [1365] исследовали ИК-спектр молекул GaF3, изолированных
в матрицах из Ne, Аг, Кг и N2. В табл. 22.6 приведены частоты GaF3, полученные в [1365] при
использовании матрицы из наиболее легкого газа—неона. Ненаблюдавшаяся экспериментально
частота vx вычислена по уравнениям поля валентных сил и значениям v3 и v4 в предположении, что
для GaF3 справедливо соотношение /r//rr=17 (как это имеет место в галогенидах А1). Погрешности
принятых частот v2, v3 и v4, с учетом возможного матричного сдвига, оцениваются в 15—20 см,
погрешность в значении vx может достигать 70 см. Сведения о возбужденных электронных
состояниях GaF3 в литературе отсутствуют, но можно предполагать по аналогии с A1F3, что их энергии
превышают 20 000 см.
Термодинамические функции GaF3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных таким образом функций определяются неточностью
молекулярных постоянных и приближенным характером расчета и составляют 2, 5 и 8 Дж-К-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций GaF3 (г) публикуется впервые.
Константа равновесия реакции GaF3 (r) = Ga (r)+3F (г) вычислена с использованием значения
ДГЯ° @)=1420,716+30 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации GaF3 (к). Этим величинам также соответствует
bfH°{GaFz, г, 0) = —917,891 ±29 нДж-моль.
GaCl (г). Термодинамические свойства газообразного хлорида галлия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 692.
Молекулярные постоянные 69Ga36Cl, принятые в справочнике на основании результатов
изучения электронного и вращательного спектров этой молекулы, приведены в табл. 22.4. В
электронном спектре GaCl в работах [1854, 2027, 2028, 2220] наблюдались переходы а3П0+** ХгЛ+ж
а3Пх ^ X1 S+ и АШ <- X1 Е+. Кроме того, в области 2419 А в поглощении наблюдался континуум,
который вероятно связан с переходом в нестабильное состояние [2027, 2028, 2220]. Так же как
у молекулы GaF, все три электронные состояния GaCl должны коррелировать с атомами в их
основных состояниях; поэтому подсостояния а3П0- и а3П2, переходы в которые не наблюдались
экспериментально, у GaCl должны быть связанными состояниями с энергиями 29 500 —
30 000 см. Состояние Хх?+ коррелирует с пределом Ga BР1/г)+С1 B/\), а состояние АгП—
с пределом СаBРз<г)-|-С1B.Рз/2). Если молекула GaCl имеет другие связанные электронные
состояния, их энергии должны превышать 40 000 — 45 000 см.
Принятые в табл. 22.4 колебательные постоянные GaCl в состояниях X1 ?+, а3П0+ и a3nt
найдены Мишером и Верли [2028] по Р-кантам полос систем а3П0+ — X1 ?+ A0 полос, v'^A, z/'<T2),
а3Пх — Хх?+ G полос, z/<13, г/'<^1). Хотя они хорошо согласуются с постоянными, полученными
позднее в работе [1854], их точность невелика. Вращательные постоянные GaCl в состоянии
Хх?+ приняты по работе Тиманна и др. [2699], которые исследовали в микроволновом спектре
этой молекулы переходы, включающие уровни с /<^1, у^З. Они хорошо согласуются с
результатами других исследований микроволнового спектра [584, 1932], но существенно отличаются от
постоянных, найденных Левиным и Винаусом [1854] из анализа вращательной структуры трех
полос в электронном спектре GaCl. Хьюбер и Герцберг [1491а] с использованием вращательных
постоянных GaCl в состоянии Х1!", найденных в работе [2699], обработали данные [1854]
для оценки вращательных постоянных в состояниях а3П0+ и а3Пх; полученные ими значения
приведены в табл. 22.4. Состояние 4'П у GaCl является слабосвязанным; в системе ^41П*-'Х12+
наблюдались только переходы на уровни с v'=0 и 1, причем полосы с z/=l предиссоциированы.
Термодинамические функции GaCl (г) были рассчитаны по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения QBa и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом
состояния а3П в предположении, что QW вр = 6(?^вр. Энергия возбуждения состояния а3П была
принята равной энергии возбуждения подсостояния а3^. Статистическая сумма по
колебательно-вращательным уровням энергии состояния X1 ?+ и ее производные находились по
уравнениям A.73)—A.75) непосредственным суммированием по значениями и интегрированием по /
183
GaCI (г)
Глава 22
с помощью уравнений типа A.82). В расчетах учитывались все уровни энергии с значениями
х „ где /шах „ определялись из условий A.81), в табл. 22.5 приведены соответствующие зна-
рассчитывались
' шах, р
чения ушах и /ljm. Колебательно-вращательные уровни энергии состояния Xх
по уравнениям A. 65) и A. 61). Значения коэффициентов Ykl в этих уравнениях для изотопической
модификации молекул GaCI, соответствующей природной смеси изотопов обоих элементов,
приведены в табл. 22.5. Они были рассчитаны по соотношениям A. 66) и постоянным молекулы
69Ga35Cl из табл. 22.5. Коэффициенты Yk0, в том числе Y30 и Yi0 были получены с учетом условий
A.50), коэффициент Y03 —из условий A.59).
Погрешности рассчитанных термодинамических функций GaCI (г) при низких температурах
обусловлены неточностью колебательных постоянных GaCI в состоянии X1 Е+, а также принятых
значений фундаментальных постоянных. Выше 1500—2000 К к этому добавляются погрешности
из-за отсутствия экспериментальных данных о колебательно-вращательных уровнях состояния
Xх Е+ с энергией свыше 3000 см, а также приближенного учета вклада состояния а3И.
Пренебрежение слабосвязанным состоянием 4'Пи более высокими электронными состояниями GaCI
практически не влияет на точность расчета. Погрешности в значениях Ф° (Т) при 298,15, 3000 и
6000 К оцениваются в 0,03, 0,05 и 0,2 Дж-К-моль.
Термодинамические функции GaCI (г) при Г^2000 К вычислялись ранее Келли [1656, 1658}
и Шауловым и Мосиным [441]. Расхождения данных этих авторов и настоящего справочника
достигают 1 и 2 Дж-К~1-моль в значениях Ф° (Т) и S° (T) уже при 2000 К и обусловлены
приближенным характером расчетов [441, 1656, 1658].
Константа равновесия реакции GaCI (г) =Ga(r)+Cl(r) вычислена с использованием значения
Д0 @)=D0 (GaCl)=460,62+7,3 кДж-моль-
в справочнике энтальпии образования
500+600 см
-1
соответствующего принятой
A/7°(GaCl, г, 0) = —70 + 6 кДж • моль.
Эта величина основана на результатах исследований равновесия реакций с участием GaCI (г)
(табл. 22.11). Во всех этих работах авторы приводят только уравнения, аппроксимирующие
полученные экспериментальные величины. Приведенные в табл. 22.11 погрешности включают как
воспроизводимость измерений, так и неточность использованных в расчетах термодинамических
функций и энтальпий образования. При вычислении констант равновесия соответствующих
реакций авторы приведенных в табл. 22.11 работ исходили из разных предположений о
молекулярном составе равновесной смеси. Цугель [2908а] и Кирван [1716а] учитывали в расчетах только
GaCl(r) и GaCl3(r), Куния и Хосака [1785а] дополнительно учитывали Ga2Cl4 и Ga2Cl6, а Комши-
лова [173а] — еще Ga2Cl2. В литературе нет прямых экспериментальных исследований молеку-
Таблица 22.11. Результаты определений Д/2Г° (GaCI, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (GaCI, г, 0)
II закон
III закон
Булевич и др., 1961 [787, 788] Спектрофотометрия пламен, Ga(r)+HCl(r)= — —86+20
= GaCl (г)+Н (г), 1500—2600 К
Цугель, 1965 [2908а] Статический, GaCl3 (r)+2Ga ()K)=3GaCl (г), 693— —63 —68,6+3,0
903 К
Кирван, 1970 [1716а] Статический, 3GaCl (г)+0,5Р4 (r)=2GaP (к) + —78 —61+8
+GaCl3(r), 1000—1250 К
Статический, 3GaCl (r)+0,5As4 (r)=2GaAs (к) + —81 —69+16
+GaCh (г), 900-1200 К
Статический, GaCl3(r)+H2(r)=GaCl(r)+2HCl(r), -55 —77+8
900—1330 К
Статический, GaCl3 (r)+2Ga (ж)=ЗСаС1 (г), 720— —65 —68,5+3,0
920 К
Комшилова, 1971 [173а] Статический, GaCls (r)+2Ga (jK)=3GaCl (г), 675— —73 —71,9+2,0
938 К
Куния, Хосака, 1975 [1785а] То же, 709-870 К —72 —67,6±3,0
184
Галлий и его соединения GaCl2 (г)
лярного состава системы Ga—С1 (например, масс-спектрометрическим методом). Несмотря на
удовлетворительное согласие общих давлений, экспериментально измеренных и вычисленных
в предположении о различном молекулярном составе, вопрос о молекулярном составе в условиях
обсуждаемых измерений следует считать открытым. В лучшем положении в этом отношении
оказываются исследования равновесия реакций с участием Р4, As4 и Н2, проводившиеся при более
высоких температурах в работе [1716а]. Однако как раз эти измерения имеют худшую
воспроизводимость и основывать на них выбор энтальпии образования GaCl (г) нецелесообразно.
Исследования равновесия GaQ3(r)+2Ga^)=3GaCl(r), несмотря на различающиеся предположения
о молекулярном составе, приводят к более точным и достаточно близким значениям. По-видимому,
это свидетельствует, что в рассматриваемых условиях GaCl и GaCl3 действительно являются
основными компонентами. Принятое в справочнике значение является средним из результатов этих
измерений. Обращает на себя внимание наличие несогласованности между работами [1716а, 1785а,
2908а] и [173а], так как учет дополнительных компонентов в работе Комшиловой привел к более
отрицательной энтальпии образования GaCl, в то время как из общих соображений следовало
бы ожидать обратного. Погрешность принятой величины увеличена с учетом неопределенности
молекулярного состава пара над GaCl3(K, ж).
Уровень у=1 состояния А1!! молекулы GaCl предиссоциирован (см. выше), по-видимому,
за счет взаимодействия с состоянием, имеющим общий диссоциационный предел с состоянием
X1 Е+. Предиссоциация имеет место при энергии около 40 250 см над уровнем v=0, /=0
состояния X1 Е+, и это значение может рассматриваться как верхний предел для величины D0(GaCl).
GaCl2 (г). Термодинамические свойства газообразного дихлорида галлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 693.
Молекулярные постоянные, использованные для расчета термодинамических функций GaCl2,
приведены в табл. 22.6. В литературе отсутствуют экспериментальные данные о
геометрических параметрах и частотах колебаний GaCl2. Произведение моментов инерции I/JbIg в основном
электронном состоянии ХгАу вычислено для нелинейной молекулы (группа симметрии С2о) с
параметрами г (Ga—С1)=2,10+0,05А (как в молекуле GaCl3) и ^Cl—Ga—Cl=120+5° (как в ВН2,
BF2, А1Н2). Погрешность IaIbIc составляет 5-Ю14 г3-см6. Основные частоты молекулы GaCla,
принятые в справочнике, вычислены по уравнениям поля валентных сил и силовым постоянным
молекулы GaCl3 (/r=2,63-105, /„.=0,20-105, /а/г2=0,08-105 дин-см), которые соответствуют ее
частотам, приведенным в табл. 22.6. Погрешности принятых частот GaCla оцениваются в 15%.
По аналогии с другими молекулами дигалогенидов элементов III группы можно ожидать, что
молекула GaCl2 имеет стабильное возбужденное состояние Л2ВХ. Приведенное в табл. 22.6
значение То для этого состояния оценено по принятым в справочнике постоянным для других
дигалогенидов, его погрешность составляет 5000 см.
Термодинамические функции GaCl2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом возбужденного электронного состояния AiB1 по уравнениям A. 168)—A. 170).
Погрешности вычисленных таким образом функций определяются в основном неточностью
оцененных молекулярных постоянных, а также приближенным характером расчета и составляют
2, 5 и 8 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции GaCl2 (г) рассчитывались в работе Шаулова и Мосина
[441] до 2000 К, расхождение результатов этого расчета с данными, приведенными в табл. 693,
составляют 1 Дж-К-моль~1 в значении Ф° B000 К) и объясняются некоторым различием в
принятых значениях постоянных.
Константа равновесия реакции GaCl2 (r) = Ga (г)+2С1 (г) вычислена с использованием
принятого в справочнике значения
ДгД ° @) = 750 + 50 кДж • моль.
Это значение было оценено на основании энтальпий атомизации молекул GaCl, GaCl3, A1C1,
А1С12 и А1С13, принятых в справочнике, ему соответствует
,, г, 0) = —239,760+50 кДж • моль.
185
GaCl3 (к, ж)
Гяава 22
GaCl3 (к, ж). Термодинамические свойства кристаллического и жидкого трихлорида галлия
в стандартном состоянии при температурах 298,15—1000 К приведены в табл. Д. 52.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 22.1. В справочнике за стандартное состояние GaCl3 (к) принимается триклинная
модификация [2792а]. Ввиду отсутствия экспериментальных данных по теплоемкости GaCl3 при
Г<298,15 К значения S0 B98,15 К) и Н° B98,15 К)—Н° @), приведенные в табл. 22.1, были
оценены при помощи сравнительных методов, погрешности этих величин составляют
10 Дж-К~1-моль и 1,5 кДж-моль соответственно. При Т ^> 298,15 К измерения теплоемкости
GaCl3 проводились Шемуни и др. [841а] на дифференциальном сканирующем калориметре. Для
GaCl3 (к) в интервале 310—330 К и для GaCl3 (ж) в интервале 370 — 390 К авторы [841а]
определили значения средней теплоемкости 100,8+2,5 и 109+2 Дж-К-моль соответственно.
В справочнике для теплоемкости GaCl3 (к) в интервале 298,15—351 К принято уравнение (см.
табл. 22.1), выведенное с учетом данных [841а 1. Температура плавления GaCl3 C51,0+0,2 К)
принята по хорошо согласующимся между собой данным, полученным в работах Шемуни и др.
[841а] C50,95 + 0,10 К), Фишера и др. [1096] C51,05+0,20 К), Гринвуда и др. [1258а]
C50,9 + 0,5 К) и Палкина и Остриковой [306а] C51,15 К). Энтальпия плавления A1,13 +
+0,13 кДж-моль) и теплоемкость GaCl3 (ж) приняты по калориметрическим измерениям
Шемуни и др. [841а]. Для энтальпии плавления GaCl3 в литературе известно также менее точное
значение A1,5+1,0 кДж-моль) [406], рассчитанное по измерениям давления пара твердого и
жидкого GaCl3 в работах [1096, 1823].
Погрешность вычисленных значений Фс (Г) при 298,15, 500 и 1000 К оценивается в 8, 10 и
18 Дж-К-моль соответственно. Расхождения между термодинамическими функциями
GaCl3 (к, ж), приведенными в табл. Д. 52 и в справочнике Барина и Кнакке [578], составляют
~10 Дж-К-моль при 500 К, они обусловлены тем, что авторы [578] не учли работу Шемуни
и др. [841а] по теплоемкости GaCl3 (к, ж).
Для энтальпии образования GaCl3 (к) в справочнике принимается значение:
kfH°(GaC\3, к, 298,15 К) = —525 + 4 кДж • моль,
основанное на выполненных Клеимом и Якоби [1723] измерениях энтальпии реакций Ga (к),
С12 (г) и GaCl3 (к) с раствором, содержащим КВг3. В работе Тилка [2709], ссылка на которую
приводится авторами [1723], получено значение —523 ккал-моль, совпадающее в пределах
погрешностей с принятым.
Для энтальпии сублимации трихлорида галлия в справочнике принимается значение
к, 0) = 96 + 6 кДж ¦ моль,
Таблица 22.12. Результаты
Авторы
эпределений LSH° (GaCb, к, 0) (в кДж-моль
Метод
\П° (GaCl3, к, 0)
II закон
III закон
Фишер, Юберманн, 1936 [1096] Статический, 341—350 К, 4 точки, GaCl3 (к) 87+27 96,4+0,2
Тоже, 351-472 К, 19 точек, GaCl3 (ж) 85,8+1,0 98,1±0,6
Лаубенгайер, Ширмер, 1940 [1823] Статический, 323—348 К, 6 точек, GaCl3 (к) 85,3+1,8 96,0±0,3
То же, 353—473 К, 13 точек, GaCl3 (ж) 86,2+1,0 98,4±0,8
Комшилова и др., 1970 [173в] Статический, 353—403 К, данные представ- 85,2 97,3+1,1
лены в виде уравнения, GaCl3 (ж)
Думас, Потье, 1970 [991а] Статический, 320—337 К, 21 точка, 88,0+2,0 95,9+0,1
GaCl3 (к)
Агафонов и др., 1974 [2] Маес-спектрометрический, 321—378 К, 125 —
GaCl3 (ж)
Куния, Хосака, 1975 [17856] Статический, 352—420 К, данные пред- 83,2 97,2+1,7
ставлены в виде уравнения, GaCle (ж)
186
Галлий и его соединения GaCl3 (г), Ga2Cle (г)
основанное на приведенных в табл. 22.12 результатах определений этой величины.
Приведенные в таблице величины соответствуют испарению трихлорида галлия в виде мономерных
молекул. В равновесном паре над GaCl3 (к, ж) присутствуют в основном димерные молекулы Ga2Cle (г).
В работах [1096, 1738, 1785в, 1823] было исследовано равновесие реакции Ga2Cl6 (r)=2GaCl3 (г)
(см. текст по Ga2Gl6 (г)). На основании констант равновесия этой реакции, рассчитанных по
принятым в справочнике данным и измеренному в работах [991а, 1096, 1738, 1785в, 1823] общему
давлению насыщенного пара, было вычислено парциальное давление мономерных молекул и
соответствующие энтальпии сублимации.
Как видно из табл. 22.12 (приведенные в таблице погрешности характеризуют только
воспроизводимость измерений), результаты всех исследований (за исключением работы [2])
находятся в хорошем соответствии. Однако для всех работ имеют место систематические расхождения
между величинами, рассчитанными по методам II и III законов, а также полученными из данных
по давлению пара над кристаллическим и жидким GaCl3. По-видимому, это обусловлено
неточностью термодинамических функций GaCl3 в конденсированном, в частности жидком, состоянии.
Принятое значение является средним из вычисленных по методу III закона, причем
предпочтение отдавалось данным, соответствующим измерениям над GaCl3 (к). Погрешность принятого
значения Д4 Н° @) учитывает неточность термодинамических функций трихлорида галлия в
конденсированном и газообразном состояниях E кДж-моль) и неточность оценки концентрации
Ga2Cle в паре C кДж-моль).
GaCl3 (г). Термодинамические свойства газообразного трихлорида галлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 694.
Молекулярные постоянные, использованные для расчета термодинамических функций GaCl3,
приведены в табл. 22.6. Спектроскопические исследования [368, 633, 636, 2246] показали, что
молекула GaCl3 в основном электронном состоянии ХХА имеет плоскую симметричную структуру
и принадлежит к точечной группе симметрии D3k. Произведение моментов инерции IaIbIc
вычислено по значению г (Ga—С1)=2,10 +0,05 А, оцененному по межатомному расстоянию r(Ga—С1ВНОШН)
в молекуле Ga2Cle (см. текст по Ga2Cl6). Погрешность значения IaIbIc не превышает
Г)-10~113 г3-см6. Колебательные спектры молекулы GaCl3 исследовались в парах [368, 636, 983]
и в матрице из Аг [633, 2246]. Приведенные в табл. 22.6 значения основных частот vlt v3, v4,
активных в спектре КР, приняты по работе Дрейка и Розенблатта [983], результаты которой
согласуются с исследованиями в работах [368, 633, 636, 2246]. Для частоты v2 приведено значение,
полученное Селивановым и Мальцевым [368] при исследовании ИК-спектра газообразного GaCl3.
Погрешности принятых частот оцениваются в 10 см для v2 и 3—5 см для остальных частот.
Сведения о возбужденных электронных состояниях GaCl3 в литературе отсутствуют, но так же,
как для других тригалогенидов элементов группы ША, можно предполагать, что их энергии
превышают 15 000—20 000 см.
Термодинамические функции GaCl3 (г) рассчитаны в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A.122)—A. 124), A. 128),
A. 130). Погрешности рассчитанных величин определяются в основном приближенным
характером расчета и составляют 2, 6 и 9 Дж-К -моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции GaCl3 (г) вычислялись в работах Шаулова, Мосина [441 ]
и Дрейка, Розенблатта [983] до 2000 К, а также Юшина и др. [475] до 1500 К. Расхождения
результатов расчета [983] и данных табл. 694 не превышают 0,4 Дж -К -моль в значениях Ф° (Т).
Расчеты [441, 475] были выполнены по оцененным постоянным GaCl3 и расхождения с данными
табл. 694 достигают 4 Дж-К-моль для [475] и 9 Дж-К-моль для [441] в значениях Ф° (Т).
Константа равновесия реакции GaCl3 (r) = Ga (г)+ЗС1 (г) вычислена с использованием
значения ДГЯ° @)=1059,518+8,2 кДж-моль"*1, соответствующего принятым энтальпиям
образованиями сублимации GaCl3 (к). Этим величинам также соответствует
A/F°(GaGl3, г, 0) = —429,658+-7,2 кДж • моль.
Ga2Cl6 (г). Термодинамические свойства газообразного гексахлорида дигаллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 695.
Молекулярные постоянные, использованные для расчета термодинамических'функций Ga2Cl4,-
приведены в табл. 22.6. Электронографические исследования [26, 2506] и исследования спектров
187
Ga2Cl6 (г) Глава 22
[573а, 633, 634а, 636, 2246, 2666а] показали,что молекула Ga2Cle в основном электронном
состоянии Х^А имеет мостиковую структуру и принадлежит к точечной группе симметрии D2h.
Приведенное в табл. 22.6 произведение моментов инерции IjJbIc вычислено по структурным параметрам
г (Ga-GlMemJ = 2,099 + 0,002 А, г (Ga-G]M0CI) = 2,300 + 0, 003 A, ^ClOTemiI-Ga-ClBBeiIIH == 124,6 +
+ 1,8° и -^С1М0СТ-—Ga—С1МООТ = 88,3 + 0,8°, найденным Шеном [2bO6] в результате электронографи-
ческого исследования. Погрешность значения IaIbIo составляет 2-Ю12 г3-см6. Колебательные
спектры молекулы Ga2Gle исследовались в газовой [634а, 636], жидкой [573а, 634а], твердой фазах
[634, 2666а], а также в матрице [633, 2246]. Приведенные в табл. 22.6 значения основных частот
Ga2Cl6, за исключением v5, v7, v10 и v15, получены Битти с соавторами при исследованиях ИК-
спектра [634а] и спектра КР [636] в газовой фазе. Эти значения хорошо согласуются с
результатами измерений авторов [573а, 633, 2246, 2666а]. Исключение составляет частота v4, для которой
расхождение с данными других работ составляет около 10 см. Отнесение к v6 одной из двух
полос C10 см и 344 см), зарегистрированных в работе Битти и ^Сордера [636], было сделано
в настоящем справочнике, на основании близости значений v6 и v13 в молекулах А12С16 D38,
420 см), А12Вг6 C50, 350 см) и In2Ie A50, 115 см). Для полосы 344 см на основании
имеющихся экспериментальных данных нельзя, по-видимому, дать разумное отнесение. Значения не
наблюдавшихся экспериментально основных частот v5, v7, v10, v15 1 были оценены на основании
соответствующих данных для молекул А12С]6 и 1п2С16. Погрешности принятых значений
основных частот составляют до 30 см для v7 и v15, 10 и 5 см для v6 и v10 и 5 см для остальных частот.
Термодинамические функции Ga2Cl6 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A.10), A. 122)—A. 124), A.128),
A. 130), их погрешности обусловлены главным образом приближенным характером расчета,
а также неточностью принятых молекулярных постоянных. Они составляют 12, 25 и
35 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно (в том числе
до 5Дж-К"х -моль из-за неточности молекулярных постоянных).
Ранее таблица термодинамических функций Ga2Cl6 (г) не публиковалась.
Константа равновесия реакции Ga2Cle (r)=2Ga (г)+6С1 (г) вычислена с использованием
значения АГН° @)=2214,72+22 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д//° (Ga2Cl6, г, 0) = —955 + 20 кДж • моль.
Эта величина основана на результатах измерений константы равновесия реакции Ga2Cle (г) =
=2GaCl3 (г), приведенных в табл. 22.13; погрешности соответствующих значений учитывают
Таблица 22.13. Результаты определений Do (GaCl3—GaCb) (в кДж-моль)
Авторы
Фишер, Юберман, 1936 [1096]
Лаубенгайер, Ширмер, 1940 [1823]
Комшипова л др., 1970 [173в]
Куния, Хосака, 1975 [17856]
Метод
Статический, 015—635 К, 3 точки
Статический, 450—771 К, 11 точек
Статический, 458—811 К а
Статический, 423—823 К а
а Экспериментальные данные представлены только в виде уравнения.
Do(GaCl
II закон
94+5
100+7
90
96
в-GaCls)
III закон
96,0+0,1
96,3±0,6
95,6±2,2
95,6±0,3
только воспроизводимость данных. Неточность термодинамических функций приводит к
дополнительной погрешности в 10 кДж-моль в энтальпии диссоциации Ga2Cle.
Как видно из таблицы, результаты всех четырех работ хорошо согласуются, значению
D0(GaCl3—GaCl3)=96 +10 кДж-моль соответствует принятая в справочнике энтальпия
образования Ga2Cl6 (г).
1 Биттц п др. [634а] отнесли к частотам v7, v16 и ve полосы при 128, 215 и 231 см * соответственно. Однако
по данным более поздней работы Бптти и Хордера [636] эти полосы не принадлежат молекуле Ga2Cls.
Глава 23
ИНДИЙ И ЕГО СОЕДИНЕНИЯ
In (к, ж), In, In+, InO, In2O, In2O3 (к, ж), InH, InOH, InF, InF2, InF3 (к, ж), InF3, 1пС1(к, ж),
InCl, InCl2, InCIg (к, ж), InCl3, In2CI6
В настоящей главе рассматриваются 14 веществ — индий и его соединения с кислородом,
водородом, фтором и хлором. Для пяти веществ приведены данные о термодинамических свойствах
в конденсированном состоянии и для 13 веществ — в газообразном состоянии. Термодинамические
функции газообразных индия и его иона рассчитаны для температур до 10 000 К, для остальных
газов — до 6000 К. Для элементарного индия в Приложении 3.9 приведены сведения о критических
постоянных.
Термодинамические свойства всех веществ приведены для природной изотопической смеси
индия (атомный вес 114,82) и других элементов. Однако различия в молекулярных постоянных
индивидуальных изотопических модификаций веществ в большинстве случаев не учитывались
в расчетах из-за отсутствия необходимых данных и их несущественного влияния на
термодинамические функции соединений индия.
In (к, ж). Термодинамические свойства кристаллического и жидкого индия в стандартном
состоянии при температурах 100—3800 К приведены в табл. 696.
Значения постоянных, использованных для расчета термодинамических функций, приведены
б табл. 23.1. В справочнике за стандартное состояние In (к) в интервале 0—429,784 К принята
тетрагональная модификация (структурный тип у-Mn) г.
Таблица 23.1. Принятые значения термодинамических величин для индия и его соединений
в кристаллическом и жидком состояниях
Вещество
In
In2O
InF3
InCl
InCl
Состояние
к, тетр.
ж
5 К, Куб.
ж
к, гекс.
ж
к11, куб.
к1, ромб.
ж
) К, МОНОКЛ.
ж
СО "—'
О5 о
кДжХ
X моль
6,610
—
17,3
—
19,4
—
12,4
—
—
21,0
—
Примечание. Ср=а~\-ЬТ—с
In: a
d. 106=0,502
5,15 К)
^^
О
Дж • К-*
57,65
—
108,0
—
110,0
—
100,0
—.
—
142.0
—
S.15K)
о а,
• лоль
Коэффициенты
нении для С
а
26,90 18,297
— 27,456
100,20 124,539
- 160
92,0 81,081
- 134
51,46 42,936
— 56
— 67
100,0 90,914
— 117
(в Дж-К^-моль)
Ь -103
24,172
—1,092
6,397
—
36,62
—
28,59
—
—
30,474
—
в урав-
р(Т)
с ¦ Ю-5
—1,241
—4,234 а
23,331
—
.—
—
—
—
—
Интервал
температур
К
298,15—429,784
429,784—3800
298,15—2186
2186—3500
298,15—1445
1445—3000
298,15-387
387-484
484—2000
298,15-856
856-1200
Ttr или
Т
1 m
К
429,784
—
2186
—
1445
—
387
484
—
856
—
или ДтЯ
кДжХ
X моль
3,283
—
105
—
64
—
0,52
21,3
—
27
—
Термодинамические функции кристаллического индия при Т<4,2 К вычислены по
результатам измерений теплоемкости индия в сверхпроводящем и нормальном состояниях [776, 858,:
1092, 2163], в интервале 4—21 К — по данным Клемента и Квинелла [858], а в интервале
21—298,15 К — по результатам измерений теплоемкости индия в работах Клузиуса и Шахин-
1 Температура перехода в сверхпроводящее состояние по данным [328а, 776, 858, 930, 1092, 2034, 2070, 2163, 2321,
2504] равна 3,400+0,002 К.
189
In (к, ж) Глава 23
гера [860] A2—273 К) и Крамера и Нолтинга [1770] B00—429 К) с учетом данных по
энтальпии индия, полученных Ротом и др. [2383] B73—428 К). Принятое значение энтропии индия
при 298,15 К на 0,17 Дж ¦ К-моль ниже значения, рекомендованного в справочниках [406]
и [1503] без учета данных [1770] и [2383]. Погрешности принятых значений S° B98,15 К) и
Н° B98,15 К)—Н° @) оцениваются в 0,4 Дж-К-моль и 0,04 кДж-моль соответственно.
В интервале 298,15 К—Тт термодинамические функции In (к) вычислены по уравнению для
теплоемкости, выведенному методом Шомейта при совместной обработке данных пяти работ по
энтальпии и теплоемкости индия [1271а, 1647, 1770, 2383, 2461]. При этом погрешности
результатов весьма точных измерений теплоемкости индия (99,9999% In), выполненных Грёнволлем
[1271а] C02—430 К), оценены в 0,3%, погрешности определений теплоемкости In (к) в работах
Казнова и др. [1647] C54—430 К) и Крамера и др. [1770] B98—350 К) оценены в 1%, а
погрешности измерений энтальпии In (к), проведенных Ротом и др. [2383] B73—428 К) и Шнейдеров
и др. [2461] C73—428 К), которые имеют относительно большой разброс, принимались
равными 2%.
Температура плавления индия D29,784 +0,03 К) принята по рекомендации положения
«Международная практическая температурная шкала 1968 г.» [255а]. Это значение подтверждено
недавно Грёнволлем [1271а], который для чистого образца индия (99,9999% In) нашел Тт=429,77 +
±0,01 К. Результаты других измерений [311, 1928, 2383, 2642, 2751] лежат в пределах 429 —
429,6 К. Энтальпия плавления индия C,283+0,010 кДж-моль) принята по данным Грёнволля
[1271а]. С этим значением согласуются результаты менее точных измерений [498, 739, 1928,
2156, 2335, 2385, 2461, 2851]. Данные работ [256, 345, 1110, 1878, 2154, 2266] неточны и не
принимались во внимание при расчетах.
Экспериментальные данные по теплоемкости жидкого индия [276, 416а, 1271а, 1647]
противоречивы. Анализ этих работ показывает, что наиболее надежными являются результаты измерений
теплоемкости очень чистого образца индия, полученные Грёнволлем [1271а] в интервале 431 —
981 К на калориметрической установке, точность которой ( —0,5%) была проверена по измерениям
теплоемкости корунда. Измерения теплоемкости жидкого индия D30—801 К), выполненные
Казновым и др. [1647], удовлетворительно согласуются с данными Грёнволля вблизи точки
плавления и приводят к значению С°р D30 К)=29,37 Дж-К-моль. Однако при более высоких
температурах данные [1647] лежат значительно выше полученных Грёнволлем [1271а] (при 800 К
на 6%). Очевидно в работе [1647] имеется систематическая погрешность, поскольку
аналогичное расхождение наблюдается также для теплоемкости жидкого олова (при 750 К на 3 %, см.
[1271а]). Поэтому данные [1647] не учитывались при построении уравнения для теплоемкости
жидкого индия. В интервале температур 1100—2100 К измерения теплоемкости жидкого индия
проведены Банчила и др. [276] методом радиальных температурных волн (с погрешностью —7%).
Результаты этих измерений представлены авторами [276] в виде линейного уравнения для
теплоемкости, согласно которому она уменьшается от Ср A100 К)=29,1 Дж-К-моль до С°р B100 К) =
=25,7 Дж-К-моль. Сравнение данных [276] с результатами измерений Грёнволля [1271а],;
а также с данными по теплоемкости жидкого галлия (см. выше),5 олова и свинца (см. т. 2, кн. 1),
приводит к выводу, что характер зависимости С°р (In, ж) от температуры найден в работе [2763
неверно *.
Принятое в справочнике уравнение для теплоемкости жидкого индия (см. табл. 23.1) выведено
методом Щомейта с использованием следующих данных: С°, D29,784 К) =29,37 Дж-К-1-моль-1
[1271а, 1647]; 12 значений энтальпий Н° (Т) - Н° D29, 784 К) D50-1000 К, через 50 К),
вычисленных по данным Грёнволля [1271а], их погрешность оценена в 0,5%; 5 значений энтальпии
A200—2000 К, через 200 К), вычисленных по работе Банчила и др. [276] с использованием
данных [1271а] ниже 1000 К, погрешность этих значений оценена в 5%; значений Н° (Т)—Н° @) при
2500 и 3000 К, вычисленных с учетом данных [1271а, 276] и оценки теплоемкости In (ж) по теп-
лоемкостям Ga, Sn, Pb и щелочных металлов. Согласно принятому уравнению теплоемкость In (ж)
имеет плоский минимум A200—1600 К, С°р=27,1 Дж-К~1-моль~1), а затем возрастает с
ускорением (С; B000 К)=27,4 Дж-К^-моль, С°р D000 К)=31,1 Дж-К^-моль'1).
1 Теплоемкость жидкого индия, так же как и теплоемкость жидких щелочных металлов, галлия и олова, должна
иметь минимум, расположенный (с учетом характера изменения теплоемкости In (ж), найденного в работе [1271а])
вблизи 1200 К.
190
Индий и его соединения
In (г)
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,25, 0,6, 1,5 и 3 Дж-К~1-моль~1 соответственно.
Термодинамические функции In (к), приведенные в работах [1503, 1656], незначительно
отличаются от данных табл. 696 (в пределах 0,5 Дж -К -моль в значениях Ф° (Т)). Для In (ж)
в связи с тем, что в настоящем справочнике учтены данные [276, 1271а], соответствующие
расхождения увеличиваются и достигают 2 Дж-К-моль при 2500 К.
В справочнике за стандартное состояние индия принят In (к, тетр.), в соответствии с'этим
AfHo(In, к) = 0.
Давление пара индия In (ks ж)=1п (г) вычислено с использованием принятой в справочнике
энтальпии сублимации:
ДД°Aп, к, 0) = 240,8 + 2,0 кДж • моль.
Эта величина является средневзвешенной из данных, полученных в работах [97, 339, 490,
1067, 1390, 1903, 27496] и приведенных в табл. 23.2. В качестве погрешностей в таблице указана
Таблица 23.2. Результаты определений ASH° (In, к, 0) (в кДж-моль)
Авторы
Андерсон, 1943 [514]
Кольмейер, Шпандау, 1945 [1742]
Любимов, Любитов, 1957 [227]
Приселков и др., 1960 [339]
Гурвич, 1960 [97]
Хворостухива и др., 1962 [422]
Киршенбаум и др., 1962, 1963,
[1716, 1992]
Норман и др., 1964 [2149]
Херрик, 1964 [1390]
Алкок и др., 1966 [490]
Мекар и др., 1966 [1903]
Фарроу, 1974 [1067]
Валдеррама, Джакоб, 1978 [27496
Метод
Эффузиошшй, 1000—1348 К, 8 точек
Точек гашения, 2273 К, 1 точка
Масс-спектрометрический, испчрение с открытой
поверхности, 640—1065 К, 8 точек
Эффузионный, эффективный размер отверстия —
1,54-10-4 см2, 1061—1318 К, 9 точек
Обработка данных [305а] по аномальной
дисперсии света в парах индия ,
Эффузионный, 1073—1186 К, 13 точек
Точек кипения, 2285 К, 1 точка
Эффузионный, 829—1049 К, 2 точки
То же, 1000—1100 К, 4 точки
Торзионно-эффузионный, 1102—1422 К, 88 точек
Эффузионный, 1180—1301 К, 7 точек
Протока, 1228—1346 К, 3 точки
Эффузионный, 1196—1472 К, 9 точек
Масс-спектрометрический, 806—943 К, 40 точек
Эффузионный, 1095—1308 К, 10 точек
Д.Я° (
II закон
241+9
—
237+5
225+9
239+2,5
232+7
—
234+2
248+3
—
250+10
262
249+15
242+3
236+3
1а, к, 0)
III закон
244,4+0,9
235,8+1
239,5±0,8
—
235,4+0,4
237,0+1,5
—
—
244,0+0,8 а
240,5+0,4
240,0+4,0
237,7+1,0
243,4+0,1
240,9+0,3
а Работа не опубликована, приведенное значение получено пересчетом данных из справочника [1503].
воспроизводимость измерений. При оценке погрешности принятой энтальпии сублимации были
приняты во внимание расхождения результатов измерений разных авторов и неточность
термодинамических функций индия в конденсированном состоянии. Как видно из таблицы, измерения,
на которых основывается принятое значение, находятся в хорошем соответствии.
In (г). Термодинамические свойства газообразного индия в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 697.
Уровни энергии атома In, использованные при расчете термодинамических функций, приведены
в табл. 23.3, в которую включены валентные состояния, относящиеся к электронным
конфигурациям . . .5s25p, . . .5s25d, . . .5s25/, . . .5s5p2 и лежащие ниже ионизационного предела. Энергии
состояний . . .5s25Z приняты по результатам спектрального исследования Йоханссона и Лицена
[1572], а состояний . . .5s5p2 (*Р) — по рекомендации Мур [2053]. Валентные состояния,
принадлежащие другим электронным конфигурациям In, по-видимому, лежат выше ионизационного
предела, и они не представлены в таблице.
191
In+ (г)
Глава 23
Таблица 23.3. Уровни энергии атомов In и 1п+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Состояние
In ...5s25p гР,и
...5s25d %D,k
/2
...5s5p2 *Р1/г
Tt
см
0
2 212,598
32 892,21
32 915,54
34 977,66
36 020,80
37 451,90
Pi
2
4
4
6
2
4
6
Атом
Электронная
конфигурация
In ...5s24/
...5^:5/
In+ ... 5s2
... bsbp
... Ьвэр
...5p*
Состояние
2F
гр
1S
3 p
3 p
1P
Ti
CM
39 707,59
42 220,25
0
42 275
43 349
45 827
63 033,81
97 623,61
Pi
14
14
1
1
3
5
3
5
Термодинамические функции In (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленГ^ЗООО К 002 Д11
ных значений при
р р р
К не превышают 0,02 Дж-К-моль~1 и обусловлены только
неточноП
р ^ р Д
стью принятого значения R. При более высоких температурах погрешности увеличиваются,
в особенности в S° (Т) и С°р (Т), из-за пренебрежения ридберговскими состояниями и могут
достигать 0,8 и 5 Дж-К -моль в значениях S° A0 000 К) и Ср A0 000 К) соответственно.
Ранее таблицы термодинамических функций In (г) вычислялись в ряде работ, в том числе
в работах Гурвича [97] до 3500 К, Кольского и др. [1749] до 8000 К, Хилзенрата и др. [1436]
до 10 000 К, а также в справочнике Сталла и Зинке [2632] до 3000 К. Результаты всех расчетов,
за исключением [1749], совпадают при всех температурах с данными настоящего справочника
в пределах 0,02—0,03 Дж-К~1-моль~1. Расчет [1749] выполнен с неточными значениями
физических постоянных и соответствующие расхождения составляют 0,05 Дж-К~1-моль~1.
Энтальпия образования In (г)
Д//°Aп, г, 0) = 240,8 ± 2,0 кДж • моль
соответствует принятой в справочнике величине А3Н° (In, к, 0) (см. выше).
1п+ (г). Термодинамические свойства газообразного положительного иона индия в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 698.
Уровни энергии иона 1п+, использованные^ при расчете термодинамических функций,
приведены в табл. 23.3, в которую включены валентные состояния, относящиеся к электронным
конфигурациям . . .5s2, . . . 5s5p, . . .5р2 и имеющие энергии не выше 100 000 см. Энергии этих
состояний приняты по рекомендации Мур [2053].
Термодинамические функции 1п+ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A.10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных значений во всем интервале температур не превышают 0,02 Дж-К-моль г и обусловлены
главным образом неточностью принятого значения R.
Ранее таблицы термодинамических функций 1п+ (г) вычислялись в работах Грина и др. [1246]
до 50 000 К и Хилзенрата и др. [1436] до 10 000 К. Вычисленные в этих работах значения
совпадают с данными настоящего справочника в пределах указанных выше погрешностей.
Константа равновесия реакции 1п+ (г)+е (г)=1п (г) вычислена на основании значения
Ь.ГИ° @)= —558,299 + 0,001 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации атома индия
Io (In)=46 670,11 + 0,05 см-х=558,299 + 0,001 кДж-моль.
Эта величина была найдена как предел ридберговской серии . . .5s2nf BF), изученной Йоханс-
соном и Лиценом [1572] в спектре In. Практически совпадающие, но менее точные значения
найдены в работах [316, 1175]. Принятому значению /0 (In) соответствует также
Д^0 Aп+, г, 0) == 799,099 + 2,0 кДж • моль.
192
Индий и его соединения
InO (г)
InO (г). Термодинамические функции газообразного оксида индия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 699.
Молекулярные постоянные InO, принятые для расчета термодинамических функций этого
газа, приведены в табл. 23.4. Экспериментальные данные о молекулярных постоянных InO
отрывочны и ненадежны. В ИК-спектре продуктов реакции In+N2O, изолированных в матрице из
Таблица
Молекула
23.4. Молекулярные постоянные InO, InH, InF и InCl
Состояние
Те
Ве
ах • 103
D. ¦ Ю'
СМ
ге
к
InO Х22+ 0 700 4,7а 0,31 2,9 2,4б 1,97
А2П{ 3 000в — — — — —
В2 2+ 23 500 630 — 0,30 1,8 2,5 2,00
InH X12+ 0 1476,04 25,61 а 4,994 5 142,8б 2 231в 1,838 0
о3П0_ 16 230 1467 82 5,349 326,7 2 810г 1,776
а3П0+ 16 278,15 1458,57 61,03» 5,329 246,8 е 2 770ж 1,779
а3П1 16 941,61 1415,11 43,553 5,399 6 235,6 й 3 210к 1,768
а3П2 17 810 1440 69,5 5,489 332 3 460л 1,753
441 22 654,37 202,65 30,45 5,336 3 318 м 38 900н 1,778
n19F Л?2+ 0 535,3 2,6 0,262 324 1,879 8a 2,5 1,985 396
а3П0+ 30 445,8 575,2 3,68 0,273 4 2,0 2,5б 1,944 8
о3П) 31255,7 572,3 2,63в 0,273 8 2,0 2,5б 1,943 3
441 42 809,2 463,9 7,35 г 0,267 0 5,0 3,65 Д 1,967 2
И51п35С1 X12+ 0 317,0 1,00 0,109 058 0,517 8a 0,514 2,401 169
а3П0+ 27 764,7 340,4 2,02 0,115 5 0,654 0,65 2,333
а3Щ 28 561,95 337,5 1,95 0,115 3 0,6 0,54 б 2,334
Am 37 484,4 179,3 13,9 — — _ _
Примечание. Ниже все постоянные даны в см.
InO: а вычислено по соотношениям A.67) и значению Do=26 000; б вычислено по соотношению A.68); в оценка,
InH: а и>еУе=0,308; б а2=1,23.10-3; в приведено значение Da\ Я0=4,63-10-9; г приведено значение Do,
На=— 2,9-10-8; д шА=-б,83; е «2=—38,95-Ю; ж приведено значение ?>0; Яо= —3,76-Ю-8;
з Шеуе=—13,14; и а2=—56,7-Ю-3; к приведено значение Do, Яо=—3,6-10-8; л приведено значение Do,
Яо=— 4,7-10-8; м а2=691,5-10-3; н приведено значение Do.
InF: а а2=5-10-6; б вычислено по соотношению A.68); в шеуе=—0,095; г и>еуе=—0,05; Д р=0,5.10-'.
InCl: a а2=8,4.10-7; б вычислено по соотношению A.68).
аргона, в работе [304а] наблюдалась полоса в области 707 см, отнесенная к основной частоте
InO. В электронном спектре InO в области 3900—4700 А в работах [304а, 444, 1301, 1826, 2819]
наблюдались перекрывающиеся группы полос, однозначный анализ которых не выполнен до сих
пор. На основании данных, полученных в работах [304а, 1826], можно предполагать, что в этой
области лежат полосы, принадлежащие переходу 52Е +—Х2Е+, а также переходу B2t+—A2U( и,
возможно, С2П—Х22 + и С2П —А2Т1.. В табл. 23.4 включены три электронные состояния этой
молекулы (Х22+, А*П{, 52?+), причем энергия состояния А%П. оценена в предположении, что
в: наблюдавшемся электронном спектре InO наиболее длинноволновая область соответствует
переходу Б22+ -> AZYL. Погрешность принятого значения Те оценивается в 2000 см. Возможно,
что "эта молекула имеет другие связанные валентные состояния, однако их энергии должны
превышать 25 000 см.
Попытки анализа колебательной структуры электронного спектра InO предпринимались
в работах Уотсона и Шамбо [2819], Шевелькова и др. [444] и Осина [304а], однако полученные
в них данные вызывают сомнение, причем Осин отмечает невозможность однозначной
интерпретации собственных экспериментальных данных, а авторы [444] — невозможность описания
полученного ими спектра 1п18О на основании колебательных постоянных, полученных в работе [2819].
Значения постоянной ше молекулы InO в состоянии Х2?+, полученные в работах [304а, 443а,
2819], лежат в интервале 660—703 см. Приведенное в табл. 23.4 значение этой постоянной
принято на основании этих данных и основной частоты, наблюдавшейся Осиным [304а] в ИК-
193
1иО (г)
Глава 23
спектре молекул InO, изолированных в матрице. Ее погрешность оценивается в 20 см. Анализ
вращательной структуры двух полос перехода В2 Е+—X2 ?+ был выполнен Лавенди и Жакино
[1826], однако авторы этой работы не смогли установить колебательную нумерацию
проанализированных полос, что не позволило им определить значения Ве в комбинирующих состояниях.
Приведенные, в табл. 23.4 постоянные Ве и at в состоянии X2 Е+ основаны на данных работы [182611,
авторы которой нашли значения В"„=0,306 55 см и Z?i'+1=0,303 63 см; погрешность принятого
значения Ве оценивается в 0,01 см. Постоянные InO в состоянии i?2E+ приняты по
результатам исследований, выполненных в работах [1826, 2819].
Термодинамические функции InO (г) были вычислены по уравнениям A. 3)—A. 6)г A. 9),
A. 10), A. 93)—A. 95). Значения QBK и ее производных рассчитывались по уравнениям A. 0
A.92) с учетом двух возбужденных состояний в предположении, что @*ол ъ^=(р{/рх)
Колебательно-вращательная статистическая сумма для состояния Х2Е+ и ее производные
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням энергии с помощью соотношений типа A. 82).
В расчетах учитывались все уровни энергии с значениями /^/шах „ последние определялись из
условий A. 81). Колебательно-вращательные уровни состояния Х2?+ находились по уравнениям
Таблица 23.5. Коэффициенты в уравнениях, описывающих уровни энергии (в см), а также значения
*Wx и t/iim, принятые для расчета термодинамических функций InO, InH, InF и InCl
(
90)
i
Молекула
Электронное
состояние
2>10-4
. Ую-Ю-3
Уго-Ю-1
Узв-Ю1
У40-Ю4
Ум
Уи-102
У21-104
Узх-104
Уо2-Ю6
Уоз-Ю11
Ув4-Ю12
(a0=De)-10
ai
а2-10*
аз-Ю8
ymax
Aim
Примечание.
InO: a Т„{АЧ\
InO
0
0,700
—0,47
—
—
0,31
—0,29
—
—
—0,24
—0,064 86
—
-4 _
—
—
—
73
401
Ниже все величины даны
;)=3000, 2\, (?2 2+) = 23
InH
Х1 +
0
1,477 398
—2,659 500
5,629 020
—214,879
4,995 61
—14,596 6
26,221 6
—1,695 50
-223,1
463,0
—0,424 8
2,079 212
0,280 817
0,372 244
0,303 918
23
88
В СМ.
500. InF: a
InCl: a
а3П
1,694 161
1,415 11
—4,355
—131,4
—
5,399 6
—23,56
-567,0
—
—320,0
—3600,0
—
0,385 025
2,307 426
1,259 964
5,508 743
3
50
Те (а3Щ = 31 255
Те (аЧ1) = 28 561
InF
0
0,535 826
-0,273 046
0,079 719
—0,120 068
0,262 324
—0,187 980
0,05
—
-0,250
0,013243
—
—
—
—
—
209
497
,6.
,95.
InCl
Yly+a
0
0,315 481
—0,101 658
0,014 952
—0,009 435
0,107 937
—0,051 002
0,008 228
—
—0,050 347
—0,000 717
. .
—
—
—
—
321
788
A. 65), A. 62), мультиплетность уровней учитывалась статистическим весом. В табл. 23.5
приведены значения коэффициентов в этих уравнениях, тождественные молекулярным постоянным,
принятым в табл. 23.4, в той же таблице приведены значения ушах и /Iim.
Основные погрешности рассчитанных термодинамических функций InO (г) при 298,15 К
обусловлены неточностью принятых постоянных этой молекулы (главным образом величины Ве)
1 Найденные в работе [1826] значения постоянных Dv превышают в два раза рассчитанные по уравнению A. 68),
что вызывает некоторые сомнения в надежности проведенного анализа.
194
Индий и его соединения
1п2О
в состоянии Xs ?+. Выше 500 К становятся существенными погрешности из-за отсутствия
экспериментальных данных о состоянии А2И, а выше 5000 К — о других электронных состояниях.
Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,5, 4 и 6 Дж-К-моль.
Термодинамические функции InO (г) вычислялись ранее Бруэром и Розенблаттом [745]
(Ф° (Т) при Г<;3000 К). Расхождения данных [745] и табл. 699 составляют 1—6 Дж-К^-моль
и обусловлены разницей в значениях Ве, принятых в расчетах, а также пренебрежением
состоянием А2П( в работе [745].
Константа равновесия реакции InO (r) = In (г)-|-0 (г) рассчитана с использованием принятой
в справочнике энергии диссоциации
\Н°Щ = D0-(InO) = 315 + 25 кДж • моль« 26 000 ± 2000 см.
Принятая величина основана на результатах масс-спектрометрического исследования
испарения 1п2О8 (к) в работе Ббрнса и др. [794]. Измерения проводились в интервале 1280—1540 К
(около 30 точек). Авторы вычислили парциальные давления In, In2O и О2 (эти величины
представлены только в виде графика), однако для InO было получено только приближенное условие
р AпОХ1,2-10~9 атм при 1482 К, так как ион 1пО+ мог частично образовываться за счет
диссоциативной ионизации. Из данных работы [794] при подготовке настоящего справочника были
найдены предельные значения констант равновесия двух реакций
1п2О (г) = 1пО (г)+1п (г), К° A482 К) < 6,7-Ю-10,
InO (г) =1п (г) +V2Oa (г), К° A482 К) > 1.
Принятое значение Do (InO) основано на этих величинах. Ему соответствует
A//°(InO, г, 0) = 172,583 + 25 кДж • моль.
1п2О (г). Термодинамические свойства газообразного оксида дииндия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 700.
Молекулярные постоянные, использованные для расчета термодинамических функций 1п2От
приведены в табл. 23.6. В настоящее время не существует однозначной интерпретации
экспериментальных данных по строению и частотам колебаний 1п2О. Рамбиди и Толмачев [341] из элек-
тронографического исследования паров 1п2О установили, что средней в условиях эксперимента
(Г=1300 К) является нелинейная структура с Д1п—О—1п=150 + 10°. Однако позднее
теми же авторами [340, 2708] было показано, что электронографические данные не
противоречат линейной или близкой к линейной равновесной структуре 1п2О, если эта молекула
имеет низкую частоту деформационного колебания. Мальцев и Шевельков [240] в ИК-спектре
газообразного 1п2О наблюдали только частоту va, однако авторы не делают вывода о
Таблица 23.6. Значения молекулярных постоянных, а также о и р^, принятые для расчета
термодинамических функций 1н2О, InOH, InF2, I11F3, InCI2, I11CI3 и In2CI6
Молекула
Электронное
состояние
In2O %*A
InOH lM
InF2 l^A^
InF3 %ХА
InCl2 ХМ/
InCl3 %?A
In2Cle X1 A
InOH: a Приведено значение /•
InF2: a A*Bi, Го=15 000 см,
InCl2: a A2B\, Г0=10 000 см,
442
520
630
600
370
350
368
1039 г-
о=2,
о=2,
v2
160
300 B)
100
150
80
НО
298
см2.
Р1=2.
V
CM
722
3600
640
640
390
394
131
In2Cl6: a
I
B)
B)
v5=25,
^10=16,
v15=50,
—
130 B)
—
95B)
70 a
ve=267,
vu=400,
vw=350,
Г3 • CMS
1,8-10*
H a
2,0-10s
1,4-10*
2,5-10*
2,0-106
2,0-10'
v7=118,
vi2=110,
vi,=250
Q117
a
2
1
2
6
2
6
4
v8=400,
vls=270,
v18=100
Pi
1
1
2
1
2
1
1
v9=180,
v1,=100,
(в емг1).
195
In2O3 ( г) Глава 23
линейной структуре этой молекулы, так как полоса vx может не наблюдаться из-за ее слабой
интенсивности. Хинчклифф и Огден [1439] в ИК-спектре молекул 1п2О, изолированных в матрице
из N2, наряду с интенсивной полосой v3 наблюдали слабую полосу 442 см, отнесенную ими
к частоте vx (в аргоновой матрице наблюдалась только полоса v3). Поводом для такого отнесения
послужило изучение зависимости интенсивности полос от температуры, хотя подтвердить это
количественно из-за слабой интенсивности полосы 442 см не удалось По той же причине оказалось
невозможным изучить изотопные сдвиги полос. Поэтому отнесение vx Хинчклифф и Огден
рассматривают как предварительное. Из отношения значений частоты v3 разных изотопических
модификаций молекулы 1п2О авторы [1439] оценили Д1п—О—1п=135°, но указали, что эта
величина является нижней границей, а учет ангармоничности колебаний может привести к углу,
значительно более близкому к 180°.
На основании результатов электронографического исследования [341, 2708] и интерпретации
ИК-спектров молекул 1п2О, изолированных в матрицах [1439], в справочнике принято, что
в основном электронном состоянии Х1А молекула 1п2О имеет нелинейную структуру
симметрии С2„. Приведенное в табл. 23.6 произведение моментов инерции вычислено для значений
г (In—О)=2,00+0,03 А и -3ln—О—1п=144 + 10°, найденных Толмачевым и Рамбиди [2708],
его погрешность составляет 1-Ю13 г3-смв.
ИК-спе.ктр 1п2О исследовался как в матрицах инертных газов и азота [304а, 321, 1437, 1439,
1927], так и в паре [239, 240], однако надежно определена только частота антисимметричного
колебания v3. Поскольку в ИК-спектре паров 1п2О наблюдаются широкие полосы, приведенное
в табл. 23.6 значение v3 принято по результатам исследования Хинчклиффом и Огденом [1439]
ИК-спектра молекул 1п2О, изолированных в матрице из N2. По результатам той же работы
принято значение vx. Частота v2 вычислена по уравнениям поля валентных сил в предположении,
что /а/г2=0,05/г, как у молекулы А12О *. Погрешности в значениях v2, v2 и v3 оцениваются в 30—
40 см. Возбужденные электронные состояния 1п2О в справочнике не рассматриваются, так
как согласно предварительным исследованиям электронного спектра они лежат выше 30 000 см
[446].
Термодинамические функции 1п2О (г) были вычислены по уравнениям A.3)—A.6), A.9),
A. 10), A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета; они могут достигать
3—8 Дж-К^-моль-1 в значениях Ф° (Т) в интервале 100—6000 К.
Термодинамические функции 1п2О (г) вычислялись ранее в работах Шевелькова и Мальцева
[447] (Г<2000 К) и Бернса и др. [794] (ф° (Г) для 7=1000-М600 К). Расхождения
результатов этих расчетов и данных табл. 700 не превышают 0,5 и 2 Дж-К~1-моль~1 в значениях Ф° (Т)
и обусловлены некоторым отличием принятых молекулярных постоянных.
Константа равновесия реакции 1п2О (г)=2 In (г)+О (г) вычислена с использованием
значения &ГН° @) =766,383+20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д7Я° Aп2О, г, 0) = —38 + 20 кДж • моль.
Эта величина основывается на приведенных в табл. 23.7 данных. Приведенные в таблице
погрешности учитывают воспроизводимость измерений, неточность использованных в расчетах
термодинамических функций и других термохимических величин. Из приведенных данных видно,
что расхождения между результатами измерений разных авторов гораздо больше, чем можно
объяснить их погрешностями. В справочнике принимается среднее из значений,
рассчитанных по III закону. Его погрешность оценена с учетом расхождений приведенных величин.
1 Маковецкий и др. [1927] отнесли к частотам ч1 и v2 полосы 555 и 404 см г, наблюдаемые в ИК-спектре молекул
1п2О, изолированных в матрице из Аг. Однако в работе [1438] было показано, что эти полосы не принадлежат
1п2О. Мальцев и Шевельков [239] отнесли полосу 360 см в ИК-спектре газообразного 1п2О к частоте vx, но
позднее в работе [240] они пришли к выводу, что эта полоса, по-видимому, является обертоном 2v2.
196
Индий и его соединения
1п2О3 (к, ж)
Таблица 23.7. Результаты определений Ду>Н° Aп2О, г, 0) (в кДж-моль *)
Авторы
Метод
AfH° (In2O, г, 0)
II закон
III закон
Берне и др., 1963 [794] Масс-спектрометрический, 1п2О (г)=21п (г)+О (г), —5 —10+8
1300—1538 К, данные представлены в виде
графика
Берне, 1966 [793] То же, 1300—1500 К, 10 точек —84+90 —14+10
Щукарев и др., 1969 [470] Масс-спектрометрический, 1п2Оз (к)=1п2О (г)+ —5+24 —60,6+6,0
+О2 (г), 1539-1631 К, 8 точек
Масс-спектрометрический, 4/з1п (ж)+1/з1п2Оз(к)= —65+.40 —56,4+5,0
=1п2О (г), 1025—1101 К, 6 точек
Чаплыгин, 1975 [432] Протока, 21п (ж)+Н20 (г)=1п2О (г)+Н2 (г), 1068- —73+32 -59,4+6,0
1285 К, 21 точка
Валдеррама, Джакоб, 1977 [2749а] Эффузионный, */з1п(ж)+1/з1п203 (к)=-1п2О (г), —30+6 —28,2+5,0
946—1215 К, 9 точек
1п2О3 (к, ж). Термодинамические свойства кристаллического и жидкого триоксида дииндия
в стандартном состоянии при температурах 298,15—3500 К приведены в табл. 701.
Значения постоянных г принятые для расчета термодинамических функций, приведены
в табл. 23.1. В справочнике за стандартное состояние 1п2О3 принята кубическая модификация
(структурный тип С [2464]).
В литературе отсутствуют экспериментальные данные по теплоемкости 1п2О3 (к) при Т<С
<298,15 К. Термодинамические функции кристаллического 1п2О3 при 298,15 К рассчитаны на
основании значений теплоемкости, оцененных с использованием низкотемпературных данных
для |3-Ga2O3 (см. выше). Значение S° B98,15 К), приведенное в табл. 23.1, рассчитано Ньюн-
сом и др. [2138] с использованием значения A^G° B98,15 К), найденного в результате измерения
ЭДС по реакции 21п (ж)+ЗШ0 (к)=1п2О3 (к)+3№ (к) в гальванической ячейке. Значение
Су B98,15 К) получено из измерений энтальпии 1п2О3 (к) при Т ^ 298,15 К, выполненных Ца-
гарейшвили и Гвелесиани [430]. Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—
—Н°@) оцениваются в 3 Дне-К-моль и 0,4 кДж-моль соответственно.
В интервале 298,15—2186 К для С°р Aп2О3, к) принято уравнение (см. табл. 23.1), выведенное
методом Шомейта по результатам измерений энтальпии образца чистотой 99,99% в работе [430]
B98—1605 К); погрешность этих данных оценивается в 0,5%. Температура плавления 2186 +
+ 10 К (с учетом поправки на 3 К по шкале МПТШ-68) принята по данным Шнейдера [2464],
полученным для образца с суммарным содержанием примесей ~0,08%. Энтальпия плавления
A05 + 10 кДж-моль) и теплоемкость 1п2О3 (ж) A60 + 30 Дж-К-моль~1) были приняты равными
соответствующим величинам для А12О3.
Погрешности вычисленных значений Ф° (Т) при 298,15,1000, 2000 и 3000 К оцениваются в 3,
4, 6 и 12 Дж-К~1-моль~1. Термодинамические функции, вычисленные в справочнике Барина
и Кнакке [578] при Г^2000 К, близки к приведенным в табл. 701 (расхождения не превышают
1,5 Дж-К'^моль).
Константа равновесия реакции 1п2О3 (к, ж)=21п (г)+30 (г) вычислена с
значения ДГЯ° @)=2139,306+4,3 кДж-моль, соответствующего принятой в
тальпии образования
Д/Я"вAпяО8, к, 298,15 К) = —926,3 + 1,7 кДж . моль.
Результаты определений энтальпии образования 1п2О3 (к) приведены в табл. 23.8. Принятая
величина основана на наиболее точных калориметрических измерениях, выполненных Холли
и др. [1462]. Погрешность приведенных в таблице величин, вычисленных из данных.по
равновесиям, помимо воспроизводимости измерений учитывают также неточность термодинамических
функций. Как видно из табл. 23.8, многочисленные исследования равновесия реакций с участием
1п2О3 (к) приводят к несколько меньшим значениям, что, вероятно, свидетельствует о неточности
термодинамических функций 1п2О3 (к).
использованием
справочнике эн-
197
InH (г)
Глава 23
Таблица 23.8. Результаты определений
Aп2Оз, к, 298,15 К) (в кДж-моль х)
Авторы
Метод
Д/Я°Aп203, к, 298,15 К)
II закон
III закон
Дитте, 1871 [957] (см. [638]) Калориметрическое измерение энтальпии раство- —920,5
рения In и 1п2Оз в серной кислоте
Беккер, Рот, 1932 [638] Калориметрическое измерение энтальпии сгора- —930,9+_3,0
ния индия, 293 К, 3 опыта
Холли и др., 1952 [1462] То же, 298,15 К, 14 опытов —926,3±1,7
Стабс и др., 1952 [2629] Исследование равновесия реакции 1/з1п203 (к)+ —926±10 —917,4±5
+Н2 (г)=2/з1п (ж)+Н20 (г) динамическим
методом, 823—1120 К, 100 точек
Торопова, Погорелый, 1961 [407] Исследование равновесия реакции 1пгОз (к)+ —873+60 —922+.12
+ЗСО (г)=21п (ж)+ЗСО2 (г) динамическим
методом, 873—1173 К, 4 точки
Хочгешвендер, Инграм, 1967 [1450] Исследование равновесия реакции 1п3Оз (к)= —922 —918±5
=21п (ж)+3/2О2 (г) динамическим методом,
430—850 К а
Бёрд и др., 1975 [6936] То же, 633—1085 Ка —919 —917±8
Шефер, 1971 [2443] ЭДС ячейки с твердым электролитом, 21п (жL- — —927
+3Cua0 (к)=1п2О3 (к)+6Си (к), 790—1230 К 6
Клайндинст, Стивенсон, 1973 [1729] ЭДС ячейки с твердым электролитом, 21п (ж)+ —921 ±6 —915,8+5,5
+3/аО2 (г) = 1п2О3 (к), 873—1273 К, 9 точек
Чаттерджи, Вест, 1972 [840а] То же, 823-1073 К а- б —904 —915+8
Чаттерджи, Смит, 1973 [840] То же, 823—1073 Ка — 920±6 —917,4+5,5
Ныонс, Пелмор, 1968 [2138] ЭДС ячейки с твердым электролитом 21п (ж)+ —927 —918+8
+ЗШО (к)=1п2О3 (K)+3Ni (к), 723—973 К а
Катояма, Козука, 1973 [1635] То же, 1013—1233 К а —928 —917 + 8
Катояма и др., 1975 [1636] То же, 959—1284 К, 40 точека —922 —914,6±5,5
Андерсон, Донахью, 1977 [517, 519] То же, 951—1128 К, 11 точек —940±6 —916,2+5,5
а Результаты измерений представлены только в виде уравнения.
6 Приведено по реферату.
InH (г). Термодинамические свойства газообразного гидрида индия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 702.
Молекулярные постоянные InH, принятые в справочнике на основании результатов
исследований электронного спектра InH, представлены в табл. 23.4. В спектре InH наблюдались 5 систем
3 1 [8] 3 1 3^ X1?* [1217 1297
р р рд
полос, связанных с переходами: а3П0- -> Х1!,* [1218], а3П0+ «* X1!.
1] 3 1 [] Ш Х [ 11]
X1?,* [1217, 1297,
р 0
2120, 2121], а3П2 -» X1!:* [1217] и АШ
, 0 ^
Е+ [1217, 2120, 2121]. Все наблюдаемые в спектре
BР) B5)X1
, , 2 , ,
состояния должны коррелировать с двумя диссоциационными пределами: In BРу2)+Н B5)—X1
а3П0-', а3Пх и In BP3/2)+H BS)~а3П0+, а3П2 и АгИ. Со вторым пределом должно также
коррелировать состояние 3S+, которое не наблюдалось в спектрах InH и как у других гидридов элементов
группы ША, по-видимому, является отталкивательным состоянием х. По аналогии с молекулами
ВН и А1Н можно предполагать, что InH имеет также связанные валентные и ридберговские
состояния с энергиями свыше 30 000 см; полосы, наблюдавшиеся Гартманом [1173] в спектре
поглощения InH в области 2330—2500 А, по-видимому, связаны с этими состояниями, которые не
включены в табл. 23.4.
Приведенные в табл. 23.4 постоянные InH в состояниях Х1!", а3П0+ и а3Пх найдены Гинте-
ром [1217] на основании анализа структуры 12 полос перехода а3П0+—ХХЕ+ (г/ <J! 3, v" ^ 5) и
8 полос перехода а3Пх—Xх И* (v', v" <J 3); вращательные постоянные в состоянии а3Пх являются
средними из соответствующих значений, найденных для компонент 3ПХ+ и 3П!~. Принятые постоян-
1 Следует отметить, что экспериментальные данные Гинтера [1217] не подтверждают такую схему корреляции.
Попытка Везета и Лофтуса [2771, 2772] объяснить аномальную величину Л-удвоения в состоянии а3!^ молекулы
InH взаимодействием со связанным состоянием 32+ представляется сомнительной.
198
Индий и его соединения InH (г)
ные в состоянии а3П0- основаны на результатах анализа структуры полос 0—0, 1—0, 0—1 и 1—1
перехода а3П0—X1 Е+ в работе Гинтера [1218], а постоянные в состоянии а3П2 — на результатах
анализа полос 0—0и1—1 перехода а3П2—XxL + b работе [1217]. При обработке данных [1217,1218]
принималось, что величины G @) в состояниях 3П0+ и 3П0- одинаковы, а разность энергии
состояний SII1 и 3П2 в минимуме потенциальных кривых равна 870 см (при v=Q и 1 эта разность равна
875,91 и 891,48 см соответственно).
Приведенные в табл. 23.4 значения постоянных InH в состоянии ЛХП найдены Нейхаузом [2120]
при анализе 6 полос перехода АгИ—X1 ?+ (z/, v" ^ 2); во всех полосах наблюдалась предиссоциа-
ция при малых значениях / (см. ниже).
Термодинамические функции InH (г), приведенные в табл. 702, были вычислены по уравнениям
A. 3)—A. 10), A. 93)—A. 95). Значения QBn и ее производных рассчитывались по уравнениям
A. 76)—A. 78) непосредственным суммированием по уровням энергии состояний X1 ?+ и а3П, при
этом состояния а3П0-, а3П0+, а3Пх и а3П2 рассматривались как компоненты одного состояния со
средней энергией возбуждения, а мультиплетности и вырождение колебательно-вращательных
уровней учитывались статистическим весом 6. В расчете учитывались все
колебательно-вращательные уровни, ограниченные предельными кривыми диссоциации состояний X1 ?+ и а3П.
Колебательно-вращательные уровни состояния Х1Е+ вычислялись по уравнениям A. 65), A. 60), а
состояния а3П — по уравнениям A. 65), A. 61), значения коэффициентов Ykl в этих уравнениях
приведены в табл. 23.5; для состояния Х1^ они получены с учетом условий A. 50), A. 57) и
A. 59), для состояния а3П —• по постоянным InH в подсостоянии а.3Пх. В той же таблице приведены
значения величин ушах и /11Ш, а также коэффициентов ai в уравнениях, аппроксимирующих
предельные кривые диссоциации.
Принятые постоянные InH хорошо описывают энергию колебательно-вращательных уровней
состояния X1 Е+ вплоть до 7000 см, поэтому для Т ^ 1500-^-2000 К основные погрешности
вычисленных термодинамических функций обусловлены неточностью фундаментальных постоянных.
При более высоких температурах становятся существенными погрешности из-за отсутствия
экспериментальных данных о высоких колебательно-вращательных уровнях состояния Xх 2+,
пренебрежения состояниями с энергией выше 30 000 см, а также обусловленные приближенным учетом
составляющих компонент состояния а3П и пренебрежением состоянием А1П, имеющим менее 50
связанных колебательно-вращательных уровней. Погрешности в значениях Ф° (Т) при 298,15,
3000 и 6000 К составляют 0,02, 0,03 и 0,05 Дж-К-моль. Погрешности из-за приближенного
учета состояния а3П и пренебрежения вкладом состояния АгЛ достигают при 6000 К 0,02, 0,05 и
0,08 Дж-К^-моль в значениях Ф° (Г), S0 (Т) и С% (Т).
Ранее термодинамические функции InH (г) вычислялись в справочнике [1336] (Т ^ 5000 К).
В работе [2463] рассчитаны значения (?„„ и In QEm этого газа и приведены полиномы,
аппроксимирующие эти величины для 2'=1000—6000 К. Расхождения данных [1336] и табл. 702 достигают
0,5, 1,4 и 2 Дж-К^-моль в значениях Ф° E000 К), S° E000 К) и С°р E000 К) и обусловлены
пренебрежением в расчете [1336] вкладом состояния а3П и использованием приближенной методики;
расхождения с данными [2463] имеют такие же величины.
Константа равновесия реакции InH (r)=In (г)+Н (г) вычислена по энергии диссоциации
-1
ДД° @) = Do (InH) == 20 060 + 35 см = 239,97 + 0,40 кДж • моль
Это значение принято в справочнике на основании результатов исследований предиссоциации
InH в состояниях А1!! и а3П. Предиссоциация в состоянии А1!! была изучена Нейхаузом [2120,
2121], который показал, что обрыв вращательной структуры имеет место при у=0» /=9, v=ls
/=8 и v=2, /=5. Форма предельной кривой диссоциации, построенной по этим данным,
свидетельствует об отсутствии максимума на потенциальной кривой состояния А1]! для невращающейся
молекулы, короткая экстраполяция этой кривой дает для энергии диссоциационного предела
состояния А1]! значение 22 270 см. Принятая энергия диссоциации основана на этом значении.
Следует отметить возможную неточность определения последних наблюдавшихся уровней
состояния ^11П и предела этого состояния, которая учтена погрешностью, приписанной принятому
значению. Нейхауз [2121 ] допустил ошибку при расчете энергии предела над уровнем v=0, /=0
состояния Х1!!"; полученное им значение 21 250 см воспроизведено в справочнике [1491а].
Энергия последнего наблюдавшегося колебательного уровня состояния ^П (Too-\-Go B)=
199
InOH (г)
Глава 23
=22 239 см х) позволяет оценить нижний предел для энергии диссоциации молекулы InH
B0 026 см).
Несмотря на то что подсостояния а3П0+, а3П2, с одной стороны, и подсостояние а3Пх, с другой,
коррелируют с разными диссоциационными пределами, их предельные кривые диссоциации,
построенные Гинтером [1217] из данных по предиссоциации InH, практически совпадают и имеют форму,
характерную для состояний, имеющих максимум на потенциальной кривой при отсутствии
вращения. Энергия максимума на потенциальной кривой состояния, коррелирующего с пределом
In BРчг)-{-Н BS), полученная Гинтером экстраполяцией предельных кривых, равна 20 НО см
и несколько превышает величину Do (InH).
Принятому значёййю энергии диссоциации соответствует
AfH°(lnE, г, 0) = 216,864 + 2,0 кДж-моль.
InOH (г). Термодинамические свойства газообразного гидроксида индия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 703.
Молекулярные постоянные, использованные при расчете термодинамических функций InOH (г),
приведены в табл. 23.6. Спектр и структура InOH экспериментально не исследовались. По
аналогии с АЮН в справочнике принимается, что в основном электронном состоянии X1 ? эта
молекула имеет линейную структуру. Момент инерции, приведенный в табл. 23.6, рассчитан по
оцененным значениям г (In—О)=2,00 +0,05 А (как в 1п2О) и г (О—Н)=0,96 +0,02 А, его погрешность
составляет 1-Ю9 г-см2. Частота v1=vIn_0 оценена сопоставлением значения шв (InF)=535,4 см
со значением vx=483 см, вычисленным для InOH по силовой постоянной связи молекулы 1п2О
(/in-o=2,04-105 дин-см). Частота v2 = vIn_0_H оценена по частотам деформационных колебаний
молекул гидрооксидов щелочных металлов. Для v3=v0_h принято характеристичное значение.
Погрешности принятых значений частот vx, v2 и v3 составляют 50, 60 и 100 см. На основании
известных данных для двухатомных галогенидов индия можно предполагать, что возбужденные-
электронные состояния молекулы InOH имеют энергии свыше 25 000 см.
Термодинамические функции InOH (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)т
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
значений молекулярных постоянных и приближенным характером расчета, они составляют 2, 4 и
6 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций InOH (г) публикуется впервые.
Константа равновесия реакции InOH (г) =1п (г)+О (г)+Н (г) вычислена с использованием
значения АГН° @)=803,617+10 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
\fH°(InOH, г, 0) = —100 + 10 кДж • моль.
Результаты определения Д^0 (InOH) методом спектрофотометрии пламен приведены
в табл. 23.9, где в качестве погрешностей дана воспроизводимость измерений. Наиболее тщатель-
Таблица 23.9. Результаты определений &/Н° (InOH, г, 0) (в кДж-моль)
Авторы
Метод
AfH° (InOH, г, 0)
II закон
III закон
Булевич, Сагден, 1958 [790] Спектрофотометрияпламен, H2+O2+N2,In(r)+ — 79±30 —99+40
+Н2О(г)=1пОН(г)+Н (г), 1770-2450 К, 16 точек
Гурвичидр., 1969 [100,101, 1334] Спектрофотометрия пламен, H,+O2+N2, In (г)+ —100±40 —119,8+4,0
+Н2О (г)=1пОН (г)+Н (г), 1670—2200 К, 8 точек
Масс-споктрометрический, 1п2О (г)+Н2О (г)= —115+18 —123±1,0
=21пОН (г), 817—951 К, 15 точек
Келли, Педли, 1971 [1665] Спектрофотометрия пламен, Н2+О2+СО2 и Н2+ —97,0+5,8 —97,8+3,0
+O2+N2, In (r)+H2O (r)=JnOH (г)+Щг),
1950—2750 К, 32 точки
200
Индий и его соединения InF (г)
ные измерения были выполнены Келли и Педли [1665] и принятая величина основывается на них.
В связи с дополнительной неопределенностью, вносимой неточностью термодинамических
функций InOH (г) (см. выше), погрешность в принятом значении энтальпии образования увеличена
до 10 кДж-моль.
InF (г). Термодинамические свойства газообразного фторида индия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 704.
Молекулярные постоянные InF, принятые в справочнике на основании результатов изучения
электронного и вращательного спектров этой молекулы, приведены в табл. 23.4. В электронном
спектре InF наблюдались переходы а3П0+ ^ X1 Е+, а3Пг ^ X1 ?+ [597, 598, 1351, 1357, 2829, 2830]
и АгП^ Хг?+ [597, 598, 1351, 1357, 2101, 2829, 2830]; кроме того, в спектре поглощения InF
были обнаружены неидентифицированные диффузные полосы в области 2103—2187 А [598]. Так же
как и у других галогенидов элементов группы IIIA, состояния X1 2+, а3П и ЛХП молекулы InF
коррелируют с атомами в основных состояниях, причем подсостояния а3П0- и а3П2, не
наблюдавшиеся в спектрах, должны иметь общий предел с Xх ?+ состоянием — In BPi/2)+F Bi\) и быть
связанными состояниями. Состояние А1!!, по-видимому, коррелирует с пределом In BРг^)-\-? B/\).
В табл. 23.4 представлены данные только для трех электронных состояний.
Принятые в справочнике колебательные постоянные в состоянии X1 2+ получены Барроу и
др. [597] по началам полос 0—0, 0—1, 1—0 и 1—2 системы а3П0+ =?s X1 ?+ с учетом данных Барроу
и др. [598] по (^-кантам большого числа полос систем а3Пх—X1!,* и у1хП—X1!,* (для v" ^ 11)
и Р-кантам полос перехода а3П0+—X1 ?+. Эти постоянные близки к найденным ранее по кантам
полос в работах [2829, 2830]. Приведенные в табл. 23.4 вращательные постоянные 115In19F найдены
в работе Хоэфта и др. [1452] при исследовании микроволнового спектра, соответствующего
переходам с v ^ 3, / ^ 1. Эти постоянные определялись также в работах [597, 2101] на основании
изучения вращательной структуры полос 0—0, 0—1, 1—0 и 1—2 системы а3П0+—ХХЕ+, полос
0—0, 1—0 и 0—1 системы а3Лг—ХХЕ+ и полос 0—0, 1—0, 2—2, 3—3, 4—4, 2—4, 3—5, 4—6 и
5—7 системы АгП—Х1!", а также в работе [1890] при исследовании микроволнового спектра;
полученные в этих работах постоянные хорошо согласуются с принятыми в
справочнике.
Колебательные и вращательные постоянные молекулы InF в состояниях а3П0+, а3^ и
колебательные постоянные в состоянии А1!! приняты по работе [597], вращательные постоянные в
состоянии А1!! — по данным Нампури и др. [2101]. Согласно [597] состояние А1!! имеет
неглубокий минимум (~3700 см) и систему быстро сходящихся колебательных уровней.
Термодинамические функции InF (г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9)t
A.10), A.93)—A.95). Значения QBB и ее производных вычислялись по уравнениям A.90)—
A.92) с учетом одного возбужденного состояния а3П в предположении, что Q$z вр = 6 Q^J вР-
Статистическая сумма по колебательно-вращательным уровням энергии состояния X1 ?+и ее
производные находились по соотношениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням энергии и интегрированием по значениям / с помощью уравнений типа A. 82). В
расчете учитывались все уровни энергии с значениями / ^ /шах, „> последние определялись из
условий A. 81). Уровни энергии состояния X1 ?+ вычислялись по уравнениям A.65) и A. 61), значения
коэффициентов в этих уравнениях, а также величин ^шахи/Иш приведены в табл. 23.5,
коэффициенты Yk0, включая Yso. и Yi0, были получены с учетом условий A. 50), а коэффициенты У03 —
из условий A. 59).
Погрешности рассчитанных термодинамических функций InF (г)принизких температурах
практически обусловлены только неточностью фундаментальных постоянных, выше 2000 К становятся
существенными отсутствие экспериментальных данных о колебательно-вращательных уровнях
состояния Хх?+ с энергией выше 5600 см, а также приближенный учет вклада состояния а3П.
Пренебрежение вкладом состояния А1!!, которое имеет примерно на порядок меньше связанных
колебательно-вращательных уровней энергии, чем состояние X1 Е+, практически не влияет на
точность расчета. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К составляют 0,02, 0,04 и
0,2 Дж-К^-моль.
Таблица термодинамических функций InF (г) публикуется впервые. В работе [2082] были
рассчитаны значения Ф° (Т) для Г=1400-^1700 К, расхождения данных [2082] и табл. 704 не
превосходят 0,4 Дж-К"х-моль"х.
201
InF2 (r), InF3 (к, ж) Глава 23
Константа равновесия реакции InF (r)=In (r)+F (г) вычислена с использованием принятой
в справочнике энергии диссоциации
\Н° @) = Do (InF) = 510 + 10 кДж • моль« 42 600 ± 850 ом.
Эта величина была найдена на основании результатов масс-спектрометрического исследования
равновесия реакции CaF (г)+1п (г)=Са (r)+InF (г) A593—1693 К, 9 точек), выполненного Мура-
дом и др. [2082]. Обработка данных [2082] приводит к значениям 487 +150 кДж -моль (II закон)
и 507+10 кДж-моль (III закон), погрешность последней величины принята по рекомендации
авторов [2082]. Булевичидр. [788] исследовали равновесие реакции In (r)+HF (r)=InF (г)+Н (г)
в пламенах H2+O2+N2 фотометрическим методом и нашли Do (InF)=525+30 кДж-моль
(III закон), что в пределах погрешности совпадает с величиной, полученной из данных [2082].
Короткая экстраполяция колебательных уровней состояния А1!! (на 350 см) свидетельствует,
что они сходятся в области 46 200 см над уровнем z;=0, /=0 состояния X1 2+. Соответствующее
значение энергии диссоциации InF равно 44 000+300 см. Расхождение между принятой
величиной и этим значением может быть объяснено (см. [589, 2082]) наличием максимума на
потенциальной кривой состояния А1]! высотой в 1400 +900 см.
Принятой энергии диссоциации соответствует
bfH°(lnF, г, 0) = —191,925 + 10 кДж • моль.
InF2 (г). Термодинамические свойства газообразного дифторида индия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 705.
Молекулярные постоянные, использованные для расчета термодинамических функций InF3,
приведены в табл. 23.6. В литературе отсутствуют экспериментальные данные о структурных
параметрах и частотах колебаний InF2. По аналогии с молекулами дигалогенидов других элементов
группы ША в справочнике принимается, что молекула InF2 в основном электронном состоянии
Х2АХ имеет нелинейную структуру симметрии C2v с углом <^F—In—F = 120+10°. Приведенное
в табл. 23.6 произведение моментов инерции IaIbIg вычислено для межатомного расстояния
г (In—F)=2,00+0,05 А, принятого равным соответствующему расстоянию в молекуле InF3.
Погрешность IaIbIc составляет 1-1014 г3-см". Основные частоты InF2 вычислены по уравнениям
поля валентных сил и силовым постоянным, принятым равными силовым постоянным InF3 x.
Погрешности в рекомендуемых частотах не превышают 10—15 %. Энергия возбужденного
электронного состояния А2В1 молекулы InFa принимается равной 15 000 +5000 см. Это значение
получено грубой оценкой, основанной на сравнении с энергией соответствующего состояния молекулы
A1F2, принятой в настоящем справочнике.
Термодинамические функции InF2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9)—A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом возбужденного электронного состояния А2Вг по уравнениям A. 168)—A. 170).
Погрешности вычисленных термодинамических функций определяются в основном неточностью
молекулярных постоянных, при высоких температурах становятся существенными погрешности,
обусловленные приближенным характером расчета. Погрешности в значениях Ф° (Т) оцениваются
в 2, 4 и 6 Дж-К^-моль'1 при 298,15, 3000 и 6000 К.
Таблица термодинамических функций InF2 (г) публикуется впервые.
Константа равновесия реакции InF2 (r)=In (r)+2F (г) вычислена с использованием принятого
в справочнике значения
ДгЯ° @) = 870 + 50 кДж • моль.
Эта величина оценена на основании сопоставления энергий атомизации молекул InF, InF2,
InF3, A1F, A1F2 и A1F3, ей соответствует
t\fH° (InF2, г, 0) = —474,65 + 50 кДж • моль.
InF3 (к, ж). Термодинамические свойства кристаллического и жидкого трифторида индия
в стандартном состоянии при температурах 298,15—3000 К приведены в табл. 706.
1 /г=3,90, /„.=0,23, /а/г2=0,05 (в 105 дин-см"*).
202
Индий и ere соединения . InF, (к, ж)
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 23.1. В справочнике за стандартное состояние InF3 (к) в интервале 0—4445 К принята
гексагональная модификация (структурный тип VF3 [2008a]).1 В литературе отсутствуют
экспериментальные данные о теплоемкости и энтальпии InF3 (к, ж) как при низких, так и при высоких
температурах.
Значения S0 B98,15 К) и Н° B98,15 К)—Н° @), приведенные в табл. 23.1, оценены при
подготовке справочника с помощью эмпирических методик, предложенных в работах [216, 427, 814а],
и с использованием экспериментальных данных по теплоемкостям A1F3 и CeF3 при низких
температурах; точность этих значений оценивается в 8 Дж-К" -моль и 1,5 кДж-моль" соответственно.
В интервале 298,15—1445 К для теплоемкости InF3 (к) принято уравнение (см. табл. 23.1),
выведенное по значениям теплоемкости СрB98,15 К)=92 Дж-К~1-моль~1 и С°р A445 К) =
=134 Дж-К~1-моль~1, оцененным сравнением экспериментальных данных по теплоемкости A1F3,
YF3, LaF3 и CeF3. Температура плавления InF3 A445 +5 К) принята по хорошо согласующимся
между собой результатам измерений, проведенных Робертсом и др. [2349а] A445+5 К) и Ханне-
боном и Клеимом [1347] A443+10 К) на достаточно чистых образцах, содержащих>99,5% InF3.
Энтальпия плавления InF3 F4+15 кДж-моль) оценена в предположении о равенстве энтропии
плавления InF3 и A1F3. Для теплоемкости InF3 (ж) принято значение 134+15 Дж-К-моль~1,
равное теплоемкости CeF3 (ж).
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 8,
15, 30 и 40 Дж-К-моль соответственно.
Термодинамические функции InF3 (к), вычисленные в работе Стейгера и Маргрейва [2710]
C00—1400 К), отличаются от приведенных в табл. 706 в пределах 3 Дж-К~1-моль~1 в значениях
Ф° (Т).
В справочнике принята энтальпия образования
AfH°(lnFs, к, 298,15К) = —1190 + 25 кДж • моль,
основанная на исследовании равновесия реакции 4/3 InF3 (k) + SiO2 (кварцевое стекло)=2/31п2О3 (к)-}-
+SiF4 (г), выполненном Сониным [391] мембранным методом. Было найдено, что давление SiF4
составляет ~0,13 атм при 783 К. Расчеты проводились как с функциями SiO2 (стекл.), так и SiO2
(кварц.) и привели к значениям Д/Я° (InF3, к), совпадающим в пределах 3 кДж-моль.
Проведенные в этой же работе аналогичные измерения с A1F3 (к) привели к значению Д/#° (A1F3, к),
совпадающему в пределах 7 кДж -моль с принятым в справочнике. Однако в отличие от A1F3
равновесие с InF3 устанавливалось очень медленно, что могло привести к завышенному (по
абсолютной величине) значению Д///° (InF3, к).
Имеющаяся в литературе (см. [2678]) оценка этой величины (—1046 кДж-моль) основана на
неверных значениях энтальпии образования A1F3 и ScF3.
Давление пара трифторида индия InF3 (к, ffl)=InF3 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
Д8Я° (InF3, к, 0) = 335 + 20 кДж • моль.
Принятая величина основана на измерениях давления пара, выполненных Стейгером и Маргрей-
вом [2610] (масс-спектрометрический метод, 1030—1150 К, 19 точек, Д8Я° @)=330+8 кДж-моль
по II закону и 339,8+0,3 кДж-моль по III закону) и Сониным [391] (статический метод, 1414—
1646 К, 21 точка, ^SH° @) =381 кДж-моль по II закону и 315+4 кДж-моль по III закону).
Приведенные погрешности отражают только воспроизводимость измерений. Неточность
термодинамических функций приводит к дополнительным погрешностям в энтальпии сублимации, равным
17 кДж-моль [2610] и 33 кДж-моль [391]. При выборе рекомендованного значения больший
вес придавался работе [2610],где было получено лучшее соответствие расчетов noil и III законам и
меньше погрешность, связанная с неточностью термодинамических функций.
1 Бек, Куглер и Хендлер [637а] сообщили недавно о существовании метастабильной тетрагональной
модификации (Y-InF3). Монотропное превращение y-InF3 в кубическую модификацию (a-InF3) авторы [637а] наблюдали
при 648 К.
203
InF3 (r), InCl (к, ж) Глава 23
InF3 (г). Термодинамические свойства газообразного трифторида индия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 707.
Молекулярные постоянные, использованные для расчета термодинамических функций InF3 (г),
приведены в табл. 23.6. Структура молекулы InF3 экспериментально не исследовалась. По
аналогии с молекулами ВХ3, А1Х3, GaX3 (X=F, Cl, Br, I) в настоящем справочнике принимается, что
в основном электронном состоянии ХХА молекула InF3 имеет плоскую симметричную структуру и
принадлежит к точечной группе DSh. Произведение главных моментов инерции, приведенное в таб
лице, вычислено для межатомного расстояния г (In—F)=2,00+0,05 А, оцененного на основании
сравнения величины г (М—F) в молекулах MF и MF3, где М=В, Al, Ga, Sc, Y, La. Погрешность
значения IaIbIc составляет 5 -1014 г3 -см6. Приведенные в табл. 23.6 основные частоты v2, v3 и v4 приняты
по работе Хауге, который исследовал ИК-спектр молекул InF3, изолированных в
низкотемпературной матрице (см. [2610]). Частота vx вычислена по уравнениям поля валентных сил и значениям
v3 и v, в предположении, что в молекуле InF3 /r//rr=17 (аналогичное соотношение имеет место для
молекул А1Х3). Погрешности в экспериментально наблюдавшихся частотах составляют 10—15 см,
погрешность значения vx может достигать 10—15%. Сведения о возбужденных электронных
состояниях InF8 в литературе отсутствуют ипо аналогии с A1F3 в справочнике принимается, что их
энергии превышают 20 000 см'.
Термодинамические функции InF3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности рассчитанных величин определяются неточностью молекулярных
постоянных (до 3 Дж-К-моль) и приближенным характером расчета и составляют 2, 6 и
8 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции InF3 (г) вычисляли Стейгер иМаргрейв [2610] (Т ^ 1000 К).
Расхождения результатов расчета [2610] с данными, приведенными в табл. 707, не превышают
0,5 Дж-К-моль в значениях Ф° (Т) и объясняются незначительными различиями в принятых
значениях молекулярных постоянных.
Константа равновесия реакции InF3 (r)=In (r)+3F (г) вычислена с использованием значения
Ь.ГН° @)=1327,178+32 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации InF3 (к).
Этим величинам также соответствует
&fH° (InFs, г, 0) = —854,553 + 32 кДж • моль.
InCl (к, ж). Термодинамические свойства кристаллического и жидкого хлорида индия в
стандартном состоянии при температурах 298,15—2000 К приведены в табл. 708.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 23.1. В справочнике за стандартное состояние InCl (к) в интервале 0—387 К принята
кубическая (желтая) модификация, а в интервале 387—484 К — ромбическая (красная)
модификация [655а, 1719, 2753а]. В литературе отсутствуют данные по термодинамическим свойствам
InCl (к, ж), за исключением измерений температур и энтальпий превращения и плавления, а
также энтальпии образования.
Значения S0 B98,15 К) и Н° B98,15 К)—Н° @), приведенные в табл. 23.1, оценены с помощью
различных эмпирических методик (см. [216, 273, 427, 814а, 1781]) и экспериментальных данных по
теплоемкости Т1С1 при низких температурах (см. гл. 24). Погрешности принятых значений
оцениваются в 5 Дж-К-моль~1 и 1,5 кДж-моль соответственно.
В интервале 298,15—387 К для теплоемкости а-InCl принято уравнение (см. табл. 23.1),
выведенное по значениям ѰРB98,15 К) =51,5 Дж-К^-моль и С°р C87 К) =54 Дж-К^-моль'1,
оцененным с использованием данных по теплоемкости Т1С1 [907 ]; таким же способом оценена
теплоемкость MnCl в интервале 387—487 К E6+6 Дж-К-моль~1). Температура полиморфного
превращения C87+10 К) и энтальпия превращения @,52+0,04 кДж-моль) найдены Смирновым
и др. [382] при исследовании весьма чистого образца InCl (не менее 99,9% основного вещества).
Большая погрешность Ttr связана с термическим гистерезисом полиморфного перехода (при
нагревании — 417 К, при охлаждении — 354 К). С принятым значением Тtr удовлетворительно
согласуются данные Клемма [1719], полученные для менее чистого образца InCl (интервал превращения
408-353 К).
204
Иидий и его соединения InCl (г)
Температура плавления InCl D84+3 К) также принята по данным Смирнова и др. [382],
согласно которым область инконгруэнтного плавления InCl лежит в узких пределах 482,5—486 К.
Результаты измерений, проведенных другими авторами [169а, 307, 382, 414, 1719], по-видимому,
на менее чистых образцах InCl, приводят к значениям 488—498 К г. Измерения энтальпий
плавления и кристаллизации InCl, проведенные в работе [382] методом количественной термографии,
приводят к значениям 20,9 и 21,6 кДж-моль соответственно. В справочнике принимается
среднее значение (ДШЯ=21,3+1,0 кДж-моль). Теплоемкость жидкого InCl F7+10 Дж-К-моль~1)
оценена по эмпирическому соотношению Ср =га-33,5 Дж-К-моль~1.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 5, 14 и
22 Дж-К~1-моль~1 соответственно.
Расхождения в термодинамических функциях InCl (к, ж), приведенных в справочнике Барина
и Кнакке [578] B98—881) и табл. 708, достигают 17 Дж-К^-моль.
В справочнике принята энтальпия образования хлорида индия
AfH° (InCl, к, 298,15 К) = —186,0 + 4,0 кДж • моль,
основанная на измеренных Клеимом и Брейтигамом [1721] энтальпиях реакций растворения
металлического индия, монохлорида индия и хлора в растворе, содержащем КВг и Вг2. Измерения
были проведены в ледяном калориметре. Позднее Смит и Барроу [25526] провели повторные
измерения основной реакции этого термохимического цикла — реакции растворения металлического
индия, и получили величины, совпадающие в пределах 1 кДж-моль с данными работы [1721].
Давление пара InCl (к, ж) = 1пС1 (г) вычислено с использованием принятой в справочнике
энтальпии сублимации
AsH°(InC\, к, 0) = 117,0 + 3,0 кДж1
Эта величина основана на результатах измерений давления пара InCl (к, ж), приведенных
в табл. 23.10. Погрешности приведенных значений характеризуют воспроизводимость измерений
Таблица 23.10. Результаты определений Д„-Н~° (InCl, к, 0) (в кДж-моль)
Авторы
Роберт, 1936 [2348]
Смит, Барроу, 1958 [25526]
Фадеев, Федоров, 1964 [411]
Поляченок, Комшилова, 1970 [3316]
Куния и др., 1974 [1785 в]
а Результаты измерений представлень
Метод
Статический, 573—863 К,
Торзиояный, 400—500 К,
Статический, 583—713 К,
Статический, 749—924 К
Статический, 643—923 К
i в виде уравнения.
15
50
13
а
а
точек
точек а
точек
AgH°
II закон
124 ±1
112
124+3
124
123
(InCl, к, 0)
III закон
109,9±7,0
117,4±3,0
115,0±7,0
112,9±10
113,8±10
е неточность термодинамических функций InCl (к); последняя вносит особенно большой вклад
в погрешности величин, вычисленных из данных [3316, 1785в, 2348]. При обработке
экспериментальных данных предполагалось, что пары состоят только из мономерных молекул, как это
следует из выполненных Фадеевым и Федоровым [411 ] измерений среднего молекулярного веса паров.
Величины, полученные по методу III закона по данным разных авторов, имеют значительный
разброс, достигающий 7 кДж-моль. Для большинства работ характерно также систематическое
расхождение между результатами расчетов по II и III законам, что, возможно, свидетельствует
0 неточности термодинамических функций InCl (ж), использованных в расчетах.
InCl (г). Термодинамические свойства газообразного хлорида индия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 709.
Молекулярные постоянные InCl, принятые в справочнике на основании результатов изучения
электронного и вращательного спектров этой молекулы, приведены в табл. 23.4. В электронном
1 Расхождение данных по температуре плавления InCl возможно также связано с отклонением состава образцов
от стехиометрического.
205
InCl (г) Глава 23
спектре InCl наблюдались переходы а3П0+ ^ XXS + [540, 1134, 2220, 2297, 2825, 2826, 2832, 2860,
2889], а3^ <± Хх? + [540, 542, 1134, 2220, 2825, 2826, 2832, 2860, 2889] и АШ *» Хх? + [1134, 1350,
1351, 2026, 2220, 2825, 2826, 2860]. Кроме того, в поглощении наблюдались неидентифицированные-
континуумы в области 2613 А [1134, 2220, 2826] и 2100 А [1134]. Так же как у других галогенидов
элементов группы IIIA, состояния Х1!!4", а3П и АЩ молекулы InCl коррелируют с атомами в их
основных электронных состояниях. Сопоставление энергий возбуждения подсостояний а3П0+ и
asli1 с величиной Do (InCl) позволяет предполагать, что не наблюдавшиеся в спектрах подсостоя-
ния а3П0- иа3П2, которые должны иметь общий предел In BPi/2)+Cl BP^2) с состоянием Xх ?+,
являются связанными. В табл. 23.4 представлены данные только для трех электронных состояний
этой молекулы, другие состояния должны иметь энергии не ниже 40 000 см и не рассматриваются
в справочнике.
Колебательные постоянные 1151п35С1 в состоянии Х1Л+ приняты по работе Ашрафунниза и
др. [540], где они были получены на основании анализа колебательной структуры переходов
а3П0+—Х1Е+иа3П1—Хх? + (свыше 200 Р-кантов для г;" <; 12и(?-кантов для г;" <; 9). Этипостоян-
ные практически совпадают с данными Верли и Мишера [2826] и близки к полученным
авторами [1134, 2026, 2825, 2860]. Приведенные в табл. 23.4 значения вращательных постоянных
InCl в состоянии X1!!* приняты по работе Дельвина и Де-Вейна [940], где они были получены при
исследовании в микроволновом спектре вращательных переходов с v <^ 3, / ^ 4. Вращательные
постоянные определялись в работах [542, 1134, 2297, 2826, 2860] при исследовании тонкой
структуры электронных переходов и в работах [583, 584, 1451] при изучении микроволнового спектра
этой молекулы; найденные в этих работах значения постоянных близки к приведенным в таблице,
за исключением данных [1134, 2860], которые могут рассматриваться лишь как оценочные.
Колебательные постоянные в состояниях а3П0+ и а3!^ приняты по данным [540], в состоянии
АгЛ найдены Верли и Мшпером [2826]; вращательные постоянные в возбужденных состояниях
InCl приняты по работам [542, 2889]. Состояние А1!! молекулы InCl имеет неглубокий минимум
(~600 см), уровни с v=i и 2 предиссоциированы, а общее число связанных колебательных
уровней, по-видимому, не превышает 7.
Термодинамические функции InCl (г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производных вычислялись по уравнениям A. 90)—A. 92)
с учетом вклада состояния а3П, выполненного в предположении, что Qi^l. ад = 6 QifJ.IV-
Статистическая сумма по колебательно-вращательным уровням энергии состояния ХХЕ+ и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В
расчете учитывались все колебательно-вращательные уровни со значениями / <С /ma3t v, последние
определялись из условий A. 81). Энергия уровней состояния Xх ? + вычислялась по уравнениям
A. 65) и A. 61). Значения коэффициентов Ykl в этих уравнениях, полученные с помощью
соотношений A. 66) для изотопической модификации InCl, соответствующей природной смеси обоих
элементов, на основании молекулярных постоянных 1161п35С1, приведены в табл 23.5. Коэффициенты
YM, включая Y30 и Yi0, получены с учетом условий A. 50), коэффициент Y03 — из условий A. 59).
В табл. 23.5 приведены также величины vmax и /lim.
Погрешности рассчитанных термодинамических функций InCl (г) при низких температурах
обусловлены неточностью принятых фундаментальных постоянных, а также колебательных
постоянных в состоянии Xх ?+, которые определены по кантам полос. Выше 1500—2000 К становятся
заметными погрешности из-за отсутствия экспериментальных данных об уровнях состояния Х*Е +
с энергией более 3700 см, а также из-за приближенного учета вклада состояния й3П.
Пренебрежение состоянием А1!! и другими более высокими состояниями практически не влияет на точность
расчета. Погрешности в значениях Ф° (Т) оцениваются в 0,03, 0,05 и 0,3 Дж-К~1'моль при
298,15, 3000 и 6000 К.
Термодинамические функции InCl (г) ранее вычислялись Келли [1656, 1658] до 2000 К,
а также в работе [231 ] до 1100 К приближенным методом по неточным значениям постоянных.
Расхождения данных [1656,. 1658] и табл. 709 достигают 2 Дж-К-моль в значении S0 B000 К).
Константа равновесия реакции InCl (r)=In (г)+С1 (г) вычислена с использованием значения
ДД° @) = Do (InCl) = 430,62 + 5,8 кДж • моль д* 36 000 ± 500 см'1,
соответствующего принятым в справочнике энтальпиям образования и сублимации InCl (к). Это
206
Индий и его соединения In€l, (r), InCL, (к, ж)
значение хорошо согласуется с полученным короткой экстраполяцией колебательных уровней
состояния ЛШ C5 750 см) [2826]. Сопоставление этих двух значений позволяет предполагать, что
у молекулы InCl на потенциальной кривой состояния А1!! максимум отсутствует.
Спектрофотометрическое исследование равновесия реакции In (г)+НС1 (г) = 1пС1 (г)+Н (г),
выполненное Булевич и др. [787] A500—2000 К), также привело к близкому,, но менее точному
значению энергии диссоциации 435 + 12 кДж-моль.
Принятым значениям энтальпий образования и сублимации InCl (к) соответствует
bfH° (InCl, г, 0) = —70,2 + 5,0 кДж • моль.
InCI2 (г). Термодинамические свойства газообразного дихлорида индия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 710.
Молекулярные постоянные, использованные для расчета термодинамических функций 1пС12,
приведены в табл. 23.6. В литературе отсутствуют экспериментальные данные о структурных
параметрах и частотах колебаний 1пС12. По аналогии с молекулами ВН2, BF2 и АШ2, структура
которых исследовалась экспериментально и определялась на основании квантовомеханических
расчетов, в справочнике принимается, что молекула 1пС12 в основном электронном состоянии Х2Аг
имеет нелинейную структуру (симметрия C2v) с углом ^Cl—In—Cl = 120 + 10°. Произведение
главных моментов инерции в основном электронном состоянии Х2Аг вычислено для межатомного
расстояния г (In—С1)=2,30+ 0,05 А, принятого равным соответствующему расстоянию в молекуле
InGlg, см. ниже. Погрешность значения IaIbIc составляет 1 • 10~113 г3-см6. Основные частоты
молекулы 1пС12, приведенные в таблице, вычислены по уравнениям поля валентных сил и силовым
постоянным, принятым равными соответствующим силовым постоянным 1пС13 1. Погрешности в этих
значениях оцениваются в 10—15% от их величины. По аналогии с А1С12 можно предполагать, что
у молекулы 1пС12 должно быть стабильное возбужденное состояние типа А2Вг с энергией
возбуждения 10 000+5000 см.
Термодинамические функции 1пС12 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), 5A. 122)—A. 124), |A. 128),
A. 130) с учетом возбужденного электронного состояния АгВх по уравнениям A.168)—A. 170),
Погрешности рассмотренных величин составляют 3, 5 и 8 Дж-К~1-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К. Они определяются неточностью молекулярных постоянных, а также
приближенным характером расчета.
Таблица термодинамических функций 1пС12 (г) публикуется впервые.
Константа равновесия реакции 1пС12 (г)=1п (г)+2С1 (г) вычислена с использованием принятого
в справочнике значения
ДД° @) = 700 + 50 кДж • моль.
Эта величина оценена на основании сопоставления энергий атомизации молекул InCl, InCl2,
InCl3, A1C1, A1C12 и А1С13; ей соответствует
г, 0) = —219,96 + 50 кДж • моль.
InCl3 (к, ж). Термодинамические свойства кристаллического и жидкого трихлорида индия
в стандартном состоянии при температурах 298,15—1200 К приведены в табл. Д.53.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 23.1. В справочнике за стандартное состояние 1пС13 (к) в интервале 0—856 К принимается
моноклинная модификация (структурный тип А1С13) [2498а].
В литературе отсутствуют экспериментальные данные о термодинамических свойствах
InClg (к, ж), за исключением температуры плавления и энтальпии образования. Значения
S0 B98,15 К) и Н° B98,15 К)—Я0 @), приведенные в табл. 23.1, оценены с помощью эмпирических
методик [216, 427, 814а] с использованием экспериментальных данных по теплоемкости А1С13
и LaCl3 при низких температурах; погрешности оцененных значений составляют 10 Дж-К^-моль
и 1,5 кДж-моль соответственно.
/г=2,32, /гг=0,12, /а/г?=0,05 (в 106 дин-см).
207
InCl3 (к, ж)
Глава 23
В интервале 298,15—856 К для теплоемкости 1пС13 (к) принято уравнение (см. табл. 23.1),
выведенное с использованием значений С°р B98,15 К)=100 Дж-К~1-моль~1 и С°р (856 К) =
=117 Дж-К-моль~1, оцененных на основании экспериментальных данных по теплоемкости
А1С13 (к) и GaCl3 (к).
Температура плавления 1пС13 (856 + 3 К) принята по согласующимся между собой данным
Клемма [1719], Палкина и др. [306] и Федорова и Фадеева [414]. Энтальпия плавления 1пС13
B7+5 кДж-моль) оценена при энтропии плавления 1пС13, равной энтропии плавления GaCl3.
Теплоемкость 1пС13 (ж) A17+15 Дж-К~1-моль~1) принята равной среднему значению из тепло-
емкостей А1С13 (ж) A26 Дж-К'^моль) и GaCl3 (ж) A07 Дж-К^-моль).
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 1000 К оцениваются в 10, 15 и
20 Дж-К-моль соответственно. Термодинамические функции 1пС13 (к), приведенные в
справочнике Барина и Кнакке [578] (Г^850 К), согласуются с данными табл. Д.53 в пределах 1 Дж X
X К-моль.
Энтальпия образования 1пС13 (к) . ¦
A^^InClj, к, 298,15 К) = — 537 + 4 кДж • моль
принята по результатам калориметрических измерений, выполненных Клеммом и Брейтингамом
[1721] в ледяном калориметре. В этой работе были измерены энтальпия растворения
металлического индия, 1пС13 (к) и хлора в растворе, содержащем КВг и Вг2. Смит и Барроу [2552а] провели
повторные измерения реакции с металлическим индием и подтвердили (в пределах 1 кДж-моль)
результаты измерений [1721]. Кампбелл и др. [810] измерили энтальпию растворения 1п2О3 в
соляной кислоте. Совместно с данными Рота и Бюхнера [2380] по энтальпии растворения 1пС13
эти измерения дают для ^fH° (InCl3, к, 298,15 К) значение —523 кДж-моль, которое
существенно меньше принятого.1
В справочнике принята энтальпия сублимации трихлорида индия
Д,# ° AпС13, к, 0) = 162 + 6 кДж • моль.
Эта величина основана на результатах измерений давления пара 1пС13 (к), приведенных
в табл. 23.11. В состав продуктов испарения 1пС13 (к) входят как мономерные, так и димерные мо-
Таблица 23.11. Результаты определений &SH° AпС1з, к, 0) (в кДж-моль")
Авторы
Метод
\Н° (InClg, к, 0)
II закон
III закон
Роберт, 1936 [2348] Статический, 663—759 К, 15 точек 160+9 158,4±0,9
Смит, Барроу, 1958 [25526] Торзионный, пять серий, 482—564а 158 162,1+0,5
Фадеев, Федоров, 1964 [411] Статический, 613—723 К, 12 точек 144+2 160,0+0,6
Срывцев, Петров, 1969 [396а] Статический, 600—820 К а 153,0 160,0+1,5
Полячёнок, Комшилова, 1970 [3316] Статический, 647—792 К а 166 160,8±0,8
Куния и др., 1974 [1785в] Статический и адсорбционно-спектрометриче- 159 165,0+0,9
скип, 623—773 К а
* Результаты измерений представлены в виде уравнений.
лекулы. Приведенные в табл. 23.11 значения относятся к процессу испарения с образованием
мономерных молекул, для этого экспериментальные данные были соответствующим образом
пересчитаны. Приведенные в табл. 23.11 погрешности характеризуют только воспроизводимость
измерений. Неточность термодинамических функций (в первую очередь 1пС13 (к)) приводит к
дополнительной погрешности в энтальпии сублимации около 4 кДж-моль при 600 К. Принятая
энтальпия сублимации 1пС13 (к)=1пС13 (г) является средневзвешенной из приведенных в табл. 23.11
1 В работе [810] из-за ошибки при расчете энтальпии образования раствора 1пС13 было получено неверное
значение Afff° (InCl3, к, 298,15 К) = — 645 кДж-моль.
208
Индий и его соединения 1пС13 (г), In2Cle (г)
величин, причем наибольший вес придавался измерениям Смита и Барроу [25526], так как эти
измерения проводились при наиболее низкой температуре, когда доля димерных молекул в паре
и погрешности из-за неточности термодинамических функций 1пС13 (к, ж) минимальны.
InCI3 (г). Термодинамические свойства газообразного трихлорида индия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 711.
Молекулярные постоянные, использованные для расчета термодинамических функций 1пС13,
приведены в табл. 23.6. Спектроскопические исследования [368, 633, 636, 2245] показывают, что
молекула 1пС13 в состоянии ХгА имеет плоскую симметричную структуру (группа симметрии D3h).
Приведенное в табл. 23.6 произведение главных моментов инерции вычислено для межатомного
расстояния г (In—С1)=2,30>+0,05 А, оцененного на основании сопоставления межатомных
расстояний в молекулах MX и МХ3, где М=А1, Ga и Х=С1, Вг, I г. Погрешность IaIbIo составляет
МО'112 г3-см6.
Приведенные в табл. 23.6 значения частот v2, v3 и v4 получены Селивановым и Мальцевым [368]
при исследовании ИК-спектра газообразного 1пС13. Для частоты симметричного валентного
колебания vx, принято значение, найденное Битти и Хордером [636] при исследовании спектра КР
газообразного 1пС13. Близкие значения частот были получены в работах [633, 12206, 2245] при
исследованиях колебательных спектров молекул 1пС13, изолированных в матрицах из Аг и Кг
(в [12206]) 2. Погрешности в значениях приведенных в таблице частот оцениваются в 5—10 см.
Сведения о возбужденных электронных состояниях 1пС13 в литературе отсутствуют, по аналогии
с тригалогенидами других элементов группы IIIA можно предполагать, что их энергии превышают
15 000-20 000 см.
Термодинамические функции 1пС13 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A.10), A.122)—A.124), A.128), A.130).
Погрешности функций определяются в основном приближенным характером расчета и составляют
2, 6 и 8 Дж-К~1-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности,
обусловленные неточностью молекулярных постоянных, не превышают 2 Дж-К~1-моль~1.
Термодинамические функции 1пС13 (г) вычисляли Гиван и Левеншус [12206] (Т ^ 2000 К),
расхождения данных [12206] и табл. 711 составляют ~2 Дж-К-моль и обусловлены тем, что
эти авторы использовали завышенное значение г (In—С1).
Константа равновесия реакции 1пС13 (г)=1п (г)+ЗС1 (г) вычислена с использованием значения
к„Н° @)=975,280 + 7,5 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д//°AпС13, г, 0) = —375,62 +7,2 кДж • моль.
Эта величина является суммой энтальпий образования и сублимации 1пС13 (к).
IiioCI6 (г). Термодинамические свойства газообразного гексахлорида дииндия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 712.
Молекулярные постоянные, использованные для расчета термодинамических функций 1п2С16,
приведены в табл. 23.6. Структура молекулы 1п2С16 экспериментально не изучалась. По аналогии
с молекулами А12С1в и Ga2Cl6, а также на основании результатов исследования колебательных
спектров In2Cle [636, 785а], в настоящем справочнике принимается, что в основном электронном
состоянии ХХА молекула In2Cle имеет мостиковую структуру симметрии D2h. Приведенное в табл. 23.6
произведение главных моментов инерции IaIbIg вычислено по структурным параметрам:
гAп-С1)внешн = 2,3 ± 0,1 A, r(Ia-CL)MOBT = 2,5 + ОД А, ^С1ЕШШВ-1п-С1внвшн = 125 ± 5° и ^С1М00Т-
—In—С1чост=85 + 5°. Эти значения были оценены по соответствующим структурным параметрам
молекул А12С1в, Ga2Cle, InCl3 с учетом результатов теоретического расчета молекул А12С12, Ga2Cl2 и
1п„С1, в работе Соломоника [388]. Погрешность значения IaIbIg оценивается в 1-1010 г3-см6.
1 Значение г (In—С1), принятое в справочнике, существенно отличается от полученного в электронографическом
исследовании Стивенсона и Шомейкера [2615] (г (In—С1)средж=2,46 А). Возможно, это различие объясняется
наличием в исследуемом паре смеси димерных и мономерных молекул треххлористого индия.
- В работе Битти и Хордера [633] при исследовании спектра КР 1пС13 в матрице из Аг было получено значительно
более высокое значение v4=119 см, существенно отличающееся от значений, полученных в [368] (95 см),
[636] (94 см), [2245] A00,5 см) к [12206] (98 см).
209
In2Cle (г) Глава 23
Колебательные спектры молекулы In2Cle исследовались в газовой фазе Битти и Хордером [6361
и в расплаве Бусом и др. [785а]. Значения частот, измеренных в обеих работах, хорошо
согласуются между собой. Приведенные в табл. 23.6 значения частот vx, v2, v4, v7, vu приняты по работе
Битти и Хордера [636]. Частоты v3, v6, v15 основаны на данных, полученных Бусом и др. [785а],
при этом для частоты v15 отнесение взято по работе [636]. Значения остальных частот оценены
исходя из соответствующих данных для молекул А12С1в, Ga2Cl6, In2Bre и In2Ie так, чтобы изменение
величин частот колебаний в рядах А12С16, Ga2Cle, In2Cl6 и In2Cle, In2Br6, In2Ie было закономерным Ч
Погрешности экспериментально измеренных частот колебаний не превышают 2—10 см,
погрешности оцененных частот могут достигать 20% от их величины.
Термодинамические функции 1п2С16 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности рассчитанных значений определяются приближенным характером расчета,
а также неточностью молекулярных постоянных и составляют 15, 25, 40 Дж>К~1-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций 1п2С16 (г) публикуется впервые.
Константа равновесия реакции 1п2С16 (г)=21п (г)+6С1 (г) вычислена с использованием
значения А,!!0 @)=2081,32+ 20 кДж-моль, соответствующего принятой в справочнике энталышн
образования
AjH° Aп2С16, г, 0) = —882 + 20 кДж • моль.
Эта величина основана на исследованиях равновесия реакции In2Cle (г)=21пС13 (г),
выполненных статическим методом Поляченком, Комшиловой [3316] G53-—1216 К) и Куния и др. [1785в]
G63—1113 К). В этих работах были измерены общие давления над 1пС13 (к, ж) и приведены
уравнения, представляющие зависимость константы диссоциации димера от температуры. По этим
уравнениям для энтальпии диссоциации димера при Т=0 были получены значения 131,0^
+ 0,3 кДж-моль (III) и 132 кДж-моль (II) по работе [3316] и 132,4+_1,6 кДж-моль (III) и
139 кДж-моль (II) по работе [1785в]. Результаты работ [3316] и [1785в] находятся в хорошем
соответствии.
При вычислении рекомендованного значения энтальпии образования 1п2С16 принималась
Do (InCl3—InCl3)=131+18 кДж-моль; погрешность этой величины определяется главным
образом неточностью термодинамических функций 1п2С10 (г).
1 Бус и др. [785а] зарегистрировали полосу при 131 см х и отнесли ее к v12. Такое отнесение представляется
сомнительным, поскольку при этом нарушается закономерное изменение соответствующих частот колебаний в ряду
молекул А12С16, Ga2Cle и 1п2С16.
Глава 24
ТАЛЛИЙ И ЕГО СОЕДИНЕНИЯ
Т1(к, ж), Т1, Т1+, ТЮ, Т12О (к, ж), Т12О, Т12О3 (к, ж), Т1Н, Т1ОН, T1F(k, ж), TIF, T12F2,
Т1С1 (к, ж), Т1С1, Т12С12
В настоящей главе рассматриваются 11 веществ — таллий и его соединения с кислородом,
водородом, фтором и хлором. Для пяти веществ приведены данные о термодинамических свойствах
в конденсированном состоянии и для 10 веществ — в газообразном состоянии. Термодинамические
функции газообразных таллия и его иона рассчитаны для температур до 10 000 К, для остальных
газов — до 6000 К. Для элементарного таллия в Приложении 3.9 приведены сведения о
критических постоянных.
Термодинамические свойства всех веществ приведены для природной изотопической смеси
таллия (атомный вес 204,37) и других элементов. Однако различие в молекулярных постоянных ин<-
дивидуальных изотопических модификаций веществ в большинстве случаев не учитывалось в
расчетах из-за отсутствия необходимых данных и их несущественного влияния на термодинамические
функции соединений таллия.
Т1 (к, ж). Термодинамические свойства кристаллического и жидкого таллия в стандартном
состоянии при температурах 100—3000 К приведены в табл. 713.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 24.1. В справочнике за стандартное состояние Т1 (к) в интервале 0—507 К принята
гексагональная модификация (структурный тип Mg), а в интервале 507—576 К — кубическая
модификация (структурный тип a-Fe).
Таблица 24.1. Принятые значения термодинамических величин для таллия и его соединений
в кристаллическом и жидком состояниях
Вещество
Т1
Т1,0
Т12О3
T1F
Т1С1
Состояние
к11, гекс.
к1, куб.
ж
к, монокл.
ж
к, куб.
ж
к11, ромб.
к1, тетр.
ж
к, куб.
ж
1
К
СО ¦—'
кДжХ
X моль
6,832
—
15,1
—
20,2
—
11,89
—
—
12,68
—
1,15
OS
О
Дж-К
64,30
—
146,0
—
160,0
—
95,69
—
.—
111,24
—
Примечание. С1=а+ЪТ-сТ-*+аТ*
Т1С1:
а <М06=-34,019.
GO
аз
CN]
• МОЛЬ
Коэффициенты в
уравнении для С (Т)
а
26,32 25,017
— 115,265
— 25,844
79,75 69,664
— ИЗ
108,2 128,519
— 160
53,39 27,668
— 51,028
— 67,3
50,92 59,431
— 75,6
(в Дж-К^-моль)
6-Ю3
с ¦ Ю-5
7,360 0,794
—91,630 98,36
1,673 —12,251
28,004 —1,541
— _
8,602 20,344
— —
74,546 —3,105
16,577 4,293
— —
—23,221 4,099а
— —
Интервал
температур
к
298,15—507
507—576
576—3000
298,15-852
852—4000
298,15-1107
1107—3000
298,15—356,3
356,3—599
599—2300
298,15—704
704—2200
Ttr или
Т
К
507
576
—
852
—
1107
—
356,3
599
—
704
—
Д/г# или
m
кДжХ
X моль
0,36
4,20
—
30,3
—
53
—
0,336
13,87
—
15,56
—
Термодинамические функции Т1 при низких температурах вычислены: при Т <С 4,2 К по р&-
зультатам измерений теплоемкости в сверхпроводящем и нормальном состояниях 11455, 1648,
2034, 2572] \ в интервале 4,2—298,15 К — по данным Клузиуса и Бона [861] A0—200 К) и
1 Температура перехода Т1 в сверхпроводящее состояние 2,38+0,02 К принята по данным работ [930, 1455, 1648,
2034, 2572].
211
Т1(к, ж) Глава 24
Хикса [1403] A4—300 К). Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—Я0 @)
оцениваются в 0,2 Дж-К~1-моль~1 и 0,02 кДж-моль соответственно.
В интервале 298,15—507 К для теплоемкости Т1 (к) принято уравнение (см. табл. 24.1),
выведенное методом Шомейта при совместной обработке результатов измерений энтальпии,
выполненных Умино [2743] C73—773 К) и Ротом и др. [2383] C73—628 К), и измерений теплоемкости,
проведенных Зеекампом [2489] B91—773 К). При этом погрешность измерений [2743] оценена
в 0,5%, [2383] - в 1% и [2489] - в 2%.
В интервале 507—576 К для теплоемкости Т1 (к) принято уравнение (см. табл. 24.1),
выведенное по трем значениям теплоемкости: Ср E07 К)=30,54 Дж-К~1-моль~1 [2461]; С°р E33 К) =
=31,8 Дж-К^-моль и С; E63 К)=32,6 Дж-К^-моль (по данным [2743] и [2489]).
Температура полиморфного превращения E07 + 1 К) принята по согласующимся между собой данным
11928, 2510, 2651, 2750]. Результаты измерений Т,г [332, 415, 2260, 2333, 2461, 2489, 2743] менее
надежны и не учитывались в справочнике. Энтальпия превращения @,36 + 0,04 кДж-моль) принята
в результате усреднения данных [216, 1400, 1928, 2155, 2156, 2384, 2489, 2651 ]. Температура
плавления E76 + 1 К) выбрана по согласующимся измерениям [1928, 2333, 2383, 2461, 2743, 2750].
Энтальпия плавления D,20+0,08 кДж-моль) получена усреднением данных [1400, 2155, 2156,
2383, 2961]. Измерения [1928, 2743] привели к более низким значениям C,14 и 4,07 кДж-моль)
и не принимались во внимание.
Экспериментальные измерения энтальпии ([2153] E76—753 К), [2383] E76—628 К), [2461]
<576-673 К), [2743] E76-773 К)) и теплоемкости ([8] E76-773 К) и [28] A000-1700 К))
жидкого таллия плохо согласуются между собой г. Уравнение для теплоемкости Т1 (ж), принятое
в справочнике (см. табл. 24. 1), выведено с учетом характера изменения теплоемкости жидких
галлия, индия, олова и свинца. При выводе уравнения было использовано экспериментальное
значение теплоемкости жидкого таллия в точке плавления С°р E76 К)=30,5 Дж-К~1-моль
(полученное усреднением данных Эльсена и др. [2153] и Шнейдера и др. [2461 ]) и два оцененных
значения теплоемкости Т1 (ж) Ср A600 К)=29 Дж-К^-моль и С; C000 К)=31 Дж^К^-моль.
Последние получены сопоставлением принятых в настоящем справочнике данных для
температурной зависимости теплоемкости Ga, In, Sn и Pb, выраженных как функции от аргумента Т/Ть.
Согласно принятому уравнению кривая теплоемкости Т1 (ж) имеет минимум при 1150 К
¦(С; = 28,7 Дж-К^-моль).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
1, 3 и 5 Дж-К-моль~1 соответственно.
Термодинамические функции Т1 (к, ж), приведенные в табл. 713, отличаются от данных
справочников [1503] и [1656] в пределах 2 Дж-К~1-моль~1 в значениях Ф° (Т).
В справочнике за стандартное состояние таллия принят Т1 (к, гекс); в соответствии с этим
Д//°(Т!, к) = 0.
Давление пара таллия в реакции Т1 (к, ж)=Т1 (г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Д,#°(Т], к, 0) = 181,4 + 1,5 кДж-моль-1.
Эта величина является средней из данных [86, 493, 870, 945, 1093, 1552], приведенных
в табл. 24.2, где в качестве погрешностей указаны воспроизводимости измерений. Погрешности
в рассчитанных значениях энтальпии сублимации, обусловленные неточностью
термодинамических функций Т1 (к, ж), изменяются от 1 кДж-моль для измерений, выполненных при 1000 К,
до 6 кДж-моль для измерений, выполненных при 2000 К. Это значительно увеличивает
погрешность данных [718, 1843, 2455]. Данные шести работ, на которых основывается рекомендуемая
величина, имеют минимальные погрешности из-за разброса измерений и из-за неточности
термодинамических функций, они также удовлетворительно согласуются между собою.
Измерения Банчила и др. [28] методом радиальных температурных волн в интервале 1000—1700 К привели
к выводу, что с ростом температуры теплоемкость жидкого таллия резко уменьшается от 38 Дж-К~1-моль~1
при 1000 К до 26 Дж-К^-моль при 1700 К. Это находится в противоречии с результатами исследований
теплоемкости Т1 (ж) вблизи Тт, а также теплоемкости ряда других жидких металлов (Ga, Sn, щелочные металлы),
для которых экспериментально установлено, что с ростом температуры теплоемкость проходит через минимум.
212
Таллий и его соединения
Таблица 24.2. Результаты определений Д8Н° (TI, к, 0) в (кДж-моль
АвторЪ1
Гибсон, 1911 [1198]
Вартенберг, 1913 [2799]
Ляйтгебель, 1931 [1843]
Мюллер, 1935 [2074]
Колман, Эджертон, 1935 [870]
Фишер, 1935 [1093]
Гурвич, 1960 [97]
Генов и др., 1961 [86]
Щукарев и др., 1962 [469]
Олдред, Пратт, 1963 [493]
Бохдански, Шине, 1967 [718]
Ядзава и др., 1966 [1552]
Дезре и др., 1968 [945]
Шине и др., 1971 [2455]
Наппи и др., 1978 [2103а]
Метод
Статический, 1223—1473 К, 6 точек
Протока, 907—1243 К, 3 точки
Точек кипения, 1730 К, 1 точка
Измерение интенсивности спектра поглощения,
623—773 К, 6 точек
Эффузионный, 846—905 К, 11 точек
Эффузионный, 801—927 К, 19 точек
Точек кипения, 1247—1400 К, 17 точек
Обработка данных [160] по аномальной дисперсии
света в парах таллия, 885—1336 К, 13 точек
Эффузионный, 719—760 К, 19 точек
Испарение с открытой поверхности, 519—757 К,
10 точек
Эффузионный, 701—973 К, 14 точек
Торзионно-эффузионный, 783—914 К, 12 точек
Тепловой трубы, 1312—1892 К, 8 точек
Протока, 1176—1483 К, результаты измерений
представлены в виде уравнения
Торзпонно-эффузионный, 931—996 К, 10 точек
Тепловой трубы, 1670—2050 К, 22 точки
Масс-спектрометрический, 783—878 К, 69 точек
Торзионный, 914—1024 К, 46 точек
Д8Я°(
II закон
132 ±5
181 ±2
—
182 ±10
179 ±7
182+4
181 ±5
180+2
179+4
177 ±4
181 ±4
185 ±3
192±3
175
184±7
181 ±5
178+3
180+4
Т1, к, 0)
III закон
179,5+3,5
170,9+4,0
180,4±1
182,6±0,2
183,1 ±0,2
181,4±0,2
_
180,2±0,4
180,3±0,5
187,7±0,6
179,9±0,2
180,6±1,4
181,7 ±0,8
182,5±0,1
179,9±0,2
184,2+4,5
177,9±2,5
Tl (г). Термодинамические свойства газообразного таллия в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 714.
Уровни энергии атома Т1, использованные в расчете термодинамических функций, приведены
в табл. 24.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. . .6s26p, . . .6s26d, . . .6s25/, . . .6s26/, . . .6s6p2 и лежащие ниже ионизационного предела. Энергии
этих состояний приняты по рекомендации Мур [2053]. Валентные состояния, принадлежащие
другим электронным конфигурациям Т1, по-видимому, лежат выше ионизационного предела и не
учитывались в расчете.
Термодинамические функции Т1 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
значений при Т ^ 3000 К не превышают 0,02 Дж-К~1-моль и обусловлены только неточностью
значения R. При более высоких температурах погрешности в S° (Т)ш С°р (Г) увеличиваются из-за
пренебрежения ридберговскими состояниями (практически не изменяясь в Ф° (Т)) и могут
достигать 1 и 6,5 Дж-К^-моль в значениях S° A0 000 К) и С], A0 000 К) соответственно.
Ранее таблицы термодинамических функций Т1 (г) вычислялись в ряде работ, в том числе в
работе Гурвича [97]доЗООО К, Поландаидр. [2241 ] до 7000 Ки Хилзенратаи др. [1436] до 10 000 К.
Результаты этих расчетов при всех температурах в пределах 0,02—0,03 Дж-К-моль~1 совпадают
с данными настоящего справочника.
Энтальпия образования Т1 (г)
t±jH°{T\, г, 0) = 181,4 ± 1,5 кДж-моль
соответствует принятой в справочнике величине Д,ЯС (Т1, к, 0) (см. Т1 (к, ж)).
Т1+ (г). Термодинамические свойства газообразного положительного иона таллия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 715.
Уровни энергии иона Т1+, использованные в расчете термодинамических функций, приведены
в табл. 24.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
213
*10 (г)
Глава 24
Таблица 24.3. Уровни энергии атомов Т1 и Т1+, принятые для расчета термодинамических функций
Атом
Tl
Электронная
конфигурация
... 6s26p
...6s26d
...6s25/
...6s26/
2 p
2 p
2?4
2DS,
гр
гр
т,
см
0
7 792,7
36117,9
36199,9
42 318,4
44 823,5
Pi
2
4
4
6
14
14
Tl
T1+
Электрон-
фигурация
...6s6p2
...6s2
.. . 6s6p
15
3D
3D
3 P
CM
45 220
0
49 451
52 393
61725
75 660
Pi
2
1
1
3
5
3
4 . .6s2 и . . .Qs6p и имеющие энергии не выше 100 000 см х. Энергии этих состояний приняты по
рекомендации Мур [2053].
Термодинамические функции Т1+ (г) вычислены по уравнениям A. 3)—A. 6), A.9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
значений во всем интервале температур не превышают 0,02 Дж-К~1-моль и обусловлены главным
образом неточностью принятого значения R.
Ранее таблицы термодинамических функций Т1+ (г) вычислялись Грином и др. [1246] до 5000 К
и Хилзенратом и др. [1436] до 10 000 К. Вычисленные в этих работах значения совпадают с
данными настоящего справочника в пределах указанных выше погрешностей.
Константа равновесия реакции Т1+ (г)+е (г)=Т1 (г) рассчитана на основании значения
Д^Р @) = —589,361+0,001 кДж-моль, соответствующего потенциалу ионизации атома таллия
70(Т1)=49 266,7 + 0,1 см-1=589,361±0,001 кДж-моль.
Это значение было принято на основании расчета предела ридберговской серии . . .Gs2ns BS)
(га=7+43) атома Т1, изученной в работе Ривса и др. [2322]. Принятому значению /„ (Т1)
соответствует также
&fH°(n+, г, 0) = 770,761 + 1,5 кДж-моль.
ТЮ (г). Термодинамические свойства газообразного оксида таллия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 716.
Молекулярные постоянные ТЮ, использованные для расчета термодинамических функций,
приведены в табл. 24.4. Экспериментальные данные о спектре, структуре и стабильности молекулы
ТЮ фактически отсутствуют. Единственным прямым экспериментальным подтверждением
существования у молекулы ТЮ связанного основного состояния является ИК-спектр этой молекулы,
зарегистрированный Осиным и др. [304а] при изучении реакцпи T1+N2O в матрице из аргона.
По аналогии с молекулами АЮ, GaO и InO в справочнике принимается, что молекула ТЮ имеет
основное состояние X2S+ (конфигурация . . ,т?о) и низкорасположенное состояние А2И{
(конфигурация. . . л3о2), хотя нельзя исключить, что у ТЮ эти состояния расположены в обратном порядке.
Безуспешность попыток получения электронного спектра ТЮ в видимой области и ближнем
ультрафиолете позволяет предполагать, что эта молекула не имеет других связанных валентных
состояний с энергиями меньше 25 000—30 000 см.
Молекулярные постоянные ТЮ в состоянии Х2?+, приведенные в табл. 24.4, основаны на
значении основной частоты F32 см), наблюдавшейся в ИК-спектре молекул ТЮ, изолированных
в матрице из аргона [304а], а также на оцененных значениях энергии диссоциации этой молекулы
(см. ниже) и ее вращательной постоянной. Последняя была получена из сопоставления
межатомных расстояний в молекулах МО и М2О, где М=А1, Ga, In, и значения г (Т1—О) в молекуле Т12О.
Погрешности приведенных в табл. 24.4 постоянных и>ви Ве оцениваются в 20 и 0,02 см. Энергия
состояния А2Л оценена по соотношению A.89) с некоторой корректировкой ее величины по
имеющимся экспериментальным данным для других молекул, ее погрешность не превышает 3000 см.
Термодинамические функции ТЮ (г) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн# и ее производных рассчитывались по уравнениям A. 90)—
214
Таллий и его соединения
ТЮ (г)
Таблица
Молекула
24.4. Молекулярные постоянные ТЮ, Т1Н, T1F, Т1С1
Состояние
Т.
«Л
в.
аг ¦ 10а
D, • 10»
СМ"
г.
А
ТЮ Х22+ 0 640б 5,3 е 0,26а 0,2в 0,2 г 2,10а
Л2П< 2000а ______
205ТШ Х1^ 0 1390,7 22,7 4,806 15,4а 325г 1,870
а3П0+ 17 723,4 1043,3 142,1 4,97б 71б 568г 1,839
о3П2 23 654,6в — — 1,53 Д — 3500г 3,32 е
Am 24181 140 21 2,027 Д — 2700г 2,88 е
2o;,T1ieF х12+ 0 477,3 2,3 0,223 149 9 0,150 353 a 0,179б 2,084 44
а3Д0+ 35186,0 436,3 7,1 в 0,2309 0,274г 0,281 Д 2,049
а3Лг 36 863,1 366,6 10,22 е 0,2249 0,307ж 0,3353 2,076
Am 45 430 й 345к — — — — —
205Тр5С1 х1 S+ 0 283,75 0,818 0,0913 966 6 0,039 793 2a 0,037 53 2,484 83
а3П0+ 31 049,4 223,1 11,4б 0,092 27 0,131 0,075 2,473
Примечание. Ниже все постоянные даны в см".
ТЮ: а опенка; ° вычислено по значениям Д(?,д =630 и Do= 19000; в вычислено по соотношению A.69); г
вычислено по соотношению A.68).
Т1Н: а я2=4,4-10-3; б постоянные описывают только значения So, Bi, i52; B3=2,98, JS4=2,86, S5=2,78, Вв—2,69;
в приведено значение Го; г приведено значение Do; д приведено значение Во', е приведено значение г0,
T1F: а а2_3,0Ы0-6; б Pi=4,8-10-»; в ^eye=0,i; г а2——1,35-Ю; Д рх=—2,3-10"8; е швг/е__1,15;
ж otj——9-10~4; 3 pi——1,09-Ю, В2=1,05-10~8; и приведено значение Го; к приведено значение шо-
Т1С1: а а2=4,17-10-7; й и>еуе—\,ЪЪ, ше2в—0,115, в работу [2023], по-видимому, вкралась опечатка, и приведено
значение <¦>„?„=—0,115, воспроизведенное в ряде справочных изданий.
A.92) с учетом состояния А2И в предположении, что QK^.BV = ^Qr^,,n-
Колебательно-вращательная статистическая сумма'состояния Х22+ и ее производные вычислялись по уравнениям A. 73) —
A. 75) непосредственным суммированием по колебательным уровням и интегрированием по
вращательным уровням энергии с помощью уравнения типа A. 82). В расчетах учитывались все
уровни энергии со значениями /^/тах> „, где /шах>„ находились из условий A. 81). Колебательно-
вращательные уровни состояния Х2Е+ вычислялись по уравнениям A. 65), A. 62), значения
коэффициентов в этих уравнениях, тождественные постоянным из табл. 24.4, а также величины иша1
и /Иш приведены в табл. 24.5. Мультиплетность вращательных уровней состояния
Х22+учитывалась статистическим весом.
Погрешности рассчитанных термодинамических функций ТЮ (г) при низких и умеренных
температурах обусловлены в основном отсутствием данных об относительном расположении состояний
3?+ и 2П<5 а также неточностью принятого значения Ве; при высоких температурах становятся
существенными неточность оценок всех постоянных и отсутствие данных о других возбужденных
состояниях ТЮ. Погрешности табулированных значений Ф° (Т) могут достигать 8—10 Дж-К~хХ
Хмоль во всем интервале температур.
Термодинамические функции ТЮ (г) раньше вычислялись в работе Бруэра и Розенблата [745]
(значения Ф° (Т) для Т ^ 3000 К) по оцененным постоянным; расхождения данных [745] и
табл. 716 составляют 4—5 Дж-К~1-моль~1.
Константа равновесия реакции ТЮ (г)=Т1 (г)+О (г) вычислена на основании принятой в
справочнике энергии диссоциации
Дг#° @) — Do (ТЮ) = 230 + 40 кДж • моль« 19 000 ± 3500 см.
Эта величина была оценена на основании сопоставления энергии диссоциации молекул МО и
MX, а также энергий разрыва связей М—ОН и МО—М, где М=А1, Ga, In и Tl, X=F, G1, Вг.
Принятому значению соответствует
г, 0) = 198,183 ± 40 кДж • моль.
215
Т120 (к, ж)
Глава 24
Т12О (к, ж). Термодинамические свойства кристаллического и жидкого оксида диталлия в
стандартном состоянии при температурах 298,15—4000 К приведены в табл. 717.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 24.1. В справочнике за стандартное состояние Т12О в интервале 0—852 К принята
моноклинная модификация [2420, 2710] х. В литературе отсутствуют экспериментальные данные по
Таблица 24.5. Коэффициенты в уравнениях, описывающих уровни энергии (в см), а также значения
''max и t/lim, принятые для расчета термодинамических функций ТЮ, Т1Н, T1F и Т1С1
Молекула
Электронное
состояние
ТЮ
тш
+ a
7>10~4 0 0
Ую-Ю-3 0,640 1,391602
Узо-Ю —0,53 —2,345 003
Узо-Ю1 - 2,288 140
У40-Ю4 — —230,177
У5О-Ю9 - -
Ум 0,26 4,807 93
Уц-Ю2 —0,2 —16,022 3
У21-Ю4 - 80,327 5
Уз1-Ю4 — —5,304 04
Уо2-106 -0,2 —325,0
Уоз-Ю11 -0,0165 2196,89
уо4.101* _ —0,724 555
(an=De)-l0-i — 1,659 997
ах — 0,325 287
а2-Ю4 — 0,915 072
аз-10е — 0,027 168
Ушах 59 19
JUm 379 81
Примечание. Ниже все величины даны в см'1.
ТЮ: а Те (Л2П) = 2000. Т1Н: а Те (а3П0+) = 17 723,
АШ
2,418 1
0,140
-2,1
—
—
2,25
—45,0
—
—
—2 700,0
324 000,0
-358 899,0
0,024 0
0,218 819
6,908 643
412,996
3
14
4. TIF: a Te {(
T1F
0
0,477 350
—0,233 131
0,053 115
—0,049 941
—
0,223 150
—0,150 353
0,030 1
—
—0,179
0,014 359
—
—
.—
—
—
239
554
ДП) = 36 863,1.
Т1С1
Х1
0
0,282 139
—0,080 586
—0,002 183
0,051 723
—7,172 155
0,090 036 98
-0,039 125
0,004 077
—
—0,036 69
—0,001 511
—
—
—
—
—
322
787
теплоемкости Т12О при Т ^ 298,15 К. Значения термодинамических величин при 298,15 К,
приведенные в табл. 24.1, оценены при помощи различных эмпирических методик, предложенных
в работах [216, 427, 814а], с использованием экспериментальных данных для теплоемкости Ag,0 (к)
и Си2О (к) при низких температурах. Погрешности принятых значений S° (X98,15 К) п
Н° B98,15 К)—Н° @) оцениваются в 8 Дж-К~1-моль и 1 кДж-моль соответственно.
Термодинамические функции Т12О при Т ]> 298,15 К вычислены по результатам измерений
энтальпии, выполненных Кубиччотти и Эдиктом [909] в интервале 298—914 К для образца Т1„О,
содержащего в виде примеси 0,3% металлического таллия. Уравнение для теплоемкости Т12О (к)
(см. табл. 24.1) выведено методом Шомейта по данным [909] C72—811 К), погрешность которых
оценивается в 1%. Температура (852+10 К) и энтальпия C0,3 + 1,0 кДж-моль) плавления Т12О
приняты также ло данным [909]. Результаты измерений [1380] E73 К) и [2719] (863 К и 36 кДж
Х) Т Т1О () A133 ДК1
Хмоль) менее надежны
принята на основании
Эдинга [909].
р ( ) (
и не учитывались. Теплоемкость Т1аО (ж) A13+3 Дж-К-моль)
измерений энтальпии в интервале 857—914 К в работе Кубиччотти и
1 Согласно данным [2719, 2720] Т12О (к) в интервале 568—633 К претерпевает полиморфное превращение, однако
характер превращения и структура высокотемпературной модификации не установлены. В справочнике при
расчете термодинамических функций Т12О (к, ж) это превращение не учитывалось.
216
Таллий и его соединения
Т12О (it
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются в 8Г.
10, 15 и 25 Дж-К~1-моль~1. Термодинамические функции Т12О (к, ж) вычислялись ранее в
справочнике Барина и Кнакке [578] до 1000 К. Результаты, приведенные в [578], значительно (до
15 Дж-К-моль~1 в Ф° A000 К)) отличаются от данных табл. 717, так как эти авторы не учитывали
работу [909].
В справочнике принята энтальпия образования
Д^СДД к, 298,15 К) = —167,4 + 2,0 кДж . моль.
Это значение является средневзвешенным из приведенных в табл. 24.6 результатов измерений
в работах [905, 2382, 2686] (без учета данных [2382], полученных при растворении Т12О (к) ш
Т1 (к) в H2SO4).
Таблица 24.6. Результаты
Авторы
Томсен, 1882 [2686]
Рот, Мейхснер, 1932 [2382]
Кубиччотти, 1969 [905]
определений AfH° (Т12О, к, 298,15 К) (в кДж-мол!
Метод
Измерение энтальпии растворения Т12О (к) в воде,
291—293 К, 3 измерения
То же, 293 К, 7 измерений
Измерение энтальпии растворения Т12О (к) и Т1 (к)
в серной кислоте, 293 К, 4 измерения
Измерение энтальпии растворения Т12О (к) и Т1 (к)
в растворе (Вг2+НВг), 9 измерений
Д,Я° (Т12О, к,
298,15 К)
-167,8±3,0
-167,0+2,0
—180,7+10
—169,0+6,0
Наиболее точные величины были получены на основании измерений энтальпии растворения
Т12О (к) в воде и принятых по [406] энтальпий разведения растворов ТЮН и энтальпии
образования Tl+ (aq). Измерения энтальпии растворения Т12О (к) в воде также проводились в работах [545,
1120], однако их авторы не смогли получить надежные данные для измеряемой величины. Давление
пара оксида диталлия в реакции Т12О (к)=Т12О (г) вычислено с использованием значения
Д8#° @)=200,495 + 7,3 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования Т12О в кристаллическом и газообразном состояниях.
Т12О (г). Термодинамические свойства газообразного оксида диталлия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 718.
Молекулярные постоянные, использованные для расчета термодинамических функций Т12О,
приведены в табл. 24.7. Толмачев и Рамбиди [2708] на основании электронографического
исследования Т12О установили, что средней в условиях эксперимента (Г=1000 К) является нелинейная
конфигурация с -^Т1—О—Т1 = 133+5°, но в случае низкой частоты деформационного колебания
равновесной структуре Т12О может отвечать ^Т1—О—Tl = 150-f-180°. Недавно Спиридонов и др.
[2595г] провели новое электронографическое исследование Т12О, использовав предложенную ими
методику структурного анализа, позволяющую определять одновременно равновесную струк-
Таблица 24.7. Значения молекулярных постоянных, а также а и pj, принятые для расчета
термодинамических функций Т!2О, ТЮН, T12F2 и Т12С12
Молекула
Электронное
состояние
vi
¦'в
СМ
Т12О Х1А 400
ТЮН 11Z 460
T12F2 lM 297
Т12С12 ХМ 190,4
ТЮН: а приведено значение I-103?
120
300 B)
93
61
г-см2.
643
3600
226
131
81
40
316
189,6
257,5
170,6
111 1ПП7
'А1 В1 С IU
г3 ¦ см»
76-Ю3
13 а
6,8-106
3,7-106
а
2
1
4
4
Рх
1
1
1
1
I
217
Т120 (г) Глава 24
туру и силовое поле молекулы. Авторы [2595г] пришли к выводу, что равновесная структура
молекулы Т12О является нелинейной с -$Т1—О—Т1=145°.
В ИК-спектрах газообразного Т12О [240, 445] и молекул Т12О, изолированных в матрицах из
инертных газов [759], наблюдались две полосы, отнесенные к частотам валентных колебаний
Vj и v3. Это привело авторов [240, 445, 759] к заключению о нелинейной структуре Т12О. Из
соотношения значений v3 разных изотопических модификаций этой молекулы Бром и др. [759]
рассчитали значения валентного угла в пределах 77—104° в зависимости от матрицы. Несколько
большее значение угла A31 + 11°) получили Хинчклифф и Огден Ц437] в результате изучения
ИК-спектра Т12О в матрице из N2.
На основании совокупности этих данных в справочнике принято, что в основном электронном
состоянии ХХА молекула Т12О имеет нелинейную структуру симметрии С2в. Приведенное
в табл. 24.7 произведение моментов инерции вычислено для значений re (TI—О)=2,10+0,02 А
п ^„Tl—О—Т1=145+5°, найденных Спиридоновым и др. [2595г]. Погрешность значения IaIbIc
не превышает 2-1013 г3-см6.
ИК-спектр Т12О исследовался как в матрицах инертных газов и азота [759, 1437, 1927], так и
в газовой фазе [240, 445], однако достаточно надежно определена только частота v3. Приведенное
•в табл. 24.7 значение v3 принято по результатам исследования ИК-спектра молекул Т12О,
изолированных в матрице из Аг [759, 1927]. Его погрешность с учетом возможного матричного сдвига
оценивается в 30 см. В работе Маковецкого и др. [1927] к частотам Vj^ и v2 отнесены полосы 510
и 382 см, наблюдаемые в ИК-спектре молекул Т12О, изолированных в Аг-матрице. Однако
позднее было показано, что эти полосы не принадлежат молекуле Т12О [1438]. Мальцев и Шевельков
[240, 445] в ИК-спектре газообразного Т12О наряду с самой интенсивной полосой ~620 см
(v,) наблюдали полосу 480 см, которую на основании изучения зависимости интенсивности
полос от температуры отнесли к частоте симметричного колебания ух. С этим отнесением не согласны
Бром и др. [759], которые полагают, что полоса 480 см принадлежит димеру Т14О2. Бром и др.
[759] исследовали ИК-спектры молекул Т1216О и Т1218О, изолированных в матрицах из Аг, Кг и
N.2, п изучили зависимость интенсивности полос от температуры.
Из пяти полос средней интенсивности, наблюдаемых в ИК-спектре молекул Т1216О,
изолированных в матрице из Аг, только интенсивность полосы 571 см при нагревании падала так же
быстро, как интенсивность полосы v3. Аналогичным было поведение полос 545 и 609 см в спектре
TL1SQ. Это и послужило основанием для отнесения полос 571 см (Т1218О) и 545 см (Т1218О) к
частоте vL 1. Однако следует отметить, что в качестве полос, которые можно было бы отнести к
молекуле Т12О, авторы [759] не рассматривали слабые полосы 362 и 447 см, интенсивность которых
также уменьшалась при нагреванип. Спиридонов и др. [2595г] при обработке результатов
электронографического эксперимента, в предположении, что v3 = 643 см, рассчитали силовое поле
молекулы Т12О и нашли vx=134 см и v2=121 см.
Таким образом, имеющиеся данные о величине v1 весьма противоречивы и в настоящее время
трудно сделать однозначный выбор. По сравнению с принятыми в справочнике величинами \
молекул Ga2O и 1п2О, значение Vj, определенное Бромом и др. [759], выглядит завышенным, а
вычисленное Спиридоновым с сотрудниками [2595г] — слишком низким. Поэтому в справочнике
частота vx, а также не наблюдавшаяся в спектре частота v2 рассчитаны по уравнениям поля
валентных сил, на основании значения v3 и соотношений /rr=0,l fr и /а/г2 = 0,05 /,. Первое соотношение
соответствует наблюдаемым частотам vx и v3 молекул Ga2O и 1п2О, второе — принято из сравнения
с молекулами типа XY2. Погрешности в частотах vx и v2 оцениваются в 100 и 40 см соответственно.2
Возбужденные электронные состояния Т12О в справочнике не рассматриваются, так как согласно
предварительным результатам исследования электронного спектра [445] они лежат в области
~30 000 см.
Термодинамические функции Т12О (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены неточностью молекулярных постоян-
1 В ИК-спектре молекул Т12О, изолированных в матрице из N2, Бром и др. [759], так же как Хинчклифф и
Огден [1437], наблюдали только полосу v3.
2 В работе [943а] Спиридонов с сотр. уточнили анализ результатов электронографического исследования
Т!2О и нашли v1 = 324, v2 = 71 и v3 = 654 см".
218
Таллий и его соединения Т12О3 (к, ж)
ных и приближенным характером расчета; они могут составлять 5—10 Дж-К~1-моль в значениях
Ф° (Т) в интервале 100—6000 К.
Термодинамические функции Т12О (г) вычислялись ранее в работах [447, 906] до 1000 и 2000 К.
Расхождения результатов этих расчетов и данных табл. 718 не превышают 2,5 Дж-К"а-моль
в значениях Ф° (Г), они обусловлены некоторой разницей в принятых молекулярных постоянных.
Константа равновесия реакции Т12О (г)=2Т1 (г)+О (г) вычислена с использованием значения
A,J7O @)=573,583+ 10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^-Я^Т^О, г, 0) = 36 + 10 кДж-моль.
Это значение основано на измерениях Кубиччотти и Кенеши [909а] (метод протока, реакция
Т12О3 (к)=Т1аО (г)+О2(г),803—948 К, 6 точек, Д^0 @)=9 кДж-моль при расчете по II закону
и 36+4,6 кДж-моль при расчете по IIIзакону).Погрешность принятого значения Д^Я0 помимо
воспроизводимости результатов измерений учитывает также погрешность, обусловленную
неточностью термодинамических функций (~8 кДж-моль). К значению 40 кДж-моль, близкому
к принятому, приводит исследование испарения Т12О (к), проведенное Малфордом (см. [906]).
Аналогичные измерения, выполненные Шахтахтинским и Кулиевым [443] D70—590 К, 8 точек),
а также изучение сублимации Т12О3 (к) в атмосфере кислорода в работе Щукарева и др. [468]
(915—1030 КIпривели к существенно отличающимся значениям, равным —36 и +10 кДж-моль
•соответственно.
Т12О3 (к, ж). Термодинамические свойства кристаллического и жидкого триоксида диталлия
в стандартном состоянии при температурах 298,15—3000 К приведены в табл. 719.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 24.1. В справочнике за стандартное состояние Т12О3 (к) в интервале 0—1107 К принята
кубическая модификация (структурный тип а-Мп2О3) [2179] 2. Ввиду отсутствия данных по
теплоемкости Т12О3 (к) при низких температурах, значения термодинамических величин при 298,15 К,
приведенные в табл. 24.1, оценены на основании экспериментальных данных для теплоемкости
Ga2O3 (к) с использованием эмпирических методик [216, 427, 814а]. Погрешности принятых
значений S° B98,15 К) и Я0 B98,15 К)—Я0 @) оцениваются в 8 Дж^К^-моль и 1 кДж-моль
соответственно.
В интервале 298,15—1107 К для теплоемкости Т12О3 (к) принято уравнение (см. табл. 24.1),
выведенное методом Шомейта при совместной обработке результатов измерений энтальпии Т12О3,
проведенных Милсом [2030] B98—800 К) и Кубиччотти и Эдингом [909] B98—1000 К). При этом
погрешности данных [2030] оценивались в 0,5%, а данных [909] — в 1%. Принятая температура
плавления Т12О3 A107 + 10 К) найдена Кубиччотти и Эдингом [909]. В работах [326, 992, 1178]
были получен^ более низкие значения (в пределах 968—1043 К), которые представляются менее
надежными и не учитывались в настоящем справочнике. Энтальпия плавления Т12О3 E3+10 кДж X
Х^юль) оценена в предположении о равенстве энтропии плавления Т12О3 и А12О3. Теплоемкость
Т12О3 (ж) A60+30 Дж-К-моль~1) принята равной теплоемкости А12О3 (ж) (см. гл. 21).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 8,
10, 20 и 30 Дж-К~1-моль~1 соответственно.
Термодинамические функции Т12О3 (к), приведенные в справочнике Барина и Кнакке [578],
существенно (до 23 Дж-К-моль при 1000 К) отличаются от данных табл. 719.
Принятая в справочнике энтальпия образования Т12О3 (к)
Д//° (Т13О8, к, 298,15 К) = —387,0 + 4,0 кДж • моль
основана на калориметрических измерениях энтальпий растворения Т12О3 (к), Т1 (к), Н2О (ж)
в растворе брома в бромистоводородной кислоте, выполненных Кубиччотти [905]. В литературе
известны также исследования реакции испарения Т12О3 с разложением [407, 468, 909а, 992, 2301а],
1 Авторы [468] полагали, что испарение происходит по уравнению Т12О3 (к)=Т12О3 (г). Поскольку Т12О3 в
газовой фазе, по-видимому, не существует, результаты работы [468] пересчитаны в предположении, что продуктами
испарения являются Т12О (г) и О2 (г).
я Согласно данным [2420а] при нагревании Т12О3 в вакууме A0~5 мм рт. ст.) при ~673 К образуется нестехио-
метрическая фаза приближенного состава Т12О2|96 с измененным параметром кубической решетки.
219
ТШ (г) Глава 24
однако результаты этих работ противоречивы, поскольку авторы предполагают различные
уравнения реакции разложения. Измерения растворимости Т12О3 (к) в растворе NaC104 [2453a] привели
к значению энтальпии образования —410 кДж-моль.
Константа равновесия реакции Т12О3 (к)=2Т1 (г)+30 (г) вычислена с использованием значения
Aji/0 @) = 1483,662+5,0 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования Т12О3 (к), Т1 (г) и О (г).
ТШ (г). Термодинамические свойства газообразного гидрида таллия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 720.
Молекулярные постоянные ТШ, принятые на основании результатов исследований
электронного спектра этой молекулы, представлены в табл. 24.4. В спектре ТШ наблюдались только три
системы полос a'U^^tX1^* [1299, 1813, 2123], а3П2 «-Х1!- [1813] и Л 41 «- ХХЕ+ [1813, 1814].
По аналогии с молекулами GaH и InH, в справочнике принимается, что основное электронное
состояние ХХ2+ коррелирует с диссоциационным пределом Т1 (?Рч)-\-Н B5), а состояния а3П0+г
а3П2 и 441 — с пределом Tl BP»/2)+H BS) г. В спектрах ТШ не наблюдались переходы, связанные
с состояниями 3П0- и 3П15 которые также должны коррелировать с первым диссоциационным
пределом и соответствовать конфигурации . . .а2атг (см. GaH и InH). Ha основании сопоставления
энергий состояний а3П0+ и а3П2 (табл. 24.4) с величиной Do (ТШ)=15 910+30 ем (см. ниже)
в справочнике принимается, что состояния а3Пс- и asH1 молекулы ТШ являются отталкиватель-
ными. В спектре поглощения ТШ в области XX 4000—4400 А наблюдается несколько континуумов,
возможно связанных с этими состояниями.
Приведенные в табл. 24.4 значения постоянных ТШ в состоянии Х1Е+ найдены Грундстрёмом
и Вальбергом [1299] из анализа структуры 16 полос перехода а3П0+—Хх? + (v' ^ 6, v" ^4).
Колебательно-вращательные уровни состояния а3П0+ сильно возмущены и не могут быть описаны
уравнениями обычного вида, в связи с чем авторы [1299] дали ошибочное отнесение верхних
уровней изученного перехода. Приведенные в табл. 24.4 постоянные ТШ в этом состоянии получены
Нейхаузом и Мульдом [2123] и Ларссоном и Нейхаузом [1813] в результате исследования
переходов а3П0+—X1 Е+ в спектре T1D (г/ ^ 6) и совместной обработки экспериментальных данных для
двух изотопических модификаций этой молекулы. Соответствующие вращательные постоянные
удовлетворительно описывают значения Bv только для i;'=0-f-2. Значения Те, ше и шехе,
рассчитанные авторами [2123], имеют невысокую точность и не описывают значения Go (v) при v ^> 2
(по данным [2123] ДбЦ'3—¦Д&"/, равны 393,0, 402,2, 416,3 и 426,1 см). Исследование перехода
а8П2—Х1!!* в работе Ларссона и Нейхауза [1813], которые проанализировали структуру полосы
0—0, показало, что у молекулы ТШ состояние а3П2 является связанным только при v=6.
Найденные авторами [1813] постоянные ТШ в этом состоянии приведены в таблице. Постоянные ТШ в
состоянии Л 41 определены Ларссоном и Нейхаузом в результате анализа полос 0—0, 1—0 и 2—О
перехода Л41—Х1!^ в спектрах молекул ТШ и T1D [1813, 1814].
Термодинамические функции ТШ (г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись по уравнениям A. 90)—A.92)
с учетом возбужденных состояний а3По+ и Л1П. Колебательно-вращательные составляющие для
состояний Xх S + и А1!! находились непосредственным суммированием по уровням энергии каж~
дого состояния с помощью соотношений A. 72)—A. 75), в расчете учитывались все уровни,
ограниченные предельными кривыми диссоциации этих состояний. Составляющие состояния а3П(.+
вычислялись в предположении, что ()<°>_ вР = (?^ вр, это упрощение было сделано в связи с
невозможностью аппроксимировать колебательно-вращательные уровни этого состояния с v ^> 2
постоянными, полученными из данных для уровней с v ^ 2 (см. выше). Сопоставление энергии
последнего колебательного уровня этого состояния (Гоо+С?о F) = 19 797 см) с энергией его диссоциа-
ционного предела B3 716 см) и величиной Д6?г-р/> ПРИ *"=2-^5 (~400 см) позволяет
предполагать, что общее число связанных и квазисвязанных колебательно-вращательных уровней состояния
а3П0+ по крайней мере не меньше, чем в состоянии X1!!*. Колебательно-вращательные уровни
состояний ХХЕ+ и А1!! вычислялись по уравнениям A. 65) и A. 60), значения коэффициентов Ykl
в этих уравнениях, величин i;raax и /Иш, а также коэффициентов а( в уравнениях, описывающих
предельные кривые диссоциации обоих состояний, приведены в табл. 24.5. Коэффициенты Y30>,
Yt0 и Yn, Y31 для состояния Хх?+ получены из условий A. 50) и A. 57) соответственно, коэффи--
1 В работах [1219, 1814] отмечается неоднозначность корреляции электронных состояний Т1Н.
220
Таллий и его соединения Т1ОН (г)
циенты YQ3 и Yoi для обоих состояний — из условий A. 59). Вырождение уровней состояния А1!!
учитывалось статистическим весом 2.
Молекулярные постоянные Т1Н в состоянии Xх ?+ известны с хорошей точностью и
погрешности рассчитанных термодинамических функций при Т ^ 1500^-2000 К обусловлены только
неточностью фундаментальных постоянных и не превышают 0,02 Дж-К-моль. При более
высоких температурах становятся существенными отсутствие данных об уровнях энергии этого
состояния ci?)>4, приближенный учет составляющих состояния а3По+, а также возможность
существования у молекулы Т1Н связанных состояний с энергиями 30 000—40 000 см. Пренебрежение
в расчете состоянием а3П2, имеющим только один связанный колебательный уровень, не влияет
на точность вычисленных величин во всем температурном интервале. Погрешности в значениях
Ф° (Г) при 3000 и 6000 К оцениваются в 0,04 и 0,3 Дж-К^-моль.
Термодинамические функции Т1Н (г) вычислялись ранее в справочниках [1336] (Т ^ 5000 К),
[1656, 1658] (Т ^ 2000 К). В работе [2463] приведены коэффициенты полиномов,
аппроксимирующих значения Qm и In Qm для Т=1000—3700 К. Расхождения данных Хаара и др. [1336]
и табл. 720 достигают 0,3, 0,7 и 2 Дж-К^-моль в значениях Ф° D000 К),6ГОD000 К) и С), D000 К),
они обусловлены использованием в [1336] приближенной методики и пренебрежением всеми
возбужденными состояниями молекулы Т1Н, хотя эти два источника ошибок частично компенсируют
друг друга. Расхождения с данными [1656, 1658] достигают 1—2 Дж-К~1-моль~1 при У=1000 —^~
—2000 К. Результаты расчета [2463] и табл. 720 при Т <^ 3000 К согласуются в пределах
0,3 Дж-К^-моль.
Константа равновесия реакции Т1Н (г)=Т1 (г)+Н (г) вычислена на основании значения
энергии диссоциации
Ar#°@) = Do(TlH)= 15 925 + 15 см == 190,50 + 0,18 кДж ¦ моль.
Эта величина получена из анализа предиссоциации при вращении молекул Т1Н и T1D в
состоянии ЛШ, которая исследовалась в работах Ларссона и Нейхауза [1813, 1814]. Предельная кривая
диссоциации, построенная по точкам начала предиссоциации в спектрах Т1Н (i>=l, /=6 и v=2,
/ = 3) и T1D (v=2, / = 8), имеет вид типичный для состояний, потенциальные кривые которых не
имеют максимума при отсутствии вращения. Короткая экстраполяция предельной кривой
диссоциации дает для энергии диссоциационного предела состояния А1!! значение 23 716 см над
уровнем v=0 состояния X1!" молекулы Т1Н. Источником неточности этого значения может быть
туннельный эффект, способный приводить к распаду ТШ при / <^/тах и заниженной величине Do,
а также то, что начало предиссоциации определялось только по спектру поглощения, т. е.
методом менее чувствительным, чем обрыв структуры в спектре испускания, и способным давать
завышенное значение Do. За верхний предел этой величины можно принять энергию уровня v=2,
/=8 состояния 441 молекулы T1D над уровнем v=0, /=0 состояния X1!,* молекулы Т1Н
B3 732 см), за нижний предел — энергию уровня v=0, 7 = 1 состояния А1!! молекулы ТШ
B3 710 см).
Спектрофотометрическое исследование равновесия реакции образования ТШ (г) в пламенах,
выполненное Булевич и Сагденом [790], приводит к близкому, но существенно менее точному
значению A84+30 кДж-моль).
Принятому значению энергии диссоциации соответствует
\fH°{TlH, г, 0) = 206,934+1,5 кДж • моль.
ТЮН (г). Термодинамические свойства газообразного гидроксида таллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 721.
Молекулярные постоянные, использованные в расчете термодинамических функций ТЮН,
приведены в табл. 24.7. Спектр и структура молекул ТЮН экспериментально не исследовались, и
приведенные постоянные были оценены при подготовке справочника. Момент инерции в основном
состоянии ХХ2+, приведенный в таблице, соответствует структурным параметрам ^Т1—О—Н =
=180°, г (Т1—О)=2,25 + 0,05 А, г (О—Н)=0,96 + 0,02 А. Его погрешность оценивается в 2х
Х1О9 г-см2. Линейная структура принята по аналогии с молекулой АЮН, хотя нельзя полностью
исключить возможность угловой структуры ТЮН в основном состоянии. Величина г(Т1—О)
перенесена из Т12О, а для г (О—Н) принято такое же значение, как в других гидроксидах. Частота
\ оценена по значению о>е (T1F)=477,3 см, частота v2 — по частотам молекул гидроксидов щелоч-
221
T1F (к, ж) Глава 24
ных металлов. Для v3 принято характеристическое значение. Погрешности частот vx, v2 и v3 не
превышают 60, 60 и 100 см. Сведения о возбужденных электронных состояниях ТЮН отсутствуют,
по аналогии с молекулами T1F и Т1С1 можно предполагать, что их энергии превышают 25 000 см.
Термодинамические функции ТЮН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении модели «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных термодинамических функций обусловлены главным
образом неточностью принятых значений молекулярных постоянных, в том числе структуры ТЮН,
и в меньшей степени приближенным характером расчета, они составляют 5—10 Дж-К~1-моль
в значениях Ф° (Т).
Таблица термодинамических функций ТЮН (г) публикуется впервые.
Константа равновесия реакции ТЮН (г)=Т1 (r)+O (r)-f-H (г) вычислена с использованием
значения Дг Н° @)=724,217 + 20 кДж-моль, соответствующего принятой в справочнике
величине
Д//°(ТЮН, г, 0) = —80+20 кДж.моль.
Принятая величина основывается на значении энергии связи Т1—ОН ^ 300 кДж-моль,
полученном в работах Гурвича и др. [96, 98], Булевич и Сагдена [790] и Келли и Педли [1665].
T1F (к, ж). Термодинамические свойства кристаллического и жидкого фторида таллия в
стандартном состоянии при температурах 100—2300 К приведены в табл. 722.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 24.1. В справочнике за стандартное состояние T1F (к) в интервале 0—356,3 К принята
ромбическая а-модификация [1675а], а в интервале 356,3—599 К тетрагональная [3-модификация [2232].
При Т <^ 298,15 К термодинамические функции T1F (к) вычислены по результатам измерений
теплоемкости, проведенных Лайоном и др. [1898] F—445 К) в адиабатическом калориметре на
образце, содержащем 91,57 + 0,08% Т1 (теоретическое содержание 91,49%). Погрешности этих
измерений оцениваются в 3% при 6 К, 0,5% при 10 К и ~0,1% выше 20 К. Погрешности принятых
значений S0 B98,15 К) иН° B98,15 К)—Я0 @) составляют 0,20 Дж-К^-моль и 0,02 кДж-моль
соответственно.
В интервале 298,15—356,3 К для теплоемкости a-TIF принято уравнение, выведенное методом
Шомейта по результатам измерений энтальпии, проведенных Кубиччотти и Эдингом [908]
B98—850 К), и измерений теплоемкости, выполненных Лайоном и др. [1898]; погрешности
данных [1898] и [908] оценивались в 0,5 и 1% соответственно. Температура C56,3 + 0,5 К) и
энтальпия @,336+0,002 кДж-моль) полиморфного превращения приняты по наиболее точным
калориметрическим измерениям [1898]; результаты менее надежных определений [580, 817, 837, 866,
908, 2232 и 2724] приводят к значениям 7^=354+358 К и ДггЯ=0,385 кДж-моль [908].
В интервале 356,3—599 К для теплоемкости C-T1F принято уравнение (см. табл. 24.1),
выведенное совместной обработкой данных [1898] и [908], их погрешности оценены в 0,5 и 2%
соответственно. Температура плавления T1F E99 + 3 К) выбрана в результате усреднения данных [141,
837, 908, 1373, 2724]. Энтальпия плавления A3,87 + 0,40 кДж-моль) принята по работе
Кубиччотти и Эдинга [908], это значение более точное, чем найденное в работе [2724] A3,56 кДж-моль).
Теплоемкость жидкого T1F F7,3+0,2 Дж-К~1-моль~1) основана на измерениях энтальпии в
работе [908] E99—850 К).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 0,2, 1,5 и
5 Дж'К-моль~1 соответственно.
Термодинамические функции T1F (к, ж), приведенные в справочнике [578] (Т<^1000 К),
незначительно (до 0,5 Дж-К~1-моль) отличаются от данных табл. 722.
В справочнике принята энтальпия образования фторида таллия
bfH°(T\F, к, 298,15 К) — — 329,3+ 1,5 кДж • моль.
Это значение является средним взвешенным из величинш полученных при измерении
энтальпий растворения T1F в воде [890] (три точки, AfH° (T1F, к, 298,15 К) = — 330,2 + 1,5 кДж-моль)
и [910] (четыре точки, —327,9 + 1,2 кДж-моль) * и калориметрических измерений энтальпии реак-
1 Результаты измерений графически экстраполированы к бесконечному разведению. В расчетах использованы
значения стандартных энтальпий образования ионов Т1+(ад) [406] и F~(aq) по работе [864].
222
Таллий и его соединения T1F (г).
ции T1F (к)+К1 (р-р)=ТИ (k)+KF (р-р) и энтальпии образования ТИ (к) [910] (Д JT (T1F, к,
298,15 К) = -330,9,+1,7 кДж-моль).
Давление пара фторида таллия в реакции T1F (k)=T1F (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
/±SHO{TIF, к, 0) = 146,0+ 3,0 кДж-моль.
Это значение основано на измерениях давления насыщенного пара T1F (к) в работе Барроу
и др. [598а] (торзиоиный метод, 488—587 К, 55 точек) и измерениях парциального давления
мономера, выполненных Кенеши и Кубиччотти [1670] (статический метод и метод протока, 800 К,
1 точка), а также на определении Do (T1F) с помощью фотоионизационных измерений в работе
Берковица и Уолтера [666]. Обработка данных [598а] с учетом присутствия в парах над
фтористым таллием молекул T1F и TI2F2 и данных [1670] приводит к совпадающим значениям 147 +
+4 кДж-моль (II закон), 145,7 + 1,5 кДж-моль (III закон). Основную неточность в эти
значения вносят погрешности термодинамических функций T1F (к, ж), а также расчет парциального
давления TIF в парах из данных [598а]. Энергии диссоциации TlF, найденной Берковицем и
Уолтером [666] D41 + 2 кДж-моль), соответствует ^fH° @) = 147,6 + 2,1 кДж-моль в хорошем
согласии с величинами, рассчитанными из данных [598а, 1670]. Исследования Do (T1F) в работах [787,
2082] рассмотрены ниже (см. T1F (г)).
T1F (г). Термодинамические свойства газообразного фторида таллия в стандартном состоянии
при температурах 100—6000 К приведены в табд. 723.
Молекулярные постоянные T1F, принятые в справочнике на основании результатов исследо^
ваний электронного и вращательного спектров этой молекулы, представлены в табл. 24.4. В
электронном спектре T1F наблюдались переходы а3П0+^Х1Е+, а'П^ГЕ* [593, 720, 1484, 1970]
i44I ->¦ Х1!* [593, 720, 1484]. Кроме того, в работе [1484] в спектре поглощения наблюдались,
неидентифицированные континуумы в области 11 2630, 2200 и 2000 А, возможно принадлежащие
переходам в несвязанные состояния. В системе АЩ ¦*-Х1Е+ наблюдались переходы только на
уровни v'=0 и 1, причем соответствующие полосы диффузны и, по-видимому, предиссоциированы.
Если состояние АгТ[ у T1F коррелирует, так же как у других галогенидов группы IIIA, с пределом
М BРз,2)+Х BPsj), энергия которого у этой молекулы составляет приблизительно 45 000 см,
состояние АгИ должно быть слабосвязанным. Корреляция подсостояний а3По+и а3!^неоднозначна;
можно предполагать, что они связаны с пределом Tl BPi/2)+F BА/2), причем подсостояние 3Па
имеет небольшое число F—7) связанных колебательных уровней. Какие-либо экспериментальные
данные о подсостояниях а3По- п asU2 отсутствуют. Оба они, вероятно, коррелируют с нижним
пределом Tl BPi/,)-j-F B-Р'/2), тогда подсостояние а8По- должно быть малостабильным и иметь не более
20 связанных колебательных уровней. Подсостояние а3П2 у молекулы T1F, в отличие от фторидов
других элементов этой группы, вероятно, является отталкивательным состоянием, так как при
г~ге (Х1!!*) оно должно иметь энергию примерно на 1000 см выше соответствующего диссоциа-
щюнного предела.
Приведенные в табл. 24.4 колебательные постоянные T1F в состояниях Х1Е+, а3П0+ и а3И1
приняты по краткому сообщению Барроу и др. [593]. Авторы [593] выполнили анализ
вращательной структуры полос 0—0, 1—1 и 2—2 перехода а?П1—ХХЕ+ и полос 0—0, 2—2 и 3—3 перехода
а3Пг.+—Х1Е+. Рекомендованные ими постоянные основаны на их собственных данных и
результатах исследования спектра T1F в работе Хауэлла [1484], который выделил большое число кантов полос
для обоих переходов (соответственно v' <C 3, v" ^4и»'^8, v" <C13) и определил приближенные
значения волновых чисел начал ряда полос. В работе [593] отмечается невысокая точность полученных
колебательных постоянных, поскольку канты полос отстоят далеко от их начал, а оттенение даже
С-ветвей меняется в некоторых полосах. Вращательные постоянные T1F в состоянии Х1!!",
принятые в справочнике, получены Хозфтом и др. [1452] при исследовании вращательных переходов
(/ ^ 4, v г=С 5) в микроволновом спектре этой молекулы. Они хорошо согласуются с найденными
в работах [584, 593, 1931]. Вращательные постоянные T1F в состояниях а3П0+ и ааПх приняты по-
работе [593], постоянные в состоянии АХИ — по работе Хауэлла [1484]; Барроу [589] отметил»
что данные для состояния АгЛ неоднозначны и что соответствующее значение То может иметь
величину ~45 010 см.
Термодинамические функции T1F (г) были рассчитаны по уравнениям A. 3—A. 6), A. 9).
A. 10), A. 93)—A. 94). Значения QTn и ее производных вычислялись по соотношениям A. 90) —
223
JI2F2 (г) Глава 24
A. 92) с учетом трех компонент CП0+, 3П0- и 3П1) возбужденного состояния о3П, в предположении,
что Qioi.Bp = ^Qifz вр- Статистическая сумма по колебательно-вращательным уровням энергии
состояния ХХЕ+ и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью
соотношений типа A. 82). В расчете учитывались все уровни энергии со значениями / ^ /шах, „, последние
определялись из условий A. 81). Энергия колебательно-вращательных уровней состояния XXS+
вычислялась по уравнениям A. 65) и A. 61). Значения коэффициентов Ykl в этих уравнениях,
а также величин ушах и/1!шприведены в табл. 24.5; коэффициенты Yk0, включая Y30 и У40, были
получены по колебательным постоянным из табл. 24.4 с учетом условий A. 50), коэффициент У03 —
из условия A. 59).
Погрешности рассчитанных термодинамических функций T1F (г) до 1000—1500 К обусловлены
главным образом неточностью колебательных постоянных этой молекулы (см. выше), а также
принятых фундаментальных постоянных. При более высоких температурах становится существенным
отсутствие надежных экспериментальных данных о колебательно-вращательных уровнях энергии
состояния XXL+ си )>5, а также приближенный учет вклада возбужденных состояний этой
молекулы. Погрешности в значениях Ф°(Г) составляют 0,03, 0,05 и 0,2 Дж-К-моль при 298,15,
3000 и 6000 К.
Термодинамические функции T1F (г) ранее вычислялись в справочнике [305] (Т ^ 5000 К),
а также в работе [2082] (значения Ф° (Т) для Г=1500-|-1600 К). Оба расчета выполнены
приближенным методом, расхождения данных [305] и табл. 723 достигают 1,5, 3 и 5 Дж-К~1-моль
в значениях Ф° (Г), S0 (Г) и С), (Т).
Константа равновесия реакции T1F (г)=Т1 (r)+F (г) вычислена на основании энергии
диссоциации Дг#° @)=D0 (T1F)=442,621±3,6 кДж-моль^? 000 + 300 см.
Это значение соответствует принятым в справочнике энтальпиям образования и сублимации
T1F (к). Принятая величина учитывает также результаты измерений энергии диссоциации T1F
в работе Берковица и Уолтера C6 850 + 160 см) [666] (см. T1F (к)), масс-спектрометрическое
исследование Мурада и др. [2082] (Tl (r)+MgF(r)=TlF (r)+Mg (г), 1546—1585 К, 4 точки) и спектро-
фотометрическое исследование равновесия реакции в пламенах Булевич и др. [787] (Tl (r)+HF (г) =
= T1F (г)+Н (г), 1500—2000 К), которые приводят к значениям, согласующимся с принятым в
пределах их погрешностей D32 + 10 и 460+40 кДж-моль соответственно). Короткая экстраполяция
уровней состояния а3Л1 (примерно на 600 см) свидетельствует, что они сходятся в области
38 300 + 200 см над уровнем р=0, /=0 состояния -Х"х?+; поскольку у T1F состояние а3Пх
коррелирует с пределом, лежащим на 400 см выше предела состояния ХХ2+, можно предполагать, что
на потенциальной кривой этого состояния имеется максимум высотой ~900+400 см. Диффузный
характер полос, связанных с переходами на уровни состояния АгП с энергией ~45 500 см, и
ъ-онтинуум в области 2630 А свидетельствуют, что Do > 36 600 см и < 38 400 см, соответственно.
Принятым энтальпиям образования и сублимации T1F (к) соответствует также
г, 0) = —183,946 + 3,4 кДж • моль.
T12F2 (г). Термодинамические свойства газообразного дифторида диталлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 724.
Молекулярные постоянные, использованные для расчета термодинамических функций T12F2,
приведены в табл. 24.7. Исследования ИК-спектров и спектров КР [1848], фотоэлектронного
спектра [2625], изучение отклонения молекулярного пучка T12F2 в электрическом поле [2071 ], а также
электронографические исследования [388, 389а] показывают, что молекула T12F2 в основном
электронном состоянии %гА имеет плоское ромбическое строение и принадлежит к точечной группе
симметрии Dih. Приведенное в табл. 24.7 произведение главных моментов инерции IaIbIc
соответствует структурным параметрам г (Tl—F)=2,305 + 0,014 Аи -4 Tl—F—Т1=105,7 + 1,3°, найденным
Соломоником при электронографическом исследовании T12F2 [388]. Погрешность 1А1в1а не
превышает 5-Ю13 г3-см6. Значения основных частот, приведенные в табл. 24.7, получены Лисицким и
Найблером [1848] при исследовании ИК-спектра и спектра КР молекул T12F2, изолированных
в матрице из Аг. Отнесение наблюдаемых частот, предложенное авторами [1848], подтверждается
расчетом, выполненным Соломоником [388] для ионной модели. Погрешности рекомендуемых
частот колебаний с учетом возможного матричного сдвига оцениваются в 10—15 см. С принятыми
значениями vs и v6 практически совпадают частоты, найденные Бромом и Франценом [760] в ИК_
224
Таллий и его соединения Т1С1 (к, ж)
спектре поглощения T12F2, изолированного в матрицах из Аг и Кг. Сведения о возбужденных
электронных состояниях T12F2 в литературе отсутствуют.
Термодинамические функции T12F2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128)^
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных величин
определяются неточностью молекулярных постоянных и приближенным характером расчета и
составляют 2, 6 и 8 Дж-К~1-моль"г в значениях Ф° (Т) при 298,15, 3000 и 6000 К, погрешности
из-за неточности молекулярных постоянных не превышают 2 Дж-К~1-моль~1.
Ранее термодинамические функции T12F2 (г) вычисляли Краснов и Соломоник [196] (до 3000 K)t
расхождения результатов этого расчета с данными табл. 724 достигают 10 Дж-К-моль~1 в
значениях Ф° (Т) при 3000 К из-за использования оцененных значений постоянных.
Константа равновесия T12F2 (г)=2Т1 fr)+2F(r) вычислена с использованием значения Д,^0 @)=
=1027,35+7,6 кДж-моль"^ соответствующего принятой в справочнике энтальпии образования
AfH° (T12F2, г, 0) = —510 + 7 кДж • моль.
Это значение основано на принятой в справочнике энтальпии образования T1F (г) и энтальпии
реакции T12F2 (r)=2TlF (г). Последняя была рассчитана по результатам изучения процесса
испарения T1F (к) квазистатическим методом F90—965 К) и методом протока (800 К) в работе Кенеши
и Кубиччотти [1670]. Сопоставляя эти измерения, авторы определили давление мономера при
800 К и его энтальпию сублимации (см. выше), которая была использована ими для определения
доли мономерных молекул в насыщенном паре. Рассчитанному парциальному давлению димерных
молекул при температурах 690—965 К соответствует энтальпия сублимации 149,3+1,0 кДж-моль
и энтальпия диссоциации димера на мономер A.JS0 @) =141,5+4,0 кДж-моль. При оценке
погрешности последней величины учтена неточность термодинамических функций T12F2 (г),
приводящая к дополнительной погрешности в энтальпии диссоциации в 3 кДж-моль.
Т1С1 (к, ж). Термодинамические свойства кристаллического и жидкого хлорида таллия в
стандартном состоянии при температурах 100—2200 К приведены в табл. 725.
Значения постоянных» использованных для расчета термодинамических функций, приведены
в табл. 24.1. В справочнике за стандартное состояние Т1С1 (к) в интервале 0—704 К принята
кубическая модификация (структурный тип CsCl) [18906, 2530а].
При Т < 298,15 К термодинамические функции Т1С1 (к) вычислены по результатам измерений
теплоемкости в работе Бартки и Джиока [608] A3—310 К) на образце чистотой 99,9% Т1С1. При
Т <С 13 К значения теплоемкости определены экстраполяцией с использованием функций Дебая и
Эйнштейна. Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—Н° @) оцениваются
в 0,4 Дж-К-моль и 0,05 кДж-моль соответственно.
В интервале 298,15—704 К для теплоемкости Т1С1 (к) принято уравнение (см. табл. 24.1),
найденное методом Шомейта при обработке измерений энтальпии, выполненных Кубиччотти и Эдин-
гом [907] B98—1250 К), их погрешности оцениваются в 0,5%.
Температура плавления G04+1 К) принята по работе [907], она не противоречит данным [306,
308, 412, 413Г 453, 2541, 2801] G03 К). Измерения [38,142,143, 386, 410, 1338, 2366, 2639,; 2783]
G00—702 К) менее надежны и не учитывались в справочнике. Энтальпия плавления A5,56 +
+0,80 кДж-моль) также принята по работе [907]. Значение, найденное авторами [2366]
A6,8 Дж-К^-моль), менее надежно. Теплоемкость Т1С1 (ж) G5,6+0,8 Дж-К-моль)
вычислена по результатам измерений энтальпии, выполненных Кубиччотти и Эдингом [907] в интервале
температур 704—1250 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 0,3, 1 и
4 Дж-К-моль соответственно. Термодинамические функции Т1С1 (к, ж), приведенные в
справочнике Барина и Кнакке [578] (Т <^ 1100 К), отличаются на 5 Дж-К-моль от данных табл. 725.
В справочнике принята энтальпия образования хлорида таллия
Д,Яв(ПС1, к, 298,15 К) = —204,1 + 0,5 кДж • моль,
основанная на измерениях растворимости Т1С1(к) и на значениях термодинамических свойств
водных ионов Т1+ [406] и С1~ [864]. Измерения растворимости Т1С1 в воде и в растворах различных
солей были выполнены в работах [210, 512, 726, 800, 1431, 1743, 2144, 2150, 2151, 2289, 2553].
225
Т1С1 (г)
Глава 24
Все эти измерения находятся в хорошем соответствии между собой. Также в хорошем
соответствии находятся результаты расчетов по уравнениям II и III законов термодинамики по данным
работ [512, 800, 1743], в которых измерения проводились в достаточно широком интервале
температур. Наиболее точными из перечисленных работ являются измерения [512, 800, 1431, 1743, 2289],
для которых не были необходимы пересчеты на нулевую ионную силу и на переход к стандартной
температуре. Принятая величина является средней из этих работ, ее погрешность определяется
в первую очередь погрешностью термодинамических свойств иона Tl+ (aq). В работе Лонги и
др. [1884а] были проведены измерения ЭДС ячейки с электродом из амальгамы таллия, которые
.привели к значению —203,3 кДж-моль, достаточно хорошо согласующемуся с величинами,
вычисленными на основании измерений растворимости.
Давление пара хлорида таллия в реакции T1G1 (к, ж)=ТЮ1 (г) вычислено с использованием
ринятой в справочнике энтальпии сублимации
Д,Я°(Т1С1, к, 0) = 139,9 ± 1,5 кДж-моль.
Эта величина основана на результатах исследований давления пара Т1С1 (к, ж), приведенных
в табл. 24.8, и определениях Do (T1G1) в работах Берковица и Уолтера [666] и Булевич и др. [787].
Таблица
Вартенберг,
24.8. Результаты расчета AgH° (T1C1, к, 0) (в кДж-моль)
Авторы
Боссе, 1922 [2801]
Волмер, 1929 [2783]
Нива и др.,
1939 [2146]
Барроу и др., 1955 [598а]
Большаков
Кубиччотти
Комшилова,
и др., 1958 [46]
1964 [904]
Поляченок, 1969 [1736
Драпчо, Розеиблатт, 1979 [983а]
Метод
Точек кипения, 938—1080 К, 11 точек
Протока, 599—633 К, 7 точек
Эффузионный, 543—633 К, 9 точек
Торзионный, 501—608 К, 8 точек
Точек кипения и протока, 895—1090 К, 30 точек
Статический и протока, 714—1110 К, данные
представлены в виде уравнения
| Статический, 800—1089 К, данные представлены
в виде уравнения
Масс-спектрометрический, 543—606 К, 44 точки
ASB°
II закон
142+1
130±2
124±1
137±3
154 ±5
138+6
142+19
136+.5
(Т1С1, к, 0)
III закон
139,8±0,1
139,5+0,2
143,1+0,8
139,8±0,3
141,7±0,6
140,5+0,7
140,8±0,3
—
Пары хлорида таллия содержат молекулы T1G1 и Т12С12 (см. [1736, 904]), поэтому
экспериментальные данные, полученные в работах [598а, 2146, 2783, 2801 ], были пересчитаны с учетом
образования мономеров и димеров в насыщенном паре. В работах [46, 1736, 904] были определены
парциальные давления Т1С1 на основании сопоставления измерений двумя методами [46], измерений
давления насыщенного и ненасыщенного пара [1736], а также в результате определения состава
пара масс-спектрометрическим методом [904]. Приведенные в табл. 24.8 погрешности
характеризуют только воспроизводимость измерений. Неточность термодинамических функций Т1С1 (к, ж)
вносит дополнительную погрешность около 1 кДж-моль в данные [46, 1736, 904, 983а, 2801]
и 0,3—0,5 кДж-моль в данные [598а, 2146, 2783]; погрешности, связанные с учетом образования
димеров в насыщенных парах, в отличие от случая с T1F невелики, так как доля димера в паре
относительно мала. Принятая величина основана в первую очередь на измерениях Барроу и
др. [598а], выполненных при низких температурах, имеющих хорошую воспроизводимость и
согласие результатов обработки по II и III законам; она практически совпадает со
средневзвешенной из всех данных табл. 24.8, за исключением работы [2146]. Погрешность принятого значения
энтальпии сублимации учитывает воспроизводимость разных измерений и неточность
термодинамических данных, использованных в расчетах.
Исследования энергии диссоциации Т1С1 (г) в работах Берковица и Уолтера [666] и Булевич
и др. [787] хорошо согласуются с данными, приведенными в табл. 24.8. Им соответствуют значения
Д,Я° (Т1С1, к, 0), равные 137,7+2,5 и 142+12 кДж-моль.
Т1С1 (г). Термодинамические свойства газообразного хлорида таллжя в стандартном состоянии
при температурах 100—6000 К приведены в табл. 726.
226
Таллий и его соединения Т1С1 (г)
В табл. 24.4 представлены молекулярные постоянные 205Т1ЗБС1, принятые в справочнике на
основании результатов исследований электронного и вращательного спектров этой молекулы. В
электронном спектре Т1С1 исследована только одна система полос а3П0+ ^I'E* [587, 798, 1486, 2023,
2124]. В области 2510 А в спектре поглощения Т1С1 наблюдается континуум с примыкающими
к нему со стороны больших длин волн диффузными полосами, этот континуум в работах [931а„
1486] был отнесен к переходу в слабосвязанное состояние АХП; зарегистрированы также
континуумы с несколькими максимумами в области 3156—2870 А, вероятно, наиболее длинноволновый из
них связан с переходом в состояние azTix. В спектре испускания T1G1 в области 4000 А Рао [2298]
наблюдал сложную систему полос, предположительно отнесенную к переходу D ->• а3П0+ из
состояния с энергией ~56 000 см.
В табл. 24.4 приведены данные только для состояний XlL+ и а3П0-г, эти состояния коррелируют
с пределами Tl BPi/s)+Cl BР»/2) и Tl BPi/s)+Cl BА/2) соответственно. Из сопоставления энергии
диссоциации T1G1 (см. ниже) со значением Те (<г3П0+) и величиной расщепления подсостояний
а3По+, аъЛг и а3П2 у молекулы T1F и молекул гидридов и галогенидов других элементов подгруппы
1ПА, а также принятой в справочнике для других галогенидов элементов этой подгруппы
корреляции этих подсостояний с диссоциационными пределами очевидно, что у Т1С1 подсостояния аЧ\г
и а3П2 должны быть отталкивательными. Подсостояние а3П0-, которое имеет общий предел с
состоянием Х12+, по-видимому, также является отталкивательным или имеет минимум глубиной не
больше 100 см.
Колебательные постоянные Т1С1, приведенные в табл. 24.4, найдены Мишером [2023] при
исследовании перехода а3По+**Х1?+ (v" ^ 22, v' ^ 7). Они существенно отличаются (на 3,7 и
0,42 см в значениях ш", и ш"ех"е) от полученных ранее Хауэллом и Коулсоном [1486] при
исследовании той же системы полос \v" <J 15, v' ^ 5). В обеих работах отмечается трудность определения
надежных значений колебательных постоянных Т1С1 из-за больших и изменяющихся от полосы
к полосе интервалов между кантами и началами полос; поскольку интенсивности линий в области
малых значений / малы, авторы [1486, 2023] постарались оценить положения нулевых
промежутков и проводили анализ колебательной структуры по ним. В справочнике предпочтение отдано
данным Мишера, который получил для Д6?у2 значение, близкое к найденному позднее Барроу [5871
по началам полос 0—0, 1—0 и 1—1 B82,1 и 281,88 см соответственно) из анализа вращательной
структуры этих полос. Кроме того, Мишер наблюдал переходы, связанные с бблыпим числом
колебательных уровней состояния ХХЕ+ по сравнению с авторами [1486].
Вращательные постоянные Т1С1 в состоянии ХХЕ+приняты по работе Де Вина [2852],
который исследовал в микроволновом спектре вращательные переходы с v ^ 12, / ^ 11. Эти
постоянные хорошо согласуются с результатами исследований микроволнового спектра Т1С1 в работах [584,
1931, 2704], а также анализом вращательной структуры трех полос перехода а3П0+—Хг1>+ в
работе Барроу [587] х. Вращательные постоянные в состоянии а3П0+ приняты по этой работе Барроу.
Следует отметить, что как было показано в работах [1486, 2023], состояние а3П0+ имеет небольшое
число связанных колебательных уровней, общее количество которых не превышает 10.
Термодинамические функции Т1С1 (г), приведенные в табл. 726, были рассчитаны по
уравнениям A. 3)—A. 6), A. 9), A. 10), A. 93)—A. 95) без учета возбужденных электронных состояний
этой молекулы (Qsn = @W вр). Значения $№ вр и ее производных вычислялись по уравнениям
A. 73)—A. 75) непосредственным суммированием по колебательным уровням энергии и
интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались все уровни
энергии со значениями / ^ /Шах, »> последние определялись из условий A. 81); соответствующие
значения уши и /Иш приведены в табл. 24.5. Колебательно-вращательные уровни энергии состояния
Х1!" находились по уравнениям A. 65) и A. 61), значения коэффициентов YM в этих уравнениях,
рассчитанные по уравнениям A. 66) и постоянным молекулы 2°6'Г136С1 для изотопической
модификации молекулы Т1С1, соответствующей природной смеси изотопов таллия и хлора, приведены
в табл. 24.5. Коэффициенты Yk0, включая Y30 и У40, были получены с учетом условий A. 50),
коэффициент У03 найден из условий A. 59).
Погрешности рассчитанных термодинамических функций Т1С1 (г) при низких температурах
обусловлены неточностью фундаментальных постоянных, принятых в справочнике, а также извест-
1 В работах [1610, 1611] при попытке анализа вращательной структуры полос были допущены ошибки, что
привело к противоречиям с данными других авторов.
227
Tl,Clt (г) Глава 24
ных колебательных постоянных. Выше 2000 К начинает сказываться отсутствие
экспериментальных данных о колебательно-вращательных уровнях состояния ХгЕ+ с энергией свыше 5500 см.
Пренебрежение вкладом состояния а3П0+, имеющего не больше 10 колебательных уровней, а также
другими еще не наблюдавшимися состояниями, практически не влияет на точность расчета.
Погрешности в значениях Ф° {Т) оцениваются в 0,05, 0,1 и 0,3 Дж-К^-моль при 298,15, 3000 и 6000 К.
Термодинамические функции Т1С1 (г) ранее вычисляли Келли [1656,1658] (до 2000 К) и Эдинг и
Кубиччотти [10093 (S° (T) до 1250 К). Расхождения в значениях функций, приведенных в табл. 726
и в перечисленных выше работах, достигают 0,8 Дж-К~1-моль.
Константа равновесия реакции Т1С1 (г)=Т1 (г)+С1 (г) вычислена с использованием значения
ДГЯ° @) = Do (T1C1) = 366,478 + 2,2 кДж • моль & 30 640 + 200 см,
соответствующего принятым в справочнике значениям энтальпии образования и сублимации
Т1С1 (к). Оно хорошо согласуется с энергиями диссоциации, полученными Берковицем и
Уолтером [666] при фотоионизационных измерениях (Т1С1 (r)+fcv=Tl + (г)+С1 (г)+ е (г), Do (T1G1) =
=368,6+2,0 кДж-моль) и Булевич и др. [787] на основании спектрофотометрического
изучения равновесия реакции образования T1G1 (г) в пламенах (Т1 (г)+НС1 (г)=Т1С1 (г)+Н (г),
1500-2000 К, DO(T1C1)=364±12 кДж-моль).
Короткая экстраполяция (на 70—100 см) уровней состояния а3П0+ по постоянным, принятым
выше (см. примечание к табл. 24.4), приводит к схождению колебательных уровней в области
32 000 см при tW=8-f-9. Эта энергия примерно на 700 см выше предела состояния а3П0+,
соответствующего принятым в справочнике величинам. По-видимому, также как у T1F, потенциальная
кривая состояния а*П0+ у молекулы T1G1 имеет небольшой максимум, его высота составляет 700 +
+200 см.
Принятым энтальпиям образования и сублимации T1G1 (к) соответствует также
ДД0 (Т1С1, г, 0) = —65,458 + 1,6. кДж • моль.
Т12С12 (г). Термодинамические свойства газообразного дихлорида диталлия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 727.
Молекулярные постоянные, использованные для расчета термодинамических функций Т12С1г,
приведены в табл. 24.7. Для молекулы Т1аС1а в основном состоянии ХгА на основании
спектроскопического исследования [1848], а также по аналогии с молекулой T12F2, принята плоская
ромбическая структура (симметрия D2h). Приведенное в табл. 24.7 произведение главных моментов инерции
IaIbIg) соответствует структурным параметрам г (Т1—С1)=2,76+0,03 Аи^ Т1—С1—Т1=95+5°,
оцененным Соломоником [388] в результате расчета по модели поляризующихся ионов.
Погрешность 1\1в1е составляет 5-Ю12 г3-см6. Принятые значения основных частот, приведенные
в табл. 24.7, получены Лесицким и Найблером [1848] при исследовании спектра КР и ИК-спектра
молекул Т1гС12, изолированных в низкотемпературной аргоновой матрице. Деформационная
частота, связанная с изгибом цикла (v4), экспериментально не наблюдалась. Возможный интервал
ее значений C1—46 см) был вычислен авторами работы [1848] при значениях для
соответствующей силовой постоянной, оцененных сопоставлением силовых постоянных T12F2 и Т12С12. Среднее
из полученных авторами значений частоты v4 находится в хорошем согласии с результатами
расчета Соломоника [388] C9 см) и принимается в настоящем справочнике. Погрешности принятых
значений частот с учетом матричного сдвига оцениваются в 15 см. Частоты, наблюдавшиеся
Бромом и Франценом [760] в ИК-спектре поглощения молекул Т12С12, изолированных в матрицах
из Аг и Кг (соответственно 171,189 см и 168,184 см), в пределах погрешности согласуются с
принятыми значениями v5 и v6. Сведения о возбужденных электронных состояниях Т12С12 в литературе
отсутствуют, и в справочнике они не учитываются.
Термодинамические функции Т12С12 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности рассчитанных таким образом функций определяются как неточностью
молекулярных постоянных, так и приближенным характером расчета и составляют 4, 8 и
10 Дж-К-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности, обусловленные
неточностью молекулярных постоянных, не превышают 3 Дж-К-моль~1.
Ранее таблицы термодинамических функций Т12С12 (г) рассчитывались Красновым и
Соломоником [196] до 3000 К и Драпчо и Розенблаттом [983а] до 2000 К. Молекулярные постоянные
228
Таллий и его соединения
Т12С13 (г)
Т12С12, принятые в настоящем справочнике, существенно отличаются от оцененных Красновым и
Соломоником [196]. Расхождения результатов расчета [196] и данных, приведенных в табл. 727,
достигают 11 Дж-К-моль в значениях Ф° (Т) при3000 К, данные [983а] итабл. 727 согласуются
в пределах 3 Дж-К-моль~1.
Константа равновесия реакции Т12С12 (г)=2Т1 (г)+2С1 (г) вычислена с использованием
значения ArH" @) =862,04+8,5 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(Tl2Cl2, г, 0) = — 260 + 8 кДж • моль-1.
Эта величина получена на основании принятого в справочнике значения энтальпии образования
T1G1 (г) и энтальпии диссоциации Т12С12 (г)=2Т1С1 (г), АГН° @)=107 + 7 кДж-моль. Результаты
измерений энтальпии диссоциации приведены в табл. 24.9, где в качестве погрешностей дана
воспроизводимость измерений. Наиболее полные и тщательные измерения были выполнены Кубич-
чотти [904] и Комшиловой, Поляченком [1736]. Результаты работы [983а] не являются
независимыми, так как ее авторы проводили калибровку прибора, принимая энтальпию сублимации
мономера по работе [904]. В работе Большакова [46] было принято, что ниже 793 К в парах образуется
Таблица 24.9. Результаты определения Do (T1C1—Т1С1)
Авторы
Большаков и др., 1958
Кубиччотти, 1964 [904]
Комшилова, Поляченок,
Бенневис, Шефер, 1974
[46]
1969 [1736,
[693а]
Драпчо, Розенблатт, 1979 [983а]
а Учтена погрешность,
Метод
Статический, 767—860 К, 4
Статический и протока, 714-
ставлены в виде уравнения
Статический, 973—1232 К,
в виде уравнения
(в нДж
точки
-1111 К
данные
Масс-спектрометрический, системы
ThCl4 и CdCl2, по 1 точке
Масс-спектрометрический, 543—615
обусловленная неточностью принятых
•МОЛЬ Х)
, данные пред-
предетавлены
Т1С1 с VC14,
К, 44 точки
D,(T1C1-T1C1)
II закон
285
83
123
—
91±15
значений сечений ионизации.
III закон
129,5
109,7 ±5,0
108,3±3,0
110±10
103,5±3,0 *
в основном димер, а выше 893 К — мономер. Это противоречит данным более поздних работ [1736,
904], в том числе прямым масс-спектрометрическим исследованиям, проведенным в работах [904,
983а]. Расхождения данных [983а], с одной стороны, и [1736, 904], с другой, не превышают суммы
их погрешностей, и использованное в справочнике значение &ГН° @) основано на трех этих
работах. При оценке погрешности принятого значения была учтена неточность термодинамических
функций, соответствующая погрешность в энергии диссоциации для измерений, выполненных при
1000 К, равна 6 кДж-моль.
Глава 25
БЕРИЛЛИЙ И ЕГО СОЕДИНЕНИЯ
Be (к, ж), Be, Ве+, ВеО (к, ж), ВеО, Ве2О, Ве2О2, Ве3О3, Ве4О4, ВеН, ВеН2 (аморф.), ВеОН,
Ве(ОНJ(к), Ве(ОНJ, BeF, BeF2 (к, ж), BeF2, Be2F4, BeCl, ВеС12 (к, ж), р-ВеС12(к), ВеС12,
Ве2С14, ВеВг, ВеВг2 (к, ж), ВеВг2, Bel, Bel2 (к, ж), Ве12, ВеБ(к), BeS, BeSO4 (к, ж), Ве31Чг (к, ж),
ВеСО3 (к)
В настоящей главе рассматриваются 25 веществ, включающих бериллий и его соединения. Для
восьми веществ приведены свойства в конденсированном и газообразном состояниях, для четырех
(ВеН2Ж BeSO4, Be3N2 и ВеСО3) — только в конденсированном и для тринадцати — только в
газообразном состоянии. Для трех газов (Be, Be+ и ВеО) термодинамические свойства рассчитаны
вплоть до 10 000 К, для всех остальных — до 6000 К. Для ВеС12 (к), помимо свойств в
стандартном (равновесном) состоянии, приведены свойства высокотемпературной модификации |3-ВеС12.
Соответствующая таблица термодинамических свойств, а также таблицы свойств ВеН2 (аморф.),
Ве(ОНJ (к) и ВеСО3 (к), у которых равновесное давление диссоциации превышает 100 атм при
1000 К, представлены в Дополнении второй книги этого тома.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов
бериллия (атомный вес 9,01218) и других элементов. Однако отличие постоянных разных изотопических
модификаций молекул соответствующих газов, как правило, не учитывалось в расчетах ввиду
его несущественного влияния на термодинамические функции большинства веществ по
сравнению с другими источниками ошибок, а также из-за отсутствия соответствующих данных.
В справочнике, в отличие от таблиц JANAF, не включены такие газообразные ионы, как ВеО+,
ВеОН+, BeF+ и др., поскольку стабильность соответствующих нейтральных соединений
недостаточно велика, а их потенциалы ионизации слишком высоки для того, чтобы соответствующие
ионы присутствовали в заметных количествах в равновесных условиях вплоть до 6000 К. Не
рассматривается также, в отличие от предыдущих изданий настоящего справочника, газообразный
нитрид бериллия (BeN), поскольку неэмпирический расчет с учетом конфигурационного
взаимодействия, выполненный в работе [257а], показал, что основное 4? состояние этой молекулы
является .слабосвязанным.
В Приложении 3.9 для дихлорида бериллия приведены данные, позволяющие учесть влияние
межмолекулярного взаимодействия на термодинамические свойства этого газа, а также сведения
0 критических постоянных Be.
Be (к, ж). Термодинамические свойства кристаллического и жидкого бериллия в стандартном
состоянии при температурах 100—4400 К приведены в табл. 728.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В справочнике за стандартное состояние Be (к) в интервале 0—1550 К принята
гексагональная модификация (а-Ве, структурный тип Mg); в узком интервале температур 1550—1560 К
существует высокотемпературная модификация с кубической объемно-центрированной решеткой
(Р-Ве, структурный тип a-Fe).
При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работах Хилла и Смита [1433] D—300 К) на образце, содержащем 99,5% Be, и Алерса
[488] A,4—30 К). Данные Кристеску и Симона [903] A0—300 К) не учитывались, поскольку они
лежат выше полученных в работе [1433]; соответствующие расхождения достигают —10% х.
Погрешности принятых значений S° B98,15 К) и Н° B98,15 К)—Н° @), которые совпадают с
рекомендованными КО ДАТА—МСНС [864] в качестве ключевых термодинамических величин,
оцениваются в 0,08 Дж-К~1-моль и 0,02 кДж-моль соответственно. В интервале 298,15—
1550 К для теплоемкости Be (к) принято уравнение (см. табл. 25.1), выведенное совместной
обработкой методом Шомейта результатов измерений энтальпии, проведенных Гиннингсом и др. [1211]
B73-1173 К), Кантор и др. [1461 F00-2200 К), Уокером и др. [2791] C03-1073 К), Магнусом
и Хольцманом [1910] C73—1173 К) и Егером и Розенбомом [1538] C73—1338 К). При обработке
погрешности измерений [1211] оценивались в 0,5%, [146, 2791] — в 2%, а данных [1538, 1910] —
в 5%. Значение теплоемкости f>-Be оценено равным 30+1 Дж-К~1-моль~1.
1 Найденная Кристеску и Спмоном [903] аномалия теплоемкости вблизи 11 К является, по-видимому, следствием
загрязнения образца. Более поздние работы [488, 1433] не подтвердили ее существования.
230
Бериллий и его соединения
Be (к, ж)
Таблица 25.1. Принятые значения термодинамических величин для бериллия и его соединений
в твердом и жидком состояниях
Вещество
Be
BeO
BeH2
Be(OH)
BeF2
ВеС12
ВеС12
ВеВг2
Ве12
BeS
BeSO,
BesN2
ВеСОз
Состояние
кП, гекс. (а)
к1, куб. (Р)
ж
кП, гекс. (а)
к1, тетр. (Р)
ж
аморф.
2 к, ромб. (Р)
к11, гекс. (а)
к1, гекс. (Р)
ж
кП, ромб, (а
к1(р)
ж
к1 (р)
к, ромб,
ж
к, ромб,
ж
к, куб.
1
К
кДжХ
Хмоль
1,95
2,837
4,2
8,293
8,468
11,420
11,98
14,9
15,2
5,5
кШ, тетр. (а) 12,97
кП, ромб, (р) —
к1, куб. (г) —
ж —
кП, куб. (а)
к1, гекс. (C)
ж
к
7,124
9,2
S0 B98,15 К)
К
ю
00*
оз
eq
о е.
Дж-К-^моль
9,50 16,443
13,77 25,564
24,1 в 29,85
45,48 62,13
53,35 51,84
75,81 62,42
82,68 64,85
108 69,45
121 71,13
34 34
77,91 85,69
34,4 64,76
52 65
Коэффициенты в
НИИ ДЛЯ С°}
а
18,736
30
30
7,171
56
84
12,036
99,638
-4,610
46,998
102,64
71,480
66,99
92
75,199
79,35?
100
83,796
100
40,542
71,069
150
150
150
й-103
9,293
114,336
59,743
16,920
155,821
33,149
—1,527
11,720
20,92
14,126
. 7,456
2,80с
11.34С
102,19?
21,084 255,184
145 -
167 —
87,92
a C°p(T)=a+hT—cT-i+dT2+eT3+fTt (в Дж-К-^моль); б
в приведено значение .5° B98,15 К)—S" @).
38,83
d=—118
уравне-
с-10
4,501
Интервал
температур
К
298,15—1550
1550—1560
1560—4400
5,840б 298,15—2373
— 2373—2851
— 2851—6000
—
37,825
—8,882
156,482
11,159
12,944
> 10,780
12,004
8,823
S 14,090
i 13,297
30,670
373-10-8,
298,15—500
298,15-500
298,15—493
493—823
823—2100
298,15—676
676—688
688—1200
298,15-688
298,15—781
781—1300
298,15—763
763-1300
298,15—3000
298,15-861
861—912
912—1400
1400—2000
298,15—1673
1673—2473
2473—5000
298,15—1000
е=55,425-10-8,
или
т
" m
К
1550
1560
2373
2851
—
—
493
823
676
688
688
781
763
—
861
912
1400
1673
2473
—
/--9,
или
кДжХ
Хмоль
2,1
12,6
0,265
86
—
—
0,175
4,77
6.230
8,66
—
18
18
—
5
2,1
6
16
НО
—
25Ы0-12;
Температура полиморфного превращения A550 +5 К) принята как средневзвешенное
значение из данных Лоусби и Диарден [1879] A552+3 К) и Мартина и Мура [1955] A545+5 К).
В этих работах методом ДТА исследовались образцы Be с содержанием примесей 0,3 и 0,1%
соответственно. В работе [1879] установлено, что содержание примесей до 1,5% в пределах
погрешности не влияет на определяемое значение Ttr. Результаты работ [16, 1129, 2673], в которых
определены более низкие значения Tir A527—1536 К), не учитывались, поскольку измерения
температуры превращения проводились в них с меньшей точностью. Энтальпия полиморфного
превращения 2,1+0,2 кДж-моль принята на основании измерений Лоусби и Диарден [1879].
Значение b.irH=l ,Ь кДж -моль, полученное Франсуа и Контр [1129] расчетом из р—Г-диаграмм,
является ошибочным.
Температура плавления 1560 +5 К принята на основании хорошо согласующихся между
собой данных работ [146, 725» 802, 1879, 1955]. Данные, полученные авторами [2673, 1184],
занижены и не учитывались при выборе этой величины. В работе Кантор и др. [146] получена энталь-
231
Be (г)
Глава 25
пия плавления 14,7+0,3 кДж-моль г, которая является суммой значений LtrH (Be) и &тН (Be).
Принятая в настоящем справочнике энтальпия плавления 12,6+0,3 кДж-моль является
разностью между полученным Кантор и др. значением Дот# и величиной Д/г#, 'принятой по
работе [1879]. Теплоемкость Be (ж) принята равной 30,0+1 Дж-К-моль~1 по результатам
измерений энтальпии в интервале 1566—2200 К в работе Кантор и др. [146].
Погрешности вычисленных значений Ф° (Г) при 298,15,1000, 2000 и 4000 К оцениваются в 0,05,
0,15, 0,5 и 2 Дж-К-моль~1 соответственно. Термодинамические функции Be (к, ж),
приведенные в табл. 728 и в справочниках [105, 1543, 1656, 2216], удовлетворительно согласуются между
собой. Расхождения в значениях Ф° (Т) составляют до 0,4 Дж -К -моль при 1000 К и 1 ДжХ
XК-моль при 4000 К.
В справочнике за стандартное состояние бериллия принят Be (к) и в соответствии с этим
AfH°(Be, к) = 0.
Давление пара бериллия Be (к, ж)=Ве (г) вычислено на основании значения АГН° @) =
= Д,Я° (Bet к, 0) =319,753+5 кДж-моль, соответствующего принятой в справочнике энтальпии
сублимации
Д,#° (Be, к, 298,15 К) = 324 + 5 кДж • моль.
Эта величина является средневзвешенной из приведенных в табл. 25.2 результатов измерений
давления пара бериллия. В качестве погрешностей в табл. 25.2 указана воспроизводимость
измерений. Погрешности из-за неточности термодинамических функций незначительны, в
интервале 1000—1300 К они не превышают 0,3 кДж-моль, а при 2000 К — 0,8 кДж-моль. При
Таблица 25.2. Результаты определений Д,Л°(Ве, к, 298,15 К) (в кДж-жоль)
Авторы
Баур, Бруннер, 1934 [627]
Шуман, Гарретт, 1944 {2478, 2479]
Холден и др., 1948 [1459]
Гулбрансен, Эндрю, 1950 [1306]
Ковтун и др., 1964 [169]
Ансара, Бонье, 1965 [525]
Метод
Точек кипения, 1850—2331 К, 6 точек
Лэнгагора, 1174—1336 К, 19 точек
Эффузионный, 1320—1552 К, 7 точек
Лэнгмюра, 1172—1365 К, 22 точки
Лэнгмюра, 1103—1223 К, 25 точек
Эффузионный, 1228—1373 К, 5 точек
Лэнгмюра, 1178—1309 К, 5 точек
Эффузионный, 1578—1765 К, 11 точек
Д, Н° (Be,
II закон
248±15
342
324+25
341 ±20
340±25
351 ±50
319 ±40
289 ±100
к, 298,15 К)
III »акон
333 ±8
337,6±1,5
324,1 ±1,2
326,8±0,8
327,5±0,8
320,1 ±1,1
318,3±2,3
320,2±1,9
выборе принятого значения Ь.,Н° наряду с воспроизводимостью измерений учитывалось
отклонение данных каждой работы от средней величины. Принятое значение основывается в первую
очередь на работах Холдена и др. [1459] и Гулбрансена, Эндрю [1306], данные которых имеют
минимальный разброс, удовлетворительное согласие расчетов по II и III законам и близки к
среднему значению из всех работ; оно совпадает с рекомендацией КОДАТА—МСНС [864].
Be (г). Термодинамические свойства газообразного бериллия в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 729.
Уровни энергии атома Be, использованные в расчете термодинамических функций,
приведены в табл. 25.3, в которой даны валентные состояния, относящиеся к электронным
конфигурациям . . .2s2 и . . .2s2p. Энергии этих состояний приняты по результатам спектральных измерений
Йоханссона [1574], позволившим уточнить рекомендации справочника Мур [2053].
Термодинамические функции Be (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25) непосредственным суммированием по уровням энергии атома Be. Погрешности
вычисленных значений Ф° (Т) во всем интервале температур не превышают 0,02—0,04 Дж-К~1Х
Хмоль и обусловлены главным образом неточностью фундаментальных постоянных; в значениях
S° (Т) и в особенности С°р (Т) из-за пренебрежения ридберговскими состояниями погрешности
Т 6000 б 03 1 ДК11
при Т
р () рр рр
6000 К существенно больше и достигают 0,3 и 1 Дж-К-моль~1 соответственно.
232
Бериллий и его соединения
Ве+ (г)
Таблица 25.3. Уровни энергии атомов Be и Ве+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Состояние
CM
Pi
Be lsa2sa iSo 0 1
lsa2*2p 3Po 21 978,28 1
«Pi 21978,92 3
3P2 21 981,27 5
Is22s2p JPi 42 565,35 3
Atom
Электронная
конфигурация
Состояние
Tt
CM
Be+ ls*2s aSy, 0 2
lsa2p aPv, 31 928,76 2
aP'/2 31 935,34 4
Ранее таблицы термодинамических функций Be (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106]t в справочниках [1543, 1904, 1974] до 6000 К, в работе
Хилзенрата [1436] до 10 000 К, в работе Кольского и др. [1749] до 8000 К, а также в справочнике
[2632] до 3000 К и в работе Каца и Маргрейва [1639] до 2000 К. Все эти расчеты, за
исключением [1904], выполнены непосредственным суммированием по уровням энергии атома Be,
рекомендованным в справочнике Мур [2053]; различия в значениях термодинамических функций,
вычисленных в большинстве работ и в настоящем издании, не превышают 0,01—0t02 Дж-К-1Х
Xмоль во всем интервале температур. Для расчетов [1749, 2632] соответствующие различия
несколько больше (около 0,05 Дж-К~1-моль), а результаты расчета Хилзенрата и др. [1436] при
температурах около 10 000 К уже заметно превышают данные настоящего издания вследствие
учета ридберговских состояний в работе [1436]. Расчет [1904] выполнен в грубом приближении;
ошибки в этой работе возрастают с температурой и достигают 2,5 Дж-К-моль~1 в значении
5° F000 К).
Энтальпия образования Be (г)
AfH° (Be, г, 298,15 К) = 324+5 кДж • моль
принята по рекомендации КО ДАТА—МСНС [864] (см. Be (к, ж)).
Ве+ (г). Термодинамические свойства газообразного положительного иона бериллия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 730.
Уровни энергии иона Ве+, использованные в расчете термодинамических функций,
приведены в табл. 25.3, в которой даны валентные состояния, относящиеся к электронным
конфигурациям . . .2s и . . .2р. Энергии этих состояний приняты по спектральным измерениям Йоханссона
[1573],, которые практически совпадают с рекомендацией Мур [2053].
Термодинамические функции Ве+ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),,
A. 22)—A. 25) непосредственным суммированием по уровням энергии иона Ве+. Погрешности
вычисленных значений функций во всем интервале температур не превышают 0,02 Дж-К~1-моль~1
и обусловлены только неточностью принятого значения R.
Ранее таблицы термодинамических функций Ве+ (г) вычислялись в справочнике [1543], а также
в работах Грина и др. [1246] до 50000К и Хилзенрата и др. [1436] до 10 000 К. Результаты
этих расчетов и настоящего издания хорошо согласуются между собой (максимальное
расхождение составляет 0,01 Дж-К^-моль).
Константа равновесия реакции Be + (г)-\-е (г)=Be (г) вычислена с использованием значения
Дг#° @) =—899,502+0,001 кДж-моль,, соответствующего потенциалу ионизации атома Be
/0 (Be)=75192,5+0,1 см=8993502+0,001 кДж-моль,
принятому в справочнике.
Эта величина найдена Ситоном [2487] на основании анализа схождения различных серий
в спектре Be, измеренных для я^8в работах Йоханссона [1574] и Хольмстрема и Йоханссона
[1465]. Она несколько выше значения 75192,07+0,10 см, полученного в [1574], и лучше согла-
233
ВеО (к, ж) Глава 25
суется с рекомендацией Мур [20533, сделанной на основании анализа более старых работ
G5192,29 см). Принятому значению соответствует также
^Н°(Ве+, г, 0) = 1219, 255 + 5,0 кДж . моль.
ВеО (к, ж). Термодинамические свойства кристаллического и жидкого оксида бериллия в
стандартном состоянии при температурах 100—6000 К приведены в табл. 731.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В справочнике за стандартное состояние ВеО (к) в интервале 0—2373 К принята
гексагональная модификация (а-ВеО), а в интервале 2373—2851 К — тетрагональная
модификация (Р-ВеО) [2552]. При Т < 298,15 К термодинамические функции вычислены на основании
измерений теплоемкости ВеО в работе Фурукава и Рейлли [1155] A5—300 К) на образце,
содержание примесей в котором не превышало 0,1%. Точность этих измерений оценивается в 0,2%,
а погрешности принятых по [1155] значений 5° B98,15 К) и Н° B98,15 К)—Я0 @) — в 0,04 ДжХ
ХК-моль~1 и 0,008 кДж-моль соответственно. Принятые значения совпадают с
рекомендованными КОДАТА—МСНС [864]. Измерения теплоемкости ВеО в работах [1224, 1315, 1653] менее
точны и не учитывались в справочнике.
В интервале 298,15—2373 К для теплоемкости ВеО (к) принято уравнение, выведенное методом
Шомейта по результатам измерений энтальпии в работах Родигиной и Гомельского [348] B98—
1128 К), Виктора и Дугласа [2774] B73-1173 К), Кандыбы и др. [144] A142-2334 К) и Шпиль-
райна, Кагана и Бархатова [460] B023—2266 К). Погрешность данных [348, 2774] принята
равной 0,5%, данных [460] — 1%, а данных [144] — 1 и 1,5% при Т < 2100 К и Т > 2100 К
соответственно. Измерения энтальпии ВеО (к), выполненные Гринбаумом и др. [1252] B270—
2525 К), не учитывались ввиду их систематического занижения (примерно на 13%) по сравнению
с данными [144, 460]. Температура полиморфного превращения принята по хорошо согласующимся
между собой данным [877] B373+10 К), [1821] B371 ±10 К) и [2433] B378+8 К). Менее точные
данные [187, 563, 1032, 2552, 2668], приводящие к значениям в интервале 2295—2358 К, не
учитывались. Энтальпия превращения @,265 кДж-моль) вычислена на основании выбранных
уравнений для энтальпии а- и Р-модификаций ВеО. Теплоемкость Р-ВеО в интервале 2373—2851 К
принята равной 56+2 Дж-К~1-моль на основании измерений энтальпии в работах [460] B373—
2828 К) и [144] B400—2820 К). Температура плавления ВеО B851+12 К) найдена Бархатовым
и др. [30]; с ней удовлетворительно согласуются данные [1252] B853 К), [52] B843 К), [304]
B835 К), [2339] B838 К). Измерения Тт (ВеО) в работах [144, 1032, 1186, 2404, 2803] приводят
к более низким значениям B720—2830 К) и не учитывались в настоящем справочнике. Энтальпия
плавления ВеО (86 +6 кДж -моль) и значение теплоемкости жидкого ВеО (84 +8 Дж -К -моль)
приняты по данным Шпильрайна и др. [460], которые исследовали энтальпию ВеО (ж) в
интервале 2850-3159 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 6000 К оцениваются
в 0,02, 0,2, 1 и 8 Дж-К-моль~1 соответственно. Расхождения между термодинамическими
функциями ВеО (к), приведенными в табл. 731 и в справочниках [105, 106, 1543], не превышают
0,5 Дж-К~1-моль" в значениях Ф° (Г). Для ВеО (ж) соответствующие расхождения достигают
4 Дж-К~1-моль благодаря существенному уточнению энтальпии плавления и теплоемкости
жидкого ВеО в работе Шпильрайна и др.
В справочнике принята энтальпия образования кристаллического оксида бериллия
AfH° (ВеО, к, 298,15 К) = —609,4 + 2,5 кДж • моль.
Это значение, совпадающее с рекомендацией КОДАТА—МСНС [864], основано на
приведенных в табл. 25.4 измерениях [171, 630, 694, 1144, 1706, 2684]. Полученные в этих работах методом
растворения данные пересчитаны и скорректированы в обзорной работе Паркер [2186]. В
литературе известны также значения к*Н° (ВеО, к), полученные сожжением бериллия в кислороде
[883, 2019, 2057, 2126, 2129, 2379]. Есть основания предполагать, что в этих работах сгорание
бериллия протекало не полностью, так как даже в лучшей из них — работе [883] — не был
проведен анализ на полноту сгорания бериллия, что привело к заниженным значениям энтальпии
образования. Обработка результатов измерений энтальпии растворения ВеО (к) в плавиковой
кислоте с использованием данных [536] приводит к менее точному значению (—602,1 +
+6 кДж-моль), так же как исследование равновесия образования ВеО (к) в работе [383].
234
Бериллий и его соединения ВеО (г)
Таблица 25.4. Результаты определений КгН° (ВеО, к, 298,15 К) (в кДж-моль")
Авторы
Метод
Д/Я°(ВвО. к, 298,15 К)
Нейман и др., 1934 [2126, 2129] Калориметрическое измерение энтальпии сгора- —607,9
ния Be (к) в N2 и ВезМ2 (к) в О2
Косгров, Снайдер, 1953 [883] Калориметрическое измерение энтальпии сгора- —598,7
ния Be (к) в О2
Фрикке, Вульхорста, 1932 [1144] Калориметрическое измерение энтальпии реак- —608,8+6,0
ции растворения ВеО (к) в плавиковой кислоте
Колесов и др., 1959 [171]; Калориметрическое измерение энтальпии реак- —609,6+2,5
Килди и др., 1973 [1706] ции растворения ВеО (к) в плавиковой кислоте
Килди и др., 1973 [1706]; Килди, Калориметрическое измерение энтальпии раство- —608,8+5,0
Чарни б рения ВеО (к) и BeF2 (к) в плавиковой кислоте
Томпсон и др., 1962 [2684]; Блачник Калориметрическое измерение энтальпии раство- —609,6+2,5
и др., 1968 [694]; Килди и др., 1973 рения ВеО и Be в соляной кислоте
[1706]
а В расчете использованы данные Бэра и Тернбулла [630] по растворению Be (к) в плавиковой кислоте.
6 Результаты этих измерений известны по работе Паркер [2186].
Давление пара оксида бериллия в реакции ВеО (к, ж)=ВеО (г) вычислено на основании
значения АГН° @) =737,482 +11 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования кристаллического и газообразного оксида бериллия.
ВеО (г). Термодинамические свойства газообразного оксида бериллия в стандартном состоянии
при температурах 100—10 000 К приведены в табл. 732.
В табл. 25.5 приведены молекулярные постоянные ВеО, принятые на основании спектральных
исследований и квантовомеханических расчетов. В электронном спектре ВеО исследованы три
«истемы полос, связанные с переходами между тремя валентными состояниями АгП—X1 ?+т
В1!,*—АгП и B1i:+—X1I,+ [1397, 1790, 2660, 2697]. Неэмпирические квантовомеханические
расчеты с учетом корреляции электронов [579, 1512, 1601, 2164, 2201, 2441, 2766] показали, что
помимо трех состояний, известных из спектральных исследований, молекула ВеО должна иметь
еще два связанных низколежащих триплетных состояния: а3П (конфигурация . . Ап?5 а), которое
коррелирует с атомами Be С1^) и О CРд), и 63S+ (конфигурация . . ,4а1я*5о), коррелирующее
с атомами Be CР,) и О CРд). О существовании этих состояний свидетельствуют также возмущения
в системах А1!!—Х1Е+ и В1 ?+—X1 Е+, отмеченные Лагерквистом [1790] и частично
проанализированные Хуо, Фридом и Клемперером [1512]. По аналогии с молекулой MgO, для которой
имеется детальный квантовомеханический расчет всех связанных электронных состояний,
образованных валентными состояниями атомов [2446], следует ожидать существование у молекулы ВеО
ряда синглетных и триплетных состояний с энергиями выше 25 000 см. С этими состояниями
связаны, по-видимому, многочисленные полосы в области 2600—3600 А (см. [1790]), которые,
однако, не были проанализированы из-за сложности их структуры.
В табл. 25.5 приведены данные для пяти валентных состояний ВеО. Молекулярные постоянные
в состояниях X1 ?+, А1!! и В1 S+ приняты по результатам анализа указанных систем полос
{v (Х1!*) <! 9, v (А1!!) «^ 11, v (В1!,*) ^ 11), проведенного Лагерквистом [1790]. В состояниях
а3П и Ъ3 2+ постоянные Вв и а>й приняты равными соответствующим величинам в изоконфигура-
ционных состояниях АХИ и #Х2+. Энергия возбуждения состояния а3П получена в работе [1512]
на основании приближенного анализа возмущений в системе АгИ—X1 S+. Принятое значение
хорошо согласуется с квантовомеханическими расчетами [1512, 2164, 2201, 2441] и
соответствующими данными для оксидов других щелочноземельных металлов [1080]. Величина Te(b3L+)
принята на основании расчетов [2164, 2201, 2441]. Погрешность значений Т, (а3И) и Те (Ь3 Е+)
не превышает 2000 см.
Термодинамические функции ВеО (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QB1[ и ее производных вычислялись по соотношениям A. 90)—A. 92) с учетом четырех
возбужденных электронных состояний. Статистическая сумма по колебательно-вращательным
235
ВеО (г)
Глава 25
Т а б л и
Молекула
ВеО
ВеН
BeF
Ве85С1
Ве"Вг
Bel
Be82S
i
Ча 25.5. Молекулярные постоянные ВеО, ВеН, BeF, ВезбС1, Be79Br, Bel,
Состояние
441
Л82+
Ха2+
лт
xas+
4аП
Ва2+
С22+
xas+
4'П
BaS+
zas+
4аП
Ха2+
4аП
хч:+
о3!!
Ь3?+
Примечание. Ниже вес
ВеО:
ВеН:
BeF:
Be^Cl:
Ве7»Вг:
Bel:
Be^S:
г
г
г
г
8
s
t
^^=0,02235
соотношению
У,
0
8 000
9 405,43
15 000
21 253,43
0
20 033,19
27 600
0
33 234,3 б
49 529,3
50 376,3
0
27 992,9 а
48 827,6 б
0а
26 444,2 в
0а
23 724,8 Д
0
7 000
7 960,1
18 700
25 941,8
; постоянные
6 Я0=12,6
1487,323
1140
1144,238
1370
1370,817
2060,78
2088,58
1270
1265,9
1171,1
1356,4
1408,2
847,19
822,11
925,5 в
715,06
702,29
611,7
603,8
997,94
—
762,46
851,35
даны в см"
СМ
11,8297 а
11,1 в
8,4145 в
10,6 3
7,7455 и
36,31 а
40,41 г
—
9,35»
8,53
15,75 в
4,94 ж
5,14
5,24
—
4,30б
3,98 б
1,6
2,1
6,137
4,12
_
4,85
Be
1,651 0
1,366
1,366 1
1,58 у
1,575 8
10,316 4
10,456 7
3,6
1,488 9
1,420 2
1,558 г
1,570
0,728 5
0,709 4
0,775 1
0,548 0 в
0,535 3 ж
0,422 7
0,422 7 в
0,790 59
0,659 0
_
0,728 94
• 10~12; в оценено по соотношению A
A.69); Д оценено по соотношению A.68); е ш.у
по соотношению A.67) и Do
«¦>.Sf.=-0,38;
г ш<Уе=_о,4
й,вувг=0,033;
б «2=_2,7.
7; Д а2=—4
6 4=21,8;
(bs 2+)=21 400; и <*еу,=-
Ю-8; в р^З.Э-Ю-", Н.=
,2-10.
8 ».у.=0,5;
г рассчитано
A.69); е оценено по соотношению A.68); ж ъ>вуе=
«4=52,8-1,4
Г=0,0242; б
в рассчитано
ж рассчитано
т=0,0459; б
6 рассчитано
1 а2=—2-Ю-8;
(у+1/г); приведено Го;
в уравнения
в приведено
-0,0027; к f
=8,75-Ю-8, h
через Во=1
= —1,1.
Дбу,.
<vio2
1,0
1,6 г
1,628 ж
1,7 г
1,54
30,3 б
32,22 Д
—
1,76
1,75
2,3 Д
1,4
0,69
0,68
—
0,43 Т
0,42'
0,17 в
0,22°
0,664»
—
0,605 б
—
0,604
Be32S
D..10»
8,195 б
7,7 Д
7,7
8,4 Д
8,4 к
1022,1 в
1038
—
8,28
8,30
8,2 е
7,8 е
2,5
2,3
3,5
1,31 Д
1,31 Д
0,82 г
0,73 г
2,02
—
2,00
—
2,12
г.
А
1,331
1,463
1,4632
1,36
1,3623
1,34264
1,33359
2,27
1,3609
1,3935
1,330
1,325
1,7970
1,8211
1,7422
1,9489
1,9719
2,1770
2,1771
1,7415
—
1,9075
—
1,8137
67) и D0(asII)=28 400; г оценено по
.=0,0338;
ж a2=5,5-10~B; 3 оценено
!1=0,07.10-в;Яв=27-10-«
1=7,3-10"9
,547 и сч;
, Ье=3,8-10-1а
Д оценено по
для кантов полос введен коррегирующий множитель 6=1,2 (у'+!
из So=O,5459 и ах; г <
из Я0=0,5332 и oi.
рассчитано из Bq= 0,4219
из 5о=О,4216 и ох для
6 «.-2,5.1<
эценено по соотношению A.69); д приведено Do',
и ai; B оценено по A.69);
подсостояния
4гП1д, Вй(А
, yi=2-10 ls.
т z1=6.io~12;
соотношению
/г)-(г/'+1/2');
е 4=198+6;
г приведено DB; Д 4=361,1+0,05;
12П,/5)=0,41
82.
уровням энергии всех состояний и их производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по колебательным уровням энергии и интегрированием по J
с помощью соотношений типа A. 82). В расчете учитывались все колебательно-вращательные
уровни состояний X1S+, а3П, А1!!, Ь3Е+ и S1?+ со значениями / ^ ^т»1,0'> последние
находились из условий A. 81). Колебательно-вращательные уровни энергии указанных состояний
вычислялись по уравнениям A. 65), A. 60), A. 61) со значениями YkI, полученными с учетом
условий A. 50). Последние были обеспечены для состояний X1 Е+, А1!! и В1 ?+ введением дополнитель-
236
Таблица 25.6. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см '), а также Значения v max и/11Ш,
принятые для расчетов термодинамических функций ВеО, ВеН, BeF, BeCl, ВеВг, Bel, BeS
Коэффициенты
Те
Ую-
X 20*
Узо-
Ум-
У5о
У«
Уц
у«
У31
^02
у»
Уоз-
Ум
(а„=?
«1
а2
аз
ю-*
10
ю-1
10
103
10
103
10*
• ю5
ю7
1010
ю11
>.vio-
ю4
•ю8
шах
Jam
ХЧ+
0
1,487 338
—1,183 688
0,235 082
—0,059 377
—
1,651 0
—0,190
—
—
-0,819 5
—
0,126
—
—
—
—
74
238
а3П
0,800
1,144
ВеО
Am
0,940 543
1,144 407
-1,11 -0,848 456
—
—
1,366
0,434 306
-0,410 253
—
1,366 1
—0,16 —0,162 8
—
—
—0,77
—
—
—
—
—
—
—
51
196
Примечание. Ниже все постоянные
ВеН
BeF
: а Состояние Л2П
Те 20 033,
: а Состояние А2И
Те 33 234
19 27 600
Ва2+
3 49 529,
0,055
—
-0,77
—
—
—
—
—
—
—
70
242
даны в см
С8 2+
3 50 376,3
63Х+
1,500
1,372
-1,06
—
—
—
1,585
-0,17
—
—
ВЧ+
ВеН
2,125 343 0
1,372 073 2,069 997
-0,826 848 -4,399 872
0,712 529 19,719 9
-3,085 93 —237,139
_ _
8 1,575 8 10,323 5
-0,154 -3,23916
— 12,6501
- -32,168 4
-0,84 -0,841 -102,21
—
—
—
—
—
—
—
64
224
-X
0,7 39,0
0,27 -
-0,2
— 1,706 40
— 0,558 261
— 7,204 627
— —7,201515
43 12
219 56
BfiCl: a Тв(А*П)=21 992,9
ВеВг: а Г,(Л*П)=26 444,2
Bel: a Г,(Л2П)=23 724
BeS: a Состояние а'П
Тв 7000
BeF
X.S+.
0
1,266 343
-0,948 688
0,464 573
-0,413 290
—
1,488 9
-0,176
—
—
-0,828
—
0,094 24
—
—
—
—
—
71
243
Am
7960,
BeCl
0
0,845 664
—0,503 524
—0,058 846
,
—
0,726 5
—0,068 7
—
—
—0,249
—
0,046 8
—
—
—
—
—
73
286
ВеВг
X2S+*
0
0,715 002
-0,427 426
—0,029 731
—
—
0,548 0
-0,043 0
—
—
—0,131
—
—
—
—
—
—
—
76
303
b» 2+ B1 2+
t 18700 25941,8
Bel
X*V
0
0,611
—0,136
-0,391
—
—
0,439
—0,017
—
—
—0,175
—
0,031
—
—
—
—
—
60
349
a
327
817
929
4
2
9
0
-0
0
0
0
—0
0
—0
-0
BeS
-)
997
605
135
a
517
645
118
118 827
—
790
,067
011
—
,201
—
,021
_
—
—
—
—
80
296
213
639
686
756
655
ВеО (г)
Глава 25.
ных членов с коэффициентами Yi0 в уравнение A. 65). Соответствующие значения Ykl, а также-
ушах и Jlim приведены в табл. 25.6. Мультиплетность и вырождение уровней в состояниях а*П,
Л41, Ъ3 ?+ учитывались статистическими весами 6, 2 и 3 соответственно.
Погрешности вычисленных термодинамических функций при температурах до 1000 К
обусловлены неточностью фундаментальных постоянных, при более высоких температурах —
неточностью принятых энергий возбуждения состояний а3П и Ъ3 ?+, а также отсутствием данных о
валентных электронных состояниях с энергией выше 25 000 см. Общие погрешности в значениях
Ф° (Т) при 298,15, 3000, 6000 и 10 000 К могут достигать 0,02, 2, 3 и 5 Дж-К^-моль
соответственно.
Ранее термодинамические функции ВеО (г) вычислялись в предыдущих изданиях настоящего
справочника [105, 106], а также в справочниках JANAF [1543, 1544], Мак-Брайд и др. [1974]»
Мадера [1904] и в работах Брюэра и Розенблатта [745] (до 3000 К) и Шнейдера [2463]. Данные
[1544] и табл. 732 удовлетворительно согласуются во всем интервале температур; имеющиеся
расхождения, достигающие 0,5, 1 и 0,8 Дж-К-моль~1 в значениях Ф° F000 К),: 5° F000 К)
и С\ F000 К), обусловлены использованием в [1544] упрощенной методики расчета и учетом
шести электронных состояний с энергиями от 37 000 до 47 000 см'1. Результаты остальных
расчетов, за исключением [745], при Т ^ 1000 К также практически совпадают с приведенными
в табл. 732. При более высоких температурах расхождения монотонно возрастают, достигая
6—9 Дж-К~1-моль~1 в значении Ф° F000 К), и объясняются тем, что в [1543] не учитывались^
а в [105, 106, 1904, 1974, 2463] учитывались только частично низколежащие возбужденные
состояния ВеО. Значительные расхождения данных [745] с приведенными в табл. 732 обусловлены
неверной оценкой энергии состояний а3П и Ъ3 2+.
Константа равновесия реакции ВеО (г)=Ве (г)+О (г) вычислена на основании принятой
в справочнике энергии диссоциации
ДГЯ° @) = Do (ВеО) = 435 + 10 кДж ¦ моль т=а 36 400 ± 800 см.
Это значение является средним из приведенных в табл. 25.7 величин.
Таблица 25.7. Результаты определений D0(BeO) (в кДж-мошГ1)
Авторы
Метод
Чапка и др., 1959 [852] Мйсс-спектрометрический, ВеО (г)=Ве (г)+О (г),
1914-2304 К, 6 точек
То же, ВеО (г)+О (г)=Ве (г)+О, (г), ДГЯ° @)=
=-48,7+1,9 (III)
Тоже, ВеО(к)=ВеО(г), Д,#°@)=737,3±1,3(Ш)
Серд, Хилденбранд, 1964 [2679] Масс-спектрометрический, ВеО (г)—Be (г)+О (г),
2380 К, 1 точка
То же, ВеО (г)+О (г)=Ве (г)+О2 (г), Дг#°@)=
=—63,3 (III)
То же, ВеО (к)=ВеО (г), Д8ЯВ @)=--738,9 (III)
D,
II закон
441 ±22
455 ±50
440 ±24
—
—
—
(ВеО)
III закон
434,9±7,0
444,9±7,0
435,2+10
432,3+8
430,3+8
433,6+11
Как видно из таблицы, результаты измерений находятся в хорошем соответствии. Основной
вклад в погрешность приведенных величин дает неточность использованных в расчетах сечений
ионизации. Результаты измерений давления пара над оксидом бериллия, проведенных эффузион-
ным методом и методом испарения с поверхности [276, 1039], не могут служить для выбора
энтальпии сублимации ВеО, так как масс-спектрометрические измерения [852, 2679] и
термодинамические расчеты [253] показали, что основными компонентами пара являются Be (г) и О (г).
Линейная экстраполяция колебательных уровней состояния ЛХП по данным [1790] приводит к
завышенному значению Do (ВеО) =465 кДж-моль.
Принятому значению энергии диссоциации соответствует
Д/?Г°(Ве0, г, 0) = 131,536 ± И кДж • моль.
238
Бериллий и его соединения
Ве2О (г), Ве2О2 (г)
Ве2О (г). Термодинамические свойства газообразного оксида дибериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 733.
Молекулярные постоянные, использованные для расчета термодинамических функций Ве2О,
приведены в табл. 25.8. Экспериментальные данные о строении и основных частотах молекулы
Таблица 25.8. Значения молекулярных постоянных, а также а и р%, принятые для расчета
термодинамических функций Ве2О, ВеОН, BeF2, BeCI2, ВеВг2 и Ве12
Молекула
Ве2О
ВеОН
BeF2
ВеС12
ВеВг2
Ве12
Состояние
х*и-
Х22+
Jf * 2+
х*ц
620
1200
. 730
410
250
175
М2)
см
320
320
345
250
220
190
^3
1200
3700
1530
1110
990
870
7.1039
г-см2
5,9
2,2
12,4
36,1
97
186
а
О
1
2
2
2
2
Pi
3
2
1
1
1
1
Вег0 в литературе неизвестны. Оценки соответствующих величин выполнены в справочнике
JANAF [1543] и в работе Серда и Хилденбранда [2679]. В справочнике, как и в [1543, 2679],
принята линейная симметричная структура молекулы Ве2О (точечная группа D^^. Основное
электронное состояние Ве2О принято триплетным CЕ~), а межатомное расстояние Be—О равным
1,4 А, как в ВеОН. Соответствующее значение момента инерции (см. табл. 25.8) имеет погрешность
около 1-109 г-см2. Основные частоты v2 и v3 оценены по частотам молекулы ВеОН; для vx оценка
выполнена по величине v3 в приближении простого силового поля. Погрешности принятых
значений оцениваются в 60—100 см. Оценка частот в таблицах [1543] привела к более высоким
значениям, оценка в [2679] — к более низким. Молекула Ве2О, по-видимому, имеет по крайней
мере одно низкорасположенное синглетное возбужденное состояние.
Термодинамические функции Ве2О (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126)
и A. 129) без учета возбужденных состояний. Погрешности вычисленных значений определяются
главным образом неточностью молекулярных постоянных, в том числе, отсутствием данных о
возбужденных электронных состояниях, а также приближенным характером расчета; они
составляют 3, 5 и 8 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Ве2О (г) вычислялись в справочнике [1543] и в работе
[2679] (для Г=2300, 2400 и 2500 К). Расхождения результатов расчета [1543] и табл. 733
возрастают с температурой от 3 до 13 Дж -К -моль для Ф° (Г); они обусловлены в основном разной
оценкой частот колебаний. Расчет [2679] выполнен в предположении, что молекула Ве2О имеет
синглетное основное состояние. При указанных температурах это частично компенсирует низкие
значения основных частот и приводит к удовлетворительному согласию данных [2679] и табл. 733
Константа равновесия реакции Ве2О (г)=2Ве (г)+О (г) вычислена с использованием значения
\ГН° @)=926,289±27 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH° (Ве2О, г, 0) — —40 + 25 кДж ¦ моль.
Эта величина является средней из значений —49+25, —28+35 и —45 кДж-моль,
рассчитанных на основании данных по константам равновесия реакций 2ВеО (к)=Ве2О (г)+О (г), 2ВеО (г) =
=Ве2О (г)+О (г) и Ве2О2 (г)=Ве2О (г)+О (г) в интервале температур 2345—2497 К. Эти данные
получены при масс-спектрометрическом исследовании испарения оксида бериллия в работе [2679].
Ве2О2 (г). Термодинамические свойства газообразного диоксида дибериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 734.
Молекулярные постоянные, используемые для расчета термодинамических функций Ве2Оа,
приведены в табл. 25.9. Экспериментальные или расчетные данные о строении и основных
частотах молекулы Ве2О2 в литературе отсутствуют. Оценки этих величин выполнены в справочниках
239
(г)
Глава 25
Таблица 25.9.
Значения молекулярных постоянных, а также о и р%, принятые для расчета
термодинамических функций Ве2О2, ВезОз, Ве*О4, Ве(ОНJ, Be2F4 и Ве2С1<
Ве2О2
ВезОз
Ве4О*
Ве(ОНJ
Be2F4
Be2CL,
Ве(ОНJ: а 1
frAi
&А9
11 А 9
1200
1200 C)
1200 (8)
700
1275
860
1ривецено значение I-
Be2F4: a v7=260 см,
Ве2С14: а >
'7=180 см-1,
vs=158 (
1100
1000
600
320
541
450
10" r
v8=80 см,
С1
1000
B) 700 B)
(8) 300 B)
B) 1400
336
200
•смг.
ve=842 см,
v»=608 см
г1
800
650
—
3700 B)
806
600
мы=173
, vlo=130
600
450 B)
—
320 D)
242
180
СМ,
СМ,
и
200
250 B)
—
—
170 а
130 а
vu=1251 см,
vH=857 см,
1В1с-№
Г3 • СМ6
180
9,1.10s
1,1.10е
12 а
1,8-10*
4,1.10е
v12=503 см-1.
v12=450 см
4
6
8
2
4
4
РХ
1
1
1
1
1
1
JANAF [1543] и Мак-Брайд и др. [1974] для расчета термодинамических функций. В настоящем
справочнике принимается, что в основном состоянии ХгАд эта молекула имеет ромбическую
структуру симметрии ZJA с -^ О—Be—0=55° и г (Be—О)=1,63 А. Соответствующее значение IaIbIo
(см. табл. 25.9) имеет погрешность около 5 -Ю16 г3-смв. Оценка основных частот Ве2О2 выполнена
по основным частотам Li2Oa, известным из квантовомеханического расчета [2881]. Погрешности
принятых значений составляют 50—100 см.
Термодинамические функции Ве2О2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности вычисленных значений определяются
главным образом неточностью молекулярных постоянных, а при высоких температурах
приближенным характером расчета и составляют 4, 6 и 10 Дж -К -моль в значениях Ф° (Г) при 298t15,
3000 и 6000 К.
Ранее таблицы термодинамических функций Ве2О2 (г) вычислялись в справочниках [1543,
1974]. Расхождения результатов расчета [1543] с данными табл. 734 возрастают с температурой
и составляют от 1 до 13 Дж-К^-моль в значениях Ф° (Т); они обусловлены в основном
различием принятых в расчетах основных частот Ве2О2. Результаты расчета [1974] согласуются с
данными настоящего справочника в пределах 1 Дж-К-моль. Приведенные в табл. 734 значения
энтропии согласуются в пределах погрешностей с полученными по результатам масс-спектрометри-
ческого изучения равновесия реакций с участием Ве2Оа (г) [852].
Константа равновесия реакции Ве2О2 (г)=2Ве (г)+2О (г) вычислена с использованием
значения АГН° @)=1518,072+27 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(ВваОя,
г, 0) = —385 + 25 кДж
моль
Эта величина является средней из результатов масс-спектрометрических исследований
испарения оксида бериллия в работах Серда и Хилденбранда [2679] и Чапки и др. [852]. Серд и Хил-
денбранд провели измерение при 2380 К, ему соответствует значение А^Н0 @)=—400 кДж-моль.
Чапка и др. провели исследование в интервале 1914—2304 К, однако парциальные давления
приведены в этой публикации только для температуры 2242 К. По данным этой работы вычислено
значение —364 кДж-моль.
Ве3О3 (г). Термодинамические свойства газообразного триоксида трибериллпя в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 735.
Молекулярные постоянные, использованные для расчета термодинамических функций Ве3О3,;
приведены в табл. 25.9. Экспериментальные данные о строении и основных частотах молекулы
Ве3О3 в литературе отсутствуют. Оценки этих величин выполнены в справочниках JANAF [1543]
и Мак-Брайд и др. [1974]. На основании этих оценок в настоящем справочнике для молекулы
Ве3О3 в основном состоянии XхА[ принимается структура плоского шестичленного кольца (точеч-
240
Бериллий и его соединения Ве4О4 (г)
ная группа D3h) с величиной г (Be—О)=1,63 А. Соответствующее значение 1А1в1с (см. табл. 25.9)
имеет погрешность 2-Ю14 г3-см6. Основные частоты Ве3О8 в справочнике [1543] оценены по
аналогии с молекулами (НВОK и (НВО2K, однако при этом не введена поправка на замену атомов В
в кольце атомами Be. В настоящем справочнике принимаются значения основных частот, скоррели-
рованные соответствующим образом. Оценка [1974] представляется весьма приближенной.
Погрешность принятых значений оценивается в 100—150 см.
Термодинамические функции Ве3О3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности вычисленных значений определяются главным образом неточностью
молекулярных постоянных, а также приближенным характером расчета; они составляют 5, 10 и
15 Дж-К-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций Ве3О3 (г) вычислялись в справочниках [1543,;
1974]. Результаты расчетов [1543, 1974] близки между собой, но заметно отличаются от данных
табл. 735. Соответствующие расхождения возрастают с температурой и составляют от 4—6 до 26—
27 Дж-К -моль в значениях Ф° (Г). Рассчитанные в настоящем справочнике значения
5° (Ве3О3, г) согласуются в пределах погрешностей с вычисленными по результатам масс-спектро-
метрического изучения равновесия реакций с участием Ве3О3 (г) [852].
Константа равновесия реакции Ве3О3 (г)=ЗВе (г)+30 (г) вычислена с использованием
значения Дг#° @)=2719,608+34 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/HBegOg, г, 0) ——1020 + 30 кДж.моль.
Эта величина является средней из полученных по уравнению III закона на основании
результатов масс-спектрометрического исследования процесса сублимации оксида бериллия при 2380 К
в работе Серда и Хилденбранда [2679] (—1016+16 кДж-моль) и при 2242 К|в работе Чапки
и др. [852] (—1028+16 кДж-моль). В работе [852] измерения выполнены в интервале
температур 1914—2304 К и по методу II закона получено Д,# (Ве3О„, г, 298,15 К)=674+25 кДж-моль
для реакции ЗВеО (к)=Ве3О3 (г), откуда следует &fH° (Ве8О8, г, 0)=—1101+25 кДж-моль
(см. Ве2О2 (г)).
Ве4О4 (г). Термодинамические свойства газообразного тетроксида тетрабериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 736.
Молекулярные постоянные, использованные для расчета термодинамических функций Ве4О4>
приведены в табл. 25.9. Экспериментальные данные о строении и основных частотах молекулы
Ве4О4 в литературе отсутствуют. Оценки соответствующих величин выполнены в справочниках
JANAF [1543] и Мак-Брайд и др. [1974]. В настоящем справочнике для молекулы Ве4О4 в
состоянии ^гА1р принимается структура плоского правильного 8-членного цикла (точечная группа
Dih) с г (Be—О)=1,63 А. Соответствующее значение IaIbIc (cm. табл. 25.9) имеет погрешность
около 4-1013 г3-cm6. Значения основных частот приведены в табл. 25.9 по оценке Хилденбранда,
выполненной для справочника [1974]. Эти значения согласуются с основными частотами
циклических молекул Ве2О2 и Ве3О3. Погрешности принятых значений могут достигать 100—200 см.
Попытка авторов [1543] выполнить более детальную оценку основных частот Ве4О4 не
представляется убедительной, хотя ее результаты не противоречат принятым значениям частот.
Термодинамические функции Ве4О4 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Погрешности вычисленных значений
определяются главным образом неточностью молекулярных постоянных, а также приближенным
характером расчета; они составляют 6, 15 и 25 Дж-К-моль~1 в значениях Ф° (Т) при 298,15,
3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций Ве4О4 (г) вычислялись в справочниках [1543,
1974]. Результаты расчета [1974] совпадают в пределах 0,1 Дж-К~1-моль~1 с данными табл. 736;
расхождения с данными [1543] достигают 7 Дж-К~1-моль~1 в значениях Ф° (Т) и обусловлены
разной оценкой основных частот Ве4О4.
Константа равновесия реакции Ве4О4 (г)=4Ве (г)+40 (г) вычислена с использованием
значения Дг#° @)=3906,144+32 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(Ве4О4, г, 0) = —1640 + 40 кДж • моль.
241
ВеН (г) Глава 25
Эта величина является средней из вычисленных по методу III закона на основании
результатов масс-спектрометрического исследования сублимации оксида бериллия при 2380 К в работе
Серда и Хилденбранда [2679] (—1638 + 25 кДж-моль") и при 2242 К в работе Чапки и др. [8521
(—1643 кДж-моль). В работе [852] измерения выполнены в интервале температур 1914—
2304 К и по уравнению II закона получено AHS° (Ве4О4, г, 2150 К)=741+35 кДж-моль для
реакции 4ВеО (к)=Ве4О4 (г). Этому значению соответствует величина Д^0 (Ве4О4, г, 0) =
= —1623+35 кДж-моль, хорошо согласующаяся с принятой в справочнике.
ВеН (г). Термодинамические свойства газообразного гидрида бериллия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 737.
В табл. 25.5 приведены молекулярные постоянные ВеН, принятые на основании результатов
исследований спектров и квантовомеханических расчетов. В спектрах ВеН наблюдалось шесть
систем полос (см. [649, 872, 1480, 2161, 2813, 2814]), связанных с переходами между семью
электронными состояниями, однако только одно из шести возбужденных состояний является
валентным (А2Щ; остальные состояния должны рассматриваться как ридберговские и имеют энергии
свыше 50000 см. Квантовомеханические расчеты [833., 1029, 1839] показали, что из трех
состояний DП, 4Е+ и2Е+), имеющих общий диссоциационный предел с состоянием А2П (Be (SP)+H (*S))r
только состояние С22 + является связанным.
Приведенные в табл. 25.5 постоянные ВеН в состояниях Х2? + и А2П найдены Хорном и
Коленом [1480] из анализа системы полос А2П -» Х2?+ с учетом данных Ульссона [2161] по спектру
поглощения ВеН. Постоянные ВеН в состоянии С22+ приняты по результатам неэмпирического
расчета Лефевр-Брион и Колена [1839].
Термодинамические функции ВеН (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения (?вн и ее производных по температуре вычислялись по уравнениям A. 90)—A. 92) с
учетом двух возбужденных состояний в предположении, что (?<*>, ве=^(р{!Рх) QifJ BP- Статистическая
сумма по колебательно-вращательным уровням состояния Х2?+ и ее производные находились по
уравнениям A. 70)—A. 75) непосредственным суммированием по колебательно-вращательным
уровням энергии. В расчетах учитывались все уровни энергии, ограниченные предельной кривой
диссоциации. Колебательно-вращательные уровни состояния Х2?+ вычислялись по уравнениям
A. 62), A. 65). Значения коэффициентов YM и величин ршах и /Иш, использованные в расчетах,,
приведены в табл. 25.6. Коэффициент У40 определялся из условий A. 50), а коэффициент Y31—
из условий A. 57). Мультиплетность и вырождение вращательных уровней энергии учитывались
соответствующими статистическими весами.
Постоянные ВеН в состоянии Х2Б+ известны с достаточной точностью, поэтому погрешности
вычисленных термодинамических функций при Т ^ 3000 К обусловлены лишь неточностью
фундаментальных постоянных и не превышают 0,02 Дж'К~1-моль~1. При более высоких температурах
становятся существенными погрешности из-за неточности принятого значения энергии
диссоциации ВеН и пренебрежения всеми возбужденными электронными состояниями, кроме состояний
А2И и C2S+. Общая погрешность в значении Ф° F000 К) может достигать 0,15 Дж-К-моль.
Термодинамические функции ВеН (г) вычислялись ранее в предыдущих изданиях настоящего-
справочника [105, 106], таблицах JANAF [1543], справочниках Мак-Брайд и др. [1974], Мадера
[1904] и Хара и др. [1336] до 5000 К. Кроме того, в работе Шнейдера [2463] выполнен расчет Qm
и приведены полиномы, аппроксимирующие значения QBK и In QBB в интервалах 1000—4300 К ir
1000—4600 К. Пренебрежение необходимостью корректного ограничения числа колебательно-
вращательных уровней энергии и возбужденными состояниями ВеН, а также использование
несколько отличающихся постоянных этой молекулы привело к тому, что при Т ^ 4000 К
результаты предыдущих расчетов существенно отличаются от данных табл. 737. Соответствующие
расхождения достигают 6 Дж-К-моль в С°р F000 К) [1543, 1974] и 0,7 Дж-К^-моль
в Ф° F000 К) [105].
Константа равновесия реакции ВеН (г)=Ве (г)+Н (г) вычислена на основании значения
ДД° @) = Do (ВеН) = 16 040 + 400 см ?«191 + 5 кДж • моль.
Эта величина принята по результатам квантовомеханического расчета, выполненного Багусом
и др. [556] по методу ЛКАО МО KB с учетом в качестве верхнего предела значения, найденного
Коленом и Де-Грифом [872] при интерпретации предиссоциации в состоянии 2?2П. Она согласу-
242
Бериллий и его соединения ВеН2 (аморф.), ВеОН (г)
ется с результатами других квантовомеханических расчетов [805, 1863, 2015], а также с энергией
диссоциации, оцененной Гейдоном [1183] графической экстраполяцией колебательных уровней
состояний А2П и .Х22+.
Принятой энергии диссоциации соответствует
г, 0) = 344,787 ± 7,1 кДж ¦ моль.
ВеН2 (аморф.). Термодинамические свойства аморфного дигидрида бериллия при температурах
100—500 К приведены в табл. Д.54.
Значения постоянных, принятые для расчета термодинамических функций ВеН2 (аморф.),
приведены в табл. 25.1. В литературе отсутствуют сведения о существовании кристаллического
ВеН2. Все описанные методы получения дигидрида бериллия по обменным реакциям (например,
по реакции в эфирном растворе BeCl2+2LiH=BeH2+2LiCl) приводят к образованию белого
твердого рентгеноаморфного вещества, которое при обычном давлении диссоциирует на элементы при
нагревании до ~400 К. Экспериментальные измерения энтальпии образования аморфного ВеН2
(см. ниже), а также данные [365] по теплоемкости аморфного ВеН2 и вычисленные по ним значения
термодинамических функций приводят к положительным значениям Ду,(? (BeH2, аморф., 298,15 К),
что свидетельствует о его термодинамической нестабильности по отношению к Be (к) и Н2 (г) Ч
При Т <С 298,15 К термодинамические функции ВеН2 (аморф.) вычислены на основании изме^
рений теплоемкости, проведенных Саморуковым, Кострюковым и др. [365] в интервале 14^
306 К для образца, содержащего 97,6% ВеН2, 1,59% Ве(ОНJ, 0,27% ВеО, 0,04% Ве2С, 0,05%Н2О
и 0,45% прочих примесей. Точность измерений оценивается в 0,5% при Т ^> 50 К и 14% у нижней
границы измерений; поправка на примеси не превышала 1%. Экстраполяция теплоемкости ниже
14 К проводилась по уравнению С°р=аТ2'16 и привела к значению S° A4 К)=0,088 Дж-К-моль~1,
Погрешности принятых значений S0 B98,15 К)—S° @) и Н° B98,15 К)— Н° @) (см. табл. 25.1)
оцениваются в 0,4 Дж-К~1-моль~1 и 0,035 кДж-моль соответственно. При Т > 298,15 К для
теплоемкости ВеН2 (аморф.) принято уравнение, выведенное на основании приведенного
в табл. 25.1 значения С°р B98,15 К) и значения С°р (800 К)=60 Дж-К~1-моль~1, оцененного
сравнением данных по теплоемкости ВеН2 [365] и LiH [463].
Погрешности вычисленных значений Ф° (Г) при 298,15 и 500 К оцениваются (не принимая
во внимание ошибку, связанную с пренебрежением остаточной энтропией) в 0,2 и 1 Дж-К~1-моль
соответственно. Таблица термодинамических функций ВеН2 (аморф.) в интервале 5—300 К при-*
ведена в работе [365].
В справочнике принята энтальпия образования аморфного дигидрида бериллия
Д7Я° (ВеН2, аморф., 298,15 К) = —19,0 + 1,0 кДж • моль.
Эта величина является округленной средней из результатов измерений Ахачинским и др. [22]
энтальпии растворения ВеН2 (аморф.) в соляной кислоте (—19,3 + 1,0 кДж-моль) и измерений
Ивановым и Карповой [139] энтальпии разложения ВеН2 (аморф.) в калориметрической бомбе
с электрическим нагревом образца (—18,9 + 1,0 кДж-моль). Менее точные величины ранее
получены Уагманом [2786] (—4+20 кДж-моль) и Дюкро и др. [990] (—44 кДж-моль, метод
сожжения в кислороде).
ВеОН (г). Термодинамические свойства газообразного гидроксида бериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 738.
Молекулярные постоянные, использованные в расчете термодинамических функций ВеОН,
приведены в табл. 25.8. В литературе отсутствуют экспериментальные сведения о структурных
параметрах и основных частотах молекулы ВеОН. Неэмпирические квантовомеханические расчеты
с учетом конфигурационного взаимодействия, выполненные в [1а, 1439а], показали, что эта шь-
лекула имеет линейную структуру (симметрия С тв) и основное состояние Х2?+. Этот вывод со?
гласуется с результатами исследования спектра ЭПР молекул ВеОН, изолированных в матрице
из Аг, проведенного Бромом и Велтнером [763]. Приведенный в табл. 25.8 момент инерции вычисг
1 Расчет в двух крайних приближениях, основанных на предположении, что остаточная энтропия ВеН2 (аморф.)
равна 0 или Я In 2, приводит к значениям A^G (ВеН2, аморф., 298,15 К), равным 15,76 и 13,88 кДж-моль"*; соотт
ветствующие равновесные давления диссоциации ВеН2 (аморф.) равны 540 и 270 атм при 298,15 К.
243
Йе(ОНJ (к) Глава 25
лен по оцененным значениям г (Be—О) = 1,40+0,05 А (среднее между величинами re (BeF, )
и ге (ВеО, а3П), см. табл. 25.5) и г (О—Н)=0,96 + 0,02 А. Его погрешность составляет 0,2-109 г-см2.
Значения основных частот приняты на основании приближенных оценок. Частота ^ве-о оценена
так же, как величина г (Be—О), а частоты v2 и v3 по значениям этих постоянных в молекулах гидро-
ксидов щелочных металлов (см. [480, 1783] и т. 4 настоящего издания). Погрешности принятых
значений частот составляют 50—100 см. В работе [1а], выполненной после завершения
подготовки данной главы, в результате квантовомеханического расчета получены значения г (Be—О) =
*=1,399 A, v1=1540, v2=689 и v3=1275 см. Возбужденные электронные состояния ВеОН по
аналогии с изоэлектронными молекулами BeF и ВеС1 должны иметь энергии свыше 25 000 см.
Термодинамические функции ВеОН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических, фуггал^тай. обусловпмш поточностью одененных молекулярных постоянных, в том числе
некоторой неопределенностью структуры этой молекулы, а также иРибдКжвннИ„ хаР„ктаром
расчета; они составляют 2, 5 и 8 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К 1.
Ранее термодинамические функции ВеОН (г) вычислялись в таблицах JANAF [1544];
соответствующие расхождения с данными, приведенными в табл. 738, составляют 3—11 Дж-К-моль
в значениях Ф° (Т) и S° (T).
Эти расхождения обусловлены различием оцененных молекулярных постоянных, особенно v2.
Константа равновесия реакции ВеОН (г) = Ве (г)+О (г)+Н (г) вычислена с использованием
йначения &.ГН° @)=882,57+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(ВеОН, г, 0) = —100 + 40 кДж • моль.
Равновесие реакций с участием ВеОН (г) исследовалось Хилденбрандом и др. [1427] и Ко и др.
11734]. Хилденбранд (работа известна только по данным, приведенным в [1543]) исследовал масс-
спектрометрическим методом равновесие реакции Ве2О (г)+Н2О (г)=2ВеОН (г), пересчет
которого, выполненный авторами [1543], привел к значению &j,H° @) = —105+40 кДж-моль. Ко
и др. [1734] исследовали методом протока B107—2368 К, 24 точки) реакцию ВеО (к)+1/2 Н2 (г) =
=ВеОН (г). Обработка данных [1734] при подготовке настоящего справочника привела к
существенно отличающимся значениям —218 + 30 кДж-моль (II закон) и —205 + 10 кДж-моль
(III закон). Сопоставление энергий разрыва связей Be—X в молекулах ВеХ и средних энергий
связей Be—X в молекулах ВеХ2 (где X=F, C1 и ОН) позволяет отдать предпочтение данным
11427].
Ве(ОНJ (к). Термодинамические свойства кристаллического дигидроксида бериллия в
стандартном состоянии при температурах 100—500 К приведены в табл. Д.55.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В справочнике за стандартное состояние Ве(ОНK (к) принята ромбическая [3-моди-
фикация, устойчивая при комнатной и повышенных температурах 2. При Т < 298,15 К
термодинамические функции |3-Ве(ОНJ вычислены на основании измерений теплоемкости, проведенных
Кострюковым, Саморуковым и др. [185] в интервале 14—310 К на образце, содержащем 98,9%
Ве(ОНJ и примеси NaOH @,45%), стекла @,32%) и Na2GO3 @,22%). Авторы [185] оценивают
точность измерений теплоемкости Ве(ОНJ в 10% при 14 К, 3% при 50 К и 0,2% при 250 К.
Экстраполяция теплоемкости ниже 14 К проводилась по уравнению Дебая и привела к значению
?° A4 К)=0,02 Дж-К^-моль. Погрешность принятых по [185] значений 5° B98,15 К) и
Исследование равновесия реакции образования ВеОН (г) в работе Ко и др. [1734] позволяет рассчитать по
методу II закона значение S° B244 К)=315±15 Дж-К^-моль, находящееся в хорошем согласии с данными
табл. 738 C22±5 Дж-К'^моль). Следует отметить, что расчет этой величины для угловой структуры молекулы
ВеОН с параметрами ^ Be—О—Н=110°, г (Be—О)=1,40 А, г (О—Н)=0,96 A, vx=v2=1200 см"*, v3=3700 см"*
приводит к значению S" B244 К)=314+ 5 Дж-К^-моль.
Согласно [1305] кристаллическая а-модификация и аморфная форма при нагревании в автоклаве до температур
200—300° С переходят в более устойчивую при этих условиях C-Ве(ОНJ. Какие-либо данные по теплоемкости
о-Ве(ОНJ и аморфной формы Ве(ОНJ в литературе отсутствуют.
244
Бериллий и его соединения Be (OH)« (г)
Я0 B98,15 К)—Н° @) оценивается с учетом загрязненности образца и точности измерений теило»-
емкости в 0,4 Дж-К-моль и 0,02 кДж-моль соответственно. Какие-либо экспериментальны*
данные по термодинамическим свойствам Ве(ОНJ при Т ^> 298,15 К в литературе отсутствуют,
Для теплоемкости f3-Be(OHJ в интервале 298,15—500 К принято уравнение (см. табл. 25.1), оот
лученное экстраполяцией данных [185] выше 310 К, при использовании значений С°р E00 К) =
=93 Дж-К-моль~1 и Ср G00 К) = 104 Дж-К-моль, которые оценены на основании сравнения
теплоемкостей Mg(OHJ, MgO, Ca(OHJ, CaO и ВеО при этих температурах. Данные о температуре
и энтальпии плавления Ве(ОНJ отсутствуют; по-видимому, температура плавления Ве(ОН)г
превышает 480 К, когда давление диссоциации этого соединения достигает 100 атм.
Погрешность вычисленных значений Ф° (Т) при 298,15 и 500 К оценивается в 0,2 я
2 Дж-К-моль~1. Расхождения между термодинамическими функциями Ве(ОНJ (к),
приведенными в табл. Д.55 и в справочнике JANAF [1543], составляют ~0,5 Дж-К~1-моль~1 в значении
Ф° E00 К) и обусловлены учетом данных [185] по теплоемкости |3-Ве(ОНJ в настоящем изданни,
В справочнике принята энтальпия образования
&fH° (Ве(ОНJ, к, ромб., 298,15 К) = —905,7 + 2,0 кДж • моль,
основанная на анализе результатов измерений, представленных в табл. 25.10. Наиболее точные
измерения выполнены Бэром и Тернбуллом [630]. Практически совпадающее значение найдено
в работе Фрикке и Вульхорста [1144]. Хотя исследования равновесий [624, 625, 1143] менее точны,
Таблица 25.10. Результаты определения А/Н° (Ве(ОНJ, к, 298,15 К) (в кДж-моль)
Авторы
Метод
(Р()
298,15 К)
к.
Фрикке, Вульхорст, 1932 [1144] Калориметрическое измерение энтальпии раство- —905,7±2,5
рения ВеО (к) и р-Ве(ОНJ (к) в плавиковой
кислоте
Бэр, Тернбулл, 1965 [630] То же —905,7+2,1
Фрикке, Северин, 1932 [1143] Исследование равновесия реакции Ве(ОНJ (к)= —917±1О (III)
=ВеО (к)+Н2О (г), статический метод, 378 К, 1 точка
Баур, Лекок, 1963 [624, 625] Исследование равновесия реакции р-Ве(ОНJ (к) = —915±10(Ш)
=ВеО (к)+Н2О (г) методом ТГА, 496 К, 1 точка
полученные в них значения согласуются с принятым в пределах их погрешности. В работах
[1966, 1967, 2073, 2213, 2686] измерены энтальпии реакций с участием дигидроксида бериллия.
Однако они относятся к аморфному состоянию дигидроксида и поэтому не могут быть использо»
ваны для выбора энтальпии образования кристаллических модификаций Ве(ОНJ. В работах
[630, 1144] определена также энтальпия образования а-Ве(ОНJ, равная —902,6+2,0 кДж-мояь,
Ве(ОНJ (г). Термодинамические свойства газообразного дигидроксида бериллия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 739.
Молекулярные постоянные, использованные в расчете термодинамических функций Ве(ОНJ,
приведены в табл. 25.9. В литературе отсутствуют сведения об исследованиях структуры молекулы
Ве(ОНJ. В справочнике на основании принятых параметров молекул BeF2 и ВеОН принимается,
что молекула Ве(ОНJ в основном состоянии Х1^ также имеет линейную структуру. Приведенный
в табл. 25.9 момент инерции рассчитан на основании значений -$ О—Be—О=180° и г (Be—О) =
= 1,40±0,06 А, ^Ве—О—Н=180° и г (О—Н)=0,96+0,02 А (перенесены из ВеОН); его
погрешность составляет 2-109 г-см2. В литературе имеется сообщение (см. [1543, 1544]), что Фарбер и др..
наблюдали в ИК-спектре молекул Ве(ОНJ, изолированных в низкотемпературной матрице, частоту
649 см, отнесенную к симметричному колебанию связи Be—О. Это отнесение представляется
сомнительным, так как симметричное колебание линейной группы О—Be—О неактивно в ИК-
спектре, а антисимметричное должно иметь существенно большую величину. Маловероятно и
отнесение этой частоты к одному из деформационных колебаний Ве(ОНJ, поскольку частоты таких
колебаний должны быть меньше 650 см и иметь меньшую интенсивность, чем валентные
колебания. Приведенные в табл. 25.9 значения частот оценены на основании постоянных молекул
245
Be (ОНЬ (г)
Глава 25
BeF2 и ВеОН. Их погрешности оцениваются в 50—150 см 1. Сведения о возбужденных состояниях
Ве(ОНJ отсутствуют; так же как у молекул BeF2 и ВеС12, их энергии должны существенно
превышать 25 000 см.
Термодинамические функции Ве(ОНJ (г) вычислены по уравнениям A. 3)—A. 10), A. 122) —
A. 124), A. 126) и A. 129) в приближении «жесткий ротатор —гармонический осциллятор».
Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
молекулярных постоянных, в том числе, отсутствием сведений о структуре Ве(ОНJ, а также
приближенным характером расчета; они составляют 6, 10 и 15 Дж-К~1-моль~1 в значениях Ф° (Г)
при 298,15, 3000 и 6000 К К
Ранее термодинамические функции Ве(ОНJ (г) вычислялись в таблицах JANAF [1543, 1544],
справочнике Мак-Брайд и др. [1974] и в работе Альтмана [500] (Т ^ 2500 К). Расхождения в
значениях Ф° (Т), вычисленных в [1544] и настоящем издании, составляют- 6—21 Дж-К-моль~1,
а В; значении Н° B98,15 К)—Н° @) составляют 2,64 кДж-моль. Они обусловлены главным
образом различием в принятых значениях v5 (в [1544] принято 606 см). Расхождения с данными
[500, 1974] существенно больше (до 55 Дж-К~1-моль) из-за различия принятых структурных
параметров и основных частот Ве(ОНJ; в [500], кроме того, допущена ошибка в расчете.
Константа равновесия реакции Ве(ОНJ (г) = Ве (г)+2О (г)+2Н (г) вычислена с
использованием значения АГН° @) = 1880,387+16 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д//°(Ве(ОНJ, г, 0) = —635 + 15 кДж • моль.
Эта величина основана на приведенных в табл. 25.11 результатах исследования равновесия
реакции ВеО (к)+Н2О (г)=Ве(ОНJ (г). Кроме того, в работе Хилденбранда и др. [1427],
известной только по таблицам JANAF [1543, 1544], масс-спектрометрическим методом исследовано
Таблица 25.11. Результаты определений А/Н° (Ве(ОНJ, г, 0) (в кДж-моль х)
/\ п м f\ г\т X
/авторы
Хатчйсон. Мальм, 1949 [1367а] (см.
пересчет в [1287])
Гросвейнер, Зейферт, 1952 [1287]
Эллиотт, 1952 (см. [1543])
Ливи, Меррей, 1956 [1878а]
Янг, 1960 [2888]
Стюарт, Прайс, 1964 [2628а]
Блауэр и др., 1966 [706]
Мориц и др.
Л it Q Т1 /^ ТТ
1VI0 Т ОД
Протока, 1523—1823 К, 4 точки
То же, 1472—1873 К, 36 точек
То же, 1527—1583 К, 6 точек
Эффузионный, 1273—2273 К, 6 точек
Протока, 1576—1839 К, результаты измерений
представлены в виде уравнения
Протока, 1338—1653 К, 11 точек
Молекулярной эффузии, 1567—1808 К, 4
температуры, 22 точки
Результаты работы известны по [1543]
Af Н° (Be(OHJ, г, 0)
II закон
—662 ±13
—678+20
—682±15
—677
—684+10
—624±50
—644
III закон
-611+16
—636+16
—631+15
—631 +20
—638+16
—642+14
—664+18
—637
равновесие реакции ВеО (г)+Н2О (г) = Ве(ОНJ (г) при 2327 К и найдено, что энтальпия
образования меньше (по абсолютному значению) величины —635+20 кДж-моль. Работы [1287, 1878а,
2628а, 2888] приводят к хорошо согласующимся результатам АуН° @)=—639+15 кДж-моль.
В работе [706] показано, что определяемое значение константы равновесия зависит от количества
ВеО (к), так как равновесие исследуемой реакции не достигается при небольшом количестве
образца, а истинное значение константы может быть найдено только экстраполяцией данных, полу-
х Исследования равновесия реакции образования Ве(ОНJ (г) (см. табл. 25.11) позволяют рассчитать по методу
II закона значения энтропии (в Дж-К^-моль): S° A470 К)=361+13 [2628а], 5° A700 К)=380±8 [1287]
и 384+13 [1878а]. Данным табл. 739 соответствуют при этих двух температурах значения 389±12 и 404±12.
Таким образом имеется систематическое расхождение между величинами, вычисленными из экспериментальных
данных и по принятым молекулярным постоянным, которое превышает их погрешности. Следует отметить, что
соответствующие расхождения со значениями энтропии, рассчитанными для молекулы Ве(ОНJ с -4 Be—О—
—Н=110° или для линейной модели с v5=500+700 см, существенно меньше.
246
Бериллий и его соединения BeF (г)
ченных для разных образцов. Авторы [706] получили значения константы равновесия, близкие
к данным [1287], при использовании таких же по весу образцов ВеО.
В справочнике отдано предпочтение величинам, основанным на данных [1287, 1878а, 2628а,
2888]; они подтверждаются также работой Хилденбранда [1427], в которой в равновесии
участвовал ВеО (г) и обнаруженные в [706] эффекты не могли исказить результаты измерений.
BeF (г). Термодинамические свойства газообразного фторида бериллия в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 740.
В табл. 25.5 приведены молекулярные постоянные BeF, принятые на основании результатов
исследований электронных спектров этой молекулы. В спектрах BeF наблюдались переходы между
шестью электронными состояниями: А2П «а Х2? + [294, 296, 405, 929, 1126, 1556, 1570, 2077, 2078,
2295, 2792], ?2?+^Х22+, С22+ *± Х22+, D BП) 5±Х2? + и Е BП) ^Х22 + [294, 296], а также
С2 2+ -> А2И [294, 296, 2295]; энергии состояний D2U ж Е2И превышают 63 000 см. Аналогично
молекулам других галогенидов щелочноземельных металлов состояния Х2? + и42П молекулы BeF
коррелируют с атомами Be AS)-\-F BР). С пределом Be CP)+F BP) должно коррелировать
большое число возбужденных состояний BeF, в том числе квартетные состояния типа 4Д, 4П, *2+ и
4Е~. Эти состояния не рассматриваются в справочнике, поскольку можно предполагать, что они
являются слабосвязанными или отталкивательными состояниями.
Приведенные в табл. 25.5 колебательные постоянные BeF в состояниях Х2? + и А2П приняты
по работе Новикова [294], где они были получены на основании анализа колебательной структуры
системы А2П -> Х2? + (с 0 ^ v" <^ 16, 0^у'^ 15), проведенного по (^-кантам полос. Расчет
колебательных постоянных по началам полос для v', v" ^ 1 привел к значениям, согласующимся
в пределах 0,3 см с полученными из анализа по ()-кантам. Принятые значения постоянных в
состоянии Х2? + удовлетворительно согласуются также с найденными по кантам полос системы
42П-Х2? + в работах [405, 1126, 1556, 1570, 2077] и систем 52?+-Х2Е+, С3Е+—Х2Е+, D2U—
Х2?+, Е2И—ХаЕ + в работах [294, 296], за исключением постоянных, полученных из анализа
системы 2?2?+—Х2Е+, проведенного по Р-кантам, значительно удаленным от начала полос.
Вращательные постоянные молекулы BeF в этих состояниях приняты по работе Уокера и Барроу [2792],
где они были получены из анализа вращательной структуры полос 0—0, 1—0 и 0—1.
Колебательные и вращательные постоянные в состояниях В2%+ и С22+, приведенные в табл. 25.5, найдены
Новиковым и Гурвичем [294, 296].
Термодинамические функции BeF (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBU и ее производных по температуре рассчитывались по уравнениям A. 90)—A. 92)
с учетом трех возбужденных состояний в предположении, что Q^\_ вр === (pjpx) QifJ. вр- Величина
QifJ ъ Для основного состояния Х22 + и ее производные по температуре вычислялись по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82). В расчетах
учитывались все уровни энергии со значениями /^j/maXj „, где /шах> „ находились из условий A. 81).
Колебательно-вращательные уровни состояния Х2Е + вычислялись по уравнениям A.65), A.61).
Значения коэффициентов Ykl, полученные с учетом условия A. 59), а также величин vmsx и /lfm
приведены в табл. 25.6. Мультиплетность и вырождение всех электронных состояний учитывались
статистическими весами.
Молекулярные постоянные BeF в основном состоянии известны с удовлетворительной точностью,
а энергия диссоциации этой молекулы достаточно высока. Энергии возбуждения не включенных
в расчет квартетных и дублетных состояний также высоки. Поэтому погрешности рассчитанных
термодинамических функций BeF (г) во всем диапазоне температур определяются главным
образом неточностью фундаментальных постоянных и в значениях Ф° B") не превышают
0,02 Дж-К-^моль вплоть до 4000 К и 0,03 Дж-К-^моль при 6000 К.
Ранее таблицы термодинамических функций BeF (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106], таблицах JANAF [1543, 1544], справочниках Мак-Брайд и др.
11974], Мадера [1904] и Дугласа и Бекетта [977]. Все расчеты выполнены с использованием
приближенных методов и практически одинаковых молекулярных постоянных. Благодаря высокой
энергии возбужденных электронных состояний, которые не учитывались в большинстве работ,
а также большой величине Do (BeF) термодинамические функции, приведенные в табл. 740 и в
247
Глава 1а
гих справочниках, достаточно близки. Расхождения с данными [1543, 1904 и 1974] в значении
Ф° F000 К) составляют 0,1—0,2 Дж-К^-моль, а в значении 5° F000 К) составляют 0,4—
0,6 Дж-К~1-моль; термодинамические функции BeF, приведенные в 1-м, 2-м и настоящем
изданиях справочника, практически совпадают. В работе Климова и др. [167] приведены
коэффициенты полинома, аппроксимирующего значения Ф° (Т) для 293,15 ^ Т ^ 5000 К, полученные
грубым методом и существенно отличающиеся от данных табл. 740.
Константа равновесия реакции BeF (r)=Be (r)+F (г) вычислена на основании энергии
диссоциации
ДгЯ° @) = Do (BeF) = 575 + 6 кДж • моль «* 48 000 ± 500 см,
принятой по результатам исследований равновесия реакций с участием BeF (г), приведенных
в табл. 25.12. Приведенные в табл. 25.12 погрешности энергий диссоциации вычислены с учетом
воспроизводимости экспериментальных данных, погрешностей термодинамических функций,
Таблица 25.12. Результаты определений D0(BeF) (в кДж-моль)
Авторы
Метод
Do(BeF)
II закон
III закон
Гринбаум и др., 1963 [1253] Эффузионный, BeF2(г)+Ве (к, Ht)=2BeF (г), 1420— 596±14 621,2+5,4
1675 К, 17 точек, Д.ЛГ3 @)=348,1±1,8 (III)
Хилденбраяд, Мурац, 1966 [1419] Масс-спектрометрический, BeF2 (r)+Be (r)=2BeF 564±10 - 574,6+4,0
(г), 1321—1609 К, 18 точек, Д^0 @)=121,6+1,1
(III)
То же, Be (r)+AlF (r)=BeF (г)+А1 (г), 1423— 619±40 569,8±5,7
1443 К, 2 точки, ДГЯ° @)=100,8±1,0 (III)
То же, Be (r)+CaF (r)=BeF (г)+Са (г), 1321— 549±47 564,9±9,0
1503 К, 8 точек, Ь,Л0 @)=—32,9±1,4 (III)
Фарбер, Сривастава, 1974 [1055] Масс-спектрометрический, BeF2 (г)+Ве (г)= 612±17 602,0+4,2
=2BeF (г), 1415—1592 К, 10 точек, АгНа @)=
=66,7±1,4 (III)
сечений ионизации и других величин, использованных в расчетах. Как полагают авторы [1544],
результаты измерений в работе [1253] могут быть искажены вследствие димеризации BeF.
Принятая в справочнике величина является средневзвешенной из работ [1055 и 1419], причем при оценке
веса измерений, помимо указанных в табл. 25.12 погрешностей, учитывалось также отклонение
результата данной работы от среднего.
Принятой энергии диссоциации соответствует
г, 0) = —177,972 + 7,8 кДж • моль.
BeF2 (к, ж). Термодинамические свойства кристаллического и жидкого дифторида бериллия
в стандартном состоянии при температурах 100—2100 К приведены в табл. 741.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В справочнике за стандартное состояние BeF2 (к) в интервале 0—823 К принята квар-
цеподобная гексагональная модификация BeF2. При 493 К эта модификация имеет фазовый
переход II родаг аналогичный а—Р переходу кварца Ч
1 Имеется далеко идущая аналогия кристаллических форм и полиморфных превращений дифторида бериллия
и диоксида кремния. Кроме кварцеподобной формы ((?-BeF2), которая является термодинамически стабильной
модификацией при комнатной температуре, известна кристобалитоподобная кубическая форма (C-BeF2),
имеющая фазовый переход при 403 К [164, 222] с энтальпией превращения 5,3 кДж-моль [222], и еще одна
высокотемпературная форма — ромбическая тридимитоподобная G-BeF2), устойчивая по данным [176, 301] в интервале
723—953 К. Фазовые переходы между различными кристаллическими формами BeF2 затруднены, как и в
случае SiO2, а надежные данные о температурах равновесных переходов Q-, С- и Г-форм в литературе отсутствуют.
248
Бериллий и его соединения BeFz (к, ж)
При Т <^ 298,15 К термодинамические функции вычислены по данным о теплоемкости кварце-
подобной модификации BeF2, полученным в работе Тейлора и Гарднер [2662а] (8—304 К).
Значение S° (8 К), полученное экстраполяцией, равно 0,025 Дж-К~1-моль. Однако исследованный:
в [2662а] образец BeF2 содержал ~0,5% примесей (в том числе 0,3% кислорода в основном в виде
адсорбированной воды), поэтому погрешности S° B98,15 К) и Н° B98,15 К)— Н° @) оцениваются
в 0,4 Дж-К-моль и 0,04 кДж-моль.
При Т ^> 298,15 К для теплоемкости а- и ^-модификаций кварцеподобной формы BeF2
приняты уравнения (см. табл. 25.1), выведенные методом Шомейта по результатам измерений
энтальпии в работе Тейлора и Гарднер [2662а]. Эти уравнения не описывают аномальный ход
теплоемкости BeF2 в области фазового перехода второго рода; для их согласования с данными [2662а]
принимается значение изотермической энтальпии превращения AtrH=0,l75 кДж-моль,
отнесенное к температуре перехода 493+10 К [47, 176, 222, 301, 2396, 2662а]. Температура плавления
(823 + 10 К) принята по согласующимся между собой данным [48, 165, 1250, 2394, 2662а, 2681].
Энтальпия плавления D,77+0,30 кДж-моль) вычислена по результатам измерений энтальпий
растворения кварцеподобной и стеклообразной форм BeF2 при 298,15 К [2656, 2662а] с
последующим пересчетом к температуре плавления 823 К. Эта величина находится в согласии с
результатами измерений [2662а], согласно которым AmH (BeF2, к) > 4,2 кДж-моль. Уравнение для
теплоемкости жидкого BeF2 (см. табл. 25.1) выведено методом Шомейта по измерениям энтальпии в
работе Тейлора и Гарднер [2662а] (841—1183 К); согласно этому уравнению С°р (BeF2, ж) монотонно
растет от 78,28 Дж-К^-моль при 823 К до 89,94 и 95,67 Дж^К^-моль при 1200 и 2000 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 0,3, 1,0 и
2,5 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями
BeF2 (к, ж), приведенными в табл. 741 и во втором издании настоящего справочника [105],
достигают 10 Дж-К~1-моль в значении Ф° A000 К) и объясняются тем, что в настоящем издании
учтены данные [2662а]. Соответствующие расхождения с данными JANAF [1543] не превышают
1 Дж-К^-моль (до 2000 К).
Принятое в справочнике значение энтальпии образования
A/ff°;(BeF2, к, гекс, 298,15[К) = —1027 + 3 кДж-моль
основано на данных, приведенных в табл. 25.13. При вычислении энтальпии образования BeF2 (к,
гекс.) по данным работ [535, 854] и [536] использовано значение энтальпии перехода AtrH (BeF2,.
Таблица 25.13. Результаты определений А^Н° (BeF2, к, гекс, 298,15 К) (в кДж-моль)
Авторы
Метод
AfH° (BeF2, к, гекс
298,15 К)
Чарни, Армстронг, 1965 [535, 854] Измерение энтальпии реакции Be (k)+F2 (r)=BeF2 —1027,0+3,3
(стекл.) методом фторной калориметрии, 4 опыта,
AfH° (BeF2, стекл.)=—1022,3+3,3
Армстронг, Койл, 1965 [536]; Калориметрические измерения энтальпии растворе- —1027,7+4,0
Килди, Чарни а ния Be (к1) [536, 630] и BeF2 в плавиковой кислоте;
ДуЯ0 (BeF2, стекл.)=—1023±4,0
Гросс и др., 1971 [1274, 1278] Калориметрическое измерение реакции Be (к)+ —1028,0±4,0
+PbF2 (K)=BeF2 (к)+РЬ (к); в продуктах реакции
BeF2 образовывался в стекловидном и
кристаллическом состояниях в различных отношениях.
Экстраполяцией вычислено значение для
кристаллического состояния
Матьюс, Бес, 1968 [1961] Исследование равновесия реакции BeF2 (к)+Н2О (г)= —1041A1)
=ВеО (k)+2HF (г) методом протока, 773—973 К,
расчет по уравнению —1033 (III)
а Результаты этих измерений известны по обзорной работе Паркер [2186].
Известны температуры плавления Q-Be?2 и C-BeF2 A076 К). По данным ряда работ [32, 164, 176, 301, 302,
303, 2662а] при нагревании расплава кварцеподобной формы до —880 К происходит его кристаллизация с
образованием кристобалитоподобной формы.
249
BeF2 (г)
Глава 25
стекл. -> BeF2, к, гекс.) = —4,7 + 0,2 кДж-моль, полученное Тейлором, Гарднер [2662а] и
Тамару, Иококава [2656] из измерений энтальпии растворения этих модификаций в растворе ацетата
натрия в уксусной кислоте. Результаты калориметрических измерений [535, 536, 854, 1274, 1278]
находятся в хорошем соответствии между собой. Принятое в справочнике значение является
округленным средневзвешенным из полученных в этих работах.
Давление пара дифторида бериллия BeF2 (к, ж)=ВеР2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
Д4Я°(ВеР2, к, 0) = 228,2 + 2,5 кДж • моль-1.
Это значение основано на приведенных в табл. 25.14 результатах измерений давлений пара
дифторида бериллия, где в качестве погрешностей указана воспроизводимость измерений. Оно
Таблица 25.14. Результаты определений
Г° (BeF2, к, 0) (в кДж-моль х)
Авторы
Метод
Д,,Я° (BeF2, к, 0)
II закон
III закон
Сенс и др., 1954 [2492] Протока, 1019—1341 К, 33 точки 237+4 227,8+0,3
Новоселова и др., 1958 [301] Протока, 1040—1376 К, 25 точек 222+6 228,8+0,5
Сенс, Стоун, 1958 [2493] Протока, 1075—1298 К, 21 точка 226±4 228,0±0,2
Хандамирова и др., 1959 [420] Эффузионный, 846—950 К, 15 точек 256+26 230,2+0,9
Гринбаум и др., 1963 [1250] Протока, 1123—1223 К, 5 точек 231 ±47 223,5+0,6
Эффузионный, 823—1053 К, 26 точек 233+7 225,1+0,5
Блауэр п др., 1965 [705] Торзионный, 713—795 К, 38 точек 242+4 220,9±0,3
Кантор, 1965 [814] Квазистатический и точек кипения, 1146—1372 К, 232 227,8±0,1
19 точек
Хилденбранд, Серд, 1965 [1425] Торзионный, 821—942 К, 33 точки 234+4 227.7+0,2
Белоусов и др., 1967 [33] Эффузионный, 800—1000 К, число точек неиз- — 229,2±0,4
вестно
Масс-снектрометрический, 815—977 К, число точек 225+3 —
неизвестно
Ефименко, 1968 [1017] Масс-спектрометрический, Гср=755 К, число то- 232+2 —
чек неизвестно
является средневзвешенным из данных [33, 301, 420, 814, 1425, 2492, 2493], которые совпадают
в пределах 2,5 кДж-моль. Заметно меньшие величины получены тремя разными методами в
работах [705, 1250], выполненных в одной лаборатории. Погрешность принятого значения учитывает
неточность термодинамических функций BeF2 (к, ж, г).
BeF2 (г). Термодинамические свойства газообразного дифторида бериллия в стандартном
состоянии в интервале температур 100—-6000 К приведены в табл. 742.
Молекулярные постоянные, использованные для расчета термодинамических функций BeF2,
приведены в табл. 25.8. Исследования дифракции электронов [5, 12], инфракрасного спектра
[23, 25, 779, 2565, 2566] и отклонения молекулярного пучка в неоднородном электрическом
поле [783] показали, что молекула BeF2 в основном электронном состоянии 1 SJ имеет линейное
строение (симметрия Dmh). Приведенный в табл. 25.8 момент инерции вычислен на основании
значения г (Be—F)=l,40+0,03 А, найденного Акшпиным и др. [5, 12] в результате электронографи-
ческого исследования BeF2; его погрешность составляет 0,5 -109 г -см2. Принятые значения
основных частот v2 и v3 получены Байковым [23, 25] при исследовании инфракрасного спектра паров
BeF3. Бюхлер и Клемперер [779] в аналогичном исследовании нашли близкую величину v3, но
•сделали ошибочное отнесение v2 [2565]. Близкие значения получены Снелсоном [2565, 2566] из
ИК-спектров молекул BeFa, изолированных в матрицах. Погрешности частот v2 и v3 оцениваются
в 10 и 20 см. Частота vx экспериментально не наблюдалась. Приведенное в табл. 25.8 значение
{рассчитано для поля валентных сил на основании величины v3 и в предположении, что для BeF,,
так же как для дигалогенидов магния, справедливо отношение fr/frr=15. Его погрешность оцени-
250
Бериллий и его соединения Be2F4 (г)
вается в 70 см. Результаты оценок vx в работах [23, 105, 194, 748, 1543, 2565, 2566] согласуются
с принятой величиной в этих пределах. Возбужденные электронные состояния BeF2 согласно
расчету [1228] лежат выше 50 000 см.
Термодинамические функции BeF2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных функций
обусловлены неточностью принятых молекулярных постоянных, а также приближенным характером
расчета и составляют 0,5, 3 и 5 Дж-К-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К
соответственно, в том числе из-за неточности постоянных — не более 1 Дж-К-моль.
Ранее таблицы термодинамических функций BeF2(r) рассчитывались в справочниках [105, 977,
1543, 1904, 1974], а также в работах Байкова [23] (Т < 5000 К), Брюэра и др. [748], Краснова и
Светцова [195] (Т ^ 2500 К). Расхождения между значениями Ф° (Т), вычисленными в настоящем
справочнике и в [1543] и [23], не превышают 0,7 и 0,4 Дж-К-моль соответственно и
объясняются некоторыми различиями в принятых значениях частот. Расхождения с остальными расчетами
увеличиваются с ростом температуры и достигают 12—14 Дж-К~1-моль главным образом из-за
того, что в них приняты неверные значения частоты v2.
Константа равновесия реакции BeF2 (r) = Be (r)+2F (г) вычислена с использованием значения
Дг#° @)=1270,796+6,4 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации BeF2 (к). Этим величинам также соответствует
&fH° (BeF2, г, 0) = —796,493 ± 3,9 кДж • моль.
Be2F4 (г). Термодинамические свойства тетрафторида дибериллия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 743.
Молекулярные постоянные, использованные для расчета термодинамических функций Be2F4,
приведены в табл. 25.9. Произведение моментов инерции вычислено для мостиковой структуры
этой молекулы (симметрия Du) с параметрами -^FM00T—Be—FMOC.r=108+3°, г (Be—FBnei,ni)=l,40 +
+0,05 А и r (Be—FM0CT)=1,58+0,05 А, которые оценены на основании сравнения структурных
параметров молекул BeF2, ВеС12 и Ве2С14. Погрешность значения IaIbIc составляет 5-1014 г3-см6.
Приведенные в табл. 25.9 значения частот v9, vu и v12 найдены Снелсоном и др. [2571а] в ИК-
спектре молекул Be2F4, изолированных в матрице из Ne. Для частот vx—v5, v8 и v10 приняты
значения, предложенные в этой же работе из анализа нормальных координат. Они удовлетворительно
согласуются со значениями частот соответствующих колебаний в молекулах Li2F2 и Al2Fe, Mg2F4,
Mg2Cl4, Mg2Br4, Mg2I4 (см. [1848а]) и Fe2Cl4 (см. [1140а]). Предложенные Снелсоном и др. [2571а]
значения основных частот внеплоскостных колебаний v6=1086 см и v7=827 см представляются
завышенными: они выше частот валентных мостиковых колебаний, а частота v7 существенно больше
частот v7) наблюдавшихся Лесицким и Ниблером [1848а] в ИК-спектре молекул Mg2F4 и Mg2Gl4,
изолированных в матрице из Аг B40 и 140 см соответственно). Поэтому значения v6 и v7 оценены
на основании значений частоты v7 молекулы Mg2F4, частот v6, v7, v5, и v10 молекулы Fe2Ci4 [1140a]
и частот v5 и v10 молекулы Be2F4. Погрешности принятых значений основных частот Be2F4
оцениваются в 10—15% от их величины.
Термодинамические функции Be2F4 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности вычисленных термодинамических
функций обусловлены неточностью принятых значений молекулярных постоянных и приближенным
характером расчета, они составляют 4, 10 и 15 Дж-К-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К.
Таблица термодинамических функций Be2F4 (г) публикуется впервые.
Константа равновесия реакции Be2F4 (r)=2Be (r)+4F (г) вычислена с использованием
значения 1\ГН° @) =2682,606 +18 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^^Ве^, г, 0) = —1734 + 15 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании реакции 2BeF2 (K)=Be2F4 (г),
выполненном Белоусовым и др. [33] (815—977 К, 5 точек, b.fH° @) = —1737 +15 кДж-моль
по II закону и —1731 +15 кДж-моль по III закону). Погрешность ее оценена с учетом расхожде-
251
BeCl (г) Глава 25
ния результатов расчетов по уравнениям II и III законов и неточности термодинамических
функций. В работе Хилденбранда и Серда [1425] масс-спектрометрическим методом исследован
молекулярный состав пара над BeF2 (к, ж). Количество димерных молекул составляло только около 1 % г
и авторы не вычисляли констант равновесия.
BeCl (г). Термодинамические свойства газообразного хлорида бериллия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 744.
В табл. 25.5 представлены молекулярные постоянные Ве3бС1, принятые в справочнике на
основании результатов исследований электронных спектров этой молекулы. В спектрах BeCl
наблюдались переходы между четырьмя электронными состояниями: АЧ1 <^ X2 ?+ [294, 299, 300, 871,1136],
52?+->- Х2?+ [818] и D2R*± X2 2+ [294]. Состояния X2 ?+ и А2П коррелируют с пределом
Be A5)+С1 BР). По аналогии с молекулами других галогенидов группы ПА со следующим дис-
социационным пределом Be CjP)+C1 BP) должно коррелировать большое число возбужденных
состояний молекулы BeCl, в том числе квартетных с энергиями от 50 000 до 60 000 см. Энергии этих
состояний должны быть достаточно велики, что позволяет не рассматривать их в настоящем
справочнике.
Колебательные постоянные BeCl в состояниях X2 ?+ и A2TL определялись в работах [294,300,
871, 1136] из анализа полос системы Л2П ** Х22+; значения, найденные в этих работах, хорошо
согласуются. Приведенные в табл. 25.5 значения найдены Коленом и др. [871] на основании
обработки собственных данных и данных Новикова и Туницкого [300] D0 полос с v' <J. 9 и v" ^ 10).
Вращательные постоянные в этих состояниях также приняты по работе [871], где они были
получены из анализа тонкой структуры полос 0—0,1—1, 1—0 и 2—1 системы А2Л -> X2 ?+.
Колебательные постоянные в состоянии В2 ?+ приняты по работе Карлеера и др. [818].
Термодинамические функции BeCl (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QB1S и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом
состояния А2П. в предположении, что Q^fJ вР = (paIPx) Qi^J вр- Величина Q[^J вр и ее производные
находились по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82).
В расчетах учитывались все уровни энергии со значениями / <^ •/„««,,,, где /тах„ определялись из
условий A. 81). Колебательно-вращательные уровни состояния X2 ?+ вычислялись по уравнениям
A. 65), A. 64). Значения коэффициентов в этих уравнениях для «эффективной» изотопической
модификации BeCl, а также величин vmlix и /Иш, использованных в расчетах, приведены в табл. 25.6.
Коэффициент Y03 получен из условий A. 59). Мультиплетность и вырождение уровней состояний
X2 ?+ и А2И учитывались соответствующими статистическими весами.
Постоянные молекулы BeCl в состоянии X2 ?+ известны с удовлетворительной точностью, ее
энергия диссоциации достаточно велика, а энергии связанных электронных состояний, не
учитывающихся в расчетах, не ниже 45 000 см. Поэтому погрешности рассчитанных
термодинамических функций BeCl (г) вплоть до 4000—5000 К определяются только неточностью
фундаментальных постоянных (до 0,02 Дж-К-моль). Погрешность в значении Ф° F000 К) оценивается
в 0,05 Дж-К^-моль.
Термодинамические функции BeCl (г) вычислялись раньше в предыдущих изданиях настоящего
справочника [105, 106], а также в справочниках JANAF [1543], Мак-Брайд и др. [1974],
Дугласа и Бекетта [977] и Мадера [1904]. Все расчеты выполнены приближенными методами с
использованием оцененных значений вращательных постоянных, в работе [977] не учитывалось
состояние АШ. Расхождения в значениях термодинамических функций, приведенных в этих изданиях и
в настоящем справочнике, обусловлены в основном различием вращательных постоянных. При
высоких температурах возникают также расхождения из-за применения разных методов расчета.
Расхождения достигают 1,7 Дж-К-моль в значениях Ф° (Г) и 2,5 Дж-К~1-моль в значениях
S° (T). В работе Климова и др. [167] представлены коэффициенты полинома, аппроксимирующего
значения Ф° (Т) в интервале 293,15—5000 К; соответствующие данные отличаются от
представленных в табл. 744 на величины до 1,9 Дж-К~1-моль в основном из-за использования в этой работе
приближенного метода расчета.
Константа равновесия реакции BeCl (г)=Ве (г)+С1 (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (BeCl) = 385 + 5 кДж • моль« 32 200 + 400 см
252 ¦
Бериллий и его соединения
ВеС12 (к, ж)
Это значение основано на результатах исследований равновесия реакций с участием ВеС1 (г),
приведенных в табл. 25.15, где в качестве погрешностей даны величины, учитывающие
воспроизводимость измерений, неточность сечений ионизации и других величин, использованных в расчетах.
Таблица 25.15. Результаты определений D0(BeCl) (в кДж-моль)
Авторы
Гринбаум и др., 1964 [1248]
Хилденбранд, Серд, 1969 [1426]
Фарбер, Сривастава, 1974 [1055]
Метод
Эффузионный, ВеС1, (г)+Ве (ж)=2ВеС1 (г),
1573-1724 К, 10 точ'ек, ДгЯд @)=373,6±2,8 (III)
Масс-спектрометрический, ВеС12 (г)+Ве (г)=
=2ВеС1 (г)г 1382—1538 К, 14 точек, ДГЯ° @)=
= 157,0±2,5 (III)
То же, Be (г)+А1С1 (г)=ВеС1 (г)+А1 (г), 17 точек,
АГН° @)=110,4±0,8 (III)
Масс-спектрометрический, ВеС12 (г)+Ве (г)=
=2ВеС1 (г), 1415—1592 К, 10 точек, АГН° @)=
=-72,5+0,9 (III)
Do
II закон
425+40
387+40
382+25
435+10
(ВеС1)
III закон
433,7+5,0
382,1+3,9
388,3+7,0
424,3+3,0
Значения энергий диссоциации, полученные в результате исследования в работе Хилденбранда и
Серда [1426] двух разных реакций, хорошо совпали между собой. Возможно (см. [1543]), что эф-
фузионные измерения [1248] искажены вследствие образования молекул димеров. Причина
расхождения данных [1426] и [1055] остается неясной. Принятое значение является средним из
вычисленных для двух реакций, изученных в работе [1426].
Принятой энергии диссоциации соответствует
Д//°(ВеС1, г, 0) = 54,373 ± 7,1 кДж • моль.
ВеС12 (к, ж). Термодинамические свойства кристаллического и жидкого дихлорида бериллия
в стандартном состоянии при температурах 100—1200 К приведены в табл. 745.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 25.1. В справочнике за стандартное состояние ВеС12 (к) в интервале 0—676 К принимается
а-модификация, имеющая ромбическую решетку, а в узком интервале от 676 до 688 К—E-модифи-
кация ВеС12 г. При Т < 298,15 К термодинамические функции ВеС12 (к) вычислены по значениям
теплоемкости, измеренным Мак-Доналдом и Эттингом [1989] A3—304 К) на образце, содержавшем
99,72% ВеС12 и 0,28% Be. Экстраполяция теплоемкости ниже 13 К по уравнению Дебая привела
к значению S° A3 К)=0,27 Дж-К~1-моль~1. Погрешность принятых значений 5° B98,15 К) и
Н° B98,15)—Я0 @) оценивается в 0,2 Дж-К-моль~1 и 0,04 кДж-моль соответственно. Для
теплоемкости в интервале 298,15—676 К принято уравнение (см. табл. 25.1), выведенное методом
Шомейта из данных по энтальпии ВеС12, полученных Мак-Доналдом и Эттингом [1989] C25—
654 К). По той же работе приняты уравнение для теплоемкости Р-ВеС12 (см. ниже), значения
температур превращения F76+2 К) и плавления F88+2 К), энтальпий превращения F,23 +
+0,30 кДж-моль) и плавления (8,66+0,30 кДж-моль). По данным [1989] в узком интервале
температур 688—713 К теплоемкость ВеС12 (ж) равна 121 Дж-К-моль. Это значение находится
в противоречии с надежными экспериментальными данными, полученными для дихлоридов
других элементов подгруппы ПА, а также для BeF2 (ж). Ввиду этого в справочнике на основании
сопоставления данных для теплоемкостей дихлоридов и дифторидов элементов подгруппы НА
принимается С°р (ВеС12, ж)=92 +10 Дж-К^1
^
моль
°
р
р B )Д
Погрешности вычисленных значений Ф° B") при 298,15, 700 и 1200 К оценены в 0,2, 0,6 и
5 Дж-К " ~ ""
1-моль~1 соответственно. Термодинамические функции ВеС12 (к), приведенные в табл. 745
1 Литературные данные о полиморфизме ВеС12 противоречивы. Кувыркин и др! [205] указывают на существование
в интервале 523—613 К кубической а'-модификации ВеС12, однако при измерениях энтальпии ВеС12(к) [1989]
фазовые превращения при указанных температурах не были замечены. Обозначение модификаций ВеС12 (а-
и J3-) в справочнике принято по аналогии с BeF2(K), хотя в литературе обычно используются обратные
обозначения.
253
р-ВеС12 (к)
Глава 25
и в справочнике JANAF [1543], практически совпадают; для ВеС12 (ж) расхождения достигают
4 Дж-К~1-моль~1 в значении Фс A200 К), что вызвано различием принятых величин для
теплоемкости жидкого ВеС12.
Принятое в справочнике значение энтальпии образования дихлорида бериллия
Д^0 (а-ВеС12, к, 298,15 К) = — 496,2 + 2,5 кДж • моль
основано на измерениях энтальпии растворения бериллия и смеси двух форм ВеС12 (к) в соляной
кислоте в работе Томпсона и др. [2684]. В расчете использовано значение l\lrH (<х-ВеС12 -* C-
ВеС12)=5,4+0,6 кДж-моль, вычисленное для 298,15 К по результатам исследования
теплоемкости а- и р-форм ВеС12 в работе [1989]. Энтальпия образования дихлорида бериллия определялась
также Гроссом и др. [1279] и Джонсоном и Джиллилендом [1588] при сожжении бериллия в хлоре;
найденные этими авторами значения равны —490,0+2,0 кДж-моль и —493,8+2,5 кДж-моль
соответственно. В работе [1279] на основании кристаллографического анализа образца,
полученного в условиях, совпадающих с условиями калориметрических опытов, эта величина была
отнесена к E-форме. Таким образом данные [1279] находятся в полном соответствии с работой [2684].
В работе [1588] рентгенофазовый анализ образующегося ВеС12 не проводился. Энтальпия сгорания
бериллия в хлоре измерялась также в более старых работах [2019, 2520], однако полученные в них
величины недостаточно точны. В работе Маркова и Делимарского [243] методом ЭДС найдено
b.fG (ВеС12, к, 500 К) = —383,3 кДж-моль, чему соответствует значение &/Н° (ВеС12, к,
298,15 К) = —499,2 кДж-моль, близкое к принятому.
Давление пара дихлорида бериллия ВеС12 (к, ж)=ВеС12 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
Д8#° (а-ВеС12, к, 0) = 134,2 + 2,0 кДж • моль.
Эта величина основана на исследованиях давления пара над ВеС12 (к, ж), результаты которых
приведены в табл. 25.16, где в качестве погрешностей указана воспроизводимость измерений.
Таблица 25.16. Результаты
Авторы
определении ASH (ВеС12, к, 0) (в кДж-моль J)
Метод
Д„Я°(ВеС12, к, 0)
II закон
III закон
Ральфе, Фишер, 1933 [1098] Точек кипения, 613—733 К, число точек неиз- 125+30 127,2+0,9
вестно, расчет по уравнению
Рябчиков, Тихинский, 1960 [362] Масс-спектрометрический, 510—580 К, 7 точек, 145+10 —
расчет по уравнению
Шейко, Фещенко, 1962 [450] Метод протока, 573—753 К, 10 точек 127+6 129,1+0,2
Гринбаум и др., 1963 [1254] Эффузионный, 441—600 К, 28 точек 141 ±3 133,3+0,4
Торзионный, 471—510 К, 8 точек 142+5 134,9+0,2
Фишер, Петцель, 1964 [1097] Точек кипения, 638—753 К, 69 точек, расчет по 146+4 127,0+1,4
уравнению
Хилдеибранд, Серд, 1969 [1426] Торзионяый, 460—504 К, 45 точек, расчет по урав- 138 134,3+0,2
нению
В работах [1426 и 1254] получены близкие величины, имеющие минимальный разброс, и принятое
значение основано на этих данных. В работе Ко и др. [1735] сообщалось, что основным
компонентом пара дихлорида бериллия является Ве2С14. Этот вывод, по-видимому, ошибочен, так как масс-
спектр ометрические [362, 1426] и торзионно-эффузионные измерения [1254] показали, что димер
практически отсутствует в парах. Погрешность принятого значения учитывает неточность
термодинамических функций ВеС12 (к, ж) и ВеС12 (г).
?3-ВеС12 (к). Термодинамические свойства высокотемпературной модификации дихлорида
бериллия (р-ВеС12) при температурах 100—688 К приведены в табл. Д. 56.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. Как и в случае диоксида кремния, фазовые переходы между различными модификаци-
254
Бериллий и его соединения ВеС12 (г), Ве2С14 (г)
ями ВеС12 затруднены. Это позволило провести измерения теплоемкости и энтальпии Р-ВеС12,
за пределами его равновесного перехода в а-ВеС12 (при 676 К). При Т < 298,15 К
термодинамические функции C-ВеС12 вычислены по измерениям теплоемкости, проведенным Мак-Доналдом и Эт-
тингом [1989] A3—304 К) на образце чистотой не менее 99,7%. Экстраполяция теплоемкости
ниже 13 К привела к значению S° A3 К)=0,67 Дж-К-моль. Погрешности принятых по
работе [1989] значений S° B98,15 К) и Н° B98,15 К)—Я0 @) (см. табл. 25.1) оцениваются
в 0,3 Дж-К-моль и 0,04 кДж-моль соответственно.
Для теплоемкости Р-ВеС12 в интервале 298,15—688 К принято уравнение (см. табл. 25.1),
выведенное по значениям С°р' B98,15 К), С°р D00 К)=72,76 Дж-К-^моль и С°р F00 К)=80,08 ДжХ
XК-моль; последние два получены авторами справочника JANAF [1544] на основании
измерений теплоемкости и энтальпии |3-ВеС12 в работе Мак-Доналда и Эттинга [1989]. Погрешности
вычисленных значений Ф° (Г) при 298,15, 500 и 688 К оцениваются в 0,2, 0,6 и 1 Дж-К-моль
соответственно. Термодинамические функции Р-ВеС12, приведенные в справочнике JANAF [1544]
и в табл. Д.56, практически совпадают.
Принятое в справочнике значение энтальпии образования
&fH° (C-ВеС12, к, 298,15 К) = —490,8 ± 2,5 кДж • моль
основано на измерениях Томпсона и др. [2684] (см. текст по ВеС12 (к, ж)).
ВеС12 (г). Термодинамические свойства газообразного дихлорида бериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 746.
Молекулярные постоянные, использованные для расчета термодинамических функций ВеС12,.
приведены в табл. 25.8. Исследования дифракции электронов [5, 12], инфракрасного спектра [778,.
779, 2565, 2566] и отклонения молекулярного пучка в неоднородном электрическом поле [783]
показали, что молекула ВеС12 в основном электронном состоянии г S* имеет линейное строение
(симметрия Dmh). Приведенный в табл. 25.8 момент инерции вычислен на основании значения
г (Be—С1)=1,75 +0,02 А, найденного Акишиным и др. [5, 12] при электронографическом
исследовании ВеС12; его погрешность составляет 1-Ю9 г-см2. Принятое значение частоты v3
измерено Бюхлером и Клемперером [778, 779] в ИК-спектре газообразного ВеС12. Близкие значения
получены Снелсоном [2565, 2566] в матрицах из Ne, Аг и Кг. Значение v2 оценено Снелсоном [2566]
для газовой фазы на основании частоты v2=238 см, найденной им в матрице из Аг. В работах [778,
779] Бюхлер и Клемперер ошибочно отнесли к v2 полосу 482 см. Частота vx оценена, так же как
для BeF2 (см. выше). В пределах 10% она согласуется с оценками, выполненными ранее [24, 105,
194, 748, 1543, 2565, 2566]. Погрешности принятых значений vx, v2 и v3 оцениваются в 40, 10 и
20 см соответственно. Сведения о возбужденных состояниях ВеС12 отсутствуют.
Термодинамические функции ВеС12 (г) вычислены по уравнениям A. 3)—A. 6), A. 9)—A. 10),.
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных величин обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета и составляют 1, 3 и 5 Дж-К-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К, в том числе 0,5, 1 и 2 Дж-К-моль из-за неточности;
молекулярных постоянных.
Ранее таблицы термодинамических функций ВеС12 (г) вычислялись в справочниках [105, 977^
1543, 1904], а также в работах Краснова и Светцова [195] {Т ^ 2500 К) и Брюэра и др. [748]
(Т <С 2000 К). Расхождения между значениями Ф° (Т) в настоящем издании и в таблицах [15431
не превышают 2,2 Дж-К-моль и объясняются некоторыми различиями в принятых значениях
vx и v2. Расхождения с остальными расчетами увеличиваются с ростом температуры и достигают
10—13 Дж-К~1-моль главным образом из-за неверного значения v2, принятого в старых работах.
Константа равновесия реакции ВеС12 (г)=Ве (г)+2С1 (г) вычислена е использованием
значения &ГН° @) =921,281 + 5,9 кДж -моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дихлорида бериллия. Этим величинам также соответствует
•A^°(BeGl2, г, 0) = — 362,288 + 3,2 кДж • моль.
Ве2С14 (г). Термодинамические свойства тетрахлорида дибериллия в стандартном состоянии!
в интервале температур 100—6000 К приведены в табл. 747.
255
ВеВг (г)
Глава 25
Молекулярные постоянные, использованные для расчета термодинамических функций Ве2С14,
приведены в табл. 25.9. Произведение моментов инерции вычислено для мостиковой структуры
этой молекулы (симметрия DM)c^tClM0CT—Be—С1МООТ=108 +3°, г (Be—С1внепга)=1,77 + 0,05А, г (Be—
G1MOOI)=1,95+0,05 А. Эти структурные параметры получены Мак-Кордиком и др. [1899а] в
результате квантовомеханического расчета, причем величина г (Be—С1внешн) перенесена из молекулы
ВеС12 и в дальнейшем не варьировалась. Погрешности принятых структурных параметров Ве2С14
оценены на основании сопоставления их со структурными параметрами молекулы Mg2Cl4 (см.
[1304а]). Погрешность приведенного значения IaIbIc составляет 1-1012 г3-см6. Принятые в
справочнике значения частот v9 и уи найдены Бюхлером и Клемперером [779] в ИК-спектре паров ди-
хлорида бериллия при 500° С. Две полосы в этой же области F40 и 860 см) наблюдал Снел-
сон [2566] в ИК-спектре молекул ВеС12, изолированных в матрице из Ne, и отнес их к полимеру
(ВеС12)„. Значения всех остальных частот Ве2С14 оценены при подготовке справочника на
основании соответствующих частот молекул Be2F4, Li2F2, Li2Cl2, A12F6 и A12G16, а также Mg2F4, Mg2Cl4,
MgaBr4, Mg2I4 [1848a] и Fe2Cl4 [1140a]. Погрешности принятых значений основных частот
оцениваются в 10—15% от их величины.
Термодинамические функции Ве2С14 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых значений молекулярных постоянных и приближенным характером расчета;
они составляют 8, 15 и 20 Дж-К^-моль'1 в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции Ве2С14 (г) вычислялись в таблицах JANAF [1543] с
использованием молекулярных постоянных, существенно отличающихся от принятых в настоящем
справочнике. В частности, авторы [1543] приняли очень низкие значения частот, связанных с
валентными колебаниями Ве2С14. В связи с этим термодинамические функции Ве2С14 (г), вычисленные
в справочнике [1543], больше приведенных в табл. 747; расхождения в значениях Ф° (Т)
достигают 20 Дж-К-моль~1.
Константа равновесия реакции Ве2С14 (г)=2Ве (г)+4С1 (г) вычислена с использованием
значения Дг#° @)=d937,986 +22 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH° (Ве2С14, г, 0) == —820 + 20 кДж • моль.
Эта величина является средневзвешенной из приведенных в табл. 25.17 результатов
исследований равновесия реакции 2ВеС12 (г)=Ве2С14 (г), причем больший вес придавался работам [362,
Таблица 25.17. Результаты определений AjH° (ВегС14, г, 0) (в кДж-моль)
Авторы
Метод
ДуЯ0 (Ве2С14, г, 0)
II закон
III закон
Ральфе, Фишер, 1933 [1098] Измерение плотности паров, 835 К, 1 точка — —830
Рябчиков, Тихинский, 1960 [362] Масс-спектрометрический, 524—568 К, 3 точки —845 —811 а
Ко и др.. 1967 [1735] Метод протока, 623—683 К, 10 точек (см. критику —823±16 —837±11
этих измерений в работах [1418, 1426])
Хилденбранд, Серд, 1969 [1426] Масс-спектрометрический, 503 К, 1 точка — —826 а
Шефер и др., 1974 [2283а, 2443а] Масс-спектрометрический, 533 К, 1 точка — —817 ,
а Абсолютные значения констант равновесия были вычислены Шефером и Бинневисом [2443а] на основании
приведенных в этой работе интенсивностей ионных токов.
1426, 2283а, 2443а]. Ее погрешность оценена с учетом расхождения результатов отдельных
измерений и неточности термодинамических функций.
ВеВг (г). Термодинамические свойства газообразного бромида бериллия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 748.
В табл. 25.5 представлены молекулярные постоянные Ве79Вг, принятые на основании
результатов исследований спектра этой молекулы и приближенных оценок. В спектре ВеВг исследована
256
Бериллий и его соединения ВеВг2 (к, ж)
только одна система полос АШ -> X2 2+ [2313, 2318, 2320]. Оба комбинирующих состояния должны
коррелировать с атомами Be A5)+Br BР). Аналогично молекулам других галогенидов группы
ПА с диссоциационным пределом Be CР)+Вг BР) должно коррелировать большое число
возбужденных состояний ВеВг, часть которых, в том числе квартетные состояния, могут быть связанными
состояниями. Сопоставление с другими галогенидами Be и Mg свидетельствует, что энергии этих
состояний должны превышать 40 000 см.
Приведенные в табл. 25.5 колебательные постоянные молекулы Ве79Вг в состояниях X2 Е+ и
АЧ1 приняты по данным [2313], где они были рассчитаны по ф-кантам полос с и' ^ 5, v" ^ 5;
они хорошо согласуются с постоянными, найденными в работах [2318, 2320]. Вращательные
постоянные получены в работе Карлеера и др. [820] и основаны на результатах анализа
вращательной структуры полосы 0—0 системы А2И—X2 ?+.
Термодинамические функции ВеВг (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95),
Значения (?вн и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом
возбужденного состояния А2П. в предположении, что Q^\ Ер = 2 @№ вр. Величина Q^fJ вр и ее производные
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82);
мультиплетность уровней учитывалась статистическим весом. В расчетах учитывались все уровни
энергии со значениями /^ /шах> „, где Jma> v находились из условий A. 81).
Колебательно-вращательные уровни состояния Х2?+ вычислялись по уравнениям A. 65), A. 62). Значения
коэффициентов Ykl, а также величин vmS/% и /1Ш, использованные в расчетах, приведены в табл. 25.6;
они соответствуют изотопической модификации Be79Br. Значение Yso получено из условий] A. 50).
Основные погрешности рассчитанных термодинамических функций ВеВг (г) при низких
температурах обусловлены неточностью фундаментальных постоянных, молекулярных постоянных
в состоянии X2 2+ и пренебрежением различия постоянных молекул Ве79Вг и Ве81Вг. Они не
превышают 0,03 Дж -К -моль в значениях Ф° (Т) и S° (T) при 298,15 К. При высоких температурах
становятся существенными, особенно в значениях Ср (Т), погрешности из-за пренебрежения в
расчете всеми возбужденными состояниями ВеВг, кроме состояния А2Л (до 0,5 Дж-К~1-моль~1
в Ср F000 К)). Погрешности в значениях Ф° (Т) при 3000 и 6000 К составляют 0,05 и 0,1 ДжХ
X К-моль соответственно.
Термодинамические функции ВеВг (г) публиковались ранее в таблицах JANAF [1543] и работе
Климова и др. [167]. В [1543] расчет выполнен по оцененным молекулярным постоянным, заметно
отличающимся от принятых в настоящем справочнике. Соответствующие расхождения с данными
табл. 748 составляют 0,7 и 1,5 Дж-К-моль в значениях Ф° (Г) и S° (T). В работе [167]
результаты расчета представлены аппроксимирующим полиномом для Ф° (Т) в интервале 293,15 ^ Т ^
^5000 К; расхождения с данными табл. 748 не превышают 1,5 Дж-К~1-моль.
Константа равновесия реакции ВеВг (г)=Ве (г)+Вг (г) вычислена на основании энергии
диссоциации
Дг#° @) = D0.(BeBr) = 300 + 20 кДж • моль ?» 25 000 ± 1600 см.
Эта величина принята в результате сопоставления значений Do (MX), Do (MX2) и Do (MX)/
/Do (MX2) в рядах галогенидов (X=F, C1, Вг, I) щелочноземельных металлов. Она практически
совпадает с оценкой в работе [294]. Расчет с использованием ионной модели [193] приводит к
неудовлетворительным результатам для соединений бериллия.
Принятому значению соответствует
г, 0) = 137,676 ± 20 кДж • моль.
ВеВг2 (к, ж). Термодинамические свойства кристаллического и жидкого дибромида бериллия
в стандартном состоянии при температурах 298,15—1300 К приведены в табл. 749.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В литературе отсутствуют какие-либо экспериментальные данные о теплоемкости и
фазовых переходах ВеВг2 (к), за исключением температуры плавления. Недостаточно изучен
возможный полиморфизм ВеВг2 (к) и отсутствуют надежные данные об области стабильности
ромбической модификации. Последняя согласно исследованиям Семененко и Наумовой [371] и Ла-
зарини [1829] изоструктурна с а-модификацией ВеС12 и относится к структурному типу SiS2.
Согласно [371 ] свежевозогнанный образец дибромида бериллия не претерпевает полиморфных пре-
257
ВеВг2 (г) Глава 25
вращений при нагревании до Гт=781 К. В справочнике за стандартное состояние ВеВг2 (к) в
интервале 0—781 К принята ромбическая модификация. Значения S° (ВеВг2, к, 298,15 К) и
Н° B98,15 К)—Н° @) оценены методом Латимера с использованием экспериментальных данных по
теплоемкости и энтропии а-ВеС12. Погрешности этих значений, приведенных в табл. 25.1,
составляют 10 Дж -К -моль и 1,5 кДж -моль соответственно. Для теплоемкости ВеВг2 (к) в интервале
298,15—781 К принято уравнение, аппроксимирующее данные из таблиц JANAF [1543];
последние оценены сравнением данных по теплоемкости BeF2 и а-ВеС12. Температура плавления ВеВг2
G81 + 5 К) принята по данным Семененко и Наумовой [371]. В таблицах JANAF [1543]
принимается более низкое значение G61 К), полученное Фишером и Ральфсом [1099] на образце ВеВга,
структура которого не была установлена. Энтальпия плавления A8 + 4 кДж-моль) оценена на
основании предположения о равенстве энтропии плавления ВеВг2 и ZnBr2 (структурная близость
дигалогенидов Be и Zn отмечена в [371]). Близкое значение для &тН (ВеВг2) получено в [1543]
A9 кДж-моль) при сопоставлении суммы энтропии превращения и плавления в ряду BeF2—
ВеВг2.
Теплоемкость жидкого ВеВг2 оценена равной 100 + 10 Дж-К-моль; в [1543] на
основании ненадежных данных [1989] для теплоемкости ВеС12 (ж) принято, по-видимому, завышенное
значение 117 Дж-К~1-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 1300 К оцениваются в 8, 15 и
18 Дж-К-моль~1 соответственно. Термодинамические функции ВеВг2 (к, ж), приведенные
в табл. 749 и в справочнике JANAF [1543], различаются в пределах 2 Дж-К~1-моль из-за
разной оценки значений S° B98,15 К) и С°р (ВеВг2, ж).
В справочнике принята энтальпия образования дибромида бериллия
AfH°(BeBr2, к, 298,15 К) = — 358 + 8 кДж • моль,
основанная на измеренных Бильтцем и Мессеркнехтом [688] и Бильтцем и др. [689] энтальпиях
растворения ВеС12 (к) и ВеВг2 (к) в соляной кислоте.
Давление пара дибромида бериллия ВеВг2 (к, ж) = ВеВг2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
-1
ASH° (BeBr2, к, 0) = 130 + 15 кДж • моль
основанной на измерениях давления пара, выполненных Ральфсом и Фишером [1098]. Измерения
проводились методом точек кипения F24—695 К), результаты представлены только в виде
уравнения, которому соответствуют энтальпии сублимации, равные 134 кДж-моль (II закон)
и 123 кДж-моль (III закон). Использованные в этой работе сосуды из кварца оказались
нестойкими к ВеВг2, что могло привести к некоторому занижению вычисляемой энтальпии сублимации.
Из аналогичных измерений с ВеС12 этими же авторами (см. выше) получена величина,
заниженная примерно на 6 кДж-моль. В связи с этим для ВеВг2 (к, ж) принято значение несколько
увеличенное по сравнению с вычисленным из экспериментальных данных. При оценке его
погрешности учтена неточность термодинамических функций, которая вносит погрешность
в 10 кДж-моль в значение энтальпии сублимации.
ВеВг2 (г). Термодинамические свойства газообразного дибромида бериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 750.
Молекулярные постоянные, использованные для расчета термодинамических функций ВеВг2,
приведены в табл. 25.8. В соответствии с результатами электронографического исследования
[5, 12] и исследования ИК-спектра [2566] в справочнике принято, что молекула ВеВг2 имеет
линейную структуру симметрии Z^^ в основном состоянии A'1 EJ. Момент инерции рассчитан на
основании значения г (Be—Вг)=1,91 +0,02 А, найденного Акшпиным и др. [5, 12] при электроно-
графическом исследовании ВеВг2; его погрешность составляет 2-Ю"9 г-см2. Частота v3 принята
средней между измеренными Снелсоном [2566] в ИК-спектре молекул ВеВг2, изолированных в
матрицах из Ne и Аг (993 и 985 см соответственно), поскольку полученные таким образом частоты
v3 для BeF2 и ВеС12 хорошо согласуются -с найденными в газовой фазе (Снелсон принял для газа
v3=1010 см). Для v2 принята оценка Снелсона для газовой фазы (в матрице из Ne наблюдалась
частота 207 см). Значение \ рассчитано для поля валентных сил; так же как в случае BeF2,
оно согласуется в пределах 10% с оцененными в работах [748, 1543, 2566]. Погрешности при-
258
Бериллий и его соединения Bel (г)
нятых значений частот оцениваются в 10—20 см. Сведения о возбужденных состояниях ВеВг2
отсутствуют, по-видимому, их энергии превышают 25 000 см.
Термодинамические функции ВеВг2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)Л
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический осцилля-
тор». Погрешности рассчитанных величин обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета; они составляют 1, 4 и 6 Дж. К-моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно, в том числе 1—2 Дж-К-моль из-за
неточности молекулярных постоянных.
Ранее термодинамические функции ВеВг2 (г) рассчитывались в таблицах [1544], а также в
работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). В [1544] все
функции больше, чем в настоящем справочнике (на 0,4—0,6 Дж-К-моль~1 в значениях Ф° (Т)\,
что объясняется в основном разницей в оцененной величине vx. Расхождения с расчетами [7481
и [195] достигают 10 и 6 Дж-К-моль~1 соответственно в основном из-за того, что в них приняты
завышенные значения частоты v .
Константа равновесия реакции ВеВг2 (г)=Ве (г)+2Вг (г) вычислена с использованием
значения АГН° @)=772,029 +18 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дибромида бериллия. Этим величинам также соответствует
bfH°(BeBr2, г, 0) = —216,430 + 17 кДж • моль.
Bel (г). Термодинамические свойства газообразного иодида бериллия в стандартном со*
стоянии при температурах от 100 до 6000 К приведены в табл. 751.
В табл. 25.5 представлены молекулярные постоянные Bel, принятые в справочнике. В спектре
Bel наблюдалась только одна система полос А2П -> X2 ?+ [819, 2084, 2085, 2086]. По аналогии
с другими галогенидами элементов группы ПА состояния X2 ?+ и АЩ должны коррелировать с
атомами Be A5')+I BP)- Co следующим диссоциационным пределом Be CР)+1 (*Р) должно
коррелировать большое число возбужденных состояний Bel, часть из которых может быть связанными
состояниями с энергиями выше 35 000 см.
Колебательные постоянные Bel в состояниях X2 Е+ и Л2П приняты по работе Мурти и Рао
[2084], которые нашли их по ^-кантам девяти полос (i/ <CJ 3, v" ^2). Вращательные постоянные
приняты на основании результатов анализа тонкой структуры двух подполос 0—0 системы А2П ->
X2 ?+, выполненного Карлеером и Коленом [819]. Авторы [819] показали, что анализ
вращательной структуры спектра Bel в работах [2085, 2086] проведен некорректно в связи с неверной
нумерацией вращательных линий.
Термодинамические функции Bel (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95),
Значения (?ЕН и ее производных по температуре рассчитывались по уравнениям A. 90)—A. 92)
с учетом возбужденного состояния Л2П в предположении, что C^1Jp = Wfe)ffi)..p' Величину
QifJ ъ и ее производные вычислялись по уравнениям A.73) ¦—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с
помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями /^
^ /шах ¦ v, где /тзх „находились из условий A.81). Колебательно-вращательные уровни
состояния X2 ?+ вычислялись по уравнениям A. 65), A. 61). Значения коэффициентов Ybl> а также
величин ушах и /]!ш, использованные в расчетах, приведены в табл. 25.6. Коэффициенты Yso и Yo$
получены из условий A. 50) и A. 59). Мультиплетность и вырождение уровней обоих состояний
учитывались соответствующими статистическими весами.
Погрешность рассчитанных термодинамических функций Bel (г) при низких и умеренных тем?-
пературах определяется неточностью фундаментальных постоянных и постоянных Bel в
состоянии X2 ?+. При высоких температурах становится существенным пренебрежение рядом высоких
электронных состояний, не наблюдавшихся в спектрах этой молекулы (до 0,5 Дж-К~1-моль"^
в значении С°р F000 К)). Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К оцениваются
в 0,03, 0,05 и 0,2 Дж-К~1-моль соответственно.
Термодинамические функции Bel (г) вычислялись ранее в таблицах JANAF [1543] и работе
Климова и др. [167]. В [1543] расчет выполнен по оцененным молекулярным постоянным и без
учета возбужденного состояния; расхождения в значениях термодинамических функций,
приведенных в [1543] и табл. 751, достигают 0,3 и 0,5 Дж-К-моль в значениях Ф° (Г) и $° (Т) сог
259
Bet2 (к, ж) Глава 25
ответственно. В работе [167] расчет выполнен более грубым методом, и расхождение в значении
Ф°E000К) достигает 1,4 Дж-К^-моль.
Константа равновесия реакции Bel (г)=Ве (г)+1 (г) вычислена на основании принятой в
справочнике энергии диссоциации
ДД°@) = Do(Bel) = 220.+ 20 кДж • моль« 19000 + 1600 см.
Эта величина оценена сопоставлением значений Do (MX), Do (МХа) и Do (MX)/D0 (MX2), где
X=F, Cl, Br, I, для галогенидов Be, Mg, Ga, Sr и Ва. Близкие значения получены в работе [294].
Расчет с использованием ионной модели [193] приводит к неудовлетворительным результатам для
соединений бериллия.
Принятой энергии диссоциации соответствует
A^°(BeI, г, 0) = 2Q6,916 + 20 кДж • моль.
Ве12 (к, ж). Термодинамические свойства кристаллического и жидкого дииодида бериллия
в стандартном состоянии при температурах 298,15—1300 К приведены в табл. 752.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В литературе отсутствуют данные о теплоемкости или энтальпии твердого и жидкого
Ве12. Слабая изученность Ве12 (так же как и ВеВг2) объясняется его гигроскопичностью и
высоким давлением пара вблизи температуры плавления. Данные по полиморфизму Ве12 [371, 1585]
свидетельствуют о существовании нескольких модификаций; однако их термодинамическая
стабильность и характер взаимных превращений остаются не вполне выясненными г. В настоящем
справочнике при расчете термодинамических функций Ве12 (к) его полиморфизм не принимался
во внимание и в интервале 0—763 К за стандартное состояние принята ромбическая модификация.
Значения S° B98,15 К) и Н° B98,15 К)—Н° @) оценены при помощи приближенных методов,
как и для ВеВг2. Принятые значения (см. табл. 25.1) имеют погрешности 10 Дж-К-моль
й 1,5 кДж-моль соответственно. Уравнение для теплоемкости Ве12 (к) в интервале 298,15—
763 К выведено по данным, приведенным в таблицах JANAF [1543]; последние оценены
сравнением значений теплоемкости BeF2 и а-ВеС12. Температура плавления G63 +5 К) принята по
термографическим измерениям Семененко и Наумовой [371], полученным для хорошо
охарактеризованного образца Ве12. Более низкое значение G53 К) получено Ральфсом и Фишером [1098] для
образца с неизвестной структурой.
Энтальпия плавления Ве12 оценена в 18 +4 кДж -моль в предположении о равенстве энтропии
плавления дибромидов и дииодидов Be и Zn, кристаллические структуры которых очень близки
[371]. В таблицах JANAF [1543] принято более высокое значение B1 кДж-моль), метод оценки
которого не указан. Теплоемкость жидкого Ве12 оценена равной 100+10 Дж-К-моль~1; в
справочнике JANAF [1543] на основании ненадежных данных [1989] о теплоемкости ВеС12 (ж) принято
существенно более высокое значение, равное 117 Дж-К~1-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 1300 К оцениваются в 7, 15
и 18 Дж -К -моль соответственно. Различия между термодинамическими функциями Ве12 (к, ж),
приведенными в табл. 752 и в [1543], не превышают 1—2 Дж-К~1-моль~1.
В справочнике принята энтальпия образования дииодида бериллия
Д^0 (Ве12, к, 298,15[К) = —191 ±|8 кДж • моль,
основанная на измеренныхБильтцем и Мессеркнехтом [688] и Бильтцем и др. [689] энтальпиях
растворения ВеС12 (к) и Ве12 (к) в соляной кислоте.
Давление пара дииодида бериллия Ве12 (к, ж)=Ве12 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
Д,Я°(Ве!2, к, 0) = 130 + 15 кДж • моль,
Согласно Семененко и Наумовой [371 ] образующаяся при возгонке ромбическая а-модификация (структурный
тип SiS2) при нагревании до 563 К переходит в р'-модификацию, а при 643 К — в ^-модификацию (Р'- и р-моди-
фикации имеют сложную объемно-центрированную решетку). Последняя при 743 К (за 20° до плавления)
переходит в модификацию с неизвестной структурой.
260
Бериллий и его соединения Ве12 (г), BeS (к)
основанной на измерениях давления пара методом точек кипения E78—703 К) в работе Ральфса
и Фишера [1098]. В [1098] результаты измерений представлены только в виде уравнения,
которому соответствуют энтальпии сублимации, равные 123 кДж-моль (II закон) и 125 кДж-моль
(III закон). Взаимодействие исследуемого вещества с кварцем могло увеличить вычисленное из
этих данных значение энтальпии сублимации (см. ВеС12 и ВеВг2), поэтому принятое значение
несколько увеличено по сравнению с рассчитанным из данных [1098]. При оценке погрешности
учтена неточность термодинамических функций Ве12 (к, г) A0 кДж-моль).
Ве12 (г). Термодинамические свойства газообразного дииодида бериллия в стандартном со<
стоянии в интервале температур 100—8000 К приведены в табл. 753.
Молекулярные постоянные, использованные для расчета термодинамических функций Ве12>
приведены в табл. 25.8. На основании результатов электронографического исследования [5,_12]
и аналогии с другими дигалогенидами бериллия в справочнике принято, что в основном Xх 1^
состоянии молекула Ве12 имеет линейную структуру. Момент инерции Ве12 рассчитан по значению
г (Be—I) =2,10 +0,02 А, найденному Акишиным и др. [5, 12] при электронографическом
исследовании Ве12; его погрешность составляет 4-Ю9 г-см2. Частота v3 принята средней между
измеренными Снелсоном [2566] в матрицах из Ne и Аг (878,872 и 877,867 см), поскольку оцененные
таким образом частоты v3 BeF2 и ВеС12 хорошо согласуются с измеренными в газовой фазе (Снел-
сон оценил для газа v3=873 см). Частота vx рассчитана так же, как для BeF2 (см. выше); она
согласуется с величинами, оцененными в работах [748, 2566], в пределах 20 см. Частота v2 рассчи*
тана в предположении, что /г//а=45, т. е. имеет примерно такую же величину, как в молекулах
BeF2, ВеС12 и ВеВг2. Погрешности принятых значений частот оцениваются в 20—30 см.
Термодинамические функции Ве12 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных функций обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета
и составляют 2, 5 и 8 Дж -К -моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций Ве12 (г) вычислялись в таблицах [1544], а также
работах Брюэра и др. [748] (при Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Из-за
того что в этих расчетах приняты неверные значения частот, расхождения с табл. 753 достигают
в значениях Фс (Т) 2, 7 и 10 Дж-К~1-моль для [1544, 195, 748] соответственно.
Константа равновесия реакции Ве12 (г)=Ве (г)+21 (г) вычислена с использованием значения
Дг#° @)=595,133+18 кДж-моль, соответствующего принятым в справочнике энтальпиям обра»
зования и сублимации дииодида бериллия. Этим величинам также соответствует
Д//° (Ве1.2, г, 0) = —61,054 + 17 кДж • моль.
BeS (к). Термодинамические свойства кристаллического сульфида бериллия в стандартном
состоянии при температурах 298,15—3000 К приведены в табл. 754.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. В справочнике за стандартное состояние BeS (к) принята кубическая модификация
(структурный тип ZnS, сфалерит) [1005а]. В литературе отсутствуют экспериментальные данные
о термодинамических свойствах BeS (к). Принятые значения S° B98,15 К), Н° B98,15 К)—Н° @)
и СI B98,15 К) оценены с использованием эмпирических методов оценки и экспериментальных
данных для сульфидов других элементов подгруппы ПА. При этом для оценки энтропии BeS (к)
использованы эмпирические методики и учтены результаты оценок, выполненных авторами работ
[160а, 2031]; теплоемкость BeS (к) оценена по методу, использованному в работе [1781а], а
изменение энтальпии — сопоставлением данных по сульфидам Mg, Ca, Sr и Ва. Погрешности принятых
значений S° B98,15 К) и Н° B98,15 К)—Я0 @) оцениваются в 4 Дж^К^-моль и 0,6 кДж-моль
соответственно. В интервале 298,15—3000 К для теплоемкости BeS (к) принято уравнение (см,
табл. 25.1), выведенное по трем оцененным значениям теплоемкости: С°р B98,15 К)=34+3 ДжХ
ХК^-моль, С; A000 К)=51+5Дж-К-мольиС;B000К)=63+8 Дж-К^-моль; последние
два значения оценены с учетом данных по теплоемкости оксидов и сульфидов элементов под^
группы НА.
Данные по температуре плавления BeS отсутствуют; в справочнике термодинамические функ»
ции BeS (к) рассчитаны до 3000 К, при этой температуре давление диссоциации BeS (к) достн»
гает 3 атм.
261
6eS (г) Глава 25
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 4,
7, 10 и 15 Дж-К~1-моль соответственно. Термодинамические функции BeS (к), приведенные
в справочнике [578] для Т <^ 1800 К, отличаются от данных табл. 754 в пределах 4 Дж -К -моль.
Принятая в справочнике энтальпия образования
, к, 298,15 К) = — 236 + 10 кДж • мош
-1
основана на измерениях энтальпий растворения BeS (к) и Be (к) в серной кислоте, выполненных
Вартенбергом [2800а]. В справочнике Миллса [2031] сообщается о неопубликованном
исследовании равновесия реакций BeO (k)+H2S (r)=BeS (к)+Н2О (г) и 2ВеО (k)+GS2 (r)=2BeS (к)+
+СО2 (г), проведенном Блюмом в 1948 г. По уравнению II закона Миллс [2031] нашел из данных
Блюма энтальпию образования BeS (к) равной —300 кДж-моль~\ отметив плохое согласие
соответствующих значений ArS для этих реакций с рассчитанными по таблицам термодинамических
функций. По-видимому, это свидетельствует о невысокой точности измерений Блюма, результаты
Которых остались недоступными при подготовке справочника и не могли быть обработаны
повторно.
Давление пара сульфида бериллия в реакции BeS (K)=BeS (г) вычислено на основании
значения ДГЯ° @) = Д8Я° (BeS, к)=479,676+23 кДж-моль, соответствующего принятым в
справочнике энтальпиям образования BeS (к) и BeS (г).
BeS (г). Термодинамические свойства газообразного сульфида бериллия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 755.
В табл. 25.5 приведены молекулярные постоянные BeS, принятые на основании анализа
результатов исследований электронных спектров [841, 1220а], квантовомеханического расчета [2766а 1
и оценок. В электронном спектре BeS исследованы две системы полос В1 2+ <* X1 Е+ [841, 1220а]
и 441 <± X1 Е+ [841]. Квантовомеханический расчет в приближении Хартри—Фока показал
наличие у молекулы BeS низколежащих связанных триплетных состояний а3П и Ь3?+, изоконфи-
гурационных состояниям АгП и В1 ?+ соответственно. Существование состояния а3П
подтверждается возмущениями, обнаруженными в системе АХП—X1 ?+ [841 ] *. По аналогии с оксидами и
сульфидами других элементов подгруппы ПА молекула BeS должна обладать еще рядом связанных
электронных состояний, соответствующих конфигурациям . . .o27t37r C'ХД, 3'1Е+, х ?"") и . . .зк4тс
(»П, 41).
Молекулярные постоянные BeS в состояниях XXS+, А1!! и i?xS+, приведенные в табл. 25.5,
найдены Чизамом и др. [8411, которые выполнили анализ вращательной структуры ряда полос
систем A1!!—XXZ+ (i/ < 11, v" < 3) и B1L+—X1I,+ (i/ < 3, v" < 2). Энергии состояний а3П
и 63Е+ оценены сопоставлением результатов квантовомеханического расчета [2766а] с
соответствующими величинами для молекулы ВеО, их погрешности составляют 1000 и 3000 см.
Термодинамические функции BeS (г) вычислены по уравнениям A. 3)—A. 10), и A. 93)—
A. 95). Величина QBK и ее производные рассчитывались по соотношениям A.90)—A.92) с учетом
четырех возбужденных электронных состояний в предположении, что (?<*?_ вр = (pjpx) Qif2 „P-
Величина QK03 вр состояния X1 ?+ и ее производные вычислялись непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью уравнений типа
A. 82). В расчете учитывались все уровни со значениями / ^ /шах „, последние находились по
соотношению A. 81). Энергия колебательно-вращательных уровней рассчитывалась по
уравнениям A. 65) и A. 63) с коэффициентами Уы, приведенными в табл. 25.6 вместе со значениями vmtx
и /цш. Значения Ykl получены по постоянным из табл. 25.5 с учетом условий A. 50) и A. 59),
для соблюдения которых в уравнение A. 65) введены дополнительные члены с коэффициентами
^з<м Yi0 и Y03. Постоянная Y21 изменена из условия .В(,шах+1=0. Все постоянные пересчитаны по
соотношениям A.66) для изотопической модификации BeS, соответствующей природной смеси
изотопов.
Погрешности вычисленных термодинамических функций при температурах до 1000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах основной вклад в погрешность вносит неопределенность в энергии триплетных электрон-
1 Авторы [841] предположили, что возмущения обусловлены состоянием JA (Te < 13048 см х), однако в работе
[9316] показана неточность такой интерпретации.
262
Бериллий и его соединения BeSO4 (к, ж)
ных состояний а3П и Ъ3 Е+. Общие погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К
могут составить 0,05, 0,5 и 2 Дж-К~1-моль~1 соответственно.
Ранее таблица термодинамических функций BeS (г) не публиковалась.
Константа равновесия реакции BeS (r)=Be (r)+S (г) вычислена по принятой в справочнике
энергии диссоциации
АГН° @) = Do (BeS) = 350 ± 20 кДж • моль ж 29 250 + 1700 см
Это значение оценено на основании сопоставления энергий диссоциации сульфидов и оксидов
кальция, стронция и бария, а также величины Do (BeO), принятых в справочнике. Линейная
экстраполяция уровней состояния Xх ?+ приводит к близкому значению C71 кДж-моль).
Принятой энергии диссоциации соответствует
г, 0) = 244,538 ±21 кДж • моль.
BeSO4 (к, ж). Термодинамические свойства кристаллического и жидкого сульфата бериллия
в стандартном состоянии при температурах 100—2000 К приведены в табл. 756.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 25.1. Для твердого BeSO4 известны три кристаллические модификации —
низкотемпературная тетрагональная (а) и две высокотемпературные — ромбическая (f>) и кубическая (у) [49].
При Т < 298,15 К термодинамические функции BeSO4 (к) вычислены на основании данных
о теплоемкости, полученных Тейлором и др. [2663] A2—301 К) на образце чистоты 99,9%.
Экстраполяция теплоемкости к 0 приводит к значению S° A2,56 К) =0,142 Дж-К~1-моль~1.
Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—Я° @) составляют 0,5 Дж-К^-моль
и 0,07 кДж-моль соответственно. При Т > 298,15 К для теплоемкости BeSO4 (к) принято
уравнение (см. табл. 25.1), выведенное методом Шомейта по результатам измерений энтальпии в работе
Тейлора и др. [2663] C66—864 К), погрешности которых оценены в 0,8%.
Температуры полиморфных превращений сульфата бериллия (861 +5 К и 912 +5 К) приняты
на основании измерений Бэра и Тернбулла [631], выполненных методом количественного ДТА.
Результаты измерений авторов [49, 55, 416, 439] менее точны, хотя и не противоречат данным [631 ].
Энтальпии превращения BeSO4, равные соответственно 5 и 2,1 кДж-моль, также
приняты по измерениям [631], точность которых оценивается в 0,5 кДж-моль. * В литературе
отсутствуют данные о температуре и энтальпии плавления BeSO4, поскольку при —-1100 К и
атмосферном давлении наблюдается диссоциация BeSO4 [4391 2. В справочнике принято оценочное
значение Гот=1400+200 К, близкое к известной температуре плавления MgSO4 (см. ниже). Эта
оценка основывается на аналогии в температурах плавления в рядах сульфатов элементов
подгрупп IA и ПА и близости температур плавления Li2SO4 и NaaSO4. Энтальпия плавления BeSO4
оценена в предположении, что сумма энтропии полиморфных превращений и плавления BeS04
и MgSO4 одинакова и близка к значению 12,5 Дж-К-моль~1. Оценка теплоемкости жидкого
BeSO4 A50+20 Дж-К" 1-моль~1) выполнена с учетом характера изменения значений теплоемкости
жидких сульфатов в ряду MgSO4—BaSO4. Теплоемкость J3- и -f-^eSO4 также принята равной
150 Дж • К • моль.
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000 и 2000 К составляют 0,3, 2
и 13 Дж-К-моль соответственно. Расхождения между термодинамическими функциями
BeSO4 (к), приведенными в табл. 756 и в справочнике JANAF [1543], не превышают 1 Дж-К-1Х
X моль. Сведения о термодинамических функциях BeSO4 (ж) в справочной литературе отсутствуют.
Константа равновесия реакции BeSO4 (к)=Ве (r)+S (г)+4О (г) вычислена с использованием
значения АГН° @) =2770,914 +5,0 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
L}H° (a-BeSO4, к, тетр., 298,15 К) =—1200,0 + 0,5 кДж • моль.
Это значение основано на комбинации измеренной Навратилом и Эттингом [2111] энтальпии
растворения бериллия в серной кислоте и значениях энтальпии растворения BeSO4 (к) в серной
1 В работе Бэра и Тернбулла [632] определена разность энтальпий образования а- и ^-модификаций BeS04 при
1028 К. Значение AirH=13,8 кДж-моль почти в 2 раза больше суммы энтальпий переходов, найденных в [631].
2 Согласно [2611] при 1200 К равновесные парциальные давления продуктов диссоциации BeSO4 равны: p(SO2)=
= 1,17 атм, р(О2)=0,585 атм, p(SO3)=0,221 атм, 2р=1,98 атм.
263
Be3N2 (к, ж) Глава 25
кислоте и энтальпиях других реакций, измеренных Тейлором и др. [2664]. Близкое, но несколько
менее точное значение —1198,1 +2,0 кДж-моль вычислено на основании данных об энтальпиях
растворения Be и BeSO4 в плавиковой кислоте, полученных Бэром и Тернбуллом [630, 632].
Be3N2 (к, ж). Термодинамические свойства кристаллического и жидкого динитрида три-
бериллия в стандартном состоянии при температурах 100—5000 К приведены в табл. 757.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
25.1. За стандартное состояние Be3N2 (к) в интервале 0—1673 К в справочнике принята
кубическая модификация (a-Be3N2), а в интервале 1673 — 2473 К — высокотемпературная
гексагональная модификация (|3-Be3N2) [1342] г. При Т < 298,15 К термодинамические функции вычислены
по результатам измерений теплоемкости Be3N2 (к) в работе Фурукава и Рейлли [1156] B0—315 К),
выполненных на образце, содержащем 99,23% Be3N2, 0,73% ВеО и 0,04% Be, и
скорректированных на содержание примесей. Погрешности принятых значений S° B98,15 К) и Н° B98,15 К)—
Н° @) (см. табл. 25.1) оцениваются в 0,3 Дж-К-моль и 0,06 кДж-моль соответственно.
Измерения теплоемкости образца, содержащего 96,7% Be3N2, 2,7% ВеО, 0,2% Be, 0,1% Си 0,3%
Fe, в работе [1608] в интервале 100—298 К удовлетворительно согласуются с данными Фурукава
и Рейлли [1156] (в этом интервале они в среднем на 0,5% ниже данных [1156]), однако при Т <
< 100 К расхождения быстро увеличиваются, достигая 5% при 60 К и 75% при 30 К. Причины
этих расхождений неясны. В справочнике расчет выполнен на основании данных [1156], поскольку
только для образца Be3N2, исследованного в [1156], имеются измерения энтальпии при высоких
температурах [980]. В интервале 298,15 —1673 К для теплоемкости Be3N2 (к) принято уравнение
(см. табл. 25.1), выведенное методом Шомейта по результатам измерений энтальпии Be3N2 в
интервале 273—1124 К, проведенным с точностью ~0,5% Дугласом и Пейном [980]. Температура
превращения Be3N2 A673+50 К) принята по работе [1007], а энтальпия полиморфного
превращения A6+5 кДж-моль) принята как разность энтальпий образования кубической и
гексагональной модификаций при 298,15 К (предполагая равными изменение энтальпий этих
модификаций в интервале 298—1673 К). Теплоемкость высокотемпературной модификации Be3N2 A45 +
+15 Дж-К~1-моль) и жидкого Be3N2 A67+20 Дж-К-моль), а также энтальпия плавления
Be3N2 A10+20 кДж-моль) оценены с использованием соответствующих данных для оксида
бериллия. Температура плавления Be3N2 B473 +100 К) определена в работе Фихтера и Брун-
нера [1078].
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 3000 и 5000 К оцениваются 0,2,
0,5, 10 и 23 Дж-К-моль~1 соответственно. Термодинамические функции a-Be3N2 ниже 1673 К,
приведенные в табл. 757, практически совпадают с вычисленными в справочнике JANAF [1543].
Для P-Be3N2 и Be3N2 (ж) расхождения увеличиваются с ростом температуры, достигая
17 Дж-К~1-моль в значении'Ф0 D000 К), что обусловлено учетом энтальпии превращения Be3N2
при 1673 Кв настоящем справочнике и различием принятых значений теплоемкости Be3N2 (ж).
Константа равновесия реакции Be3N2 (к)=ЗВе (r)+2N (г) вычислена с использованием
значения &ГН° @)=2481,500+15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^Я" (a-Be3N2, к, куб., 298,15 К) = —588,0 + 1,3 кДж • моль.
Эта величина основана на результатах измерений, приведенных в табл. 25.18, погрешности
соответствующих значений учитывают воспроизводимость измереций и неточность использованных
постоянных. Наиболее точные измерения с хорошо охарактеризованными образцами (в том числе
кристаллографически) выполнены Гроссом и др. [1279] 2.
Исследования равновесия реакции диссоциации динитрида трибериллия [1454, 2882] приводят
к значениям, совпадающим с принятым в пределах их погрешностей. Большую погрешность
в эти данные вносит неточность значения Д^Я° (Be, г) и термодинамических функций Be3N2
F кДж-моль при 1800 К). Калориметрические измерения в старых работах [18, 2126, 2129]
выполнены с ^охарактеризованными образцами, и их результаты (—559-f—565 кДж-моль)
представляются ненадежными.
1 .Данные Эккерлина и др. [1007] о температуре A673 К) и характере полиморфного превращения Be3N2 (к) нельзя
считать достоверными.
* Данные [1279] дают также bfH°(p-BesN2, к, гекс, 298,15 К)=—572,0+6,3 кДж^моль.
264
Бериллий и его соединения ВеСО3
Таблица 25.18. Результаты измерений А/Я0 (a-Be3N2, к, 298,15 К) (в кДжмоаь)
Авторы
Метод
A,/7°(Be3N2, к,
298,15 К)
Гросс и др., 1962 [1279] Измерение энтальпий реакций в калориметре с микропечью:
1. Be (к)+С12 (г) = а-ВеС12 (к), 1
2. a-Be3N2(K)+3Cl2(r)=3a-BeCl,(K)+N2(r), J °h
3. ЗВе (k)+2NH3 (г)= a-Be3N2 (к)+ЗН„ (г) —587,9+1,3
Иейтс и др., 1964 [2882] Исследование равновесия реакции BesN2 (к)=ЗВе (r)+N2 (г) —470+30 (II)
торзионным методом, 1450—1650 К, 58 точек, —597+.15 (III)
АГН° B98,15 К)=1626,8+4,2 (III)
Хёниг, Сирей, 1967 [1454] То же, эффузионный метод, 1648—1946 К, 34 точки, —576+20A1)
Дг#° B98,15 К)=-1575,8+2,4 (III). -604+15 (III)
ВеСО3 (к). Термодинамические свойства кристаллического карбоната бериллия в стандартном
состоянии в интервале 298,15—1000 К приведены в табл. Д. 57.
Значения постоянных, принятые в расчетах термодинамических функций, представлены
в табл. 25.1. В литературе отсутствуют какие-либо экспериментальные данные о
термодинамических свойствах ВеСО3 (к), в том числе о кристаллической структуре и температуре плавления
ВеСО3- Поэтому все приведенные в табл. 25.1 величины оценены при подготовке справочника.
Оценка теплоемкости ВеСО3 (к) как при низких температурах @—298,15 К), так и при Т >
> 298,15 К выполнена на основании сравнительного расчета из данных о теплоемкостях BeSO4,
MgCO3 и MgS04 в предположении, что QBeCO,,) = CJ (BeSO4)-f Гр (MgCO3) — C°P (MgSO4).
Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—Н° @) оцениваются
в 7 Дж-К моль и 1 кДж-моль соответственно. Предложенное в работе [273] значение
5°(ВеСО3, к, 298,15 К)=67+13 Дж-К~1-моль~1 оценено по методу Латимера и представляется
завышенным, поскольку оно превышает соответствующее значение для MgCO3 (к)
F5,09 Дж-К^-моль). Погрешности значений теплоемкости ВеСО3 при Г > 298,15 К могут
достигать 20%, поскольку для их оценки использовались оцененные данные для теплоемкости MgSO4.
Уравнение для теплоемкости ВеСО3 (к) (см. табл. 25.1) выведено по оцененным значениям
теплоемкости при 298,15, 400 и 600 К, равным 65, 85 и 103 Дж-К-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 1000 К оценены в 5, 8 и
15 Дж-К-моль соответственно. Таблица термодинамических функций ВеСО3 (к) публикуется
впервые.
Константа равновесия реакции ВеСО3(к)=Ве (г)+С (г)+30 (г) вычислена с использованием
значения Ь.ГН° @) =2809,464 +30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
к, 298,15 К) = —1045 + 30 кДж ¦ моль.
Это значение основано на оценке энтальпии реакции ВеСО3 (к)=ВеО (к)+СО2 (г) методом.
Карапетьянца по имеющимся данным для карбонатов Mg, Са, Sr, Ва, а также энтальпиях реакций
М(ОНJ (к)=МО (к)+Н2О (г) и MSO4 (к)=МО (k)+SO3 (г), где М=Ве, Mg, Са, Sr, Ва.
Соответствующие значения равны 40—50 кДж-моль.
Равновесие реакции ВеСО3 (к)=ВеО (к)+СО2 (г) исследовалось Капустинским и Стахановой
[152] методом протока E23—723 К, 10 точек). В качестве исходного образца использовался
ВеСО3-ЗВе(ОНJ-ВеО. Авторы [152] в отдельных опытах установили, что нагреванием до 150° С
в течение 6 часов можно полностью удалить влагу и получить ВеСО3-4ВеО. С таким образцом
и проводились измерения. Обработка этих измерений по уравнению II закона дает
Дг#° B98,15 К) =23,9+5,0 кДж-моль. Однако обработка тех же данных по уравнению-
Ill закона выявила большой систематический ход вычисляемых величин (от 112 до 145 кДж-моль),
среднее значение, которых составило 132 +10 кДж -моль. Столь большое различие расчетов по
уравнениям II и III законов, по-видимому, указывает на серьезную систематическую ошибку
измерений, возможно связанную с неполной осушкой образца. Неточность термодинамических
функций ВеСО3 при расчете по уравнению III закона приводит к погрешности в 8 кДж-моль в
энтальпии реакции.
Глава 26
МАГНИЙ И ЕГО СОЕДИНЕНИЯ
JYIg (к, ж), Mg, Mg+, Mg2, MgO (к, ж), MgO, MgH, MgH2 (к, ж), MgOH, Mg(OHJ (к), Mg(OHJ,
MgF, MgF2 (к, ж), MgF2, MgCl, MgCl2 (к, ж), MgCl2, MgBr, MgBr2 (к, ж), MgBr2, Mgl, Mgl2 (к, ж),
Mgl2, MgS (к, ж), MgS, MgSO4 (к, ж), Mg3N2 (к), MgCO3 (к, ж)
В настоящей главе рассматриваются 20 веществ — магний и его соединения с кислородом,
водородом, галогенами, серой, азотом и углеродом; для 12 веществ приведены сведения о
термодинамических свойствах в конденсированном состоянии, для 16 — в газообразном состоянии.
Термодинамические свойства трех газов (Mg, Mg+ и MgO) рассчитаны для температур до 10 000 К, одного
(Mg2) — до 3000 К и остальных до 6000 К. Для Mg в Приложении 3.9 представлены сведения о
критических параметрах.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов магния
(атомный вес 24,305) и других элементов, однако различие молекулярных постоянных
изотопических модификаций веществ в большинстве случаев в расчетах не учитывалось из-за отсутствия
соответствующих данных и его несущественного влияния на значения термодинамических
функций. В тех случаях, когда различие постоянных учитывалось в расчетах, это оговорено в
соответствующих текстах.
В справочнике не рассматриваются многоатомные окислы магния типа MgO2 и димерные
соединения типа Mg2Cl4 ввиду их низкой стабильности. Не рассматриваются также ионизированные
соединения типа MgO+, MgOH+, MgF+ и т. п., поскольку относительно высокие потенциалы
ионизации соответствующих нейтральных соединений (свыше 7 эВ) и их невысокие энергии
диссоциации делают маловероятным образование таких ионов в заметных количествах в равновесных
условиях. Не рассматривается также нитрид магния в газообразном состоянии (MgN), поскольку
установлено (см. гл. 25), что молекула BeN имеет слабосвязанное основное состояние.
Mg (к, ж). Термодинамические свойства кристаллического и жидкого магния в стандартном
состоянии при температурах 100—2400 К приведены в табл. 758.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние Mg (к) принято состояние, соответствующее
гексагональной плотнейшей упаковке. Сведения о полиморфизме Mg отсутствуют. При
Т < 298,15 К термодинамические функции вычислены по данным о теплоемкости, полученным
в работах Клузиуса и Вогена [861 ] A0—230 К), Крейга и др. [898] A2—320 К) и Манхена и Борн-
кесселя [1938] A2—300 К). Ниже 10 К использованы согласующиеся между собой измерения
авторов [309, 1884, 2304, 2558]. Погрешности принятых значений S° B98,15 К) и Н° B98,15 К) -
Н° @), которые совпадают с рекомендованными КО ДАТА—МСНС [864] в качестве ключевых
величин, оцениваются в 0,10 Дж-К-моль~1 и 0,03 кДж-моль^1 соответственно. В интервале
298,15—923 К для теплоемкости принято уравнение (см. табл. 26.1), выведенное совместной
обработкой по методу Шомейта результатов измерений энтальпии Mg в работах Истмана и др.
[1006] B93-888 К), Зекампа [2489] B91-773 К), Сталла и Мак-Доналда [2631] G00-1100 К),
Мак-Доналда [1988] D04—1300 К), а также значений энтальпий, рассчитанных по данным об
истинной теплоемкости, которые получены Саба и др. [2415] B98—420 К). При обработке
погрешности измерений [1988, 2415] оценивались в 0,3%, [2489, 2631] — в 1 % и [1006] — в 2%. Данные
Аубери, Гриффитса [549] B89—1023 К) не учитывались, так как они менее надежные.
Температура плавления (923+1 К) принята по измерениям Сталла и Мак-Доналда [2631].
С этим значением в пределах указанной погрешности согласуются данные [830, 846, 1289, 1302,
1370, 2838а], приводящие к значениям 922—924 К. Теплота плавления (8,5+0,2 кДж-моль'1) и
теплоемкость жидкого Mg C4,3+0,8 Дж-К^-моль) приняты по измерениям энтальпии
жидкого Mg, выполненным Мак-Доналдом [1988] (923—1300 К). Данные, полученные ранее Сталлом
и Мак-Доналдом [2631] (Д,ВЯ=8,95+0,2 кДж-моль), не учитывались, поскольку для энтальпии
Mg (ж) они примерно на 1% превышают результаты измерений [1988]. Определение hmH методом
ДТА в работе [846] (8,2+0,1 кДж-моль) также не принималось во внимание.
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000 и 2000 К оцениваются в 0,05,
0,2 и 1 Дж-К-моль~1 соответственно. Термодинамические функции Mg, приведенные в табл. 758,
незначительно отличаются от табулированных в справочниках [105, 1503, 1543, 1656]. При тем-
266
Магний и его соединения
Mg (к, ж)
Таблица 26.1. Принятые значения термодинамических величин для магния и его соединений
в кристаллическом и жидком состояниях
Вещество
Mg
MgO
MgH2
Mg(OHJ
MgF2
MgCl,
MgBr2
Mgl,
MgS
MgSO4
MgsN2
MgCOs
Состояние
К)-
ю
ос'о;
см о
ЕЕ
кДжХ
Хмоль'1
к, гекс. 5,00
ж —
к, куб. 5,16
ж —
к, тетр. 5,31
ж —
к, гекс. 11,41
к, тетр. 9,92
ж —
к, гекс. 13,77
ж —
к, гекс. 15,5
к, гекс. 17,0
к, куб. 8,33
ж —
кП, ромб, (а) 15,4
к1, ромб. (Р) —
ж —
к, куб. 10,5
к, гекс. 11,63
ж —
оо"
го
см
о
со
Дж-К-»
32,68
26,95
31,1
63,18
57,2
89,62
117
134
50,33
91,6
85
65,09
Примечание. C°p (T)=a+bT—cT-2+dT2
Mg: a <M06=0,544.
MgO: a d-106=—4,046, e-109=l,042.
MgSO4: a d-106= —19,825.
К
ю
оо"
СП
см
о е.
to
• МОЛЬ
24,90
37,24
35,35
77,11
61,59
71,38
73,22
74,48
45,56
96,40
92,05
76,11
+ el* (в
Коэффициенты б
для С°р
а
22,030
34,3
45,816
84
24,831
75
100,062
77,810
94,8
75,655
88,144
75,868
100
68,940
100
45,544
67
85,180
155
155
120,470
73,335
150
Ь-103
10,226
9,029
35,282
18,314
4,050
9,975
12,000
18,564
8,757
87,575
8,611
63,990
Цж-К^-моль).
уравнении
Т)
С ¦ Ю-6
0,202 а
9,724 а
—
25,256
15,495
6,440
—59,200
5,535
—
2,307
11,671 а
27,547
14,495
Интервал
температуры
К
298,15—923
923—2400
298,15—3100
3100—5100
298,15—600
600—1000
298,15—1000
298,15—1536
1536—4200
298,15—987
987—2900
298,15-984
984-2700
298,15—906 .
906—2000
298,15—2500
2500—4000
298,15—1283
1283—1410
1410—2000
298,15—2500
298,15-1263
1263-1500
или
Т
К
923
3100
600
—
1536
987
984
906
2500
1283
1410
2500
1263
или
АтН
кДжХ
Хмоль
8,5
77
14
—
58,5-
43,1
39,3
26 .
63
3
14,6
НО
59
пературах 2000 и 2400 К различия в значениях Ф° (Т) между расчетом [1543] и данными табл. 758
составляют 0,12 и 0,8 Дж-К-моль соответственно, а между[105] и табл. 758 при 2000 и 2400К—
0,14 и 0,24 Дж-К^-моль-1.
В справочнике за стандартное состояние магния принят Mg (к) и в соответствии с этим
&fH°(Mg, k)eee0.
Давление пара магния Mg (к, ж)=Mg (г) вычислено с использованием значения ДГ^Г° @) =
= Д8#° (Mg, к, 0)=145,903+0,80 кДж-моль, соответствующего принятой в справочнике
энтальпии сублимации
Д//° (Mg, к, 298,15 К) = 147,10 +¦ 0,80 кДж • моль.
Эта величина вычислена как средневзвешенная из результатов измерений давления пара
магния, представленных в табл. 26.2. Приведенные в таблице погрешности отражают только
воспроизводимость измерений. Результаты измерений в работах [251, 361, 1201, 1247, 1981] опубликованы
только в виде уравнений или в сильно сокращенном виде, что не позволяет определить их вос-
267
Mg (г)
Глава 26
Таблица 26.2. Результаты определений Д8?Г° (Mg, к, 298,15 К) (в кДж-моль г)
Авторы
Руфф, Хартман, 1924 [24051
Хартман, Шнейдер, 1929 [1360]
Баур, Бруннер, 1934 [627]
Колман, Эджертон, 1935 [870]
Шнейдер, Эш, 1939 [2459]
Шнейдер, Штоль, 1941 [2462]
Феттер, Кубашевский, 1953 [2773]
Приселков, 1957 [335]
Смит, Смит, 1959 [2555]
Шмаль, Зпбен, 1960 [2458]
Рябчиков, Мпкулинскпй, 1963 [361]
Машовец, Пучков, 1965 [251]
Гринбанк, Арджент, 1965 [1247]
Гилбрет, 1965 [1201]
Мак-Крири, Торн, 1971 [1981]
Прасад п др., 1978 [2264а]
Метод
Точек кипения, 911—1395 К, 11 точек
То же, 1009—1380 К, 16 точек
То же, 926—1283 К, 8 точек
Эффузионный, 700—738 К, 8 точек
Точек кипения, 1367 К, 1 точка
Протока, 817—1067 К, 3 точки
То же, 973—1193 К, 4 точки
Эффузионный, 668—781 К, 18 точек
То же, 626—818 К, 9 точек
Протока, 863—909 К, 6 точек
Тоже, 927—973 К, 6 точек
Д.я ° (Mg,
II закон
124 + 17
150+5
133+12
147±5
—
155+7
140+3
149+3
145+12
139+1
139+1
Эффузионный, 685—791 К, расчет по уравнению 147
Протока, 970—1220 К, расчет по уравнению
Торзионный, 600—800 К, расчет по уравнении
148
з 143
Испарения с поверхности, 496—658 К, расчет 146
по уравнению
Торзионный, 765 К, 1 точка, 8 измерений
Эффузионный, 765 К, 1 точка, 8 измерений
Протока, 953—1123 К, 5 точек
—
—
145+1
к, 298,15 К)
III закон
147,1+3,0
149,4+0,3
147,5+1,7
147,6+0,1
148,0
147,6 + 1,6
147,7+1,0
146,6+0,2
146,9+0,9
146,2+0,2
146,7+0,2
146,7
148,3
143,5
147,9
147,6
148,2
146,9+0,2
производимость. Как видно из таблицы, за исключением двух крайних значений, полученных в
работах [1247, 1360] и не учитывавшихся при вычислении средневзвешенного, остальные величины
находятся в хорошем соответствии. Принятое в справочнике значение совпадает с рекомендацией
КО ДАТА—МСНС [864], его погрешность учитывает неточность термодинамических функций
Mg (к, ж), которая приводит к погрешности в энтальпии сублимации ~0,2 кДж-моль~х при
обработке данных, полученных в области 1000 К.
Mg (г). Термодинамические свойства газообразного магния в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 759.
Уровни энергии атома Mg, использованные в расчете термодинамических функций, приведены
в табл. 26.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
Т а б л
Атом
Mg
[ и ц а 26
Электронная
конфигурация
. . .3s2
. . .ЪэЪр
. . .3s3p
.3 Уровни энергии атомов].
Состояние
т<
см"
Х5о 0
аРо 21 850,405
3Pi 21870,464
3Р2 21 911,178
xPi 35 051,264
Р»
1
1
3
5
3
Атом
Mg
Mg и Mg
Электронная
конфигурация
. . .3s3d
. . .3s3d
. . .Зр2
+, принятые для расчета термодинамических функций
Состояние
ч>,
3D
3Ро
3Pi
*Р2
т<
СМ
Pi
46 40fe,065 5
47 957,04 15
57 812,77 1
57 833,40 3
57 873,94 5
Атом
Mg+
Электронная
конфигурация
. . .3s
...Зр
. . .3d
Состояние
•
2рч.
см
0
35 669,31
35 760,88
71 490,19
71 491,06
Pi
2
2
4
6
4
. . .3s2, . . .3s3p, . . .3s3dn. . .Зр2 (SP). Энергии этих состояний приняты по результатам
спектральных измерений Рисберга [2344], они практически совпадают с рекомендацией Мур [2053]; энергия
состояния . . .3s3d ^D^) измерена в работе [2344] впервые.
268
Магний и его соединения Mg+ (г), Mg2 (г)
Термодинамические функции Mg (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1. 22)—A. 25). Значения Q3JS и ее производных вычислялись непосредственным суммированием
по уровням энергии. Погрешности вычисленных значений Ф° (Т) во всем интервале температур
не превышают 0,02 Дж-К~1-моль~1 и обусловлены главным образом неточностью принятого
значения R. Погрешности в значениях S0 (Т) и в особенности Ср' (Т) при Т > 5000 К становятся
больше из-за пренебрежения ридберговскими состояниями, а также из-за отсутствия данных
о состояниях XSO и \D2 конфигурации . . .Зр2 и о состояниях конфигурации . . ,3p3d, лежащих
ниже первого потенциала ионизации Mg. Эти погрешности могут достигать 0,1 и 1 Дж -К -моль
в значениях S° A0 000 К) и Ср A0 000 К).
Ранее таблицы термодинамических функций Mg (г) вычислялись в предыдущих изданиях
настоящего справочника [106, 105], в справочниках [1543, 1904, 1974, 2632], а также в работах
Хилзенрата и др. [1436] (до 10 000 К), Кольского и др. [1749] (до 8000 К), Вейц и др. [63] (до
3500 К), Каца и Маргрейва [1639] (до 2000 К). Различия в значениях Ф° (Т) при Т < 6000 К,
вычисленных в большинстве работ и в настоящем издании, не превышают 0,02 Дж-К~1-моль~1.
Расхождения с данными [1749, 2632] несколько больше @,05—0,07 Дж-К-моль), а расчет
[1904] выполнен в весьма грубом приближении, что приводит к ошибкам, возрастающим с
температурой до 2,5 Дж-К^-моль в значении 5° F000 К). Данные Хилзенрата и др. [1436] и
Кольского и др. [17491 при Т > 6000 К уже заметно превышают функции (до 0,4 Дж-К-моль~1
в Ф° A0 000 К)), приведенные в табл. 759, из-за того, что в [1436, 1749] учтены ридберговские
состояния.
Энтальпия образования Mg (г)
A//°(Mg, г, 298,15 К) = 147,10+80,80 кДж • моль
принята в соответствии с величиной Д8#° (Mg, к) (см. выше).
Mg+ (г). Термодинамические свойства газообразного положительного иона магния в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 760.
Уровни энергии Mg+, использованные в расчете термодинамических функций, приведены
в табл. 26.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. . .3s, . . .Зр и . . .3d. Энергии этих состояний приняты по результатам спектральных измерений
Рисберга [2346]; они практически совпадают с рекомендацией Мур [2053].
Термодинамические функции Mg+ (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25). Значения Q3t и ее производных вычислялись непосредственным суммированием
по уровням энергии. Погрешности вычисленных термодинамических функций во всем интервале
температур не превышают 0,02 Дж-К-моль~1 и обусловлены в основном неточностью принятого
значения R; только в С°р (Т) при Т > 8000 К погрешности могут быть больше (до 0,1 Дж -К -моль)
из-за пренебрежения ридберговскими состояниями.
' Ранее таблицы термодинамических функций Mg+ (г) вычислялись в предыдущем издании
настоящего справочника [105], в справочнике [1543], а также в работах Грина и др. [1246] (до
50 000 К) и Хилзенрата и др. [1436] (до 10 000 К). Результаты этих расчетов и настоящего издания
согласуются между собой в пределах 0,01—0,02 Дж-К~1-моль~1.
Константа равновесия реакции Mg+(г)+е (г)—Mg (г) вычислена с использованием значения
АГН° @) = —737,749+0,001 кДж-моль, соответствующего принятому потенциалу ионизации
Io (Mg)=61 671,0+0,1 см=737,749±0,001 нДж-моль.
Эта величина основана на результатах трех работ: Рисберга [2344] по исследованию в спектре
испускания серии . . .3snf CF и \F) для п < 13 F1 671,02+0,02 см), Брауна и др. [768] по
исследованию в спектре поглощения серии . . .3snp (ХР) для п ^ 59 F1 671,20+0,18 см) и Камю
[813] по исследованию в спектре поглощения серий 3sns CS) для га^ 20 и 3snd CD) для п ^ 19
F1 670,9+0,1 см). Принятое значение /0 (Mg) близко к полученному Кодлингом [865] из менее
полных измерений F1 670,6 см) и рекомендации Мур [2053], основанной на более старых работах
F1 669,14 см). Принятому значению также соответствует
bfH°(Mg+, г, 0) = 883,652 + 0,80 кДж • моль.
Mg2 (г). Термодинамические свойства газообразного димагния в стандартном состоянии при
температурах 100—3000 К приведены в табл. Д. 58.
269
Mg2 (г)
Глава 26-
Таблица 26.4 Молекулярные постоянные Mg2, MgO, MgH, MgF, MgCl, MgBr, Mgl и MgS
TV/j f\ ТТЛТЛ ¦** ТГП
iVIUJlOK V Jld
р
24Mg2 X12J
24Mg0 X12+
a3a
Am
ba 2+
^2+
c32+
d3A
D1A
е3П
Fm
fll
24MgH X2 2+
42П
B22+
«4n
Я2 2+
cm
24MgF X2 2+
42II
B22+
C22+
24Mg35Cl X2 2+
42П ^
B22+
c2n
24Mg'»Br X2 2+
42II
B2 2+
C22+
Mgl X2 2+
42П
[В2 2+]
24MgS2S Xх 2+
В1 2+ a
Примечание. Ниже все
Г,
ше
">ехе
Ве
<*! ¦ 10*
De ¦ 10°
см
0
0
2 623
3 563,27
13 700 6
19 984,0
28 000 б
29 493б
29 775,32 и
30 004,22 и
37 683,5 и
36 900 б
37 879,1 и
38 700 б
39 868,6 и
0
19 234,5
22 410
31400
35 550,6 3
41 120
0
27 815,Зб
37 168,3
42 538,1
0
26 496
37 006
40 848
0
25 820,7 г
27 000 ж
39 285,9
0
24 477,8 Д
25 612,2
0
23 052,59
постоянные j
м„ . а Ш(,У(=1,6448.10-2, о)А=6,'
=2,1344-10-8,
Ее= -2,6235
51,121
785,06
664
664,44
824 в
824,08
632 в
632 в
632,4
632,5
—
—
—.
—
1495,20
1599,50
827,79
—
1445 й
1749
718,5
747,2
757,8
821,9
465,4
491,5
—
—
373,2
393,8
—
271,9
316
323
295
528,74
497,34
^аны в см.
106-Ю-4; б
МО1, yi=!
1,6448 a
5,18
3,9
3,91
4,8 в
4,76
5,0 в
5,0 в
5,2
5,3
—
—
—
—
31,889 a
32,536 г
11,74 е
—
—
56
4,10 a
4,16
6,24
4,84
2,05
2,52
—
—
1,34
2,05 Д
—
5,2
0,5
1,0
— ¦
2,704
2,333
0,092 866
0,574 3
0,50
0,505 6
0,58 в
0,582 2
0,5 в
0,5 в
0,501 4
0,500 8
0,524 9 ж
0,56
0,559 0 ж
0,52
0,522 4 ж
5,803 0
6,191 3
2,594 3
__
6,23
6,161
0,519 2
0,520 0
0,538 5
0,551 0
0,245 0 а
0,251 05
—
—
0,164 9 а
0,169 0 е
—
—
0,132 а
—
—
0,267 97
0,255 18
з2= 1,0679 -Ю-4, а3=—?
3,5415-102,
0,37758 5
0,5
0,5
0,46
0,5 в
0,45
0,5 в
0,5 в
0,48
0,48
—
—
—
—
14,93 б
19,31 Д
-1,50 ж
—
0,3
0,144
0,47
0,327
0,511
0,449
0,16
0,18
—
—
0,081 б
0,108б
—
—
0,031 б
—
—
0,176
0,155
),6831-10-в;
7^=—1/7914-Ю-15.
MgO: а Pi=2-10"8; б квантовомеханический расчет; в оценка; г Bi=5
значение Dt>; H
MgH: a a)f^e=0,384, ш
е ш(!!/в=0,099,
значение Во.
MgF: a шв!/е=0,008; б
! приведено значение Во;
А=0,113;. б
^,=0,001;
4=36,4.
а2= — 9.10
ж а2=-3,6
3 приведено значение гс
-з; в p1=l,
1,2166 в
1,22 а
1,2
1,184 г
1,1 в
1,14 Д
1,3 в
1,3 в
1,26 е
1,27 е
1,14 е
1,4 в
1,424
2,0 в
2,2
3,53 в
3,6
1,2
—
3,31 к
3,1 к
1,08
3,2
1,08
0,99
0,25
0,22
—
—
0,13 в
0,13 в
—
—
0,09 в
—
—
0,276
0,269
в р1=3,0763
• Ю-9; Д Pi=—2,5-10-8;
; и приведено значение !
4-Ю-5, р2=5-10-°; г ШеУ
е= —0,146;
10; 3 приведено значение Го; и приведено Дб^;
ге
А
3,8915
1,749
1,86
1,864
1,74 в
1,737
1,87 в
1,87 в
1,872
1,873
1,829 3
1,77 в
1,772 3
1,83 в
1,833 3
1,7331
1,6778
2,592
2,51
1,673
1,682
1,7500
1,7486
1,7185
1,6988
2,1986
2,174
—
—
2,354
2,325
—
—
2,50 г
—
—
2,143
2,196
• Ю-8; рз=
е приведено
го.
ч а2=5-10-4;
к приведено
270
Магний и его соединения MgO (к, ж)
Таб л
MgCl
MgBr
Mgl:
MgS:
И I
a
a
a
a
(a 26.4 (окончание)
рассчитано через Bo= 0,244 23
состояний А2Ни и Л2Пг,.
/2 /2
рассчитано из Z?o=O,1645 и оц;
д шег/е= —0,04; е рассчитано
и ах; " Л = 53,94; все постоянные приняты средними
0 рассчитано по соотношению A.69); в рассчитано по
из Во=О,1685 и а,; ж
отнесенному к переходу В22+—Х22+ в работе [1354].
рассчитано по соотношению A.
Состояние а3П ЛХП
Те 1500 2500
38); б рассчитано по A.
17 000 28
определено Епо континууму в
69);
^, 34,
000
в рассчитано по A.68); !
ij- iji зц
29 000
из данных
A.68); г
области X
р оценка;
для под-
Л=111,1;
^3700 А,
В табл. 26.4 приведены молекулярные постоянные Mg», принятые на основании исследования
электронного спектра этой молекулы в работе Балфура и Дугласа [571]. Молекула Mg2 имеет
основное состояние X1 EJ, коррелирующее с атомами магния в основном состоянии. Образование
молекулы Mg2 обусловлено только вандерваальсовским взаимодействием атомов Mg, поэтому
энергия диссоциации ее очень мала (см. ниже). Возбужденные электронные состояния Mg2 имеют
энергии выше 25 000 см'1 [571 ] и в справочнике не рассматриваются. Постоянные в состоянии
X1 EJ, приведенные в табл. 26.4, получены обработкой 290 колебательно-вращательных термов
с у=0—12 и /=0—76, выделенных в результате анализа около 100 полос системы X1 Е^-^-Л1 Х?
[571]. '
Термодинамические функции Mg2 (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения <?вн и ее производных вычислялись по соотношениям A. 90)—A. 92) без учета
возбужденных электронных состояний. Статистическая сумма по колебательно-вращательным уровням
основного состояния и ее производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по уровням энергии. Колебательно-вращательные уровни энергии
вычислялись по уравнениям A. 63) и A. 65) с коэффициентами Yk!, приведенными в табл. 26.5. Для
обеспечения условий A. 50) и A. 59) в уравнение A. 65) был включен член с Y50, а коэффициент YOi
изменен по сравнению со значением Le, приведенным в табл. 26.4. В расчете учитывались все уровни
состояния X1 EJ, ограниченные его предельной кривой диссоциации; в табл. 26.5 приведены коэф^
фициенты ai (%=0) в уравнении, аппроксимирующем эту кривую, вместе со значениями уша„
Погрешности вычисленных термодинамических функции обусловлены неточностью принятого
значения энергии диссоциации Mg2 и оценкой общего числа связанных колебательно-вращатель-*
ных состояний, основанной на методиках, принятых в справочнике (см. т. 1, раздел 1.4). Общие
погрешности в значениях Ф° (Т) и S° (Г) можно оценить в 0,4—0,8 Дж-К~1-моль~1 при Т ^
> 298,15 К.
Ранее термодинамические функции Mg2 (г) вычислялись в работах Климова и др. [167] (до
4500 К), Бретта и Балфура [740] (до 1000 К). Расхождения данных [167] с приведенными
в табл. Д.58 достигают 6 Дж-К~1-моль в значениях Ф° B000 К) из-за использования оцененных
молекулярных постоянных и грубой методики расчета в [167]. Расчет [740] проводился по методике щ
постоянным, практически не отличающимся от принятых в настоящем справочнике, и существен^
ные расхождения в значениях Ф° (Т), составляющие около 14 Дж-К-моль во всем интервале,
температур, можно объяснить только ошибкой в расчетах [740].
Значение энергии диссоциации
D0(Mg2) =404,1+0,5 см-^4,834+0,006 кДж-моль
принято по работе Ли и Стволли [1858], которые получили ее короткой экстраполяцией (на 19 см)
колебательных уровней Mg2, используя потенциал U (r)=De—С6/г8—С8/г8.
Ей соответствует
A//°(Mg2, г, 0) = 286,972 + 1,6 кДж • моль.
MgO (к, ж). Термодинамические свойства кристаллического и жидкого оксида магния в стан^
дартном состоянии при температурах 100—5100 К приведены в табл. 761.
271
Т а 6 л и ц а 26.S. Значения коэффициентов в' уравнениях, описывающих уровни анергии (в ем 1), а также значения «шах и t/lim, принятые
для расчетов термодинамических функций Mg2, MgO, MgH, MgF, MgCl, MgBr, Mgl, MgS
Коэффициенты
Mg2
MgO
MgH
MgF
MgCl
MgBr
Mgl
MgS
XIV4
У10
Ум
Y3a
У40
y5J
Yol
У„
У21
ю-'
• Ю-3
• 10-'
¦ 101
• 103
• 10s
• 10'
• 10s
У31 • 10s
Yn
У2!
Ym
У„
105
Ю7
10»
1O10
¦ 10-11
(a, = De) X
X M-<
a2 • 103
a, • 107
a, • 10"
«max
о о
0,05150 0,783 058
—0,185 04 —0,525 703.
0,572 11 0,234 9843
-4,027 4 -0,058 517
0,101 C5 —
0,092 866 0,571 18
-0,037 758 -0,049 59
0,106 79 0,008 705
-0,968 31 —
—0,12166 —0,12167
0,307 68 —
-2,134 4 -
-0,262 38 -
-0,85415 —
-0,145 36 -
0,042 942 —
0,010146 —
-0,015 612 —
0,008 989 —
0,262 3 0,356 327 1,3700 1,998 40
0,646 237 0,662 754 0,821898 0,821825
—0,387 880 -0,398 407 —0,477 194 —0,472 603
0,041460 0,213 004 0,003 722 —0,036 591)
- -0,072 928 - -
0,499 471 0,502 852 0,579 036 0,579 036
-0,041658 -0,045 626 -6,044 634 -0,044 634
— 0,008 012 — —
-0,119 69 -0,116 72 -0,113 75 -0,113 75
17
93
149
418
98
325
139
430
109
350
83
326
Примечание. Ниже все постоянные даны в см~
MgO: а Состояние с3Е+ #Д
Те 28 000 29 493
0
1,495 20
-3,1889
3,84
-113,0
5,840 57
—2,116 02
13,4119
-216,351
-35,3
140,0
-500,0
-7,610 304
0
0,716 423
—0,408 115
0,084188
-0,014 913
0,516 2
-0,046 59
0,032 1
-
-0,107
—
-
-0,002 52
0
0,462 28
—0,202 260
—
-
0,241 8
—0,015 680
—
-
-0,024 3
—
-
-0,001 78
0 0 0
0,371264 0,314 044 0,526 516
—0,131021 —0,041551 —0,273 026
-0,016 117 -0,078 432 0,084 772
_ _ —0,023172
0,163 2 0,130 5 0,265639
—0,007 977 —0,003 049 —0,017 371
-0,012 7 -0,009 1 -0,027 122
0,001 935
-0,004 262
1,169 860
1,096 541
3,572 409
4,508 685
11
63
—
—
-
118
370
ИЗ
442
115
507
98
588
141
475
Д'Д СЧ- E'S+ е>П F41 /3П СП
29 838 30 067 37 683,5 36 900 37 879,1 38 700 39 868,6
MgH: а Состояние АЩ B2S+ а'П Е2?+ С2П
Те 19 234,5 22 410 31 400 35 544 41 120
MgCl:
MgBr:
Mgl:
Состояние А2П В!Е+ СП
Те 36 396 37 000 40 848
Te (А2П) = 25 821
Те (А2П) = 24 478
MgF:
Состояние А2П B2S+ C2?+
Те 27 815,3 37 168,3 42 538,1
MgS: а Состояние а3п А'П Ь3Я+
Te 1500 2 500 17 000 23 052,59
+, 3S+, 'А, 3Д, 'В- 3П, "П
28 000 29 000
Магний и его соединения MgO (г)
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние MgO (к) в интервале 0—3100 К принята
кубическая модификация (структурный тип NaCl) х. При Т < 298,15 К термодинамические
функции вычислены на основании данных о теплоемкости MgO, полученных Барроном и др. [585]
C—270 К) при измерениях образца, содержание примесей в котором не превышало 0,1%.
Погрешность измерений теплоемкости MgO в [585] оценивается в 0,5% при 10 К и 0,2% при Т > 20 К,
а погрешности принятых по [585 ] значений S° B98,15 К) и Н° B98,15 К)—Я0 @) (см. табл. 26.1) —
в 0,2 Дж-К-моль и 0,02 кДж-моль соответственно. Результаты измерений теплоемкости
микрокристаллических образцов MgO (к) в работах [1315] B1—84 К), [2188] (94—291 К), [1225,
1226] A,2—340 К) менее точны и не учитывались в расчетах. При 298,15—3100 К для
теплоемкости MgO (к) принято уравнение, выведенное методом Шомейта по результатам измерений изменения
энтальпии в работах Виктора и Дугласа [2774] B73—1173 К), Кантор и др. [147] A182—2488 К),;
Цагарейшвили и Гвелесиани [429] C70—1600 К), а также с учетом табличных значений энтальпий^
приводимых в [1543]. При этом погрешности данных [2774] оценены в 0,5%, а данных [147, 429,
1543] — в 1,0—1,5%. Результаты менее точных измерений в работах [538, 1907, 2853] не
учитывались. Температура плавления MgO C100+25 К) принята по измерениям Мак-Нелли и др. [1995]
на образце, содержавшем не менее 99,95% MgO. Данные [2339] C063 К), [1627, 2803] C073 К)
относятся к менее чистым образцам и, по-видимому, менее точны. Энтальпия плавления MgO
G7+6 кДж-моль) принята по расчетам Келли [1650] по диаграмме состояния системы MgO—
ZrO2. Теплоемкость жидкого MgO (84+8 Дж-К-моль~1) принята равной теплоемкости ВеО (ж),
измеренной в работе Шпильрайна и др. [460]. Расчет Ср (MgO, ж) методом «существенных структур»
11850а], приведший к среднему значению 99 Дж-К-моль'1 (в интервале3100—3900 К),
представляется ненадежным вследствие отсутствия данных по коэффициенту линейного расширения и
сжимаемости MgO (ж), которые необходимы для расчета составляющей Ср—Cv.
Погрешность вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 5000 К оценивается в 0,ls
0,4,.2 и 6 Дж-К~1-моль~1. Расхождения между термодинамическими функциями MgO (к),
приведенными в табл. 761 и в справочниках [105, 1543], не превышают 2 Дж-К-моль в значениях
Ф° (Г). Соответствующие расхождения для MgO (ж) достигают 7 Дж-К~1-моль (при 5000 К),,
что объясняется различием оценки С^ (MgO, ж) в настоящем справочнике и в [105, 1543].
В справочнике для энтальпии образования MgO (макрокристаллическая форма) принято
значение
A//°(MgO, к, 298,15 К) = —601,5 + 0,3 кДж • моль.
Эта величина основана на измеренных Шомейтом и Хафманом [2470] энтальпиях растворения
магния и макрокристаллической формы MgO в соляной кислоте и совпадает с рекомендацией
КОДАТА—MCHG [864]. В работах [74, 465, 1461, 2057, 2088, 2355, 27981 измерялась энтальпия
сгорания магния в кислороде, однако в них не учитывалась зависимость результатов измерений
от размеров образующихся кристаллов оксида магния. В наиболее совершенных измерениях
Воробьева, Скуратова [74] и Холли, Хубера [1461] получено значение —601,8 кДж-моль, что,
видимо, свидетельствует об образовании в условиях этих опытов макрокристаллической
модификации MgO.
Энтальпия перехода макро- в микрокристаллическую модификацию оксида магния определена
по разности энтальпий растворения этих модификаций в соляной кислоте: 3,7 кДж-моль [1196,
2470] и 4,0 кДж-моль [2666], чему соответствует AfH° (MgO, микрокрист., 298,15 К) =
= —597,7+0,4 кДж-моль.
Давление пара оксида магния в реакции MgO (к, ж)=MgO (г) вычислено с использованием
значения ДГЯ° @) =630,005+15 кДж-моль", соответствующего принятым в справочнике энтальпии
образования MgO (к) и энергии диссоциации MgO.
MgO (г). Термодинамические свойства газообразного оксида магния в стандартном состоянии
при температурах 100—10 000 К приведены в табл. 762.
Имеется в виду микрокристаллическое состояние MgO. Для микрокристаллических образцов MgO измерениям
теплоемкости при низких температурах [1196] B0—301 К), [1606] E0—300 К) и [1865] A,5—4 К) соответствуют
S" B98,15 К)=27,87+0,2 Д ж-ВТ1-моль и Н° B98,15 К)—#°@)=5,30+0,04 нДж-моль.
273
MgO (г) Глава 26
В табл. 26.4 приведены молекулярные постоянные MgO, принятые на основании результатов
исследований спектров и квантовомеханических расчетов. В электронном спектре MgO
исследованы девять систем полос, обусловленных переходами между восемью синглетными состояниями:
В1?,*** Хх?+ [1789, 1802, 1916], бШ -> ХХЕ+ [2534], Е1Е+ -> Хх?+ [528, 2532], FXU -* Хх?+
[2533], ЯХЕ+ -* А1!! [1803,1916, 1920], Сх?" -> ЛЧ1 [749, 2722, 2723], Z?XA -* A4L [2445, 2723],
G1!! -> -АШ [2533] и Я12+ -> Л1!! [527] х. Неэмпирические квантовомеханические расчеты [1519,
2334, 2446] показали, что, помимо состояний, известных из спектральных исследований, молекула
MgO должна иметь еще восемь стабильных триплетных состояний, коррелирующих с валентными
состояниями атомов (а8П, Ь3?+, с3?+, с23Д, 8Е", е3П, /3П и ^Е+J. Состояния 3?" и ^Е* не
рассматриваются в справочнике, поскольку согласно расчету Скамп и Лефевр-Брион [2446] первое
из них является слабосвязанным, а второе имеет энергию свыше 50 000 см.
В табл. 26.4 приведены данные для 14 валентных состояний MgO. Молекулярные постоянные
в состояниях X1 Е+, /1ХП и В1 ?+ приняты по результатам анализа колебательно-вращательной
структуры систем полос В1 ?+-* X1 2+ (i/ < 2, v" < 2) и В1 Е+ -* АШ (i/ < 2, v" < 2) в
работах Лагерквиста и Улер [1802, 1803]. Постоянные в состояниях С1 ?~ и Х^Д получены Траймаром
и Юингом [2723] при исследовании трех полос секвенции Ду=0 системы D1 к—Л1]! и четырех
полос секвенции Др=0 системы С1!"—А1!!. Принятые колебательные постоянные состояния
2IД хорошо описывают канты полос секвенции Ду=—1 (y=0-f-5) системы #ХД—.441, выделенные
Скампом и др. [2445]. Постоянные в состояниях ЕгТ.+, F1!! и G1]! получены Сингхом [2532, 2534J
в результате анализа полос 0—0 в системах E1H+—X1I,+, ^41—Хх?+ и G4I—ХХЕ+.
В спектре MgO не наблюдались интеркомбинационные переходы, однако молекулярные
постоянные состояния а3И достаточно точно определены из анализа возмущений в системах В1 Е+ —
ХХЕ + и Бг? +—А1!!. [1526]. Приведенное в табл. 26.4 значение Те (а3П) удовлетворительно
согласуется с результатами квантовомеханического расчета [2446] B400+1000 см),
экспериментальной оценкой Те (а3П) =3200 ±1000 см, полученной Эвансом и Макки [1043] по измерению
отношения интенсивностей триплетных и синглетных переходов в спектре поглощения MgO, а также
с наблюдаемой разницей в энергии Те (<Р&) — Те (а3П)=26 870 см [2444], из которой с учетом
данных о состоянии 2?ХД следует Те (а3П) <[ 2900 см. Погрешность принятого значения Те (аЩ
не превышает 10 см. Энергии возбуждения состояний 63? +, с3? +, е3П и /3П оценены на основании
величин расщепления изоконфигурационных триплетных и синглетных состояний, вычисленных
Скампом и Лефевр-Брион [2446], и приведенных в табл. 26.4 значений Те для соответствующих
синглетных состояний. Значение Те (d3A) принято по работе [2444]. Погрешности энергий
возбуждения триплетных состояний не превышают 1000 см. Колебательные и вращательные
постоянные в состояниях Ь3?+, с3?+, Й3Д, е3П и/3П приняты приблизительно равными соответствующим
постоянным изоконфигурационных синглетных состояний. Их погрешности согласно [1043] не
превышают 20% в значениях ше и 5% в значениях Ве.
Термодинамические функции MgO (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения Q3X и ее производных вычислялись по соотношениям A. 90)—A. 92) с учетом 13
возбужденных электронных состояний в предположении, что для состояний с энергией больше
20 000 см Q?o\. вр = (pJPa) Qit). вр* Статистические суммы по колебательно-вращательным
уровням энергии состояний Хх?+, а3П, .А41, Ъ3 Е+ и В1!** и их производные находились по уравнениям
A. 73)—A. 75) непосредственным суммированием по колебательным уровням и интегрированием
по/с помощью соотношений типа A. 82). В расчете учитывались все колебательно-вращательные
уровни перечисленных состояний со значениями / =^/maXi „; последние находились по соотношениям
A. 81). Уровни энергии этих состояний вычислялись по уравнениям A. 62) и A. 65) с
коэффициентами Ykl, приведенными вместе со значениями ь>шах и Jlim в табл. 26.5. Величины Ykl для изо-
1 На основании расчета [2446] две системы полос сложной структуры, наблюдавшиеся в спектре MgO в области
3721 и 3860 А [594, 744, 749, 1043, 2208—2210, 2444, 2697], отнесены к переходам с32+—а3П и d?A—а3П. Скамп
и Гайдара [2444] путем приближенного анализа структуры полос подтвердили это отнесение последней системы.
2 Вопрос о систематике электронных состояний MgO был предметом длительной дискуссии (см. [59, 105, 594, 741,
744, 745, 2209, 2210]). В настоящее время исследования спектра поглощения MgO [1043],
квантовомеханические расчеты [1519, 2334, 2446], а также данные по окислам других щелочноземельных элементов [821, 1080,
1512] позволяют сделать однозначное заключение о типе основного электронного состояния (XS+) и об
относительном положении низколежащих электронных состояний этой молекулы.
274
Магний и его соединения MgH (г)
топической модификации, соответствующей природной смеси изотопов Mg и О, получены по
формулам A. 66) исходя из постоянных молекулы 24Mg160 (табл. 26.4). Для обеспечения условий
A. 50) для состояний ХХЕ+, ЛЧ1 и В1 Е+ в уравнение A. 65) включен член с коэффициентом YM,
а для состояний а3П и Ь31> + — с коэффициентом Y3Q. Значение коэффициента У21 для состояний
X1 Е+ и А1!! получено из условий Б„тах+1=0. Мультиплетность и вырождение вращательных
уровней в состояниях а3П, .441, Ъ3 Е+ учитывались соответствующими статистическими весами.
Погрешности рассчитанных термодинамических функций MgO (г) при низких температурах
обусловлены главным образом неточностью принятых фундаментальных постоянных и энергий
возбуждения триплетных состояний, а выше 1000 К — отсутствием данных об уровнях энергии
основного состояния с^]>2. Общие погрешности в значениях Ф° (Т) оцениваются в 0,02, 0,1,
0,3 и 0,5 Дж-К^-моль при 298,15, 3000, 6000 и 10 000 К соответственно.
Термодинамические функции MgO (г) ранее вычислялись в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF [1543, 1544], справочниках Мак-Брайд и др. [1974],
Мадера [1904], Келли [1655], а также в работах Вейц и др. (Т ^ 6000 К) [63], Брюэра и Розен-
блатта [745] (Т < 3000 К) и Шнейдера [2463] (<?вн при 1000 < Т < 6000 К). Основная причина
существенных расхождений данных предыдущих расчетов с настоящим изданием, в особенности
выше 1000 К, обусловлена принятой в них неверной систематикой низколежащих электронных
состояний MgO. В расчетах [63, 105, 106, 1543, 1904, 1974, 2463] не учитывались триплетные
состояния, что обусловило расхождения от 0,1 до 10 Дж-К^-моль в значениях Ф° (Т).
Расхождения данных [1544] и табл. 762 не превышают 1—1,5 Дж-К~1-моль в значениях Ф° (Т), S° (T)
и С; (Г).
Константа равновесия реакции MgO (r)=Mg (г)+О (г) вычислена на основании принятого
в справочнике значения
-1
ДгЯ° @) = Do (MgO) = 360 + 15 кДж • моль да 30 100 + 1300 см
Несмотря на большое число работ по определению Do (MgO), данные об этой величине остаются
противоречивыми. В табл. 26.6 представлены значения, вычисленные по результатам измерений
разных авторов. Приведенные погрешности учитывают только воспроизводимость измерений,
за исключением работ [987, 1061, 2254], для которых учтена неточность сечений ионизации.
Результаты спектрофотометрических измерений в богатых пламенах водорода [58, 789, 886],
а также в пламенах С2Н2 [58, 1496, 2899], основанные только на определении величины р (Mg),
оказались завышенными из-за образования в этих условиях больших количеств гидроксидов
магния, особенно Mg(OHJ. Результаты аналогичных исследований в бедных метановых пламенах,
проведенных Старовойтовым и др. [398], а также результаты исследования в пламени СО, вьшолг
ненного авторами [1162], должны быть в меньшей степени осложнены этим эффектом. Кроме того,
авторы [398], так же как Зегерс и др. [2899], попытались учесть образование гидроксидов магни»
в условиях их эксперимента.
Принятое в справочнике значение основано на данных [398, 494, 987, 1061], полученных
разными методами. Оно удовлетворительно согласуется с величинами, найденными в работах [1043,
2254, 28991. Причины, приведшие к резкому расхождению в разные стороны данных [501, 1162 j
и принятого значения, остаются невыясненными.
Принятому значению соответствует
A//°(MgO, г. 0) = 32,686 + 15 кДж-моль-1.
MgH (г). Термодинамические свойства газообразного гидрида магния в стандартном состоянии
при температурах 100—6000 К приведены в табл. 763.
В табл. 26.4 представлены постоянные MgH, выбранные на основании исследований
спектров и квантовомеханических расчетов этой молекулы. В электронном спектре MgH
наблюдалось десять переходов: АШ ^ X2 ?+ [566, 569, 1318, 1321, 1680, 2815, 2816], В2 Е+ -> X2 Е*
[568, 569, 570], Е2 ?+ ^ X2 Е+ [1677, 1683], В'2 ?+ ^ X2 Е+ [1295, 1320, 1321, 1683], С2П ^ X2 Е*
[1125, 1317,1319, 1321,2200, 2739], G2E+«-X2?+, Я2 Е+«-Х2Е+, /2П«-Х2Е+ [1679], СЩ-+АЩ
[1316, 1319] и Z>2?- -* A2U [1318]. Балфур и Картрайт [568] при рассмотрении возмущений и
предиссоциаций в спектре MgH предположили, что система полос В'2 Е+—X2 Е+ представляет
собой переходы с последующих колебательных уровней состояния Е2 Е+ на основное состояние
молекулы. В табл. 26.4 приведены электронные состояния MgH, которые коррелируют с валентными
275
(г)
Глава 26
Таблица 26.6 Результаты измерений Do(MgO) (в кДж-моль
Авторы
Метод
Do (MgO)
II закон
III закон
Хульдт, Лагерквист, 1950 [1496] Спектрофотометрическое исследование реак- — 478+.12
ции MgO (г)—Mg (г)+О (г), на основании
измерения р (Mg) в пламени C2H2-f воздух
B410 К, 1 точка)
Вейц, Гурвич, 1956 [58] То же в пламенах СЛ2+воздух B366—2530 К) — 404+12
и Н2+О2 C080-3210 К, 3 точки)
Булевич, Сагден, 1959 [789] То же в пламенах Ha+O2+N2 A754—2410 К), — 410±8
результаты даны в виде графика
Де Галан, Вайнфорднер, 1967 [1162] То же в пламени СО+воздух B450 К) — 455
Зегерс и др., 1969 [2899] То же в пламени С2Н2+воздух — 367+10
Коттон, Дженкинс, 1969 [886] То же для реакции Mg (г)+ ОН (r)=MgO (г)+ 465+50 399+20
+Н (г) в пламенах H2+O2+N2 A570—2370 К,
4 точки)
Старовойтов и др., 1977 [398] То же для реакции Mg (г)+О2 (r)=MgO (г)+ — 342+.12
+ О (г) в бедных пламенах CH4+O2+N2
B370—2815 К, 6 точек)
Альтман, 1963 [501] Метод протока, MgO (K)=MgO (г). 2033— — ^313+15
2175 К, 8 точек, АГН° @)=677±15 (III)
Александер и др., 1963 [494] То же, 1780—2010 К, 7 точек, Дг#° @)= 432+50 374+4
= 616+3 (III)
Маеда и др., 1978 [1905а, 2432а] То же, в атмосфере кислорода, 1825—1977 К, 450+100 388+10
10 точек, &ГН° @)=602±10 (III)
Портер и др., 1955 [2254] Масс-спектрометрический, MgO (n)=MgO (г), — <389+8
A950 К, 1 точка), АГН° @)=601+8 (III)
Дроварт и др., 1964 [987] То же, MgO (г)+О (r)=Mg (г)+О„ (г), B026— 445+100 340+8
2274 К, 8 точек), АГН° @)=—153*6±8 (III)
Фарбер, Сривастава, 1976 [1061] То же, MgO (K)=MgO (г). B020—2160 К, 11 то- 378+30 369+7
чек), Дг#°@)= 620,6+7 (III)
Эванс, Макки, 1974 [1043] Граница спектра поглощения MgO в ударных 390+.35
волнах
состояниями атомов: X2 2+ (предел Mg A5)+H {2S)), А2Л и 52 Б+ (предел Mg CP)+H {2S))f
С2П и Е21,+ (предел Mg (Ч^+Н BS)). С пределом Mg CР)+Н BS) связаны также квартетные
состояния 4П и * ?. Согласно неэмпирическому расчету [834] состояние 4? отталкивательное, а
состояние а4П имеет минимум при больших межъядерных расстояниях.
Постоянные MgH в состояниях X2 Е+, А2П и В2 ?+ приняты по работе Балфура и Картрайта
[569], где они получены при совместной обработке систем полос А2П—X2 ?+ A3 полос с v' ^ 3,
v" ^5)и52 2+—X2 Е+ C7 полос с v' ^ 9, v" ^ 6), или рассчитаны на основании значений д?е+1/г,
Bv и Dv, приведенных в этой работе. Постоянная спин-орбитального взаимодействия для
состояния А2П найдена Хана [1680]. Постоянные в состоянии Е2 Е+ рассчитаны на основании данных,
приведенных в работах Хана [1677, 1683], а постоянные в состоянии С2П приняты по
рекомендации Герцберга [1392]. Постоянные MgH в состоянии а4П приняты по результатам^квантовомеха-
яического расчета Чена и Дейвидсона [834].
Термодинамические функции MgH (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения Qvs и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом пяти
возбужденных электронных состояний в предположении, что Q^JX вр = (pjpx) QifJ Bp# Значения
QifJ. вр и ее производных находились по уравнениям A.70) — A. 73) непосредственным
суммированием по колебательно-вращательным уровням энергии. В расчетах учитывались все уровни
энергии, ограниченные предельной кривой диссоциации состояния X2 Е+.
Колебательно-вращательные уровни состояния X2 Е+ вычислялись по уравнениям A. 65) и A. 64а). Значения коэффициентов
Yk! в этих уравнениях, использованные в расчетах, приведены в табл. 26.5; они получены из
постоянных, принятых в табл. 26.4, с учетом условий A. 59) для Y03 и A. 57) для У22. В табл. 26.5
276
Магний и его соединения MgEfo (к, ж)
приведены также значения vmax, /Пш и а( — коэффициентов уравнения, описывающего
предельную кривую диссоциации (вх=0). Мультиплетность и вырождение электронных состояний
учитывались соответствующими статистическими весами.
Постоянные молекулы MgH известны с достаточной точностью, поэтому погрешности
вычисленных термодинамических функций при Т ^ 3000 К обусловлены в основном неточностью
фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль в значениях Ф° (Т). При Т >•
Р> 3000 К становятся существенными неточность экстраполяции высоких
колебательно-вращательных уровней состояния X2 ?+ и приближенный учет возбужденных состояний; погрешность в зна~
чении Ф° F000 К) может достигать 0,2 Дж-К^-моль.
Ранее термодинамические функции MgH (г) вычислялись в предыдущих изданиях настоящего,
справочника [106, 105]. таблицах JANAF [1543], Мак-Брайд и др. [1974], Мадера [1904] и Хара
и др. [1336], а также в работах Вейц и др. [63] (Т < 3500 К) и Латимера [1819] B000 < Г <
^ 5000 К). В работе [2463] приведены результаты расчета в виде полиномов, аппроксимирующих
значения QVB и In (?вн A000 ^ Т ^ 4500 К). При низких температурах результаты всех расчетов
хорошо согласуются с приведенными в табл. 763. Однако использование в них приближенных
методов расчета, не учитывающих необходимость ограничения числа колебательно-вращательных,
уровней состояния X2 Е+, привело к большим ошибкам, особенно в значениях С°р (Т).
Соответствующие расхождения достигают 0,6 и 14 Дж-К-моль~1 в С\ (Т) при 2000 и 6000 К с данными [1336,
1543, 1974] и 1—3 и 6—9 Дж-К^-моль в значениях Ф° (Т) и 5° (Т) с данными [105, 1336, 1543,
1904, 1974].
Константа равновесия реакции MgH (r)=Mg (г) + Н (г) вычислена на основании принятой
в справочнике энергии диссоциации
'ЛгН°@) = D0(MgH) = И 000 + 500 см» 131,6 + 6 кДж • моль.
Это значение получено экстраполяцией колебательных уровней состояний X2 Е+ (b>=0-f-5)
и В2 Е+ (и=0—9) по уравнениям с постоянными, приведенными в табл. 26.4 и в предположении,
что состояние В2 ?+ имеет предел диссоциации Mg CP)+H BS). Соответствующие величины
совпадают в пределах 100 см'1, погрешность принятого значения оценена как 10% от разности энергий
последних наблюдавшихся колебательных уровней и соответствующих диссоциационных пределов.
С принятой величиной хорошо согласуются значение De (MgD), оцененное Балфуром и Карт-
райтом [567] экстраполяцией колебательных уровней основного состояния (у=0-|-6), и результаты
квантовомеханического расчета Мейера и Росмуса [2015].
Принятому значению соответствует
AfH°(MgE, г, 0) = 230,337+ 6,1 кДж.моль.
MgH2 (к, ж). Термодинамические свойства кристаллического и жидкого дигидрида магния
в стандартном состоянии при температурах 100—1000 К приведены в табл. Д. 59.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние принята тетрагональная модификация
MgH2 (к) [1362], устойчивая до температуры плавления. При Т < 298,15 К термодинамические
функции вычислены в таблицах JANAF [1543] по результатам неопубликованных измерений
теплоемкости MgH2 (к), выполненным Зинке и Хилденбрандом B5—300 К). Экстраполяция
теплоемкости ниже 25 К приводит к значению S° B5 К) =0,15 Дж-К^-моль; погрешности принятых
по [1543] значений 5° B98,15 К) и Я0 B98,15 К)—#° @) (см. табл. 26.1) оцениваются
в 0,8 Дж-К~1-моль~1 и 0,10 кДж-моль соответственно. При Т > 298,15 К для ^теплоемкости
MgH2 (к) принято уравнение (см. табл. 26.1), выведенное на основании значений С°р B98,15 К) и
С), F00 К)=63 Дж-К~1-моль; последнее оценено на основании экспериментальных данных по
теплоемкостям LiH [727 ], LiF [2606 ] и MgFa [2707]. Температура плавления F00 +10 К) определена
в работе Гивеля [1221], который исследовал образцы MgH2, полученные взаимодействием магния
с водородом и с бензальдегидом; при быстром нагревании плавление этих образцов наблюдалось
в интервале 598—603 К, одновременно с началом разложения на Mg (к) и Н2 (г)^ Энтальпия
плавления A4+5 кДж-моль) оценена в предположении, что &mS° (MgH2) = Д^0 (LiH).
Теплоемкость жидкого MgH2 принята равной 75+15 Дж-К"-моль с учетом экспериментальных данных
по теплоемкости жидкого LiH [463, 1362].
277
(г)
Глава 26
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 1000 К оцениваются в 0,4, 1 и
7 Дж-К-моль~1. Расхождения между термодинамическими функциями MgH2 (к), приведенными
в таблицах JANAF [1543] и настоящем издании, не превышают 0,3 Дж-К-моль~1 в значениях
Ф° (Т). Значения термодинамических функций MgH, (к) в [977] при Т <; 300 К на 1% выше
значений, приведенных в настоящем справочнике и [1543]. Таблица термодинамических функций
MgH2 (ж) публикуется впервые.
Давление диссоциации MgH2 (ж) достигает 100 атм при 900 К.
В справочнике принята энтальпия образования
A/F°(MgHa, к, 298,15 К) = —75,7 + 2,0 кДж • моль-1,
йайденная на основании анализа результатов исследований давления диссоциации дигидрида
магния, приведенных в табл. 26.7. В качестве погрешностей в табл. 26.7 указана
воспроизводимость измерений. Результаты калориметрических измерений Пепекина и др. [318] и Зинке, Стаяла
Таблица 26.7. Результаты определений
(MgEh, к, 298, 15 К) (в кДж-моль)
Авторы
Метод
A/^°(MgH2, к, 298,15 К)
II закон
III закон
Эллингер и др., 1955 [1026] Измерение давления диссоциации MgH2 (к) —70±9 —75,9±0,8
статическим методом, 611—753 К, 5 точек
(давление до 65 атм)
Стемпфер и др., 1960 [2606] То же, 587—849 К, 129 точек, (давление до —70,5+0,3 —75,9+0,1
849 атм)
Кеннелли и др., 1960 [1673] Тоже, 500—570 К, данные представленыурав- —70,4 —75,1±0,4
нением
Моррисон, 1964 (см. [1543]) То же, 610—690 К —68,2 —75,46±0,6
Рейлли, Висволл, 1967 [2324] То же, с катализатором Mg2Cu, 546—648 К, —74,7±1 —75,7±0,1
5 точек
Рейлли, Висволл, 1968 [2325] То же, с катализатором Mg2Ni, 549—623 К, —83±4 —75,4±0,1
4 точки
Буске и др., 1969 [727] Тоже, 643—749 Кт 9 точек (давление до 71 атм) —83,1 ±3,5 —76,3±0,3
Пепекин и др., 1964 [318] Калориметрическое измерение энтальпии его- —90,6+1,7
рания MgH2 (к), два образца, 12 измерений
Зинке, Сталл, 1965 (см. [1543]) Калориметрическое измерение энтальпии гид- —80±8
ролиза MgH2 (к)
(см. [1543]) недостаточно точны, плохо согласуются между собой и с измерениями давления
диссоциации MgHa (к); в то же время последние хорошо согласуются между собой. Неточность
термодинамических функций MgH2 (к, ж) приводит к погрешности в энтальпии образования
—1 кДж-моль. При оценке погрешности принятого значения учтена воспроизводимость
измерений, неточность термодинамических функций, а также образование в продуктах диссоциации
магния, содержащего растворенный водород.
MgOII (г). Термодинамические свойства газообразного гидроксида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 764.
Молекулярные постоянные, использованные в расчете термодинамических функций MgOHr
представлены в табл. 26.8. Бром и Велтнер [761] на основании изучения спектра ЭПР молекул
MgOH, изолированных в матрицах из Аг и Ne, а также квантовомеханических расчетов пришли
к выводу, что в основном электронном состоянии X2 ?+ молекула MgOH имеет линейную структуру.
Этот вывод подтверждается аналогией с молекулами ВеОН и гидроксидами щелочных металлов
(см. также [1439а]). Экспериментальные данные о молекулярных постоянных MgOH отсутствуют,
их оценка выполнена так же, как для ВеОН (см. гл. 25). Приведенный в табл. 26.8 момент инерции
рассчитан в предположении, что г (О—Н)=0,96+0,02 А и г (Mg—О)=1,80+0,05 А; его
погрешность составляет 1 -Ю9 г-см2. Погрешности принятых значений частот vMg_0,; vMg_o_H и vo_H
оцениваются в 50, 50 и 100 см. Энергия первого возбужденного электронного состояния MgOH
278
Магний и его соединения
Mg (OHJ (к)
Таблица 26.8. Значения молекулярных постоянных, а также з и р^-, принятые для расчета
термодинамических <pyHKnira|MgOH, Mg(OHJ, MgF2, MgCl2, MgBr2, Mgl2
Молекула
MgOH
Mg(OHJ
MgF2
MgCl2
MgBr2
Mgl2
MgOH: a
Электронное
состояние
l4±
9
Am, Го=26 00
690
500
540
320
190
140
0 см-i, Pi
v2
310 B)
150 B)
165 B)
110 B)
90B)
70B)
=4, o=l.
va
CM
3700
800
825
595
490
440
-4
/ • 1039
г-сма
3700 B) 310 D) 20
— — 19,8
— — 56
— — 145
— — 268
0
1
2
2
2
2
2
Pi
2
1
1
1
1
1
принята на основании результатов исследований электронного спектра MgOH в области 3800—
3850 А и 3680—3730 А в работах Пешича и Гейдона [2210], Брюэра и Траймара [749] и Гейдона
[1182] и аналогии с изоэлектронной молекулой MgF.
Термодинамические функции MgOH (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126), A. 129) и A. 168)—A. 170) в приближении «жесткий ротатор —
гармонический осциллятор» с учетом возбужденного электронного состояния; погрешности
рассчитанных термодинамических функций обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета и составляют 1, 3 и 5 Дж-К-моль в
значениях Ф° (Г) при 298,15, 3000> 6000 К.
Ранее термодинамические функции MgOH (г) вычислялись в справочниках [1544, 1974] по
оцененным постоянным; расхождения между данными настоящего справочника и расчетами [1544,
1974] достигают 8 и 12 Дж-К^-моль соответственно в значениях Ф° (Т).
Константа равновесия реакции MgOH (r)=Mg (г)+О (г)+Н (г) вычислена для значения
Дг#° @)=753,72 +20 кДж-моль, соответствующего принятой в справочйике энтальпии
образования
, г, 0)==—145 + 20 кДж
моль
Эта величина принята на основании оцененной энергии связи Do (Mg—OH)?«330 +20 кДж-моль
Оценка выполнена из сопоставления значения Do (MgCl)=320+6 кДж-моль и величин Do (MG1)
и Do (М—ОН), где М=Ве, Са, Sr и Ва. Для четырех последних элементов разность Do (M—ОН)—
Do (MCI) (см. соответствующие разделы) находится в пределах 8—25 кДж-моль. В работе Буле-
вич и Сагдена [789] при фотометрическом исследовании в пламенах получено существенно более
низкое значение B34+20 кДж-моль), которое представляется заниженным.
Mg(OHJ (к). Термодинамические свойства кристаллического дигидроксида магния в
стандартном состоянии при температурах 100—1000 К приведены в табл. Д.60.
Значения постоянных, принятые для расчета термодинамических функций, представлены
ев табл. 26.1. В справочнике за стандартное состояние Mg(OHJ (к) принята гексагональная
модификация (структурный тип Cdl2), минерал брусит х. При температурах Т < 298,15 К
термодинамические функции Mg(OHJ вычислены на основании данных о теплоемкости, полученных Джио-
ком и Арчибалдом [1196] B2—321 К) на образце, содержавшем не более 0,2% примесей.
Экстраполяция теплоемкости ниже 22 К приводит к значению S° B2 К)=0,17 Дж-К-моль.
Погрешности принятых по [1196] значений S0 B98,15 К) и Н° B98,15 К)—Н° @) (см. табл. 26.1)
оцениваются в 0,4 Дж-К-моль и 0,06 кДж-моль соответственно.
Для теплоемкости Mg(OHJ в интервале 298,15—1000 К принято уравнение, выведенное
методом Шомейта по результатам измерений энтальпии Mg(OHJ (к) в работах Кинга и др. [1713]
Согласно данным [2877] при высоких давлениях линия перехода (разложения) бруснта на периклаз и пары воды
проходит через точки 1083 К и 10 кбар, 1218 К н 20 кбар, 1248 К и 40 кбар.
279
Mg (ОН)Я (г) Глава 2g
C50—699 К) и Лащенко и Компанского [221] C93—667 К). При этом погрешности данных [1713}
оценены в 0,5%, а данных [221] — в 3%. Экспериментальные данные о температуре плавления
Mg(OH)a в литературе отсутствуют; по-видимому, она выше 800 К, температуры, при которой
давление диссоциации Mg(OHJ достигает 100 атм.
Погрешность вычисленных значений Ф° (Г) при 298,15, 500 и 1000 К оценивается в 0,3, 0,6-
и 3 Дж-К-моль. Расхождения между термодинамическими функциями Mg(OHJ (к),
приведенными в табл. Д.60 и в таблицах JANAF [1543], составляют не более 0,5 Дж-К-моль и
обусловлены некоторым различием выбора данных по теплоемкости Mg(OH)a при Т ]> 298,15 К.
В справочнике принята энтальпия образования
A^°(Mg(OHJ, к, 298,15 К) = — 924,35 ±0,30 кДж • моль.
Эта величина основана на приведенных в табл. 26.9 результатах измерений энтальпии
растворения дигидроксида и оксида магния. Приведенные в табл. 26.9 данные находятся в хорошем
Таблица 26.9. Результаты определений AfH° (Mg(OHJ, к, 298,15 К) (в кДжмоль)
Авторы
Метод
A,ff°(Mg(OH),, к
298,15 К)"
Уэлс, Тейлор, 1937 [2828] Калориметрическое измерение энтальпии раство- —925,03+1,00а
рения MgO и Mg(OHJ в соляной кислоте, 4 опыта
Джиок, Арчибалд, 1937 [1196] Тоже, 3 опыта. Использовался микрокристалли- —924,32±0,35
ческий MgO. Результаты исправлены с учетом
разности энтальпии образования макро- и
микромодификаций MgO
Тейлор, Уэлс, 1938 [2666] То же, 4 опыта. Использовался микрокристал- —924,32±0,66
лический MgO
Торгесон, Сахама, 1948 [2716] То же, в расчетах использованы данные [2513] —924,35±0,30
по растворению MgO (макрокрист.) в соляной
кислоте той же концентрации
Беннингтон, 1956 [653] Калориметрическое измерение энтальпии раство- —924,32±0,60
рения MgO (макрокрист.) и Mg(OHJ (брусит)
в плавиковой кислоте; учтена разность энтальпии
образования синтетического Mg(OHJ и брусита
по данным [2666]
а Погрешность увеличена по сравнению с рекомендацией [2828] @,40 кДж-моль), так как в работе нет прямых
указаний на кристаллическую структуру MgO. По методу получения можно предполагать, что образец имел
структуру, близкую к микрокристаллической.
соответствии. Принятое в справочнике значение является средневзвешенным из этих величин.
Энтальпия растворения в соляной кислоте измерялась также в более ранних работах [1142, 2381].
Энтальпии взаимодействия Mg(OHJ и MgO с перекисью натрия измерил Микстер [2040], им
соответствует Д^0 (Mg(OH)a, к)=—926,7+5,0 кДж-моль.
В литературе известно большое число работ, в которых исследовалось равновесие реакции
Mg(OHJ (K)=MgO (к)+Н2О (г) [36, 129, 211, 221, 728, 797, 1157, 1196, 1522, 1592, 1671, 1984,
2393, 2395, 2416]. Значения энтальпии образования Mg(OHJ, вычисленные по этим данным, имеют
широкий разброс. Наиболее детальным измерениям [129, 1196, 1671, 2393] соответствуют
значения —927, —927, —927 и —922 кДж -моль, совпадающие в пределах их погрешностей с
принятой величиной.
Mg(OHJ (г). Термодинамические свойства газообразного дигидроксида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 765.
Молекулярные постоянные, использованные в расчете термодинамических функций Mg(QHJ,
представлены в табл. 26.8. Сведения об исследованиях структуры и спектров молекулы Mg(OHL
отсутствуют и приведенные постоянные оценены при подготовке справочника. Момент инерции
в основном состоянии X1 2J рассчитан при -^О—Mg—О=180°, г (Mg—О)=1,80+0,05 А (оценены
по молекуле MgF2), ^Mg—О—Н=180°, г (О—Н)=0,96+0,02 А (перенесены из молекулы»
280
Магний и его соединения MgF (г)-
MgOH); его погрешность составляет 2-Ю9 г-см2. Частоты vx,'va и v3 фрагмента О—Mg—О
оценены по частотам MgF2, частоты v4 и v5 фрагментов Mg—О—Н перенесены из молекулы MgOH.
Погрешности принятых значений частот оцениваются в 50—100 см. По аналогии с MgF2 можно
предполагать, что возбужденные электронные состояния молекулы Mg(OHJ имеют энергии свыше
30 000 см".
Термодинамические функции Mg(OHK (г) вычислены по уравнениям A.3)—A. 10), A. 122)—
A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический осциллятор»; их
погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 4, 8 и 12 Дж-К-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К (в том числе 2—4 Дж-К-моль из-за неточности молекулярных постоянных) г.
Ранее термодинамические функции Mg(OHJ (г) вычислялись в таблицах JANAF [1544];
расхождения между термодинамическими функциями, приведенными в [1544] и настоящем
справочнике, достигают 16 Дж-К-моль~1 в значениях Ф° (Т) и обусловлены различием принятых в
расчетах структуры и частот молекулы Mg(OHJ.
Константа равновесия реакции Mg(OHJ (r)=Mg (г)+2О (г)+2Н (г) вычислена с
использованием значения hrH° @)=1618,537 +20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A/F°(Mg@HJ, г, 0) = —547 + 20 кДж • моль.'
Это значение основано на результатах исследований равновесия реакции MgO (к)+Н2О (г) —
=Mg(OHJ (г) методом протока в работах Александера и др. [494] A780—2010 К, 7 точек) иМаед»
и др. [1905а, 2432а] A668—1948 К, 24 точки), а также равновесия реакции Mg (г)+2Н2О (г) =
=Mg(OHK(r)+2H (г) методом спектрофотометрии в пламенах в работе Старовойтова и др. [398]
B370—2815 К, 6 точек). Данным [494] соответствуют значения энтальпии образования, равные
—546+15 кДж-моль (II закон) и —546+16 кДж-моль (III закон), а данным [1905а]
соответственно —559+25 и —548+18 кДж-моль". Результаты измерений [398] приводят к значению
энтальпии образования Mg(OHJ, равной —541 кДж-моль. Погрешность принятого значения
оценена с учетом воспроизводимости измерений, погрешности термодинамических функций и
других термохимических величин, использованных в расчетах.
MgF (г). Термодинамические свойства газообразного фторида магния в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 766.
В табл. 26.4 представлены молекулярные постоянные MgF, принятые на основании анализа
результатов исследований электронных спектров этой молекулы. В спектрах MgF исследованы,
семь систем полос, связанных с переходом между восемью электронными состояниями: А2П ^
<±Х2?+ [294, 298, 592, 928, 1126, 1557, 1570, 1583, 2795], В2 ?+ ^ X2 ?+ [294, 298, 592, 928,
1557, 1570, 1583, 2795], С2?+^Х2?+ [298, 592, 1126], ОгП ^ X2 ?+, ?2П:?Х2Е+, F2 ?+ -*
-> X2 ?+, G2?+ <=fc X2 ?+ [294, 298]. Все состояния, кроме четырех нижних, приведенных в табл. 26.4,
имеют энергии свыше 50 000 см и не рассматриваются в справочнике. Так же как молекулы
других галогенидов элементов группы НА, молекула MgF может иметь связанные квартетные
состояния, однако их энергии, по-видимому, должны превышать 45 000—50 000 см".
Колебательные постоянные молекулы MgF в состоянии X2 ?+ определялись в работах [1294,
298, 592, 1126, 1570, 1583] и находятся в удовлетворительном согласии. Принятые в справочнике
значения этих постоянных получены Новиковым и Гурвичем [294, 298] на основании анализа
по (^-кантам колебательной структуры 64 полос системы А2П—X2 ?+ (и', г/'^С 16); вращательные
постоянные найдены Барроу и Билом [592] из анализа структуры полос 0—0, 1—0 и 0—1
системы АЧ1—X2 ?+, полос 0—0 и 1—1 системы В2 ?+—X2 ?+ и полос 0—0 и 1—1 системы С2 ?+—
Х2?+. По работе [592] приняты также вращательные постоянные MgF в состояниях А2П, В2 ~? +
и С2 ?+. Колебательные постоянные в этих состояниях, приведенные в табл. 26.4, найдены
соответственно Новиковым и Гурвичем [294, 298], Джевонсом [1570] и Фаулером [1126].
Обработка данных Александера и др. [494] и Маеда и др. [1905а] (см. ниже) по уравнению II закона приводит
к значениям S° A895 К)=433+8 Дж-К^-моль" и 5° A812 К)=433+12 Дж-К^-моль соответственно.
Данным табл. 765 при этих температурах соответствуют значения 444+7 и 439+7 Дж-К -моль; хотя
расхождения находятся в пределах погрешностей, возможно, что в справочнике для частоты v5 принята заниженная
величина.
281
MgF (г) Глава 26
Термодинамические функции MgF (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—
{1. 95). Значения Qm и ее производных по температуре рассчитывались по уравнениям A. 90)—
A.92) с учетом трех возбужденных состояний в предположении, что Qii\,BV^={Pilpx)Q{f^BV-
Величина Q^fJ вр и ее производные вычислялись по уравнениям A.73) — A-75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням
энергии с помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями
/ ^ Jmsx,vi гДе Jmia v находились из условий A. 81). Соответствующие значения i>max и /Ит
приведены в табл. 26.5. Колебательно-вращательные уровни состояния X2 ?+ вычислялись по
уравнениям A. 65), A. 64) со значениями коэффициентов Ykl, приведенными в табл. 26.5 и
полученными для природной изотопной смеси молекул MgF на основании принятых постоянных молекулы
^4MgF. Коэффициенты Yk0 уточнены для выполнения условия A. 59), а коэффициент У21 получен
из условия 5„шах +1 = 0. Мультиплетность и вырождение колебательно-вращательных уровней
основного и возбужденных состояний учитывались соответствующими статистическими весами.
Постоянные молекулы MgF в основном состоянии известны с удовлетворительной точностью,
а энергия диссоциации MgF и энергии неучитывавшихся возбужденных электронных состояний
достаточно высоки. Поэтому погрешности рассчитанных термодинамических функций почти во
всем интервале температур определяются лишь неточностью фундаментальных постоянных и не
превышают 0,02—0,03 Дж-К-моль~1 в значениях Ф° (Т), 0,05 и 0,1 Дж-К^-моль в
значениях S° F000 К) и С; F000 К) соответственно.
Термодинамические функции MgF (г) ранее вычислялись в предыдущих изданиях настоящего
справочника [105, 106], таблицах JANAF [1543], справочниках Мак-Брайд и др. [1974], Дугласа
и Бекетта [977] и Мадера [1904]. Климов и др. [167] рассчитали коэффициенты уравнения,
аппроксимирующего значения Ф° (Т) B93,15—5000 К). Наилучшее согласие с данными табл. 766
имеют результаты расчетов [105, 106] (в пределах 0,2 и 0,35 Дж -К -моль в значениях Ф° (Т) и
5° (Т)). Наибольшие расхождения — с данными JANAF [1543] — до 0,5, 1,5 и 2,3 Дж-К-моль
в значениях Ф° (Г), S° (Т) и Ср (Г).
Константа равновесия реакции MgF (r)=Mg (r)+F (г) вычислена с использованием принятого
в справочнике значения
Дг#° @) = Do (MgF) = 455 + 5 кДж • моль «г 38 040 + 400 ом.
В табл. 26.10 представлены результаты исследований равновесия реакций с участием MgF (r),
на основании которых принято это значение. Приведенные погрешности включают
воспроизводимость измерений, неточность сечений ионизации, термодинамических функций и других
термохимических величин, использованных в расчетах. Полученные величины различаются несколько
Таблица 26.10. Результаты определений Do(MgF) (в кДж• моль)
Авторы
Метод
Do (MgF)
II закон
III закон
Элерт и др., 1964 [1020] Масс-спектрометрический, MgF2 (K)+Mg (r)=-- 573+130 446,6+3,5
=2MgF (г), 1205—1253 К, 9 точек, АГН° @)=
=527,6+5 (III)
То же. 2Mg (r)+AlF3 (r)=AlF (r)+2MgF (г), 414+200 447,5+4,4
1238—1301 К, 5 точек, АГЯ° @)==199,3+8 (III)
Хилденбранд, 1968 [1406] То же, MgF2 (r)+Mg (r)=2MgF (г), 1280—1493 К, 474+10 458,3+3,7
11 точек, &гН°@)= 119,1+5 (III)
больше, чем это можно объяснить их погрешностями. Учитывая лучшее согласие результатов
обработки данных [1406] по уравнениям II и III законов, им приписан больший вес при выборе
принятого значения.
.Принятой энергии диссоциации соответствует энтальпия образования
Af#°(MgF, г, 0) = —231,822 + 5,1 кДж • моль.
282
Магний и его соединения MgF2 (к, ж)
MgF2 (к> ж)- Термодинамические свойства кристаллического и жидкого дифторида магния
в стандартном состоянии при температурах 100—4200 К приведены в табл. 767.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В настоящем справочнике за стандартное состояние MgF2 (к) принята тетрагональная
«-модификация, устойчивая от 0 до 1536 К (температуры плавления a-MgF2) *. При Т < 298,15 К
термодинамические функции вычислены на основании измерений теплоемкости, выполненных
Тоддом [2707] E4—297 К) на весьма чистом образце. Экстраполяция теплоемкости проводилась
по выведенному в [2707] уравнению: C°P=D C26/Т)+2Е E53/Т), которое аппроксимирует
экспериментальные данные с точностью —1% и приводит к значению S° E1 К)=2,26 Дж-К^-моль.
Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К)—Н° @) оцениваются в 0,5 Дж -К^Х
Хмоль и 0,06 кДж-моль соответственно.
Для теплоемкости MgF2 (к) от 298,15 К до температуры плавления принято уравнение,
полученное методом Шомейта на основании данных об энтальпии MgF2, полученных Нейлором
12112] в результате измерений в интервале 298,15—1760 К. По работе [2112] приняты также
приведенные в табл. 26.1 энтальпия плавления и теплоемкость жидкого MgF2. Погрешности этих
величин оцениваются в 1,5 кДж-моль и 2 Дж-К~1-моль~1 соответственно. Температура
плавления MgF2 A536+3 К) принята на основании хорошо согласующихся измерений [1290, 1747,
1753, 2112, 2308, 2712]. В работах [173, 994, 2219, 2358] получены более низкие значения,
которые не учитывались при выборе этой величины.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1500, 3000 и 4000 К составляют 0,4,
1, 5 и 10 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
MgF2 (к, ж), приведенными в табл. 767 и в справочниках [105, 1543], не превышают 0,3 и
1,0 Дж-К-моль~1 в значениях Ф° (Т) соответственно.
В справочнике принята энтальпия образования
AfH°(MgF2, к, 298,15 К) = —1124,2 + 1,2 кДж • моль.
Эта величина основана на измерении энтальпии реакции Mg (k)+F2 (r)=MgF2 (к) в работе
Рудзитиса и др. [2401]. В пределах погрешностей она согласуется с результатами
калориметрического исследования энтальпии реакции Mg(OHJ (k)+2HF (p-p 4,4 H2O)=MgF2 (к)+2Н2О (р-р
HF-4,4 H2O) в работе Торгесона и Сахама [2716] (—1121,9+2,0 кДж-моль) и исследования ЭДС
ячеек с твердым электролитом 1,5 Mg (k)+A1F3 (k)=A1 (K)+l,5MgF2 (к) в работе Резухиной и
др. [346] (—1122,3+2,4 кДж-моль при расчете по методу III закона). Практически совпадающее
значение (—1122+6 кДж-моль) получено в результате исследования равновесия реакции
MgF2 (к)+Н2О (r)=MgO (k)+2HF (г) методом протока [971]. Данные [151, 1743, 2800] уступают
по точности перечисленным выше, но не противоречат им. Причины получения существенно
отличающегося значения (—1134,7+5 кДж-моль) в работе Гросса и др. [1281] при
калориметрическом изучении реакции Mg (K)-fPbF2 (K)=MgF2 (к) и РЬ (к) не установлены.
Давление пара дифторида магния MgF2 (к, ж)=MgF2 (г) вычислено на основании принятой
в справочнике энтальпии сублимации
As#°(MgF2, к, 0) = 385 + 5 кДж • моль,
полученной в результате обработки данных, представленных в табл. 26.11. В качестве
погрешностей в таблице приведены воспроизводимости измерений. Для работы [1245] при расчете по III
закону учтена погрешность из-за неточности сечений ионизации. Причина отклонений данных [1251 ]
от результатов измерений других авторов остается непонятной. Принятое значение является
средневзвешенным из величин, вычисленных по уравнению III закона и приведенных в табл. 26.11;
при определении весов учитывалась также разность между величиной, полученной из данной
работы, и средней величиной. Погрешность полученного таким образом средневзвешенного
значения A,0 кДж-моль") увеличена с учетом неточности термодинамических функций MgF2 (к, ж)
и MgF2 (г).
1 Установлено (см. [50, 173, 655, 1441]) существование другой тетрагональной модификации (C-MgF2) с
температурой плавления 1673 К [50], а также модификации с неустановленной структурой (a'-MgF2), которая при
нагревании до ИЗО К переходит в p-MgF2;
283
MgF2 (г)
Глава 2&
Таблица 26.11. Результаты определений A.gH° (MgF2, к, 0) (в кДж-моль *)
Авторы
Руфф и др., 1934 [2407]
Гюнтер, 1958 [1314]
Евсеев и др., 1958 (см. [105])
Берковиц, Маркварт, 1962 [665]
Гринбаум и др., 1964 [1251]
Хаммер, Паск, 1964 [1346]
Хилденбранд и др., 1964 [1417]
Грин и др., 1964 [1245]
Метод
Точек кипения, 1934—2026 К, 6 точек
Эффузионный, 1284—1555 К, 20 точек
То же, 1282—1462 К, 10 точек
То же, 1450 К, 1 точка
Масс-спектрометрический, 1300—1400 К, S
Торзионный, 1273—1513 К, 40 точек
То же, 1413—1613 К, 25 точек
Эффузионный, 1451—1613 К, 10 точек
Торзионный, 1425—1606 К, 55 точек
Масс-спектрометрический, 1241—1492 К
д„я°
II закон
420±20
348 ±11
355 ±9
) точек 378
328 ±8
374+12
367 ±13
427 ±5
386±10
(MgF2, к, 0)
III закон
384,8 ±1,3
385,1 ±0,6
377,1 ±0,6
381,2
—
368,2±0,8
386,8+0,5
387,2±0,4
385,1 ±2
387,5±5
MgF2 (г). Термодинамические свойства газообразного дифторида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 768.
Молекулярные постоянные, использованные для расчета термодинамических функций
MgF2 (г), представлены в табл. 26.8. Исследования дифракции электронов [5, 13, 1045],
инфракрасного спектра [23, 24, 1369, 1626, 1849, 2565], спектра комбинационного рассеяния [2565}
и отклонения молекулярного пучка в неоднородном электрическом поле [783] показали, что в
основном электронном состоянии X1 EJ молекула MgF2 имеет линейное строение (симметрия Dmh).
Отнесение в работах [495, 808, 1934] полосы 478 см в спектре MgF2 к колебанию vx этой
молекулы, рассматривавшееся как свидетельство ее угловой структуры, было ошибочным, как это
показали Хауге и др. [1369]. Приведенный в табл. 26.8 момент инерции вычислен для значения
г (Mg—F)=1,77+0,02 А, найденного Акшшшым и др. [5, 133 при электронографическом
исследовании паров MgF2. Результаты повторного электронографического исследования, выполненного
Каспаровым и др. [1586, 1046] (г (Mg—F)=l,77 +0,01 А), согласуются сданными [5,13].
Погрешность принятого значения / составляет l-lO9 г-см2. Приведенные в табл. 26.8 значения v2 и vs
получены Байковым [23, 24] при исследовании инфракрасного спектра паров MgF2. С принятой
величиной v2 хорошо согласуется частота 160 +15 см, найденная по эффекту сокращения в
электронографическом исследовании [1586, 1046]. К частоте v2, вероятно ошибочно, отнесены
наблюдаемые в матрицах полосы 220—255 см [495, 808, 1369, 1849, 1934, 2565]. Найденные в
разных матрицах [495, 808, 1369, 1626, 1849, 1934, 2565] величины v3 колеблются от 730 см в
матрице из СО до 867 см в матрице из Ne. Для частоты vl7 так же как для vs, принята величина
несколько меньшая, чем полученные Лесицким и Ниблером [1849] в спектре комбинационного
рассеяния молекул MgF2, изолированных в матрицах из Аг и Кг E50 и 544,5 см), а также
измеренная Хауге и др. [1369] в инфракрасном спектре комплексов MgF2-X,(, изолированных в
матрице из Аг E52 см). Погрешности принятых частот MgF2 оцениваются в 10—20 см. Сведения
о возбужденных электронных состояниях MgF2 отсутствуют; можно предполагать, что их энергии
превышают 30 000 см.
Термодинамические функции MgF2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9)—A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных термодинамических функций обусловлены неточностью
принятых значений молекулярных постоянных, а также приближенным характером расчета и
составляют 1, 3 и 5 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно (в том
числе 1—2 Дж-К~1-моль из-за неточности молекулярных постоянных).
Ранее таблицы термодинамических функций MgF2 вычислялись в предыдущих изданиях
справочника [105, 106], таблицах JANAF [1543], справочниках [977, 1904, 1974], а также в работах
Байкова [23] (Т < 5000 К), Брюэра и др. [748], Краснова и Светцова [195] (Т < 2500 К). В
расчетах [105, 106, 748, 977, 1904, 1974] для v2 принято значение, в 2—3 раза большее, чем в
настоящем издании справочника, поэтому вычисленные в них термодинамические функции меньше
приведенных в табл. 768 на 7—18 Дж-К^-моль в значениях Ф° (Т). В работах [23, 1406, 15431
284
Магний и его соединения MgCl (г)
принята нелинейная структура молекулы MgF2; соответствующие расхождения уменьшаются
с увеличением температуры и составляют 8—17 Дж-К~1-моль в значениях Ф° (Т) и S° (T).
Расхождения с результатами [195] не превышают 1,3 Дж-К~1-моль в значениях Ф° (Т) и
объясняются некоторым различием в принятых частотах.
Константа равновесия реакции MgF2 (r)=Mg (r)+2F (г) вычислена с использованием
значения ДГЯ° @)=1035,748 +5,2 кДж -моль, соответствующего принятым в справочнике энтальпиям
¦образования и сублимации MgF2 (к). Этим величинам также соответствует
!±fH°(UgF2, г, 0) = —735,295 + 5,1 кДж • моль.
MgCl (г). Термодинамические свойства газообразного хлорида магния в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 769.
В табл. 26.4 представлены молекулярные постоянные MgCl, принятые на основании
результатов исследования электронного спектра этой молекулы. В спектре MgCl изучено пять систем
полос: Am ^ X2 ?+ [294, 1354, 2059, 2061, 2159, 2184, 2190, 2191, 2795], В2 ?+ <- X2 Е+ [1354, 2059,
2184], СаП«-Х2?+ [1354, 2059], ?>2?+-*Л2П [2300], E2Ii ~> X2 Е+ [294]. Состояния D2 Е+
и Е2П имеют энергии 47 619 и —53 000 см и не включены в табл. 26.4. Так же как другие галоге-
ниды элементов группы ИА, молекула MgCl может иметь связанные квартетные состояния, однако
их энергии, по-видимому, превышают 40 000 см.
Колебательные постоянные MgCl в состояниях X2 2+ и А2П определялись в работах [1354,
2059, 2184] и хорошо согласуются между собой. Принятые в табл. 26.4 значения этих постоянных
найдены Харрингтоном [1354] на основании анализа колебательной структуры полос системы
^42П—Х2?+ (г/, v' ^ 8). Вращательные постоянные в этих состояниях получены Морганом и
Барроу [2061 ] из анализа вращательной структуры полос 0—0 и 0—1 системы АЧЪ,2_ з/г—X2 ?+;-
приведенные в табл. 26.4 постоянные в состоянии А2И являются средними из найденных для двух
подсостояний.
Значения Те для состояний В2 ?+ и С2П приняты по данным Харрингтона [1354].
Термодинамические функции MgCl (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBa и ее производных по температуре рассчитывались по уравнениям A. 90)—A. 92)
с учетом трех возбужденных состояний в предположении, что QQ\ вр = (pjpx) Qi*J вр- Величина
*?1S вр и ее производные вычислялись по уравнениям A.73) — A-75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с
помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями / ^ /тах, „,
где /таХ; „ находились из условий A.81); соответствующие значения итлх и /lim приведены
в табл. 26.5. Колебательно-вращательные уровни состояния X2 2+ вычислялись по уравнениям
A. 65), A. 64). Значения коэффициентов Ykl в этих уравнениях для природной изотопической смеси
молекул MgCl, приведенные в табл. 26.5, получены на основании постоянных молекулы 24Mg35Cl
и методик, изложенных в разделе 1.4. Коэффициент Y03 получен из условия A. 59). Мультиплет-
ность и вырождение колебательно-вращательных уровней основного и возбужденных электронных
состояний учитывались соответствующими статистическими весами.
Погрешности вычисленных термодинамических функций MgCl при Т <1 3000 К обусловлены
главным образом неточностью фундаментальных постоянных, а при более высоких температурах —
отсутствием экспериментальных данных об энергии высоких колебательно-вращательных уровней
в основном состоянии, а также пренебрежением частью возбужденных электронных состояний
этой молекулы. Общие погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются
в 0,02, 0,03 и 0,05 Дж-К~1-моль~1 соответственно, -^погрешность в значении 5° F000 К) —
в 0,1 Дж-К^-моль.
Таблицы термодинамических функций MgCl публиковались ранее в предыдущих изданиях
настоящего справочника [105, 106], таблицах JANAF [1543], справочниках Мак-Брайд и др.
[1974], Дугласа и Бекетта [977], Мадера [1904] и работе Климова и др. [167] B93,15—5000 К).
Расхождения результатов этих расчетов и данных табл. 769 обусловлены различием
использованных постоянных, методов расчета и числа учитываемых электронных состояний; они составляют
до 0,5—1,3 Дж-К^-моль-1 в значениях Ф° (Т) и до 0,9—2 Дж-К^-моль-1 в значениях S° (T)
ж С; (Т).
285
MgCl2 (к, ж) Глава 26
Константа равновесия реакции MgCl (r)=Mg (r)-fCl (г) вычислена по принятой в справочники
энергии диссоциации
ДД0 @) = Do (MgCl) = 320 ± 6 кДж • моль ж 26 750 + 500 см.
Эта величина является средней из приведенных в табл. 26.12 результатов исследований
равновесия реакций с участием MgCl (г). Приведенные в таблице погрешности включают
воспроизводимость измерений, неточность сечений ионизации и использованных в расчетах термохимических
Таблица 26.12. Результаты определений Do(MgCl) (в кДж-моль)
Авторы
Метод
Do (MgCl)
II закон
III закон
Хилденбранд, 1970 [1407] Метод электронного удара, MgCl,=MgCl+Cl, 331+15
ДГЯ° @)=472±14
Хилденбранд, 1970 [1409] Масс-спектрометрический, MgCl2 (г)+Ме (г)= 331+20 315,1+3,7
=2MgCl (г), 1214-1396 К, АГЯ° @)=152,7±5 (III)
Фарбер, Сривастава, 1976 [1058] То же, 982—1243 К, 11 точек, АГН° @)= 316+6 324,5+3,7
= 134,0+5 (III)
величин (основной вклад вносит неточность значения AfE° (MgCl2, г)). Погрешность принятого.
значения оценена с учетом расхождения результатов измерений [1058 и 1409].
Принятой величине соответствует энтальпия образования
&fH°(MgCl, г, 0) = — 54,477 + 6,1 кДж • моль.
MgCl2 (к, ж). Термодинамические свойства кристаллического и жидкого дихлорида магния
в стандартном состоянии при температурах 100—2900 К приведены в табл. 770.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние принята гексагональная модификация MgCla
(слоистая структура типа CdCl2), устойчивая от 0 до 987 К. При Т < 298,15 К термодинамические
функции вычислены по результатам измерений теплоемкости MgCl2 в работе Келли и Мур [16591
E4—295 К) на образце, содержавшем 0,2% MgO. Авторы [1659] экстраполировали теплоемкость
по уравнению С"р (T)=D A94/Г)+2? C49/Т), которое приводит к значению S° E1 К)=7,3 ДжХ
ХК^-моль. Погрешности значений S° B98,15) К) и #° B98,15 К)-#° @), приведенных
в табл. 26.1 и принятых по работе [1659], оцениваются в 0,8 Дж-К-моль и 0,08 кДж-моль.
Для теплоемкости в интервале 298,15—987 К принято уравнение, выведенное методом Шомейта
по результатам измерений энтальпии MgCl2 в работе Мур [2054] B98—980 К). С данными [20541
согласуются измерения энтальпии, выполненные Холмом и Грёнволлем [1464] в узком интервале
температур (906-1072 К). Данные [229] B92-1025 К) и [21] B73-1073 К) менее надежны и не
учитывались в настоящем справочнике. Температура плавления (987 +1 К) принята по хорошо
согласующимся между собой данным [ИЗ, 1270, 1722, 2054]; измерения в [395, 869, 1624а, 1963,
2429] менее точны. Энтальпия плавления D3,1 +0,8 кДж-моль) принята по данным Мур [2054],
а уравнение для теплоемкости MgCl3 (ж) выведено в результате совместной обработки данных па
энтальпии MgCl2 (ж), полученных Мур [2054] (987—1428 К) и Холмом и Грёнволлем [1464] (980—
1072 К).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1500 и 2500 К оценены в 1, 2 и
6 Дж-К~1-моль~1. Расхождения между термодинамическими функциями MgCl2 (к, ж),
приведенными в табл. 770 и в справочниках [105, 1543], не превышают 0,6 Дж-К~1-моль~1 в значениях
Ф° {Т) до 2900 К.
В справочнике принята энтальпия образования
A/i^MgCL,, к, 298,15 К) = —644,3 + 0,7 кДж • моль.
Эта величина основана на совпадающих результатах измерений в работах [75, 262, 1262,
2509а], приведенных в табл. 26.13. В работах [2513] и [2509а] конечные концентрации раствороа
286
Магний и его соединения MgCb (к, ж)-
Таблица 26.13. Результаты определений Ду//° (MgCb, к, 298,15 К) (в кДж-моль)
Авторы
Шомейт, Хаффман, 1943 [2513]
Шин, Крисе, 1979 [2509а]
Грейсон, Снелл, 1969 [1262];
Воробьев и др., 1974 [75]; Шин, Крисе,
1979 [2509а]
Монаенкова и др., 1971 [262];
Воробьев и др., 1974 [75]
Метод
Измерение энтальпии растворения
в соляной кислоте
То же
Mg и
Измерения энтальпии растворения MgCL, в
ДГЯ° B98,15 К)=-156,4+0,8
Измерение энтальпии растворения
в хлорной кислоте и энтальпии
MgClg в воде
Mg и
MgCl2
воде,
MgO
растворения
AfH°(MgCl2, к,
298,15 К)
—641,6
—644,28 ±0,69
—644,2 ±1,3
—644,8±1,3
после растворения Mg и MgCl3 в соляной кислоте были близки, что позволило вычислить
приведенные в таблице величины. Однако в работе [2513] был, по-видимому, использован образец MgCl2r
загрязненный MgO, что привело к искажению полученного в этой работе значения (см. [2509а]).
Авторы [2509а] измерили только энтальпию растворения MgCl2 в соляной кислоте, а энтальпию
растворения Mg приняли по данным [2513]. Измерения энтальпии растворения MgCl2 в воде в
работах [75, 1262, 2509а] привели к значениям, различающимся больше, чем это можно объяснить
воспроизводимостью измерений (десятые кДж-моль), что, видимо, свидетельствует о
недостаточной чистоте образцов. Принятое значение энтальпии растворения является средним из этих трех
величин.
При расчете энтальпии образования MgCl2 (к) из этих данных использовано значение Ду#°
(Mg+2, р-р Н2О, станд. с.)=—466,4+1,0 кДж-моль, среднее из совпадающих в пределах
погрешностей результатов измерений Коффи, Олафссон [866а] и Шин, Крисе [2509а]. Это же значение
используется ниже при выборе энтальпий образования MgBr2 (к), Mgl2 (к), MgSO4 (к) и MgCO3 (к).
Монаенкова и др. [262] нашли при измерении энтальпий растворения в хлорной кислоте несколько
более отрицательное значение b.fH° (Mg+2, р-р Н2О, станд. с.) = —468,1 кДж-моль, однако
измеренная этими же авторами [75] энтальпия растворения MgCl2 в воде (—157,5 кДж-моль)
также оказалась больше, чем измеренная в работах Грейсона, Снелла [1262] (—156,0 кДж-моль)
и Шин, Крисе [2509а] (—155,8 кДж-моль). Благодаря этому значения энтальпии образования
MgCl2 (к), вычисленные по данным разных авторов, находятся в хорошем соответствии.
В литературе известны многочисленные дополнительные данные по энтальпии растворения
MgCl2 в воде и Mg в соляной кислоте (см. [406]). Однако эти измерения уступают по точности
приведенным в табл. 26.13 или проведены в условиях, когда концентрации конечных растворов,
образующихся при растворении Mg и MgCl2 в соляной кислоте, были различны.
Исследования равновесия различных реакций с участием MgCl2 (к) [184, 244, 258, 344, 677,
2048, 2430, 2431, 2647, 2686, 2727, 2736] приводят к значениям энтальпии образования MgCl2 (к)
около —642+3 кДж-моль, близким к принятому в справочнике на основании более точных
калориметрических измерений.
Давление пара дихлорида магния в реакции MgCl2 (к, ffi)=MgCl2 (г) вычислено с использовав
нием принятой в справочнике энтальпии сублимации
Ь,Н°{Щ(\, к, 0) = 246 + 5 кДж • моль-1.
Принятое значение является средневзвешенным из представленных в табл. 26.14 величин,
рассчитанных по результатам измерений давления пара MgCl2 (к, ж). При этом больший вес
придавался прецизионным измерениям Хилденбранда [1417] и работам Топор, Молдовяну [2714]
и Вагнера, Шефера [2789а], в которых достаточно точно учтено образование в паре полимерных
молекул. Ранее на основании данных [665] предполагалось, что в насыщенных парах MgCl2 (к)
количество димеров составляет около 2,3% при 919 К. Однако более поздние и детальные
измерения [2714] (комбинация результатов измерений статическим методом и методом переноса) и
[2789а] (масс-спектрометрические измерения) показали, что при 923 К количество полимерных
молекул значительно больше и составляет 7,11% для Mg2Cl4, 0,125% для Mg3Cl6 и 6,2 ЛО'3 для
Mg4Cl8 [2789a], а при 1340 К достигает 30% для Mg2Cl4 [2714]. Принимая эти данные о составе
287
MgCl2 (г)
Глава 26
Таблица 26.14. Результаты определений ASH° (MgCb, к, 0) (в кДж-моль *)
Авторы
Майер, 1929 [1923]
Берковиц, Маркварт, 1962 [665]
Шрайер, Кларк, 1963 [2473]
Фишер, Петцель, 1964 [1097]
Хилденбранд и др., 1964 [1417]
Лукашенко, Курбатов, 1970 [225]
Топор, 1972 [2713]
Топор, Молдовяну. 1972 [2714]
Вагнер, Шефер, 1979 [2789а]
Метод
Статический, 1056—1400 К, 11 точек
Эффузиошшй, 920,7 К, 1 точка
Масс-спектрометрический, —720—930 К
Точек кипения, 1208—1413 К, 18 точек а
То же, 1136—1435 К, 41 точка а
Торзиояный, 802—985 К, 36 точек а
Эффузионный, 923—1023 К, 4 точки
Статический, 1159—1321 К, 28 точек а
Переноса, 1290—1377 К, 5 точек а (в расчетах:
использованы также данные работы [2713])
Масс-спектрометрический, 758—934 К, 5 серий а
а Результаты измерений представлены только в виде уравнения.
Д8Я° (MgCl,, к, 0)
II закон
210±4
240
247
245
256
260
233
226
251,4
III закон
239,2±1,6
236,5
—
246,3
245,8
244,5
240,5 ±2,5
247,7
248,7
—
насыщеннго пара, вычислены соответствующие поправки и в таблице 26.14представлены значения,
исправленные на присутствие полимерных молекул в соответствии с данными [2714]. Эти поправки
составляют 0,6—0,9 кДж -моль при 900 К и 4,0 кДж -моль при 1300 К. Погрешность принятой
величины учитывает неточность термодинамических функций MgCl2 (к, ж, г).
MgCI2 (г). Термодинамические свойства газообразного дихлорида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 771.
Молекулярные постоянные, использованные в расчетах термодинамических функций MgGl2,
приведены в табл. 26.8. Исследования дифракции электронов [5, 13, 1046], инфракрасного спектра
[779, 863, 2291, 2842], спектра комбинационного рассеяния [1849] и отклонения молекулярного
лучка в неоднородном электрическом поле [783] показали, что в основном электронном состоянии
X1 Е* молекула MgCl2 имеет линейное строение симметрии Dmh. Приведенный в табл. 26.8 момент
инерции вычислен для значения г (Mg—С1)=2,18 +0,03 А, найденного Акишиным и др. [5, 13]
при электронографическом исследовании пара MgCl2 и подтвержденного в исследовании Каспа-
рова и др. [1586, 158в, 1046] (г (Mg—С1)=2,18 +0,01 А). Погрешность принятого значения
составляет 2-109 г-см2. Частота v3 принята по работам Бюхлера и Клемперера [779] и Рандолла и др.
[2291], в которых исследован инфракрасный спектр паров MgCl2 и найдены для v3 значения 597
и 588 см соответственно. В ИК-спектрах молекул MgGl2, изолированных в матрицах [863, 1849,
2842], получены близкие значения. Для vx принята величина несколько более низкая, чем
измеренная Лесицким и Ниблером [1849] в спектре КР молекул MgCl2, изолированных в матрицах
из Аг C26,5 см). Принятое значение v2 найдено Каспаровым и др. [1586, 158в, 1046] в
электронографическом исследовании MgCIa. В ИК-спектрах молекул MgCl2, изолированных в матрицах
из Кг и Аг, к частоте v2 отнесены полосы 87,7 см [2842] и 93 см [1849]. Бюхлер и Клемперер
[779] ошибочно отнесли к v2 (MgCl2) полосу 295 см, наблюдавшуюся ими в ИК-спектре паров MgCl2.
Погрешности принятых значений частот оцениваются в 10—20 см. Сведения о возбужденных
электронных состояниях MgCl2 отсутствуют, но можно предполагать, что их энергии превышают
30 000 см.
Термодинамические функции MgCl2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор»; их погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 2, 4 и 6 Дж-К-моль в значениях Ф°(Г)
при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций MgCl2 вычислялись в справочниках [105, 106,
977, 1543, 1904, 1974], а также в работах Краснова и Светцова [195] (Т < 2500 К) и Брюэра
и др. [748] (Т < 2000 К). В расчетах [105, 106, 748, 977, 1904, 1974] использованы неверные
значения основных частот, в связи с чем приведенные в них термодинамические функции суще-
288
Магний и его соединения MgBr (г)
ственно занижены (на 10—16 Дж-К-моль в Ф° (Т)). Расхождения данных [1543] и табл. 771
составляют 3—5 Дж-К-моль~1 в значениях Ф° (Т). Расхождения данных [195] и табл. 771
достигают 28 Дж-К~1-моль~1 из-за ошибки, допущенной в этой работе.
Константа равновесия реакции MgCl2 (r)=Mg (г)+2С1 (г) вычислена с использованием
значения &ГН° @)=783,033+5,1 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации MgCl2 (к). Этим величинам также соответствует
AfH° (MgCl2, г, 0) = —397,890 ± 5,0 кДж ¦ моль.
MgBr (г). Термодинамические свойства газообразного бромида магния в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 772.
В табл. 26.4 представлены молекулярные постоянные 24Mg'9Br, принятые в справочнике на
основании анализа результатов исследований электронных спектров этой молекулы. В спектрах
MgBr отождествлены три системы полос: А2П *± X2 Е+ [1354, 2059, 2159, 2192, 2193, 2277, 2759],
В2 ?+ <- X2 2+ [1354] иС2?+?± X2 ?+ [1354, 2309]. Так же как у молекул BeBr, Bel и Mgl,
состояние 4аПу молекулы MgBr расположено выше первого диссоциационного предела М (х5)-|-ХBРз/2)„
с которым коррелирует основное состояние X2 ?+. По-видимому, состояние 2П, коррелирующее
с этим пределом (или по крайней мере его компонента 2ГЬ/2), у всех четырех молекул является
несвязанным состоянием и обусловливает предиссоциацию MgBr в состоянии А2П (v ]> 3),
отмеченную Харрингтоном [1354]. По аналогии с другими молекулами галогенидов элементов
группы ПА молекула MgBr может иметь и другие связанные состояния, в том числе квартетные,,
коррелирующие с пределом Mg C.Р)+Вг BР). Энергии этих состояний должны быть близки
к 35 000 см или превышать эту величину, и они не рассматриваются в справочнике.
Приведенные в табл. 26.4 колебательные постоянные MgBr в состояниях X2 ?+ иА2П приняты
по работе Моргана [2059], где они были получены на основании анализа по <?-кантам 21 полосы
системы ^42П1/2 <- X2 2+ (v' ^ 5, v" ^ 6). Близкие значения этих постоянных найдены в работах
[1354, 2277, 2309]. Вращательные постоянные MgBr в этих состояниях основаны на результатах
анализа тонкой структуры полос 0—0 двух подсистем -42Пз/211/2, -> X2 2+ в работах Патела и Па-
тела [2192, 2193]. Колебательные постоянные в состоянии С2 ?+ найдены Редди и Рао [2309].
Термодинамические функции MgBr (г) вычислены по уравнениям A. 3)—A. 6), A. 93) —A. 95).
Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом одного
возбужденного состояния в предположении, что (?•$,. вр = (PilPx)QifJ. m- Величина QVO вр и ее
производные по температуре находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с помощью
уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями / <I /maij „;
/Ш|, определялись из условий A.81), в табл. 26.5 приведены соответствующие значения vmxx
и /j';m. Колебательно-вращательные уровни энергии состояния X2 Е+ вычислялись по уравнениям
A. 65), A. 64); коэффициенты YM в этих уравнениях для природной изотопической смеси молекул
MgBr, рассчитанные по постоянным молекулы 24Mg79Br, приведены в табл. 26.5. Коэффициенты
Yk0 получены с учетом условия A. 59). Мультиплетность и вырождение
колебательно-вращательных уровней всех состояний учитывались соответствующими статистическими весами.
Погрешности рассчитанных термодинамических функций MgBr (г) обусловлены неточностью
известных молекулярных постоянных в состоянии X2 ?+, а при высоких температурах — тем^
что в расчете было учтено только одно возбужденное состояние этой молекулы. Погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,03, 0,1 и 0,3 Дж -К'1 -моль, в значениях
5° F000 К) и С°р F000 К) — в 0,3 и 1 Дж-К'^моль соответственно.
Ранее термодинамические функции MgBr (г) вычислялись в таблицах JANAF [1543] и работе
Климова и др. [167], где приведены коэффициенты полинома, аппроксимирующего значения
Ф° (Т) для 293,15 < Т < 5000 К. Расхождения данных [1543] и табл. 772 составляют
до 0,4 Дж-К-моль в Ф° (Т) и 0,7 Дж-К-моль в S° (Г). Из-за различия постоянных и
неточности расчета [167] его результаты отличаются от приведенных в табл. 772 на величины до
1,2 Дж-К^-моль.
Константа равновесия реакции MgBr (r)=Mg (г)+Вг (г) вычислена на основании принятой
в справочнике энергии диссоциации
ДД° @) = Do (MgBr) = 250 ± 20 кДж • моль ж 20 900 +1800 см.
289
MgBr3 (к, ж) Глава 26
Экспериментальные определения энергии диссоциации MgBr отсутствуют. Принятая величина
оценена на основании сопоставления известных энергий диссоциации связей MX и MX—X, где
М=Ве, Mg, Ca, Sr и Ва, X=F, C1, Вг и I. Практически совпадающее значение оценено Хилден-
брандом [1408]. Оно не противоречит предиссоциации MgBr в состоянии А2И, отмеченной Хар-
рингтоном [1354]. Более низкое значение B30+30 кДж-моль) нашли Краснов и Карасева [193]
из расчета для ионной модели, однако для MgBr такой расчет должен дать заниженную величину.
Принятой энергии диссоциации соответствует
Д/nMgBr, г, 0) = 13,826+ 20 кДж-моль-1.
MgBr2 (к, ж). Термодинамические свойства кристаллического и жидкого дибромида магния
в стандартном состоянии при температурах 298,15—2700 К приведены в табл. 773.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние MgBr2 (к) принята гексагональная
модификация (структурный тип Cdl2). В литературе отсутствуют какие-либо экспериментальные данные
о теплоемкости и фазовых переходах MgBr2, за исключением температуры плавления. Значения
5° B98,15 К) и Н° B98,15 К)—Н° @), приведенные в табл. 26.1, получены при помощи
приближенных оценок; точность этих значений оценивается в 6 Дж-К~1-моль и 1,0 кДж-моль
соответственно. Принятое значение S0 B98,15 К) совпадает с оцененным в [1543]. Уравнение для
теплоемкости MgBr2 (к) в интервале 298,15—984 К выведено по данным, приведенным в таблицах
JANAF [1544]; последние оценены сравнением с экспериментальными данными для MgCl2 и Са12.
Температура плавления (984+15 К) принята по данным Кельнера [1662, 1663], а энтальпия
плавления C9,3+4 кДж-моль) оценена [1543] в предположении, что энтропия плавления MgBr,
равна энтропии плавления Са12, поскольку оба вещества относятся к одному тому же
структурному типу. Теплоемкость жидкого MgBr2 A00+10 Дж-К~1-моль~1) также оценена с учетом
экспериментальных данных для других дигалогенидов щелочноземельных металлов; в [1543]
принимается несколько более высокое значение A05 Дж-К~1-моль~1).
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000 и 2500 К оценены в 6, 12 и
30 Дж-К-моль. Расхождения между термодинамическими функциями MgBr2 (к, ж),
приведенными в табл. 773 и в справочнике [1544], не превышают 5 Дж-К~1-моль в значениях Ф° (Г) до
2000 К.
В справочнике принята энтальпия образования
t\fH° (MgBr2, к, 298,15 К) = —526,0 + 2,5 кДж • моль.
Это значение принято по совпадающим результатам измерений Финча и др. [1087] и Бекетова
[645]. Финч и др. [1087] измерили энтальпии растворения MgO (к) и MgBr2 (к) в бромистоводо-
родной кислоте, откуда для реакции MgO (к)+2НВг (p-p)=MgBr2 (к)+Н2О (ж) было найдено
значение АГН° B98,15 К)=32,5+2,1 кДж-моль, которому соответствует Д/Я° (MgBr2) =
=—526,0+2,5 кДж-моль. Бекетов 1645] измерял энтальпию растворения MgBra (к) в воде
(—181,2+5,0 кДж-моль). Вместе с энтальпией разведения этого раствора, найденной Ланге
и Штреком [1810], данные [645] приводят к значению Д^0 (MgBr2, к, 298,15 К) = — 526,5 +
+ 5,0 кДж • моль.
Давление пара дибромида магния MgBr2 (к, ж) = MgBr2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
\H°(MgBv2, к, 0) = 225 + 15 кДж • моль.
Эта величина основана на результатах измерений давления пара MgBr2, выполненных Берко-
вицем, Марквартом [665] и Топор, Молдовяну [2715]. В работе [665] проведено эффузионное
измерение при 842 К, которому соответствует энтальпия сублимации 222,2 кДж-моль (III
закон), а также масс-спектрометрическое исследование зависимости интенсивности ионного тока
MgBrJ от температуры в интервале 630—860 К, которое приводит к ASH° @)=218 кДж-моль
(II закон). Топор и Молдовяну [2715] провели исследования методом переноса A101—1206 К,
6 точек) и статическим методом. Комбинацией этих результатов они нашли парциальные
давления мономера и димера дибромида магния. Приведенным в работе [2715] парциальным давлениям
мономера соответствуют значения энтальпии сублимации 184+13 кДж-моль (II закон) и 231,4 +
290
Магний и его соединения MgBr2 (г), Mgl (г)
+ 1,3 кДж-моль (III закон). Однако масс-спектрометрическое исследование [665] показало, что
пары дибромида магния состоят практически полностью из мономерных молекул. Пересчет
результатов статических измерений [2715] в предположении о существовании в паре только
мономерных молекул приводит к значениям 211 кДж -моль (II закон) и 226,3 +0,7 кДж -моль (III
закон). Измерения [2715] выполнены при существенно более высоких температурах, когда
систематическая ошибка в значении Д8# из-за неточности термодинамических функций MgBr2 (к, ж)
и MgBr2 (г) существенно больше, чем для данных [665] (соответственно 16 и 8 кДж-моль).
Основным источником погрешности принятого значения является неточность термодинамических
функций (8 кДж-моль).
MgBr2 (г). Термодинамические свойства газообразного дибромида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 774.
Молекулярные постоянные, использованные для расчета термодинамических функций MgBr2J
приведены в табл. 26.8. Исследования дифракции электронов [5, 13], инфракрасного спектра
[863, 2291] и спектра комбинационного рассеяния [1849] показали, что в основном электронном
состоянии ХгЛд + молекула MgBr2 имеет линейное строение симметрии Ою4. Приведенный
в табл. 26.8 момент инерции вычислен для значения г (Mg—Вг)=2,34+0,03 А, найденного Аки-
шиным и др. [5, 13] при электронографическом исследовании паров MgBr2. Погрешность этой
величины составляет 5 -109 г-см2. Частота v3 найдена Рэндоллом и др. [2291], которые исследовали
инфракрасный спектр паров MgBr2. Близкие значения получены в ИК-спектре молекул MgBr2s
замороженных в матрице из Аг, Лесицким и Ниблером [1849] D97 см) и Коуком и др.
[863] D95—526 см). Для частоты vx по аналогии с частотой v3 принята величина, которая
несколько ниже найденной Лесицким и Ниблером [1849] в спектре комбинационного рассеяния
MgBr2 в матрице из Аг A97,9 см). Частота v2 оценена на основании величины, измеренной в
матрице из Аг (81,5 см) [1849], с учетом тенденции к увеличению v2, наблюдавшейся у BeF2 и MgCl2,
дри переходе от тяжелых к легким матрицам (Кг—Аг—Ne) и далее к пару. Принятое значение
v2 не согласуется с другими оценками G0 см [1544] и 220 см [748]). Погрешности принятых
значений частот оцениваются в 10—15 см. Сведения о возбужденных состояниях MgBr2
отсутствуют; по-видимому, их энергии превышают 20 000 см.
Термодинамические функции MgBr2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)„
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых постоянных, а также приближенным характером расчета; они составляют
3, 6 и 8 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции MgBr2 (г) рассчитывались в таблицах JANAF [1544],
а также работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К).
В таблицах [1544] приняты меньшие, чем в настоящем справочнике, значения vx и v2 и
рассчитанные термодинамические функции превышают приведенные в табл. 774 на 4—6 Дж-К-моль.
Расхождения с данными [748] и [195] достигают 14 и 8 Дж-К~1-моль в основном из-за
использования в этих работах неверных значений v2.
Константа равновесия реакции MgBr2 (r)=Mg (г)+2Вг (г) вычислена с использованием
значения Дг//°@)=668,729+15 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дибромида магния. Этим величинам соответствует также
AfH°(MgBi.2, г, 0) = —286,980 ± 15 кДж • моль.
Mgl (г). Термодинамические свойства газообразного иодида магния в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 775.
В табл. 26.4 представлены молекулярные постоянные, принятые в справочнике на основании
анализа результатов исследований спектров этой молекулы. В электронном спектре Mgl изучена
только одна система полос АШ <* X2 Е+ [2059, 2159, 2280, 2795, 2879]. Кроме того, в работах
[2280, 2879] в области 3750—3900 А наблюдались слабые полосы, предположительно отнесенные
к переходам, связанным с тремя возбужденными состояниями Mgl. Сопоставление со схемами
термов молекул галогенидов элементов группы ПА позволяет ожидать для Mgl в этой области только
одно состояние В2 ?+. Состояние А2И имеет энергию существенно выше энергии диссоциационного
предела Mg (x5)-fl BP*i2), с которым коррелирует основное состояние Mgl и должно коррелиро-
291
Mgl2 (к, ж) Глава 26
вать другое состояние 2П. В справочнике принимается, что, так же как у MgBr, это ненаблюдав-
шееся состояние 2П является несвязанным. Так же как другие молекулы этой группы, молекула
Mgl может иметь, кроме состояний А2И и В2 ?+, другие состояния, в том числе квартетные,
коррелирующие с пределом Mg CP)+I BР)- Эти состояния должны быть несвязанными или иметь
энергии — 30 000 см. В связи с отсутствием каких-либо данных они не рассматриваются в
справочнике.
Колебательные постоянные молекулы Mgl в состояниях X2 Е+ и А2П определялись в работах
[2059, 2280, 2879], однако сложная структура спектра затрудняла получение надежных величин.
Приведенные в табл. 26.4 значения этих постоянных рассчитаны при подготовке справочника по
данным, приведенным в работах Ямдагни [2879] и Пури и Мохана [2280] A9 полос системы
А2Щ, i/3 — X2L+). В расчете изменена нумерация полос в подсистеме Л21Ь/2—Х22+ и за полосу
0—0 принята полоса v=24 640 см, отнесенная ранее к полосе 1—0. Полученная при этом
величина постоянной спин-орбитальной связи состояния А2П оказалась равной 318 см, что в хорошем
согласии с оценкой этой величины из сравнения соответствующих постоянных в ряду галогенидов
элементов группы ПА.
Вращательные постоянные Mgl в состоянии X2 Е+ получены на основании значения re(MgI)=
=2,50 +0,05 А, оцененного по величине г (Mg—I) в молекуле Mgl2 (см. ниже) и сопоставления
величин re (MX) и г (М—X) в молекулах моно- и дигалогенидов элементов группы ПА. Для
состояния Б2Е+ в табл. 26.4 приведены постоянные, найденные в работе Ямдагни [2879].
Термодинамические функции Mgl (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBS и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом одного
возбужденного состояния в предположении, что Q$>^ вР = (pJpx)QifJ. вр- Величина Q№ вр и ее
производные вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по вращательным уровням энергии с помощью
уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями / ^ /тах г, где /msX]t
находились из условий A. 81). Колебательно-вращательные уровни состояния X2 ?+ вычислялись
по уравнениям A. 65), A. 62). Значения коэффициентов Yk, в этих уравнениях, а также величин
ь>шах и /1Ш, использованных в расчетах, приведены в табл. 26.5. Коэффициент У03 получен с
учетом условий A. 59). Мультиплетность и вырождение колебательно-вращательных уровней
электронных состояний учитывались соответствующими статистическими весами.
Погрешности вычисленных термодинамических функций Mgl (г) при температурах до 2000—
3000 К обусловлены главным образом отсутствием экспериментальных данных о вращательных
постоянных в состоянии X2 Е+. При более высоких температурах становится существенным
отсутствие данных о возбужденных электронных состояниях и энергии диссоциации этой молекулы.
Общие погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,2, 0,5 и
1 Дж-К^-моль, в значениях S° F000 К) и С°р F000 К) — до 2 Дж-К^-моль.
Таблица термодинамических функций Mgl (г) ранее не публиковалась. В работе Климова и др.
[167] приведен полином для аппроксимации значений Ф° (Т) в интервале 293,15 — 5000 К.
Расхождения в значениях Ф° (Г), рассчитанных по данным [167] и приведенных в табл. 775, не
превышают 0,7 Дж-К-моль.
Константа равновесия реакции Mgl (r)=Mg (r)+I (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ°»@) = DO (Mgl) = 190 +520 кДж ¦ моль« 15 900 + 1800 см.
Это значение оценено на основании энергии атомизации Mgl2 (г) (см. ниже) и сопоставления
величин Do (MX) и Do (MX—X) в молекулах моно- и дигалогенидов элементов группы ПА.
Близкое значение предложено Хилденбрандом [1408]. Расчет по ионной модели в работе Краснова и
Карасевой [193] привел к существенно более низкому значению A60+30 кДж-моль), так как для
Mgl такой расчет, по-видимому, должен давать заниженную величину.
• Принятой энергии диссоциации соответствует
А///° (Mgl, г, 0) = 63,066 + 20 кДж • моль.
Mgl2 (к, ж). Термодинамические свойства кристаллического и жидкого дииодида магния в
стандартном состоянии при температурах 298,15—2000 К приведены в табл. 776.
292
Магний и его соединения MglU (г)
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. За стандартное состояние в интервале 0—906 К принята гексагональная модификация
Mgl3 (к) (структурный тип Cdl2), при Т .]> 906 К — жидкий Mgl2. В литературе отсутствуют какие-
либо экспериментальные данные о теплоемкости и фазовых переходах Mgl2 (кроме температуры
плавления). Значения S° B98,15 К) и Н° B98,15 К)—Н° @), приведенные в табл. 26.1, получены
при помощи различных приближенных оценок; их погрешности оцениваются в 6 Дж-К-моль
и 1,5 кДж-моль соответственно. Для теплоемкости в интервале 298,15—906 К принято
уравнение, выведенное по оцененным значениям С°р' (Mgl2, к) при 298,15 К и в точке плавления (86 ДжХ
ХК-моль). Температура плавления (906+3 К) принята по работе Бокриса и Крука [713]s
которые исследовали образец с содержанием примесей ~ 0,01%. Близкое значение (903 К)
получено Софроновой и Колобовой [394] для образцов с большим содержанием примесей. Данные
Клемма и др. [1722] (927 К), по-видимому, ошибочны. Энтальпия плавления Mgl2 B6+4 кДжХ
Xмоль) оценена исходя из того, что энтропия плавления Mgl2 равна энтропии плавления Cdl2,
поскольку оба вещества относятся к одному и тому же структурному тину. Теплоемкость жидкого
Mgl2 A00+10 Дж-К~1-моль) оценена на основании экспериментальных данных для теплоем-
костей других дигалогенидов щелочноземельных металлов в жидком состоянии. Погрешности
вычисленных значений Ф° (Г) при 298,15, 1000 и 2000 К оцениваются в 6, 16 и 27 Дж-К-моль
соответственно. Расхождения между значениями Ф°(Т) и S° (T), приведенными в [1544] и
табл. 776, не превышают 4Дж • К • моль.
В справочнике принимается энтальпия образования
^"(Mglj, к, 298,15К) = —367,1 ±1,5 кДж • моль.
Это значение основано на результатах измерений энтальпий растворения MgO (к) и Mgl2 (к)
в иодистоводородной кислоте в работе Финча и др. [1087] (Дг#° B98,15 К) =—60,9+1,3 кДжХ
Xмоль). В расчетах использованы данные Вандерзее и Гира [2755] по энтальпии растворения
HI в воде и по энтальпии разведения растворов иодистоводородной кислоты. Выполненное в
прошлом веке Бекетовым [31 ] исследование энтальпии растворения Mgla (к) (—208 +4 кДж -моль),,
совместно с принятыми в справочнике энтальпиями образования ионов, приводит к значению
—370+5 кДж-моль, совпадающему в пределах погрешностей с принятым в справочнике.
Давление пара дииодида магния Mgl2 (к, ж)=Мд12 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
A,#°(MgI2, к, 0) = 205 + 15 кДж • моль.
Это значение основано на работе Берковица и Маркварта [665], в которой эффузионным
методом было измерено давление пара Mgl2 при 757 К. Близкое значение 198 кДж-моль (II закон)
получено этими же авторами в результате масс-спектрометрического исследования зависимости
интенсивности ионного тока MglJ от температуры. Погрешность принятой величины оценена
с учетом неточности термодинамических функций Mgl2 (к, ж) и Mgl2 (г) A0 кДж -моль) и
погрешности измерений в работе [665].
Mgl2 (г). Термодинамические свойства газообразного дииодида магния в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 777.
Молекулярные постоянные, использованные для расчета термодинамических функций Mgl2 (r)r;
приведены в табл. 26.8. Исследования дифракции электронов [5, 6], инфракрасного спектра
и спектра комбинационного рассеяния [1849] показали, что молекула Mgl2 в основном
электронном состоянии X1 Е* линейна (симметрия Dmh). Момент инерции, приведенный в таблице,
соответствует значению г (Mg—1)=2,52+0,03 А, найденному Акишиным и др. [5, 6] при электроно-
графическом исследовании паров Mgl2. Погрешность приведенной величины составляет
1-108 г-см2. Для частот vx и v3 молекулы Mgl2 по аналогии с MgF2, MgCl2 и MgBr2 приняты
значения, которые несколько ниже найденных Лесицким и Ниблером [1849] в ИК-спектре и спектре
КР молекул Mgl2, изолированных в матрице из Аг A47,6 и 444,9 см). Для частоты v2 по аналогии
с BeF2 и MgCl2 принято значение, которое несколько больше измеренного Лесицким и Ниблером
[1849] (v2=55,8 см). Погрешности принятых значений частот оцениваются в 10—15 см.
Сведения о возбужденных состояниях Mgl2 отсутствуют; можно предполагать, что их энергии пре-
293
MgS (к, ж) Глава 26
Термодинамические функции Mgl2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор1— гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета;
они составляют 4, 7 и 10 Дж-К'1 -моль в значениях Ф° {Т) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Mgl2 (г) вычислялись в таблицах JANAF [1544] и работах
Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Данные [1544]
и табл. 777 согласуются в пределах 4 Дж-К-моль. Расхождения с данными [195, 748]
составляют в значениях Ф° (Т) 8—10 и 12—16 Дж-К-моль~1 соответственно и обусловлены тем, что
в обеих работах приняты неправильно оцененные заниженные значения v2.
Константа равновесия реакции Mgl2 (r)=Mg (г)+21 (г) вычислена на основании значения
Д^0 @)=521,133+15 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации Mgl2 (к). Этим величинам соответствует также
-1
s, г, 0) = —160,904 + 15 кДж • моль
MgS (к, ж). Термодинамические свойства кристаллического и жидкого сульфида магния
в стандартных состояниях при температурах 100—4000 К приведены в табл. 778.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 26.1. В справочнике за стандартное состояние MgS (к) в интервале 0—2500 К принята
кубическая модификация (структурный тип NaCl) [1459a]. При Г<298,15 К термодинамические функции
MgS (к) вычислены на основании измерений теплоемкости, выполненных Сталлом и др. [2630а]
A5—310 К) на образце чистотой 99,7%. Погрешности измерений оценены авторами [2630а] в
интервале 80—310 К в 0,3%, при более низких температурах они возрастают до 1% при 20—30 К
и до 3% при Т <[ 20 К. Экстраполяция теплоемкости ниже 15 К выполнена авторами [2630а]
по закону Дебая и приводит к значению S° A5 К)=0,04 Дж-К~1-моль. Погрешности принятых
значений S° B98,15 К) и Н° B98,15 К)—#° @) (см. табл. 26.1) составляют 0,4 Дж-К^-моль
и 0,05 кДж-моль соответственно. В литературе отсутствуют сведения об измерениях
теплоемкости или энтальпии MgS (к, ж) при Т > 298 К. В интервале 298,15—2500 К для теплоемкости
MgS (к) принято уравнение (см. табл. 26.1), выведенное по значениям Ср B98,15 К), Ср E00 К)=
=49 Дж-К~1-моль~1 и С°р B000 К)=63 Дж-К-моль; два последних значения оценены
сопоставлением экспериментальных данных по теплоемкости окислов и сульфидов щелочноземельных
металлов. Погрешности оцененных значений теплоемкости могут достигать 10% при 1000 К и 15%
при Т > 2000 К. Какие-либо данные по температуре плавления MgS и по диаграмме состояния
системы Mg—S отсутствуют, за исключением указания в работе [365а] на то, что Tm (MgS) >
> 2273 К. В справочнике принимается, что MgS имеет такую же температуру плавления, как
сульфиды стронция и бария, погрешность этого значения составляет 200—300 К.
Энтальпия плавления F3+12 кДж-моль) оценена по эмпирическому правилу [1781], согласно
которому энтропия плавления ионных соединений с решеткой типа NaCl примерно равна
25 Дж-К^-моль. Для теплоемкости MgS (ж) принято значение 67+15 Дж-К~1-моль,
полученное по соотношению Ср (ж)=33,5га Дж-К-моль~1 (см. т. 1, кн. 1, с. 79).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются в 0,3,
4, 8 и 15 Дж-К~1-моль соответственно. Термодинамические функции MgS (к), рассчитанные
в справочниках [578, 2031, 2298], отличаются от данных табл. 778 из-за разной оценки
теплоемкости MgS (к, ж) при высоких температурах, в пределах 4 Дж-К~1-моль~1 в значениях энтропии.
Принятая в справочнике энтальпия образования
-1
bfH°(MgS, к, 298,15 К) = — 348 + 5 кДж • моль
является средневзвешенным значением из приведенных в табл. 26.15 данных (за исключением
работы Блюма, известной по справочнику [2031]). Указанные погрешности включают
воспроизводимость измерений, а также погрешности использованных в расчетах термохимических величин
и термодинамических функций (около 6 кДж-моль'1 для измерений при 1400 К). Результаты
калориметрических измерений и исследований равновесия реакций с участием MgS (к) совпадают
в пределах их погрешности.
294
Магний и его соединения MgS (г)
Таблица 26.15. Результаты определений h.fH° (MgS, к, 298,15 К) (в кДжмоль)
Авторы
Метод
b.fH°(MgS, к, 298,15 К)
II закон
III закон
Сабатье, 1881 [2417] Калориметрический, MgS (к)+3,1НС1 (р-р, -348+10
221H2O)=MgCl2 (р-р, 1,1НС1+636Н2О)+
+1.ШС1 (р-р, 623H2O)+H2S (р-р), при 13° С,
3 измерения
Капустинский, Коршунов, 1939 [1628а] То же, Mg (k)+S (K)=MgS (к) —353,1+4,0
Вартенберг, 1943 [2800а] То же, MgS (к)+12,4НС1 (р-р, 18H2O)=MgCl2 -349,7±4,0
(р-р, 10,4НС1+226Н2О)+10,4НС1 (р-р, 22Н2О)+
+H2S (г) а, комнатная температура, 8 измерений
Блюм, 1948 (см. [2031 ]) Исследование равновесия реакции MgO (к)+ —330 —
+H2S (r)=MgS (к)+Н2О (г)
Карлук, Пиджен, 1958 [913в] То же, протока, 1180—1483 К, 6 точек —341 ±10 —340±6
Дыоинг, Ричардсон, 1960 [949а] То же, циркуляционный, 1290—1768 К, 6 точек —356±6 —342±6
а В работе 12800а] измерения приближенно отнесены к реакции MgS(K)+2HCl (p-p,-30H2O)=MgCl2(p-p, 30Н2О)+
+H2S(r), которой соответствует A^ff°(MgS, к, 298,15 К)=—343,6 кДж-моль. Однако имеющиеся в этой работе
данные позволяют найти приведенное в таблице более точное уравнение.
Давление пара сульфида магния в реакции MgS (к, m)=MgS (г) вычислено с использованием
значения А,ДО @) = A,/7°(MgS, к)=467,606 +30 кДж -моль, соответствующего принятым в
справочнике энтальпии образования MgS (к) и энергии диссоциации молекулы MgS.
MgS (г). Термодинамические свойства газообразного сульфида магния в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 779.
В табл. 26.4 приведены молекулярные постоянные MgS, принятые на основании анализа
результатов исследования электронного спектра [19426] и приближенных оценок. В спектре MgS
исследована только одна система полос В1 Е+ <- X1 Е+ [19426]. По аналогии с MgO, оксидами и
сульфидами других щелочноземельных элементов молекула MgS должна иметь ряд синглетных и триплет-
ных состояний, соответствующих конфигурациям . . .о2л3о (а3П, Л 41), . . .апАа (Ь3Е+, .B1S+),
. . . ААс (ЗДД, 1S~, 1Е+, 32+) и . . .атс4тгCП, 41). Энергии ненаблюдавшихся состояний оценены
на основании соответствующих данных для молекулы MgO и соотношения этих величин у
молекул BaS и ВаО; погрешности оцененных значений составляют 1000 см для состояний а3П и
А1И и 2000—4000 см для остальных состояний.
Молекулярные постоянные состояний X1 S+ и В1 ?+, приведенные в табл. 26.4, приняты по
работе Маркано и Барроу [19426], которые выполнили анализ вращательной структуры ряда полос
системы В1 2+ «- X1 S+, (v" < 2, i/ < 2).
Термодинамические функции MgS (г) вычислены по уравнениям A. 3)—A. 10) и A. 93)—A. 95).
Значения QBS и ее производных рассчитывались по соотношениям A. 90)—A. 92) с учетом 11
возбужденных электронных состояний в предположении, что Q^. BV = {piIPx)Ql-Kf]}BV-
Статистическая сумма по колебательно-вращательным уровням состояния ХХЕ+ и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием по 7 с
помощью уравнений типа A. 82). В расчете учитывались все уровни со значениями 7 ^ 7шах> „,
последние находились по соотношению A. 81). Энергия колебательно-вращательных уровней
рассчитывалась по уравнениям A. 63а) и A. 65) с коэффициентами Ykl, приведенными в табл. 26.5
вместе со значениями fmax и 71;ш. Значения Ykl получены по постоянным из табл. 26.4 с учетом
условий A. 50) и A. 59), для соблюдения которых в уравнение A.65) введены дополнительные
члены с коэффициентами Yao, ^40 и ^оз- Постоянная У31 определена из условия ^»ши+1==0- Все
постоянные пересчитаны по соотношениям A.66) для изотопической модификации,
соответствующей природной смеси изотопов Mg и S.
Погрешности вычисленных термодинамических функций во всем интервале температур
обусловлены главным образом неточностью оцененных энергий возбуждения состояний а3П и А1!!.
295
MgS04 (к,Тж) Глава 2&
Общие погрешности в значениях Ф° (Т) составляют 3—5 Дж-К-моль при всех температурах.
Таблицы термодинамических функций MgS (г) ранее вычислялись в справочниках JANAF
[1544] и Мак-Брайд и др. [1974]. Расхождения данных [1974] с приведенными в табл. 779
достигают 16 и 20 Дж-К~1-моль~1 в значениях Ф° F000 К) и S° F000 К) соответственно и обусловлены
тем, что авторы [1974] не учли в расчете возбужденные электронные состояния MgS. В таблицах
JANAF [1544] приняты существенно завышенные значения энергий возбуждения состояний
а3П и .^П (более высокие, чем в молекулах MgO и BeS), это привело к значительным
расхождениям с данными табл. 779, которые достигают 12 и 16 Дж-К~1-моль в значениях Ф° (Т) и 5° (Г)
при 2000-3000 К.
Константа равновесия реакции MgS (r)=Mg (r)+S (г) рассчитана по принятой в справочнике
энергии диссоциации
г-1
ДГЯ° @) = Do (MgS) = 300 + 30 кДж • моль «* 25 000 ± 2500 см'
Это значение оценено при подготовке справочника на основании сопоставления энергий
диссоциации молекул окислов и сульфидов элементов подгруппы ПА и величины D0(MgO),
принятых в справочнике. Линейная экстраполяция уровней состояния X1 Е+ приводит, так же как для
молекулы MgO, к существенно заниженному значению (~200 кДж-моль). Принятой величине
соответствует
г, 0) = 120,688 + 30 кДж • моль.
MgSO4 (к, ж). Термодинамические свойства кристаллического и жидкого сульфата магния
в стандартном состоянии при температурах 100—2000 К приведены в табл. 780.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 26.1. Термодинамические функции MgSO4 (к) вычислены в интервале 0—1283 К для ромбичее-
кой модификации (oc-MgS04), а в интервале 1283—1410 К — для высокотемпературной
ромбической модификации (|3-MgSO4) х. При Т < 298,15 К термодинамические функции
вычислены по данным о теплоемкости, полученным Мур и Келли [2056] E3—295 К). Экстраполяция
теплоемкости к Г=0 проводилась в [2056] по уравнению Ср (Г)=2Э B42/Г)+2Я C78/Г)+
+2?(866/Г)+? A689/77) и привела к значению S° E0,12 К) =4,52 Дж -К -моль. Погрешности
принятых значений S0 B98,15 К) и Н° B98,15 К) — Н° @) оцениваются в 0,8 Дж-К^-моль и
0,08 кДж-моль соответственно.
Экспериментальные данные по теплоемкости и энтальпии MgSO4 (к, ж) при Т > 298,15 К
в литературе отсутствуют. Уравнение для теплоемкости ромбической модификации MgS04 в
интервале 298,15—1283 К (см. табл. 26.1) выведено по значению С\ B98,15 К) [2056] и значениям
энтальпии, оцененным в справочнике JANAF [1543] с помощью соотношения С°р (MgSO4)=
—С°р (CaSO4)+Cp (MgO)—CJ, (СаО). Согласно полученному уравнению теплоемкость MgSO4 (к)
изменяется от 96,4 Дж -К -моль при 298,15 К до 164,2 Дж -К -моль при 1283 К; погрешности
оцененных значений составляют 5—10%. Температура превращения A283+20 К) принята на
основании согласующихся между собой данных Дьюинга и др. [949] A283 +10 К), Шаргородского
[439] A273—1298 К) и Ямагути и Като [2876] A301 К). Данные Роу и др. [2390] приводят к очень
широкому интервалу A165—1368 К) и не учитывались. Энтальпия превращения MgS04 C +1 кДжХ
X моль) оценена при подготовке справочника по результатам исследования безводных сульфатов
MgS04, ZnSO4 и CdSO4 методом ДТА в работе Шаргородского [439] и данных по энтальпиям
полиморфных превращений двух последних солей (см. [406]) 2.
Температура плавления A410+5 К) принята по данным Роу и др. [2390]; результаты
измерений [266, 489, 1213, 1244, 1546] менее точны. Энтальпия плавления MgS04 A4,6 +2,0 кДж -моль)
рассчитана Келли [1650] из данных по диаграмме состояния системы MgSO4—K3SO4 [1244].
Теплоемкости р-модификации MgSO4 A283—1410 К) и жидкого MgSO4 (при Т > 1410 К) оценены по
аддитивной схеме и данным о теплоемкостиMgCl2, KC1 и K2SO4 равными 155+20 Дж-К~1-моль.
По данным [2876] имеется еще одна ромбическая модификация MgSO4, которая при нагревании до 868 ±5 К,
по-видимому, претерпевает монотропный переход в a-MgSC^.
В работе [493] опыты методом ДТА проводились с одинаковыми количествами (по объему) исследуемых
веществ; при. обработке этих измерений учитывались плотности веществ.
296
Магний и его соединения
Mg3N2 (к)
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К составляют 1, 7 и
18 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями MgSO4
(к, ж), приведенными в табл. 780 и в справочнике [1543], не превышают 0,5 Дж-К-моль~1 в
значениях Ф° (Т). Они обусловлены тем, что в настоящем справочнике учтено полиморфное
превращение MgSO4 (к) при 1283 К и приняты другие значения постоянных выше этой температуры.
Константа равновесия реакции MgSO4 (K)=Mg (r)+S (г)+40 (г) вычислена с использованием
значения &ГН° @)=2685,244 +1,0 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
ДД° (a-MgSO4, к, 298,15 К) = —1288,8 + 0,6 кДж • моль.
Результаты определений энтальпии образования MgSO4 (к) представлены в табл. 26.16, их
погрешности учитывают воспроизводимость измерений и неточности других величин, используемых
Таблица 26.16. Результаты определений
(MgSO4, к, 298,15 К) (в кДжмоль)
Авторы
Метод
A/5°(MgSOt, к, 298,15 К)
II закон
III закон
Фавр, Валсон, 1871 [1071] Калориметрическое измерение энтальпии раство- —1284,1
рения M?SO4(k) в воде, 400 Н»О, 16е С; Д.Я °
B98,15 К)=—91,9
Томсен, 1882 [2686] То же, 440 Н2О, 17° С; Дг#°B98,15 К)=—91,3 -1284,7
Бертло, Илосвай, 1882 [676, 677] Тоже, 200 НаО, 15—19°С, ДГЯ°B98,15К)=—91,4 —1284,6
Пиккеринг, 1885 [2226, 2227] То же, 440 Н2О, 2-22° С; ДГЯ°B98,15 К)=—91,5 —1284,6
Томсен, 1882 [2686] То же для реакции растворов MgCl2 и Mg(NO3J —1284,3
с серной кислотой ;
Ко, Даут, 1980 [1732а] То же для реакции растворения MgSO4 (о), —1288,8+0,6 а
MgSO4(P) и MgO в серной кислоте —1285,0±0,6 б
Маршаль, 1925 [1943] Исследование равновесия реакции MgSO4(K)= —1303±15 —1297±10
=MgO(K)+SO2(r)+1/2O2(r) статическим
методом, 1223—1463 К, 26 точек, Дг#°B98,15 К)=
-402,5±0,4(Ш)
Дьюинг, Ричардсон, 1959 [949] То же, метод ДТА, 1483—1638 К, результаты из- —1306 —1290+18
мерений представлены в виде уравнения, ЬГН°
B98,15 К)=395,7±0,8(Ш)
Лау и др., 1977 [1822] То же, реакция MgS04(K)=Mg0(K, микро- —1358±10 —1300,0±7,0
крист.)+ SO2(r)+1/2O2(r), торзионный метод,
904—993 К, 42 точки (четыре серии измерений
с разными размерами эффузионных отверстий),
ДГЯ°B98,15 К)=405,5±2,0(Ш)
То же, MgSO4(K)=MgO (к, микрокрист.)+8О3(г), —1312±10 —1297,0±7,0
947—1012 К, 34 точки (три серии измерении
с разными размерами эффузионных отверстий),
ДГЯ°B98,15 К)=303,6±2,0(Ш)
а a-MgSO4; б P-MgSO4.
в расчетах, в том числе термодинамических функций MgSO4 (к, ж). Авторы калориметрических
измерений [676, 677, 1071, 2226, 2227, 2686 ] не указывают, какие модификации MgSO4 были
исследованы. Наиболее точные калориметрические измерения были выполнены Ко и Даутом [1732а]
и принятая величина основывается на этой работе.
Исследования равновесия диссоциации MgSO4, представленные в табл. 26.16, приводят к
большим и значительно менее точным величинам, что, по-видимому, обусловлено недостаточной
точностью термодинамических функций MgSO4 (к, ж).
Mg3N2 (к). Термодинамические свойства кристаллического динитрида тримагния в стандартном
состоянии при температурах 298,15—2500 К приведены в табл. 781.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 26.1. В справочнике за стандартное состояние Mg3N2 (к) в интервале 298,15—2500 К при-
297
Mg3N2 (к) Глава 26
нята кубическая модификация (a-Mg3Na, структурный тип анти-Мп2О3 [931, 2705а]). При
температурах Т < 298,15 К экспериментальные данные по теплоемкости Mg3N2 в литературе
отсутствуют. Оценки величины S° B98,15 К), проведенные по различным эмпирическим методам [216,
427, 814а], приводят к значениям в интервале 84—96 Дж-К-моль. В справочнике принято
значение 85+8 Дж-К~1-моль~1, вычисленное Койлом и Сирей [896] из данных по равновесию
диссоциации Mg3N2 A100—1400 К). Значения Н° B98,15 К)—Н° @) и С° B98,15 К),
приведенные в табл. 26.1, оценены с учетом данных по энтальпии Mg3N2 [2438] B73—691 К) и [2038] B98—
1273 К). Уравнение для теплоемкости Mg3N2 (к) в интервале 298 — 2500 К выведено обработкой
методом Шомейта результатов 32 измерений энтальпии, проведенных Митчеллом [2038] в
интервале 298—1273 К на образце, содержавшем 99,1% Mg3Na и 0,9% MgO. Митчелл [2038] на
основании своих данных предположил существование у Mg3N2 двух полиморфных превращений при
823+3 К и 1061+5 К с небольшими энтальпиями перехода @,92 и 1,09 кДж-моль
соответственно). Однако это предположение является недостаточно обоснованным ввиду большой
погрешности данных [2038] (в среднем 1,3%, 4 точки отклоняются на 2—3%) и незначительных средних
отклонений результатов измерений энтальпии для «Р-фазы» и «у-фазы», составляющих 0,3—0,4%.
Наличие полиморфизма у Mg3N2 не было подтверждено другими исследованиями, в том числе
структурными. Измерения энтальпии Mg3N2, проведенные Сато [2438] на образце состава 78,9%
Mg3N2 и 21,1% MgO в интервале 273—691 К, резко завышены (на —5%) и не учитывались. Данные
по температуре плавления Mg3N2 в литературе отсутствуют. По-видимому, температура
плавления Mg3N2 лежит выше температуры, при которой давление диссоциации Mg3Na достигает 100 атм
{2500 К).
Погрешности вычисленных значений Фс (Т) при 298,15, 1000 и 2500 К оцениваются в 5, 8
и 20 Дж-К-моль~1 соответственно. Термодинамические функции Mg3N2 (к), приведенные в
таблицах JANAF [1543], отличаются от данных табл. 781 на 3,3 и 4,5 Дж-К~1-моль в значениях
S° (T) при 1000 и 2500 К соответственно; расхождения обусловлены различием принятых значений
S° B98,15 К) и разной интерпретацией данных Митчелла [2038].
Константа равновесия реакции Mg3N2 (K)=3Mg (r)+2N (г) вычислена с использованием
значения АГН° @)=1827,476 +3,2 кДж-моль, соответствующего принятой в справочнике энталь-
лии образования
A//°(Mg3N2, к, 298,15К) = —461,3 + 2,0 кДж • моль.
Эта величина основана на результатах измерений, приведенных в табл. 26.17. Из приведенных
в таблице калориметрических работ наиболее надежной является работа Митчелла [2038],
которая положена в основу выбора рекомендованной величины. В хорошем соответствии с ней
находится работа Апина и др. [18] (см. также более старые работы [1965, 2065]). Исследования
равновесия реакции диссоциации динитрида тримагния приводят к недостаточно точным значениям
энтальпии образования, что связано со значительными экспериментальными трудностями (см.
Т а б л нц а 26.17. Результаты определений &fH° (Mg3N2, к, 298,15 К) (в кДж
Авторы
Метод
• моль 1)
bfH° (Mg3N2, к, 298,15 К)
II закон
III закон
Нейман и др., 1932 [2128] Калориметрическое измерение энтальпии раство- —472,0
рения Mg и Mg3N2 в соляной кислоте
Митчелл, 1949 [2038] То же для растворения Mg3N2 в соляной кислоте —461,3+2,0
(использованы результаты измерений
энтальпии растворения Mg в соляной кислоте [2513])
Нейман и др., 1932 [2126] Калориметрический, реакция Mg(K)+N2(r) —485±8
Апин и др., 1958 [18] То же для реакции Mg(K)+PbN6(K) —465,7
Соулен и др., 1955 [2586] Исследование равновесия реакции Mg3N2(K)= — —568±35
=3Mg(r)+N2(r) эффузионным методом, 1118—
1254 К, 7 точек
То же, методом протока, 1337—1506 К, 6 точек — —442
Койл, Сирей, 1973 [896] То же, 1100—1400 К, расчет по уравнению —465 —459±10
298
Магний и его соединения
MgCO3 (к, ж)
[896]) и с неточностью термодинамических функций Mg3N2 (к). В наиболее детальном
исследовании [896] предприняты меры для достижения равновесных условий и получено значение,
находящееся в хорошем соответствии с принятым (см. также [698, 1424, 2787]).
MgCO3 (к, ж). Термодинамические свойства кристаллического и жидкого карбоната магния
в стандартном состоянии в интервале 100—1500 К приведены в табл. 782.
Значения постоянных, принятые в расчетах термодинамических функций, приведены в табл.
26.1. В интервале от Г=0 до температуры плавления A263 К) 1 карбонат магния известен в виде
гексагональной модификации (структурный тип кальцита). При Т < 298,15 К термодинамические
функции вычислены на основании измерений теплоемкости образца MgCO3, содержащего 0,3%
СаСО3, в работе Андерсона [510] E6—292 К). Экстраполяция к Т=0 проводилась в [510] по
уравнению С; {T)=D C54/Г)+2? D68/Г)+2? A944/Г), приводящему к значению 5° E6,2 К) =
=2,38 Дж-К^-моль. Погрешности принятых по [510] значений S° B98,15 К) и Я° B98,15 К)—
Н° @) (см. табл. 26.1) оценены в 0,8 Дж-К~1-моль и 0,08 кДж-моль с учетом чистоты
исследованного образца и существенного вклада в значение энтропии составляющей, обусловленной
экстраполяцией теплоемкости к Г=0. При Т >¦ 298,15 К для теплоемкости MgCO3 (к) принято
уравнение, выведенное методом Шомейта на основании результатов неопубликованных измерений
энтальпии, выполненных Шомейтом B98,15—743 К) и известных по справочнику Келли [1656].
Погрешность этих данных оценивается в 1 %.
Температура плавления A263+20 К) найдена Велденом [2761] при исследовании равновесия
диссоциации карбоната магния в расплавах со щелочными металлами в атмосфере СО2. По тем же
данным для теплоты плавления MgC03 получено значение 59+8 кДж-моль. Теплоемкость
MgC03 (ж) принята равной 155+20 Дж-К-моль на основании сравнения данных по
теплоемкости жидких Na2GO3, Na2SO4 и MgSO4.
Погрешность вычисленных значений Ф° (Т) оценена при 298,15, 1000 и 1500 К равной 1,
1,5 и 5 Дж-К~1-моль. Приведенные в справочниках [1543, 1656] значения термодинамических
функций для MgCO3 (к) близки к приведенным в табл. 782. Термодинамические функции MgCO3 (ж)
публикуются впервые.
Константа равновесия реакции MgCO3 (K)=Mg (г)+С (г)+30 (г) вычислена с использованием
значения Дг/?° @)=2685,994+3,1 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
&fH°(MgCOs, к, 298,15 К) = —1096 + 3 кДж ¦ моль.
В табл. 26.18 приведены результаты некоторых измерений этой величины. Из
калориметрических измерений энтальпии образования лучшей является работа Капустинского и Стахановой
Т а б л и ц а 26.18. Результаты определений Ду?Г° (MgCO3, к, 298,15 К) (в кДжмоль)
Авторы
Рот, 1941 [2370]
Капустпнский, Стаханова, 1954 [154]
Фолдвари и др., 1958 [1113]; Томас-
сон и др., 1964 [2683]
Марк, Шимек, 1913 [1942]
Лангмюр, 1965 [1811]
Метод
Калориметрическое измерение энтальпии
реакции растворения MgO и MgCOa в соляной
кислоте при 90° С
То же, реакция MgCO3 (K)=MgO(K)+COa(r) при
сожжении парафинового масла с добавкой
МвСО<,(к)
То же, реакция растворения MgCO3(K) и MgO(K)
в соляной кислоте при 25° С
Измерение энтальпии реакции MgCO3(K)=
=MgO(K)+CO2(r) методом высокотемпературной
калориметрии
Исследование равновесия реакции MgCO3(K)=
= MgO(K)+CO2(r), статический метод, 671—
782 К, 23 точки
Анализ данных ло давлению диссоциации и
растворимости MgCO3, М?СОз-ЗН2О и др. данных
bfH° (MgCOs, к,
298,15 К)
—1105
—1090
—1093,5
—1110
—1097+30A1)
—1113,1 ±1,5A11)
—1096±3
1 В точке плавления MgCO3 равновесное давление СО2(г) составляет ~1 атм.
299
MgCO8 (к, ж) Глава 26*
Г1543- Однако есть основания сомневаться в том, что исследованный в этой работе образец MgCO3.
был достаточно чистый — судя по методу его синтеза он мог содержать дигидроксид магния
(см. [1811]). Исследования равновесий диссоциации MgCO3 (K)=MgO (к)+СО2 (г) затруднены
крайне медленным^установлением равновесия. В лучшей из этих работ, Марка и Шимека [1942],.
для ускорения достижения равновесия магнезит смешивался с нитратом калия, что приводило
к дополнительным осложнениям (коррозия, собственное давление пара KNO3, возможное
снижение активности). Исследования растворимости магнезита в реакции MgCO3 (K)=Mg+2 (aq)-\~CO^2 (aq)
в работах [1341,1343, 1479,1646, 1811, 2100, 2307, 2518] и в реакции MgCO3 (к)+СО2 (г, 1 атм)+
+Н2О ^)=Mg+2 (ag)+2HCO~ (aq) в работах [477, 1341, 1811 ] приводят к противоречивым
результатам (—1084 и —1113 кДж-моль).
Наиболее полный анализ данных по термодинамическим свойствам ряда карбонатных
соединений магния проведен Лангмюром [1811], который, помимо приведенных в табл. 26.18 данных,
использовал также результаты исследования равновесий реакций с участием MgCO3, MgC03-3H2O
и 3MgCO3-Mg(OHJ-3H2O. Принятое в справочнике значение основывается на работе [1811].
Глава 27
КАЛЬЦИЙ И ЕГО СОЕДИНЕНИЯ
Са (к. ж), Са, Са+, Са2, СаО (к, ж), СаО, СаО+, СаН, СаН2 (к, ж), СаОН, СаОН+, Са(ОНJ (к, ж),
€а(ОМ)о, CaF, CaF+, CaF2 (к, ж), CaF2, CaCl, СаС1+, СаС12 (к, ж), СаС12, СаВг, СаВг2 (к, ж), СаВг2,
Cal, Cal2 (к, ж), Cal2, CaS (к, ж), CaS, GaS04 (к, ж), СаСО3 (к, ж)
В настоящей главе рассматриваются 23 вещества — кальций и его соединения с кислородом,
водородом, галоидами, серой и углеродом, для 11 веществ приведены данные о термодинамических
-свойствах в конденсированном состоянии и для 20 веществ — в газообразном состоянии.
Термодинамические свойства четырех газов (Са, Са+, СаО, и СаО+) рассчитаны для температур до
10 000 К, для одного газа (Са2) — до 3000 К, для остальных — до 6000 К. Для Са в
Приложении 3.9 представлены сведения о его критических параметрах.
Термодинамические свойства всех веществ приведены для природной изотопической смеси
кальция (атомный вес 39,098) и других элементов. Однако различия в молекулярных постоянных
индивидуальных изотопических модификаций веществ в большинстве случаев не учитывались
в расчетах из-за отсутствия необходимых данных и их несущественного влияния на
термодинамические функции соединений кальция.
В справочник не включены многоатомные окислы кальция типа СаО2 и Са2О2 ввиду их низкой
•стабильности, а также ионы СаВг+ и Са1+, поскольку в равновесных условиях присутствие их
маловероятно из-за невысокой стабильности соответствующих нейтральных соединений.
Са (к, ж). Термодинамические свойства кристаллического и жидкого кальция в стандартном
состоянии при температурах 100—3300 К приведены в табл. 783.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние Са (к) в интервале 0—716 К принимается
кубическая плотноупакованная модификация (а-Са, структурный тип Си), а в интервале 716—
1115 К — кубическая объемно-центрированная модификация (у-Са, структурный тип a-Fe) x. При
Т < 298,15 К термодинамические функции вычислены по результатам измерений теплоемкости
в работах Клузиуса и Вогена [861] A0—201 К), Робертса [2350] A,5—20 К) и Агарвала и Веттер-
тона [487] A,1—4,2 К). Данные [2350] в интервале 5—10 К скорректированы на —20%, поскольку
они завышены по сравнению с полученными в работах [487, 861 ]. Сведения о теплоемкости кальция
в интервале 200—298 К отсутствуют; соответствующие значения Ср (Т) получены интерполяцией
с выделением решеточной и электронной составляющих и величин С —С„. Значения 5° B98,15 К)
и Н° B98,15 К)—Н° @), приведенные в табл. 27.1, имеют погрешности в 0,4 Дж-К-моль~1
и 0,04 кДж-моль соответственно, они совпадают с величинами, рекомендованными КОДАТА—
МСНС [864] в качестве ключевых термодинамических величин.
В интервале 298,15—716 К для теплоемкости принято уравнение, выведенное совместной
обработкой по методу Шомейта результатов измерений энтальпии в работах Истмана и др. [1006]
{373—878 К) и Яуха [1550] C00—720 К). При этом погрешности данных [1550] оценивались
в 1,5%, а данных [1006] — в 3%. В интервале 716—1115 К для теплоемкости принято уравнение
(см. табл. 27.1), выведенное Яухом [1550] на основании проведенных им измерений энтальпии
кальция при температурах 753—1023 К.
Температура полиморфного превращения G16 +3 К) принята на основании согласующихся
измерений Кайзера и Содерквиста [1645] (99,96% Са, 716+2 К), Питерсона, Фаттора [2215]
(99,9% Са, 715—721 К) и Яуха [1550] G13 К). Данные других авторов [1551] G21 К) и [1637]
G01 К), [2340] G23 К), [2476] G23 К), [2554] G37 К) менее надежны из-за присутствия
значительных примесей в исследованных образцах. Энтальпия превращения @,93+0,20 кДж-моль)
принята но работам [846] @,93 кДж-моль), [1550] A,00 кДж-моль) и [2476] @,89 кДж-моль).
Температура плавления A115+3 К) выбрана на основании измерений Кайзера и Содерквиста
[1645] A115+2 К) и Питерсона, Фаттора [1650] A112+2 К), выполненных для образцов с содер-
1 Данные [1241а, 2476] о существовании в интервале 573—723 К промежуточной модификации кальция ((З-Са)
со сложной (неустановленной) структурой не подтвердились в более поздних исследованиях. Смит и др. [2554],
¦исследуя образцы Са с различным содержанием примесей, показали, что промежуточная модификация образуется
только в случае присутствия в Са примесей водорода.
301
Са (к, ж)
Глава 27
Таблица 27.1. Принятые значения термодинамических величин для кальция и его соединений
и жидком состояниях
в твердом
Вещество
Са
СаО
СаН3
Са(ОН)а
CaF2
СаС12
СаВг2
Cal2
CaS
CaSCU
СаСОз
Состояние
КН, Куб. (а
к1, куб. (ч)
ж
к, куб.
ж
Я0 B98,15 К) —
- 11° @)
кДжХ
Хмоль
) 5,73
6,75
к11,ромб. (о) 6,77
к1, куб. (р) -
ж —
к, гекс.
ж
14,16
KlI, куб. (а) 11,64
КИ, куб. (а) —
к1, тетр. (р) —
к, ромб,
ж
к, ромб,
ж
к, гекс.
ж
к, куб.
ж
кП, ромб.
к1, куб.
ж
к, гекс.
ж
15,30 i
17 i
17,97 1
9,01
17,2 i
14,48
Примечание. С° (Т)=а+ЬТ—
CaF2: a d=81,731.10-e; б d=
оо"
CN1
о
С° B98,15 К)
Дж-K-iX
Хмоль
41,6
38,1
41,4
83,4
68,45
08,4
30
145,3
56,5
07
91,71
Коэффициенты в уравнении
для с;(Г)
а
25,94 16,314
— 6,276
— 31
42,05 51,858
— 84
41,00 29,930
— 69
— 75
87,49 89,264
— 153
67,03 122,478
— —100 858,297
— -1428,10
— 99
72,85 71,877
— 89,66
75,06 73,771
— 100
76,99 71,944
— 103
47,45 46,290
- 67,0
99,66 113,05
- 165
— 165
83,47 99,546
— 160
Ь ¦ 10s
22,209
32,384
. 2,439
37,129
33,112
-110,307
173 424,666
655,520
12,719
13,192
18,995
8,370
48,70
27,137
с ¦ Ю-5
Интервал
температур
К
—2,672 298,15—716
—10,460 716—1115
— 1115—3300
9,366 298,15—2900
— 2900—6000
— 298,15—1053
— 1053—1273
— 1273—2000
10,355 298,15—1023
— 1023—1500
26,515 а 298,15—1000
—135 337,356 6 ЮОО-1424
—12 829,927 1424—1691
— 1691—4500
2,510 298,15—1048
—221,29 1048—3900
2,350 298,15—1015
— 1015—3800
0,552 298,15—1056
— 1056—4500
1,188 298,15-2800
— 2800—5500
24,81 298,15—1473
— 1473—1733
— 1733—3000
21,481 298,15—1603
— 1603-2000
:T~*+dT2+eT3 (в Дж-К^-моль).
= —111 377,511-10-6, е=253 68,666.10-".
Т,г
или
Т
1 m
К
716
1115
2900
1053
1273
1023
1424
1691
1048
1015
1056
2800
1473
1733
1603
или
кДжХ
Хмоль
0,93
8,54
52
6,7
22
29
0
30
28,05
29,1
41,8
70
5
28
36
жанием —99,9% кальция. Данные работ [530, 1550, 1551, 2554, 2711, 2891а] менее надежны и
приводят к значениям Тт от 1076 до 1124 К. Энтальпия плавления кальция (8,54+0,40 кДж-моль)
найдена Шиотти и др. [846]. Для теплоемкости жидкого Са принято постоянное значение 31 +
+1 Дж-К-моль, полученное Яухом [1550] из измерений энтальпии в интервале 1122—1223 К.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,3, 1, 2 и 3 Дж-К~1-моль~1 соответственно. При Т < 1000 К термодинамические функции Са,
приведенные в справочниках [105, 1503, 1543, 1656], незначительно (до 0,5 Дж-К~1-моль)
отличаются от данных табл. 783. При 2000 и 3300 К расхождения в значениях Ф° (Т) для [1051
и табл. 783 составляют 0,8 Дж-К~1-моль, для [1543] при 3000 К это расхождение достигает
1,2 Дж-К-моль~1, что обусловлено различием принятых значений теплоемкости Са (ж).
В справочнике ва стандартное состояние кальция принят Са (к) и в соответствии с этим
Д^^Са, к) = 0.
302
Кальций и его соединения
Са(г>
Давление пара кальция Са (к, ж)=Са (г) вычислено для значения ЬГН° (O) — ASH° (Са, к, 0) =
=177,333+0,8 кДж-моль, соответствующего принятой в справочнике энтальпии сублимации
Д8/7°(Са, к, 298,15К) = 177,8±0,8 кДж • моль.
Эта величина является средневзвешенной из найденных по результатам измерений давления
пара кальция и приведенных в табл. 27.2. Она совпадает с рекомендованной КО ДАТА—MCHG
Таблица 27.2. Результаты определений A.SH° (Са, к, 298,15 К) (в кДж-моль)
Авторы
Метод
Д8Я°(Са, к, 298,15 К)
II закон
III закон
Пиллинг, 1921 [2229] Испарение с поверхности, 776—973 К, 6 точек 192±30 176,9±4,0
Хартман, Шнейдер, 1929 [1360] Точек кипения, 1254—1546 К, 11 точек 177±1 177,2+3,0
Приселков, Несмеянов, 1954 [336] Эффузионный, 748—943 К, 11 точек . 169±5 176,9+1,5
Дуглас, 1954 [976] Эффузионный, 807—918 К, 13 точек 199+7 177,1+1,5
Томлин, 1954 [2710] Эффузионный, 801—877 К, 6 точек 192+12 176,9±1,7
Кочеров, Гепьд, 1959 [188, 189, Эффузионный, 823—993 К, 13 точек, расчет по 196 176,4+4
190] уравнению
Смит, Смит, 1959 [2555] Эффузионный, 730—965 К, 9 точек 178+4 179,6±3
Мурадов, Гельд, 1967 [268] Эффузионный, 837-920 К, 4 точки 179+20 177,9±1,7
Мурадов и др., 1965 [267, 269] Эффузионный, 641—837 К, 9 точек 177+8 178,0±1,7
Боданский, Шине, 1967 [718] Точек кипения, 1313—2060 К, 8 точек 177+2 178,2±3,0
Богословский и др., 1968 [41] Эффузионный, 771—1026 К, 14 точек 177+7 174,4+4
Шине и др., 1971 [2455] Точек кипения, 1558—2115 К, 25 точек 175±3 178,2+4,0
Де Мария, Пиаценте, 1974 [943] Протока, 1126—1300 К, 31 точка 160+5 178,9+3,0
[864]. Приведенные в табл. 27.2 погрешности учитывают воспроизводимость экспериментальных
данных, неточность термодинамических функций Са (к, ж) (эта составляющая погрешности
изменяется от 0,6 кДж-моль,для измерений, выполненных при 700—900 К, до 2,6 кДж-моль для
данных, полученных при 1800 К) и отклонение от среднего. Давление пара Са (к, ж) измерялось
также в работах [251, 2399, 2405], однако полученные в них данные имеют плохую
воспроизводимость и не принимались во внимание.
Са (г). Термодинамические свойства газообразного кальция в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 784.
Уровни энергии атома Са, использованные в расчете термодинамических функций,
представлены в табл. 27.3, в которой даны валентные состояния, относящиеся к электронным
конфигурациям . . .4s2, . . AsAp, . . .4s4d, . . .4s4/, . . As3d, . . .3d2 (aP, W, 3F), . . .3dip и . . Ар2. Энергии
этих состояний приняты по результатам спектральных измерений Рисберга [2345]; они
практически совпадают с рекомендацией Мур [2053].
Термодинамические функции Са (г) рассчитаны по уравнениям A.3)—A.6), A.9), A.10),
A. 22)—A. 25). Значения Qai и ее производных находились непосредственным суммированием
по уровням энергии. Погрешности вычисленных значений Ф° (Т) при Т ^ 3000 К не превышают
0,02 Дж-К-моль~1 и обусловлены только неточностью принятых фундаментальных постоянных.
При более высоких температурах погрешности возрастают из-за пренебрежения ридберговскими
состояниями и составляют 1 и 3 Дж-К-моль в Ф° (Т) при 6000 и 10 000 К.
Ранее таблицы термодинамических функций Са (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106], таблицах JANAF [1543], справочнике [2632] (до 3000 К), в
работах Вейц и др. [63] (Т < 3500 К), Хилзенрата и др. [1436] (Т < 10 000 К), Кольского и др.
[1749] (Т < 8000 К), Каца и Маргрейва [1639] до 2000 К. При температурах ниже 5000—6000 К
результаты всех расчетов удовлетворительно согласуются между собой и с данными настоящего
издания (расхождения в значениях Ф° (Т) не превышают 0,02 Дж-К-моль, за исключением
данных [1749, 2632], для которых они достигают 0,05 Дж-К-моль~1). При более высоких тем-
303
Са+ (г)
Глава 27
Таблица 27.3. Уровни энергии атомов Са и Са+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Са . . ,4s2
. . ,4s4p
. . .4s3d
. . ,4s4p
. . .3d4p
. . AsAd
. . .3dAp
. . -4p2
. ..3d4p
Состояние
3Po
8PI
sp2
3Di
xPi
3Fa
1Di
3Ft
ID 2
3Di
3D.2
3D3
3Di
3D2
3DS
3Po
3Pi
8pI
3P\
3P2
V.
CM
0
15 157,901
15 210,063
15 315,943
20 335,360
20 349,260
20 371,000
21 849,634
23 652,304
35 730,454
35 818,713
35 835,413
35 896,889
37 298,287
37 748,197
37 751,867
37 757,449
38 192,392
38 219,118
38 259,124
38 417,543
38 464,808
38 551,558
39 333,382
39 335,322
39 340,080
40 537,893
Pi
1
1
3
5
3
5
7
5
3
5
7
5
9
5
3
5
7
3
5
7
1
3
5
1
3
5
7
Atom
Электронная
конфигурация
Состояние
Ca . . .4p2 4>a
'So
. . .4s4/ 3F2
SFS
3Ft
. . .3d2 3^2
3^4
. . .3d4p xPi
. . .3d2 xDi
3Po
3d
3D
Ca+ . . .4s 25'1/
/2
. . .3d 2DS/
*D4t
¦ ¦ АР 2*Ч
2p3
. . .4d W
/2
2D
At 2F
Tt
CM
40 719,847
41 786,276
42 170,214
42 170,558
42 171,026
42 343,587
43 474,827
43 489,119
43 508,098
43 933,477
47 449,083
48 524,093
48 537,623
48 563,522
0
13 650,19
13 710,88
25 191,51
25 414,40
56 839,25
56 858,46
68 056,91
Pi
5
1
5
7
9
7
5
7
9
3
5
1
3
5
2
4
6
2
4
4
6
14
пературах расхождения возрастают (До 0,5 Дж-К 1-моль~1 в значении Ф° (8000 К) с данными
[1749]), что обусловлено учетом в последней работе ридберговских состояний Са.
Энтальпия образования Са (г)
Д/Дгс
(Са, г, 298,15 К) = 177,8 ± 0,8 кДж • молхГ
соответствует принятой энтальпии сублимации Са (к).
Са+ (г). Термодинамические свойства газообразного положительного иона кальция в
стандартном состоянии в интервале температур 298,15—10000 К приведены в табл. 785.
Уровни энергии иона Са+, использованные в расчете термодинамических функций, приведены
в табл. 27.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. .4s, . . .3d, . . Лр, . . Ad и . . .4/. Энергии этих состояний приняты по результатам
спектральных измерений Эдлена и Рисберга [1011 ], они практически совпадают с рекомендацией Мур [2053].
Термодинамические функции Са+ (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25). Значения (?Э1 и ее производных находились непосредственным суммированием
по уровням энергии, приведенным в табл. 27.3. Погрешности вычисленных термодинамических
функций ниже 9000 К не превышают 0,02 Дн?-К~1-моль~1 и обусловлены в основном неточностью
принятых фундаментальных постоянных. При более высоких температурах возникают
погрешности из-за пренебрежения ридберговскими состояниями, которые достигают 0,1 Дж-К~1-моль
для с; (ю ооо к).
304
Кальций и его соединения
Са2 (г)
Ранее таблицы термодинамических функций Са+ (г) вычислялись в предыдущих изданиях
настоящего справочника [105, 106], таблицах JANAF [1543], а также в работах Грина и др. [1246]
до 50 000 К и Хилзенрата и др. [1436] до 10 000 К. Результаты этих расчетов и данные табл. 785
хорошо согласуются между собой (в пределах 0,01—0,02 Дж-К~1-моль во всем интервале
температур).
Константа равновесия реакции Са+(г)+е (г)=Са (г) вычислена на основании значения
&ГН° @) =—589,831+0,001 кДж-моль, соответствующего принятому потенциалу ионизации Са
/0 (Са)=49 305,99+0,12 см-х=589,831 ±0,001 кДж-моль.
Эта величина принята по работе Брауна и др. [769], которые измерили в поглощении
протяженную (п ^ 79) серию Asnp AJPi); она находится в хорошем согласии с неопубликованными
данными Томкина для этой же серии (п ^ 76), цитированными в работе Камю [813]. Значения
/0 (Са), найденные в других спектральных работах [317, 813, 1176, 2345], хотя и близки к
принятому в настоящем издании, но уступают ему по точности; в справочнике Мур [2053] на
основании более ранних работ рекомендовано заниженное значение этой величины. Принятому
значению соответствует
bfE°(Ga.+, г, 0) = 767,164 + 0,8 кДж • моль.
Са2 (г). Термодинамические свойства газообразного дикальция в стандартном состоянии при
температурах 100—3000 К приведены в табл. Д. 61.
В табл. 27.4 приведены молекулярные постоянные Са2, принятые на основании результатов
исследований [572, 573] системы полос vl1 ??-»• X1 ??. Образование молекулы Са2 в состоянии
Таблица 27.4. Молекулярные постоянные Саг, СаО, СаО+, СаН и CaS
Молекула
Са2
СаО
СаО+
СаН
CaS
Состояние
о3П
АЧ1
В1 2+
с3П
cm
Х2П
Л2 2+
Л 41
а4П е
С2 2+
ХХ2+
Те
ве
ах • 103
De ¦ 10е
см
0
0
8 094°
8 433
10 500 г
11 555,5
25 500
25 991
26 000 г
28 858,19
0
3 000
0а
14 415 в
15 760,7
20 416,5
22 603,7
28 278
0
15 220,79
64,928
733,4
545,7 в
545.7
720 г
718,9
570 г, Д
574,4 Д
560 г
560,9
500
700
1298,34
1333
1285
1248,6
1147,5
1450
462,23
409,04
1,0651 а 0,046113
5,28а 0,444 523
2,54 в 0,337 2 в
2,54 0,337 2
2,1 г 0,406 г
2,11 0,405 92
— 0.39 г
— 0,388 2
4 г 0,33 г
4,0 0,3731
2,1 0,33
4,7 0,40
19,10 4,276 6
20 4,34
20 4,492
21,8 4,284 4 г
32,3 2,51
22 4,58г
1,780 0,176 67
0,818 0,166 66
0,070 277 б
0,337 5
0,21 в
0,21
0,14 г
0,137
0,5 г
0,55
0,3 г
0,32
0,20 ^
0,33 г
9,70
9,5
11,6
1
0,083 7
0,060 5
0,095 214 в
0,657 б
0,52 в
0,52
0,5 г
0,54
0,7 г
0,7
0,7 г
0,7
0,57
0,52
183,7 б
236
216
100 ж
168
0,102 0
0,108 7
Те
к
4,277
1,822
2,092 в
2,092
1,91 г
1,907
1,95 г
1,950
2,11 г
1,989
2,11
1,92
2,002
1,9878
1,954
2,001 Д
2,61
1,935 Д
2,3178
2,3863
Примечание. Ниже все постоянные даны в см~1.
Са2: а шег/е=2,5902-10-3; б «2= —0,73486-10~5; в ft= —0,43146-10"8, ?,=0,10655-lO"9, #„= —0,1365-102
Г1=0,61987-Ю-13, Le= -0,84572-10-».
СаО: a ше«„=4,4-10-2; *> Pi=—3-Ю"8; в см. текст; Г оценка; Д приведено значение ДС,/.
СаН: а у=0,0424; б приведено De, #о=5,49.10-9; в 4=80; г приведено Вв; Д
постоянные компоненты *n_i/s; ж #о=1,4-1О-6.
CaS: a
г3П, АгП (Г,=7000); Ъ3 2
M7W4 000
); iZ+, iZ-, «Z+, 1Д, Ч
ь (Г,=25 000
приведено го;
6 приведены
; »П, Щ (Ге=29 000).
305
Са2 (г)
Глава 27
X1 T,g обусловлено только вандерваальсовским взаимодействием атомов кальция в состоянии
( ) Вб С
g у р
поэтому ее энергия диссоциации мала (см. ниже). Возбужденные электронные состояния Са2 имеют
энергии выше 14000 см [572, 573] и в справочнике не рассматриваются. Приведенные в табл. 27.4
постоянные Са2 в состоянии X1 ?? получены обработкой 430 колебательно-вращательных
термов этого состояния с i?=0-f-7 и /=20-|-162, выделенных Балфуром и Уитлоком [573] при
анализе 47 полос системы А1 ?„—X1 Щ 1.
Термодинамические функции Са2 (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)— A. 95).
Значения <?вн и ее производных вычислялись по соотношениям A. 90)— A. 92) без учета
возбужденных состояний. Величина Qj-f' находилась непосредственным суммированием по всем коле-
Таблица 27.5.
Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см 1),
а также значения г>ша1 и J\im, принятые для расчета термодинамических функций Саг, СаО,
СаО+, СаН, CaS
Коэффициенты
Са2
СаО
а3П
CaO+
CaH
CaS
7V10
Ую-Ю-3
Ует-101
У4о-Ю3
Ум
Уи-Ю1
У21.103
Ум.10» -
У03-10в —0,009 521-
У«.1О7 —0,043146
УИ.1О8 —0,010 655
Уоз-1010 —0,001365
Уи-Ю11 —0,006 199
—0,004 043
0,107 879
0 0 0,809 4 0,843 3 1,050 0 1,155 55 О
0,064 928 0,733193 0,545 703 0,545 566 0,718 815 0,718196 0,500
¦0,106 51 -0,529 200 -0,254 182 -0,250 674 -0,205 720 —0,193 654-0,21
0,025 902 0,504 236 0,003 076-0,026 175-0,089 232—0,103 945 —
— —0,30016 —
— 0,610 604 —
0,046 ИЗ 0,444 523 0,337 2
-0,007 028 -0,03375 -0,021
-0,007 349 0,003 8 —
0,067167 —
-0,162 34 -
0,337 2 0,405 92
—0,021 —0,0137
0,002 8 -
0 0
1,294 535 0,461 603
—1,612 247—0,179 147
—6,616 740 0,028 039
— —0,004174
0,405 92 0,33
—0,013 7 —0,02
4,276 60 0,176 171
—0,973 983 —0,008 335
0,829 762 —
П OOQ ОТО
0,065 9 —0,052 —0,052 -0,054 —0,054 —0,057 -18,37 -0,010 142
— —0 001422
Х10-"
0,-Ю1
а2-105
0,076 133
0,016 467
0,007 298
34
209
157
538
109
392
224
513
103
455
101
457
119
400
78.9
-1 388,5
1,424 315
5,174 678
8,774 071
-3,174 440
18
82
—0,000 900
175
595
Примечание. Ниже все постоянные даны в см.
СаО: !
СаО+:
СаН:
1 Состояние
Те
а Состояние
Те
а Состояние
25 500
3000
Л 41
14 415,1
cm
25 991
15 760,7
' 26000
20 416,5 22 603,7
28 858,19
28 278
CaS:
Состояние
Т,
а8П, Am
7000
14 000
BXX+
15 220,8
32+, JS-,
25 000
41, m
29 000
1 Исследования спектра молекул Са2, изолированных в матрицах азота и инертных газов [522а, 20296, 2029в],
а также спектра лазерной флуоресценции [2425а] не привели к уточнению молекулярных постоянных.
306
Кальций и его соединения СаО (к, ж)
бательно-вращательным уровням энергии, ограниченным предельной кривой диссоциации.
Уровни энергии вычислялись по уравнениям A. 63) и A. 65) с коэффициентами YM,
приведенными в табл. 27.5 вместе со значениями ушах, /lim, и коэффициентами а( в уравнении,
аппроксимирующем предельную кривую диссоциации состояния Xх t^. Значения Yk0 идентичны
соответствующим постоянным из табл. 27.4, поскольку последние удовлетворяют с необходимой точностью
условиям A.50) и коэффициент Yoi вычислен из условия A. 59).
Логрешности вычисленных термодинамических функций обусловлены главным образом
неточностью значения энергии диссоциации Са2 и связанной с ней неопределенностью в общем числе
связанных колебательно-вращательных состояний этой молекулы. Общие погрешности в
значениях Ф° (Г) и S° (T) можно оценить в 1—3 Дж-К^-моль-1.
Ранее термодинамические функции Са2 (г) вычислялись Климовым и др. [167] (до 4500 К)
в приближении «жесткий ротатор — гармонический осциллятор» по оцененным постоянным.
Расхождения в значениях Ф° (Т), приведенных в [167] и табл. Д.61, достигающие 8 Дж -К -моль
при 3000 К, обусловлены использованием в работе [167] приближенного метода расчета и
отличающихся молекулярных постоянных.
Значение энепгии диссоциации
| Do (Ca2)=1040 ±150 см ~ 12,4+2,0 кДж-моль
принято по работе Бальфура и Уитлока [573], которые получили эту величину на основании
экстраполяции колебательных уровней состояния X1 2J на 600 см по уравнению A. 46) с
постоянными из табл. 27.4. Ей соответствует
ДуЯ^Сая^г, 0) = 342,266|+,2,6 кДж • моль.
СаО (к, ж). Термодинамические свойства кристаллического и жидкого оксида кальция в
стандартном состоянии при температурах 100—6000 К приведены в табл. 786.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
27.1. В справочнике за стандартное состояние СаО (к) принята кубическая модификация
(структурный тип NaCl), устойчивая от 0 до температуры плавления 2900 К 2. Принятые значения
5° B98,15 К) и Н° B98,15 К)—Я° @) (см. табл. 27.1) вычислены Гмелиным на основании
выполненных им измерений теплоемкости СаО A—65 К) [1224] и A—300 К) [1226] на образцах высокой
чистоты с точностью —1 % при Т ^> 4 К и 2% при 2 К. Они имеют погрешности 0,4 Дж -К -моль
и 0,06 кДж-моль" соответственно и совпадают со значениями, рекомендованными
КОДАТА—МСНС в качестве ключевых термохимических величин. Результаты других измерений
теплоемкости СаО [2115, 2188] менее точны и не учитывались. Для теплоемкости СаО (к) в
интервале 298,15—2900 К принято уравнение (см. табл. 27.1), выведенное методом Шомейта по
результатам измерений энтальпии в работах Ландера [1808] E64—1176 К), Гроиова и Швите [1271]
C73-1773 К), Эссера и др. [1040] D73-1473 К), Магнуса [1907] C73-1037 К) и Рота и
Бертрама [2375] F90—1125 К). При этом погрешности данных [1808] оцениваются в 0,5%, данных
[1271, 1907] — в 1,5%, а данных [1040, 2375] — в 3%.
Данные по температуре плавления СаО противоречивы. Результаты измерений, проведенных
рядом авторов с разными образцами СаО в различных газовых средах (воздух, О2, Na, H2 и др.),
дали значения Тт в пределах 2840—2900 К. Из этих измерений предпочтение можно отдать
согласующимся между собой данным Домана и др. [970] B898+15 К), Ольшанского [304] B893 +
+30 К) и Панека [2175а] B890+30 К). Результаты других определений [1627, 2072, 2148, 2404»
2477, 2734] приводят к несколько более низким значениям (в пределах 2840—2880 К). Однако
в опубликованных в 1966—1969 гг. работах Фоэкса и др. [1112, 2726] получены значения
температуры плавления СаО в пределах 3173 —¦ 3223 К, т. е. примерно на 300 К более высокие. Фоэкс
проводил измерения температуры кристаллизации жидкого СаО на воздухе термографичееким
методом с использованием солнечной печи, исследовались весьма чистые образцы СаО.
Провести однозначный выбор температуры плавления СаО в настоящее время затруднительно.
Учитывая, что результаты измерений Фоэкса пока не получили подтверждения в последующих
1 Имеющиеся в литературе данные (см. [1899]) о фазовых переходах в СаО(к) при 700—1000 К не были
подтверждены в последующих работах. Согласно рентгенографическому исследованию [807] в этом интервале
температур имеет место только термическое расширение кубической решетки.
307
СаО (г) Глава 27
работах (Ногучи [2148], 2858+20 К; Панек [2175а], 2890+30 К), в справочнике для температуры
плавления принято значение 2900 К, выбранное по работам [304, 970, 2175а]. Пределы
погрешности этого значения могут составлять —50 К и -j-ЗОО К. Энтальпия плавления СаО E2 +
+5 кДж-моль) принята по результатам расчетов [1531,1650] изданных по диаграммам
состояния двойных систем с СаО. Теплоемкость СаО (ж) оценена на основании экспериментальных
данных по теплоемкости ВеО (ж); ее погрешность может достигать 8 Дж-К~1-моль~1.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 6000 К оценены в 0,3,
0,6, 4 и 12 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
СаО (к), приведенными в справочниках [105 и 1543] и табл. 786, не превышают 1 Дж-К~1-моль
в значениях Ф°(Г). Для СаО (ж) соответствующие расхождения возрастают до 3 Дж-К~1-моль~1
ввиду разных оценок теплоемкости СаО (ж).
Принятая в справочнике энтальпия образования
AfN°(CaO, к, 298,15 К) = —635,09 + 0,90 кДж • моль
основана на измерениях энтальпии сгорания кальция в кислороде в работе Хубера и Холли [1490].
Она совпадает с величиной, рекомендованной КОДАТА—МСНС [864]. Менее точные значения
энтальпии сгорания кальция в кислороде получены Мутманом и др. [2089] (—600,5 кДж-моль)
и Кучеровым и др. [191 ] (—640,2 кДж -моль). Энтальпия образования СаО (к) может быть также
вычислена из термохимического цикла, включающего энтальпии растворения СаО (к) и Са (к)
в соляной кислоте. Эти измерения проведены соответственно в работах [554, 670, 878, 879, 888,
1179, 1964, 2381, 2483, 2666, 2695, 2717, 2828, 2887] и [191, 685, 686, 692, 1022, 1187, 1323, 1324,
1325, 1326]. Критический анализ и пересчеты результатов этих измерений проводились авторами
справочников [406, 681, 2368], а также при подготовке настоящего издания и привели к значению
—635,5 кДж-моль, которое совпадает в пределах погрешностей измерений с полученным в
работе [1490], но уступает последнему по точности.
Давление пара оксида кальция в реакции СаО (к, ж)=СаО (г) вычислено с использованием
значения ЬГН° (О) = Д,?Г° (СаО, к, 0) =675,885 +15 кДж-моль, соответствующего принятым
в справочнике значениям энтальпии образования СаО (к) и энергии диссоциации СаО (г).
СаО (г). Термодинамические свойства газообразного оксида кальция в стандартном состоянии
при температуре 100—10 000 К приведены в табл. 787.
В табл. 27.4 приведены молекулярные постоянные СаО, принятые на основании спектральных
исследований и квантовомеханических расчетов. В электронном спектре СаО исследованы четыре
системы полос, связанные с пятью синглетными состояниями: АгП ~> X1 ?+ [1082], В1!,*-*¦
-+Х1 Е+ [743, 757, 758,1080, 1508,1509,1916, 1999], СХП <* X1 Е+ и D1 Е+ ^ Xх ?+ [62, 757, 758,
1793, 1916, 1999] *. Квантовомеханический расчет в приближении Хартри—Фока с эмпирической
оценкой корреляционной энергии [821] показал, что молекула СаО должна иметь низколежащие
триплетные состояния а3П и bs ?+, изоконфигурационные состояниям ЛШиВ'Е* соответственно.
По аналогии с молекулой MgO (см. гл. 26) следует ожидать, что состояние с3П и группа
состояний ХД, 3Д, 1 2~ и d3 ?+, изоконфигурационных состояниям СХП и D1 Е+ соответственно, также
должны быть связанными. Наличие связанных состояний а3П и d3 ?+ у молекулы СаО
подтверждено анализом возмущений в системах В1!,*—Хх?+ [1080] и С1]!—Х*Е+ [1793].
В табл. 27.4 приведены данные для 12 валентных состояний молекулы СаО 2. Колебательные
постоянные в состояниях X1 ?+ и В1 ?+ приняты по работе Брюэра и Хауга [743], которые
рассчитывали их на основании результатов собственных измерений и данных Хультина и Лагерквиста
[1509] по началам полос системы В1!,*—Хх?+ (v" ^14, v' <^ 12). Вращательные постоянные
1 Система полос в зеленой области около 5500 А, отнесенная в ряде работ [68, 1844] к молекуле СаО, согласно
[1498, 1541] принадлежит молекуле СаОН. В справочнике принята символика электронных состояний молекулы
СаО, отличающаяся от используемой в литературе и аналогичная принятой в справочнике для MgO, SrO и ВаО.
? Тип основного электронного состояния СаО (ХХ2+) не вызывает сомнения [60, 62, 546, 1080], однако в ряде
работ на основании неверной интерпретации спектров [750] или из анализа данных по равновесию реакций с
участием СаО (г) [1621 ] делался ошибочный вывод, что эта молекула имеет триплетное основное состояние C2 или
3П).
308
Кальций и его соединения СаО (г)
в основном состоянии X1 Е+, приведенные в табл. 27.4, получены Филдом [1080] совместной
обработкой величин Вв, определенных в работах [1509] (у=0-|-3) и [743] (v=5-^-8]\ они хорошо
согласуются с результатами исследования микроволнового спектра СаО [901 ] B?0=0,442 796 406 см»
2^=0,439 483 748 см). Вращательные постоянные в состоянии В1!,* приняты по данным [1509].
Колебательные постоянные в состоянии AxYi приняты по работе Филда и др. [1082], которые
выполнили анализ 20 полос системы АЩ—ХХЕ+ (i/=9-^21); вращательные постоянные получены
авторами [1082] из анализа некоторых полос указанной системы и данных по возмущениям в
системе 51?+--Х1Е + [1080, 1509]. Молекулярные постоянные в состоянии а3П получены Филдом
[1080] путем анализа возмущений в системе 5ХЕ+—Х1Е+ [1509] G*0=8225+25 см'1, ше=556,2 +
±10 см, ю,^=3,30+0,22 см, Яе=0,3353 +0,0068 см, 0^=0,0015+0,0007 см). В табл. 27.4
для состояния а3П приведены те же колебательные и вращательные постоянные, что для
состояния ЛХП. Для последнего они получены Филдом и др. [1082] из прямых экспериментальных
данных, причем оказалось, что они заметно отличаются от найденных Филдом ранее в работе [1080]
из возмущений. В то же время анализ Филда [1080] и квантовомеханический расчет Карлсона
и др. [821] показали, что постоянные в обоих состояниях должны практически совпадать. Энергия
состояния а3П, полученная Филдом в работе [1080], скорректирована с учетом принятого
значения Те {А1!!) и величины Те (А1!!)—Те (а3П)=339 см, найденной в [1080]. Колебательные и
вращательные постоянные СаО в состояниях С1!! и D1 Е+ приняты на основании анализа четырех
полос (i/ < 1, v" < 3) системы СШ—X1 ?+ и шести полос (i/ < 3, v" <; 4) системы D1 ?+—X1 Е+
в работе Лагерквиста [1793]. Энергия возбуждения состояния Ъ3 Е+ оценена по величине
расщепления изоконфигурационных состояний 5XE+ и Ъ3 Е+ A050 см) [821] и принятого значения
Те (В1 ?+); ее погрешность не превышает 1000 см. Энергии возбуждения состояния с3П и группы
состояний ХД, 3Д, х ?", d3 E+ оценены из сопоставления величин расщепления
изоконфигурационных состояний в молекулах MgO [2446] и СаО [821], а также на основании наличия
возмущений в системе СЧЗ—ХХЕ+ [1793]. Погрешности оцененных значений могут достигать 2000—
3000 см. Колебательные и вращательные постоянные состояний Ь3 Е+ и с3П приняты равными
постоянным изоконфигурационных состояний.
Термодинамические функции СаО (г) вычислены по уравнениям A. 3)—A. 10) и A. 93)—A. 95).
Величина QBS и ее производные находились по соотношениям A. 90)—A. 92) с учетом 11
возбужденных электронных состояний. Для состояний с Т\ > 25 000 см принималось, что Q$x- вр =
—(PilPa)Qm]t вр- Статистические суммы по колебательно-вращательным уровням пяти состояний
(Х1Е+, а3П, А1!!, Ь3 Е+, ВХЕ+) и их производные рассчитывались непосредственным
суммированием по колебательным уровням и интегрированием по / с помощью уравнений типа A. 82). В
расчете учитывались все уровни этих состояний со значениями / ^ Jms.%< „", последние находились
по соотношению A. 81). Энергия колебательно-вращательных уровней рассчитывалась по
уравнениям A. 62) и A. 65) с коэффициентами, приведенными в табл. 27.5 вместе со значениями vmgx
ж /j,m. Значения Yk0 получены по постоянным из табл. 27.4 с учетом условий A. 50), для
соблюдения которых в уравнение A. 65) для состояний X1 Е+ и А1!! введены дополнительные члены
с коэффициентами У40 и У50, а для состояний а3П, Ъ3 Е+ и В1 Е+ — члены с коэффициентами У3о-
Постоянные Y2i Для состояний Хх?+ и АЩ определены из условия Bv _1_1 = 0.1Мультиплетность
и вырождение состояний а3П, АХП и Ъ3 Е+ учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при температурах до 1000 К в
основном обусловлены неточностью фундаментальных постоянных. При более высоких температурах
становятся существенными погрешности, связанные с неточностью оцененных энергий ряда
состояний (главным образом Ъ3 Е+) и пренебрежением ридберговскими состояниями. Общие
погрешности в значениях Ф° (Т) при 298,15, 3000, 6000 и 10 000 К могут достигать 0,02, 0,1, 0,5
и 1 Дж-К-моль" соответственно.
Ранее термодинамические функции СаО (г) вычислялись в предыдущем издании настоящего
справочника [105], таблицах JANAF [1544], а также в работах Вейц и др. [63] (до 6000 К), Брю-
эра и Розенблатта [745] (до 3000 К) и Шнейдера [2463] (<?1Ш для 1000 < Т < 6000 К). Данные
[1544] и табл. 787 согласуются в пределах 0,1 Дж-К^-моль. Данные [63, 105 и 2463] до 1000 К
практически совпадают с приведенными в табл. 787; при более высоких температурах расхождения
монотонно возрастают, достигая 10 Дж-К-1-моль~1 в значении Ф° F000 К), что связано с
пренебрежением в расчетах [63, 105 и 2463] состояниями а3П, АЩ и 63Е+. Расхождения значений
309
СаО (г)
Глава 27
Ф° (Т), приведенных в [745] и табл. 787, составляют 16 Дж-К 1-моль -1 во всем интервале
температур.
Константа равновесия реакции СаО (г)=Са (г)+О (г) вычислена на основании принятого
в справочнике значения энергии диссоциации
ДД° @) = Do (СаО) = 380 + 15 кДж • моль *» 31 800 ± 1300 см.
Энергия диссоциации СаО в течение многих лет была предметом многочисленных измерений,
приводивших к противоречивым результатам (см. [105, 1183]). В табл. 27.6 представлены
соответствующие данные, пересчитанные с использованием вспомогательных величин, принятых в
справочнике; приведенные погрешности характеризуют воспроизводимость измерений, а также
неточности сечений ионизации (для масс-спектрометрических работ), вероятностей переходов (для
спектрофотометрических измерений и работы [1034]) и других термохимических величин.
В исследованиях скорости испарения с открытой поверхности [855, 2203] не учитывалась
диссоциация СаО (к) с образованием Са (г)+О2 (г), которая должна была доминировать в этих
условиях (см. [105]). От этих ошибок может быть свободна работа [551], однако отсутствие в
публикации первичных данных и большая погрешность значения, найденного по II закону,
существенно обесценивают ее результат. Спектрофотометрические исследования в пламенах [58, 59,
60, 1497, 1619, 1621] являются недостаточно надежными, так как в работах [58, 59, 60, 1497]
Таблица 27.6. Результаты определений Do (СаО) (в кДж-моль)
Авторы
Метод
Do(СаО)
II закон
III закон
Классен, Вилеманс, 1933 [855] Испарение с открытой поверхности, СаО (к)= — 478+10
=СаО(г), 1617—1728 К, Д,Я° @)=578±10 (III)
Пельхович, 1954 [2203] То же, 1550-1800 К, Д8#° @)=728 (II) 328 —
Бабельёвский, 1963 [551] Эффузионный, с масс-спектрометрической реги- 445+60 —
страцией, 2165—2259 К, Д„#° @)=611±60 (II)
Хульдт, Лагерквист, 1954 [1497] Спектрофотометрическое исследование равновесия — 462
СаО (г)=Са (г)+О (г) на основании измерения
р (Са) в пламенах ацетилена с воздухом, 2240—
2430 К
Вейц, Гурвич, 1956 [58, 59, 60] То же, в пламенах С2Н2, СО и Н2 с воздухом и — 460+20
кислородом, 2452—3210 К, 10 точек
Калфф и др., 1965 [1621] Тоже, в пламенах CO+N2+O2, 2156—2450 К, 404±160 420+7
6 точек
Калфф и др., 1973 [1619] Тоже, в пламенах CO+N2O+HaO; пересчитано 750±200 414±20
с учетом образования СаОН (г) и Са(ОНJ, 2684—
2857 К, 12 точек
Беляев и др., 1979 [34а] Спектрофотометрическое исследование реакции 376±20 —
Са (г)+О2 (г)=СаО (г)+О (г) по отношению
интенсивности ИК-полос СаО и линия Са в
спектрах бедных пламен CH4+O2+N2, 2160—2900 К,
48 точек
Колен и др., 1964 [873] Масс-спектрометрический, Са (r)-!-SO (г)= — 405+35
=СаО (r)+S (г), 2180-2386 К, 3 точки,
ДгЯ°@)=112+30(Ш)
Дроварт и др., 1964 [987] Масс-спектрометрический,Са (г)+О3 (г)= — 378+13
=СаО (г)+О (г), 2168—2410 К, 4 точки, Дг#°@)=
=—116,1+8,5 (III)
Фарбер, Сривастава, 1976 [1060] Масс-спектрометрическпй, СаО (г)=^Са (г)+ 387+16 385+7
+1/!,О,(г), 2080—2206 К, 8 точек, Дг#° @)=
=138,6+7 (III)
То же, СаО (к)=СаО (г), 2080 К, 1 точка, — 369+7
Д„Я° @)=687±7 (III)
Эягелке и др., 1976 [1033а, 1034] Исследование хемилюминесценции при образова- >460±15
нии СаО в скрещенных пучках Са и СЮ2
310
Кальций и его соединения СаО+ (г)
не были приняты меры для устранения градиента температуры по сечению пламени и, кроме того,
во всех работах, включая [59 и 1619], в большинстве пламен образовывались значительные
количества гидроксидов кальция (приведенные в табл. 27.6 данные для работ [58, 59, 60 и 1619]
пересчитаны с учетом этих соединений). От указанных недостатков свободно исследование Беляева
и др. [34а], в котором непосредственно измерялась зависимость от температуры величины,
пропорциональной константе равновесия соответствующей обменной реакции. Данные [34а]
находятся в хорошем согласии с результатами масс-спектрометрических исследований Дроварта и др.
[987] и Фарбера и Сривастава [1060], они также не противоречат данным Колена и др. [873].
Причины, приведшие к резкому расхождению величины, полученной при исследовании
образования СаО в пучках [1033а, 1034] с данными [34а, 987, 1060], остаются непонятными.
Принятое значение основывается на данных [34а, 987, 1060]; ему соответствует
Д/й'°(СаО, г, 0) = 44,116±15 кДж . моль,
СаО+ (г). Термодинамические свойства газообразного положительного иона оксида кальция
в стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 788.
Спектры молекулы СаО+ экспериментально не исследовались. Приведенные в табл. 27.4
молекулярные постоянные СаО+, основаны на приближенных оценках. По аналогии с молекулами
MgO+ [2446] и КО [2577] в справочнике принимается, что СаО+ имеет основное электронное
состояние Х2П (конфигурация . . . о2тс3) и низколежащее возбужденное состояние А2 ?+
(конфигурация . . . аи4). Энергия возбуждения состояния А2 Е+ оценена на основании энергий состояний
3П, *П (. . . <з2тс3а) и 3 ?, х S (. . . ап*а) в молекуле СаО. Эти состояния являются первыми членами
ридберговских серий, сходящихся соответственно к Х2П и А2 ?+ состояниям СаО+. Погрешность
принятого значения Тв (А2 Е+) может составлять 2000 см. Постоянные и>е и Ве, приведенные
в табл. 27.4, оценены по постоянным соответствующих возбужденных состояний молекулы СаО;
их погрешности могут достигать 50 и 0,02 см соответственно.
Термодинамические функции СаО+ (г) рассчитаны по уравнениям A. 3)—A. 10) и A. 93)—
A. 95). Величина (?вн и ее производные вычислялись по соотношениям A. 90)—A. 92) с учетом
одного возбужденного состояния Л22+ в предположении, что Q[fJ_ вр = 1/2<?Й). вр- Статистическая
сумма по колебательно-вращательным уровням состояния Х2П и ее производные рассчитывались
непосредственным суммированием по колебательным уровням и интегрированием по / с помощью
уравнений типа A. 82). В расчете учитывались все уровни этого состояния со значениями / <1
<; /шах> „; последние находились по уравнению A. 81). Энергия колебательно-вращательных
уровней вычислялась по уравнениям A. 64а) и A. 65) с коэффициентами YM, приведенными вместе
со значениями vmi7L и /1Ш в табл. 27.5. Величина Y03 вычислялась из условия A. 59). Мультиплет-
ность и вырождение колебательно-вращательных уровней обоих состояний учитывались
статистическими весами.
Погрешности вычисленных термодинамических функций во всем интервале температур
определяются главным образом неопределенностью значения энергии возбуждения состояния А2 Е+
и отсутствием экспериментальных данных о молекулярных постоянных СаО+, при низких
температурах — дополнительные погрешности обусловлены отсутствием данных о постоянной спин-
орбитального взаимодействия состояния Х2П. Общие погрешности в значениях Ф° (Т)
оцениваются в 1, 3, 4 и 5 Дж-К^-моль при 298,15, 3000, 6000 и 10 000 К соответственно.
Ранее таблица термодинамических функций СаО+(г) не публиковалась.
Константа равновесия реакции СаО+ (г)+е (г)=Са (г)+О (г) вычислена для значения Дг#° @)=
= —250+54^кДж-моль, соответствующего принятому в справочнике потенциалу ионизации
/0|(СаО)=630 +50|кДж -моль.
|Эта величина принята на основании измерений, выполненных методом электронного удара
в работах Дроварта и др. [987] и Фарбера и Сривастава [1060], в которых получено совпадающее
значение F,5 эВ).
Принятой величине соответствуют
Д/Я°(СаО+, г, 0) = 674,116 + 52 кДж моль,
Do (СаО+) = 339,831 ± 52 кДж • молья* 28 400 ± 4500 см.
311
СаН (г) Глава 27
СаН (г). Термодинамические свойства газообразного гидрида кальция в стандартном состоянии
при температурах 100—6000 К приведены в табл. 789.
В табл. 27.4 представлены молекулярные постоянные СаН, принятые на основании
результатов исследований электронных спектров этой молекулы. В спектре СаН наблюдались 13 систем
полос: А2П <* X2 ?+ [656, 657, 1292, 1504, 1859], В2 Е+ ^ X2 ?+ [656, 657, 657а, 1504, 1859, 2811],
?2П->Х2Е+ [2820], D* ? DП) -* X2 Б+ [1292, 1293], С2Е+^Х2Е+ [1292, 1294, 2075, 2811],
G2S+<- Х22+ [1644, 1684, 1689], F2Z,+ <- Х2?+ [1013, 1680, 1689], К2 Е+ -* X2 Е+ [1643, 1690],
?2П->Х2Е+ [1643], /2П«-Х2?+ [1644, 1690], М2А <- Х2Е+ [1644], #2?+<-Х2?+
[1689] и /2Е+<- Х2Е+ [1689]. В работах [1643, 1644] состояния К2 Е+, /Л1, G2?+, /2П и МЧ
интерпретированы как ридберговские состояния, состояния F2 Е+ GТО=36797 см), Я2Е+(Г0=
=38798 см) и /2 Е+ G10=39477 см), по-видимому, также не могут быть скоррелированы с
валентными состояниями атома Са, соответствующими конфигурациям . . .4s2, . . Asip и . . .4s3d.
Поэтому все они не представлены в табл. 27.4. Состояние X2 Е+ коррелирует с пределом Са A*?)+
-f-H BiS), а состояния -42П и В2 Е+ с пределом Са CР)+Н BS). С последним должны быть связаны
также квартетные состояния 4П и *Е+. Грундстром [1292, 1294, 1296] показал, что Л-состояшге
СаН также должно быть связано с пределом Са C.Р)+Н (S2), что невозможно для состояния 2 Е+,
и предположил [1298], что оно является компонентой 4IL/2 квартетного состояния. Поскольку
молекула MgH (см. главу 26) имеет связанное 4П состояние с гв-> ге (X2 Е+) и такое же
соотношение справедливо для D-состояния СаН, в справочнике это состояние рассматривается как
квартетное состояние а4П.
Молекулярные постоянные СаН в состоянии X2 Е+ приняты по работе Берга и Клининга [657];
они получены в результате совместной обработки полос 0—0, 1—1, 1—0, 2—2 и 2—1 системы
АШ—X2 Е+ и полос 0—0, 0—1, 1—1, 1—2, 2—2 системы В2 Е+—X2 Е+. Эти постоянные
удовлетворительно согласуются с найденными Грундстромом [1292] из анализа системы а — X2 Е+ (v" ^ 3).
Колебательные постоянные в состоянии А2П получены Грундстромом [1292] по кантам полос
с v' ^2; вращательные постоянные рассчитаны из данных, найденных Хультеном [1504] на
основании анализа вращательной структуры полос 0—0 и 1—1 системы A2tl—X2 ?+. Вращательные
постоянные в состоянии В2 Е+ приняты по работе Уотсона [2811], колебательные — по работе
Грундстрома [1292]; постоянные в состоянии 2?2П получены Уотсоном и Вебером [2820].
Молекулярные постоянные в состоянии я4П_ч2 найдены Грундстромом [1292] из анализа
вращательной структуры семи полос системы а—X (i/ ^ 2). Постоянные СаН в других компонентах
состояния 4П приняты равными постоянным в состоянии а4П_1/2. Мультиплетное расщепление
состояния 4П оценено на основании того, что в структуре полос а—X с i/=l и 2 наблюдались
дополнительные вращательные линии, интерпретированные как переходы из неизвестного
состояния. Предположение, что это неизвестное состояние является компонентой 4П1/2, позволяет
принять постоянную спин-орбитального взаимодействия равной Д(?1/2 состояния 4П_1/2. В табл. 27.4
для я-состояния приведена энергия центра мультиплета, рассчитанная в этом предположении.
Постоянные СаН в состоянии С2 Е+ приняты по работе Грундстрома [1292, 1294]; это состояние
предиссоциировано, обрыв вращательной структуры в испускании наблюдается при г;=0 и К=11.
В спектре поглощения наблюдаются диффузные полосы с v ^ 5.
Термодинамические функции СаН (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBB и ее производных находились по уравнениям A. 90)—A. 92) с учетом пяти
возбужденных электронных состояний в предположении, что Q^o\, вр =¦ (PilPx)Qif^. „р- Значения Q[fJ. вр и ее
производных вычислялись по уравнениям A. 70)—A. 75) непосредственным суммированием по
колебательно-вращательным уровням энергии. В расчетах учитывались все уровни энергии,
ограниченные предельной кривой диссоциации этого состояния. Колебательно-вращательные
уровни энергии состояния X2 Е+ вычислялись по уравнениям A. 65), A. 61). Значения
коэффициентов Ykl в этих уравнениях, использованные в расчетах, приведены в табл. 27.5. Коэффициент
У3о определялся из условий A. 50), коэффициенты У21 и Y31 — из условий A. 57), а коэффициенты
Y03 и У04 — из условий A. 59). В табл. 27.5 приведены также коэффициенты а{ в уравнении,
аппроксимирующем предельную кривую диссоциации состояния X2 Е+, величины ушах и /lim. Муль-
типлетность и вырождение электронных состояний СаН учитывались соответствующими
статистическими весами.
Постоянные молекулы СаН известны с удовлетворительной точностью, поэтому погрешности
вычисленных термодинамических функций при Т < 3000 К обусловлены лишь неточностью
фундаментальных постоянных. Погрешности из-за приближенного учета возбужденных электронных
312
Кальций и его соединения СаЩ (к, ж)
состояний и неполноты данных об этих состояниях существенны только выше 4000 К. Общие
погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 0,02, 0,03 и 0,2 Дж -К -моль
соответственно.
Ранее термодинамические функции СаН (г) вычислялись в справочнике Хара и др. [1336]
и работе Вейц и др. [63] (до 3500 К). В работе Климова и др. [167] приведены коэффициенты
полинома, аппроксимирующего значения Ф° (Т) в интервале 500—4500 К, а в работе Шнейдера
[2463] — полиномы, аппроксимирующие значения QB1I и In QBB в интервале 1000—3700 К.
Наилучшее согласие с данными настоящего справочника имеет место для работы [63] (в пределах 0,06 и
0,15 Дж-К~1-моль в значениях Ф° (Т) и S° (T)). Расхождения с данными [1336] значительно
больше и достигают 0,7 Дж-К^-моль в S0 C000 К) и 0,9 Дж-К^-моль в С°р B000 К) из-за
использования этими авторами приближенного метода расчета и пренебрежения возбужденными
состояниями. Расхождения с данными [2463] и особенно [167] значительны и достигают
2 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции СаН (г)=Са (г)+Н (г) вычислена на основании принятой
в справочнике энергии диссоциации
Ar#°@) = D0(CaH) = 13 600 + 150 см да 162,7 + 1,8 кДж • моль.
Эта величина получена в результате совместной обработки данных по предиссоциации молекул
СаН [1292, 1294, 1296, 2075] и CaD [1294, 2811] в состоянии С2 Е+ (v=0, Л=11 и v=0, Д = 18
соответственно) и предположения, что предиссоциация связана с пределом СаC/I)+Н BS) или
с пределом Са CР2)+Н B5).
Принятое значение энергии диссоциации подтверждается результатами исследования процесса
испарения дигидрида кальция [1019], согласно которым Do (СаН) ^ 16 800 см, и согласуется
с величинами, предложенными ранее Гейдоном [1183] и Герцбергом [1392]. Ему соответствует
ЬуН°(СаЕ, г, 0) = 230,667 + 2,0 кДж • моль.
СаН3 (к, ж). Термодинамические свойства кристаллического и жидкого дигидрида кальция
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 790.
Значения постоянных, принятых для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние СаН2 (к) в интервале 0—1053 К
принимается ромбическая модификация (а), структурный тип РЬС12 [661], а при 1053—1273 К —
кубическая модификация (р), структурный тип CaF2 [2014]. Какие-либо экспериментальные данные
по термодинамическим свойствам СаН2 (к, ж) в литературе отсутствуют, за исключением весьма
ненадежных данных по теплоемкости в узком интервале температур G0—86 К) [1315] и данных
по температурам превращения и плавления Cali2 [2215]. При Т <^ 298,15 К термодинамические
функции СаН2 (к) вычислены по значениям теплоемкости, оцененным при подготовке
справочника; принятые значения теплоемкости в интервале 70—86 К примерно на 10% выше
экспериментальных данных Гюнтера [1315], поскольку именно такую величину имеют расхождения данных
[1315] для MgO и СаО с результатами более точных измерений.
Погрешности принятых значений S° B98,15 К) и #° B98,15 К) — Н° @) (см. табл. 27.1)
оцениваются в 4 Дж-К-моль и 0,3 кДж-моль соответственно. При Т > 298,15 К для
теплоемкости а-СаН2 принято уравнение, выведенное по оцененным значениям Cv B98,15 К) и С°р A053 К)
(см. табл. 27.1).
Температура превращения A053+10 К) принята по данным Питерсона и Фаттора [2215] для
образца чистоты 99,94% СаН2, а энтальпия превращения F,7+2 кДж-моль) — по расчетам
Мюллера и др. [2069] из данных по давлению диссоциации а- и Р-СаН2. Температура плавления
СаН2 A273 К) принята по рекомендациям [17, 2069], а энтальпия плавления B2+5 кДж-моль)
оценена в предположении равенства между энтропией плавления MgH2 и суммой энтропии
полиморфного превращения и плавления СаН2. Значения теплоемкостей |3-СаН2 и СаН2 (ж) оценены
с учетом экспериментальных данных для теплоемкости твердого и жидкого гидрида лития [661,
2782]; их погрешности составляют 10 и 15 Дж-К-моль соответственно.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 4, 10
и 20 Дж-К-моль соответственно. Расхождения между термодинамическими функциями
а-СаН2, приведенными в табл. 790 и в справочнике [578], не превышают 3 Дж-К"'
313
СаОН (г)
Глава 27
нии Ф° A000 К). Расчеты термодинамических функций E-СаН2 и СаНа (ж) в справочной литературе
отсутствуют.
Константа равновесия реакции СаН2 (к)=Са (г)+2Н (г) вычислена с использованием значения
ДГЯ° @)=778,973+8,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(CaE2, к, 298,15 К) = —177 + 8 кДж • моль.
Эта величина вычислена как средневзвешенное из приведенных в табл. 27.7 результатов тер-
М01и1Ш§СК11 ЮМереПШ И 1ШеД9Рании равновесия реакции диссоциации СаН2 (к). При оценке
погрешности данных [764, 1022, 1323, 1625], помимо указанной в табл. 27.7 воспроизводимости
Таблица 27.7. Результаты исследований А/-Н"" (СаНг, к, 298,15 К) (в кДж-моль)
Авторы
Метод
bfH° (CaH2, к, 298,15 К)
II закон
III закон
Гунц, 1905 [1323]
Бренстед, 1914 [764]
Каменский, 1926 [1625]
Эрлик и др., 1963 [1022]
Мольденхауэр, Ролл-Хансея, 1913
[2049]
Бронстед, 1914 [764]
Краус, Хард, 1923 [1771]
Реми-Женнет, 1929 [2326]
Херд, Уокер, 1931 [1513]
Джонсон и др., 1939 [1587]
Тредуэлл, Стичор, 1953 [2729]
Кертис, Кпотти, 1963 [914]
Калориметрическое измерение энтальпии
растворения Са (к) и СаН2 (к) в соляной кислоте,
283К
То же, 291 К
То же, 290 К
То же, 298 К
Исследование равновесия СаН2 (к)—Са (к)+
+Н2 (г) статическим методом, 1053—1300 К,
8 точек
То же, 914—1020 К, 19 точек
То же, 1007—1258 К, 4 точки
То же, 1088—1238 К, 10 точек
То же, 991—1210 К, 17 точек
То же, 1023—1167 К, 27 точек
То же, 1053—1133 К, 992—1214 К, 31 точка
То же, 851—1053 К, расчет п# уравнению
То же, 1053—1173 К, расчет по уравнению
-193,1
-176,
,7+5,5
—176,2
—186.
—175+23
194,9+5,5
194,8+4.3
—230±15
—215+6
_
—181+10
—178,4
—170,8
,2+2,1
—177,9±1,4
—178,2±0,3
-178,0+0,7
—173,7±1,7
-176,1 ±0,8
—172,8+3,5
—175,4+5,0
—179,4+0,1
-180,2+1,0
измерений, учитывались также оцененные систематические погрешности определения энтальпии
реакции растворения кальция и гидрида кальция, составляющие (по аналогии с ВаН2) около
8 кДж-моль. При анализе данных, полученных при исследованиях равновесия реакции с
участием СаН2 (к, ж), учтена неточность термодинамических функций СаН2 (к); соответствующая
погрешность в энтальпии образования составляет 10—15 кДж -моль. Все приведенные в табл. 27.7
данные, за исключением работы [1323], согласуются между собой в пределах их погрешностей.
СаОН (г). Термодинамические свойства газообразного гидроксида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 791.
Молекулярные постоянные, использованные для расчета термодинамических функций СаОН,
приведены в табл. 27.8. Экспериментальные данные об этих постоянных отсутствуют. По
аналогии с ВеОН, MgOH и гидроксидами щелочных металлов для молекулы СаОН в основном
электронном состоянии X2 ?+ принята линейная конфигурация (Гейдон [1181] отметил, что структура
электронного перехода в спектре СаОН позволяет предполагать, что в одном из комбинирующих
состояний эта молекула имеет нелинейную структуру). Оценка геометрических параметров и
частот СаОН выполнена, так же как и для ВеОН (см. гл. 25). Приведенный в табл. 27.8 момент
инерции рассчитан в предположении, что г (О—Н)=0,96+0,02 А и г (Са—О)=2,00+0,05 А; его
погрешность составляет 1 • 10~39 г -см2. Принятая величина ^са-о согласуется со значением 587 см,
полученным Гейдоном [1181] при изучении системы ло^с' cli^muyMZ^55Z6"ATf[^^mm^m
принятых значений частот оцениваются в 50—100 см. На основании работ Гейдона [1181, 1182]
и Хумана, Зегерса [1510], а также по аналогии с изоэлектронной молекулой CaF, в справочнике
314
Кальций и его соединения
СаОН+ (г)
Таблица 27.8. Значения молекулярных постоянных, а также о и pg, использованных для расчета
термодинамических функций СаОН, СаОН*, Са(ОН)г, CaF2, СаС12, СаВгг, Са12
Молекула
СаОНа
СаОН+
Са(ОНJ
CaF2
СаС12
СаВг2
Cals
СаОН: а
Са(ОНJ: а
CaF2: a
Электронное
состояние
v
1
Z22+ 570
Х^+ 530
Х1А1 475
ХЫ-у 490
Х1Ц 260
Х''Ц 160
Х*Ц 115
'
305 B)
305 B)
110
120
75B)
60B)
50B)
Го (Л)=1Ъ 000 см, pi=4: To (B)=iS
1рпведено значение IjJ^t
Приведено значение 1AIB
v
СМ
3700 — —
3700 — —
550 3700 B) 305 D)
575 — —
395 — —
330 — —
290 — —
000 гаг1, рв=2.
Гс-10115 г3-см6.
Гс-10115 r3-cs
Iе.
/ • 1039
г -см2
8,5
9
6а
8,7 а
74
1,9-Ю2
3,5-Ю2
о
1
1
2
2
2
2
2
Рх
2
i
1
1
1
1
1
принимается, что СаОН имеет два дублетных возбужденных электронных состояния; поскольку
анализ структуры соответствующих систем полос не проводился, энергии этих состояний могут
иметь погрешность в 1000 см.
Термодинамические функции СаОН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных термодинамических функций обусловлены неточностью принятых молекулярных
постоянных, в том числе структуры этой молекулы, а также приближенным характером расчета. Они
составляют 2, 4 и 6 Дж^К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции СаОН (г) вычислялись в таблицах JANAF [1544],
соответствующие расхождения составляют 2—7 Дж-К-моль в значениях Ф° (Г) и S° (T); они
обусловлены главным образом различием в оценке величины v2.
Константа равновесия реакции СаОН (г)=Са (г)+О (г)+Н (г) вычислена с использованием
значения Д,.'#° @)=822,15 +20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
О г, 0) = —182 + 20 кДж • моль.
Эта величина рассчитана по методу II закона из результатов исследования в работах Гурвича
и др. [424, 1333] равновесия реакции Са (г)+Н2О (г)=СаОН (г)+Н (г) в пламенах Ha-f-O2+Na
A860—2580 К, 16 точек) на основании измерений интенсивности полос СаОН и линий Са в спектрах
пламен (Do (Са—ОН)=395+15 кДж-моль'1). Близкое значение D05+20 кДж-моль) получено
в тех же работах при изучении равновесия реакции СаС1 (г)+Н2О (г)=СаОН (г)+НС1 (г) по
интенсивностям полос СаОН и СаС1. Коттон и Дженкинс [885] из измерений концентрации атомов
Са в трех группах пламен H2+O2+N2 оценили Do (Са—ОН)яь431 +15 кДж-моль, чему
соответствует Д^Я° (СаОН, г) =—215+20 кДж-моль. Косвенные измерения в работах Вейц и Гурвича
[57, 61], Рябовой и Гурвича [357, 358], Сагдена и Скофилда [2635] и Калффа и Алькемаде [1619,
1620] приводят к значениям энтальпии образования от —185 до —212 кДж-моль.
СаОН+ (г). Термодинамические свойства газообразного положительного иона гидроксида
кальция в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 792.
Молекулярные постоянные, использованные в расчете термодинамических функций СаОН+ (г),
представлены в табл. 27.8. Спектр и структура СаОН+ не исследовались, и приведенные
молекулярные постоянные оценены на основании постоянных молекул СаОН и КОН (последняя изо-
электронна СаОН+). Значение момента инерции в основном электронном состоянии г Е+ рассчитано
для параметров ^ Са—О—Н=180э, г (Са—О)=2,05 +0,10 А, г (О—Н)=0,96+0,02 А; его
погрешность составляет 2-Ю9 г-см2. Погрешности принятых значений частот оцениваются в 50—
100 см.
315
Са @НJ (к, ж)
Глава 27
Термодинамические функции СаОН + (г) вычислены по формулам A.3)—A.10), A. 122)—A. 124),
A. 126) и A. 129) в приближении «жесткий ротатор-гармонический осциллятор». Погрешности
вычисленных величин обусловлены неточностью принятых молекулярных постоянных и в
меньшей степени приближенным характером расчета, они составляют 3, 5 и 8 Дж-К-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции СаОН+(г) рассчитывались в таблицах JANAF [1544|.
Расхождения между термодинамическими функциями, вычисленными в [1544] и настоящем
справочнике, составляют 3—8 Дж-К-моль в значениях Ф° (Т) и S° (T) и обусловлены
различиями в оценках молекулярных постоянных, особенно v2.
Константа равновесия реакции СаОН+ (г)+е (г)=Са (г) + О (г)+Н (г) вычислена с
использованием значения Дг Н° @)=297,15 +36 кДж-моль, соответствующего принятому в справочник»
потенциалу ионизации
/0(СаОН)=525+30 кДж-моль.
Эта величина основана на обработке по уравнению III закона приведенных в табл. 27.91 данных
[1374, 1375, 1564].
Таблица 27.9. Результаты определений 10 (СаОИ) (в кДж • моль)
Авторы
Метод
/0(СаОН)
II закон
III закон
Енсен, 1968 [1564] Спрктрофотометрия в пламенах, равновесие реак- 491 536+20
ции Са+ОН=СаОН++е, 2020—2575 К, данные
приведены в виде уравнения, АГН" @)=143 +
+11 (III)
Келли.Педли, 1971 [1664, 1666] То же, равновесие реакции Са(ОНJ+ЗН=СаОН++ 554+27 —
+Н2О+Н2+», температурных! интервал и число
точек не указаны, Д,.Я° B300 К)=77 (II)
Хейхёрст, Кительсон, 1074 [1374, Масс-спектрометрическое определение концент- 526+77 514+18
1375] рации понов р пламенах, Са++Н.,0=Са0Н++Н,
—1200—1500 К, 3 точки, Д,.Я° @)=-25,0+6,4 (III)
Принятому значению соответствует
Д//°(СаОН+, г, 0) = 343 ± 36 кДж • моль.
Са (ОНJ (к, ж). Термодинамические свойства кристаллического и жидкого дигидроксида
кальция в стандартном состоянии при температурах 100—1500 К приведены в табл. 793.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние Са(ОНJ (к) принята гексагональная
модификация (структурный тип СсЛ2), устойчивая в интервале от 0 до Гт=1023 К. При температурах
Т <; 298,15 К термодинамические функции получены по данным Хаттона и др. [1368] A9—
330 К), которые выполнили измерения теплоемкости на образце, содержавшем 99,75% Са(ОН)^
и примеси СаСО3 и Н2О. Погрешности измерений [1368] оцениваются в 0,3% при Т > 50 К и
~1% при более низких температурах. Экстраполяция теплоемкости ниже 20 К приводит к
значению 5°B0К)=0,3 Дж-К^-моль-1. Погрешности принятых по [1368] величин S° B98,15 К)
и Н° B98,15 К) - Я° @) (см. табл. 27.1) составляют 0,4 Дж-К^-моль-1 и 0,06 кДж-моль
соответственно.
Для теплоемкости в интервале 298,15—1023 К принято уравнение, выведенное методом Шо-
мейта по результатам измерений энтальпии Са(ОНJ (к) в работах Кобояши [1737] B98—800 К)
и Лащенко и Компанского [221] [386—778 К). При этом погрешности данных [1737] оценены
в 0,5%, а данных [221] — в 3% ввиду того, что в работе [221] исследовался недостаточно чистый
образец Са(ОНJ B,6% примесей), а полученные значения энтальпии имеют существенный разброс.
Температура плавления Са(ОНJ A023+30 К) принята по данным [2857а], а энтальпия
плавления B9+3 кДж-моль) оценена с учетом данных [2262] по энтропиям плавления Sr(OHJ и
316
Кальций и его соединения Са (ОНJ (г)
Ва(ОНJ. Теплоемкость жидкого Са(ОНJ принята равной С°р (Sr(OH2), ж), измеренной
экспериментально (см. табл. 28.1); ее погрешность может составлять до 20 Дж-К~1-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 1500 К оценены в 0,3, 3 и
8 Дж -К -моль соответственно. Расхождения между термодинамическими функциямиСа(ОНJ (к),
приведенными в табл. 793 и в справочниках [578, 1544], не превышают 1 Дж-К-моль~1 в
значениях Ф° (Т) до 1000 К; они обусловлены некоторым различием выбора данных по теплоемкости
Са(ОНJ при Т > 298,15 К.
В справочнике принята энтальпия образования
Д//° (Са(ОНJ, к, 298,15 К) = — 985,9 ± 1,0 кДж • моль.
Эта величина основывается на хорошо согласующихся между собой результатах
калориметрических измерений энтальпии растворения СаО (к) и Са(ОНJ(к)(в кДж-моль) в работах Томсена
[2686] (—985,9), Бертло [672] (—984,1), Рота и Чолла [2381] (—986,5), Торвальдсона [2693,
2694, 2695] (—986,4+1,5), Швите и др. [2482, 2483] (—986,1 ±1,5), Тейлора и Уэлса [2666]
(—985,9+1,0), энтальпии растворения Са(ОНJ (к) в воде в работе Гопкинса [1478] (—985,2+1,5),
энтальпии гидратирования СаО (к) в работах Торвальдсона и др. [2694] (—985,8+1,5), Тейлора
и Уэлса [2666] (—985,5+2,5) (см. также [982, 1745, 1746, 1939]). К близким значениям приводят
также исследования равновесия диссоциации Са(ОНJ (к) на СаО (к) и Н2О (г) статическим методом
в работах Тамару и Сиоми [2653], Берга и Рассонекой [36], Холстеда и Мура [1345] (обработка
этих данных по методу III закона дает соответственно —985,7+2,0, —988,3+4,0 и —985,0+2,0)
и растворимости Са(ОНJ в воде в работах Гринберга и Копленда [1255] и Ейтс и Маршалла
[2883] (—983,7+2,0) х. Несколько отличающиеся значения (на 5—20 кДж-моль") получены
авторами [1521, 1592, 2090, 2427].
Давление пара дигидроксида кальция в реакции Са(ОНJ (к)=Са(ОНJ (г) вычислено с
использованием значения \ГН° @) = Ь.„Н° (Са(ОНJ, к, 0)=377,18+20 кДж-моль, соответствующего
принятым в справочнике энтальпиям образования дигидроксида кальция в кристаллическом и
газообразном состояниях.
Са(ОНJ (г). Термодинамические свойства газообразного дигидроксида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 794.
Молекулярные постоянные, использованные в расчете термодинамических функций,
представлены в табл. 27.8. Исследования структуры и спектров молекулы Са(ОНJ неизвестны.
Произведение моментов инерции в основном электронном состоянии XхАх вычислено в предположении.
что ^ О-Са-О=150+10°, г (Са-О) =2,10+0,05 А (оценены из Са?а), -4 Са-О-Н=180°,
г (О—Н)=0,96+0,02 А (перенесены из СаОН); его погрешность оценивается в 4-10~115 г8-см6.
Частоты Vj, v2, v3 фрагмента О—Са—О также оценены на основании данных для CaF2, частоты
v4 и v5 фрагментов Са—О—Н перенесены из СаОН. Погрешности принятых значений оцениваются
в 30 см для v2 и в 50—100 см для других частот.
Термодинамические функции Са(ОНJ (г) вычислены по формулам A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический осциллятор»;
их погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 5, 10 и 15 Дж -К -моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К.
Ранее термодинамические функции Са(ОНJ (г) вычислялись в таблицах JANAF [1544],
соответствующие расхождения составляют 4—14 Дж-К-моль в значениях Ф° (Т) и S° (Г); они
обусловлены разной оценкой молекулярных постоянных, особенно значений ^ О—Са—О, v2 и v5.
Константа равновесия реакции Са(ОНJ (г)=Са (г)+2О (г)+2Н (г) вычислена с использованием
значения АГН° @)=1702,967+20 кДж-моль, соответствующего принятой в справочнике энталь
пии образования
t±fH°(Ca (ОН)Я, г, 0) = —600 + 20 кДж • моль.
Эта величина основывается на результатах спектрофотометрических измерений равновесия
реакций образования Са(ОНJ в пламенах H2+O2+N2 в работе Коттона и Дженкинса [885] A570—
1 Для обработки данных по растворимости Са(ОНJ использованы сведения, приведенные в Приложении 5.
317
CaF (г) Глава 27
2030 К), в пламенах CH4+O2+N2 в работах Старовойтова и др. [397,399] B265—2815 К, 7 точек)
и в пламенах обоих типов в работах Старовойтова и др. [359, 400] A860—2815 К, 23 точки).
Энтальпии образования Са(ОНJ, вычисленные из данных этих авторов по методу III закона, равны
соответственно —603 +12, —588 +8 и —596,2 +2,6 кДж -моль. В погрешности принятой величины,
помимо воспроизводимости измерений и погрешностей использованных в расчетах
термохимических величин, учтена неточность значений Ф° (Са(ОНJ, г), которая вносит погрешность
—15 кДж-моль при обработке данных, полученных при 2000 К.
CaF (г). Термодинамические свойства газообразного фторида кальция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 795.
Молекулярные постоянные CaF, принятые в справочнике на основании результатов
исследований электронных спектров этой молекулы, представлены в табл. 27.10. В спектрах CaF
наблюдались шесть систем полос, связанных с переходами между семью электронными состояниями:
А*П^Х*Х+ [928, 1084, 1361, 1384а, 1583, 2044, 2097а, 2634, 2795], ?2?+^*Х2?+ [928, 1084,
1361, 1384а, 1583, 1695, 2103, 22856, 2528, 2795], С2П *s Х2?+ [928, 1126, 1583], D2L+<~ X2 Е+
[1126], Е2 ?+ *а X2 ?+ [1126, 2265] и F2U ^ X2 ?+ [1126, 2265]. Состояния X2 ?+ и А2П у молекулы
CaF, так же как у молекул других галогенидов Са, Sr и Ва, коррелируют с атомами в их основных
состояниях 1<S'+2P. Все эти молекулы должны иметь большое число валентных электронных
состояний (дублетных и квартетных), коррелирующих с пределами М CР)+Х BР), М (sD)-\-X (%P),
М (XD)+X BР) и М AР)-\-Х BР), причем у соединений с Вг и I пределы, соответствующие разным
компонентам состояния X {2Р), могут быть разделены пределами, связанными с другими
состояниями атома металла. Благодаря низким потенциалам ионизации атомов металлов и большому
сродству к электрону атомов галогенов диссоционный предел М+ B5)+Х~ ^S) расположен
относительно близко (от 13 000 до 24 000 см) от предела М AlS')+X BР). Коррелирующее с этим
пределом состояние 2?+ должно оказывать существенное влияние на увеличение стабильности
молекул MX в других состояниях этой симметрии. Наконец, первые ридберговские состояния молекул
MX должны иметь относительно низкие энергии, близкие по величине к энергии ридберговских
состояний соответствующих атомов (ниже 32 000 см). Взаимодействие нижних ридберговских
состояний с валентными состояниями может приводить, как у NO, к тому, что отталкивательные
или слабосвязанные валентные состояния приобретают характер ридберговских связанных
состояний. Такой характер, по-видимому, имеет состояние Е2 ?+ у молекулы CaF. Таким образом,
можно предполагать, что все 12 молекул этой группы имеют большое число связанных состояний,
только небольшая часть из которых наблюдалась экспериментально. Ввиду трудности оценки
числа, стабильности и энергии ненаблюдавшихся состояний, а также того, что квартетные
состояния, по-видимому, являются отталкивательными или слабосвязанными состояниями,
в табл. 27.10, так же как в аналогичные таблицы глав 28 и 29, включены только
наблюдавшиеся в спектрах дублетные состояния.
Колебательные постоянные CaF в состоянии 2Х? + определялись из анализа систем полос АШ ¦*
^Х2И+ [1361, 1384а, 1583, 2044, 2634], ?2?+^Х2? + [1361, 1583, 1695, 2097а, 22856, 2528],
C2n<*X2S+ [1126, 1583], ?2?+ч_Х2? + [1126], а также ?2?+^Х2? + и ffl<±B+ [1126,
2265]. Они удовлетворительно согласуются между собой, за исключением постоянных,
полученных в работах [1361, 1583, 1695, 2528] из анализа системы 52?—Х2?+, проводившегося по Р-
кантам полос. Анализ этой системы, выполненный Рамом и др. [22856] по рассчитанным началам
полос, устранил это расхождение. Приведенные в табл. 27.10 колебательные постоянные CaF
в состоянии Х2? + найдены Прасадом и Нарайяна [2265]. Вращательные постоянные в состоянии
Х2?+ приняты по работе Накагава и др. [2097а] на основании анализа тонкой структуры полос
0—0 и 1—1 системы А2П—Х2?+.
Значения вращательных постоянных, полученные в работах [1084, 1361, 2044], содержали
ошибки (см. [2097а]).
Колебательные постоянные в состоянии А2П приняты по данным Харви [1361 ] и Накагава
с соавторами [2097а], в состоянии 52?+ — по работе Сингха и Мохана [2528], в состояниях СаП
и 2J?+ — по данным Фаулера [1126], в состояниях Е% ?+ и F2U — по работе Прасада и Нарайяна
[2265]. Вращательные постоянные CaF в состоянии А2П найдены Накагава и др. [2097а], в
состоянии 5г?+ — Рамом и др. [22856].
Термодинамические функции CaF (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QMB и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом шести
318
Кальций и его соединения
CaF (г)
Таблица
Молекула
27.10. Молекулярные постоянные моногалогенидов CaF, CaF+
40Ca18F
CaF+
40Ca3sCl
СаС1+
«Ca79Br
40Ca127I
Примечание.
CaF: a yo==1
CaCl: a To=O
Состояние
Х22+
А2П
J522+
С2П
?22+
Е21+
F2n
XJ2+
X2 2+
АЧ1^
А2Щ
B2Z+
С2П
D22+
Я2 2+
ЯП
С2Д
А2И
/2
С2П
ZJ2+
Е2Т+
Y V +
А2Щ
А2Щ2
В22+2
С2П е
П2 2+
т.
0а
16 526,85
18 854,5
30 270
30 772,4
34 135,0
37 547,8
0
0а
16 093,5
16 164,0
16 856.,69 б
26 536
31 107.8
34 266,4
35 688
36 708
0
0
15 922,5
15 985,8
16 380
25 426
31 190,6
33 942,2
36 798,1 Д
0
15 586,20
15 645,57
15 713,5
23 315,51
31 011,4
587,3
593,4
566,7
481,7
650,7
645,5
682,2
680
369,43
372,2
372,1
366,4
337,1
423,3
413,3
432,5
434
460
285,15
288,1
384,6
265,2
327,64
318
343,4
239,0
242,65
241,65
239,95
229,75
265,0
Ниже все постоянные даны в см.
31 -Ю-3;
00136; б
СаВг: а рассчитано по
6 4=71,475.
То= —0,0652;
В ш « — 1
«А
2,77
3,11
2,82
2,02
2,89
3,12
3,68
2,4
1,22
1,38
1,33
1,42
1,12
1,61
1,68
0,8 в
1,1
1,4
0,865
0,92
0,92
0,97
1,245
1,2
1,0
0,628 а
0,62
0,83
0,62
0,633
0,8
),06.
в.
СМ
0,343 704
0,348 744
0,341 767
—
—
—
—
0,362
0,152 233
—
—
0,154 700
0,142 42
—
—
—
—
0,177
0,090 5 а
—
—
—
0,068 1 б
—
—
—
—
—
соотношению A.38); б оценено по соотношению A.69);
г оценка (см. текст); Д рассчитано из v00
Cal: a шеУе=
=0,008; °
ношению A.68)
рассчитано по
; Д оценки (см
соотношению
. текст); е з
'F21—А2Щ.
A.38); в
1риведены
, CaCl, СаС1+,
ах ¦ 103
2,436
2,529
2,6
—
—
—
1,8
0.800
—
_
0,889
0,747
—
—
—
—
0,75
0,4 б
—
—
—
0,24 в
—
—
—
—
—
СаВг, Cal
De. 10'
4,5
4,4
4,58
—
—
4,1
1,027
—
—
1,097
1,0
—
—
1,0
0,36 в
_
—
0,22 г
—
—
.—
—
—
Те
к
1,950 7
1,936 6
1,956 3
—
1,90
2,330 6
—
.—
2,421 06
2.516
—.
,
.
—
2,25
2,65 г
,
—
2,85 Д
—
—
.
—
в оценено по соотношению A.68);
оценено по соотношению A.69)
постоянные для
подсос тояния
; г оценено по соот-
cm,.
возбужденных состояний, приведенных в табл. 27.10, в предположении, что Q^ BV = (pt[px)Qi$ „•
Величина @W вр и ее производные находились по уравнениям A.73) — A-75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии
с помощью уравнения типа A. 82). В расчетах учитывались все колебательно-вращательные уровни
319
CaF (г)
Глава 27
со значениями / ^ ^max, t>' гДе ^та.,* определялись из условий A. 81). Колебательно-вращательные
уровни энергии состояния Х2? + вычислялись по уравнениям A. 65), A. 61); значения
коэффициентов Ykl в этих уравнениях, а также величин итлх и /Пш, использованные в расчетах, приведены
Таблица 27.11. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см),
а также значения wmax и </цт, принятые для расчета термодинамических функций CaF,
CaF+, CaCl, СаС1+, СаВг и Cal.
Коэффициенты
CaF
г.. ю-* о
Ую-Ю-3 0.587 185
у20.Ю-1 -0,273 407
Узо-Ю1 —0,036 295
Ую-103 0,120 892
У5о-Ю6 —0,419 166
У01 0,338 85
Ун-103 -2,6
У2,-10в 3,4
V Jrt! / /.
^02-1" —*,*
Уоз-Ю13 0,303
Ушак 165
Aim 502
CaF+
Х>2+
0
0,680
—0,24
—
—
—
0,362
-1,8
—
-4,1
—
140
484
Примечание. Ниже все постоянные даны в см.
CaF: a Состояние A-U
Те 16 526,8
CaCl: a Состояние А2П
Те 16 130
СаВг: а Состояние Л2П
Те 15 955
Cal: a Состояние Am
Те 15 615
В2 2+
18 854,5
В2 2+
16 850,6
В2 2+
16 380
В2 2+
15 713,5
CaCl
X2S+a
0
0,367 955
—0,123 847
0,016 344
—0,002 152
—
0,150 2
—0,772
0,091
-0,98
0,176
197
633
cm d
30 270 30
cm d
26 536 31
cm d
25 426 31
cm d
23 530 31
CaCl+
СаВг
X22+a
0 0
0,460 0,285 165
—0,14 -0,087 039
— 0,004 739
— —
— —
0,177 0.090 5
—0,75 -0,4
— —.
—1,0 —0,36
- 0,088
161 194
610 711
2 2+ Ег 2+ Fan
772,4 34 135,0 37 547,8
2 2+ p* 2+ pm
107,8 34 266,4 35 688
2 2+ e2 2+ F2 2+
190,6 33 942,2 36 798,1
2 2 +
011,4
Cal
X22-
0
0,239 112
-0,0659 191
0,107 487
—0,075 522
—
0,068 1
—0,24
—
—0,22
0,039
122
754
G2A
36 708
в табл. 27.11; коэффициент Y03 получен из условия A. 59), a Y21 — из условия #,шзх+1=0. Муль-
типлетность и вырождение вращательных уровней всех электронных состояний учитывались
соответствующими статистическими весами.
Основные погрешности вычисленных термодинамических функций CaF (г) связаны с невысокой
точностью известных колебательных постоянных в состоянии Х2?, с отсутствием
экспериментальных данных о высоких колебательно-вращательных уровнях этого состояния и о квартетных
состояниях CaF, а также с приближенным расчетом составляющих возбужденных состояний.
При Т <^ 3000 К эти факторы не влияют на термодинамические функции CaF (г) и погрешности
в значениях Ф° (Т) и S° (Г) не превышают 0,02—0,03 Дж-К^-моль. При более высоких
температурах они возрастают, особенно в значениях S° (Т) и С°р (Г), и могут достигать0,5 Дж-К-моль
в значениях Ф° F000 К).
Ранее термодинамические функции CaF (г) публиковались в таблицах JANAF [1543] и в
справочнике [305], где воспроизведены данные [1543]. В работе Климова и др. [167] приведены
коэффициенты полинома, аппроксимирующего значения Ф° (Т). Данные [1543] и табл. 795 из-за
различия в принятых значениях вращательных постоянных расходятся при 298,15 К на
0,4 Дж-К-моль~1. Различие в постоянных и методиках расчета приводит к расхождениям
разного знака, поэтому с ростом температуры расхождения в Ф° (Т) уменьшаются по абсолютной
320
Кальций и его соединения CaF+ (г)
величине, проходят через минимум и достигают 1 Дж-К-моль при 6000.К. Данные [167]
отличаются существенно больше из-за использования других значений постоянных и приближенного
метода fрасчета.
Константа равновесия реакции CaF (r)=Ca (r)-f-F (г) вычислена на основании принятой в
справочнике энергии диссоциации
Дг#° @) = Do (CaF) = 530 + 5 кДж ^моль^ 44 300 +'400 см.
Это значение основано на масс-спектрометрических исследованиях равновесия реакции
A1F (г)+Са (r)=CaF (г)+А1 (г) в работе Блу и др. [711] A271—1351 К, 11 точек) и реакции
CaF2 (г)+Са (r)=2CaF (г) — в работах Блу и др. [711] A271—1351 К, 9 точек) и Хилденбранда
[1420] A583—1730 К, 15 точек). Расчеты по II и III законам и этим данным приводят к значениям
526+35, 471+40 и 529+43 кДж-моль и 534 ±7f 526+4 и 531+4 кДш-моль (приведенные
погрешности учитывают воспроизводимость измерений, неточность сечений ионизации и других
величин, использованных в расчетах). Исследования хемилюминесценции CaF, образующегося
при реакции Са и F2 в пучках, привели к близким величинам (^ 537+8 кДж-моль [2007] и
^ 523 +8 кДж •моль[1033б]). Спектрофотометрическое исследование равновесия реакции Са (г)+
+HF (г) =CaF (г)+Н (г) в пламенах [355а] привело к завышенному значению E65 +30 кДж -моль)
из-за методической ошибки (см. [1333]).
Принятому значению соответствует
A^°(CaF, г, 0) = — 275,392 + 5,1 кДж • моль.
CaF+ (г). Термодинамические свойства газообразного положительного иона фторида кальция
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 796.
В табл. 27.10 приведены молекулярные постоянные CaF+, принятые на основании
теоретического расчета [1707] и оценок. Так же как у изоэлектронных молекул KF и СаО, основным
электронным состоянием CaF+ является ХХБ+ — состояние (конфигурация . . .о2 л4). Вращательная
постоянная, приведенная в табл. 27.10, вычислена на основании значения ге (Са—F)=l,90 +0,05 Af
полученного в квантовомеханическом расчете потенциальной функции взаимодействия ионов
Саа+ и F" [1707]. По тем же данным [1707] вычислена частота колебания. Погрешности принятых
значений Ве и ше могут достигать 0,02 и 70 см соответственно. Энергии возбуждения связанных
электронных состояний, соответствующих конфигурациям . . .<?я?о, . . .<?i?n я . . . . ая*гс,
оценить трудно. Можно предполагать, что в ряду изоэлектронных молекул СаО -*¦ GaF+ -» KF
энергии состояний, принадлежащих возбужденным электронным конфигурациям, возрастают по мере
увеличения полярности связи в молекулах, как это имеет место в ряду Щ -*¦ CN -*¦ СО+. Поэтому
в справочнике принимается, что эти энергии существенно больше, чем у аналогичных состояний
молекулы СаО, и составляют 20 000—25 000 см.
Термодинамические функции CaF+ (г) вычислены по уравнениям A. 3)—A. 10), A.93)—A. 95).
Величина QBS и ее производные вычислялись без учета возбужденных состояний CaF+ по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням состояния ZXS+ с помощью уравнений типа A. 82). В расчетах
учитывались все уровни энергии со значениями /^/max,»i причем последние находились по
соотношениям A. 81). Колебательно-вращательные уровни вычислялись по уравнениям A. 65) и
A. 62). Значения коэффициентов Ykl в этих уравнениях, идентичные соответствующим
постоянным из табл. 27.10, а также величины ушах и /Иш приведены в табл. 27.11.
Погрешности вычисленных термодинамических функций CaF+ (г) обусловлены неточностью
оценок колебательно-вращательных постоянных, а также пренебрежением возбужденными
электронными состояниями CaF+. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут
достигать 0,5, 1 и 3 Дж-К'^моль соответственно.
Ранее таблицы термодинамических функций CaF+(r) не публиковались.
Константа равновесия реакции CaF+ (г)+е (r)=Ca (r)+F (г) вычислена с использованием
значения ДГЯ° @)=*—30,000+30 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
Io (CaF)=560+30 кДж-моль.
321
CaF2 (к, ж) Глава 27
Принятая величина основана на измерениях методом электронного удара, проведенных Блу
и др. [711 ] E60 +30 кДж -моль) и Мурадом, Хилденбрандом [1419, 2081 ] E80 +50 кДж -моль).
Принятой величине соответствуют
Д//0 (CaF+, г, 0) = 284,608 + 30 кДж • моль,
D0(CaF+) —559,831 ± 30 кДж • моль я* 46 800 + 2500 см.
CaF2 (к, ж). Термодинамические свойства кристаллического и жидкого дифторида кальция
в стандартном состоянии при температурах 100—4500 К приведены в табл. 797.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 27.1. В справочнике за стандартное состояние CaF2 (к) в интервале 0—1424 К принята
кубическая модификация (a-CaF2), а в интервале 1424—1671 К — высокотемпературная разупо-
рядоченная модификация (P-CaF2), которая согласно данным [2033] относится к тетрагональной
сингонии. При температурах Т г^ 298,15 К термодинамические функции вычислены на основании
данных о теплоемкости CaF2 (к), полученных Тоддом [2707] E3—297 К) на образце природного
флюорита (состав близок к стехиометрическому) и Хафманом и Норвудом [1494] C,6—30 К)
на образце искусственного монокристалла. Погрешности значений S° B98,15 К) и Н° B98,15 К)—
—Н° @), приведенных в табл. 27.1, составляют 0,30 Дж-К-моль~1 и 0,05 кДж-моль
соответственно.
При Т ^> 298,15 К для теплоемкости а- и Р-модификаций CaF2 приняты уравнения
(см. табл. 27.1), полученные методом Шомейта по результатам измерений энтальпии в работе
Нейлора [2112] B98—1789 К). Ввиду того что у CaF2 имеется Х-аномалия теплоемкости в широком
интервале температур, связанная с а ^± Р-переходом CaF2, теплоемкость a-CaF2 описана двумя
уравнениями. Второе уравнение передает быстрый рост теплоемкости в левой ветви ^-кривой
(от С D000 К)=91 Дж-К-^моль ДО С° A424 К)=177 Дж-К^-моль'1). Температура фазового
перехода A424+10 К), соответствующая максимуму Vi\^wm ^жхолмкдет, основана на работах
Нейлора [2112] и Бредига [738], энтальпия фазового перехода второго рода у CaF2 принята равной
нулю. Данные по энтальпии CaF2 (к), полученные в работах [202, 203] B88—1273 К) и [229]
B91—1490 К), менее точны и не учитывались. Температура плавления CaF2 A691 +3 К) принята
на основании измерений, выполненных в работах [550, 1335, 1463, 1747, 2072, 2112]. Данные
[51, 54, 117, 939, 1024а, 1290, 1629, 1980, 2251, 2356, 2358] приводят к более низким значениям
A600—1683 К), по-видимому, из-за использования недостаточно чистых образцов. Энтальпия
плавления CaF2 C0+1,5 кДж-моль) и теплоемкость жидкого CaF2 (99+3,0 Дж-К^-моль) приняты
в результате обработки данных Нейлора [2112] по энтальпии CaF2 (ж) в интервале 1691—1789 К-
Погрешности вычисленных значений Ф° (Т) при 298,15, 1500, 3000 и 4500 К оцениваются в 0,3,
1, 5 и 13 Дж-К^-моль соответственно. Расхождения между термодинамическими функциями
CaF2 (к, ж), приведенными в табл. 797 и в таблицах JANAF [1543], не превышают
1,5 Дж-К~1-моль в значениях Ф° (Т).
В справочнике принята энтальпия образования
-1
AyH°(CaF2t к, 298,15 К) = —1228 + 3 кДж • моль
Это значение основано на результатах измерений, приведенных в табл. 27.12. Оно является
средневзвешенным из полученных в наиболее точных измерениях [64, 346, 384, 1087, 2628, 2716L
При вычислении среднего учитывалось отклонение результата каждой из этих работ от средней
величины. Погрешность принятой величины оценена с учетом неточности отдельных измерений
и их согласованности между собой. Исследования равновесия реакций с участием CaF2 (к) в
работах [330, 971, 2453] приводят к неверным значениям Д7#° (CaF2, к).
Давление пара дифторида кальция CaF2 (к, ж)=СаР2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
Д,#° (CaF2, к, 0) = 436 + 6 кДж • моль.
Эта величина является средневзвешенной из данных, представленных в табл. 27.13.
Приведенные в таблице погрешности характеризуют только воспроизводимость измерений. При вычислении
средневзвешенного наибольший вес придавался работам [1982, 2474]. Погрешность принятой ве-
322
Кальций в его соединения
CaF2 (г)
Таблица 27.12. Результаты определений AjH° (CaF2, к, 298,15 К) (в кДж-моль)
Авторы
Метод
AfB° (CaF2, к, 298,15 К)
II закон
III закон
Гущ, 1884 [1328] Калориметрический, Са(ОЮ2 (ag)+2HF (ag)= —1231+10
=CaF2 (к)+2Н2О (ж)
Торгесон, Сахама, 1948 [2716] То же, CaO (k)+2HF (p-p, 4,444H2O)=CaF, (к)+ —1224,8+1,7
+Н2О (р-р)
Финч и др., 1968 [1087] То же, СаС12 (K)+2NaF (ад)= CaF2 (K)+2NaCl (ад) —1220,8+2,0
Смышляев, Эделева, 1962 а Измерение растворимости CaF2 (к) в воде при — —1229,0+2,0
[384] 298,15 К
Штрюбел, 1965 а [2628] То же, 298—573 К. Пересчет этих данных про- — —1231,9+2,0
веден Ходаковским и др. [425]
Братленд и др., 1970 [735] Протока, равновесие реакции 3SiO2 (к)+ —1224 —1238+20
+2CaF2 (K)=2CaSi0:i (K)+SiF* (г), 1380-1913 К,
расчет но уравнению
Вечер, Вечер, 1967 [64] Измерение ЭДС реакции CaO (K)+MgF2 (к)= — —1231,1+3,0
=CaF2 (K)+MgO (к), 973—1213 К, ArG° A200 К)=
=—73,7 "кДж-моль
Резухина и др., 1973 [346] Измерение ЭДС реакции Са (k)+Ms?F2 (к) = —1229,6+2,0 —1224,3+2,5
=-CaF2(K)+Mg (к), 710—800 К, расчет по
уравнению
а Эти измерения подтверждаются более ранними исследованиями Енсена [1562] и Кольрауша [1743]. В расчетах
использованы данные, приведенные в Приложении 5.
Таблица 27.13. Результаты определений Asfi"° (CaF2, к, 0) (в кДж-моль х)
Авторы
Метод
ASH° (CaF2, к, 0)
II закон
III 8ЭК0Н
Руфф, Ле Буше, 1934 [2407] Точек кипения, 2088—2208 К, 7 точек 445±50 440,3±0,9
Шульц, Сирей, 1963 [2474] Торзионный, 1421—1689 К, 34 точки? 427±3 434,8±0,2
Блу и др., 1963 [711] Испарение с поверхности, 1246—1499 К, 6 точек 417+4 442,8±0,5
Масс-спектрометрический, 1242—1669 К, 19 точек 440+.4 442,8±0,5
Мак-Крири, Торн, 1973 [1982] Эффузионный, 7 серий; расчет по уравнению 426+18 435,8±1,9
III закона проводился из данных при 1475 К,
полученных в каждой из этих серий (более
детальные данные в работе не приведены)
личины оценена с учетом расхождения результатов отдельных измерений и неточности
термодинамических функций CaF2 (к, ж) и CaF2 (г) E кДж-моль).
СаГ2 (г). Термодинамические свойства газообразного дифторида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 798.
Молекулярные постоянные, использованные для расчета термодинамических функций CaF2 (г)я
приведены в табл. 27.8. Исследования инфракрасного спектра [23, 24, 808, 1626, 2565] и
отклонения молекулярного пучка в неоднородном электрическом поле [782, 783, 2839] показали, что
в основном электронном состоянии молекула CaF2 имеет нелинейное строение (симметрия С2г).
Приведенное в табл. 27.8 произведение моментов инерции соответствует оцененной величине
-$ F—Са—F=145±10° и значению г (Са—F)=2,10+0,03 А, найденному Акишиным и др. при
электронографическом исследовании паров CaF2 [5,11 ]; его погрешность составляет 5 -1015 г3 -см'.
Принятая величина -? F—Са—F является средней из рекомендуемых в работах [24,808, 2565].
Приведенные в табл. 27.8 значения v2 и v3 найдены Байковым [23, 24] при исследовании
инфракрасного спектра паров CaFa. Как показывают работы Снелсона [2565], Колдера и др. [808],
323
CaCl (г) Глава 27
Кан'ана и др. [1626], в ИК-спектре молекул CaF2» изолированных в матрицах, имеет место
значительный сдвиг этих частот; так, в матрице из Аг v2=163,4 см, v3=553,7 см [808]. Частота vlr
которая не наблюдалась в спектре паров, вычислена по формулам простого поля валентных сил
в предположении, что /r//rr=9, как это имеет место для CaF2 в криптоновой матрице. Погрешности
принятых частот оцениваются в 25 см. Сведения о возбужденных состояниях молекулы CaFa
отсутствуют, можно предполагать, что их энергии превышают 25 000 см.
Термодинамические функции CaF2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 2, 3 и
4 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции CaF2 (г) вычислялись в таблицах JANAF [1543], а также
в работах Байкова [23] (Г < 5000 К), Брюэра и др. [748] (Т < 2500 К), Краснова и Светцова
[195] (Г ^ 2500 К). В настоящем издании справочника и в [1543] приняты близкие значения
молекулярных постоянных CaF2, поэтому расхождения в значениях Ф° (Т) не превышают 0,1 —
0,5 Дж-К~1-моль, расхождения с данными [23] достигают 3 Дж-К-моль~1, а с [195, 748]
составляют 13—16 Дж-К-моль~1 (в двух последних работах для молекулы GaF2 принята
линейная структура).
Константа равновесия реакции CaF2 (r)=Ca (r)+2F (г) вычислена с использованием значения
ДДГ3 @)=1120,968+7,0 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации GaF2 (к). Этим величинам также соответствует
AfH°(GeiF2, г, 0) = —789,085 +6,7 кДж • моль.
CaCl (г). Термодинамические свойства газообразного хлорида кальция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 799.
Молекулярные постоянные CaCl, принятые в справочнике на основании результатов
исследований электронных спектров этой молекулы, приведены в табл. 27.10. В спектрах CaCl
наблюдались девять систем полос, связанных с переходами между восемью электронными состояниями:
Am ^ Х*Е + [543, 752а, 772а, 1354, 1378, 1384а, 1697, 2184], 52Е + *ъ Х2? + [543, 752а, 926, 965,
966, 1354, 1378, 1384а, 1697, 2184, 2795], С2п - Х2Е + [1354, 1697, 2060, 2184], ?>2?+,* Х2Е +
[658, 658а, 1354, 1697], ?2Е + ^ Х2Е + [6586, 1354, 1697], Fm—A2U, ^2П-52Е+, G2A-A2H
[2480], jf?*?+ ->Z?S? + [9656, 1696]. Так же как у молекул других галогенидов Са, Sr и Ва,
состояния Х2? + и АЩ молекулы CaCl коррелируют с основными состояниями атомов 1S-\-2P. Co
следующим диссоциационным пределом Са CР)+С1 BР) коррелирует большое число валентных
состояний, в том числе квартетные состояния. По крайней мере часть этих состояний может быть
связанными состояниями (см. CaF (г)).
Колебательные постоянные в состоянии X2 ?+ определялись по кантам полос А2Н—X2 Е+
[543, 1354, 1696, 1697, 2184], С2П-Х2? + [1354, 1696, 1697], ?>2Е+-Х2?+ [658, 658а, 1354, 1696,
1697], i?2?+—Х2? + [9656, 1697] и удовлетворительно согласуются между собой. Близкие
значения получены в работах Домейла и др. [965, 9656, 966] по началам полос. Приведенные
в табл. 27.10 колебательные постоянные CaCl в состоянии Х2?+ получены Харрингтоном [1354]
из анализа систем полос А—X, С—X, D—X и Е—X и подтверждены позднее в работах [965,
9656, 966, 1696, 1697]. Вращательные постоянные приняты по работам Домейля и др. [965,
966], которые исследовали структуру полос 0—0, 1—1 и 2—2 системы В2Е+—Х22+.
Результаты этого анализа хорошо согласуются с исследованием структуры полос 0—0, 0—1 и 1—0
системы С2П—Х2Е+ в работе Моргана и Барроу [2060], а также с результатами анализа Бергом и др.
[658, 658а] полос системы ?2Е+-Х22+.
% Колебательные постоянные CaCl в состояниях A2U, Б2Е+, С2П, D2I>+, Z?22+ приняты по
работам Кханна и Дьюби [1696, 1697], в состояниях F2U. и G2A — по работе Шютте [2480].
Вращательные постоянные в состояниях 52Е + и ?2? + найдены Домейлем и др. [965, 9656, 966], в состоянии
С2П — Морганом и Барроу [2060], в состоянии ZJ? + — Бергом и др. [958, 958а].
1 ¦ Термодинамические функции CaCl (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QBS и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом семи
возбужденных состояний в предположении, что Q$x_ яр = (PilPx)QlJf2. вр- Величина (?№ вр и ее
производные вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебатель-
324
Кальций и его соединения
СаС1 (г)
ным уровням и интегрированием по вращательным уровням с помощью уравнения типа A. 82).
В расчетах учитывались все уровни энергии со значениями / ^ /шах,„, где /шах „ находились из
условий A. 81). Соответствующие значения i>max и ^1т приведены в табл. 27.11. Колебательно-
вращательные уровни состояния Х2?+ вычислялись по уравнениям A. 65) и A. 62), для
зависимости Dv от v использовалось уравнение A. 58). Коэффициенты Ykl в этих уравнениях,,
пересчитанные на природную изотопическую смесь молекул СаС1 по соотношениям A. 66) и постоянным
из табл. 27.10, приведены в табл. 27.11. Коэффициент Y03 получен с учетом условий A. 59), a Y21 —
из условия 5,ши+1=0. Мультиплетность и вырождение уровней электронных состояний
учитывались статистическими весами.
Погрешности рассчитанных термодинамических функций в основном связаны с тем,, что
колебательные постоянные СаС1 найдены по кантам полос и отсутствуют экспериментальные данные
о высоких колебательно-вращательных уровнях состояния Х2Е+. При Т > 3000 К становится
существенной неполнота данных о возбужденных электронных состояниях этой молекулы.
Погрешности в значениях Ф° (Г) могут достигать 0,03, 0,1 и 1 Дж-К~1-моль при 298,15, 3000 и
6000 К.
Термодинамические функции СаС1 (г) вычислялись ранее в таблицах JANAF [1543] и работе
Климова и др. [167] (приведен полином, аппроксимирующий Ф° (Т) при Т ^ 5000 К).
Расхождения термодинамических функций, приведенных в[1543] и в табл. 799, не превышают 0,1 Дж-К~хХ
Хмодь при 298,15 К и растут с температурой, достигая 0,6, 1,4 Дж-К^-моль в значениях ФС(Г),:
S° (T) при 6000 К, главным образом из-за того что в [1543] использован приближенный метод
расчета. Данные [167] отличаются от табл. 799 еще больше.
Константа равновесия реакции СаС1 (г)=Са (г)+С1 (г) вычислена е использованием принятой
в справочнике энергии диссоциации
AJI" @) = Do (CaCJ) = 400 + 6 кДж • моль «* 33 400 ± 500 см,
основанной на исследованиях равновесия реакций с участием СаС1 (г), представленных в табл. 27.14.
Приведенные погрешности учитывают воспроизводимость измерений, неточность сечений
ионизации, термодинамических функций и термохимических величин, использованных при обработке
Таблица 27.14.
Авторы
Змбов, 1969 [2908]
Хилденбранд, 1970
Поттер и др., 1970
Хилденбранд, 1970
Гурвич и др., 1973
Результаты определений Do (CaCl) (в кДжмоль *)
[1409]
[2261]
[1409]
[1333]
Метод
Масс-спектпометрическнй, СаС12 (г)+Са (г)=2СаС1 (г),
976-1043 К, 11 точек, ДГЯ° @)=108,8±1,7 (III)
То же, СаС12(г)+Са(г)=2СаС1(г), 1219-1353 К,
11 точек, ДГЯ° @)=95,5+0,7 (III)
То же, Са (г)+А1С1 (г)=CaCl (г)+А1 (г), 11 точек,
ДгЯ°@)=100,5+0,5 (III)
То же, CaCl. (г)+Са (г)=2СаС1 (г), 1235—1540 К,
18 точек, ДгЯ«"@)=94,0±1,1 (III)
То же, Са(г)+А1С1(г)=СаС1(г)+А1(г), ДгЯ°@)=
=85,3±3,4 (III)
Электронный удар, СаС12 (г) +е(г)=СаС1+ (г)+С1 (г)+
+2е Д Я0 С0)=482+20
Фотометрия в пламенах, Са (г)+НС1 (г)=СаС1 (г)+
+Н(г), 1745-2450 К, 14 точек, Д^" @)=33,9 +
±6,7 (II)
D
II закон
425 ±45
402±11
401 ±17
418±6
474 ±55
394±10
о(СаС1)
III закон
396,5+3,7
403±3,6
398+7
404 ±4
413±8
420±20
экспериментальных данных. Из таблицы видно, что результаты всех измерений равновесия
реакций с CaCl (г) хорошо согласуются между собой, хотя данные [2261 ] и приводят, так же как для
SrCl, к несколько более высоким значениям. Принятая величина основана на данных [1409, 1333
и 2908]. Исследование порога реакции образования CaCl по хемилюминесценции в работе [2007],
325
СаС1+ (г), СаС12 (к, ж) Глава 27
так же как для других молекул, привело к резко завышенной величине (^ 481 +6). Принятому
значению соответствует
AyfHCaCl, г, 0) = —103,047 + 6,1 кДж • моль-1.
СаС1+ (г). Термодинамические свойства газообразного положительного иона хлорида кальция
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 800.
В табл. 27.10 приведены молекулярные постоянные СаС1+, принятые на основании
теоретического расчета [1707] и оценок. Так же как у изоэлектронных молекул КС1 и CaS, основным
электронным состоянием СаС1+ должно быть состояние ХгЕ+ (конфигурация . . .о2тс4). Приведенная
в табл. 27.10 вращательная постоянная в этом состоянии вычислена на основании значения
ге (Са—С1)=2,25+0,05 А, полученного в квантовомеханическом расчете потенциальной функции
взаимодействия ионов Са2+ и С1~ [1707]. Частота колебания ше также вычислена по данным [1707]
и хорошо согласуется с соответствующими величинами в ридберговских состояниях молекулы
CaGl. Погрешности принятых значений Ве и ше не превышают 0,01 и 50 см соответственно.
Энергии возбуждения электронных состояний, соответствующих конфигурациям . . .о2л3а, . . .ойт?%
и ... а714тс, оценить трудно. Можно ожидать (см. CaF+ (г)), что они лежат существенно выше
аналогичных состояний в молекуле CaS и имеют энергии 15 000—20 000 см.
Термодинамические функции СаС1+ (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95)
без учета возбужденных электронных состояний. Величина <?кож.вр и ее производные по температуре
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням с помощью уравнений типа A. 82). В
расчетах учитывались все уровни энергии со значениями / <^/maiil,, причем последние находились по
соотношению A. 81). Колебательно-вращательные уровни энергии вычислялись по уравнениям
A. 65) и A. 62). Значения коэффициентов Ykl в этих уравнениях, идентичные соответствующим
постоянным из табл. 27.10, а также величины уши.и /lim приведены в табл. 27.11.
Погрешности вычисленных термодинамических функций обусловлены отсутствием
экспериментальных данных о молекулярных постоянных СаС1+, а также пренебрежением возбужденными
электронными состояниями. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут
достигать 0,5, 2 и 4 Дж-К-моль~1 соответственно.
Ранее таблицы термодинамических функций СаС1+ не публиковались.
Константа равновесия реакции СаС1+ (г)+е (г)=Са (г)+С1 (г) вычислена с использованием
значения ДГЯ° @) =—180,000 +12 кДж -моль, соответствующего принятому в справочнике
потенциалу ионизации
Io (CaGl)=580 +10 кДж-моль.
Измерения этой величины выполнены методом электронного удара в работах Змбова [2908]
E30+50 кДж-моль), Хилденбранда [1407] E80+10 кДж-моль), [1409] E40+50 кДж-моль),
Поттера и др. [2261] E60+50 кДж-моль). Наиболее точное значение получено в детальном
исследовании Хилденбранда [1407], и оно принято в справочнике, ему соответствуют
i!fH° (СаСГ, г, 0) = 476,953 + 12 кДж • моль,
Do (СаСГ) = 409,831 + 12 кДж • моль.
СаС12 (к, ж). Термодинамические свойства кристаллического и жидкого дихлорида кальция
в стандартном состоянии при температурах 100—3900 К приведены в табл. 801.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние СаС12 (к) в интервале 0—1045 К принята
ромбическая модификация СаС12. При Т ^ 298,15 К термодинамические функции вычислены на
основании измерений теплоемкости СаС12, выполненных Келли и Муром [1659] E3—-295К) на образце,
содержавшем примесь 0,6% MgCl2. Экстраполяция теплоемкости ниже 50 К проведена с учетом
данных по низкотемпературной теплоемкости SrCl2 [2551] G—30 К) и ВаС12 [1231] F—346 К)
и привела х к значению S° E0 К) = 14,0 Дж-К-моль~1. Погрешности значений 5° B98,15 К) и
1 В работе [1659] экстраполяция теплоемкости СаС13 ниже 50 К по уравнению С°р =¦ D A01/Г) + 2Е B94/Т)
привела к резко завышенному значению S" E0 К) = 19,2 Дж • К ¦ моль.
326
Кальций и его соединения СаСЬ (к, ж)
Н° B98,15 К)—Н° @), приведенных в табл. 27.1, оцениваются в 3,0 Дж-К~1-моль~1 и
0,2 кДж -моль соответственно. При Т >¦ 298,15 К для теплоемкости СаС12 (к) принято уравнение
(см. табл. 27.1), найденное Муром [2054] в результате измерений энтальпии образца СаС12,
исследованного в [1659]. С данными Мура [2054] в пределах 1% согласуются результаты измерений
энтальпии вблизи точки плавления, выполненные Дворкиным и Бредигом [1003] для определения
энтальпии плавления СаС12. Температура плавления A048 +3 К) принята по результатам
измерений, выполненных Грётхеймом и др. [1269] A044,85+0,5 К), Токаревой [406а] A045 К),
Майло и др. [1926] A048,4 + 0,6 К) и Дворкиным и Бредигом [1003] A045 К). Данные других
многочисленных измерений [54, 113, 263, 449, 537, 1031, 1075а, 1291, 1563, 1629, 1760а, 2004,
2127, 2236, 2440] лежат в пределах 1040—1055 К и, по-видимому, менее точны. Энтальпия
плавления B8,05+0,8 кДж-моль) вычислена как разность принятых по данным Мур [2054]
значений энтальпии кристаллического и жидкого СаС12. С этой величиной удовлетворительно
согласуются измерения Дворкина и Бредига [1003] B8,4 кДж-моль), Эмонса и др. [1031]
B9,0 кДж-моль) и Мартынова [247] B8,9 кДж-моль). Уравнение для теплоемкости жидкого
СаС12 (см. табл. 27.1) выведено по данным Мура [2054], который измерил энтальпию СаС12 (ж)
в интервале 1045—1667 К. Согласно этому уравнению теплоемкость в указанном интервале
уменьшается от 110 до 98 Дж-К~1-моль~1. В работе [2054] для теплоемкости СаС12 (ж) рекомендовано
постоянное значение 102 Дж-К~1-моль~1. В работах [1003] и [247] на основании измерений
энтальпии СаС12 (ж) в узких интервалах температур получены менее точные значения
99 Дж-К-моль~1 и 105 Дж-К-моль соответственно.
Погрешности вычисленных значений Ф° (Т) при 298,15, 2000 и 3500 К оцениваются в 3, 5 и
10 Дж-К~1-моль~1 соответственно.
Термодинамические функции СаС12 (к, ж), приведенные в табл. 801 и в таблицах JANAF
11543], различаются на 4 Дж-К-моль~1 в значениях Ф° (Т), ввиду того что они основаны на
разных значениях энтропии при 298,15 К и теплоемкости жидкого СаС12.
В справочнике принята энтальпия образования
bfH° (СаС12, к, 298,15 К) = —795,8 + 1,0 кДж • моль.
Эта величина основана на результатах калориметрических измерений, представленных
в табл. 27.15. При вычислении &fH° (СаС12, к) из данных по энтальпии растворения СаС12 (к)
Т аблица 27.15. Результаты определений А^Н° (СаСЬ, к, 298,15 К) (в кДж-моль)
Авторы
Метод
Д,Я°(СаС]2, к, 298,15 К)
Симонсен, 1951 [2520] Измерение энтальпии сгорания Са в С12 —786
Эрлик и др., 1963 [1022] Измерение энтальпий растворения Са и СаС12 в соля- —799,3+5,0
ной кислоте
а Измерение энтальпии растворения СаО в соляной —794,9 + 2,0
кислоте
" Измерения энтальпии растворения СаС12 в воде до —795,9+_1,0
состояния бесконечно разбавленного раствора,
Дг Н° B98,15 К)=— 81,4+0,3 кДж-моль-1
а Измерения проведены в работах Торгесона, Шомейта [2717], Тейлора, Уэлса [2666, 2828], Торвальдсена и др.
[2695], а также [554, 670, 878, 888, 1179, 1964, 2686, 2887].
*> Наиболее точные измерения выполнены Грейсоном, Снеллом [1262]. Результаты измерений [206, 223, 620, 677,
958, 1119, 1840, 2225, 2520, 2686] менее точны и в основном совпадают с данными [1262]. Энтальпия
разведения растворов СаС12 (к) принята по работам Ланге, Штрека [1810] и Ричардса, Дола [2331] (см. также
работы [223, 1356], где даны энтальпии разведения для высоких концентраций). В работах [56, 149, 364, 1022]
измерены энтальпии растворения СаС12 (к) в соляной кислоте.
в воде использованы принятые в справочнике энтальпии образования водных ионов (см.
Приложение 5). Как видно из таблицы, этот путь расчета привел к наиболее точному значению.
Принятая величина является средневзвешенной из данных, приведенных в таблице (за исключением
12520]).
327
СаС12 (г)
Глава 27
Давление пара дихлорида кальция СаС12 (к, ж)=СаС12 (г) вычислено с использованием
принятого значения энтальпии сублимации
Д,#°(СаС12, к, 0) = 311 + 5 кДж-моль-1.
Эта величина основана на результатах измерений давления пара СаС12, представленных в
таблице 27.16. Приведенные погрешности характеризуют воспроизводимость измерений. Неточность
Таблица 27.16. Результаты определений A,-ff° (СаСЬ, к, 0) (в кДж-модь)
Авторы
Вартенберг, Боссе, 1922 [2801 ]
Дьюинг (см. [1423])
Хилденбранд, Поттер, 1963 [1423]
Баутиста, Маргрейв, 1963 [628]
Новиков, Гаврюченков, 1964 [291]
Фрид и др., 1964 [419]
Лукашенко, Реутова, 1970 [226]
Эмонси др., 1976 [1030а]
Метод
Точек кипения, 1819 К, 1 точка
Эффузионный, 1069—1225 К, 4 точки
То же, 1142—1281 К, 29 точек
Испарения с открытой поверхности, 952—
993 К, 8 точек
Точек кипения, 1591—1701 К, 5 точек
Точек кипения, 1323—1423 К, 2 точки
Эффузионный, 1073—1273 К, 6 точек
Испарения с открытой поверхности, 973—
1023 К, 6 точек
Эффузионный, 1063—1213 К, расчет по
уравнению
Д8Я° (СаС12, к, 0)
II закон
270±30
309±3
340±70
320+30
321
278 ±2
350±40
248
III закон
306
311 ±4
311,0±0,1
321,7 ±0,8
311,7±0,5
318,3±1,4
305 ±2,3
303±1,3
329 ±6
термодинамических функций вносит дополнительную погрешность в энтальпию сублимация
—7 кДж-моль. Принятое значение основывается на данных Хилденбранда, Поттера [1423],
которые достигли хорошей воспроизводимости измерений и согласия расчетов по уравнениям II
и III законов. Близкие значения получены в работах [291] и неопубликованной работе
Дьюинга.
В работе [213] получены слишком низкие значения этой величины (~270 кДж-моль).
Измерения методом испарения с открытой поверхности в работе [628] привели к значительно большей
энтальпии сублимации, что, возможно, связано с низким значением коэффициента испарения.
Резкое расхождение результатов обработки данных [1030а] по уравнениям II и III законов,
по-видимому, свидетельствует об ошибках в измерениях.
СаС12 (г). Термодинамические свойства газообразного дихлорида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 802.
Молекулярные постоянные, использованные для расчета термодинамических функций СаС12,
приведены в табл. 27.8. Исследования дифракции электронов [5, И], инфракрасного спектра [24,
1364, 2842] и отклонения молекулярного пучка в неоднородном электрическом поле [2839]
показали, что в основном электронном состоянии молекула СаС12 имеет линейное строение
(симметрия Dmh). Приведенный в табл. 27.8 момент инерции вычислен при г (Са—С1)=2,51+0,03 А
в соответствии с электронографическим исследованием пара СаС1г, выполненным Акишиным и др.
[5, 11]. Погрешность значения / составляет 5-Ю9 г-см2. Приведенное в табл. 27.8 значение v8
найдено Байковым [24] в инфракрасном спектре паров СаС12. Близкие величины получены Хасти
и др. [1364] и Уайтом и др. [2842] в ИК-спектре молекул СаС12, замороженных в матрицах. Не-
наблюдавшаяся в паре частота vx вычислена по формулам поля валентных сил на основании
принятого значения v3 и предположения, что по аналогии с дигалогенидами магния в молекуле СаС1,
/г//„.=15. Значения ч±, оцененные в работах [24, 194, 748, 2842], согласуются с приведенным
в табл. 27.8 в пределах 10%. Для частоты v2 по аналогии с MgCl2 принята величина, которая
несколько больше измеренной Уайтом и др. [2842] в ИК-спектре молекул СаС12 в матрице из Кг
F3,6 см). Отнесение к v2 полосы 122 см в матрице из Аг, сделанное Хасти и др. [1364], явно
328
Кальций и его соединения СаВг (г)
ошибочно х. Погрешности принятых значений частот оцениваются в 20—40 см. Сведения о
возбужденных электронных состояниях СаС12 отсутствуют, можно предполагать, что их энергии
превышают 20 000—25 000 см.
Термодинамические функции СаС12(г) вычислены по уравнениям A. 3)—A. 6), A. 9),A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор»; их погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 3, 5 и 7 Дж-К~1-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К соответственно 2.
Ранее термодинамические функции СаС12 (г) рассчитывались в таблицах JANAF [1543],
работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Расхождения
с данными [1543], [748] и [195] достигают соответственно 4, 3 и 6 Дж-К-моль в значениях
Ф° (Т), они вызваны использованием в этих работах менее надежных [1543] или оцененных [195,
748] значений основных частот.
Константа равновесия реакции СаС12 (г)=Са (г)+2С1 (г) вычислена с использованием значения
Д^0 @)=901,763+5,2 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации СаС12 (к). Этим величинам также соответствует
г, 0)=:—485,190 +5,1 кДж • моль.
СаВг (г). Термодинамические свойства газообразного бромида кальция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 803.
В табл. 27.10 приведены молекулярные постоянные СаВг, принятые в справочнике на основании
анализа результатов исследований электронных спектров этой молекулы. В спектрах СаВг
наблюдались переходы между семью электронными состояниями: A2R spt Х2? + 11354, 1378, 2159, 2795],
52Е+_>Х2?+ [1354, 1378, 2795], С2П ^ Х2? + [1354, 1378, 1600, 1622, 2281], Z>2? + ** Х2? +
[1354, 2281, 2319, 2499], E*L+ -* Х2Е + [2319] и F2Z + -* А2П [2312]. Так же как у молекул
других галогенидов щелочноземельных металлов, состояния Х2? + и АЩ молекулы СаВг коррелируют
с основными состояниями атомов 1S+*P. С первым возбужденным пределом СаCР)+ВтBР)
должно коррелировать большое число валентных состояний молекулы СаВг, в том числе
квартетные состояния. По крайней мере часть этих состояний могут быть связанными, см. текст по CaF.
Колебательные постоянные СаВг в состоянии Х2? + определялись по кантам полос систем Л2П—
Х2? + [1354, 1378], Я2Е+-Х2? + [1354], С2П-Х2? + [1354, 1378, 1600, 2281], ?2Е+-Х2? + [1354,
2281, 2319, 2499] и удовлетворительно согласуются между собой. Приведенные в табл. 27.10
колебательные постоянные получены Ша [2499] из анализа системы ?J? +—Х2? + @ <^ у' ^8, 0 ^
^ v" ^ 7). Вращательная структура электронных переходов СаВг до настоящего времени не
проанализирована. Приведенная в табл. 27.10 вращательная постоянная в состоянии Х2? + вычислена
при ге (СаВг)=2,65+0,05 А на основании величины г (Са—Вг) в молекуле СаВг2. Ранее эта
постоянная определялась Камаласананом [1622] из анализа структуры полосы 0—0 системы С2П—
Х2?+ молекул 40Са81Вг и 40Са79Вг. Однако полученное в этой работе значение re (Х2?+)=2,799 A
существенно больше межатомного расстояния в молекуле СаВг2B,67+0,03 А) и не согласуется
с известными значениями г. (MX) в молекулах других галогенидов щелочноземельных металлов.
В работе Альтман [15а], выполненной после завершения подготовки настоящего тома, в результате электроно-
графического исследования были получены следующие постоянные шести дигалогенидов щелочноземельных
металлов:
г.
л
*«
v2,
(Me—X), А
еХ—Me—X, град
, МДИН-А
, мдин• А
, СМ
, СМ
, см
СаС12
2,451
180
0,0308
0,00013
310
52
393
СаВг2
2,630
180
0,0153
0,0044
180
40
330
SrCl2
2,642
142
-0,0150
0,005
286
42
310
SrBp2
2,775
180
0,0206
0,0052
162
35
219
BaCI2
2,771
127
—0,030
0,0049
260
36
265
BaBr2
2,953
139
—0,0115
0,0018
145
22
163
Расхождения термодинамических функций СаС12 (г), СаВг2 (г), SrCl2 (г), SrBr2 (г), ВаС1а (г) и ВаВг2 (г),
рассчитанных по постоянным, полученным в работе [15а] с учетом ангармоничности деформационного колебания, с
функциями, приведенными в табл. 802, 805, 832, 835, 862 и 865, достигают 2—12 Дж-К~>-моль в значениях Ф° (Т).
329
CaBr2 (к, ж) Глава 27
По-видимому, в анализе [1622] допущены какие-то ошибки, однако краткая форма публикации
этой работы не позволяет проверить анализ, сделанный ее автором. Колебательные постоянные
в состояниях А2И, В2 ?+ и С2П приняты по работе Харрингтона [1354], в состояниях D2?+, ?2E +
и F2Z — по работам [2312, 2319, 2499] соответственно.
Термодинамические функции СаВг (г) вычислены по уравнениям A. 3)—A.10), A. 93)—A. 95).
Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом шести
возбужденных состояний в предположении, что Q^o\_ вр = (pJPx)QiS. вр- Величина (?№ вр и ее
производные вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по вращательным уровням энергии с помощью уравнений типа
A. 82). В расчетах учитывались все уровни энергии со значениями / ^/ша1)г,, где /тиг,
находились из условий A. 81). Колебательно-вращательные уровни состояния Х2? + находились по
уравнениям A. 65) и A. 61). Значения коэффициентов Ykl, а также величин ушах и /11ш, использованных
в расчетах, приведены в табл. 27.11. Коэффициент F03 получен из условия A. 59). Мультиплетность
и вырождение уровней электронных состояний учитывались соответствующими статистическими
весами. Погрешности вычисленных термодинамических функций СаВг (г) при низких температурах
связаны с отсутствием экспериментальных данных о вращательных постоянных в состоянии
Х2?+, а при высоких температурах — данных о квартетных состояниях этой молекулы.
Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,3, 0,5 и 2 Дж-К^-моль
соответственно.
Ранее термодинамические функции СаВг (г) вычислялись в таблицах JANAF [1544] и Красно^
вым и др. [167] (даны коэффициенты полинома, аппроксимирующего значения Ф° (Т) при
Т <^ 5000 К). Расхождения данных [1544] и табл. 803 составляют от 0,5 до 1,5 Дж-К~1-моль,
расхождения с данными [167] несколько больше.
Константа равновесия реакции СаВг (г)=Са (г)+Вг (г) вычислена с использованием принятой
в справочнике энергии диссоциации
АгН° @) = Do (СаВг) = 318 ± 10 кДж • моль да 26 580 + 850 см.
Это значение основано на результатах спектрофотометрического исследования в работе Гур-
вича и др. [1333] равновесия реакции Са (г)+НВг (г)=СаВг (г)+Н (г) в пламенах H2+O2+N2
по зависимости / (СаВг)// (Са) от температуры пламен A745—2430 К, 12 точек, расчет по II
закону дает 316+25 кДж-моль) и масс-спектрометрического исследования в работе Хилденбранда
[1411] равновесия реакции Са (г)+А1Вг (г)=СаВг (г)+А1 (г) A972—2158 К, 6 точек, расчеты
по II и III законам дают 346+70 и 318+9 кДж-моль). Изучение хемилюминесценции СаВг
в работе Менцингера [2007] привело к неверному и резко завышенному значению D08 +
+7 кДж-моль"), по-видимому, из-за наличия систематических ошибок в измерениях.
Приведенные погрешности учитывают воспроизводимость измерений, а также неточность сечений
ионизации, термодинамических функций и энтальпий образования, использованных в расчетах.
Принятому значению соответствует
&fH°(CaBr, г, 0) = —22,744 + 10 кДж • молзГ1.
СаВг3 (к, ж). Термодинамические свойства кристаллического и жидкого дибромида кальция
в стандартном состоянии при температурах 298,15—3800 К приведены в табл. 804.
Значения постоянных, принятых для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние СаВг2 (к) принята ромбическая
модификация СаВг2 (структурный тип СаС12) [731], устойчивая в интервале температур от 0 до 1015 К
(температура плавления СаВг2). При Т <^ 298,15 К данные по теплоемкости СаВг2 в литературе
отсутствуют. Принятые в справочнике (см. табл. 27.1) значения S0 B98,15 К) и Н°- B98,15 К)—Я0 @)
оценены, их погрешности составляют 4 Дж-К~1-моль~1 и 1,0 кДж-моль. Уравнение для
теплоемкости СаВг2 (к) в интервале 298,15—1015 К основано на результатах измерений энтальпии
СаВг2 (к) в работе Дворкина и Бредига [1003] вблизи точки плавления. Эти измерения привели
к значению Н° A015 К) — Н° B98,15 К)=58,6 кДж-моль. Температура плавления A015 +
+3 К) принята по хорошо согласующимся между собой данным [1003] A015 К), [1549] A013 К),
[1031] A018 К) и [2454] A016 К), а энтальпия плавления — по данным Дворкина и Бредига
[1003] B9,1+0,4 кДж-моль). С последним значением согласуются результаты менее точных
измерений, проведенных Джанзом и др. [1549] B8,3 кДж-моль) и Эмонсом и др. [1031]
330
Кальций и его соединения СаВгг (г)
<B9,4 кДж-моль). Значения теплоемкости СаВг2 (ж), найденные в работах [1003] и [1549] A13
и 114,5 Дж-К-моль соответственно) на основании измерений энтальпии в узком интервале
температур (~100 К), представляются сильно завышенными. Сопоставление более надежных
экспериментальных данных, полученных в широком интервале температур для СаС12 (ж), SrCl2 (ж),
ВаВг2 (ж) (см. табл. 27.1, 28.1 и 29.1), а также наблюдавшееся уменьшение значений С" (ж) в рядах
талогенидов Ва—Be1 дают основание принять для СаВга (ж) значение 100+5 Дж-К -моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 4, 5,
10 и 15 Дж-К~1-моль~1. Расхождения между термодинамическими функциями СаВг2 (к, ж),
приведенными в табл. 804 и в таблицах JANAF [1544], составляют 1,5 Дж-К-моль~1 в значениях
Ф°B000К).
В справочнике принята энтальпия образования
Д/Я°(СаВг2, к, 298,15 К) = —685 + 5 кДж • моль'1.
Эта величина основана на результатах измерений энтальпии растворения Са и СаВг2 в броми-
стоводородной кислоте (—685,5+5,0 кДж-моль), выполненных Эрликом и др. [1022].
Исследования энтальпии растворения СаВг2 в воде вместе с принятыми в справочнике энтальпиями
образования ионов (Приложение 5) приводят к величинам: —681,7+5,0 кДж-моль (Томсен [2686]),
—687,1 +5,0 кДж-моль (Пиккеринг [2228]) и —673,7 кДж-моль (Варэ [2757]). Эти измерения
энтальпий растворения выполнены еще в прошлом веке. В пределах погрешностей они приводят
к величинам, совпадающим между собой и с работой Эрлика и др. [1022] (кроме работы [2757],
которая, по-видимому, содержит ошибки).
Давление пара дибромида кальция СаВг2 (к, ж)=СаВг2 (г) вычислено на основании принятой
в справочнике энтальпии сублимации
Д,Я°(СаВг2> к, 0) = 298 + 8 кДж • моль.
Эта величина является средней из рассчитанных по результатам эффузионных измерений Пи-
терсона и Хатчисона [2217] A160—1320 К, 19 точек, данные представлены уравнением) и торзион-
ных измерений Хилденбранда [1412] (934—1287 К, 35 точек, данные представлены уравнением).
Соответствующие значения равны 293 и 292+6 кДж-моль (расчет по II закону) и 299,4+1 и
297,5+1 кДж-моль (расчет по III закону), где приведенные погрешности характеризуют
воспроизводимость измерений. Существенно менее надежные данные получены из старых работ Штока,
Хайнемана [2617] A точка, 285 кДж-моль) и Вартенберга, Боссе [2801] B точки, 287 +
+3 кДж-моль). Погрешность принятой величины учитывает неточность термодинамических
•функций СаВг2 (к, ж) и СаВг2 (г).
СаВг2 (г). Термодинамические свойства газообразного дибромида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 805.
Молекулярные постоянные, использованные для расчета термодинамических функций СаВг2,
представлены в табл. 27.8. Момент инерции в основном электронном состоянии Х1^ рассчитан
лри г (Са—Вг)=2,67 +0,03 Аи4 Вг—Са—Вг=180° в соответствии с результатами электроногра-
фического исследования паров СаВг2 в работе Акишина и др. [5, 11]. Исследование отклонения
молекулярного пучка СаВг2 в неоднородном электрическом поле подтверждает линейную
структуру этой молекулы [2839]. Погрешность приведенного значения / составляет 1-Ю8 г-см2.
Частота v3 найдена Байковым [24] при исследовании инфракрасного спектра паров СаВг2. Частота
\ рассчитана по формуле поля валентных сил на основании принятого значения v3 и в
предположении, что по аналогии с дигалогенидами магния для молекулы СаВг2 справедливо соотношение
/r//rr=15. Значение v2 также вычислено по уравнениям поля валентных сил для /г//а=65; эта
величина принята на основании того, что в молекулах СаС12 и Са12 соответствующее отношение равно
60 и 70. Погрешности принятых значений частот оцениваются в 20—25 см (см. текст по СаС12 (г)).
Сведения о возбужденных электронных состояниях СаВг2 отсутствуют, можно предполагать,
что их энергии превышают 20 000—25 000 см.
1 Исключениями из этой закономерности, крэмэ СаВг3, являются такжэ ВеС12 и SrBr2, значения теплоемкости
которых получены из измерений энтальпии в относительно узких интервалах температур.
331
Cal (г) . Глака 27
Термодинамические функции СаВг2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных величин обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета и составляют 4, 6 и 8 Дж-К~1-моль
в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно (в том числе 3—4 Дж-К~1-моль
из-за неточности молекулярных постоянных).
Ранее термодинамические функции СаВг2 (г) рассчитывались в таблицах JANAF 11544] в
работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К).
Расхождения в термодинамических функциях, вычисленных в настоящем издании и в работах [195, 748,
1544], объясняются тем, что в последних приняты менее надежные значения частот, в частности
v2; эти расхождения достигают 6, 2 и 5 Дж -К -моль в значениях Ф° (Г) соответственно (см. текст
по СаС1г(г)).
Константа равновесия реакции СаВга (г)=Са (г)+2Вг (г) вычислена с использованием
значения Дг#° @)=786,929+9 кДж-моль, соответствующего принятым в справочнике энтальпия»
образования и сублимации СаВгг (к). Этим величинам также соответствует
2, г, 0) = —373,75 + 9 кДж • моль.
Cal (г). Термодинамические свойства газообразного иодида кальция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 806.
В табл. 27.10 приведены молекулярные постоянные Cal, принятые в справочнике на основании*
анализа результатов исследований электронных спектров этой молекулы. В спектрах Cal
наблюдались переходы между пятью электронными состояниями: 4!П**Х22+, 52Б+*^Х2? + [927,
1378, 1694, 2645, 2795], С2П <* X2 2+ [1378, 1921, 2011, 2087, 2645, 2795], Z>2?+^ X2S + [1693„
2645, 2795]. Состояния X2 2+ и А2П молекулы Cal коррелируют с атомами Са A5)+1 BР), а три
остальные — с пределом Са (аР)+1 BР). С последним должен также коррелировать ряд других*
в том числе квартетных состояний, среди которых могут быть связанные состояния (см. CaF).
Колебательные постоянные Cal в состоянии X2 2+ определялись из анализа систем ^42П—Х%Ъ*~
[927, 2645], ?2?+-Х2Е+ [927, 1694, 2645] и Я2?+-Х22 + [1693, 2645] и удовлетворительно
согласуются между собой. Приведенные в табл. 27.10 постоянные приняты по работе Кханна в
Дьюби [1693], где они получены из анализа системы ZJ2+—X2S + G2 полосы, v' <; 20, v" <J 18).
Анализ вращательной структуры спектров Cal не проводился; приведенная в табл. 27.10
вращательная постоянная рассчитана при гв (Cal) =2,85+0,05 А на основании величины г (Са—1)=
=2,88+0,03 А в молекуле Са1а. Колебательные постоянные в состояниях А2Н и 2?2?+ приняты
по работе Даржи и Вайдиа [927], в состояниях С2П и D2E + найдены Марти и др. [2087] и Кханна
и Дьюби [1693].
Термодинамические функции Cal (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения <?вн и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом четырех
возбужденных состояний в предположении, что (?(j>J# вр = (pilpx)QifI] вр- Величина Qi*2 вр и ее ПР°"
изводные вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по вращательным уровням энергии с помощью уравнения
типа A. 82). В расчетах учитывались все уровни энергии со значениями / <С /тах,„, где /ши>,
находились из условий A. 81). Колебательно-вращательные уровни состояния Х2? + вычислялись
по уравнениям A. 65), A. 61). Значения коэффициентов Ykl в этих уравнениях, а также величин
ушах и /lim, использованных в расчетах, приведены в табл. 27.11. Коэффициент Y03 получен из
условия A. 59). Мультиплетность и вырождение уровней электронных состояний учитывались
соответствующими статистическими весами.
Погрешности вычисленных термодинамических функций Cal (г) при Т ^ 2000 К обусловлены
главным образом отсутствием экспериментальных данных о вращательных постоянных в
состоянии Х2Е +, а при более высоких температурах неполнотой сведений о возбужденных электронных
состояниях этой молекулы. Общие погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
могут достигать 0,3, 0,5 и 2 Дж-К~1-моль соответственно.
Ранее термодинамические функции Cal (г) вычислялись в таблицах JANAF [1544] и в работе
Краснова и др. [167] (приведен полином, аппроксимирующий Ф° (Т) для Т <^ 5000 К).
Расхождения данных [1544] и табл. 806 не превышают 1 Дж-К^-моль в значениях Ф° (Т) и ?° (Т);
расхождения с данными [167] достигают 2 Дж-К~1-моль.
332
Кальций и его соединения Cala (к, ж)
Константа равновесия реакции Cal (г)=Са (г)+1 (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ° @) = Do (Gal) = 270 + 8 кДж • моль» 22 600 + 700 см.
Это значение основано на выполненном Клейншмидтом и Хилденбрандом [17166] масс-спектро-
метрическом исследовании равновесия реакции Са (г)+АП (г)=Са1 (г)+А1 (г) A541—1674 К,
10 точек). Обработка результатов этих измерений привела к значениям Do (Cal) =270 +
±30 кДж-моль (II) и 270+8 кДж-моль" (III). Погрешности включают воспроизводимость
измерений, неточность сечений ионизации, термодинамических функций и используемого в
расчете значения Do (All). Исследование хемилюминесценции, возникающей в ходе реакции Са (г)+
+HI (r)=CaI(r)+H (г) в молекулярных пучках [2032], приводит к значению Do (Cal)^272 кДж-
•моль, согласующемуся с принятой величиной, ей соответствует
AfIIo(CaI, г, 0) = 14,496 + 8 кДж • моль.
Са12 (к, ж). Термодинамические свойства кристаллического и жидкого дииодида кальция
в стандартном состоянии при температурах 100—4500 К приведены в табл. 807.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние Са12 (к) принята гексагональная модификация
(структурный тип Cdl2), устойчивая в интервале температур от 0 до 1056 К (температура
плавления Са12). При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости, проведенных Пауковым и др. [314] A3—309 К) на чистом образце Са12 (сумма
примесей <^0,01%). Точность данных [314] оценивается в 1% при Т < 30 К
и 0,2% при Т > 50 К. Экстраполяция к Т=0 по закону Дебая приводит к S° A3 " К) =
=2,04 Дж-КГ1-моль. Погрешности принятых значений 5° B98,15 К) и Н° B98,15 К)—Н° @)
оцениваются в 0,4 Дж -К -моль и 0,04 кДж -моль соответственно. Уравнение для теплоемкости
Са12 (к) в интервале 298,15—1056 К выведено по результатам измерений энтальпии в работе Двор-
кина и Бредига [1003], приводящих к значению Н° A052 К)—Я0 B98,15 К)=63,2 кДж-моль.
Температура плавления A056+3 К) получена Мак-Крири [1980] для спектрально чистого
образца Са12. Результаты измерений Тт в работах [1031] A053 К) и [1003] A052 К) относятся к
образцам, состав которых не охарактеризован. Энтальпия плавления D1,8+1 кДж-моль") принята
по данным Дворкина и Бредига. Эмонс и Лёфельхольц [1031] вычислили из данных по
диаграммам состояния Са12 с Nal и KI гораздо более низкую величину B8+2 кДж-моль). Возможно,
что причиной этого расхождения является не установленный до настоящего времени полиморфизм
Са12. В этом случав принятая энтальпия плавления Са1г представляет сумму энтальпий плавле-
ния и полиморфного превращения Са12. Теплоемкость жидкого Са12 A03 +5 Дж -К -моль'1) также
принята по данным Дворкина и Бредига, которые измерили энтальпию Са12 (ж) в узком
интервале температур A056—1100 К). Погрешности вычисленных значений Ф° (Т) при 298,15, 1000,
2000 и 4000 К оцениваются в 0,3, 1, 6 и 18 Дж-К~1-моль~1 соответственно. Расхождения между
термодинамическими функциями Са12 (к, ж), вычисленными в справочнике JANAF [1544] и
приведенными в табл. 807, не превышают 0,5 Дж-К~1-моль~1 в значении S° B500 К).
В справочнике принята энтальпия образования
Д//°(Са12, к, 298, 15 К) = —538 + 8 кДж • моль,
основанная на измерениях энтальпии растворения Са12 (к) и металлического кальция в растворах
иодистоводородной кислоты в работе Эрлика и др. [1022]. Погрешность этих измерений связана
с тем, что реакции проводились в негерметичном калориметре, а также с возможностью
взаимодействия образующегося водорода с медной сеткой, в которую были завернуты образцы металла
(см. текст по ВаН2 (к, ж)).
Давление пара дииодида кальция Са12 (к, ж)=Са12 (г) вычислено на основании принятой
в справочнике энтальпии сублимации
Д//°(Са12, к, 0) = 278 + 5 кДж • моль.
Это значение получено при обработке результатов эффузионных измерений давления пара
Са1г, выполненных Питерсоном и Хатчисоном [2217] A075—1294 К, 18 точек) и Хилденбрандом
333
CaI2(r), CaS (к, ж) Глава 27
[1413] (970—1035 К, 14 точек). Результаты этих измерений представлены только в виде
уравнений. Обработка соответствующих данных привела к величинам 300 и 286 кДж-моль (II закон)
и 278+2 и 278,7+0,5 кДж-моль (III закон) соответственно. Погрешность принятого значения
определяется в первую очередь неточностью термодинамических функций Са12 (к, ж) и особенно-
Са12 (г).
Са12 (г). Термодинамические свойства газообразного дииодида кальция в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 808.
Молекулярные постоянные, использованные для расчета термодинамических функций Са12,.
приведены в табл. 27.8. Момент инерции в основном электронном состоянии Х1!** соответствует
значениям -^1—Са—1=180°, г (Са—I)=2,88+0,03 А, найденным Акишиным и др. [5, 11]вэлек-
тронографическом исследовании паров Са12. Данные [5, 11] согласуются с результатами
повторного электронографического исследования Са12, выполненного Каспаровым и др. [1586, 158г,
1046] (-$ I—Са—1=180°, г (Са—1)=2,87+0,01 А). Погрешность принятого значения /
составляет 1-10~38 г-см2. Частота v3 найдена Байковым [24] при исследовании инфракрасного спектра
паров Са12. Значение v2 вычислено Каспаровым и др. [1586, 158г, 1046] по эффекту сокращения»
наблюдавшемуся при электронографическом исследовании Са12. Частота vx рассчитана по
уравнениям поля валентных сил на основании принятой величины v3 и предположения, что по аналогии
с дигалогенидами магния для молекулы Са12 справедливо соотношение /г//гг=15. Погрешности
принятых значений всех частот оцениваются в 10—20 см. Сведения о возбужденных электронных
состояниях Са12 отсутствуют, можно предполагать, что их энергии превышают 20 000 см.
Термодинамические функции Са12 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных функций обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета и могут составлять 5, 7 и:
10 Дж-К-моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно (в том числе
3—5 Дж-К-моль из-за неточности молекулярных постоянных).
Ранее термодинамические функции Са12 (г) рассчитывались в таблицах JANAF [1544] и
работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Расхождения
между термодинамическими функциями, вычисленными в работах[748, 1544] и данными табл. 808,
не превышают 1 Дж-К-моль в значении Ф° (Т). В [195] принято неверное значение v2 и
расхождения достигают 9 Дж-К-моль.
Константа равновесия Са12 (г)=Са (г)+21 (г) вычислена с использованием значения &ГН° @) =
=650,703+9,4 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации Са12 (к). Этим величинам также соответствует
\fB° (Cal2, г, 0) = —259,044 + 9,4 кДж ¦ моль.
CaS (к, ж). Термодинамические свойства кристаллического и жидкого сульфида кальция
в стандартном состоянии при температурах 100—5500 К приведены в табл. 809.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 27.1. В справочнике за стандартное состояние CaS (к) принята кубическая модификация
(структурный тип NaCl) [1459а]. При Т ^ 298,15 К термодинамические функции вычислены
по данным о теплоемкости CaS, полученным Андерсоном [508а] E8—295 К) на образце, состав
которого совпадал с теоретическим в пределах погрешности химического анализа (+0,1 %).
Экстраполяция теплоемкости ниже 58 К выполнена в работе [508а] по уравнению Сp—D B84/Г)+
+Е C69/Г) и привела к значению 5° E8 К) =4,6 Дж-К-моль~1. Погрешности принятых по
[508а] значений. S° B98,15 К) и Н° B98,15 К)—Н° @) (см. табл. 27.1) оцениваются в 1 Дж-К'хХ
Хмоль и 0,1 кДж-моль соответственно.
В литературе отсутствуют какие-либо данные по теплоемкости или энтальпии CaS (к, ж) выше
298,15 К. В интервале 298,15—2800 К для теплоемкости CaS (к) принято уравнение (см. табл. 27.1),
выведенное по значениям Ср B98,15 К), С°р E00 К)=50 Дж-К^-моль и С°р B000 К) =
=63 Дж-К~1-моль~1, последние два значения оценены с учетом данных по теплоемкости оксидов
других щелочноземельных металлов. Погрешности оцененных значений теплоемкости CaS (к)
составляют —10% при 1000 К и —15% при 2000 К. Температура плавления B800 +50 К) принята
по работе Юза и Бунзена [1608а], где она была определена методом термического анализа. Дан-
334
Кальций и его соединения
CaS (к, ж)
ные Фогеля и др. [2781] B723 К) менее надежны, так как получены далекой экстраполяцией
кривой ликвидуса системы CaS—FeS. Энтальпия плавления G0+15 кДж-моль) оценена по
приближенному эмпирическому правилу [178П, согласно которому энтропия плавления ионных
соединений с решеткой типа NaCl составляет около 25 Дж-К~1-моль. Теплоемкость CaS (ж) F7 +
+15 Дж-К~1-моль) оценена по эмпирическому правилу (см. т. 1, гл. 2).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 5000 К оцениваются в 1,
5, 12 и 20 Дж ¦К~1-моль~1 соответственно. Термодинамические функции CaS (к), вычисленные
в справочниках [578, 2031 и 2298], согласуются с приведенными в табл. 809 в пределах
2 Дж-К-моль в значениях S° B000 К); имеющиеся расхождения обусловлены разной оценкой
теплоемкости CaS (к) при высоких температурах.
В справочнике принята энтальпия образования
AfH
(CaS, к, 298,15 К) = —475 + 8 кДж • моль.
Эта величина вычислена как средневзвешенная из значений, приведенных в табл. 27.17, их
погрешности включают воспроизводимость измерений, неточности использованных в расчетах
сечений ионизации и термодинамических свойств. Результаты калориметрических измерений и
исследований равновесия реакций с участием CaS (к) находятся в удовлетворительном согласии. В
работе [873] отсутствуют измеренные константы равновесия. В таблице даны величины, вычисленные
на основании найденных в работе [873 ] интенсивностей ионных токов и принятых в
справочнике сечений ионизации и распространенности изотопов.
Таблица 27.17. Результаты определений AfH° (CaS, к, 298,15 К) (в кДжмоль ]
Авторы
Метод
)
AfH° (CaS, к, 298,15 К)
II закон
III закон
Сабатье, 1881 [2417] Калориметрический, CaS (к)+6,6НС1 (р-р, —474+10
221Н2О)=СаС12 (р-р, 4,6НС1+1460Н2О)+4,6НС1
(р-р, 317H2O)+H2S(p-p), 283,7 К, 7 измерений
Вартенберг, 1943 [2800а] Калориметрический, CaS (к)+11НС1 (р-р, —482,7+4,0
18Н2О)=СаС12 (р-р, 9НС1+198Н2О)+9НС1 (р-р,
22H2O)+H2S (г) а, комнатная температура, 9
измерений
Завадзкий, Сиричинский, 1930 [2898] Статический, исследование равновесия реакции —623+40 —480+30
0,25CaS (K)+0,75CaSO4 (к)=СаО (k)+SO2 (t),
1172—1343 К, 11 точек
Шенк, Хаммершмидт, 1933 [2451] Протока, CaS (k)+2SO2 (r)=CaSO4 (k)+S2 (г), —501+15 —475+12
1124—1348 К, 6 точек
Фикс, 1956 [1158] То же, 1304—1373 К, 4 точки —497±15 —477±14
Розенквист, 1951 [2362а] То же, CaS (к)+Н2О (г)=СаО (k)+H2S (г), 1031— —475+10 —473+8
1698 К, 12 точек
Уно, 1951 [2743а] То же, 1273—1373 К, 3 точки —474±30 —473+8
Филиповска, Белл, 1975 [1084а] То же, 1019—1704 К, 15 точек —479+10 —474±9
Сандер, Хейли, 1968 [2428а] То же, CaO (k)+V2S2 (r)=CaS (к)+1/М2 (г), —499+15 -478+14
1673—1923 К, 7 точек
Беркович Маркварт, 1963 [665а] Масс-спектрометрический, CaS (к)=Са (г)+ — —465+15
+1/2S2(r), 1709 К, 1 точка
Колен и др., 1964 [873] То же, 2/3Са (r)+4/3S2 (r)=2/3CaS (k)+2S (г), —442+50 -485+15
1849—2155 К, 7 точек
То же, 2072—2319 К, 7 точек (CaS окисленный) —470+100 —508±15
а В работе [2800а] измерения отнесены к реакции CaS (к)+2НС1 (р-р, 15Н2О)=СаС12 (р-р, 15H2O)+H2S (г), что
привело к значению AfH° (CaS, к, 298,15 К)=—476,9 кДж-моль-1.
Давление пара сульфида кальция в реакции CaS (к, ж)=СаЭ (г) вычислено с использованием
значения Дг#° @)= Д8Я° (CaS, к, 0)=595,986+17 кДж-моль, соответствующего принятым
в справочнике энтальпии образования CaS (к) и энергии диссоциации молекулы Caiv
335
CaS(r), CaSO4 (к, ж) Глава 27
CaS (г). Термодинамические свойства газообразного сульфида кальция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 810.
В табл. 27.4 приведены молекулярные постоянные CaS, принятые на основании анализа
результатов исследований электронного спектра этой молекулы и приближенных оценок. В спектре
CaS исследована только одна система полос 5Х2+—Хх?+ [711а]. По аналогии с молекулами
оксидов и сульфидов других щелочноземельных металлов молекула CaS должна иметь еще ряд низко-
лежащих синглетных и триплетных состояний, соответствующих конфигурациям . . .о2к3а (a3JJf
42П), . . .аА (Ь3?+), . . . ААг (»Дд, Х2-, 3S+) и . . .ои4иCП, ХП).
В табл. 27.4 приведены данные для 12 валентных состояний молекулы CaS. Молекулярные
постоянные в состояниях Х1'^ + и 5XS+ приняты по результатам анализа полос системы S1S+—
Х1!,*, (г/, v" ^ 2) в работе Блу и Барроу [711а]. Энергии возбуждения остальных электронных
состояний оценены по данным для молекул СаО, ВаО и BaS с погрешностью 2000—4000 см,
причем изоконфигурационные состояния объединены в группы с одинаковой энергией.
Термодинамические функции CaS (г) вычислены по уравнениям A. 3)—A. 10), и A. 93)—A/95).
Величина Qm и ее производные рассчитывались по соотношениям A. 90)—A. 92) с учетом 11
возбужденных электронных состояний в предположении, что G^ sp=(PilPx)Ql?2 „p* Статистическая
сумма по колебательно-вращательным уровням энергии состояния Х1^* и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием
по / с помощью уравнений типа A. 82). В расчете учитывались все уровни со значениями / ^
.<! /шах_ „, последние находились по соотношению A.81). Энергия колебательно-вращательных
уровней рассчитывалась по уравнениям A. 65) и A. 64а) с коэффициентами Ykl, приведенными,
в табл. 27.5 вместе со значениями ушах и /1Ш. Значения Y м получены по постоянным из табл. 27.4
с учетом условий A. 50) и A. 59), для соблюдения которых в уравнение A. 65) введены
дополнительные члены с коэффициентами Y30, Yi0 и Y03. Постоянная Y31 определена из условия Bv +i=0.
Все постоянные Ykl пересчитаны по соотношениям A. 66) для изотопической модификации
молекулы CaS, соответствующей природной смеси изотопов.
Погрешности вычисленных термодинамических функций при температурах до 1000 К в
основном обусловлены отсутствием данных о колебательно-вращательных уровнях состояния ZLS +
со значениями v ]> 2. При более высоких температурах погрешности обусловлены главным
образом неточностью оцененных энергий возбужденных состояний (особенно а3П и Ь32+), а также
пренебрежением ридберговскими состояниями CaS. Погрешности в значениях Ф° (Г) при 298,15,
3000 и 6000 К оцениваются в 0,1, 1 и 3 Дж-К^-моль.
Таблица термодинамических функций GaS (г) публикуется впервые. В работе Колина и др.
[873] приводятся значения Ф° (Т) в интервале 1700—2300 К, отличающиеся от данных табл. 810
на 8 Дж-К-моль~1. Расхождения обусловлены тем, что авторы [873] провели расчет в
предположении, что эта молекула имеет основное электронное состояние 32.
Константа равновесия CaS (г)=Са (r)+S (г) |вычислена с использованием принятого в
справочнике значения
,-1
АгН° @) = Do (CaS) = 330 + 15 кДж . моль «* 27 600 + 1200 см
Эта величина основана на масс-спектрометрических исследованиях равновесия реакции Са (гL-
+S2 (r)=CaS (r)+S (г), выполненных Марквартом, Берковицем [1951а] A709 и 1962 К, 2 точки)
и Колином и др. [873] B058—2319 К, 9 точек). По данным этих работ найдены близкие значения
энергии диссоциации 330+15 (III) и 328A1) [1951а] и 334+15 (III) и 328+30 (II) кДж-моль
[873]. Линейная экстраполяция уровней состояния X1!! приводит к значению около
250 кДж-моль.
Принятой величине соответствует
A/ff°(CaS, г, 0) = 122,118 + 15 кДж • моль.
CaSO4 (к, ж). Термодинамические свойства кристаллического и жидкого сульфата кальция
р стандартном состоянии при температурах 100—3000 К приведены в табл. 811.
336
Кальций и его соединения CaSO4 (к, ж)
Значения постоянных, принятых для расчета термодинамических функций, представлены
в табл. 27.1. За стандартное состояние CaSO4 (к) в интервале 0—1473 К принята ромбическая
модификация, а в интервале 1473—1733 К высокотемпературная кубическая модификация 1.
При Т ^ 298,15 К термодинамические функции CaSO4 (к) вычислены на основании измерений
теплоемкости, выполненных Келли и др. [1660] E0—296 К) на образце природного
ангидрита (CaSO4), содержавшего —0,09% гипса. Экстраполяция теплоемкости проводилась
по уравнению Ср {T)=D B08/Г)+2? C00/Г)+2? (815/Г) и привела к значению S° E0 К) =
=6,3 Дж -К -моль. Погрешности табулированных значений S° B98,15 К) и Н° B98,15 К)—Н° @)
составляют 2 Дж-К-моль~1 и 0,2 кДж-моль соответственно. Данные Коппа [1755]
по средней теплоемкости CaSO4 в интервале 290—320 К выше данных [1660] приблизительно
на 1%.
При высоких температурах измерения энтальпии GaS04 (к) проведены в работе Лащенко и Ком-
панского [220] C73—1393 К) на образце, чистота которого не была характеризована.
Ввиду несовершенства методики измерений данные [220] имеют разброс до 10% и приводят, по-
видимому, к резко завышенным значениям энтальпии и теплоемкости CaSO4. В справочнике в
интервале 298,15—1473 К для теплоемкости приняты результаты расчетов, проведенных по методу
Сакамото [2424, 2425] с учетом различных составляющих теплоемкости по уравнению: Ср =
=22) {277/Т)+_Е C56/Г)+B/3) Е F56/Г)+?(916/Г)+A/3) Е A465/Г)+? A629/Г)+24,85-10-6 С/Т
(С°р в Дж-К-моль~1) 2. Уравнение для теплоемкости CaSO4 (см. табл. 27.1) выведено
на основании С'р B98,15 К) =99,66 [1660], С°р E00 К)=127 и С\ A000 К)=160 Дж-К^-моль;
последние рассчитаны по приведенному уравнению. Температура превращения A473+10 К)
принята на основании согласующихся данных [88, 642, 949, 1109, 1244, 2231]. Результаты измерений
[439, 1300, 1959, 2134] менее точны и не принимались во внимание. Энтальпия превращения E +
+2 кДж-моль) оценена при подготовке справочника по термографическим данным,
полученным Шаргородским [439], аналогично соответствующей оценке для MgSO4. Меньшая точность
значения энтальпии превращения CaSO4 (по сравнению с MgS04) обусловлена тем, что в работе
[439] измерения выполнены для гипса (CaSO4-2H2O), а не безводного CaSO4. Температура
плавления A733+10 К) принята по данным Роу и др. [2389, 2390] A735 К), Голубевой и др. [88]
A723 К), Гривзона и др. [1263] A738 К), флерке [1109] A723 К) и Грамана [1244] A723 К).
Данные [642, 803, 1300, 1740, 2735] менее точны и не учитывались. Энтальпия плавления B8 +
+3 кДж-моль) рассчитана из диаграмм состояния двойных систем CaSO4 с сульфатами
щелочных металлов [1233, 1650].
Теплоемкости высокотемпературной модификации A473—1733 К) и GaSO4 (ж) оценены по
аддитивной схеме с учетом данных по теплоемкости хлоридов и сульфатов щелочных и
щелочноземельных металлов равными 165+20 Дж-К-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000,2000 и 3000 К составляют 2,
7, 20 и 30 Дж-К-моль соответственно. Расхождения между термодинамическими функциями
CaSO4 (к), приведенными в справочниках [578, 1656] и табл. 811, достигают 19 и 3 Дж-К-моль
соответственно в значениях Ф° A000 К) и вызваны тем, что в [578, 1656] приняты завышенные
значения энтальпии CaSO4 по работе [220]. Расчеты термодинамических функций CaSO4 (ж)
в справочной литературе отсутствуют.
Константа равновесия реакции CaSO4 (к, ж)=Са (r)+S (г)+4О (г) вычислена на основании
значения \ГН° @)=2863,444 +1,8 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
^fH°(GъSOv к, ромб., 298,15 К) = —1434,5 + 1,5 кДж • моль.
Принятая величина основана на приведенных в табл. 27.18 результатах измерений. Помимо
приведенных в табл. 27.18 работ, известны еще исследования [753, 838, 1115, 1169, 1386, 1398,
1 Известны еще две метастабилыше модификации гексагональной сингонии (а- и ^-«растворимые»
модификации CaSO4, получаемые при термической дигидратации a-H^-GaS04 ¦ 0,5 Н2О соответственно). Монотропный
переход «растворимых» модификаций в ромбическую наблюдался в интервале 493—643 К.
2 Использованные в этом уравнении параметры и постоянная в выражении для составляющей С°р—С„ оценены на
основании данных [1660] по теплоемкости CaS04 в интервале 50—298 К с учетом аналогичных параметров для
BaSO4 и PbSO4.
337
СаСО3 (к, ж)
Глава 27
1651, 1944, 2018, 2040, 2447, 2451, 2686, 2721, 2725, 2738, 2898], которые>озволяют вычислить
энтальпию образования CaSO4, однако в них нет указаний, какая из кристаллических
модификаций CaSO4 была исследована, или они уступают по точности работам, приведенным в таблице.
Как видно из таблицы, большое число исследований посвящено измерению^энтальпии дегидратации
Та блица 27.18. Результаты определений
(CaSO4, к, ромб., 298,15 К) (в кДж-моль)
Авторы
Метод
A,.ff°(CaSO4, к, ромб.,
298,15 К)
II закон
III закон
Форкран, 1906 [1115] Калориметрическое измерение энтальпии реак- —1437,3
ции CaSO4-2HaO (к, nrac)=GaS04 (к, ромб.)+
+2Н2О (ж)
Ньюмен, Уэлс, 1938 [2135, 2136] То же —1435,4
Саузард, 1940 [2587] То же —1434,9±1,3
Леви, 1948 [1851] То же —1434,9±1,3
Ейтс, Маршалл, 1969 [2884] Измерение растворимости CaSO4 (к, ромб.) и —1428,4 —1433,6±1,8
обработка литературных данных [1430, 1743,
1905, 2260, 2359]
Завадзкий, 1932 [2896], Исследование равновесия реакции CaSO4 —1430+25 —1435+17
(к, ромб.)=СаО (k)+SO2 (r)+VgO2 (г),
статический метод, 1453—1643 К, 11 точек
Чаппат, Пис, 1956 [2735] То же, 1425—1700 К, результаты измерений —1378 —1430±17
представлены в виде уравнения
Дьюинг, Ричардсон, 1959 [949] То же, метод ДТА, 1173—1423 К —1423 —1439±15
Лау и др., 1977 [1822] То же, эффузионный метод, 1143—1266 К, —1454±20 —1437,1+9
31 точка, три разных эффузионных отверстия
гипса CaSO4-2H2O. В расчетах использовано значение b.fH° (CaSO4-2H2O, к, гипс, 298,15 К) =
=—2023,4+1,3 кДж-моль, принятое по справочнику [406] с учетом отличающихся энтальпий
образования ионов Са+2 (aq) и SOj2 (aq), приведенных в Приложении 5. Принятое в [4063 значени&
основано на энтальпии растворения гипса, измеренной Ланге и Дюрром [1809], и энтальпиях
разведения, измеренных Ланге и Штреком [1810а]. Приведенные в табл. 27.18 погрешности
значений b.fH° (CaSO4, к, ромб.), найденных калориметрическим методом [1851, 2587], определяются
неточностью использованного в расчетах значения энтальпии образования гипса.
При оценке погрешностей величин, вычисленных из данных по равновесию реакции
диссоциации, учтена неточность термодинамических функций CaSO4 (к, ромб.); соответствующие вклады
составляют 7 и 17 кДж-моль для измерений, выполненных при 1000 и 1500 К соответственно.
В работе [1822] получено бблыпее значение энтальпии образования (—1442,6 кДж-моль), что
объясняется различием использованных в расчетах термодинамических функций.
Как видно из табл. 27.18, наиболее точные данные получены при исследовании растворимости
[2884] и в калориметрических измерениях [1851, 2587]. Принятое значение является средневзве-
шенным из этих работ. Имеющиеся в работах [1851, 2587] данные по энтальпии растворения
гексагональных а- и |3-растворимых модификаций CaSO4 позволяют найти их энтальпии
образования, равные —1426,0+1,5 кДж-моль и —1421,6+1,5 кДж-моль соответственно.
СаСО3 (к, ж). Термодинамические свойства кристаллического и жидкого карбоната кальция
в стандартном состоянии в интервале 100—2000 К приведены в табл. 812.
Значения постоянных, принятые в расчетах термодинамических функций СаС08 (к, ж),
представлены в табл. 27.1. За стандартное состояние СаСО3 (кальцит) принята гексагональная
модификация — единственная равновесная модификация вплоть до температуры плавления. Для
карбоната кальция известны также ромбическая модификация (арагонит) и псевдогексагональная
модификация (ватерит), которые при нагревании претерпевают монотропные переходы в кальцит
(при 600—700 и —840 К соответственно). При Т ^ 298,15 К термодинамические функции
вычислены на основании измерений теплоемкости кальцита в работе Стейфели и Линфорда [26071
338
Кальций и его соединения
СаСО3 (к, ж)
A1—303 К). Погрешности этих измерений оцениваются в 0,1—0,2% (при Т > 30 К), а
погрешности принятых по [2607] значений 5° B98,15 К) и Н° B98,15 К)-#° @) (см. табл. 27.1)-,
в 0,3 Дж -К -моль и 0,04 кДж -моль соответственно. Результаты измерений теплоемкоети
кальцита в более ранних работах [509, 2115, 2524] менее точны и выполнены в более узком
интервале температур; данные Андерсона [509] C7—296 К) приводят к значению S° B98,15 К) —
=93 Дж-К~1-моль, которое на 1,5% выше приведенного в табл. 27.1.
В интервале 298,15—1603 К для теплоемкости кальцита принято уравнение, выведенное на ос*
новании табличных данных по энтальпии СаСО3, выбранных Келли [1656] по результатам измерен
ний Гронова и Швите [1271] B93-1173 К), Кобаяши [1738] B98-1053 К) и Магнуса [1907] B89-^
1029 К). Погрешности этих данных по энтальпии составляют 0,5%. Температура конгру^
энтного плавления A603+10 К) принята по работе Бейкера [560], где она была определена под
внешним давлением СО2 в 1000 атм. Данные работ [1121, 2564] менее надежны и приводят к
величинам в пределах 1520—1612 К. Температура инконгруэнтного плавления СаСО3 в тройной точке
(рсо =40 атм) находится в пределах 1460—1515 К. Энтальпия плавления СаСО3 C6±
+4 кДж-моль) принята по результатам исследования термического разложения СаСО3 в рас^
плаве карбонатов щелочных металлов; расчетные данные Келли [1650] E3 кДж-моль) и данные
работы [1108] A4 кДж-моль), по-видимому, ошибочны. Теплоемкость СаСО3 (ж) оценена равной
160+20 Дж-К-моль~1 с учетом экспериментальных данных по теплоемкости расплавленных
соединений щелочноземельных и щелочных металлов.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К составляют 0,3, 1,5 и
8 Дж-К-моль соответственно. Расхождения между термодинамическими функциями СаСО3 (к),;
приведенными в табл. 812 и в справочниках [578, 1543, 1656], составляют 3 Дж-К^-моль"*
в значениях Ф° (Г). Расчеты термодинамических функций СаСО3 (ж) в литературе отсутствуют.
Константа равновесия реакции СаСО3 (к, ж)=Са (г)+С (г)+30 (г) вычислена с
использованием значения ДГ?Г° @)=2830,144 +1,4 кДж -моль, соответствующего принятой в справок
нике энтальпии образования
Д^ЧСаСОд, к, кальцит, 298,15 К) = —1206,6 + 1,0 кДж • моль.
Эта величина основана на результатах измерений, приведенных в табл. 27.19, где в качестве
погрешностей указана их воспроизводимость. В таблице представлены только результаты
обработки по III закону данных, полученных в наиболее детальных исследованиях равновесия реакции
СаСО3 (к)=СаО (к)+СО2 (г).
Т а б л и и, а 27.19. Результаты определений A/Jff° (CaCO3, к, 298,15 К) (в кДж-моль)
Авторы
Бекстрём, 1925 [553, 554]
Рот, Чолл, 1928 [2381]
Бильтц и др., 1934 [690]
Рот и др., 1941 [2374]
Келли, Андерсон, 1935 [1657]
Якобсон, Лангаюр, 1974 [1536]
[1339, 1432, 1591, 1593, 2564, 2589]
[524, 1548, 2655]
Метод
Калориметрическое измерение энтальпии
растворения СаО и СаСОз (кальцит) в соляной кислоте,
25° С
То же, 50° С
То же, 90° С
То же, 20° С
Анализ результатов измерений [1134, 1434, 1667]
растворимости СаСОз
То же, по работам [464. 476, 1134, 1172, 1261,
1344, 1434, 1667, 2099], с учетом образования
СаСОз (ад) и СаНСОз+ (ад)
Статический, равновесие реакции СаСОз (к)=
—СаО CkI+COo (г)
То же
Д,Я°(СаСО„ к,
298,15 К)
—1206,8
—1206,4±1,0
—1207,0+1,0
—1205,6+1,0
-1206,5+2,0
—1207,3+1,5
-1208,0+2,0 (Ш)
-1206,0+2,0 (III)
339
СаСО3 (к, ж) Глава 27
Как видно из табл. 27.19, результаты калориметрических измерений [553, 554, 690, 2374,
2381], исследований растворимости (см. [1536, 1657]) и исследований равновесия [524, 1339, 1432,
1548, 1591, 1593, 2564, 2589, 2655, 2763] оказались в прекрасном соответствии и принятое значение
является средневзвешенным из этих работ. При оценке погрешностей величин, вычисленных
по результатам исследований равновесия диссоциации СаСО3 (к), учтена неточность
термодинамических функций этого соединения (около 1,2 кДж-моль для измерений при 1000 К).
Равновесие реакции диссоциации СаСО3 (к) изучалось также в работах [140, 555, 560, 565,
1001, 1107, 1108, 1121, 1434, 1523, 1760, 1886, 1969, 2240, 2423, 2847, 2897]. Известны также более
старые калориметрические работы [669, 1070, 1502, 2686, 2828] и работа [529], где исследовалась
зависимость энтальпии растворения СаСО3 от степени его измельчения.
Арагонит по сравнению с кальцитом является термодинамически менее устойчивой
модификацией (свободная энергия его образования менее отрицательна). Однако энтальпия образования
арагонита, вычисленная с использованием энтальпии перехода кальцита в арагонит по данным
[553, 554, 2381], более отрицательна и равна Af #°(СаСО3, к, арагонит, 298,15 К) =—1206,8 кДжХ
Хмоль-
Глава 28
СТРОНЦИЙ И ЕГО СОЕДИНЕНИЯ
Sr (к, ж), Sr, Sr+, Sr2, SrO (к, ж), SrO, SrO+, SrH, SrH2 (к, ж), SrOH, SrOH% Sr(OHJ (к, ж).
Sr(OHJ, SrF, SrF+, SrF2 (к, ж), SrF2, SrCl, SrCl+, SrCla (к, ж), SrCl2, SrBr, SrBr2 (к, ж)
SrBr2, Sri, Srl2 (к, ж), Srl2, SrS (к, ж), SrS, SrSO4 (к, ж), SrCO3 (к, ж)
В настоящей главе рассматриваются свойства 23 веществ — стронция и его соединений с кисло»
родом, водородом, галоидами, серой и углеродом, для 11 веществ приводятся данные о
термодинамических свойствах в конденсированном состоянии и для 20 веществ — в газообразном состоянии.
Для четырех газов (Sr, Sr+, SrO и SrO+) термодинамические свойства рассчитаны до 10 000 К,
для одного (Sr2) — до 3000 К и для всех остальных — до 6000 К. В Приложении 3.9 приведены
сведения о критических постоянных стронция.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов стронция
(атомный вес 87,62) и других элементов. Однако различие молекулярных постоянных
индивидуальных изотопических модификаций соединений стронция, как правило, не принималось во
внимание из-за отсутствия соответствующих данных и их несущественного влияния на
термодинамические функции соответствующих газов х.
В справочнике не рассматриваются многоатомные окислы стронция, такие, как SrO2, и
полимерные соединения типа Sr2O2 и Sr2F4, ввиду их низкой стабильности. Не рассматриваются также
ионы типа SrBr+, Srl+ и SrFJ ввиду невысоких энергий диссоциации соответствующих
нейтральных двухатомных молекул и высоких потенциалов ионизации трехатомных молекул. Образование
таких соединений в равновесных условиях в рассматриваемом интервале температур в заметных
количествах представляется маловероятным.
Sr (к, ж). Термодинамические свойства кристаллического и жидкого стронция в стандартном
состоянии при температурах 100—3400 К приведены в табл. 813.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 28.1. В справочнике за стандартное состояние Sr (к) в интервале 0—828 К принимается
кубическая плотноупакованная модификация (a-Sr, структурный тип Си), а в интервале 828—
1041 К — кубическая объемно-центрированная модификация (y-Sr, структурный тип a-Fe) 2,
При Т <[ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Боэрио и Веструма [717] E—350 К). Исследованный образец Sr был тщательно
очищен фракционной сублимацией и длительным прокаливанием в вакууме при 1200 К, суммарное
содержание примесей не превышало 0,2%; точность измерения теплоемкости оценена в 0,1%
при Т > 25 К. Погрешности приведенных в табл. 28.1 значений S° B98,15 К) и Н° B98,15 К)—
Н° @) оцениваются в 0,2 Дж-К~1-моль и 0,02 кДж-моль соответственно. Данные Паукова и
Хриплович [426] по теплоемкости образца Sr E—300 К) чистотой 99,4% Sr в интервале 5—130 К
ниже данных Боэрио и Веструма [717], а в интервале 130—300 К — выше (при 298,15 К
расхождения составляют 8%); авторы [717] объяснили эти расхождения возможным содержанием примеси
SrO в образце, исследованном в работе [426]. Данные [426], так же как и менее точные данные
Робертса [2350] A,5—20 К) и Гласкока [1221а] B93 К), не учитывались в расчете.
При Т > 298,15 К экспериментальные сведения о теплоемкости, изменении энтальпии, а также
энтальпиях превращения и плавления Sr в литературе отсутствуют (за исключением измерений
[717] в интервале 298—350 К). При оценке этих величин учитывались экспериментальные данные
1 Получение чистых образцов стронция встречает большие трудности, причем основные трудности вызывает
анализ на содержание в них кислорода, водорода и азота и освобождение от соответствующих примесей. Поэтому
исследования термодинамических свойств стронция в конденсированном состоянии (за исключением работы [717])
и измерения энтальпии его реакций могут содержать значительные систематические ошибки, как правило не
учитывавшиеся авторами измерений, особенно в старых работах. Этим, по-видимому, объясняются имеющиеся
резкие противоречия в термохимии соединений стронция.
2 Данные [1443, 2342, 2472, 2505] о существовании в интервале 485—880 К еще одной промежуточной модифнка»
ции стронция (|3~Sr) не подтвердились в более поздних исследованиях. Петерсон и Колбурн [2214], изучая
образцы с различным содержанием примесей, показали, что промежуточная модификация стронция образуется
только в присутствии примесей водорода.
341
f а^бли
О
m
Вещест]
Sr
SrO
SrH2
Sr(OHJ
SrF2
SrCl2
SrBr2
Srl2
SrS
SrSO,,
SrCOs
Ha 28.1. Принятые значения термодинамических велячйн для
Состояние
кП, куб. (а)
к1, куб. (у)
ж
к, куб.
ж
Kl, ромб, (а)
к1(Р)
ж
кП, тетр. (Р)
К1(а)
ж
кП , куб. (а)
кП, куб. (а)
к1 (Р)
ж
КП, куб. (а)
КП, Куб. (а)
к1 (Р)
ж
кН, тетр. (а)
к1 (В)
ж
к, ромб.
ж
к, куб.
ж
КН, ромб, (а)
к1, гекс. (Р)
ж
кШ, ромб, (а
кП, гекс. (р)
к1, куб. (у)
ж
Примечание. C°p (T) —
SrF2: a e=112,052-10-»
SrCl2: a e=107 334,361 •
о.
о
аз
i
и
ю
оо" •
оз
о
аз
6,57
8,66
8,0
15,7
13,075
16,23
18,06
18,92
10,04
18,4
) 15,12
К
ю
оо"
см
о
55,7
55,44
52
94
82,13
114,9
143,44
159,1
68,2
121
97,2
а+ Ь Т—сТ"«+ dT*+ e T3
10"».
ю
во"
аэ
о R,
26,79
44,98
44
92,06
70,00
75,61
76,53
77,40
48,70
102
82,42
(в Дж-К-1
( стращия и
его соединений в твердом и жидком состояниях
Коэффициенты в уравнении для С°р (Т)
а
19,795
24,234
36
47,409
84
34,301
71
75
72,295
122
153
71,774
755,405
—99 525,372
99
80,081
—142 907,561
—5 103,108
106,298
70,504
100
100
72,531
110
47,533
67
109,691
170
170
152,093
146
146
165
моль).
6-Ю3
16,196
14,435
10,904
32,530
66,275
15,652
-831,256
8 202,188
7,619
353 767,359
3 081,265
20,761
24,029
7,746
53,431
—111,650
с ¦ 10-5
..10»
Интервал
температуры
К
—1,922 — 298,15—828
_ _ 828-1041
_ _ Ю41-3400
4,931 -1,514 298,15-2930
_ _ 2930-6000
— — 298,15—1128
_ _ 1128—1323
_ _ 1323—2000
— — 298,15—753
_ _ 753—808
__ _ 808—2000
5,727 — 298,15—800
1 028,041 150,407 а 800—1484
—435 411,224 —18 980,89 1484—1750
_ _ 1750—4300
5,998 — 298,15—700
—92 501,224 —327 222,233а 700—990
—22 055,203 — 990—1147
134,850 - 1147-3800
0,148 - 298,15-918
_ _ 918—930
- — 930-3900
2,037 — 298,15-811
- — 811-5300
1,016 — 298,15—2500
_ _ 2500—5200
20,993 — 298,15—1430
_ _ 1430—1880
_ _ 1880—4000
39,392 89,249 298,15—1198
_ _ 1198-1689
' _ _ 1689-1767
_ _ 1767-2500
или
Т
1 m
К
или
хё
Ш 2
828 0,75
1041 8,2
2930 70
1128 7,2
1323 23
753 0
808 23
1484 0
1750 28,5
990 1,0
1147 17,5
918 12,2
930 10,1
811 19,7
2500 63
1430 10
1880 36
1198 16,7
1689 0
1767 40
Стронций и его соединения
Sr (к, ж)
для кальция и бария, а также результаты расчета различных составляющих теплоемкости a-Sr
с использованием данных [717]. Уравнение для теплоемкости a-Sr (см. табл. 28.1) выведено по
значениям Ср B98,15 К) и Ср C50 К), полученным в этой работе, и оцененному значению Ср (828 К) =
=33,5 Дж-К~1-моль~1. Погрешность теплоемкости a-Sr, вычисленной по этому уравнению,
увеличивается с возрастанием температуры до 3—5% при 828 К. Уравнение для теплоемкости f-Sr
{828—1041 К) оценено с учетом данных для теплоемкости -[-Са; погрешности значений
теплоемкости, полученных по этому уравнению, составляют 10%.
Температура полиморфного превращения (828 +2 К) принята по измерениям Петерсона и Кол-
бурна [2214], выполненным методом ДТА. Она согласуется с данными авторов [1996] (830 К).
Работы [1443, 1637, 2342, 2472, 2505] (813—878 К) менее надежны из-за наличия большого
количества примесей в исследованных образцах, что приводило к появлению промежуточной
модификации |3-Sr. Энтальпия полиморфного превращения @,75+0,20 кДж-моль) принята по оценке,
сделанной Ринком [2342]. Температура плавления A041 +2 К) принята по согласующимся между
собой данным работ [1443, 2214, 2342, 2505]. Измерения [2472] A047 К) и [1075] A031 К) менее
надежны и не учитывались. Энтальпия плавления (8,2+2,0 кДж-моль) оценена по энтропии
плавления кальция, поскольку оба металла имеют одинаковую кристаллическую структуру и
близкие температуры плавления. Для теплоемкости жидкого Sr принято значение 36+5 Дж-К X
Хмель, полученное как среднее из теплоемкости жидких Ва и Са.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,2, 1,5, 4 и 6 Дж-К-моль. Расхождения между термодинамическими функциями,
приведенными в табл. 813 и в справочниках [105, 1544], при Т < 1000 К не превышают 2,5 и 3 Дж-К~1Х
Хмоль соответственно в значениях Ф° (Г). При 3000 К различия в значениях Ф° (Т)
составляют 4 и 1,5 Дж-К-моль~1, имеющиеся расхождения обусловлены разной оценкой теплоемкости
твердого и жидкого стронция.
В настоящем справочнике за стандартное состояние стронция принят Sr (к) и в соответствии
с этим
(Sr, к) =
AfH°
Давление пара стронция в реакции Sr (к, ж)=Эг (г) вычислено с использованием значения
ArH° @) = AsH° (Sr, к, 0)=160,873+2,0 кДж-моль, соответствующего принятой в справочнике
энтальпии сублимации
As#°(Sr, к, 298,15 К) = 160,5 + 2,0 кДж • моль
Это значение является средневзвешенным из рассчитанных по результатам измерений
давления пара стронция в работах, представленных в табл. 28.2. Приведенные в таблице погрешности
Таблица 28.2. Результаты определений &SH° (Sr, к, 298,15 К) (в кДж-моль)
Авторы
Руфф, Хартман, 1924 [2405]
Хартмая, Шнейдер, 1929 [1360]
Приселков, Несмеянов, 1954 [336]
Бурбом и др., 1963 [716]
Боданский, Шине, 1967 [718]
Шине и др., 1971 [2455]
Де Мария, Пьяченте, 1974 [943]
Асано и др., 1978 [538а]
Метод
Точек кипения, 1245—1412 К, 4 точки
То же, 1200—1380 К, 8 точек
Эффузионный, 673—873 К, 9 точек
Масс-спектрометрический, 773—931 К, расчет
по уравнению
Точек кипения, 1218—1983 К, 8 точек
То же, 1218—1979 К, 26 точек
Протока, 1086—1310 К, 26 точек
Масс-спектрометрический, 494—660 К, расчет
по уравнению
Д,Я° (Sr,
II закон
380±70
167±6
149±5
151
167 ±5
167±3
181+7
163,8+1,6
к, 298,15 К)
III закон
156±19
161,7±0,2
159,3±0,8
160,7+2,0
160,0+1,2
160,1 ±0,5
161,7±0,4
161,9+2,0
отражают воспроизводимость измерений, а также неточность сечений ионизации для масс-спектро-
метрических измерений. При вычислении средневзвешенного учитывались отклонение результата
измерений в данной работе от средней величины и погрешности, обусловленные неточностью термо-
343
Sr(r)
Глава 28
динамических функций Sr (к, ж). Последние являются основным источником погрешностей,
особенно для измерений выше 1200 К. Они приводят к погрешности в энтальпии сублимации от
1 кДж -моль для величин, вычисленных из данных о давлении пара при 700—800 К, до 4 кДжХ
Хмоль при обработке данных, полученных при 1700 К.
Sr (г). Термодинамические свойства газообразного стронция в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 814.
Уровни энергии атома Sr, использованные в расчете термодинамических функций, приведены
в табл. 28.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
Т а б л
Атом
и ц a 28.3. Уровни энергии атомов Sr и
Электронная
конфигурация
Sr . . .5s2
.. ,5s5p
. . .5*4rf
• . .5*4^
. . .5s5p
. . Ad5p
. . Adbp
. . Adbp
. . .5s5d
. . .5s5d
. . .5pa
. . .4d5p
. . .Ър*
. . .5p2
. . .4d5p
Состояние
CM
1S 0
3Ра 14 317,520
3Pi 14 504,351
3Рг 14 898,563
3Di 18159,056
3D2 18 218,795
3D3 18 319,267
W2 20 149,7
xPi 21 698,482
3F2 33 266,872
3F3 33 589,724
ID2 33 826,927
3Ft 33 919,33
ID2 34 727,483
sDi 35 006,943
3D2 35 022,015
3Da 35 045,055
3Po 35 193,47
3Pi 35 400,138
aP2 35 674,668
3Di 36 264,181
3D2 36 381,769
3D3 36 559,514
4>2 36 960,881
^o 37 160,278
3Po 37 292,106
3Pi 37 302,76
SP2 37 336,616
Sr+, i
Pi
1
1
3
5
3
5
7
5
3
5
7
5
9
5
3
5
7
1
3
5
3
5
7
5
1
1
3
5
принятые для расчета
Атом
Электронная
конфигурация
Sr . . Ad5p
. . .5*4/
. . .5*4/
. . .5*5/
. . .5*5/
. . Ad*
Sr+ . . .5.s
. . Ad
. . .bp
. . .bd
. . Af
...5/
..Ag
...5?
i термодинамических функции
Состояние
3F2
3F3
3F*
*F3
3F2
3F3
sFi
lFs
sPo
3Pi
3P2
%S4z
*D,h
a?>5/
2p/
*p
w3J2
2 J?
CM
38 008
38 750,454
38 752,44
38 755,199
39 539,04
41 364,614
41 365,532
41 366,695
41 518,91
44 525,88
44 595,97
44 729,67
0
14 555,9
14 836,24
23 715,19
24 516,65
53 286,31
53 372,97
60 991,7
71 065,8
71 357,8
76 737,7
Pi
7
5 ¦
7
9
7
5
7
9
7
1
3
5
2
4
6
2
4
4
6
14
14
18
18
. . .5s2, . . .5s5p, . . .5s5d, . . .5*5/, . . .5sAd, . . .5s4/, . . Adbp (кроме *Р), . . Ад? CР) и . . .5p2..
Энергии этих состояний приняты по рекомендации Мур [2053].
Термодинамические функции Sr (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),.
A. 22)—A. 25). Значения <?эл и ее производных вычислялись непосредственным суммированием
по уровням энергии. Погрешности вычисленных значений Ф° (Г) при Т <; 2000 К не превышают
0,02 Дж-К-моль~1 и обусловлены главным образом неточностью принятых фундаментальных
постоянных. При более высоких температурах погрешности возрастают из-за пренебрежения рид-
берговскими состояниями, а также из-за отсутствия данных о ряде высокорасположенных
валентных состояний. Эти погрешности составляют 0,04, 1 и 3 Дж-К~1-моль~1 в значениях Ф° (Г) при
3000, 6000 и 10 000 К.
344
Стронции и его соединения Sr+ (г), Sr2 (г)*
Ранее таблицы термодинамических функций Sr (г) вычислялись в предыдущем издании
настоящего справочника [105] и таблицах JANAF [1543], в работах Хилзенрата и др. [1436] (до
10 000 К) и Кольского и др. [1749] (до 8000 К), в справочнике [2632] (до 3000 К) и работе Каца и
Маргрейва [1639] (до 2000 К). При Т ^ 4000 К результаты всех расчетов удовлетворительно
согласуются между собой и с данными настоящего издания (расхождения в значениях Ф° (Т) не
превышают 0,02 Дж-К-моль~1, за исключением расчетов [2632, 1749], для которых они достигают
0,05 Дж-К-моль). При более высоких температурах расхождения возрастают (до 3,5 Дж-К^Х
Хмоль в значении Ф° (8000 К) с данными [1749], где были учтены ридберговские состояния).
Исключение составляет расчет Хилзенрата и др., в котором учитывались те же электронные
состояния, что в настоящем издании, поэтому термодинамические функции Sr (г), вычисленные
в [1436], практически совпадают с приведенными в табл. 814.
Энтальпия образования Sr (г)
bfH°(Sr, г, 298,15К)=:160,5±2,0 кДж • моль
принята в соответствии с энтальпией сублимации Sr (к).
Sr+ (г). Термодинамические свойства газообразного положительного иона стронция в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 815.
Уровни энергии иона Sr+, использованные в расчете термодинамических функций, приведены
в табл. 28.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
. . .5s—. , ,5g и , . Ad— . . Ag. Энергии этих состояний приняты по рекомендации Мур
[2053].
Термодинамические функции Sr+ (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25). Значения Qax и ее производных вычислялись непосредственным суммированием
по уровням энергии. Погрешности вычисленных значений при Т <^ 8000 К не превышают
0,02 Дж-К-моль и обусловлены в основном неточностью принятых фундаментальных
постоянных. При более высоких температурах возникают погрешности из-за пренебрежения ридбергов-
скими состояниями, достигающие 0,03 и ОД Дж-К-моль^1 в значениях Фс (Т) и С°р (Т).
Ранее таблицы термодинамических функций Sr+ (г) вычислялись в предыдущем издании
настоящего справочника [105] до 6000 К, а также в работах Хилзенрата и др. [1436] и Грина и др.
[1246] до 10 000 К. Результаты этих расчетов и настоящего издания согласуются между собой
в пределах 0,01—0,02 Дж-К-моль во всем интервале температур, за исключением области
около 10 000 К, где сказывается различие в числе учитываемых в расчетах уровней.
Константа равновесия реакции Sr+(r)+e(r)=Sr (г) вычислена с использованием значения
ДГЯ° @) =—549,470+0,001 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Sr)=45932,l+0,l см-^549,470 +0,001 кДж-моль.
Эта величина получена в работе Камю [813] из измерений в спектре поглощения серий 5sns CS)
для п <; 19 и bsnf CF) для п ^ 16; она хорошо согласуется с результатами неопубликованного
анализа серии 5snp (sPi) для п <^ 89 D5932,2 см), выполненного Томкилсом (известно по [813]),
а также измерениями Пенкина и Шабановой [317] D5 932,3 +0,3 см). В справочнике Мур [20531
на основании более старых работ рекомендовано менее точное значение.
Принятому потенциалу ионизации соответствует
AfH°(Sr+, г, 0) = 710,343 + 2,0 кДж • моль.
Sr2 (г). Термодинамические свойства газообразного дистронция в стандартном состоянии
в интервале температур 100—3000 К приведены в табл. Д. 62.
В табл. 28.4 приведены молекулярные постоянные Sr2, которые из-за отсутствия
экспериментальных данных оценены при подготовке справочника. По аналогии с молекулами Mg2 и Са2
принимается, что молекула Sr2 в состоянии ХгЩ образуется за счет вандерваальсовского
взаимодействия атомов стронция в основном состоянии и имеет небольшую энергию диссоциации (см.
ниже). Возбужденные состояния молекулы Sr2 должны иметь энергии выше 15 000 см и могут
не учитываться при расчете термодинамических функций. Приведенное в табл. 28.4 значение-
ге (Sr2)=4,60+0,2 А оценено из сопоставления соответствующих величин в молекулах Mg2, Саг
345
Srs (г)
Глава 28
Таблица 28.4. Молекулярные постоянные Sr2, SrO, SrO+, SrH и SrS
Молекула
Sr2
SrO
SrO+
SrH
SrS
Состояние
Те
X1!!" 0
XJS+ 0
а32+ 9 0006
Ь3П 9 121
Am 9 400
fi!2+ 10 886,8
с3П 24 000 б
СгП 24 701,1
1д>Т+ 2650°б
D1 2+ 28 632,9
Х2П 0
Л2 2+ 2 000
X2 2+ 0 а
Am 13 467 в
В2 2+ 14 356 Д
Е2П 18 897
а*П 20 847,6
С2 2+ 26 230
Х*2+ 0
<?2+а 26 531,21
СО
36,0
653,2
620 б
472,8
472,8
619,58
520 б
519,91
480 б
480,2
450
600
1206,2
1275 г
1205 е
1230 г
1014,1
1347
388,38
286,81
Примечание. Ниже все постоянные даны в см.
SrO: a
SrH: a
SrS: a
?i=2,4-10-8; б оценка; в ш,^
7=0,10; 6 постоянная для у=0
ний2Ш, nil,,; Дт=— 3,81;
=1153.
г>П, Am (Гв=9000), Ь3 2+ (Ге=
=-0,054;
; В ^4 = 146; '
е вычислено
= 12 0004 В1
==28 000); & приведено значение Do.
а,
0,40
3,92
0,9б
2,06
2,06
0,89 в
3,2 б
3,24
—
2,6
1,7
3,3
17
—
26,4 е
—
15,4
23,5
1,310
0,843
г р1==—6-
Вв
Г1
0,018
0,337 980
0,305 б
0,256 5
0,256 5
0,304 71
0,294 б
0,293 77
0,274б
0,274 2
0,26
0,30
3,674 3
3,668 3 б
3,878 8
3,638 8 б
1,925
4,008
0,120 72
0,105 66
10"8.
ах ¦ 102
0,02
0,2194
0,1 б
0,17
0,17
0,112
0,16 б
0,155
—
0,21
0,14
0,21
8,14
—
9,30
—
2,4
13,2
0,0440
0,0320
D, ¦ 106
0,018
0,342 а
0,3 б
0,30
0,30
0,32
0,5 б
0,45 г
—
0,35
0,35
0,30
134 б
133 б
ЮЗ6
30 б
140 б
0,0479 6
0,0575 б
'среднее из вычисленных по соотношению A.69) для
по соотношениям A.67)
2+ {Te—li.
при Do (В2
000), !2-, 1Д, 32+, 3Д
г.
к
4,60
1,920
2,02
2,204
2,204
2,022
2,06
2,059
2,13
2,131
2,19
2,04
2,146
2,148 б
2,088
2,156 б
2,965
2,055
2,4405
2,609
подсостоя-
2+)=13 010±200и AGl;.=
(Т. ==25 000), 3П
, 1П (?У=
и Ne2, Ar2 и Кг2 [662]. Частота колебания ше оценена из соотношения <л\12ВeDere=const,
справедливого для двухатомных молекул, образованных элементами одной группы периодической системы
(см. [2579]). Значение const=9,8 А получено как среднее значение из соответствующих величин
для молекул Mg2 и Са2.
Термодинамические функции Sr2 (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения <?ш и ее производных вычислялись по соотношениям A. 90)—A. 92)
без учета возбужденных состояний. Величина <?^од вр и ее производные находились
непосредственным суммированием по колебательным уровням и интегрированием по вращательным уровням
энергии с помощью уравнений типа A. 82). В расчете учитывались все уровни энергии со
значениями / ^ /шах> „, где /maXj „ находились из условий A. 81). Колебательно-вращательные уровни
энергии вычислялись по уравнениям A. 64а) и A. 65), коэффициенты Ykl в которых, а также
величины vmai и /1шприведены в табл. 28.5. Значение У03 вычислено из условия A. 59).
Погрешности вычисленных термодинамических функций обусловлены отсутствием
экспериментальных данных об энергии диссоциации и молекулярных постоянных Sr2 и трудностью
оценить их с точностью, большей чем 10—20%. Погрешности в значениях Ф° (Т) могут достигать
5—10 Дж-К^-моль.
346
Стронций и его соединения
SrO (к, ж)
Ранее термодинамические функции Sr2 (г) вычислялись Климовым и др. [167] (от 500 до 4500 К)
!В приближении «жесткий ротатор — гармонический осциллятор», расхождения данных [167] и
табл. Д.62 в значениях Ф° (Т) достигают 10 Дж-К^-моль при 3000 К.
Принятая в справочнике энергия диссоциации
D0(Sr2)=800+200 см-1=9,6+2,5 нДж-моль
оценена в предположении, что отношение C6/Der6e, где Св — постоянная вандерваальсовского
взаимодействия, одинаково для молекул Mga, Ca2 и Sr2, как это имеет место для молекул
благородных газов (см. [667]). На основании этого предположения, значений Св для атомов Mg, Ca и Sr [436 ]
и принятых в справочнике значений De для Mg2 и Са2 (см. табл. 26.5 и 27.5) получена эта
величина. Ей соответствует
bfH°(Sr2, г, 0) — 312,146 + 4,7 кДж • моль.
SrO (к, ж). Термодинамические свойства кристаллического и жидкого оксида стронция в
стандартном состоянии при температурах 100 — 6000 К приведены в табл. 816.
Таблица 28.5. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см),
а также значения wmax и </цт, принятые для расчета термодинамических функций Sr2, SrO,
SrO+, SrH и SrS
Коэффициенты
г,, ю-*
Ую-Ю-3
У.,0-10-1
Узо-Ю2
Ую-10*
Уох-Ю1
Уи-Ю2
Ун-10*
Уз1-Ю5
У02.10в
Уоз-Юп
Уо,-Ю12
(ao=D,);
xlO
щ-101
й2-105
а3-1010
ишах
Sr2
А'^
0
0,036 0
—0,040
—
—
0,18
—0,022
—
—
-0,020
-0,008 047
—
—
—
—
44
297
Примечание. Ниже
SrO: »
SrO+: a
SrH: a
SrS: a
Состояние
Г„D22+) =
Состояние
т.
Состояние
т.
0
0,653
—0,414
1,634
—0.285
3,379
—0,219
0,03
—
—0,354
—
—
—
—
—
—
216
515
97с
495
14
058
8
4
0,900
0,619
-0,081
—1,262
3,047
-0,112
—
—
—0,32
—
—
—
—
—
—
107
490
SrO
0 0,915
460 0,463
529 —0,158
48 —0,372
—
1 2.584
-0,2
—
—
-0,32
—,
—
—
—
—
—
105
427
все постоянные даны в см.
с3П
24 000
= 2000
42П
13 467
а3П, А
9000
m
cm
24 701,1
В22+
14 356
63S+
12 000
12-, 3?+,
441
0 0,942
465 0,463
797 —0,163
255 0,193
-0,010
2,584
—0,2
0,037
—
-0,32
—
—
—
—
—
—
204
538
1Д, 3Д 1
9
577
43S
07S
793
B^
1,088
0,619
i —0,081
-1,305
—
3,047
—0,112
—
—
—0,32
—
—
—
—
—
—
106
486
26 500 28 632,9
ЕЧ1
18 897
512+
13 000
20 847,6
25 000
С2
2+
26 230
>Д С1
а
68
456
274
63
1
S+
26 531,2
SrO+
xm*
0
0,450
t -0,17
—
—
2,60
—0,14
—
—
—0,35 -
__
—
—
—
—-
—
132
447
41, 41
28 000
SrH
X^
0
1,207
—1,786
32,132
-401,655
36,761
—8,523
23,987
—40,778
-133.9
487,7
—1,529
1,360
3,462
8,088
-4,261
17
85
a
084
356
4
7
56
2
2
911
934
997
746
SrS
X^
0
0,388
-0,131
0,146
—0,001
1,205
—0,043
—
—
-0,046
-0,003
—
—
—
—
—
239
710
a
172
714
159
523
822
925
494
012
347
SrO (г) Глава 2&
Значения постоянных, принятые для расчета термодинамических функций, приведение
в табл. 28.1. В справочнике за стандартное состояние SrO (к) принята кубическая модификация
(структурный тип NaCl), устойчивая от 0 до температуры плавления. При Т < 298,15 К
термодинамические функции SrO (к) вычислены на основании данных по теплоемкости, полученных
в работах Гмелина [1226] A — 300 К) и Андерсона [511] E8—298 К). Погрешности измерений
[1226] и [511] можно оценить в 0,5% и 1,5%, а погрешность полученных по ним значений S"
B98,15 К) и Н° B98,15 К)-#° @) (см. табл. 28.1) - в 0,5 Дж -К -моль и 0,05 кДж -моль
соответственно. Для теплоемкости в интервале 298,15—2930 К принято уравнение, выведенное
методом Шомейта по результатам измерений энтальпии SrO (к) в работе Ландера [1808] D06—1266 К)
и с учетом оцененных значений энтальпии при более высоких температурах. Погрешность
описания теплоемкости этим уравнением оценивается в 1% при 1000 К, 5% при 2000 К и до 10% при
температуре плавления SrO. Температура плавления B930+30 К) принята по согласующимся
между собой результатам измерений Фоэкса [1112] B923 К) и Траверса и Фоэкса [2726] B933 К)
для образцов 99,99% чистоты. Результаты измерений Шумахера [2477] B703 К) на образце SrO
неисследованного состава и Ногути [2148] B872 К) на образце состава 99% SrO не учитывались.
Энтальпия плавления SrO G0+7 кДж-моль) принята по оценкам, проведенным в [1781] и
[105]. Теплоемкость SrO (ж) (84+8 Дж-К~1-моль~1) принята равной теплоемкости ВеО (ж),
которая была определена экспериментально [460].
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 3000 и 6000 К оценены в 0,5,.
1, 5 и 13 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями,,
приведенными в табл. 816 и в справочниках [105, 1543], достигают 1,5 и 5 Дж-К~1-моль~1 в
значениях Ф° (Г) для SrO (к) и SrO (ж) соответственно. Для SrO (ж) эти расхождения вызваны
различием оценок теплоемкости.
В справочнике принята энтальпия образования оксида стронция
*1
A^°(SrO, к, 298,15 К) = — 590,5 + 1,0 кДж • моль
Эта величина получена в работе Бринка и Холли [752] в результате измерения энтальпии
реакции растворения SrO (к) и Sr (к) в растворе соляной кислоты одинаковой концентрации. В
работе [752] измерения проводились в негерметичном калориметре, однако поправки,
учитывающие испарение раствора при выделении водорода, были тщательно учтены авторами работы.
Измерения энтальпий растворения SrO (к) и Sr (к) в соляной кислоте также выполнены авторами [261 г
408, 418, 481, 670, 754, 1116, 2686] и [1022, 1325, 1327] соответственно. В некоторых из этих работ
измерения проведены на высоком экспериментальном уровне, однако конечные концентрации
в них не совпадали, что не позволяет провести достаточно точный расчет энтальпии образования
SrO (к) по этим данным, так как в солянокислых растворах стронция, по-видимому, образуются
комплексы, учесть образование которых в настоящее время не представляется возможным. Крома
того, чистота использованных образцов Sr (к) вызывает сомнение (см. примечание на с. 341)-
Измеренная Ма [1913] энтальпия сгорания стронция приводит к значению —604,3 кДж-моль,,
противоречащему результатам измерений энтальпий растворения и уступающему им по точности^
(см. текст по ВаО).
Давление пара оксида стронция в реакции SrO (к, ж)=ЭгО (г) вычислено с использованием1
значения Д8#° @)=575,905+15 кДж-моль, соответствующего принятым в справочнике
энтальпии образования SrO (к) и энергии диссоциации SrO (г).
SrO (г). Термодинамические свойства газообразного оксида стронция в стандартном состоянии
при температурах 100—10 000 К приведены в табл. 817.
В табл. 28.4 приведены молекулярные постоянные SrO, принятые на основании анализа
результатов исследований оптического и микроволнового спектров этой молекулы. В электронном спектре-
SrO исследованы четыре системы полос, связанные с переходами между пятью синглетными
состояниями: A1n-X1S+ [815, 1376], S1E+-X1S+ [502, 503, 743, 1801, 1922], С1П<*Х12+ [62, 937,
1765, 1766, 1916, 1997] и ОХЪ+**Х1Ъ+ [62, 504, 1794]. По аналогии с молекулами MgO и СаО у SrO-
следует ожидать наличие еще ряда низколежащих синглетных и триплетных стабильных
состояний, одно из которых (Ь3П) было идентифицировано при анализе возмущений в системе В1!!"—
X1S+ [503, 1080].
В табл. 28.4 приведены данные для 12 валентных состояний SrO. Колебательные постоянные
в основном состоянии приняты по результатам анализа полос с v" ^ 9 системы ВХЕ+—X1S+V
348
¦Стронций и его соединения SrO (г)
^проведенного Брюэром и Хауге [743]. Вращательные постоянные рассчитаны Филдом [1080]
совместной обработкой данных, полученных при исследовании электронного [502, 503] и
микроволнового [1641] спектров SrO. Молекулярные постоянные в состоянии АХП. найдены авторами [815]
при анализе системы АЧ1—ХХЕ+ E ^ v' <C 9, v" ^ 1). Для состояния 63П приняты те же
значения постоянных, что для изоконфигурационного состояния АХТ1, поскольку анализ возмущений
в системе S1?+—ХХП+ [108011 показал их практическое совпадение. Энергия возбуждения
состояния b3U, полученная Филдом [1080], скорректирована с учетом разницы Те (ЛЧ1)—Te(b3R) =
=279 см и принятого значения Те (А1!!). Молекулярные постоянные в состоянии С1!! приняты
по результатам совместной обработки данных [937, 1765, 1766] для девяти полос системы С1!!—
X1 ?+ (i/ ^ 2, v" ^ 6). Постоянные в состоянии D1t+ найдены Альмквистом и Лагерквистом [504,
1794] из анализа шести полос системы D1!!4"—XXI> +(v' ^ 4). Энергии состояний a3S + и с3П оценены
исходя из того, что расщепление триплетных и синглетных изоконфигурационных состояний в
молекуле SrO приблизительно в 4 раза меньше, чем в молекуле MgO, для которой был выполнен
квантовомеханический расчет [2446]; точность такой оценки составляет 1000—2000 см. Энергия
группы близколежащих состояний 3S+, 3Д, х?" и ХД оценена так же, как для молекулы СаО.
Погрешность оцененного таким образом значения, по-видимому, не превышает 4000 см.
Колебательные и вращательные постоянные в состояниях а3?+, с3П, 3Д, 3Е+, 1^~, 1Д приняты равными
соответствующим постоянным изоконфигурационных состояний.
Термодинамические функции SrO (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qss и ее производных вычислялись по соотношениям A. 90)—A. 92)
с учетом 11 возбужденных электронных состояний в предположении, что Q^ Bt=(pJpb)Q(Kl[
для состояний с Те ]> 20 000 см. Статистические суммы по колебательно-вращательным уровням
энергии состояний X1I>+, а3?+, Ь3И, i1!!,' BJS+ и их производные находились по уравнениям
A. 73)—A. 75) непосредственным суммированием по колебательным уровням и интегрированием
по / с помощью соотношений типа A. 82). В расчете учитывались все
колебательно-вращательные уровни перечисленных состояний со значениями / ^/max> 0, последние находились по
соотношению A. 81). Колебательно-вращательные уровни энергии вычислялись по уравнениям A. 65)
и A. 62) с коэффициентами Ykl, приведенными вместе со значениями vm&JL и /Ит в табл. 28.5. Для
обеспечения условий A. 50) для состояний X1 ?+, Ь3П и АХИ в уравнение A. 65) был включен
член с коэффициентом У3о> а Для состояний а3 ?+ и Z?1 Е+ — с коэффициентом Yi0. Значения
коэффициента Yn для состояний Х1И+иу11П вычислены из условия В, +i=0. Мультиплетность и
вырождение вращательных уровней учитывались статистическими весами.
Погрешности рассчитанных термодинамических функций SrO (г) при температурах до 1000 К
•обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах основной вклад в погрешность дает неточность энергии возбуждения состояния
»а3?+ и группы состояний с Те~26 500 см, а также пренебрежение ридберговскими состояниями
SrO. Общие погрешности в значении Ф° (Т) могут достигать 0,02, 0,1, 0,5 и 3 Дж-К-моль при
:298,15, 3000, 6000 и 10 000 К соответственно.
Термодинамические функции SrO (г) ранее вычислялись в предыдущем издании настоящего
-справочника 1105], таблицах JANAF [1544], а также в работах Вейц и др. [63] (до 3500 К), Брюэра
и Розенблатта [745] (до 3000 К) и Шнейдера [2463] (<?вн при 1000 < Т < 7800 К). При
температурах до 1000 К результаты всех расчетов (за исключением [745]) практически совпадают с данными
табл. 817. При более высоких температурах расхождения данных [63, 105, 2463] и табл. 817
возрастают, достигая 9 Дж-К-моль в значениях Ф° (Т) при 6000 К, что обусловлено
пренебрежением в этих работах триплетными состояниями SrO. Расхождения с данными [1544] не
превышают 0,1 Дж-К-мольвФ° (Т) и в С°р (Т), поскольку оба расчета проведены по близким
значениям молекулярных постоянных, а различие в использованных методах расчета мало влияет на их
результаты при Т ^ 6000 К. В работе [745] из-за ошибки в оценке энергии состояния Ь3П
получены резко завышенные значения Ф° (Г), превышающие данные табл. 817 на 16—18 Дж -К -моль.
л Из анализа возмущений в системе ВХ2+—Х12+ Филд [1080] получил для состояния Ь3П (в см"): Г0=9055+5,
№,,=463,5+0,8, шЛ=1,61±0,05, Ве=0,2584+0,0005, ае=0,0020 +0,0002, однако рассчитанные таким образом
.постоянные в состоянии А1П несколько отличаются от данных [815].
349
SrO (г)
Глава 28.
Константа равновесия реакции SrO (r)=O (r)+Sr (г) вычислена на основании принятой в
справочнике энергии диссоциации
ДГЯ° @) = Do (SrO) = 420 ± 15 кДж • моль = 35 100 + 1300 см.
Энергия диссоциации SrO (г) и теплота сублимации SrO (к) были предметом многочисленных
измерений, приводивших к противоречивым результатам, хотя расхождения между величинами,,
полученными разными авторами и разными методами, были меньше, чем в случае СаО.
Соответствующие данные представлены в табл. 28.6, их погрешности характеризуют воспроизводимость
Таблица 28.6. Результаты определений Do(SrO) (в кДж-моль)
Авторы
Метод
Do (SrO)
И закон
III закон
Классен, Винеманс, 1933 [855] Сублимация с открытой поверхности, SrO (к)= 391+180 465,6+5,7
-SrO (г), 1495—1635 К, 8 точек, Д.Я° @)=
= 530±5 (III)
Пельхович, 1954 [2203] То же, с масс-спектрометрической регистрацией, 352 —
1400-1750 К, Д8Я°@)=644(И)
Портер и др., 1955 [2254] То же, 2100 К, Д,Я° @)=628 (III) — 368±12
Асано и др., 1972 [539] То же, 1747—1850 К, 23 точки, Д8Я°@)=590± 434 406+7
±2,0 (III), результаты измерений даны в виде
уравнения
Хульдт, Лагерквист, 1954 [1497] Спектрофотометрическое"исследование равновесия — 466
SrO (r)=-Sr (г)+О (г) на основании измерения
р (Sr) в пламенах С2Н2 с воздухом, 2240—2430 К
Вейц, Гурвич, 1956 [58, 59. 60] То же, в пламенах С2Н„,Н2 и СО с воздухом и 477+35 465+20
кислородом, 2310—3210 К, 12 точек
Калфф и др., 1965 [1621] То же, в пламенах СО с кислородом, 270+160 416+15
2156—2450 К, 6 точек
Калфф и др., 1973 [1619] Тоже, в пламенахCO+N2O+H2O, 2684-2857 К, 141±115 416±20
12 точек
Беляев и др., 1979 [34а] То же, для реакции Sr (г)+Н2О (г)—SrO (г)+ 414±15 —
+Н2 (г) по интенсивности линии Sr и полос SrO
в пламенах СН4+ О2+ N2,2147—2900 К, 33 точки
Колен и др., 1964 [873] Мясс-спектрометрический.вг (r)+SO (r)=SrO (r)+ 602+88 426+10
+S(r), 1745-2170 К, 6 точек, ДГЯ° @)=90,8±
±7,6 (III)
Дроварт и др., 1964 [987] То же, Sr (г)+О„ (r)=SrO (г) + О (г), 2156- 842±450 424+23
2321 К, 4 точки, "ДгЯ°@)=69±22 (III)
Фарбер, Сривастава, 1976 [1060] То+же, SrO (r)=-Sr (r)+V.Oa (г), 2010—2102 К, 391+26 416±7
9 точек, ДГЯ° @)=--168,7±1",0 (III)
Тоже, SrO (к)=SrO (г), 2010—2102, 9 точек, 391+25 420±7,0
Д,Я° @)=575+2,1 (III)
Батали—Космовпчи, Мйхель, 1972 Определение порога реакции Sr (г)+О2 (г)= 471 ±15
[614] =SrO (г)+О (г) при исследовании столкновений
в молекулярных пучках
Энгелке и др., 1976 [1034] То же, для реакции Sr (г)+СЮ, (r)=SrO (г)+ >440+1О
4 СЮ (г)
измерений, а также неточность величин, использованных в расчетах. Принятое значение основано»
на исследованиях равновесия обменных реакций с участием молекул SrO с помощью масс-спектро-
метрических измерений в работах Дроварта и др. [987] и Колена и др. [873] и спектрофотометри-
ческих измерений в работе Беляева и др. [34а]. Близкие значения получены в масс-спектрометри-
ческом исследовании диссоциации SrO (г) и сублимации SrO (к) Фарбером и Сривастава [1060 ]»
а также рассчитаны (после внесения поправок на образование молекул SrOH и Sr(OHJ) из спектро-
фотометрических измерений концентрации Sr (г) в различных пламенах,, выполненных Калффом
и др. [1619, 1621].
350
Стронций и его соединения SrO+ (г), SrH (г)>
Определение порога реакции образования молекул SrO в пучках в работах [614, 1034],
измерения концентрации атомов Sr в незащищенных пламенах ацетилена [58, 59, 60, 1497] и
исследования сублимации SrO (к) [2203, 2254], по-видимому, содержали методические ошибки.
Спектрофотометрические измерения в работе [1162] привели, так же как для других молекул,.
к резко завышенному значению E15 кДж-моль), по-видимому, за счет каких-то систематических
ошибок; данные [855] и [2055] по интегральной скорости испарения SrO (к) не учитывались, так
как в условиях опытов должна была происходить диссоциация оксида на Sr (г) и О2 (г).
Принятому значению энергии диссоциации соответствует
i\fH° (SrO, г, 0) = —12,344+15 кДж • моль.
SrO+ (г). Термодинамические свойства газообразного положительного иона оксида стронция
в стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 818.
В табл. 28.4 приведены молекулярные постоянные SrO+, оцененные при подготовке
справочника из-за отсутствия экспериментальных данных. По аналогии с MgO+ и изоэлектронной
молекулой RbO [2577] принимается, что ион SrO+ имеет основное электронное состояние Х2П
(конфигурация . . . Аг3) и низколежащее возбужденное состояние А2!** (конфигурация . . .отг4). Энергия
возбуждения последнего оценена на основании разности энергий состояний 3П, 43 (. . . тААз) и
32, гЕ (. . .%*оа) молекулы SrO. Эти состояния SrO являются первыми уровнями ридберговских
серий, сходящихся соответственно к состояниям Х2П и А2Т>+. Погрешность этой величины
составляет 2000 см. Значения ше и Ве, приведенные в табл. 28.4, оценены по соответствующим
постоянным возбужденных состояний молекулы SrO; их погрешности составляют около 50 и 0,02 см
соответственно.
Термодинамические функции SrO+ (г) вычислены по уравнениям A.3)—A.6), A. 9), A. 10) и
A. 93)—A. 95). Величина (?вн и ее производные вычислялись по соотношениям A. 90)—A. 92)
с учетом одного возбужденного состояния и в предположении, что Qi?l.3t=0,5 QS.Bp.
Статистическая сумма по колебательно-вращательным уровням состояния Х2П и ее производные
рассчитывались непосредственным суммированием по колебательным уровням и интегрированием по
значениям / с помощью уравнений типа A. 82). В расчете учитывались все уровни этого состояния
со значениями / <^ /max, vi последние находились по соотношению A. 81).
'Колебательно-вращательные уровни состояния Х2П вычислялись по уравнениям A. 65) и A. 62) с коэффициентами Yklf
идентичными с постоянными из табл. 28.4 и приведенными в табл. 28.5 вместе со значениями ршах
и /1Im. Мультиплетность состояния Х2П учитывалась статистическим весом 4.
Погрешность вычисленных термодинамических функций во всем интервале температур
определяется главным образом неопределенностью в энергии возбуждения состояния Л2Е+, а также
отсутствием экспериментальных данных о молекулярных постоянных SrO+. Погрешности в
значениях Ф° (Т) могут достигать 5—10 Дж-К-моль во всем интервале температур.
Ранее таблицы термодинамических функций SrO+ (г) не публиковались.
Константа равновесия реакции SrO+ (г)+е (r)=Sr (г)+О (г) вычислена на основании
значения ДГЯ° @) =—190+52 кДж-моль, соответствующего потенциалу ионизации
/0 (SrO) = 610 + 50 кДж-моль.
Эта величина принята на основании измерений методом электронного удара, выполненных
Дровартом и др. [987] и Фарбером, Сривастава [1060], которые нашли соответственно 590+50
и 630+95 кДж-моль.
Принятому значению соответствуют
AfB°(SrO+, г, 0) = 597,656+ 52 кДж • моль,
Do (SrO+) = 359,47 + 52 кДж • моль да 30 800 + 4000 см.
SrH (г). Термодинамические свойства газообразного гидрида стронция в стандартном
состоянии при температурах 100—6000 К приведены в табл. 819.
351
.SrH (г) Глава 28
В табл. 28.4 представлены молекулярные постоянные, принятые в справочнике на основании
анализа результатов исследования спектра SrH. В электронном спектре SrH наблюдалось 11
систем полос, связанных с переходами между 12 электронными состояниями: Л2П -> Х2? + [2817],
?2?+-н>Х2? + [2817, 2818], Е2П -* Х2? + [1137],?2? + (а4П) -^ Х2? + [1137, 2058, 2818], С2?+-Х2? +
[1137, 1511, 1682, 2058, 2818], /Я1«-Х2? + [1692], K2Z+ <~ X2?+ [1692], G2?+-X2?+ [1685,
1686], /2П<- X2? + [1691], ^2?+^ X2? + [1012, 1013, 1681, 1685, 1686], #2?+<- X2?+ [1686].
Состояния L2n и K2U [1692], J4J и G2?+ [1691] интерпретированы как ридберговские состояния,
состояния F2 ?+ (Г0=34 190 см) и Нг ?+ (Г0=41 361 см), по-видимому, также являются ридбер-
говскими и не рассматриваются в справочнике.
Система электронных состояний SrH аналогична системе состояний молекулы СаН и в
справочнике принята одинаковая корреляция электронных состояний обеих молекул с образующими их
атомами, а состояние Z)a?+ рассматривается как компонента состояния а4П.
Приведенные в табл. 28.4 колебательные постоянные в состоянии X2 ?+ (и состоянии а4П)
получены Мором и Корнеллом [2058] по началам полос системы а4П—Х2?+ (и" ^ 3, и' ^ 4),
вращательные постоянные найдены Эдвинссоном и др. [1013] из анализа полосы 0—0 системы F2L+—
Х2?+ и Уотсоном и др. [2818] из анализа полос 0—0, 1—1 системы 52?+—Х2?+, полос 0—2 и
О—3 системы а4П—Х2?+и полосы 0—0 системы С2? +—Х2?+. Вращательные постоянные в
состоянии А2П приняты по работе Уотсона и Фредриксона [2817], которые провели анализ двух под-
лолос 0—0 системы А2П—Х2?+, а в состоянии 52?+ — по работе Уотсона и др. [2818], где они
были получены из анализа полос 0—0 и 1—1 системы B2L+—Х2?+. Экспериментальные данные
о колебательных постоянных в состояниях А2И, 52?+, а также Е2И отсутствуют; значения,
приведенные в таблице, оценены при подготовке справочника, их погрешности могут составлять
50—70 см в значениях ше. Вращательные постоянные SrH в состоянии 2?2П приняты по работе
Фредриксона и Хогана [1137], постоянные в состояниях а4П B32?+) и С2?+ — по работе Мора и
Корнелла [2058]. Состояние С2?+ предиссоциировано, и в спектре испускания наблюдаются
только переходы с z/=0 и 1 [1682].
Термодинамические функции SrH (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
i(l. 93) —A. 95). Значения (?вн и ее производных находились по уравнениям A. 90)—A. 92) с
учетом пяти возбужденных электронных состояний в предположении, что QiOi.Bv=(Pi/Px)QmJ BP-
Величина QSLw и ее производные вычислялись по уравнениям A. 70)—A. 75) непосредственным
суммированием по колебательно-вращательным уровням энергии. В расчетах учитывались все
уровни этого состояния, ограниченные его предельной кривой диссоциации; в табл. 28.5 приведены
коэффициенты at в уравнении, аппроксимирующем эту кривую, и значения fmax и /]Jm.
Колебательно-вращательные уровни состояния Х2?+ вычислялись по уравнениям A. 65) и A. 62),
значения коэффициентов Ykl в этих уравнениях приведены в табл. 28.5. Коэффициенты У3о и ^40
.определялись из условий A. 50), коэффициенты У21, Y31 — из условий A. 57), а коэффициенты
Y03 и YOi — из условий A. 59). Мультиплетность и вырождение вращательных уровней основного
зИ возбужденных электронных состояний учитывались соответствующими статистическими весами.
Постоянные молекулы SrH известны с удовлетворительной точностью, и погрешность
вычисленных термодинамических функций при Т < 3000 К обусловлена практически только неточностью
фундаментальных постоянных. При более высоких температурах становятся существенными
погрешности из-за приближенного учета возбужденных электронных состояний, отсутствия данных
ю высоких колебательно-вращательных уровнях энергии состояния Х2?+ и неточности энергии
диссоциации SrH. Общие погрешности в значениях Ф° (Т) при 3000 и 6000 К могут достигать 0,03
и 0,15 Дж-К~1-моль~1 соответственно.
Термодинамические функции SrH (г) вычислялись ранее в справочнике Хара и др. [1336]
до 5000 К и работе Вейц и др. [63] до 3500 К; в работах Климова и др. [167] и Шнейдера [2463]
приведены коэффициенты полинома, аппроксимирующего значения Ф° (Т), ()вн и In QBS.
Расхождения данных табл. 819 и справочника [1336] составляют до 0,4 и 1,2 Дж-К-моль в значениях
Ф° (Г) и С°р (Т), расхождения с данными [167] достигают 1,6 Дж-К-моль. Наилучшее согласие
имеет место с работой [63] — в пределах 0,1 Дж-К~1-моль.
Константа равновесия реакции SrH (r)=Sr (г)+Н (г) вычислена на основании принятой в спра-
зочнике энергии диссоциации
ш~1 = 155,6 ± 2,4 кДж • моль.
352
Стронций и его соединения SrH4 (к, ж), SrOH (г)
Это значение найдено из анализа данных по предиссоциации молекул SrH и SrD в состоянии
С* S+ (v=0, 1), полученных в работах [1137, 1682, 2058, 2818] в предположении, что предиссоциа-
ция связана с пределом Sr CРХ)+Н B?) или с пределом Sr C.Р2)+Н BS).
Рекомендованные ранее значения энергий диссоциации [1183, 1392] рассчитывались по одной
точке обрыва в полосе 0—0 системы С2 2+—X2 2+ молекулы SrH. В пределах погрешностей они
согласуются со значением, принятым в настоящем справочнике.
Принятому значению соответствует
A/HSrH, г, 0) = 221,307 ±3,2 кДж-моль.
SrH2 (к, ж). Термодинамические свойства кристаллического и жидкого дигидрида стронция
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 820.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. В справочнике за стандартное состояние SrH2 (к) в интервале 0—1128 К принимается
ромбическая модификация (a-SrH2, структурный тип РЬС12), а в интервале 1128—1323 К — Р-мо-
дификация с неустановленной структурой. Экспериментальные данные по термодинамическим
свойствам SrH2 (к, ж) в литературе отсутствуют (за исключением данных по Т(г и Тт). При
Т < 298,15 К термодинамические функции SrH2 (к) вычислены по значениям теплоемкости^
оцененным с помощью различных эмпирических методов. Точность значений 5° B98,15 К) и Н°
B98,15|К)—Н° @), приведенных в табл. 28.1^ оценивается в 4 Дж-К^-моль и 0,8 кДж-моль
соответственно. При Т > 298,15 К теплоемкость SrH2 аппроксимирована уравнением,
выведенным по значениям С°р B98,15 К)*и Ср A128 К)=71 Дж«К-моль; для теплоемкости |5-SrH8
принято значение 71 + 10 Дж-К^-моль» а для Ср (SrH2S ж) — значение 75+15 Дж-К^-моль'1.
Температура полиморфного превращения A128+20 К) основывается на данных Петерсона и Кол-
берна [2214], полученных методом ДТА для образца SrH2 с суммарным количеством примесей
металлов не более 0,3%. Энтальпия превращения G,2 + 1,5 кДж-моль) оценена в
предположении о равенстве энтропии превращения СаНа и SrH2 ввиду идентичности структур
ос-модификаций этих дигидридов и близости температур их превращения. Энтальпия плавления SrH2 B3 ±
+5 кДж-моль)|оценена так же,; как для СаН2 (см. выше). Температура плавления SrH2 A323±
+50 К) определена из данных по фазовой диаграмме системы Sr—SrH2t полученных в работе
Петерсона и Колберна [2214].
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000 и 2000 К оцениваются в 4, 10 и
20 Дж -К -моль соответственно. Расхождения между термодинамическими функциями a-SrHa (к),
приведенными в справочнике [578] и табл. 820, достигают 5 Дж -К -моль в значении Ф° A000 К)
ввиду того, что в [578] на основании оценок приняты более низкие значения S0 B98,15 К) и Ср (Г).
Расчеты термодинамических функций P-SrH2 (к) и SrH2 (ж) в справочной литературе отсутствуют.
**** Константа равновесия реакции SrH2 (K)=Sr (г)+2Н (г) вычислена с использованием
значения ДГЯ° @)=765,903+ 10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
к, 298,15JK) = —180 + 10 кДж.моль.
Эта величина основана на измеренных Эрликом и др. [1022] энтальпиях растворения стронция
и дигидрида стронция в соляной кислоте. Ее погрешность увеличена по сравнению с оценкой
авторов [1022], так как выделяющийся водород мог вступать в реакцию с медной сеткой, в которую
был завернут образец стронция (см. текст по ВаН2 (к, ж)). Более ранние аналогичные измерения»
выполненные Гунцем и Бенуа [1322, 1325], привели к близкой величине —177 кДж-моль.
Исследования равновесия диссоциации SrH8 [575, 1035, 1330, 2709] приводят к менее надежным
результатам (около —197 кДж'МОль по данным работ [1330, 27091) *.
SrOH (г). Термодинамические свойства газообразного гидроксида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 821.
^Молекулярные постоянные,; использованные в расчете термодинамических функций SrOHs
приведены в табл. 28.7. Экспериментальные данные о молекулярных постоянных SrOH
отсутствуют. По аналогии с молекулами Be ОН и гидроксидов щелочных металлов для SrOH принята
линейная конфигурация (основное электронное состояние л2 2+). Оценка геометрических парамет-
См. примечание 1 на с. 341.
353
SrOH+ (г)
Глава 2в
Таблица 28.7. Значения молекулярных постоянных, а также и и р$, принятые для расчета термодинамических,
функций SrOH, SrOH+, Sr(OHJ, SrF2, SrCl2, SrBr2, Srl2
Молекула
SrOHa
SrOH+
Sr(OHJ
SrF2
SrCl.,
SrBr2
Srl2
SrOH: a
Sr(OH)a:
Электронное
состояние
X2S+
1*A
^A-l
ХЧ+
To (A) = 15 000
1
480
450
450
450
270
140
105
CM,
= 16 500 см, pB = 2.
a приведено значение /
v
2
305 B)
305 B)
80
90
50
35B)
30B)
РЛ==4, Г»
3
CM
3700
3700
450
455
300
225
190
(B) =
r'-CM6.
__
—
3700
SrF.,:
3rCl2:
v
5
B) 305 (-4)
—
—
—
a приведено
a приведено
I ¦ 1039
г -см2
11,7
12
3,4 a
4a
5a
211
387
значение /
значение /
a
1
1
2
2
2
2
2
2
1
1
1
1
1
1
г^см*.
I^-CM*.
ров и частот выполнена так же, как для ВеОН (см. гл. 25). Приведенный в табл. 28.7 момент
инерции рассчитан на основании значений г (О—Н)=0,96+0,02 А и г (Sr—О)=2,15+0,05 А, его
погрешность составляет 1-Ю9 г-см2. Погрешности принятых значений частот оцениваются
в 50—100 см. На основании работ Гейдона [1182] и Хёрка [1514], наблюдавших полосатый
спектр SrOH, а также по аналогии с молекулой SrF, которая изоэлектронна SrOH, в справочнике
принимается, что молекула гидроксида стронция имеет два возбужденных электронных состояния;
их энергии, приведенные в табл. 28.7, известны с точностью не менее 1000 см.
Термодинамические функции SrOH (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных величин обусловлены неточностью принятых молекулярных постоянных, а также-
приближенным характером расчета, они составляют 2, 4 и 6 Дж-К~1-моль~1 в значениях Ф° (Т\
при 298,15, 3000 и 6000 К.
Термодинамические функции SrOH (г) ранее вычислялись в таблицах JANAF [1544];
расхождения данных [15441 и табл. 821 составляют 3—7 Дж-К~1-моль и обусловлены главным
образом разной оценкой величины v2.
Константа равновесия реакции SrOH (r)=Sr (г)+О (г)+Н (г) вычислена с использованием
значения ЬГН° @) =813,69 +20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(SrOH, г, 0) ——190 + 20 кДж • моль'1.
Эта величина получена по методу II закона из результатов исследования равновесия реакции
Sr (г)+Н2О (r)=SrOH (г)+Н (г) в работах Гурвича и др. [424, 1333] на основании измерений
интенсивности полос SrOH и линий Sr в спектрах пламен H2+-O2+N2 A860—2580 К, 16 точек,
Do (Sr—OH)=389 + 15 кДж-моль). К близкому значению (—195 + 20 кДж-моль) приводит
исследование равновесия реакции SrCl (г)+Н2О (г) = SrOH (r)-j-HCl (г) в тех же пламенах,
выполненное в работах [424, 1333] по интенсивности полос SrCl и SrOH. Коттон и Дженкинс [885];
из измерений концентрации атомов Sr в трех группах пламен H2-j-O2+N2 оценили Do (Sr—С
424+15 кДж-моль х, чему соответствует
Д//
(SrOH,
^—224 + 20 кДж-моль. Косвенные
измерения в работах Вейц, Гурвича [57, 611, Рябовой, Гурвича [357, 358], Сагдена, Скофилда
[2635] и Калффа, Алкемаде [1619, 16201 приводят к значениям от —180 до —225 кДж-моль..
SrOH+ (г). Термодинамические свойства газообразного положительного иона гидроксида
стронция в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 822.
Молекулярные постоянные, использованные в расчете термодинамических функций SrOH+,
представлены в табл. 28.7. Исследования структуры и спектров SrOH+ не проводились, поэтому
принятые молекулярные постоянные оценены на основании постоянных SrOH и молекулы RbOH,.
354
Стронций и его соединения Sr (OHJ (к, ж)
которая изоэлектронна SrOH+ (см. тексты по SrOH и RbOH). Моменту инерции в состоянии X1 Е+
соответствуют ^Sr-O-H=180°, r (Sr-O)=2,20 + 0,10 A, r (O-H) =0,96±0,03 А; его
погрешность составляет 2-Ю9 г-см2. Погрешности принятых значений частот оцениваются в 50—
100 см.
Термодинамические функции SrOH+ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),,
A. 122)—A. 124), A. 126) и A.129) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний; погрешности вычисленных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета и
составляют 4, 7 и 10 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Термодинамические функции SrOH+ (г) вычислялись ранее в таблицах JANAF [1544];
расхождения данных [1544] и табл. 822 составляют 3—6 Дж-К~1-моль~1 и обусловлены главным
образом разной оценкой величины v2.
Константа равновесия реакции SrOH+ (r)+e (r)=Sr (г)+О (г)+Н (г) вычислена с
использованием значения ДГЯ° @)=328,69+28 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(SrOH, г)=485+20 кДж-моль.
Эта величина основывается на результатах обработки по III закону спектрометрического
исследования процесса Sr+OH=SrOH++e в пламенах в работе Енсена [1564] D89+20 кДж-моль)
и масс-спектрометрического исследования реакции Sr++H2O=SrOH++H в пламенах в работе
Хейхерста и Кительсона [1374, 1375] D80+20 кДж-моль"). Она не противоречит данным,
полученным Келли и Педли [1664, 1666] при спектрофотометрическом исследовании реакции Sr(OHJ+
+3H=SrOH++H2O-fH2+? D94+27 кДж-моль), хотя интерпретация первичных
экспериментальных данных в этой работе была затруднительна.
Принятому значению соответствует
bfE°(SrOE+, г, 0) = 295 + 28 кДж • моль.
Sr (OHJ (к, ж). Термодинамические свойства кристаллического и жидкого дигидроксида
стронция в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 823.
Значения постоянных, принятые для расчета термодинамических функций,, приведены
в табл. 28.1. В справочнике за стандартное состояние Sr(OHJ (к) в интервале 0—753 К принята
тетрагональная модификация P-Sr (ОНJ [955], а при 753—808 К — высокотемпературная
модификация <x-Sr(OHJ с неизученной структурой. Ввиду отсутствия в литературе данных о
теплоемкости Sr(OHJ при низких температурах термодинамические функции P-Sr@HJ при Т < 298,15 К
вычислены по оцененным значениям теплоемкости, которым соответствуют величины S° B98,15 К)
и Н° B98,15 К)—Я° @), приведенные в табл. 28.1, их погрешности составляют 7 Дж-К^-моль
и 1 кДж-моль соответственно. При Т > 298,15 К для теплоемкости P-Sr(OHJ принято
уравнение (см. табл. 28.1), полученное по результатам измерений энтальпии Sr(OHJ (к) в работе
Пауэрса и Блелока [2262] 1. В работе [2262] не было замечено полиморфное превращение Sr(OH)a
при 753 + 10 К, установленное позднее Динеску и Преда [955], что, по-видимому, свидетельствуе»
о небольшой величине теплоты полиморфного превращения. В справочнике принято
Д<г# G53 К)=0. Теплоемкость a-Sr(OHJ принята равной теплоемкости P-Sr(OHJ в точке
превращения A22 Дж-К~1-моль), а температура плавления — равной 808 + 5 К на основании
согласующихся между собой данных Вольфа [2867] (808+2 К) и Динеску и Преда [955] (803 + 5 К).
Измерения Бергрена и Брауна [660] G23 К) и Брчича и др. [737] G98 К) привели к заниженным
величинам и не принимались во внимание. Энтальпия плавления B3,0+0,3 кДж-моль) и
теплоемкость жидкого дигидроксида стронция A53 + 10 Дж-К~1-моль~1) основаны на исследовании
энтальпии Sr(OHJ до 1187 К в работе Пауэрса и Блелока [2262].
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 7, 9 и
20 Дж-К^-моль соответственно. Термодинамические функции Sr(OHJ (к, ж), приведенные
в табл. 823 и в справочнике [1656], практически совпадают.
Давление пара дигидроксида стронция в реакции Sr(OHJ (к, ж)=8г@НJ (г) вычислено с
использованием значения Дг#° @) = Д,#° (Sr(OHJ, к, 0)=374,98 + 20 кДж-моль, соответствую-
1 Данные [2262] известны по справочнику Келли [1656], их погрешности оценены в 0,5%.
355
Sr (ОН)* (г)
Глава 28
Таблица 28.8. Результаты определений t±fH° (Sr(OHJ, к, 298,15 К) (в кДжмоль)
Авторы
Бертло, 1875 [670, 672]
Томсен, 1882 [2686]
Форкран, 1908 [1118]
Джонстон, 1908 [1592]
Тамару, Сиомн, 1934 [2654]
Сано, 1939 [2432]
Метод
Калориметрическое измерение энтальпии
растворения SrO и Sr(OHJ в соляной кислоте
То же, 2 опыта
То же
Измерение константы равновесия реакции
Sr(OHJ(K)=--SrO(K)+H2O(r) статическим
методом, 703—870 К, 5 точек
То же, 743—962 К, 10 точек
То же, 667—942 К, 16 точек
То же, 613—743 К, 5 точек
а Расчет из термохимического цикла, включающего растворение SrO (к) и Sr (ОН
6 Расчет из термохимического цикла
растворов SrCl2 (см. [406]).
Д/Я° (Sr(OHJ, к, 298,15 К)
II закон
III закон
—948 а
—966 б
—950+4 а
—958±6 б
—958+10 а
—965 ±10 6
—919+10 —967 ±8
-960±5 —970±8
—948 ±5 —965 ±8
_943 ±5 —963 ±8
2 (К).
, включающего растворение Sr(OHJ (к)иданныепоэнтальшт образования
щего принятым в справочнике энтальпиям образования Sr(OHJ в кристаллическом и
газообразном состояниях. Значение
A/ff°(Sr(OH)a, к, 298,15 К) = —965 + 8 кДж • моль
основано на приведенных в табл. 28.8 результатах калориметрических измерений и исследований
равновесия диссоциации Sr(OHJ (к). Указанные в таблице погрешности отражают
воспроизводимость измерений, а также погрешности других величин, использованных при обработке
экспериментальных данных. Как видно из таблицы, расчет AfH° (Sr(OHJ, к) из термохимических
измерений [670, 672, 1118, 2686] приводит к заметно отличающимся величинам в зависимости от
выбранного цикла. Сравнение рассчитанных таким образом величин с результатами обработки
данных, полученных при исследованиях равновесия диссоциации Sr(OHJ, свидетельствует о большей
надежности расчетов, исключающих реакции с оксидом стронция. Как видно из таблицы, в этом
случае величины, полученные из термохимических циклов и исследований равновесия реакции
диссоциации, оказались в полном соответствии. Принятое значение является средним из
соответствующих значений, приведенных в табл. 28.8.
Sr(OHJ (г). Термодинамические свойства газообразного дигидроксида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 824.
Молекулярные постоянные, использованные в расчете термодинамических функций Sr(OHJ,
приведены в табл. 28.7. Спектры и структура молекулы Sr(OHJ экспериментально не
исследовались, и приведенные постоянные основаны на приближенных оценках, выполненных при
подготовке справочника. Произведение моментов инерции в основном электронном состоянии
вычислено на основании структурных параметров -$О—Sr—0=115+5°, г (Sr—О) =2,20+0,05 А
(оценены по молекуле SrF2), -^Sr—О—Н=180°, г (О—Н)=0,96+0,02 А (перенесены из SrOH),
^го погрешность составляет 2-Ю1* г3-см6. Частоты v1? v2, v3 фрагмента О—Sr—О оценены из
данных для молекулы SrF2, частоты v4 и v5 фрагментов Sr—О—Н перенесены из молекулы SrOH.
Погрешности принятых значений частот оцениваются в 20 см для v2 и 50—100 см для
остальных частот. Сведения о возбужденных состояниях Sr(OHJ отсутствуют; можно предполагать, что
их энергии превышают 20 000 см.
*¦ Термодинамические функции Sr(OHJ (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены не-
356
Стронций и его соединения SrF (г)
точностью принятых молекулярных постоянных, а также приближенным характером расчета и
могут достигать 6, 10 и 12 Дж-К^-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Термодинамические функции Sr(OHJ (г) вычислялись ранее в таблицах; JANAF [1544],
расхождения данных [1544] и табл. 824 составляют 4—11 Дж-К~1-моль и обусловлены главным
образом разной оценкой величины v5.
Константа равновесия реакции Sr(OHJ (r)=Sr (г)+2О (г)+2Н (г) вычислена с
использованием значения Ь.ГН° @)=1668,507 +20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д//° (Sr @НJ, г, 0) = —582 ± 20 кДж ¦ моль'1.
Эта величина основана на спектрофотометрических измерениях равновесия реакции Sr (г)+
+2Н2О (r)=Sr(OHJ (г)+2Н (г) в пламенах, выполненных Коттоном, Дженкинсом [885]
(пламена H2+O2+N2, 1570—2030 К, 3 точки) и Рябовой и др. [359] (пламена H2+O2+N2, 1860—
2580 К, 16 точек). Результаты этих работ оказались близкими. Совместная обработка полученных
в них значений константы равновесия приводит для изученной реакции к значениям ДГЯ° @) =
=110+30 кДж-моль (II закон) и ДГЯ° @)=166,6 +5,5 кДж-моль (III закон). Основной вклад
в погрешность принятой энтальпии образования вносит неточность термодинамических функций
Sr(OHJ (г) (—20 кДж-моль). Сагден и Скофилд [2635] также исследовали равновесие
приведенной выше реакции. Однако результаты работы [2635] представлены только в виде графиков, что
не позволяет провести пересчет с использованием принятых в справочнике термодинамических
функций. Методом II закона в работе [2635] для энтальпии реакции получено значение Дг#°=
=125 кДж-моль. Расхождение с данными работ [359, 885] может быть частично объяснено тем,,
что авторы [2635] не учитывали образование SrO и SrOH в условиях экспериментов.
SrF (г). Термодинамические свойства газообразного фторида стронция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 825.
Молекулярные постоянные SrF, принятые в справочнике по результатам исследований
электронных спектров этой молекулы, приведены в табл. 28.9. В спектре SrF экспериментально
наблюдались переходы между восемью электронными состояниями: А2П ^ X2 ?+ [294, 297, 928, 1126,
1361, 1583, 1845, 26106, 2795], В2 Z+** X2 Е+ [294, 297, 928, 965а, 1126, 1361, 1583, 1845, 2610аг
2795], Cm *ь X2 S+ [294, 297, 928, 1126, 1583, 1845, 2795], D2 Е+ +* X2 ?\ E2U *± X2 ?+, F2 Е+ **
*±Х2Е+ и С2П^Х2?+ [294, 297, 1126].
Как и у молекул других галогенидов щелочноземельных металлов, состояния X2 Е+ и А2Л
молекулы SrF коррелируют с основными состояниями атомов Sr A5)+F BP). Go следующим дис-
социационным пределом Sr CP)+F BР) коррелирует большое число состояний, в том числе
квартетные состояния. В справочнике эти состояния не рассматриваются, хотя часть из них может
быть связанными (см. текст по BeF и CaF). Частично это компенсируется учетом ряда дублетных
состояний типа D2 2+ и Е2П, имеющих как и у CaF ридберговский характер (см. текст по CaF).
Колебательные постоянные SrF в состоянии X2 ?+ определялись по кантам полос систем
А2П«* Х2?+, 522+-Х2Е+ и С2П-Х2 Е+ в работах [294, 297, 1361, 1583] и удовлетворительно
согласуются между собой. Приведенные в таблице постоянные найдены Новиковым и Гурвичем
[294, 297] при исследовании системы А2П *±: X2 Е+ . (i/ < 29, v" <; 29).
Вращательные постоянные SrF в состояниях X2 ?+, А2П и В2 Е+ приняты по работам Домейла,
Стеймла и Харриса, которые выполнили анализ тонкой структуры полос 0—0, 1—1, 2—2 системы
В2 ?+—X2 Е+ [2610а], полос 1—0, 2—1 системы 42П—X2 Е+ [261061 и чисто вращательного спектра
этой молекулы [965а 1 г.
Колебательные постоянные в состояниях А2И и С2П приняты по работам Новикова и Гурвича
[294, 297], в состояниях В2 2+ и D2 ?+, ?2П, F2 Е+ и G2II по работам Джонсона [1583] и Фаулера
11126].
Термодинамические функции SrF (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBB и ее производных рассчитывались по уравнениям A. 90)—A.92)
1 В работе [2610а] по началам трех полос найдены также значения колебательных постоянных в состояниях
Х22+ (<ов=502,4 см, а>ехе=2,27 см) и ?22+ (а>е=495,8 см, шеже=2,34 см). В справочнике отдано
предпочтение постоянным, полученным в работе [294, 297], так как значения постоянных, предложенные авторами
{2610а], плохо описывают колебательные уровни состояния Х22+ с v > 20.
357
SrF (г)
Глава 28
Таблица 28.9. Молекулярные постоянные 88Sr19F, SrF+, Sr35Cl, SrCl+, SrBr и Sri
Молекула
Состояние
Т.
ве
аг ¦ 10*
D, ¦ 108
см~х
83SrlsF X2 2+ 0a
SrF+
Sr^Cl
SrCl+
SrBr
Sri
АШ1и 15 067,7
42П5/' 15 348,3
В2 2+ 17 267,42
С2П 27 383,7
D% 2+ 28 296,6
Em 31 528,7
Я 2+ 32 820,3
G2n 34 759,2
ХЧ+ 0
X22+ 0
,42n7i 14 818
Атз'г 15 112,8
В2 2+ 15 779,5
С2П,А 25 244,4
C2ns/i 25 399,6
В2 2+* 28 822,9
?22+ 32 201,8
ЯП 32 940 Д
G2n 34 072 Д
IP 2+ 34 256,7
XX2+ 0
X22+ 0
Л2П71 14 699,4
А*П,'г 15 000,7
В2 2+' 15 352
СгП1Л 24 504
Z>2 2+ 28 958,2
Я2 2+ 32 052,5
ЯП 33 174 Д
^Д 34 270 Д
Н% 2+ 34 358
Х22+ 0
Am4i 14 422,7
Ат,'ы 14 815,90
В2 2+' 14 748,83
С2П 22 945 е
D2 2+ 28 944
ЯП 34 485 е
499,8
507,9
507,3
495,8
450,52
552,1
564,4
598,5
573,9
600 а
301,8
309,1
306,4
282,1
283,4
344,8
346,3
354,3
356,7
364,6
370 а
216,5
222,18
222,07
222,0
205,2
247,8
248,0
256,7
256,6
267,7
173,8
182,3
182,2
179,5
170,9
200,4
210,4
Примечание. Ниже все постоянные даны в см~
SrF: a
SrF+:
SrCl: a
r=2,49-10; 6 o),,Ve=3,9-l
2,14 0,250 533
2,206б 0,252 12
2,18
2,34 0,249 396
1,72 —
2,15 —
3,20 —
3,42 —
1,28 —
1,9 б 0,270 в
0,92 0,099 8 а
0,98 —
—
0,98 —
0,89 —
0,92
1,04
1,10 —
1,09 —
1,00 —
1,08 —
0,91б 0,107 в
0,51 0,051 8 а
0,53 —
0,53 —
0,55 —
0,49 —
0,55 —
0,65 —
.
— —
_ _
0,285 а 0,036 1 б
0,54 —
0,37 —
0,315 —
0,36 —
0,5 —
0,4 -
15,46
15,6
15,57
—
—
—
12,0 г
4,03 б
—
—
—
—
_
—
3,5 г
1,6 б
—
—
—
—
—
0,81 в
—
—
—
—
г.
к
24,9 2,0754
22 2,0689
25,2 2,0801
—. —
— —
— —
—
_ _
22,0 Д 2,00 а
4,4 " 2,60 г
— —
— —
— —
_ _
—
— —
— —
_ _
3,6 Д 2,50 а
1,2 в 2,80 г
_ _
_ _
—
— . —
— —
—
— —
_ _
0,62 г 3,00 Д
— —
— —
— —
— —
а оценка, см. текст; б вычислено по соотношению A.67) и значению Do (SrF +)=49 300; в рассчитано
по соотношению A.38); г
рассчитано по соотношении
нено по соотношению A.68
оценено по соотношению A.69); д оценено по соотношению A.68).
> A.38) и принятому значению ге; б оценено по соотношению A.69); в оце-
; г оценка (
см. текст); Д приведена энергия центра дублета.
358
Стронций и его соединения
SrF (г)
Таблица 28.9 (окончание)
SrCl+: а оценка, см. текст; ° вычислено по соотношению A.67) п значению Do(SrCl+)----36 300; в рассчитано
по соотношению A.38); г оценено по соотношению A.69); Д оценено по соотношению A.68).
SrBr: а рассчитано по соотношению A.38) и принятому значению ге; б оценено по соотношению A.69); в
оценено по соотношению A.68); г оценка (см. текст); Д приведена энергия центра дублета.
Sri: а шеув—0,00015; 6 вычислено по соотношению A.38) и принятому значению re; B оценено по
соотношению A.69); г оценено по соотношению A.68); Д оценка (см. текст); е приведена энергия центра дублета.
с учетом восьми возбужденных электронных состояний в предположении, что Q(*\^ вр = (pjpx) QifJ. вр.
Величина <?S,Bp и ее производные вычислялись по уравнениям A.73)—A.75) непосредственным
¦суммированием по колебательным уровням и интегрированием по вращательным уровням
энергии с помощью уравнений типа A. 82). В расчетах учитывались все уровни со значениями
J ^ Jmat „, где /ш1|, находились из условий A. 81). Колебательно-вращательные уровни
состояния X2 ?+ вычислялись по уравнениям A. 65), A. 61). Значения коэффициентов Ykl, а также
величин fmax и /llm, использованных в расчетах, приведены в табл. 28.10. Коэффициент Уоз
получен из условия A. 59), a Y21 — из условия Bv x=0. Мультиплетность и вырождение
колебательно-вращательных уровней электронных состояний учитывались статистическими весами.
Погрешности рассчитанных термодинамических функций SrF (г) при низких температурах
обусловлены главным образом неточностью колебательных постоянных в состоянии X2 Е+, а при
высоких температурах — неполнотой сведений о возбужденных электронных состояниях этой
Таблица 28.10. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см),
а также значения vmax и >Тит, принятые для расчета термодинамических функций SrF,
SrF+, SrCl, SrCl+, SrBr и Sri
Коэффициенты
Г.-Ю
Ую-Ю-3
У2о- Ю
Узо-Ю2
У40-Ю4
yso-ioe
Уо1-1О1
Уп-Ю»
У02-Юв
Уоз-1014
"та.
¦^llm
SrF
0
0,499 374
—0,205 891
—0,483 827
0,962 994
—0,273 926
2,505 33
—0,154 6
—0,249
3,127
197
589
SrF+
0
0,600
-0,19
—
—
—
2,70
—0,12
—0,22
—
160
561
Примечание. Ниже все постоянные даны в см
SrF: a Состояние
Т.
SrCl: a Состояние
Т.
SrBr: a Состояние
Т.
Sri: a Состояние
Т.
лт,, А*щг
15 067,5 15 348,3
лтч а2щ
14818 15 112,8
i!4Iv А*Щг
14 699,4 15 000,7
а*пч% лт,1г
14 422,7 14 815,9
SrCl
Х22+»
0
0,300
—0,089
0,096
0,175
—0,040
0,989
—0,039
—0,043
0,660
244
772
cm
17 267,42 27 383,7
15 779,5
15 352
Ва2+
14 748 Д
cm
25 320
cm
24 504
cm
I 22 945
365
448
171
426
001
8
28 296,6
ZJ2+
28 822,9
Д22+
28 958,2
Я2 2+
28 944
SrCl+
ХЧ+
0
0,370
—0,091
—
—
—
J,07
-0,035
—0,036
—
203
786
Em
31 528,7
32 201,8
i?2E+
32 052,5
Fm
34 485
SrBr
X2S+
0
0.216
—0,051
0,035
—
—
0,518
—0,016
—0,012
—0,151
295
954
32 820,3
Fm
32 940
ЯП
33 174
Sri
X2S+a
0
551 0,173 812
866 —0,028 710 6
021 0,026 227
—0,018 489
—
0,361
—0,008 1
-0,006 2
0,023
224
1075
вт
34 759,2
62П Яа 2+
34 072 34 256,7
С2Д Я8 2+
34 270 34 358
359
SrF+ (г) Глава 28
молекулы. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К могут достигать 0,03, 0,1
и 1,0 Дж-К-моль'1 соответственно.
Ранее термодинамические функции SrF (г) публиковались в таблицах JANAF [1544],
справочнике [305] (до 5000 К) и работе Краснова и др. [167] (приведен полином, аппроксимирующий
значения Ф° (Г) до 5000 К).
Расхождения данных [1544] и табл. 825 составляют 0,4 и 1,3 Дж -К -моль в значениях Ф° (Т)
и S° (T) при 6000 К и обусловлены различием в методиках расчета и небольшой разницей в
принятых молекулярных постоянных. Расхождения с расчетами [167, 305] существенно больше, так как
они выполнены без учета возбужденных электронных состояний и по грубой методике.
Константа равновесия реакции SrF (r)=Sr (r)+F (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Д.Л0 @) = Do (SrF) = 540 + 6 кДж • моль ^ 45 140 + 500 см.
Эта величина основана на результатах исследований равновесия реакций с участием SrF (г),
представленных в табл. 28.11. Погрешности значений Do (SrF), приведенных в таблице, включают
Таблица 28.11. Результаты определений Do (SrF) (в кДж-моль)
Авторы
Метод
D0(SrF)
II закон
III г а кон
Элерт и др., 1964 [1020] Масс-споктрометрический, SrF, (K)+Sr (r)=2SrF (г), 503+13 527,6±4
1125—1292 К, 10 точек, Дг Н° @)=487,0±1,9 (III)
То же, Sr (r)+BaF (г)=Ва (r)+SrF (г), 1174— 546±13 535,6±7
1290 К, 9 точек, ДГЯ° @)=44,4±0,5 (III)
Хилденбранд, 1968 [1406] То же, SrF,, (r)+Sr (r)=2SrF (г), 1436—1525 К, 9 то- 540±30 544,0±4,2
чек, ДгЯ°@)=11,0±1IAП)
То же, 2Sr (r)+BF8 (r)=BF (r)+2SrF (г), 1476— 542±40 538,4±6
1585 К, 10 точек, Дг#° @)=101,9±1,4 (Ш)
Меннингер, 1974 [2007] Исследование хемилюминесценщга SrF при образо- >533±6
вании в скрещенных молекулярных пучках
Энгелке, 1979 [10336] То же >531±8
воспроизводимость измерений, неточность сечений ионизации, термодинамических функций и
энтальпий образования, использованных в расчетах. Принятое значение является средневзвешенным
из полученных в работах [1020] и [14061, причем при оценке веса данного измерения учитывалось
его отклонение от среднего значения. В работе [355а] на основании спектрофотометрического
определения изменения интенсивности линии Sr в пламенах Н2+Ог+1Ч.2 при введении в пламена
соединений фтора получено завышенное значение Do (SrF) из-за того, что авторы не учитывали
образование в пламенах молекул SrOHF.
Принятой энергии диссоциации соответствует
gA^B (SrF.gr, 0) = — 301,852 + 6,3 кДж • моль.
SrF+ (г). Термодинамические свойства газообразного положительного иона фторида стронция
в стандартном состоянии в интервале температур 298,15 — 6000 К приведены в табл. 826.
В табл. 28.9 приведены молекулярные постоянные SrF+, принятые из-за отсутствия
экспериментальных данных на основании оценок. По аналогии с изоэлектронными молекулами RbF и SrO
основным состоянием иона SrF + должно быть состояние X1 Б+ (конфигурация . . . а2 л4).
Вращательная постоянная, приведенная в табл. 28.9, вычислена по оцененному значению re(Sr—F)=2,00 +
+0,05 а, полученному сопоставлением межатомных расстояний в молекулах CaF, CaF+ и SrF.
Значение ше оценено из сравнения соответствующих величин в тех же молекулах и хорошо
согласуется с колебательными постоянными SrF в ридберговских состояниях. Погрешности принятых
значений Ве и <ое, по-видимому, не превышают 0,015 и 60 см. Энергии возбуждения
электронных состояний SrF+, соответствующих конфигурациям . . .<?ч?а,1 .-. ,^п3п и . . .отт:4тс, оценить
360
Стронций и его соединения SrFa (к, )
трудно. Можно ожидать (см. текст по CaF+ (г)), что они больше энергий аналогичных состояний
молекулы SrO и лежат в области 20 000 см.
Термодинамические функции SrF+ (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A.93)—A. 95) без учета возбужденных состояний. Значения Qmi вр и ее производных вычислялись
по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням
и интегрированием по вращательным уровням с помощью уравнений типа A.82). В расчетах
учитывались все уровни энергии со значениями / ^ «^тах,»? последние находились по формуле A. 81).
Колебательно-вращательные уровни состояния Xх Е+ вычислялись по уравнениям A. 65) и A. 62).
Значения коэффициентов Ykl в этих уравнениях, идентичные соответствующим постоянным из
табл. 28.9, а также величины vmax и Jlm приведены в табл. 28.10.
Погрешности вычисленных термодинамических функций при низких температурах обусловлены
неточностью принятых постоянных SrF+ в состоянии X1 2+, а при умеренных и высоких
температурах — пренебрежением возбужденными электронными состояниями. Общие погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,5, 1 и 3 Дж -К -моль соответственно.
Ранее термодинамические функции SrF+ (г) вычислялись в таблицах JANAF [1544] по
постоянным, близким к принятым в настоящем справочнике, но с неоправданным учетом большого числа
возбужденных электронных состояний с энергиями 10 000—35 000 см. Расхождения данных
[1544] и настоящего издания, небольшие при низких температурах, быстро растут, достигая 5
и 11 Дж-К^-моль в значениях Ф° F000 К) и S° F000 К) соответственно.
Константа равновесия реакции SrF+ (т)-\-е (r)=Sr (r)+F (г) вычислена с использованием
значения Дг#° @) =40,000+40 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
J0(SrF)=500+40 кДж-моль.
Эта величина основана на результатах измерений методом электронного удара, выполненных
Грином и др. [1245] D73 +30 кДж-моль-1), Элертом и др. [1020] E02 +30 кДж-моль) и Хилден-
брандом [1406] D82+30 кДж-моль). Предпочтение отдано более высокому значению, поскольку
оно лучше согласуется с принятыми в справочнике потенциалами ионизации молекул CaF, BaF
и CaCl, SrCl, BaCl. Оно приводит также к значению Do (SrF+), лучше согласующемуся с величиной
Do (SrF). Принятой величине соответствуют
bfH° (SrF+, г, 0) = 198,148 ± 40 кДж • моль,
Do (SrF+) = 589,470 + 40 кДж • моль да 49 300 + 3300 см.
SrF2 (к, ж). Термодинамические свойства кристаллического и жидкого дифторида стронция
в стандартном состоянии при температурах 100—4300 К приведены в табл. 827.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. В справочнике за стандартное состояние SrF2 (к) в интервале 0—1484 К принята
кубическая а-модификация (структурный тип флюорита CaF2), а в интервале 1484—1750 К —
высокотемпературная разупорядоченная Р-модификация. При Т < 298,15 К термодинамические
функции SrF2 вычислены по значениям теплоемкости, найденным Смитом, Гарднером и др. [2551]
A1—300 К) для образца SrF2, который содержал менее 0,02% примесей металлов и небольшую
примесь некристаллического SrF2. Экстраполяция теплоемкости ниже 11 К по уравнению Дебая
дает 5° A1 К)=0,10 Дж-К^-моль. Погрешности принятых по [2551 ] значений S° B98,15 К)
и Н° B98,15 К)—Я° @) оцениваются в 0,4 Дж-К-моль~1 и 0,04 кДж-моль соответственно.
При Т > 298,15 К расчет термодинамических функций SrF2 (к) основан на результатах измерений
энтальпии в работе Ефремовой и Матизена [127]-, выполненных в интервале 813—2126 К на об-
вазце, содержавшем около 0,03% примесей. Погрешности этих измерений оцениваются в 0,5%.
Фазовый переход у SrF2 (к) сопровождается ^-аномалией теплоемкости в интервале 1000—1600 К
с максимумом при 1484 К. Теплоемкость SrF2 (к) в интервале 298,15—1750 К аппроксимирована
тремя уравнениями, выведенными по данным [127]; одно уравнение описывает медленное
возрастание теплоемкости в интервале 298,15—800 К (от 70,0 до 83,4 Дж-К~1-моль), а^ два других
аппроксимируют восходящую и нисходящую ветви Х-кривой теплоемкости (рост С°р от 83,4 до
172,6 Дж-К^-моль в интервале 800—1484 К и затем снижение до 101,5 Дж-К'1-моль в точке
плавления). Величина Tm (SrF2)=1750+3 К принята по хорошо согласующимся между собой дан-
361
SrF2 (г)
Глава 28
ным Ефремовой и Матизена [127] A750+2 К) и Коздима и др. [1747] A746 К). Портер и Браун
[2251] получили более низкое значение A736+5 К), по-видимому, из-за недостаточной чистоты
исследованного образца. Значения АтН (SrF2) =28,5 +0,6 кДж-моль" и С°р (SrF2, ж)=99 +
+2 Дж-К-моль приняты на основании измерений энтальпии SrF2 в работе Ефремовой и
Матизена [127].
Погрешности вычисленных значений Ф° (Г) при 298,15, 2000 и 4000 К составляют 0,3, 1,5 и
7 Дж-К-моль. Расхождения между термодинамическими функциями SrF2 (к, ж),
приведенными в справочнике JANAF [1544] и табл. 827, не превышающие 1 Дж-К~1-моль~1 в значениях
Ф° (Т) до 3000 К, обусловлены различной аппроксимацией данных [127] в [1544] и в настоящем
издании.
В справочнике принята энтальпия образования
AfH°(SrF2, к, 298,15 К) = —1228 ±4 кДж • моль.
Эта величина основана на калориметрических исследованиях энтальпии реакции SrO (к)+
-f 2HF (p-p)=SrF2 (к)+Н2О (р-р) в работе Барань и др. [576] (—1223,1 +4 нДж-моль) и реакции
SrCl2 (р-р 300 H2O)+2NaF (K)=0,995SrF2(K) +0,005SrF2 (p-p coH2O)+2NaCl (p-p 150H2O) в работе
Финча и др. [1087] (—1228,1+4 кДж-моль'1), а также определениях растворимости SrF2 (к)
в воде в работах Кольрауша [1743] (—1229,2+ЗкДж-моль'1) иТалипова, Хадеева [403] (—1229,2 +
+2 кДж-моль). Использованные для обработки этих данных термодинамические свойства ионов
см. в Приложении 5. Исследование растворимости SrF2 (к) авторами [826], по-видимому, содержит
систематическую ошибку, так же как измерение растворимости BaF2 (к) этими же авторами.
Расчет энтальпии образования из данных [330] по равновесию реакции 3SrCl2 (ж)+2А1Р3 (ж) =
=3SrF2 (ж)+2А1С13 (г) приводит к неверному значению (—1260 кДж-моль).
Давление пара дифторида стронция SrFa (к, ж)=8гР2 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
A,#°(SrF2, к, 0) = 443 + 5 кДж • моль.
В табл. 28.12 приведены результаты обработки данных по давлению пара SrF2 (к, ж),
полученных разными авторами. В качестве погрешностей в таблице приведена воспроизводимость
измерений. Дополнительную погрешность вносит неточность термодинамических функций SrF2 (к, ж)
и SrF2 (г) (—7 кДж-моль для данных [2407] и 3—4 кДж-моль для данных из других работ),
Таблица 28.12. Результаты определений Д,Н° (SrF2, к, 0) (в кДж-моль)
Авторы
Руфф, Ле Буше, 1934 [2407]
Грин и др., 1964 [1245]
Баутиста, Маргрейв, 1965 [629]
Хилденбранд и др., 1965 [1422]
Метод
Точек кипения, 2095—2232 К, 6 точек
Масс-спектрометрический, 1207—1563 К, 30 точек
Испарение с открытой поверхности, 1235—1319 К,
9 точек
Торзионный, 1420—1710 К, 24 точки, расчет
по уравнению
As#°(SrF2, к, 0)
II закон
III закон
490±80 443,9+1,8
447±6 449,1 ±0,4
437 ±37 450,0±0,8
434 443±1,7
а также неточность сечений ионизации для данных [1245] G кДж-моль *). Несколько завышенное
значение энтальпии сублимации, полученное методом испарения с открытой поверхности [629],
может быть объяснено отличием коэффициента испарения от 1. Наиболее достоверные данные
получены в работе Хилденбранда и др. [1422], и принятое значение основано на этой работе.
SrF2 (г). Термодинамические свойства газообразного дифторида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 828.
Молекулярные постоянные, использованные для расчета термодинамических функций SrF2,
приведены в табл. 28.7. Исследования инфракрасного спектра [23, 24, 808, 1626, 2565] и
отклонения молекулярного пучка в неоднородном электрическом поле [2839] показали, что в основном
362
Стронций и его соединения SrCI (г)
электронном состоянии молекула SrF2 имеет нелинейную симметричную структуру. Приведенное
в табл. 28.7 произведение моментов инерции вычислено при ДР—Sr—F=110+5° (среднее из
значений, рассчитанных Колдером и др. [808] по изотопическому расщеплению полос SrFa) и
г (Sr—F)=2,20+0,03 А (найдено Акишиным и др. [5, 14] при электронографическом
исследовании паров SrF2). Погрешность величины IaIbIc составляет 1-1014 г3-см6. Значение v3 принято
по данным Байкова [24], который исследовал инфракрасный спектр газообразного SrF2. Оно
хорошо согласуется с частотами, наблюдавшимися Снелсоном [2565], Колдером и др. [808] и
Кан'аном и др. [1626] в ИК-спектре молекул SrF2, изолированных в матрицах. Для \х принято
значение, которое на 10 см больше, чем частота, наблюдавшаяся в матрице из криптона в
работах [808, 2565], в соответствии с разницей между величиной v3 в ИК-спектре пара [24] и в матрице
из Кг [808, 2565]. Для v2 принято значение среднее между измеренными в ИК-спектре пара [23]
A05 см) и в матрице из Кг [808] (82 см), так как Байков в области 90—160 см наблюдал не
всю полосу, а только ее высокочастотную часть. Погрешности принятых значений частот
оцениваются в 10—15 см. Сведения о возбужденных состояниях SrF2 отсутствуют; можно
предполагать, что их энергии превышают 20 000 см.
Термодинамические функции SrF2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор». Погрешности рассчитанных термодинамических функций обусловлены неточностью
молекулярных постоянных и приближенным характером расчета, они составляют 2,4 и 6 Дж -К -моль
в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно (в том числе 1—2 Дж-К-моль~1
из-за неточности молекулярных постоянных).
Ранее термодинамические функции SrF2 (г) рассчитывались в справочниках [1544] и [305],
а также в работах Байкова [23] (Т < 5000 К), Брюэра и др. [748] (Т < 2500 К), Краснова и
Светцова [195] (Т ^ 2500 К). В настоящем издании и в расчетах [23, 305, 1544] приняты близкие
значения молекулярных постоянных, поэтому расхождения в значениях Ф° (Т) не превышают 0,5,
5 и 1 Дж-К-моль для [23, 305, 1544] соответственно. В расчетах [195, 748] для молекулы SrF2
принята линейная структура и расхождения с настоящим изданием достигают 18—20 Дж-К"хХ
Хмоль в значениях Ф° (Т).
Константа равновесия реакции SrF2 (r)=Sr (r)+2F (г) вычислена с использованием значения
АГН° @)=1098,103 +6,7 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дифторида стронция. Этим величинам также соответствует
bfH°(STF2, г, 0) = —782,68 + 6,4 кДж • моль.
SrCI (г). Термодинамические свойства газообразного хлорида стронция в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 829.
Молекулярные постоянные SrCI, принятые в справочнике на основании исследований
электронных спектров этой молекулы, приведены в табл. 28.9. В спектре SrCI наблюдались 13 систем
полос, связанных с переходами между девятью электронными состояниями: -42П<*Х2 Е+ [294, 295,
1354, 1378, 2159, 2184, 2795], В2 ?+ *а X2 Е+ [294, 295, 1354, 2159, 2795], С2П ** X2 2+ [294,
295, 1354, 2159, 2184, 2795], D2 2+ *ь X2 2+, Е2 S+ ** X2 2+ и Я2 ?+ т* X2 2+ [294, 295, 1354],
а также F2U. -> A2U, 62П -* A2tt, D2 ?+ -> В2 Е+, D2 2+ -> А2П, Н2 2+ -+ В2 Е+, Е2 2+ -* АЧ\
и F2!! -> В2 ?+ [294, 295]. Как у других молекул галогенидов щелочноземельных металлов,
состояния X2 И+ и A2IL молекулы SrCI коррелируют с основными состояниями атомов Sr 0?)+
+С1 BР). Со следующим диссоциационным пределом Sr CP)+C1 BР) должно коррелировать
большое число состояний, в том числе квартетных; часть из них может быть связанными
состояниями. В то же время часть наблюдавшихся дублетных состояний, приведенных в таблице, имеют
характер ридберговских состояний (см. текст по CaF).
Колебательные постоянные SrCI в состоянии X2 Е+ определялись по кантам полос систем А2Л—
X2 Е+ [294, 295, 1354, 1378, 2184], В2 ?+—X2 ?+ [1354], С2П—X2 Е+ [294, 295, 1354, 2184], D2 Е+—
X2 Е+, Е2 ?+—X2 Е+ и Н2 Е+—X2 2+ [1354]. Приведенные в табл. 28.9 постоянные основаны на
данных [294, 295, 1354] и близки к рекомендованным в справочнике [2645]; можно предполагать, что
погрешность значения а>в не превышает 1—2 см. Вращательные постоянные SrCI
экспериментально не определялись. Приведенное в табл. 28.9 значение re(SrCl)=2,60+0,05 А в состоянии
X2 Е+ оценено на основании величины г (Sr—С1) в молекуле SrCl2 (см. ниже) и сопоставления
величин rt (MX) и г (М—X) в других галогенидах и дигалогенидах щелочноземельных металлов.
363
SrCl+ (г) Глава 28
Колебательные постоянные в состояниях Л2П, СЧ1, F2U и G4I приняты по работе Новикова
и Гурвича [294, 295], в состояниях ?2?+, ?>2Е+, ?2Е+ и #2?+ — по данным Харрингтона [1354].
Термодинамические функции SrCl (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производные рассчитывались по уравнениям A. 90)—A.92 )
с учетом девяти возбужденных состояний в предположении, что Qlil.BS=(Pi/px) QmJ.vy Величина
*?S. Вр и ее производные вычислялись по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с
помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями
/ ^ /ш „, где /шах „находились из условия A. 81). Колебательно-вращательные уровни
состояния Х2?+ вычислялись по уравнениям A. 65), A. 64). В табл. 28.10 приведены коэффициенты Ykl
в этих уравнениях, рассчитанные для природной изотопической смеси молекул SrCl по
соотношениям A. 66) и постоянным из табл. 28.9. Коэффициент Yos получен с учетом условий A. 59).
В табл. 28.10 приведены также значения i;max и JUm, использованные в расчетах. Мультиплетность
и вырождение колебательно-вращательных уровней электронных состояний учитывались
статистическими весами.
Погрешности вычисленных термодинамических функций при Т ^ 2000 К обусловлены
главным образом отсутствием данных о вращательных постоянных, а при более высоких
температурах — неполнотой сведений о возбужденных электронных состояниях этой молекулы. Общие
погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,3, 0,5 и 1 Дж- К- моль
соответственно.
Термодинамические функции SrCl (г) рассчитывались ранее в таблицах JANAF [1544] и работе
Климова и др. [167] (приведен полином, аппроксимирующий значения Ф° (Т) для Т <^ 5000 К).
Расхождения данных [1544] и табл. 829 растут с температурой и достигают 0,8 и 1,7 Дж-КХ
Хмоль в значениях Ф° (Т) и S° (T) при 6000 К, что связано с различием в методиках расчета и
некоторой разницей в использованных значениях постоянных. Расхождения с данными [167]
существенно больше, так как этот расчет выполнен по грубой методике.
Константа равновесия реакции SrCl (r)=Sr (r)-f-Cl (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДГЯ° @) = Doj(SrCl) = 405| + 10 кДж • моль да 33 900 + 800 см.
Это значение основано на результатах масс-спектрометрического исследования равновесия
реакций SrCl2 (r)+Sr (r)=2SrCl (г) A305-1406 К, 9 точек) и Sr (г)+А1С1 (r)=SrCl (г)+А1 (г) в работе
Хилденбранда [1409] и спектрофотометрического исследования равновесия реакции Sr (г)+
+НС1 (r)=SrCl (г)+Н (г) в пламенах A745—2580 К, 16 точек) в работе Гурвича и др. [1333].
Обработка данных [1409] по уравнениям II и III законов дает 411+16 и 410+4 кДж-моль
для первой реакции и 427 +35 и 405 +7 кДж-моль для второй реакции; обработка данных [1333]
по зависимости логарифма отношения интенсивности полосы SrCl к интенсивности линии Sr в
спектрах пламен от Т'1 дает 400+11 кДж-моль (погрешности учитывают воспроизводимость
измерений, неточность использованных в расчетах термодинамических величин и сечений ионизации).
Данные [1333 и 1409] удовлетворительно согласуются с полученными Хилденбрандом [1407]
методом электронного удара D14+15 кДж-моль). Поттер и др. [2261] в масс-спектрометриче-
ских измерениях, аналогичных выполненным в [1409], получили несколько более высокие
значения D12+4 и 421+7 кДж-моль при расчете по III закону). Определение нижней границы
величины Do (SrCl) по хемилюминесценции SrCl в экспериментах с молекулярными пучками привело
к значениям >414+8 кДж-моль [1594] и >518+13 кДж-моль [2007].
Принятой величине соответствует
A/Jff°(SrCl, г, 0) = —124,507 + 10 кДж • моль.
SrCl+ (г). Термодинамические свойства газообразного положительного иона хлорида стронция
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 830.
В табл. 28.9 приведены молекулярные постоянные SrCl+, принятые из-за отсутствия
экспериментальных данных на основании оценок. По аналогии с изоэлектронными молекулами RbCl и
SrS основным состоянием SrCl+ должно быть состояние X1 Е+ (конфигурация . . . о2тс4).
Вращательная постоянная SrCl+ в этом состоянии вычислена на основании значения re (Sr—С1)=2,5+0,05 А,
364
Стронций и его соединения SrCb (к, ж)
оцененного из сопоставления межатомных расстояний в молекулах СаС1, СаС1+ и SrCl. Значение ше
оценено сравнением соответствующих величин в тех же молекулах и хорошо согласуется с
частотами колебаний молекулы SrCl в ридберговских состояниях. Погрешности принятых значений Вв
и и>в не должны превышать 0,01 и 50 см соответственно. Так же как у GaF+, возбужденные
электронные состояния SrCl+, принадлежащие конфигурациям . . .в*т?о, . . . ая*аи . . . атс4тс,
по-видимому, имеют энергии —20 000 см и выше и не рассматриваются в справочнике.
Термодинамические функции SrCl+ (г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95) без учета возбужденных электронных состояний. Значения <?ЕШГ. вр и ее производных
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по / с помощью уравнений типа A. 82). В расчетах учитывались все
уровни энергии со значениями / ^ ^Шах, *> последние находились по соотношениям A. 81).
Колебательно-вращательные уровни состояния ХХЕ+ вычислялись по уравнениям A.65) и A.62);
значения коэффициентов Yы в этих уравнениях, идентичные соответствующим постоянным из
табл. 28.9, а также величины t>max и /lim приведены в табл. 28.10.
Погрешности вычисленных термодинамических функций обусловлены неточностью оцененных
постоянных в состоянии Xх ?+, а также пренебрежением возбужденными электронными
состояниями; погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,5, 2 и 4 ДжХ
X К ~1 • моль соответственно.
Ранее таблица термодинамических функций SrCl+ (г) не публиковалась.
Константа равновесия реакции SrCl+ (т)-\-е (r)=Sr (г)+С1 (г) вычислена с использованием
значения Дг#° @) =—115,0 +22 кДж -моль, соответствующего принятому в справочнике потенциалу
ионизации
Io (SrCl)=520 +20 кДж-моль.
Эта величина основана на измерениях методом электронного удара, выполненных Хилденбран-
дом [1407] E39+10 кДж-моль), и методом поверхностной ионизации молекулярного пучка
SrCl2 (SrCl (r)+Sr + (r)=SrCl+ (r)+Sr (г), 1616—1900 К, результаты измерений представлены в виде
графика, /0=492+6 кДж-моль (II закон) и 498+2 кДж-моль (III закон)), выполненных
Логиновым, Митцевым [224]. Близкие значения получены Поттером [2261] E40 кДж-моль")
и Хилденбрандом [1409] E11 +50 кДж-моль) методом электронного удара. Принятой величине
соответствуют
AfH° (SrCl+, г, 0) == 395,493 + 22 кДж • моль,
Do (SrCl+) = 434,470 + 22 кДж • моль ж 36 300 + 1800 см.
SrCl2 (к, ж). Термодинамические свойства кристаллического и жидкого дихлорида стронция
в стандартном состоянии при температурах 100—3800 К приведены в табл. 831.
Зпачения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. В справочнике за стандартное состояние SrCla (к) в интервале 0—990 К принимается
кубическая а-модификация (структурный тип флюорита CaF2), а в интервале 990—1147 К —
высокотемпературная разупорядоченная (S-модификация.
При Т <С 298,15 К термодинамические функции SrCl2 (к) вычислены на основании измерений
теплоемкости, выполненных Смитом и др. [2551] G—300 К) на образце высокой чистоты (примеси
металлов в сумме —0,01%). Экстраполяция теплоемкости по уравнению Дебая приводит
к S° G,38 К)==0,05 Дж-К-моль~1. Погрешности принятых по этой работе значений S° B98,15 К)
и Н° B98,15 К)—Н° @) оцениваются в 0,4 Дж-К-моль и 0,04 кДж-моль соответственно.
При Т ^> 298,15 К приведенные в табл. 28.1 уравнения для теплоемкости и значения энтальпии
фазовых переходов основываются на результатах хорошо согласующихся между собой измерений
энтальпии SrCl2 (к) в работах Ефремовой и Матизена [127] (805—1797 К) и Дворкина и Бре-
дига [1002] D18—1204 К) г. Фазовый переход второго рода у SrCl2 сопровождается широкой
Х-аномалией теплоемкости в интервале —^800-^-1100 К, с максимумом при 990 +10 К [127]. Тепло-
1 Энтальпия SrCl2 измерена также Янцем и др. [1549] D33—1287 К). В работе [1549] не приведены
экспериментальные данные и даны только уравнения для Ср (Т) и Н° (Т)—Н° B98,15 К). Величины, вычисленные по
этим уравнениям, существенно отличаются от полученных в работах [127, 1002], поэтому данные [1549]
не учитывались при выводе уравнений для С (SrCl2, к).
365
SrCl2 (к, ж) Глава 28
емкость SrCl2 (к) аппроксимирована тремя уравнениями (см. табл. 28.1). Для интервала
298,15 —700 К уравнение выведено по данным [1002] и описывает плавное возрастание
теплоемкости (от 75,6 до 84,2 Дж>К~1-моль~1). Два других уравнения получены по данным [127] и
[1002] и аппроксимируют восходящую и нисходящую ветви Х-кривой (теплоемкость возрастает
от 84,2 до 196 Дж-К^-моль в интервале 700—990 К и снижается от 198 до 107,5 Дж-К^-моль
в интервале 990—1147 К). Ввиду того что вблизи максимума теплоемкости эти уравнения
описывают ход теплоемкости со значительной погрешностью, для согласования соответствующих
уравнений с экспериментальными данными, полученными в работах [127] и [1002], эффективное
значение энтальпии перехода, отнесенное к 990 К, принято равным 1,0 кДж-моль.1
Температура плавления A147 +2 К) найдена Ефремовой и Матизеном [127] на образце с
содержанием основного вещества не менее 99,9%. С этим значением в пределах указанной погрешности
согласуются данные [37, 537, 1002, 2127, 2421]. В работах [53, 1030, 1031, 1954, 2236] получены
более низкие значения (в пределах 1139—1144 К), по-видимому, из-за наличия примесей в
исследованных образцах. Уравнение для теплоемкости жидкого SrCl2 (см. табл. 28.1) получено
совместной обработкой данных Ефремовой и Матизена [127] A147—1797 К) и Дворкина, Бредига [1002]
A147—1204 К). Согласно этому уравнению С°р (SrCl2, ж) с ростом температуры увеличивается
от 96,0 A147 К) до —,102 Дж -К -моль A800 К); погрешности этих данных оцениваются в 1—2%.
Энтальпия плавления A7,5+0,6 кДж-моль) рассчитана из данных для энтальпии |3-SrCl2 и
SrCl2 (ж) в точке плавления.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1500 и 3000 К оцениваются в 0,3, 1
и 6 Дж-К~1-моль~1. Расхождения между термодинамическими функциями SrCl2 (к, ж),
приведенными в таблицах J AN AF [1544] и в табл. 831, не превышают 2 Дж-К-моль в значениях Ф°(Т)
до 3000 К и вызваны только различной аппроксимацией данных по теплоемкости в [1544] и в
настоящем справочнике.
В справочнике принята энтальпия образования
к, 298,15 К) = —833,2 + 3,0 кДж • моль.
Эта величина основана на данных, приведенных в табл. 28.13. При вычислении Д^0 (8гС12,к)
по результатам измерений энтальпии растворения SrCl2 (к) в воде использованы энтальпии
образования ионов, приведенные в Приложении 5. Этот путь, по-видимому, привел к наиболее точному
из приведенных в табл. 28.13 значений, которое также близко к результатам измерений [1022].
Таблица 28.13. Результаты определений А^Н° (SrCl2, к, 298,15 К) (в кДж-моль)
Авторы
Метод
fi° (SrC]2 к, 298,15 К)
Спмонсеи, 1951 [2520] Измерение энтальпии сгорания Sr в С12 —879
Эрлих и др., 1963 [1022] Измерение энтальпии растворения Sr и SrC]2 в со- —830+5
ляной кислоте
а Измерение энтальпии растворения SrO и SrCl, в со- —826+8
ляной кислоте
6 Измерение энтальпии растворения SrCl2 в воде до со- —834,2+2,0
стояния бесконечно разбавленного раствора
ДгЯ°B98,15)=50,4+0,3 кДж-моль-1
Наиболее точные измерения энтальпии растворения SrO проведены Монаенковой и Воробьевым [261].
Энтальпии разбавления приняты по справочнику [406[. Измерения в работах [107, 754, 1116] менее надежны.
6 Измерения проведены Грейсоном, Снеллом [1262], Дэдгером, Тахерианом [919], Мишенко, Стагиеом [259] и
Самойловым [364]. Значения энтальпии разведения приняты по работе Ланге, Штрека [1810].
Цикл, включающий энтальпии растворения SrO (к) и SrCl2 (к) в соляной кислоте, дает заметно
отличающуюся величину 2, что свидетельствует о несогласованности имеющихся термохимических
1 Энтальпия фазового перехода II рода, определяемая как интеграл аномальной части теплоемкости SrCl2,
составляет около 7 кДж-моль.
г Погрешности величин, вычисленных по этому циклу, увеличены, поскольку отсутствуют данные по энтальпии
разведения солянокислых растворов SrCl2, в которых возможно комплексообразование.
366
Стронций и его соединения
SrCb
данных для Sr+2 (ag), SrCl2 (к) и SrO (к). Принятое в справочнике значение является
средневзвешенным из приведенных в табл. 28.13 величин (за исключением работы [2520]).
Давление пара дихлорида стронция SrCl2 (к, jK)=SrCl2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
А,Н° (SrCl2, к, 0) = 348+6 кДж ¦ моль.
Это значение основано на результатах измерений давления пара дихлорида стронция,
приведенных в табл. 28.14. В таблице в качестве погрешностей дана воспроизводимость измерений»
Таблица 28.14. Результаты определений ASH° (SrCb, к, 0) (в кДж-моль)
Авторы
Новиков, Гаврючснков, 1964 [291]
Фрид и др., 1964 [419]
Ван-Уэстенбург, 1964 [2756]
Леман и др., 1965 [1882]
Хилденбранд и др., 1965 [1422]
Нарышкин и др., 1968 [272]
Новиков и др., 1970 [292]
Эмонс и др., 1976 [1030а]
Метод
Точек кипения, 1620—1710 К, 3 точки
То же, 1323 и 1423 К, 2 точки
Эффузионный, 1175—1287 К
То же, 1195—1307 К, 19 точек
Торзионный, 1220—1425 К
Эффузионный, 893—1141 К, расчет по
уравнению
То же, 1337—1422 К, 5 точек
То же, 1153—1233 К, расчет по уравнению
Д,Я° (SrGla. к, 0)
II закон
318
346
340±2
351 ±40
352
319
315
341 ±15
III закон
357,8+4,0
352,7 ±3,0
346,1 ±0,8
352,0±0,9
348,0±0,3
326,4±1,0
344,4±0,9
377,5±1,6
Принятое значение вычислено как средневзвешенное из приведенных в таблице величин, кроме
найденных в работах [272, 1030а]. При определении веса каждой работы учитывались
воспроизводимость измерений и отклонение результата от среднего. Наибольший вес был придан работе
Хилденбранда [1422]. При оценке погрешности рекомендованного значения учтена неточность
термодинамических функций E кДж-моль).
SrCl2 (г). Термодинамические свойства газообразного дихлорида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 832.
Молекулярные постоянные, использованные для расчета термодинамических функций SrCl2,
приведены в табл. 28.7. Исследования инфракрасного спектра [24, 1363, 2842] и отклонения
молекулярного пучка в неоднородном электрическом поле [783, 2839] показали, что в основном
электронном состоянии молекула SrCl2 имеет нелинейное строение (симметрия C2s). Приведенное
в табл. 28.7 произведение моментов инерции вычислено при -4 С1—Sr—С1=125 +10° (среднее иа
рассчитанных Уайтом и др. [2842] A18+5°) иоХасти и др. [1363] A30+8°) из изотопных
расщеплений полос vx и vs) и г (Sr—С1)=2,67 +0,03 А (найдено Акишиным и др. [5, 14] при электроно-
графическом исследовании паров SrCl2). Погрешность величины IaIbIc составляет 2-Ю13 г3-см6..
Приведенное в табл. 28.7 значение v3 получено Байковым [24] в инфракрасном спектре
газообразного SrCl2. Оно хорошо согласуется с частотами, найденными Хасти и др. [1363] и Уайтоад
и др. [2842] в ИК-спектре молекул SrCl2, изолированных в матрицах (соответственно 308 см*
в матрице из Аг и 299,5 см в матрице из Кг). Частота vx принята по ИК-спектру SrCl2 в матрице
из Кг [1363, 2842], поскольку v3 в ИК-спектре пара [24] и в матрице из Кг [1363, 2842] почти
одинаковы. Для частоты v2 принято значение, среднее между измеренным Уайтом и др. [2842]
в матрице D4 см) и оцененным Хасти и др. [1363] для газовой фазы F0+15 см). Погрешности
принятых значений частот оцениваются в 10—15 см. Сведения о возбужденных электронных
состояниях SrCl2 отсутствуют; можно предполагать, что их энергии превышают 20 000 см.
Термодинамические функции SrCl2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор»; их погрешности обусловлены неточностью принятых значений молекулярных постоянных,
а также приближенным характером расчета. Они составляют 2, 5 и 7 Дж-К~1-моль в значениях,
Ф° (Т) при 298,15, 3000 и 6000 К соответственно (в том числе 2—3 Дж-К~1-моль~1 из-за неточности,
молекулярных постоянных) (см. текст по СаС1а (г)).
367
SrBr (г) Глава 28
Ранее термодинамические функции SrCl2 (г) рассчитывались в таблицах JANAF [1544],
а также в работах Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Г < 2500 К).
Расхождения с данными [1544] не превышают 1,4 Дж-К-моль в значениях Ф° (Г) и
объясняются небольшими отличиями в принятых величинах *$ С1—Sr—С1 и v3. В работах [195, 748] для
молекулы SrCl2 принята линейная модель и расхождения с данными настоящего издания достигают
18—22 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции SrCl2 (r)=Sr (г)+2С1 (г) вычислена с использованием значения
Д^йГ0 @) =885,793 +7,0 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дихлорида стронция. Этим величинам также соответствует
AfH°(STC}2, г, 0) = —485,680+ 6,7 кДж • моль.
SrBr (г). Термодинамические свойства газообразного бромида стронция в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 833.
Молекулярные постоянные SrBr, принятые в справочнике по результатам исследований
электронных спектров этой молекулы, приведены в табл. 28.9. В спектре SrBr наблюдались девять
систем полос, связанных с переходами между девятью состояниями: А2П *^ X2 Е+ [1354, 1378, 2159,
2795], 522+<- Х22+ [1354], С2П «* X2 2+ [1354, 1378, 1600, 2159, 2795], Я2 2+«- X2 2+ [1354],
Ег 2+ -нк X2 2+ [2317], а также ЯП -^ В2 2+, ЯП-*Л2П, б2Д-»ЛаП и Я2 Е+ -> А2П [2311].
Как и у других молекул галогенидов щелочноземельных металлов, состояния X2 Е+ и Л2П
молекулы SrBr коррелируют с основными состояниями атомов SrA5)+Br BP). Со следующим дис-
социационным пределом Sr CР)+Вг BР) должно коррелировать большое число состояний, в том
числе квартетных; по крайней мере часть из этих ненаблюдавшихся состояний может быть
связанными состояними. Значительная часть наблюдавшихся в работе [2317] высоких возбужденных
состояний, по-видимому, является ридберговскими состояниями, хотя они и могут коррелировать
с валентными состояниями атомов (см. текст по CaF).
Колебательные постоянные SrBr в состоянии X2 S+ определялись из анализа систем полос
A2U—X2 Е+ [1354, 1378], В2 S+—X2 S+ [1354], С2П-Х2 Е+[1354, 1600], D2 2+-Х2 Е+ [1354],
Е2 Е+—X2 Е+ [2317]; результаты этих работ хорошо согласуются между собой. Приведенные
в табл. 28.9 колебательные постоянные в состоянии X3 Е+ получены Джоши и Гопала [1600] по
кантам 129 полос системы С2П—X2 2+ (v" ^ 17); они практически совпадают с найденными Хар-
рингтоном [1354]. Вращательная структура полос SrBr не анализировалась; приведенная
в табл. 28.9 постоянная Ве рассчитана по оцененному в справочнике значению re (SrBr)=2,80 +
+0,05 А. Оценка сделана на основании величины г (Sr—Br) в молекуле SrBr2 и сопоставления
значений т0 (MX) и г (М—X) в других соединениях этой группы. Колебательные постоянные в
состояниях А2Л, В2 Е+, D2 Е+ приняты по данным Харрингтона [1354], в состоянии С2П — по работе
Джоша и Гопала [1600], в состояниях Е2 S\ F2U, 02Д иЯ2 2+ — по работам Редди иРао [2317] и
Редди и др. [2311].
Термодинамические функции SrBr (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBK и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с|учетом девяти возбужденных состояний в предположении, что (?'*]. в —(Pilpx)Qi$ в • Величина
Qw вр и ее производные вычислялись по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с
помощью уравнений типа A.82). В расчетах учитывались все уровни энергии со значениями/^/msXo,;
где /шах< „ находились из условий A. 81). Колебательно-вращательные уровни состояния Х2Е +
вычислялись по уравнениям A. 65), A. 61). Значения коэффициентов Yk! в этих уравнениях,
а также величин ушаз. и /1Ш, использованных в расчетах, приведены в табл. 28.10. Коэффициент У03
получен с помощью условий A. 59). Мультиплетность и вырождение колебательно-вращательных
уровней электронных состояний учитывались статистическими весами. Различие молекулярных
постоянных изотопических модификаций SrBr в расчете не учитывалось.
Погрешности вычисленных термодинамических функций при Т <^ 2000 К связаны в основном
с отсутствием экспериментальных данных о вращательных постоянных SrBr, а при более высоких
температурах — с неполнотой сведений о связанных электронных состояниях этой молекулы.
Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К могут достигать 0,3, 0,5 и 1 Дж-К-1Х
Хмоль соответственно.
368
Стронций и его соединения SrBra (к, ж)
Ранее термодинамические функции SrBr (г) вычислялись в справочнике JANAF [1544] и работе
Климова и др. [167] (приведены коэффициенты полинома, аппроксимирующего Ф° (Т) при
Т ^ 5000 К). Расхождения данных [1544] с табл. 833 заметны во всем диапазоне температур и
составляют от 0,5 до 1 Дж-К~1-моль в значениях Ф° (Т) и до 2 Дж-К-моль в значениях
S° (T). При низких и средних температурах они обусловлены различием принятых
вращательных постоянных, при высоких температурах к этому добавляется различие методик расчета.
Расхождения с данными [167] большие, так как в этой работе использовались грубая методика расчета
и отличающиеся значения постоянных SrBr.
Константа равновесия реакции SrBr (r)=Sr (г)+Вг (г) вычислена с использованием принятой
в справочнике энергии диссоциации
\Н° @) = Do (SrBr) = 335 + 10 кДж . моль да 28 000 + 800 см.
Это значение основано на результатах масс-спектрометрического исследования равновесия
реакции Sr (г)+А1Вг (r)=SrBr (г)+А1 (г) в работе Хилденбранда [1411] A819—2005 К, 7 точек,
расчет по II и III законам дает 360 +26 и 342 +10 кДж-моль) и спектрофотометрического
исследования равновесия реакции Sr (г)+НВг (r)=SrBr (г)+Н (г) в пламенах в работе Гурвича и др. [1333]
A745—2320 К, 13 точек, расчет по II закону из данных по интенсивности полос SrBr и линий Sr
дает 328+20 кДж-моль). Приведенные погрешности включают воспроизводимость измерений,-
а также неточность сечений ионизации и термодинамических величин, использованных в расчетах.
Исследование Менцигером [2007] порога хемилюминесценции SrBr при образовании этих
молекул в реакциях в пучках привело к существенно более высокому значению (^ 391 кДж X
Хмоль), по-видимому, из-за систематической ошибки в измерениях.
Принятой величине соответствует
A^°(SrBr, г, 0) = —56,204 + 10 кДж • моль.
SrBr2 (к, ж). Термодинамические свойства кристаллического и жидкого дибромида стронция
в стандартном состоянии при температурах 100—3900 К приведены в табл. 834.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. В справочнике за стандартное состояние SrBr2 (к) в интервале 0—918 К принята
тетрагональная модификация a-SrBr2 [2542], а в узком интервале 918—930 К — ^-модификация с
неисследованной структурой. Исходные данные для расчета термодинамических функций SrBr2 (к)
при Т < 298,15 К заимствованы из таблиц JANAF [1544]. В интервале 60—245 К принятые
в [1544] значения теплоемкости SrBr2 (к) основаны на измерениях Тейлора и Смита [26653
F0—302 К). При температурах 245—298,15 К данные [2665] существенно исправлены (увеличены,
при 298,15 К — на 2,5%) с учетом измерений энтальпии SrBr2 (к) в работе Хюттига и Сло-
нима [1525] (85—368 К). При Т < 60 К авторы [1544] провели экстраполяцию теплоемкости,
существенно отличную от выполненной Тейлором и Смитом [2665]; при этом они использовали
известные экспериментальные данные для теплоемкости SrCl2 G—60 К), ВаС12 F—60 К)?и Са18
A3-60 К). Погрешности принятых значений S° B98,15 К) и Я° B98,15 К)-#° @) (см. табл. 28.1)
оцениваются в 4 Дж-К-моль и 0,5 кДж-моль соответственно. В работе [2665] рекомендовано
значение S° B98,15 К), которое на 8,3 Дж-К-моль~1 меньше приведенного в табл. 28.1.
Уравнение для теплоемкости a-SrBr2 при Т > 298,15 К выведено с учетом значения Н° (918 К)—
—Н° B98,15 К) =51,7 кДж-моль, найденного в работе Дворкина и Бредига [1003]. Данные
Тейлора и Смита [2665] по энтальпии SrBr2 (к) имеют большой разброс (—3-^-4%) и не учитывались.
Температуры полиморфного превращения (918+3 К) и плавления (930+2 К) приняты по
хорошо согласующимся между собой данным Дворкина и Бредига [1003], Эмонса и Лефель-
хольца [1031] и Риккарди и др. [2329]. Энтальпия превращения A2,2+0,2 кДж-моль) и
энтальпия плавления A0,1 +0,2 кДж-моль) приняты по работе [1003], близкое значение для энтальпии
плавления A0,9+0,5 кДж-моль) получено криоскопическим методом авторами [1031]. Тейлор
и Смит [2665] нашли, что сумма значений &trH и kmH равна 22 кДж-моль в удовлетворительном
согласии с принятыми величинами. Энтальпия SrBr2 (ж) измерялась в узких интервалах
температур (927—1118 К) и (931—1002 К) в работах [2665] и [1003]; эти данные приводят к неточным и,;
по-видимому, сильно завышенным значениям теплоемкости A19 и 116 Дж-К~1-моль~1
соответственно). В справочнике принимается значение Ср (SrBr2, ж)=100 +10 Дж-К~1-моль1 оцененное
369
SrBr* (г) Глава 28
с учетом достаточно падежных данных по теплоемкости жидких SrCl2 (99 Дж-К~1-моль~1) и ВаВга
A05 Дж-К~1-моль~1). Теплоемкость ($-SrBr2 также оценена равной 100+10 Дж-К~1-моль.
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 4, 6„
12 и 20 Дж -К -моль. Термодинамические функции SrBr2 (к), приведенные в табл. 834 и таблицах
JANAF [1544], практически совпадают. Соответствующие расхождения в значениях Ф° (Т) для
SrBr2 (ж) превышают 4 Дж-К-моль~1 при 3000 К, что вызвано различием принятых значений
С°р (SrBr2, ж).
Принятая в справочнике энтальпия образования
Д/f (SrBr2, к, 298,15 К) = — 717 + 5 кДж • моль
основана на результатах измерений энтальпии реакций растворения Sr (к) и SrBr2 (к) в бромисто-
водородной кислоте, выполненных Эрлихом и др. [1022]. Измерения энтальпии растворения
SrBr2 (к) в воде [1524, 1925, 2659, 2686] приводят к более отрицательным значениям (—725 +
+5 кДж -моль). В справочнике отдано предпочтение измерениям Эрлиха и др. [1022], так как они
представляют термохимический цикл, в который не входят никакие дополнительные величины^
кроме Д^#° (HI, aq). В расчеты из данных по энтальпии растворения SrBr2 (к) в воде входит
значение ДуЯ° (Sr+2, ag) (см. Приложение 5), сведения о котором весьма противоречивы.
Давление пара дибромида стронция SrBr2 (к, ж)=8гВг2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
A,ff°(SrBra, к, 0)==317 + 10 кДж • моль.
Эта величина основана на измерениях давления пара, выполненных Хилденбрандом [14121
(торзионный метод, 1080—1251 К, 19 точек, результаты измерений представлены только в виде
уравнения), Питерсоном, Хатчисоном [2217] (эффузионный метод, 1150—1300 К, 15 точек,
результаты измерений представлены только в виде уравнения) и Штоком, Хейнеманом [2617]
(эффузионный метод, 1040 К, 1 точка). Обработка этих данных по уравнению III закона дает 317,1 +
+0,6, 316,9+1,5 и 298 кДж-моль соответственно. Принятое значение основано на совпадающих
результатах работ [1412] и [2217]. Работа [2617] выполнена в 1909 г. и, по-видимому, содержит
систематическую ошибку. Погрешность принятой величины определяется главным образом
неточностью термодинамических функций SrBr2 (к, ж) и SrBr2 (г).
SxBr2 (г). Термодинамические свойства газообразного дибромида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 835.
Молекулярные постоянные, использованные для расчета термодинамических функций SrBr2,
приведены в табл. 28.7. Исследования дифракции электронов [5, 14] и отклонения молекулярного
пучка в неоднородном электрическом поле [2839] показывают, что молекула SrBr2 линейна в
основном электронном состоянии. Приведенный в табл. 28.7 момент инерции соответствует значению
г (Sr—Вг)=2,82+0,03 А, найденному Акишиным и др. [5, 14] при электронографическом
исследовании паров SrBr2. Его погрешность оценивается в 1-Ю8 г-см2. Спектр SrBr2 не исследован.
Принятые значения частот рассчитаны по формулам валентного силового поля. Силовая
постоянная /г=0,89-105 дин-см принята по оценке Байкова [25], выполненной по установленной им
корреляции между величиной /г и длиной связи в молекулах дигалогенидов элементов подгруппы
ПА. Силовая постоянная frr вычислена в предположении, что в SrBr2, так же как в дигалогенидах
магния, выполняется соотношение /г//„.=15. Силовая постоянная fa вычислена в предположении,
что /г//я=80, так как в SrCl2 и Srl2 это отношение равно 72 и 86. Погрешности принятых частот
оцениваются в 15—30 см (см. текст по СаС12 (г)). Сведения о возбужденных электронных
состояниях SrBr2 отсутствуют, по-видимому, их энергии не превышают 20 000 см.
Термодинамические функции SrBr2 (г) вычислены по уравнениям A. 3)—A.6), A.9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор»; их погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 6, 8 и 10 Дж-К^-моль в значениях Ф° G")
при 298,15, 3000 и 6000 К соответственно (в том числе 5—6 Дж-К-моль~1 из-за неточности молекуг
лярных постоянных) (см. текст но СаС12 (г)).
Ранее термодинамические функции SrBr2 (г) вычислялись в таблицах JANAF [1544], в работах
Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Расхождения между
370
Стронций и его соединения Sri (г)
термодинамическими функциями, приведенными в этих работах и настоящем справочнике,
достигают 8, 3 и 3 Дж-К-моль в значениях Ф° (Т) для [195, 748, 1544] и объясняются различиями
в оценках частот этой молекулы.
Константа равновесия реакции SrBr2 (r)=Sr (r)+2Br (г) вычислена с использованием значения
АГН° @)=783,689+11 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации дибромида стронция. Этим величинам также соответствует
г, 0) = —386,970 +11 кДж • моль.
Sri (г). Термодинамические свойства газообразного иодида стронция в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 836.
Молекулярные постоянные Sri, принятые в справочнике на основании анализа результатов
исследований электронных спектров этой молекулы и приближенных оценок, представлены
в табл. 28.9. В спектре Sri наблюдалось пять систем полос, связанных с переходами между шестью
электронными состояниями: А2П ^ Х22 + [541, 2159, 2315, 2516, 2795],.Б2Е + <> Х2? + [541, 2159,
2315, 2795], С2П«* Х2? + [2011, 2159, 2314, 2795], D2Z+ ** Х2? + [541, 2501, 2795], F2IL =&
^±52?+[1623]. Как и у других молекул галогенидов щелочноземельных металлов, состояния Х2? +
ш А2И молекулы Sri коррелируют с основными состояниями атомов Sr (г5)+1 BP). Co следующим
диссоциационным пределом Sr (sP)+l BР) должны коррелировать несколько состояний, в том
числе не наблюдавшиеся экспериментально дублетные и квартетные состояния; часть из них,
по-видимому, может быть связанными состояниями (см. текст по GaF).
Колебательные постоянные Sri в состоянии X2 Е+ определялись из анализа систем полос
Л2П-Х2? + [541, 2315, 2516], ?2S+-X2? + [541, 2315], С2П-Х2? + [2011,2314], 1>2?+-Х2? + [541,
2501); найденные в этих работах значения хорошо согласуются между собой. Приведенные
в табл. 28.9 постоянные найдены по кантам полос Ашрафуниза и др. [541 ] из анализа
колебательной структуры системы A2H>k -> Х2? + (v" ^ 17) и системы Б2?+^.Х2?+ (г/'<;32). Вращательная
структура спектральных переходов Sri не анализировалась. Приведенное в табл. 28.9 значение
r (sr—1)==3,00+0,05 А оценено по величине г (Sr—I) в молекуле Srl2 (см. ниже). Использование
других методов оценки приводит к близким значениям. Колебательные постоянные в состояниях
А2Л и ?2? + приняты по работе [541], в состояниях С2П, Z>2? + и F2U — по работам [2314, 2501„
1623] соответственно.
Термодинамические функции Sri (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом шести возбужденных состояний в предположении, что Q^. „p = (P,-//>x)<?S. вр. Величина
QW в и ее производные вычислялись по уравнениям A.73)—• A.75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням энергии с помощью
уравнений типа A. 82). В расчетах учитывались все уровни энергии со значениями / <I Jmu%,v,
где /ш31 „ находились из условий A. 81). Колебательно-вращательные уровни состояния Х2?+
вычислялись по уравнениям A. 65), A. 61). Значения коэффициентов Ykl, а также величин vmia
и /Iim, использованных в расчетах, приведены в табл. 28.10. Коэффициент Y03 получен из условия
A. 59). Мультиплетность и вырождение колебательно-вращательных уровней электронных
состояний учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при Т -< 2000 К обусловлены
отсутствием экспериментальных данных о вращательных постоянных Sri, а при более высоких
температурах — неполнотой информации о возбужденных состояниях этой молекулы. Погрешности в
значениях Ф° (Г) при 298,15, 3000 и 6000 К могут достигать 0,4, 0,8 и 2 Дж- К- моль соответственно.
Ранее термодинамические функции Sri (г) вычислялись в таблицах JANAF [1544] и работе
Климова и др. [167] (приведен полином, аппроксимирующий Ф° (Т) для Т <] 5000 К).
Расхождения данных [1544] и настоящего издания обусловлены использованием разных постоянных Sri
в состоянии Х2Е+, а также различием методик расчета; они составляют 0,5 и 1,5 Дж-К~1-моль"г
в значениях Ф° (Т) и S° (T). Расчет [167] сделан по упрощенной методике, и его результаты
отличаются от приведенных в табл. 836 на 0,6—1,6 Дж-К-моль~1.
Константа равновесия реакции Sri (r)=Sr (r)+I (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ° @) = Do (Sri) = 271 + 8 кДж • моль« 22 650 + 650 см.
371
Srl4 (к, ж), Srl2 (г) Глава 28
Это значение основано на масс-спектрометрическом исследовании равновесия реакций Sr (г) +
+SrI2 (r)=2SrI (г) A626-1787 К, 8 точек) [Шбб^иЭг (г)+АП (r)=SrI (г)+А1 (г) A626-1810 К,
13 точек), выполненном |Клейшпмидтом и Хилденбрандом [1413, 17166]. По результатам этих
исследований вычислены значения энергий диссоциации 264 + 50 и 267+15 кДж-моль по
уравнению II закона и 266+8 и 276+8 кДж-моль по уравнению III закона. Принятое значение
подтверждается исследованием хемилюминесценции молекул Sri, возникающих в реакции Sr (г)+
+HI (r)=SrI (г)+Н (г) в скрещенных молекулярных пучках [2032]; эти данные приводят
к значению Do (Sri) ^ 272+8 кДж-моль".
Принятой энергии диссоциации соответствует
bfH°(STl, г, 0) = —2,964 + 8,2 кДж • моль.
Srl2 (к, ж). Термодинамические свойства кристаллического и жидкого дииодида стронция
в стандартном состоянии при температурах 100—5300 К приведены в табл. 837.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. В справочнике за стандартное состояние Srl2 (к) принимается ромбическая
модификация, устойчивая в интервале температур от 0 до 811 К. При Т <^ 298,15 К термодинамические
функции вычислены по результатам измерений теплоемкости, проведенных Пауковым и др. [313]
A3—301 К) на образце, содержавшем ~1% Ва и 0,03% других примесей. Погрешности
данных [313] оцениваются в 1% при Т <^ 30 К и ~ 0,3% при Т > 50 К. Экстраполяция теплоемкости
к Г=0 дает S° A3 К)=2,4 Дж- К^-моль'1. Погрешности принятых по [313]]значений S° B98,15 К)
и Н° B98,15 К)—Я° @) оцениваются в 0,8 Дж-К~1-моль~1 и 0,08 кДж-моль соответственно.
Уравнение для теплоемкости Srl2 (к) при Т j> 298,15 К выведено по данным, приведенным в
таблицах JANAF [1544]; последние основаны на найденном в работе Дворкина и Бредига [1003]
значении Я0 (811 К)—Я0 B98,15 К) =43,5 кДж-моль. Температура плавления (811+10 К)
принята по данным [1003]; измерения, выполненные в работах [1031] (802 К) и [1520] (973 К),
по-видимому, менее точны и не учитывались. Принятые значения энтальпии плавления A9,7 +
+0,4 кДж-моль) и теплоемкости жидкого Srl2 A10+6 Дж-К~1-моль~1) получены Дворкиным
и Бредигом в результате измерений энтальпии в узком интервале температур. Погрешности
вычисленных значений Ф" (Т) при 298,15, 1000, 2000, 3000 и 5000 К оцениваются в 0,6, 1,5, 6, 13 и
20 Дж-К-моль~1 соответственно. Термодинамические функции Srl2 (к, ж), приведенные в
справочнике JANAF [1544] и табл. 837, практически совпадают.
В справочнике принята энтальпия образования
&fE° (Srl2) к,?298,15 К) — —562 + 10 кДж • моль,
вычисленная по данным Эрлиха и др. [1022]. В этой работе измерены энтальпии растворения
металлического стронция и Srl2 (к) в растворах иодистоводородной кислоты. Значительная погрешность,
приписанная результатам этих измерений, связана с тем, что измерения проводились в
негерметичном калориметре и возможна побочная реакция выделяющегося водорода с медной сеткой,
в которую был завернут стронций (см. ВаН2 (к, ж)).
Давление пара дииодида стронция Srl2 (к, ж)=Эг12 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
A,#°(SrI2, к, 0) = 292 + 7 кДж • моль.
Это значение получено в результате обработки эффузионных измерений Питерсона и Хатчи-
сона [2217] A110—1310 К, 19 точек) и Хилденбранда [1413] (958—1003 К, 9 точек). Результаты
обеих работ представлены только в виде уравнений. Проведенные расчеты привели к величинам
290 и 281 кДж-моль (II закон) и 291,4+0,1 и 292,2+0,3 кДж-моль (III закон) соответственно.
При оценке погрешности принятой величины учтена ошибка из-за неточности термодинамических
функций Srl2 (к, ж) и Srla (г), составляющая около 7 кДж-моль".
Srl2 (г). Термодинамические свойства газообразного дииодида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 838.
Молекулярные постоянные, использованные для расчета термодинамических функций Srl2,
приведены в табл. 28.7. Исследования дифракции электронов [5, 14, 1048] и отклонения
молекулярного пучка в неоднородном электрическом поле [2839] показали, что в основном электронном
372
Стронций и его соединения SrS (к, ж)
состоянии молекула Srl2 линейна (симметрия D ГОЛ). Момент инерции, приведенный в таблице,
вычислен для значения г (Sr—1)=3,03+0,03 А, найденного Акишиным и др. [5, 14] в электроногра-
фическом исследовании паров Srl2. Близкое значение C,01 +0,01 А) получено позднее Каспаровым
и др. [1586, 158г, 1046]. Погрешность принятого момента инерции оценивается в 2-108 г-см2.
Спектр молекулы Srl2 не исследован. Принятые значения v1 и v3 рассчитаны по формулам
поля валентных сил. Для постоянной fr использовано значение 0,74-105 дин-см,
предложенное Байковым [25] на основании сопоставления величин /г и г в молекулах дигало-
генидов элементов подгруппы ПА.Постоянная /„. вычислена в предположении, что для
молекулы Srl2 так же, как для дигалогенидов магния, справедливо соотношение /,.//,.,.=15. Частота v2
получена Каспаровым и др. [1586, 158г, 1046] из обнаруженного ими при электронографическом
исследовании паров Srl2 эффекта сокращения межатомного расстояния I—I. Погрешности
принятых значений частот оцениваются в 10—20 см. Сведения о возбужденных электронных состояниях
молекулы Srl2 отсутствуют, можно предполагать, что их энергии не превышают 20 000 см.
Термодинамические функции Srl2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический осциллятор»
без учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 7, 9 и 10 Дж-К~1Х
Хмоль в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно (см. примеч. на с. 329).
Ранее термодинамические функции Srl2 (г) вычислялись в таблицах JANAF [1544], в работах
Брюэра и др. [748] {Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Данные [748, 1544]
и табл.838 согласуются в пределах 2 Дж-К-моль~1 в значениях Ф° (Т), расхождения с
работой [195] достигают 9 Дж-К^-моль из-за неточной оценки частот ее авторами.
Константа равновесия реакции Srl2 (r)=Sr (г)+21 (г) вычислена с использованием значения
A^ff0 @)=644,353 +12 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации Srl2 (к). Этим величинам соответствует также
AfH°(STl2, г, 0) = —269,154 + 12 кДж ¦ моль.
SrS (к, ж). Термодинамические функции кристаллического и жидкого сульфида стронция
в стандартном состоянии при температурах 100—5200 К приведены в табл. 839.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 28.1. В справочнике за стандартное состояние SrS (к) принята кубическая модификация
(структурный тип NaCl) [1459а]. При Т < 298,15 К термодинамические функции вычислены по
результатам измерений теплоемкости в работе Кинга и Уиллера [1713а] E1 — 297 К) на образце
состава 99,16% SrS, 0,83% SrSO4 и 0,01% Si, погрешности этих данных составляют —0,5%.
Экстраполяция ниже 51 К по уравнению Cp=D B08/Г)+1? C11/Г) привела к значению 5° E1 К) =
=6,7 Дж-К^-моль [1713а]. Погрешности принятых по [1713а] значений 5° B98,15 К) и
Н° B98,15 К) — #° @) (см. табл. 28.1) оцениваются в 0,8 Дж-К^-моль и 0,06 кДж-моль'1
соответственно.
В литературе отсутствуют данные о теплоемкости или изменении энтальпии SrS (к, ж) при
Т > 298,15 К. В интервале 298,15 — 2500 К для теплоемкости SrS (к) принято уравнение (см.
табл. 28.1), аппроксимирующее значения С;B98,15 К), С;E00К)=51 Дж-К^-моль^и С;B000К)=
= 63 Дж- К"-моль. Два последних значения оценены, как и в случае CaS (к), с учетом данных
по теплоемкости окислов щелочноземельных металлов. Погрешности оцененных значений
теплоемкости SrS (к) составляют —10% при 500 К и ~15% при 2000 К. Данные по температуре
плавления сульфида стронция в литературе отсутствуют, за исключением указаний, что Тm (SrS) ^>
!> 2300 К (см. [365а, 2031]). В справочнике принимается оцененное значение B500+200 К), при
выборе которого учитывались данные по температурам плавления SrO, CaO и CaS. Энтальпия
плавления F3+15 кДж-моль) оценена по приближенному эмпирическому соотношению, как
и в случае CaS.
Теплоемкость SrS (ж) F7+15 Дж-К~1-моль) также оценена по приближенному соотношению.
Погрешности вычисленных значений Фс (Т) при 298,15, 1000, 2000, 3000 и 5000< Kf оцениваются
в0,8, 5, 9,12 и 20 Дж- К"-моль соответственно. Термодинамические функции SrS (к), вычисленные
в справочнике [2031], отличаются от приводимых в табл. 839 на 6 Дж-К-моль в значении
S° B000 К), что вызвано разной оценкой теплоемкости SrS (к) при Т ]> 298,15 К.
373
SrS (г)
Глава 28
В справочнике принята энтальпия образования
bfH°(SrS, к, 298,15К) = — 480 + 10 кДж • моль-1,
основанная на результатах измерений, представленных в табл. 28.15. Приведенные в таблице
погрешности включают воспроизводимость измерений и неточность использованных в расчетах
Таблица 28.15. Результаты определений Д^?Г° (SrS, к, 298,15 К) (в кДж-моль)
Авторы
Метод
Ayff^SrS, к, 298,15 К)
II закон
III закон
Сабатье, 1881 [2417] Калориметрический, SrS (к)+5,0НС1 (р-р, —480+10
221H,O)=SrCL (р-р, 3,0НС1+Ш5НаО)+3,0НС1
(р-р, 368Н2О), при 10,5° С, 4 измерения
Мурло, 1899 [2067а] То же, детали измерений не известны —479±10
Шенк, Хаммершмидт, 1933 [2451а] Исследование равновесия реакции SrS (к)+ —501+100 —479+15
+2SO2 (r)==SrS04 (k)+S3 (г) методом протока,
1237—1383 К, 3 точки
Берковиц, Маркварт. 1963 [665а] Исследование равновесия реакции SrS (к) ~Sr (г)+ — —460±15
+1/2S.1 (г) масе-спектрометрическим методом,
1809 К, 1 точка
Тоже, SrS (K)=Sr(r)+S(r), 1809 К, 1 точка - —455±15
Колен и др., 1964 [S73] То же, 2/3Sr (r)+V3S2 (r)=2/3SrS (k)+2S (г) масс- —400±100 —502±20
спектрометрическим методом, 1745—2170 ;К,
11 точек
Катер, Джонсон, 1967 [827а] Тоже, SrS (к)=вг (r)+S (г) масс-спектрометри- —477 —445+20
ческим и эффузиошшм методами, 1900—2100 К,
2 точки
термохимических величин, сечений ионизации и термодинамических функций G и 16 кДж-моль *
для SrS (к) при измерениях при 1300 и 2000 К соответственно и 15 кДж- моль для SrSO4 (к) — при
1300 К).
Величина, вычисленная по данным работы Сабатье [2417 ], включает ряд неточностей, связанных
с недостаточно надежной методикой, отсутствием данных о проведении измерений и тем, что
измерения относятся к 10,5° Си в литературе нет сведений, позволяющих провести пересчет к
стандартной температуре. Тем не менее эта работа учтена при выборе рекомендованной величины, так как
полученные в ней -данные для MgS (к) и CaS (к) оказались в хорошем соответствии с другими
калориметрическими измерениями и исследованиями равновесия реакций (см. соответствующие тексты).
Это является косвенным подтверждением отсутствия в работе [2417] грубых ошибок. Детали
измерений в работе Мурло [2067а] неизвестны; вычисленная по этой работе величина совпадает с
найденной по измерениям, выполненным Сабатье [2417]. В работе [873] не приведены измеренные
константы равновесия. В таблице даны величины, вычисленные по константам равновесия,
найденным на основании приведенных в [873] ионных токов и принятых в справочнике сечений
ионизации и распространенностей изотопов.
Принятая величина является средневзвешенной из данных [665а, 827а, 873, 2067а, 2417, 2451а],
результаты которых совпадают в пределах их погрешностей.
Давление пара сульфида стронция SrS (к, ж)=ЗгЗ (г) вычислено с использованием значения
/° @) = Д,#° (SrS, к, 0) =584,716+18 кДж-моль~\ соответствующего принятым в справочнике
энтальпиям образования SrS (к) и SrS (г).
SrS (г). Термодинамические свойства газообразного сульфида стронция в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 840.
В табл. 28.4 приведены молекулярные постоянные SrS, принятые на основании анализа
результатов исследования электронного спектра [1942а] этой молекулы и приближенных оценок. В
электронном спектре SrS исследована одна система полос, связанная с переходом СХЕ+ «— X1S+. По
аналогии с SrO, оксидами других щелочноземельных металлов, а также с молекулой BaS молекула
SrS должна иметь ещэ ряд иизколежащих синглетных и триплетных состояний, соответствующих
374
Стронций и его соединения SrS (г)
Таблица 28.16. Результаты определений D0(SrS) (в кДж-моль)
Авторы
Маркварт, Берковиц,
Колен
Катер,
а См.
1963
и др.. 1964 [873]
[1951а
Джонсон, 1967 [S27a]
текст по Д/А'с
(SrS,
к).
Метод
Масс-сиектрометрический, Sr
+S
То
То
То
То
(г),
же,
же,
же,
же,
1809—1934 К, 2 точки
2010—2170 К, 4 точки
1900—2100 К, 2 точки
SrS (r)=Sr (r)+S (г)
SrS (K)=SrS (г), AtHa
(r)+S2 (r)=SrS (r)+
a
@)=552±17
Do
II закон
277
352
329
333
363
(SrS)
III закон
333+15
337±16
327+15
327 ±15
360±25
конфигурациям . . . о2и3с (а3П, ^441), . . ,оя4а (Ь3Е+, В1?!*), . . . а2тс3т: C> ХД, гЛ~, 3?+) и . . .<згЛп
CП, Ш).
В табл. 28.4 приведены данные для 12 валентных состояний молекулы SrS. Молекулярные
постоянные в состояниях Х1!!* и C1S+ приняты по результатам исследования Маркано и Барроу
[1942а] системы CXS+ — XXS+ (г/ ^ 2, z/<C 2) в спектре поглощения. Энергии возбуждения
остальных электронных состояний оценены по данным для молекул SrO, BaO и BaS с погрешностью
1000 — 3000 см, причем близколежащие состояния объединены в группы.
Термодинамические функции SrS (г) вычислены по уравнениям A.3) — A-6), A.9), A. 10)
и A. 93) —A. 95). Величина <?вн и ее производные рассчитывались по соотношениям A. 90) — A.92)
с~учетом 11 возбужденных электронных состояний в предположении, что ()$ж.вр=(рAрх) (???!. вР*
Статистическая сумма по колебательно-вращательным уровням состояния Х1Е+ и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием
по / с помощью уравнений типа A. 82). В расчете учитывались все уровни со значениями /^ Jm^,e,
последние находились по соотношению A. 81). Энергия колебательно-вращательных уровней
рассчитывалась по уравнениям A. 65) и A. 64а) с коэффициентами Ykl, приведенными в табл. 28.5
вместе со значениями ушах и /Иш. Значения Ykl получены по постоянным из табл. 28.4 с учетом
условий A. 50) и A. 59), для соблюдения которых в уравнение A. 65) введены дополнительные
члены с коэффициентами YSQ, Yi0 и F03. Постоянная YS1 определена из условия Z?emax+1 = 0. Все
постоянные Ykl пересчитаны по соотношениям A. 66) для изотопической модификации SrS,
соответствующей природной смеси изотопов Sr и S.
Погрешности вычисленных термодинамических функций при температурах до 1000 К в
основном обусловлены отсутствием данных о колебательно-вращательных уровнях основного
электронного состояния с v >¦ 2. При более высоких температурах становятся существенными погрешности,
связанные с неточностью оценки энергии ряда возбужденных состояний (главным образом asH
и Ь3?+). Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,05, 0,5 и
2 Дж-К~1-моль~1 соответственно.
Таблицы термодинамических функций SrS (г) ранее не публиковались. В работе Колина и др.
[873] приводятся значения Ф° (Г) при Г=1700, 2000 и 2300 К, отличающиеся от данных табл. 840
на 8 Дне-К"-моль. Это расхождение обусловлено главным образом тем, что в работе [873] за
основное состояние SrS принято состояние 32.
Константа равновесия реакции SrS (r)=Sr (r)+S (г) вычислена с использованием принятого
в справочнике значения
' @) = Do (SrS) = 330 + 15 кДж ¦ моль« 27 600 + 1200 см.
Эта величина основана на результатах исследования равновесия реакций, представленных
в табл. 28.16. Указанные в таблице погрешности включают воспроизводимость измерений,
неточность использованных в расчетах термохимических величин, термодинамических функций и
сечений ионизации. Принятая величина является средней из приведенных данных, за исключением
выпадающей величины, вычисленной по измерениям давления пара SrS (к) в работе [827а].
Линейная экстраполяция уровней состояния Хх^+ приводит к существенно более низкому значению
B40 кДж-моль).
375
SrSO4 (к, ж) Глава 28
Принятой величине соответствует
A/T°(SrS, г, 0I=105,658 + 15 кДж • моль.
SrSO4 (к, ж). Термодинамические свойства кристаллического и жидкого сульфата стронция
в стандартном состоянии при температурах 298,15—4000 К приведены в табл. 841.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 28.1. За стандартное состояние SrSO4 (к) в интервале 0—1430 К принята ромбическая
а-модификация, а в интервале 1430 — 1880 К—высокотемпературная гексагональная [3-модифи-
кация. Экспериментальные данные по теплоемкости и энтальпии SrS04 (к) в литературе
отсутствуют, за исключением единичного измерения С°р C07 К)=24,8 Дж- К-моль, проведенного в 1865 г.
Коппом [1755]. В справочнике для теплоемкости SrSO4 при Т <^ 298,15 К приняты значения,
рассчитанные Сакамото [2424, 2425] на основании анализа различных составляющих теплоемкости
сульфатов кальция, стронция и бария. В расчетах [2424, 2425] использованы данные этих авторов
по теплоемкости CaSO4 и BaS04 при низких температурах, оценены соответствующие параметры
для SrSO4 (температуры Дебая, Эйнштейна и другие параметры) и получено уравнение для
теплоемкости С°р (T)=2D B20/Т)+Е C08/Т)+2/3Е F62/Т)+Е (915/r)+V3? A439/Г)+^ A645/Г)+
+26,36-10 С2рТ. По этому уравнению рассчитаны значения теплоемкости, 5° B98,15 К) и
Н° B98,15 К) — Н° @) (см. табл. 28.1); погрешности двух последних величин оцениваются
в 4 Дж> К- моль и 0,4 кДж- моль соответственно. По тому же уравнению оценена теплоемкость
при высоких температурах и на основании значений С°р B98,15 К), С°р E00 К)=128 Дж-К~1-моль~1
и Ср A000 К)=161 Дж-К-моль выведено уравнение для теплоемкости a-SrSO4 (см. табл. 28.1)
в интервале 298,15—1430 К. Погрешности оцененных величин составляют 5 — 8%.
Температура полиморфного превращения A430+10 К) принята по совпадающим между собой
измерениям Батлера и Сорреля [801 ] A429 +5 К) и Грамана [1244] A425 — 1429 К) с учетом
поправки на переход к шкале МПТШ-68. Данные авторов [19, 439, 1109, 1300, 1959, 2584],
по-видимому, менее надежны, хотя и не противоречат в пределах их|погрешности принятому значению.
Энтальпия превращения A0+3 кДж-моль) выбрана на основании термографических данных
Шаргородского [439 ] так же, как это было сделано для MgSO4 (см. гл. 26). Температура плавления
A880+20 К) принята по единственным известным в литературе измерениям Грамана [1244].
Энтальпия плавления C6+8 кДж-моль") оценена из сопоставления суммы энтропии превращения
и плавления в ряду сульфатов щелочноземельных элементов. Теплоемкость высокотемпературной
модификации A430—1880 К) и SrS04 (ж) оценена по аддитивной схеме (см. гл. 3) равной 170 +
+20 Дж-К^-моль". Погрешности вычисленных значений Ф° G) при 298,15, 1000, 2000 и 4000 К
оцениваются в 4, 10, 20 и 40 Дж-К-моль соответственно. Расхождения между
термодинамическими функциями SrSO4 (к), приведенными в справочнике [578] и табл. 841, составляют до
10 Дж- К- моль в значениях Ф° (Т); это объясняется тем, что в [578] приняты существенно более
низкие значения теплоемкости SrSO4.
Константа равновесия реакции SrSO4 (к)=Бг (r)+S (г)+40 (г) вычислена с использованием
значения ДДГ0 @)=2871,344+3,6 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A/TtSrSO^ к, 298,15 К) = —1458,5 + 3,0 кДж • моль.
Эта величина основана на результатах измерений, приведенных в табл. 28.17. При обработке
экспериментальных данных использованы термодинамические свойства ионов, представленные
в Приложении 5. Растворимость SrSO4 (к) измерялась также в более старых работах [1402, 1460,
1743, 1744, 2869], которые менее точны по сравнению с приведенными в таблице. Однако
вычисленные по ним энтальпии образования отличаются от данных [114,172] не более чем на 1,2 кДж-моль.
Калориметрические исследования реакции осаждения SrSO4 (к) выполнены еще в прошлом веке
в работах [850, 2686]. Рассчитанные по этим данным энтальпии образования SrS04 (к) близки к
результатам измерений [369, 454].
Из таблицы видно, что определения энтальпии образования SrS04 (к) из исследования
растворимости и калориметрическим методом находятся в хорошем соответствии. В справочнике принято
среднее значение, погрешность которого оценена с учетом расхождения результатов различных
работ и их погрешностей.
376
Стронций и его соединения
SrCO3 (к, ж)
Таблица 28.17. Результаты определений h.fH° (SrSO4, к, 298,15 К) (в кДжмоль
Авторы
Галло, 1935 [1164]; Селиванова,
Зубова, 1965 [369]
Колосов, 1965 [172]
Дирссен и др., 1969 [114]; Дзвис,
Коллинз, 1971 [932]
Селиванова, Зубова, 1956 [369]
Шидловскпй, Воскресенский, 1963,
[454]
Метод
Измерение растворимости SrSO4 (K)=Sr+2 (яд)+
+SO4-2H), Ar Я0 B98,15 К)=— 3,3±0,6
(III закон)
То же, АГЯ° B98,15 К)=—1,1±0,6 (III закон)
То же, АГН° B98,15 К)=— 2,7±0,5 (III закон)
Калориметрическое измерение янтальпии
реакции SrCl2 (p-p)+K2SO4 (p-p)=SrSO4 (к)+2КС1 (р-р)
То же, SrCla-6H2O (k)+K2SO4 (p-p)=SrSO4 (к)+-
+2КС1 (р-р)+6Н2О (р-р)
То же, Sr(NO3J (p-p)+Na2SO4 (p-p)=SrSO4 (к)+
+2NaNO3 (р-р)
A,#°(SrS04, к,
298,15 К)
-1457,7±1,0 (III)
—1460,0±1,0 (III)
—1458,4±1,0 (III)
—1459,7±1,5
-1456,2+3,0
—1459,2±1,5
SrCO3 (к, ж). Термодинамические свойства кристаллического и жидкого карбоната стронция
в стандартном состоянии в интервале 100—2500 К приведены в табл. 842.
Значения постоянных, принятые в расчетах термодинамических функций SrCO3 (к, ж),
приведены в табл. 28.1. В интервале 0—1198 К за стандартное состояние SrCO3 принята ромбическая
а-модификация (структурный тип арагонита СаСО3), в интервале 1198 — 1689 К—гексагональная
[3-модификация (структурный тип кальцита СаСО3), а от 1689 К до точки плавления A767 К) —
Т-модификация, предположительно кубическая [561 ].
При Т <i 298,15 К термодинамические функции вычислены на основании измерений
теплоемкости SrCO3 в работе Андерсона [509] E5—291 К) на образце, содержащем ~7% СаСО3.
Экстраполяция теплоемкости ниже 55 К проводилась Андерсоном по уравнению С°р (T)=D B10/2")+
+2Е C04/Т)+2Я A344/Г), которое приводит к значению S° E4,7 К)=8,3 Дж-К^-моль.
Найденные Андерсоном значения теплоемкости в интервале 260—298,15 К скорректированы
(увеличены при 298,15 К примерно на 1,2%) для лучшего согласования с данными Ландера [1808]
по энтальпии SrCO3 (к) при высоких температурах. Погрешности принятых значений S° B98,15 К)
и Н° B98,15 К) — Н° @) (см. табл. 28.1) с учетом загрязненности образца,
далекой,?экстраполяции теплоемкости и погрешности данных [509] оцениваются в 2 Дж-К~1-моль~1и 0,2 кДж-моль
соответственно.
В интервале 298,15—1198 К для теплоемкости принято уравнение (см. табл. 28.1), выведенное
методом Шомейта по результатам измерений энтальпии SrCO3 в работе Ландера [1808] D56 —1187 К);
погрешность этих измерений составляет 1 %. Температура полиморфного (а —*¦ (В) превращения
A198+5 К) принята по данным 11 работ, в которых получены значения в интервале 1193—1203 К
[715, 964,1024,1604,1605,1808,1971,1954, 2005, 2301, 2907]. В работах [440,1168а, 1807] найдены
более низкие значения A183—1188 К), которые не учитывались в справочнике. Энтальпия
превращения A6,7+0,6 кДж-моль) выбрана на основании данных Ландера [1808] по энтальпии
а- и C-модификаций SrCO3 (к), данных Евстигнеевой и др. [118] по давлению диссоциации а- и
[3-SrCO3 (к) и данных Джадда и Попа [1604], полученных методом количественного ДТА.
Теплоемкость гексагональной модификации A46+3 Дж-К-моль~1) основана на измерениях энтальпии
в работе [1808] A198—1543 К). Теплоемкость высокотемпературной ^-модификации A689—
1767 К) также принята равной 146 Дж-К~1-моль~1. Температура Р -> т превращения A689+15 К)
принята по данным Бейкера [561 ], полученным под давлением СО2 примерно в 20 атм. Температура
конгруэнтного плавления A767 +10 К) принята по данным Бейкера [561] A767 К) и Беке [715]
A770 К); измерения[561 ] более предпочтительны, поскольку проводились в атмосфере СО2 с
использованием термографического и термогравиметрического методов, а также по измерениям
электропроводности образца при плавлении. Энтальпия плавления D0+8 кДж-моль) оценена с учетом
экспериментального значения энтропии плавления MgCO3. Теплоемкость! SrCO3 (ж) оценена
в 165+20 Дж-К~1'Моль~1 по аддитивной схеме с учетом экспериментальных данных по
теплоемкости расплавленных соединений щелочноземельных и щелочных металлов.
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000 и 2500 К составляют 2, 3 и
12 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями SrCO3 (к),
377
SrCO3 (к, ж)
Глава 28
приведенными в справочниках [578, 1656] и в табл. 842, достигают 3 Дж-К-моль х в значениях
Ф° (Т). Расчеты термодинамических функций SrCO3 (ж) в справочной литературе отсутствуют.
Константа равновесия реакции SrCO3 (к)=Эг (г)+С (г)+30 (г) вычислена с использованием
значения AJI0 @)=2833,884+2,8 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH°(SxCOs, к, 298,15 К) = —1227,0 + 2,0 кДж • моль.
Эта величина основана на результатах измерений, приведенных в табл. 28.18. Измерения
энтальпии растворения SrCO3 (к) в соляной кислоте были также проведены Бертло [672] и Томсе-
ном [2686]. Из калориметрических работ наиболее точными являются измерения Адами и Конуэя
Таблица 28.18. Результаты определений KfH° (SrCO3, к, 298,15 К) (в кДж-моль)
Авторы
Метод
AfH° (SrCO3, к, 298,15 К)
II вакон
III закон
Форкран, 1908 [1117] Калориметрическое измерение энтальпии расхво- —1224
рения SrCOa в соляной кислоте
Рот, 1941 [2370] То же —1214
Капустинский, Дезидерева, 1946 Тоже —1218,6
[1628]
Адами, Конуэй, 1966 [481] То же для растворения SrCCb и SrO в соляной —1218,7±1,5
кислоте
Джонс, Беккер, 1927 [1596] Измерение константы равновесия SrCO3 (к)-= —1180±15 —1216,4+ДО
—SrO (к)+СО3 (г) статическим методом, 962—
1376 К
Тамару, Сиоми, 1932 [2652] То же, 1073—1723 К, 16 точек —1218±4 —1229±4
Капустинский, Стаханова, 1947 То же методом протока, 1073—1323 К, 12 точек — Ш5±20 —1225+6
[153]
Ландер, 1951 [1808] Тоже, статическим методом, 969—1364 К, 19 точек —1222±7 —1228±5
Бейкер, 1963 [561] То же, 1422—1532 К, 5 точек —1213±10 —1231 ±9
Евстигнеева и др., 1969 [118, То же, 1203—1273 К, данные представлены в виде —1214 —1228+7
119] уравнения
Чипман, 1933 [847] Анализ литературных данных [1596, 2652] и др. —1206 —1220,4±4,0
Ходаковский и др., 1980 [4246] Анализ литературных данных по растворимости — —1227,2+2,0
SrCO3 (к)=3г+2 (ag)+COs-2 (aq); Д<? B98,15 К)=
=- 52,7 ±1,0
[481 ], которые исследовали реакции, составляющие замкнутый термохимический цикл. Измерения
Капустинского и Дезидеревой [1628] привели к совпадающей величине. Однако расчет по данным
[481 ] энтальпии реакции SrCO3 (к) с соляной кислотой (без учета данных по растворению SrO)
приводит к значению около —1227 кДж-моль. В пользу более отрицательного значения
свидетельствуют также результаты многочисленных исследований равновесий и расчеты [4246],
основанные на изучении растворимости SrCO3. Принятое в справочнике значение основано в первую
очередь на исследованиях растворимости SrCO3 (к); для их обработки использовались
термодинамические свойства ионов, приведенные в Приложении 5. При оценке погрешностей величин,
вычисленных из исследований равновесия диссоциации SrCO3 (к), учтена неточность
термодинамических функций, которая приводит к погрешности в 6 кДж-моль" для измерений при 1200 К.
Глава 29
БАРИЙ И ЕГО СОЕДИНЕНИЯ
Ва (к, ж), Ва, Ва+, Ва2, ВаО (к, ж), ВаО, ВаО+, ВаН, ВаН2 (к, ж), ВаОН, ВаОН+, Ва(ОНJ(к, ж),
Ва(ОНJ, BaF, BaF+, BaF2 (к, ж), BaF2, BaCl, ВаС1+, ВаС12 (к, ж), ВаС12, ВаВг, ВаВг2 (к, ж),
ВаВг2, Bal, Bal2 (к, ж), Bal2, BaS (к, ж), BaS, BaSO4 (к, ж), ВаСО3 (к, ж)
В настоящей главе рассматриваются свойства 23 веществ —• бария и его соединений с кислородом,
водородом, галогенами, серой и углеродом; для 11 веществ приводятся данные о термодинамических
свойствах? конденсированном состоянии и для 20—в газообразном состоянии. Для четырех газов
(Ва, Ва+, ВаО и ВаО+) термодинамические свойства рассчитаны для температур до 10 000 К, для
одного (Ва2) — до 3000 К, для остальных — до 6000 К. Для бария в Приложении 3.9 приведены
сведения о его критических постоянных.
Термодинамические свойства всех веществ приведены для природной изотопической смеси Ва
(атомный вес 137,34) и других элементов. Однако различия в молекулярных постоянных
индивидуальных изотопических модификаций, как правило, в расчетах не учитывались из-за отсутствия
соответствующих данных и их несущественного влияния на термодинамические функции
соединений бария 1.
В справочнике не рассматриваются многоатомные окислы бария типа ВаО2 и такие полимерные
соединения, как Ва2О2 и Ba2F4, ввиду их низкой стабильности. Не рассматриваются также ионы
ВаВг+, Bal+, BaFJ и т. п. ввиду невысоких энергий диссоциации соответствующих нейтральных
двухатомных соединений и высоких потенциалов ионизации трехатомных молекул. Образование
таких соединений в заметных количествах в равновесных условиях представляется маловероятным
в интервале 100 — 6000 К.
Ва (к, ж). Термодинамические свойства кристаллического и жидкого бария в стандартном
состоянии при температурах 100—4000 К приведены в табл. 843.
Значения постоянных, принятых для расчета термодинамических функций, приведены в табл. 29.1.
В справочнике за стандартное состояние Ва (к) принимается кубическая объемно-центрированная
модификация (структурный тип a-Fe). Эта модификация является единственной стабильной при
обычном давлении (см. обзор [4066 ]) 2. Имеющиеся в литературе противоречивые сведения о
характере изменения теплоемкости бария в области 500—800 К, по-видимому, обусловлены тем, что
исследовались образцы бария, содержавшие значительные количества примесей (см. ниже) 3.
При Т <^ 298,15 К термодинамические функции бария вычислены по измерениям теплоемкости
бария, выполненным Фурукавой и Исихарой [1154] A8—370 К), с учетом данных Робертса [2350]
A,5— 20 К) и Рахменкулова и др. [343] A3—300 К). Измерения теплоемкости в работе [1154]
выполнены на образце, очищенном вакуумной дистилляцией; после очистки образец содержал
4,2% примесей C,4% ВаО, 0,7% Sr, 0,06% Са, 0,005% Mg), на которые авторы[1154] вводили
поправки. В работах [343] и [2350] детальный химический анализ исследованных образцов не
проводился. Данные Робертса [2350] лежат несколько ниже данных [1154] и [343] и использовались
для расчета термодинамических функций бария ниже 10 К. Результаты измерений [343, 1154]
при Т <^ 200 К удовлетворительно (в пределах 1%) согласуются между собой. Выше 200 К наблю-
1 Следует отметить, что получение чистых образцов металлического бария встречает большие трудности, главным
образом при определении содержания кислорода, водорода и азота и удалении соответствующих примесей. Эти
трудности не преодолены до настоящего времени. Поэтому результаты исследований термодинамических
свойств Ва (к, ж), а также измерения энтальпии реакций бария могут содержать значительные систематические
ошибки, как правило не учитывавшиеся авторами работ. Этим, видимо, обусловлены имеющиеся противоречия
в термохимии соединений бария, сделавшие невозможным для Рабочей группы КОДАТА—МСНС обоснованный
выбор ключевых термохимических величин для этого элемента.
- При высоких давлениях у бария известны два фазовых перехода — при 55,3 кбар B95 К) образуется
гексагональная модификация бария (структурный тип Mg), которая при давлении около 120 кбар B93 К) превращается
в другую модификацию с неисследованной структурой [4066].
3 Ринк [2341] на основании измерения электропроводности бария пришел к выводу о наличии полиморфного
превращения при 648 К; Шпильрайн и Каган [459] по измерениям энтальпии (см. ниже) определили энтальпию
этого перехода равной 0,86 кДж-моль. Дитмарс и Дуглас [956] не обнаружили превращения при 648 К,
но установили наличие аномалий теплоемкости при 583 и 768 К.
379
Ва (к, ж)
Глава 29*
Т а б л и
Вещество
Ва
ВаО
ВаН2
Ва(ОНM
BaF2
ВаС12
BaBr3
Bal»
BaS
BaSO<
ВаСОз
ц а 29.1. Принятые значения термодинамических величин для (
и жидком состояниях
Состояние
1
К
оо~о
стэ *~^"
cq о
'&
о ^
ЕЧ 1
кДжХ
Хмоль
к, куб. 6,91
ж —
к, куб. 9,96
ж —
кП, ромб, (а) 9,1
к1 ромб. (Р), —
кИ, ромб, (р) 17,8
к1, ромб, (а) —
ж —
кШ, куб. (а) 14,44
кИ(Р)
к1 (Т) -
ж —
кИ, ромб, (а) 16,7
к1, куб. (Р) -
ж —
к, ромб. 19,0
ж —
к, ромб. 19,23
ж —
к, куб. 10,82
ж —
кН, ромб, (а) 19,18
к1, куб. (Р) -
ж —
кШ, ромб, (а) 16,51
кП, гекс. (Р) —
к1, куб. (f) —
ж —
К
1Л
оо"
аз
si
о
со
Дж ¦ К-1
62,5
72,0
63
108,5
96,36
123,7
150
165,2
78,45
132,1
112,1
Примечание. C°p (T) = o+ 6 T—e T~2+ ёП (в
ВаО: а d=2,083-10-6.
ВаСОз: а d=124,210-10-«.
И
ю
оо"
о R,
С)
• МОЛЬ
28,10
46,99
46
98
71,13
75,14
75,94
77,49
49,37
102,10
85,98
Дж- К-1
>ария и его соединений
Коэффициенты в уравнении
для с;(Т)
а
42,888
45,105
32,2
48,117
84
37,672
71
75
70,681
118
138
64,275
126
129
105
69,37
131
109
70,117
105
71,569
113
49,229
67,0
112,420
170
170
157,778
158
160
165
МОЛЬ).
Ь- 103
3,665
—4,309
11,527
27,931
91,630
29,830
19,129
21,991
19,953
6,907
51,550
-134,210
с • Ю-5
Интервал
температуры
К
14,117 298,15—1000
— 1000-3000
— 3000-4000
3,896 а 298,15—2290
— 2290—6000
— 298,15—871
— 871-1473
— 1473—2000
— 298.15—519
— 519—681,5
— 681,5—3000
1,813 298,15—1240
— 1240—14S0
— 1480—1641
— 1641-4300
-0,059 298,15—1198
— 1198—1234
— 1234—4100
0,650 298.15—1130
— 1130—4000
0,027 298,15—984
— 984-4400
1,705 298,15—2500
— 2500—6000
22,840 298,15—1423
— 1423—1853
— 1853—4000
38,066 а 298,15—1083
— 1083—1233
— 1233— 182S
— 1828-3000
в твердом
Ttr
или
Т
К
1000
2290
871
1473
519
681,5
1240
1480
1641
1198
1234
ИЗО
984
2500
1423
1853
1083
1233
1828
или
хк
7,12
59
5,6
25,0
3,6
16
5,49
2,55
17,8
17,4
15,85
32,2
26,5
E3
10
40
16,3
2,9
40
даются небольшие систематические расхождения, которые при Г?»300К достигают 2%; в этом
интервале температур данные [343] не учитывались. Погрешности принятых значений S° B98,15 К)
и Н° B98,15 К) — Н° @) (см. табл. 29.1) оцениваются в 1,0 Ды-К^-моль и 0,1 кДж-моль
соответственно. Измерения теплоемкости образца бария с неохарактеризованным составом в работе
Тини и Моруцци [2672] A5—300 К) не учитывались, поскольку они приведены только в виде
мелкомасштабного графика и значительно отклоняются (до 20%) от данных [343, 1154, 2350].
Расчет термодинамических функций Ва (к) при Т ]> 298,15 К основан на результатах
измерений энтальпии в работах Дитмарса и Дугласа [956] B73—873 К, 27 измерений) и Шпильрайна
380
Барий и его соединения • Ва (г)
и Кагана [459] D84—979 К, 8 измерений). Между данными этих авторов имеются существенные
расхождения (см. примечание на с. 379), достигающие при 500—600 К ~10%. Дитмарс и Дуглас
J956J измеряли энтальпию того же образца бария, который исследовался в работе [1154];
по-видимому, влиянием примесей можно объяснить аномалии теплоемкости, найденные ими при 583
и 768 К. Воспроизводимость измерений [956] можно оценить примерно в 3%. В работе [459]
исследовался образец бария с неизвестным содержанием кислорода; данные [459] имеют разброс
около 5%. Принятое в справочнике уравнение для теплоемкости бария (см. табл. 29.1) получено
методом Шомейта в результате совместной обработки данных [956] и [459], которым были
приписаны погрешности в 3% и 5% соответственно. Это уравнение при комнатных температурах плохо
согласуется с измерениями теплоемкости в работе [1154], которые имеют более медленное
изменение с ростом температуры.
Температура плавления бария A000 +5 К) выбрана на основании согласующихся между собой
измерений Миллера и др. [2029] (999,3 К), Келлера и др. [1661] (998 К) и Питерсона и Хинкбей-
на [2216] A002 К). Данные [2341] (983 К) и [1443] (987 К) менее надежны и не учитывались.
Энтальпия плавления G,12 +0,50 кДж- моль) определена как разность между значениями энтальпии
Ва (к) и Ва (ж), найденными в работах [459, 956].
Для теплоемкости жидкого бария в интервале 1000—3000 К принято линейное уравнение,
основанное на измерениях, выполненных Шпильрайном и др. [2515а ] импульсно-дифференциальным
методом в интервале 1000—2000 К с погрешностью около 4%. Это уравнение в пределах
указанной точности согласуется с результатами измерений энтальпии жидкого бария Каганом [459]
(ЮОО—1253 К, 4 измерения, С'р (ж) =39,2 Дж-К'^моль'1) и Дитмарсом, Дугласом [956] A003—
1173 К, 12 измерений, С, (ж) = E5,682 — 12,355- 10Г) Дж-К^-моль). При Т > 3000 К
принято постоянное значение теплоемкости Ва (ж), равное С°р (Ва, ж, 3000 К)=32,2 Дж-К~1-моль~1.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются
в 0,7, 1,5, 3 и 6 Дж-К-моль~1 соответственно.
Термодинамические функции Ва (к), табулированные в справочниках [1503, 1543 ], близки к
приведенным в табл. 843 (расхождения не превышают 0,5 Дж- К- моль). Соответствующие
расхождения для Ва (ж) увеличиваются с ростом температуры и достигают 5 Дж- К- моль для S° C000 К),
что обусловлено в основном учетом новых экспериментальных данных по теплоемкости жидкого
бария. Расхождения в Ф° (Т) с данными [105], где расчет был выполнен на основании оцененных
величин, составляют 4 и 10 Дж-К-моль при 1000 и 3000 К.
В настоящем справочнике за стандартное состояние бария принят Ва (к) и в соответствии
с этим
AfH°(Ba, к)==0.
Давление пара в реакции Ва (к, ж)=Ва (г) вычислено с использованием значения Д^0 @) =
= &,Н°(В&, к, 0)=179,713 +3,0 кДж-моль, соответствующего принятой в справочнике
энтальпии сублимации
Д,#° (Ва, к, 298,15 К) = 179,0 + 3,0 кДж • моль.
Принятая величина является средневзвешенной из приведенных в табл. 29.2 результатов
обработки измерений давления насыщенного пара бария, выполненных в работах [158, 462, 718, 1440,
17846, 2455, 2895]. Приведенные в табл. 29.2 погрешности отражают только воспроизводимость
измерений. При вычислении средневзвешенного учитывались отклонение результатов измерений
от средней величины и неточность термодинамических функций бария в конденсированном
состоянии, которая приводит к погрешности в энтальпии сублимации от 1,5 кДж-моль (для измерений
при 900 К) до 5 кДж-моль (для измерений при 1700 К). При вычислении средневзвешенного
наибольший вес придавался работам [158, 462, 2895]. Наблюдающиеся большие расхождения
найденных величин, вероятно, в значительной степени объясняются различием использованных образцов
бария и имевшимися в них примесями.
Ва (г). Термодинамические свойства газообразного бария в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 844.
Уровни энергии атома Ва, использованные в расчете термодинамических функций, приведены
в табл. 29.3, в которой представлены валентные состояния, относящиеся к электронным
конфигурациям . . ,6s2, . . Шр, . . .6s6d, . . .6s6/, . . .6s5d, . . .6s5/, . . .6s4/, . . .6p\ . . .5d6p, . . .5dQd
381
Ва+ (г)
Глава 29
Таблица 29.2. Результаты определений Д« Н° (Ва, к, 298,15 К) (в кДж-моль х)
Авторы
Метод
ДаЯ°(Ва, к, 298,15 К)
II закон
III закон
Руфф, Хартман, 1924 [2405] Точек гашения, 1204—1412 К, 10 точек 376+10 154,3+10
Хартман, Шнейдер, 1929 [1360] То же, 1334—1413 К, 7 точек 172±40 170,4±0,8
Рудберг, Лемперг, 1935 [2400] Эффузионный, 798—998 К, 14 точек 179±10 191,7+1,0
Баранова, Гаврилов, 1966 [29] Протока, 1344—\475 К, 11 точек 160±30 168,9±1,0
Зайцева, 1965 [132] Эффузгтошшй, 767—996 К, 13 точек . 260±25 193,7±3,2
Боданский, Шине, 1967 [718] Точек кипения, 1493—2027 К, 6 точек 197±5 178,7±2,2
Завитсанос, 1968 [2895] Эффузпоняый, 1120—1210 К, 8 точек 257±40 182,9+1,1
Торзионный, 1003—1216 К, 18 точек 151 ±90 181,3±1,8
Хинов, Олендорф, 1969 [1440] Измерение интенсивности резонансной флюо- 181 —
ресценции бария, 730—1200 К, расчет по
уравнению
Шпильрайн и др., 1969 [462] Статический, 1255—1380 К, 17 точек 192±3 175,4±0,3
Шине и др., 1971 [2455] Точек кипения, 1492—2035 К, 69 точек 231±2 184,3±1,2
Кармышин и др., 1974 [158] Статический, 1309—1412 К, 12 точек 178+4 177,3+0,3
То же, 1346-1540 К, 43 точки 179±2 177,1 ±0,3
Паршина, Ковтуненко, 1974 [312] Протока, 983—1408 К, расчет по уравнению 177 168,5±2,0
Кулкарни, Дадап, 1976 [17846] Протока, 1160-1330 К, 6 точек 182±25 181,1+3,0
(кроме XS), . . .5d? и . . .5dAfCF). Энергии этих состояний приняты по рекомендации Мур [2053],
а также результатам спектральных измерений Палениуса [2171] для состояний . . 5d? CF, XS)
и оценки [2171] для состояния . . .бе^Ч^) (точность оценки 300 см).
Термодинамические функции Ва (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 22)—A. 25). Значения Qax и ее производных находились непосредственным суммированием по
уровням энергии. Погрешности вычисленных значений Ф° (Т) при Т ^ 2000 К не превышают
0,02 Дж-К-моль и обусловлены главным образом неточностью принятых фундаментальных
постоянных. При более высоких температурах погрешности возрастают из-за пренебрежения рид-
берговскими состояниями, а также из-за отсутствия данных о ряде высоких валентных состояний
(включая неточность оценки состояния . . . ЪсР i^G^}. Эти погрешности составляют 0,05, 2
и 4 Дж- К- моль'1 в значении Ф° (Т) при 3000, 6000 и 10 000 К.
Ранее таблицы термодинамических функций В а (г) вычислялись в предыдущем издании
настоящего справочника [105] и таблицах JANAF [1543], в работах Вейц и др. [63] (до 3500 К), Хилзен-
рата и др. [1436] (до 10 000 К) и Кольского и др. [1749] (до 8000 К), а также в справочнике [26321
(до 3000 К). Результаты расчетов [63, 105, 1436, 1543] при Т < 3000 К удовлетворительно (в пре-
делах^ 0,01—0,02 Дж-К~1-моль) согласуются между собой и с величинами, табулированными
в настоящем издании (для расчетов [1749, 2632] соответствующие расхождения достигают
0,05 Дж-К-моль при этих температурах). При более высоких температурах расхождения
возрастают и достигают сданными [1436] 0,9 Дж- К~1-моль~1 в значении Ф° A0 000 К). Эти расхождения
обусловлены тем, что в работе [1436] были частично учтены ридберговские состояния атома Ва.
Энтальпия образования Ва (г)
Д^/^Ва, г, 298,15 К) = 179,0 + 3,0 кДж • моль
соответствует принятой энтальпии сублимации Ва (к).
Ва+ (г). Термодинамические свойства газообразного положительного иона бария в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 845.
Уровни энергии иона Ва+, использованные в расчете термодинамических функций, приведены
в табл. 29.3, в которой даны валентные состояния, относящиеся к электронным конфигурациям
ш . .6s — . . .Qg, . . .5d — 5gw . . Af. Энергии этих состояний приняты по рекомендации Мур [2053].
Термодинамические функции Ва+(г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A. 22)—A. 25). Значения Q3X и ее производных находились непосредственным суммированием па
382
Барий и его соединения
Ва+ (г)
Таблица 29.3. Уровни энергии атомов Ва и Ва+, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Ва . . .6sa
. . .6s5d
.6*5rf
. . .6s6p
. . .Ыр
.5Й2
. . .5d6p
.5d*
. . .5d6p
. . .50"
. ..5dbp
.5d3
.5d6p
. . .5<22
.5d6p
. . .5<76p
. . .5d2
. . .5d6p
. . .5d6p
. . .usod-
. . .Qsod
.. .6^2
. . .6s4/
. . .6s4/
. . .6p2
::%
Состояние
1Л
3Di
"D2
3D3
гог
3Pa
3Pi
1Pi
3F<,
3F~s
V*
sps
1?J
1?J
3P»
3 77
3d
3Di
3D2
3Di
3Po
3Pi
iS0
1F3
Юг
3Dn
3dI
1So
3Z
>fI
*Ft
1F3
3Pi
Tt
CM
0
9 033,985
9 215,518
9 696,551
11 395,382
12 266,021
12 636,616
13 514,738
18 060,264
20 934,04
21 250,25
21 623,7
22 064,661
22 947,438
23 062,06
23 074,416
23 209,11
23 480,01
23 787,077
23 918,44
24 192,057
24 300
24 531,536
24 979,859
25 642,157
25 704,14
25 956,549
26 757,4
26 816,293
28 554,257
30 236,815
30 695,594
30 750,669
30 818,11
34 370,78
34 493,898
34 602,802
34 616,683
34 630,809
34 736,423
34 823,420
35 344,423
35 616,947
Pi
1
3
5
7
5
1
3
5
3
5
7
9
5
7
5
5
1
3
9
5
3
9
5
7
1
3
5
1
7
3
5
3
5
7
1
1
5
7
9
7
3
5
5
Atom
Электронная
конфигурация
В a . . .5dQd
. . .5<Ш
. . .5d6d
. . .5d&d
. . .ЫЫ
. . .5d6d
. . ,5сШ
. . .5сШ
. . .ЫЫ
. . .bdQd
. . .6s5/
. . .6s5/
. . .5d&d
. . .6s5/
. . .5dGd
. . ,5d6d
. . ,5dQd
. . .5d6d
. . ,5<Ш
. . .5сШ
. . .6s6/
. . .6s6/
. . .5<24/
Ba+ . . .6s
. . .Ы
...6p
. . .Ы
...if
...5/
. . .6/
...5?
Состояние
3G3
*Di
XF3
*D,
*Gt
3Si
3Da
3Gb
1 P_
3 E1
*Fi
3F,
*F3
3Ft
3P*
3Fi
XD%
3Pi
2G4
*Pi
%Fi
3F%
1Рг
3F
25w
*D'l
2 p
2Fb *
2 E1
2 E1
2рч
'/j
3G
CM
35 894,28
35 933,825
36 165,311
36 200,423
36 348,98
36 446,583
36 628,87
36 837,5
36 902,55
37 088,844
37 282,175
37 394,925
37 418,967
37 504,02
37 524,184
37 675,84
37 732,23
37 837,4
38 023,22
38 177,10
38 267,67
38 815,735
38 818,418
38 825,265
38 884
39 765,31
0
4 873,85
5 674,824
20 261,562
21 952,422
45 949,496
46 154,89
48 258,59
48 483,29
57 390,93
57 631,64
64 596,31
64 697,08
63 026,53
68 425,87
Pi
7
3
7
5
9
3
7
11
3
5
7
5
7
7
9
1
9
5
3
9
5
5
7
9
7
21
2
4
6
2
4
4
6
6
8
6
8
6
8
18
18
уровням энергии. Погрешности вычисленных термодинамических функций при Т <[ 7000 К не
превышают 0,02 Дж-К~1-моль и обусловлены в основном неточностью принятых значений
фундаментальных постоянных. При более высоких температурах возникают погрешности из-за
пренебрежения ридберговскими состояниями, но даже в 5° (Т) и Ср (Т) эти погрешности не превышают
0,1 Дж-К-моль.
383
Ва+ (г)
Глава 29
Ранее таблицы термодинамических функций Ва+ (г) вычислялись в предыдущем издании
настоящего справочника [105] и работах Хилзенрата и др. [1436] и Грина и др. [1246] до 50 000 К.
Результаты этих расчетов и настоящего издания хорошо согласуются между собой (в пределах
0,01—0,02 Дж-К~1-моль~1) во всем интервале температур.
Константа равновесия реакции Ва+ (г)+в (г)—Ва (г) вычислена с использованием значения
Дг#°@) =—502,852+0,001 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Ва) =42 035,14+0,05 см-х=502,852 ±0,001 кДж-моль.
Эта величина получена Гартоном и Томкинсом [1177] в результате анализа двух наименее
подверженных возмущениям серий bdnp (п ^ 50) и 5dnf (п ^ 29) в спектре поглощения атома Ва,
сходящихся к состоянию 2Z)s/2 иона Ва+. Принятое значение /0 (Ва) существенно точнее величин,
Т а б л и
Ц
Молекула
Ва2
В;Ю
ВаО+
ВаН
BaS
a 29.4. Молекулярные постоянные Вая
Постоянно
Х*2+
а3 2+
А1 2+
Ь3П
вт
с3П
cm
12-, 1Д 1
3Д, 32+ J
Dl2+
X2 2+ 1
а2п }
Х32+
Л'аД
А*П1/г
Ат%
В2 2+'
Етч>
?'2Пз/г
D4Hu
С22+*
ХХ2+
а3П
ЛЧ1
В1 2+
С!2+Д
Примечание. Ниже
ВаО: а
ВаО+: а
ВаН: а
BaS: a
«>«?/<,=— 4,5-
Т
е
0
0
15 000 в
16 807,16
17 506
17 702
32 000 в
32 866,4
37 000 в
40 000 в
0
0
7 930
9 457,45
9 939,82
11090
14 602
15 057
21885
23 675
0
11744
12 186
14 450 в
26 997,74
все постоянные даны
со
е
26,5
669,81
500 в
499,7
442,45
442,45
490 в
488
—
—
450 а
1168,31
1107
1110,55
1109,98
1088,9
1226,3
428
1282
379,42
.
294,3
254,10
в см.
, ВаО,
ш X
е е
0,24
2,054
1,6 в
1,64
1,652
1,652
3,6 в
3,6
—
—
1,6б
14,50
14
15,29
13,59
15,32
17,36
4,5
15
ВаО+, ВаН, BaS
вв
СМ
0,009 6
а 0,312 614 1
0,258 в
0,258 32
0,223 5
0,223 5
0,27 в
0,272
—¦
—
0,24 а
3,382 85
3,068 в
3,278 87
3,322
3,267
3,485 5
3,559 7
1,62
3,59
0,8842 0,103 314 212
3,08
0,438
Ю-3; б а2 = —4,19-Ю-6; в оценка;
0,093 53
0,086 04
ох • 103
0,084
1,392 52б
1В
1,07
1,196
1,196
1 в
—
—
—
1,2»
65,99
96 г
72,83
82
70
72
75
17
64
0,315 091а
0,715
0,44
г вычислено по соотношению A
De ¦ 10'
0,05
2,68
Зв
2,8
2,3
2,3 г
3,4 в
3,4 г
—
—
2,7 г
1126,7 а- б
940 Д
1218 е
1322 а> ж
1110
1170 а- 3
—
1000
1000
0,306 401
_
0,38 г
0,40 г
.68).
оценка; б вычислено по соотношению A.67), принимая Do (ВаО+)=32 400; в вычислено по
нию A.69);
г вычислено по соотношению
приведено Do; б #о=2,89-10-9;
нию A.69);
а2=-4,4441
ДЬ«2+(Г.=
A.68).
в рассчитано по
Д рассчитано по соотношению
10-'; б pi=—5,184
13 000); 12-, 1Д,
• Ю1; в
зд з 2+
A.68);
значениям Во=3,
6 #о=4.1О-»; *
приведено значение Го!
5 000); 3П, m
02 и од г рассчитано по
к #о=6,6.10-
»; 3 #о=2,О
А
5,0
1,9397
2,13
2,134
2,294
2,294
2,08
2,08
—•
—
2,2
2,2317
2,36
2,267
2,252
2,271
2,199
2,176
3,22
2,17
6 2,5067
2,635
2,7468
соотяоше-
соотноше-
• ю-».
г вычислено по соотношению A.68);
(Г, =30 000)
384
Барий н его соединения
Ва,(г)
измеренных в работах [317, 1174], а также величины, рекомендованной Мур [2053] на основании
более ранних работ. Ему соответствует
Д/Я°(Ва+, г, 0) = 682,565 + 3,0 кДж • моль.
Ва2 (г). Термодинамические свойства газообразного дибария в стандартном состоянии при
температурах 100—3000 К приведены в табл. Д.63.
В табл. 29.4 приведены молекулярные постоянные Bas, которые из-за отсутствия
экспериментальных данных оценены при подготовке справочника. По аналогии с молекулами Mg2 и Cas
в справочнике принимается, что молекула Ваа в основном электронном состоянии X1 EJ образуется
за счет вандерваальсовского взаимодействия атомов бария в основном состоянии и имеет
небольшую энергию диссоциации (см. ниже). Возбужденные электронные состояния Ваа должны иметь
энергии выше 10 000 см и могут не учитываться при расчетах термодинамических функций. При-
Таблица 29.5. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в си),
а также значения vmax и t/ijm, принятые для расчета термодинамических функций Bas,
BaO, BaO\ BaH, BaS
Коэффициент
ге. ю-*
Ую-10"»
Уго-10
Узо-Ю2
У40-Ю4
У5в-10в
У01-101
Уи-Юа
У.1.10*
Уз1-108
Уог-10в
У12-101°
Уоз-ЮИ
Ув4-Ю13
(ao=De)X
XlO
aj-101
ar106
яз ¦ 101°
^'гпах
Ва2
X4J
0
0,027 0
—0,024
—
—
—
0,098
—0,008 4
—
—
—0,005 2
:—
—0,001 326
—
—
—
—
55
394
0 1,5000
0,669 727 0,4997
—0,202 95 —0,164 -
—0,682 873 —
0,720 695 —
—0,187 016 —
3,126141 2,5832
—0,139 252 —0,107 -
—0,0419 —
1,3 -
—0,268 —0,28
—
— —
— —
— —
— —
— —
174 151
561 520
Примечание. Ниже все постоянные даны в
ВаО: а
ВаО+: а
ВаН: а
BaS: a
Состояние
Те
Состояние
Те
Состояние
те
Собтояние
Те
сзп cm !?-
32 000 32 866,4
АШ
0
А'Ч 4ЧЦ АЩ
ВаО
Л12+
1,680 716
0,499 718
-0,164 804
0,092 753
—
_
2,583 2
-0,107
—
—
-0,28
—
—
—
,—
—
—
—
177
530
СМ.
1,750 6
0,442 45
—0,169 3
—
—
—
2,235
—0,119 6
—
—
-0,23
—
—
—
—
—
—
—
130
476
, 1Д, *2+, 3Д D41+
37 000
31,
7930 9457,45 9939,82 11090
аШ АШ 632+
И 744 12186 13 00(
) 14 450
40 000
ЕШЧг
14 602
12-, 3S+,
В1П»
1,770 2
0,442 59
ВаО+
Х*2-
0
0,450
—0,175 44 —0,16
0,231 981
—
—
2,235
—0,119 6 -
0,011
—
—0,23
—
—
—
—
—
—
—
234
554
—
—
—
2,40
-0,12
—
—
-0,27
—
—
—
—
—
—
—
140
486
15 057 21 885
!A, 3Д С
2.5 000 26
397,7
на
ВаН
0
1,166 667
—1,321 433
—28,570 4
—
_
33,824 07
—6,35 935
—22,330 2
—
—112,67
289,0
1,680
1,644 999
2,889 603
5,069 330
—1,380 884
24
98
С*Х+
23 675
8П, Щ
30 000
BaS
и-
0
0,379 148
-0,088 410
0,009 604
0,008 836
—
1,031 642
—0,031 441
—0,004 431
0,054 457
-0,030 551
—0,516 521
—0,000 729
—
—
—
—
260
861
385
ВаО (к, ж) Глава 29
веде иное в табл. 29.4 значение ге (Ва2)=5,0 +0,2 А оценено на основании сопоставления межатомных
расстояний в молекулах Mg2, Ca2 и Ne2, Ar2 и Хе2 [662]. Частота колебаний юе оценена из условия
©г/ 2BeD/e=9,8 A [2579], как и для молекулы Sr2.
Термодинамические функции Ва2 (г) рассчитаны по уравнениям A. 3)—A. 10), A. 93)—A. 95)
без учета возбужденных состояний. Значения Q^2 вр и ее производных находились по уравнениям
A. 73)—A. 75) непосредственным суммированием по колебательным уровням энергии и
интегрированием по вращательным уровням с помощью уравнений типа A. 82). В расчете учитывались все
уровни со значениями / ^ «/max, vi гДе ^тах,» находились из условий A. 81). Уровни энергии
вычислялись по уравнениям A. 64а) и A. 65), коэффициенты Ykl в которых, а также значения г>т!а
и /lim приведены в табл. 29.5. Коэффициент Yo3 вычислялся из условия A. 59).
Погрешности вычисленных термодинамических функций обусловлены отсутствием
экспериментальных данных об энергии диссоциации и молекулярных постоянных Ва2 и трудностью
оценить их с точностью, большей чем 10—20%. Общие погрешности в значениях Ф° (Т) могут
достигать 5—10 Дж-К-моль.
Ранее термодинамические функции Ва2 (г) вычислялись Климовым и др. [167] (Ф° (Т) до
4500 К) в приближении «жесткий ротатор — гармонический осциллятор»; резкие расхождения
между данными [167] и табл. Д.63, достигающие 10 Дж-К~1-моль в значении Ф° C000 К),
обусловлены грубой методикой, примененной авторами [167].
Принятая в справочнике энергия диссоциации
Do (Ваа)=750+200 см-1=9,0+2,5 нДж-моль
оценена на основании энергии диссоциации молекул Mg2 и Са2, постоянных вандерваальсовского
взаимодействия (Св) атомов Mg, Ca и Ва [436] и предположения, что величина C6lDerue
приблизительно постоянна в ряду аналогичных молекул, как это имеет место для благородных газов [662].
Принятому значению соответствует
Д^^Ваз, г, 0) = 350,426 + 6,5 кДж • моль.
ВаО (к, ж). Термодинамические свойства кристаллического и жидкого оксида бария в
стандартном состоянии при температурах 100—6000 К приведены в табл. 846.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 29.1. В справочнике за стандартное состояние ВаО (к) принята кубическая модификация
(структурный тип NaCl), устойчивая от 0 до температуры плавления 2290 К.
При Т ^ 298,15 К термодинамические функции вычислены на основании данных по
теплоемкости, полученных в работе Гмелина [1226] A—300 К) на образце, содержавшем не более 0,3%
примесей, и в работе Андерсона [511] E5—299 К) на образце ВаО фирмы Кальбаум. Погрешности
измерений [1226] и [511] оценены в 0,5% и 1,5%, а погрешности вычисленных по этим данным
значений S° B98,15 К) и Н° B98,15 К)—Н° @) (см. табл. 29.1) - в 0,5 Дж-К-моль и
0,05 кДж-мОль соответственно. Для теплоемкости ВаО (к) в интервале 298,15—2290 К принято
уравнение, выведенное методом Шомейта по результатам измерений энтальпии ВаО (к) в работе
Ландера [1808] C91—1299 К). Учитывая недостаточно высокую чистоту исследованных Ландероад
образцов и погрешности измерений энтальпии (—4%), теплоемкость ВаО может быть вычислена
по этому уравнению с точностью в 1,5% при 1000 К, 3% при 1500 К и 8% при температуре
плавления ВаО. Температура плавления B290+30 К) принята по измерениям Фоэкса [1111, 1112],
выполненным на образце чистотой 99,8%, с учетом поправки на пересчет к температурной шкале
МПТШ-68. Данные Шумахера [2477] занижены B196 К), вероятно вследствие взаимодействия ВаО
с вольфрамовой ампулой. Энтальпия плавления E9+6 кДж-моль) принята по оценкам,
выполненным в справочниках [105] и [1543]. Теплоемкость ВаО (ж) (84+8 Дж-К~1-моль) принята
равной теплоемкости ВеО (ж) (см. выше).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,4,
0,8, 6 и 14 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
ВаО, приведенными в справочниках [105,1543] и табл. 846, достигают 2 и 8 Дж-К -моль в
значениях Ф° (Т) для ВаО (к) и ВаО (ж) соответственно. Для ВаО (ж) эти расхождения обусловлены
раз ной оценкой теплоемкости ВаО (ж).
386
Барий и его соединения ВаО (г)
В справочнике принята энтальпия образования оксида бария
к, 298,15 К) = — 548,0 + 5,0 кДж • моль,
основанная на измерениях Фицгиббона и др. [1103]. В этой работе измерены энтальпии
растворения образцов ВаО (к) и Ва (к) в соляной кислоте одинаковой концентрации. В литературе
известен ряд других измерений энтальпий растворения ВаО (к): [408, 418, 481, 670, 1116, 2686] и
Ва (к): [71, 1022, 1322, 1523] в соляной кислоте, в том числе выполненные на прецизионной
калориметрической аппаратуре и предположительно с хорошими образцами. Однако конечные
концентрации в них не совпадают, что может привести к большим погрешностям при вычислении по
этим данным энтальпии образования ВаО (к), так как в солянокислых растворах хлорида бария
можно ожидать образование комплексных ионов. Прямое измерение энтальпии образования
ВаО (к) методом сожжения бария в кислороде, выполненное Ма [1913], привело к значению
—581,8 кДж-моль, которое, по-видимому, искажено побочными реакциями (образованием
перекиси бария или растворением образующегося оксида в оксиде магния, использованном для фут?-
ровки калориметрической бомбы).
Давление пара оксида бария в реакции ВаО (к, ж)=ВаО (г) вычислено на основании значения
krH° @) = Д8#° (ВаО, к, 0)=421,205+10 кДж-моль, соответствующего принятым в
справочнике энтальпии образования ВаО (к) и энергии диссоциации ВаО.
ВаО (г). Термодинамические свойства газообразного оксида бария в стандартном состоянии
при температуре 100—10 000 К приведены в табл. 847.
В табл. 29.4 приведены молекулярные постоянные ВаО, принятые на основании анализа
результатов исследований электронных [62, 836, 1080, 1081, 1489, 1542, 1767, 1768, 1796, 1797, 1917,
1997, 2187, 2276, 2426, 2875], колебательных [1870, 2682] и вращательных [766, 1453, 2700, 2840,
2841] спектров этой молекулы. В электронном спектре ВаО изучены три системы полос, связанные
с переходами между четырьмя синглетными состояниями: ЛхЕ+<^ Х*? + [1542, 1767, 1768, 1796,
1797, 1917, 1997], ?ХП -* Хх?+ [1489, 2276, 2875] и C4L *> Х^ + [62, 2187]. По аналогии с
оксидами других щелочноземельных металлов молекула ВаО, помимо экспериментально
исследованных синглетных состояний, должна иметь связанные триплетные состояния а3?+, Ь3П и с3П, иза-
конфигурационные соответственно состояниям Л1!!4" (конфигурация. . . 12о5я*13з), В1!!
(конфигурация . . .12а2 бтгНЗа) и СХП (конфигурация . . .12а5тс46тс), а также группу состояний C> *Д,
1Е", *¦ 3И+), соответствующих конфигурации . . .12а25л36тс. Из всех.этих состояний только
состояние &3П идентифицировано по возмущениям в системе Аг1>+—Хх? + [1080, 1797].
В табл. 29.4 приведены данные для 12 валентных состояний молекулы ВаОх. Колебательные
постоянные ВаО в основном состоянии X1!! + приняты по результатам исследования спектра
лазерной флуоресценции (система 41?+— Хх?+) в работе Сакураи и др. [2426], которые наблюдали
переходы на колебательные уровни с v" ^ 17. Вращательные постоянные получены ТиМанном
и др. [2700] из анализа переходов /=1 —>¦ 2 для у=0-^-4 в микроволновом спектре ВаО и хорошо
согласуются (за исключением значения а2) с менее точными данными, найденными из анализа
тонкой структуры полос в электронном спектре [1797, 2426]. Колебательные постоянные в состояния
^41Е+приняты на основании анализа системы А1Л+—Х111+ A1 полос, v' <С[5), проведенного Ла-
герквистом и др. [1797], а вращательные постоянные — по результатам исследования той же
системы методом двойного резонанса Филдом и др. [1081, 1083]. Колебательные постоянные
состояния В1]! получены по кантам 80 полос системы 5*П—ХгЛ+ (v' <1 40) из анализа, проведенного
Уиссом и Бройда [2875]. Приведенные в табл. 29.4 вращательные постоянные ВаО в состоянии
В1!! получены Пруэттом и Зэра [2276] из анализа полос 12—0, 17—0 и 18—0 этой же системы.
Молекулярные постоянные в состоянии Ъ3П определялись Филдом [1080] из анализа возмущений
в системе Л1!!*—ХЧ^ [1797] (Го=17 372+10, «о,=448,3 +0,7, 0)^=2,39+0,13, ?„=0,2244 +
+0,0005 и а!=0,0014 +0,0002 см). Однако в табл. 29.4 для состояния 63П приняты те же
постоянные, что и для состояния В1]!, поскольку последние были получены непосредственно из
экспериментальных данных [2276, 2875]. Эти постоянные ВаО в состоянии В1!! несколько отличаются от
рассчитанных по возмущениям, а анализ возмущений показал, что в изоконфигурационных еостоя-
1 Тип основного электронного состояния молекулы ВаО не вызывает сомнения [62, 766, 1870, 2187, 2682., 2840,
2841], однако в ряде работ по исследованию равновесия реакций с участием ВаО (см. [1618, 2132]) принималось,
что основным является состояние 3IL
387
ВаО (г) Глава 29
ниях Ь3П и В1!! постоянные должны практически совпадать. Энергия возбуждения состояния Ь3П,
полученная Филдом [1080], скорректирована с учетом принятого значения Те EХП) [2875] и
разницы в энергиях Те (B1U)—Te (Ь3П)=196 см, найденной Филдом [1080]. Колебательные
постоянные в состоянии СШ определены Паркинсоном [2187], который исследовал систему СХП— Х1^*,,
а вращательная постоянная Вв оценена в предположении, что отношение re (XlL+)lre (СШ) =
=1,15, как в молекуле SrO. Энергии возбуждения состояний а3Е + и с3П приняты на основании
оценки величины расщепления изоконфигурационных состояний в работе Филда [10801, который
проанализировал соответствующие данные для молекул MgO [2446] и СаО [821]. Колебательные
и вращательные постоянные этих состояний приняты близкими к соответствующим постоянным
изоконфигурационных состояний. Для группы состояний ХЕ", ХД, 3Д, х?+ и 3? + средняя энергия
возбуждения оценена по данным для молекулы SrO и соотношению Т\ (BaO)/^ (SrO)=d,4,
которое справедливо для состояний Л1Е+ и СХП. Погрешность оцененных энергий возбуждения,
по-видимому, не превышает 2000 см.
Термодинамические функции ВаО (г) вычислены по уравнениям A. 3)—A. 10) и A. 93)—A. 95).
Величина Qm и ее производные рассчитывались по соотношениям A. 90)—A. 92) с учетом 11
возбужденных электронных состояний в предположении, что Q[•'_ вр = (р{[ра) (?<>1. вР Для всех состояний
с Т\ >¦ 30 000 см. Статистические суммы по колебательно-вращательным уровням состояний
Хх?+, а3?+, А1!,*, 63П и ВЧ1 и их производные вычислялись непосредственным суммированием по
колебательным уровням и интегрированием по / с помощью уравнений типа A. 82). В расчете
учитывались все уровни этих состояний со значениями / ^ •Лши, v последние находились по
соотношению A. 81). Энергия колебательно-вращательных уровней этих состояний рассчитывалась по
уравнениям A. 65) и A. 62) с коэффициентами Ykl, приведенными в табл. 29.5 вместе со
значениями уши и /Иш. Значения Yk0 получены по постоянным из табл. 29.4 с учетом условий A. 50),
для соблюдения которых в уравнение A. 65) для состояния Хх?+ введены дополнительные члены
с коэффициентами Yi0 и У60, а для состояний Лх?+ и ВЩ — с коэффициентом У30. Постоянные YS1
в состоянии Х1!!4" и У21 в состоянии ВгЛ определены из условия 5Сшах+1 = 0. Мультиплетность и
вырождение колебательно-вращательных уровней состояний учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при температурах до 2000 К в основ-
йом обусловлены неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль.
При более высоких температурах становятся существенными погрешности, связанные с
неточностью оцененных энергий возбуждения ряда состояний (главным образом состояния 3? +), а также
пренебрежением ридберговскими состояниями ВаО. Погрешности в значениях Ф° (Т) при 3000,
6000 и 10 000 К могут достигать 0,05, 0,3 и 2 Дж-К-моль соответственно.
Ранее термодинамические функции ВаО (г) вычислялись в предыдущем издании настоящего
справочника [105], таблицах JANAF [1544], а также работах Вейц и др. [63] (Т ^ 3500 К),
Брюэра и Розенблатта [745] (Ф° (Т) до 3000 К) и Шнейдера [2463] (QBB для 2000 < Т < 6000 К).
Расхождения результатов этих расчетов с данными табл. 847 становятся существенными при
Т ]> 2000 К и обусловлены главным образом различием в энергиях возбуждения и числе
учитываемых электронных состояний. Различие значений Ф° (Г), приведенных в [63, 105] и табл. 847,
достигает 3 Дж-К~1-моль при 6000 К и объясняется тем, что в этих работах учитывалось только
одно возбужденное состояние. В расчете [1544] для состояний с конфигурацией (. . .12о25те36тс)
приняты заниженные энергии, соответствующие расхождения достигают 0,7, 3 и 7 Дж-К~1*моль
в значениях Ф° (Т), S° (Т) и С°р (Т).
Константа равновесия реакции ВаО (г)=Ва (г)+О (г)вычислена на основании принятой в
справочнике энергии диссоциации
ДгЯ° @) = Do (ВаО) = 552 + 8 кДж • моль да 46150 ± 650 см.
В табл. 29.6 представлены результаты экспериментальных исследований энергии диссоциации
этой молекулы и энтальпии сублимации ВаО (к), рассмотренных при выборе этой величины;
погрешности приведенных значений учитывают воспроизводимость измерений, а также неточности
термодинамических функций, сечений ионизации и термохимических величин, использованных
в расчетах. В скобках приведены значения Do или Д8#°, соответствующие величине,
непосредственно определявшейся в данной работе. Как видно из таблицы, в отличие от других окислов
щелочноземельных металлов результаты масс-спектрометрических, спектрофотометрических и
других измерений в случае ВаО находятся в удовлетворительном согласии. Принятое значение
388
Барий и его соединения
ВаО (г)
Таблица 29.6. Результаты
Авторы
Классен, Винеманс, 1933 [855]
Герман, 1937 [1391]
Блуэтги др., 1939 [707]
Инграм и др., 1955 [1528]
Щукарев, Семенов, 1957 [467]
Никонов, Отмахова, 1961 [286]
Ньюбери и др., 1968 [2132]
Семенов и др., 1972 [378]
Хилперт, Герадс, 1975 [1435]
Фарбер, Сривастава, 1975
[1057]
Хульдт, Лагерквист, 1954
[1497]
Вейц, Гурвич, 1956—1957
[58—60]
Гурвич, Рябова, 1965 [103,
355]
Калфф, Ллкемаде, 1973 [1619]
Старовойтов, 1977 [397]
Старовойтов, 1977 [397]
Колен и др., 1964 [873]
Ионах и др., 1972 [1595]
Херм и др., 1973 [1389]
Дагдиджан и др., 1974 [920]
Энгелке и др., 1976 [1034]
Панчеяков и др., 1973 [310]
измерении энтальпии сублимации ВаО (к)
Метод
Испарение с открытой поверхности, 1223—
1475 К, 8 точек
То же, 1250—1580 К, расчет по уравнению
Эффузионный, 1526—1800 К, 12 точек
Масс-спектрометрический, 1530—1758 К,
4 точки
Испарение с открытой поверхности, 1173—
1351 К, расчет по уравнению
Эффузионный, 1100—1600 К, расчет по
уравнению
То же, 1553—1862 К, 40 точек
Масс-спектрометрйческин, 1365—1917 К,
78 точек
Эффузпонньтй, 1423—1723 К, расчет по
уравнению
Масс-спектрометрический, 1332—1681 К,
расчет по уравнению
То же, 1643—1803 К, 10 точек
Спектрофотометрическое исследование
реакции ВаО=Ва+О па основе измерения
р (Ва) в пламенах ацетилена с воздухом,
2240—2430 К
То же, в пламенах C2H2-j-воздух и Н2+О2,
2452-3150 К, 12 точек
То же, в пламенах СО+О2, 2965 К
То же, в пламенах CO+N2O+H2O, 2684—
2857 К, 12 точек
То же, в бедных пламенах CHi+Oo+N2,
2370—2815К, 6 точек
Исследование равновесия реакции Ва+Оа=
=ВаО+О на основании измерения интен-
сивностей полосы ВаО и линии Ва в
спектрах бедных пламен CHi+O2+N2, 2180—
2815 К, 8 точек
Масс-спектрометрический, Ba+SO=BaO+
+S, 1934—2069 К, 3 точки
Исследование порога реакции Ba+NO2=
=BaO-4-NO
Исследование порога реакции Ba+SO2=
=BaO+SO по хемилюминесценции
Исследование лазерной флуоресценции при
взаимодействии Ва+СО2=ВаО+СО в
скрещенных пучках
Исследование порога реакции Ва (г)+
+СЮ2 (г)=ВаО (г)+СЮ (г) в скрещенных
пучках по хемилюминесценции
Электронный удар
и энергии диссоциации ВаО (г)
Do (ВаО, г, 0)
кДж • 1
E73 ±23)
E43+29)
D50)
E50 ±12)
E40+38)
E37 ±6)
E65+170)
E31 ±10)
E83)
E49 ±27)
E65)
E55 ±29)
E09 ±8)
E46 ±6)
E07 ±6)
E46 ±6)
E20)
E45±7)
E57)
E42 ±8)
E29 ±25)
E42 ±9)
540±20 (III)
574±8 (III)
566±12 (III)
548,6±3,4 (III)
589+6 (II)
567,9±2,0 (III)
562 ±10 (II)
636 (И)
550±12 (III)
>550
>546
558,6 ±5,4
>550±8
554 ±14
Д8#° (ВаО, к, 0)
ноль
400+22 (II)
431+28A11)
522 (II)
424±10 (III)
433 ±38 (И)
436,0±2,8 (III)
408+170 (ID
442,0±8,6 (III)
390 (II)
423,8±26 (III)
408 (II)
418±28(Ш)
464+5 (II)
427,3±2,8 (III)
466+3 (II)
427,1 ±2,8 (III)
453 (II)
428,7±3,0 (III)
416 (II)
431,2±5,5 (III)
433 ±24 (II)
. 431,3±6,6 (III)
D33)
C99 ±10)
D07)
D24,6±6,7)
C84+8)
D05 ±6)
D11 ±12)
C37)
D23 ±13)
«423)
«427)
D14,6 ±8,0)
(<423±10)
D19 ±15)
389
ВаО+ (г), ВаН (г) Глава 29
осйовывается на измерениях энергии диссоциации в работах Колена и др. [873], Старовойтова и
др. [397] (по зависимости In/Ва0//ва от Г), Гурвича и Рябовой [103, 355], Панченкова и др. [310],
которые подтверждаются исследованиями порога хемилюминесценции в работах [920, 1034, 1389,
1595] и согласуются в пределах погрешностей измерений с результатами исследований теплоты
сублимации ВаО (к) в работах [378, 467, 855, 1057, 1391, 1435, 2132]. Предпочтение отдано
прямым измерениям энергии диссоциации, поскольку значения, рассчитанные через Д,Д° (ВаО, к),
содержат дополнительные систематические погрешности из-за неточности термодинамических
функций ВаО (к) и величины \fH° (ВаО, к).
Принятому значению энергии диссоциации соответствует
г, 0) = —125,504 + 8,5 кДж • моль.
ВаО+ (г). Термодинамические свойства положительного иона оксида бария в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 848.
В табл. 29.4 приведены молекулярные постоянные ВаО+, принятые из-за отсутствия
экспериментальных данных на основании оценок. Ион ВаО+ имеет два близколежащих нижних
электронных состояния 2Е+ (конфигурация . . .тс4а) и 211 (конфигурация . . .а2л3), относительное
расположение которых оценить трудно, поэтому в справочнике для обоих состояний принята нулевая
энергия. Этот вывод следует из рассмотрения изоэлектронной молекулы CsO [1867], [2577], а также из
энергии возбуждения состояний 3П, ХП (. . . тг3а2а) и 3Е, х? (. . ,п*оа) молекулы ВаО, которые
являются первыми уровнями ридберговских серий, сходящихся к 2П- и 22-состояниям ВаО+. Из
этих же данных следует, что возможное различие в энергиях состояний 2П и 2 2 не превышает
2000 см. Значения <ое и Ве, приведенные в табл. 29.4, оценены на основании значений этих
постоянных в возбужденных состояниях молекулы ВаО, их погрешности могут достигать 50 и
0,02 см соответственно.
Термодинамические функции ВаО+ (г) вычислены по уравнениям A. 3)—A.10) и A. 93)—A. 95).
Величина QBS и ее производные вычислялись по соотношениям A. 90)—A. 92) с учетом двух
электронных состояний, указанных в табл. 29.4, в предположении, что состояния 2Е и 2П имеют
одинаковую энергию и суммарный статистический вес, равный 6. Статистическая сумма по
колебательно-вращательным уровням и ее производные рассчитывались непосредственным суммированием
по колебательным уровням и интегрированием по/с помощью уравнений типа A. 82). В расчете
учитывались все уровни этого состояния со значениями / <^ /шах, последние находились по
соотношениям A. 81). Энергия колебательно-вращательных уровней рассчитывалась по уравнениям
A. 65) и A. 62) с коэффициентами Ykl, идентичными постоянным из табл. 29.4 и приведенными
вместе со значениями ь>шах и JUm в табл. 29.5.
Погрешности вычисленных термодинамических функций во всем интервале температур
определяются главным образом неопределенностью в расположении низколежащих электронных
состояний, а также отсутствием экспериментальных данных о молекулярных постоянных молекулы ВаО+.
Погрешности в значениях Ф° (Г) оцениваются в 5—10 Дж-К-моль~1.
Таблица термодинамических функций ВаО+ (г) публикуется впервые.
Константа равновесия реакции ВаО+ (г)+е (г)=Ва (г)+О(г) вычислена на основании значения
LrH° @) =—115+13 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0 (ВаО)=667+10 кДж-моль.
Это значение принято по результатам измерений, выполненных методом электронного удара
в работах Панченкова и др. [3101 F73+10 кДж-моль) и Рауха, Аккермана [2302а] F61 +
+10 кДж-моль). Близкие, но существенно менее точные значения получены Стаффордом, Бер-
ковицем [2604], Фарбером, Сривастава [1057], Хилпертом и др. [1435а] и Менаром [2011а].
Принятому значению соответствуют
kfH°{B&O+, г, 0) = 541,496+ 13 кДж • моль,
Do (ВаО+) = 387,852 + 13 кДж • моль^ 32 400 + 1100 см.
ВаН (г). Термодинамические свойства газообразного гидрида бария в стандартном состоянии
При температурах 100—6000 К приведены в табл. 849.
В табл. 29.4 представлены молекулярные постоянные, принятые на основании анализа
результатов исследования спектра ВаН. В электронном спектре ВаН наблюдались девять систем полос,
390
Барий и его соединения ВаН (г)
«вязанные с переходами между 10 электронными состояниями: АЩ ** Х2? + [1754, 1758, 2812],
В2 2+ -* Х*1,+ [1754, 2810], Е2П -* Х22 + 11150, 2440а], ?2? ** X2?,* [1756], С2? =** X2 2+ [1150a,
1296, 1756], Н2Е *- Х2? + [1687], F2Z ^Х2!* [1012, 1013, 1686], G2? «- Х2? + [1686] и /2S <-
*- X22 + [557a]. Анализ возмущений в состоянии А2П показал [1758,1759а, 2770], что первым
возбужденным состоянием ВаН является состояние 4'2Д, которое, так же как состояния А2П и 2?2Е+,
коррелирует с атомами Ва CZ>)+H B<S). Состояния #2Б (Го=27 080см), F2Z (T0=30 747,9 см),
<G2E {7'0=31 645 см) и/2? (Го=33 416 см), по-видимому, являются ридберговскими, как у
молекул СаН и SrH, и не включены в табл. 29.4.
Молекула ВаН, подобно молекулам гидридов других щелочноземельных металлов, должна
иметь ряд квартетных состояний, коррелирующих с атомом Ва в состояниях aD и 3Р. В работе
Кодпа и др. [1756] отмечено, что распределение интенсивности вращательной структуры системы
D -» Х22 отличается от ожидаемого для перехода 2Е—а?, а минимум потенциальной кривой
сдвинут в сторону больших г, что приводит к пересечению потенциальных кривых этого состояния
и состояния В2 Е+. В связи с этим в справочнике состояние D рассматривается как компонента
2=х/2 состояния 4П и принимается, что остальные компоненты имеют такую же энергию. Состояние
41, по-видимому, обнаружено Коппом и др. [1758] по возмущениям уровней состояния D, его
энергия составляет около 22 300 см, и оно не включено в табл. 29.4 из-за скудности
соответствующей информации и низкой энергии диссоциации ВаН в этом состоянии « 3000см), также как
в состоянии 4П. Молекулярные постоянные ВаН в состояниях Х2Е+и.42П, приведенные в табл. 29.4,
получены Коппом и др. [1758] из анализа полос системы А2Н—X2 ?+ (z/, v" ^ 2). Постоянные в
состоянии А'2Ь. рассчитаны на основании значений AG и Bv, найденных Коппом и др. [1758], и
нумерации колебательных уровней, предложенной Везетом [2770] по возмущениям в спектрах ВаН
и BaD.
Вращательные постоянные ВаН в состояниях 52?+ и Е2П приняты по работам Уотсонэ и др.
11754, 2810], которые исследовали вращательную структуру четырех полос системы 2?2Е+—
-Х2Е+, и Функе [1150], который исследовал структуру полос системы 1?2П—Х2? + (v' ^ 2).
Колебательные постоянные и значения Те в этих работах не определялись и были рассчитаны на
основании соответствующих постоянных молекулы BaD в этих состояниях, найденных Коппом и Вир-
хедом [1759а]. Постоянные в состояниях D4S. и С2? приняты по работе Коппа и др. [1756], где они
были получены в результате совместной обработки данных для систем полос D—Х2? + A <Ii/ ^ 9)
и С2Е-Х2?+ (i/ <3).
Термодинамические функции ВаН (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения ()вн и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом восьми
возбужденных электронных состояний (подсостояния А2Ии2 и A2U^J2, а также -Е2П1/2 и ?2Пз/2
рассматривались как отдельные состояния) в предположении, что QHl,-BV:=(piiPx) (?S.bP- Величина Q^.Bf
и ее производные вычислялись по уравнениям A. 70)—A. 75) непосредственным суммированием
по колебательно-вращательным уровням энергии. В расчетах учитывались все уровни энергии,
ограниченные предельной кривой диссоциации состояния Х2?+; в табл. 29.5 приведены значения
vm3jX, Jlim и коэффициентов at в уравнении для предельной кривой диссоциации.
Колебательно-вращательные уровни состояния Х2?+ вычислялись по уравнениям A. 65) и A. 60). Значения
коэффициентов Ykl в этих уравнениях приведены в табл. 29.5. Коэффициент Yso определен из условий
.A. 50), коэффициент Г21 — из условий A. 57), а коэффициенты YQ3 и YOi — из условий A. 59).
Мультиплетность и вырождение колебательно-вращательных уровней всех электронных состояний
учитывались соответствующими статистическими весами.
Постоянные молекулы ВаН известны с удовлетворительной точностью, поэтому погрешности
вычисленных термодинамических функций при Т ^ 3000 К обусловлены главным образом
неточностью фундаментальных постоянных. При более высоких температурах становятся
существенными отсутствие экспериментальных данных об уровнях энергии состояния Х2?+ с v ^> 2Я
неточность постоянных в состоянии А'2А и пренебрежение рядом электронных состояний ВаН.
Общая погрешность в значениях Ф° (Т) при 298,15, 3000 и 6000 К может составлять 0,02, 0,05 и
0,3 Дж-К~1-моль соответственно.
Термодинамические функции ВаН (г) вычислялись ранее в справочнике Хара и др. [1336] (до
5000 К), работах Вейц и др. [63] (до 3500 К), Климова и др. [167] и Шнейдера [2463] (приведены
коэффициенты полиномов, аппроксимирующих значения Ф° (Г) и In QBX). Данные [63]
согласуются с приведенными в табл. 849 в пределах 0,7 и 2,5 Дж -К -моль в значениях Ф° (Т) и S° (T).
391
ВаН2 (к, ж) Глава 29
Расхождения с данными [1336] достигают 3, 9, и 12 Дж-К-моль" в значениях Ф° (Г), 5° (Г) и
Ср (Т) главным образом из-за пренебрежения в [1336] возбужденными состояниями ВаН.
Константа равновесия реакции ВаН (г)=Ва (г)+Н (г) вычислена на основании принятой в
справочнике энергии диссоциации)
@) = Do (ВаН) = 15 870 + 270 см да 189,8 + 3,2 кДж • моль.
Это значение найдено в результате анализа данных по предиссоциации молекул ВаН и BaD
в состоянии С2? (р=1, К=10 и 23 соответственно), полученных в работах Грудстрома и др. [1150а,
1296] и Коппа и др. [1756]. Погрешность учитывает невозможность построения предельной
кривой диссоциации состояния С2 ? по двум точкам и неопределенность того, с каким состоянием атома
Ва CZ>2 или 8DS) коррелирует состояние, вызывающее лредиссоциацию. Близкое значение
«45770 см) найдено в работе [1756] без учета данных по предиссоциации BaD. Принятому
значению соответствует
A^°(BaH,«r, 0) = 205,947 ± 4,4 кДж • моль.
ВаН2 (к, ж). Термодинамические свойства кристаллического и жидкого дигидрида бария
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 850.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. B9.1. В справочнике за стандартное состояние ВаН2(к) в интервале 0—871 К принята
ромбическая модификация [1362] (<*-ВаН2, структурный тип СаН2), а в интервале 871—1473 К —
Р-ВаН2 с неустановленной структурой. Экспериментальные данные о термодинамических
свойствах ВаН2 (к, ж) фактически отсутствуют. Приведенные в табл. 29.1 значения термодинамических
свойств при 298,15 К оценены при подготовке справочника. • Приведенные в табл. 29.1
значения 5° B98,15 К) и Я0 B98,15 К) — Я°@) имеют погрешности 6 Дж • К^-моль и
1 кДж • моль соответственно. Уравнение для теплоемкости а-ВаН„ в интервале 298,15—871 К
(см. табл. 29.1) выведено на основании значений С? B98,15 К) и С, (871 К) = 62 Дж-К^-моль;
последнее выбрано по значениям С°р в точке перехода для SrH2(K) и СаН2(к), которые в свою
очередь оценены. Погрешности величин, вычисляемых по этому уравнению, могут достигать 10%.
Значение Ср(|3-ВаН2) принято постоянным и равным 71 + 10 Дж-К^-моль.
Температура полиморфного превращения ВаН2 (871 +20 К) получена Питерсоном и Инди-
гом [2218] методом ДТА. Энтальпия полиморфного превращения E,6+1,0 кДж-моль) оценена
в предположении, что энтропии превращения ВаН2, SrH2 и СаН2 близки. Температура плавления
ВаН2 A473+50 К) найдена из данных по фазовой диаграмме, полученной в работе [2218].
Энтальпия плавления ВаНа B5,0 ±5,0 кДж-моль) оценена на основании предположения, что энтропии
плавления LiH и ВаН2 близки. Для теплоемкости жидкого ВаНа принято значение G5 +
+15 Дж-К-моль~1), полученное оценкой с учетом экспериментальных данных по теплоемкости
жидкого LiH [463, 2782]. '
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 6, 13 и
30 Дж-К-моль соответственно. Расхождения между термодинамическими функциями а-ВаН2,
приведенными в справочнике [578] и табл. 850, достигают 8 Дж-К-^моль^в значении Ф° (800 К),
ввиду того что в [578] на основании оценок приняты более низкие значения S° B98,15 К) и С°р (Т).
Таблицы термодинамических функций ?-ВаН2 и ВаН2 (ж) в справочной литературе отсутствуют.
Константа равновесия реакции ВаН2 (к, ж)=Ва (г)+2Н (г) вычислена на основании значения
Дг#° @) =795,603 +3,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
ДуГ (ВаН2, к, 298,15 К) = —190,1 + 0,5 кДж • моль.
Эта величина получена Воробьевым и др. [71, 72] измерением энтальпии реакции
гидрирования бария в герметичной калориметрической бомбе с электрическим подогревом образца. Менее
надежным является метод растворения бария и дигидрида бария в соляной кислоте,
использованный в работах Эрлиха и др. [1022] (—178 кДж-моль), Гунца и Бенуа [1325] (—171 кДж-моль)
и Гунца [1322] (—157 кДж-моль), поскольку в этих работах были использованы негерметичные
калориметры и выделяющийся водород мог вступить в побочную реакцию с материалом сетки (медь,
платина), в которой помещался образец (см. [69]). Исследование равновесия диссоциации ВаН2 (к>
[1035] привело к значению —128 кДж-моль"^ которое, очевидно, является неверным.
392
Барии и его соединения
ВаОН (г>
ВаОН (г). Термодинамические свойства газообразного гидроксида бария в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 851.
Молекулярные постоянные, использованные в расчете термодинамических функций ВаОН (г),
представлены в табл. 29.7. Экспериментальные данные о структуре и спектрах молекулы ВаОН
Таблица 29,7.' Значения молекулярных постоянных, а также а и р^, принятых для расчета
Молекула
термодинамических функции ВаОН, ВаОН+,
Состояние
ВаОНа . J?22+
ВаОН+ J^S*
Ва(ОНJ l^t
BaF2 iMj
ВаС12 1*Ах
ВаВг2 1>АХ
Ва13 КХАХ
v2
450 305 B)
420 305B)
430 70
440 80
260 45
155 30
120 25
ВаОН: а То (Л) = 11 000 см, pj=i; То (В)=12 000
ВаОН+: а приведено значение /-Ю39 г-см2.
CM
3700
3700
430
415
265
170
145
см, ps=2;
Ba(OHJ,
BaF2, BaCl2, BaBr2, Bal
v3
_
— _
3700 B) 305 D)
— —
— —
_ _
— —
г3 ¦ см6
12,9 б
14 а
5,3-103
6,8-Ю3
1,06-105
1.10е
4.10s
6 приведено значение /.1039 г-см2
C
1
1
2
2
2
2
2
PS
2
1
1
1
1
1 ',
1
отсутствуют, в связи с этим приведенные постоянные оценены при подготовке справочника. По
аналогии с ВеОН, MgOH и молекулами гидррксидов щелочных металлов в справочнике принимается,,
что в основном электронном состоянии .X'2 2+ молекула ВаОН имеет линейную конфигурацию.
Оценка геометрических параметров и частот ВаОН выполнена, так же как для молекулы ВеОН
(см. гл. 25). Приведенный в табл. 29.7 момент инерции соответствует значениям г (О—Н)=0,96 +
+0,02 А и г (Ва—О)=2,20+0,05 А, а его погрешность оценивается в 1-109 г(-см2. Погрешности,
принятых значений основных частот составляют 50—100 см. На основании данных Гейдона [1182],
исследовавшего электронный спектр ВаОН, и аналогии с изоэлектронной молекулой BaF в
справочнике принимается, что молекула ВаОН имеет два возбужденных электронных состояния.
Приведенные в таблице энергии этих состояний имеют погрешности, не превышающие 1000 см.
Термодинамические функции ВаОН (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)г
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных величин обусловлены неточностью принятых молекулярных постоянных, а также
приближенным методом расчета и составляют 2,4 и 6 Дж-К-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К.
Ранее термодинамические функции ВаОН (г) вычислялись в таблицах JANAF [1544];
расхождения данных [1544] и настоящего издания составляют 2,5—6 Дж-К-моль и обусловлены тем,,
что авторы таблиц JANAF приняли в расчете более высокое значение v2.
Константа равновесия реакции ВаОН (г)=Ва (г)+О (г)+Н (г) вычислена на основании
значения Д^0 @) =887,53+20 кДж-моль, соответствующего принятой в справочнике энтальпии;
образования
Д fH° (ВаОН, г, 0) = — 245 + 20 кДж • моль.
В табл. 29.8 представлены результаты исследований равновесия реакций с участием ВаОН (г);
погрешности приведенных значений энтальпий образования включают воспроизводимость
измерений и неточность величин, использованных при обработке экспериментальных данных. Наиболее
детальные измерения выполнены Гурвичем и др. [424, 1333]. ;В работе [1333] дополнительно
изучено равновесие реакции ВаС1(г)+Н2О (г)=ВаОН (г)+НС1 (г) по интенсивности полос ВаС1
и ВаОН в тех же пламенах, причем результаты, полученные для обеих реакций, практически
совпали. Данные [1333] подтверждены в работе Старовойтова [397] и удовлетворительно согласуются;
с найденными в работах [885, 2604]. Следует отметить, что масс-спектрометрические измерениа
393
ВаОН+ (г)
Глава 29
Таблица 29.8. Результаты определений Д/-Н"° (BaOH, г, 0) (в кДжмоль ')
Авторы
Метод
dfH° (ВаОН, г, 0)
II закон
III закон
Стаффорд, Берковиц, 1964 [2604] Масс-спектрометрическое исследование равновесия — —223
реакции 2ВаО (г)+Н20 (г)=2ВаОН (г)+О (г), 1785 К
То же, ВаО (к)+ВаО (г)+Н2О (г)=2ВаОН (г)+ — -201
+1/2О2 (г), 1755—1785 К, 2 точки
Ыьюбери, 1965 [2131] То же, ВаО И+'/Д (г)=ВаОН (г), 1851 К — —210
Коттон, Дженкинс, 1968 [885] СпектросЬотометрическое исследование равновесия — —257+20
реакцшГВа (г)+Н2О (г)=ВаОН (г)+Н (г) в
пламенах H»+O2+N2 на основании определения
концентраций Ва, 1570—1800 К, 2 точки
Гурвич и др., 1973 [424, 1333] Спектрофотометрическое исследование равновесия —245+20 —
реакции Ва (г)+НгО (г)=ВаОН (г)+Н (г) на
основании измерений интенсивности полос ВаОН и Ва
в пламенах H2+O2+N2, 1860—2580 К, 16 точек
Старовойтов, 1977 [397] То же, в пламенах CH4+O2+N2, 2370—2815 К, —240+20 —
6 точек
То же, в пламенах СН4+O2-j-N2 для реакции —225±20 —
ВаО (г)+ОН (г)=ВаОН (г)+О (г), по полосам ВаО
и ВаОН, 2180—2815 К, 8 точек
[2131, 2604] включают малое число точек, могут быть осложнены диссоциативной ионизацией
молекул Ва(ОНJ и неравновесными условиями в [2604]. Косвенные измерения в работах [57, 61, 357,
358, 1619, 1620, 2635] приводят к значениям от —240 до—257 кДж-моль, Принятое значение
основано на данных [1333].
ВаОН+ (г). Термодинамические свойства газообразного положительного иона гидроксида
бария в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 852.
Молекулярные постоянные, использованные в расчете термодинамических функций ВаОН+ (г),
представлены в табл. 29.7. В литературе отсутствуют сведения об исследованиях спектров и
структуры ВаОН+; приведенные постоянные оценены на основании постоянных изоэлектронной
молекулы CsOH и молекулы ВаОН. Приведенный в табл. 29.7 момент инерции в состоянии XXS+
соответствует значениям ^Ва—О—Н=180°, г (Ва—О)=2,25+0,10 А, г (О—Н) =0,96 +0,02 А; его
погрешность составляет 2-Ю9 г-см2, погрешности принятых основных частот оцениваются в 50—
100 см. Возбужденные электронные состояния ВаОН+, по-видимому, имеют энергии,
превышающие 20 000 см.
Термодинамические функции ВаОН+ (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 126) и A. 129) в приближении «жесткий ротэтор—гармонический осциллятор» без
учета возбужденных электронных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета и
могут достигать 4, 6 и 10 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Термодинамические функции ВаОН+ (г) ранее вычислялись в таблицах JANAF [1544]; расхож-
.дения данных [1544] и настоящего издания составляют 3—7 Дж -К" -моль и обусловлены разной
оценкой постоянных ВаОН+, главным образом величины v2.
Константа равновесия реакции ВэОН+ (г) +е (г)=Ва (г)+О (г)+Н (г) вычислена с
использованием значения Дг Н° @)=417,53 +36 кДж-моль, соответствующего принятому в
справочнике потенциалу ионизации
/0 (ВаОН) =470+30 кДж-моль.
Принятое значение основано на спектрофотометрических исследованиях в пламенах реакций
Ва (г)+ОН (г)=ВаОН+ (г)+е(г) [1564] D88+20кДж-моль^ иВа (ОНJ (г)+ЗН (г)=ВаОН+ (г)+
+Н2О (г)+Н2 (г)+е (г) [1664, 1666] E18+27 кДж-моль), а также измерениях методом
электронного ударе [2604] D34 +96 кДж-моль). Сопоставление потенциалов ионизации молекул фто-
394
Барий и его соединения Ва (ОН)а (к, ж), Ва(ОНJ (г)
ридов, хлоридов и гидроксидов Са, Sr и Ва показывает, что для ВэОН эта величина должна быть
близка к 470 кДж-моль в согласии с данными [1564] и средним арифметическим из трех
приведенных значений.
Принятому значению соответствует
Д^^ВаОИ*, г, 0) = 225 + 36 кДж • моль.
Ва(ОНJ (к, ж). Термодинамические свойства кристаллического и жидкого дигидроксида бария
в стандартном состоянии при температурах 298,15—3000 К приведены в табл. 853.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. В справочнике за стандартное состояние Ва(ОНJ (к) в интервале 0—519 К принята
низкотемпературная ромбическая ^-модификация, а при 519—681,5 К — высокотемпературная
ромбическая а-модификация 1785]. Экспериментальные данные о теплоемкости Ва(ОНJ при
Т <^ 298,15 К отсутствуют. Значения термодинамических функций при 298,15 К, приведенные
в таблице, оценены с помощью различных эмпирических методов и с учетом значения С B98,15 К),
найденного в работе [2262]. Погрешности значений S° B98,15 К) и Н° B98,15 К)—Н° @)
оцениваются в 10 Дж-К~1-моль~1 и 1 кДж-моль соответственно. При Т > 298,15 К для теплоемкости
|3-Ва(ОНJ принято уравнение (см. табл. 29.1), основанное на результатах измерений энтальпии
Ва(ОНJ (к) в работе Пауэрса и Блейлока [2262], погрешности этих измерений оцениваются
в ~0,5%. В работе [2262] не было замечено полиморфное превращение при 519 К,
установленное позднее Мишо [2016, 2017]. Энтальпия этого превращения 3,6+1,5 кДж-моль оценена при
подготовке справочника как разность между энтальпией плавления, измеренной методом
смешения в работе [2262] A9,2+1,0 кДж-моль), и энтальпией плавления, вычисленной из данных
по термическому анализу [2017] A6+1 кДж-моль). С последней величиной удовлетворительно
согласуется значение 14,2 кДж-моль, найденное Сьюардом [2497] по результатам термического
анализа образца Ва(ОНJ чистотой 99,3%. Температура полиморфного превращения E19+5 К)
принята по данным Мишо [2016, 2017], а температура плавления F81,5+2 К) — по данным
[2016, 2017, 2497]. В работах [564, 1381 ] получены менее точные значения Тт, равные 648 и 660 К.
Пауэре и Блейлок [2262] рекомендуют более высокое значение F90 К), погрешность которого
нельзя оценить, поскольку работа [2262] осталась неизвестна в оригинале. Теплоемкость
высокотемпературной а-модификации принята равной теплоемкости C-Ва(ОНJ в точке превращения,
а величина С (Ва(ОНJ, ж)=138+10 Дж-К-моль~1 основывается на измерениях энтальпии
Ва(ОНJ (ж) до 1181 К в работе [2262].
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 3000 К оцениваются в 10, 12
и 25 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
Ва(ОНJ (к), приведенными в справочнике [578] и в табл. 853, составляют 8 Дж-К-моль~1 в
значениях Ф° (Т) вследствие различия оценок величины S0 (Ва(ОНJ, к, 298,15 К); данные для
Ва(ОНJ (ж) публикуются впервые. >
Давление пара дигидроксида бария в реакции Ва(ОНJ (к, ж)=Ва@НJ (г) вычислено с
использованием значения АГН° @) = Д8#° (Ва(ОНJ, к, 0)=320,34+21 кДж-моль, соответствующего
принятым в справочнике энтальпиям образования Ва(ОНJ в кристаллическом и газообразном
состояниях.
Значение
Д/Я°(Ва(ОНJ, к, 298,15К) = —941,6 ±5,0 кДж • моль
основано на измерениях энтальпии растворения Ва(ОНJ (к) в соляной кислоте, выполненных
Щукаревым [466], и аналогичных измерениях с ВаО (к), выполненных Фицгиббоном и др. [1103].
Более старые калориметрические измерения [670, 672, 1118, 2686] приводят к достаточно близким
значениям (—935 кДж-моль [1118]). В хорошем соответствии с принятой величиной находятся
результаты исследований равновесия реакции Ва(ОНJ (к)=ВаО (к)+Н2О (г), проведенных
статическим методом Джонстоном [1592] (918—1203 К, 11 точек, —953+14 кДж-моль по II и
—940+13 кДж-моль по III закону) и Тамару, Сиоми [2654] G80—1018 К, 7 точек, —937 +
+10 кДж-моль по II и —945+10 кДж-моль по III закону).
Ва(ОНJ (г). Термодинамические свойства газообразного дигидроксида бария в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 854.
395
Ва(ОНJ (г)
Глава 2»
Молекулярные постоянные, использованные в расчете термодинамических функций Ва(ОНJг
представлены в табл. 29.7. Сведения об исследованиях спектров и структуры молекулы Ва(ОН)г
отсутствуют, поэтому приведенные молекулярные постоянные оценены при подготовке
справочника на основании принятых постоянных молекул BaF3 и ВаОН. Произведение моментов инерции
в основном электронном состоянии вычислено по структурным параметрам ДО—Ва—0=110+10°,.
г (Ва—О)=2,30+0,10 А (оценены из BaF2), i?Ba—О—Н=180°, г (О—Н)=0,96+0,02 А
(перенесены из ВаОН), его погрешность составляет 2-10~114 г3- см6. Частоты vl7 v2) v3 фрагмента О—Ва—О'
оценены по частотам молекулы BaF2, частоты v4 и v. фрагментов Ва—О—Н перенесены из ВаОН.
Погрешность принятого значения v2 оценивается в 15 см, остальных частот — в 50—100 см.
Сведения о возбужденных состояниях молекулы Ва(ОНJ отсутствуют; можно предполагать, что
их энергии превышают 15 000—20 000 см.
Термодинамические функции Ва(ОНJ (г) вычислены по уравнениям A. 3)—A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор» без
учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным методом расчета и составляют 4, 8 и 12 Дж-К~1-моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К г.
Термодинамические функции Ва(ОНJ (г) ранее вычислялись в таблицах JANAF [1544];
расхождения данных [1544] и настоящего издания составляют 3—8 Дж-К~1-моль~1 и обусловлены
разной оценкой молекулярных постоянных Ва(ОНJ.
Константа равновесия реакции Ва(ОНJ (г)=Ва (г)+2О (г)+2Н (г) вычислена с
использованием значения Ь.ГН° @)=1720,347+20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
ДуЕГ" (Ва (ОНJ, г, 0) = —615 + 20 кДж • моль.
Принятая величина основана на результатах измерений, приведенных в табл. 29.9. Укаган-
ные в таблице погрешности учитывают воспроизводимость измерений и неточность
использованных в расчетах термохимических величин и сечений ионизации (для работ [2131, 2604]). Некоторые
методические недостатки измерений в работе [2604] отмечены Ньюбери [2131]. Наиболее деталь-
Таблица 29.9. Результаты определений
(Ва(ОНJ, г, 0) (в кДжмоль"
Авторы
Метод
Д/Я°(Ва(ОНJ, г, 0)
II закон
III закон
Стаф*орд, Берковиц, 1964 [2604] Масс-спектрометрический, ВаО (к)+Н20 (г)= —545+25 —581,0+7,3
=Ва(ОНJ (г), 1485—1727 К, 23 точки, ДГЯ° @)=
=204,6±1.6 (III)
То же, 1755-1800 К, 3 точки, ДгЯ°@)=211+ — —574+13
±11 (III)
Ньюбери, 1965 [2131] То же, 1428—1899 К, 44 точки, пять серий изме- —621+7 —606,2±7,3
рений, ДгЯ°@)=179,4+0,3 (III)
Коттон, Дженкинс, 1968 [885] Фотометрия в иламенах H2+O,+ N,, Ва (г)+ — —625±10
+2Н,0 (г)=Ва(ОНJ (г)+2Н (г),' 1570-1800 К,
2 точки, ДГЯ° @)=105±10 (III)
Рябова и др., 1977 [359] То же, 1860—2580 К, 16 точек, ДГЯ° @)= —561 ±25 —623±6
= 107,3±4,7 (III)
Старовойтовидр.,1977 [397, 399] То же, пламя СН4 с воздухом, 2370—2460 К, — —588±15
2 точки, ДГЯ° @)=142,fi±15 (III)j
То же, ВаО (г)+Н,0 (г)=Ва(ОНJ (г), 2180- — —576±6
2460 К, 4 точки, \Н° @)=—214,8±4,4 (III)
1 Исследования равновесия реакций с участием Ва(ОНJ(г) позволяют вычислить значения S°(T) по II закону.
Наиболее точные измерения, выполненные Ньюбери [2131] (см. табл. 29.9) приводят к значению 5° A712 К)=>
=471 ±3|Дж-К~1-моль'~1, вто время как данным табл. 854 соответствует 479+8 Дж-К'^моль. Хотя
расхождения находятся в пределах погрешностей, возможно, что, так же как для дигидроксидов других элементов под»
группы ПА, они свидетельствуют о неточности оценки постоянных этой молекулы,
396
Барий и его соединения BaF (r)
яые измерения выполнены в работах Ньюбери [2131 ] и Рябовой и др. [359], с которыми
удовлетворительно согласуются данные [885]. Принятое значение основано на данных [359, 885 и 2131],
•его погрешность учитывает неточность термодинамических функций Ва(ОНJ (г).
BaF (г). Термодинамические свойства газообразного фторида бария в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 855.
Молекулярные постоянные BaF, принятые в справочнике на основании результатов
исследований электронных спектров этой молекулы, приведены в табл. 29.10. В спектре BaF наблюдались
10 систем полос, связанных с переходами между 11 электронными состояниями: АШ ** X2 ?+
[591, 1558, 2130], S2?+<*X22+ [591, 928, 1192, 1558, 1584, 2130, 2526, 2795], С2П«*Х2?+
J591, 1192, 1558, 1584, 1786, 1787, 2039, 2526, 2795], ZJ 2-^ X2 2+ [591, 928, 1558, 2526,2527],
Я'аЕ+**Х8Е+ [558, 591, 928, 2526, 2527], ?22+^X22+ [591, 1126, 2526, 2527], F2 ?+<* X2 ?+
[1126, 2526, 2527], G2E+^X22+ [1126, 2526, 2527], Я22+:<±Х22+ [1126, 2526, 2527], /2Е+->
-»-Х2 Е+ [2526, 2527]. Так же как у м.олекул других галогенидов щелочноземельных металлов,
состояния X2 Е+ и А2П. молекулы BaF коррелируют с основными состояниями атомов х5-(-2Р. Со
следующими диссоциационными пределами 3D+2P и W+2P должно коррелировать большое число
состояний, в том числе дублетные и квартетные состояния, по крайней мере часть этих состояний может
5ыть связанными состояниями. В то же время большая часть известных дублетных состояний
(D2 2, D'2 2 и последующие) имеет ридберговский характер, что не исключает возможность их
корреляции с валентными состояниями атомов. Поэтому в табл. 29.10 включены все известные
состояния BaF.
Колебательные постоянные BaF в основном состоянии определялись из анализа систем полос
АШ—Х22+ [1558, 2130], JS2?+—Х22+ [1558, 1584, 2130, 2526], С2П—Х22+ [1558, 1584, 1786,
1787, 2526], #22+—Х22+ [1558, 2526, 2527], Z>'22+—Х22+ [2526, 2527], ?2Е+—Х22+ [1126,
2526, 2527], /?22+-Х22+ [1126, 2526, 2527], G22+-X2?+ [1126, 2526, 2527] и #22+-Х22+ [Ц26,
2526, 2527]. Приведенные в табл. 29.10 колебательные постоянные в состоянии Х22+ найдены
Кушаваха [1786] по началам полос системы С21Ь/г—Х22+ (у" ^ 18), которые были рассчитаны
по измеренным кантам полос с помощью вращательных постоянных. Вращательные постоянные,
принятые в справочнике, основаны на результатах анализа полосы 0—0 в системах А2И—X2 Е+,
В22+—Х22+, D2E+—Х22+, ?2Е+-Х22+ и полосы 1—0 системы D'22+—Х22+ в работе Барроу
и др. [591] и оценке величины В'[ по разности Д1кант—QlKinT в полосах 0—0, 0—1 и 1—1 системы
•С2П.,2—Х22+ в работе Кушаваха [1786]. Постоянные BaF в состояниях А2И, ?2?+, Z>2?+,
?>'22+ и ?22+ приняты по данным [591, 1558], в состояниях С2П, F2 2+, GaE+, H2 2+ по работам
Кушаваха [1786], Фаулера [11263 и Сингха и Мохана [2527].
Термодинамические функции BaF (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения <JBB и ее производные рассчитывались по уравнениям A. 90)—A.92) с учетом 10
возбужденных состояний в предположении, что Q^,l.B9 = (pJpx)Qifl,-B9- Величина ^1риеепроизводные
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82).
В расчетах учитывались все уровни энергии с значениями / ^ -Лих, »> гДе «Ллах * находились из
условий A.81). Колебательно-вращательные уровни состояния X2 2+ вычислялись по уравне
ниям A. 65), A. 61). Значения коэффициентов Ykl, а также величин vmax и /нт, использованные
в расчетах, приведены в табл. 29.11. Коэффициент Y03 получен из условий A. 59), а У21 — из
условия Вр +1=0. Мультиплетность и вырождение колебательно-вращательных уровней
электронных состояний учитывались статистическими весами.
Погрешности рассчитанных термодинамических функций при Т ^ 2000 К обусловлены
главным образом невысокой точностью принятых постоянных BaF в состоянии X2 ?+, а при более
высоких температурах неполнотой данных о возбужденных электронных состояниях этой молекулы.
Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 0,03, 0,3 и 1,0 Дж-К-1Х
Хмель соответственно.
Ранее термодинамические функции BaF (г) вычислялись в таблицах JANAF[1544],
справочнике [305] (до 5000 К) и работе Климова и др. [167] (приведен полином, аппроксимирующий
значения Ф° (Т) для Т ^ 5000 К). Расхождения данных [1544] и табл. 855 растут с температурой,
но не превышают 0,3 и 1 Дж-К-моль~1 в значениях Ф° (Т) и S° (T), они обусловлены только
различием методов расчета. Остальные расчеты выполнены без учета возбужденных состояний
и по-грубым методикам, соответствующие расхождения в значениях Ф° (Т) и S° (T) превышают 1
и 5 Дж-К-моль~1.
397
BaF (г)
Глава 29
Таблица 29.10. Молекулярные постоянные BaF, BaF+, Ва35С1, ВаС1+, Ва7эВг и Bal
Соединение
Состояние
138Ba19F X2 2+
А2Щ2
В22+2
с2п1/з
с2п328
В2 2+
Z)'22+
\Е22+
F2 2+
' G2 2+
Я2 2+
BaF+ X12+
138Ваз5С1 х22+
A2tl
A2U%
В2 2+
с2п1/2
D22+
Я2 2+
Я 2+
G22+
BaGl+ XJ2+
138Ва7 9Вг X2 2+
л2п1/2
л2п3/2
В2 2+
с2п1/г
с2гц
В% 2+2
Я2 2+
138Ва1 Х22+
Л2П,,
а2п3/2
В2Е+2
с2п1/г
С2Пз/2
Л?22+2
?2Х+
Те
0
И 647,47
12 278,8
14 063,1
19 993,72
20194
24 157,4
26 223,5
28 140,33
29 412,2
31 452,6
31 584,7
0
0
10 363
10 885,3
И 880,0
19 062,9
19 450,1
25 471,5
27 064,6
29 493,6
32 511,4
0
0
9 980Д
10 604 Д
И 325 Д
18 650,9
19 192,5
25 670,9
26 865,9
0
9 268Д
9 921Д
10 417 Д
17 819,5
18 572,6
25 777,4
26 764,5
Ше
470,07
435,5
436,7
, 424,4
457,91
508,4
504,9
538,4
529,9
510,4
508,8
540 а
279,3
256,35
255,25
280,2
285,0
304,6
311,5
331,8
331,3
340 а
193,8
—
—
—
197,9
196,9
209,1
219,9
151,4
—
—
156,88
156,82
160,81
177,0
ве
СМ
1,717 а 0,2164
1,68 0,2124б
1,82 0,2125б
1,88 0,2077б
1,559 Д 0,2136
1,88 0,2279б
1,54 0,2283б
1,90 0,2295
2,00 —
0,83 —
2,00 —
1,4б 0,229 в
0,89 0,0799 а
0,73 _
0,83 —
0,79 —
0,79 —
1,04 —
0,93 —
1,30 —
1,29 —
0,74 б 0,082 в
0,42 0,0386а
— —
— —
— —
0,41 —
0,41 —
0,53 —
0,35 —
0,46 0,0258а
— —
— —
0,48 —
0,31 —
0,31 -
0,65 —
ах ¦ 10*
12
12 в
12 в
12 в
12
12 в
10 в
И в
—.
¦ —
—
8,7 г
3,2 б
—
—
—
—
—
—
—
—
2,4 г
1,1 б
—
—
—
—
—
—
—
0,85 б
—
—
—
—
—
—
#е • Ю8
17,5
20 г
20 г
19
19 г
17 г
18 г
17 г
—
—
—
16 Д
2,6 в
—
—
—
—
—
—
—
1,9Д
0,61 в
—
—
—
—
—
—
—
0,3 в
—
—
—
—
—
А
2,1599
2,1802
2,1797
2,2047
2,1741
2,1047
2,1030
2,097
—
—
—
2,10 а
2,75 г
—
—
—
—
—
—
—
—
2,70 а
2,95 г
—
—
—
—
—
—
—
3,15 г
— ч
—
—
—
—
398
Барий и его соединения
BaF (г)!
Таблица 29.10. (окончание)
Примечание.
BaF: a
Ниже все величины даны в см *.
уе=—0,03297, швг(!=0,009357; *> вычислено по значениям Во и
рассчитано по
соотношению A.69); Г рассчитано по соотношению A.68); Д и>еуе= —0,03689, <1>егв=0,0016.
BaF+: а оценка; б вычислено по соотношению (.1.67) и значению Do=51 200; в вычислено по
соотношению A.38); Г вычислено по соотношению A.69); Д вычислено по соотношению A.68),
BaCl: а вычислено по соотношению A.38); ° вычислено по соотношению A.69); в вычислено по
соотношению A.68); г оценка.
ВаС1+: а оценка; б вычислено по соотношению A.67) и значению Do=38 700; в вычислено по
соотношению A.38); г вычислено по соотношению A.69); Д вычислено по соотношению A.68).
BaBr, Bal: а вычислено по соотношению A.38); б вычислено по соотношению A.69);
¦ нию A.68); г оценка; Д приведено значение То.
вычислено по соотноше-
Таблица 29.11. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см'1),
а также значения г>Шах и J\im, принятые для расчета термодинамических функций BaF,
BaF+, BaCl, BaCI+, BaBr, Bal
Коэффициенты
г.. ю-*
Ую-Ю-3
Узо-Ю2
У40-Ю*
Убо-Ю6
Убо-Ю9
Уох-Ю1
Уп-102
У21-Ю*
Уо2-10'
Уоз-Ю14
Ушах
Примечание. Ниже
BaF: a Состояние
Те
Состояние
Те
BaCl: a Состояние
Те
BaBr. a Состояние
Те
ВаГ. а Состояние
Те
BaF
Z22+a
0
0,470 824
—0,218 060
1,348 68
0,767 082
0,214 410
-0,219 208
2,164
—0,12
0,016 4
—1,75
2,807
311
718
BaF+
ХЧ+
0
0,540
-0,14
—
—
—
—
2,29
—0,087
—
-1,6
—
188
631
BaCl
X2S+a
0
0,277 483 '
—0,083 196
—0,181 834
0,224 926
—0,042 871
—
0,791
—0,031 5
0,001 67
—0,255
0,846
293
930
все постоянные даны п см.
А2Щ
И 647,5
F22+
29 412,2
А2Щ
10 363
А2Щ
9980
АЧЧ
9268
А2Щ2
12 278,8
G2S+
31 452,6
А2Щ
10 885,3
А2Щ
10 604
А2ПЧ
99212
В2 Z+ С2П
14 063,1 20 100
Я2 2+
31 584,7
в2 s+ cm
11 880 19 257
в2 х+ cm
11 325 18 922
В22+ С2П
10 417 18 196
ВаС1+
ХЧ+
0
0,340
—0,074
—
—
—
—
0,820
—0,024
—
—0,19
—
229
916
?22+
24 157,4
?>22+
25 471,5
D2 Б+
25 670,9
25 777,4
BaBr
0
0,193 955
—0,043 6544
0,041 219
—0,001 444
_
—
0,386
-0,011
0,000 540
—0,061
—0,082
439'
1180
D '2 2+ jp
26 223,5 28
Е2г+ f
27 064,6 29
?2 2+
26 865,9
/?2 2+
26 764,5
Bal
*2S+.a
0
0,151 423
-0,046 913
0,091 646
—0,007 746
,
0,258
—0,008 5
0,000 630
-0,030
0.053
459
1303
'2 2+
140,3
'2 s+ G2 2+
493,6 32 511,4
399
BaF+ (г) Глава 9
Константа равновесия реакции BaF (r)=Ba (r)+F (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (BaF) = 580 ±[6 кДж • моль да 48 500 + 500 см.
Это значение основано на результатах масс-спектрометрических исследований равновесия
реакции Ва (r)+AlF (г)=А1 (r)+BaF (г) в работе Элерта и др. [1020] A175—1255 К, 8 точек) и
реакций BaF2 (г)+Ва (r)=2BaF (г) и 2Ва (r)+BF3 (r)=BF (r)+2BaF (г) в работе Хилденбранда [1406]
A418—1512 К, 8 точек для каждой реакции). Обработка этих данных по уравнению II закона
приводит к значениям 563 +13, 573 +13 и 573 +45 кДж- моль, а по уравнению III закона — к
значениям 567+7, 577 ±6 и 584+6 кДж-моль. Приведенные погрешности учитывают
воспроизводимость измерений, неточность сечений ионизации, термодинамических функций и термохимических
величин, использованных в расчетах. В работе [1020] изучено также равновесие реакции BaF2 (к)+
+Ba(r)=2BaF (г); полученным данным соответствует более низкое значение энергии
диссоциации E14 ±24 и 561+6 кДж-моль), по-видимому, из-за понижения активности BaF2 (к) в
условиях эксперимента (испарялась смесь BaF2+Al). В работе Гурвича и Рябовой [102] при спектро-
фотометрическом исследовании в пламенах получено существенно более высокое значение
{625 кДж-моль) из-за того, что при обработке экспериментальных данных авторы не учли
образование молекул BaOHF (см. [1333]).
Принятое значение подтверждается результатами изучения хемилюминесценции BaF при
образовании этой молекулы в скрещенных пучках (^=572 кДж-моль [10336]). Принятой величине
соответствует
° (BaF, г, 0) = —323,012 + 6,7 кДж • моль.
BaF+ (г). Термодинамические свойства газообразного положительного иона фторида бария
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 856.
В табл. 29.10 приведены молекулярные постоянные BaF+, принятые из-за отсутствия
экспериментальных данных на основании оценок. Основным электронным состоянием BaF+, как у изо-
электронных молекул CsF и ВаО, должно быть состояние XXS+ с конфигурацией . . . а2ц4.
Вращательная постоянная, приведенная в табл. 29.10, вычислена по значению гв (Ва—F)=2,10+.0,05 A,
оцененному сопоставлением межатомных расстояний в молекулах CaF, CaF+ и BaF. Из сравнения
частот колебаний в тех же молекулах принято значение <ое, которое хорошо согласуется с
частотами колебаний молекулы BaF в ридберговских состояниях. Погрешности принятых постоянных
уцениваются в 0,02 и 50 см соответственно. В справочнике принимается, что также, как у
молекулы GaF+, состояния, принадлежащие возбужденным конфигурациям . . . o27i3a, . . .an4o
и . . .а2тс3тс, должны иметь у BaF+ энергии около 20 000 см или выше.
Термодинамические функции BaF+ (г) рассчитаны по уравнениям A. 3)— A. 10), A. 93)—
A. 95) без!учета возбужденных электронных состояний. Значения QU?> вр и ее производных
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В расчетах учитывались
все колебательно-вращательные уровни энергии со значениями / ^/шах> „, причем последние
находились по формуле A. 81). Колебательно-вращательные уровни состояния Z1S+ вычислялись
по уравнениям A. 65) и A. 62). Значения коэффициентов Ykl в этих уравнениях, идентичные
соответствующим постоянным из табл. 29.10, а также величины i?max и /цш, приведены в табл. 29.11.
Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
постоянных BaF+ в состоянии Х1Б+, а также пренебрежением возбужденными электронными
состояниями; погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,5, 1 и
3 Дж-К-моль~1 соответственно.
Ранее термодинамические функции BaF+(г) вычислялись в таблицах JANAF [1544] по
постоянным, близким к принятым в настоящем справочнике, но с неоправданным учетом большого
числа возбужденных электронных состояний с низкими энергиями. Расхождения данных [1544]
и приведенных в табл. 856, относительно небольшие при низких температурах, быстро растут,
достигая 6 и6 Дж-К-моль в значениях Ф° F000 К) и S° F000 К) соответственно.
Константа равновесия реакции BaF+ (т)-\-е (г)=Ва (r)+F (г) вычислена на основании
значения &ГН° @)=110,0+31 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0 (BaF)=470+30 кДж-моль.
400
Барий и его соединения BaFa (к« ж)
Эта величина является средней из найденных методом электронного удара в работах Элерта
и др. [1020] D73+30 кДж-моль") и Хилденбранда [1406] D63+30 кДж-моль). Ей
соответствуют
A/#°(BaF+, г, 0) = 146,988 + 31 кДж • моль,
Do (BaF+) = 612,852.+|31|кДж • моль «* 51J200 + 2600 см".
BaF.2 (к, ж). Термодинамические свойства кристаллического и жидкого дифторида бария
в стандартном состоянии в интервале температур 100—4300 К приведены в табл. 857.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. В справочнике за стандартное состояние BaF2 (к) в интервале 0—1240 К принята
кубическая а-модификация BaF2 (структурный тип GaF2), в интервале 1240—1480 К — разупоря-
доченная р-модификация и в интервале 1480—1615 К — у-модификация с неизвестной
структурой. При Т < 298,15 К термодинамические функции вычислены по данным о теплоемкости BaF2,
полученным Питцером и др. [2233] A3—300 К) на образце, содержавшем 99,7% BaF2.
Экстраполяция теплоемкости приводит к Sa A3,34 К)=0,25 Дж-К~1-моль~1 [2233]. Погрешности
принятых по работе [2233] значений S° B98,15 К) и Н° B98,15 К)-#° @) оцениваются в 0,4 Дж-К^Х
Хмоль и 0,05 кДж-моль соответственно. При Т > 298,15 К измерения энтальпии BaF2 (к)
проведены Ефремовой и Матизеном [127] G57—1631 К) и Крестовниковым и др. [202] E73—
1273 К). Измерения [202] ненадежны и не учитывались, поскольку они приводят к существенно
более низким величинам, чем данные [127] (на 3—4% при Т <^ 1200 К), и не отражают
аномального'изменения теплоемкости, связанного с фазовым переходом II рода. В работе Ефремовой и
Матизена [127] на основании 37 измерений энтальпии трех модификаций BaF2 (к), проведенных
с погрешностями, не превышающими 0,5%, установлено наличие А-кривой теплоемкости в области
фазового перехода второго рода A100—1240 К) и получены уравнения, описывающие
теплоемкость В- и "f-модификаций. В справочнике принята упрощенная трактовка данных [127]. Для
теплоемкости a-BaF2 в интервале 298,15—1240 К получено трехчленное уравнение, не
отражающее ^-зависимости; для C-BaF2 и T-BaF2 приняты постоянные значения теплоемкости (см.
табл. 29.1). Эти величины согласованы со значениями энтальпий превращений: «эффективной»
энтальпией фазового перехода второго рода E,49 кДж-моль), отнесенной к температуре
максимума кривой теплоемкости 1240 К, и энтальпией изотермического перехода при 1480+5 К [127]
B,55 кДж-моль).
Температура плавления A641+10 К) также принята по работе [127], где она была получена
в измерениях с молибденовыми тиглями. Данные других авторов [1004, 1358, 1747, 2219],
приводящие к более низким значениям A593—1628 К), найдены при использовании тиглей из графита
и тантала, которые взаимодействуют с BaF2 уже в области 1400 К. Энтальпия плавления A7,8 +
+0,60 кДж-моль) и С° (BaF2, ж)=105+2 Дж-К-моль~1 приняты на основании измерений
энтальпии BaF2 (ж) в работе Ефремовой и Матизена [127] A752—2126 К). Ивамото вГдрГЧшГ]
методом высокотемпературной криометрии нашли близкое значение &тН, равное 19,1 кДж-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются
в 0,4, 1, 2 и 7 Дж- К- моль соответственно. Расхождения между термодинамическими функциями
BaF2 (к, ж), приведенными в таблицах JANAF [1544] и настоящем издании, не превышают
1 Дж-К^-моль.
В справочнике принята энтальпия образования
^"(BaFj,, к, 298,15К) = —1194 ± 10 кДж • моль.
Эта величина основывается на результатах измерений, приведенных в табл. 29.12, где в
качестве погрешностей дана воспроизводимость соответствующих данных. Следует отметить, что
величины, полученные Барань и др. [576] в калориметрическом исследовании и Талиповым,
Хадеевым [402] и Бутом, Бидвеллом [723] (см. [425]) при измерении растворимости BaF2 (к) 1
отличаются больше, чем это может быть объяснено приписанными им погрешностями. Это
расхождение свидетельствует о несогласованности использованного при обработке данных [576]
значения энтальпии образования ВаО (к) и значений энтальпии образования и энтропии Ва+2 (aq)
(см. Приложение 5), которые входят в расчеты по данным [402, 425, 723]. Принятое значение ос-
Измерения растворимости BaF2 (к) в воде в работах Кольрауша [1743] и Картера [826] подтверждают эти данные.
401
BaFs (г)
Глава 29
Таблица 29.12. Результаты определений АуН° (BaF2, к, 298,15 К) (в кДжмоль
Авторы
Гущ, 1884 [1328]
Петерсен. 1889 [2212]
Барань и др., 1957 [576]
Доманж, 1937 [971]
Худ, Войский, 1951 [1476]
Талипов, Хадеев, 1950 [402]
Бут, Бидвелл, 1950 [723]
Метод
Калориметрическое измерение энтальпии реакции
Ва(ОНJ (р-р, 660H2O)+2HF (р-р, 110Н,О)=
=BaF2 (к)+2Н2О (р-р)
То же, ВаС13 H)+2AgF (ag)=BaF2 (K)+2AgCl (к)
То же, Ва(ОНJ (a?)+2HF (ag)=BaF2 (к)+2Н2О (ж)
То же, BaO (k)+2HF (р-р, 4,444H2O)=BaF2 (к)+
+Н2О (р-р)
Протока, BaF2 (к)+Н2О (г)=ВаО (k)+2HF (г),
1173—1373 К, 3 точки
То же, ВаС12 (k)+2HF (r)=BaF2 (к)+2НС1 (г), 477-
1033 К, 9 точек
Измерение растворимости BaF2 (к) в воде при
298,15 К
То же, 473—668 К, экстраполяция к 298,15 К
проведена Ходаковским и др. [425]
ДуЯ° (BaF2) к, 298,15 К)
-1200
—1199
-1192
—1213,0±3,0
-1023+40 (II)
—1116±20 (III)
—1156+8 (II)
—1157±8 (III)
—1194,8±3,0 (III)
—1194,2±3,0 (III)
новано на измерениях растворимости, так как при этом обеспечивается его согласованность с вн-
тальпией образования Ва+2 (aq) — основной ключевой величиной для соединений бария.
Давление пара дифторида бария BaF2 (к, jK)=BaF2 (г) вычислено с использованием
принятой в справочнике энтальпии сублимации
ДД° (BaF2, к, 0) = 395 + 5 кДж • моль.
Эта величина принята на основании результатов измерений Хилденбранда и др. [1422] и Харта
и Сирей [1358], приведенных в табл. 29.13, где в качестве погрешностей дана только
воспроизводимость измерений. Погрешности из-за неточности термодинамических функций BaF2 (к, ж)
Таблица 29.13. Результаты определений А.гШ° (BaF2, к, 0) (в кДж-моль)
Авторы
Метод
Руфф, Ле Буше, 1934 [2407] Точек кипения, 1960—2206 К, 9 точек
Грин и др., 1964 [1245] Масс-спектрометрический, 1207—1563 К, 20 точек
Баутиста, Маргрейв, 1965 [629] Испарения с поверхности, 1130—1250 К, 10 точек
Хилденбранд и др., 1965 [1422] Торзионный, 1265—1550 К, расчет по уравнению
Харт, Сирей, 1966 [1358] То же, 1261—1548 К, 46 точек
Торзионный, вариант метода Люнгмюра, 1315—
1492 К, 25 точек
\Н° (BaF2, к, 0)
II закон
445±16
406±7
400±20
396
379±7
391 ±10
III закон
395±1,4
407,8+5
406,1 ±0,5
393,3±0,5
399,1 ±0,5
400,6±0,4
и BaF2 (г) составляют —7 кДж-моль для данных [2407] и 3—5 кДж-моль для остальных
работ. При выборе принятого значения не учитывались работа [1245], так как неточности в сечениях
ионизации приводят к большой дополнительной погрешности этих данных, и работа [629], в
которой заниженное давление пара BaF2 (к) могло быть получено вследствие отличия коэффициента
испарения от единицы. Погрешность принятого значения определяется в основном неточностью
термодинамических функций BaF2 (к, ж) и BaF2 (г).
BaF2 (г). Термодинамические свойства газообразного дифторида бария в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 858.
Молекулярные постоянные, использованные для расчета термодинамических функций BaF2,
приведены в табл. 29.7. Исследования инфракрасного спектра [23, 24, 808, 2565 ] и отклонения мо-
402
Варил и его соединения ВаС1 (г)
лекулярного пучка в неоднородном электрическом поле [783, 2839] показали, что молекула BaF2
в основном электронном состоянии ХхАг имеет нелинейное строение (симметрия С2в).
Приведенному в табл. 29.7 произведению моментов инерции соответствуют оцененная величина Д F—Ва—F=
=100+10° и г (Ва—F)=2,32+0,03 А, найденное Акишиным и др. [5, 15] при электронографиче-
ском исследовании паров BaF2. Значение ДР—Ва—F оценено Колдером и др. [808] по
зависимости величины валентного угла в ряду молекул MF2 (M=Be, Mg, Ca, Sr, Ва) от массы атома М.
Оценки этой величины в работах [23, 2839] A15 и 120°) представляются менее достоверными.
Погрешность принятого значения IaIbIc может достигать 2-Ю14 г3-см6. Приведенное в табл. 29.7
значение v3 найдено Байковым [24] в инфракрасном спектре паров BaF2. Для не наблюдавшейся
в спектре паров BaF2 частоты \ принято значение, превышающее на 25 см частоту v3, так как
примерно такая разница между частотами \ и v3 наблюдалась Снелсоном [2565] в инфракрасном
спектре молекул BaF2, изолированных в матрицах из Ne, Ar и Кг. Отнесение частот \ и v3
выполнено Снелсоном на основании их интенсивностей. Колдер и др. [808] при исследовании ИК-спектра
молекул BaF2, изолированных в матрице из Кг, сделали противоположное отнесение этих полос,
предполагая, что частота v3 должна быть больше vx. Для частоты v2 в табл. 29.7 приведено
значение, промежуточное между найденным Байковым в ИК-спектре пара [23] A00 см) и оцененным
Колдером и др. [808] на основании силовой постоянной /О=0,02-105 дин-см F4 см); такой
выбор обусловлен тем, что Банков наблюдал только высокочастотную часть полосы v2.
Погрешности принятых значений частот оцениваются в 20 см.
Сведения о возбужденных электронных состояниях молекулы BaF2 отсутствуют, можно
предполагать, что их энергии превышают 20 000 см.
Термодинамические функции BaF2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных величин
обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета; они составляют 2, 4 и 6 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000 и
6000 К соответственно (см. примеч. на с. 329).
Ранее термодинамические функции BaF2 (г) рассчитывались в таблицах JANAF [1544],
справочнике [305], работах Байкова [23] (Т <; 5000 К), Брюэра и др. [748] (Т <; 2500 К), Краснова
и Светцова [195] (Т ^ 2500 К). Расхождения между результатами расчетов [23, 305, 1544]
и данными табл. 858 не превышают 2—3 Дж'К~1-моль~1 в значениях Ф° (Т). В работах [195,
748] для молекул BaF2 принята линейная структура и расхождения с табл. 858 достигают 18—
20 Дж-К-^моль в значениях Ф° (Т).
Константа равновесия реакции BaF2 (r)=Ba (r)+2F (г) вычислена с использованием
значения b.JH0 @) = 1131,968+11 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации BaF2 (к). Этим величинам также соответствует
bfH°\(BaF2, г, 0) = —797,705 + И кДж • моль.
ВаС1 (г). Термодинамические свойства газообразного хлорида бария в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 859.
Молекулярные постоянные ВаС1, принятые в справочнике на основании результатов
исследований электронных спектров этой молекулы, приведены в табл. 29.10. В спектрах ВаС1
наблюдались семь'^систем полос, связанных с переходами между восемью электронными состояниями*
42П 2 X2S+ [595, 733, 1354, 2645], ?2?+-* Х2? + [595, 733, 2645], С2П <* Х2Е+ [1354, 1378,
1709, 2159, 2183, 2795], ?>2?+ ^ Х2Е+, ?2?+ *± Х2Е+ [1354, 2183], F2Z+ <- Х2Е+, G2E+ <- Х2Е+
[1354]. Так же как у молекул других галогенидов щелочноземельных металлов, состояния Х2Е +
и А2И молекулы ВаС1 коррелируют с основными состояниями атомов Ва A6')+С1 BР). Со
следующими диссоциационными пределами SD+*P, W+2P и SP+*P должно коррелировать большое
число валентных состояний ВаС1, в том числе квартетные состояния. По крайней мере часть не-
наблюдавшихся состояний может быть связанными состояниями, в то время как некоторые
известные дублетные состояния ф2?, Я2? и другие) имеют ридберговский характер (см. выше
CaF и BaF). -
Колебательные постоянные ВаС1 в состоянии Х2Е + определялись по кантам полос из анализа
разных систем и удовлетворительно согласуются между собой. Приведенные в табл. 29.10
колебательные постоянные в состоянии Х2? + получены Харрингтоном [1354]. Анализ вращательной
структуры спектров ВаС1 не проводился. Приведенное в табл. 29.10 значение г (ВаС1)~2,754"=-
403
ВаС1+ (г) _______ Глава 29
+0,05 А оценено на основании величины г (Ва—G1) в молекуле ВаС12 и сопоставления величин
г, (MX) и г (М—X) в молекулах галогенидов и дигалогенидов щелочноземельных металлов.
Колебательные постоянные Bad в состояниях Л2П, Б2? + приняты по работам Барроу и Крау-
форда [595[ и Лагерквиста [2645[, в состояниях С2П, D2E+, ?2?+, F2Z+ и G22+ - по работе
Харрингтона [1354].
Термодинамические функции Bad (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения Qm и ее производные рассчитывались по уравнениям A. 90)—A. 92) с учетом
восьми возбужденных состояний (подсостояния A2Iii/2 и А2Щг рассматривались как разные
состояния) в предположении, что QHl.tt = (Pilpx)QiflBV- Величина (?™-Бр и ее производные
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82).
В расчетах учитывались все уровни со значениями / ^ Jmax, «,> гДе Лпах, „находились из условий
A. 81). В табл. 29.11 приведены значения ушах и /lim, использованные в расчетах. Колебательно-
вращательные уровни состояния Ха ?+ вычислялись по уравнениям A. 65), A. 64); коэффициенты
Ykl в этих уравнениях для изотопической модификации молекулы ВаС1, соответствующей
природной смеси изотопов, рассчитанные по соотношениям A. 66) и постоянным из табл. 29.10,
представлены в табл. 29.11. Коэффициент Y03 получен с учетом условий A. 59), коэффициент Yn —
из условия Bv +i=0. Мультиплетность и вырождение колебательно-вращательных уровней
электронных состояний учитывались статистическими весами.
Основные погрешности рассчитанных термодинамических функций связаны с отсутствием
экспериментальных данных о вращательных постоянных и с невысокой точностью колебательных
постоянных молекулы Bad в состоянии Х2Е+, а также неполнотой информации о возбужденных
электронных состояниях этой молекулы. Погрешности в значениях Ф° (Т) могут достигать 0,2,
0,5 и 2 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции ВаС1 (г) вычислялись ранее в таблицах JANAF [1544] и работе
Климова и др. [167] (приведен полином, аппроксимирующий Ф° (Т) для Т <^ 5000 К).
Расхождения данных [1544] и табл. 859 обусловлены различием принятых вращательных постоянных
(—0,6 Дж-К-моль при 298,15 К), а также различием использованных методов расчета. При
6000 К расхождения достигают 1 и 3 Дж-К~1-моль в значениях Ф° (Т) и S° (Г). Расхождения
с данными [167] существенно больше из-за ряда упрощений, использованных в этой работе.
Константа равновесия реакции Bad (г)=Ва (г)+С1 (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (Bad) = 440 + 6 кДж • моль яй 36 800 + 500 см.
В табл. 29.14 представлены результаты измерений этой величины. Приведенные погрешности
учитывают воспроизводимость измерений, неточность сечений ионизации и термохимических
величин, использованных в расчетах. Принятое значение основано на масс-спектрометрических
измерениях Хилденбранда [1409, 1411] и спектрофотометрическом исследовании Гурвича и др.
[13331, в котором была определена зависимость / (ВаС1)//(Ва) в широком интервале температур.
Данные Змбова представляются заниженными и, по-видимому, содержат систематическую
ошибку, так же как измерения Погтера и др. (см. SrCl (г)). Исследования хемилюминесценции
Bad в экспериментах с молекулярными пучками [923, 1594, 2007, 2008] приводят к более
высоким значениям, лежащим в интервале 460—506 кДж-моль.
Принятому значению соответствует
г, 0) = —140,667 +6,7 кДж • моль.
ВаС1+ (г). Термодинамические свойства газообразного положительного иона хлорида бария
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 860.
В табл. 29.10 приведены молекулярные постоянные ВаС1+, оцененные при подготовке
справочника из-за отсутствия экспериментальных данных. По аналогии с изоэлектронными молекулами
GsGl и BaS в справочнике принимается, что ВаС1+ имеет основное состояние Х^+,
соответствующее конфигурации . . . а2п;4. Вращательная постоянная в этом состоянии вычислена на основании
значения ге (Ва—С1)=2,70+0,10 А, оцененного из сопоставления межатомных расстояний в
молекулах CaCl, CaCl+, ВаС1. Значение со, оценено сравнением частот колебаний тех же молекул;
оно хорошо согласуется с частотами колебаний молекулы Bad в ридберговских состояниях. По-
404
Барий и его соединения
ВаС1+ (г)
грешности принятых значений Ве и we не превышают 0,01 и 50 см соответственно. В справочнике
принимается, что у молекулы ВаС1+, так же как у CaF+ и BaF+, возбужденные состояния,
принадлежащие конфигурациям . . .о^к^а, . . .атс4а и . . . а2я;37г, имеют энергии выше 15 000 см.
Таблица 29.14.
Результаты
Авторы
Змбов, 1969 [2908]
Хилденбранд, 1970
Поттер и др., 1970
Хилденбранд, 1970
Гурвич и др., 1973
Хилденбранд, 1977
Кулкарни, Дадап,
[1409]
[2261 ]
[1407]
[1333]
[1411]
1976 [17846]
определении Do (BaCl) (в кДж-моль х)
Метод
Масс-спектрометрический, ВаС12 (г)+Ва (г)=
=2ВаС1 (г), 974—1052 К, 11 точек, Дг Н° @)=
=-90,4+1,9 (III)
То же, ВаС12(г)+-Ва(г)=2ВаС1(г), 1234—
1380 К, 13 точек, Дг Н° @)-34,0±1,1 (III)
То же, Ва (г)+А1С1 (г)=ВаС1 (г)+А1 (г),
13точек, АГН° @)=55,2+1,2 (III)
То же, ВаС1»(г)+Ва(г)=2ВаС1 (г), 1507—
1702 К, 7 точек; Дг#9 @)=61,9±1,3 (III)
То же, Ва (г)+А1С1 (г)=ВаС1 (г)+А1 (г\ 7
точек, Аг Н° @)=75,0±1,0 (III)
Электронного удара, ВаС12=ВаС1++С1+е,
АГЫ° @)=434±14
Спектрофотометрическое исследование
равновесия реакции Ва (г)+НС1 (г)=ВаС1 (г)+Н (г)
в пламенах, 1745—2580 К, 16точек, АГН° @)=
==-19,2±9,2 (II)
Масс-спектрометрпческий, Ва (г)-ЬА1С1 (г)=
=ВаС1 (г)+А1 (г), 2039-2186 К, 6 точек,
ДгЯ°@)=64,2±0,3(Ш)
Протока, Ba(r)f ВаС12 (к)=2ВаС1 (г), 1210—
1305 К, 6 точек, АГН° @)=327,9±2,8 (III)
D0(BaCl)
II закон
407 ±44
445+18
461 +40
428+17
427+17
III за коп
408+6
436±6
444+7
422+6
424 ±7
424+.17
447±12
442+9
436+100
435±7
465+10
Термодинамические функции BaCl+ (г) рассчитаны по уравнениям A.3)—A.10), A.93)—
A. 95) без учета возбужденных электронных состояний. Величина <??^вр и ее производные
вычислялись по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по / с помощью уравнений типа A. 82). В расчетах учитывались все
уровни энергии со значениями / ^ /mwCi „, последние находились из соотношений A. 81).
Колебательно-вращательные уровни энергии в состоянии ХХ2 + вычислялись по уравнениям A. 65) и
A. 62). Значения коэффициентов Yы в этих уравнениях, идентичные соответствующим постоянным
из табл. 29.10, а также величины ушах и /Иш приведены в табл. 29.11.
Погрешности вычисленных значений термодинамических функций обусловлены неточностью
принятых постоянных ВаС1+, а также пренебрежением возбужденными электронными состояниями.
Погрешностив значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 1, 2 и4 Дж-К~1-моль
соответственно.
Ранее таблицы термодинамических функций ВаС1+ (г) не публиковались.
Константа равновесия реакции ВаС1+ (г)+е (г)=Ва (г)+С1 (г) вычислена с использованием
значения ДГЯ° @) = —40,000+12 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0 (ВаС1)=480+10 кДж-моль.
Эта величина основывается на детальном исследовании, выполненном Хилденбранд ом [1407]
методом электронного удара D83+10 кДж-моль). Совпадающие в пределах их погрешностей
значения получены этим же методом Змбовым [2908] E31 +50 кДж -моль), Хилденбрандом [1409]
D82+50 кДж-моль), Поттером и др. [2261] D82 кДж-моль) и Хилденбрандом [1411] D82 +
+30 кДж-моль). Принятой величине соответствуют также
Д^ЧВаСГ, г, 0) = 339,333+12 кДж ¦ моль,
Do (ВаСГ) = 462,852 + 12 кДж • моль« 38 700 + 1000 см.
405
ВаСЦ (к, ж) Глава 29
ВаС12 (к, ж). Термодинамические свойства кристаллического и жидкого дихлорида бария
в стандартном состоянии в интервале температур 100—4100 К приведены в табл. 861.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. В справочнике за стандартное состояние ВаС12 (к) в интервале 0—1198 К принята
ромбическая а-модификация ВаС12, а в интервале 1198—1234 К — кубическая ^-модификация [732].
При Т <^ 298,15 К термодинамические функции вычислены по измерениям Гудмана и Вест-
рума [1231] F—346 К), проведенным на весьма чистом образце (> 99,9% ВаС12). Экстраполяция
ниже 6 К приводит к значению 5° F К)=0,05 Дж-К~1-моль~1. Погрешности принятых по [1231]
значений S° B98,15 К) и Я° B98,15 К)—Н° @) оцениваются в 0,3 Дж-К^-моль-1 и 0,04 кДжХ
Хмоль соответственно. При Т ^> 298,15 К для теплоемкости а-ВаС12 принято уравнение,
выведенное методом Шомейта по результатам измерений энтальпии ВаС12 (к, ж) в работе Гарднера и
Тейлора [1170] C40—1347 К), погрешности которых оцениваются в 0,3%. Измерения энтальпии
ВаС12 в работах [1003] A100-1200 К), [1549] (892-1200 К) и [1168] D06-1200 К) менее точны,
и полученные в них величины отклоняются от данных [1170] на 2—3% в разные стороны.
Теплоемкость ВаС12 в интервале 443—973 К определялась в работе [333], соответствующие
экспериментальные данные отличаются от рассчитанных по уравнению, приведенному в табл. 29.1,
в пределах 5%.
Температура полиморфного превращения A198+2 К) принята по данным [1170]; она хорошо
согласуется с результатами измерений в работах [1549, 1776а]. Значения Д^0 (ВаС12)=17,4 +
±0,2 кДж-моль, Дт#° (ВаС12)=15,85+0,4 кДж-моль и С°< (|5-ВаС12)=131+10 Дж-К^-моль
также приняты на основании измерений энтальпии, выполненных Гарднером и Тейлором [1170].
Близкие величины найдены в работах [1003, 1549] (AtrH°=17,l и 17,4 кДж-моль, Дт#о=16,3 и
17,3 кДж-моль соответственно). Температура плавления A234+2 К) принята по данным [1003,
1168, 1170, 1549], с ней согласуются менее точные измерения в ряде других работ. Значение
С (ВаС12, ж)=109+3 Дж-К-моль~1 найдено авторами [1170]; результаты измерений энтальпии
ВаС12 (ж) в работах [1003, 1168, 1549] согласуются с этой величиной в пределах 1—3%.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оцениваются в 0,2,
1, 3 и 12 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
ВаС12 (к, ж), приведенными в таблицах JANAF [1543] и настоящем издании, не превышают
0,1 Дж-К^-моль-1 в значениях Ф° (Т).
В справочнике принята энтальпия образования
к, 298,15 К) = — 844 + 8 кДж • моль.
Это значение вычислено с использованием приведенных в Приложении 5 энтальпий образования
ионов Ва+2 и С1~ и энтальпии растворения ВаС12 до состояния бесконечно разбавленного водного
раствора (—13,7+0,4 кДж-моль). Последняя величина получена как средняя из измерений
Воробьева и др. [70] (—14,3 кДж-моль), Крисса, Кобла [902] (—13,3 кДж-моль) и Дэдгера,
Тахериана [919] (—13,5 кДж-моль). Результаты более старых работ [364, 677, 2471, 2481, 2508,
2686] менее точны. Основным источником погрешности принятого значения является неточность
величины Д^Я"° (Ва+2, aq). Нужно отметить, что наблюдается плохое соответствие между
приведенным выше значением и результатами расчета энтальпии образования ВаС12 (к) из
термохимического цикла, включающего энтальпии растворения ВаС12 (к) и Ва (к) в соляной кислоте, найденные
Эрлихом и др. [1022] (—859 кДж -моль), и из термохимического цикла, включающего энтальпию
растворения ВаО (к) в соляной кислоте, измеренную Фицгиббоном и др. [1103] (—851 кДж-моль).
Это свидетельствует о противоречивости и ненадежности имеющихся в настоящее время данных по
энтальпиям образования ВаС12 (к, aq), ВаО (к), Ва+2 (aq) (см. примечание на с. 379).
Симонсен [2520] измерил энтальпию образования ВаС12 путем сожжения бария в хлоре
(—918 кДж-моль). Однако эти данные, по-видимому, еще менее надежды, чем рассмотренные
выше, о чем свидетельствуют неверные значения, полученные для ряда других хлоридов этим
автором на той же аппаратуре.
Давление пара дихлорида бария ВаС12 (к, ж)=ВаС12 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
\Н° (ВаС12, к, 0) = 358 + 5 кДж • моль.
Это значение основано на измерениях давления пара ВаС12 (к, ж) в работах Хилденбранда [1422]
(торзионный метод, 1235—1440 К, 83 точки), Ван-Уэстенбурга [2756] (эффузионный метод, 1244—
406
Барий и его соединения ВаС1»(г), ВаВг (г)
1277 К, 3 точки) и Новикова, Гаврюченкова [291] (метод точек кипения, 1588—1710 К, 5 точек).
Обработка этих данных по методам II и III законов приводит соответственно к значениям
(погрешности характеризуют воспроизводимость измерений): 357, 329 +_7, 320+50 кДж -моль и 358,4 +
+0,3, 356,7+0,9, 359,4+1,8 кДж-моль. Измерения Эмонса и др. [1030а] (эффузионный метод,
1238—1333 К) приводят (расчет по уравнению) к неверным величинам, равным 306 и
388+5 кДж-моль. Принятая величина основана в первую очередь на данных [1422], ее
погрешность учитывает неточность термодинамических функций ВаС12 (к, ж) и ВаС12 (г).
ВаС12 (г). Термодинамические свойства газообразного дихлорида бария в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 862.
Молекулярные постоянные, использованные для расчета термодинамических функций ВаС12,
приведены в табл. 29.7. Исследования инфракрасного спектра [1363, 2842], отклонения
молекулярного пучка в неоднородном электрическом поле [2839] и эффекта Штарка [922] показали, что
молекула ВаС12 в основном электронном состоянии Х1А1 имеет нелинейную структуру
(симметрия С2г>). Приведенному в табл. 29.7 произведению моментов инерции соответствуют
Д С1—Ва—01=110+10° (средний из оценок Хасти и др. [1363] A20+10°) по относительной
интенсивности полос vx и v8(h Уайта и др. [2842] A00°) по формулам простого поля валентных сил)
и г (Ва—С1)=2,82+0,03 А (найдено Акишиным и др. [5,15] в электронографическом исследовании
паров ВаС12). Погрешность принятого значения 1А1в1с оценивается в 3-Ю13 г3-см6. Приведенная
в табл. 29.7 частота v3 найдена Байковым [24] в инфракрасном спектре паров ВаС12. Она хорошо
согласуется с данными Хасти и др. [1363] и Уайта и др. [2842] по ИК-спектру молекул ВэС12,
изолированных в матрицах благородных газов B68 и 260 см). Для частоты vx принято значение,
которое на 5 см меньше величины v3, так как такая разница наблюдалась между этими частотами
в ИК-спектрах в матрицах авторами [1363, 2842]. Для частоты v2 принято значение, среднее между
оцененным для газа Хасти и др. [1363] F0 +10 см) и оцененным Уайтом и др. [2842]
экстраполяцией значений этой частоты у молекул других дихлоридов, полученных из их ИК-спектров в
матрицах из Кг C6 см). Погрешности принятых значений оцениваются в 10—20 см, см. текст по
СаС12 (г).
Сведения о возбужденных электронных состояниях молекулы ВаС12 отсутствуют, можно
предполагать, что их энергии превышают 20 000 см.
Термодинамические функции ВаС12 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор»
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета; они
составляют 3, 5 и 7 Дж-К~1-моль в значениях Ф° (Т) при 298,15, 3000 и 6000 К соответственно
(в том числе 3—4 Дж-К-моль~1 из-за неточности молекулярных постоянных), см. текст по СаС12(г).
Ранее термодинамические функции ВаС12 (г) рассчитывались в таблицах JANAF [1544],
работах Брюэра и др. [748] (Г < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Расхождения
с данными [1544] не превышают 2,5 Дж-К-моль~1 и объясняются тем, что в этом расчете
были использованы несколько отличающиеся значения постоянных ВаС12. Расхождения с
данными [195, 748] достигают 19—22 Дж-К-моль и обусловлены тем, что в этих работах для
молекул ВаС12 была принята линейная структура.
Константа равновесия реакции ВаС12 (г)=Ва (г)+2С1 (г) вычислена с использованием значения
&ГН° @)=905,563+9,9 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации ВаС12 (к). Этим величинам также соответствует
"^S^BaCl^-r, ) = — 486,610]±|9,4|кДж -"моль.
ВаВг (г). Термодинамические свойства газообразного бромида бария в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 863.
Молекулярные постоянные ВаВг, принятые в справочнике на основании результатов
исследований спектров этой молекулы, приведены в табл. 29.10. В спектре ВаВг наблюдались пять систем
полос, связанных с переходами между шестью электронными состояниями: -42П ->¦ X2 ?+ и
В2 Е+ -> X2 Е+ [733], C2U ^ X2 2+ [1354, 1378, 1600, 2159, 2279, 2360, 2795], D2 Е ^ X2 Е+ [1354,
1600, 2035, 2279], ^S^TS1 [1354, 1600, 2035]. Так же как у молекул других галогенидов
щелочноземельных металлов, состояния X2 ?+ и А2П молекулы ВаВг коррелируют с основными
состояниями атомов BaAiS)+Br BP). Со следующими диссоциациошшми пределами 3D-\r2P,
407
ВаВгг (к, ж) Глава 29
*D+*P и 3Р+2Р коррелирует большое число валентных состояний, в том числе квартетные
состояния. По крайней мере часть ненаблюдавшихся состояний может быть связанными состояниями,
в то время как состояния D2 Е+ и Е2 ?+, по-видимому, имеют ридберговский характер (см. тексты
по BaF и CaF).
Колебательные постоянные ВаВг в состоянии Хг Е+ определялись по кантам полос из анализа
систем С2П-Х2Е+ [1354, 1378, 1600], D2 ?+-Х2 2+ и Е2 S+-X2 E+ [1354, 1600, 2035]; найденные
значения хорошо согласуются между собой х. Приведенные в табл. 29.10 постоянные найдены Хар-
рингтоном [1354] по кантам полос трех систем. Вращательная структура спектра ВаВг не
исследовалась. Приведенное в табл. 29.10 значение ге (ВаВг)=2,95+0,05 А оценено на основании
величины г (Ва—Вг) в молекуле ВаВг2 с учетом различия в значениях re (MX) и г (М—X) в молекулах
галогенидов и дигалогенидов щелочноземельных металлов. Постоянные ВаВг в состояниях А2И и
В2 Е+ приняты по работе Брадфорда и др. [733], а в состояниях C2U, D2 2+ и Е2 Б+ — по данным
Харрингтона [13541.
Термодинамические функции ВаВг (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95).
Значения QB4 и ее производных рассчитывались по уравнениям A. 90)—A. 92) с учетом шести
возбужденных состояний (подсостояния ^42П1/2 и -42Пз/г учитывались независимо) в предположении,
что Qiol.vP = (pJpx) <??.вР- Величина (?^.EP и ее производные по температуре вычислялись по
уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82). В расчетах
учитывались все уровни со значениями / ^ /щах, „> гДе •Лпах, v находились из условий A. 81).
Колебательно-вращательные уровни состояния X2 Е+ вычислялись по уравнениям A. 65) и A. 61).
Значения коэффициентов Ykl, а также величин 1>шах и /1Ш, использованные в расчетах, приведены
в табл. 29.11. Коэффициент У03 получен из условий A. 59). Различие молекулярных постоянных
изотопических модификаций ВаВг в расчетах не учитывалось. Мультиплетность и вырождение
колебательно-вращательных уровней учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при Т ^ 2000 К связаны в основном
с отсутствием данных о вращательных постоянных и неточностью колебательных постоянных
молекулы ВаВг в состоянии X2 2+, а при более высоких температурах — с ограниченной
информацией о возбужденных состояниях этой молекулы. Общие погрешности в значениях Ф° (Г) при
298,15, 3000 и 6000 К могут достигать 0,3, 0,5 и 2 Дж-К^-моль соответственно.
Ранее термодинамические функции ВаВг (г) вычислялись в справочнике JANAF [1544] и
в работе Климова и др. [167] (приведены коэффициенты полинома, аппроксимирующего Ф° (Т)
при Т ^5000 К). Расхождения в термодинамических функциях, приведенных в [1544] и
в табл. 863, составляют 0,4 Дж-К~1-моль при 298,15 К из-за различия в оценке вращательной
постоянной. G ростом температуры расхождения увеличиваются из-за неверной оценки энергии
низколежащих электронных состояний ВаВг авторами [1544]; они достигают 2 и 4 Дж-К -моль
в значениях Ф° (Т) и S°(T) при 6000 К. Расхождения с данными [167] еще больше из-за
использования авторами этой работы упрощенной методики расчета и отличающихся значений постоянных.
Константа равновесия реакции ВаВг (г)=Ва (г)+Вг (г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#° @) = Do (ВаВг) = 375 + 15 кДж • моль «* 31 400 + 1300 см.
В табл. 29.15 приведены результаты измерений этой величины. Погрешности, указанные в
таблице, учитывают воспроизводимость измерений, а также неточность сечений ионизации,
термодинамических функций и термохимических величин, использованных в расчетах. Принятая величина
является средней из полученных в работах [90, 1333, 1411]; данные [2007], так же как для
других соединений, завышены, по-видимому, из-за методических ошибок (см. [1040а]).
Принятой энергии диссоциации соответствует
г, 0) = —77,364+15 кДж • моль.
ВаВг2 (к, ж). Термодинамические свойства кристаллического и жидкого дибромида бария
в стандартном состоянии при температурах 298,15—4000 К приведены в табл. 864.
1 В работе Шульца и Сигела [2473а] в спектре лазерной флуоресценции в системе С2П—Х22+ наблюдались
переходы на уровни с v" <, 74; найденные в этой работе значения ш,= 192,47 и (о'^'=0,4312 см"
удовлетворительно согласуются с принятыми в табл. 29.10.
408
Барий и его соединения
ВаВгг (к, ж)
Таблица 29.15. Результаты определений Do (BaBr) (в кДж-моль
Авторы
Метод
Do (BaBr)
II закон
III закон
Гурвич я др., 1973 [1333] Спектрофотометрическое исследование реакции 3R5+12 —
Ва (r)+HBr(r)=--BaBr (г)+Н (г) в пламрнах по
зависимости /(ВаВг)//(ВаЧ, 1745—2580 К, 16
точек, ДГЯ° @)=- 2,9+11 (II)
Хилденбранд, 1977 [1411] Масс-сиектрометрический, Ва (г)->-А1Вг (г)= 348+30 370+10
=-ВаВг(г)+А1(г), 1962—2186 К, 13 точек,
АгЯ°@)=С2,2±1,2(Ш)
Горохов, Московская, 1975 [901 То же, Ва (г)+А1Вг (г)=ВаВг (г)+А1 (г), 1243— 397+30 390+10
1469 К, 18 точек, Arff' @)=41,6+1,3 (IID
То же, Ва (г)+КВг(г)=К (г)+ВаВг(г), 1143— 413+50 392+7
1250 К, 19 точек. ЛГН° @)=—12,0+1,1 (III)
Тоже. ВаВг,(г)+ВаГг)=2ВаВг(г>, 1179— 434+50 384+6
1233 К, 9 точек, ДгЯ°'@)=50,0±2',1 (III)
Менцингер, 1974 [2007] Исследование хемштамипесцешпш ВаВг в скре- >438+9
щенных молекулярных пучках
Эстлер, Заре, 1978 [1040а] То же, в реакции ВаC#)+Вг2= ВаВг*+Вг 359+8
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. В справочнике за стандартное состояние ВаВг2 (к) принимается ромбическая
модификация (структурный тип РЬС12 [732]). Экспериментальные данные по теплоемкости ВаВг2 (к)
при Т < 298,15 К в литературе отсутствуют. Значения S° B98,15 К) и Н° B98,15 К)—Н° @),
приведенные в табл. 29.1, получены с помощью приближенных оценок, их точность оценивается
в 8 Дж-К~1-моль и 1 кДж-моль. Уравнение для теплоемкости ВаВг2 (к) B98,15—-1130 К)
выведено методом Шомейта по результатам 27 измерений энтальпии, проведенных Ефремовой и Мати-
зеном [127] на образце ВаВг2, содержавшем не более 0,13% примесей (главным образом ВаО).
Погрешности этих измерений составляют около 0,5%. В пределах 0,7% с принятыми величинами
согласуются измерения энтальпии в работах Дворкина и Бредига [1003] (Н° (ИЗО К)—
Н° B98,15 К)=70,7 кДж-моль'1) и Джанза и др. [1549] D87-1126 К, погрешности -1%).
Температура плавления ВаВг2 A130+2 К) принята по хорошо согласующимся данным [127, 1003,
2329], а энтальпия плавления C2,2+0,5 кДж-моль"} — по данным Ефремовой и Матизена [127].
Близкое значение энтальпии плавления получено в работе [1003] C1,9 кДж-моль"). Теплоемкость
жидкого ВаВг2 A05+2 Дж-К-моль~1) рассчитана из измерений энтальпии в интервале 1134—
1963 К в работе [127] х. Менее точные значения A08 и 130 Дж-К~1-моль~1) получены из измерений
энтальпии ВаВг2 (ж) в более узких интервалах температур в работах [1003, 1549].
Погрешности вычисленных значений Ф° (Г) при 298,15, 1000, 2000 и 4000 К оценены в 8, 9,,
10 и 20 Дж-К-моль~1 соответственно. Термодинамические функции ВаВг2 (к, ж) вычислялись
ранее в таблицах JANAF [1544] для Т ^ 3000 К; расхождения данных [1544] и табл. 864
составляют 1,5 Дж-К-моль и обусловлены разной оценкой величины 5° B98,15 К).
Принятая в справочнике энтальпия образования
Д//°(ВаВг2, к, 298,15 К) = — 757 + 5 кДж • моль
основана на результатах измерений энтальпии растворения Ва (к) и ВаВг2 (к) в бромистоводород-
ной кислоте, проведенных Эрлихом и др. [1022]. Измерения энтальпии растворения ВаВг2
в воде [2686, 2757] приводят к значению около —747 кДж-моль. Их надежность невелика, так
как эти работы были выполнены еще в прошлом веке, а для расчета энтальпии образования
необходимо значение Д^ Н° (Ва+2, ад), данные о котором противоречивы (см. Приложение 5).
1 Авторы [127] из этих данных получили для С°р (ВаВг2, ж) линейную зависимость от температуры, согласно
которой теплоемкость в интервале ИЗО—2000 К возрастает от 99 до 115 Да;-К-моль.
409
ВаВг2 (г), Bal (г) Глава 29
Давление пара дибромида бария ВаВг2 (к, ж)=ВаВг2 (г) вычислено на основании принятой
в справочнике энтальпии сублимации
Д,#°(ВаВгя, к, 0)=:343 + 10 кДж • моль.
Принятое значение является средним из вычисленных по результатам измерений давления
пара в работе Хилденбранда [1412] (торзионный метод, 1048—1275 К, 39 точек) и в работе Питер-
сона, Хатчисона [2217] (эффузионный метод, 1180—1320 К, 16 точек). В обеих работах данные
представлены только в виде уравнений; им соответствуют энтальпии сублимации 343 кДж-моль
¦(II закон), 343,6+0,8 кДж-моль-1 (III закон) [1412] и 345 кДж-моль (II закон), 342,7 +
+0,3 кДж-моль (III закон) [2217]. В пределах погрешности с принятой величиной согласуется
значение 340+4,0 кДж-моль (III закон), найденное Гороховым и Московской [90]
(эффузионный метод, 1199 и 1233 К, 2 точки). Погрешность принятой величины определяется в первую
очередь неточностью термодинамических функций ВаВг2 (к, ж) и ВаВг2 (г).
ВаВг2 (г). Термодинамические свойства газообразного дибромида бария в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 865.
Молекулярные постоянные, использованные для расчета термодинамических функций ВаВг2,
приведены в табл. 29.7. Экспериментальные исследования структурных параметров и спектров
молекулы ВаВг2 неизвестны. Изучение отклонения молекулярного пучка в неоднородном
электрическом поле в работе [2839]_привело к выводу о нелинейной структуре этой молекулы в основном
электронном состоянии XXAX. Приведенной в таблице величине IAIBIC соответствуют
Д Вг—Ва—Вг=120+10° и г (Ва—Вг) =2,99+0,03 А. Величина 4 Вг—Ва—Вг оценена на
основании изучения изменения угла X—М—X в ряду MF2—МС12—МВг2—М12 (где М=Са, Sr, Ва).
Значение г (Ва—Вг) найдено Акишиным и др. [5, 15] в электронографическом исследовании
паров ВаВг2. Погрешность принятого значения IaIbIc составляет 3-1012 г3-см6. Частоты vx и v3,
приведенные в табл. 29.7, вычислены по уравнениям поля валентных сил. Силовая постоянная
/г=0,81-105 дин-см принята по оценке Байкова [25], основанной на корреляции между
величинами fr и г (М—X) в молекулах дигалогенидов подгруппы ПА. Отношение /,.//„.=15 принято
по аналогии с молекулами BaF2 и ВаС12, для которых оно равно 13 и 19 соответственно. Частота v2
оценена по графикам va=/ (Zx) и v2=/ (ZM). Погрешности принятых значений частот оцениваются
в 10—30 см, см. текст по СаС12 (г).
Сведения о возбужденных электронных состояниях молекулы ВаВг2 отсутствуют, можно
предполагать, что их энергии превышают 15 000—20 000 см.
Термодинамические функции ВаВг2 (г) вычислены по уравнениям A. 3)—A. 6), A. 9)—A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор»
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены неточностью
принятых молекулярных постоянных, а также приближенным характером расчета; они составляют
4, 7 и 10 Дж-К-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно, см. текст
по СаС12 (г).
Ранее термодинамические функции ВаВг2 (г) вычислялись в таблицах JANAF [1544], работах
Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Т < 2500 К). Расхождения
данных [1544] и табл. 865 составляют 5—6 Дж -К -моль и обусловлены разной оценкой постоянных
этой молекулы. Расхождения с данными [195, 748] достигают 21—22 Дж-К-моль~1 в значениях
Ф° (Т) из-за того, что авторы этих работ принимали для молекулы ВаВг2 линейную структуру.
Константа равновесия реакции ВаВг2 (г)=Ва (г)+2Вг (г) вычислена с использованием
значения ДГЛ° @) =817,129+12 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации ВаВг2 (к). Этим величинам также соответствует
Д/Я°|(ВаВг2,|г,д0) = -401,57 + |112<кДж • моль.
Bal (г). Термодинамические свойства газообразного иодида бария в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 866.
Молекулярные постоянные Bal, принятые в справочнике на основании результатов
исследований электронных спектров этой молекулы, приведены в табл. 29.10. В спектре Bal
экспериментально наблюдались пять систем полос, связанных с переходами между шестью электронными
состояниями: АЩ -+ X2 Е+ и В2 ?+ -* X2 Е+ [733], С2П ** X2 2+ [733, 2011, 2159, 2194, 2645,
410
Барий и его соединения ^lial (г)
2795], Z>2?+^X2?+ [733, 2159, 2194, 2310, 2500, 2645, 2795], Е% ?+ ^ X2 ?+ [733, 2194, 2310,
2500, 26451, а также система полос в области 22 000 см [733]. Так же как у других молекул гало-
генидов щелочноземельных металлов, состояния X2 ?+ и А2П молекулы Bat коррелируют с
основными состояниями атомов Ва A5)+1 BР). Со следующими диссоциационными пределами 3D-\-2P,
D1Jr2P, sP-\-2P коррелирует большое число валентных состояний, в том числе квартетных; по
крайней мере часть ненаблюдавшихся состояний может быть связанными состояниями. В то же
время такие состояния, как D2 ? и Е2 ?, имеют ридберговский характер (см. текст по CaF и BaF).
Колебательные постоянные Bal в состоянии X2 ?+ определялись по кантам полос из анализа
систем C2U-X2 ?+ [2011, 2194, 2645], D2 ?+-Х2 ?+ и Е2 ?+-Х2 ?+ [2194, 2310, 2500, 2645].
Приведенные в табл. 29.10 значения колебательных постоянных найдены в работе Патела и
Шаха [2194] из анализа колебательной структуры систем полос С—X, D—X и Е—Х. Они близки
к рекомендованным в справочнике [2645] на основании данных, полученных Морганом в
неопубликованном исследовании спектра поглощения Bal. Существенно отличающиеся значения
постоянных найдены Рао и Редди [2310], однако в этой работе анализ колебательной структуры спектра,
по-видимому, выполнен некорректно.
Вращательная структура спектра Bal не исследовалась. Приведенное в табл. 29.10 значение
ге(Ва1)=3,15+0,05 А оценено на основании величины г (Ва—I) в молекуле Ва12 и сопоставления
величин re (MX) и /' (М—X) в молекулах галогенидов и дигалогенидов щелочноземельных металлов.
Энергии состояний А2И и В2 ?+ приняты по работе Брандфорда и др. [733], значения
постоянных в остальных состояниях — по работам Шаха и др. [2194, 2500]. Следует отметить невысокую
точность последних; существенно отличающиеся значения получены в других работах (см.
[2645]).
Термодинамические функции Bal (г) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 951.
Значения QBU и ее производные рассчитывались по уравнениям A. 90)—A. 92) с учетом шести
возбужденных состояний (подсостояния A2Ili/2 и ^12Пз/2 рассматривались как отдельные состояния)
в предположении, что (?(„].вр = (р{1рх) QlS.w Величина <?S.Bp и ее производные вычислялись
по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью уравнений типа A. 82). В расчетах
учитывались все уровни энергии со значениями / ^ Jma.x, „> где Jimx „ находились из условий
A. 81). Колебательно-вращательные уровни состояния X2 ?+ вычислялись по уравнениям A. 65),
A. 61). Значения коэффициентов Ykl, а также величин fmax и /1Ш, использованные в расчетах,
приведены в табл. 29.11. Коэффициент У03 получен из условий A. 59). Мультиплетность и
вырождение колебательно-вращательных уровней учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при Т <^ 2000 К связаны в основном
с отсутствием данных о вращательных постоянных и с неточностью колебательных постоянных
молекулы Bal в состоянии X2 Е+, а при более высоких температурах — с неполнотой
экспериментальных данных о возбужденных электронных состояниях этой молекулы. Погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,5, 0,8 и 2 Дж-К~1-моль соответственно.
Ранее таблицы термодинамических функций Bal (г) вычислялись в таблицах JANAF [1544]
и работе Климова [167] (приведен полином, аппроксимирующий значения Ф° (Т) для Т ^ 5000 К).
Данные [15441 и табл. 866 отличаются в значениях Ф° (Т) на величины от 0,2 до 4 Дж-К~1-моль~1,
расхождения обусловлены пренебрежением рядом электронных состояний в таблицах JANAF.
Расчет [167] выполнен упрощенным методом и без учета возбужденных состояний Bal.
Константа равновесия реакции Bal (г)=Ва (г)+1 (г) вычислена с использованием принятой
в справочнике энергии диссоциации
г-1
ДгЯ° @) = Do (Bal) = 302 + 7 кДж • моль «* 25 250 + 600 см'
Эта величина основана на результатах масс-спектрометрического исследования равновесия
реакции Ва (г)+Ва12 (г)=2Ва1 (г) и Ва (r)+GaI (r)=BaI (r)-j-Ga (г) в работе Клейншмидта и
Хилденбранда [17166] A873—2011 К, 6 точек). Обработка этих измерений по III закону приводит
к значениям 304,3 +4 и 299,6+ ЗкДж-моль, где приведенные погрешности характеризуют только
воспроизводимость экспериментальных данных. Принятое значение подтверждается изучением
хемилюминесценции молекул Bal, образующихся в реакции Ва (sZ))+I2=BaI*+I, протекающей
в скрещенных пучках C05+8 кДж-моль [1040а]) (см. также [2032]). В работе [952] из-за
методической ошибки получено существенно более высокое значение (^424 кДж-моль).
411
Bal2 (к, ж) Ва12 (г) Глава 2»
Принятой величине соответствует
Д/Я°(Ва1, г, 0) = —15,124 + 7,6 кДж • моль.
Ва12 (к, ж). Термодинамические свойства кристаллического и жидкого дииодида барпя в
стандартном состоянии при температурах 100—4400 К приведены в табл. 867.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. В справочнике за стандартное состояние Ва12 (к) принята ромбическая модификация
Ва12 (структурный тип РЬС12 [732]). При Т <^ 298,15 К термодинамические функции вычислены
по результатам измерений теплоемкости, проведенных Пауковым и др. [315] A3—300 К) на весьма
чистом образце Ва12; их погрешности составляют около 0,3% при Т ^> 50 К. Экстраполяция
теплоемкости приводит к значению S° A3 К)=2,6 Дж-К-моль~1. Погрешности значений S° B98,15 К)
и Н° B98,15 К)—Н° @), принятых по работе [315], оцениваются в 0,6 Дж-К-моль~1 и
0,06 кДж-моль соответственно. Уравнение для теплоемкости Ва12 (к) B98,15—984 К) выведено
на основании измеренного в работе Дворкина и Бредига [1003] значения энтальпии Ва12
Н° (984 К)—Я° B98,15 К)=58 кДж-моль. Температура плавления Ва12 (984+2 К) принята по
данным Дворкина и Бредига [1003] и Хатчисона [1520]; другие измерения [1549] A013 К),
{1031] (981 К) менее точны и не учитывались. Энтальпия плавления B6,5+0,5 кДж-моль)
также принята по данным Дворкина и Бредига; несколько менее точные значения определены в
работах [1549] B7,3 кДж-моль) и [1031] B5,3 кДж-моль). Теплоемкость жидкого Bal2 A13 +
+3 Дж-К~1-моль~1) принята на основании измерений энтальпии Ва12 (ж) в узком интервале
температур в работе [1003].
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К оценены в 0,6,
2, 7 и 20 Дж-К~1-моль~1. Термодинамические функции Ва12 (к, ж), приведенные в справочнике
JANAF [1544] и в табл. 867, практически совпадают.
В справочнике принята энтальпия образования
A^°(BaI3, к, 298,15 К) = — 607 + 10 кДж • моль,
вычисленная по результатам измерений энтальпии реакций взаимодействия металлического бария
и Ва12 (к) с растворами иодистоводородной кислоты, выполненных Эрлихом и др. [1022].
Значительная погрешность принятой величины обусловлена тем, что в работе [1022] был использован
негерметичный калориметр, а также была возможна побочная реакция образующегося водорода
с медной сеткой, в которую был завернут металл.
Давление пара дииодида бария Ва12 (к, ж) = Ва12 (г) вычислено с использованием принятой
в справочнике энтальпии сублимации
Д,#°(Ва12, к, 0) = 321 + 7 кДж - моль.
Это значение основано на результатах масс-спектрометрических измерений Уинчелла [2861 ]
A019—1278 К) и эффузионных измерений Питерсона и Хатчисона [2217] A150—1300 К, 18 точек)
и Хилденбранда [1413] (985—1051 К, 8 точек). В работах [1413, 2217] результаты измерений
представлены в виде уравнений. Расчеты по этим уравнениям приводят к значениям 318 и 285 кДж-моль
(II закон) и 321,0+0,1 и 322,5+2,3 кДж-моль (III закон) соответственно. В публикации [2861]
дано только вычисленное ее автором значение энтальпии сублимации, равное 283+20 кДж-моль'1
(II закон) и 325+20 кДж-моль (III закон). Погрешность принятой по данным Хилденбранда
величины определяется в основном неточностью термодинамических функций Ва12 (к, ж) и
Ва12 (г).
Ва12 (г). Термодинамические свойства газообразного дииодида бария в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 868.
Молекулярные постоянные, использованные для расчета термодинамических функций Ва12,
приведены в табл. 29.7. Исследования отклонения молекулярного пучка в неоднородном
электрическом поле, выполненные Вартоном и др. [2839], показали, что в основном электронном
состоянии Х1А1 молекула Ва12 имеет нелинейную структуру (симметрия С2„). Произведение моментов
инерции, приведенное в таблице, вычислено для Д I—Ва—1=125+10° (оценен в результате
анализа изменения угла X—М—X в рядах дигалогенидов металлов подгруппы ПА) и г (Ва—1) =
=3,20+0,03 А (найдено Акишиным и др. [5, 15] в электронографическом исследовании паров
412
Барвй и его соединения BaS (к, ж)
Ва12). Погрешность значения IaIbIc составляет 1-1011 г3-смв.Спектр Ва12 не исследован.
Основные частоты Ва12 оценены при подготовке справочника на основании закономерностей в изменении
постоянных молекул дигалогенидов элементов подгруппы ПА. Частоты \ и v3 рассчитаны по
уравнениям поля валентных сил. Силовая постоянная /г=0,68-105 дин-см принята по оценке Бай-
кова [25], выполненной на основании корреляции величин /г и г (М—X). Значение frr вычислено
в предположении /Г//ГР=15. Частота v2 оценена графически по зависимостям ^2=/(Zx) и v2=/ (ZM).
Погрешности принятых значений частот оцениваются в 10—20 см.
Сведения о возбужденных состояниях Ва12 в литературе отсутствуют, по-видимому их энергии
превышают 15 000 см.
Термодинамические функции Ва12 (г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближений «жесткий ротатор—гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности рассчитанных термодинамических
функций обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета и составляют 5, 9 и 12 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000
и 8000 К соответственно х.
Ранее термодинамические функции Ва12 (г) вычислялись в таблицах JANAF [1544] и в работах
Брюэра и др. [748] (Т < 2000 К) и Краснова и Светцова [195] (Г < 2500 К). Расхождения
результатов этих расчетов и данных табл. 868 достигают 14—20 Дж-К-моль из-за того, что в них
принималась линейная [195, 748] или близкая к линейной [1544] структура молекулы Ва12.
Константа равновесия реакции Ва12 (г) = Ва (г)+21 (г) вычислена с использованием значения
Ar#° @)—679,163+13 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации Ва12 (к). Этим величинам также соответствует
&.fH°(BaI2, г, 0) = —285,124+13 кДж • моль.
BaS (к, ж). Термодинамические свойства кристаллического и жидкого сульфида бария в
стандартном состоянии при температурах 100—6000 К приведены в табл. 869.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 29.1. В справочнике за стандартное состояние BaS (к) принята кубическая модификация
(структурный тип NaCl) [1459a]. При Т <^ 298,15 К термодинамические функции вычислены по
данным о теплоемкости, полученным Кингом и Уиллером [1713а] E1—297 К) на образце состава
99,53% BaS, 0,22% BaSO4 и 0,04% Si; погрешности этих измерений составляют ~0,5%.
Экстраполяция теплоемкости ниже 51 К проводилась по уравнению CP=D A59/Г)+?' B67/71) и привела
к значению S0 E1 К) = 10,9 Дж-К~1-моль~1. Погрешности принятых по работе [1713а] значений
S0 B98,15 К) и Я° B98,15 К)—Н° @) (см. табл. 29.1) оцениваются в 1 Дж-К^-моль и 0,1 кДжХ
Хмоль соответственно.
Данные по теплоемкости или энтальпии BaS (к, ж) при высоких температурах отсутствуют.
В интервале 298,15 — 2500 К для теплоемкости BaS (к) принято уравнение (см. табл. 29.1),
выведенное по значениям Ср B98,15К), С;E00 К)=52Дж-К-1-мольиС; B000К)=63 Дж-К^-моль;
последние два значения оценены с учетом данных о теплоемкости оксидов щелочноземельных
металлов. Погрешности оцененных значений могут достигать 10% при 1000 К и 15% при 2000 К.
Температура плавления BaS B500+200 К) оценена с учетом указаний (см. [365а, 2031]), что
она превышает 2473 К. Энтальпия плавления F3+15 кДж-моль) получена так же, как для CaS
и SrS, в предположении, что энтропия плавления равна 25 Дж-К-моль~1 [1781]. Теплоемкость
BaS (ж) принята равной 67+15 Дж-К-моль на основании приближенной оценки (см. т. 1,
гл. 1).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 6000 К оцениваются в 1,
5, 13 и 25 Дж-К^-моль соответственно.
1 После завершения расчета термодинамических функций Ва12(г) для настоящего справочника Спиридонов и др.
[2595 д] провели новое эпектронографическое исследование этой молекулы, которое позволило определить ее
равновесные структурные параметры, а также значения частот Vj,v3 и постоянных ангармонического потенциала,
характеризующего деформационное колебание Ва12. Термодинамические функции Ва12(г), рассчитанные по этим
постоянным (Д I—Ва—1 = 148±9°, г (Ва—1)=3,150+0,007 A, vx=106±12 см, v3=145±21 см, 7F)=
= —468 62+774 6* (см) с приближенным учетом ангармоничности деформационного колебания по уравнениям
типа A5.2), согласуются с приведенными в табл. 868 в пределах 1,7—5,3 Дж• К • моль в значениях Ф°(Т),
3—7 Дж-К^-моль в значениях S°(T) и 1,3—1,7 Дж-К^-моль в значениях С°р(Т).
413
BaS (г)
Глава 29
Термодинамические функции BaS (к), приведенные в справочниках [578, 2031 ] для Т ^ 2000 К,
отличаются от данных табл. 869 на 4 Дж-К-моль~1 в значениях S° B000 Ю; расхождения
обусловлены разной оценкой теплоемкости BaS (к) при высоких температурах.
В справочнике принята энтальпия образования
AfH°(BaS, к, 298,15 К) — — 453 + 10 кДж ¦ моль,
основанная на измерениях, приведенных в табл. 29.16. Провести пересчет данных [2417] и [2067а]
к стандартной температуре не удается вследствие отсутствия в литературе необходимых для этого
Т а б л и ц а 29.16. Результаты определений AfH° (BaS, к, 298,15 К) (в кДж• моль)
Авторы
Метод
AfH° (BaS, к, 298,15 К)
II закон
III закон
Сабатье, 1881 [2417] Калориметрический, BaS (к) + 5,2НС1 (р-р, —452+10
22Ш2О)=-ВаС12 (р-р, 3,2НС1+1150Н2О)+3.2НС1
(р-р, 360H2O)+H2S (р-р), 10,5° С, 5 измерений
Мурло, 1899 [2067а] То же, детали измерений не известны —450+10
Шенк, Хаммершмидт, 1933 [2451а] Исследование равновесия реакции BaS (к)+ —496+20 —475 + 25
+2SO2 (r)=BaSC>4 (k)+S2 (г) методом протока,
1355-1399 К, 2 точки
Окуно, 1935 [21576] То же, BaSO4 (к)+4СО (r)=BaS (к)+4СО2 (г), —408+100 —450+20
1073—1173 К, 3 точки
Кальвер, Хемдорф, 1955 [913а] То же, BaSO4 (к)+4Н2 (r)=BaS (к)+4Н2О (г), —455 —455+20
1073—1373 К, расчет по уравнению
Колен и др., 1964 [873] То же, */sBa (r)+4/3S2 (r)=2/sBaS (k)+2S (г) масс- —584±100 —492+25
спектрометрическим методом, 1846—2120 К,
10 точек
сведений (см. текст по SrS (к)). В работе [873] не приведены найденные константы равновесия.
В таблице даны величины, вычисленные по константам равновесия, найденным на основании
приведенных в этой работе интенсивностей ионных токов и принятых в справочнике сечений
ионизации и распространенности изотопов. Аналогичным образом вычислены константы
равновесия реакций с участием BaS (г) (см. ниже).
Приведенные в таблице погрешности включают воспроизводимость измерений, погрешности
использованных в расчетах термохимических величин, термодинамических функций и сечений
ионизации. Принятая величина является средневзвешенной из приведенных в таблице величин, за
исключением данных [873].
Давление пара сульфида бария в реакции BaS (к, ж)=Ва8 (г) вычислено с использованием
значения ДГЯ° @)=Д<Д° (BaS, к, 0) =486,996 +18 кДж-моль, соответствующего принятым
энтальпиям образования BaS (к) и BaS (г).
BaS (г). Термодинамические свойства газообразного сульфида бария в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 870.
В табл. 29.4 приведены молекулярные постоянные BaS, принятые на основании анализа
результатов исследований электронного [592а, 596а, 858а, 9316] и микроволнового [1385, 2700а] спектров
этой молекулы и оценок. В электронном спектре BaS исследованы две системы полос, связанные
с переходами между основным и двумя возбужденными состояниями 5*? + — Х12+ [592а, 9316],
СХЕ+ — X1S+ [592а, 596а, 858а, 9316]. По аналогии с ВаО и оксидами других щелочноземельных
металлов молекула BaS должна иметь еще ряд низколежащих синглетных и триплетных состояний,
соответствующих конфигурациям . . .а2тс3о (а3П, А1!!), . . .an* a (bsL+), . . . а2^3^ C> ХД, 1S~,
3Е+) и . . . атт47г CП, 43). Из всех этих состояний только состояния а3П и А1!! идентифицированы
по возмущениям в системе 5ХЕ+ — Хх? + [592а, 9316].
В табл. 29.4 приведены данные для 12 валентных состояний молекулы BaS. Колебательные
постоянные BaS в основном состоянии Х1^ +, а также колебательные и вращательные постоянные в
состояниях В1!,* и С1!!* приняты по результатам исследования Барроу и др. [592а] систем В1!,* —
414
Барий и его соединения
BaS
(v" <; 3, v' <; 2) и СХЕ+ — ХХ11+ (v" <; 7, i/ <I 4) в спектре поглощения. Вращательные
постоянные основного состояния получены Хелмсом и др. [1385] из анализа переходов / -> /+1
(/ <; 54) для v=0, 1 и 2 во вращательном спектре BaS и хорошо согласуются с менее точными
данными, найденными при исследованиях вращательного [2700а] и электронного [592а ] спектров.
Энергии возбуждения состояний АШш а3И приняты по данным Камминса и др. [93161, которые провели
детальный анализ возмущений в системе 51S + — Х1^ +. Погрешности этих значений не превышают
10 см. Энергии возбуждения остальных состояний оценены по аналогии с оксидами
щелочноземельных металлов, их погрешности составляют 1000 — 4000 см.
Термодинамические функции BaS (г) вычислены по уравнениям A. 3)—A. 10) и A. 93)—A. 95).
Величина QBS и ее производные рассчитывались по соотношениям A. 90)—A. 92) с учетом 11
возбужденных электронных состояний в предположении, что QiH.BV==(pilpx)Qi^^r Статистическая
сумма по колебательно-вращательным уровням энергии состояния X1!!+ и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием по / с
помощью уравнений типа A. 82). В расчете учитывались все уровни со значениями J ^ /шах v,
последние находились по соотношению A. 81). Энергия колебательно-вращательных уровней
рассчитывалась по уравнениям A. 65) и A. 64а) с коэффициентами Ykl, приведенными в табл. 29.5
вместе со значениями vmax и /1]ш. Значения Ykl получены по постоянным из табл. 29.4 с учетом
условий A. 50) и A. 59), для соблюдения которых в уравнение A. 65) введены дополнительные члены
с коэффициентами Ybt>, Yi0 и Y03. Постоянная Ysl определена из условия 5»тах+х=0. Все постоянные
Ykl пересчитаны с помощью уравнений A. 66) для изотопической модификации молекулы BaS,
соответствующей природной смеси изотопов.
Погрешности вычисленных термодинамических функций при температурах до 1500 К в
основном обусловлены неточностью фундаментальных постоянных. При более высоких температурах
становятся существенными погрешности, связанные с неточностью оцененных энергий возбуждения
ряда состояний (главным образом 32+), а также пренебрежением ридберговскими состояниями
BaS. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К оцениваются в 0,02, 0,05 и
0,5 Дж-К-моль~1 соответственно.
Ранее термодинамические функции BaS (г) вычислялись в справочнике Миллса [2031 ] и работах
Цителаури и Самуйлова [431а] (до 3000 К) и Колина и др. [873] (значения Ф° (Г) при 1700, 2000
и 2300 К). Расхождения данных [2031 ] и табл. 870 не превышают 0,5 Дж -К -моль; данные [431а,
873] отличаются от приведенных в табл. 870 приблизительно на 8 Дж -К -моль в значениях Ф° (Т)
и S° (T) при всех температурах. Расхождения обусловлены главным образом тем, что авторы [431а,
873] приняли за основное состояние молекулы триплетное состояние SS+.
Константа равновесия реакции BaS (г)=Ва (r)+S (г) вычислена с использованием принятого
в справочнике значения
ДД0 @) = Do (BaS) = 420 + 15 кДж • моль« 35 100 + 1300 ем.
Эта величина основана на приведенном в табл. 29.17 исследовании равновесия реакции с
участием BaS (г), выполненном Коленом и др. [873]. Измерения давления пара над BaS (к) в работах
Таблица 29.17. Результаты определений Do (BaS) (в кДжмоль)
Авторы
Метод
D0(BaS)
II закон
III закон
Гретидж, Джон, 1952 [1244а] Масс-спектрометрический, испарение с открытой 449 —
поверхности, BaS (к) =- BaS (г), Т (средн.) = 1600 К,
д8Я° @)=460
Никонов, Отмахова, 1961 [286] Эффузионный, BaS (к)=BaS (г), 1400—1600 К, 392 373+30
данные представлены в виде уравнения, A,#° @)=
=533 ±30
Колен и др., 1964 [873] Масс-спектрометрический, Ва (r)+S3 (г)= 430+100 420+15
=BaS (r)+S (г), 1846—2120 К, 10 точек а
а См. текст по выбору АуН° (BaS, к).
415
BaSO4 (к, ж) Глава 29
[286, 1244а] приводят к менее надежным значениям. Указанные в таблице погрешности включают
воспроизводимость измерений, неточность термодинамических функций, термохимических величин
и сечений ионизации. Линейная экстраполяция уровней состояния X1S+ приводит к несколько
более низкому значению 380 кДж-моль.
Принятой величине соответствует
AfH°(BaS, г, 0) = 34,498 + 15 кДж • моль.
BaSO4 (к, ж). Термодинамические свойства кристаллического и жидкого сульфата бария
в стандартном состоянии при температурах 100—4000 К приведены в табл. 871.
Значения постоянных, принятые для расчета термодинамических функций, представлены
в табл. 29.1. За стандартное состояние BaSO4 (к) в интервале 0—1423 К принята ромбическая
а-модификация, а в интервале 1423 — 1853 К — высокотемпературная кубическая |5-модифи-
кация. При Т < 298,15 К расчет термодинамических функций проведен по данным о
теплоемкости, полученным в работе Латимера и др. [1820] A5 — 298 К). Экстраполяция теплоемкости
ниже 15 К по кубическому закону приводит к S° A5 К)=0,70 Дж-К~1-моль. Погрешности
принятых значений S° B98,15 К) и Н° B98,15 К) — Н° @) оцениваются в 0,8 Дж-К^-моль
и 0,08 кДж-моль соответственно. При Т > 298,15 К измерения энтальпии BaSO4 (к)
выполнены в единственной работе Лащенко [219] D23 — 1323 К) в 1908 г. с использованием весьма
несовершенной методики измерений. Эти данные имеют большой разброс (до 10%), резко занижены
по сравнению с данными для других сульфатов щелочноземельных металлов и не учитывались в
справочнике при расчете термодинамических функций BaSO4 (к). Для аппроксимации теплоемкости в
интервале 298,15 — 1423 К использованы значения С\ B98,15 К), С°р E00 К)=30,8 Дж-К^-моль
и С°р A000 К)=38,6 Дж-К^-моль. Два последних рассчитаны по уравнению Cp=2D A86/Г)+
;
выведенному Сакамото [2424] на основании анализа результатов низкотемпературных измерений
теплоемкости BaSO4 в работе Латимера и др. [18201. Погрешность оценки значений С (Т) поэтому
уравнению составляет 3 — 5%. Температура полиморфного превращения A423+10 К) принята
на основании согласующихся между собой результатов [439, 801, 1244, 2584]. Данные [1109,1300,
1959] менее точны и не принимались во внимание. Энтальпия превращения A0+3 кДж-моль)
выбрана при подготовке справочника на основании анализа результатов исследования сульфатов
щелочноземельных металлов, цинка и кадмия методом ДТА [439]. Температура плавления A853 +
+ 30 К) принята по измерениям Каролевы [158а]; результаты других измерений [807а, 926а] менее
надежны и приводят к заниженным значениямA623 — 1723 К). Энтальпия плавления D0+8 кДж-
•моль), теплоемкость высокотемпературной |3-модификации BaS04 A423—1853 К) и жидкого
BaSO4 (Т >• 1853 К) оценены так же, как для SrSO4; последние приняты равными 170 +
+ 20 Дж-К-моль.
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 2000 и 4000 К составляют 0,5,
5, 15 и 35 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
BaSO4 (к), приведенными в табл. 871 и в справочнике [578], достигают 7 Дж -К -моль в значении
Ф° A500 К); они обусловлены тем, что в [578] приняты неверные данные [219] по энтальпии BaSO4 (к)
при высоких температурах.
Константа равновесия реакции BaS04 (к)=Ва (r)+S (г)+4О (г) вычислена с использованием
значения ДГЯ° @)=2891,124+3,9 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A//°(BaSO4, к, 298,15 К) = —1459,0 + 2,5 кДж • моль.
Эта величина основана на экспериментальных данных, приведенных в табл. 29.18. Как видно из
таблицы, результаты определений энтальпии образования BaSO4 (к) разными методами находятся
в хорошем соответствии. Погрешность значения, вычисленного из данных по равновесию реакции
диссоциации, определяется в основном неточностью термодинамических функций BaS04 (к).
Наиболее надежные значения получены в исследованиях растворимости и в калориметрическом
исследовании [2509]. Принятое значение основано на результатах этих измерений.
416
Барий и его соединения ВаСО3 (к, ж)
Таблица 29.18. Результаты определений LjH° (BaSO4, к, 298,15 К) (в кДжмоль)
Авторы
Метод
A,#°(BaS04, к
298,15 К)
Томсен, 1882 [2686] Калориметрическое измерение энтальпии реакции —1457±6
осаждеиия BaSO4 (к) из разных растворов
Шибата, Тэрасаки, 1936 [2509] Калориметрическое измерение энтальпии реакции —1458,0±3,0
ВаС1г (p-p)+K2SO« (p-p)=BaS04 (к)+2КС1(р-р)
а Исследование растворимости BaSO4 (к) —1459,2+2,5 (III)
Мохацаби, Сирсп, 1976 [20451 Исследование равновесия реакции BaSO« (к)= —1476 (II)
=ВаО (lO+SOjfiO+VaO, (г), эффузионный ме- —1461,5 ±15 (III)
тод, 1422—1540 К, результаты измерений
представлены в виде уравнения, h^" @)=- 616,7±
±0,6 (III)
а Исследования растворимости BaSO4 (к) проведены разными методами в работах [370, 710а, 867, 932, 1331, 1460,
1501, 1597, 1744, 1750, 1820, 2002, 2102, 2125, 2367, 2522, 26741 и хорошо согласуются между собой. Для
обработки этих измерений использованы данные из Приложения 5. Погрешность приведенного значения
определяется в основном неточностью величины Ду//° (Ва+2, ад).
ВаСО3 (к, ж). Термодинамические свойства кристаллического и жидкого карбоната бария
в стандартном состоянии в интервале температур 100—3000 К приведены в табл. 872.
Значения постоянных, принятые в расчетах термодинамических функций ВаСО3 (к, ж),
приведены в табл. 29.1. Для карбоната бария известны три кристаллические модификации —
низкотемпературная ромбическая (а) и две высокотемпературные — гексагональная (|3) и кубическая G)
модификации. При Т < 298,15 К термодинамические функции ВаСО3 вычислены на основании
данных о теплоемкости ВаСО3 (к), полученных Андерсоном [509] E5 — 296 К) на образце,
имевшем 0,1% примесей. Экстраполяция теплоемкости ниже 55 К проводилась Андерсоном по
уравнению С' (T)=D (№/Т)+2Е B74/Г)+2Я A730/Г) и дала S0 E4,8 К)=14,56 Дж-К^-моль
-1
Значения теплоемкости при температурах 270—298,15 К скорректированы (увеличены при 298,15К
на —0,8%) для улучшения согласования с данными Ландера [1808] по энтальпии ВаСО3 (к)
при высоких температурах. Погрешности принятых значений S0 B98,15 К) и Н° B98,15 К) — Н° @)
(см. табл. 29.1) оцениваются в 2 Дж-К~1-моль~1 и 0,15 кДж-моль соответственно. В интервале
298,15 — 1083 К для теплоемкости а-ВаСО3 принято уравнение (см. табл. 29.1), выведенное
методом Шомейта по данным об энтальпии, полученным Ландером [1808] D32—1072 К) с погрешностью
около 0,7%. На основании измерений энтальпии Р- и j-модификаций ВаСО3 в работе Ландера для
их теплоемкостей приняты значения 158+3 Дж-К~1-моль и 160+3 Дж-К~1-моль.
Температуры полиморфных превращений A083 +3 К и 1233 +10 К) приняты на основании
анализа многочисленных литературных данных. При этом температура а—Р-превращения
основывается на весьма надежных измерениях Вильмана [2858]. Она подтверждается результатами
измерений [714, 715, 1024, 1603, 1973, 2907] и не противоречит данным менее точных работ [218,
440, 964, 1168а, 1602, 1972, 2145а, 2301, 2762, 2794а]. Температура Р — т-превращения принята
по измерениям [440, 1168а, 2145а, 2762, 2794а]; данные [715, 1340, 1807, 1808, 1973] менее точны
и не учитывались. Энтальпии превращения A6,3 +1,0 кДж -моль и 2,9 +0,5 кДж -моль) получены
в работе Ландера; первая из них не противоречит менее точным измерениям, выполненным Джаддом
и Попом [1604] A6,6 кДж-моль), Лащенко [1815] A5,7 кДж-моль) и Нисино и др. [2145а1
A5 кДж -моль). Температура плавления A828 +15 К) принята по работе Бейкера [562], в которой
найдено, что конгруэнтное плавление при 1828 К происходит при внешнем давлении СО2 в 450 атм.
Данные других авторов приводят к более низким значениям A423 К [1340], 1621 К [2762]). Это
объясняется тем, что соответствующие измерения проводились при внешнем давлении в 1 атм СОа
и относятся к инконгруэнтному плавлению ВаСО3. Энтальпия плавления D0+8 кДж-моль)
и теплоемкость жидкого ВаСО3 A65 +20 Дж -К -моль) оценены так же, как для SrCO3 (см. выше).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000 и 3000 К оцениваются в 2, 3 и
25 Дж -К •моль соответственно. Расхождения между термодинамическими функциями ВаСО3 (к),
приведенными в справочниках [578,1656] и в табл. 872, не превышают 2 Дж-К-моль в
значениях Ф° (Т). Таблица термодинамических функций ВаСО3 (ж) публикуется впервые.
417
ВаСО3 (к, ж)
Глава 2»
Таблица 29.19. Результаты определений ДуЯ° (ВаСО3, к, 298,15 К) (в кДжмоль)
Авторы
Метод
° (BaCO3, к,
298,15 К)
Томсен, 1882 [2686]
Рот, 1941 [2370]
Капустинский, Стаханова, 1954 [154]
Адами, Конуэй, 1966 [481]
Финкельштейн, 1906 [1090]
Ландер, 1951 [1808]
Евстигнеева и др., 1969 [110, 120]
Басу, Сирей, 1976 [613]
Калориметрическое измерение энтальпии реакции —1206
ВаС1? (р-рУ с Na2CO3 (р-р) и К2СОз (р-р)
То же, для знтальшш растворения ВаСОз в азот- —1190
пой кислоте
То же, для растворения ВаСОз в соляной кислоте —1189
(использованы данные [1103] по энтальпии
растворения ВаО)
То же, для растворения ВаСОз и ВаО в соляной —1210,8±5,0
кислоте
Изучение диссоциации ВаСОз (к)^ВаО (к)+ —119fi±15 (III)
+СО2 (г), статический метод, 1623 К, 1 точка
То же, 1073—1477 К, 19 точек —1229 (II)
—1214 (III)
То же, 1203—1273 К, данные представлены в виде —1202 (И)
уравнения —1213+10 (III)
То же, 1031—1099 К, торзионно-эффузионный ме- —1204 (II)
тод, данные представлены в виде уравнения —1217+8 (III)
То же, 1160—1211 К, испарение с поверхности —1199 (Н)
(торзионный) —1300 (III)
Константа равновесия реакции ВаСО3 (к)=Ва (г)+С (г)+ЗО (г) вычислена с использованием
соответствующего принятой в справочнике энталь-
значения ДгЯ°~@)=2837,774 +5,8 кДж-моль
пин образования
-1
Д7Я° (ВаСО3) к, 298,15 К) = —1211,0 ± 5,0 кДж • моль
Эта величина основана на результатах измерений, представленных в табл. 29.19. При оценке
погрешности результатов исследований равновесия диссоциации ВаСО3 (к) учтена неточность
термодинамических функций, приводящая к погрешности 3 и 10 кДж-моль для измерений,
выполненных при 1000 и 1500 К соответственно. Наиболее точным в методическом отношении и по
полученным данным является калориметрическое исследование реакций, составляющих замкнутый
термохимический цикл, в работе Адами и Конуэя [481]. Принятое значение основывается на этой
работе. Более старые калориметрические измерения и результаты исследований диссоциации
уступают им по точности. Известны также измерения методом ДТА [1379] и ТГА [2762], которые
приводят к менее точным значениям, также согласующимся в пределах их погрешности с данными,
полученными другими методами.
В работе Хорлингса и др. [1478а] исследовано равновесие реакции BaSO4 (к)+СО~32 (aq) =
=ВаСО3 (k)+SC>42 (aq) в интервале 293—466 К. Найденным в этой работе константам равновесия
соответствует разность энтальпий образования ВаСО3 (к) и BaSO4 (к) при 298,15 К, равная 257,7 +
+1,0 кДж-моль при расчете по уравнению III закона и 255,5+1,0 кДж-моль при расчете
по уравнению II закона. Вместе с принятой в справочнике величиной \*Н° (BaSO4, к,
298,15 К) это приводит к значениям AfH° (ВаСО3, к, 298,15 К), равным —1201,3 +3,0 и —1203,5 +
+3,0 кДж-моль соответственно,, т. е. отличающимся от принятого. Принятое в справочнике
значение энтальпии образования BaSO4 основано на энтальпии образования Ва+2 (aq), а энтальпия
образования ВаСО3 — на значении AfH° (ВаО, к). Выше (см. ВаС12 (к)) отмечалось несоответствие
данных по энтальпиям образования Ва+2 (ад) и ВаО (к), делающее невозможным выбор
согласованных значений этих величин, а также создание согласованной системы термохимических данных
для соединений бария. Отмеченная разница в значении к^Н° (ВаСО3, к) при его определении из
разных термохимических циклов иллюстрирует эту ситуацию.
ПРИЛОЖЕНИЯ
Приложение 3*
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ГАЗОВ
ПРИ ПОВЫШЕННОМ ДАВЛЕНИИ
П3.9. Обзор данных для веществ III тома
В2Н6. рУТ-свойства B2Hg в газовой и жидкой фазе изучены Смитом и Миллером [2559] в диапазоне
температур 243,6—292,5 К придавлениидо7,6МПаиПаридономидр. [2182] от нормальной точки
кипения до 300 К при давлении до 5,9 МПа. В работе [2559] также измерены давление пара и
ортобарические плотности на линии насыщения в диапазоне температур от 243,6 К до критической
точки. Второй вириальный коэффициент В2Нв при Г=275,16 К измерен в работе [822].
Рифкин и др. [2337] измерили давление пара и теплоемкость жидкости на линии насыщения
в интервале температур 170—282 К, рассчитали энтропию и энтальпию вдоль линии
насыщения и оценили критический объем.
Галбрейт и Мази [11631 обработали рУТ-данные [2182], по уравнению состояния Мартина—Хоу
и рассчитали термодинамические свойства В2Н6 в интервале температур 130—1500 К и давлений
0,001—10 МПа. Ими также обработаны литературные данные по давлению пара и плотности
жидкости, равновесной с паром, и рассчитаны термодинамические свойства на линии насыщения от
тройной точки до критической.
Аппроксимационное уравнение для второго вириального коэффициента В2Нв (табл. Д.64)
принято по данным 11163]. Критические температура и давление измерены Ньюкирком [2133].
Критические постоянные (табл. Д. 65) приняты по работам [2133, 2337].
BF3. рУГ-свойства BF3 исследованы Смитом [2544, 2545] в узком температурном интервале 273—
348 К при давлении до 25 МПа и Ваксманом и др. [2821 ] в интервале температур 273—498 К при
давлении до 25 МПа. Pay [2303] измерил второй вириальный коэффициент BF3 в диапазоне
температур 293—343 К и определил параметры потенциала Леннарда—Джонса (гIk=178 К, а=4,38А).
Смит [2544] воспользовался уравнением состояния Су-Чанга [2633], основанным на теории подобия.
Отклонения рассчитанных таким образом рУГ-данных от экспериментальных составляют 3—5%.
Висванат [2777] обработал рVГ-данные, полученные Смитом [2544, 2545], и рассчитал второй
вириальный коэффициент, поправки к энтальпии на неидеальность и параметры потенциала Леннарда —
Джонса. Сешадри [2495] на основе тех же рУГ-данных построил уравнения состояния Мартина—
Хоу и Бенедикта—Вебба—Рубина в диапазоне температур 273—328 К и давлений 2,7—15,70 МПа.
Критические температура и давление BF3 измерены в работе Бута и Картера [724], критический
объем определен Смитом [2545]. Критические постоянные BF3 (табл. Д.65) приняты по работам
[724, 2545].
Для расчета второго вириального коэффициента принят потенциал Леннарда—Джонса с
параметрами, определенными в работе [2303]. Аппроксимационное уравнение (табл. Д. 64) принято
по работе Зубарева и др. [137] (см. т. 1, кн. 1, табл. П3.6).
ВС13, ВВг3, В13. Нисельсон и др. [290] измерили ортобарическую плотность ВС13 в интервале
температур от 259 до 455 К, ВВг3 — от 273 до 581 К, В13 — от 319 до 773 К. По максимуму кривой
р (Г) на линии насыщения ими определены критические температуры для этих веществ, а по правилу
прямолинейного диаметра определены критические плотности.
Критическое давление ВС13 оценено в работе [2630] экстраполяцией данных по давлению
пара. Хоз и др. [1371, 2303а] на основании измерения вязкости ВС13 определили параметры
потенциала Леннарда—Джонса (s/A;=346 К, а=5,10 А).
Аппроксимационное уравнение для второго вириального коэффициента ВС13 (табл. Д.64)
принято по работе Зубарева и др. [137] (см. т. 1, кн. 1, табл. П3.6) с использованием параметров
потенциала Леннарда—Джонса из работы [2303а]. Для оценки второго вириального коэффициента
* Продолжение. Начало Приложения 3 см. в т. 1, кн. 1, с. 397 и в т. 2, кн. 1, с. 352.
420
Термодинамические свойства газов при повышенном давлении Приложение 3
ВВг3 и В13 использована корреляция Мартина (см. т. 1, кн. 1, раздел П3.5). Необходимые для
расчета значения критического давления ВВг3 и В13 определены в предположении термодинамического
подобия всех трех веществ: ZKp=0,255.
Значения Гкр и 7кр для ВС13 (табл. Д.65) приняты по работе [290], Ркр получено экстраполяцией
данных [2630] по давлению паров ВС13 к Гкр. Для ВВг3 и В13 критические параметры (табл. Д.65)
приняты по работе [290].
Галогениды алюминия. Галогениды алюминия представляют собой диссоциирующие смеси,
состоящие из мономеров А1Х3 и димеров А12Хв, где Х=С1, Вг или I. Такие системы обладают всеми
свойствами индивидуального вещества, имеют постоянные точки плавления и кипения,
критические параметры и единую линию насыщения. Краткий обзор физико-химических свойств галоге-
нидов алюминия представлен в работе [279]. В настоящем справочнике приведены данные по
константам равновесия реакций диссоциации А12Хв и сделан выбор значений энтальпий
диссоциации. На основе этих данных может быть рассчитан состав и свойства идеальной
диссоциирующей смеси.
Для хлорида алюминия в широком интервале температур имеются экспериментальные данные
по давлению насыщенного газа и ортобарической плотности. В работахЧНО, 111 ] измерено давление
пара от тройной D67 К) до критической точки. В работе [2776] измерено давление пара над
твердой и жидкой фазами в диапазоне температур от 387 до 529 К и определено значение температуры
тройной точки. Ортобарическая плотность хлорида алюминия во всем диапазоне жидкого состояния
измерена в работах [110, 289, 1580, 1714, 2488]. На основании полученных данных авторы этих
работ определили значения критических постоянных (Тщ и ркр). В работе [110] также определено
значение критического давления. В ряде работ [110, 2561, 2775а, 2784] выполнены измерения pVT-
свойств ненасыщенных паров хлорида алюминия. В работе [2775а] проведен большой объем
измерений в низкотемпературной области D40—550 К) при давлении от 0,02 до 0,4 МПа и получены
для уравнения Ван-дер-Ваальса (p-\-a/V2) (V— b)=RT, где V — мольный объем, определенный как
произведение удельного объема на молекулярный вес А12С16, значения констант а=56,382 атм • дм6 X
Хмоль, 6=0,1799 дм3 -моль. Это уравнение эффективно учитывает как диссоциацию, так и
«физическую» неидеальность смеси. В работе [278] выполнен расчет константы равновесия реакции
диссоциации хлорида алюминия и степени диссоциации в диапазоне температур 460—2000 К и
давлений 0,1—15 МПа в предположении идеальности диссоциирующей смеси.
Для бромида алюминия имеются данные по давлению пара и ортобарической плотности в
широком интервале температур. В работах Джонсона и др. [1581, 1582] выполнены измерения
ортобарической плотности в интервале температур от 374,3 К до критической точки и давления пара в
интервале температур 536—761 К, а также определены критические постоянные. Олсон и др. [2160]
получили данные по ортобарической плотности в диапазоне температур 365—592 К и оценили
значение критической плотности. Нисельсон и др. [290] измерили ортобарическую плотность бромида
алюминия в интервале температур 385—768 К и определили критические температуру и
плотность. В той же работе приведены данные по ортобарической плотности иодида алюминия в
интервале температур] 484—990 К, также определены критические температура и плотность.
Для оценки второго вириального коэффициента галогенидов алюминия (табл. Д.64) принято
уравнение Ван-дер-Ваальса. Константы уравнения для хлорида алюминия приняты по работе
[2775а]. В этой же работе показано, что грубая оценка констант через критические постоянные
(TKV и FKp) дает неплохое согласие с результатами, полученными из /?УГ-данных. Этот способ оценки
применен для бромида и иодида алюминия. При этом второй вириальный коэффициент отнесен
к 1 молю А12Х6 (pvMAi2xJRT=l-\-B/V, где V=vMAbxe) и учитывает отклонения от
термодинамических свойств идеальных паров А12Хв> связанные как с диссоциацией, так и межмолекулярным
взаимодействием.
Критические постоянные (табл. Д.65) приняты для хлорида алюминия по работе [110], для
бромида алюминия—по работам [1581, 1582] и для иодида алюминия — по работе [290].
Оценки критических постоянных элементов. Для подавляющего большинства элементов
отсутствуют экспериментальные данные о параметрах критического состояния, поскольку оно
реализуется при высоких температурах (Т ^> 3000 К), обычно недостижимых в условиях эксперимента.
Поэтому в литературе используются оценки, основанные на корреляционной связи критических
параметров и других физических свойств, измеряемых в низкотемпературной области. Существуют
421
Приложение 3
Термодинамические свойства газов при повышенном давлении
корреляции между критической температурой и точкой кипения, теплотой испарения, поверхностным
натяжением и т. д. Используются также некоторые физические модели: принципы соответственных
состояний [4186, 1286г, 2064а], модель Ван-дер-Ваальса [2886а], закон прямолинейного диаметра
[12866]. Обзор различных методов оценки критических постоянных приведен в работах [4186,
2157а]. Однако для элементов с высокотемпературным критическим состоянием, в особенности
для металлов, эти методы дают резко расходящиеся оценки. В табл. П3.17 приведены для
иллюстрации значения критических параметров Be и Mg, оцененные различными методами.
Т а б л и г
Элемент
{ а
Be
Mg
П3.17.
V к
6153
8800
3590
8080
2535
3850
3500
3590
Сводка различных
Ржр, МПа
дм3
4,8
922
—
1170
25,7
175
146,9
198
оценок
• моль"
3,115
0,025
—
0,016
0,239
0,059
0,064
0,043
критических параметров Be и Mg
Литература
1177а]
2064а]
418а]
41861
[1177а]
[1992а]
[2064а]
[4186]
Метод
Ивязь с параметрами в точке кипения
Принцип соответственных состояний
Модель Ван-дер-Ваальса
Обобщенный принцип соответственных состояний
Связь с параметрами в точке кипения
Закон прямолинейного диаметра
Принцип соответственных состояний
Обобщенный принцип соответственных состояний
Для металлов наибольшее распространение получил метод Гроссе [12866, 1286г], который
развил специальный вариант принципа соответственных состояний. В этом методе принимается, что
мольная энтропия испарения всех металлов является универсальной функцией приведенной
температуры Гг=77Ткри тогда Ткр определяется по данным об энтальпии испарения. Критическую
плотность Гроссе оценивает по правилу прямолинейного диаметра, т. е. полагая, что (р' + р")/2 является
линейной функцией температуры, а критическое давление определяет экстраполяцией
экспериментальных данных по давлению пара к ТкГ
Юнг и Альдер [2886а] использовали для оценки критических параметров большой группы
металлов обобщенную модель Ван-дер-Ваальса для жидкости жестких сфер. Эта модель
позволила выразить критические постоянные через два параметра: диаметр жестких сфер и константу
а Ван-дер-Ваальса, которые определяются из данных по сжимаемости при низких температурах.
Фортов, Дремин и Леонтьев [4186], рассмотрев существующие методы оценки критических
постоянных металлов, предложили улучшенный вариант принципа соответственных состояний. Для
определения критической температуры использовалась, так же как в методе Гроссе, зависимость
энтропии испарения от Тг, причем оценки проводились по кривой цезия. Критическая плотность
определялась по графику зависимости безразмерной величины 2TrpKV/(p'-\-p") от Тг. Давление
в критической точке оценивалось из условия
0,367 для элементов II группы,
= \ 0,22 для В, Se, S,
0,285 для остальных элементов.
В табл. Д.65 приведены для 10 элементов (В, Al, Ga, In, Tl, Be, Mg, Ga, Sr, Ba) оценки
критических параметров, полученные в работе [4186].
Приложение 5
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ВОДНЫХ ИОНОВ
В СТАНДАРТНОМ СОСТОЯНИИ
В III и IV том справочника входит большое число кристаллических солей, являющихся сильными
электролитами. Во многих случаях их энтальпии образования вычисляются на основании
измерений их энтальпии растворения. Энтальпии растворения экстраполируются к бесконечному
разведению, и полученные таким образом величины &.aqH° (со Н2О) позволяют по уравнениям типа
ДД°(СаС1я, к, 298,15К) = —ДД1й-°(соН20, 298,15 К)+ A^°(
станд. сост.) -(- 2AjH° (СГ, р-р Н2О, станд. сост.)
2, р-р Н2О,
вычислять энтальпии образования соответствующих кристаллических солей.
При расчетах энтальпии образования веществ на основании измерений их растворимости
помимо энтальпии образования используются также значения энтропии ионов в водном растворе
в стандартном состоянии (см. обсуждение связанных с этим вопросов в [253а]).
В справочнике для обеспечения внутренней согласованности приводимых в нем данных
использованы единые значения необходимых для этих расчетов термодинамических свойств водных ионов
в стандартном состоянии. Ниже дана таблица этих величин (табл. П5.1). Большинство их принято
по рекомендации КО ДАТА—MCHG [864] (см. также [253а]). Часть величин, не вошедших в эти
рекомендации, основываются на данных справочника «Термические константы веществ» [406].
Эти величины отмечены звездочкой. Выбор энтальпии образования иона Mg+2 (aq) обсуждается
в тексте по MgCl2 (к).
Энтальпии разведения растворов, необходимые для вычисленияДаг#°(ооН20), принимались как
правило по справочнику [406]. Значения теплоемкостей водных ионов в стандартном состоянии
принимались по данным приложения V к X выпуску справочника [406], а изменения теплоемкости
при переходе от стандартного состояния к конечным концентрациям — по работе Паркер [2185].
Таблица П5.1. Принятые значения термодинамических свойств водных ионов в стандартном состоянии
ион
Н+
он-
F-
С1-
Вг
I-
SO;2
HS-
HSOj
NOj
NHf
COJ*
HCOf
Sn+*
Pb+*
Ga-1*
In*»
AfH° B98,15 K)
кДж • моль
0
—230,025±0,045
-335,35±0,65
—167,0S0±0,088
—121,50±0,15
—56,90+0,10
—909,60 ±0,40
—16,8±0,9
—887,2±1,4
—206,85±0,40
—133,26±0,25
—675,23±0,25
—689,93±0,20
—8,9±0,8
0,92±0,25
—214,2 •
—132,6 ¦
S° B98,15 K)
Дж • К • моль
0
-10,71 ±0,20
—13,18±0,54
56,73±0,16
82,84±0,20
106,70±0,20
18,83±0,50
65,8±4,0
—131,8±1,4
146,60±0,40
lll,17±0,75
—50,0±l,0
98,43±0,60
—15,8+4,0
17,7±0,8
—330,4 »
—252,6 •
JfiOH
Tl+
Zn+2
Cd+a
Hg+2
Cu+2
A»+
Be+2
Ca+a
Sr+*
Ba+a
Li+
Na+
K+
Rb+
Cs+
AfH° B98,15 K)
кДж • моль
5,52±0,25 *
—153,39 ±0,20
—75,88±0,60
170,16±0,20
65,69+0,80
105,750±0,085
—377,4±1,3 *
—466,4±l,0
—543,10±0,80
—551,5±0,60 *
—524,0±2,0 *
—278,455±0,090
—240,300±0,065
—252,17±0,10
—251,12±0,13
—258,04±0,13
S° B98,15 K)
Дж • К • моль
126,3±l,0 *
—109,6±0,7
—72,8±1,2
-36,32±0,80
97,1 ±1,2
73,38±0,40
-126,637 ±5,0*
-128,4 ±4,5
—56,4±0,4
—33,1 ±3,0*
8,4±1,3 *
ll,30±0,35
58,41 ±0,20
101,04±0,25
120,46±0,40
132,84±0,40
ЛИТЕРАТУРА
1. Абаулина д. И., Заварицкий Н. В. — Журн. экспе-
рим. и теорет. физ., 1955, 28, с. 250.
1а.Абашкин Ю. Г., Дементьев А. И. — Журн.
структур, хим. (в печати).
16.Абрикосов Н. X., Лян-Цзунъ-У, Шишков Ю. М. —
Изв. АН СССР. Отд. техн. наук, 1960, № 4, с. 156.
2. Агафонов И. Л., Николаева Л. Г., Аглиулов Н. X. —
Журн. физ. хим., 1974, 48, с. 1065.
2а.Акишин П. А., Наумов В. А., Татевский В. М. —
Вестн. МГУ. Сер. хим., 1959, № 1, с. 229.
26.Акишин П. А., Наумов В. А., Татевский В. М. —•
Кристаллография, 1959, 4, с. 194.
3. Акишин П. А., Никитин О. Т., Горохов Л. Н. —
Докл. АН СССР, 1959, 129, с. 1075.
4. Акишин П. А., Рамбиди Н. Г., Засорин Е. 3. —
Кристаллография, 1959, 4, с. 186.
5. Акишин П. А., Спиридонов В. П. —
Кристаллография, 1957, 2, с. 475.
6. Акишин П. А., Спиридонов В. П. — Журн. физ.
хим., 1*658, 32, с. 1682.
7. Акишин П. А., Спиридонов В. П. — Докл.
АН СССР, 1959, 129, с. 1317.
8. Акишин П. А., Спиридонов В. П. — Докл.
АН СССР, 1960, 131, с. 557.
9. Акишин П. А., Спиридонов В. П. — Журн.
структур, хим., 1961, 2, с. 63.
10. Акишин П. А., Спиридонов В. П. — Журн.
структур, хим., 1962, 3, с. 267.
11. Акишин П. А., Спиридонов В. П., Соболев Г. А. —-
Журн. физ. хим., 1957, 31, с. 648.
12. Акишин Н. А., Спиридонов В. П., Соболев Г. А. —
Докл. АН СССР, 1958, 118, с. 1134.
13. Акишин П. А., Спиридонов В. П., Соболев Г. А.,
Наумов В. А. — Журн. физ. хим., 1957, 31, с. 461.
14. Акишин П. А., Спиридонов В. П., Соболев Г. А.,
Наумов В. А. — Журн. физ. хим., 1957, 31, с. 1871.
15. Акишин П. А., Спиридонов В. П., Соболев Г. А.,
Наумов В. А. — Журн. физ. хим., 1958, 32, с. 58.
15а.Альтман А. К. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1981.
16. Амоненко В. М., Иванов В. Е., Тихвинский Т. Ф.,
Финкелъ В. А., Шпагин И. В. — Физ. металлов
и металловедение, 1961, 12, с. 865.
17. Антонова М. М. Свойства гидридов металлов:
Справочник. Киев: Наукова думка, 1975.
18. Апин А. Я., Лебедев Ю. А., Нефедова О. И. —
Журн. физ. хим., 1958, 32, с. 819.
19. Арифов П. А., Сиражиддинов Н. А. — В кн.:
Физико-химические исследования глинистых
минералов и силикатных материалов. Ташкент: Фан,
1970, с. 61.
20. Ария С. М., Морозова М. П., Семенов Г. А.,
Рыбакова Г. А. — Журн. физ. хим., 1971, 45, с. 181.
21. Аужбикович А. 9. — Легкие металлы, 1936, 5,
№ 7, с. 52.
22. Ахачинский В. В., Иопытин Л. М., Сенин М. Д. —
Атомная энергия, 1970, 28, с. 245.
23. Байков В. И. — Оптика и спектроскопия, 1968, 25,
с. 356.
24. Вайков В. И. — Оптика и спектроскопия, 1969, 27,
с. 923.
25. Байков В. И. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1973.
26. Баймаков А. Ю. ¦— Труды/Ленингр. политехи,
ин-т, 1957, № 188, с. 156.
27. Банашек Е. И., Соколов В. А., Рубинчик С. М.,
Фомин А. И. — Изв. АН СССР. Неорган, матер.,
1965, 1, с. 698.
27а.Банчила С. Н. Автореф. дис. . . . канд. физ.-мат.
наук. М.: МГУ, 1970.
276. Банчила С. Н. — Теплофизика высоких
температур., 1979, 17, с. 507.
28. Банчила С. Н., Палчаев Д. К. — В кн.: Прикладная
физика твердого тела. Махачкала: Дагестанское
книжное изд-во, 1973, с. 162.
29. Баранова М.К.,Гаврилов Г. М. — Научные труды/
Гиредмет. Москва, 1966.
30. Бархатов Л. С, Каган Д. Н., Кенисарин М. М.,
Чеховский В. Я., Шпилърайн Э. Э. — Теплофизика
высоких температур, 1975, 13, с. 525.
30а.Бархатов Л. С, Каган Д. Н., Цыцаркин А. Ф.,
Шпилърайн Э. 9., Якимович К. А. — Теплофизика
высоких температур, 1973, И, с. 1188.
31. Бекетов Н. — Bull. Acad. Sci. Russ., 1892, 34,
p. 291.
32. Белоусов В. И., Сидоров Л. Н. — Журн. физ. хим.,
1970, 44, с. 254.
33. Белоусов В. И., Сидоров Л. Н., Комаров С. А.,
Акишин П. А. — Журн. физ. хим., 1967, 41, с. 2969.
34. Беляев А. И., Фирсанова Л. А. — Изв. вузов.
Цвет, мет., 1958, № 1, с. 116.
35. Белых Л. П., Несмеянов А. Н. — В кн.: Физико-
химические основы производства стали. М.:
Изд-во АН СССР, 1961, с. 342.
36. Берг Л. О., Рассонская И. С. — Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1953, 22, с. 140.
37. Бергман А. Г., Бухалова Г. А. — Журн. общ. хим.,
1949, 19, с. 603.
38. Бергман А. Г., Генке Т. А. — Журн. Рус. физ.-
хим. об-ва, 1926, 58, с. 83.
39. Бобров В. С. — В кн.: Физико-химические
свойства растворов. Л.: ЛГУ, 1964, с. 40.
40. Богданов В. И., Векилов Ю. X., Цагарейшвили Г. В.,
Жгенти Н. М. — Физ. твердого тела, 1970, 12,
с. 3333.
41. Богословский С. С, Крестовников А. Н. — Изв.
АН СССР. Металлы, 1968, № 1, с. 226.
42. Бодров Н. В., Николаев Г. И. — Журн.
спектроскопии, 1974, 21, с. 400.
43. Боженко К. В., Чаркин О. П. — Журн. структур,
хим., 1977, 18, с. 219.
44. Болгар А. С, Гордиенко С. П., Рыклис Э. А.,
Фесенко В. В. —-В кн.: Химия и физика нитридов.
Киев: Наукова думка, 1968, с. 151.
45. Болотин А. Б., Ляш А. В., Литинский А. О. —
Литовск. физ. сб., 1972, 12, с. 253.
424
Литература
46. Большаков К. А., Федоров П. И., Шахов М. Н. —
Научн. докл. высш. школы. Хим. и хим. технол.,
1958, № 3, с. 408.
47. Ворвенкова М. П., Галина В. Н., Новоселова А. В. —
Изв. АН СССР. Неорган, матер., 1970, 6, с. 25.
48. Борзенкова М. П., Новоселова А. В., Симанов Ю. П.,
Черных В. И., Ярембаш Е. И. — Журн. неорган,
хим., 1956, 1, с. 2071.
49. Босик И. И., Новоселова А. В., Симанов Ю. П. —
Журн. неорган, хим., 1961, 6, с. 2563.
50. Бреусов О. II. — Журн. структур, хим., 1961, 2,
с. 173.
51. Будников П. П., Тресвятский С. Г. — Докл.
АН СССР, 1953, 89, с. 479.
52. Будников П. П., Черепанов А. М. — Вопросы
петрографии и минералогии, 1953, 2, с. 241.
53. Бурлакова В. М., Бухалова Г. А. — Изв. вузов.
Хим. и хим. технол., 1968, 11, с. 259.
54. Бухалова Г. А., Бергман А. Г. — Журн. общ.
хим., 1951, 21, с. 1571.
55. Васильев В. Г., Ершова 3. В. — Журн. неорган,
хим., 1972, 17, с. 631.
56. Веденеев А. В., Казарновская Л. И.,
Казарновский И. А. — Журн. физ. хим., 1952, 26, с. 1808.
57. Вейц И. В. — Автореф. дис. . . . канд. хим. наук.
М.: ИГИ, 1957.
58. Вейц И. В., Гурвич Л. В. — Оптика и
спектроскопия, 1956, 1, с. 22.
59. Вейц И. В., Гурвич Л. В. — Журн. физ. хим.,
1957, 31, с. 2306.
60. Вейц И. В., Гурвич Л. В. — Оптика и
спектроскопия, 1957, 2, с. 145.
61. Вейц И. В., Гурвич Л. В. — Оптика и
спектроскопия, 1957, 2, с. 274.
62. Вейц И. В., Гурвич Л. В. — Докл. АН СССР, 1967,
173, с. 1325.
63. Вейц И. В., Гурвич Л. В., Ртищева Н. П. — Журн.
физ. хим., 1958, 32, с. 2532.
63а.Верещагин Л. Ф., Ададуров Г. А., Бреусов О. Н.,
Бурдина К. П., Буренкова Л. Н., Дремин А. Н.,
Зубова Е. В., Рогачева А. И. — Докл. АН СССР,
1968, 182, с. 301.
64. Вечер Д. А., Вечер А. А. — Журн. физ. хим.,
1967, 41, с. 2103.
65. Воздвиженский В. М. — Журн. физ. хим., 1970,
44, с. 317.
66. Войтович Б. А., Барабанова А. С. — Укр. хим.
журн., 1963, 29, с. 1264.
67. Вол А. Е. Строение и свойства двойных
металлических систем. М.: Физматгиз, 1959, т. 1; 1962, т. 2.
68. Волынец Г. А., Коваленок Г. В., Соколов В. А. —¦
Оптика и спектроскопия, 1974, 36, с. 1034.
69. Воробьев А. Ф., Монаенкова А. С. — Вестн. МГУ.
Сер. хим., 1972, 13, с. 182.
70. Воробьев А. Ф., Монаенкова А. С,
Капитонова Т. А., Скуратов С. М. — Журн. неорган,
хим., 1966, И, с. 738.
71. Воробьев А. Ф., Монаенкова А. С,
Скуратов С. М. — Вестн. МГУ, Сер. хим., 1967, 22,
№ 6, с. 3.
72. Воробьев А. Ф., Монаенкова А. С, Скуратов С. М.—
Докл. АН СССР, 1968, 179, с. 1129.
73. Воробьев А. Ф., Привалова Н. М., Монаенкова А. С,
Скуратов С. М. — Докл. АН СССР, 1960, 135,
с. 1388.
74. Воробьев А. Ф., Скуратов С. М. — Журн. физ.
хим., 1958, 32, с. 2580.
75. Воробьев А. Ф., Умярова Р. С, Урусов В. С. —
Журн. общ. хим., 1974, 44, с. 979.
76. Воронин В. А., Сандулова А. В. — В кн.: Сборник
трудов по полупроводниковым материалам,
приборам и их применению. Воронеж, 1968, с. 65.
77. Гаджиев С. #"., Шарифов К. А. — В кн.: Вопросы
металлургии и физики полупроводников: Тр. 4-го
совещания. М.: Изд-во АН СССР, 1961, с. 43.
78. Галицкий Н. В., Минина К. П. —-В кн.: Общая
и прикладная химия. Минск: Вышэйш. школа,
1969, вып. 1, с. 35.
79. Галкин Н. П., Туманов Ю. Н., Бутылкин Ю. П.
Термодинамические свойства неорганических
фторидов. М.: Атомиздат, 1972.
80. Гальченко Г. Л. Автореф. дис. . . . докт. хим. наук.
М.: МГУ, 1972.
81. Гальченко Г. Л., Корнилов А. Н., Скуратов С. М. —
Журн. неорган, хим., 1960, 5, с. 2141.
82. Гальченко Г. Л., Корнилов А. Н., Скуратов С. М. —
Журн. неорган, хим., 1960, 5, с. 2651.
83. Гальченко Г. Л., Корнилов А. Н., Тимофеев Б. И.,
Скуратов С. М. — Докл. АН СССР, 1959, 127,
с. 1016.
84. Гальченко Г. Л., Тимофеев Б. И., Макаренко Г. Н.,
Самсонов Г. В. — Журн. физ. хим., 1970, 44, с. 2469.
85. Гальченко Г. Л., Тимофеев Б. И., Скуратов С. М. —
Журн. неорган, хим., 1960, 5, с. 2645.
86. Генов Л. X., Несмеянов А. Н., Приселков Ю. А. —
Докл. АН СССР, 1961, 140, с. 159.
87. Герцберг Г. Электронные спектры и строение
многоатомных молекул. М.: Мир, 1969.
87а.Гершиков А. Г. Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1979.
88. Голубева М. С, Бергман А. Г. — Журн. общ. хим.,
1956, 26, с. 328.
89. Гомельский К. 3. — Журн. общ. хим., 1958, 32,
с. 1859.
90. Горохов Л. Н., Московская М. Ф. — В кн.:
Институт высоких температур АН СССР: Важнейшие
результаты научно-исследовательских работ за
1974 год. М.: Наука, 1975, с. 34.
91. Гринберг Я. X., Жуков 9. Г., Коряжкин В. А. —
Докл. АН СССР, 1969, 184, с. 847.
92. Гринберг Я. X., Жуков Э. Г., Коряжкин В. А. —
Докл. АН СССР, 1970, 190, с. 589.
93. Гринберг Я. X., Жуков Э. Г., Коряжкин В. А.,
Медведева 3. С. — Журн. неорган, хим., 1969, 14,
с. 2583.
94. Гринберг Я. X., Лужная Н. П., Медведева 3. С. —
Изв. АН СССР. Неорган, матер., 1966, 2, с. 2130.
95. Гринберг Я. X., Лужная Н. П., Медведева 3. С. —
В кн.: Химия фосфидов с полупроводниковыми
свойствами. Новосибирск: Наука, 1970, с. 11.
96. Гурвич Л. В. Автореф. дис. . . . канд. хим. наук.
М.: ИГИ, 1957.
97. Гурвич Л. В. — Журн. физ. хим., 1960, 34, с. 1691.
98. Гурвич Л. В., Вейц И. В. — Изв. АН СССР. Сер.
физ., 1958, 22, с. 673.
99. Гурвич Л. В., Карачевцев Г. В., Кондратьев В. Н.,
Лебедев Ю. А., Медведев В. А., Потапов В. К.,
Ходеев Ю. С. Энергии разрыва химических связей.
Потенциалы ионизации и сродство к электрону.
М.: Наука, 1974.
100.Гурвич Л. В., Новиков М. М., Рябова В. Г. —
В кн.: Труды комиссии по спектроскопии АН СССР,
1964, вып. 1, с. 560.
101.Гурвич Л. В., Новиков М. М., Рябова В. Г. —
Оптика и спектроскопия, 1965, 18, с. 132.
102.Гурвич Л. В., Рябова В. Г. — Теплофизика
высоких температур, 1964, 2, с. 401.
425
Литература
103. Гурвич Л. В., Рябова В. Г. — Оптика и
спектроскопия, 1965, 18, с. 143.
104. Гурвич Л. В., Рябова В. Г., Хитрое А. И., Ста-
ровойтов Е. М. — Теплофизика высоких
температур, 1971, 9, с. 290.
105. Гурвич Л. В., Хачкурузов Г. А., Медведев В. А.,
Вейц И. В., Бергман Г. А., Юнгман В. С,
Ртищева И. Я., Куратова Л. Ф., Юрков Г. Я.,
Кане А. А., Юдин В. Ф., Броунштейн Б. И.,
Байбуз В. Ф., Квливидзе В. А., Прозоровский Е. А.,
Воробьев Б. А. Термодинамические свойства
индивидуальных веществ / Под ред. В. П. Глушко.
М.: Изд-во АН СССР, 1962, т. 1—2.
106. Гурвич Л. В., Юнгман В. С, Хачкурузов Г. А.,
Бергман Г. А., Медведев В. А., Ртищева Я. П.,
Броунштейн Б. И., Вейц И. В., Коробов В. В.,
Куратова Л. Ф. Термодинамические свойства
компонентов продуктов сгорания. М.: Изд-во
АН СССР, 1956, т. 1-3.
107. Гуркова Г. В., Ковтуненко П. В., Бундаль А. А. —
Труды Моск. хим.-технол. ин-та им. Д. И.
Менделеева, 1962, вып. 39, с. 72.
107а.Гусаров А. В., Пятенко А. Т., Горохов Л. Я. —
Теплофизика высоких температур, 1980, 18, с. 961.
108. Густавсон Г. — Журн. Рус. физ.-хим. об-ва,
1886, 17, с. 57.
108а.Девина О. А., Ходаковский И. Л., Ефимов М. Е.,
Медведев В. А. — VII Всесоюз. конф. по
калориметрии. Москва, янв. 1977 (расширенные тез.
докл.). Черноголовка, 1977, с. 231.
109. Девятых Г. Г., Юшин А. С. — Журн. неорган,
хим., 1969, 14, с. 2015.
109а.Демиденко А. Ф., Кощенко В. И., Сабанова Л. Д.,
Гран Ю. М. — ВИНИТИ. Деп. 610-75. М., 1975.
110. Денисова Я. Д., Баскова А. П. — Журн. физ.
хим., 1969, 43, с. 2353.
111. Денисова Я. Д., Сафронов Е. К. — Докл. АН СССР,
1968, 183, с. 648.
112. Денисова Я. Д., Сафронов Е. К., Быстрова О. Н. —
Журн. неорган, хим., 1966, 11, с. 2412.
113. Десятник В. Я., Катышев С. Ф., Мельников Ю. Т.,
Раепопин С. П. — В кн.: Физическая химия
и электрохимия расплавов солей и твердых
электролитов. Свердловск, 1973, ч. 1, с. 6.
114. Дирссен Д., Иванова Е. К., Орен К. — Вестн.
МГУ. Сер. хим., 1969, № 1, с. 41.
115. Долгов Е. Л. Автореф. дис. . . . канд. хим. наук.
Львов: Львов, полит, ин-т, 1970.
116. Евсеев А. М., Пожарская Г. В., Несмеянов А. П.,
Герасимов Я. И. — Журн. неорган, хим., 1959,
4, с. 2196.
117. Евсеев П. П., Крючкова Р. А., Уваров В. А.,
Филиппов А. Ф. — Изв. вузов. Черн. мет., 1969,
12, № 9, с. 47.
118. Евстигнеева М. М., Бунделъ А. А. — Труды
Моск. хим.-технол. ин-та им. Д. И. Менделеева,
1969, вып. 62, с. 34.
119. Евстигнеева М. М., Бундель А. А.,
Кондаков Б. В. — Журн. физ. хим., 1969, 43, с. 2613.
120. Евстигнеева М. М., Бундель А. А.,
Кондаков Б. В. — Труды Моск. хим.-технол. ин-та
им. Д. И. Менделеева, 1969, вып. 62, с. 291.
121. Егоров Л. П., Беляев А. И. — Докл. АН СССР,
1966, 170, с. 1356.
122. Ежов Ю. С, Комаров С. А. — Журн. структур,
хим., 1975, 16, с. 662.
123. Ежов Ю. С, Толмачев С. М., Рамбиди Н. Г. .—
Журн. структур, хим., 1970, 11, с. 527.
124. Ежов Ю. С, Толмачев С. М., Рамбиди П. Г. —
Журн. структур, хим., 1972, 13, с. 972.
125. Ежов Ю. С, Толмачев С. М., Спиридонов В. П.,
Рамбиди Н. Г. — Теплофизика высоких
температур, 1968, 6, с. 68.
126. Ерохин Е. В., Жегулъская Н. А., Сидоров Л. Я.,
Акишин П. А. — Изв. АН СССР, Неорган, матер.,
1967, 3, с. 873.
126&.Ефимов М. Е., Кислова Г. Н., Медведев В. А. —
Тезисы докл. VIII Всесоюз. конф. по
калориметрии. Иваново; Ивановский хим.-технол. ин-т,
1979.
127. Ефремова Р. И., Матизен Э. В. — Изв.
СО АН СССР. Сер. хим. наук, 1970, № 1, с. 3.
128. Ефремова Р. И., Матизен Э. В. — Пятая Всесоюз.
конф. по калориметрии, 21—25 июня 1971
(расширенные тезисы докл.). М.: МГУ, 1971, с. 344.
129. Жаброва Г. М., Каденаци Б. М. — Журн. общ.
хим., 1954, 24, с. 1135.
130. Жданов Г. С, Севастьянов Н. Г. — Журн. физ.
хим., 1943, 17, с. 326.
130а. Жегулъская Н. А., Шольц В. Б., Сидоров Л. Н. —
Журн. физ. хим., 1972, 46, с. 1889.
131. Журавлев Н. Я., Кострова Н. А. — Журн. техн.
физ., 1937, 7, с. 1626.
132. Зайцем С. А. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1965.
133. Засорим, Е. 3. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1965.
134. Засорин Е. 3., Рамбиди Н. Г. — Журн. структур,
хим., 1967, 8, с. 391.
135. Здановский А. Б., Дерябина Л. Д. — Журн.
физ. хим., 1965, 39, с. 678.
136. Зиновьев А. В., Кулешов Г. Г. — Изв. АН БССР.
Сер. физ.-энерг. наук, 1970, № 2, с. 47.
137. Зубарев В. Я., Козлов А. Д., Спиридонов Г. А. —
Теплофизика высоких температур, 1971, 9, с. 943.
137а. Зюбина Т. С, Чаркин О. П., Гурвич Л. В. —
Журн. структур, хим., 1979, 20, с. 3.
1376. Зюбина Т. С, Чаркин О. П., Гурвич Л. В. —
Журн. структур, хим., 1979, 20, с. 12.
138. Иванов А. А., Толмачев С. М., Ежов Ю. С,
Спиридонов В. П., Рамбиди Н. Г. — Журн.
структур, хим., 1973, 14, с. 917.
139. Иванов М. И., Карпова Т. Ф. — ВИНИТИ. Деп.
№ 2967—71. М., 1971.
140. Икорникова Н. Ю., Шептунов В. М. — В кн.:
Исследование процессов кристаллизации в
гидротермальных условиях. М.: Наука, 1970, с. 103.
141. Икрами Д. Д., Хайтова М. — Журн. неорган,
хим., 1977, 22, с. 1822.
142. Ильясов И. И. — Журн. неорган, хим., 1968,
13, с. 1659.
143. Ильясов И. И., Рожковская А. В., Бергман А. Г. —
Журн. неорган, хим., 1957, 2, с. 1883.
144. Кандыба В. В., Кантор П. Б., Красовицкая Р. М.,
Фомичев Е. Н. — Докл. АН СССР, 1960, 131, с. 566.
145. Кантор П. Б., Кандыба В. В., Кан Л. С,
Красовицкая Р. М., Фомичев Е. Я. — Укр. физ.
журн., 1962, 7, с. 205.
146. Кантор П. Б., Красовицкая Р. М., Кисель А. П. —
Физ. металлов и металловедение, 1960, 10, с. 835.
147. Кантор П. Б., Фомичев Е. Я. — В кн.: Тепло-
физические свойства твердых тел при высоких
температурах. М., 1969, т. 1, с. 406.
148. Капустинский А. Ф., Голу теин Ю. М. — Изв.
АН СССР. Отд. хим. наук, 1951, с. 192.
426
Литература
449. Капустинский А. Ф., Самойлов О. Я. — Изв.
АН СССР. Отд. хим. наук, 1950, с. 337.
150. Капустинский А. Ф., Самойлов О. Я. — Изв.
АН СССР. Отд. хим. наук, 1952, с. 218.
151. Капустинский А. Ф., Самплавская К. К. —
Журн. неорган, хим., 1961, 6, с. 2237.
152. Капустинский А. Ф., Стаханова М. С. — Докл.
АН СССР, 1947, 57, с. 575.
153. Капустинский А. Ф., Стаханова М. С. — Изв.
АН СССР. Отд. хим. наук, 1947, с. 11.
454. Капустинский А. Ф., Стаханова М. С. — Изв.
АН СССР. Отд. хим. наук, 1954, с. 587.
155. Капустинский А. Ф., Яцимирский К. Б. — Журн.
физ. хим., 1948, 22, с. 1271.
156. Карапетьянц М. X. — Журн. физ. хим., 1953,
27, с. 775.
157. Карапетьянц М. X. Методы сравнительного
расчета физико-химических свойств. М.: Наука, 1965.
158. Кармышин Ю. В., Тощий Е. Е., Шпиль-
райн Э. 9. — Теплофизика высоких температур,
1974, 3, с. 519.
158а. Каролева В. Д. — Химия и индустрия, 1973,
45, с. 258.
1586.Каспаров В. В. Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1980.
А58в.Каспаров В. В., Ежов Ю. С, Рамбиди II. Г. —
Журн. структур, хим., 1979, 20, с. 260.
158г.Каспаров В. В., Ежов Ю. С, Рамбиди Н. Г. —
Журн. структур, хим., 1979, 20, с. 341.
159. Катаев Д. И. — Письма журн. эксперим. и теор.
физ., 1976, 23, с. 152.
160. Кватер Г. С. — Журн. эксперим. и теор. физ.,
1941, 11, № 4, с. 421.
Ш&.Киреев В. А. — Журн. общ. хим., 1946, 16,
с. 1569.
161. Киреев В.А. — Журн. физ. хим., 1948, 22, с. 847.
162. Кириллин В. А., Шейндлин А. Е., Чеховской В. Я. —
Докл. АН СССР, 1960, 135, с. 125.
463. Кириллин В. А., Шейндлин А. Е., Чеховской В. Я.—
Инж.-физ. журн., 1961, 4, с. 3.
464. КиркинаД. Ф., Новоселова А. В., Симанов Ю. П. —
Докл. АН СССР, 1956, 107, с. 837.
165. КиркинаД. Ф., Новоселова А. В., Симанов Ю. Л. —
Журн. неорган, хим., 1956, 1, с. 125.
166. Кирпичев Е. П., Рубцов Ю. И., Манелис Г. В. —
Журн. физ. хим., 1971, 45, с. 1526.
167. Климов В. Л., Кочина Е. А., Краснов К. С,
Морозов Е. В., Данилова Т. Г. — Изв. вузов. Хии.
ж хим. технол., 1970, 13, с. 1104.
468. Князева И. М., Васильев В. П. — Журн. физ.
хим., 1972, 46, с. 2401.
169. Ковтун Г. П., Круглых А. А., Павлов В. С. —
Изв. АН СССР. Металлургия и горное дело, 1964,
JNI 2, с. 177.
169а.Кокове А. Н., Малюгин А. С, Шерешкова В. И. —
Журн. прикл. хим., 1970, 43, с. 683.
170. Колесов В. П., Мартынов А. М., Скуратов С. М.—
Журн. неорган, хим., 1961, 6, с. 2623.
171. Колесов В. П., Попов М. М., Скуратов С. М. —
Журн. неорган, хим., 1959, 4, с. 1233.
172. Колосов И. В. — Журн. неорган, хим., 1965, 10,
е. 2200.
173. Комиссарова Л. Н., Покровский Б. И. — Докл.
АН СССР, 1963, 149, с. 599.
173а. Комшилова О. Н. Автореф. дис. . . . канд. хим.
наук. Минск: БГУ, 1971.
1736. Комшилова О. Н., Поляченок О. Г. — Журн.
физ. хим., 1969, 43, с. 2676.
173в.Комшилова О. П., Поляченок О. Г.,
Новиков Г. И. — Журн. неорган, хим., 1970, 15, с. 251.
174. Кондаков Б. В., Ковтуненко П. В., Бун-
дель А. А. — Журн. физ. хим., 1964, 38, с. 190.
175. Копылова Е. А., Ни Л. П., Захарова М. В.,
Ключников Ю. Ф. — Журн. прикл. хим., 1974,
47, с. 2336.
176. Коренев Ю. М., Новоселова А. В. — Докл.
АН СССР, 1963, 149, с. 1337.
177. Коршунов И. А. — Журн. физ. хим., 1939, 13,
с. 703.
178. Коряжкин В. А. Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1967.
179. Коряжкин В. А., Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1966, № 1, с. 6.
180. Коряжкин В. А., Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1968, № 4, с. 92.
181. Коряжкин В. А., Матвеев В. К. — Вестн. МГУ.
Сер. хим., 1976, № 4, с. 490.
182. Коряжкин В. А., Саламонова А. А. — ВИНИТИ.
Деп. № 3608-75. М., 1975.
183. Коряжкин В. А., Саламонова А. А. — Вестн.
МГУ. Сер. хим., 1975, № 4, с. 487.
184. Коснырев Г. Т., Савинкова Е. И., Вильнян-
ский Я. Е. — Изв. вузов. Цвет, мет., 1966, № 5,
с. 57.
185. Кострюков В. Н., Костылев Ф.А., Саморуков О. П.,
Саморукова Н. X., Чесалина Л. А. — ВИНИТИ.
Деп. № 4332—76. М., 1976.
186. Кострюков В. Н., Стрелков П. Г. — Журн. физ.
хим., 1954, 28, с. 1825.
187. Кочержинский Ю. А., Шишкин Е. А.,
Юпко Л. М. — Огнеупоры, 1969, 34, с. 50.
188. Кочеров П. В., Гелъд П. В. — Труды Уральск,
политехи, ин-та, 1959, сб. 92, с. 141.
189. Кочеров П. В., Гелъд П. В. — Журн. неорган,
хим., 1960, 5, с. 1774. -
190. Кочеров П. В., Гелъд Л. В. — Изв. вузов. Черн.
мет., 1960, 2, с. 15.
191. Кочеров П. В., Гертман Ю. М., Гелъд П. В. —
Журн. неорган, хим., 1959, 4, с. 1106.
191а. Кочеткова Н. М., Реаухина Т. Н. — В кн.:
Вопросы металлургии и физики
полупроводников: Тр. IV совещания по полупроводниковым
материалам. М.: Изд-во АН СССР, 1961, с. 34.
192. Краснов К. С. — Изв. вузов. Хим. и хим. технол.,
1965, 8, с. 871.
193. Краснов К. С, Карасева Н. В. — Оптика и
спектроскопия, 1965, 19, с. 30.
194. Краснов К. С, Максимов А. И. — Журн.
структур, хим., 1962, 3, с. 703.
195. Краснов К. С, Светцов В. И. — Изв. вузов.
Хим. и хим. технол., 1963, 6, с. 167.
196. Краснов К. С, Соломоник В. Г. — Теплофизика
высоких температур, 1972, 10, с. 973.
197. Краснов К. С, ТимошининВ. С, Данилова Т. Г.,
Хандожко С. В. Молекулярные постоянные
неорганических соединений. Л.: Химия, 1968.
198. Красовицкая Р. М., Кантор П. Б., Кан Л. С,
Кандыба В. В., Куцина Л. М., Фомичев Е. Н. —
Журн. физ. хим. 1961, 35, с. 1499.
199. Кренев В. А., Бунин В. М., Евдокимов В. И. —
Изв. АН СССР. Неорган, матер., 1969, 4, с. 801.
200. Кренев В. А., Бунин В. М., Евдокимов В. И. —
Изв. АН СССР. Неорган, матер., 1970, 6, с. 1052.
201. Кренев В. А., Евдокимов В. Л., Бунин В. М. —
В кн.: Термодинамика и кинетика процессов
восстановления металлов. М.: Наука, 1972, с. 124.
202. Крестовников А. Н., Каретников Г. А. — Легкие
металлы, 1934, 3, № 4, с. 29.
203. Крестовников А. Н., Каретников Г. А. — Легкие
металлы, 1935, 4, № 1, с. 16.
427
Лнтература<
204. Крягова А. И. — Журн. прикл. хим., 1948, 21,
с. 561.
205. Кувыркин О. Л., Бреусов О. Н., Новоселова А. В.,
Семененко К. Н. — Журн. физ. хим., 1960, 34,
с. 343.
206. Кудряшова 3. П. — Вестн. ЛГУ. Сер. физ. и хим.,
1965, вып. 4, № 22, с. 172.
207. Кузяков Ю. Я., Татевский В. М., Туниц-
кий Л. Н. — Оптика и спектроскопия, 1960, 9,
с. 156.
208. Кулифеев В. К., Ухлинов Г. А. — Изв. вузов.
Цвет, мет., 1968, 11, № 6, с. 43.
209. Кулифеев В. К., Ухлинов Г. А. — Изв. вузов.
Цвет, мет., 1969, 12, № 2, с. 72.
210. Кулъба Ф. Я., Миронов В, Е., Федоров В. А. —
Журн. неорган, хим., 1961, 6, с. 1586.
211. Курнаков Н. С, Жемчужный С. Ф., Агеева В. А. —
Журн. прикл. хим., 1929, 2, с. 651.
212. Кутпыркин В. Н., Пейзулаев Ш. И., Ту-
ницкий Л. Н. — Физ. сб. Львовского ун-та,
1957, вып. 3, № 8, с. 486.
213. Кушкин Б. И., Родякин В. В., Кузнецов С. И. —
Журн. неорган, хим., 1967, 12, с. 1657.
214. Ландия Н. А. Расчет высокотемпературных тепло-
емкостей твердых неорганических веществ по
стандартной энтропии. Тбилиси: Изд-во АН ГрузССР,
1962.
215. Ландсберг Г. С, Барышанская Ф. С. — Изв.
АН СССР. Сер. физ., 1946, 10, с. 509.
216. Лариков Л. Н., Фальченко В. М., Коблова Е. А. —
Укр. физ. журн., 1966, 11, с. 212.
217. Латимер В. М. Окислительные состояния
элементов и их потенциалы в водных растворах.
М.: Изд-во иностр. лит., 1954.
218. Лащенко П. Н. — Сообщения Донск. политехи,
ин-та, 1913, 2, с. 8.
219. Лащенко П. Н. — Журн. Рус. физ.-хим. об-ва,
1910, 42, с. 1604.
220. Лащенко П. Н., Компанский Д. И. — Журн.
Рус. физ.-хим. об-ва, 1928, 60, с. 579.
221. Лащенко П. Н., Компанский Д. И. — Журн.
прикл. хим., 1935, 8, с. 628.
222. Левина М. Е. — Изв. вузов. Хим. и хим. технол.,
1965, 8, с. 177.
223. Лилич Л. С, Могилев М. Е. — В кн.: Физико-
химические свойства растворов. Л.: ЛГУ, 1964,
с. 90.
224. Логинов М. В., Митцев М. А. — Журн. техн.
физ., 1971, 41, с. 709.
225. Лукашенко Э. Е., Курбатов В. Л. — Журн.
физ. хим., 1970, 44, с. 331.
226. Лукашенко Э. Е., Реутова Г. А. — Журн. физ.
хим., 1970, 44, с. 600.
227. Любимов А. П., Любитов Ю. II. — В кн.:
Обработка стали и сплавов. М.: Ин-т стали, 1957,
№ 36, с. 191.
228. Лютый А. И. Автореф. дис. . . . канд. физ.-мат.
наук. Днепропетровск: Днепропетровский ун-т,
1963.
229. Ляшенко В. С. — Металлург, 1935, 10, № 11, с. 85.
230. Макаров А. В., Никитин О. Т. — Вестн. МГУ.
Сер. хим., 1976. 17, № 6, с. 749.
231. Малкова А. С, Пашинкин А. С. — ВИНИТИ.
Деп. № 950—77. М., 1977.
232. Мальцев А. А. Автореф. дис. . . . докт. хим. наук.
М.: МГУ, 1965.
233. Мальцев А. А., Катаев Д. И. — Вестн. МГУ.
Сер. хим., 1967, № 1, с. 92.
234. Мальцев А. А., Катаев Д. И., Татевский В. М. —
Оптика и спектроскопия, 1960, 9, с. 713.
235. Мальцев А. А., Катаев Д. И., Татевский В. М. —
В кн.: Физические проблемы спектроскопии. М.:-
АН СССР, 1962, т. I, с. 194.
236. Мальцев А. А., Кузяков Ю. Я., ТатевскийВ. М. —
Материалы X Всесоюз. совещания по
спектроскопии. Молекулярная спектроскопия. Львов::
Львовск. ун-т, 1957, т. 1, с. 475.
237. Мальцев А. А., Матвеев В. К., ТатевскийВ. М. —
Докл. АН СССР, 1961, 137, с. 123.
238. Мальцев А. А., Москвина Е. Н., ТатевскийВ. М.—
Материалы X Всесоюз. совещания по
спектроскопии. Молекулярная спектроскопия. Львовг
Львовск. ун-т, 1957, т. 1, с. 465.
239. Мальцев А. А., Шевелъков В. Ф. — Теплофизика
высоких температур, 1964, 2, с. 650.
240. Мальцев А. А., Шевелъков В. Ф. — В кн.:
Колебательные спектры в неорганической химии. М.:-
Наука, 1971, с. 89.
241. Мальцев А. А., Шевелъков В. Ф., Крупников Э. Д. —
В кн.: Оптика и спектроскопия. М.; Л.: АН СССР,
1963, т. 2, с. 7.
242. Мамыкин П. С, Дроздова Т. А. — Журн. прикл.
хим., 1969, 42, с. 2829.
243. Марков Б. Ф., Делимарский Ю. К. — Журн. физ.
хим., 1957, 31, с. 2589.
244. Марков Б. Ф., Делимарский Ю. К., Пан-
ченко И. Д. — Журн. физ. хим., 1955, 29, с. 51.
245. Мартынкевич Г. М. — В кн.: Строение и свойства!
жидких металлов. М., 1960, с. 210.
246. Мартынкевич Г. М. — Журн. физ. хим., 1970,
44, с. 325.
247. Мартынов Ю. М. — Журн. физ. хим., 1971, 45,.
с. 2734.
248. Матвеев В. К., Мальцев А. А., ТатевскийВ. М. —
Вестн. МГУ. Сер. хим., 1961, Л? 1, с. 51.
24Й. Матерн Г., Сапожников Ю. А., Хорджосуканто С.,,
Приселков Ю. А. — Изв. АН СССР. Металлы,
1969, № 3, с. 210.
250. Маткович В. И., Экономи Д., Смит В. Д. —
В кн.: Бор. Получение, структура и свойства:
Материалы IV Междунар. симпоз. по бору. М.:
Наука, 1974, с. 183.
251. Машовец В. П., Пучков Л. В. — Журн. прикл.-
хим., 1965, 38, с. 949.
251а. Машовец В. П., Ревозян А. А. — Журн. прикл..
хим., 1957, 30, с. 1006.
252. Медведев В. А. — Журн. физ. хим., 1958, 32,
с. 1690.
253. Медведев В. А. — Журн. физ. хим., 1961, 35,
с. 1481.
253а. Медведев В. А., Ходаковский И. Л. — Усп.
химии, 1979, 48, с. 2164.
254. Медведева 3. С, Борякова В. А., Гринберг Я. X.,
Жуков Э. Г. — Журн. неорган, хим., 1968, 13,
с. 1440.
255. Меерсон Г. А., Самсонов Г. В. — Изв. сектора^
физ.-хим. анализа ИОНХ АН СССР, 1952, 22,
с. 92.
255а. Международная практическая температурная
шкала 1968 г. Редакция 1975 г. М.: Изд-во
стандартов, 1976.
256. Мечковский Л. А., Вечер А. А. — Журн. физ.
хим., 1969, 43, с. 1346.
257. Микулинский А. С, Умова М. А. — В кн.:
Теория и практика рудной электротермии. Свердловск;
Москва: Уральский НИИхим, 1948.
258. Мищенко К. П., Резников И. Л., Клюева М. Л.,
Соколов В. В., Поляков Ю. А. — Журн. прикл. хим.,
1965, 38, с. 1939.
428
•Литература
259. Мищенко К. П., Стагис А. Я. — Журн. общ.
хим., 1970, 40, с. 2537.
260. Мищенко К. П., Яковлев И. Ф. — Журн. общ.
хим., 1959, 29, с. 1761.
'261. Монаенкова А. С, Воробьев А. Ф. — Изв. вузов.
Хим. и хим. технол., 1972, 15, с. 191.
262. Монаенкова А. С, Пашилова Е. В., Воробьев А.Ф.—•
Докл. АН СССР, 1971, 199, с. 1332.
263. Морозов И. С, Шевцова 3. Н., Клюкина Л. В. —
Журн. неорган, хим., 1957, 2, с. 1639.
64. Морозова М. П., Рыбакова Г. А. — Вестн. ЛГУ.
Сер. физ. хим., 1968, № 22, с. 161.
265. Морозова М. Я., Рыбакова Г. А. — Труды/ ГИПХ,
1970, вып. 66, с. 35.
266. Муку мое С. И., Крылова Н. И., Бергман А. Г. —
Труды/ Ин-т химии АН УзССР, 1942, № 2, с. 94.
267. Мурадов В. Г., Гельд П. В. — Заводская
лаборатория, 1965, 31, с. 820.
268. Мурадов В. Г., Гельд П. В. — Журн. физ. хим.,
1967, 41, с. 2133.
269. Мурадов В. Г., Кочеров П. В. — Изв. вузов.
Цвет, мет., 1966, 9, с. 85.
270. Нарышкин И. И. — Журн. физ. хим., 1939, 13,
с. 528.
271. Нарышкин Н. И. — Журн. физ. хим., 1939, 13,
с. 690.
272. Нарышкин И. И., Глазов В. И., Харламов В. М. —
Журн. прикл. хим., 1968, 41, с. 1329.
273. Наумов Г. Б., Рыженко Б. Н., Ходаковский И. Л.
Справочник термодинамических величин (для
геологов). М.: Атомиздат, 1971.
274. Нахмансон М. С, Барановский В. И. — Теор. и
эксперим. хим., 1971, 7, с. 15.
275. Несмеянов А. Н. Давление пара химических
элементов. М.: Изд-во АН СССР, 1961.
276. Несмеянов А. Н., ФирсоваЛ. П. — Изв. АН СССР.
Отд. техн. наук, 1959, № 3, с. 150.
277. Несмеянов А. Н., Фирсова Л. П. — Журн. физ.
хим., 1960, 34, с. 1032.
278. Нестеренко В. В., Зиновьев А. В., Бажин М. А. —
Изв. АН БССР. Сер. физ.-техн. наук, 1967, № 2,
с. 32. /
279. Нестеренко В. Б., Тимофеев Б. Д. — В кн.:
Диссоциирующие газы, как теплоносители и рабочие
тела энергетических установок. Минск: Наука и
техника, 1970, с. 166.
280. Никитин В. С. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1967.
281. Никитин В. С, Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1966, № 3, с. 40.
282. Никитин В. С, Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1966, № 6, с. 12.
283. Никитин В. С, Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1970, 11, с. 22.
284. Никитин В. С, Мальцев А. А., Пчелкина М. А.,
Виноградова 3. Ф. — Вестн. МГУ. Сер. хим.,
1963, № 3, с. 14.
285. Никитин О. Т., Акишин П. А. — Докл. АН СССР,
1962, 145, с. 1294.
286. Никонов Б. П., Отмахова Н. Г. — Журн. физ.
хим.., 1961, 35, с. 1494.
287. Нисельсон Л. А., Петрусевич И. В. — Журн.
неорган, хим., 1961, 6, с. 748.
288. Нисельсон Л. А., Пустылъник А. И., Гаврилов О. Р.,
Родин В. А. — Журн. неорган, хим., 1965, 10,
с. 2339.
289. Нисельсон Л, А., Соколова Т. Д. — Журн. неорган,
хим., 1965, 10, с. 1516.
290. Нисельсон Л. А., Соколова Т. Д., Николаев Р. К. —
В кн.:: Теплофжзические свойства веществ и
материалов. М.: Изд-во стандартов, 1970, вып. 2,
с. 246.
291. Новиков Г. И., Гаврюченков Ф. Г. — Журн. неорган,
хим., 1964, 9, с. 475.
292. Новиков Г. И., РатьковскийИ. А., КрисъкоЛ. Я.—
Докл. АН БССР, 1970, 14, с. 918.
293. Новиков И. И., Орехова С. Е. — В кн.: Химия
и химическая технология. Минск: Выш.эйш. школа,
1974, вып. 7, с. 12.
294. Новиков М. М. — Автореф. дис. . . . канд. хим.
наук. М.: ИВТАН, 1969.
295. Новиков М. М., ГурвичЛ. В. — Оптика и
спектроскопия, 1965, 19, с. 143.
296. Новиков М. М., Гурвич Л. В. — Оптика и
спектроскопия, 1967, 23, с. 323.
2&7. Новиков М. М., Гурвич Л. В. — Оптика и
спектроскопия, 1967, 22, с. 720.
298. Новиков М. М., Гурвич Л. В. — Журн. прикл.
спектроскопии, 1971, 14, с. 1113.
299. Новиков М. М., Туницкий Л. Н. — В кн.:
Физические проблемы спектроскопии. М.: Изд-во
АН СССР, 1962, 1, с. 192.
300. Новиков М. М., Туницкий Л. Н. — Оптика и
спектроскопия, 1960, 8, с. 752.
301. Новоселова А. В., Левина М. Е., Савельева М. П. —
Журн. неорган, хим., 1958, 3, с. 2562.
302. Новоселова А. В., Левина М. Е., Симанов Ю. П.,
Жасмин А. Г. — Журн. общ. хим., 1944, 14,
с. 385.
303. Новоселова А. В., Муратов Ф. Ш.,
Решетникова Л. П., Гордеев И. В. — Вестн. МГУ. Сер.
мат. механ., 1958, №. 6, с. 181.
304. Ольшанский Я. И. — Докл. АН СССР, 1948, 59,
с. 1105.
304а. Осин С. Б. Автореф. дис. . . . канд. хим. наук.
М.: МГУ, 1978.
3046. Осин С. Б., Серебренников Л. В., Шевельков В. Ф.,
Мальцев А. А. — Вестн. МГУ. Сер. хим., 1978,
19, с. 229.
305. Основные свойства неорганических фторидов:
Справочник/ Под ред. Н. П. Галкина. М.:
Атомиздат, 1976.
305а. Островский А. И., Пенкин Н. П. — Оптика и
спектроскопия, 1958, 4, с. 719.
306. Палкин А. П., Вигутова Т. Н., Глотова Н. И. —
Журн. неорган, хим., 1963, 8, с. 253.
306а. Палкин А. П., Острикова Н. В. — Журн.
неорган, хим., 1964, 9, с. 2043.
307. Палкин А. П., Острикова Н. В., Вигутова Т. Н, —
Журн. неорган, хим., 1964, 9, с. 381.
308. Палюра И. П., ПалкинаА. П. — Журн. неорган,
хим., 1964, 9, с. 2668.
309. Панова Г. X., Самойлов Б. Н. — Журн. эксперим.
и теорет. физ., 1965, 49, с. 456.
310. Панченков И. Г., Гусаров А. В., Горохов Л. Н. —
Журн. физ. хим., 1973, 47, с. 101.
311. Панчук О. Е., Фейчук П. П., Панчук И. Е. —
Изв. АН СССР. Неорган, матер., 1973, 9, с. 1437.
312. Паршина М. П., Ковтуненко П. В. — Журн.
физ. хим., 1974, 48, с. 483.
313. Пауков И. Е., Рахменкулов Ф. С, Врублев-
ская С. С. — ВИНИТИ. Деп. № 6108—73. М.,
1973.
314. Пауков И. Е., Хриплович Л. М., Коротких А. М. —
Журн. физ. хим., 1969, 43, с. 774.
315. ПауковИ. Е., ХрипловичЛ. М., СмирноваО. М. —
Журн. физ. хим., 1971, 45, с. 1204.
316. Пенкин Н. П., Шабанова Л. Н. — Оптика и
спектроскопия, 1965, 18, с. 425.
429
Литература.
317. Пенкин Н. П., Шабанова Л. Н. — Оптика и
спектроскопия, 1965, 18, с. 749.
318. Пепекин В. Л., Дымова Т. И., Лебедев Ю. А. —
Журн. физ. хим., 1964, 38, с. 1024.
319. Перевозчиков В. И., Грибов Л. А. — Оптика и
спектроскопия, 1975, 38, с. 510.
320. Перов П. А., Недяк С. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1974, 15, с. 201.
321. Перов П. А., Шевельков В. Ф., Мальцев А. А. —
Вестн. МГУ. Сер. хим., 1975, 16, с. 109.
322. Першина Е. В., Раскин Ш. Ш. — Оптика и
спектроскопия, 1962, 13, с. 272.
323. Першина Е. В., Раскин Ш. И. — Оптика и
спектроскопия, 1962, 13, с. 488.
324. Петропавловский И. А., Торочешников Н. С. —
Журн. Всес. хим. об-ва им. Д. И. Менделеева,
1969, 14, с. 466.
325. Пинчук Ю. М., Беляев А. И. — Изв. вузов. Цвет,
мет., 1964, № 1, с. 108.
326. Плоткин С. С, Плющев В. Е. — Порошковая
металлургия, 1972, № 1, с. 83.
327. Плотников В. А., Якубсон С. И. — Зап. ин-та
химии АН СССР, 1937, 4, с. 115.
328. Плотников В. А., Якубсон С. И. — Журн. физ.
хим., 1938, 12, с. ИЗ.
328а. Погорелова О. Ф., Кытин Г. А., Орлова М. П. —
Журн. физ. хим., 1977, 51, с. 1065.
329. Половинкина Р. А., Заболоцкий Т. В. — Изв.
СО АН СССР. Сер. хим. наук, 1963, 2, с. 34.
330. Поляченок О. Г. — Журн. неорган, хим., 1967,
12, с. 851.
331. Поляченок Л. Д., Дудчик Г. П., Поляченок О. Г. —
Журн. физ. хим., 1976, 50, с. 387.
331а. Поляченок О. Г., Комшилова О. Н. —
Теплофизика высоких температур, 1972, 10, с. 195.
3316. Поляченок О. Г., Комшилова О. Н. — Изв.
АН СССР. Сер. физ.-энерг. наук, 1970, № 2, с. 90.
332. Понятовский Е. Г., Зазаров А. И. —
Кристаллография, 1962, 6, с. 461.
333. Попов М. М., Галъченко Г. Л. — Журн. общ.
хим., 1951, 21, с. 2220.
334. Портной К. И., Левинский Ю. В., Ромашов В. М.,
Мордовии О. А., Левинская М. X. — Изв.
АН СССР. Металлы, 1967, № 4, с. 171.
335. Приселков Ю. А. Автореф. дис. . . . канд. хим.
наук, М.: МГУ, 1954.
336. Приселков Ю. А., Несмеянов А. Л. — Докл.
АН СССР, 1954, 95, с. 1207.
337. Приселков Ю. А., Сапожников Ю. А., Цеп-
ляева А. В. — Изв. АН СССР. Отд. техн. наук.
Металлургия и топливо, 1959, № 1, с. 106.
338. Приселков Ю. А., Сапожников Ю. А., Цепля-
ева А. В. — Изв. АН СССР. Отд. техн. наук.
Металлургия и топливо, 1960, № 1, с. 134.
339. Приселков Ю. А., Сапожников Ю. А., Цепля-
ева А. В., Карелин В. В. — Ивв. вузов. Хим. и
хим. технол., 1960, № 3, с. 447.
340. Рамбиди Н. Г., Ежов Ю. С. — Журн. структур,
хим., 1968, 9, с. 363.
341. Рамбиди Н. Г., Толмачев С. М. — Теплофизика
высоких температур, 1965, 3, с. 487.
342. Ратьковский И. А., Семенов Г. А. — Изв. вузов.
Хим. и хим. технол., 1970, 13, с. 168.
343. Рахменкулов Ф. С, Фролов Г. И., Пауков И. Е. —
ВИНИТИ. Деп. № 735-76. М., 1976.
344. Резников И. Л. Термодинамика хлорирования
окиси магния в расплавах. М.: ЦНИИцветмет,
1958.
345. Резницкий Л. А., Холлер В. А., Филиппов С. Е. —
Журн. физ. хим., 1970, 44, с. 534.
346. Резухина Т. П., Сысоева Т. Ф., Холохонова Л. И.—
В кн.: Шестая Всесоюз. конф. по калориметрии.
1973 (расширенные тезисы докл.). Тбилиси: Мец-
ниереба, 1973, с. 324.
346а. Рихтер Л. Я., Свердлов Л. М. — Журн. физ*
хим., 1971, 45, с. 1579.
3466. Рихтер Л. Я., Свердлов Л. М. — Журн. физ.
хим., 1975, 49, с. 2712.
347. Родигина Э. Н., Гомельский К. 3. — Журн. физ.
хим., 1955, 29, с. 1105.
348. Родигина Э. Н., Гомельский К. 3. — Журн. физ.
хим., 1961, 35, с. 1828.
349. Романовский В. А., Тарасов В. В. — Физ.
твердого тела, 1960, 2, с. 1294.
350. Рубцов Ю. И., Кирпичев Е. П., Манелис Г. В. —
Журн. физ. хим., 1969, 43, с. 1415.
351. Русин А. Д., ТатевскийВ. М. — Журн. физ. хим.,
1963, 37, с. 716.
352. Русин А. Д., Татевский В. М. — Теплофизика
высоких температур, 1965, 3, с. 547.
353. Русин А. Д., Татевский В. М. — Теплофизика
высоких температур, 1969, 7, с. 62.
354. Рябов М. А., Клименко И. М., Чаркин О. П., Зай
цев Б. Е., Молодкин А. К. — В кн.: Современные
задачи в точных науках. М., 1976, вып. 2, с. 131.
355. Рябова В. Г. Автореф. дис. . . . канд. хим. наук.
М.: ИВТАН, 1965.
355а. Рябова В. Г., Гурвич Л. В. — Теплофизика
высоких температур, 1964, 2, с. 834.
356. Рябова В. Г., Гурвич Л. В. — Теплофизика
высоких температур, 1965, 3, с. 284.
357. Рябова В. Г., Гурвич Л. В. — Теплофизика
высоких температур, 1965, 3, с. 318.
358. Рябова В. Г., Гурвич Л. В. — В кн.: Прикладная
спектроскопия. М.: Наука, 1969, т. 1, с. 258.
359. Рябова В. Г., Гурвич Л. В., Хитрое А. Л., Наза-
ренко И. И., Старовойтов Е. М. — В кн.:
Седьмая Всесоюз. конф. по калориметрии, 31 янв. —
3 февр. 1977. Черноголовка, 1977, 2, с. 293.
360. Рябова В. Г., Хитрое А. Н., Гурвич Л. В. —
Материалы Всесоюз. конф. по спектроскопии,
24—26 мая 1973. Минск, 1973, с. 171.
361. Рябчиков И. В., Микулинский А. С. — Изв.
вузов. Цвет, мет., 1963, № 1, с. 95.
362. Рябчиков Л. Н., Тихинский Г. Ф. — Физ.
металлов и металловедение, 1960, 10, с. 635.
363. Савельев Б. А., Кренов В. А., Евдокимов В. И. —
Журн. неорган, хим., 1973, 18, с. 1413.
364. Самойлов О. Я. — Изв. АН СССР. Отд. хим.
наук, 1952, Л» 4, с. 627.
365. Саморуков О. П., Кострюков В. Н., Сенип М. Д.,
Михаленко И. П. — Журн. физ. хим., 1974, 48,
с. 2437.
365а. Самсонов Г. В., Дроздова С. В. Сульфиды. М.:
Металлургия, 1972.
366. Самсонов Г. В., Цагарейшвили Г. В, — В кн.: Бор.
Получение, структура и свойства: Материалы
IV Междунар. симпоз. по бору. М.: Наука, 1974,
с. 5.
367. СандуловаА. В., Воронин В. А., Прохоров В. А. —
Тр. по химии и хим. технол. (Горький), 1969,
вып. 3, № 24, с. 62.
367а. Секачев Ю. Н. Автореф. дис. канд. хим. наук.
М.: МГУ, 1980.
3676. Секачев Ю. П., Серебренников Л. В.,
Никитин В. С, Мальцев А. А. — ВИНИТИ. Деп.
№ 2059—79. М., 1979.
368. Селиванов Г. К., Мальцев А. А. — Журн. структур,
хим., 1973, 14, с. 943.
430
Литература
369. Селиванова И. М., Зубова Г. А. — Труды Моск. 392.
хим.-технол. ин-та им. Д. И. Менделеева, 1956,
вып. 22, с. 38. 393.
370. Селиванова Н. М., Капустинский А. Ф. — Журн.
физ. хим., 1953, 27, с. 565. 394.
371. Семененко К. Н., Наумова Т. Н. — Журн.
структур, хим., 1963, 4, с. 67. 395.
372. Семененко К. В., Наумова Т. Н. — Журн. неорган,
хим., 1964, 9, с. 1316. 396.
373. Семененко К. Н., Савченкова А. П., Ильина Т. С,
Суров В. Н. — Журн. неорган, хим., 1971, 16, 396а
с. 2939.
374. Семенкович С. А. — Журн. прикл. хим., 1957, 397.
30 с 933
375. Семенкович С. А. — Журн. прикл. хим., 1960, 33, 398.
с. 1281.
376. Семенкович С. А. —Труды Всесоюз. алюминиево-
магниевого ин-та, 1960, № 44, с. 113.
377. Семенкович С. А. — Труды Всесоюз. алюминиево- 399.
магниевого ин-та, 1960, № 44, с. 120.
378. Семенов Г. А., Попков О. С, Соловейчик А. И.,
Персиянинов С. Н. — Журн. физ. хим., 1972, 46, 400.
с. 1568.
378а. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1975, 16, с. 250. 401.
379. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1975, 16, с. 363. 402.
379а. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим. (в печати). 403.
380. Серебренников Л. В., Мальцев А. А. — В кн.:
XI Менделеевский съезд по общей и прикладной 404.
химии: Рефераты докл. и сообщений, № 3. М.:
Наука, 1975, с. 87. 405.
380а. Серебренников Л. В., Осин С. Б., Секачев Ю. Н.,
Мальцев А. А. — Спектроскопия
комбинационного рассеяния света: Материалы II Всесоюз. 406.
конф. М., 1978, с. 243.
3806. Серебренников Л. В., Осин С. В., Секачев Ю. Н.,
Мальцев А. А. — Вестн. МГУ. Сер. хим., 1979, 406а
20, с. 179.
380в. Сидоров Л. Н., Колосов Е. Н., Давыдов В. А., 4066
Шолъц В. Б. — Вестн. МГУ. Сер. хим., 1973, 14,
с. 35. 406в
381. Сидоров Т. А., Соболев Н. Н. — Оптика и
спектроскопия, 1956, 1, с. 393.
382. Смирнов В. А., Половое В. М., Бронников А. Д. —
ВИНИТИ. Деп. Л» 984—75. М., 1975. 407.
383. Смирнов М. В., Чупреев Н. Я. — Журн. неорган,
хим., 1958, 3, с. 2445.
384. Смышляев С. И., Эделева И. П. — Изв. вузов. 407а
Хим. и хим. технол., 1962, 5, с. 871.
385. Соколов В. А., Банашек Е. И., Рубинчик С. М. — 408.
Журн. неорган, хим., 1963, 8, с. 2017.
386. Соколов С. И. — Журн. Рус. физ.-хим. об-ва, 1930,
62, с. 2319. 409.
387. Соколова Н. Д., Скуратов С. М., Шемонаева А. М., 410.
Юлдашева В. М. — Журн. неорган, хим., 1961,
6, с. 774. 411.
388. Соломоник В. Г. Автореф. дис. . . . канд. хим.
наук. Иваново: Ивановский хим.-техн. ин-т, 1977. 412.
389. Соломоник В. Г., Бобкова В. А., Данилова Т. Г.,
Краснов К. С. — Журн. физ. хим., 1975, 49, 413.
с. 2721.
389а. Соломоник В. Г., Засорин Е. 3., Гиричев Г. В., 414.
Краснов К. С. — Изв. вузов. Хим. и хим. технол.,
1974, 17, с. 136. 415.
390. Соломоник В. Г., Краснов К. С. — Журн. прикл.
спектроскопии, 1974, 21, с. 360.
391. Сонин В. И. Автореф. дие. . . . канд. хим. наук. 416.
Минск: БГУ, 1975.
Сонин В. И., Лоляченок О. Г. — Журн. физ. хим.,
1973, 47, с. 1612.
Сорокин В. П., Веснина Б. И., Климова Н. С. —
Журн. неорган, хим., 1963, 8, с. 66.
Софронова А. В., Колобова Л. В. — Сб. научн.
тр. Пермского политехи, ин-та, 1972, № 120, с. 30.
Сперанская Е. И. — Изв. АН СССР. Сер. хим.,
1938, с. 463.
Спиридонов В. П., ЕрохинЕ. В. — Журн. неорган.
хим., 1969, 14, с. 636.
СрывцевВ.А.,ПетровЕ.С. — Изв. СО АН СССР.
Сер. хим. наук, 1969, № 4, вып. 2, с. 7.
Староеойтов Е. М. Автореф. дис. . . . канд. хим.
наук. Иваново: Ивановский хим.-техн. ин-т, 1977.
Староеойтов Е. М., Беляев В. Н., Краснов К. С,
Гурвич Л. В. — 7-я Всесоюз. конф. по
калориметрии, 31 янв. 1977 (расширенные тез. докл.).
Черноголовка, 1977.
Староеойтов Е. М., Беляев В. Н., Краснов К. С,
Лебедева Н. Л. — Труды Ивановен, хим.-технол.
ин-т.а, 1976, вып. 20, с. 90.
Староеойтов Е. М., Рябова В. Г., Гурвич Л. В.,
Хитрое А. Н., Назаренко И. И., Беляев В. И. —
Теплофизика высоких температур, 1977, 15, с. 909.
Суворов А. В., Малкова А. С, Аврорина В. И. —
Журн. неорган, хим., 1969, 14, с. 1374.
Талипов Ш. Т., Хадеев В. А. — Журн. общ. хим.,
1950, 20, с. 774.
Талипов Ш. Т., Хадеев В. А. — Журн. общ. хим.,.
1950, 20, с. 783.
Татевский В. М., Коптев Г. С, Мальцев А. А. —
Оптика и спектроскопия, 1961, 11, с. 724.
Татевский В. М., Туницкий Л. П.,
Новиков М. М.—Оптика и спектроскопия, 1958, 5, с.
520.
Термические константы веществ: Справочник /
Отв. ред. В. П. Глушко. М.: ВИНИТИ, 1965—
1979, вып. 1—9.
. Токарева М. В. — Журн. неорган, хим., 1957, 2,
с. 1591.
. Тонкое Е. Ю. Фазовые диаграммы элементов при
высоком давлении. М.: Наука, 1979.
. Торопов Н.А., Барзаковский В. П., Бондарь И. А.,
Удалое Ю. П. Диаграммы состояния
силикатных систем: Справочник. Л.: Наука, 1970,
вып. 2.
Торопова Т. Г., Погорелый А. Д. — Труды Сев.-
Кавказского горно-металлургич. ин-та, 1961,
вып. 17, с. 38.
, Турдакин В. А., Тарасов В. В. ¦— Журн. неорган,
хим., 1966, 11, с. 931.
Туркова Г. В., Ковтуненко П. В., Бунделъ А. А. —
Труды Моск. хим.-технол. ин-та им. Д. И.
Менделеева, 1962, вып. 39, с. 72.
Урусов В. В. — Докл. АН СССР, 1957, 116, с. 97.
Фаворский Л. И. — Изв. сектора физ.-хим.
анализа ИОНХ АН СССР, 1940, 13, с. 281.
Фадеев В. Н., Федоров П. И. — Журн. неорган,
хим., 1964, 9, с. 381.
Фадеев В. Н., Федоров П. Л. — Журн. неорган,
хим., 1964, 9, с. 2028.
Федоров П. И., Ильина Н. И. — Журн. неорган,
хим., 1964, 9, с. 1207.
Федоров П. И., Фадеев В. И. — Журн. неорган,
хим., 1964, 9, с. 378.
Фейчук Л. И., Панчук О. Е., Щербаков Л. П.,
Антипин Л. Н. — Изв. АН СССР. Неорган,
матер., 1977, 13, о. 164.
Фиалков Я. И., Шаргородский С. Д. — Журн. общ.,
хим., 1948, 18, с. 1747.
431
Литература
416а.Филиппов Л. Л., Благонравов Л. А.,
Алексеев В. А. — В кн.: Физика и физическая химия
жидкостей. М.: МГУ, 1976, с. 152.
417. Фирсова Л. П., Несмеянов А. И. — Журн. физ.
хим., 1960, 34, с. 2615.
418. Флидлидер Г. В., Ковтуненко П. В., Бун-
дельА.А. —Журн. физ. хим., 1966, 40, с. 2168.
418а. Фокеев В. М. — Изв. вузов. Сер. геол. развед.,
1969, 12, с. 45.
4186. Фортов В. Е., Дремин А. Н., Леонтьев А. А. —
Теплофизика высоких температур, 1975, 13,
с. 1072.
419. Фрид С. А., Поляченок О. Г., Новиков Г. И. —
Журн. неорган, хим., 1964, 9, с. 1017.
419а. Хаит Ю. Г., Барановский В. И. — Журн.
структур, хим., 1980, 21, с. 153.
420. Хандамирова Н. 9., Евсеев А. М., Пожарская Г. В.,
Борисов Е. А., Несмеянов А. Л., Герасимов Я. И. —
Журн. неорган, хим., 1959, 4, с. 2192.
420а. Харгиттаи М., Харгиттаи И. Геометрия
молекул координационных соединений в
парообразной фазе. М.: Мир, 1976.
421. Хворостухина Н. А. — Труды Вост.-Сиб. филиала
СО АН СССР, 1962, вып. 41, с. 78.
422. Хворостухина Н. А., Румянцев Ю. В., Скобеев
И. К. — Труды Вост.-Сиб. филиала СО АН СССР,
1962, вып. 41, с. 67.
423. Хвостенко В. И., Султанов А. Ш. — Журн.
физ. хим., 1965, 39, с. 475.
424. Хитрое А. Н. Автореф. дис. . . . канд. хим. наук.
М.: ИВТАН, 1976.
424а. Ходаковский И. Л., Каторча Л. В., Куюнко
Н. А. —Геохимия, 1980, № 11, с. 1606.
4246. Ходаковский И. Л., Машнин А. А., Дра-
кин С. И. — Геохимия, 1980, N 11, с. 1606.
425. Ходаковский И. Л., Мишин И. В., Жогина В. В. —
Геохимия, 1966, № 7, с. 861.
426. Хриплович Л. М., Пауков И. Е. — ВИНИТИ.
Деп. № 1586—76. М., 1976.
427. Цагарейшвили Д. El. Методы расчета термических
и упругих свойств кристаллических
неорганических веществ. Тбилиси: Мецниереба, 1977.
428. Цагарейшвили Д. ЛГ., Гвелисиани Г. Г. — Изв.
АН СССР. Неорган, матер., 1973, 9, с. 1936.
429. Цагарейшвили Д. Ш., Гвелисиани Г. Г. —
Теплофизика высоких температур, 1974, 1, с. 208.
430. Цагарейшвили Д. Ш., Гвелисиани Г. Г. —
Теплофизика высоких температур, 1975, 13, с. 847.
431. Цемехман Л. Ш., Немойтин М. А., Вайсбург
С. Е. — Журн. прикл. хим., 1969, 42, с. 1402.
431а. Цителаури Н. Н., Самуилов Е. В. — В кн.:
Физическая газодинамика ионизированных
химически реагирующих газов. М.: Наука, 1968, с. 8.
432. Чаплыгин Г. В. — Журн. физ. хим., 1975, 49,
с. 2767.
433. Чеховской В. Я. — Инж. физ. журн., 1962, 5,
с. 62.
434. Чеховской В. Я. — Теплофизика высоких
температур, 1964, 2, с. 296.
435. Чиркин В. С. Теплофизические свойства
материалов. М.: Физматгиз, 1959.
436. Шабанова Л. Н. — Оптика и спектроскопия, 1969,
27, с. 383.
437. Шаповалов А. М., Шевелъков В. Ф.,
Мальцев А. А. — Весты. МГУ. Сер. хим., 1975, 16,
с. 153.
438. Шаповалов А. М., ШевельковВ. Ф., Мальцев А. А.—
В кн.: XI Менделеевский съезд по общей и
прикладной химии: Рефераты докл. и сообщений.
М.: Наука, 1975, № 1.
439. Шаргородский С. Д. — Укр. хим. журн., 1950,
16, с. 310.
440. Шаргородский С. Д., Шор О. И. — Укр. хим.
журн., 1954, 20, с. 357.
441. Шаулов Ю. X., Мосин А. М. — Журн. физ. хим.,
1973, 47, с. 1131.
442. Шаулов Ю. X., Шмырева Г. О., Тубянская В. С. —
Журн. физ. хим., 1966, 40, с. 122.
443. Шахтахтинский М. Г., Кулиев А. А. — Докл.
АН СССР, 1958, 123, с. 1071.
443а. Шевельков В. Ф. Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1967.
444. Шевельков В. Ф., Катаев Д. Л., Мальцев А. А. —
Вестн. МГУ. Сер. хим., 1969, № 4, с. 108.
445. Шевельков В. Ф., Клюев Н. А., Мальцев А. А. —
Вестн. МГУ. Сер. хим., 1969, 24, с. 32.
446. Шевельков В. Ф., Мальцев А. А. — Теплофизика
высоких температур, 1965, 3, с. 486.
447. Шевельков В. Ф., Мальцев А. А. — ВИНИТИ.
Доп. № 6897—73. М., 1973.
448. Шевельков В. Ф., Рябов Ю. С, Мальцев А. А. —
Вестн. МГУ. Сер. хим., 1972, № 6, с. 645.
449. Шевцова 3. Н., Морозов И. С, Приходкина Л. Н. —
Труды Моск. ин-та тонкой хим. технол., 1958,
вып. 7, с. 117.
450. Шейко И. Н., Фещенко В. Г. — Укр. хим. журн.,
1962, 28, с. 478.
451. Шейндлин А. Е., Белевич И. С., Кожевников И. Г.—
Теплофизика высоких температур, 1972, 2, с. 421.
452. Шейндлин А. Е., Чеховской В. Я., Петров В. А. —
Инж. физ. журн., 1964, 7, с. 63.
453. Шека И. А. — Журн. общ. хим., 1955, 25, с. 2401.
454. Шидловский А. А., Воскресенский А. А. — Журн.
физ. хим., 1963, 37, с. 2062.
455. Шифрин Ф. Ш. — Докл. АН СССР, 1956, 106,
с. 233.
456. Шишкин П. Т., Нудельман Б. И. — В кн.:
Исследования по технологии строительных
материалов. Ташкент: Фан, 1969, вып. 4, с. 35.
457. Шмидт Н. Е. — Журн. неорган, хим., 1966, 11,
с. 441.
458. Шмидт Н. Е., Соколов В. А. — Журн. неорган,
хим., 1960, 5, с. 1641.
459. Шпилърайн 9. 9., Каган Д. Н. — Теплофизика
высоких температур, 1969, 7, с. 577.
460. Шпилърайн Э. 9., Каган Д. Н., Бархатов Л. С. —
Теплофизика высоких температур, 1971, 9, с. 926.
461. Шпилърайн Э. Э., Каган Д. Н., Бархатов Л. С. —
Теплофизика высоких температур, 1972, 10, с. 193.
461а. Шпильрайн 9. 9., Каган Д. Н., Ульянов С. Н. —
Теплофизика высоких температур, 1980, 18,
с. 1184.
462. Шпилърайн 9. 9., Тоцкий Е. Е., Кармышин
Ю. В. — Докл. научно-техн. конф. МЭИ по
итогам научно-исслед. работ за 1968—1969 гг.
Подсекция Теплофиз. М.: МЭИ, 1969, ч. 1.
463. Шпилърайн Э. 9., Якимович К. А. Гидрид лития.
М.: Изд-во стандартов, 1972.
464. Штернина Е. Б., Фролова Е. Б. — Изв. сектора
физ.-хим. анализа ИОНХ АН СССР, 1952, 21,
с. 271.
465. Щукарев С. А., Васильева Л. В., Шарупин Б. П. —
Вестн. ЛГУ. Сер. физ. хим., 1960, вып. 2, № 10,
с. 112.
466. Щукарев С. А., Лилич Л. С, Латышева В. А. —
Докл. АН СССР, 1953, 91, с. 237.
467. Щукарев С. А., Семенов Г. А. — Журн. неорган.
хим., 1957, 2, с. 1217.
432
Литератур*
468. Щукарев С. А., Семенов Г. А., РатьковскийИ. А.—
Журн. неорган, хим., 1961, 6, с. 2817.
469. Щукарев С. А., Семенов Г. А., Ратьковский И'. А.—
Журн. неорган, хим., 1962, 7, с. 469.
470. Щукарев С. А., Семенов Г. А., Ратьковский И. А.—
Журн. неорган, хим., 1969, 14, с. 3.
471. Эпелъбаум В. А., Старостина М. И. — В кн.:
Бор: Тр. конф. по химии бора и его соединений.
М.: Госхимиздат, 1958.
472. Юдин В. Ф. — Труды Ленингр. технол. ин-та
им. Ленсовета, 1960, вып. 61, с. 9.
473. Юшин А. С. — Тр. по химии и хим. технол.
(Горький), 1968, вып. 2, № 20, с. 45.
474. Юшин А. С, Осипова Л. И., Слегина В. И. —
Журн. физ. хим., 1973, 47, с. 1828.
474а. Юшин А. С, Слегина В. И., Осипова Л. И. —
ВИНИТИ. Деп. № 4742-72. М., 1972.
475. Юшин А. С, Слегина В. И., Осипова Л. И. —
Журн. физ. хим., 1973, 47, с. 278.
476. Янатъева О. К. — Докл. АН СССР, 1954, 96,
с. 777.
477. Янатъева О. К., Рассонская И. С. — Журн.
неорган, хим., 1961, 6, с. 730.
477а. Abashkin Yu. G., Dementjev A. I. — Chem.
Phys. Lett, (in press).
478. Abramowitz S., Armstrong G. Т., Beckett С W.,
Churney K. L., Dibeler V. H., Douglas Т. В.,
Berron J. Т., Krause R. F. Jr., McCulloh K.E.,
Reilly M. L., Rosenstock H. M., Tsang W. New
Ideal-Gas Thermochemical Tables (In the Format
of the JANAF thermochemical tables). AFOSB.
Sci. Rept AFOSR-IR-72-2004. NBS Rept 10904.
Wash., 1972.
479. Ackerman R. /., Thorn R. J. — 3. Amer. Chem.
Soc, 1956, 78, p. 4169.
480. Acquista N., Abramowitz S., Lide D. R. — 3. Chem.
Phys., 1968, 49, p. 780.
481. Adami L. H., Conway К. С — U. S. Bur. Mines.
Rept Invest., 1966, N 6822.
482. Adams D. M., Churchill R. G. — J. Chem. Soc.
A, 1968, p. 2141.
483. Adams D. M., Churchill R. G. — J. Chem. Soc.
A, 1970, p. 697.
484. Adams G. В., Johnston H. L., Kerr E. С — 3.
Amer. Chem. Soc, 1952, 74, p. 4784.
485. Adamsky R. F., Wleeler С. Н. — J. Phys. Chem.,
1954, 58, p. 225.
486. Adlassnig K. — Planseeber. Pulvermet., 1958, 6,
N 3, S. 92.
487. Agarwal K. L., Betterton J. O. — 3. Low Temp.
Phys., 1974, 17, p. 509.
488. Ahlers G. — Phys. Rev., 1966, 145, p. 419.
489. Ahlrichs R., Keil F., Lischka H., Kutzelnigg W.,
Staemmler V. — 3. Chem. Phys., 1975, 63, p. 455.
490. Alcock С. В., Cornish J. В., Grieveson P. — Proc.
IAEA Symp. on Thermodyn. Vienna, 1966,1, p. 211.
491. Alcock С. В., Cornish J. В., Grieveson P. — Proc.
IAEA Symp. on Thermodyn. Vienna, 1966, 1, p. 367.
492. Alcock С. В., Grieveson P. — In: Thermodynamics
of Nuclear Materials. Vienna: IAEA, 1962, p. 563.
493. Aldred А. Т., Pratt J. N. — J. Chem. and Eng.
Data, 1963, 8, p. 429.
493a. Alexander M. H. — J. Chem. Phys., 1978, 69,
p. 3502.
494. Alexander C. A., Ogden J. S., Levy A. — J. Chem.
Phys., 1963, 39, p. 3057.
495. Allavena M., Rysnik R., White D., Colder G. V.,
Mann D. E. — 3. Chem. Phys., 1969, 50, p. 3399.
496. Allen D. R. — J. Amer. Chem. Soc, 1953, 75,
p. 3582.
497. Almy G. M., Horsfall R. — Phys. Rev., 1937, 51,
p. 491.
498. Alpant О., Неитапп Т. — Acta met., 1965, 13,
p. 543.
499. Altman R. L. — J. Chem. Phys., 1959, 31, p. 1035.
500. Altman R. L. — J. Chem. and Eng. Data, 1963,
8, p. 534.
501. Altman R. L. — J. Phys. Chem., 1963, 67, p. 366.
502. Almkvist G., Lagerqvist A. — Ark. fys., 1949, 1,
s. 477.
503. Almkvist G., Lagerqvist A. — Ark. fys., 1950, 2,
s. 233.
504. Almkvist G., Lagerqvist A. — Nature, 1952, 170,
p. 885.
505. Altschuller A. P. — 3. Amer. Chem. Soc., 1955,
77, p. 6187.
506. Ananthakrishnan R. — Proc. Indian Acad. Sci.,
1936, 4, p. 74.
506a. Ananthakrishnan R. — Proc. Indian Acad. Sci.,
1937, A5, p. 200.
507. Anderson A. B. — 3. Chem. Phys., 1973, 58, p. 381.
508. Anderson А. В., Parr R. G. — 3. Chem. Phys.,
1970, 53, p. 3375.
508a. Anderson С. Т. — J. Amer. Chem. Soc, 1931,
53, p. 476.
509. Anderson С. Т. — 3. Amer. Chem. Soc, 1934, 56,
p. 340.
510. Anderson С. Т. — J. Amer. Chem. Soc, 1934, 56,
p. 849.
511. Anderson С. Т. — 3. Amer. Chem. Soc, 1935, 57,
p. 429.
512. Anderson E. L. — Thesis. State Coll. Washington,
1955.
513. Anderson H. C, Belz L. H. — J. Amer. Chem.
Soc, 1953, 75, p. 4828.
514. Anderson J. S. — 3. Chem. Soc, 1943, p. 141.
515. Anderson T. F., Burg A. B. — J. Chem. Phys.,
1938, 6, p. 586.
516. Anderson T. F., Lassettre E. N., Yost D. — 3. Chem.
Phys., 1936, 4, p. 703.
517. Anderson T. J. — Nucl. Sci. Abstrs, 1976, 33, N 9,
N 20743.
518. Anderson T. J., Donaghey L. F. — 3. Chem.
Thermodyn., 1977, 9, p. 603.
519. Anderson T. J., Donaghey L. F. — 3. Chem.
Thermodyn., 1977, 9, p. 617.
520. Anderson W. E., Barker E. F. — J. Chem. Phys.,
1950, 18, p. 698.
520a. Andon R. J. L., Connett J. E., Martin J. F. —
Nat. Phys. Lab. Rept Chem. Taddington, 1979,
N 101: 11 p.
521. Andrews L. — 3. Chem. Phys., 1969, 50, p. 4288.
521a.Andrews L. — J. Phys. Chem., 1969, 73, p. 3922.
5216.Andrews L. — 3. Chem. Phys., 1971, 54, p. 4935.
521b. Andrews L. — 3. Chem. Phys., 1973, 58, p. 2258.
522. Andrews L. — Appl. Spectrosc. Revs, 1976, 11,
p. 125.
522a.Andrews L., Duley W. W., Brewer L. — 3. Mol.
Spectrosc, 1978, 70, p. 41.
5226. Andrews L., Hwang J. Т., Trindle С — 3. Phys.
Chem., 1973, 77, p. 1065.
523. Andrews L., Pimentel G. C. — J. Chem. Phys.,
1967, 47, p. 3637.
524. Andrussov L. — Z. phys. Chem. Leipzig, 1925, 116,
S. 81.
525. Ansara I., Bonnier E. — In: Conf. Intern. Met.
Beryllium. Grenoble, 1965. P., 1966, p. 17.
526. Anthoney M. E., Finch A., Gardner P. J. — J.
Chem. Soc. Dalton Trans., 1973, p. 659.
433
Литература
527. Antic-Jovanovic A., Bojovic V., Pesic D. S. — J. 559.
Phys. B:Atom. and Mol. Phys., 1976, 9, p. L575.
528. Antic-Jovanovic A., Pesic D. S., Bo<ovic V. — J. 560.
Mol. Spectrosc, 1976. 60, p. 416. 561.
529. Antolini R., Gravelle P., Tramboure Y. — J. chim. 562.
phys. et phys.-chim. biol., 1962, 59, p. 715. 563.
530. Antropoff A., Falk E. — Z. anorg. und allg. Chem.,
1930, 187, S. 405. 564.
530a. Armstrong D. R., Clark D. T. — Theor. chim.
acta, 1972, 24, p. 307. 565.
531. Armstrong D. Д., Perkins P. G. — J. Chem. Soc.
Chem. Communs, 1969, N 15, p. 856. 566.
532. Armstrong D. R., Perkins P. G. — J. Chem. Soc.
A, 1969, p. 1044. 567.
533. Armstrong D. R., Perkins P. G. — Theor. chim.
acta, 1969, 15, p. 413. 568.
534. Armstrong D. R., Perkins P. G., Sorbie K. S. —
Rev. roum. chim., 1974, 19, p. 747. 569.
535. Armstrong G. T. — In: NASA Accession N 65-31317,
Rept N AD467028, 1965, p. 81. 570.
536. Armstrong G. Т., Coyle C. F. — U. S. AECAED-
Conf-65-358-1, 1965, 35 p. 571.
537. Arndt K. — Z. Elektrochem.. 1906, 12, S. 337.
537a. Arthur J. R. — J. Phys. Chem. Solids, 1967, 28, 572.
p. 2257.
538. Arthur J. S. — J. Appl. Phys., 1950, 21, p. 732. 573.
538a. Asano M., Kubo K. — J. Nucl. Sci. and Technol.,
1978, 15, p. 765. 573a
539. Asano M., Yamamoto Y., Sasaki JV., Kubo K. —
Bull. Chem, Soc. Jap., 1972, 45, p. 82. 5736
540. Ashrafunnisa, Rao D. V. K., Murthy A. A. N.,
Rao P. T. — Physica, 1974. 73, p. 421. 574.
541. Ashrafunnisa, Rao D. V. K., Rao P. T. — J. Phys.
B: Atom, and Mol. Phys., 1973, 6, p. 1503. 575.
542. Ashrafunnisa, Rao D. V. K., Rao R. T. — Spectrosc.
Lett., 1976, 9, p. 9. 575a
543. Asundi R. K. — Proc. Indian Acad. Sci., 1935,
Al, p. 830. 576.
543a. Atchayya M., Dadape V. V. — High Temp. Sci.,
1971, 3, p. 456. 577.
544. Attwood В., Shelton R. A. J. — J. Less-Common
Metals, 1970, 20, p. 131. 578.
545. Aufenast F., Terrey H. — J. Chem. Soc, 1926,
p. 1546.
546. Ault B. S., Andrews L. — J. Chem. Phys., 1975,
62, p. 2320.
547. Awbery J. H. — Philos. Mag., 1938, 26, p. 776. 579.
548. Awbery J. H., Griffiths E. — Proc. Phys. Soc. 580.
London, 1926, 38, p. 378.
549. Awbery J. H., Griffiths E. — Proc. Phys. Soc. 581.
London, 1926, 38, p. 395.
550. Baak T. — Acta chem. scand., 1955, 9, p. 1350. 582.
551. Babeloiowsky T. — J. Chem. Phys., 1963, 38,
p. 2036. 583.
552. Bach M.-Ch., Crasnier F., Labarre J.-F. — J. Mol.
Struct., 1973, 16, p. 89. 584.
553. Backstrom H. L. — J. Amer. Chem. Soc, 1925,
47, p. 2432. 585.
554. Backstrom H. L. — J. Amer. Chem. Soc, 1925,
47, p. 2443. 586.
555. Backstrom H. L. — Z. phys. Chem. Leipzig, 1926, 587.
121, S. 289.
556. Bagus P. S., Moser С. М., Goethals P., Verhae- 588.
gen G. — J. Chem. Phys., 1973, 58, p. 1886.
557. Bahr F. — Z. anorg. und allg. Chem., 1911, 71, 589.
S. 79. 590.
557a. Baig M. A., Rafi M., Khan M. A. — Nuovo 591.
cim., 1977, B40, p. 365.
558. Bailey C. R., Hale J. В., Thompson J. W. — J. 592.
Chem. Phys., 1937, 5, p. 275
Bailey C. R., Hale J. В., Thompson J. W. — Proc
Roy. Soc. London, 1937, A161, p. 107.
Baker E. H. — J. Chem. Soc, 1962, p. 464.
Baker E. H. — J. Chem. Soc, 1963, p. 339.
Baker E. H. — J. Chem. Soc, 1964, p. 699.
Baker T. W., Baldock P.. J. — J. Nucl. Mater.,
1966, 19, p. 210.
Balarew D.—Z. anorg. und allg. Chem., 1924, 134,
S. 121.
Balarew D., Lukowa N. — Kolloid-Z., 1930, 52,
S 222
Balfour W. J. — J. Phys. B: Atom, and Mol.
Phys., 1970, 3, p. 1749.
Balfour W. J., Cartwright H. M: — Canad. J.
Phys., 1975, 53, p. 1477.
Balfour W. J., Cartwright H. M. — Chem. Phys.
Lett., 1975, 32, p. 82.
Balfour W. J., Cartwright H. M. — Astron. and
Astrophys. Suppl. Ser., 1976, 26, p. 389.
Balfour W. J., Cartwright H. M., Hugh M. —
Canad. J. Phys., 1976, 54, p. 1898.
Balfour W. J., Douglas A. E. — Canad. J. Phys.,
1970, 48, p. 901.
Balfour W. J., Whitlock R. F. — J. Chem. Soc.
Chem. Communs, 1971, p. 1231.
Balfour W. J., Whitlock R. F. — Canad. J. Phys.,
1975, 53, p. 472.
. Balls A., Downs A. J., Greenwood N. N., Straug-
han B. P. — Trans. Faraday Soc, 1966, 62, p. 521.
. Baltayan P., Nedelec O. — J. Chem. Phys., 1979,
70, p. 2399.
Bandorawalla J. S., Altekar V. A. — Trans. Indian
Inst. Metals, 1968, 21, p. 68.
Banus M. D., Bragdon R. W. U. S. AEC Rept
CF-52-2-212. Metal Hydrides, Inc., 1952, Febr. 1.
. Barany R., Kelley K. K. — U. S. Bur. Mines.
Rept Invest., 1961, N 5825.
Barany R., King E. G., Todd S. S. — Z. Amer.
Chem. Soc, 1957, 79, p. 3639.
Barber W. R., Boynton C. F., Gallacher P. E. —
J. Chem. and Eng. Data, 1964, 9, p. 137.
Barin I., Knacke O. Thermochemical properties
of inorganic substances. B. etc.: Springer-Verl.,
1973. Barin I., Knacke O., Kubaschewski O.
Thermochemical properties of inorganic substances.
Supplement. B. etc.: Springer-Verl., 1977.
Barlocher M. — Helv. chim. acta, 1963, 46, p. 2920.
Barlow M., Meredith С — Z. Kristallogr., 1969,
130, S. 304.
Barnes J. — Philos. Trans. Roy. Soc London.
1909, A209, p. 447.
Barnighausen H., Rietschel E. T. — Z. anorg. und
allg. Chem., 1967, 354, S. 23.
Barrett A. H., Mandel M. — Bull. Amer. Phys.
Soc, 1955, 30, p. 66.
Barrett A. H., Mandel M. — Phys. Rev., 1958,
109, p. 1572.
Barren T. H. K., Berg W. Т., Morrison J. A. —
Proc. Roy. Soc. London, 1959, A250, p. 70.
Barrow R. F. — J. Chem. Phys., 1954, 22, p. 573.
Barrow R. F. — Proc. Phys. Soc. London, 1957,
A70, p. 622.
Barrow R. F. — Proc. Phys. Soc. London, 1960,
A75, p. 933.
Barrow R. F. — Trans. Faraday Soc, 1960, 56,p. 952.
Barrow R. F. — Nature, 1961, 189, p. 480.
Barrow R. F., Bastin M. W., Longborough L. B. —
Proc. Phys. Soc. London, 1967, 92, p. 518.
Barrow R. F., Beale J. R. — J. Chem. Soc. Chem.
Communs, 1967, p. 606.
434
Литература
592а. Barrow R. F., Burton W. G., Jones P. A. — Trans.
Faraday Soc, 1971, 67, p. 902.
593. Barrow R. F., Cheele H. F. K., Thomas P. M.,
Zeeman P. B. — Proc. Phys. Soc. London, 1958,
A71, p. 128.
594. Barrow R. F., Crawford D. V. — Proc. Phys. Soc.
London, 1945, 57, p. 12.
595. Barrow R. F., Crawford D. V. — Nature, 1946,
157, p. 33.
596. Barrow R. F., Dodworth P. G., Zeeman P. B. —
Proc. Phys. Soc. London, 1957, A70, p. 34.
596a. Barrow R. F., Gissane W. J. M., Rose G. V. M. —
Proc. Phys. Soc. London, 1964, A84, p. 1035.
597. Barrow R. F., Glasser D. V., Zeeman P. B. — Proc.
Phys. Soc. London, 1955, A68, p. 962.
598. Ba rrow R. F., Jacquest I. А. Т., Thompson E. W. —
Proc. Phys. Soc. London, 1954, A67, p. 528.
598a. Barrow R. F., Jeffries E. A. N. S., Swin-
stead J. M. — Trans. Faraday Soc, 1955, 51,
p. 1650.
599. Barrow R. F., Johns J. W. C, Smith F. J. — Trans.
Faraday Soc, 1956, 52, p. 913.
600. Barrow R. F., Kopp I., Malmberg С — Phys.
scr., 1974, 10, s. 86.
601. Barrow R. F., Kopp I., Scullman R. — Proc.
Phys. Soc. London, 1963, A82, p. 635.
602. Barrow R. F., Premaswarup D., Winternitz /.,
Zeeman P. B. — Proc. Phys. Soc. London, 1958,
A71, p. 61.
603. Barrow R. F., Rowlinson H. С — J. phys. et
radium, 1954, 15, p. 499.
604. Barrow R. F., Rowlinson H. С — Proc. Roy.
Soc. London, 1954, A224, p. 134.
605. Bartell Lf S., Carrol B. L. — J. Chem. Phys.,
1965, 42, 'p. 1135.
606. Bartky I. R. — J. Mol. Spectrosc, 1960, 5, p. 206.
607. Bartky I. R. — J. Mol. Spectrosc, 1961, 6, p. 275.
608. Bartky I. R., Giauque W. F. — J. Amer. Chem.
Soc, 1959, 81, p. 4169.
609. Barton В., Claxton T. A., Hamshere S. J.,
Marshall H. E., Overill R. E., Symons M. С R. —
J. Chem. Soc. Dalton Trans., 1976, N 23, p. 2446.
610. Basquin О, Н. — Astrophys. J., 1901, 14, p. 8.
611. BassC.D.,LyndsL.,WolframT.,DeWamesR.E. —
Inorg. Chem., 1964, 3, p. 1063.
612. Bass С D., LyndsL., Wolfram Т., De WamesR. E, —
J. Chem. Phys., 1964, 40, p. 3611.
612a. Bassler J. M., Timms P. L., Margrave J. L. —
J. Chem. Phys,, 1966, 45, p. 2704.
613. Basu Т. К„ Searcy A. W. — J. Chem. Soc.
Faraday Trans. I, 1976, 72, p. 1889.
614. Batalli-Cosmovici C, Michel K. — Chem. Phys.
Lett., 1972, 16, p. 77.
615. Bates J. B. — J. Chem. Phys., 1971, 55, p. 489.
616 Bates J. В., Quis A. S. — Spectrochim. acta, 1975,
A31, p. 1317.
617 Bates J. В., Quis A. S., Boyd G. E. — J. Chem.
Phys., 1971, 54, p. 124.
618. Battat D., Faktor M. M., Garett I., Moss R. H. —
J. Chem. Soc. Faraday Trans. I, 1974, 70, p. 2280.
618a. Battat D., Faktor M. M., Garett I., Moss R. H. —
J. Chem. Soc. Faraday Trans. I, 1974, 70, p. 2302.
619. Baud E. — С. г. Acad. sci., 1902, 135, p. 1103.
620. Baud E. —Ann. chim. phys., 1904, 1, p. 8.
621 Bauer S. H. — J. Amer. Chem. Soc, 1938, 60,
p. 524.
622 Bauer S. H. — J. Amer. Chem. Soc, 1956, 78,
p. 5775.
623. Bauer S. H., Herzberg G., Johns J. W. C. — J.
Mol. Spectrosc, 1964, 13, p. 256.
624. Baur A., Lecocq A. — С. г. Acad. sci., 1963, 257,
p. 1445.
625. Baur A., Lecocq A. — Kept CEA-R2611. Com.
Energie Atom. Sacklay, France, 1964, p. 17.
626. Baur E., Brunner R. — Z. Elektrochem., 1934,
40, S. 154.
627. Baur E., Brunner R. — Helv. chim. acta, 1934,
17, p. 958.
628. Bautista R. G., Margrave J. L. — J. Phys. Chem
1963, 67, p. 2411.
629. Bautista R. G., Margrave J. L. — J. Phys. Chem.,
1965, 69, p. 1770.
629a. Baylis A., Pressley G., Stafford F. — J. Amer,
Chem. Soc, 1966, 88, p. 2428.
630. Bear I. /., Turnbull A. G. — J. Phys. Chem
1965, 69, p. 2828.
631. Bear I. J., Turnbull A. G. —Austral. J. Chem
1966, 19, p. 751.
632. Bear I. J., Turnbull A. G. — J. Phys. Chem
1966, 70, p. 711.
633. Beattie I. R., Blayden H. E., Hall S. M.,
Jenny S. N., Ogden J. S. — J. Chem. Soc. Dalton
Trans., 1976, p. 666.
634. Beattie I. Д., Blayden H. E., Ogden J. S.—l
Chem. Phys., 1976, 64, p. 909.
634a. Beattie I. R., Gilson Т., Cocking P. — J. Chem
Soc A, 1967, p. 702.
635. Beattie I. R., Gilson Т., Ozin G. A. — J. Chem"
Soc. A, 1968, p. 813.
636. Beattie I. R., Harder J. R. — J. Chem. Soc A
1969, p. 2655.
бзба. Becart M., Decler F. — С. г. Acad. sci., 1960
251, p. 2153.
бзбб. Becart M., Mahieu J. M. — J. phys. (France),
1964, 25, p. 873.
637. Becher H. J., Schnockel H. — Z. anorg. und allg
Chem., 1970, 379, S. 136.
637a. Beck L. K., Kugler B. H., Haendler H. M. —
J. Solid State Chem., 1973, 8, p. 312.
638. Becker G., Roth W. A. — Z. phys. Chem. Leipzig,
1932, A161, S. 69.
639. Begun G. M., Boston С R., Torsi G., MamantovG. ¦—
Inorg. Chem., 1971, 10, p. 886.
640. Bell R. P., Longuet-Higgins H, С — Proc. Roy.
Soc. London, 1945, A183, p. 357.
641. Bell S.. McLean M. L. — l. Mol. Spectrosc.,
1976, 63, p. 521.
642. Bellanca A. — Period, miner., 1942, 13, p. 21.
643. Bellanca A. — G. sci. nat. e econ., Palermo, 1948,
45, N 3.
644. Bellanca A., Sgarlata F. — Rend. soc. ital. miner*
e petrol., 1951, 20, p. 257.
645. Beketoff N. N. — Bull. Acad. sci. Russ., 1892,
34, p. 291.
646. Bender С F., Davidson E. R. — J. Chem. Phys.,
1967, 46, p. 3313.
647. Bender С F., Schaefer H. F. — J. Mol. Spectrosc,
1971, 37, p. 423.
648. Bengtsson E. — Z. Phys., 1928, 51, S. 889.
649. Bengtsson E. — Nature, 1929, 123, p. 529.
650. Bengtsson E. — Nova acta Regiae soc. scient. up^
saliensis. Ser. IV, 1932, 8, s. 1.
651. Bengtsson E., Hulthen E. — Z. Phys., 1928, 52,
S 275
652. Bengtsson E., Rydberg R. — Z. Phys., 1930, 59,
S. 540.
653. Bennington R. O. — J. Geol., 1956, 64, p. 558.
654. Benrath A. — Z. anorg. und allg. Chem., 1942,
249, S. 245.
435
Литература
655. Berac /., Tomczak I. — Rocz. chem., 1965, 39,
s. 519.
655a. Berg J. M. — Acta crystallogr., 1966, 20, p. 905.
656. Berg L. E., Klynning L. — Astron. and Astrophys.
Suppl. Ser., 1974, 13, p. 325.
657. Berg L. E., Klynning L. — Phys. scr., 1974, 10,
s. 331.
657a. Berg L. E., Klynning L., Martin H. — Opt. Com-
muns, 1976, 17, p. 320.
658. Berg L. E., Klynning L., Martin H. — Phys.
scr., 1978, 18, s. 61.
658a. Berg L. E., Klynning L., Martin H. — Phys. scr.,
1978, 18, s. 149.
6586. Berg L. E., Klynning L., Martin H., Pereira A.,
Boyen P. — Chem. Phys. Lett., 1978, 54, p. 357.
659. Berg W. T. — Phys. Rev., 1968, 167, p. 583.
660. Berggren G., Brown A. — Acta chem. scand., 1971,
25, p. 1377.
661. Bergsma J., Loopstra B. O. — Acta crystallogr.,
1962, 15, p. 92.
662. Berber J.A. — In: Rare gas solids./ Ed. M. L. Klein,
J. A. Venables. L.: Acad. Press, 1976, vol. 1, p. 212.
663. Berkowitz J. — J. Chem. Phys., 1972, 56, p. 2766.
664. Berkowitz /., Chupka W. A. — J. Chem. Phys.,
1966, 45, p. 1287.
665. Berkowitz J., Marquart J. R. — J. Chem. Phys.,
1962, 37, p. 1853.
665a. Berkowitz J., Marquart J. R. — J. Chem. Phys.,
1963, 39, p. 275.
666. Berkowitz /., Walter T. A. — J. Chem. Phys.,
1968, 49, p. 1184.
667. Bernal J. D., Megaw H. D. — Proc. Roy. Soc.
London, 1935, A151, p. 384.
668. Bernstein E. R., Reily J. P. — J. Chem. Phys.,
1972, 57, p. 3961.
669. Berihelot M. — Ann. chim. phys., 1875, 4, p. 160.
670. Berthelot M. — Ann. chim. phys., 1875, 4, p. 531.
671. Berthelot M. — Ann. chim. phys., 1878, 15, p. 185.
671a. Berthelot M. — C. r. Acad. sci., 1878, 86, p. 786.
672. Berthelot M. Thermochemie. P.: Gauthiet-Villar-
set Fils, 1897. T. 2.
673. Berthelot M. — Ann. chim. phys., 1901, 22, p. 464-
674. Berthelot M. — Ann. chim. phys., 1901, 22, p. 479.
675. Berthelot M. — С. г. Acad. sci., 1901, 132, p. 281.
676. Berthelot M., Ilosvay J. N. — C. r. Acad. sci.,
1882, 94, p. 1487.
677. Berthelot M., Ilosvay J. N. — Ann. chim. phys.,
1883, 29, p. 295.
678. Bethell D. E., Sheppard N. — Trans. Faraday Soc,
1955, 51, p. 9.
679.: Bezzi S. — Gazz. chim. ital., 1935, 65, p. 766.
680. Bhaduri B. N., Fowler A. — Proc. Roy. Soc. Lon-
, don, 1934, A145, p. 321.
681. Bichowsky F. R., Rossini F. S. The thermochemistry
of the chemical substances. N. Y.: Reinhold Publ.
Corp., 1936.
682. Bilir N:, Phillips W. A., Geballe T. H. — Proc.
Intern. Conf. Low Temp. Phys., 14th. Otaniemi,
Finland, 1975, 3, p. 9.
683. Bills J. L., Cotton F. A. — J. Phys. Chem., 1960,
64, p. 1477.
684. Biltz W., Caspari F.Z. — Z. anorg. und allg. Chem.,
1950, 261, S. 248.
685. Biltz W., Hohorst G. — Z. anorg. und allg. Chem.,
1922, 121, S. 1.
686. Biltz W., Hohorst G. — Z. anorg. und allg. Chem.,
1924, 140, S. 261.
687. Biltz W., Kennecke E. L. — Z. anorg. und allg.
Chem., 1925, 147, S. 171.
688. Biltz W., Messerknecht C. — Z. anorg. und allg.
Chem., 1925, 148, S. 157.
689. Biltz W., Klatte K., Rahlfs E.—Z. anorg. und
allg. Chem., 1927, 166, S. 339.
690. Biltz W., Rohlffs G., Vogel H. U.—Z. anorg.
und allg. Chem., 1934, 220, S. 113.
691. Biltz W.. Voigt A. — Z. anorg. und allg. Chem.,
1923 126 S. 39.
692. Biltz W., 'Wagner W. — Z. anorg. und allg. Chem.,
1924, 134, S. 1.
693. Binford J. S., James /., Strohmenger M., He-
bert Т. Н. — J. Phys. Chem., 1967, 71, p. 2404.
693a. Binnewies M., Schdfer H. — Z. anorg. und allg.
Chem., 1974, 407, S. 327.
6936. Bird J. M., Bryant A. W., Pratt J. N. — J.
Chem. Thermodyn., 1975, 7, p. 577.
694. Blachnik R. O. G., Gross P., Hay man C. — Ful-
mer Res. Inst. sci. Rept N 5, Contract AF 61
@52)-863, 1968.
695. Blachnik R. O. G., Gross P., Hayman C. — Trans.
Faraday Soc, 1970, 66, p. 1058.
696. Blackburn P. E., Buckler A. — J. Phys. Chem.,
1965, 69, p. 4250.
697. Blank B. A. H. — U. S. Atom. Energy Commis.,
1965, UCRL-16018.
698. Blank B. A. H., Searcy A. W. — J. Phys. Chem.,
1968, 72, p. 2241.
699. Blauer J. A., Farber M. — J. Chem. Phys., 1963,
39, p. 158.
700. Blauer J. A., Farber M. — J. Phys. Chem., 1964,
68, p. 2357.
701. Blauer J. A., Farber M. —Trans. Faraday Soc,
1964, 60, p. 301.
702. Blauer J. A., Greenbaum M. A., Arshadi M.,
Farber M. The thermodynamics of several light metal
oxides, halides and oxyhalides: NASA Doc.
N 63-18886, 1963.
703. Blauer J. A., Greenbaum M. A.,Farber M. The
thermodynamics of several light metal oxides, halides
and oxyhalides. Pasadena (Cal.): Rocket Power,
Inc. Res. Lab., 1963, AD400935.
704. Blauer J. A., Greenbaum M. A., Farber M. — J.
Phys. Chem., 1964, 68, p. 2332.
705. Blauer J. A., Greenbaum M. A., Farber M. —
J. Phys. Chem., 1965, 69, p. 1069.
706. Blauer J. A., Greenbaum M. A., Farber M. — J.
Phys. Chem., 1966, 70, p. 973.
707. Blewett J. P., Liebhafsky H. A., Hennelly E. F. —
J. Chem. Phys., 1939, 7, p. 478.
708. Blick K. E., Dawson J. W., Niedenzn K. — Inorg.
Chem., 1970, 9, p. 1416.
709. Blint R. J., Goddard W. A. — Chem. Phys., 1974,
3, p. 297.
710. Bloem J., Bosman B. — Philips Res. Repts, 1975,
30, p. 2066.
710a. Blount C. — Amer. Miner., 1977, 62, p. 942.
711. Blue G. D., Green J. W., Bautista R. G.,
Margrave J. L. — J. Phys. Chem., 1963, 67, p. 877.
711a. Blues R. C., Barrow R. F. — Trans. Faraday Soc,
1969, 65, p. 646.
712. Blustin P. H., Linnet J. W. — J. Chem. Soe.
Faraday Trans. II, 1975, 71, p. 1058.
713. Bockris J. O. M., Crook E. H. — Chem. and Ind.,
1959, N 37, p. 1162.
714. Boeke H. E. — Z. anorg. und allg. Chem., 1906,
50, S. 244.
715. Boeke H. E. — Mitt. Naturforsch. Ges. Halle,
1913, 3, S. 13.
716. Boerboom A. J. H. — Vak.-Techn., 1963, 12,
S. 205.
436
Литература
717. Boerio J., Westrum E. F. — J. Chem. Thermodyn.,
1978, 10, p. 1.
718. Bohdansky J., Schins H. E. J. — J. Phys. Chem.,
1967, 71, p. 215.
719. Bohr С — Ann. Phys., 1899, 68, S. 500.
719a. BoitardE., Bros J. P., Laffitte M. — J. chim.
phys. et phys.-chim. biol., 1969, 66, p. 166.
720. Boitzowa Z. V., Butkow K. V. — Phys. Z. Sowjet-
union, 1934, 5, S. 765.
721. Bonadeo H. A., Silberman E. — J. Mol. Spectrosc,
1969, 32, p. 214.
722. Bonadeo H. A., Silberman E. — Spectrochim.
acta, 1970, A26, p. 2337.
723. Booth H. S., Bidwell R. M. — J. Amer. Chem.
Soc, 1950, 72, p. 2567.
724. Booth H. S., Carter J. M. — J. Phys. Chem.,
1932, 36, p. 1359.
725. Borches H. — Metallwirtschaft, 1931, 10, S. 863.
726. Bottger W. — Z. phys. Chem. Leipzig, 1903, 46,
S. 521.
727. Bousquet J., Blanchard J. M., Bonnetot В.,
Claudy P. — Bull. Soc. chim. France, 1969, p. 1841.
728. Bowen N. L., Tuttle O. F. — Bull. Geol. Soc. Amer.,
1949, 60, p. 439.
729. BoydR. J., Frost D. С —Chem. Phys. Lett., 1968,
1, p. 649.
730. Boyd R. J., Whitchead M. A. — J. Chem. Soc.
Dalton Trans., 1972, 1, p. 73.
731. Brackett E. В., Brackett T. F., Sass R. L. — J.
Inorg. and Nucl. Chem., 1963, 25, p. 1295.
732. Brackett E. В., Brackett T. F. Sass R. L. — J.
Phys. Chem., 1963, 67, p. 2132.
733. Bradford R. S., Jones C. R., Southall L. A.,
Broida H. P. — J. Chem. Phys., 1975, 62, p. 2060.
734. Bradley R. S., Munro D. C, All S. I. — J. Inorg.
and Nucl. Chem., 1970, 32, p. 2513.
735. BratlandD., FekriA., Grjotheim K., Motzfeldt K. —
Acta chem. scand., 1970, 24, p. 864.
736. Braune #., Pinnoro P. — Z. phys. Chem. Leipzig,
1937, B35, S. 239.
737. Brcic B. S., Jernejcic J. — Vestn. Slov. kem.
drust., 1962, 9, s. 65.
738. Bredig M. A. — Colloq. intern. CNRS, 1972,
N 205, p. 183.
739. Brennan W. P., Gray A. P. Thermal analysis
applications study. N 9. Norwalk (Conn.): Perkin-Elmer
Corp., 1973.
740. Brett A. C, Balfour W. J. — J. Chem. Phys.,
1971, 54, p. 3240.
740a. Brewer F. M., Garton G., Goodgame D. M. L. —
J. Inorg. and Nucl. Chem., 1959, 9, p. 56.
741. Brewer L. — Chem. Rev., 1953, 52, p. 1.
742. Brewer L., Bromley L. A., Gilles P. W., Lof-
gren N. L. — In: The chemistry and metallurgie
of miscelaneous materials. Thermodynamics/Ed.
L. L. Quill. N. Y., etc., 1950, Pap. 3—7, p. 74.
743. Brewer L., Hauge R. — J. Mol. Spectrosc., 1968,
25, p. 330.
744. Brewer L., Porter R. F. — J. Chem. Phys., 1954,
22, p. 1867.
745. Brewer L., Rosenblatt G. M. — In: Advances in high
temperature chemistry/Ed. L. Eyring. N. Y.; L.:
Acad. Press, 1969, vol. 2.
746. Brewer L., Searcy A. W. — J. Amer. Chem. Soc,
1951, 73, p. 5308.
747. Brewer L., Searcy A. W. — Annu. Rev. Phys.
Chem., 1956, 7, p. 259.
748. Brewer L., Somayajulu G. R., Brackett E. — Chem.
Rev., 1963, 63, p. 111.
749. Brewer L., Trajmar S. — J. Chem. Phys., 1962
36, p. 1585.
750. Brewer L., Wang J. L.-F. — J. Chem. Phys
1972, 56, p. 4305.
751. Briggs A. G., Piercy R. — Spectrochim. acta, 1973
A29, p. 851.
752. Brink I. /., Holley C. E.,Jr. — l. Chem.
Thermodyn., 1978, 10, p. 259.
752a. Brinkmann U., Telle H. — J. Phys. B: Atom
and Mol. Phys., 1977, 10, p. 133.
753. Brinner E., Pamm G., Paillard H. — Helv. chim
acta, 1948, 31, p. 2220.
754. Brisi C, Abbatista F. — Ann. chim., 1960, 50,
p. 165.
755. Broadhead P., Newman G. A. — J. Mol. Struct
1971, 10, p. 157.
756. Brode H. — Ann. Phys., 1940, 37, p. 344.
757. Brodersen P. H. — Z. Phys., 1932, 76, S. 613.
758. Brodersen P. H.—Z. Phys., 1936, 104, S. 135.
759. Brom J. M., Jr., Devore Т., Franzen H. F. — J
Chem. Phys., 1971, 54, p. 2742.
760. Brom J. M., Jr., Franzen H. — J. Chem. Phys.,
1971, 54, p. 2874.
761. Brom J. M., Jr., Weltner W., Jr. — J. Chem.
Phys., 1973, 58, p. 5322.
762. Brom J. M., Jr., Weltner W., Jr. — J. Mol.
Spectrosc, 1973, 45, p. 82.
763. Brom J. M., Jr., Weltner W., Jr. — J. Chem,
Phys., 1976, 64, p. 3894.
764. Bronsted J. N. — Z. Elektrochem., 1914, 20, S. 81.
765. Brooks C. R., Bingham R. E. — J. Phys. and Chem.
Solids, 1968, 29, p. 553.
766. Brooks R., Kaufman M. — J. Chem. Phys., 1965,
43, p. 3406.
767. Brotchie D. A., Thompson С — Chem. Phys. Lett.,
1973, 22, p. 338.
767a. Brower F. M., Matzek N. E., Reigler P. F.,
Rinn H. W., Roberts С. В., Schmidt D. L., Sno*
ver J. A., Terada K. — J. Amer. Chem. Soc, 1976,
98, p. 2450.
768. Brown С M., Naber R. H., Tilford S. G., Gin-
ter M. L. — Appl. Opt., 1973, 12, p. 1858.
769. Brown C. M., Tilford S. G., Ginter M. L. — J. Opt.
Soc. Amer., 1973, 63, p. 1454.
770. Brown C. M., Tilford S. G., Ginter M. L. — J,
Opt. Soc. Amer., 1974, 64, p. 877.
771. Brown C. W., Overend J. — Can. J. Phys., 1968,
46, p. 977.
772. Brown C. W., Overend J. — Spectrochim. acta,
1969, A25, p. 1535.
772a. Brown J. M., Martin H., Wayne F. D. — Chem.
Phys. Lett., 1978, 55, p. 67.
7726. Brown H., Bartholomay H., Taylor M. — J.
Amer. Chem. Soc, 1944, 66, p. 435.
773. Brown R. D., Williams G. R. — Mol. Phys., 1973,
25, p. 673.
774. Brown R. D., Williams G. R. — Chem. Phys.,
1974, 3, p. 19.
775. Brunner R. — Z. Elektrochem., 1932, 38, S. 55.
776. Bryant С A., Keesom P. H. — Phys. Rev. Lett..
1960, 4, p. 460.
777. Buchler A., Berkowitz-Mattuck J. B. —¦ J. Chem,
Phys., 1963, 39, p. 286.
778. Buchler A., Klemperer W. — Spectrochim. acta,
1957, A10, p. 218.
779. Buchler A., Klemperer W. — J. Chem. Phys.,
1958, 29, p. 121.
780. Buchler A., Marram E. P. — J. Chem. Phys.,
1963, 39, p. 292.
437
Литература,
781. Buckler A., Marram E. P., Stauffer J. L. — J.
Phys. Chem., 1967, 71, p. 4139.
782. Buckler A., Stauffer J. L., Klemperer W. — J.
Chem. Phys., 1964, 40, p. 3471.
783. Buckler A., Stauffer J. L., Klemperer W. — J.
Amer. Chem. Soc, 1964, 86, p. 4544.
784. Buckler A., Stauffer J. L., Klemperer W., Whar-
ton L. — J. Chem. Phys., 1963, 39, p. 2299.
785. Buck P., Barnighausen H. — Acta crystallogr.,
1968, 24, p. 1705.
785a. Bues W., Akhras Z., Окоп G. — Z. anorg. und
allg. Chem. 1976, 425, S. 193.
786. Bulewicz E. M. — Nature, 1956, 177, p. 670.
787. Bulewicz E. M., Phillips L. F., Sugden Т. М. —
Trans. Faraday Soc, 1961, 57, p. 921.
788. Bulewicz E. M., Phillips L. F., Sugden Т. М. —
Trans. Faraday Soc, 1961, 57, p. 4540.
789. Bulewicz E. M., Sugden T. M. — Trans. Faraday
Soc, 1959, 55, p. 720.
790. Bulewicz E. M., Sugden T. M. — Trans. Faraday
Soc, 1958, 54, p. 830.
791. Burg А. В., Fu Yuan-Chin. — J. Amer. Chem.
Soc, 1966, 88, ]. 1147.
792. Burnelle L., Kan man J. J. — J. Chem. Phys.,
1965, 43, p. 3540.
793. Burns R. P. — J. Chem.Phys., 1966, 44, p. 3307.
794. Burns R. P., De Maria G., Drowart /., Ing-
hram M. G. — J.^Chem. Phys., 1963, 38, p. 1035.
795. Burns R. P., Jason A. J., Inghram M. G. — J.
¦ Chem. Phys., 1967, 46, p. 394.
796. Burton В., Claxton T. A., Hamshere S. /.,
Marshall H. E., Overill R. E., Symons M. C. R. —
J. Chem. Soc. Dalton Trans., 1976, N 23, p. 2446.
797. Bussem W., Koberich F. — Z. phys. Chem.
Leipzig, 1932, B17, S. 315.
798. Butkov K. — Z. Phys., 1929, 58, S. 232.
799. Butkov K., Terenin A.—Z. Phys., 1928, 49, S.
865.
800. Butler J. A. V., Hiscocks E. S. — J. Chem. Soc,
1926, p. 2554.
801. Butler J. G., Sorrell С A. — High Temp. Sci.,
1971, 3, p. 389.
801a. Buyco E. H., Davis F. E. — J. Chem. and Eng.
Data, 1970, 15, p. 518.
802. Buzzard R. W. — J. Res. NBS, 1953, 50, p. 63.
803. Bye J., Kiehl J. G. — Bull. Soc. chim. France,
1948, 15, p. 847.
804. Cabrerizo J. L. — An. quim. Real soc. exp. fis.
у quim., 1976, 72, p. 315.
805. Cade P. F., Huo W. M. — J. Chem. Phys., 1967,
47, p. 614.
806. Cade P. E., Huo W. M. — J. Chem. Phys., 1967,
47, p. 649.
807. Cakajdova I. — Chem. zvest., 1972, 26, s. 41.
807a. Calcagni G. — Atti Accad. naz. Lincei. Mem. Cl.
sci. fis., mat. e natur. Sez. II, 1912, 21, p. 483.
808. Colder G. V., Mann D. E., Seshadri K. S., Alla-
vena M., White D. — J. Chem. Phys., 1969, 51,
p. 2093.
809. Colder G. V., Ruedenberg K. — J. Chem. Phys.,
1968, 49, p. 5399.
810. Campbell A. N., Kartzmark E. M., Bhatna-
gar O. N. — Canad. J. Chem., 1974, 52, p. 954.
811. Campbell С S. — Metall Soc. Conf., 1961, 7,
p. 412.
812. Campbell J. E., Powell С F., Nowicki D. #., Gon-
ser B. W. — J. Electrochem. Soc, 1949, 96, p. 318.
813. Camus P. — J. Phys. B: Atom, and Mol. Phys;
1974, 7, p. 1154.
814. Cantor S. — J. Chem. and Eng. Data, 1965, 10,
p. 237.
814a. Cantor S. — Inorg. and Nucl. Chem. Lett., 1973,
9, p. 1275.
815. Capelle G. A., Broida H. P., Field R. W. — J.
Chem. Phys., 1975, 62, p. 3131.
816. Carabedion M. E., Benson S. W. — J. Amer. Chem.
Soc, 1964, 86, p. 176.
817. Caranoni C, Tavier R., Capella L., Tranquard A. —
С r. Acad. sci., 1970, C270, p. 1795.
818. Carleer M., Burtin В., Colin R. — Canad. J.
Phys., 1977, 55, p. 582.
819. Carleer M., Colin R. — Canad. J. Phys., 1977,
55, p. 129.
820. Carleer M., Herman M., Colin R. — Canad. J.
Phys., 1975, 53, p. 1321.
821. Carlson K. D., Kaiser K., Moser C, Wahl A. C. —
J. Chem. Phys., 1970, 52, p. 4678.
822. Carr E. M., Clarne J. Т., Johnston H. L. — J.
Amer. Chem. Soc, 1949, 71, p. 740.
823. Carreira L. A., Maroni V. A., Swaine J. M., Jr.,
Plumb R. С — J. Chem. Phys., 1966, 45, p. 2216.
824. Carreira L. A., Odom J. D., Durig J. R. — J.
Chem. Phys., 1973, 59, p. 4955.
825. Carsky P., Malek J. — Collect. Czech. Chem.
Communs, 1977, 42, s. 2758.
826. Carter R. H. — Ind. and Eng. Chem., 1928, 20,
p. 1195.
827. Castellari C. — Atti. Accad. naz. Lincei. Rend.
Cl. sci. fis., mat. e natur., 1969, 45, p. 533.
827a. Cater E. D., Johnson E. W. — J. Chem. Phys.,
1967, 47, p. 5353.
828. Caton R. В., Douglas A. E. —Canad. J. Phys.,
1970, 48, p. 432.
829. Centola G. — Congr. intern, quim. pura alicacha.
Madrid, 1934, 3, p. 230.
830. Chadwick R. — J. Inst. Metals, 1928, 39, p. 285.
831. Chai B.J.,KoH.C, Greenbaum M.A., Farber M. —
J. Phys. Chem., 1967, 71, p. 3331.
832. Challacombe C. N., Almy G. M. —}Phys. Rev.,
1937, 51, p. 930.
833. Chan A. C. H., Davidson E. R. — J. Chem. Phys.,
1968, 49, p. 727.
834. Chan A. C. H., Davidson E. R. — J. Chem. Phys.,
1970, 52, p. 4108.
835. Chang С H., Porter R. F., Bauer S. H. — Inorg.
Chem., 1969, 8, p. 1689.
835a. Chang S. S. — In: Seventh Symp. on Thermophys.
Properties, U. S. NBS. Gaithersburg (Md), 1978,
p. 83.
836. Charton M. M., Gaydon A. G. — Proc. Phys. Soc.
London, 1956, A69, p. 520.
837. Chassaing J. — Rev. chim. miner., 1972, 9, p. 265.
838. Chassevent L. — С. г. Acad. sci., 1924, 179, p. 44.
839. Chatillon Ch., Allibert M., Pattoret A. — С. г.
Acad. sci., 1975, C280, p. 1505.
840. Chatterji D.. Smith J. V. — J. Electrochem. Soc,
1973, 120, p. 770.
840a. Chatterji D., Vest R. W. — J. Amer. Ceram.
Soc, 1972, 55, p. 575.
8406. Chaudhry A. K., Upadhya K. N. — Indian J.
Phys., 1968, 42, p. 544.
841. Cheetham C. J., Gissane W. J. M., Barrow R. F. —
Trans. Faraday Soc, 1965, 61, p. 1308.
841a. Chemouni E., Maglione M. H., Potier A. — Bull.
Soc. chim. France, 1970, N 2, p. 489.
8416. Chen H. S., Turnbull D. — Acta met., 1968,
16, p. 369.
842. Chen Horng-yih, ConardB. R., GillesP. W. — Inorg.
Chem., 1970, 9, p. 1776.
438
Литература
843. Chen Horng-yih, Gilles P. W. — J. Phys. Chem.,
1972, 76, p. 2035.
844. Chervonnyi A. D., Piven V. A., Kashireninov О. Е.,
Manelis G. B. — High Temp. Sci., 1977, 9, p. 99.
845. Chiang J. F., Whitman D. R. — Theor. chim.
acta, 1970, 17, p, 155.
846. Chiotti P., Gartner G. J., Stevens E. R., Saito Y. —
J. Chem. and Eng. Data, 1966, 11, p. 571.
847. Chipman J. — Trans. Faraday Soc, 1933, 29,
p. 1266.
848. Chretien M. — Helv. phys. acta, 1950, 23, p. 259.
849. Chretien M., Miescher E. — Helv. phys. acta,
1949, 22, p. 588.
850. Chroustchoff M., Martinoff A. — Ann. chim. phys.,
1887, 11, p. 234.
851. Chu S. Y., Frost A. A. — J. Chem. Phys., 1971,
54, p. 764.
852. Chupka W.A., Berkowitz J., Giesse С F. — J. Chem.
Phys., 1959, 30, p. 827.
853. Chupka W. A., Berkowitz J., Giese С F., Ingh-
ram M. G. — J. Phys. Chem., 1958, 62, p. 611.
854. Churney K. L., Armstrong G. T. — J. Res. NBS,
1969, A73, p. 281.
855. Claassen A., Veenemans С — Z. Phys., 1933, 80,
S. 342.
856. Clark R. I., Griswold E., Kleinberg I. — J. Amer.
Chem. Soc, 1958, 80, p. 4764.
857. Claudy P., Bonnetot В., Etienne J., Turck G. —
J. Therm. Anal., 1975, 8, p. 255.
857a. Claudy P., Bonnetot В., Letoffe J. M. — Thermo-
chim. acta, 1978, 27, p. 205.
858. Clement J. R., Quinnell E. H. — Phys. Rev.,
1953, 92, p. 258.
858a. Clements R. M., Barrow R. F. — J. Chem. Soc.
Chem. Communs, 1968, N 22, p. 1408.
859. Cline C. F. — J. Electrochem. Soc, 1959, 106,
p. 322.
859a. Clusius K., Harteck P. — Z. phys. Chem., 1928,
134, p. 243.
860. Clusius K., Schachinger L. — Z. angew. Phys.,
1952, 4, S. 442.
861. Clusius K., Vaughen J. V. — J. Amer. Chem.
Soc, 1930, 52, p. 4684.
862. Cochran С N., Foster L. M. — J. Electrochem.
Soc, 1962, 109, p. 144.
863. Соске D. L., Change C. A., Gingerich K. A. — Appl.
Spectrosc, 1973, 27, p. 260.
864. CODATA recommended key values for
thermodynamics: COD ATA Bull., 1971, N 5; CODATA
Bull., 1973, N 10; CODATA Bull., 1975, N 17;
CODATA Bull., 1976, N 22.
865. Codling K. — Proc. Phys. Soc. London, 1961,
77, p. 797.
866. Coffy G. — These doct. sci. phys. Univ. Claude
Bernard, Lyon, 1972.
866a. Coffy G., Olofsson G. — J. Chem. Thermodyn.,
1979, 11, p. 141.
867. Cohen E., Blekkingh J. J. A. — Z. phys. Chem.
Leipzig, 1940, A186, S. 257.
867a. Coheur F. P., Rosen B. — Bull. Soc. roy. sci.
Liege, 1941, 10, p. 405.
868. Cole S. S., Taylor N. W. — J. Amer. Ceram. Soc,
1935, 18, p. 82.
869. Coleman D. S., Pollitt R. — Inst. Min. Metall
Trans., Sect. C, 1969, 78, p. 233.
870. Coleman F. F., Egerton A. C. — Philos. Trans.
Roy. Soc. London, 1935, A234, p. 177.
871. Colin R., Carleer M., Prevot F. — Canad. J. Phys.,
1972, 50, p. 171.
872. Colin R., De Greef D. — Canad. J. Phys., 1975,
53, p. 2142.
873. Colin R., Goldfinger P., Jeunehomme M. — Trans.
Faraday Soc, 1964, 60, p. 306.
874. Collins J. В., von Schleuer P. R., Binkley J. S.,
Pople J. A. — J. Chem. Phys., 1976, 64, p. 5142.
875. Comeford J. J., Abramowitz S., Levin I. W. — J.
Chem. Phys., 1965, 43, p. 4536.
876. Connerade J. P. — Astrophys. J., 1972, 172, p. 213.
877. Conway J. В., Hein R. A. — Nucleonics, 1964,
22, p. 71.
878. Copaux H., Philips Ch. — С. г. Acad. sci., 1920,
171, p. 630.
879. Copaux H., Philips Ch. — С. г. Acad. sci., 1923,
176, p. 579.
880. Coppens P., Smoes S., Drowart J. — Trans.
Faraday Soc, 1968, 64, p. 630.
881. Corbett /., Gregory N. — J. Amer. Chem. Soc,
1954, 76, p. 1446.
882. Corson M. D. Aluminium, the metall and its
alloys. L.: Chapman and Hall, 1926.
883. Cosgrove S. A., Snyder P. E. — J. Amer. Ghem.
Soc, 1953, 75, p. 3102.
884. Cote G. L., Thompson H. W. — Proc. Roy. Soc.
London, 1952, A210, p. 217.
885. Cotton D. H., Jenkins D. R. — Trans. Faraday
Soc, 1968, 64, p. 2988.
886. Cotton D., Jenkins D. — Trans. Faraday Soc,
1969, 65, p. 376.
887. Coughlin J. P. — Bull. Bur. Mines (USA), 1954,
N 542.
888. Coughlin J. P. — J. Amer. Chem. Soc, 1956, 78,
p. 5479.
889. Coughlin J. P. — J. Phys. Chem., 1958, 62, p. 419.
890. Caunt A. D., Barrow R. F. — Trans. Faraday Soc,
1950, 46, p. 154.
891. Cousseins J. C, Freundlich W., Erb A. — C. r.
Acad. sci., 1969, C268, p. 717.
892. Cowley J. M. — Acta crystallogr., 1953, 6, p. 615.
893. Cowley J. M. — Nature, 1953, 171, p. 440.
894. Cowley A. H., White W. D., Damasco M. C. —..
J. Amer. Chem. Soc, 1969, 91, p. 1922.
895. Cox J. H., Pidgeon L. M. — Canad. J. Chem.,
1963, 41, p. 671.
896. Coyle R. Т., Searcy A. W. — High Temp. Sci.,
1973, 5, p. 335.
897. Coyle T. D., Ritter J. J., Farrar Т. С — Proc.
Chem. Soc. London, 1964, p. 25.
898. Craig R. S., Krier С A., Coffer L. W., Bates E. A.,
Wallace W. E. — J. Amer. Chem. Soc, 1954,
76, p. 238.
899. Crawford B. L., Edsall J. T. — J. Chem. Phys.,
1939, 7, p. 223.
900. Crawford F. #., Folliott С. Е. F. — Phys. Rev.,
1933, 44, p. 953.
901. Creswell R. A., Mocking W. H., Pearson E. F. —
Chem. Phys. Lett., 1977, 48, p. 369.
902. Criss С M., Cobble С — J. Amer. Chem. Soc,
1961, 83, p. 3233.
903. Cristescu S., Simon F. — Z. phys. Chem. Leipzig,
1934, B25, S. 273.
904. Cubicciotti D. — J. Phys. Chem. 1964, 68, p. 1528.
905. Cubicciotti D. — High Temp. Sci., 1969, 1, p. 11.
906. Cubicciotti D. — High Temp. Sci., 1970, 2, p. 213.
907. Cubicciotti D., Eding H. — J. Chem. and Eng.
Data, 1964, 9, p. 524.
908. Cubicciotti D., Eding H. — J. Chem. and Eng.
Data, 1965, 10, p. 343.
909. Cubicciotti D., Eding H. — J. Chem. and Eng.
Data, 1967, 12, p. 548.
439
Литература
909а. Cubicciotti D., Keneshia F. J. — J. Phys. Chem.,
1967, 71, p. 808.
910. Cubicciotti D., Withers G. L. — J. Phys. Chem.,
1965, 69, p. 4030.
911. Cueilleron M. G. — C. r. Acad. sci., 1944, 219,
p. 209.
912. Cueilleron M. G. — Ann. chim., 1944, 19, p. 459.
913. Culbert H., Huebener R. P. — Phys. Lett., 1967,
24, p. 530.
913a. Culver R. V., Hamdorf C.J. — J. Appl. Chem.,
1955, 5, p. 383.
9136. Cummins P. G., Field R. W., Renhorn I. — in
press.
913b. Curlook W., Pidgeon L. M. — Trans. АШЕ,
1958, 212, p. 671.
914. Curtis R. W., Chiotti P. — J. Phys. Chem., 1963,
67, p. 1061.
914a. Curtiss L. A. — Intern. J. Quant. Chem., 1978,
14, p. 709.
915. Cyvin S. J. — Spectrosc. Lett., 1974, 7, p. 255.
915a. Cyvin S. J., Cyvin B. N., Snelson A. — J. Phys.
Chem., 1971, 75, p. 2609.
916. Cyvin S. J., Phongsatha A. — Speotrosc. Lett.,
1974, 7, p. 523.
917. Cyvin S. J., Torset 0. — Rev. chim. miner., 1972,
9, p. 179.
918. Czarny J., Felenbok P. — Chem. Phys. Lett., 1968,
2, p. 533.
919. Dadgar A., Taherian M. R. — J. Chem. Thermo-
dyn., 1977, 9, p. 711.
920. Dagdigian P. J., Cruse H. W., Schultz A.,
Zare R. N. — J. Chem. Phys., 1974, 61, p. 4450.
921. Dagdigian P. /., Cruse H. W., Zare R. N. — J.
Chem. Phys., 1975, 62, p. 1824.
922. Dagdigian P. J., Wharton L. — J. Chem. Phys.,
1973, 58, p. 1243.
923. Dagdigian P. J., Zare R. N. — J. Chem. Phys.,
1974, 61, p. 2464.
924. Danielson D. D., Patton J. V., Hedberg K. — J.
Amer. Chem. Soc, 1977, 99, p. 6484.
925. Dareis R. E., Murthy A. S. — Tetrahedron, 1968,
24, p. 4595.
926. Darji А. В., Shah S. G. — Indian J. Pure and
Appl. Phys., 1975, 13, p. 187.
927. Darji А. В., Vaidya S. P. — Indian J. Pure and
Appl. Phys., 1973, 11, p. 923.
927a. Datta R., Datta M. K., Ghosh D. C. — Indian
J. Chem., 1977, A15, p. 259.
928. Datta S. — Proc. Roy. Soc. London, 1921, A99,
p. 436.
929. Datta S. — Proc. Roy. Soc. London, 1922, A101,
p. 187.
930. Daunt J. G., Horseman A., Mendelssohn K. —• Phil.
Mag., 1939, 27, p. 754.
930a. David D. J. — Anal. Chem., 1964, 36, p. 2162.
931. David J., Laurent Y., Lang J. — Bull. Soc. franc,
miner, et cristallogr., 1971 A972), 94, p. 340.
931a. Davidovitz P., Bellisio J. A. — 1. Chem. Phys.,
1969, 50, p. 3560.
932. Davis J. W., Collins A. G. — Environ. Sci. and
Technol., 1971, 5, p. 1039.
933. Dazord J., Mongeot H. — Bull. Soc. chim. France,
1971, N 1, p. 51.
934. Dear D., Eley D. — J. Chem. Soc, 1954, p. 4684.
935. Decius J. C, Becker С R., Fredericks W. J. — J.
Chem. Phys., 1972, 56, p. 5189.
936. Decius J. C, Becker С. Д., Fredericks W. J. — J.
Chem. Phys., 1973, 58, p. 1782.
937. Deezsi J., Koczkas E., Matrai T. — Acta phys.
Acad. sci. hung., 1953, 3, p. 95.
938. Dehmer J. L., Berkowitz J., Cusachs L. С — J..
Chem. Phys., 1973, 58, p. 5681.
939. Delbove F. — С. г. Acad. sci., 1961, 252, p. 2192.
940. Delvigne G. A. L., De Wijn H. W. — J. Chem.
Phys., 1966, 45, p. 3318.
941. De Maria G., Drowart J., Inghram M. G. — J.
Chem. Phys., 1959, 30, p. 318.
942. De Maria G., Gingerich K. A., Piacente V. — J.
Chem. Phys., 1968, 49, p. 4705.
943. De Maria G., Piacente V. — J. Chem. Thermodyn.T
1974, 6, p. 1.
944. Denison G. W. — Diss. Abstrs, 1962, 23, p. 85.
945. Desre P. J., Hawkins D. Т., Hultgren R. — Trans.
Met. Soc. AIME, 1968, 242, p. 1231.
946. Dessaux O., Goudmand P., Pannetier G. — C. r.
Acad. sci., 1967, 265, p. 480.
947. Dessaux O., Goudmand P., Pannetier G. — Bull.
Soc. chim. France, 1969, p. 447.
948. Dewar M. J. S., McKee M. L. — J. Amer. Chem.
Soc, 1977, 99, p. 5231.
949. Dewing E. W., Richardson F. D. — Trans. Faraday
Soc, 1959, 55, p. 611.
949a. Dewing E. W., Richardson F. D. — J. Iron and
Steel Inst., 1960, 195, p. 56.
950. Dibeler V. H., Listen S. K. — Inorg. Chem., 1968,
7, p. 1742.
951. Dibeler V. H., Walker J. A. — Inorg. Chem., 1969r
8, p. 50.
952. Dickson С R., Kinney J. В., Zare R. N. — Chem.
Phys., 1976, 15, p. 243.
952a. Diercks H., Krebs B. — Angew. Chem., 1977, 89,
S 327
953. Dijkerman H., Flegel W., Graff G., Monter B. —
Z. Naturforsch., 1972, A27, S. 100.
954. Dill J. D., Schleyer P. v. R., Pople J. A. — J.
Amer. Chem. Soc, 1975, 97, p. 3402.
955. Dinescu R., Preda M. — J. Therm. Anal., 1973,
5, p. 465.
956. Ditmars D. A., Douglas T. B. — U. S. NBS, 1970,
Rept N 10326.
957. Ditte A. — С. г. Acad. sci., 1871, 72, p. 858.
958. Ditte A. — С. г. Acad. sci., 1877, 85, p. 1103.
959. Dixon M., Hoare F. E., Holden Т. М.,
Moody D. M. — Proc. Roy. Soc. London, 1965,
A 285, p. 561.
960. Dixon R. N., Field D., Noble M. — Chem. Phys.
Lett., 1977, 50, p. 1.
961. Dodsworth P. G., Barrow R. F. — Proc. Phys.
Soc. London, 1954, A67, p. 94.
962. Dodsworth P. G., Barrow R. F. — Proc Phys.
Soc. London, 1955, A68, p. 824.
962a. Doeltz F. O., Mostowitsch W.—Ъ. anorg. und
allg. Chem., 1913, 81, p. 257.
963. Doescher R. N. — J. Chem. Phys., 1951, 19, p. 1070.
964. Dollimore D., Griffiths D. L. — J. Therm. Anal.,
1970, 2, p. 229.
964a. Dolloff W. — U. S., WADD Tech. Rept 60-143,
1960.
965. Domaille P. /., Steimle T. C, Harris D. O. — J.
Mol. Spectrosc, 1977, 66, p. 503.
965a. Domaille P. J., Steimle T. C, Harris D. O. —
J. Mol. Spectrosc, 1977, 68, p. 146.
9656. Domaille P. /., Steimle T. C, Harris D. O. —
J. Chem. Phys., 1978, 68, p. 4977.
966. Domaille P. /., Steimle Т. С, Wong Ning Bew,
Harris D. O. — J. Mol. Spectrosc, 1977, 65, p. 354.
967. Domalski E. S., Armstrong G. T. — J. Res. NBS,
1965, A69, p. 137.
440
Литература
968. Domalski Е. S., Armstrong G. Т. — J. Res. NBS,
1967, A71, p. 195.
969. Domalski E. S., Armstrong G. T. — J. Res. NBS,
1968, A72, p. 133.
970. Doman R. S., Barr J. В., McNally R. N., Al-
per A. M. — J. Amer. Ceram. Soc, 1963, 46,
p. 313.
971. Domange L. — Ann. chim., 1937, 7, p. 225.
972. Donoghue J. J., HubbardD. — J. Res. NBS, 1941,
27, p. 371.
972a. Donahue P. C. — J. Solid State Chem., 1970,
2, p. 6.
973. Douglas A. E. — Canad. J. Res., 1941, A19, p. 27.
974. Douglas A. E., Herzberg G. — Canad. J. Res.,
1940, A18, p. 165.
975. Douglas A. E., Herzberg G. — Canad. J. Res., 1940,
A18, p. 179.
976. Douglas P. E. — Proc. Phys. Soc. London, 1954,
B67, p. 783.
977. Douglas Т. В., Beckett С W. Preliminary report
on the thermodynamic properties of selected light-
element compounds: NBS Rept 6928. Wash., 1960.
978. Douglas Т. В., Ditmart D. A. — J. Res. NBS,
1967, A71, p. 185.
979. Douglas T. 5., Furukawa G. Т., Saba W. G.,
Victor A. C. — J. Res. NBS, 1965, A69, p. 423.
980. Douglas Т. В., Payne W. H. — J. Res. NBS, 1969,
A73, p. 471.
981. Dows D. A., Porter R. F. — J. Amer. Chem. Soc,
1956, 78, p. 5165.
982. Drdgert W. — Dissertation. В., 1914.
983. Drake M. C, Rosenblatt G. M. — J. Chem. Phys.,
1976, 65, p. 4067.
983a. Drapcho D. L., Rosenblatt G. M. — J. Chem.
Thermodyn., 1979, II, p. 335.
984. Dreger L. H., Dadape V. V., Margrave J. L. — J.
Phys. Chem., 1962, 66, p. 1556.
985. Drowart J. — Faraday Symp. Chem. Soc, 1973,
N 8, p. 165.
986. Drowart J., De Maria G., Burns R. P., Ingh-
ram M. G. — J. Chem. Phys., 1960, 32, p. 1366.
987. Drowart /., Exsteen G., Verhaegen G. — Trans.
Faraday Soc, 1964, 60, p. 1920.
988. Drowart /., Honig R. E. — Bull. Soc. chim. belg.,
1957, 66, p. 411.
989. Drowart J., Pattoret A., Smoes S. — Proc. Brit.
Ceram. Soc, 1967, N 8, p. 67.
990. Ducros M., Levy R., Meliava G. — Bull. Soc. chim.
France, 1970, p. 2763.
991. Duke F. R., Carfinkel H. M. — J. Phys. Chem.,
1961, 65, p. 461.
991a. Dumas Y., Potier A. — Bull. Soc. chim. France,
1970, p. 1319.
992. Duncan A. B. F. — J. Amer. Chem. Soc, 1929,
51, p. 2697.
993. Duncan J. L. — J. Mol. Spectrosc, 1967, 22,
p. 247.
994. Duncanson A., Stevenson R. W. H. — Proc. Phys.
Soc London, 1958, 72, p. 1001.
995. Dunn T. M., Hanson L: — Canad. J. Phys., 1969,
47, p. 1657.
996. Dunne T. G., Gregory N. W. — J. Amer. Chem.
Soc, 1958, 80, p. 1526.
996a. Dupuis M., Liu B. — J. Chem. Phys., 1978,
68, p. 2902.
997. Durand J. F. — Bull. Soc. chim. France, 1924,
35, p. 1207.
998. Durig J. R., Green W. H., Marston A. L. — J.
Mol. Struct.,ч 1968, 2, p. 19.
998a. Durig J. R., Li Y. S., Odom J. D. — J. Mol.
Struct., 1973, 16, p. 443.
999. Durig J. R., Sounders J. E., Odom J. D~. — J.
Chem. Phys., 1971, 54, p. 5285.
1000. Durig J. R., Thompson J. W., Witt J. D.,
Odom J. D. — J. Chem. Phys., 1973, 58, p. 5339.
1001. Dutoit W. — J. chim. phys. et phys.-chim. biol.r
1927, 24, p. 110.
1001a. Duval C, Lecompte J. — Bull. Soc. chim. France,
1941, 8, p. 713.
1002. Dworkin A. S., Bredig M. A. — J. Chem. and
Eng. Data, 1963, 8, p. 416.
1003. Dworkin A. S., Bredig M. A. — J. Phys. Chem.,.
1963, 67, p. 697.
1004. Dworkin A. S., Bredig M. A. — J. Phys. Chem.,
1971, 75, p. 2340.
1005. Dworkin A. S., Sasmor D. /., Artsdalen E. R.
van. — J. Phys. Chem., 1954, 22, p. 837.
1005a.Easley W. C, Weltner W. — J. Chem. Phys.,
1970, 52, p. 1489.
10056.Eastman E.. D., Brewer L. — J. Amer. Chem.
Soc, 1950, 72, p. 2248.
1006. Eastman E. D., Williams A.M., Young T. F. —
J. Amer. Chem. Soc, 1924, 46, p. 1178.
1007. Eckerlin P., Rabenau A. — Z. anorg. und allg>
Chem., 1960, 304, S. 218.
1008. Eckstein B. H., Artsdalen E. R. van. — J. Amer.
Chem. Soc, 1958, 80, p. 1352.
1009. Eding H., Cubicciotti D. — J. Chem. and Eng.
Data, 1964, 9, p. 524.
1010. Edlen В., dime A., Herzberg G., Johns J. W. C. —
J. Opt. Soc. Amer., 1970, 60, p. 889.
1011. Edlen В., Risberg P. — Ark. fys., 1966,10, p. 553.
1012. Edvinsson G. — Naturwissenschaften, 1962, 49,
S. 418.
1013. Edvinsson G., Kopp I., Lindgren В., Aslund N. —
Ark. fys., 1963, 25, p. 95.
1014. Edwards J. G., Leitnaker J.M., Wiedemeier Я.,
Gilles P. W. — J. Phys. Chem., 1971, 75, p. 2410.
1015. Edwards J. O., Morrison G., Ross V. F.,.
Schultz J. W. — J. Amer. Chem. Soc, 1955, 77,
p. 266.
1016. Edwards J. W., Kington G. L. ~ Trans. Faraday
Soc, 1962, 58, p. 1313.
1017. Efimenko J. — J. Res. NBS, 1968, A72, p. 75.
1017a. Efimov M. E., Kislova G. N., Medvedev V. A.
Thesises of papers of 34 Annual calorimetry
conference. Kent (Ohio), 1979.
1018. Eggersgluess W., Monroe A. G., Parker W. G. —
Trans. Faraday Soc, 1949, 45, p. 661.
1019. Ehlert Т. С — J. Inorg. and Nucl. Chem., 1968,
30, p. 3112.
1020. Ehlert T. C, Blue G. D., Green J. W.,
Margrave J. L. — 3. Chem. Phys., 1964, 41, p. 2250.
1021. Ehlert T. C, Margrave J. L. — J. Amer. Chem.
Soc, 1964, 86, p. 3901.
1022. Ehrlich P., Peik K., Koch E. — Z. anorg. und
allg. Chem., 1963, 324, S. 113.
1022a. Eichenauer W., Schulze M. — Z. Naturforsch.,
1959, A14, S. 962.
1023. Einecke E. — Angew. Chem., 1942, 55, p. 40.
1024. Eitel W. — Z. Kristallogr., 1925, 61, S. 596.
1024a. Eitel W. — Zement, 1938, 27, S. 455, 469.
1025. Eley D. D., Watts H. — J. Chem. Soc, 1954r
p. 1319.
1026. Ellinger F. H., Ilolley C. E., Mclnteer В. В.,
Pavone D., Potter R. M., Staritzky E., Zacharia-
sen W.H. — J. Amer. Chem. Soc, 1955, 77, p. 2647.
441
Литература
1027. Ellis В., Mortland M. M. — Amer. Miner., 1962,
47, p. 371.
1028. Ellisson F. 0. — J. Phys. Chem., 1962, 66, p. 2294.
1029. Ellisson F.O. — J. Chem. Phys., 1965, 43, p. 3654.
1030. Emons H. H. — Z. anorg. und allg. Chem., 1963,
323, S. 114.
1030a. Emons H. H., Brautigam G., Thomas R. —
Chem. zvest., 1976, 30, S. 773.
1031. Emons H. #., Loffelholz B. — Wiss. Z. Techn.
Hochsch. Chem. Leuna-Merseburg, 1964, 6, S. 261.
1032. Engberg С I., Zehms E. H. — J. Amer. Ceram.
Soc, 1959, 42, p. 300.
1033. Engel T. K. — J. Nucl. Mater., 1969, 31, p. 211.
1033a. Engelke F. — Ber. Bunsenges. phys. Chem.,
1977, 81, S. 135.
10336. Engelke F. — Chem. Phys., 1979, 39, p. 279.
1034. Engelke F., Sander R., Zare R. — J. Chem. Phys.,
1976, 65, p. 1146.
1035. Ephraim F., Michel E. — Helv. chim. acta, 1921,
4, p. 900.
1036. Eriksson G., Hulthen E. — Z. Phys., 1925, 34,
5. 786.
1037. Eriksson K. B. S., Isberg H. B. S. — Ark. fys.,
1963, 23, p. 527.
1038. Eriksson K. B. S., Isberg H. B. S. — Ark. fys.,
1967, 33, p. 593.
1039. Erway N. D., Seifert R. L. — J. Electrochem.
Soc, 1951, 98, p. 83.
¦1040. Esser H., Averdieck R., Grass W. — Arch. Eisen-
hiittenw., 1933, 6, S. 289.
1040a. Estler R. C, Zare R. N. — Chem. Phys., 1978,
28, p. 253.
1041. Eucken A., Schroder E.—Z. phys. Ghem.
Leipzig, 1938, B41, S. 307.
1042. Evans M. G., Warhurst E., Whittle E. — J. Chem.
Soc, 1950, p. 1524.
1043. Evans P. J., Mackie J. C. — Chem. Phys., 1974,
5, p. 277.
1044. Evans W. H. - U. S. NBS, Rept N 7093, 1961.
1045. Evans W. H., Prosen E. /., Wagman D. D. Ther-
modynamic and transport properties of gases,
liquids and solids. N. Y., 1959.
1045a. Evans W. H., Wagman D. D., Prosen E. J. —
U. S. NBS, Rept. N 4943, 1956.
1046. Ezhov Y. S., Kasparov V. V. — In: VII Austin
Symp. on gas phase molecular structure. Austin
(Tex.), 1978.
1046a. Farber M. — 1960—1962 (см. [1543]).
1047. Farber M. — J. Chem. Phys., 1962, 36, p. 661.
1048. Farber M. — J. Chem. Phys., 1962, 36, p. 1101.
1049. Farber M., Blauer J. — Trans. Faraday Soc,
1962, 58, p. 2090.
1050. Farber M., Buyers A. — In: Proc. Symp. Thermo-
phys. Properties, 5th, N. Y., 1970, p. 483.
1051. Farber M., Frisch M. A. — In: Proc. First Intern.
Conf. calorimetry and thermodynamics. Warsaw,
1969, p. 443.
1051a. Farber M., Frisch M. A., Grenier G., Ко H. C.
Space sciences: Inc., Final Report, under USAF
contract F04611—67—C—0010, AFRPL—TR—
67—244. 1967, nov., (Цит. по: JANAF 1971).
1052. Farber M., Harris S. P. — High Temps. Sci.,
1971, 3, p. 231.
1053. Farber M., Pelersen H. L. — Trans. Faraday Soc,
1963, 59, p. 836.
1054. Farber M., Petersen H. L., Blauer J. A., Brown D.,
Davis J. Thermodynamics of reactions involving
light metal oxides and propeliant gases: Rocket
Power, Inc., AD284428. Pasadena (Calif.), 1962.
1055. Farber M., Srivastava R. D. — J. Chem. Soc.
Faraday Trans. 1, 1974, 70, p. 1581.
1056. Farber M.. Srivastava R. D. — In: 4eme Conf.
intern, thermodyn. chim. Montpellier, 1975,
vol. 8, p. 34.
1057. Farber M., Srivastava R. D. — High Temp. Sci.,
1975, 7, p. 74.
1058. Farber M., Srivastava R. D. — Chem. Phys. Lett.,
1976, 42, p. 567.
1059. Farber M., Srivastava R. D. — Combust, and
Flame, 1976, 27, p. 99.
1060. Farber M., Srivastava R. D. — High Temp. Sci.,
1976, 8, p. 73.
1061. Farber M., Srivastava R. D. — High Temp. Sci.,
1976, 8, p. 195.
1062. Farber M., Srivastava R. D., Frish M. A.,
Harris S. P. — Faraday Symp. Chem. Soc, 1973,
N 8, p. 121.
1063. Farber M., Srivastava R. D., Uy О. М. — J. Chem.
Phys., 1971, 55, p. 4142.
1064. Farber M., Srivastava R. D., Uy О. М. — J. Chem.
Soc. Faraday Trans. 1, 1972, p. 249.
1065. Farkas L. — Z. Phys., 1931, 70, S. 733.
1066. Farkas L., Levy S. — Z. Phys., 1933, 84, S. 195.
1067. Farrow R. F. C. — J. Phys. D: Appl. Phys.,
1974, 7, p. 2436.
1068. Fasolino L. G. — In: Abstracts of Papers 18th
Calorimetry Conf. Bartlesille (Oklh), 1963.
1069. Fasolino L. G. — J. Chem. and Eng. Data, 1965,
10, p. 373.
1070. Favre P. A., Silberman J. T. — Ann. chim. phys.,
1852, 36, p. 5.
1071. Favre P. A., Valson C. A. — C. r. Acad. sci.,
1871, 73, p. 1147.
1071a. Feather D. H., Buckler A., Searcy A. W. — High
Temp. Sci., 1972, 4, p. 290.
1072. Feber R. C., Herrik С. С An improved calculation
of the ideal gas thermodynamic functions of
selected diatomic molecules: Rept LA—3597. Los
Alamos, 1966/67.
i073.' Fehlner T. P., Koski W. S.—J. Amer. Chem.
Soc, 1964, 86, p. 2733.
1074. Fehlner T. P., Koski W. S. — J. Amer. Chem.
Soc, 1965, 87, p. 409.
1075. Feller-Kniepmeier M., Heumann T. — Z. Metallk.,
1960, 51, S. 404.
1075a. Ferrari A., Inganni A. — Atti Accad. naz.
Lincei. Rend. Cl. sci. fis., mat. e natur., 1929,
10, p. 253.
1076. Ferrier A., Olette M. — С. г. Acad. sci., 1962,
254, p. 4293.
1077. Ficalora P. J.. Hastie J. W., Margrave J. L. —
J. Phys. Chem., 1968, 72, p. 1660.
1078. Fichter F., Brunner E. — Z. anorg. und allg.
Chem., 1915, 93, S. 84.
1079. Fichter F., Jenny E. — Helv. chim. acta, 1922,
5, p. 448.
1080. Field R. W. — J. Chem. Phys., 1974, 60, p. 2400.
1081. Field R. W., Bradford R. S., Harris D. O., Bro-
ida H. P. — J. Chem. Phys., 1972, 56, p. 4712.
1082. Field R. W., Capelle G. A., Jones С R. — J. Mol.
Spectrosc, 1975, 54, p. 156.
1082a. Field R. W., Capelle G. A., Revelli M. A. —
J. Chem. Phys., 1975, 63, p. 3228.
1083. Field R. W., English A. D., Tanaka Т.,
Harris D. O., Jennings D. A. — J. Chem. Phys.,
1973, 59, p..2191.
1084. Field R. W., Harris D. O., Tanaka T. — J. Mol.
Spectrosc, 1975, 57, p. 107.
442
Литература
1084а. Filipovska N. J., Bell Н. В. — Гласник Хем.
друшт, Београд, 1975, 40, р. 491.
1085. Finch А. — Recueil trav. chim., 1964, 83, p. 1325.
1086. Finch A., Gardner.P. J., Hyams I. J. —Trans.
Faraday Soc, 1965, 61, p. 649.
1087. Finch A., Gardner P. J., Steadman С J. — Canad.
J. Chem., 1968, 46, p. 3447.
1088. Finch A., Hyams I. J., Steele D. —Trans.
Faraday Soc, 1965, 61, p. 398.
1089. Finch A., Hyams I. J., Steele D. — Spectrochim.
acta, 1965, 21, p. 1423.
1090. Finkelstein S. — Berichte, 1906, 39, S. 1586.
1091. Finn P. A., Gruen D. M., Page D. — Adv. Chem.
Ser., 1976, 158, p. 30.
1092. Finnemore D. K., Mapother D. E. — Phys. Rev.,
1965, A140, p. 507.
1093. Fischer J. — Fest. Techn. Hochschule. Breslau,
1935, S. 172.
1094. Fischer W. — Z. anorg. und allg. Chem.. 1931,
200, S. 332.
1095. Fischer W. — Z. anorg. und allg. Chem., 1933,
211, S. 321.
1096. Fischer W., Jiibermann O. — Z. anorg. und allg.
Chem., 1936, 227, S. 227.
1097. Fischer W., Petzel Th. — Z. anorg. und allg.
Chem., 1964, 333, S. 226.
1098. Fischer W., Rahlfs O. — Z. anorg. und allg. Chem.,
1933, 211, S. 349.
1099. Fischer W., Rahlfs O. — Z. Elektrochem., 1932,
38, S. 592.
1100. Fischer W., Rahlfs O., Benze B. — Z. anorg. und
allg. Chem., 1932, 205, S. 1.
1101. Fisher H. D., Kiehl J., Cane A. — U. S. Dept.
Com. Office Techn. Serv., AD 274, 243, 1961.
1102. Fisher H. D., Lehmann W. /., Shapiro J. —
J. Phys. Chem., 1961, 65, p. 1166.
1103. Fitzgibbon G. C, Huber E. J., Jr., Holley С. Е.,
Jr. — J. Chem. Thermodyn., 1973, 5, p. 577.
1104. Fitzpatrick N. J. — Inorg. and Nucl. Chem. Lett.,
1973, 9, p. 965.
1105. Flahaut J. — Ann. chim. Ser. 12, 1952, 7, p. 632.
1106. Fleischhauer J., Beckers M. — Tetrahedron Lett.,
1973, 43, p. 4273.
1107. Fleissner H. — Montan-Rdsch., 1925, 17, p. 523.
1107a. Flood E., Gropen O. — J. Organometal. Chem.,
. 1976, 110, p. 7.
1108. Flood H., Forland Т., Roald B. — J. Amer. Chem.
Soc, 1949, 71, p. 572.
1109. Florke O. W. — Naturwissenschaften, 1952, 39,
S. 478.
1110. Flynn J. H. — Thermochim. acta, 1974, 8, p. 69.
1111. Foex M. — Solar Energy, 1965, 9, p. 61.
1112. Foex M. — Rev. intern, hautes temp, et refract.,
1966, 3, p. 309.
1113. Foldvari M., Kliburszky B. — Acta geol. Acad.
sci. hung., 1958, 5, p. 187
1114. Fontijn A., Felder W. — J. Chem. Phys., 1977,
67, p. 1561.
1115. Forcrand de R. — Bull. Soc. chim. France, 1906,
35, p. 781.
1116. Forcrand de R. —Ann. chim. phys., 1908, 15,
p. 457.
1117. Forcrand de R. — C. r. Acad. sci., 1908, 146, p. 512.
1118. Forcrand de R. — С. г. Acad. sci., 1908,147, p. 165.
1119. Forcrand de R. — С. г. Acad. sci., 1911, 152, p. 27.
1120. Forcrand de R. — С. г. Acad. sci., 1923, 176, p. 873.
1121. Forland T. — J. Phys. Chem., 1955, 59, p. 152.
1122. Foster L. M. — J. Amer. Chem. Soc, 1950, 72,
p. 1902.
1123. Foster L. M., Long G., Hunter M. S. — J. Amer.
Ceram. Soc, 1956, 39, p. 1.
1124. Foster L. M., Russel A. S., Cochran C. N. —
J. Amer. Chem. Soc, 1950, 72, p. 2580.
1125. Fowler A. — Philos. Trans. Roy. Soc. London,
1909, A209, p. 447.
1126. Fowler C. A. — Phys. Rev., 1941, 59, p. 645.
1127. Fraenkel W. — Z. Elektrochem., 1913, 19, S. 362.
1128. Fraga S., Ransil B. J. — J. Chem. Phys., 1962,
36, p. 1127.
1129. Francoi S. M., Centre M. — In: Conf. Intern.
Met. Beryllium, Grenoble, 1965. P., 1966, p. 201.
1130. Frank P., Krauss L. — Z. Naturforsch., 1974,
A29, S. 742.
1131. Frank P., Krauss L. — Z. Naturforsch., 1976, A31,
S. 1193.
1132. Frank W. B. — J. Phys. Chem., 1961, 65, p. 2081.
1133. Franklin J. L., Dillard J. G., Rosenstock H. M.,
Herron J. Т., Draxe K. Ionization potential,
appearance potentials and heats formation of gaseous
positive ions. NSRDS-NBS-26. Wash.: NBS, 1969.
1134. Fraslie H. M., Winaus J. G. — Phys. Rev., 1947,
72, p. 481.
1135. Frear G. L., Johnston J. — J. Amer. Chem. Soc,
1929, 51, p. 2082.
1136. Fredrickson W. R., Hogan M. E. — Phys. Rev.,
1934, 46, p. 454.
1137. Fredrickson W. R., Hogan M. E. — Phys. Rev.,
1935, 48, p. 602.
1138. FreedmanP. A., Jones W. J. — J. Mol. Spectrosc,
1975, 54, p. 182. '
1139. Freund F., Sperling V. — Mater. Res. Bull.,
1976, 11, p. 621.
1140. Freund I., Halford R. S. — J. Chem. Phys., 1965,
43, p. 3795.
1140a. Frey R. A., Werder R. D., Gunthard Hs. H. —
J. Mol. Spectrosc, 1970, 35, p. 260.
1141. Fricke R. — Z. Elektrochem., 1929, 35, S. 631.
1142. Fricke R., Luke J. — Z. Elektrochem., 1935, 41,
S. 174.
1143. Fricke R., Severin H. ¦— Z. anorg. und allg. Chem.,
1932, 205, S. 287.
1144. Fricke R., Wullhorst B.—Z. anorg. und allg.
Chem., 1932, 205, S. 127.
1145. Friederich E., Sittig L. — Z. anorg. und allg.
Chem., 1925, 143, S. 293.
1146. Frisch M. A., Greenbaum M. A., Farber M. —
J. Phys. Chem., 1965, 69, p. 3001.
1147. Frosch С J., Thurmond C. D. — J. Phys. Chem.,
1962, 66, p. 877.
1148. Frost A. A. — Theor. chim. acta, 1970, 18, p. 156.
1149. Fu С. М., Burns R. P. — High Temp. Sci., 1976,
8, p. 353.
1149a. Fugate R. Q., Swenson C. A. — J. Appl. Phys.,
1969, 40, p. 3034.
11496. Fujimoto H., Kato S., Yamabe S., Fukui K. —
J. Chem. Phys., 1974, 60, p. 572.
1150. Funke G. W. — Z. Phys., 1933, 84, S. 610.
1150a. Funke G. M., Grundstrom B. — Z. Phys., 1936,
100, S. 293.
1151. Furukawa G. T. - J. Res. NBS, 1974, A78, p. 477.
1152. Furukawa G. Т., Douglas Т. В., McCoskey R. E.,
Ginnings D. С — J. Res. NBS, 1956, 57, p. 67.
1153. Furukawa G. Т., Douglas Т. В., Saba W. G.,
Victor A. C. — J. Res. NBS, 1965, A69, p. 423.
1154. Furukawa G. Т., Ishihara S. — U. S. NBS,
Rept N 10326, 1970.
1155. Furukawa G. Т., Reilly M. L. — U. S. NBS,
Rept N 9905, 1968.
443
Литература
1156. Furukawa G. Т., Reilly M. L. — J. Res. NBS,
1970, A74, p. 617.
1157. Fyfe W. S. — Amer. J. Sci., 1958, 256, p. 729.
1158. Fykse O. — Tidsskr. kjemi. bergv. og met., 1956,
16, p. 203.
1159. Gage D. M.. Barker E. F. — J. Chem. Phys.,
1939, 7, p. 455.
1160. Gaines A. F., Page F. M. — Trans. Faraday Soc,
1966, 62, p. 3086.
1161. Galan L. — Physica, 1965, 31, p. 1286.
1162. Galan L., Winefordner J. D. — J. Quant. Spectrosc.
and Radiat. Transfer, 1967, 7, p. 251.
1163. Galbraith H. I., Masi I. F. — In: Thermodynamic
and transport properties of gases, liquids and solids:
Symp. on Thermal Properties. N. Y.: McGraw-
Hill, 1959, p. 90.
1164. Gallo G. — Ann. chim. appl., 1935, 25, p. 628.
1165. Ganguli P. S., Gordon L., McGee H. A. — 1. Chem.
Phys., 1970, 53, p. 782.
1166. Ganguli P. S., McGee H. A. — J. Chem. Phys.,
1969, 50, p. 4658.
1167. Ganguli P. S., McGee H. A. — J. Chem. Phys.,
1970, 53, p. 782.
1167a. Gani M. S. /.. McPherson R. — Thermochim.
acta, 1973, 7, p. 251.
1168. Gant F. A. — Diss. Abstrs, 1967, B28, p. 133.
1168a. Garcia-Clavel E.. Burriel-Marti F., Rodrigues
de la Репа M. — Inform, quim. anal., 1970, 24,
p. 31.
1169. Gardet J. /., Gruilot В., Soustelle M. — Bull.
Soc. chim. France, 1970, N 10, p. 3377.
1170. Gardner T. E.. Taylor A. R. — J. Chem. and Eng.
Data, 1969, 14. p. 281.
1171. Garnett S. H. — Ph. D. Thes. Prinston (N. J.):
Prinston Univ., 1968.
1172. Garrels R. M., Thompson M. E. — Amer. J. Sci.,
1962, 260, p. 57.
1173. Garton W. R. S. — Proc. Phys. Soc. London, 1951,
A64, p. 509.
1174. Garton W. R. S., Codling K. — Proc. Phys. Soc.
London, 1960, 75, p. 87.
1175. Garton W. R. S.. Codling K. — Proc. Phys. Soc.
London, 1961, 78, p. 600.
1176. Garton W. R. S., Codling K. — Proc. Phys. Soc.
London, 1965, 86, p. 1067.
1177. Garton W. R. S., Tomkins F. S. — Astrophys.
J., 1969, 158, p. 1219.
1177a. Gates D. S., Thodos Z). — Amer. Inst. Chem.
Eng. J., 1960. 6, p. 50.
1178. Gaultier M., Pannetier G. — Rev. chim. miner.,
1972, 9, p. 271.
1179. Gautier H. — С. г. Acad. sci., 1899, 128, p. 939.
1180. Gaydon A. G. Dissociation energies and spectra
of diatomic molecules. 2 ed. L.: Chapman and
Hall, 1953.
1181. Gaydon A. G. — Proc. Roy. Soc. London, 1955,
A231, p. 437.
1182. Gaydon A. G. — Mem. Soc. roy. sci. Liege, 1957,
18, p. 507.
1183. Gaydon A. G. Dissociation energies and spectra
of diatomic molecules. 3 ed. L.: Chapman and
Hall, 1968.
1184. Gayler M. L. V. — Metallwirtschaft, 1930, 9, S. 677.
1185. Gayles J. N., Self /. — J. Chem. Phys., 1964,
40, p. 3530.
1186. Geach G. A., Harper M. E. — Metallurgia, 1953,
47, p. 269.
1187. Geiseler G., Buchner W. — Z. anorg. und allg.
Chem., 1966, 343, S. 286.
1188. Geller R. Г., Bunting E. N. — J. Res. NBS, 1943,.
31, p. 255.
1189. Geller R. Т., Jaworsky R. I. — J. Res. NBS, 1945,
34, p. 395.
1190. Geller S. — J. Solid State Chem., 1977, 20, p. 209.
1191. Gelus M., Kutzelnigg W. — Theor. chim. acta,.
1973, 28, p. 103.
1192. George H. — Z. wiss. Photogr., 1913, 12, S. 237.
1193. Gerdins, H., Smit E. — Z. phys. Chem. Leipzig,
1941, B50, S. 171.
1194. Gerding #., Smit E. — Z. phys. Chem. Leipzig,.
1942, B51, S. 217.
1195. Chosh ?>., Kay D. A. R. — J. Electrochem. Soc,
1977, 124, p. 1836.
1196. Giauque W. F., Archibald R. C. — J. Amer.
Chem. Soc, 1937, 59, p. 561.
1197. Giauque W. F., Meads P. F. — J. Amer. Chem.
Soc, 1941, 63, p. 1897.
1198. Gibson G. E. — Dissertation. Breslau, 1911.
1199. Gilbert В., Mamantov G., Begun G. M. — Inorg.
and Nucl. Chem. Lett., 1974, 10, p. 1123.
1200. Gilbert L. F., Levi M. — J. Chem. Soc, 1929V
p. 527.
1201. Gilbreath W. P. — Rept NASA-TN-D-2723. Ac-
cossion N 65—18506. Washington, 1965.
1202. Gilde W. — Metallurgie und G-iessereitechnik,
1953, 3, S. 324.
1203. Gimarc В. М. — 3. Amer. Chem. Soc, 1973, 95,
p. 1417.
1204. Gingerich K. A. — J. Chem. Soc. Chem. Communs,
1970, N 10, p. 580.
1205. Gingerich K. A. — J. Cryst. Growth, 1971, 9, p. 31.
1205a. Gingerich K. A. — J. Chem. Phys., 1972, 56,
p. 4239.
1206. Ginn S. G. W., Brown G. W., Kenney J. K.,
Overend J. — J. Mol. Spectrosc, 1968, 28, p. 509.
1207. Ginn S. G. W., Johansen D., Overend J. — J. Mol.
Spectrosc, 1970, 36, p. 448.
1208. Ginn S. G. W., Kenney J. K., Overend J. — J.
Chem. Phys., 1968, 48, p. 1571.
1209. Ginn S. G. W., Reichman S., Overend J. ~ Spectro-
chim. acta, 1970, A26, p. 291.
1210. Ginnings D. C, Corruccini B. J. — J. Res. NBS,.
1947, 38, p. 593.
1211. Ginnings D. C, Douglas Т. В., Ball A. F. — .T.
Amer. Chem. Soc, 1951, 73, p. 1236.
1212. Ginnings D. C., Furukawa G. T. — J. Amer.
Chem. Soc, 1953, 75, p. 522.
1213. Ginsberg A. S. — Z. anorg. und allg. Chem., 1909,.
61, S. 122.
1214. Ginsberg #.. Sparwald V. — Aluminium, 1965,
41, p. 219.
1215. Ginter D. S. — Diss. Abstrs, 1965, 26, p. 1934.
1216. Ginter D. S., Ginter M. L., Innes К. К. —
Astrophys. J., 1964, 139, p. 365.
1217. Ginter M. L. — J. Mol. Spectrosc, 1963, 11, p. 301.
1218. Ginter M. L. — J. Mol. Spectrosc, 1966, 20,
p. 240.
1219. Ginter M. L., Battino R. — J. Chem. Phys., 1965,
42, p. 3222.
1220. Ginter M. L., Innes К. К. — J.Mol. Spectrosc,
1961, 7, p. 64.
1220a. Gissane M., Barrow R. F. — Proc. Phys. Soc.
London, 1963, 82, p. 1065.
12206. Givan A., Loewenschuss A. — J. Mol. Struct.,
1979, 55, p. 163.
1221. Givele M. — С. г. Acad. sci., 1968, C267, p. 881.
1222. Glaser F. M., Moskowitz D., Post В.. — J. AppL
Phys., 1953, 24, p. 731.
444
Литература
1223. Gmelin Е. — С. г. Acad. sci., 1964, 259, p. 3459.
1224. Gmelin E. — С. г. Acad. sci., 1966, C262, p. 1452.
1225. Gmelin E. — Cryogenics, 1967, 7, p. 225.
1226. Gmelin E. — Z. Naturforsch., 1969, A24, S. 1794.
1227. Gmelins Handbuch der anorganischen Chemie.
В.: Verl. Chemie, 1957, Bd. 28, S. 303.
1228. Gole J. L. — J. Chem. Phys., 1973, 58, p. 869.
1229. Gole J. L., Zare K. N. — J. Chem. Phys., 1972,
57, p. 5331.
1230. Good W. D.,Mansson M. — J. Phys. Chem., 1966,
70, p. 97.
1230a. Goodlett V. W., Innes К. K. — Nature, 1959,
183, p. 243.
1231. Goodman R. M., Westrum E. F. — J. Chem. and
Eng. Data, 1966, 11, p. 294.
1232. GoorvitchD., Valero F. P. — Astrophys. J., 1972,
171, p. 643. •
1233. Goranson R. W. — Geol. Soc. Amer. Spec. Pap.,
1942, N 36, p. 223.
1234. Gordon M. S., England W. — Chem. Phys. Lett.,
1972, 15, p. 59.
1235. Gordon J. S. — ARS J., 1959, 29, p. 455.
1235a. Gottscho R. A. — J. Chem. Phys., 1979, 70, p. 3554.
12356. Gottscho R. A., Koffend J. R., Field R. W. —
J. Mol. Spectrosc, 1980,82, p. 310.
1235b. Gottscho R. A., Koffend J. R., Field R. W.,
Lombardi J. R. — J. Chem. Phys., 1978, 68,
p. 4110.
1235г. Gottscho R. A., Weiss P. S., Field R. W., Pru-
ett J. G. — J. Mol. Spectrosc, 1980, 82, p. 283.
1236. Goubeau J., Rues W. — Z. anorg. und allg. Chem.,
1952, 268, S. 221.
1237. Goubeau /., Hummel D. — Z. phys. Chem. Neue
Folge, 1959, 20, S. 15.
1238. Goubeau /., Keller H. — Z. anorg. und allg. Chem.,
1953, 272, S. 303.
1239. Goubeau /., RichterD. E., Becker H. J. — Z. anorg.
und alig. Chem., 1955, 278, S. 12.
1239a. Goubeau /., Richer E.— Z. anorg. und allg.
Chem.. 1961, 310, S. 123.
1240. Goutier D., Burnelle L. A. — Chem. Phys. Lett.,
1973, 18, p. 460.
1241. GrabeB., Hulthen E. — Z. Phys., 1939, 114, S. 470.
1241a. Graf L. — Metallwirtschaft, 1933, 12, S. 649.
1242. Graff G., Paul W., Schlier С — Z. Phys., 1958,
153, S. 38.
1243. Graham W. R. M., Weltner W., jr. — J. Chem.
Phys., 1976, 65, p. 1516.
1244. Grahmann W. — Z. anorg. und allg. Chem., 1913,
81, S. 257.
1244a. Grattidge W., John II. — J. Appl. Phys., 1952,
23, p. 1145.
1245. Green J. W., Blue G. D., Ehlert Т. С,
Margrave J. L. — J. Chem. Phys., 1964, 41, p. 2245.
1246. Green J. W., Poland D. E., Margrave J. L. —
J. Chem. Phys., 1960, 33, p. 35.
1247. Greenbank J. C, Argent B. B. — Trans. Faraday
Soc, 1965, 61, p. 655.
1247a. Greenbaum M. A., Arin M. L., Farber M. —
J. Phys. Chem., 1963, 67, p. 1191.
1248. Greenbaum M. A., Arin M. L., Wong M.,
Farber M. — J. Phys. Chem., 1964, 68, p. 791.
1248a. Greenbaum M. A., Blauer J. A., Arshadi M. R.,
Farber M. — Trans. Faraday Soc, 1964, 60,
p. 1592.
1249. Greenbaum M. A., Farber M. — In: Perform.
High-Temp. Syst. N. Y. etc., 1968, vol. 1, p. 1.
1250. Greenbaum M. A., Foster J. N., Arin M. L.,
Farber M. — l. Phys. Chem., 1963, 67, p. 36.
1251. Greenbaum M. А., Ко #., Wong M., Farber M. —
J. Phys. Chem. 1964, 68, p. 965.
1252. Greenbaum M.A., Weiher J., Farber M. — J. Phys.
Chem., 1965, 69, p. 4035.
1253. Greenbaum M. A., Yates R. E., Arin M. L.,
Arshadi M., Weiher J., Farber M. — J. Phys.
Chem., 1963, 67, p. 703.
1254. Greenbaum M. A., Yates R. E., Farber M. —
J. Phys. Chem., 1963, 67, p. 1802.
1255. Greenberg S. A., Copeland L. E. ¦— J. Phys. Chem.,
1960, 64, p. 1057.
1256. Greene F. Т., Gilles P. W. — J. Amer. Chem. Soc,
1964, 86, p. 3964.
1257. Greene F. Т., Margrave J. L. — J. Amer. Chem.
Soc, 1959, 81, p. 5555.
1258. Greene F. Т., Margrave J. L. — J. Phys. Chem.,
1966, 70, p. 2112.
1258a. Greenwood N. N., Wade K. — J. Chem. Soc,
1956, p. 1527.
1259. Gregg A. H., Hampson G. C, Jenkins G. I.,
Jones P. L. F., Sutton L. E. —Trans. Faraday
Soc, 1937, 33, p. 852.
1260. Gregory N. W. — J. Phys. Chem., 1977, 81, p. 1854.
1261. Grezes G., Basset M. — С. г. Acad sci., 1965,
260, p. 869.
1262. Greyson /., Snell H. — J. Phys. Chem., 1969,
73, p. 3208.
1263. Grieveson P., Turkdogan E. T. — Trans. AIME,
1962, 224, p. 1086.
1264. Griff ing K. M., Simons J. — J. Chem. Phys.,
1976, 64, p. 3610.
1265. Griffiths E. H., Griffiths E. — Proc. Roy. Soc,
1914, A90, p. 557.
1266. Grimaldi F., Lecourt A., Lefebvre-Brion H., Mo-
ser С. М. — J. Mol. Spectrosc, 1966, 20, p. 341.
1267. Grimm A., Warton L., Porter R. F. — Inorg.
Chem., 1968, 7, p. 1309.
1268. Grjotheim K., Herstad O., Stahl-J'ohannessen K. —
Z. anorg. und allg. Chem., 1964, 327, S. 267.
1269. Grjotheim K., Holm J. L., Malmo J. — Acta
chem. scand., 1970, 24, p. 77.
1270. Grjotheim K., Holm J. L., R0tnes M. — Acta chem.
scand., 1972, 26, p. 3802.
1271. Gronow H. E., Schwiete H. E. — Z. anorg. und
allg. Chem., 1933, 216, S. 185.
1271a. Gr0nvold F. — J. Therm. Anal., 1978, 13, p. 419.
1272. Gropen O., Johansen R. — J. Mol. Struct., 1975,
25, p. 161.
1273. Gropen O., Nilssen E. W. — J. Organometal. Chem.,
1976, 111, p. 257.
1274. Gross P. — In: III Intern. Conf. on Chem. Thermo-
dyn. Austria, 1973, vol. 1, p. 107.
1275. Gross P., Campbell С S., Kent P. J. C,
Levi D. L. — Discuss. Faraday Soc, 1948, 4,
p. 206.
1275a. Gross P., Hayman С — Fulmer Res. Inst.
Rept, 1970 (Chem. Absts, 1970, 73, N 92306).
1276. Gross P., Hayman С — U. S. Clearinghouse Fed.
sci. Techn. Inform., 1967, AD 661565. Avail
CFSTI.
1277. Gross P., Hayman С — Trans. Faraday Soc,
1970, 66, p. 30.
1278. Gross P., Hayman C, Ringham H. T. — Fulmer
Res. Inst. Ltd Sci. Rept N 7. Contract AF 61
@52)-447, 1971.
1279. Gross P., Hayman C, Green P.D., Ringham J. T. —
Trans. Faraday, Soc, 1966, 62, p. 2719.
1280. Gross P., Hayman C, Joel H. A. — Trans.
Faraday Soc, 1968, 64, p. 317.
445
Литература
1281. Gross P., Hayman С, Levi D. L. — Trans.
Faraday Soc, 1954, 50, p. 477.
1282. Gross P., Hayman C, Levi D. L. — Trans.
Faraday Soc, 1955, 51, p. 626.
1283. Gross P., Hayman C, Levi D. L. — Metall. Soc.
Conf., 1961, 8, p. 903.
1284. Gross P.. Hayman C, Levi D. L., Lewin R. H. —
Fulmer Res. Inst. Ltd Sci. Rept N 154/12, 1963,
Aug.
1285. Gross P., Hayman C, Stuart M. С — Proc.
Brit. Ceram. Soc, 1967, N 8, p. 39.
1286. Gross P., Hayman C, Stuart M. С — Fulmer
Res. Inst. Ltd Sci. Rept 163/SR. 1971, July.
1286a. Gross P., Lewin R. H. — Fulmer Res. Inst.
Ltd Sci. Rept, 1965.
12866. Grosse A. V. — J. Inorg. and Nucl. Ghem., 1961,
22, p. 23.
1286b. Grosse A. V. — J. Inorg. and Nucl. Chem., 1962,
24, p. 147.
1286г. Grosse A. V., McGonical P. J. — J. Phys. Chem.,
1964, 68, p. 414.
1287. Grosweiner L., Seifert R. L. — J. Amer. Ghem.
Soc, 1952, 74, p. 2701.
1288. Grotewold I., Lissi E. A., Villa A. E. — J. Chem.
Soc. A., 1966, p. 1038.
1288a. Grow D. Т., Pitzer R. M. — J. Chem. Phys.,
1977, 67, p. 4019.
1289. Grube G., Bornhan R.—Z. Elektrochem., 1934,
40, S. 141.
1290. Grube G., Henne H. — Z. Elektrochem., 1930,
36, S. 129.
1291. Grube G., Rudel W. — Z. anorg. und allg. Chem.,
1924, 133, S. 375.
1292. Grundstrom B. — Z. Phys., 1931, 69, S. 235.
1293. Grundstrom B. — Z. Phys., 1932, 75, S. 302.
1294. Grundstrom B. — Z. Phys., 1935, 95, S. 574.
1295. Grundstrom B. — Nature, 1936, 137, p. 108.
1296. Grundstrom B. — Z. Phys., 1936, 99, S. 595.
1297. Grundstrom B. — Z. Phys., 1939, 113, S. 721.
1298. Grundstrom B. — Z. Phys., 1940, 115, S. 120.
1299. Grundstrom В., Volberg P. — Z. Phys., 1938,
108, S. 326.
1300. Gruver R. M. — J. Amer. Ceram. Soc, 1951,
34, p. 353.
1301. Guernsey M. L. — Phys. Rev., 1934, 46, p. 114.
1302. Guertler W., Pirani M. — Z. Metallk., 1919, 11,
S. 1.
1303. Guest M. F., Hillier I. H. — Z. Chem. Soc.
Faraday Trans. II, 1974, 70, p. 398.
1304. Guggi D. /., Neubert A., Zmbov K. F. — In:
4eme Conf. Intern, thermodyn. chim. Montpel-
lier, 1975, vol. 3, p. 124.
1304a. Guido M., Gigli G. — J. Chem. Phys., 1977, 66,
p. 3920.
1305. Guillemaut A., Lecocq A. — С. г. Acad. sci., 1963,
257, p. 1260.
1306. Gulbransen E. A., Andrew K. F. — J. Electro-
chem. Soc, 1950, 97, p. 383.
1307. Gunn S. R. — J. Phys. Chem., 1965, 69, p. 1010.
1308. Gunn S. Д., Green L. G. — J. Phys. Chem., 1960,
64, p. 61.
1309. Gunn S. R., Green L. G. — J. Phys. Chem., 1961,
65, p. 178.
1310. Gunn S. R., Green L. G. — J. Phys. Chem., 1961,
65, p. 779.
1311. Gunn S. R., Green L. G., Egidy A. von. — J.
Phys. Chem., 1959, 63, p. 1787.
1312. Gunn S. R., Sanborn R. H. — J. Chem. Phys.,
1960, 33, p. 955.
1313. Gunnvald P., Minnhagen L. — Ark. fys., 1962,
22, p. 327.
1314. Gunther K. G. — Glastechn. Ber., 1958, 31, S. 9.
1315. Gunther P. — Ann. Phys., 1916, 51, S. 828.
1316. Guntsch A. — Z. Phys., 1934, 87, S. 312.
1317. Guntsch A. — Z. Phys., 1935, 93, S. 534.
1318. Guntsch A. — Z. Phys., 1937, 104, S. 584.
1319. Guntsch A. — Z. Phys., 1937, 107, S. 420.
1320. Guntsch A. — Z. Phys., 1938, 110, S. 549.
1321. Guntsch A. — Dissertation. Stockholm, 1939.
1322. Guntz A.— Ann. chim. phys., 1905, 4, p. 5.
1323. Guntz A. — C. r. Acad. sci., 1905, 140, p. 863.
1324. Guntz A., Bassett N. — J. chim. phys. et phys. —
Chim. biol., 1906, 4, p. 1.
1325. Guntz A., Benoit F. — Ann. chim., 1923, 20, p. 5.
1326. Guntz A., Benoit F. — C. r. Acad. sci., 1923, 176,
p. 219.
1327. Guntz A., Roedere A. — Bull. Soc. chim. France,
1906, 35, p. 512.
1328. Guntz M. — Ann. chim. phys., 1884, 3, p. 5.
1329. Guntz M. — C. r. Acad. sci., 1884, 98, p. 819.
1330. Guntz M. — С. г. Acad. sci., 1902, 134, p. 838.
1330a. Gupta S. K., Porter R. F. — J. Phys. Chem.,
1963, 67, p. 1286.
1331. Gupta Т. С, Bhattacharya A. K.—Z. anal.
Chem., 1958, 161, S. 321.
1332. Gurr G. E., Mantgomery P. W., Knutson C. D.,
Gorres B. T. — Acta crystallogr., 1970, B26, p. 906.
1333. Gurvich L. V., Ryabdva V. G., Khitrov A. N. —
Faraday Symp. Chem. Soc, 1973, N 8, p. 83.
1334. Gurvich L., Ryabova V., Semenov G., Ratkov-
ski I. — In: Proc. First Intern. Conf. Calori-
metry and Thermodynamics. Warsaw, 1969, F-2.
1335. Gutt W., Chatterjee A. K., Zhmoidin G. T. —
J. Mater. Sci. 1970, 5, p. 960.
1336. Haar L., Fridman A. S., Beckett Ch. M. Ideal gas
thermodynamic functions and isotope, exchange-
functions for the diatomic hydrides, deuterides,
and tritides. Wash.: NBS, 1961.
1337. Haber F., Fleischman F. — Z. anorg. und allg..
Chem., 1906, 51, S. 336.
1338. Hachmeister K. — Z. anorg. und allg. Chem.»
1920, 109, S. 145.
1339. Hackspill L., Caillat R., Cheutin A. — С. г. Acad.
sci., 1944, 218, p. 838.
1340. Hackspill L., Wolf G. — С. г. Acad. sci., 1937,
204, p. 1820.
1341. Haehnel O. — J. prakt. Chem., 1924, 108, S. 61.
1342. Hall D., Gurr G. E., Jeffrey G. A.—Z. anorg.
und allg. Chem., 1969, 369, S. 108.
1343. Halla F., Tassel R. van. — Radex Rdsch., 1966„
S. 356.
1344. Halla F., Tassel R. van. — Radex Rdsch., 1968,
S. 27.
1345. Halstead P. E., Moore A. E. — J. Chem. Soc,
1957, p. 3873.
1346. Hammer R. R., Pask J. A. — Z. Amer. Ceram.
Soc, 1964, 47, p. 264.
1347. Hannebohn O., Klemm W. — Z. anorg. und allg,
Chem., 1936, 229, S. 331.
1348. Hanst P. L., Farly V. H., Klemperer W. — J. Chem.
Phys., 1965, 42, p. 1097.
1349. Haraguchi H., Fuwa K. — Chem. Lett., 1972,
N 10, p. 913.
1350. Haraguchi H., Fuwa K. — Bull. Chem. Soc,
Jap., 1975, 48, p. 3056.
1351. Haraguchi H., Fuwa K. — Spectrochim. acta,
1975, B30, p. 535.
1352. Hargittai J., Hargittai M. — J. Chem. Phys.,
1974, 60, p. 2563.
446
Литература
1353. Harker J. W., Tuttle O. F. — Amer. J. Sci.,
1955, 253, p. 209.
1354. Harrington R. E. Dissertation. Berkley, Univ.
Calif., 1942.
1355. Harrison J. F., Allen L. C. — J. Mol. Spectrosc,
1969, 29, p. 432.
1356. Harrison W. R., Perman E. P. — Trans. Faraday
Soc, 1927, 23, p. 1.
1357. Harshbarger W., Lee G., Porter R. F., Bauer S. H.~
Inorg. Chem., 1969, 8, p. 1683.
1358. Hart P. E., Searcy A. W. — J. Phys. Chem., 1966,
70, p. 2763.
1359. Harteck P. — Z. phys. Chem. Leipzig, 1928, 134,
S. 1.
1360. Hartmann H., Schneider R. — Z. anorg. und allg.
Chem., 1929, 180, S. 275.
1361. Harvey A. — Proc. Roy. Soc, 1931, A133, p. 336.
1362. Haschke J. M., Clark M. R. — High Temp. Sci.,
1975, 7, p. 152.
1363. Hastie J. W., Hauge R. H., Margrave J. L. —
High Temp. Sci., 1971, 3, p. 56.
1364. Hastie J. W., Hauge R. H., Margrave J. L. —
High Temp. Sci., 1971, 3, p. 257.
1365. Hastie J. W., Hauge R. H., Margrave J. L. —
J. Fluor. Chem., 1973, 3, p. 285.
1366. Hastie J. W., Margrave J. L. — Dep. of Chem.
Rice Univ. Houston (Tex.), 1968, Rept 77001, p. 1.
1367. Hastie J. M., Margrave J. L. — J. Phys. Chem.,
1969, 73, p. 1105.
1367a. Hutchison С A., Malm J. G. — J. Amer. Chem.
Soc, 1949, 71, p. 1338.
1368. Hatton W. E., Hildenbrand D. L., Sinke G. C,
Stull D. R. — J. Amer. Chem. Soc, 1959, 81,
p. 5028.
1369. Hauge R. H., Margrave J. L., Kana'an A. S. —
J. Chem. Soc. Faraday Trans. II, 1975, 71, p. 1082.
1370. Haughton J. L., Payne R. J. M. — J. Inst. Metals,
1934, 54, p. 278.
1371. Hawes J., Mackenzie A. E., Raw С J. G. — J. S.
Afr. Chem. Inst., 1955, 8, p. 21.
1372. Haworth D., Hohnstedt L. F. — Chem. and Ind.,
1960, N 20, p. 559.
1373. Hayek E. — Z. anorg. und allg. Chem., 1935, 47,
S 225
1374. Hayhurst A. N., Kittelson D. В.— Combust, and
Flame, 1972, 19, p. 306.
1375. Hayhurst A. N., Kittelson D. B. —Proc. Roy.
Soc. London, 1974, A338, p. 155.
1376. Hecht J. — 1. Chem. Phys., 1976, 65, p. 5026.
1377. Hedberg K., Schomaker V. — J. Amer. Chem.
Soc, 1951, 73, p. 1482.
1378. Hedfeld K. - Z. Phys., 1931, 68, S. 610.
1379. Hedvall A. J. — Z. anorg. und allg. Chem., 1916,
98, S. 47.
1380. Hedvall J. A., Garping E., Lindekrantz N.,
Nelson L. — Z. anorg. und allg. Chem., 1931, 197,
S. 419.
1381. Hedvall J. A., Heuberger J. — Z. anorg. und allg.
Chem., 1924, 140, S. 250.
1382. Heimgartner R. — Schweiz. Arch, angew. Wiss.
und Techn., 1952, 18, S. 241.
1383. Heise M., Wieland K. — Chimia, 1950, 4, p. 262.
1384. Heise M., Wieland K. — Helv. chim. acta, 1951,
34, p. 2182.
1384a.Hellwege К. Н. — Z. Phys., 1936, 100, S. 644.
1385. Helms D. A., Winnewisser M., Winnewisser G. —
in press.
1385a. Hemingway B. S., Robie R. A. — J. Res. U. S.
Geol. Surv., 1975, 5, p. 413.
13856. Hemingway B. S., Robie R. A., Fisher J. R.,
Wilson W. H. — J. Res. U. S. Geol. Surv., 1977,
5, p. 797.
1386. Henning O., Stroebel U. — Wiss. Z. Hochsch.
Archit. und Bauw. Weimar, 1969, 16, S. 173.
1387. Henrion G., Marquardt D. — Z. Chem., 1977,
17, H. 1, S. 28.
1388. Hensen K. — Theor. chim acta, 1969, 14, S. 273.
1389. Herm R., Lin-Shen-Maw M. C. — J. Phys. Chem
1973, 77, p. 293.
1390. Herrick С. С. — Trans. Met. Soc. AIME, 1964.
230, p. 1439.
1391. Herrmann G. — Z. phys. Chem. Leipzig, 1937,
B35, S. 298.
1392. Herzberg G. — Molecular spectra and moleeular
structure I. Spectra of diatomic molecules. 2ed.
Toronto etc., 1950.
1393. Herzberg G. — Mem. Soc. roy. Sci. Liege, 1957,
18, p. 397.
1394. Herzberg G., Hushley W. — Canad. J. Res., 1941,
A19, p. 127.
1395. Herzberg G., Johns J. W. C. — Proc. Roy. Soc.
London, 1967, A298, p. 142.
1396. Herzberg G., Mundie L. C. — J. Chem. Phys.v
1940, 8, p. 263.
1397. Herzberg L. — Z. Phys., 1933, 24, S. 571.
1398. Hess G. I. — Ann. Phys., 1840, 50, p. 384.
1399. Hesser J. E., Dressier K. — J. Chem. Phys.,
1967, 47, p. 3443.
1400. Heumann Т., Predel B. — Z. Metallk., 1960, 51,
S. 509.
1401. Heus R. /., Egan J. J. — Z. phys. Chem., Leipzig,
1966, 49, S. 38.
1402. Heyrowsky /., Berezicky S. — Collect. Czech.
Chem. Communs, 1929, 1, S. 19.
1402a. Hibben J. H. — Amer. J. Sci., 1938, A35, p. 113.
1403. Hicks J. F. G. — J. Amer. Chem. Soc, 1938,
60, p. 1000.
1404. Higgins T. H. S., Leisegang E. C, Raw С J. G.,.
Rossow A. J. — J. Chem. Phys., 1955, 23, p. 1544.
1405. Higuchi J. — Bull. Chem. Soc. Jap., 1953, 26, p. 1.
1406. Hildenbrand D. L. — J. Chem. Phys., 1968, 48,
p. 3657.
1407. Hildenbrand D. L. — Intern. J. Mass Spectrom.
and Ion Phys., 1970, 4, p. 75.
1408. Hildenbrand D. L. — J. Elecktrochem. Soc. 1979
126, p. 1396.
1409. Hildenbrand D. L. — J. Chem. Phys., 1970, 52,
p. 5751.
1410. Htldenbrand D. L. — Chem. Phys. Lett., 1973,
20, p. 127.
1411. Hildenbrand D. L. — J. Chem. Phys. 1977, 66t
p. 3526.
1412. Hildenbrand D. L. Preprint. Stanford Res. Inst.,
1976.
1413. Hildenbrand D. L. Preprint. Stanford Res. Inst.,
1977.
1414. Hildenbrand D. L., Hall W. F. — J. Phys. Chem.,
1963, 67, p. 888.
1415. Hildenbrand D. L., Hall W. F. — J. Phys. Ghem ,
1964, 68, p. 989.
1416. Hildenbrand D. L., Hall W. F., Potter N. D. —
J. Chem. Phys., 1963, 39, p. 296.
1417. Hildenbrand D. L., Hall W. F., Ju F.,
Potter N. D. — J. Chem. Phys., 1964, 40, p. 2882.
1418. Hildenbrand D. L., Knight D. T. — J. Chem.
Phys., 1969, 51, p. 1260.
1419. Hildenbrand D. L., Murad E. — J. Chem. Phys.,
1966, 44, p. 1524.
447
Литература
1420. Hildenbrand D. L., Murad E. — J. Chem. Phys.,
1965, 43, p. 1400.
1421. Hildenbrand D. L., Murad E. — J. Chem. Phys.,
1969, 51, p. 807.
1422. Hildenbrand D. L., Murad E., Potter N. ?>.,
Theard L. P., Hall W. F. — Aeronutronic Rept N
U-3183. Ford Motor Co., 1965, June 30.
1423. Hildenbrand D. L., Potter N. D. — J. Phys.
Chem., 1963, 67, p. 2231.
1424. Hildenbrand D. L., Theard L. P. — ASTIA
Unclassified Rept 258410 Aeronutronic Rept U-1274, 1961.
1425. Hildenbrand D. L., Theard L. P. — J. Chem.
Phys., 1965, 42, p. 3230.
1426. Hildenbrand D. L., Theard L. P. — J. Chem.
Phys., 1969, 50, p. 5350.
1427. Hildenbrand D. L.. Theard С. Р., Ju F.
Aeronutronic Rept U-2231. Ford Motor Co., 1963, July, 31.
1428. Hildenbrand D. L., Theard L. P., Ju F. —
Aeronutronic Rept U-2055. Ford Motor Co., 1963,
Mar., 15.
1429. Hildenbrand D. L., Theard L. P., Saul A. M. —
J. Chem. Phys., 1963, 39, p. 1973.
1430. Hill A. E. — J. Amer. Chem. Soc, 1937, 59,
p. 2242.
1431. Hill A. E., Simmons J. P. — Z. phys. Chem.
Leipzig, 1909, 67, S. 594.
1432. Hill K. J., Winter E. R. — J. Phys. Chem., 1956,
60, p. 1361.
1433. Hill R. W., Smith P. L. — Phil. Mag., 1953, 44,
p. 636.
1434. Hills A. W. D. — Inst. Min. Metall. Trans., 1967,
C76, p. 241.
1435. Hilpert K.7 Gerads H. — High Temp. Sci., 1975,
7, p. 11.
1435a. Hilpert K., Naomidis A.. Wolff G. — High Temp.
Sci., 1975, 7, p. 1.
1436. Hilsenrath /., Messina С G., Evans W. H. Tables
of ideal gas thermodynamic functions for 73 atoms
and their first and second ions to 10000° K: NBS
AD 606163. Wash., 1964.
1437. Hinchcliffe A. J., Ogden J. S. — J. Chem. Soc.
Chem. Communs, 1969, 18, p. 1053.
1438. Hinchcliffe A. J., Ogden J. S. — J. Phys. Chem.,
1971, 75, p. 3908.
1439. Hinchcliffe A. J., Ogden J. S. — J. Phys. Chem.,
1973, 77, p. 2537.
1439a. Hinchcliffe A. — J. Mol. Struct,, 1980,64, p. 289.
1440. Hinnov E,, Ohlendorf W. — J. Chem. Phys., 1969,
50, p. 3005.
1441. Hinz W., Kunth P. O. — Amer. Miner., 1960,
45, p. 1198.
1442. Hirschkind W. — Z. anorg. und allg. Chem., 1910,
67, S. 113.
1443. Hirst R. G., King A. J., Kanda F. A. — J. Phys.
Chem., 1956, 60, p. 302.
1444. Hisatsune J.C., Suarez N. H. — Inorg. Chem.,
1964, 3, p. 168.
1444a. Ho P., Burns R. P. — High Temp. Sci., 1980,
12, p. 31.
1445. Hoard J. L., Blair V. — J. Amer. Chem. Soc,
1935, 57, p. 1985.
1446. Hoard J. L., Newkirk A. E. — J. Amer. Chem.
Soc, 1960, 82, p. 70.
1447. Hoch M., Johnston H. L. — J. Phys. Chem., 1961,
65, p. 855.
1448. Hoch M., White D. The vaporization of boron
nitride and aluminium nitride: Techn. Res. Rept
MCC-1023-TR-214. Ohio State Univ., 1956, Oct.
1449. Hoch M., White D. The vaporization of boron
nitride and aluminium nitride: ASTIA Unclassified
Rept 142616. 1956, Oct., 29.
1450. Hochgeschwender K., Ingraham T. R. — Canad.
Met. Quart., 1967, 6, p. 293.
1451. Hoeft J. — Z. Phys., 1961, 163, S. 262.
1452. Hoeft I., Lovas F. /., Tiemann E., Torring T. —
Z. Naturforsch., 1970, A25, S. 1029.
1453. Hoeft /., Lovas F. /., Tiemann E., Torring T. —
Z. Naturforsch., 1970, A25, S. 1750.
1454. Hoenig C, Searcy A. W. — J. Amer. Ceram. Soc,
1967, 50, p. 460.
1455. Hoeven B. J. C, Keesom P. H. — Phys. Rev.,
1964, 135, p. 631.
1455a. Hoffmann R. — J. Chem. Phys., 1964, 40,
p. 2474.
1456. H0jendahl K. — Forksn. Nord. Kemikemmde.,
1939, 5, p. 202.
1457. H0jendahl K. — Kgl. danske vid. selskab. Mat.-
fys. medd., 1946, 24, p. 1. ,
1458. Holcombe C. E., Smith D. D., Lore J. I., Duerk-
sen W. K., Carpenter D. A. — High Temp. Sci.,
1973, 5, p. 349.
1459. Holder R. В., Speiser R., JohnstonH. L. — J. Amer.
Chem. Soc, 1948, 70, p. 3897.
1459a. Holgersson S. — Z. anorg. und allg. Chem., 1923,
126, S. 179.
1460. Holleman A. F. — Z. phys. Chem. Leipzig, 1893,
12 S 125
1461. Holley С E., Huber E. J. — J. Amer. Chem.
Soc, 1951, 73, p. 5577.
1462. Holley С E., Huber E. /., Meierkord E. H. —
J. Amer. Chem. Soc, 1952, 74, p. 1084.
1463. Holm J. L. — Acta chem. scand., 1968, 22, p. 1004.
1464. Holm J. L., Gr0nvold F. — Acta chem. scand.,
1973, 27, p. 370.
1465. Holmstrom J.-E., Johansson L. — Ark. fys., 1969,
40, p. 133.
1466. Hoist W. — Nature, 1933, 132, p. 207.
1467. Hoist W. — Z. Phys., 1933, 86, S. 338.
1468. Hoist W. — Z. Phys., 1934, 89, S. 40.
1469. Hoist W. — Z. Phys., 1934, 89, S. 47.
1470. Hoist W. — Z. Phys., 1934, 90, S. 728.
1471. Hoist W. — Z. Phys., 1934, 90, S. 735.
1472. Hoist W. — Z. Phys., 1934, 93, S. 55.
1473. Hoist W. — Thesis Stockholm, 1935.
1474. Hoist W., Hulthen E. — Z. Phys., 1934, 90, S. 712.
1475. Hong K. C, Kleppa O. J. — High Temp. Sci.,
1976, 8, p. 299.
1475a. Hong K. C, Kleppa O. J. — J. Chem. Thermo-
dyn., 1978, 10, p. 797.
1476. Hood G. C, Woyski M. M. — J. Amer. Chem.
Soc, 1951, 73, p. 2738.
1477. Hopkins D. С — Diss. abstrs, 1963, 23, N 4399.
1478. Hopkins H. P., Wulff C. A. — J. Phys. Chem.,
1965, 69, p. 6.
1478a. Horlings H., Scott D. S., Wynnyckyj J. R. —
Can. J. Chem., 1976, 54, p. 3872.
1479. Horn G. — Radex Rdsch., 1969, p. 439.
1480. Home R., Colin R. — Bull. Soc. chim. belg.,
1972, 81, p. 93.
1481. Horng-yih Chen, Gilles P. W. — J. Phys. Chem.,
1972, 76, p. 2035.
1481a. Hornig D. F., Plumb R. C. — J. Chem. Phys.,
1957, 26, p. 637.
1482. Houtgraaf H., Roos A. de — Recueil trav. chim.,
1953, 72, p. 963.
1482a. Hovdan H., Huglen R., Oeye H. A. — In: High
Temp. Mater. Phenom.: Proc. Nord. High Temp.
448
Литература
Symp., 4th, Helsinki Univ. Technol., Finland,
1975, vol. 1, p. 105.
1483. Howell H. G. — Proc. Roy. Soc. London, 1935,
A148, p. 696.
1484. Howell II. G. — Proc. Roy. Soc. London, 1937,
A160, p. 242.
1485. Howell H. G. — Phys. Rev., 1945, 57, p. 32.
1486. Howell II. G., Coulson N. — Proc. Roy. Soc.
London, 1938, A166, p. 238.
1487. Howell J. M., Wazer J. R. — J. Amer. Chem.
Soc, 1974, 96, p. 7902.
1488. Howson E. E. — Astrophys. J., 1912, 36, p. 286.
1489. Hsu C. J., Krugh W. D., Palmer H. В., Obe-
nauf R. H., Aten C. F. — J. Mol. Spectrosc,
1974, 53, p. 273.
1490. HuberE.J.,jr.,HolleyC. E.,jr. — J. Phys. Chem.,
1956, 60, p. 498.
1491. HuberH., Klotzbucher W., Ozin G. A., VoetA. V. —
Canad. J. Chem., 1973, 51, p. 2722.
1491a. Huber K. P., Herzberg G. Molecular Spectra and
Molecular Structure. IV. Constants of Diatomic
Molecules. N. Y. etc.: Van Nostrand Reinhold
Co., 1979.
1492. Hudson A., Treweek R. P., Wiffen J. T. — Theor.
chim. acta, 1975, 38, p. 355.
1493. Huff V. N., Gordon S., Morrell V. E. General
method and thermodynamic tables for
computation of equilibrium composition and temperature
of chemical reactions: Rept. 1037. Cleveland
(Ohio): NASA, 1951.
1494. Huffman D. R., Norwood M. H. — Phys. Rev.,
1960, 117, p. 709.
1495. Hughes E. W. — J. Amer.. Chem. Soc, 1956, 78,
p. 502.
1495a. Huglen R. — Dissertation. Trondheim, 1976.
1496. Huldt L., Lagerqvist A. — Ark. fys., 1950, 2,
p. 333.
1497. Huldt L., Lagerqvist A. — Z. Naturforsch., 1954,
A9, S. 991.
1498. Huldt L., Lagerqvist A. — Naturwissenschaften,
1955, 42, S. 365.
1499. Huldt L., Lagerqvist A. — Ark. fys., 1956, 11,
p. 347.
1500. Iluler E., Silberman E., Jones E. A. — Spectro-
chim. acta, 1970, A26, p. 2241.
1501. Hulett G. A.—Z. phys. Chem. Leipzig, 1901,
37, S. 396.
1502. HullH., TurnbullA.G. — Geochim. et cosmochim.
acta, 1973, 37, p. 685.
1503. Hultgren R., Desai R. D., Hawkins D. Т., Glei-
ser M., Kelley K. K., Wagman D. D. Selected
values of the thermodynamic properties of the
elements. Metals Park (Ohio): Amer. Soc. for
Metals, 1973.
1504. Hulthen E. — Phys. Rev., 1927, 29, p. 97.
1505. Hulthen E. — Z. Phys., 1939, 113, S. 126.
1506. HulthenE.,RydbergR. — Nature, 1933,131, p. 470.
1507. Hulthen E., Zumstein R. V. — Phys. Rev., 1926,
28, p. 13.
1508. Hultin M., Lagerqvist A. — Nature, 1950, 166,
p. 190.
1509. Hultin M., Lagerqvist A. — Ark. fys., 1951, 2,
p. 471.
1510. Human H. G. C, Zeegers P. J. Th. — Spectro-
chim. acta, 1975, B30, p. 203.
1511. Humphreys R. F., Fredrlckson W. R. — Phys.
Rev., 1936, 50, p. 543.
1512. Huo W. M., Freed K. F., Klemperer W. — J.
Chem. Phys., 1967, 46, p. 3556.
1513. Hurd С. В., Walker К. Е. — J. Amer. Chem.
Soc, 1931, 53, p. 1681.
1514. Hurk J. van der, Hollander Т., Alkemade С Th.
J. — J. Quant Spectrosc. and Radiat. Transfer,
1974, 14, p. 1167.
1515. Hurley A. C. —Proc. Roy. Soc. London, 1958,
A248, p. 119.
1516. Hurley A. C. — Proc. Roy. Soc. London, 1959,
A249, p. 402.
1517. Hurley A. C. — Proc. Roy. Soc. London, 1961,
A261, p. 237.
1518. Huron B. — Physica, 1969, 41, p. 58.
1519. Huron В., Rancarel P. — Chem. Phys. Lett.,
1972, 13, p. 515.
1520. Hutchison J. F. — U. S. Atom. Energy Commis.,
Repts, IS-T-50, 1965.
1521. Huttig G. F., Arbes A. — Z. anorg. und alls.
Chem., 1930, 191, S. 161.
1522. Huttig G. P., Frankenstein W. — Z. anorg. und
allg. Chem., 1930, 185, S. 403.
1523. Huttig G. F., Rosenkranz E. — Z. Elektrochem.,
1929, 35, S. 308.
1524. Huttig G. F., Slonim Ch. — Z. anorg. und allg.
Chem., 1929, 181, S. 65.
1525. Huttig G. F., Slonim Ch. — Z. anorg. und allg.
Chem., 1929, 181, S. 74.
1526. Ikeda Т., Wong N. В., HarrisD. O., Field R. W. —
J. Mol. Spectrosc, 1977, 68, p. 452.
1527. Ilschner В., Humbert J.—Z. Metallk., 1960,
51, S. 626.
1528. Inghram M. <?., Chupka W., Porter R,— 1.
Chem. Phys., 1955, 23, p. 2159.
1529. Inghram M. G., Drowart J. — In: Proc. Symp.
High Temp. Technol. N. Y., 1960.
1530. Inghram M. G., Porter R. F., Chupka W. A. —
J. Chem. Phys., 1956, 25, p. 498.
1531. Inoue J. — J. Chem. Soc. Jap. Industr. Chem.
Sec, 1949, 52, p. 135.
1532. Irmann F. — Helv. chim. acta, 1950, 33, p. 1449.
1533. Isbekow W. — Z. anorg. und allg. Chem., 1925,
143, S. 80.
1534. Iwamoto N., Sulto H., Satoh I. — Trans. JWRI,
1974, 3, p. 33.
1535. Jacob K. T.,Alcock С. В. — Met. Trans., 1973, 4,
p. 2483.
1536. Jacobson. R. L., Langmuir D. — Geochim. et
cosmochim. acta , 1974, 38, p. 301.
1537. Jacquinot D., Lavendy H. — C. r. Acad. sci.,
1975, B281, p. 397.
1538. Jaeger, F. M. Rosenbohm E. — Recueil trav.
chim. 1934, 53, p. 451.
1539. Jakes J., Papousek D. — Collect. Czech. Chem.
Communs, 1961, 26, S. 2110.
1540. James С G., Sugden Т. М. — Nature, 1953, 171,
p. 428.
1541. James C. G., Sugden Т. М. — Nature, 1955, 175,
p. 333.
1542. James J. A. — Dissertation. Oxford, 1947.
1543. J ANAF thermochemical tables. 2ed/Ed. D. R. Stull,
H. Prophet, Wash.: NSRDS—NBS 1971, N 37.
1544 — 1545. JANAF thermochemical tables.
Supplements. Wash., 1974—1978.
1546. Janecke E. — Z. phys. Chem. Leipzig, 1912, 80,
S. 1.
1547. Jander G., Zschaage W. — Z. anorg. und alls.
Chem., 1953, 272, S. 53.
1548. Janjic D., Brlner E., Paillard K. — Helv. chim,
acta, 1955, 38, p. 355.
1549. Janz G. /., Kelly F. J., Perano J. L. — Trans.
Faraday Soc, 1963, 59, p. 2718.
449
Литература
1550. Jauch R. — Diplomarbeit. Techn. Hochschule.
Stuttgart, 1946.
1550a. Jaulmes P., Galhak E. — Bull. Soc. chim. France,
1937, 4, N 1, p. 149.
1551. Jayaraman A., Element W., Kennedy G. C. —
Phys. Rev., 1963, 132, p. 1620.
1552. Jazawa A., Azakami Т., Kawashima T. — Met.
Inst. Jap., 1966, 82, p. 519.
1553. Jeffes J. H. F., McKerrell H. — J. Iron and Steel
Inst., 1964, 202, p. 666.
1554. Jenc F. — J. Mol. Spectrosc, 1967, 24, p. 284.
1555. Jenkins F. A. — Proc. Nat. Acad. Sci. USA, 1927,
13, p. 496.
1556. Jenkins F. A.— Phys. Rev., 1930, 35, p. 315.
1557. Jenkins F. A., Grinfeld R. — Phys. Rev., 1934,
45, p. 229.
1558. Jenkins F. A., Harvey A. — Phys. Rev., 1932,
39, p. 922.
1559. Jenkins F. A., McKellar A. — Phys. Rev., 1932,
42, p. 464.
1560. Jennergren С G. — Nature, 1948, 161, p. 315.
1561. Jennergren С G. — Ark. Mat. Astron. Phys.,
1948, A35, N 22, p. 1.
1562. Jensen А. Т.—Ъ. phys. Chem. Leipzig, 1937,
A180, S. 93.
1563. Jensen А. Т., Lannung A. — Kgl. danske vid. sel-
skab. Mat.-fys., medd., 1943, 20, N 15, p. 1.
1564. Jensen D. E. — Combust, and Flame, 1968, 12,
p. 261.
1565. Jensen D. E. — Trans. Faraday Soc, 1969, 65,
p. 2123.
1566. Jensen D. E. — J. Chem. Phys., 1970, 52, p. 3305.
1567. Jensen D. E., Jones G. A. — J. Chem. Soc.Faraday
Trans. I, 1972, 68, p. 259.
1568. Jerding #., Smit E. — Z. phys. Chem. Leipzig,
1941, 50, S. 171.
1569. Jevons W. — Proc. Roy. Soc. London, 1924, A106,
p. 174.
1570. Jevons W. — Proc. Roy. Soc. London, 1929, A122,
p. 211.
1571. Johansen H. L. — Diss. Abstrs, 1971, B31, p. 7221.
1572. Johansson I., Litzen U. — Ark. fys., 1967, 34,
p. 573.
1573. Johansson L. — Ark. fys., 1961, 20, p. 489.
1574. Johansson L. - Ark. fys., 1962, 23, p. 119.
1575. Johns J. W. С - Can. J. Phys., 1961, 39, p. 1738.
1576. Johns J. W. C, Grimm F. A., Porter R. F. —
J. Mol. Spectrosc, 1967, 22, p. 435.
1577. Johnson G. K., Feder H. M., Hubbard W. N. —
J. Phys. Chem., 1966, 70, p. 1.
1578. Johnson G. K., Hubbard W. N. — J. Chem.
Thermodyn., 1969, 1, p. 459.
1579. Johnson G. K., Smith P. N., Hubbard W. N. —
J. Chem. Thermodyn., 1973, 5, p. 793.
1580. Johnson J. W., Cubicciotti D., Silva W. J. —
High Temp. Sci., 1971, 3, p. 523.
1581. Johnson J. W., Silva W. J., Cubicciotti D. —
J. Phys. Chem., 1968, 72, p. 1669.
1582. Johnson J. W., Silva W. J., Cubicciotti D. —
J. Phys. Chem., 1968, 72, p. 1664.
1583. Johnson R. С — Proc. Roy. Soc. London, 1929,
A72,p. 161.
1584. Johnson R. C. — Proc. Roy. Soc. London, 1929,
A122, p. 189.
1585. Johnson R. E., Starizky E., Douglass R. M. —
J. Amer. Chem. Soc, 1957, 79, p. 2037.
1586. Johnson R. G., Hudson D. E., Caldwell W. C,
Spedding F. H., Savage W. R. — J. Chem. Phys.r
1956, 25, p. 917.
1586a. Johnson S. E., Capelle G., Broida H. P. —
J. Chem. Phys., 1972, 56, p. 663.
1587. Johnson W. C, Stubbs M. F., Sidwell A. E.,
Pechukas A. — J. Amer. Chem. Soc, 1939, 61,
p. 318.
1588. Johnson W. H., Gilliland A. A. — J. Res. NBS,
1961, A65, p. 59.
1589. Johnson W. H., Miller R. G., Prosen E. J. — J.
Res. NBS, 1959, 62, p. 213.
1590. Johnston H. L., Hersch H. N., Kerr E. С — J.
Amer. Chem. Soc, 1951, 73, p. 1112.
1591. Johnston J. — J. Amer. Chem. Soc, 1908, 30,
p. 1357.
1592. Johnston J. — Z. phys. Chem. Leipzig, 1908, 62,
S 330
1593. Johnston J. — J. Amer. Chem. Soc, 1910, 32,
p. 938.
1594. Jonah С D., Zare R. N. — Chem. Phys. Lett.,
1971, 9, p. 65.
1595. Jonah С D., Zare R. N., Ottinger Ch. — J. Chem.
Phys., 1972, 56, p. 263.
1596. Jones E. O., Becker M. L. — J. Chem. Soc, 1927,
p. 2669.
1597. Jones E. V., Lietzke M. H., Marshall W. L. —
J. Amer. Chem. Soc, 1957, 79, p. 267.
1598. Jones L. H., Ryan R. R. — J. Chem. Phys.r
1972, 57, p. 1012.
1599. Jonston H. L., Kerr E. C. — J. Amer. Chem.
Soc, 1950, 72, p. 4733.
1600. Joshi M. M., Gopal R. — Pramana, 1975, k,
p. 276.
1601. Joshimine M. — J. Chem. Phys., 1964, 40, p. 2970.
1602. Judd M. D., Pope M. J. — J. Appl. Chem. and
Biotechnol., 1970, 20, p. 384.
1603. Judd M. D., Pope M. J. — J. Appl. Chem. and
Biotechnol., 1971, 21, p. 149.
1604. Judd M. D., Pope M. J. — J. Appl. Chem. and
Biotechnol., 1972, 21, p. 285.
1605. Judd M. D., Pope M. J. — Thermochim. acta,
1973, 7, p. 247.
1606. Jura G., Garland С W. — J. Amer. Chem. Soc,
1952, 74, p. 6033.
1607. Justice В. Н. — J. Chem. and Eng. Data, 1969,
14, p. 4.
1608. Justice В. Н. — J. Chem. and Eng. Data, 1969,
14, p. 384.
1608a. Juza R., Bunzen K. — Z. phys. Chem. (BRD),
1958, 17, S. 82.
1609. Kablukov I. — Chem. Zbl., 1908, 11, p. 486.
1610. Kadesch R. R. — Diss. Abstrs, 1956, 16, p. 1155.
1611. Kadesch R. R., Moldehauer J. C. — Spectrochim.
acta, 1956, 8, p. 192.
1612. Kadesch R. R., Moldehauer J. C, Winaus J. G. —
Spectrosc. Mol., 1956, 5, p.. 18.
1612a. Kahovec L. — Z. phys. Chem. Leipzig, 1938,
B40, S. 135.
1613. Kaiser E. W., Muenter J. S., Klemperer W. — J.
Chem. Phys., 1968, 48, p. 3339.
1614. Kakubari H., Konaka S., Kimura M. — Bull.
Chem. Soc. Jap., 1974, 47, p. 2337.
1615. Kaldor A. — J. Chem. Phys., 1971, 55, p. 4641.
1616. Kaldor A., Porter R. F. — Inorg. Chem., 1971,
10, p. 775.
1617. Kaldor A., Porter R. F. — J. Amer. Chem. Soc,
1971, 93, p. 2140.
1618. Kalff P. J., Alkemade С Th. J. — J. Chem.
Phys., 1970, 52, p. 1006.
450
Литература
1619. Kalff P. /., Alkemade С. Th. J. — J. Chem.
Phys., 1973, 59, p. 2572.
1620. Kalff P. /., Alkemade C. Th. J. — J. Chem.
Phys., 1974, 60, p. 1698.
1621. Kalff P. /., Hollander Т., Alkemade С Th. J. —
J. Chem. Phys. 1965, 43, p. 2299.
1622. Kamalasanan M. N. — Curr. Sci., 1974, 43, p. 540.
1623. Kamalasanan M. N. — Indian J. Pure and Appl.
Phys., 1975, 13, p. 124.
1624. Kameyama N., Yoshida F. — Denki Kagaku,
1944, 12, p. 171.
1624a. Kamibayashi K. — Pat. 6257/1953 (Japan), Dec. 7.
1625. Kamienski B. — Bull, intern. Acad. pol. sci.
Litters Ser. sci. natur., 1926, s. 109.
1626. Капа'an A. S., Hauge R. H., Margrave J. L. —
J. Chem. Soc. Faraday Trans. II, 1976, 72, p. 1991.
1627. Kanolt С W. — Z. anorg. und allg. Chem., 1914,
85, S. 1.
1627a. Kao J. — J. Mol. Spectrosc, 1979, 56, p. 147.
1628. Kapustinsky A. F., Dezideryeva I. P. — Trans.
Faraday Soc, 1946, 42, p. 69.
1628a. Kapustinsky A. F., Korshunov J. A. — Acta
physicochim. USSR, 1939, 10, p. 259.
1629. Karandeeff B. — Z. anorg. und allg. Chem., 1910,
68, S. 188.
1630. Kartha V. В., Krishamachari S. L. N. G., Sub-
ramaniam С R. — J. Mol. Spectrosc, 1967, 23,
p. 149.
1630a. Kashireninov О. Е., Chervonnyi A. D., Pi-
ven V. A. — High Temp. Sci., 1981 (in press).
1631. Kaskan W. E., Mackenzie J. D., Millikan R. С —
J. Chem. Phys. 1961, 34, p. 570.
1632. Kaskan W. E., Millikan R. С — J. Chem. Phys.,
1960, 32, p. 1273.
1633. Kassner G., Stempel B. — Z. anorg. und allg.
Chem., 1929, 181, S. 83.
1634. Kasuya Т., Lafferty W. J., LideD. R. — J. Chem.
Phys., 1968, 48, p. 1.
1635. Katayama /., Kozuka Z. — Techn. Rept Osaka
Univ., 1973, 23, p. 411.
1636. Katayama I., Shibata J., Kozuka Z. — Nippon
Kinzoku Gakkaishi, 1975, 39, p. 990.
1637. KaterbergJ., NiemeyerS., PenningD., ZytveldJ.B.
van. — J. Phys. F: Met. Phys., 1975, 5, p. L74.
1638. Katz C. — Thesis Cornell Univ., 1949.
1639. Katz. T. J., Margrave J. L. — J. Chem. Phys.,
1955, 23, p. 983.
1640. Kaufman J. J., Koski W. S., Anacreon R. P. —
J. Mol. Spectrosc, 1963, 11, p. 1.
1641. Kaufman M., Wharton L., Klemperer W. — J.
Chem. Phys., 1965, 43, p. 943.
1642. Kaufmann L. —Acta Met., 1961, 9, 896.
1643. Raving В., Lindgren B. — Phys. scr., 1974, 10,
p. 73.
1644. Kaving В., Lindgren B. — Phys. scr., 1974, 10,
p. 81.
1644a. Kawashima Y., Takeo H., Matsumura Chi. —
Chem. Phys. Lett. 1978, 57, p. 145.
16446. Kawashima Y., Takeo II., Matsumura Chi. —
J. Mol. Spectrosc, 1979, 78, p. 493.
1645. Kayser F. X., Soderquist S. D. — J. Phys. and
Chem. Solids, 1967, 28, p. 2343.
1646. Kazakov A. V., Tikhomirova M. M., Plotni-
kova V. I. — Intern. Geol. Rev., 1959, 1, p. 1.
1647. Kaznoff A. J., Orr R. L., Hultgren R. — In:
Thermodynamic Symp. Heidelberg, 1967, vol. 4,
p. 3.
1648. Keesom W. ff., Kok J. A. - Physica, 1934, 1,
p. 175.
1649. Kelley К. К. — Bull. Bur. Mines (USA), 1932,
N 350.
1650. Kelley К. К. Contributions to the data on
theoretical metallurgy. V. Evalution of the heats of
fusion of metals from freezing point lowering
(equilibrium diagrams). — Bull. Bur. Mines (USA),
1936, N 393.
1651. Kelley К. К. — Bull. Bur. Mines (USA), 1937,
N 406.
1652. Kelley К. К. — Bull. Bur. Mines (USA), 1937,
N 407.
1653. Kelley К. К. — 1. Amer. Chem. Soc, 1939, 61,
p. 1217.
1654. Kelley K. K.—Z. Amer. Chem. Soc, 1941, 63,
p. 1137.
1655. Kelley К. К. — Bull. Bur. Mines (USA), 1949,
N 476.
1656. Kelley К. К. Contributions to the data on
theoretical metallurgy. XIII. High—temperature heat-
content, heat capacity and entropy data for the
elements and inorganic compounds. — Bull. Bur.
Mines (USA), 1960, N 584.
1657. Kelley K. K., Anderson С. Т. — Bull. Bur. Mines
(USA), 1935, N 384.
1658. Kelley K. K., King E. G. Contributions to the
data on theoretical metallurgy. XII. Entropies
of the elements and inorganic compounds. — Bull.
Bur. Mines (USA), 1961, N 592.
1659. Kelley K. K., Moore G. E. — J. Amer. Chem.
Soc, 1943, 65, p. 1264.
1660. Kelley K. K., Southard J. G., Anderson С. Т. —
U. S. Bur. Mines. Tech. Pap., 1941, N 625.
1661. Keller D. V., Kanda F. A., King A. J. — J.
Phys. Chem., 1958, 62, p. 732.
1662. Kellner G. — Z. anorg. und allg. Chem., 1917,
99, S. 137.
1663. Kellner G. — J. Chem. Soc, 1917, 112, p. 469.
1664. Kelly R., Padley P. J. — J. Chem. Soc. Chem.
Communs, 1970, N 23, p. 1606.
1665. Kelly Д., Padley P. J. — Trans. Faraday Soc,
1971, 67, p. 740.
1666. Kelly R., Padley P. J. — Trans. Faraday Soc,
1971, 67, p. 1384.
1667. Kendall J. — Philos. Mag. Ser. 6, 1912, 23, p. 958.
1668. Kendall /., Crittenden E. D., Miller H. K. — J.
Amer. Chem. Soc, 1923, 45, p. 963.
1669. Keneshea F. J., Cubicciotti D. — J. Phys. Chem.,
1965, 69, p. 3910.
1670. Keneshea F. /., Cubicciotti D. — J. Phys. Chem.,
1967, 71, p. 1958.
1671. Kennedy G. C. — Amer. J. Sci., 1956, 254, p. 567.
1672. Kennedy G. C, La Mori P. N. — J. Geophys.
Res., 1962, 67, p. 851.
1673. Kennelley J. A., Varwig J. W., Myers H. W. —
J. Phys. Chem., 1960, 64, p. 703.
1674. Kerr E. C, Hersch H. N., Johnston H. L. —
J. Amer. Chem. Soc, 1950, 72, p. 4738.
1675. Kerr E. C, Johnston H. L., Hallett N. C. —
J. Amer. Chem. Soc, 1950, 72, p. 4740.
1675a. Ketelaar J. A. A.—Z. Kristallogr., 1935, 92,
S. 30.
1676. Khan M. A. — Proc Phys. Soc. London, 1958,
71, p. 65.
1677. Khan M. A. — Proc. Phys. Soc. London, 1961,
77, p. 1133.
1678. Khan M. A. — Proc. Phys. Soc. London, 1962,
79, p. 745.
1679. Khan M. A. — Proc. Phys. Soc. London, 1962,
80, p. 209.
451
Литература
1680. Khan M. A. — Ргос. Phys. Soc. London, 1962,
80, p. 523.
1681. Khan M. A. — Proc. Phys. Soc. London, 1963,
81, p. 1047.
1682. Khan M. A. — Proc. Phys. Soc. London, 1963,
82, p. 564.
1683. Khan M. A. — Proc. Phys. Soc. London, 1963,
82, p. 572.
1684. Khan M. A. — Proc. Phys. Soc. London, 1966,
87, p. 569.
1685. Khan M. A. — Proc. Phys. Soc. London, 1966,
89, p. 165.
1686. Khan M.A. — Z. Phys. B: Atom, and Mol. Phys.,
1968, 1, p. 985.
1687. Khan M. A. — J. Sci. Phys. Soc, 1972, 1, p. 17.
1688. Khan M. A., Alfridi M. K. — J. Phys. B: Atom,
and Mol. Phys., 1968, 1, p. 260.
1689. Khan M. A., Butt M. R. — J. Phys. B: Atom,
and Mol. Phys., 1968, 1, p. 745.
1690. Khan M. A., Hasnain S. S. — Nuovo cim.,
1973, B18, p. 384.
1691. Khan M. A., Rafi M., Hussainee S. J. A. — J.
Phys. B: Atom, and Mol. Phys., 1976, 9, p. 1953.
1692. Khan M. A., Rafi M., Khan I. A., Baig M. A. —
J. Phys. B: Atom, and Mol. Phys., 1976, 9,
p. 2313.
1693. Khanna L. K., Dubey V. S. — Indian J. Pure
and Appl. Phys., 1973, 11, p. 286.
1694. Khanna L. K., Dubey V. S. — Indian J. Pure
and Appl. Phys., 1973, 11, p. 375.
1695. Khanna L. K., Dubey V. S. — Indian J. Pure
and Appl. Phys., 1973, 11, p. 444.
1696. Khanna L. K., Dubey V. S. — Indian J. Pure
and Appl. Phys., 1973, 11, p. 510.
1697. Khanna L. K., Dubey V. S. —Indian J. Pure and
Appl. Phys., 1975, 13, p. 603.
1698. Kieffer R., Gugel E., Leimer G., Ettmayer P. —
Ber. Dtsch. keram. Ges., 1971, 48, S. 385.
1699. Kikuchi Т., Kurosawa Т., Yagihaski T. — J.
Jap. Inst. Metal. 1964, 28, p. 497.
1700. Kikuchi Т., Kurosawa Т., Yagihashi T. — Rept
Nat. Res. Inst. Metals 1964, 7, p. 328.
1701. Kikuchi Т., Kurosawa Т., Yagihashi T. — Trans.
Jap. Inst. Metals, 1964, 5, p. 122.
1702. Kikuchi Г., Kurosawa Т., Yagihashi T. — Trans.
Jap. Inst. Metals, 1972, 13, p. 355.
1703. KildayM. V., Johnson W. H., Prosen E. J. — J.
Res. NBS, 1961, A65, p. 101.
1704. Kilday M. V., Prosen E. J. — J. Amer. Chem.
Soc, 1960, 82, p. 5508.
1705. Kilday M. V., Prosen E. J. — J. Res. NBS, 1964,
A68, p. 127.
1706. Kilday M. V., Prosen E. J., Wagman D. D. —
J. Res. NBS, 1973, A77, p. 217.
1707. Kim Y. S., Gordon R. G. — J. Chem. Phys.,
1974, 60, p. 4332.
1708. Kimpel R. F., Moss R. G. — J. Chem. and. Eng.
Data, 1968, 13, p. 231.
1709. King A. S. — Astrophys. J., 1905, 21, p. 236.
1710. King E. G. — Ind. and Eng. Chem., 1949, 41,
p. 1298.
1711. King E. G. — J. Amer. Chem. Soc, 1957, 79,
p. 2056.
1712. King E. G. — J. Amer. Chem. Soc, 1958, 80,
p. 1799.
1713. King E. G., Ferrante M. J., Pankratz L. B. —
U. S. Bur. Mines. Rept Invest., 1975, N 8041.
1713a. King E. G., Weller W. W. — NBS Rept.
Invest., 1960, N 5590.
1714. King L. A., Seegmiller D. W. — J. Chem. and
Eng. Data, 1971, 16, p. 23.
1715. King R. C, Armstrong G. T. — J. Res. NBS,
1964, A68, p. 661.
1716. Kirschenbaum A. D., Cahill J. A. — J. Inorg.
and Nucl. Chem., 1963, 25, p. 232.
1716a. Kirwan D. J. — J. Electrpchem. Soc, 1970,
117, p. 1572.
17166. Kleinschmidt P. D., Hildenbrand D. L. — J .
Chem. Phys., 1978, 68, p. 2819.
1717. Kleman B. — Ark. fys., 1953, 6, p. 407.
1718. Kleman В., Lagercrantz A., Uhler U. — Ark. fys.,
1950—1951, 2, p. 359.
1719. Klemm W. — Z. anorg. und allg. Chem., 1926,
152 S 252
1720. Klemm W.—Z. anorg. und allg. Chem., 1942,
249, S. 23.
1721. Klemm W., Brautigam M. — Z. anorg. und allg.
Chem., 1927, 163, S. 225.
1722. Klemm W., Beyersdorfer K., Oryschkewitsch J. —
Z. anorg. und allg. Chem., 1948, 256, S. 25.
1723. Klemm W., Jacobi H. — Z. anorg. und allg.
Chem., 1932, 207, S. 117.
1724. Klemm W., Jacobi H. — Ъ. anorg. und allg.
Chem., 1932, 207, S. 186.
1725. Klemm W., Schnick I. — Z. anorg. und allg.
Chem., 1936, 226, S. 353.
1726. Klemm W., Тапке Е. — Z. anorg. und allg. Chem.,
1931, 200,. S. 343.
1727. Klemperer W. — J. Chem. Phys., 1956, 24, p. 353.
1728. Klinedinst K. A., Stevenson D. A. — J. Chem.
Thermodyn., 1972, 4, p. 565.
1729. Klinedinst K. A., Stevenson D. A. — J. Chem.
Thermodyn., 1973, 5, p. 21.
1730. Knight L. В., Jr., Weltner W., Jr. — J. Chem.
Phys., 1971, 55, p. 5066.
1731. Knopf H. J., Staude H. — Z. phys. Chem.
Leipzig, 1955, 204, S. 265.
1732. Knudsen M. The kinetic theory of gases. L., 1958.
1732a. Ко H. C, Daut G. E. — U. S. Bur. Mines. Rept
Invest., 1980, RI 8409. 8 p.
1733. Ко H. C, Greenbaum M. A., Blauer J. A., Far-
ber M. — J. Phys. Chem., 1965, 69, p. 2311.
1734. Ко H. C, Greenbaum M. A., Farber M. — J.
Phys. Chem., 1967, 71, p. 1875.
1735. Ко H. C, Greenbaum M. A., Farber M.r
Selph С. С. — J. Phys. Chem., 1967, 71, p. 254.
1736. Ко H. C, Stuve J. M., Brown R. R. — U. S.
Bur. Mines. Rept Invest., 1976, N 8203.
1737. Koboyashi K. — Sci. Repts Tohoku Univ., 1950,.
34, p. 153.
1738. Koboyashi K. —Sci. Repts Tohoku Univ., 1951,.
35, p. 103.
1739. Kockel B. — Z. Naturforsch., 1970, A25, S. 595.
1740. Kohlmeyer E. J., Lohrke G. — Z. Erzberghau und
Metallhuttenwesen, 1956, 2, S. 326.
1741. Kohlmeyer E. /., Petzlaff H. W. — Z. anorg. und
allg. Chem., 1950, 261, S. 248.
1742. Kohlmeyer E. J., Spandau H.—Z. anorg. und
allg. Chem., 1945, 253, S. 37.
1743. Kohlrausch F. — Z. phys. Chem. Leipzig, 1908,
64, S. 129.
1744. Kohlrausch F., Rose F. — Z. phys. Chem.
Leipzig, 1893, 12, S. 234.
1745. Kohlschutter V., Feitknecht W. — Helv. chim.
acta, 1923, 6, p. 337.
1745a. Kohn J. A., Katz G., Broder J. D. — Amer .
Miner., 1957, 42, p. 398.
1746. Kohnert W. — Dissertation. В., 1914.
452
Литература
1747. Kojima #., Whiteway S. G., Masson С R. — Canad.
J. Chem., 1968, 46, p. 2968.
1748. Kok J. A., Keesom W. II. — Physica, 1937, 4,
p. 835.
1748a. Kolb C. E., Gersh M. E., Herschbach D. R. —
Combust, and Flame, 1975, 25, p. 31.
1749. Kolsky H. G., Gilmer R. M., Gilles P. W. —
J. Chem. Phys., 1957, 27, p. 494.
1750. Kolthoff I. M., Vogelenzang E. H. — Z. anal.
Chem., 1919, 58, S. 49.
1750a. Komas M., Golik L., KolarD. — J. Less-Common
Metals, 1971, 24, p. 121.
1751. Kanaka S., Ito Т., Morino G. — Bull. Chem.
Soc. Jap., 1966, 39, p. 1146.
1752. Kanaka S., Murata Y., Kuchitsu K., Morino Y. —
Bull. Chem. Soc. Jap., 1966, 39, p. 1134.
1753. Konishi У. — J. Chem. Soc. Jap. Industr. Chem.
Sec, 1936, 39, suppl. Binding, p. 209.
1754. Koontz P. G., Watson W. W. — Phys. Rev.,
1935, 48, p. 937.
1755. Kopp H. — Philos. Trans. Roy. Soc. London,
1865, 155, p. 171.
1756. Kopp /., Aslund N., Edvinsson G., Lindgren B. —
Ark. fys., 1965, 30, p. 321.
1757. Kopp I., Barrow R. F. — J. Phys. B: Atom,
and Mol. Phys., 1970, 3, p. L118.
1758. Kopp /., Kronekvist M., Guntsch A. — Ark. fys.,
1966, 32, p. 371.
1759. Kopp I., Lindgren В., Malmberg С — Phys.
scr., 1976, 14, p. 170.
1759a. Kopp I., Wirhed R. — Ark. fys., 1967, 38, p. 277.
1760. Koref F. —Ann. Phys., 1911, 36, S. 49.
1760a. Korreng E. — Neues Jahrb. Mineral. Geol. Bei-
lage, 1914, 37, p. 51.
1761. Koski W. S., Kaufman J. J., Pachucki С F. —
J. Amer. Chem. Soc, 1959, 81, p. 1326.
1762. Koski W. S., Kaufman J. J., Pachucki С F.,
Shipko F. J. — J. Amer. Chem. Soc, 1958, 80,
p. 3202.
1762a. Kostomaroff V., Rey M. — Silicates Ind., 1963,
28, p. 9.
1763. Kouba J. E., uhrn Y. — J. Chem. Phys., 1970,
53, p. 3923.
1764. Koutnik V., Benes J. — Chem. prumysl., 1958,
8, N 4, s. 187.
1765. Kovacs I., Budo A. — Acta phys. Acad. sci.
hung., 1952, 1, p. 469.
1766. Kevacs /., Budo A. — Ann. Phys., 1953, 12, S. 17.
1767. Kovacs I., Lagerqvist A. — Ark. fys., 1950, 2,
p. 411.
1768. Kovacs I., Lagerqvist A. — J. Chem. Phys., 1950,
18, p. 1683.
1769. Kracek F. C, Morey G. W., Merwin H. E. —
Amer. J. Sci., 1938, A35, p. 143.
1770. Krames W., Nolting J. — Acta met., 1972, 20,
p. 1353.
1771. Kraus C. A., Hurd С. В. — J. Amer. Chem. Soc,
1923, 45, p. 2559.
1772. Krause R. F., Douglas Т. В. — J. Phys. Chem.,
1968, 72, p. 475.
1772a. Krause R.IF., Douglas Т. В. — J. Phys. Chem.,
1968, 72, p. 3444.
1773. Kreuzer H. — Z. Natiirforsch., 1957, A12, S. 519.
1773a. Krishnamachari S. L., Narasimham N. A.,
Singh M. — Canad. J. Phys., 1966, 44, p. 2513.
1774. Krishnamachari S. L. N. G., Narasimham N. A.,
Singh M. — In: Proc Intern. Conf. Spectrosc.
Bombay, 1967, 1, p. 181.
1775. Krishnamachari S. L. N. G., Singh M. — Curr.
Sci., 1965, 34, p. 655.
1775a. Krishnan K. — Proc. Indian Acad. Sci., 1963,
A53, p. 103.
1776. Kristiansen L. A., Mooney R. W., Cyvin S. J.,
Brunvoll J. — Acta chem. scand., 1965, 19,
p. 1749.
1776a. Krohn C. — Acta chem. scand., 1966, 20, p. 255.
1777. Kronekvist M., Lagerqvist A.— Ark. fys., 1969,
39, p. 133.
1778. Kronekvist M., Lagerqvist A., Neuhaus II. — J.
Mol. Spectrosc, 1971, 39, p. 516.
1779. Kronen W., Boehm A. — Glas-Email-Keramo-
Techn., 1972, 23, S. 319.
1780. Kubas Z. — Arch, hutn., 1964, 9, s. 269.
1781. Kubaschewski O., Evans E. L., Alcock С. В.
Metallurgical thermochemistry. 4ed. Oxford;
London: Pergamon Press, 1967.
1781a. Kubaschewski O., Unal II. — High Temps. —
High Pressur., 1977, 9, p. 361.
1782. Kuchitsu K., Konaka S. — J. Chem. Phys., 1966,
45, p. 4342.
1783. Kuczkowski R. L., Lide D. R. — J. Chem. Phys.,
1966, 44, p. 3131.
1784. Kuhn H. — Z. Phys., 1930, 63, S. 458.
1784a. Kujumzelis T. G. — Phys. Z., 1938, 39, S. 665.
17846. Kulkarni M. P., Dadape V. V. — Indian J.
Chem., 1976, AM, p. 964.
1785. Kulkarni K. S., Sarma С R., Murthy J. S. —
Indian J. Pure and Appl. Phys., 1973, 11, p. 431.
1785a. Kuniya Y., Hosaka M. — J. Cryst. Growth,
1975, 28, p. 385.
17856. Kuniya Y., Hesaka M. — Denki Kagaku, 1975,
43, p. 372.
1785b. Kuniya Y., Hosoda S., Hosaka M. — Denki
Kagaku, 1974, 42, p. 20.
1786. Kushawaha V. S. — Spectrosc. Lett., 1973, 6,
p. 633.
1787. Kushawaha V. S., Asthana B. P., Shanker P.,
Pathak С. М. — Spectrose. Lett., 1972, 5, p. 407.
1788. Lacher J. R., Scruby R.-E., Park J. D. — 3.
i'Amer. Chem. Soc, 1952, 74, p. 5292.
1789/ Lagerqvist A. — Ark. mat. astr. fys., 1943, A29,
p. 1.
1790. Lagerqvist A. — Dissertation. Uppsala, 1948,
1791. Lagerqvist A. — Natiirwissenschaften, 1953, 40, S.
268.
1792. Lagerqvist A. —Ark. fys., 1954, 7, p. 473.
1793. Lagerqvist A. — Ark. fys., 1954, 8, p. 83.
1794. Lagerqvist A. — Ark. fys., 1954, 8, p. 481.
1795. Lagerqvist A., Huldt L. — Natiirwissenschaften,
1955, 42, S. 365.
1796. Lagerqvist A., Lind E., Barrow R. F. — Nature,
1949, 164, p. 922.
1797. Lagerqvist A., Lind E., Barrow R. F. — Proc.
Phys. Soc, London, 1950, A63, p. 1132.
1798. Lagerqvist A., Lundh L. E., Neuhaus H. — Phys.
scr., 1970, 1, p. 261.
1799. Lagerqvist A., Nilsson N. E. L., Barrow R. F. —
Ark. fys., 1957, 12, p. 543.
1800. Lagerqvist A., Nilsson N. E. L., Wigarz K. — Ark.
fys., 1958, 13, p. 379.
1801. Lagerqvist A., Selin L. E. —Ark. fys., 1956, 11,
p. 323.
Uhler U. — Ark, fys., 1949, 1,
Uhler U. — Nature, 1949, 164,
1802. Lagerqvist A.
p. 459.
1803. Lagerqvist A.
p. 665.
1804. Lafferty W. J., Maki A. G., Coyle Th. D. —
J. Mol. Spectrosc, 1970, 33, p. 345.
1805. Lakshminarajana A., Haranath P. B. V. — Curr.
Sci., 1970, 39, p. 228.
453
Литература
1806. Lakshminarajana A., Haranath Р. В. — Curr Sci.,
1970, 39, p. 344.
1807. Lander J. J. — J. Chem. Phys., 1949, 17, p. 892.
1808. Lander J. J. — J. Amer. Chem. Soc, 1951, 73,
p. 5794.
1809. Lange E., Durr F. — Z. phys. Chem. Leipzig,
1925, 118, S. 129.
1810. Lange E., Streeck H. — Z. phys. Chem. Leipzig,
1931, A152, S. 1.
1810a. Lange E., Streeck H. — Z. phys. Chem. Leipzig,
1931, A157, S. 1.
1811. Langmuir D. — J. Geol., 1965, 73, p. 730.
1812. Lannung A., Tovborg-Jensen A. — Kgl. danske
vid. selskab. Mat.-fys. medd., 1949, 25, p. 36.
1813. Larsson Т., Neuhaus H. — Ark. fys., 1963, 23,
p. 461.
1814. Larsson Т., Neuhaus H. — Ark. fys., 1966, 31,
p. 299.
1815. Laschchenko P. N. — С. г. Acad. sci., 1908, 147,
p. 58.
1816. Latimer В., Devlin J. P. — Spectrochim. acta,
1965, 21, p. 1437.
1817. Latimer В., Devlin J. P. —Spectrochim. acta,
1967, A23, p. 81.
1818. Latimer W. M. — J. Amer. Chem. Soc, 1951,
73, p. 1480.
1819. Latimer W. M. Tables of free energy functions
for Elements and compounds in the temperature
range 2000—5000 K: U. S. Atom. Energy Commis.,
Rept MMDC-1462, 1947.
1820. Latimer W. M., Hicks J. F. E., Schutz P. W. —
Z. chem. Phys. Leipzig, 1933, 1, S. 620.
1821. Latta R. E., Duderstedt E. G., Fryxell R. E. —
J. Nucl. Mater., 1970, 35, p. 350.
1822. Lau K.H., Cubicciotti D., Hildenbrand D. L.—
J. Chem. Phys., 1977, 66, p. 4532.
1822a. Lau K. H., Hildenbrand D. L. — J. Chem.
Phys., 1980, 72, p. 4928.
18226. Lau S., Schlott P. — In: 11 Sess. Comm.
consult, thermom. Com., intern, poids et mes. 1976.
P., 1977, p. 161.
1823. Laubengayer A. W., Schirmer F. B. — J. Amer.
Chem. Soc, 1940, 62, p. 1578.
1824. Laubengayer A. W., Sears D. С — J. Amer.
Chem. Soc, 1945, 67, p. 164.
1825. Laud В. В. — Indian J. Phys., 1962, 36, p. 639.
1826. Lavendy H., Jacquinot D. — С. г. Acad. sci.
1975, B17, p. 397.
1827. Lavendy H., Mahieu J. M., Becart M. — Canad.
J. Spectrosc, 1973, 18, p. 13.
1828. Law R. W., Margrave J. L. — J. Chem. Phys.,
1956, 25, p. 1086.
1829. Lazaarini F. — J. Appl. Crystallogr., 1975, 8,
p. 568.
1830. Leadbetter A. J.— J. Phys. C: Solid State Phys.
Ser. 2, 1968, 1, p. 1481.
1831. Leadbetter A. /., Wycherley К. Е. — Phys. and
Chem. Glasses, 1971, 12, p. 41.
1832. LeBoucher. — An. quim. Real. soc. exp. fis.
у quim., 1934, 219, p. 316.
1832a. Lebreton J. — J. chim. phys. et phys.-chim.
biol., 1973, 70, p. 738.
1833. Lebreton J., Ferran J., Chatalic A., Lacocca D.,
Marsigni L. — J. chim phys. et phys.-chim.
biol., 1974, 71, p. 587.
1834. Lebreton J., Ferran /., Marsigny L. — J. Phys.
B: Atom, and Mol. Phys., 1975, 8, L465 .
1835. Lebreton J., Marsigny L., Basser G. — G. r. Acad.
sci., 1970, C271, p. 1113.
1836. Lebreton J., Marsigny L., Ferran J. — C. r. Acad.
sci., 1971, C272, p. 1094.
1837. Lecompte /., Duval C, Wadier С. — С. г. Acad.
sci., 1959, 249, p. 1991.
1837a. Lee G. H, — Dissertation. N. Y.: Rensselaer
Polytechnic Inst., 1965.
1838. Lee G. H., Bauer W. H., Wiberley S. E. — J.
Phys. Chem., 1963, 67, p. 1742.
1839. Lefebvre-Brion H., Colin R. — J. Mol. Spectrosc,
1977, 65, p. 33.
1840. Lehtonen L. — Soc. sci. Fennica Commentationes,
Phys.-Math., 1921, 1, N 13, p. 1.
1840a. Leibovici C. — J. Mol. Struct., 1972, 14, p. 459.
1841. Leibovici C, Labarre J. F. — J. chim. phys.
et phys.-chim. biol., 1971, 68, p. 726.
1842. Leibovici C, Labarre J. F. — J. chim phys. et
phys.-chim. biol., 1972, 69, p. 1571.
1843. Leitgebel W. — Z. anorg. und allg. Chem., 1931,
202, S. 305.
1844. Lejeune J. M. — Bull. Soc. roy. sci. Liege., 1945,
14, p. 318.
1845. Leopold P. — Z. wiss Photogr., 1913, 11, S. 105.
1846. Leroi G. E. — Ph. D. Thesis Harward Univ.,
Cambrige, 1960.
1847. Lescocur H. — Ann. chim. phys., 1890, 19, p. 35.
1848. Lesiecki M. L., Nibler J. W. — J. Chem. Phys.,
1975, 63, p. 3452.
1848a. Lesiecki M. L., Nibler J. W. — J. Chem. Phys.,
1976, 64, p. 871.
1849. Lesiecki M. L., Nibler J. W. — J. Chem. Phys.,
1976, 64, p. 871.
1850. Lesiecki M. L., Shirk J. S. — J. Chem. Phys.,
1972, 56, p. 417.
1850a. Leu An-Lu, Ma Shao-Mu, Eyring H. — Proc
Nat. Acad. Sci. USA, 1975, 72, p. 1026.
1851. Levi J. P. — Rev. mater, constr. et trav. publies,
1948, N 388, p. 12.
1852. Levin I. W., Abramowitz S. A. — J. Chem. Phys.,
1965, 43, p. 4213.
1853. Levin I. W., Abramovitz S. A. — Chem. Phys.
Lett., 1971, 9, p. 247.
1854. Levin F. K., Winaus J. G. — Phys. Rev., 1951,
84, p. 431.
1855. Levy H. A., Brockway L. O. — J. Amer. Chem.
Soc, 1937, 59, p. 2085.
1856. Lewis G. N., Randall M., Pitzer K. S., Brewer L.
Thermodynamics. — N. Y. etc., 1961.
1857. Ley R., Schauer W. — Z. Naturforsch., 1972,
A27, S. 77.
1858. Li K. C., Stwalley W. С — J. Chem. Phys.,
1973, 59, p. 4423.
1859. Liberate G., Weniger S. — Physica, 1969, 41,
p. 47.
1860. Lide D. R. — J. Chem. Phys., 1963, 38, p. 2027.
1861. Lide D. R. — In: Proc. Meeting Interagency
Chem. Rocket. Propulsion Group Thermochem.
1-st. N. Y., 1963, 1, p. 1. (Publ. 1964).
1862. Lide D. R. — J. Chem. Phys., 1965, 42, p. 1013.
1863. Lie G. C, Clementi E. — J. Chem. Phys., 1974,
60, p. 1275.
1864. Lie G. C, Clementi E. — J. Chem. Phys., 1974,
60, p. 1288.
1865. Lien W. H., Phillips N. E. — J. Chem. Phys.,
1958, 29, p. 1415.
1866. Lindeman L. P., Wilson K. — J. Chem. Phys.,
1956, 24, p. 242.
1867. Lindsay D. M., HerschbachD. R., KwiramA. L. —
J. Chem. Phys., 1974, 60, p. 315.
454
Литература
1868. Linevsky M. J. Spectroscopic studies of the
vaporization of refractory materials: Final Rept
AD 609121. Philadelphia, 1964, Nov., 61 pp.
1869. Linevsky M. J. The infrared spectra of
magnesium and aluminium fluorides by matrix
isolation: Proc. of the Second Meeting of the Working
Group on Thermochemistry. Chem. Propulsion
Inform. Agency Publ. N 54, 1964.
1870. Linevsky M. J. — Techn. Rept RADC-TR-70-212.
General Electric Co, 1970.
1871. Linevsky M. J., Shull E. R., Mann D. E., War-
tik T. — J. Amer. Chem. Soc, 1953, 75, p. 3287.
1872. Linevsky M. J., Wartik T. — J. Phys. Chem.,
1958, 62, p. 1146.
1873. Linevsky M. J., White D., Mann D. E. — J.
Chem. Phys., 1964, 41, p. 542.
1874. Linke R., Rohrmann W. — Z. phys. Chem.
Leipzig, 1937, B35, S. 256.
1875. Lippert E. L., Lipscomb W. N. — J. Amer. Chem.
Soc, 1956, 78, p. 503.
1876. Lipson H., Stokes A. R. — Nature, 1941, 148,
p. 437.
1877. Lisiecki M. L., Shirk J. S. — J. Chem. Phys.,
1972, 56, p. 4171.
1878. Lisy J. M., Kytei J. — In: Terminal 76: Proc.
Celostatna Conf. Term. Anal., 7th, 1976, p. 5.
1878a. Livey D. Т., Murrey P. — J. Nucl. Energy,
1956, 2, p. 202.
1879. Loasby R. G., Dearden D. — J. Less-Common
Metals, 1977, 52, p. 137.
1880. Lochte-Holtgreven W., Vleugel E. S. van der. —
Nature, 1931, 127, p. 235.
1881. Lochte-Holtgreven W., Vleugel E. S. van der. —
Z. Phys., 1931, 70, S. 188.
1882. Loehman R. E., Kent R. A., Margrave J. L. —
J. Chem. and Eng. Data, 1965, 10, p. 296.
1883. Loewenschuss A. — Spectrochim. acta, 1975, A31,
p. 679.
1884. Logan J. K., Clement J. R., Jeffers H. R. —
Phys. Rev., 1957, 105, p. 1435.
1884a. Longhi P., Mussini Т., Rondinini S., Sala B. —
J. Chem. Thermodyn., 1979, 11, p. 73.
1885. Lord R. C, Nielsen E. — J. Chem. Phys., 1951,
19, p. 1.
1886. Lorenz M. R., Janz G. L. — J. Chem. Educ,
1963, 40, p. 611.
1887. Lorenz R., Wolcock J. — Z. anorg. und allg.
Chem., 1928, 176, S. 289.
1888. Lory E. R., Porter R. F. — J. Amer. Chem. Soc,
1971, 93, p. 6301.
1889. Lovas F. J., Johnson D. R. — J. Chem. Phys.,
1971, 55, p. 41.
1889a. Lovas F. J., Tiemann E- — J. Phys. and Chem.
Ref. Data, 1974, 3, p. 609.
1890. Lovas F. J., Torring T. — Z. Naturforsch., 1969,
A24, S. 634.
1890a. Lowings M. G., McCurdy J. G., Hepler L. G. —
Thermochim. acta, 1978, 23, p. 365.
18906. Lunde G. — Norsk geol. tidsskr., 1925, 8, s. 217.
1891. Lunch D. A., Jr., Zehe J., Carlson K. D. — J.
Phys. Chem., 1974, 78, p. 236.
1892. Lynds L. — J. Chem. Phys., 1965, 42, p. 1124.
1893. Lynds L. — J. Chem. Phys., 1966, 44, p. 1721.
1894. Lynds L. — Spectrochim. acta, 1966, 22, p. 2123.
1895. Lynds L., Bass CD.— Inorg. Chem., 1964, 3,
p. 1147.
1896. Lynds L., Bass C. D. — J. Chem. Phys., 1964,
40, p. 1590.
1897. Lynds L., Bass С D. — J. Chem. Phys., 1965,
43, p. 4357.
1898. Lyon W. G., Westrum E. F., Jr., Charvet M. —
J. Chem. Thermodyn., 1971, 3, p. 571.
1899. Macarovici D. — Rev. roum. chim., 1966, 11,
p. 233.
1899a. MacCordick J., Choplin F., Kaufmann G. —
Theor. chim. acta, 1973, 32, p. 183.
1900. MacKinney С N., Innes K. K. — J. Mol. Spec-
trosc, 1959, 3, p. 235.
1901. Macleod A. C. — Trans. Faraday Soc, 1967, 63,
p. 300.
1902. MacNeil K. A. G., Thynne J. С J. — J. Phys.
Chem., 1970, 74, p. 2257.
1903. Macur G. J., Edwards R. K., Wahlbeck R. G. —
J. Phys. Chem., 1966, 70, p. 2956.
1904. Mader C. L. Ideal gas thermodynamic properties
of detonation product: U. S. Atom. Energy Com-
mis., Rept AECU-4508, 1959.
1905. Madgin W. M., Swales D. A. — J. Chem. Soc,
1956, p. 196.
1905a. Maeda E., Sasamoto Т., Sata T. — J. Ceram.
Soc. Jap., 1978, 86, p. 491.
1906. Magec E. N. — J. Inorg. and Nucl. Chem., 1961,
22, p. 155.
1907. Magnus A. — Phys. Z., 1913, 14, S. 5.
1908. Magnus A., Danz H. — Ann. Phys., 1926, 80,
S. 808.
1909. Magnus A., Danz H. — Ann. Phys., 1926, 81,
S. 407.
1910. Magnus A., Holzmann H. — Ann. Phys., 1929,
3, S. 585.
1911. Mah A. D. — J. Phys. Chem., 1957, 61, p. 1572.
1912. Mah A. D. — U. S. Bur. Mines. Rept Invest.,
1962, N 5965.
1913. Mah A. D. — U. S. Bur. Mines. Rept Invest.,
1963, N 6171.
1914. Mah A. D. — U. S. Bur. Mines. Rept Invest.,
1964, N 6415.
1915. Mah A. D., King E. G., Weller W. W., Chri-
stensen A. U. — U. S. Bur. Mines. Rept Invest.,
1961, N 5716.
1916. Mahanti P. C, — Phys. Rev., 1932, 42, p. 609.
1917. Mahanti P. С — Proc. Phys. Soc. London, 1934,
46, p. 51.
1918. Mahanti P. С — Z. Phys., 1934, 88, S. 550.
1919. Mahanti P. C. — Indian J. Phys., 1935, 9, p. 369.
1920. Mahanti P. C. — Indian J. Phys., 1935, 9, p. 455.
1921. Maheshwari R. C, Shukla M. M., Singh L. D. —
Indian J. Pure and Appl. Phys., 1971, 9, p. 327.
1921a. Mahieu J. M., Becart M. — Canad. J. Spec-
trosc, 1968, 13, p. 95.
1922. Mahla K. — Z. Phys., 1933, 81, S. 625.
1923. Maier С G. — U. S. Bur. Mines. Techn. Pap.,
1929, N 360.
1924. Maier С G., Anderson С. Т. — J. Chem. Phys.,
1934, 2, p. 513.
1925. Maier O. — Dissertation. Prague, 1927.
1926. Maillot J. F., Morris D. R. — Canad. J. Chem.,
1972, 50, p. 839.
1927. Makowiecki D. M., Lynch D. A., Jr.,
Carlson K. D. — J. Phys. Chem., 1971, 75, p. 1963.
1928. Malaspina L., Gigli R., Piacente F. — Rev.
intern, hautes temp, et refract., 1971, 8, p. 211.
1929. Maloney K. M., Gupta S. K., Lynch D. A.,
Jr. — J. Inorg. and Nucl. Chem., 1976, 36, p. 49.
1930. Maloney K. M., Lynch D. A., Jr. — Intern. J.
Mass. Spectrom. and Ion Phys., 1974, 14, p. 415.
1931. Mandel M., Barrett A. H. — Phys. Rev., 1955,
98, p. 1159.
1932. Mandel M., Barrett A. H. — Bull. Amer. Phys.
Soc. Ser. II, 1956, 1, p. 284.
455
Литература
1933. Mandirola de 0. Brieux, Westerkamp J. F. — Spec- 1961. Mathews A. L., Baes С F., Jr. — Inorg. Chem.,
trochim. acta, 1964, 20, p. 1633. 1968, 7, p. 373.
1933a. Mangum B. W., Thornton D. D. — Metrologia, 1962. Mathews С M.r Innes K. K. — J. Mol. Spectrosc,
1979, 15, p. 201.
1934. MannD. E., CalderG. V., Seshadri K. S., White D.,
Linevsky M. J. — J. Ghem. Phys., 1967, 46,
p. 1138.
1935. Mann D. E., Fano L. — J. Chem. Phys., 1957,
26, p. 1665.
1936. Mann J. B. — J. Chem. Phys., 1967, 46, p. 1646.
1937. Mann J. B. — In: Recent Developments in Mass
1965, 15, p. 199.
1963. Matiasowsky K. — Chem. zvesti, 1959, 13, s. 69.
1964. Matignon C. — Ann. chim. phys., 1906, 8, p. 242.
1965. Matignon С — С. г. Acad. sci., 1912, 154, p. 1351.
1966. Matignon C, Marchal G. — Bull. Soc. chim.
France, 1926, 39, p. 167.
1967. Matignon C, Marchal G. — C. r. Acad. sci.,
1926, 183, p. 927.
Spectroscopy /Ed. K. Ogata, T. Hayakawa. To- 1967a. Matossi F., Bluschke H. — Z. Phys., 1938, 108,
kyo: Univ. Tokyo Press., 1970, p. 814.
1938. Mannchen W., Bornkessel K. — Z. Naturforsch.,
1959, A14, S. 925.
1939. Mannheimer E. — Z. phys. Chem. Ulterricht,
1924, 37, S. 45.
1940. Mar R. W. — Thermochim. acta, 1972, 4, p. 367.
1941. Mar R. W., Bedford R. G. — High Temp. Sci.,
1976, 8, p. 365.
1942. Marc R., Simek A. — Z. anorg. und allg. Ghem.,
1913,, 82, S. 17.
1942a. Marcano M., Barrow R. F. — Trans. Faraday
Soc, 1970, 66, p. 1917.
19426. Marcano M., Barrow R. F. — Trans. Faraday
Soc, 1970, 66, p. 2936.
1943. Marchal G. — J. chim. phys. et phys.-chim.
biol., 1925, 22, p. 493.
1944. Marchal G. — J. chim phys. et phys.-chim. biol.,
1926, 23, p. 38.
1944a. Marchidan D. I., Pandele L., Nicolescu A. —
Rev. roum. chim., 1972, 17, p. 1493.
1945. Margrave J. L. — J. Chem. Phys., 1954, 58,
p. 258.
1946. Margrave J. L. — Physico-chemical measurements
at high temperatures. L., 1959.
1947. Margrave J. L., Soulen J. R., Leroi G. E.,
Greene F. Т., Randall S. P. — In: XVI Congr.
IUPAC. P., 1957.
1948. Maria H. J., McDonald J. R., McGlynn S. P. —
J. Amer. Chem. Soc, 1973, 95, p. 1050.
S. 295.
1968. Matsushima Т., Ito Т., Опо К. — Sci. Rept Res.
Inst. Tohoku Univ., 1964, A16, p. 195.
1969. Mauras H. — Bull. Soc. chim. France, 1959,
N 1, p. 16.
1970. Maya J., Nordine P. С — J. Chem. Phys., 1976,
64, p. 84.
1971. McAdie H. G. — J. Inorg. and Nucl. Chem., 1966,
28, p. 2806.
1972. McAdie H. G. — Thermal Anal. Rept Comm.
Stand. Intern. Confederat. N. Y.: Acad. Press,
1969, Append. N 3.
1973. McAdie H. G. — J. Thermal Anal., 1971, 3, p. 79.
1974. McBride B. J., Heimal S., Ehlers J. G., Gordon S.
Thermodynamic Properties to 6000 К for 210
substances involving the first 18 elements, NASA
SP-3001. Wash., 1963.
1975. McCain D. C, Palke W. E. — J. Chem. Phys.,
1972, 56, p. 4957.
1976. McCarty L., Carpenter D. — J. Electrochem. Soc,
1960, 107, p. 38.
1977. McCopy L. D., Paule R. C, Margrave J. L. —
J. Phys. Chem., 1963, 67, p. 1086.
1978. McCoy H. N., Smith H. J. — J. Amer. Chem.
Soc, 1911, 33, p. 468.
1979. McCoy R. E., Bauer S. H. — J. Amer. Chem.
Soc, 1956, 78, p. 2061.
1980. McCreary W. J. — J. Amer. Chem. Soc, 1955,
77, p. 2113.
1949. Marino С P., White D. J. — J. Phys. Chem., 1981. McCreary J. R., Thorn R. J. — High Temp.
1973, 77, p. 2929.
1950. Markin T. L., Bones R. /., Wheeler V. J. — Proc.
Brit. Ceram. Soc, 1967, 8, p. 51.
1951. Maroni V. A., Gruen D. M., McBeth R. L.,
Cairns E. J. — Spectrochim. acta, 1970, A26,
p. 418.
Sci., 1971, 3, p. 300.
1982. McCreary J. R., Thorn R. J. — High Temp.
Sci., 1973, 5, p. 365.
1983. McCulloch L. — J. Amer. Chem. Soc, 1937, 59,
p. 2650.
1984. McDonald G. J. F. — J. GeoL, 1955, 63, p. 244.
1951a. Marquart J. R., Berkowitz J. — J. Chem. Phys., 1985. McDonald J. K., Innes K. K. — J. Mol. Spectrosc.,
1963, 39, p. 283.
1969, 29, p. 251.
1952. Marriott J., Craggs J. D. — J. Electron. Control, 1986. McDonald J. K., Innes К. К. — J. Mol. Spectrosc,
1957, 3, p. 194.
1953. Marsh F. J., Gordon M. S. — Chem. Phys. Lett.,
1977, 45, p. 255.
1954. Marte A. M., Pauillen P. — G. r. Acad. sci.,
1966, C263, p. 1477.
1955. Martin A. J., Moore A. — J. Less-Common
Metals, 1959, 1, p. 85.
1955a. Martin E., Barrow R. F. — Phys. scr., 1978,
17, p. 501.
1956. Marsi F. N. — J. Mol. Spectrosc, 1972, 43,
p. 168. .
1957. Masse J. L., Bdrlocher M. — Helv. chim. acta,
1964, 47, p. 314.
1958. Massey A., Zwolenik J. — J. Ghem. Soc, 1963,
p. 5354.
1959. Masuda M. — Proc. Imp. Acad. Jap., 1932, 8,
p. 436.
1960. Mathews C. W. — J. Mol. Spectrosc, 1966, 19,
p. 203.
1969, 32, p. 501.
1986a. McDonald J. K., Innes K. K., Goodlett V. W.,
Tolbert T. W. — J. Mol. Spectrosc., 1969, 32,
p. 511.
1987. McDonald R. A. — J. Chem. and Eng. Data,
1987, 12, p. 115.
1988. McDonald R. A. — J. Chem. and Eng. Data,
1967, 12, p. 131.
1989. McDonald R. A., Oetting F. L. — J. Phys. Chem.,
1985, 69, p. 3839.
1990. McDonald R. A., Stull D. R. — J. Phys. Chem.,
1961, 65, p. 1918.
1991. McDonald R. A., Stull D. R. — J. Ghem. and
Eng. Data, 1962, 7, p. 84.
1992. McGonigal P. I., Cahill J. A., Kirschen-
baum A. D. — J. Inorg. and Nucl. Ghem., 1962,
24, p. 1012.
1992a. McGonigal P. /., Kirshenbaum. A. D.,
Grosse A. V. — J. Phys. Chem., 1962, 66, p. 737.
456
Литература
1993. Melntosh D., Ozin G. A. — Inorg. Chem., 1976,
15, p. 2869.
1994. McKeanD. С — J. Chem. Phys., 1956, 24, p. 1002.
1995. McNalley R. N., Peters F. /., Ribble A. H. —
J. Amer. Ceram. Soc, 1961, 44, p. 491.
1996. McWhan D. В., Jayaraman A. — Appl. Phys.
Lett., 1963, p. 129.
1996a. Mecke R. — Phys. Z., 1925, 26, S. 217.
1997. Mecke R., Guillery M. — Phys. Z., 1927, 28,
s. 514.
1998. Medvedeva Z. S., Greenberg J. #., Zukov E. G.,
Luznafa N. P. — Krist. und Techn., 1967, 2,
N 4, S. 523.
1999. Meggers W. F. — J. Res. NBS, 1933, A10, p. 669.
2000. Mehta S. M., Kantak K. V. — Curr. Sci., 1946,
15, p. 129.
2001. Meichsner A., Roth W. А.—Ъ. Elektrochem.,
1934, 40, S. 19.
2002. Melcher A. C. — J. Amer. Chem. Soc, 1910,
32, p. 50.
2003. Melrose M. P. — J. Chem. Phys., 1971, 55, p. 470.
2004. Menge O. — Z. anorg. und allg. Chem., 1911,
72, S. 162.
2005. Menis O., Sterling J. T. — NBS U. S. Spec. Publ.,
1973, N 338, p. 61.
2006. Menzel #., Schulz #., Deckert H. — Z. anorg.
und allg. Chem., 1934, 220, S. 49.
2007. Menzinger M. — Can. J. Chem., 1974, 52, p. 1688.
2008. Menzinger M., Wren D. J. — Chem. Phys. Lett.,
1973, 18, p. 431.
2008a. Mermant G., Belinski C, Lalau-Keraly F. —
С r. Acad. sci. 1967, C265, p. 1447.
2009. Meschi D. J., Chupka W. A., Berkovitz J. — J.
Chem. Phys., 1960, 33, p. 530.
2010. Meschi D. J., Searcy A. W. — J. Phys.
Chem., 1959, 63, p. 1175.
2011. Mesnage P. — Ann. phys. (France), 1939, 12, p. 5.
2011a. Mesnard G., Uzan R., Cabaud B. — Cah. phys.,
1963, 17, p. 333.
2012. Mesrobian G., Rolin M., Pham H. — Rev. intern,
hautes temp, et refract., 1972, 9, p. 131.
2013- Messer С E. — U. S. Atomic Energy Commiss.
Repts. 1960, NYO-8028.
2014. Messer С. Е. — J. Less-Common Metals, 1972,
27, p. 371.
2015. Meyer W., Rosmus P. — J. Chem. Phys., 1975,
63, p. 2356.
2015a. Mezaki R., Tilleux E. W., Jambois T. F.,
Margrave J. Z. — In: Symp. Thermophys. Properties.
Pap. 3rd. Lafayette (USA), 1965, p. 138.
2016. Michaud M. — С r. Acad. sci., 1966, C262, p. 1143.
2017. Michaud M. — Rev. chim. miner., 1968, 5, p. 89.
2018. Michel M. — С. г. Acad. sci., 1955, 241, p. 1462.
2018a. Michels H. В., Harris R., Lillis J. — In:
Electron. Transit. Lasers. Cambrige: Mass. MIT Press.,
1976, p. 278.
2019. Mielenz W., Wartenberg H. — Z. anorg. und allg.
Chem., 1921, 116, S. 267.
2020. Miescher E. — Helv. phys. acta, 1934, 7, p. 462.
2021. Miescher E. — Helv. phys. acta, 1935, 8, p. 279.
2022. Miescher E. — Helv. phys. acta, 1936, 9, p. 693.
2023. Miescher E. — Helv phys. acta, 1941, 14, p. 148.
2024. Miescher E., Chretien M. — Nature, 1949, 163,
p. 996.
2025. Miescher E., Rosenthaler E. — Nature, 1940, 145,
p. 624.
2026. Miescher E., Wehrli M. — Helv. phys. acta, 1933,
6, p. 256.
2027. Miescher E., Wehrli M. — Helv. phys. acta, 1933,
6, p. 458.
2028. Miescher E., Wehrli M. — Helv. phys. acta, 1934,
7, p. 331.
2029. Miller E., Komare K., Codaff I. — Trans. Metal.
Soc. AIME, 1961, 218, p. 978.
2029a. Miller F. A., Wilkins С. Н. — Anal. Chem.,
1952, 24, p. 1253.
20296. Miller J. C, Andrews L. — Chem. Phys. Lett.,
1977, 50, p. 315.
2029b. Miller J. C, Aull B. S., Andrews L. — J. Chem.
Phys., 1977, 67, p. 2478.
2029г. Millet J. P., Pham H., Rolin M. — Rev. intern.
hautes temp, et refract., 1974 A975), 11, N 4,
p. 277.
2030. Mills К. С — High Temp. — High Pressur.,
1972, 4, p. 371.
2031. Mills К. С Thermodynamic Data for Inorganic
Sulphides, Selenides and Tellurides. L.: Butter-
worths and Co, 1974.
2032. Minis Ch. A., Lin S.-W., Herm R. R. — J. Chem.
Phys., 1972, 57, p. 3099.
2033. Mirwald P. W. — Naturwissenschaften, 1977, 64,
S. 221.
2034. Misener A. D.\ Wills H. H. — Proc. Roy. Soc.
London, 1940, A174, p. 262.
2035. Mishra R. K., Majumdar K. — Proc. Nat. Inst.
Sci. India, 1966, A36, p. 369.
2036. Mitani H., Nagai H. — J. Jap. Inst. Metals,
1967, 31, p. 1296.
2037. Mitchell A. — Trans. Faraday Soc, 1965, 61, p. 5.
2038. Mitchell D. W. — Ind. and Eng. Chem., 1949,
41, p. 2027.
2038a. Mitra M. — Indian J.Phys., 1938, 12, p. 9.
2039. Mitscherlich A. — Pogg. Ann., 1864, 121, p. 459.
2040. Mixter W. G. — Amer. J. Sci., 1915, 40, p. 23.
2041. Mizuno M., Yamada Т., Noguchi T. — J. Ceram.
Soc. Jap., 1975, 83, p. 175.
2042. Moffat J. B. — J. Mol. Struct., 1973, 16, p. 307.
2043. Mohan N., Mueller A. — Canad. J. Spectrosc,
1972, 17, p. 132.
2044. Mohanty B. S., Upadhya K. N. — Indian J.
Pure and Appl. Phys., 1967, 5, p. 523.
2045. Mohazzabi P., Searcy A. W. — J. Chem. Soc.
Faraday Trans. I, 1976, 72, p. 290.
2046. Moireau M.-Cl., Veillard A. — Theor. chim. acta,
1968, 11, p. 344.
2047. Moissan H. — С. г. Acad. sci., 1892, 115, p. 201.
2048. Moldenhauer W. — Z. anorg. und allg. Chem.,
1906, 51, S. 369.
2049. Moldenhauer W., Roll-Hansen C. — Z. anorg. und
allg. Chem., 1913, 82, S. 130.
2050. Moles E., Bareia J. G. de. — An. quim. Real,
soc. exp. fis. у quim., 1936, 34, p. 802.
2051. Moles E., Vian A. — Bol. Acad. cienc. exactas
fis. quim. у natur (Madrid), 1930, 2, p. 2.
2052. Moolenaar R. J., Evans J. C, McKeever L. D. —
J. Phys. Chem., 1970, 74, p. 3629.
2053. Moore Ch. E. — Atomic Energy Levels NSRDS-
NBS 35. Wash., 1971, vol. 1—3.
2054. Moore G. E. — J. Amer. Chem. Soc, 1943, 65,
p. 1700.
2055. Moore G. E., Allison #., Struthers J. — J. Chem.
Phys., 1950, 18, p. 1572.
2056. Moore G. E., Kelley К. К. — J. Amer. Chem.
Soc, 1942, 64, p. 2949.
2057. Moose J. E., Parr S. W. — J. Amer. Chem. Soc,
1924, 46, p. 2656.
2058. More K. R., Cornell S. D. — Phys. Rev., 1938,
53, p. 806.
457
Литература<
2059. Morgan F. — Phys. Rev., 1936, 50, p. 603. 2095.
2060. Morgan E., Barrow R. F. — Nature, 1960, 185,
p. 754. 2096.
2061. Morgan E., Barrow R. F. — Nature, 1961, 192, 2097.
p. 1182.
2062. Morgan H. W., Staats P. A. — Spectrochim. 2097a
acta, 1972, A28, p. 600.
2063. Morikofer W. — Dissertation. Bale, 1925.
2064. Morney J. R., Johnson А. В., Fu Y.-C, 2098.
Hill G. R. — Adv. Chem. Ser., 1961, N 32, p. 157.
2064a. Morris E. An Application of the theory of cor- 2099.
responding states to the prediction of the critical 2100.
constants of metals: AWRE Rept 0-67/6. L.:
UKAEA, 1964. 2101.
2065. Moser L., Herzner R. — Monatsh. Chem., 1923,
44, S. 115.
2066. Mosher O. A., Frosch R. P. — J. Chem. Phys., 2102.
1970, 52, p. 5781.
2067. Motzfeldt K. — Tekn. ukebl., 1962, s. 1137. 2103.
2067a. MourlotA. — Ann. chim. phys., 1899, 17, p. 527.
2068. Mozer H.t Otto /., Thomas W. — Z. Phys., 1967, 2103a
206, S. 223.
2069. Mueller W. H., Blackledge J. P., Libowitz G. G. 2104.
Metal hydrides. N. Y.; L.: Acad. Press., 1968.
2070. Muench N. L. — Phys. Rev., 1955, 99, p. 1814. 2105.
2071. Muenter J. S. — Chem. Phys. Lett., 1974, 26,
p. 37. 2106.
2071a. Mukaibo Т., Takahashi Y., Yamada K. — In:
Proc. First. Intern. Conf. on calorimetry and ther- 2107.
modynamics. Warsaw: Pol sci. publ., 1969, s. 375.
2072. Mukerji J. — Mem. sci. rev. metall., 1963, 60, 2108.
p. 785.
2073. Mulert 0. — Z. anorg. und allg. chem., 1912, 2109.
75, S. 198.
2074. Muller F. — Helv. phys. acta, 1935, 8, p. 152. 2110.
2075. Mulliken R. S. — Phys. Rev., 1925, 25, p. 509.
2076. Mulliken R. S. — Phys. Rev., 1925, 25, p. 259. 2111.
2077. Mulliken R. S. — Phys. Rev., 1925, 26, p. 571.
2078. Mulliken R. S. — Phys. Rev., 1931, 38, p. 836. 2112.
2079. Mulliken R. S. — Chem. Rev., 1947, 41, p. 207.
2080. Muner Z. A., Searcy A. W. — J. Electrochem. 2113.
Soc, 1964, 111, p. 1170.
2081. Murad E., Hildenbrand D. L. — J. Chem. Phys., 2114.
1966, 45, p. 4751.
2082. Murad E., Hildenbrand D. L., Main R. P. — 2115.
J. Chem. Phys., 1966, 45, p. 263.
2083. Murib J. H., Horvitz D., Bonecutter Ch. A. —
Ind. and Eng. Chem. Prod. Res. and Develop., 2116.
1965, 4, p. 273. 2117.
2084. Murty P. S., Rau P. T. — Curr. Sci., 1969, 38,
p. 187. 2118.
2085. Murty P. S., Rao P. T. — Curr. Sci., 1969, 38,
p. 537. 2119.
2086. Murty P. S., Rao P. T. — Proc. Roy. Irish. 2120.
Acad., 1972, A72, N 5, p. 71. 2121.
2087. Murty P. S., Reddy Y. P., Rao P. T. — J. Phys. 2122.
B: Atom, and Mol. Phys., 1970, 3, p. 425. 2123.
2088. Muthmann W., Weiss L. — Justus Liebigs Ann.
Chem., 1904, 331, S. 1. 2124.
2089. Muthmann W., Weiss L., Metzger J. — Justus
Liebigs Ann. Chem., 1907, 355, S. 137. 2125.
2090. Nacken R. — Zement, 1930, 19, S. 818.
2091. Nagarajan G. — Bull. Soc. chim. belg., 1962, 2126.
71, p. 65.
2092. Nagarajan G. — Bull. Soc. chim. belg., 1962, 2127.
71, p. 73.
2093. Nagarajan G. — Z. phys. Chem. (BRD), 1962, 2128.
31, S. 347.
2094. Nagarajan G. - Z. phys. Chem. (DDR), 1963, 2129.
223, S. 27.
Nagarajan G. — Bull. Soc. chim. belg., 1964,
73, p. 811.
Nagarajan G. — Acta phys. pol., 1966, 29, p. 841.
Nair K. P. R., Ammini Amma R. — Indian J.
Pure and Appl. Phys., 1972, 10, p. 93.
, Nakagawa J., Domaile P. J., Steimle T. C.,.
Harris D. O. — J. Mol. Spectrosc, 1978, 70,
p. 374.
Nakatsuji H. — J. Amer. Chem. Soc, 1973, 95,
p. 354.
Nakayama F. S. — Soil. Sci., 1968, 106, p. 429.
Nakayama F. S. — J. Chem. and Eng. Data,
1971, 16, p. 178.
Nampoori V. P. N., Kamalasanan M. N., Patel
M. M. — J. Phys. B: Atom, and Mol. Phys.
1975, 8, p. 284.
Nancollas G. H., Purdie N. — Trans. Faraday
Soc, 1963, 59, p. 735.
Nanda D. P., Mohanty B. S. — Curr. Sci., 1970,
39, p. 300.
. Nappi B. M., Pelino M., Ferro D., Piacente V. —
J. Chem. and Eng. Data, 1978, 23, p. 47.
Naray-Szabo G. — Intern. J. Quant. Chem., 1973,
7, p. 569.
Nathan С. С. — Ph. Thesis. Univ. Pittsburgh,
1948.
Naude S. M., Hugo T. J. — Canad. J. Phys.,
1953, 31, p. 1106.
Naude S. M., Hugo T. J. — Phys. Rev., 1953,
90, p. 318.
Naude S. M., Hugo T. J. — Canad. J. Phys.,
1954, 32, p. 246.
Naude S. M., Hugo T. J. — Canad. J. Phys.,
1955, 33, p. 573.
Naude S. M., Hugo T. /. — Canad. J. Phys.,
1957, 35, p. 64.
Navratil J. D., Oetting F. L. — J. Inorg. and
Nucl. Chem., 1973, 35, p. 3943.
Naylor B. F. — J. Amer. Chem. Soc, 1945. 67,
p. 150. .
Nelson L. S., Ramsay D. A. — J. Chem. Phys.,
1956, 25, p. 372.
Nelson W., Gordy W. — J. Chem. Phys., 1969,
51, p. 4710.
Nernst W., Schwers F. — Sitzungsber. kgl. preuss.
Akad. Wiss. Math.-naturwiss. Kl. Abt. II, 1914,
S. 355.
Nesbet R. K. — J. Chem. Phys., 1965, 43, p. 4403.
Nespital W. — Z. phys. Chem. Leipzig, 1932,
B16, S. 153.
Neugebauer С A., Margrave J. L. — Z. anorg.
und allg. Chem., 1957, 290, S. 82.
Neuhaus H. — Nature, 1957, 180, p. 433.
Neuhaus H. — Z. Phys., 1958, 150, S. 4.
Neuhaus H. — Z. Phys., 1958, 152, S. 402.
Neuhaus H. — Ark. fys., 1959, 14, p. 551.
Neuhaus H., Muld V. — Z. Phys., 1959, 153,
S. 412.
Neujmin H. — Phys. Z. Sowjetunion, 1934, 5,
S. 580.
Neuman E. W. — J. Amer. Chem. Soc, 1933,
55, p. 879.
Neumann В., Kroger C, Haebler H. — Z. anorg.
und allg. Chem., 1932, 204, S. 81.
Neumann В., Kroger C, Jutterer H. — Z. Elek-
trochem., 1935, 41, S. 725.
Neumann В., Kroger C, Kunz H. — Z. anorg.
und allg. Chem., 1932, 207, S. 133.
Neumann В., Kroger C, Kunz H. — Z. anorg.
und allg. Chem., 1934, 218, S. 379.
458
Литература
2130. Nevin Т. Е. — Ргос. Phys. Soc. London, 1931,
43, p. 554.
2131. Newbury R. S. — Dissertation. Berkeley, Univ.
Calif., 1965.
2132. Newbury R. S., Barton G. W., Searcy A. W. —
J. Chem. Phys., 1968, 48, p. 793.
2133. Newkirk A. E. — J. Amer. Ghem. Soc, 1948,
70, p. 1978.
2134. Newman E. S. — J. Res. NBS, 1941, 27, p. 191.
2135. Newman E. S. — J. Res. NBS, 1958, 61, p. 75.
2136. Newman E. S., Wells L. S. — J. Res. NBS, 1938,
20, p. 825.
2137. Newman R. N., Page F. M. — Combust, and
Flame, 1970, 15, p. 317.
2138. Newns G. R., Pelmore J. M. — J. Chem. Soc,
A, 1968, p. 360.
2139. Niedenzu K., Savodny W., Watanabe ff., Daw-
son J. W., Totani Т., Weber W. — Inorg. Chem.,
1967, 6, p. 1453.
2140. Nielsen A. H. — J. Chem. Phys., 1954, 22, p. 659.
2141. Nilson L. F., Pettersson O. — Berichte, 1880, 13,
S. 1459.
2142. Nilson L. F., Pettersson O. — С. г. Acad. sci.,
1880, 91, p. 232.
2143. Nilsson B. E. —Ark. mat. astr. fys., 1948, A35,
p. 1.
2144. Nilsson R. O. — Ark. kemi mineral, geol., 1957,
10, p. 363.
2145. Nimon L. A., Seshardi K. S., Taylor R. C,
White D. — J. Chem. Phys., 1970, 53, p. 2416.
2145a. Nishino Т., Nishiyama S. — J. Ceram. Assoc.
Jap., 1967, 75, p. 292.
2146. Newa K., Sato M., Yosiyama M. — J. Chem.
Soc. Jap., 1939, 60, p. 918.
2147. Newa K., Sato M., Yosiyama M. — J. Fac. Sci.
Hokkaido Univ. Ser. Ill, 1940, N 1, p. 17.
2148. Noguchi T. — Adv. High Temp. Chem., 1969, 2,
p. 235.
2149. Norman J. H., Winchell P., Staley H.G.— J. Chem.
Phys., 1964, 41, p. 60.
2150. Noyes A. A. — I. phys. Chem. Leipzig, 1892,
9, S. 603.
2151. Noyes A. A., Abbot С G. — Ъ. phys. Chem.
Leipzig, 1895, 16, S. 125.
2152. O'Brien C. J., Kelley K. K. — J. Amer. Chem.
Soc, 1957, 79, p. 5616.
2153. Oelsen W. — Arch. Eisenhtittenw., 1955, 26, S. 519.
2154. Oelsen W. — Arch. Eisenhtittenw., 1957, 28, S. 1.
2155. Oelsen W., Oelsen O. Thiel D. — Z. Metallk.,
1955, 46, S. 555.
2156. Oelsen W., Rieskamp K. H., Oelsen O. — Arch.
Eisenhtittenw., 1955, 26, S. 253.
2157. Ohno K. — J. Phys. Soc. Jap., 1957, 12, p. 938.
2157a. Ohse R. W., Tippelskirch H. — High Temp. —
High Pressur., 1977, 9, p. 367.
21576. Okuno T. — J. Chem. Soc. Jap. Chem. and
Ind. Chem., 1935, 38, p. 421.
2158. Olbrich W. — Dissertation. Breslau, 1924.
2158a. Olme A. — Phys. scr., 1970, 1, s. 256.
2159. Olmsted С. М. — Z. wiss. Photogr., 1906, 4,
S. 255.
2160. Olson D. S., Kibler F. C, Seegmiller D. W., Fan-
nin A., King L. A. — J. Chem. and Eng. Data,
1974, 19, p. 27.
2161. Olsson E. — Z. Phys., 1932, 73, S. 732.
2162. Ohaka R. — J. Chem. Phys., 1957, 27, p. 374.
2163. O'Neal H. R., Phillips N. E. — Phys. Rev.,
1965, A137, p. 748.
2164. O'Neil S. V., Pearson P. K., Schaefer H. F. —
Chem. Phys. Lett. 1971, 10, p. 404.
2164a. O'Neil S. V., Schaefer H. F., Bender С F. —
J. Chem. Phys., 1973, 59, p. 3608.
2165. Onishi Т., Shimanouchi T. — Spectrochim. acta,
1964, A20, p. 325.
2166. Onodera Natsuo, Kimoto Arata, Sakiyama Ml*-
noru, Seki Syuzo. — Bull. Chem. Soc. Jap., 1971,
44, p. 1463.
2167. Osberghaus O. — Z. Phys., 1950, 128, S. 366.
2168. Otvas J. W., Stevenson D. P. — J. Amer. Chem,
Soc, 1956, 78, p. 546.
2169. Ownby P. D., Gretz R. D. — Surface Sci., 1968,
9, p. 37.
2170. Padgett A. A., Griff ing V. — J. Chem. Phys.,
1959, 30, p. 1286.
2171. Palenius H. P. — Phys. Lett., 1976, A56, p. 451.
2172. Palke W. E. — J. Chem. Phys., 1972, 56, p. 5308.
2173. Palke W. E., Lipscomb W. N. — J. Chem. Phys,
1966, 45, p. 3498.
2174. Palke W. E., Lipscomb W. N. — J. Amer. Chem.
Soc, 1966, 88, p. 2384.
2175. Palmer K. J., Elliott N. — J. Amer. Chem. Soc.,
1938, 60, p. 1852.
2175a. Panek Z. — Silikaty, 1979, 23, S. 97.
2176. Panish M. В., Arthur J. R. — J. Chem. Thermor
dyn., 1970, 2, p. 299.
2177. Pankratz L. В., Kelley K. K. — U. S. Bur. Mines,
Rept Invest., 1963, N 6198.
2178. Pannetier G., Goudmand P.,Dessaux O., Arditi I. —
С r. Acad. sci., 1964, C258, p. 1201.
2179. Papamantellos P. — Z. Kristallogr., 1968, 126,
S. 143.
2180. Papatheodorou G. N., Capote M. A., Goates S. -r
(Preprint).
2181. Papousek D. — Collect. Czech. Chem. Communs,,
1961, 26, s. 1901.
2182. Paridon L. J., MacWood G. E., Ни J. H. — J.
Phys. Chem., 1959, 63, p. 1998.
2183. Parker A. — Phys. Rev., 1934, 46, p. 301.
2184. Parker A. — Phys. Rev., 1935, 47, p. 349.
2185. Parker A. — Thermal properties of aqueous unir
univalent electrolytes. Washington, NSRDS —
NBS 2, 1965.
2186. Parker V. B. — J. Res. NBS, 1973, A77, p. 227.
2187. Parkinson W. H. — Proc Phys. Soc. London.
1961, 78, p. 705.
2188. Parks G. S., Kelley К. К. — J. Phys. Chem.,
1926, 30, p. 47.
2189. Parson J. L. — J. Chem. Phys., 1960, 33, p. I860,
2189a. Pasternack L., Dagdigian P. J. — J. Chem.1
Phys., 1977, 67, p. 3854.
2190. Patel M. M., Patel P. D. — Curr. Sci., 1967,,
36, p. 571.
2191. Patel M. M., Patel P. D. — Indian J. Phys.,
1968, 42, p. 254. r
2192. Patel M. M., Patel P. D. — Indian J. Phys.,
1968, 42, p. 419.
2193. Patel M. M., Patel P. D. — J. Phys. B: Atom.
and Mol. Phys., 1969, 2, p. 515.
2194. Patel M. M., Shah N. R. — Indian J. Pure
and Appl. Phys., 1970, 8, p. 681.
2195. Paton R. F., Almy G. M. — Phys. Rev., 1931,
37, p. 1710.
2196. Paul F. W., Knauss H. P. — Phys. Rev., 1938,
54, p. 1072.
2197. Paule R. С — High Temp. Sci., 1976, 8, p. 257.
2198. Paule R. C., Margrave J. L. — J. Phys. Chem.,
1963, 67, p. 1368.
2199. Pawlenko S. — Z. anorg. und allg. Chem., 1965,
340, S. 201.
459
Литература
2200. Pearse R. W. В. — Proc. Roy. Soc, 1928, A122,
p. 442.
2201. Pearson P. K., O'Neil S. V., Schaefer H. F. - J.
Chem. Phys., 1972, 56, p. 3938.
2202. Pearson W. B. A Handbook of Lattice Spacinos
and Structures of Metals and Alloys. L.: Pergamon
Press, 1958.
2203. Pelchowitch J. — Phillips Res. Repts, 1954, 9,
p. 42.
2203a. Pelissier M., Malrien J. P. — J. Chem. Phys.,
1977, 67, p. 5963.
22036. Pelissier M., Malrien J. P. — J. Mol. Spectrosc,
1979, 77, p. 322.
2204. Pendred D., Richards R. E. — Trans. Faraday
Soc, 1955, 51, p. 468.
2205. Perec M., Веска L. N. — J. Chem. Phys., 1965,
43, p. 721.
2206. Perec M., Веска L. N. — J. Chem. Phys., 1966,
44, p. 3149.
2207. Person С. С — Ann. chim. phys., 1851, 33, p. 448.
2208. Pesic D. S. — Proc. Phys. Soc. London, 1960,
76, p. 844.
2209. Pesit D. S. — Proc. Phys. Soc. London, 1964,
83, p. 885.
2210. Pesit D. S., Gaydon A. G. — Proc. Phys. Soc.
London, 1959, 73, p. 244.
2211. Peters C. R., Milberg M. E. — Acta crystallogr.,
1964, 17, p. 229.
2212. Petersen E. — Z. phys. Chem. Leipzig, 1889, 4,
S. 384.
2213. Petersen E. — Z. phys. Chem. Leipzig, 1890, 5,
S. 259.
2214. Peterson D. Т., Colburn R. P. — J. Phys. Chem.,
1966, 70, p. 468.
2215. Peterson D. Т., Fattore V. G. — J. Phys. Chem.,
1961, 65, p. 2062.
2216. Peterson D. Т., Hinkebein J. A. — J. Phys. Chem.,
1959, 63, p. 1360.
2217. Peterson D. Т., Hutchison J. F. — J. Chem.
and Eng. Data, 1970, 15, p. 320.
2218. Peterson D. Т., Indig M. — J. Amer. Chem. Soc,
1960, 82, p. 5645.
2219. Petit G., Delbove F. — С. г. Acad. sci., 1962,
254, p. 1388.
2220. Petrikaln A., Hochberg J. — Z. Phys., 1933, 86,
S. 214.
2221. Petty F., Wang J. L. F., Steiger R. P., Harland
P. W., Franklin J. L., Margrave J.L.— High Temp.
Sci., 1973, 5, p. 25.
2222. Peyerimhoff S. D., Buenker P. J. — J. Chem.
Phys., 1968, 49, p. 312.
2222a. PhanfXuan D., Castanet R., Laffitte M. —
Rev. interu. hautes temp, et refract., 1974, 11,
p. 285.
2223. Phillips N. E. — Phys. Rev., 1959, 114, p. 676.
2224. Phongsatha A., Cyvin S. — Spectrosc. Lett., 1974,
7, p. 365.
2224a. Phongsatha A., Cyvin S. J. — Spectrosc. Lett.,
1975, 8, p. 91.
2225. Pickering L. — J. Chem. Soc, 1888, 53, p. 865.
2226. Pickering S. U. — J. Chem. Soc, 1885, 47, p. 100.
2227. Pickering S. U. — J. Chem. Soc, 1886, 49, p. 260.
2228 Pickering S. U. — J. Chem. Soc, 1888, 53, p. 865.
2229. Pilling N. B. -Phys. Rev., 1921, 18, p. 362.
2230. Pistorius С W. F. T. — J. Chem. Phys., 1959,
31, p. 1454.
2231. Pistorius С W. F. Т., Boeyens J. C. A., Clark
J. В.— High Temp. — High Pressur., 1969, 1,
p. 41.
2232. Pistorius С W. F. Т., Clark J. B. — Phys. Rev.,
1968, 173, p. 692.
2233. Pitzer K. S., Smith W. V., Latimer W. M. — J.
Amer. Chem. Soc, 1938, 60, p. 1826.
2234. Planckaert A. A., Sauvagean P., Sandorfy C. —
Chem. Phys. Lett., 1973, 20, p. 170.
2235. Plante E. R., Schreyer C. H. — J. Res. NBS,
1966, A70, p. 253.
2236. Plato W. — Z. phys. Chem. Leipzig, 1907, 58,
S. 350.
2237. Ploog K. — J. Cryst. Growth, 1974, 24—25, p. 197.
2238. Podszus E. — Z. anorg. und allg. Chem., 1933,
211, S. 41.
2239. Pohland E. — Z. anorg. und allg. Chem., 1931,
201, S. 282.
2240. Pokol Gy., Gal S., Tomor K., Domokos L. — In:
Thermal Anal. Proc. 4th Intern. Conf. Thermal.
Anal. Budapest, 1975, vol. 1, p. 479.
2241. Poland D. E., Margrave L., Green J. W. — J.
Chem. and Eng. Data, 1962, 7, p. 389.
2242. Pollitzer F. — Z. Elektrochem., 1913, 19, S.
513.
2242a. Pomeroy W. — Phys. Rev., 1927, 29, p. 59.
2243. Pomposiello M. C, Mondirola O'Brieux de. —
J. Mol. Struct., 1972, 11, p. 191.
2244. Pong R. G. S., Shirk A. E., Shirk J. S. — In:
Intern. Conf. Matrix. Isolat. Spectrosc. West
Berlin, 1977, June, 21—24, p. 161.
2245. Pong R. G. S., Shirk A. E., Shirk J. S. — J.
Mol. Spectrosc, 1977, 66, p. 35.
2245a. Pong R. G. S., Shirk A. E., Shirk J. S. — J.
Chem. Phys., 1979, 70, p. 525.
2246. Pong R. G. S., Stachnik R. A., Shirk A. E., Shirk
J. S. — J. Chem. Phys., 1975, 63, p. 1525.
2247. Popkie H. F. — J. Chem. Phys., 1971, 54, p. 4597.
2248. Pople J. A., Beveridge D. L. Approximate
molecular orbital theory. N. Y.: McGraw-Hill, 1970.
2249. Pople J. A., Beveridge D. L., Dobosh P. A. — J.
Chem. Phys., 1967, 47, p. 2026.
2250. Pople J. A., Segal G. A. — J. Chem. Phys., 1966,
44, p. 3289.
2251. Porter В., Brown E. A. — J. Amer. Ceram. Soc,
1962, 45, p. 49.
2252. Porter R. F. — J. Chem. Phys., 1960, 33, p. 951.
2253. Porter R. F., Bidinosti D. В., Watterson V. F. —
J. Chem. Phys., 1962, 36, p. 2104.
2254. Porter R. F., Chupka W., Inghram M. G. — J.
Chem. Phys., 1955, 23, p. 1347.
2255. Porter R. F., Gupta S. K. — J. Phys. Chem.,
1964, 68, p. 280.
2255a. Porter R. F., Gupta S. K. — J. Phys. Chem.,
1964, 68. p. 2732.
2256. Porter R. F., Schissel P., Inghram M. G. — J.
Chem. Phys., 1955, 23, p. 339.
2257. Porter R. F., Sholette W. P. — J. Chem. Phys.,
1962, 37, p. 198.
2258. Porter R. F., Wason S. K. — J. Phys. Chem.,
1965, 69, p. 2208.
2259. Porter R. F., Zeller E. E. — J. Chem. Phys.,
1960, 33, p. 858.
2260. Posnjak E. — Amer. J. Sci., 1938, 35, p. 247.
2261. Potter N. D., Boyer M. H., Ju F., iHildenbrand
D. L., Murad E., Hall W. F. — U. S. Aeronutro-
nic Division PHILCO, Final Techn. Rept
AD-715567, N U-4859., 1970, Aug, 31.
2262. Powers W. D., Blalock G. C. Enthalpies and
specific heats of alkali and alkaline earth hydroxides
at high temperatures. U. S. Atomic Energy Comm.,
1954. ORNL-1653. 48 p. (см. Kelley К. К. — Bull.
Bur. Mines (USA), I960, N 584).
460
Литература
2263. Poynor P. С. — Thesis. Nashville (Tenn.), 1967. 2292.
2264. Poynor P. C, Innes K. K., Ginter M. L. — J.
Mol. Spectrosc, 1967, 23, p. 237. 2292a,
2264a. PrasadR.,VenugopalV.,IyerP.N., SoodD.D. —
J. Chem. Thermodyn., 1978, 10, p. 135. 2293.
22646. Prasad S. C, Narayan M. K. — Indian J. Pure
and Appl. Phys., 1969, 7, p. 413И 2294.
2265. Prasad S. C, Narayan M. K. — Indian J. Phys.,
1969, 43, p. 205. 2295.
2266. Predel B. — Z. Metallk., 1964, 55, S. 97.
2267. Prescott C. #., Hincke W. B. — J. Amer. Chem. 2296.
Soc, 1927, 49, p. 2753.
2268. Prescott C. H., Hincke W. B. — J. Amer. Chem. 2297.
Soc, 1928, 50, p. 3228.
2269. Prewitt С. Т., Shanon R. D. — Acta crystallogr., 2298.
1968, B24, p. 869. 2299.
2270. Price W. С — J. Chem. Phys., 1948, 16, p. 894.
2271. Price W. C, Fraser R. D. В., Robinson T. S., 2300.
Longuet-Higgins H. C. — Discuss. Faraday Soc,
1950, 9, p. 131. 2301.
2271a. Price W., Passmore Т., Roessler D. — Discuss.
Faraday Soc, 1963, 35, p. 201. 2301a
2272. Pringle W. C, Meizer A. L. — J. Chem. Phys., 2302.
1972, 57, p. 2920.
2273. Prophet U., Stull D. R. — J. Chem. and Eng. 2302a
Data, 1963, 8, p. 78.
2274. Prosen E. J., Johnson W. H., Pergiel F. Y. — J. 2303.
Res. NBS, 1958, 61, p. 247. 2303a
2275. Prosen E. J., Johnson W. II., Pergiel F. Y. — J.
Res. NBS, 1959, 62, p. 43. 2304.
2276. Pruett J. G., Zare R. W. — J. Chem. Phys., 1975,
62, p. 2050. 2305.
2277. Puri S. N., Mohan H. — Curr. Sci., 1974, 43, p. 442.
2278. Puri S. N., Mohan H. — Curr. Sci., 1975, 44, 2306.
p. 152.
2279. Puri S. N., Mohan H. — Acta Cienc. Indica, 2307.
1976, 2, p. 369.
2280. Puri S. TV., Mohan H. — Pramana, 1975, 4, p. 171. 2308.
2281. Puri S. TV., Mohan H. — Indian J. Pure and
Appl. Phys., 1976, 14, p. 512. 2309.
2282. Quist A. S., Bates J. В., Boyd G. E. — J. Chem.
Phys., 1971, 54, p. 4896. 2310.
2283. Quist A. S., Bates J. В., Boyd G. E. — 3. Chem.
Phys., 1971, 55, p. 2836. 2311.
2283a. Rabeneck H., Schdfer H. — Z. anorg. und allg.
Chem., 1973, 395, S. 69. 2312.
2284. Radovanov S., Simic M., Vukanovic V. — Beitr.
Plasmaphys., 1976, 16, S. 345. 2313.
2285. Ram R. S. — Spectrosc. Lett., 1976, 9, p. 435.
2285a. Ram R. S. — Opt. pura у appl., 1973, 6, p. 38. 2314.
22856. Ram R. S., Rai S. В., Upadhya K. N. —
Pramana, 1979, 13, p. 149. 2315.
2286. Ramaswamy K. L. — Proc. Indian Acad. Sci.,
1935, A2, p. 364. 2316.
2287. Ramaswamy K. L. — Proc. Indian Acad. Sci.,
1935, A2, p. 630. 2317.
2288. Ramaswamy K., Jayaraman L. — Indian J. Phys.,
1975, 49, p. 1. 2318.
2288a. Rambidi N. G., Stepanov N. F., Simkin V. Y.,
Zhilinskii B. I., Dement'ev A. I. — In: Eight 2319.
Austin Symp. on gas phase molecular structure.
Austin (Tex.), 1980. 2320.
2289. Randall M., Chang K. S. — J. Amer. Chem. Soc,
1928, 50, p. 1535. 2321.
2289a. Randall S. P. — Ph. D. Thesis. Univ. Wiscon- 2322.
sin, 1968; Diss. Abstrs., 1969, B30, p. 107.
2290. Randall S. P., Margrave J. L. — J. Inorg. and 2323.
Nucl. Chem., 1960, 16, p. 29.
2291. Randall S. P., Greene F. Т., Margrave J. L. — 2324.
J. Phys. Chem., 1959, 63, p. 758.
Rao B. D., Motzfeldt K. — Acta chem. scand.,
1970, 24, p. 2796.
Rao B. S., Dadape V. V. — Indian J. Chem.
1970, 8, p. 1004.
Rao D. В., Dadape V. V. — J. Phys. Chem.,
1966, 70, p. 1349.
Rao J., Rao P. T. — Indian J. Phys., 1955,
29, p. 20.
Rao K. M., Rao P. T. — Indian J. Pure and
Appl. Phys., 1965, 3, p. 177.
Rao K. V. R. В., Suryanarayana V. — Indian J,
Phys., 1972, 46, p. 479.
Rao M. L. P., Rao D. V. K., Rao P. T. —
Spectrosc. Lett., 1976, 9, p. 101.
Rao P. T. — Indian J. Phys., 1949, 23, p. 393.
Rao P. Т., Tiruvenganna P. — Indian J. Phys.,
1950, 24, p. 434.
Rao V. S., Rao P. T. — Indian J. Phys., 1963,
37, p. 640.
Rapoport E., Pistorius С W. — J. Geophys,
Res., 1967, 12, p. 6353.
. Rassa M. — Ann. Chim., 1953, 8, p. 755.
Ratkje S. K., Rytter E. — J. Phys. Chem., 1974,
78, p. 1499.
. Raugh E. G., Ackermann R. J. — J. Chem,
Phys., 1976, 64, p. 1862.
Raw С J. G. — J. Chem. Phys., 1961, 34, p. 1452,
. Raw C. J. G. — J. S. Afr. Chem. Inst., 1955,
8, p. 25.
Rayne J. A. — J. Phys. and Chem. Solids, 1958,
7, p. 268.
Raziunas V., Macur G. J., Katz S. — J. Chem,
Phys., 1963, 39, p. 1161.
Raziunas V., Macur G. /., Katz S. — J. Chem,
Phys., 1965, 42, p. 2634.
Reardon E. J., Langmuir D. — Amer. J. Sci.,
1974, 274, p. 599.
Recker K., Leckebusch R. — J. Cryst. Growth,
1969, 5, p. 125.
Reddy B. R. K., Rao P. T. — Curr. Sei., 1970,
39, p. 509.
Reddy B. R. K., Rao P. T. — J. Phys. B: Atom,
and Mol. Phys., 1970, 3, p. 1008.
Reddy B.R. K., Reddy Y.P., Ashrafunnisa R. P. —
Curr. Sci., 1971, 40, p. 317.
Reddy B. R. K., Reddy Y. P., Rao P. T. — Curr,
Sci., 1970, 39, p. 485.
Reddy B. R. K., Reddy Y. P., Rao P. T. — J.
Phys. B: Atom, and Mol. Phys., 1970, 3, L 1.
Reddy B. R. K., Reddy Y. P., Rao P. T. — J.
Phys. B: Atom, and Mol. Phys., 1971, 4, p. 574.
Reddy B. R. K., Reddy Y. P., Rao С G. R.,
Rao P. T. — Curr. Sci., 1971, 40, p. 186.
Reddy S. P., Rao P. T. — Canad. J. Phys., 1957,
35, p. 912.
Reddy Y. P., Rao P. T. — Indian J. Pure and
Appl. Phys., 1966, 4, p. 251.
Reddy Y. P., Rao P. T. — Curr. Sci., 1967, 36,
p. 399.
Reddy Y. P., Rao P. T. — Indian J. Pure and
Appl. Phys., 1968, 6, p. 181.
Reddy Y. P., Rao P. T. — J. Phys. B: Atom,
and Mol. Phys., 1968, 1, p. 482.
Reeber M. D. — Phys. Rev. Lett., 1960, 4, p. 198,
Reeves E. M., Garton W. R. S., Bass A. — Proc.
Phys. Soc. London, 1965, 86, p. 1077.
Reid R. W., Sugden Т. М. — Discuss. Faraday
Soc, 1962, 33, p. 213.
Reilly J. /., Wiswall R. H. — Inorg. Chem.,
1967, 6, p. 2220.
461
Литература
2325. Reilly J. J., Wiswall R. H. — Inorg. Chem., 2361.
1968, 7, p. 2254.
2326. Remy—Cennete P. — C. r. Acad. sci., 1929, 189, 2361a
p. 579. 23616
2326a. Renes R. A., MacGillavry С. Н. — Recueil trav. 2362.
chim., 1945, 64, p. 275.
23266. Renner T. — Z. anorg. und allg. Chem., 1959, 2362a
298, S. 22. 2363.
2327. Rentzepis P., White D., Walsh P. N. — J. Phys. 2364.
Chem., 1960, 64, p. 1784. 2364a
2327a. Rey M. — С. г. Acad. sci., 1965, 260, p. 5528.
2328. Reynaud С - С. r. Acad. sci., 1967, C264, p. 1115. 2365.
2329. Riccardi R., Sinistri C, Campari G. V., Magi-
stris A. — Z. Naturforsch., 1970, A25, S. 781. 2366.
2330. Richards T. W., Burgess L. L. — J. Amer. Chem.
Soc, 1910, 32, p. 431. 2367.
2331. Richards T. W., Dole M. — J. Amer. Chem.
Soc, 1929, 51, p. 794. 2368.
2332. Richards T. W., Rowe A. W., Burgess L. L. — J.
Amer. Chem. Soc, 1910, 32, p. 1176.
2333. Richards T. W., Smyth С P. — J. Amer. Chem. 2369.
Soc, 1922, 44, p. 524.
2334. Richards W. G., Verhaegen G., Moser С. М. — J. 2370.
Chem. Phys., 1966, 45, p. 3226. 2371.
2335. Richardson M. J., Savill N. G. — Thermochim. 2372.
acta, 1975, 12, p. 221. 2373.
2336. Ridgeway E. E. — Trans. Amer. Electrochem.
Soc, 1934, 66, p. 117. 2374.
2337. Rifkin E. В., Kerr E. C, Johnston II. L. — J.
Amer. Chem. Soc, 1953, 75, p. 785. 2375.
2338. Rigden J. S., Koski W. S. — J. Amer. Chem.
Soc, 1961, 83, p. 3037. 2376.
2339. Riley B. — Rev. intern, hautes temp, et refract.,
1966, 3, p. 327. 2377.
2340. Rinck E. - С. г. Acad. sci., 1931, 192, p. 421.
2341. Rinck E. — Ann. chim. (France), 1932, 18, p. 510. 2378.
2342. Rinck E. — C. r. Acad. sci., 1952, 234, p. 845.
2343. Ring M. A., Donnay J. D. II., Koski W. S. — 2379.
Inorg. Chem., 1962, 1, p. 109.
2344. Risberg G. — Ark. fys., 1965, 28, p. 381. 2380.
2345. Risberg G. —Ark. fys., 1967, 37, p. 231.
2346. Risberg P. — Ark. fys., 1955, 9, p. 483. 2381.
2347. Ritchie R. K., Lew 11. — Canad. J. Phys., 1965,
43, p. 1701. 2382.
2348. Robert C. — Helv. phys. acta., 1936, 9, p. 405.
2349. Robert C, Wehrli M. — Helv. phys. acta, 1935, 2383.
8, p. 322.
2349a. Roberts J. S., Laubengayer A. W. — J. Amer. 2384.
Chem. Soc, 1957, 79, p. 5895.
2350. Roberts L. M. — Proc. Phys. Soc London, 1957, 2385.
B70, p. 738.
2351. Robertson J. A. — Ph. D. Thesis. Cornell. Univ., 2385a
1944.
2352. Robinson D. W. — J. Mol. Spectrosc, 1963, 11, 2386.
p. 275.
2353. Robson H. E., Gilles P. W. — J. Phys. Chem., 2387.
1964, 68, p. 983.
2354. Rochester G. D. — Phys. Rev., 1939, 56, p. 305. 2388.
2354a. Roeser W. F., Hoffman J. I. — J. Res. NBS,
1934, 13, p. 673. 2388a
2355. Rogers I. —Amer. J. Sci., 1892, 43, p. 301.
2356. Rogers P. S., Tomlinson J. W., Richardson F. D. — 2389.
Metallurg. Soc. Conf., 1961, 8, p. 909.
2357. Roig R. A., Tondello G. — J. Phys. B: Atom. 2390.
and Mol. Phys., 1976, 9. p. 2373.
2358. Rolin M., Clausier M. — Rev. intern, hautes 2391.
temp, et refract. 1967, 4, p. 39.
2359. Roller P. S. — J. Phys. Chem., 1931, 35, p. 1132. 2392.
2360. Rommel M.,iSchultz A. — Ber. Bunsenges. phys.
Chem., 1977, 81, S. 139. 2392a
Rong R. С S., Stachnik R. A. — J. Chem. Phys.,
1975, 63, p. 1525.
Rosen B. — Phys. Rev., 1945, 68, p. 124.
Rosen B. — Physica, 1946, 12, p. 184.
Rosenbaum E. D. — J. Chem. Phys., 1940, 8,
p. 643.
. Rosenqvist T. — Trans. AIME, 1951, 191, p. 535.
Rosenthaler E. — Helv. phys. acta, 1940, 13, p. 355.
Rosenwaks S. — J. Chem. Phys., 1976, 65, p. 3668.
. Rosenwaks S., Stecle R. E., Broida H. P. — J.
Chem. Phys., 1975, 63, p. 1963.
Rosenwaks S., Steele R. E., Broida H. P. —
Chem. Phys. Lett., 1976, 38, p. 121.
Rosicky /., Kmonickova S. — Collect. Czech.
Chem. Communs, 1976, 41, p. 3350.
Rosseinsky D. R. — Trans. Faraday Soc, 1958,
54, p. 116.
Rossini F.D., Wagman D. D., Evans W. H., Le-
vine S., Jafje J. Selected values of chemical thermo-
dynamic properties: Circ. 500. Wash.: NBS, 1952.
Rotenberg S., Schaefer H. F. — J. Amer. Chem.
Soc, 1973, 95, p. 2095.
Roth W. A. — J. prakt. Chem., 1941, 158, p. 117.
Roth W. A. — Z. Elektroohem., 1942, 48, S. 267.
Roth W. A. — Z. Naturforsch., 1946, 1, S. 574.
Roth W. A., Becker G. — Z. phys. Chem.
Leipzig, 1932, A159, S. 1.
Roth W. A., Berendt H., Wirths G. — Z. Elektro-
chem., 1941, 47, S. 185.
Roth W. A., Bertram W. W. — Z. Elektrochem.,
1929, 35, S. 297.
Roth V/. A., Borger E. — Berichte, 1937, B70,
S. 48.
Roth W. A., Borger E. — Z. Elektrochem., 1938,
44, S. 540.
Roth W. A., Borger E., Bertram A. ~ Berichte,
1937, B70, S. 971.
Roth W. A., Borger E., Siemonsen II. — Z. anorg.
und allg. Chem., 1938, 239, S. 321.
Roth W. A., Buchner A. — Z. Elektrochem.,
1934, 40, S. 87.
Roth W. A., Chall P. — Z. Elektrochem., 1928,
34, S. 185.
Roth W. A., Meichsner A.—Z. Elektrochem.,
1932, 38, S. 87.
Roth W. A., Meyer I., Zeumer H. — Z. anorg.
und allg. Chem., 1933, 214, S. 309.
Roth W. A., Meyer I., Zeumer H. — Z. anorg.
und ailg. chem., 1934, 216, S. 303.
Roth W. A., Mutter D. — Z. phys. Chem.
Leipzig, 1929, A144, S. 253.
. Roth W. A., Troitzsch H. — Z. angew Chem.,
1936, 49, S. 198.
Roth W. A., Werths G., Berendt H. — Z.
Elektrochem., 1942, 48, S. 264.
Roth W. A., Wolf U. — Z. Elektrochem., 1940,
46, S. 232.
Roth W. A., Wolf U., Fritz O. — Z. Elektrochem.,
1940, 46, S. 42.
. Roussel В., Chapput A., Fleury G.—J. Mol.
Struct., 1976, 31, p. 371.
Rowe J. J., Morey G. W., Hansen J. D. — J.
Inorg. und Nucl. Chem., 1965, 27, p. 53.
Rowe J. /., Morey G. W., Silber С. С. — J. Inorg.
and Nucl. Chem., 1967, 29, p. 925.
Rowlinson H. C, Barrow R. F. — Proc. Phys.
Soc. London, 1953, 66, p. 437.
Rowlinson H. C, Barrow R. F. — Proc. Phys.
Soc. London, 1953, A66, p. 771.
. Roy D. — Indian J. Phys., 1939, 13, p. 231.
462
Литература
2393. Roy D. M., Roy R. — Amer. J. Sci., 1957, 255,
p. 574.
2394. Roy D. M., Roy R., Osborn E. F. — J. Amer.
Ceram. Soc, 1950, 33, p. 85.
2395. Roy D. M., Roy R., Osborn E. F. — Amer. J.
Sci., 1953, 251, p. 337.
2396. Roy D. M., Roy R., Osborn E. F. — J. Amer.
Ceram. Soc, 1953, 36, p. 185.
2397. Roy N. N., Steward E. G. — Nature, 1969, 224,
p. 905.
2398. Roy R., Hill V. G., Osborn E. F. — J. Amer. Chem.
Soc, 1952, 74, p. 719.
2399. Rudberg E. — Phys. Rev., 1934, 46, p. 763.
2400. Rudberg E., Lempert J. — J. Chem. Phys., 1935,
3, p. 627.
.2401. Rudzitis E., Feder H. M., Hubbard W. N. — J.
Phys. Chem., 1964, 68, p. 2978.
2402. Rudzitis E., Feder H. M., Hubbard W. N. — Inorg.
Chem., 1967, 6, p. 1716.
Z403. Ruff O. — Z. Elektrochem., 1918, 24, S. 157.
2404. Ruff O. — Z. anorg. und allg. Chem., 1933, 46,
S. 1.
2405. RuffO., Hartmann H. — Z. anorg. und allg. Chem.,
1924, 133, S. 29.
2406. Ruff O., Jellinek E. — Z. anorg. und allg. Chem.,
1916, 97, S. 312.
2407. Ruff O., LeRoucher L. — Z. anorg. und allg.
Chem., 1934, 219, S. 376.
2408. Runow P. — J. Mater. Sci., 1972, 7, p. 499.
2409. Russell A. S., Martin K. E., Cochran С N. —
J. Amer. Chem. Soc, 1951, 73, p. 1466.
2410. Russell D. K., Kroll M., Reaudet R. A. — J.
Chem. Phys., 1977, 66, p. 1999.
2411. Russell D. K., Kroll M., Dows D. A., Beaudet
R. A.— Chem. Phys. Lett., 1973, 20, p. 153.
2412. Ryan R. R. — Diss. Abstrs, 1966, 26, N 3656.
2413. Ryan R. R., Hedberg K. — J. Chem. Phys., 1969,
50, p. 4986.
2414. Rytter E., Ratkje S. K. — Acta chem. scand.,
1975, A29, p. 565.
2414a. Saalfeld H. — Neues Jahrb. Mineral. Abh.,
1960, 95, S. 1.
2415. Saba W. G., Sterrett K. F., Craig R. S.,
Wallace W. E. — J. Amer. Chem. Soc, 1957, 79,
p. 3637.
2416. Sabatier G. — Bull. Soc. franc, miner, et crystal-
logr., 1954, 77, p. 1077.
2417. Sabatier M. — Ann. chim. phys., 1881, 22, p. 5.
2418. Sabatier M. — Bull. Soc. chim. France, 1891, 6,
p. 215.
2419. Sabatier M. — С. г. Acad. sci., 1891, 112, p. 862.
2420. Sabrowsky H. — Z. anorg. und allg. Chem., 1971,
381, S. 266.
2420a. Sabrowsky H., Murza J. — Naturwissenschaf-
ten, 1977, 64, S. 270.
2421. Sackur O. — Z. phys. Chem. Leipzig, 1912, 78,
S. 551.
2422. Sahni R. С — J. Chem. Phys., 1956, 25, p. 332.
2423. Saito H. — Sci. Repts Tohoku Imp. Univ. Ser.
I, 1927, 16, p. 37.
2424. Sakamoto Y. — J. Sci. Hiroshima Univ., 1954,
A17, p. 397.
2425. Sakamoto Y. — J. Sci. Hiroshima Univ., 1954,
A17, p. 407.
2425a. Sakurai K., Broida H. P. — J. Chem. Phys.,
1976, 65, p. 1138.
2426. Sakurai K., Johnson S. E., Broida H. P. — J.
Chem. Phys., 1970, 52, p. 1625.
2427. Samms J. A. C, Evans В. Е, -=• J. Appl. Chem.,
1967, 18, p. 5,
2428. Sandaram S. — Z. phys. Chem. (BRD), 1963,
36, S. 376.
2428a. Sander L. F., Healy G. M. — Trans. AIME,
1968, 242, p. 1039.
2429. Sandonnini С — Gazz. chim. ital., 1914, 44,
p. 290.
2430. Sana K. — Sci. Repts Tohoku Imp.' Univ., 1935,
24, p.' 240.
2431. Sano K. — Sci. Repts Tohoku Imp. Univ., 1936,
25, p. 745.
2432. Sano K. — J. Chem. Soc. Jap., 1939, 60, p. 758.
2432a. Sata Т., Sasamoto Т., Lee H. L., Maeda E. —
Rev. intern, hautes temp, et refract., 1978, 15,
p. 237. (Publ. 1979).
2433. Sata Т., Takahashi T. — In: Colloq. intern.
Centra Nat. Rech. Sci., P., 1972, N 205, p. 331.
2434. Sato S. — Bull. Inst. Phys. and Chem. Res.
Tokyo, 1935, 14, p. 862.
2435. Sato S. — Sci. Pap. Inst. Phys. and Chem. Res.,
1936, 29, p. 19.
2436. Sato S. — Sci. Pap. Inst. Phys. and Chem. Res.,
1936, 29, p. 53.
2437. Sato S. — Sci. Pap. Inst. Phys. and Chem. Res.,
1938, 34, p. 50.
2438. Sato S. — Sci. Pap. Inst. Phys. and Chem. Res.,
1938, 34, p. 399.
2439. Sato S. — Sci. Pap. Inst. Phys. and Chem. Res.,
1938, 34, p. 751.
2440. Sato Т., Атапо Т. — Kinzoku no kenkyu, 1934,
11, p. 305.
2440a. Sohaafsma A. — Z. Phys., 1932, 74, S. 254.
2441. Schaefer H. F. — J. Chem. Phys., 1971, 55, p. 176.
2442. Schaefer H. F., Ней Т. G. — J. Chem. Phys.,
1971, 54, p. 2573.
2443. Schaefer S. С — U. S. Nat. Techn. Inform.
Serv., PB Rept N 203731, 1971.
2443a. Schafer H., Binnewies M. — Z. anorg. und allg.
Chem., 1974, 410, S. 251.
24436. Schafer H., Schafer G., Weiss A.—Z. anorg.
und allg. Chem., 1963, 325, S. 77.
2443b. Schamps J. — Chem. Phys., 1973, 2, p. 352.
2444. Schamps J., Gandara G. — J. Mol. Spectrosc,
1976, 62, p. 80.
2445. Schamps J., Gandara G., Becart M. — Canad. J.
Spectrosc, 1969, 14, p. 13.
2446. Schamps J., Lefebvre-Brion H. — J. Chem. Phys.,
1972, 56, p. 573.
2447. Schedling J. A., Wein J. — Sitzungsber. Osterr.
Akad. Wiss. Math.-naturwiss. Kl. Abt. II, 1955,
164, S. 175.
2448. Scheer M. D. — J. Phys. Chem., 1957, 61, p. 1184.
2449. Scheer M.D. — J. Phys. Chem., 1958, 62, p. 490.
2450. Scheib W. — Z. Phys., 1930, 60, S. 74.
2451. Schenck R., Hammerschmidt F. — Z. anorg. und
allg. Chem., 1933, 210, S. 305.
2451a. Schenck R., Hammerschmidt F. — Z. anorg. und
allg. Chem., 1933, 210, S. 313.
2452. Schick H. L. Thermodynamics of Certain
Refractory Compounds. N. Y.; L.: Acad. Press, 1966.
Vol. 1/2.
2453. Schiemann G., Immel O., Braunling E. — Z.
phys. Chem. (BRD), 1963, 38, S. 56.
2453a. SchindlerP. — Helv. chim. acta, 1959, 42, p. 577.
2454. Schinke H., Sauerwald F. — Z. anorg. und allg.
Chem., 1960, 304, S. 25.
2455. Schins H. E., Van Wijk R. W. M., Dorpema B. —
Z. Metallk., 1971, 62, S. 330.
2456. Schissel P. O., Trulson О. С — J. Phys. Chem,
1962, 66, p. 1492.
463
Литература
2457. Schissel P. 0., Williams W. S. — Bull. Amer.
Phys. Soc, Ser. II, 1959, 4, N .3, p. 139.
2458. Schmahl N. G., Sieben P. — In: Phys. Chem.
Metallic. Solution and Intermettalic Compounds.
Chem. Publ. Co, Inc., 1960, vol. 1, N 4, p. 268.
2459. Schneider A., Esch U. — Z. Elektrochem., 1939,
45, S. 888.
2460. Schneider A., Gattow G. — Z. anorg. und allg.
Chem., 1954, 277, S. 41.
2461. Schneider A., Hilmer O. — Z. anorg. und allg.
Chem., 1956, 286, S. 97.
2462. Schneider A., Stoll E. K. — Z. Elektrochem., 1941,
47, S. 519.
2463. Schneider J. — Z. phys. Chem. (DDR), 1974,
255, S. 986.
2464. Schneider S. J. — J. Res. NBS, 1961, A65, p. 429.
2465. Schneider S. J. Compilation of the Melting
Points of the Metal Oxides. Monogr. N 68. Wash.:
NBS, 1963.
2466. Schneider S. J., Waring J. L. — J. Res. NBS,
1965, A67, p. 19.
2467. Schnockel Hg. — Z. anorg. und allg. Chem.,
1976, 424, S. 203.
2468. Schnockel Hg. — Z. Naturforsch.. 1976, B31,
S. 1291.
2468a. Schnockel Eg. — J. Mol. Struct., 1978, 50, p. 275.
24686. Schnockel Hg. — J. Mol. Struct., 1978, 50, p. 267.
2469. Schofield K. — Chem. Rev., 1967, 67, p. 707.
2470. Schomate C. H., Huffmann E. H. — J. Amer.
Chem. Soc, 1943, 65, p. 1625.
2471. Schottky W. F. — Z. phys. Chem. Leipzig, 1908,
64, S. 415.
2472. Schottmiller J. C, King A. J., Kanda T. A. —
J. Phys. Chem., 1958, 62, p. 1446.
2473. Schrier E. E., Clark H. M. — 3. Phys. Chem.,
1963, 67, p. 1259.
2473a. Schultz A., Siegel A. — J. Mol. Spectrosc, 1979,
77, p. 235.
2474. Schulz D. A., Searcy A. W. — J. Phys. Chem.,
1963, 67, p. 103.
2475. Schulze A. — Z. Metallk., 1930, 22, S. 308.
2476. Schulze A. —Phys. Z., 1935, 36, S. 595.
2477. Schumacher E. E. — J. Amer. Chem. Soc, 1926,
48, p. 396.
2478. Schuman R., Garrett A. B. — J. Amer. «hem.
Soc, 1944, 66, p. 442.
2479. Schuman R., Garrett A. B. — J. Amer. Chem.,
Soc, 1945, 67, p. 2279.
2480. Schutte M. — Naturwissenschaften, 1953, 40,
S. 528.
2481. Schwartz E., Coblans H. — Z. phys. Chem.
Leipzig, 1936, A176, S. 430.
2482. Schwiete H. E., Hey H. —Z. anorg. und allg.
Chem., 1934, 217, S. 396.
2483. Schwiete H. E., Pranschke A. B. — Zement, 1935,
24, S. 593.
2484. Scruby R. E., Lacher J. Д., Park J. D. — J.
Chem. Phys., 1951, 19, p. 386.
2485. Searcy A. W. — U. S. Univ. CaL, Rad. Lab.
Rept, UCRL-1404, 1951.
2486. Searcy A. W., Myers С. Е. — J. Phys. Chem.,
1957, 61, p. 957.
2487. Seaton M. J. — J. Phys. B: Atom, and Mol.
Phys., 1976, 9, p. 3001.
2488. Seegmiller D. W., Fannin A. A., Olson D. S.,
King L. A. — J. Chem. and Eng. Data, 1972,
17, p. 295.
2489. Seekamp H. — Z. anorg. und allg. Chem., 1931,
195, S. 345.
2490. Segel S. L., Barnes K. G. — Bull. Amer. Phys.
Soc, 1956, 1, p. 282.
2490a. Seidel G., Keesom P. H. — Phys. Rev., 1958,
112, p. 1083.
2491. Sen M. K. — Indian J. Phys., 1936, 10, p. 429.
2491a. Sen M. K. — Indian J. Phys., 1937, 11, p. 251.
2492. Sense K. A., Snyder M. J., Clegg J. W. — J.
Phys. Chem., 1954, 58, p. 223.
2493. Sense K. A., Stone R. W. — J. Phys. Chem.,
1958, 62, p. 453.
2493a. Serebrennikov L. V., Osin S. В., Sekachev Yu. N.,
Maltsev A. A. — In: XIHth Eur. Congr.
Molecular Spectroscopy. Wroclaw, 1977, Abstrs, p. 521.
24936. Serebrennikov L. V., Sekachev Yu. N.,
Maltsev A. A. — High Temp. Sci. (in press.).
2494. Servons R. R., Clark H. M. — J. Ghem. Phys.,
1957, 26, p. 1175.
2495. Seshadri D. N. — Indian J. Technol., 1975, 13,
p. 19.
2496. Seshadri K. S., Nimon L. A., White D. — J. Mol.
Spectrosc, 1969, 30, p. 128.
2497. Seward R. — J. Amer. Chem. Soc, 1945, 67,
p. 1189.
2498. Seybolt A. U. — J. Electrochem. Soc, 1964,
111, p. 697.
2498a. Seyed S. M. A., Vitse P. — J. Appl. Crystal-
logr., 1978, 11, p. 292.
2499. Shah S. G. — Indian J. Pure and Appl. Phys.,
1970, 8, p. 118.
2500. Shah S. G., Patel M. M., Darji A. B. — J. Phys.
B: Atom, and Mol. Phys., 1972, 5, p. 191.
2501. Shan S. G., Patel M. M., Darji A. B. — J. Phys.
B: Atom, and Mol. Phys., 1973, 6, p. 1344.
2502. Sharma D. — Astrophys. J., 1951, 113, p. 210.
2503. Sharma D. — Astrophys. J., 1951, 113, p. 219.
2504. Shaw R. W., Mapother D. E., Hopkins D. С —
Phys. Rev., 1960, 120, p. 88.
2505. Sheldon E. A., King A. J. — Acta crystallogr.,
1953, 6, p. 100.
2506. Shen Q. — Diss. Abstrs, 1974, B34, N 3735.
2507. Shepp A., Bauer S. H. — J. Amer. Chem. Soc,
1954, 76, p. 265.
2508. Shibata L., Terasaki Y. — J. Chem. Soc. Jap.
Pure Chem. Sec, 1936, 57, p. 208.
2509. Shibata Z., Terasaki Y. — J. Chem. Soc. Jap.
Pure Chem. Sec, 1936, 57, p. 1212.
2509a. Shin C, Criss С M. — J. Chem. Thermodyn.
1979, 11, p. 663.
2510. Shiozaki I., Sato T. — J. Phys. Soc. Jap., 1970,
29, p. 259.
2511. Shirk J. S., Shim A.E. — J. Chem. Phys., 1976,
64, p. 910.
2512. Sholotte W. P., Porter R. F. — J. Chem. Phys.,
1963, 67, p. 177.
2512a. Shomate С. Н., Cook О. К. — J. Amer. Chem.
Soc, 1964, 68, p. 2140.
2513. Shomate C. H., Huffman E. H. — J. Amer. Chem.
Soc, 1943, 65, p. 1625.
2514. Shomate С H., Naylor B. F. — J. Amer. Chem.
Soc, 1945, 67, p. 72.
2515. Shor A. J., Smith W. Т., Jr., Bredig M. A. — J.
Phys. Chem., 1966, 70, p. 1511.
2515a. Shpil'rain E. E., Fomin V. A., Kagan D. N.,
Sokol G. F., Kachalov V. V., Uljanov S. N. —
High Temp.—High Pressur., 1977, 9, p. 49.
25156. Spil'rain E. E., Kagan D. N., Barkhatov L. S. —
High Temp. — High Pressur., 1972, 4, p. 605.
2516. Shukla M. M. — Indian J. Pure and Appl. Phys.,
1970, 8, p. 855.
464
Литература
2517. Shurman E., Trager H. — Arch. Eisenhuttenw.,
1961, 32, S. 397.
2517a. Sidorov L. N., Zhegulskaya N. A. — Intern. J.
Mass Spectrom. and Ion Phys., 1975, 17, p. 111.
2518. Siebert R. M., Hostetler P. B. — Amer. J. Sci.,
1977, 277, p. 716.
2519. Siegel B. — Inorg. chim. acta. Revs, 1968, 2,
p. 137.
2520. Siemonsen H. — Z. Elektrochem., 1951, 55, S. 327.
2521. Silberman E. — Spectrochim. acta, 1967, A23,
p. 2021.
2522. Sillen L. G. Stability Constants of Metal-Ion
Complexes. L.: 1964.
2523. Simmons J. D., McDonald J. K. — J. Mol. Spec-
trosc, 1972, 41, p. 584.
2524. Simon F., Swain R. S. — Z. phys. Chem.
Leipzig, 1935, B28, S. 189.
2525. Sinanoglu O., Pamuk H. O. — S. Amer. Chem.
Soc, 1973, 95, p. 5435.
2526. Singh J., Mohan H. — J. Phys. B: Atom, and
Mol. Phys., 1971, 4, p. 1395.
2527. Singh J., Mohan H. — Indian J. Pure and Appl.
Phys., 1973, 11, p. 918.
2528. Singh J., Mohan H. — Spectrosc. Lett., 1973,
6, p. 139.
2529. Singh /., Nair K. P. R., Rai D. K. — J. Mol.
Struct., 1970, 6, p. 328.
2530. Singh J., Nair K. P. R., Rai D. K. — J. Quant.
Spectrosc. and Radiat. Transfer, 1971, 11, p. 1577.
2531. Singh J., Tewari D. P., Mohan H. — Indian J.
Pure and Appl. Phys., 1971, 9, p. 269.
2531a. Singh M. — Proc. Indian Acad. Sci., 1970, A71,
p. 82.
2532. Singh M. — J. Phys. B: Atom, and Mol. Phys.,
1971, 4, p. 565.
2532a. Singh M. — J. Phys. B: Atom, and Mol. Phys.,
1973, 6, p. 521.
2533. Singh M. — J. Phys. B: Atom, and Mol. Phys.,
1973, 6, p. 1339.
2534. Singh M. — J. Phys. B: Atom, and Mol. Phys.,
1973, 6, p. 1917.
2534a. Singh M., Narasimham N. A. — J. Phys. B:
Atom, and Mol. Phys., 1969, 2, p. 119.
25346. Singh M., Saksena M. D. — Proc. Indian Acad.
Sci., 1973, A77, p. 139.
2535. Singh R. В., Rai D. K. — J. Quant. Sepctrosc.
and Radiat. Transfer, 1965, 5, p. 723.
2536. Sinke E. J., Pressley G. A., Baylis А. В.,
Stafford F. E. — J. Chem. Phys., 1964, 41, p. 2207.
2537. Sinke G. C, Walker L. C, Oetting F. L., Stu.ll
D. R. — J. Chem. Phys., 1967, 47, p. 2759.
2538. Skinner H. A., Bennett J. E., Pedley J. B. —
Pure and Appl. Chem., 1961, 2, p. 17.
2539. Skinner H. A., Smith N. B. — Trans. Faraday
Soc, 1953, 49, p. 601.
2540. Skinner H. A., Smith N. B. — Trans. Faraday
Soc, 1955, 51, p. 19.
2540a. Smakula A., Kalnajs J. — Phys. Rev., 1955,
99, p. 1737.
2541. Smakula A., Kalnajs J., Sils V. — J. Opt. Soc.
Amer., 1953, 43, p. 698.
2542. Smeggil J. G., Eick H. A. — Inorg. Chem.,
1971, 10, p. 1458.
2543. Smisko J., Mason L. S. — J. Amer. Chem. Soc,
1950, 72, p. 3679.
2544. Smith C. R. F. — U. S. Atom. Intern. Rept
NAA-SR-MEMO 2308, 1958.
2545. Smith C. R. F. — U. S. Rept Congr. Atom. Energy
Comm., NAA-SR-5286, 1960.
2546. Smith D., Dworkin A. S., Artsdalen E. R. van. —
J. Amer. Chem. Soc, 1955, 77, p. 2564.
2547. Smith D., Laurence R. W. — U. S. Aerojet Rept
N 1952. Aerojet-General Corp., 1961.
2548. Smith D. F., Jr. — J. Chem. Phys., 1973, 58,
p. 4776.
2549. Smith D. F., Jr. — Spectrochim. acta, 1974, A30,
p. 875.
2550. Smith D. F., Jr. — Spectrochim. acta, 1976, A32,
p. 319.
2551. Smith D. F., Gardner T. E., Letson В. В.,
Taylor A. R. — U. S. Bur. Mines. Rept Invest., 1963,
N 6316.
2552. Smith D. K., Cline С F., Austerman S. B. — Acta
crystallogr., 1965, 18, p. 393.
2552a. Smith F. J., Barrow R. F. — Trans. Faraday
Soc, 1955, 51, p. 1478.
25526. Smith F. /., Barrow R. F. — Trans. Faraday
Soc, 1958, 54, p. 826.
2553. Smith G. F. — Trans. Faraday Soc, 1962, 58,
p. 350.
2554. Smith J. F., Carlson O. N., Vest R. W. — J.
Electrochem. Soc, 1956, 103, p. 409.
2555. Smith J. F., Smythe R. L. — Acta met., 1959,
7, p. 261.
2556. Smith J. V., Chatterji D. — J. Amer. Ceram.
Soc, 1973, 56, p. 288.
2557. Smith /., Seshadri K. S., White D. — J. Mol.
Spectrosc, 1973, 45, p. 327.
2558. Smith P. L. — Philos. Mag., 1955, 46, p. 744.
2559. Smith S. H., Miller R. R. — J. Amer. Chem.
Soc, 1950, 72, p. 1452.
2560. Smith W. L., Mills I. M.—J. Chem. Phys.,
1964, 41, p. 1479.
2561. Smith A., Meyering J. L. — Z. Phys. Chem.
Leipzig, 1938, B41, S. 98.
2562. Smith A., Meyering J. L., Kamermans M. A. —
Proc. Kon. ned. akad. wetensch., 1931, 34, s. 1327.
2563. Smith A., Meyering J. L., Kamermans M. A. —
Proc. Kon. ned. akad. wetensch., 1932, 35, s. 193.
2564. Smyth F. II., Adams L. H. — J. Amer. Chem.
Soc, 1923, 45, p. 1167.
2565. Snelson A. — J. Phys. Chem., 1966, 70, p. 3208.
2565a. Snelson A. — J. Phys. Chem., 1967, 71, p. 3202.
2566. Snelson A. — J. Phys. Chem., 1968, 72, p. 250.
2567. Snelson A. — J. Phys. Chem., 1969, 73, p. 1919.
2568. Snelson A. — J. Phys. Chem., 1970, 74, p. 2574.
2569. Snelson A. — High Temp. Sci., 1972, 4, p. 141.
2570. Snelson A. — High Temp. Sci., 1972, 4, p. 318.
2571. Snelson A. — High Temp. Sci., 1973, 5, p. 77.
2571a. Snelson A., Cyvin B. N., Cyvin S. J. — Z. anorg.
und allg. Chem., 1974, 410, S. 206.
2572. Snider J. L., Nicol J. — Phys. Rev., 1957, 105,
p. 1242.
2573. Snipes H. P., Manly C, Ensor D. D. — J. Chem.
• and Eng. Data, 1975, 20, p. 287.
2573a. Snowden B. S., Jr. — Ph. D. Thesis. Vander-
bilt Univ., 1963; Diss. Abstrs, 1964, 24, p. 5030.
2574. Snyder P. E., Seltz H. — J. Amer. Chem. Soc.
1945, 67, p. 683.
2575. So S. P. — J. Mol. Struct., 1977, 39, p. 127.
2576. So S. P., Richards W. G. — J. Phys. B: Atom.
and Mol. Phys., 1974, 7, p. 1973.
2577. So S. P., Richards W. G. — Chem. Phys. Lett.,
1975, 32, p. 227.
2578. So S. P., Richards W. G. — Chem. Phys. Lett.,
1975, 32, p. 231.
2579. Somayajulu G. — J. Chem. Phys., 1960, 33, p. 1541.
2580. Sommer A. — Diss. Abstrs, 1963, 24, p. 98.
465
Литература
2581. Sommer A., Walsh P. N., White D. — J. Chem. 2609.
Phys., 1960, 33, p. 296.
2582. Sommer A., White ?>., Linevsky M. J., Mann 2609a,
D. E. — Spectrochem. acta, 1962, 18, p. 1376.
2583. Sommer A., White D., Linevsky M. J., Mann 2610.
D. E. — J. Chem. Phys., 1963, 38, p. 87.
2584. Sorrell С. А. — Amer. Miner., 1962, 47, p. 291. 2610a.
2585. Soulen J. R., Margrave J. L. — J. Amer. Chem.
Soc, 1956, 78, p. 2911. 2611.
2586. Souler J. R., Sthapitanonda P., Margrave J. L. —
J. Phys. Chem., 1955, 59, p. 132.
2587. Southard J. С — Ind, and. Eng. Chem., 1940, 2612.
32, p. 442.
2588. Southard J. С — J. Amer. Chem. Soc, 1941, 2612a,
63, p. 3147.
2589. Southard J. C, Royster P. H. — 1. Phys. Chem.,
1936, 40, p. 435. 2613.
2590. Spurr R. A., Chang S. — J. Chem. Phys., 1951,
19, p. 528. | 2614.
2591. Speiser R., Johnston H. L. — J. Amer. Chem.
Soc, 1953, 75, p. 1469. 2615.
2592. Speiser R., Naiditch S., Johnston H. L. — J. Amer.
Chem. Soc, 1950, 72, p. 2578. 2616.
2593. Spencer C, Lipscomb W. N. — J. Chem. Phys., 2617.
1958, 28, p. 355.
2594. Spencer H. M. — J. Chem. Phys., 1946, 14, 2618.
p. 729. 2619.
2595. SperosD. W., Woodhouse R. L. — J. Phys. Chem.,
1963, 67, p. 2164. 2620.
2595a. Spiridonov V. P., Gershikov A. G., Zasorin E. Z.,
Ivanov A. A., Ermolaeva L. I. — High Temp. 2621.
Sci. (in press).
25956. Spiridonov V. P., Zasorin E. Z. — In: Proc. 2622.
10th Mater. Res. Symp. Characterization High
Temp. Vapors and Gases/ Ed. J. W. Hastie. Wash.: 2623.
NBS Spec. Publ., 1979, N 561.
2596. Srivastava R. D., Farber M. — J. Phys. Chem., 2624.
1971, 75, p. 1760.
2597. Srivastava R. D., Farber M. — Trans. Faraday 2625.
Soc, 1971, 67, p. 2298.
2597a. Srivastava R. D., Farber M. — Chem. Rev., 2626.
1978, 78, p. 627.
2598. Srivastava R. D., Uy O. M., Farber M. — Trans. 2627.
Faraday Soc, 1971, 67, p. 2941.
2599. Srivastava R. D., Uy O. M., Farber M. — J. 2628.
Chem. Soc. Faraday Trans. II, 1972, 68, p. 1388.
2600. Srivastava R. D., Uy O. M., Farber M. — J. 2628a
Chem. Soc. Faraday Trans. 11, 1974, 70, p. 1033.
2601. Stackelberg M., Quairam F., Dressel J. — Z. 2629.
Elektrochem., 1937, 43, S. 14.
2602. Stackelberg M., Schorrenberg E., Paulus R., 2630.
Spiess K. F. — Z. phys. Chem. Leipzig, 1935,
A175, S. 127. 2630a
2603. Stackelberg M., Spiess K. F. — Z. phys. Chem.
Leipzig, 1935, A175, S. 140.
2604. Stafford F. E., Berkowitz J. — J. Chem. Phys.,
1964, 40, p. 2963.
2605. Stafford F. E., Pressley G. A., Baylis A. B. — 2632.
In: Mass Spectrometry in Inorganic Chemistry.
Advances in Chemistry, Series 72/Ed. R. F. Gould.
Wash.: Amer. Chem. Soc, 1968, p. 137. 2633.
2606. Stampfer J. F., Holley С E., Suttle T. F. — J.
Amer. Chem. Soc, 1960, 82, p. 3504. 2634.
2607. Staveley L. A. K., Linford R. G. — J. Chem.
Thermodyn., 1965, 1, p. 1. 2635.
2608. Steam A. E., Smith G. — J. Amer. Chem. Soc,
1920, 42, p. 16. 2636.
5608a. Stearns С A., Kohl F. J. — J. Phys. Chem., 2637.
1973, 77, p. 13.
Stearns C. A., Kohl F. /. — High Temp. Sci.,
1973, 5, p. 113.
, Steck S., Pressley G., Stafford F. — J. Phys.
Chem., 1969, 73, p. 1000.
Steiger R. P., Margrave J. L. — High Temp. —
High Pressur., 1973, 5, p. 471.
Steimle T. C, Domaille P. J., Harris D. O. —
J. Mol. Spectrosc, 1977, 68, p. 134.
Stern K. H., Weise E. L. High temperature
properties and decomposition of inorganic salts. Part
I. Sulfates. Wash., 1966, NSRDS-NBS-7.
Stern P. S., Kaldon U. — J. Chem. Phys., 1976,
64, p. 2002.
, Sterrett K. F., Blackburn D. #., Bestul А. В.,
Chang S. S., Herman J. — J. Res. NBS, 1965,
C69, p. 19.
Sterten A., Haugen S., Hamberg K. — Electro-
chim. acta, 1976, 21, p. 589.
Stevenson D. P. — J. Chem. Phys., 1940, 8,
p. 898.
Stevenson D. P., Schomaker V. — J. Amer. Chem.
Soc, 1942, 64, p. 2514.
Stitt F. — J. Chem. Phys., 1941, 9, p. 780.
Stock A., Heynemann H. — Berichte, 1909, 42,
S. 4088.
Stock A., Kuss E. — Berichte, 1914, 47, S. 3114.
Stock A., Pohland E. — Berichte, 1926, 59,
S. 2216.
Stock A., WibergE., Martini H. — Berichte, 1930,
63, S. 2927.
Stock A., Wierl R. — Z. anorg. und allg. Chem.,
1931, 203, S. 208.
Storms E., Mueller B. — J. Phys. Chem., 1977,
81, p. 318.
Stout J. W., Robil R. A. — J. Phys. Chem.,
1963, 67, p. 2248.
Stout N. D., Mar R. W., Boo O. J. W. — High
Temps Sci., 1973, 5, p. 241.
Streets D. G., Berkowitz J. — Chem. Phys. Lett.,
1976, 38, p. 475.
Stromme K. O. — Acta chem. scand., 1975, A29,
p. 105.
Strong M., Knauss H. — Phys. Rev., 1936, 49,
p. 740.
Striibel G. —¦ Neues Jahrb. Mineral. Monatsch.,
1965, N 3, p. 83.
. Stuart W. J., Price G. H. — J. Nucl. Mater.,
1964, 14, p. 417.
Stubbs M. E., Schufle J. A., Thompson A. J. —
J. Amer. Chem. Soc, 1952, 74, p. 6201.
Stull D. R. — Ind. and Eng. Chem., 1947, 39,
p. 517.
. Stull D. R., Hildenbrand D. L., Oetting F. L.,
Sinke G. С — J. Chem. and Eng. Data, 1970,
15, p. 52.
Stull D. R., McDonald R. A. — J. Amer. Chem.
Soc, 1955, 77, p. 5293.
Stull D. R., Sinke G. C. Thermodynamic
properties of the elements: Advances in chemistry. Wash.
Amer. Chem. Soc, 1956, Ser. N 18.
Su G. /., Chang С. Н. — J. Amer. Chem. Soc,
1946, 68, p. 1080.
Subbaram K. V., Rao D. R. — Indian J. Phys.,
1969, 43, p. 312.
Sugden T. M., Schofield K. — Trans. Faraday
Soc, 1966, 62, p. 566.
Sugden S. — J. Chem. Soc, 1929, p. 328.
Sulzmann K. G. P. — J. Electrochem. Soc. , 1974,
121, p. 1239.
466
Литература
2637а. Summers N. L., Tyrrell /. — J. Amer. Chem. 2668.
Soc, 1977, 99, p. 3960.
2638. Sundaram S. — Z. phys. Chem. (BRD), 1963, 2669.
36, S. 376. 2670.
.2639. Sutra G. — С. г. Acad. sci., 1952, 234, p. 1283.
2640. Sutton P., Bertoncini P., Das G., Gilbert F. L., 2671.
Wahl A. C. — Intern. J. Quant. Chem. Symp., 2672.
1970, N 3, pt2, p. 479.
2641. Sutton P., Bertoncini P., Das G., Gilbert F. L., 2673.
Wahl A. C. — Intern. J. Quant. Chem., 1970,
4, p. 633. 2674.
2642. Svirbely W. J., Selis S. M. -~ J. Phys. Chem.,
1954, 58, p. 33. 2675.
2643. Switkes E., Stevens R. M., Lipscomb W. N. —
J. Chem. Phys., 1969, 51, p. 5229. 2676.
2644. Syvernd A. TV. — J. Inorg. and Nucl. Chem.,
1976, 38, p. 2163. 2677.
2645. Tables internationales de constantes selectionnes.
17. Donnes specrtroscopiques relatives aux mole- 2678.
cules diatomiques/Ed. B. Rosen. Oxford etc.:
Pergamon Press, 1970.
2646. Takahashi G. — J. Chem. Soc. Jap. Chem. and 2679.
Ind. Chem. 1954, 57, p. 337.
2647. Takahashi M. — J. Electrochem. Soc. Jap., 1961, 2680.
29, p. 54.
2648- Takahashi T. — J. Chem. Soc. Jap., 1954, 57, 2681.
p. 337.
2649. Takahata K. — Chem. Phys. Lett., 1974, 26, 2682.
p. 557.
2650. Takeo #., Curl R. F. — J. Chem. Phys., 1972, 2683.
56, p. 4314.
.2651. Takezawa Т., Shimakura K., Ono A., Hishiyuma 2684.
Y. — J. Inst. Metals, 1968, 56, p. 94.
2652. Tamaru S., Siomi K. — Z. phys. Chem. Leipzig, 2685.
1932 A159 S 227.
2653. Tamaru S.,' Siomi K. — Z. phys. Chem. Leipzig, 2685a
1932, A161, S. 421.
2654. Tamaru S., Siomi K. — Z. phys. Chem. Leipzig, 2686.
1934, A171, S. 221.
2655. Tamaru S., Siomi K., Adati M. — Z. phys. Chem. 2686a
Leipzig, 1931, A157, S. 447. 2687.
2656. Tamaru S., Yokokawa T. — Bull. Chem. Soc. Jap.,
1975, 48, p. 2542. 2688.
2657. Tanabe I., Konno H., Sawada Y., Takahashi T. —
J. Electrochem. Soc. Jap. (Denki Kagaku), 1964, 2689.
32, p. 285.
2658. Tanaka H., Yamaguchi A., Morivama J. — J. 2690.
Jap. Inst. Metals,, 1971, 35, p. 1161.
2659. Tassilly E. — Ann. chim. phys., 1899, 17, p. 38. 2691.
2659a. Tarte P., Preudhomme J. — Spectrochim. acta,
1970, A26, p. 747. 2692.
2660. Tawde N. R., Patankar V. S. — Curr. Sci., 1936, 2692a
4, p. 869.
2661. Tazaki H. — J. Sci. Hiroshima Univ, 1940, A10, 2693.
p. 37.
2662. Tazaki H. — J. Sci. Hiroshima Univ., 1940, A10, 2694.
p. 109.
2662a. Taylor A. R., Gardner Т. Е. — U. S. Bur. Mines. 2695.
Rept Invest., 1965, N 6664.
2663. Taylor А. Д., Gardner T. E., Smith D. F. — U. S. 2696.
Bur. Mines. Rept Invest., 1963, N 6240. 2697.
2664. Taylor A. R., Letson В. В., Smith D. F. — U. S. 2698.
Bur. Mines. Rept Invest., 1966, N 6724. 2699.
2665. Taylor A. R., Smith D. F. — U. S. Bur Mines.
Rept Invest., 1962, N 5967. 2700.
2666. Taylor K., Wells L. S. — J. Res. NBS, 1938,
21, p. 133. 2700a
2666a. Taylor M. /. — J. Chem. Soc. A, 1970, p. 2812.
2667. Taylor N. W., Cole S. S. — J. Amer. Chem. 2701.
1934, 56., p. 1648.
Taylor R. — J. Amer. Ceram. Soc, 1962, 45,
p. 74.
Taylor R. С — Adv. Chem. Ser., 1964, 42, p. 59.
Taylor R. C, Emery A. R. — Spectrochim. acta,
1958, 10, p. 419.
Taylor W. J. — J. Chem. Phys., 1958, 28, p. 625.
TeaneyD. Т., Morussi V. L. — In: Colloq. Intern.
Centre Nat. Rech. Sci., P., 1970, N 180/2, p. 131.
Teital R. J., Kohen M. — J. Metals, 1949, 185,
p. 285.
Templeton С. С. — J. Chem. and Eng. Data, 1960,
5, p. 514.
Terenin A., Popov B. — Phys. Z. Sowjetunion,
1932, 2, p. 299.
Thakur R. L., Rock E. J., Pepinsky R. — Amer.
Miner., 1952, 37, p. 695.
Thakur S. N. — Indian J. Pure and Appl. Phys.,
1971, 9, p. 293.
The Chemistry and Metallurgy of Miscelaneous
Materials Thermodynamics./Ed. L. L. Quill. N. Y.
etc., 1950, Pap. 3—7.
Theard L. P., Hildenbrand D. L. — J. Chem.
Phys., 1964, 41, p. 3416.
Thiel A., Siebeneck H. — Z. anorg. und allg.
Chem., 1934, 220, S. 236.
Thoma R. E., Insley H., Fridman H. A.,
Weaver С F. — J. Phys. Chem., 1960, 64, p. 865.
Thomas D. M., Andrews L. — J. Mol. Spectrosc,
1974, 50, p. 220.
Thomasson C. V., Cunningham D. A. —• J. Sci.
Instrum., 1964, 41, p. 308.
Thompson С J., Sinke G. C, Stull D. R. — J.
Chem. and Eng. Data, 1962, 7, p. 380.
Thompson K. R. — High Temp. Sci., 1973, 5,
p. 62.
. Thompson M. K., Seyl R. G. — Trans.
Electrochem. Soc, 1933, 64, p. 321.
Thomsen J. Thermochemische Untersuchungen.
Leipzig: Verl. von J. A. Barth, 1882—1886.
, Thomson С — J. Chem. Phys., 1973, 58, p. 216.
Thomson C. — Chem. Phys. Lett., 1977, 46,
p. 138.
Thomson C, Brotchie D. A. — Chem. Phys.
Lett., 1972, 16, p. 573.
Thomson C, Brotchie D. A. — Theor. chim. acta,
1973, 32, p. 101.
Thomson C, Brotchie D. A. — Mol. Phys., 1974,
28, p. 301.
Thomson C, Wishart B. J. — Theor. chim. acta,
1974, 35, p. 267.
Thonstad J. — Canad. J. Chem., 1964, 42, p. 2739.
, Thonstad J. — Electrochim. acta, 1968, 13,
p. 449.
Thorvaldson Т., Brown W. G. — J. Amer. Chem.
Soc, 1930, 52, p. 80.
Thorvaldson Т., Brown W. G., Peaker C. R. —
J. Amer. Chem. Soc, 1930, 52, p. 910.
Thorvaldson Т., Brown W. G., Peaker C. R. — J.
Amer. Chem. Soc, 1929, 51, p. 2678.
Thrush B. A. — Nature, 1960, 186, p. 1044.
Thrush B. A. — Proc. Chem. Soc, 1960, p. 339.
Thunberg S. F. — Z. Phys., 1936, 100, S. 471.
Tiemann E., Grasshoff M., Hoeft J. — Z. Natur-
forsch., 1972, A27, S. 753.
Tiemann E., Bojaschewsky M., Sauter-Servaes Ch.,
Torring T. — Z. Naturforsch., :1974, A29, S. 1692.
. Tiemann E., Ryzlewicz Ch., Torring T. — Z.
Naturforsch., 1976, A31, S. 128.
Tiensuu V. H. — Diss. Abstrs, 1962, 23, N 5,
p. 1535.
467
Литератур»
2702. Tilk W. — Dissertation. Hannover, 1930. 2736.
2703. Tinland B. — J. Mol. Struct., 1969, 3, p. 244. 2737.
2704. Tttzky H. G. — Z. Phys., 1958, 151, S. 351. 2738.
2705. Tlahaut J. — Ann. chim. Ser. 12, 1952, 7, p. 632.
2705a. Tobisch J., Hase W. — Wiss. Z. Techn. Univ. 2738a
Dresden, 1971, 20, S. 441.
2706. Todd B. /., Miller В. В. — J. Amer. Chem. 2739.
Soc, 1946, 68, p. 530.
2707. Todd S. S. — J. Amer. Chem. Soc, 1949, 71, 2739a
p. 4115. 27396
2708. Tolmachev S. M., Bambidi N. G. — High Temp.
Sci., 1973, 5, p. 385. 2740.
2709. Tomkinson E. — Chem. News, 1921, 122, p. 243. 2741.
2710. Tomlin D. H. ~ Proc. Phys. Soc. London, 1954,
B67, p. 787. 2741a.
2711. Tomlinson J. W., Richardson F. D. — Metallurg.
Soc. Conf., 1961, 8, p. 909. 2742.
2712. Tomlinson J. W., Welch B. J. — J. Inorg. and
Nucl. Chem., 1966, 28, p. 2131. 2743.
2713. Topor L. — Rev. roum. chim., 1972, 17, s. 1503.
2714. Topor L., Moldoveanu I. — Rev. roum. chim., 2743a
1972, 17, s. 1705.
2715. Topor L., Moldoveanu I. — Rev. roum. chim., 2744.
1974, 19, s. 985.
2716. Torgeson D. В., Sahama T. G. — J. Amer. Chem. 2745.
Soc, 1948, 70, p. 2156.
2717. Torgeson D. В., Shomate С. H. — I. Amer. Chem. 2746.
Soc, 1947, 69, p. 2103.
2718. Torring Т., Tiemann E. — Z. Naturforsch., 1973, 2747.
A28, S. 1062.
2719. Touboul M., Bouaziz B. — С. г. Acad. sci., 1970, 2748.
C270, p. 1235.
2720. Touboul M., Marchand В., Tournoux M. — Bull. 2749.
Soc. chim. France, 1972, N 2, p. 570.
2721. Tournadre M. — С. г. Acad. sci., 1958, 246, 2749a
p. 947.
2721a. Tournoux M., Marchand R., Bouchama M. — 27496
С r. Acad. sci., 1970, C270, p. 1007.
2722. Trajmar S., Ewing G. E. — J. Chem. Phys., 2750.
1964, 40, p. 1170. 2751.
2723. Trajmar S., Ewing G. E. — Astrophys. J., 1965,
142, p. 77. 2752.
2724. Tranquard A., Coffy G., Boinon M. J. — Bull.
Soc. chim. France, 1969, N 8, p. 2608. 2753.
2724a. Tranquille M., Foussier M. — J. Chem. Soc.
Faraday Trans. II, 1980, 76, p. 26.
2725. Trautz M., Paschwer S. — J. prakt. Chem., 1929, 2754.
122, S. 147. 2755.
2726. Traverse J. P., Foex M. — High Temp.—High
Pressur., 1969, 1, p. 409. 2755a
2727. Treadwell W. D., Cohen A. — Helv. chim. acta,
1939, 22, p. 433. 2756.
2728. Treadwell W. D., Gyger A. — Helv. chim. acta,
1933, 16, p. 1214. 2757.
2729. Treadwell W. D., Sticker J. — Helv. chim. acta, 2758.
1953, 36, p. 1820.
2730. Treadwell W. D., Terebesi L. — Helv. chim. acta, 2759.
1932, 15, p. 1053. 2760.
2730a. Treadwell W. D., Terebisi L. — Helv. chim.
acta, 1933, 16, p. 922. 2761.
2731. Trefonas L., Lipscomb W. N. — J. Chem. Phys.,
1958, 28, p. 54. 2761a
2732. Troost L., Hautefeuille P. — C. r. Acad. sci., 2762.
1870, 70, p. 252.
2733. Troost L., Hautefeuille P. — Ann. chim. phys., 2763.
1876, 9, p. 70.
2734. Trzebiatowski W., Horyn B. — Bull. Acad. pol. 2764.
sci. Ser. sci. chim., 1965, 13, s. 315.
2735. Tschappat Ch., Pied R. — Helv. chim. acta, 1956, 2764a
39, p. 1427.
Tsuchiya R. — Denki Kagaku, 1948, 16, p. 162".
Tully J. С — J. Chem. Phys., 1973, 58, p. 1396.
Turkdogan E. Т., Bice В. В., Vinters J. V. — Met.
Trans., 1974, 5, p. 1527.
, Turley J. W., Rinn H. W. — Inorg. Chem.,.
1969, 8, p. 18.
Turner L. A., Harris W. T. — Phys. Rev., 1937,
52, p. 626.
Tyte D. C. — Nature, 1964, 202, p. 383.
Tyte D. С — Proc. Phys. Soc. London, 1967,
92, p. 1134.
Ulmer W. — Z. Naturforsch., 1972, A27, S. 133-
Ultee Casper J. — J. Chem. Phys., 1964, 40..
p. 3746.
Umeyama H., Morokuma K. — J. Amer. Слет-
вое, 1976, 98, p. 7208.
Umino S. — Sci. Repts Tohoku Univ., 1926, 15,.
p. 597.
Umino S. — Sci. Repts Tohoku Univ., 1927, 16,.
p. 775.
Uno T. — J. Iron and Steel Inst. Jap., 1951,.
37, N 1, p. 14.
Urry G., Wartik Т., Moore В., Schlesinger H. I. —
J. Amer. Chem. Soc, 1954, 76, p. 5293.
Uy О. М., Drowart J. — High Temp. Sci., 1970,
2, p. 293.
Uy O. M., Drowart J. — Trans. Faraday Soc.
1971, 67, p. 1293.
Uy O. M., Srivastava B. D., Farber M. — High
Temp. Sci., 1971, 3, p. 462.
Uy O. M., Srivastava R. D., Farber M. — High.
Temp. Sci., 1972, 4, p. 227.
Vajda E., Hargittai I., Tremmel J. — Inorg.
chim. acta, 1977, 25, p. 143.
. Valderrama N. /., Jacob K. T. — Thermochim..
acta, 1977, 21, p. 215.
. Valderrama N. /., Jacob К. Т. — J. Inorg. and:
Nucl. Chem., 1978, 40, p. 993.
Valentiner S. — Z. Metallk., 1940, 32, S. 244.
Valentiner S. — Z. anorg. und allg. Chem., 1954,.
277, S. 201.
Van Artsdalen E. R., Anderson K. P. — J. Amer..
Chem. Soc, 1951, 73, p. 579.
Van der Hurk J., Hollander Т., Alkemade C. —
J. Quant. Spectrosc. and Radiat. Transfer, 1973r
13, p. 273.
Vanderryn J. — J. Chem. Phys., 1959, 30, p. 331.
Vanderzee С E., Gier L. J. — J. Chem. Thermodyn.,
1974, 6, p. 441.
. Vanpee M., Kineyko W. R., Caruso R. —
Combust, and Flame, 1970, 14, p. 381.
Van Westenburg J. A. — Ph. D. Thesis. Iowa
State Univ., 1964.
Varet R. — Ann. chim. phys., 1896, 8, p. 240.
Vasko A., Srb J. ~ Czech. J. Phys., 1967, B17,.
p. 1110.
Veillard A. — Chem. Phys. Lett., 1969, 3, p. 128..
Veillard A., Levy В., Daudel B.,Gallais F. — Theor.
chim. acta, 1967, 8, p. 312.
Velden P. E. — Trans. Faraday Soc, 1967, 63t
p. 175.
. Venkateswaran С S. — Nature, 1937, 140, p. 151.
Verdonk A. H., Broersma A. — Thermochim. acta,
1973, 6, p. 95.
Verdonk A. H., Nedermeijer J., Laverman J. W. —
J. Chem. Thermodyn., 1975, 7, p. 1047.
Venkateswarlu K., Nagarajan A. — Acta phys.
pol., 1967, 32, s. 205.
. Verhaegen G., Colin В., Exsteen G., Drowart J. —-
Trans. Faraday Soc, 1965, 61, p. 1372.
468
Литература
2765. Verhaegen G., Drowart J. — J. Chem. Phys., 2795.
1962, 37, p. 1367.
2766. Verhaegen G., Richards W. G. — J. Chem. Phys., 2796.
1966, 45, p. 1828.
2766a. Verhaegen G., Richards W. G. — Proc. Phys.
Soc. London, 1967, 90, p. 579. 2797.
2767. Verhaegen G., Richards W. G., Moser С. М. — J.
Chem. Phys., 1967, 46, p. 160.
2768. Verhaegen G., Stafford E. F., Ackerman M., Dro- 2798.
wart J. — Nature, 1962, 193, p. 1280.
2769. Verhaegen G., Stafford F. E., Drowart J. — J. 2799.
Chem. Phys., 1964, 40, p. 1622.
2770. Veseth L. — Mol. Phys., 1973, 25, p. 333. 2800.
2771. Veseth L. — J. Mol. Spectrosc, 1976, 59, p. 51.
2772. Veseth L., Lofthus A. — J. Mol. Spectrosc, 1974, 2800a
49, p. 414.
2773. Vetter F. A., Kubashewski O. — Z. Elektrochem., 2801.
1953, 57. S. 243.
2774. Victor A. C, Douglas Т. В. — J. Res. NBS, 1963, 2802.
A67, p. 325.
2775. Villars D. S., Condon E. U. — Phys. Rev., 1930, 2803.
35, p. 1028.
2775a. Viola J. Т., Fannin A. A., King L. A., Seeg- 2804.
miller D. W. — J. Chem. and Eng. Data, 1978,
23, p. 118. 2805.
2776. Viola J. Т., Seegmiller D. W., Fannin A. A., 2806.
King L. A. — J. Chem. and. Eng. Data, 1977,
22, p. 367. 2807.
2777. Viswanath D. S. — J. Chem. and Eng. Data,
1966, 11, p. 453. 2808.
2777a. Viswanathan R. — J. Appl. Phys., 1975, 46,
p. 4086. 2809.
2778. Vizi B. — Veszpremi vegyipari egyet. kozl., 1966, 2810.
10, s. 205. 2811.
2779. Vizi B. — Veszpremi vegyipari egyet. kozl., 1966, 2812.
10, s. 349. " 2813.
2780. Vizi В., Cyvin S. — Acta chim. Acad. sci. hung.,
1973, 75, s. 377. 2814.
2781. Vogel R., Heumann T. — Arch. Eisenhiittenw.
1841, 15, S. 195. 2815.
2782. Vogt J. W. U. S. NASA N 63—22167. Cleveland
(Ohio): Thompson Ramo Wooldridge Inc., 1962. 2816.
2783. Volmer F. — Phys. Z., 1929, 30, S. 590.
2784. Vrieland G., Stull D. — J. Chem. and. Eng. Data, 2817.
1967, 12, p. 532.
2785. Vuillard G. — С. г. Acad. sci., 1963, 257, p. 3927. 2818.
2786. Wagman D. D. U. S. NBS Rept N 7437, 1962.
2787. Wagman D. D., Evans W. H., Parker V. В., Hal- 2819.
low J., Bailey S. M., Schumm В. Н. — In:
Selected values of chemical thermodynamic proper- 2820.
ties. Wash.: NBS Techn. Notes, 1965, N 270—1;
1966, N 270-2; 1968, N 270-3; 1969, N 270-4; 2821.
1971, N 270—5; 1971, N 270—6; 1973, N 270—7.
2788. Wagner E. L. — Theor. chim. acta, 1974, 32, p. 295. 2822.
2789. Wagner J. — Z. phys. Chem. Leipzig, 1943, 193,
S. 55. 2823.
2789a. Wagner K., Schdfer H. — Z. anorg. und allg.
Chem., 1979, 451, p. 67. 2824.
2790. Walker B. E., Grand J. A., Miller R. R. — J. Phys.
Chem., 1956, 60, p. 231. 2825.
2791. Walker B. E., Ewing С. Т., Miller R. R. — J.
Chem. and. Eng. Data, 1962, 7, p. 595. 2826.
2792. Walker T. E. H., Barrow R. F. — J. Phys. B:
Atom, and Mol. Phys., 1969, 2, p. 102. 2827
2792a. Wallwork S. C, Worrall I. J. — J. Chem. Soc,
1965, p. 1816. „„„„
2793. Walsh A. D.-J. Chem. Soc, 1953, p. 2260. 2828-
2794. Walsh A. D. — J. Chem. Soc, 1953, p. 2288.
2794a. Walter-Levy L., Laniepce J. — С. г. Acad. sci., 2829.
1965, 261, p. 3789.
Walters О. H., Barratt S. — Proc. Roy. Soc,
1928, A118, p. 120.
Ward J. J., Hussey M. A. — In: Third Symp.
on Combustion and Flame and Explosion
Phenomena. Baltimore, 1949, p. 599.
Warrier K. M., Girijavallabhan С. Р., Warrier
M. K. R. — Indian J. Pure and Appl. Phys.,
1973, 11, p. 221.
Wartenberg H. von. — Z. Elektrochem., 1909, 15,
S. 866.
Wartenberg H. von, — Z. Elektrochem., 1913, 19,
S. 482.
Wartenberg H. von. — Z. anorg. und allg. chem.,
1942, 249, S. 100.
. Wartenberg H. von. — Z. anorg. und allg. Chem.,
1943, 252, S. 136.
Wartenberg H. von, Bosse O. — Z. Elektrochem.,
1922, 28, S. 384.
Wartenberg H. von, Reusch H. J. — Z. anorg.
und allg. Chem., 1932, 207, S. 1.
Wartenberg H. von, Reusch H. J., Saran E. —
Z. anorg. und allg. Chem., 1937, 230, S. 257.
Wartenberg H. von, Witzel G. — Z. Elektrochem.,
1919, 25, S. 209.
Wason S. K. — Diss. Abstrs, 1966, 26, p. 5749.
Wason S* K., Porter R. F. — J. Phys. Chem.,
1964, 68, p. 1443.
Watanabe H., Kubo M. — J. Amer. Chem. Soc,
1960, 82, p. 2428.
Watanabe H., Totani Т., Nakagawa Т., Kubo M. —
Spectrochim. acta, 1960, 16, p. 1076.
Watson W. W. — Phys. Rev., 1928, 32, p. C00.
Watson W. W. — Phys. Rev., 1933, 43, p. 9.
Watson W. W. — Phys. Rev., 1935, 47, p. 27.
Watson W. W. — Phys. Rev., 1935, 47, p. 213.
Watson W. W., Humphreys R. F. — Phys. Rev.,
1937, 52, p. 318.
Watson W. W., Parker A. S. — Phys. Rev., 1931,
37, p. 167.
Watson W. W., Rudnick P. — Astrophys. J.,
1926, 63, p. 20.
Watson W. W., Rudnick P. — Phys. Rev., 1927,
29, p. 413.
Watson W. W., Fredrickson W. R. — Phys. Rev.,
1932, 39, p. 765.
Watson W. W., Fredrickson W. R., Hogan M. E. —
Phys. Rev., 1936, 49, p. 150.
Watson W. W., Shambon A. — Phys. Rev., 1936,
50, p. 607.
Watson W. W., Weber R. L. — Phys. Rev., 1935,
48, p. 732.
Waxman M., Hilsenrath /., Chen W. T. — J.
Chem. Phys., 1973, 58, p. 3692.
Weaver J. R., Shore S. G., Parry R. W. — J.
Chem. Phys., 1958, 29, p. 1.
Webb A. N., Neu J. Т., Pitzer K. S. — J. Chem.
Phys., 1949, 17, p. 1007.
Webb D. U., Justice B. H., Prophet H. — J.
Chem. Thermodyn., 1969, 1, p. 227.
Wehrli M., Miescher E. — Helv. phys. acta,
1933, 6, p. 457.
Wehrli M., Miescher E. — Helv. phys. acta, 1934,
7, p. 298.
Weiss P. — Z. Erzbergbau und Metallhiittenwesea,
1950, 3, S. 241.
Wells L. S., Taylor K. — J. Res. NBS, 1937,
19, p. 215.
Welti D., Barrow R. F. — Nature, 1951, 168,
p. 161.
469
Литератур»)
2830. Welti D., Barrow R. F. — Proc. Phys. Soc.
London, 1952, A65, p. 629.
2831. Weltner W., Jr., Warn J. R. W. — J. Chem.
Phys., 1962, 37, p. 292.
2832. Wenk W. — Helv. phys. acta, 1941, 14, p. 355.
2833. Wentink Т., Jr., Tiensuu V. H. — J. Chem.
Phys., 1958, 28, p. 826.
2834. WentorfR. H. — J. Phys. Chem., 1959, 63, p. 1934.
2835. Werli M., Miescher E. — Helv. phys. acta, 1934,
7, p. 298.
2836. West E. D., Ginnings D. С — J. Phys. Chem.,
1957, 61, p. 1573.
2837. West E. D., Ginnings D. С — J. Res. NBS, 1958,
60, p. 309.
2838. Westrum E. F. In: Thermodynamic and Transport
Properties of Gases, Liquids and Solids. N. Y.,
1962, p. 275.
2838a. Wetherill J. P. — Metals and Alloys, 1935, 6,
p. 153.
2839. Wharton L., BergR. A., Klemperer W. — J. Chem.
Phys., 1963, 39, p. 2023.
2840. Wharton L., Kaufman M., Klemperer W. — J.
Chem. Phys., 1962, 37, p. 621.
2841. Wharton L., Klemperer W. — J. Chem. Phys.,
1963, 39, p. 240.
2842. White D., Colder G. V., Hemple S. — J. Chem.
Phys., 1973, 59, p. 6645.
2843. WhiteD., MannD. E., WalshP. N., Sommer A. —
J. Chem. Phys., 1960, 32, p. 481.
2844. White D., Mann D. E., Walsh P. N., Sommer
A. — J. Chem. Phys., 1960, 32, p. 488.
2845. White D., Walsh P. TV., Goldstein II. W., Dever
D. F. — J. Phys. Chem., 1961, 65, p. 1404.
2846. WhiteD., Walsh P. TV., MannD. E. — J. Chem.
Phys., 1958, 28, p. 508.
2847. Whiting G. #., Turnes W. E. S. — J. Soc. Glass
Technol., 1930, 14, p. 409.
2848. Wiberg E. — Naturwissenschaften, 1948, 35, S. 182.
2849. Wiberg E. — Naturwissenschaften, 1948, 35, S. 218.
2850. Wiberg E., Bolz A. — Berichte, 1940, 73, S. 209.
2851. Widmann G. — Thermochim. acta, 1975, 11, p. 331.
2851a. Wight C. A., Willner H., Andrews L. — J. Mol.
Spectrosc, 1978, 72, p. 332.
2852. Wijn H. W. de. — Physica, 1965, 31, p. 1193.
2853. Wilkes G. B. — J. Amer. Ceram. Soc, 1932, 15,
p. 72.
2854. Wilkins R. L., Altmann R. L. — J. Chem. Phys.,
1959, 31, p. 337.
2855. Wilkins R. L., Lodvig R. M., Greene S. A. —
In: Symp. Combust., 8th, Pasadena (Calif.), 1960.
p. 375-388. (Publ. 1962).
2856. Wilkinson P. G. — Astrophys. J., 1963, 138,
p. 778.
2857. Will G., Ploog K. — Nature, 1974, 251, p. 406.
2857a. Wille P. J., Tittle O. F. — J. Petrol., 1960,
1, p. 1.
2858. Willmann G. — Z. anal. Chem., 1973, 266, S. 21.
2859. Wilson J. H., McGee H. A., Jr. — J. Chem.
Phys., 1967, 46, p. 1444.
2860. Winaus J. G., Fraslie H. M. — Phys. Rev., 1947,
71, p. 137.
2861. Winchell P. — Nature, 1965, 206, p. 1252.
2862. Wise S. S., Margrave J. L., Altman R. L. —
J. Phys. Chem., 1960, 64, p. 915.
2863. Wise S. S., Margrave J. L., Feder H. M., Hub-
bard W. N. — J. Phys. Chem., 1966, 70, p. 7.
2864. Witt W. P., Barrow R. F. — Trans. Faraday
Soc, 1959, 55, p. 730.
2865. Witting К. Е. — Z. Metallk., 1952, 43, S. 158.
2866. Wohler L., Hofer K. — Z. Elektrochem., 1934f
40, S. 19.
2867. Wolf G. — С. г. Acad. sci., 1935, 200, p. 1203.
2868. Wolfe D. E,, Humphrey G. L. — J. Mol. Struct.,
1969, 3, p. 293.
2869. Wolfman J. — Osterr.-Ungar. Z. Zuckering, 1896,
25, S. 988.
2870. Wu C. H. — Ber. Kernforschungsanlage Jiilich,.
1971, N 739.
2871. Wast F., Meuthen A., Durrer R. — Forsch. Arb.
Ver. Deut. Ing., 1918, 204, S. 41.
2872. Wyse F. С — Diss. Abstrs В., 1973, 33, p. 4439.
2873. Wyse F. C, Gordy W. — J. Chem. Phys., 1972,
56, p. 2130.
2874. Wyse F. C, Gordy W., Pearson E. F. — J. Chem.
Phys., 1970, 52, p. 3887.
2875. Wyss J. C, Broida H. P. — J. Mol. Spectrosc,
1976, 59, p. 235.
2875a. Yadav В. Д., Лаг 5. В., Rai D. K. — Canada
J. Phys., 1979, 57, p. 496.
2876. Yamaguchi A., Kato E. — .T. Ceram. Soc. Jap.,
1972, 80, p. 337.
2877. Yamaoka S., Fukunage O., Saito S. — J. Amer..
Ceram. Soc, 1970, 53, p. 179.
2878. Yamdagni R. — D. Ph. Thesis. Univ. of
Allahabad, India, 1954.
2879. Yamdagni R. — Curr. Sci., 1970, 39, p. 34.
2880. Yang Y. S., Shirk J. S. — J. Mol. Spectrosc
1975, 54, p. 39.
2881. Yates J. H., Pitzer R. M. — J. Chem. Phys.,
1977, 66, p. 3592.
2882. Yates R. E., Greenbaum M. A., Farber M.—
J. Phys. Chem., 1964, 68, p. 2682.
2883. Yeatts be Roy В., Marshall W. L. — J. Phys-
Chem., 1967, 71, p. 2641.
2884. Yeatts Le Roy В., Marshall W. L. — J. Phys.
Chem., 1969, 73, p. 81.
2885. Yeh H. I. C, Ragle J. L. — .T. Phys., Chem.,
1968, 72, p. 3688.
2885a. Yokohama Т., Kleppa O. J. — J. Phys., Chem.,
1964, 68, p. 3246.
2886. Yost D. M., De Vault D., Anderson Т. Е., Las-
settre E. N. — J. Chem. Phys., 1938, 6, p. 424..
2886a. Young D. A., Alder B. J. — Phys. Rev., 1971,
A3, p. 364.
2887. Young F. E. — J. Amer. Chem. Soc, 1944, 66,
p. 773.
2888. Young W. A. — J. Phys. Chem., 1960, 64, p..
1003.
2889. Younger P., Winaus J. G. — J. Mol. Spectrosc,
1960, 4, p. 23.
2890. Zachariasen W. H.—Z. Kristallogr., 1934, 88,.
S. 150.
2891. Zachariasen W. H. — Acta crystallogr., 1954, 7,
p. 305.
2891a. Zachariasen W. H. — Acta crystallogr., 1963,.
16, p. 385.
28916. Zalesinski E., Zulinski R. — Bull, intern. Acad.
pol., A, 1928, s. 479.
2892. Zare R. N. — Ber. Bunsenges. phys. Chem., 1974,.
78, S. 153.
2893. Zaucer M., Azman A. — Croat, chem. acta, 1971,
43, p. 139.
2894. Zauli C. — In: 16th Intern. Congr. Pure and Appl.
Chem., Paris, 1957, Sec. on Inorg. Chem., p. 447.
2895. Zavitsanos P. D. — General Electric Techn.
Inform., 1968. NTIS Doc. N AD 713715.
2896. Zawadzki J. — Z. anorg. und allg. Chem., 1932,,
205, S. 180.
470
Литература
2897.
2898.
2899.
2900.
2901.
2902.
2903.
Zawadzki /., Bretsznajder S. — Z. phys. Chem.
Leipzig, 1933, B22, S. 79.
Zawadzki /., Syryczynski Z. — Rocz. chem., 1930,
10, s. 715.
Zeegers P., Townsend W., Winefordner J. — Spec-
trochim. acta, 1969, B24, p. 243.
Zeeman P. B. — Phys. Rev., 1950, 80, p. 902.
Zeeman P. B. — Canad. J. Phys., 1951, 29, p. 336.
Zeeman P. B. — Canad. J. Phys., 1954, 32, p. 9.
~'hys.,
Zeeman P. В., Ritter G. J. — Canad. J. Phys.
1954, 32, p. 555.
2904. Zehe M. J. — Diss. Abstr., 1977, B37, p. 6162.
2904a. Zehe M. J., Lynch D. A., Kelsall B. J.,
Carlson K. D. — J. Phys. Chem., 1979, 83, p. 656.
2905. Zeise H. Thermodynamic. Bd. HI/1. Tabellen;
Bd. Ш/2, Grafische Darstellungen und Literatur.
Leipzig: S. Hirzel Verl., 1954—1957.
2906. Zhidomirov G. M., Chuvylkin N. D. — Chem.
Phys. Lett., 1972, 14, p. 52.
2907. Zimens К. Е. — Z. phys. Chem. Leipzig, 1937,
B37, S. 241.
2908. Zmbov K. F. — Chem. Phys. Lett., 1969, 4, p. 191.
2908a. Zuegel M. A. — J. Electrochem. Soc, 1965,
112, p. 1153.
Дополнительная литература
1096. Дементьев А. И., Павловский И. В.,
Степанов Н. Ф. и др. (в печати).
257а. Митин А. В., Дементьев А. И., Сафонов А. А.,
Хрустов В. Ф. — Журн. физ. хим., 1981 (в
печати).
340а. Рамбиди Н. Г., Степанов Н. Ф. и др. — Журн.
структур, хим., 1981, 22, с. 29.
943а. Demidov А. V., Zasorin E.Z., Gershikov A. G.
et al. — J. Mol. Struct, (in press).
1221a. Glascock B. L. — J. Amer. Chem. Soc, 1910, 32,
p. 1222.
2595b. Spiridonov V. P., Gershikov A. G. et al. — High.
Temp. Sci. (in press).
2595г. Spiridonov V. P., Gershikov A. G. et al. — Eighth
Austin Sympos. onMolec. Structure. Austin (USA).
1980, p. 97.
2595д. Spiridonov V. P., Gershikov A. G., Altman А. В.,
Romanov G. V., Ivanov A. A. — Chem. Phys.
Lett., 1981, 77, p. 41.
2753a. Van der Vorst C. P. J. M., Verschoor G. C,
Maaskant W. J. A. — Acta crystallogr., 1978,
R34, p. 3333.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ИНДИВИДУАЛЬНЫХ ВЕЩЕСТВ
Том III, Книга I
Утверждено к печати Институтом высоких температур Академии наук СССР
Редакторы издательства В. П. Сироткина, Н. Н. Лезнова. Художник В. Г. Виноградов
Художественный редактор Т. П. Поленова. Технический редактор Э. Л. Кунина
Корректоры Н. Б. Габасова, Н. И. Казарина
ИБ № 12591
Сдано в набор 04.11.80. Подписано к печати 13.07.81. Т—22112. Формат 84ХЮ81/16. Бумага типографская № 1.
Гарнитура обыкновенная. Печать высокая. Усл. печ. л. 49,56. Усл. кр.-отт. 51,24. Уч.-изд. л. 56,5. Тираж 7100 экз.
Тип. зак. 1928. Цена III т. (кн. 1 п 2) 8 р. 70 к.
Издательство «Наука». 117864 ГСП-7. Москва В-485, Профсоюзная ул., 90.
Ордена Трудового Красного Знамени Первая типография издательства «Наука»
199034, Ленинград, В-34, 9-я линия, д. 12.
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
к «Термодинамическим свойствам индивидуальных веществ»
Страница
34
55
70
1S2
183
199
244
325
391
Строка
т.
22 св.
Табл. 1.7
5 стб.
11 сн.
Табл. 9.2
5 стб.
8 стб.
3 св.
11 сн.
18 св.
Табл. 10.2
5 стб.
6 стб.
7 стб.
8 стб.
Табл. 12.4
6 стб.
7 стб.
Табл. 13.22
7 стб.
Табл. III.2
5 стб.
Напечатано
1, кн. 1
807
СсоА
12 ,
126,5802
298,15
298,15
298
298,15
—133,0993
423,24
-5,44816
298,15
740
780
1470,0
10,129 385
Должно быть
870
DodA
126,971
310
310
320
320
—6,976
154,055
—1,3351
320
74
78
—1470,0
10,0129 385
Страница
7
251
258
259
289
294
308
321
328
338
346
Строка
т.
24 сн.
7 св.
26 св.
7 сн.
15 сн.
Табл. 17.13
4 стб.
Табл. 18.1
3 стб.
4 стб.
5 стб.
6 стб.
7 стб.
18 св.
Табл. 19.1
8 стб.
»
» -
9 стб.
13 св.
28 сн.
2 св.
Напечатано
2, кн. 1
Фирсовой
100
6000
802,005
GeS, г
74,8
17,0
134,0
82,0
71,25
36,06
100
0,533
-1,134 а
6537,505б
—2923,933
2500
2000
100
2500
Должно быть
Фирсановой
298,15
5800
802,884
GeS, к
-74,8
17,33
134,1
178,05
62,005
53,817
298,15
0,553
-1,134
6537,505 а
—2923,933 6
3000
3200
298,15
3000