/






























































































































































































































































































Author: Маурах М.А. Митин Б. С
Tags: цветные металлы в целом химия химическая промышленность обработка металлов издательство металлургия
Year: 1979




Text
УДК 669.2-31
Рецензент: докт. техн. наук, профессор Б. X. Хан
УДК 669.2-31
Жидкие тугоплавкие окислы. М а у р а х М. А., М и т и н Б. С.
«Металлургия», 1979. 288 с.
В книге впервые обобщены, систематизированы и критически
рассмотрены данные о физико-химических и теплофизических свойствах
жидких тугоплавких окислов. Дан теоретический анализ процессов
взаимодействия жидких окислов с твердыми высокотемпературными
материалами. Описаны методики, аппаратура и результаты
оригинальных исследований. Особое внимание уделено рассмотрению
свойств окиси алюминия, широко применяющейся в
микроэлектронике, ракетной и ядерной технике, оптике высокого разрешения,
приборостроении.
Книга рассчитана на научных работников институтов и
предприятий черной и цветной металлургии, занятых исследованием процессов
с участием жидких тугоплавких окислов, а также на инженеров-кри-
сталлохимиков и специалистов по созданию и использованию
высокотемпературных и оптических материалов. Ил. 78. Табл. 30. Библи-
огр. список: 375 назв.
31002—194
М 73—79 2605000000
040(01)—79
© Издательство «Металлургия», 1979
ПРЕДИСЛОВИЕ
За последние 10—15 лет в связи с развитием техники
высоких температур значительно возрос интерес к
таким процессам с участием жидких тугоплавких окислов,
как фасонное литье, получение окисных монокристаллов,
нанесение окисных покрытий и др. Особую роль в
современной технике играют процессы образования жидких
окислов в продуктах горения ракетного топлива с
металлическими добавками, частичное расплавление окислов
при производстве керметов методами порошковой
металлургии. Анализ и усовершенствование этих процессов
требует надежных данных по свойствам окислов в
жидком состоянии.
В настоящей книге предпринята попытка обобщить и
систематизировать экспериментальные данные по
физико-химическим (включая смачивание) и теплофизичес-
ким свойствам жидких тугоплавких окислов,
выполненные советскими и зарубежными исследователями.
Широко представлены материалы многолетних исследований
тугоплавких окислов, проведенных авторами на кафедре
«Высокотемпературные материалы» Московского
института стали и сплавов. Последовательно изложены
сведения о температуре плавления, плотности, поверхностном
натяжении, вязкости, а также об электрических,
диффузионных и теплофизических характеристиках жидких
тугоплавких окислов. Критически рассмотрены методики
определения указанных свойств, даны некоторые
теоретические обобщения, касающиеся изменения свойств в
результате межфазного взаимодействия.
Авторы использовали в тексте те системы единиц,
которые пока широко распространены в соответствующих
областях науки. Для пересчета метрических и
внесистемных единиц в единицы Международной системы (СИ)
следует пользоваться следующей таблицей перевода:
Метрические и внесистемные единицы Единицы СИ
Единицы силы
J килограмм-сила (кгс) ....... 9,80665Н
1 дина (дин) ^ ]0-5 Ц
Единицы давления
1 атмосфера физическая (ат) 101325 Па
1 мм Рт- ст 133,322 Па
1*
3
Продолжение
Единицы работы и энергии
1 кал физическая 4Д87 Дж
1 эрг . Ю-7 Дж
Единицы удельной энтропии
1 кал/(г-К) 4,1868 Дж/(кг-К)
1 кал/(моль-К) 4,1868 Дж/
/(моль-К)
Единицы удельной теплоемкости
1 ккал/(кг-К) 4186,8 ДжДкг-К)
Единицы динамической вязкости
1 пуаз (П) 0,1 Па-с
В проведении исследований принимали участие
студенты и аспиранты Московкого института стали и
сплавов И. И. Богданов, С. А. Лисовский, Ю. А. Нагибин,
В. В. Самотейкин, Е. Ф. Гриц, Ю. С. Анисимов и др.,
которым авторы приносят благодарность.
Все главы книги написаны авторами совместно; в
работе над главами III и V принимали участие канд. физ.-
мат. наук С. А. Лисовский и канд. техн. наук В. В.
Самотейкин.
Авторы с благодарностью приняли советы и
замечания докт. техн. наук, проф. Б. X. Хана, сделанные им при
рецензировании рукописи.
ВВЕДЕНИЕ
Для современной научно-технической революции
характерно как бурное развитие новой техники, так и
интенсификация технологических процессов в
традиционных отраслях промышленности. Успехи в том и в другом
направлениях чаще всего определяются наличием
материалов, выдерживающих постоянно возрастающие
параметры процессов — температуру, давление, скорость,
механические усилия и т. п. Особенно заметно возросли
требования к высокотемпературным материалам. В
области высокотемпературной техники научная и
конструкторская мысль постоянно обгоняла и обгоняет
реальные возможности существующих
высокотемпературных материалов; отсутствие материалов,
удовлетворяющих высоким современным требованиям, сдерживает
реализацию многих научных достижений в новой
технике, энергетике, металлургии.
Интенсивное исследование высокотемпературных
материалов в течение последних десятилетий привело к
возникновению нового направления —
высокотемпературного материаловедения, — основная цель которого
состоит в разработке и изучении материалов,
предназначенных для эксплуатации при температурах,
превышающих 1200° С. Рассмотрим, какое место занимают
окислы в высокотемпературном материаловедении.
В настоящее время существует четыре класса
высокотемпературных материалов.
1. Однородные высокотемпературные материалы,
подразделяемые на металлические (сплавы на основе
тугоплавких металлов) и неметаллические (окислы,
графит, карбиды и другие тугоплавкие соединения).
2. Композиционные высокотемпературные
материалы, включающие керметы (в том числе типа окисел —
металл, графит — металл); металлы, упрочненные окис-
ными и другими волокнами; материалы с защитными
(в том числе окисными) покрытиями; пористые
материалы, пропитанные металлами; материалы, армированные
тонкой проволокой; слоистые материалы. Качество и
свойства композиционных материалов определяются не
только индивидуальными свойствами упрочняющих и
связывающих фаз, но и в значительной мере
взаимодействием между ними.
5
3. Абляционные теплозащитные
высокотемпературные материалы, теряющие во время эксплуатации при
высоких температурах часть своей массы в виде газа или
пара. Сюда входят и окисные материалы, например те,
которые имеют большую величину теплоты плавления и
испарения, или те, которые при нагреве реагируют с
другими составляющими со значительным поглощением
тепла.
4. Охлаждаемые высокотемпературные материалы —
материалы с внутренним или внешним охлаждением.
Если материалы с внутренним охлаждением трудно
представить себе из окислов, так как они должны
прежде всего обладать высокой теплопроводностью, то
материалы с внешним охлаждением могут быть изготовлены
из пористых тугоплавких окислов. В этом случае
надежность материалов при эксплуатации зависит от
характера взаимодействия окисной основы материала с жидким
охлаждающим реагентом, в качестве которого обычно
выступают расплавленные металлы.
Даже из такого краткого рассмотрения
высокотемпературных материалов следует, что роль окислов в
современном высокотемпературном материаловедении
достаточно велика: тугоплавкие окислы уже используются или
принципиально могут использоваться в производстве
высокотемпературных материалов всех классов. И это
естественно, поскольку окислы представляют собой
единственный материал, обладающий исключительно
высокой стойкостью против окисления при высоких
температурах. Все другие материалы имеют недостаточную
стойкость при работе в окислительной среде или на воздухе.
К сожалению, окислам присущи и недостатки такие,
как хрупкость, низкая теплопроводность, низкая
термостойкость, что часто ограничивает их применение. Здесь
необходимы дальнейшие исследования, направленные
как на улучшение комплекса свойств окислов, так и на
расширение областей их применения при высоких
температурах.
К тугоплавким окислам принято относить окисные
соединения, температура плавления которых превышает
1800° С. Всего простых соединений кислорода с
металлами типа МехОУ1 в которых металл проявляет высшую
валентность, насчитывается более 40. Среди них окислы
некоторых очень редких лантанидов и актинидов,
окислы металлов, расположенных в периодической таблице
б
элементов за лантанидами и актинидами, а также
окислы, которые при температурах, близких к температуре
плавления, сублимируют или диссоциируют с
интенсивным испарением продуктов диссоциации. Изучение
таких окислов затруднено и не получило достаточного
развития.
Если рассматривать тугоплавкие окислы с позиции
их практической значимости, то среди них наиболее
важными оказываются следующие 20 окислов металлов:
двуокись тория Th02; двуокись гафния НЮ2; окись
магния MgO; двуокись урана U02; двуокись церия Се02;
двуокись циркония Zr02; окись кальция СаО; окись
бериллия ВеО; окись хрома Сг203; окись скандия Sc203;
окись иттрия Y203; окись лантана La203; окись самария
Sm203; окись гадолиния Gd203; окись алюминия А1203;
окись бария ВаО; закись никеля NiO; окись цинка ZnO;
пятиокись тантала Та205; закись кобальта СоО.
Учитывая возрастающую роль окислов в
высокотемпературном материаловедении, можно выделить три
научно-технические области, связанные с жидкими
окислами и требующие изучения.
Первая включает производство изделий из
тугоплавких окислов. К числу процессов получения изделий из
окислов можно отнести фасонное литье, выращивание
окисных монокристаллов направленной
кристаллизацией, получение окисных волокнистых материалов,
нанесение окисных защргтных покрытий, спекание порошковых
изделий в присутствии жидкой фазы. Во всех
перечисленных технологических процессах участвуют
тугоплавкие окислы в жидком состоянии.
Вторая рассматривает свойства изделий
(механические, теплофизические, оптические и др.), зависящие от
структуры вещества. Если изделие формируется через
жидкую фазу, то свойства жидкой фазы и параметры
процесса перехода окислов из жидкого состояния в
твердое будут во многом определять протекание
кристаллизационных процессов и в итоге структуру изделия.
Наконец, третья устанавливает совместимость
высокотемпературных материалов с жидкими тугоплавкими
окислами. Совместимость материалов приходится
учитывать при эксплуатации окисных покрытий на
металлических или графитовых материалах; металлоокисных кер-
метов и композиционных материалов, упрочненных окис-
■1ыми волокнами; высокотемпературных материалов в
7
ракетных двигателях, когда горючее содержит добавки
металлов. Исследование совместимости или, шире,
взаимодействия высокотемпературных материалов с
жидкими тугоплавкими окислами имеет особое значение еще и
потому, что при высоких температурах основной процесс
может протекать в зависимости от структуры и свойств
жидких окислов по нескольким механизмам, может
сопровождаться одним или несколькими побочными
процессами, что существенно усложняет понимание и
управление процессом взаимодействия.
Таким образом, для решения широкого круга задач в
этих трех направлениях требуется информация о
физических и физико-химических свойствах жидких
тугоплавких окислов и о параметрах межфазного взаимодействия
жидких окислов с твердыми тугоплавкими веществами
на границе их соприкосновения. Именно эти вопросы
рассматриваются в предлагаемой читателю книге.
В главе I приводится обзор данных по температурам
плавления тугоплавких окислов. Целесообразность
введения такой главы определяется стремлением не только
собрать известные данные об этой характеристике, но
также и тем, и это главное, что при проведении физико-
химических измерений в некотором температурном
интервале перегрев жидкости выше температуры
плавления можно строго оценить только в том случае, если
точно установлена величина последней.
Представленный в главе I обзор опубликованных
сведений является попыткой критически рассмотреть
экспериментальные данные по всем тугоплавким окислам.
В ранее опубликованных обзорах Бревера и Ришке-
вича отсутствует критический анализ данных. В обзорах
Кенисарина и Чеховского приводятся данные лишь для
нескольких тугоплавких окислов. В современной
справочной литературе данные по температурам плавления
окислов даются, на наш взгляд, без надлежащего
анализа. По некоторым тугоплавким окислам данные,
приводимые в разных справочниках, отличаются друг от
друга на чрезмерно большую величину: например, на
800° С для Се02, на 450° С для Сг203, на 350° С для
Th02, на 120° С для НЮ2 и ТЮ2, на 75° С для Zr02. Это
затрудняет использование указанных окислов в науке и
технике.
Из тугоплавких окислов наиболее точные данные по
температуре плавления известны для окиси алюминия
8
(2054±6°С), что объясняется стремлением включить
температуру плавления окиси алюминия в число
вспомогательных фиксированных точек температурной
шкалы, а также широким использованием этого окисла в
промышленности. В последние годы в связи с
появлением более совершенной измерительной аппаратуры
(например, фотоэлектрический оптический пирометр),
использованием более чистых по химическому составу
окислов для исследования и обеспечением более строгих
условий проведения опытов (например, имитация
абсолютно черного тела), сведения о температуре плавления
некоторых окислов подверглись существенному
уточнению.
Опираясь на результаты критического анализа
экспериментальных данных, авторы рекомендуют для
большинства из рассмотренных тугоплавких окислов
наиболее достоверную температуру плавления.
Значительное место в книге (главы II и III)
занимают изложение методики измерения, данные по физико-
химическим и теплофизическим свойствам жидких
тугоплавких окислов и их анализ. Проведение
физико-химических измерений при температурах 2000—2700° С
сопряжено с преодолением больших экспериментальных
трудностей. Не только получение высоких температур,
но и измерение размеров или усилий, выбор материалов
для измерительной аппаратуры, предотвращение
конвективного перемешивания в объеме образца,
предупреждение интенсивного испарения жидкости, выбор
атмосферы печи, рабочего газа и ряд других вопросов
решаются для таких высоких температур с несравненно
большими затратами труда и времени, чем для комнатной
или несколько повышенной температуры.
Авторы книги вместе с группой студентов и
аспирантов кафедры «Высокотемпературные материалы»
МИСиСа выполнили исследования, потребовавшие
тщательной, кропотливой и длительной работы.
Результатом этих исследований явилось создание серии
высокотемпературных экспериментальных установок,
разработка методик измерения и получение большого объема
новых данных по свойствам жидких тугоплавких окислов.
Перспективными методами для
высокотемпературных физико-химических исследований являются те, в
которых исследуемая тугоплавкая и, как правило,
химически активная жидкость не соприкасается при проведе-
9
нии опыта с деталями измерительной аппаратуры.
Нельзя считать, что жидкие окислы имеют такую же высокую
химическую активность, как жидкие тугоплавкие
металлы или соединения, но тем не менее загрязнение их
примесями и искажение результатов измерений вполне
возможны при недостаточно хорошо продуманной методике
исследований. В частности, авторы отметили, что во
время измерений плотности и поверхностного натяжения
методом максимального давления в пузырьке газа,
образующемся на конце молибденового или вольфрамового
капилляра, присутствие в рабочем газе даже малой
концентрации кислорода или паров воды вызывает
заметное искажение экспериментальных данных. Механизм
этого явления заключается во взаимодействии
кислорода в рабочем газе с материалом капилляра, образовании
легколетучих окислов молибдена или вольфрама,
которые диссоциируют на поверхности жидкой окиси
алюминия с выделением металла, создающего на ней тонкую,
иногда сплошную металлическую пленку.
К числу методов, в которых отсутствует опасность
загрязнения объекта исследования, относятся методы
«летящей капли» и «падающей струи», разработанные
авторами; целесообразно дальнейшее развитие этих
методов, а также методов, основанных на нагреве твердых и
жидких образцов во взвешенном состоянии.
Применительно к жидким окислам часто
оказывается возможным использование обычных методов
измерения свойств, которые применяются при комнатной и
умеренно повышенных температурах. К ним относятся
методы максимального давления, гидростатического
взвешивания, крутильных колебаний, лежащей капли,
отрыва кольца или диска и некоторые другие. Все эти
методы пригодны для работы с жидкими тугоплавкими
окислами при одном непременном условии: на всех стадиях
проведения эксперимента необходима оценка влияния
высокой температуры.
Таким образом, разработка методики требует
проведения предварительных широких и нередко весьма
сложных методических исследований. Этим исследованиям
авторы уделили должное внимание. Их результаты
послужили основанием для внесения в конструкции
экспериментальных установок и в методику измерений ряд
усовершенствований, повышающих качество работы и
надежность получаемых данных.
10
Начало исследований свойств жидких окислов на
кафедре «Высокотемпературные материалы» относится к
1957 г. В то время данные о свойствах жидких
тугоплавких окислов практически полностью отсутствовали.
Имелась лишь единственная публикация Вартенберга с сотр.
(1936 г.), в которой содержались ориентировочные
сведения о плотности и поверхностном натяжении жидкой
окиси алюминия. Вероятно, в это же время (1956—
1958 гг.) начались аналогичные исследования в
лаборатории профессора Кинджери из Массачусетского
технологического института (г. Бостон, США), однако в
опубликованной им в 1958 г. книге1 данные по свойствам
жидких тугоплавких окислов не приводятся.
В дальнейшем в статьях Кинджери, Киршенбаума,
Кэхилла, Козакевича были опубликованы некоторые
сведения по физико-химическим свойствам, но эти сведения
требовали дополнительного подтверждения. Лишь после
1965 г. в мировой литературе появилось значительное
число публикаций по свойствам жидких тугоплавких
окислов, в том числе окиси алюминия и двуокиси титана.
В последние годы опубликованы данные по свойствам
более тугоплавких окислов таких, как окиси бериллия,
иттрия, скандия.
В книге собраны практически все известные сведения
о плотности, поверхностном натяжении, вязкости,
электро- и теплопроводности жидких тугоплавких окислов,
опубликованные до 1978 г. Авторы, книги принимали
активное участие в разработке этого научного
направления. Так, исследования физико-химических свойств
чистой жидкой двуокиси титана, плотности и
поверхностного натяжения расплавов на основе окиси алюминия,
влияния газовой атмосферы на поверхностное натяжение
жидкой окиси алюминия, а также исследования
диффузионных и теплофизических характеристик жидкой
окиси алюминия проведены авторами впервые.
На основе сравнительного анализа данных о
конкретных свойствах жидких тугоплавких окислов и после
тщательного отбора наиболее достоверных из них авторы
рекомендуют величины, характеризующие то или иное
свойство жидких окиси алюминия и двуокиси титана в
широком температурном интервале. Итогом комплексно-
у\ин^жеРи В- Д- Измерения при высоких температурах. Пер. с
cm реА" Б* В' Линчевск°го. М., Металлургиздат, 1963. 466 с.
И
го рассмотрения данных по свойствам с теоретических
позиций явились выводы о строении указанных окислов,
о размерах структурных составляющих в них и об
абсорбции газовых молекул на их поверхности.
Из работ авторов, касающихся изучения
физико-химических свойств жидких тугоплавких окислов, хотелось
бы выделить исследования по теплопроводности
окислов (глава III). Трудности, возникающие при измерении
теплопроводности окисных материалов как в твердом,
так и особенно в жидком состоянии, при высокой
температуре обусловлены прозрачностью объекта
исследования, т. е. наличием радиационного теплопереноса
излучением, не подчиняющимся в общем случае закону Фурье.
Последнее обстоятельство не позволяет использовать
известные теоретические обоснования методики и
известные экспериментальные методы измерения
теплопроводности. Для оценки теплопроводности полупрозрачных
материалов нужен метод, позволяющий одновременно
определять коэффициент теплопроводности и параметр,
учитывающий внутренний радиационный теплоперенос.
Авторы дали теоретическое решение задачи о
распространении плоских температурных волн в
полупрозрачной пластине, в которой разделены фононная и
фотонная составляющие теплопроводности. На основе
полученного решения создан оригинальный измерительный
стенд для определения теплофизических свойств
полупрозрачных твердых и жидких веществ при
температурах до 2500° С. Методические исследования
продемонстрировали хорошее совпадение экспериментальных
данных, полученных этим методом, с литературными для
температурного интервала, в котором фотонная
теплопроводность не получает заметного развития. Показано
faкжe, что если не учитывать фотонную составляющую
(это имеет место в исследованиях теплопроводности
окислов при температурах 600°С и выше), то полученные
экспериментальные данные оказываются существенно
заниженными.
В книге приведены впервые полученные сведения о
величинах коэффициента фотонной теплопроводности и
эффективного параметра радиационного теплопереноса
для монокристаллической окиси алюминия при
температурах до 1400° С и, что представляет особую ценность,
для жидкой окиси алюминия при температуре,
превышающей 2050° С.
12
Материал, изложенный в главах II и III, имеет не
только теоретическое, но и практическое значение, так
как данные по свойствам жидких окиси алюминия и
двуокиси титана, а также расплавов на основе окиси
алюминия могут широко использоваться конструкторами
при разработке новой техники.
Выше отмечалось, что взаимодействие на границе
между жидкими окислами и твердыми
высокотемпературными материалами встречается не только в процессе
производства изделий из окислов, но и во время их
эксплуатации (совместимость). Более того, можно сказать,
что управляемое межфазное взаимодействие
(смачивание) лежит в основе создания высококачественных
композиционных окисных материалов с оптимальным
сочетанием их характеристик. Поэтому вопросы смачивания
и частично совместимости в системах жидкий
тугоплавкий окисел — высокотемпературный материал нашли
отражение в большой по объему и достаточно глубокой по
содержанию главе IV.
Развитие авторами теоретических представлений о
смачивании твердых материалов жидкими окислами
базируется главным образом на работах ученых советской
школы: А. А. Жуховицкого, В. Н. Еременко, Ю. В. Най-
дича, О. А. Есина, А. А. Аппена, Ю. Б. Горюнова,
Б. Д. Сумма и др.
Среди теоретических вопросов, рассмотренных в этой
главе, обращают на себя внимание несколько иное, но,
вероятно, более полное толкование уравнения Еременко
для описания процесса смачивания, протекающего с
высокой степенью интенсивности; вопросы, касающиеся
кинетики смачивания в зависимости от плотности фазового
слоя жидкости; введение в теоретические расчеты
процесса смачивания динамической межфазной энергии,
которая, как показали исследования А. А. Жуховицкого и
В. А. Григоряна, может существенно отличаться от
равновесной величины; развитие теории адсорбции в
системах жидкость — твердое тело. Во всех случаях
теоретические выводы авторы стремились подтвердить
экспериментально, для чего ими были выполнены исследования
жидких окислов, которые не относятся к тугоплавким,
жидких хлоридов и т. п. Результаты этих исследований
показывают не только правильность теоретических
заключений, но и то, что они носят обобщающий
характер.
13
Из новых экспериментальных данных, приведенных в
этой главе и имеющих непосредственное отношение к
высокотемпературному материаловедению, особенно
ценными для практических целей являются сведения о
смачивании жидкой окисью алюминия графита и
тугоплавких металлов, о смачивании тугоплавких металлов
двойными расплавами на основе окиси алюминия; о влиянии
реакционно-активных добавок на смачивание
тугоплавких металлов жидкой окисью алюминия. Использование
этих сведений позволяет реально управлять процессом
смачивания.
В заключительной (V) главе книги рассмотрены
закономерности процесса образования тугоплавких
окислов в результате окисления и горения частиц малого
размера химически активных металлов. В качестве
модельного металла выбран алюминий.
Различные теоретические представления о процессе
окисления мелкодисперсного алюминия объединены
единой концепцией, основным положением которой
является уменьшение диффузионной проницаемости твердой
окисной пленки в связи с релаксационным изменением ее
структуры. Введение в теорию окисления
количественного параметра времени релаксации, зависящего от
температуры, привело к хорошему совпадению расчетных и
известных экспериментальных данных в широком
температурном интервале. Развитые в этой главе
теоретические представления имеют особое значение для тех
условий, когда нельзя пренебрегать в общем тепловом
балансе эффектом саморазогрева нагревающейся
металлической частицы. Что касается экспериментального
изучения процесса, то здесь следует выделить
оригинальное исследование окисления жидкого алюминия при
температурах до 1700° С, выполненное авторами с
использованием метода капли, взвешенной в высокочастотном
электромагнитном поле.
В заключение следует сказать, что авторами
выполнена большая и своевременная работа, а материал,
представленный в книге, имеет теоретическое и
практическое значение.
Заведующий кафедрой
«Высокотемпературные материалы» МИСиСа
чл.-корр. АН СССР проф. В. /7. Елютин
Глава I
ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
ТУГОПЛАВКИХ ОКИСЛОВ
1. ТОЧНОСТЬ ИЗМЕРЕНИЯ
ВЫСОКИХ ТЕМПЕРАТУР
Температура плавления тугоплавких окислов находится
в пределах 1800—3300° С. Для точного измерения таких
температур требуется учитывать возможное влияние
ряда факторов, которые могут внести значительную
погрешность в полученные результаты. Объективные
причины, снижающие точность измерения температуры
плавления тугоплавких окислов, можно сгруппировать
следующим образом:
1) погрешности, связанные с несовершенством
измерительной аппаратуры, ошибками при ее градуировке
и т. п.;
2) погрешности, вызываемые изменением состава
окисных образцов во время нагрева и выдержки при
температуре плавления;
3) погрешности, возникающие из-за конструктивных
недостатков экспериментальных установок.
Первые попытки измерения температуры плавления
тугоплавких окислов относятся к концу прошлого века,
когда вплоть до 1927 г. не существовало единой и
общепринятой температурной шкалы в области высоких
температур. В лучших работах того времени приводятся
поэтому данные об использованной температурной шкале,
сведения о градуировочных точках и способах
градуировки измерительных приборов. Однако во многих
ранних публикациях о температуре плавления тугоплавких
окислов такие сведения, к сожалению, отсутствуют.
В 1927 г. на VII Генеральной конференции по мерам
и весам была принята первая Международная
практическая температурная шкала (МПТШ-27). В области
высоких температур шкала построена с помощью реперной
точки золота (температура равновесия между твердым
" *\идким золотом), которой было присвоено значение
ШЬЗ С, закона излучения Стефана — Больцмана и
уравнения Вина о распределении энергии по спектру с кон-
15
стантой С2= 1,432 см-К. В качестве средства измерения
высокой температуры рекомендован оптический
пирометр с исчезающей нитью.
На IX Генеральной конференции по мерам и весам,
состоявшейся в 1948 г., Международная температурная
шкала подверглась некоторым изменениям. Во-первых,
вместо уравнения Вина высокотемпературную часть
шкалы предложено определять по закону теплового
излучения Планка, который является следствием
квантовой теории:
Из закона Планка уравнение Вина вытекает только
для случая, когда величина кТ мала по сравнению с С2.
Надо заметить, что до сих пор во многих работах
используется уравнение Вина, а не закон Планка.
Объясняется это тем, что, если величина К (/+273)
оказывается меньше 0,3 см-град, то ошибка, вызванная
использованием уравнения Вина, составляет менее 1°. Кроме
того, уточнена величина С2: предложено использовать
С2 = 1,438 см-К вместо С2 = 1,432 см-К. В результате
этого изменились численные значения некоторых репер-
ных точек, однако очень важная для
высокотемпературной части шкалы температура равновесия между
твердым и жидким золотом не изменилась.
К 1948 г. уже достаточно хорошо были разработаны
такие новые методы измерения высокой температуры,
как измерение с помощью высокотемпературных
термопар, цветовая пирометрия. Тем не менее IX Генеральная
конференция по мерам и весам подтвердила, что
лучшим инструментом для измерения высоких температур
остается визуальный оптический пирометр с
исчезающей нитью в монохроматическом свете.
Для определения температуры / более высокой, чем
температура плавления золота /ди> применяется
соотношение:
ImT+TojJ
где Го=273,15 К.
70 (0 .__
'•('ли) ~ Г С2 1 , • О'2)
ехр ' '
16
Уточнение МПТШ-48 привело к тому, что данные о
температуре плавления тугоплавких окислов,
полученные в период с 1927 по 1948 гг., оказались несколько
завышенными. В области высоких температур (1800—
3200° С) разница между температурами по МПТШ-27 и
МПТШ-48 составляет 5—20°. Эта разница близка к
величине обычной ошибки при измерении высокой
температуры. Однако измерения температуры плавления
тугоплавких окислов, проведенные до 1927 г., когда
каждый исследователь использовал свою температурную
шкалу, отличались от температуры по МПТШ-48
нередко на 50° С и более.
Это отличие было устранено в принятой в 1968 г.
новой Международной практической температурной шкале
(МПТШ-68) [58, 59]. Согласно этой шкале,
температура затвердевания золота составляет 1064,3° С, а
величина константы в уравнении Планка С2= 1,4388 см-К
при температурах выше температуры затвердевания
золота. Основные реперные и вспомогательные
фиксированные точки Международной температурной шкалы
1968 г. приведены ниже.
Фиксированная точка Температура °С
Равновесие между твердым и жидким:
серебром 961,93
золотом 1064,3
Затвердевание:
меди 1084,5
никеля 1455,0
кобальта 1494,0
палладия 1554,0
платины 1772,0
родия 1963,0
иридия 2447,0
Плавление вольфрама 3387,0
Величины поправок для перевода температур
МПТШ-48 в МПТШ-68 (°С) приведены ниже.
0 100 200 300 400 500 600 700 800 900 1000
in^ — 1,5 1,7 1,8 2,0 2,2 2,4 2,6 2,8 3,0 3,2
SX2 3'2 3>5 3>7 4'° 4'2 4>5 4>8 5'° 5>3 5>6 5>9
d°00 5,9 6,2 6,5 6,9 7,2 7,5 7,9 8,2 8,6 9,0 9,3
2-894
17
Законы излучения предполагают, что излучающее
тело представляет собой абсолютно черное тело,
которое поглощает все падающие на него лучи без их
отражения и обладает максимальной излучающей
способностью при данной температуре. Лучшей моделью
абсолютно черного тела, которая была предложена еще
Кирхгофом, является небольшое отверстие в стенке
равномерно нагретой полости.
Нечерное или серое тело, как и реальные вещества с
открытой поверхностью, не только излучает энергии
меньше, чем абсолютно черное тело при одной и той же
температуре, но может также отражать часть
излучения, падающего на него. Таким образом, температура
поверхности серого тела, которая измеряется
оптическим пирометром, так называемая кажущаяся, или ярко-
стная температура, может быть меньше истинной из-за
пониженной интенсивности излучения или больше
истинной, если на нее падают и частично отражаются лучи от
других тел, нагретых до более высокой температуры.
Для расчета истинной температуры серого тела по
его яркостной температуре необходимо использовать
поправочный безразмерный коэффициент — степень
черноты, который представляет собой отношение количества
энергии, испускаемой единицей поверхности серого тела,
к аналогичной величине для абсолютно черного тела при
одинаковой температуре.
Степень черноты зависит от природы вещества и
температуры, а также от степени шероховатости
поверхности, ее конфигурации, наличия на поверхности
различного сорта загрязнений и т. п. Кроме того, степень черноты
зависит от длины волны излучения. Различают поэтому
степень черноты вещества, которая характеризует
излучение непрозрачного образца с оптически гладкой и
плоской поверхностью, не имеющей загрязнений, и степень
черноты тела или предмета. В последнем случае
учитывается состояние и конфигурация поверхности,
прозрачность тела, направленность излучения. Кроме того,
различают степень черноты полного излучения,
спектральную или монохроматическую степень черноты для
узкого интервала длин волн спектра и интегральную степень
черноты для некоторого участка длин волны спектра
излучения.
Учитывая недостаточную надежность данных о
степени черноты при точном измерении температуры образ-
18
ца, например, при исследовании температуры плавления,
надо стремиться создать условия для образца,
максимально приближающие его к абсолютно черному телу.
Для этого образец помещают в систему равномерно
нагретых экранов, измеряют температуру дна
цилиндрической или клиновой полости в образце или дна тонкой
трубочки с запаянным концом, погруженной в жидкий
образец. Для выбора лучшей модели абсолютно черного
тела проводят тщательные методические исследования,
так как в области высоких температур неучтенное
отклонение степени черноты от единицы на 0,1—0,2
вызывает ошибку в определении температуры на 50—250° С.
Значительная ошибка возникает, если серое тело —
образец — окружено предметами, имеющими более
низкую или, что еще хуже, более высокую температуру.
Последний случай встречается, например, при излишне
быстром нагреве образцов с помощью радиационного
нагревателя и проведении измерений температуры раньше,
чем установится стационарное состояние. При разнице
температуры образца и нагревателя в 100° С ошибка при
измерении температуры образца со степенью черноты
0,75 может достигать 40° С. Окружающая обстановка не
оказывает влияния на точность измерения температуры
лишь тогда, когда образец находится в условиях,
имитирующих абсолютно черное тело [56].
Из-за значительного разброса данных по степени
черноты для твердых окислов и практически полного их
отсутствия для тугоплавких окислов в жидком
состоянии нельзя считать точными сведения о температуре
плавления окислов, если исследуемый образец не
находился в условиях, обеспечивающих его максимальное
приближение к абсолютно черному телу.
При измерении температуры плавления тугоплавких
окислов яркость нити оптического пирометра, которая
нагревается примерно до 1300° С, оказывается
значительно меньше яркости объекта. В таких случаях на
пути светового потока от образца к пирометру
устанавливается поглощающий светофильтр. Лучшим
нейтральным светофильтром является быстро вращающийся диск
из непрозрачного материала с вырезанными, одним или
несколькими секторами. Если суммарный угол
вырезанного сектора а рад, то коэффициент пропускания такого
фильтра будет равен а/(2л). Использование
вращающегося секторного светофильтра несколько усложняет про-
2*
19
цесс измерения высокой температуры и поэтому часто
вместо него применяют стеклянные поглощающие
светофильтры, коэффициент пропускания которых
определяется экспериментально.
Применение поглощающих светофильтров
увеличивает ошибку при измерении температуры, так как,
во-первых, ошибка при градиуровке стеклянного светофильтра
должна включаться в суммарную ошибку опыта и, во-
вторых, при использовании поглощающего светофильтра
в работе с монохроматическим излучением нарушается
соответствие в цвете образца и пирометрической нити:
образец становится более светлым. Последнее
затрудняет работу исследователя, так как не происходит полного
исчезновения нити, что повышает субъективную ошибку
и ухудшает воспроизводимость результатов опытов.
Влияние газовой среды на пути световых лучей
обычно преувеличивается. Если измерение проводится в
монохроматическом свете, влияние избирательного
поглощения газами не должно оказываться на интенсивности
его излучения: такие газы, как водяной пар, окись
углерода, двуокись углерода, совершенно не поглощают,
например, излучение красного участка спектра. Но парили
дым, находящийся даже в виде тонкого слоя вблизи
поверхности образца, температура которого измеряется,
может заметно снизить как полную, так и спектральную
интенсивность излучения. Особенно нежелателен пар
или дым, когда он заполняет небольшую полость в
образце, выполненную как модель абсолютно черного тела.
Ко второй группе причин, снижающих точность
измерения температуры плавления тугоплавких окислов,
можно отнести: 1) низкую или неустановленную степень
чистоты исходных образцов; 2) изменение состава
образцов во время проведения опыта; 3) использование
давления, отличающегося от 1 ат; 4) погрешности при
определении момента плавления.
Примеси, присутствующие в тугоплавких веществах,
снижают температуру их плавления, причем
проявляется это тем более резко, чем выше температура
плавления вещества [57]. Чистые тугоплавкие окислы не
являются исключением из этого правила: наиболее
чистые образцы являются наиболее тугоплавкими.
Интенсивность влияния примеси на температуру
плавления тугоплавких окислов определяется природой
примеси. Так, 1 % (мол.) La203 уменьшает температуру
20
плавления окиси бериллия на 100° С, а ВеО в свою
очередь сильно понижает температуру плавления Th02 (на
83° С). Многие примеси снижают температуру
плавления тугоплавких окислов на 30—50° С на 1% (мол.)
примеси.
Оценивая влияние примесей, следует учитывать
возможность их удаления из образца во время нагрева и
плавления. Достаточно полно при этом удаляются
легколетучие примеси. Таким образом, образцы для
измерения температуры целесообразно изготавливать из
предварительно переплавленного материала; определять
химический состав образцов следует не только до опыта,
по и после него.
Изменение состава образца во время опыта может
наступить вследствие развития таких процессов, как
диссоциация окислов, их взаимодействие с газовой
окружающей атмосферой, взаимодействие с парами
материалов, которые вместе с окисным образцом находятся в
зоне высоких температур, твердофазное взаимодействие
с материалом нагревателя, печи, контейнера, в котором
находится образец, и т. п. Выбор условий
эксперимента должен сокращать до минимума возможность
изменения состава образца во время его нагрева и
плавления. В ряде случаев, однако, полностью
предотвратить развитие отмеченных выше процессов не
представляется возможным. Тогда единственным средством
уменьшить их вредное влияние остается быстрота
проведения опыта, особенно в области высоких температур.
Но при этом приходится тщательно следить за тем,
чтобы повышенная скорость нагрева не привела бы к
нарушению условий, имитирующих абсолютно черное тело:
в любых обстоятельствах измерение температуры
оптическим пирометром должно проводиться при
максимальном приближении к стационарному тепловому
состоянию при минимальных градиентах температуры.
При измерении температуры плавления тугоплавких
окислов исследователи часто помещают образец в
атмосферу различных газов: воздуха, азота, водорода,
углекислого газа, кислорода, аргона, гелия, находящихся
под давлением от 10~5 мм рт. ст. (вакуум) до 2—3 ат.
Такое многообразие условий проведения опытов
объясняется стремлением обеспечить постоянство состава
образца во время измерения температуры плавления.
Наиболее подходящей оказалась среда кислорода, однако
21
использование сильно окислительной атмосферы
затрудняет проведение высокотемпературного эксперимента.
Кроме того, следует учитывать влияние давления на
температуру плавления, так как плавление — процесс
фазового превращения 1-го рода, протекающий с
изменением объема. Влияние давления описывается
уравнением Клаузиуса — Клапейрона:
dTJdP = Тил (vx - vTH)/LnJl, (1-3)
где vm и vTB—удельные объемы жидкой и твердой фаз
при температуре плавления;
/,пл— удельная теплота плавления.
Отклонение давление от 1 ат в отмеченных выше
пределах незначительно изменяет температуру
плавления металлов (несколько градусов), но в случае
окислов, особенно тугоплавких, влияние давления на
температуру может оказаться заметным.
Уменьшение давления или проведение опытов в
вакууме интенсифицируют процессы испарения,
диссоциации, восстановления, что ухудшает условия измерения
температуры из-за появления около поверхности образца
экранирующей оболочки из пара или дыма.
Очень важным является правильное определение
момента плавления окисных образцов. Чаще всего начало
плавления образца определяют визуально по изменению
состояния и цвета его поверхности, по началу растекания
образца по материалу опоры, на которой он установлен,
по изменению формы образца, изготовленного,
например, в виде конуса или пирамиды. Некоторые
исследователи судят о температуре плавления или
затвердевания по температурой остановке на кривых
нагрева или охлаждения образцов. Иногда проводят
микроскопическое исследование структуры окислов,
предварительно нагретых до различной температуры, близкой к
температуре плавления. Появление в окисле участков с
измененной структурой указывает на начало процесса
плавления. Известен [33] и такой прием определения
температуры плавления, когда небольшие частицы
окисла остроугольной формы свободно падают в равномерном
температурном поле, имеющем достаточную
протяженность. Температуру поля повышают от опыта к опыту,
приближая ее к температуре плавления. За температуру
плавления принимают ту, при которой частицы окисла
приобретают округлую, шаровидную форму со сглажен-
22
пыми углами и выступами. Исследование формы
образцов проводятся после охлаждения.
При оценке правильности выбора того или иного
способа определения момента плавления следует иметь
в виду, что окислы очень медленно прогреваются,
особенно при высокой температуре. Для большинства
тугоплавких окислов (за исключением ВеО)
коэффициент теплопроводности составляет 2—4 ккал/(м-ч-град).
Эта величина в 30—40 раз меньше, чем для тугоплавких
металлов, в 20—30 раз меньше, чем для многих
тугоплавких соединений. Правильнее, однако, связывать
скорость нагрева материала не с его теплопроводностью,
а с температуропроводностью
а = Щсу), (1-4)
где Я— теплопроводность;
с— удельная теплоемкость;
7 — плотность,
так как именно температуропроводность является
коэффициентом пропорциональности в уравнении
теплопроводности, которое определяет скорость изменения
температуры тела в нестационарном режиме.
Тугоплавкие окислы имеют температуропроводность
при высокой температуре в пределах 0,001—0,006 м2/ч,
что почти на два порядка ниже, чем
температуропроводность металлов. Низкая температуропроводность
окислов сохраняется и в жидком состоянии.
Отмеченная особенность окислов затрудняет
получение в образцах температурного поля, близкого к
стационарному: при скоростях нагрева, принятых во
многих исследованиях, в образцах сохраняется заметный
градиент температуры. Это может явиться причиной
ошибки как в определении момента плавления, так и при
измерении температуры. Например, при радиационном
нагреве образца и измерении температуры пирометром
на дне углубления, имитирующего абсолютно черное
тело, измеренная температура может оказаться заметно
меньше в зависимости от скорости нагрева, чем истинная
температура плавления, несмотря на то, что на
поверхности образца уже наблюдается процесс плавления.
Из-за низкой температуропроводности окислов
считаются мало пригодными динамические методы определения
температуры плавления, такие как анализ кривых
нагрева или охлаждения.
23
Необходимо учитывать также, что даже у весьма
чистых тугоплавких окислов проявляется различие между
температурой плавления и температурой затвердевания,
т. е. наблюдается гистерезис фазового перехода твердое
тело — жидкость [55]. Гистерезис наблюдается при
сравнительно небольших скоростях нагрева и
охлаждения. Величина гистерезиса может достигать нескольких
десятков градусов и его нельзя не учитывать при
анализе экспериментальных данных по температуре
плавления тугоплавких окислов. Причинами гистерезиса,
который изучен совершенно недостаточно, могут быть
склонность жидких окислов к переохлаждению, изменение
структуры этих окислов при плавлении, влияние
примесей и т. п.
Для повышения точности измерения температуры
плавления окислов надо: 1) снизить скорость нагрева в
районе температур, близких к измеряемой температуре
плавления, 2) использовать микроскопические способы
определения момента плавления и 3) использовать
методы нагрева, при которых тепло генерируется в массе
образца.
Третья группа причин может оказаться наиболее
обширной, если в ее пределах рассматривать
разнообразные конкретные недостатки в технике эксперимента
при измерении температуры плавления тугоплавких
окислов. Однако можно выделить несколько общих
факторов, влияющих на точность измерения температуры
плавления окислов в зависимости от конструкции
экспериментальной установки. В современных установках для
определения температуры плавления образец, как
правило, помещают в герметичную камеру, где создается
атмосфера нужного состава и давления. На пути лучей
от образца к пирометру находятся стекло смотрового
окна и иногда зеркало или призма, если требуется изменить
направление луча. Нередко в оптическую систему
установки включается один или несколько цветных и
поглощающих светофильтров.
Уменьшение яркости образца из-за поглощения в
оптической системе установки учитывают либо путем
градуировки пирометра на данной установке, либо
путем предварительной оценки степени поглощения
(коэффициента пропускания) каждого элемента или
оптической системы в целом и введения поправки в
результат определения температуры.
24
В процессе работы следует систематически проверять
коэффициент пропускания системы, а также тщательно
защищать ее элементы от конденсации на их
поверхности паров материалов, находящихся в зоне высокой
температуры. Чаще всего элементы оптической системы
закрывают непрозрачными экранами, нередко со
специальным охлаждением, которые отодвигают в сторону
только на короткое время измерения температуры.
Иногда внутри камеры рядом со смотровым окном
устанавливают поворачивающийся стеклянный диск,
находящийся в охлаждаемом кожухе с вырезом. Вырез в
кожухе расположен против смотрового окна. Перед
измерением температуры диск поворачивают так, чтобы
против смотрового окна находилась незагрязненная
часть диска. Газы в установку обычно вводят через
отверстие около смотрового окна. Это ограничивает
приближение паров к стеклу смотрового окна, уменьшая
тем самым его загрязнение вследствие конденсации. Эта
мера, однако, полностью не предупреждает опасности
загрязнения элементов оптической системы.
Нежелательным является воздействие температуры
на элементы оптической системы. Из-за повышения
температуры элементов оптической системы возможно
помутнение стекла, изменение структуры отражающего
слоя зеркала и т. п., что увеличивает степень поглощения
системы.
Недостаточно тщательное выполнение требований,
которые предъявляются к оптической системе, может
привести к серьезным ошибкам в измерении
температуры, достигающих десятков градусов. В большинстве
случаев причины, вызывающие ошибку, приводят к
тому, что зафиксированная пирометром температура
оказывается ниже истинной.
Для измерения температуры плавления тугоплавких
окислов применяли разнообразные методы нагрева
образцов: от кислородно-ацетиленового пламени до
солнечных печей. Важным является не столько природа
источника тепла, сколько то, в какой степени метод
нагрева позволяет обеспечить равномерный нагрев
образцов с регулируемой скоростью в условиях, имитирующих
абсолютно черное тело. Не исключено, однако, что метод
нагрева может также оказывать влияние на темпера-
ТУРУ плавления веществ. Бомбардировка поверхности
образцов электронами, имеющими скорость 60000—
25
80 000 км/с, при электронном нагреве может явиться
причиной изменения структуры поверхностных слоев,
образования в ней многочисленных дефектов, снижающих
температуру плавления.
Пламенные печи с любым видом топлива (водород,
ацетилен, углеводороды и др.), когда пламя
непосредственно воздействует на образец, являются наименее
пригодными. Отражение лучистой энергии пламени
поверхностью образца, значительный градиент
температуры между пламенем и образцом, трудность
регулирования температуры пламени, отсутствие возможности
изменять атмосферу печи — основные недостатки печей
этого типа. Отмеченные недостатки сохраняются и в том
случае, если окисный образец помещают в специальную
камеру, куда не проникают пламенные газы: камеру
практически невозможно изготовить герметичной,
температура стенок камеры не регулируется, они
оказываются более яркими, чем образец при температуре
плавления.
Отражение лучистой энергии в пламенных, а также
в световых печах приводит к завышению температуры.
Широко применяющиеся в научно-исследовательской
практике печи сопротивления с нагревательными
элементами из графита, тугоплавких окислов, соединений
или металлов обладают определенными достоинствами.
Они позволяют получать высокую температуру (до
3000° С), регулировать ее в достаточно узких пределах,
создавать в рабочем пространстве печи требуемую
атмосферу, в том числе окислительную, если печь имеет
окисные нагревательные элементы, или высокий вакуум
при использовании металлических нагревателей.
Большое распространение в высокотемпературных печах
сопротивления получили трубчатые и ленточные
нагревательные элементы. Но в печах сопротивления обычно не
удается получить равномерное температурное поле, что
затрудняет создание для исследуемого образца условий,
при которых его излучение будет близко к излучению
абсолютно черного тела. Этот недостаток печей
сопротивления оказывается настолько серьезным, что многие
специалисты отказываются сейчас от их применения для
точных измерений температуры плавления.
Лучшим методом нагрева образцов следует считать
индукционный, с помощью которого тепло генерируется
частично и в объеме образца. Соответствующим подбо-
26
ром плотности энергии электромагнитного поля можно
полностью ликвидировать температурный градиент,
возникающий, например, на торцах контейнера с
образцом. Тонким регулированием мощности поля можно
поддерживать требуемую температуру в очень узких
пределах и обеспечить равномерный нагрев образца с
любой заданной скоростью. Для нагрева окислов
индукционным методом требуется стартовый подогрев,
увеличивающий их электропроводность. Другие
современные способы нагрева — электродуговой, электронный
и плазменный — не имеют преимуществ по сравнению с
индукционным: трудность регулирования температуры и
скорости нагрева в первом, поверхностный и
обязательно в вакууме нагрев во втором, очень высокая и
нерегулируемая температура в третьем — не дают оснований
считать эти способы перспективными для
научно-исследовательских установок, где требуется равномерный
нагрев всего объема образца со строго определенной
скоростью.
Квалифицированный обзор по методам определения
температуры плавления тугоплавких веществ и
экспериментальных данных по температурам плавления
тугоплавких металлов и некоторых окислов выполнен Ке-
нисариным и Чеховским [2].
2. ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
ПО ТЕМПЕРАТУРЕ ПЛАВЛЕНИЯ
ТУГОПЛАВКИХ ОКИСЛОВ
ДВУОКИСЬ ТОРИЯ
В справочной литературе [3—6, 15, 16] для температуры
плавления двуокиси тория приводятся значения от 2950 до 3300° С. Эти
данные в основном базируются на результатах исследований,
выполненных Ламберстоном с сотр. [7, 8] и Гейча с Харпером [9].
Ламбсрстон и Мюллер [7] измеряли температуру плавления
■ и*уокиси тория с помощью оптического пирометра. Образцы из
двуокиси тория с содержанием основного вещества 99,7% нагревали в
печи сопротивления с вольфрамовым нагревателем. Момент
расплавления определяли путем исследования образцов после охлаждения.
Полученное ими значение температуры плавления двуокиси тория
составляет по МПТШ-48 3220±50°С.
Аналогичная методика была использована в работе [8] при
исследовании фазового равновесия в системе Th02—U02. Измерение
температуры проводили на модели абсолютно черного тела с точно-
27
Температура плавления
ния
Год измере
1
*пл. °С, по
МПТШ-48
2
оригинальной шкале
3
Точность
измерения, °С
Характеристика
оригинальной
шкалы
4 | 5
Метод
измерения
температуры
6
Определение
момента
плавления
7
1932
1954
1959
—
2758
2900
2770
2812
2774
2900
2770
+ 25 1
±25
±25
—
МПТШ-27
МПТШ-48
— МПТШ-48
1913
1914
1961
- С0
2150-
2599*
2784
2825
2120—
2550
2880
2825
—
±20
1,46;
-1755°С
*пл. Аи
=968,5° С;
Wpt ~
= 1755°С
МПТШ-48;
*пл. А120,=
=2044 °С
Оптический
пирометр
Оптический
пирометр,
образец
с полостью,
АЧТ
Оптический
пирометр
То же
Оптический
пирометр
Оптический
пирометр;
образец
с
отверстием; АЧТ
Оптический
пирометр,
направленный на дно
графитового
полого
цилиндра, АЧТ|
Двуокись
Начало образо
вания жидкой
фазы на
плоской
поверхности образца
Начало
растекания образца
Начало
образования
жидкой фазы на
плоской
поверхности
образца
Окись
Начало
изменения формы
образца при
нагреве
Анализ кривых
нагрева
Появление
капли при
нагреве образца
Таблица 1
некоторых тугоплавких окислов
Условия опыта
тип печи
8
среда
9
материал
тигля,
контейнера,
подложки
10
Характеристика объекта
исследования
11
Библиография
12
гафния
Сопротивления с
вольфрамовым
нагревателем
Сопротивления с
ленточным
вольфрамовым
нагревателем
Пламенная
ацетилен-
кислородная
Сопротивления с
ленточным
нагревателем из
вольфрама или
графита
магния
Водород
или аргон
Водород
Воздух
Нейтральная
—
—
Образец
в виде
стержня
—
Теоретически чистая
нюя
Примесь 1 %
Zr02
99,8% НЮ2
Примеси, %:
Zr5;
Fe 0,5-1,0;
Si 0,5—1,0
Хенинг [13]
Клаузинг
[Ю]
Картис,
Дани,
Джонсон
[17]
Марк
[И]
Сопротивления с
графитовым
нагревателем
То же
Индукционная
с графитовым
концентратором
Азот
Водород
или СО
Азот,
воздух
Графитовая
подложка
Графитовый
тигель
Графитовый
тигель или
подвешенный образец
в виде
стержня
Примеси, %:
Со 0,13;
С1 0,275;
Fe 0,0005;
А1203 0,0002
Примеси, %
(не более):
W03 0,1;
С03О4 0,1;
Si02 0,05;
Fe2O30,05
Рафф и др.
171]
Канольт
[18]
Мак-Нейли
[19]
29
1
1964
2
2790—
2820
3
2790—
2820
4
±20
МПТШ-48
1952
1956
1956
1958
1964
1964
2878
2405
2760
2860
2805
2775-
2785
2878
2405
2760
2860
2805
2775—
2785
±22
±25
±30
±45
±15
±15
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48;
*iw.Pt=
= 1769°С
МПТШ-48
МПТШ-48
Оптический
пирометр,
АЧТ
Оптический
пирометр;
АЧТ
Оптический
пирометр
Оптический
пирометр
Оптический
пирометр;
АЧТ
Оптический
пирометр;
пирометр
полного
излучения,
АЧТ
Визуальное,
без нарушения
условий АЧТ
Двуокись
Начало
образования
жидкой фазы на
плоской
поверхности
образца
Начало
образования
жидкой фазы на
плоской
поверхности
образца
Начало
образования жид
кой фазы на
плоской
поверхности
образца
Анализ кривых
охлаждения
Визуальное без
нарушения
условий
АЧТ
1934
2765
2715
±20
С2= 1,457;
Wpt~
= 1755°С
Оптический
пирометр
Двуокись
Наблюдение
за формой
конусного
образца при нагреве
30
Продолжение табл. 1
1 *.
Сопротивления с
трубчатым
графитовым нагрева-
] тел ем
урана
Сопротивления с
вольфрамовым
нагревателем
—
Сопротивления с U-образ-
пым
вольфрамовым
нагревателем
Индукционная с
графитовым
концентратом
Индукционная с
вольфрамовым
концентратором
Сопротивления с
графитовым
нагревателем
Циркония
Пламенная
«'Щетил ен-
кислородная
9
~-~
Гелий
Вакуум
Гелий,
водород,
аргон,
вакуум
Вакуум
Вакуум 10—4
мм. рт. ст.
Вакуум,
инертные
газы
Воздух
10
Вольфрам; 1
графит;
образец
в виде
стержня
Вольфрамовый тигель
Вольфрамовый
контейнер
Подставка
из
вольфрамового
стержня
Подвеска из
вольфрамовой
проволоки
Вольфрамовая капсула
Вольфрам,
графит
и
Химически
чистая MgO
Очень
чистая U02
—
Чистая 1Ю2
Примеси
в сумме
0,035%;
^^2,002
Химически
чистая U02
12
Райли
[20, 21]
Ламберстон,
Мюллер
17]
Акерман
[22]
Уисни, Пья-
новский
[24]
Элерт,
Маргрейв
[25]
Хауснер
[26]
Райли
[20, 21]
Жирнова
[28]
31
1
1952
1954
1956
1959
1965
2
2710
2850
2710
2690
2710
3
2710
2850
2710
2690
2710
4
±15
±25
±10
—
±10
МПТШ 48
МПТШ-48
МПТШ-48
—" МПТШ-48
МПТШ-48;
^пл.А12Оа—
=2042 °С
Оптический
пирометр
То же
1916
1930
1932
1937
2410
2557
2518
2508
2410
2557
2530
2520
—
—
—
±30
С,= 1,437,
^пл.Аи ==
= 1062,4 °С
— МПТШ-27
МПТШ-27
МПТШ-27
Фотоэлектрический
оптический
пирометр;
АЧТ
Оптический
пирометр
То же
Исследование
после нагрева
Начало обра
зования
жидкой фазы на
плоской по
верхности об
разца
То же
Анализ
кривых
охлаждения
Окись
Начало
образования
жидкой фазы на
плоской
поверхности
образца
Начало
изменения формы
стержневого
образца при
нагреве
То же
32
Продолжение табл. 1
8
Сопротивления с
вольфрамовым
нагревателем
Пламенная,
ацетилен-
кислородная
Сопротивления с
вольфрамовым
нагревателем
Сопротивления с
нагревателем из
вольфрама или
графита
Солнечная
9
Гелий
Воздух
Высокий
вакуум,
гелий
Нейтральная
Воздух
10
Контейнер
из
вольфрама
Гарнисаж-
ный тигель
п
Примеси, %:
НЮ2 2,03;
прочие 0,03
99,9% Zr02;
ppm НЮ2
99,96%
Zr02
12
Ламберстон,
Мюллер,
Гун цель
[8]
Картис,
Дани,
Джонсон
[17]
Эрли,
Ламберстон
[30]
Марк
[14]
Фойкс
[31]
бериллия
Сопротивления с
графитовым
нагревателем
Пламенная,
углеводород-
кислородная
То же
» »
Вакуум
15 мм рт. ст.
Воздух
» »
» »
Графит
Образец
подвешен в
контейнере
из Zr02
То же
» »
Чистая
ВеО
Технически
чистая ВеО
—
Технически
чистая ВеО
Рафф,
Лошке
[35]
Вартенберг,
Верт
[34]
Вартенберг
[36]
Вартенберг,
Раш, Саран
[37]
3-894
33
1
1948
1953
1956
1959
1960
1964
1965
1970
1975
2
2570
2810
2452
2550
2550
2555
2580
2434
2578
3
2570
2810
2452
2550
2550
2555
2580
2434
2578
4
±30
Значительная
погрешность
^пл.тьо2=
=3530°С
±20
±50
±9
±10
±10
±10
±9
5
МПТШ-48
МПТШ-48
МПТШ-48;
Wai2o, ~
=2035 °С
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-68
6
Оптический
пирометр,
измерение
температуры
печи
Оптический
пирометр,
направленный на
границу
твердой и
жидкой фаз
Оптический
пирометр
—
Оптический
пирометр
Оптический
пирометр;
АЧТ
Оптический
пирометр
Фотоэлектрический
пирометр
Оптический
пирометр
7
Исследование
образцов после
нагрева
Начало
образования жидкой
фазы на
плоской
поверхности образца.
Анализ линий
ликвидуса
Начало
изменения формы
пирамидального образца
при нагреве
—
Исследование
образца после
нагрева
Визуальное без
нарушения
условий АЧТ
—
Термический
анализ
То же
Продолжение табл. /
8
Сопротивления с
графитовым
нагревателем.
Бесконтактный метод
нагрева
Дуговая,
вольфрамовый
электрод
Сопротивления с
графитовым иагревате-1
л ем
—
Сопротивления с
вольфрамовым
нагревателем
Сопротивления с
графитовым
нагревателем
Электронная
Высокочастотный нагрев
вольфрамового тигля
Вольфрамовая печь
сопротивления
1
9
Г
Азот
Аргон,
200 мм рт. ст.
Аргон
—
Вакуум
Вакуум,
инертные
газы
Вакуум
10-4— Ю-6
мм рт.ст.
Вакуум 10—^
мм рт. ст.
10
Взвешенное
состояние L
Водоохлаж-
даемая
подложка
Бесконтактный способ
—
Молибденовая и
вольфрамовая
ампулы
Вольфрам,
графит
Капсула
из рения
Капсула из
вольфрама
То же
П
99,9%
ВеО
—
99,9%
ВеО
Химически
чистая ВеО
99,5%
ВеО
99,9%
ВеО
>99,88%
ВеО
12
Ольшанский
[33]
Гейч,
Харпер
[9]
Ланг и др.
[38]
Мак-
Дональд,
Ренсли
[39]
Кандыба
и др.
[40]
Райли
[20, 21]
Гринбаум
и ДР.
[41]
Латт и др.
[42]
Бархатов
и др.
[43]
3*
35
1 2
19561
1959
1959
1960
1960
1962
1963
1964
1964
1965
2049
2060
2020
2025
2043
2042
2044
2070
2050
2042
=t 10
zt4
±6
:10
МПТШ-48,
*пл.ВеО=
=2510 °C
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
Оптический
пирометр
Радиационный
пирометр
Оптический
пирометр
Оптический
пирометр
То же
Оптический
пирометр,
условия АЧТ|
Термопара
100 1г/601г—
40Rh
Оптический
пирометр,
условия АЧТ
Фотоэлектрический
оптический
пирометр
Окись
Начало изме
нения формы
пирамидального образца
при нагреве
Анализ кривых
нагрева
Начало обра
зования
жидкой фазы на
плоской
поверхности
образца
Начало
кристаллизации
образца при
охлаждении
Начало
изменения формы
образца при
нагреве
Исследование
образца после
нагрева
Анализ кривых
нагрева и
охлаждения
Визуальное без
нарушения
условий АЧТ
Анализ кривых
охлаждения
Продолжение табл. 1
ю
алюминия
Сопротивления с
графитовым
нагревателем
Дуговая
Сопротивления с
графитовым
нагревателем
Солнечная
Индукционная с
графитовым
концентратором
Сопротивления с
графитовым
нагревателем
Сопротивления с
вольфрамовым
нагревателем
Сопротивления с
иридиевым
нагревателем
Сопротивления с
графитовым элементом
Солнечная
Аргон
Воздух
Нейтральная
Воздух
Воздух,
азот, аргон
Аргон
10—20 мм
рт. ст.
Аргон
Аргон
Аргон
Воздух
Вольфрам
—
Образец
укреплен в
виде
стержня
Образец
подвешен
Ампула из
молибдена
Молибденовая капсула
Вольфрамовый тигель
Гарнисаж-
ный тигель
99,9%
А!203
А1203
высокой
чистоты
99,9%
А1203
99,99%
А1203
До 0,2%
примесей;
после опыта
0,4-0,9%
Мо
Химически
чистая А!203
99,97%
А1203
Ланг и др.
[38]
Элдред,
Уайт
[44]
Марк
114]
Даймонд,
Шнейдер,
[45]
Мак-Нейли,
Питере,
Риб
[19]
Кантор,
Лазарева
и др.
[46]
Чеховской,
Петров
[47]
Гут
[48]
Райли
[20, 21]
Фойкс
[31]
37
2041
2049,7
2037
2047
а)
2051; I
б)
2045;
в)
2041
а)
2046;
б)
2036;
в)
2038
(лик-I
|видус)
1660
1640
±10
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
МПТШ-48
Са=1,46;
^пл.Р/ ™
= 1755°С
Оптический
пирометр
Оптический
пирометр
Термопара
100W/74W-
-26Re
Оптический
пирометр
Фотоэлектрический
пирометр,
условия АЧТ|
Фотоэлектрический
пирометр,
условия АЧТ
Анализ кривых
нагрева
Анализ
кривых нагрева и
охлаждения
То же
Исследования
образца после
нагрева
Исследования
образца после
нагрева
Двуокись
1857
=50
Оптический
пирометр
То же
Начало
изменения формы
конусного
образца при
нагреве
То же
Продолжение табл. 1
Сопротивления с
графитовым
нагревателем
Сопротивления с
графитовым
нагревателем
То же
Индукционная
Иидукцион-
Аргон
400 мм рт. ст.
Вакуум
Ю-4 мм
рт. ст.; аргон|
20 мм рт. ст.
Аргон
30 мм рт. ст.
Аргон
а) Вакуум;
б) аргон;
в) гелий
а) Вакуум;
б) гелий;
в) воздух
ю
Вольфрам
Вольфрамовый
контейнер
Иридиевый
контейнер
>99,70/0
А1203
99,99%
А1203
«Чистая»
А1203
До 0,3%
примесей
Состав см.
в табл. 9
То же
12
Гитлесен,
Мольцфельд
[49]
Кантор и др.
[50]
Сата
[51]
Урбайн,
Раннет
[52]
Шнейдер,
Мак-Даниэл
[53]
Шнейдер,
Мак-Даниэл
[53]
Сопротивления с
графитовым
нагревателем
То же
Азот 1 ат.
Азот,
водород
Графит
Образец
в виде
стержня
«Чистая»
Ti02 рутил
Частично
восстановленная
Ti02
Рафф,
(Зифферфельд
[71]
Фридерик,
Ситтинг
[73]
39
1931
1932
1933
1937
1949
1951
1952
1952
1953
1845
1820
1820
1850
1825
1716
1840
1845
1839
1850
1825
1825
1855
1825
1720
1840
—
—
±20
±20
±25
±20
—
—
±10
—
±10
МПТШ-27
МПТШ-27
МПТШ-27
МПТШ-27
МПТШ-48;
4Ui.Au~
= 1065 °С;
пл. MgTi2Otl=|
=1660°С
МПТШ-27;
'пл. Pt=
= 1774 СС;
= 1555°С
МПТШ-48
МПТШ-48
МПТШ-48
'пл. Pt=
= 1769°С
Оптический
пирометр
То же
Оптический
пирометр
направлен
на
нагреватель
Оптический
пирометр
Оптический
пирометр
направлен
на образец
То же
Начало обра-|
зования
жидкой фазы на
плоской по-|
(верхности
образца
То же
Исследование
образца после |
нагрева
Начало обра-|
зования
жидкой фазы на
плоской
поверхности об-1
разца
То же
Начало
изменения формы
пирамидального образца|
при нагреве
|То же
40
Продолжение табл. 1
8
Пламенная
ацетилен-
кислородная
Пламенная
дород-кислородная или
газо-воздуш-
ная
Индукционная
с иридиевым
нагревателем
Пламенная,
ацетилен-
кислородная
Сопротивления
Сопротивления с
молибденовым
ленточным
нагревателем
Индукционная
Сопротивления с окисны-
ми (Th02) на-
гревателями
То же
9
Воздух
То же
»
»
Окислительная
Вакуум
Воздух
То же
»
10
Образец
подвешен
То же
Иридиевый
тигель
Образец
подвешен
—
—
—
—
П
«Чистая»
Ti02
«Чистая»
Ti02
Технически
чистая
Ti02
«Чистая»
Ti02
—
Химически
чистая Ti02
ТЮ2с
0,04% Si,
0,02% Mg,
0,01% Ca
99,9%
Ti02
99,9%
Ti02
12
Вартенберг,
Гурр
[74]
Вартенберг,
Профст
[75]
Бантинг
[76]
Вартенберг
[361
Сигурдсон
[77]
Статтон
[72]
Пирр
[78]
Ланг,
Филмор,
Максвелл
[79]
Кохинор,
де Прос
[80]
41
1
1955
1960
1960
2
1870
1840
3
1830
4
=Ы5
±5
5
'пл.Сао— SiOa-
= 1698 °C
МПТШ-48
МПТШ-48
6
Оптический
пирометр
направлен
на
нагреватель
Радиационный
пирометр
Оптический
пирометр
7
Начало
образования
жидкой фазы на
плоской
поверхности
образца
То же
Наблюдения за
формой
образца при нагреве
* В зависимости от степени восстановления.
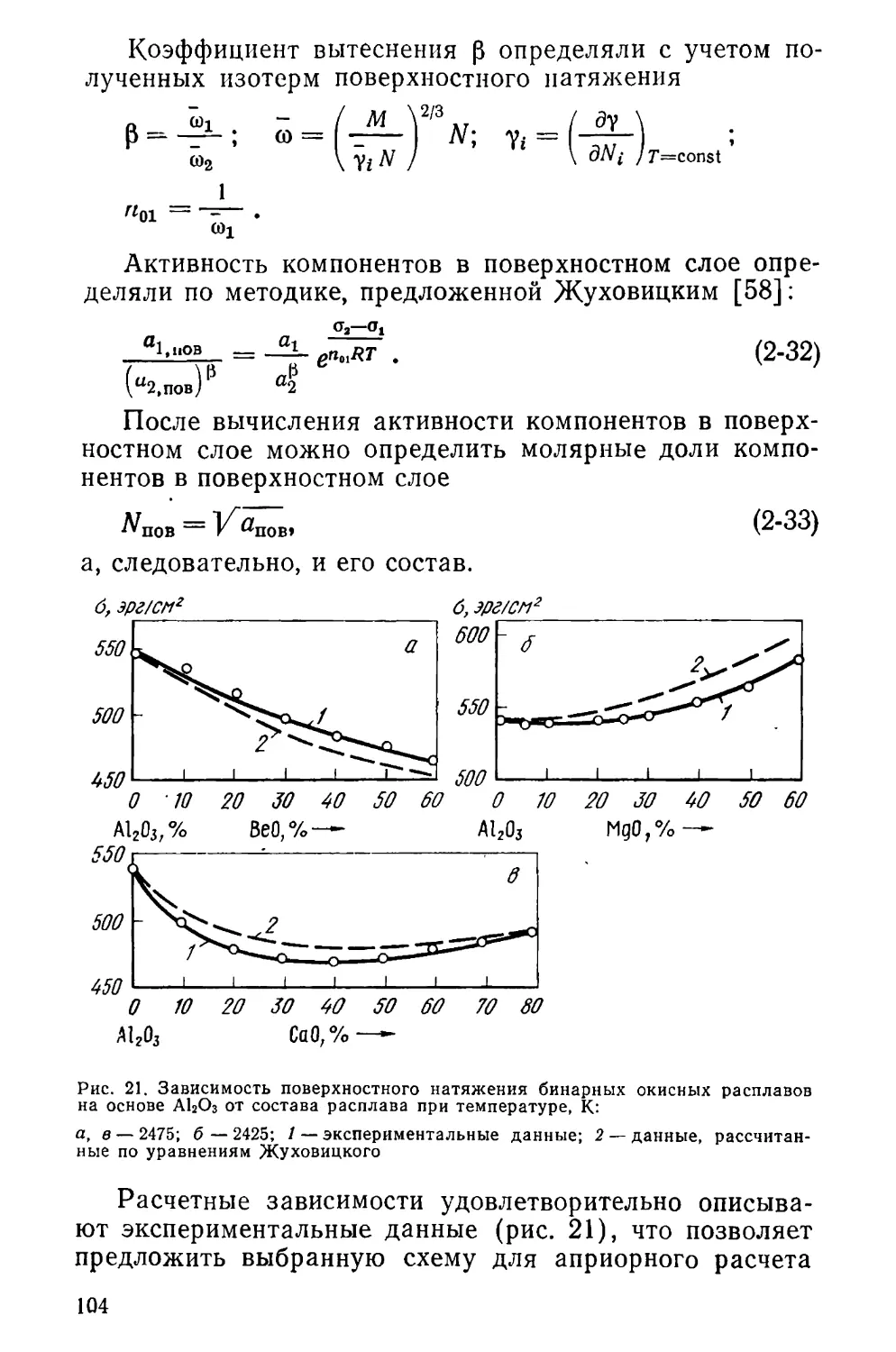
* Температура плавления сплава 15% СаО — 85% S1O2.
стью ±10° С в интервале 2000—3000° С. Однако образец чистой
двуокиси тория, нагретый до 3232° С в печи сопротивления, не имел
следов расплавления. Более высокую температуру в этой печи получить
практически невозможно. Поэтому авторы изменили метод нагрева
и исследование при более высокой температуре проводили в
электродуговой аргоновой печи с вольфрамовым электродом и вогнутым
вольфрамовым поддоном, на котором находился образец. Такую
технику эксперимента нельзя считать удовлетворительной в
исследованиях для измерения температуры с высокой точностью, поскольку
условия, в которых находится образец, далеки от условий излучения
абсолютно черного тела; данные о степени черноты двуокиси тория
при высоких температурах отсутствуют; невозможно оценить
влияние отраженного образцом света от электрической дуги на
измеряемую температуру.
Свидетельством низкой точности измерения является также
следующее: при дуговом нагреве и совершенно одинаковом режиме
полученные авторами данные для температуры плавления (3285, 3344,
3390 и 3555° С) отличаются друг от друга почти на 300° С. В одном
опыте с дуговым нагревом была зафиксирована температура 3287° С,
но образец из двуокиси тория не расплавился, а в другом
аналогичном по режиму опыте при 3285° С было обнаружено полное
расплавление образца.
Таким образом, данные Ламберстона с сотр. нельзя считать
достоверными в отношении измерения температуры плавления двуокиси
тория. Их можно рассматривать лишь как указание на то, что
температура плавления двуокиси тория выше, чем 3220° С.
42
Продолжение табл. 1
8
Сопротивления с
U-образным
нагревателем из
расплава
Солнечная
Солнечная
9
Воздух
Кислород
1 — 1,5 ат
Воздух
10
Образец
в виде
стержня
То же
и
>99,8%
Ti02
99,8%
Ti02
12
Райе, Рой
[81]
Брауэр,
Литке
[83]
Даймонд,
Шнейдер
[82]
ДВУОКИСЬ ГАФНИЯ
В справочной литературе для температуры плавления двуокиси
гафния приводятся значения от 2780 до 2900° С (табл. 1). Надо
заметить, что величина 2780° С — округленный результат работы Клау-
зинга [10] — приводится по МПТШ-27.
Кроме данных о температуре плавления двуокиси гафния,
собранных в табл. 1, известно, что специалистами американской фирмы
«Карборундум» [11] установлена температура плавления двуокиси
гафния, равная 2838±50° С, а по сведениям Национального Совета
США по исследованиям, температура плавления двуокиси гафния
составляет 2810° С [12].
Из работ, перечисленных в табл. 1, к сожалению, ни одна не
удовлетворяет необходимым требованиям по точности определения
температуры плавления. В работе Хенинга [13], которая является
первой попыткой измерения температуры плавления чистой двуокиси
гафния, не указаны характеристики использованной температурной
шкалы, а также не приведен химический состав исследованной
двуокиси (смысл оценки степени чистоты окиси термином «теоретически
чистая» остается неясным).
Исследования Клаузинга [10] и Марка [14] являются более
совершенными с позиций экспериментального оформления и техники
измерения температуры. Однако и в этих исследованиях авторы
изучали не достаточно чистую двуокись гафния. Полученное ими
примерно одинаковое значение для температуры плавления двуокиси
гафния: 2760—2770° С является заниженным из-за наличия примесей.
43
В работе Марка [14], например, суммарное содержание примесей
6-7%.
В исследовании Картиса, Дани и Джонсона [17] исходная
двуокись гафния имела высокую степень чистоты: суммарное содержание
примесей менее 0,2%. Но применение кислородно-ацетиленового
пламени для нагрева образцов дает основания предполагать, что
полученное в этой работе значение температуры плавления 2900° С
несколько завышено из-за влияния отраженного излучения. Сведения о
степени черноты, которые можно было бы использовать для
определения поправки на отраженное излучение, отсутствуют. Таким
образом, за температуру плавления двуокиси гафния нельзя принять ни
величину 2760—2770° С, ни величину 2900° С. До появления новых,
более точных данных, вероятно, следует считать, что температура
плавления двуокиси гафния составляет около 2850° С.
ОКИСЬ МАГНИЯ
Окись магния отличается летучестью, легкой восстановимостью
и активным взаимодействием со многими огнеупорными материалами
при высоких температурах, особенно в жидком состоянии. Эта окись
является одной из наиболее трудных в отношении изучения ее
свойств. Тем не менее, температура плавления окиси магния
установлена достаточно надежно. В справочной литературе для нее
приводятся данные, которые лежат в весьма узких пределах: 2800—2826° С.
Правда, величина 2800° С, полученная Канольтом [18] в 1914 г.,
попала в современные справочники без исправления в соответствии с
Международной практической температурной шкалой 1948 г., но эта
поправка составляет 16° С.
Результаты отдельных исследований температуры плавления
окиси магния, представленные в табл. 1, показывают, что они очень
близки. Pie считая работы Раффа [71] и еще нескольких ранних
работ, в которых получены значения 2685 и 2645° С, во всех остальных
работах измеренная температура плавления окиси магния
незначительно колеблется около значения 2800° С [19—21]. Наиболее
достоверное определение температуры плавления окиси магния провел
Мак-Нейли с сотр. [19]. Образец окиси магния в виде стержня
подвешивали внутри концентратора тепла; в работе использовали
индукционный нагрев, температуру образца измеряли в условиях,
имитирующих условия абсолютно черного тела. Однако наличие в зоне
высокой температуры графитового концентратора тепла привело
к появлению паров углерода в атмосфере, окружающей образец
окиси магния и к его восстановлению, что может явиться причиной
занижения экспериментальных данных, так как это отмечено у Раффа.
Кроме значений, перечисленных в табл. 1, известно значение
температуры плавления окиси магния, равное 2826±22°С [11], которое
хорошо совпадает со значением, полученным Мак-Нейли.
ДВУОКИСЬ УРАНА
Особый интерес к двуокиси урана проявился в конце 50-х годов
нынешнего столетия. Если до 1950 г. были известны два значения
температуры плавления двуокиси урана, то после 1950 г.
опубликовано более 15 исследований, посвященных определению температуры
ее плавления. Результаты этих работ показали, что температура
плавления двуокиси урана оказалась значительно выше, чем считалось
ранее: по последним данным температура плавления двуокиси урана
44
составляет 2800° С, т. е. на 600° С превышает ранее опубликованные
значения.
В современной справочной литературе температура плавления
двуокиси урана оценивается в 2815—2860° С [4—6].
В оригинальных работах, выполненных после 1952 г. (см. табл. 1),
данные о температуре плавления двуокиси урана различаются
больше. Так, в исследовании Акермана [22], относящемся к 1956 г.,
температура плавления двуокиси урана определена в 2405±22° С, тогда
как в работе Андерсона и Сойера
[23], опубликованной в 1960 г., для
температуры плавления U02
получено значение 2900+100° С, т. е.
различие достигает 500° С.
Двуокись урана достаточно
устойчива и с трудом
восстанавливается даже при высоких температу-
Рнс. 1. Схема установки [26]
для определения
температуры плавлния UO2:
/ — концентратор тепла;
2 — вольфрамовая
трубочка-имитатор АЧТ; 3 —
капсула из вольфрама; 4 —
образец U02; 5 — подставка
капсулы; 6 — вольфрамовый
стержень
2900
2800
2700
tL ^>P
- %
-
i
1
п""
. 1
o^t»
XI?
1 1
ГоС1
1
20 60 WO ПО /80 220 т, с
Рис. 2. Кривые охлаждения капсулы с
образцом U02 после нагрева до 2940° С
[26]
рах. При нагреве в окислительной атмосфере и на воздухе она
окисляется до ЫзОв, но при температурах, превышающих 1360° С,
разлагается и вновь превращается в двуокись. Трудности измерения
температуры плавления двуокиси урана связаны главным образом с ее
высокой летучестью при температурах, близких к температуре
плавления. По упругости пара и скорости испарения двуокись урана
лишь немного уступает окиси магния. Высокая летучесть двуокиси
урана может явиться причиной занижения данных о ее температуре
плавления.
Хауснер [26] сделал попытку ограничить интенсивное испарение
двуокиси урана, поместив образец двуокиси в герметичную
вольфрамовую капсулу, в центре которой имеется тонкая (диаметр 1,3 мм)
вольфрамовая трубочка с заваренным дном; трубочка погружена в
исследуемый образец, имитирует абсолютно черное тело и служит
для измерения температуры. Схема капсулы с образцом показана на
рис. 1.
Загрузка образца в форме таблетки с центральным отверстием в
капсулу и сварка капсулы проводятся в атмосфере аргона. Герметич-
45
ность капсулы проверяется непосредственно после сварки и после
опыта на масс-спектрографическом течеискатсле. Капсулу с образцом
нагревают индукционным методом. Если капсула сохраняет
герметичность, нагрев и плавление образца происходят под равновесным
парциальным давлением паров двуокиси урана, которое вблизи
температуры плавления составляет около 40 мм рт. ст. Однако
необходимо отметить, что нагрев герметичной капсулы, половина
объема (1,3 см3) которой занята образцом и половина объема заполнена
аргоном, должен привести к значительному росту общего давления
в ней. Хауснер заметил, что некоторые капсулы теряют
герметичность: в них появляется течь во время нагрева. Так как нагрев
проводится в вакууме, выход газа из ампулы сопровождается
непродолжительным коронным разрядом в электромагнитном поле
индукционной печи.
Эта методика при сохранении герметичности ампулы в
значительной степени ограничивает скорость испарения двуокиси урана,
подавляет диссоциацию и уменьшает в результате этого опасность
изменения состава образца во время опыта.
Исследование проведено Хауснером с большой тщательностью.
Использована двуокись урана высокой степени чистоты: содержание
в ней азота составило 0,09%, кремния 0,1% и суммарное
содержание остальных примесей 0,16%; отношение кислород — уран в
исходной окиси 2,002; период кристаллической решетки двуокиси урана
равен до опыта 5,4710±0,002 А, после опыта 5,4718±0,0004 А.
Изменение химического состава двуокиси урана после плавления
по сравнению с исходным спектрографическим анализом не
обнаружено. Однако среди большого числа анализируемых элементов
отсутствует вольфрам — единственный элемент, с которым двуокись
урана входит в соприкосновение во время опыта. Взаимодействие
двуокиси урана с вольфрамом при высокой температуре и особенно,
когда двуокись урана находится в жидком состоянии, в той или иной
степени должно происходить Данные о высокотемпературном
взаимодействии двуокиси урана с вольфрамом отсутствуют, но известно,
например, что двуокись циркония, имеющая примерно такие же
термодинамические свойства, как двуокись урана, начинает заметно
реагировать с вольфрамом при 2100° С.
Измерение температуры оптическим пирометром проводилось с
погрешностью ± 10° С.
Температуру плавления или точнее температуру кристаллизации
Хауснер определял по термическим кривым охлаждения жидкой
двуокиси урана после ее перегрева до 2940° С. Две такие кривые
показаны на рис. 2. Отчетливо выраженные изотермические площадки
на этих кривых располагаются при 2780 и 2805° С. Данные Хауснера
по определению температуры плавления двуокиси урана
представлены ниже [26].
Номер капсулы 1 2 3
*пл. °С 2790; 2810; 2805; 2800; 2800;2790
2830 2815 2780
/пл.ср,°С 2802ztl2
Хауснер поставил контрольные опыты, в которых две капсулы
нагревались одна до 2780° С, а другая до 2810° С с выдержкой при
этих температурах в течение 3 мин. После охлаждения образцы двуо-
46
кнси урана подвергались визуальному осмотру и микроскопическому
анализу. После нагрева до 2780° С никаких следов плавления
двуокиси урана обнаружено не было, тогда как образец, нагретый до
2810° С, имел структуру, свидетельствующую о полном его
плавлении.
В работах Элерта и Маргрейва [25], Ламберстона и Мюллера
[7] получены значения температуры плавления более высокие, чем
у Хауснера. Они равны соответственно 2860±45° С и 2878±22° С.
Во всех работах использовались образцы из чистой или очень чистой
двуокиси урана. Некоторые методические недостатки, имеющие место
в этих работах, позволяют думать, что полученные данные являются
завышенными. Это возможное увеличение яркостной температуры
образца, подвешенного на вольфрамовой проволоке, из-за
отраженной энергии концентратора тепла в работе Элерта и Маргрейва [25]
и визуальное определение момента плавления образца по изменению
состояния его поверхности в работе Ламберстона и Мюллера [7].
ДВУОКИСЬ ЦЕРИЯ
Из-за недостаточного количества экспериментальных данных
выбрать надежное значение температуры плавления двуокиси церия
трудно. Различие между результатами оригинальных исследований
и данными, приводимыми в справочной литературе [4—6],
составляет 600—800° С.
Очевидно, нужны дополнительные измерения температуры
плавления этого окисла. До тех пор наиболее вероятной температурой
плавления двуокиси церия можно считать температуру 2750° С,
рекомендуемую Ришкевичем [27]. Эта величина получена Хармоном,
Лохом и Остиным в институте им. Баттела в 1948 г. [15], сведения
об условиях эксперимента не известны.
ДВУОКИСЬ ЦИРКОНИЯ
В справочной литературе [3—6] для температуры плавления
двуокиси циркония приводятся данные от 2690 до 2765° С, т. е. со
сравнительно небольшим различием в 75° С. В то же время даже в
современных справочниках иногда рассматриваются данные о
температуре плавления без соответствующей поправки, связанной с
переводом оригинальной температурной шкалы в МПТШ-48 (например,
данные Жирновой [28] в справочнике [3], для которых поправка
составляет 50°С), а также нередко рекомендуются данные [11],
полученные в работах, не лишенных методических недостатков.
Значения температуры плавления двуокиси циркония,
полученные Жирновой [28], Картисом с сотр. [17] и Раффом с сотр. [29],
на 50—150° С выше результатов, полученных в других работах, в
которых для нагрева образцов использовалось ацетиленкислородное
пламя, приводящее к завышению экспериментальных данных из-за
отражения образцов лучистой энергии пламени.
Из остальных работ, в которых получены результаты, близкие
к 2700° С, наибольшего внимания заслуживают исследования
Ламберстона — Мюллера и Гунцеля [8], а также Эрли и Ламберстона
[30]. В этих работах зафиксирована температура плавления
двуокиси циркония, равная соответственно 2710±15°С и 2710 + 10° С.
Авторы проводили опыты r вакууме и в атмосфере гелия, использовали
совершенный способ определения момента начала плавления, темпе-
47
ратуру измеряли с высокой точностью. Однако две причины —
исследование двуокиси циркония, содержащей 2%НЮ2, а также
определенная вероятность загрязнения двуокиси циркония вольфрамом в
процессе опыта — могут вызвать заметную систематическую ошибку,
направленную в сторону занижения экспериментальных данных.
Итак, основываясь главным образом на работах Ламберстона
с сотр., можно предположить, что температура плавления двуокиси
циркония составляет величину, близкую к 2730° С.
ОКИСЬ КАЛЬЦИЯ
Окись кальция имеет высокую летучесть, достаточно легко
восстанавливается углеродом, образуя менее тугоплавкий карбид или
оксикарбид кальция, энергично взаимодействует с другими
тугоплавкими окислами и металлами.
Применительно к температуре плавления эти особенности окиси
кальция могут вызвать занижение экспериментальных данных как
из-за ослабления интенсивности излучения, так и в результате
изменения состава образца при нагреве и плавлении.
Несмотря на отмеченные трудности экспериментирования с
окисью кальция при высокой температуре, результаты оригинальных
исследований температуры ее плавления очень близки друг к другу.
Если не учитывать работу Шумахера [32], в которой изучалась окись
кальция технической чистоты, различие между опубликованными
данными по температуре плавления окиси кальция составляет всего 45° С.
Вероятно, такое положение можно объяснить тем, что даже в ранних
исследованиях объект изучения представлял собой материал
достаточно высокой степени чистоты, так как технология получения чистой
окиси кальция давно известна, эффективна и проста.
Таким образом, результаты, приведенные в работах Ольшанского
[33], Домана с сотр. [63] и др. [64], позволяют принять температуру
плавления окиси кальция 2630° С.
В справочной литературе [3—6] приводятся заниженные данные,
которые укладываются в пределы 2572—2587° С. Эти значения
температуры плавления окиси кальция получены в ранних работах Ка-
нольта [18], Раффа [29, 35], а также в работе Шумахера [32] для
технической окиси кальция.
Следует заметить, что в справочниках можно встретить значение
температуры плавления окиси кальция 2572° С, полученное Каноль-
том в 1914 г. с использованием оригинальной температурной шкалы.
Поправка для перевода этой температуры в МПТШ-48
составляет 42° С.
ОКИСЬ БЕРИЛЛИЯ
В большинстве справочников для температуры плавления ВеО
приводится одно и то же значение 2550° С [3—6]. Такое постоянство
в оценке температуры плавления ВеО имеет основание: температура
2550° С зафиксирована по крайней мере в 5 исследованиях из 13
известных (см. табл. 1).
Окись бериллия — вещество, с которым очень трудно работать
экспериментатору при высокой температуре. Окись бериллия имеет
достаточно высокую собственную летучесть, но в присутствии паров
воды летучесть окиси бериллия катастрофически возрастает из-за
образования газообразной гидроокиси бериллия. Во время нагрева,
48
начиная с 2000° С, окись бериллия активно взаимодействует с твердым
углеродом, его парами и углеродсодержащими газами с
образованием карбида бериллия. При температуре, превышающей 2100° С,
карбид бериллия разлагается; если при этом присутствует окись
бериллия, то одновременно протекает ее частичное восстановление с
изменением состава образца и выделением окиси углерода и пара
бериллия, упругость которого при 2100° С составляет около 0,1 ат.
Скорость разрушения окиси бериллия по такому механизму выше,
чем скорость ее испарения.
Очевидно, что при выборе условий эксперимента по измерению
температуры плавления окиси бериллия требуется особая
осторожность. Необходимо избегать углеродсодержащей атмосферы и
атмосферы с парами воды, нельзя использовать прямой нагрев
образца пламенем с помощью графитового нагревателя сопротивления,
нельзя применять графит для изготовления аппаратуры,
находящейся в зоне высокой температуры. Нарушение этих требований
приводит к изменению состава образца во время нагрева и к занижению
данных о температуре плавления окиси бериллия. Относительное
отклонение состава образца от стехиометрического зависит от
размеров образца, продолжительности нагрева, содержания паров и газов
в окружающей атмосфере и т. п.
Как следует из данных табл. 1, не всегда исследователи
учитывали влияние указанных факторов. Так, в работах Вартенберга [34,
36, 37] использовался пламенный нагрев в углерод-кислородной
атмосфере. Размещение образца в капсуле из двуокиси циркония лишь
в некоторой степени предупреждало воздействие атмосферы на
образец. В тех работах, в которых используется атмосфера воздуха
наряду с графитовым нагревателем или графитовой подставкой,
измеренная температура плавления окиси бериллия оказывается
заниженной. Так, в работе Раффа и Ло.шке [35] температура плавления
окиси бериллия определена в 2410° С. Графитовую подставку для
образца применяли и в некоторых современных работах [21].
Подробный анализ исследований по определению температуоы
плавления окиси бериллия приведен в работе [2]. Заслуживает
внимания работа Латта с сотр. [42], в которой температуру плавления
определяли на окиси бериллия чистотой 99,9%, плавление
осуществляли в вольфрамовом тигле при использовании индукционного
нагрева. Определение температуры проводили с помощью
фотоэлектрического пирометра, проградуированного в Национальном бюро
стандартов США. Скорость нагрева не превышала 35° С/мин. Образец
подвергали четырехкратному нагреву, проходящему через область
плавления. Определение температуры плавления и затвердевания
осуществляли с помощью анализа кривых нагрева и охлаждения.
Среднее значение температуры плавления оказалось равным
2434±10°С. Вероятно, следует признать, что в этой работе была
допущена систематическая ошибка, так как данные многих
исследований, выполненных в течение разных лет, при разном уровне
экспериментальной техники, достаточно хорошо совпадают друг с другом и
дают величину, более чем на 100° С превышающую 2434° С.
Последнее определение температуры плавления окиси бериллия
выполнено Бархатовым и др. [43]. Окись бериллия чистотой выше
99,88% помещали в вольфрамовый тигель, который затем заваривали
в вакууме с помощью электроннолучевой плавки. Температуру
измеряли оптическим пирометром, который периодически градуировали по
эталонным лампам. Исследование проводили методом термического
4—894
49
Таблица 2
Результаты определения температуры плавления окиси
бериллия [2]
Чистота
образца, %
99,9
99,8
Число
измерений
3
3
11 1
Среднее взвешенное
значение
W °с
2559
2562
2565
2578
2566
Погрешность,
°С
±9
±30
±10
±9
±12
Библиография
Кандыба и
др. [38]
Ольшанский
[32]
Райли и др.
[20, 21]
Бархатов и
ДР. [41]
анализа. Скорость измерения температуры 2—4° С/мин.
Зарегистрированные температуры плавления окиси бериллия лежали в
пределах 2577—2580° С. Результаты наиболее достоверных работ
приведены в табл. 2; сведения о температуре плавления в ней приводятся
по МПТШ-68.
ОКИСЬ ХРОМА
В некоторых современных справочниках для температуры
плавления окиси хрома приводится очень широкий интервал температуры.
Так, в справочнике [4] дается интервал 1988—2438° С, в
справочнике [3] — интервал 2270—2400° С.
Это связано с тем, что определения температуры плавления
окиси хрома, выполненные в основном 40—50 лет назад, проводились с
использованием несовершенных методик эксперимента и недостаточно
чистых материалов. Наиболее тщательно температуру плавления
окиси хрома определил Мак-Нейли и др. [19]. В этой работе
исследовалась окись хрома с суммарным содержанием примесей около
0,6%. Стехиометричность окиси хрома не определяли. Образец из
порошка окиси хрома прессовали в виде стержня и подвешивали в
графитовом тигле или плавили в нем. Тепло подводили с помощью
индукционного нагрева. Эксперименты проводили на воздухе или в
атмосфере азота, за температуру плавления принимали температуру
в тот момент, когда на образце появлялась первая капля жидкой
фазы. Согласно данным этой работы, температура плавления окиси
хрома составляет по МПТШ-48 в азоте 2315±20°С, на воздухе
2330 ±20° С.
ОКИСЬ АЛЮМИНИЯ
По измерению температуры плавления окиси алюминия известно
несколько десятков экспериментальных работ, что связано не только
с расширением области применения окиси алюминия в технике, но
50
также со стремлением исследователей использовать температуру
плавления окиси алюминия в качестве вспомогательной
фиксированной точки в температурной шкале. Ряд определений температуры
плавления окиси алюминия выполнен предельно точно в специальных
пирометрических лабораториях.
Результаты экспериментальных работ по определению
температуры плавления окиси алюминия, выполненных в период с 1956 по
1967 г., приведены в табл. 1. Данные различных авторов
сравнительно мало отличаются друг от друга, однако наиболее строго
температуру плавления окиси алюминия определили Шнейдер и Мак-Да-
ниэл[53], Мак-Нейли с сотр. [19] и Райли [20, 21].
В исследовании Шнейдера и Мак-Даниэла [53], а также в
работах Райли [20, 21] особое внимание было уделено выяснению
причин, искажающих данные о температуре плавления окиси
алюминия. Из ряда факторов, которые принципиально могут оказывать
влияние на ее температуру плавления, были изучены следующие:
структура исходной окиси алюминия; ее химический состав и
изменение химического состава при нагреве и плавлении; взаимодействие
с материалом измерительной ячейки; взаимодействие с окружающей
атмосферой; степень отклонения условий измерения от идеальной
модели абсолютно черного тела.
Так, Шнейдер и Мак-Даниэл определяли температуру плавления
образцов, приготовленных из четырех различных монокристаллов
окиси алюминия и из ее порошка. Исходный порошок представлял
собой аморфное вещество; во время нагрева он превращался в
кристаллическую а-А1203. Суммарное содержание металлических
примесей в монокристалле I и в порошковой окиси алюминия одинаковое
(табл. 3). Образцы из этих материалов изготавливали с предельной
тщательностью. От монокристалла образцы отрезали алмазной пилой,
затем промывали спиртом, кипятили в НС1, промывали
дистиллированной водой и сушили при 140° С. Из порошка образцы таких же
размеров (диаметр 0,8 и длина 0,8 мм) получали прессованием с
добавкой в шихту дистиллированной воды.
Результаты исследования (табл. 4) показали, что окись
алюминия максимальной степени чистоты независимо от структуры —
монокристаллической или поликристаллической — плавится при одной
и той же температуре: 2051 и 2050,5° С соответственно в вакууме и
в вольфрамовом контейнере.
Как видно из табл. 3, в наиболее загрязненном исходном
монокристалле IV суммарное содержание металлических примесей
составляет около 0,3%, эта величина в 15 раз больше, чем содержание
примесей в монокристалле I. Тем не менее, образцы этих
монокристаллов показали одинаковую температуру плавления 205ГС (в
вакууме, в вольфрамовом контейнере).
Представление об изменении состава окиси алюминия во время
опыта дает сравнение данных спектрографического и химического
анализов монокристалла I в исходном состоянии, а также после
нагрева до 2047° С (ниже температуры плавления) и до 2054° С (выше
температуры плавления). Данные табл. 3 показывают, что при этом
возрастает содержание кремния, хрома, магния, титана и др., а
суммарное содержание металлических примесей увеличивается
соответственно до 0,15 и 0,26%, но не выходит за пределы содержания
примесей в монокристалле IV. На высокое содержание вольфрама в
образце после плавления можно не обращать внимания, так как, по
утверждению авторов, оно является следствием механического за-
4*
51
Таблица 3
Состав образцов А1203, %, не более [53]
Примесный
элемент
Ag
В
Са
Сг
Си
Fe
Mg
Mn
Ni
Pb
Si
Sn
Ti
W
Сумма
Монокристалл I
в исходном
состоянии
0,001
0,0001
0,0001
0,0001
0,01
0,001
0,0001
0,01
0,0224
после нагрева
до 2047 °С*1
0,0001
0,01
0,01
0,001
0,001
0,001
0,0001
0,1
0,001
0,01
0,1522
после нагрева
до 2054 °С*2
0,0001
0,01
0,01
0,001
0,1
0,01
0,0001
0,001
0,01
0,1
0,01
0,01
(10,0)*4
0,2622
Монокристалл в исходном состоянии
II
0,001
0,001
0,01
0,01
0,001
0,001
0,01
0,001
0,001
0,1
0,01
0,01
0,1561
III
0,001
0,001
0,01
0,01
0,001
0,001
0,01
0,001
0,001
0,1
0,01
0,01
0,156
IV
0,001
0,001
0,001
0,01
0,001
0,01
0,1
0,001
0,1
0,01
0,325
Порошок
поликристаллический
исходный
0,0001
0,0001
0,001
0,001
0,01
0,01
0,0222
после
прессования и
плавления*1
0,001
0,0001
0,01
0,01
■ 0,01
0,0411
*' Нагрев без плавления.
*2 Нагрев с плавлением в вакууме.
*3 Плавление в вакууме бесконтактным методом.
*4 Результат механического загрязнения образца материалом подложки.
Таблица 4
Температура плавления А1203 [53]
Образец
Монокристалл:
I
II
III
IV
Порошок
поликристаллический
после переплавки в
вакууме
Газовая среда1
Вакуум
8-Ю-5 мм рт. ст.
Гелий
Аргон
Гелий
Аргон
Воздух
Гелий
Вакуум
7,5-10-5 мм рт. ст.
Вакуум
З'Ю-5 мм рт. ст.
Гелий
»
Аргон
Вакуум
7- Ю-5 мм рт. ст.
Воздух
Материал
контейнера
Вольфрам
»
»
»
»
Иридий
1 Вольфрам
»
Иридий
Вольфрам
Иридий
Вольфрам
»
Иридий
(МПТШ-48)
2051
2041
2045
2039,5
2045
Ликвидус
2038
Солидус
<2015
2040,5
2051
2045,5
2041
2036
2045
2050,5
Ликвидус
2037
Солидус
2026
При давлении 760 мм рт. ст.
грязнения при удалении образца окиси алюминия с вольфрамовой
подложки. Как уже было отмечено, температура плавления
монокристалла IV не отличается от температуры плавления наиболее
чистых монокристаллов I и поликристаллического порошка окиси
алюминия.
Эти данные имеют большое значение, так как изменение состава
во время опыта наиболее часто приводит к ошибке при измерении
температуры плавления окислов. Они показали, что незначительное
изменение состава образца, не превышающее в данном случае 0,3%,
вызывает снижение температуры плавления окиси алюминия,
которое не улавливается современными приборами для измерения
температуры. Авторы полагают, что снижение температуры
плавления окиси алюминия не превышает Г С при наличии в ней примесей
в количестве до 0,2% (мол.).
Взаимодействие исследуемого окисла с материалом контейнера
или тигля — еще одна часто встречающаяся причина искажения
данных о температуре плавления окислов. В работе [53] независимо от
53
использованного материала — вольфрама или иридия — при
отсутствии влияния других факторов окись алюминия плавится конгруэнтно,
что свидетельствует о плавлении в условиях равновесия фаз
одинакового химического состава. Тем не менее, различие в величине
температуры плавления окиси алюминия при использовании
измерительной ячейки из вольфрама или иридия имело место.
Поэтому для решения вопроса, происходит ли взаимодействие
окиси алюминия с вольфрамом и иридием, авторы приготовили смеси
из порошков окиси алюминия и тугоплавкого металла с содержанием
последнего 10; 33,3 и 50% (мол.). После перемешивания образцы из
этих смесей прессовали аналогично образцам из порошка чистой
окиси алюминия. Затем образцы нагревали до высокой температуры,
проводили рентгеноструктурный анализ и определяли температуру
начала плавления для каждого состава.
Результаты исследования подтвердили, что твердофазного
взаимодействия в системах AI2O3—W и А1203—1г не происходит вплоть
до температуры на 15° С ниже температуры плавления окиси: в смеси
обнаружены только а-А1203 и соответственно вольфрам или иридий.
Температура начала плавления всех исследованных составов
практически совпадает с температурой плавления окиси алюминия, в этом
случае температура конца плавления смеси должна совпадать с
температурой начала плавления вольфрама или иридия, а количество
жидкой фазы в интервале температуры плавления не должно
изменяться.
Результаты исследований Шнейдера и Мак-Даниэла [53]
свидетельствуют о необходимости соблюдения условий, обеспечивающих
минимальный контакт между образцом и тиглем во время опыта.
Влияние атмосферы, окружающей образец окиси алюминия, на
температуру его плавления, изученное Шнейдером и Мак-Даниэлом,
заключается в том, что использование любой газовой атмосферы —
гелия, аргона, воздуха — приводит к уменьшению температуры
плавления окиси алюминия по сравнению с результатами опытов, когда
образец находится в вакууме. Основанием для такого заключения
могут служить данные, представленные в табл. 4. Не исключено, что
снижение температуры плавления окиси алюминия в газовой среде
связано с бомбардировкой поверхности образца атомами, молекулами
или ионами газа, имеющими при высокой температуре достаточно
большую скорость и вызывающими нарушение кристаллической
структуры в поверхностных слоях вещества с соответствующим
снижением температуры плавления. Это подтверждается результатами
работ Козловского и Шляковой [54], а также Барбера и Тиха [55],
в которых экспериментально установлен факт изменения структуры
в поверхностных слоях образцов из окиси алюминия после их
нагрева до высокой температуры в газовой среде, тогда как
аналогичный нагрев образцов в вакууме не вызывает изменения структуры.
В работе Шнейдера и Мак-Даниэла [53] также проведено
сравнение вольфрамовых и иридиевых контейнеров для установления
степени их соответствия модели абсолютно черного тела. Устройство
измерительной ячейки показано на рис. 3. Образец размещается на
подставке, которая находится на дне внутреннего тигля. В
подставке диаметром 8 мм имеется углубление диаметром 0,75X0,7 мм для
образца диаметром 0,75X0,75 мм. Центр углубления и,
следовательно, образца, находится на расстоянии 1,2 мм от центровой оси ячейки.
Используя метод Коффи [56], проверенный позднее де
Боссом [60], степень черноты измерительной ячейки рассчитывали по
54
ее геометрическим размерам и оптическим свойствам материала. Для
измерительной ячейки, изготовленной из вольфрама, степень черноты
оказалась равной 0,999 при комнатной температуре и 0,998 при
2700° С. Отклонение степени черноты от единицы на 0,002 вызывает
расхождение между истинной и яркостной температурой не более чем
на 1° С. Степень черноты для ячейки из иридия составляет 0,998 при
комнатной температуре. Для расчета степени черноты при высоких
температурах отсутствуют необходимые данные.
Проведенные авторами
расчеты справедливы лишь в тех
случаях, когда в пределах
измерительной ячейки сохраняется
равномерная температура и когда
внутренняя поверхность ячейки не
является зеркальной, а полностью
рассеивает падающие на нее лучи.
Однородность температуры
измерительной ячейки
обеспечивается выбором правильного
положения ячейки в тепловом поле,
использованием тепловых экранов и
тепловой изоляции ячейки, а
также за счет высокой
теплопроводности вольфрама и иридия,
характерной для металлов. Измерения
оптическим пирометром яркостной
температуры на поверхности
внешнего тигля не обнаружили
градиентов температуры, превышающих
точность измерения. Условия же
работы внутреннего тигля
являются более благоприятными:
образцы монокристаллической окиси
алюминия, расположенные в
разных частях внутреннего тигля,
ведут себя при нагреве совершенно
одинаково.
Для создания рассеивающей
поверхности тигли как из
вольфрама, так и из иридия, подвергали
пескоструйной обработке. Ее
эффективность строго не определена,
но отмечено, что после
однократного нагрева до высокой температуры поверхность вольфрама
остается матовой, а поверхность иридия становится зеркально-блестящей.
Последнее может вызвать большее отклонение степени черноты
измерительной ячейки от единицы, чем получено расчетом.
Из сравнения вольфрама и иридия следует, что более
предпочтительным материалом для изготовления измерительной ячейки
является вольфрам. Для иридиевой измерительной ячейки, единственным
преимуществом которой является возможность проведения опытов в
атмосфере воздуха, степень черноты оказывается ниже, чем для
вольфрамовой, что приводит к занижению данных о температуре
плавления. Анализ результатов работы Шнейдера и Мак-Даниэ-
ла[53] по определению температуры плавления окиси алюминия под-
W//////////\/////////"/t
Рис. 3. Конструкция
измерительной ячейки для определения
температуры плавления окиси
алюминия:
/ — верхние радиационные
экраны; 2 — концентратор тепла,
вольфрамовый; 3 —
вольфрамовый контейнер; 4 — образец;
5 —вольфрамовая подложка
образца; 6 — нижние
радиационные экраны
55
Результаты совместного определения температуры
Чистота объекта
после опыта
Тип печи
Материал I
контейнера
Определение
момента
плавления
Прибор или
метод
измерения
0,01% Са;
0,02% Si
Не опр.
0,006—
0,01%Мо
0,01% Fe203;
210—б% N2
<0,005%
щелочных
металлов;
<0,0002%
Са, Mg;
<0,01% W
Не опр.
0,001% Fe;
0,0001% Si
Сопротивления с
графитовой трубой
Вольфрамовая
печь
сопротивления
То же
Ir, W
или Та
Графитовая
сопротивления
Сопротивления с
нагревателем из
стабилизированной Zr02
Вольфрамовая печь
сопротивления
Сопротивления с
танталовым
нагревателем
Солнечная
W
Мо
Мо
W
или
Мо
W
Начало
образования
жидкой фазы
на плоской
поверхности
образца
То же
Анализ:
а) кривых
нагрева;
б) кривых
охлаждения
Начало
образования
жидкой фазы
на плоской
поверхности
образца
Анализ
кривых
охлаждения
Анализ
кривых нагрева
Анализ
кривых
охлаждения
Оптический
пирометр,
АЧТ
термопара
Pt/Pt— tfh
до 1700°С
Термопара
W—5% Re
W—26% Re
вблизи
образца
Оптический
пирометр,
АЧТ
То же
Оптический
пирометр,
АЧТ
То же
Оптический
пирометр,
направленный
на образец.
Проведена
коррекция
на излучение
В скобках указаны минимальные и максимальные значения tn
56
Таблица 5
плавления окиси алюминия [61]
Системы
калибровки
Атмосфера
печи;
давление,
мм рт. ст.
Число
опытов;
погрешность, °С
*пл.ср» °С
МПТШ-48*
МПТШ-68
МПТШ-48
t _ =1083° С
пл. Си
t A =1063° С
пл. А и
пл. Pf'
•■ 1769° С
/ \ = 1063° С
пл. Аи
t _. = 1552° С
пл. Pd
t _, = 1769° С
пл. Р/
t D. =1960° С
пл. Rh
t =: 1063° С
пл. Аи
'пл.Аи = 1063° с
t п. = 1769° С
пл. Pt
t 0. = 1960° С
пл. Rh
t
пл.
<пл
t
пл.
t
пл.
л =1063° С
Аи
Pd = 1552°C
pt = 1769oC
я = 1063° С
Аи
—
Аргон,
760
Вакуум,
10~4-
ю-6
Воздух,
760
Аргон,
760
Вакуум,
2. Ю-4;
Аргон,
100
Вакуум,
2.10-6;
Аргон,
10-20
Аргон,
20
Воздух
оздух
-760
10; d=2
4; ±6
11; =Ь4,5
10; rt3,7
14; =ЬЗ,6
10; =t7
24; ±6
8; ±6
30; ±15
2040
(2037—
2043)
2042
(2040—
2044)
2051,4
(2049,9—
2054,4)
2050,8
(2050,1—
2051,2)
2050,6
(2049,5—
2052,1)
2051
(2042—
2056)
2051
(2047—
2056)
2052
(2050—
2055)
2070
(2043—
2086)
2043
2045
2054
2054
2054
2054
2054
2055
2073
определенные по шкале МПТШ-48.
57
тверждает это положение: если образец окиси находится в иридиевой
ячейке, то результат измерения ее температуры плавления
оказывается, как видно из табл. 4, на 5—7° С ниже по сравнению с
данными, полученными при использовании вольфрамовой
измерительной ячейки.
В 1965 г. была создана рабочая группа по определению
вспомогательных температурных реперных точек [61]. Первым объектом
исследования послужила окись алюминия. Она была выбрана из-за
высокой химической стойкости, широкой распространенности,
сравнительно простых и недорогих способов получения ее с повышенной
степенью чистоты.
Девяти лабораториям в разных странах были выделены
унифицированные образцы окиси алюминия, переплавленной в солнечной
печи [61]. Применение таких унифицированных образцов исключало
ошибку, связанную с различием состава объекта исследования. После
переплава в солнечной печи чистота окиси алюминия 99,994%-
Лабораториями использовались различные методы определения
температуры плавления, различные типы нагревательных устройств;
материалами для изготовления измерительных ячеек служили
вольфрам, молибден и иридий. Результаты определения температуры
плавления окиси алюминия приведены в табл. 5. На основе анализа
этих данных Шнейдер [61] установил, что температура плавления
окиси алюминия составляет 2054±6°С (МПТШ-68). С этими
рекомендациями согласны и авторы обзора [2].
окись иттрия
Упомянутая выше рабочая группа комиссии по высоким
температурам и огнеупорам произвела также совместное определение
температуры плавления окиси иттрия [2]. Окись иттрия в качестве
объекта исследования была выбрана из-за того, что имеет высокую
химическую стойкость, обладает низкой упругостью пара и относительно
слабо взаимодействует с вольфрамом, который может служить мате-
Результаты совместного определения темпера
Определение момента плавления
Начало образования жидкой
фазы на плоской поверхности
образца
Термический анализ
Исследование образца после
охлаждения
Термический анализ
То же
» »
» »
Ступенчатый нагрев
Материал тигля
или подложки
Вольфрам
»
Вольфрам,
герметичный тигель
То же
Вольфрам
»
Вакуум, мм
рт. ст.,
газовая среда
МО-*
10-2—Ю-з
10-а—Ю-8
Гелий
ю-*
58
риалом для измерительной ячейки. Помещенная в вольфрамовый
тигель окись иттрия после 2-ч выдержки при 2407° С в вакууме имеет
состав, близкий к Y202,997 [84].
При совместном определении температуры плавления окиси
иттрия использовались образцы, переплавленные в солнечной печи,
имевшие чистоту 99,999%. Результаты измерений приведены в
табл. 6. Работы Кандыбы, Саты и Ота могут быть исключены из
рассмотрения при установлении наиболее точного значения
температуры плавления окиси иттрия. Среднее из остальных измерений
составляет величину 2436±8° С.
ДВУОКИСЬ ТИТАНА
В современной справочной литературе [3—6] для температуры
плавления двуокиси титана приводятся значения от 1800 до 1920° С.
Между тем анализ экспериментальных данных показывает, что
если из перечисленных в табл. 1 не учитывать работы Раффа с сотр.
[71] и Статтона [72], то результаты остальных работ располагаются
в диапазоне 1820—1870° С, причем величина температуры плавления
двуокиси титана в пределах 1840—1850° С получена в шести
работах (см. табл. 1).
В исследовании Брауэра и Литке [83] показано, что температура
плавления двуокиси титана заметно падает даже при незначительной
потере кислорода. Они измеряли температуру плавления двуокиси
титана в атмосфере, в которой парциальное давление ее
составляющих (кислорода и аргона) изменяли в широких пределах. Условия
опытов и полученные результаты представлены в табл. 7. Изменение
состава двуокиси титана во время плавления наблюдается, если
парциальное давление кислорода в атмосфере, окружающей образец,
составляет менее 300 мм рт. ст.; соответственно изменяется
температура плавления. Если давление кислорода находится в пределах от
300 до 600 мм рт. ст., то химический анализ не улавливает изменения
состава двуокиси титана, однако температура плавления изменяется
Таблица 6
туры плавления окиси иттрия [2]
Число
измерений
6
17
16
6
t °с
2434
2436
2409
2430
2438
2446
2466
2422
Погрешность, °С
±10
=1=6
±9
±8
=t:10
±25
1 ±15
±4
Библиография
Аккерман, Раух [84] (США)
Джонс [85] (Австралия)
Кандыба (СССР)
Кенисарин, Чеховской [86, 87]
(СССР)
Кутюр, Фойкс (Франция)
Ногучи (Япония)
Сэта. Ота (Япония)
Шнейдер, Мак-Даниэл [88] (США)
59
Таблица 7
Влияние газовой среды на температуру плавления двуокиси титана
Давление газа,
мм рт. ст.
кислорода
ю-3
(вакуум)
15
159
(воздух)
аргона
780
745
С °с
1795
1800
1820
1830
х**
1,969
1,975
1,990
1,996
Давление газа,
мм рт. ст.
кислорода
300
! 500
600
760
1140
аргона
460
260
160
*
t , °с
1840
1860
1870
1870
1870
X**
2,000
2,000
2,000
2,000
2,000
* Точность измерения ±15° С.
** Значение х в соединении TiOx, определенное после плавления по
химическому анализу.
на 30° С, что в два раза превышает максимальную ошибку
измерения температуры. Температура плавления двуокиси титана
становится постоянной только при давлениях кислорода, превышающих
600 мм рт. ст. В этих условиях
температура плавления
двуокиси титана составляет 1870° С.
Таким образом, Брауэр и
Литке опытным путем
установили причину занижения
температуры плавления двуокиси
титана, которая заключается в
изменении состава образцов
вследствие диссоциации при
нагреве в вакууме, на воздухе
и в атмосфере аргона. Ни в
одной из других
экспериментальных работ (см. табл. 1), кроме
работы Сигурдсона и Гола
[77], окислительная
(кислородная) атмосфера не
применялась. Не удивительно поэтому,
что в них была получена
температура плавления Ti02 в
пределах 1820—1850° С.
Следует отметить, что
Брауэр и Литке использовали для
нагрева образцов солнечную
печь, а для измерения
температуры — радиационный
пирометр. Выбор этих методов нагрева и измерения температуры не
является рациональным; он, вероятно, вызвал появление значительной
погрешности (±15°С) при измерении температуры.
Тем не менее из известных работ исследование Брауэра и Литке
представляется наиболее надежным, что дает основание принять ре-
Рис. 4. Линии ликвидуса в системе
Ti-O:
сплошная — по данным Брауэра и
Литке [83], полученным в атмосфере
аргона (1 ат), темные точки — температура
плавления Ti02 в атмосфере кислорода
под давлением 600 (/); 150 (2) и 15 (3)
мм рт. ст.; пунктирная — по данным
де Вриеза и Роя [64]
60
комендованную ими температуру (1870° С) за температуру
плавления двуокиси титана.
Брауэр и Литке [83] измерили также температуру плавления
(ликвидус) окисных соединений титана в интервале концентраций от
TiOi,45 до ТЮ2. Эти измерения были проведены в атмосфере аргона.
Результаты исследования, представленные на рис. 4, показывают, что
соединение Ti203(TiOi,5) плавится конгруэнтно при температуре
1820±15°С, а между составами Ti203 и ТЮ2 образуется эвтектика
при температуре 1660±15°С.
ОКИСЛЫ РЕДКОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Большую группу тугоплавких окислов составляют окислы
скандия, иттрия, лантана и лантанидов, т. е. окислы редкоземельных
металлов (РЗА1).
Из справочной литературы по температуре плавления окислов
РЗМ следует, что для полуторных окислов РЗМ она находится в
пределах 1900—2600° С [89]. Расхождение по температуре
плавления отдельных окислов оказывается чрезмерно большим даже, если
сравнить данные из справочников, вышедших из печати в одно и то
же время. Для некоторых окислов, например для Eu203 и Nd203,
расхождение достигает величины в 250—400° С. Данные
экспериментальных определений температур плавления окислов редкоземельных
металлов приведены в табл. 8.
Из известных исследований по измерению температуры плавления
окислов РЗМ почти всем требованиям, которые предъявляются к
таким исследованиям, удовлетворяет работа Шнейдера и Варинга
[86]. К сожалению, в ней изучено только два окисла — Sc203 и
Eu203.
В работе Ногучи и Мизуко [90] для нагрева образцов
использовали солнечную печь; при измерении температуры не были
обеспечены условия абсолютно черного тела, а принятая авторами величина
степени черноты для жидких окислов РЗМ вряд ли является
надежной. Результаты этой работы, по-видимому, могут содержать
систематическую ошибку.
Аналогичные недостатки методики свойственны также работе
Картиса и Джонсона [91], в которой для нагрева образцов
использовали ацетилен-кислородную печь. Попытки авторов приблизить
образцы по условиям их излучения к абсолютно черному телу
оказались малоуспешными. Главным образом из-за этой причины
погрешность при измерении температуры оказалась достаточно
большой: ±50° С.
Чтобы избежать отмеченных методических недостатков, Фойкс
[31, 92] измерял температуру образца при выключенном световом
нагреве, т. е. он фиксировал температуру затвердевания окислов во
время их охлаждения. Фойкс предпринял также попытку создать
оригинальную модель абсолютно черного тела. В экспериментальной
установке с солнечной печью горизонтально расположенный водо-
охлаждаемый гарнисажный тигелек с исследуемым окислом
приводится во вращение со скоростью до 1400 об/мин. При этом в тигле
на поверхности заранее отформованной полости глубиной 15—30 и
диаметром 6—7 мм образовалась пленка жидкой окиси толщиной
в несколько миллиметров на каждой стороне. Пирометр направля-
61
Таблица 8
Температура плавления окислов редкоземельных металлов
(экспериментальные данные)
Окисел
Se203
Y203
La203
Ce02
Рг203
Nd203
Sm203
т .
1 пл'
°С
2405
2410
2458
2435
2376
2436
1859
2307
2210
2300
2256
2217
1973
2600
2395
2749
2296
2183
2127
2272
2310
I 2233
2211
2300
2350
2320
2269
2262
2310
Библиография
[89]
[35]
[951
[31,92]
[901
[2]
[95]
[74]
[71
[30,92]
[90]
[931
[951
[74]
[931
[15]
[921
[90]
[93]
[71
[92]
[90]
[93]
[24]
[91]
[92]
[90]
[93]
[94]
Окисел
Еи2Оэ
Gd203
Tb203
Dy203
Ho203
Ег203
Tm203
Yb203
Lu203
T
ПЛ*
°C
2050
2240
2320
2291
2002
2330
2350
2395
2330
2322
2390
2303
2292
2340
2391
2228
2352
2396
2330
2367
2400
2344
2387
2341
2392
2411
2333
2372
2373
2467
Библиография
[24]
[89]
[92]
[90]
[93]
[24]
[91]
[92]
[90,94]
[93]
[92]
[90]
[931
[24]
[92]
[90]
[93]
[921
[90]
[93]
[92]
[90]
[93]
[90]
[93[
[92]
[90]
[93]
[901
[93]
ется па донную часть этой полости, имитирующей абсолютно черное
тело. Измерение температуры затвердевания, которая может быть
ниже температуры плавления, и одновременно недостаточно строгая
модель абсолютно черного тела позволяют предположить
возможность некоторого занижения экспериментальных данных по
температуре плавления. Однако рис. 5 показывает, что занижение данных
у Фойкса отсутствует. Кривые охлаждения окислов, которые
приводит Фойкс в своих работах, подтверждают отсутствие
переохлаждения жидкой фазы для всех изученных окислов — Zr02, CaO, ВаО,
SrO, окислов РЗМ и др., кроме А1203, несмотря на высокую
скорость охлаждения (до 120 град/с).
Мордовии, Тимофеева и Дроздова [93] определили температуру
плавления почти всех окислов РЗМ в условиях, предотвращающих
62
загрязнение образцов окислов и изменение их состава во время
опытов. Однако измерение температуры они проводили оптическим
пирометром, направляя его на открытую поверхность образца. Таким
образом, измерялась яркостная температура; кроме того, авторы не
указывают, при выключенном или при работающем нагревателе
проводились измерения температуры. Данные о степени черноты жидких
тугоплавких окислов отсутствуют. Поэтому для определения ис-
*ПЛ; О
2500
2400 V
2300
2200
2100
Г ODD П й g \^L
_ а А * л Л Л а
И а а УК-г *
о. *S* •V/ • 1
L ? • □ j
Г Д ж*
57 58 59 60 61 62 65 64 65 66 67 68 69 70 71
La Се Рг U Pm 5m Eu Gd Tb Щ Но Ег Tu Yb Lu
Рис. 5. Температура плавления окислов редкоземельных металлов по данным:
/ — Мордовина с сотр. [93]; 2 — Ногучи и Мизука [90]; 3 — Фойкса [92]; 4 —
других авторов; 5 — средневзвешенные значения
тинной температуры авторы градуировали оптический пирометр по
известным данным о температуре плавления таких тугоплавких
окислов, как А1203, Сг203, Y203, СаО, Zr02, Hf02 и MgO.
К сожалению, при использовании этой методики могут
возникнуть ошибки. Во-первых, оптический пирометр следует градуировать
только по рекомендованным фисированным точкам. Во-вторых,
авторы не указывают, какая именно температура плавления была
принята для каждого из окислов, использованных для градуировки
пирометра. А эти сведения очень важны, так как расхождение
справочных данных по температуре плавления окислов, использованных для
градуировки, достигает не только десятков, а иногда сотен градусов.
Далее, использованный авторами прием предполагает равенство
степени черноты для всех окислов в жидком состоянии, что также
нельзя признать правильным. Имеющиеся данные о степени черноты
твердых тугоплавких окислов [3] свидетельствуют о том, что эта
величина колеблется от 0,2 до 0,8 в зависимости от природы окисла
и его температуры.
И наконец, полученная авторами поправка, т. е. разница между
яркостной и истинной температурами, составляет 5° С при 2000° С и
8° С при 2800° С, что, с одной стороны, в 4—6 раз меньше, чем
погрешность измерения температуры (±30°С), и, с другой стороны,
практически предполагает равенство степени черноты градуировочных
и исследуемых окислов единице. В работе отмечается, что момент
63
начала плавления авторы определяли по исчезновению граней на
конце образца, но для этого образец должен быть видимым, т. е.
степень его черноты должна заметно отличаться от единицы.
Авторы работы [93] показали, что температура плавления
полуторных окислов РЗМ растет с увеличением атомного номера
соответствующего РЗМ. На рис. 5 представлена эта связь, не только по
данным работы О. А. Мордовина с сотр., но и по данным других
известных исследований. Она дает возможность предсказать температуру
плавления окиси прометия, для которой экспериментальные данные
отсутствуют, а также уточнить температуру плавления других
окислов.
В работе Уисни и Пъянковского [24] возможно занижение
экспериментальных данных вследствие, по крайней мере, двух причин.
Во-первых, не исключено изменение состава окислов во время
опытов и, во-вторых, принятое авторами значение степени черноты (0,9—
0,95) является недостаточно обоснованным. Эти причины, учитывая
различную склонность окислов РЗМ к диссоциации и восстановлению,
обусловили разную погрешность при изменении температуры
плавления отдельных окислов. Так, авторы определили величину ошибки
при измерении температуры плавления:
SmA в ± 50 °С; Eu203 в ± 30 °С;
Gd203 в ± 20 °С и Dy203 в ± 10 °С.
Данные Ламберстона с сотр. [7] также могут оказаться ниже,
чем действительная температура плавления, так как в их работах
объект исследования не всегда имел достаточно высокую степень
чистоты. Надо отметить, что в остальных исследованиях изучались
весьма чистые окислы: содержание основного вещества в исходных
образцах составляло не менее 99,0%, а для многих окислов 99,6—
99,9%. Ногучи и Мизуко [90] исследовали исключительно чистые
окислы, содержание примесей в которых не превышало 0,05%.
Г л а в а II
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
жидких тугоплавких окислов
Систематических исследований физико-химических
свойств жидких тугоплавких окислов выполнено
сравнительно мало, хотя аналогичных работ по свойствам
шлаков, петрургического сырья, стекла, легкоплавких
жидких окислов известно достаточное количество. Это
связано, вероятно, с трудностями, которые приходится
преодолевать исследователям, работающим в области
высоких температур. Жидкие тугоплавкие окислы,
однако, характеризуются меньшей химической
активностью, чем тугоплавкие металлы и многие тугоплавкие
соединения в расплавленном состоянии. Наличие мате-
64
риалов, слабо взаимодействующих с жидкими
тугоплавкими окислами, позволяет достаточно часто
использовать для измерения их свойств обычные
физико-химические методы, применяющиеся в исследованиях
легкоплавких жидкостей.
Из известных работ, посвященных изучению физико-
химических свойств жидких тугоплавких окислов,
значительную часть составляют исследования жидкой
окиси алюминия. Объясняется это, главным образом, тем,
что исследования жидкой окиси алюминия приобрели в
последнее время особую актуальность в связи с
использованием порошкообразного алюминия в твердом
ракетном топливе [1].
1. ПЛОТНОСТЬ ЖИДКИХ
ТУГОПЛАВКИХ ОКИСЛОВ
ОКИСЬ АЛЮМИНИЯ
Первое упоминание об экспериментальном
определении плотности жидкой окиси алюминия относится к
1936 г. (табл. 9), когда Вартенберг, Венер и Саран [2]
Таблица 9
Плотность жидкой окиси алюминия при температуре плавления
Год
1936
1959
I960
1968
1969
1969
1971
1973
1973
1973
Плотность,
г/см3
2,5
2,97
3,014
3,05
2,7
3,00
3,01
3,03
3,01
3,05
Погрешность
измерения
г/см1 | % J
±0,1
±0,06
=1=0,09
±0,135
±0,1
±0,06
±0,07
±0,02
±4
±2
±3
±5
±3,5
±2
±2,5
±0,7
Метод измерения
Лежащая капля
То же
Гидростатическое
взвешивание
Летящая капля
Лежащая капля
Гидростатическое
взвешивание
Плавление в
капилляре
Давление газа в
пузырьке
1 Лежащая капля
Давление газа в
пузырьке
Библиография
Вартенберг, Венер,
Саран [2]
Кинджери [3]
Киршенбаум,
Кэхилл [7]
Маурах и др. [5]
Зубарев, Митин
и др. [8]
1 Митин, Нагибин
Расмуссен Нельсон
Елютин, Митин,
Анисимов [10]
Митин, Семененко
и др. [12]
Бархатов и др.
|[П1
5—894
65
провели измерение с помощью метода лежащей
капли. Образец из «чистой» окиси алюминия
расплавляли ацетилен-кислородным пламенем на подложке из
молибдена. Температуру окисного образца авторы не
измеряли, считая ее равной температуре плавления окиси
алюминия. Полноту
расплавления образцов, их
состав и структуру после
опыта, а также состав
атмосферы печи не
контролировали. Наблюдение за
формой капли жидкой
окиси проводили только с
одной стороны. Это не
позволяет считать
достаточно обоснованным
предположение авторов о
том, что капля жидкой
окиси на плоской
молибденовой подложке
является совершенным телом
вращения. Можно назвать
несколько причин —
наклон подложки,
ориентированная шероховатость
подложки, градиент температуры в плоскости подложки
и др., которые могут вызвать деформацию капли и
отклонение ее формы от формы совершенного тела
вращения. Объем капли, лежащей на плоской
молибденовой подложке, рассчитывали, используя таблицы Баш-
форта и Адамса. В работе [2] получена величина
плотности жидкой окиси алюминия при температуре
плавления, равной 2,5 г/см3 при погрешности ±0,1 г/см3, или
±4%.
Спустя 23 года измерение плогности жидкой окиси
алюминия провел Кинджери [3]: он использовал для
этого тот же метод капли, лежащей на молибденовой
подложке; проводил наблюдение за формой капли
только с одной стороны; не измерял температуру опыта, счи-
тая>ее «несколько превышающей температуру
плавления» окиси алюминия. Для расчета объема капли
Кинджери использовал таблицы Башфорта и Адамса.
Погрешность определения по оценке автора составляет
±2% без учета влияния погрешности при измерении
2000 210022002300240025002600 t,°C
Рис. 6. Температурная зависимость
плотности жидкой окиси алюминия по
данным:
/-[Ю]; 2 -[9]; 5-[7]; 4 -[11]
66
температуры. Величина плотности, полученная Киндже-
ри, составляет 2,97 г/см3, что на 16% превышает
величину плотности, полученную Вартенбергом.
Когда для оценки плотности вещества в жидком
состоянии при температуре его плавления используется
метод лежащей капли,
момент расплавления
образца определяется
визуально по изменению его
формы. Затем после
небольшой выдержки,
необходимой для полного
расплавления образца и
выравнивания температуры,
измеряют размеры капли
для расчета ее объема.
При этом наиболее
вероятными являются две
ошибки: измерение
размеров неполностью
расплавленной капли, что
приводит к завышению
экспериментальных
данных, и перегрев жидкой
капли выше температуры
плавления, что вызывает
занижение данных.
Определение плотности жидкости при температуре
плавления твердой фазы достигается при использования
метода летящей капли. По этому методу измеряется
объем капли, образовавшейся при нагреве твердого
стержня из исследуемого вещества после ее отрыва от
стержня. Перегрев жидкой капли здесь практически
исключается, так как капля до момента ее отрыва
находится в контакте с твердым веществом и тепло, которое
к ней подводится, тратится на плавление твердой фазы.
Для измерения плотности по этому методу образец в
виде стержня, спрессованного из порошка чистого
окисла, помещается в вертикальный нагревательный элемент
печи сопротивления с герметичным корпусом [4, 5].
Схема установки показана на рис. 7. Образец укрепляется в
молибденовом держателе. Нагреватель печи имеет
узкую продольную щель, через которую с помощью
кинокамеры фиксируется процесс образования, отрыва и по-
Рис. .7. Схема установки для
измерения плотности жидких тугоплавких
веществ по методу летящей капли:
/ — нагреватель; 2 — кинокамера; 3 —
молибденовый стаканчик — изложница;
4 — каплеобразователь; 5 — образец
5*
67
лета капли. Скорость киносъемки должна обеспечивать
получение снимков с четким контуром капли. Скорость
капли после отрыва возрастает; одновременно в капле
Рис. 8. Фотографии капли жидкого окисла в полете после отрыва через, с:
/ — 0; 2 — 0,07; ^ — 0,1; 4 — 0,13; 5—0,16; 5 — 0,19; 7 — 0,24; 8 — 0,28; 9 — 0,33
затухают инерционные перемещения массы, особенно
опасные, если они происходят в поперечном направлении.
Обычно капля на расстоянии от места отрыва, равном
4—6 ее диаметрам, приобретает вид совершенного тела
вращения. Скорость же движения капли в этот момент
68
составляет 60—80 см/с. При такой скорости движения
объекта для получения четкого снимка требуется
повышенная скорость киносъемки, например не менее 96 кад-
ров в секунду.
В рассматриваемой установке стационарно
закрепленная кинокамера расположена с таким расчетом,
чтобы фотографирование проводить на расстоянии от места
отрыва капли, в несколько раз превышающем ее
диаметр. Тогда капля в момент отрыва окажется на
снимках в несколько искаженном виде, но ряд изображений
капель, по которым рассчитывается объем, будет строго
передавать ее форму и размеры (рис. 8).
Капля жидкости в полете может иметь форму
эллипсоида вращения, шарообразную, яйцевидную,
грушевидную и т. п. Но во всех случаях удается достаточно
точно рассчитать объем капли, если допустить, что
капля представляет собой тело вращения относительно
своей вертикальной оси. Если контур капли не
описывается элементарным уравнением (эллипс, окружность),
то для подсчета объема капли можно использовать,
например, один из способов, приведенных ниже.
Убедившись предварительно, что ордината у делит
изображение капли строго на две части, надо разделить
ординату на п произвольных частей, для каждой
ординаты найти соответствующее значение абсциссы х и
рассчитать объем по уравнению:
(2-1)
Этот простой способ расчета дает удовлетворительные
результаты, когда форма капли близка к эллиптической
и когда п принимается равной 10 или более.
По другому способу, основанному на использовании
метода наименьших квадратов, для каждого сектора
контура капли, заключенного между максимальным
диаметром капли и вертикальной осью подбирается
уравнение, которое в общем виде можно представить как
у = ахь или х = (у/а)1/ь9 (2-2)
где х, у — координаты точек контура капли;
Ь— коэффициент;
а— размерный коэффициент.
69
Для определения коэффициентов используется
система уравнений:
п п \
(2-3)
1=1 1=1 1=1 j
где п — число условных ординат, по которым
определяются численные значения х и у; обычно п
принимается равным 5—6.
Объем каждой части капли определяется
интегрированием выражения
V» х(у)
V = я j dy \ xdx% (2-4)
где у0— координата пересечения максимального
диаметра капли с вертикальной осью.
Полный объем капли получают суммированием
объема ее частей. Для повышения точности измерения
объема летящей капли рассчитывается объем одной и той же
капли по нескольким, чаще трем-четырем
последовательным кадрам. Если полученные величины отличаются
друг от друга не более чем на 1%, расчет можно считать
выполненным удовлетворительно. Для определения
плотности берется средняя величина объема капли.
Капля жидкого окисла падает на наклонную,
внутреннюю стенку молибденового стаканчика, что
предотвращает ее разбрызгивание при ударе.
Экспериментальная ошибка определения плотности
жидкой окиси алюминия и других тугоплавких окислов
при температуре плавления с помощью этого метода
составляет не более ±3%. Средняя величина плотности
жидкой окиси алюминия, измеренная этим методом,
равна 3,05 г/см3 [5]. Содержание примесей в исходном
порошке не превышает 0,2%. Загрязнение окиси во
время опыта молибденом по данным спектрального анализа
не происходит.
Метод летящей капли с расплавлением конца
стержня непригоден ни для измерения плотности чистых
веществ при температуре более высокой, чем
температура плавления, ни для определения плотности веществ,
70
не имеющих фиксированной температуры плавления
или плавящихся в интервале температуры. В последнем
случае отрыв капли может происходить до ее полцого
расплавления, что вносит большую неопределенность в
условия опыта.
Оригинальную методику измерения плотности жидкой
окиси алюминия использовали Расмуссен и Нельсон
[6], которые проводили радиографическую съемку рас-
Рис. 9. Схема установки Рас-
муссена и Нельсона [6] для
измерения плотности и
поверхностного натяжения жидких окис-
/ — источник рентгеновского
излучения (300 кВ); 2 —
оптический микропирометр; 3 —
указатель температуры; 4 — тигель
или капилляр из тугоплавкого
металла; 5 — смотровое окно из
плавленого кварца; 6—
микропечь; 7 — нагревательный
элемент из вольфрамовой сетки; п
8 — радиационные экраны; 9 —
водоохлаждаемый медный кор- 8
пус печи; 10, 13 — изображение у
капилляра с жидким окислом;
// —телевизионная камера,
фиксирующая изображение тигля
в рентгеновских лучах; 12 —
видеомагнитофон
плавленной окиси внутри молибденового капилляра.
Схема экспериментальной установки Расмуссена
показана на рис. 9. Для проведения опытов образец из
монокристалла окиси алюминия помещали в тонкостенный
молибденовый капилляр; капилляр запаивали в вакууме
и нагревали в вакуумной печи сопротивления до
расплавления окиси. Для измерения уровня жидкости капилляр
просвечивали рентгеновскими лучами и фиксировали его
изображение с помощью видеомагнитофона. Объем
жидкой окиси алюминия в капилляре рассчитывали,
учитывая тепловое расширение молибдена.
Расмуссен и Нельсон определили, что плотность
жидкой окиси алюминия при температуре плавления
равна 3,01 г/см3. Температуру в этой работе измеряли
оптическим пирометром с погрешностью ±10° С.
В отличие от метода летящей капли метод
заполнения капилляра можно применять для измерения
плотности жидких окислов при температуре, превышающей
температуру плавления. Температурную зависимость
71
плотности можно получить также с помощью метода
лежащей капли, различных вариантов
гидростатического взвешивания, пикнометрических методов и т. п.
Зависимость плотности жидкой окиси алюминия от
температуры впервые определили Киршенбаум и Кэхилл
[7] в 1960 г. взвешиванием вольфрамового шарика,
погруженного в жидкость. Образец окиси алюминия они
плавили в молибденовом тигле. Исследовали окись
алюминия с суммарным содержанием примесей не более
0,1%. Погрешность определения плотности в работе не
указана. Авторы выразили полученную температурную
зависимость плотности жидкой окиси алюминия
уравнением:
V = 5,632 — 1,127- Ю-3 Г, (2-5)
где у — плотность, г/см3;
Т — температура, К.
Из уравнения следует, что при температуре
плавления плотность жидкой окиси алюминия составляет
3,014 г/см3.
Оценка температурной зависимости плотности
жидкой окиси алюминия приведена в работе [8], авторы
которой применили метод лежащей капли. Образец,
содержащий 99,9% А1203, помещали на пластинку изпиро-
литического графита, которую нагревали пропусканием
электрического тока.
До начала опыта в установке создавали вакуум
~10~2 мм рт. ст. Впоследствии было показано, что из-за
взаимодействия окиси алюминия с графитом измерения
фактически проводились в разреженной атмосфере
окиси углерода. Процесс плавления окисного образца и его
дальнейшего нагрева снимали киноаппаратом со
скоростью 24 кадра в секунду. Размеры капли определяли
по изображению ее контура на кинопленке с помощью
инструментального микроскопа. Эталоном для
установления масштабного фактора являлась пластинка из пи-
рографита, изменение размеров которой при нагреве до
2500°С не превышает 1%.
Оригинальным в этой методике является способ
определения температуры, которую рассчитывали по
степени почернения кинопленки, принимая почернение при
температуре плавления в качестве базисной точки для
расчета более высоких температур.
72
Степень почернения светочувствительной пленки
подчиняется зависимости
s = a\gHkx + aK, (2-6)
где а— коэффициент, характеризующий
контрастность эмульсии пленки;
Н— освещенность;
Дт— продолжительность экспозиции;
К— постоянная, зависящая от вида пленки.
Освещенность пропорциональна энергетической
светимости нагретого тела:
Я = р#э, (2-7)
где Р — коэффициент.
Светимость в свою очередь пропорциональна
четвертой степени абсолютной температуры тела по закону
Стефана—Больцмана:
«э = теГ\ (2-8)
где а— коэффициент Стефана;
s— степень черноты.
Следовательно:
Н = раеГ4. (2-9)
Объединив последнее уравнение с первым, получим
для степени почернения
s = ct(lgAT+ Igpe + lg<7+ К) + 4alg7\ (2-10)
При неизменных условиях опыта, спектральной
характеристики пленки и продолжительности экспозиции
величины а, р, /С и Дт можно считать постоянными.
Постоянными являются также степень черноты е и
коэффициент Стефана а. Тогда это уравнение приводится к
виду:
s = 4 + 4alg7\ (2-11)
где А — суммарный коэффициент.
Если температура на /-том кадре пленки
S; = ,4 + 4alg7\., (2-12)
известна, например Т{ = ТПЛ, то для определения
искомой температуры на /г-ном кадре надо сравнить степень
почернения эмульсии пленки на f-том и я-ном кадрах,
так как
*,-*„ = 4^(777^. (2-13)
73
Погрешность определения температуры по этому
способу составляет 3% при разрешающей способности
10° С.
Инструментальная погрешность при измерении
размеров капли в рассматриваемой работе составляет
менее 2%, таким образом, суммарная погрешность при
определении температурной зависимости плотности жидкой
окиси алюминия в работе [8] не превышает ±5%.
По данным, полученным в работе [8], плотность
жидкой окиси алюминия уменьшается от 2,7 г/см3 при
температуре плавления до 2,4 г/см3 при 2425° С. В
работе подтверждена прямолинейность температурной
зависимости плотности жидкой окиси алюминия, но
абсолютные значения плотности заметно занижены.
Причиной этого явилось взаимодействие между окисью
алюминия и углеродом пирографитовой подложки,
приводящее к увеличению объема жидкости. Однако авторы
работы [8] не обнаружили газовой пористости в
образце окиси алюминия при ее микроскопическом
исследовании после охлаждения и не нашли следов углерода в
нем по данным химического анализа.
В работе [9] плотность жидкой окиси алюминия
определяли с помощью метода гидростатического
взвешивания. Схема экспериментальной установки приведена
на рис. 10. Взвешивание осуществляли с помощью тен-
зометрических датчиков, укрепленных на консольной
детали 12, изготовленной из бериллиевой бронзы.
Через держатель 13 и шток 5 деталь 12 соединяется с
винтовой парой 1, 2, посредством которой эту деталь
можно перемещать на расстояние до 50 мм. На деталь 12
через теплоизоляционную втулку 8 с молибденовыми
экранами и нить подвеса передается усилие — вес
погружаемого тела 10. Нить подвеса—это вольфрамовая
проволочка диаметром 0,2 мм. Для нагрева образцов
использовали вольфрамовый элемент сопротивления,
длина зоны постоянной температуры которого
составляет 70 мм. Опыты проводили в вакууме 10~4 мм рт. ст.
или в атмосфере инертных газов (аргон, гелий).
К консоли прикреплялись два тензометрических
датчика: один сверху —он испытывал растягивающие
напряжения и другой снизу — он испытывал напряжения
сжатия. Выводы от тензодатчиков через вакуумные
уплотнения подавались на измерительные приборы, где
сигналы тензодатчиков суммировались и измерялись по
74
Н20
— -9
10
Рис. 10. Схема установки для измерения плотности жидкостей при высоких
температурах путем гидростатического взвешивания (корпус вакуумной
высокотемпературной печи не показан):
/, 2 —винтовой механизм для подъема и опускания тела погружения; 5 —
сильфон; 4 — неподвижный шток; 5 — шток подвижной системы; 6 —
держатель нити подвеса; 7 — корпус измерительной приставки; 5 — тепловая
изоляция; 9 — исследуемая жидкость; 10 — тело погружения; // — призма; 12 —
консоль с тензодатчиками; 13 — держатель консоли; 14 — вакуумное
уплотнение; 15 — регистрирующая аппаратура
мостовой схеме. Для питания тензодатчиков
использовали переменный ток частотой 300 Гц; силой 300 мА и
напряжением 6 В.
75
Температуру печи установки измеряли оптическими
пирометрами ОППИР-017 и ОМП-21, которые
тарировали по вспомогательным реперным точкам
температурной шкалы, а также дополнительно по температурам
плавления окислов титана и алюминия. Излучение от
стенки тигля попадает на кварцевую призму полного
отражения 11 и выходит через смотровое окно к
пирометру. Погрешность при измерении температуры порядка
2000° С составляет ±10° С для микропирометра ОМП-21
и ±25° С для пирометра ОППИР-017.
Учитывая значительную продолжительность опыта,
большое значение приобретает вопрос об интенсивности
растворения материала тигля и погружаемого тела в
жидкой окиси алюминия. Для выяснения возможной
степени загрязнения жидкую окись выдерживали в
молибденовом или вольфрамовом тигле при температуре
2200° С в течение 50 ч (суммарное время) и затем
подвергали химическому и рентгеновскому анализу. Эти
опыты показали, что взаимодействие жидкой окиси
алюминия с молибденом хотя и имеет место, но
развивается с крайне низкой скоростью: содержание молибдена
возрастает за 50 ч до 0,06%. На скорость
взаимодействия атмосфера печи (вакуум или аргон) не оказывает
заметного влияния. Рентгеноструктурный анализ
подтвердил, что как до плавки, так и после нее образцы
целиком состоят из а-А120з; изменения параметров
кристаллической решетки CZ-AI2O3 не отмечено ни в
одном из опытов.
Взаимодействие жидкой окиси алюминия с
вольфрамом в этих опытах с помощью указанных методов не
было обнаружено. Однако вид внутренней поверхности
вольфрамового тигля свидетельствует о том, что и в
этом случае слабое взаимодействие жидкой окиси
алюминия с материалом тигля имеет место. При этом
образуются летучие окислы вольфрама. Таким образом,
можно считать, что и молибден, и вольфрам являются
вполне пригодными материалами для изготовления
деталей измерительной аппаратуры при работе с
расплавленной окисью алюминия в нейтральной по отношению
к этим металлам атмосфере. В работе [9] использовали
молибденовые тигли и тела погружения из вольфрама.
Исследованию подвергали окись алюминия марки
ч. д. а. с содержанием 99,8% А1203 в исходном веществе.
Вакуум в установке поддерживали равным 10~4 мм рт.
76
ст., что способствовало глубокому рафинированию оки
си от примесей. По данным спектрального анализа
суммарное содержание примесей в окиси алюминия
после ее переплавки составляло 0,01—0,03%.
Температурную зависимость плотности жидкой
окиси алюминия определяли двумя способами: по
первому— тело погружения находилось в жидкости во время
подъема или снижения температуры; по второму —
тело погружения извлекали из тигля во время изменения
температуры.
Для расчета плотности жидких окислов
использовали уравнение:
= Мх — M2 + 2nrolg ' ,2_14,
V 1/(1+ За)3 ;
где Мх— масса тела погружения на воздухе, г;
М2—масса тела погружения в жидкости, г;
г — радиус нити подвеса, см;
а — поверхностное натяжение, дин/см;
V— объем тела погружения при комнатной
температуре, см3;
а— коэффициент термического расширения
вольфрама.
В это уравнение введены поправки, учитывающие
влияние поверхностного натяжения жидкости и
термического расширения материала погружаемого тела.
При температуре плавления плотность жидкой окиси
алюминия равна 3,00±0,1 г/см3 (табл. 9). С ростом
температуры плотность прямолинейно падает до
2,40 г/см3 при 2530° С (рис. 6). Уравнение температурной
зависимости плотности, полученной по методу
наименьших квадратов, имеет вид:
у = 3,00 — 1,15- Ю-3 (t - 2054), (2-15)
где t — температура, ° С.
Суммарная погрешность при определении плотности
жидкой окиси алюминия составляет ±3%.
В последние годы выполнены исследования плотности
жидкой окиси алюминия с использованием метода
максимального давления в газовом пузырьке [10, 11].
Устройство измерительной камеры, использованной
в работе [10], показано на рис. 11. Капилляр 9
закрепляется во втулке грибкового вакуумного
уплотнения и перемещается с помощью микровинтов / и 2\ глу-
77
бина погружения
капилляра в расплав
контролируется
микрометрическим
датчиком 14 с погреш
ностью ±0,01 мм.
В качестве
рабочего газа выбран
гелий,
подвергнутый дополнительной
очистке. Экспери-
Рис. П. Схема установки для измерения
плотности и поверхностного натяжения жидкости
методом максимального давления в газовом
пузырьке:
1,2 — винтовой механизм для перемещения
капилляра; 3 — сильфон; 4 — неподвижный
шток; 5 — подвижный шток; 6 — корпус
измерительной приставки; 7 — вакуумное
уплотнение; 8 — наклонный манометр; 9 — капилляр;
10 — исследуемая жидкость; // — термопара
в защитном колпачке; 12 — призма; 13 —
вывод термопары; 14— микрометр
Рис. 12. Схема установки
для определения плотности
методом максимального
давления в газовом
пузырьке [11]:
/ — капилляр; 2 —
нагреватель; 3 — тигель с жидкой
окисью; 4 — тепловые
экраны; 5 — корпус печи; 6 —
модель АЧТ; 7 — оптический
пирометр; 5 —подача газа
ментально показано, что присутствие в рабочем газе
кислорода или паров воды наиболее заметно влияет на
получаемые результаты. Даже при незначительном
количестве кислорода в рабочем газе на поверхности
расплавленной окиси алюминия обнаруживается налет металлов,
из которых изготовлен капилляр, т. е. молибдена или
вольфрама. При взаимодействии их с жидкой окисью
алюминия образуются их легколетучие окислы. При
выходе пузырька газа, содержащего пары
легколетучих окислов, на поверхность расплава часть их дис-
78
социирует с выделением металла, который не
растворяется в жидкой окиси алюминия и собирается на ее
поверхности. Это явление особенно заметно при
использовании капилляров, изготовленных из молибдена.
Расчет плотности проводили по методике,
изложенной в работе [13]. Отклонение экспериментальных
данных от прямой не превышает 1%. Погрешность
определения плотности методом максимального давления в
газовом пузырьке составляет ±2%-
Полученная величина плотности жидкой окиси
алюминия при температуре плавления составляет 3,03±
±0,06 г/см3, что хорошо согласуется с данными других
исследователей (см. табл. 9).
Температурная зависимость плотности жидкой окиси
алюминия, полученная в работе [10], выражается
уравнением
7=3,03— 1,28-10~3(* — 2054).
Примерно в это же время в Институте высоких
температур АН СССР с использованием метода
максимального давления в газовом пузырьке была измерена
плотность жидкой окиси алюминия при температурах от
температуры плавления до 2700° С [11]. В этой работе
более точно по сравнению с предыдущей работой
измерялась температура благодаря использованию модели
черного тела. Кроме того, была повышена точность
определения глубины погружения капилляра (рис. 12).
Капилляры и тигли для плавления окиси алюминия
изготовлены из молибдена или вольфрама. Максимальная
погрешность определения плотности составляет ±0,7%.
Значения плотности в изученном интервале температур
описываются уравнением
Y = 5,035 —0,965- Ю-31, (2-16)
которое дает для температуры плавления величину
3,05 г/см3. Значение температурного коэффициента
плотности здесь значительно занижено по сравнению со
значениями, полученными в других исследованиях (см.
рис. 6). Авторы работы [11] не указывают,
исследовалось ли возможное влияние кислорода и паров воды,
содержащихся в рабочем газе, на результаты
измерений.
Температурная зависимость плотности жидкой окиси
алюминия была определена также по методу лежащей
79
капли (графитовая подложка), но в условиях
подавления взаимодействия окиси алюминия с графитом [12].
Величину плотности рассчитывали с помощью графиков
Хантадзе по экспериментальным данным о краевом
угле, геометрическим размерам и массе капли. Погрешность
определения плотности жидкой окиси алюминия этим
методом с учетом ошибки измерения температуры
составляет ±2,5%. В исследовании установлено, что
плотность жидкой окиси алюминия при температуре
плавления равна 3,01+0,07 г/см3 (см. табл. 9).
Температурная зависимость плотности жидкой окиси
алюминия описывается уравнением
Y = 3,01 — 1,16-10-3(* — 2050). (2-17)
Основываясь на работах, выполненных в последние
годы, без учета тех, где результаты заведомо искажены
вследствие взаимодействия с деталями измерительной
аппаратуры, можно предложить результирующее
уравнение для температурной зависимости жидкой окиси
алюминия в виде:
7 = 3,03 — 1,08- Ю-3 (t — 2054). (2-18)
Пунктирная прямая, соответствующая этому
уравнению (см. рис. 6), располагается между прямыми,
полученными в рассмотренных выше исследованиях.
РАСПЛАВЫ НА ОСНОВЕ ОКИСИ АЛЮМИНИЯ
Значительный практический интерес представляет
изучение концентрационной и температурной
зависимостей плотности двойных расплавов на основе окиси
алюминия. Плотность жидких окисных расплавов
А1203—СаО, А1203—MgO, A1203—ВеО, А1203—Сг203 и
А1203—Si02 измеряли с помощью метода максимального
давления в газовом пузырьке [14—15]. Тигли для
расплавов изготавливали из молибдена, а капилляры —
из вольфрама. В работе использовали материалы с
содержанием основного компонента, %: окись алюминия
99,8, окись бериллия 99,5, окись магния 99,5, окись
кальция 97,5, окись хрома 99,0 и окись кремния 98,3.
Смешанные в соответствующих пропорциях окислы
подвергали прессованию и прокаливанию на воздухе при
1200° С; затем их расплавляли в тигле в вакуумной
печи; смеси, содержащие окислы кальция или магния,
из-за их сравнительно высокой летучести плавили при
80
небольшом избыточном давлении гелия. Контроль
состава образцов проводили по массе, считая, что ее
постоянство обеспечивает неизменность состава. В
образцах после опытов фотокалориметрическим методом
определяли содержание молибдена и вольфрама.
Результаты этих анализов приведены в табл. 10: вольфрам в
Таблица 10
Содержание молибдена в окисных образцах1 А1203—МеО
после опытов
Добавляемый
окисел
ВеО
MgO
СаО
Сг203
Содерж
окисла в
образце
5—60
5-70
20
30
50
60
70
10
20
ание, %
молибдена
Нет
Следы
»
»
0,38
1,73
2,88
Следы
»
Добавляемый
окисел
Сг203
Si02
Содержание, %
окисла в
образце
30
50
70
80
28
40
50
70
90
молибдена
»
0,15
2,92
3,54
Следы
»
0,20
1,95
2,48
1 Вольфрам не обнаружен.
окисных образцах не обнаружен, а молибден
присутствует в заметных количествах в образцах с высоким
содержанием окислов кальция, хрома и кремния.
Микроскопический анализ окисных образцов после опытов
показал, что молибден в виде самостоятельной фазы
находится в основном в донной части образца по
границам зерен.
Методика измерения плотности окисных расплавов
аналогична методике, описанной для жидкой окиси
алюминия [9]; погрешность измерения плотности
составляет ±2,5%. Экспериментальные данные по
определению плотности жидких окисных расплавов приведены в
табл. И. Опубликованные в работах [16—19] сведения
по плотности отдельных расплавов удовлетворительно
согласуются с приведенными в табл. 11 данными.
Изотермы изменения плотности монотонны для
расплавов А1203—ВеО, А1203—Сг203 и A1203—Si02
независимо от наличия особых точек на диаграммах состояния
6-894
81
Таблица 1]
Зависимость плотности расплавов А1203—МеО от температуры
Содержание
добавки в окиси
алюминия,
% (по
массе)
5
10
20
25
30
40
50
60
70
10
20
30
40
50
60
ПЛ
д /**, °с
гсм~3-град~~ Н
А1203—MgO
1925
(эвтектика)
• 1950—2000
2040—2120
2135
(MgO-Al203)
2030—2130
2030—2080
2030—2150
2030—2200
2030—2220
2050—2350
2050—2350
2120-2350
2135-2350
2140-2350
2120-2350
2080-2350
2120-2350
2240—2350
А120з-Е
1890-1900
(эвтектика)
1870
(2А1203Х
ХВеО)
1835-1900
1835-1970
1835—2000
1900—2200
1910—2300
1890—2300
1920—2300
2000—2300
2100—2300
2220—2300
—0,9
—0,7
—0,5
-0,5
-0,5
-0,55
—0,6
—0,75
—0,9
teO
—0,60
—0,50
-0,50
-0,65
—0,65
—0,90
V=f (0
2,9—0,9-Ю-3
(^—1950)
2,86—0,7*10-3
(/—2020)
2,76-0,5.10~3
(/—2100)
2,76—0,5-10-3
(/—2140)
2,76—0,5-Ю-з
(/—2110)
2,77—0,55-Ю-з
(/-2100)
2,8—0,6-10-3
(/—2150)
2,85—0,75-10-3
(/—2200)
2,9—0,9.10-3
(/-2220)
2,86—0,6-Ю-з
(/-1900)
2,74—0,5-Ю-з
(/—1870)
2,67—0,5-Ю-з
(/—1900)
2,59-0,65-Ю-з
(/—1970)
2,47—0,65-Ю-з
(/—2000) *
2,35—0,90-Ю-з
(/-2200)
82
Продолжение табл. 11
Содержание
добавки в окиси
алюминия, %
(по^ массе)
10
20
30
40
50
60
70
10
20
40
50
70
90
28 1
22
50
70
80
* По фа
** Темпе
1 #
<пл« °С
А ***, °С
(dy/dt)\o\
г см-3 град-
А1203— СаО
[ 1785—1920
1785—1875
1580—1650
1400—1520
1380
(эвтектика)
1380—1520
1540—1750
1920—2200
1910—2200
1700—2200
1540—2200
1400—2200
1540—2200
1770—2200
| —1,05
—0,80
—0,60
—0,45
—0,31
-0,28
—0,30
А1203—Сг203
2075—2085
2085—2100
2125-2175
2150—2200
2175—2225
2225-2275
2100—2350
2125—2350
2200—2350
2200—2350
2250—2350
2300—2350
A1203-S
1850 1
(эвтектика)
1910
(ЗА1203Х
XSi02)
1850
1770
1700
1875—2200 1
1935-2200
1875—2200
1800—2200
1725—2200
зовой диаграмме [20].
ратурный интервал измерения.
—0,7
—0,3
+0,2
+0,4
+0,65
+0,85
>Ю2
+0,5
+0,35
+0,25
+0,25
+0,15
V4 (0
I 2,9—1,05.10-3
(/—1920)
2,67—0,80.10-3
(/—1875)
2,56—0,60.10-3
(/—1675)
2,63—0,45.10-3
(/—1520)
2,63—0,ЗЫ0-3
1 (/—1380)
2,65—0,28.10-3
(/—1520)
2,77-0,30-10-3
(/—1750)
3,05—0,70-10-3
(/—2085)
3,11—0,30-Ю-з
(/—2100)
3,25+0,20-Ю-з
(/—2175)
3,31+0,40.10-3
(/—2200)
3,48+0,65-Ю-з
(/—2225)
3,72+0,85-Ю-з
(/—2275)
2,6+0,50-10-3
(/-1870)
2,50+0,35-10-3
(/—1910)
2,45+0,25-Ю-з
(/-1850)
2,25+0,25.10-3
(/—1770)
2,20+0,15-10-3
(/—1700)
6*
83
[20]. Для расплавов А1203--СаО и А1203—MgO на
изотермах плотности наблюдается четко выраженный
минимум, соответствующий эквнмоляриым соединениям
в этих системах.
Характер изменения температурных коэффициентов
4f~MJ, г/(см3град)
Рис. 13. Зависимость температур- Рис. 14. Зависимость изменения мо-
ного коэффициента плотности би- лярного объема бинарных окисных
парных окисных расплавов от со- расплавов от состава
става
плотности расплавов (см. табл. 11) показан на рис. 13.
Для всех расплавов, за исключением расплавов
системы А1203—Сг203, концентрационная зависимость
температурного коэффициента плотности имеет
экстремальный вид. Экстремум соответствует составу, при котором
образуется наиболее прочное для данной системы
химическое соединение. Значения температурных
коэффициентов плотности расплавов с ростом содержания в
них окиси кремния или окиси хрома изменяют знак с
отрицательного на положительный. Для изученных
расплавов это происходит при 10% Si02 и при 30% Сг203.
84
Для расплавов с окисью кремния этот факт обясняется
диссоциацией трехмерной силикатной сетки на
комплексные ионы [21]. Однако маловероятно, чтобы этой же
причиной можно было бы объяснить аномальный
характер концентрационной зависимости температурного
коэффициента плотности для расплавов окиси
алюминия с окисью хрома. Возможно, что для этих расплавов
с ростом температуры происходит увеличение
координационного числа первой главной сферы.
Концентрационные зависимости молярных объемов
изученных окисных расплавов отклоняются от прямых,
построенных по правилу аддитивности (рис. 14). Для
систем А1203—Сг203 и AI2O3—Si02 эти отклонения
положительны во всем интервале изученных
концентраций, для других систем отклонения от аддитивности
носят переменный характер. Характер отклонения
молярного объема от аддитивности для систем с окисью
кремния или окисью хрома типичен для ассоциированных
жидкостей. Положительные отклонения в системах с
окислами магния, бериллия и кальция при
превалирующем содержании окиси алюминия связаны, вероятно, с
некоторым уменьшением степени взаимодействия
разноименных частиц по сравнению с одноименными. Когда
определяющим становится содержание добавляемых
окислов, то отклонения становятся отрицательными
вследствие того, что заметно усиливается
взаимодействие разноименных частиц. При этом степень отклонения
увеличивается с уменьшением силы поля катиона
в.ряду BeO-^MgO->-CaO. Аналогичные закономерности были
обнаружены при исследовании силикатов и боратов
щелочноземельных металлов [22, 23].
Большие положительные отклонения реального
молярного объема от объема, рассчитанного по правилу
аддитивности при содержании в окиси алюминия 0,3—
0,5 молярной доли окиси кальция, определяются,
вероятно, присутствием в жидкости комплексных ионов,
образующихся при диссоциации химического
соединения, существующего в данной области диаграммы
состояния системы АЬОз—СаО.
ОКИСЬ БЕРИЛЛИЯ
Плотность жидкой окиси бериллия в интервале от
температуры плавления (2550° С) до 2830° С изучали
сотрудники Института высоких температур АН СССР в
85
1974 г. Они использовали метод максимального
давления в газовом пузырьке; методика проведения опытов и
расчета плотности аналогична той, которая была
использована этими исследователями в работе с окисью
алюминия [11]. Содержание основного вещества в окиси
бериллия составляло 99,37—99,85%. Детали
измерительной части экспериментальной установки были
изготовлены из вольфрама. Авторы работы [11] не
обнаружили на границе контакта образца с вольфрамовым
тиглем присутствия каких-либо новых фаз, что, по их
мнению, свидетельствует об отсутствии заметного
взаимодействия жидкой окиси бериллия с материалом
тигля за время опыта (3 ч). Погрешность измерения
плотности жидкой окиси бериллия, по данным авторов,
составляет ±1%. Плотность жидкой окиси бериллия при
высоких температурах составляет:
t, °С 2602 2629 2744 2831
7Ве0, г/см3 .... 2,380 2,352 2,287 2,226
В аналитической форме эти результаты описываются
уравнением
Y = 4,070 — 0,651 • 10~3 /, (2-19)
из которого следует, что при температуре
плавления плотность жидкой окиси бериллия
составляет 2,39 г/см3. Эта величина выше, чем полученное в
работе [10] значение для плотности расплава 60%
ВеО—40% А1203 при температуре плавления, хотя
плотность окиси алюминия выше, чем плотность окиси
бериллия. Не исключено, что полученное авторами
работы [24] значение плотности жидкой окиси бериллия
несколько завышено из-за неучтенного ими
взаимодействия жидкости с вольфрамом, которое, как будет
показано в гл. IV, обнаруживается при контроле состава
по массе.
ДВУОКИСЬ ТИТАНА
Несмотря на то что плотность жидких
высокотитанистых окисных расплавов определяли многие
исследователи [25—30], плотность чистой двуокиси титана в
жидком состоянии определена совсем недавно.
Плотность жидкой двуокиси титана при
температуре ее плавления была измерена с помощью метода
летящей капли и оказалась равной 4,0±0,12 г/см3 [5]. Ис-
86
следовалась двуокись титана в модификации анатаза с
суммарным содержанием примесей не более 0,2%.
Рентгеноструктурный анализ показал, что после
экспериментов образцы двуокиси титана представляли собой
рутил. Плотность анатаза при комнатной температуре
составляет 3,8 г/см3, а плотность рутила 4,2 г/см3. Если
сравнить с этими данными плотность жидкой двуокиси
титана, то окажется, что она выше, чем плотность
анатаза и только на 5% ниже плотности рутила. В работе
[5] определяли плотность двуокиси титана только в
модификации анатаза, поэтому вопрос о том, влияет ли
исходная модификация окиси на ее плотность в жидком
состоянии остался невыясненным.
При плавлении и особенно при перегреве выше
температуры плавления двуокись титана частично
диссоциирует с образованием низших окисных соединений
титана. В образцах двуокиси титана после нагрева в
среде аргона до 2200° С и охлаждения до комнатной
температуры обнаружены в заметных количествах такие
окислы, как Ti305 и Ti203. Степень диссоциации зависит
от величины парциального давления кислорода в
газовой фазе. Упругость диссоциации двуокиси титана при
температуре плавления составляет примерно 10_б мм
рт. ст., а при 2200° С — около Ю-3 мм рт. ст. Было
отмечено, что диссоциация жидкой двуокиси титана в
вакууме протекает более полно, чем в атмосфере аргона.
По-видимому, в атмосфере аргона сравнительно быстро
достигается парциальное давление кислорода, близкое
к упругости диссоциации, тогда как в вакууме
атмосфера печи постоянно обновляется.
Взаимодействие жидкой двуокиси титана с
вольфрамом и молибденом протекает менее интенсивно, чем в
случае жидкой окиси алюминия. После выдержки
жидкой двуокиси титана в тиглях из тугоплавких металлов
при температуре 2200° С в течение 50 ч, вольфрам в
ней не обнаруживается, а содержание молибдена
составляет в среднем 0,04%. Следовательно, в физико-
химических исследованиях жидкой двуокиси титана
тигли и измерительную аппаратуру можно
изготавливать из молибдена или вольфрама.
Температурная зависимость плотности жидкой
двуокиси титана была определена с помощью метода
гидростатического взвешивания на тензометрической
измерительной установке в интервале от температуры
87
плавления до 2350° С [9]. Методика этого исследования
аналогична методике, описанной выше для случая
окиси алюминия. Исследованию подвергали двуокись
титана марки ЧДА при содержании основного вещества не
менее 99,5%. Рентгеноструктурныи анализ исходной
окиси показал, что она представляет собой анатаз.
Расчетная погрешность измерения плотности жидкой
двуокиси титана ±4,0%. Значения плотности приведены
ниже.
Год определения .
Ytkv г/см3* • ' •
1968
4,0
1970
3,65
Погрешность измерения:
г-см~э
%
Метод измерения . . .
Библиография ....
±0,12 ±0,15
±3,0 ±4,0
Летящая Гидростатическое
капля взвешивание
[5] [9]
При повышении температуры плотность жидкой
двуокиси титана прямолинейно снижается от 3,65 г/см3
при температуре плавления до 3,1 г/см3 при 2300° С
(рис. 15). Зависимость плотности жидкой двуокиси
титана от температуры описывается уравнением
у = 3,65 — 1,12- Ю-3 (*— 1870),
где t — температура, °С.
Величину плотности
жидкой двуокиси титана,
полученную с помощью
летящей капли, следует
признать завышенной.
Очевидно, вследствие
склонности двуокиси
титана к разложению при
плавлении с
образованием низших более
тугоплавких окислов титана
капля жидкости,
оторвавшаяся от образца,
содержит некоторое
количество твердой фазы, что
увеличивает плотность.
1800 1900 2000 2100 2200 23001}°С
Рис. 15. Температурная зависимость
плотности жидкой двуокиси титана,
полученная с помощью метода
гидростатического взвешивании
Плотность других тугоплавких окислов в жидком
состоянии неизвестна, за исключением плотности жидкой
пятиокиси тантала, которая измерена с помощью
метода летящей капли и равна при температуре плавления
6,55 ±0,2 г/см3 [5].
2. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
ЖИДКИХ ТУГОПЛАВКИХ ОКИСЛОВ
окись алюминия
Вартенберг с сотр."[2] измеряли поверхностное
натяжение жидкой окиси алюминия при температуре,
близкой к температуре ее плавления, методом веса
капель, сформированных на прутках диаметром 1—2 мм.
Диаметр шейки после отрыва капли измеряли в
твердом состоянии после остывания образца. Полученные
ими значения колебались от 502 до 678 эрг/см2. Среднее
значение поверхностного натяжения, рекомендуемое
авторами, составляет 580 эрг/см2. Но Вартенберг с сотр.
использовали для расчетов заметно заниженную
величину плотности окиси алюминия, полученную в этой же
работе. Расмуссен и Нельсон [6] пересчитали исходные
данные Вартенберга, Венера и Сарана по
поверхностному натяжению, использовав свое значение плотности
жидкой окиси алюминия (3,01 г/см3) и получили
величину, равную 690 эрг/см2 (табл. 12).
Кинжери [3] измерил поверхностное натяжение
жидкой окиси алюминия при температуре ее плавления
методом формы капли, сформированной на молибденовом
прутке после его погружения в расплавленную окись.
Среднее значение, полученное им, составило 690 эрг/см2
при погрешности измерения ±3%.
Бартлетт и Холл [31] для определения
поверхностного натяжения жидкой окиси алюминия использовали
метод лежащей капли. Они определили, что величина
поверхностного натяжения жидкой окиси алюминия при
температуре 2200° С равна 551 эрг/см2 (см. табл. 12).
Погрешность измерения авторами не указывается.
В 1968 г. Мак-Нейли с сотр. [32] использовал метод
веса капель и получил для поверхностного натяжения
жидкой окиси алюминия при температуре плавления
величину, равную 600 эрг/см2 (см. табл. 12). Затем
Якобашвили с сотр. [33] измерил поверхностное натяжение
89
Таблица 12
Поверхностное натяжение жидкой окиси алюминия
при температуре плавления
определе-
о к
1936
1959
1965
1968
1968
1968
1968
1969
1970
1971
1973
1973
ст, эрг/см2
580 (690)*
690
551
(при
2200° С)
600
660**
670
680
690
660
360 1
638
574
670
570
о к
О 5
—
—
±5
±5
—
±5,5
±10
±15
±11,5
±1,5
±4
Метод измерения
Вес капель
Форма висящей
капли
Лежащая капля
Вес капель
Лежащая капля
Вес капель
Форма висящей
капли
Капля, лежащая на
пирографите
Отрыв диска или
кольца
Форма мениска в
молибденовом
капилляре
Форма мениска в
вольфрамовом
капилляре
Форма висящей
капли
Метод максимального
давления в газовом
пузырьке
То же
Библиография
Вартенберг, Венер,
Саран [2]
Кинджери [3]
Бартлет, Холл [31]
Мак-Нейли с сотр.
[321
Якобашвили с сотр.
[33]
Маурах с сотр. [5,
35]
Маурах с сотр. [5,
35]
Зубарев с сотр. [8]
Митин, Нагибин [9]
Расмусссн, Нельсон
[6]
Шпильрайн, Якимо-
вич, Цицаркин [47]
Елютин, Митин, Ани-
симов [10]
* С учетом поправки на плотность.
* При температуре на ~30° С выше температуры плавления.
90
жидкой окиси алюминия с помощью метода лежащей
капли (табл. 12). Значение поверхностного натяжения
жидкой окиси алюминия при температуре,
превышающей приблизительно на 30° С температуру плавления
окиси, составляет 625 эрг/см2.
В это же время было определено поверхностное
натяжение жидкой окиси алюминия с применением
расчетов по форме висящей капли и по весу капель [5, 35].
Поверхностное натяжение по форме висящей капли
вычисляли, пользуясь формулой Фордхема [34]:
a=(l/H)ygd^ (2-20)
где у— плотность:
de— экваториальный диаметр висящей капли;
табличная функция [13, 34] — = f\-f-) » где
Н \ Н \ае J
ds—диаметр висящей капли на расстоянии de от
ее нижней точки].
Для расчета поверхностного натяжения по весу
капли применяли формулу Лонштейна [13]:
o = F(mgir), (2-21)
где т— масса оторвавшейся части капли;
г— радиус капли в плоскости отрыва;
F— корректирующий коэффициент,
связывающий массу жидкости с массой
оторвавшейся ее части [36, 37] [F=/(V/r3)];
V =т/у— объем оторвавшейся капли.
Анализ данных табл. 12 показывает, что в обоих
случаях получены близкие результаты.
Исследование, выполненное Зубаревым и др.
результаты которого частично опубликованы в работе [8],
интересно тем, что в нем поверхностное натяжение жидкой
окиси алюминия измеряли с помощью метода лежащей
капли в различной атмосфере: азота, окиси и двуокиси
углерода, а также в низком вакууме (Ю-1 мм рт. ст.).
Подложку изготавливали из пиролитического графита и
нагревали пропусканием тока. Образцы окиси
прессовали из порошка с содержанием не менее 99,9% А1203.
Результаты этого исследования показывают, что
состав атмосферы оказывает влияние на величину
поверхностного натяжения жидкой А1203. Наиболее низкое
значение поверхностного натяжения получено для
91
АЬОз в атмосфере СО; наиболее высокое значение,
приведенное в табл. 12, получено в вакууме.
Температурная зависимость поверхностного натяжения жидкой
окиси алюминия, по данным этой работы, описывается
уравнением
о = 690 — 0,50 (t — 2054). (2-22)
Поверхностное натяжение жидкой окиси алюминия,
полученное Мак-Нейли и Дженом с помощью метода
веса капель [48] составляет 600 эрг/см2 при температуре
плавления.
Температурную зависимость поверхностного
натяжения жидкой окиси алюминия исследовали методом
отрыва диска или кольца [9]. Теория метода
разработана Тихоновским [38] и развита Фрейдами [39], Харкин-
сом, а также Клячко и Куниным [40, 41].
Обычно этот метод используется как
относительный — в расчетное уравнение вводится /(-постоянная
измерительной установки:
Р '= fas cos 6 или а = P/(Ks cos 8),
где Р— усилие отрыва;
s— поверхность соприкосновения;
9— краевой угол смачивания.
Величина К определяется по жидкости с известным
натяжением.
Относительный метод измерения упрощает способ
расчета поверхностного натяжения, так как в этом
случае имеет место прямая пропорциональная
зависимость между усилием отрыва и величиной
поверхностного натяжения.
Для расчета по абсолютному методу отрыва диска
(цилиндра с плоским торцом) Кунин [41] рекомендует
уравнение
Р = nRgy
R*[l& 2aVR2 -0,5772)+а*
(2-23)
где a=2a/(gy)—капиллярная постоянная.
Широко используется вариант метода отрыва
кольца, т. е. твердого тела, имеющего форму полого
цилиндра с наружным радиусом R и толщиной стенки 26 [41],
42]. Для расчета поверхностного натяжения в этом
случае Фершафельт [43] предложил уравнение
92
Pg
4nR
1 — 2,8224 — 0,6095
VhR
+
+ 3 + 2,585 l/ — + 0,371 — —
' \ V R R hR
(2-24)
или
a
Pg
4nR
Рф>
где h=P/(nR2y)\
Рф—поправка Фершафельта, которая
рассчитывается аналитическим [44] или графическим [45]
путем.
Установлено [46], что при хорошем смачивании, т. е.
при небольших значениях краевого угла смачивания
поправка Фершафельта практически не зависит от его
величины.
Поверхностное натяжение жидких тугоплавких
окислов измеряли по методу отрыва кольца или диска на
тензометрической установке, конструкция которой
(рис. 16) аналогична установке для измерения
плотности. Различие заключается в том, что к концу
измерительной консоли через асбоцементную втулку с
экранами подвешивают пе тело погружения, а сплошной или
полый цилиндр. Из методических соображений
цилиндры изготавливали различных размеров (диаметр от 14
до 16 мм) из двух металлов — вольфрама и молибдена.
Торцовую поверхность цилиндров, т. е. поверхность
касания с жидкостью, полировали. Диаметр цилиндров
измеряли с точностью до 0,01 мм по крайней мере в
восьми направлениях и определяли среднее значение.
Во всех опытах проверяли горизонтальность нижней
поверхности цилиндров, так как нарушение
горизонтальности вызывает занижение экспериментальных данных.
При проведении опытов для выравнивания
температуры цилиндр выдерживали в горячей зоне печи в
течение не менее 10 мин, затем его погружали в жидкость
на глубину 0,5—1,0 мм. Максимальное усилие отрыва
цилиндра от поверхности жидкости фиксировали во
время его медленного извлечения из жидкости.
Экспериментальные данные описываются
уравнением, полученным с помощью метода наименьших
квадратов:
93
ZZZZZZZZZJ
zzzzz
Рис. 16. Схема установки для измерения поверхностного натяжения с помощью
метода отрыва кольца или диска:
/, 2 — механизм для перемещения измерительной системы; 3 — сильфон; 4—
неподвижный шток; 5 — подвижный шток; 6 — держатель нити подвеса; 7 —
корпус; 8 — тепловая изоляция; 9 — тело отрыва — кольцо или диск; 70 —
исследуемая жидкость; 11 — призма в системе измерения температуры; 12 —
консоль с тензодатчиками; 13 — держатель консоли; 14— вакуумное
уплотнение для вывода проводов от измерительной системы; 15 — регистрирующая
аппаратура
а = 660 — 0,195 (t — 2054), (2-25)
где t — температура, °С.
Это уравнение действительно для интервала от
температуры плавления до 2550° С. Из него следует, что при
94
температуре плавления величина поверхностного
натяжения равна 660 эрг/см2, а температурный коэффициент
поверхностного натяжения жидкой окиси алюминия
равен 0,195 эрг/(см2-град). Ошибка измерения составляет
±6%.
Температурную зависимость поверхностного
натяжения жидкой окиси алюминия определили авторы работы
[10] с помощью метода максимального давления в
газовом пузырьке. Схема установки описана в главе I.
Поверхностное натяжение рассчитывали по методу Сагде-
на. Погрешность измерения составляет ±4%.
Полученные экспериментальные данные описываются
уравнением
о = 575 — 0,187 (t — 2054). (2-25а)
Согласно этому уравнению поверхностное натяжение
жидкой окиси алюминия при температуре плавления
примерно на 100 эрг/см2 ниже, чем данные, полученные
многими другими исследователями и в том числе
самими авторами, когда они использовали иные методы
измерения. По мнению авторов, такое различие связано с
влиянием взаимодействия жидкой окиси с материалом
измерительной аппаратуры, а также с наличием в
рабочем газе примеси кислорода. Следует отметить, что
значения поверхностного натяжения не изменялись,
независимо от того, какой материал (молибден или вольфрам)
использовали для изготовления капилляра.
Этим же методом максимального давления в газовом
пузырьке поверхностное натяжение жидкой окиси
алюминия при температурах до 2750° С измерили Шпиль-
райн, Якимович и Цицаркин [47]. Авторы уделили
большое внимание влиянию кривизны поверхности жидкости
в тигле на величину поверхностного натяжения. С
учетом этого влияния авторы предложили следующее
расчетное уравнение:
гэф
2
mg 1 / Г 1 /о cos 9
■rl/Rl)l/l
nR2(l-r2K/R20)J/ [ Rod-rK/R0)
(2-26)
где Р—экстраполированное значение максимального
давления газа, когда срез капилляра
находится на дне тигля;
гк и го — внешний и внутренний радиусы капилляра
при комнатной температуре;
95
R0 и R—внутренний радиус тигля при комнатной и
рабочей температуре;
т — масса жидкости в тигле;
0—краевой угол смачивания жидкостью
материала измерительной аппаратуры;
гэф—эффективный радиус капилляра,
вычисленный по методу Сагдена на основе
усовершенствованной методики, предложенной Шпиль-
райном и др. [48].
Погрешность измерения в этой работе по оценке ее
авторов не превышает ±1,5%. Экспериментальные
данные описываются уравнением
а= 1560,6 — 522,2-10-**+ 43,2-10~6/2. (2-27)
По этому уравнению поверхностное натяжение
жидкой окиси алюминия при температуре плавления
составляет 670 эрг/см2.
Для сравнения все известные уравнения
температурной зависимости поверхностного натяжения жидкой
окиси алюминия приведены в табл. 13.
Таблица 13
Зависимость поверхностного натяжения жидкой окиси алюминия
от температуры
Метод
исследования
Лежащей
капли
Отрыва капли
Максимального давления
в газовом
пузырьке
То же
Л /, °С
2054—2700
2054—2300
2054—2400
2054-2750
б=/ (/), эрг/см2
690—0,46 (/—2054)
660—0,195 (/—2054)
575—0,187 (/—2054)
670—0,30 (/—2054)
Библиография
Зубарев и др.
[8]
Митин,
Нагибин [9]
Елютин,
Митин, Анисимов
[10]
Шпильрайн
и др. [47]
При анализе этих данных следует обратить внимание
на аномально высокие значения температурного
коэффициента поверхностного натяжения, полученные в
работах [8] и [47]. В первой работе завышенные значения
можно объяснить адсорбционным понижением
поверхностного натяжения из-за образования окиси углерода при
96
взаимодействии пирографита с жидкой окисью
алюминия. Во второй работе резкое снижение поверхностного
натяжения также связано с тем, что недостаточно полно
учитывалось влияние взаимодействия между жидкой
окисью алюминия и материалом деталей
измерительной аппаратуры; а как известно [6], это взаимодействие
приводит к существенному уменьшению поверхностного
натяжения.
Значительный практический и теоретический интерес
представляют сведения о влиянии окружающей газовой
атмосферы на поверхностное натяжение жидкой окиси
алюминия. С этой целью было проведено исследование
поверхностного натяжения жидкой окиси алюминия в
атмосфере азота и окиси углерода при давлениях от 1
до 760 цы рт. ст. [12]. В работе использовали метод
лежащей капли. Опыты проводили в вакуумной печи
сопротивления с графитовым нагревательным элементом.
В качестве подложки использовали пиролитический
графит. Вакуумная система позволяла получать
разрежение до 5-Ю"5 мм рт. ст. Рабочее пространство
печи заполняли газами через систему очистки, состоящую
из колонок с ангидроном магния, синтетическим
цеолитом и фильтров для улавливания пыли. Окись углерода
получали в результате реакции между муравьиной и
серной кислотами; в применяемом азоте суммарное
содержание кислорода и паров воды не превышало 0,1%.
Загрязнение окиси алюминия во время опыта не
происходило; более того, наблюдалось некоторое ее
рафинирование во время плавления и выдержки в
расплавленном состоянии. Спектральный анализ образцов
окиси алюминия после опыта показал, что в них
содержится <0,01%Fe; <0,001% Mg и <0,001% Si. Контур
капли определяли на пленке после киносъемки капли
во время опыта.
Это исследование подтвердило, что величина
поверхностного натяжения жидкой окиси алюминия зависит
как от природы, так и от давления окружающего газа.
На рис. 17 показано влияние окиси углерода и
азота на поверхностное натяжение жидкой окиси
алюминия: при увеличении внешнего давления до 760 мм
рт. ст. поверхностное натяжение жидкой окиси
алюминия при температуре плавления снижается от 680 до
200 эрг/см2 в среде окиси углерода и до 300 эрг/см2 в
среде азота. Зависимость поверхностного натяжения от
7—894
97
температуры в изученном интервале давления газовой
фазы остается линейной. Изотермы зависимости
поверхностного натяжения жидкой окиси алюминия от дав-
6, дин/см
I 1 1 1 i I I i i i i I
О 0,2 ОА 0,6. 0,8р,am 0 0,2 0,4 0,6 0,8р,am
Рис. 17. Изотермические зависимости поверхностного натяжения жидкой
окиси алюминия от давления окиси углерода и азота при температуре, К:
/ — 2325; 2 — 2500; 3 — 2700
ления удовлетворительно аппроксимируются уравнением
Шишковского:
<7 = <т0 — z0RT\n(l +ЬР),
где а0— поверхностное натяжение в вакууме;
Р — давление газа;
z0 и Ь— постоянные, значения которых приведены
ниже.
Температура, К . . . .2325 2500 2700
г0, моль-см-2 . . . .11,0/11,5 10,8/11,2 10,5/10,9
Ьу 1/ат 13,2/6,0 9,0/4,2 5,5/2,6
Примечание. В числителе приведены значения коэффициентов
для расчета О" в среде окиси углерода, в знаменателе — для расчета в
среде азота.
Причиной снижения поверхностного натяжения
жидкой окиси алюминия в атмосфере реакционноспособных
газов является их адсорбция поверхностью окисла. На
рис. 18, а приведены изотермы адсорбции окиси
углерода и азота на поверхности жидкой окиси алюминия,
полученные графическим дифференцированием кривых
рис. 17 с использованием адсорбционного уравнения
Гиббса. Изотермы адсорбции в свою очередь хорошо
спрямляются в линейных координатах уравнения Лэнг-
мюра (рис. 18,6):
98
P/z=l/(zQb)+P/z0i (2-28)
где z0—максимальное количество адсорбированного
газа при условии образования
мономолекулярного слоя;
b— постоянная Лэнгмюра.
10 Z, моль/см2
101гр/1
9
7
5
3
1
N2 _-с
1)/*Р^
- /rs*
- СО
1J?
- / ff
-Iff
^J
"\T ]
a
0 0,2 0,4 0 0,2 p,am 0 0,2 0,4 0 0,2 0,4 p, am
Рис. 18. Изотермы адсорбции газов СО и N2 на поверхности жидкой окиси
алюминия в обычных координатах (с) и в линейных координатах уравнения
Лэнгмюра (б) при температуре, К:
/ — 2325; 2 — 2500; 3 — 2700
Значения z0 и 6, рассчитанные по
экспериментальным данным для уравнения (2-28), приведены ниже.
Температура, К . .
20, МОЛЬ-СМ-2 . .
6, ат-1
2325
11,3/12,2
12,8/6,1
2500
11,2/11,9
8,3/4,1
2700
11,1/12,1
5,3/2,7
Примечание. В числителе приведены значения коэффициентов
для расчета о* в среде окиси углерода, в знаменателе — для расчета в
среде азота.
Экспериментальные данные располагаются на
прямой в координатах Лэнгмюра, что свидетельствует о
мономолекулярной адсорбции. Поэтому, используя
полученные данные, можно рассчитать размер
адсорбированных молекул по уравнению
d=U(z0Noyi\ (2-29)
где d — диаметр молекулы,
Af0 — число Авогадро.
Расчет по уравнению (2-29) дает значения
диаметра молекул d«3,9'10-8 см (в среде окиси углерода)
и d^3,?M0-8 см (в среде азота), что удовлетворитель-
7* ' 99
но согласуется с опубликованными данными: dco=
= (3,174-3,79) • Ю-8 см и d*2=3,5-10-8 см [49] и
является дополнительным подтверждением мономолекуляр-
ности адсорбции окиси углерода и азота на поверхности
жидкой окиси алюминия.
РАСПЛАВЫ НА ОСНОВЕ ОКИСИ АЛЮМИНИЯ
С помощью метода максимального давления в
газовом пузырьке изучена температурная зависимость по-
6, эрг/см2 о, зрг/сн2 о, эрг/см2
О 10 20 30 40 50 60 О 10 20 50 40 50 60 70 О 10 20 30 40 50 60 70 80
Л1203 ВеО,% — Мг03 МдО,%— А1203 CaQ% —
б, эрг/см2 6, эрг/см2
Рис. 19. Изотермические
зависимости
поверхностного натяжения
бинарных окисных
расплавов от состава при
температуре, К:
/ — 2175; 2—2275; 3 —
2375; 4-2475; 5 — 2575;
6—2525; 7 — 2625; 8 —
2675; 9 — 2575; 10 — 2075
верхностного натяжения ряда бинарных систем на
основе окиси алюминия:
А1203—ВеО, А120з—MgO, A1203—Сг203, А1203—СаО и
Д1203—Si02 [10, 15]. Состав изученных окисных
расплавов, экспериментальная установка и методика
измерений описаны выше. Полученные результаты
представлены в табл. 14т
050
600
550*
*пп
д 1
8
*к\
14
< i i i i
550 {
500
450
400
оЧ
Л
О 20 40 60 80100 10 30 50 70 90
AT П Гр.Л, %-*- Al П Ы(\л.°/п-т.
100
Таблица 14
Зависимость поверхностного натяжения окисных расплавов
на основе окиси алюминия от температуры
Содержание
добавки в
окиси алюминия,
% (по массе)
0
10
20
30
40
50
60
5
10
20
25
30
40
50
60
70
10
20
30
40
50
60
70
80
10
20
40
50
70
80
20
28
50
70
80
А Г, К
2330—2675
2155-2675
2135—2675
2175—2675
2205—2675
2355—2675
2475—2675
2300—2725
2295-2725
2365—2725
2385-2725
2385—2725
2375—2725
2325—2725
2375—2725
2495—2725
2175-2475
2160—2475
1950—2475
1795—2475
1655—2475
1795—2475
2025—2475
2375—2475
2350—2625
2365—2625
2420—2625
2450—2625
2500—2625
2545—2625
2225—2575
2185—2575
2075—2575
2045—2575
2050—2575
d o/dT
А120з—BeO
—0,187
-0,065
—0,060
—0,070
—0,075
—0,10
—0,12
А120з—MgO
-0,16
—0,12
—0,08
.—0,06
—0,06
—0,07
—0,08
—0,09
—0,10
А120з—CaO
—0,16
—0,12
—0,09
-0,06
—0,06
—0,07
—0,10
—0,13
Al2Os—Cr20
+0,023
+0,037
+0,06
+0,07
+0,095
+0,12
AI2O3—Si02
+0,085
+0,07
+0,025
+0,02
0
o=}(T)
575—0,187(7—2327)
540—0,065(7—2155)
525—0,060(7—2135)
505-0,070(7—2175)
490—0,075(7—2205)
480—0,10(7—2355)
460—0,12(7—2475)
565—0,16(7-2300)
560—0,12(7—2295)
555-0,08(7—2365)
550-0,06(7-2385)
555-0,06(7—2385)
565-0,07(7—2375)
575-0,08(7—2325)
585—0,09(7—2375)
595-0,10(7—2495)
545—0,16(7—2175)
525—0,12(7—2160)
520—0,09(7—1950)
515—0,06(7—1795)
515-0,06(7—1655)
525—0,07(7—1795)
530—0,10(7—2025)
540—0,13(7—2375)
3
f 570+0,023(7—2350)
575+0,037(7—2365)
585+0,06(7—2420)
j 590+0,07(7—2450)
605+0,095(7—2500)
615+0,12(7—2545)
475+0,085(7—2225)
440+0,07(7—2185)
415+0,025(7—2075)
410+0,02(7—2045)
405
Исследования поверхностного натяжения расплавов
окиси алюминия с окислами бериллия, магния, хрома
и кремния неизвестны. Имеются данные по
поверхностному натяжению для расплава эвтектического
состава 55% СаО—45% А1203 системы А1203—СаО
[49—55], которые показывают, что эта величина
колеблется, по данным
различных
исследователей, от 525 до
670 эрг/см2 при
температуре плавления.
В наших
исследованиях для этого
сплава получено
значение поверхностного
натяжения, равное
575 эрг/см2.
Изотермы
поверхностного
натяжения для
расплавов систем А1203—
MgO и А1203—ВеО
представляют собой
плавные кривые
(рис. 19, а, б).
Изотермы для системы
А1203—СаО имеют
ряд кривых с
отчетливо выраженным
минимумом,
расположение которого
соответствует составу алюмината кальция на диаграмме
состояния СаО-А1203 (рис. 19, в). На изотермах
поверхностного натяжения расплавов А1203—Сг203 и А1203—
Si02 имеются участки, где наблюдается резкое
изменение кривизны в небольшом интервале концентраций
(рис. 19, г, д).
Политермы поверхностного натяжения во всех
исследованных системах линейны. Однако, если при
добавлении к окиси алюминия окислов кальция, магния
и бериллия поверхностное натяжение с ростом
температуры понижается, то значения температурных
коэффициентов поверхностного натяжения для расплавов
А1203—Si02 и А1203—Сг203 переходят из области от-
d6
dT
0,10
0,06
0,02
О
-0,02
~0,06\
~0,Ю
-0,14
-0,18
X
/ х4^
id
\Ч
Mr / •
\f° ' /
W
\ I i
х^Чч*ч-
йй\
i
Сг20з^
v*Si02
—
ХсаО
1
10
30
А120х
50 70
90
Рис. 20. Зависимость коэффициента
поверхностного натяжения бинарных окис-
ных расплавов от состава расплава
102
рицательных в область положительных значений.
Причиной этого для расплавов, содержащих БЮг, является
диссоциация при добавке других окислов трехмерной
сетки, образованной ионами SiOT [56, 57]. Для
расплавов АЬОз—СГ2О3 положительные значения da/dt связаны,
вероятно, с диссоциацией образующихся при плавлении
ионных комплексов на ионы. Зависимость
температурного коэффициента поверхностного натяжения от
состава для всех исследованных расплавов (кроме А1203 —
Сг20з) имеет четко выраженный максимум, который
соответствует образованию наиболее прочных для данной
системы соединений (рис. 20).
С помощью полученных результатов авторы
проверили возможность теоретического расчета
поверхностного натяжения бинарных окисных расплавов по
методу, предложенному Жуховицким [58]. Этот метод
позволяет рассчитывать поверхностное натяжение сложных
расплавов, используя значения величин поверхностного
натяжения компонентов и значения их активностей в
расплаве. Основой расчета служат уравнения:
о = аг + п01 RT In ai9TloJ(hi
^пов. = /fW\p exp V^ (2-30)
где р — Юх/юг;
ог и а2 — поверхностное натяжение компонентов;
а№ов и at — активность компонента в поверхностном
слое и в объеме;
со—парциальная молярная площадь
компонента;
п01—удельное число молей первого
компонента в поверхностном слое.
Трудность практических расчетов заключается в том,
что неизвестны значения активностей компонентов в
изученных окисных расплавах. Очевидно, что применять к
рассматриваемым расплавам законы идеальных
растворов нельзя. Расчет для рассматриваемых систем
проводили в предположении, что эти растворы
соответствуют модели, предложенной Темкиным [59]:
аг=(-1*—)*=1Рг (2-31)
\ Щ + mj J l
103
Коэффициент вытеснения р определяли с учетом
полученных изотерм поверхностного натяжения
р=
'01
(Ох
(О
со2
М
2/3
ЛГ; 7i =
ду
dNi )т=соп${ '
1
Активность компонентов в поверхностном слое
определяли по методике, предложенной Жуховицким [58]:
Л1,иОВ __
аг
0ПЬ1ЯТ
(2-32)
Кпов)Р 4
После вычисления активности компонентов в
поверхностном слое можно определить молярные доли
компонентов в поверхностном слое
AUb = V^o~b, (2-33)
а, следовательно, и его состав.
6, эрг/смг
550 [
6, эре/сп2
600 \
0 10 20 J0 40 50 60
А1203;% ВеО,% —
5501
О 10 20 30 40 50 60
Al203 MgO,% —
О 10 20 30 40 50 60 70
Al203 CaO,%~—
Рис. 21. Зависимость поверхностного натяжения бинарных окисных расплавов
на основе А1203 от состава расплава при температуре, К:
а, в — 2475; б—2425; / — экспериментальные данные; 2 — данные,
рассчитанные по уравнениям Жуховицкого
Расчетные зависимости удовлетворительно
описывают экспериментальные данные (рис. 21), что позволяет
предложить выбранную схему для априорного расчета
104
поверхностного натяжения бинарных окисных
расплавов. Расчет состава поверхностного слоя
исследованных расплавов показывает, что по отношению к окиси
алюминия окислы бериллия, кальция и кремния
являются поверхностно-активными, а окислы хрома и
магния — инактивными.
ОКИСЬ БЕРИЛЛИЯ
Поверхностное натяжение жидкой окиси бериллия
при температуре, несколько превышающей температуру
плавления, измеренное Бартлетом и Холлом [31]
методом лежащей капли, составляет 300 эрг/см2.
Температурная зависимость поверхностного натяжения жидкой
окиси бериллия была определена Шпильрайном с сотр.
[24] методом максимального давления в газовом
пузырьке. Чистота исходной окиси составляла 99,4—
99,8%. Методика эксперимента и расчета аналогична
методике, использованной в работе [47]. Результаты
экспериментального определения температурной
зависимости (до 3000° С) поверхностного натяжения
жидкой окиси бериллия описываются уравнением
о = 762,7— 139,3- Ю-3/. (2-34)
Расчет по этому уравнению для температуры
плавления окиси бериллия (2550° С) дает величину
415 эрг/см2, что выше, чем было получено в работе [31].
ДВУОКИСЬ ТИТАНА
Исследование зависимости поверхностного
натяжения жидкой двуокиси титана от температуры было
проведено Елютиным с сотр. [9]; температурная
зависимость имеет вид
а = 355 — 0,1740— 1870). (2-35)
Поверхностное натяжение жидкой двуокиси титана
измеряли с помощью описанного выше метода отрыва
цилиндра. Экспериментальные данные (рис. 22) не
зависят от того, какой формы (цилиндр или пластина) и
из какого металла (вольфрам или молибден)
изготовлено тело отрыва. Погрешность определения составляет
±6%.
105
6, эрг/c/i2
Рис. 22. Температурная
зависимость поверхностного натяжения
жидкой двуокиси титана,
полученная с помощью метода
отрыва:
/ — вольфрамовый цилиндр
диаметром 16 мм; 2 —
молибденовый цилиндр диаметром 16 мм;
3 — вольфрамовая пластина со
стороной 14 мм
1800 /000 2000 2100 2200 t, °C
Величину поверхностного натяжения жидкой
двуокиси титана при температуре плавления определил Мау-
рах с сотр. [5, 35]: по форме капли а=380 эрг/см2; по
весу капли а=360 эрг/см2.
Погрешность определения составляет ±5%.
3. ВЯЗКОСТЬ ЖИДКИХ ТУГОПЛАВКИХ
ОКИСЛОВ
В литературе сведения о вязкости чистых
тугоплавких окислов практически отсутствуют, имеется лишь
несколько публикаций по вязкости чистой окиси
алюминия.
ОКИСЬ АЛЮМИНИЯ
Впервые оценку величины вязкости жидкой окиси
алюминия дал Казакевич [60] в 1960 г. С помощью
метода коаксиальных цилиндров он измерил вязкость
жидкой окиси алюминия при температуре, близкой к
температуре плавления. Казакевич проводил опыты в печи
сопротивления с графитовым нагревателем в среде
азота. В контакте с жидкой окисью алюминия находились
молибденовые цилиндры. Казакевич установил, что
жидкая окись алюминия имеет коэффициент динамической
вязкости при температуре плавления, равный 0,6 П, а
при температуре 2100° С—равный 0,5 П (рис. 23).
Вязкость жидкой окиси алюминия в более широком
температурном интервале измерил Гофмайер [61] на
ротационном вискозиметре. Окись алюминия он расплав-
106
лял в молибденовом тигле в печи сопротивления с
вольфрамовым нагревателем в среде аргона.
Гофмайер получил для вязкости жидкой окиси
алюминия при температуре плавления величину 0,41 П, что
на 30% ниже, чем у Казакевича (см. рис. 23). С ростом
температуры вязкость
снижается, достигая 0,15 П при
2370° С, т. е. при перегреве
на 300° С вязкость
уменьшается примерно в три раза.
Изменение вязкости носит
экспоненциальный
характер: зависимость lg ц—1/Г
представляет собой прямую
линию. По этим данным
экспериментальное значение
энергии активации вязкого
течения Е ц = 39,8 ± 6,2
ккал/моль.
Елютин с сотр. [62]
измерил вязкость жидкой
окиси алюминия с помощью
метода крутильных колебаний
тигля, наполненного
жидкостью. Конструкция установки приведена на рис. 24.
Тигли изготавливали из молибдена с
завинчивающейся крышкой, что необходимо для подавления
взаимодействия окисла с материалом тигля. Параметры
подвесной системы выбирали на основе методики Швид-
ковского [63]; наиболее удачными оказались такие
параметры, для которых пригоден аппарат расчета,
предложенный Швидковским для сильновязких
жидкостей.
Результаты измерения вязкости жидкой окиси
алюминия приведены на рис. 25. Суммарная ошибка
определения вязкости ±5%.
Вязкость жидкой окиси алюминия вблизи
температуры плавления составляет 0,57 П, но резко падает при
повышении температуры. Эти значения вязкости близки
к данным Казакевича, но заметно отличаются от
данных Гофмайера.
Полученные экспериментальные данные,
выраженные в полулогарифмических координатах lg т]—1/Г
(рис. 26), не подчиняются линейной зависимости. От-
107
2000 2100 2200230024002500t,°C
Рис. 23. Температурная
зависимость коэффициента
динамической вязкости жидкой окиси
алюминия по данным:
/ — Казакевича [60J; 2 —
Гофмайера [61]; 3 — Елютина с
сотр. [62]
Рис. 24. Схема установки для измерения вязкости жидких окислов с помощью
метода крутильных колебаний:
/ — механизм поворота подвесной системы; 2 — вакуумное уплотнение штока
подвесной системы; 3 — верхний и нижний зажимы нити подвеса; 4 — нить
подвеса; 5 — система автоматической записи колебаний подвесной системы;
С — потенциометр ЭПП-09-4; 7 — корпус вискозиметрической приставки к
высокотемпературной печи; 5—тепловая изоляция; 9 — тигель с крышкой; 10 —
нагреватель печи; 11— водяное охлаждение; 12 — смотровое окно; 13 — рам?са
на подвесной системе; 14 — приборы питания ферродинамического датчика
клонение от линейной зависимости в предкристаллиза-
ционный период для жидкой окиси алюминия
позволяет предположить наличие структурных изменений в
ней в этот период. О возможности структурных изме-
v, Cm
2000 2/00 2200 2300 2400 t, °C
Рис. 25. Температурная зависимость
кинематической вязкости жидкой
окиси алюминия, полученная в
различных условиях:
/—диаметр тигля 33 мм; 2,3 —
34 мм; 4, 5 — 26 мм; /, 3, 5 —
вакуум; 2, 4 — аргон
нений в жидкой окиси алюминия свидетельствует
также и отклонение полученных результатов от расчетных,
полученных по формуле Бачинского [63]:
ц = c/(v — b)9
где с и b — постоянные;
v — удельный объем жидкости.
Согласно теории активированного комплекса,
развитого Эйрингом [64], вязкость жидкости определяется
выражением
4=^.«p(-^L). (2-36)
где N— число Авогадро;
h—постоянная Планка;
V— молярный объем жидкости;
AFt, — свободная энергия активации вязкого течения.
На основе этого выражения вычислена свободная
энергия вязкого течения для жидкой окиси алюминия.
В интервале температур 2054—2250° С имеет место от-
3,4 3,6 3,8 4,0 4,2
(1/Т)'Ю\ К'1
Рис. 26. Зависимость Ign — 1/74 для
жидкой окиси алюминия
109
клонение этой величины от прямолинейной зависимости
(рис. 27). Это отклонение от линейности также
свидетельствует о возможных структурных изменениях в
области температур, близких к
температуре кристаллизации.
Изменение энтропии вязкого
течения в этом интервале
температур оценивали на основе
выражения:
ASn=—dAFJ]/dT. (2-37)
Величина Д5Л в интервале от
температуры плавления до
2500 К изменяется от —1 до
—10 ккал/(град-моль),
оставаясь далее постоянной. Так как
энтальпия вязкого течения равна:
ДЯп = £„ — Д7\ (2-38)
где Ек] — энергия активации
вязкого течения,то
AF^^E^ — RT — TAS^
и
т, = -^- exp (|L) ехр (- 1) ехр (- -^-). (2-39)
Для температурного интервала, где изменение AF^
при повышении температуры происходит по линейному
закону, можно записать
т1 = Лехр(^). (2-40)
Если пренебречь незначительным изменением
константы А в уравнении (2-40) при повышении
температуры, то на основе этого уравнения можно определить
энергию активации вязкого течения для
высокотемпературной области. Энергия активации вязкого течения
жидкой окиси алюминия уменьшается от 44 ккал/моль
при температурах, близких к температуре ее плавления,
до 17 ккал/моль при температурах, близких к 2550 К,
оставаясь в дальнейшем постоянной.
л f7, мал/моль
42
41
40
39
^ 1
1
_ ^А1203 /
_J
l" /I i 1
2275 2475 2675 Т, /Г
Рис. 27. Температурная
зависимость свободной
энергии активации вязкого
течения жидкой окиси
алюминия
ПО
Нелинейность соотношения lgrj—l/Т в предкристал-
лизационном периоде для жидкой окиси алюминия
свидетельствует о том, что это — ассоциированная
жидкость, а уменьшение энергии активации вязкого
течения — о том, что степень ассоциации быстро
уменьшается при повышении температуры.
Используя значения поверхностного натяжения для
жидкой окиси алюминия по формуле Фюртца [65]
Уд = 0,68
kT \3/2
(2-41)
где У*
•объем образующейся дырки при плавлении
жидкости,
можно оценить радиус дырок, возникающих при
плавлении окисла. Полученное значение радиуса дырок
при температуре плавления окиси алюминия равно
1,20-Ю-8 см.
Если предположить, что вся энергия активации
вязкого течения расходуется только на образование
вакансий, то можно вычислить радиус этих вакансий по
уравнению
Еп = 4nrl а.
Т} В
(2-42)
Результаты расчетов приведены в табл. 15.
Таблица 15
Размеры вакансий и дырок, образующихся при плавлении
окиси алюминия и двуокиси титана
Окисел
А1203
г, к
Т'пл
2500
2800
г-108, см
вакансий гв
2,28
1,52
1,40
дырки
ГД
1,20
Окисел
тю2
г, к
7*пл
2300
2600
г-108, см
вакансий гв
2,26
2,36
2,59
дырки
1,56
Из анализа данных следует, что размер вакансий,
необходимых для обеспечения вязкого течения жидкой
окиси алюминия, больше, чем размер дырок,
полученных по уравнению Фюртца. Следовательно, единицей
вязкого течения в жидкой окиси алюминия, по крайней
мере, при температурах, близких к темпреатуре плав-
111
V,0m
ления, являются не элементарные ионы, как у неассо-
циированных жидкостей, а некоторые ассоциации ионов,
возникающие в результате существенной доли кова-
лентной составляющей связи между металлом и
кислородом.
ДВУОКИСЬ ТИТАНА
Результаты измерения вязкости жидкой двуокиси
титана в интервале от температуры плавления (1870° С)
до 2350° С, полученные с помощью метода крутильных
колебаний, представлены
на рис. 28. Исследовали
двуокись титана марки
ч. д. а. с содержанием
основного компонента не
менее 99,5%.
Вязкость жидкой
двуокиси титана вблизи
температуры плавления
равна 1,1 П, т. е. жидкая
двуокись титана при
температуре плавления в два
раза более вязкая, чем
окись алюминия.
Температурная
зависимость вязкости жидкой
двуокиси титана в
изученном температурном
интервале
удовлетворительно подчиняется
уравнению (2-40): в
координатах Igrj—1/Г эта зависимость выражается прямой
линией (рис. 29,а). Энергия активации вязкого
течения в этом случае является величиной постоянной и
равной 33,0±1,5 ккал/моль. Уравнение, выражающее
температурную зависимость динамической вязкости
жидкой двуокиси титана, имеет вид
0,30
0,25
020
0,15
0,10
п
■ д
I I I I
о /
• 2
A 3
А 4
1800 1900 2000 2100 2200 2300t,°0
Рис. 28. Температурная зависимость
кинематической вязкости жидкой
двуокиси титана, полученная в различных
условиях:
/, 2 — диаметр тигля 26 мм; 3, 4—
30 мм; /, 3 — аргон; 2, 4 — вакуум
tl=:5-l04exp(:
ззооо
~RT~
(2-43)
Свободная энергия активации вязкого течения
жидкой двуокиси титана линейно возрастает при
повышении температуры (рис. 29, б). Линейный рост свобод-
П2
igq
0,1
0
0,1
0,J
0,4
0,5
-
" cr
® i
39
I I |__
4,1 4,3 4,5
WH04,K'r '
a
4J
AF, икал/моль '
39,0
38,5
38,0
37,5
37,0
A
У
" Jf
~ <f 5
1 1 1 1 1
2175227523752475 T,K
Рис. 29. Зависимость IgT] — 1/Г (а) и температурная зависимость
свободной энергии активации вязкого течения (б) для жидкой двуокиси
титана
ной энергии активации вязкого течения при повышении
температуры объясняется происходящим при нагреве
жидкости увеличением расстояний между частицами,
ослаблением сил связи между частицами, увеличением
числа вакансий в жидкости.
В этом случае можно использовать выражение
Д/5,Л = ЛЯЧ — TASn
(2-44)
и определить AS^. Энтропия активации вязкого течения
для жидкой двуокиси титана ASn=—4 кал/(град-моль).
Если использовать известное значение
поверхностного натяжения для жидкой двуокиси титана, то по
формуле Фюртца (2-41) можно рассчитать объем и радиус
дырок, образующихся при ее плавлении. Такой расчет
дает для радиуса дырки в жидкой двуокиси титана
величину, равную 1,56- Ю-8 см (табл. 15).
Однако, если вычислить для жидкой двуокиси
титана радиус вакансий по уравнению (2-42), то окажется,
что размер вакансий (табл. 15), обеспечивающих
вязкое течение, больше, чем размер дырок. Следовательно,
в случае двуокиси титана, как и в случае окиси
алюминия, единицей вязкого течения являются более
крупные структурные образования, чем элементарные ионы.
8-894
113
4. ЭЛЕКТРОПРОВОДНОСТЬ
жидких тугоплавких окислов
ОКИСЬ АЛЮМИНИЯ И РАСПЛАВЫ НА ЕЕ ОСНОВЕ
Фей [66] определил электропроводность жидкой
окиси алюминия с помощью переменного тока в иридиевом
тигле с иридиевым центральным электродом.
Полученное им значение составляет при 2400 К величину
3,84 Ом-1 «см"1. Окись алюминия плавили на воздухе,
но локальная среда из-за пламени горелки могла быть
восстановительной, поэтому автор считает, что
полученное им значение электропроводности жидкой окиси
алюминия возможно несколько завышено. Феи считает, что
характер проводимости жидкой окиси алюминия
ионный.
Рассмотрим здесь некоторые из исследований
электропроводности сложных окисных расплавов [30, 67], в
состав которых входит окись алюминия. Бокрис с сотр.
[67] изучал электропроводность расплавов А1203—Si62
с помощью моста Уитстона переменного тока.
Защитной атмосферой служил осушенный водород или аргон.
Удельная электропроводность расплавов А120з — Si02
при содержании окиси алюминия порядка 0,08
молярной доли составила 0,004 Ом-1 «см-1, а при увеличении
содержания окиси алюминия возрастала. Невысокая
электропроводность этих расплавов объясняется
склонностью ионов А13+ и Si44- к комплексообразованию.
Характер проводимости их в основном ионный {d%ldt>Q).
Филиппов с сотр. [68] измерял электропроводность
расплавов А1203—СаО методом вольтметра-амперметра
с применением молибденовых измерительных ячеек в
атмосфере аргона. Точность измерений ±6%.
Проводимость расплава А1203—СаО, содержащего 47,25%
А1203, изменяется от 0,35 до 2,3 Ом-1 • см~1 при
повышении температуры от 1500 до 1800° С. Энергия активации
проводимости равна 36,0 ккал/моль. При добавлении в
эти расплавы окислов титана и магния их
электропроводность увеличивается. При введении в расплав окиси
магния увеличение электропроводности происходит в
области высоких, а при добавлении двуокиси титана —
в области низких температур. При введении в расплав
окислов кремния и циркония электропроводность его
114
снижается. Подобное влияние окисных добавок
объясняется процессами комплексообразования в окисных
расплавах.
Елютин с сотр. [71] определил температурную
зависимость электропроводности жидкой окиси алюминия с
помощью метода амперметра-вольтметра. Схема
установки представлена на рис. 30. В качестве
измерительной ячейки применяли молибденовый тигель,
являющийся одним электродом; вторым электродом
служил молибденовый стержень, погруженный в
окисный расплав. Измерения проводили в атмосфере
аргона или в вакууме не ниже 10~4 мм рт. ст.
Погрешность измерения ±5%. Исходная окись алюминия
содержала 99,5% основного элемента, однако после
вакуумного переплава в молибденовом тигле количество
примесей в образцах снижалось до сотых долей
процента.
Результаты измерения электропроводности жидкой
окиси алюминия приведены на рис. 31:
электропроводность повышается от 0,71 Ом-1-см-1 при температуре,
близкой к температуре плавления окиси, до 1,0 Ом_1Х
Хсм-1 при 2525° С. Температурный коэффициент
электропроводности для исследованного температурного
интервала имеет положительный знак. При переходе окиси
Таблица 16
Скачок электропроводности при переходе из твердого состояния
в жидкое для веществ с разной природой проводимости
Вещество
NaCl
CdCl
Pb
Mg
Ge
Si
50% CaO—50% Si02
50% MnO—50% Si02
A1203
BeO
Y203
Sc203
TiQ2
*ж/хтв ПРИ 'пл
ЗЛО3
2-Ю3
0,5
0,2
-И
-20
2-102
3-102
7-103
3,4-102
0,5-102
0,9-102
<20
Характер проводимости
Ионная
»
Электронная
»
Полупроводники с
электронной проводимостью
То же
Ионная
»
»
»
»
Смешанная ионно-элек-
тронная
8*
115
УУ////////Л
алюминия из твердого состояния в жидкое наблюдается
значительный скачок электропроводности, характерный
для веществ с ионной проводимостью (табл. 16),
Температурная зависимость электропроводности жидкой
окиси алюминия в координатах lgx—\/Т отклоняется
от прямолинейной (рис.
32): энергия активации
изменяется от 20,5 ккал/
/моль при температурах,
близких к температуре
плавления, до 14 ккал/
/моль при 2500° С.
Энергия активации
электропроводности жидкой
окиси алюминия оказалась
ниже, чем энергия
активации ее вязкого течения.
Особенно большая
разница между ними
наблюдается при температурах,
близких к температуре
кристаллизации. Это
означает, что частицы,
являющиеся «единицами» вязкого течения, отличаются
от частиц, определяющих электропроводность. Различие
в энергиях активации вязкого течения и проводимости,
а также характер изменения свободной энергии
активации вязкого течения жидкой окиси алюминия
свидетельствует о наличии комплексных ионов в жидкости
вблизи температуры кристаллизации.
По Френкелю [72], для жидкостей, состоящих из
существенно различных сортов ионов при Ец /£х = п>1,
выполняется эмпирическое правило %пц = const.
Для жидкой окиси алюминия зависимость lgx—lgii
не является линейной (рис. 33), что подтверждает
предположение о том, что при повышении температуры
происходит изменение структуры расплавленной окиси
алюминия.
Рис. 30. Схема установки для измерения электропроводности жидких окислов:
1, 2 — винтовой механизм перемещения центрального электрода; «? — сильфон;
4— неподвижный шток; 5 — держатель центрального электрода; 6 — вакуумное
уплотнение; 7 — ввод второго электрода; 5 — второй электрод; 9 —
расплавленный окисел; 10 — центральный электрод; // — призма полного отражения;
12— корпус измерительной приставки к высокотемпературной печи; 13 —
подвод тока к центральному электроду; 14 — указатель глубины погружения
центрального электрода
x,0v'c/i-f
2000 2/00 2200 2300 2^00 t. °C
Рис. 31. Температурная зависимость
удельной электропроводности
жидкой окиси алюминия, полученная в
различных условиях:
1,2 — диаметр центрального
электрода 4 мм; 3, 4 — 2 мм; /, 3 —
вакуум; 2, 4 — аргон
117
Александров с сотр. [73] измерил электропроводность
жидкой окиси алюминия на воздухе при температурах
до 2225° С с помощью метода вольтметра — амперметра.
3,5 3,7 3,9 4,1 4,3 3,8 4,0 4,2 4,4 4,6 4,8
(1/V-104, К'1
Рис. 32. Зависимость lgx — \/T для жидких окиси алюминия (а) и двуокиси
титана (б)
Для расплавления окиси алюминия использовали
высокочастотный нагрев окиси в холодном контейнере.
Авторы [73] установили, что электропроводность жидкой
окиси алюминия
составляет 10 Ом-1-см-1 и не
зависит от температуры в
указанном температурном
интервале. Погрешность
измерения они оценивают
как ±3%,
действительная погрешность,
по-видимому, значительно
выше, поскольку
использованный метод нагрева
приводит к значительным
температурным
градиентам в расплаве, которые
могут существенно влиять
на результаты измерений.
Шпильрайн с сотр. [74, 75] измерил
электропроводность жидких окислов и в том числе жидкой окиси
алюминия при температурах до 2700° С. Для измерений
был использован метод вольтметра-амперметра; одним
электродом являлся тигель, в котором расплавляли оки-
Рис. 33. Зависимость
жидкой окиси алюминия
0,4 0,2 Щ
lgx— lgr] для
118
3000 2700 2500 2300
2100 Т, К 32003000
lgx\
2700 2500 2300 Г, /Г
т
3,8 4,4 5,0 3,1 3,5 3,9
WT)'W*K-1 (1/Г)104,К'}
Рис. 34. Зависимость Inx— ЦТ для жидких окислов:
/-4-АЬОа, по данным [73] (/), [66] (2), [74] (3, 4); 5 - ВеО [75]; 6-
[75]; 7 — Sc203 [75]
-YA,
си, другим — стержень, погружаемый в расплав.
Эксперимент проводили таким образом, что измерялась
электропроводность не только жидкой, но и твердой окиси.
Измерительную ячейку — тигель и центральный
стержень—изготавливали из молибдена для измерений при
температурах до 2300° С и из вольфрама —при
измерениях в более высокотемпературной области.
Использовали окись алюминия чистотой 99,9%- Измерения
проводили как в среде аргона, так и в вакууме; в обоих
случаях получены одинаковые значения электропроводности
при соответствующих температурах. Результаты
экспериментов представлены на рис. 34, а. Погрешность
измерений не превышает ±5%. Значения удельной
электропроводности жидкой окиси алюминия при различных
температурах приведены ниже.
т, к
2342
2383
2408
2465
2512
2537
к, Ом 1 см 1
2,69
3,12
3,36
4,06
4,77
5,02
т, к
2554
2559
2579
2598
2681
2697
Ом- L <
5,60
5,45
6,03
6,20
7,42
7,69
Г, К
2747
2835
2891
2935
2991
3008
х, Ом 1ci
8,61
10,13
10,75
11,52
12,51
12,66
И9
Энергия активации электропроводности жидкой
окиси алюминия, по данным этих авторов, снижается от
42,5 ккал/моль при 2125° С до 21,2 ккал/моль при
2725° С.
Значения электропроводности жидкой окиси
алюминия и ее энергия активации, определенные в работах
[74, 76], сильно отличаются от значений, полученных
Елютиным с сотр. [71]. Следует отметить, что значения
электропроводности жидкой окиси алюминия,
полученные Шпильрайном с сотр., выше обычных для ионных
расплавов.
ОКИСЬ БЕРИЛЛИЯ
Электропроводность жидкой окиси бериллия изучали
в атмосфере аргона при температурах от 2560 до 2725° С
[74, 76]. В экспериментах использовали окись бериллия
чистотой 99,9%. При проведении экспериментов
наблюдалось значительное испарение окиси бериллия с
открытой поверхности. Результаты измерений
электропроводности в полулогарифмических координатах
представлены на рис. 34, а.
Значения удельной электропроводности жидкой
окиси бериллия при различных температурах приведены
ниже:
г, к
2844
2851
2859
2867
2873
2880
И, Ом"™1 •см"""1
11,18
11,11
11,13
11,41
11,66
11,63
г, к
2888
2899
2902
2911
2917
2922
и, Ом~~хсм~х
11,46
11,53
11,79
12,06
11,93
11,43
г, к
2931
2948
2970
3002
>i9 Ом 1-см х
12,30
12,41
12,93
13,27
Энергия активации электропроводности жидкой
окиси бериллия постоянна и равна 20 ккал/моль.
ОКИСИ ИТТРИЯ И СКАНДИЯ
Электропроводность жидких окислов иттрия и
скандия определил Шпильрайн с сотр. [74—76]. В первом
случае измерения были осуществлены при температурах
до 2925° С, а во втором — при температурах до 2825° С.
Результаты измерений электропроводности в
координатах 1пх—1/7" представлены на рис. 34, б.
120
Характер температурного изменения
электропроводности этих окислов, а также величина скачка
электропроводности при переходе из твердого состояния в
жидкое позволяет считать эти расплавы ионными
жидкостями.
Значения удельной электропроводности жидких
окиси иттрия и окиси скандия при различных температурах
приведены ниже:
т, к
2716
2748
2768
2783
2791
2795
2782
2826
2840
2854
х, О**-1 о,,-1
18,43
19,08
19,51
19,69
20,23
19,83
16,70
17,96
18,15
18,47
т* к и. Ом-1 см"-1
Окись
2826
2858
2885
2906
2926
2950
Окись
2867
2895
2909
2932
иттрия
20,13
22,35
20,94
22,13
22,47
22,01
скандия
18,18
18,05
19,24
19,14
г, к
2981
2989
3052
3062
3075
3182
2953
2982
3014
3056
х, Ом*"1 см-1
22,74
24,21
24,75
26,41
24,38
26,17
19,42
19,61
20,20
20,88
Энергия активации электропроводности для обоих
окислов постоянна во всем интервале температурных
измерений и составляет 13,7 ккал/моль для окиси иттрия
и 12 ккал/моль для окиси скандия (рис. 34,6).
ДВУОКИСЬ ТИТАНА
Проводимость титановых шлаков в широкой области
изменения содержания в них двуокиси титана была
исследована Резниченко с сотр. [25, 26, 69, 70]. Измерения
проводили по методике неуравновешенного моста
переменного тока с использованием молибденовой
измерительной ячейки в атмосфере очищенного азота.
Согласно этим исследованиям, электропроводность титановых
шлаков при содержании двуокиси титана в них до 90%
слабо зависит от температуры и ее характер и величина
не изменяются при переходе из твердого состояния в
жидкое. Такой характер проводимости свидетельствует
о том, что с увеличением в шлаке содержания двуокиси
титана все меньшую роль играют ионы, проводимость
становится в основном электронной.
121
Электропроводность чистой жидкой окиси титана
выше, чем у рассмотренных выше окислов и изменяется от
21,6 Ом-1-см"1 при температуре, близкой к температуре
плавления, до 31 Ом^-см"1 при 2325°С (рис. 35).
Скачок проводимости при
переходе из твердого
состояния в жидкое
относительно невелик (см. табл.
16). Принимая во
внимание это, а также
положительное значение
температурного коэффициента
электропроводности,
можно предположить, что
проводимость двуокиси
титана в жидком
состоянии носит смешанный
ионно-электронный
характер.
При нагреве до 2100—
2200° С диссоциация
жидкой двуокиси титана в
атмосфере аргона
относительно невелика, поэтому и увеличение
электропроводности незначительно. При более высоких температурах
происходит резкое увеличение электропроводности,
обусловленное развивающимся процессом диссоциации.
Подобный эффект ранее был обнаружен Есиным [29] при
измерении электропроводности жидкой пятиокиси
ванадия.
Зависимость lgx—1/Г для жидкой двуокиси титана
линейна при температурах до 2450° С (см. рис. 32,6).
Энергия активации для этого участка равна 46 ккал/моль,
что выше, чем энергия проводимости двуокиси титана в
твердом состоянии. Подобное соотношение наблюдалось
ранее при исследовании проводимости пятиокисей
ванадия и ниобия [77].
5. ДИФФУЗИЯ В ЖИДКОЙ ОКИСИ АЛЮМИНИЯ
Диффузионные процессы в расплавленных
индивидуальных тугоплавких окислах не изучались, однако
работы но определению коэффициента диффузии
алюминия в расплавах А1203—Саб—Si02 известны.
х, онч сп-
то 1900 2000 2/00 2200 t. °C
Рис. 35. Температурная зависимость
удельной электропроводности жидкой
двуокиси титана, полученная в
различных условиях:
/ — центральный электрод диаметром
2 мм; 2 — 3 мм; 3, 4 — 4 мм; /, 3 —
аргон; 4 — вакуум
122
В работе [78] методом капилляра-резервуара с
помощью радиоактивного изотопа 26А1 определяли
коэффициент диффузии алюминия в расплаве. Для расплава,
содержащего 47,2% СаО; 47% Si02 и 6% А1203,
температурная зависимость коэффициента диффузии ионов
алюминия описывается уравнением
D = 4,3.10-exp(-8-^), (2-45)
а для расплава, содержащего 43,8% СаО; 43,7% Si02
и 12,5% А1203 — уравнением
D = 5,4.10-<exp(-6-^). (2-46)
Предполагается, что в этих расплавах алюминий
входит в состав алюминатных и алюмосиликатных
ионов.
Для изучения параметров процессов диффузии в
жидкой окиси алюминия Митин и др. [79, 80]
использовали капиллярный метод. Капилляры изготавливали из
молибдена; диаметр внутренней полости 2 мм, длина
порядка 40 мм. Величина диаметра обеспечивала
подавление конвекции при исследовании.
Капилляры заполнялись жидкой окисью алюминия
с помощью вакуумного всасывания. Анализ продольных
шлифов капилляров показал, что они заполняются
полностью и без раковин. Затем верхнюю часть стержня из
окиси алюминия в капилляре удаляли (на глубину 3—
4 мм), помещали туда активную окись алюминия
(изотоп 26А1), закрывали капилляр молибденовой крышкой
и в вакууме приваривали крышку к капилляру
электронным лучом. Заряженные капилляры вводили в
горячую зону печи и после установленной выдержки
извлекали из нее.
Условия проведения экспериментов соответствовали
диффузии из плоского непостоянного источника в
полубесконечный образец. Изменение концентрации
(активности) в этом случае описывается уравнением второго
закона Фика
-^ = D-^ (2-47)
дт дх2
при начальных и граничных условиях:
с (0 < х < К 0) = с0;
123
с {х > ft, 0) = 0; (2-48)
дх
Уравнение (2-47) имеет решение
с = —&— ехр (- —), (2-49)
где Q0 — начальное количество диффундирующего
вещества, г/см2.
Построив кривую распределения изотопа вдоль
образца в координатах \gc—lgx2, можно найти
коэффициент диффузии по тангенсу угла наклона касательной:
D = lgc/(4rtga). (2-50)
Для получения достоверных результатов необходимо
соблюдение начальных и граничных условий. Условие
бесконечно тонкого слоя и полубесконечной длины
образца будет выполнено, если отношение высоты слоя
изотопа к длине образца будет много меньше
единицы, т. е.
/1«2]/7>г7 (2-51)
Для увеличения чувствительности метода время
диффузионного отжига (2—3 ч) выбирали таким, чтобы
небольшие изменения а вызывали значительные
изменения tga.
После опыта капилляр сжигали в окислительной
атмосфере при 600° С, освобождая таким образом образец
из окиси алюминия.
Для анализа распределения активности в образце
его механически разделяли на отдельные части. Так как
невозможно обеспечить строгую параллельность границ
отдельных слоев, то в уравнении (2-49) целесообразно
заменить
с на J/P или сг ж -^- , (2-52)
где Pt — масса t-того слоя;
Jt— интенсивность излучения i-того слоя;
ct = kCll3 (Сиз — концентрация изотопа в f-том слое;
k — константа).
124
Тогда ^P = ymnr2xt
где ут— плотность расплава;
xt — координата i-того слоя;
г— радиус капилляра.
Подставив значения d и xt в (2-49), получим
h
Pi
0 exp
VnDx
(i=n
i=\
(y^nr^Wx
(2-53)
Построив зависимость IgJJP i—(2Р^)2, можно
найти угол наклона tga, связанный с коэффициентом
диффузии соотношением
D = — . (2-54)
Судить о том, насколько хорошо выполняются
граничные условия, можно на основе зависимости \gJ/P—
SP)2: если она линейна,
то граничные условия
выполнены. В указанном
исследовании эта зависимость
является линейной.
Погрешность измерения
коэффициента диффузии
описанным методом
составляет +6%.
Параметры диффузии в
жидкой окиси алюминия
исследованы в интервале
2060—2300° С; результаты
исследования приведены на
рис. 36.
Зависимость IgD—1/Г
для диффузии в жидкой
окиси алюминия хорошо аппроксимируется прямой
(рис. 36); рассчитанное по этим данным уравнение
зависимости коэффициента диффузии в жидкой окиси
алюминия от температуры имеет вид:
D = 2,3.10-3exp(-|A). (2-55)
Величина энергии активации процесса диффузии
3,9 4,0 4,1
Рис. 36. Зависимость \gD — [JT для
жидкой окиси алюминия (разные
точки соответствуют трем сериям
эксперимента)
125
Таблица 17
Связь между электропроводностью и коэффициентом
диффузии в жидкой окиси алюминия
7\ К
2335
2375
2400
2425
D-10',
см2/с
1,95
2,05
2,15
2,35
Электропроводность х, Ом .см х
ментальная
0,71
0,78
0,91
1,03
расчетная при диссоциации по типу
А12037±
-2А1++30-
4,5
5,2
5,95
6,0
2А12Оа^
^А1++ЗАЮо-
2,25
2,6
3,0
3,25
А1г03^
-АЮ++АЮ2-
0,25
0,29
0,33
0,36
примерно равна величине энергии активации процесса
электропереноса. Это обстоятельство позволяет
полагать, что и в том и в другом случаях перенос
осуществляется однотипными носителями, являющимися
наиболее подвижными в расплавленной окиси алюминия.
Линейная зависимость IgD—\jT для жидкой окиси
алюминия свидетельствует о том, что реализуемый
механизм диффузии соответствует активационной теории
Эйринга [64]. Поэтому, используя уравнение Эйринга
Для случая диффузии в чистом веществе, можно
определить радиус частиц, обеспечивающих диффузионный
перенос:
гч = kT/(2DY\), (2-56)
где гч— радиус частицы,
Г[— ВЯЗКОСТЬ.
Результаты расчетов размера частиц,
осуществляющих диффузионный перенос в жидкой окиси алюминия,
приведены ниже. В исследованной области температур
величина радиуса этих частиц удовлетворительно
совпадает с радиусом иона А10+.
Температура, К . . - .2335 2375 2400 2425
D-105, см2/с ..... 1,95 2,05 2,15 2,35
т),П • -0,58 0,52 0,45 0,39
'ион-Ю8, см 1,50 1,52 1,70 1,82
Примечание. гАЮ+=1,82- Ю~8 СМ; /-А1+=0,53- Ю""8 СМ.
Используя данные по электропроводности и
диффузии в жидкой окиси алюминия по соотношению
Эйнштейне [29]
126
y. = Dz*F*cl{NRT),
(2-57)
где D— коэффициент диффузии;
z — заряд иона;
F — число Фарадея;
с— концентрация ионов,
можно оценить наиболее вероятный характер
диссоциации окиси алюминия при плавлении. В табл. 17
представлены результаты расчетов, из которых следует, что
наиболее вероятными реакциями диссоциации при
плавлении А1203 являются А120з^А10;Г4-А10+ и 2А1203^
^А1++ЗА10Г.
Глава III
ТЕПЛОПРОВОДНОСТЬ ТВЕРДОЙ
И ЖИДКОЙ ОКИСИ АЛЮМИНИЯ
ПРИ ВЫСОКОЙ ТЕМПЕРАТУРЕ
Определение теплофизических свойств, в том числе
теплопроводности, жидких тугоплавких окислов сопряжено
с преодолением значительных теоретических и
экспериментальных трудностей. В этом, видимо, скрывается
причина полного отсутствия данных по
теплопроводности жидких тугоплавких окислов, несмотря на большую
потребность в них.
1. ТЕОРЕТИЧЕСКАЯ ОЦЕНКА
ТЕПЛОПРОВОДНОСТИ
жидких диэлектриков
Теоретические расчеты теплопроводности
чрезвычайно сложны даже для кристаллических тел. Эти расчеты
для кристаллических тел при высоких температурах
справедливы с точностью до порядка величины и
отражают лишь общий вид температурной зависимости
теплопроводности. Теория носит полуфеноменологический
характер и ее параметры определяются из сравнения
теоретических зависимостей с экспериментальными
данными.
127
Определение теплопроводности в жидком состоянии
еще более осложняется: в жидкости отсутствует
трансляционная симметрия и обычный подход к расчету
теплопроводности, применяемый для кристаллических тел,
здесь непригоден. Теплосопротивление в жидкостях
обусловливается в основном двумя факторами: аморфиза-
цией структуры и возникновением многочисленных
центров рассеяния типа дырок. Теории теплопроводности
жидкости, базирующиеся на простых газовых или
квазикристаллических моделях, не могут дать
сколько-нибудь четкого ответа даже на вопрос о механизме
переноса тепла [1—11].
Бриджмен [12] ввел в формулу Дебая скорость
звука v8 и межмолекулярное расстояние, а теплоемкость
для всех жидкостей предположил постоянной Су = 3&в
и получил формулу для расчета теплопроводности
жидкостей:
% = 3kbvJVV\
где kb — постоянная Больцмана;
V — атомный (молекулярный) объем.
Расчеты по этой формуле, по оценке Миснара [13],
дают значения коэффициента теплопроводности,
отличающиеся от экспериментальных данных на 30—50%.
Дальнейшее развитие это направление получило в
работе [3].
Осида [2] вместо скорости звука вводит частоту
актов энергетического обмена v. В этом случае
теплопроводность описывается уравнением вида:
X = 4kBv/V. (3-2)
При v, равной дебаевской частоте твердого тела при
температуре плавления, определяемой по правилу Лин-
демана, уравнение (3-2) преобразуется к виду
где 5Ж— эмпирическая постоянная;
М—молекулярная масса.
Формула (3-3) дает расхождение с экспериментом
до 50%. В последующих работах [9] эту теорию
использовали лишь для описания зависимости
теплопроводности от температуры и давления.
128
(3-1)
(3-3)
Филиппов [14] рассмотрел корреляцию между
теплопроводностью и другими свойствами жидкости и
сделал вывод о значительной общности механизмов
переноса тепла в твердых телах и жидкостях. Это дает
возможность использовать модель Дебая о переносе тепла
гиперакустическими колебаниями, частота которых
лежит в пределах от 0 до сотах- Для T^QDf учитывая
процессы поглощения и рассеяния гиперзвука,
теплопроводность определяется формулой:
X = 2,l2ot{cvP)Wk\P(yJyy*, (3-4)
где у и 7нр— плотности при данной и критической
температурах соответственно.
Для 19 органических веществ среднеквадратичное
отклонение расчетных данных от действительных
значений составляет 4,5%, а максимальное отклонение не
превышает 9% [14]. Кроме того, уравнение дает
правильную температурную зависимость коэффициента
теплопроводности. В принципе, оно должно
удовлетворительно описывать и теплопроводность жидких
диэлектриков при средних и высоких температурах.
Более строгий подход к проблеме теплопроводности
возможен в статистической теории [10, 11, 15—22].
Однако использование этих методов для практических
расчетов затруднено из-за недостаточной
разработанности теории жидкого состояния, вследствие чего
приходится прибегать к различного рода модельным
представлениям о характере движения молекул в жидкости,
потенциале межмолекулярного взаимодействия, функции
распределения и т. п.
Так, например, в работах Энскога и др. [7—10]
выполнены расчеты для модели жидкости в виде набора
твердых сфер, взаимодействующих между собой только
соударениями (двойными и тройными). С
использованием этого метода была получена правильная
зависимость коэффициента теплопроводности жидкости от
давления [3, 6]. Однако этот метод не учитывает
принципиальную особенность жидкостей — взаимодействия
коллектива молекул. Более последовательное решение
задачи дает метод исследования кинетических
коэффициентов в неупорядоченных системах на основе формул
Кубо — Мори, связывающих корреляционные функции
потоков с соответствующими кинетическими
коэффициентами [17—19]. На основе этих формул в ряде работ
9-894
129
был рассмотрен вопрос о коэффициенте
теплопроводности в неупорядоченных твердых телах и жидких
диэлектриках. Однако расшифровка формул Кубо — Мори
требует специальных, иногда весьма сложных расчетов и
пока может быть выполнена лишь для малых величин
степени неупорядоченности.
Другие теоретические представления о
теплопроводности жидкостей можно найти в обзорной статье Мак-
Лафлина [20], работах Филиппова [14], Рида и
Шервуда [21], Миснара [13]. Однако известные
феноменологические и полуэмпирические теории имеют
принципиальный недостаток — они не отражают специфику
жидкого состояния. Поэтому нельзя распространять
результаты теоретических разработок, справедливые для
одного класса жидкостей (например, органические
диэлектрики при температурах, близких к комнатной), без
предварительной экспериментальной проверки на другие
классы жидкостей (например, неорганические жидкие
диэлектрики при высоких температурах). Следует
заметить также, что жидкости имеют самый низкий
коэффициент теплопроводности среди конденсированных тел и,
следовательно, роль радиационного теплопереноса в
жидкостях значительно возрастает [23]. Поэтому при
рассмотрении теплопереноса в области средних и
высоких температур особенно важно знать величину и
молекулярной, и радиационной составляющих.
2. фононная теплопроводность
диэлектриков
при высоких температурах
При температурах выше комнатной, когда можно
пренебречь рассеянием фононов на границах зерен,
примесях и изотопах, теплопроводность чистых
кристаллических диэлектриков определяется рассеянием фононов
на фононах. В этом случае тепловое сопротивление
кристаллической гранецентрированной кубической решетки,
которое определяется трехфононными процессами,
описывается выражением [24]:
Ш9Ь?у1т
Wm = — - * (3-5)
зМз^1'3
где Т — температура, К;
130
hy k& — постоянные Планка и Больцмана
соответственно;
уг — постоянная Грюнайзена;
М — средняя атомная масса;
QD — температура Дебая;
V— атомный объем.
Несмотря на то что все параметры, входящие в (3-5),
непосредственно измеряются или несложно
вычисляются по экспериментальным данным, полученные
результаты справедливы с точностью до порядка величины.
Возможно, что это связано с тем, что в
теплопроводности способ усреднения по фононному спектру и
направлениям волновых и поляризационных векторов весьма
специфичен и величины QD и -уг М0ГУТ не совпадать
с аналогичными параметрами, определяемыми другими
путями. Для удовлетворительного соответствия
результатов расчета с экспериментально определяемыми
значениями постоянную Грюнайзена приходится принимать
в 1,5—3 раза большую, чем дают оценки этого
параметра, полученные любым из доступных способов
(измерение упругих постоянных третьего порядка,
термодинамические соотношения, эмпирические зависимости и т.д.)
[24].
Из сравнения времен релаксации при Г>8я
находится температурная зависимость четырехфононной
составляющей Wiv полного теплосопротивления [25]:
wlvwlu~T/Tnjl. (з-б)
Приближенные оценки коэффициента
пропорциональности для гранецентрированных решеток при ле-
нард-джонсоновском потенциале приводят к величине
порядка единицы, откуда следует, что четырехфононное
рассеяние при достаточно высоких температурах
сравнимо с трехфононным. В приближении аддитивности
температурная зависимость полного
теплосопротивления имеет вид
W = Wm + Wlv = aT + ЪТ\ (3-7)
где а и Ь — константы.
В общем случае без детального анализа нельзя
установить, какие члены — третьего или четвертого
порядка — играют более важную роль в том или ином
процессе.
9*
131
Эти обстоятельства заставляют прибегать при
расчете коэффициента теплопроводности к различным
полуфеноменологическим и эмпирическим закономерностям
[13, 26—29, 35—38]. Так, например, Кейс [27], выражая
температуру Дебая в уравнении (3-5) по правилу Лин-
демана
где d— постоянная решетки;
е — относительная амплитуда тепловых
колебаний атомов (в точке плавления е«0,1);
Тпл — температура плавления,
получил формулу вида:
*кТ — R пл (3-9)
"~ М1/2 V2'3 '
в которой ВТв — постоянная, зависящая от типа
химической связи в кристалле. Для расчета коэффициентов
теплопроводности многих кристаллов с чисто ковалент-
ной связью следует принять ВТВ=3,Ы0~2, а для
кристаллов ионной связи Втв=3,6-10~3 [при этом Я
измеряется в кал/(см-с-град)].
Используя соотношение Грюнайзена
yr = (V/cf)(a/% (3-10)
где а— коэффициент объемного расширения;
Р— коэффициент сжимаемости;
Cf— полная теплоемкость;
можно получить ряд полуэмпирических соотношений.
Наибольшей точностью обладают эмпирические
формулы, полученные Миснаром [13]: по оценке автора
погрешность не хуже 20%.
Величины фононной теплопроводности диэлектриков
при средних и высоких температурах нельзя
рассчитывать по полуфеноменологическим и эмпирическим
зависимостям, так как в большинстве случаев они основаны
на экспериментальных данных по теплопроводности,
представляющих собой эффективный коэффициент
совместного теплопереноса теплопроводностью и
излучением.
В работах [30, 31] определение коэффициента
фононной теплопроводности кристаллических диэлектриков
132
при высоких температурах проводится экстраполяцией
экспериментальных данных для низких температур по
зависимости вида
К^ 1/7\ (3-11)
Как указано выше, вместо этой зависимости
необходимо использовать зависимость вида (3-7). Кроме того,
важно выяснить, на какие экспериментальные данные
можно опираться при экстраполяции. Использование
зависимостей (3-7) и (3-11) корректно лишь при T>QD.
Для многих кристаллов температура Дебая имеет столь
высокие значения (1280 К для ВеО, 1048 К для А1203,
930 К для MgO), что при TttQD радиационный теплопе-
ренос дает существенный вклад в экспериментально
определяемый коэффициент теплопроводности.
3. РАДИАЦИОННЫЙ ТЕПЛООБМЕН
В ДИЭЛЕКТРИКАХ
ПРИ ВЫСОКИХ ТЕМПЕРАТУРАХ
Появление дополнительного механизма
теплопроводности диэлектриков — внутреннего радиационного
теплообмена тепловым электромагнитным излучением —
связано с их прозрачностью в инфракрасной области
спектра. Величина радиационного вклада зависит от
оптических свойств, геометрии и температурного поля
образца и граничных поверхностей. Во многих работах
учет радиационного переноса производится введением
«радиационной теплопроводности» по формуле Россе-
ланда [32]:
XR=\6on*TV(3kp)y (3-12)
где a— постоянная Стефана — Больцмаиа;
п— показатель преломления;
&р—осредневдшй по спектру коэффициент
поглощения — «росселандово среднее».
Однако применение этой формулы дает корректные
результаты лишь для оптически толстых образцов1 в
состоянии, близком к радиационному равновесию.
1 Т. е. тогда, когда характеристические размеры образца
значительно провышают k~l *
133
Для расчета радиационного теплообмена в других
условиях применяются другие приближенные методы.
Различные аспекты теории радиационного теплоперено-
са рассмотрены в работах [22, 23, 32—36]. Количество
приближенных методов велико, поэтому большое
значение имеет математическое описание обмена
электромагнитным излучением в наиболее общей физической
постановке. Оно позволяет проводить наиболее точные
и детальные исследования этих процессов и в то же
время является основой для построения приближенных
аналитических методов расчета.
Проблему теоретического расчета радиационной
составляющей при совместном теплопереносе
теплопроводностью и излучением изучали многие исследователи
[38—47]. Хотя пока не найдено общего решения
проблемы, но можно достаточно надежно рассчитать процесс
радиационно-кондуктивного теплообмена (РКТ) в тех
условиях, для которых справедливы используемые
приближения.
Однако для аналитического расчета необходимо
располагать спектральными радиационными
характеристиками среды и граничных поверхностей в исследуемом
интервале температур. К сожалению, радиационные
характеристики такие, как спектральные коэффициенты
поглощения, рассеяния, ослабления и т. п., известны
лишь для ограниченного круга полупрозрачных
материалов [52—55]. К тому же коэффициент рассеяния, а
следовательно, и ослабления, является
структурно-чувствительной величиной. Вопрос о достоверности и
точности экспериментально определенных коэффициентов
ослабления, рассеяния и поглощения, особенно для
высоких температур, очень сложный вопрос. Так,
например, значения спектральных коэффициентов ослабления
даже такого хорошо изученного вещества, как
плавленый кварц, значительно различаются в области слабого
поглощения [48]. Это вынуждает при исследовании
теплопроводности полупрозрачных веществ
одновременно измерять их эффективную теплопроводность и
спектральные характеристики [56].
Таким образом, аналитические методы расчета
радиационной составляющей дают достаточно высокую
точность, но требуют значительного объема
математических вычислений и наличия надежных спектральных
данных. Поэтому в практике теплофизических йсследо-
134
ваний получили распространение приближенные
методы экспериментального определения радиационной и
фононной составляющих теплопереноса.
4. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
РАЗДЕЛЕНИЯ ФОНОННОЙ И РАДИАЦИОННОЙ
СОСТАВЛЯЮЩИХ ТЕПЛОПЕРЕНОСА
Вектор радиации является функционалом
температурного поля, геометрии и радиационных свойств
образца, поэтому их контролируемое изменение дает
принципиальную возможность определения составляющих РКТ.
Существуют следующие методы разделения
составляющих теплопереноса:
методы, основанные на изменении геометрии
системы, радиационных свойств среды и граничных
поверхностей;
методы, основанные на изменении теплового поля
образца — изменение величины теплового потока,
проходящего через образцы;
методы регулярных тепловых режимов.
В работах [26—28], в которых рассматривается
первая группа методов, величину вклада радиационного
теплопереноса определяли из сопоставления данных по
теплопроводности двух образцов, радиационный тепло-
перенос в одном из которых подавлен за счет
увеличения коэффициента ослабления.
Для серой среды1 с изотропным распределением
излучения вектор потока излучения равен:
9r = — — grad Т. (3-13)
о к
При прочих равных условиях вектор qR можно
получить как угодно малым, увеличивая значение
коэффициента ослабления k. Увеличение коэффициента
ослабления достигается увеличением коэффициента
рассеяния. Рассеяние происходит на микротрещинах, границах
зерен, включениях, зонах сегрегации и т. п.
В работе [77] был исследован теплоперенос в
монокристаллических и спеченных образцах из ТЮг, А1203,
1 Для среды, в которой коэффициент ослабления не зависит от
длины волны.
135
CaF2 (рис. 37,а). На основе сопоставления данных для
моно- и поликристаллических образцов рассчитана
величина «радиационного» коэффициента
теплопроводности (рис. 37,6). Однако анализ температурной
зависимости теплопроводности спеченных образцов позволяет
заключить, что в них радиационный теплообмен
подавлен неполностью.
W, м-к/Вт
0,5
ОА
0,3
0,2
0,1
а
у^^Шг
л1>-
400
800 1200 Т,К
500 100 О 2000 Т, К
Рис. 37. Температурные зависимости теплосопротивления монокристаллических
(/) и спеченных (2) образцов (а) и «радиационной» теплопроводности
монокристаллических образцов (б) [57]
Следует отметить, что при уменьшении температуры
длина волны, соответствующая максимуму теплового
излучения, увеличивается. Это вызывает уменьшение
эффективности рассеивающих центров [58].
Кроме того, центры рассеяния могут изменить не
только оптические, но и локальные механические и
другие характеристики решетки, определяющие
коэффициент фононнои теплопроводности. В некоторых случаях,
например в жидкостях, плазменно-напыленных
образцах и других аналогичных системах, невозможно
увеличить коэффициент ослабления таким путем. Поэтому
данный метод выделения фононнои теплопроводности
является недостаточно универсальным, но позволяет в
отдельных случаях с не слишком большой точностью
разделять фононную и фотонную составляющие
теплового потока.
Изменение радиационных свойств граничных
поверхностей иногда позволяет изменить величину вектора ра-
136
диации и тем самым оценить вклад радиационной
составляющей в общий теплоперенос. Область
применимости данной методики легко определить,
воспользовавшись результатами работ [13, 37, 48]. Так, для плоско;
го слоя серой среды с непрозрачными границами общий
теплоперенос через образец определяется
выражением [37]:
, = Я-
(3-14)
/
где
Т0 и Ti— температура граничных поверхностей;
I — толщина
слоя;
Аэф—
эффективная
радиационная
функция
параметров
среды и
граничных
поверхностей.
Семейство кривых ЛЭф (а',
/, г) показано на рис. 38
(а' — осредненный
коэффициент поглощения; a'=kv)-
Аналогичный характер
зависимости получен в
работе Польтца [48] при решении этой задачи с
использованием линеаризации четвертой степени температуры:
^-(Ш2Г3Ф(а',/,г), (3-15)
12Kvl
Рис. 38. Эффективная функция
радиационных параметров среды и
граничных поверхностей [37] при:
= 10; 2 — 0,8; 3 — 0,6; 4 — 0,4;
-0,2; 6-
h =
где %ц — радиационная составляющая
теплопроводности.
Функция Ф имеет вид:
ф = -±^\1-^—(1-АЕ6) Зг
а' I [
коэффициента
4а7
а'/
X
X
J l+re-*'ls *б J'
(3-16)
где
Еь(а\ /)— интегрально-экспоненциальная функция
пятого порядка
137
[£*(«',/)
\e^'l\
'dsls\
(3-17)
В несколько ином виде получена функция Ф в
работе Филиппова [14]в дифференциально-разностном
приближении:
Ф =
1 —
а' /
JL
(3-18)
Следует отметить, что результаты расчетов по всем
этим формулам близки между собой и близки к
результатам численного решения
А,Вт/(^К) задачи, полученного в
работах [39—41].
Данные, представленные
на рис. 38, свидетельствуют
о том, что степень черноты
стенок оказывает заметное
влияние на величину
эффективной теплопроводности
при толщинах а'/^2, a при
а'/^5 влияние стенок
практически незаметно. Отсюда
следует, что изменение
степени черноты границ
позволяет определить наличие
радиационного переноса в
образцах, оптическая
толщина которых меньше 2.
В работе [43] исследовали теплоперенос через
пластину из плавленого кварца толщиной 20 и диаметром
50 мм. В первом случае шлифованные поверхности
нагревателя и холодильника притирались к шлифованным
поверхностям образца. Во втором случае образец,
нагреватель и холодильник имели полированные
поверхности. Результаты эксперимента показаны на рис. 39.
Как видно, данные [60], оцениваемые авторами, как
наиболее достоверные, совпадают с
экспериментальными данными для обоих образцов при температурах до
300° С. При более высоких температурах результаты
эксперимента для двух образцов резко расходятся, а
литературные данные лежат между ними. При темпера-
200 400 600 800 /ООО Т,/Г
Рис. 39. Влияние степени черноты
поверхности на величину
эффективной теплопроводности кварцевого
стекла:
/ — поверхность шлифованная [43];
2 — поверхность полированная [43];
3 — данные [59]; 4 — данные [60]
138
туре 700° С эффективная теплопроводность второго
образца в два раза выше, чем у первого, хотя и в нем
вектор радиации отличен от нуля.
Следует отметить, что данная методика позволяет
лишь качественно судить о наличии радиационного
переноса в полупрозрачных образцах. Кроме того, степень
черноты материала границ в контакте с исследуемой
средой может отличаться от значений, измеренных в
вакууме или газах.
Метод разделения радиационной и кондуктивной
составляющих теплового потока, основанный на
изменении геометрических характеристик образца, можно
пояснить, переписав формулу (3-14) в виде
q =— (к + 4оп*АТ31) Д77/. (3-19)
При а^<1 второе слагаемое в скобках,
определяющее радиационный вклад в эффективный коэффициент
теплопроводности, стремится к нулю при уменьшении /.
Измеряя эффективный коэффициент теплопроводности
на нескольких образцах разной толщины и
экстраполируя на нулевую толщину, можно получить коэффициент
фононной теплопроводности. Но так как величина А
нелинейно зависит от толщины, то точность подобной
экстраполяции невелика.
Для плоского слоя этот метод использован в
работах [48, 61, 62]. Математический аппарат для
цилиндрического слоя разработан в работе [51]. Измерения по
этому методу проведены в работах [63, 64].
Польтц [48, 61] установил, что теплопроводность
семи органических жидкостей в интервале температур
10—80° С зависит от толщины слоя. Изменение
теплопроводности при изменении толщины слоя составляет
3—4% и находится в хорошем согласии с
теоретическими оценками. Подробное обсуждение этих работ можно
найти у Филиппова [14].
Методы, основанные на изменении
характеристического размера образца, во многих случаях позволяют с
удовлетворительной точностью разделять радиационную
и кондуктивную составляющие теплопереноса. Для
исследования требуются несколько идентичных образцов
разной толщины. Легче всего это сделать при
исследовании жидкости, где метод нашел широкое применение.
В работах [42, 44] анализируются регулярные
тепловые режимы в полупрозрачных средах. В работе Ме-
139
ня [42] рассмотрен регулярный тепловой режим
второго рода применительно к симметричной задаче нагрева
чисто поглощающей пластины, температура границ
которой изменяется по линейному закону
Т = Тн + 1п, (3-20;
где b — константа.
Вектор радиации определяется в интегральном
приближении [44]. Числовой расчет показал, что
распределение температуры близко к параболическому. При
использовании этого приближения
температуропроводность вещества определяется выражением
а = —У- ^/, (3-21)
'Л^тах УС
где
jnl ^п.|j^ _|\[к(|(V)_*<_/,6,v)ldSJdv;
0 -/
/—толщина образца;
К(х, |, v) — функция радиационных свойств среды и
граничных поверхностей;
Ец — функция Планка;
v — частота;
у и с — плотность и удельная теплоемкость.
Для вычисления величины / пришлось измерить
величины спектрального коэффициента поглощения в
интервале температур 20—800° С.
В практике для определения теплофизических
свойств полупрозрачных материалов предложенный
метод не выявляет каких-либо существенных преимуществ
регулярного теплового обмена второго рода перед
другими тепловыми режимами.
Филиппов с сотр. [65] отмечает, что регулярный
тепловой режим третьего рода позволяет варьировать
температурное поле среды путем изменения частоты
температурных колебаний. Проведение экспериментов на
различных частотах может сыграть весьма
существенную роль в исследовании влияния процессов излучения.
В работе Шатса и Симонса [66] предложен
оригинальный метод определения коэффициента
температуропроводности и эффективного коэффициента ослабления
методом плоских температурных волн. Идея метода "за-
140
ключается в следующем. Тепловое излучение,
испускаемое полупрозрачной пластиной с прозрачными
границами, регистрируется фотоэлектрическим датчиком,
сигнал которого равен:
Rt (t) = Г Sv dv j ccv £nv (x,t) exp [— ^ (/ — x)] dx, (3-22)
о о
где Sv — спектральная чувствительность датчика;
&v и av — спектральные коэффициенты ослабления и
поглощения;
£nv— функция Планка;
I — толщина образца.
При регулярном тепловом режиме третьего рода
температура каждой точки образца колеблется вокруг
своего среднего значения Т с амплитудой в, зависящей
от координаты, с частотой со (0 — величина
комплексная). В этом случае тепловое излучение, а
следовательно, и сигнал датчика имеют переменные составляющие,
которые колеблются с той же частотой со. Фаза
колебаний зависит от температуропроводности и
коэффициентов ослабления и поглощения. Величина переменной
составляющей сигнала датчика для малых 0
определяется выражением
R = const f 0 (jc,t) exp [— k' (I — xj\ dx. (3-23)
о
Эффективный коэффициент ослабления k
определяется из уравнения
|avSvEBvexp [—k'v(1-х)] dv
exp[—V{1 — *)] = " . (3-24)
f avSv£Bvdv
Уравнения (3-22) — (3-24) являются основой метода
определения составляющих теплопереноса. Далее
авторы предполагают, что в полупрозрачных средах
температурная волна имеет тот же вид, что и в непрозрачных:
0 = и ехр [— §х + i (сот -f 6)] -f a ехр [$х + i (ют + р)],
(3-25)
где и, v, 6, р— действительные величины;
р—величина комплексная.
141
Константы и, t>, б, р определяются из граничных
условий.
Прих=0 F^-K^ + q^ (3-26)
при х = I О = %эф ^ + qR% (3-26а)
где F— переменная составляющая теплового потока;
ЯЭф— эффективная теплопроводность
Кф = Х + ^И1; (3-27)
1+3/4 k I
о — постоянная Стефана—Больцмана;
п— показатель преломления;
Чн — тепловое излучение с поверхности образца.
Необходимо отметить, что граничные условия (3-26)
записаны для непрозрачных границ. Для прозрачных
границ вместо (3-26) следовало бы использовать при
х = 0 выражение
dx
и при х=1
О = dQ/dx. (3-28)
Величину радиационного потока можно определить,
используя результаты работы [67]:
оо
qR~^dv f IVi+n(7)cos(7n)U^-r(s))d<*$), (3-29)
О (2я+л)
где IVi+n(s)—спектральная интенсивность падающего
излучения на граничной поверхности в
направлении 5;
+/г — внешняя по отношению к образцу
нормаль;
r(s) — спектральная отражательная
способность для направления падающего
излучения s;
dco- — дифференциал телесного угла в
направлении s.
Интегрирование распространяется на внешнюю
полусферу.
142
Эта неточность в определении граничных условий
снижает ценность последующих рассуждений и
полученных результатов. Необходимо сделать еще несколько
замечаний относительно постановки работы.
Уравнения (3-22), (3-23) не учитывают рассеяния и
применимы только для чисто поглощающих сред, хотя
авторы этим методом проводят исследование спеченной
окиси алюминия, обладающей значительным
рассеянием.
Вид осреднения (3-24) ставит такую характеристику
материала, как коэффициент ослабления, в зависимость
от спектральной чувствительности регистрирующей
системы. Применимость такого осреднения, даже в случае
использования более широкодиапазонных датчиков, чем
фотосопротивление из PbS, весьма проблематична.
Температурная волна в полупрозрачных средах
имеет более сложный характер, чем определяемый
уравнением (3-25).
Авторы используют лишь часть информации,
даваемой регулярным тепловым режимом третьего рода.
Вопрос об амплитудах температурных волн не
рассматривается, это вынуждает авторов пользоваться методом
подбора с соответствующим значительным снижением
точности определения.
Регулярный тепловой режим третьего рода,
применительно к симметричному случаю распространения
температурных волн в плоскопараллельной
полупрозрачной пластине с непрозрачными зеркально
отражающими границами, рассмотрен в работе Меня [45].
Уравнение распространения температурных волн в
полупрозрачных средах записывается в виде
_*£+^ = -',f. (3-30)
дх* дх дх
где <7я — вектор радиации;
ср —теплоемкость единицы объема.
Уравнение, описывающее вектор радиации, взято из
более ранней работы [42]. Граничным условием для
уравнения (3-30) является условие
б(± /,т)=Лехршт. (3-31)
Комплексная амплитуда температурных колебаний
Э(я) находится на основе решения системы двух
интегральных уравнений второго рода типа Фредгольма. Эта
143
система является формально точным описанием
распространения температурных волн в полупрозрачной
среде при данной постановке задачи. В силу сложности
ядра уравнений и свободной функции решение
уравнения в общем виде провести невозможно, поэтому
проведено решение для серой среды (численными методами).
Из этого решения сделан вывод, что температурные
поля при регулярном тепловом режиме третьего рода в
полупрозрачных и непрозрачных телах существенно
различны, и в полупрозрачных телах температурная
волна может не установиться.
Практически применить полученные результаты для
определения теплофизических свойств весьма сложно,
однако это не снижает теоретического значения данной
работы.
В заключение следует отметить, что лишь методы
теоретического расчета радиационной составляющей
(при наличии соответствующих радиационных
характеристик) и метод измерения эффективного коэффициента
теплопроводности на образцах различной толщины
дают удовлетворительные результаты. Результаты
исследования теплофизических свойств остальными
методами носят ориентировочный характер.
5. МЕТОД ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ
ТЕПЛОФИЗИЧЕСКИХ И РАДИАЦИОННЫХ
СВОЙСТВ ПОЛУПРОЗРАЧНЫХ МАТЕРИАЛОВ
В связи с изложенными выше соображениями
была рассмотрена задача о распространении
температурных волн в полупрозрачных средах и на основе ее
решения разработана методика и создана установка для
измерения теплофизических свойств диэлектриков в
твердом и жидком состояниях при температурах до 2500° С.
РАСПРОСТРАНЕНИЕ ТЕМПЕРАТУРНЫХ ВОЛН
В ПОЛУПРОЗРАЧНЫХ СРЕДАХ
Задача о распространении температурных волн в
полупрозрачной теплопроводной среде сходна со случаем
радиационно-кондуктивного теплообмена (РКТ) в
среде с объемными источниками или стоками тепла, но
вместо объемной плотности источников (стоков) тепла
144
фигурирует величина ср —- , где ср— теплоемкость еди-
дх
ницы объема при постоянном давлении.
Решение задачи может быть проведено с помощью
разных приближений: дифференциально-разностного,
диффузионного, тензорного и т. д. При пользовании
любого из этих приближений получается приблизительно
одинаковое дифференциальное уравнение.
Ниже эта задача рассмотрена для одномерного
случая. Рассмотрим полупрозрачную теплопроводную
плоскопараллельную пластину толщиной I, одна из сторон
которой нагревается переменным тепловым потоком, т. е.
как в обычном варианте определения теплофизических
свойств непрозрачных материалов методом плоских
температурных волн. Материал пластины характеризуется
коэффициентом теплопроводности X и спектральными
коэффициентами поглощения ccv, рассеяния pv и
преломления nv. Вид индикатрисы рассеяния
характеризуется коэффициентом 6V [35].
Граничные поверхности непрозрачны со
спектральной поглощательной способностью av. Теплообмен
граничных поверхностей характеризуется критерием Био.
Для описания распространения температурных волн
в полупрозрачной среде была использована система
уравнений диффузионного приближения РКТ для
плоского слоя ослабляющей и рассеивающей среды с
внутренними источниками тепла, полученная в работе Ад-
рианова [35, 37].
Так как плотность источников тепла мы заменили на
дТ
величину —ср — , то:
^-^-Т^-,р^; (3-32)
дх дх* р дх * х '
^- = 4an я2 оТ* -a'cU; (3-33)
_±djcui {ЗЩ
ч* W дх У v ;
где <7# — вектор потока излучения;
kf—эффективный коэффициент ослабления среды,
учитывающий вид индикатрисы рассеяния и
распределения спектральной интенсивности
излучения /v (s) по различным направлениям,
10—894 145
| (оц, + 6V Pv) dv [ /v (T) cos (sx) da>s
К = ^ &> ; (3-35)
| dv f /v (s) cos (s x) d(x)s
v=0 (4л)
an—осредненныи коэффициент поглощения среды
в состоянии термодинамического равновесия
(«планковский средний» коэффициент
поглощения; здесь Е0 — поверхностная плотность
равновесного излучения);
оо
j" kv [ 1 - exp (- h\lkB T) ] п\ Е^ dv
«*= — ; (3-36)
v=o
a'—эффективный коэффициент поглощения для
рассматриваемой точки, равный
оо
J av [1 — exp (— hv/kB Т)] cv i/v dv
a' = 2=2- ; (3-37)
oo
J cvUvdv
v=0
/i — осредненныи коэффициент преломления;
c0 — скорость света в вакууме;
U0—объемная плотность энергии полного
излучения в вакууме;
Cv>Uv—спектральная скорость света и объемная
плотность в среде соответственно;
£ — коэффициент, учитывающий изменение
интенсивности излучения по различным
направлениям [35];
A, a, kB — постоянные Планка, Стефана — Больцмана
и Больцмана соответственно.
Граничными условиями для уравнений (3-32) — (3-34)
служат уравнения:
при х=0 (обогреваемая сторона пластины)
- * g" |,=0 + f*U + ? Vo ~ G <3-38>
при х=1
7?tL + ,*--^-°- <3-39)
146
Здесь
, _ _i
Р
(CV)x=0,t __j_T4
2т а
*=°» I.
(340)
где qR — вектор радиации на границах пластины;
т — коэффициент распределения интенсивности
плотного излучения по всем направлениям у
рассматриваемой точки граничной
поверхности [35];
р = яг1—0,5 — полное радиационное
сопротивление границы раздела;
qr — результирующая плотность теплового потока
при теплообмене образца с окружающей
средой;
е— интегральная полусферическая излучательная
способность.
Плотность поверхностного источника тепла
периодически изменяется во времени
Q = Qo + Я ехР t(0T> (3-41)
где Q0 и q— величина постоянной и амплитуда
переменной составляющих;
со—угловая скорость колебаний.
Периодические колебания мощности нагрева
поверхности образца вызывают периодические колебания
температуры, причем фаза и амплитуда температурных
колебаний в каждой точке различны и зависят от тепло-
физических характеристик материала. В этом случае
температуру можно представить в виде суммы двух
составляющих [69]:
Т = Т(х) + Q{x) exp for, (3-42)
первая из которых зависит только от координаты, а
вторая—и от координаты, и от времени.
Ограничимся рассмотрением случая 6<C7V Это
позволяет нам линеаризовать все операторы, включающие
температуру, и разделить уравнения (3-32) — (3-34) и
граничные условия (3-38) — (3-40) для постоянной и
переменной составляющих. Уравнения (3-32), (3-34) для
переменной составляющей приводятся к одному
дифференциальному уравнению четвертого порядка:
d29 , л d49 lc 2 d4T*Q) .
— шсгу — + % — = 16an an2 — +
р dx2 dx* n dx2
+ а £й' (— icon 0 + X —). (3-43)
\ р dx2)
10*
147
Введем безразмерные параметры:
у ^ xll\ О = е/90; х = УШР;
Виа = 2ал/; Виа. = 2сс' /; Яил, = 2*'/;
1/ = 4аГ31/Х = 4/и>; Si = 4еаТ3 /А, (3-44)
где у и ft—безразмерные координата и
комплексная амплитуда
температурных колебаний;
а — температуропроводность;
Виа , Виа** Buk— критерии Бугера,
определяемые по осредненным
коэффициентам поглощения ап и а!
и рассеяния k'\
Iw — радиационно-кондуктивный
критерий Иванцова;
Э0—амплитуда пульсаций
температуры
e0 = <7/(fcp<D). (3-45)
которая имела бы место в
случае, если процесс происходил
бы очень медленно (со-Я)),
а граничные условия
обеспечивали бы отсутствие
градиентов.
В безразмерных координатах уравнение (3-43)
преобразуется к виду:
*? _ 2BUa Л.*№ + t ь, _ _L виа, iBuk,\ х
dy* an dy2 \ 4 а k }
Х— + —Ви, lBuk, 6 = 0. (3-46)
Граничные условия для уравнения (3-46)
получаются путем совместного решения уравнений (3-32), (3-33),
(3-38), (3-39) и (3-40). Для переменной составляющей
они имеют вид:
для у=0
1 /r»D„ Т/„2 .-..2ч 6 ЛГ..2
Виа, т
(2Bu„ Vn2 — iK2) — — Vn2
+ BL\Q-
_d±+ I g»nxg, (347)
dy рД«а' m d#2
148
для /у = 1
б"а'
■ (2Выап Уя2 — /х2) — — КяЧ — Bi}
—Bt,U —
_«+_J_«. = 0. (3-48)
Полученное уравнение (3-46) с граничными
условиями (3-47) и (3-48) является общим и формально точным
описанием процесса распространения температурных
волн в плоскопараллельной полупрозрачной
теплопроводной пластине с непрозрачными границами. Однако в
общем виде решение уравнения (3-46) получить
невозможно, так как в него входит неизвестный параметр V,
для определения которого необходимо решить систему
уравнений (3-32) — (3-34) с граничными условиями
(3-38) и (3-39) относительно постоянной составляющей
температуры, что в общем виде не удается. Для
приближенного решения задачи в качестве первого
приближения будем считать постоянную составляющую
температуру неизменной по толщине слоя. Практически этого
можно добиться с помощью охранных нагревателей,
экранов и т. п. В этом случае параметр V выносится из-
под знака дифференциала и уравнение (3-46)
преобразуется к линейному дифференциальному уравнению
четвертого порядка с постоянными коэффициентами
^- - (2Виа nW + fx2 -f 4 Ви. lBuk) X
x^ + i^! Bua,lBuft.# = 0, (3-49)
ayr 4
общее решение которого имеет обычный вид [68]
*(0=2с/етрР/*' (3"50)
где Cj — постоянные коэффициенты, определяемые из
граничных условий;
Pj—корни характеристического уравнения,
равные:
Р, = ± {я» VBuan + -L Виа. lBuk, + ^±
± [»vb4+ -!■&»„, бв«4. + ^)1-
-^Виа,1Вик.]'У- (3-51)
149
Уравнение (3-49) с граничными условиями (3-4?) и
(3-48) является формально точным для случая, когда
отсутствует постоянный тепловой поток через пластину.
Решение (3-50) зависит от параметров Виа' , Виа^
Btik'\ и; n2V\ m\ p; Bi\ е/а,
из которых четыре первых подлежат определению, а
остальные задаются с некоторой погрешностью. Эти
неизвестные можно определить из системы уравнений:
$(Виа,; Виа ; Вик,хх) =Flexpi%\
•& [Виа,\ Виа ; Buk/y х2) = F2expn|>2;
(3-52)
Xi/Xa = Ксох/(02 >
где F и г|)—измеренные значения модуля и аргумента
переменной составляющей температуры
при у=1.
Решение подобных систем уравнений возможно лишь
численными методами, и остается открытым вопрос об
области взаимно-однозначного соответствия и
устойчивости полученного решения по всем параметрам. Это
затрудняет практическое применение системы
уравнений (3-47) — (3-49) в качестве основы метода измерения
фононной и фотонной составляющей теплопроводности.
МЕТОД ОПРЕДЕЛЕНИЯ ТЕПЛОФИЗИЧЕСКИХ СВОЙСТВ
СЛАБОПОГЛОЩАЮЩИХ ВЕЩЕСТВ
Аналитическое решение задачи можно получить,
уменьшив число независимых переменных. Для
полупрозрачных образцов в состоянии, близком к
радиационному равновесию, коэффициенты поглощения а' и ап
примерно равны между собой, т. е. Виап = Buaf = Виа .
Как следует из уравнения (3-51), при
Buk » 8n*V (3-53)
и
у? » (2м2 V + Buh/4) Bua (3-54)
влиянием объемного испускания и поглощения
излучения на процесс распространения температурной волны
можно пренебречь. Неравенство (3-53) соответствует
сильно ослабляющим материалам, в которых вклад
радиационного переноса пренебрежимо мал.
150
Практический интерес представляет случай (3-54),
когда уравнение распространения температурной волны
в полупрозрачных средах приобретает тот же вид, что
и в непрозрачных:
сРШу2 = «Л>. (3-55)
Это допущение основывается на том, что при малых
коэффициентах поглощения (а, следовательно, и
испускания) основным источником излучения являются
граничные поверхности, и хотя сам вектор потока
излучения может быть велик, его дивергенция мала
вследствие малости а. Физически это означает, что при
рассмотрении процесса распространения температурных
волн в полупрозрачных средах мы ограничиваемся
случаем, когда радиационным теплообменом граничных
поверхностей со средой и внутри самой среды можно
пренебречь. Это справедливо как для оптически тонких
слоев, когда Buy <Cl, так и для систем с большим
значением Buk', но основную роль в механизме их
ослабления играет рассеяние. К подобным системам можно
отнести, например, керамику из А1203, BeO, MgO и плаз-
менно-напыленные покрытия из этих материалов.
В слабопоглощающих образцах вектор радиации
определяется температурами граничных поверхностей, их
радиационными свойствами и радиационными
свойствами среды:
дк = п*Ао(Ц-Т*), (3-56)
где А— эффективная радиационная функция
оптических параметров среды и граничных
поверхностей (рис. 38).
Для рассеивающих сред величина Л, определяемая
из уравнений (3-32) — (3-34) с граничными условиями
(3-40), равна
A = 4rml(Buk,a). (3-57)
Граничными условиями для уравнения (3-55)
являются уравнения (3-38), (3-39), в которых вектор
радиации задается выражением (3-56). Переменная
составляющая вектора радиации имеет вид:
qR = AAn*o{Tl%-T)Q^ (3-58)
где Г0 и Ti\ 0о и 6/ — постоянные и переменные
составляющие температур на границах х=0 и х = 1.
\Ы
Таким образом, задача сводится к решению
уравнения
_£* + &«(> = О
с граничными условиями:
при */=0 (нагреваемая поверхность; S=n2A)
~\ + SVi (6*0 - OJ + Bi0 d0 = х2; (3-59)
ау |^=ю
при t/=l
— — I + SV± (6fl0 — *,) — Bix Gx = 0. (3-60)
При высоких температурах наиболее важным
является случай, когда теплообмен на границах
осуществляется излучением. В этом случае критерии Био равны:
Bt± = 4аеф/Т?А ■= eq>Vlf ^^
Bi0 = 4аеф/ГоД = ефб^,
где е—полусферическая интегральная степень
черноты граничных поверхностей;
Ф—коэффициент облученности.
Параметр &=Vq/V\ (Vq и V\—значения параметра
V на границах у=0 и у=\ соответственно) можно
определить, решив уравнения (3-39) и (3-56) относительно
постоянной составляющей температуры. В случае
теплообмена излучением б определяется как
8=1+-±_*!-. (3-62)
4 \+SV V ;
Расчеты показывают, что величина б близка к
единице, а снизив Bi\ путем введения дополнительных
экранов или охранных нагревателей, эту величину можно
еще более приблизить к единице. Поэтому в
дальнейшем будем предполагать 6=1.
Решение системы уравнений (3-55), (3-59), (3-60) в
случае с граничными условиями (3-61) для
безразмерной амплитуды температурных колебаний на необогре-
ваемой стороне имеет вид:
Ф = *2(g + sy6shg)
(1) (\+6)(SV+eV)Z sh g+[gi+eVa(2SV+eV)] sh g—5Vg(l+6)'
(3-63)
152
Это уравнение является основным расчетным
соотношением метода определения теплофизических
характеристик полупрозрачных веществ.
АНАЛИЗ РЕШЕНИЯ УРАВНЕНИЯ
РАСПРОСТРАНЕНИЯ ТЕМПЕРАТУРНЫХ ВОЛН
В ПОЛУПРОЗРАЧНОЙ ПЛАСТИНЕ
В уравнении (3-63) гиперболические функции от
комплексного аргумента £ можно заменить
произведениями гиперболических и тригонометрических функций
действительного аргумента. По полученному уравнению
был проведен расчет значений модуля F и аргумента г|)
комплексной амплитуды температурной волны на
границе у=1 первоначально для массива:
S = 0,01;...; 100,0 с логарифмическим шагом 0,5;
V = 0,01;...; 10,0 с тем же шагом;
8 = 0,1; 0,2; 0,3; 0,4; 0,5; 0,75; 1,00;
х = 0,1; 0,2; 0,3; 0,4; 0,5; 0,7; 1,0; 1,5; 2,0;
3,0; 4,0; 5,0; 6,0,
а затем более подробно для практически наиболее
важной области:
S = 0,01;...; 6,31 с логарифмическим шагом 0,2;
е = 0,1;...; 0,631 с тем же шагом;
К = 0,1; 1,0; 10,0;
х = 0,7; 1,0; 1,5; 2,0;...; 8,0; 8,5; 9,0.
Приводить раздельный анализ зависимости модуля
и аргумента температурной волны от переменных
параметров нецелесообразно, так как подобное разделение
резко затрудняет уяснение физической стороны
волнового процесса.
По результатам расчета были построены годографы
векторов О (и, SV, Bi) для переменных % и SV при
фиксированных значениях критерия Bi (рис. 40). На рис. 40
параметры обозначены в машинных переменных ф=г|);
M=F\ по вертикали даны значения параметра SV;
значения х указаны около соответствующих кривых.
Годограф вектора Ф при 51^0,0025 совпадает со
153
значениями, получаемыми для непрозрачных
образцов [69].
Качественное объяснение зависимости Ф от SV
можно дать, исходя из следующей модели.
Допустим, что вектор комплексной амплитуды Ф яв-
Рис. 40. Годографы вектора
a — Bt=0,0631;
ляется суммой двух векторов, один из которых $ь
обусловлен действием только кондуктивного механизма теп-
лопереноса и его зависимость от % имеет обычный вид.
Второй вектор ^я, обусловленный непосредственным
радиационным теплообменом между граничными
поверхностями, может быть определен из следующих сообра-
154
комплексной амплитуды:
о - Вi=0,398
жений. При достаточно больших к фаза переменной
составляющей температуры на границе г/=1 (при
отсутствии теплообмена с окружающей средой) стремится к
величине л/4, а амплитуда 60 пропорциональна со"~1/2—
как в случае распространения температурных волн в
полубесконечной среде [69]. Связанная с Э0 перемен-
155
ная составляющая вектора радиации индуцирует
температурные колебания на границе */=1 с фазой я/4
относительно 0О и амплитудой, пропорциональной
90SW~1/2. Обратное влияние границы */=1 на границу
*/=0 можно не принимать во внимание.
Учитывая соотношение 9»ft/co, получаем, что Он не
зависит от х, имеет величину, пропорциональную SV
и постоянный сдвиг фаз, равный я/2.
Таким образом, влияние внутреннего радиационного
переноса проявляется в изменении вектора
комплексной амплитуды температурной волны в непрозрачной
среде Ох(х) на вектор ^(SK). Графически это
выглядит так — годограф вектора #(xSV) получается в
результате параллельного переноса годографа вектора
#ь(х) на величину Он {SV).
При малых х основную роль в процессе
распространения температурных волн играет процесс
установления теплового равновесия с окружающей средой.
В этом случае образец ведет себя как единое целое
независимо от характера внутреннего теплообмена, и
модуль вектора О является слабой функцией параметров
х и SV. При малых значениях критерия Био (Bi^.
^0,064) это дает возможность по величине амплитуды
тепловых пульсаций определять эффективную
теплоемкость полупрозрачных образцов по методике,
разработанной для непрозрачных материалов [69].
Область больших значений критерия Био (0,1 ^Bi)
малопригодна для измерения теплоемкости из-за
резкого уменьшения модуля F вектора Ф с увеличением
критерия Био. Так как функция ft(x, SV, Bi) в некоторой
области является непрерывной и однозначной, то в этой
области каждому вектору ft при известном критерии Bi
соответствует единственная пара значений х и SV.
Определяя экспериментальные значения F и ^ (ij)—
аргумент вектора Ф), находим соответствующие им
х и SV, а с их помощью — температуропроводность
а и эффективную функцию радиационных
параметров А.
Значения х и SV определяются из годографов
вектора ft, так как линии x=const и SV = const представ-
156
ляют собой сетку криволинейных координат. Определив
координаты точки ft в этой системе координат, мы
однозначно определяем теплофизические параметры
среды.
Таким образом, метод плоских температурных волн
позволяет в ходе одного эксперимента определять
температуропроводность, теплоемкость и эффективный
радиационный параметр, определяющие РКТ в плоском
слое теплопроводной, слабопоглощающей и
рассеивающей среды. При этом объем расчетов для прозрачных
сред не превышает объем расчетов для непрозрачных
сред. Зная аппаратурную погрешность измерения F
и г|з, значения погрешностей определения х и 5У, а
следовательно, а и Л, легко находятся графически.
6. УСТАНОВКА ДЛЯ ИССЛЕДОВАНИЯ
ТЕПЛОФИЗИЧЕСКИХ СВОЙСТВ
ПОЛУПРОЗРАЧНЫХ МАТЕРИАЛОВ
КОНСТРУКЦИЯ УСТАНОВКИ
Для реализации разработанной методики измерения
теплофизических характеристик полупрозрачных
материалов была создана установка, отличающаяся от
известных следующим.
Для измерения переменной составляющей
температуры применяется система, состоящая из монохромато-
ра инфракрасного спектрометра ИКС-11 и
фотоэлектронного умножителя ФЭУ-62, работающего в области
0,4—1,2 мкм, что позволяет исследовать спектральные
коэффициенты ослабления в данном оптическом
интервале.
Для измерения сдвига фаз используется фазочувст-
вительный вольтметр типа В5-1, измеряющий сдвиг фаз
только между первыми гармониками переменных
составляющих. Его применение резко сокращает объем
расчетов, причем точность получаемых результатов не ниже>
чем при использовании других методов регистрации.
Для измерения амплитуды переменных составляющих
используется вольтметр повышенной точности ВЗ-7,
имеющий выход с усилителя, что позволяет с помощью
осциллографа визуально контролировать форму
измеряемого сигнала.
157
Усилитель мощности на лампе ГУ-80 позволяет
получать большую величину переменной составляющей
мощности нагрева, имеющей практически
синусоидальную форму.
Исследуемый образец (рис. 41) в виде диска
диаметром 10—20 мм, помещенный на вольфрамовые
проволочки диаметром 0,08 мм, натянутые в виде
треугольника в держателе, снизу
нагревается электронной
бомбардировкой. Катод
изготовлен из танталовой
проволоки в виде
плоской спирали. Расстояние
между катодом и
образцом составляет 10—20 мм.
Электронный пучок
формируется двумя
фокусирующими электродами.
Сверху над образцом
стоит тепловой экран,
одновременно выполняющий
роль диафрагмы. В
качестве изолирующих
элементов используется
керамика из окислов
алюминия и бериллия. Теп-
лоотвод от всех частей
измерительной ячейки
происходит за счет излучения. Измерительная ячейка
устанавливается в вакуумную водоохлаждаемую
камеру. Вакуумная система, состоящая из вакуумного
агрегата ВА-01-1 с форнасосом ВН-461, обеспечивает
вакуум не ниже 3 • 10—5 мм рт. ст.
Анодное напряжение высоковольтного выпрямителя
(рис. 42) модулируется декадным генератором звуковых
и инфразвуковых колебаний ГЗ-39 с помощью
усилителя мощности на лампе ГУ-80. Мощность усилителя
позволяет расширить диапазон используемых % до 5—7, что
повышает точность измерения теплофизических
параметров. Напряжение смещения на управляющую сетку и
питание экранирующей сетки ГУ-80 подается от УИП-1.
Температурные колебания необогреваемой границы
образца регистрируются с помощью оптической системы,
состоящей из зеркального объектива с поворачивающей
Рис. 41. Схема измерительной ячейки:
/ — тепловой экран; 2 — образец; 3 —
вольфрамовые проволочки диаметром
0,08 мм; 4 — держатель образца; 5 и
6 — фокусирующие электроды; 7 — тан-
таловый катод; 8 — держатель катода;
9 — токоподводы; 10 — алундовые
изоляторы; И — основание
158
системой, монохроматора инфракрасного спектрометра
ИКС-11 (призма Ф-1, диапазон 0,4—2,5 мкм) и
фотоэлектронного умножителя ФЭУ-62 с областью
спектральной чувствительности 0,4—1,2 мкм. Проверка спектро-
р
J N
W 1
il 11
\2 /
." Д"
гг
р
м
[7]
3 У
/
iTfiil
/■-Чу [~^2—
\ш
ji
[71
_^_
«
| ,
1а иа 1
г
П
1Q | 4/
1 14
il
~£/ • £IU У \~и0
\l6 {-
Лп
\18
tfiOH I
\20
\21
п I
\~
\¥
ш
8
9
11 1
1
15 I
15 I
19 I
22 I
17
Рис. 42. Блок-схема установки для исследования теплофизических свойств:
1 — катод; 2 — образец; 3 — монохроматор ИКС-11; 4 ~ ФЭУ-62; 5 — блок
питания ФЭУ; 6 — магазин емкостей; 7, 12—магазины сопротивлений Р-517М и
Р-4001 соответственно; 8 — гальванометр М-95; 9 — потенциометр ПП-63; 10 —
блок коммутации; 11 — вольтметр ВЗ-7; 13 — осциллограф; 14 — накальный
выпрямитель; 15 — фазочувствительный вольтметр В5-1; 16— стабилизатор
СЗ-С; 17 — PHO-250-5; 18 — анодный выпрямитель; 19 — частотомер 43-28;
20 — УИП-1; 21 — усилитель мощности; 22 — генератор ГЗ-39
метра осуществляется по спектральным линиям ртути.
Использование ближнего ИК диапазона позволило
расширить рабочую область установки в сторону низких
температур до 700—900° С и измерять спектральные
коэффициенты ослабления полупрозрачных веществ в
области 0,4—1,2 мкм.
Для повышения точности измерения применена
система регистрации, позволяющая измерять величину и
159
фазы переменных и постоянных составляющих тока ФЭУ
и анодных тока и напряжения одними и теми же
приборами. Система состоит из блока коммутации с
безреактивными магазинами сопротивлений Р-517 м и
Р-4001 и магазина емкостей Р-583. Измерение
постоянных составляющих производится потенциометром
ПП-63, переменных — вольтметром повышенной
точности ВЗ-7.
Форма измеряемого сигнала, наличие шумов и
наводок контролируется с помощью осциллографа
«Duoskop». Сдвиг фаз между эталонным сигналом
генератора и переменными составляющими измеряется
фазочувствительным вольтметром В5-1
(чувствительность 15 мВ при входном сопротивлении 2 МОм и
емкости 10 пФ) и избирательность (ослабление
шумов и гармоник на 40 дб), которого позволяют измерять
фазу первой гармоники сигнала непосредственно с ФЭУ.
Применение фазочувствительного вольтметра по
сравнению с другими методами измерения сдвига фаз
значительно облегчит обработку результатов.
Частота генератора ГЗ-39 контролируется
электронно-счетным частотомером 43-28 с точностью не хуже
0,1%.
Примененная схема регистрации позволяет измерять
постоянные составляющие с точностью 0,2% и
исключить влияние систематической погрешности вольтметра
ВЗ-7.
Постоянная составляющая температуры измеряется
оптическим пирометром ОППИР-017, градуировку
которого проводили по температурам затвердевания серебра,
меди, никеля, платины. Точность измерения
температуры составляет ±1,5%.
Работоспособность установки проверялась серией
контрольных измерений температуропроводности и
теплоемкости образцов технического молибдена толщиной
1,20 мм. Эксперименты показали хорошую
воспроизводимость результатов. Полученные значения
температуропроводности в интервале 1000—1900 К на 3,5—5,5%
ниже рекомендованных [70]. Это расхождение,
возможно, связано с низкой чистотой образца. Данные по
теплоемкости на 2—5% ниже рекомендованных [71].
Наиболее вероятная причина занижения значений —
погрешность определения критерия Био и влияние шума
фотоэлектронного умножителя.
160
ТОЧНОСТЬ МЕТОДА
Точность метода определяется тремя факторами:
точностью экспериментального определения величин
F и гр;
точностью оценки связи между параметрами F, г|) и и,
SV, 60;
ошибками, вносимыми переходом от параметров к
температуропроводности, теплоемкости,
теплопроводности и эффективной радиационной функции Л.
Величина сдвига фаз между генератором и
переменными составляющими тока ФЭУ и анодным током и
напряжением измеряется фазочувствительным
вольтметром В5-1, погрешность градуировки которого
составляет 3° (по паспорту). Для повышения точности измерения
прибор периодически градуировали по мостовой схеме,
два плеча которой — эталонные катушки сопротивлений
Р-321 (10 Ом) класса 0,01, а другие два — магазин
безреактивных сопротивлений Р-517М класса 0,05 и
магазин емкости Р-583 класса 0,2. Электронный частотомер
43-28 обеспечивает точность ±1 последнего знака и при
градуировке обеспечивает точность не ниже 0,1%.
Отсюда систематическая погрешность градуировки
вольтметра равна 0,37%, что составляет 0,5°. Так как прибор
измеряет фазу одной из переменных составляющих
относительно синусоидального сигнала от генератора ГЗ-39,
подаваемого на эталонный вход, то погрешность
измерения сдвига фаз между самими переменными
составляющими удваивается. Таким образом, аргумент г|)
измеряется с точностью 1°.
Величина модуля F определяется по формуле
f = ez/e0> (3-64)
где QQ=qJlcv(u имеет тот же смысл, что и в формуле
(3-45). Амплитуда температурных пульсаций 0/
определяется из выражения [69]:
где Г и Г—постоянная и переменная составляющие
тока ФЭУ.
Максимальная систематическая ошибка,
возникающая при подобном методе определения 8/, составляет
11—894
161
3,8%, причем основной вклад дает ошибка измерения
постоянной составляющей температуры пирометром.
Систематическая погрешность определения
переменной составляющей мощности складывается из
аппаратурной погрешности, связанной со вторичной
электронной эмиссией, и погрешности, связанной с высшими
гармониками.
Аппаратурная погрешность измерения мощности
составляет 0,7%. При этом на сдвиг фаз между анодным
током и напряжением, равный 2,5°, необходимо вносить
поправку 0,1%. Применение относительно небольшого
анодного напряжения 800—1500 В и фокусирующих
электродов сводит влияние вторичной электронной
эмиссии к минимуму. Как показывает гармонический анализ
осциллограмм анодных тока и напряжения, амплитуда
третьей гармоники имеет относительную величину
0,03—0,10, а амплитуды второй, четвертой, пятой и т.д.
гармоник в пределах ошибки равны нулю. Для учета
высших гармоник необходимо вносить поправку,
точность которой около 0,5%. Таким образом,
систематическая погрешность измерения переменной
составляющей мощности равна 1,2%. Систематическая ошибка
вычисления 0О складывается из ошибки определения q,
погрешности использованных значений теплоемкости
(2%), погрешности определения ш (0,2%) и погрешности
определения массы образца (0,2%) и составляет 3,6%.
Таким образом, систематическая ошибка измерения
величины F весьма велика и составляет 7,6%.
В некоторых случаях (например, когда Bi^0,01)
модуль целесообразней определять по формуле
f = ег/е*, (3-66)
где 8* — амплитуда тепловых пульсаций при малых
значениях х. В этом случае систематическая погрешность
может быть снижена до 3—5%.
Аналитическое выражение, описывающее связь
погрешности величин F, г|э и х, SV, получить в общем
виде весьма сложно. Значительно легче сделать это
графически. Графически погрешность вектора Ф
определяется областью, ограниченной кольцевым сегментом
размерами ±Дгр по окружности и ±AF по радиусу.
Погрешность параметров х и SV определяется тем же
сегментом, но в координатах х и SV и при неизменных величи-
162
пах б/7, бг|) эта погрешность существенным образом
зависит от вектора ft. Так, при 0=0,5 ехр(—£Зя/4)
погрешностям 8F=7,4% и б<ф = 1% соответствуют
погрешности бх=2,6% и 6(51/) =5,6%. В то же время при 0 =
=0,8 ехр(—йя/4) погрешность 6к=2,4%, а
погрешность S(SV)=100%. (В обоих случаях В* = 0,01).
Параметр Bi определяется со значительной (~20%)
погрешностью, которую необходимо учитывать при
определении погрешностей бх и 6(SV). Легче всего эти
погрешности определять из графиков с разными значениями Bi,
причем при ££=^0,63, погрешностью можно пренебречь,
поскольку она меньше 0,5%. В некоторых случаях
погрешность может превосходить 10%.
При переходе от параметров к и SV к коэффициенту
температуропроводности а и эффективной радиационной
функции п2А возникают дополнительные погрешности
Ьа = 8(0 + 26/ + 28х = 0,004 + 28х. (3-67)
Используя найденное значение а для определения
коэффициента теплопроводности, входящего в параметр
Vy находим
ЬА = 38Г + 6р + 6СР + 8© + б/ + b(SV) + 8х =
=■ 0,074 + 8 (SV) + 8х. (3-68)
Большой вклад в погрешность 6Л вносит ошибка
измерения постоянной составляющей температуры.
Численные значения систематических ошибок [при
6х=2,6%, и 8(S10=5,6%] равны ба = 5,6%; 8Х=8,6%;
6(п2Л)=18,2%.
Погрешность определения теплоемкости
определяется выражением
8ср = 8F + 89, (3-69)
где 80—погрешность, определенная нами ранее,
составляет величину 3,8%;
8F' — погрешность определения теоретического
значения F из-за неточности определения
значений параметров Bi, и, SV.
При малых х величина F слабо зависит от х и SV.
В этом случае при В£^0,063 погрешность 6F<1%.
Таким образом, при В£^0,063 систематическая
погрешность определения теплоемкости не превышает
4,8%. При больших значениях Bi погрешность
возрастает.
11*
163
МЕТОДИКА ПРИГОТОВЛЕНИЯ ОБРАЗЦОВ ^^
Для определения теплофизических свойств твердой
окиси алюминия были использованы
монокристаллические образцы лейкосапфира сорта «Экстра», вырезанные*
в базисной плоскости (0001), полученные из Института1
кристаллографии АН СССР. Образцы имели толщину
0,94 мм, диаметр 15 мм, оптически гладкую поверхность,
высокую плоскопараллельность. На поверхность
образца вакуумным распылением наносился тонкий (около
1 мкм) слой молибдена.
Теплофизические свойства окиси алюминия
определяли на образцах, полученных плазменным напылением.
Известно [72], что наибольшее влияние на свойства
плазменно-напыленных образцов оказывает дистанция
напыления. Поэтому изготавливали три серии образцов,
плазменное напыление которых проводили с расстояния
150, 180 и 220 мм. По данным рентгеноструктурного
анализа плазменно-напыленные образцы имеют
кубическую решетку типа 7-АЬ03, содержащую до 20%
а-А1203. При увеличении дистанции напыления
пористость образцов возрастает от 6 до 11%. Это связано с
тем, что при плазменном напылении существует
оптимальная дистанция (120—150 мм), где распыляемые
частицы имеют максимальную температуру и,
следовательно, пластичность. При увеличении дистанции
напыления частицы быстро охлаждаются и достигают
поверхности в малопластичном состоянии, что уменьшает
размер поверхности контакта частиц друг с другом и
приводит к увеличению пористости образцов.
Измерение теплофизических свойств жидкой окиси
алюминия проводили в плоских молибденовых тиглях
высотой 0,75 мм и диаметром 15 мм. Толщина стенок
тигля 0,15—0,20 мм. Крышку тигля изготавливали из
молибденовой фольги толщиной 0,05 мм.
Монокристаллические образцы загружали в тигли и расплавляли..
7. ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА
ОКИСИ АЛЮМИНИЯ
ПРИ ВЫСОКОЙ ТЕМПЕРАТУРЕ
МОНОКРИСТАЛЛИЧЕСКАЯ ОКИСЬ АЛЮМИНИЯ
Теплофизические свойства лейкосапфира
исследованы в интервале 1193—1663 К. На рис. 43, а показаны
результаты измерения температуропроводности. Указаны
Ш
доверительные интерзалы для надежности 0,95. С
использованием литературных данных по теплоемкости и
плотности лейкосапфира [70] были рассчитаны
значения коэффициента фононной теплопроводности (рис.
43,6) и теплосопротивления (рис. 43, б). Использование
литературных данных, безусловно, увеличивает
систематическую погрешность.
а, сп2/с
0,015
0,010
W, м-К/Вт
0,20
0,005
1100 1200 1300 то 1500 1600 Т, К
<4
9,7W~6T+1,28W8TZ
W=9,7WSH,28W'8T^:
Угч—f—»д
1200 1500 /400 /500 /600 Г, К 1000110012001300 /4001500 Г, К
Рис. 43. Температурные зависимости температуропроводности {а),
теплопроводности (б) и теплового сопротивления (в) сапфира:
/ — данные [66]; 2 — данные [73]; 3 — экспериментальные данные
Температурная зависимость теплосопротивления
лейкосапфира аппроксимируется уравнением
U/ = 9,70.10~5Г + 1,28-10"8Г2.
(3-70)
Из этого уравнения можно вычислить коэффициент
пропорциональности в уравнении (3-6), определяющий
отношение вкладов четырех- и трехфононного рассеяния
в общее теплосопротивление решетки:
W
IV
_^=0,31 —
W
(3-71)
ш
Величина этого коэффициента находится в разумном
соответствии с приближенными теоретическими
оценками [60], дающими его величину порядка единицы.
Теоретически рассчитать эту величину для окиси алюминия
очень сложно, так как даже в случае затвердевших бла-
165
городных газов приходится из-за математических
сложностей прибегать к приближенным методам.
Соотношение (3-71) показывает, что вклад четырех-
фононных процессов в суммарное теплосопротивление
решетки велик. Экстраполяция низкотемпературных
данных по закону Я^Г"1 (т. е. в пренебрежении четы-
рехфононным рассеянием) должна привести к
завышению значений теплопроводности при температурах,
близких к температуре плавления, примерно на 30%.
1 *
5***-
Ь
.1
о
1_
А = 1,55-6,55-10~*Т
-/
оо4^!
2
1
_1_ 1 1 1
1100 1200 1J00 1400 1500 1600 Т, К 1100 1200 1500 1400 1500 1600 Т, К
Рис. 44. Температурные зависимости эффективного параметра радиационного
переноса (а) и эффективной радиационной функции (б) в образце лейкосап-
фира (границы из молибдена) толщиной I мм:
/ — экспериментальные данные; 2 — расчетные данные
Данные по теплопроводности лейкосапфира,
полученные в работах Черванта и Кинджери [77] и Ли и Киндже-
ри [73], лежат намного выше значений, полученных по
изложенной выше методике, и имеют другой характер
температурной зависимости (рис. 43,6). Подобные
расхождения объясняются тем, что в работах Кинджери
фактически определялся эффективный суммарный
коэффициент теплопереноса теплопроводностью и
излучением. Появление дополнительного механизма
теплопереноса изменило характер температурной зависимости и
привело к увеличению абсолютной величины измеряемого
коэффициента теплопроводности.
Измеренные значения эффективного радиационного
параметра SV (рис. 44, а) и эффективной радиационной
функции А (рис. 44, б) для образца толщиной 0,94 мм
позволяют рассчитать величину суммарного
коэффициента теплопроводности образцов, лейкосапфира (с
непрозрачными границами из молибдена) разной толщины
166
по формуле
где SVj—величина радиационного параметра для
слоя толщиной /.
Для определения границ допустимой экстраполяции
необходимо иметь осредненный по спектру коэффициент
поглощения лейкосапфира. Для не слишком больших
оптических толщин осреднение предпочтительнее
проводить по Планку, т. е. по уравнению (3-36).
Значения ап, полученные при использовании данных
для av лейкосапфира из работы [78], увеличиваются
от 14,4 м-1 при 1170° С до 18,9 м-1 при 1500° С.
Для образцов лейкосапфира толщиной 0,94 мм
величина критерия Бугера (Виа =2an/) соответственно
равна 0,026 и 0,035. Это позволяет считать, что измерения
SV и А проводились при Виа=0. Величина А при Виа =
= 0 однозначно определяет значения А (и
соответственно SV) при других Виа (см. рис. 37), что позволяет
проводить экстраполяцию в пределах применимости
осреднения по Планку:
Виа<1. (3-73)
Это условие выполняется для слоев лейкосапфира
толщиной до 35 мм при 1170° С и до 27 мм при 1500° С.
Для слоев толщиной до 5—7 мм величина А практически
не отличается от значения для нулевой оптической
толщины, что позволяет в этих пределах проводить
линейную экстраполяцию
Kv^b^+SVf^y (3-74)
где I — толщина слоя, для которого измерено значение
SVi;
b — толщина слоя, на которую производится
экстраполяция.
Экспериментально полученные значения функции А
(рис. 44) лежат выше теоретических оценок,
проведенных по формуле Кристиансена
а=■»,+■'*.-■ ■ (з-7б>
Это расхождение связано с тем, что, во-первых,
радиационные характеристики молибдена, напыленного в
вакууме Ю-5 мм рт. ст., отличаются от значений для
компактного молибдена, использованных при оценке А,
167
и, во-вторых, степень черноты металла в контакте с
диэлектриком может отличаться от значений, полученных
при измерении в вакууме.
ПЛАЗМЕННО-НАПЫЛЕННАЯ ОКИСЬ АЛЮМИНИЯ
На рис. 45, а представлены значения
температуропроводности образцов из А1203 в зависимости от
дистанции напыления, а на рис. 45,6 — вычисленные значения
12 \ 1
900 то то mot;с
Рис. 45. Температуропроводность (а) и теплопроводность (б) образцов из
окиси алюминия, полученных плазменным напылением с расстояния, мм:
/ — 150; 2—180; 5 — 220; светлые точки — отожженные образцы
(температура отжига 1500° С, выдержка 100 мин); темные точки — образцы в исходном
состоянии
168
теплопроводности этих покрытий. Значения,
определенные при 900° С, оказались близкими к полученным в
работе [76]. Однако в этой работе не указан метод
определения теплопроводности и режим напыления, что
затрудняет анализ некоторых расхождений.
Как видно из данных, приведенных на рис. 45,
величины а и Я плазменно-напыленных образцов в исходном
состоянии возрастают при увеличении температуры.
Такой характер температурной зависимости объясняется
двумя причинами.
При температурах 900—1500° С в окиси алюминия
происходит фазовое превращение у-АЬОз-нх-АЬОз [54].
Теплопроводность а-А1203 в 2—2,5 раза выше
теплопроводности y-A1203 [74], что приводит к увеличению
теплопроводности материала.
Процесс фазового превращения и резко
неравновесная форма пор интенсифицируют протекание процесса
спекания. При этом увеличивается размер пятен
контакта частиц друг с другом, в результате чего
увеличивается теплопроводность гетерогенной системы в целом.
Таким образом, подтверждено экспериментально, что
разработанный метод определения теплофизических
свойств полупрозрачных диэлектриков вполне корректен
и может быть рекомендован для исследований этих и
других веществ в широком температурном интервале.
ЖИДКАЯ ОКИСЬ АЛЮМИНИЯ
Теплофизические свойства жидкой окиси алюминия
определяли при небольшом перегреве выше температуры
плавления. Экспериментально установлено, что
температуропроводность жидкой окиси алюминия имеет
величину (4,8±0,4) -10-7 м2/с. С использованием известных
значений теплоемкости и плотности жидкой окиси
алюминия [74] было вычислено значение ее
теплопроводности, которое оказалось равным (2,05±0,15) Вт/(м-К).
Теплопроводность твердой окиси алюминия при
температуре плавления, полученная экстраполяцией
температурной зависимости теплопроводности лейкосапфира,
равна 3,4 Вт/(м-К).
Величина отношения теплопроводностей окиси
алюминия в твердом и жидком состоянии А,ТвАж = 1,65, что
хорошо согласуется с теоретическими оценками измене-
№
ния теплопроводности при плавлении твердых тел с ион-
но-ковалентным характером связи [4].
Эффективный радиационный параметр SV для слоя
жидкого окисла толщиной 0,75 мм равен 0,61+0,08.
Литературные данные по коэффициенту преломления
жидкой окиси алюминия отсутствуют, поэтому при оценке
величины эффективной радиационной функции А были
использованы значения коэффициента преломления для
твердой окиси алюминия [21]. Вычисленное значение
Л, равное 0,136, близко к расчетным значениям А
(0,130) для оптически тонкого слоя с границами из
молибдена.
Для определения допустимых границ экстраполяции
величины SV нами были проведены оценки величины
«планковского среднего» коэффициента поглощения. По
данным работы [75], коэффициент поглощения жидкой
окиси алюминия имеет тот же характер зависимости от
длины волны, что и у лейкосапфира, а его величина при
плавлении увеличивается примерно в 20 раз.
Вычисленное значение ап равно примерно 50 м-1, а интервал
допустимой линейной экстраполяции составляет ~2 мм.
Вклад радиационного теплопереноса в общий
теплообмен весьма значителен и для слоев окисла толщиной
более 1,25 мм превышает вклад теплопроводности.
Вычисленные значения эффективного коэффициента
теплопроводности для слоев жидкой окиси алюминия
(границы из молибдена) толщиной 0,75 и 2,0 мм равны 3,3 и
5,4 Вт/(м-К) соответственно. Оценки теплопроводности
слоя толщиной 20 мм дают величину порядка
40 Вт/(м-К).
Приближенную оценку теплопроводности жидкой
окиси алюминия при температурах ~ 2500° С можно
провести, используя наиболее вероятные значения
плотности жидкого окисла:
у = 3,03 — 1,08 • 10"3 М, (3-76)
где А/ — перегрев выше температуры плавления.
По разным данным показатель степени в
зависимости %~уп изменяется от 0,6 до 2,0. В работе Филиппова
[14] показано, что температурная зависимость
теплопроводности большого числа жидкостей описывается
зависимостью с показателем п=4/3.
170
Предполагая, что это справедливо и для жидкой
окиси алюминия, температурную зависимость
теплопроводности жидкой окиси алюминия можно выразить
уравнением
Я, = 2,05 (1 — 5,3- 1(Г4 Д*). (3-77)
Глава IV
СМАЧИВАНИЕ ТУГОПЛАВКИХ ВЕЩЕСТВ
И ВЫСОКОТЕМПЕРАТУРНЫХ
МАТЕРИАЛОВ
жидкими окислами
1. ПРОЦЕССЫ СМАЧИВАНИЯ И РАСТЕКАНИЯ
Жидкость на поверхности твердого тела или образует
каплю с конечным краевым углом, или растекается по
ней до образования краевого угла, близкого к нулю.
Поведение жидкости на поверхности твердого тела
определяется краевым углом смачивания [1]:
COS 9= ат-г-°т-ж ^ (4_j}
аж-г
где ат_-г, (Ут-ж, <тж_г— поверхностное натяжение на
границе раздела
соответствующих фаз.
Когда краевой угол смачивания близок к нулю,
смачивание количественно характеризуют конечной
площадью растекания при постоянных массе или объеме
жидкой фазы или скоростью растекания жидкости. По
Харкинсу, интенсивность процесса растекания жидкости
по поверхности твердого тела определяется
коэффициентом растекания
K = Wa-Wn, (4-2)
где W&—работа адгезии;
WH—работа когезии.
Для системы твердое тело — жидкость
^. = *т_г + аж_г-огт_ж; (4-3)
^к = 2<тж_г. (4-4)
171
Тогда
К = о --{о +о ). (4-5)
v т—г V ж—г ' т—ж)' v '
Коэффициент Харкинса рассматривается как
выигрыш свободной энергии системы, возникающий в
результате процесса растекания. Чем он более положителен,
тем более полно должен протекать процесс растекания.
Из уравнения (4-5) следует, что коэффициент
растекания возрастает при увеличении поверхностной энергии
твердого тела, а также при уменьшении межфазной
поверхностной энергии и поверхностной энергии жидкости.
Коэффициент растекания Харкинса статичен, для его
расчета используют равновесные величины, и он не
определяет момента прекращения процесса растекания.
Общим условием растекания жидкости по гладкой
поверхности твердого тела является стремление системы к
уменьшению свободной энергии за счет увеличения
площади контакта между ними. Если пренебречь
объемными эффектами, то это условие означает, что Да<0, где
Да — изменение свободной поверхностной энергии
системы. Если а— свободная поверхностная энергия на
какой-либо границе раздела, a S — площадь поверхности
раздела на соответствующих границах, то
Aa=AS a + AS о + AS a .
^ж—г ж—г • ^т—ж т—ж ' ^т—г т—г*
Если принять Д5Ж_Г =А5т-ж = —AST-r,
что справедливо для случая полного смачивания, то
получим
Да == а + а — а . (4-6)
ж—г ' т—ж т—г* ^ /
Процесс растекания начинается при соприкосновении
капли с твердой поверхностью, т. е. когда
неравновесный краевой или так называемый контактный угол
близок к 180°. При этом движущая сила процесса
растекания, которая пропорциональна уменьшению свободной
поверхностной энергии, является максимальной. Затем
при уменьшении контактного угла абсолютна^ величина
ее падает и при достижении равновесия становится
равной нулю. Поэтому для определения движущей силы
растекания используется выражение [3, 4]
Да = а* +а cosG* —а , (4-7)
т—ж ' ж—г т—г» \ж ' /
где а*_ж и cos0* — величины, изменяющиеся в процессе
растекания.
172
В условиях равновесия: выражение* (4-7)
представляет собой уравнение Юнга.— Неймана, а для случая
полного смачивания (cos 6 = 1)
Д0 = -/С, (4-8)
где К — коэффициент растекания Харкинса.
Величину Да определяли Есин с сотр. [4—5],
которые рассматривали растекание с позиций
термодинамики необратимых процессов. Скорость растекания
рассматривается как поток свойства; окончательное
выражение имеет вид:
v^A
о —а —а
т_г т_ж ж-г, ^_ж
dS
(4-9)
где А— является сложной функцией времени и
физических свойств (главным образом вязкости)
растекающейся жидкости.
Щукин с сотр. [6] при расчете движущей силы
растекания учитывал шероховатость поверхности твердой
фазы:
Да = аж_г (£cos 6— 1), (4-10)
где k — коэффициент шероховатости.
С помощью этого соотношения им была рассчитана
движущая сила растекания для системы ртуть — цинк.
Расчетные данные удовлетворительно согласуются с
экспериментальными. Для этой системы характерно
отсутствие сколько-нибудь заметного химического
взаимодействия растекающейся жидкости с подложкой; не
происходит также заметного растворения твердой фазы в
жидкости.
В настоящее время считается общепринятым, что во
время смачивания и растекания определяющую роль
играет уменьшение свободной межфазной энергии на
границе твердое тело — жидкость [8]. Одним из
процессов, приводящих к снижению межфазной поверхностной
энергии, является адсорбция примесей жидкой фазы на
поверхности раздела жидкость — твердое тело.
Предполагают, что именно этот процесс приводит к понижению
межфазной поверхностной энергии в системах Fe—А1203
и Ni—А1203 при добавке титана, хрома или кремния к
железу или никелю [4—7]. Принципиальная возмож-
173
ность понижения межфазной поверхностной энергии
следует из адсорбционного уравнения Гиббса:
Г---±—^-, (4-П)
RT d\na
где а — активность.
Другой причиной снижения межфазной
поверхностной энергии является массоперенос вещества, например,
диффузия через межфазную границу независимо от
направления. При контакте жидкости с твердым телом,
имеющим значительную растворимость в ней,
межфазная поверхностная энергия при этом изменяется при
изменении состава жидкости. Количественно процесс
понижения межфазной поверхностной энергии вследствие
массопереноса на межфазной границе описан Жуховиц-
ким, Григоряном и Михаликом [8]. Градиент
химического потенциала Ар, непосредственно определяет
изменение межфазной поверхностной энергии:
Aa^^AlAi, • (4-12)
где М — число поверхностного переноса.
Практическое применение соотношения (4-12)
затруднено, так как определить число поверхностного
переноса можно только в некоторых простейших случаях.
Гак, для случая идеальный раствор — газ предложено
выражение
М = zb&IQ, (4-13)
а для случая совершенный раствор — газ — выражение
M=£p_Z^L_f£# (4.i4)
RT Q v '
В приведенных уравнениях используются следующие
обозначения:
г—число адсорбционных центров на
поверхности;
Ь — отношение констант скорости адсорбции и
скорости десорбции (b=KJKp)\
ap— равновесная межфазная поверхностная
энергия; ■----..
ох— поверхностная энергия промежуточного
поверхностно-активного соединения;
Й = I+K1/K2 {Ki — скорость прямой реакции;
#2 — скорость обратной реакции).
174
Экспериментальная проверка полученных
соотношений, выполненная авторами на системах водные
растворы— газ и шлак — газ, подтвердила их правильность.
При интенсивном химическом взаимодействии
твердой и жидкой фаз явления смачивания носят
необратимый характер. Однако Найдич [9] считает, что иногда
возможно использование законов классической
термодинамики для описания смачивания, и в этом случае
принимается, что химический процесс в приконтактном слое
достигает псевдоравновесных условий. По мнению
автора, такой подход не является вполне строгим, но
позволяет дать качественное объяснение капиллярных и
адгезионных явлений в большинстве систем, а для ряда из
них допускает численные приближенные расчеты. При
этом предполагается, что работа адгезии состоит из двух
величин:
"а = **а(неравн) •"" **а(равн)' (4-15)
где Т^а(неравн) определяется работой реакций на
контактной границе в зависимости от механизма ее протекания.
Величина №а(равн) состоит из энергии образующихся на
контактной границе химических равновесных связей и
энергии ван-дер-ваальсова взаимодействия на той же
границе. Найдич отмечает, что непосредственно из
уравнения (4-3) следует
т—ж т—г ■ ж—г а»
а с учетом (4-15)
т—ж т—г •"" ж—г а (равн) а(неравн)
ИЛИ
а =а —Да . (4-16^
т—ж равн неравн Vх 1U/
При достаточно интенсивной химической реакции на
контактной границе величина ДсгНеравн может стать
больше, чем аравн, тогда межфазное поверхностное
натяжение примет отрицательное значение и система окажется
термодинамически неустойчивой: возникнет, например,
сильное стремление к диспергированию.
Приведенные соотношения в большей части
описывают поведение жидкости на идеально гладкой
поверхности. В реальных условиях каждая твердая
поверхность обладает определенным, свойственным ей
микрорельефом, и для правильного описания поведения жид-
175
кости на поверхности твердого тела необходимо наряду
с физическими свойствами системы такими, как
поверхностное натяжение жидкости, межфазная поверхностная
энергия, свободная поверхностная энергия твердого
тела, учитывать также и геометрические особенности
твердой поверхности. Так, по гладкой, свободной от
окиси, поверхности золота ртуть не растекается, в то же
время наблюдается распространение ртути по
поверхности золота, если используется крупнозернистый
исходный материал, обработанный в холодном состоянии [13].
Установлено, что геометрия твердой поверхности
заметно влияет на краевой угол оттекания жидкости и слабо
влияет на краевой угол натекания [4—10]. Связь между
шероховатостью поверхности твердого тела и его
смачиваемостью впервые определил Венцель [11]. Дерягин
[12] термодинамически обосновал формулу Венцеля.
Количественной оценкой шероховатости твердой
поверхности служит коэффициент шероховатости (/Сш),
определяемый как отношение фактической площади
поверхности к геометрической
/?ш = 5<А- (4-17)
Для идеально гладкой поверхности /(ш = 1, по мере
увеличения шероховатости Кш растет. Величина его
зависит от вида механической и термической обработки
данной поверхности. Определяющим фактором
возникновения микрорельефа поверхности является
анизотропия свободной поверхностной энергии твердых тел,
благодаря чему минимум свободной энергии достигается не
на гладкой поверхности, а на поверхности с
определенным микрорельефом.
Величина краевого угла смачивания на шероховатой
поверхности определяется уравнением Венцеля — Деря-
гина
cosQ = /Cmcose0, (4-18)
где 0О— краевой угол смачивания на идеально гладкой
поверхности.
Из уравнения (4-18) следует, что при значительной
шероховатости, соответствующей условию
/Сш> l/cos0ot (4-19)
возможен переход от ограниченного смачивания к
растеканию, если 0О<9О°. Такой переход наблюдали,
например, в системе ртуть — цинк [14].
176
Все же роль микрорельефа в процессах смачивания
и растекания еще нельзя считать полностью выясненной.
При одинаковом значении коэффициента растекания
поверхность твердого тела может иметь совершенно
различную структуру [14].
Объясняется это, вероятно, тем, что коэффициент
шероховатости представляет собой интегральную
характеристику поверхности и учитывает только изменение
поверхности при переходе от идеально гладкой к реальной.
При исследовании смачивания кварца сплавом меди с
титаном [9] наблюдался эффект, противоположный
теоретическому: при переходе от полированной поверхности
к шлифованной смачиваемость ухудшалась независимо
от того, острым или тупым был краевой угол
смачивания на гладкой поверхности. Подобная картина
наблюдается и при смачивании платины жидким стеклом [2].
Для объяснения этих эффектов Найдич [9, 15]
выдвинул предположение о том, что микрогребешки могут
задерживать растекание жидкости, и поэтому краевой
угол, измеряемый при натекании, должен быть больше
угла на гладкой поверхности.
Аналогичный подход к этой проблеме развили
Джонсон и Детре [16], которые профиль поверхности
аппроксимировали синусоидой. Достоверность расчетных
уравнений, полученных этими авторами, подтверждается
экспериментальными данными при исследовании процесса
смачивания парафина водой, гексаденом и йодистым
метиленом. Предполагают также, что растекание по
шероховатой поверхности происходит лишь вдоль канавок с
достаточно острым двугранным углом при вершине и
возможность растекания зависит не столько от
интегрального коэффициента шероховатости, сколько от
скопления на поверхности микродефектов определенной
геометрии [17].
2. КИНЕТИКА
И КОНЕЧНАЯ ПЛОЩАДЬ РАСТЕКАНИЯ
Исследование кинетики растекания жидкости по
поверхности твердого тела позволяет определить величину
движущей силы растекания и соотношения между
величинами, ее определяющими. Именно по этой причине
такое широкое развитие получили исследования законо-
12-894
177
мерностей кинетики растекания жидкостей по
поверхности твердых тел.
Обобщение результатов исследований, выполненных
в этой области, сделано Горюновым и Суммом [18—20].
В основу теоретического рассмотрения процесса
растекания положена зависимость радиуса растекающейся
капли г от времени т. Решение соответствующей задачи
на основе уравнений Навье — Стокса связано с
большими математическими трудностями. Поэтому при выводе
аналитической зависимости г=г{%) обычно делают
различные упрощающие допущения о характере течения
жидкости.
Процесс растекания можно условно разделить на три
стадии. Первая — быстрое растекание под совместным
действием капиллярных сил, сил тяжести и
динамического удара. За ней следует вторая, основная стадия
процесса с квазистационарным течением жидкости, и затем
наступает третья стадия, на которой течение жидкости
резко замедляется и прекращается. На второй стадии
процесса растекания общие, характерные для любых
систем закономерности растекания проявляются в
большей степени, чем на первой или последней стадии.
Поэтому при теоретическом рассмотрении процесса
растекания обычно ограничиваются рассмотрением лишь
второй стадии.
Щукин, Горюнов с сотр. [6] исследовали и
математически описали процесс растекания ртути по поверхности
цинка под действием поверхностных сил в
предположении, что:
1) движущая сила растекания, определяющая
перемещение фронта фазового слоя жидкости,
пропорциональна убыли свободной поверхностной энергии;^
2) на основной стадии процесса масса жидкости не
изменяется;
3) пленка жидкости по мере растекания утонынается
равномерно;
4) градиент скорости по толщине пленки постоянен;
5) сила тяжести не оказывает влияния на параметры
процесса растекания.
Решая линейную задачу растекания жидкости от
точечного источника конечной мощности в обе стороны от
него, авторы получили:
х = (Зт12)1/3т1/3 = Лт1/3; (4-20)
178
ntx = /n/(2a); 2 = Да/(лт),
где m — масса капли;
a — ширина полосы растекания;
х — координата фронта течения;
т], Y — вязкость и плотность жидкости
соответственно;
Да—движущая сила растекания.
Аналогичная задача о круговом (двумерном)
растекании капли имеет решение:
r^-lm^'V'4. (4-21)
Если отказаться от допущения о равномерном утонь-
шении пленки, то необходимо сделать какие-либо другие
допущения о профиле растекающейся капли. В случае
аппроксимации истинной формы капли шаровым
сегментом в уравнения (4-20) и (4-21) вводится множитель
1/х, где х — коэффициент формы. Для случаев одно- и
двумерного растекания к определяется из следующих
выражений:
«-■»(£)-!: (4-22,
, / 3m \ 3
х = 1п
Ыг2 у J 2
В этих формулах коэффициент X — величина порядка
атомных размеров. Для квазистационарной стадии
процесса растекания коэффициент формы обычно
принимается постоянным и равным 7—11, тогда уравнения (4-20)
и (4-21) примут вид:
* = р^у/Зт1/з; ^
Различия в показателях степени для линейной и
двумерной задачи связано с тем, что при линейном
растекании толщина пленки жидкости убывает
пропорционально расстоянию, а при двумерном — пропорционально
квадрату расстояния, поэтому при двумерном
растекании вязкое сопротивление увеличивается быстрее, и,
следовательно, скорость растекания со временем
убывает также быстрее.
12*
179
Елютин с сотр. [21], решая задачу о растекании
Жидкости по поверхности твердого тела, исходил из
уравнения
2пгАо — яг2т1— =0 (4-24)
dz
и получил для случая кругового растекания капли:
\ ■
X
8mAa\i/4^i/4 (4-25)
Кинетические уравнения Щукина—Горюнова (4-23),
Елютина (4-25) применимы только в случае отсутствия
заметного химического взаимодействия растекающейся
жидкости с твердым телом, т. е. при условии, что
свойства растекающейся жидкости сохраняются
неизменными. В реальных условиях растекания, и особенно при
высоких температурах, состав и, следовательно, физико-
химические свойства жидкости заметно изменяются.
Учет изменения физико-химических свойств жидкости
приводит к значительному усложнению задачи,
нахождение решения которой в общем виде вряд ли
целесообразно. Поэтому приходится ограничиваться учетом
наиболее важных факторов.
Елютин с сотр. [21] при изучении процесса
распространения жидкого титана по поверхности графита с
учетом их взаимодействия получил кинетические
уравнения, учитывающие изменение вязкости жидкой фазы
в процессе растекания. Предварительно был исследован
и математически описан процесс науглероживания
жидкого титана при непосредственном контакте с графитом
[22]. Кроме того, было исследовано изменение вязкости
титана в зависимости от концентрации в нем углерода
[23]. Использование этих данных для расчета
кривых растекания по уравнению (4-25) обеспечивает
лучшее совпадение с экспериментальными данными.
Задача значительно упрощается, если допустима
аппроксимация изменения свойств растекающейся
жидкости степенными функциями от времени или от радиуса
капли. При этом уравнение всегда интегрируется в
элементарных функциях и получается степенная
зависимость радиуса растекающейся капли от времени. В
работе [21] этот подход использован при исследовании
кинетики растекания жидких титана и циркония по
поверхности поликристаллического графита. Изменение
180
вязкости жидкого металла достаточно хорошо
описывается уравнением:
v = Nv0xir\ (4-26)
где N— коэффициент науглероживания;
v0—кинематическая вязкость чистого металла.
В этом случае кинетическое уравнение, расчеты по
которому находятся в хорошем соответствии с
экспериментальными данными, имеет вид:
г=Ат1/8. (4-27)
При выводе уравнений (4-23), (4-25) вопрос о
допустимости принятых предположений о характере течения
жидкости не рассматривался. Границы применимости
уравнений установлены в работе [24] посредством
анализа растекания ньютоновской жидкости по
горизонтальной поверхности на основе более общих
представлений, чем использованные ранее. В этой работе при
определенных допущениях решались система уравнений
Навье — Стокса и уравнение непрерывности. Так как
растекание — процесс квазистационарный, то
производной компоненты скорости по времени можно пренебречь
по сравнению с членом, учитывающим вязкость, при
выполнении условия
т » X б2, (4-28)
где 6 — толщина слоя жидкости.
При #>6 толщина 8 = т/(2аху) и (4-28) можно
записать в виде
т»Р^с%, (4-29)
гАе Рф.с— плотность фазового слоя жидкости, т. е.
масса жидкой фазы, приходящаяся на
единицу поверхности твердого тела
Рф.с(^) =т/(2ха).
В результате решения системы уравнений Навье —
Стокса при выполнении условия (4-28) выражение для
профиля скорости в растекающейся капле в
зависимости от ординаты у имеет вид
v = ^L{2by-y% (4-30)
цт
181
а для перемещения фронта текущей жидкости в случае
одномерного растекания
/_8_ Доту/з т,/3
\ 4 т\уа ]
и в случае двумерного (кругового) растекания
_ /4_ Дату/4 1/4
(4-31)
(4-32)
Полученные соотношения с точностью до числовых
множителей совпадают с предложенными ранее (4-23),
(4-25). Примененный метод решения задачи позволяет
также рассмотреть случай, когда движение жидкости
происходит в основном за счет уменьшения
потенциальной энергии при понижении центра тяжести слоя.
Такой подход приводит соответственно для одномерного
и кругового растекания к уравнениям:
х =
' 5m3g
, 32т|а3у2,
V'\vs.
U3 y\l
Выражение (4-31)
неравенства
(4-33)
справедливо при выполнении
С-£Г«,
(4-34)
Экспериментальная проверка полученных
соотношений, проведенная при изучении смачивания твердого
свинца ртутью, подтвердила их достоверность [24].
В некоторых системах имеет место растекание
жидкости по поверхности твердого тела под слоем твердых
окислов. В этом случае растекание происходит как бы
между плоскопараллельными пластинами и
зависимость имеет вид [25]:
г = Лт1/2.
(4-35)
Полученные кинетические уравнения не позволяют
установить расстояние, на которое распространяется
жидкость при неограниченном времени растекания. Это
182
объясняется тем, что при выводе указанных уравнений
рассматривалась только одна стадия —
квазистационарное течение жидкости и не учитывались факторы,
приводящие к прекращению растекания. К основным
причинам прекращения растекания жидкости в
системах без химического взаимодействия относятся
следующие [25, 26]:
1) возрастание поверхностного натяжения
жидкости в тонких слоях (10~5 см) до таких значений, когда
неравенство, определяющее растекание Дат-г>(Тт-ж+
+сгж-г, превратится в равенство [27];
2) уменьшение толщины слоя жидкости до
величины, соизмеримой с высотой микрогребешков при
достаточно большом коэффициенте шероховатости [28];
3) изменение других, кроме поверхностного
натяжения, физико-химических свойств в тонком слое по
сравнению со свойствами массивных образцов (так,
например, в тонких слоях резко возрастает вязкость
жидкости [29].
В системах с заметным химическим
взаимодействием прекращение растекания чаще всего определяется
изменением физико-химических свойств жидкости,
таких, как вязкость, поверхностное натяжение,
определяемое изменением состава жидкости. Кроме того,
растекание жидкости может прекратиться из-за ее расхода
на химическое взаимодействие с твердым телом. Если
продуктами химической реакции являются при данной
температуре твердые вещества, то они, выпадая в
осадок, затрудняют и в конце концов прекращают
процесс растекания. Аналогичную роль могут играть
продукты диссоциации сложной по составу растекающейся
жидкости. Причиной прекращения растекания может
явиться процесс растворения твердого тела в жидкости,
если в результате их взаимодействия образуются
сплавы или соединения, имеющие более высокую
температуру плавления, чем температура, при которой
протекает растекание.
Очень часто, особенно при высоких температурах,
действуют сразу несколько причин, обусловливающих
прекращение растекания жидкости.
Горюнов с сотр. [19, 21] определил зависимость
конечной площади растекания от массы жидкости для
систем, в которых имеется конкуренция процессов
растекания и расхода жидкости на диффузию в твердое
183
тело. Радиус смоченного жидкостью круга г в этом
случае определяется формулой
г = ВтЪ1\ (4-36)
где В — функция начальной концентрации,
коэффициента диффузии, вязкости, плотности
жидкости и движущих сил растекания.
Экспериментальную проверку полученных
соотношений проводили при изучении смачивания твердого
цинка галием.
Елютин с сотр. [30] рассмотрел закономерности
прекращения растекания для химически интенсивно
взаимодействующих систем, полагая, что растекание
капли прекращается тогда, когда ее масса
израсходована на пропитку пористого твердого тела и на
кристаллизацию тугоплавких продуктов взаимодействия. Эти
причины прекращения растекания возникают в
системах графит — жидкий карбидообразующий металл
IV группы (титан, цирконий).
3. ЗАКОНОМЕРНОСТИ СМАЧИВАНИЯ
И РАСТЕКАНИЯ В СИСТЕМАХ С ОКИСЛАМИ
Анализ накопленного экспериментального
материала по смачиванию жидкими металлами твердых
окислов и теоретическое рассмотрение этого явления
выполнил Найдич [9]. Он показал, что смачиваемость
твердого окисла жидким металлом возрастает при
повышении сродства к кислороду металла, входящего в
состав жидкой фазы. Так как поверхность окислов
образована, как правило, анионами кислорода, то
химическое взаимодействие жидкого металла с кислородом
твердого окисла описывается уравнением
Me" + Me' О ^ Me' + Me" О, (4-37)
для которого AG = AG" — AG',
где AG" и AG"— термодинамические потенциалы
реакций образования окислов
металла (Ме')у входящего в состав
твердого тела, и жидкого металла
{Me"),
Уменьшение величины AG и ее переход в
отрицательную область должны приводить к уменьшению
184
краевого угла смачивания, что и наблюдается
экспериментально. В соответствии с изложенными
представлениями обнаружено заметное снижение межфазного
натяжения на границе металл — окисел под влиянием
добавок кислорода к жидкому металлу (рис. 46).
На контактной поверхности раздела твердый
окисел — жидкий металл происходит по представлениям
Найдича следующее. Положительно заряженные ионы
жидкого металла адсорбируются на поверхности твер-
02,%(ат.)
Рис. 46. Межфазное натяжение на
границе раздела металл с кислородом —
окисел [9]:
/ — медь с кислородом — MgO, 1150° С;
2— медь с кислородом — АЬОз. 1150° С;
3 — никель с кислородом — А1203,
1500° С
WA, мДж/м2
1000 х
*^~
0г%(ат.)
Рис. 47. Работа адгезии в системах
жидкий металл с кислородом —
окисел [0]. Обозначения см. на
рис. 46
дого окисла, при этом они связываются прочной
химической связью с кислородными анионами твердого
окисла, достраивая его решетку. Ионы кислорода,
растворенные в жидком металле, закрепляются на этих
положительно заряженных ионах, и таким образом,
возникает слой окисла металла жидкой фазы.
Для образования прочной связи на границе атомы
жидкого металла должны отдать свои электроны
атомам растворенного кислорода. Оценка энергии
взаимодействия окисленного жидкого металла с поверхностью
твердого окисла, полученная Найдичем на основе учета
сил электростатического притяжения, по порядку
величины составляет 103 мДж/м2. Эта величина
соответствует работе адгезии (рис. 47), установленной
экспериментально. Численную оценку энергии связи металла
185
и окисла (работу адгезии) можно осуществить, вычисляя
энергию контактной химической реакции. Если реакция
протекает по типу
Me" + Me' О ^ Me" О + Ме\ (4-38)
то
ДС = AG" - AG' = - RT In ^ г, (4-39)
где пг и п"—количества молей соответствующих
окислов, умещающихся на 1 см2 в виде
моноатомной пленки;
а0—степень диссоциации при равновесии.
Дисперсионная энергия связи определяется по
уравнению
£^JL?^_ZiA. (4-40)
где аг и а2— поляризуемости;
Ji и /2 — первые ионизационные потенциалы
взаимодействующих атомов;
7? — расстояние между атомами, которое
принимается равным сумме радиусов
атома металла и иона кислорода.
Результаты этих расчетов показывают, что при
достаточно больших отрицательных значениях ДО
вычисленная химическая часть работы адгезии сравнима с
определяемой экспериментально. При положительных
AG вычисленная химическая часть энергии связи мала
и уступает по величине энергии ван-дер-ваальсового
взаимодействия.
Найдич отмечает, что большей частью расчет в
предположении полного окисления дает заниженные
результаты по сравнению с экспериментальным значением
величины работы адгезии. Это обстоятельство
определяется более вероятным восстановлением до низшего
окисла твердой фазы и окислением также до низшего
окисла металла жидкой фазы, в этом случае выигрыш
энергии может быть больше. Возможен также
дополнительный выигрыш энергии вследствие образования
соединений между возникшим и имевшимся окислами.
Рассматривая механизм образования связи на
границе раздела твердый металл — жидкий окисел, Аппен
[3] также полагает, что в данном случае возникает хи-
186
мическая связь по схеме Мет — О — Мет, а адгезия
жидких окислов к металлу должна определяться
величиной энергии химической связи Мет — О.
Эти положения подтверждаются
экспериментальными данными, полученными при исследовании влияния
содержания компонентов силикатных расплавов на
величину их адгезии к стали. В структурном мостике
Мет — О—Меж упрочнение связи с одной стороны от
кислорода сопровождается ослаблением связи с
другой стороны. Действительно, добавление к силикатным
расплавам окислов с высокой энергией связи металла
с кислородом (V2O5; Мо03; Сг203; В203) ухудшает их
адгезию к стали и, наоборот, добавление окислов с
невысокой энергией связи (Na20; MnO; СоО) ее
улучшает.
Очевидно, на основе такого подхода можно
объяснить некоторые известные факты, но трудно
прогнозировать характер смачивания в системах металл —
окисел, поскольку существующая методика расчета
энергии связи в сложных кристаллах еще далека от
совершенства. Следует учитывать также, что связь
металла с кислородом в окислах не является чисто ионной,
а всегда содержит ковалентную составляющую. Кроме
того, в методе оценки адгезии по Аппену не участвуют
поверхностные свойства фаз, что вряд ли справедливо.
Подход к рассматриваемым явлениям,
предложенный Найдичем [9], по нашему мнению, более
рационален. Однако в его методе имеется некоторая
неопределенность, вызванная тем, что процесс рассматривается
в нефиксированном приповерхностном объеме.
Наиболее удобен метод Найдича при расчетах систем с
ограниченной степенью смачивания.
Для систем с полным смачиванием можно
использовать термодинамический расчет межфазной энергии
и движущей сшщ растекания химической реакции на
границе раздела, который успешно использовался
Еременко, Аппеном, Найдичем и др. Изменение свободной
энергии системы при смачивании в общем случае
выражается уравнением
До = a AS +0 AS — а AS ,
"v ж—г ^ж—г ' т—ж ^т—ж т—г т—г»
а для систем с полным смачиванием
Да = а -\-g —а , (4-41)
ж—г ' т—ж т—г' х '
187
так как в этом случае можно принять, что Д5Ж_Г=
=== /д*Ь т—ж == —Д*^ т—г*
Если растекание сопровождается химической
реакцией на границе контакта, то в выражение (4-41)
вместо статической ат-ж следует подставлять динамическую
межфазную энергию (о*__ж) и таким образом
учитывать энергию, выделяющуюся при контактной
химической реакции. Эту оценку можно произвести, если
принять, что контактная реакция протекает в
мономолекулярном слое, а образующиеся продукты реакции резко
затормаживают ее развитие в направлении,
перпендикулярном поверхности контакта. Такой случай
реализуется, например, при полном смачивании жидкими
окислами поверхности твердого металла, когда
образуются твердые продукты взаимодействия и диффузия
через них значительно ниже скорости растекания. При
этом жидкость практически неизменного состава
реагирует с поверхностью твердого тела и эффект
химической реакции определяет в основном изменение
межфазной поверхностной энергии. Другими видами рассеяния
энергии при малых временах растекания можно
пренебречь. Тогда
К_ж dS = <т;_ж dS + AGp dn, (4-42)
где AGP — парциальное изменение свободной энергии
контактной химической реакции (r = const);
dn—количество вещества жидкой фазы в молях
при увеличении площади контакта на
величину dS.
Уравнение изменения поверхностной энергии
системы запишется в виде:
do = аж_г dS + <_ж dS - AGp dn - <тт_г dS. (4-43)
Поскольку используются динамическая величина
межфазной поверхностной энергии и представление
о мономолекулярном характере реакции, то можно
принять, что на контактной границе соблюдается
квазиравновесные условия, т.е. da=0. Окончательно
решение имеет вид:
а* =а —а + AG AS, (4-44)
т—ж т—г ж—г ' р » х /
где S— площадь, занимаемая одним молем жидкости
в виде мономолекулярной пленки.
188
Полученное выражение по форме весьма близко к
выражению, ранее предложенному Еременко [32].
Отличия заключаются в том, что оговаривается механизм
реакции, протекающей на контактной границе, и,
кроме того, в величину а*_ж вкладывается иной смысл. Эта
величина принимается неравновесной, действующей
только в период растекания.
С использованием уравнения (4-44) можно
рассчитать динамическую межфазную энергию и движущую
силу растекания для случаев смачивания, например,
поверхности тугоплавких металлов жидкими окислами.
При проведении этих расчетов учитывалось то, что
многие окислы (Та205, ТЮ2, Nb205; Mo03; W03; V205)
легко образуют дефектную по кислороду структуру типа
МеОя-х, где х изменяется от нескольких сотых до
нескольких десятых единицы [33]. Поэтому для этих
окислов принималось, что контактная химическая реакция
протекает до образования устойчивого в данных
условиях окисла твердого металла (подложки), так как
отщепление кислорода от окисла и образование окисла
нестехиометрического состава происходят при весьма
малых затратах энергии. Если же окислы не образуют
дефектных по кислороду структур (ВеО; Сг203; А1203),
то контактную реакцию рассчитывали при условии
восстановления жидкого окисла до низшего окисла или
металла и окисления твердого металла (подложки) до
термодинамически устойчивого в рассматриваемых
условиях окисла.
Значения поверхностной энергии твердого металла
вычисляли по формуле:
<тт_г = 1,15<гж_г+ 0,1 [tmMe - tnji окисла), (4-45)
где (Тж_г— поверхностное натяжение жидкого
металла подложки при температуре плавления.
Значения поверхностного, натяжения жидких
окислов и металлов взяты из работ, приведенных в главе II,
а также из работ [4, 36].
Движущую силу растекания рассчитывали по
формуле
Аа = аж_т + АОуд. (4-46)
Из результатов расчета (табл. 18) вытекает, что
жидкие окислы Сг203; Ti02; Ta205; Nb205; W03; Мо03;
189
V2O5 должны полностью смачивать поверхность
вольфрама, молибдена, тантала и ниобия, а жидкие окислы
ВеО и АЬОз — образовывать на поверхности этих
металлов каплю с конечным краевым углом смачивания.
В ряде случаев значения динамической межфазной
поверхностной энергии становятся отрицательными, что
свидетельствует о термодинамической неустойчивости
системы и возможности самопроизвольного
диспергирования.
Таблица 18
Расчетные значения межфазной динамической
поверхностной энергии и движущей силы растекания
при смачивании поверхности тугоплавких металлов
жидкими окислами
Окнеел
ВеО
А12Оэ
Сг208
тю2
Та205
Nb2Oe
W03
MoO,
v2o5
Молибден
•
°т-ж
_
6450
1400
650
600
300
150
—
-1150
Да
5400
—1000
—1850
—1950
—2400
—2700
—
I -3550
Вольфрам
*
а
т—ж
6800
6800
1100
—150
—200
—350
—
—2550
—2650
Ла
5000
5400
—1700
—2950
—3000
—3600
—
—5550
—5700
Ниобий
*
а
т—ж
_
4650
—
—4900
—5100
—
—5900
—8150
—8000
Ла
3 900
__
—7 250
—7 400
—
—8 500
—10 750
—10 900
Тантал
•
о
т—ж
4 750
3 700
—8 100
—9 500
—
-10 100
—10 800
—13 850
-13 950
Ла
4 250
2 800
—10 400
—11 650
—
—12 800
—13 400
—16 450
—16 600
Экспериментальное исследование смачивания
вольфрама, молибдена, тантала и ниобия жидкими
окислами [35] осуществляли при нанесении капли жидкого
окисла на поверхность металла в изотермических
условиях при температуре, несколько превышающей
температуру плавления окисла. Исследования проводили на
катаных поликристаллических металлических
пластинах с шероховатостью поверхности V3 для вольфрама
и молибдена, и V5 для ниобия и тантала. Перед
опытом металлические пластины протравливали в
соответствующих травителях, а затем промывали в
дистиллированной воде и ацетоне. Окислы расплавляли на
молибденовой подставке, расположенной на высоте 5 мм
от пластины; при плавлении капля окисла попадала на
образец твердого металла. Процесс смачивания
фиксировали с помощью киносъемки. В тех случаях, когда
окислы на поверхности тугоплавких металлов образо-
190
вывали каплю с острым конечным краевым углом, его
определяли по методике, предложенной в работе [34]:
6 = arcsinf x\ \% (4-47)
где у — высота капли;
х — диаметр основания капли.
Результаты исследования приведены в табл. 19, из
которой следует, что данные расчетов удовлетворитель-
Таблица 19
Смачивание твердых тугоплавких металлов жидкими окислами
Окисел
ВеО*
Сг203
А1203
Ti02
Та2Об
Nb205
wo3
Мо03
v2o6
т, к
2855
2655
2325
2075
2105
1735
1765
1100
975
Краевой угол смачивания, град,
Мо | W
0
15
0
0
0
0
1 °
0
0
0
7
0
0
0
0
0
0
Nb
_
—
40
0
5—7
10
0
0
0
Та
0
0
30
0
5-7
10
0
10
0
* Получено Бартлетом и Холлом [34] в условиях расплавления окисла на
поверхности твердого тела.
но совпадают с экспериментом, за исключением
случаев с окисью бериллия.
Однако следует отметить, что условия опытов с
окисью бериллия отличались от остальных тем, что
нагревали окись, находящуюся в контакте с тугоплавким
металлом. Такая методика содержит в себе
возможность ошибки вследствие протекания твердофазных
реакций на границе контакта во время нагрева.
4. КИНЕТИКА СМАЧИВАНИЯ
ТУГОПЛАВКИХ МЕТАЛЛОВ
ЖИДКИМИ ОКИСЛАМИ
Исследование кинетики растекания не только
позволяет решить некоторые практические вопросы,
связанные с организацией окисных покрытий на металлах, но
и дает возможность оценить движущие силы растека-
191
ния. При теоретическом рассмотрении процессов
растекания жидких окислов по поверхности тугоплавких
металлов следует учитывать, что реальные процессы,
протекающие на контактной границе, намного сложнее,
чем в схеме, предложенной авторами выше. На
характер и кинетику растекания сильно влияют не только
процессы контактного взаимодействия, но и те
изменения, которые претерпевают окислы в процессе
растекания: диссоциация с образованием твердых продуктов,
летучесть и т. д.
С помощью киносъемки капли жидкого окисла,
нанесенной на поверхность металла, изучали
закономерности процесса смачивания и растекания жидких
окислов и тугоплавких металлов при температуре
плавления соответствующего окисла [35]. Процесс
осуществляли в вакууме (~10~3 мм рт. ст.) и среде аргона при
исследовании окислов титана, тантала, ванадия и
ниобия и только в среде аргона при исследовании окислов
вольфрама и молибдена, которые характеризуются
повышенной летучестью.
Так как растекание жидкости по поверхности
твердого тела зависит от плотности фазового слоя рф.с, то
те или иные закономерности процесса будут
наблюдаться только в определенном интервале ее значений.
Основные закономерности распространения жидких
окислов по поверхности тугоплавких металлов изучали
с использованием двуокиси титана; в качестве
подложек использовали вольфрам, молибден и тантал.
Исследования проводили при изменении плотности
фазового слоя жидкости от 7-10-1 до 5-Ю-5 г/см2. Из
опытов получали временную зависимость координаты
движения фазового слоя жидкости при круговом
растекании г или при растекании по одному направлению х
[58—60].
В исследованном интервале значений рф. с
экспериментальные точки хорошо укладываются на прямые в
координатах \gr(x)—lgx, что свидетельствует о
возможности описания процессов растекания
зависимостью вида г(х)—Ахп. Оказалось, что каждому
интервалу значений рф. с соответствует определенное
значение показателя п (п принимает значения 0,2; 0,33;
0,5; 1 и снова 0,5 при уменьшении рф. с для
одномерного растекания). При достаточно больших плотностях
фазового слоя [(7—1,5)-Ю-1 г/см2] показатель п
192
Рис. 48. Характер растекания жидкой
двуокиси титана по поверхности
молибдена при плотности фазового слоя
(7,0—1,5) -10 г/см2. Масса капли, г:
/ — 0,59; 2 — 0,51; 3 — 0,25
близок к 0,2 для одномерного (х) и 0,125 для кругового
(г) растекания, что характерно для растекания
жидкости под действием силы тяжести, описываемого
выражениями (4-33).
Изучение растекания под действием силы тяжести
позволяет определить величину коэффициента формы
х, расчет которой по данным геометрических
измерений затруднен. При данной плотности фазового слоя
жидкости показатель я,
определенный по
экспериментальным данным
(рис. 48), равен 0,125—
0,14, т.е. в некоторых
случаях несколько выше, чем
следует из соотношений
(4-33). Это связано с тем,
что в некоторых случаях
помимо силы тяжести,
определенную роль
начинают играть поверхностные
силы. В случае
совместного действия сил
тяжести и поверхностных сил
значения п должны лежать в пределах 0,125О<0,25
ближе к 0,125 или 0,25 в зависимости от того, какие
силы вносят больший вклад в движущую силу растекания.
Влияние поверхностных сил на кинетику растекания
в данном интервале плотностей фазового слоя
подтверждается также характером зависимости величины
А от массы капли. Теоретически, согласно (4-33),
зависимость lgi4 — lgm должна иметь вид .прямой линии
с угловым коэффициентом, равным 0,375. Фактически
в области малых масс в данном интервале плотностей
фазового слоя жидкости наблюдается отклонение от
прямой линии. Величина же углового коэффициента
для прямолинейного участка оказывается равной 0,34,
т. е. несколько ниже теоретической.
Величина коэффициента формы х, определенная с
помощью соотношений (4-33), также оказывается
зависящей от плотности фазового слоя жидкости. При
плотностях фазового слоя, соответствующих
прямолинейному характеру зависимости \gA—lgm коэффициент
формы х=8, что хорошо согласуется с результатами
работы [6]. При плотностях фазового слоя, при которых
13-894
193
зависимость IgA— lgm отклоняется от
прямолинейной, х=1, что связано с влиянием поверхностных сил.
Поэтому истинные значения к следует определять по
прямолинейному участку зависимости lgЛ — lgm.
В вакууме значения А оказываются выше, чем
в атмосфере аргона, вследствие увеличения движущей
силы растекания, вызванного двумя причинами: во-
первых, поверхность металла менее окислена и,
во-вторых, удаление легколетучих окислов вольфрама и
молибдена способствует более интенсивному протеканию
контактных реакций.
На тантале — наиболее химически активном
металле из всех исследованных — растекание под действием
силы тяжести не наблюдается ни в атмосфере аргона,
ни в вакууме при плотностях фазового слоя жидкости
вплоть до 0,7 г/см2.
При плотности фазового слоя жидкости в интервале
(15—2,0)-Ю-2 г/см2 реализуется закон растекания
с показателями 0,33(х) и 0,25(г)—растекание под
действием поверхностных сил по механизму
равномерного утоныыения текущего фазового слоя.
Аналитические выражения процессов растекания в этом случае
даются уравнениями (4-20) и (4-21). Этот тип
растекания наблюдается в атмосфере аргона как при
совместном, так и при раздельном нагреве твердого металла
и жидкого окисла. При исследованиях в вакууме во
избежание твердофазного взаимодействия применяли
только раздельный нагрев окисла и металла. Кинетика
растекания жидких окислов по тугоплавким металлам
в логарифмических координатах описывается прямыми
с угловыми коэффициентами, определяемыми
уравнениями (4-20) и (4-21) (рис. 49). Исключение
составляет случай растекания двуокиси титана по танталу,
когда угловой коэффициент равен 0,5 как для
одномерного, так и для кругового (двумерного) растекания.
При плотностях фазового слоя жидкости (15—
—2,0) • 10-2 г/см2 зависимость IgA —lgm является
линейной (рис. 50) и экспериментально определенный
угловой коэффициент (0,22) достаточно хорошо совпадает
с расчетным (0,25), что указывает на правильность
выбранного подхода. Из графика lg Л — lgm можно
рассчитать величину Л, а с ее помощью определить
движущую силу растекания по формулам (4-20)
и (4-21). Необходимые для расчета значения коэффи-
194
циента формы берутся из эскпериментальных данных
по растеканию жидких окислов под действием силы
тяжести. Результаты расчетов движущей силы растека-
1дА
О 0,4 0,8 12 1дт
0,2
0,1
N.
No
j i i 1—
0,2 0,4 0,6 0,8 -Igm
Рис. 50. Зависимость ]gA — \gm при
кой двуокиси титана по поверхно- растекании жидкой двуокиси тита-
Рис. 49. Характер растекания жид-
сти молибдена при плотности
фазового слоя (15—2,0)-Ю-2 г/см2.
Масса капли, г:
/ — 0,48; 2 — 0,32; 3 — 0,19; 4-0,11
на по поверхности молибдена при
плотности фазового слоя (15—2,0) X
Х10~2 г/см2
ния и динамической межфазной поверхностной энергии
приведены в табл. 20 и находятся в хорошем
качественном согласии с результатами расчетов, приведенных
Таблица 20
Движущая сила растекания и динамическая межфазная
энергия при растекании жидкой двуокиси титана
по поверхности тугоплавких металлов
Рф.с , г/см8
(15-2,0). Ю-2
(20—1,5). Ю-з
ч
СП
СУ
2
Мо
W
Nb
Та
Мо
W
Nb
Та
,
§8
о я
—390
—220
—480
—
Вакуум
двумерное
Ло
—380
—200
—480
—
1 —2000
•
ат—ж
1850
2300
1390
i —
! —во
5S
О X
-55
-55
— 110
—290
Аргон
двумерное
Да
-50
-40
—110
[-300
—440
1—310
"
*
ат—ж
2180
2450
1760
1560
1780
2100
"
13*
195
в табл. 18. Практически при любой плотности фазового)
слоя жидкости при растекании двуокиси титана по
танталу в вакууме показатель п = 0,5 (рис. 51). Этой же
закономерностью описывается и кинетика растекания:
двуокиси титана по вольфраму и молибдену в аргоне
при плотностях фазового слоя жидкости (20—1,5) X
Х10~3 г/см2. Известно, что кинетика распространения
жидкости по поверхности твердого тела при п = 0,5-
характерна либо для
случая поверхностной
диффузии, либо для; случая:
распространения
жидкости в зазоре между
двумя твердыми
поверхностями: например,
распространение жидкости по
металлической
поверхности под слоем его
твердых окислов. При
плотностях фазового слоя
жидкости (20—1,5)-10~3 г/см2'
диффузионный механизм
распространения
двуокиси титана по поверхности
исследованных
тугоплавких металлов не
наблюдается. Это доказывается
опытами по растеканию
двуокиси титана по гори-
и наклонной
металлическим
пластинкам. При диффузионном
распространении жидкости показатель А в уравнении
х=А%°'ъ должен оставаться постоянным независимо от
угла наклона пластины. В наших же исследованиях
показатель А изменялся от 30 до 6 при изменении угла
наклона на пластины от 0 до 45°, что доказывает
распространение жидкости в виде фазового слоя. Не обеспечивает
реализацию такого механизма процесса и растекание
жидкости в зазоре, так как при образовании фазового
слоя окисла кинетика растекания описывается
уравнением, полученным при допущении о равномерности
утоньшения фазового слоя жидкости. Математическое
описание процесса распространения жидкости в этом
1дх
W
1,0
0,6
о,б4
ш/ф
j*y
Sp0'4
Mcf
S <f 0,2
l i
\
9
X
Jt
**
X
,x
x'
' v>/
ijy
ШУХ
-
i ( I
-0,8 -0,4
0,4 0,8 Igr
Рис. 51. Характер растекания жидкой
двуокиси титана по поверхности
тугоплавких металлов при плотности
фазового слоя (20— 1,5) -10 г/см2. Условия
растекания:
/—тантал, вакуум, масса капли ЗОНТЗЛЬНОЙ
0,312 г; 2 — молибден ,аргон, масса
капли 0,04 г; 3 — вольфрам, аргон, масса
капли 0,1 г
196
Рис. 52. Схема растекания жидкости
при учете инерционных эффектов
случае проводится без учета инерционных эффектов,
что возможно при выполнении условия [24]:
v « цх/(уЬ2), (4-48)
где к], у— вязкость и плотность жидкости;
х— координата фронта текущего слоя;
8 — толщина текущего слоя.
В случае же растекания жидкой двуокиси титана по
танталу в вакууме это соотношение не выполняется,
поэтому при описании
процесса необходимо
учитывать инерционные
эффекты, влияющие на
геометрию растекающейся
капли [60]. Ввиду того
что «тянущая сила» при
растекании приложена в
непосредственной
близости к контактной линии,
естественно предположить,
что из-за действия инерционных сил слой
растекающейся жидкости будет неравномерным по толщине (рис.52).
Возможность подобного течения жидкости
подтверждается профильной съемкой растекающейся капли [35].
Точное решение кинетического уравнения возможно при
условии, что, начиная с некоторого расстояния,
допустима аппроксимация вида
е-аЛ
«Тянущая» сила растекания
/т = 2ягДа. (4-49)
Сила вязкого сопротивления определяется путем
интегрирования в пределах от 0 до г выражения
2:rtEdg, (4-50)
#с = Л-
I
1-<7 dx 1±2
^
где
г1-*
ti-я
tf
градиент скорости по толщине слоя
на расстоянии \ от центра.
197
Тогда
J+Q dr
fc = 2jiY]—гг- . (4-51)
В условиях квазистационарного течения /m=/c.
После интегрирования полученного выражения в
пределах от 0 до т и от 0 до т и некоторых преобразований
окончательное выражение приобретает вид:
г, х =
2Aa^+*(2?+l)(l+-2-)
Г)Х
1
2+<7
1
т2+*. (4-52)
В случае течения слоя постоянной толщины (^=0)
уравнение (4-52) переходит в уравнение
г = 1^-ух^. (4-53)
Необходимо выяснить, при каких условиях может
осуществляться течение жидкости слоем слабоменяю-
щейся толщины из источника конечной мощности.
Случай, когда не выполняется неравенство (4-48) и
приходится учитывать инерционные силы, описан выше.
Однако он не может быть использован, например, для
описания растекания двуокиси титана по вольфраму и
молибдену при плотности фазового слоя жидкости
(20—1,5)'Ю-3 г/см2, так как в этом случае неравенство
(4-48) выполняется.
Геометрия растекания может определяться
начальными условиями формирования растекающегося слоя,
в зависимости от которых могут осуществляться случаи
течения с линиями тока, показанными на рис. 53.
Воздействие динамического удара, возникающего при
нанесении жидкости на поверхность твердого тела, может
привести к реализации течения жидкости в виде
«цилиндрического блина» (рис. 53, а). При отсутствии
динамического удара возможно возникновение течения по
схеме, изображенной на рис. 53, б, продолжительность
существования которого будет определяться
устойчивостью резервуара. На резервуар действуют силы
тяжести и силы поверхностного натяжения: эти силы
будут приводить к размытию резервуара. Если скорость
размытия достигнет скорости распространения фронта
198
жидкости, то течение перейдет в течение с
равномерным утоньшением слоя. При малых плотностях
фазового слоя действием сил тяжести по сравнению с
действием сил поверхностного натяжения можно пренебречь.
&-^?ЕЕ_Ёж /fe^«
a F
Рис. 53. Схемы растекания жидкости в зависимости от начальных условий
образования текущего слоя:
а —в условиях динамического удара; б —в условиях устойчивости резервуара
Полагая, что резервуар имеет форму шарового
сегмента с краевым углом меньше 60°, получим
rp=vW^-r)°V\ (4_54)
где V— объем резервуара,
гр—радиус основания резервуара.
Для устойчивого резервуара необходимо, чтобы,
начиная с достаточно малого времени, выполнялось
соотношение
drv У/ dr
или
А «"Г (4"55)
d% dx
уо.з (^У'Х т-°.9 « 5 (*^l)°'S х-*-\
В расчетные формулы входит толщина слоя б,
экспериментальное определение которой затруднительно.
Она может быть определена расчетным путем: для
кругового (двумерного) растекания
8J-*
__ (2 — д)т
ZJl/KOH Г
для одномерного
SH*=IIz^, (4-56)
где а — ширина слоя жидкости.
199
По кинематическим данным с использованием
формул (4-52) и (4-53) можно определить движущую силу
растекания. Зная движущую силу растекания, с
помощью ранее описанной методики можно рассчитать
межфазную поверхностную энергию (см. табл. 20).
Результаты, приведенные в табл. 20, довольно
хорошо согласуются между собой, хотя исходные данные
для расчетов получены из разных опытов, что
свидетельствует о правильности выбранного подхода к
рассмотрению кинетики распространения жидкости в
данном интервале плотности фазового слоя.
При изменении плотности фазового слоя жидкости
от 15-Ю-4 до 5-Ю-4 г/см2 растекание носит смешанный
характер и значение показателя изменяется от 0,5 до 1.
При плотности фазового слоя (5—1)-10~4 г/см2
наблюдается растекание жидкости с показателем га, близким
к 1. Причем, если в тех случаях, когда га<1 скорость
растекания со временем убывает, то в указанном
интервале фазового слоя при п=\ скорость растекания
остается в течение всего процесса постоянной.
Подобная же закономерность наблюдалась при растекании
ртути по свинцу [36], когда силы вязкого
сопротивления сосредоточены в слое жидкости, находящемся
между подложкой и окисной пленкой. Не исключена
реализация этого же механизма распространения
жидкой двуокиси титана по поверхности исследованных
тугоплавких металлов при плотности жидкости
(5—1)-10-4 г/см2. Возможны и другие механизмы,
обеспечивающие распространение жидкости на
поверхности твердого тела с постоянной скоростью. Равенство
показателя га единице в квазистационарном режиме при
постоянной величине движущей силы растекания и
постоянстве свойств жидкости означает постоянство силы
сопротивления растеканию, т.е. сопротивление
растеканию не может определяться внутренним трением.
Возможность постоянства силы сопротивления растеканию
показана Ребиндером [37]. При течении слоя
жидкости, толщина которого соизмерима с высотой гребешков
микрорельефа на поверхности твердого тела, что имеет
место в исследуемом случае, возникает дополнительная
сила трения по контуру, величина которой будет
определяться соотношением толщины слоя и размером
неровностей поверхности. Если сила трения намного
больше силы вязкого сопротивления, то суммарная сила
200
сопротивления будет слабо зависеть от расстояния, на
которое распространилась жидкость, и показатель п
будет близок к единице. При плотности фазового слоя
жидкости меньше Ю-4 г/см2 наблюдается
распространение двуокиси титана с показателем п, близким к 0,5.
Такой характер растекания жидкости в значительной
мере обусловлен диффузионными процессами,
посредством которых создается новый микрорельеф поверх-
hytiti ь,мм
Рис. 54. Зависимость высоты поднятия жидкой двуокиси титана
между параллельными пластинами (а) или в капиллярах (б) из
тугоплавких металлов от расстояния между пластинами или
диаметра капилляра соответственно
ности. Аналогичный характер наблюдается для
распространения ртути по поверхности цинка [19].
Дополнительную информацию о движущих силах
растекания возможно получить путем исследования
процесса поднятия жидкой двуокиси титана в
капиллярах, а также в зазоре между параллельными
пластинами из вольфрама, молибдена, ниобия и тантала
[62]. Исследования проводили в вакууме 10~4 мм
рт. ст., диаметр капилляров и величина зазора
составляли от 1 до 5 мм. Капилляры или пластины
приводили в соприкосновение с жидкой окисью, находящейся
в молибденовом тигле при температуре плавления
окиси. Глубина погружения капилляра 2—3 мм;
продолжительность контакта капилляра или пластин с
жидкостью составляла до нескольких десятков секунд.
Высоту поднятия жидкой окиси определяли на
продольном разрезе капилляра после быстрого охлаждения
печи, в которой проводили нагревание. Полученные
результаты (рис. 54) хорошо спрямляются в координа-
201
тах h—1/d, что соответствует теории капиллярного
поднятия:
4аж__г
пк = для капилляров;
VgdK
1 2(Уж-г
ппл = для пластинок.
ygdua
(4-57)
(4-58)
Высота капиллярного поднятия является функцией
свойств расплава и от свойств материала капилляра
или пластинок при равенстве углов смачивания
твердого тела жидкостью зависеть не должна. Однако,
как следует из приведенных ниже данных, при
поднятии жидкой двуокиси титана в капиллярах и между
пластинами из тугоплавких металлов такая
зависимость имеет место, хотя краевые углы смачивания
в обоих случаях равны нулю.
Л(Т, кгс/мм2 . . .
а*т—ж, кгс/мм2 . .
Аа, кгс/мм2 . . ,
а*т—ж, кгс/мм2 . ,
Мо
—640/—800
1600/1420
W
, —450/—590
1980/1910
Nb
—600/—825
1270/1060
Продолжение
Та
—900/—1300
1080/680
Примечание. В числителе приведены данные для поднятия жидкости
в капиллярах, в знаменателе— между пластинами.
Кроме того, если использовать ранее полученные
значения поверхностного натяжения жидкой двуокиси
титана для расчета высоты поднятия, то окажется, что
расчетные значения значительно ниже экспериментальных.
Полученные экспериментальные значения высоты
капиллярного поднятия не являются равновесными. В процессе
длительной выдержки (т=2 ч) в капиллярах из
молибдена и вольфрама, т. е. в тех случаях, когда химическое
взаимодействие в основном ограничивается контактной
областью, наблюдается оттекание жидкой окиси на
высоту, определяемую ее поверхностным натяжением.
Проведение аналогичных опытов в капиллярах из ниобия и
тантала невозможно из-за сильного химического
взаимодействия.
202
Для расчетов в условиях осуществленных
экспериментов можно использовать уравнения, предложенные
Елютиным с сотр.;
"к.неравн= ~ » (4-59)
ygdK
„2К-г-<У-ж) (4.ш
"пл.неравн— " » V* uu/
Jgdun
где dK— диаметр капилляра;
dUJl— расстояние между пластинами.
Значения движущих сил растекания и динамической
межфазной энергии, рассчитанные с помощью этих
уравнений, представлены выше. Из этих данных следует, что
расчетные значения движущей силы растекания и
динамической межфазной энергии, полученные на основе
экспериментов по капиллярному поднятию,
удовлетворительно совпадают с расчетными данными,
полученными на основе исследований кинетики растекания
(табл. 20).
5. СМАЧИВАНИЕ
ВЫСОКОТЕМПЕРАТУРНЫХ МАТЕРИАЛОВ
жидкими тугоплавкими окислами
И ИХ РАСПЛАВАМИ
Бартлет и Холл [34] изучали смачивание вольфрама,
тантала и др., перечисленных в табл. 21 материалов,
жидкими окислами алюминия и бериллия в вакууме,
атмосфере водорода, окиси углерода или азота.
Результаты этих исследований, проведенных в условиях
совместного нагрева окисла и подложки (табл. 21),
свидетельствуют о том, что жидкая окись алюминия на вольфраме
образует краевой угол смачивания порядка 50° в
вакууме, 35° в атмосфере азота, 16—18° в атмосфере окиси
углерода и водорода. Отмеченное снижение краевого угла
смачивания может произойти вследствие адсорбционного
понижения поверхностного натяжения жидкой окись
алюминия, что, как показано в гл. 2, получает заметное
развитие в атмосфере окиси углерода и азота. Нельзя
не учитывать также возможности протекания
межфазных реакций с участием окружающего газа. Об этом сви-
203
Таблица 21
Смачивание поверхности тугоплавких материалов
жидкими окислами алюминия и бериллия [34]
Материал
подложки
Вакуум
Водород
Окись
углерода
Азот
Окись алюминия
W
Та
90 % Та—
10 % W
ТаС
4ТаС — ZrC
ZrC
TiN
ZrB2
51/49
38/12
42/—
7/5
17/16
43/35
40/33
32/22
29/23
13/13
16/13
21/12
22/18
H
н
36/23
28/21
15/15
Н
21/13
37/35
Н
н
41/30
23/16
10/9
36/31
17/11
W
Та
90 % Та —
10% W
Окись бериллия
0/0
0/0
о/о
о/о
11/0
16/11
12/12
0/0
12/0
20/20
0/0
0/0
Примечания: 1. В числителе приведены значения 0 , в
знаменателе -е°
кон
2. Н — данные недостоверны.
3. ZrB2, ZrCuTiN смачиваются жидкой окисью бериллия с краевым углом
в, равным нулю.
детельствует характер смачивания тантала жидкой
окисью алюминия: начальный краевой угол смачивания,
равный 38°, по Бартлету и Холлу, заметно снижается в
процессе эксперимента, продолжительность которого
составляет 30 мин.
Жидкая окись бериллия, по данным Бартлета и
Холла [34], полностью смачивает поверхность вольфрама и
тантала как в вакууме, так и в атмосфере водорода.
Августинникссотр. [40] изучал смачивание
тугоплавких металлов жидкой окисью алюминия при совместном
нагреве в вакууме Ю-4 мм рт. ст. или в атмосфере
инертных газов — гелия или аргона. В качестве подложек
использовали компактные листовые или прессованные из
204
порошков металлы, которые перед опытом полировали и-
очищали^от поверхностных загрязнений. Окись алюминия
имела чистоту 99,99%. Результаты опытов иллюстрирует
рис. 55. Краевой угол смачивания молибдена снижается
от 52 до 36° в течение 40 мин и затем остается
постоянным. Краевой угол смачивания ниобия и тантала также
снижается во время изотермической выдержки. В случае
тантала он достигает нулевого значения после 50-мин
выдержки, что
свидетельствует о межфазном
взаимодействии между
жидкой окисью алюминия и
тугоплавкими металлами.
Следует заметить, что
данные, полученные в этой
работе, не совпадают с
данными, полученными
ранее в работах [34, 35].
Значения краевого
угла смачивания жидкой
окисью алюминия,
полученные в работе [35],
ниже значений, полученных
Августинником с сотр.
Это' вызвано более интенсивным протеканием
межфазной реакции из-за применения метода исследования с
раздельным нагревом окиси и подложки, причем нельзя
исключить и влияние динамического удара, который
возникает при соприкосновении капли с подложкой и
действует в сторону уменьшения краевого угла. Авторы
[35] так же, как и Августинник с сотр., наблюдали
достаточно интенсивное взаимодействие жидкой окиси
алюминия с танталом и ниобием во время изотермической
выдержки.
Митин с сотр. [39, 58—66] изучал смачивание
вольфрама, молибдена и ниобия жидкой окисью алюминия,
а также бинарными расплавами окиси алюминия с ВеО,
MgO, Сг203, СаО, V205, Zr02, Si02, В203, ТЮ2 и Nb205
при содержании добавки от 5 до 80—100%. В опытах
использовали предварительно протравленные плавленые
металлы. Методика заключалась в совместном нагреве
образцов металла и окисла вплоть до расплавления
последнего в атмосфере гелия. Измерения краевого угла
смачивания начинали сразу же после формирования кап-
Рис. 55. Временная зависимость
краевого угла смачивания тугоплавких
металлов жидкой окисью алюминия [40]:
/ — ниобии; 2 — молибден; 3 — тантал
205
е\
О Ю 20 30 40 50 60 70 80 90
А1203 МеО, % —*-
Рис. 56. Зависимость краевого угла смачивания вольфрама (а), молибдена
на основе А12Оэ от концентрации в расплаве окисла-добавки
ли и продолжали в течение 30 мин. Изучение кинетики
растекания показало, что заметное изменение краевого
угла смачивания происходит в первые 5—7 мин, но при
дальнейшей выдержке он остается практически
постоянным. Авторы цитированных работ не обнаружили раз-
206
личия в смачивании жидкой окисью алюминия изученных
тугоплавких металлов: для всех металлов краевой угол
смачивания составляет 33—35°, что лежит в пределах
ошибки опыта.
При смачивании поверхности тугоплавких металлов
двойными окисными расплавами наблюдается общая
закономерность, заключающаяся в том, что краевые углы
смачивания возрастают при добавлении к окиси
алюминия любого из перечисленных выше окислов, проходят
через максимум и затем падают до уровня, который
определяется природой окисла-добавки (рис. 56). Для
многих из исследованных окисных систем не представляется
возможным получить зависимость краевого угла
смачивания от состава окисной фазы во всем интервале
концентраций от 0 до 100% окиси-добавки. Это объясняется
или интенсивным химическим взаимодействием,
например, для системы А1203—MgO, или тем, что при
достаточно больших содержаниях окиси-добавки наступает
состояние полного смачивания металла (системы А1203—
Сг203; А1203—Ti02; А1203—V2O5). В некоторых системах
например, А1203—Zr02, A1203—BeO, достигается второй
50
40
30
20
10
ВеО
^Г^МдО%у\
14 \Ч°
1 ^ч>£
Ysi02
1 F 1 L-
6
4-VvCaO
•
1 L_
A1203
10 20 30 40 50 60
MeO,%
(б) и ниобия (в) окисными расплавами
0
А1203
v2o5
/О
20 31
Nb205
CuO?% —
Рис. 57. Зависимость краевого угла
смачивания ниобия расплавами
А1?03—Nb205 (/) и меди
расплавами V2O5—CuO (2) от концентрации
в расплаве окисла
металла-подложки
максимум краевого угла смачивания, т. е. наблюдается
симметричный эффект изменения краевого угла
смачивания в зависимости от концентрации добавки. Эффект
207
проявляется и в тех случаях, когда к окиси алюминия
добавляется окисел металла-подложки, например пяти-
окиси ниобия при смачивании ниобия (рис. 57).
Изучение смачивания поверхности тугоплавких металлов
бинарными расплавами на основе других окислов
(см. рис. 57) подтвердило универсальность этой
закономерности, которая не зависит ни от кислотности или
основности окисла, ни от вида фазовой диаграммы. Дейст-
10 20 30 40 50
МеО,% -*-
Рис. 58. Зависимость межфазной
поверхностной энергии при
смачивании вольфрама (а), молибдена
(б) и ниобия (в) окисными
расплавами на основе Al203 от
концентрации окисла-добавки
вительно, изучены окислы-добавки MgO и СаО —
основные окислы, Сг203 — амфотерный окисел, и V2O5 и
Si02 — кислотные окислы.
Исследованные окисные системы охватывают
широкий круг диаграмм состояния: от непрерывного ряда
твердых и жидких растворов (А1203 — Сг203) до
диаграмм с образованием большого числа химических
соединений (А1203—ВеО; А1203—СаО). Максимумы и
минимумы на кривых смачивания никак не связаны с
особыми точками диаграммы состояния. Не имеет значения
и соотношение свободных энергий основного и
добавляемого окислов, а также окислов, образующих расплав,
и окисла металла-подложки. Не может быть объяснено
это явление и изменением поверхностного натяжения
расплава при добавлении другого окисла, так как, согласно
уравнению Юнга — Неймана, краевой угол смачивания
растет при увеличении поверхностного натяжения
жидкости. В действительности, в некоторых случаях
(см. рис. 56) краевой угол увеличивается, а поверхност-
208
ное натяжение расплавов уменьшается (см. гл. 2);
величина поверхностной энергии твердого тела остается
постоянной.
Обнаруженный характер изменения краевых углов
смачивания можно, однако, понять с позиций изменения
межфазной энергии и ее влияния на процесс смачивания.
На рис. 58 приведена зависимость свободной
поверхностной энергии на границе тугоплавкий металл — окисный
расплав от состава расплава. Значения межфазной
энергии рассчитывали по уравнению Юнга — Неймана, а по-
поверхностную энергию твердого тела — по методике,
изложенной в работе [35]. Межфазная энергия имеет
максимум при содержании добавляемого окисла от 10 до 20%,
и изменение ее достаточно велико (до 200 мДж/м2).
В свою очередь подобное изменение межфазной
поверхностной энергии может быть обусловлено особенностями
адсорбционных процессов на межфазной границе.
6. ВЛИЯНИЕ
РЕАКЦИОННО-АКТИВНЫХ ДОБАВОК
НА СМАЧИВАНИЕ ТУГОПЛАВКИХ МЕТАЛЛОВ
ЖИДКОЙ ОКИСЬЮ АЛЮМИНИЯ
Качество окисных покрытий на тугоплавких металлах
во многом определяется характером смачивания
металла окисным расплавом и величиной работы адгезии.
Поэтому особое значение имеет выявление путей
воздействия на характер процессов смачивания в желаемом
направлении. Эффективным путем улучшения смачивания,
что обеспечивает получение покрытий более высокого
качества, являются, очевидно, способы, вызывающие
снижение межфазной энергии границы раздела
тугоплавкий металл — окисный расплав. Существенное влияние
на межфазную энергию оказывает изменение
химического потенциала на соответствующей границе раздела фаз,
поэтому, регулируя интенсивность химического
взаимодействия, можно изменять величину межфазной энергии
и, следовательно, управлять процессом смачивания.
Снижение межфазной энергии будет определяться
изменением свободной энергии при протекании химической
реакции на границе раздела фаз.
Естественным способом повышения интенсивности
химического взаимодействия на границе раздела является
14—894
209
введение, по крайней мере, в одну из фаз веществ,
химически активных к другой фазе. Практически проще
вводить добавки в наносимый расплав, чем в
покрываемое изделия, хотя второй способ также допустим,
особенно если применять легирование приповерхностного
слоя изделия, например посредством
химико-термической обработки.
Результаты исследования влияния различных
реакционно-активных добавок к окиси алюминия на смачивание
тугоплавких металлов — вольфрама, молибдена и
ниобия— окисными расплавами иллюстрируют рис. 59—61.
В качестве реакционно-активных добавок были выбраны
различные карбиды, нитриды, бориды, силициды, а также
чистый бор.
Для исследования влияния нитридных добавок на
смачивание вольфрама, молибдена и ниобия окисью
алюминия были выбраны ZrN — нитрид переходного
металла с преобладающей долей металлической связи, A1N —
нитрид непереходного металла с меньшей долей
металлической связи и со значительной долей ионной связи и
BN — нитрид с ковалентной связью. Эти нитриды
охватывают типы связей, существующие не только в
нитридах, но и в большинстве других реакционно-активных
добавок.
Система А1203—A1N интересна также тем, что в ней
в отличие от двойных окисных систем одинаковые
катионы, но различные анионы.
Выбор в качестве реакционно-активных добавок
вышеперечисленных нитридов основан на том, что, хотя
термодинамический потенциал образования нитридов
молибдена и вольфрама при температуре плавления окиси
алюминия положителен, тем не менее, следует ожидать
протекания химической реакции, так как образующиеся
при этом Mo2N и W2N обладают высокой летучестью
(давление паров составляет 1 мм рт. ст. уже при
температурах 319 и 344° С соответственно для Mo2N и W2N)
и удаляются из зоны реакции.
При исследовании влияния реакционно-активных
добавок на процесс смачивания применяли совместный
нагрев металла и окисла. Измерения краевых углов
проводили обычно в течение 30 мин, в отдельных случаях
выдержку расплава на металле увеличивали до нескольких
часов. Примерно в течение 2—3 мин после расплавления
и формирования капли происходит уменьшение краево-
210
го угла смачивания, связанное, по-видимому, с
возрастанием интенсивности химического взаимодействия.
В дальнейшем краевой угол либо остается в течение
всего времени выдержки постоянным (так, например, в
системе AI2O3—BN краевой угол практически не меняется
в течение 3 ч), либо несколько возрастает, вероятно, из-
за того, что в результате химического взаимодействия
снижается содержание нитрида в расплаве.
Полученные зависимости краевых углов смачивания
от содержания добавляемого нитрида приведены на
рис. 59. При весьма малых содержаниях нитрида имеет
место некоторое увеличение краевого угла смачивания,
который проходит через максимум, а затем заметно
уменьшается, причем до значений, которые меньше
величины краевого угла смачивания чистой окиси
алюминия на тех же металлах, т. е. характер зависимости
краевой угол — состав для систем окисел — нитрид такой же,
как и для бинарных окисных расплавов. Величину
межфазной энергии границы раздела тугоплавкий металл —
оксинитридный расплав можно представить как сумму,
состоящую из двух слагаемых, а именно: а$!__ж в
отсутствие химического взаимодействия плюс Дат-Ж,
обусловленная протеканием химической реакции на границе
раздела фаз. Величина последнего слагаемого определяется
уравнением Жуховицкого (4-12). Из-за отсутствия
методов определения или расчета входящего в это уравнение
числа поверхностного переноса М не представляется
возможным количественно оценить величину Аат-ж. Тем
не менее, можно сделать качественный вывод о
характере изменения Дат-Ж в зависимости от концентрации
реакционно-активной добавки в расплаве: абсолютная
величина Аат-ж будет монотонно возрастать с увеличением
концентрации добавки до какого-то определенного
предела. Следовательно, добавление к <т$!_ж изменяющегося
таким образом слагаемого не окажет влияния на общий
вид кривой в координатах межфазная энергия — состав
расплава (а следовательно, и кривой в координатах
краевой угол — состав), а только приведет к изменению
положения характеристических точек, например изменит
положение максимума. При этом максимум краевого
угла должен смещаться влево, т. е. в сторону меньших
концентраций добавки, что и наблюдается на самом
деле. Так, например, при смачивании тугоплавких металлов
двойными окисными расплавами положение первого
14*
211
Рис. 59. Зависимость краевого угла смачивания вольфрама (а), молибдена (б)
и ниобия (в) жидкой окисью алюминия от концентрации в ней нитридов бора,
алюминия и цирконии
212
максимума краевого угла соответствует 5—20%
добавляемого окисла. В случае же систем окисел — нитрид
максимум краевого угла наблюдается при концентрации
добавки 1—3%, а иногда при концентрации, равной
десятым долям процента. Образование максимума
краевого угла смачивания наиболее отчетливо выражено для
в° в°
0 12 3 4 5 6ZrSiz°/o
Рис. 60. Зависимость краевого угла смачивания вольфрама, молибдена и
ниобия жидкой окисью алюминия от концентрации в ней карбида циркония (а),
диборида циркония (б) и дисилицида циркония (в)
системы А1203—ZrN, далее следуют системы А1203—BN
и А120з—A1N.
При достаточно больших концентрациях
добавляемого нитрида (~5%) краевой угол смачивания
уменьшается до значений ~10° и менее, т. е. достигается
хорошее смачивание и работа адгезии становится
практически равной когезии жидкости. Действительно, cos 10°=
=0,985 и, следовательно, работа адгезии составляет
99,2% от работы когезии. Наибольший эффект
улучшения смачивания дают добавки нитрида бора, далее
следуют нитриды циркония и алюминия. Среди
исследованных металлов эффект улучшения смачиваемости за счет
нитридных добавок примерно одинаков в случае молиб-
дена и вольфрама, а в случае ниобия несколько ниже,
хотя из всех этих металлов образование нитрида ниобия
сопровождается большей убылью свободной энергии. Это
объясняется, вероятно, тем, что нитриды молибдена и
вольфрама более летучи при температуре опыта
плавления окиси алюминия, чем нитрид ниобия, что и
обусловливает более интенсивное
0°i z 1 химическое
взаимодействие в случае молибдена
и вольфрама.
При изучении влияния
нитридных добавок на
смачивание тугоплавких
металлов двойными окис-
ными расплавами (90%
А120з+Ю% Cr203-BN)
было обнаружено, что
влияние нитрида на
краевой угол смачивания в
этой системе такое же,
как и в случае одноком-
понентного окисного рас-
2 84С;% плава, т. е. краевой угол
смачивания проходит
через максимум, а затем
имеет место значительное
улучшение смачивания.
Рассмотренные выше
особенности смачивания тугоплавких металлов окисны-
ми расплавами с нитридными добавками сохраняются
также, если в качестве реакционно-активных добавок
применяются карбиды, бориды, силициды. Во всех
случаях при введении добавки краевой угол смачивания
сначала незначительно увеличивается, достигает
максимума, а затем уменьшается: при содержании добавки
порядка нескольких процентов наступает полное
смачивание.
На рис. 60 приведены зависимости краевого угла
смачивания вольфрама, молибдена и ниобия расплавами
окиси алюминия с добавками карбида, борида и
силицида циркония. Наиболее эффективно смачивание
улучшается при добавлении борида и карбида, далее следуют
добавки нитрида и, наконец, силицида циркония.
Поскольку эффект улучшения смачивания при добавке к
Рис. 61. Зависимость краевого угла
смачивания вольфрама, молибдена и
ниобия жидкой окисью алюминия от
концентрации в ней карбида бора
214
окиси алюминия боридов и карбидов обусловлен, по всей
вероятности, действием бора и углерода, интересно
оценить их совместное действие. В связи с этим было
изучено влияние добавок В4С на смачивание тугоплавких
металлов расплавами А1203 и установлено, что В4С
является наиболее сильнодействующей
реакционно-способной добавкой: уже при содержании 1% В4С в окиси
алюминия имеет место полное смачивание этим расплавом
поверхности молибдена, при содержании 2% В4С —
поверхности ниобия и при содержании 2,5% В4С —
поверхности вольфрама (рис.61).
Выше было показано, что весьма эффективной
добавкой является нитрид бора. Эксперименты по применению
чистого бора в качестве добавки, улучшающей
смачивание тугоплавких металлов окисными расплавами, дали
положительный результат уже при весьма малом
содержании бора (~1%).
Наиболее эффективными добавками, улучшающими
смачивание тугоплавких металлов окисными расплавами
на основе окиси алюминия, являются соединения бора,
углерода, азота и кремния между собой.
7. ФИЗИКО-ХИМИЧЕСКИЕ ПРОЦЕССЫ
НА МЕЖФАЗНОИ ГРАНИЦЕ
Для понимания процессов, происходящих на
межфазной границе, при смачивании тугоплавких металлов
сложными расплавами, необходимо рассмотреть
возможный характер адсорбции на этой границе.
Общее уравнение для адсорбции из растворов на
поверхности твердых тел имеет вид [41]:
*2*1 = 72 ?1 с НТ 1Л 2 2) VI UJ ^ -4egj
х2 х\ Yi 72
где х—концентрация компонента раствора;
у — коэффициент активности;
ц°— химический потенциал в стандартном
состоянии.
Индексы 1 и 2 относятся соответственно к первому
и второму компонентам в поверхностном растворе,
значения со штрихами относятся к компонентам в объеме.
Величины, входящие в данную формулу, обозначим
215
следующим образом: y2/y\—Kv— коэффициент,
учитывающий влияние молекулярных полей объемного
раствора, аналогично 72 /у\=Ка учитывает влияние
молекулярных полей поверхностного раствора и ехр | U\i°2'—
— Vb) — (НчГ — Iх?)] \=К* — влияние силового поля
твердого тела.
Для изотермы адсорбции из двойных растворов
справедливо выражение
. = кч (4 62)
TJXeK = ^Ks.
Совместное решение уравнений (4-61) и (4-62)
позволяет определить состав поверхностного раствора,
поверхностную концентрацию каждого из компонентов,
гиббсовскую адсорбцию и т. д. Однако в эти уравнения
входят коэффициенты активностей компонентов в
объемном и поверхностном растворах, что значительно
затрудняет их практическое применение. Поэтому
представляется целесообразным получить выражение, описывающее
адсорбцию, но содержащее вместо коэффициентов
активности другие величины.
Задача упрощается, если рассматривать отдельно
влияние твердой поверхности (Ks) и влияние
соотношения молекулярных полей объемного и поверхностного
раствора (Kv/Ka)-
Для соотношения Kv/Ka Семенченко [42] на основе
приближенного выражения для коэффициентов
активности у=е~е!кт дает выражение:
Kv/Ka = e
kT
(4-63)
В этом выражении каждая из разностей в
отдельности представляет приближенное значение работы
перехода молекулы из объема на поверхность. Эту работу
будем определять как разность работ удаления одной
молекулы из объема и из поверхностного слоя расплава,
приняв, что окисный расплав является регулярным
раствором. Жуховицкий [43] получил следующие
выражения для работы удаления молекулы из;
216
поверхностного слоя регулярного раствора
(4-64)
iv г + Р Д , ,Х9 п Д
объема регулярного раствора
"i = f («„-4 А);
(4-65)
^=f («fc-afA).
где (/— энергия удаления одной молекулы;
г—координационное число молекулы в
поверхностном слое;
Р—число связей молекулы в поверхностном
слое с молекулами в объеме раствора;
q—координационное число молекул в объеме.
Д=ец+е22—2б12;
еи, е12, е22 — энергии взаимодействия между
молекулами первого, первого и второго, второго
компонентов соответственно.
Подставляя (4-64) и (4-65) в (4-63) с учетом того,
что ЛГ1 = 1—х2 и х[ =1—х'2, получим
- А \2х2 + (Р -q)x2- '-^f13
Анализ этого выражения показывает, что при
(4-66)
Хо =
1 = х9 отношение — = 1,
2 \ Д
т. е. без учета влияния Ks данной концентрации второго
компонента соответствует азеотропная точка.
Перепишем (4-66) в виде
*v . _._ А
= ехр
Ка , kT
(r + P-q)x"2-
(4-67)
217
Пусть *2>*2, тогда (г + Я — q) <(r—+ P — q J.
Для того чтобы Kv/Ka было больше единицы, нужно,
чтобы показатель экспоненты был больше нуля,
следовательно, если Д>0, то необходимо выполнение
неравенства х2<,х"2\ если Д<0, тогда х2>х\ . Отрицательная
адсорбция второго компонента наблюдается при значениях
концентрации Х2<х1 при Д<0 и *2>*2 при Д>0.
Следовательно, характер адсорбции будет
определяться знаком теплоты смешения данного раствора.
В случае отрицательных теплот смешения (Д<0) при
добавлении второго компонента сначала имеет место его
отрицательная адсорбция, при концентрации х"2
наблюдается адсорбционная азеотропия, и при больших
концентрациях наблюдается положительная адсорбция
второго компонента. Если теплота смешения данного
раствора положительная (Д>0), то знак адсорбции
меняется на противоположный. Величина К8 не зависит от
состава раствора, и показатель экспоненты в выражении,
определяемый соответствующими разностями
стандартных химических потенциалов компонентов в объемном и
поверхностном растворах, сохраняет знак во всем
интервале концентраций. Физически это означает, что все
время будет иметь место положительная адсорбция того
компонента, изменение свободной энергии реакции
которого с твердым телом будет более отрицательно. Если
Ks<Kv/Ka или Ks&Kv/Ka, характер зависимости
адсорбции от концентрации, обусловленный влиянием
отношения Kv/Ka, не изменится, Ks будет оказывать
только количественное влияние, что приведет к сужению
области отрицательной адсорбции химически более
активного по отношению к твердому телу компонента. К
сожалению, для анализа конкретных случаев смачивания
в высокотемпературных системах отсутствуют
необходимые экспериментальные данные по теплотам смешения
исследуемых расплавов. Есть определенные основания
считать, что энергия связи разнородных молекул ei2
больше энергии связи однородных молекул ец и егг- Этот
вывод следует из измерений коэффициента термического
расширения окисных композиций [44]: как правило,
зависимость коэффициента термического расширения от
состава имеет минимум при каком-то содержании добав-
218
ляемого окисла, что свидетельствует об увеличении
связи между разнородными молекулами.
Выше рассматривались регулярные растворы, однако
характер адсорбции, обусловленный степенью
взаимодействия одноименных и разноименных молекул в
расплаве, не изменится и в
случае отклонения окисных
расплавов от регулярных
растворов.
Изложенные представления
об особенности адсорбции на
межфазной границе, вероятно,
могут быть распространены не
только на смачивание
тугоплавких металлов окисными
расплавами, но и на другие
ионные расплавы.
Действительно, подобное явление
наблюдается при смачивании
графита расплавами NaCl—
КС1 (рис. 62).
is
С помощью коэффициента К=-^- Ks можно вычис-
20 40 60
КС1,% —
80 /00
Рис. 62. Зависимость краевого
угла смачивания графита
расплавами NaCl—KCI в
зависимости от состава жидкой фазы
лить гиббсову адсорбцию:
Ка
"**)
i + C^i-'K
(4-68)
где атг— предельная концентрация плотного монослоя
второго компонента am2 = l/com2, где
соШ2—площадь, приходящаяся на одну молекулу второго
компонента в плотном монослое;
p2,i — коэффициент поверхностного вытеснения
второго и первого компонентов (P2,i=c°m2/comi).
Исходя из уравнения (4-68), можно найти положение
минимума адсорбции, а следовательно, и максимума
межфазной энергии и краевого угла смачивания:
df2
dx9
дх.
дК
дК
дх2
(4-69)
Если рассматривать положение минимума адсорбции
для достаточно большого количества систем, то можно
осуществить усреднение в данной формуле. При этом
дК/дх2 обратится в нуль и будет получена зависимость
219
положения экстремума от какого-либо параметра
системы. Такая зависимость будет выполняться тем точнее,
чем слабее К отклоняется от единицы. Таким образом',
если
дх.>
= *-,(*■
i-2x2 + 4 + Kfi214 0
то концентрация, отвечающая экстремуму, равна
1 1
ЛГ2Э —
1 + (ATPai)
1/2
1 +
К®„
1/2
(4-70)
(4-71)
МеО,%
Аналогичная
формула приводится в
работе [41] для случая
независимости К от х2.
Для случая
смачивания тугоплавких
металлов двойными окис-
ными расплавами на
основе окиси
алюминия зависимость х2 от
сот2 представлена на
рис. 63.
Экстремальные точки достаточно
хорошо ложатся на
кривые, рассчитанные
по уравнению (4-71).
Оклонения
наблюдаются только для
систем А1203—Si02 и А1203—ТЮ2, что связано с
повышенной склонностью двуокиси кремния и двуокиси титана к
комплексообразованию, в результате чего величина сот2
возрастает. Для каждого из тугоплавких металлов
получается своя кривая, что определяется различием их
силовых полей, выраженных через /Cs.
50 M/f
Рис. 63. Зависимость экстремальной
концентрации добавки к жидкой А1203 от
размерного фактора при смачивании
тугоплавких металлов окисными расплавами
на основе А1?Оз
8. ВЗАИМОДЕЙСТВИЕ ЖИДКИХ ОКИСЛОВ
С ТУГОПЛАВКИМИ МЕТАЛЛАМИ
Взаимодействие жидких окислов с твердыми
металлами начинается непосредственно при смачивании.
Наряду с процессом химического взаимодействия жидкости
220
с твердым телом может происходить процесс частичной
диссоциации жидкой фазы. При рентгеноструктурном
анализе окислов ванадия, молибдена, вольфрама и
титана после опытов по смачиванию и растеканию, когда
время взаимодействия не превышало существенным
образом времени растекания, обнаружены как высшие, так
и промежуточные окислы этих металлов. При
растекании пятиокиси ниобия по молибдену обнаружены NbO и
Мо4Оц, а на рентгенограммах продуктов после
растекания жидкой пятиокиси тантала по вольфраму и
молибдену обнаружены неидентифицированные
дифракционные пики. Металлографический анализ, однако, не
показал наличия переходных зон в металле подложки после
смачивания его окислами: микротвердность металла —
подложки в зоне контакта такая же, как и в объеме
металла [35].
Исследование взаимодействия жидких окислов
алюминия и бериллия с тугоплавкими металлами и
соединениями во время смачивания (30 мин) выполнено Барт-
летом и Холлом [34]. Результаты исследования,
представленные в табл. 22 и 23, показывают, что вольфрам и
нитрид титана не реагируют заметным образом ни с
одним из изученных окислов.
Тантал и сплав 90% Та — 10% W реагируют с окисью
алюминия с образованием изоморфной рутилу фазы
ТаАЮ4. Согласно рентгеновским данным, эта фаза
является главной составляющей в нейтральной атмосфере,
но ее количество резко уменьшается в атмосфере
водорода.
Обнаружена также переходная зона между окисью
алюминия и сплавом 90% Та — 10% W, которая
особенно заметна после смачивания в вакууме. Дифракционная
картина после контакта окиси бериллия с танталом или
сплавом 90% Та — 10% W является весьма сложной.
Наиболее сильными оказались линии вещества со
структурой типа Ta02F. Вполне возможно, что наблюдаемая
структура представляет собой дефектную структуру
окисла ТаОз- В атмосфере окиси углерода основной продукт
реакции — карбид тантала. Продукты реакции,
образующиеся при контакте окислов алюминия и бериллия с ТаС,
подобны тем, которые образуются при их
взаимодействии с чистым танталом или сплавом 90% Та — 10% W.
При контакте окислов с карбидом циркония образуется
Zr02 как моноклинной, так и кубической формы, соотно-
221
Таблица 22
Конденсированные продукты взаимодействия жидкой окиси алюминия с высокотемпературными материалами [34]
Вакуум или инертный газ
Азот
Водород
Окись углерода
ТаАЮ/ (гл. с)
НДП (м.с)
ТаА104 (гл. с)
НДП (м.с)
ZrO *2 (гл- с)
Та2С (гл. с)
ТаА104 (м.с)
НДП (м.с)
ТаА104 (м. с)
НДП (м.с)
AITi (?) (гл. с) I AlTi3 (?) (ел.)
НДП (ел.) НДП (ел.)
Zr02 (гл. с) Zr02 (гл. с)
ZrB (ел.;
Примечание, гл. с — главная составляющая; м. с -
дифракционные пики.
ТаАЮ4 (м. с)
НДП (м.с)
Ta(W)N (м.с)
НДП (м.с)
Zr02 (гл. с)
НДП (м.с)
Та2С (гл. с)
НДП (м.с)
НДП
Не обнаружены
ТаА104 (м. с)
НДП (ел.)
ТаА104 (м. с)
НДП (ел.)
ZrC>23 (м.с)
Та2С*3 (ел.)
НДП (м.с)
Та (м. с)
Zr (м. с)
Не обнаружен
Zr02 (гл. с)
меньшая составляющая;
ТаС (гл. с)
ТаАЮ4 (ел.)
НДП (ел.)
ТаС (гл. с)
НДП (м.с)
Zr02 (м.с)
Не обнаружена
TiC (ел.)
ZrB (м. с)
ел. — следы; НДП — неидентифицируемые
*' Структура изоморфна рутилу и предположительно имеет состав ТаАЮ4.
*2 Во всех случаях Zr02 присутствует как в кубической, так и в моноклинной форме.
*3 Наблюдаются также пики 100% интенсивности ТаН и ZrH.
Таблица 23
Конденсированные продукты взаимодействия жидкой окиси бериллия с высокотемпературными материалами [34]
Материал
подложки
Вольфрам
Тантал
90% Та—
10 % W
ZrC
ТаС
4ТаС—
ZrC
TiN
ZrB2
Вакуум или инертный газ
Азот
Не обнаружены
ТаО?(гл. с)
НДП (м.с.)
—
Zr02 (гл. с)
(м.с)*2
НДП (гл. с)
Та205 (м.с)
ТаОз (м. с)
Та2С (м. с)
Та2С (м. с)
Ti02 (сл.)
Zr02 (м.с)
Та02 (гл. с)
НДП (м.с)
Та02 (гл. с)
Н. П. (м. с)
Zr02 (гл. с)
(м.с)*2
Та03 (гл. с)
TaN (м.с)
— ;
Не обнаруж
Zr02 (м.с)
Водород
НДП
Та02 (гл. с)
Н. П. (м. с)
ТаОз (м.с)
Та205 (м. с)
НДП (м.с)
Zr02 (гл. с)
Та03 (м. с)
Та2С (м. с)
Та205 (м.с)
НДП (м.с)
Та2С (м. с)
вны
Zr02 (м.с)
Окись углерода
НДП
ТаС (гл. с)
НДП (гл. с)
ТаС (гл. с)
НДП (гл. с)
Zr02 (гл. с)
Та205 (м. с)
Та03 (м.с)
Zr02 (сл.)
TiC (гл. с)
Zr02 (гл. с)
Примечание. Обозначения см. в примечании к табл. 22.
*1 Структура идентична структуре Ta02F.
ю *2 Структура изоморфна структуре Na2BeF.
шение между которыми колеблется для различных
условий
ZrC (т) + А1203 (ж) -> Zr02 (т) + 2A1 (г) + СО.
Жидкая окись алюминия взаимодействует с боридом
циркония также с образованием Zr02:
3ZrB2 (т) + 5А1203 (ж) -* 3Zr02 (т) + ЗВ203 (г) + 10A1 (г).
Вероятно, Вг03 испаряется при температуре
эксперимента, так как она не была обнаружена в оставшейся
капле.
Заметного взаимодействия молибдена с жидкой
окисью алюминия Августинник и его сотр. не
обнаружили [40]. На металлографическом шлифе наблюдается
четкая граница раздела между металлической и окисной
фазами, микротвердость металла у границы раздела и
вдали от нее одинакова. Тантал же заметно реагирует с
жидкой окисью алюминия. В окисной фазе после
часовой выдержки образуются, по крайней мере, три
соединения, отличающиеся друг от друга по микротвердости.
Образование новых фаз происходит на поверхности
раздела АЬОз (ж)/Та (т) с последующей их диффузией в
тантал. Одной из стадий механизма взаимодействия, по
предположению Августинника с сотр., является
образование летучих субокислов тантала, так как, согласно
термодинамическим расчетам, образование Та205 в
условиях опыта мало вероятно. Наблюдаемые новые фазы,
по-видимому, представляют собой продукты реакции
субокислов тантала с окисью алюминия.
Взаимодействие между окисью алюминия и ниобием
протекает с образованием структур, напоминающих
эвтектические сплавы, причем характер взаимодействия
окиси алюминия с компактным и пористым металлом
одинаковый: проникновение окисла в металл происходит
по границам зерен. Механизм взаимодействия
рассматривается на основе модели, предложенной Юдиным
с сотр. [46]: процесс реализуется за счет кислорода,
выделяющегося при диссоциативном испарении окиси
алюминия. Процесс этот идет до тех пор, пока давление
кислорода не достигнет равновесного, отвечающего
образованию NbO. Вероятно, этот окисел и дает фазу
эвтектического вида с А1203 или субокислами алюминия,
возникающими при ее диссоциации.
224
При взаимодействии Si02 с танталом образуются
фазы с разной микротвердостью [45]:
Фаза Нм, кгс/мм2
Светлая (тантал) 160—230j
Светло-серая 290
Серая I 530
Серая II 960—1050
Микротвердость Si02 после переплавки в аргоне
составляет 825—860 кгс/мм2.
Взаимодействие тантала с расплавленной Si02 идет
с выделением газообразных продуктов. Это
подтверждается различием в величинах краевого угла смачивания
в вакууме и в аргоне и ухудшением вакуума при
повышении температуры выше 2000 К. Авторы считают, что
в системе Та—Si02 в интервале температур 2000—
2300 К определяющей реакцией является реакция
2Та (T)+5Si02 (ж)-^Та205 (»0+5SiO (г). Но эта
реакция не конечная, так как на рентгенограммах
расплава и металла после изотермической выдержки линии,
характерные для Та2Об, не обнаружены. Очевидно,
образующаяся Та2Об вступает в реакцию с Si02 с
образованием силикатов тантала. Здесь следует отметить работу
Армстронга и др. [47], которые, исследуя взаимодействие
тантала с твердым Si02 при температурах до 1900 К,
зафиксировали образование в этой системе нескольких фаз
и рентгенографически выявили среди них Ta2Os- При
взаимодействии расплавленной Si02 с вольфрамом и
молибденом продукты взаимодействия представляют собой
прозрачное кварцевое стекло. Микроскопический анализ
не выявил изменений ни в металле, ни в окисной фазе.
Рентгенографически подтверждется отсутствие
кристаллических фаз в расплаве и неизменность состава
металла-подложки. Считается, что в результате
взаимодействия двуокиси кремния с молибденом и вольфрамом
образуются летучие низшие окислы вольфрама и
молибдена, а также моноокись кремния.
Елютин с сотр. изучал [39, 62—66] взаимодействие
А120з и двойных окисных расплавов на ее основе с
вольфрамом, молибденом и ниобием при температуре
плавления окислов в вакууме и в атмосфере гелия при
выдержках до 30 мин. В качестве добавок к окиси
алюминия использовали BeO; MgO; CaO; Сг203; V205; Ti02 и
Zr02.
15—894
225
С помощью металлографического анализа
установлено, что окисный расплав при взаимодействии с ниобием
как бы просачивается через поверхность металла, в
результате чего образуется прослойка окисла между
основной массой металла и его поверхностным слоем.
Просачивание наблюдается в отдельных участках
поверхностного слоя металла-подложки, а затем окисел
распространяется в горизонтальном направлении. На
фотографиях микрошлифов четко видны выделения светлой
фазы по границам зерен ниобия. Рентгенографически
установлено, что это фаза NbO. Для некоторых
систем (А1203 — ВеО) наиболее интенсивное
взаимодействие наблюдается для чистой АЬОз, а для других систем
(А120з—MgO) интенсивность взаимодействия возрастает
при увеличении содержания окисла-добавки.
Растворение кислорода в ниобии, вероятно, имеет место, но за
незначительный период контакта расплава с металлом
диффузионные процессы в твердом теле протекают в малой
степени; микротвердость ниобия по направлению от
границы раздела в глубь металла не меняется и для
различных образцов колеблется в пределах 370—
390 кгс/мм2. Микротвердость ниобия, отожженного в
соответствующей атмосфере при температуре опытов,
составляет 280—300 кгс/мм2. Микротвердость окисной
фазы, несмотря на некоторый разброс значений, несколько
уменьшается с увеличением количества окисла,
добавляемого к окиси алюминия.
Явление просачивания окисла через поверхностный
слой металла с последующим распространением в
горизонтальном направлении для молибдена и
вольфрама выражено в более слабой степени, чем для
ниобия.
При рентгенографическом исследовании поверхности
металлических образцов после удаления окисной фазы на
ниобии всегда обнаруживается NbO и линии неиденти-
фицированных соединений, которых особенно много
после опытов в вакууме. На поверхности вольфрама и
молибдена рентгенографически не обнаружено образования
каких-либо соединений, хотя при взаимодействии этих
металлов с окисными расплавами, несомненно, имеет
место окисление металла-подложки. Однако
образующиеся при этом окислы вольфрама и молибдена
возгоняются, проходят через слой окисла и разлагаются на
поверхности расплава. Выделяющийся молибден или
226
вольфрам располагается на поверхности капли в виде
металлического налета.
Кинетику взаимодействия жидких окислов с
вольфрамом изучали в атмосфере очищенного аргона при
температурах 2300—2700° С [35]. Окисный образец
помещали на вольфрамовую подложку, которую
разогревали пропусканием через нее электрического тока.
В результате взаимодействия выделяется настолько
большое количество легколетучей окиси вольфрама, что
измерение температуры оптическим пирометром
оказывается невозможным. Поэтому для каждого образца
металла с помощью пирометра определяли температуру в
холостом опыте, а в ходе эксперимента устанавливали
ее по электрическим параметрам питающего устройства.
Кинетику взаимодействия оценивали по убыли массы
металлического образца вместе с каплей окисла.
Кинетику взаимодействия А1203 и Si02 с вольфрамом
иллюстрирует рис. 64.
В процессе взаимодействия образуется большое
количество трехокиси вольфрама, которая затем оседает на
холодных частях установки. С помощью спектрального
анализа в этом осадке обнаружены не только вольфрам,
но также алюминий и кремний. При этом W03
диссоциирует частично на поверхности жидкого окисла, а
частично в его объеме.
Взаимодействие А1203 и ВеО с вольфрамом в
атмосфере аргона при температурах от 1500 до 3200° С
изучали по аналогичной методике с той разницей, что окись
с целью локализации зоны взаимодействия помещали в
вольфрамовые трубочки, а кинетику взаимодействия
оценивали не по убыли общей массы металла и окисла, а по
убыли массы металлического образца-подложки.
Помимо нагрева образца металла прямым пропусканием
тока, подогрев зоны контакта осуществляли с помощью
оптической печи. Результаты исследования кинетики
взаимодействия указанных окислов с вольфрамом приведены
на рис. 65. Удельная убыль массы выражается линейной
зависимостью от времени, т. е. dAP=Kd%, где К — кон^
станта скорости взаимодействия.
Наиболее целесообразным для оценки характера
взаимодействия в указанных системах является подход,
предложенный Юдиным с сотр. [46], согласно которому
взаимодействие осуществляется за счет диссоциативного
испарения окиси алюминия, и наиболее активным реа-
15*
227
АР, г/см2 АР, г/см2
О 10 20 JO г, мин О /О 20 г, мин
Рис. 64. Взаимодействие жидких Si02 (а) и Al203 (б) с вольфрамом при
температуре, °С:
1 - 2300; 2 — 2500; 3 — 2700
1 3 5 7 9 11 13 т,мин 1 3 3 7 9 11 13 т,мин
Рис. 65. Взаимодействие жидких А1203 (о) и ВеО (б) с вольфрамом в
условиях локализации зоны взаимодействия при температуре, °С:
J — 2000; 2-2200; 5-2400; 4-2600; 5-2800; 5-3000; 7-^3200
228
гентом является кислород. Исходя из этих позиций,
можно представить себе механизм взаимодействия
следующим образом. Согласно данным работы [49], основными
компонентами пара окиси алюминия при высоких
температурах являются кислород и алюминий, вследствие
этого суммарную реакцию взаимодействия окиси алюминия
с вольфрамом можно записать в виде:
W + А1203+ V3 (W03)3 + 2A1.
Рассматриваемые условия не являются стандартными
и изменение изобарно-изотермического потенциала
реакции выражается уравнением:
AGrs = ЫРп + RT\n [pjf-PT^]. (4-72)
Давление пара алюминия над окисью алюминия
определяется выражением [49]
кРм = -^^- + е,8. (4-73)
Поскольку исследования проводили в атмосфере
аргона с предварительной откачкой до 10~4 мм рт. ст., то
можно принять остаточное давление кислорода в
системе, равным 2,3-Ю-6 мм рт. ст. Изобарно-изотермический
потенциал в стандартных условиях реакции образования
(W03)3 [49,50] равен:
AG™™ ч = 15Г — 120 000 кал/г-атом.
В условиях равновесия
^|fr = 21gpA1 + V3lgp(WOiV (4-74)
Рассчитанная на основе этого уравнения температура
начала взаимодействия вольфрама с окисью алюминия
оказалась равной 2000 К. Это значение хорошо
согласуется с данными работы [46], в которой для давления
Ю-3—Ю-4 мм рт. ст., температура начала
взаимодействия с вольфрамом определена в 2100—2400 К.
Константа скорости взаимодействия вольфрама с
окисью алюминия независимо от ее агрегатного
состояния описывается уравнением:
К = 9(5.10Чхр(-7-^), (4-75)
229
т. е. взаимодействие осуществляется по одному и тому
же механизму.
Взаимодействие вольфрама с окисью алюминия
происходит следующим образом [47, 52]: диссоциативное
испарение окисла, транспорт кислорода к вольфраму;
химико-адсорбционный акт; десорбция и миграция
продуктов реакции.
Согласно данным работы [53], последние три стадии
процесса осуществляются с гораздо меньшей энергией
активации, чем первая. Теплота диссоциативного
испарения в системе А120з—А1 равна 66 ккал/моль [48].
Величину энергии активации при взаимодействии
одноатомного вещества с двухатомной молекулой можно
рассчитать по эмпирическому уравнению [53]
£ = 4-D*"> (4-76)
где Dxy — энергия диссоциации молекулы ху. Молекула
А1203 имеет следующее строение:
А1 —О —А1
II II
О 0.
В первом приближении реакцию взаимодействия
вольфрама с А1203 можно представить в виде:
W + А1203=0-> 1/3 (W08)3 + 2A1, (4-77)
где А12О3=0 —«двухатомная» молекула.
Так как Da\2o, =740 ккал/моль [50], то на одну
связь кислорода с алюминием приходится в среднем
123 ккал, следовательно, Аи2оя = О составляет
250 ккал/моль, и, исходя из (4-76), следует, что Е=
=63 ккал/моль. Это значение близко к
экспериментально полученному значению энергии активации процесса
(70 ккал/моль). Таким образом, лимитирующей стадией
взаимодействия вольфрама с окисью алюминия следует
считать процесс ее диссоциативного испарения
независимо от агрегатного состояния.
Аналогично для реакции взаимодействия окиси
бериллия с вольфрамом можно записать:
W + ЗВеО-* V3 (W03)3 + ЗВе.
Изобарно-изотермический потенциал реакции
AGr, = AGO s + RT In [pi; p$0>),], (4-78)
230
так как при высоких температурах основными
компонентами пара над ВеО являются кислород и бериллий [48].
В условиях равновесия
^g- = ilgP(wo,b + 31gpBe. (4-79)
Давление пара бериллия над ВеО выражается
уравнением:
1 3-Ю4 , -
•gPBe = T~~ + ly
а, как установлено выше,
i„ ч ю о . 2,6-Ю4
,gP(W = -I3'8 + -^— •
По этим данным, была рассчитана температура
начала взаимодействия окиси бериллия с вольфрамом,
которая оказалась равной 2200 К, что вполне
удовлетворительно согласуется с данными работы [48].
Константа скорости взаимодействия окиси бериллия
с вольфрамом в интервале температур 2500—3500 К
описывается уравнением
К = 4,Ь10*ехр(-^). (4-80)
Как и в случае с окисью алюминия, взаимодействие
ВеО с тугоплавкими металлами протекает по одному и
тому же механизму независимо от агрегатного состояния
окиси бериллия. Поскольку энергия разрыва связи в
молекуле ВеО составляет 70 ккал/моль, а энергия
активации процесса 80 ккал/моль, то лимитирующей стадией
взаимодействия окиси бериллия с вольфрамом также
является диссоциативное испарение этой окиси.
9. СМАЧИВАНИЕ ГРАФИТА
И ТУГОПЛАВКИХ КАРБИДОВ
ЖИДКИМИ ОКИСЛАМИ
Елютин с сотр. исследовал смачивание и
взаимодействие графита и некоторых углеродсодержащих
материалов с жидкими окислами в атмосфере аргона и в
вакууме при нанесении капли жидкого окисла на нагретый
образец-подложку. Экспериментальные данные приведе-
231
ны в табл. 24. Как и следовало ожидать, окислы,
взаимодействующие с углеродом, растекаются по
поверхности графита. Однако растекание не происходит, если в
результате восстановления некоторых окислов
образуются вещества, не смачивающие графит в чистом виде
(медь) или в науглероженном состоянии (кобальт,
никель, марганец, железо).
Таблица 24
Смачивание графита жидкими окислами в среде аргона
Окисел
А12Оя
ВаО
Ti02
Та2Об
v2o5
Nb206
W03
М0О3
Mns04
Fe203
CoO
NiO
t, °C
2100
1950
1800
1850
700
1470
1500
820
1620
1720
1830
1960
Характер
смачивания или 6
0=1204-150°
6-150°
Энергичное
растекание
Медленное полное
растекание
Энергичное
растекание
9=100-120°
Медленное полное
растекание
То же
0 = 150°
0-150°
6~50-г-60°
0«50~70°
Продукты взаимодействия
на поверхности
графита
А1*
Не обн.
TiC
ТаС
Не обн.
NbC
W; W2C; WC
Mo; Mo2C
в объеме
окисла
Не обн.**
» »
» »
» »
v2o3
Не обн.
» »
» »
Окисел восстанавливается
до металла
* Обнаружен спектральным анализом.
** Веществ, отличных от исходных, не обнаружено.
При изучении растекания окислов алюминия и бария
при более высоких температурах, чем температура
плавления окисла, обнаружено растекание А1203 по графиту
с образованием карбида алюминия при температуре
подложки 2150° С и выше. Окись бария при температуре
графитового образца до 2300° С не растекается по его
поверхности, хотя взаимодействие на границе имеет место:
капля как бы «бегает» по поверхности образца. Окись
ниобия начинает растекаться по поверхности графита
при достижении температуры подложки 1830° С.
232
Исследование растекания окислов алюминия,
ванадия, ниобия, тантала и титана проводили как в вакууме,
так и в атмосфере аргона. Существенного различия в
характере смачивания в зависимости от атмосферы опыта
не наблюдается. Растекание окислов вольфрама,
молибдена, железа, кобальта, марганца, никеля, бария и меди
исследовали только в атмосфере аргона.
При оценке степени возможного взаимодействия угле-
родсодержащих высокотемпературных материалов с
жидкими окислами обнаружено, что восстановление
термодинамически прочных окислов (окислы алюминия,
бария, ниобия, титана, тантала) карбидами при
температуре плавления соответствующего окисла не происходит.
Остальные окислы легко восстанавливаются изученными
карбидами.
Кроме окиси бария, почти все исследованные окислы,
в том числе окислы, которые не растекались по графиту,
например, окислы алюминия и ниобия, хорошо
растекаются по поверхности тугоплавких карбидов циркония,
ниобия и кремния (табл. 25).
Таблица 25
Смачивание карбидов циркония, ниобия и кремния жидкими
окислами
Окись
А120*
ВаО
Ti02
Та2Об
Nb205
v2o5
W03
М0О3
Fe«A
МП3О4
СоО
NiO
tr °с
2050
1950
1800
1850
1470
700
1500
820
1620
1620
1830
1960
Характер смачивания
или G
Энергичное
кание
0=904-120°
Растекание
То же
>■> :>
» >
» »
Медленное
растекание
Энергичное
кание
Медленное
кание
0 = 504-60°
9 = 504-60°
расте-
полное
расте-
расте-
Продукты взаимодействия
на поверхности
образца
А14С3; Si02; Zr02;
NbO*
Не обн.
TiC; TiO
ТаС
NbC; NbO
He обн.
W2C; WC
Mo2C
Fe3C
Окисел восстанав
металл
в объеме
окисла
Не обн.**
» »
» »
» »
» »
v2o3
Не обн.
» »
» »
ливался до
а
* В зависимости от состава образца-подложки.
** Веществ, отличных от исходных, не обнаружено.
233
Окислы железа, марганца также хорошо
растекаются по поверхности этих карбидов, что связано с менее
энергичным их восстановлением карбидами по
сравнению с восстановлением графитом. Окислы кобальта и
никеля и в этих условиях легко восстанавливаются до
металла, которые и образуют каплю с краевым углом
смачивания порядка 50—60°.
Для определения факторов, влияющих на смачивание
графита жидкими окислами, было исследовано
растекание жидкой окиси алюминия по графиту при добавке в
нее других окислов (табл. 26)—окислов карбидообра-
зующих элементов (кремния, титана, циркония, бора,
ниобия, хрома, железа, церия, бериллия и тория) и
окислов элементов, не образующих карбиды (бария,
кальция, магния, олова, цинка, кобальта, никеля и меди).
При добавке в окись алюминия окислов карбидообра-
зующих элементов всегда имеет место растекание окиси
алюминия по поверхности графита с восстановлением
окиси до карбида алюминия. Причем при добавке в окись
алюминия кремния, титана, ванадия, ниобия, хрома и
железа интенсивное растекание происходит при
содержании очень небольших (~1%) количеств окислов. При
добавке окислов церия, тория, бериллия, которые
труднее восстанавливаются до карбида, растекание
наступает при содержании больших (5% и более) количеств
окислов. При добавке в окись алюминия окислов некар-
бидообразующих элементов растекание не наблюдается.
Такое влияние окисных добавок на растекаемость
окиси алюминия по поверхности графита позволяет
выдвинуть предположение, что реакция, протекающая на
границе контакта, интенсифицируется за счет
образования на границе карбидов. Дополнительным основанием
для такого предположения является хорошая
растекаемость жидкой окиси алюминия по поверхности
карбидов.
Если предложенный механизм правилен, то при
нанесении чистой окиси алюминия на карбид при
температуре плавления А1203, промежуточным продуктом
реакции должен быть окисел металла, образующего карбид
подложки. Для проверки этого предположения жидкую
окись алюминия без добавок наносили на поверхность
силицированного графита, а также на поверхность
карбидов циркония, титана и ниобия. Сразу же после
нанесения капли установку резко охлаждали и продукты ре-
234
Таблица 26
Влияние добавок окислов на растекаемость
окиси алюминия по поверхности графита
в изотермических условиях
Содержание добавки
в жидкой А1203, %
1 Si02
7 Si02
29 Si02
50 Si02
1 Ti02
10 ТЮ2
30 Ti02
40 Ti02
1 Zr02
1 B203
1 Nb205
1 Fe203
1 Cr203
5 Ce02
1 BeO
5 BeO
5 Th02
1 BaO
5 BaO
1 CaO
5 CaO
1 MgO
5 MgO
1 Sn02
5 Sn02
1 ZnO
5 ZnO
1 CoO
5 CoO
1 NiO
5 NiO
1 Cu20
5 Cu20
Характер смачивания*1 или в
Энергичное растекание '
То же
Смачивание, 0 = 35°
То же
Энергичное растекание
То же
Медленное растекание
Смачивание, 0 = 15°
Энергичное растекание
То
»
же
»
»
» »
Медленное растекание
Расстекания нет,
G = 100-М 20°*2
Медленное растекание
То же
Растекания нет
То
»
»
»
»
»
»
»
»
»
»
же
»
»
»
»
»
»
»
»
»
»
»
»
»
»
Продукты
взаимодействия на поверхности
графита
АЦС3
АЦС3; SiC
SiC
SiC
А14С3
А14С3; TiC
А14С3; TiC
TiC
А14С3
А14С,
АЦСЯ
А14С3
А14Са
А14С3
АЦСз
А14Сз
А14С3
А14С3
АЦСз
АЦС3
АЦС„
АЦСз
АЦСз
АЦС3
Sn*3, А14С3
АЦСз
АЦС,
АЦС3
Со*3, АЦС3
АЦС3
Ni*3, A14C3
Си*3, АЦС3
Си*3, АЦС3
Примечание. Состав окисла не изменился.
*' Визуальная оценка.
*2 Краевой угол при отсутствии растекания всюду составлял величину
порядка 90—120°.
*3 Обнаружено спектральным анализом.
235
акции исследовали с помощью рентгенографического
анализа. При анализе продуктов реакции на карбидах
титана и ниобия были обнаружены карбиды
соответствующих металлов, карбид алюминия и низшие окислы
титана и ниобия (см. табл. 25).
При рентгенографическом анализе продуктов
взаимодействия жидкой окиси алюминия с карбидом циркония
и силицированным графитом также обнаружены окислы
кремния и циркония. Анализ продуктов взаимодействия
окиси алюминия с силицированным графитом показал,
что нельзя делать строго определенное заключение о
присутствии двуокиси кремния, так как многие линии Si02
перекрываются более интенсивными линиями SiC или
А1203, т. е. линиями фаз, которые неизбежно должны
присутствовать в продуктах реакции. При анализе же
продуктов взаимодействия жидкой окиси алюминия с
карбидом циркония присутствие двуокиси циркония
доказано строго.
Эти результаты хорошо подтверждаются данными
работы [34]: при плавлении окиси алюминия на карбиде
циркония и бориде циркония на контактной границе
обнаружена двуокись циркония, при плавлении на
карбиде тантала — окись тантала Ta2Os, при плавлении на
нитриде титана — окись титана Ti62.
Термодинамические расчеты показывают, что
восстановление окиси алюминия до карбида в присутствии
окислов карбидообразующих элементов может
происходить при более низкой температуре, чем температура
плавления окиси алюминия. При добавке окиси кремния
эта температура снижается до 2000° С, а при добавке
окиси титана — до 1850° С.
Действительно, при добавке в окись алюминия
окислов кремния до 15% (температура плавления смеси
порядка 2000° С) и титана до 30% (температура плавления
смеси около 1850° С) все же происходит растекание
окиси по поверхности графита с восстановлением ее до
карбида алюминия. При более высоких содержаниях
окислов-добавок, когда температура смеси еще более
снижается, восстановления окиси алюминия до карбида и ее
полного растекания не происходит, но имеет место
восстановление добавляемого окисла до карбида.
Возможно термодинамически рассчитать равновесное
давление СО для выбранных реакций по уравнению
AG = — «Г In/Ср.
236
Подобные расчеты были сделаны для случаев
восстановления углеродом жидкой окиси алюминия,
легированной окислами кремния, титана и циркония. Результаты
расчетов приведены ниже:
Система А1203—Si02—С AIaOj~ Zr02—С A1203—ТЮа— С
рс0 (равн.),ат . . 2,4 2,6 10
Для экспериментальной проверки результатов
проведено исследование изотермического смачивания и расте-
каемости в атмосфере окиси углерода жидкой окиси
алюминия, легированной указанными окислами по
поверхности графита, и жидкой окиси алюминия по
поверхности карбида циркония, карбида титана и силицирован-
ного графита. Результаты этих исследований
качественно согласуются с результатами расчетов (табл. 27).
Взаимодействие окиси алюминия с графитом
изучалось многими исследователями, однако определенно
установившихся взглядов на механизм и кинетику этого
процесса до настоящего времени нет. Подробный обзор
имеющейся литературы и собственные результаты
исследований в этой области приведены в работах Елютина
с сотр. [55]. Термодинамический анализ системы
А1203—С позволяет утверждать, что в ней на первой
стадии взаимодействия должен образовываться оксикарбид
алюминия (А1404С), который затем взаимодействует с
углеродом и образует карбид алюминия (АЦСз).
Экспериментальное исследование карботермического
восстановления окиси алюминия при пониженном
давлении (температура 1700—2000 К) [56] показало, что в
процессе восстановления действительно образуются
промежуточные продукты оксикарбиды и карбид
А1203 -* А12ОС -+АЩС -£ А14С3,
которые затем взаимодействуют друг с другом по
реакции
А1404С + А14Сз-+ 8А1 + 4СО
с образованием металлического алюминия.
Авторы считают, что первая стадия процесса
протекает преимущественно за счет диссоциативного
испарения окиси алюминия. При более низких температурах
(1300—1600 К) в вакууме идет процесс образования
карбида алюминия по суммарной реакции [57]
2А1203 + ЗС-^ А14Сз + 6СО.
237
Скорость этой реакции подчиняется параболическому
закону; контролирующей стадией является диффузия
через промежуточный слой оксикарбида или карбида
алюминия. Энергия активации этого процесса составляет
316 ккал/моль.
Та
блица 27
Растекаемость жидких окислов по графиту и карбидам
в атмосфере
Система
А1203+5% SiQ2
графит
Al203+5%Zr02
графит
А12Оэ+5%ТЮ2
графит
А1203
ZrC
А1203
SiC
А1203
TiC
* При давлени
Растекаемость при давлении СО*, ат
i
0,8 | 1,2
Медленное
растекание с
образованием АЦС3
То же
Энергичное
растекание с
образованием А14Сз
Медленное
растекание с
образованием АЦС3
То же
Энергичное
растекание с
образованием А14С3
Растекания нет;
9^90°; на
поверхности графита
обнаружен SiC
Растекания нет;
8 = 90°; на
поверхности графита
обнаружен ZrC
Растекание с
образованием АЦС3
Растекания нет,
0 = 60°
То же
Растекание с
образованием А14С3
2; 5
—
—
Растекания
нет; 6=90°;
на
поверхности
графита
обнаружен TiC
-—
—
Растекания
нет; 0=60°
и СО, равном 0,4 ат, происходит энергичное растекание с
образованием АЦС3.
Взаимодействие порошка окиси алюминия с сажей
при температурах 1600—2400° С протекает через стадии
образования оксикарбидов, а образующийся в итоге
карбид алюминия диссоциирует [55]. Эффективная энергия
активации процесса составляет 94 ккал/моль.
238
-расплавленная окись алю-
АРЮ\ г/ммг
Рис. 66. Схемы установок для исследования взаимодействия графита
ми окиси алюминия (а) и жидкой окисью алюминия (6):
/ — графитовая подвеска; 2 — образец графита; 3 -
мииия; 4— молибденовый тигель
С целью
выявления механизма и
кинетики
взаимодействия окиси алюминия
с углеродом, а
также для
установления энергетических
характеристик
процесса, авторами этот
процесс был
исследован в интервале
температур 2120—
2350° С в вакууме
при раздельном
расположении окисла и
углерода. Причем
конструкция
установки обеспечивала
взаимодействие
графитового образца с
парами окиси
алюминия только на
одной плоскости (рис.
66, а).
Образцы из гра-
т,мин
Рис. 67. Взаимодействие графита с парами
окиси алюминия в вакууме при температуре,
°С:
/ — 2050; 2 — 2100; 3 — 2200; 4 — 2400
239
АР'Ю4,г/пмг
W 20 JO 40 50 60 70 60 90 т,мин
Рис. 68. Взаимодействие графита с парами окиси
алюминия в вакууме при периодическом удалении
образующегося покрытия при температуре, °С:
/ — 2050; 2 — 2100; 3 — 2200; 4 — 2400
фита пористостью 1—2% перед каждым опытом
отжигали в вакууме при 2100° С в течение 30 мин. Образцы
располагали над поверхностью жидкой окиси алюминия
на расстоянии 5 мм. Уста-
1дДР/Дт
М
4,0 4,1 4,2
Зависимость lg(AP/At) —
реакций взаимодействия
с жидкой А120з и парами
Рис. 69.
\/Т для
графита
А!203:
/ — травленый образец; 2 —
нетравленый образец; 3 — начальная
стадия; 4 — установившаяся стадия
новлено, что на поверхности
графита образуется
покрытие, в состав которого
входят оксикарбид алюминия,
карбид алюминия и окись
алюминия. Несмотря на то
что происходит нарастание
покрытия на графите, идет
убыль массы
образующейся системы от времени по
линейному закону (рис.67).
Известно, что пары окиси
алюминия в основном
состоят из кислорода и
алюминия [48], а также
субокислов алюминия [49].
Очевидно, убыль массы
графитового образца
происходит в результате взаимодей-
240
ствия с кислородом, а также субокислами алюминия,
причем в последнем случае образуются оксикарбид и
карбид алюминия, становящиеся впоследствии центрами
конденсации вещества из паровой фазы. Одновременно
происходят вторичные реакции между всеми
компонентами возникающего покрытия. Для оценки убыли массы
графита покрытие стравливали после каждой выдержки
и в этом случае убыль
массы описывалась ли- ьр'10>г/м"2
нейной зависимостью от /7f
времени (рис. 68).
Характерно, что энергия
активации в обоих случаях
одинакова (рис. 69) и
составляет 190 кка л/моль.
Величина энергии
активации и линейный характер
кинетической зависимости
процесса позволяют
считать, что лимитирующей
стадией взаимодействия
является процесс
диссоциации карбида алюминия.
Взаимодействие
жидкой окиси алюминия с
графитом в условиях их
непрерывного фазового
контакта изучали при
погружении графитового
образца в расплав окиси (рис. 66, б) и непрерывной про-
дувке печного пространства аргоном для удаления
газообразных продуктов взаимодействия.
Образец, свободно перемещающийся в вертикальном
направлении, опускали в расплав, по истечении
определенного времени извлекали из расплава и определяли
убыль его массы. После каждого опыта расплав в тигле
обновляли. В первые минуты происходит интенсивное
взаимодействие жидкой окиси алюминия с графитом, а
затем через 2—3 мин процесс описывается практически
линейной зависимостью, соответствующей
установившемуся (для данного расхода аргона) псевдоравновесному
давлению СО (рис. 70). Энергия активации для
начального линейного участка и для установившегося
линейного участка одинакова (см. рис. 69) и составляет 75—
Рис. 70. Взаимодействие графита с
жидкой окисью алюминия при
температуре, °С:
i — 2050; 2-2100; 5-2400
16-894
241
80 ккал/моль, что хорошо соответствует развитым выше
представлениям о том, что определяющей стадией
взаимодействия жидкой окиси алюминия с
высокотемпературными материалами является процесс ее
диссоциативного испарения. Следует отметить, что при рентгено-
структурном исследовании расплава после опыта в нем
обнаружен оксикарбид алюминия, который в результате
конвективного перемешивания жидкой окиси алюминия
не оказывал заметного влияния на процесс
взаимодействия окиси с графитом.
Таким образом, при взаимодействии жидкой окиси
алюминия с графитом процесс идет с образованием ок-
сикарбида и карбида алюминия. Лимитирующей стадией
процесса при температурах до 2000 К является диффузия
углерода через образующийся слой карбида, при более
высоких температурах — диссоциация карбида
алюминия. Если в результате взаимодействия создаются
условия, исключающие образования сплошного покрытия, то
процесс определяется диссоциативным испарением окиси
алюминия.
Г л а в а V
ОБРАЗОВАНИЕ ОКИСИ АЛЮМИНИЯ
ПРИ ОКИСЛЕНИИ И ГОРЕНИИ
АЛЮМИНИЯ
1. ОКИСЛЕНИЕ АЛЮМИНИЯ
Алюминий взаимодействует с кислородом чрезвычайно
медленно, что обусловлено защитными свойствами
образующейся на поверхности металла твердой окисной
пленки. Контролирующей стадией гетерогенной реакции
окисления алюминия является транспорт реагентов в
зону реакции. Подробный обзор работ по исследованию
образования тонких пленок на алюминии составлен
Хартом [1]. В условиях низких температур и
отсутствия влаги рост пленки на алюминии вначале идет
быстро, а затем значительно замедляется. Критическая
толщина пленки, при которой наблюдается резкое
замедление скорости роста, составляет от 20 до 30 А.
242
Рис. 71. Окисление алюминия при
температуре, °С:
1 — 450; 2 — 500;
5 -600; 6 — 400
3 — 650; 4 — 550;
Присутствие паров воды способствует росту пленки
окисла. Окисление в сухом кислороде идет но
обратному логарифмическому закону, а во влажном — сначала
по прямому, а через 10 ч — по обратному
логарифмическому закону.
Если процесс окисления протекает при повышенных
температурах, то обнаруживается сильная зависимость
не только скорости, но и характера окисления от
чистоты исходного
металла и способа подготов- SO \
ки образцов;
зависимость скорости
окисления от давления
кислорода в интервале
температур 350—840° С
отсутствует [2—10].
Интерпретировать
зависимость окисления
алюминия одним
известным законом во
всем температурном
диапазоне не
представляется возможным. На
кривых окисления
алюминия при температурах выше 500° С выявляются две
области (рис.71): область, где происходит быстрое
окисление и образуется значительное количество окиси
алюминия на образце за сравнительно небольшой
промежуток времени (ее обычно интерпретируют либо линейной
[2, 6], либо параболической [2, 5, 9J зависимостью), и
область чрезвычайно медленного окисления,
количественные параметры которой пока еще никто не
рассматривал. В табл. 28 для сравнения представлены
результаты, полученные различными авторами.
В отдельной колонке этой таблицы приведены
значения констант окисления К при 600° С. Значения
констант на начальном и среднем участках кривой окисления
различаются у Смелтцера в два —три раза, а от
данных Гульбрансена [2], экстраполированных к 600° С,
отличаются на порядок и больше. Эти факты связаны,
по-видимому, с тем, что при определении
параболических констант не учитывалась начальная толщина окис-
ной пленки. Поэтому, когда толщина образующейся
пленки несущественно отличается от исходной, констан-
16*
243
Таблица 28
Зависимость константы скорости окисления твердого алюминия от температуры
Рекомендуемая зависимость для константы
окисления К, г2-см"~4с
К при 600 °С
Диапазон
измерений,
°С
Примечание
Библиография
/ 22,8-103\
/Спар = (0,39-* 1,03).10-^ ехР [- ^ )
/ 37,9-103\
/Спар==1,б.10-3ехр(— ~^—J
^nap=8.10~3exp
Кпар=1,7-Ю-2ехр
4Ы03
RT
40,5-103
#пар=6 ехР
/Спар=6,33.10-бехр
RT
f 50,7-103
RT
33, Ы03
~ RT
, 51,9-10» \
(0,82 f-2,3)- Ю-13
6,8-Ю-13
5-Ю-13
1,35-Ю-13
1,5-Ю-12
3,7-Ю-13
1,6- Ю-9
350-450
400—600
400—600
400—600
400—600
400—600
400—650
Начальный участок
То же
Толстые пленки
То же
Данные Гульбран-
сена [2]
Гульбрансен, Вай-
сон [2]
Смелтцер [5]
Эйлмор и др. [61
Примечание. Кпат)— константа параболического окисления, /Слин — константа линейного окисления, г-см -с
vnap
* Данные, полученные после предварительного обжига при 600° С в течение I ч.
та параболического окисления имеет заниженные
значения.
Слеппи [9] исследовал окисление расплавленного
алюминия в сухом кислороде при температурах 660—
825° С. В опытах была обнаружена сильная зависимость
скорости процесса от первоначального состояния
поверхности. Окисление алюминия при температурах до
700° С, по мнению автора, удовлетворяет
логарифмическому закону, а в области температур выше 700° С —
параболическому. Энергия активации этого процесса,
измеренная Слеппи составляет 99 ккал/моль.
Сложность и низкая точность измерений при высоких
температурах связаны с растрескиванием окисной
пленки вследствие разницы термических коэффициентов
расширения окисла и металла. Между тем наблюдение
за состоянием алюминиевых проволочек перед
воспламенением [11] свидетельствует о сохранении прочности
окисной пленки вплоть до точки плавления окисла,
несмотря на существенно неизотермические условия ее
образования. Поэтому необходимо знать параметры
процесса окисления в условиях, при которых пленка
сохраняет свои защитные свойства. Прямые измерения
скорости окисления массивных образцов при
температурах 950—1300° С проводили в работе [12]. Окисление
вели в замкнутом объеме, поэтому скорость процесса
определяли манометром по расходу кислорода.
Использование метода индукционного нагрева образца во
взвешенном состоянии позволило избежать
растрескивания защитной пленки вплоть до 1300° С. Было
обнаружено, что происходит «старение» окисной пленки,
причем скорость этого процесса возрастает вместе с ростом
температуры окисления. Параболический закон,
описывающий кривую окисления, свидетельствует о
диффузионной природе явлений, контролирующих процесс
окисления. Полученное в работе [12] значение энергии
активации (100±20) ккал/моль находится в хорошем
согласии с результатом Слеппи [9], полученным для
интервала 700—850° С (рис. 72).
Таким образом, кинетические параметры,
описывающие окисление алюминия, изменяются при переходе от
сравнительно низких температур (450—700° С) к
высоким (выше 750° С). Более того, значения этих
параметров зависят от предыстории окисной пленки, в
частности от времени, в течение которого пленка находилась
245
4 h
Jb
1 Y-
Ъ\ж К,
\\\
\>^*
1k\v
L v4.
x\
Г \
I 1 II L_
V
'^\
4Л
VI
Lftl
при той или иной температуре. Все это свидетельствует
0 значительной роли структурных превращений при
объяснении кинетических зависимостей окисления
алюминия.
Броукер [13] получал окисные пленки при окислении
образцов в пламени воздушной горелки или в открытой
печи, затем отделял их от подложки и изучал
полученные с пленок электронограм-
tyX/r мы. В зависимости от
температуры окисления и
времени выдержки образцов
дифракционная картина
указывала на аморфную или
кристаллическую у- или
кристаллическую а-модифика-
цию окиси алюминия.
Пленки с аморфной структурой
преобладали на образцах,
окислявшихся при низких
температурах, однако и в
этих условиях при
увеличении времени окисления на
фотографиях появлялись
следы колец
кристаллической 7-фазы.
При 400° С
кристаллическая у-фаза появлялась
после 6-ч выдержки, при
500° С —после 1-ч выдержки, а при 700° С —через
1 мин. После нагрева до 1300° С наблюдался переход
7-фазы в а-фазу окиси алюминия, но и в этом случае
после 15-мин выдержки еще обнаруживались следы
7-фазы. Структурные исследования пленок, отделенных
от поверхности анодированного алюминия [14], также
показали, что при нагреве до 700° С на изображении из
диффузных колец, которое давала первоначальная
пленка, возникали резкие кольца 7-AI2O3. Мальцев и др. [15],
изучавшие структуру отделенных от металла пленок,
полученных при окислении алюминия вблизи 700° С,
также обнаружили появление фазы 7-AI2O3 при нагреве
выше 700° С.
Хантер и Фаул [4] исследовали состав окисной
пленки электрохимическим методом в динамике. Образцы
алюминия, которые окислялись на воздухе в течение
0,75 0,8
0,9
0,95
а/т)-ю3,к-
Рис. 72. К расчету энергии
активации высокотемпературного
окисления алюминия по данным:
/, Г — [9]; 2, 2', 2" — [12]
246
определенного времени при известной температуре,
после этого помещали в раствор электролита,
нейтральный по отношению к алюминию, и по значению
барьерного потенциала определяли толщину барьерного слоя.
Полученная в работе [4] зависимость запирающего
напряжения от времени окисления при различных
температурах представлена
на рис. 73. При
небольших выдержках
величина барьерного
потенциала растет и с
течением времени
достигает предельного
значения. Для образцов,
окисляющихся в
течение 24ч при 450° С,
наблюдается один
уровень предельного
значения, а для образцов,
окислявшихся при 475
и 500° С, — два уровня.
При температурах
окисления 550—600° С
снова наблюдается
только одно предельное значение барьерного
потенциала, что связано с невозможностью в условиях данных
экспериментов разделить процессы роста пленки и ее
кристаллизации.
Уровень предельного значения барьерного
потенциала не связан с толщиной пленки, а определяется ее
фазовым составом. На нижнем уровне насыщения
структурный анализ показывает существование аморфной
пленки, а на верхнем — у-кристаллической. Время
перехода с одного уровня насыщения на другой, иными
словами, время смены аморфной модификации окисла на
кристаллическую, уменьшается с ростом температуры и
составляет при 475; 500 и 550° С соответственно 15; 6
и 1 ч. Это свидетельствует о постепенном характере
превращений, происходящих в окисной пленке.
Доерти и Девису [10] удалось показать, что
кристаллизация окисла при повышенных температурах не
является следствием поликристалличности структуры
материала образца: свои исследования они проводили на
различных гранях монокристалла. Путем прямых на-
20 Т, Ч
Рис. 73. Изменение барьерного
потенциала У в кислороде (/) и на воздухе (//)
при различных температурах °С [4]:
/ — 600; 2 — 550; 3 — 500; 4 — 475; 5 — 450;
6 — 300; 7 — 200; 8 — 75
247
блюдений за направлением роста пленки им удалось,
кроме того, установить, что пленка растет в
направлении к металлу, что свидетельствует о преимущественной
диффузии кислорода в результате его взаимодействия с
алюминием. Кристаллы окисла появились на внутренней
поверхности окисных пленок во время окисления при
600° С.
Анализ отмеченных работ позволил Елютину с сотр.
[16] предположить, что кристаллическая фаза в окисной
пленке на поверхности алюминия появляется не только
при высоких температурах, но присутствует и при
относительно низких (450—500° С) температурах, правда, в
незначительных количествах. Была предложена модель
для количественного окисления алюминия в атмосфере
сухого кислорода.
Процесс окисления алюминия контролируется
диффузией одного из компонентов через окисный слой.
Структура первоначально образующегося окисла
аморфна и является метастабильной. Поэтому с
течением времени она превращается в свою более
устойчивую модификацию, из-за чего свойства окисной пленки
изменяются по глубине: вновь образующийся аморфный
слой характеризуется объемным коэффициентом
диффузии D0. В остальной части переменный по глубине
коэффициент диффузии D определяется долями
метастабильной и устойчивой модификации, т. е.
D = D0v0+D„v„=v0[D0-DJ) + D„, (5-1)
где Doo — объемный коэффициент диффузии
устойчивой модификации;
v0 и Voo — объемные доли, соответствующие
метастабильной и устойчивой модификациям
окисла (v0-\-Voo=\).
Далее в предположении релаксационного механизма
фазового превращения, когда скорость образования
новой фазы пропорциональна объему старой, т. е. когда
dvoo/dx=vo/xo9 было получено выражение для значения
объемного коэффициента диффузии в слое, с момента
образования которого прошло время т:
D = (D0-D^)<rT/Tp+Zb (5-2)
где тр — время релаксации.
246
При решении задачи о законе движения границы в
диффузионной задаче Стефана авторы работы [16]
получили выражение для координаты границы g в
зависимости от времени, начальной скорости движения
rfg/rfr | т=^о , времени релаксации то и коэффициентов
диффузии D0 и Doo:
Д у = у arctg
-/('+i
exp (29/Y) - 1 - 1
a0 +
l+-7lexP(2e/Y)-l
(5-3)
Здесь введены безразмерные переменные
dx
в = т/т/- a.-- dl л! т° dy ■
Т/Т' a dx У DQ-D„ -d9.
X =
1-Л-Д>«
В работе [17] проведено сравнение
экспериментальных кривых окисления с расчетными данными,
полученными с помощью выражения (5-3). Для облегчения
сравнения была составлена таблица функции у/у<х> для
различных ао в зависимости от Э/v (табл. 29). Данные,
Таблица 29
Таблица функции у/уоо
э/v
0,1
0,2
0,3
0,4
0,5
0,7
1,0
1,25
1,5
2,0
2,5
3,0
3,5
4,0
5,0
6,0
г
0,01 |
0,09517
0,1813
0,2592
0,3297
0,3935
0,5034
0,6321
0,7135
0,7769
0,8647
0,9179
0,9502
0,9698
0,9817
0,9933
0,9975
0,1
0,09543
0,1817
0,2597
0,3303
0,3941
0,5040
0,6326
0,7139
0,7772
0,8649
0,9188
0,9503
0,9697
0,9817
0,9933
0,9975
У/Уоо Аля значений а0
0,5
0,1015
0,1915
0,2717
0,3433
0,4075
0,5170
0,6435
0,7229
0,7844
0,8694
0,9208
0,9520
0,9709
0,9823
0,9935
0,9976
1 1»°
0,1160
0,2139
0,2980
0,3713
0,4356
0,5432
0,6649
0,7403
0,7983
0,8780
0,9261
0,9552
0,9728
0,9836
0,9939
1 0,9978
5.0
0,2053
0,3214
0,4080
0,4778
0,5362
0,6297
0,7313
0,7927
0,8394
0,9031
0,9413
0,9644
0,9784
0,9869
0,9952
0,9982
10
0,2383
0,3528
0,4366
0,5037
0,5596
0,6487
0,7453
0,8035
0,8478
0,9082
0,9444
0,9663
0,9796
0,9876
0,9954
, 0,9983
249
представленные на рис. 74 [17], согласуются с
расчетными, а полученная температурная зависимость времени
релаксации определяется выражением:
YTp = 6,7.10-*exp(^3
Прямым доказательством существования
релаксационного механизма является изменение величины
диффузионного потока только за счет изменения свойств
пленки, через которую
S/U
1,0
0,8
0,6
ол
0,2
/_Wj$
-£ря
р i
^~~гыаЛ&<&&тФшт*0
«^•gssss04* д 1
• 2
о J
*4
v 5 !
4 Г/jTTp
Рис. 74. Сравнение расчетных кривых
окисления с экспериментальными данными
при температурах, °С:
У—520; 5 — 550; 3 — 570; 4 — 600; 5-650;
/ —а0=Ю; //— ао=1
осуществляется
диффузия. Для
подтверждения релаксационного
механизма в
рассматриваемом случае было
проведено
исследование окисления
алюминия с прерыванием и
промежуточным
отжигом.
Образцы
предварительно отжигали в
вакууме в течение
времени тОТж,о. При этом
изменялись свойства пленки толщиной Л0,
первоначально имевшейся на образце. Затем образцы окисляли
в течение времени п, длительность которого подбиралась
так, чтобы скорость окисления изменялась
незначительно. Далее снова проводили отжиг в вакууме в течение
времени тотж,ь в результате которого изменялись
свойства первоначальной пленки толщиной Л0 и пленки
толщиной hu образовавшейся при первом окислении и т. д.
Поскольку в этих условиях диффузионный поток не
изменяется и определяется толщиной ранее
образовавшейся пленки, то можно записать
F =п —• F - Ah • - - -
1 00 h0 ' 2 (h0/Dol) + (kxIDn) '
Ft+i
Ah
(5-4)
(h0IDQt)+..-+(h{/Dij)
где Л/г — разность концентраций диффундирующего
компонента на границах металл — окисел и
окисел — газ;
Ft—значение диффузионного потока при t-том
окислении, который можно считать постоян-
250
ным, если за время окисления т* свойства
уже имеющихся пленок изменяются
незначительно, а сопротивлением диффузии вновь
образующихся пленок можно пренебречь;
DtJ—коэффициент диффузии в пленке,
образовавшейся при t-том окислении к тому времени,
когда идет /-тое окисление.
В предположении релаксационного механизма
изменения коэффициента диффузии получено соотношение
между скоростями окисления:
k
к 2
2 V Тр /
X
X
1 + -~ ехр
^0
т -I-
отж.о '
0,5т,
#3 \ Тр /
X
X
+ -2-1 ехр
h0kx
(-
^отж,1+°'5т2
—~ = ехр
•-отж,!
-Т.
отж, 2
Тр
+ гх + т2
(5-6)
X
X
1 +
тотж.1 + °-5т1
Тр
+ ^х
х ехр
тотж.0 + Т1+Татж.1 + °'5т2
На рис. 75 представлены результаты исследования
окисления с прерыванием при температуре 570° С после
предварительного отжига в течение 50 или 35 мин. Ниже
приведены для сравнения экспериментальные и
расчетные значения отношения скоростей окисления с
прерыванием и промежуточным отжигом.
V
мин
Эксперимент 1 . , —
Расчет 56,6
Эксперимент 2 . . —
Расчет 60,2
М2
2,37
f2Z
2,09
34
2,98 2,09
2,07
1,96
0,278 0,117
0,278 0,117
0,350 0,117
0,056
0,056
0,056
0,027
Примечание. Условия эксперимента
0,350 0,117 0,056 0,027
1: /=570° С, р^ = 40 мм рт.
Л = 50 мин, т
0 отж,
отж
римента 2: t = 570'
. = т _ = 30 мин;
1 отж, 2
С, р~ = 40 мм рт. ст., т
1
р0 =40 мм
= т = 10 мин. Условия экспе-
— т 0 = 30 мин; т, =т_ =т0 = 10 мин.
отж, 3 12 3
=35 мин, т
отж, 0 отж.
251
Здесь /12 = kx/k2\ /23 = k2lk6\ /34 = V*4-
Время релаксации, определенное путем
сопоставления начальных скоростей окисления при температуре
570° С с различными значениями времени
предварительного отжига, дало значение т=65 мин [17].
Согласие между расчетными и экспериментальными
значениями свидетельствует, по-видимому, о хорошем со-
Ат, пкг/спг
Рис. 75. Окисление алюминия с
прерыванием и промежуточным
отжигом при 570° С, давлении
кислорода 40 мм рт. ст. и времени, мин
[17]:
/—тотж,0 =35; 2 —^ = 10, тотж>1 =
= 30; 3 — 12=10, т0ТЖ2 =3; 4 — т3 =
-10, Тотж>3 =30; /' — тОТЖ|о =50;
2' — Xi = \0, Тотж>1 -30; У — т2=10,
тотж,2 =30
О 5 10
Т, MUH
ответствии между заложенной в основу расчета
зависимостью D(x) и наблюдаемым явлением.
Физическая картина окисления алюминия в этих
условиях может быть интерпретирована на основе
представлений Архарова [18]. Согласно этим
представлениям этапу образования новой фазы в твердофазной
химической реакции предшествует «переходная» стадия, в
которой диффузионным путем происходит объединение
атомов компонентов исходных сред — металла и газа —
в некоторой зоне у поверхности их раздела. В общем
случае можно говорить о зоне сочленения двух
кристаллических фаз, различающихся составом и
пространственным расположением атомов.
Переход от такой метастабильной к физически
устойчивой фазе может идти по бездиффузионному
механизму (типа «мартенситных превращений»), аналогичному
механизму образования зон ГП при распаде твердых
252
растворов. Образующаяся при этом решетка химического
соединения может отличаться от старой не только
кристаллографически, но и типом связей. При окислении
алюминия кислород из газовой фазы адсорбируется на
поверхности образца и начинает диффундировать через
слой уже имеющегося соединения к поверхности
металла. Это — этап объединения компонентов диффузионным
путем, который, согласно Архарову, представляет собой
лишь переходную стадию твердофазной химической
реакции. Кислород внедряется в граничную область
металла, но в отличие от обычного растворения решетка
металла настолько искажается, что при этом теряется
дальний порядок и образуется аморфная структура,
состоящая из хаотически расположенных молекулярных
групп А1406. Между металлом и кислородом в этих
группах может преобладать ковалентная связь, чем
объясняется возможность возникновения структуры типа
AS2O3. Эта структура не характерна для окиси
алюминия и является метастабильной, причем неустойчивым в
ней является положение атомов кислорода. Этим,
по-видимому, и объясняется малая энергия активации при
движении кислорода через аморфную пленку по
сравнению с а-кристаллической, в которой, по данным Кинже-
ри [19], энергия активации составляет 152 ккал/моль.
В дальнейшем аморфная структура как всякая
неустойчивая система «расстекловывается» и переходит в
кристаллическую у-Фазу. Скорость перехода растет с
увеличением температуры, так как для образования
зародыша новой фазы требуется преодолеть энергетический
барьер. Поскольку образование зародыша связано с
перемещением кислорода, становится объяснимой
близость энергий активации окисления и процессов
релаксации.
Зародышами, вероятно, являются области
когерентности, которыми ограничивается «мартенситное»
превращение, начавшееся в одной из точек окисла. Поэтому
скорость образования новой кристаллической фазы
определяется скоростью появления зародышей.
Позднейшие рентгеноструктурные и электронномик-
роскопические исследования свидетельствуют о
достоверности этой картины. В пленках окисла, полученных в
результате анодного окисления [20], после нагрева в
вакууме электронным пучком до температур порядка
800° С наряду с аморфной структурой наблюдались по-
253
ликристаллические агрегаты, аналогичные по
морфологии y-A1203. При фокусировке электронного пучка на
небольшом участке образца или при нагреве на воздухе
до 1200° С кристаллы укрупняются и появляется а-фаза.
При изучении превращения у-^а методом
дифференциального термического анализа [21] скорость
превращения регистрировали по тепловому эффекту в
интервале 600—1500° С. Полученные при этом экзотермические
пики при 1085 и 1270° С авторы связывают с
образованием у-А1203 и а-А1203 соответственно. Указанные
температуры не следует рассматривать как точки фазовых
превращений, так как в условиях проведения
исследований наблюдалась массовая скорость перехода,
которая падает с уменьшением количества первоначальной
фазы, несмотря на рост самой скорости превращения.
Обычно структуре окиси алюминия приписывают
преимущественно ионный характер связи, что, с одной
стороны, противоречит существованию аморфной
модификации окиси алюминия, поскольку склонность к стекло-
образованию связана с преобладанием направленных
короткодействующих ковалентных связей [22], а, с
другой стороны, запрещает образование молекулярных
групп А1406 [23], обнаруженных Вилсдорфом [24].
Исследования электронной структуры алюминия в окиси
алюминия [25] привели к выводу, что при переходе
А1->А1203 происходит не полная передача внешних
электронов алюминия кислороду, а лишь частичная, причем
остается значительная связь этих электронов со своими
атомами, т. е. доля ковалентности довольно большая.
К аналогичному выводу приходят и авторы работы [26],
в которой сравнивались формы /С-полосы испускания
кислорода в а- и у-фазах окиси алюминия.
При исследовании с помощью микроскопа условий
возникновения первых выделений №3А1 при
диффузионном взаимодействии алюминия с никелем [27] авторы
интерпретировали полученную зависимость времени
появления №3А1-фазы от температуры выдержки
эмпирической формулой
о in 7 /27000 \
т = 2-10~7ехр мин
со значением энергии активации процесса, характерным
для бездиффузионных превращений. Этот результат МО'
жно рассматривать как непосредственное доказательств
254
во существования метастабильного состояния при
твердофазной реакции, переход которого в более устойчивое
не требует разрыва основных связей.
Превращение 7-AI2O3 в а-А1203, наблюдавшееся при
нагреве свыше 1000° С [13, 20, 28], свидетельствует о ме-
тастабильности кристаллического состояния 7-AI2O3.
Исследование образования метастабильных фаз в
частицах А120з, получавшихся в пламени и плазме,
проведено в работе [28]. При затвердевании жидких
капель в условиях значительного переохлаждения
образуется у~, а не а-А1203 вследствие более низкой
критической свободной энергии зародышеобразования. У частиц
с размером меньше 10 мкм неустойчивое состояние
сохраняется, а у частиц с размером больше 10 мкм
происходит переход в а-фазу. Авторы работы [28] не придают
значения скорости охлаждения, но причина
наблюдаемого явления заключается, по-видимому, именно в ней:
первоначально образующаяся -у-фаза в быстро остывающих
маленьких частицах не успевает превратиться в а-А120з,
а в медленно остывающих крупных частицах такой
переход происходит.
2. ВОСПЛАМЕНЕНИЕ И ГОРЕНИЕ
АЛЮМИНИЯ
Методы экспериментального исследования процессов
воспламенения и горения алюминия достаточно
подробно рассмотрены в обзорах Гордона, Дрю [29] и Похила
и Фролова [30, 31].
Исследования воспламенения и горения по
характеру подхода к проблеме и полученным результатам
можно разделить на две группы. В первую группу входят
исследования влияния металлизированных добавок на
характеристики горения жидкого или твердого топлива,
как, например, работы [32—38]. При этом изучаются не
собственно горение металла, а изменения в горении
жидкого или твердого топлива, связанные с внесением в его
состав того или иного количества алюминия той или
иной степени дисперсности.
Вторую группу составляют исследования механизма
воспламенения и горения частиц алюминия, получение
математической модели и количественных характеристик
в зависимости от контролируемых условий. Поэтому
методика исследований в этом случае предусматривает на-
255
блюдение за отдельными частицами, причем
контролируются температура, размер и другие параметры на
разных стадиях процесса. Часто в целях удобства
моделирования используются проволочки с нагревом за счет
пропускания электрического тока.
Правильность построения теоретических моделей
проверяется на модельных двигательных установках.
Однако при интерпретации получающихся при этом
результатов возникают трудности, связанные с тем, что на
процесс горения отдельных частиц накладываются
«коллективные» эффекты. Например, изменяется
эффективный излучательный теплообмен, увеличивается скорость
расхода окислителя, сами частицы могут образовывать
агломераты как в процессе горения, так и на стадии
воспламенения. Поэтому часто ответить, скажем, на вопрос,
в какой мере наблюдаемый эффект объясняется
изменившейся дисперсностью исходных частиц, а в какой —
их коллективным взаимодействием, бывает
затруднительно. Тем не менее, в итоге многолетней работы
советских и зарубежных исследователей было установлено
несколько следующих общепринятых в настоящее время
положений.
Воспламенение алюминия происходит в момент,
когда температура на его поверхности достигнет точки
плавления окисла. Этот факт был впервые установлен
в работе [39], авторы которой объясняли его резким
возрастанием скорости окисления в момент плавления
защитной окисной пленки.
Время воспламенения пропорционально квадрату
диаметра частицы независимо от температуры и
давления окружающей среды [39]. Это свидетельствует о том,
что большую часть времени разогрева частица ведет
себя как пассивный элемент, когда скорость разогрева
определяется газодинамичным теплообменом.
Температура газа, при которой возможно
воспламенение алюминия, может быть меньше температуры
плавления окисла [11, 40, 41]. Объяснение этого факта
кроется в интенсивном взаимодействии частицы с
окислителем, тепловой эффект которого приводит к
саморазогреву частицы до температуры плавления окисла, причем
время саморазогрева настолько мало по сравнению с
общим временем воспламенения, что квадратичная
зависимость времени воспламенения от размера частицы
сохраняется.
256
Температура воспламенения совокупности частиц
меньше температуры воспламенения одиночной
частицы [37].
Увеличение концентрации частиц приводит к
уменьшению времени их воспламенения [42].
Время воспламенения малочувствительно к составу
окисляющей среды и давлению, но сильно зависит от
температуры газового потока [43].
Три последних положения характеризуют
интенсивность взаимодействия алюминия с окислителем на
стадии саморазогрева.
Интересно отметить, что как для одиночной частицы,
так и для совокупности частиц время воспламенения
малочувствительно к содержанию окислителя в
окружающей среде. Отсюда, казалось бы, можно сделать вывод
о незначительной роли теплового эффекта. Однако
увеличение концентрации алюминиевых частиц в топливе
заметно уменьшает время воспламенения. Противоречия
здесь, по-видимому, нет: первое обстоятельство говорит
лишь о слабой концентрационной зависимости скорости
окисления от содержания окислителя, самим же
взаимодействием, как показывает второе обстоятельство,
пренебрегать нельзя.
Воспламенение алюминия противоречит
общепринятой тепловой теории воспламенения, согласно которой
критическая температура газа, когда еще возможно
воспламенение, уменьшается по мере роста размера
частицы. На зависимости критической температуры
воспламенения алюминия от диаметра частиц есть минимум [41,
44, 45].
Тепловая теория воспламенения, созданная для
капель жидкого топлива [46] и с успехом применявшаяся
для объяснения воспламенения многих твердых топлив,
предполагает, что скорость реакции экспоненциально
возрастает с температурой. Поэтому в системе,
разогревающейся благодаря тепловому эффекту окислительной
реакции, происходит тепловой взрыв, как только в
системе достигается некоторая критическая температура,
зависящая от дисперсности и содержания окислителя в
объеме. Поскольку относительная эффективность тепло-
отвода от частицы в окружающую среду уменьшается с
увеличением ее размера, это приводит к уменьшению
критической температуры воспламенения. Аномальный
ход этой зависимости у алюминия объясняется присут-
17-894
257
ствием плотной окисной пленки на поверхности
частицы, резко изменяющей кинетику реакции.
Горение алюминия в кислороде — это процесс его
окисления в паровой фазе. Пары алюминия
диффундируют с незакрытой окислом части поверхности в
пространство и сгорают, соединяясь с кислородом, который
диффундирует к частице из окружающего объема.
Степень удаленности зоны горения от поверхности зависит
от относительной скорости диффузии компонентов.
Поэтому при повышении давления зона горения
приближается к поверхности, а при понижении — удаляется от нее.
Максимальная температура горения алюминия не
превосходит температуры кипения его окисла (~ 3800 К)
[47].
Время горения практически не зависит от давления
и температуры (при давлении более 20 ат и
температуре выше 2000° С) окружающей среды, но сильно зависит
от концентрации в ней активных кислородсодержащих
продуктов [43]. Это, вероятно, является следствием
того, что режим внутри зоны горения зависит лишь от ее
радиуса, который определяется только содержанием
окислителя, а не общим давлением. Чем меньше
окислителя, тем больше радиус зоны горения, меньше
температура поверхности частицы, а следовательно, и скорость
испарения, и наоборот.
Время горения частицы пропорционально ее
диаметру в степени п (л=1,5-т-1,8) [39, 43, 48]. При изучении
горения раскаленного жидкого топлива было
установлено, что скорость горения определяется скоростью
испарения капель [49]. Еще Максвелл показал, что в
изотермических условиях испарение сферической капли
происходит с постоянной скоростью изменения величины
поверхности
rdrldx = const,
откуда для полного времени испарения (и,
следовательно, горения жидкого топлива) следует квадратичная
зависимость от начального размера %~R2.
По такому же закону горят и частицы магния [11,
48]. Значит, в течение большей части времени горения
капли жидкого топлива и магния условия испарения с
поверхности остаются неизменными. Этот факт,
очевидно, и объясняет успех квазистационарного приближения
258
при построении диффузионной теории горения этих
веществ в дисперсном состоянии [48—50].
Установленный закон для времени горения
алюминия свидетельствует о том, что скорость изменения
поверхности испарения зависит от радиуса частицы и,
следовательно, условия на поверхности изменяются.
Поэтому использование квазистационарного приближения в
теории горения алюминия следует всякий раз
обосновывать.
Рост концентрации частиц окислителя в топливе
увеличивает время горения [42]. Это положение является
следствием того, что время горения сильно зависит от
содержания окислителя в среде. Увеличение концентрации
горящих частиц ведет к усиленному расходу
окислителя и к увеличению времени горения. Отсюда можно
сделать вывод о существовании оптимальной (для данного
содержания окислителя) концентрации частиц
определенной дисперсности, способствующей получению
максимальной скорости горения всей совокупности
частиц.
При горении происходит агломерация частиц и
продуктов их сгорания, причем с увеличением размера и
концентрации частиц процесс интенсифицируется [37].
Агломерация металлических частиц в период индукции
и горения приводит к увеличению времени задержки
воспламенения и ухудшению условий горения,
следствием чего являются потери в импульсе из-за неполного
сгорания топлива (полнота сгорания алюминия в
модельном двигателе по оценкам [35] составила 94%).
Агломерация продуктов горения является причиной
двухфазных потерь, которые, по оценкам [35], могут
достигать 3—5%.
Наряду с перечисленными достаточно надежно
установленными закономерностями имеются неясные
вопросы, составляющие предмет дальнейших исследований, в
частности вопросы легирования алюминия другими
металлами с целью интенсификации процесса горения.
Остается открытым вопрос о том, какие свойства сплава —
его способность менять скорость окисления в период
индукции, интенсифицировать процесс самого горения,
повышать склонность частиц к фрагментации, уменьшать
агломерационную способность частиц продуктов
горения— должны считаться определяющими в процессе
поиска.
J 7*
259
В результате сравнительного рентгеноспектрального
и электронно-графического анализа продуктов сгорания
чистого алюминия и его сплавов обнаружено [37], что
добавки 1—2% Zn и 5—10% Mg значительно повышают
дисперсность продуктов сгорания. Причиной этого
является не столько дробление горящих частиц, сколько
улучшение условий образования критических зародышей.
Влияние больших добавок магния (>10%) на
горение алюминиево-магниевого порошка изучали на
одиночных частицах [38] и была установлена двустадий-
ность процесса. На первой стадии выгорает более
летучий магний, причем пленка окисла на частице
находится в твердом состоянии. На второй стадии горит
алюминий, пленка расплавляется, а частица при достаточно
интенсивном горении часто дробится. Для перехода
горения во вторую стадию необходимо, чтобы содержание
магния превышало 30%. Это условие связано с
необходимостью расплавления защитного слоя окисла за
время горения магния и определяется скорее размером
частиц и условиями теплоотвода (частицы диаметром 100—
600 мкм горели на вольфрамовой игле) нежели
свойствами самого сплава.
Общепринятую диффузионную модель горения в
квазистационарном приближении нельзя считать вполне
удовлетворительной, так как неквадратичная
зависимость времени горения от размера частицы
свидетельствует об изменении условий на ее поверхности. Под этим
понимается изменение температуры и, возможно,
количества окисла на поверхности.
По мнению Клячко [48], слой окисла на поверхности
частицы может со временем нарастать за счет
конденсации части паров окисла, образующихся в зоне горения.
Гуревич с сотр. [37] при учете конденсации окисла
пришел к заключению, что поверхность конденсации, во
всяком случае для магния, должна находиться за зоной
горения и потоком окисла на частицу можно пренебречь.
В этой модели по мере выгорания металла должна, по-
видимому, меняться температура поверхности. Оценка
правильности модели требует довольно детального
знания параметров среды в зоне горения и даже в случае
ее пригодности для объяснения горения в чистом
кислороде, она не описывает горения во влажной среде,
когда слой окисла на частице со временем нарастает.
Является ли этот процесс следствием поверхностного окисле-
260
ния, как предполагает Бартле [51], или горение
по-прежнему обеспечивается испарением металла с
поверхности, но интенсифицируется процесс поверхностной
конденсации окисла, как считают [52, 53], сказать
трудно. Во всяком случае, следует стремиться к тому, чтобы
в реальных условиях большая часть алюминия сгорала
именно по этому пути, так как в этом случае процесс
сопровождается фрагментацией частиц, что способствует
его интенсификации.
3. МОДЕЛИ ВОСПЛАМЕНЕНИЯ
МЕЛКОДИСПЕРСНОГО ПОРОШКА АЛЮМИНИЯ
Одним из следствий тепловой теории воспламенения является
уменьшение критической температуры по мере роста размера частиц.
Алюминий при воспламенении ведет себя иначе: начиная с
некоторого размера, который зависит от состава исходного материала и
окружающей частицу среды, кри-
tnp у О
1200 х
1100Y-
1000
тическая температура
возрастает одновременно с ростом
размера частиц [41] (рис.76).
Тепловая теория в своей
основе учитывает два канала
подвода энергии для
разогрева частиц. Первый —
теплообмен с окружающей средой —
создает положительный или
отрицательный тепловой поток
на частицу, если температура
среды соответственно выше или
ниже температуры частицы.
Второй ■— тепловой эффект
химической реакции окисления —
в обычных предположениях
создает поток, растущий с
ростом температуры быстрее, чем
теплообмен. Аномалии закона
воспламенения алюминия
объясняются невыполнением этих
условий.
При объяснении этого
явления важную роль отводят процессам высокотемпературного
окисления, в частности, причину столь необычного поведения связывают
с резким уменьшением скорости окислительного процесса с
течением времени. Применяют различные способы интерпретации процесса
на основе той или иной теоретической схемы.
В основе одной из попыток лежит [11, 30, 31, 44, 51, 55]
предположение о преимущественном влиянии толщины окисного слоя на
скорость высокотемпературного окисления.
Выбирая для качественных оценок степенную зависимость
скорости окисления от толщины слоя окисла б
20 ti; мкм
Рис. 76. Зависимость предельной
температуры воспламенения частиц
алюминия па воздухе (/) и в водяном парс
(2) от их размеров
261
• exp —
2300
d% ""' 6n ~"r \ RT .
где К—константа реакции;
с— концентрация окислителя;
т— порядок реакции по окислителю;
Е— энергия активации окислителя,
авторы [44] приходят к выводу, что для п>1 на кривой
зависимости предельной температуры вос-
Т пламенения от размера частицы
может наблюдаться минимум.
Сторонники этого объяснения
исследовали воспламеняемость
алюминиевой проволоки диаметром
30 и 50 мкм, нагревая образцы
током [45] (рис. 77). Мощность,
выделяющаяся на проволоке, со
временем не изменялась. Вводя
понятие критической мощности по
аналогии с критической
температурой в условиях нагретого газа
окружающей среды, авторы не
обнаружили заметного влияния
начальной толщины пленки окисла
на величину критической
мощности, хотя и подтвердили ее
зависимость от продолжительности
предварительного отжига
проволоки при 600° С. Результаты этих
исследований подтверждают
предположение о том, что в период
окисления до воспламенения
прирост толщины окисла значительно
превышает ее начальное значение. Не имея возвожности прямо
измерять скорость окисления при высоких температурах, авторы
работы [54] на основе своей модели рассчитали температурную
зависимость приращения толщины слоя окисла (см/с):
46 1,9-Ю-5 / 17 000 \
л = Т~ехр Г -W) ■
Не говоря уже о том, что полученное значение показателя
степени в зависимости от скорости приращения толщины слоя окисла
не обеспечивает оптимального по условиям воспламенения размера
частиц, само значение энергии активации окисления в интервале
1600—2000° С противоречит известным результатам.
Причиной этого, возможно, служит то обстоятельство, что в
выбранной зависимости неявно предполагается более сильное влияние
на скорость окисления изменения толщины слоя, а не температуры.
Такой подход к описанию кинетики окисления является
справедливым, строго говоря, лишь в изотермическом случае. Воспламенение
частиц при низких температурах среды носит характер теплового
взрыва с того момента, когда достигаются условия саморазогрева.
В этот момент изменением количества тепла за счет теплообмена со
средой по сравнению с тепловыделением саморазогрева можно
пренебречь. Более того, известная зависимость времени индукции от
г, мкм
Рис. 77. Зависимость относительно]"!
температуры воспламенения
алюминиевой проволоки от се радиуса
[451 при:
262
температуры среды позволяет считать, что, пока частица нагревается
средой, можно пренебречь тепловым вкладом реакции окисления.
Тогда легко можно оценить основной прирост окисла, исходя из
условия разогрева частиц от температуры среды до температуры
плавления окисла.
Пусть частица имеет объем V, теплоемкость С=0,6 кал/(см3X
Хград), а тепловой эффект окисления #=20 ккал/см3. Пусть за
время саморазогрева доля окислившегося объема равна |. Тогда
g=CATIH
или для сферической частицы
г м зьок
^~ V ~" гф '
где Лок — толщина окисленного слоя, ср — отношение окисленного
объема окисла к объему металла h0K— 1,3|г/3.
Выбрав с запасом ДГ=103, получим
0,6-103
Это означает, что толщина окисленного слоя составляет ~ 1 % от
радиуса частицы. Для частиц размером 1—10 мкм этот слой имеет
о
толщину 100—1000 А, что либо меньше, либо сравнимо с толщиной
начальной окисной пленки.
В работах Гуревича с сотр. [41, 57] уменьшение проницаемости
пленки связывается с кристаллизацией первоначально аморфного
окисла. Авторы предполагают, что окисление алюминия определяется
диффузией реагентов через аморфную часть пленки, покрывающей
частицу. С течением времени происходит ее кристаллизация, причем
скорость процесса пропорциональна диффузионному потоку.
Поскольку проницаемость кристаллизованного слоя значительно меньше
проницаемости аморфного слоя, окислением той части поверхности,
которая покрыта кристаллической окисью алюминия, авторы
пренебрегают. В зависимости от скорости, с которой происходит разогрев
частицы, происходит практически полная или частичная
кристаллизация аморфного окисла. В первом случае саморазогрев частицы
отсутствует и, если достигнутая температура меньше температуры
плавления окисла, воспламенения не следует. Во втором случае
частица продолжает разогреваться за счет реакции окисления вплоть до
воспламенения.
Из-за отсутствия необходимых экспериментальных данных по
окислению алюминия в высокотемпературной области в работе
Гуревича с сотр. константы окисления определялись в ходе численного
решения задачи о теплообмене. Решалась обратная задача о
нахождении параметров температурной зависимости константы окисления
на основе экспериментальных данных для предельных температур
воспламенения частиц разного размера. Однако расхождение между
вычисленными таким образом значениями и известными
экспериментальными данными оказалось значительным.
В табл. 30 для сравнения представлены константы линейного
окисления при 600° С, полученные разными авторами.
263
Таблица 30
Константа скорости окисления алюминия при 600° С
г/(см2-с)
Примечание
Библиография
5-Ю-8
2-10-е
6,2.Ю-9
3,Ы0-9
1,6-Ю-9
30
ью-8
Оценка авторов на начальном
участке
Оценка авторов
Оценка авторов
Оценка Бартле [57]
Оценка по зависимости
/ 51,9-103\
/Ci = l,28-10'exp(- RT j
Оценка по полученной
авторами зависимости
/ 20-Ю3
Ялин=18,5.105 ехр - RT
Смелтцер [5]
Блекборн, Гульбрансен
[7]
Коран, Слеппи [8]
Эйлмор и др. [6]
Гуревич и др. [57]
Елютин и др. [16]
Для зависимости предельной температуры воспламенения от
размера частицы и содержания кислорода авторы работы [41]
рекомендуют эмпирическое выражение:
Г™ = Тдл.ок- 0,6^1 ехр (-0,85/d)»
[ пр •
X
где d — диаметр частицы, мкм.
Отмечаемая рядом исследователей [6, 39] зависимость
предельной температуры воспламенения от содержания кислорода служит
признаком того, что существенное влияние на воспламенение
оказывает также высокотемпературное окисление, константа которого, по
нашей оценке, пропорциональна Pq4 • Пренебрежением окисления
через кристаллический слой окисла при высоких температурах
объясняются завышенные по сравнению с экспериментальными значения
констант окисления через аморфный окисел, вычисленные в
работе [37] на основе экспериментов по воспламенению.
Полученные Елютиным с сотр. [12, 16] сведения об окислении
алюминия в интервале 450—1700° С послужили основой модели
саморазогрева мелкой частицы перед воспламенением [45]. Поскольку
исследовали сравнительно массивные образцы, приложение
полученных результатов к мелкодисперсным частицам требует определенных
допущений.
Тождественность механизмов окисления крупных и мелких
частиц в соответствующих температурных интервалах сомнения не
вызывает. Но надо учитывать влияние различия временных масштабов
процесса воспламенения частиц. Мелкие частицы до воспламенения
окисляются в течение миллисекунд в неизотермических условиях, а
данные о константах окисления получены из экспериментов,
длившихся в течение нескольких десятков или сотен минут в
изотермических или почти изотермических условиях, причем было показано,
264
что временной масштаб существенно влияет на относительную долю
окисления через аморфную и кристаллическую фазы. Для 650° С в
течение ~100 мин окисление определялось диффузией через
аморфную фазу, а после 120 мин —через у-кристаллическую фазу. Для
750° С влияние кристаллической фазы должно сказываться уже
через несколько минут. Поэтому, как и в исследованиях Слеппи, так
и в исследованиях Елютина с сотр., этап окисления через аморфную
пленку не регистрировался.
Если рассматривать аморфную фазу как метастабильное
состояние, через которое проходит окисел после его образования при всех
температурах, то можно экстраполировать выражение для
характеристического времени жизни тк вплоть до температуры воспламенения
и оценить его значение в точке плавления окисла (1,6-10~5 с) и для
предельных температур воспламенения (3-10~3 с для 1300° С, 4Х
ХЮ~4 с для 1600° С). Эти величины оказались сравнимыми со
временем индукции. Это означает, что для мелких частиц в условиях
воспламенения в нагретом газовом потоке окислением через
аморфную фазу окисла пренебрегать нельзя.
Данные скоростной киносъемки частицы до ее воспламенения
свидетельствуют о том, что пленка окисла на мелких частицах в
период индукции сохраняет свои защитные свойства. Поэтому
наблюдавшееся в экспериментах Елютина с сотр. [45] растрескивание при
температурах выше 1300° С представляет собой особенность толстой
окисной пленки, которую не следует учитывать в модели окисления
мелкодисперсной частицы перед воспламенением.
Наконец, в ходе разогрева частицы можно пренебречь
уменьшением скорости окисления из-за изменения толщины окисла,
связанного с ростом температуры. Поэтому в выражении для скорости
окисления можно ограничиться линейными константами, а именно:
^ = ^exp^-^-J-J + [,-exp^J-||X
4 о /
где £"1 = 30 ккал/моль; £2=100 ккал/моль; Ki = 0,35 и /С2=4,ЗХ
Х1012г/(см2-с).
Здесь первое слагаемое учитывает окисление при диффузии
компонентов через аморфную фазу, а второе — через
кристаллическую -у-фазу. Выражение в квадратных скобках определяет долю
кристаллизованного окисла; интегралы в показателях экспоненты
определяют имеющиеся к моменту т доли аморфной и кристаллической
фаз. Окисление через а-А1203 не учитывается, так как ее
проницаемость более чем на пять порядков ниже проницаемости у-А1203.
Уравнение теплового баланса на поверхности частицы радиуса г,
находящейся в среде с температурой Th записывается как
roc dT
3 •■57==МТ,т)-МГ,Т1), (5-7)
где /i ■— тепловая мощность реакции окисления [функция
саморазогрева fi(Ty т) =/(#];
265
/2 — функция теплообмена со средой
/,(Т,ТЛ)=—(Т-Гл)
р, с и Т —плотность, удельная теплоемкость и температура
частицы соответственно;
Я — коэффициент теплопроводности среды [Я=Я0 (Т1Т0) ];
/(■_скорость окисления, определяемая выражением (5-6);
Н — тепловой эффект реакции окисления по кислороду (#=
= 8,3-103 кал/г);
Т —начальная температура частицы (Г=300 К).
Интегрирование
уравнения (5-7) для
различных значений Ti и г
совместно с условием (5-6)
выполнялось численным
методом. Особенностью
уравнения (5-7) является то,
что fi(T, х)—неизвестная
функция. Совместное
решение уравнений (5-6) и (5-7)
дает зависимость х от Т
и значения функции /i(7\
т) с заданной точностью,
которая используется при
вычислении времени
индукции:
грс
Хинд — о *
т
dT
X
70 т,мс
Рис. 78. Зависимость температуры
воспламеняющихся частиц алюминия радиусом
I мкм от времени при температуре
окружающей среды, °С:
/-1795:2-1555; 3-1285 л .) /х (Т, 7\) - /, (Т, Tj'
(5-8)
Это значение отличается от полного времени индукции на время,
необходимое для расплавления металла частицы при 660° С:
Хлл —
r2pL
ЗМ^-Гдл)
(5-9)
где L — удельная теплота плавления металла.
На рис. 78 показана зависимость температуры
воспламеняющейся частицы от времени для ряда значений температуры
среды. Начальный участок кривой определяется чисто
газодинамическим теплообменом, поэтому его наклон растет с ростом Т\, точнее
пропорционален (Ti—Tq). Затем становится заметным саморазогрев
и скорость роста температуры увеличивается. Релаксационные
процессы в пленке снижают скорость разогрева и на кривой появляется
более или менее пологий участок. Если время индукции (без учета
Хил) мало, что имеет место при достаточно высоких температурах
среды, то пологий участок укорачивается или вообще исчезает. Если
же температура среды оказывается ниже предельной, то разогрев
прекращается и воспламенения не происходит. Вслед за пологим
участком температура частицы достигает точки воспламенения практи-
266
чески мгновенно, поэтому время воспламенения фактически
определяется моментом окончания пологого участка.
Вычисление предельной температуры среды требует
установления необходимых и достаточных условий воспламенения частицы.
Необходимым и достаточным условием является выполнение
неравенства
МТ.ТЛ)>/Я(Т, 7\) (5-10)
при всех 7\ < Т < 2330/С. (5-11)
Множество значений температур среды Т\ при которой
выполняется условие (5-11), соответствует температуре воспламенения, а
минимальный член этого множества — предельной температуре
воспламенения.
Если бы отсутствовал механизм уменьшения диффузионной
проницаемости окисной пленки, то критерием воспламенения служило бы
условие касания функций /i и /2 в некоторой точке Т*
-^-=-^-1 А (Т*,ТЛ) =f2(T*,T1) при Т = Т*
и температура среды, определенная из этих условий, была бы
предельной температурой воспламенения. Она уменьшалась бы с
увеличением размера частицы из-за относительного ухудшения
теплопередачи по сравнению с саморазогревом. Это наблюдается, например,
при воспламенении магния. Результаты расчетов представлены на
рис. 77. Точки взяты из экспериментальной работы, выполненной
Гуревичем с сотр. [37, 41]). Таким образом, получено
удовлетворительное совпадение расчета, выполненного с использованием
реальных констант окисления алюминия, и экспериментальных данных
для предельных температур воспламенения мелких частиц.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
К главе I
1. «Metrologia», 1969, v. 5, № 2, p. 35—38.
2. Кенисарин AT. М., Чеховской В. #. Теплофизические свойства
веществ. Обзорная информация № 4. М., ВИНИТИ, 1976. 73 с. с ил.
3. Особо тугоплавкие элементы и соединения. Справочник. М.,
«Металлургия», 1969. 372 с. с ил. Авт.: Р. Б. Котельников, С. Н.
Башлыков, 3. Г. Галикбаров, А. И. Каштанов.
4. Огнеупоры для космоса. Справочник. Пер. с англ. М.,
«Металлургия», 1967. 266 с. с ил.
5. Brewer Leo — «Chemical Reviews», 1952, v. 52, p. 1—75.
6. Уикс К. E.j Блок Ф. E. Термодинамические свойства 65
элементов, их окислов, галогенидов, карбидов и нитридов. М.,
«Металлургия», 1965. 240 с. с ил.
7. Lambersion W. A., Muller М. Я.— «J. Am. Ceram. Soc», 1953, v.
36, K° 10, p. 329—334; 1953, v. 36, № 11, p. 365—368.
8. Lamberston W. A., Muller M. H., Gunzel F. H.~ «J. Am. Ceram.
Soc.» 1953, v. 36, № 12, p. 397—403.
9. Geach C. A., Harper M. E,— «Metallurgia», 1953, v. 47, №283,
p. 269—272.
10. Clausing P.— «Z. Anorg. u. Allgem. Chem.,», 1932, v. 204, p. 33—38.
11. Хейг Дж. Р., Линч Дж. Ф„ Хоулден Ф. С— В кн.: Огнеупоры для
космоса. Пер. с англ. М., «Металлургия», 1967, с. 133—138 с ил.
12. Data on chemical for Ceram Use. Bull. Nat. Res. Consul, 1949, p.
118, 193—195; Ceram; Abstr. Feb. 1951, p. 40—43-
13. Henning F.— «Naturwissenschaften», 1925, Bd 13, S. 661—666.
14. Mark S. D.— «J. Am. Ceram Soc», 1959, v. 42, № 4, p. 208.
15. Хармон С. Г,, Лох Л. Д., Остин С. Р. — В кн.: Огнеупоры для
космоса. Пер. с англ. М., «Металлургия», 1967, с. 211—234 с ил.
16. Физико-химические свойства окислов. Справочник. Под ред.
Г. В. Самсонова. М., «Металлургия», 1969. 455 с. с ил.
17. Cur ties С Е., Doney L. M„ Johnson С. R.— «J. Am. Ceram. Soc»,
1954, v. 37, № 10, p. 458—473.
18. Kanolt W.— «Bull NBS», 1914, Bd 10, S. 295; «Z. Anorg. u. All-
gem.— Chem.», 1914, Bd 85, S. 1—19; «J. Wash. Acad. Sci.», 1913,
v. 3, p. 315—318.
19. McNally R. N., Peters F. S., Ribbe P. H.— «J. Am. Ceram. Soc»,
1961, v- 44, p. 491—496.
20. Riley В.—«J. Sci. Instrum.», 1964, v. 41, p. 504—511.
21. Riley В.—«Rev. Hautes Temper, at Refract.», 1966, №3, p.
327 332
22. Ackermann R. S.-«J. Chem. Phys.», 1956, v. 25, №6, p.
1089—1097.
23. Anderson J. S„ Sawyer /. O,—«Nature» (London), 1960, v. 185,
№4717, p. 915—916.
24. Wisnyi L. G., Pijanowski S. Nuclear reactor fuel elements;
Metallurgy and Fabrication. Ed A. R. Kaufmann, NY — L, 1962, p.
312 326.
25. Ehlert 7\ C, Margrave S. L.— «J. Am. Ceram. Soc», 1958, v. 41,
p. 330—333.
26. Hausner tf.— «J. Nuclear Materials», 1965, v. 15, № 3, p. 179—183.
27. Ryshkewitch £. —«Oxide Ceramics, Physical Chemistry and
Technology», Academic Press, N. Y., 1960. 472 с
268
28. /Карпова Я. Л.—ЖПХ, 1939, т. 12, № 9, с. 1269—1270.
29. Ruff О., Ebert F., Woitinek Я— «Z. Anorg. u. Allgem. Chem.»,
1929, Bd 180, S. 252—256.
30. Техника высоких температур. Пер. с англ. М., ИЛ, 1959, 436 с.
с ил.
31. Foex Af.— «Solar Energy», 1965, v. 9, № 1, p. 61—66.
32. Schumacher E. £.—«J. Amer. Chem. Soc», 1926, v. 48, p. 396—405.
33. Ольшанский Я. Я.—ДАН СССР, 1959, т. IX, № 6, с. 1105—1109.
34. Wartenberg Я., Werth Я.— «Z. Anirg. u. Allgem. Chem.», 1930,
Bd 190, S. 178—180.
35. Ruff О., Laushke G—«Z. Anorg. Chem.», 1916, Bd 97, S. 73—76.
36. Wartenberg Я.— «Z. Anorg. u. Allgem. Chem.», 1932, Bd 207, S.
5-9.
37. Wartenberg H.t Reusch Я. J., Saran E. — «Z. Anorg. u. Allgem.
Chem.», 1937, Bd. 230, № 3, S. 257—260.
38. Lang S. Л1, Knudsen F. P., Filmore C. L.t Roth R. S. — «NBS
Circ», 1956, № 568, p. 1—32.
39. MacDonald N. F., Ransley C. £.— «Powder Metallurgy», 1959, v. 3,
p. 172—176.
40. Кандыба В. В., Кантор П. Б., Красовицкая Р. М., Фомичев Е. Я.—
«Изв. АН СССР», 1960, т. 131, № 1—6, с. 265—266; ДАН СССР,
I960,, т. 131, №3. с. 566—567.
41. Greenbaum M„ Weiner Л, Farber Ж.— «J. Phys. Chem.», 1965, v. 69,
№11, p. 4035—4039.
42. Latia R, E., Duderstedt E. C, Fryxell R. E.— «J. Nucl; Mater.»,
1970, v. 35, p. 350—352.
43. Бархатов Л. С, Каган Д. Я., Кенисарин М. М., Чеховской В. Я-,
Шпильрайн Э. Э. — «Теплофизика высоких температур», 1975,
№ 19 с 525 531
44. Aldred F. H., White Я. Е. 5.— «Trans. Brit. Ceram. Soc», 1959,
v. 58, p. 199—202.
45. Diamond /. S., Schneider S. S.— «J. Am. Ceram. Soc», 1960, v. 43,
p. 1—19.
46. Кантор П. Б., Лазарева Л. С., Кандыба В. В., Фомичев Е. М.—
«Укр. физич. журн.», 1962, т. VII, № 2, с. 206—210.
47. Чеховской В. Я., Петров В. Л.— «Измерительная техника», 1963,
№ 9, с. 26—28.
48. Gutt UP.—«J. Sci. Instrum.», 1964, v. 41, p. 393—398.
49. Gitlesen G., Motzfeld /(.— «Acta Chem. Scand.», 1965, v. 19, p.
661—667.
50. Кантор П. Б., Фомичев Е. М., Кандыба В. В.— «Измерительная
техника», 1966, № 5, с. 27—32.
51. Sata Г.—«Rev. Hautes Temer. et Refract.», 1966, v. 3, p. 337—343.
52. Urbain G.t Rouannet M.— «Rev. Haures Temer. et Refract», 1966,
v. 3, p. 363—369.
53. Schneider S. S., McDaniel C. L.— «J. Res. of NBS. A. (Phys. a.
Chem.)», 1967, v. 71A, № 317, p. 4—12.
54. Козловский Л. Л., Шлякова /С. С.— «Труды ВНИИ автогенной
обработки металлов». Сб. № 6. М., изд. ВНИИ автогенной
обработки металлов, 1960, с. 136—139.
55 Barber D. J.,Tighe N. /.— «J. Res. of NBS. (Phys. a. Chem.)»,
1965, v. 69A, p 23-27.
56. Gouffe Л.— «Rev. Opt.», 1945, № 1, p. 24—26.
57. Уббелоде Л. Плавление и кристаллическая структура. М., «Мир»,
1969. 420 с с ил.
269
58. Излучательные свойства твердых материалов. Справочник. Под
ред. А. Е. Шейндлиыа. М., «Энергия», 1974. 471 с. с ил.
59. Международная практическая температурная шкала МПТШ-68.
М., Изд-во стандартов, 1971. 33 с.
60. De Voss /. С—«Physica», 1954, v. 20, p. 669—689.
61. Schneider S, /.— «Pure and Applied Chemistry», 1970, v. 21, p.
117—122.
62. Foex M.— «Rev. Hautes Temper, et Refract», 1966, v. 3, p. 3—6.
63. Doman R. C, Ban Л В., McNally N. R., Alper A. M.— «Amer. Ce-
ram. Soc. Bull.», 1962, v 41, p. 584.
64. De Vries R. C, Roy R. — «Amer. Ceram. Soc. Bull.», 1954, v. 33,
№ 12, p. 370.
65. Кенисарин М. М., Кузичкин В. А., Чеховской В. Я.—
«Метрология», 1972, №8, с. 15—17.
66. Фомичев Е. Н., Кантор П. В., Кандыба В. В.— «Труды
метрологических институтов СССР. Исследования в области тепловых
измерений». Л.— М., ВНИИМ Комитет по стандартам СССР,
1969, № 111 (171), с. 31—36; 1971, № 129 (189), с. 43—45.
67. Фомичев Е. H.t Кантор П. Б., Кандыба В. В.—В кн.: Теплофизи-
ческие свойства твердых веществ. М.. «Наука», 1971, с. 147—150.
68. Gorski W., Dietzel Л.—«Rev. Hautes Temper et Refract», 1969, v.
6, p. 105—109.
69. Urbain G., Rouannet M.— «Rev. Hautes Temper, et Refract», 1969,
v. 6, p. 363—365.
70. Jones T. P.—«J. Austral. Ceram. Soc», 1969, v. 5, p. 41—44.
71. Ruff O., Sieferfeld H., Suda /.—«Z. Anorg. u. Allgem. Chem.»,
1913, Bd 82, S. 373-375.
72. Station W. 0.— «J. Chem. Phys.», 1951, v. 19, p. 33—34.
73. Friderikck £., Sitting L.—«Z. Anorg. u. Allgem. Chem.», 1925, Bd
145, S. 127—140.
74. Wartenberg H„ Gurr W.,— «Z. Anorg. u. Allgem. Chem.», 1931,
Bd 196, S. 374—378.
75. Wartenberg H., Prophet £.—«Z. Anorg. u. Allgem. Chem.», 1932,
Bd 208, S. 369—373.
76. Bunting E. N,— «J. of NBS», 1933, № 11, p. 713—715.
77. Sigurdson H.t Cole S. 5.—«J. Metals», 1949, v. 1, p. 905—909.
78. Pierre P. D. 5.—«J. Am. Ceram. Soc», 1952, v. 35, p. 118—122.
79. Lang S. M., Fillmore С L., Maxwell L. M. — «J, Res. of NBS»,
1952, Bd 48, S. 298—303.
80. Coughanour L W., De Prosse V. A.—«J. Res. of NBS», 1953, Bd
5 j $ g£ Q2
81. Rase D. E.} Roy R.— «J. Am. Ceram. Soc.» 1955, v. 38, p. 102—113.
82. Diamond J. S., Schneider 5. 5.—«J. Am. Ceram. Soc», 1960, v. 43,
p. 1—21.
83. Brauer G.> Littke W.— «J. Inorg. Nucl. Chem.», 1960, v. 16, p.
67—76.
84. Ackermann R. S., Rauh E. G.— «J. Chem. Thermodyn.», 1973, v. 5,
p. 331—340.
85. Jones T. P.—«J. Austral. Ceram. Soc», 1974, v. 10, p. 21—22.
86. Шейндлин А. Е., Кенисарин М. Н., Чеховской В. Я. — ДАН СССР,
1974, т. 216, № 3, с. 582—594.
87. Kenisarin M. H., Chekhouskoij V. У. (Кенисарин М. Н., Чеховской
В. Я0 —«Rev. Inst. Hautes Temper, et Refract», 1975, v. 12, p.
329—336.
270
88. Schneider S. J„ McDaniel CL— «Canad. Metallurg. Quart.», 1974,
v. 13, №2. p. 365—371.
89. Гордиенко С. П., Феночка Б. В., Фесенко В. £. Редкоземельные
металлы и их тугоплавкие соединения. Справочник. Киев. «Нау-
кова думка», 1971. 168 с. с ил.
90. Noguchi Т., Mizuko М. — «Solar Energy», 1967, v. 2, № 11, p. 90.
91. Curtis С. E.f Johnson J. R.— «J. Am. Ceram. Soc», 1957, v. 40,
p. 15—19.
92. Foex M. — «Z. Anorg. Allgem. Chem.», 1965, Bd 337, S. 313—324.
93. Мордовии О. А., Тимофеева H. И.,. Дроздова Л. //.— Изв. АН
СССР. Неорганические материалы», 1967, т. 3, № 1, с. 187—189.
94. Тресвятский С. Г., Кушаковский Б. И., Белеванцев Б. С—
«Атомная энергия», 1960, т. 9, вып. 1, с. 54—59.
95. Ruff О.— «Z. Angen. Chem.», 1933, Bd 46, S. 1—8.
К главе II
1. Горение порошкообразных металлов в активных средах. М.,
«Наука», 1972. 293 с. с ил. Авт.: П. Ф. Похил, А. Ф. Беляев, Ю. В.
Фролов, В. С. Логачев, А. И. Короткое; Мошкин Е. К- Развитие
отечественного ракетного двигателестроения. М., «Машиностроение»,
1973. 255 с. с ил.
2. Wartenberg Н„ Wahner G., Saran E. Nachr. Acad. Wiss. Gottin-
gen. Math —Phys. Klass. Fashgruppe, 1936, p. 65—71.
3. Kingery W. D.— «J. Amer. Ceram. Soc», 1959, v. 42. № 1, p. 6—11.
4. Maypax M. А., Митин Б. С, Ройтберг М. Б.— «Заводская
лаборатория», 1967, № 5, с. 964—966.
5. Костиков Б. Я., May pax M. А., Митин Б. С, Пеньков И. А.,
Свердлов Г. М.— В кн.: Высокотемпературные материалы. М.,
«Металлургия», 1968, с. 106—114.
6. Rasmussen /. /., Nelson R. P.—«J. Amer. Ceram. Soc», 1971, v.
54, № 8, p. 398—404.
7. Kirshenbaum A. D.t Cahil J. A.— «J. Inorg. Nucl. Chem.», 1960, v.
14, p. 283—292.
8. Зубарев Ю. Б., Костиков Б. И., Митин Б. С, Нагибин Ю. А.,
Нищета Б. В.— «Изв. АН СССР. Неорганические материалы», 1969,
т. 5, № 9, с. 1563.
9. Митин Б. С, Нагибин Ю. А.— «Журнал физической химии», 1970,
-~, т. XLIV, № 5, с. 1325—1326.
;10.Елютин Б. П., Митин Б. С, Анисимов Ю. С—«Изв. АН СССР,
V' Неорганические материалы», 1973, т. 9, № 9, с. 1585—1587.
11. Бархатов Л. С, Каган Д. Н.} Цыцаркин А. Ф., Шпильрайн Э. Э.,
Якимович /С Л.—«Теплофизика высоких температур», 1973, т. И,
№6, с 1188—1191.
12. Зубарев Ю. Б., Митин Б. С, Нищета Б. Б., Семененко Ю. П.—
«Журнал физической химии», 1976, т. XX, № 2, с 461—463.
13. Семенченко Б. К. Поверхностные явления в металлах и сплавах.
М., Гостехтеориздат, 1957. 490 с. с ил.
14. Анисимов Ю. С, Елютин В. П., Митин Б. С— «Изв. вуз.,
Цветная металлургия», 1974, № 4, с 42—44.
15. Анисимов Ю. С., Гриц Е. Ф., Митин Б. С— «Изв. АН СССР».
Неорганические материалы», 1977, т. 13, № 8, с. 1444—1447.
16. Смоляренко Б. Д., Якушев А. М., Еднерал В. П.— «Изв. вуз.
Черная металлургия», 1965, № 1, с. 55—60.
17 Евсеев П. П., Филиппов А. Ф.,— «Изв. вуз. Черная металлургия»,
1965, № 3, с. 55—59.
271
18. Дымов В. В., Байдов В. В. — «Специальные стали и сплавы». М.,
«Металлургия», 1968 (ЦНИИЧМ. Сб. № 61), с. 78—80.
19. Евсеев П. П., Филиппов А. Ф.— «Изв. вуз. Черная металлургия»,
1967. №3, с. 41—45.
20. Черепанов А. М., Тресвятский С. Т. Высокоогнеупорные
материалы и изделия из окислов. М., «Металлургия», 1964. 400 с. с ил.
21. Bockris I. О. М., Tomlinson J. M., White I. L. — «Transactions
Faraday Society», 1956, v. 52, № 3, p. 299—310.
22. Tomlinson /. M„ Heynes M. S. R., Bockris L О. M.— «Transactions
Faraday Society», 1958, v. 54, № 12, p. 1822—1833.
23. Shartsis L. Spinner S., Capps W.— «J. of the American ceramic
society», 1952, v. 35, p. 169—172.
24. Шпильрайн Э. Э., Якимович К А., Цицаркин А. Ф.,—
«Теплофизика высоких температур», 1974, т. 12, N° 2, с. 277—281.
25. Карязин И. А., Резниченко В. Л.—В кн.: Титан и его сплавы.
Вып. IV. М., Изд-во АН СССР, 1960, с. 73—88 с ил.
26. Резниченко В. А. Электротермия титановых руд. М., «Наука»,
1969. 205 с. сил.
27. Карязин И. А., Морозов А. А., Резниченко В. А. — «Изв. АН
СССР. Металлы», 1969, № 5, с. 28—34.
28. Морозов А. А., .Карязин И. А., Меняйлова Т. А., Резниченко
В. А.— В кн.: Процессы производства титана и его двуокиси. М.,
«Наука», 1973, с. 18—24 с ил.
29. Есин О. А., Гельд П. В. Физическая химия пирометаллургических
процессов. Ч. П. М., «Металлургия», 1966. 703 с. с ил.
30. Денисов С. И. Электротермия титановых шлаков. М.,
«Металлургия», 1970. 165 с. с ил.
31. Bartlett R. W., Hall J. К. — «Amer. Ceram. Bull.», 1965, v. 44, №5,
p. 444—451.
32. McNally R. #., Jen H. C, Balasubramanian N.— «J. Material Scien-
se», 1968, v. 3, p. 136—138.
33. Якобашвили С. В.. Хорунов В. X., Несмих В. С, Синица М. И.—
В кн.: Поверхностные явления в металлах. Кишинев, изд.
Кишиневского ГУ, с. 183—188.
34. Смирнова В. И., Ормонт Б. Ф.— ДАН СССР, 1952. т. LXXXI, № 5;
с. 751—753; Fordham 5.—«Proc. Roy Soc», 1948, А-194, р. 1—16.
35. Елютин В. П., Костиков В. И., Маурах М. А.. Митин Б. С,
Пеньков И. А.— В кн. Поверхностные явления в расплавах. Киев, На-
укова думка», 1968, с. 155—159.
36. Harkins W. D., Brown P. £.— «J. Am. Chem. Soc», 1919, v. 41,
p. 499—519.
37. Namba S., fsobe Т.— «Proc. 4-th Symp. of Electron Beam. Techn.»,
1960, p. 29—30.
38 Tihanovski I. I. (Тихановский И. И.) — «Phusikalisch Z.», 1924,
Bd 25, S. 299; 1925, Bd 26, S. 522.
39 Frend В. В., Frend H. Z. — «J. Amer. Chem. Soc», 1930, v. 52, p.
1772—1781.
40. Клячко Ю. Л.—«Заводская лаборатория», 1937, т. 6, № 11, с.
1376—1382 с ил.
41. Кунин Л. Л. Поверхностные явления в металлах. М., Металлург-
издат, 1955. 304 с. с ил.
42. Куколев Г. В. Химия кремния и физическая химия силикатов. М.,
«Высшая школа», 1966. 463 с. с ил.
43. Champion F. С, Davy #.— «Properties of matter», Prentice — Hall.
Inc. N. Y., 1937, p. 126.
272
44. King Т. В—«J. Soc. Glass Technology», 1951, v. 35, p. 241—247.
45. Shartsis L., Spinner S., Smock A. W.~ «J. Res. NBS», 1948, v. 40,
p. 61—73.
46. Shartsis L., Shermer H. F., Bestul А £k— «J. Amer- Ceram. Sec »,
1958, v. 41, № 12, p. 507—516.
47. Шпильрайн Э. Э., Якимович К А., Цицаркин А. Ф. —
«Теплофизика высоких температур». 1973, т. 11, № 5, с. 1001—1009.
48. McNally R. #., Jen Н. С, Balasubramanian N.— «J. Mater. Sci.»,
1968, v. 3, p. 136—138.
49. Таблицы физических величин. Справочник. Под ред. акад И. К.
Кикоина. М., Атомиздат, 1976. 1006 с- с ил.
50. Евсеев П. Я., Филиппов Л. Ф.— «Изв. вуз. Черная металлургия»,
1965, № 3, с. 70—76.
51- Попель СИ. — В кн.: Металлургические шлаки и применение их в
строительстве. М., Госстройиздат, 1962, с. 97—127 с ил.
52. Дерябин А. А., Попель С. Я.— «Изв. вуз. Черная металлургия»,
1964, №8, с. 5—11.
53. Смоляренко В. Д., Якушев А. М., Еднерал Ф. Я.— «Изв. вуз.
Черная металлургия», 1965, № 1, с. 55.
54. Попель С. И., Есин О. А.—«Журнал неорганической химии»,
1957, т. 2, вып. 2, с. 632—641 с ил.
55. Бобкова О. С, Пастухов В. С, — В кн.: Поверхностные явления
в расплавах и процессах порошковой металлургии». Киев. Изд-во
АН УССР, 1963, с. 212—221 с ил.
56. Есин О. А. —«Успехи химии», 1957, № 6, с. 1374—1387 с ил.
57. Есин О. А. — «Изв. вуз. Черная металлургия», 1960, № 8, с. 5—
14 с ил.
58,; Жуховицкий А. А. — «Журнал физической химии», 1945, т. 19.
' вып. 7, с. 337—349 с ил.
59. Темкин М. Я.— «Журнал физической химии», 1946, т. 20, вып. 1,
с. 105—110.
60. Kazakevitch P. P.— «Revue de Metallurgie», 1960, v. 57, № 2,
p. 149—162.
61. Hofmaier <?. — «Berg, und Huttenmannische Monatschefte», 1968,
v. 113, №7, p. 271—278.
62. Елютин В. П., Костиков В. И,, Митин Б. С, Нагибин Ю. А. —
«Журнал физической химии», 1969, т. XLIII, № 3, с. 579—583.
63. Швидковский £". Г. Некоторые вопросы вязкости расплавленных
металлов. М., ГИНТЛ, 1955. 207 с. с ил.
64. Глестон С, Лейдлер К, Эйринг Г. Теория абсолютных скоростей
реакций. М., ИЛ, 1948, 584 с. с ил.
65. Furthtz R. — «Proc. Camb. Phil. Soc», 1941, v. 37, p. 255—264.
66. Fay Я. —«J. Phys. Chem.», 1966, v. 70, № 3, p. 890—893.
67. Bockris /. O. H., Kitschener J., Ignatowich J. S., Tomlinson J. —
«Trans Farad. Soc», 1952, v, 48, № 1, p. 75—91.
68 Евсеев П. Я., Филиппов А. Ф. — «Изв. вуз. Черная металлургия»,
1967, № 1,с 55—60.
69. Бардин Я. П., Резниченко В. А. — В кн.: 40 лет Советской черной
металлургии. М., «Металлургиздат», 1957, с. 31—37.
70. Резниченко В. Л., Рапопорт М. Б., Ткаченко В. А. Исследование
электроплавки титановых шлаков. М., Изд-во АН СССР, 1963.
200 с. с ил.
71. Елютин В. П., Митин Б. С, Нагибин Ю. Л. —«Изв. АН СССР.
Неорганические материалы». 1971, т. 7, № 5, с. 880—882.
18—894
273
72. Белащенко Д. К. Явления переноса в жидких металлах и
полупроводниках. М., Атомиздат, 1970. 397 с. с ил.
73. Александров В. #., Осико В. В., Татаринцев В. М. — «Изв. АН
СССР. Неорганические материалы», 1972, т. 8, № 5, с. 956—957.
74. Шпильрайн Э. Э., Каган Д. Н., Бархатов А. С, Жмакин Л. И. —
«Теплофизика высоких температур», 1976, т. 14, № 5, с. 948—952.
75. Шпильрайн Э. Э., Каган Д. Н., Бархатов А. С, Жмакин Л. И. —
«Теплофизика высоких температур», 1977, т. 15, № 2, с. 423—424.
76. Shpilrain Е. Е., Kagan D. N., Barkhatov A. S., Zhmakin L. /. —
(Шпильрайн Э. Э., Каган Д. Н., Бархатов А. С, Жмакин Л. И.) —
Symposium on Thermophysical Propterties; Abstracts, Gaithersburg
USA, 1977, p. 55.
77. Манаков А. И., Есин О. A.t Ленинских Б. Ж —ДАН СССР. 1962,
т. 142, №5, с. 1124—1127.
78. Degre J., Pang L„ Henderson Л —«Trans. Metal Soc. AJME», 1961,
v. 221, p. 56.
79. Анисимов Ю. С, Елютин В. П., Митин Б. С, Солдатов Е. А.—
«Изв. вуз. Цветная металлургия», 1974, № 3, с. 60—62.
80. Митин Б. С.—В кн.: Физическая химия поверхности расплавов.
Тбилиси, «Мецниереба», 1977, с. 311—319.
К главе III
1. Боровик К. — ЖЭТФ, 1948, № 186. с. 48—56.
2. Osida /. — «Ргос. Phis. Math; Soc. Japan.», 1939, v. 21, p. 3353.
3. Horrocks J. K., McLaghlin E. — «Trans. Faraday Soc», 1963, v. 59,
p. 1709—1716.
4. Horrocks J. К*, McLaghlin E. — «Trans. Faraday Soc», 1960, v. 56,
p. 206—212.
5. Kamal /., McLaghlin E. — «Trans. Faraday Soc», 1964, v. 60,
p. 809—816.
6. Чепмен С, Каулинг Т. Математическая теория неоднородных
газов. М., ИЛ., I960. 510 с с ил.
7. Longuet-Higgins Н. С, Pople J. A, — «J. Chem. Phis.», 1956, v. 25,
p. 884—889.
8. Мак-Кой Б. Д., Даллер Д. С. —«Механика», 1970, т. 1, с. 79—95.
9. Longuet-Higgins F. С, Pople К-— «J- Mol. Phis.», 1958, v. 1,
p. 284—294.
10. Green H. S. Moleqular Theory of Fluids. Interscience. N. Y.—L.,
1952, 200 p.
11. Irving J. H., Kikwood J. С — «J- Chem. Phis.», 1950, v. 18, p. 818—
823.
12. Бриджмен В. П. Физика высоких давлений. М., ОНТИ, 1935.
402 с. с ил.
13. Миснар А. Теплопроводность твердых тел, жидкостей, газов и
их композиций. М., «Мир», 1968. 464 с. с ил.
14 Филиппов Л. П. Исследование теплопроводности жидкости. М.,
Изд. МГУ, 1970. 239 с. с ил.
15 Zwanzig R. W., Kirkwood J- G., Stripp К F., Oppenheim /- — «J.
Chem. Phis.», 1953, v. 21, p. 2050—2055.
16. Уленбек Дж., Форд Дж. Лекции по статистической механике.
Пер. с англ. М., «Мир», 1965. 307 с. с ил.
17. Честер Дж. Теория необратимых процессов. Пер. с англ. М.,
«Наука», 1966. 111 с с ил.
18. Кубо Р. Термодинамика необратимых процессов. Пер. с нем. М.,
«Мир», 1970. 304 с. с ил.
274
19. Зубарев Д.*Н. Неравновесная статистическая термодинамика.
«Наука», 1971. 415 с. с ил.
20. McLaughlin Е. — «Chin. Rev.», 1964, v. 64, p. 389.
21. Pud P., Шервуд Т. Свойства газов и жидкостей. Л., «Химия»,
1971. 702 с. с ил.
22. Чандрасекар С. Перенос лучистой энергии. Пер. с англ. Под ред.
Е. С. Кузнецова. М., ИЛ. 1953. 432 с. с ил.
23. Соболев В. В. Перенос лучистой энергии в атмосферах звезд
и планет. М., Гостехиздат, 1956, 391 с. с ил.
24. Крупская Я. H.t Манжелин В. Г. — ЖЭТФ, 1968, т. 55, № 6,
с. 2075—2082.
25. Dukdald /. S., MacDonald D. К. S. — «Phis. Rev.», 1955, v. 98,
№6, p. 1751-1756.
26. Lawson A. W. — «J. Phys. Chem. Soc», 1957, v. 3, p. 154—161.
27. Keyes R. W,— «Phys. Rev.», 1959, v. 115, № 3, p. 565—567.
28. Wits G. K., Woods S. B. — «Phil. Mag», 1958, v. 3, p. 785—792.
29. Ромашин А. Г. — В кн.: Теплофизические свойства твердых тел.
Киев, «Наукова думка», 1971. 218 с. с ил.
30. Shack G. A., Austerman S. В.— «J. Appl. Phis.», 1971, v. 42, № 12,
p. 4713—4718.
31. Физическая акустика. Т. III. Ч. Б. Пер. с англ. Под ред.
Л. Д. Розенберга. М., «Мир», 1968. 319 с. с ил.
32. Росселанд С. Астрофизика на основе теории атома. М.—Л.,
ОНТИ, 1936. 302 с. с ил.
33. Невский А. С. Теплообмен излучением в металлургических печах
и топках котлов. Свердловск, Металлургиздат, 1958. 368 с. с ил.
34. Спэрроу Е. М., Сесс Р. Д. Теплообмен излучением. Пер. с англ.
Л., «Энергия», 1971. 294 с. с ил.
35. Адрианов В. Н. Основы радиационного и сложного теплообмена.
М., «Энергия», 1972. 464 с. с ил.
36. Кейз К-, Цвайфель П. Линейная теория переноса. Пер. с англ.
Под ред. В. М. Масленникова. М., «Мир», 1972. 384 с. с ил.
37. Адрианов В. Н. — В кн. Тепло- и массоперенос. Т. 2. Минск,
«Наука и техника», 1965. 454 с. с ил.
38. Белов Г. #. — ТВТ, 1972, т. 10, № 6, с. 1268—1276.
39. Висканта Р. «Теплопередача, сер. С», 1965, т. 87, № 1, с. 171—
180.
40. Viskanta R., Grosh R. J. — «Trans. ASME. Ser. C», 1962, v. 84, № 1,
p. 63—72.
41. Viskanta R., Grosh R. /. — «Appl. Mech. Rev.», 1964, v. 17, № 2.
p. 91—100.
42. Мень А. А., Сеттарова 3. С. — «Теплофизика высоких
температур», 1972, т. 9, № 2, с. 279—284 с ил.
43. Мень А. Л., Сергеев О. А., Чечельницкий А. 3. — В кн.: Тепло-
и массоперенос. Т. 7. Минск, «Наука и техника». 1972, с. 448—451.
44. Мень А. А, — «Теплофизика высоких температур», 1972, т. 10,
№ 5, с. 1073—1079.
45 Мень А. А., Сергеев О. А. — «Труды метрологических институтов
СССР». Сб. № 171. Вып. III. M., Изд-во стандартов, 1969, с. 18—
20.
46. Kellet В. 5. — «J. Opt. Soc. Amer.», 1952, v. 42, № 5, p. 339-343.
47. Висканта Р., Мерриан Р. А. — «Теплопередача, сер. С», 1968,
т 90 № 2 с 71—81 с ил.
48 Poltz H. ~ «Int. J. Heat Mass Transfer», 1965, v. 8, № 4, p. 609—620.
49. Хрусталев Б. Л. — ИФЖ, 1970, т. 18, № 4, с. 740—762 с ил.
18*
275
50. Филимонов С. С, Хрусталев Б. А. — В кн.: Теплообмен,
гидродинамика и теплофнзнческке свойства веществ. М., «Наука», 1968.
279 с. с ил.
51. Пигальская Л. А. Теплофизические свойства твердых тел. Киев,
«Наукова думка», 1971. 218 с. с ил.
52. Оптические материалы для инфракрасной техники. М., «Наука»,
1965. 335 с. с ил. Авт.: Е. М. Воронков, Г. И. Дистлер, Б. Н. Гре-
чушникова, И. П. Петров.
53. Излучательные свойства твердых материалов. Справочник. Под
ред. А. Е. Шейндлина. М., «Энергия», 1974. 471 с. с ил.
54. Инфракрасные спектры неорганических стекол и кристаллов. Л.,
«Химия», 1972. 304 с. с ил. Авт.: А. Г. Власов, В. А. Флоринская,
А. А. Венедиктов, К. П. Дутова, В. Н. Морощов, Е. В. Смирнова.
55. Hulton А. Л —«Electron. Mater.», 1973, v. 2, p. 211—215.
56. Мень А, А., Чечельницкий А. 3., Соколов В. А., Холодова Л. А. —
«Неорг. матер.», 1974. т. 10, № 3, с. 548—549.
57. Charvant F. R., Kingery W. D. — «J. Am. Cer. Soc», 1957, v. 40,
№5, p. 306—315.
58. Plass G. N. — «Appl. Opt.», 1965, т. 4, № 12, p. 1616—1619.
59. О скотский В. С, Смирнов И. А. Дефекты в кристаллах и
теплопроводность. Л., «Наука», 1972. 160 с. с ил.
60. Девяткова Е. Д., Петров А. В., Смирнов В. А., Мойжес Б. #. —
ФТТ, 1960, т. 2, № 4, с. 738—744.
61. Poltz П., Jugel R. — «Int. J. Heat mass Transfer.», 1967, v. 10,
p. 1075—1084.
62. Гуренкова Т. В., Норден П. А., Усманов А. Г. — В кн.:
Теплофизические свойства жидкостей. М., «Наука», 1973. с. 57—59.
63. Геллер В. 3., Парамонов И. А., Татевосов Г. Д. — В кн.: Тепло-
физические свойства жидкостей. М., «Наука», 1973, с. 93—98.
64. Расторгеев Ю. А., Богатое Г. Ф., Григорьев Б. А. — В кн.:
Теплофизические свойства жидкостей. М., «Наука», 1970, с. 88—92.
65. Теплопроводность газов и жидкостей. М., Изд-во стандартов.
1970. 155 с. с ил. Авт.: Н. Б. Варгафтик, Л. П. Филиппов,
А. А. Тарзиманов, Р. И. Юрчак.
66. Schatz J. E.t Simmons G. J. — «Appl. Phys.», 1972, v. 43, № 6,
p. 2586—2591.
67. Поляк Г. Л. — В кн.: Конвективный и лучистый теплообмен. М.,
Изд-во АН СССР, 1960, с. 255—263.
68. Камке Э. Справочник по обыкновенным дифференциальным
уравнениям. М., «Наука», 1965. 703 с. с ил.
69. Филиппов Л. П. Измерение тепловых свойств твердых и жидких
металлов при высоких температурах. М., Изд. МГУ, 1967, 325 с.
с ил.
70. Thermophysical Properties of Matter. V. 6. Ed. J. S. Toulouksain.
N. Y—W., 1970. 388 p.
71. Thermophysical Properties of Matter. V. 1, Ed. J. S. Toulouksain.
N. Y.—W., 1970. 418 p.
72. Соколова Т. В., Козлова И. А., Дерко X., Кийко А. В.— «Неорг.
матер.», 1973, т. IX, № 4, с. 611—617.
73. Lee D W., Kingery W. D. — «J. Amer. Cer. Soc», 1960, v. 43, № 1,
p. 13—18.
74. Кантор Н. Б., Лазарева Л. С, Кандыба В. В., Фомичев Е. Н. —
УФЖ, 1962, т. VII, № 2, с. 210—215.
75. Gryvnak D. A., Burch D. Е. — «J. Opt. Soc. Amer.», 1965, vc oo,
№ 6, p. 625.
276
76. Соколова Т. В., Бузовкина Т. Б., Кийко ЛГ В., Успенская Р. И.—
В кн.: Тепло- и массоперенос. Т. 7. Минск. «Наука и техника»,
1972, с. 506—609.
К главе IV
1. Neuman F. Vorlesungen uber die Theorie der Kapillaritat. Leipzig:,
1894. 118 S.
2. Иarkitis W. D. — «Phys- Chem. of Surface Films», Reinhold Publ.
Corp. N. Y., 1952, 413 p.
3. Лппен А. А. Температуроустойчивые неорганические покрытия.
Л., «Химия», 1967. 240 с. с ил.
4. Костиков В, И., Митин Б. С. — «Высокотемпературные
материалы». М., «Металлургия», 1968 (МИСиС. Сб. № 49), с. 114—124.
5. Сорокин Ю. В., Хлынов В. В.. Есин О. А. — ЖФХ, 1966, т. 40,
№7, с. 1598—1603.
6. Щукин Е. Д., Горюнов Ю. В., Деньщикова Г. И., Перцев Н. В.,
Сумм Б. Д. — «Коллоидный журнал», 1963, т. 108,, № 1, с. 25—33.
7. Humenic M., Klngery W. D. — «J. Amer. Ceram. Soc», 1954, v. 18,
p. 37—44.
8. Жуховицкий А. А., Григорян Б. Л., Михалик Е. — ДАН СССР,
т. 155, № 2, с. 392—394.
9. Найдич Ю. В. Контактные явления в металлических расплавах.
Киев, «Наукова думка», 1972. 196 с. с ил.
10. Neuman Л. W., Renzow D., Reumath Н., Rlchter /. Е. — «Forschvit-
tsher Koloide und Polym.», 1961. Bd 49, S. 55—64.
11. Wenzel R. N. — «Ind. Eng. Chem.», 1936, v. 988, p. 26—32.
12. Дерягин Б. В.—ДАН СССР, 1946, т. 51, с. 357—362.
13. Горюнов Ю. В., Перцов И. В., Сумм Б. Д., Щукин Е. Д. —ДАН
СССР, 1962, т. 146, с. 638—641.
14. Shuttleworth R., Bailey G. Z. — «Disc. Faraday Soc», 1948, v. 16,
p. 3—11.
15. Найдич Ю. В. — В кн.: Поверхностные явления в расплавах и
возникающих из них твердых фазах. Нальчик,
Кабардино-Балкарское книжное изд-во, 1965, с. 30—39 с ил.
16. Johnson R. E.t Deffre R, Н. — «Adv. in Chemistry Series», 1964,
v. 112, p. 43—52.
17. Blkerman I. /. — «Proc. 2-nd Int. Congress, of Surface Activity»
(London), 1957, v. 125, p. 2—9.
18. Горюнов Ю. В., Сумм Б. Д.— «Вестник МГУ. Хим. В», 1973,
с. 259—270.
19. Сумм Б. Д., Горюнов Ю, В. — В кн.: Физико-химические основы
смачивания и растекания. М., «Химия», 1976. 232 с. с ил.
20. Горюнов Ю. В. — «Успехи химии», 1964, т. 33, № 3, с. 1062—1067.
21. Елютин В. П., Костиков В. И., Маурах М. А. — В кн.:
Поверхностные явления в расплавах и возникающих из них твердых
фазах. Нальчик, Кабардино-Балкарское книжное изд-во, 1965,
с. 352—356.
22 Елютин В. П., Маурах М. А. — «Высокотемпературные материа-
" лы». М., «Металлургия», 1968 (МИСиС. Сб. № 49), с. 139—151.
23 Григорьев Г. А., Елютин В. П., Маурах М, Л. — «Изв. АН СССР.
' ОТН», 1957, № 8, с. 139—151.
24 Рауд Э. А,, Сумм Б. Д., Щукин Е. Д. —ДАН СССР, 1972, т. 205,
№5, с. 1134-1138.
25. Левин В. Г. Физико-химическая гидродинамика. М., Физматгиз,
1959. 699 с. с ил.
277
26. Сумм В. Д., Горюнов Ю. В., Щукин В. Д. — В кн.: Физическая
химия поверхностных явлений при высоких температурах. Киев,
«Наукова думка», 1971, с. 133—138.
27. Фрумкин А. Я. — ЖФХ, 1938, т. 12, с. 337- -342.
28. Деньщикова Л Я., Горюнов Ю. В, Сумм Б. Д., Травкин В. Д. —
«Физико-химическая механика материалов», 1965, т. 1, № 2,
с. 350—355.
29. Френкель Я. Я. Кинетическая теория жидкостей. М., Изд-во АН
СССР. 1945. 424 с. с ил.
30. Елютин В. П., Костиков В. И., Маурах М. А. — В кн.:
Поверхностные явления в расплавах и возникающих из них твердых
фазах. Нальчик, Кабардино-Балкарское книжное изд-во, 1965,
с. 345—351 с ил.
31. Аппен А. А., Глушкова В. Б., Каялова С. С.— «Изв. АН СССР.
Неорганические материалы», 1965, т. 1, № 3, с. 7—12.
32. Еременко В. Н., Найдич Ю. В. — ЖФХ, 1959, т. 33, № 2, с. 339-
347.
33. Третьяков Ю. Д. Химия нестехиометрических окислов. М., Изд-
во МГУ, 1974. 364 с. с ил.
34. Bartlett R. W., Hall J. К — «Ceram. Bull.», 1965, v. 44, № 5,
p. 444—448.
35. Костиков В. И., Левин В. Я., Маурах М. А., Митин Б. С. —
«Высокотемпературные материалы». М., «Металлургия», 1968
(МИСиС. Сб. № 49), с. 125—138.
36. Быковский А. Я., Пролесковская Ю. В. — В кн.: Поверхностная
диффузия и растекание. М., «Наука», 1969, с. 193—200.
37. Физико-химия флотационных процессов. М., ГОНТИ, 1933, 320 с.
с ил. Авт.: П. А. Ребиндер, М. Е. Липец, М. М. Римская, А. Б. Та-
убман.
38. Елютин В. П., Костиков В. Я., Маурах М. А. — «Изв. вуз.
Черная металлургия», 1965, № 3, с. 5—9.
39. Елютин В. П., Митин Б. С. Гриц Е. Ф. — «Изв. АН СССР.
Неорганические материалы», 1974, т. 10, № 5, с. 844—847.
40. Августинник А. И., Журавлев Г. И., Матусов И. А. — ЖПХ,
1969, т. 42, № 1,с. 62—68.
41. Курс физической химии. Т. 1. Под ред. Я. П. Герасимова. М.,
«Химия», 1969. 592 с. с ил.
42. Семенченко В. К. Поверхностные явления в металлах и сплавах.
М., Техтеоретиздат, 1957. 491 с. с ил.
43. Жуховицкий Л. Л. —ЖФХ, 1946, т. 19, с. 337—343.
44. Тресвятский С. Т., Черепанов Л. М. Высокоогнеупорные
материалы и изделия из окислов. М., «Металлургия», 1964. 246 с. с ил.
45. Августинник Л. Я., Журавлев Г. Я., Матусов Я. Л. —ЖПХ,
1969, т. 42, № 3, с. 689—691.
46. Юдин Б. Ф., Марколия Г. П., Воронин Я. Я. — «Труды
Всесоюзного государственного института научно-исследовательских
работ огнеупорной промышленности», Ленинград, Стройиздат, 1965,
с. 204—238 с ил.
47. Armstrong W. M., Chaklader А. С. D.t Declane M. Z. Л. —
«J. Amer. Ceram. Soc», 1962, v. 45, № 9, p. 1107—1113.
48. Куликов Я. С. Термическое разложение соединений. М.,
«Металлургия», 1969. 250 с. с ил.
49. Уикс К. Е., Блок Ф. Е. Термодинамические свойства 65
элементов и их соединений. М., «Металлургия», 1965. 240 с. с ил.
278
50. Термодинамические свойства индивидуальных веществ. В 2-х т.
Под ред. В. П. Глушкова. М., Академиздат, 1962, 2078 с. с ил.
51. Кофстад П.—В кн.: Реакции между газами и металлами при
высоких температурах. Под ред. В. А. Кириллина. М., «Наука»,
1967, с. 183—205.
52. Защита вольфрама от окисления при высоких температурах. М.,
Атомиздат, 1968. 159 с. с ил. Авт.: В. Е. Иванов, Е. П. Нечипо-
ренко. Л. Н. Ефименко, М. И. Юрченко.
53. Глестон С, Лейдлер К, Эйринг Г. Теория абсолютных
скоростей реакций. М., ИЛ., 1948, 535 с. с ил.
54. Елютин В. Я., Павлов Ю. А. Высокотемпературные материалы.
Ч. I, M., «Металлургия», 1972. 264 с. с ил.
55. Елютин В. П., Павлов Ю. А., Челноков В. С. — «Изв. вуз.
Цветная металлургия», 1972, № 4, с. 43—48.
56. Сох J. H.t Ridgeen Z. М. — «Canad. Journ. of Chem.», 1963, v. 41,
№ 4, p. 671—678.
57. Kamarek K. Z., Cahdes A., Klinger N. — «Journ. of the Electrochem.
Soc. 2», 1963, v. 110, № 7, p. 763—769.
58. Гриц Е. Ф., Митин Б. С, Резников А. Д. — «Физика и химия
обработки материалов», 1975, № 3, с. 70—74.
59. Гриц Е. Ф., Митин Б. С, Резников А. Д. — «Коллоидный
журнал», 1975, т. 37, № 5, с. 956—958.
60. Гриц £. Ф., Митин Б. С, Резников А. Д. — «Изв. АН СССР.
Неорганические материалы», 1977, т. 13, № 2, с. 271—274.
61. Гриц Е. Ф., Митин Б. С. — «Физическая химия», 1974, т. 49, № 10,
с. 2664—2666.
62. Гриц Е. Ф., Елютин В. П., Митин Б. С.—«Изв. АН СССР.
Неорганические материалы», 1974, т. 10, № II, с. 2093—2095.
63. Гриц Е. Ф., Елютин Б. П., Митин Б. С. — «Изв. АН СССР.
Неорганические материалы», 1974, т. 10, № 12, с. 2171—2174.
64. Митин Б. С, Гриц Е. Ф., — В кн.: Адгезия расплавов. Киев,
«Наукова думка», 1974, с. 80—84.
65. Гриц Е. Ф., Митин Б. С. Деп. ВИНИТИ № 2344—76, 1976.
66. Анисимов Ю. С, Гриц Е. Ф., Митин Б. С. Деп., ВИНИТИ,
№ 2759—76, 1976.
К г л а в е V
1. Hart R. К. — «Ргос. Roy. Soc», 1956, v. 236A, p. 68—73.
2. Gulbrusen E. A., Wysoug W. S. — «J. Phys. Chem.», 1947, v. 51,
№ 12, p. 1087—1091.
3. Gulbrasen E.A. — «Ann. N. Y. Acad. Scb, 1954, v. 58, p. 830—837.
4. Hunter M. S., Fowle D. — «J. Electrochem. Soc», 1956, v. 103, №9,
p. 482—485.
5. Smeltzer W. W. — «J. Electrochem. Soc», 1956, v. 103, № 4,
p. 209—214.
6 Aylmore D. W., Gregg S. /., Jepson W. B. — «J. of Institute of
Metals», 1960, v. 88, № 1, p. 205—208.
7 Blackburn P. E., Gulbransen E. A, — «J. Electrochem. Soc», 1960,
' v. 107, № 12, p. 944—950.
8. Cohran C. N„ Sleppy W. S. — «J. Electrochem. Soc», 1961, v. 108,
№ 4, p. 322—327.
9. Sleppy W. 5. —«J. Electrochem. Soc», 1961, v. 108, № 12,
p Ю97—1102.
10. Doherty P. E., Davis R. S. — «J. Appl. Phys.» 1963, v. 34, № 3,
p. 619—628.
279
11. Мержанов А. Г., Чальченко Ю. А., Григорьев Ю. М., Машка-
нов Л. Б. — В кн.: Горение и взрыв. М., «Наука», 1972, с. 245—
249.
12. Митин Б. С, Самотейкин В. В. — Деп. ВИНИТИ № 2374—70,
1970.
13. L. De Brouckere. — «J. Inst. Metalls», 1953, v. 66, p. 317—322.
14. Kerr J. 5. —«Acta Cryst.», 1956, v. 9, p. 879—886.
15. Мальцев М. В., Чистяков Ю. Д., Ципин М. Я. — ДАН СССР,
1954, т. 99, с. 813—814.
16. Елютин Е. П., Митин Б. С, Самотейкин В. В. — «Журнал
физической химии», 1971, т. 45, с. 2342—2344.
17. Елютин В. П., Митин Б. С, Самотейкин В. В. — «Журнал
физической химии», 1971, т. 45, с. 2340—2342.
18. Архаров В. Я., Кичигина 3. 77.— В кн.: Механизм
взаимодействия металлов с газами. М., ИФХ АН СССР, 1964, с. 93—116.
19. Oishi Y.t Kingery W. D. — «J. Chem. Phys.», 1960, v. 33, № 2,
p. 480—486.
20. Neufeld P., Nagpaul N. K., Ashdown R., Akbar M. — «Electrochem.
Acta», 1972, v. 17, № 9, p. 1543—1546.
21. Cohen A. S., Rossini A. R. — «Bol. Soc. esp. ceram. у vidrio», 1972,
v. 11, №3, p. 183—186.
22. Мюллер Р. Л. Электропроводность стеклообразных веществ. Л.,
«Химия», 1968. 251 с. с ил.
23. Шишаков Н. А., Андреева В. В., Андрущенко Я. К. Строение и
механизм образования окисных пленок на металлах. М., Академ-
издат, 1959. 195 с. с ил.
24. Wilsdorf Я. G. F.— «Nature», 1951, v. 168, p. 600—601.
25. Каральник С. М., Домашевская Д. П. — В кн.: Химическая связь
в кристаллах. Минск, «Наука и техника», 1969, с. 161—166.
26. Chun Я. V., Klein G. — «Physics Letters», 1968, v. A 28, p. 334—
343.
27. Бочарова Т. Т., Кример Б. Я., Лоцманов С. Я.— «Физика
металлов и металловедение», 1971, т. 31, с. 1093—1094.
28. McPherson — «J. Mat Sci.», 1973, v. 8, № 6, p. 851—854.
29. Гордон Д. А., Дрю Ч., Прентис Дж., Найп Р. — «Ракетная
техника и космонавтика», 1968, т. 6, № 4, с. 3—11.
30. Горение порошкообразных металлов в активных средах. М.,
«Металлургия», 1972. 290 с. с ил. Авт.: П. Ф. Похил, А. Ф. Беляев,
Ю. В. Фролов, В. С. Логачев, А. И. Короткое.
31. Фролов Ю. В., Похил Я. Ф., Логачев В. С. — «Физика горения
и взрыва», 1972, т. 8, № 2, с. 213—236 с ил.
32. Watermeier L. A. and oth. — «Ninth Sympos (Intern.) on
Combust». New York, 1963, p. 703—715.
33. Инами Я. Х„ Шенфилд X. — «Ракетная техника и космонавтика»,
1964, т. 7, с. 196—204.
34. Crump /. — I. С. R. P. G. 2-nd Combustion Conference; Chemical
propulsion information agency publication 105, May 1966, p. 321—
329.
35. Dean L. E., Keith R. C. and oth. —«J. Spacecraft and Rockets»,
1965, v. 2, p. 753—757.
36. Зенин Л. А., Гладкова А. П., Бобылев В. К-, Лейпунский О. И.—
ДАН СССР, 1968, т. 181, с. 637—639.
37. Фролов Ю. В., Короткое А. И., Лейпунский О. И., Похил Я. Ф.—
IX Украинская республиканская межвузовская конф. по вопросам
280
испарения, горения и газовой динамики дисперсных систем,
Материалы конференции. Одесса, изд. Одесского ГУ, 1969, с. 45—49.
38. Попов Е. И., Кашпоров Л. Я., Мальцев В. М., Брейтер А. Л. —
«Физика горения и взрыва». 1973, т. 9, с. 240—246.
39. Friedman R., Machek Л. —«Combust and Flame», 1962, v. 6,
p. 9—19.
40. Кюл Д. К.— «Ракетная техника и космонавтика», 1965, т. 12,
с. 83—95 с ил.
41. Гуревич М. А., Лапкина К. И., Озеров Е. С.—«Физика горения
и взрыва», 1970, т. 6, с. 172—175.
42. Беляев А. Ф., Ермолаев Б. С, Короткое Л. #.. Фролов Ю. В. —
«Физика горения и взрыва», 1969, т. 5, № 25, с. 207—217.
43. Беляев А. Ф., Фролов Ю. В., Короткое А. И. — «Физика горения
и взрыва», 1968, т. 4, № 3, с. 323—329.
44. Хайкин Б. И., Блашенко В. #., Мерханов А. Г.— «Физика
горения и взрыва», 1970, т. 6, № 4, с. 474—488 с ил.
45. Елютин В. Л., Митин Б. С, Самотейкин В. В. — В кн.: Горение
и взрыв. М., «Наука», 1972, с. 241—244.
46. Семенов Н. //. — «Успехи физических наук», 1940, т. 23, вып. 3,
с> 249—292.
47. Rautenberg Т. #., Johnson P. ZX — «J. Opt. Soc. Amer.», 1960,
v. 50, p. 602-606.
48. Клячко Л. А. — «Физика горения и взрыва», 1969, т. 5, № 3,
с. 404—413.
49. Варшавский Г, А. — «Труды Одесского государственного
университета, Сер. физическ.». Т. 150. Вып. 7. Одесса, изд. Одесского
ГУ, 1960, с. 15-18.
50. Brzustowski Т. A., Classman /. —AIAA, 1964, v. 15, p. 75—117.
51. Bartlett R. W., Oug J. N. — «Comb, and Flame», 1963, v. 7,
p. 227—231.
52. Prentice /. L. — «Comb, and Flame», 1965, v. 9, p. 209—212.
53. Drew С. М. — «Comb, and Flame», 1965, v. 9, p. 205—208.
54. Григорьев Ю. М., Чальченко Ю. А., Мержанов А. Г. — «Физика
горения и взрыва», 1973, т. 9, вып. 2, с. 191—199.
55. Чальченко Ю. А., Григорьев Ю. М., Мержанов А. Г.—«Физика
горения и взрыва», 1975, т. 11, вып. 1, с. 115—118 с ил.
56. Алексеева Т. И., Гуревич М. А., Озеров Е. С. — «Труды
Ленинградского политехнического института». Сб. № 280. Л., изд. ЛПИ,
1967, с. 98—103.
57. Bartlett R. W. — «J. Electrochem. Soc», 1964, v. Ill, p. 903—908.
58. Маркштейн Дж. Г.—«Ракетная техника и космонавтика», 1963,
№ 3, с. 3—20 с ил.
предметный
указатель
А
Абсолютно черное тело (АЧТ) 18
Кирхгофа модель 18
условия, имитирующие АЧТ
18, 19, 45, 61
Алгомерация частиц:
металлических в топливе 259
окисных в продуктах сгорания
259
Адсорбция:
газов на поверхности жидкой
А120з изотермы 98, 99
постоянные в
адсорбционных уравнениях 99
размер
адсорбированных молекул 99, 100
примесей жидкой фазы на
межфазной границе 173, 215
Активность компонента в
поверхностном слое 104, 105
Амплитуда тепловых колебаний 147
Антипова формула 79
Б
Бачинского формула 109
Био критерий 145, 152
Бриджмена формула 128
Бугера критерий 148
В
Вакансии:
размер в жидкой А1203 111
ТЮ2 111
Взаимодействие химическое:
жидких А1203 и ВеО с
вольфрамом, кинетика 227, 230
константа скорости
227, 229, 231
лимитирующая
стадия 230, 231
механизм 229
тугоплавкими
соединениями (карбидами, нитридами,
боридами) 221
— двойных расплавов на основе
А12Оз с тугоплавкими металлами
225
— металлов с окислами 221, 224,
225
— окислов с тугоплавкими
металлами 225
— титана и циркония с графитом
184.
жидкой А1203 с графитом,
кинетика 81
механизм 241,
как причина прекращения
растекания 184
паров А120з с графитом,
кинетика
механизм 239,
растекающейся жидкости с
подложкой
Вина уравнение 15
Волна температурная 140, 143, 144,
149
Воспламенение:
алюминия 255, 256
тепловая теория 261
условия 256
Вязкое течение:
энергия активизации ПО, 112
— свободная 109
энтропия ПО
Вязкость:
жидких окислов, измерение
методом коаксиальных цилиндров
106
крутильных колебаний
тигля 107
ротационным 107
погрешность при измерении
107
температурная зависимость
109, 112
жидкой А1203 107, 109, 119
— Ti02 112
Г
Гиббса уравнение 98, 174
Гистерезис процесса плавление —
затвердевание окислов 24
Горение:
алюминия 261
диффузионная теория 260
Граница (стенка) непрозрачная
142
— прозрачная 141, 143, 149
— полупрозрачная 141, 143
Грюнайзена формула 132
Д
Дебая температура 131, 133
Дебая формула 128
Диспергирование 175, 190
Диффузионные параметры в
жидкости, капиллярный метод
измерения 123
погрешность измерения 125
Диффузия:
в жидкой А1203 125
— расплавах АЬОз—СаО—02 123
— энергия активизации 126
Дырки:
размер в жидкой АЬОз 111
Ti02 111, 113
Е
Елютина формулы 203
Ж
Жидкость в виде множества
твердых сфер, модель 11
Жуховицкого формула 216
Жуховицкого — Григоряна — Ми-
халика формула 174
И
Иванцова критерий 148
Испарение А1203 диссоциативное
224, 227, 230, 231, 242
К
Капля жидкости в полете 68
— растекающаяся, коэффициент
формы 193, 195
профиль 197, 199
Капиллярная постоянная 92
Капиллярное поднятие,
неравновесная высота 202
Кейса формула 132
282
Комплексообразование в окисных
расплавах 85
Коэффициент:
диффузии в жидкой А12Оз 123
— температурная зависимость 125
науглероживания 181
ослабления 134, 141, 145
поглощения 134, 140, 141
— жидкой А120з 170
— осредненный по спектру («рос-
селандово среднее») 133
— среды в состоянии
термодинамического равновесия,
осредненный («планковское среднее») 146,
170
преломления 142, 146, 170
— осредненный 146
рассеяния 134,
сжимаемости 132
температуропроводности 140, 148
теплопроводности 132, 137, 170
шероховатости 7
Кристиансена формула 167
Куба — Мори формула 129
Кунина уравнение 92
Л
Линдемана правило 132
Лонштейна формула 91
Лэнгмюра уравнение 99
М
Материал:
непрозрачный 151
полупрозрачный (чисто
ослабляющий) 139, 140, 143, 151
рассеивающий 151
Международная практическая
температурная шкала (МПТШ):
МПТШ-27 15
МПТШ-48 17
МПТШ-68 17
Механизм образования связи на
границе между:
жидким металлом и твердым
окислом 185
— окислом и твердым металлом
186
Миснара формулы 132
Момент плавления 23, 28—43
хЧодель воспламенения 261
— горения, диффузионная 260
Н
Навье — Стокса формула 178, 181
Нагрев высокотемпературный:
в печах сопротивления 26, 29,
31, 33, 35, 37, 39, 41, 56
индукционный 26, 29, 31, 37, 39,
41
плазменный 27.
пламенный 25, 29, 3!, 33, 41
световой (солнечный) 25, 33, 37.
43, 56, 60. 61
электродуговой 27, 35, 37, 42
электронный 25, 35
Натяжение поверхностное:
влияние атмосферы 91, 97
измерение методом веса капли
(взвешивания капли) 90, 91
лежащей капли 89. 90.
максимального давления
в газовом пузырьке 90
отрыва диска или
кольца 93,
формы капли (висящей
капли) 91
мениска в капилляре
90
погрешности при измерении
89, 90, 95
жидкой А1203 89, 90
— ТЮ2 105
— ВеО 105
коэффициент температурной
зависимости жидкой А1203 92,95,96
ТЮ2 105
ВеО 105
расплавов системы А120з—
ВеО 100, 104
А1203—СаО 100, 104
А12Оз—Сг2Оз 100
А12Оз—MgO 100, 104
А1203—Si02 100
расплавов системы А12Оз—ВеО 100
А1203—СаО 100
А12Оз—Сг2Оз 100
АЬОз-MgO 100
Al203-Si02 100
Окисление мелкодисперсного
алюминиевого порошка модели 248,
261
Окислы поверхностно-активные 105
инактивные 105
Ослабление, эффективный
коэффициент 140, 141, 145
П
Перепое поверхностный, число 174,
211
Пирометр оптический:
визуальный 16, 28, 32, 34, 36, 38,
-<П. 42. •% 7К
фотоэлектрический 32, 34, 36, 38,
49
Планка закон 16
Плотность:
жидких окислов 86
измерение методом
гидростатического взвешивания 74
лежащей капли 66, 72
летящей капли 67
- максимального
давления в газовом пузырьке 77, 85
плавления в капилляре
71
погрешность при измерении
65. 66, 70, 77, 79, 86
температурная зависимость
66, 72. 77, 79, 80, 82, 83, 88
жидкой А1203 65
— ТЮ2 86
— ВеО 85
источника тепла 145
коэффициент температурной
зависимости жидкой А1203 66, 77, 79,
80
ТЮ2 86
расплавов системы А1203—
ВеО 81, 82
Al2Oj— СаО 83, 84
А1203—Сг2Ог, 83. 84
Al203-MgO 82
А12Оз—Si02 83, 85
теплового потока 147
283
фазового слоя жидкости 192—200
влияние на кинетику
растекания 192
Поглощение, эффективный
коэффициент 146
Поднятие жидкой ТЮ2 в
капилляре 201
между параллельными
пластинами 201
— капиллярное, неравновесная
высота 202
Потери импульса, двухфазные 259
Превращение в пленке А1203:
структурное 246, 248
релаксационный механизм
фазового превращения 248
Р
Работа адгезии 171, 175, 185
— когезии 171
Радиационно-иидуктнвный
теплообмен (РКТ)
Рассеяние фононов:
трехфононные 180
центры 180
четырехфононные 181
Растекание:
жидкого титана по графиту 180,
184
жидкой Ti02 по тугоплавким
металлам 192
жидкости по наклонной
подложке 196
поверхности твердого тела в
квазистационарном режиме 179.
181, 183, 200
движущая сила 173,
178, 189, 197
■ — — — кинетика 177
круговое (двумерное)
178
линейное
(одномерное) 179
площадь конечная 183
по наклонной
подложке 196
причины прекращения
183, 184
стадии процесса 178
Росссланда формула 133
С
Светофильтр:
коэффициент пропускания 19. 20
секторный 19
Свойства теплофизичеекч'-».
погрешности при измерении .161
Семенченко формула 216
Скорость:
звука 128
растекания 171
света в вакууме 146
спектральная 146
Смачивание:
графита, влияние атмосферы СО
237
добавок окислов карбидооб-
разующих элементов 234
не образующих
карбидов 234
— жидкими окислами 231
— механизм процесса 234
поверхности твердого тела
жидкостью 171
тугоплавких карбидов жидким:
окислами 233
управление процессом 236
Способность:
излучательная 18
отражательная 142
Среда серая 135
Степень черноты:
границ вещества 18, 19, 63
измерительной ячейки 51, 55
стенок
Стефана — Больцмана закон 15
Тело серое 18, 19
Температура:
измерение, влияние газовой среды
20, 22, 45, 51, 59
конструкции установки 24
■ отклонения от условий ЛЧТ
19, 22, 51
дыма 20, 22
• пара 20, 22
— по почернению кинопленки 72
плавления, влияния давления 22
изменения состава образца
20, 51, 59
примесей 20, 51
— окислов А1203 36, 50. 53, 56
ВеО 32, 48
СаО 48
Се02 47
СгУОэ 50
Y203 58
НЮ, 29, 43
MgO 29, 41
Th02 27
Ti02 38, 59
Zr02 30, 44
U02 30, 47
—— редкоземельных металлов 61
тройной точки воды 16
яркостпая 18
Температуропроводность:
жидкой А120;1 169
лейкосапфира 164
плазменно-напыленной АЬО^ 1G8
спеченной А1203
Теплоперенос:
внутренний радиационный 133
гиперакустическими
колебаниями, модель Дебая 129
теплопроводностью 132
экспериментальные методы
разделения фононной и
радиационной составляющих 135
Теплопроводность:
диэлектрической жидкости,
теория 128
жидкой А1оО:<. температурная
зависимость 171
измерение методом плоских
температурных воли 140, 144, 151.
157
радиационная 134, 136, 144
расчет по Кейсу 132
эмпирическим формулам
Миспара 132
скачок при плавлении А120з 169
лейкосапфира 166
Теплосопротивление:
жидкости 128
кристаллической решетки 130, J66
284
лейкосапфира, температурная
зависимость 165
полное 131
Толщина оптическая 138
Точки вспомогательные,
фиксированные 17
— реперные 17
У
Угол смачивания:
контактный 172
краевой 171
— влияние шероховатости
поверхности твердого тела 175
— концентрационная зависимость
с двумя симметричными
максимумами 207
— тугоплавких металлов, влияние
окислов тугоплавких металлов
реакционно-аттивпых
добавок 209
жидкими окислами 203, 204
— расплавами на основе
А1Й03 206
— расчетный для чистых
окислов 189
Установка экспериментальная для
измерения:
вязкости методом крутильных
колебаний тигля 108
натяжения поверхностного
методом плавления внутри капилляра
71
веса капли 67
максимального давления
в газовом пузырьке 77, 95
—отрыва кольца или диска
93
формы (висящей) капли
67
плотности методом
гидростатического взвешивания 74
плавления внутри капилляра
71
летящей капли 67,
максимального давления в
газовом пузырьке 77,
температуры плавления 45, 55
теплофизических свойств
полупрозрачных материалов 157
электропроводности методом
амперметра — вольтметра 115
Ф
Фершафельта формула 92
Филиппова фоомула 129, 138
Фика закон 123
Фононы:
рассеяние на границах зерен 180
изотопах 180
■ примесях 180
фононах 180
Фордхема формула 91
Френкеля правило 117
Фюртца формула 111, 113
X
Харкинса коэффициент растекания
171, 173
Частота дебаевская твердого тела
128
Ш
Швидковского метод расчета 107
Шероховатость поверхности 173, 176
Шишковского уравнение 98
Щ
Щукина — Горюнова формула 178
Э
Эйнштейна формула 126
Эйринга формула 109
Электропроводность:
жидких окислов, измерение
методом амперметра — вольтметра
114, 115, 118
— мостика сопротивления
114
погрешность измерения 115,
118, 119
температурная зависимость
жидкой АЬОз П7, 119
— ВеО 120
— Y203 121
— Sc203 121
— Ti02 122
ионная 115
ионно-электронная 115, 121
коэффициент температурной
зависимости жидкой А12Оз 115
ТЮ2 122
скачок при плавлении АЬОз И»
системы 50% СаО — 50%
Si02 115
50% МпО — 50% Si02
115
ТЮ2 115, 122
энергия активации для
АЬОз 117
ВеО 120
Y20, 121
Sc203 121
ТЮ2 122
Энергия:
межфазная, динамическая 188.
189, 195, 203
в системах жидкий окисел —
тугоплавкий металл 190, 195
■ методика расчета 188, 194
— снижение за счет адсорбции
примесей 173
— массоперепоса вещества
174
-- — количественная оценка
поверхностная на границе
жидкость — газ 171
твердое тело — газ 171
■ твердое тело —
жидкость 171
Ю
Юнга формула 171, 173
ОГЛАВЛЕНИЕ
Стр.
Предисловие 3
Введение 5
ГЛАВА I. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ ТУГОПЛАВКИХ ОКИСЛОВ 15
1. Точность измерения высоких температур 15
2. Экспериментальные данные по температуре плавления
тугоплавких окислов 27
Двуокись тория 27
Двуокись гафния 43
Окись магния 44
Двуокись урана 44
Двуокись церия 47
Двуокись циркония 47
Окись кальция 48
Окись бериллия 48
Окись хрома 50
Окись алюминия 50
Окись иттрия 58
Двуокись титана * . 59
Окислы редкоземельных металлов 61
ГЛАВА П. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ЖИДКИХ
ТУГОПЛАВКИХ окислов 64
1. Плотность жидких тугоплавких окислов 65
Окись алюминия 65
Расплавы на основе окиси алюминия 80
Окись бериллия 85
Двуокись титана 86
2. Поверхностное натяжение жидких тугоплавких
окислов 89
Окись алюминия 89
Расплавы на основе окиси алюминия 100
Окись бериллия 105
Двуокись титана 105
3. Вязкость жидких тугоплавких окислов 106
Окись алюминия 106
Двуокись титана 112
4. Электропроводность жидких тугоплавких окислов . 114
Окись алюминия и расплавы на ее основе .... 114
Окись бериллия 120
Окиси иттрия и скандия 120
Двуокись титана 121
5. Диффузия в жидкой окиси алюминия 122
ГЛАВА III. ТЕПЛОПРОВОДНОСТЬ ТВЕРДОЙ И ЖИДКОЙ ОКИСИ
АЛЮМИНИЯ ПРИ ВЫСОКОЙ ТЕМПЕРАТУРЕ ... I27
1. Теоретическая оценка теплопроводности жидких
диэлектриков 127
2. Фононная теплопроводность диэлектриков при
высоких температурах - . • 130
286
Стр.
3. Радиационный теплообмен в диэлектриках при
высоких температурах 133
4. Экспериментальные методы разделения фононной и
радиационной составляющих теплопереноса .... 135
5. Метод одновременного определения теплофизических
и радиационных свойств полупрозрачных материалов 144
Распространение температурных волн в
полупрозрачных средах 144
Метод определения теплофизических свойств сла-
бопоглощающих веществ 150
Анализ решения уравнения распространения
температурных волн в полупрозрачной пластине . . . 153
6. Установка для исследования теплофизических свойств
полупрозрачных материалов 157
Конструкция установки 157
Точность метода 161
Методика приготовления образцов 164
7. Теплофизические свойства окиси алюминия при
высокой температуре 164
Монокристаллическая окись алюминия 164
Плазмепно-напыленная окись алюминия 168
Жидкая окись алюминия 169
ГЛАВА IV. СМАЧИВАНИЕ ТУГОПЛАВКИХ ВЕЩЕСТВ И
ВЫСОКОТЕМПЕРАТУРНЫХ МАТЕРИАЛОВ ЖИДКИМИ
ОКИСЛАМИ 171
1. Процессы смачивания и растекания 171
2. Кинетика и конечная площадь растекания .... 177
3. Закономерности смачивания и растекания в системах
с окислами 184
4. Кинетика смачивания тугоплавких металлов жидкими
окислами 191
5. Смачивание высокотемпературных материалов
жидкими тугоплавкими окислами и их расплавами . . . 203
6. Влияние реакционно-активных добавок на смачивание
тугоплавких металлов жидкой окисью алюминия . . 209
7. Физико-химические процессы на межфазной границе 215
8. Взаимодействие жидких окислов с тугоплавкими
металлами 220
9. Смачивание графита и тугоплавких карбидов
жидкими окислами 231
ГЛАВА V. ОБРАЗОВАНИЕ ОКИСИ АЛЮМИНИЯ ПРИ ОКИСЛЕНИИ
И ГОРЕНИИ АЛЮМИНИЯ 242
1. Окисление алюминия 242
2. Воспламенение и горение алюминия 255
3. Модели воспламенения мелкодисперсного порошка
алюминия 261
Библиографический список 268
Предметный указатель 282
ИБ № 1003
Михаил Александрович Маурах
Борис Сергеевич Ми*ин
ЖИДКИЕ ТУГОПЛАВКИЕ ОКИСЛЫ
Редактор издательства В. П. Молокова
Художественный редактор В. В. Баталова
Технический редактор И. А. Коровина
Корректоры Н. И. Шефтель, И. П. Собко
Переплет художника К. И. Милаева
Сдано в набор 16.02.79. Подписано в печать 08.08.79. Т-14066. Формат бумаги
84ХЮ87з2. Бумага типографская № 1. Гарнитура литературная. Печать
высокая. Усл. печ. л. 15,12. Уч.-изд. л. 16,85. Тираж 1500 экз. Заказ № 894.
Цена 2 р. 80 к. Изд. № 3249
Издательство «Металлургия», 119034, Москва, Г-34, 2-й Обыденский пер., д. 14
Владимирская типография «Союзполиграфпрома»
при Государственном комитете СССР по делам издательств, полиграфии
и книжной торговли
600000, г. Владимир, Октябрьский проспект, д. 7